Job Results:
Ligand
Structure
Job ID
57b30c77c1e7b273b1e911481965e4cd
Job name
NA
Time
2025-10-13 17:21:20
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 81 | Cytochrome c | 3ZOO | 6.54 | |
Target general information Gen name CYCS Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms CYC Protein family Cytochrome c family Biochemical class Oxidoreductase Function Electron transporter, transferring electrons from CoQH2-cytochrome c reductase complex and cytochrome c oxidase complex activity.Heme binding.Metal ion binding. Related diseases Thrombocytopenia 4 (THC4) [MIM:612004]: A form of thrombocytopenia, a hematologic disorder defined by a decrease in the number of platelets in circulating blood, resulting in the potential for increased bleeding and decreased ability for clotting. {ECO:0000269|PubMed:18345000}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB11638; DB03317; DB03366; DB01017; DB02110; DB03977; DB03934; DB04249 Interacts with O14727; P05067; Q6XD76; Q9NSI6-4; Q3SXR2; Q96BR5; Q9UKG9-2; O00303; Q8IZU1; Q3SYB3; P06241; Q8N5Z5; Q6A162; Q1L5Z9; P02750; Q8IYG6; Q6FHY5; A0A0A0MR05; Q9BUL5; Q6ZMI0-5; Q66K80; Q9NTN9-3; P37840; Q13573; Q92797-2; O43829; Q9FKS5 EC number NA Uniprot keywords 3D-structure; Acetylation; Apoptosis; Direct protein sequencing; Disease variant; Electron transport; Heme; Iron; Metal-binding; Mitochondrion; Phosphoprotein; Proteomics identification; Reference proteome; Respiratory chain; Transport Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 11601.4 Length 104 Aromaticity 0.09 Instability index 12.21 Isoelectric point 9.61 Charge (pH=7) 9.01 2D Binding mode Binding energy (Kcal/mol) -8.92  Molscript Map 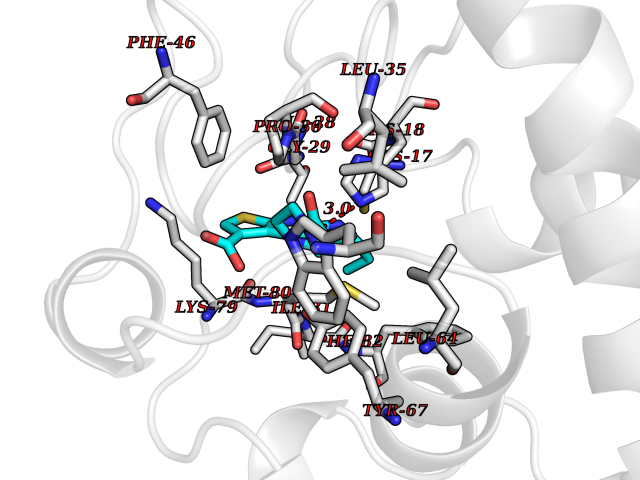 Pymol Map 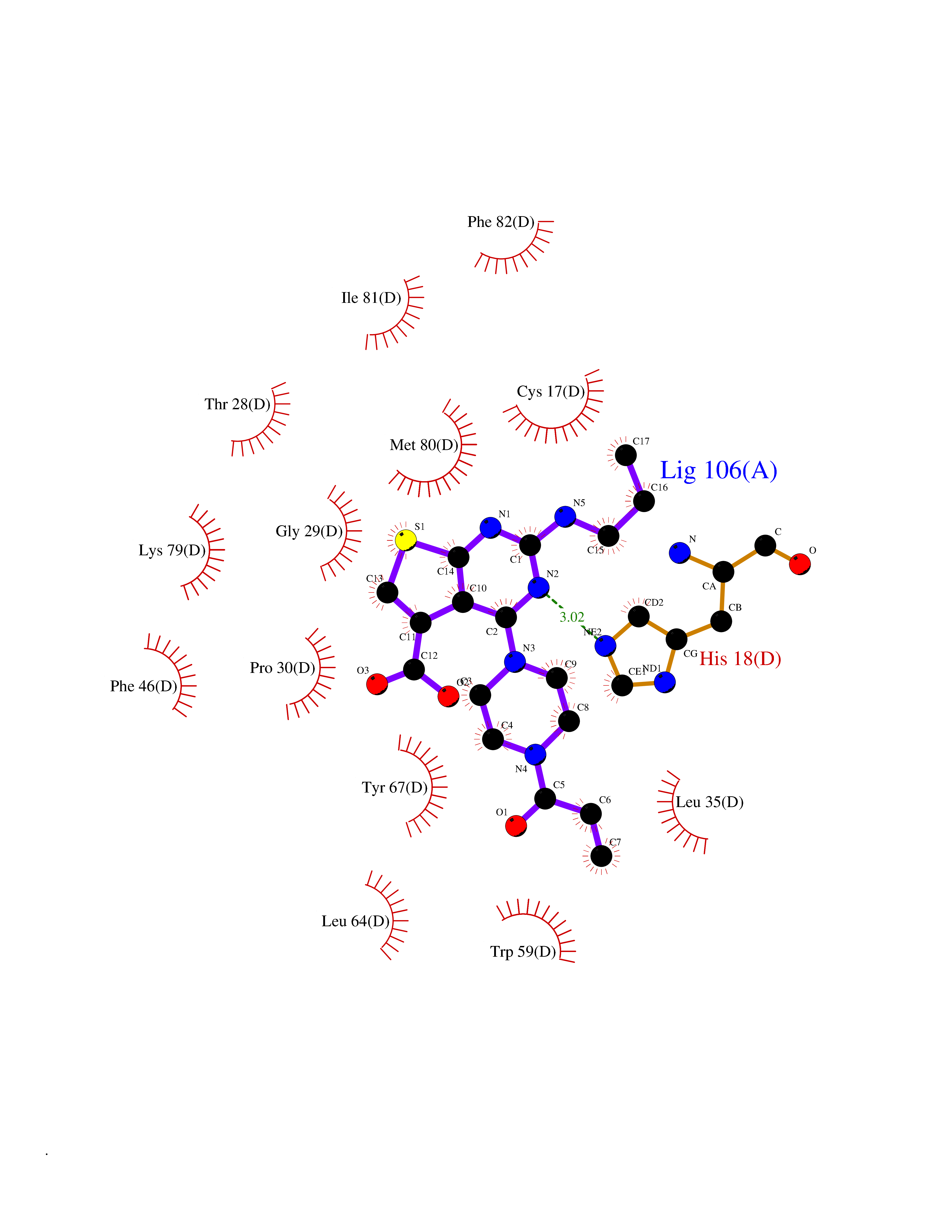 Ligplot Map 3D Binding mode Sequence GDVEKGKKIFIMKCSQCHTVEKGGKHKTGPNLHGLFGRKTGQAPGFSYTAANKNKGIIWGEDTLMEYLENPKKYIPGTKMIFVGIKKKEERADLIAYLKKATNE Hydrogen bonds contact Hydrophobic contact | ||||
| 82 | Bile acid receptor | 4QE6 | 6.54 | |
Target general information Gen name NR1H4 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms HRR1;BAR;RIP14;FXR Protein family Nuclear hormone receptor family, NR1 subfamily Biochemical class Transcription Function Bile acid binding.Bile acid receptor activity.Chenodeoxycholic acid binding.Ligand-dependent nuclear receptor binding.Retinoid X receptor binding.RNA polymerase II distal enhancer sequence-specific DNA binding.RNA polymerase II regulatory region sequence-specific DNA binding.RNA polymerase II transcription factor activity, ligand-activated sequence-specific DNA binding.Sequence-specific DNA binding.Steroid hormone receptor activity.Thyroid hormone receptor activity.Transcriptional activator activity, RNA polymerase II core promoter proximal region sequence-specific binding.Transcriptional activator activity, RNA polymerase II transcription factor binding.Transcription coactivator activity.Transcription corepressor activity.Transcription factor activity, RNA polymerase II distal enhancer sequence-specific binding.Transcription factor activity, sequence-specific DNA binding.Transcription regulatory region sequence-specific DNA binding.Zinc ion binding. Related diseases May be involved in intrahepatic cholestasis of pregnancy. {ECO:0000305|PubMed:17681172}.; DISEASE: May be involved in cholesterol cholelithiasis. {ECO:0000305|PubMed:17931734}.; DISEASE: Cholestasis, progressive familial intrahepatic, 5 (PFIC5) [MIM:617049]: A disorder characterized by early onset of cholestasis that progresses to hepatic fibrosis, cirrhosis, and end-stage liver disease before adulthood. PFIC5 is an autosomal recessive, severe form characterized by onset of intralobular cholestasis in the neonatal period. {ECO:0000269|PubMed:26888176}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08220; DB00132; DB04557; DB06777; DB02659; DB03619; DB02509; DB02545; DB11605; DB16255; DB05990; DB04348; DB16343; DB01586 Interacts with Q15788; O75151; Q8WTS6; P28702; P28702-3; P48443; Q15788; P78527; P03372 EC number NA Uniprot keywords 3D-structure; Acetylation; Activator; Alternative promoter usage; Alternative splicing; Disease variant; DNA-binding; Immunity; Inflammatory response; Innate immunity; Intrahepatic cholestasis; Isopeptide bond; Metal-binding; Methylation; Nucleus; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Repressor; Transcription; Transcription regulation; Ubl conjugation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A Molecular weight (Da) 28271.2 Length 241 Aromaticity 0.08 Instability index 47.06 Isoelectric point 5.39 Charge (pH=7) -11.24 2D Binding mode Binding energy (Kcal/mol) -8.92  Molscript Map 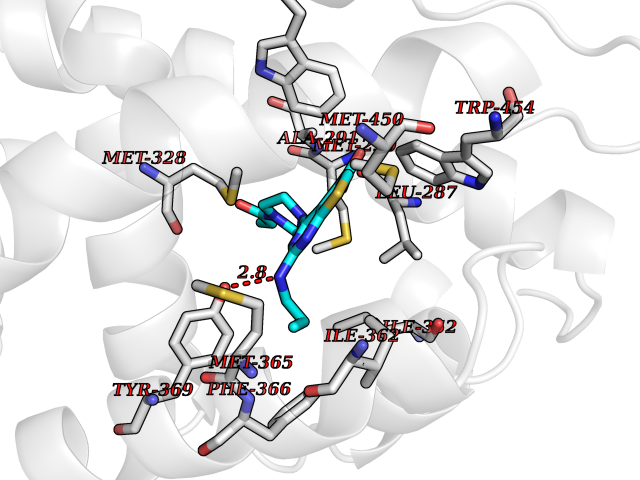 Pymol Map 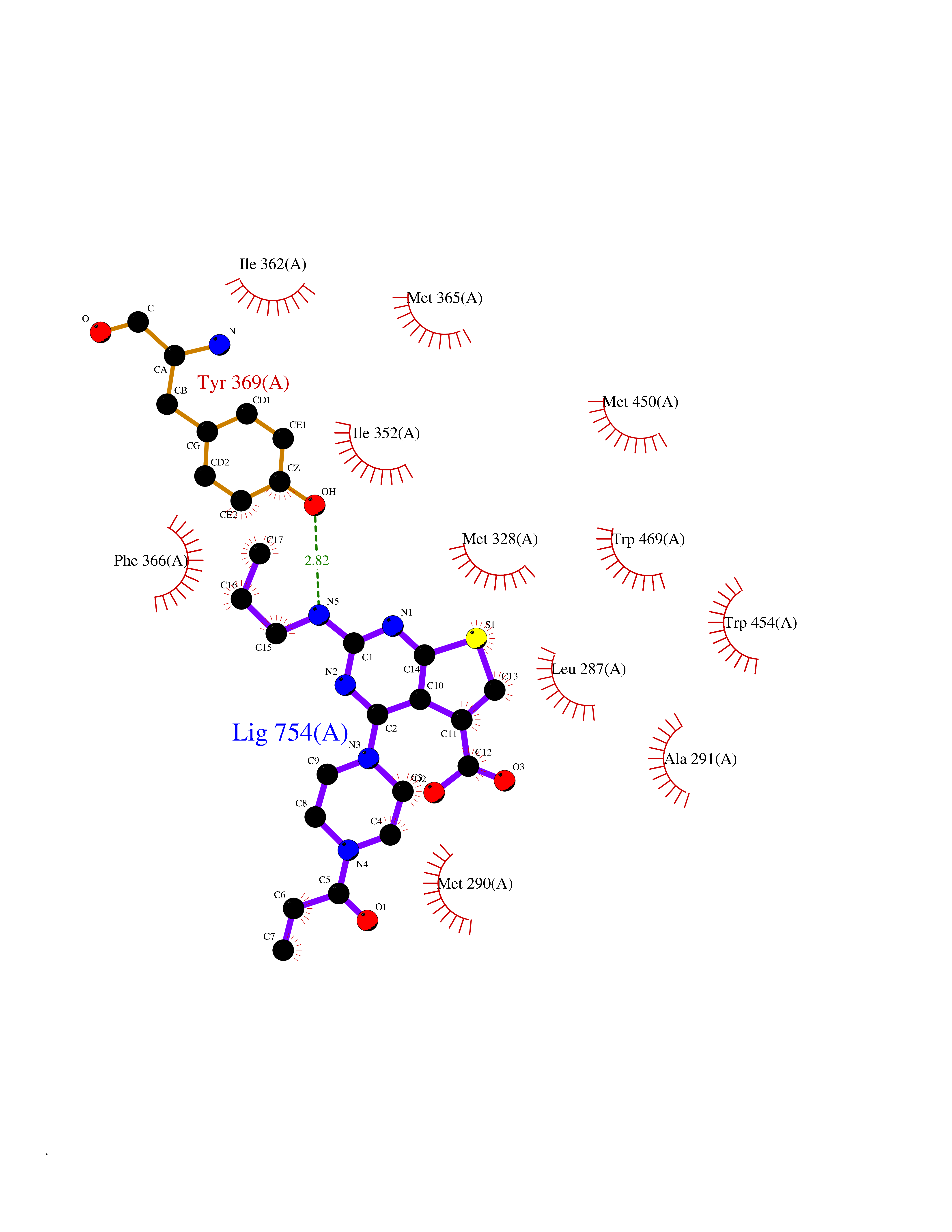 Ligplot Map 3D Binding mode Sequence HMELTPDQQTLLHFIMDSYNKQRMPQEITNKILKEEFSAEENFLILTEMATNHVQVLVEFTKKLPGFQTLDHEDQIALLKGSAVEAMFLRSAEIFNKSGHSDLLEERIRNSGISDEYITPMFSFYKSIGELKMTQEEYALLTAIVILSPDRQYIKDREAVEKLQEPLLDVLQKLCKIHQPENPQHFACLLGRLTELRTFNHHHAEMLMSWRVNDHKFTPLLCEIWDVQKENALLRYLLDKD Hydrogen bonds contact Hydrophobic contact | ||||
| 83 | Beta-arrestin-1 (ARRB1) | 6TKO | 6.54 | |
Target general information Gen name ARRB1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Non-visual arrestin-2; Betaarrestin1; Arrestin beta1; Arrestin beta-1; ARR1 Protein family Arrestin family Biochemical class Arrestin protein Function During homologous desensitization, beta-arrestins bind to the GPRK-phosphorylated receptor and sterically preclude its coupling to the cognate G-protein; the binding appears to require additional receptor determinants exposed only in the active receptor conformation. The beta-arrestins target many receptors for internalization by acting as endocytic adapters (CLASPs, clathrin-associated sorting proteins) and recruiting the GPRCs to the adapter protein 2 complex 2 (AP-2) in clathrin-coated pits (CCPs). However, the extent of beta-arrestin involvement appears to vary significantly depending on the receptor, agonist and cell type. Internalized arrestin-receptor complexes traffic to intracellular endosomes, where they remain uncoupled from G-proteins. Two different modes of arrestin-mediated internalization occur. Class A receptors, like ADRB2, OPRM1, ENDRA, D1AR and ADRA1B dissociate from beta-arrestin at or near the plasma membrane and undergo rapid recycling. Class B receptors, like AVPR2, AGTR1, NTSR1, TRHR and TACR1 internalize as a complex with arrestin and traffic with it to endosomal vesicles, presumably as desensitized receptors, for extended periods of time. Receptor resensitization then requires that receptor-bound arrestin is removed so that the receptor can be dephosphorylated and returned to the plasma membrane. Involved in internalization of P2RY4 and UTP-stimulated internalization of P2RY2. Involved in phosphorylation-dependent internalization of OPRD1 ands subsequent recycling. Involved in the degradation of cAMP by recruiting cAMP phosphodiesterases to ligand-activated receptors. Beta-arrestins function as multivalent adapter proteins that can switch the GPCR from a G-protein signaling mode that transmits short-lived signals from the plasma membrane via small molecule second messengers and ion channels to a beta-arrestin signaling mode that transmits a distinct set of signals that are initiated as the receptor internalizes and transits the intracellular compartment. Acts as signaling scaffold for MAPK pathways such as MAPK1/3 (ERK1/2). ERK1/2 activated by the beta-arrestin scaffold is largely excluded from the nucleus and confined to cytoplasmic locations such as endocytic vesicles, also called beta-arrestin signalosomes. Recruits c-Src/SRC to ADRB2 resulting in ERK activation. GPCRs for which the beta-arrestin-mediated signaling relies on both ARRB1 and ARRB2 (codependent regulation) include ADRB2, F2RL1 and PTH1R. For some GPCRs the beta-arrestin-mediated signaling relies on either ARRB1 or ARRB2 and is inhibited by the other respective beta-arrestin form (reciprocal regulation). Inhibits ERK1/2 signaling in AGTR1- and AVPR2-mediated activation (reciprocal regulation). Is required for SP-stimulated endocytosis of NK1R and recruits c-Src/SRC to internalized NK1R resulting in ERK1/2 activation, which is required for the antiapoptotic effects of SP. Is involved in proteinase-activated F2RL1-mediated ERK activity. Acts as signaling scaffold for the AKT1 pathway. Is involved in alpha-thrombin-stimulated AKT1 signaling. Is involved in IGF1-stimulated AKT1 signaling leading to increased protection from apoptosis. Involved in activation of the p38 MAPK signaling pathway and in actin bundle formation. Involved in F2RL1-mediated cytoskeletal rearrangement and chemotaxis. Involved in AGTR1-mediated stress fiber formation by acting together with GNAQ to activate RHOA. Appears to function as signaling scaffold involved in regulation of MIP-1-beta-stimulated CCR5-dependent chemotaxis. Involved in attenuation of NF-kappa-B-dependent transcription in response to GPCR or cytokine stimulation by interacting with and stabilizing CHUK. May serve as nuclear messenger for GPCRs. Involved in OPRD1-stimulated transcriptional regulation by translocating to CDKN1B and FOS promoter regions and recruiting EP300 resulting in acetylation of histone H4. Involved in regulation of LEF1 transcriptional activity via interaction with DVL1 and/or DVL2 Also involved in regulation of receptors other than GPCRs. Involved in Toll-like receptor and IL-1 receptor signaling through the interaction with TRAF6 which prevents TRAF6 autoubiquitination and oligomerization required for activation of NF-kappa-B and JUN. Binds phosphoinositides. Binds inositolhexakisphosphate (InsP6). Involved in IL8-mediated granule release in neutrophils. Required for atypical chemokine receptor ACKR2-induced RAC1-LIMK1-PAK1-dependent phosphorylation of cofilin (CFL1) and for the up-regulation of ACKR2 from endosomal compartment to cell membrane, increasing its efficiency in chemokine uptake and degradation. Involved in the internalization of the atypical chemokine receptor ACKR3. Negatively