Job Results:
Ligand
Structure
Job ID
13aa62e81038093cc0a5aa14a89b9a62
Job name
NA
Time
2026-01-10 22:45:10
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 1 | Leukotriene A-4 hydrolase (LTA4H) | 3U9W | 7.77 | |
Target general information Gen name LTA4H Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Leukotriene A4 hydrolase; Leukotriene A(4)Leukotriene A-4 hydrolase hydrolase; Leukotriene A(4) hydrolase; LTA4; LTA-H; LTA-4hydrolase; LTA-4 hydrolase Protein family Peptidase M1 family Biochemical class Ether bond hydrolase Function Has also aminopeptidase activity. Epoxide hydrolase that catalyzes the final step in the biosynthesis of the proinflammatory mediator leukotriene B4. Related diseases Pigmentary disorder, reticulate, with systemic manifestations, X-linked (PDR) [MIM:301220]: An X-linked recessive disorder characterized by recurrent infections and sterile inflammation in various organs. Diffuse skin hyperpigmentation with a distinctive reticulate pattern is universally evident by early childhood. This is later followed in many patients by hypohidrosis, corneal inflammation and scarring, enterocolitis that resembles inflammatory bowel disease, and recurrent urethral strictures. Melanin and amyloid deposition is present in the dermis. Affected males also have a characteristic facies with frontally upswept hair and flared eyebrows. Female carriers have only restricted pigmentary changes along Blaschko's lines. {ECO:0000269|PubMed:27019227}. The disease is caused by variants affecting the gene represented in this entry. XLPDR is caused by a recurrent intronic mutation that results in missplicing and reduced POLA1 expression. This leads to a decrease in cytosolic RNA:DNA hybrids and constitutive activation of type I interferon responses, but has no effect on cell replication. {ECO:0000269|PubMed:27019227}.; DISEASE: Van Esch-O'Driscoll syndrome (VEODS) [MIM:301030]: An X-linked recessive syndrome characterized by different degrees of intellectual disability, moderate to severe short stature, microcephaly, hypogonadism, and variable congenital malformations. {ECO:0000269|PubMed:31006512}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07102; DB06917; DB07258; DB07094; DB07259; DB02352; DB07292; DB07104; DB06828; DB08466; DB01197; DB05177; DB03366; DB08040; DB06851; DB02062; DB07099; DB07260; DB07196; DB11781; DB03424; DB07237 Interacts with Q9BSI4 EC number EC 3.3.2.6 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Cytoplasm; Direct protein sequencing; Hydrolase; Leukotriene biosynthesis; Lipid metabolism; Metal-binding; Metalloprotease; Phosphoprotein; Protease; Proteomics identification; Reference proteome; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 68927 Length 608 Aromaticity 0.1 Instability index 38.84 Isoelectric point 5.87 Charge (pH=7) -9.86 2D Binding mode Binding energy (Kcal/mol) -10.6 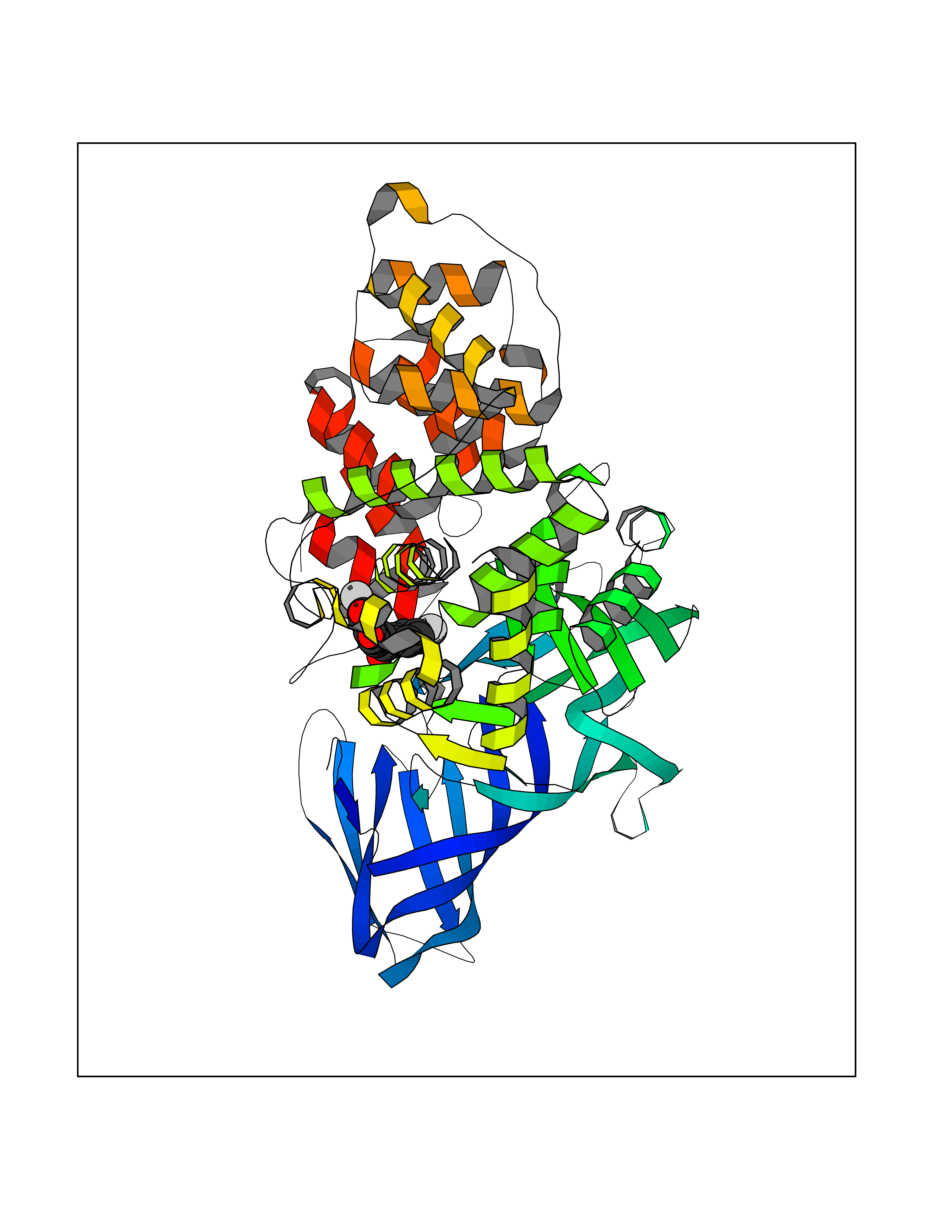 Molscript Map 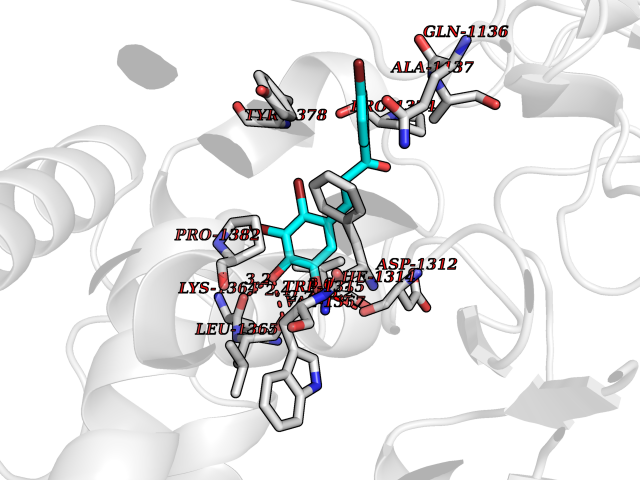 Pymol Map 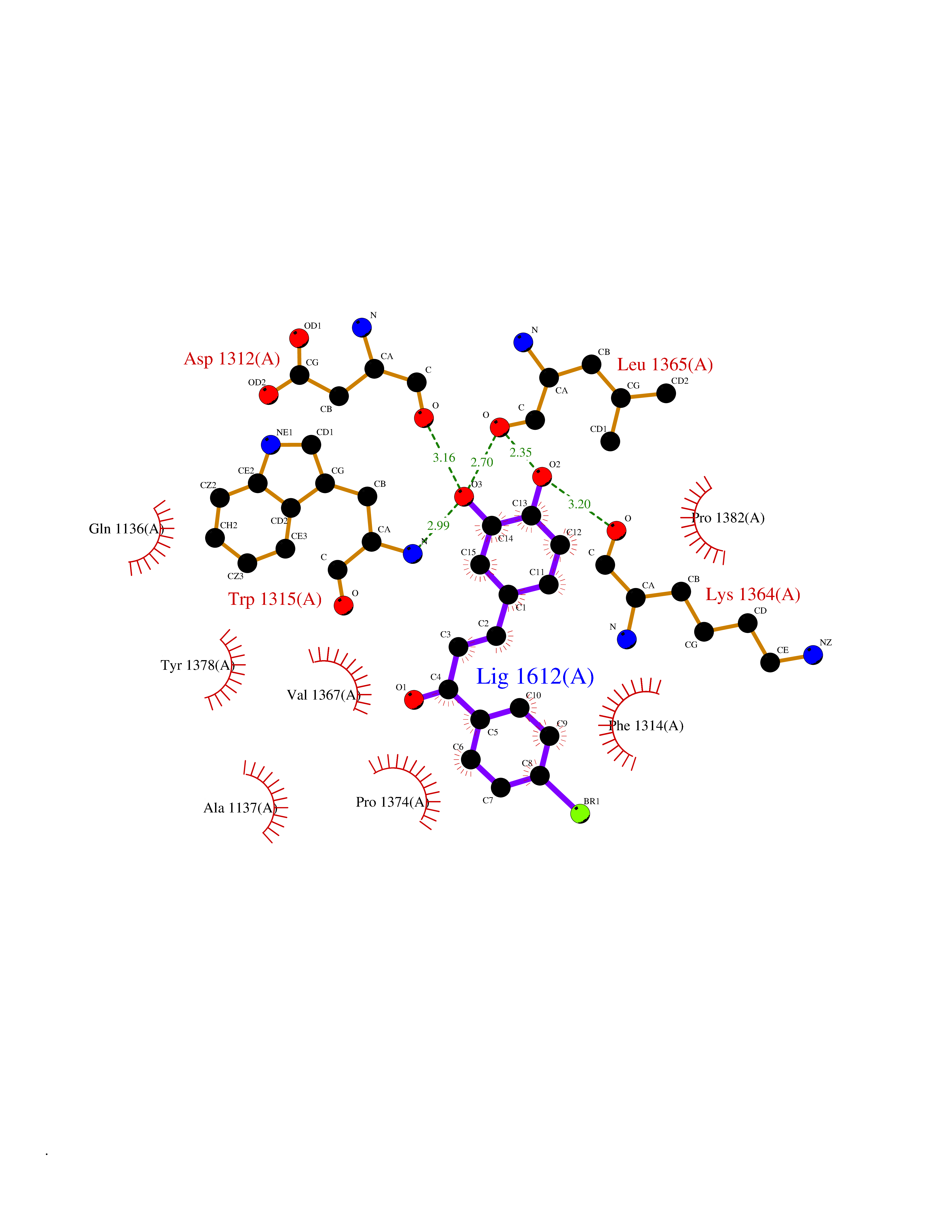 Ligplot Map 3D Binding mode Sequence IVDTCSLASPASVCRTKHLHLRCSVDFTRRTLTGTAALTVQSQEDNLRSLVLDTKDLTIEKVVINGQEVKYALGERQSYKGSPMEISLPIALSKNQEIVIEISFETSPKSSALQWLTPEQTSGKEHPYLFSQCQAIHCRAILPCQDTPSVKLTYTAEVSVPKELVALMSAIRDGETPDPEDPSRKIYKFIQKVPIPCYLIALVVGALESRQIGPRTLVWSEKEQVEKSAYEFSETESMLKIAEDLGGPYVWGQYDLLVLPPSFPYGGMENPCLTFVTPTLLAGDKSLSNVIAHEISHSWTGNLVTNKTWDHFWLNEGHTVYLERHICGRLFGEKFRHFNALGGWGELQNSVKTFGETHPFTKLVVDLTDIDPDVAYSSVPYEKGFALLFYLEQLLGGPEIFLGFLKAYVEKFSYKSITTDDWKDFLYSYFKDKVDVLNQVDWNAWLYSPGLPPIKPNYDMTLTNACIALSQRWITAKEDDLNSFNATDLKDLSSHQLNEFLAQTLQRAPLPLGHIKRMQEVYNFNAINNSEIRFRWLRLCIQSKWEDAIPLALKMATEQGRMKFTRPLFKDLAAFDKSHDQAVRTYQEHKASMHPVTAMLVGKDLKVD Hydrogen bonds contact Hydrophobic contact | ||||
| 2 | Neuronal acetylcholine receptor subunit alpha-3 | 4ZK4 | 7.55 | |
Target general information Gen name CHRNA3 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NACHRA3 Protein family Ligand-gated ion channel (TC 1.A.9) family, Acetylcholine receptor (TC 1.A.9.1) subfamily, Alpha-3/CHRNA3 sub-subfamily Biochemical class Acetylcholine binding protein Function Acetylcholine binding.Acetylcholine-gated cation-selective channel activity.Acetylcholine receptor activity.Ligand-gated ion channel activity.Serotonin-gated cation-selective channel activity. Related diseases Bladder dysfunction, autonomic, with impaired pupillary reflex and secondary CAKUT (BAIPRCK) [MIM:191800]: An autosomal recessive disease characterized by impaired innervation and autonomic dysfunction of the urinary bladder, hydronephrosis, vesicoureteral reflux, small kidneys, recurrent urinary tract infections, and progressive renal insufficiency. Additional autonomic features are impaired pupillary reflex and orthostatic hypotension. The disease manifests in utero or early childhood. {ECO:0000269|PubMed:31708116}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00915; DB01156; DB00237; DB00565; DB09028; DB00514; DB07720; DB00898; DB00472; DB05710; DB01227; DB00848; DB00333; DB00184; DB01090; DB00202; DB01273 Interacts with NA EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disease variant; Disulfide bond; Endoplasmic reticulum; Glycoprotein; Golgi apparatus; Ion channel; Ion transport; Ligand-gated ion channel; Membrane; Phosphoprotein; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Signal; Synapse; Transmembrane; Transmembrane helix; Transport; Ubl conjugation Protein physicochemical properties Chain ID A,B,C,D,E Molecular weight (Da) 46391.5 Length 408 Aromaticity 0.12 Instability index 30.23 Isoelectric point 4.6 Charge (pH=7) -22.73 2D Binding mode Binding energy (Kcal/mol) -10.3 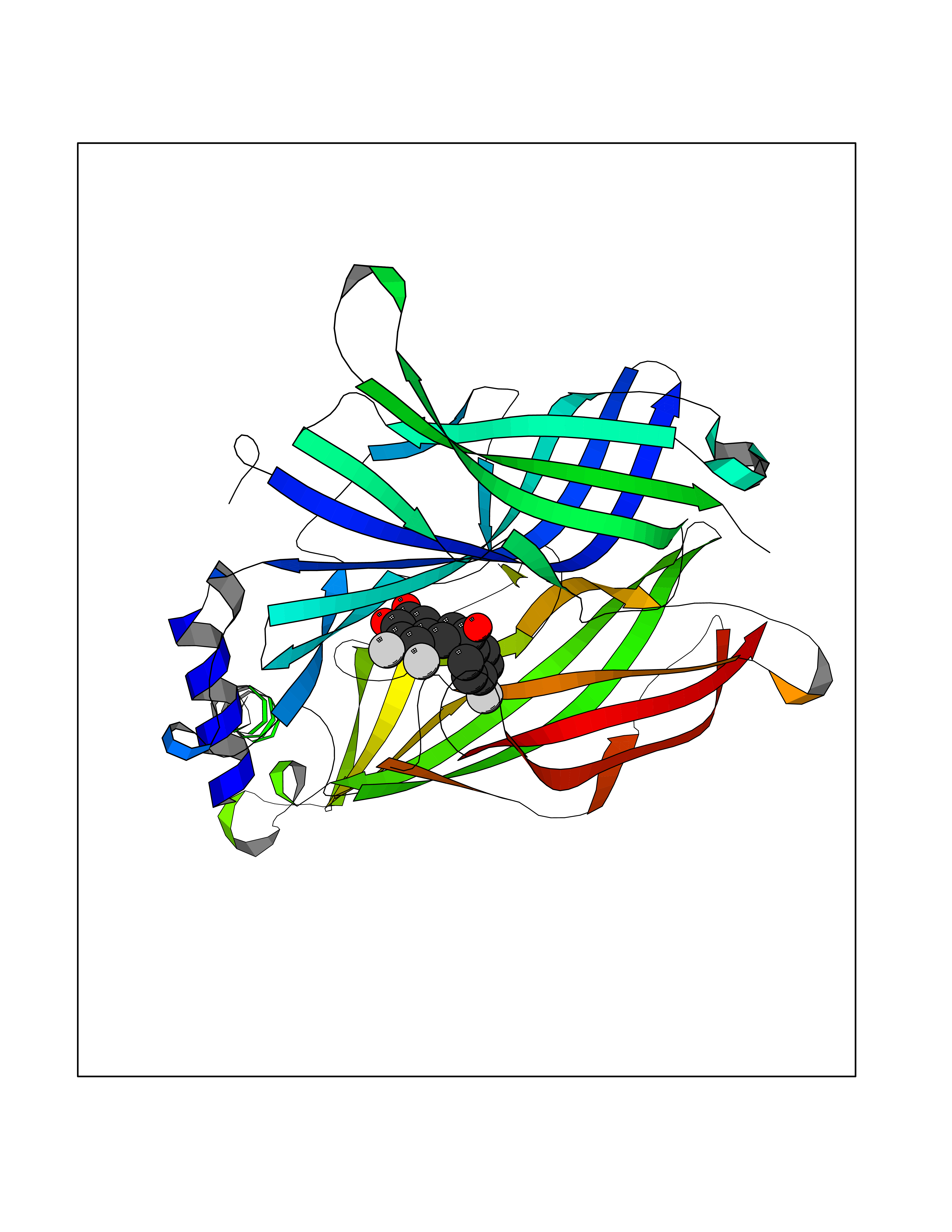 Molscript Map 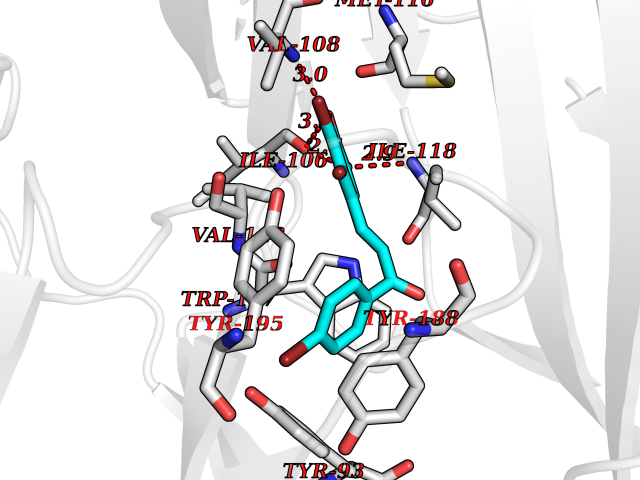 Pymol Map 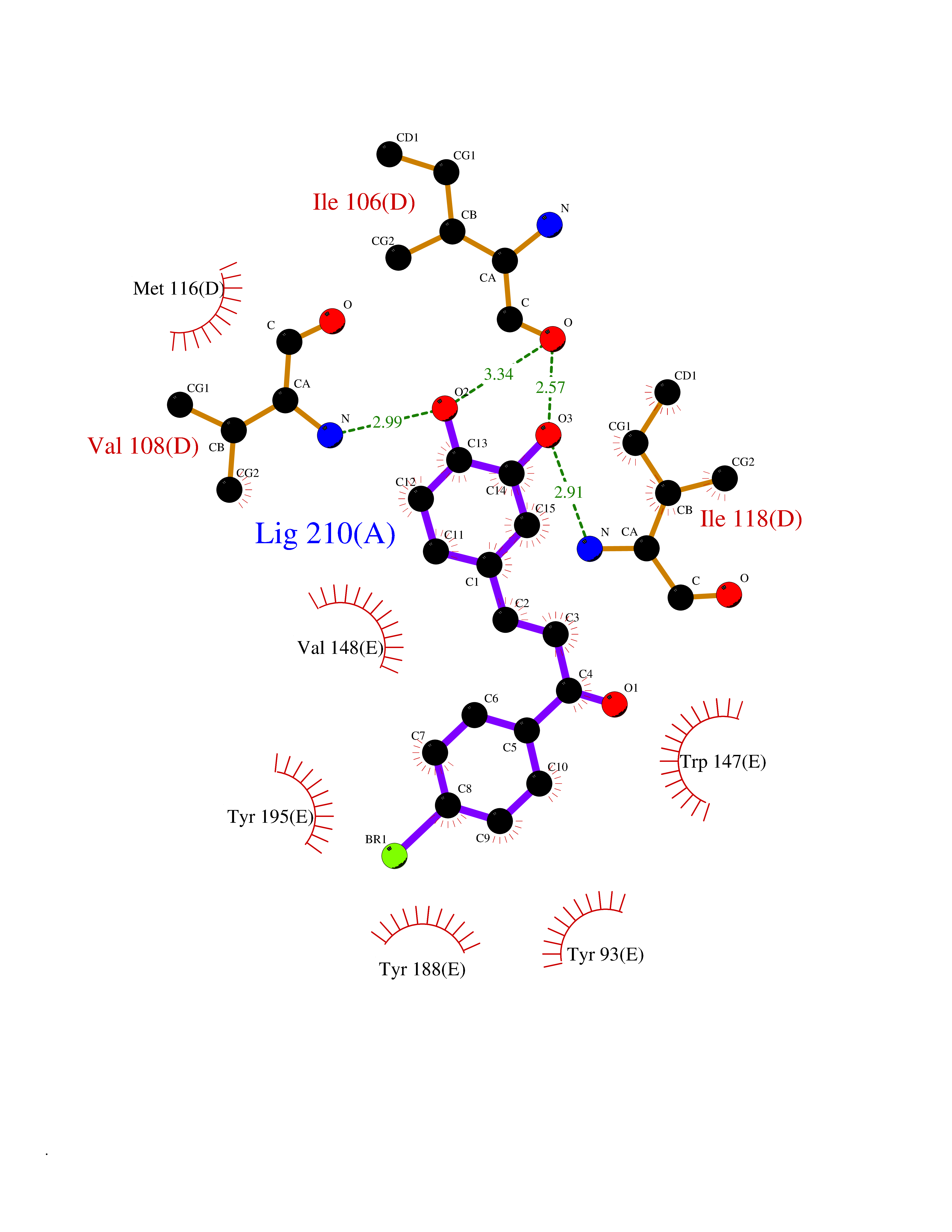 Ligplot Map 3D Binding mode Sequence LHSQANLMRLKSDLFYPGPTKDDPLTVTLGFTLQDIVKADSSTNEVDLVYWEQQRWKLNSLMWDPNEYGNITDFRTSAADIWTPDITAYSSTRPVQVLSPQIAVVTHDGSVMFIPAQRLSFMCDPTGVDSEEGATCAVKFGSWVYSGFEIDLKTDTDQVDLSSYYASSKYEILSATQYKHDIKYNCCEEIYPDVVLVVKFRERRLHSQANLMRLKSDLFNRYPGPTKDDPLTVTLGFTLQDIVKADSSTNEVDLVYWEQQRWKLNSLMWDPNEYGNITDFRTSAADIWTPDITAYSSTRPVQVLSPQIAVVTHDGSVMFIPAQRLSFMCDPTGVDSEEGATCAVKFGSWVYSGFEIDLKTDTDQVDLSSYYASSKYEILSATQYKHDIKYNCCEEIYPDVVLVVKFRE Hydrogen bonds contact Hydrophobic contact | ||||
| 3 | Glucose-dependent insulinotropic receptor (GPR119) | 7XZ6 | 7.55 | |
Target general information Gen name GPR119 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms GPR119; G-protein coupled receptor 119 Protein family G-protein coupled receptor 1 family Biochemical class GPCR rhodopsin Function Receptor for the endogenous fatty-acid ethanolamide oleoylethanolamide (OEA) and lysophosphatidylcholine (LPC). Functions as a glucose-dependent insulinotropic receptor. The activity of this receptor is mediated by G proteins which activate adenylate cyclase. Seems to act through a G(s) mediated pathway. Related diseases Developmental and epileptic encephalopathy 24 (DEE24) [MIM:615871]: A disease characterized by early-onset seizures, intellectual disability of varying degrees, and behavioral disturbances or autistic features in most individuals. {ECO:0000269|PubMed:24747641, ECO:0000269|PubMed:27864847, ECO:0000269|PubMed:30351409}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Generalized epilepsy with febrile seizures plus 10 (GEFSP10) [MIM:618482]: An autosomal dominant neurologic disorder with incomplete penetrance, characterized by variable types of seizures including