Job Results:
Ligand
Structure
Job ID
13aa62e81038093cc0a5aa14a89b9a62
Job name
NA
Time
2026-01-10 22:45:10
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 81 | Carbamoyl-phosphate synthetase I (CPS1) | 5DOU | 7.13 | |
Target general information Gen name CPS1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Carbamoyl-phosphate synthase [ammonia], mitochondrial; CPSase I; CPS1 Protein family NA Biochemical class Carbon-nitrogen ligase Function Involved in the urea cycle of ureotelic animals where the enzyme plays an important role in removing excess ammonia from the cell. Related diseases Carbamoyl phosphate synthetase 1 deficiency (CPS1D) [MIM:237300]: An autosomal recessive disorder of the urea cycle causing hyperammonemia. It can present as a devastating metabolic disease dominated by severe hyperammonemia in neonates or as a more insidious late-onset condition, generally manifesting as life-threatening hyperammonemic crises under catabolic situations. Clinical features include protein intolerance, intermittent ataxia, seizures, lethargy, developmental delay and intellectual disability. {ECO:0000269|PubMed:11388595, ECO:0000269|PubMed:11474210, ECO:0000269|PubMed:12655559, ECO:0000269|PubMed:12955727, ECO:0000269|PubMed:15164414, ECO:0000269|PubMed:15617192, ECO:0000269|PubMed:16737834, ECO:0000269|PubMed:17310273, ECO:0000269|PubMed:20578160, ECO:0000269|PubMed:21120950, ECO:0000269|PubMed:22173106, ECO:0000269|PubMed:23649895, ECO:0000269|PubMed:24813853, ECO:0000269|PubMed:26440671, ECO:0000269|PubMed:9711878}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Pulmonary hypertension, neonatal (PHN) [MIM:615371]: A disease characterized by elevated pulmonary artery pressure. Pulmonary hypertension in the neonate is associated with multiple underlying problems such as respiratory distress syndrome, meconium aspiration syndrome, congenital diaphragmatic hernia, bronchopulmonary dysplasia, sepsis, or congenital heart disease. {ECO:0000269|PubMed:11407344}. Disease susceptibility is associated with variants affecting the gene represented in this entry. CPS1 variants influence the availability of precursors for nitric oxide (NO) synthesis and play a role in clinical situations where endogenous NO production is critically important, such as neonatal pulmonary hypertension, increased pulmonary artery pressure following surgical repair of congenital heart defects or hepatovenocclusive disease following bone marrow transplantation. Infants with neonatal pulmonary hypertension homozygous for Thr-1406 have lower L-arginine concentrations than neonates homozygous for Asn-1406 (PubMed:11407344). {ECO:0000269|PubMed:11407344}. Drugs (DrugBank ID) DB11118; DB06775 Interacts with P10398; P04049; P63104 EC number EC 6.3.4.16 Uniprot keywords 3D-structure; Acetylation; Allosteric enzyme; Alternative splicing; ATP-binding; Cell membrane; Disease variant; Glycoprotein; Ligase; Membrane; Mitochondrion; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Transit peptide; Urea cycle Protein physicochemical properties Chain ID A Molecular weight (Da) 156194 Length 1422 Aromaticity 0.08 Instability index 33.61 Isoelectric point 5.78 Charge (pH=7) -19.83 2D Binding mode Binding energy (Kcal/mol) -9.73 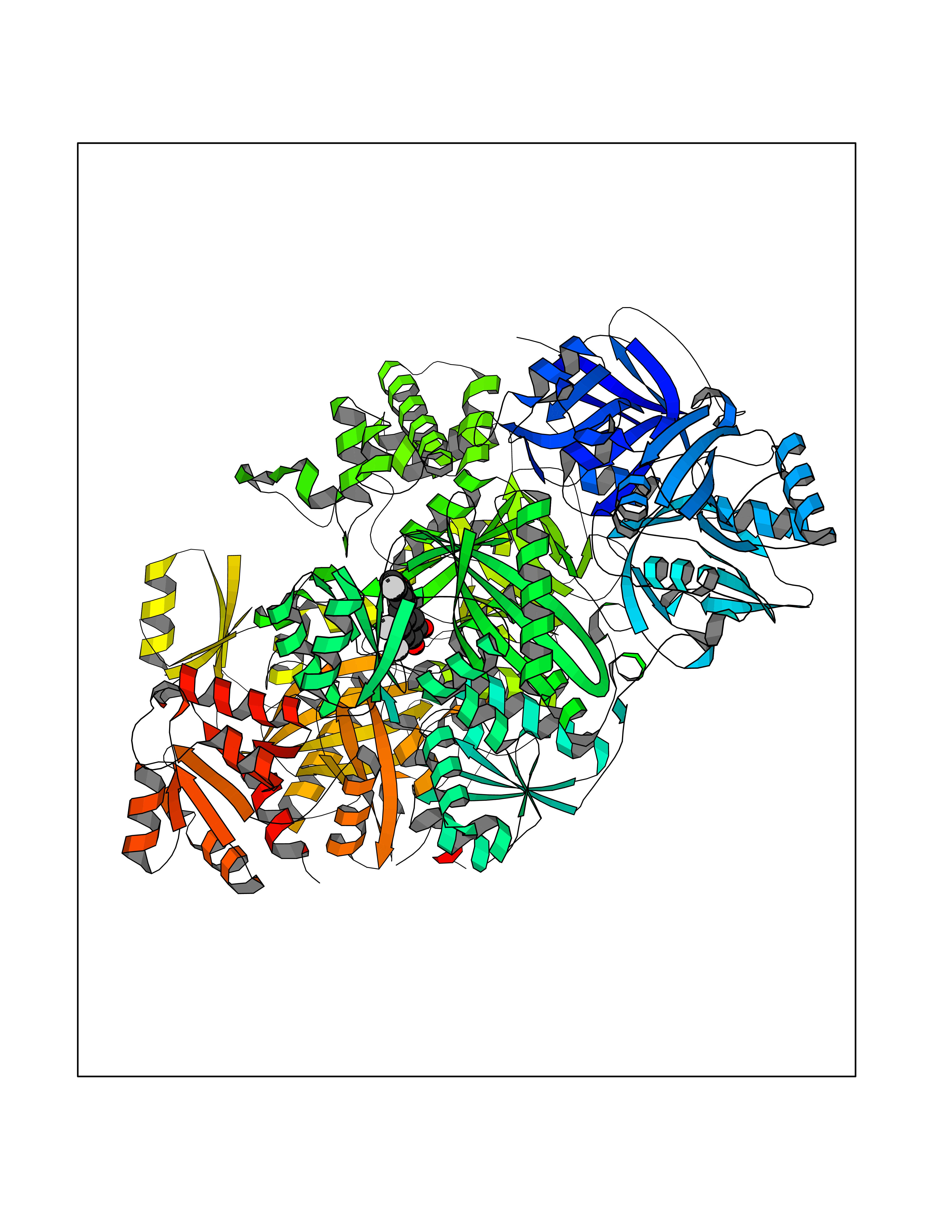 Molscript Map 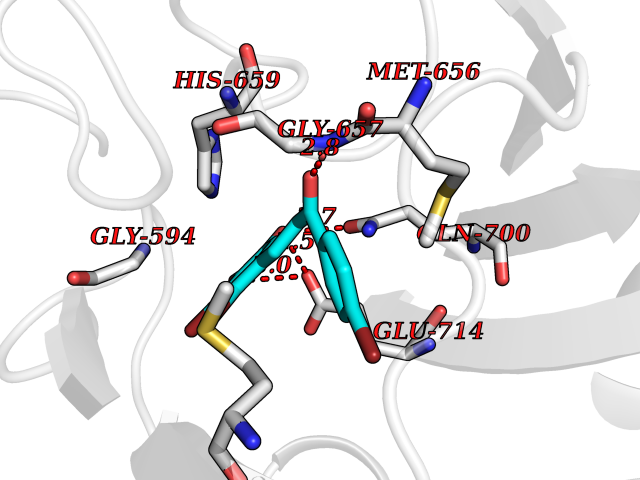 Pymol Map 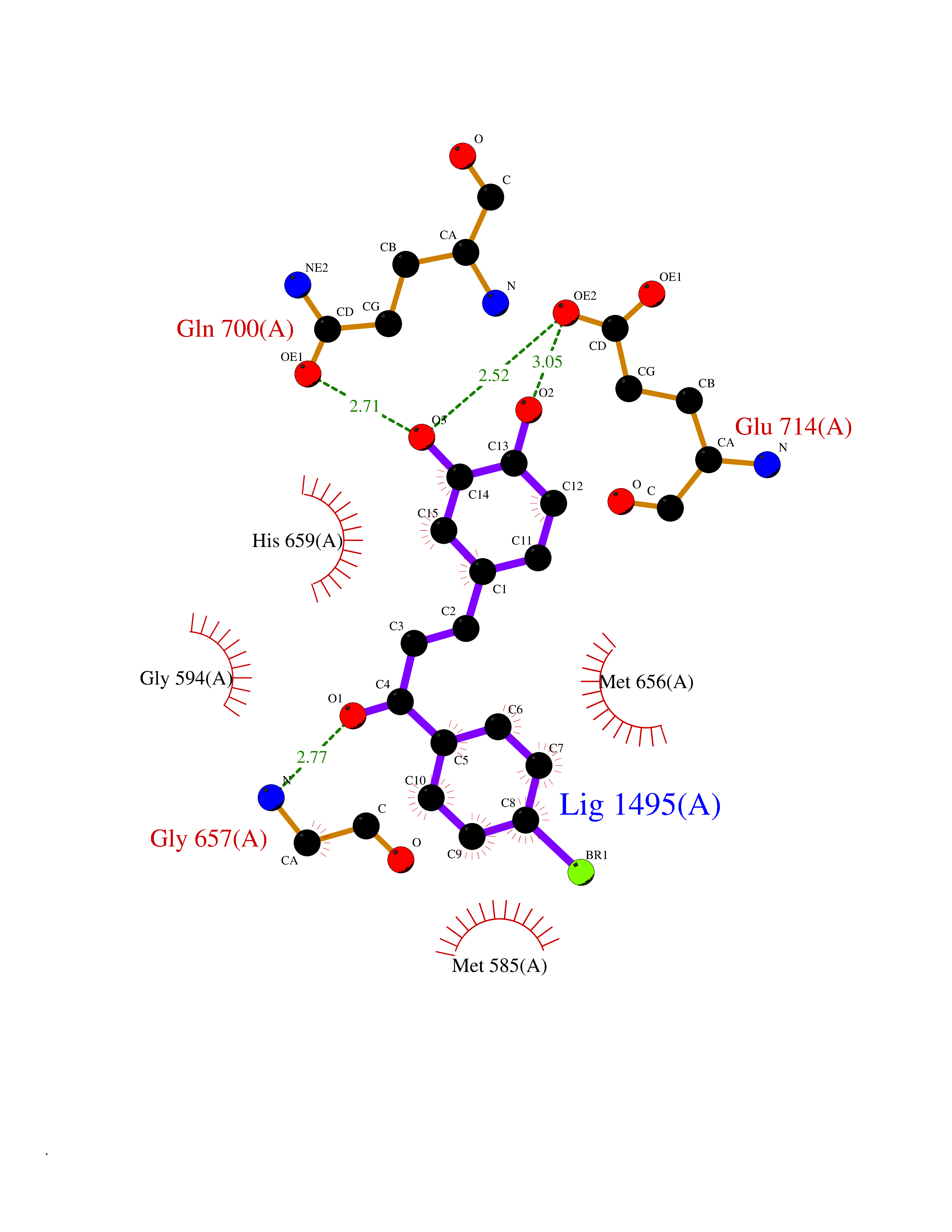 Ligplot Map 3D Binding mode Sequence AQTAHIVLEDGTKMKGYSFGHPSSVAGEVVFNTGLGGYPEAITDPAYKGQILTMANPIIGNGGAPDTTALDELGLSKYLESNGIKVSGLLVLDYSKDYNHWLATKSLGQWLQEEKVPAIYGVDTRMLTKIIRDKGTMLGKIEFEGQPVDFVDPNKQNLIAEVSTKDVKVYGKGNPTKVVAVDCGIKNNVIRLLVKRGAEVHLVPWNHDFTKMEYDGILIAGGPGNPALAEPLIQNVRKILESDRKEPLFGISTGNLITGLAAGAKTYKMSMANRGQNQPVLNITNKQAFITAQNHGYALDNTLPAGWKPLFVNVNDQTNEGIMHESKPFFAVQFHPEVTPGPIDTEYLFDSFFSLIKKGKATTITSVLPKPALVASRVEVSKVLILGSGGLSIGQAGEFDYSGSQAVKAMKEENVKTVLMNPNIASVQTNEVGLKQADTVYFLPITPQFVTEVIKAEQPDGLILGMGGQTALNCGVELFKRGVLKEYGVKVLGTSVESIMATEDRQLFSDKLNEINEKIAPSFAVESIEDALKAADTIGYPVMIRSAYALGGLGSGICPNRETLMDLSTKAFAMTNQILVEKSVTGWKEIEYEVVRDADDNCVTVCNMENVDAMGVHTGDSVVVAPAQTLSNAEFQMLRRTSINVVRHLGIVGECNIQFALHPTSMEYCIIEVNARLSRSSALASKATGYPLAFIAAKIALGIPLPEIKNVVSGKTSACFEPSLDYMVTKIPRWDLDRFHGTSSRIGSSMKSVGEVMAIGRTFEESFQKALRMCHPSIEGFTPRLPMNKEWPSNLDLRKELSEPSSTRIYAIAKAIDDNMSLDEIEKLTYIDKWFLYKMRDILNMEKTLKGLNSESMTEETLKRAKEIGFSDKQISKCLGLTEAQTRELRLKKNIHPWVKQIDTLAAEYPSVTNYLYVTYNGQEHDVNFDDHGMMVLGCGPYHIGSSVEFDWCAVSSIRTLRQLGKKTVVVNCNPETVSTDFDECDKLYFEELSLERILDIYHQEACGGCIISVGGQIPNNLAVPLYKNGVKIMGTSPLQIDRAEDRSIFSAVLDELKVAQAPWKAVNTLNEALEFAKSVDYPCLLRMNVVFSEDPVVLTKFVEGAREVEMDAVGKDGRVISHAISEHVEDAGVHSGDATLMLPTQTISQGAIEKVKDATRKIAKAFAISGPFNVQFLVKGNDVLVIECNLRASRSFPFVSKTLGVDFIDVATKVMIGENVDEKHLPTLDHPIIPADYVAIKAPMFSWPRLRDADPILRCEMASTGEVACFGEGIHTAFLKAMLSTGFKIPQKGILIGIQQSFRPRFLGVAEQLHNEGFKLFATEATSDWLNANNVPATPVAWPSQEGQNPSLSSIRKLIRDGSIDLVINLPNNNTKFVHDNYVIRRTAVDSGIPLLTNFQVTKLFAEAVQKDSKSLFHYRQ Hydrogen bonds contact Hydrophobic contact | ||||
| 82 | NT-3 growth factor receptor (TrkC) | 4YMJ | 7.13 | |
Target general information Gen name NTRK3 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms TrkC tyrosine kinase; Trk-C; TRKC; Neurotrophic tyrosine kinase receptor type 3; NT3 growth factor receptor; GP145TrkC; GP145-TrkC Protein family Protein kinase superfamily, Tyr protein kinase family, Insulin receptor subfamily Biochemical class Kinase Function Upon binding of its ligand NTF3/neurotrophin-3, NTRK3 autophosphorylates and activates different signaling pathways, including the phosphatidylinositol 3-kinase/AKT and the MAPK pathways, that control cell survival and differentiation. Receptor tyrosine kinase involved in nervous system and probably heart development. Related diseases Defects in NTRK3 are associated with susceptibility to congenital heart defects (CHD). A disease characterized by congenital developmental abnormalities involving structures of the heart. CHD are the most common major birth defects and the leading cause of death from congenital malformations. {ECO:0000269|PubMed:25196463}. Drugs (DrugBank ID) DB11986; DB12010; DB14723; DB15822; DB16826 Interacts with Q16832; Q6PKX4; Q15375; P08581; P04629; Q16620; P18031; P61619; Q9UBV2; P31947; Q99523; P62258 EC number EC 2.7.10.1 Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Developmental protein; Differentiation; Direct protein sequencing; Disulfide bond; Glycoprotein; Immunoglobulin domain; Kinase; Leucine-rich repeat; Membrane; Neurogenesis; Nucleotide-binding; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Repeat; Signal; Transferase; Transmembrane; Transmembrane helix; Tyrosine-protein kinase Protein physicochemical properties Chain ID A Molecular weight (Da) 31111.9 Length 271 Aromaticity 0.1 Instability index 46.88 Isoelectric point 8.65 Charge (pH=7) 4.69 2D Binding mode Binding energy (Kcal/mol) -9.72 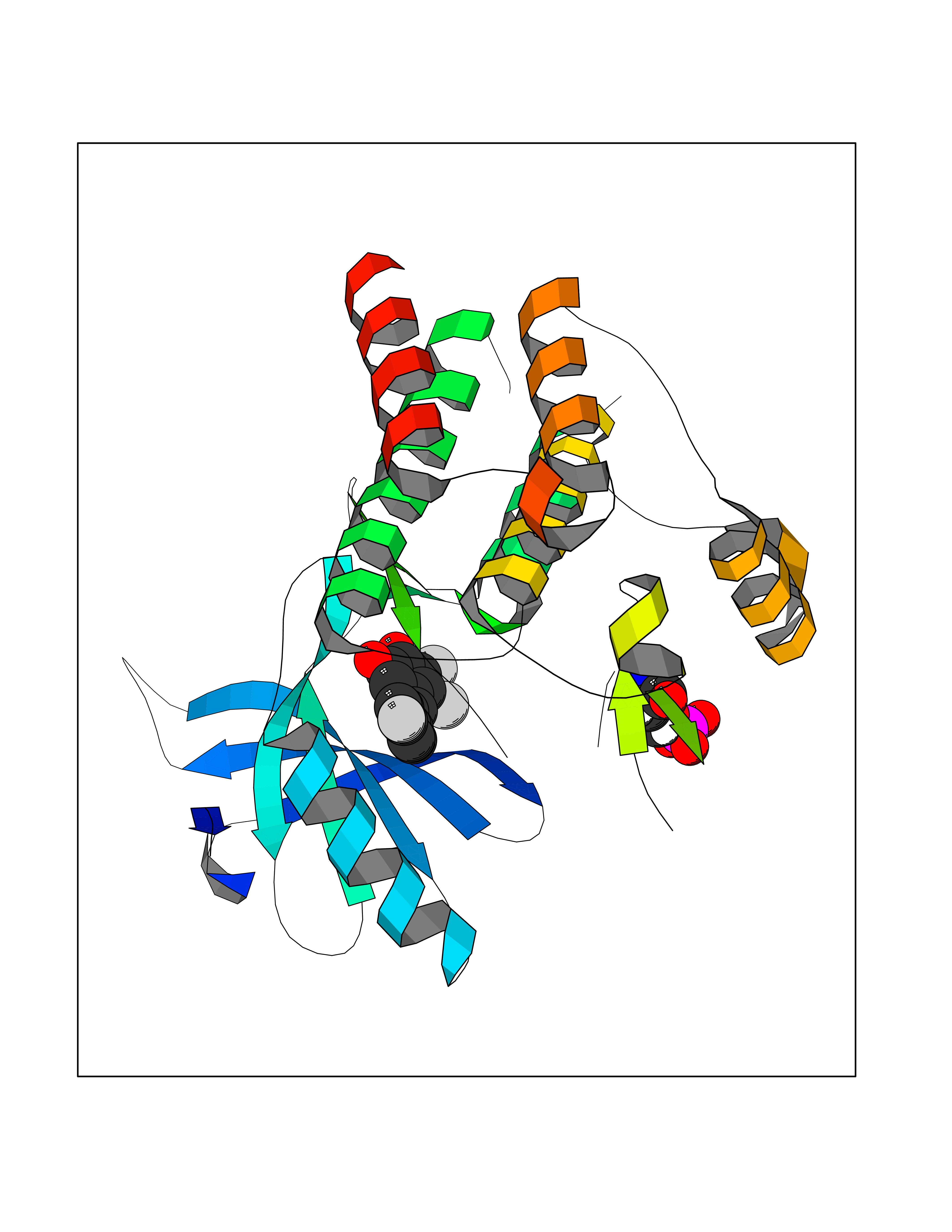 Molscript Map 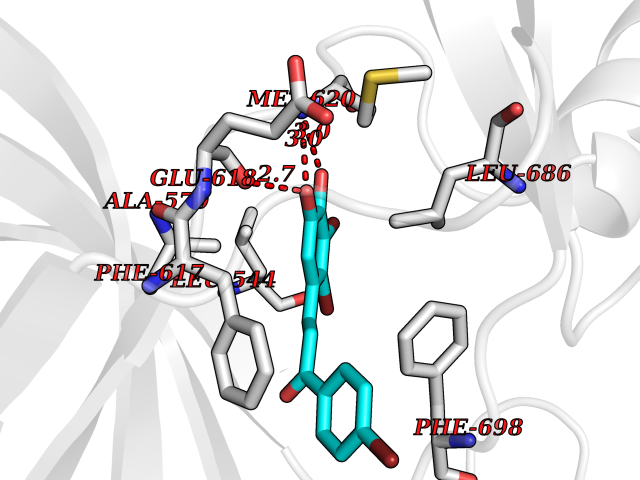 Pymol Map 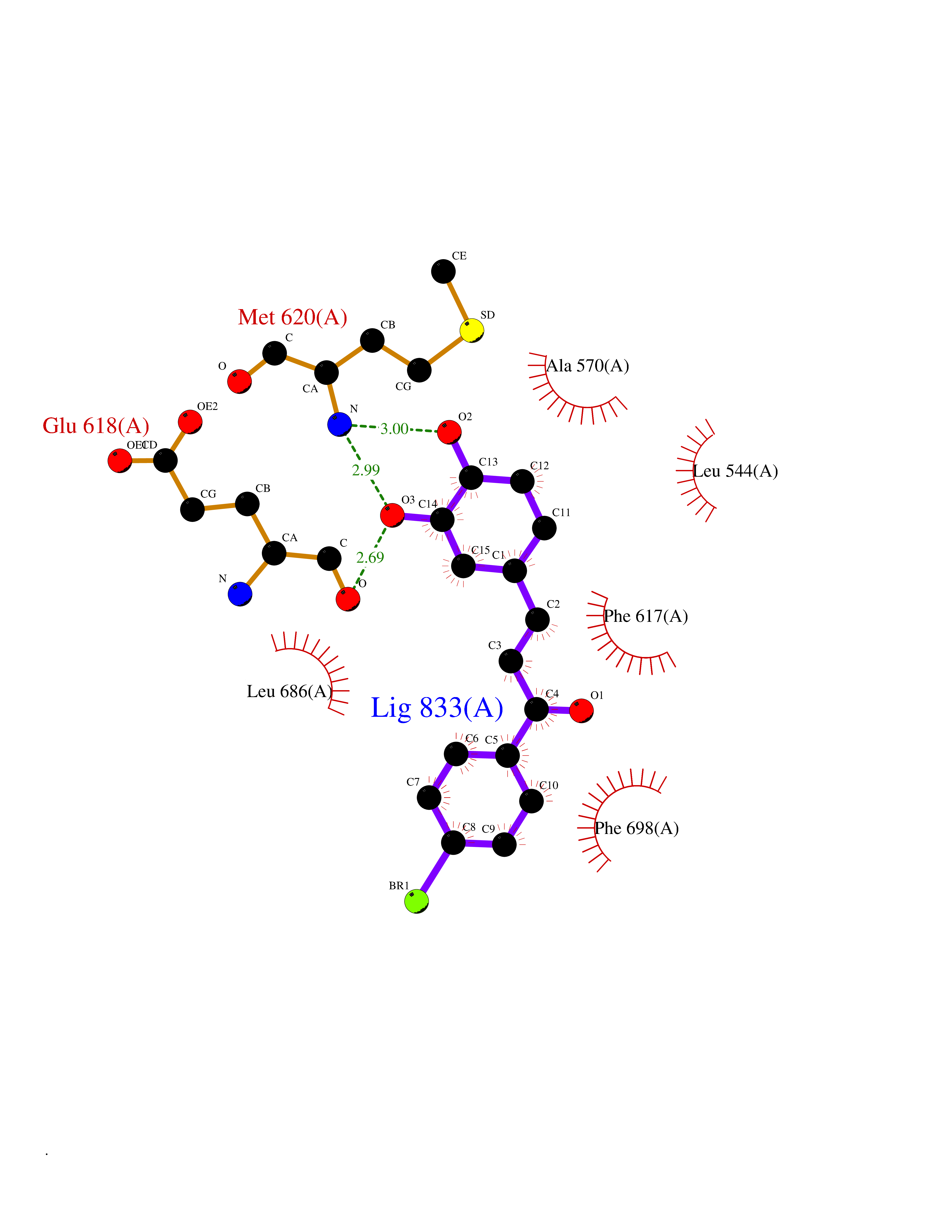 Ligplot Map 3D Binding mode Sequence GIHVQHIKRRDIVLKRELGEGAFGKVFLAECYNLSPTKDKMLVAVKALKDPTLAARKDFQREAELLTNLQHEHIVKFYGVCGDGDPLIMVFEYMKHGDLNKFLRAHGPGELGLSQMLHIASQIASGMVYLASQHFVHRDLATRNCLVGANLLVKIGDFGDVYXTDYYRHTMLPIRWMPPESIMYRKFTTESDVWSFGVILWEIFTYGKQPWFQLSNTEVIECITQGRVLERPRVCPKEVYDVMLGCWQREPQQRLNIKEIYKILHALGKAT Hydrogen bonds contact Hydrophobic contact | ||||
| 83 | Histamine H3 receptor (H3R) | 7F61 | 7.13 | |
Target general information Gen name HRH3 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Histamine receptor 3; HH3R; GPCR97; G-protein coupled receptor 97; G protein-coupled receptor 97 Protein family G-protein coupled receptor 1 family Biochemical class GPCR rhodopsin Function Signals through the inhibition of adenylate cyclase and displays high constitutive activity (spontaneous activity in the absence of agonist). Agonist stimulation of isoform 3 neither modified adenylate cyclase activity nor induced intracellular calcium mobilization. The H3 subclass of histamine receptors could mediate the histamine signals in CNS and peripheral nervous system. Related diseases Immunodeficiency 48 (IMD48) [MIM:269840]: A form of severe immunodeficiency characterized by a selective absence of CD8+ T-cells. {ECO:0000269|PubMed:11123350, ECO:0000269|PubMed:11412303, ECO:0000269|PubMed:18509675, ECO:0000269|PubMed:8124727, ECO:0000269|PubMed:8202713}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Autoimmune disease, multisystem, infantile-onset, 2 (ADMIO2) [MIM:617006]: An autosomal recessive, autoimmune disorder characterized by systemic manifestations including blistering skin disease, uncontrollable bullous pemphigoid, inflammatory colitis, autoimmune hypothyroidism, proteinuria and nephrotic syndrome. {ECO:0000269|PubMed:26783323}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01238; DB06698; DB05381; DB17087; DB05080; DB00768; DB11642 Interacts with NA EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disulfide bond; G-protein coupled receptor; Glycoprotein; Membrane; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 34321.1 Length 301 Aromaticity 0.16 Instability index 32.22 Isoelectric point 9.63 Charge (pH=7) 15.11 2D Binding mode Binding energy (Kcal/mol) -9.72 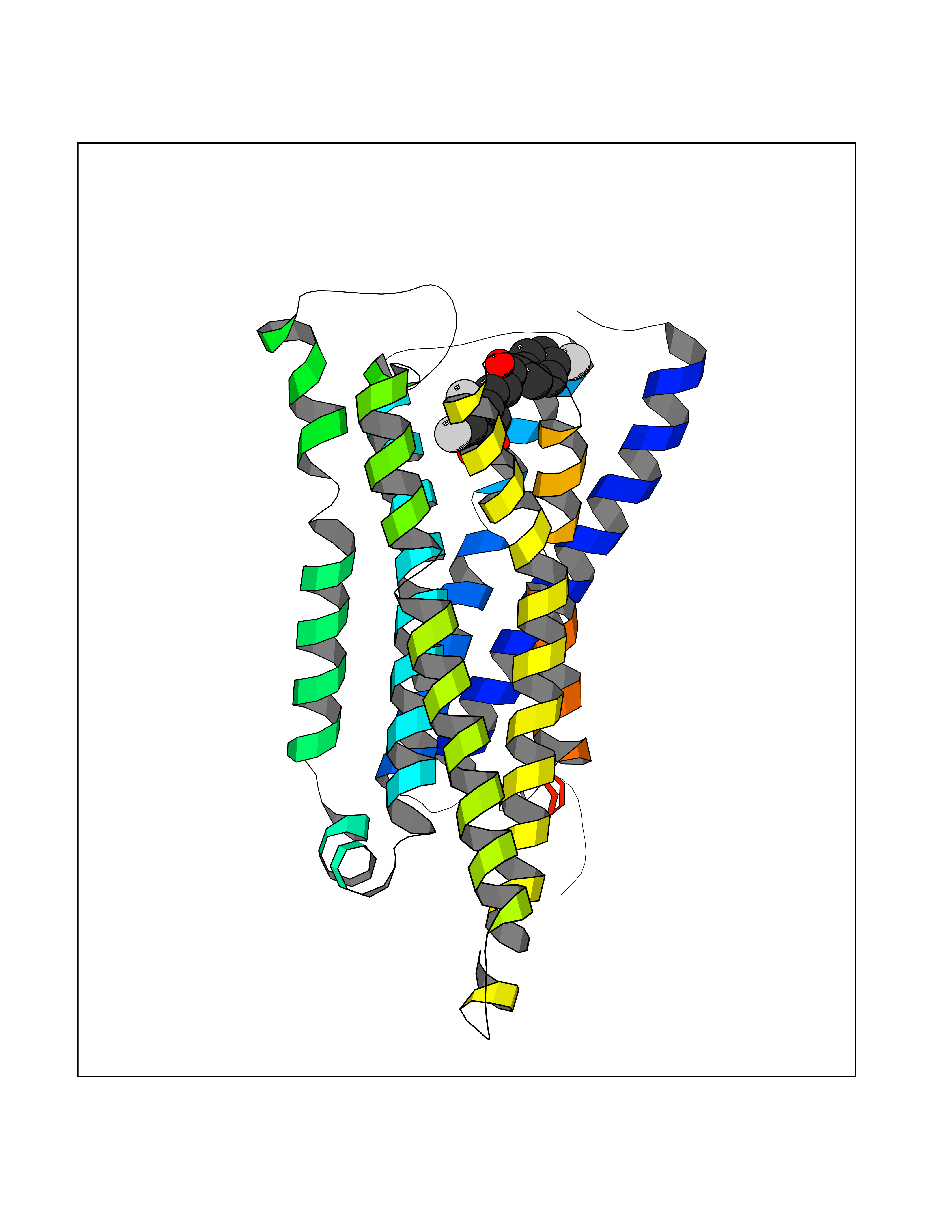 Molscript Map 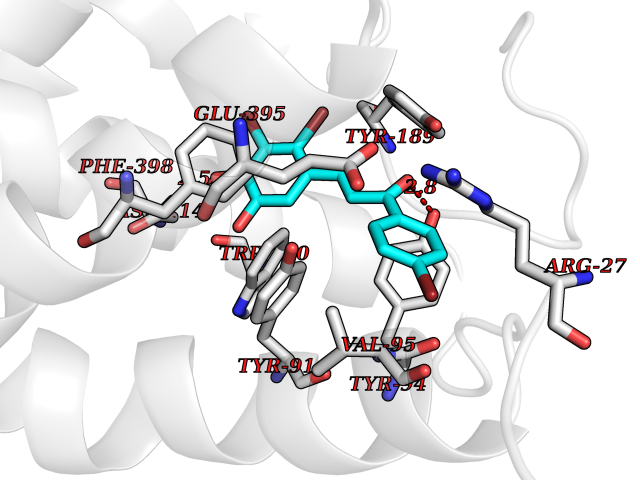 Pymol Map 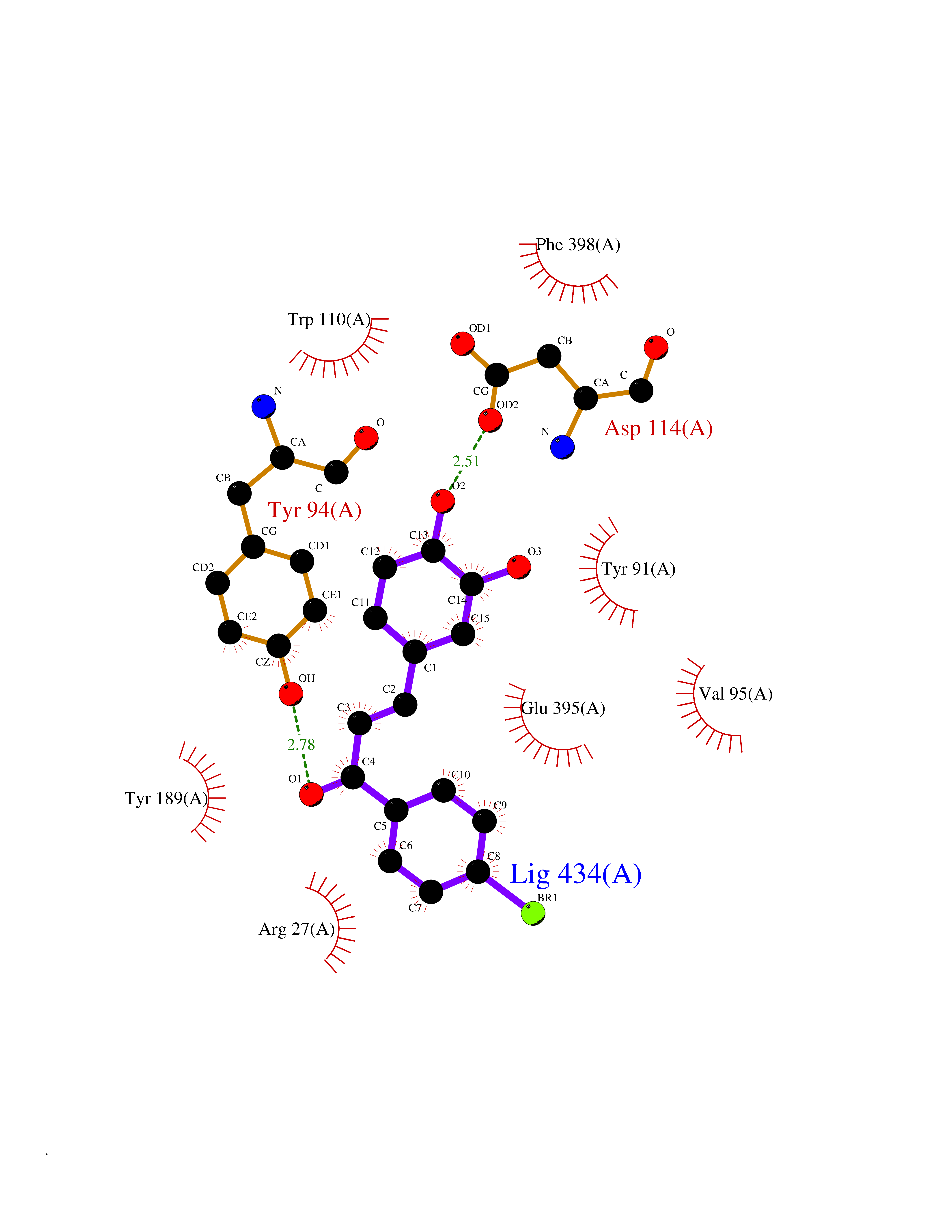 Ligplot Map 3D Binding mode Sequence RGFSAAWTAVLAALMALLIVATVLGNALVMLAFVADSSLRTQNNFFLLNLAISDFLVGAFCIPLYVPYVLTGRWTFGRGLCKLWLVVDYLLCTSKAFNIVLISYDRFLSVTRAVSYRAQQGDTRRAVRKMLLVWVLAFLLYGPAILSWEYLSGGSSIPEGHCYAEFFYNWYFLITASTLEFFTPFLSVTFFNLSIYLNIQRRTRLRLDGAREAAGRFRLSRDRKVAKSLAVIVSIFGLCWAPYTLLMIIRAACHGHCVPDYWYETSFWLLWANSAVNPVLYPLCHHSFRRAFTKLLCPQKL Hydrogen bonds contact Hydrophobic contact | ||||
