Job Results:
Ligand
Structure
Job ID
b448ce30ce2c1c80e042f47c74d47daa
Job name
NA
Time
2026-01-10 22:40:02
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 1 | Neuronal acetylcholine receptor subunit alpha-3 | 4ZK4 | 7.37 | |
Target general information Gen name CHRNA3 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NACHRA3 Protein family Ligand-gated ion channel (TC 1.A.9) family, Acetylcholine receptor (TC 1.A.9.1) subfamily, Alpha-3/CHRNA3 sub-subfamily Biochemical class Acetylcholine binding protein Function Acetylcholine binding.Acetylcholine-gated cation-selective channel activity.Acetylcholine receptor activity.Ligand-gated ion channel activity.Serotonin-gated cation-selective channel activity. Related diseases Bladder dysfunction, autonomic, with impaired pupillary reflex and secondary CAKUT (BAIPRCK) [MIM:191800]: An autosomal recessive disease characterized by impaired innervation and autonomic dysfunction of the urinary bladder, hydronephrosis, vesicoureteral reflux, small kidneys, recurrent urinary tract infections, and progressive renal insufficiency. Additional autonomic features are impaired pupillary reflex and orthostatic hypotension. The disease manifests in utero or early childhood. {ECO:0000269|PubMed:31708116}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00915; DB01156; DB00237; DB00565; DB09028; DB00514; DB07720; DB00898; DB00472; DB05710; DB01227; DB00848; DB00333; DB00184; DB01090; DB00202; DB01273 Interacts with NA EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disease variant; Disulfide bond; Endoplasmic reticulum; Glycoprotein; Golgi apparatus; Ion channel; Ion transport; Ligand-gated ion channel; Membrane; Phosphoprotein; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Signal; Synapse; Transmembrane; Transmembrane helix; Transport; Ubl conjugation Protein physicochemical properties Chain ID A,B,C,D,E Molecular weight (Da) 46391.5 Length 408 Aromaticity 0.12 Instability index 30.23 Isoelectric point 4.6 Charge (pH=7) -22.73 2D Binding mode Binding energy (Kcal/mol) -10.06 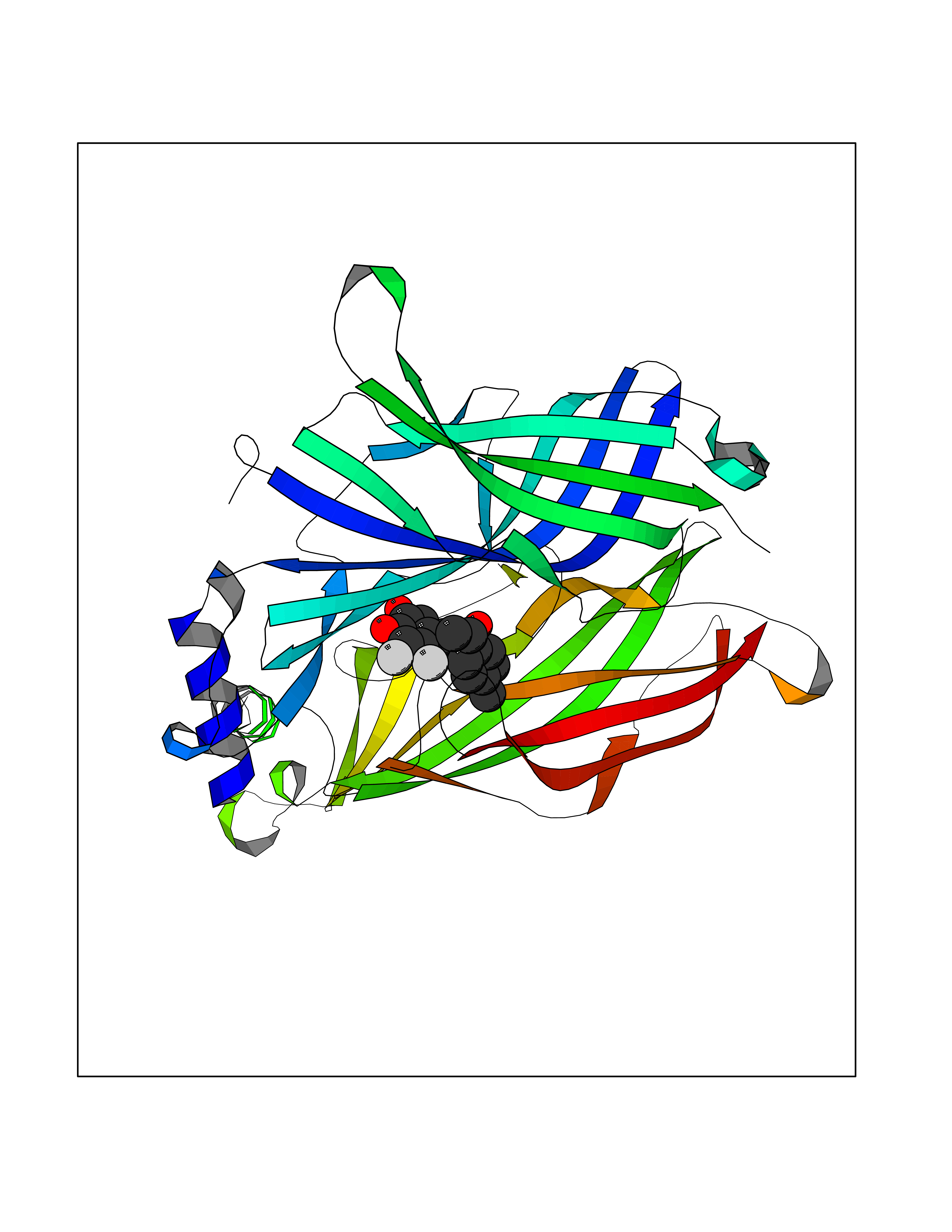 Molscript Map 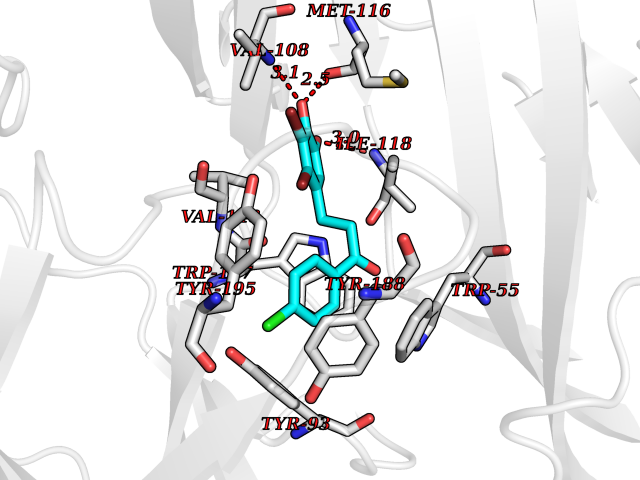 Pymol Map 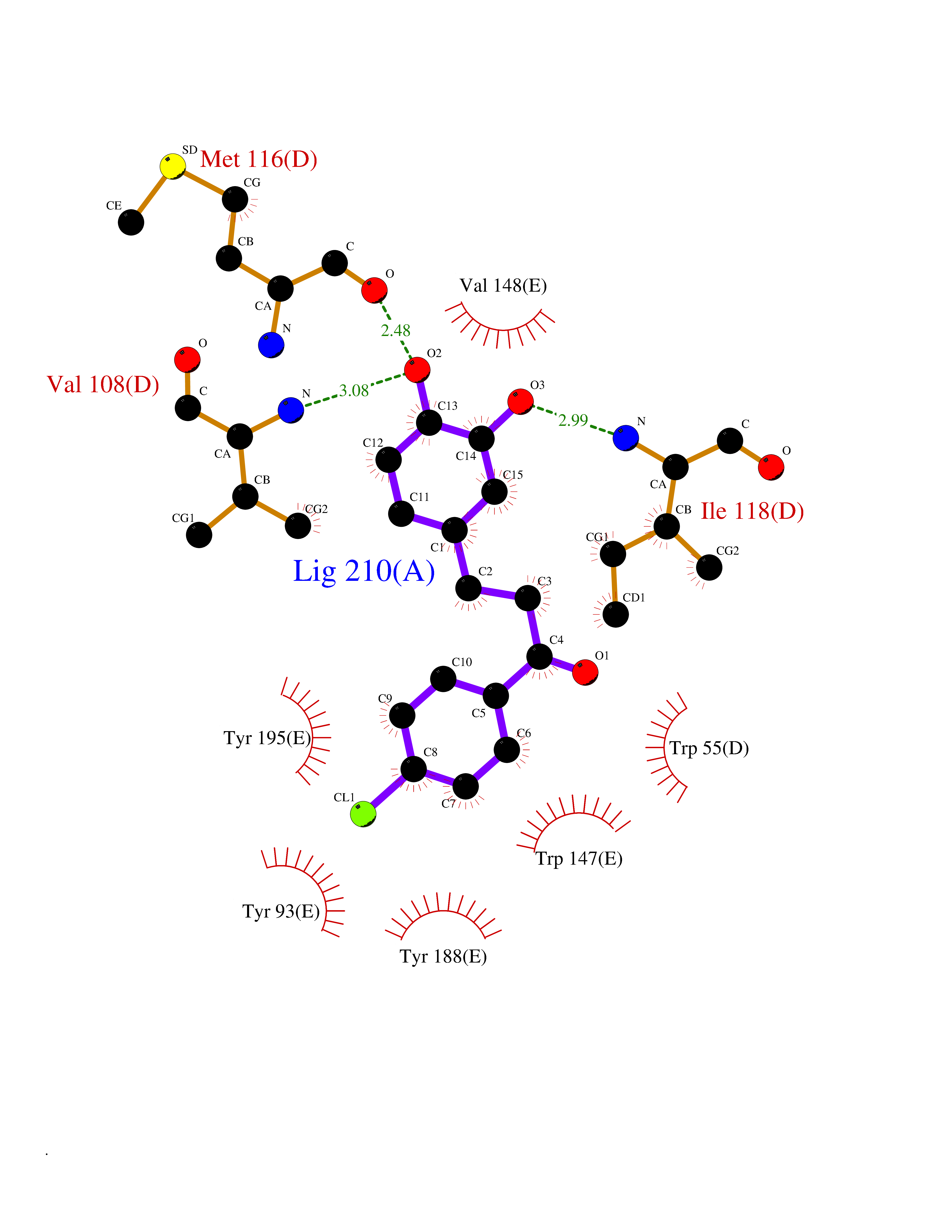 Ligplot Map 3D Binding mode Sequence LHSQANLMRLKSDLFYPGPTKDDPLTVTLGFTLQDIVKADSSTNEVDLVYWEQQRWKLNSLMWDPNEYGNITDFRTSAADIWTPDITAYSSTRPVQVLSPQIAVVTHDGSVMFIPAQRLSFMCDPTGVDSEEGATCAVKFGSWVYSGFEIDLKTDTDQVDLSSYYASSKYEILSATQYKHDIKYNCCEEIYPDVVLVVKFRERRLHSQANLMRLKSDLFNRYPGPTKDDPLTVTLGFTLQDIVKADSSTNEVDLVYWEQQRWKLNSLMWDPNEYGNITDFRTSAADIWTPDITAYSSTRPVQVLSPQIAVVTHDGSVMFIPAQRLSFMCDPTGVDSEEGATCAVKFGSWVYSGFEIDLKTDTDQVDLSSYYASSKYEILSATQYKHDIKYNCCEEIYPDVVLVVKFRE Hydrogen bonds contact Hydrophobic contact | ||||
| 2 | Leukotriene A-4 hydrolase (LTA4H) | 3U9W | 7.36 | |
Target general information Gen name LTA4H Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Leukotriene A4 hydrolase; Leukotriene A(4)Leukotriene A-4 hydrolase hydrolase; Leukotriene A(4) hydrolase; LTA4; LTA-H; LTA-4hydrolase; LTA-4 hydrolase Protein family Peptidase M1 family Biochemical class Ether bond hydrolase Function Has also aminopeptidase activity. Epoxide hydrolase that catalyzes the final step in the biosynthesis of the proinflammatory mediator leukotriene B4. Related diseases Pigmentary disorder, reticulate, with systemic manifestations, X-linked (PDR) [MIM:301220]: An X-linked recessive disorder characterized by recurrent infections and sterile inflammation in various organs. Diffuse skin hyperpigmentation with a distinctive reticulate pattern is universally evident by early childhood. This is later followed in many patients by hypohidrosis, corneal inflammation and scarring, enterocolitis that resembles inflammatory bowel disease, and recurrent urethral strictures. Melanin and amyloid deposition is present in the dermis. Affected males also have a characteristic facies with frontally upswept hair and flared eyebrows. Female carriers have only restricted pigmentary changes along Blaschko's lines. {ECO:0000269|PubMed:27019227}. The disease is caused by variants affecting the gene represented in this entry. XLPDR is caused by a recurrent intronic mutation that results in missplicing and reduced POLA1 expression. This leads to a decrease in cytosolic RNA:DNA hybrids and constitutive activation of type I interferon responses, but has no effect on cell replication. {ECO:0000269|PubMed:27019227}.; DISEASE: Van Esch-O'Driscoll syndrome (VEODS) [MIM:301030]: An X-linked recessive syndrome characterized by different degrees of intellectual disability, moderate to severe short stature, microcephaly, hypogonadism, and variable congenital malformations. {ECO:0000269|PubMed:31006512}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07102; DB06917; DB07258; DB07094; DB07259; DB02352; DB07292; DB07104; DB06828; DB08466; DB01197; DB05177; DB03366; DB08040; DB06851; DB02062; DB07099; DB07260; DB07196; DB11781; DB03424; DB07237 Interacts with Q9BSI4 EC number EC 3.3.2.6 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Cytoplasm; Direct protein sequencing; Hydrolase; Leukotriene biosynthesis; Lipid metabolism; Metal-binding; Metalloprotease; Phosphoprotein; Protease; Proteomics identification; Reference proteome; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 68927 Length 608 Aromaticity 0.1 Instability index 38.84 Isoelectric point 5.87 Charge (pH=7) -9.86 2D Binding mode Binding energy (Kcal/mol) -10.04 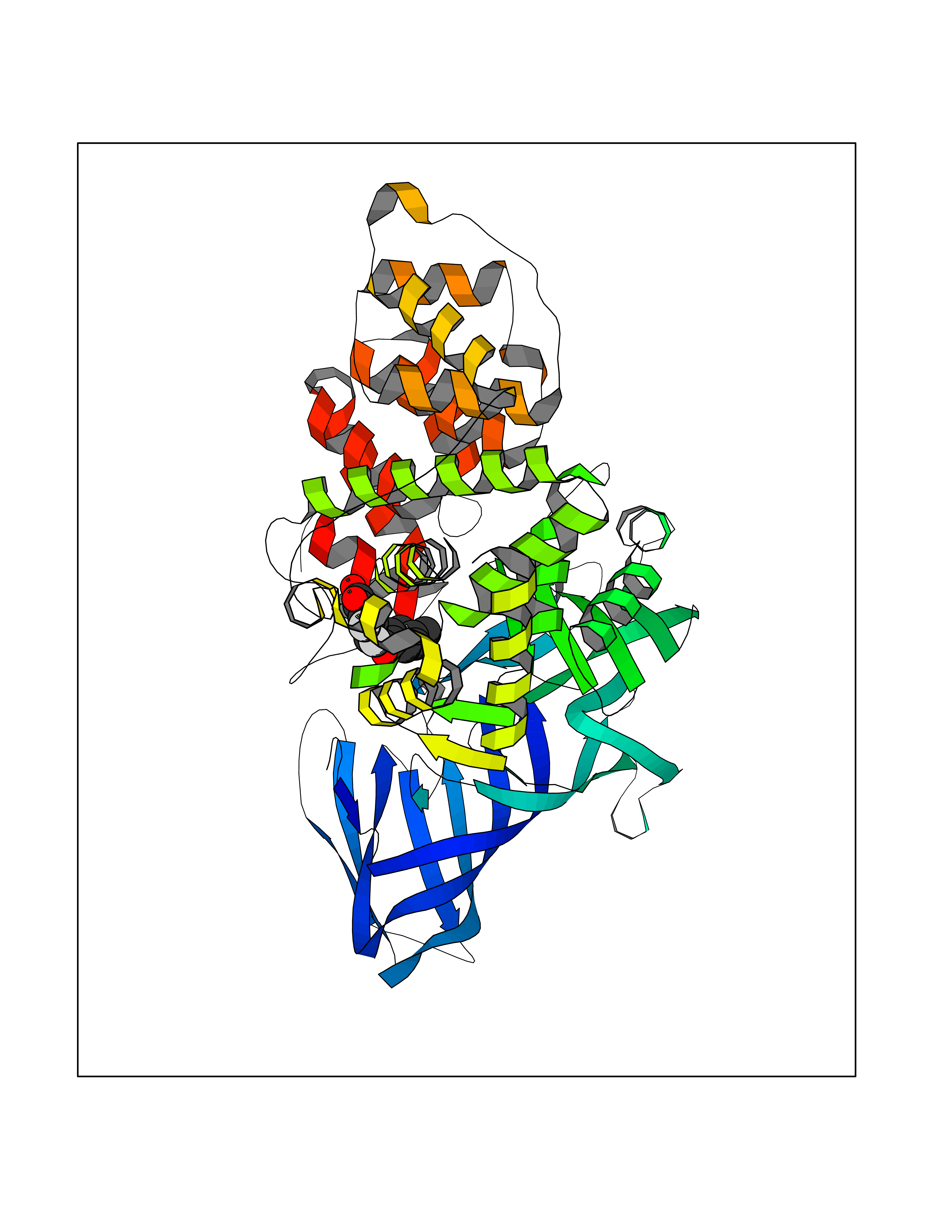 Molscript Map 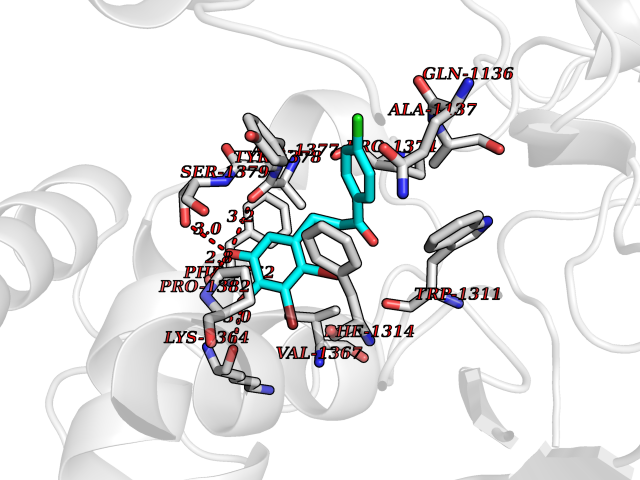 Pymol Map 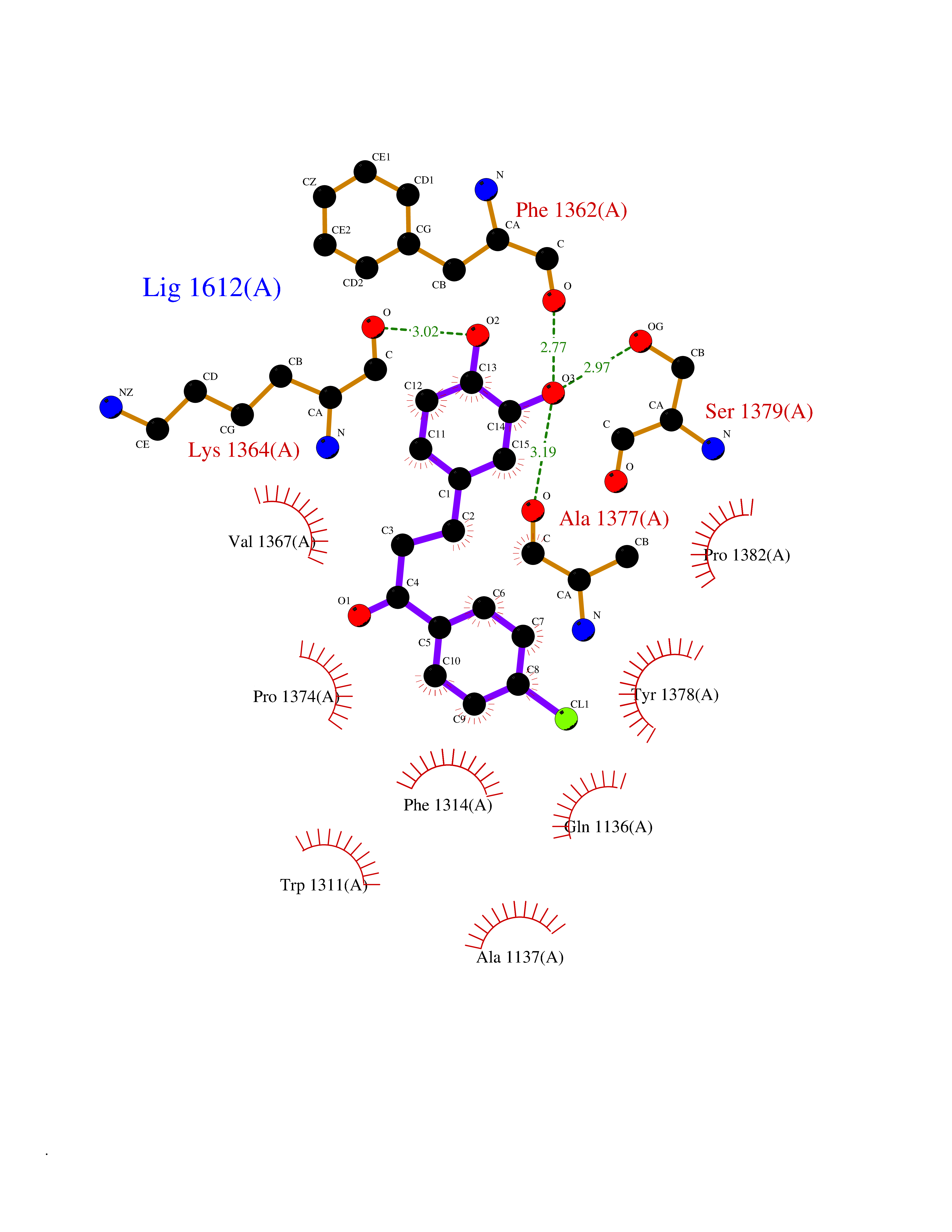 Ligplot Map 3D Binding mode Sequence IVDTCSLASPASVCRTKHLHLRCSVDFTRRTLTGTAALTVQSQEDNLRSLVLDTKDLTIEKVVINGQEVKYALGERQSYKGSPMEISLPIALSKNQEIVIEISFETSPKSSALQWLTPEQTSGKEHPYLFSQCQAIHCRAILPCQDTPSVKLTYTAEVSVPKELVALMSAIRDGETPDPEDPSRKIYKFIQKVPIPCYLIALVVGALESRQIGPRTLVWSEKEQVEKSAYEFSETESMLKIAEDLGGPYVWGQYDLLVLPPSFPYGGMENPCLTFVTPTLLAGDKSLSNVIAHEISHSWTGNLVTNKTWDHFWLNEGHTVYLERHICGRLFGEKFRHFNALGGWGELQNSVKTFGETHPFTKLVVDLTDIDPDVAYSSVPYEKGFALLFYLEQLLGGPEIFLGFLKAYVEKFSYKSITTDDWKDFLYSYFKDKVDVLNQVDWNAWLYSPGLPPIKPNYDMTLTNACIALSQRWITAKEDDLNSFNATDLKDLSSHQLNEFLAQTLQRAPLPLGHIKRMQEVYNFNAINNSEIRFRWLRLCIQSKWEDAIPLALKMATEQGRMKFTRPLFKDLAAFDKSHDQAVRTYQEHKASMHPVTAMLVGKDLKVD Hydrogen bonds contact Hydrophobic contact | ||||
| 3 | Mutated Histone H3.3 (H3F3A) | 4GUS | 7.21 | |