regulates the NOTCH signaling pathway by mediating the ubiquitination and degradation of NOTCH1 by ITCH. Participates to the recruitment of the ubiquitin-protein ligase to the receptor. Functions in regulating agonist-mediated G-protein coupled receptor (GPCR) signaling by mediating both receptor desensitization and resensitization processes. Related diseases Intellectual developmental disorder with dysmorphic facies and ptosis (IDDDFP) [MIM:617333]: An autosomal dominant neurodevelopmental disorder characterized by delayed psychomotor development, intellectual disability, delayed language, and facial dysmorphisms, most notably ptosis. Additional features may include poor growth, hypotonia, and seizures. {ECO:0000269|PubMed:27939639, ECO:0000269|PubMed:27939640}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P63010-2; O15169; P0DP25; P20963; P25101; P50148; Q5JWF2; Q14749; P06396; Q16665; P11142; Q99683; P53779; P45984; Q00987; P19338; Q14978; P14618; P14859-6; P35813; O75688; Q13523; P06702; P12931; Q15208; Q13428; P04637; P27348; P25490; O43298; O95218; Q7DB77 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Cell projection; Coated pit; Cytoplasm; Cytoplasmic vesicle; Membrane; Nucleus; Phosphoprotein; Protein transport; Proteomics identification; Reference proteome; Signal transduction inhibitor; Transcription; Transcription regulation; Transport; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 32455.6 Length 293 Aromaticity 0.12 Instability index 28.99 Isoelectric point 9.12 Charge (pH=7) 9.86 2D Binding mode Binding energy (Kcal/mol) -8.92  Molscript Map 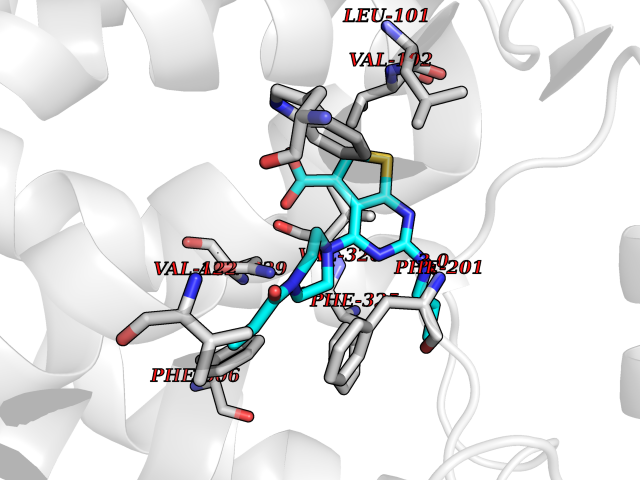 Pymol Map 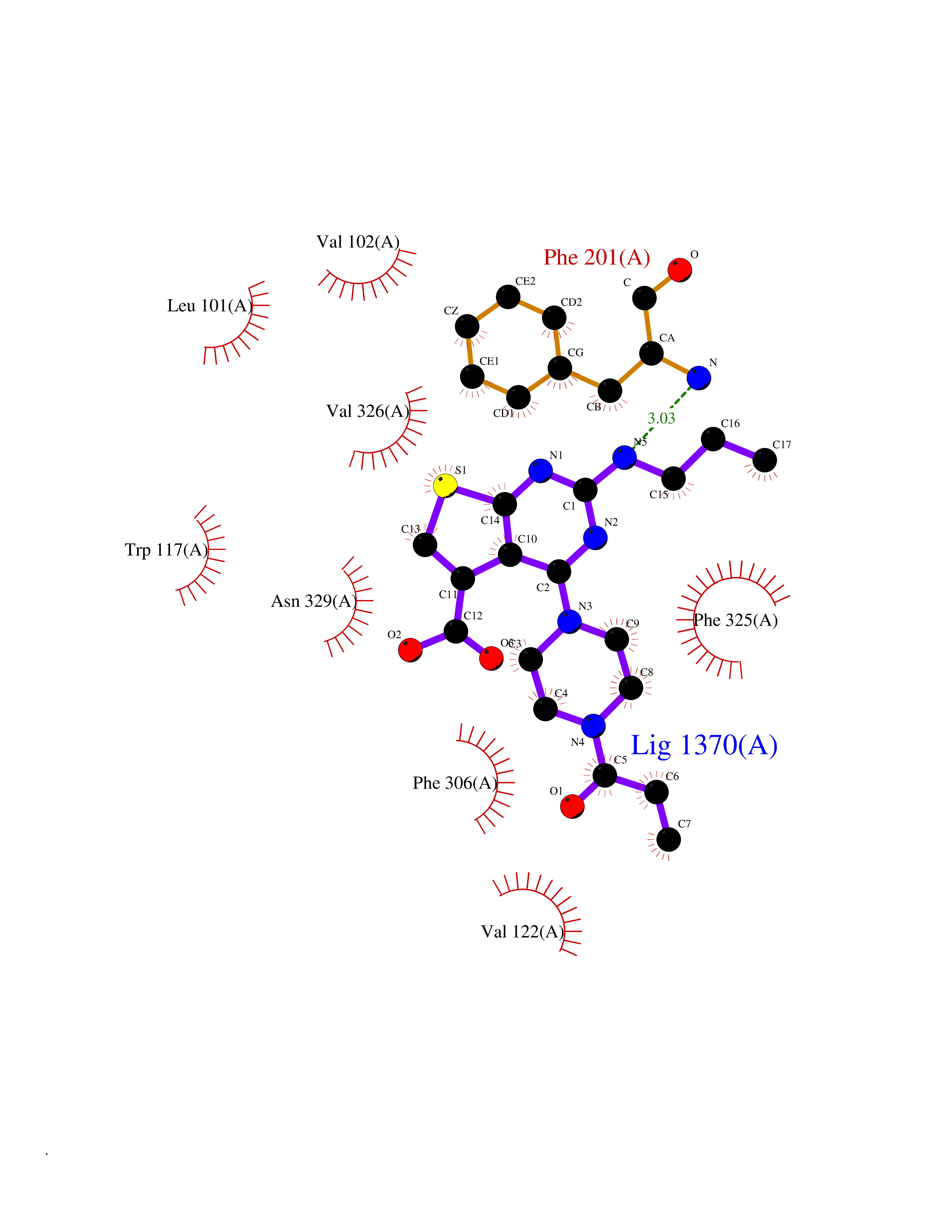 Ligplot Map 3D Binding mode Sequence AGCSLLMALVVLLIVAGNVLVIAAIGRTQRLQTLTNLFITSLACADLVVGLLVVPFGATLVCRGTWLWGSFLCELWTSLDVLCVTASIWTLCVIAIDRYLAITSPFRYQSLMTRARAKVIICTVWAISALVSFLPIMMHWWRDEDPQALKCYQDPGCCDFVTNRAYAIASSIISFYIPLLIMIFVYLRVYREAKEQIRKIDVMAMREHKALKTLGIIMGVFTLCWLPFFLVNIVNVFNRDLVPKWLFVAFNWLGYANSAMNPIIYCRSPDFRKAFKRLLAEXAXXAXXXLAKD Hydrogen bonds contact Hydrophobic contact | ||||
| 84 | Target of rapamycin complex 2 MAPKAP1 (MTORC2) | 7LC2 | 6.54 | |
Target general information Gen name MAPKAP1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms mSIN1; Target of rapamycin complex 2 subunit MAPKAP1; TORC2 subunit MAPKAP1; Stress-activated map kinase-interacting protein 1; SIN1; SAPK-interacting protein 1; Mitogen-activated protein kinase 2-ass Protein family SIN1 family Biochemical class NA Function mTORC2 is activated by growth factors, but, in contrast to mTORC1, seems to be nutrient-insensitive. mTORC2 seems to function upstream of Rho GTPases to regulate the actin cytoskeleton, probably by activating one or more Rho-type guanine nucleotide exchange factors. mTORC2 promotes the serum-induced formation of stress-fibers or F-actin. mTORC2 plays a critical role in AKT1 'Ser-473' phosphorylation, which may facilitate the phosphorylation of the activation loop of AKT1 on 'Thr-308' by PDK1 which is a prerequisite for full activation. mTORC2 regulates the phosphorylation of SGK1 at 'Ser-422'. mTORC2 also modulates the phosphorylation of PRKCA on 'Ser-657'. Within mTORC2, MAPKAP1 is required for complex formation and mTORC2 kinase activity. MAPKAP1 inhibits MAP3K2 by preventing its dimerization and autophosphorylation. Inhibits HRAS and KRAS signaling. Enhances osmotic stress-induced phosphorylation of ATF2 and ATF2-mediated transcription. Involved in ciliogenesis, regulates cilia length through its interaction with CCDC28B independently of mTORC2 complex. Subunit of mTORC2, which regulates cell growth and survival in response to hormonal signals. Related diseases Intellectual developmental disorder, X-linked, syndromic, Claes-Jensen type (MRXSCJ) [MIM:300534]: A disorder characterized by significantly below average general intellectual functioning associated with impairments in adaptive behavior and manifested during the developmental period. MRXSCJ patients manifest intellectual disability associated with variable features such as slowly progressive spastic paraplegia, seizures, facial dysmorphism. {ECO:0000269|PubMed:15586325, ECO:0000269|PubMed:16538222, ECO:0000269|PubMed:16541399, ECO:0000269|PubMed:17320160, ECO:0000269|PubMed:17468742, ECO:0000269|PubMed:23356856, ECO:0000269|PubMed:25666439}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q02156; P78527 EC number NA Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Cell membrane; Cytoplasmic vesicle; Direct protein sequencing; Membrane; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Stress response Protein physicochemical properties Chain ID A Molecular weight (Da) 18420.4 Length 167 Aromaticity 0.09 Instability index 39.36 Isoelectric point 5.24 Charge (pH=7) -5.89 2D Binding mode Binding energy (Kcal/mol) -8.92  Molscript Map 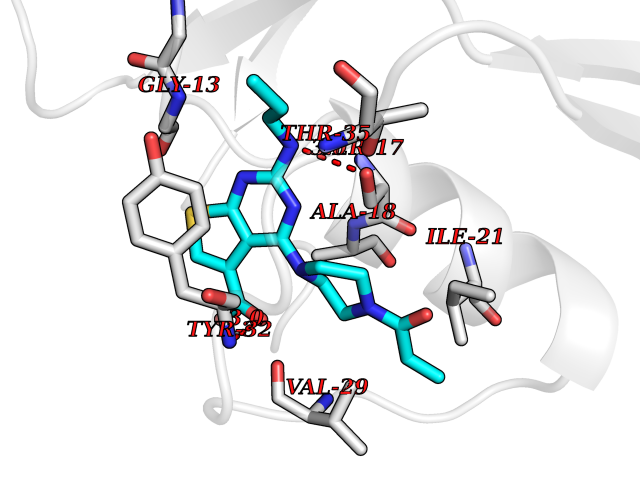 Pymol Map 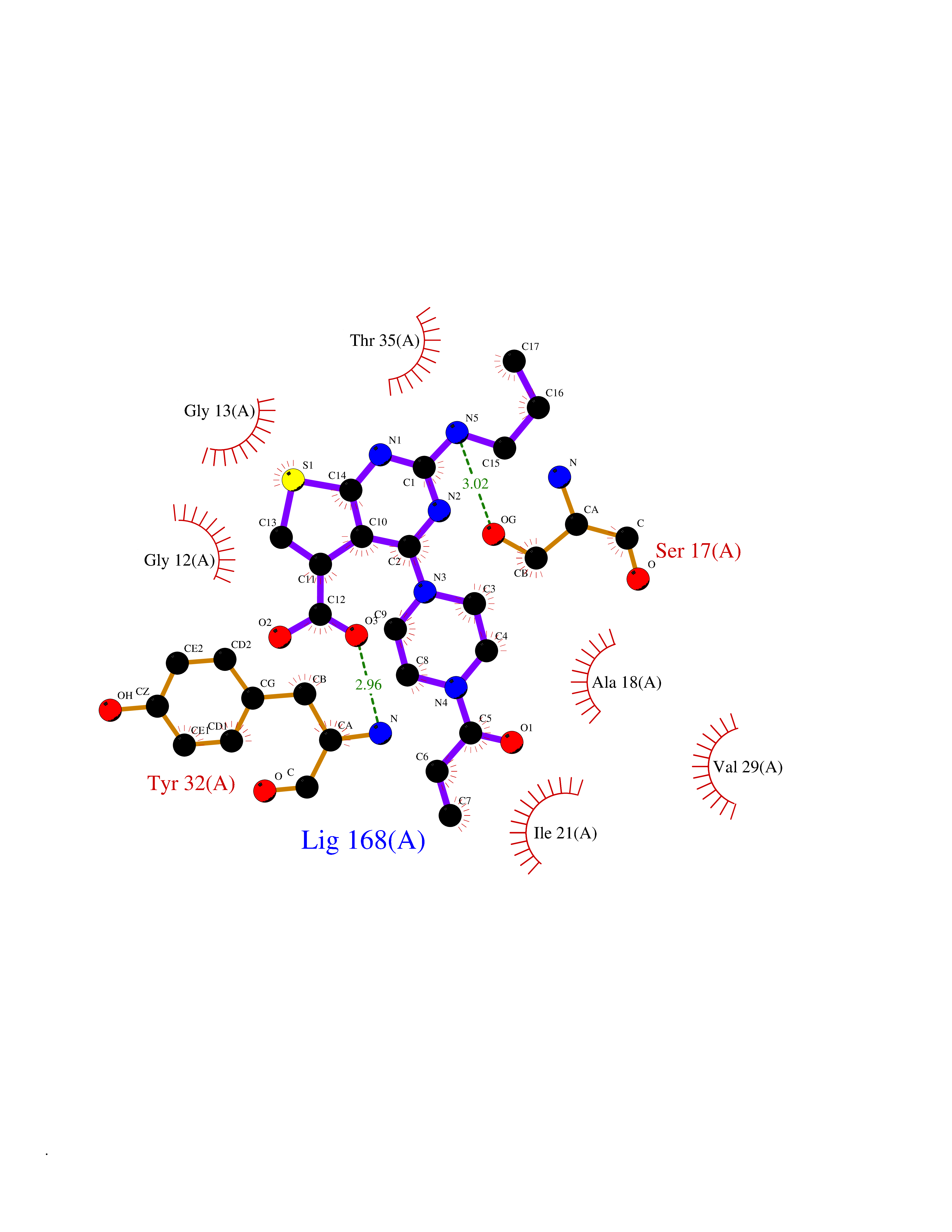 Ligplot Map 3D Binding mode Sequence GXTEYKLVVVGAGGVGKSALTIQLIQNHFVDEYDPTIEDSYRKQVVIDGETCLLDILDTAGREEYSAXRDQYXRTGEGFLCVFAINNTKSFEDIHHYREQIKRVKDSEDVPXVLVGNKCDLPSRTVDTKQAQDLARSYGIPFIETSAKTRQGVDDAFYTLVREIRKH Hydrogen bonds contact Hydrophobic contact | ||||
| 85 | Mineralocorticoid receptor (MR) | 4PF3 | 6.53 | |
Target general information Gen name NR3C2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Nuclear receptor subfamily 3 group C member 2; Mineralocorticoid receptor; MLR; MCR; Inner ear mineralocorticoid receptor; Delta Protein family Nuclear hormone receptor family, NR3 subfamily Biochemical class Nuclear hormone receptor Function Binds to mineralocorticoid response elements (MRE) and transactivates target genes. The effect of MC is to increase ion and water transport and thus raise extracellular fluid volume and blood pressure and lower potassium levels. Receptor for both mineralocorticoids (MC) such as aldosterone and glucocorticoids (GC) such as corticosterone or cortisol. Related diseases Pseudohypoaldosteronism 1, autosomal dominant (PHA1A) [MIM:177735]: A salt wasting disease resulting from target organ unresponsiveness to mineralocorticoids. PHA1A is a mild form characterized by target organ defects confined to kidney. Patients may present with neonatal renal salt wasting with hyperkalaemic acidosis despite high aldosterone levels. These patients improve with age and usually become asymptomatic without treatment. {ECO:0000269|PubMed:11134129, ECO:0000269|PubMed:12788847, ECO:0000269|PubMed:16954160, ECO:0000269|PubMed:16972228}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Early-onset hypertension with severe exacerbation in pregnancy (EOHSEP) [MIM:605115]: Inheritance is autosomal dominant. The disease is characterized by the onset of severe hypertension before the age of 20, and by suppression of aldosterone secretion. {ECO:0000269|PubMed:10884226, ECO:0000269|PubMed:15908963, ECO:0000269|PubMed:15967794}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB04630; DB01013; DB04652; DB06780; DB01134; DB01395; DB00700; DB01023; DB16165; DB00687; DB13867; DB08906; DB00588; DB02998; DB00393; DB00396; DB00421; DB02901; DB13951; DB00624; DB13943; DB13944; DB15114 Interacts with NA EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; Disease variant; DNA-binding; Endoplasmic reticulum; Lipid-binding; Membrane; Metal-binding; Nucleus; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Steroid-binding; Transcription; Transcription regulation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A Molecular weight (Da) 29012.4 Length 249 Aromaticity 0.12 Instability index 51.27 Isoelectric point 6.3 Charge (pH=7) -2.08 2D Binding mode Binding energy (Kcal/mol) -8.91 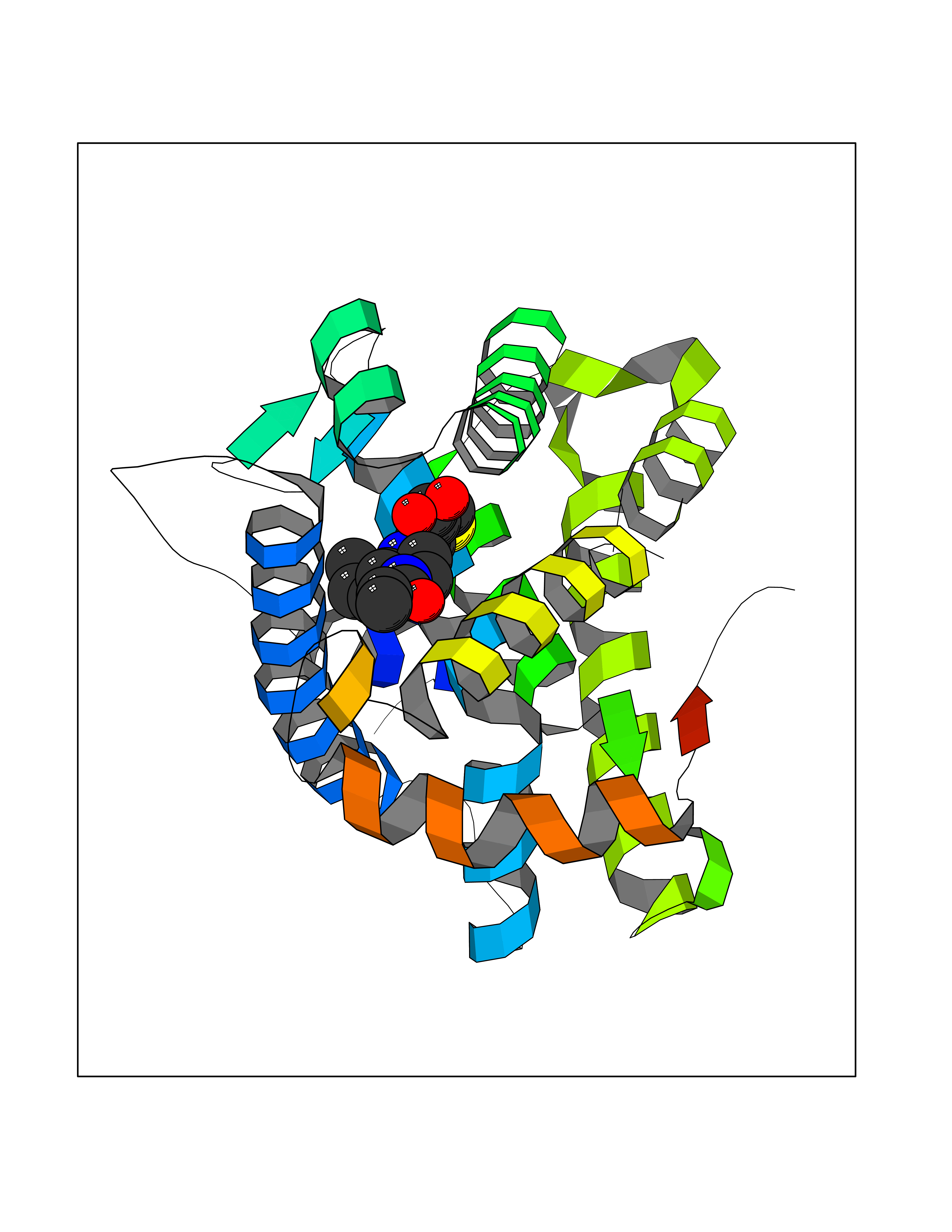 Molscript Map 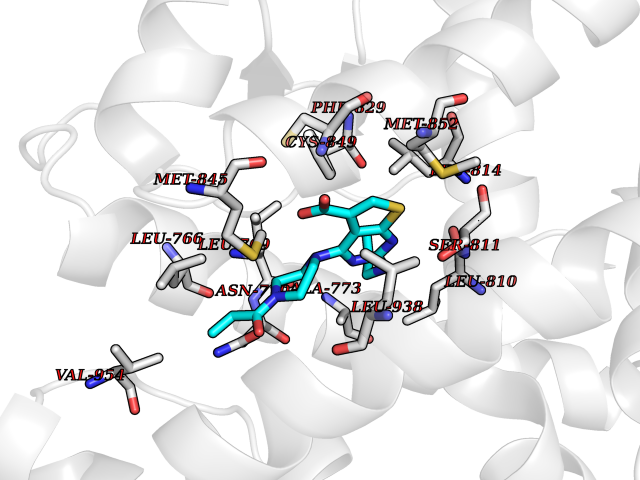 Pymol Map 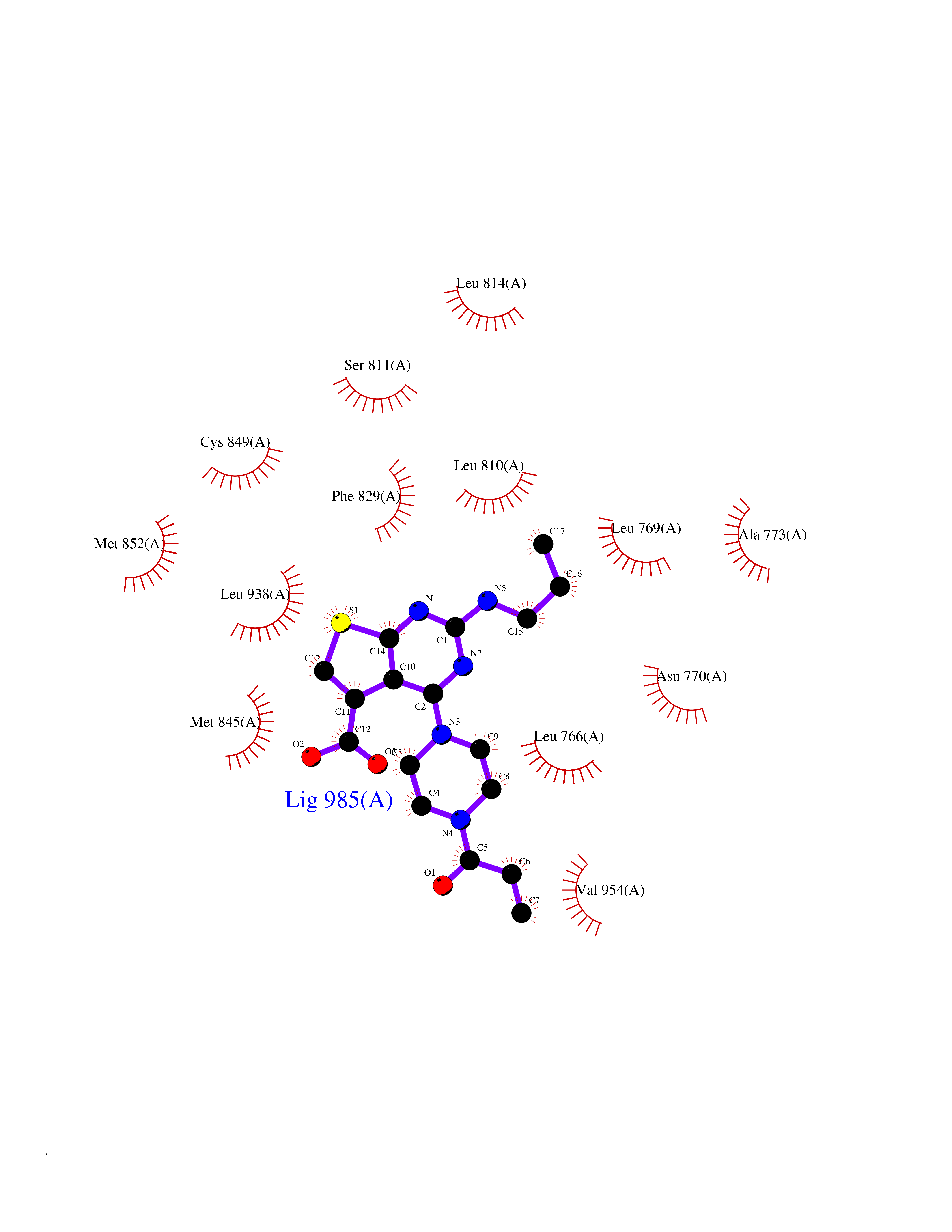 Ligplot Map 3D Binding mode Sequence TPSPVMVLENIEPEIVYAGYDSSKPDTAENLLSTLNRLAGKQMIQVVKWAKVLPGFKNLPLEDQITLIQYSWMSLLSFALSWRSYKHTNSQFLYFAPDLVFNEEKMHQSAMYELCQGMHQISLQFVRLQLTFEEYTIMKVLLLLSTIPKDGLKSQAAFEEMRTNYIKELRKMVTKCPNNSGQSWQRFYQLTKLLDSMHDLVSDLLEFCFYTFRESHALKVEFPAMLVEIISDQLPKVESGNVKPLYFHR Hydrogen bonds contact Hydrophobic contact | ||||
| 86 | Cholesterol oxidase | 4REK | 6.53 | |
Target general information Gen name choA Organism Streptomyces sp. (strain SA-COO) Uniprot ID TTD ID NA Synonyms NA Protein family GMC oxidoreductase family Biochemical class Oxidoreductase Function Cholesterol oxidase activity.Flavin adenine dinucleotide binding.Steroid delta-isomerase activity. Related diseases Bothnia retinal dystrophy (BRD) [MIM:607475]: A type of retinitis punctata albescens. Affected individuals show night blindness from early childhood with features consistent with retinitis punctata albescens and macular degeneration. {ECO:0000269|PubMed:10102298}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Rod-cone dystrophy Newfoundland (NFRCD) [MIM:607476]: A rod-cone dystrophy reminiscent of retinitis punctata albescens but with a substantially lower age at onset and more-rapid and distinctive progression. Rod-cone dystrophies results from initial loss of rod photoreceptors, later followed by cone photoreceptors loss. {ECO:0000269|PubMed:11868161}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Retinitis punctata albescens (RPA) [MIM:136880]: A form of fleck retina disease characterized by aggregation of white flecks posteriorly in the retina, causing night blindness and delayed dark adaptation. It differs from fundus albipunctatus in being progressive and evolving to generalized atrophy of the retina. {ECO:0000269|PubMed:10102299, ECO:0000269|PubMed:11453974, ECO:0000269|PubMed:9326942}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03147; DB02332 Interacts with NA EC number 1.1.3.6; 5.3.3.1 Uniprot keywords 3D-structure; Cholesterol metabolism; Direct protein sequencing; FAD; Flavoprotein; Isomerase; Lipid metabolism; Oxidoreductase; Secreted; Signal; Steroid metabolism; Sterol metabolism Protein physicochemical properties Chain ID A Molecular weight (Da) 54367.8 Length 498 Aromaticity 0.1 Instability index 30.62 Isoelectric point 6.69 Charge (pH=7) -0.71 2D Binding mode Binding energy (Kcal/mol) -8.9 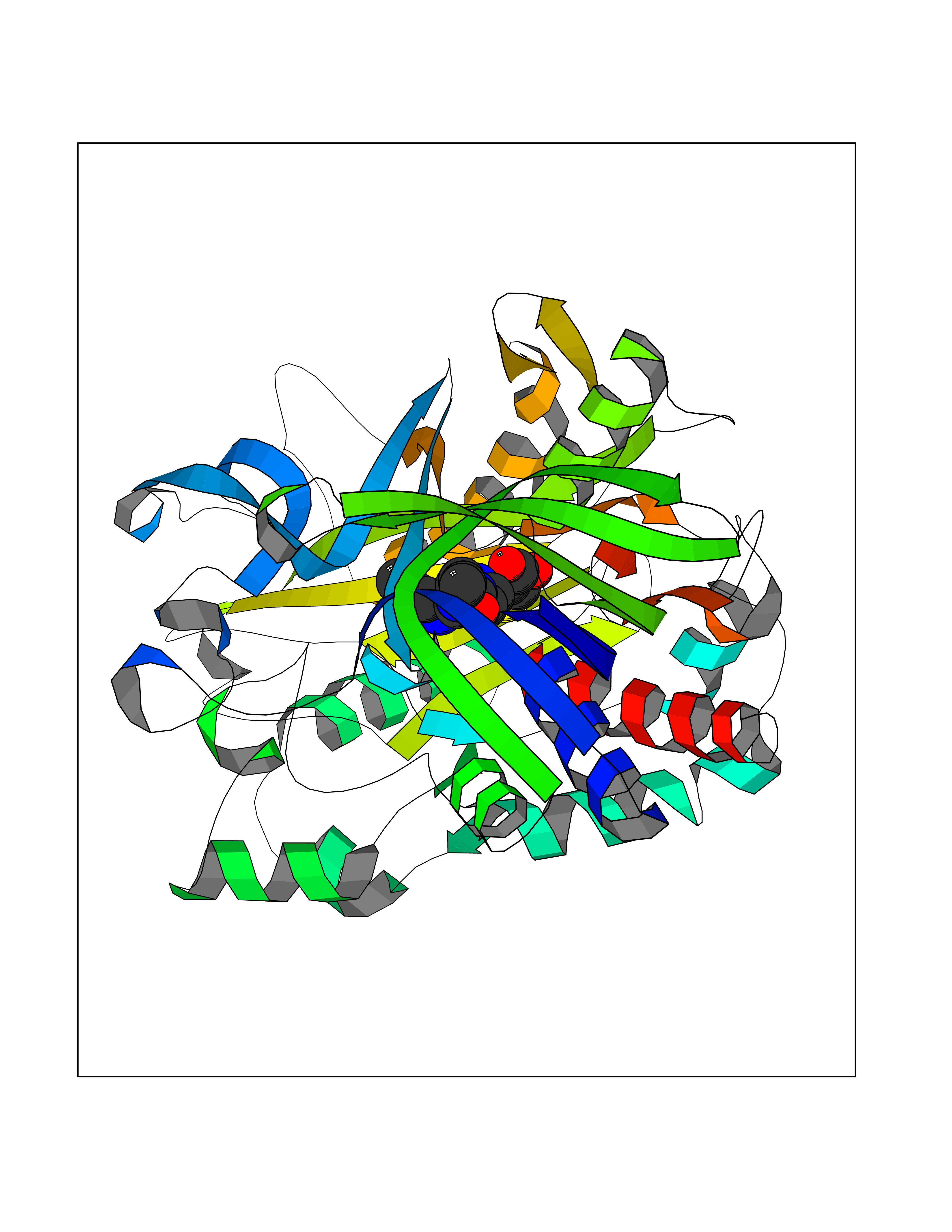 Molscript Map 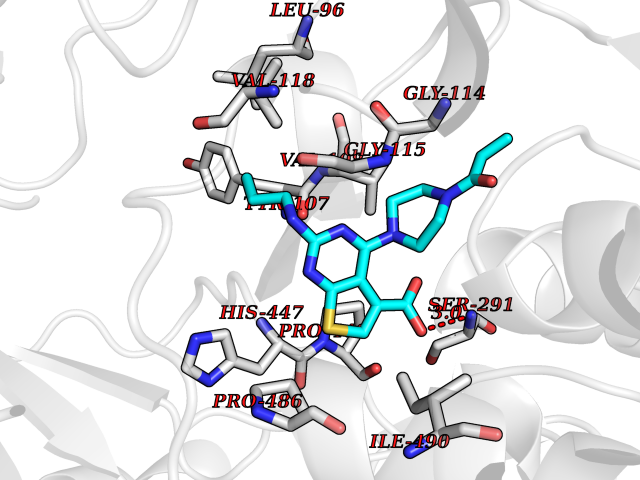 Pymol Map 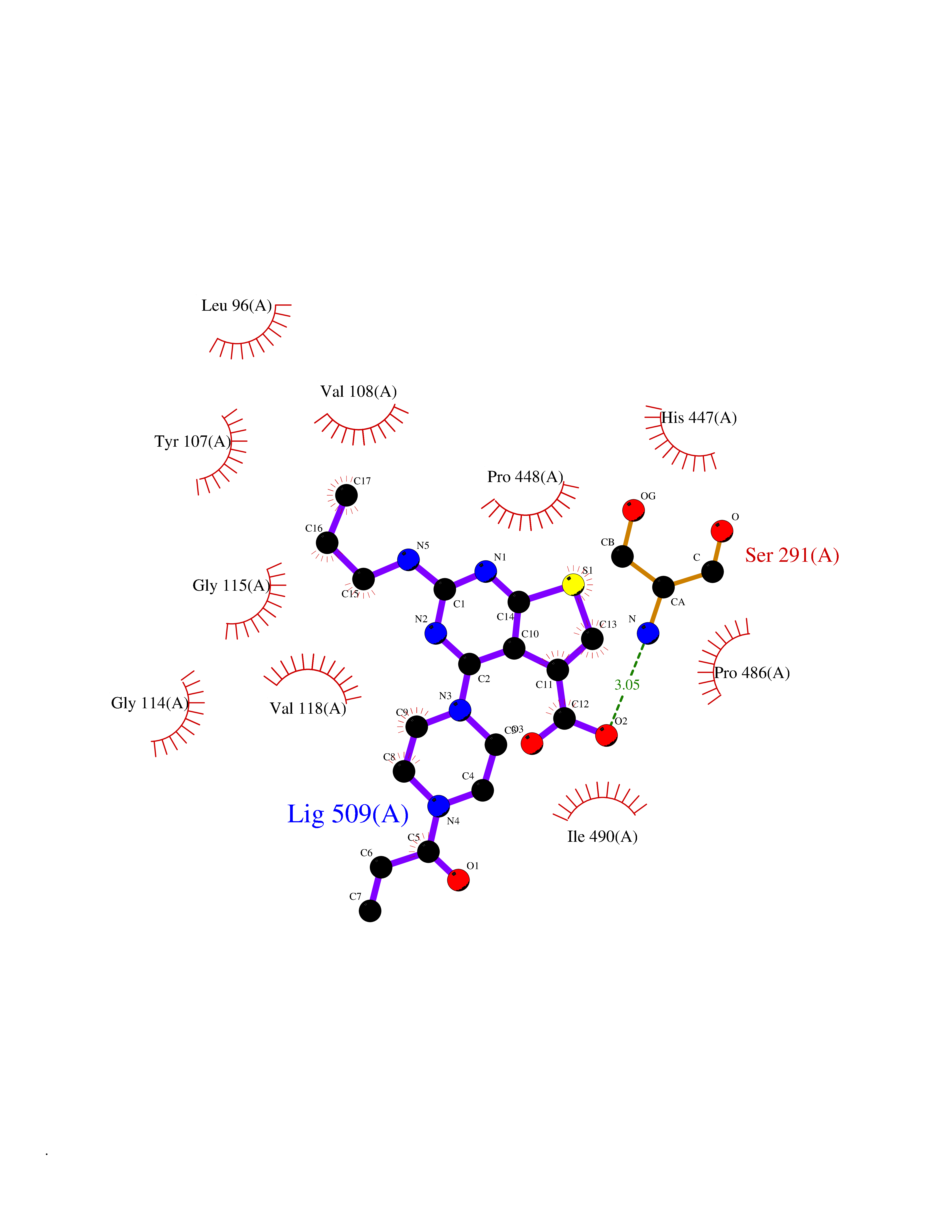 Ligplot Map 3D Binding mode Sequence GYVPAVVIGTGYGAAVSALRLGEAGVQTLMLEMGQLWNQPGPDGNIFCGMLNPDKRSSWFKNRTEAPLGSFLWLDVVNRNIDPYAGVLDRVNYDQMSVYVGRGVGGGSLVNGGMAVEPKRSYFEEILPRVDSSEMYDRYFPRANSMLRVNHIDTKWFEDTEWYKFARVSREQAGKAGLGTVFVPNVYDFGYMQREAAGEVPKSALATEVIYGNNHGKQSLDKTYLAAALGTGKVTIQTLHQVKTIRQTKDGGYALTVEQKDTDGKLLATKEISCRYLFLGAGSLGSTELLVRARDTGTLPNLNSEVGAGWGPNGNIMTARANHMWNPTGAHQSSIPALGIDAWDNSDSSVFAEIAPMPAGLETWVSLYLAITKNPQRGTFVYDAATDRAKLNWTRDQNAPAVNAAKALFDRINKANGTIYRYDLFGTQLKAFADDFCYHPLGGCVLGKATDDYGRVAGYKNLYVTDGSLIPGSVGVNPFVTITALAERNVERIIKQDV Hydrogen bonds contact Hydrophobic contact | ||||