absence, tonic-clonic, febrile, focal, and eyelid myoclonia. Some patients have normal neurologic development. Others have mild-to-moderate intellectual disability or autism spectrum disorder. {ECO:0000269|PubMed:29936235, ECO:0000269|PubMed:30351409}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB05166 Interacts with Q12797-6 EC number NA Uniprot keywords 3D-structure; Cell membrane; G-protein coupled receptor; Lipid-binding; Membrane; Receptor; Reference proteome; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID R Molecular weight (Da) 32134.1 Length 292 Aromaticity 0.12 Instability index 34.96 Isoelectric point 9.12 Charge (pH=7) 8.03 2D Binding mode Binding energy (Kcal/mol) -10.3 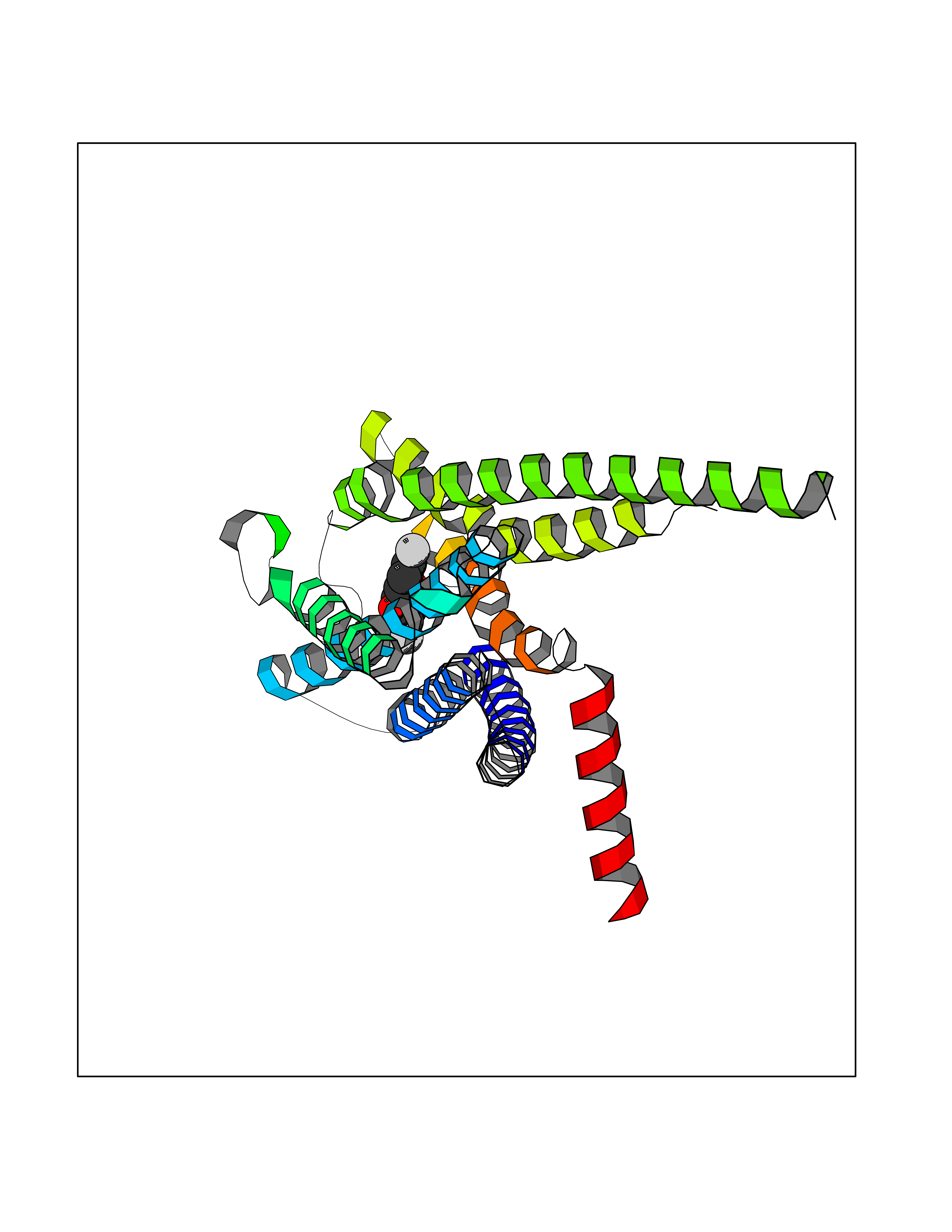 Molscript Map 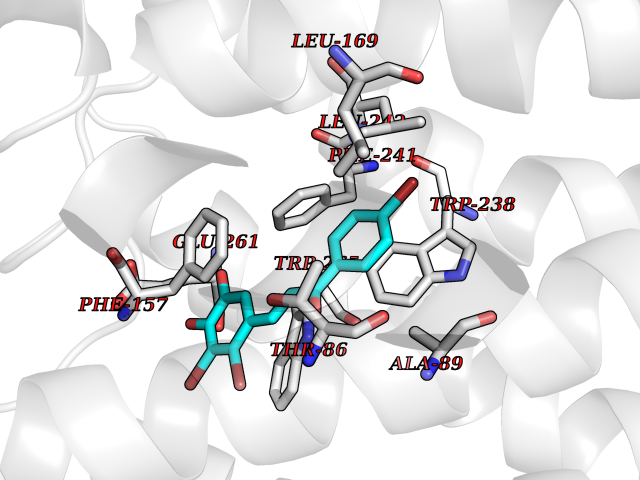 Pymol Map 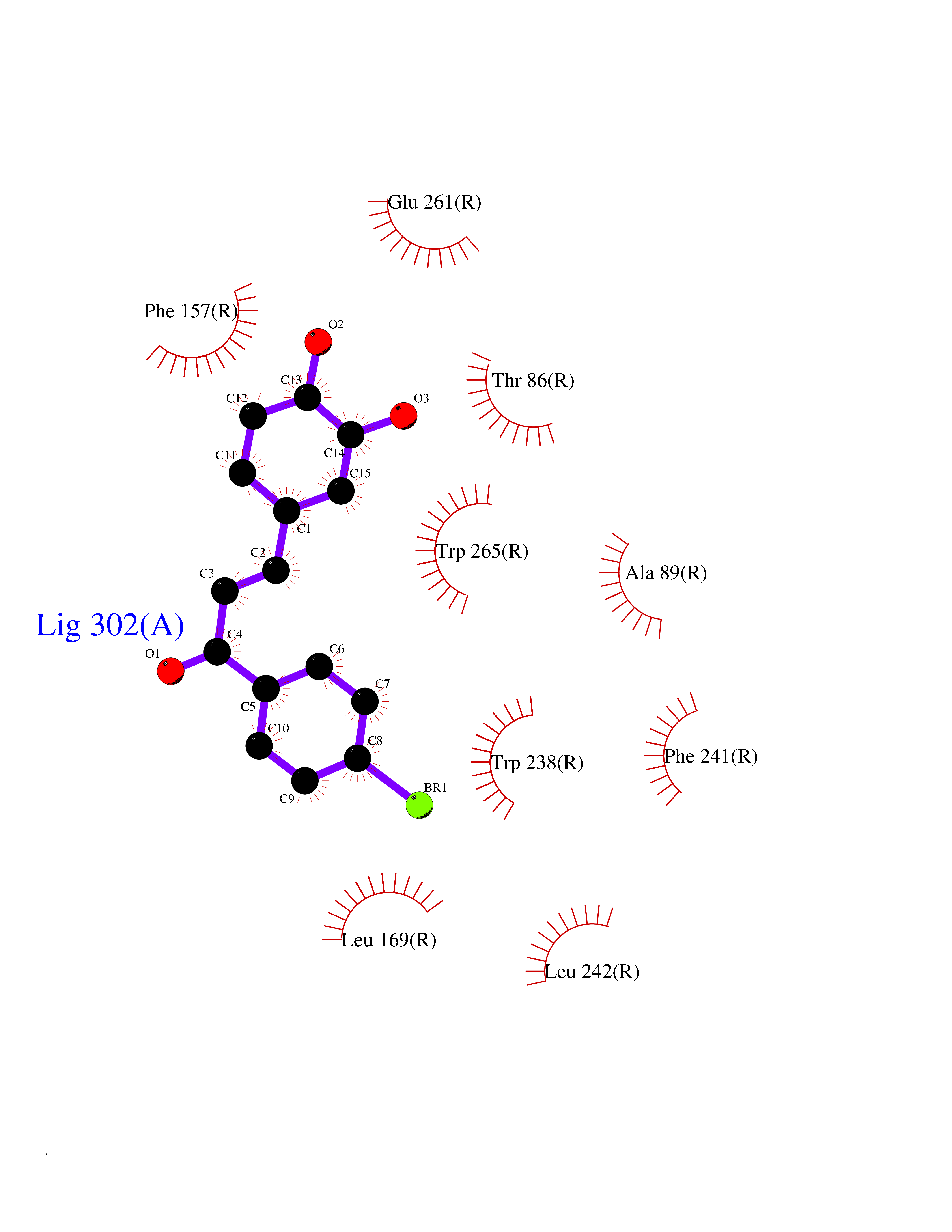 Ligplot Map 3D Binding mode Sequence MESSFSFGVILAVLASLIIATNTLVAVAVLLLIHKNDGVSLCFTLNLAVADTLIGVAISGLLTDQLSSPSRPTQKTLCSLRMAFVTSSAAASVLTVMLITFDRYLAIKQPFRYLKIMSGFVAGACIAGLWLVSYLIGFLPLGIPMFQQTAYKGQCSFFAVFHPHFVLTLSCVGFFPAMLLFVFFYCDMLKIASMHSQQIRKMEHAGAMAGSDFKALRTVSVLIGSFALSWTPFLITGIVQVACQECHLYLVLERYLWLLGVGNSLLNPLIYAYWQKEVRLQLYHMALGVKKV Hydrogen bonds contact Hydrophobic contact | ||||
| 4 | Plasmodium Dihydroorotate dehydrogenase (Malaria DHOdehase) | 1TV5 | 7.44 | |
Target general information Gen name Malaria DHOdehase Organism Plasmodium falciparum (isolate 3D7) Uniprot ID TTD ID Synonyms PFF0160c; Mitochondrially bound dihydroorotate-ubiqui oxidoreductase; Dihydroorotate oxidase of Plasmodium falciparum; Dihydroorotate dehydrogenase of Plasmodium falciparum; DHOdehase of Plasmodium fa Protein family Dihydroorotate dehydrogenase family, Type 2 subfamily Biochemical class CH-CH donor oxidoreductase Function Catalyzes the conversion of dihydroorotate to orotate with quinone as electron acceptor. Related diseases Combined oxidative phosphorylation deficiency 33 (COXPD33) [MIM:617713]: An autosomal recessive disorder caused by multiple mitochondrial respiratory chain defects and impaired mitochondrial energy metabolism. Clinical manifestations are highly variable. Affected infants present with cardiomyopathy accompanied by multisystemic features involving liver, kidney, and brain. Death in infancy is observed in some patients. Children and adults present with myopathy and progressive external ophthalmoplegia. {ECO:0000269|PubMed:28942965}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01117 Interacts with NA EC number EC 1.3.5.2 Uniprot keywords 3D-structure; Flavoprotein; FMN; Membrane; Mitochondrion; Mitochondrion inner membrane; Oxidoreductase; Pyrimidine biosynthesis; Reference proteome; Transit peptide; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 41846.8 Length 371 Aromaticity 0.1 Instability index 37.25 Isoelectric point 8.21 Charge (pH=7) 3.13 2D Binding mode Binding energy (Kcal/mol) -10.15 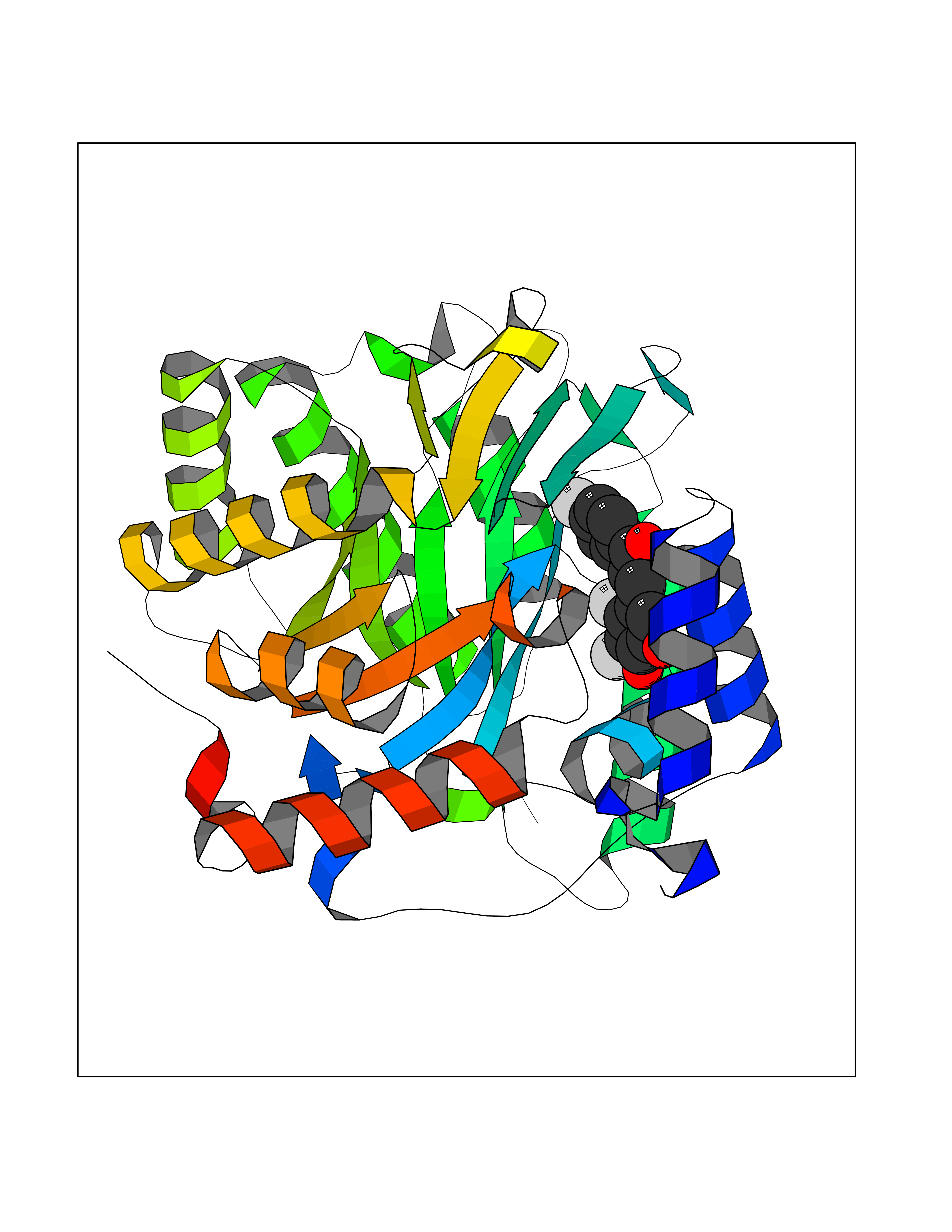 Molscript Map 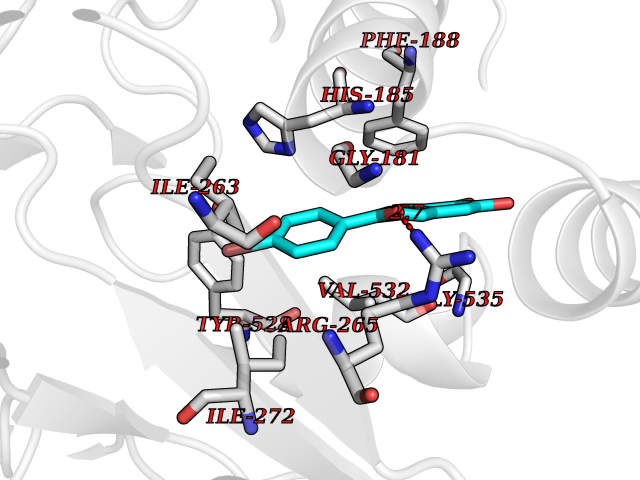 Pymol Map 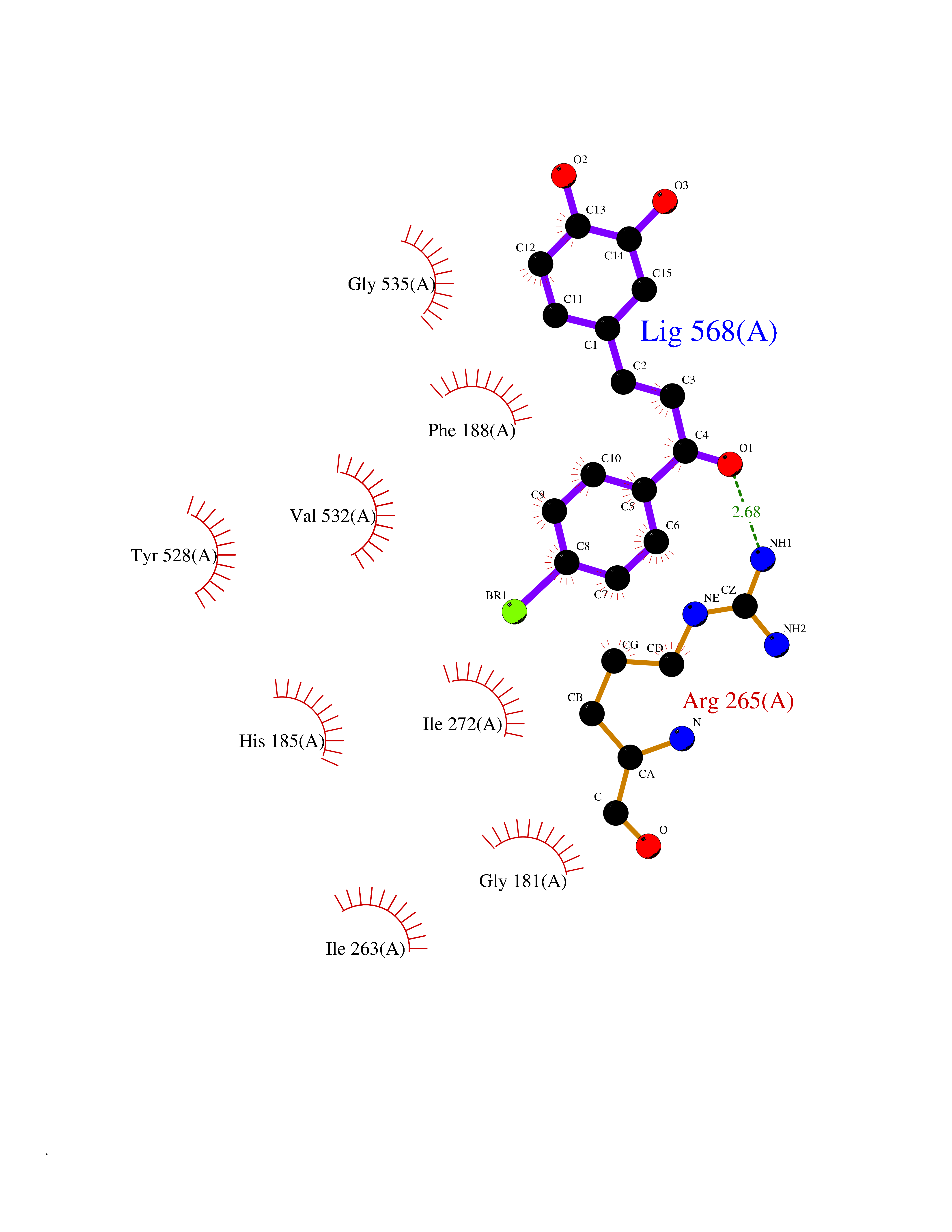 Ligplot Map 3D Binding mode Sequence FESYNPEFFLYDIFLKFCLKYIDGEICHDLFLLLGKYNILPYDTSNDSIYACTNIKHLDFINPFGVAAGFDKNGVCIDSILKLGFSFIEIGTITPRGQTGNAKPRIFRDVESRSIINSCGFNNMGCDKVTENLILFRKRQEEDKLLSKHIVGVSIGKNKDTVNIVDDLKYCINKIGRYADYIAINVSSPNTPGLRDNQEAGKLKNIILSVKEEIDNLEFLWFNTTKKKPLVFVKLAPDLNQEQKKEIADVLLETNIDGMIISNTTTQINDIKSFENKKGGVSGAKLKDISTKFICEMYNYTNKQIPIIASGGIFSGLDALEKIEAGASVCQLYSCLVFNGMKSAVQIKRELNHLLYQRGYYNLKEAIGRKH Hydrogen bonds contact Hydrophobic contact | ||||
| 5 | SEC14-like protein 3 | 4UYB | 7.43 | |
Target general information Gen name SEC14L3 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms TAP2 Protein family NA Biochemical class Transport protein Function Lipid binding.Transporter activity. Related diseases Chondrodysplasia with platyspondyly, distinctive brachydactyly, hydrocephaly, and microphthalmia (CDP-PBHM) [MIM:300863]: A disease characterized by chondrodysplasia, severe platyspondyly, hydrocephaly, and facial features with microphthalmia. Bone abnormalities include a distinctive metaphyseal cupping of the metacarpals, metatarsals, and phalanges. Affected females show a milder phenotype with small stature, sometimes associated with body asymmetry and mild intellectual disability. {ECO:0000269|PubMed:20181727}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB14003; DB14001; DB14002; DB11635; DB11251; DB00163 Interacts with NA EC number NA Uniprot keywords 3D-structure; Alternative splicing; Lipid-binding; Proteomics identification; Reference proteome; Transport Protein physicochemical properties Chain ID A Molecular weight (Da) 46148.7 Length 401 Aromaticity 0.1 Instability index 45.19 Isoelectric point 5.79 Charge (pH=7) -5.94 2D Binding mode Binding energy (Kcal/mol) -10.14 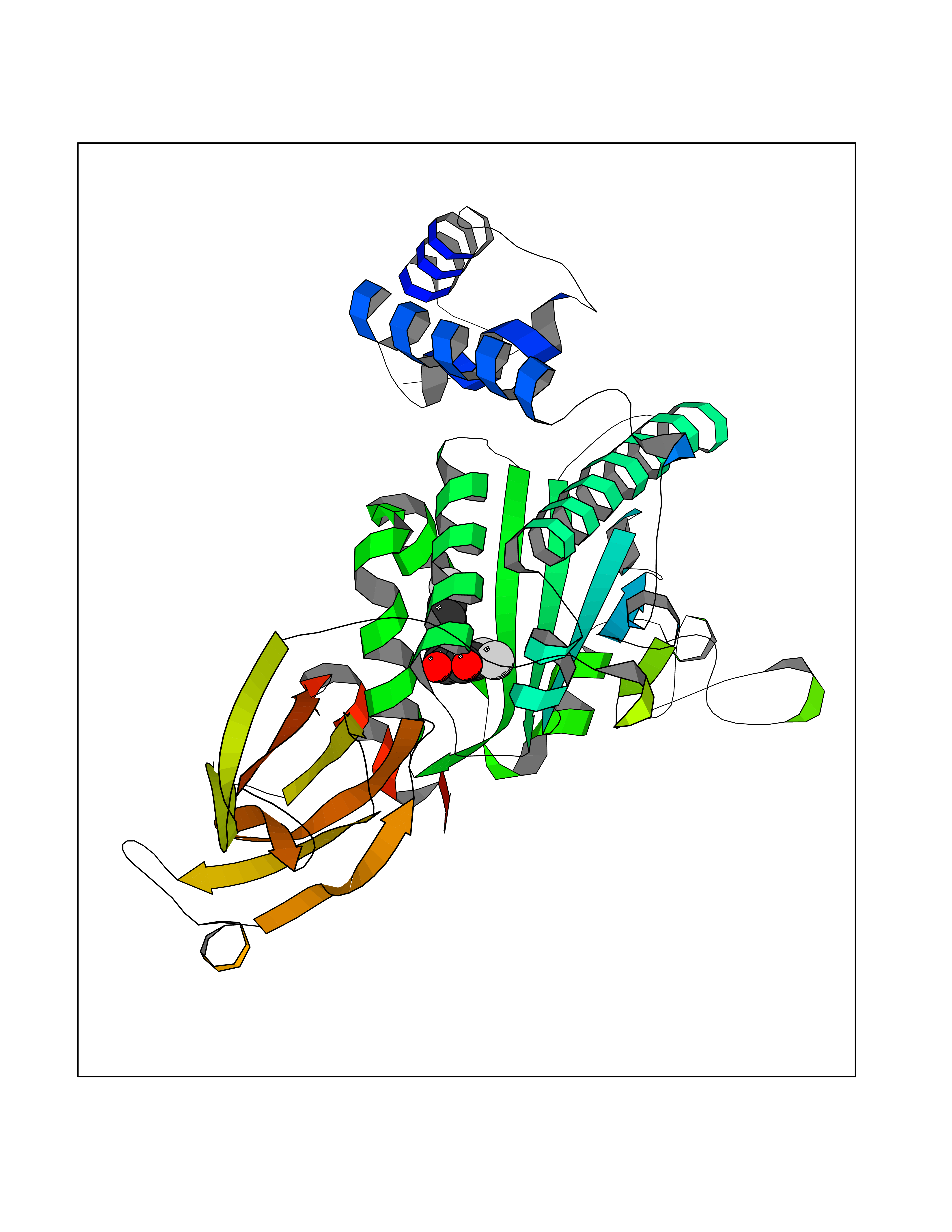 Molscript Map 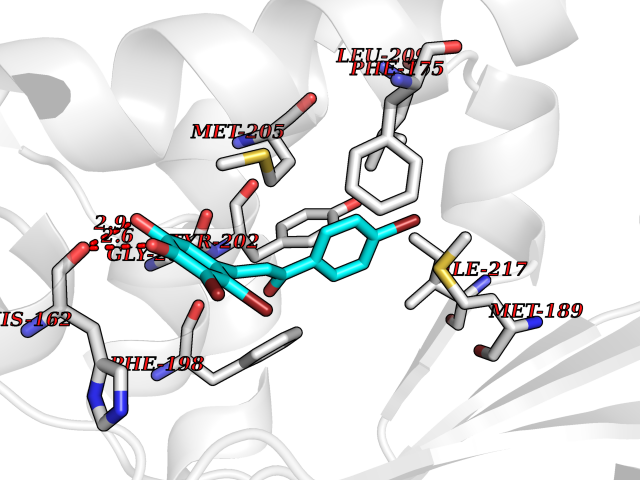 Pymol Map 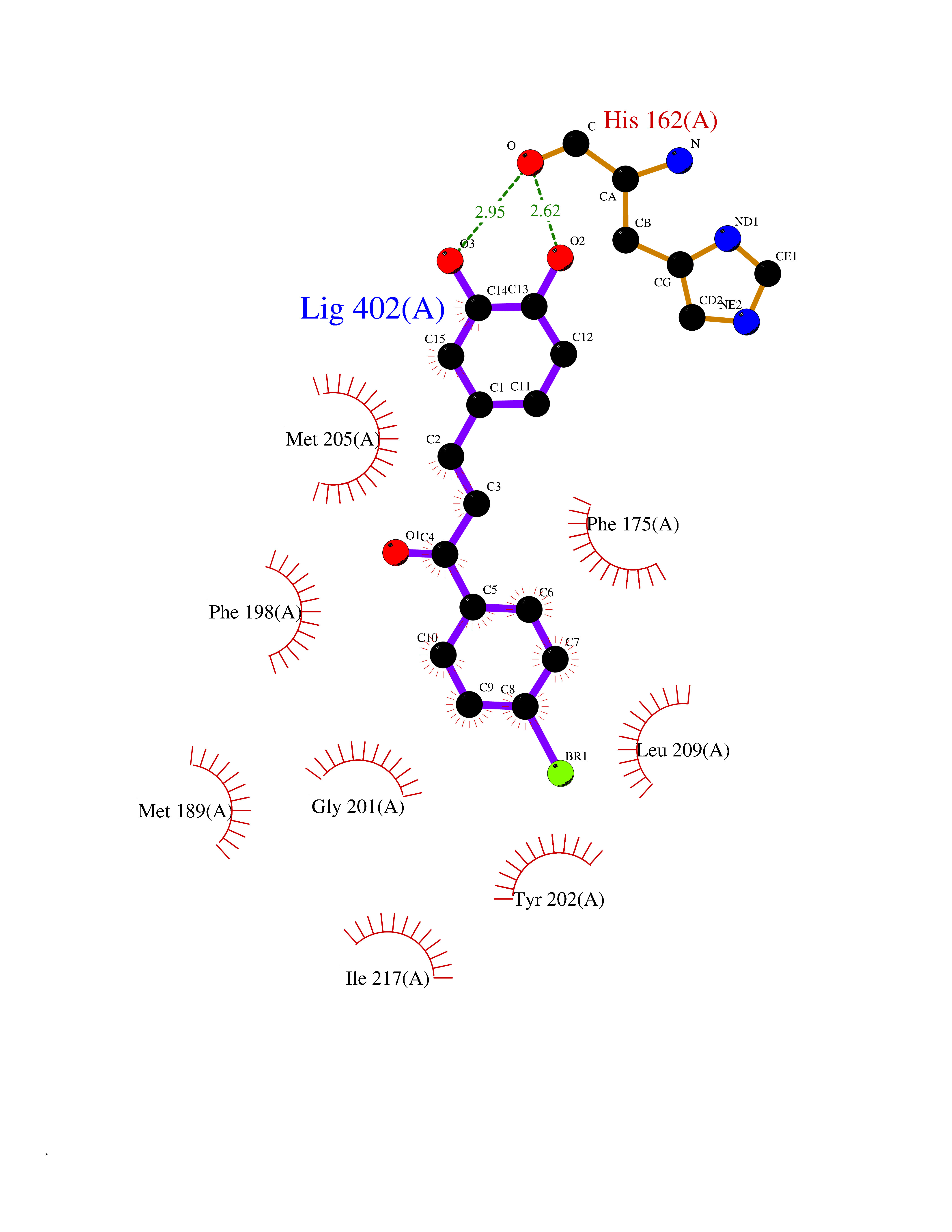 Ligplot Map 3D Binding mode Sequence SMSGRVGDLSPKQAETLAKFRENVQDVLPALPNPDDYFLLRWLRARNFDLQKSEALLRKYMEFRKTMDIDHILDWQPPEVIQKYMPGGLCGYDRDGCPVWYDIIGPLDPKGLLFSVTKQDLLKTKMRDCERILHECDLQTERLGKKIETIVMIFDCEGLGLKHFWKPLVEVYQEFFGLLEENYPETLKFMLIVKATKLFPVGYNLMKPFLSEDTRRKIIVLGNNWKEGLLKLISPEELPAQFGGTLTDPDGNPKCLTKINYGGEIPKSMYVRDQVKTQYEHSVQINRGSSHQVEYEILFPGCVLRWQFSSDGADIGFGVFLKTKMGERQRAGEMTEVLPSQRYNAHMVPEDGNLTCSEAGVYVLRFDNTYSFVHAKKVSFTVEVLLPDEGMQKYDKELTPV Hydrogen bonds contact Hydrophobic contact | ||||
| 6 | Phenylethanolamine N-methyltransferase (PNMT) | 2G72 | 7.41 | |
Target general information Gen name PNMT Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms PNMTase; PENT; Noradrenaline N-methyltransferase Protein family Class I-like SAM-binding methyltransferase superfamily, NNMT/PNMT/TEMT family Biochemical class NA Function Converts noradrenaline to adrenaline. Related diseases A chromosomal aberration involving TRIM24/TIF1 is found in papillary thyroid carcinomas (PTCs). Translocation t(7;10)(q32;q11) with RET. The translocation generates the TRIM24/RET (PTC6) oncogene. {ECO:0000269|PubMed:10439047}. Drugs (DrugBank ID) DB08129; DB08128; DB07739; DB07798; DB07747; DB03468; DB08550; DB03824; DB04273; DB07906; DB07597; DB09571; DB00968; DB08631; DB01752; DB08654 Interacts with Q9P2G9-2; Q8TBB1 EC number EC 2.1.1.28 Uniprot keywords 3D-structure; Catecholamine biosynthesis; Direct protein sequencing; Methyltransferase; Phosphoprotein; Proteomics identification; Reference proteome; S-adenosyl-L-methionine; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 29198.9 Length 264 Aromaticity 0.09 Instability index 54.33 Isoelectric point 5.91 Charge (pH=7) -3.69 2D Binding mode Binding energy (Kcal/mol) -10.11  Molscript Map 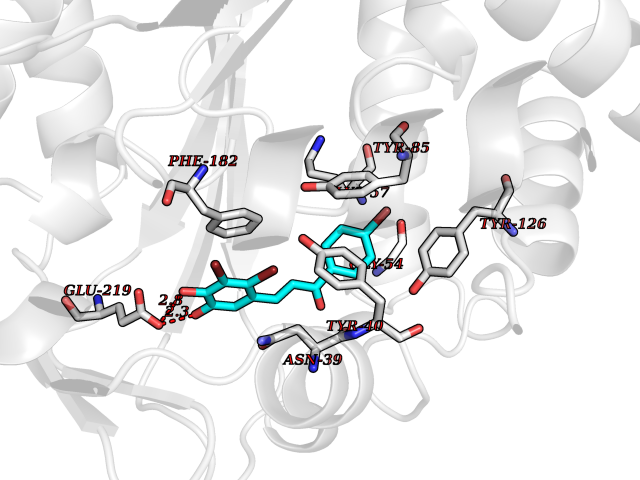 Pymol Map 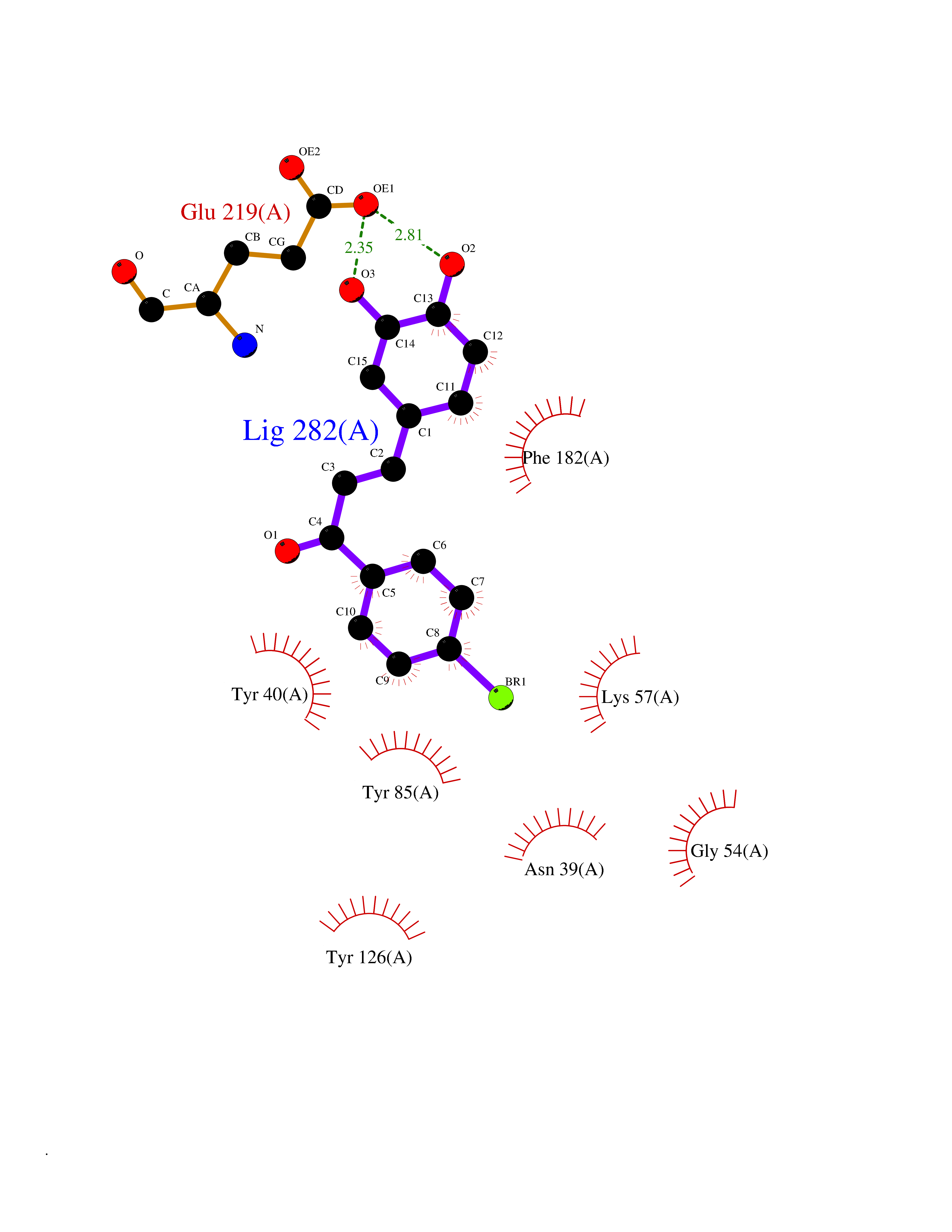 Ligplot Map 3D Binding mode Sequence APGQAAVASAYQRFEPRAYLRNNYAPPRGDLCNPNGVGPWKLRCLAQTFATGEVSGRTLIDIGSGPTVYQLLSACSHFEDITMTDFLEVNRQELGRWLQEEPGAFNWSMYSQHACLIEGKGECWQDKERQLRARVKRVLPIDVHQPQPLGAGSPAPLPADALVSAFCLEAVSPDLASFQRALDHITTLLRPGGHLLLIGALEESWYLAGEARLTVVPVSEEEVREALVRSGYKVRDLRTYIMPAHLQTGVDDVKGVFFAWAQKV Hydrogen bonds contact Hydrophobic contact | ||||
| 7 | 3-oxoacyl-[acyl-carrier-protein] synthase 2 | 3I8P | 7.39 | |
