Job Results:
Ligand
Structure
Job ID
b448ce30ce2c1c80e042f47c74d47daa
Job name
NA
Time
2026-01-10 22:40:02
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 81 | Cytochrome c oxidase subunit 1 | 3DTU | 6.91 | |
Target general information Gen name ctaD Organism Cereibacter sphaeroides (Rhodobacter sphaeroides) Uniprot ID TTD ID NA Synonyms NA Protein family Heme-copper respiratory oxidase family Biochemical class Oxidoreductase Function Copper ion binding.Cytochrome-c oxidase activity.Heme binding.Iron ion binding. Related diseases Cystathioninuria (CSTNU) [MIM:219500]: Autosomal recessive phenotype characterized by abnormal accumulation of plasma cystathionine, leading to increased urinary excretion. {ECO:0000269|PubMed:12574942, ECO:0000269|PubMed:18476726}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03619 Interacts with Q03736 EC number 7.1.1.9 Uniprot keywords 3D-structure; Cell membrane; Copper; Electron transport; Heme; Hydrogen ion transport; Ion transport; Iron; Membrane; Metal-binding; Respiratory chain; Translocase; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A,C Molecular weight (Da) 88419.8 Length 794 Aromaticity 0.15 Instability index 40.59 Isoelectric point 6.09 Charge (pH=7) -10.78 2D Binding mode Binding energy (Kcal/mol) -9.43 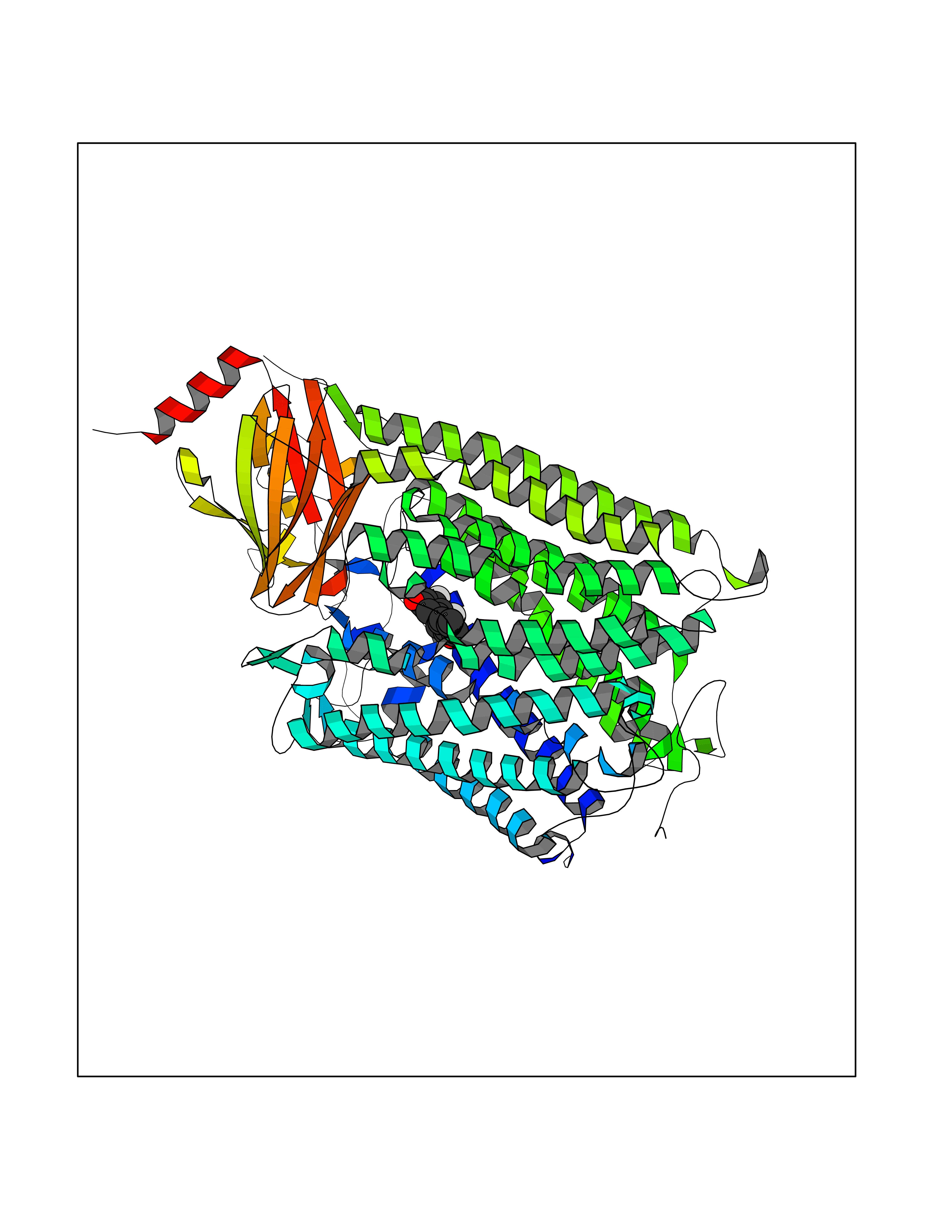 Molscript Map 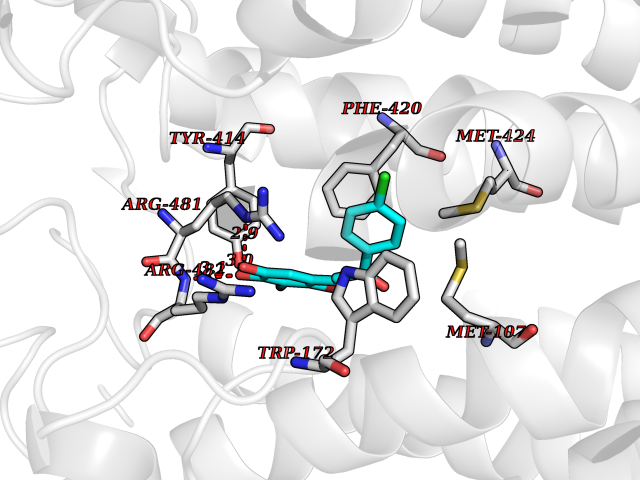 Pymol Map 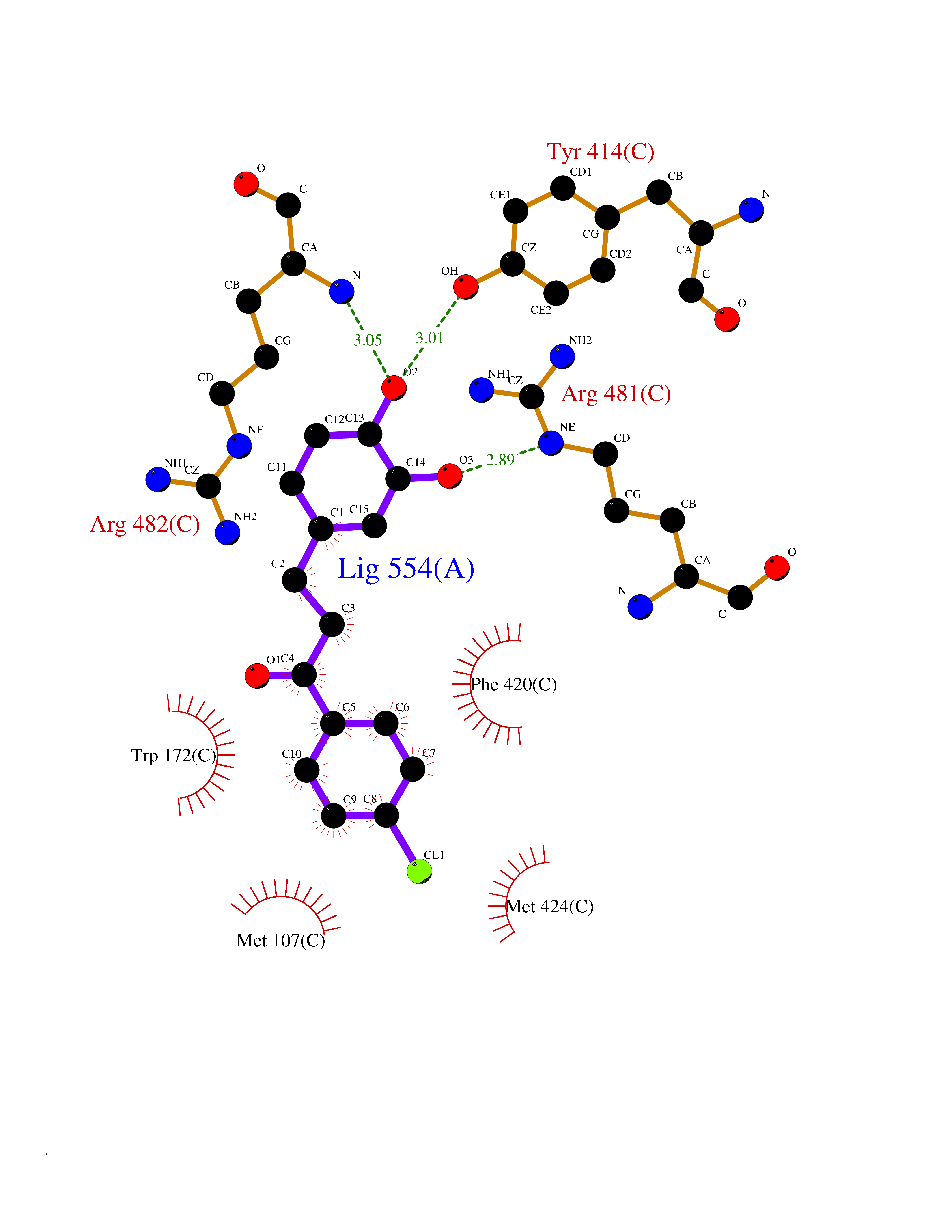 Ligplot Map 3D Binding mode Sequence FTRWFMSTNHKDIGVLYLFTGGLVGLISVAFTVYMRMELMAPGVQFMCAEHLESGLVKGFFQSLWPSAVENCTPNGHLWNVMITGHGILMMFFVVIPALFGGFGNYFMPLHIGAPDMAFPRMNNLSYWLYVAGTSLAVASLFAPGGNGQLGSGIGWVLYPPLSTSESGYSTDLAIFAVHLSGASSILGAINMITTFLNMRAPGMTMHKVPLFAWSIFVTAWLILLALPVLAGAITMLLTDRNFGTTFFQPSGGGDPVLYQHILWFFGHPEVYIIVLPAFGIVSHVIATFAKKPIFGYLPMVYAMVAIGVLGFVVWAHHMYTAGLSLTQQSYFMMATMVIAVPTGIKIFSWIATMWGGSIELKTPMLWALGFLFLFTVGGVTGIVLSQASVDRYYHDTYYVVAHFHYVMSLGAVFGIFAGIYFWIGKMSGRQYPEWAGKLHFWMMFVGANLTFFPQHFLGRQGMPRRYIDYPEAFATWNFVSSLGAFLSFASFLFFLGVIFYTLTRGARVTANNYWNEHADTLEWTLTSPPPEHTFEQSLEIIGRPQPGGTGFQPSASPVATQIHWLDGFILVIIAAITIFVTLLILYAVWRFHEKRNKVPARFTHNSPLEIAWTIVPIVILVAIGAFSLPVLFNQQEIPEADVTVKVTGYQWYWGYEYPDEEISFESYMIGSPATGGDNRMSPEVEQQLIEAGYSRDEFLLATDTAMVVPVNKTVVVQVTGADVIHSWTVPAFGVKQDAVPGRLAQLWFRAEREGIFFGQCSELCGISHAYMPITVKVVSEEAYAAWLEQHHHH Hydrogen bonds contact Hydrophobic contact | ||||
| 82 | Cytochrome c | 3ZOO | 6.91 | |
Target general information Gen name CYCS Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms CYC Protein family Cytochrome c family Biochemical class Oxidoreductase Function Electron transporter, transferring electrons from CoQH2-cytochrome c reductase complex and cytochrome c oxidase complex activity.Heme binding.Metal ion binding. Related diseases Thrombocytopenia 4 (THC4) [MIM:612004]: A form of thrombocytopenia, a hematologic disorder defined by a decrease in the number of platelets in circulating blood, resulting in the potential for increased bleeding and decreased ability for clotting. {ECO:0000269|PubMed:18345000}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB11638; DB03317; DB03366; DB01017; DB02110; DB03977; DB03934; DB04249 Interacts with O14727; P05067; Q6XD76; Q9NSI6-4; Q3SXR2; Q96BR5; Q9UKG9-2; O00303; Q8IZU1; Q3SYB3; P06241; Q8N5Z5; Q6A162; Q1L5Z9; P02750; Q8IYG6; Q6FHY5; A0A0A0MR05; Q9BUL5; Q6ZMI0-5; Q66K80; Q9NTN9-3; P37840; Q13573; Q92797-2; O43829; Q9FKS5 EC number NA Uniprot keywords 3D-structure; Acetylation; Apoptosis; Direct protein sequencing; Disease variant; Electron transport; Heme; Iron; Metal-binding; Mitochondrion; Phosphoprotein; Proteomics identification; Reference proteome; Respiratory chain; Transport Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 11601.4 Length 104 Aromaticity 0.09 Instability index 12.21 Isoelectric point 9.61 Charge (pH=7) 9.01 2D Binding mode Binding energy (Kcal/mol) -9.42 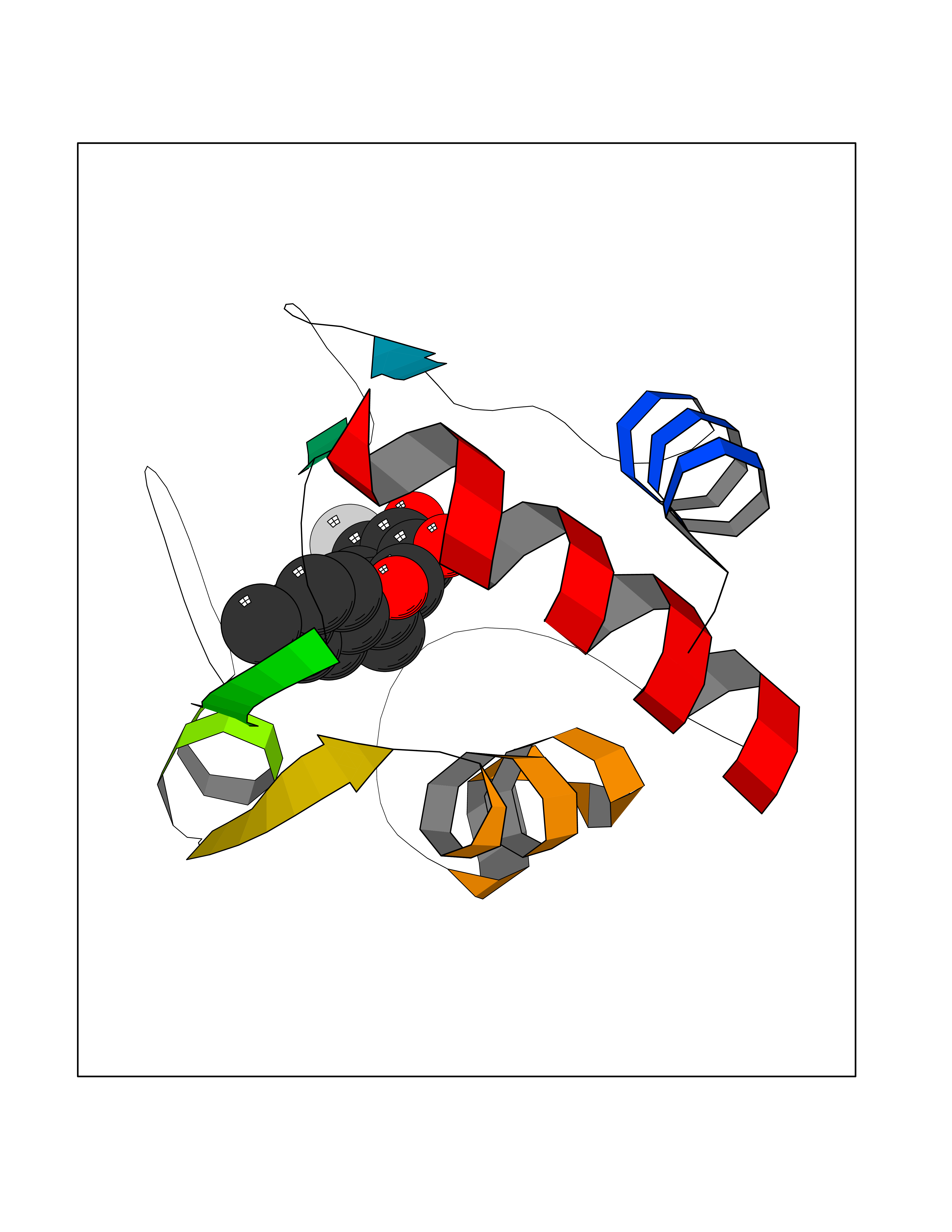 Molscript Map 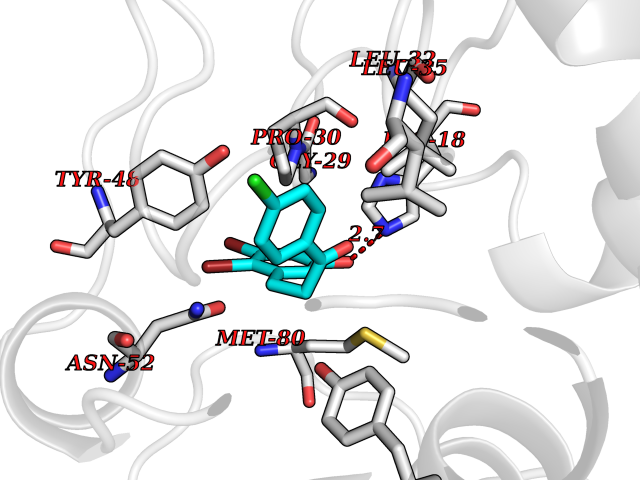 Pymol Map 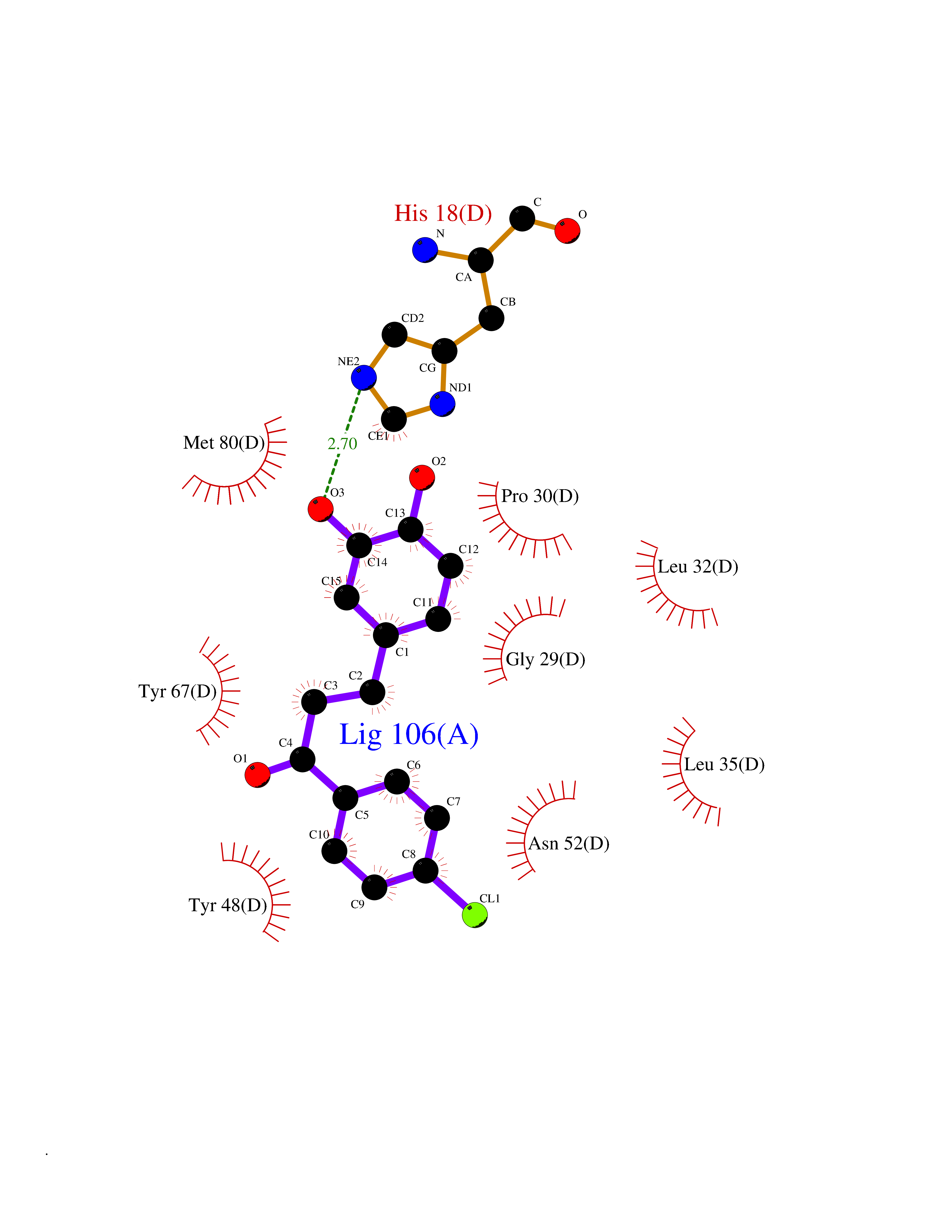 Ligplot Map 3D Binding mode Sequence GDVEKGKKIFIMKCSQCHTVEKGGKHKTGPNLHGLFGRKTGQAPGFSYTAANKNKGIIWGEDTLMEYLENPKKYIPGTKMIFVGIKKKEERADLIAYLKKATNE Hydrogen bonds contact Hydrophobic contact | ||||
| 83 | Aldo-keto reductase family 1 member C1 | 1MRQ | 6.91 | |
Target general information Gen name AKR1C1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms DDH1;DDH Protein family Aldo/keto reductase family Biochemical class Oxidoreductase Function 17-alpha,20-alpha-dihydroxypregn-4-en-3-one dehydrogenase activity.Alditol:NADP+ 1-oxidoreductase activity.Aldo-keto reductase (NADP) activity.Androsterone dehydrogenase (B-specific) activity.Bile acid binding.Carboxylic acid binding.Indanol dehydrogenase activity.Ketosteroid monooxygenase activity.Oxidoreductase activity, acting on NAD(P)H, quinone or similar compound as acceptor.Phenanthrene 9,10-monooxygenase activity.Trans-1,2-dihydrobenzene-1,2-diol dehydrogenase activity. Related diseases Fibrodysplasia ossificans progressiva (FOP) [MIM:135100]: A rare autosomal dominant connective tissue disorder resulting in skeletal malformations and progressive extraskeletal ossification. Heterotopic ossification begins in childhood and can be induced by trauma or may occur without warning. Bone formation is episodic and progressive, leading to a debilitating ankylosis of all major joints of the axial and appendicular skeleton, rendering movement impossible. {ECO:0000269|PubMed:16642017, ECO:0000269|PubMed:19085907, ECO:0000269|PubMed:19330033}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB04674; DB00945; DB07768; DB01039; DB07931; DB06077; DB00959; DB00461; DB00157; DB03467; DB03461; DB00776; DB12612; DB00936 Interacts with P51857; P26045; Q7Z699 EC number 1.1.1.-; 1.1.1.112; 1.1.1.149; 1.1.1.209; 1.1.1.210; 1.1.1.357; 1.1.1.51; 1.1.1.53; 1.1.1.62; 1.3.1.20 Uniprot keywords 3D-structure; Cytoplasm; Direct protein sequencing; Lipid metabolism; NADP; Oxidoreductase; Proteomics identification; Reference proteome Protein physicochemical properties Chain ID A Molecular weight (Da) 36784.9 Length 323 Aromaticity 0.09 Instability index 42.07 Isoelectric point 8.06 Charge (pH=7) 2.42 2D Binding mode Binding energy (Kcal/mol) -9.42 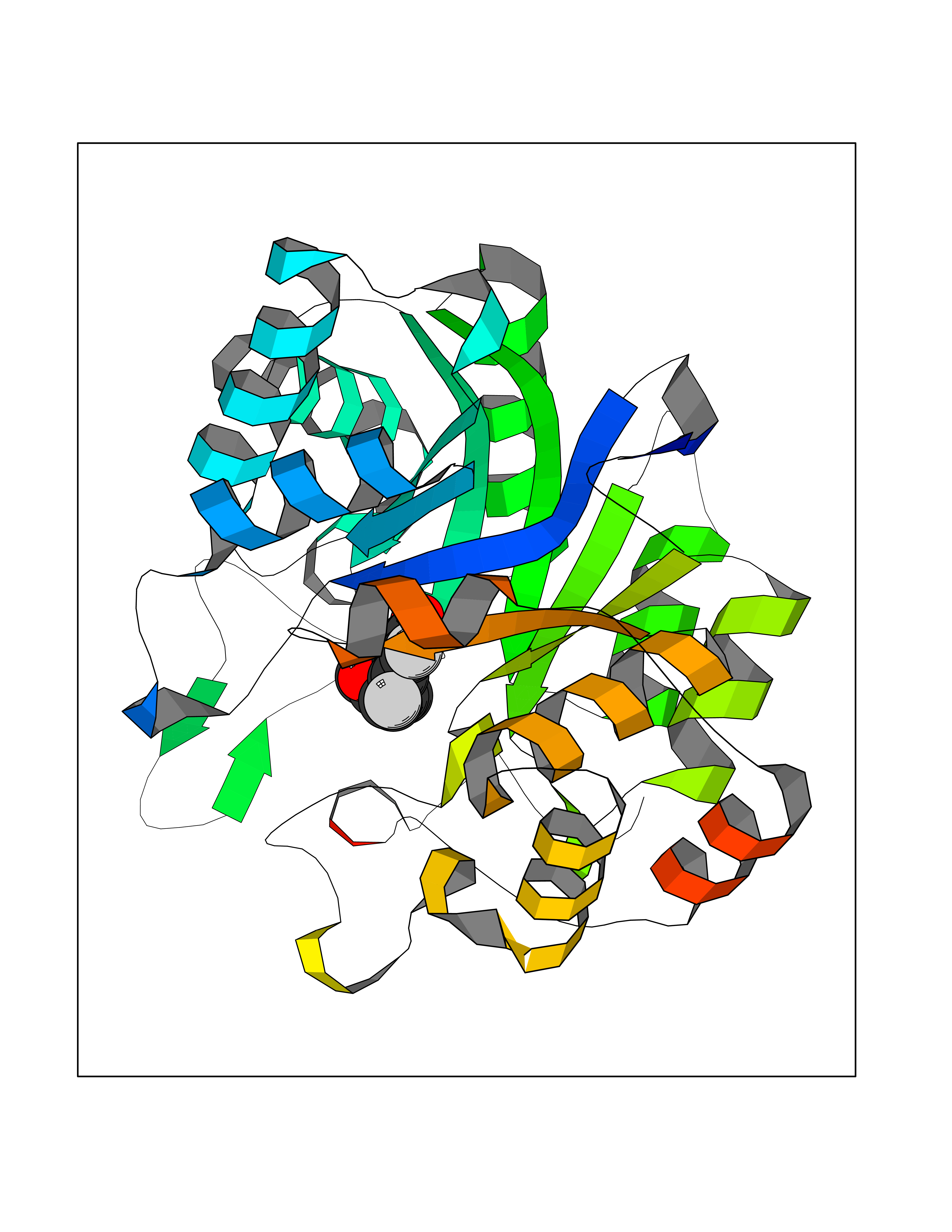 Molscript Map 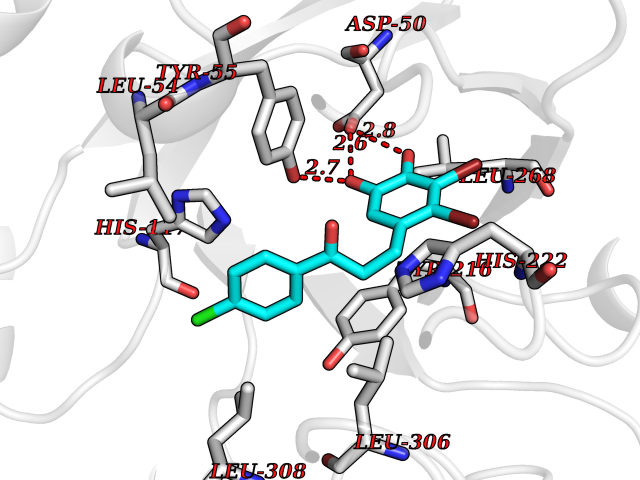 Pymol Map 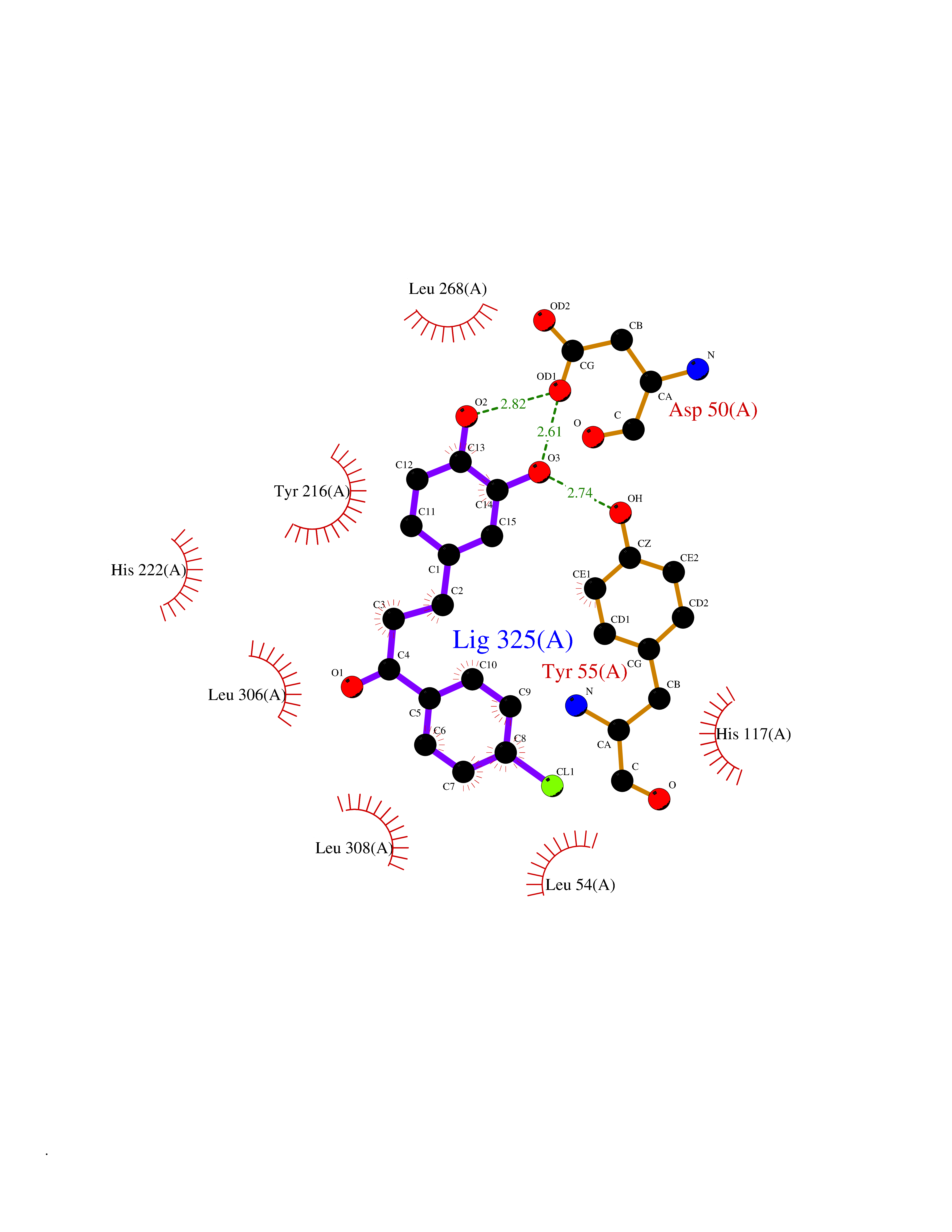 Ligplot Map 3D Binding mode Sequence QDSKYQCVKLNDGHFMPVLGFGTYAPAEVPKSKALEATKLAIEAGFRHIDSAHLYNNEEQVGLAIRSKIADGSVKREDIFYTSKLWCNSHRPELVRPALERSLKNLQLDYVDLYLIHFPVSVKPGEEVIPKDENGKILFDTVDLCATWEAVEKCKDAGLAKSIGVSNFNRRQLEMILNKPGLKYKPVCNQVECHPYFNQRKLLDFCKSKDIVLVAYSALGSHREEPWVDPNSPVLLEDPVLCALAKKHKRTPALIALRYQLQRGVVVLAKSYNEQRIRQNVQVFEFQLTSEEMKAIDGLNRNVRYLTLDIFAGPPNYPFSDEY Hydrogen bonds contact Hydrophobic contact | ||||
| 84 | Sodium/glucose cotransporter 2 (SGLT2) | 7VSI | 6.91 | |
Target general information Gen name SLC5A2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Solute carrier family 5 member 2; Na(+)/glucose cotransporter 2; Low affinity sodium-glucose cotransporter Protein family Sodium:solute symporter (SSF) (TC 2.A.21) family Biochemical class Solute:sodium symporter Function Has a Na(+) to glucose coupling ratio of 1:1. Sodium-dependent glucose transporter. Related diseases Renal glucosuria (GLYS) [MIM:233100]: A disorder characterized by persistent isolated glucosuria, normal fasting serum glucose concentration, decreased renal tubular resorption of glucose from the urine, and absence of any other signs of tubular dysfunction. {ECO:0000269|PubMed:14614622}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB12236; DB08907; DB01914; DB06292; DB09038; DB11827; DB12713 Interacts with O14556; Q13113 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disease variant; Disulfide bond; Glycoprotein; Ion transport; Membrane; Metal-binding; Proteomics identification; Reference proteome; Sodium; Sodium transport; Sugar transport; Symport; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A Molecular weight (Da) 63858.9 Length 586 Aromaticity 0.12 Instability index 39.46 Isoelectric point 8.62 Charge (pH=7) 7.41 2D Binding mode Binding energy (Kcal/mol) -9.43 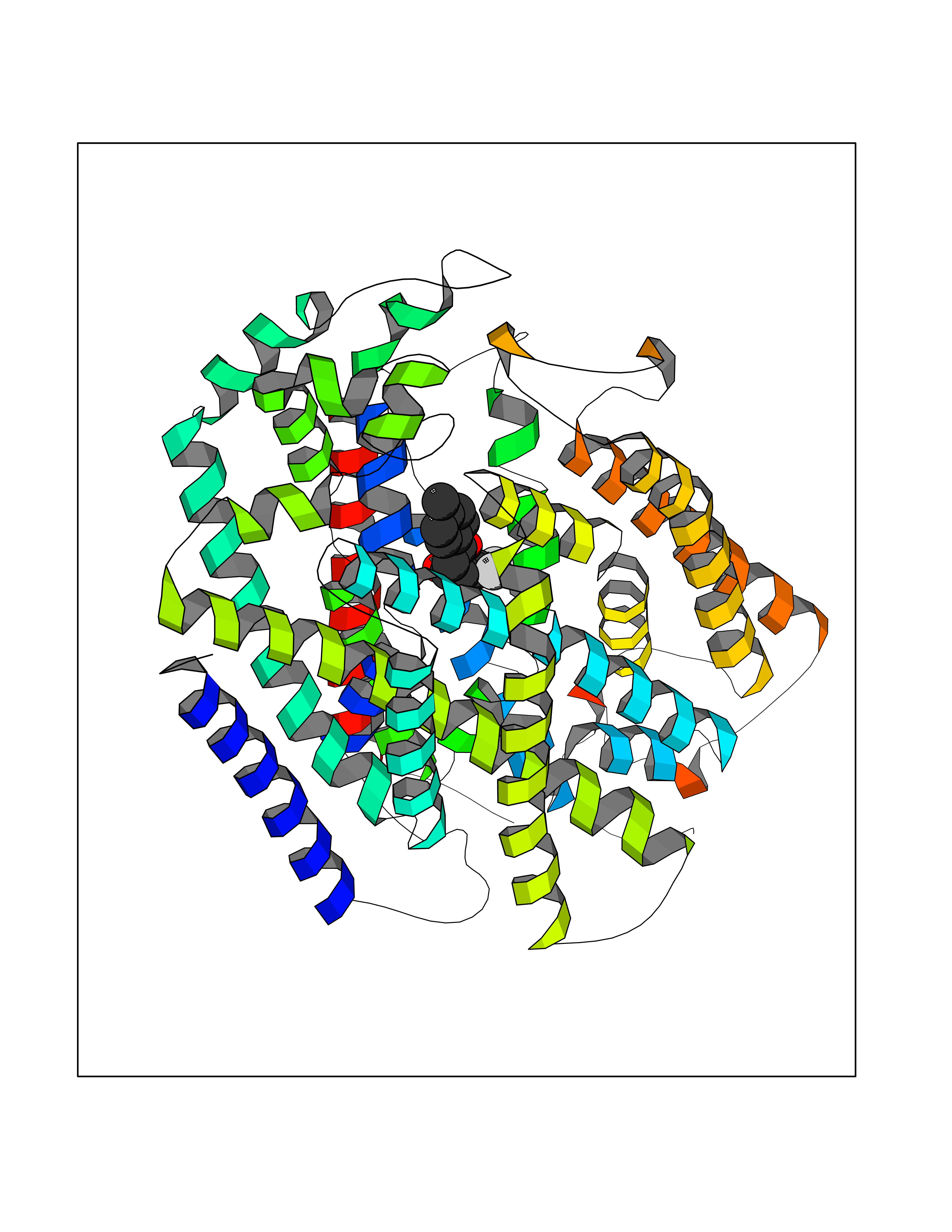 Molscript Map  Pymol Map 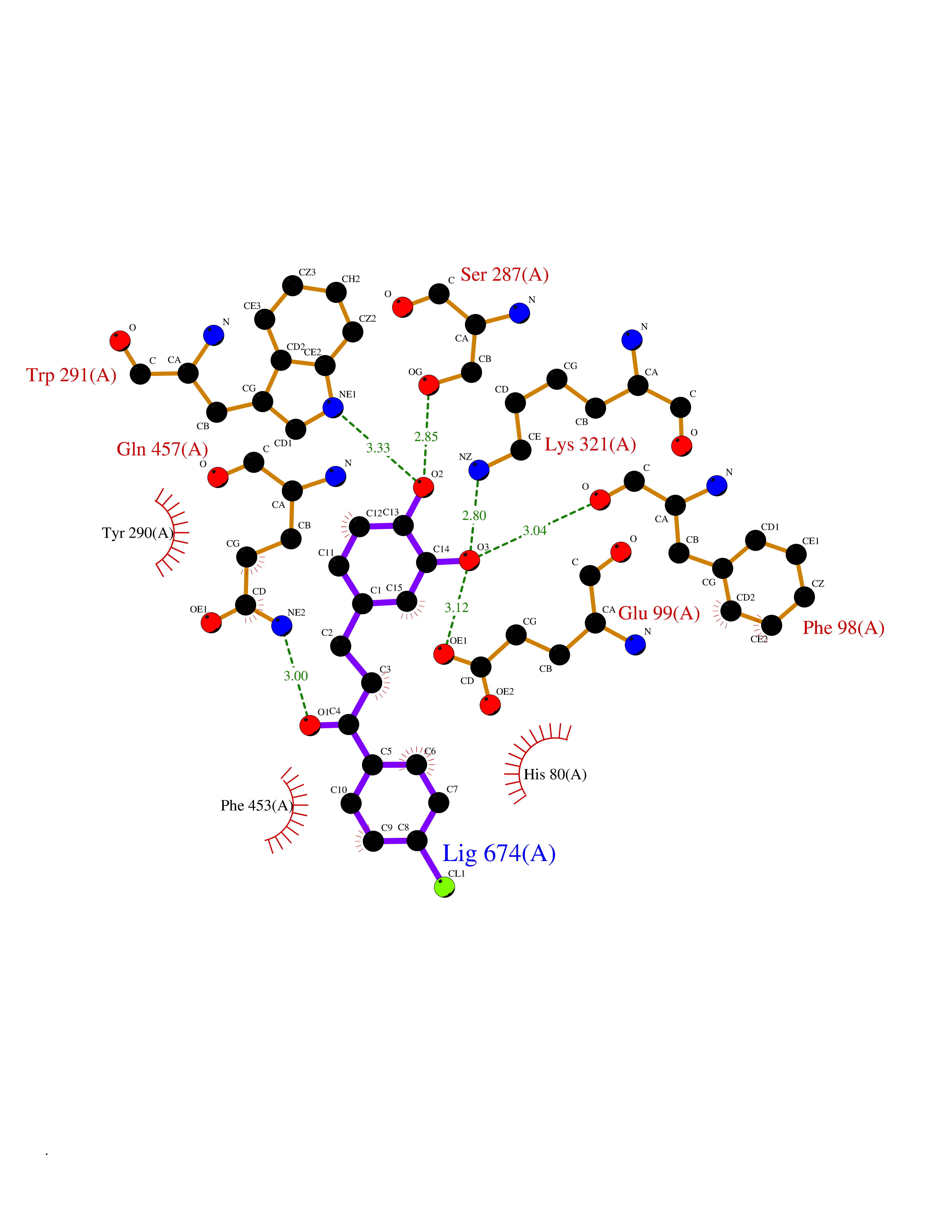 Ligplot Map 3D Binding mode Sequence DNPADILVIAAYFLLVIGVGLWSMCRTNRGTVGGYFLAGRSMVWWPVGASLFASNIGSGHFVGLAGTGAASGLAVAGFEWNALFVVLLLGWLFAPVYLTAGVITMPQYLRKRFGGRRIRLYLSVLSLFLYIFTKISVDMFSGAVFIQQALGWNIYASVIALLGITMIYTVTGGLAALMYTDTVQTFVILGGACILMGYAFHEVGGYSGLFDKYLGAATSLTVSEDPAVGNISSFCYRPRPDSYHLLRHPVTGDLPWPALLLGLTIVSGWYWCSDQVIVQRCLAGKSLTHIKAGCILCGYLKLTPMFLMVMPGMISRILYPDEVACVVPEVCRRVCGTEVGCSNIAYPRLVVKLMPNGLRGLMLAVMLAALMSSLASIFNSSSTLFTMDIYTRLRPRAGDRELLLVGRLWVVFIVVVSVAWLPVVQAAQGGQLFDYIQAVSSYLAPPVSAVFVLALFVPRVNEQGAFWGLIGGLLMGLARLIPEFSFGSGSCVQPSACPAFLCGVHYLYFAIVLFFCSGLLTLTVSLCTAPIPRKHLHRLVFSLRHSKEEREDLDEDISEDPSWARVVNLNALLMMAVAVFLWGFYA Hydrogen bonds contact Hydrophobic contact | ||||