| 84 | Neutrophil gelatinase-associated lipocalin (LCN2) | 5NKN | 7.13 | |
Target general information Gen name LCN2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms p25; Siderocalin LCN2; Oncogene 24p3; NGAL; Lipocalin-2; LCN2; 25 kDa alpha-2-microglobulin-related subunit of MMP-9 Protein family Calycin superfamily, Lipocalin family Biochemical class Calycin family Function Iron-trafficking protein involved in multiple processes such as apoptosis, innate immunity and renal development. Binds iron through association with 2,5-dihydroxybenzoic acid (2,5- DHBA), a siderophore that shares structural similarities withbacterial enterobactin, and delivers or removes iron from the cell, depending on the context. Iron-bound form (holo-24p3) is internalized following binding to the SLC22A17 (24p3R) receptor, leading to release of iron and subsequent increase of intracellular iron concentration. In contrast, association of the iron-free form (apo-24p3) with the SLC22A17 (24p3R) receptor is followed by association with an intracellular siderophore, iron chelation and iron transfer to the extracellular medium, thereby reducing intracellular iron concentration. Involved in apoptosis due to interleukin-3 (IL3) deprivation: iron-loaded form increases intracellular iron concentration without promoting apoptosis, while iron-free form decreases intracellular iron levels, inducing expression of the proapoptotic protein BCL2L11/BIM, resulting in apoptosis. Involved in innate immunity, possibly by sequestrating iron, leading to limit bacterial growth. . Related diseases Pseudovaginal perineoscrotal hypospadias (PPSH) [MIM:264600]: A form of male pseudohermaphroditism in which 46,XY males show ambiguous genitalia at birth, including perineal hypospadias and a blind perineal pouch, and develop masculinization at puberty. The name of the disorder stems from the finding of a blind-ending perineal opening resembling a vagina and a severely hypospadiac penis with the urethra opening onto the perineum. {ECO:0000269|PubMed:10718838, ECO:0000269|PubMed:10898110, ECO:0000269|PubMed:10999800, ECO:0000269|PubMed:12843198, ECO:0000269|PubMed:15064320, ECO:0000269|PubMed:1522235, ECO:0000269|PubMed:15528927, ECO:0000269|PubMed:15770495, ECO:0000269|PubMed:16098368, ECO:0000269|PubMed:16181229, ECO:0000269|PubMed:7554313, ECO:0000269|PubMed:8626825, ECO:0000269|PubMed:8768837, ECO:0000269|PubMed:9208814, ECO:0000269|PubMed:9745434, ECO:0000269|PubMed:9843052}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02710; DB01672; DB01926; DB04043; DB01631; DB04476 Interacts with P49419-2; Q9NXW9; Q8WXI3; Q12797-6; Q9BXY8; Q96LC9; P49069; P24863; Q9UKJ5; Q9H1P6; Q9H6B4; O14595; Q08426; Q6NZ36-4; B3EWG5; Q7Z4H3; Q6ISS4; Q5TA76; P80188; Q9UIQ6-2; Q9Y6Y9; Q96JG8; Q8IXL7-2; Q969H8; Q969S2; Q17RF5; P07237; P13667; Q96FA3; Q9NRD5; Q13526; Q9UGP5-2; Q12837; P54646; Q86Y79; O60895; Q9BWG6; P60059; O43765; Q96EQ0; Q8IYX1; Q9UL33-2; P20396; O43715; Q13049; Q99816; Q5W5X9-3; Q99757; P57075-2; Q969M7; Q9UMX0; Q9UHD9; P15692-12; Q14119; Q9Y6T4; Q9H0D6; O96006; A0A1U9X8X8 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Apoptosis; Cytoplasmic vesicle; Direct protein sequencing; Disulfide bond; Glycoprotein; Immunity; Innate immunity; Ion transport; Iron; Iron transport; Proteomics identification; Pyrrolidone carboxylic acid; Reference proteome; Secreted; Signal; Transport Protein physicochemical properties Chain ID A Molecular weight (Da) 19748.4 Length 172 Aromaticity 0.13 Instability index 30.73 Isoelectric point 7.71 Charge (pH=7) 0.72 2D Binding mode Binding energy (Kcal/mol) -9.73 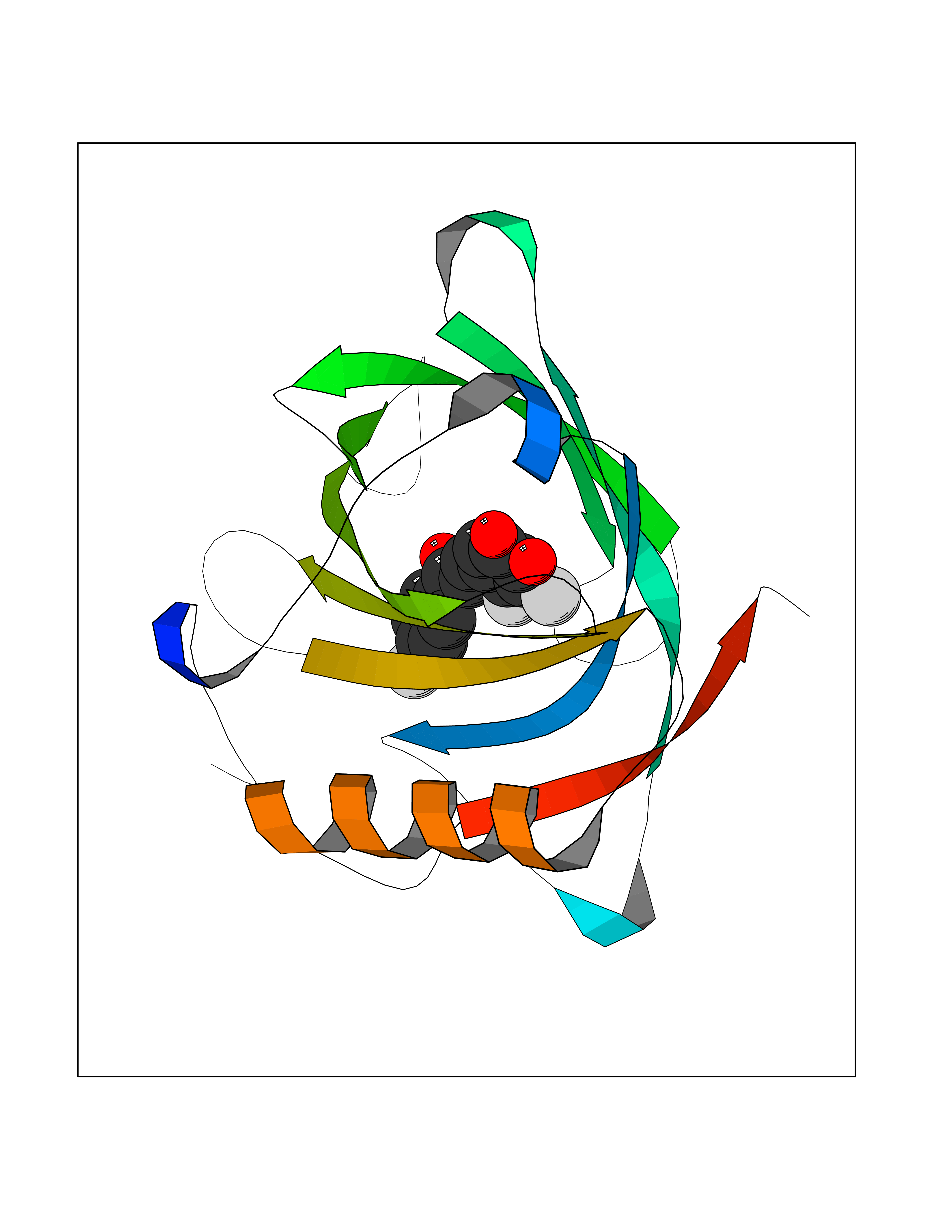 Molscript Map 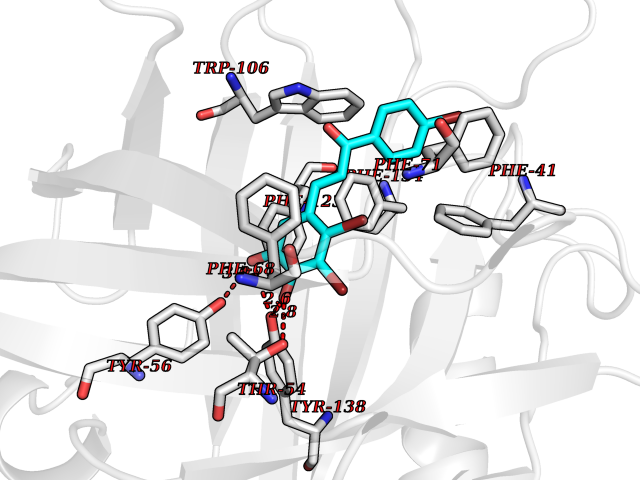 Pymol Map 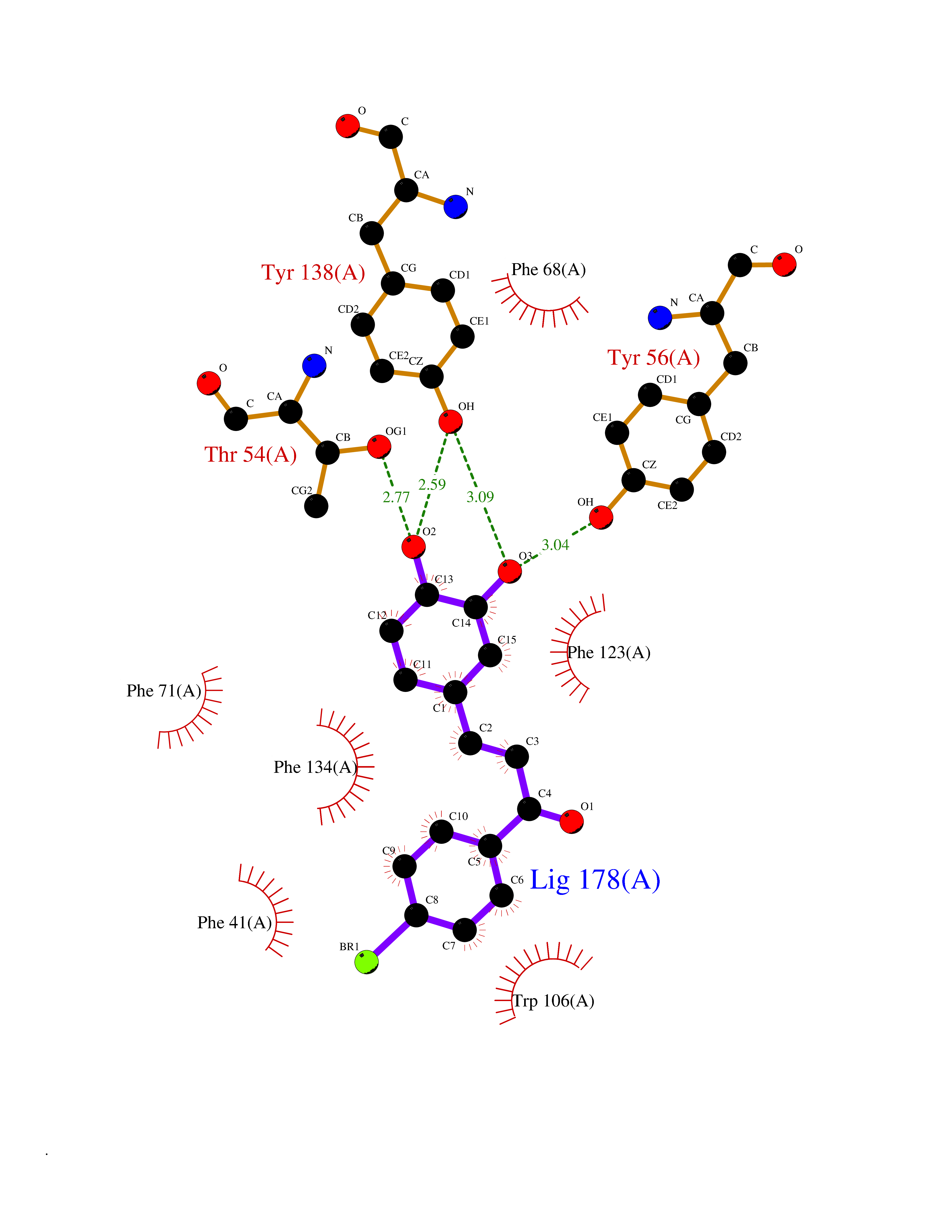 Ligplot Map 3D Binding mode Sequence SDLIPAPPLSKVPLQQNFQDNQFHGKWYVVGVAGNGFLREDKDPIKMAATIYELKEDKSYNVTFQKFPMKKCQYMTDTLVPGSQPGEFTLGNIKSEPGYTSWLVRVVSTNYNQHAMVFFKAVQQNREDFFITLYGRTKELTSELKENFIRFSKSLGLPENHIVFPVPIDQCI Hydrogen bonds contact Hydrophobic contact | ||||
| 85 | Plasmodium Adenylosuccinate synthetase (Malaria Adss) | 1P9B | 7.13 | |
Target general information Gen name Malaria Adss Organism Plasmodium falciparum Uniprot ID TTD ID Synonyms IMP--aspartate ligase; Adenylosuccinate synthase; AdSS; AMPSase Protein family Adenylosuccinate synthetase family Biochemical class Carbon-nitrogen ligase Function Plays an important role in the salvage pathway for purine nucleotide biosynthesis. Catalyzes the first committed step in the biosynthesis of AMP from IMP. Related diseases Hypertension and brachydactyly syndrome (HTNB) [MIM:112410]: A syndrome characterized by brachydactyly type E, severe salt-independent but age-dependent hypertension, an increased fibroblast growth rate, neurovascular contact at the rostral-ventrolateral medulla, and altered baroreflex blood pressure regulation. It results in death from stroke before age 50 years when untreated. Brachydactyly type E is characterized by shortening of the fingers mainly in the metacarpals and metatarsals. {ECO:0000269|PubMed:25961942}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03510; DB04315; DB02109 Interacts with NA EC number EC 6.3.4.4 Uniprot keywords 3D-structure; Cytoplasm; GTP-binding; Ligase; Magnesium; Metal-binding; Nucleotide-binding; Purine biosynthesis Protein physicochemical properties Chain ID A Molecular weight (Da) 47877.9 Length 424 Aromaticity 0.09 Instability index 31.72 Isoelectric point 7.63 Charge (pH=7) 1.58 2D Binding mode Binding energy (Kcal/mol) -9.72 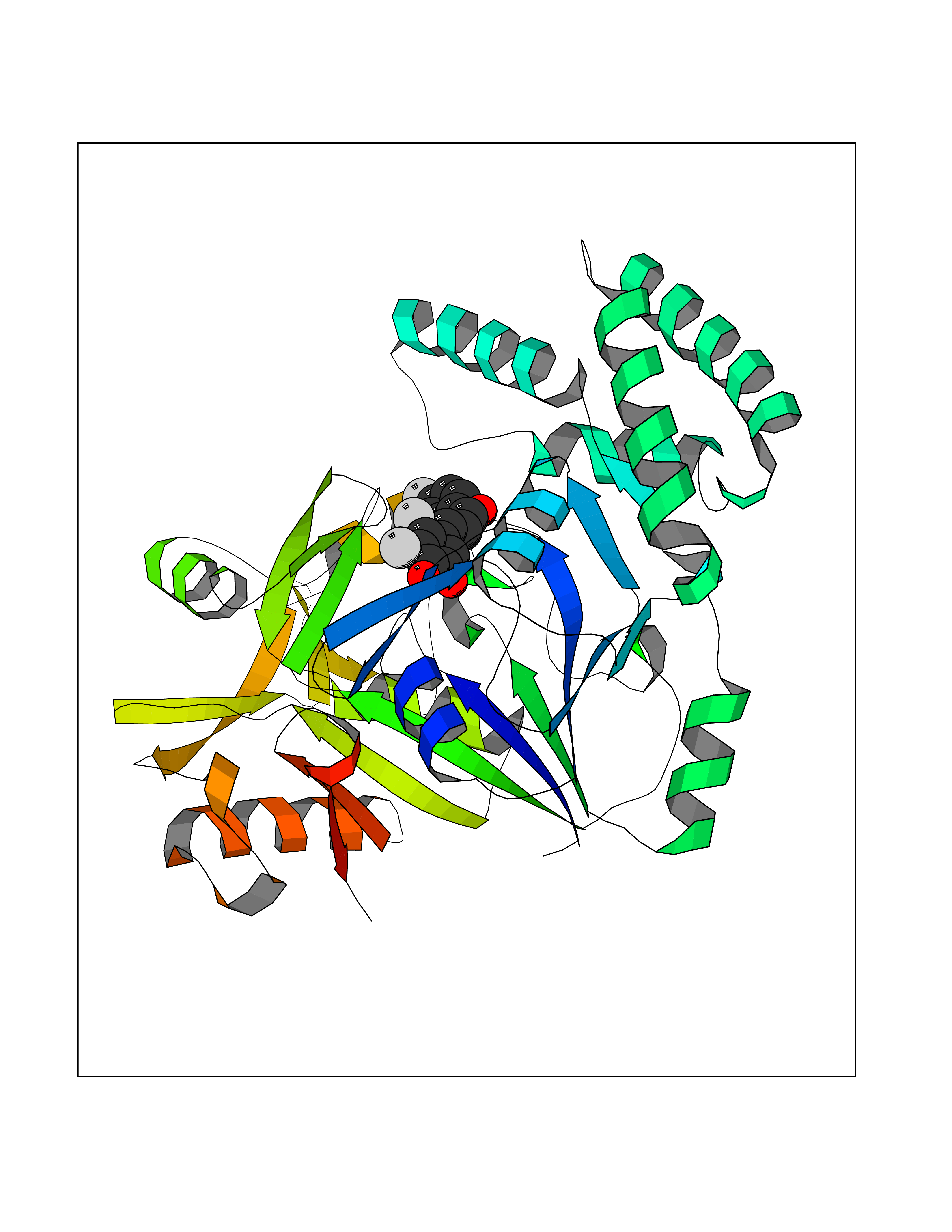 Molscript Map 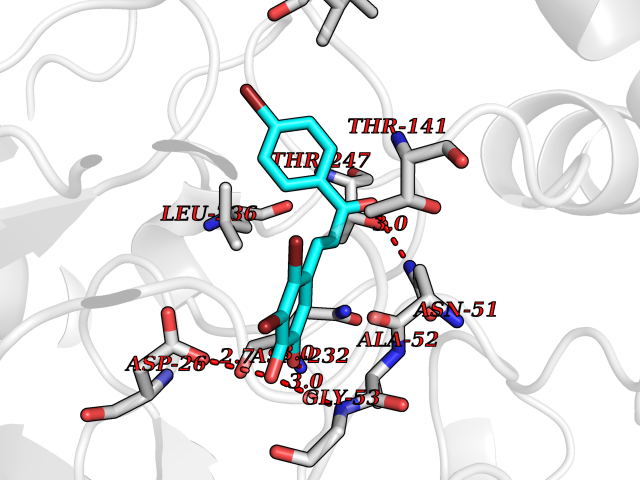 Pymol Map 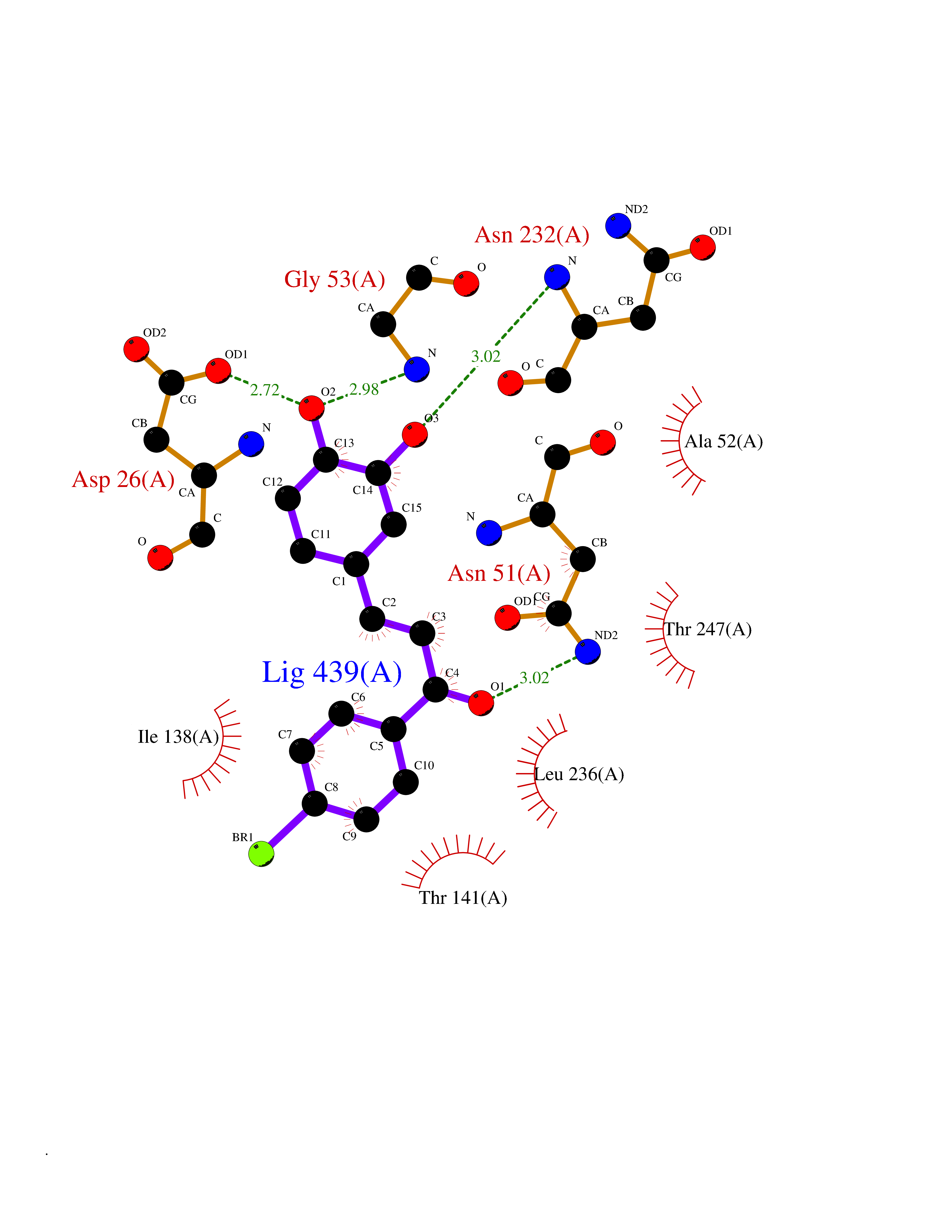 Ligplot Map 3D Binding mode Sequence GNVVAILGAQWGDEGKGKIIDMLSEYSDITCRFNGGANAGHTISVNDKKYALHLLPCGVLYDNNISVLGNGMVIHVKSLMEEIESVGGKLLDRLYLSNKAHILFDIHQIIDSIQETKKLKEGKQIGTTKRGIGPCYSTKASRIGIRLGTLKNFENFKNMYSKLIDHLMDLYNITEYDKEKELNLFYNYHIKLRDRIVDVISFMNTNLENNKKVLIEGANAAMLDIDFGTYPYVTSSCTTVGGVFSGLGIHHKKLNLVVGVVKSYLTRVGCGPFLTELNNDVGQYLREKGHEYGTTTKRPRRCGWLDIPMLLYVKCINSIDMINLTKLDVLSGLEEILLCVNFKNKKTGELLEKGCYPVEEEISEEYEPVYEKFSGWKEDISTCNEFDELPENAKKYILAIEKYLKTPIVWIGVGPNRKNMIVKK Hydrogen bonds contact Hydrophobic contact | ||||
| 86 | TRANSPORT INHIBITOR RESPONSE 1 protein | 2P1Q | 7.12 | |
Target general information Gen name IAA7 Organism Arabidopsis thaliana (Mouse-ear cress) Uniprot ID TTD ID NA Synonyms AXR2;At3g23050;MXC7.8 Protein family Aux/IAA family Biochemical class Signaling protein Function DNA binding transcription factor activity. Related diseases LTC4 synthase deficiency is associated with a neurometabolic developmental disorder characterized by muscular hypotonia, psychomotor retardation, failure to thrive, and microcephaly. {ECO:0000269|PubMed:10896305, ECO:0000269|PubMed:9820300}. Drugs (DrugBank ID) NA Interacts with Q9LW29; Q9C5W9; Q8RYC8; Q94AH6; Q9ZR12; P49677; Q38828; Q38829; Q38830; Q38831; O24407; O24408; O24409; P49678; O24410; Q8LAL2; Q9XFM0; Q38822; Q9M1R4; Q9C5X0; Q9C8Y3; Q39255; Q570C0 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Auxin signaling pathway; Nucleus; Reference proteome; Repressor; Transcription; Transcription regulation Protein physicochemical properties Chain ID B,C Molecular weight (Da) 65385.2 Length 581 Aromaticity 0.09 Instability index 47.83 Isoelectric point 7.46 Charge (pH=7) 1.23 2D Binding mode Binding energy (Kcal/mol) -9.72 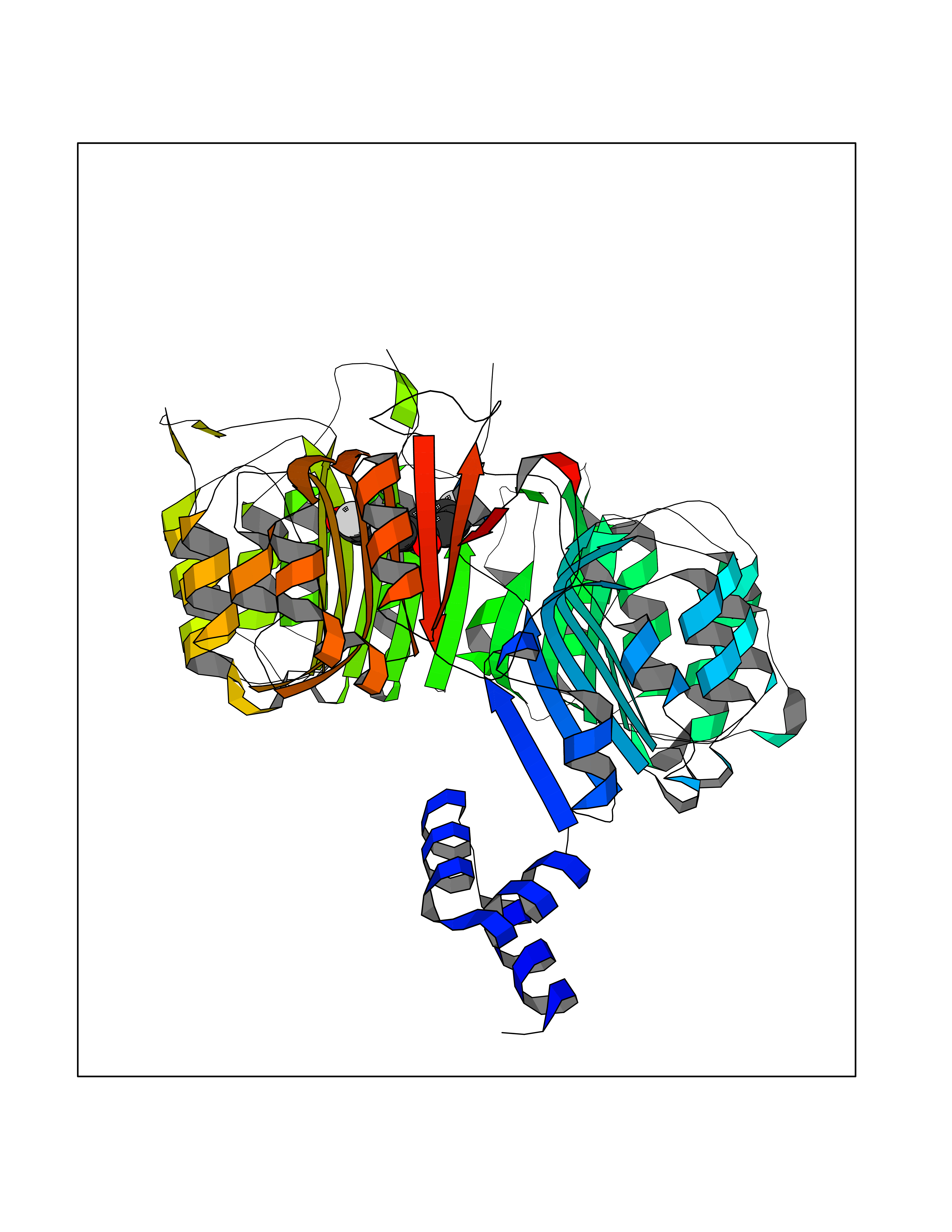 Molscript Map 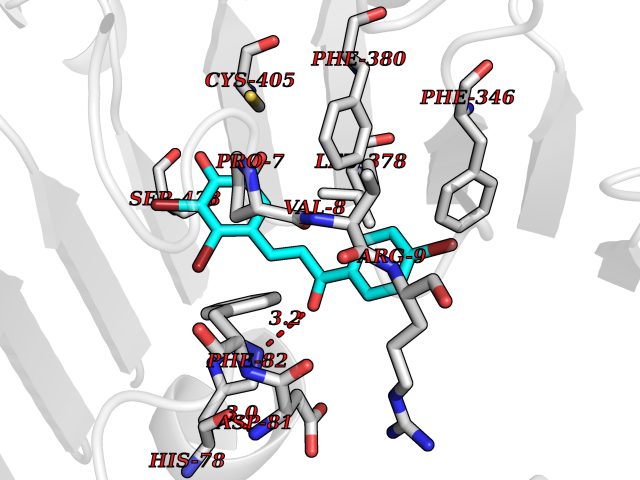 Pymol Map 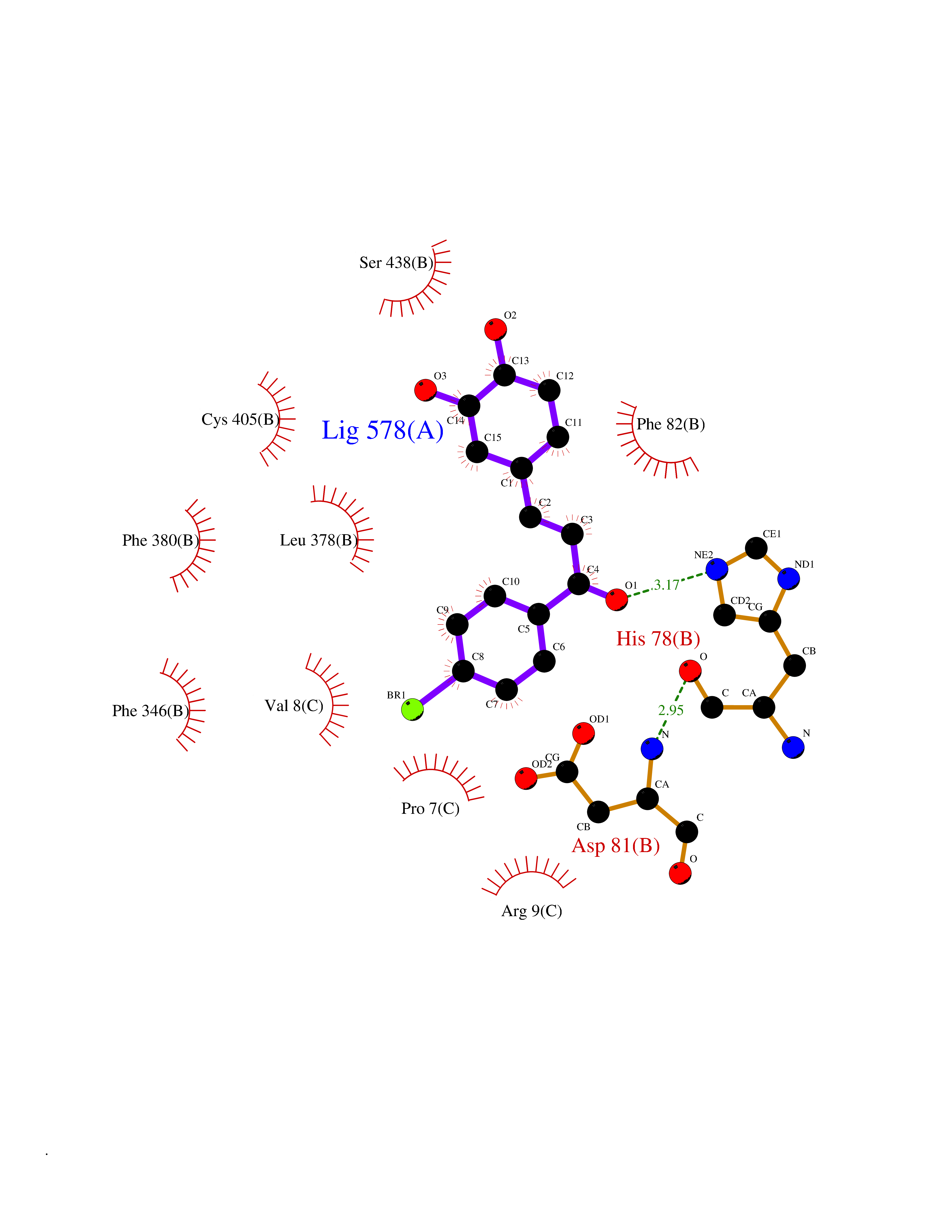 Ligplot Map 3D Binding mode Sequence QVVGWPPVRNYRKFPEEVLEHVFSFIQLDKDRNSVSLVCKSWYEIERWCRRKVFIGNCYAVSPATVIRRFPKVRSVELKGKPHFADFNLVPDGWGGYVYPWIEAMSSSYTWLEEIRLKRMVVTDDCLELIAKSFKNFKVLVLSSCEGFSTDGLAAIAATCRNLKELDLRESDVDDVSGHWLSHFPDTYTSLVSLNISCLASEVSFSALERLVTRCPNLKSLKLNRAVPLEKLATLLQRAPQLEELGTGGYTAEVRPDVYSGLSVALSGCKELRCLSGFWDAVPAYLPAVYSVCSRLTTLNLSYATVQSYDLVKLLCQCPKLQRLWVLDYIEDAGLEVLASTCKDLRELRVFPSEPFVMEPNVALTEQGLVSVSMGCPKLESVLYFCRQMTNAALITIARNRPNMTRFRLCIIEPKAPDYLTLEPLDIGFGAIVEHCKDLRRLSLSGLLTDKVFEYIGTYAKKMEMLSVAFAGDSDLGMHHVLSGCDSLRKLEIRDCPFGDKALLANASKLETMRSLWMSSCSVSFGACKLLGQKMPKLNVEVIDERGAPDSRPESCPVERVFIYRTVAGPRFDMPGFVWNM Hydrogen bonds contact Hydrophobic contact | ||||