Target general information Gen name fabF Organism Escherichia coli (strain K12) Uniprot ID TTD ID NA Synonyms JW1081;b1095;fabJ Protein family Thiolase-like superfamily, Beta-ketoacyl-ACP synthases family Biochemical class Transferase Function 3-oxoacyl-[acyl-carrier-protein] synthase activity.Beta-ketoacyl-acyl-carrier-protein synthase II activity. Related diseases Pigmentary disorder, reticulate, with systemic manifestations, X-linked (PDR) [MIM:301220]: An X-linked recessive disorder characterized by recurrent infections and sterile inflammation in various organs. Diffuse skin hyperpigmentation with a distinctive reticulate pattern is universally evident by early childhood. This is later followed in many patients by hypohidrosis, corneal inflammation and scarring, enterocolitis that resembles inflammatory bowel disease, and recurrent urethral strictures. Melanin and amyloid deposition is present in the dermis. Affected males also have a characteristic facies with frontally upswept hair and flared eyebrows. Female carriers have only restricted pigmentary changes along Blaschko's lines. {ECO:0000269|PubMed:27019227}. The disease is caused by variants affecting the gene represented in this entry. XLPDR is caused by a recurrent intronic mutation that results in missplicing and reduced POLA1 expression. This leads to a decrease in cytosolic RNA:DNA hybrids and constitutive activation of type I interferon responses, but has no effect on cell replication. {ECO:0000269|PubMed:27019227}.; DISEASE: Van Esch-O'Driscoll syndrome (VEODS) [MIM:301030]: An X-linked recessive syndrome characterized by different degrees of intellectual disability, moderate to severe short stature, microcephaly, hypogonadism, and variable congenital malformations. {ECO:0000269|PubMed:31006512}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08366; DB01034; DB03017; DB08407 Interacts with P0A6Y8 EC number 2.3.1.179 Uniprot keywords 3D-structure; Acyltransferase; Direct protein sequencing; Fatty acid biosynthesis; Fatty acid metabolism; Lipid biosynthesis; Lipid metabolism; Reference proteome; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 42794.9 Length 411 Aromaticity 0.06 Instability index 31.41 Isoelectric point 5.72 Charge (pH=7) -7.34 2D Binding mode Binding energy (Kcal/mol) -10.08  Molscript Map 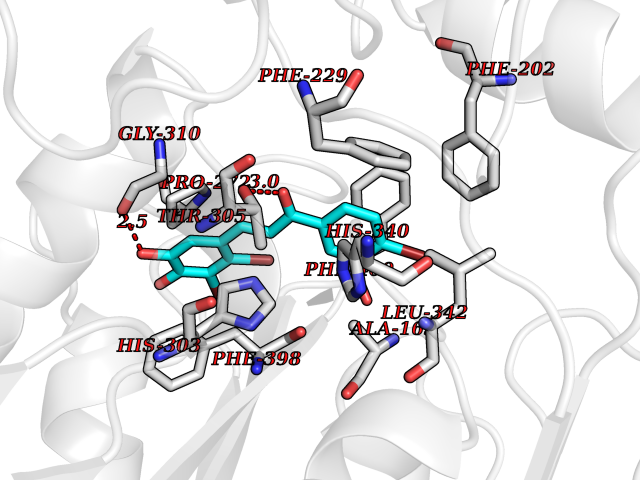 Pymol Map 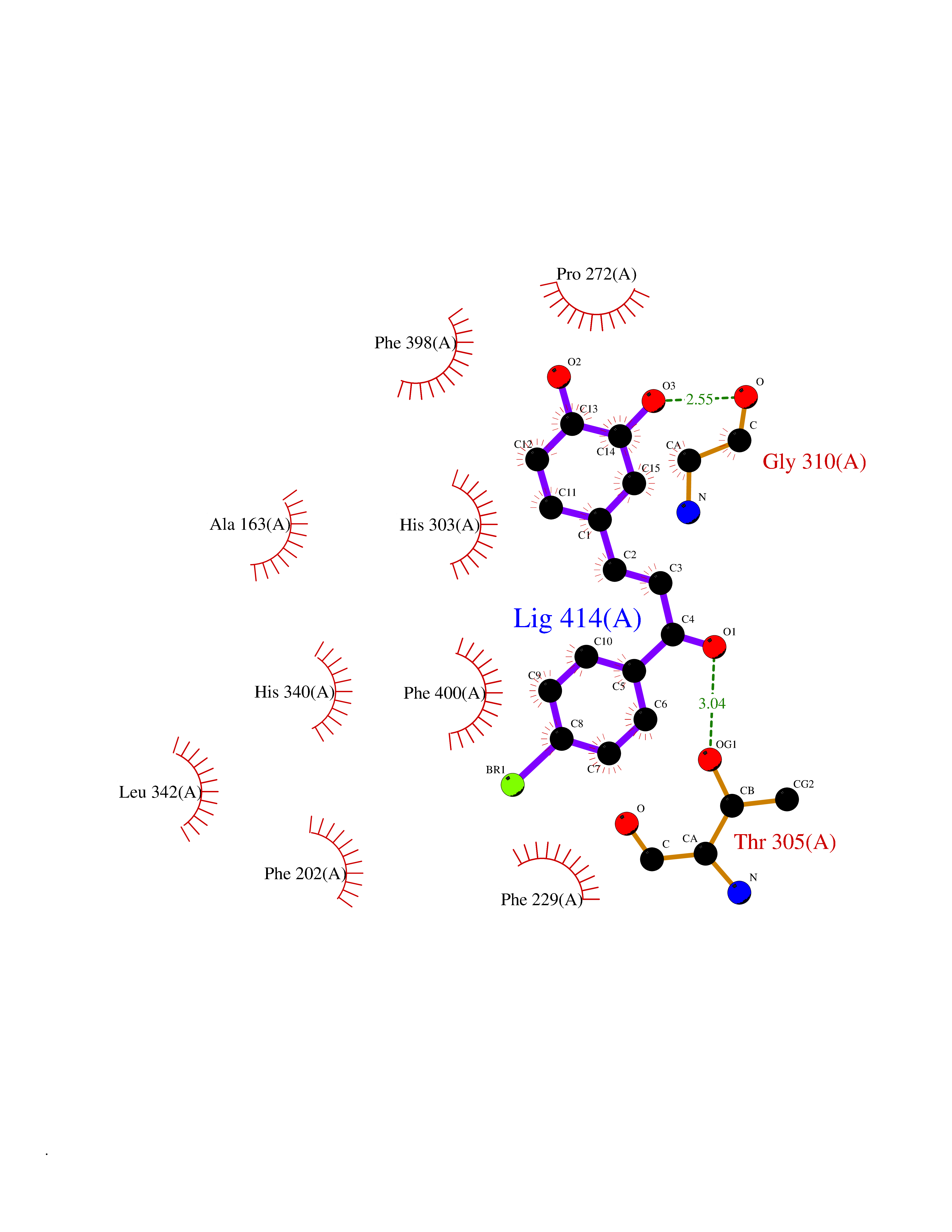 Ligplot Map 3D Binding mode Sequence KRRVVVTGLGMLSPVGNTVESTWKALLAGQSGISLIDHFDTSAYATKFAGLVKDFNCEDIISRKEQRKMDAFIQYGIVAGVQAMQDSGLEITEENATRIGAAIGSGIGGLGLIEENHTSLMNGGPRKISPFFVPSTIVNMVAGHLTIMYGLRGPSISIATAATSGVHNIGHAARIIAYGDADVMVAGGAEKASTPLGVGGFGAARALSTRNDNPQAASRPWDKERDGFVLGDGAGMLVLEEYEHAKKRGAKIYAELVGFGMSSDAYHMTSPPENGAGAALAMANALRDAGIEASQIGYVNAHGTSTPAGDKAEAQAVKTIFGEAASRVLVSSTKSMTGHLLGAAGAVESIYSILALRDQAVPPTINLDNPDEGCDLDFVPHEARQVSGMEYTLCNSFGFGGTNGSLIFKKI Hydrogen bonds contact Hydrophobic contact | ||||
| 8 | BUB1 mitotic checkpoint serine/threonine kinase (BUB1) | 6F7B | 7.36 | |
Target general information Gen name BUB1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms hBUB1; Mitotic checkpoint serine/threonine-protein kinase BUB1; BUB1L; BUB1A Protein family Protein kinase superfamily, Ser/Thr protein kinase family, BUB1 subfamily Biochemical class Kinase Function Has a key role in the assembly of checkpoint proteins at the kinetochore, being required for the subsequent localization of CENPF, BUB1B, CENPE and MAD2L1. Required for the kinetochore localization of PLK1. Required for centromeric enrichment of AUKRB in prometaphase. Plays an important role in defining SGO1 localization and thereby affects sister chromatid cohesion. Acts as a substrate for anaphase-promoting complex or cyclosome (APC/C) in complex with its activator CDH1 (APC/C-Cdh1). Necessary for ensuring proper chromosome segregation and binding to BUB3 is essential for this function. Can regulate chromosome segregation in a kinetochore-independent manner. Can phosphorylate BUB3. The BUB1-BUB3 complex plays a role in the inhibition of APC/C when spindle-assembly checkpoint is activated and inhibits the ubiquitin ligase activity of APC/C by phosphorylating its activator CDC20. This complex can also phosphorylate MAD1L1. Kinase activity is essential for inhibition of APC/CCDC20 and for chromosome alignment but does not play a major role in the spindle-assembly checkpoint activity. Mediates cell death in response to chromosome missegregation and acts to suppress spontaneous tumorigenesis. Serine/threonine-protein kinase that performs 2 crucial functions during mitosis: it is essential for spindle-assembly checkpoint signaling and for correct chromosome alignment. Related diseases Microcephaly 30, primary, autosomal recessive (MCPH30) [MIM:620183]: A form of microcephaly, a disease defined as a head circumference more than 3 standard deviations below the age, sex and ethnically matched mean. Brain weight is markedly reduced and the cerebral cortex is disproportionately small. MCPH30 is characterized by small head, poor overall growth, and global developmental delay with variably impaired intellectual development. Affected individuals may also have variable congenital anomalies, including atrial septal defect, dysmorphic facial features, tracheal stenosis, and anomalies of the skin and teeth. {ECO:0000269|PubMed:35044816}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with O95376; O60566; O43684; P46108; Q8NG31; Q8NG31-2; Q9GZQ8; P03070 EC number EC 2.7.11.1 Uniprot keywords 3D-structure; Alternative splicing; Apoptosis; ATP-binding; Cell cycle; Cell division; Centromere; Chromosome; Chromosome partition; Host-virus interaction; Intellectual disability; Kinase; Kinetochore; Mitosis; Nucleotide-binding; Nucleus; Phosphoprotein; Primary microcephaly; Proteomics identification; Reference proteome; Serine/threonine-protein kinase; Transferase; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 39564.8 Length 343 Aromaticity 0.12 Instability index 31.17 Isoelectric point 8.49 Charge (pH=7) 4.7 2D Binding mode Binding energy (Kcal/mol) -10.04 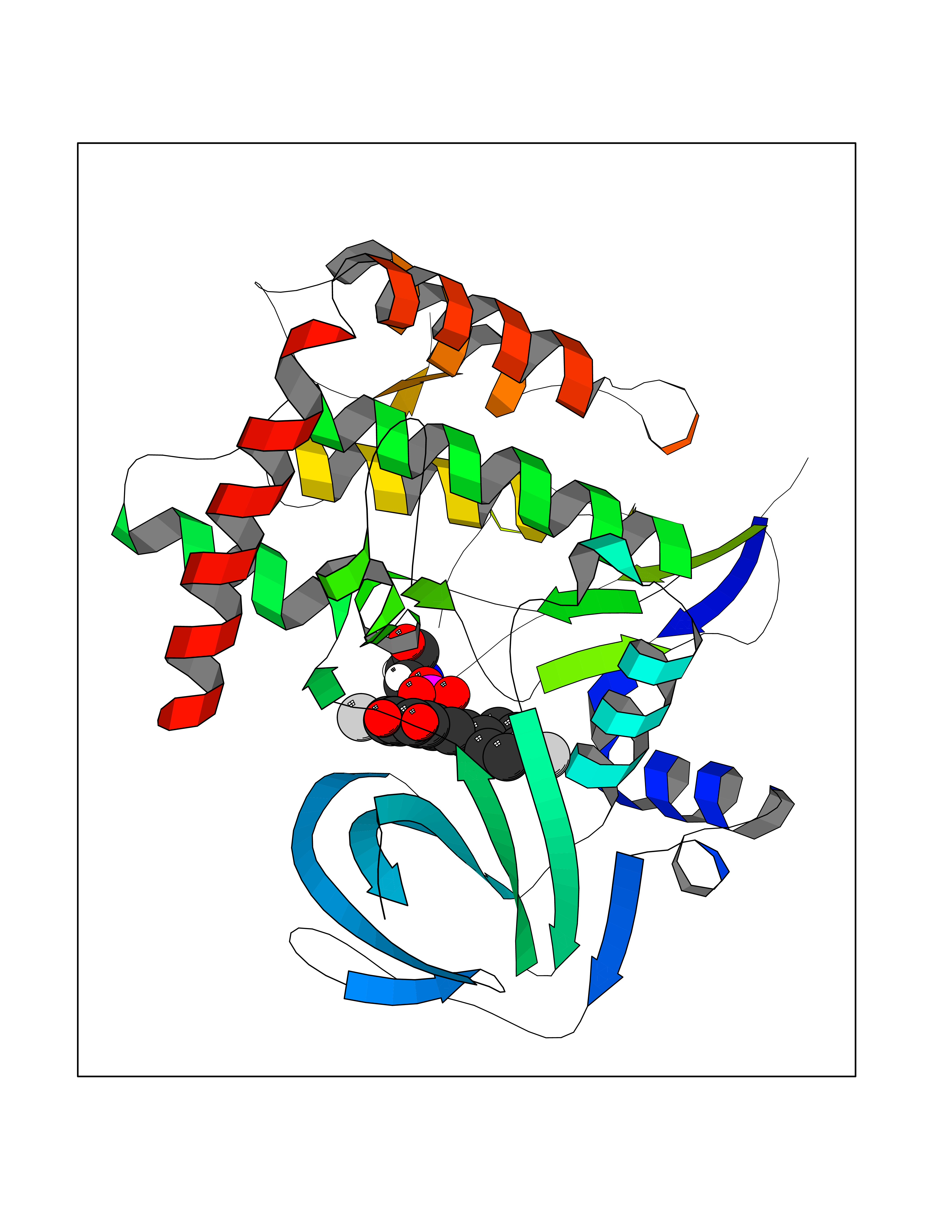 Molscript Map 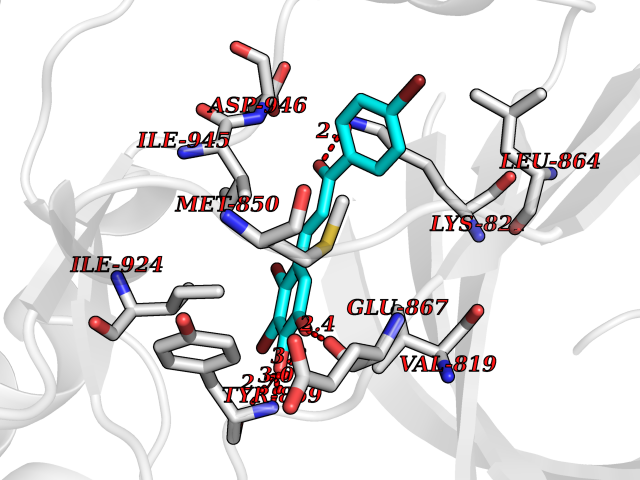 Pymol Map 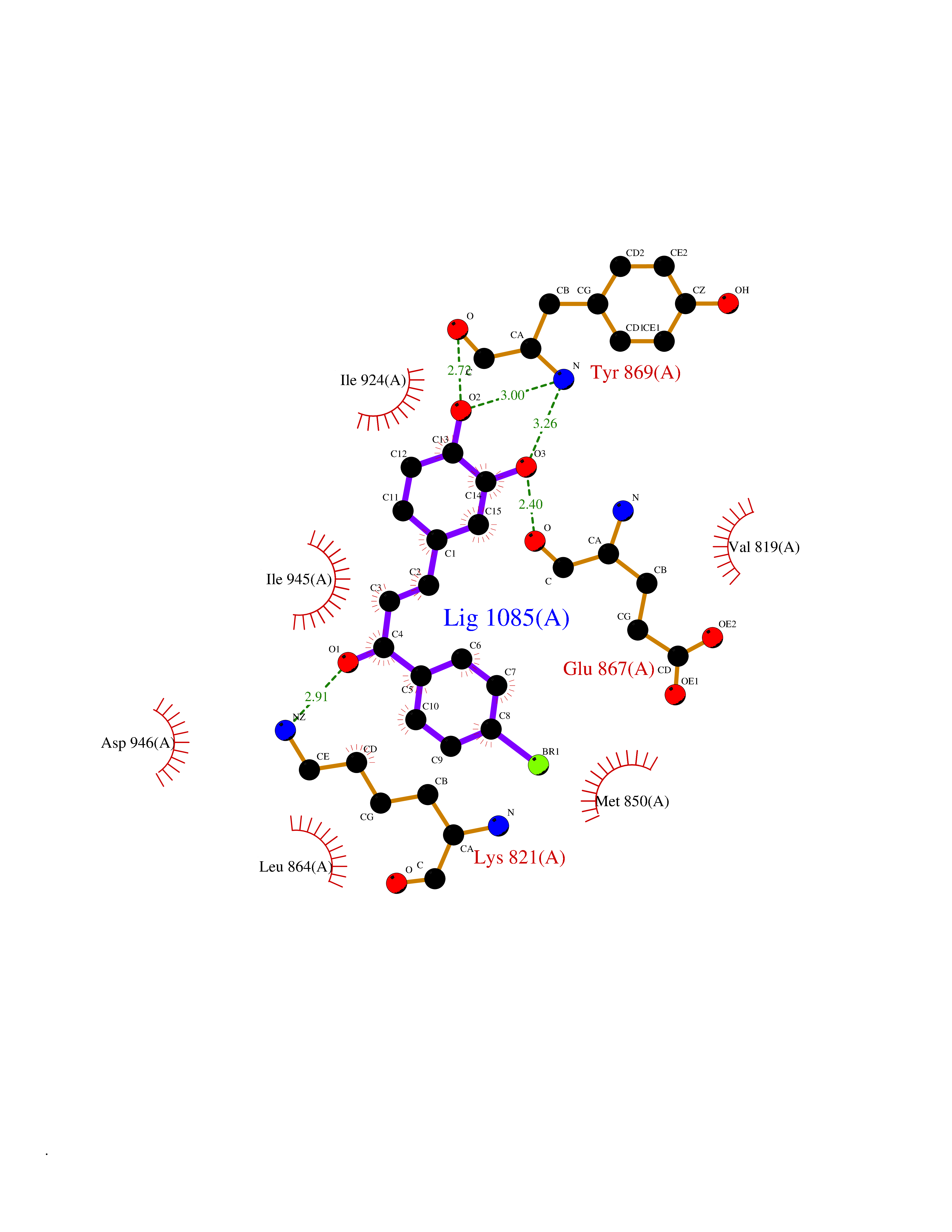 Ligplot Map 3D Binding mode Sequence APNFIVGNPWDDKLIFKLLSGLSKPVSSYPNTFEWQCKLPAIKPKTEFQLGSKLVYVHHLLGEGAFAQVYEATQKNKQKFVLKVQKPANPWEFYIGTQLMERLKPSMQHMFMKFYSAHLFQNGSVLVGELYSYGTLLNAINLYKNTPEKVMPQGLVISFAMRMLYMIEQVHDCEIIHGDIKPDNFILGNGFLEQDDEDDLSAGLALIDLGQSIDMKLFPKGTIFTAKCETXGFQCVEMLSNKPWNYQIDYFGVAATVYCMLFGTYMKVKNEECKPEGLFRRLPHLDMWNEFFHVMLNIPDCHHLPSLDLLRQKLKKVFQQHYTNKIRALRNRLIVLLLECKRS Hydrogen bonds contact Hydrophobic contact | ||||
| 9 | Cytochrome c oxidase subunit 2 | 3VRJ | 7.34 | |
Target general information Gen name MT-CO2 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms MTCO2;COXII;COII;COX2 Protein family Cytochrome c oxidase subunit 2 family Biochemical class Immune system Function Copper ion binding.Cytochrome-c oxidase activity. Related diseases Mitochondrial complex IV deficiency (MT-C4D) [MIM:220110]: A disorder of the mitochondrial respiratory chain with heterogeneous clinical manifestations, ranging from isolated myopathy to severe multisystem disease affecting several tissues and organs. Features include hypertrophic cardiomyopathy, hepatomegaly and liver dysfunction, hypotonia, muscle weakness, exercise intolerance, developmental delay, delayed motor development and intellectual disability. Some affected individuals manifest a fatal hypertrophic cardiomyopathy resulting in neonatal death. A subset of patients manifest Leigh syndrome. {ECO:0000269|PubMed:10486321}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02659; DB04464; DB05412 Interacts with Q9NZ94-2; P49281-3 EC number 7.1.1.9 Uniprot keywords 3D-structure; Copper; Disease variant; Electron transport; Magnesium; Membrane; Metal-binding; Mitochondrion; Mitochondrion inner membrane; Primary mitochondrial disease; Proteomics identification; Reference proteome; Respiratory chain; Translocase; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID C Molecular weight (Da) 21687.9 Length 189 Aromaticity 0.11 Instability index 38 Isoelectric point 5.68 Charge (pH=7) -3.26 2D Binding mode Binding energy (Kcal/mol) -10.01 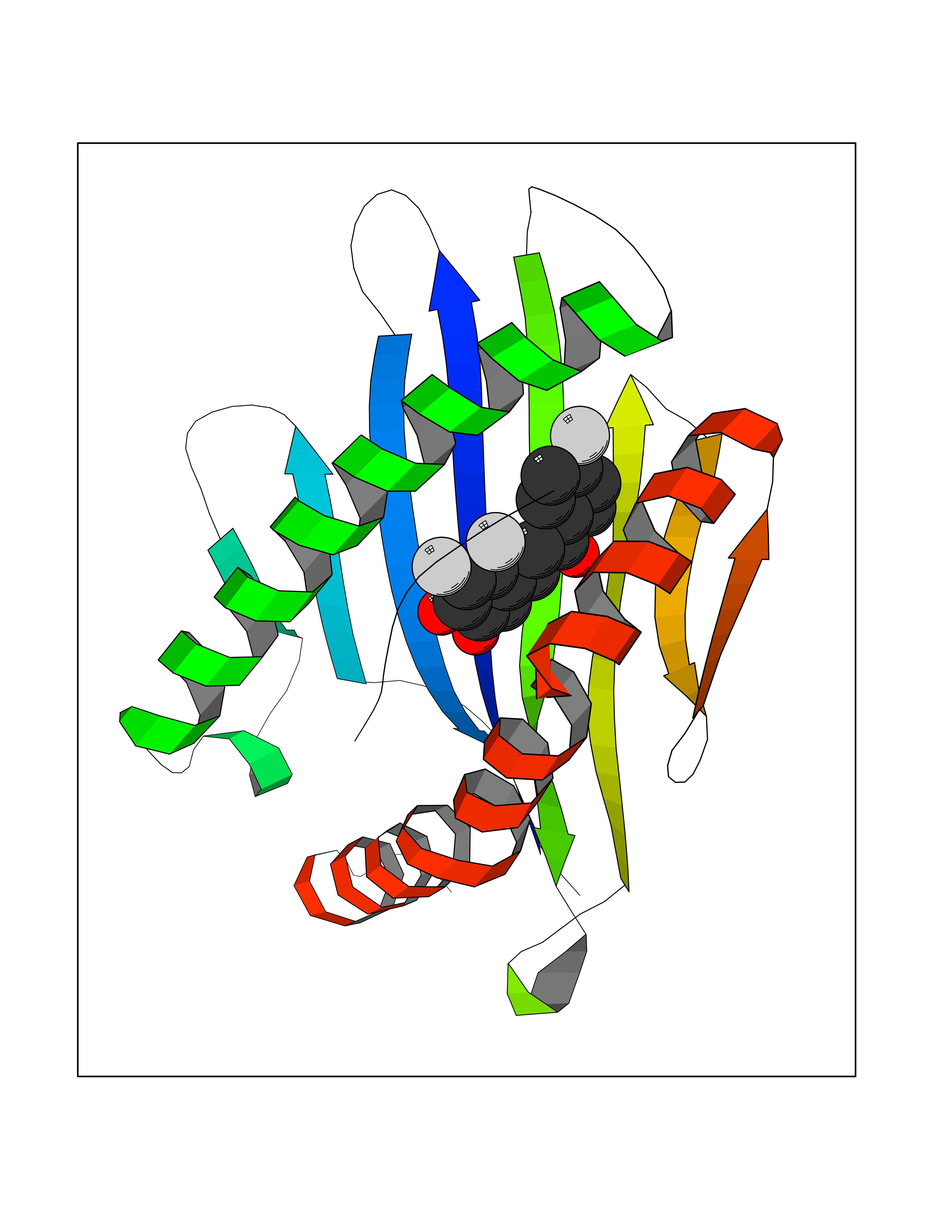 Molscript Map 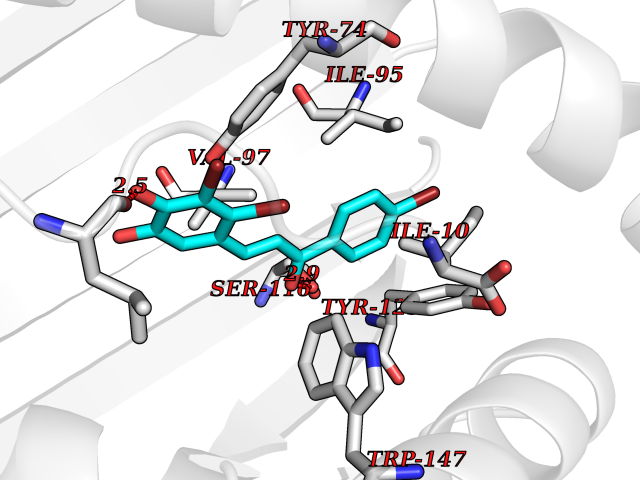 Pymol Map 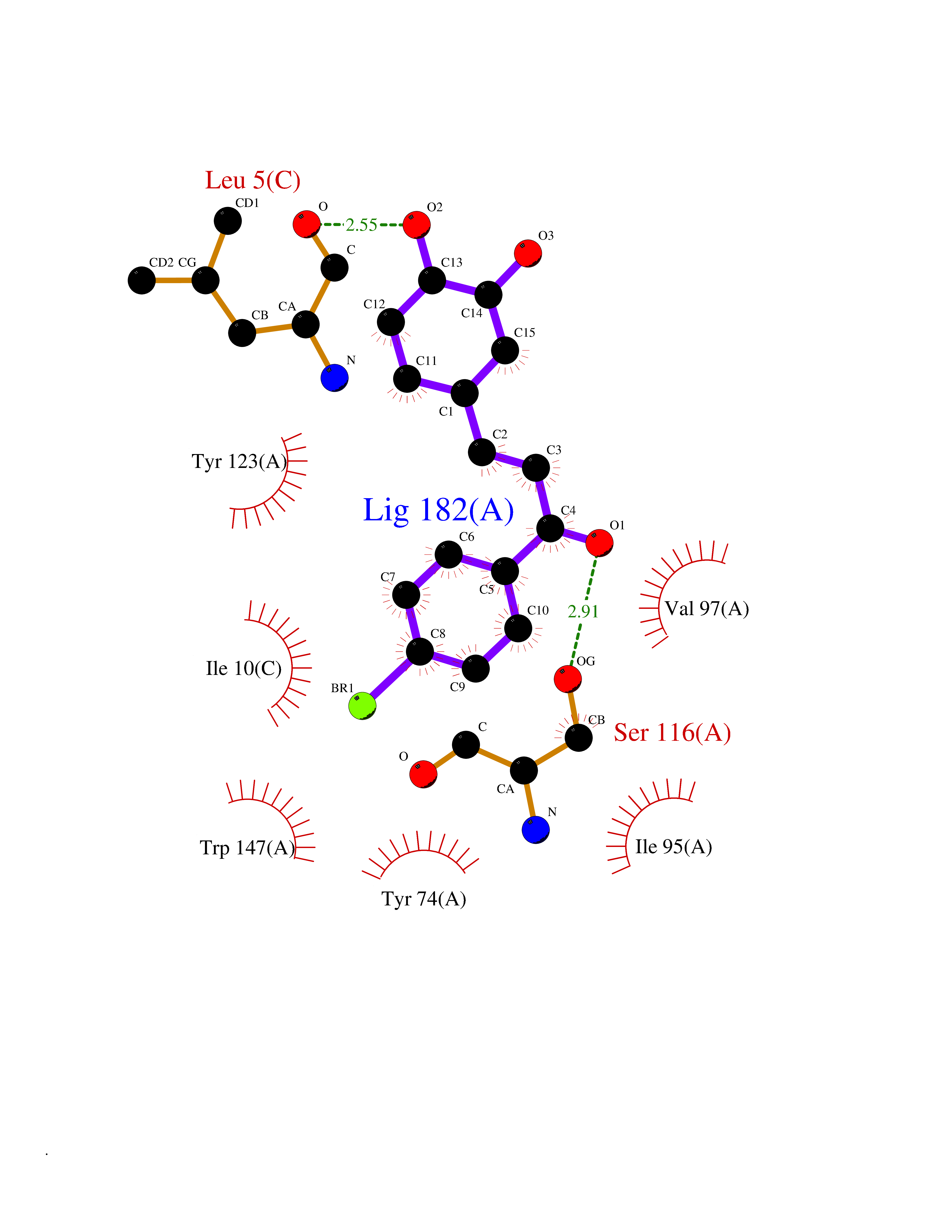 Ligplot Map 3D Binding mode Sequence SHSMRYFYTAMSRPGRGEPRFIAVGYVDDTQFVRFDSDAASPRMAPRAPWIEQEGPEYWDGETRNMKASAQTYRENLRIALRYYNQSEAGSHIIQVMYGCDVGPDGRLLRGHDQSAYDGKDYIALNEDLSSWTAADTAAQITQRKWEAARVAEQLRAYLEGLCVEWLRRYLENGKETLQLTTKLTNTNI Hydrogen bonds contact Hydrophobic contact | ||||