| 87 | Penicillin acylase | 2PVA | 6.53 | |
Target general information Gen name N/A Organism Lysinibacillus sphaericus (Bacillus sphaericus) Uniprot ID TTD ID NA Synonyms NA Protein family Peptidase C59 family Biochemical class Hydrolase Function Penicillin amidase activity. Related diseases Usher syndrome 3B (USH3B) [MIM:614504]: A syndrome characterized by progressive vision and hearing loss during early childhood. Some patients have the so-called 'Charles Bonnet syndrome,' involving decreased visual acuity and vivid visual hallucinations. USH is a genetically heterogeneous condition characterized by the association of retinitis pigmentosa with sensorineural deafness. Age at onset and differences in auditory and vestibular function distinguish Usher syndrome type 1 (USH1), Usher syndrome type 2 (USH2) and Usher syndrome type 3 (USH3). USH3 is characterized by postlingual, progressive hearing loss, variable vestibular dysfunction, and onset of retinitis pigmentosa symptoms, including nyctalopia, constriction of the visual fields, and loss of central visual acuity, usually by the second decade of life. {ECO:0000269|PubMed:22279524}. The disease may be caused by variants affecting the gene represented in this entry.; DISEASE: Charcot-Marie-Tooth disease, axonal, 2W (CMT2W) [MIM:616625]: An autosomal dominant, axonal form of Charcot-Marie-Tooth disease, a disorder of the peripheral nervous system, characterized by progressive weakness and atrophy, initially of the peroneal muscles and later of the distal muscles of the arms. Charcot-Marie-Tooth disease is classified in two main groups on the basis of electrophysiologic properties and histopathology: primary peripheral demyelinating neuropathies (designated CMT1 when they are dominantly inherited) and primary peripheral axonal neuropathies (CMT2). Neuropathies of the CMT2 group are characterized by signs of axonal degeneration in the absence of obvious myelin alterations, normal or slightly reduced nerve conduction velocities, and progressive distal muscle weakness and atrophy. CMT2W patients manifest a peripheral neuropathy mainly affecting the lower limbs and resulting in gait difficulties and distal sensory impairment. Most patients also have upper limb involvement. {ECO:0000269|PubMed:22930593, ECO:0000269|PubMed:26072516, ECO:0000269|PubMed:29235198}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01822; DB03661; DB00417 Interacts with NA EC number 3.5.1.11 Uniprot keywords 3D-structure; Antibiotic resistance; Direct protein sequencing; Hydrolase; Zymogen Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 32972.1 Length 295 Aromaticity 0.11 Instability index 30.94 Isoelectric point 5.65 Charge (pH=7) -4.18 2D Binding mode Binding energy (Kcal/mol) -8.91 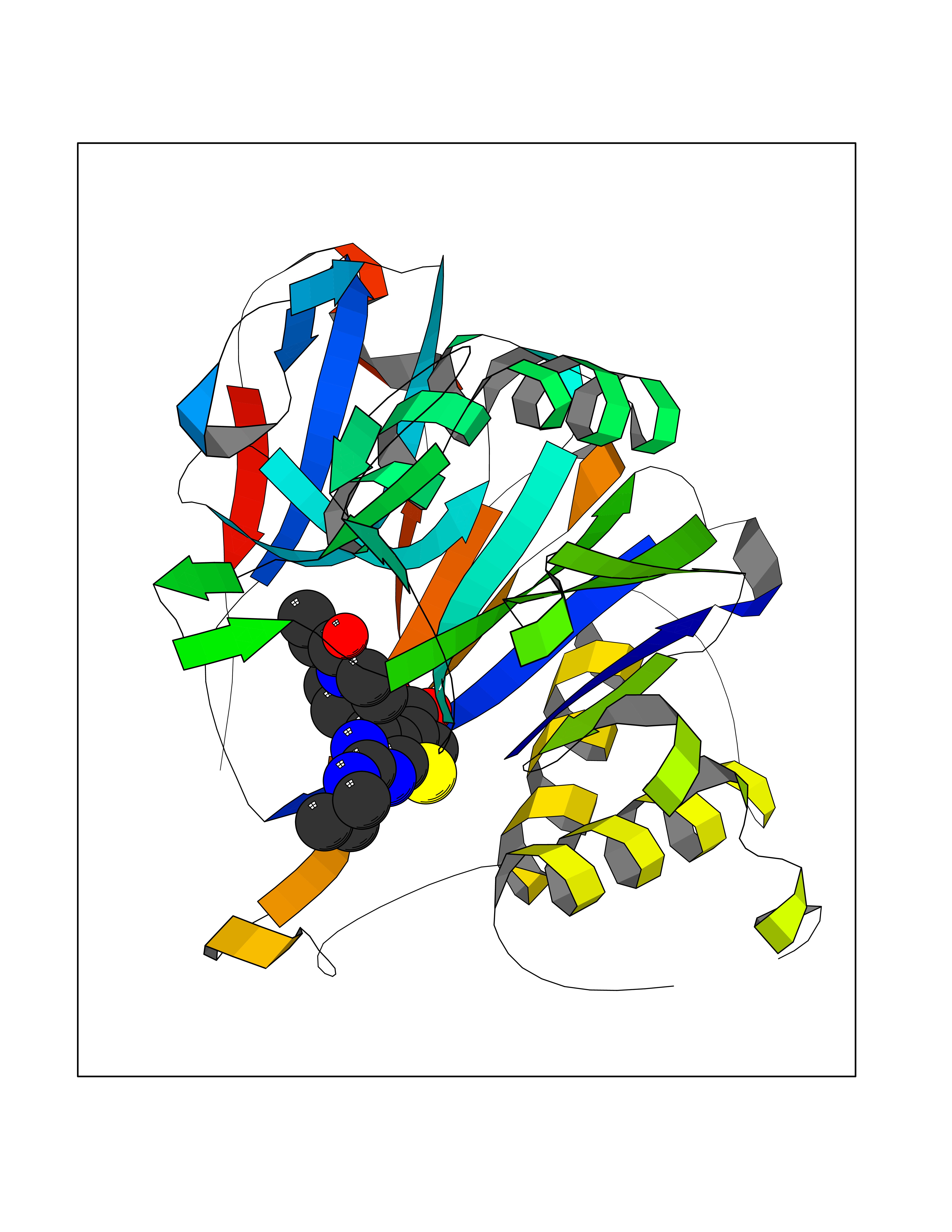 Molscript Map 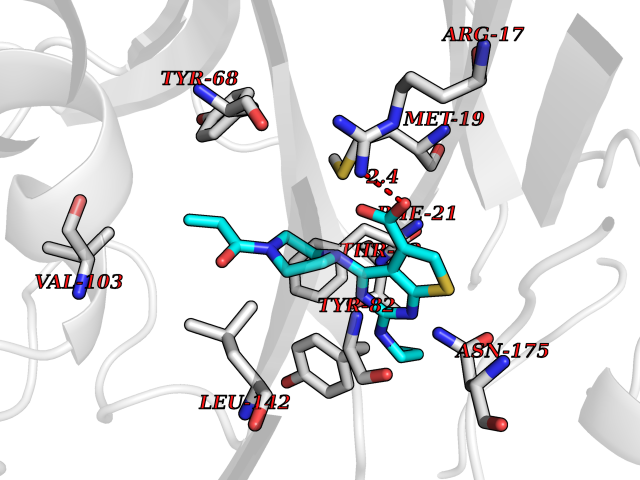 Pymol Map 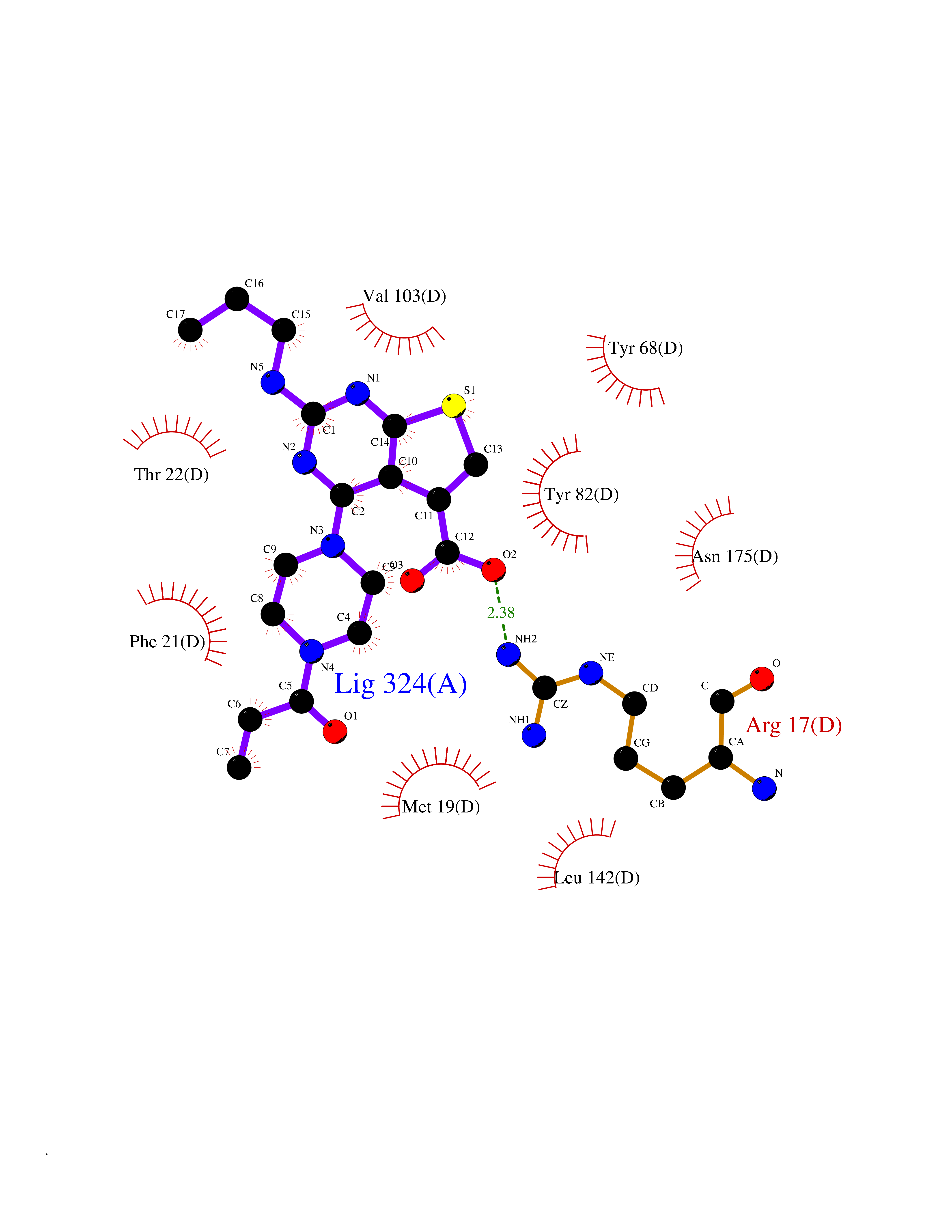 Ligplot Map 3D Binding mode Sequence SSLSIRTTDDKSLFARTMDFTMEPDSKVIIVPRNYGIRLLEKENVVINNSYAFVGMGSTDITSPVLYDGVNEKGLMGAMLYYATFATYADEPKKGTTGINPVYVISQVLGNCVTVDDVIEKLTSYTLLNEANIILGFAPPLHYTFTDASGESIVIEPDKTGITIHRKTIGVMTNSPGYEWHQTNLRAYIGVLPGDFTPSARFLRVAYWKKYTEKAKNETEGVTNLFHILSSVNIPKGVVLTNEGKTDYTIYTSAMCAQSKNYYFKLYDNSRISAVSLMAENLNSQDLITFEWDRK Hydrogen bonds contact Hydrophobic contact | ||||
| 88 | Retinoic acid receptor RXR-beta (RXRB) | 5HJP | 6.53 | |
Target general information Gen name RXRB Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Retinoid X receptor beta; Nuclear receptor subfamily 2 group B member 2; NR2B2 Protein family Nuclear hormone receptor family, NR2 subfamily Biochemical class Nuclear hormone receptor Function Retinoic acid receptors bind as heterodimers to their target response elements in response to their ligands, all-trans or 9-cis retinoic acid, and regulate gene expression in various biological processes. The RAR/RXR heterodimers bind to the retinoic acid response elements (RARE). Receptor for retinoic acid. Related diseases Noonan syndrome 13 (NS13) [MIM:619087]: A form of Noonan syndrome, a disease characterized by short stature, facial dysmorphic features such as hypertelorism, a downward eyeslant and low-set posteriorly rotated ears, and a high incidence of congenital heart defects and hypertrophic cardiomyopathy. Other features can include a short neck with webbing or redundancy of skin, deafness, motor delay, variable intellectual deficits, multiple skeletal defects, cryptorchidism, and bleeding diathesis. Individuals with Noonan syndrome are at risk of juvenile myelomonocytic leukemia, a myeloproliferative disorder characterized by excessive production of myelomonocytic cells. NS13 inheritance is autosomal dominant. There is considerable variability in severity. {ECO:0000269|PubMed:32721402}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08175; DB00459; DB00210; DB00523; DB00307; DB01393; DB03756; DB00926; DB01941; DB07929; DB02746; DB00412; DB00799; DB07080; DB00755 Interacts with Q00975; Q9HB07; F1D8P7; Q13133; Q13133-3; Q96RI1-1; P04150; Q9NRD5; P37231; P10276; P10276-2; P10826-2; P13631; Q6IQ16; Q13137; Q96B26; Q08379; Q6A162; Q9UJV3-2; Q13133-3; Q96RI1-1; O43586; P10276; P10826-2; Q8IUQ4-2; O75528; Q12800; Q9UBB9; Q05BL1; P14373; O94972; Q96S82 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; DNA-binding; Metal-binding; Methylation; Nucleus; Proteomics identification; Receptor; Reference proteome; Transcription; Transcription regulation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A,C Molecular weight (Da) 28845.8 Length 251 Aromaticity 0.08 Instability index 54.86 Isoelectric point 6.74 Charge (pH=7) -0.6 2D Binding mode Binding energy (Kcal/mol) -8.9 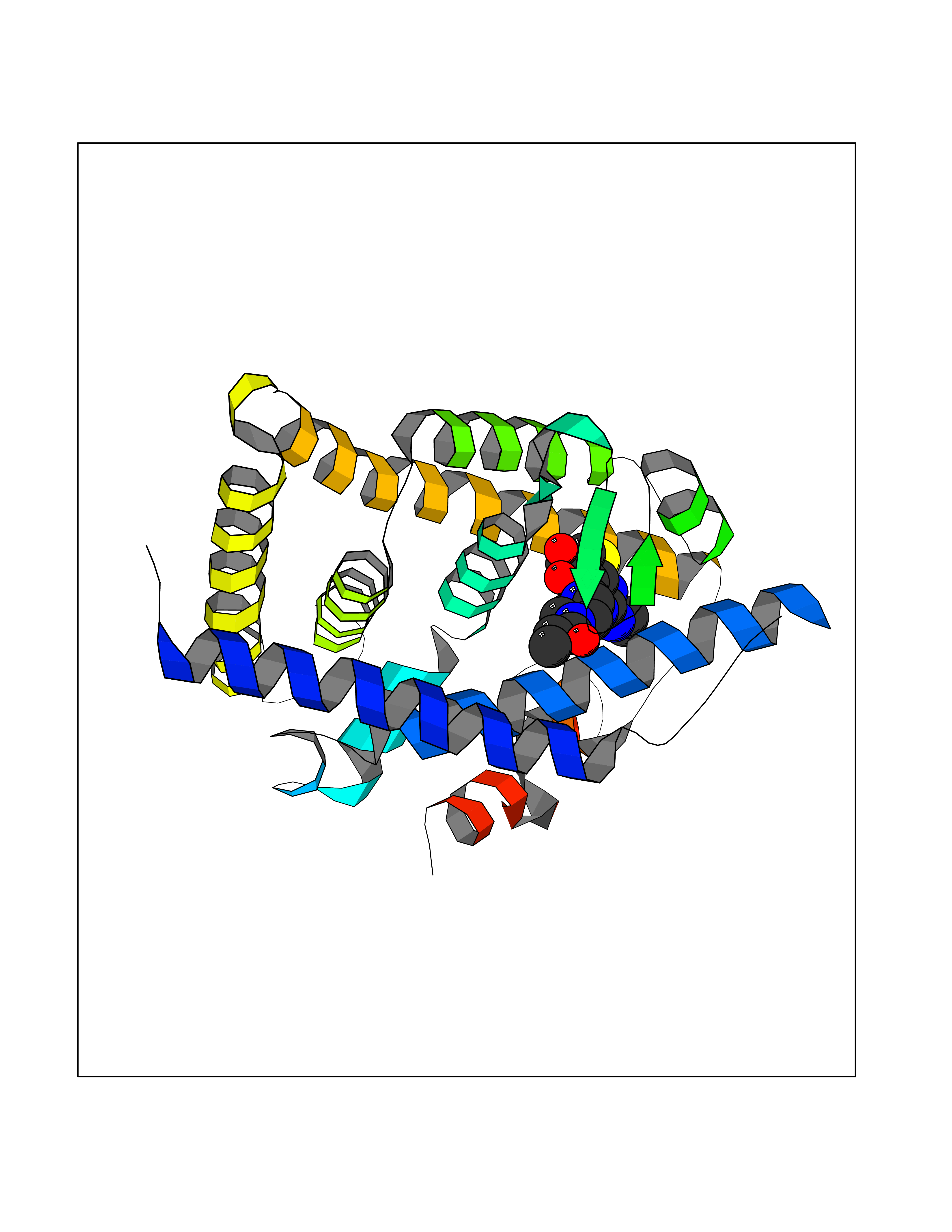 Molscript Map 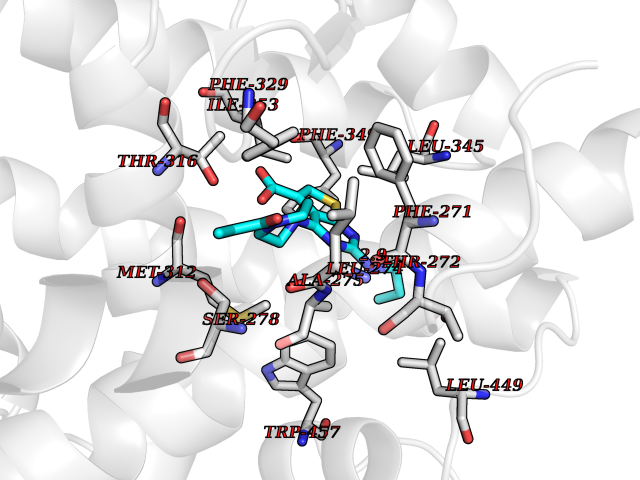 Pymol Map 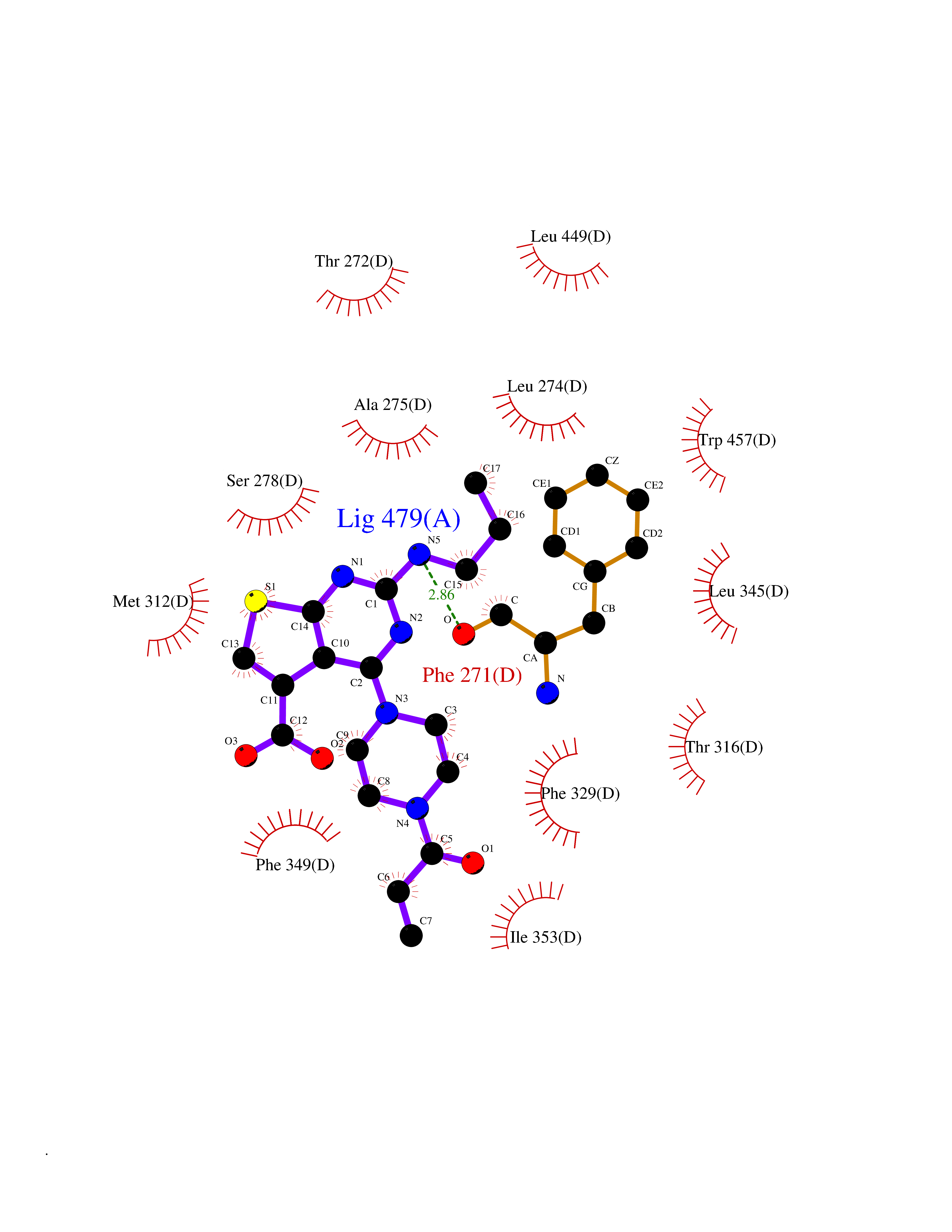 Ligplot Map 3D Binding mode Sequence QLTAAQELMIQQLVAAQLQCNKRSFSDQPKVTPWPSASQQRFAHFTELAIISVQEIVDFAKQVPGFLQLGREDQIALLKASTIEIMLLETARRYNHETECITFLKDFTYSKDDFHRAGLQVEFINPIFEFSRAMRRLGLDDAEYALLIAINIFSADRPNVQEPGRVEALQQPYVEALLSYTRIKRPQDQLRFPRMLMKLVSLRTLSSVHSEQVFALRLQDKKLPPLLSEIWDVHEGSGSGSHKILHRLLQD Hydrogen bonds contact Hydrophobic contact | ||||
| 89 | Glycolipid transfer protein | 3RZN | 6.53 | |
Target general information Gen name GLTP Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family GLTP family Biochemical class Lipid transport Function Glycolipid binding.Glycolipid transporter activity.Identical protein binding.Intermembrane lipid transfer activity.Lipid binding. Related diseases Brugada syndrome 7 (BRGDA7) [MIM:613120]: A tachyarrhythmia characterized by right bundle branch block and ST segment elevation on an electrocardiogram (ECG). It can cause the ventricles to beat so fast that the blood is prevented from circulating efficiently in the body. When this situation occurs, the individual will faint and may die in a few minutes if the heart is not reset. {ECO:0000269|PubMed:20031595}. The gene represented in this entry may be involved in disease pathogenesis.; DISEASE: Atrial fibrillation, familial, 16 (ATFB16) [MIM:613120]: A familial form of atrial fibrillation, a common sustained cardiac rhythm disturbance. Atrial fibrillation is characterized by disorganized atrial electrical activity and ineffective atrial contraction promoting blood stasis in the atria and reduces ventricular filling. It can result in palpitations, syncope, thromboembolic stroke, and congestive heart failure. {ECO:0000269|PubMed:20558140, ECO:0000269|PubMed:21051419}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03600; DB04465; DB03017; DB03203 Interacts with Q96DZ9; Q9NZD2 EC number NA Uniprot keywords 3D-structure; Acetylation; Cytoplasm; Lipid transport; Proteomics identification; Reference proteome; Repeat; Transport Protein physicochemical properties Chain ID A Molecular weight (Da) 23534.1 Length 206 Aromaticity 0.11 Instability index 36.45 Isoelectric point 7.08 Charge (pH=7) 0.1 2D Binding mode Binding energy (Kcal/mol) -8.9 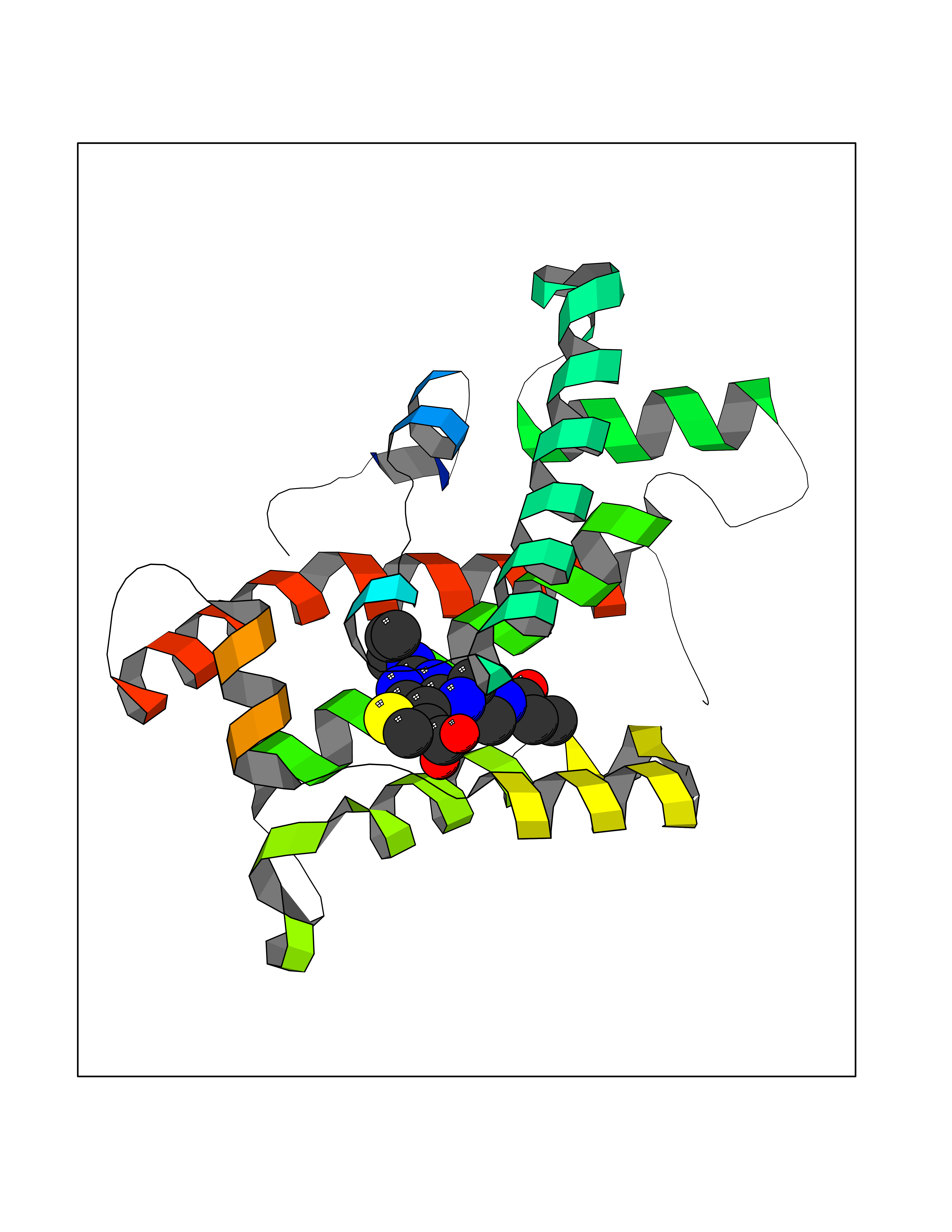 Molscript Map 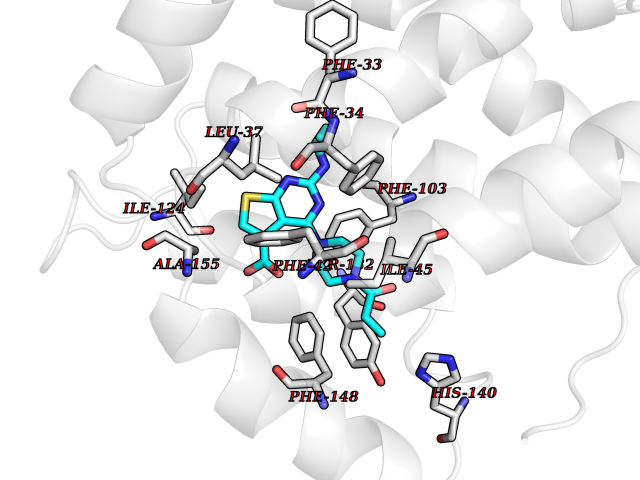 Pymol Map 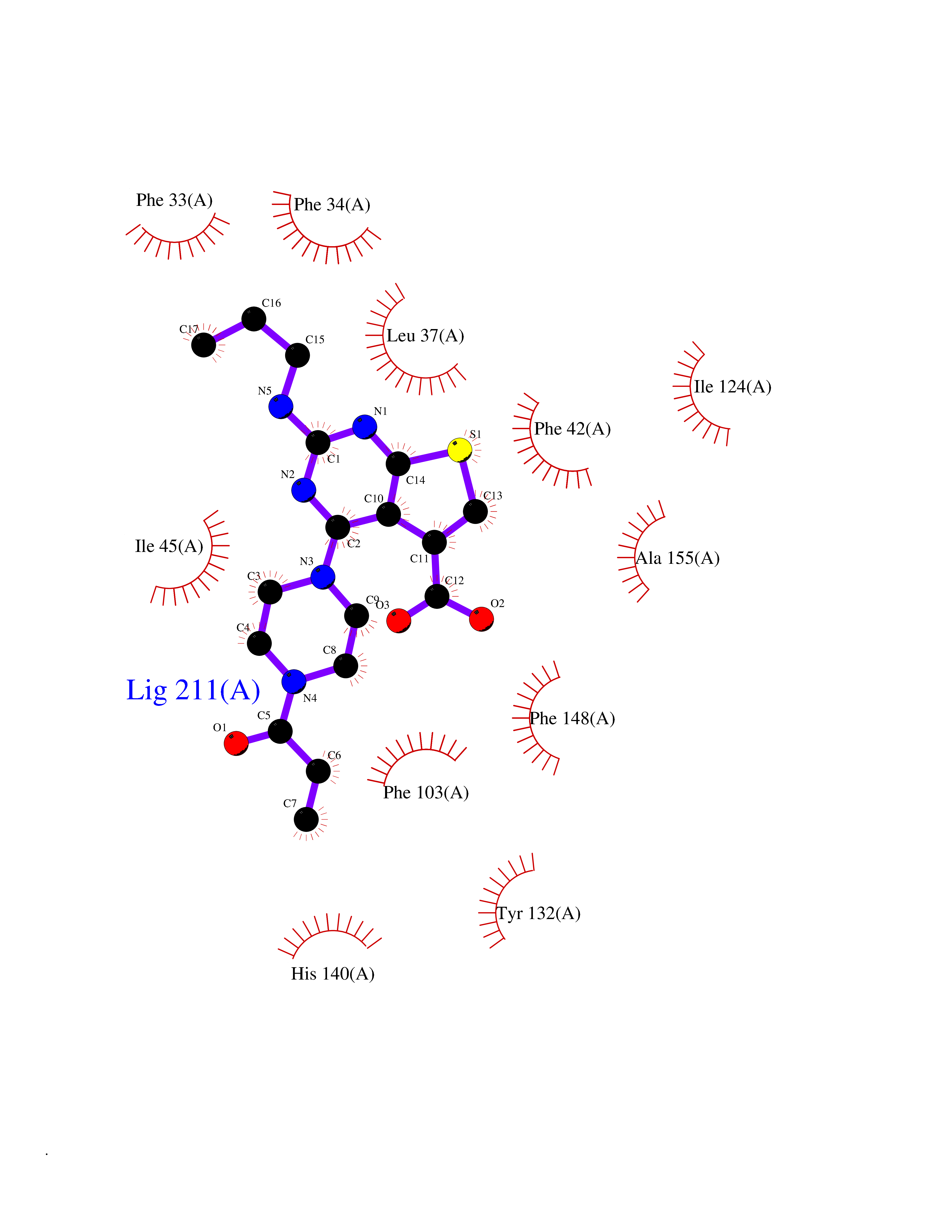 Ligplot Map 3D Binding mode Sequence LAEHLLKPLPADKQIETGPFLEAVSHLPPFFDCLGSPVFTPIKADISGNITKIKAVYDTNPAKFRTLQNILEVEKEMYGAEWPKVGATLALMWLKRGLRFIQVFLQSICDGERDENHPNLIRVNATKAYEMALKKYHGWIVQKIFQAALYAAPYKSDFLKALSKGQNVTEEECLEKIRLFLVNYTATIDVIYEMYTQMNAELNYKV Hydrogen bonds contact Hydrophobic contact | ||||
| 90 | Lysine-specific histone demethylase 1 (LSD) | 6W4K | 6.53 | |