Target general information Gen name H3F3A Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms PP781; Histone H3.3; H3F3; H3.3B; H3.3A Protein family Histone H3 family Biochemical class NA Function Variant histone H3 which replaces conventional H3 in a wide range of nucleosomes in active genes. Constitutes the predominant form of histone H3 in non-dividing cells and is incorporated into chromatin independently of DNA synthesis. Deposited at sites of nucleosomal displacement throughout transcribed genes, suggesting that it represents an epigenetic imprint of transcriptionally active chromatin. Nucleosomes wrap and compact DNA into chromatin, limiting DNA accessibility to the cellular machineries which require DNA as a template. Histones thereby play a central role in transcription regulation, DNA repair, DNA replication and chromosomal stability. DNA accessibility is regulated via a complex set of post-translational modifications of histones, also called histone code, and nucleosome remodeling. Related diseases Glioma (GLM) [MIM:137800]: Gliomas are benign or malignant central nervous system neoplasms derived from glial cells. They comprise astrocytomas and glioblastoma multiforme that are derived from astrocytes, oligodendrogliomas derived from oligodendrocytes and ependymomas derived from ependymocytes. {ECO:0000269|PubMed:22286061, ECO:0000269|PubMed:22286216, ECO:0000269|PubMed:23539269}. The gene represented in this entry is involved in disease pathogenesis. H3F3A mutations affecting residues involved in post-translational modifications of histone H3.3 are recurrent in malignant, aggressive gliomas including glioblastoma multiforme (GBM) and diffuse intrinsic pontine glioma (DIPG) (PubMed:22286061, PubMed:22286216). The mechanism through which mutations lead to tumorigenesis involves altered histones methylation, impaired regulation of Polycomb repressive complex 2 (PRC2) activity, and aberrant epigenetic regulation of gene expression (PubMed:23539183, PubMed:23539269, PubMed:23603901). {ECO:0000269|PubMed:22286061, ECO:0000269|PubMed:22286216, ECO:0000269|PubMed:23539183, ECO:0000269|PubMed:23539269, ECO:0000269|PubMed:23603901}.; DISEASE: Bryant-Li-Bhoj neurodevelopmental syndrome 1 (BRYLIB1) [MIM:619720]: An autosomal dominant disorder predominantly characterized by global developmental delay, impaired intellectual development, poor or absent speech, and delayed motor milestones. Clinical manifestations are highly variable, including abnormal head shape, dysmorphic facial features, oculomotor abnormalities, feeding problems, and non-specific brain imaging abnormalities. Additional features may include hearing loss, seizures, short stature, and mild skeletal defects. {ECO:0000269|PubMed:33268356, ECO:0000269|PubMed:34876591}. The disease is caused by variants affecting the gene represented in this entry. BRYLIB1 is caused by variants in H3-3A. {ECO:0000269|PubMed:33268356, ECO:0000269|PubMed:34876591}.; DISEASE: Bryant-Li-Bhoj neurodevelopmental syndrome 2 (BRYLIB2) [MIM:619721]: An autosomal dominant disorder predominantly characterized by global developmental delay, impaired intellectual development, poor or absent speech, and delayed motor milestones. Clinical manifestations are highly variable, including abnormal head shape, dysmorphic facial features, oculomotor abnormalities, feeding problems, and non-specific brain imaging abnormalities. Additional features may include hearing loss, seizures, short stature, and mild skeletal defects. {ECO:0000269|PubMed:33268356, ECO:0000269|PubMed:34876591}. The disease is caused by variants affecting the gene represented in this entry. BRYLIB2 is caused by variants in H3-3B. {ECO:0000269|PubMed:33268356, ECO:0000269|PubMed:34876591}.; DISEASE: H3F3A and H3F3B mutations affecting residues involved in post-translational modifications of histone H3.3 are implicated in the pathogenesis of some bone and cartilage neoplasms. Mutations have been found with high prevalence in chondroblastoma and giant cell tumors of bone, and with low frequency in osteosarcoma, conventional chondrosarcoma and clear cell chondrosarcoma. Chondroblastoma samples frequently carry a H3F3B mutation affecting residue Lys-37 (H3K36), although H3F3A is mutated in some cases. Most giant cell tumors of bone harbor H3F3A mutations affecting residue Gly-35 (H3G34). {ECO:0000269|PubMed:24162739}. Drugs (DrugBank ID) NA Interacts with Q9NVP2; P45973; Q13111; Q9UER7; Q9UER7-1; Q9Y6K1; P62805; P49321-2; Q8IZL8; Q5VWG9; Q9VK33; Q8R5C8 EC number NA Uniprot keywords 3D-structure; Acetylation; ADP-ribosylation; Chromosome; Citrullination; Direct protein sequencing; Disease variant; DNA-binding; Hydroxylation; Intellectual disability; Lipoprotein; Methylation; Nucleosome core; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Ubl conjugation Protein physicochemical properties Chain ID A,C Molecular weight (Da) 86148.9 Length 766 Aromaticity 0.1 Instability index 37.57 Isoelectric point 8.25 Charge (pH=7) 7.16 2D Binding mode Binding energy (Kcal/mol) -9.84 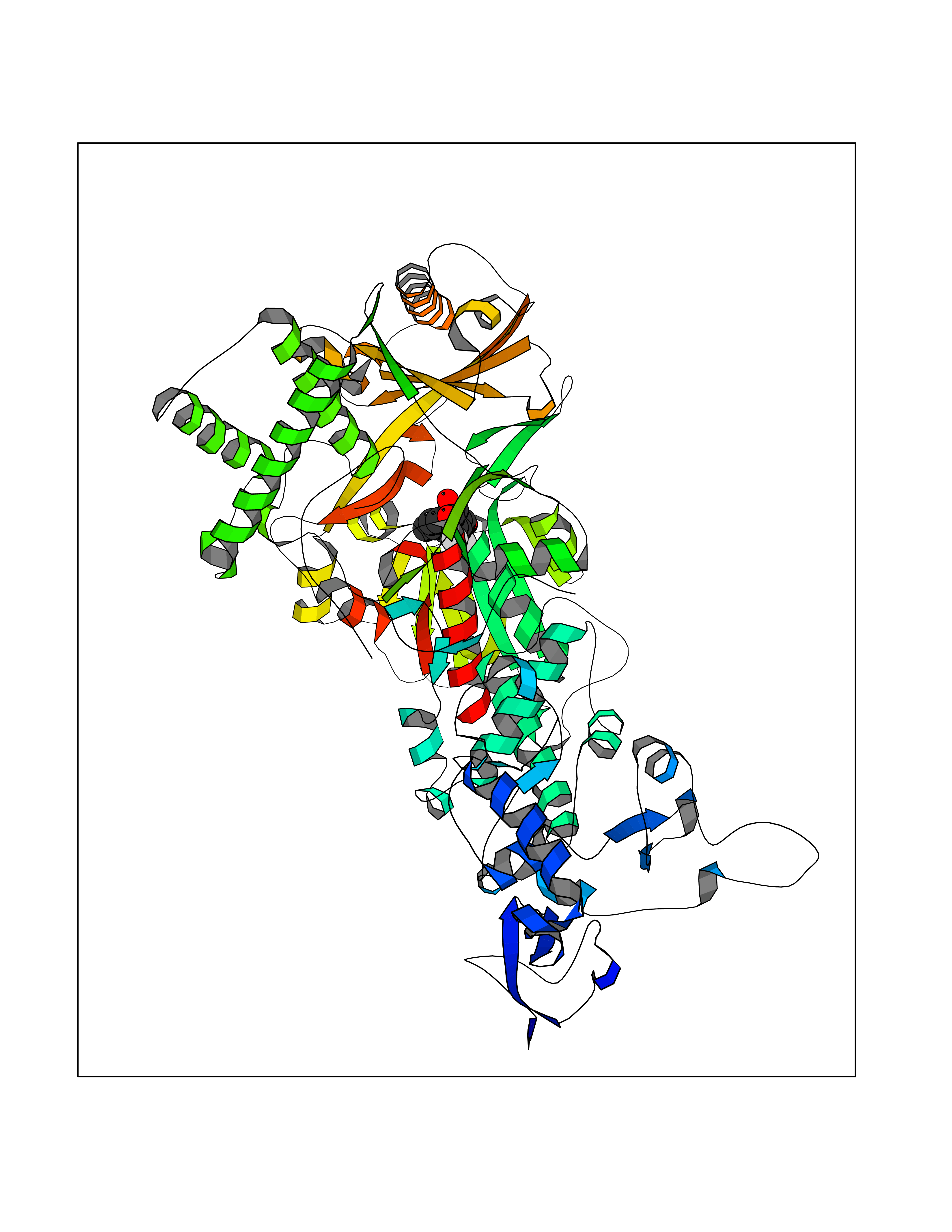 Molscript Map 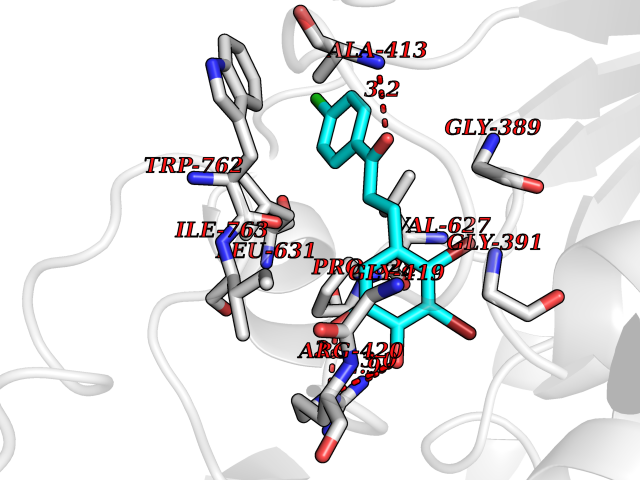 Pymol Map 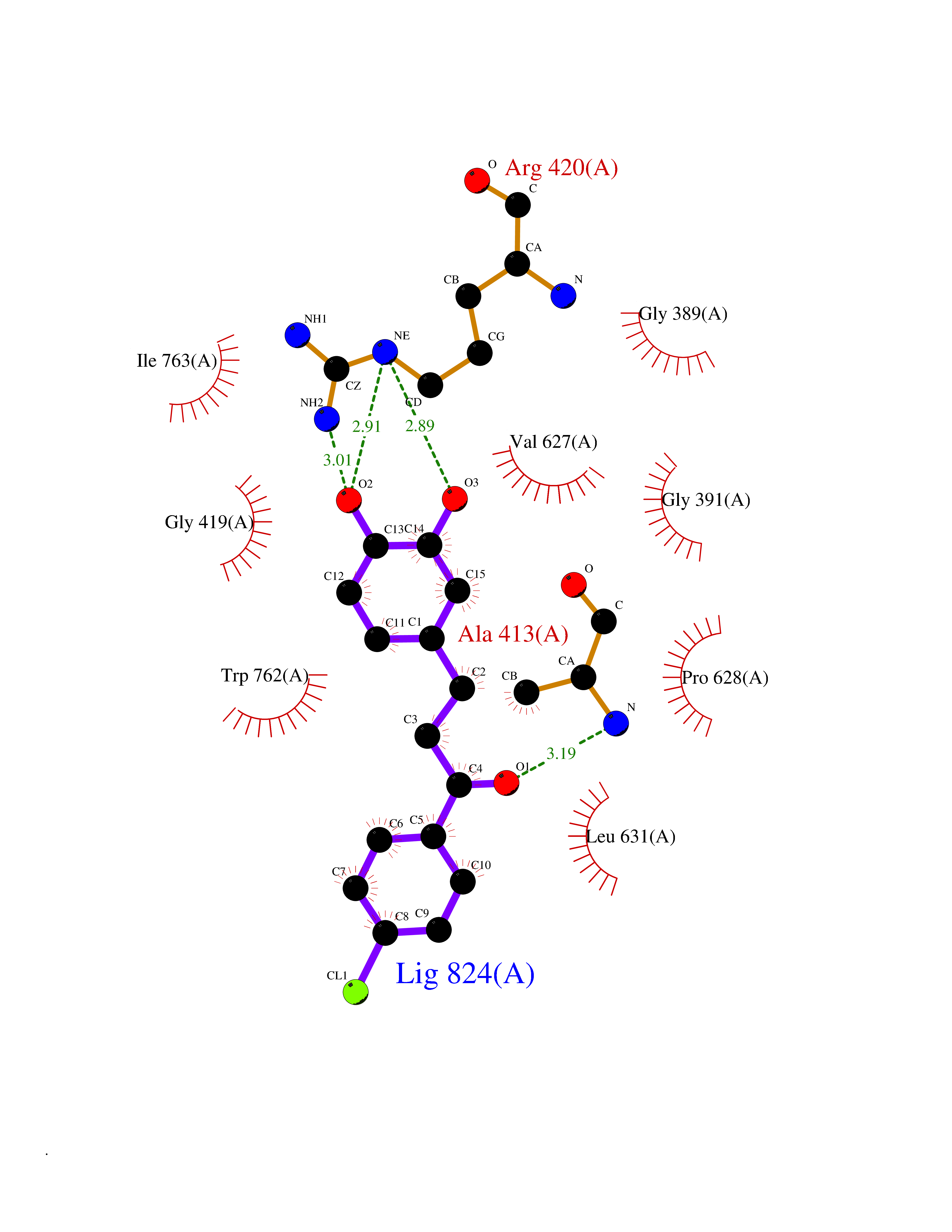 Ligplot Map 3D Binding mode Sequence GSRKCEKAGCTATCPVCFASASERCAKNGYTSRWYHLSCGEHFCNECFDHYYRSHKDGYDKYTTWKKIWTSNGKTEPSPKAFMADQQLPYWVQCTKPECRKWRQLTKEIQLTPQIAKTYRCGMKPNTAIKPETSDHCSLPEDLRVLEVSNHWWYSMLILPPLLKDSVAAPLLSAYYPDCVGMSPSCTGMNRYFQPFYQPNECGKALCVRPDVMELDELYEFPEYSRDPTMYLALRNLILALWYTNCKEALTPQKCIPHIIVRGLVRIRCVQEVERILYFMTRKGLINTGVLSVGADQYLLPKDYHNKSVIIIGAGPAGLAAARQLHNFGIKVTVLEAKDRIGGRVWDDKSFKGVTVGRGAQIVNGCINNPVALMCEQLGISMHKFGERCDLIQEGGRITDPTIDKRMDFHFNALLDVVSEWRKDKTQLQDVPLGEKIEEIYKAFIKESGIQFSELEGQVLQFHLSNLEYACGSNLHQVSARSWDHNEFFAQFAGDHTLLTPGYSVIIEKLAEGLDIQLKSPVQCIDYSGDEVQVTTTDGTGYSAQKVLVTVPLALLQKGAIQFNPPLSEKKMKAINSLGAGIIEKIALQFPYRFWDSKVQGADFFGHVPPSASKRGLFAVFYDMDPQKKHSVLMSVIAGEAVASVRTLDDKQVLQQCMATLRELFKEQEVPDPTKYFVTRWSTDPWIQMAYSFVKTGGSGEAYDIIAEDIQGTVFFAGEATNRHFPQTVTGAYLSGVREASKIAAFARTMQTARKSTGGKAPRKQL Hydrogen bonds contact Hydrophobic contact | ||||
| 4 | Pyruvate synthase | 2C42 | 7.20 | |
Target general information Gen name por Organism Desulfocurvibacter africanus (Desulfovibrio africanus) Uniprot ID TTD ID NA Synonyms NA Protein family Pyruvate:ferredoxin/flavodoxin oxidoreductase family Biochemical class Oxidoreductase Function 4 iron, 4 sulfur cluster binding.Iron ion binding.Pyruvate synthase activity.Thiamine pyrophosphate binding. Related diseases Intellectual developmental disorder, autosomal dominant 62 (MRD62) [MIM:618793]: An autosomal dominant form of intellectual disability, a disorder characterized by significantly below average general intellectual functioning associated with impairments in adaptive behavior and manifested during the developmental period. MRD62 is characterized by mild to moderately impaired intellectual development. {ECO:0000269|PubMed:27479843, ECO:0000269|PubMed:29460436}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02410; DB01987; DB00507 Interacts with NA EC number 1.2.7.1 Uniprot keywords 3D-structure; 4Fe-4S; Calcium; Cytoplasm; Direct protein sequencing; Disulfide bond; Electron transport; Iron; Iron-sulfur; Magnesium; Metal-binding; Oxidoreductase; Pyruvate; Thiamine pyrophosphate; Transport Protein physicochemical properties Chain ID A,B Molecular weight (Da) 115569 Length 1065 Aromaticity 0.09 Instability index 31.51 Isoelectric point 6.32 Charge (pH=7) -5.62 2D Binding mode Binding energy (Kcal/mol) -9.82 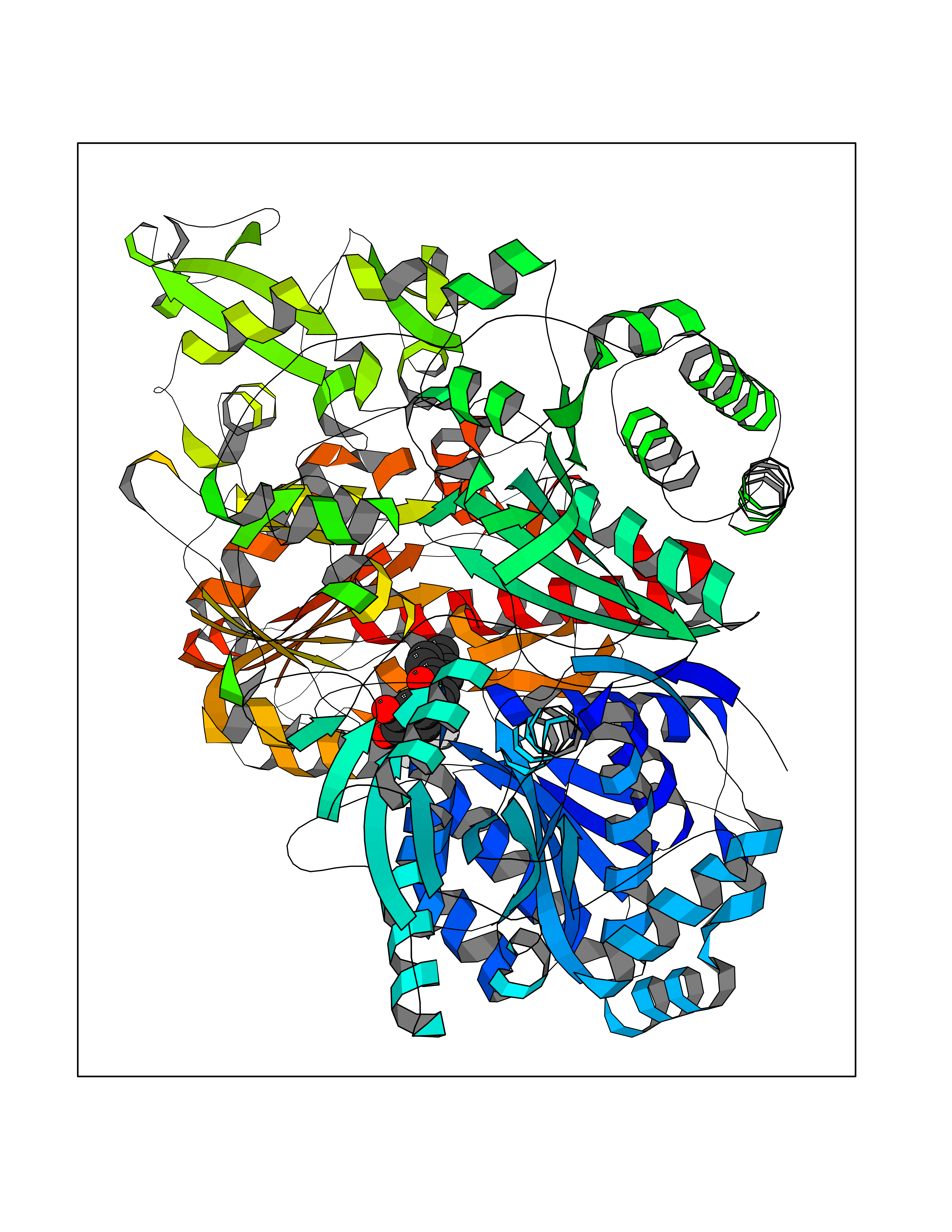 Molscript Map 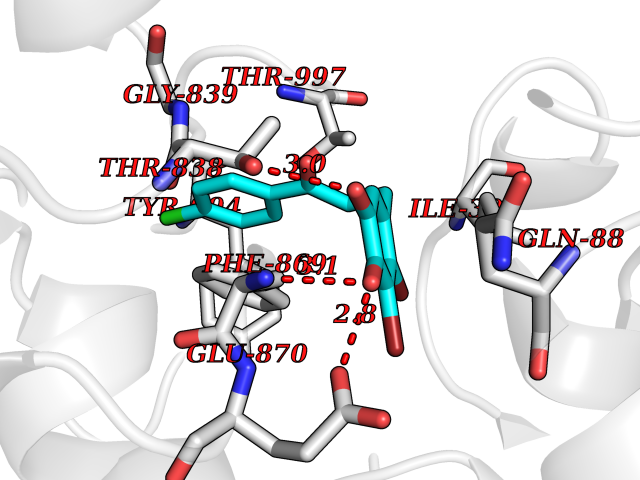 Pymol Map 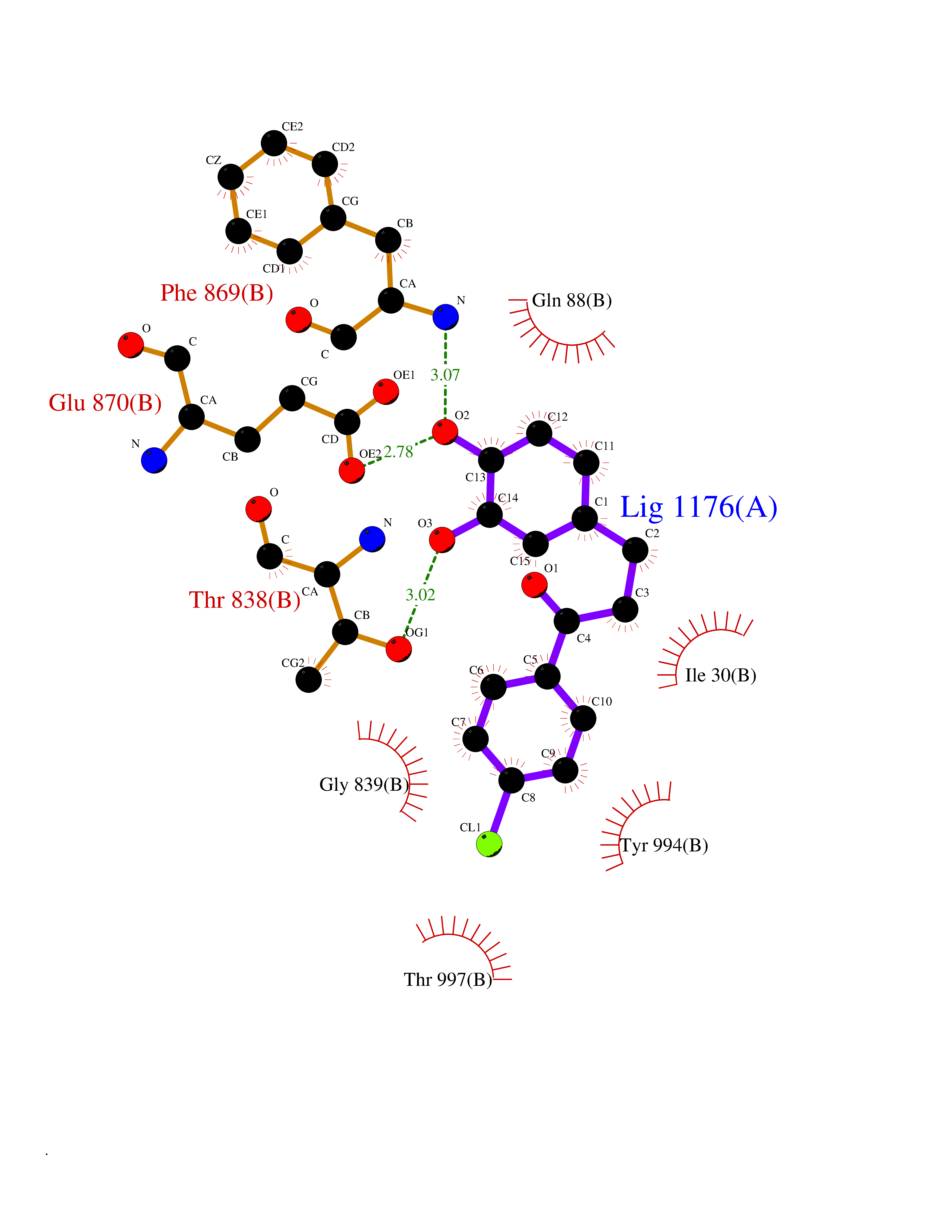 Ligplot Map 3D Binding mode