| 10 | Pseudomonas Transcriptional activator protein LasR (Pseudo LasR) | 3IX3 | 7.34 | |
Target general information Gen name Pseudo LasR Organism Pseudomonas aeruginosa (strain ATCC 15692 / DSM 22644 / CIP 104116 / JCM 14847 / LMG 12228 / 1C / PRS 101 / PAO1) Uniprot ID TTD ID Synonyms NA Protein family Autoinducer-regulated transcriptional regulatory protein family Biochemical class NA Function Transcriptional activator of elastase structural gene (LasB). Binds to the PAI autoinducer. Related diseases Growth hormone deficiency, isolated, 1A (IGHD1A) [MIM:262400]: An autosomal recessive, severe deficiency of growth hormone leading to dwarfism. Patients often develop antibodies to administered growth hormone. {ECO:0000269|PubMed:8364549}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Growth hormone deficiency, isolated, 1B (IGHD1B) [MIM:612781]: An autosomal recessive deficiency of growth hormone leading to short stature. Patients have low but detectable levels of growth hormone, significantly retarded bone age, and a positive response and immunologic tolerance to growth hormone therapy. {ECO:0000269|PubMed:12655557}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Kowarski syndrome (KWKS) [MIM:262650]: A syndrome clinically characterized by short stature associated with bioinactive growth hormone, normal or slightly increased growth hormone secretion, pathologically low insulin-like growth factor 1 levels, and normal catch-up growth on growth hormone replacement therapy. {ECO:0000269|PubMed:17519310, ECO:0000269|PubMed:8552145, ECO:0000269|PubMed:9276733}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Growth hormone deficiency, isolated, 2 (IGHD2) [MIM:173100]: An autosomal dominant deficiency of growth hormone leading to short stature. Clinical severity is variable. Patients have a positive response and immunologic tolerance to growth hormone therapy. {ECO:0000269|PubMed:11502836, ECO:0000269|PubMed:9152628}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08324 Interacts with NA EC number NA Uniprot keywords 3D-structure; Activator; DNA-binding; Quorum sensing; Reference proteome; Transcription; Transcription regulation Protein physicochemical properties Chain ID A Molecular weight (Da) 18305.5 Length 163 Aromaticity 0.12 Instability index 46.52 Isoelectric point 5.19 Charge (pH=7) -6.78 2D Binding mode Binding energy (Kcal/mol) -10.01 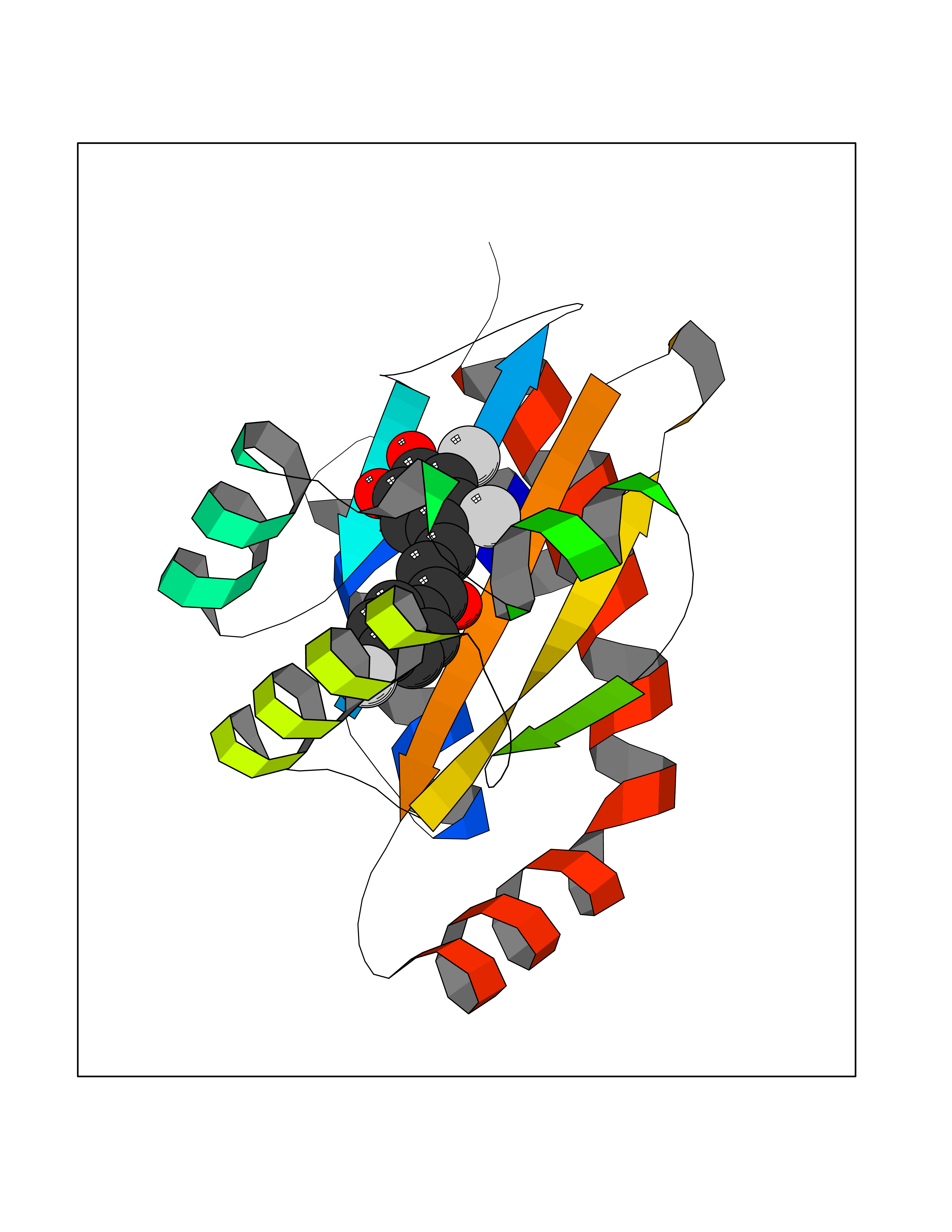 Molscript Map 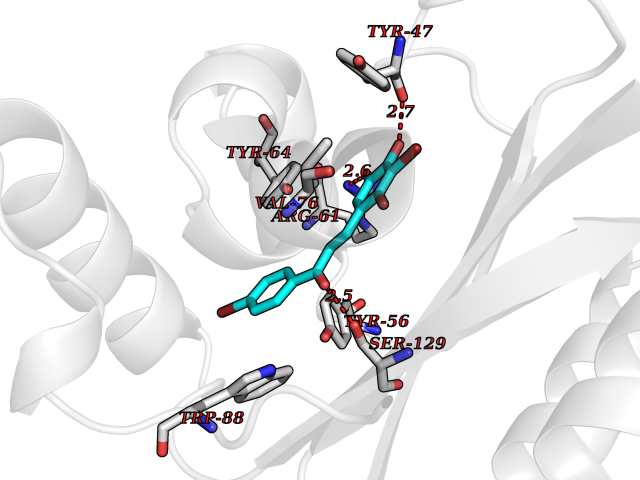 Pymol Map 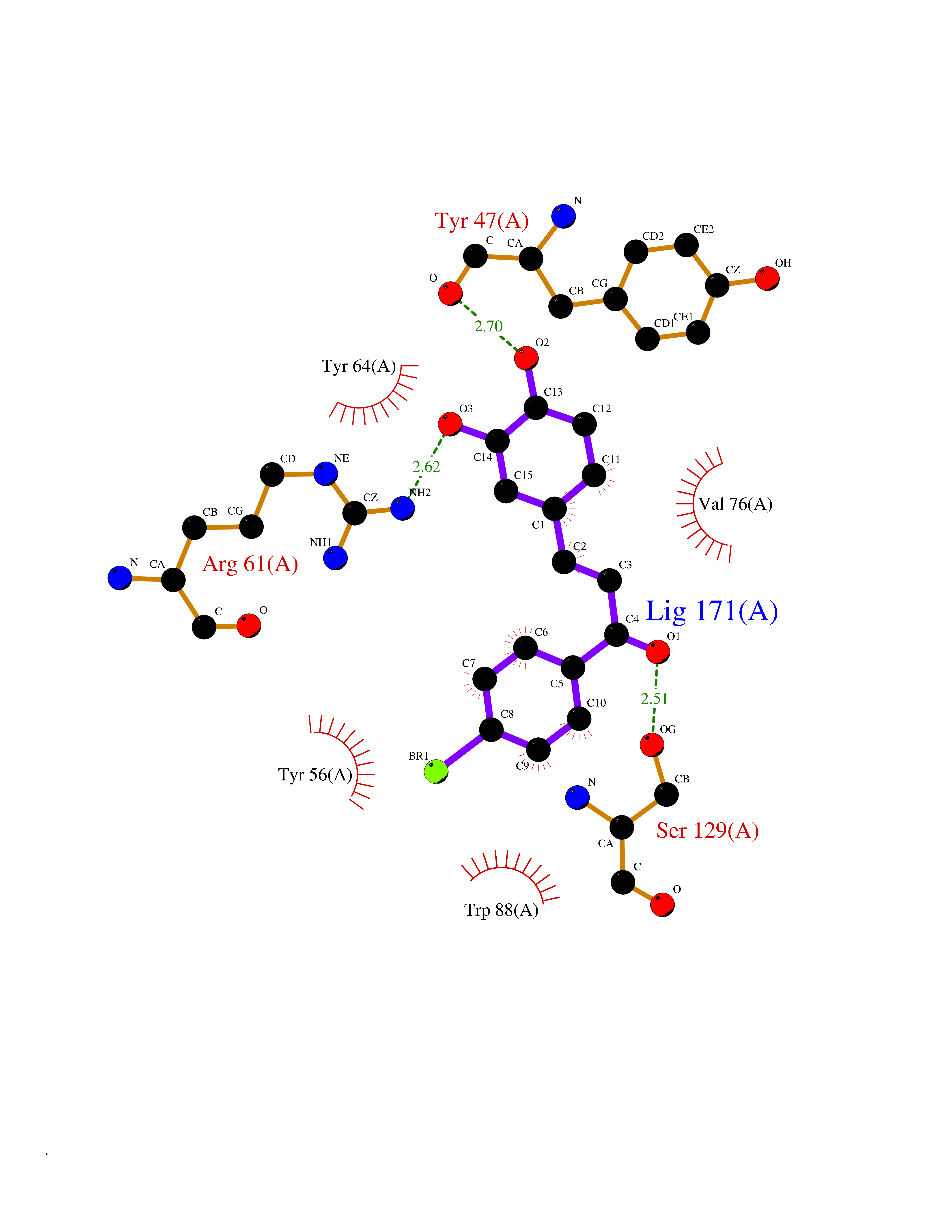 Ligplot Map 3D Binding mode Sequence FLELERSSGKLEWSAILQKMASDLGFSKILFGLLPKDSQDYENAFIVGNYPAAWREHYDRAGYARVDPTVSHCTQSVLPIFWEPSIYQTRKQHEFFEEASAAGLVYGLTMPLHGARGELGALSLSVEAENRAEANRFMESVLPTLWMLKDYALQSGAGLAFEH Hydrogen bonds contact Hydrophobic contact | ||||
| 11 | Spermine synthase | 3C6K | 7.33 | |
Target general information Gen name SMS Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Spermidine/spermine synthase family Biochemical class Transferase Function Spermine synthase activity. Related diseases Intellectual developmental disorder, X-linked, syndromic, Snyder-Robinson type (MRXSSR) [MIM:309583]: An X-linked intellectual disability syndrome characterized by a collection of clinical features including facial asymmetry, marfanoid habitus, hypertonia, osteoporosis and unsteady gait. {ECO:0000269|PubMed:14508504, ECO:0000269|PubMed:18550699, ECO:0000269|PubMed:19206178, ECO:0000269|PubMed:22612257, ECO:0000269|PubMed:23696453, ECO:0000269|PubMed:23897707}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00127 Interacts with NA EC number 2.5.1.22 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Direct protein sequencing; Intellectual disability; Phosphoprotein; Polyamine biosynthesis; Proteomics identification; Reference proteome; Transferase Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 27177.1 Length 238 Aromaticity 0.11 Instability index 41.31 Isoelectric point 4.82 Charge (pH=7) -9.19 2D Binding mode Binding energy (Kcal/mol) -10  Molscript Map 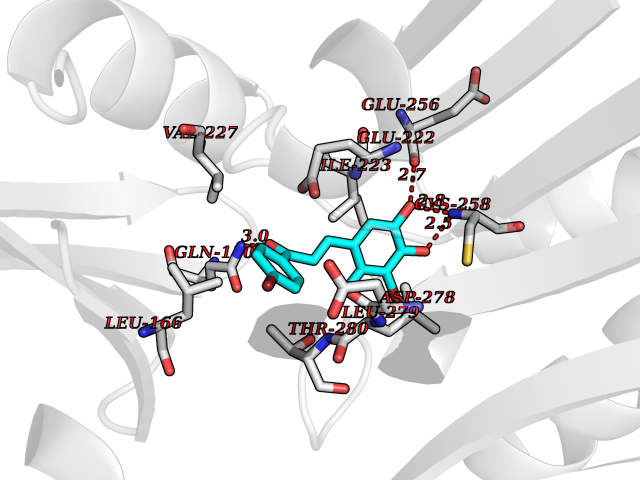 Pymol Map 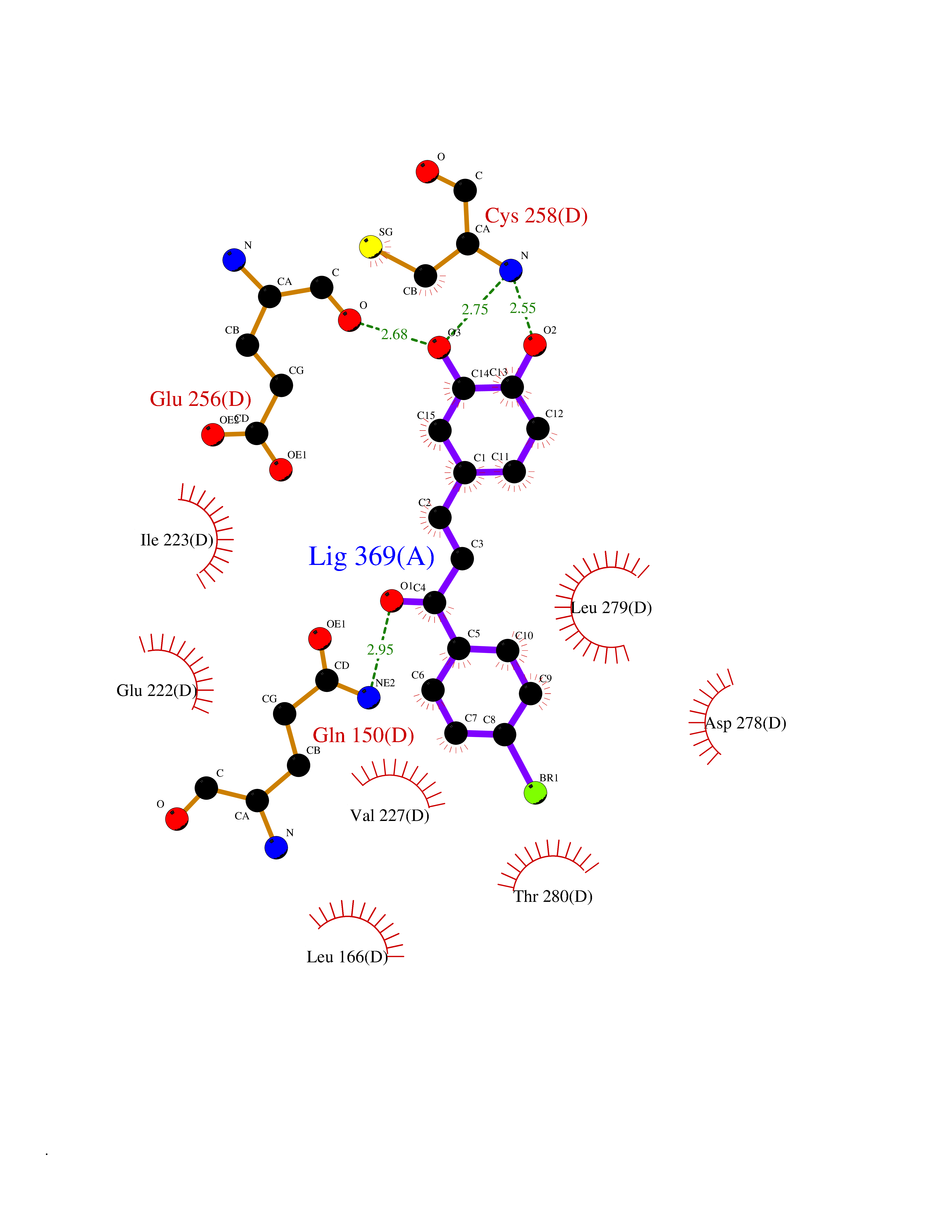 Ligplot Map 3D Binding mode Sequence RYWPTADGRLVEYDIDEVVYDEDSPYQNIKILHSKQFGNILILSGDVNLAESDLAYTRAIMGSGKEDYTGKDVLILGGGDGGILCEIVKLKPKMVTMVEIDQMVIDGCKKYMRKDVLDNLKGDCYQVLIEDCIPVLKRYAKEGREFDYVINDLTAVPISTSPSTWEFLRLILDLSMKVLKQDGKYFTQGNCVNLTEALSLYEEQLGRLYCPVEFSKEIVCVPSYLELWVFYTVWKKAK Hydrogen bonds contact Hydrophobic contact | ||||
| 12 | Metabotropic glutamate receptor 5 (mGluR5) | 4OO9 | 7.32 | |
Target general information Gen name GRM5 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms MGLUR5; GPRC1E Protein family G-protein coupled receptor 3 family Biochemical class GPCR glutamate Function G-protein coupled receptor for glutamate. Ligand binding causes a conformation change that triggers signaling via guanine nucleotide-binding proteins (G proteins) and modulates the activity of down-stream effectors. Signaling activates a phosphatidylinositol-calcium second messenger system and generates a calcium-activated chloride current. Plays an important role in the regulation of synaptic plasticity and the modulation of the neural network activity. Related diseases Charcot-Marie-Tooth disease, axonal, 2D (CMT2D) [MIM:601472]: A dominant axonal form of Charcot-Marie-Tooth disease, a disorder of the peripheral nervous system, characterized by progressive weakness and atrophy, initially of the peroneal muscles and later of the distal muscles of the arms. Charcot-Marie-Tooth disease is classified in two main groups on the basis of electrophysiologic properties and histopathology: primary peripheral demyelinating neuropathies (designated CMT1 when they are dominantly inherited) and primary peripheral axonal neuropathies (CMT2). Neuropathies of the CMT2 group are characterized by signs of axonal degeneration in the absence of obvious myelin alterations, normal or slightly reduced nerve conduction velocities, and progressive distal muscle weakness and atrophy. {ECO:0000269|PubMed:12690580, ECO:0000269|PubMed:17035524, ECO:0000269|PubMed:17101916, ECO:0000269|PubMed:17663003, ECO:0000269|PubMed:20169446, ECO:0000269|PubMed:24604904, ECO:0000269|PubMed:25168514, ECO:0000269|PubMed:26244500, ECO:0000269|PubMed:26503042, ECO:0000269|PubMed:31173493}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Neuronopathy, distal hereditary motor, autosomal dominant 5 (HMND5) [MIM:600794]: A form of distal hereditary motor neuronopathy, a heterogeneous group of neuromuscular disorders caused by selective degeneration of motor neurons in the anterior horn of the