Target general information Gen name KDM1A Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Lysine-specific histone demethylase 1A; LSD1; KIAA0601; KDM1; Flavin-containing amine oxidase domain-containing protein 2; BRAF35-HDAC complex protein BHC110; AOF2 Protein family Flavin monoamine oxidase family Biochemical class CH-NH(2) donor oxidoreductase Function Histone demethylase that can demethylate both 'Lys-4' (H3K4me) and 'Lys-9' (H3K9me) of histone H3, thereby acting as a coactivator or a corepressor, depending on the context. Acts by oxidizing the substrate by FAD to generate the corresponding imine that is subsequently hydrolyzed. Acts as a corepressor by mediating demethylation of H3K4me, a specific tag for epigenetic transcriptional activation. Demethylates both mono- (H3K4me1) and di-methylated (H3K4me2) H3K4me. May play a role in the repression of neuronal genes. Alone, it is unable to demethylate H3K4me on nucleosomes and requires the presence of RCOR1/CoREST to achieve such activity. Also acts as a coactivator of androgen receptor (ANDR)-dependent transcription, by being recruited to ANDR target genes and mediating demethylation of H3K9me, a specific tag for epigenetic transcriptional repression. The presence of PRKCB in ANDR-containing complexes, which mediates phosphorylation of 'Thr-6' of histone H3 (H3T6ph), a specific tag that prevents demethylation H3K4me, prevents H3K4me demethylase activity of KDM1A. Demethylates di-methylated 'Lys-370' of p53/TP53 which prevents interaction of p53/TP53 with TP53BP1 and represses p53/TP53-mediated transcriptional activation. Demethylates and stabilizes the DNA methylase DNMT1. Required for gastrulation during embryogenesis. Component of a RCOR/GFI/KDM1A/HDAC complex that suppresses, via histone deacetylase (HDAC) recruitment, a number of genes implicated in multilineage blood cell development. Effector of SNAI1-mediated transcription repression of E-cadherin/CDH1, CDN7 and KRT8. Required for the maintenance of the silenced state of the SNAI1 target genes E-cadherin/CDH1 and CDN7. Related diseases Cleft palate, psychomotor retardation, and distinctive facial features (CPRF) [MIM:616728]: A syndrome characterized by cleft palate, developmental delay, psychomotor retardation, and facial dysmorphic features including a prominent forehead, slightly arched eyebrows, elongated palpebral fissures, a wide nasal bridge, thin lips, and widely spaced teeth. Cleft palate is a congenital fissure of the soft and/or hard palate, due to faulty fusion. {ECO:0000269|PubMed:23020937, ECO:0000269|PubMed:24838796, ECO:0000269|PubMed:26656649}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB16446 Interacts with Q9BYF1; Q99996; Q86SG2; Q13490; Q6P047; Q8TC20-4; Q9BWT7; Q49A88-3; Q99459; Q9BXL8; Q86XR8-3; Q8NHQ1; Q8TAP6; Q12873; P38432; Q96EY1; P26378-2; Q9UPT5-1; Q3B820; Q9H8W3; Q8IZU1; Q9BQS8; O95995; Q96CN9; Q08379; Q9NYA3; Q8NEC7; Q9BX10; Q16695; Q96CS2; O15379; Q9UBX0; Q9NSC5; Q16891; O75564-2; O60341; Q9BVG8-5; Q8TBB5; P19012; Q15323; Q14525; Q92764; Q6A163; Q6A162; Q96JB6; Q9Y250; O95983; P01106; Q3BBV0; Q7Z6G3-2; P35240; Q16236; P46531; Q13133; Q9Y466; Q96F24; A5D8V7; Q8IZS5; Q9BYU1; Q8IZL8; Q99471; Q5T6S3; Q03181; P62191; Q9NS23; P50749; Q86WH2; Q06330; Q9UKL0; Q8IZ40; Q9P2K3-2; Q96P16-3; Q8N6K7; Q9UGK8; Q13435; O15198; O95863; Q96H20; Q8N0Z3; Q96BD6; Q8N4C7; Q8N6V9; Q9UBB9; Q08117-2; Q2M3C6; P45379-11; Q05BL1; Q9BUZ4; Q5W5X9-3; Q8TF42; Q9H9H4; Q9Y3C0; O96006; O15060; Q92618; O14646; Q96KQ7; Q96KQ7-1; P05771-1; Q9UKL0; P17542; P04637; P22091 EC number EC 1.-.-.- Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Chromatin regulator; Chromosome; Coiled coil; Developmental protein; Direct protein sequencing; Disease variant; FAD; Flavoprotein; Isopeptide bond; Nucleus; Oxidoreductase; Phosphoprotein; Proteomics identification; Reference proteome; Repressor; Transcription; Transcription regulation; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 72462.2 Length 651 Aromaticity 0.08 Instability index 32.07 Isoelectric point 7.01 Charge (pH=7) 0.05 2D Binding mode Binding energy (Kcal/mol) -8.9  Molscript Map 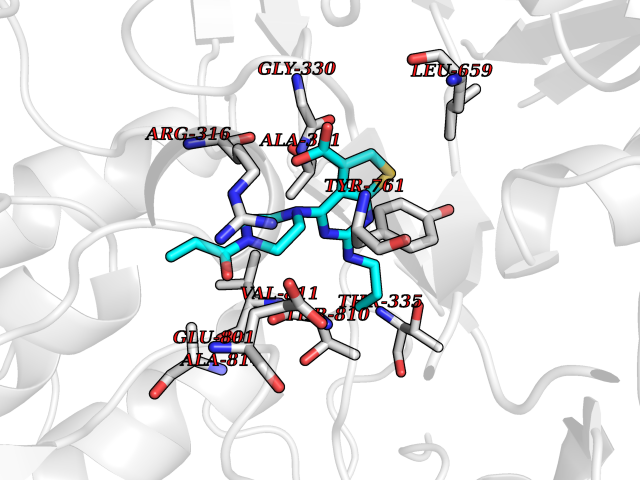 Pymol Map 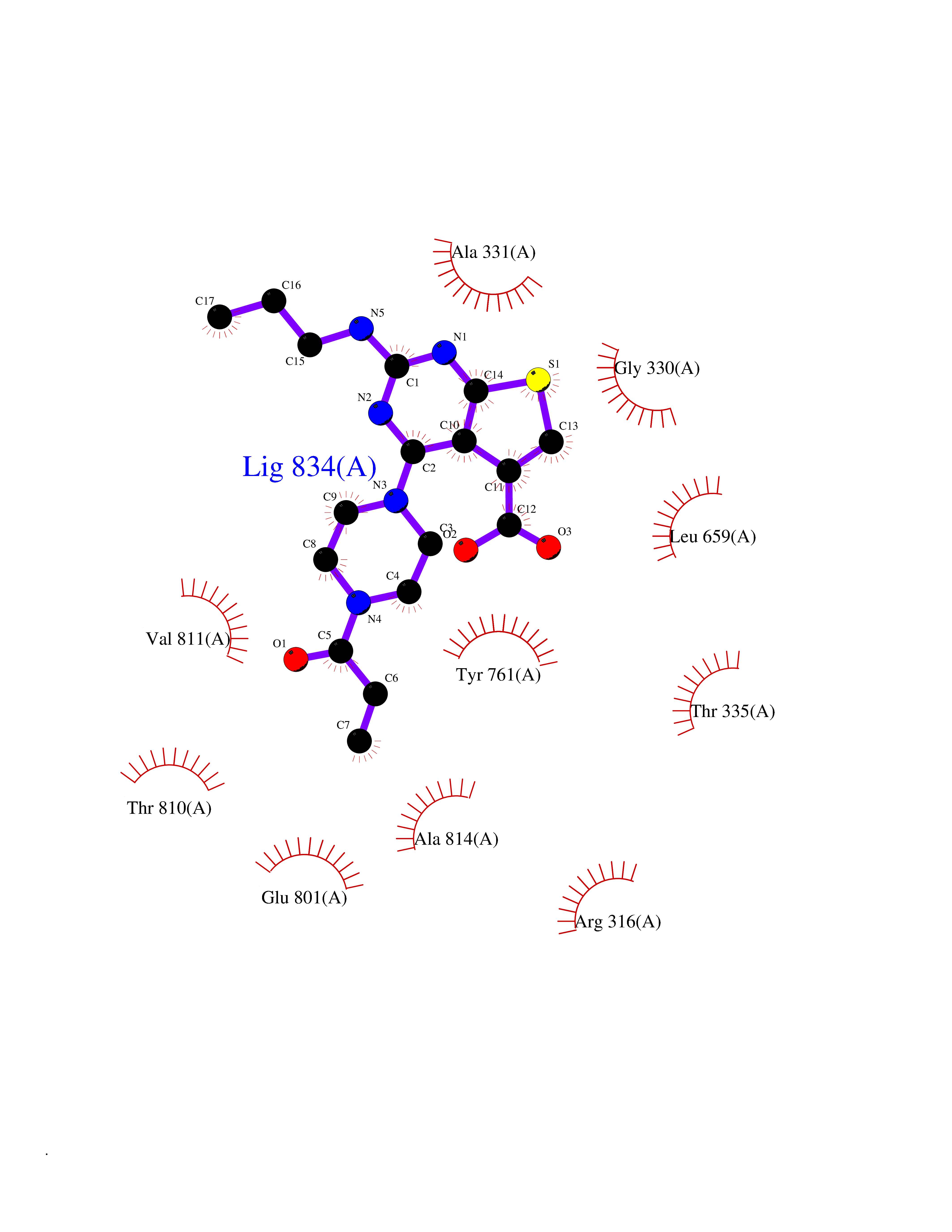 Ligplot Map 3D Binding mode Sequence VEGAAFQSRLPHDRMTSQEAACFPDIISGPQQTQKVFLFIRNRTLQLWLDNPKIQLTFEATLQQLEAPYNSDTVLVHRVHSYLERHGLINFGIYKRIKPLPTKKTGKVIIIGSGVSGLAAARQLQSFGMDVTLLEARDRVGGRVATFRKGNYVADLGAMVVTGLGGNPMMELAKIKQKCPLYEANGQAVPKEKDEMVEQEFNRLLEATSYLSHQLDFNVLNNKPVSLGQALEVVIQLQEKHVKDEQIEHWKKIVKTQEELKELLNKMVNLKEKIKELHQQYKEASEVKPPRDITAEFLVKSKHRDLTALCKEYDELAETQGKLEEKLQELEANPPSDVYLSSRDRQILDWHFANLEFANATPLSTLSLKHWDQDDDFEFTGSHLTVRNGYSCVPVALAEGLDIKLNTAVRQVRYTASGCEVIAVNTRSTSQTFIYKCDAVLCTLPLGVLKQQPPAVQFVPPLPEWKTSAVQRMGFGNLNKVVLCFDRVFWDPSVNLFGHVGSTTASRGELFLFWNLYKAPILLALVAGEAAGIMENISDDVIVGRCLAILKGIFGSSAVPQPKETVVSRWRADPWARGSYSYVAAGSSGNDYDLMAQPITPGPSIPGAPQPIPRLFFAGEHTIRNYPATVHGALLSGLREAGRIADQFLGA Hydrogen bonds contact Hydrophobic contact | ||||
| 91 | Glucagon-like peptide 1 receptor (GLP1R) | 6X1A | 6.53 | |
Target general information Gen name GLP1R Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms GLP-1R; GLP-1-R; GLP-1 receptor Protein family G-protein coupled receptor 2 family Biochemical class GPCR secretin Function Ligand binding triggers activation of a signaling cascade that leads to the activation of adenylyl cyclase and increased intracellular cAMP levels. Plays a role in regulating insulin secretion in response to GLP-1. G-protein coupled receptor for glucagon-like peptide 1 (GLP-1). Related diseases Lynch syndrome 2 (LYNCH2) [MIM:609310]: A form of Lynch syndrome, an autosomal dominant disease associated with marked increase in cancer susceptibility. It is characterized by a familial predisposition to early-onset colorectal carcinoma (CRC) and extra-colonic tumors of the gastrointestinal, urological and female reproductive tracts. Lynch syndrome is reported to be the most common form of inherited colorectal cancer in the Western world. Clinically, it is often divided into two subgroups. Type I is characterized by hereditary predisposition to colorectal cancer, a young age of onset, and carcinoma observed in the proximal colon. Type II is characterized by increased risk for cancers in certain tissues such as the uterus, ovary, breast, stomach, small intestine, skin, and larynx in addition to the colon. Diagnosis of classical Lynch syndrome is based on the Amsterdam criteria: 3 or more relatives affected by colorectal cancer, one a first degree relative of the other two; 2 or more generation affected; 1 or more colorectal cancers presenting before 50 years of age; exclusion of hereditary polyposis syndromes. The term 'suspected Lynch syndrome' or 'incomplete Lynch syndrome' can be used to describe families who do not or only partially fulfill the Amsterdam criteria, but in whom a genetic basis for colon cancer is strongly suspected. {ECO:0000269|PubMed:10323887, ECO:0000269|PubMed:10375096, ECO:0000269|PubMed:10386556, ECO:0000269|PubMed:10413423, ECO:0000269|PubMed:10480359, ECO:0000269|PubMed:10598809, ECO:0000269|PubMed:10627141, ECO:0000269|PubMed:10660333, ECO:0000269|PubMed:10671064, ECO:0000269|PubMed:10713887, ECO:0000269|PubMed:10777691, ECO:0000269|PubMed:10882759, ECO:0000269|PubMed:11139242, ECO:0000269|PubMed:11427529, ECO:0000269|PubMed:11726306, ECO:0000269|PubMed:11748856, ECO:0000269|PubMed:11754112, ECO:0000269|PubMed:11781295, ECO:0000269|PubMed:11793442, ECO:0000269|PubMed:11839723, ECO:0000269|PubMed:11870161, ECO:0000269|PubMed:12095971, ECO:0000269|PubMed:12132870, ECO:0000269|PubMed:12200596, ECO:0000269|PubMed:12362047, ECO:0000269|PubMed:12373605, ECO:0000269|PubMed:12655562, ECO:0000269|PubMed:12658575, ECO:0000269|PubMed:14635101, ECO:0000269|PubMed:14961575, ECO:0000269|PubMed:15064764, ECO:0000269|PubMed:15139004, ECO:0000269|PubMed:15365995, ECO:0000269|PubMed:15365996, ECO:0000269|PubMed:16083711, ECO:0000269|PubMed:16451135, ECO:0000269|PubMed:17301300, ECO:0000269|PubMed:17510385, ECO:0000269|PubMed:18561205, ECO:0000269|PubMed:20020535, ECO:0000269|PubMed:21120944, ECO:0000269|PubMed:22753075, ECO:0000269|PubMed:7757073, ECO:0000269|PubMed:8566964, ECO:0000269|PubMed:8571956, ECO:0000269|PubMed:8574961, ECO:0000269|PubMed:8797773, ECO:0000269|PubMed:8872463, ECO:0000269|PubMed:8993976, ECO:0000269|PubMed:9048925, ECO:0000269|PubMed:9067757, ECO:0000269|PubMed:9218993, ECO:0000269|PubMed:9272156, ECO:0000269|PubMed:9298827, ECO:0000269|PubMed:9311737, ECO:0000269|PubMed:9326924, ECO:0000269|PubMed:9399661, ECO:0000269|PubMed:9559627, ECO:0000269|PubMed:9718327, ECO:0000269|PubMed:9833759, ECO:0000269|PubMed:9927034, ECO:0000269|Ref.5}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Mismatch repair cancer syndrome 1 (MMRCS1) [MIM:276300]: An autosomal recessive form of mismatch repair cancer syndrome, a childhood cancer predisposition syndrome encompassing a broad tumor spectrum. This includes hematological malignancies, central nervous system tumors, Lynch syndrome-associated malignancies such as colorectal tumors as well as multiple intestinal polyps, embryonic tumors and rhabdomyosarcoma. Multiple cafe-au-lait macules, a feature reminiscent of neurofibromatosis type 1, are often found as first manifestation of the underlying cancer. {ECO:0000269|PubMed:11427529, ECO:0000269|PubMed:17440981, ECO:0000269|PubMed:7661930}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Muir-Torre syndrome (MRTES) [MIM:158320]: Rare autosomal dominant disorder characterized by sebaceous neoplasms and visceral malignancy. {ECO:0000269|PubMed:8751876}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Defects in MLH1 may contribute to lobular carcinoma in situ (LCIS), a non-invasive neoplastic disease of the breast.; DISEASE: Endometrial cancer (ENDMC) [MIM:608089]: A malignancy of endometrium, the mucous lining of the uterus. Most endometrial cancers are adenocarcinomas, cancers that begin in cells that make and release mucus and other fluids. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Some epigenetic changes can be transmitted unchanged through the germline (termed 'epigenetic inheritance'). Evidence that this mechanism occurs in humans is provided by the identification of individuals in whom 1 allele of the MLH1 gene is epigenetically silenced throughout the soma (implying a germline event). These individuals are affected by Lynch syndrome but does not have identifiable mutations in MLH1, even though it is silenced, which demonstrates that an epimutation can phenocopy a genetic disease.; DISEASE: Colorectal cancer (CRC) [MIM:114500]: A complex disease characterized by malignant lesions arising from the inner wall of the large intestine (the colon) and the rectum. Genetic alterations are often associated with progression from premalignant lesion (adenoma) to invasive adenocarcinoma. Risk factors for cancer of the colon and rectum include colon polyps, long-standing ulcerative colitis, and genetic family history. {ECO:0000269|PubMed:10598809, ECO:0000269|PubMed:10882759, ECO:0000269|PubMed:12132870, ECO:0000269|PubMed:12655564, ECO:0000269|PubMed:14504054, ECO:0000269|PubMed:15184898, ECO:0000269|PubMed:18033691, ECO:0000269|PubMed:8872463, ECO:0000269|PubMed:9032648, ECO:0000269|PubMed:9087566, ECO:0000269|PubMed:9611074}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB09043; DB09045; DB15650; DB01276; DB00040; DB16697; DB06655; DB09265; DB13928; DB14027; DB15171 Interacts with A8MQ03; Q07627; Q8IUG1; P60409; P60410; P60411; Q9BYP8; P26371; Q7Z3S9; P0DPK4 EC number NA Uniprot keywords 3D-structure; ADP-ribosylation; Cell membrane; Disulfide bond; G-protein coupled receptor; Glycoprotein; Membrane; Receptor; Reference proteome; Signal; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID R Molecular weight (Da) 45579.6 Length 390 Aromaticity 0.16 Instability index 39.66 Isoelectric point 6.73 Charge (pH=7) -0.68 2D Binding mode Binding energy (Kcal/mol) -8.9  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence ATVSLWETVQKWREYRRQCQRSLTEDPPPATDLFCNRTFDEYACWPDGEPGSFVNVSCPWYLPWASSVPQGHVYRFCTAEGLWLQKDNSSLPWRDLSECEESSPEEQLLFLYIIYTVGYALSFSALVIASAILLGFRHLHCTRNYIHLNLFASFILRALSVFIKDAALKWMYSTAAQQHQWDGLLSYQDSLSCRLVFLLMQYCVAANYYWLLVEGVYLYTLLAFSVFSEQWIFRLYVSIGWGVPLLFVVPWGIVKYLYEDEGCWTRNSNMNYWLIIRLPILFAIGVNFLIFVRVICIVVSKLKANLMCKTDIKCRLAKSTLTLIPLLGTHEVIFAFVMDEHARGTLRFIKLFTELSFTSFQGLMVAILYCFVNNEVQLEFRKSWERWRLE Hydrogen bonds contact Hydrophobic contact | ||||