Sequence GKKMMTTDGNTATAHVAYAMSEVAAIYPITPSSTMGEEADDWAAQGRKNIFGQTLTIREMQSEAGAAGAVHGALAAGALTTTFTASQGLLLMIPNMYKISGELLPGVFHVTARAIAAHALSIFGDHQDIYAARQTGFAMLASSSVQEAHDMALVAHLAAIESNVPFMHFFDGFRTSHEIQKIEVLDYADMASLVNQKALAEFRAKSPGIVAEYMQKVASLTGRSYKLFDYVGAPDAERVIVSMGSSCETIEEVINHLAAKGEKIGLIKVRLYRPFVSEAFFAALPASAKVITVLDRTKEPGAPGDPLYLDVCSAFVERGEAMPKILAGRYGLGSKEFSPAMVKSVYDNMSGAKKNHFTVGIEDDVTGTSLPVDNAFADTTPKGTIQCQFWGLGADGTVGANKQAIKIIGDNTDLFAQGYFSYDSKKSGGITISHLRFGEKPIQSTYLVNRADYVACHNPAYVGIYDILEGIKDGGTFVLNSPWSSLEDMDKHLPSGIKRTIANKKLKFYNIDAVKIATDVGLGGRINMIMQTAFFKLAGVLPFEKAVDLLKKSIHKAYGKKGEKIVKMNTDAVDQAVTSLQEFKYPDSWKDAPAETKAEPMTNEFFKNVVKPILTQQGDKLPVSAFEADGRFPLGTSQFEKRGVAINVPQWVPENCIQCNQCAFVCPHSAILPVLAKEEELVGAPANFTALEAKGKELKGYKFRIQINTLDCMGCGNCADICPPKEKALVMQPLDTQRDAQVPNLEYAARIPVKSEVLPRDSLKGSQFQEPLMEFSGACSGCGETPYVRVITQLFGERMFIANATGCSSIWGASAPSMPYKTNRLGQGPAWGNSLFEDAAEYGFGMSVWIFGGDGWAYDIGYGGLDHVLASGEDVNVFVMDTEVYSNTGGQSSKATPTGAVAKFAAAGKRTGKKDLARMVMTYGYVYVATVSMGYSKQQFLKVLKEAESFPGPSLVIAYATCINQGLRKGMGKSQDVMNTAVKSGYWPLFRYDPRLAAQGKNPFQLDSKAPDGSVEEFLMAQNRFAVLDRSFPEDAKRLRAQVAHELDVRFKELEHMAATNIFES Hydrogen bonds contact Hydrophobic contact | ||||
| 5 | SEC14-like protein 3 | 4UYB | 7.20 | |
Target general information Gen name SEC14L3 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms TAP2 Protein family NA Biochemical class Transport protein Function Lipid binding.Transporter activity. Related diseases Chondrodysplasia with platyspondyly, distinctive brachydactyly, hydrocephaly, and microphthalmia (CDP-PBHM) [MIM:300863]: A disease characterized by chondrodysplasia, severe platyspondyly, hydrocephaly, and facial features with microphthalmia. Bone abnormalities include a distinctive metaphyseal cupping of the metacarpals, metatarsals, and phalanges. Affected females show a milder phenotype with small stature, sometimes associated with body asymmetry and mild intellectual disability. {ECO:0000269|PubMed:20181727}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB14003; DB14001; DB14002; DB11635; DB11251; DB00163 Interacts with NA EC number NA Uniprot keywords 3D-structure; Alternative splicing; Lipid-binding; Proteomics identification; Reference proteome; Transport Protein physicochemical properties Chain ID A Molecular weight (Da) 46148.7 Length 401 Aromaticity 0.1 Instability index 45.19 Isoelectric point 5.79 Charge (pH=7) -5.94 2D Binding mode Binding energy (Kcal/mol) -9.82  Molscript Map 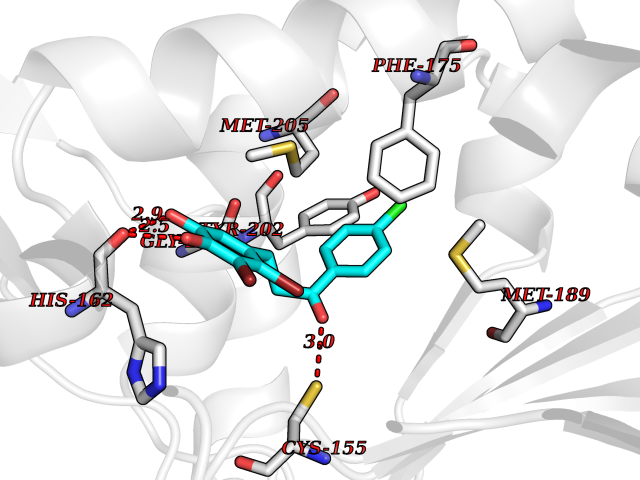 Pymol Map 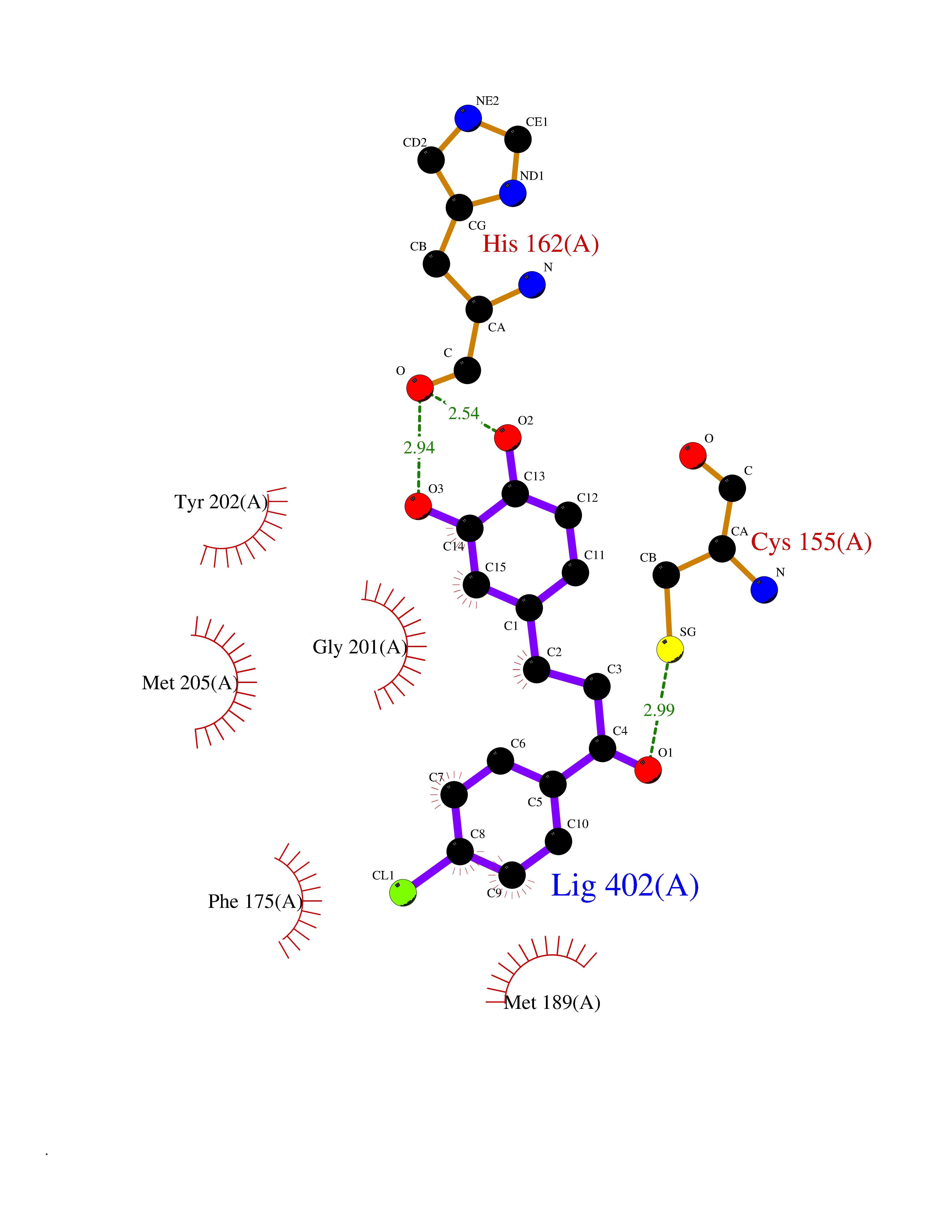 Ligplot Map 3D Binding mode Sequence SMSGRVGDLSPKQAETLAKFRENVQDVLPALPNPDDYFLLRWLRARNFDLQKSEALLRKYMEFRKTMDIDHILDWQPPEVIQKYMPGGLCGYDRDGCPVWYDIIGPLDPKGLLFSVTKQDLLKTKMRDCERILHECDLQTERLGKKIETIVMIFDCEGLGLKHFWKPLVEVYQEFFGLLEENYPETLKFMLIVKATKLFPVGYNLMKPFLSEDTRRKIIVLGNNWKEGLLKLISPEELPAQFGGTLTDPDGNPKCLTKINYGGEIPKSMYVRDQVKTQYEHSVQINRGSSHQVEYEILFPGCVLRWQFSSDGADIGFGVFLKTKMGERQRAGEMTEVLPSQRYNAHMVPEDGNLTCSEAGVYVLRFDNTYSFVHAKKVSFTVEVLLPDEGMQKYDKELTPV Hydrogen bonds contact Hydrophobic contact | ||||
| 6 | Plasmodium Dihydroorotate dehydrogenase (Malaria DHOdehase) | 1TV5 | 7.18 | |
Target general information Gen name Malaria DHOdehase Organism Plasmodium falciparum (isolate 3D7) Uniprot ID TTD ID Synonyms PFF0160c; Mitochondrially bound dihydroorotate-ubiqui oxidoreductase; Dihydroorotate oxidase of Plasmodium falciparum; Dihydroorotate dehydrogenase of Plasmodium falciparum; DHOdehase of Plasmodium fa Protein family Dihydroorotate dehydrogenase family, Type 2 subfamily Biochemical class CH-CH donor oxidoreductase Function Catalyzes the conversion of dihydroorotate to orotate with quinone as electron acceptor. Related diseases Combined oxidative phosphorylation deficiency 33 (COXPD33) [MIM:617713]: An autosomal recessive disorder caused by multiple mitochondrial respiratory chain defects and impaired mitochondrial energy metabolism. Clinical manifestations are highly variable. Affected infants present with cardiomyopathy accompanied by multisystemic features involving liver, kidney, and brain. Death in infancy is observed in some patients. Children and adults present with myopathy and progressive external ophthalmoplegia. {ECO:0000269|PubMed:28942965}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01117 Interacts with NA EC number EC 1.3.5.2 Uniprot keywords 3D-structure; Flavoprotein; FMN; Membrane; Mitochondrion; Mitochondrion inner membrane; Oxidoreductase; Pyrimidine biosynthesis; Reference proteome; Transit peptide; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 41846.8 Length 371 Aromaticity 0.1 Instability index 37.25 Isoelectric point 8.21 Charge (pH=7) 3.13 2D Binding mode Binding energy (Kcal/mol) -9.79 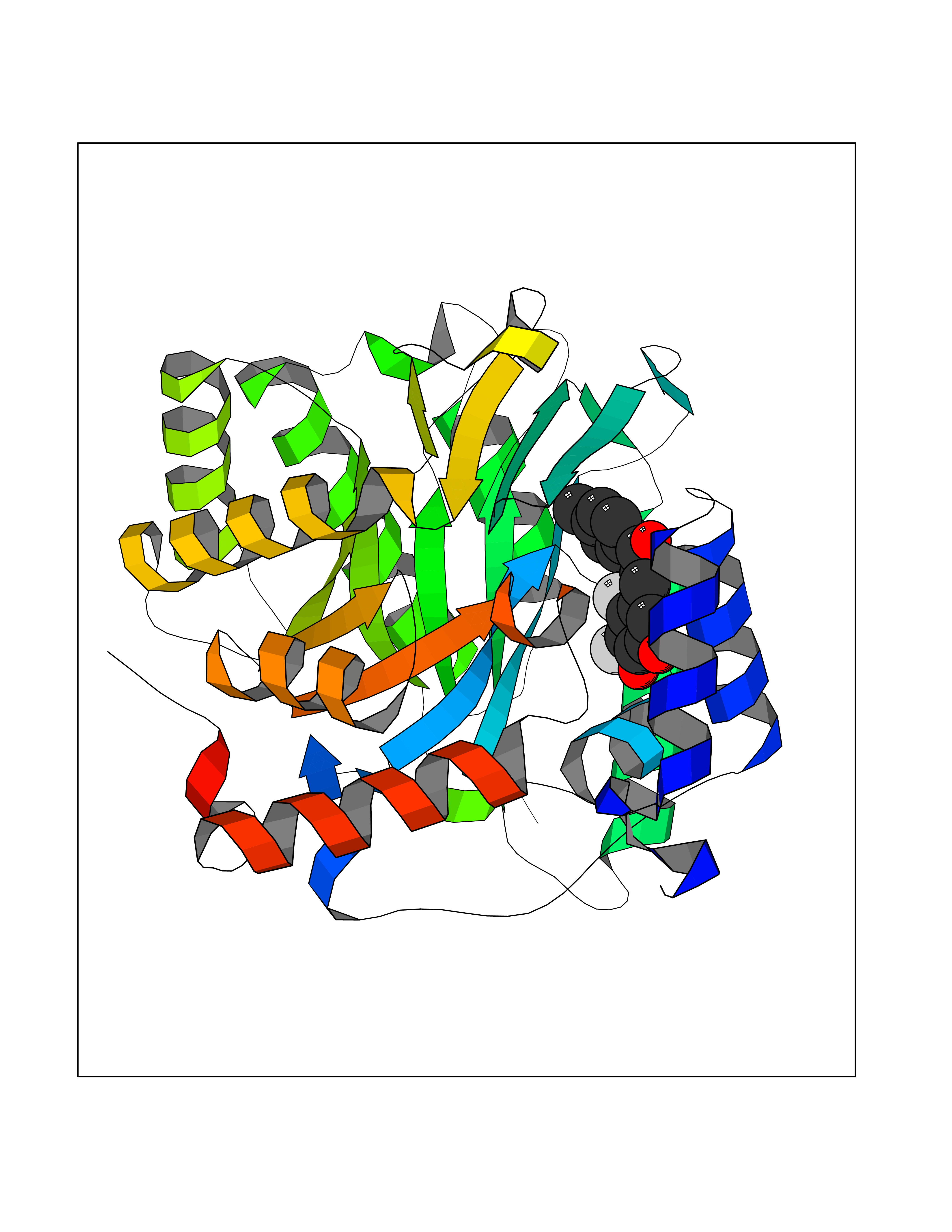 Molscript Map 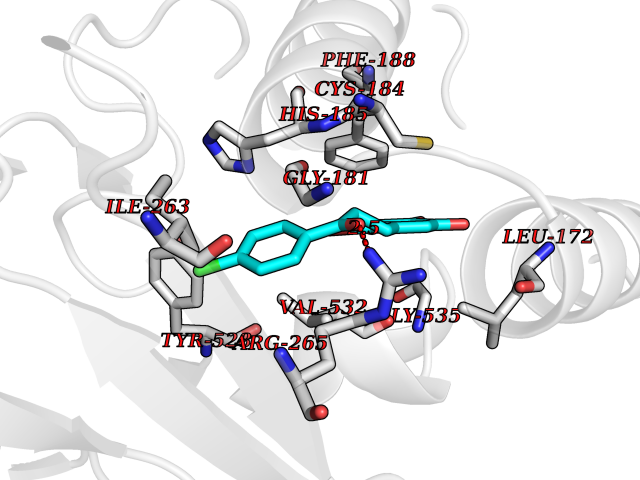 Pymol Map 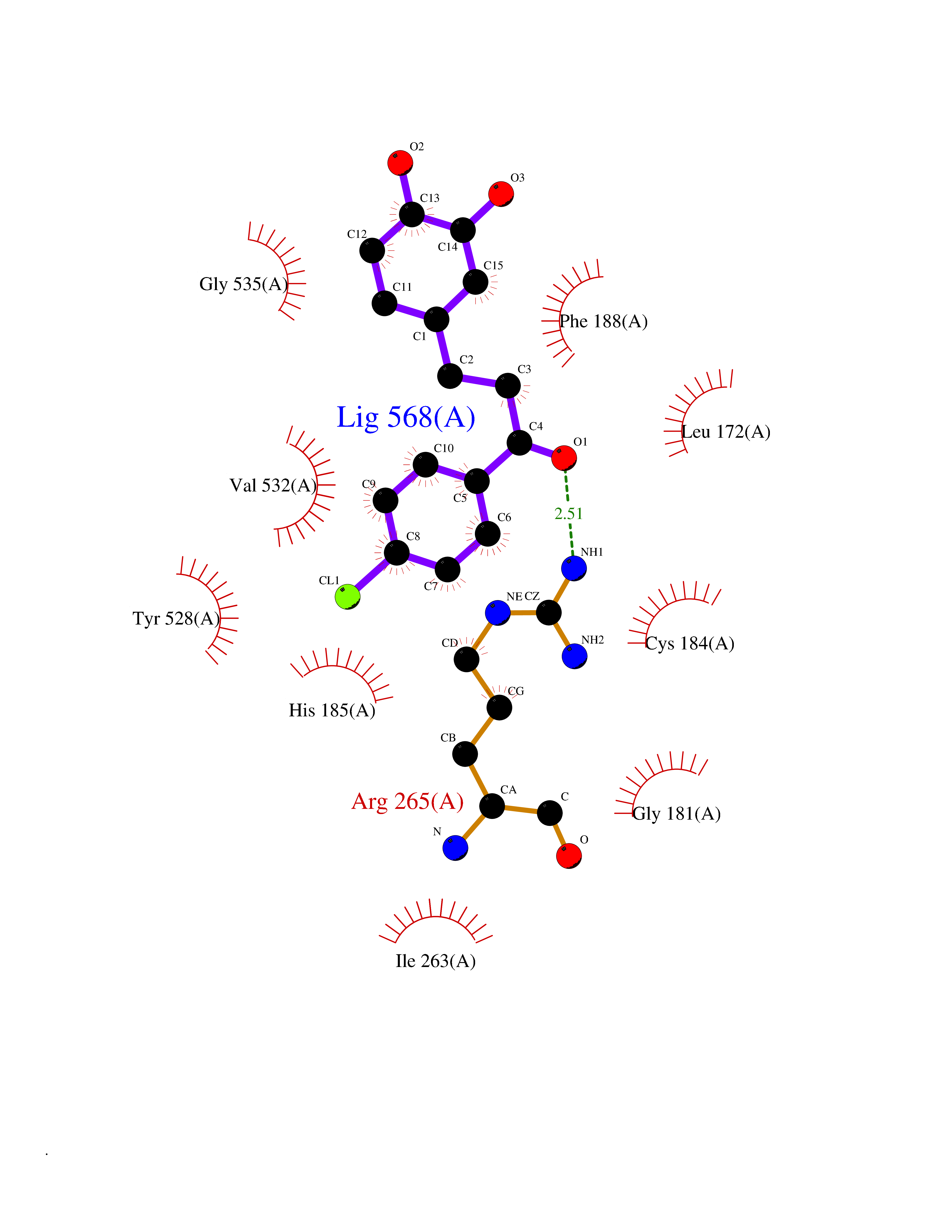 Ligplot Map 3D Binding mode Sequence FESYNPEFFLYDIFLKFCLKYIDGEICHDLFLLLGKYNILPYDTSNDSIYACTNIKHLDFINPFGVAAGFDKNGVCIDSILKLGFSFIEIGTITPRGQTGNAKPRIFRDVESRSIINSCGFNNMGCDKVTENLILFRKRQEEDKLLSKHIVGVSIGKNKDTVNIVDDLKYCINKIGRYADYIAINVSSPNTPGLRDNQEAGKLKNIILSVKEEIDNLEFLWFNTTKKKPLVFVKLAPDLNQEQKKEIADVLLETNIDGMIISNTTTQINDIKSFENKKGGVSGAKLKDISTKFICEMYNYTNKQIPIIASGGIFSGLDALEKIEAGASVCQLYSCLVFNGMKSAVQIKRELNHLLYQRGYYNLKEAIGRKH Hydrogen bonds contact Hydrophobic contact | ||||
| 7 | Glucose-dependent insulinotropic receptor (GPR119) | 7XZ6 | 7.18 | |
Target general information Gen name GPR119 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms GPR119; G-protein coupled receptor 119 Protein family G-protein coupled receptor 1 family Biochemical class GPCR rhodopsin Function Receptor for the endogenous fatty-acid ethanolamide oleoylethanolamide (OEA) and lysophosphatidylcholine (LPC). Functions as a glucose-dependent insulinotropic receptor. The activity of this receptor is mediated by G proteins which activate adenylate cyclase. Seems to act through a G(s) mediated pathway. Related diseases Developmental and epileptic encephalopathy 24 (DEE24) [MIM:615871]: A disease characterized by early-onset seizures, intellectual disability of varying degrees, and behavioral disturbances or autistic features in most individuals. {ECO:0000269|PubMed:24747641, ECO:0000269|PubMed:27864847, ECO:0000269|PubMed:30351409}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Generalized epilepsy with febrile seizures plus 10 (GEFSP10) [MIM:618482]: An autosomal dominant neurologic disorder with incomplete penetrance, characterized by variable types of seizures including absence, tonic-clonic, febrile, focal, and eyelid myoclonia. Some patients have normal neurologic development. Others have mild-to-moderate intellectual disability or autism spectrum disorder. {ECO:0000269|PubMed:29936235, ECO:0000269|PubMed:30351409}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB05166 Interacts with Q12797-6 EC number NA Uniprot keywords 3D-structure; Cell membrane; G-protein coupled receptor; Lipid-binding; Membrane; Receptor; Reference proteome; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID R Molecular weight (Da) 32134.1 Length 292 Aromaticity 0.12 Instability index 34.96 Isoelectric point 9.12 Charge (pH=7) 8.03 2D Binding mode Binding energy (Kcal/mol) -9.79 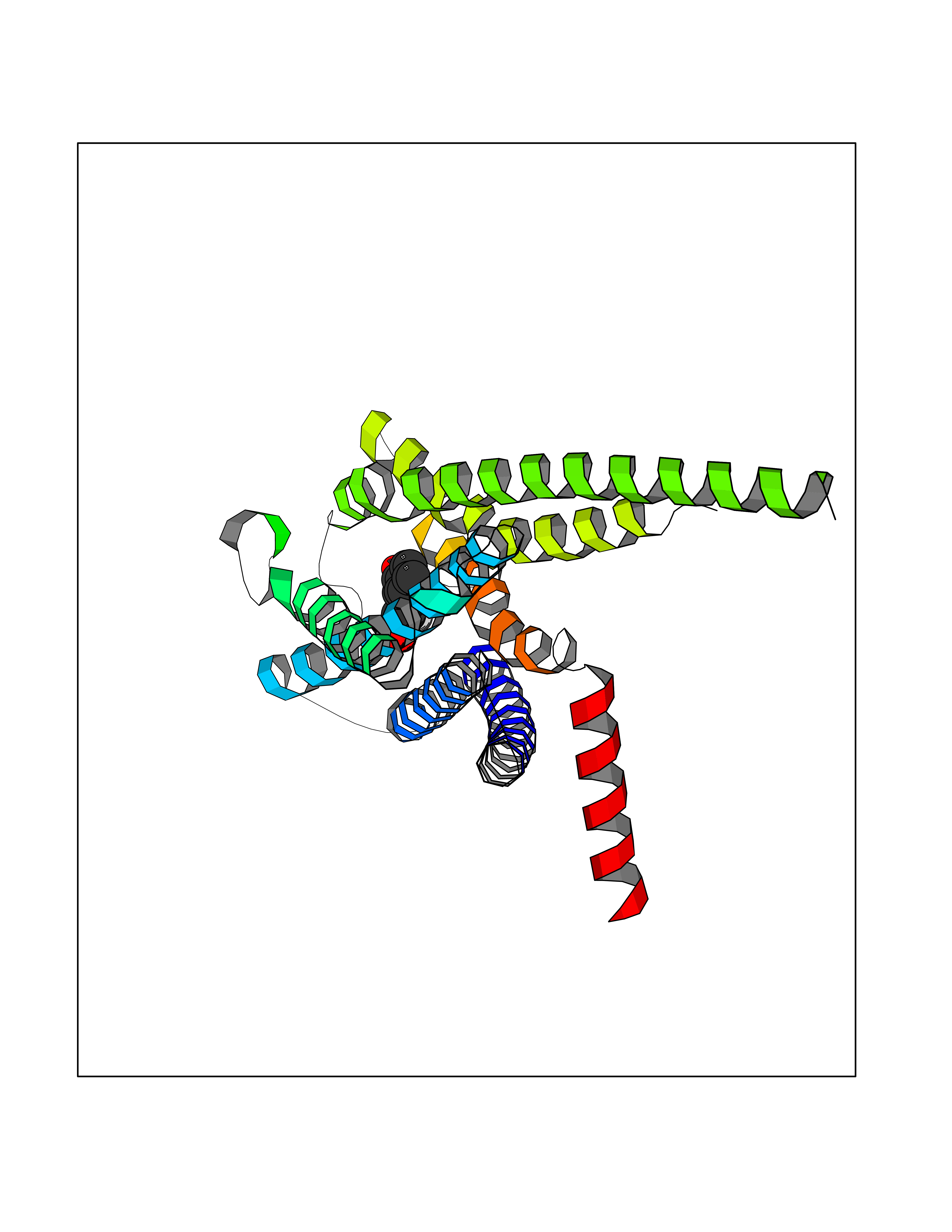 Molscript Map 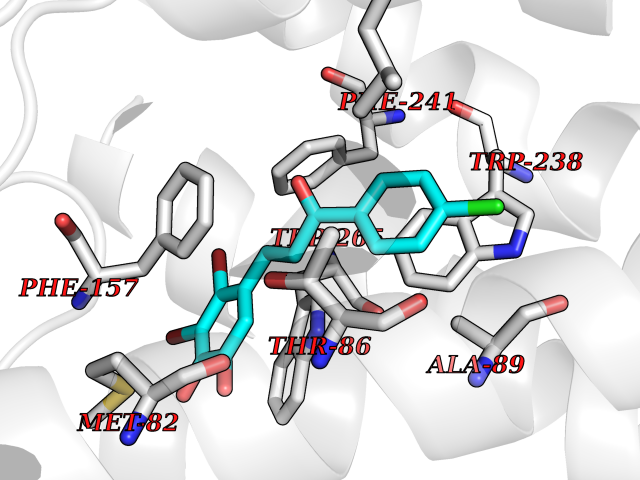 Pymol Map 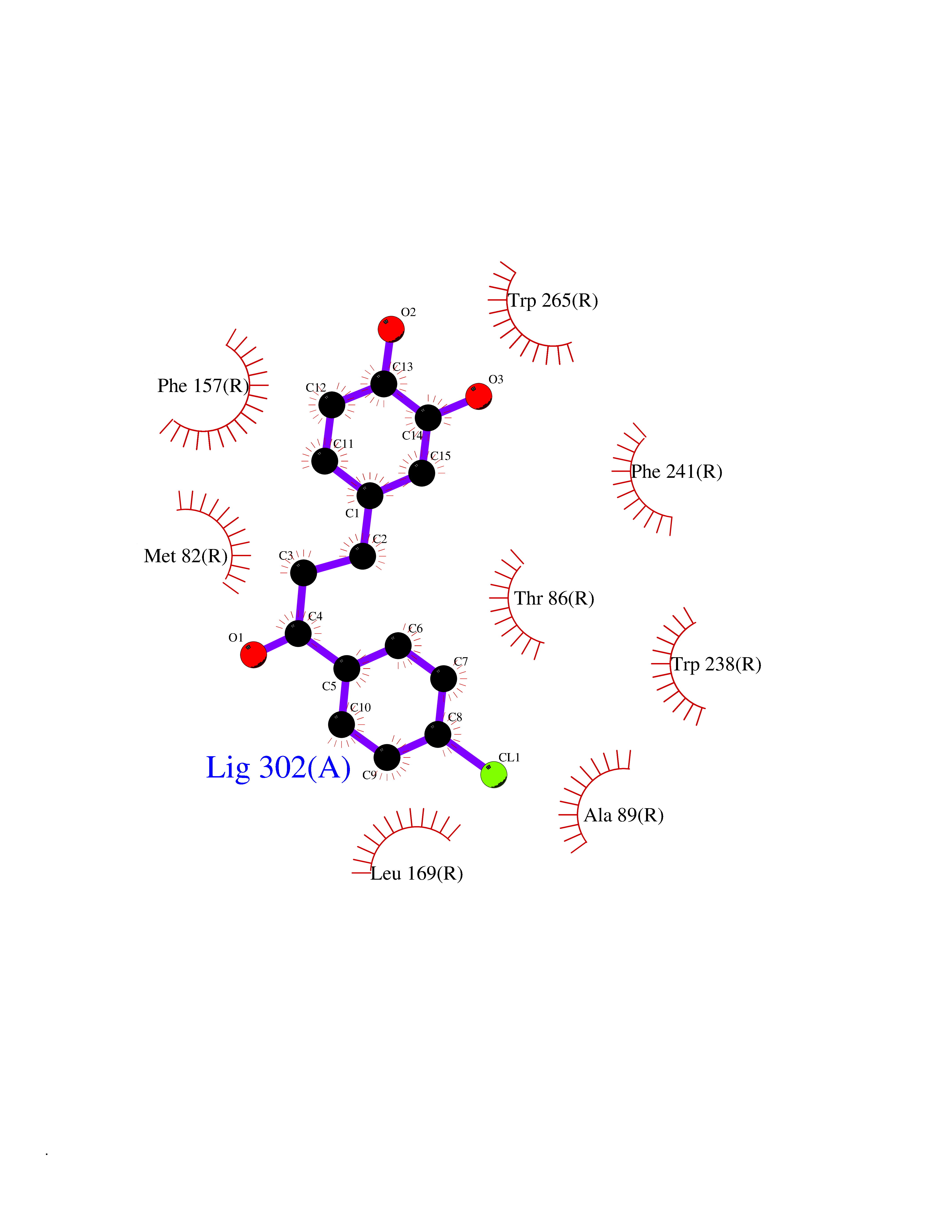 Ligplot Map 3D Binding mode Sequence MESSFSFGVILAVLASLIIATNTLVAVAVLLLIHKNDGVSLCFTLNLAVADTLIGVAISGLLTDQLSSPSRPTQKTLCSLRMAFVTSSAAASVLTVMLITFDRYLAIKQPFRYLKIMSGFVAGACIAGLWLVSYLIGFLPLGIPMFQQTAYKGQCSFFAVFHPHFVLTLSCVGFFPAMLLFVFFYCDMLKIASMHSQQIRKMEHAGAMAGSDFKALRTVSVLIGSFALSWTPFLITGIVQVACQECHLYLVLERYLWLLGVGNSLLNPLIYAYWQKEVRLQLYHMALGVKKV Hydrogen bonds contact Hydrophobic contact | ||||