| 85 | Sodium/glucose cotransporter 1 (SGLT1) | 7WMV | 6.91 | |
Target general information Gen name SLC5A1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Solute carrier family 5 member 1; Na(+)/glucose cotransporter 1; NAGT; High affinity sodium-glucose cotransporter Protein family Sodium:solute symporter (SSF) (TC 2.A.21) family Biochemical class Solute:sodium symporter Function Efficient substrate transport in mammalian kidney is provided by the concerted action of a low affinity high capacity and a high affinity low capacity Na(+)/glucose cotransporter arranged in series along kidney proximal tubules. Actively transports glucose into cells by Na(+) cotransport with a Na(+) to glucose coupling ratio of 2:1. Related diseases Congenital glucose/galactose malabsorption (GGM) [MIM:606824]: Intestinal monosaccharide transporter deficiency. It is an autosomal recessive disorder manifesting itself within the first weeks of life. It is characterized by severe diarrhea and dehydration which are usually fatal unless glucose and galactose are eliminated from the diet. {ECO:0000269|PubMed:10036327, ECO:0000269|PubMed:11406349, ECO:0000269|PubMed:2008213, ECO:0000269|PubMed:8195156, ECO:0000269|PubMed:8563765}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00766; DB01914; DB09341; DB05018; DB12713 Interacts with P00533 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disease variant; Disulfide bond; Glycoprotein; Ion transport; Membrane; Phosphoprotein; Proteomics identification; Reference proteome; Sodium; Sodium transport; Sugar transport; Symport; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A Molecular weight (Da) 66451.3 Length 602 Aromaticity 0.12 Instability index 35.84 Isoelectric point 8.3 Charge (pH=7) 4.43 2D Binding mode Binding energy (Kcal/mol) -9.43  Molscript Map 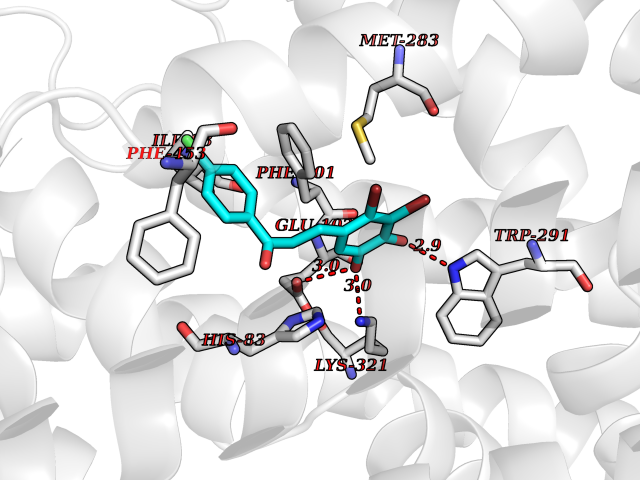 Pymol Map 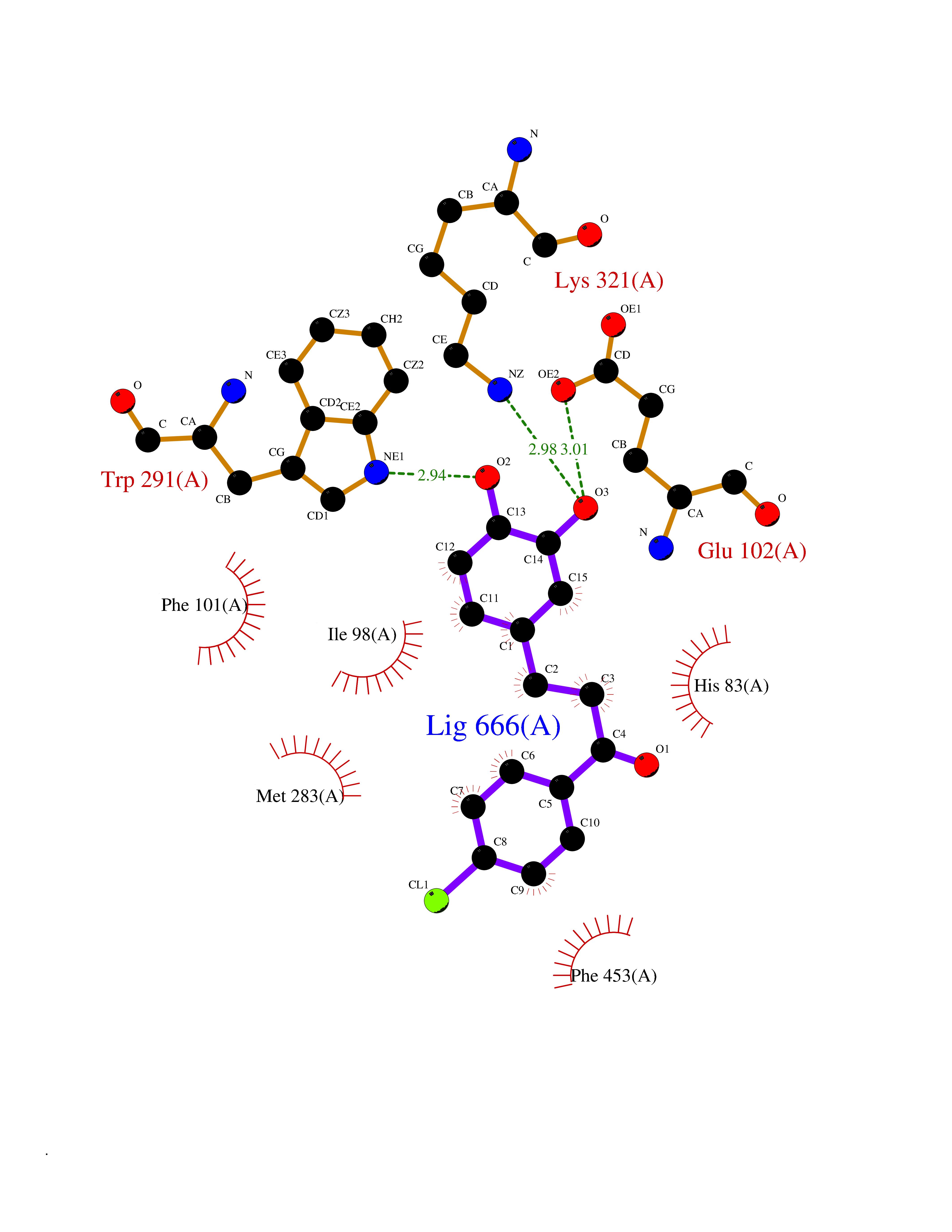 Ligplot Map 3D Binding mode Sequence ETHELIRNAADISIIVIYFVVVMAVGLWAMFSTNRGTVGGFFLAGRSMVWWPIGASLFASNIGSGHFVGLAGTGAASGIAIGGFEWNALVLVVVLGWLFVPIYIKAGVVTMPEYLRKRFGGQRIQVYLSLLSLLLYIFTKISADIFSGAIFINLALGLNLYLAIFLLLAITALYTITGGLAAVIYTDTLQTVIMLVGSLILTGFAFHEVGGYDAFMEKYMKAIPTIVSDGNTTFQEKCYTPRADSFHIFRDPLTGDLPWPGFIFGMSILTLWYWCTDQVIVQRCLSAKNMSHVKGGCILCGYLKLMPMFIMVMPGMISRILYTEKIACVVPSECEKYCGTKVGCTNIAYPTLVVELMPNGLRGLMLSVMLASLMSSLTSIFNSASTLFTMDIYAKVRKRASEKELMIAGRLFILVLIGISIAWVPIVQSAQSGQLFDYIQSITSYLGPPIAAVFLLAIFWKRVNEPGAFWGLILGLLIGISRMITEFAYGTGSCMEPSNCPTIICGVHYLYFAIILFAISFITIVVISLLTKPIPDVHLYRLCWSLRNSKEERIDLDATEEEEKAMKMKMTDTSEKPLWRTVLNVNGIILVTVAVFCHAYFA Hydrogen bonds contact Hydrophobic contact | ||||
| 86 | Thymidine phosphorylase (TYMP) | 2J0F | 6.91 | |
Target general information Gen name TYMP Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms TdRPase; TYMP; TP; Platelet-derived endothelial cell growth factor; PDECGF; PD-ECGF; Gliostatin Protein family Thymidine/pyrimidine-nucleoside phosphorylase family Biochemical class Pentosyltransferase Function Catalyzes the reversible phosphorolysis of thymidine. The produced molecules are then utilized as carbon and energy sources or in the rescue of pyrimidine bases for nucleotide synthesis. Related diseases Mitochondrial DNA depletion syndrome 1, MNGIE type (MTDPS1) [MIM:603041]: A multisystem disease associated with mitochondrial dysfunction. It is clinically characterized by onset between the second and fifth decades of life, ptosis, progressive external ophthalmoplegia, gastrointestinal dysmotility (often pseudoobstruction), diffuse leukoencephalopathy, cachexia, peripheral neuropathy, and myopathy. {ECO:0000269|PubMed:12177387, ECO:0000269|PubMed:9924029}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01101; DB00369; DB00322; DB00544; DB06433; DB09343; DB00432 Interacts with Q14696; Q9H0C1 EC number EC 2.4.2.4 Uniprot keywords 3D-structure; Alternative splicing; Angiogenesis; Chemotaxis; Developmental protein; Differentiation; Direct protein sequencing; Disease variant; Glycosyltransferase; Growth factor; Neuropathy; Phosphoprotein; Primary mitochondrial disease; Progressive external ophthalmoplegia; Proteomics identification; Reference proteome; Repeat; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 46508.2 Length 446 Aromaticity 0.03 Instability index 37.49 Isoelectric point 5.81 Charge (pH=7) -4.7 2D Binding mode Binding energy (Kcal/mol) -9.43  Molscript Map 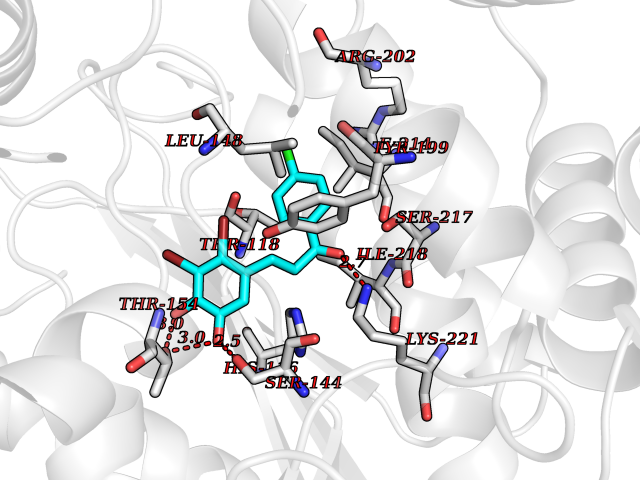 Pymol Map 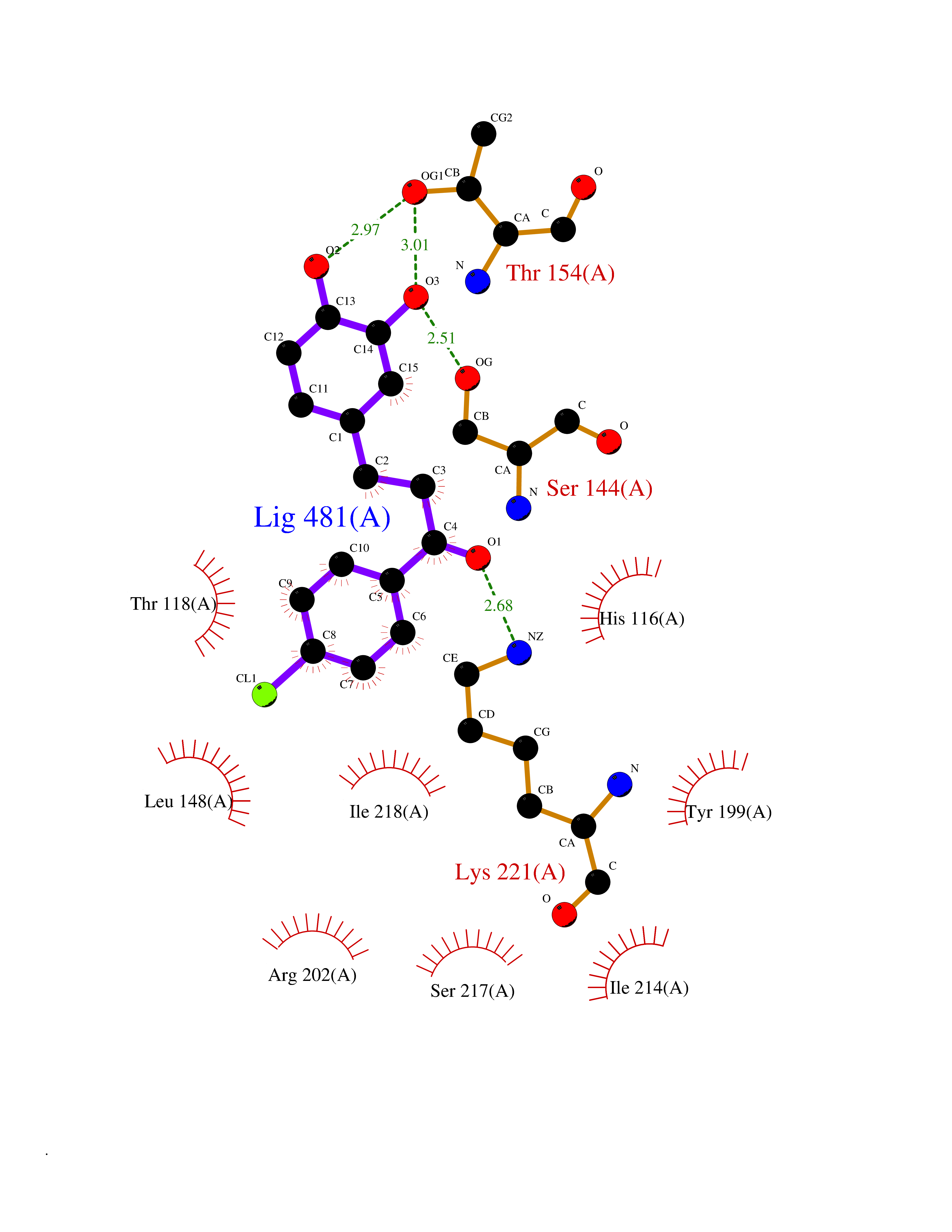 Ligplot Map 3D Binding mode Sequence KQLPELIRMKRDGGRLSEADIRGFVAAVVNGSAQGAQIGAMLMAIRLRGMDLEETSVLTQALAQSGQQLEWPEAWRQQLVDKHSTGGVGDKVSLVLAPALAACGCKVPMISGRGLGHTGGTLDKLESIPGFNVIQSPEQMQVLLDQAGCCIVGQSEQLVPADGILYAARDVTATVDSLPLITASILSKKLVEGLSALVVDVKFGAGAVFPNQEQARELAKTLVGVGASLGLRVAAALTAMDKPLGRCVGHALEVEEALLCMDGAGPPDLRDLVTTLGGALLWLSGHAGTQAQGAARVAAALDDGSALGRFERMLAAQGVDPGLARALCSGSPAERRQLLPRAREQEELLAPADGTVELVRALPLALVLHELGAGRSRAGEPLRLGVGAELLVDVGQRLRRGTPWLRVHRDGPALSGPQSRALQEALVLSDRAPFAAPLPFAELVLP Hydrogen bonds contact Hydrophobic contact | ||||
| 87 | Oxalosuccinate decarboxylase (IDH1) | 6ADG | 6.91 | |
Target general information Gen name IDH1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms PICD; NADP(+)-specific ICDH; Isocitrate dehydrogenase [NADP] cytoplasmic; IDP; IDH; Cytosolic NADP-isocitrate dehydrogenase Protein family Isocitrate and isopropylmalate dehydrogenases family Biochemical class Short-chain dehydrogenases reductase Function Catalyses the NADPH-dependent reduction of alpha-ketoglutarate to R(-)-2-hydroxyglutarate (2HG). Related diseases Glioma (GLM) [MIM:137800]: Gliomas are benign or malignant central nervous system neoplasms derived from glial cells. They comprise astrocytomas and glioblastoma multiforme that are derived from astrocytes, oligodendrogliomas derived from oligodendrocytes and ependymomas derived from ependymocytes. {ECO:0000269|PubMed:19117336, ECO:0000269|PubMed:19935646}. The gene represented in this entry is involved in disease pathogenesis. Mutations affecting Arg-132 are tissue-specific, and suggest that this residue plays a unique role in the development of high-grade gliomas. Mutations of Arg-132 to Cys, His, Leu or Ser abolish magnesium binding and abolish the conversion of isocitrate to alpha-ketoglutarate. Instead, alpha-ketoglutarate is converted to R(-)-2-hydroxyglutarate. Elevated levels of R(-)-2-hydroxyglutarate are correlated with an elevated risk of malignant brain tumors. {ECO:0000269|PubMed:19935646}.; DISEASE: Genetic variations are associated with cartilaginous tumors such as enchondroma or chondrosarcoma. Mutations of Arg-132 to Cys, Gly or His abolish the conversion of isocitrate to alpha-ketoglutarate. Instead, alpha-ketoglutarate is converted to R(-)-2-hydroxyglutarate. {ECO:0000269|PubMed:26161668}. Drugs (DrugBank ID) DB09374; DB01727; DB14568; DB03461; DB16267 Interacts with P0DP23; P27797; P36957; O75874; Q8TDX7; P16284; P17612; P50454; P37173; Q05086-3 EC number EC 1.1.1.42 Uniprot keywords 3D-structure; Acetylation; Cytoplasm; Direct protein sequencing; Glyoxylate bypass; Magnesium; Manganese; Metal-binding; NADP; Oxidoreductase; Peroxisome; Phosphoprotein; Proteomics identification; Reference proteome; Tricarboxylic acid cycle Protein physicochemical