| 92 | Bacterial Cystathionine beta-lyase (Bact metC) | 4ITX | 6.53 | |
Target general information Gen name Bact metC Organism Escherichia coli (strain K12) Uniprot ID TTD ID Synonyms Cysteine-S-conjugate beta-lyase MetC; Cysteine lyase MetC; Cysteine desulfhydrase MetC; Cystathionine beta-lyase MetC; CL; CBL; Beta-cystathionase MetC; Bacterial CD Protein family Trans-sulfuration enzymes family Biochemical class Carbon-sulfur lyases Function Primarily catalyzes the cleavage of cystathionine to homocysteine, pyruvate and ammonia during methionine biosynthesis. Also exhibits cysteine desulfhydrase activity, producing sulfide from cysteine. In addition, under certain growth conditions, exhibits significant alanine racemase coactivity. Related diseases Coronary artery disease, autosomal dominant, 2 (ADCAD2) [MIM:610947]: A common heart disease characterized by reduced or absent blood flow in one or more of the arteries that encircle and supply the heart. Its most important complication is acute myocardial infarction. {ECO:0000269|PubMed:17332414, ECO:0000269|PubMed:23703864}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Tooth agenesis, selective, 7 (STHAG7) [MIM:616724]: An autosomal dominant form of selective tooth agenesis, a common anomaly characterized by the congenital absence of one or more teeth. Selective tooth agenesis without associated systemic disorders has sometimes been divided into 2 types: oligodontia, defined as agenesis of 6 or more permanent teeth, and hypodontia, defined as agenesis of less than 6 teeth. The number in both cases does not include absence of third molars (wisdom teeth). {ECO:0000269|PubMed:26387593}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number EC 4.4.1.13 Uniprot keywords 3D-structure; Amino-acid biosynthesis; Cytoplasm; Direct protein sequencing; Lyase; Methionine biosynthesis; Pyridoxal phosphate; Reference proteome Protein physicochemical properties Chain ID A Molecular weight (Da) 42756.3 Length 391 Aromaticity 0.08 Instability index 27.11 Isoelectric point 6.01 Charge (pH=7) -6.11 2D Binding mode Binding energy (Kcal/mol) -8.91  Molscript Map  Pymol Map 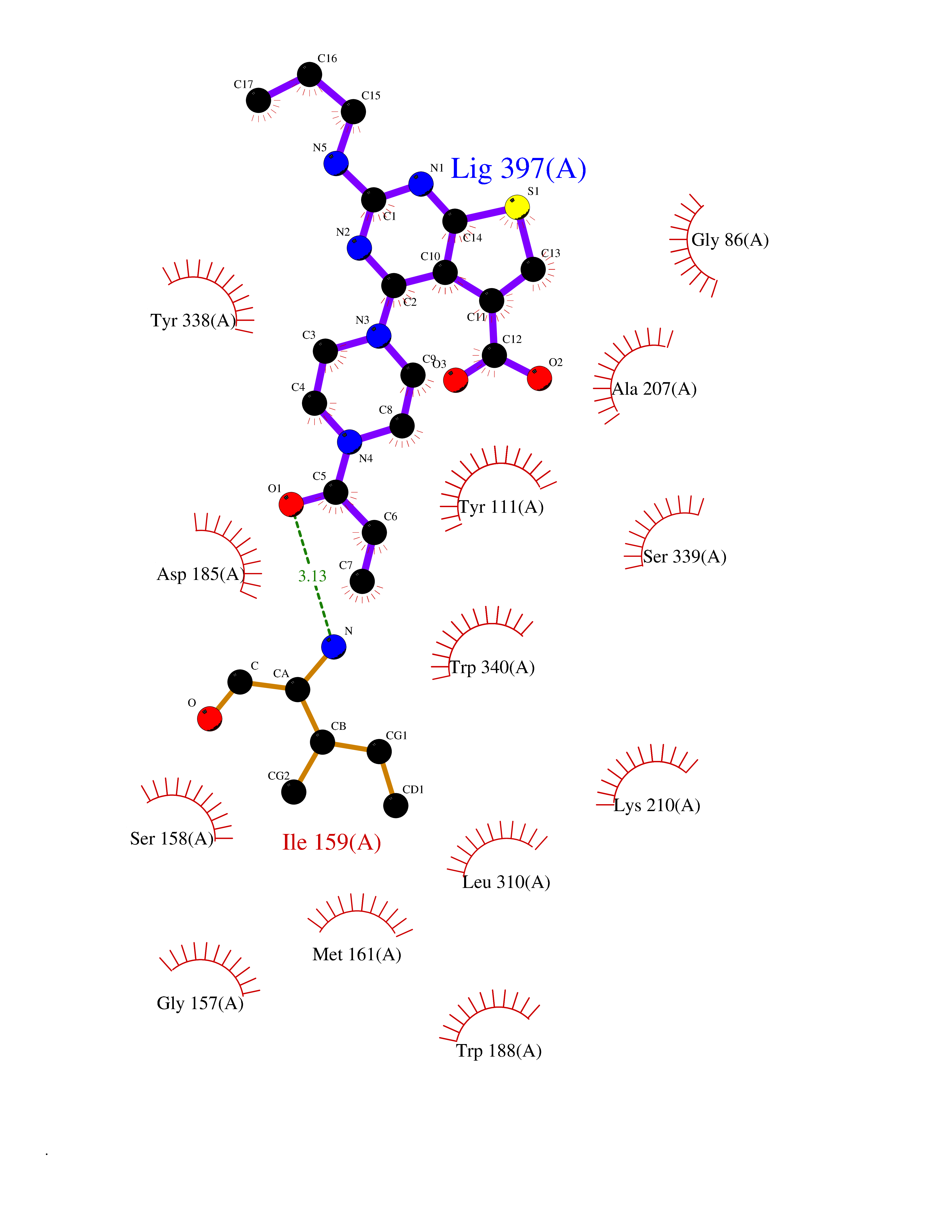 Ligplot Map 3D Binding mode Sequence KLDTQLVNAGRSKKYTLGAVNSVIQRASSLVFDSVEAKKHATRNRANGELFYGRRGTLTHFSLQQAMCELEGGAGCVLFPCGAAAVANSILAFIEQGDHVLMTNTAYESSQDFCSKILSKLGVTTSWFDPLIGADIVKHLQPNTKIVFLESPGSITMEVHDVPAIVAAVRSVVPDAIIMIDNTWAAGVLFKALDFGIDVSIQAATKYLVGHSDAMIGTAVCNARCWEQLRENAYLMGQMVDADTAYITSRGLRTLGVRLRQHHESSLKVAEWLAEHPQVARVNHPALPGSKGHEFWKRDFTGSSGLFSFVLKKKLNNEELANYLDNFSLFSMAYSWGGYESLILANQPEHIAAIRPQGEIDFSGTLIRLHIGLEDVDDLIADLDAGFARIV Hydrogen bonds contact Hydrophobic contact | ||||
| 93 | Mutated oxalosuccinate decarboxylase (mIDH1) | 6ADG | 6.53 | |
Target general information Gen name IDH1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms PICD (mutated); Oxalosuccinate decarboxylase (mutated); NADP(+)-specific ICDH (mutated); Isocitrate dehydrogenase [NADP] cytoplasmic (mutated); IDP (mutated); IDH (mutated); Cytosolic NADP-isocitrate Protein family Isocitrate and isopropylmalate dehydrogenases family Biochemical class Short-chain dehydrogenases reductase Function Catalyses the NADPH-dependent reduction of alpha-ketoglutarate to R(-)-2-hydroxyglutarate (2HG). Related diseases Glioma (GLM) [MIM:137800]: Gliomas are benign or malignant central nervous system neoplasms derived from glial cells. They comprise astrocytomas and glioblastoma multiforme that are derived from astrocytes, oligodendrogliomas derived from oligodendrocytes and ependymomas derived from ependymocytes. {ECO:0000269|PubMed:19117336, ECO:0000269|PubMed:19935646}. The gene represented in this entry is involved in disease pathogenesis. Mutations affecting Arg-132 are tissue-specific, and suggest that this residue plays a unique role in the development of high-grade gliomas. Mutations of Arg-132 to Cys, His, Leu or Ser abolish magnesium binding and abolish the conversion of isocitrate to alpha-ketoglutarate. Instead, alpha-ketoglutarate is converted to R(-)-2-hydroxyglutarate. Elevated levels of R(-)-2-hydroxyglutarate are correlated with an elevated risk of malignant brain tumors. {ECO:0000269|PubMed:19935646}.; DISEASE: Genetic variations are associated with cartilaginous tumors such as enchondroma or chondrosarcoma. Mutations of Arg-132 to Cys, Gly or His abolish the conversion of isocitrate to alpha-ketoglutarate. Instead, alpha-ketoglutarate is converted to R(-)-2-hydroxyglutarate. {ECO:0000269|PubMed:26161668}. Drugs (DrugBank ID) DB09374; DB01727; DB14568; DB03461; DB16267 Interacts with P0DP23; P27797; P36957; O75874; Q8TDX7; P16284; P17612; P50454; P37173; Q05086-3 EC number EC 1.1.1.42 Uniprot keywords 3D-structure; Acetylation; Cytoplasm; Direct protein sequencing; Glyoxylate bypass; Magnesium; Manganese; Metal-binding; NADP; Oxidoreductase; Peroxisome; Phosphoprotein; Proteomics identification; Reference proteome; Tricarboxylic acid cycle Protein physicochemical properties Chain ID A,B Molecular weight (Da) 92711.7 Length 823 Aromaticity 0.1 Instability index 26.74 Isoelectric point 6.42 Charge (pH=7) -4.48 2D Binding mode Binding energy (Kcal/mol) -8.9 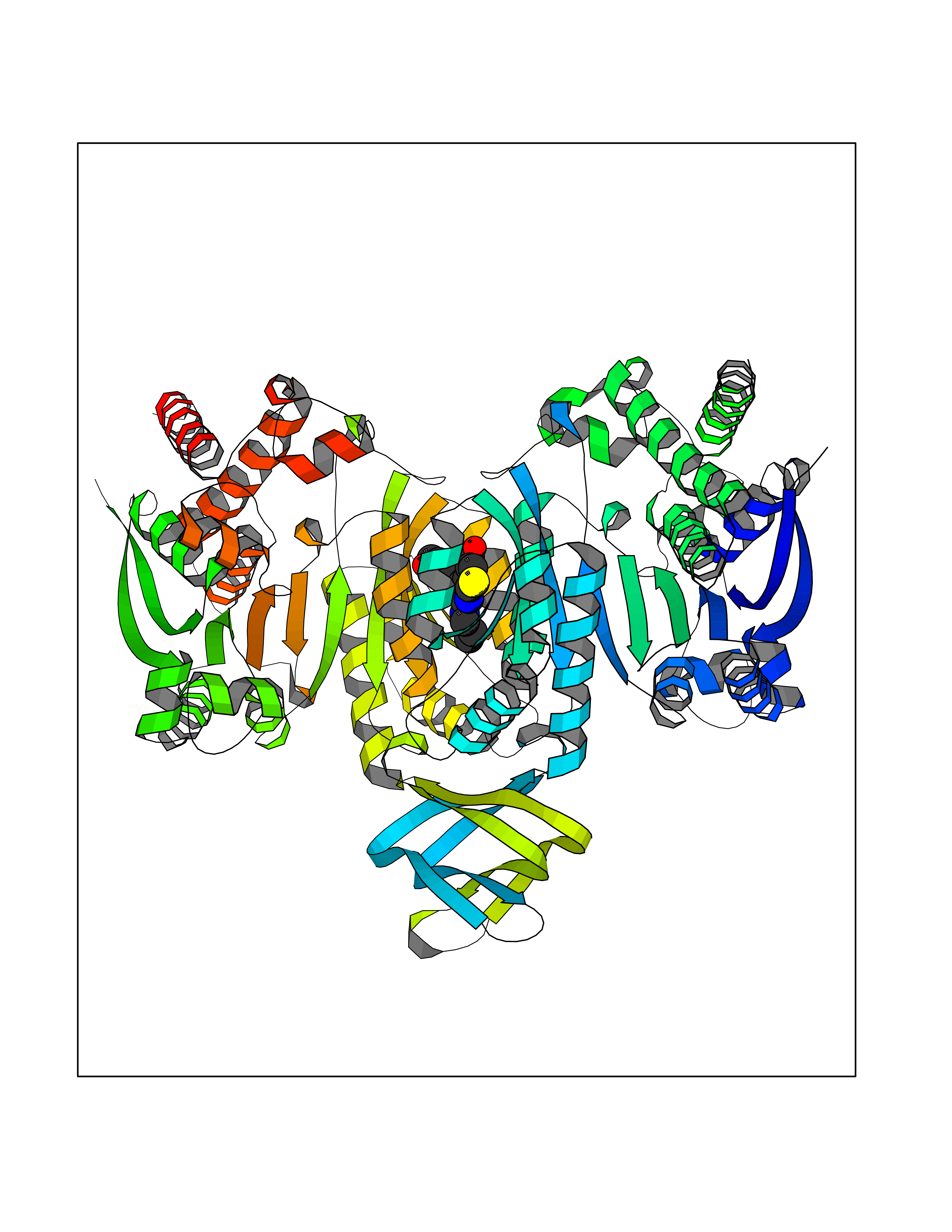 Molscript Map 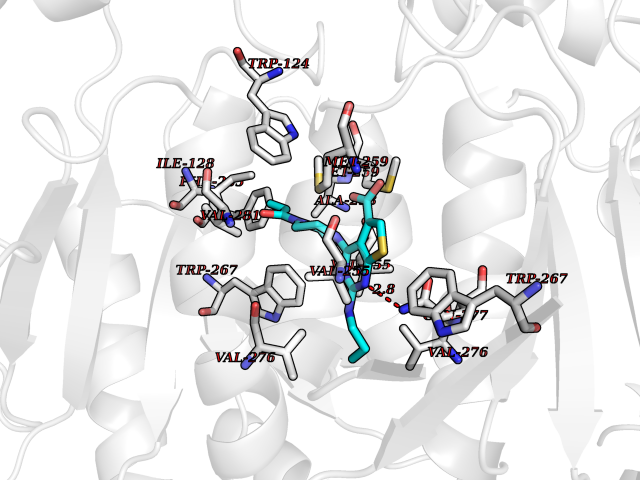 Pymol Map 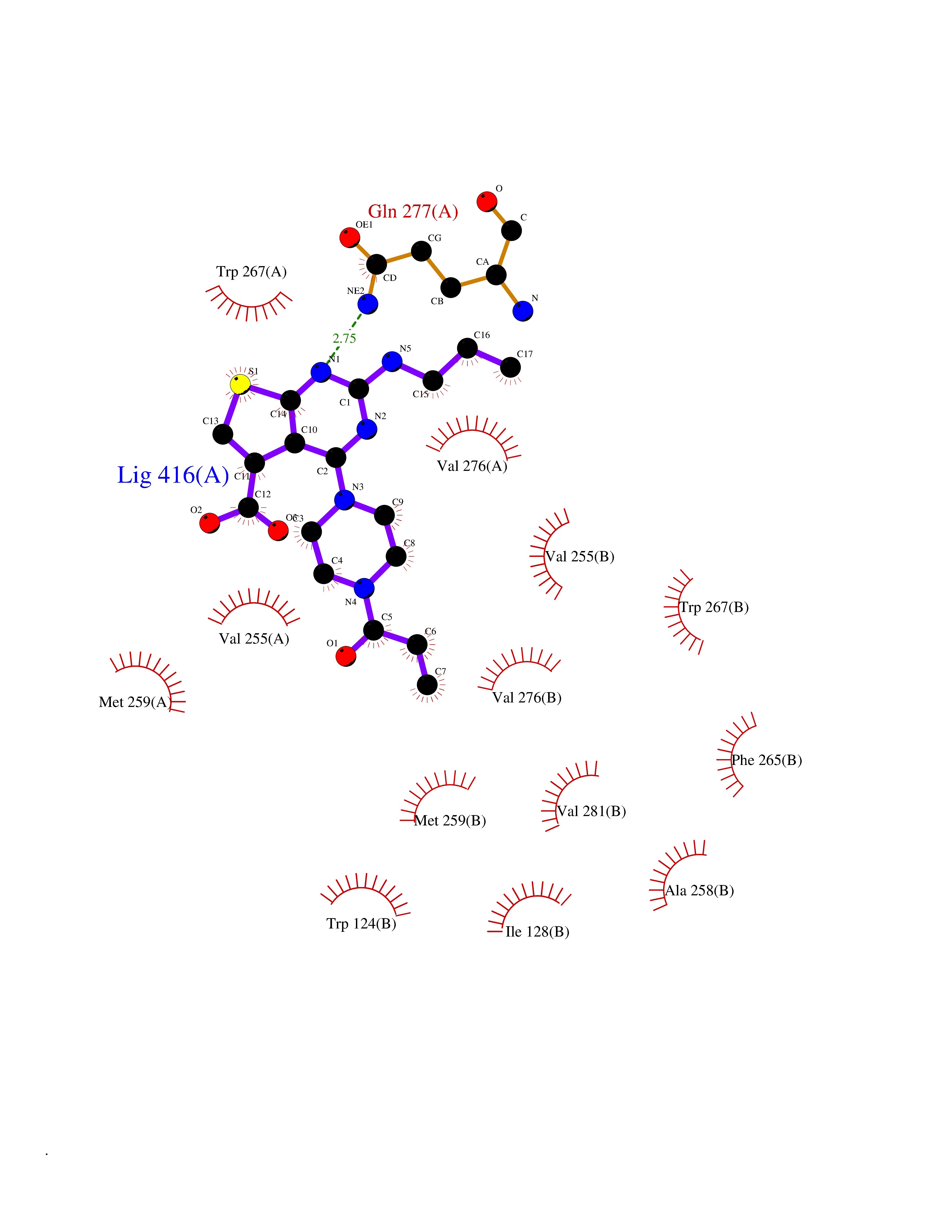 Ligplot Map 3D Binding mode Sequence KKISGGSVVEMQGDEMTRIIWELIKEKLIFPYVELDLHSYDLGIENRDATNDQVTKDAAEAIKKHNVGVKCATITPDEKRVEEFKLKQMWKSPNGTIRNILGGTVFREAIICKNIPRLVSGWVKPIIIGHHAYGDQYRATDFVVPGPGKVEITYTPSDGTQKVTYLVHNFEEGGGVAMGMYNQDKSIEDFAHSSFQMALSKGWPLYLSTKNTILKKYDGRFKDIFQEIYDKQYKSQFEAQKIWYEHRLIDDMVAQAMKSEGGFIWACKNYDGDVQSDSVAQGYGSLGMMTSVLVCPDGKTVEAEAAHGTVTRHYRMYQKGQETSTNPIASIFAWTRGLAHRAKLDNNKELAFFANALEEVSIETIEAGFMTKDLAACIKGLPNVQRSDYLNTFEFMDKLGENLKIKLAQAKLKKISGGSVVEMQGDEMTRIIWELIKEKLIFPYVELDLHSYDLGIENRDATNDQVTKDAAEAIKKHNVGVKCATITPDEKRVEEFKLKQMWKSPNGTIRNILGGTVFREAIICKNIPRLVSGWVKPIIIGHHAYGDQYRATDFVVPGPGKVEITYTPSDGTQKVTYLVHNFEEGGGVAMGMYNQDKSIEDFAHSSFQMALSKGWPLYLSTKNTILKKYDGRFKDIFQEIYDKQYKSQFEAQKIWYEHRLIDDMVAQAMKSEGGFIWACKNYDGDVQSDSVAQGYGSLGMMTSVLVCPDGKTVEAEAAHGTVTRHYRMYQKGQETSTNPIASIFAWTRGLAHRAKLDNNKELAFFANALEEVSIETIEAGFMTKDLAACIKGLPNVQRSDYLNTFEFMDKLGENLKIKLAQAK Hydrogen bonds contact Hydrophobic contact | ||||
| 94 | Fumarate reductase flavoprotein subunit | 1KF6 | 6.52 | |
Target general information Gen name frdA Organism Escherichia coli (strain K12) Uniprot ID TTD ID NA Synonyms JW4115;b4154 Protein family FAD-dependent oxidoreductase 2 family, FRD/SDH subfamily Biochemical class Oxidoreductase Function Electron carrier activity.FAD binding.Fumarate reductase (menaquinone).Succinate dehydrogenase activity. Related diseases Glycogen storage disease 11 (GSD11) [MIM:612933]: A metabolic disorder that results in exertional myoglobinuria, pain, cramps and easy fatigue. {ECO:0000269|PubMed:2334430}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07490; DB07918; DB00730 Interacts with P0AC47; P0ACB4; P76111 EC number 1.3.5.1 Uniprot keywords 3D-structure; Cell inner membrane; Cell membrane; Direct protein sequencing; Electron transport; FAD; Flavoprotein; Membrane; Nucleotide-binding; Oxidoreductase; Reference proteome; Transport Protein physicochemical properties Chain ID A,M Molecular weight (Da) 90370.7 Length 820 Aromaticity 0.08 Instability index 28.88 Isoelectric point 5.86 Charge (pH=7) -16.21 2D Binding mode Binding energy (Kcal/mol) -8.9  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence MQTFQADLAIVGAGGAGLRAAIAAAQANPNAKIALISKVYPMRSHTVAAEGGSAAVAQDHDSFEYHFHDTVAGGDWLCEQDVVDYFVHHCPTEMTQLELWGCPWSRRPDGSVNVRRFGGMKIERTWFAADKTGFHMLHTLFQTSLQFPQIQRFDEHFVLDILVDDGHVRGLVAMNMMEGTLVQIRANAVVMATGGAGRVYRYNTNGGIVTGDGMGMALSHGVPLRDMEFVQYHPTGLPGSGILMTEGCRGEGGILVNKNGYRYLQDYGMGPETPLGEPKNKYMELGPRDKVSQAFWHEWRKGNTISTPRGDVVYLDLRHLGEKKLHERLPFICELAKAYVGVDPVKEPIPVRPTAHYTMGGIETDQNCETRIKGLFAVGECSSVGLHGANRLGSNSLAELVVFGRLAGEQATERAATAGNGNEAAIEAQAAGVEQRLKDLVNQDGGENWAKIRDEMGLAMEEGCGIYRTPELMQKTIDKLAELQERFKRVRITDTSSVFNTDLLYTIELGHGLNVAECMAHSAMARKESRGAHQRLDEGCTERDDVNFLKHTLAFRDADGTTRLEYSDVKITTLPPAAEMKNLKIEVVRYNPEVDTAPHSAFYEVPYDATTSLLDALGYIKDNLAPDLSYRWSCRMAICGSCGMMVNNVPKLACKTFLRDYTDGMKVEALANFPIERDLVVDMTHFIESLEAIKPYIIGNSRTADQGTNIQTPAQMAKYHQFSGCINCGLCYAACPQFGLNPEFIGPAAITLAHRYNEDSRDHGKKERMAQLNSQNGVWSCTFVGYCSEVCPKHVDPAAAIQQGKVESSKDFLIATLKPR Hydrogen bonds contact Hydrophobic contact | ||||
| 95 | Medium-chain specific acyl-CoA dehydrogenase, mitochondrial | 4P13 | 6.52 | |