| 8 | Phenylethanolamine N-methyltransferase (PNMT) | 2G72 | 7.15 | |
Target general information Gen name PNMT Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms PNMTase; PENT; Noradrenaline N-methyltransferase Protein family Class I-like SAM-binding methyltransferase superfamily, NNMT/PNMT/TEMT family Biochemical class NA Function Converts noradrenaline to adrenaline. Related diseases A chromosomal aberration involving TRIM24/TIF1 is found in papillary thyroid carcinomas (PTCs). Translocation t(7;10)(q32;q11) with RET. The translocation generates the TRIM24/RET (PTC6) oncogene. {ECO:0000269|PubMed:10439047}. Drugs (DrugBank ID) DB08129; DB08128; DB07739; DB07798; DB07747; DB03468; DB08550; DB03824; DB04273; DB07906; DB07597; DB09571; DB00968; DB08631; DB01752; DB08654 Interacts with Q9P2G9-2; Q8TBB1 EC number EC 2.1.1.28 Uniprot keywords 3D-structure; Catecholamine biosynthesis; Direct protein sequencing; Methyltransferase; Phosphoprotein; Proteomics identification; Reference proteome; S-adenosyl-L-methionine; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 29198.9 Length 264 Aromaticity 0.09 Instability index 54.33 Isoelectric point 5.91 Charge (pH=7) -3.69 2D Binding mode Binding energy (Kcal/mol) -9.76  Molscript Map 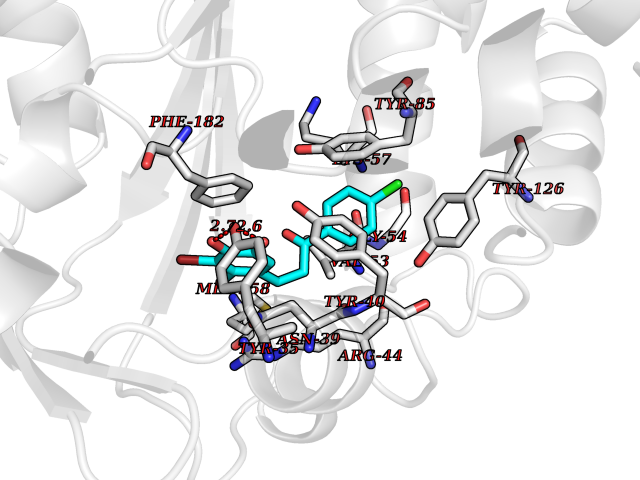 Pymol Map 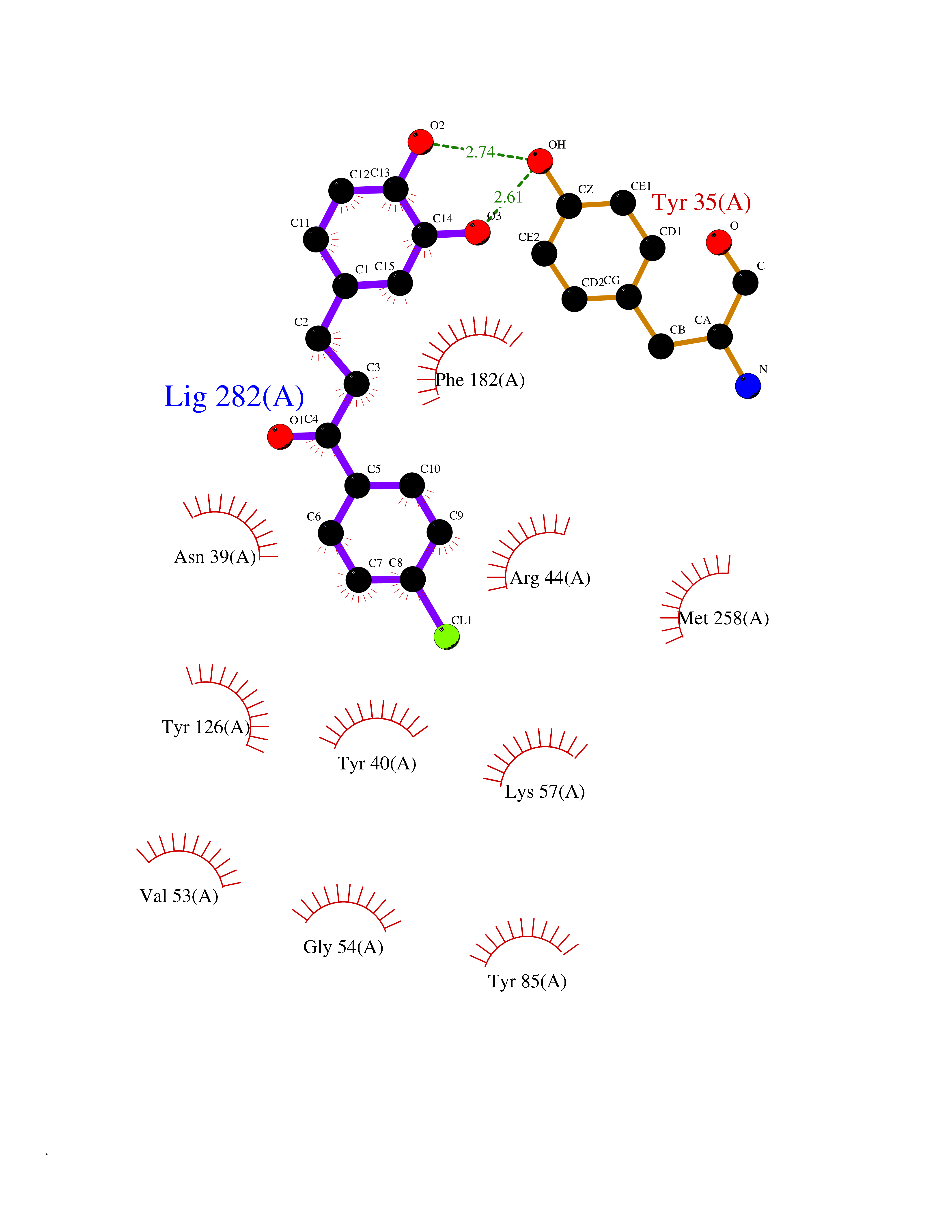 Ligplot Map 3D Binding mode Sequence APGQAAVASAYQRFEPRAYLRNNYAPPRGDLCNPNGVGPWKLRCLAQTFATGEVSGRTLIDIGSGPTVYQLLSACSHFEDITMTDFLEVNRQELGRWLQEEPGAFNWSMYSQHACLIEGKGECWQDKERQLRARVKRVLPIDVHQPQPLGAGSPAPLPADALVSAFCLEAVSPDLASFQRALDHITTLLRPGGHLLLIGALEESWYLAGEARLTVVPVSEEEVREALVRSGYKVRDLRTYIMPAHLQTGVDDVKGVFFAWAQKV Hydrogen bonds contact Hydrophobic contact | ||||
| 9 | Vascular endothelial growth factor receptor 2 (KDR) | 2XIR | 7.12 | |
Target general information Gen name KDR Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms VEGFR2; VEGFR-2; VEGF-2 receptor; Protein-tyrosine kinase receptor flk-1; Kinase insert domain receptor; Fetal liver kinase 1; FLK1; FLK-1; CD309 Protein family Protein kinase superfamily, Tyr protein kinase family, CSF-1/PDGF receptor subfamily Biochemical class Kinase Function Plays an essential role in the regulation of angiogenesis, vascular development, vascular permeability, and embryonic hematopoiesis. Promotes proliferation, survival, migration and differentiation of endothelial cells. Promotes reorganization of the actin cytoskeleton. Isoforms lacking a transmembrane domain, such as isoform 2 and isoform 3, may function as decoy receptors for VEGFA, VEGFC and/or VEGFD. Isoform 2 plays an important role as negative regulator of VEGFA- and VEGFC-mediated lymphangiogenesis by limiting the amount of free VEGFA and/or VEGFC and preventing their binding to FLT4. Modulates FLT1 and FLT4 signaling by forming heterodimers. Binding of vascular growth factors to isoform 1 leads to the activation of several signaling cascades. Activation of PLCG1 leads to the production of the cellular signaling molecules diacylglycerol and inositol 1,4,5-trisphosphate and the activation of protein kinase C. Mediates activation of MAPK1/ERK2, MAPK3/ERK1 and the MAP kinase signaling pathway, as well as of the AKT1 signaling pathway. Mediates phosphorylation of PIK3R1, the regulatory subunit of phosphatidylinositol 3-kinase, reorganization of the actin cytoskeleton and activation of PTK2/FAK1. Required for VEGFA-mediated induction of NOS2 and NOS3, leading to the production of the signaling molecule nitric oxide (NO) by endothelial cells. Phosphorylates PLCG1. Promotes phosphorylation of FYN, NCK1, NOS3, PIK3R1, PTK2/FAK1 and SRC. Tyrosine-protein kinase that acts as a cell-surface receptor for VEGFA, VEGFC and VEGFD. Related diseases Hemangioma, capillary infantile (HCI) [MIM:602089]: A condition characterized by dull red, firm, dome-shaped hemangiomas, sharply demarcated from surrounding skin, usually presenting at birth or occurring within the first two or three months of life. They result from highly proliferative, localized growth of capillary endothelium and generally undergo regression and involution without scarring. {ECO:0000269|PubMed:11807987, ECO:0000269|PubMed:18931684}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Plays a major role in tumor angiogenesis. In case of HIV-1 infection, the interaction with extracellular viral Tat protein seems to enhance angiogenesis in Kaposi's sarcoma lesions. Drugs (DrugBank ID) DB04727; DB07514; DB07528; DB06938; DB07326; DB06626; DB08875; DB04849; DB05198; DB12147; DB12307; DB12010; DB11679; DB06101; DB09078; DB06080; DB06595; DB07537; DB07183; DB07333; DB07334; DB07274; DB09079; DB08519; DB08042; DB16265; DB06589; DB05931; DB08901; DB15822; DB05984; DB05578; DB08896; DB14840; DB06436; DB00398; DB01268; DB05075; DB11800; DB04879; DB05146; DB05014 Interacts with P35916; O60565; P98160; PRO_0000391621 [P98160]; PRO_0000391622 [P98160]; P17301; P35968; P09382; P08581; P16333; O14786; O75340; P09619; P29350; Q12913; P12931; P15692; P15692-4; P49767; Q9MYV3-3 EC number EC 2.7.10.1 Uniprot keywords 3D-structure; Alternative splicing; Angiogenesis; ATP-binding; Cell junction; Cell membrane; Cytoplasm; Cytoplasmic vesicle; Developmental protein; Differentiation; Disulfide bond; Endoplasmic reticulum; Endosome; Glycoprotein; Host-virus interaction; Immunoglobulin domain; Kinase; Membrane; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Repeat; Secreted; Signal; Transferase; Transmembrane; Transmembrane helix; Tyrosine-protein kinase; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 33863.9 Length 296 Aromaticity 0.1 Instability index 40.01 Isoelectric point 8.5 Charge (pH=7) 4.59 2D Binding mode Binding energy (Kcal/mol) -9.72 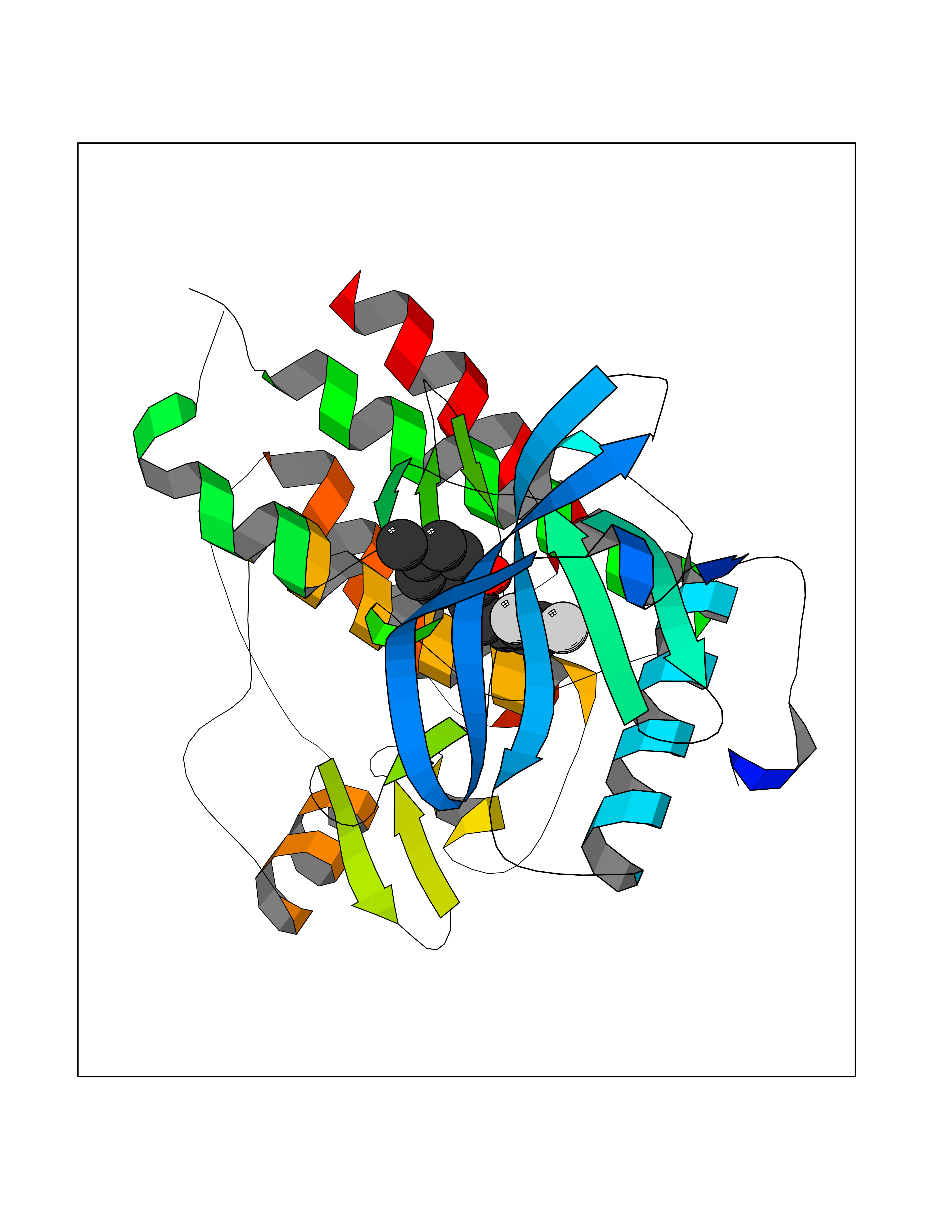 Molscript Map 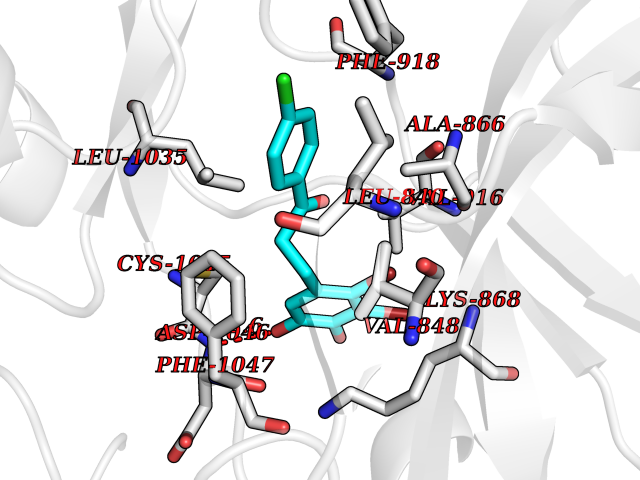 Pymol Map 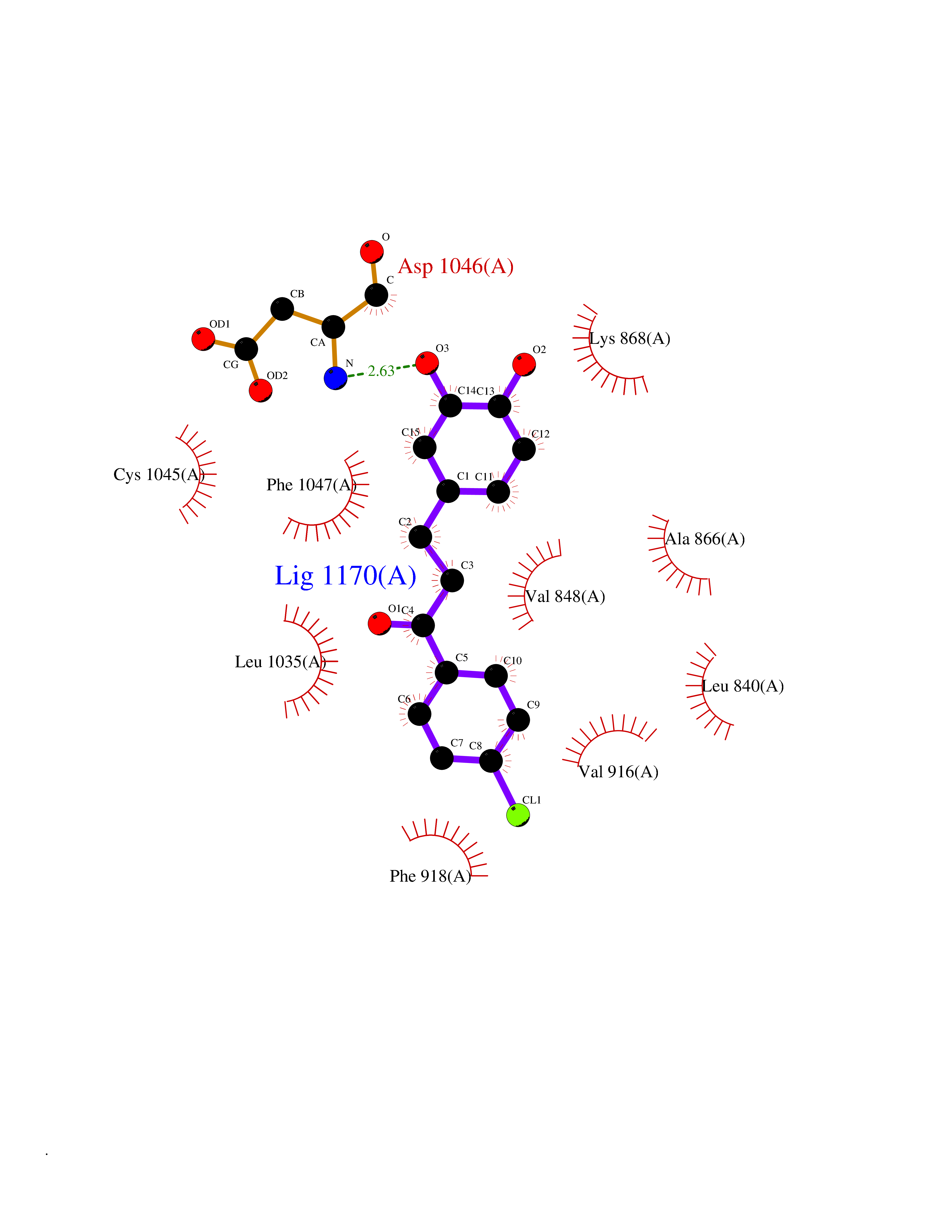 Ligplot Map 3D Binding mode Sequence HCERLPYDASKWEFPRDRLKLGKPLGRGAFGQVIEADAFGIDKTATCRTVAVKMLKEGATHSEHRALMSELKILIHIGHHLNVVNLLGACTKPGGPLMVIVEFCKFGNLSTYLRSKRNEFVPYYKDFLTLEHLICYSFQVAKGMEFLASRKCIHRDLAARNILLSEKNVVKICDFGLARDIYKDPDYVRKGDARLPLKWMAPETIFDRVYTIQSDVWSFGVLLWEIFSLGASPYPGVKIDEEFCRRLKEGTRMRAPDYTTPEMYQTMLDCWHGEPSQRPTFSELVEHLGNLLQANA Hydrogen bonds contact Hydrophobic contact | ||||
| 10 | Cytochrome c oxidase subunit 2 | 3VRJ | 7.11 | |