properties Chain ID A,B Molecular weight (Da) 92711.7 Length 823 Aromaticity 0.1 Instability index 26.74 Isoelectric point 6.42 Charge (pH=7) -4.48 2D Binding mode Binding energy (Kcal/mol) -9.42  Molscript Map 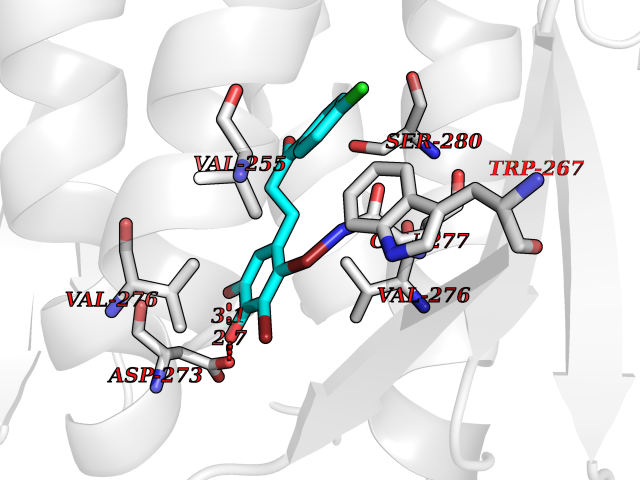 Pymol Map 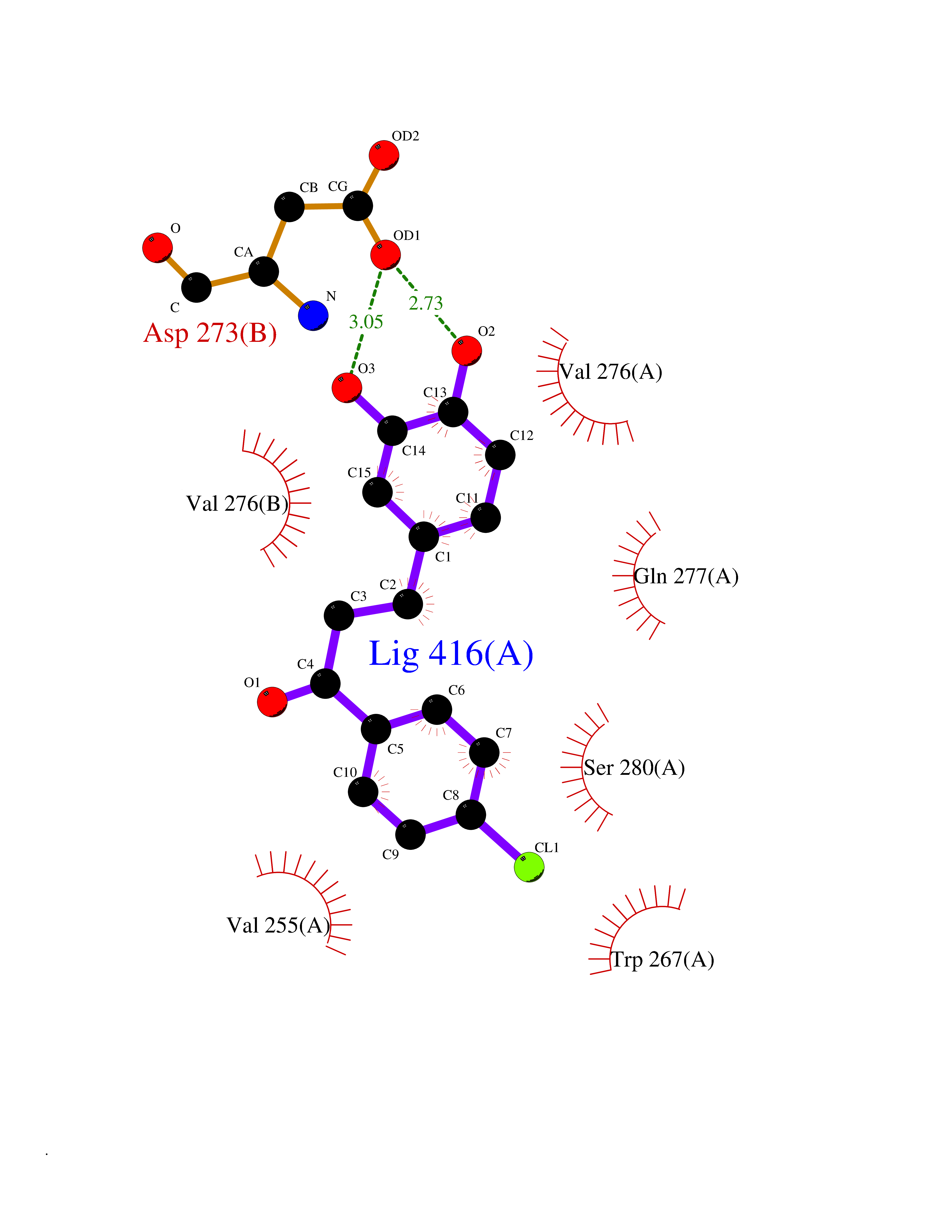 Ligplot Map 3D Binding mode Sequence KKISGGSVVEMQGDEMTRIIWELIKEKLIFPYVELDLHSYDLGIENRDATNDQVTKDAAEAIKKHNVGVKCATITPDEKRVEEFKLKQMWKSPNGTIRNILGGTVFREAIICKNIPRLVSGWVKPIIIGHHAYGDQYRATDFVVPGPGKVEITYTPSDGTQKVTYLVHNFEEGGGVAMGMYNQDKSIEDFAHSSFQMALSKGWPLYLSTKNTILKKYDGRFKDIFQEIYDKQYKSQFEAQKIWYEHRLIDDMVAQAMKSEGGFIWACKNYDGDVQSDSVAQGYGSLGMMTSVLVCPDGKTVEAEAAHGTVTRHYRMYQKGQETSTNPIASIFAWTRGLAHRAKLDNNKELAFFANALEEVSIETIEAGFMTKDLAACIKGLPNVQRSDYLNTFEFMDKLGENLKIKLAQAKLKKISGGSVVEMQGDEMTRIIWELIKEKLIFPYVELDLHSYDLGIENRDATNDQVTKDAAEAIKKHNVGVKCATITPDEKRVEEFKLKQMWKSPNGTIRNILGGTVFREAIICKNIPRLVSGWVKPIIIGHHAYGDQYRATDFVVPGPGKVEITYTPSDGTQKVTYLVHNFEEGGGVAMGMYNQDKSIEDFAHSSFQMALSKGWPLYLSTKNTILKKYDGRFKDIFQEIYDKQYKSQFEAQKIWYEHRLIDDMVAQAMKSEGGFIWACKNYDGDVQSDSVAQGYGSLGMMTSVLVCPDGKTVEAEAAHGTVTRHYRMYQKGQETSTNPIASIFAWTRGLAHRAKLDNNKELAFFANALEEVSIETIEAGFMTKDLAACIKGLPNVQRSDYLNTFEFMDKLGENLKIKLAQAK Hydrogen bonds contact Hydrophobic contact | ||||
| 88 | Catechol-O-methyl-transferase (COMT) | 3BWY | 6.90 | |
Target general information Gen name COMT Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms S-COMT; MB-COMT; Catechol-O-methyltransferase; COMT Protein family Class I-like SAM-binding methyltransferase superfamily, Cation-dependent O-methyltransferase family Biochemical class Methyltransferase Function Catalyzes the O-methylation, and thereby the inactivation, of catecholamine neurotransmitters and catechol hormones. Also shortens the biological half-lives of certain neuroactive drugs, like L-DOPA, alpha-methyl DOPA and isoproterenol. Related diseases Schizophrenia (SCZD) [MIM:181500]: A complex, multifactorial psychotic disorder or group of disorders characterized by disturbances in the form and content of thought (e.g. delusions, hallucinations), in mood (e.g. inappropriate affect), in sense of self and relationship to the external world (e.g. loss of ego boundaries, withdrawal), and in behavior (e.g bizarre or apparently purposeless behavior). Although it affects emotions, it is distinguished from mood disorders in which such disturbances are primary. Similarly, there may be mild impairment of cognitive function, and it is distinguished from the dementias in which disturbed cognitive function is considered primary. Some patients manifest schizophrenic as well as bipolar disorder symptoms and are often given the diagnosis of schizoaffective disorder. {ECO:0000269|PubMed:15645182}. Disease susceptibility may be associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07462; DB02342; DB02105; DB08049; DB00118; DB00714; DB03336; DB00286; DB00255; DB00841; DB00988; DB15488; DB00494; DB00668; DB00783; DB00977; DB01064; DB00968; DB01141; DB03907; DB04820; DB06152; DB11632; DB00252; DB01420; DB00323 Interacts with Q6P5T0; P30518; Q8NFU1; Q8NHW4; P34972; Q96BA8; P50402; Q5JX71; O14843; O00258; P08034; O75712; Q9NTQ9; O95377; Q8TDT2; Q8N6U8; O15529; P31937; Q9H2F3; O95279; Q5SR56; A6NDP7; Q0D2K0; Q7RTS5; Q9UHJ9-5; Q8IY26; Q9H6H4; Q6NTF9-3; O75783; Q99500; Q9Y6D0; Q3KNW5; O60669; P22732; Q96G79; Q5T1Q4; Q9NY26; Q9NP94; Q6P1K1; P30825; Q9UHI5; B2RUZ4; Q9UPZ6; Q96MV1; Q9NV29; A0PK00; Q9NUH8; Q9P0S9; Q14656; Q6UW68; Q9H0R3; O95807; P34981; Q15645; Q15836; O95183; O76024; P30260; Q9H816; Q92997; P29323-3; P22607; P06396; Q15323; Q6A162; P26371; O15116; P20645; O14744; Q5T160; Q9UJD0; Q2MKA7; Q8N488; O75880; Q14141; Q9UNE7; Q15645; Q9NYH9; Q8NA23-2 EC number EC 2.1.1.6 Uniprot keywords 3D-structure; Alternative initiation; Catecholamine metabolism; Cell membrane; Cytoplasm; Direct protein sequencing; Lipid metabolism; Magnesium; Membrane; Metal-binding; Methyltransferase; Neurotransmitter degradation; Phosphoprotein; Proteomics identification; Reference proteome; S-adenosyl-L-methionine; Schizophrenia; Signal-anchor; Transferase; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 23851.2 Length 214 Aromaticity 0.07 Instability index 25.99 Isoelectric point 5.25 Charge (pH=7) -7.75 2D Binding mode Binding energy (Kcal/mol) -9.41 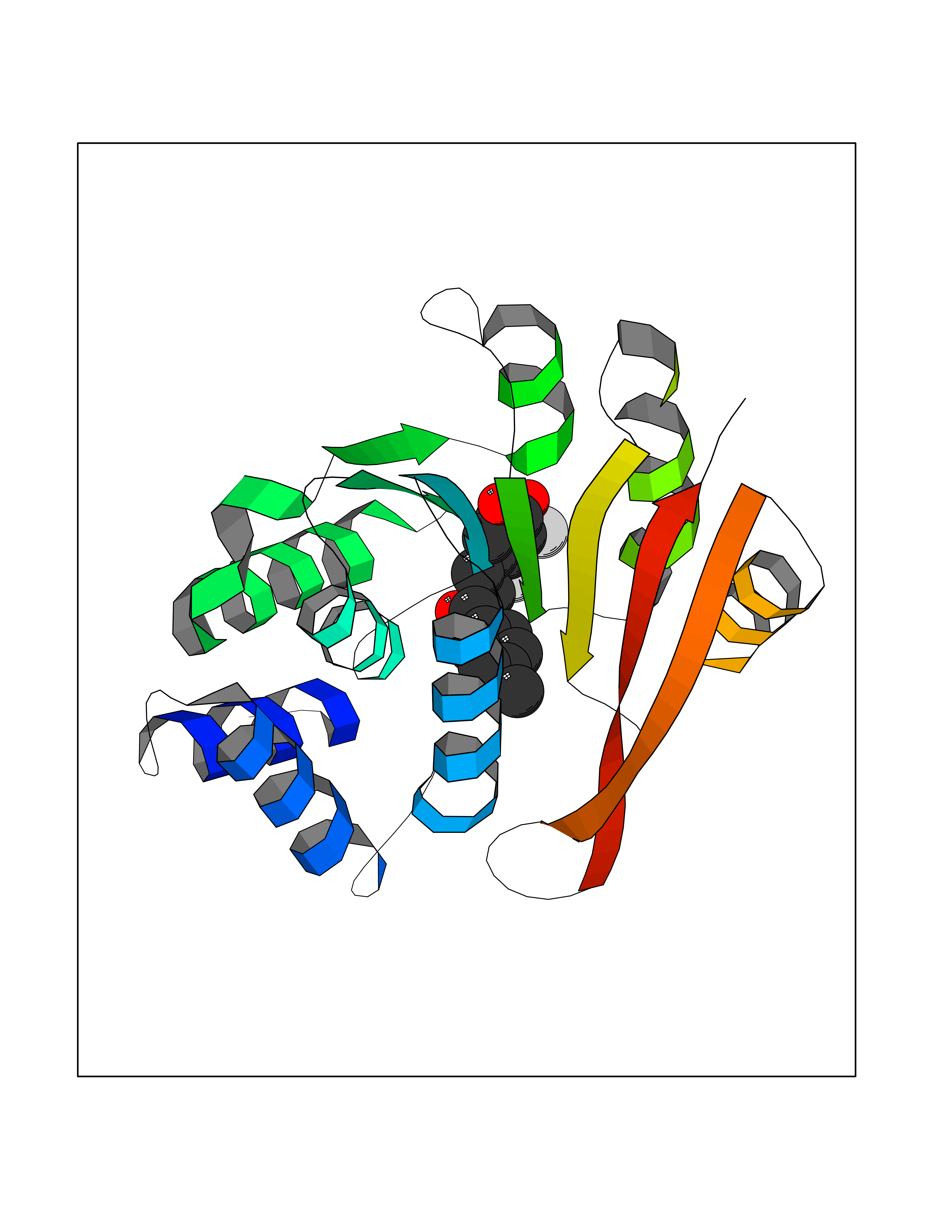 Molscript Map 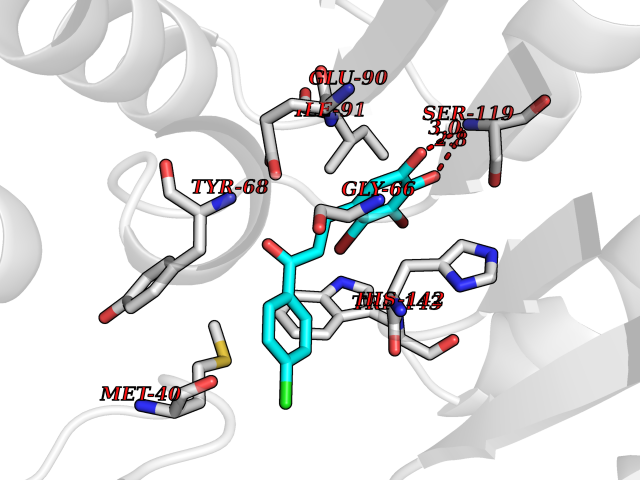 Pymol Map 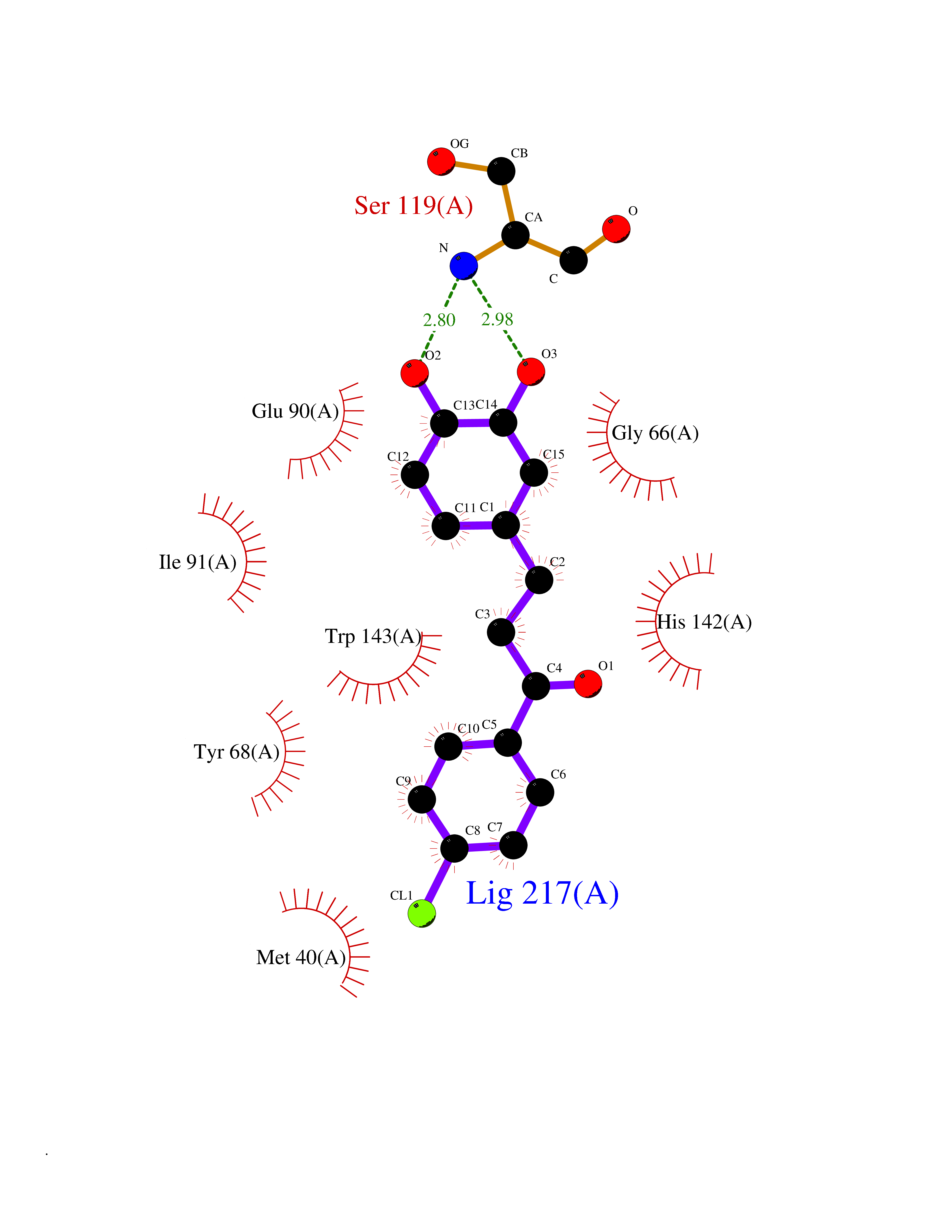 Ligplot Map 3D Binding mode Sequence GDTKEQRILNHVLQHAEPGNAQSVLEAIDTYCEQKEWAMNVGDKKGKIVDAVIQEHQPSVLLELGAYCGYSAVRMARLLSPGARLITIEINPDCAAITQRMVDFAGMKDKVTLVVGASQDIIPQLKKKYDVDTLDMVFLDHWKDRYLPDTLLLEECGLLRKGTVLLADNVICPGAPDFLAHVRGSSCFECTHYQSFLEYREVVDGLEKAIYKGP Hydrogen bonds contact Hydrophobic contact | ||||
| 89 | Serine/threonine-protein kinase cot (COT) | 4Y85 | 6.90 | |
Target general information Gen name MAP3K8 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Tumor progression locus 2; TPL-2; Proto-oncogene c-Cot; Mitogen-activated protein kinase kinase kinase 8; ESTF; Cancer Osaka thyroid oncogene; COT Protein family Protein kinase superfamily, STE Ser/Thr protein kinase family, MAP kinase kinase kinase subfamily Biochemical class NA Function Required for lipopolysaccharide (LPS)-induced, TLR4-mediated activation of the MAPK/ERK pathway in macrophages, thus being critical for production of the proinflammatory cytokine TNF-alpha (TNF) during immune responses. Involved in the regulation of T-helper cell differentiation and IFNG expression in T-cells. Involved in mediating host resistance to bacterial infection through negative regulation of type I interferon (IFN) production. In vitro, activates MAPK/ERK pathway in response to IL1 in an IRAK1-independent manner, leading to up-regulation of IL8 and CCL4. Transduces CD40 and TNFRSF1A signals that activate ERK in B-cells and macrophages, and thus may play a role in the regulation of immunoglobulin production. May also play a role in the transduction of TNF signals that activate JNK and NF-kappa-B in some cell types. In adipocytes, activates MAPK/ERK pathway in an IKBKB-dependent manner in response to IL1B and TNF, but not insulin, leading to induction of lipolysis. Plays a role in the cell cycle. Isoform 1 shows some transforming activity, although it is much weaker than that of the activated oncogenic variant. Related diseases Hyperinsulinemic hypoglycemia, familial, 2 (HHF2) [MIM:601820]: A form of hyperinsulinemic hypoglycemia, a clinically and genetically heterogeneous disorder characterized by inappropriate insulin secretion from the pancreatic beta-cells in the presence of low blood glucose levels. HHF2 is a common cause of persistent hypoglycemia in infancy. Unless early and aggressive intervention is undertaken, brain damage from recurrent episodes of hypoglycemia may occur. HHF2 inheritance can be autosomal dominant or autosomal recessive. {ECO:0000269|PubMed:10204114, ECO:0000269|PubMed:12364426, ECO:0000269|PubMed:15562009, ECO:0000269|PubMed:15579781, ECO:0000269|PubMed:15807877, ECO:0000269|PubMed:15998776, ECO:0000269|PubMed:16332676, ECO:0000269|PubMed:16357843, ECO:0000269|PubMed:18596924, ECO:0000269|PubMed:19357197, ECO:0000269|PubMed:7847376, ECO:0000269|PubMed:8923010}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Diabetes mellitus, permanent neonatal, 2 (PNDM2) [MIM:618856]: A form of permanent neonatal diabetes mellitus, a type of diabetes characterized by onset of persistent hyperglycemia within the first six months of life. Initial clinical manifestations include intrauterine growth retardation, hyperglycemia, glycosuria, osmotic polyuria, severe dehydration, and failure to thrive. Some PNDM2 patients may also have developmental delay, muscle weakness, epilepsy and dysmorphic features. PNDM2 transmission pattern is consistent with autosomal dominant inheritance. {ECO:0000269|PubMed:15115830, ECO:0000269|PubMed:15292329, ECO:0000269|PubMed:15448106, ECO:0000269|PubMed:15448107, ECO:0000269|PubMed:15580558, ECO:0000269|PubMed:15583126, ECO:0000269|PubMed:16609879, ECO:0000269|PubMed:16731833, ECO:0000269|PubMed:17213273, ECO:0000269|PubMed:17652641, ECO:0000269|PubMed:17855752, ECO:0000269|PubMed:20022885, ECO:0000269|PubMed:28842488}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Transient