| 87 | UDP-N-acetylglucosamine 1-carboxyvinyltransferase | 1C3R | 7.11 | |
Target general information Gen name murA Organism Aquifex aeolicus (strain VF5) Uniprot ID TTD ID NA Synonyms aq_1023 Protein family Histone deacetylase family Biochemical class Lyase Function UDP-N-acetylglucosamine 1-carboxyvinyltransferase activity. Related diseases Atrial fibrillation, familial, 14 (ATFB14) [MIM:615378]: A familial form of atrial fibrillation, a common sustained cardiac rhythm disturbance. Atrial fibrillation is characterized by disorganized atrial electrical activity and ineffective atrial contraction promoting blood stasis in the atria and reduces ventricular filling. It can result in palpitations, syncope, thromboembolic stroke, and congestive heart failure. {ECO:0000269|PubMed:19808477}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Genetic variations in SCN2B may be involved in Brugada syndrome (PubMed:23559163). This tachyarrhythmia is characterized by right bundle branch block and ST segment elevation on an electrocardiogram (ECG). It can cause the ventricles to beat so fast that the blood is prevented from circulating efficiently in the body. When this situation occurs, the individual will faint and may die in a few minutes if the heart is not reset. {ECO:0000269|PubMed:23559163}. Drugs (DrugBank ID) DB04297; DB02546 Interacts with NA EC number NA Uniprot keywords 3D-structure; Acetoin catabolism; Metal-binding; Reference proteome; Zinc Protein physicochemical properties Chain ID A,B Molecular weight (Da) 42338.9 Length 372 Aromaticity 0.12 Instability index 35.74 Isoelectric point 5.77 Charge (pH=7) -6.5 2D Binding mode Binding energy (Kcal/mol) -9.7 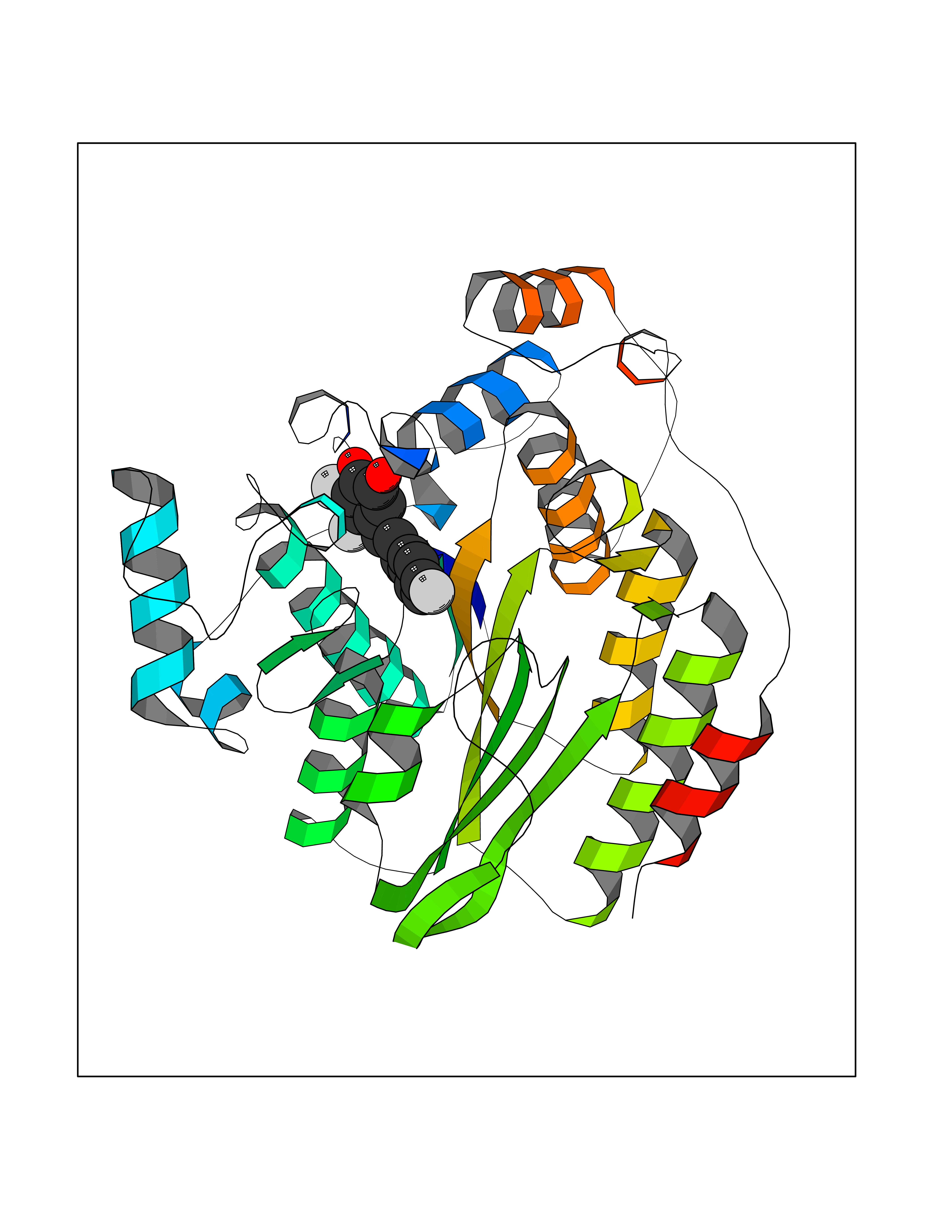 Molscript Map 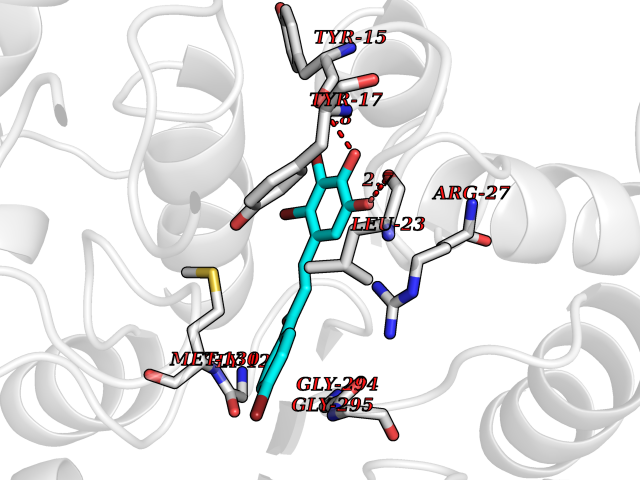 Pymol Map 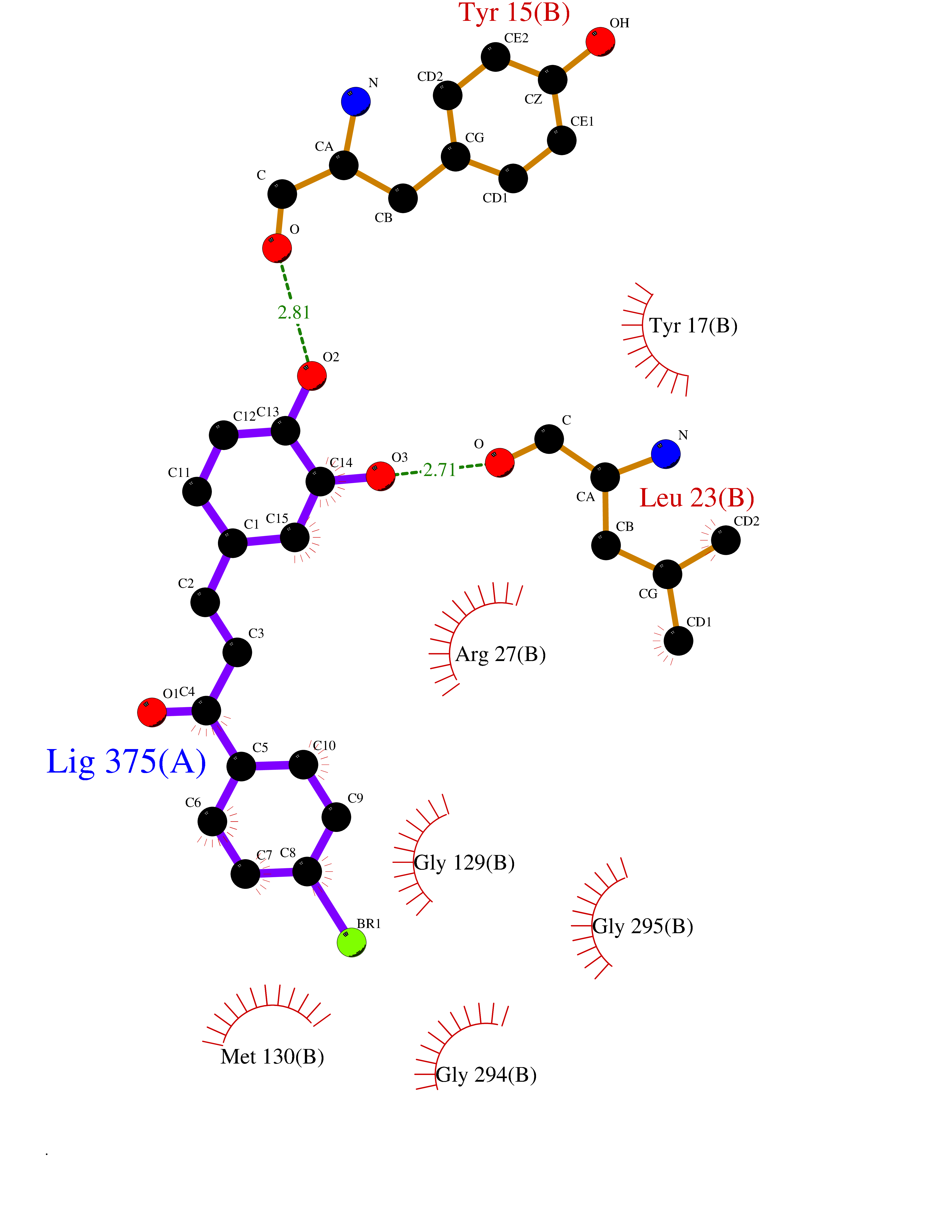 Ligplot Map 3D Binding mode Sequence KKVKLIGTLDYGKYRYPKNHPLKIPRVSLLLRFKDAMNLIDEKELIKSRPATKEELLLFHTEDYINTLMEAERSQSVPKGAREKYNIGGYENPVSYAMFTGSSLATGSTVQAIEEFLKGNVAFNPAGGMHHAFKSRANGFCYINNPAVGIEYLRKKGFKRILYIDLDAHHCDGVQEAFYDTDQVFVLSLHQSPEYAFPFEKGFLEEIGEGKGKGYNLNIPLPKGLNDNEFLFALEKSLEIVKEVFEPEVYLLQLGTDPLLEDYLSKFNLSNVAFLKAFNIVREVFGEGVYLGGGGYHPYALARAWTLIWCELSGREVPEKLNNKAKELLKSIDFEEFDDEVDRSYMLETLKDPWRGGEVRKEVKDTLEKAKA Hydrogen bonds contact Hydrophobic contact | ||||
| 88 | Progesterone receptor (PGR) | 1SQN | 7.11 | |
Target general information Gen name PGR Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms PR; Nuclear receptor subfamily 3 group C member 3; NR3C3 Protein family Nuclear hormone receptor family, NR3 subfamily Biochemical class Nuclear hormone receptor Function Depending on the isoform, progesterone receptor functions as transcriptional activator or repressor. The steroid hormones and their receptors are involved in the regulation of eukaryotic gene expression and affect cellular proliferation and differentiation in target tissues. Related diseases Butyrylcholinesterase deficiency (BCHED) [MIM:617936]: An autosomal recessive metabolic condition characterized by increased sensitivity to certain anesthetic drugs, including the muscle relaxants succinylcholine or mivacurium. BCHED results in slower hydrolysis of these drugs and, consequently, a prolonged neuromuscular block, leading to apnea. The duration of the prolonged apnea varies significantly depending on the extent of the enzyme deficiency. {ECO:0000269|PubMed:10404729, ECO:0000269|PubMed:11928765, ECO:0000269|PubMed:12881446, ECO:0000269|PubMed:1306123, ECO:0000269|PubMed:1349196, ECO:0000269|PubMed:1415224, ECO:0000269|PubMed:15563885, ECO:0000269|PubMed:15781196, ECO:0000269|PubMed:1611188, ECO:0000269|PubMed:16788378, ECO:0000269|PubMed:17700357, ECO:0000269|PubMed:18075469, ECO:0000269|PubMed:18300943, ECO:0000269|PubMed:25054547, ECO:0000269|PubMed:25264279, ECO:0000269|PubMed:2915989, ECO:0000269|PubMed:7634491, ECO:0000269|PubMed:8554068, ECO:0000269|PubMed:8680411, ECO:0000269|PubMed:9110359, ECO:0000269|PubMed:9191541, ECO:0000269|PubMed:9388484, ECO:0000269|PubMed:9543549, ECO:0000269|PubMed:9694584}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01431; DB06680; DB01406; DB12941; DB13857; DB00304; DB09123; DB01395; DB00378; DB11219; DB00823; DB00294; DB13867; DB08906; DB00588; DB06730; DB11619; DB11064; DB06789; DB00367; DB00431; DB09124; DB00603; DB00351; DB02998; DB00834; DB00648; DB00764; DB14512; DB06713; DB00717; DB00957; DB09389; DB01428; DB02746; DB00396; DB14583; DB00421; DB04787; DB05253; DB08867 Interacts with Q9H467; P03372; P06401; P40763; P03372 EC number NA Uniprot keywords 3D-structure; Alternative promoter usage; Alternative splicing; Cytoplasm; Direct protein sequencing; DNA-binding; Isopeptide bond; Lipid-binding; Lipoprotein; Membrane; Metal-binding; Mitochondrion; Mitochondrion outer membrane; Nucleus; Palmitate; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Steroid-binding; Transcription; Transcription regulation; Ubl conjugation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A,B Molecular weight (Da) 28853.6 Length 250 Aromaticity 0.09 Instability index 54.82 Isoelectric point 8.4 Charge (pH=7) 2.28 2D Binding mode Binding energy (Kcal/mol) -9.7 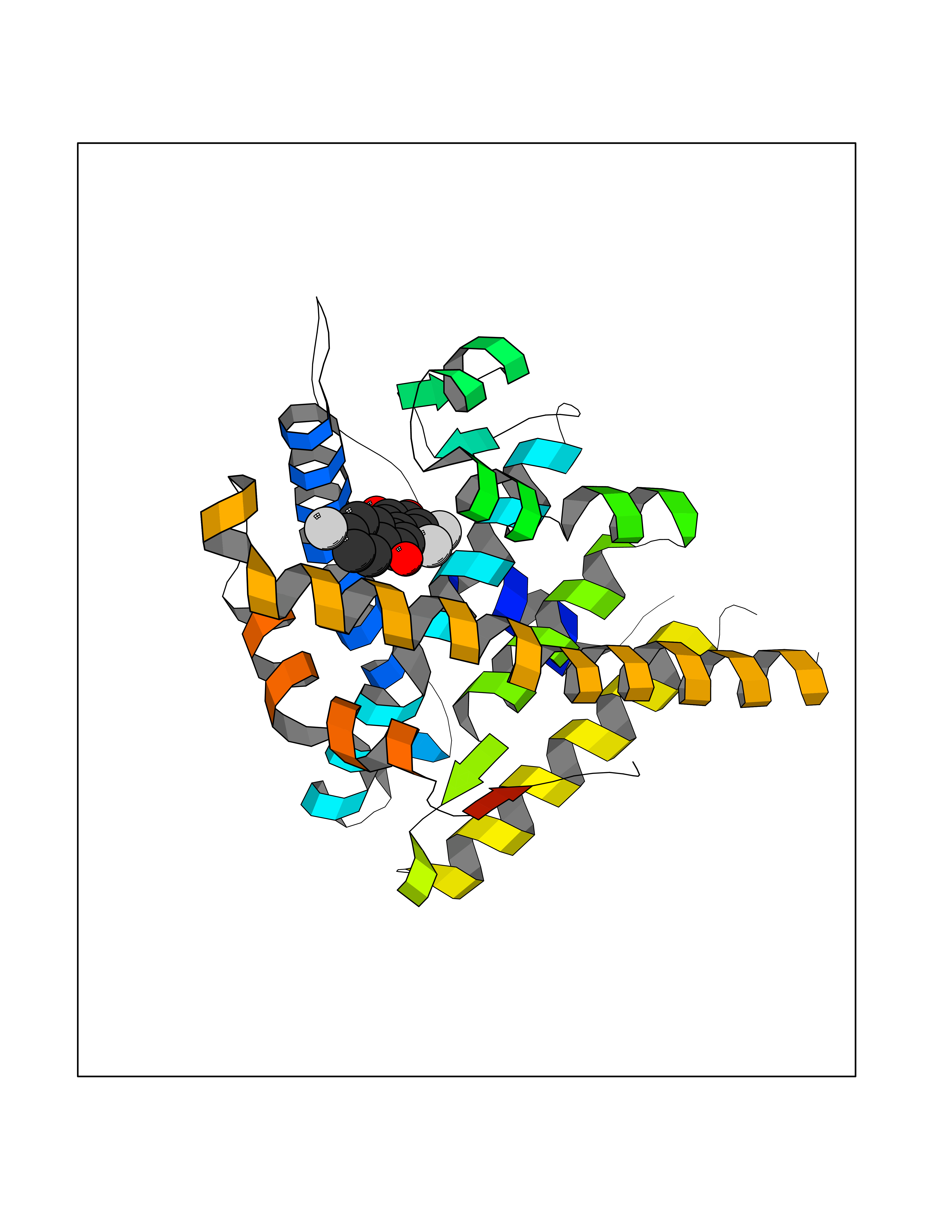 Molscript Map 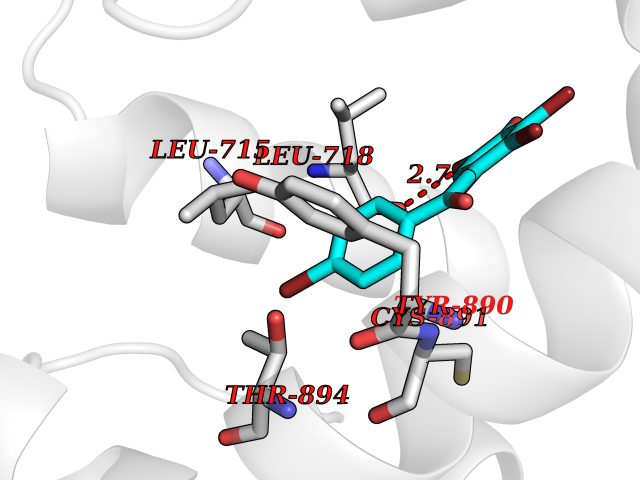 Pymol Map 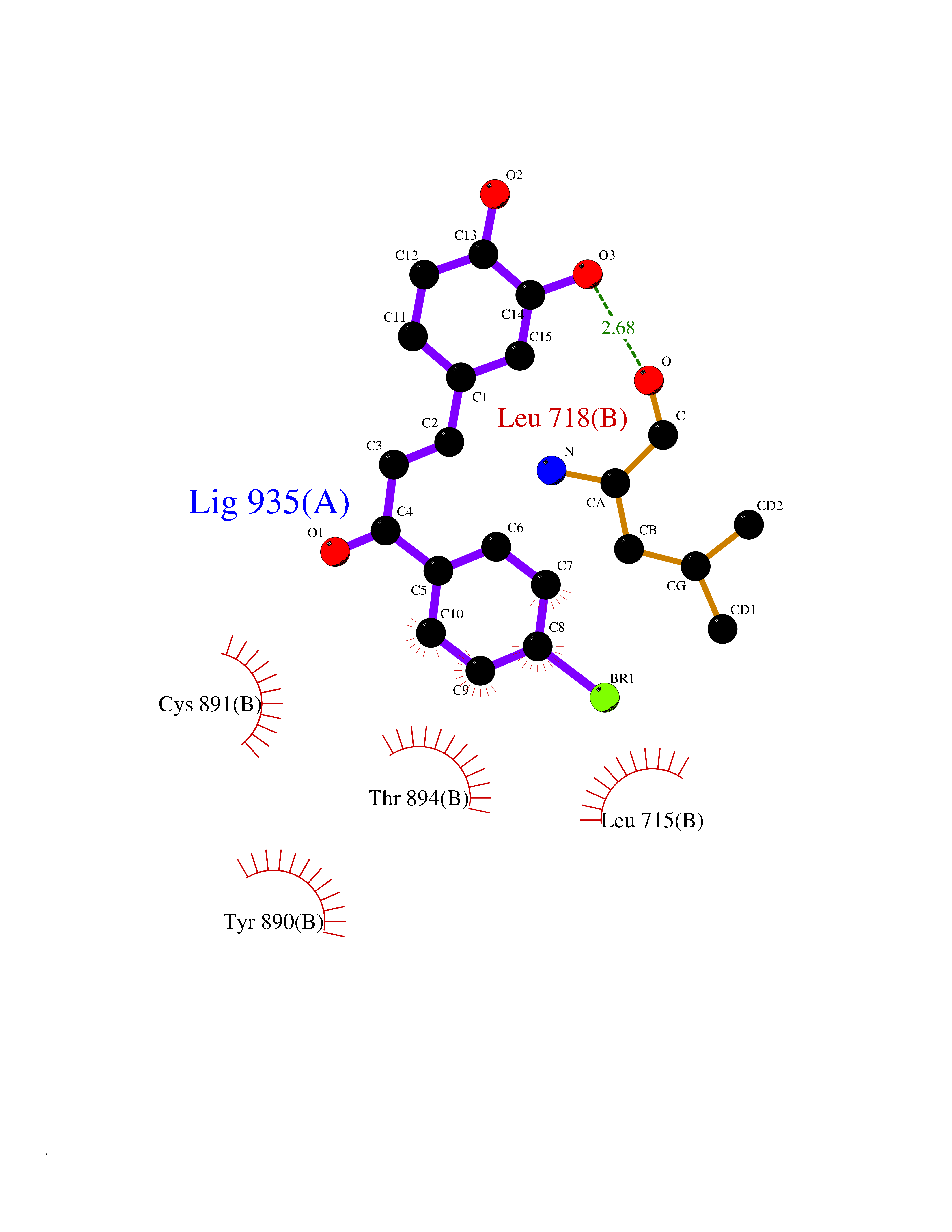 Ligplot Map 3D Binding mode Sequence LIPPLINLLMSIEPDVIYAGHDNTKPDTSSSLLTSLNQLGERQLLSVVKWSKSLPGFRNLHIDDQITLIQYSWMSLMVFGLGWRSYKHVSGQMLYFAPDLILNEQRMKESSFYSLCLTMWQIPQEFVKLQVSQEEFLCMKVLLLLNTIPLEGLRSQTQFEEMRSSYIRELIKAIGLRQGVVSSSQRFYQLTKLLDNLHDLVKQLHLYCLNTFIQSRALSVEFPEMMSEVIAAQLPKILAGMVKPLLFHKK Hydrogen bonds contact Hydrophobic contact | ||||
| 89 | Glutamate receptor ionotropic NMDA 2A (NMDAR2A) | 5I2N | 7.11 | |
Target general information Gen name GRIN2A Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms NR2A; NMDA receptor NR2A; N-methyl D-aspartate receptor subtype 2A; HNR2A; Glutamate receptor ionotropic, NMDA 2A; Glutamate [NMDA] receptor subunit epsilon-1; GluN2A Protein family Glutamate-gated ion channel (TC 1.A.10.1) family, NR1/GRIN1 subfamily Biochemical class Glutamate-gated ion channel Function Channel activation requires binding of the neurotransmitter glutamate to the epsilon subunit, glycine binding to the zeta subunit, plus membrane depolarization to eliminate channel inhibition by Mg(2+). Sensitivity to glutamate and channel kinetics depend on the subunit composition; channels containing GRIN1 and GRIN2A have higher sensitivity to glutamate and faster kinetics than channels formed by GRIN1 and GRIN2B. Contributes to the slow phase of excitatory postsynaptic current, long-term synaptic potentiation, and learning. Component of NMDA receptor complexes that function as heterotetrameric, ligand-gated ion channels with high calcium permeability and voltage-dependent sensitivity to magnesium. Related diseases Neurodevelopmental disorder with or without hyperkinetic movements and seizures, autosomal dominant (NDHMSD) [MIM:614254]: An autosomal dominant neurodevelopmental disorder characterized by severe intellectual disability and developmental delay, absent speech, muscular hypotonia, dyskinesia, and hyperkinetic movements. Cortical blindness, cerebral atrophy, and seizures are present in some patients. {ECO:0000269|PubMed:21376300, ECO:0000269|PubMed:25167861, ECO:0000269|PubMed:25864721, ECO:0000269|PubMed:27164704, ECO:0000269|PubMed:28095420, ECO:0000269|PubMed:28228639, ECO:0000269|PubMed:28389307, ECO:0000269|PubMed:38538865}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Neurodevelopmental disorder with or without hyperkinetic movements and seizures, autosomal recessive (NDHMSR) [MIM:617820]: An autosomal recessive neurodevelopmental disorder characterized by severe intellectual disability and psychomotor developmental delay, involuntary and stereotypic movements, spasticity, and inability to walk without support. Intractable seizures manifest in some patients. {ECO:0000269|PubMed:27164704, ECO:0000269|PubMed:28051072}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Developmental and epileptic encephalopathy 101 (DEE101) [MIM:619814]: A form of epileptic encephalopathy, a heterogeneous group of early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE101 is an autosomal recessive, severe form characterized by onset of seizures in early infancy. Death in infancy may occur. {ECO:0000269|PubMed:27164704, ECO:0000269|PubMed:34611970}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01931; DB00659; DB06151; DB08838; DB01238; DB00289; DB05824; DB04620; DB03929; DB00647; DB00843; DB00228; DB11823; DB13146; DB06741; DB00142; DB00874; DB08954; DB06738; DB09409; DB09481; DB01043; DB00454; DB00333; DB04896; DB01173; DB00312; DB01174; DB01708; DB00418; DB00193 