Target general information Gen name MT-CO2 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms MTCO2;COXII;COII;COX2 Protein family Cytochrome c oxidase subunit 2 family Biochemical class Immune system Function Copper ion binding.Cytochrome-c oxidase activity. Related diseases Mitochondrial complex IV deficiency (MT-C4D) [MIM:220110]: A disorder of the mitochondrial respiratory chain with heterogeneous clinical manifestations, ranging from isolated myopathy to severe multisystem disease affecting several tissues and organs. Features include hypertrophic cardiomyopathy, hepatomegaly and liver dysfunction, hypotonia, muscle weakness, exercise intolerance, developmental delay, delayed motor development and intellectual disability. Some affected individuals manifest a fatal hypertrophic cardiomyopathy resulting in neonatal death. A subset of patients manifest Leigh syndrome. {ECO:0000269|PubMed:10486321}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02659; DB04464; DB05412 Interacts with Q9NZ94-2; P49281-3 EC number 7.1.1.9 Uniprot keywords 3D-structure; Copper; Disease variant; Electron transport; Magnesium; Membrane; Metal-binding; Mitochondrion; Mitochondrion inner membrane; Primary mitochondrial disease; Proteomics identification; Reference proteome; Respiratory chain; Translocase; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID C Molecular weight (Da) 21687.9 Length 189 Aromaticity 0.11 Instability index 38 Isoelectric point 5.68 Charge (pH=7) -3.26 2D Binding mode Binding energy (Kcal/mol) -9.69 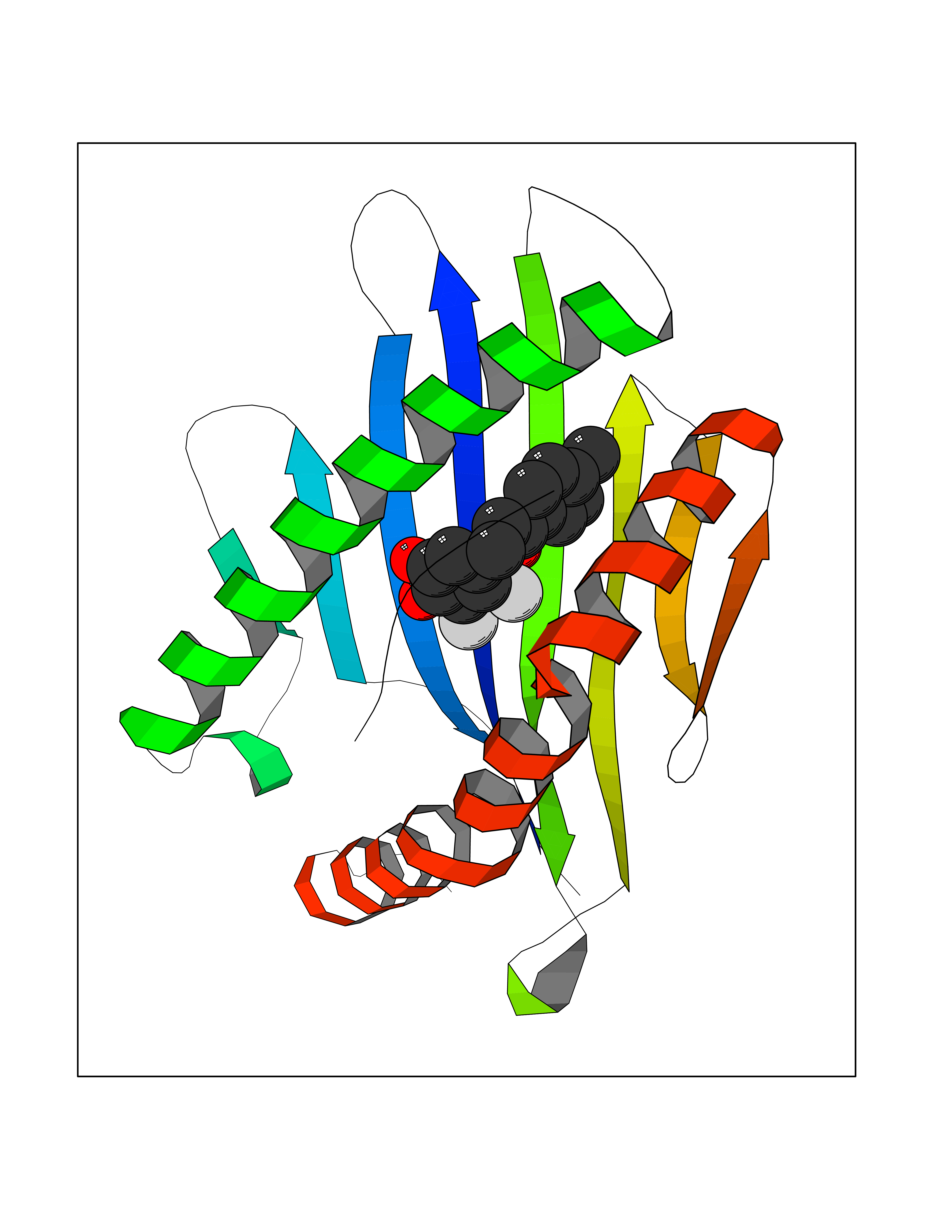 Molscript Map 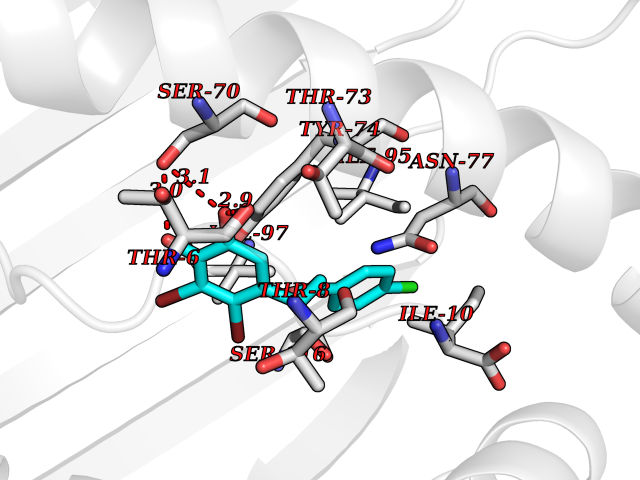 Pymol Map 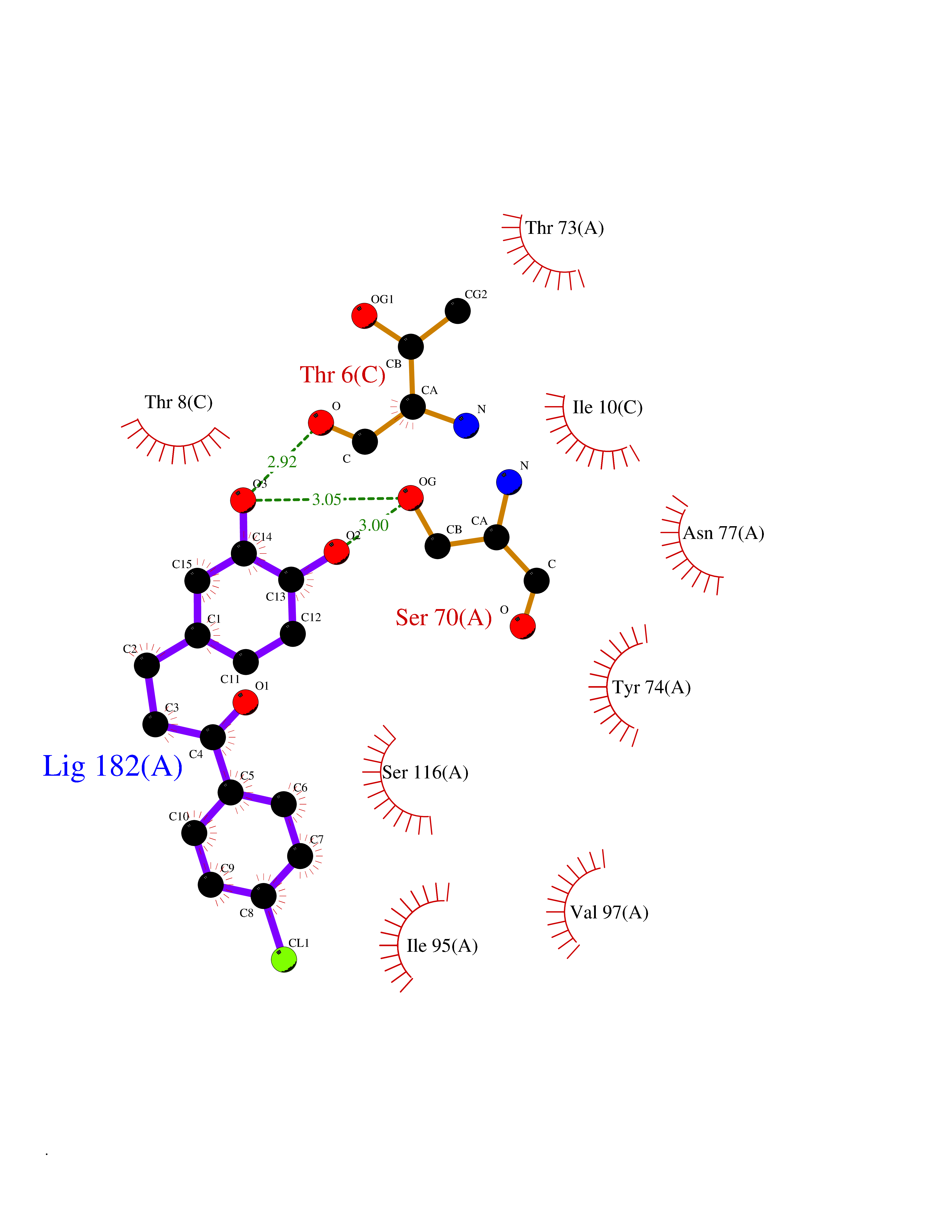 Ligplot Map 3D Binding mode Sequence SHSMRYFYTAMSRPGRGEPRFIAVGYVDDTQFVRFDSDAASPRMAPRAPWIEQEGPEYWDGETRNMKASAQTYRENLRIALRYYNQSEAGSHIIQVMYGCDVGPDGRLLRGHDQSAYDGKDYIALNEDLSSWTAADTAAQITQRKWEAARVAEQLRAYLEGLCVEWLRRYLENGKETLQLTTKLTNTNI Hydrogen bonds contact Hydrophobic contact | ||||
| 11 | Caspase-7 (CASP7) | 1SHJ | 7.11 | |
Target general information Gen name CASP7 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms MCH3; ICE-like apoptotic protease 3; ICE-LAP3; CMH-1; CASP-7; Apoptotic protease Mch-3 Protein family Peptidase C14A family Biochemical class Peptidase Function Cleaves and activates sterol regulatory element binding proteins (SREBPs). Proteolytically cleaves poly(ADP-ribose) polymerase (PARP) at a '216-Asp-|-Gly-217' bond. Overexpression promotes programmed cell death. Involved in the activation cascade of caspases responsible for apoptosis execution. Related diseases Pregnancy loss, recurrent, 3 (RPRGL3) [MIM:614391]: A common complication of pregnancy, resulting in spontaneous abortion before the fetus has reached viability. The term includes all miscarriages from the time of conception until 24 weeks of gestation. Recurrent pregnancy loss is defined as 3 or more consecutive spontaneous abortions. {ECO:0000269|PubMed:17339269}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB05408; DB03384; DB06255 Interacts with Q13490; P83105; P42858; Q8N4N3-2; P43364; Q16236; Q9GZT8; Q13177; P27986-2; P21673; Q86WV1-2; P17405; P98170 EC number EC 3.4.22.60 Uniprot keywords 3D-structure; Acetylation; Allosteric enzyme; Alternative splicing; Apoptosis; Cytoplasm; Hydrolase; Nucleus; Phosphoprotein; Protease; Proteomics identification; Reference proteome; RNA-binding; Secreted; Thiol protease; Ubl conjugation; Zymogen Protein physicochemical properties Chain ID A,B Molecular weight (Da) 47441.5 Length 417 Aromaticity 0.11 Instability index 20.98 Isoelectric point 8.38 Charge (pH=7) 6.12 2D Binding mode Binding energy (Kcal/mol) -9.7  Molscript Map 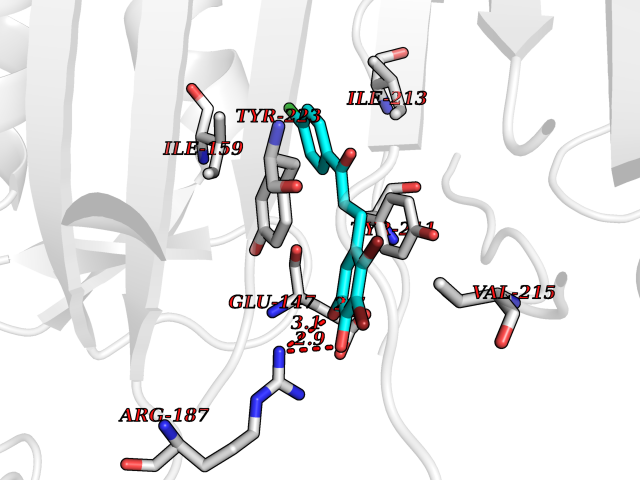 Pymol Map 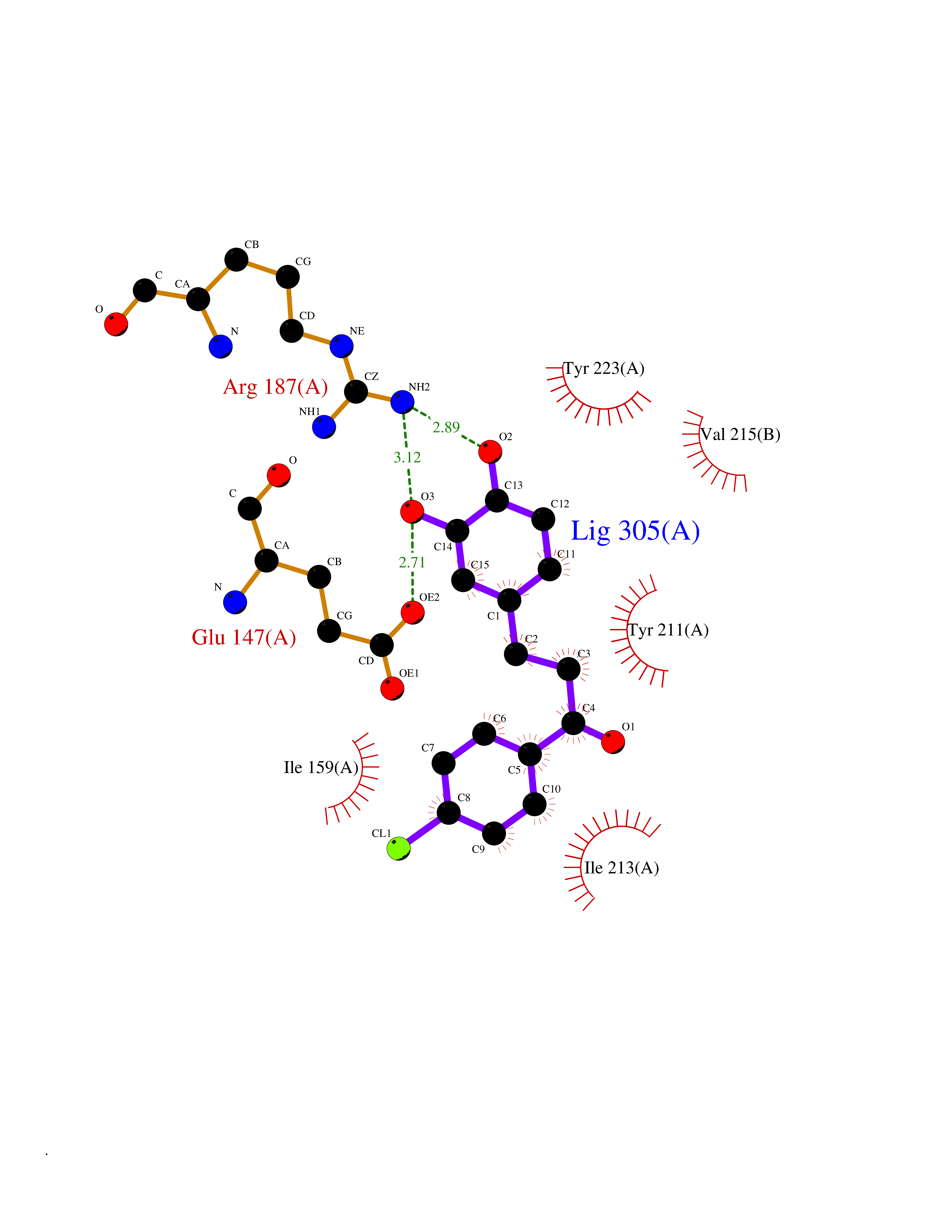 Ligplot Map 3D Binding mode Sequence TYQYNMNFEKLGKCIIINNKNFDKVTGMGVRNGTDKDAEALFKCFRSLGFDVIVYNDCSCAKMQDLLKKASEEDHTNAACFACILLSHGEENVIYGKDGVTPIKDLTAHFRGARCKTLLEKPKLFFIQACRGTEPRYKIPVEADFLFAYSTVRGSWFVQALCSILEEHGKDLEIMQILTRVNDRVARHFKKQIPCVVSMLTKELYFSQVPTYQYNMNFEKLGKCIIINNKNFDKVTGMGVRNGTDKDAEALFKCFRSLGFDVIVYNDCSCAKMQDLLKKASEEDHTNAACFACILLSHGEENVIYGKDGVTPIKDLTAHFRGARCKTLLEKPKLFFIQACRGPRYKIPVEADFLFAYSTVPGSWFVQALCSILEEHGKDLEIMQILTRVNDRVARHFESKQIPCVVSMLTKELYFSQ Hydrogen bonds contact Hydrophobic contact | ||||
| 12 | Neprilysin | 1R1H | 7.09 | |
Target general information Gen name MME Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms EPN Protein family Peptidase M13 family Biochemical class Hydrolase Function Endopeptidase activity.Exopeptidase activity.Metalloendopeptidase activity.Metallopeptidase activity.Peptide binding.Zinc ion binding. Related diseases Charcot-Marie-Tooth disease, axonal, 2T (CMT2T) [MIM:617017]: An axonal form of Charcot-Marie-Tooth disease, a disorder of the peripheral nervous system, characterized by progressive weakness and atrophy, initially of the peroneal muscles and later of the distal muscles of the arms. Charcot-Marie-Tooth disease is classified in two main groups on the basis of electrophysiologic properties and histopathology: primary peripheral demyelinating neuropathies (designated CMT1 when they are dominantly inherited) and primary peripheral axonal neuropathies (CMT2). Neuropathies of the CMT2 group are characterized by signs of axonal degeneration in the absence of obvious myelin alterations, normal or slightly reduced nerve conduction velocities, and progressive distal muscle weakness and atrophy. {ECO:0000269|PubMed:26991897, ECO:0000269|PubMed:27588448}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Spinocerebellar ataxia 43 (SCA43) [MIM:617018]: A form of spinocerebellar ataxia, a clinically and genetically heterogeneous group of cerebellar disorders. Patients show progressive incoordination of gait and often poor coordination of hands, speech and eye movements, due to degeneration of the cerebellum with variable involvement of the brainstem and spinal cord. SCA43 is a slowly progressive, autosomal dominant form. {ECO:0000269|PubMed:27583304}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08575; DB02597; DB00616; DB11623; DB05796; DB06655; DB02558; DB02062; DB00886; DB02557; DB09292; DB13928; DB08626 Interacts with P05067; P21926; Q06787-7; P08107; P04792 EC number 3.4.24.11 Uniprot keywords 3D-structure; Cell membrane; Charcot-Marie-Tooth disease; Disease variant; Disulfide bond; Glycoprotein; Hydrolase; Lipoprotein; Membrane; Metal-binding; Metalloprotease; Myristate; Neurodegeneration; Neuropathy; Phosphoprotein; Protease; Proteomics identification; Reference proteome; Signal-anchor; Spinocerebellar ataxia; Transmembrane; Transmembrane helix; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 79435.8 Length 696 Aromaticity 0.11 Instability index 37.5 Isoelectric point 5.53 Charge (pH=7) -11.46 2D Binding mode Binding energy (Kcal/mol) -9.67  Molscript Map 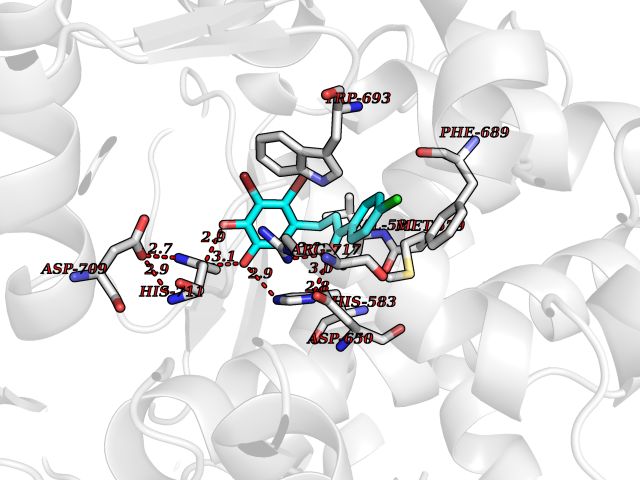 Pymol Map 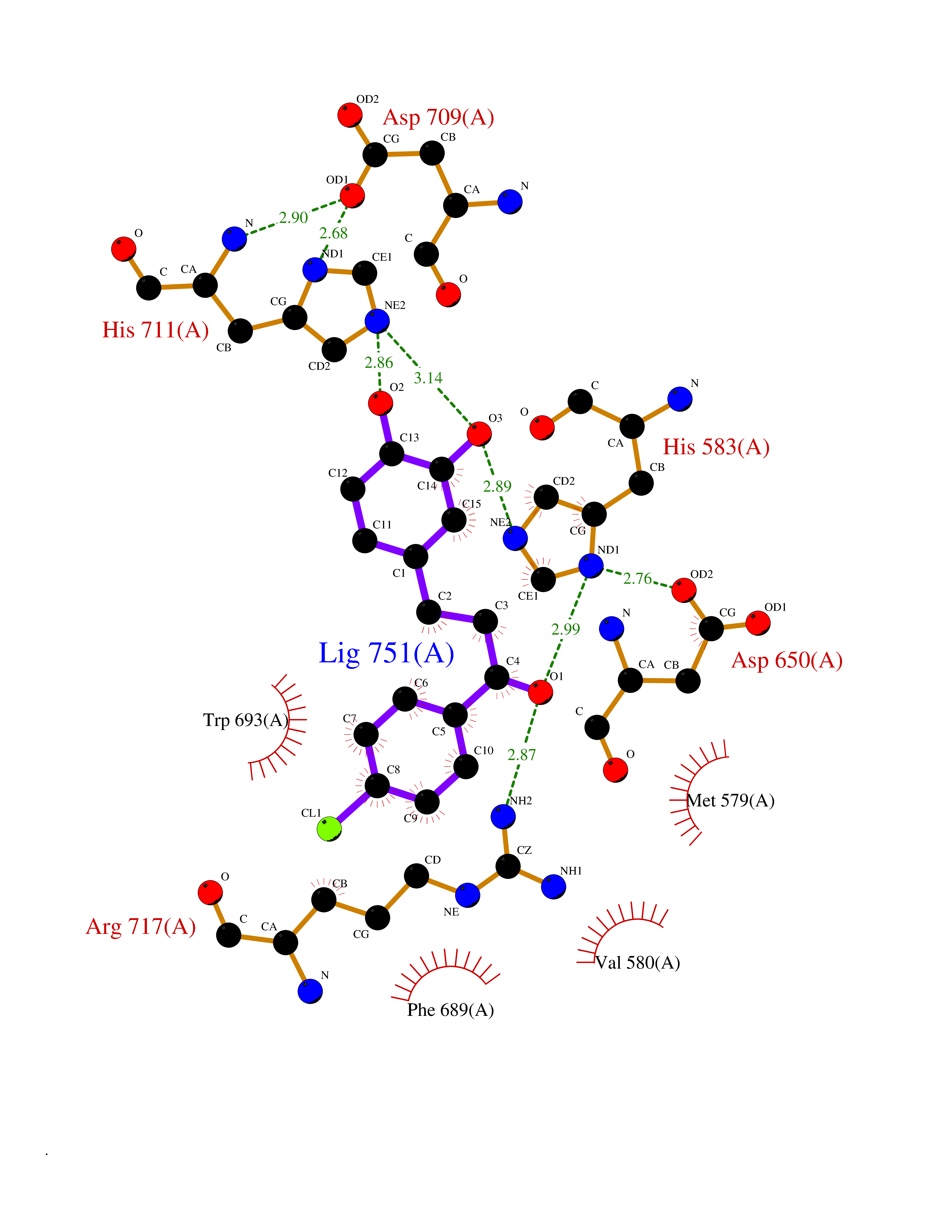 Ligplot Map 3D Binding mode Sequence GICKSSDCIKSAARLIQNMDATTEPCTDFFKYACGGWLKRNVIPETSSRYGNFDILRDELEVVLKDVLQEPKTEDIVAVQKAKALYRSCINESAIDSRGGEPLLKLLPDIYGWPVATENWEQKYGASWTAEKAIAQLNSKYGKKVLINLFVGTDDKNSVNHVIHIDQPRLGLPSRDYYECTGIYKEACTAYVDFMISVARLIRQEERLPIDENQLALEMNKVMELEKEIANATAKPEDRNDPMLLYNKMTLAQIQNNFSLEINGKPFSWLNFTNEIMSTVNISITNEEDVVVYAPEYLTKLKPILTKYSARDLQNLMSWRFIMDLVSSLSRTYKESRNAFRKALYGTTSETATWRRCANYVNGNMENAVGRLYVEAAFAGESKHVVEDLIAQIREVFIQTLDDLTWMDAETKKRAEEKALAIKERIGYPDDIVSNDNKLNNEYLELNYKEDEYFENIIQNLKFSQSKQLKKLREKVDKDEWISGAAVVNAFYSSGRNQIVFPAGILQPPFFSAQQSNSLNYGGIGMVIGHEITHGFDDNGRNFNKDGDLVDWWTQQSASNFKEQSQCMVYQYGNFSWDLAGGQHLNGINTLGENIADNGGLGQAYRAYQNYIKKNGEEKLLPGLDLNHKQLFFLNFAQVWCGTYRPEYAVNSIKTDVHSPGNFRIIGTLQNSAEFSEAFHCRKNSYMNPEKKCRVW Hydrogen bonds contact Hydrophobic contact | ||||