Target general information Gen name ACADM Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Acyl-CoA dehydrogenase family Biochemical class Oxidoreductase Function Acyl-CoA dehydrogenase activity.Flavin adenine dinucleotide binding.Identical protein binding.Medium-chain-acyl-CoA dehydrogenase activity. Related diseases Acyl-CoA dehydrogenase medium-chain deficiency (ACADMD) [MIM:201450]: An inborn error of mitochondrial fatty acid beta-oxidation which causes fasting hypoglycemia, hepatic dysfunction and encephalopathy, often resulting in death in infancy. {ECO:0000269|PubMed:10767181, ECO:0000269|PubMed:11349232, ECO:0000269|PubMed:11409868, ECO:0000269|PubMed:11486912, ECO:0000269|PubMed:1363805, ECO:0000269|PubMed:1671131, ECO:0000269|PubMed:1684086, ECO:0000269|PubMed:1902818, ECO:0000269|PubMed:2251268, ECO:0000269|PubMed:2393404, ECO:0000269|PubMed:2394825, ECO:0000269|PubMed:7603790, ECO:0000269|PubMed:7929823, ECO:0000269|PubMed:8198141, ECO:0000269|PubMed:9158144, ECO:0000269|PubMed:9882619}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03415; DB03147; DB02910 Interacts with PRO_0000000502 [P11310] EC number 1.3.8.7 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Direct protein sequencing; Disease variant; FAD; Fatty acid metabolism; Flavoprotein; Lipid metabolism; Mitochondrion; Oxidoreductase; Phosphoprotein; Proteomics identification; Reference proteome; Transit peptide Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 85080.3 Length 773 Aromaticity 0.09 Instability index 30.55 Isoelectric point 5.71 Charge (pH=7) -7.7 2D Binding mode Binding energy (Kcal/mol) -8.89  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence LGFSFEFTEQQKEFQATARKFAREEIIPVAAEYDKTGEYPVPLIRRAWELGLMNTHIPENCGGLGLGTFDACLISEELAYGCTGVQTAIEGNSLGQMPIIIAGNDQQKKKYLGRMTEEPLMCAYCVTEPGAGSDVAGIKTKAEKKGDEYIINGQKMWITNGGKANWYFLLARSDPDPKAPANKAFTGFIVEADTPGIQIGRKELNMGQRCSDTRGIVFEDVKVPKENVLIGDGAGFKVAMGAFDKTRPVVAAGAVGLAQRALDEATKYALERKTFGKLLVEHQAISFMLAEMAMEVELARMSYQRAAWEVDSGRRNTYYASIAKAFAGDIANQLATDAVQILGGNGFNTEYPVEKLMRDAKIYQIYEGTSQIQRLIVAREHIDKYKLGFSFEFTEQQKEFQATARKFAREEIIPVAAEYDKTGEYPVPLIRRAWELGLMNTHIPENCGGLGLGTFDACLISEELAYGCTGVQTAIEGNSLGQMPIIIAGNDQQKKKYLGRMTEEPLMCAYCVTEPGAGSDVAGIKTKAEKKGDEYIINGQKMWITNGGKANWYFLLARSDPDPKAPANKAFTGFIVEADTPGIQIGRKELNMGQRCSDTRGIVFEDVKVPKENVLIGDGAGFKVAMGAFDKTRPVVAAGAVGLAQRALDEATKYALERKTFGKLLVEHQAISFMLAEMAMEVELARMSYQRAAWEVDSGRRNTYYASIAKAFAGDIANQLATDAVQILGGNGFNTEYPVEKLMRDAKIYQIYEGTSQIQRLIVAREHIDKYKN Hydrogen bonds contact Hydrophobic contact | ||||
| 96 | Pyruvate kinase M2 (PKM) | 3GR4 | 6.52 | |
Target general information Gen name PKM Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms p58; Tumor M2-PK; Thyroid hormone-binding protein 1; THBP1; Pyruvate kinase muscle isozyme; Pyruvate kinase isozymes M1/M2; Pyruvate kinase PKM; Pyruvate kinase 2/3; PKM2; PK3; PK2; Opa-interacting pr Protein family Pyruvate kinase family Biochemical class Kinase Function Stimulates POU5F1-mediated transcriptional activation. Plays a general role in caspase independent cell death of tumor cells. The ratio between the highly active tetrameric form and nearly inactive dimeric form determines whether glucose carbons are channeled to biosynthetic processes or used for glycolytic ATP production. The transition between the 2 forms contributes to the control of glycolysis and is important for tumor cell proliferation and survival. Promotes in a STAT1-dependent manner, the expression of the immune checkpoint protein CD274 in ARNTL/BMAL1-deficient macrophages. Glycolytic enzyme that catalyzes the transfer of a phosphoryl group from phosphoenolpyruvate (PEP) to ADP, generating ATP. Related diseases Congenital sucrase-isomaltase deficiency (CSID) [MIM:222900]: Autosomal recessive intestinal disorder that is clinically characterized by fermentative diarrhea, abdominal pain, and cramps upon ingestion of sugar. The symptoms are the consequence of absent or drastically reduced enzymatic activities of sucrase and isomaltase. The prevalence of CSID is 0.02 % in individuals of European descent and appears to be much higher in Greenland, Alaskan, and Canadian native people. CSID arises due to post-translational perturbations in the intracellular transport, polarized sorting, aberrant processing, and defective function of SI. {ECO:0000269|PubMed:10903344, ECO:0000269|PubMed:11340066, ECO:0000269|PubMed:14724820, ECO:0000269|PubMed:16329100, ECO:0000269|PubMed:8609217}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07697; DB07692; DB02726; DB07628; DB00787; DB11638; DB09130; DB08951; DB01733; DB11263; DB00119 Interacts with P49407; P32121; Q96IK1-2; P35222; P53355; P22607; P42858; P04049; Q8N488; Q7Z699; Q9BSI4; Q9UMX0; Q9Y649; Q9WMX2; P35222; P53355; Q9H6Z9; P68431; Q16665; P27361 EC number EC 2.7.1.40 Uniprot keywords 3D-structure; Acetylation; Allosteric enzyme; Alternative splicing; ATP-binding; Cytoplasm; Direct protein sequencing; Glycolysis; Hydroxylation; Isopeptide bond; Kinase; Magnesium; Metal-binding; Methylation; Nucleotide-binding; Nucleus; Phosphoprotein; Potassium; Proteomics identification; Pyruvate; Reference proteome; S-nitrosylation; Transferase; Translation regulation; Ubl conjugation Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 112053 Length 1024 Aromaticity 0.05 Instability index 27.06 Isoelectric point 7.34 Charge (pH=7) 1.66 2D Binding mode Binding energy (Kcal/mol) -8.89  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence IQTQQLHAAMADTFLEHMCRLDIDSPPITARNTGIICTIGPASRSVETLKEMIKSGMNVARLNFSHGTHEYHAETIKNVRTATESFASDPILYRPVAVALDTKGPEIRTGLIKGSGTAEVELKKGATLKITLDNAYMEKCDENILWLDYKNICKVVEVGSKIYVDDGLISLQVKQKGADFLVTEVENGGSLGSKKGVNLPGAAVDLPAVSEKDIQDLKFGVEQDVDMVFASFIRKASDVHEVRKVLGEKGKNIKIISKIENHEGVRRFDEILEASDGIMVARGDLGIEIPAEKVFLAQKMMIGRCNRAGKPVICATQMLESMIKKPRPTRAEGSDVANAVLDGADCIMLSGETAKGDYPLEAVRMQHLIAREAEAAIYHLQLFEELRRLAPITSDPTEATAVGAVEASFKCCSGAIIVLTKSGRSAHQVARYRPRAPIIAVTRNPQTARQAHLYRGIFPVLCKDPVQEAWAEDVDLRVNFAMNVGKARGFFKKGDVVIVLTGWRPGSGFTNTMRVVPVPIQTQQLHAAMADTFLEHMCRLDIDSPPITARNTGIICTIGPASRSVETLKEMIKSGMNVARLNFSHGTHEYHAETIKNVRTATESFASDPILYRPVAVALDTKGPEIRTGLIKEVEATLKITLDNAYMEKCDENILWLDYKNICKVVEVGSKIYVDDGLISLQVDFLVTEVENGGSLGSKKGVNLPGAAVDLPAVSEKDIQDLKFGVEQDVDMVFASFIRKASDVHEVRKVLGEKGKNIKIISKIENHEGVRRFDEILEASDGIMVARGDLGIEIPAEKVFLAQKMMIGRCNRAGKPVICATQMLESMIKKPRPTRAEGSDVANAVLDGADCIMLSGETAKGDYPLEAVRMQHLIAREAEAAIYHLQLFEELRRLAPITSDPTEATAVGAVEASFKCCSGAIIVLTKSGRSAHQVARYRPRAPIIAVTRNPQTARQAHLYRGIFPVLCKDPVQEAWAEDVDLRVNFAMNVGKARGFFKKGDVVIVLTGWRPGSGFTNTMRVVPVP Hydrogen bonds contact Hydrophobic contact | ||||
| 97 | Cannabinoid receptor 1 (CB1) | 5U09 | 6.52 | |
Target general information Gen name CNR1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Cannabinoid CB1 receptor; CNR; CB-R; CANN6 Protein family G-protein coupled receptor 1 family Biochemical class GPCR rhodopsin Function Mediates many cannabinoid-induced effects, acting, among others, on food intake, memory loss, gastrointestinal motility, catalepsy, ambulatory activity, anxiety, chronic pain. Signaling typically involves reduction in cyclic AMP. In the hypothalamus, may have a dual effect on mitochondrial respiration depending upon the agonist dose and possibly upon the cell type. Increases respiration at low doses, while decreases respiration at high doses. At high doses, CNR1 signal transduction involves G-protein alpha-i protein activation and subsequent inhibition of mitochondrial soluble adenylate cyclase, decrease in cyclic AMP concentration, inhibition of protein kinase A (PKA)-dependent phosphorylation of specific subunits of the mitochondrial electron transport system, including NDUFS2. In the hypothalamus, inhibits leptin-induced reactive oxygen species (ROS) formation and mediates cannabinoid-induced increase in SREBF1 and FASN gene expression. In response to cannabinoids, drives the release of orexigenic beta-endorphin, but not that of melanocyte-stimulating hormone alpha/alpha-MSH, from hypothalamic POMC neurons, hence promoting food intake. In the hippocampus, regulates cellular respiration and energy production in response to cannabinoids. Involved in cannabinoid-dependent depolarization-induced suppression of inhibition (DSI), a process in which depolarization of CA1 postsynaptic pyramidal neurons mobilizes eCBs, which retrogradely activate presynaptic CB1 receptors, transiently decreasing GABAergic inhibitory neurotransmission. Also reduces excitatory synaptic transmission. In superior cervical ganglions and cerebral vascular smooth muscle cells, inhibits voltage-gated Ca(2+) channels in a constitutive, as well as agonist-dependent manner. In cerebral vascular smooth muscle cells, cannabinoid-induced inhibition of voltage-gated Ca(2+) channels leads to vasodilation and decreased vascular tone. Induces leptin production in adipocytes and reduces LRP2-mediated leptin clearance in the kidney, hence participating in hyperleptinemia. In adipose tissue, CNR1 signaling leads to increased expression of SREBF1, ACACA and FASN genes. In the liver, activation by endocannabinoids leads to increased de novo lipogenesis and reduced fatty acid catabolism, associated with increased expression of SREBF1/SREBP-1, GCK, ACACA, ACACB and FASN genes. May also affect de novo cholesterol synthesis and HDL-cholesteryl ether uptake. Peripherally modulates energy metabolism. In high carbohydrate diet-induced obesity, may decrease the expression of mitochondrial dihydrolipoyl dehydrogenase/DLD in striated muscles, as well as that of selected glucose/ pyruvate metabolic enzymes, hence affecting energy expenditure through mitochondrial metabolism. In response to cannabinoid anandamide, elicits a proinflammatory response in macrophages, which involves NLRP3 inflammasome activation and IL1B and IL18 secretion. In macrophages infiltrating pancreatic islets, this process may participate in the progression of type-2 diabetes and associated loss of pancreatic beta-cells. G-protein coupled receptor for endogenous cannabinoids (eCBs), including N-arachidonoylethanolamide (also called anandamide or AEA) and 2-arachidonoylglycerol (2-AG), as well as phytocannabinoids, such as delta(9)-tetrahydrocannabinol (THC). Related diseases Obesity (OBESITY) [MIM:601665]: A condition characterized by an increase of body weight beyond the limitation of skeletal and physical requirements, as the result of excessive accumulation of body