Interacts with P05067; P35637; Q12879-1; Q13224; Q62936 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Calcium; Cell membrane; Disease variant; Disulfide bond; Epilepsy; Glycoprotein; Intellectual disability; Ion channel; Ion transport; Ligand-gated ion channel; Magnesium; Membrane; Metal-binding; Phosphoprotein; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Signal; Synapse; Transmembrane; Transmembrane helix; Transport; Zinc Protein physicochemical properties Chain ID B Molecular weight (Da) 63184.8 Length 559 Aromaticity 0.1 Instability index 29.67 Isoelectric point 8.51 Charge (pH=7) 6.66 2D Binding mode Binding energy (Kcal/mol) -9.7  Molscript Map  Pymol Map 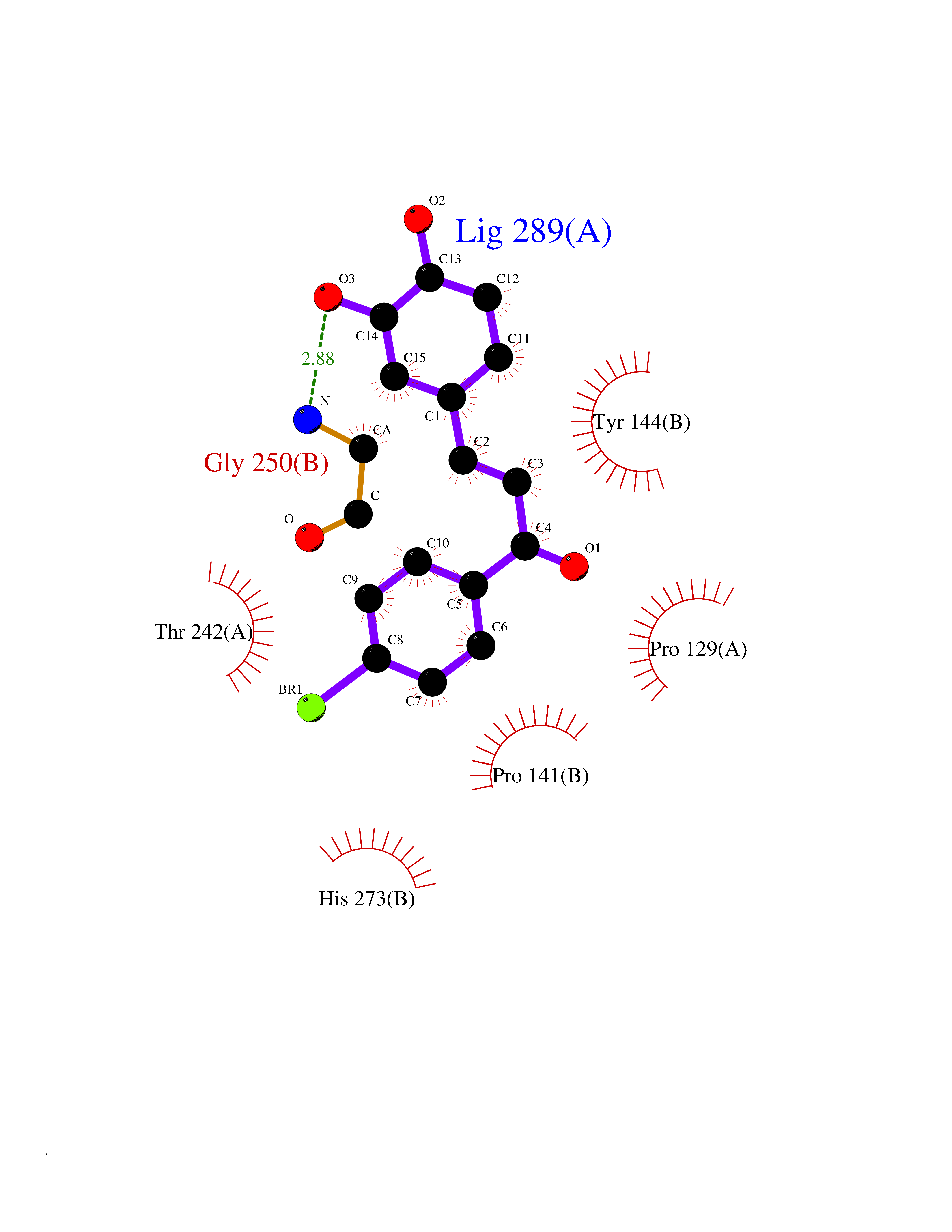 Ligplot Map 3D Binding mode Sequence DNHLSIVTLEEAPFVIVEDIDPETCVRNTVPCRKFVKINNSTNEGMNVKKCCKGFCIDILKKLSRTVKFTYDLYLVTNGKHGKKVNNVWNGMIGEVVYQRAVMAVGSLTINEERSEVVDFSVPFVETGISVMVSRGTQVTGLSDKKFQRPHDYSPPFRFGTVPNGSTERNIRNNYPYMHQYMTKFNQKGVEDALVSLKTGKLDAFIYDAAVLNYKAGRDEGCKLVTIGSGYIFATTGYGIALQKGSPWKRQIDLALLQFVGDGEMEELETLWLTGICMSTRLKIVTIHQEPFVYVKPTLSDGTCKEEFTVNGDPVKKVICTGPNDTSPGSPRHTVPQCCYGFCIDLLIKLARTMNFTYEVHLVADGKFGTQERVNKKEWNGMMGELLSGQADMIVAPLTINNERAQYIEFSKPFKYQGLTILVKKGTRITGINDPRLRNPSDKFIYATVKQSSVDIYFRRQVELSTMYRHMEKHNYESAAEAIQAVRDNKLHAFIWDSAVLEFEASQKCDLVTTGELFFRSGFGIGMRKDSPWKQNVSLSILKSHENGFMEDLDKTWVR Hydrogen bonds contact Hydrophobic contact | ||||
| 90 | Androgen receptor (AR) | 2AM9 | 7.11 | |
Target general information Gen name AR Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Testosterone receptor; Nuclear receptor subfamily 3 group C member 4; NR3C4; Dihydrotestosterone receptor; DHTR Protein family Nuclear hormone receptor family, NR3 subfamily Biochemical class Nuclear hormone receptor Function Transcription factor activity is modulated by bound coactivator and corepressor proteins like ZBTB7A that recruits NCOR1 and NCOR2 to the androgen response elements/ARE on target genes, negatively regulating androgen receptor signaling and androgen-induced cell proliferation. Transcription activation is also down-regulated by NR0B2. Activated, but not phosphorylated, by HIPK3 and ZIPK/DAPK3. Steroid hormone receptors are ligand-activated transcription factors that regulate eukaryotic gene expression and affect cellular proliferation and differentiation in target tissues. Related diseases Androgen insensitivity syndrome (AIS) [MIM:300068]: An X-linked recessive form of pseudohermaphroditism due end-organ resistance to androgen. Affected males have female external genitalia, female breast development, blind vagina, absent uterus and female adnexa, and abdominal or inguinal testes, despite a normal 46,XY karyotype. {ECO:0000269|PubMed:10022458, ECO:0000269|PubMed:10221692, ECO:0000269|PubMed:10221770, ECO:0000269|PubMed:10404311, ECO:0000269|PubMed:10458483, ECO:0000269|PubMed:10571951, ECO:0000269|PubMed:10590024, ECO:0000269|PubMed:10690872, ECO:0000269|PubMed:11587068, ECO:0000269|PubMed:11744994, ECO:0000269|PubMed:1307250, ECO:0000269|PubMed:1316540, ECO:0000269|PubMed:1426313, ECO:0000269|PubMed:1430233, ECO:0000269|PubMed:1464650, ECO:0000269|PubMed:14756668, ECO:0000269|PubMed:1480178, ECO:0000269|PubMed:1487249, ECO:0000269|PubMed:1569163, ECO:0000269|PubMed:1609793, ECO:0000269|PubMed:16129672, ECO:0000269|PubMed:16595706, ECO:0000269|PubMed:1775137, ECO:0000269|PubMed:1999491, ECO:0000269|PubMed:2082179, ECO:0000269|PubMed:2594783, ECO:0000269|PubMed:7537149, ECO:0000269|PubMed:7581399, ECO:0000269|PubMed:7633398, ECO:0000269|PubMed:7641413, ECO:0000269|PubMed:7671849, ECO:0000269|PubMed:7929841, ECO:0000269|PubMed:7962294, ECO:0000269|PubMed:7970939, ECO:0000269|PubMed:7981687, ECO:0000269|PubMed:7981689, ECO:0000269|PubMed:7993455, ECO:0000269|PubMed:8040309, ECO:0000269|PubMed:8096390, ECO:0000269|PubMed:8103398, ECO:0000269|PubMed:8162033, ECO:0000269|PubMed:8224266, ECO:0000269|PubMed:8281140, ECO:0000269|PubMed:8325950, ECO:0000269|PubMed:8339746, ECO:0000269|PubMed:8413310, ECO:0000269|PubMed:8446106, ECO:0000269|PubMed:8626869, ECO:0000269|PubMed:8647313, ECO:0000269|PubMed:8683794, ECO:0000269|PubMed:8723113, ECO:0000269|PubMed:8768864, ECO:0000269|PubMed:8809734, ECO:0000269|PubMed:8830623, ECO:0000269|PubMed:8918984, ECO:0000269|PubMed:8990010, ECO:0000269|PubMed:9001799, ECO:0000269|PubMed:9007482, ECO:0000269|PubMed:9039340, ECO:0000269|PubMed:9106550, ECO:0000269|PubMed:9160185, ECO:0000269|PubMed:9252933, ECO:0000269|PubMed:9255042, ECO:0000269|PubMed:9302173, ECO:0000269|PubMed:9328206, ECO:0000269|PubMed:9544375, ECO:0000269|PubMed:9554754, ECO:0000269|PubMed:9610419, ECO:0000269|PubMed:9627582, ECO:0000269|PubMed:9698822, ECO:0000269|PubMed:9851768, ECO:0000269|PubMed:9856504, ECO:0000269|Ref.116, ECO:0000269|Ref.182}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Spinal and bulbar muscular atrophy X-linked 1 (SMAX1) [MIM:313200]: An X-linked recessive form of spinal muscular atrophy. Spinal muscular atrophy refers to a group of neuromuscular disorders characterized by degeneration of the anterior horn cells of the spinal cord, leading to symmetrical muscle weakness and atrophy. SMAX1 occurs only in men. Age at onset is usually in the third to fifth decade of life, but earlier involvement has been reported. It is characterized by slowly progressive limb and bulbar muscle weakness with fasciculations, muscle atrophy, and gynecomastia. The disorder is clinically similar to classic forms of autosomal spinal muscular atrophy. {ECO:0000269|PubMed:15851746}. The disease is caused by variants affecting the gene represented in this entry. Caused by trinucleotide CAG repeat expansion. In SMAX1 patients the number of Gln ranges from 38 to 62. Longer expansions result in earlier onset and more severe clinical manifestations of the disease.; DISEASE: Prostate cancer, hereditary, X-linked 3 (HPCX3) [MIM:301120]: A condition associated with familial predisposition to cancer of the prostate. Most prostate cancers are adenocarcinomas that develop in the acini of the prostatic ducts. Other rare histopathologic types of prostate cancer that occur in approximately 5% of patients include small cell carcinoma, mucinous carcinoma, prostatic ductal carcinoma, transitional cell carcinoma, squamous cell carcinoma, basal cell carcinoma, adenoid cystic carcinoma (basaloid), signet-ring cell carcinoma and neuroendocrine carcinoma. {ECO:0000269|PubMed:8530589}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Defects in AR may play a role in metastatic prostate cancer. The mutated receptor stimulates prostate growth and metastases development despite of androgen ablation. This treatment can reduce primary and metastatic lesions probably by inducing apoptosis of tumor cells when they express the wild-type receptor. {ECO:0000269|PubMed:10363963, ECO:0000269|PubMed:10569618, ECO:0000269|PubMed:1562539, ECO:0000269|PubMed:16129672, ECO:0000269|PubMed:17311914, ECO:0000269|PubMed:2260966, ECO:0000269|PubMed:25091737, ECO:0000269|PubMed:8187068, ECO:0000269|PubMed:8274409, ECO:0000269|PubMed:8827083}.; DISEASE: Androgen insensitivity, partial (PAIS) [MIM:312300]: A disorder that is characterized by hypospadias, hypogonadism, gynecomastia, genital ambiguity, normal XY karyotype, and a pedigree pattern consistent with X-linked recessive inheritance. Some patients present azoospermia or severe oligospermia without other clinical manifestations. {ECO:0000269|PubMed:10022458, ECO:0000269|PubMed:10221692, ECO:0000269|PubMed:10470409, ECO:0000269|PubMed:10502786, ECO:0000269|PubMed:10543676, ECO:0000269|PubMed:11587068, ECO:0000269|PubMed:1303262, ECO:0000269|PubMed:1307250, ECO:0000269|PubMed:1316540, ECO:0000269|PubMed:1424203, ECO:0000269|PubMed:1430233, ECO:0000269|PubMed:14756668, ECO:0000269|PubMed:2010552, ECO:0000269|PubMed:7581399, ECO:0000269|PubMed:7649358, ECO:0000269|PubMed:7671849, ECO:0000269|PubMed:7909256, ECO:0000269|PubMed:7910529, ECO:0000269|PubMed:7929841, ECO:0000269|PubMed:7970939, ECO:0000269|PubMed:7981687, ECO:0000269|PubMed:8033918, ECO:0000269|PubMed:8097257, ECO:0000269|PubMed:8126121, ECO:0000269|PubMed:8205256, ECO:0000269|PubMed:8281139, ECO:0000269|PubMed:8325932, ECO:0000269|PubMed:8325950, ECO:0000269|PubMed:8446106, ECO:0000269|PubMed:8550758, ECO:0000269|PubMed:8809734, ECO:0000269|PubMed:8823308, ECO:0000269|PubMed:8824883, ECO:0000269|PubMed:9039340, ECO:0000269|PubMed:9196614, ECO:0000269|PubMed:9302173, ECO:0000269|PubMed:9329414, ECO:0000269|PubMed:9543136, ECO:0000269|PubMed:9607727, ECO:0000269|PubMed:9768671, ECO:0000269|PubMed:9856504, ECO:0000269|Ref.124}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Hypospadias 1, X-linked (HYSP1) [MIM:300633]: A