spinal cord, without sensory deficit in the posterior horn. The overall clinical picture consists of a classical distal muscular atrophy syndrome in the legs without clinical sensory loss. The disease starts with weakness and wasting of distal muscles of the anterior tibial and peroneal compartments of the legs. Later on, weakness and atrophy may expand to the proximal muscles of the lower limbs and/or to the distal upper limbs. {ECO:0000269|PubMed:12690580, ECO:0000269|PubMed:17035524, ECO:0000269|PubMed:23279345, ECO:0000269|PubMed:24627108, ECO:0000269|PubMed:26503042}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Spinal muscular atrophy, infantile, James type (SMAJI) [MIM:619042]: An autosomal dominant form of spinal muscular atrophy, a group of neuromuscular disorders characterized by degeneration of the anterior horn cells of the spinal cord, leading to symmetrical muscle weakness and atrophy. SMAJI is a severe disease characterized by hypotonia manifesting in the first weeks or months of life, delayed motor development, motor regression, and muscle weakness and atrophy primarily affecting distal muscles. Additional variable features include feeding difficulties, poor overall growth, foot deformities, kyphosis, hyperlordosis, scoliosis, vocal cord dysfunction, and respiratory insufficiency. {ECO:0000269|PubMed:32181591}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00659; DB05070; DB12733; DB06201 Interacts with P41594; Q7Z6G3 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disulfide bond; G-protein coupled receptor; Glycoprotein; Membrane; Methylation; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Signal; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 27065.4 Length 247 Aromaticity 0.13 Instability index 42.92 Isoelectric point 9.24 Charge (pH=7) 11.34 2D Binding mode Binding energy (Kcal/mol) -9.98 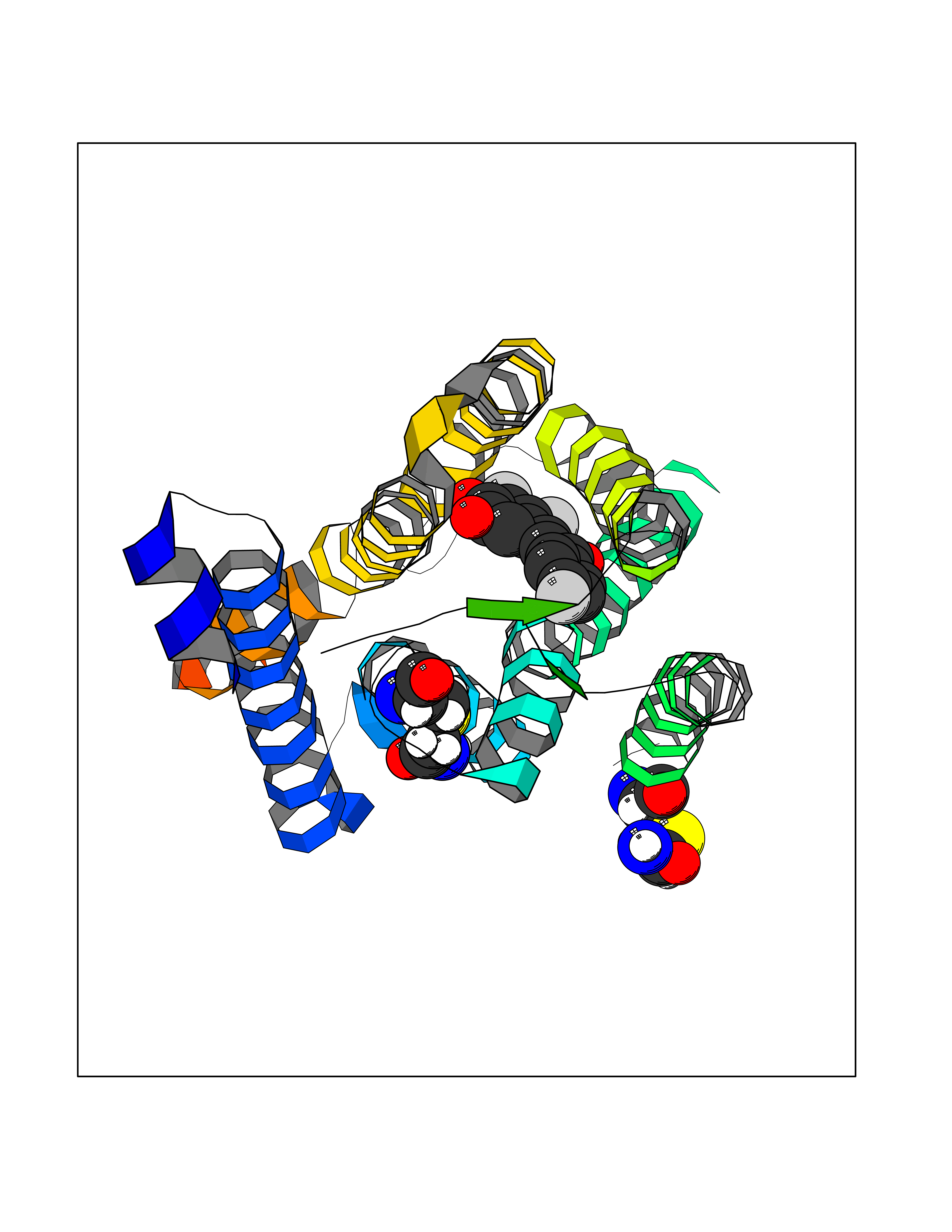 Molscript Map 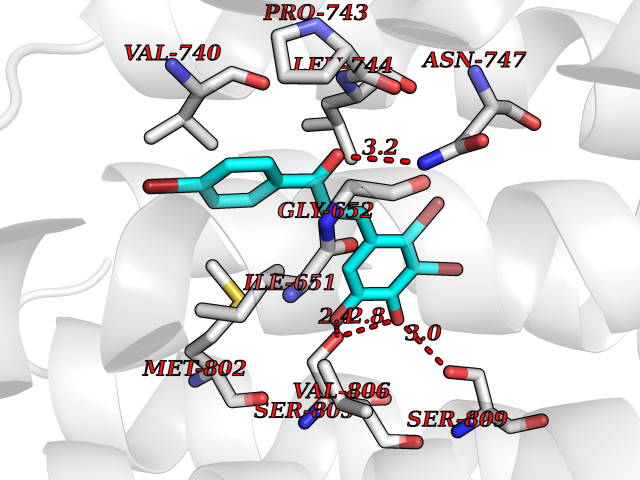 Pymol Map 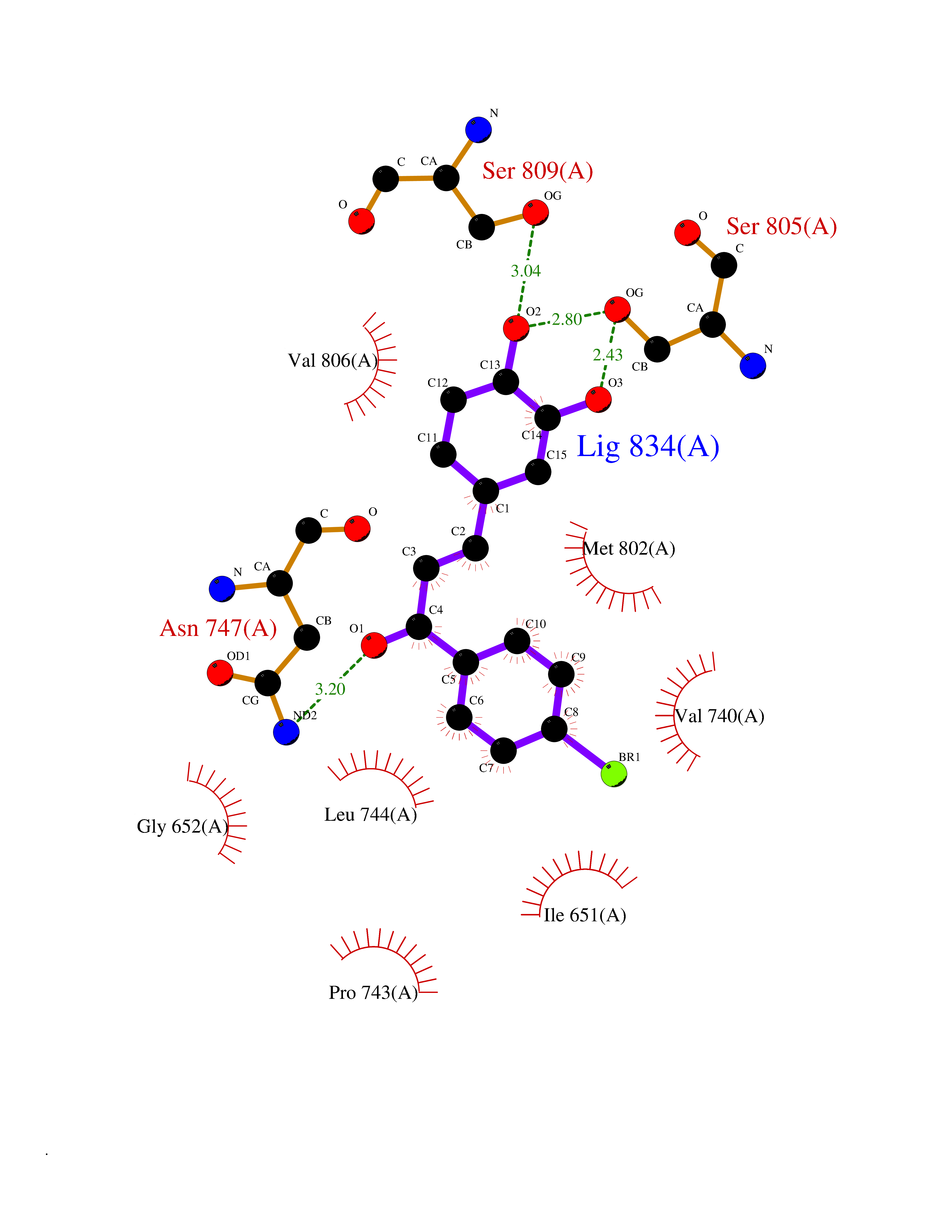 Ligplot Map 3D Binding mode Sequence SPVQYLRWGDPAPIAAVVFACLGLLATLFVTVVFIIYRDTPVVKSSSRELCYIILAGICLGYLCTFXLIAKPKQIYCYLQRIGIGLSPAMSYSALVTKTYRAARILAMSKKSAXAQLVIAFILICIQLGIIVALFIMEPPDIMVYLICNTTNLGVVAPLGYNGLLILACTFYAFKTRNVPANFNEAKYIAFTMYTTCIIWLAFVPIYFGSNYKIITMCFSVSLSATVALGCMFVPKVYIILAKPERN Hydrogen bonds contact Hydrophobic contact | ||||
| 13 | Dihydroorotate dehydrogenase (DHODH) | 4OQV | 7.32 | |
Target general information Gen name DHODH Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Dihydroorotate oxidase; Dihydroorotate dehydrogenase (quinone), mitochondrial; DHOdehase; DHODH Protein family Dihydroorotate dehydrogenase family, Type 2 subfamily Biochemical class CH-CH donor oxidoreductase Function Catalyzes the conversion of dihydroorotate to orotate with quinone as electron acceptor. Related diseases Postaxial acrofacial dysostosis (POADS) [MIM:263750]: POADS is characterized by severe micrognathia, cleft lip and/or palate, hypoplasia or aplasia of the posterior elements of the limbs, coloboma of the eyelids and supernumerary nipples. POADS is a very rare disorder: only 2 multiplex families, each consisting of 2 affected siblings born to unaffected, nonconsanguineous parents, have been described among a total of around 30 reported cases. {ECO:0000269|PubMed:19915526}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07559; DB07561; DB08172; DB08169; DB07443; DB07978; DB07975; DB04281; DB08249; DB07977; DB07976; DB04583; DB08008; DB01117; DB03523; DB03480; DB02613; DB04147; DB03247; DB01097; DB06481; DB08006; DB02262; DB05125; DB08880; DB07646 Interacts with Q6ZMZ0; P49638 EC number EC 1.3.5.2 Uniprot keywords 3D-structure; Disease variant; Flavoprotein; FMN; Membrane; Mitochondrion; Mitochondrion inner membrane; Oxidoreductase; Proteomics identification; Pyrimidine biosynthesis; Reference proteome; Transit peptide; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 38341.4 Length 353 Aromaticity 0.05 Instability index 39.27 Isoelectric point 9.28 Charge (pH=7) 5.52 2D Binding mode Binding energy (Kcal/mol) -9.98 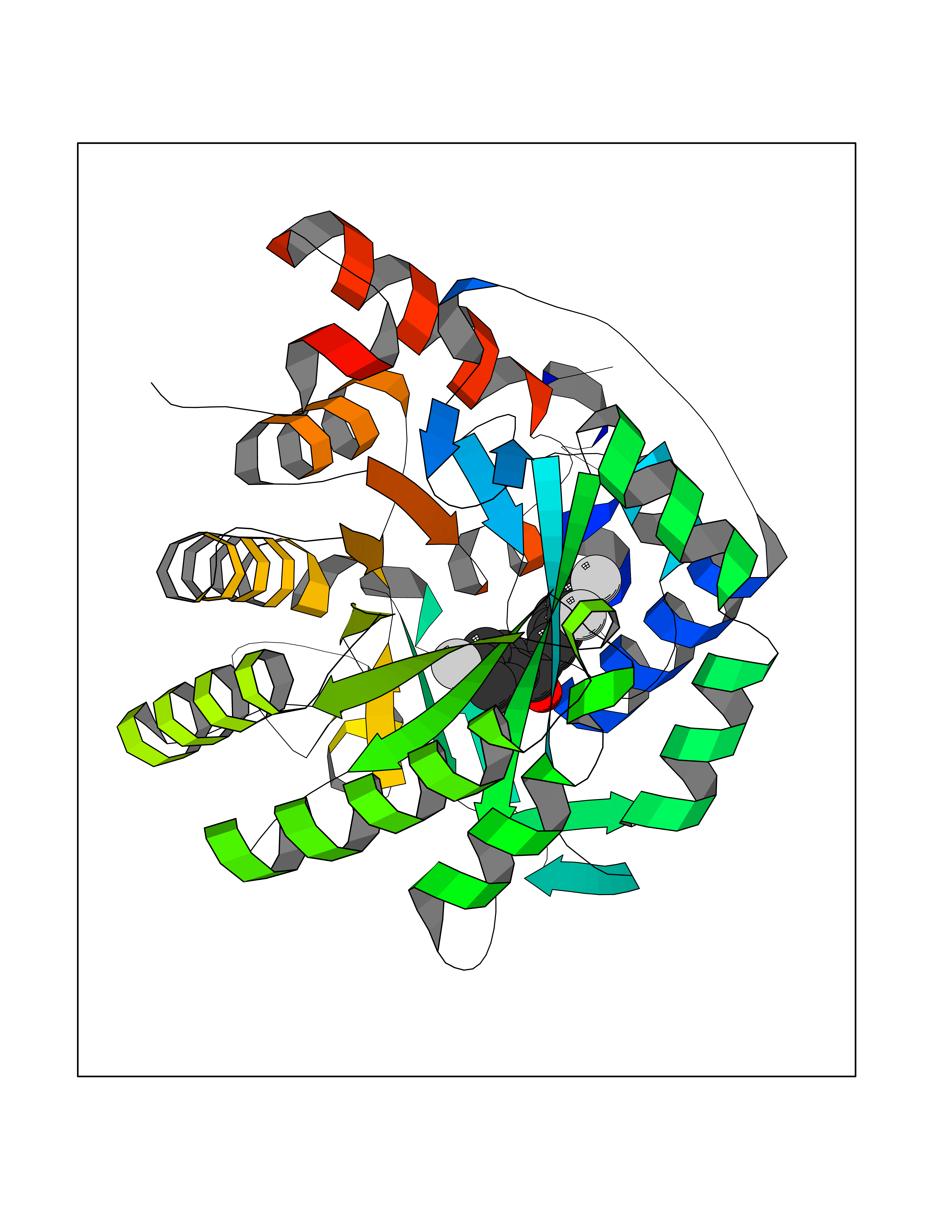 Molscript Map 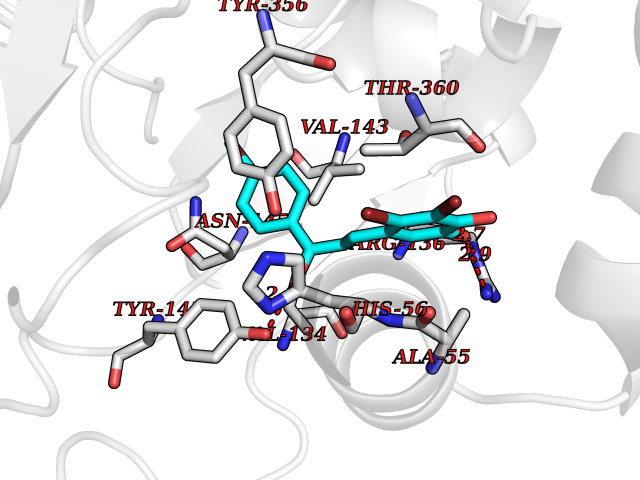 Pymol Map 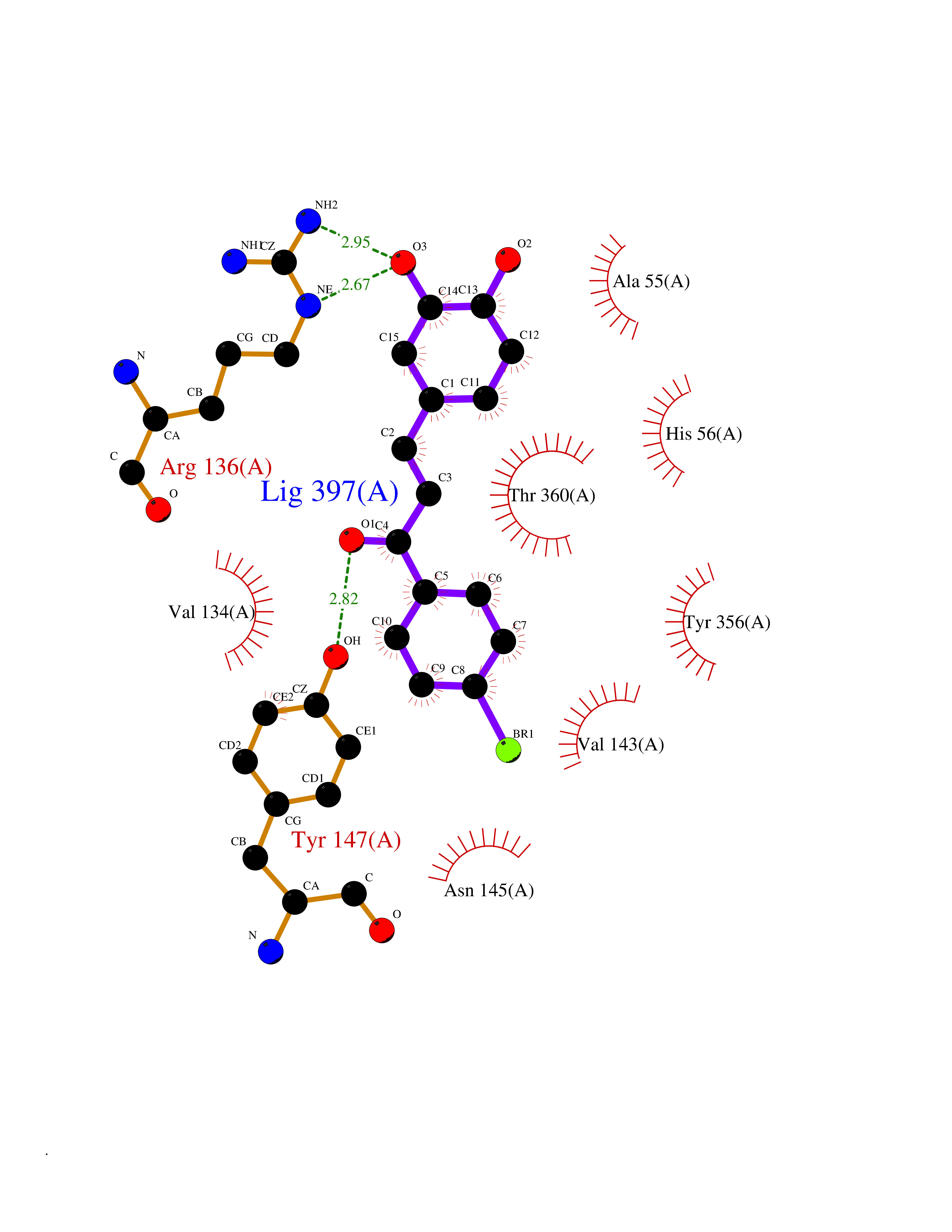 Ligplot Map 3D Binding mode Sequence DERFYAEHLMPTLQGLLDPESAHRLAVRFTSLGLLPRARFQDSDMLEVRVLGHKFRNPVGIAAGFDKHGEAVDGLYKMGFGFVEIGSVTPKPQEGNPRPRVFRLPEDQAVINRYGFNSHGLSVVEHRLRARQQKQAKLTEDGLPLGVNLGKNKTSVDAAEDYAEGVRVLGPLADYLVVNVSSPGKAELRRLLTKVLQERDGLRRVHRPAVLVKIAPDLTSQDKEDIASVVKELGIDGLIVTNTTVSRPAGLQGALRSETGGLSGKPLRDLSTQTIREMYALTQGRVPIIGVGGVSSGQDALEKIRAGASLVQLYTALTFWGPPVVGKVKRELEALLKEQGFGGVTDAIGADHR Hydrogen bonds contact Hydrophobic contact | ||||
| 14 | Rhinovirus Protease 3C (HRV P3C) | 1FPN | 7.32 | |
Target general information Gen name HRV P3C Organism Human rhinovirus 2 (HRV-2) Uniprot ID TTD ID Synonyms Rhinovirus P3C Protein family Picornaviruses polyprotein family Biochemical class NA Function Capsid protein VP1: Forms an icosahedral capsid of pseudo T=3 symmetry with capsid proteins VP2 and VP3. The capsid is 300 Angstroms in diameter, composed of 60 copies of each capsid protein and enclosing the viral positive strand RNA genome. Capsid protein VP1 mainly forms the vertices of the capsid. Capsid protein VP1 interacts with host VLDLR to provide virion attachment to target host cells. This attachment induces virion internalization. Tyrosine kinases are probably involved in the entry process. After binding to its receptor, the capsid undergoes conformational changes. Capsid protein VP1 N-terminus (that contains an amphipathic alpha-helix) and capsid protein VP4 are externalized. Together, they shape a pore in the host membrane through which viral genome is translocated to host cell cytoplasm. After genome has been released, the channel shrinks (By similarity). Related diseases Charcot-Marie-Tooth disease, axonal, 2DD (CMT2DD) [MIM:618036]: A dominant axonal form of Charcot-Marie-Tooth disease, a disorder of the peripheral nervous system, characterized by progressive weakness and atrophy, initially of the peroneal muscles and later of the distal muscles of the arms. Charcot-Marie-Tooth disease is classified in two main groups on the basis of electrophysiologic properties and histopathology: primary peripheral demyelinating neuropathies (designated CMT1 when they are dominantly inherited) and primary peripheral axonal neuropathies (CMT2). Neuropathies of the CMT2 group are characterized by signs of axonal degeneration in the absence of obvious myelin alterations, normal or slightly reduced nerve conduction velocities, and progressive distal muscle weakness and atrophy. {ECO:0000269|PubMed:29499166}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Hypomagnesemia, seizures, and impaired intellectual development 2 (HOMGSMR2) [MIM:618314]: An autosomal dominant disease characterized by generalized seizures in infancy, severe hypomagnesemia, and renal magnesium wasting. Seizures persist despite magnesium supplementation and are associated with significant intellectual disability. {ECO:0000269|PubMed:30388404}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02313; DB03017 Interacts with NA EC number EC 3.4.22.28 Uniprot keywords 3D-structure; Activation of host autophagy by virus; ATP-binding; Autocatalytic cleavage; Capsid protein; Covalent protein-RNA linkage; DNA replication; Eukaryotic host gene expression shutoff by virus; Eukaryotic host translation shutoff by virus; Helicase; Host cytoplasm; Host