neonatal diabetes mellitus 3 (TNDM3) [MIM:610582]: Neonatal diabetes mellitus, defined as insulin-requiring hyperglycemia within the first month of life, is a rare entity. In about half of the neonates, diabetes is transient and resolves at a median age of 3 months, whereas the rest have a permanent form of diabetes. In a significant number of patients with transient neonatal diabetes mellitus, diabetes type 2 appears later in life. The onset and severity of TNDM3 is variable with childhood-onset diabetes, gestational diabetes or adult-onset diabetes described. {ECO:0000269|PubMed:15718250, ECO:0000269|PubMed:15784703}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Defects in KCNJ11 may contribute to non-insulin-dependent diabetes mellitus (NIDDM), also known as diabetes mellitus type 2.; DISEASE: Maturity-onset diabetes of the young 13 (MODY13) [MIM:616329]: A form of diabetes that is characterized by an autosomal dominant mode of inheritance, onset in childhood or early adulthood (usually before 25 years of age), a primary defect in insulin secretion and frequent insulin-independence at the beginning of the disease. {ECO:0000269|PubMed:22701567}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P08238; P19838; Q00653; Q13526; Q8NFZ5 EC number EC 2.7.11.25 Uniprot keywords 3D-structure; Alternative initiation; ATP-binding; Cell cycle; Cytoplasm; Immunity; Kinase; Magnesium; Metal-binding; Nucleotide-binding; Phosphoprotein; Proteomics identification; Proto-oncogene; Reference proteome; Serine/threonine-protein kinase; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 34773.8 Length 307 Aromaticity 0.09 Instability index 39.24 Isoelectric point 6.68 Charge (pH=7) -1.2 2D Binding mode Binding energy (Kcal/mol) -9.41 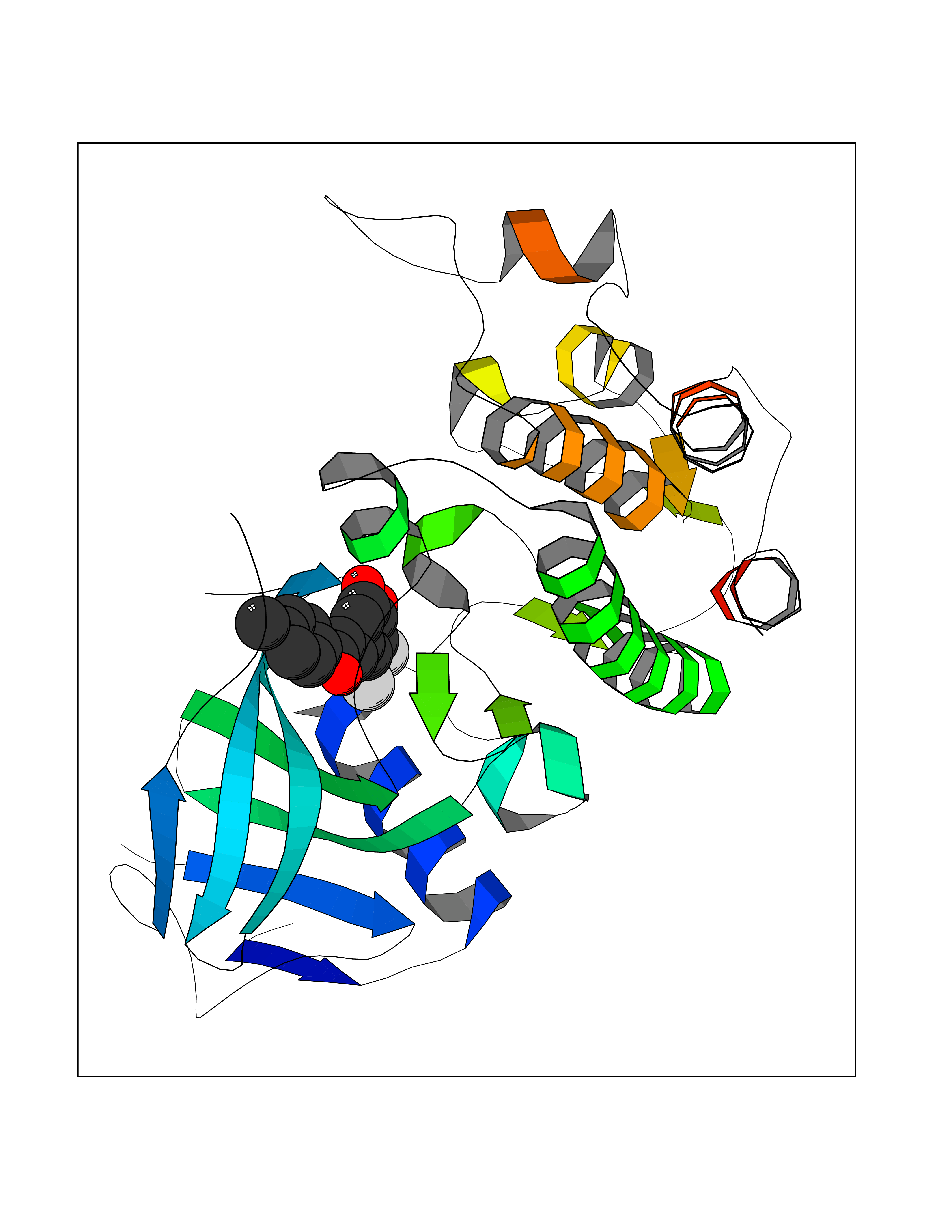 Molscript Map 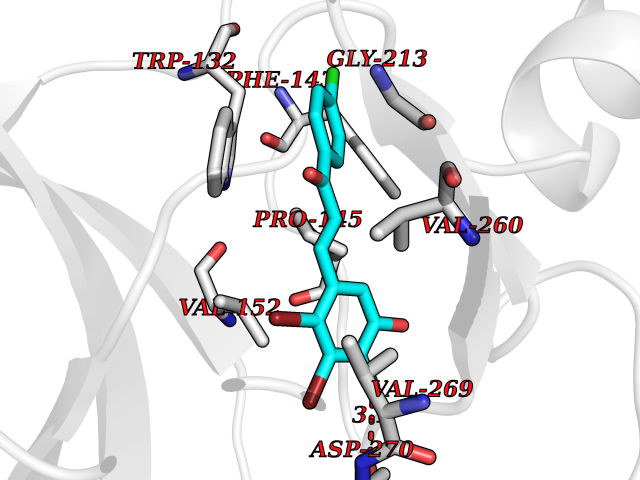 Pymol Map 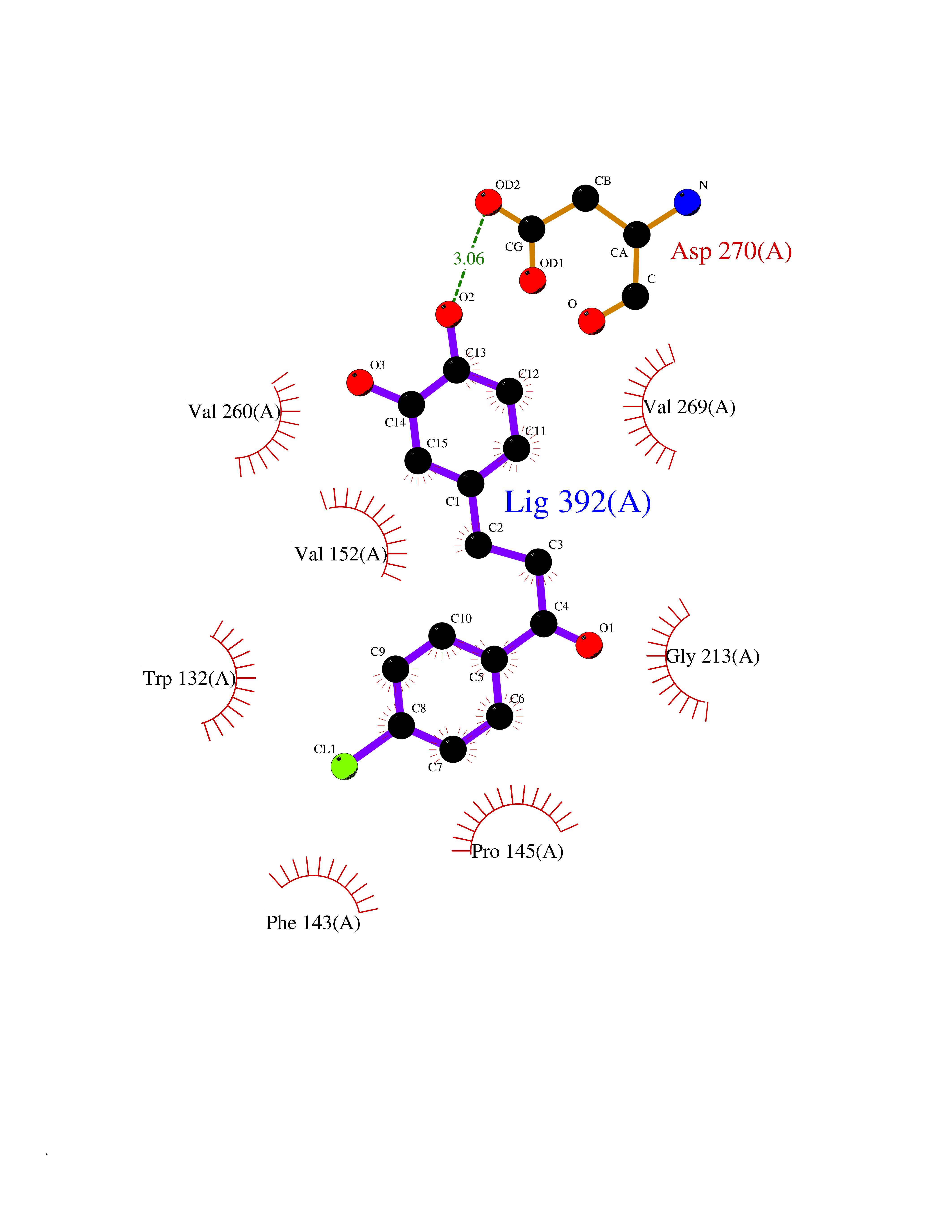 Ligplot Map 3D Binding mode Sequence LSSVRYGTVEDLLAFANHISNTPQESGILLNMVITPQNGRYQIDSDVLLIPWKLTYRNIFIPRGAFGKVYLAQDIKTKKRMACKLIPVDQFKPSDVEIQACFRHENIAELYGAVLWGETVHLFMEAGEGGSVLEKLESCGPMREFEIIWVTKHVLKGLDFLHSKKVIHHDIKPSNIVFMSTKAVLVDFGLSVQMTEDVYFPKDLRGTEIYMSPEVILCRGHSTKADIYSLGATLIHMQTGTPPWVKRYPRSAYPSYLYIIHKQAPPLEDIADDCSPGMRELIEASLERNPNHRPRAADLLKHEALNP Hydrogen bonds contact Hydrophobic contact | ||||
| 90 | ABL T315I mutant (ABL T315I) | 4TWP | 6.90 | |
Target general information Gen name ABL1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms p150 T315I; T315I Abl; Proto-oncogene tyrosine-protein kinase ABL1 T315I; Proto-oncogene c-Abl T315I; JTK7 T315I; C-ABL T315I; Abl T315I; Abelson tyrosine-protein kinase 1 T315I; Abelson murine leukem Protein family Protein kinase superfamily, Tyr protein kinase family, ABL subfamily Biochemical class Kinase Function Coordinates actin remodeling through tyrosine phosphorylation of proteins controlling cytoskeleton dynamics like WASF3 (involved in branch formation); ANXA1 (involved in membrane anchoring); DBN1, DBNL, CTTN, RAPH1 and ENAH (involved in signaling); or MAPT and PXN (microtubule-binding proteins). Phosphorylation of WASF3 is critical for the stimulation of lamellipodia formation and cell migration. Involved in the regulation of cell adhesion and motility through phosphorylation of key regulators of these processes such as BCAR1, CRK, CRKL, DOK1, EFS or NEDD9. Phosphorylates multiple receptor tyrosine kinases and more particularly promotes endocytosis of EGFR, facilitates the formation of neuromuscular synapses through MUSK, inhibits PDGFRB-mediated chemotaxis and modulates the endocytosis of activated B-cell receptor complexes. Other substrates which are involved in endocytosis regulation are the caveolin (CAV1) and RIN1. Moreover, ABL1 regulates the CBL family of ubiquitin ligases that drive receptor down-regulation and actin remodeling. Phosphorylation of CBL leads to increased EGFR stability. Involved in late-stage autophagy by regulating positively the trafficking and function of lysosomal components. ABL1 targets to mitochondria in response to oxidative stress and thereby mediates mitochondrial dysfunction and cell death. In response to oxidative stress, phosphorylates serine/threonine kinase PRKD2 at 'Tyr-717'. ABL1 is also translocated in the nucleus where it has DNA-binding activity and is involved in DNA-damage response and apoptosis. Many substrates are known mediators of DNA repair: DDB1, DDB2, ERCC3, ERCC6, RAD9A, RAD51, RAD52 or WRN. Activates the proapoptotic pathway when the DNA damage is too severe to be repaired. Phosphorylates TP73, a primary regulator for this type of damage-induced apoptosis. Phosphorylates the caspase CASP9 on 'Tyr-153' and regulates its processing in the apoptotic response to DNA damage. Phosphorylates PSMA7 that leads to an inhibition of proteasomal activity and cell cycle transition blocks. ABL1 acts also as a regulator of multiple pathological signaling cascades during infection. Several known tyrosine-phosphorylated microbial proteins have been identified as ABL1 substrates. This is the case of A36R of Vaccinia virus, Tir (translocated intimin receptor) of pathogenic E. coli and possibly Citrobacter, CagA (cytotoxin-associated gene A) of H. pylori, or AnkA (ankyrin repeat-containing protein A) of A. phagocytophilum. Pathogens can highjack ABL1 kinase signaling to reorganize the host actin cytoskeleton for multiple purposes, like facilitating intracellular movement and host cell exit. Finally, functions as its own regulator through autocatalytic activity as well as through phosphorylation of its inhibitor, ABI1. Regulates T-cell differentiation in a TBX21-dependent manner. Phosphorylates TBX21 on tyrosine residues leading to an enhancement of its transcriptional activator activity. Non-receptor tyrosine-protein kinase that plays a role in many key processes linked to cell growth and survival such as cytoskeleton remodeling in response to extracellular stimuli, cell motility and adhesion, receptor endocytosis, autophagy, DNA damage response and apoptosis. Related diseases Leukemia, chronic myeloid (CML) [MIM:608232]: A clonal myeloproliferative disorder of a pluripotent stem cell with a specific cytogenetic abnormality, the Philadelphia chromosome (Ph), involving myeloid, erythroid, megakaryocytic, B-lymphoid, and sometimes T-lymphoid cells, but not marrow fibroblasts. The gene represented in this entry is involved in disease pathogenesis.; DISEASE: A chromosomal aberration involving ABL1 has been found in patients with chronic myeloid leukemia. Translocation t(9;22)(q34;q11) with BCR. The translocation produces a BCR-ABL found also in acute myeloid leukemia (AML) and acute lymphoblastic leukemia (ALL). {ECO:0000269|PubMed:3021337}.; DISEASE: A chromosomal aberration involving ABL1 is found in a form of acute lymphoblastic leukemia (PubMed:15361874). Translocation t(9;9)(q34;q34) with NUP214 (PubMed:15361874). {ECO:0000269|PubMed:15361874}.; DISEASE: Congenital heart defects and skeletal malformations syndrome (CHDSKM) [MIM:617602]: An autosomal dominant disorder characterized by congenital heart disease with atrial and ventricular septal defects, variable skeletal abnormalities, and failure to thrive. Skeletal defects include pectus excavatum, scoliosis, and finger contractures. Some patient exhibit joint laxity. {ECO:0000269|PubMed:28288113}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08043; DB08583; DB07831; DB08350; DB12597; DB00171; DB06616; DB12267; DB01254; DB12010; DB00619; DB13749; DB08231; DB03878; DB04868; DB08339; DB08901; DB12323; DB08896; DB14989; DB05184 Interacts with Q8IZP0; Q9NYB9; O14672; P10275; Q13315; Q4KMG0; P46108; P46109; P35222; P00533; P04626; Q03468; Q14315; P36888; P05107; P10721; Q38SD2; Q92918; Q7Z434; O43196; P15941; P15941-12; P16333; O43900; Q13905; Q86UR5; Q13671; P31947; Q15464; O75751; P37840; Q9BX66; O60504-2; Q07890; P12931; P51692; Q9Y4G6; P11387; P04637; P15498; Q9Y6W5; P62258; P61981; P63104; O35158; P37840; P48165; Q15323; P37840 