fat. {ECO:0000269|PubMed:18177726}. The protein represented in this entry may be involved in disease pathogenesis. May contribute to the development of diet-induced obesity and several obesity-associated features, such as dyslipidemia and liver steatosis, regulating peripheral lipogenesis, energy expenditure and feeding behavior. CNR1 inverse agonists have been shown to reduce body weight and improve metabolic abnormalities in obese subjects, although adverse neuropsychiatric effects, including anxiety, irritability, and depressed mood, halted their therapeutic development (PubMed:18177726). In obese mice, peripherally restricted CNR1 inverse agonists have been shown to normalize metabolic abnormalities, including insulin resistance and fatty liver, and to reverse leptin resistance. {ECO:0000269|PubMed:18177726}.; DISEASE: Dysfunction of the endogenous cannabinoid system including CNR1 has been implicated in the pathogenesis of a number of central nervous system disorders, including Huntington disease, Parkinson disease, and Alzheimer disease (PubMed:32549916). In post-mortem brains from Huntington disease patients, a progressive CNR1 loss has been observed in the caudate nucleus, putamen, and substantia nigra pars reticulata, and altered expression and abnormal endocannabinoid levels precede motor symptoms in a disease mouse model (PubMed:10828533, PubMed:19524019, PubMed:8255419). In Parkinson disease, low CNR1 expression in mid-superior frontal gyrus and mid-cingulate cortex has been associated with poor mind, poor executive functioning and poor episode memory, while patients with more severe visuospatial dysfunction showed decreased receptor availability in the precuneus, mid-cingulate, supplementary motor cortex, inferior orbitofrontal gyrus and thalamus (PubMed:31342135). In an animal model for Alzheimer disease, CNR1 heterozygous deletion has been associated with decreased levels of postsynaptic density protein 95 (DLG4/PSD95) and accelerated memory impairment, suggesting synaptic dysfunction and a crucial role for CNR1 in the progression of disease symptoms (PubMed:10828533, PubMed:19524019, PubMed:30096288, PubMed:31342135, PubMed:8255419). {ECO:0000269|PubMed:10828533, ECO:0000269|PubMed:19524019, ECO:0000269|PubMed:30096288, ECO:0000269|PubMed:31342135, ECO:0000269|PubMed:32549916, ECO:0000269|PubMed:8255419}. Drugs (DrugBank ID) DB05750; DB09061; DB00470; DB14009; DB00486; DB14011; DB11745; DB09288; DB02955; DB06155; DB05077; DB11755; DB05201 Interacts with P29274; P21554 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Cell projection; G-protein coupled receptor; Glycoprotein; Lipoprotein; Membrane; Mitochondrion; Mitochondrion outer membrane; Neurodegeneration; Obesity; Palmitate; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Synapse; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 32070.3 Length 282 Aromaticity 0.13 Instability index 40.15 Isoelectric point 9.16 Charge (pH=7) 9.36 2D Binding mode Binding energy (Kcal/mol) -8.9  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence ENFMDIECFMVLNPSQQLAIAVLSLTLGTFTVLENLLVLCVILHSRSLRCRPSYHFIGSLAVADLLGSVIFVYSFIDFHVFHRKDSRNVFLFKLGGVTASFTASVGSLFLAAIDRYISIHRPLAYKRIVTRPKAVVAFCLMWTIAIVIAVLPLLGWNCEKLQSVCSDIFPHIDETYLMFWIGVTSVLLLFIVYAYMYILWKADQARMDIRLAKTLVLILVVLIICWGPLLAIMVYDVFGKMNKLIKTVFAFCSMLCLLNSTVNPIIYALRSKDLRHAFRSMF Hydrogen bonds contact Hydrophobic contact | ||||
| 98 | Cryptochrome DASH | 1NP7 | 6.52 | |
Target general information Gen name cry Organism Synechocystis sp. (strain ATCC 27184 / PCC 6803 / Kazusa) Uniprot ID TTD ID NA Synonyms phr;phrB;sll1629 Protein family DNA photolyase class-1 family Biochemical class Lyase Function DNA binding.DNA photolyase activity. Related diseases Heinz body anemias (HEIBAN) [MIM:140700]: Form of non-spherocytic hemolytic anemia of Dacie type 1. After splenectomy, which has little benefit, basophilic inclusions called Heinz bodies are demonstrable in the erythrocytes. Before splenectomy, diffuse or punctate basophilia may be evident. Most of these cases are probably instances of hemoglobinopathy. The hemoglobin demonstrates heat lability. Heinz bodies are observed also with the Ivemark syndrome (asplenia with cardiovascular anomalies) and with glutathione peroxidase deficiency. {ECO:0000269|PubMed:2833478}. The disease may be caused by variants affecting the gene represented in this entry.; DISEASE: Alpha-thalassemia (A-THAL) [MIM:604131]: A form of thalassemia. Thalassemias are common monogenic diseases occurring mostly in Mediterranean and Southeast Asian populations. The hallmark of alpha-thalassemia is an imbalance in globin-chain production in the adult HbA molecule. The level of alpha chain production can range from none to very nearly normal levels. Deletion of both copies of each of the two alpha-globin genes causes alpha(0)-thalassemia, also known as homozygous alpha thalassemia. Due to the complete absence of alpha chains, the predominant fetal hemoglobin is a tetramer of gamma-chains (Bart hemoglobin) that has essentially no oxygen carrying capacity. This causes oxygen starvation in the fetal tissues leading to prenatal lethality or early neonatal death. The loss of two alpha genes results in mild alpha-thalassemia, also known as heterozygous alpha-thalassemia. Affected individuals have small red cells and a mild anemia (microcytosis). If three of the four alpha-globin genes are functional, individuals are completely asymptomatic. Some rare forms of alpha-thalassemia are due to point mutations (non-deletional alpha-thalassemia). The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Alpha(0)-thalassemia is associated with non-immune hydrops fetalis, a generalized edema of the fetus with fluid accumulation in the body cavities due to non-immune causes. Non-immune hydrops fetalis is not a diagnosis in itself but a symptom, a feature of many genetic disorders, and the end-stage of a wide variety of disorders.; DISEASE: Hemoglobin H disease (HBH) [MIM:613978]: A form of alpha-thalassemia due to the loss of three alpha genes. This results in high levels of a tetramer of four beta chains (hemoglobin H), causing a severe and life-threatening anemia. Untreated, most patients die in childhood or early adolescence. {ECO:0000269|PubMed:10569720}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03147 Interacts with NA EC number NA Uniprot keywords 3D-structure; Chromophore; DNA-binding; FAD; Flavoprotein; Reference proteome; Repressor; Transcription; Transcription regulation Protein physicochemical properties Chain ID A,B Molecular weight (Da) 56439.5 Length 483 Aromaticity 0.13 Instability index 48.53 Isoelectric point 8.82 Charge (pH=7) 7.62 2D Binding mode Binding energy (Kcal/mol) -8.89  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence MKHVPPTVLVWFRNDLRLHDHEPLHRALKSGLAITAVYCYDPRQFAQTHQGFAKTGPWRSNFLQQSVQNLAESLQKVGNKLLVTTGLPEQVIPQIAKQINAKTIYYHREVTQEELDVERNLVKQLTILGIEAKGYWGSTLCHPEDLPFSIQDLPDLFTKFRKDIEKKKISIRPCFFAPSQLLPSPNIKLELTAPPPEFFPQINFDHRSVLAFQGGETAGLARLQDYFWHGDRLKDYKETRNGMVGADYSSKFSPWLALGCLSPRFIYQEVKRYEQERVSNDSTHWLIFELLWRDFFRFVAQKYGNKLFNRGGLLNKNFPWQEDQVRFELWRSGQTGYPLVDANMRELNLTGFMSNRGRQNVASFLCKNLGIDWRWGAEWFESCLIDYDVCSNWGNWNYTAGIGNDARDFRYFNIPKQSQQYDPQGTYLRHWLPELKNLPGDKIHQPWLLSATEQKQWGVQLGVDYPRPCVNFHQSVEARRKIE Hydrogen bonds contact Hydrophobic contact | ||||
| 99 | Metabotropic glutamate receptor 3 (mGluR3) | 4XAR | 6.52 | |
Target general information Gen name GRM3 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms mGLUR3; Group III metabotropic glutamate receptor; GPRC1C Protein family G-protein coupled receptor 3 family Biochemical class GPCR glutamate Function Ligand binding causes a conformation change that triggers signaling via guanine nucleotide-binding proteins (G proteins) and modulates the activity of down-stream effectors. Signaling inhibits adenylate cyclase activity. G-protein coupled receptor for glutamate. Related diseases Paramyotonia congenita (PMC) [MIM:168300]: An autosomal dominant channelopathy characterized by myotonia, increased by exposure to cold, intermittent flaccid paresis, not necessarily dependent on cold or myotonia, lability of serum potassium, non-progressive nature and lack of atrophy or hypertrophy of muscles. In some patients, myotonia is not increased by cold exposure (paramyotonia without cold paralysis). Patients may have a combination phenotype of PMC and HYPP. {ECO:0000269|PubMed:10369308, ECO:0000269|PubMed:10727489, ECO:0000269|PubMed:1310898, ECO:0000269|PubMed:1316765, ECO:0000269|PubMed:1338909, ECO:0000269|PubMed:15318338, ECO:0000269|PubMed:15790667, ECO:0000269|PubMed:16786525, ECO:0000269|PubMed:18166706, ECO:0000269|PubMed:18690054, ECO:0000269|PubMed:19077043, ECO:0000269|PubMed:20076800, ECO:0000269|PubMed:8242056, ECO:0000269|PubMed:8308722, ECO:0000269|PubMed:8388676, ECO:0000269|PubMed:8580427}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Periodic paralysis hypokalemic 2 (HOKPP2) [MIM:613345]: An autosomal dominant disorder manifested by episodic flaccid generalized muscle weakness associated with falls of serum potassium levels. {ECO:0000269|PubMed:10599760, ECO:0000269|PubMed:10851391, ECO:0000269|PubMed:10944223, ECO:0000269|PubMed:11558801, ECO:0000269|PubMed:11591859, ECO:0000269|PubMed:16890191, ECO:0000269|PubMed:17898326, ECO:0000269|PubMed:18162704, ECO:0000269|PubMed:19118277, ECO:0000269|PubMed:20522878, ECO:0000269|PubMed:21043388, ECO:0000269|PubMed:24549961}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Periodic paralysis hyperkalemic (HYPP) [MIM:170500]: An autosomal dominant channelopathy characterized by episodic flaccid generalized muscle weakness associated with high levels of serum potassium. Concurrence of myotonia is found in HYPP patients. {ECO:0000269|PubMed:1659668, ECO:0000269|PubMed:1659948, ECO:0000269|PubMed:20076800}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Periodic paralysis normokalemic (NKPP) [MIM:170500]: A disorder closely related to hyperkalemic periodic paralysis, but marked by a lack of alterations in potassium levels during attacks of muscle