cytoplasmic vesicle; Host gene expression shutoff by virus; Host membrane; Host mRNA suppression by virus; Host nucleus; Host-virus interaction; Hydrolase; Inhibition of host innate immune response by virus; Inhibition of host mRNA nuclear export by virus; Inhibition of host RIG-I by virus; Inhibition of host RLR pathway by virus; Ion channel; Ion transport; Lipoprotein; Magnesium; Membrane; Metal-binding; Myristate; Nucleotide-binding; Nucleotidyltransferase; Phosphoprotein; Pore-mediated penetration of viral genome into host cell; Protease; Repeat; RNA-binding; RNA-directed RNA polymerase; T=pseudo3 icosahedral capsid protein; Thiol protease; Transferase; Transport; Viral attachment to host cell; Viral immunoevasion; Viral ion channel; Viral penetration into host cytoplasm; Viral RNA replication; Virion; Virus endocytosis by host; Virus entry into host cell; Zinc; Zinc-finger Protein physicochemical properties Chain ID 1 Molecular weight (Da) 30610.1 Length 269 Aromaticity 0.1 Instability index 55.66 Isoelectric point 6.86 Charge (pH=7) -0.38 2D Binding mode Binding energy (Kcal/mol) -9.98 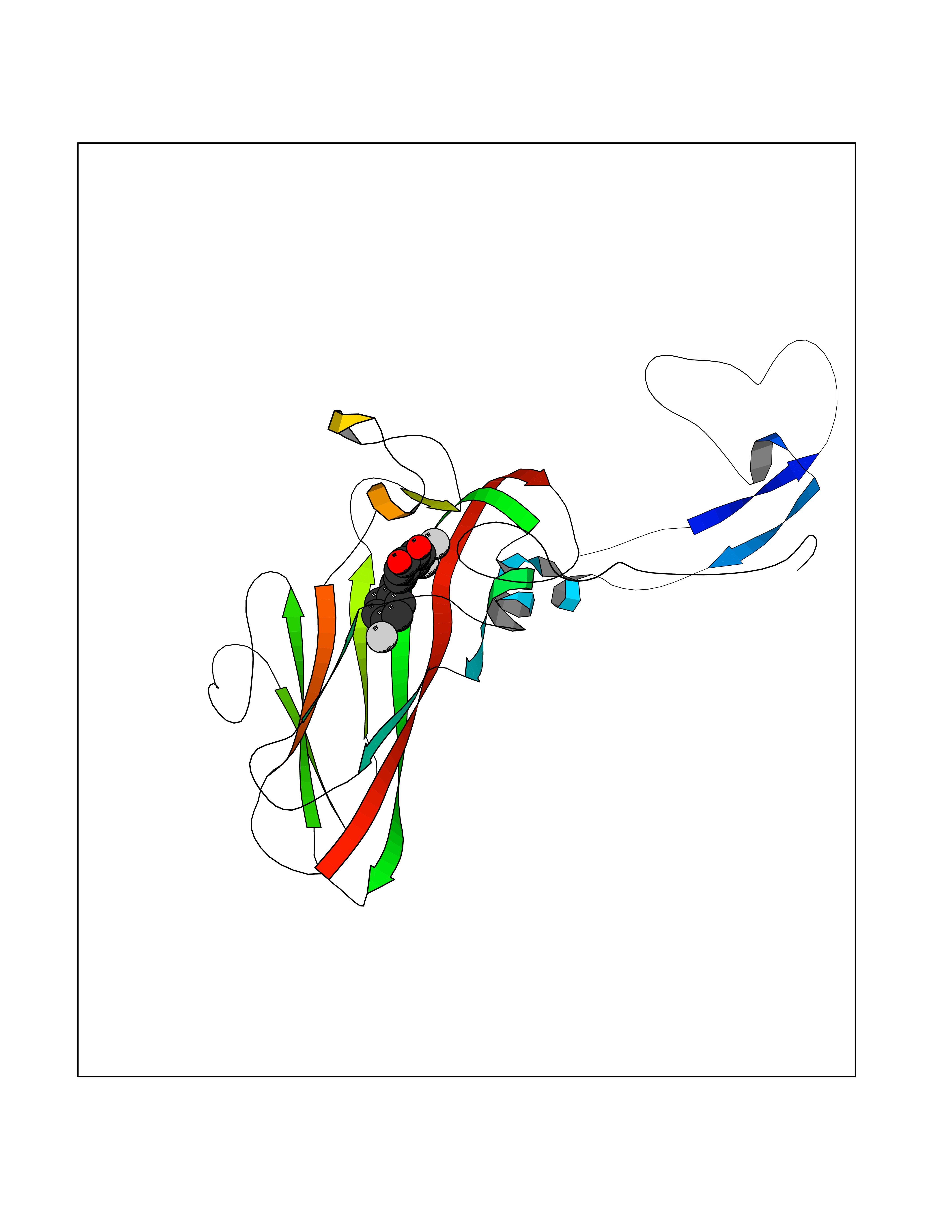 Molscript Map 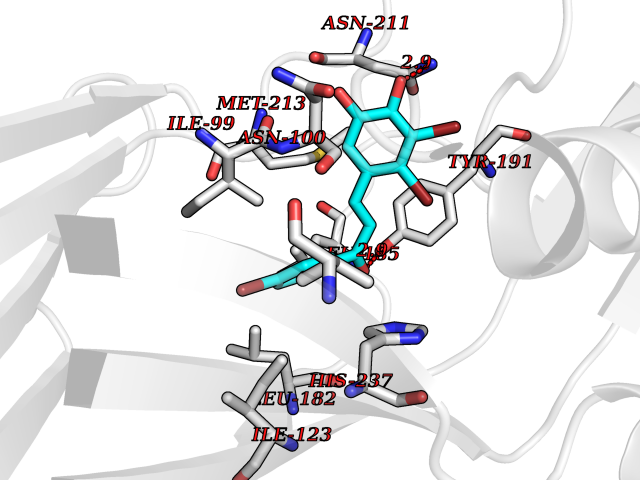 Pymol Map 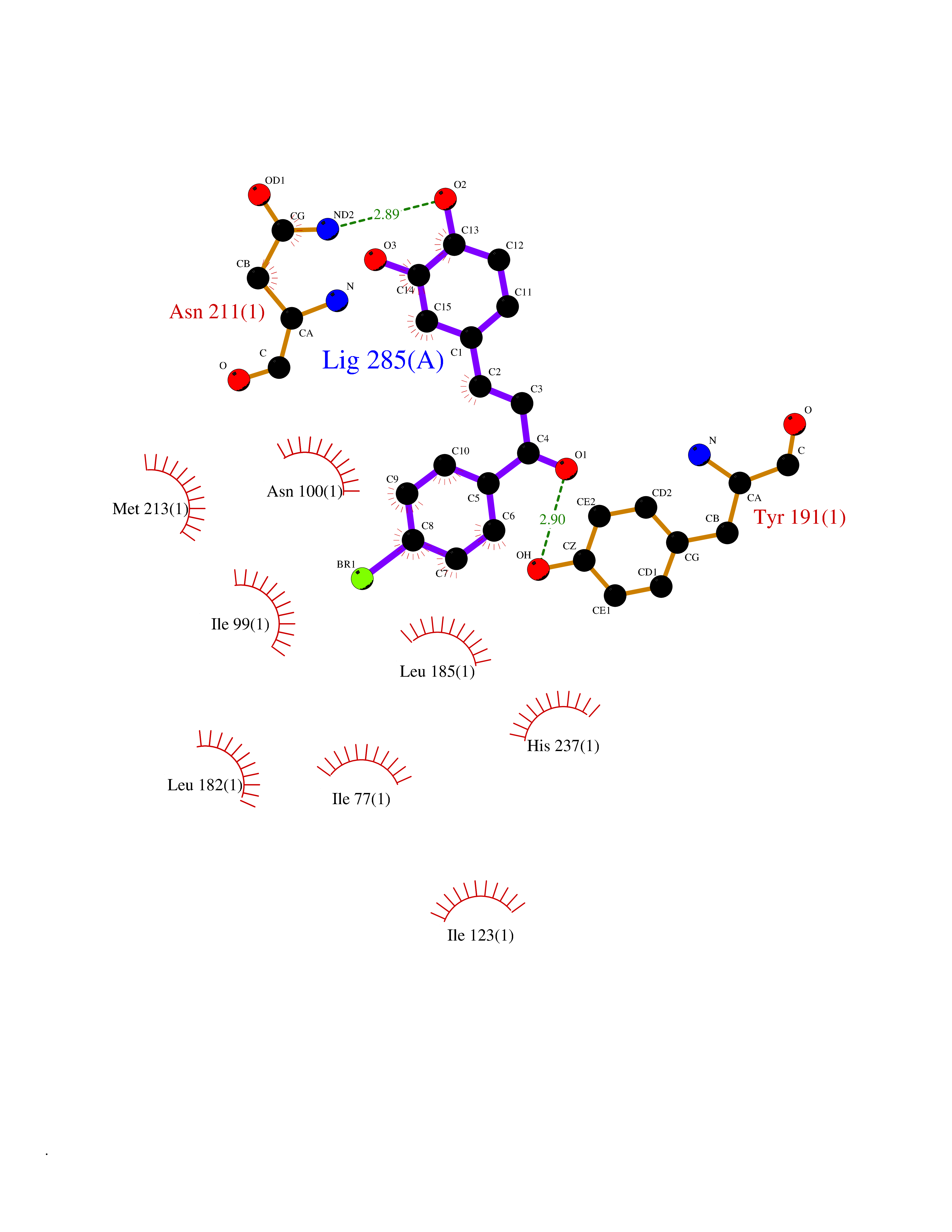 Ligplot Map 3D Binding mode Sequence LVVPNINSSNPTTSNSAPALDAAETGHTSSVQPEDVIETRYVQTSQTRDEMSLESFLGRSGCIHESKLEVTLANYNKENFTVWAINLQEMAQIRRKFELFTYTRFDSEITLVPCISALSQDIGHITMQYMYVPPGAPVPNSRDDYAWQSGTNASVFWQHGQAYPRFSLPFLSVASAYYMFYDGYDEQDQNYGTANTNNMGSLCSRIVTEKHIHKVHIMTRIYHKAKHVKAWCPRPPRALEYTRAHRTNFKIEDRSIQTAIVTRPIITTA Hydrogen bonds contact Hydrophobic contact | ||||
| 15 | Caspase-7 (CASP7) | 1SHJ | 7.32 | |
Target general information Gen name CASP7 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms MCH3; ICE-like apoptotic protease 3; ICE-LAP3; CMH-1; CASP-7; Apoptotic protease Mch-3 Protein family Peptidase C14A family Biochemical class Peptidase Function Cleaves and activates sterol regulatory element binding proteins (SREBPs). Proteolytically cleaves poly(ADP-ribose) polymerase (PARP) at a '216-Asp-|-Gly-217' bond. Overexpression promotes programmed cell death. Involved in the activation cascade of caspases responsible for apoptosis execution. Related diseases Pregnancy loss, recurrent, 3 (RPRGL3) [MIM:614391]: A common complication of pregnancy, resulting in spontaneous abortion before the fetus has reached viability. The term includes all miscarriages from the time of conception until 24 weeks of gestation. Recurrent pregnancy loss is defined as 3 or more consecutive spontaneous abortions. {ECO:0000269|PubMed:17339269}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB05408; DB03384; DB06255 Interacts with Q13490; P83105; P42858; Q8N4N3-2; P43364; Q16236; Q9GZT8; Q13177; P27986-2; P21673; Q86WV1-2; P17405; P98170 EC number EC 3.4.22.60 Uniprot keywords 3D-structure; Acetylation; Allosteric enzyme; Alternative splicing; Apoptosis; Cytoplasm; Hydrolase; Nucleus; Phosphoprotein; Protease; Proteomics identification; Reference proteome; RNA-binding; Secreted; Thiol protease; Ubl conjugation; Zymogen Protein physicochemical properties Chain ID A,B Molecular weight (Da) 47441.5 Length 417 Aromaticity 0.11 Instability index 20.98 Isoelectric point 8.38 Charge (pH=7) 6.12 2D Binding mode Binding energy (Kcal/mol) -9.99 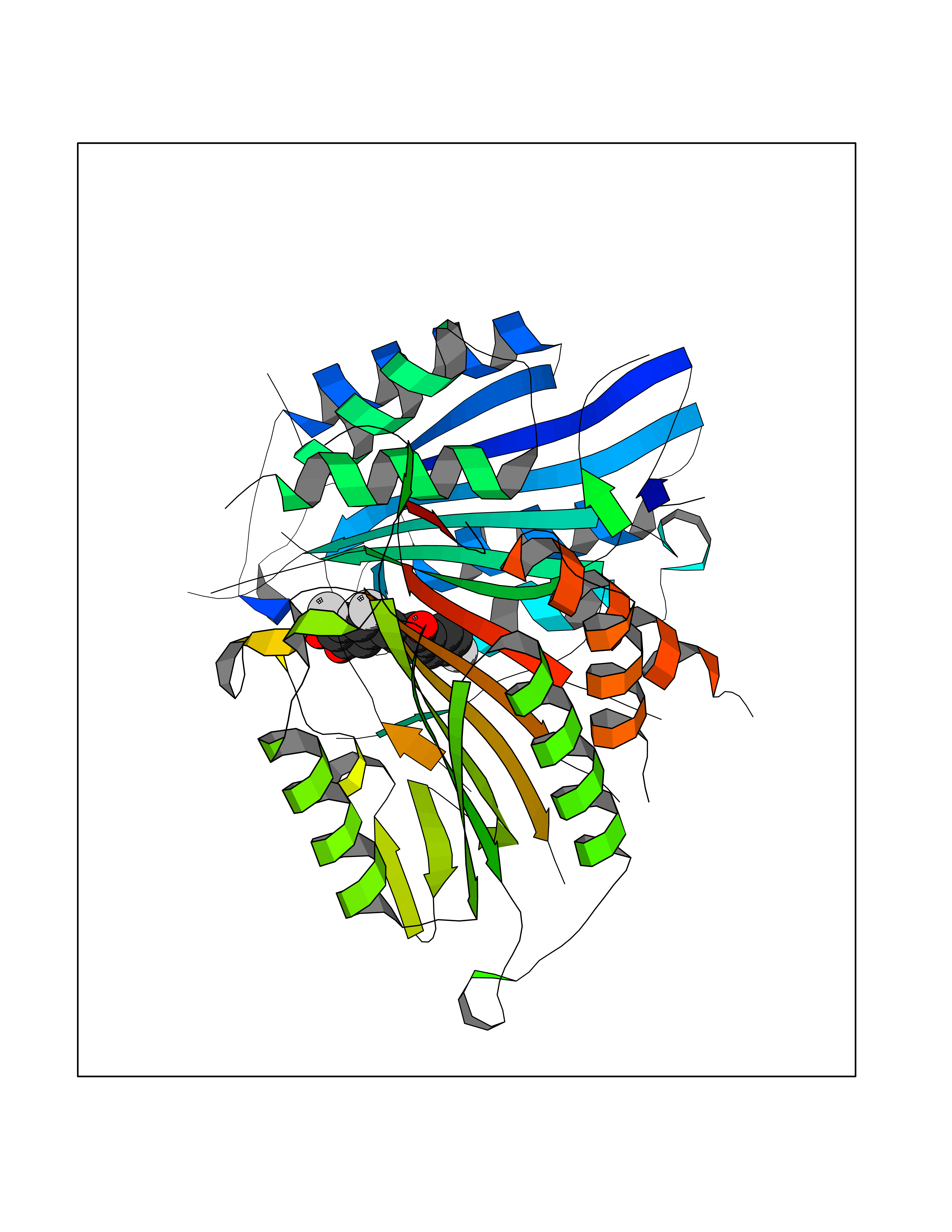 Molscript Map 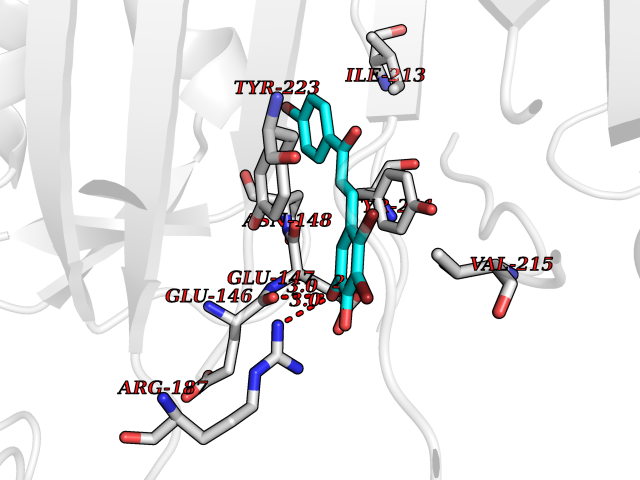 Pymol Map 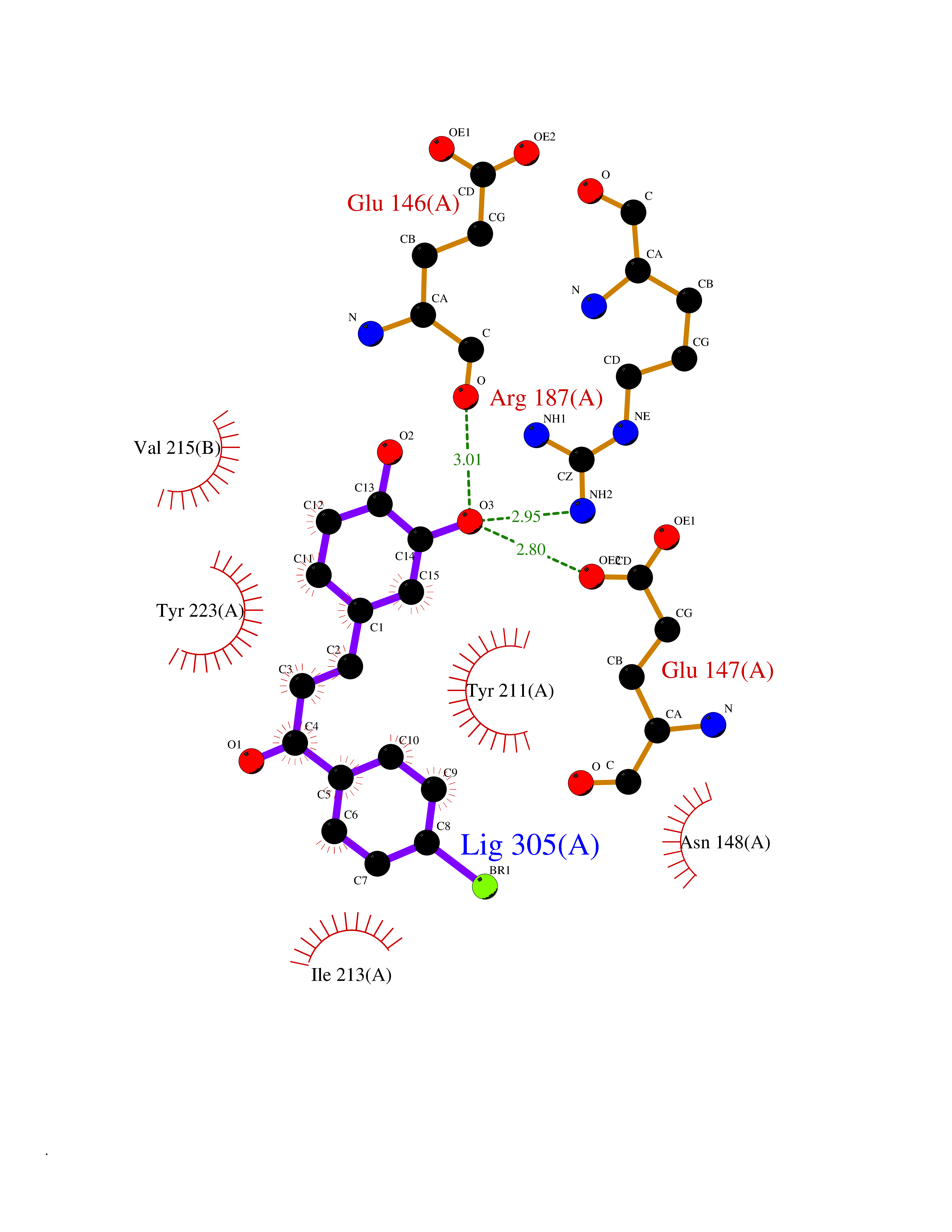 Ligplot Map 3D Binding mode Sequence TYQYNMNFEKLGKCIIINNKNFDKVTGMGVRNGTDKDAEALFKCFRSLGFDVIVYNDCSCAKMQDLLKKASEEDHTNAACFACILLSHGEENVIYGKDGVTPIKDLTAHFRGARCKTLLEKPKLFFIQACRGTEPRYKIPVEADFLFAYSTVRGSWFVQALCSILEEHGKDLEIMQILTRVNDRVARHFKKQIPCVVSMLTKELYFSQVPTYQYNMNFEKLGKCIIINNKNFDKVTGMGVRNGTDKDAEALFKCFRSLGFDVIVYNDCSCAKMQDLLKKASEEDHTNAACFACILLSHGEENVIYGKDGVTPIKDLTAHFRGARCKTLLEKPKLFFIQACRGPRYKIPVEADFLFAYSTVPGSWFVQALCSILEEHGKDLEIMQILTRVNDRVARHFESKQIPCVVSMLTKELYFSQ Hydrogen bonds contact Hydrophobic contact | ||||
| 16 | Acetyl-CoA carboxylase (ACC) (EC 6.4.1.2) (Fatty acid synthetase 3) (mRNA transport-defective protein 7) [Includes: Biotin carboxylase (EC 6.3.4.14)] | 1UYS | 7.31 | |
Target general information Gen name ACC1 Organism Saccharomyces cerevisiae (strain ATCC 204508 / S288c) (Baker's yeast) Uniprot ID TTD ID NA Synonyms MTR7;YNR016C;N3175;ABP2;FAS3 Protein family NA Biochemical class NA Function Carries out three functions: biotin carboxyl carrier protein, biotin carboxylase and carboxyltransferase. Involved in the synthesis of very-long-chain fatty acid synthesis which is required to maintain a functional nuclear envelope. Required for acylation and vacuolar membrane association of VAC8 which is necessary to maintain a normal morphology of the vacuole. {ECO:0000269|PubMed:10757783, ECO:0000269|PubMed:12730220, ECO:0000269|PubMed:6103540, ECO:0000269|PubMed:6108218, ECO:0000269|PubMed:8943372}." Related diseases A chromosomal aberration involving NFKB2 is found in a case of B-cell non Hodgkin lymphoma (B-NHL). Translocation t(10;14)(q24;q32) with IGHA1. The resulting oncogene is also called Lyt-10C alpha variant.; DISEASE: A chromosomal aberration involving NFKB2 is found in a cutaneous T-cell leukemia (C-TCL) cell line. This rearrangement produces the p80HT gene which codes for a truncated 80 kDa protein (p80HT).; DISEASE: In B-cell leukemia (B-CLL) cell line, LB40 and EB308, can be found after heterogeneous chromosomal aberrations, such as internal deletions.; DISEASE: Immunodeficiency, common variable, 10 (CVID10) [MIM:615577]: A primary immunodeficiency characterized by childhood-onset of recurrent infections, hypogammaglobulinemia, and decreased numbers of memory and marginal zone B-cells. Some patients may develop autoimmune features and have circulating autoantibodies. An unusual feature is central adrenal insufficiency. {ECO:0000269|PubMed:24140114, ECO:0000269|PubMed:25524009}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q00955 EC number 6.3.4.14; 6.4.1.2 Uniprot keywords 3D-structure; Acetylation; ATP-binding; Biotin; Cytoplasm; Direct protein sequencing; Endoplasmic reticulum; Fatty acid biosynthesis; Fatty acid metabolism; Ligase; Lipid biosynthesis; Lipid metabolism; Manganese; Membrane; Metal-binding; Multifunctional enzyme; Nucleotide-binding; Phosphoprotein; Reference proteome Protein physicochemical properties Chain ID B,C Molecular weight (Da) 145619 Length 1328 Aromaticity 0.1 Instability index 30.31 Isoelectric point 5.32 Charge (pH=7) -26.79 2D Binding mode Binding energy (Kcal/mol) -9.97  Molscript Map  Pymol Map 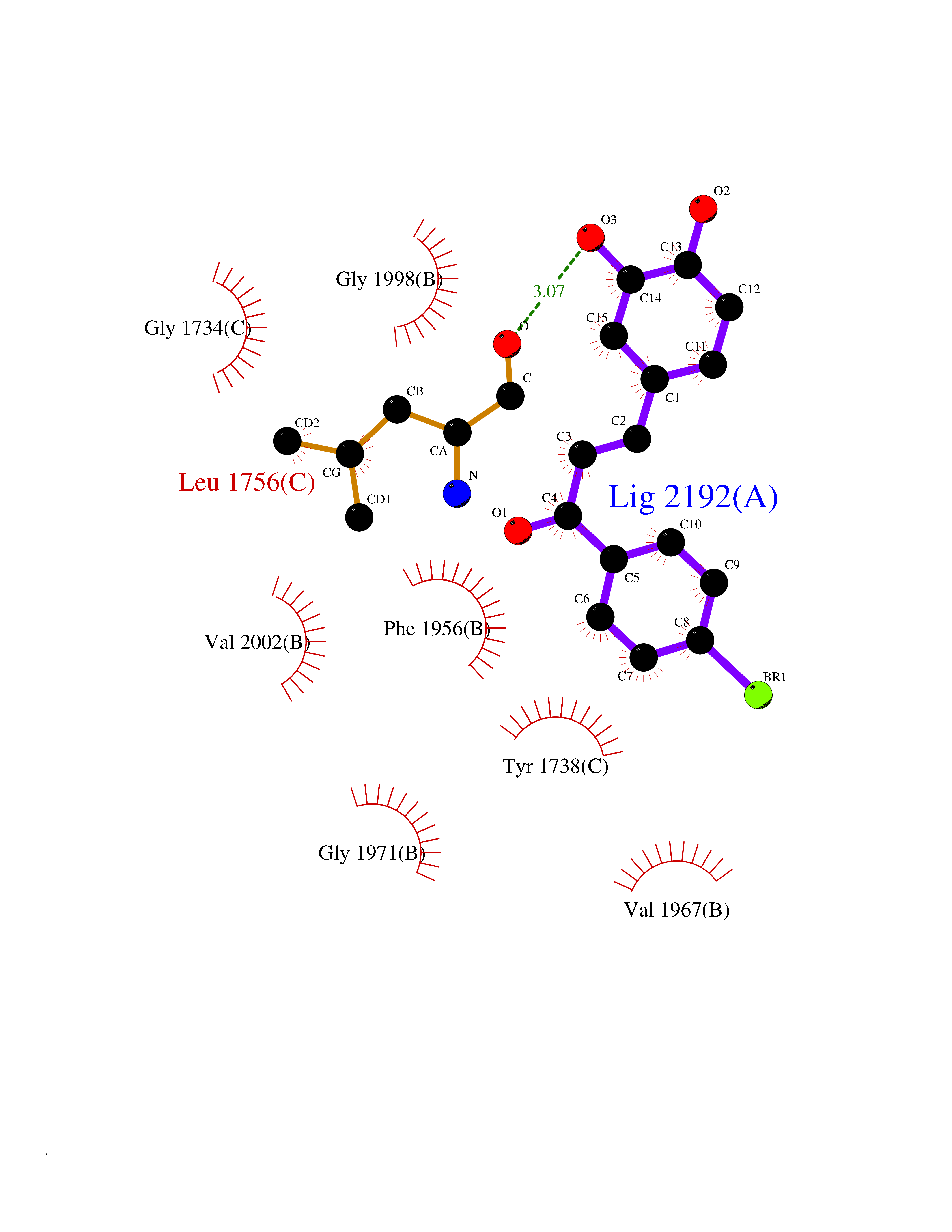 Ligplot Map 3D Binding mode Sequence WLQPKRYKAHLXGTTYVYDFPELFRQASSSQWKNFSADVKLTDDFFISNELIEDENGELTEVEREPGANAIGXVAFKITVKTPEYPRGRQFVVVANDITFKIGSFGPQEDEFFNKVTEYARKRGIPRIYLAANSGARIGXAEEIVPLFQVAWNDAANPDKGFQYLYLTSEGXETLKKFDKENSVLTERTVINGEERFVIKTIIGSEDGLGVECLRGSGLIAGATSRAYHDIFTITLVTCRSVGIGAYLVRLGQRAIQVEGQPIILTGAPAINKXLGREVYTSNLQLGGTQIXYNNGVSHLTAVDDLAGVEKIVEWXSYVPAKRNXPVPILETKDTWDRPVDFTPTNDETYDVRWXIEGRETESGFEYGLFDKGSFFETLSGWAKGVVVGRARLGGIPLGVIGVETRTVENLIPADPANPNSAETLIQEPGQVWHPNSAFKTAQAINDFNNGEQLPXXILANWRGFSGNEVLKYGSFIVDALVDYKQPIIIYIPPTGELRGGSWVVVDPTINADQXEXYADVNARAGVLEPQGXVGIKFRREKLLDTXNRLELLPIYGQISLQFADLHDRSSRXVAKGVISKELEWTEARRFFFWRLRRRLNEEYLIKRLSHQVGEASRLEKIARIRSWYPASVDHEDDRQVATWIEENYKTLDDKLKGLPIATPYPVKEWLQPKRYKAHLXGTTYVYDFPELFRQASSSQWKNFSADVKLTDDFFISNELIEDENGELTEVEREPGANAIGXVAFKITVKTPEYPRGRQFVVVANDITFKIGSFGPQEDEFFNKVTEYARKRGIPRIYLAANSGARIGXAEEIVPLFQVAWNDAANPDKGFQYLYLTSEGXETLKKFDKENSVLTERTVINGEERFVIKTIIGSEDGLGVECLRGSGLIAGATSRAYHDIFTITLVTCRSVGIGAYLVRLGQRAIQVEGQPIILTGAPAINKXLGREVYTSNLQLGGTQIXYNNGVSHLTAVDDLAGVEKIVEWXSYVPAKRNXPVPILETKDTWDRPVDFTPTNDETYDVRWXIEGRETESGFEYGLFDKGSFFETLSGWAKGVVVGRARLGGIPLGVIGVETRTVENLIPADPANPNSAETLIQEPGQVWHPNSAFKTAQAINDFNNGEQLPXXILANWRGFSGNEVLKYGSFIVDALVDYKQPIIIYIPPTGELRGGSWVVVDPTINADQXEXYADVNARAGVLEPQGXVGIKFRREKLLDTXNRLELLPIYGQISLQFADLHDRSSRXVAKGVISKELEWTEARRFFFWRLRRRLNEEYLIKRLSHQVGEASRLEKIARIRSWYPASVDHEDDRQVATWIEENYKTLDDKLKGL Hydrogen bonds contact Hydrophobic contact | ||||