EC number EC 2.7.10.2 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Apoptosis; ATP-binding; Autophagy; Cell adhesion; Chromosomal rearrangement; Cytoplasm; Cytoskeleton; Disease variant; DNA damage; DNA repair; DNA-binding; Endocytosis; Kinase; Lipoprotein; Magnesium; Manganese; Membrane; Metal-binding; Mitochondrion; Myristate; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Proto-oncogene; Reference proteome; SH2 domain; SH3 domain; Transferase; Tyrosine-protein kinase; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 30776.1 Length 266 Aromaticity 0.12 Instability index 36.47 Isoelectric point 5.39 Charge (pH=7) -7.71 2D Binding mode Binding energy (Kcal/mol) -9.41  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence DKWEMERTDITMKHKLGGGQYGEVYEGVWKKYSLTVAVKTLEVEEFLKEAAVMKEIKHPNLVQLLGVCTREPPFYIIIEFMTYGNLLDYLRECNRQEVNAVVLLYMATQISSAMEYLEKKNFIHRDLAARNCLVGENHLVKVADFGLSRLMTGDTYTAHAGAKFPIKWTAPESLAYNKFSIKSDVWAFGVLLWEIATYGMSPYPGIDLSQVYELLEKDYRMERPEGCPEKVYELMRACWQWNPSDRPSFAEIHQAFETMFQESSIS Hydrogen bonds contact Hydrophobic contact | ||||
| 91 | Rhinovirus Protease 3C (HRV P3C) | 1FPN | 6.90 | |
Target general information Gen name HRV P3C Organism Human rhinovirus 2 (HRV-2) Uniprot ID TTD ID Synonyms Rhinovirus P3C Protein family Picornaviruses polyprotein family Biochemical class NA Function Capsid protein VP1: Forms an icosahedral capsid of pseudo T=3 symmetry with capsid proteins VP2 and VP3. The capsid is 300 Angstroms in diameter, composed of 60 copies of each capsid protein and enclosing the viral positive strand RNA genome. Capsid protein VP1 mainly forms the vertices of the capsid. Capsid protein VP1 interacts with host VLDLR to provide virion attachment to target host cells. This attachment induces virion internalization. Tyrosine kinases are probably involved in the entry process. After binding to its receptor, the capsid undergoes conformational changes. Capsid protein VP1 N-terminus (that contains an amphipathic alpha-helix) and capsid protein VP4 are externalized. Together, they shape a pore in the host membrane through which viral genome is translocated to host cell cytoplasm. After genome has been released, the channel shrinks (By similarity). Related diseases Charcot-Marie-Tooth disease, axonal, 2DD (CMT2DD) [MIM:618036]: A dominant axonal form of Charcot-Marie-Tooth disease, a disorder of the peripheral nervous system, characterized by progressive weakness and atrophy, initially of the peroneal muscles and later of the distal muscles of the arms. Charcot-Marie-Tooth disease is classified in two main groups on the basis of electrophysiologic properties and histopathology: primary peripheral demyelinating neuropathies (designated CMT1 when they are dominantly inherited) and primary peripheral axonal neuropathies (CMT2). Neuropathies of the CMT2 group are characterized by signs of axonal degeneration in the absence of obvious myelin alterations, normal or slightly reduced nerve conduction velocities, and progressive distal muscle weakness and atrophy. {ECO:0000269|PubMed:29499166}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Hypomagnesemia, seizures, and impaired intellectual development 2 (HOMGSMR2) [MIM:618314]: An autosomal dominant disease characterized by generalized seizures in infancy, severe hypomagnesemia, and renal magnesium wasting. Seizures persist despite magnesium supplementation and are associated with significant intellectual disability. {ECO:0000269|PubMed:30388404}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02313; DB03017 Interacts with NA EC number EC 3.4.22.28 Uniprot keywords 3D-structure; Activation of host autophagy by virus; ATP-binding; Autocatalytic cleavage; Capsid protein; Covalent protein-RNA linkage; DNA replication; Eukaryotic host gene expression shutoff by virus; Eukaryotic host translation shutoff by virus; Helicase; Host cytoplasm; Host cytoplasmic vesicle; Host gene expression shutoff by virus; Host membrane; Host mRNA suppression by virus; Host nucleus; Host-virus interaction; Hydrolase; Inhibition of host innate immune response by virus; Inhibition of host mRNA nuclear export by virus; Inhibition of host RIG-I by virus; Inhibition of host RLR pathway by virus; Ion channel; Ion transport; Lipoprotein; Magnesium; Membrane; Metal-binding; Myristate; Nucleotide-binding; Nucleotidyltransferase; Phosphoprotein; Pore-mediated penetration of viral genome into host cell; Protease; Repeat; RNA-binding; RNA-directed RNA polymerase; T=pseudo3 icosahedral capsid protein; Thiol protease; Transferase; Transport; Viral attachment to host cell; Viral immunoevasion; Viral ion channel; Viral penetration into host cytoplasm; Viral RNA replication; Virion; Virus endocytosis by host; Virus entry into host cell; Zinc; Zinc-finger Protein physicochemical properties Chain ID 1 Molecular weight (Da) 30610.1 Length 269 Aromaticity 0.1 Instability index 55.66 Isoelectric point 6.86 Charge (pH=7) -0.38 2D Binding mode Binding energy (Kcal/mol) -9.41  Molscript Map 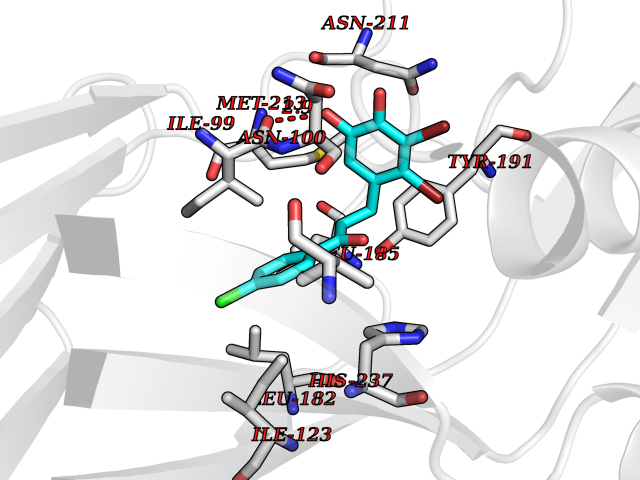 Pymol Map 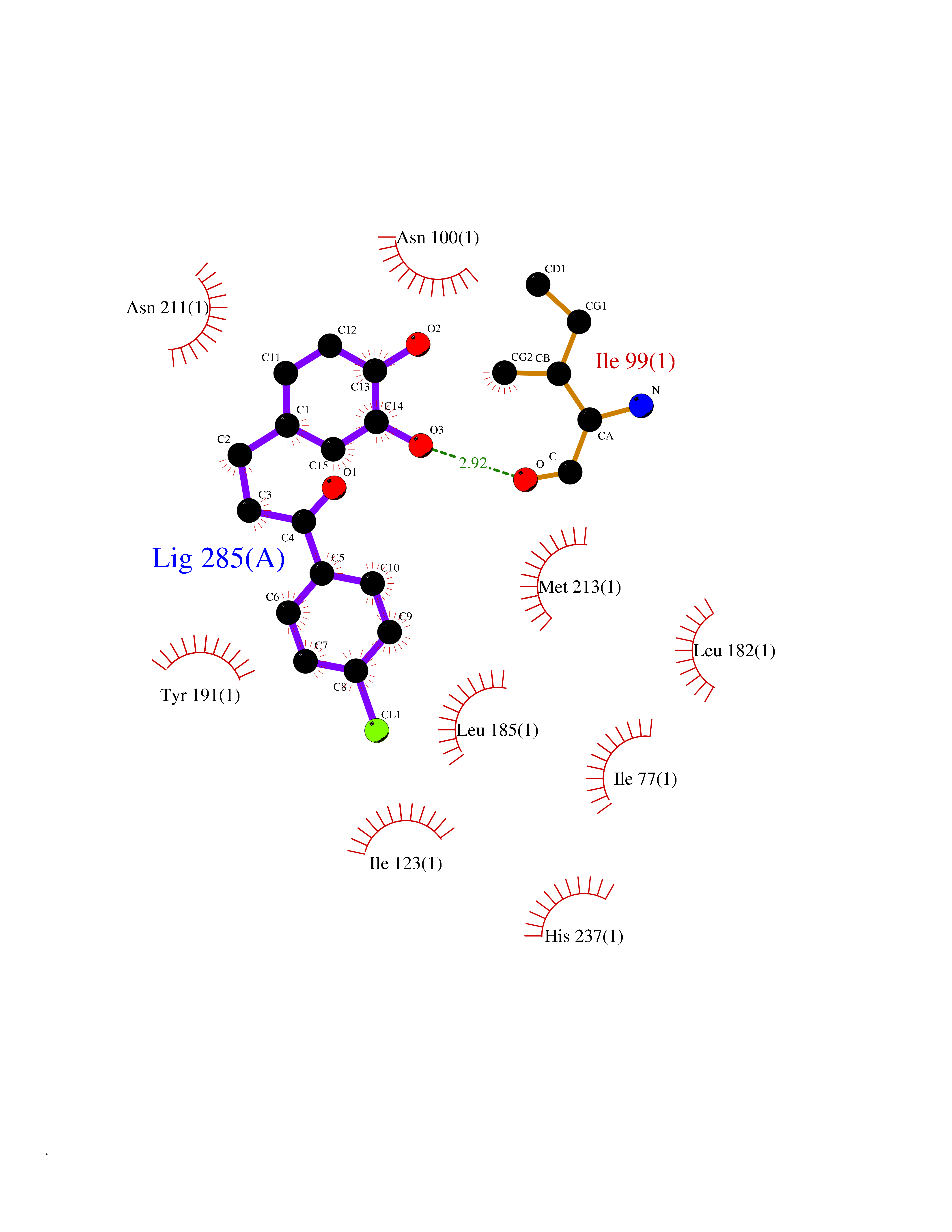 Ligplot Map 3D Binding mode Sequence LVVPNINSSNPTTSNSAPALDAAETGHTSSVQPEDVIETRYVQTSQTRDEMSLESFLGRSGCIHESKLEVTLANYNKENFTVWAINLQEMAQIRRKFELFTYTRFDSEITLVPCISALSQDIGHITMQYMYVPPGAPVPNSRDDYAWQSGTNASVFWQHGQAYPRFSLPFLSVASAYYMFYDGYDEQDQNYGTANTNNMGSLCSRIVTEKHIHKVHIMTRIYHKAKHVKAWCPRPPRALEYTRAHRTNFKIEDRSIQTAIVTRPIITTA Hydrogen bonds contact Hydrophobic contact | ||||
| 92 | Protein arginine methyltransferase 5 (PRMT5) | 7MXC | 6.90 | |
Target general information Gen name PRMT5 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Shk1 kinase-binding protein 1 homolog; SKB1Hs; SKB1 homolog; SKB1; Protein arginine N-methyltransferase 5; Jak-binding protein 1; JBP1; IBP72; Histone-arginine N-methyltransferase PRMT5; HRMT1L5; 72 k Protein family Class I-like SAM-binding methyltransferase superfamily, Protein arginine N-methyltransferase family Biochemical class Methyltransferase Function Specifically mediates the symmetrical dimethylation of arginine residues in the small nuclear ribonucleoproteins Sm D1 (SNRPD1) and Sm D3 (SNRPD3); such methylation being required for the assembly and biogenesis of snRNP core particles. Methylates SUPT5H and may regulate its transcriptional elongation properties. Mono- and dimethylates arginine residues of myelin basic protein (MBP) in vitro. May play a role in cytokine-activated transduction pathways. Negatively regulates cyclin E1 promoter activity and cellular proliferation. Methylates histone H2A and H4 'Arg-3' during germ cell development. Methylates histone H3 'Arg-8', which may repress transcription. Methylates the Piwi proteins (PIWIL1, PIWIL2 and PIWIL4), methylation of Piwi proteins being required for the interaction with Tudor domain-containing proteins and subsequent localization to the meiotic nuage. Methylates RPS10. Attenuates EGF signaling through the MAPK1/MAPK3 pathway acting at 2 levels. First, monomethylates EGFR; this enhances EGFR 'Tyr-1197' phosphorylation and PTPN6 recruitment, eventually leading to reduced SOS1 phosphorylation. Second, methylates RAF1 and probably BRAF, hence destabilizing these 2 signaling proteins and reducing their catalytic activity. Required for induction of E-selectin and VCAM-1, on the endothelial cells surface at sites of inflammation. Methylates HOXA9. Methylates and regulates SRGAP2 which is involved in cell migration and differentiation. Acts as a transcriptional corepressor in CRY1-mediated repression of the core circadian component PER1 by regulating the H4R3 dimethylation at the PER1 promoter. Methylates GM130/GOLGA2, regulating Golgi ribbon formation. Methylates H4R3 in genes involved in glioblastomagenesis in a CHTOP- and/or TET1-dependent manner. Symmetrically methylates POLR2A, a modification that allows the recruitment to POLR2A of proteins including SMN1/SMN2 and SETX. This