| 13 | 3-oxoacyl-[acyl-carrier-protein] synthase 2 | 3I8P | 7.09 | |
Target general information Gen name fabF Organism Escherichia coli (strain K12) Uniprot ID TTD ID NA Synonyms JW1081;b1095;fabJ Protein family Thiolase-like superfamily, Beta-ketoacyl-ACP synthases family Biochemical class Transferase Function 3-oxoacyl-[acyl-carrier-protein] synthase activity.Beta-ketoacyl-acyl-carrier-protein synthase II activity. Related diseases Pigmentary disorder, reticulate, with systemic manifestations, X-linked (PDR) [MIM:301220]: An X-linked recessive disorder characterized by recurrent infections and sterile inflammation in various organs. Diffuse skin hyperpigmentation with a distinctive reticulate pattern is universally evident by early childhood. This is later followed in many patients by hypohidrosis, corneal inflammation and scarring, enterocolitis that resembles inflammatory bowel disease, and recurrent urethral strictures. Melanin and amyloid deposition is present in the dermis. Affected males also have a characteristic facies with frontally upswept hair and flared eyebrows. Female carriers have only restricted pigmentary changes along Blaschko's lines. {ECO:0000269|PubMed:27019227}. The disease is caused by variants affecting the gene represented in this entry. XLPDR is caused by a recurrent intronic mutation that results in missplicing and reduced POLA1 expression. This leads to a decrease in cytosolic RNA:DNA hybrids and constitutive activation of type I interferon responses, but has no effect on cell replication. {ECO:0000269|PubMed:27019227}.; DISEASE: Van Esch-O'Driscoll syndrome (VEODS) [MIM:301030]: An X-linked recessive syndrome characterized by different degrees of intellectual disability, moderate to severe short stature, microcephaly, hypogonadism, and variable congenital malformations. {ECO:0000269|PubMed:31006512}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08366; DB01034; DB03017; DB08407 Interacts with P0A6Y8 EC number 2.3.1.179 Uniprot keywords 3D-structure; Acyltransferase; Direct protein sequencing; Fatty acid biosynthesis; Fatty acid metabolism; Lipid biosynthesis; Lipid metabolism; Reference proteome; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 42794.9 Length 411 Aromaticity 0.06 Instability index 31.41 Isoelectric point 5.72 Charge (pH=7) -7.34 2D Binding mode Binding energy (Kcal/mol) -9.67  Molscript Map 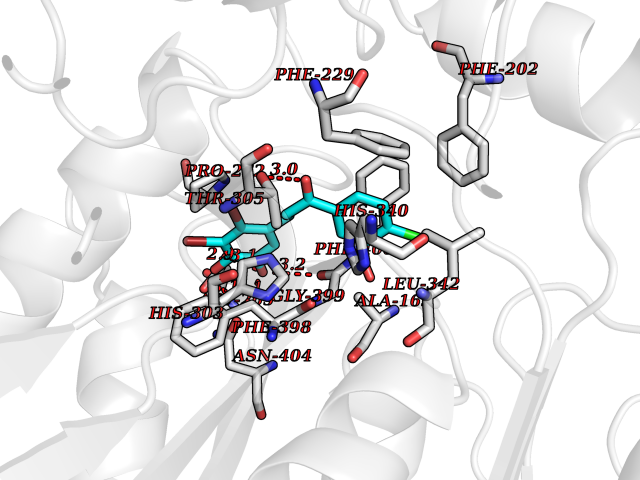 Pymol Map 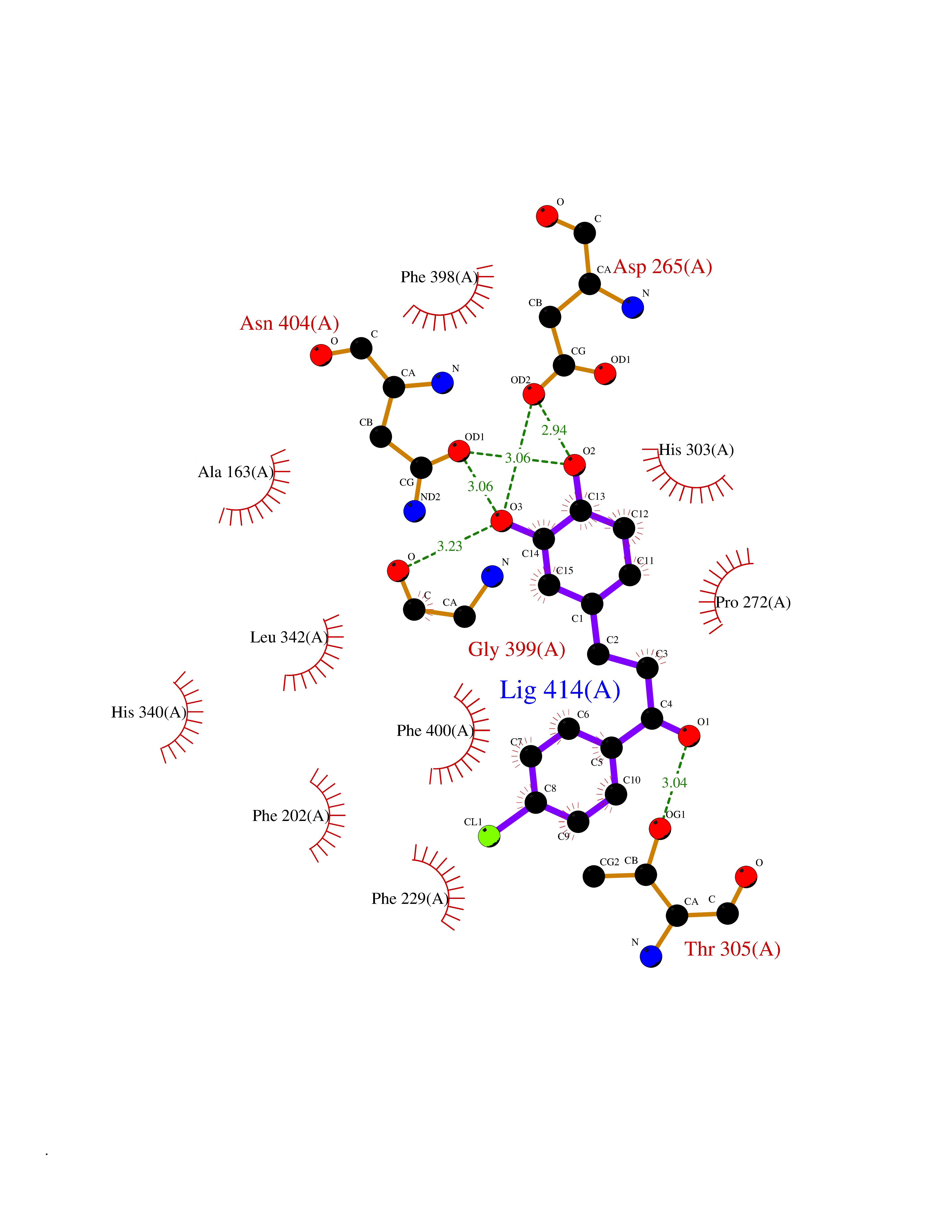 Ligplot Map 3D Binding mode Sequence KRRVVVTGLGMLSPVGNTVESTWKALLAGQSGISLIDHFDTSAYATKFAGLVKDFNCEDIISRKEQRKMDAFIQYGIVAGVQAMQDSGLEITEENATRIGAAIGSGIGGLGLIEENHTSLMNGGPRKISPFFVPSTIVNMVAGHLTIMYGLRGPSISIATAATSGVHNIGHAARIIAYGDADVMVAGGAEKASTPLGVGGFGAARALSTRNDNPQAASRPWDKERDGFVLGDGAGMLVLEEYEHAKKRGAKIYAELVGFGMSSDAYHMTSPPENGAGAALAMANALRDAGIEASQIGYVNAHGTSTPAGDKAEAQAVKTIFGEAASRVLVSSTKSMTGHLLGAAGAVESIYSILALRDQAVPPTINLDNPDEGCDLDFVPHEARQVSGMEYTLCNSFGFGGTNGSLIFKKI Hydrogen bonds contact Hydrophobic contact | ||||
| 14 | Spermine synthase | 3C6K | 7.09 | |
Target general information Gen name SMS Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Spermidine/spermine synthase family Biochemical class Transferase Function Spermine synthase activity. Related diseases Intellectual developmental disorder, X-linked, syndromic, Snyder-Robinson type (MRXSSR) [MIM:309583]: An X-linked intellectual disability syndrome characterized by a collection of clinical features including facial asymmetry, marfanoid habitus, hypertonia, osteoporosis and unsteady gait. {ECO:0000269|PubMed:14508504, ECO:0000269|PubMed:18550699, ECO:0000269|PubMed:19206178, ECO:0000269|PubMed:22612257, ECO:0000269|PubMed:23696453, ECO:0000269|PubMed:23897707}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00127 Interacts with NA EC number 2.5.1.22 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Direct protein sequencing; Intellectual disability; Phosphoprotein; Polyamine biosynthesis; Proteomics identification; Reference proteome; Transferase Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 27177.1 Length 238 Aromaticity 0.11 Instability index 41.31 Isoelectric point 4.82 Charge (pH=7) -9.19 2D Binding mode Binding energy (Kcal/mol) -9.68  Molscript Map 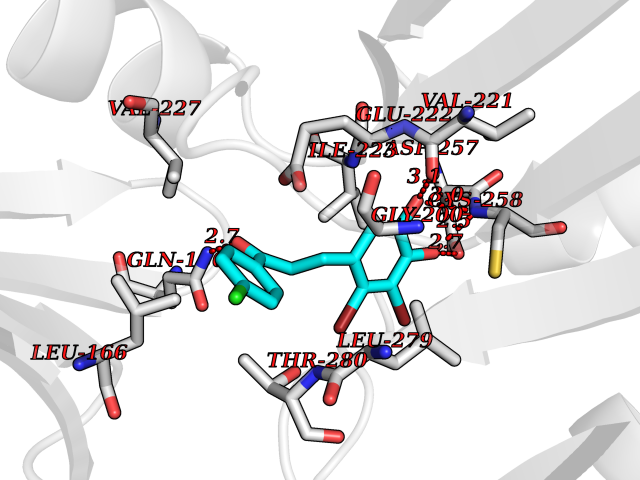 Pymol Map 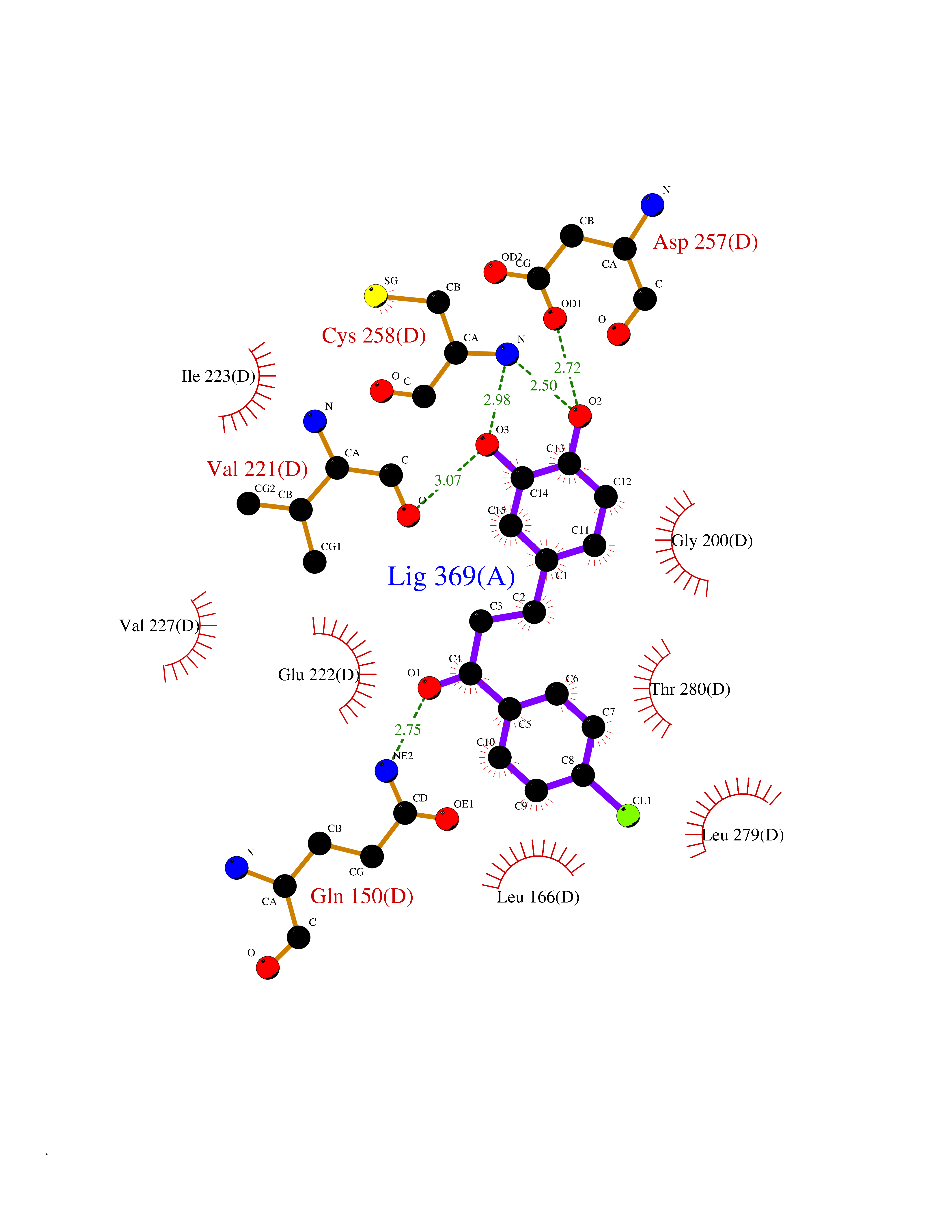 Ligplot Map 3D Binding mode Sequence RYWPTADGRLVEYDIDEVVYDEDSPYQNIKILHSKQFGNILILSGDVNLAESDLAYTRAIMGSGKEDYTGKDVLILGGGDGGILCEIVKLKPKMVTMVEIDQMVIDGCKKYMRKDVLDNLKGDCYQVLIEDCIPVLKRYAKEGREFDYVINDLTAVPISTSPSTWEFLRLILDLSMKVLKQDGKYFTQGNCVNLTEALSLYEEQLGRLYCPVEFSKEIVCVPSYLELWVFYTVWKKAK Hydrogen bonds contact Hydrophobic contact | ||||
| 15 | Monoamine oxidase type B (MAO-B) | 2V5Z | 7.08 | |
Target general information Gen name MAOB Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms MAO-B; Amine oxidase [flavin-containing] B Protein family Flavin monoamine oxidase family Biochemical class CH-NH(2) donor oxidoreductase Function Catalyzes the oxidative deamination of biogenic and xenobiotic amines and has important functions in the metabolism of neuroactive and vasoactive amines in the central nervous system and peripheral tissues. MAOB preferentially degrades benzylamine and phenylethylamine. Related diseases Microvascular complications of diabetes 5 (MVCD5) [MIM:612633]: Pathological conditions that develop in numerous tissues and organs as a consequence of diabetes mellitus. They include diabetic retinopathy, diabetic nephropathy leading to end-stage renal disease, and diabetic neuropathy. Diabetic retinopathy remains the major cause of new-onset blindness among diabetic adults. It is characterized by vascular permeability and increased tissue ischemia and angiogenesis. Disease susceptibility is associated with variants affecting the gene represented in this entry. Homozygosity for the Leu-55 allele is strongly associated with the development of retinal disease in diabetic patients. Drugs (DrugBank ID) DB08176; DB02211; DB08516; DB08480; DB01472; DB04307; DB07512; DB07513; DB00915; DB00182; DB06698; DB04889; DB00215; DB09130; DB04147; DB00988; DB01363; DB00668; DB01175; DB02509; DB03147; DB14914; DB00614; DB04818; DB02095; DB01247; DB00601; DB01577; DB01442; DB01171; DB08082; DB02643; DB04677; DB03894; DB08804; DB04820; DB00184; DB04821; DB12612; DB01626; DB00780; DB00191; DB00388; DB01132; DB00721; DB01168; DB01367; DB09363; DB06654; DB01037; DB01104; DB14569; DB09042; DB00752; DB16446; DB09185; DB04832; DB00909 