| 17 | Bacterial Oxoacyl-[acyl-carrier-protein] synthase II (Bact fabF) | 2GFX | 7.31 | |
Target general information Gen name Bact fabF Organism Escherichia coli (strain K12) Uniprot ID TTD ID Synonyms KASB; KAS II; KAS 2; FabF; Condensing enzyme FabF; Beta-ketoacyl-acyl carrier protein synthase B; Beta-ketoacyl-ACP synthase II; Beta-ketoacyl-ACP synthase 2; 3-oxoacyl-[acyl-carrier-protein] synthase Protein family Thiolase-like superfamily, Beta-ketoacyl-ACP synthases family Biochemical class Acyltransferase Function Catalyzes the condensation reaction of fatty acid synthesis by the addition to an acyl acceptor of two carbons from malonyl-ACP. Has a preference for short chain acid substrates and may function to supply the octanoic substrates for lipoic acid biosynthesis. Related diseases Pigmentary disorder, reticulate, with systemic manifestations, X-linked (PDR) [MIM:301220]: An X-linked recessive disorder characterized by recurrent infections and sterile inflammation in various organs. Diffuse skin hyperpigmentation with a distinctive reticulate pattern is universally evident by early childhood. This is later followed in many patients by hypohidrosis, corneal inflammation and scarring, enterocolitis that resembles inflammatory bowel disease, and recurrent urethral strictures. Melanin and amyloid deposition is present in the dermis. Affected males also have a characteristic facies with frontally upswept hair and flared eyebrows. Female carriers have only restricted pigmentary changes along Blaschko's lines. {ECO:0000269|PubMed:27019227}. The disease is caused by variants affecting the gene represented in this entry. XLPDR is caused by a recurrent intronic mutation that results in missplicing and reduced POLA1 expression. This leads to a decrease in cytosolic RNA:DNA hybrids and constitutive activation of type I interferon responses, but has no effect on cell replication. {ECO:0000269|PubMed:27019227}.; DISEASE: Van Esch-O'Driscoll syndrome (VEODS) [MIM:301030]: An X-linked recessive syndrome characterized by different degrees of intellectual disability, moderate to severe short stature, microcephaly, hypogonadism, and variable congenital malformations. {ECO:0000269|PubMed:31006512}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08366; DB01034; DB03017; DB08407 Interacts with P0A6Y8 EC number EC 2.3.1.179 Uniprot keywords 3D-structure; Acyltransferase; Direct protein sequencing; Fatty acid biosynthesis; Fatty acid metabolism; Lipid biosynthesis; Lipid metabolism; Reference proteome; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 42852 Length 411 Aromaticity 0.06 Instability index 31.41 Isoelectric point 5.72 Charge (pH=7) -7.34 2D Binding mode Binding energy (Kcal/mol) -9.97  Molscript Map 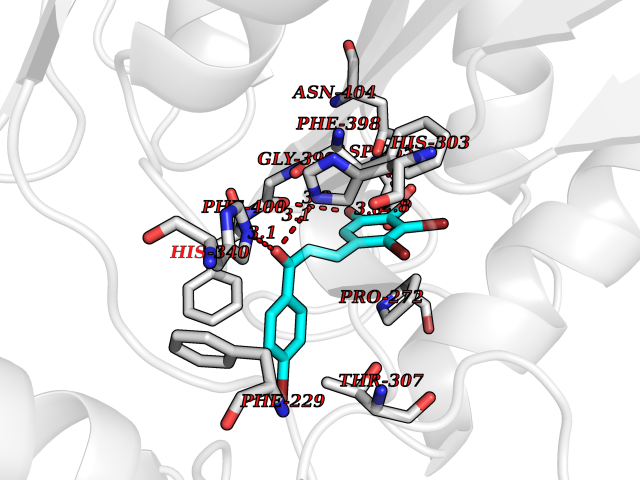 Pymol Map 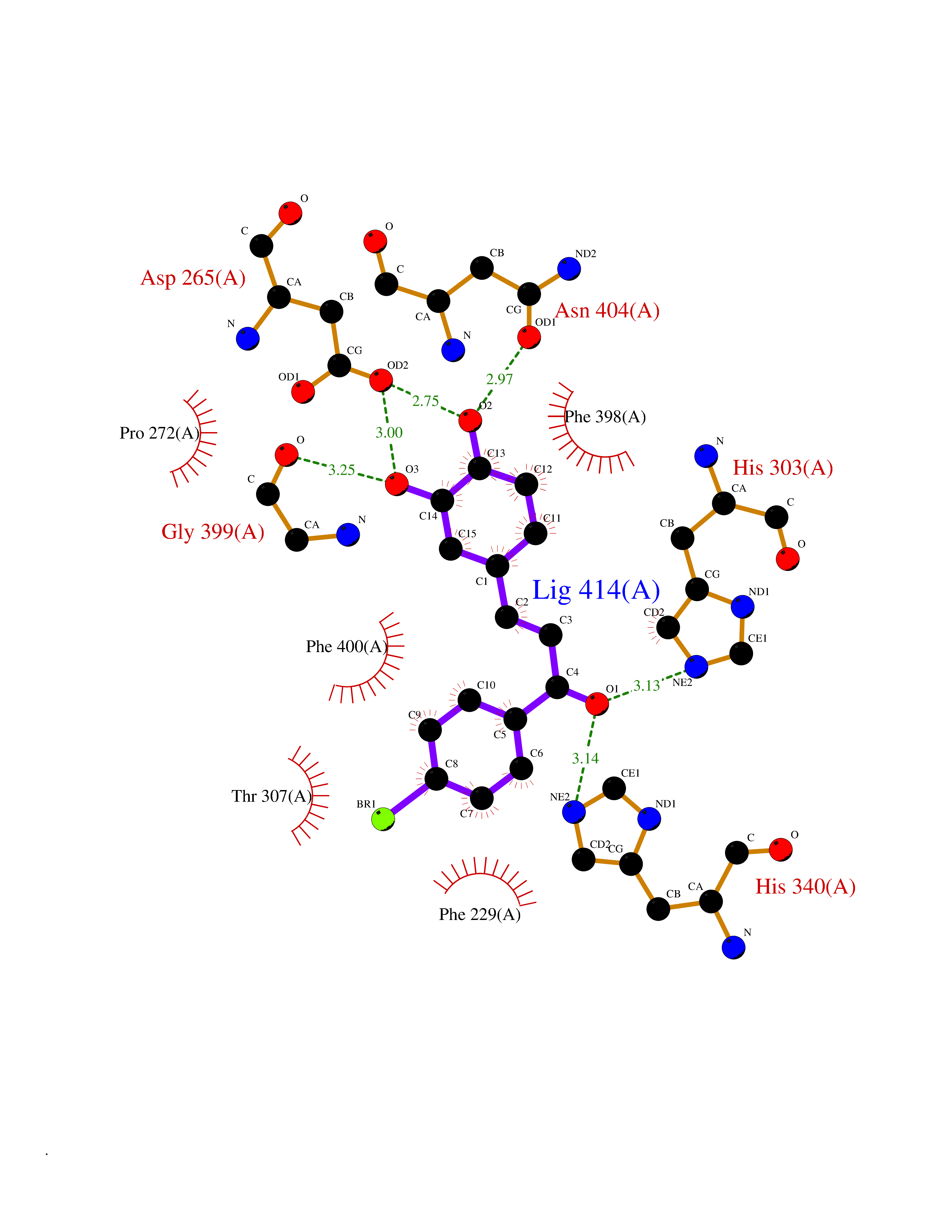 Ligplot Map 3D Binding mode Sequence KRRVVVTGLGMLSPVGNTVESTWKALLAGQSGISLIDHFDTSAYATKFAGLVKDFNCEDIISRKEQRKMDAFIQYGIVAGVQAMQDSGLEITEENATRIGAAIGSGIGGLGLIEENHTSLMNGGPRKISPFFVPSTIVNMVAGHLTIMYGLRGPSISIATAQTSGVHNIGHAARIIAYGDADVMVAGGAEKASTPLGVGGFGAARALSTRNDNPQAASRPWDKERDGFVLGDGAGMLVLEEYEHAKKRGAKIYAELVGFGMSSDAYHMTSPPENGAGAALAMANALRDAGIEASQIGYVNAHGTSTPAGDKAEAQAVKTIFGEAASRVLVSSTKSMTGHLLGAAGAVESIYSILALRDQAVPPTINLDNPDEGCDLDFVPHEARQVSGMEYTLCNSFGFGGTNGSLIFKKI Hydrogen bonds contact Hydrophobic contact | ||||
| 18 | SEC14-like protein 4 | 4TLG | 7.30 | |
Target general information Gen name SEC14L4 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms TAP3 Protein family NA Biochemical class Transport protein Function Lipid binding.Transporter activity. Related diseases Chondrodysplasia with platyspondyly, distinctive brachydactyly, hydrocephaly, and microphthalmia (CDP-PBHM) [MIM:300863]: A disease characterized by chondrodysplasia, severe platyspondyly, hydrocephaly, and facial features with microphthalmia. Bone abnormalities include a distinctive metaphyseal cupping of the metacarpals, metatarsals, and phalanges. Affected females show a milder phenotype with small stature, sometimes associated with body asymmetry and mild intellectual disability. {ECO:0000269|PubMed:20181727}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB14003; DB11635; DB11251; DB00163 Interacts with Q96LC9; O43186; P78358; Q9NYQ3; Q0VD86; Q15323; O76011; P50221; Q6FHY5; Q02548; P26367; Q9H8W4; Q04864; Q04864-2; Q9UHV2; P15884; P15884-3; Q96N21; Q9BYV2; Q8N6Y0; Q9H0C1 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Lipid-binding; Proteomics identification; Reference proteome; Transport Protein physicochemical properties Chain ID A,B Molecular weight (Da) 23947.6 Length 210 Aromaticity 0.1 Instability index 50.84 Isoelectric point 5.55 Charge (pH=7) -3.11 2D Binding mode Binding energy (Kcal/mol) -9.95  Molscript Map 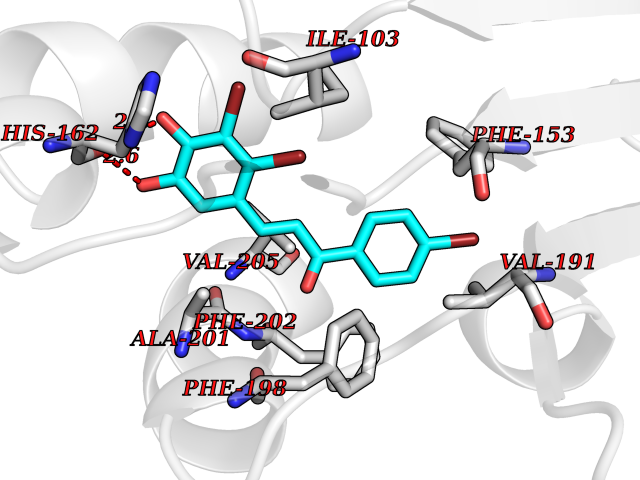 Pymol Map 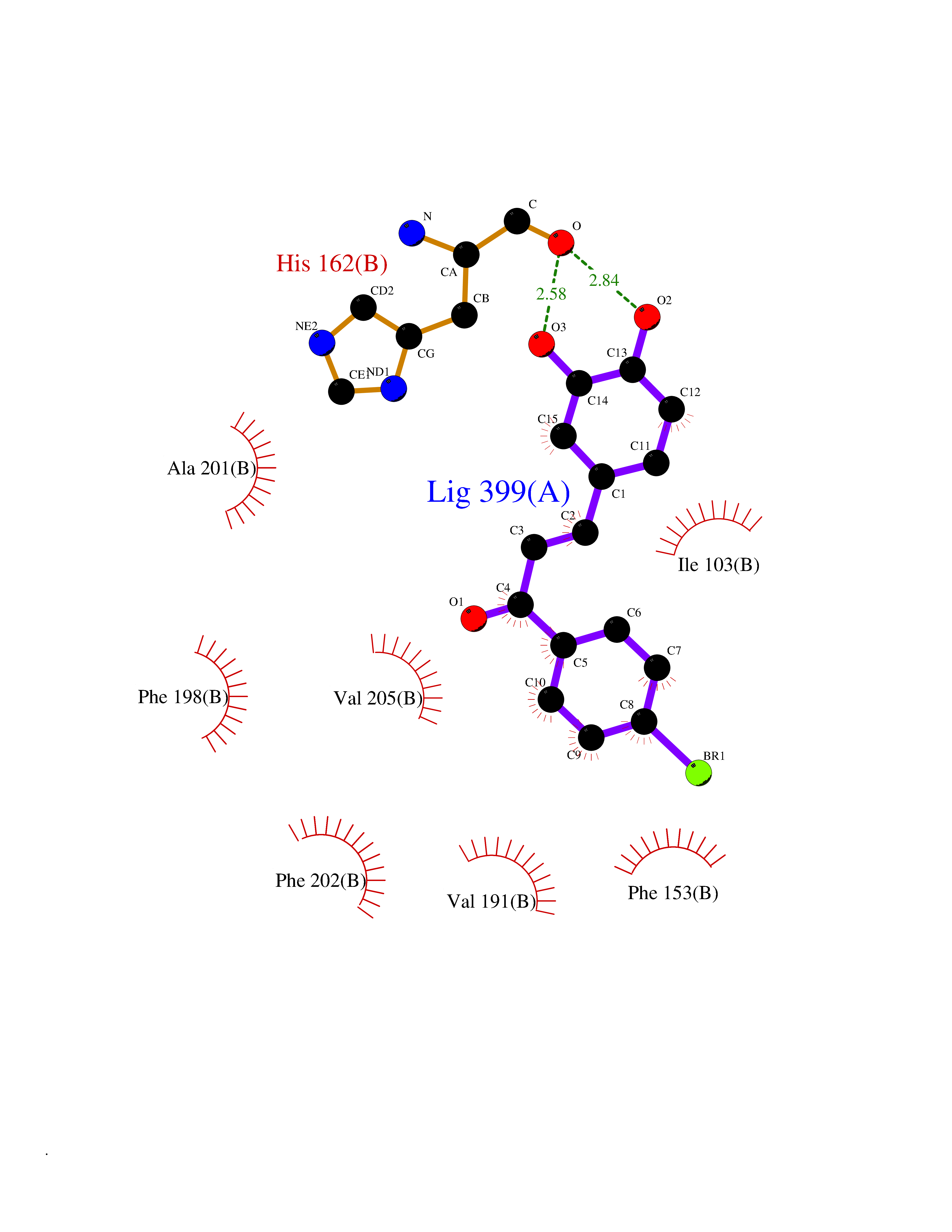 Ligplot Map 3D Binding mode Sequence VTWQPPEVIQLYDSGGLCGYDYEGCPVYFNIIGSLDPKGLLLSASKQDMIRKRIKVCELLLHECELQTQKLGRKIEMALMVFDMEGLSLKHLWKPAVEVYQQFFSILEANYPETLKNLIVIRAPKLFPVAFNLVKSFMSEETRRKIVILGDNWKQELTKFISPDQLPVEFGGTMTDPDGNPKCLTKINYGGEVPKSYYPDKASEETLQSM Hydrogen bonds contact Hydrophobic contact | ||||
| 19 | Soluble epoxide hydrolase (EPHX2) | 1ZD3 | 7.30 | |
Target general information Gen name EPHX2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Bifunctional epoxide hydrolase 2 Protein family AB hydrolase superfamily, Epoxide hydrolase family Biochemical class Ether bond hydrolase Function Bifunctional enzyme. The C-terminal domain has epoxide hydrolase activity and acts on epoxides (alkene oxides, oxiranes) and arene oxides. Plays a role in xenobiotic metabolism by degrading potentially toxic epoxides (By similarity). Also determines steady-state levels of physiological mediators. The N-terminal domain has lipid phosphatase activity, with the highest activity towards threo-9,10-phosphonooxy-hydroxy-octadecanoic acid, followed by erythro-9,10-phosphonooxy-hydroxy-octadecanoic acid, 12-phosphonooxy-octadec-9Z-enoic acid and 12-phosphonooxy-octadec-9E-enoic acid. Related diseases Leukemia, juvenile myelomonocytic (JMML) [MIM:607785]: An aggressive pediatric myelodysplastic syndrome/myeloproliferative disorder characterized by malignant transformation in the hematopoietic stem cell compartment with proliferation of differentiated progeny. Patients have splenomegaly, enlarged lymph nodes, rashes, and hemorrhages. {ECO:0000269|PubMed:17332249}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Noonan syndrome 6 (NS6) [MIM:613224]: A form of Noonan syndrome, a disease characterized by short stature, facial dysmorphic features such as hypertelorism, a downward eyeslant and low-set posteriorly rotated ears, and a high incidence of congenital heart defects and hypertrophic cardiomyopathy. Other features can include a short neck with webbing or redundancy of skin, deafness, motor delay, variable intellectual deficits, multiple skeletal defects, cryptorchidism, and bleeding