common malformation in which the urethra opens on the ventral side of the penis, due to developmental arrest of urethral fusion. The opening can be located glandular, penile, or even more posterior in the scrotum or perineum. Hypospadias is a feature of several syndromic disorders, including the androgen insensitivity syndrome and Opitz syndrome. {ECO:0000269|PubMed:8097257}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07422; DB07039; DB04709; DB07717; DB07454; DB02932; DB08035; DB01481; DB08088; DB08461; DB08087; DB07421; DB01063; DB07423; DB11901; DB01128; DB07286; DB01541; DB14639; DB01564; DB12499; DB04839; DB01406; DB12941; DB09123; DB00255; DB06133; DB01395; DB00858; DB15488; DB11219; DB08899; DB13155; DB00655; DB09086; DB02266; DB01185; DB00623; DB00499; DB11619; DB11064; DB01026; DB15647; DB00367; DB08089; DB05234; DB13934; DB11425; DB06710; DB02998; DB11429; DB00648; DB08804; DB00984; DB00665; DB06713; DB00717; DB09371; DB00957; DB09389; DB00621; DB01428; DB06412; DB01608; DB11447; DB01708; DB00396; DB07419; DB07769; DB14583; DB00421; DB02901; DB13951; DB06718; DB00675; DB00624; DB13943; DB13944; DB01420; DB13946; DB06870; DB08604; DB08867 Interacts with P00519; Q9UBL3; P51451; Q8WV28; O60885-1; P78543; Q14790; P24385; Q92793; O14595; P35222; Q9UER7; P20711; P11308; P07332; P09769; Q02790; P55317; O75593; Q14451; P06396; P56524; Q16665; Q16666; O15357; Q15652; O95251; Q9BY66; Q9BY66-3; Q03164; O14686; P06239; P07948; P20794; P42679; Q00987; Q15596; Q14686; O96028; Q99497; P27986; O00459; Q92569; P19174; P16885; Q06830; P78527; Q06124; P20936; Q9UBS8; Q9Y252; O14796; Q9NP31; P29353; Q6S5L8; Q5VZ18; Q15797; O14544; P12931; Q9ULZ2; P63165; Q9HBL0; P07947; Q9R1E0; Q06986 EC number NA Uniprot keywords 3D-structure; Activator; Alternative splicing; Cytoplasm; Disease variant; DNA-binding; Isopeptide bond; Lipid-binding; Lipoprotein; Metal-binding; Neurodegeneration; Nucleus; Palmitate; Phosphoprotein; Proteomics identification; Pseudohermaphroditism; Receptor; Reference proteome; Steroid-binding; Transcription; Transcription regulation; Triplet repeat expansion; Ubl conjugation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A Molecular weight (Da) 29137.9 Length 250 Aromaticity 0.11 Instability index 42.11 Isoelectric point 8.94 Charge (pH=7) 5.43 2D Binding mode Binding energy (Kcal/mol) -9.69  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence QPIFLNVLEAIEPGVVCAGHDNNQPDSFAALLSSLNELGERQLVHVVKWAKALPGFRNLHVDDQMAVIQYSWMGLMVFAMGWRSFTNVNSRMLYFAPDLVFNEYRMHKSRMYSQCVRMRHLSQEFGWLQITPQEFLCMKALLLFSIIPVDGLKNQKFFDELRMNYIKELDRIIACKRKNPTSCSRRFYQLTKLLDSVQPIARELHQFTFDLLIKSHMVSVDFPEMMAEIISVQVPKILSGKVKPIYFHTQ Hydrogen bonds contact Hydrophobic contact | ||||
| 91 | Renal carcinoma antigen NY-REN-64 (IRAK-4) | 6EG9 | 7.11 | |
Target general information Gen name IRAK4 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Interleukin-1 receptor-associated kinase 4; IRAK-4 Protein family Protein kinase superfamily, TKL Ser/Thr protein kinase family, Pelle subfamily Biochemical class Kinase Function Serine/threonine-protein kinase that plays a critical role in initiating innate immune response against foreign pathogens. Involved in Toll-like receptor (TLR) and IL-1R signaling pathways. Is rapidly recruited by MYD88 to the receptor-signaling complex upon TLR activation to form the Myddosome together with IRAK2. Phosphorylates initially IRAK1, thus stimulating the kinase activity and intensive autophosphorylation of IRAK1. Phosphorylates E3 ubiquitin ligases Pellino proteins (PELI1, PELI2 and PELI3) to promote pellino-mediated polyubiquitination of IRAK1. Then, the ubiquitin-binding domain of IKBKG/NEMO binds to polyubiquitinated IRAK1 bringing together the IRAK1-MAP3K7/TAK1-TRAF6 complex and the NEMO-IKKA-IKKB complex. In turn, MAP3K7/TAK1 activates IKKs (CHUK/IKKA and IKBKB/IKKB) leading to NF-kappa-B nuclear translocation and activation. Alternatively, phosphorylates TIRAP to promote its ubiquitination and subsequent degradation. Phosphorylates NCF1 and regulates NADPH oxidase activation after LPS stimulation suggesting a similar mechanism during microbial infections. Related diseases Immunodeficiency 67 (IMD67) [MIM:607676]: An autosomal recessive primary immunodeficiency characterized by recurrent, life-threatening systemic and invasive bacterial infections beginning in infancy or early childhood. {ECO:0000269|PubMed:12637671, ECO:0000269|PubMed:12925671, ECO:0000269|PubMed:16950813, ECO:0000269|PubMed:17878374, ECO:0000269|PubMed:19663824, ECO:0000269|PubMed:21057262, ECO:0000269|PubMed:24316379}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08590; DB12010 Interacts with Q9UBH0; O43187; Q99836; Q99836-1; Q96FA3; Q9HAT8; P58753; Q9C029; P0DPA2; Q96LX8; Q8K4B2 EC number EC 2.7.11.1 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; ATP-binding; Cytoplasm; Disease variant; Immunity; Innate immunity; Kinase; Magnesium; Nucleotide-binding; Phosphoprotein; Proteomics identification; Reference proteome; Serine/threonine-protein kinase; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 30948.8 Length 276 Aromaticity 0.08 Instability index 43.33 Isoelectric point 5.09 Charge (pH=7) -13.45 2D Binding mode Binding energy (Kcal/mol) -9.7  Molscript Map 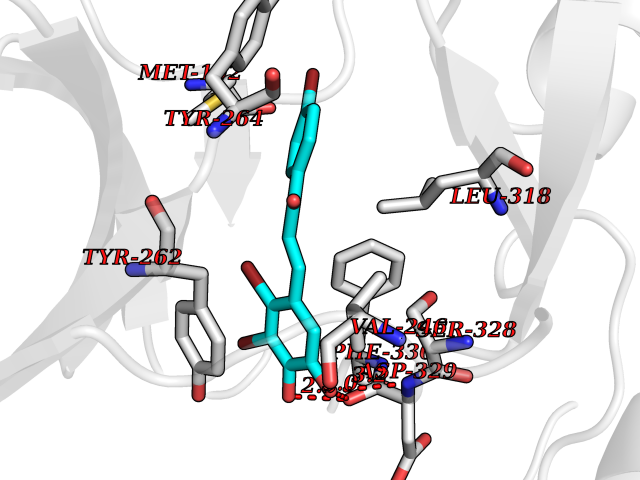 Pymol Map 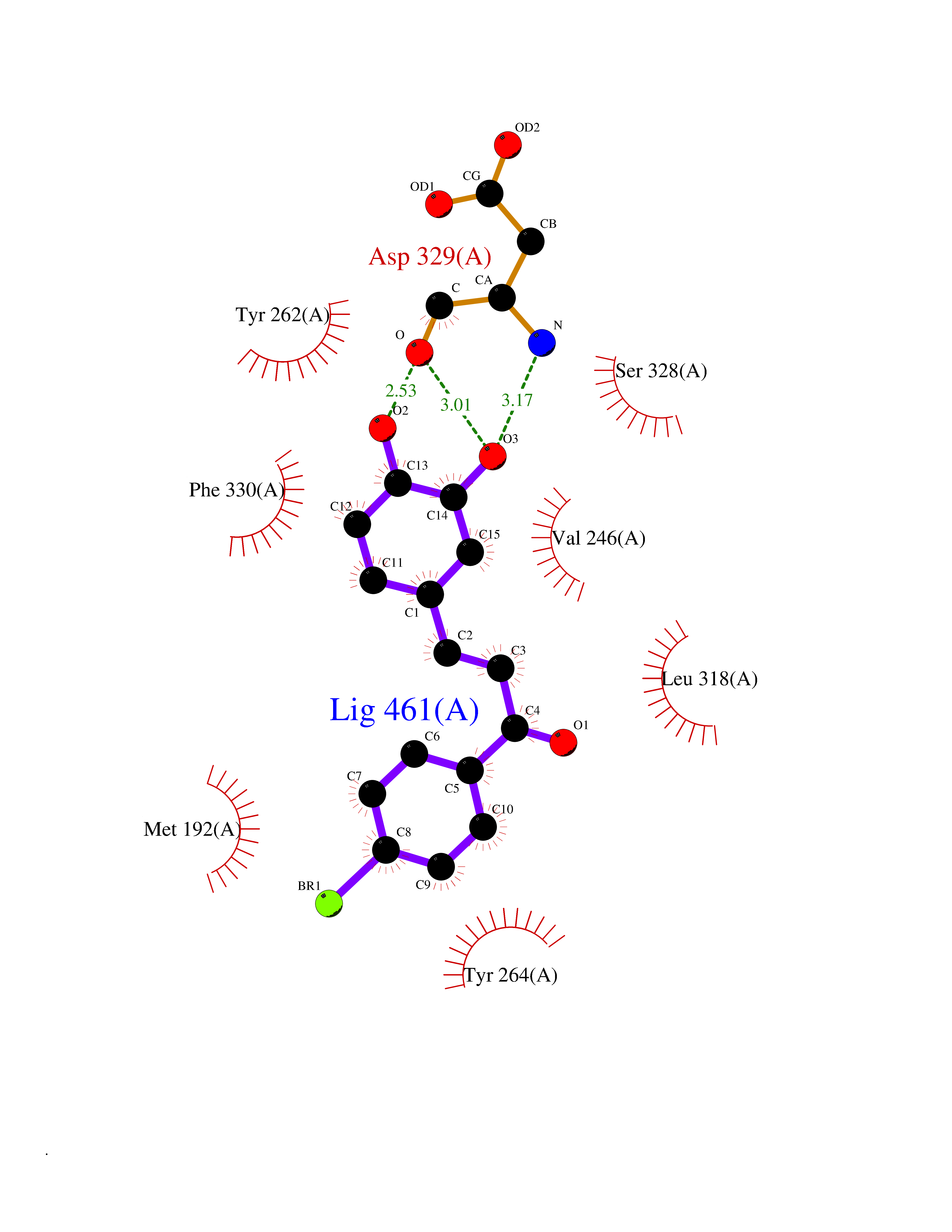 Ligplot Map 3D Binding mode Sequence RFHSFSFYELKNVTNNFDERPISVGGNKMGEGGFGVVYKGYVNNTTVAVKKLAAITTEELKQQFDQEIKVMAKCQHENLVELLGFSSDGDDLCLVYVYMPNGSLLDRLSCLDGTPPLSWHMRCKIAQGAANGINFLHENHHIHRDIKSANILLDEAFTAKISDFGVGTTAYMAPEALRGEITPKSDIYSFGVVLLEIITGLPAVDEHREPQLLLDIKEEIEDEEKTIEDYIDKKMNDADSTSVEAMYSVASQCLHEKKNKRPDIKKVQQLLQEMTA Hydrogen bonds contact Hydrophobic contact | ||||
| 92 | Phosphodiesterase 3A (PDE3A) | 7EG0 | 7.11 | |
Target general information Gen name PDE3A Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms cGMP-inhibited 3',5'-cyclic phosphodiesterase A; Phosphodiesterase 3A; Cyclic GMP-inhibited phosphodiesterase A; Cyclic GMP inhibited phosphodiesterase A; CGI-PDE A Protein family Cyclic nucleotide phosphodiesterase family, PDE3 subfamily Biochemical class Phosphoric diester hydrolase Function Cyclic nucleotide phosphodiesterase with a dual-specificity for the second messengers cAMP and cGMP, which are key regulators of many important physiological processes. Related diseases Hypertension and brachydactyly syndrome (HTNB) [MIM:112410]: A syndrome characterized by brachydactyly type E, severe salt-independent but age-dependent hypertension, an increased fibroblast growth rate, neurovascular contact at the rostral-ventrolateral medulla, and altered baroreflex blood pressure regulation. It results in death from stroke before age 50 years when untreated. Brachydactyly type E is characterized by shortening of the fingers mainly in the metacarpals and metatarsals. {ECO:0000269|PubMed:25961942}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01223; DB01427; DB00261; DB00201; DB01166; DB04880; DB05266; DB00922; DB00235; DB01303; DB00277; DB08811; DB09283 Interacts with Q9Y6D6; Q9Y6D5 EC number EC 3.1.4.17 Uniprot keywords 3D-structure; cAMP; cGMP; Cytoplasm; Direct protein sequencing; Disease variant; Hydrolase; Isopeptide bond; Manganese; Membrane; Metal-binding; Phosphoprotein; Proteomics identification; Reference proteome; Transmembrane; Transmembrane helix; Ubl conjugation Protein physicochemical properties Chain ID A,B Molecular weight (Da) 108171 Length 939 Aromaticity 0.11 Instability index 39.75 Isoelectric point 6.53 Charge (pH=7) -4.15 2D Binding mode Binding energy (Kcal/mol) -9.7 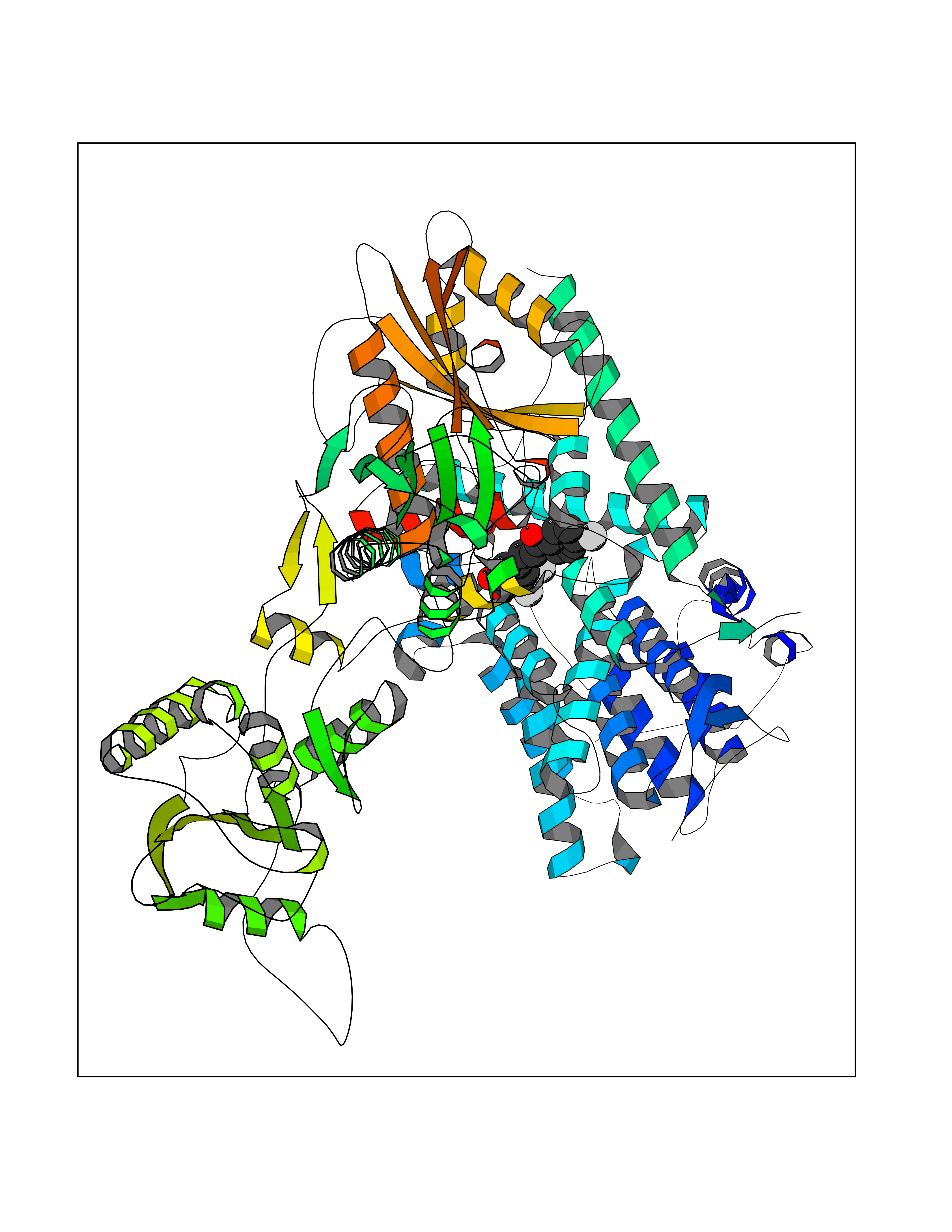 Molscript Map 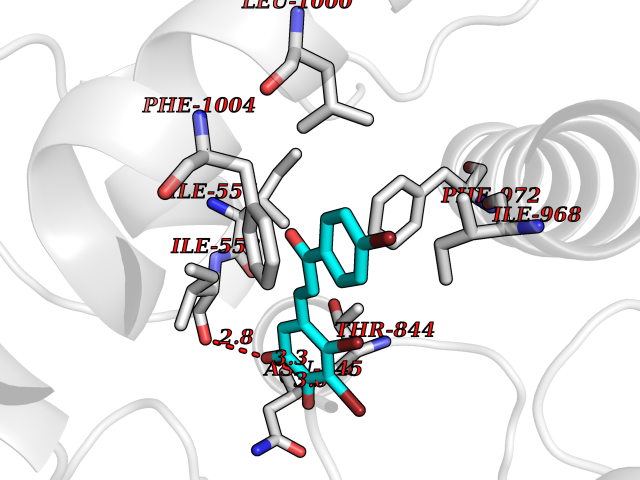 Pymol Map 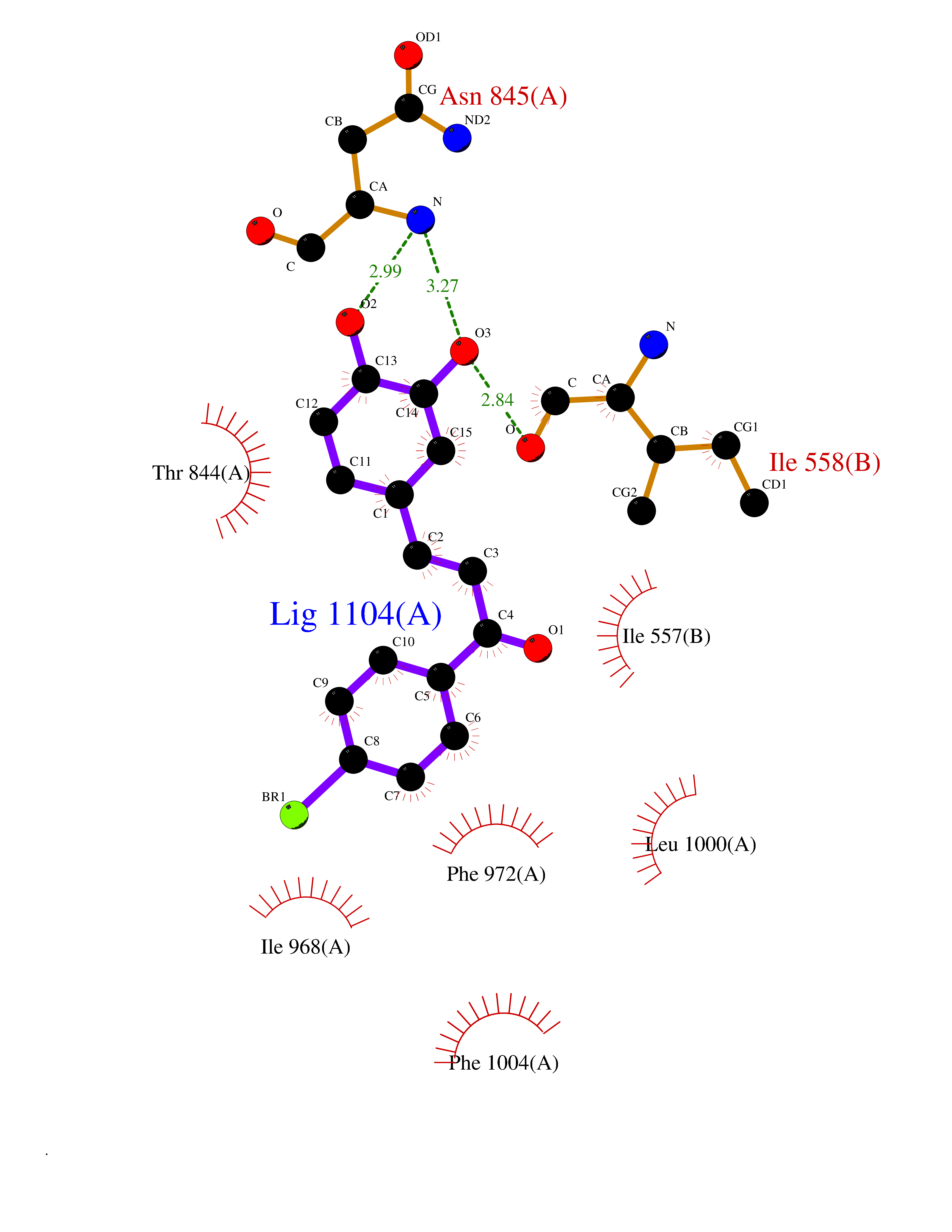 Ligplot Map 3D Binding mode Sequence KPILAPEPLVMDNLDSIMEQLNTWNFPIFDLVENIGRKCGRILSQVSYRLFEDMGLFEAFKIPIREFMNYFHALEIGYRDIPYHNRIHATDVLHAVWYLTTQPIPGLSTVGYVFSKTYNVTDDKYGCLSGNIPALELMALYVAAAMHDYDHPGRTNAFLVATSAPQAVLYNDRSVLENHHAAAAWNLFMSRPEYNFLINLDHVEFKHFRFLVIEAILATDLKKHFDFVAKFNGKVNDDVGIDWTNENDRLLVCQMCIKLADINGPAKCKELHLQWTDGIVNEFYEQGDEEASLGLPISPFMDRSAPQLANLQESFISHIVGPLCNSYDSAGLMPGKWVEKIYCQITQHLLQNHKMWKKVIEEEQRLAGIENQNISVDLETNYAELVLDVGRVTLGENSRKKMKDCKLRKKQNESVSRAMCALLNSGGGVIKAEIENEDYSYTKDGIGLDLENSFSNILLFVPEYLDFMQNGNYFLIFVKSWSLNTSGLRITTLSSNLYKRDITSAKVMNATAALEFLKDMKKTRGRLYLRPELLAKRPCVDIQEENNMKALAGVFFDRTELDRKEKLTFTESTHVEIKNFSTERLLQRIKEILPQYVSAFANTDGGYLFIGLNEDKEIIGFKAEMSDLDDLEREIEKSIRKMPVHHFCMEKKKINYSCKFLGVYDKGSLCGYVCALRVERFCCAVFAKEPDSWHVKDNRVMQLTRKEWIQFMVEAEPKFSSAYEEVISQINTSLPAPHSWPLLEWQRQRHHCPGLSGRITYTPENLCRKLFLQHEGLKQLICEEMSSVRKGSLIFSRSWSVDLGLQENHKVLCDALLISQDSPPVLYTFHMVQDEEFKGYSTQTALTLKQKLAKIGGYTKKVCVMTKIFYLSPEGMTSCQYDLRSQVIYPESYYFTRRKYLLKALFKALKRLKSLRDQFSFAENLYQIIGIDCFQKNDK Hydrogen bonds contact Hydrophobic contact | ||||
| 93 | Urea transporter 1 (SLC14A1) | 6QD5 | 7.11 | |