is required for resolving RNA-DNA hybrids created by RNA polymerase II, that form R-loop in transcription terminal regions, an important step in proper transcription termination. Along with LYAR, binds the promoter of gamma-globin HBG1/HBG2 and represses its expression. Symmetrically methylates NCL. Methylates TP53; methylation might possibly affect TP53 target gene specificity. Arginine methyltransferase that can both catalyze the formation of omega-N monomethylarginine (MMA) and symmetrical dimethylarginine (sDMA), with a preference for the formation of MMA. Related diseases Epilepsy, nocturnal frontal lobe, 3 (ENFL3) [MIM:605375]: An autosomal dominant focal epilepsy characterized by nocturnal seizures with hyperkinetic automatisms and poorly organized stereotyped movements. {ECO:0000269|PubMed:11062464, ECO:0000269|PubMed:11104662}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P01019; Q9NX04; Q8WUW1; Q08289; P78371; Q16543; Q8N8U2; P54105; P21964-2; Q9NQ92; Q16526; Q9Y6K1; Q01094; Q08426; P38919; Q14241; O15197-2; Q6ZV65; P01100; O95995; P62993; Q8TE85; Q9BX10; P62805; P31269; Q00613; Q63ZY3; P03952; Q8TBB1; P06858; Q86UQ8-1; Q96HA8; Q8WVJ2; P24928; O14744; Q86U06; Q9BRS2; O75044; Q96RU7; P31930; P40337-2; Q9BQA1; P63104; Q96E35; Q91XC0; P03418; Q6ZV65-2 EC number EC 2.1.1.320 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Biological rhythms; Chromatin regulator; Chromosome; Cytoplasm; Direct protein sequencing; Golgi apparatus; Methyltransferase; Nucleus; Proteomics identification; Reference proteome; Repressor; S-adenosyl-L-methionine; Transcription; Transcription regulation; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 71188.3 Length 621 Aromaticity 0.11 Instability index 44.6 Isoelectric point 5.95 Charge (pH=7) -9.68 2D Binding mode Binding energy (Kcal/mol) -9.41 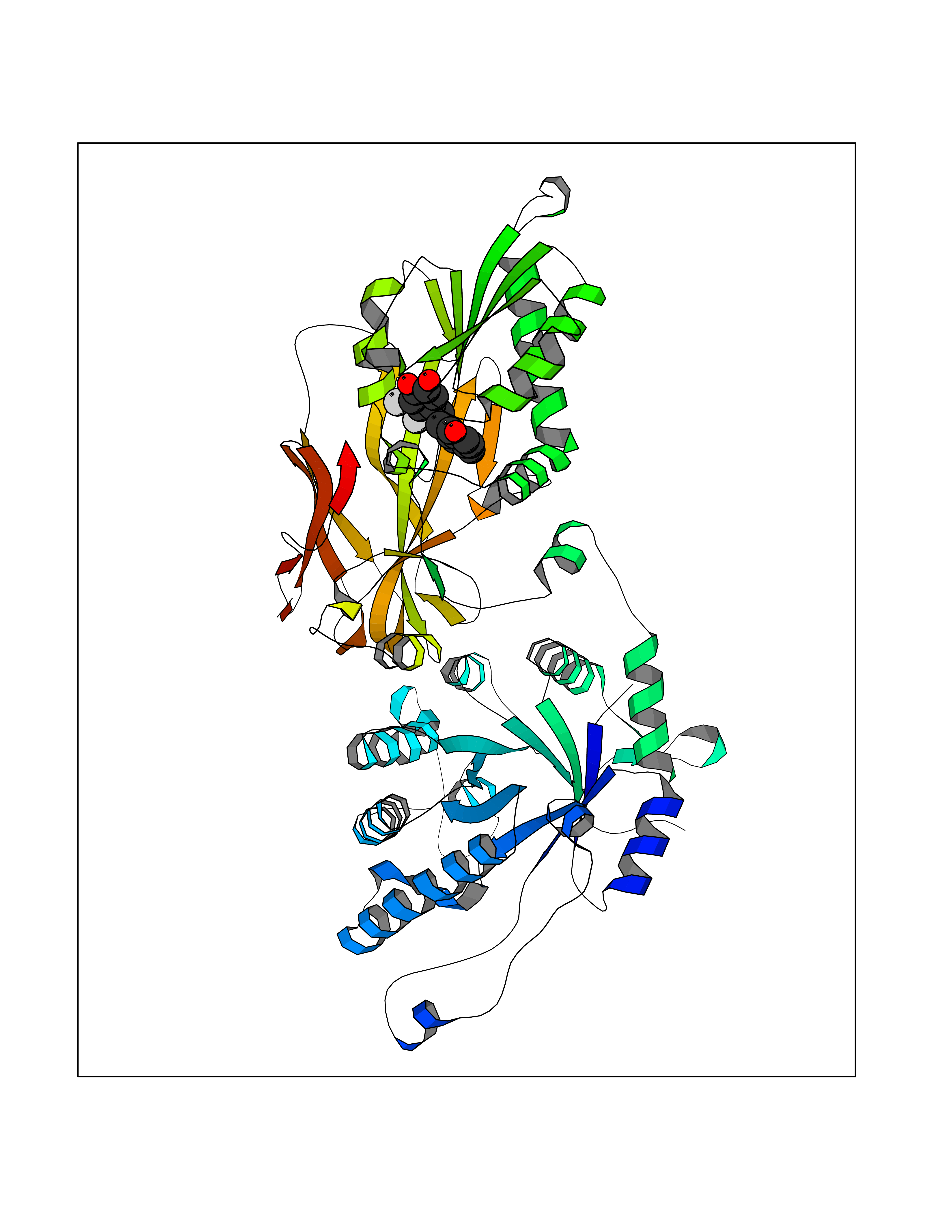 Molscript Map 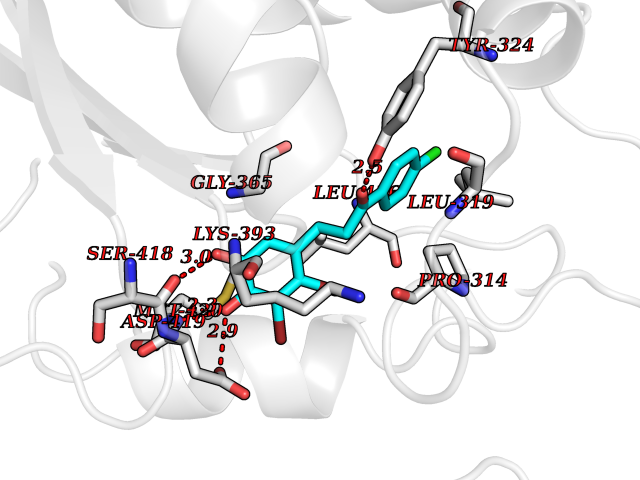 Pymol Map 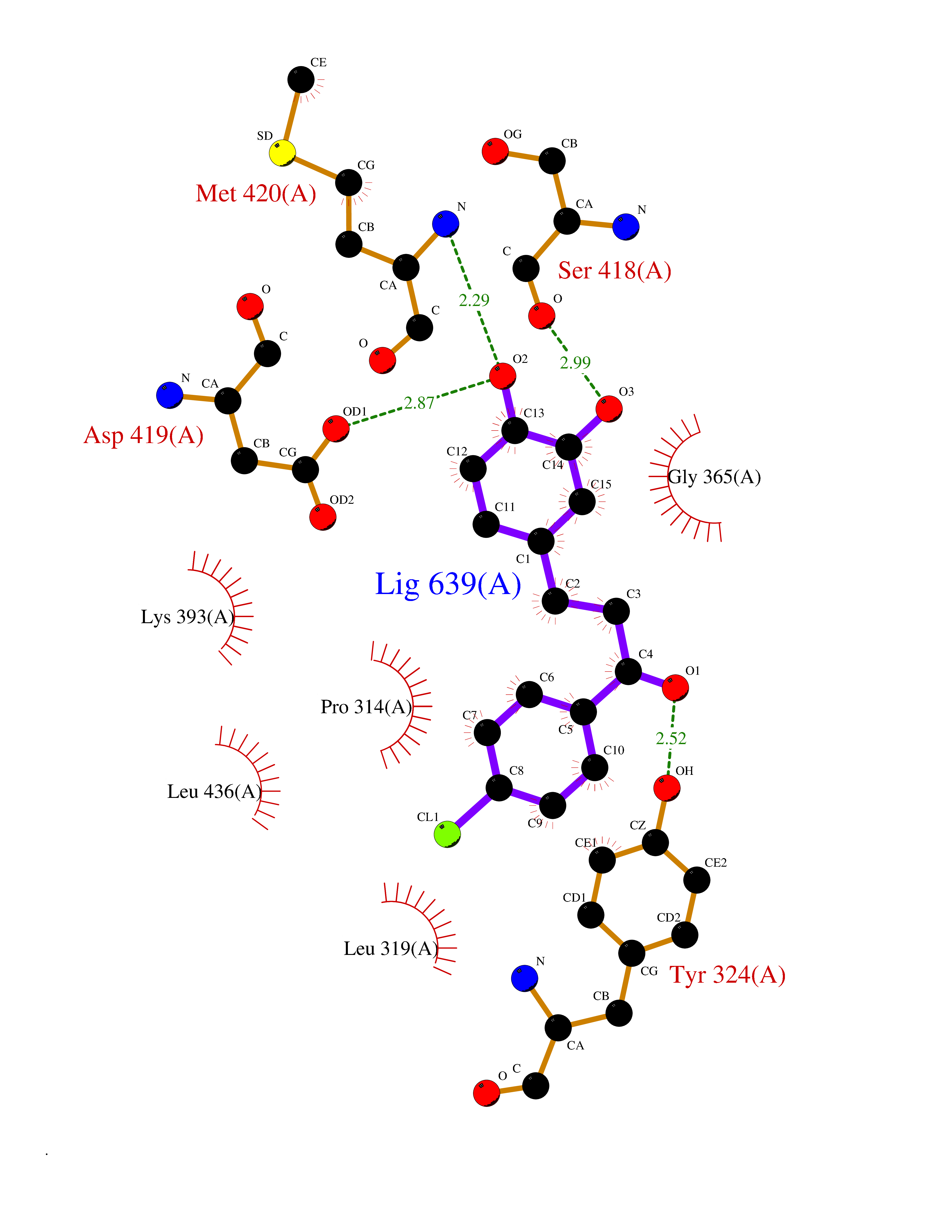 Ligplot Map 3D Binding mode Sequence RVSSGRDLNCVPEIADTLGAVAKQGFDFLCMPVFHPRFKREFIQEPAKNRPGPQTRSDLLLSGRDWNTLIVGKLSPWIRPDSKVEKIRRNSEAAMLQELNFGAYLGLPAFLLPLNQEDNTNLARVLTNHIHTGHHSSMFWMRVPLVAPEDLRDDIIENAPTTHTEEYSGEEKTWMWWHNFRTLCDYSKRIAVALEIGADLPSNHVIDRWLGEPIKAAILPTSIFLTNKKGFPVLSKMHQRLIFRLLKLEVQFIITGTNHHSCSYLQYLEYLSQNRPPPNAYELFAKGYEDYLQSPLQPLMDNLESQTYEVFEKDPIKYSQYQQAIYKCLLDRVPEEEKDTNVQVLMVLGAGRGPLVNASLRAAKQADRRIKLYAVEKNPNAVVTLENWQFEEWGSQVTVVSSDMREWVAPEKADIIVSELLGSFADNELSPECLDGAQHFLKDDGVSIPGEYTSFLAPISSSKLYNEVRACREKDRDPEAQFEMPYVVRLHNFHQLSAPQPCFTFSHPNRDPMIDNNRYCTLEFPVEVNTVLHGFAGYFETVLYQDITLSIRPETHSPGMFSWFPILFPIKQPITVREGQTICVRFWRCSNSKKVWYEWAVTAPVCSAIHNPTGRSYTIGL Hydrogen bonds contact Hydrophobic contact | ||||
| 93 | Mineralocorticoid receptor (MR) | 4PF3 | 6.89 | |
Target general information Gen name NR3C2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Nuclear receptor subfamily 3 group C member 2; Mineralocorticoid receptor; MLR; MCR; Inner ear mineralocorticoid receptor; Delta Protein family Nuclear hormone receptor family, NR3 subfamily Biochemical class Nuclear hormone receptor Function Binds to mineralocorticoid response elements (MRE) and transactivates target genes. The effect of MC is to increase ion and water transport and thus raise extracellular fluid volume and blood pressure and lower potassium levels. Receptor for both mineralocorticoids (MC) such as aldosterone and glucocorticoids (GC) such as corticosterone or cortisol. Related diseases Pseudohypoaldosteronism 1, autosomal dominant (PHA1A) [MIM:177735]: A salt wasting disease resulting from target organ unresponsiveness to mineralocorticoids. PHA1A is a mild form characterized by target organ defects confined to kidney. Patients may present with neonatal renal salt wasting with hyperkalaemic acidosis despite high aldosterone levels. These patients improve with age and usually become asymptomatic without treatment. {ECO:0000269|PubMed:11134129, ECO:0000269|PubMed:12788847, ECO:0000269|PubMed:16954160, ECO:0000269|PubMed:16972228}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Early-onset hypertension with severe exacerbation in pregnancy (EOHSEP) [MIM:605115]: Inheritance is autosomal dominant. The disease is characterized by the onset of severe hypertension before the age of 20, and by suppression of aldosterone secretion. {ECO:0000269|PubMed:10884226, ECO:0000269|PubMed:15908963, ECO:0000269|PubMed:15967794}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB04630; DB01013; DB04652; DB06780; DB01134; DB01395; DB00700; DB01023; DB16165; DB00687; DB13867; DB08906; DB00588; DB02998; DB00393; DB00396; DB00421; DB02901; DB13951; DB00624; DB13943; DB13944; DB15114 Interacts with NA EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; Disease variant; DNA-binding; Endoplasmic reticulum; Lipid-binding; Membrane; Metal-binding; Nucleus; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Steroid-binding; Transcription; Transcription regulation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A Molecular weight (Da) 29012.4 Length 249 Aromaticity 0.12 Instability index 51.27 Isoelectric point 6.3 Charge (pH=7) -2.08 2D Binding mode Binding energy (Kcal/mol) -9.4 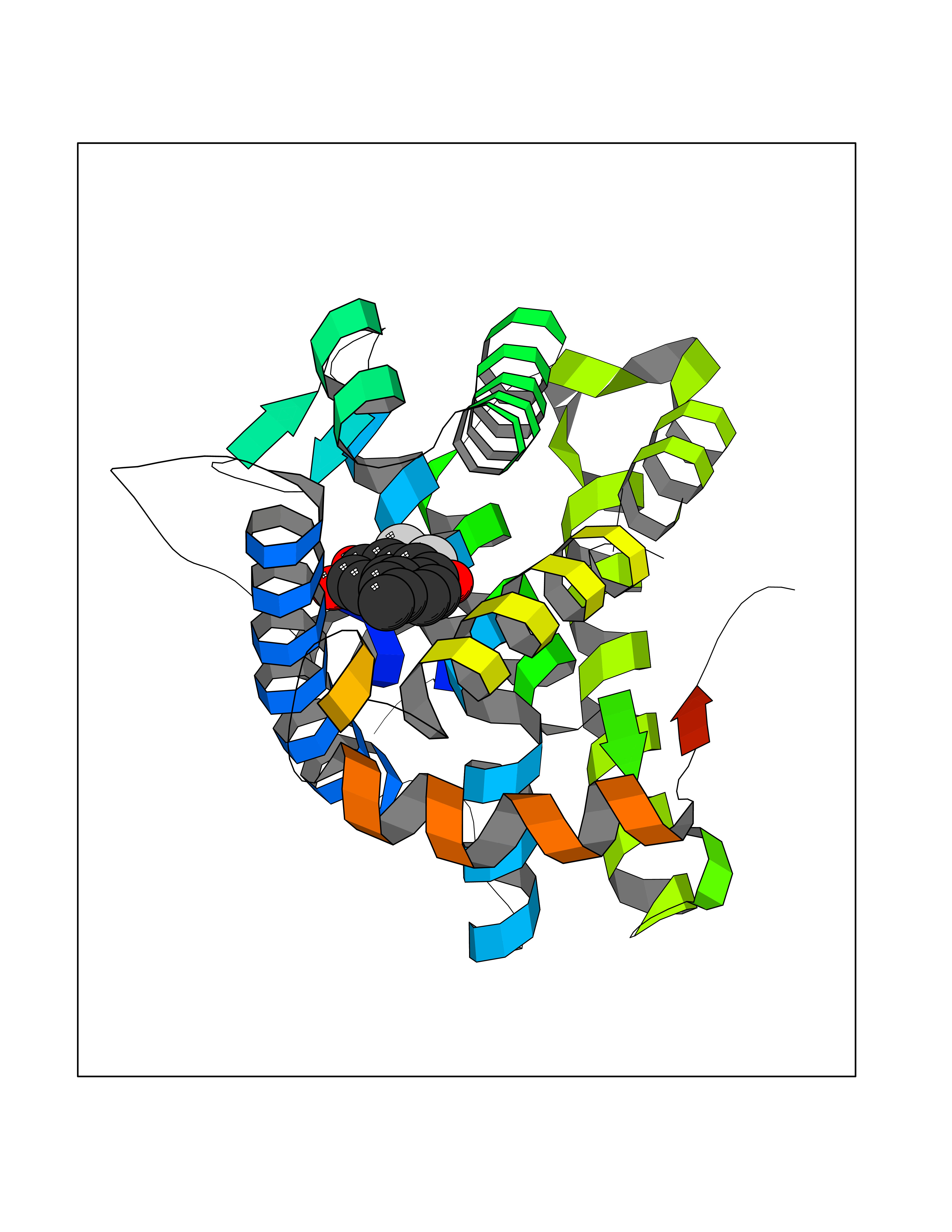 Molscript Map 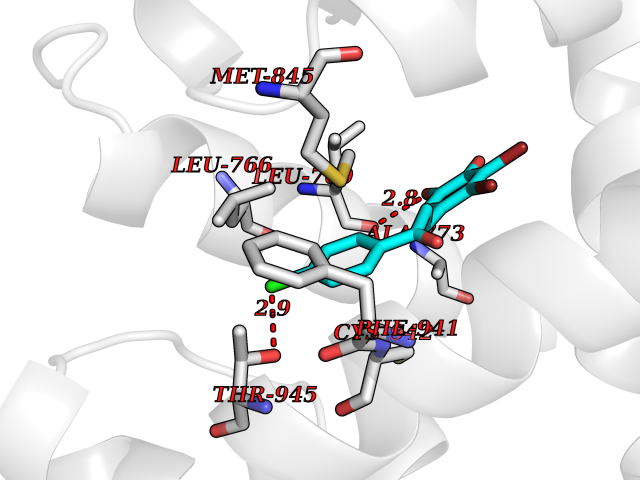 Pymol Map 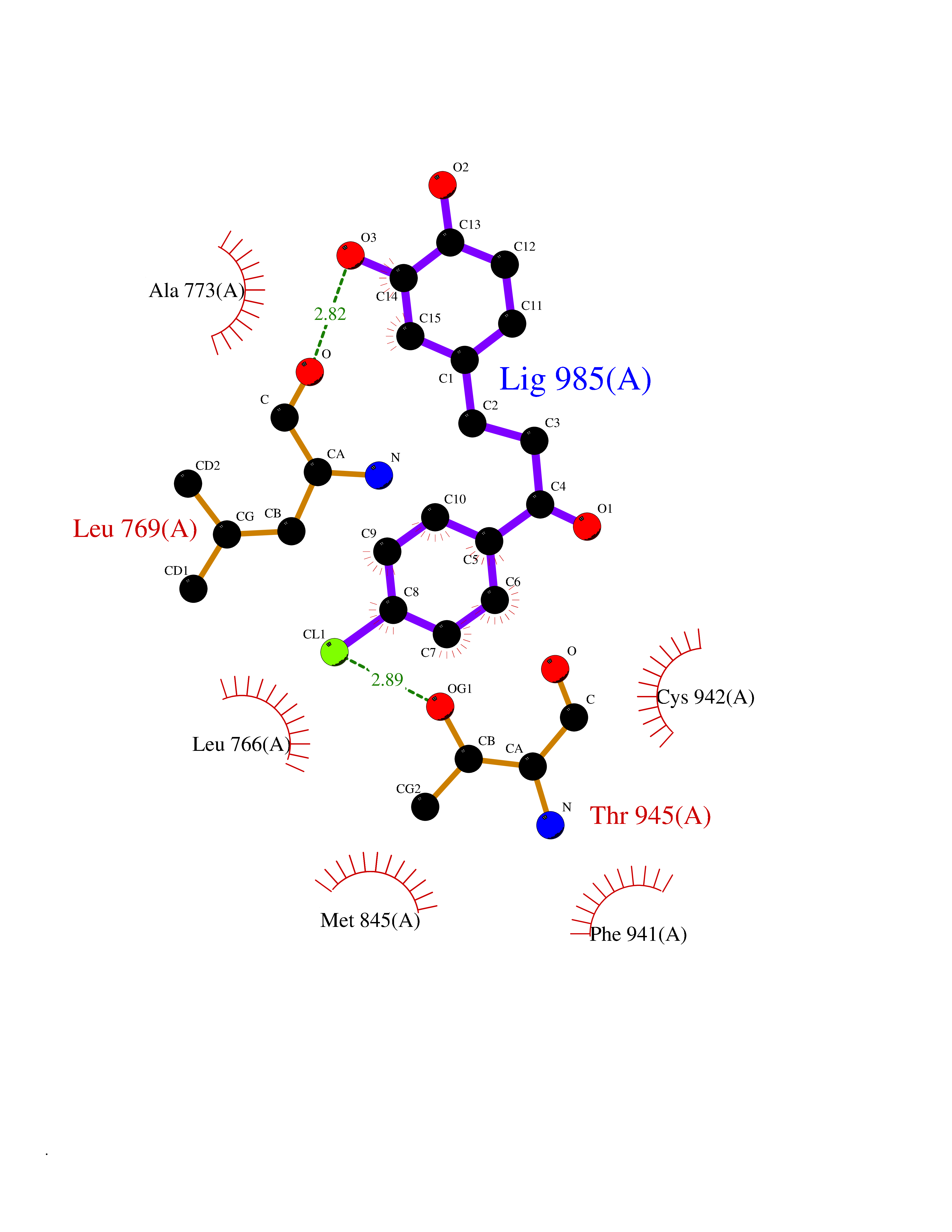 Ligplot Map 3D Binding mode Sequence TPSPVMVLENIEPEIVYAGYDSSKPDTAENLLSTLNRLAGKQMIQVVKWAKVLPGFKNLPLEDQITLIQYSWMSLLSFALSWRSYKHTNSQFLYFAPDLVFNEEKMHQSAMYELCQGMHQISLQFVRLQLTFEEYTIMKVLLLLSTIPKDGLKSQAAFEEMRTNYIKELRKMVTKCPNNSGQSWQRFYQLTKLLDSMHDLVSDLLEFCFYTFRESHALKVEFPAMLVEIISDQLPKVESGNVKPLYFHR Hydrogen bonds contact Hydrophobic contact | ||||
| 94 | Progesterone receptor (PGR) | 1SQN | 6.89 | |
Target general information Gen name PGR Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms PR; Nuclear receptor subfamily 3 group C member 3; NR3C3 Protein family Nuclear hormone receptor family, NR3 subfamily Biochemical class Nuclear hormone receptor Function Depending on the isoform, progesterone receptor functions as transcriptional activator or repressor. The steroid hormones and their receptors are involved in the regulation of eukaryotic gene expression and affect cellular proliferation and differentiation in target tissues. Related diseases Butyrylcholinesterase deficiency (BCHED) [MIM:617936]: An autosomal recessive metabolic condition characterized by increased sensitivity to certain anesthetic drugs, including the muscle relaxants succinylcholine or mivacurium. BCHED results in slower hydrolysis of these drugs and, consequently, a prolonged neuromuscular block, leading to apnea. The duration of the prolonged apnea varies significantly depending on the extent of the enzyme deficiency. {ECO:0000269|PubMed:10404729, ECO:0000269|PubMed:11928765, ECO:0000269|PubMed:12881446, ECO:0000269|PubMed:1306123, ECO:0000269|PubMed:1349196, ECO:0000269|PubMed:1415224, ECO:0000269|PubMed:15563885, ECO:0000269|PubMed:15781196, ECO:0000269|PubMed:1611188, ECO:0000269|PubMed:16788378, ECO:0000269|PubMed:17700357, ECO:0000269|PubMed:18075469, ECO:0000269|PubMed:18300943, ECO:0000269|PubMed:25054547, ECO:0000269|PubMed:25264279, ECO:0000269|PubMed:2915989, ECO:0000269|PubMed:7634491, ECO:0000269|PubMed:8554068, ECO:0000269|PubMed:8680411, ECO:0000269|PubMed:9110359, ECO:0000269|PubMed:9191541, ECO:0000269|PubMed:9388484, ECO:0000269|PubMed:9543549, ECO:0000269|PubMed:9694584}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01431; DB06680; DB01406; DB12941; DB13857; DB00304; DB09123; DB01395; DB00378; DB11219; DB00823; DB00294; DB13867; DB08906; DB00588; DB06730; DB11619; DB11064; DB06789; DB00367; DB00431; DB09124; DB00603; DB00351; DB02998; DB00834; DB00648; DB00764; DB14512; DB06713; DB00717; DB00957; DB09389; DB01428; DB02746; DB00396; DB14583; DB00421; DB04787; DB05253; DB08867 Interacts with Q9H467; P03372; P06401; P40763; P03372 EC number NA Uniprot keywords 3D-structure; Alternative promoter usage; Alternative splicing; Cytoplasm; Direct protein sequencing; DNA-binding; Isopeptide bond; Lipid-binding; Lipoprotein; Membrane; Metal-binding; Mitochondrion; Mitochondrion outer membrane; Nucleus; Palmitate; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Steroid-binding; Transcription; Transcription regulation; Ubl conjugation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A,B Molecular weight (Da) 28853.6 Length 250 Aromaticity 0.09 Instability index 54.82 Isoelectric point 8.4 Charge (pH=7) 2.28 2D Binding mode Binding energy (Kcal/mol) -9.39  Molscript Map 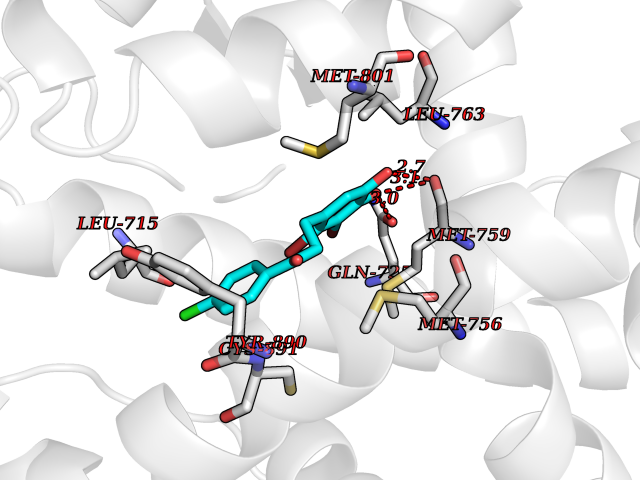 Pymol Map 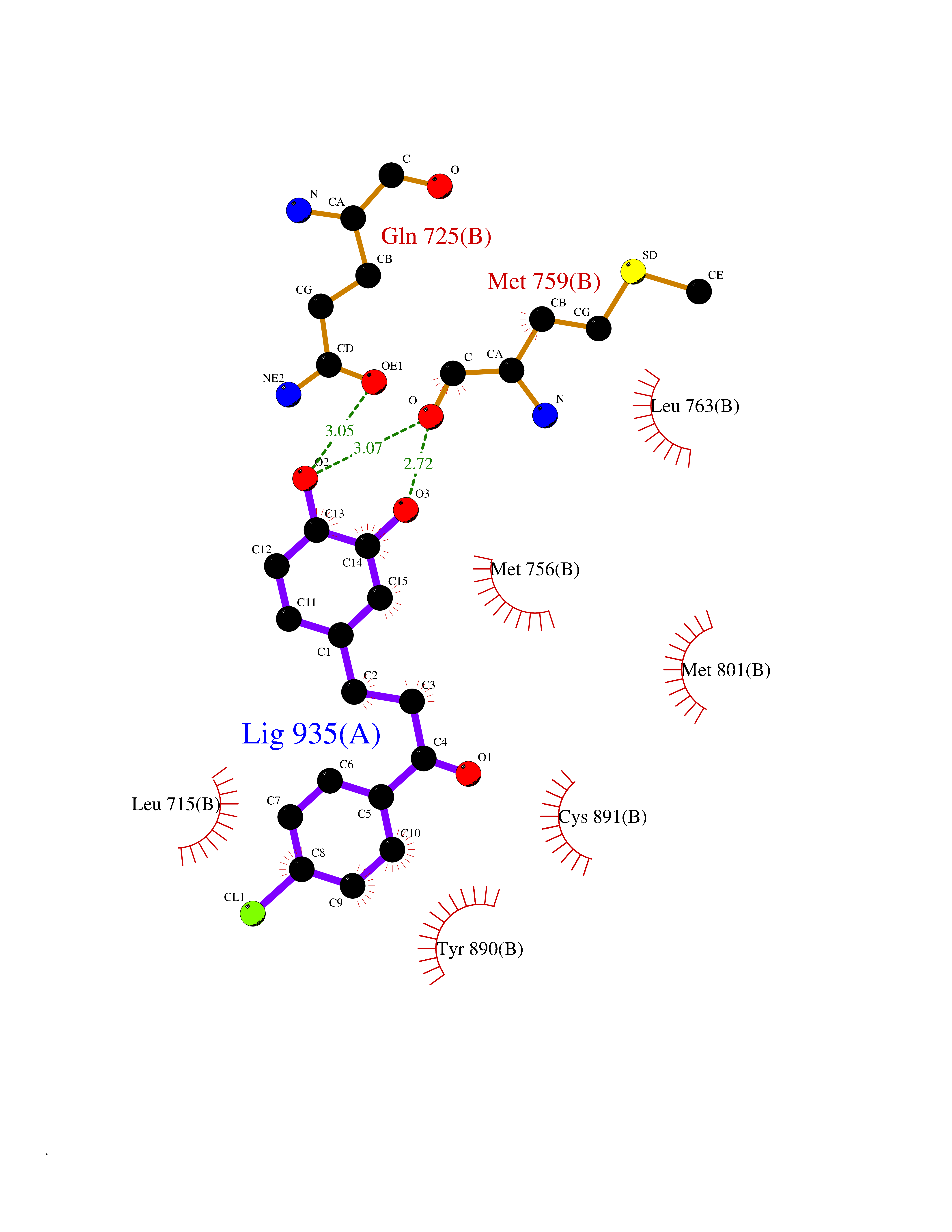 Ligplot Map 3D Binding mode Sequence LIPPLINLLMSIEPDVIYAGHDNTKPDTSSSLLTSLNQLGERQLLSVVKWSKSLPGFRNLHIDDQITLIQYSWMSLMVFGLGWRSYKHVSGQMLYFAPDLILNEQRMKESSFYSLCLTMWQIPQEFVKLQVSQEEFLCMKVLLLLNTIPLEGLRSQTQFEEMRSSYIRELIKAIGLRQGVVSSSQRFYQLTKLLDNLHDLVKQLHLYCLNTFIQSRALSVEFPEMMSEVIAAQLPKILAGMVKPLLFHKK Hydrogen bonds contact Hydrophobic contact | ||||