diathesis. Individuals with Noonan syndrome are at risk of juvenile myelomonocytic leukemia, a myeloproliferative disorder characterized by excessive production of myelomonocytic cells. {ECO:0000269|PubMed:19966803}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: RAS-associated autoimmune leukoproliferative disorder (RALD) [MIM:614470]: A disorder of apoptosis, characterized by chronic accumulation of non-malignant lymphocytes, defective lymphocyte apoptosis, and an increased risk for the development of hematologic malignancies. {ECO:0000269|PubMed:17517660}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Melanocytic nevus syndrome, congenital (CMNS) [MIM:137550]: A syndrome characterized by congenital pigmentary skin lesions which can occur at any site and can cover most of the body surface. These lesions may or may not be hairy. Congenital melanocytic nevi are associated with neuromelanosis (the presence of melanin-producing cells within the brain parenchyma or leptomeninges). Less commonly they are associated with malignant melanoma in childhood, both in the skin and the central nervous system. CMNS patients also tend to have a characteristic facial appearance, including wide or prominent forehead, periorbital fullness, small short nose with narrow nasal bridge, round face, full cheeks, prominent premaxilla, and everted lower lip. {ECO:0000269|PubMed:18633438, ECO:0000269|PubMed:23392294}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Melanosis, neurocutaneous (NCMS) [MIM:249400]: A rare congenital disease characterized by the presence of giant or multiple melanocytic nevi on the skin, foci of melanin-producing cells within the brain parenchyma, and infiltration of leptomeninges by abnormal melanin deposits. Neurologic abnormalities include seizures, hydrocephalus, arachnoid cysts, tumors, and syringomyelia. Some patients may develop malignant melanoma. {ECO:0000269|PubMed:23392294}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Keratinocytic non-epidermolytic nevus (KNEN) [MIM:162900]: Epidermal nevi of the common, non-organoid and non-epidermolytic type are benign skin lesions and may vary in their extent from a single (usually linear) lesion to widespread and systematized involvement. They may be present at birth or develop early during childhood. {ECO:0000269|PubMed:22499344}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Thyroid cancer, non-medullary, 2 (NMTC2) [MIM:188470]: A form of non-medullary thyroid cancer (NMTC), a cancer characterized by tumors originating from the thyroid follicular cells. NMTCs represent approximately 95% of all cases of thyroid cancer and are classified into papillary, follicular, Hurthle cell, and anaplastic neoplasms. {ECO:0000269|PubMed:12727991}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08257; DB08258; DB08259; DB06345; DB12610; DB08256; DB02029; DB04213; DB03677 Interacts with NA EC number NA Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Aromatic hydrocarbons catabolism; Cytoplasm; Detoxification; Direct protein sequencing; Hydrolase; Lipid metabolism; Lipoprotein; Magnesium; Metal-binding; Multifunctional enzyme; Peroxisome; Phosphoprotein; Proteomics identification; Reference proteome Protein physicochemical properties Chain ID A Molecular weight (Da) 61744.9 Length 547 Aromaticity 0.09 Instability index 43.97 Isoelectric point 5.81 Charge (pH=7) -7.76 2D Binding mode Binding energy (Kcal/mol) -9.96 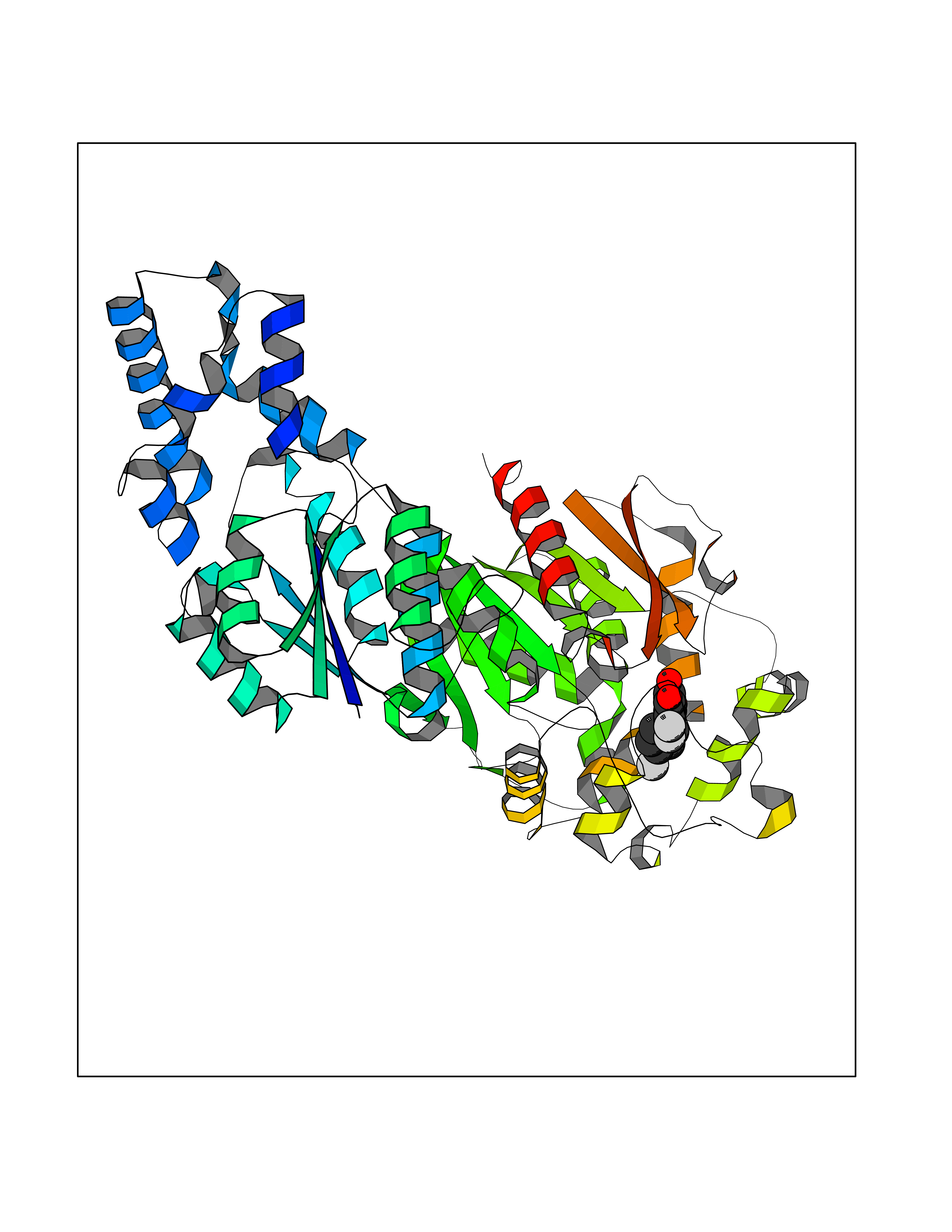 Molscript Map 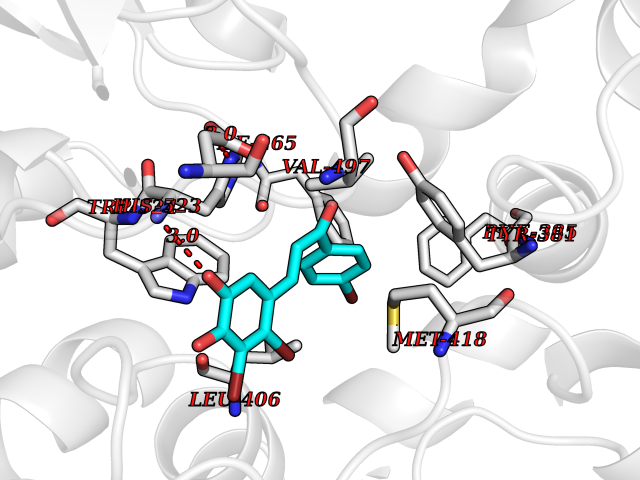 Pymol Map 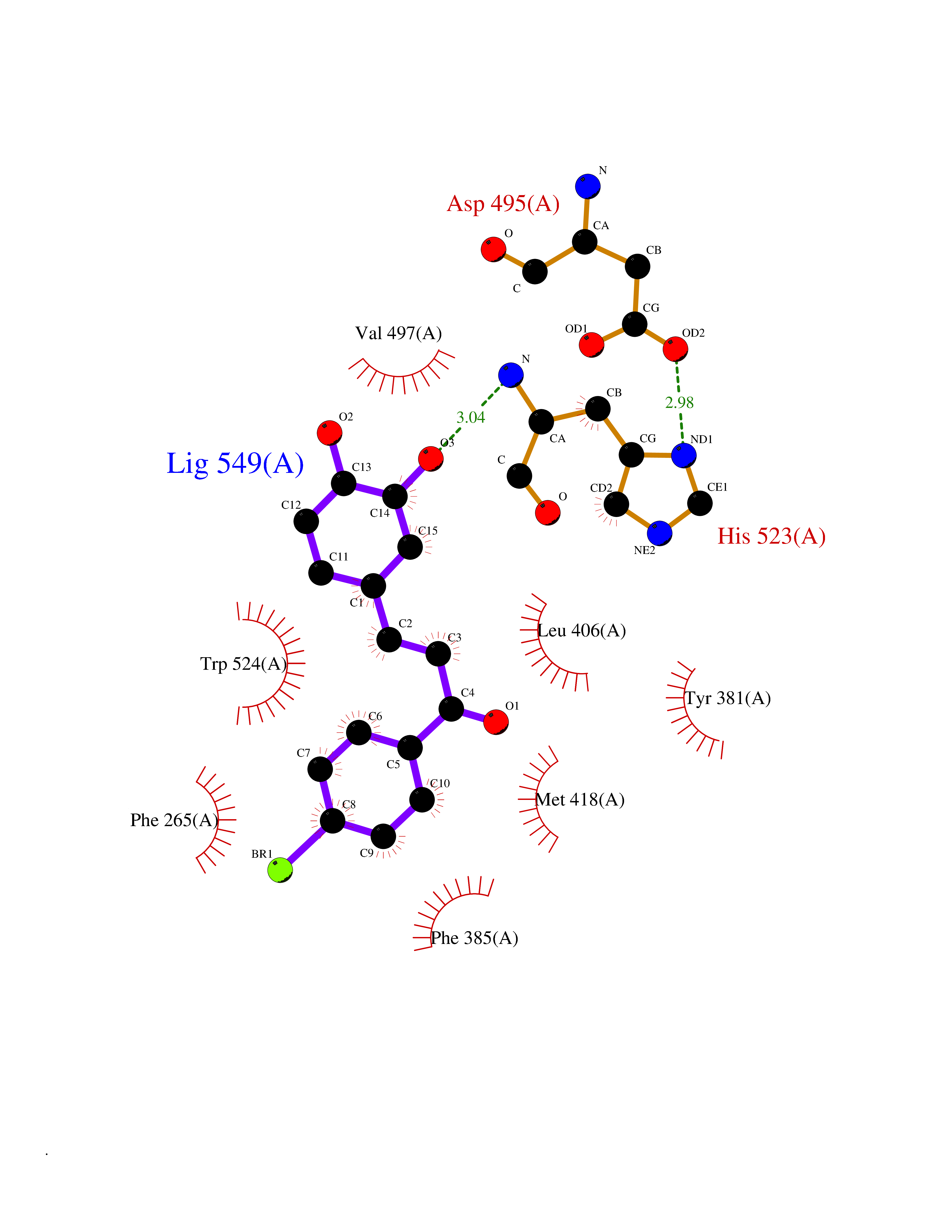 Ligplot Map 3D Binding mode Sequence TLRAAVFDLDGVLALPAVFGVLGRTEEALALPRGLLNDAFQKGGPEGATTRLMKGEITLSQWIPLMEENCRKCSETAKVCLPKNFSIKEIFDKAISARKINRPMLQAALMLRKKGFTTAILTNTWLDDRAERDGLAQLMCELKMHFDFLIESCQVGMVKPEPQIYKFLLDTLKASPSEVVFLDDIGANLKPARDLGMVTILVQDTDTALKELEKVTGIQLLNTPAPLPTSCNPSDMSHGYVTVKPRVRLHFVELGSGPAVCLCHGFPESWYSWRYQIPALAQAGYRVLAMDMKGYGESSAPPEIEEYCMEVLCKEMVTFLDKLGLSQAVFIGHDWGGMLVWYMALFYPERVRAVASLNTPFIPANPNMSPLESIKANPVFDYQLYFQEPGVAEAELEQNLSRTFKSLFRASDESVLSMHKVCEAGGLFVNSPEEPSLSRMVTEEEIQFYVQQFKKSGFRGPLNWYRNMERNWKWACKSLGRKILIPALMVTAEKDFVLVPQMSQHMEDWIPHLKRGHIEDCGHWTQMDKPTEVNQILIKWLDSDARN Hydrogen bonds contact Hydrophobic contact | ||||
| 20 | Penicillin acylase | 2PVA | 7.29 | |
Target general information Gen name N/A Organism Lysinibacillus sphaericus (Bacillus sphaericus) Uniprot ID TTD ID NA Synonyms NA Protein family Peptidase C59 family Biochemical class Hydrolase Function Penicillin amidase activity. Related diseases Usher syndrome 3B (USH3B) [MIM:614504]: A syndrome characterized by progressive vision and hearing loss during early childhood. Some patients have the so-called 'Charles Bonnet syndrome,' involving decreased visual acuity and vivid visual hallucinations. USH is a genetically heterogeneous condition characterized by the association of retinitis pigmentosa with sensorineural deafness. Age at onset and differences in auditory and vestibular function distinguish Usher syndrome type 1 (USH1), Usher syndrome type 2 (USH2) and Usher syndrome type 3 (USH3). USH3 is characterized by postlingual, progressive hearing loss, variable vestibular dysfunction, and onset of retinitis pigmentosa symptoms, including nyctalopia, constriction of the visual fields, and loss of central visual acuity, usually by the second decade of life. {ECO:0000269|PubMed:22279524}. The disease may be caused by variants affecting the gene represented in this entry.; DISEASE: Charcot-Marie-Tooth disease, axonal, 2W (CMT2W) [MIM:616625]: An autosomal dominant, axonal form of Charcot-Marie-Tooth disease, a disorder of the peripheral nervous system, characterized by progressive weakness and atrophy, initially of the peroneal muscles and later of the distal muscles of the arms. Charcot-Marie-Tooth disease is classified in two main groups on the basis of electrophysiologic properties and histopathology: primary peripheral demyelinating neuropathies (designated CMT1 when they are dominantly inherited) and primary peripheral axonal neuropathies (CMT2). Neuropathies of the CMT2 group are characterized by signs of axonal degeneration in the absence of obvious myelin alterations, normal or slightly reduced nerve conduction velocities, and progressive distal muscle weakness and atrophy. CMT2W patients manifest a peripheral neuropathy mainly affecting the lower limbs and resulting in gait difficulties and distal sensory impairment. Most patients also have upper limb involvement. {ECO:0000269|PubMed:22930593, ECO:0000269|PubMed:26072516, ECO:0000269|PubMed:29235198}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01822; DB03661; DB00417 Interacts with NA EC number 3.5.1.11 Uniprot keywords 3D-structure; Antibiotic resistance; Direct protein sequencing; Hydrolase; Zymogen Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 32972.1 Length 295 Aromaticity 0.11 Instability index 30.94 Isoelectric point 5.65 Charge (pH=7) -4.18 2D Binding mode Binding energy (Kcal/mol) -9.95  Molscript Map 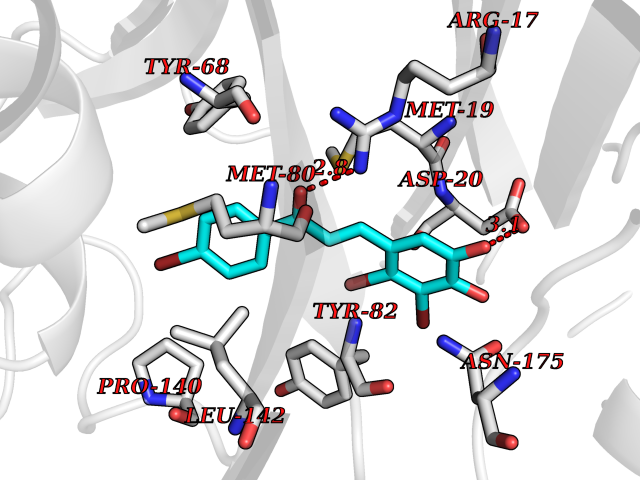 Pymol Map 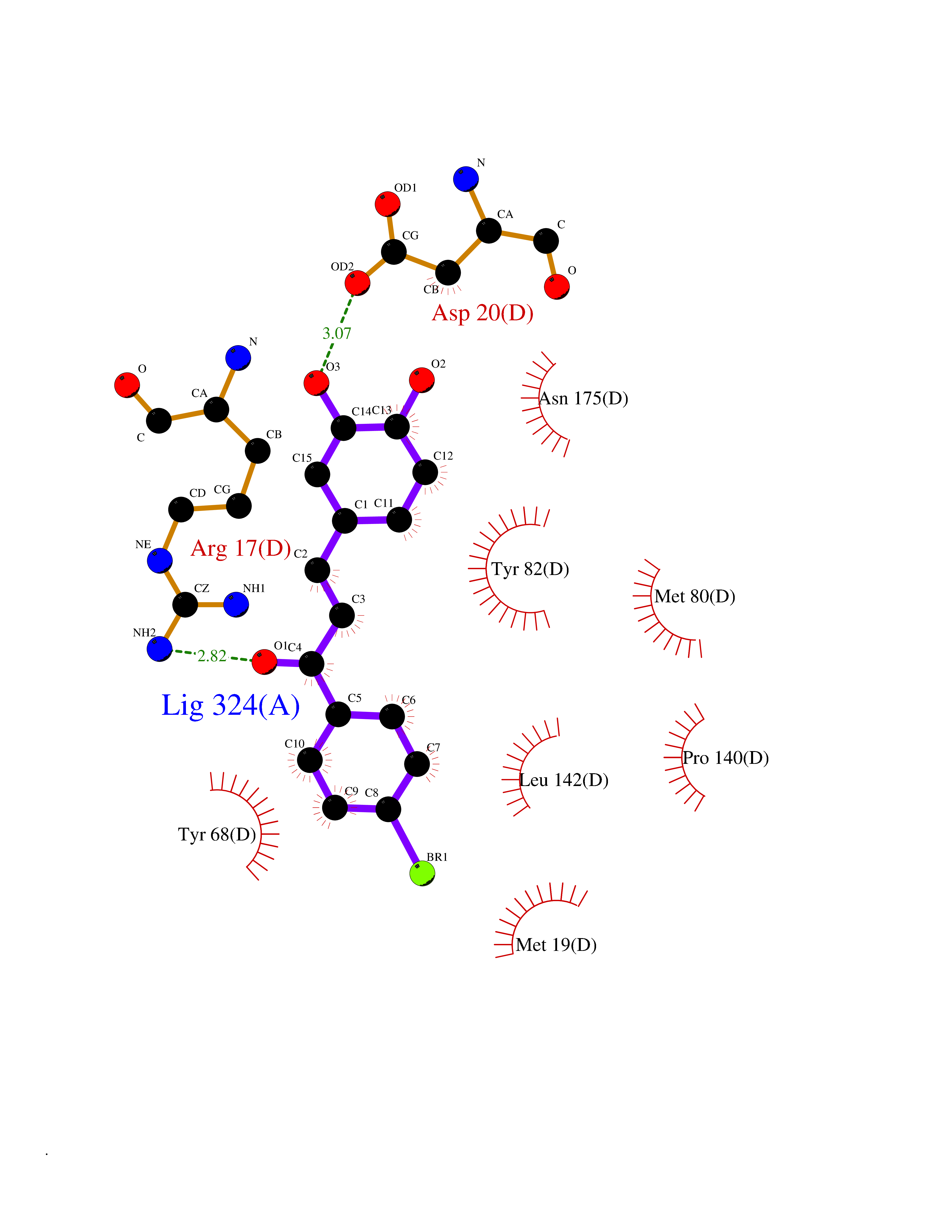 Ligplot Map 3D Binding mode Sequence SSLSIRTTDDKSLFARTMDFTMEPDSKVIIVPRNYGIRLLEKENVVINNSYAFVGMGSTDITSPVLYDGVNEKGLMGAMLYYATFATYADEPKKGTTGINPVYVISQVLGNCVTVDDVIEKLTSYTLLNEANIILGFAPPLHYTFTDASGESIVIEPDKTGITIHRKTIGVMTNSPGYEWHQTNLRAYIGVLPGDFTPSARFLRVAYWKKYTEKAKNETEGVTNLFHILSSVNIPKGVVLTNEGKTDYTIYTSAMCAQSKNYYFKLYDNSRISAVSLMAENLNSQDLITFEWDRK Hydrogen bonds contact Hydrophobic contact | ||||