Target general information Gen name SLC14A1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Urea transporter, erythrocyte; UTE; UT1; Solute carrier family 14 member 1; RACH1; JK; HUT11 Protein family Urea transporter family Biochemical class Urea transporter family Function Urea channel that facilitates transmembrane urea transport down a concentration gradient. A constriction of the transmembrane channel functions as selectivity filter through which urea is expected to pass in dehydrated form. The rate of urea conduction is increased by hypotonic stress. Plays an important role in the kidney medulla collecting ducts, where it allows rapid equilibration between the lumen of the collecting ducts and the interstitium, and thereby prevents water loss driven by the high concentration of urea in the urine. Facilitates urea transport across erythrocyte membranes. May also play a role in transmembrane water transport, possibly by indirect means. Related diseases Immunodeficiency 12 (IMD12) [MIM:615468]: A primary immunodeficiency characterized by onset in infancy of recurrent bacterial and candidal infections resulting in bronchiectasis and growth delay. Manifestations include mastoiditis, aphthous ulcers, cheilitis, gingivitis, esophagitis, gastritis, duodenitis, and meningitis. Levels of absolute lymphocytes and serum immunoglobulins are normal, but specific antibody titers are low despite immunization, and T-cells show impaired proliferative responses to mitogens. {ECO:0000269|PubMed:23727036}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: A chromosomal aberration involving MALT1 is recurrent in low-grade mucosa-associated lymphoid tissue (MALT lymphoma). Translocation t(11;18)(q21;q21) with BIRC2. This translocation is found in approximately 50% of cytogenetically abnormal low-grade MALT lymphoma. {ECO:0000269|PubMed:10339464, ECO:0000269|PubMed:10523859, ECO:0000269|PubMed:10702396, ECO:0000269|PubMed:11090634}. Drugs (DrugBank ID) DB01005; DB03904 Interacts with Q8WVV5; Q9Y3D6; Q8WWP7; P30301; Q5QGT7; Q6UX34; P0DN84; Q9C0I4; Q5BJF2 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Blood group antigen; Cell membrane; Glycoprotein; Membrane; Proteomics identification; Reference proteome; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A Molecular weight (Da) 38862.8 Length 356 Aromaticity 0.12 Instability index 40.98 Isoelectric point 7.67 Charge (pH=7) 0.95 2D Binding mode Binding energy (Kcal/mol) -9.69  Molscript Map 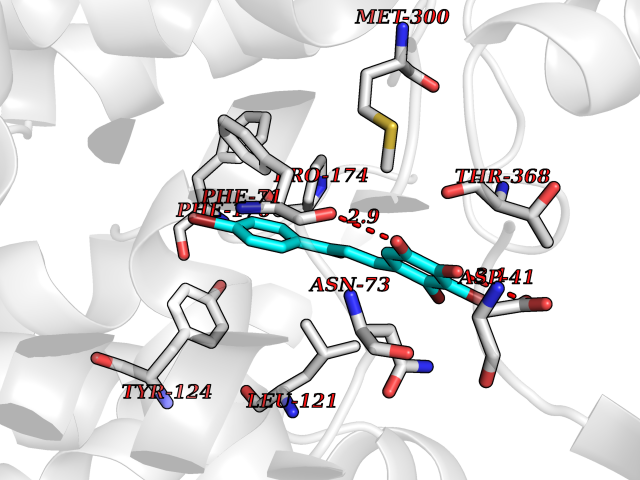 Pymol Map 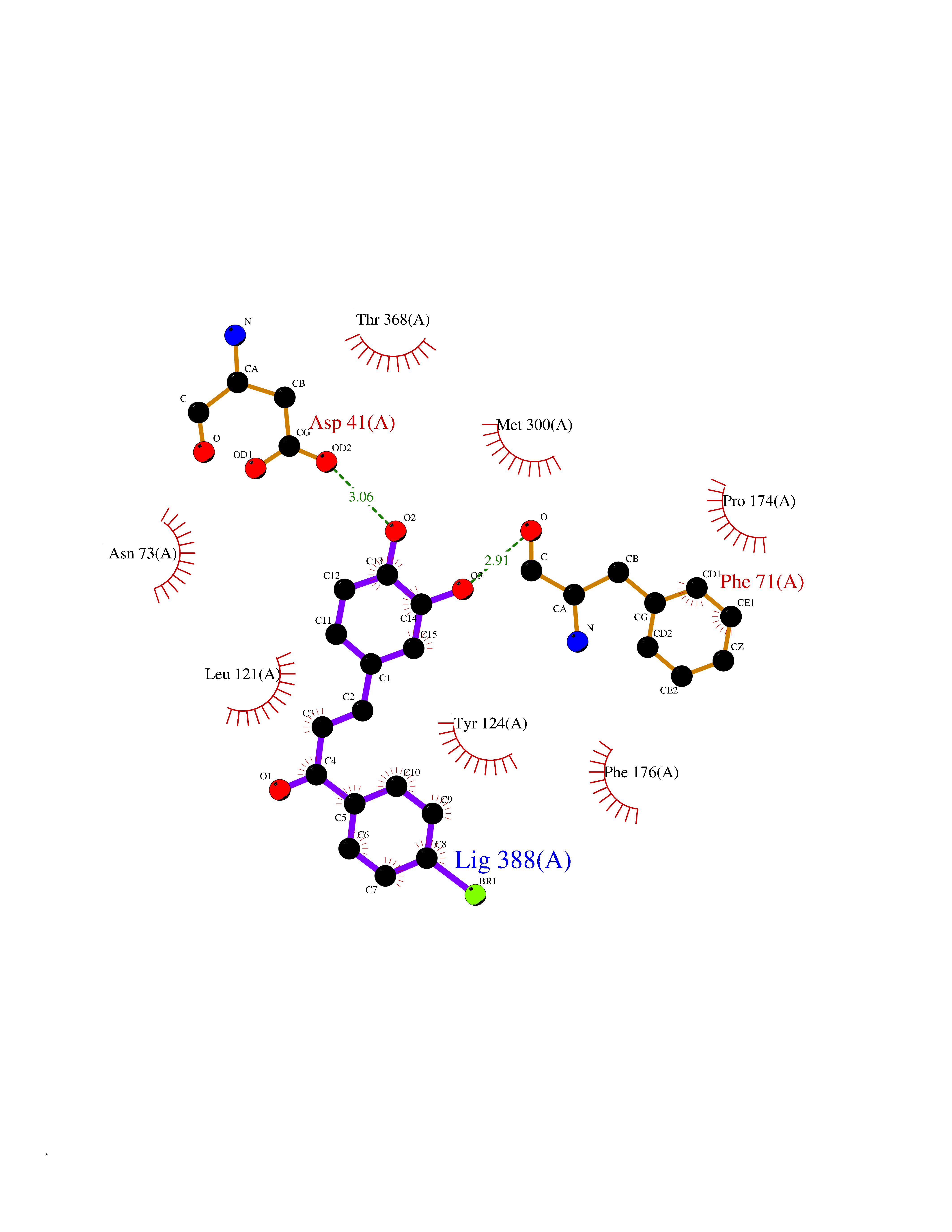 Ligplot Map 3D Binding mode Sequence FPKALGYVTGDMKELANQLKDKPVVLQFIDWILRGISQVVFVNNPVSGILILVGLLVQNPWWALTGWLGTVVSTLMALLLSQDRSLIASGLYGYNATLVGVLMAVFSDKGDYFWWLLLPVCAMSMTCPIFSSALNSVLSKWDLPVFTLPFNMALSMYLSATGHYNPFFPAKLVIPITTAPQISWSDLSALELLKSIPVGVGQIYGCDNPWTGGIFLGAILLSSPLMCLHAAIGSLLGIAAGLSLSAPFENIYFGLWGFNSSLACIAMGGMFMALTWQTHLLALGCALFTAYLGVGMANFMAEVGLPACTWPFCLATLLFLIMTTKNSNIYKMPLSKVTYPEENRIFYLQAKKRMVE Hydrogen bonds contact Hydrophobic contact | ||||
| 94 | MAP kinase signal-integrating kinase 2 (MKNK2) | 6CK6 | 7.11 | |
Target general information Gen name MKNK2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Mnk2; MAPK signal-integrating kinase 2 Protein family Protein kinase superfamily, CAMK Ser/Thr protein kinase family Biochemical class Protein kinase superfamily. CAMK Ser/Thr protein kinase family Function Serine/threonine-protein kinase that phosphorylates SFPQ/PSF, HNRNPA1 and EIF4E. May play a role in the response to environmental stress and cytokines. Appears to regulate translation by phosphorylating EIF4E, thus increasing the affinity of this protein for the 7-methylguanosine-containing mRNA cap. Required for mediating PP2A-inhibition-induced EIF4E phosphorylation. Triggers EIF4E shuttling from cytoplasm to nucleus. Isoform 1 displays a high basal kinase activity, but isoform 2 exhibits a very low kinase activity. Acts as a mediator of the suppressive effects of IFNgamma on hematopoiesis. Negative regulator for signals that control generation of arsenic trioxide As(2)O(3)-dependent apoptosis and anti-leukemic responses. Involved in anti-apoptotic signaling in response to serum withdrawal. Related diseases Charcot-Marie-Tooth disease, X-linked dominant, 6 (CMTX6) [MIM:300905]: A form of Charcot-Marie-Tooth disease, a disorder of the peripheral nervous system, characterized by progressive weakness and atrophy, initially of the peroneal muscles and later of the distal muscles of the arms. Charcot-Marie-Tooth disease is classified in two main groups on the basis of electrophysiologic properties and histopathology: primary peripheral demyelinating neuropathies characterized by severely reduced motor nerve conduction velocities (NCVs) (less than 38m/s) and segmental demyelination and remyelination, and primary peripheral axonal neuropathies characterized by normal or mildly reduced NCVs and chronic axonal degeneration and regeneration on nerve biopsy. {ECO:0000269|PubMed:23297365}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB12010 Interacts with Q16539; P46379-2; O14901; Q14696; P25786 EC number EC 2.7.11.1 Uniprot keywords 3D-structure; Alternative splicing; Apoptosis; ATP-binding; Cytoplasm; Kinase; Magnesium; Metal-binding; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Serine/threonine-protein kinase; Transferase; Translation regulation; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 30812.8 Length 271 Aromaticity 0.1 Instability index 60.57 Isoelectric point 5.48 Charge (pH=7) -10.33 2D Binding mode Binding energy (Kcal/mol) -9.69 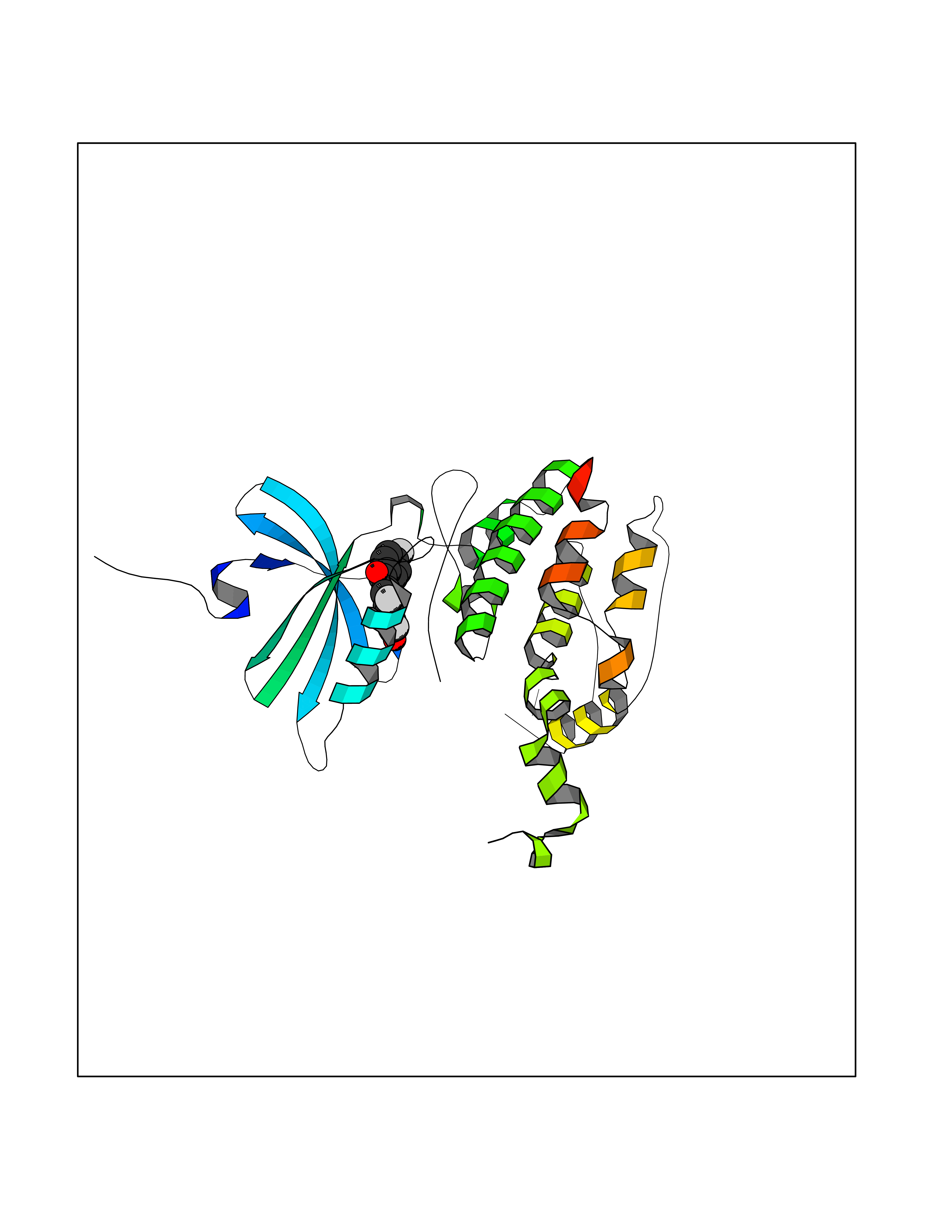 Molscript Map 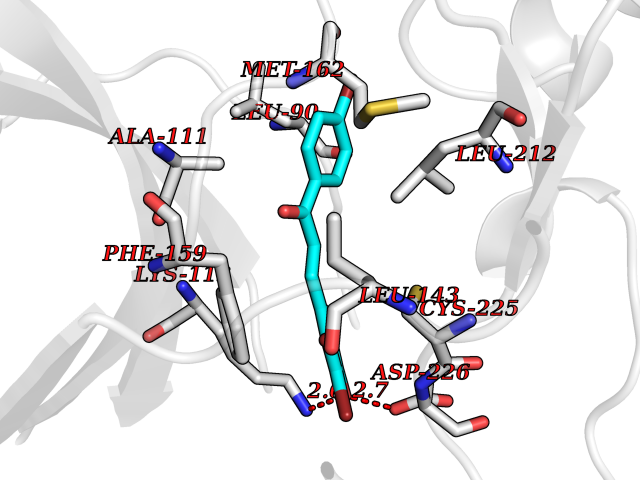 Pymol Map 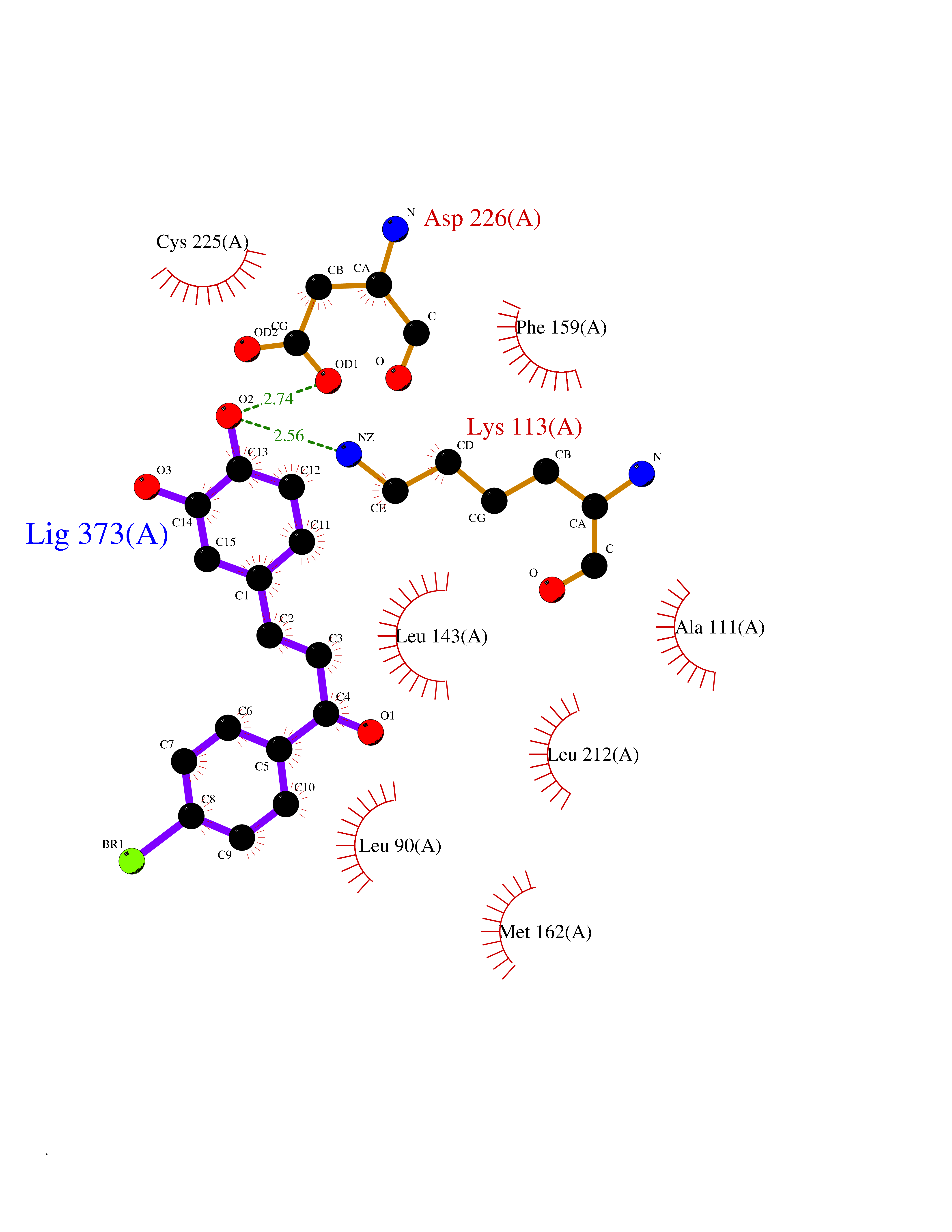 Ligplot Map 3D Binding mode Sequence GSTDSFSGRFEDVYQLQEDVLGEGAHARVQTCINLITSQEYAVKIIEKQPGHIRSRVFREVEMLYQCQGHRNVLELIEFFEEEDRFYLVFEKMRGGSILSHIHKRRHFNELEASVVVQDVASALDFLHNKGIAHRDLKPENILCEHPNQVSPVKICDFGGSAEYMAPEVVEAFSEEASIYDKRCDLWSLGVILYILLSGYPPFVGRCCGACPACQNMLFESIQEGKYEFPDKDWAHISCAAKDLISKLLVRDAKQRLSAAQVLQHPWVQGC Hydrogen bonds contact Hydrophobic contact | ||||
| 95 | Non-heme chloroperoxidase | 1A8U | 7.10 | |
Target general information Gen name cpo Organism Kitasatospora aureofaciens (Streptomyces aureofaciens) Uniprot ID TTD ID NA Synonyms cpoT Protein family AB hydrolase superfamily, Bacterial non-heme haloperoxidase / perhydrolase family Biochemical class Haloperoxidase Function Chloride peroxidase activity. Related diseases Hypervalinemia and hyperleucine-isoleucinemia (HVLI) [MIM:618850]: An autosomal recessive metabolic disorder characterized by highly elevated plasma concentrations of valine and leucine/isoleucine. Affected individuals suffer from headache and mild memory impairment. {ECO:0000269|PubMed:25653144}. The disease is caused by variants affecting the gene represented in this entry. A patient with hypervalinemia and hyperleucine-isoleucinemia was identified as compound heterozygote for Gln-170 (inherited from his father) and Lys-264 (inherited from his mother), both variants reduced the catalytic activity of the enzyme. After treatment with vitamin B6, a precursor of pyridoxal 5'-phosphate, a BCAT2 cofactor, the blood levels of branched chain amino acids, especially valine, were decreased and brain lesions were improved. {ECO:0000269|PubMed:25653144}. Drugs (DrugBank ID) DB03793 Interacts with NA EC number 1.11.1.- Uniprot keywords 3D-structure; Chloride; Oxidoreductase; Peroxidase Protein physicochemical properties Chain ID A,B Molecular weight (Da) 60428.4 Length 554 Aromaticity 0.13 Instability index 31.26 Isoelectric point 4.65 Charge (pH=7) -32.72 2D Binding mode Binding energy (Kcal/mol) -9.69 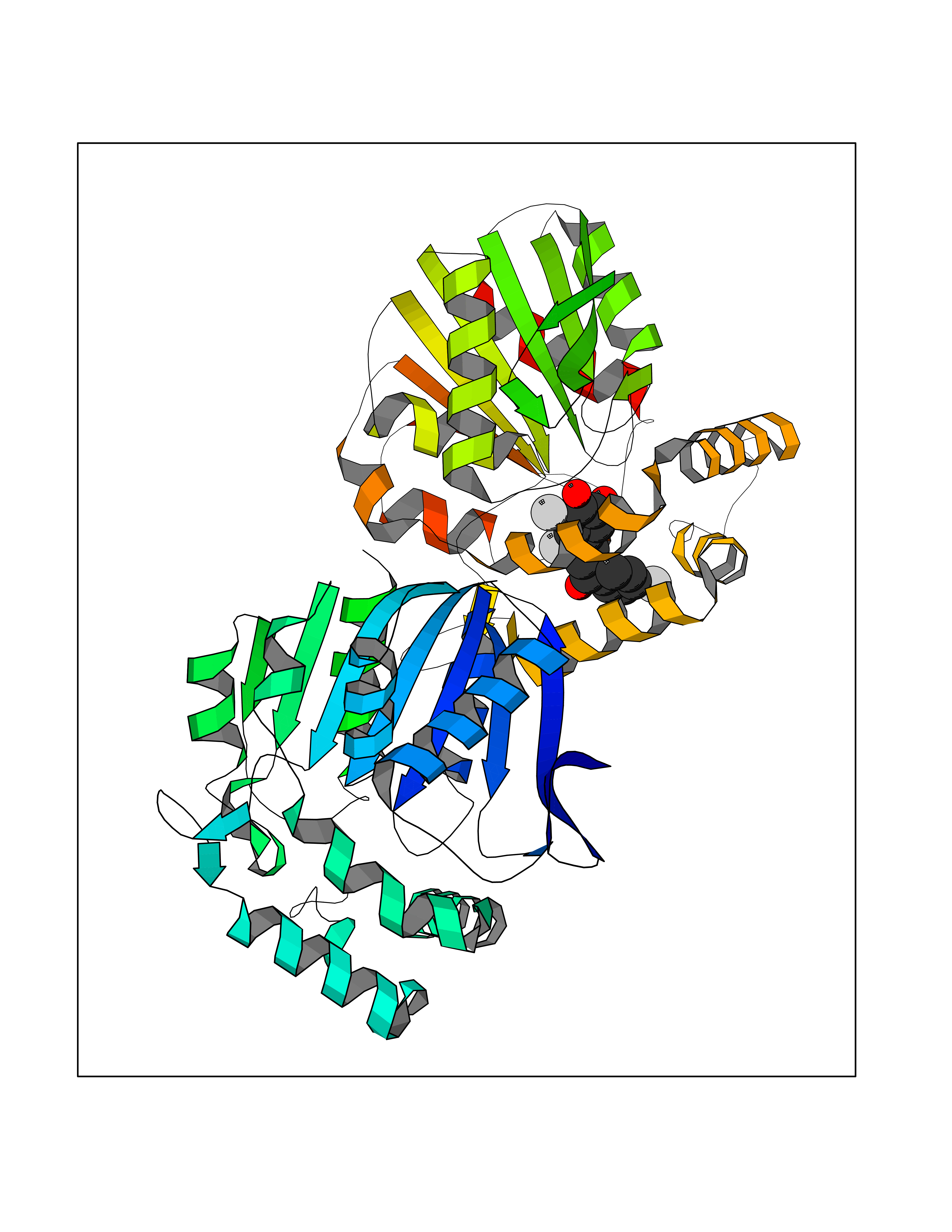 Molscript Map 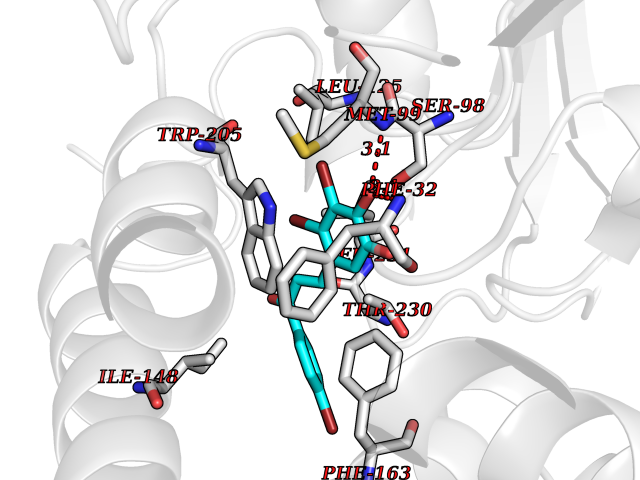 Pymol Map 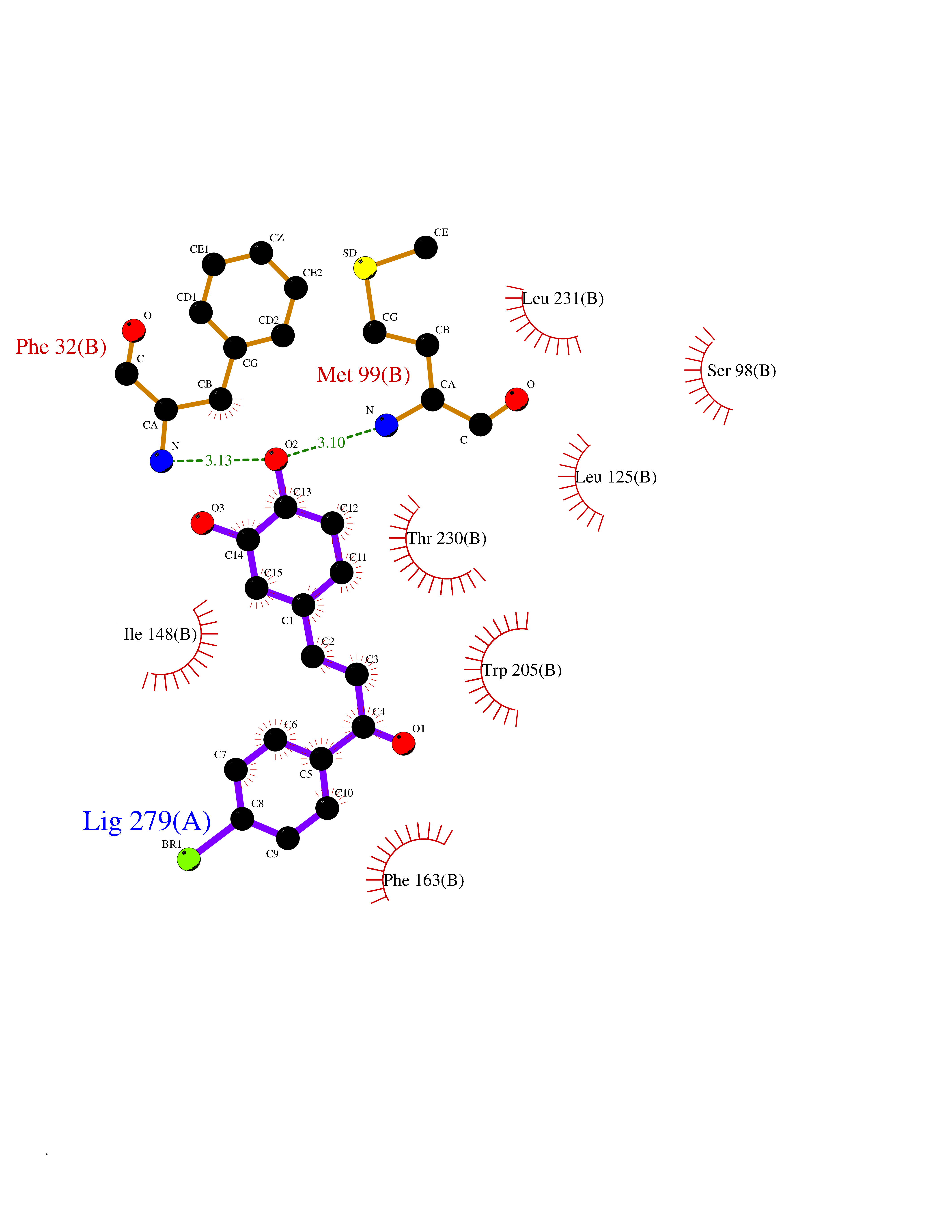 Ligplot Map 3D Binding mode Sequence PFITVGQENSTSIDLYYEDHGAGQPVVLIHGFPLSGHSWERQSAALLDAGYRVITYDRRGFGQSSQPTTGYDYDTFAADLNTVLETLDLQDAVLVGFSMGTGEVARYVSSYGTARIAKVAFLASLEPFLLKTDDNPDGAAPKEFFDGIVAAVKADRYAFYTGFFNDFYNLDENLGTRISEEAVRNSWNTAASGGFFAAAAAPTTWYTDFRADIPRIDVPALILHGTGDRTLPIENTARVFHKALPSAEYVEVEGAPHGLLWTHAEEVNTALLAFLAKPFITVGQENSTSIDLYYEDHGAGQPVVLIHGFPLSGHSWERQSAALLDAGYRVITYDRRGFGQSSQPTTGYDYDTFAADLNTVLETLDLQDAVLVGFSMGTGEVARYVSSYGTARIAKVAFLASLEPFLLKTDDNPDGAAPKEFFDGIVAAVKADRYAFYTGFFNDFYNLDENLGTRISEEAVRNSWNTAASGGFFAAAAAPTTWYTDFRADIPRIDVPALILHGTGDRTLPIENTARVFHKALPSAEYVEVEGAPHGLLWTHAEEVNTALLAFLAK Hydrogen bonds contact Hydrophobic contact | ||||
| 96 | Retinol-binding protein 1 | 5HBS | 7.10 | |