Interacts with P55212; P28329-3; Q8NI60; Q5RI15; Q92915-2; P22607; Q53GS7; P06396; P01112; O14901; P13473-2; P21397; Q9BVL2; O75400-2; P62826; Q6NTF9-3; Q9Y371; Q7Z699; Q9UMX0; Q9Y649 EC number EC 1.4.3.4 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Direct protein sequencing; FAD; Flavoprotein; Membrane; Mitochondrion; Mitochondrion outer membrane; Oxidoreductase; Proteomics identification; Reference proteome; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A,B Molecular weight (Da) 56019.9 Length 494 Aromaticity 0.09 Instability index 34.81 Isoelectric point 6.51 Charge (pH=7) -2.2 2D Binding mode Binding energy (Kcal/mol) -9.66 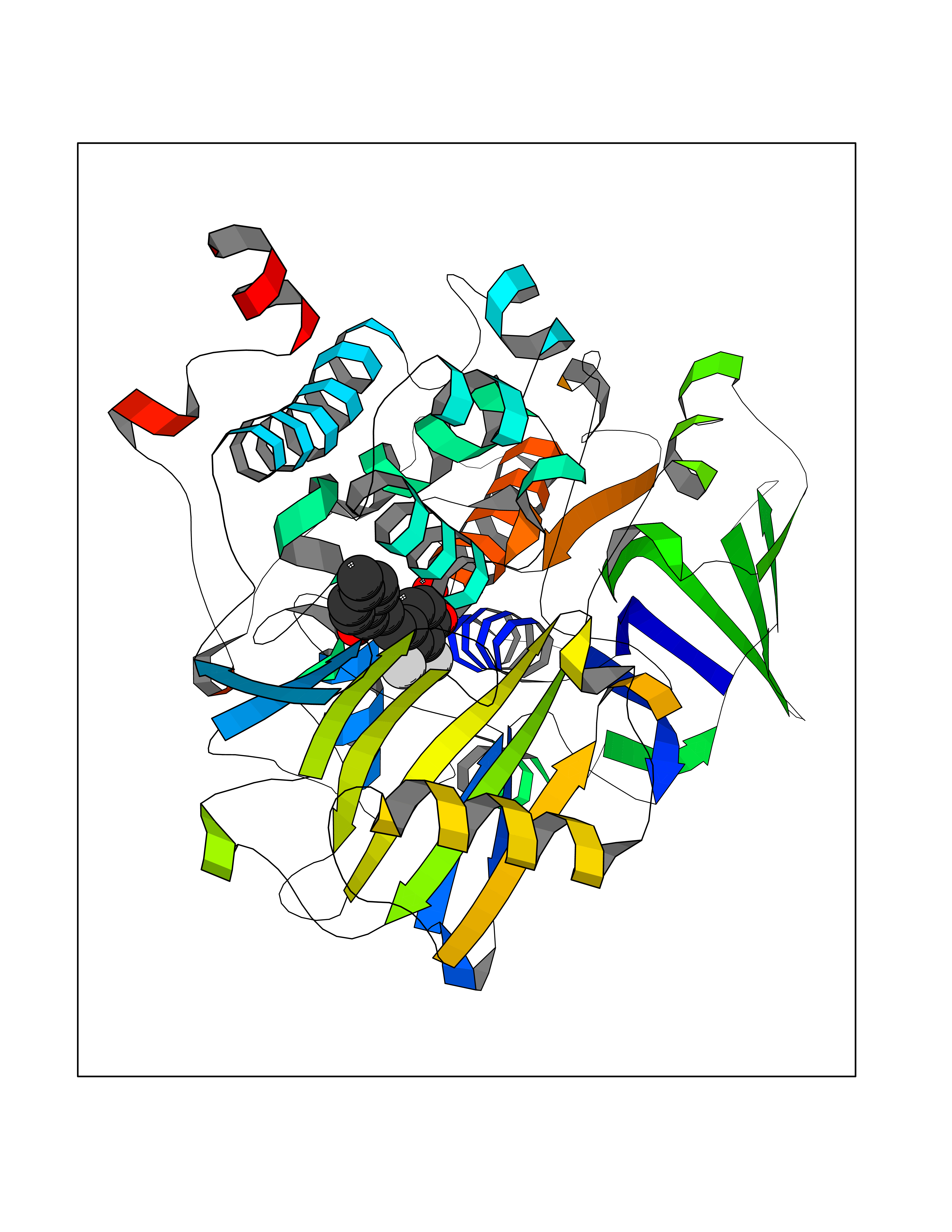 Molscript Map 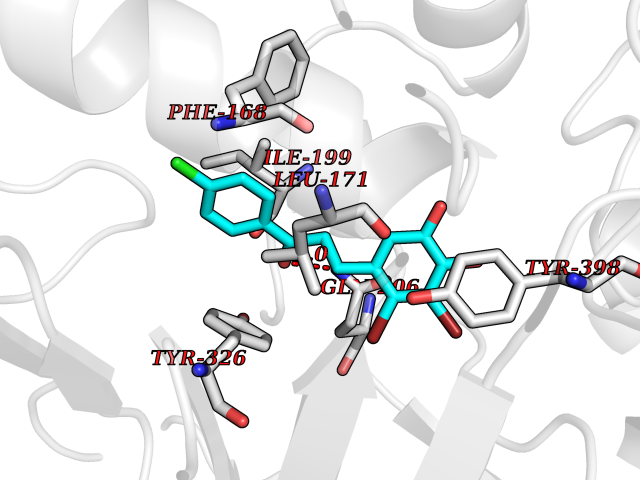 Pymol Map 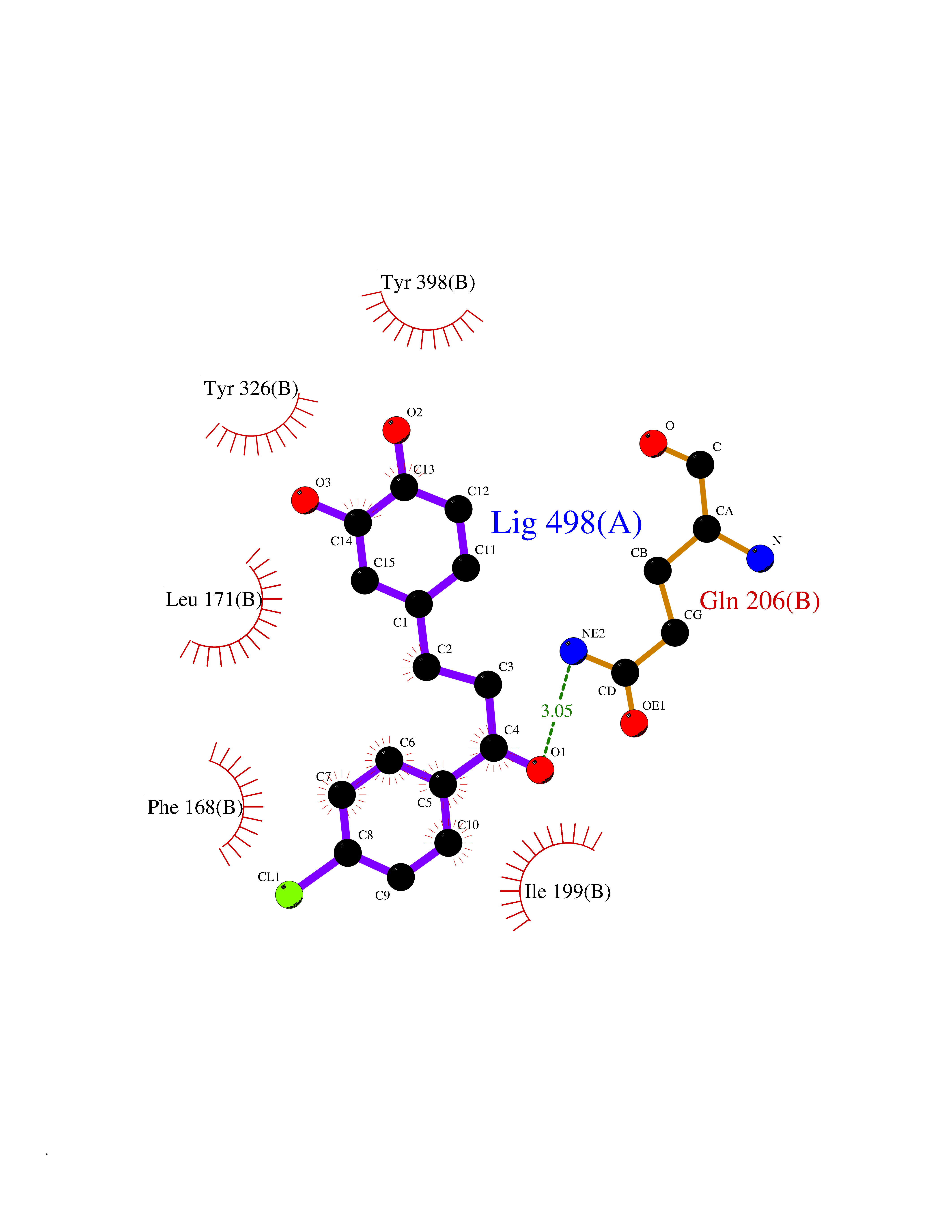 Ligplot Map 3D Binding mode Sequence NKCDVVVVGGGISGMAAAKLLHDSGLNVVVLEARDRVGGRTYTLRNQKVKYVDLGGSYVGPTQNRILRLAKELGLETYKVNEVERLIHHVKGKSYPFRGPFPPVWNPITYLDHNNFWRTMDDMGREIPSDAPWKAPLAEEWDNMTMKELLDKLCWTESAKQLATLFVNLCVTAETHEVSALWFLWYVKQCGGTTRIISTTNGGQERKFVGGSGQVSERIMDLLGDRVKLERPVIYIDQTRENVLVETLNHEMYEAKYVISAIPPTLGMKIHFNPPLPMMRNQMITRVPLGSVIKCIVYYKEPFWRKKDYCGTMIIDGEEAPVAYTLDDTKPEGNYAAIMGFILAHKARKLARLTKEERLKKLCELYAKVLGSLEALEPVHYEEKNWCEEQYSGGCYTTYFPPGILTQYGRVLRQPVDRIYFAGTETATHWSGYMEGAVEAGERAAREILHAMGKIPEDEIWQSEPESVDVPAQPITTTFLERHLPSVPGLLRLI Hydrogen bonds contact Hydrophobic contact | ||||
| 16 | Dihydroorotate dehydrogenase (quinone), mitochondrial | 4CQ8 | 7.08 | |
Target general information Gen name PFF0160c Organism Plasmodium falciparum (isolate 3D7) Uniprot ID TTD ID NA Synonyms NA Protein family Dihydroorotate dehydrogenase family, Type 2 subfamily Biochemical class Oxidoreductase Function Dihydroorotate dehydrogenase activity. Related diseases Combined oxidative phosphorylation deficiency 33 (COXPD33) [MIM:617713]: An autosomal recessive disorder caused by multiple mitochondrial respiratory chain defects and impaired mitochondrial energy metabolism. Clinical manifestations are highly variable. Affected infants present with cardiomyopathy accompanied by multisystemic features involving liver, kidney, and brain. Death in infancy is observed in some patients. Children and adults present with myopathy and progressive external ophthalmoplegia. {ECO:0000269|PubMed:28942965}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01117 Interacts with NA EC number 1.3.5.2 Uniprot keywords 3D-structure; Flavoprotein; FMN; Membrane; Mitochondrion; Mitochondrion inner membrane; Oxidoreductase; Pyrimidine biosynthesis; Reference proteome; Transit peptide; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A,B Molecular weight (Da) 42573.5 Length 378 Aromaticity 0.1 Instability index 36.63 Isoelectric point 8.21 Charge (pH=7) 3.17 2D Binding mode Binding energy (Kcal/mol) -9.65 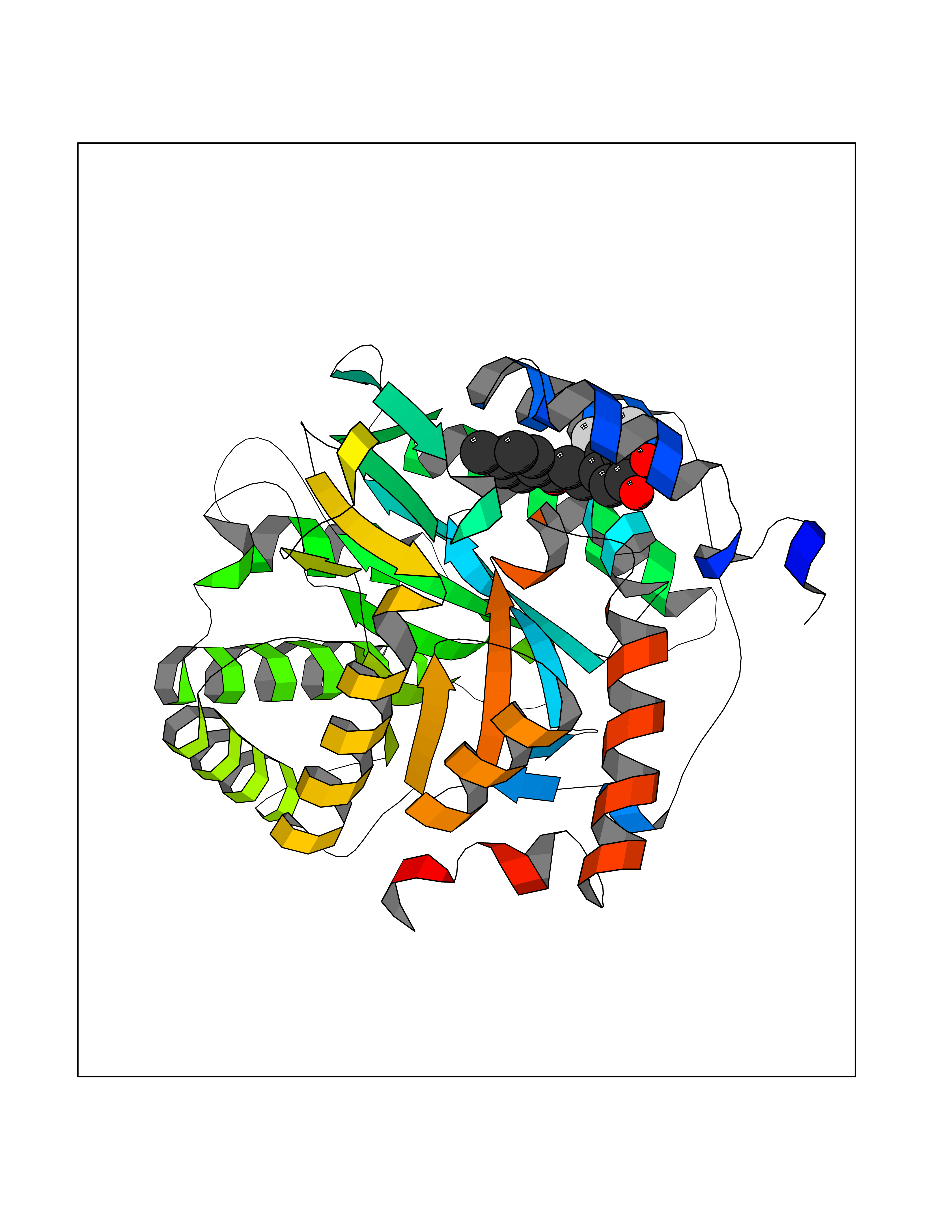 Molscript Map 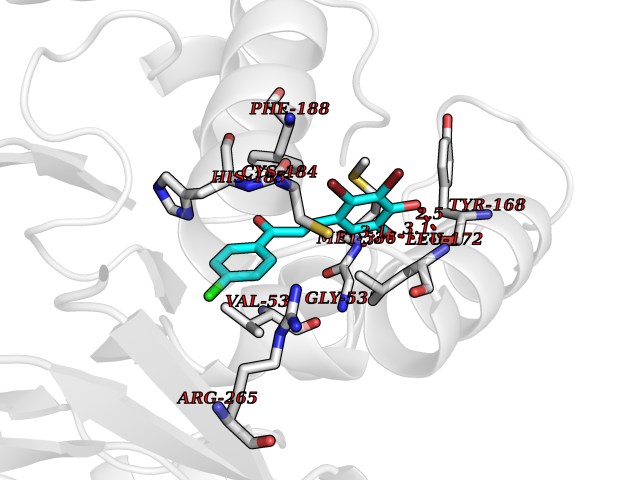 Pymol Map 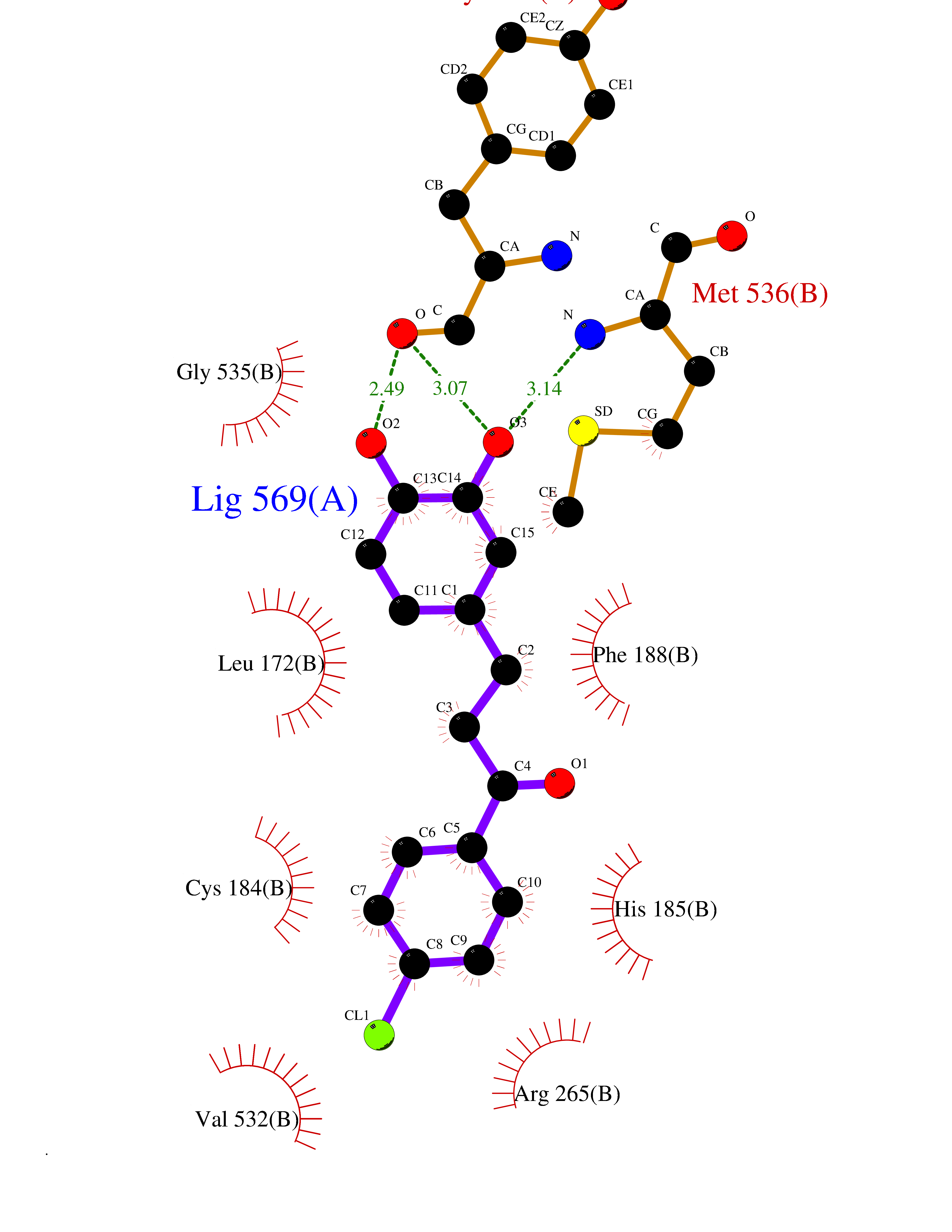 Ligplot Map 3D Binding mode Sequence ADPFESYNPEFFLYDIFLKFCLKYIDGEICHDLFLLLGKYNILPYDTSNDSIYACTNIKHLDFINPFGVAAGFDKNGVCIDSILKLGFSFIEIGTITPRGQTGNAKPRIFRDVESRSIINSCGFNNMGCDKVTENLILFRKRQEEDKLLSKHIVGVSIGKNKDTVNIVDDLKYCINKIGRYADYIAINVSSPNTPGLRDNQEAGKLKNIILSVKEEIDNLEKNNFLWFNTTKKKPLVFVKLAPDLNQEQKKEIADVLLETNIDGMIISNTTTQINDIKSFENKKGGVSGAKLKDISTKFICEMYNYTNKQIPIIASGGIFSGLDALEKIEAGASVCQLYSCLVFNGMKSAVQIKRELNHLLYQRGYYNLKEAIGRKHS Hydrogen bonds contact Hydrophobic contact | ||||
| 17 | Pseudomonas Transcriptional activator protein LasR (Pseudo LasR) | 3IX3 | 7.08 | |
Target general information Gen name Pseudo LasR Organism Pseudomonas aeruginosa (strain ATCC 15692 / DSM 22644 / CIP 104116 / JCM 14847 / LMG 12228 / 1C / PRS 101 / PAO1) Uniprot ID TTD ID Synonyms NA Protein family Autoinducer-regulated transcriptional regulatory protein family Biochemical class NA Function Transcriptional activator of elastase structural gene (LasB). Binds to the PAI autoinducer. Related diseases Growth hormone deficiency, isolated, 1A (IGHD1A) [MIM:262400]: An autosomal recessive, severe deficiency of growth hormone leading to dwarfism. Patients often develop antibodies to administered growth hormone. {ECO:0000269|PubMed:8364549}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Growth hormone deficiency, isolated, 1B (IGHD1B) [MIM:612781]: An autosomal recessive deficiency of growth hormone leading to short stature. Patients have low but detectable levels of growth hormone, significantly retarded bone age, and a positive response and immunologic tolerance to growth hormone therapy. {ECO:0000269|PubMed:12655557}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Kowarski syndrome (KWKS) [MIM:262650]: A syndrome clinically characterized by short stature associated with bioinactive growth hormone, normal or slightly increased growth hormone secretion, pathologically low insulin-like growth factor 1 levels, and normal catch-up growth on growth hormone replacement therapy. {ECO:0000269|PubMed:17519310, ECO:0000269|PubMed:8552145, ECO:0000269|PubMed:9276733}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Growth hormone deficiency, isolated, 2 (IGHD2) [MIM:173100]: An autosomal dominant deficiency of growth hormone leading to short stature. Clinical severity is variable. Patients have a positive response and immunologic tolerance to growth hormone therapy. {ECO:0000269|PubMed:11502836, ECO:0000269|PubMed:9152628}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08324 Interacts with NA EC number NA Uniprot keywords 3D-structure; Activator; DNA-binding; Quorum sensing; Reference proteome; Transcription; Transcription regulation Protein physicochemical properties Chain ID A Molecular weight (Da) 18305.5 Length 163 Aromaticity 0.12 Instability index 46.52 Isoelectric point 5.19 Charge (pH=7) -6.78 2D Binding mode Binding energy (Kcal/mol) -9.66 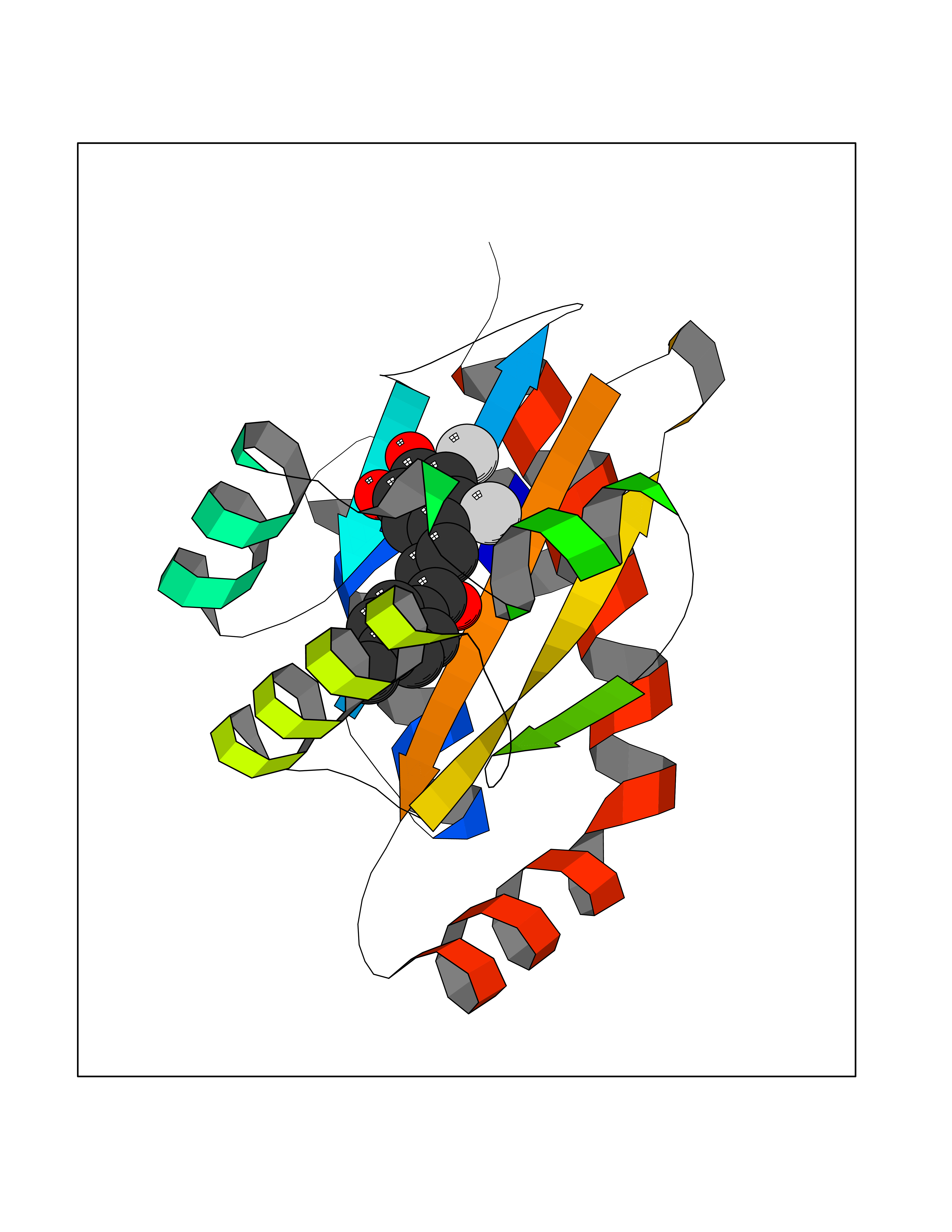 Molscript Map 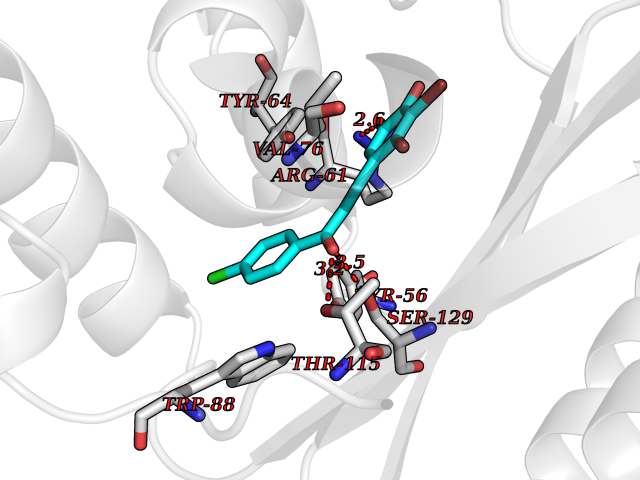 Pymol Map 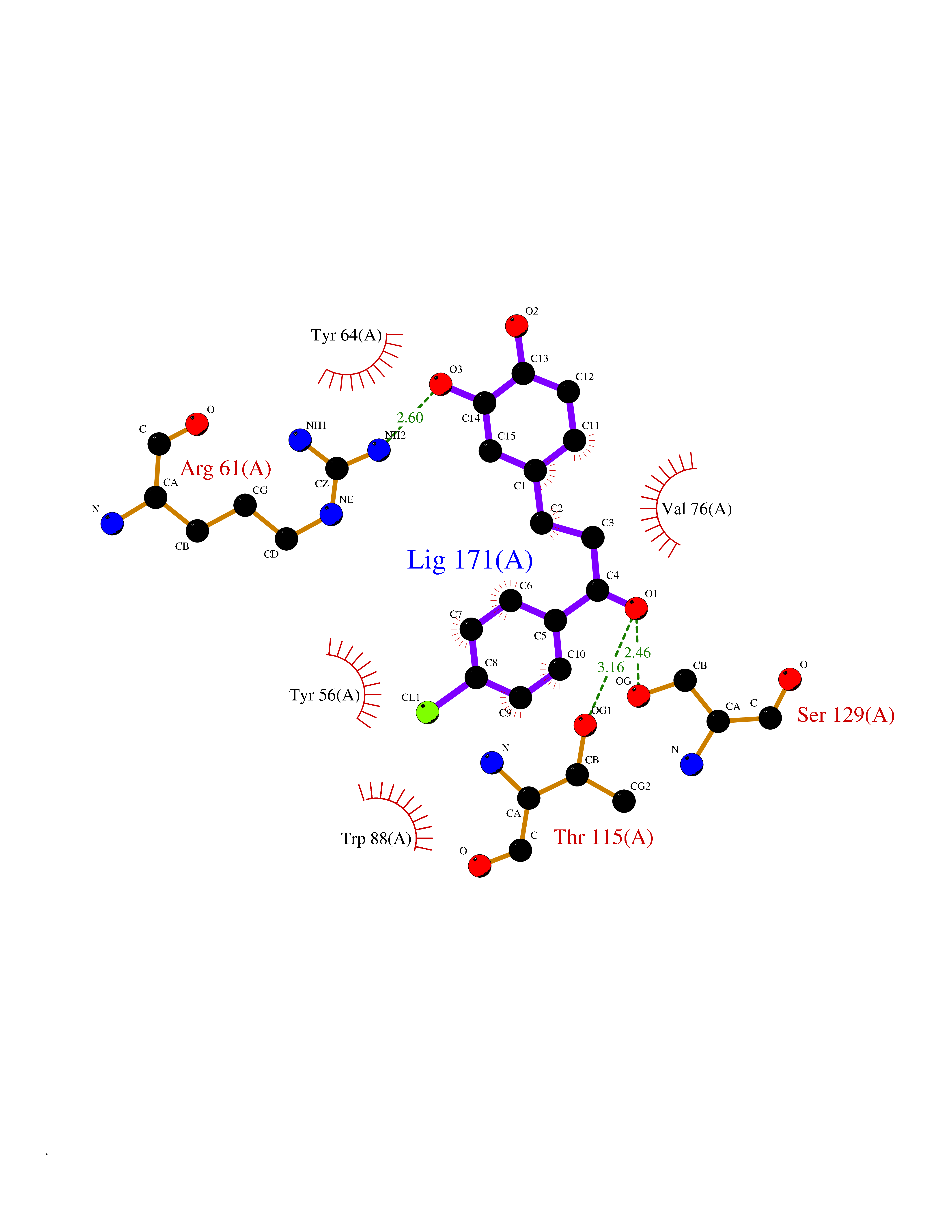 Ligplot Map 3D Binding mode Sequence FLELERSSGKLEWSAILQKMASDLGFSKILFGLLPKDSQDYENAFIVGNYPAAWREHYDRAGYARVDPTVSHCTQSVLPIFWEPSIYQTRKQHEFFEEASAAGLVYGLTMPLHGARGELGALSLSVEAENRAEANRFMESVLPTLWMLKDYALQSGAGLAFEH Hydrogen bonds contact Hydrophobic contact | ||||
| 18 | Cytosolic 10-formyltetrahydrofolate dehydrogenase | 2CFI | 7.07 | |