| 95 | Aldehyde dehydrogenase family 1 member A3 | 5FHZ | 6.89 | |
Target general information Gen name ALDH1A3 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms ALDH6 Protein family Aldehyde dehydrogenase family Biochemical class Oxidoreductase Function Aldehyde dehydrogenase (NAD) activity.Aldehyde dehydrogenase [NAD(P)+] activity.NAD+ binding.Protein homodimerization activity.Retinal dehydrogenase activity.Thyroid hormone binding. Related diseases Microphthalmia, isolated, 8 (MCOP8) [MIM:615113]: A disorder of eye formation, ranging from small size of a single eye to complete bilateral absence of ocular tissues. Ocular abnormalities like opacities of the cornea and lens, scaring of the retina and choroid, and other abnormalities may also be present. {ECO:0000269|PubMed:23312594, ECO:0000269|PubMed:23591992, ECO:0000269|PubMed:23646827, ECO:0000269|PubMed:23881059, ECO:0000269|PubMed:24024553, ECO:0000269|PubMed:24568872, ECO:0000269|PubMed:24777706}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00157; DB00162 Interacts with NA EC number 1.2.1.36 Uniprot keywords 3D-structure; Acetylation; Cytoplasm; Direct protein sequencing; Disease variant; Lipid metabolism; Microphthalmia; NAD; Oxidoreductase; Proteomics identification; Reference proteome Protein physicochemical properties Chain ID A,B,C,D,E,F,G,H Molecular weight (Da) 50635.7 Length 461 Aromaticity 0.09 Instability index 33.75 Isoelectric point 7.07 Charge (pH=7) 0.15 2D Binding mode Binding energy (Kcal/mol) -9.4  Molscript Map 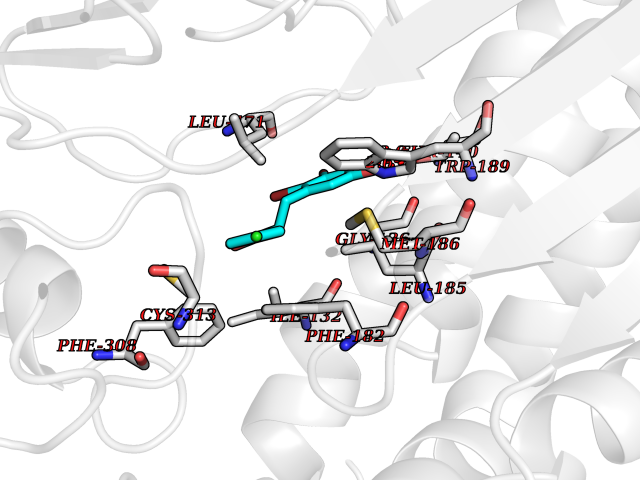 Pymol Map 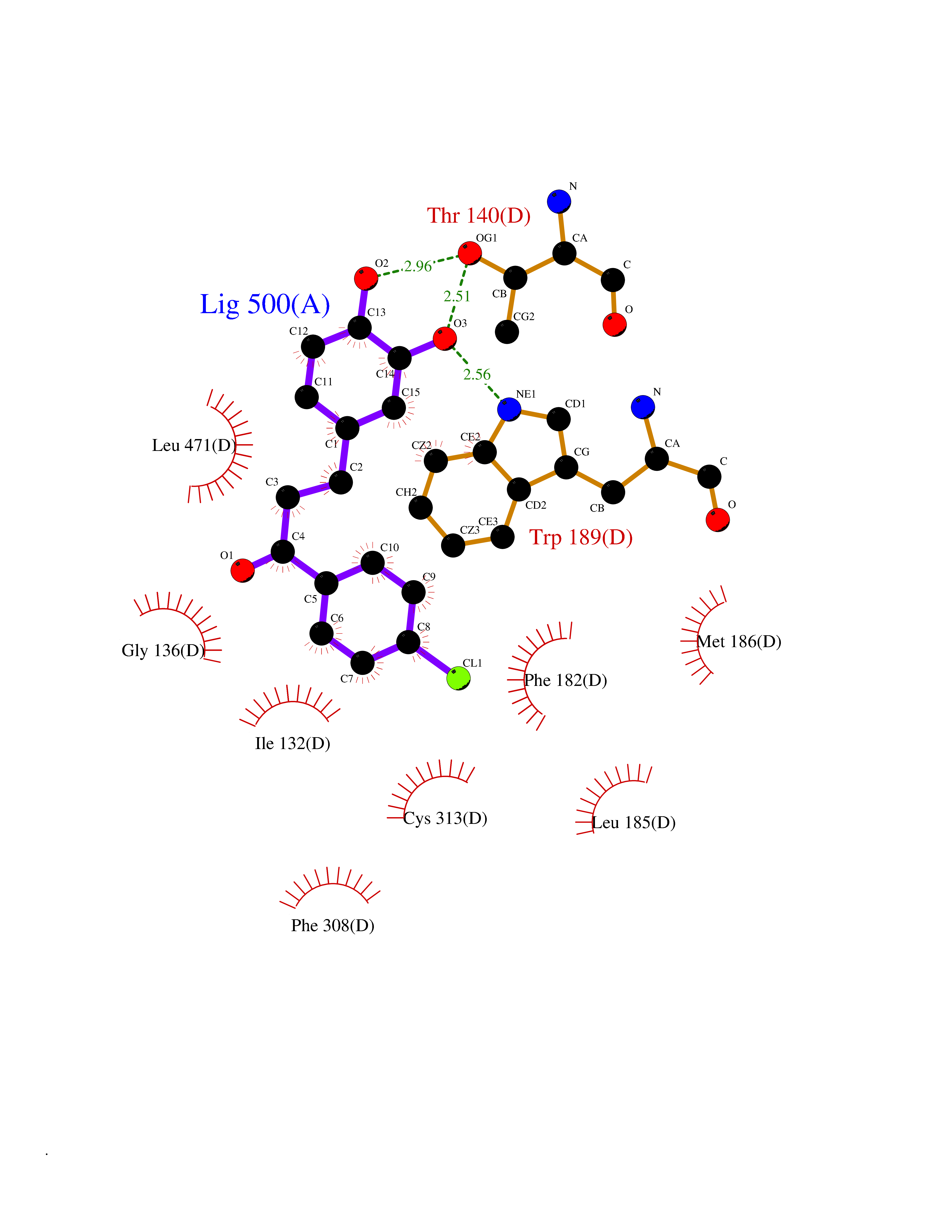 Ligplot Map 3D Binding mode Sequence LPRPIRNLEVKFTKIFINNEWHESKSGKKFATCNPSTREQICEVEEGDKPDVDKAVEAAQVAFQRGSPWRRLDALSRGRLLHQLADLVERDRATLAALETMDTGKPFLHAFFIDLEGCIRTLRYFAGWADKIPIGVCGAITPWNFPLLMLVWKLAPALCCGNTMVLKPAEQTPLTALYLGSLIKEAGFPPGVVNIVPGFGPTVGAAISSHPQINKIAFTGSTEVGKLVKEAASRSNLKRVTLELGGKNPCIVCADADLDLAVECAHQGVFFNQGQCCTAASRVFVEEQVYSEFVRRSVEYAKKRPVGDPFDVKTEQGPQIDQKQFDKILELIESGKKEGAKLECGGSAMEDKGLFIKPTVFSEVTDNMRIAKEEIFGPVQPILKFKSIEEVIKRANSTDYGLTAAVFTKNLDKALKLASALESGTVWINCYNALYAQAPFGGFKMSGNGRELGEYALAEYT Hydrogen bonds contact Hydrophobic contact | ||||
| 96 | 2-hydroxy-6-oxo-7-methylocta-2,4-dienoate hydrolase | 1UK8 | 6.89 | |
Target general information Gen name cumD Organism Pseudomonas fluorescens Uniprot ID TTD ID NA Synonyms NA Protein family NA Biochemical class Hydrolase Function Hydrolase activity. Related diseases Intellectual developmental disorder, autosomal dominant 62 (MRD62) [MIM:618793]: An autosomal dominant form of intellectual disability, a disorder characterized by significantly below average general intellectual functioning associated with impairments in adaptive behavior and manifested during the developmental period. MRD62 is characterized by mild to moderately impaired intellectual development. {ECO:0000269|PubMed:27479843, ECO:0000269|PubMed:29460436}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03741; DB03793; DB03568; DB02531; DB03750; DB02406; DB03766 Interacts with NA EC number NA Uniprot keywords 3D-structure; Hydrolase Protein physicochemical properties Chain ID A Molecular weight (Da) 30307.9 Length 271 Aromaticity 0.1 Instability index 37.49 Isoelectric point 5.02 Charge (pH=7) -11.58 2D Binding mode Binding energy (Kcal/mol) -9.4 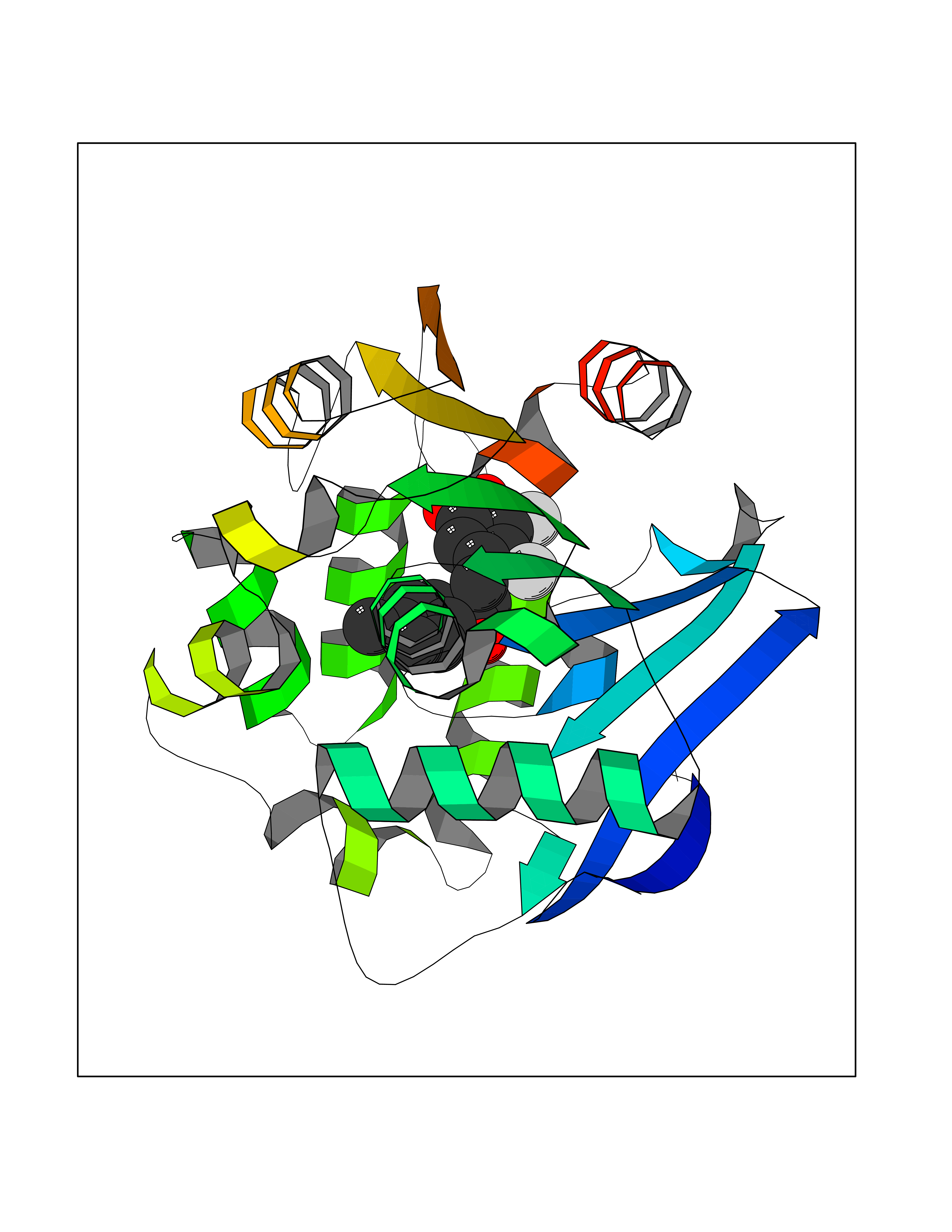 Molscript Map 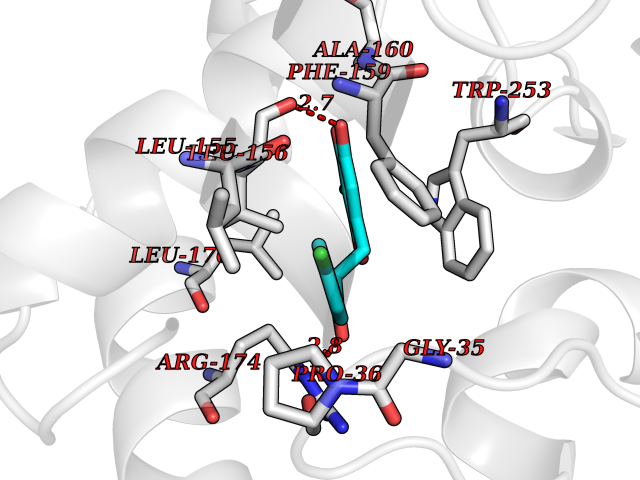 Pymol Map 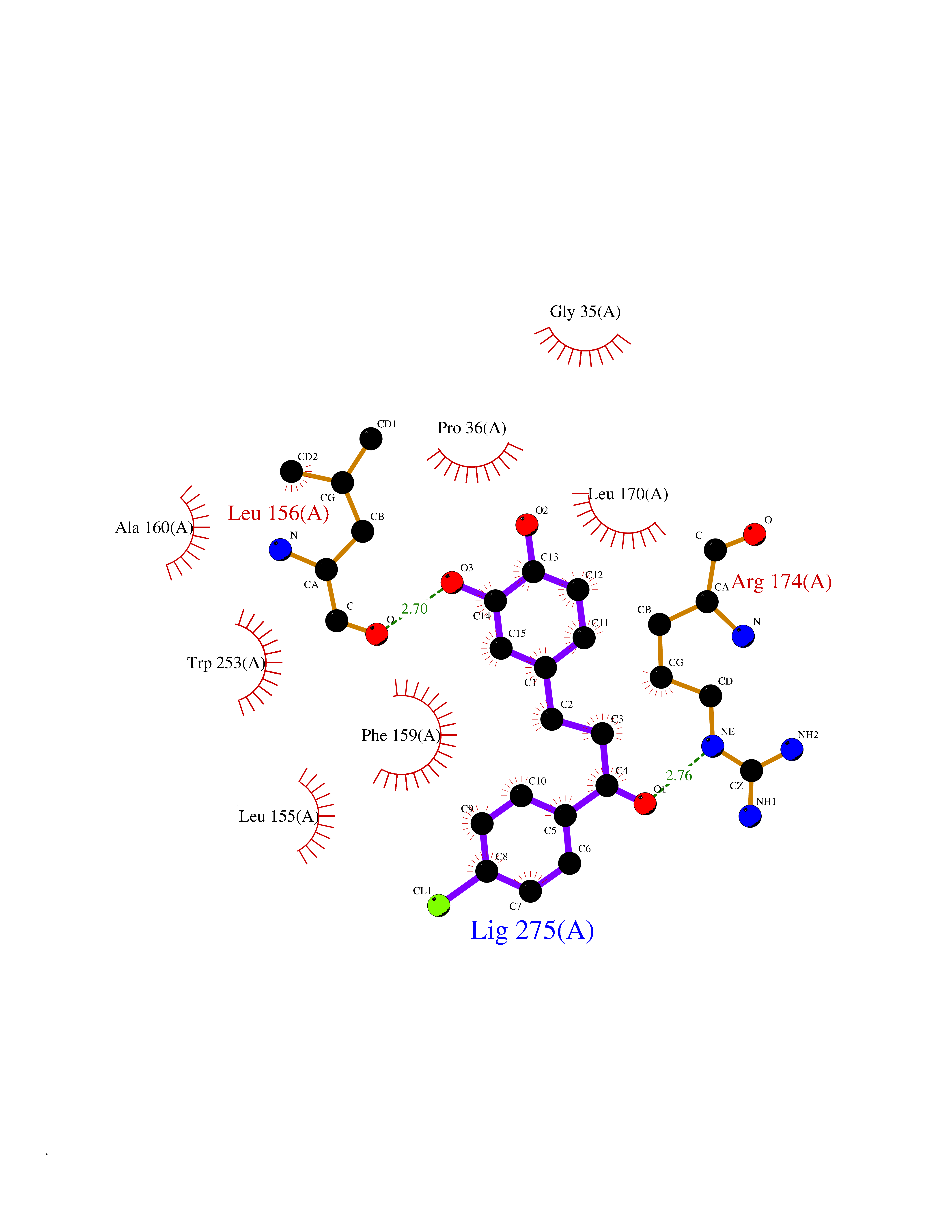 Ligplot Map 3D Binding mode Sequence NLEIGKSILAAGVLTNYHDVGEGQPVILIHGSGPGVSAYANWRLTIPALSKFYRVIAPDMVGFGFTDRPENYNYSKDSWVDHIIGIMDALEIEKAHIVGNAFGGGLAIATALRYSERVDRMVLMGAAGTRFDVTEGLNAVWGYTPSIENMRNLLDIFAYDRSLVTDELARLRYEASIQPGFQESFSSMFPEPRQRWIDALASSDEDIKTLPNETLIIHGREDQVVPLSSSLRLGELIDRAQLHVFGRCGHWTQIEQTDRFNRLVVEFFNEA Hydrogen bonds contact Hydrophobic contact | ||||
| 97 | Dihydroorotate dehydrogenase (DHODH) | 4OQV | 6.89 | |
Target general information Gen name DHODH Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Dihydroorotate oxidase; Dihydroorotate dehydrogenase (quinone), mitochondrial; DHOdehase; DHODH Protein family Dihydroorotate dehydrogenase family, Type 2 subfamily Biochemical class CH-CH donor oxidoreductase Function Catalyzes the conversion of dihydroorotate to orotate with quinone as electron acceptor. Related diseases Postaxial acrofacial dysostosis (POADS) [MIM:263750]: POADS is characterized by severe micrognathia, cleft lip and/or palate, hypoplasia or aplasia of the posterior elements of the limbs, coloboma of the eyelids and supernumerary nipples. POADS is a very rare disorder: only 2 multiplex families, each consisting of 2 affected siblings born to unaffected, nonconsanguineous parents, have been described among a total of around 30 reported cases. {ECO:0000269|PubMed:19915526}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07559; DB07561; DB08172; DB08169; DB07443; DB07978; DB07975; DB04281; DB08249; DB07977; DB07976; DB04583; DB08008; DB01117; DB03523; DB03480; DB02613; DB04147; DB03247; DB01097; DB06481; DB08006; DB02262; DB05125; DB08880; DB07646 Interacts with Q6ZMZ0; P49638 EC number EC 1.3.5.2 Uniprot keywords 3D-structure; Disease variant; Flavoprotein; FMN; Membrane; Mitochondrion; Mitochondrion inner membrane; Oxidoreductase; Proteomics identification; Pyrimidine biosynthesis; Reference proteome; Transit peptide; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 38341.4 Length 353 Aromaticity 0.05 Instability index 39.27 Isoelectric point 9.28 Charge (pH=7) 5.52 2D Binding mode Binding energy (Kcal/mol) -9.4  Molscript Map 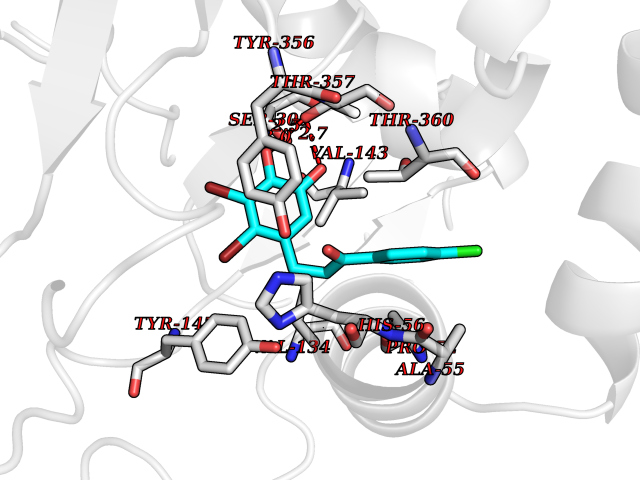 Pymol Map 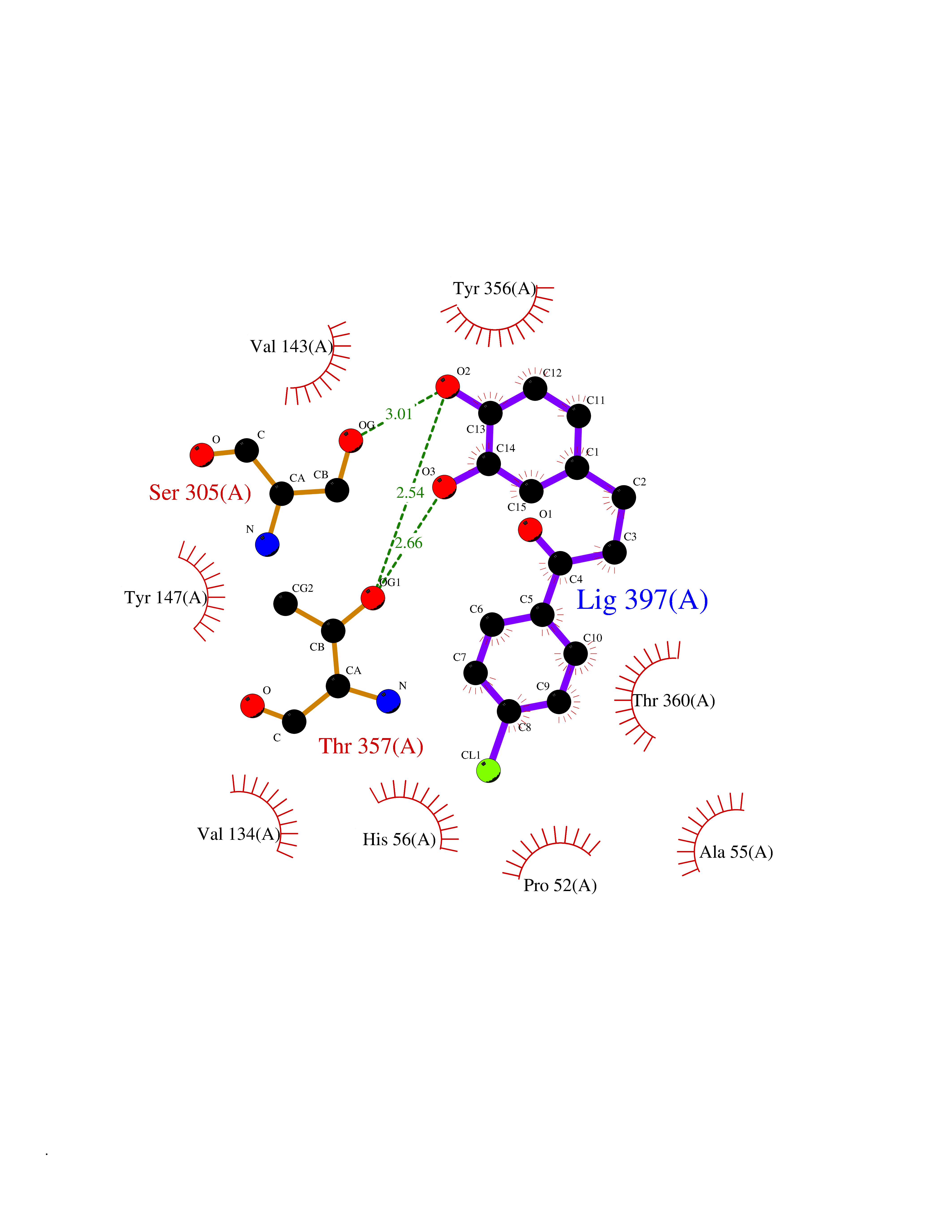 Ligplot Map 3D Binding mode Sequence DERFYAEHLMPTLQGLLDPESAHRLAVRFTSLGLLPRARFQDSDMLEVRVLGHKFRNPVGIAAGFDKHGEAVDGLYKMGFGFVEIGSVTPKPQEGNPRPRVFRLPEDQAVINRYGFNSHGLSVVEHRLRARQQKQAKLTEDGLPLGVNLGKNKTSVDAAEDYAEGVRVLGPLADYLVVNVSSPGKAELRRLLTKVLQERDGLRRVHRPAVLVKIAPDLTSQDKEDIASVVKELGIDGLIVTNTTVSRPAGLQGALRSETGGLSGKPLRDLSTQTIREMYALTQGRVPIIGVGGVSSGQDALEKIRAGASLVQLYTALTFWGPPVVGKVKRELEALLKEQGFGGVTDAIGADHR Hydrogen bonds contact Hydrophobic contact | ||||
| 98 | Aldehyde oxidoreductase | 4USA | 6.89 | |