Target general information Gen name RBP1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms CRBP1 Protein family Calycin superfamily, Fatty-acid binding protein (FABP) family Biochemical class Retinol-binding protein Function Retinal binding.Retinoid binding.Retinol binding.Transporter activity. Related diseases Orthostatic hypotension 1 (ORTHYP1) [MIM:223360]: A form of orthostatic hypotension due to congenital dopamine beta-hydroxylase deficiency. Orthostatic hypotension, also known as postural hypotension, is a finding defined as a 20-mm Hg decrease in systolic pressure or a 10-mm Hg decrease in diastolic pressure occurring 3 minutes after a person has risen from supine to standing. Symptoms include dizziness, blurred vision, and sometimes syncope. ORTHYP1 is an autosomal recessive condition apparent from infancy or early childhood and characterized by low plasma and urinary levels of norepinephrine and epinephrine, and episodic hypoglycemia. {ECO:0000269|PubMed:11857564}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00459; DB06755; DB00162 Interacts with P49366; Q9UBN6; Q6DKK2; Q8N2K1 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; Lipid droplet; Methylation; Proteomics identification; Reference proteome; Retinol-binding; Transport; Vitamin A Protein physicochemical properties Chain ID A Molecular weight (Da) 16404.5 Length 139 Aromaticity 0.09 Instability index 16.54 Isoelectric point 5.62 Charge (pH=7) -5.43 2D Binding mode Binding energy (Kcal/mol) -9.68  Molscript Map 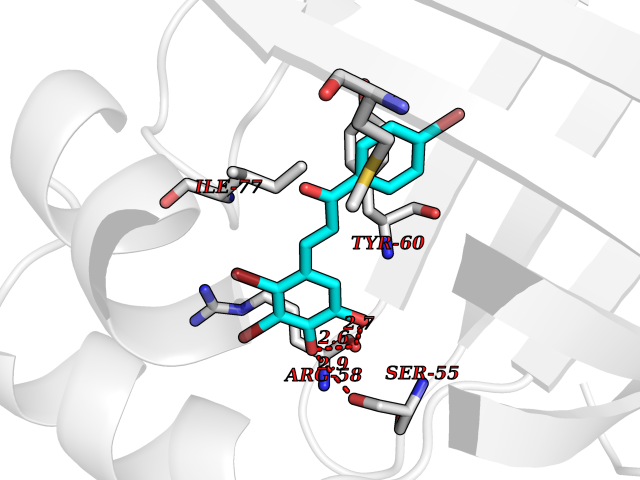 Pymol Map 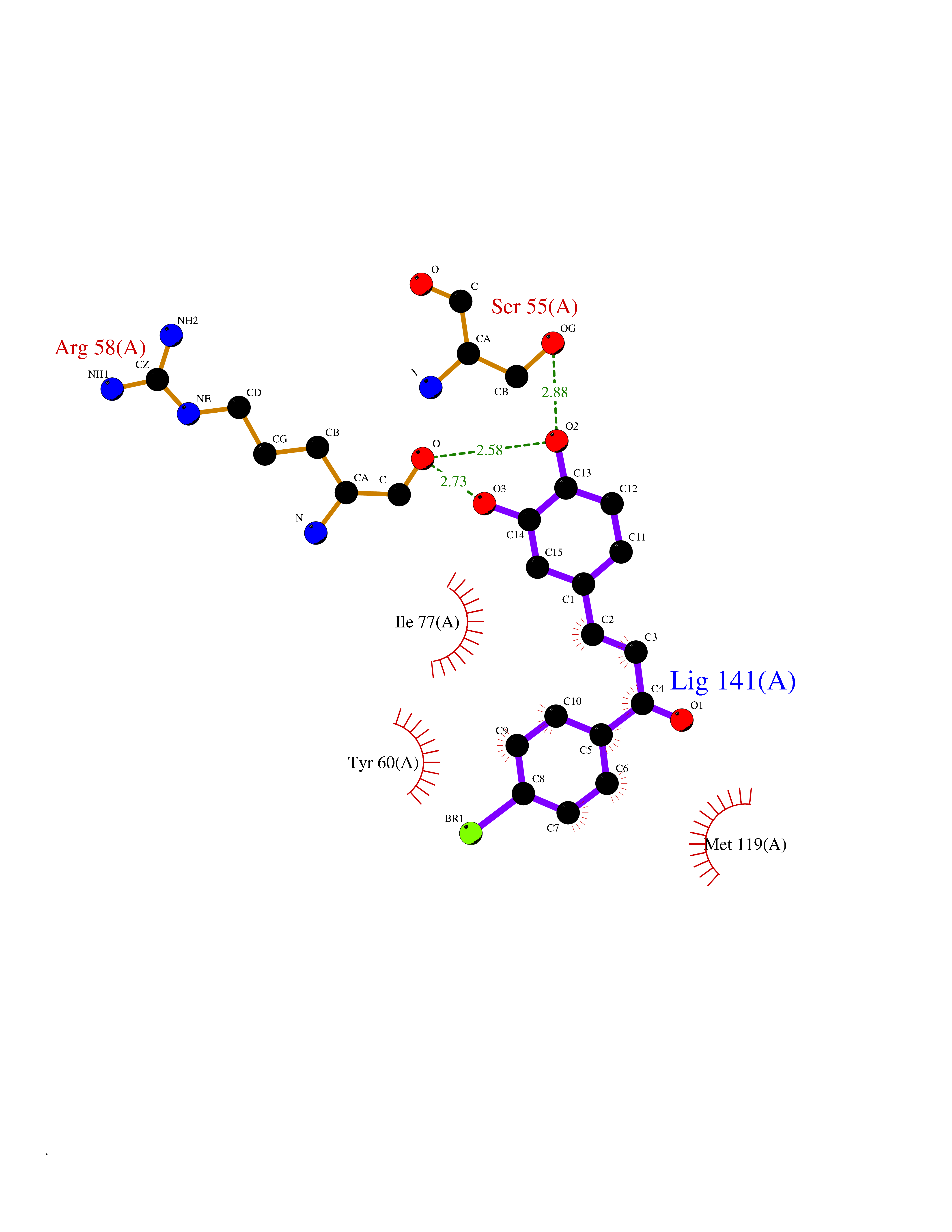 Ligplot Map 3D Binding mode Sequence PVDFTGYWKMLVNENFEEYLRALDVNVALRKIANLLKPDKEIVQDGDHMIIRTLSTFRNYIMDFQVGKEFEEDLTGIDDRKCMTTVSWDGDKLQCVQKGEKEGRGWTQWIEGDELHLEMRVEGVVCKQVFKKVQHHHHH Hydrogen bonds contact Hydrophobic contact | ||||
| 97 | Monoamine oxidase type B (MAO-B) | 2V5Z | 7.10 | |
Target general information Gen name MAOB Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms MAO-B; Amine oxidase [flavin-containing] B Protein family Flavin monoamine oxidase family Biochemical class CH-NH(2) donor oxidoreductase Function Catalyzes the oxidative deamination of biogenic and xenobiotic amines and has important functions in the metabolism of neuroactive and vasoactive amines in the central nervous system and peripheral tissues. MAOB preferentially degrades benzylamine and phenylethylamine. Related diseases Microvascular complications of diabetes 5 (MVCD5) [MIM:612633]: Pathological conditions that develop in numerous tissues and organs as a consequence of diabetes mellitus. They include diabetic retinopathy, diabetic nephropathy leading to end-stage renal disease, and diabetic neuropathy. Diabetic retinopathy remains the major cause of new-onset blindness among diabetic adults. It is characterized by vascular permeability and increased tissue ischemia and angiogenesis. Disease susceptibility is associated with variants affecting the gene represented in this entry. Homozygosity for the Leu-55 allele is strongly associated with the development of retinal disease in diabetic patients. Drugs (DrugBank ID) DB08176; DB02211; DB08516; DB08480; DB01472; DB04307; DB07512; DB07513; DB00915; DB00182; DB06698; DB04889; DB00215; DB09130; DB04147; DB00988; DB01363; DB00668; DB01175; DB02509; DB03147; DB14914; DB00614; DB04818; DB02095; DB01247; DB00601; DB01577; DB01442; DB01171; DB08082; DB02643; DB04677; DB03894; DB08804; DB04820; DB00184; DB04821; DB12612; DB01626; DB00780; DB00191; DB00388; DB01132; DB00721; DB01168; DB01367; DB09363; DB06654; DB01037; DB01104; DB14569; DB09042; DB00752; DB16446; DB09185; DB04832; DB00909 Interacts with P55212; P28329-3; Q8NI60; Q5RI15; Q92915-2; P22607; Q53GS7; P06396; P01112; O14901; P13473-2; P21397; Q9BVL2; O75400-2; P62826; Q6NTF9-3; Q9Y371; Q7Z699; Q9UMX0; Q9Y649 EC number EC 1.4.3.4 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Direct protein sequencing; FAD; Flavoprotein; Membrane; Mitochondrion; Mitochondrion outer membrane; Oxidoreductase; Proteomics identification; Reference proteome; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A,B Molecular weight (Da) 56019.9 Length 494 Aromaticity 0.09 Instability index 34.81 Isoelectric point 6.51 Charge (pH=7) -2.2 2D Binding mode Binding energy (Kcal/mol) -9.69 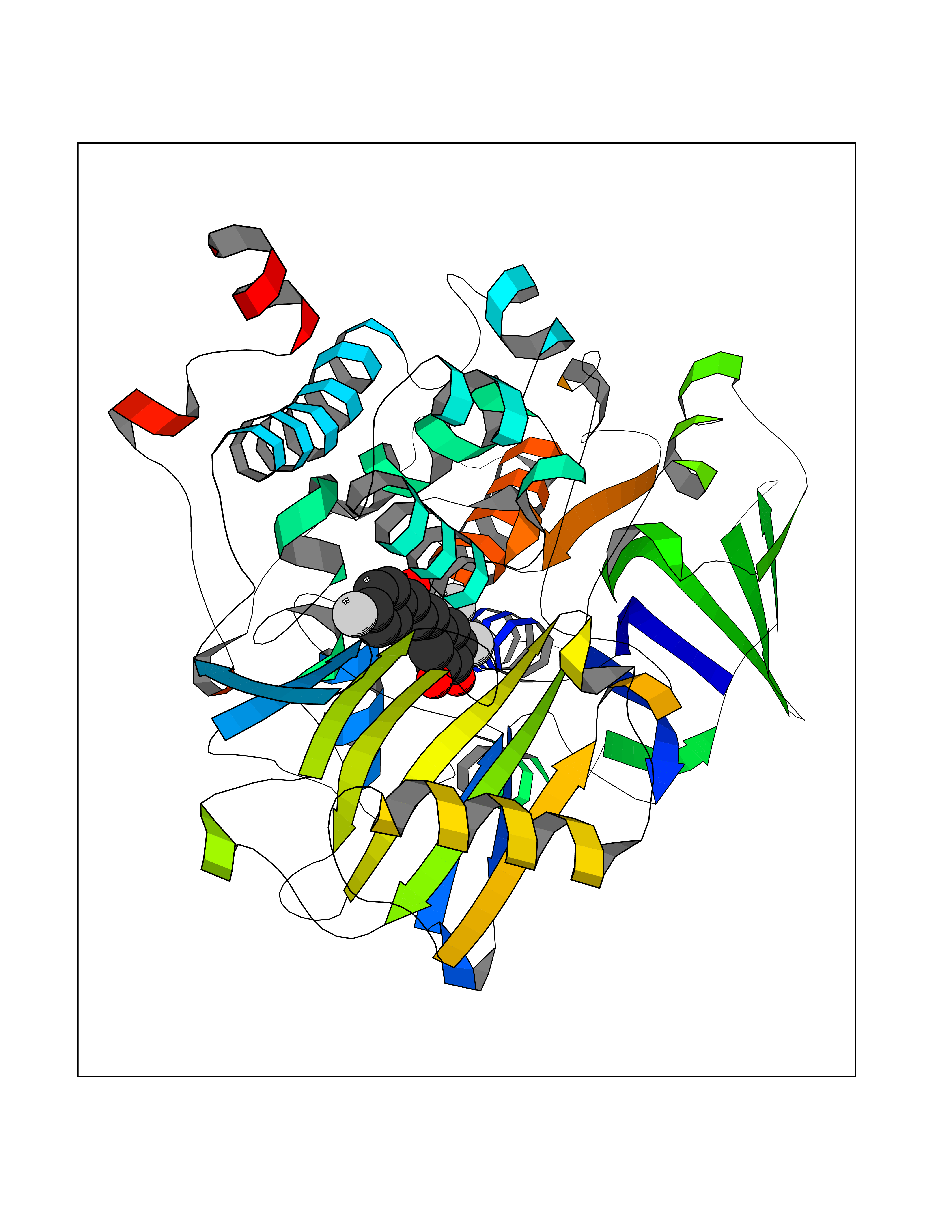 Molscript Map 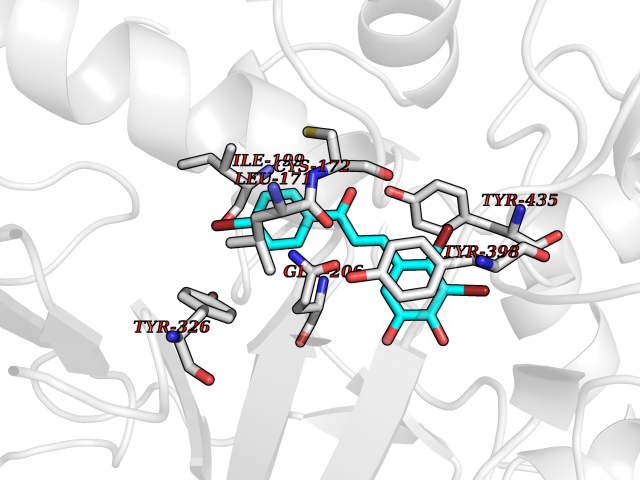 Pymol Map 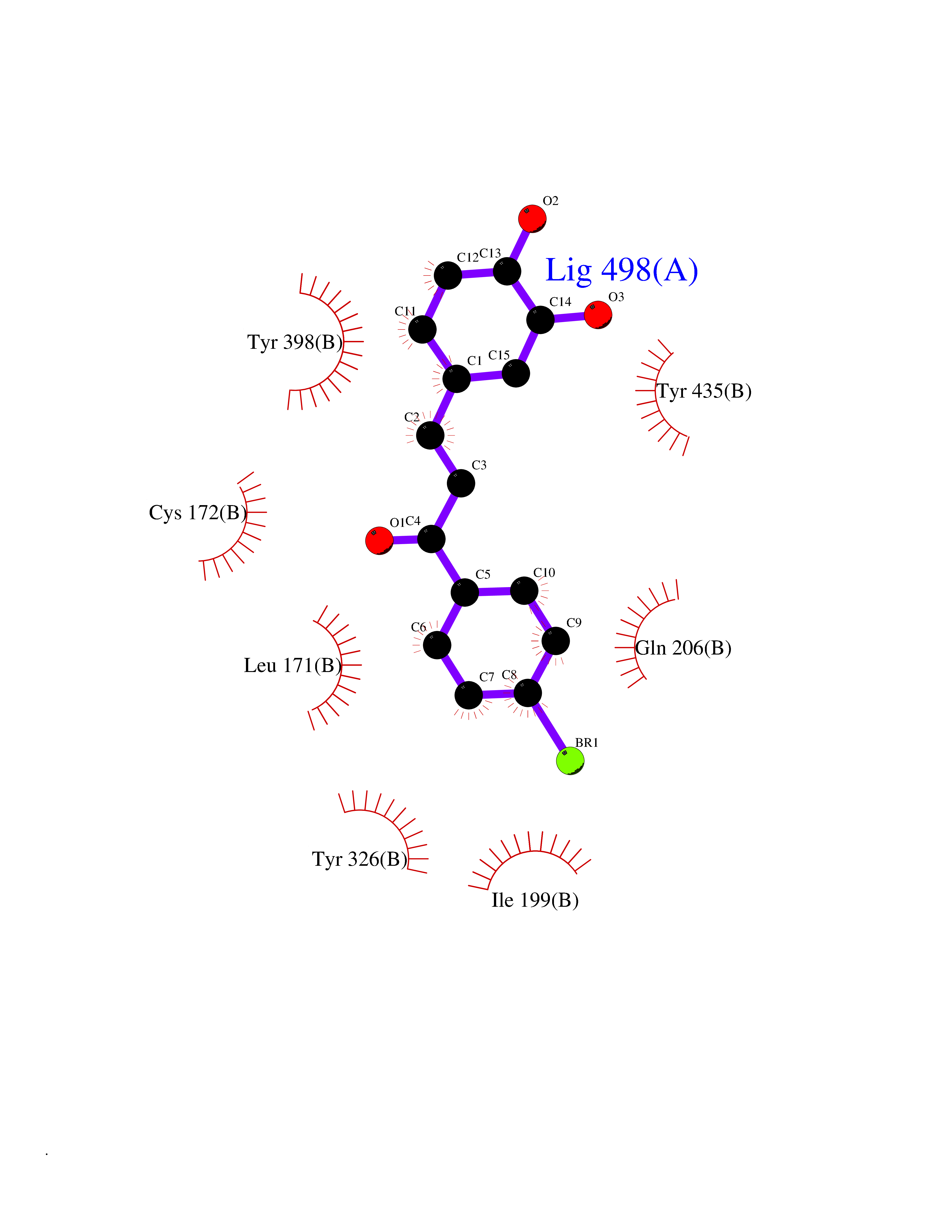 Ligplot Map 3D Binding mode Sequence NKCDVVVVGGGISGMAAAKLLHDSGLNVVVLEARDRVGGRTYTLRNQKVKYVDLGGSYVGPTQNRILRLAKELGLETYKVNEVERLIHHVKGKSYPFRGPFPPVWNPITYLDHNNFWRTMDDMGREIPSDAPWKAPLAEEWDNMTMKELLDKLCWTESAKQLATLFVNLCVTAETHEVSALWFLWYVKQCGGTTRIISTTNGGQERKFVGGSGQVSERIMDLLGDRVKLERPVIYIDQTRENVLVETLNHEMYEAKYVISAIPPTLGMKIHFNPPLPMMRNQMITRVPLGSVIKCIVYYKEPFWRKKDYCGTMIIDGEEAPVAYTLDDTKPEGNYAAIMGFILAHKARKLARLTKEERLKKLCELYAKVLGSLEALEPVHYEEKNWCEEQYSGGCYTTYFPPGILTQYGRVLRQPVDRIYFAGTETATHWSGYMEGAVEAGERAAREILHAMGKIPEDEIWQSEPESVDVPAQPITTTFLERHLPSVPGLLRLI Hydrogen bonds contact Hydrophobic contact | ||||
| 98 | Dihydroorotate dehydrogenase (DHODH) | 4OQV | 7.10 | |
Target general information Gen name DHODH Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Dihydroorotate oxidase; Dihydroorotate dehydrogenase (quinone), mitochondrial; DHOdehase; DHODH Protein family Dihydroorotate dehydrogenase family, Type 2 subfamily Biochemical class CH-CH donor oxidoreductase Function Catalyzes the conversion of dihydroorotate to orotate with quinone as electron acceptor. Related diseases Postaxial acrofacial dysostosis (POADS) [MIM:263750]: POADS is characterized by severe micrognathia, cleft lip and/or palate, hypoplasia or aplasia of the posterior elements of the limbs, coloboma of the eyelids and supernumerary nipples. POADS is a very rare disorder: only 2 multiplex families, each consisting of 2 affected siblings born to unaffected, nonconsanguineous parents, have been described among a total of around 30 reported cases. {ECO:0000269|PubMed:19915526}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07559; DB07561; DB08172; DB08169; DB07443; DB07978; DB07975; DB04281; DB08249; DB07977; DB07976; DB04583; DB08008; DB01117; DB03523; DB03480; DB02613; DB04147; DB03247; DB01097; DB06481; DB08006; DB02262; DB05125; DB08880; DB07646 Interacts with Q6ZMZ0; P49638 EC number EC 1.3.5.2 Uniprot keywords 3D-structure; Disease variant; Flavoprotein; FMN; Membrane; Mitochondrion; Mitochondrion inner membrane; Oxidoreductase; Proteomics identification; Pyrimidine biosynthesis; Reference proteome; Transit peptide; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 38341.4 Length 353 Aromaticity 0.05 Instability index 39.27 Isoelectric point 9.28 Charge (pH=7) 5.52 2D Binding mode Binding energy (Kcal/mol) -9.68  Molscript Map 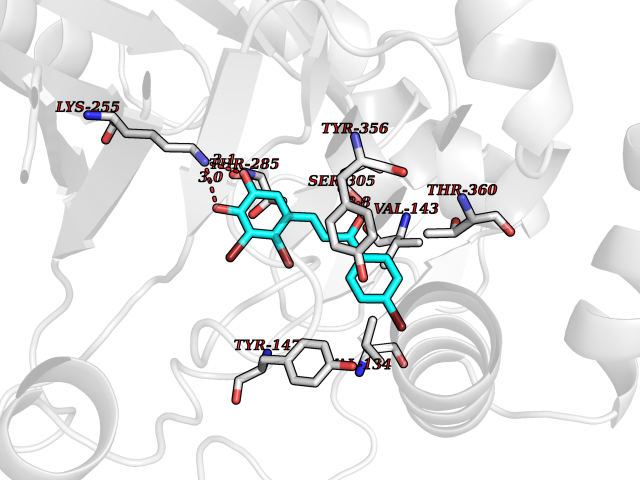 Pymol Map 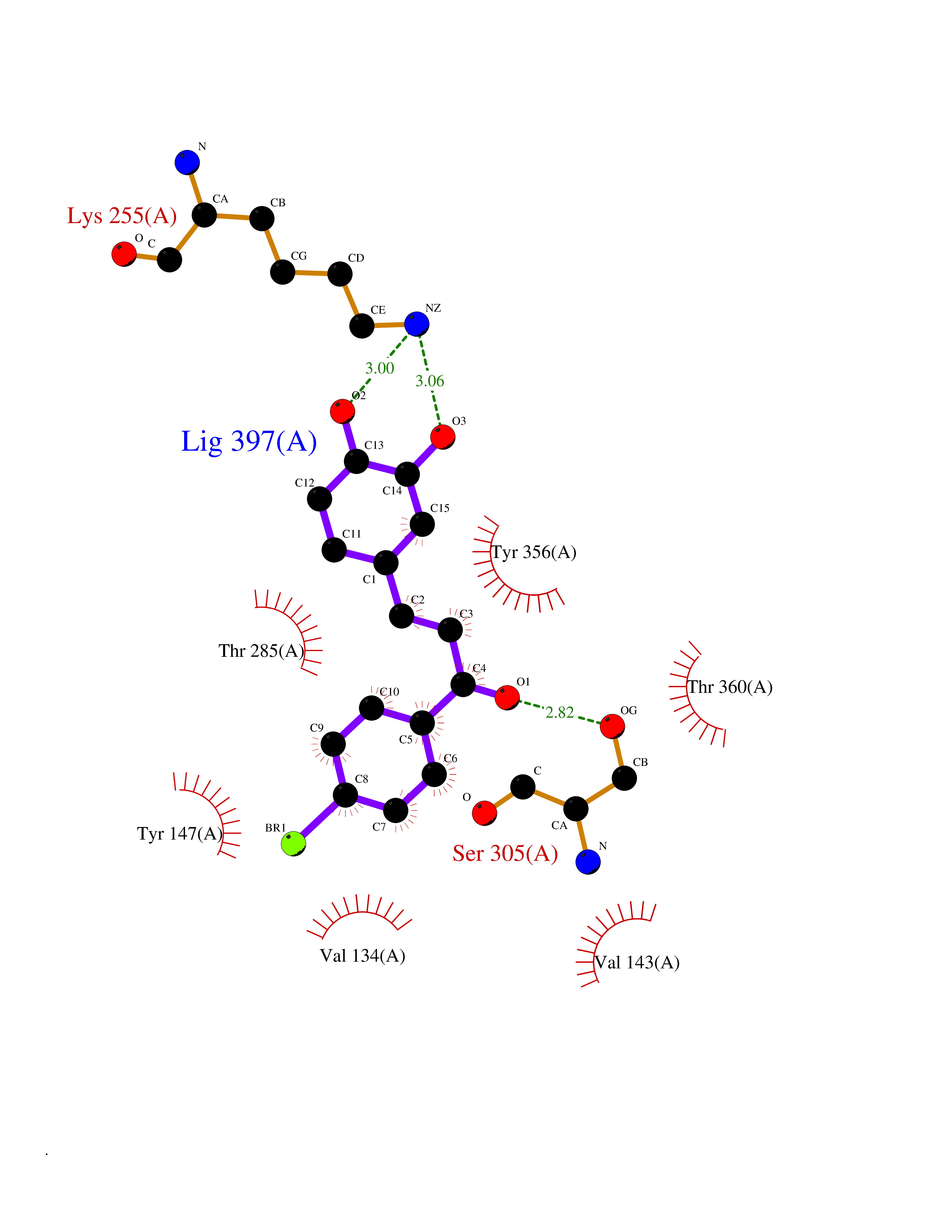 Ligplot Map 3D Binding mode Sequence DERFYAEHLMPTLQGLLDPESAHRLAVRFTSLGLLPRARFQDSDMLEVRVLGHKFRNPVGIAAGFDKHGEAVDGLYKMGFGFVEIGSVTPKPQEGNPRPRVFRLPEDQAVINRYGFNSHGLSVVEHRLRARQQKQAKLTEDGLPLGVNLGKNKTSVDAAEDYAEGVRVLGPLADYLVVNVSSPGKAELRRLLTKVLQERDGLRRVHRPAVLVKIAPDLTSQDKEDIASVVKELGIDGLIVTNTTVSRPAGLQGALRSETGGLSGKPLRDLSTQTIREMYALTQGRVPIIGVGGVSSGQDALEKIRAGASLVQLYTALTFWGPPVVGKVKRELEALLKEQGFGGVTDAIGADHR Hydrogen bonds contact Hydrophobic contact | ||||
| 99 | Lysine-specific histone demethylase 1 (LSD) | 6W4K | 7.10 | |
Target general information Gen name KDM1A Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Lysine-specific histone demethylase 1A; LSD1; KIAA0601; KDM1; Flavin-containing amine oxidase domain-containing protein 2; BRAF35-HDAC complex protein BHC110; AOF2 Protein family Flavin monoamine oxidase family Biochemical class CH-NH(2) donor oxidoreductase Function Histone demethylase that can demethylate both 'Lys-4' (H3K4me) and 'Lys-9' (H3K9me) of histone H3, thereby acting as a coactivator or a corepressor, depending on the context. Acts by oxidizing the substrate by FAD to generate the corresponding imine that is subsequently hydrolyzed. Acts as a corepressor by mediating demethylation of H3K4me, a specific tag for epigenetic transcriptional activation. Demethylates both mono- (H3K4me1) and di-methylated (H3K4me2) H3K4me. May play a role in the repression of neuronal genes. Alone, it is unable to demethylate H3K4me on nucleosomes and requires the presence of RCOR1/CoREST to achieve such activity. Also acts as a coactivator of androgen receptor (ANDR)-dependent transcription, by being recruited to ANDR target genes and mediating demethylation of H3K9me, a specific tag for epigenetic transcriptional repression. The presence of PRKCB in ANDR-containing complexes, which mediates phosphorylation of 'Thr-6' of histone H3 (H3T6ph), a specific tag that prevents demethylation H3K4me, prevents H3K4me demethylase activity of KDM1A. Demethylates di-methylated 'Lys-370' of p53/TP53 which prevents interaction of p53/TP53 with TP53BP1 and represses p53/TP53-mediated transcriptional activation. Demethylates and stabilizes the DNA methylase DNMT1. Required for gastrulation during embryogenesis. Component of a RCOR/GFI/KDM1A/HDAC complex that