Target general information Gen name ALDH1L1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms FTHFD Protein family GART family; Aldehyde dehydrogenase family, ALDH1L subfamily Biochemical class Oxidoreductase Function Aldehyde dehydrogenase (NAD) activity.Catalytic activity.Formyltetrahydrofolate dehydrogenase activity.Hydroxymethyl-, formyl- and related transferase activity. Related diseases Developmental and epileptic encephalopathy 39 with leukodystrophy (DEE39) [MIM:612949]: A form of epileptic encephalopathy, a heterogeneous group of severe early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE39 is characterized by global hypomyelination of the central nervous system, with the gray matter appearing relatively unaffected. Inheritance is autosomal recessive. {ECO:0000269|PubMed:19641205, ECO:0000269|PubMed:24515575}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00116 Interacts with Q3SY69; Q92624 EC number 1.5.1.6 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Cytoplasm; NADP; One-carbon metabolism; Oxidoreductase; Phosphopantetheine; Phosphoprotein; Proteomics identification; Reference proteome Protein physicochemical properties Chain ID A Molecular weight (Da) 33869.5 Length 308 Aromaticity 0.08 Instability index 30.42 Isoelectric point 6.09 Charge (pH=7) -3.83 2D Binding mode Binding energy (Kcal/mol) -9.64  Molscript Map 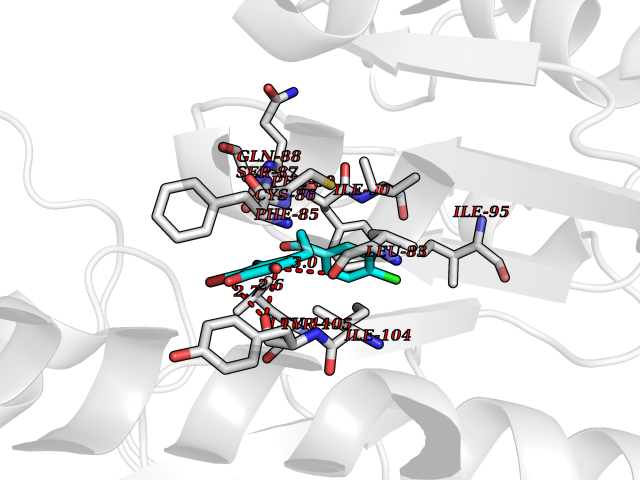 Pymol Map 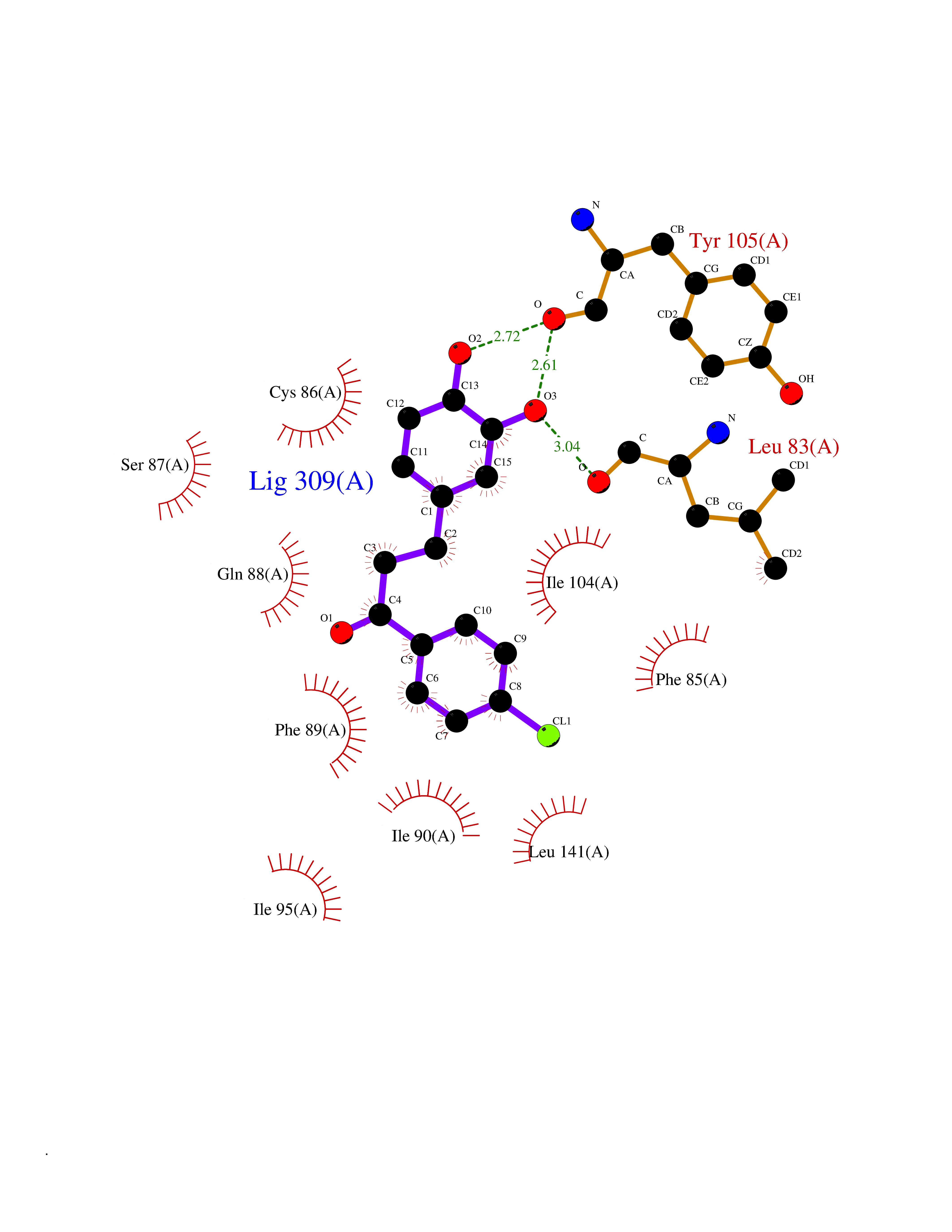 Ligplot Map 3D Binding mode Sequence SMKIAVIGQSLFGQEVYCHLRKEGHEVVGVFTVPDKDGKADPLGLEAEKDGVPVFKYSRWRAKGQALPDVVAKYQALGAELNVLPFCSQFIPMEIISAPRHGSIIYHPSLLPRHRGASAINWTLIHGDKKGGFSIFWADDGLDTGDLLLQKECEVLPDDTVSTLYNRFLFPEGIKGMVQAVRLIAEGKAPRLPQPEEGATYEGIQKKETAKINWDQPAEAIHNWIRGNDKVPGAWTEACEQKLTFFNSTLNTSGLVPEGDALPIPGAHRPGVVTKAGLILFGNDDKMLLVKNIQLEDGKMILASNFFK Hydrogen bonds contact Hydrophobic contact | ||||
| 19 | Acetyl-CoA carboxylase (ACC) (EC 6.4.1.2) (Fatty acid synthetase 3) (mRNA transport-defective protein 7) [Includes: Biotin carboxylase (EC 6.3.4.14)] | 1UYS | 7.07 | |
Target general information Gen name ACC1 Organism Saccharomyces cerevisiae (strain ATCC 204508 / S288c) (Baker's yeast) Uniprot ID TTD ID NA Synonyms MTR7;YNR016C;N3175;ABP2;FAS3 Protein family NA Biochemical class NA Function Carries out three functions: biotin carboxyl carrier protein, biotin carboxylase and carboxyltransferase. Involved in the synthesis of very-long-chain fatty acid synthesis which is required to maintain a functional nuclear envelope. Required for acylation and vacuolar membrane association of VAC8 which is necessary to maintain a normal morphology of the vacuole. {ECO:0000269|PubMed:10757783, ECO:0000269|PubMed:12730220, ECO:0000269|PubMed:6103540, ECO:0000269|PubMed:6108218, ECO:0000269|PubMed:8943372}." Related diseases A chromosomal aberration involving NFKB2 is found in a case of B-cell non Hodgkin lymphoma (B-NHL). Translocation t(10;14)(q24;q32) with IGHA1. The resulting oncogene is also called Lyt-10C alpha variant.; DISEASE: A chromosomal aberration involving NFKB2 is found in a cutaneous T-cell leukemia (C-TCL) cell line. This rearrangement produces the p80HT gene which codes for a truncated 80 kDa protein (p80HT).; DISEASE: In B-cell leukemia (B-CLL) cell line, LB40 and EB308, can be found after heterogeneous chromosomal aberrations, such as internal deletions.; DISEASE: Immunodeficiency, common variable, 10 (CVID10) [MIM:615577]: A primary immunodeficiency characterized by childhood-onset of recurrent infections, hypogammaglobulinemia, and decreased numbers of memory and marginal zone B-cells. Some patients may develop autoimmune features and have circulating autoantibodies. An unusual feature is central adrenal insufficiency. {ECO:0000269|PubMed:24140114, ECO:0000269|PubMed:25524009}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q00955 EC number 6.3.4.14; 6.4.1.2 Uniprot keywords 3D-structure; Acetylation; ATP-binding; Biotin; Cytoplasm; Direct protein sequencing; Endoplasmic reticulum; Fatty acid biosynthesis; Fatty acid metabolism; Ligase; Lipid biosynthesis; Lipid metabolism; Manganese; Membrane; Metal-binding; Multifunctional enzyme; Nucleotide-binding; Phosphoprotein; Reference proteome Protein physicochemical properties Chain ID B,C Molecular weight (Da) 145619 Length 1328 Aromaticity 0.1 Instability index 30.31 Isoelectric point 5.32 Charge (pH=7) -26.79 2D Binding mode Binding energy (Kcal/mol) -9.64  Molscript Map 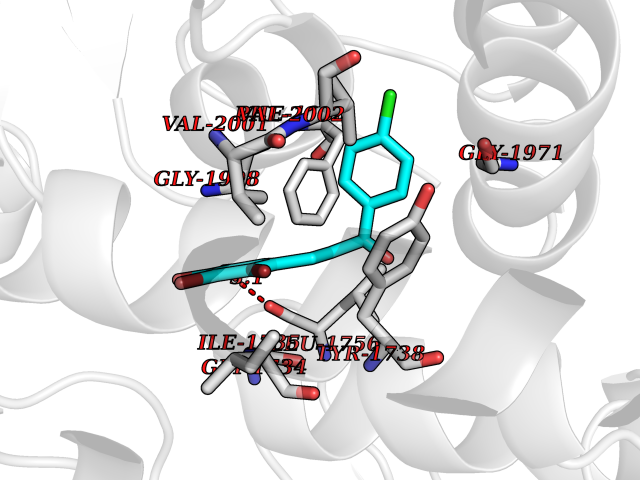 Pymol Map 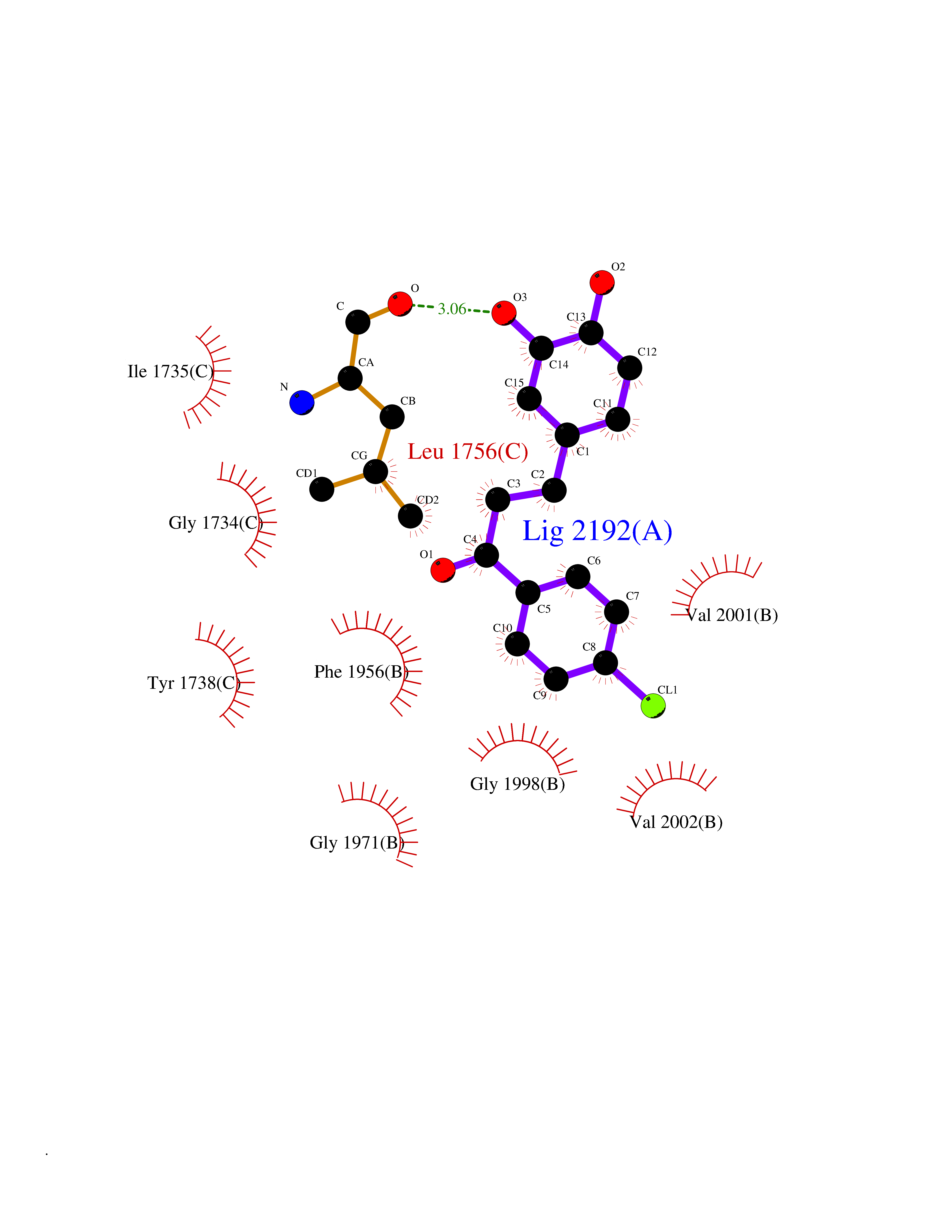 Ligplot Map 3D Binding mode Sequence WLQPKRYKAHLXGTTYVYDFPELFRQASSSQWKNFSADVKLTDDFFISNELIEDENGELTEVEREPGANAIGXVAFKITVKTPEYPRGRQFVVVANDITFKIGSFGPQEDEFFNKVTEYARKRGIPRIYLAANSGARIGXAEEIVPLFQVAWNDAANPDKGFQYLYLTSEGXETLKKFDKENSVLTERTVINGEERFVIKTIIGSEDGLGVECLRGSGLIAGATSRAYHDIFTITLVTCRSVGIGAYLVRLGQRAIQVEGQPIILTGAPAINKXLGREVYTSNLQLGGTQIXYNNGVSHLTAVDDLAGVEKIVEWXSYVPAKRNXPVPILETKDTWDRPVDFTPTNDETYDVRWXIEGRETESGFEYGLFDKGSFFETLSGWAKGVVVGRARLGGIPLGVIGVETRTVENLIPADPANPNSAETLIQEPGQVWHPNSAFKTAQAINDFNNGEQLPXXILANWRGFSGNEVLKYGSFIVDALVDYKQPIIIYIPPTGELRGGSWVVVDPTINADQXEXYADVNARAGVLEPQGXVGIKFRREKLLDTXNRLELLPIYGQISLQFADLHDRSSRXVAKGVISKELEWTEARRFFFWRLRRRLNEEYLIKRLSHQVGEASRLEKIARIRSWYPASVDHEDDRQVATWIEENYKTLDDKLKGLPIATPYPVKEWLQPKRYKAHLXGTTYVYDFPELFRQASSSQWKNFSADVKLTDDFFISNELIEDENGELTEVEREPGANAIGXVAFKITVKTPEYPRGRQFVVVANDITFKIGSFGPQEDEFFNKVTEYARKRGIPRIYLAANSGARIGXAEEIVPLFQVAWNDAANPDKGFQYLYLTSEGXETLKKFDKENSVLTERTVINGEERFVIKTIIGSEDGLGVECLRGSGLIAGATSRAYHDIFTITLVTCRSVGIGAYLVRLGQRAIQVEGQPIILTGAPAINKXLGREVYTSNLQLGGTQIXYNNGVSHLTAVDDLAGVEKIVEWXSYVPAKRNXPVPILETKDTWDRPVDFTPTNDETYDVRWXIEGRETESGFEYGLFDKGSFFETLSGWAKGVVVGRARLGGIPLGVIGVETRTVENLIPADPANPNSAETLIQEPGQVWHPNSAFKTAQAINDFNNGEQLPXXILANWRGFSGNEVLKYGSFIVDALVDYKQPIIIYIPPTGELRGGSWVVVDPTINADQXEXYADVNARAGVLEPQGXVGIKFRREKLLDTXNRLELLPIYGQISLQFADLHDRSSRXVAKGVISKELEWTEARRFFFWRLRRRLNEEYLIKRLSHQVGEASRLEKIARIRSWYPASVDHEDDRQVATWIEENYKTLDDKLKGL Hydrogen bonds contact Hydrophobic contact | ||||
| 20 | Cyclopropane mycolic acid synthase MmaA2 | 1TPY | 7.07 | |
Target general information Gen name mmaA2 Organism Mycobacterium tuberculosis (strain ATCC 25618 / H37Rv) Uniprot ID TTD ID NA Synonyms Rv0644c;mma2 Protein family CFA/CMAS family Biochemical class Transferase Function Cyclopropane-fatty-acyl-phospholipid synthase activity.Methyltransferase activity. Related diseases Oocyte/zygote/embryo maturation arrest 16 (OZEMA16) [MIM:617234]: A rare cause of female primary infertility. In affected women, ovulation and fertilization proceed normally but embryos are arrested at early stages of development. Inheritance is autosomal recessive. {ECO:0000269|PubMed:27545678}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01718; DB01752 Interacts with NA EC number 2.1.1.79 Uniprot keywords 3D-structure; Acetylation; Lipid biosynthesis; Lipid metabolism; Methyltransferase; Reference proteome; S-adenosyl-L-methionine; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 32493.6 Length 285 Aromaticity 0.1 Instability index 43.61 Isoelectric point 5.53 Charge (pH=7) -10.17 2D Binding mode Binding energy (Kcal/mol) -9.64 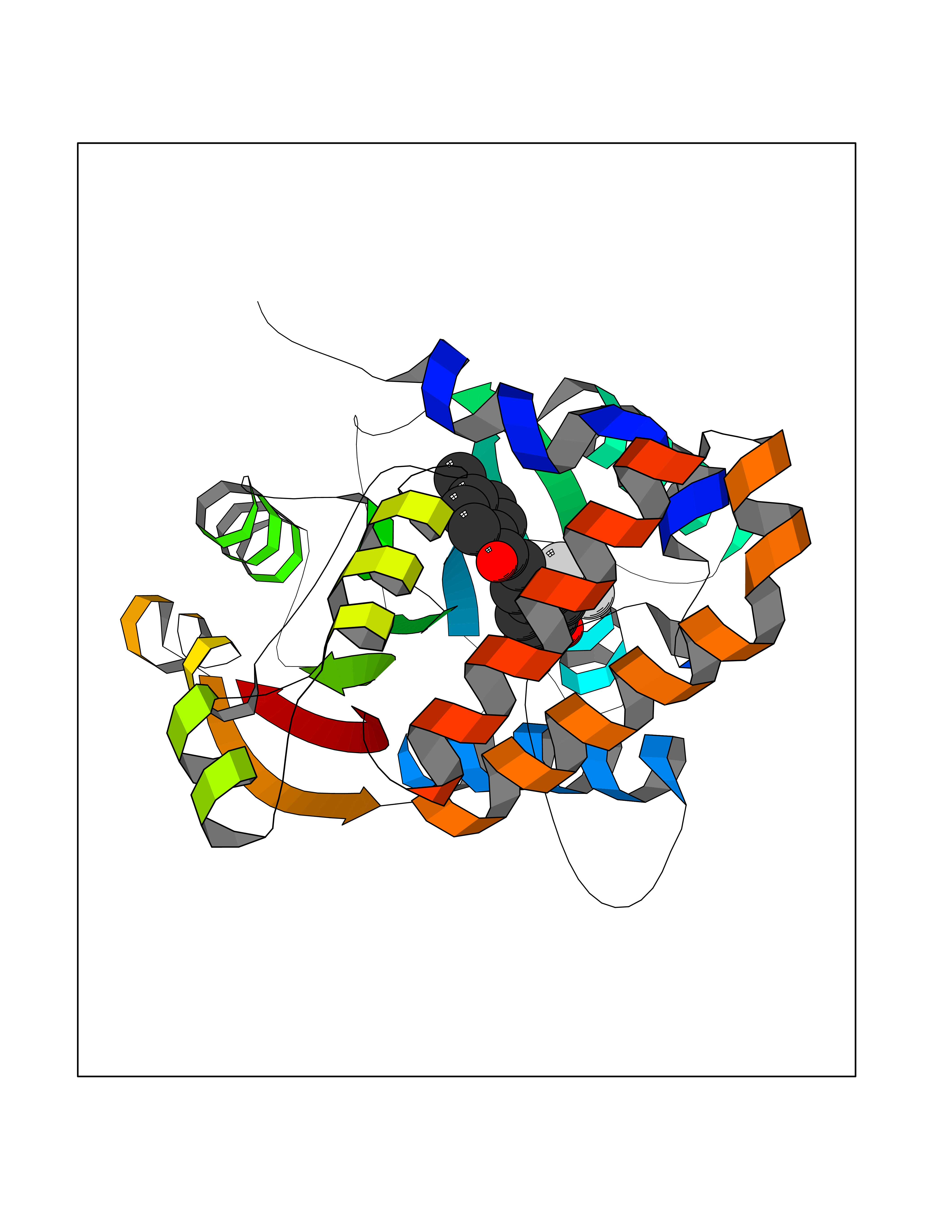 Molscript Map 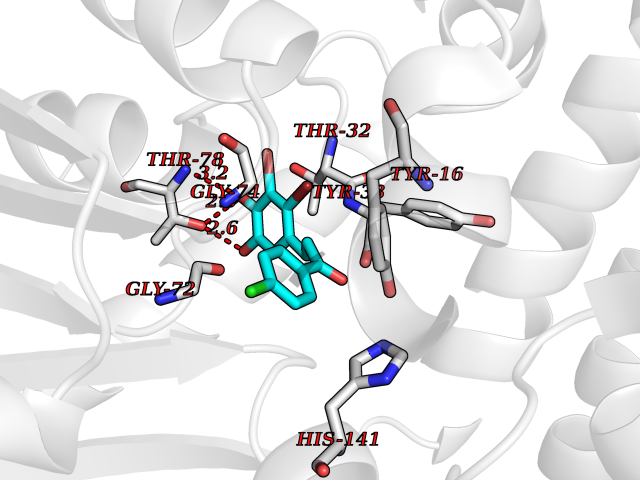 Pymol Map 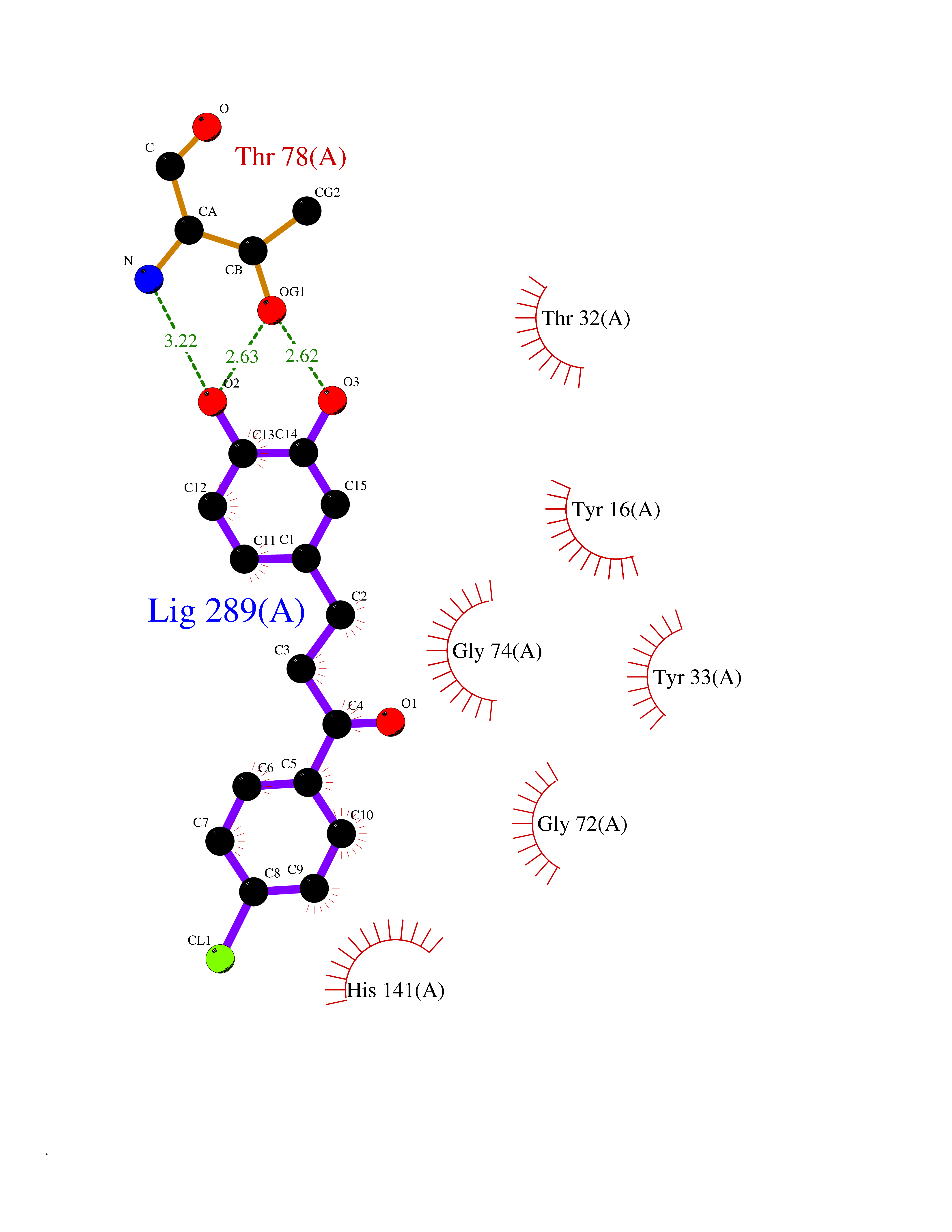 Ligplot Map 3D Binding mode Sequence NDLTPHFEDVQAHYDLSDDFFRLFLDPTQTYSCAHFEREDMTLEEAQIAKIDLALGKLGLQPGMTLLDIGCGWGATMRRAIAQYDVNVVGLTLSKNQAAHVQKSFDEMDTPRDRRVLLAGWEQFNEPVDRIVSIGAFEHFGHDRHADFFARAHKILPPDGVLLLHTITGLTRQQMVDHGLPLTLWLARFLKFIATEIFPGGQPPTIEMVEEQSAKTGFTLTRRQSLQPHYARTLDLWAEALQEHKSEAIAIQSEEVYERYMKYLTGCAKLFRVGYIDVNQFTLAK Hydrogen bonds contact Hydrophobic contact | ||||