Target general information Gen name mop Organism Megalodesulfovibrio gigas (Desulfovibrio gigas) Uniprot ID TTD ID NA Synonyms NA Protein family Xanthine dehydrogenase family Biochemical class Oxidoreductase Function 2 iron, 2 sulfur cluster binding.Aldehyde dehydrogenase (FAD-independent) activity.Electron carrier activity.Metal ion binding. Related diseases LTC4 synthase deficiency is associated with a neurometabolic developmental disorder characterized by muscular hypotonia, psychomotor retardation, failure to thrive, and microcephaly. {ECO:0000269|PubMed:10896305, ECO:0000269|PubMed:9820300}. Drugs (DrugBank ID) DB02137 Interacts with NA EC number 1.2.99.7 Uniprot keywords 2Fe-2S; 3D-structure; FAD; Flavoprotein; Iron; Iron-sulfur; Metal-binding; Molybdenum; NAD; Oxidoreductase Protein physicochemical properties Chain ID A Molecular weight (Da) 96930.4 Length 907 Aromaticity 0.07 Instability index 29.17 Isoelectric point 5.69 Charge (pH=7) -17.56 2D Binding mode Binding energy (Kcal/mol) -9.39  Molscript Map 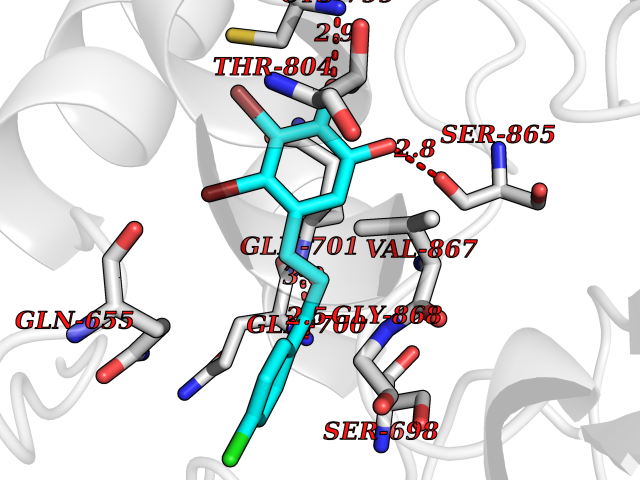 Pymol Map 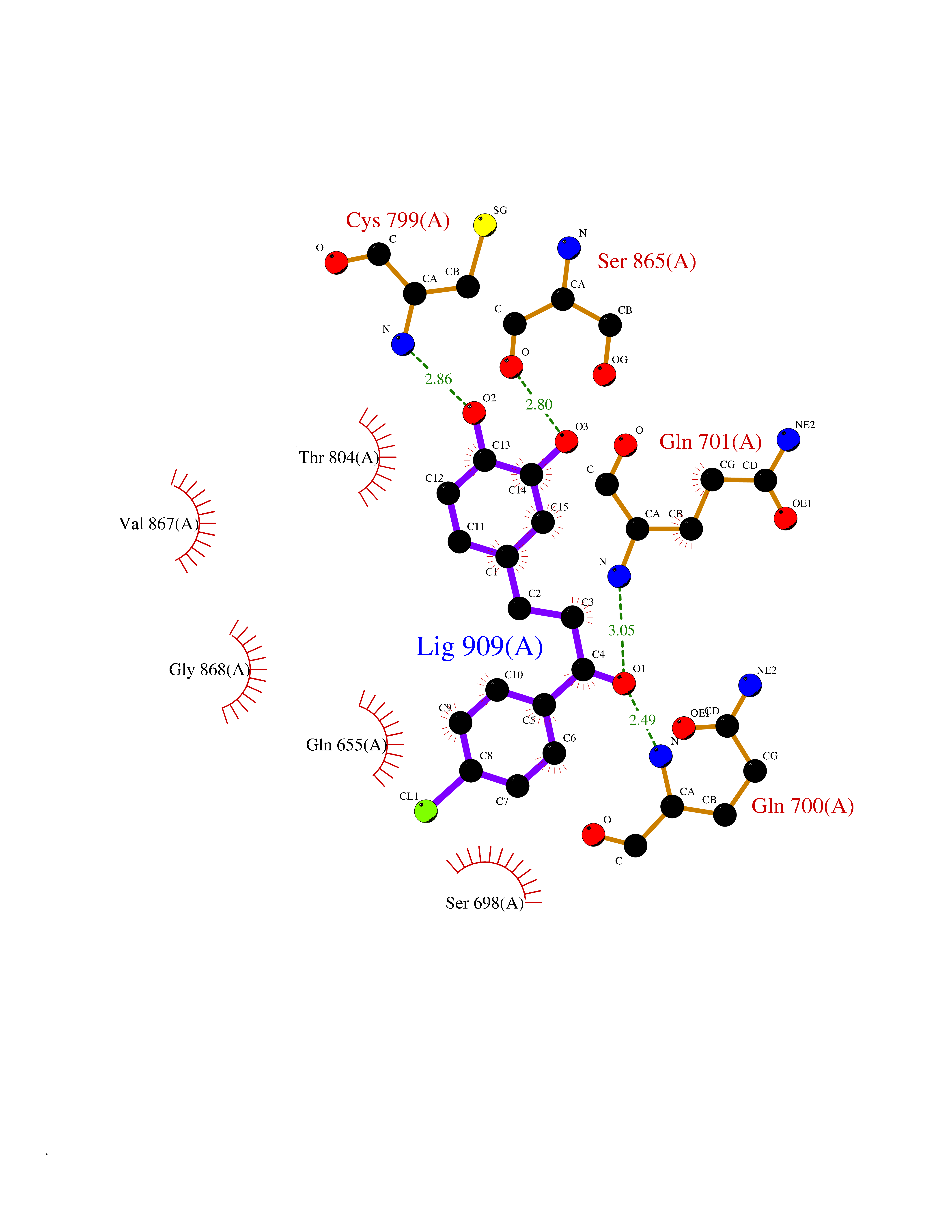 Ligplot Map 3D Binding mode Sequence MIQKVITVNGIEQNLFVDAEALLSDVLRQQLGLTGVKVGCEQGQCGACSVILDGKVVRACVTKMKRVADGAQITTIEGVGQPENLHPLQKAWVLHGGAQCGFCSPGFIVSAKGLLDTNADPSREDVRDWFQKHRNACRCTGYKPLVDAVMDAAAVINGKKPETDLEFKMPADGRIWGSKYPRPTAVAKVTGTLDYGADLGLKMPAGTLHLAMVQAKVSHANIKGIDTSEALTMPGVHSVITHKDVKGKNRITGLITFPTNKGDGWDRPILXDEKVFQYGDCIALVCADSEANARAAAEKVKVDLEELPAYMSGPAAAAEDAIEIHPGTPNVYFEQPIVKGEDTGPIFASADVTVEGDFYVGRQPHMPIEPDVAFAYMGDDGKCYIHSKSIGVHLHLYMIAPGVGLEPDQLVLVANPMGGTFGYKFSPTSEALVAVAAMATGRPVHLRYNYQQQQQYTGKRSPWEMNVKFAAKKDGTLLAMESDWLVDHGPYSEFGDLLTLRGAQFIGAGYNIPNIRGLGRTVATNHVWGSAFRGYGAPQSMFASECLMDMLAEKLGMDPLELRYKNAYRPGDTNPTGQEPEVFSLPDMIDQLRPKYQAALEKAQKESTATHKKGVGISIGVYGSGLDGPDASEAWAELNADGTITVHTAWEDHGQGADIGCVGTAHEALRPMGVAPEKIKFTWPNTATTPNSGPSGGSRQQVMTGNAIRVACENLLKACEKPGGGYYTYDELKAADKPTKITGNWTASGATHCDAVTGLGKPFVVYMYGVFMAEVTVDVATGQTTVDGMTLMADLGSLCNQLATDGQIYGGLAQGIGLALSEDFEDIKKHATLVGAGFPFIKQIPDKLDIVYVNHPRPDGPFGASGVGELPLTSPHAAIINAIKSATGVRIYRLPAYPEKVLEALKA Hydrogen bonds contact Hydrophobic contact | ||||
| 99 | Glutathione S-transferase kappa 1 | 3RPN | 6.89 | |
Target general information Gen name GSTK1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms HDCMD47P Protein family GST superfamily, Kappa family Biochemical class Transferase / transferase inhibitor Function Glutathione peroxidase activity.Glutathione transferase activity.Protein disulfide oxidoreductase activity.Receptor binding. Related diseases Dyskinesia, limb and orofacial, infantile-onset (IOLOD) [MIM:616921]: An autosomal recessive, early-onset hyperkinetic movement disorder characterized by axial hypotonia, dyskinesia of the limbs and trunk, orofacial dyskinesia, drooling, and dysarthria. The severity of the hyperkinesis is variable. {ECO:0000269|PubMed:27058446}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Striatal degeneration, autosomal dominant 2 (ADSD2) [MIM:616922]: An autosomal dominant disorder characterized by striatal degeneration and dysfunction of basal ganglia, resulting in hyperkinesis. {ECO:0000269|PubMed:27058447}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00143; DB04700 Interacts with O95273; Q8IZU0; Q60994; Q7Z3Y8 EC number 2.5.1.18 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Peroxisome; Proteomics identification; Reference proteome; Transferase Protein physicochemical properties Chain ID A,B,C,D,E,F Molecular weight (Da) 24811.7 Length 220 Aromaticity 0.08 Instability index 47.17 Isoelectric point 7.96 Charge (pH=7) 1.39 2D Binding mode Binding energy (Kcal/mol) -9.4 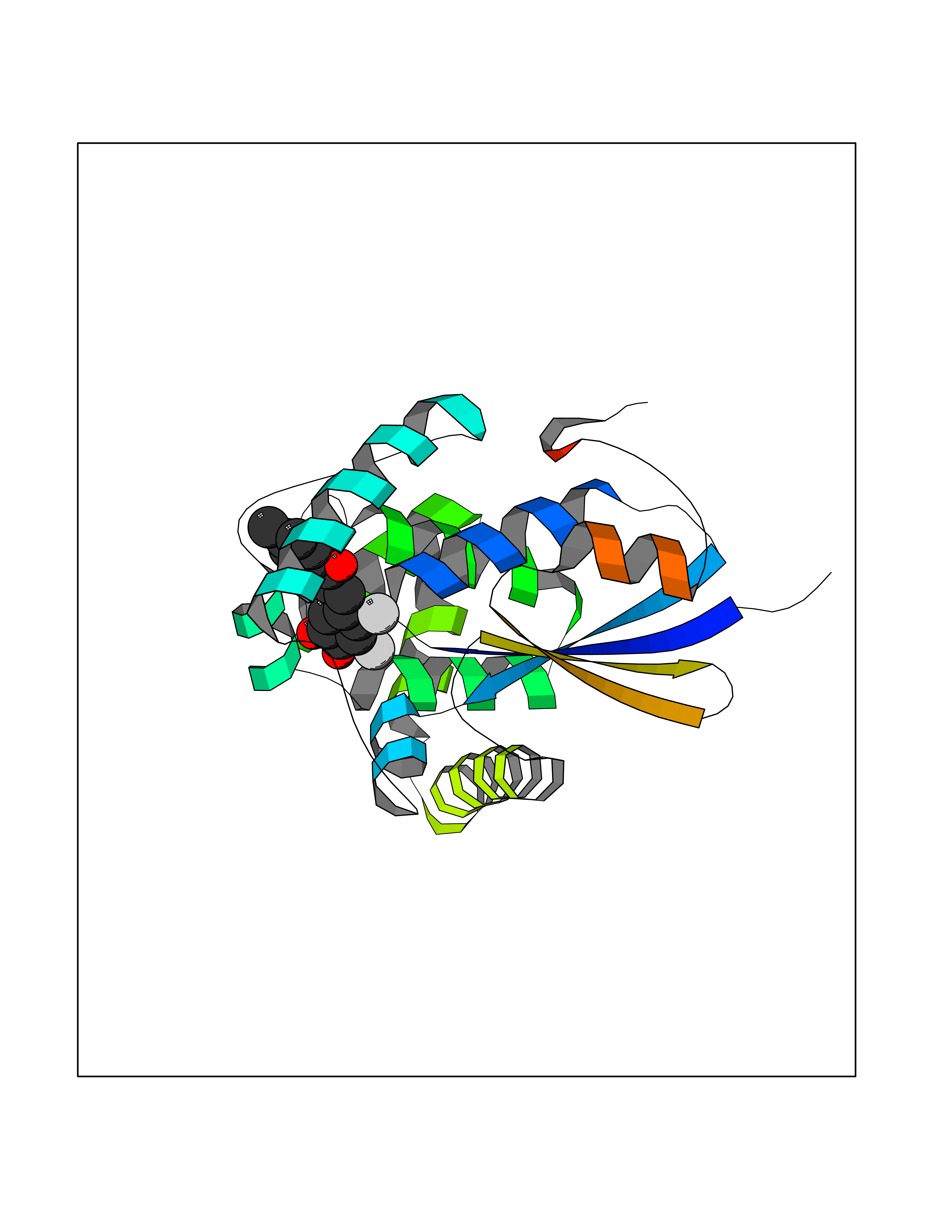 Molscript Map 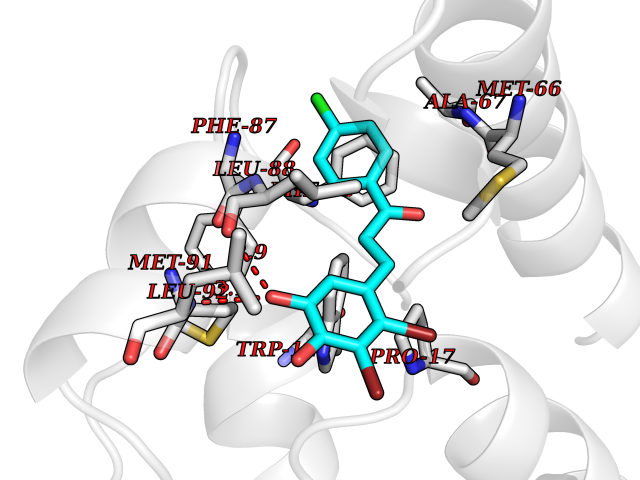 Pymol Map 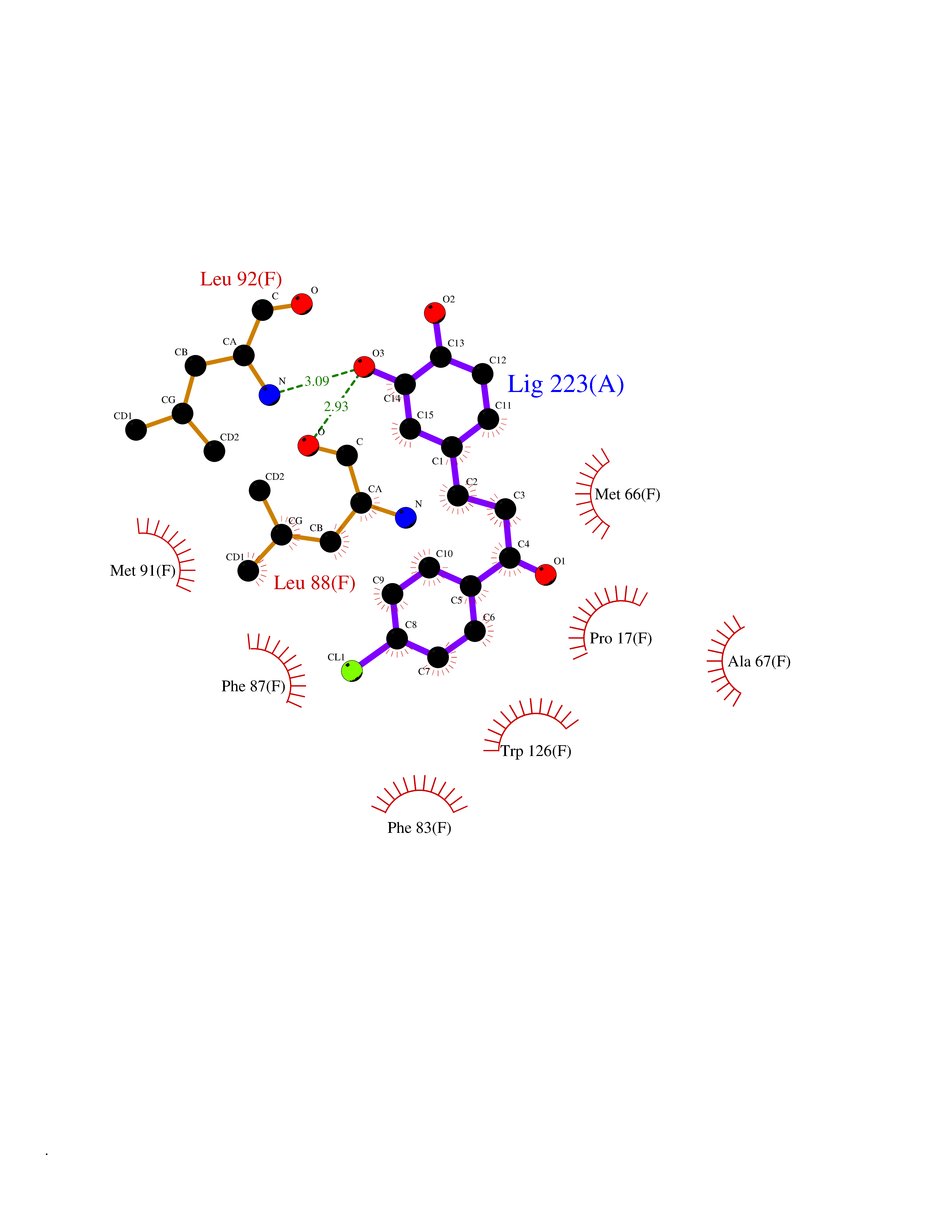 Ligplot Map 3D Binding mode Sequence GPLPRTVELFYDVLSPYSWLGFEILCRYQNIWNINLQLRPSLITGIMKDSGNKPPGLLPRKGLYMANDLKLLRHHLQIPIHFPKDFLSVMLEKGSLSAMRFLTAVNLEHPEMLEKASRELWMRVWSRNEDITEPQSILAAAEKAGMSAEQAQGLLEKIATPKVKNQLKETTEAACRYGAFGLPITVAHVDGQTHMLFGSDRMELLAHLLGEKWMGPIPPA Hydrogen bonds contact Hydrophobic contact | ||||
| 100 | Caspase-6 (CASP6) | 4NBL | 6.89 | |
Target general information Gen name CASP6 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms MCH2; Caspase-6 subunit p18; Caspase-6 subunit p11; CASP-6; Apoptotic protease Mch-2 Protein family Peptidase C14A family Biochemical class Peptidase Function Cleaves poly(ADP-ribose) polymerase in vitro, as well as lamins. Overexpression promotes programmed cell death. Involved in the activation cascade of caspases responsible for apoptosis execution. Related diseases Growth hormone deficiency, isolated, 1A (IGHD1A) [MIM:262400]: An autosomal recessive, severe deficiency of growth hormone leading to dwarfism. Patients often develop antibodies to administered growth hormone. {ECO:0000269|PubMed:8364549}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Growth hormone deficiency, isolated, 1B (IGHD1B) [MIM:612781]: An autosomal recessive deficiency of growth hormone leading to short stature. Patients have low but detectable levels of growth hormone, significantly retarded bone age, and a positive response and immunologic tolerance to growth hormone therapy. {ECO:0000269|PubMed:12655557}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Kowarski syndrome (KWKS) [MIM:262650]: A syndrome clinically characterized by short stature associated with bioinactive growth hormone, normal or slightly increased growth hormone secretion, pathologically low insulin-like growth factor 1 levels, and normal catch-up growth on growth hormone replacement therapy. {ECO:0000269|PubMed:17519310, ECO:0000269|PubMed:8552145, ECO:0000269|PubMed:9276733}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Growth hormone deficiency, isolated, 2 (IGHD2) [MIM:173100]: An autosomal dominant deficiency of growth hormone leading to short stature. Clinical severity is variable. Patients have a positive response and immunologic tolerance to growth hormone therapy. {ECO:0000269|PubMed:11502836, ECO:0000269|PubMed:9152628}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q9Y614; Q6DHV7-2; Q6UY14-3; Q96MA6; Q5T2L2; Q96Q83-2; Q9Y303-2; Q9NU02; P09525; P06727; Q8WW27; Q66PJ3-4; Q6XD76; P18848; Q9H0Y0; Q14032; P54687-4; P06276; Q9NSI6-4; Q96Q07-2; Q9H0W9-3; Q9NQ89; Q13901; Q3SXR2; Q8N1A6; P17655; P20807-4; P42574; P55212; O00257-3; P24863; Q9NNX6-10; Q9UJX2; P42773; O95674; Q494V2-2; Q8WUX9; Q9Y3D0; Q8N365; Q3SX64; Q99966; P09496-2; Q6PJW8-3; Q96BR5; P02458-1; Q9UGL9; Q9UKG9-2; P26998; P35222; Q53TN4; P61962; O60479; Q96EY1-3; Q92782-2; Q9BPU6; A0AVK6; Q658K8; O00303; Q13347; O00472; O00423; Q6NXG1-3; Q49AJ0-4; Q8N128-2; Q8IZU1; Q6ZNL6; Q9NSA1; Q06547-3; Q49A26-4; Q9HAV0; Q6NXT2; Q9BT25; Q9NRZ9-6; Q96EW2-2; P42858; Q8N6M8-2; Q92613; P0C870; Q9UK76; Q8N5Z5; Q8TBB5-2; Q9UH77; Q8N4N3-2; Q5JUW0-3; Q8N1A0; P13473-2; Q6DKI2; Q9H2C1; Q8N0U6; Q9Y234; Q8TBB1; Q1L5Z9; Q96JB6; Q16609; Q8IYG6; P0DP58-2; Q969L2; P27338; A6NJ78-4; Q96C03-3; Q8N5J2-3; A0A0A0MR05; P34949-2; Q9BV20; Q6IN84-2; A2RUH7; P01106; Q9H7X0; Q15742-2; Q9UJ70-2; Q8NDH3-5; Q96HA8; P36639-4; Q8NFH4; Q8NFH3; Q7Z3B4; Q6N063-2; Q6GQQ9-2; Q9H8K7; Q99447; P27815-4; O15534; Q9BUL5; Q00169; P48739; P61925; Q58EX7-2; O60664; Q14181; P0DPB6; P36954; Q07869; O60927; Q6ZMI0-5; P54619; Q8NCQ7-2; P41222; P29074; Q8WUD1-2; Q5R372-9; Q9HD47-3; Q09028; Q04206; P47804-3; Q15382; Q06587; Q8N5U6; P62701; Q66K80; Q01826; O15126; P22307-3; Q9BRK5; Q9NTN9-3; P01011; Q15393; Q9NR46; Q9BZQ2; O60902-3; Q86US8; P37840; Q96H20; Q13573; Q7Z6I5; Q496A3; Q9C004; Q5W111-2; Q96BD6; Q92797-2; O60506-4; O15273; Q86WV5; Q96A09; P54274-2; P22735; O43548; Q9NQ88; Q9UIK5-2; Q53NU3; P04637; Q12888; P36406; Q86WT6-2; Q13885; P49459; Q9P1Q0-4; Q9NX94; Q8NA23-2; Q9BQA1; O00755; O95070; O43829; Q8IWT0-2; Q53FD0-2; Q05CR2; Q96JL9-2; Q96LX8; Q3KNS6-3; Q5JTY5; A0A384MDV8; B7Z3E8; Q86V28 EC number EC 3.4.22.59 Uniprot keywords 3D-structure; Alternative splicing; Apoptosis; Autocatalytic cleavage; Cytoplasm; Hydrolase; Lipoprotein; Nucleus; Palmitate; Phosphoprotein; Protease; Proteomics identification; Reference proteome; Thiol protease; Zymogen Protein physicochemical properties Chain ID A,B Molecular weight (Da) 57170 Length 500 Aromaticity 0.12 Instability index 31.33 Isoelectric point 8.05 Charge (pH=7) 4.52 2D Binding mode Binding energy (Kcal/mol) -9.4 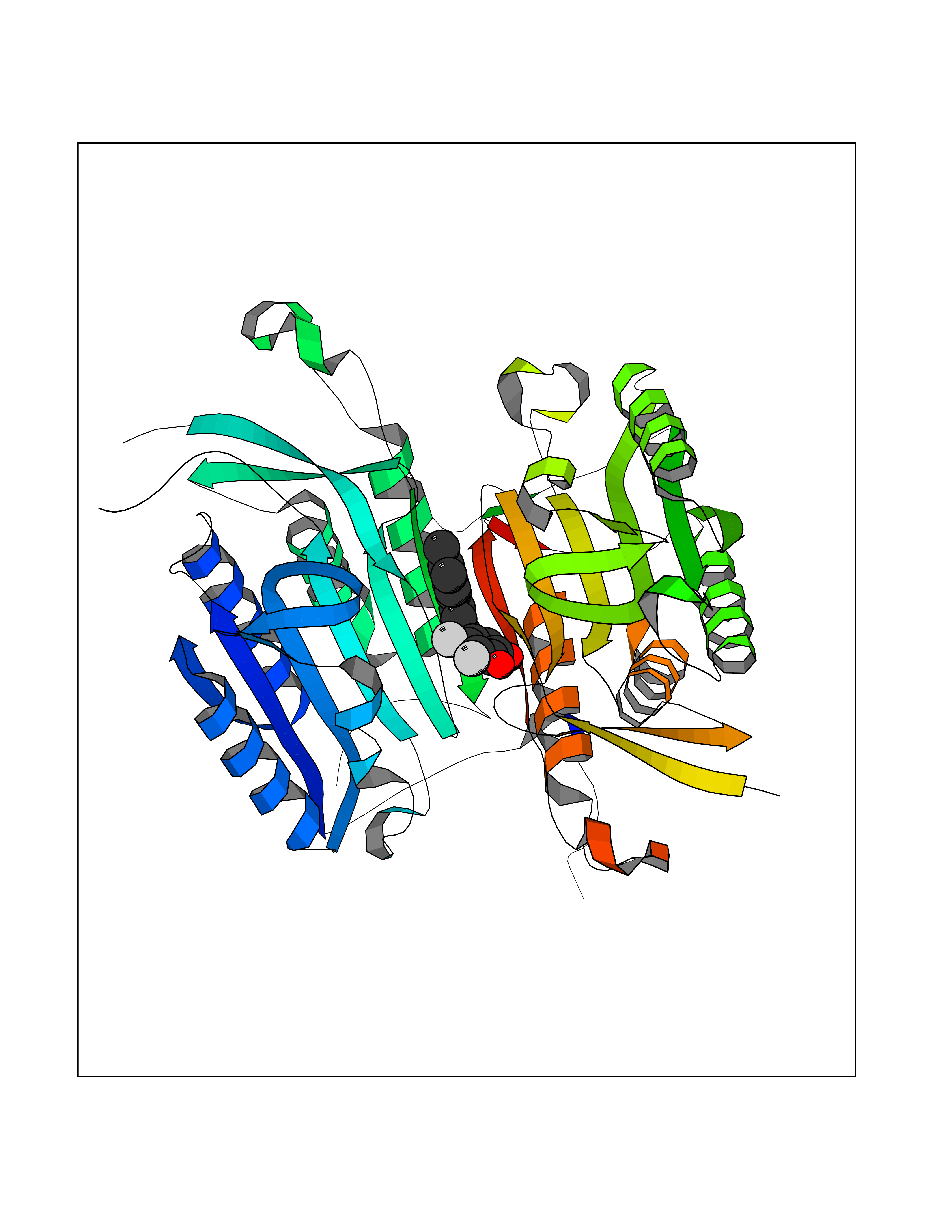 Molscript Map 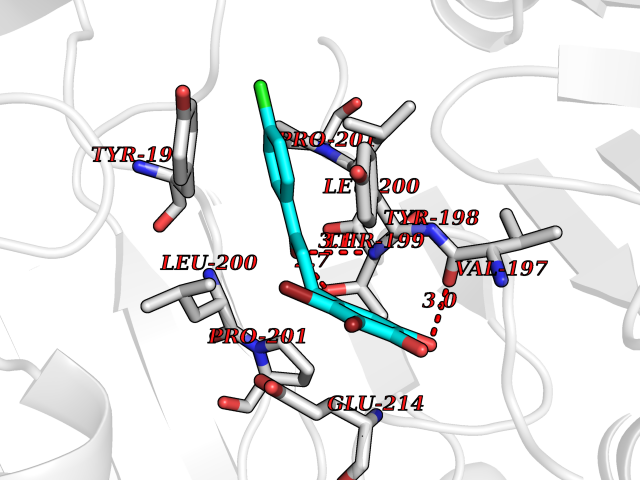 Pymol Map 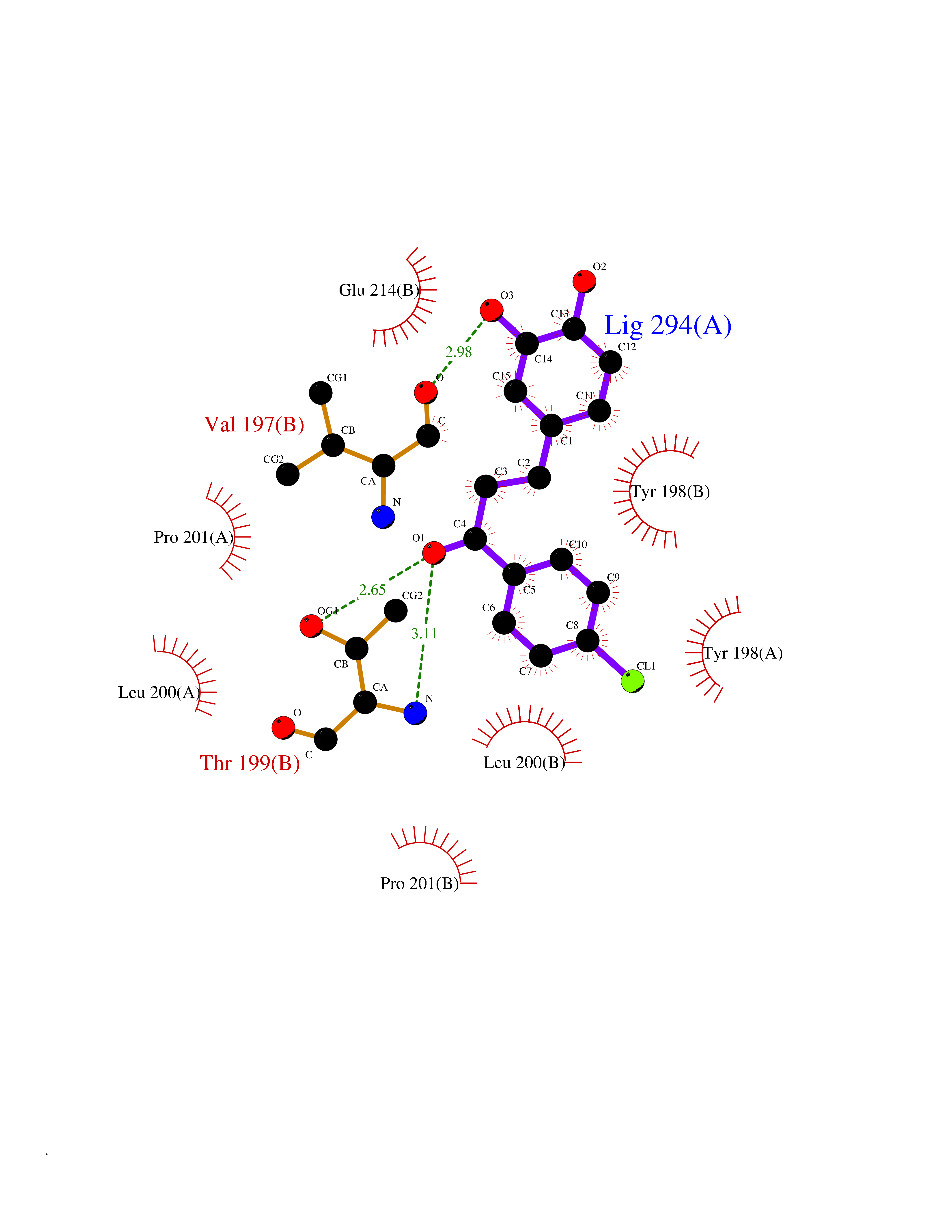 Ligplot Map 3D Binding mode Sequence AFYKREMFDPAEKYKMDHRRRGIALIFNHERFFWHLTLPERRGTCADRDNLTRRFSDLGFEVKCFNDLKAEELLLKIHEVSTVSHADADCFVCVFLSHGEGNHIYAYDAKIEIQTLTGLFKGDKCHSLVGKPKIFIIQAARGNQHDVPVIPDTNITEVDAASVYTLPAGADFLMCYSVAEGYYSHRETVNGSWYIQDLCEMLGKYGSSLEFTELLTLVNRKVSQRRVDFCKDPSAIGKKQVPCFASMLTKKLHFFPKSMFDPAEKYKMDHRRRGIALIFNHERFFWHLTLPERRGTCADRDNLTRRFSDLGFEVKCFNDLKAEELLLKIHEVSTVSHADADCFVCVFLSHGEGNHIYAYDAKIEIQTLTGLFKGDKCHSLVGKPKIFIIQAARGNTNITEVDAASVYTLPAGADFLMCYSVAEGYYSHRETVNGSWYIQDLCEMLGKYGSSLEFTELLTLVNRKVSQRRVDFCKDPSAIGKKQVPCFASMLTKKLHFFPK Hydrogen bonds contact Hydrophobic contact | ||||