suppresses, via histone deacetylase (HDAC) recruitment, a number of genes implicated in multilineage blood cell development. Effector of SNAI1-mediated transcription repression of E-cadherin/CDH1, CDN7 and KRT8. Required for the maintenance of the silenced state of the SNAI1 target genes E-cadherin/CDH1 and CDN7. Related diseases Cleft palate, psychomotor retardation, and distinctive facial features (CPRF) [MIM:616728]: A syndrome characterized by cleft palate, developmental delay, psychomotor retardation, and facial dysmorphic features including a prominent forehead, slightly arched eyebrows, elongated palpebral fissures, a wide nasal bridge, thin lips, and widely spaced teeth. Cleft palate is a congenital fissure of the soft and/or hard palate, due to faulty fusion. {ECO:0000269|PubMed:23020937, ECO:0000269|PubMed:24838796, ECO:0000269|PubMed:26656649}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB16446 Interacts with Q9BYF1; Q99996; Q86SG2; Q13490; Q6P047; Q8TC20-4; Q9BWT7; Q49A88-3; Q99459; Q9BXL8; Q86XR8-3; Q8NHQ1; Q8TAP6; Q12873; P38432; Q96EY1; P26378-2; Q9UPT5-1; Q3B820; Q9H8W3; Q8IZU1; Q9BQS8; O95995; Q96CN9; Q08379; Q9NYA3; Q8NEC7; Q9BX10; Q16695; Q96CS2; O15379; Q9UBX0; Q9NSC5; Q16891; O75564-2; O60341; Q9BVG8-5; Q8TBB5; P19012; Q15323; Q14525; Q92764; Q6A163; Q6A162; Q96JB6; Q9Y250; O95983; P01106; Q3BBV0; Q7Z6G3-2; P35240; Q16236; P46531; Q13133; Q9Y466; Q96F24; A5D8V7; Q8IZS5; Q9BYU1; Q8IZL8; Q99471; Q5T6S3; Q03181; P62191; Q9NS23; P50749; Q86WH2; Q06330; Q9UKL0; Q8IZ40; Q9P2K3-2; Q96P16-3; Q8N6K7; Q9UGK8; Q13435; O15198; O95863; Q96H20; Q8N0Z3; Q96BD6; Q8N4C7; Q8N6V9; Q9UBB9; Q08117-2; Q2M3C6; P45379-11; Q05BL1; Q9BUZ4; Q5W5X9-3; Q8TF42; Q9H9H4; Q9Y3C0; O96006; O15060; Q92618; O14646; Q96KQ7; Q96KQ7-1; P05771-1; Q9UKL0; P17542; P04637; P22091 EC number EC 1.-.-.- Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Chromatin regulator; Chromosome; Coiled coil; Developmental protein; Direct protein sequencing; Disease variant; FAD; Flavoprotein; Isopeptide bond; Nucleus; Oxidoreductase; Phosphoprotein; Proteomics identification; Reference proteome; Repressor; Transcription; Transcription regulation; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 72462.2 Length 651 Aromaticity 0.08 Instability index 32.07 Isoelectric point 7.01 Charge (pH=7) 0.05 2D Binding mode Binding energy (Kcal/mol) -9.69  Molscript Map 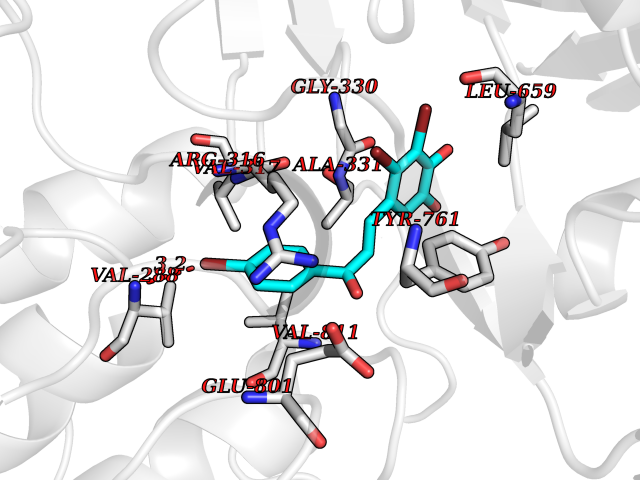 Pymol Map 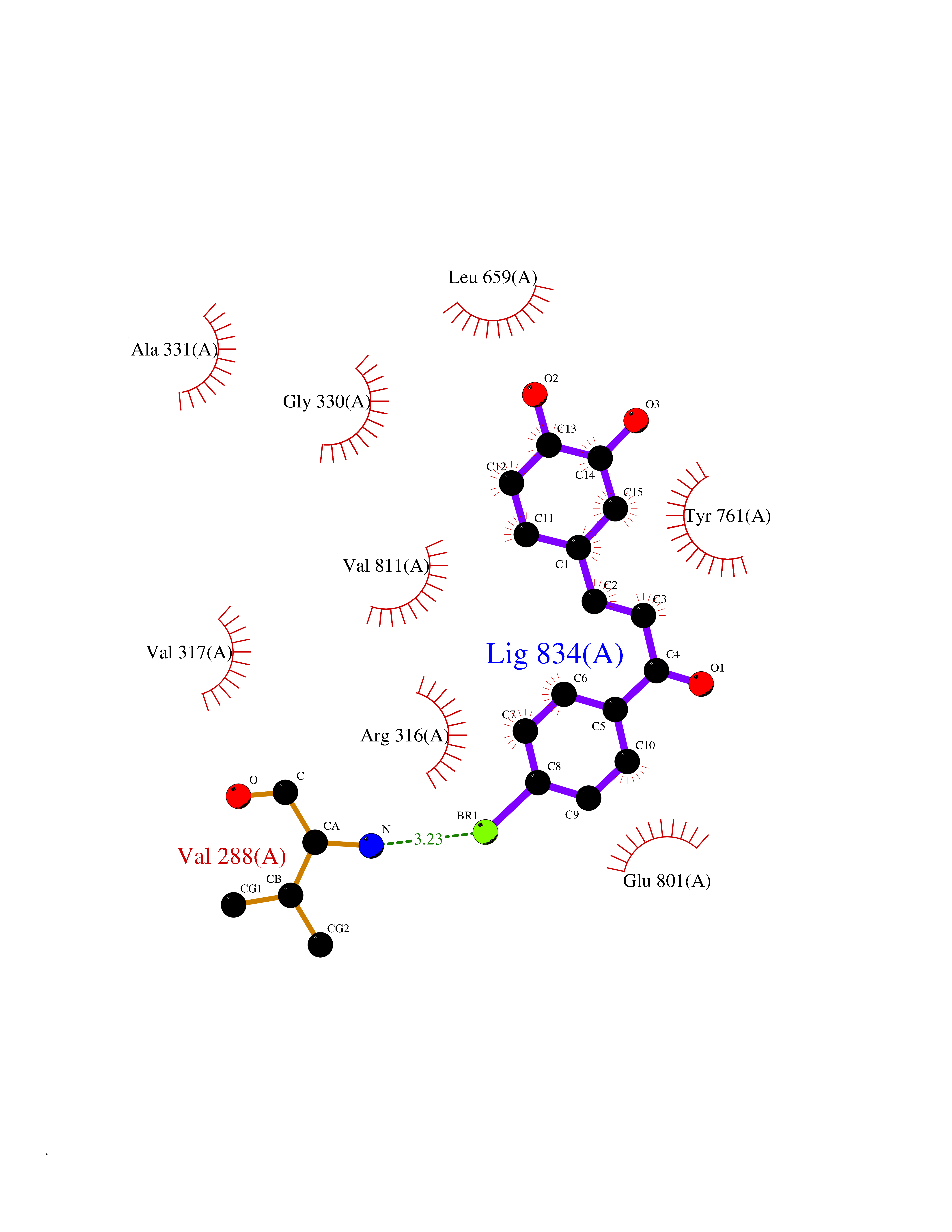 Ligplot Map 3D Binding mode Sequence VEGAAFQSRLPHDRMTSQEAACFPDIISGPQQTQKVFLFIRNRTLQLWLDNPKIQLTFEATLQQLEAPYNSDTVLVHRVHSYLERHGLINFGIYKRIKPLPTKKTGKVIIIGSGVSGLAAARQLQSFGMDVTLLEARDRVGGRVATFRKGNYVADLGAMVVTGLGGNPMMELAKIKQKCPLYEANGQAVPKEKDEMVEQEFNRLLEATSYLSHQLDFNVLNNKPVSLGQALEVVIQLQEKHVKDEQIEHWKKIVKTQEELKELLNKMVNLKEKIKELHQQYKEASEVKPPRDITAEFLVKSKHRDLTALCKEYDELAETQGKLEEKLQELEANPPSDVYLSSRDRQILDWHFANLEFANATPLSTLSLKHWDQDDDFEFTGSHLTVRNGYSCVPVALAEGLDIKLNTAVRQVRYTASGCEVIAVNTRSTSQTFIYKCDAVLCTLPLGVLKQQPPAVQFVPPLPEWKTSAVQRMGFGNLNKVVLCFDRVFWDPSVNLFGHVGSTTASRGELFLFWNLYKAPILLALVAGEAAGIMENISDDVIVGRCLAILKGIFGSSAVPQPKETVVSRWRADPWARGSYSYVAAGSSGNDYDLMAQPITPGPSIPGAPQPIPRLFFAGEHTIRNYPATVHGALLSGLREAGRIADQFLGA Hydrogen bonds contact Hydrophobic contact | ||||
| 100 | Histone-lysine N-methyltransferase SMYD3 (SMYD3) | 6P7Z | 7.10 | |
Target general information Gen name SMYD3 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms SET and MYND domain-containing protein 3; Zinc finger MYND domain-containing protein 1 Protein family Class V-like SAM-binding methyltransferase superfamily, Histone-lysine methyltransferase family Biochemical class NA Function Histone methyltransferase. Specifically methylates 'Lys-4' of histone H3, inducing di- and tri-methylation, but not monomethylation . Also methylates 'Lys-5' of histone H4. Plays an important role in transcriptional activation as a member of an RNA polymerase complex. Binds DNA containing 5'-CCCTCC-3' or 5'-GAGGGG-3' sequences. Related diseases Leukemia, juvenile myelomonocytic (JMML) [MIM:607785]: An aggressive pediatric myelodysplastic syndrome/myeloproliferative disorder characterized by malignant transformation in the hematopoietic stem cell compartment with proliferation of differentiated progeny. Patients have splenomegaly, enlarged lymph nodes, rashes, and hemorrhages. {ECO:0000269|PubMed:17332249}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Noonan syndrome 6 (NS6) [MIM:613224]: A form of Noonan syndrome, a disease characterized by short stature, facial dysmorphic features such as hypertelorism, a downward eyeslant and low-set posteriorly rotated ears, and a high incidence of congenital heart defects and hypertrophic cardiomyopathy. Other features can include a short neck with webbing or redundancy of skin, deafness, motor delay, variable intellectual deficits, multiple skeletal defects, cryptorchidism, and bleeding diathesis. Individuals with Noonan syndrome are at risk of juvenile myelomonocytic leukemia, a myeloproliferative disorder characterized by excessive production of myelomonocytic cells. {ECO:0000269|PubMed:19966803}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: RAS-associated autoimmune leukoproliferative disorder (RALD) [MIM:614470]: A disorder of apoptosis, characterized by chronic accumulation of non-malignant lymphocytes, defective lymphocyte apoptosis, and an increased risk for the development of hematologic malignancies. {ECO:0000269|PubMed:17517660}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Melanocytic nevus syndrome, congenital (CMNS) [MIM:137550]: A syndrome characterized by congenital pigmentary skin lesions which can occur at any site and can cover most of the body surface. These lesions may or may not be hairy. Congenital melanocytic nevi are associated with neuromelanosis (the presence of melanin-producing cells within the brain parenchyma or leptomeninges). Less commonly they are associated with malignant melanoma in childhood, both in the skin and the central nervous system. CMNS patients also tend to have a characteristic facial appearance, including wide or prominent forehead, periorbital fullness, small short nose with narrow nasal bridge, round face, full cheeks, prominent premaxilla, and everted lower lip. {ECO:0000269|PubMed:18633438, ECO:0000269|PubMed:23392294}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Melanosis, neurocutaneous (NCMS) [MIM:249400]: A rare congenital disease characterized by the presence of giant or multiple melanocytic nevi on the skin, foci of melanin-producing cells within the brain parenchyma, and infiltration of leptomeninges by abnormal melanin deposits. Neurologic abnormalities include seizures, hydrocephalus, arachnoid cysts, tumors, and syringomyelia. Some patients may develop malignant melanoma. {ECO:0000269|PubMed:23392294}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Keratinocytic non-epidermolytic nevus (KNEN) [MIM:162900]: Epidermal nevi of the common, non-organoid and non-epidermolytic type are benign skin lesions and may vary in their extent from a single (usually linear) lesion to widespread and systematized involvement. They may be present at birth or develop early during childhood. {ECO:0000269|PubMed:22499344}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Thyroid cancer, non-medullary, 2 (NMTC2) [MIM:188470]: A form of non-medullary thyroid cancer (NMTC), a cancer characterized by tumors originating from the thyroid follicular cells. NMTCs represent approximately 95% of all cases of thyroid cancer and are classified into papillary, follicular, Hurthle cell, and anaplastic neoplasms. {ECO:0000269|PubMed:12727991}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q9H0L4; Q9H0I2; Q13064; Q7Z3B4; Q16512; Q92529; Q15915; Q9Y2U5 EC number EC 2.1.1.354 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Chromatin regulator; Cytoplasm; Metal-binding; Methyltransferase; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; S-adenosyl-L-methionine; Transferase; Zinc; Zinc-finger Protein physicochemical properties Chain ID A Molecular weight (Da) 48694.1 Length 425 Aromaticity 0.08 Instability index 44.91 Isoelectric point 6.86 Charge (pH=7) -0.4 2D Binding mode Binding energy (Kcal/mol) -9.69  Molscript Map 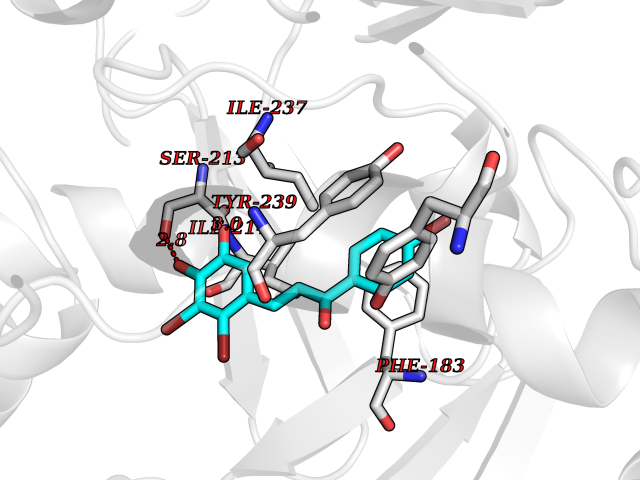 Pymol Map 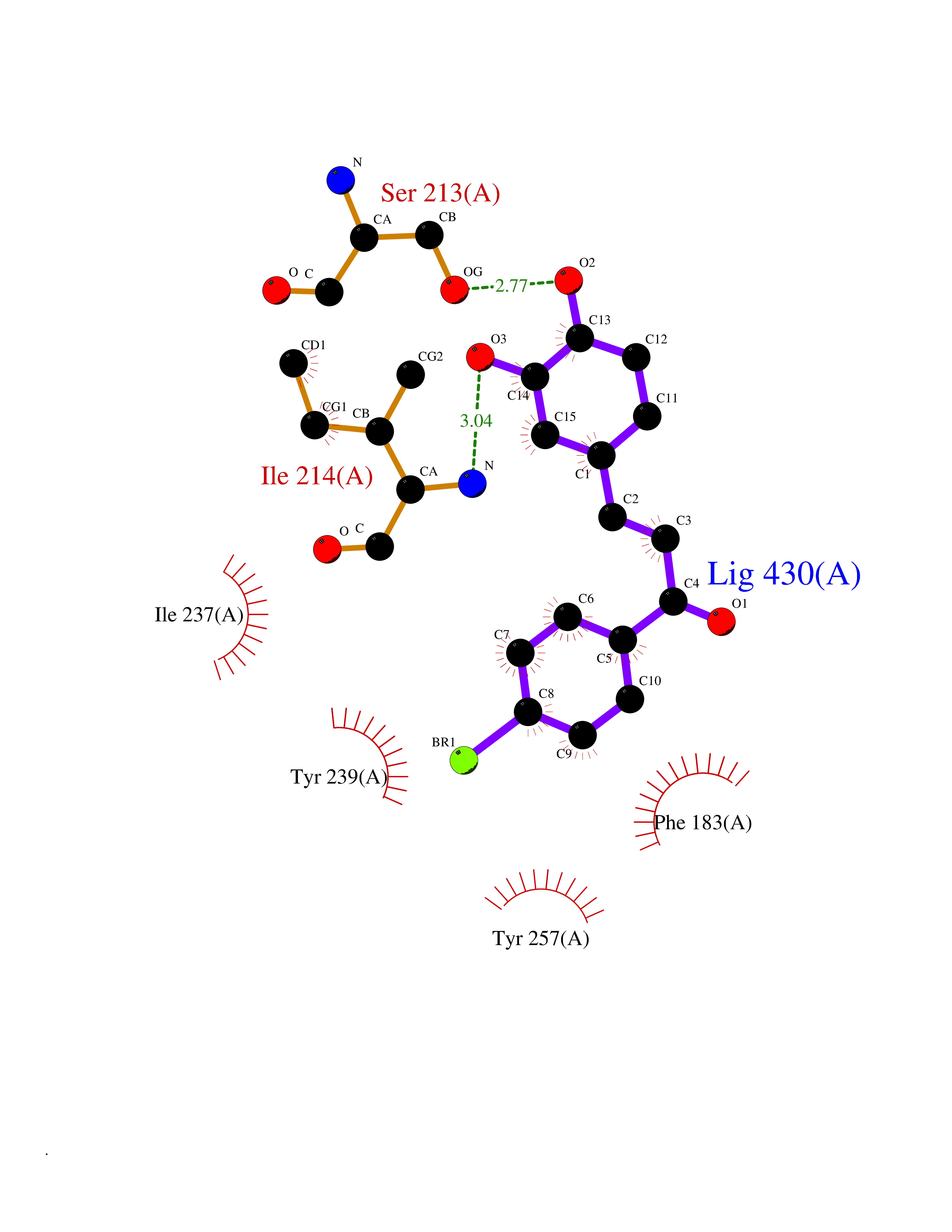 Ligplot Map 3D Binding mode Sequence PLKVEKFATANRGNGLRAVTPLRPGELLFRSDPLAYTVCKGSRGVVCDRCLLGKEKLMRCSQCRVAKYCSAKCQKKAWPDHKRECKCLKSCPRYPPDSVRLLGRVVFKLMDGAPSESEKLYSFYDLESNINKLTEDKKEGLRQLVMTFQHFMREEIQDASQLPPAFDLFEAFAKVICNSFTICNAEMQEVGVGLYPSISLLNHSCDPNCSIVFNGPHLLLRAVRDIEVGEELTICYLDMLMTSEERRKQLRDQYCFECDCFRCQTQDKDADMLTGDEQVWKEVQESLKKIEELKAHWKWEQVLAMCQAIISSNSERLPDINIYQLKVLDCAMDACINLGLLEEALFYGTRTMEPYRIFFPGSHPVRGVQVMKVGKLQLHQGMFPQAMKNLRLAFDIMRVTHGREHSLIEDLILLLEECDANIRAS Hydrogen bonds contact Hydrophobic contact | ||||