weakness. {ECO:0000269|PubMed:15596759, ECO:0000269|PubMed:18046642, ECO:0000269|PubMed:20522878}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Myotonia SCN4A-related (MYOSCN4A) [MIM:608390]: A phenotypically highly variable myotonia aggravated by potassium loading, and sometimes by cold. Myotonia is characterized by sustained muscle tensing that prevents muscles from relaxing normally. It causes muscle stiffness that can interfere with movement. In some people the stiffness is very mild, while in other cases it may be severe enough to interfere with walking, running, and other activities of daily life. Myotonia SCN4A-related includes myotonia permanens and myotonia fluctuans. In myotonia permanens, the myotonia is generalized and there is a hypertrophy of the muscle, particularly in the neck and the shoulder. Attacks of severe muscle stiffness of the thoracic muscles may be life threatening due to impaired ventilation. In myotonia fluctuans, the muscle stiffness may fluctuate from day to day, provoked by exercise. {ECO:0000269|PubMed:10218481, ECO:0000269|PubMed:16786525, ECO:0000269|PubMed:16832098, ECO:0000269|PubMed:17212350, ECO:0000269|PubMed:17998485, ECO:0000269|PubMed:18203179, ECO:0000269|PubMed:18337100, ECO:0000269|PubMed:19015483, ECO:0000269|PubMed:19347921, ECO:0000269|PubMed:20076800, ECO:0000269|PubMed:27653901, ECO:0000269|PubMed:8058156, ECO:0000269|PubMed:9392583}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Myasthenic syndrome, congenital, 16 (CMS16) [MIM:614198]: A form of congenital myasthenic syndrome, a group of disorders characterized by failure of neuromuscular transmission, including pre-synaptic, synaptic, and post-synaptic disorders that are not of autoimmune origin. Clinical features are easy fatigability and muscle weakness. CMS16 is characterized by fatigable generalized weakness and recurrent attacks of respiratory and bulbar paralysis since birth. The fatigable weakness involves lid-elevator, external ocular, facial, limb and truncal muscles and an decremental response of the compound muscle action potential on repetitive stimulation. {ECO:0000269|PubMed:12766226, ECO:0000269|PubMed:25707578, ECO:0000269|PubMed:26659129}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Congenital myopathy 22A, classic (CMYO22A) [MIM:620351]: A form of congenital myopathy, a clinically and genetically heterogeneous group of muscle disorders characterized by hypotonia and muscle weakness apparent at birth, and specific pathological features on muscle biopsy. CMYO22A is an autosomal recessive form characterized by fetal hypokinesia, polyhydramnios, and severe neonatal hypotonia associated with respiratory insufficiency. Affected individuals who survive the neonatal period have delayed motor development, difficulty walking, proximal muscle weakness of the upper and lower limbs, facial and neck muscle weakness, easy fatigability, and mild limb contractures or foot deformities. {ECO:0000269|PubMed:26700687, ECO:0000269|PubMed:28262468, ECO:0000269|PubMed:36090556}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Congenital myopathy 22B, severe fetal (CMYO22B) [MIM:620369]: A severe congenital myopathy, a clinically and genetically heterogeneous group of muscle disorders characterized by hypotonia and muscle weakness apparent at birth, and specific pathological features on muscle biopsy. CMYO22B is an autosomal recessive form characterized by onset in utero. Affected individuals show fetal akinesia, and develop fetal hydrops with pulmonary hypoplasia, severe joint contractures, and generalized muscle hypoplasia. Death occurs in utero or soon after birth. {ECO:0000269|PubMed:26700687}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB05096 Interacts with NA EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disulfide bond; G-protein coupled receptor; Glycoprotein; Membrane; Proteomics identification; Receptor; Reference proteome; Signal; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 50355.5 Length 445 Aromaticity 0.11 Instability index 38.26 Isoelectric point 6.52 Charge (pH=7) -1.53 2D Binding mode Binding energy (Kcal/mol) -8.89  Molscript Map 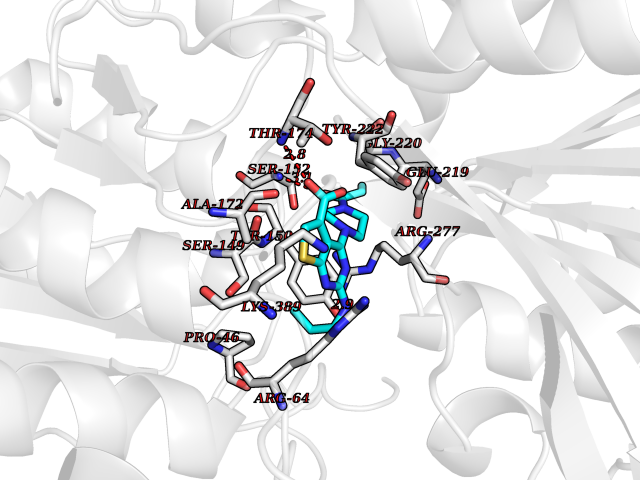 Pymol Map 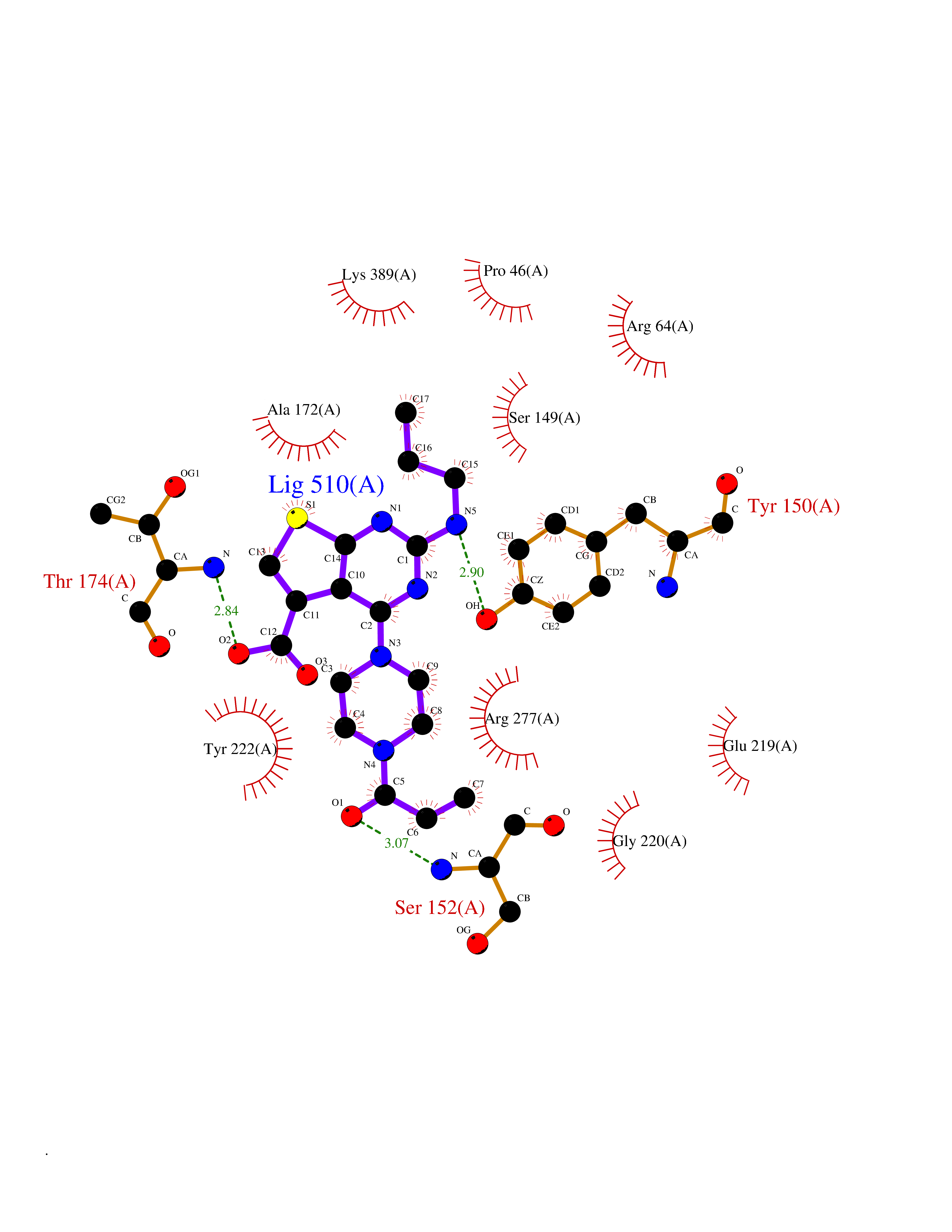 Ligplot Map 3D Binding mode Sequence RREIKIEGDLVLGGLFPINEKGTGTEECGRINEDRGIQRLEAMLFAIDEINKDDYLLPGVKLGVHILDTCSRDTYALEQSLEFVRASLLLIAGVIGGSYSSVSIQVANLLRLFQIPQISYASTSAKLSDKSRYDYFARTVPPDFYQAKAMAEILRFFNWTYVSTVASEGDYGETGIEAFEQEARLRNISIATAEKVGRSNIRKSYDSVIRELLQKPNARVVVLFMRSDDSRELIAAASRANASFTWVASDGWGAQESIIKGSEHVAYGAITLELASQPVRQFDRYFQSLNPYNNHRNPWFRDFWEQKFQCSLRVCDKHLAIDSSNYEQESKIMFVVNAVYAMAHALHKMQRTLCPNTTKLCDAMKILDGKKLYKDYLLKINFTAPDADSIVKFDTFGDGMGRYNVFNFQNVGGKYSYLKVGHWAETLSLDVNSIHWSRNSVPTSE Hydrogen bonds contact Hydrophobic contact | ||||
| 100 | Debrisoquine 4-hydroxylase (CYP2D6) | 4WNV | 6.52 | |
Target general information Gen name CYP2D6 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms P450-DB1; Cytochrome P450-DB1; Cytochrome P450 2D6; CYPIID6; CYP2DL1 Protein family Cytochrome P450 family Biochemical class Paired donor oxygen oxidoreductase Function It is involved in the metabolism of drugs such as antiarrhythmics, adrenoceptor antagonists, and tricyclic antidepressants. Responsible for the metabolism of many drugs and environmental chemicals that it oxidizes. Related diseases A chromosomal aberration involving BCL2 has been found in chronic lymphatic leukemia. Translocation t(14;18)(q32;q21) with immunoglobulin gene regions. BCL2 mutations found in non-Hodgkin lymphomas carrying the chromosomal translocation could be attributed to the Ig somatic hypermutation mechanism resulting in nucleotide transitions. {ECO:0000269|PubMed:2875799, ECO:0000269|PubMed:3285301}. Drugs (DrugBank ID) DB01562; DB01472; DB14010; DB12001; DB05812; DB01193; DB00316; DB15568; DB00918; DB06203; DB00866; DB01424; DB01118; DB00321; DB00381; DB00613; DB00543; DB00182; DB00701; DB11785; DB01435; DB01429; DB01274; DB01238; DB14185; DB09204; DB11638; DB06216; DB00637; DB11586; DB00335; DB00289; DB01076; DB00972; DB04957; DB09013; DB16703; DB01086; DB06770; DB01244; DB15982; DB00195; DB01295; DB12236; DB01128; DB04889; DB00810; DB13975; DB08807; DB00188; DB09128; DB12151; DB12752; DB06726; DB00297; DB08808; DB00921; DB01156; DB00490; DB09173; DB00201; DB09061; DB14737; DB06016; DB00521; DB01136; DB00482; DB04846; DB00439; DB00185; DB00608; DB01114; DB00477; DB00356; DB01410; DB01166; DB00501; DB01012; DB00568; DB00604; DB00215; DB12499; DB00283; DB04920; DB14025; DB00349; DB00845; DB01242; DB00575; DB13508; DB00257; DB00363; DB09065; DB05239; DB00907; DB00318; DB11672; DB14635; DB00924; DB00091; DB11963; DB06292; DB04884; DB00496; DB01264; DB09183; DB04840; DB00705; DB06512; DB01151; DB06700; DB16650; DB12161; DB13679; DB09555; DB01191; DB00633; DB01576; DB00514; DB00647; DB11994; DB01551; DB00343; DB01093; DB01075; DB00757; DB01184; DB00843; DB09167; DB00590; DB01142; DB00997; DB00470; DB04855; DB00476; DB00625; DB11979; DB00216; DB15444; DB09039; DB13874; DB01228; DB06735; DB11718; DB00494; DB13757; DB00751; DB00530; DB13443; DB01175; DB06678; DB00187; DB00330; DB01466; DB01628; DB01590; DB12500; DB01023; DB00574; DB06702; DB12265; DB01195; DB04841; DB00472; DB00623; DB01095; DB00176; DB00983; DB02703; DB15149; DB00674; DB05087; DB00317; DB08909; DB00986; DB01218; DB00502; DB00956; DB01611; DB00557; DB09053; DB01177; DB04946; DB00619; DB00458; DB08952; DB00224; DB06370; DB13293; DB04818; DB16200; DB11633; DB06636; DB00951; DB11757; DB00602; DB09570; DB01026; DB00598; DB12212; DB00448; DB11732; DB16217; DB09078; DB00528; DB12070; DB09351; DB01210; DB08918; DB00281; DB04948; DB01206; DB00836; DB01601; DB00455; DB04871; DB09195; DB06708; DB04829; DB09238; DB00934; DB14921; DB00737; DB14009; DB09224; DB00170; DB00454; DB00532; DB13530; DB06691; DB01071; DB00933; DB01577; DB00333; DB00763; DB01403; DB01028; DB09241; DB01214; DB01233; DB00264; DB00379; DB06148; DB01388; DB01110; DB00211; DB01454; DB06595; DB00834; DB00805; DB08893; DB00370; DB12523; DB01171; DB00745; DB14011; DB09049; DB00731; DB04861; DB01149; DB00220; DB09048; DB00238; DB00627; DB00622; DB00699; DB02701; DB00184; DB01115; DB04868; DB12005; DB00540; DB00334; DB14881; DB00338; DB00904; DB11130; DB04911; DB01173; DB11837; DB04938; DB01096; DB01580; DB01062; DB00497; DB06412; DB01192; DB01267; DB00377; DB06603; DB00715; DB06589; DB00022; DB01359; DB00738; DB01074; DB08922; DB00850; DB03783; DB00780; DB00914; DB00252; DB05316; DB01100; DB00960; DB00592; DB01621; DB04951; DB17472; DB11642; DB08901; DB01297; DB15822; DB01087; DB01035; DB00433; DB00396; DB01131; DB00420; DB01069; DB09288; DB01182; DB00571; DB04216; DB01224; DB00908; DB00468; DB01129; DB00863; DB00243; DB00234; DB14761; DB00409; DB06506; DB02709; DB11855; DB13174; DB11753; DB08864; DB14840; DB00734; DB12693; DB00503; DB00953; DB09291; DB15119; DB00412; DB05271; DB12332; DB11614; DB06654; DB01232; DB01037; DB06144; DB01104; DB00203; DB00641; DB01591; DB00398; DB12713; DB00489; DB06727; DB01323; DB09118; DB06820; DB06729; DB06608; DB11770; DB00675; DB00706; DB06204; DB06083; DB01079; DB12095; DB06287; DB00857; DB00342; DB13775; DB04905; DB04844; DB11712; DB00277; DB00679; DB01623; DB00208; DB00373; DB01409; DB00932; DB06137; DB01036; DB05109; DB00193; DB00752; DB00656; DB12245; DB00726; DB00792; DB00209; DB15328; DB09076; DB13609; DB15091; DB11915; DB00862; DB08881; DB00285; DB00661; DB06217; DB06684; DB09185; DB00570; DB00361; DB11739; DB09068; DB01392; DB00549; DB15688; DB00425; DB01624 Interacts with NA EC number EC 1.14.14.- Uniprot keywords 3D-structure; Alternative splicing; Cholesterol metabolism; Endoplasmic reticulum; Fatty acid metabolism; Heme; Iron; Lipid metabolism; Membrane; Metal-binding; Microsome; Monooxygenase; Oxidoreductase; Proteomics identification; Reference proteome; Steroid metabolism; Sterol metabolism Protein physicochemical properties Chain ID A Molecular weight (Da) 51898.1 Length 464 Aromaticity 0.09 Instability index 43.83 Isoelectric point 6.76 Charge (pH=7) -0.99 2D Binding mode Binding energy (Kcal/mol) -8.89  Molscript Map 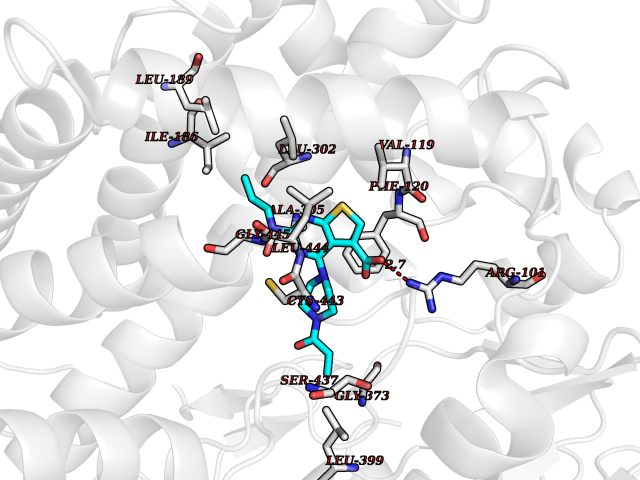 Pymol Map 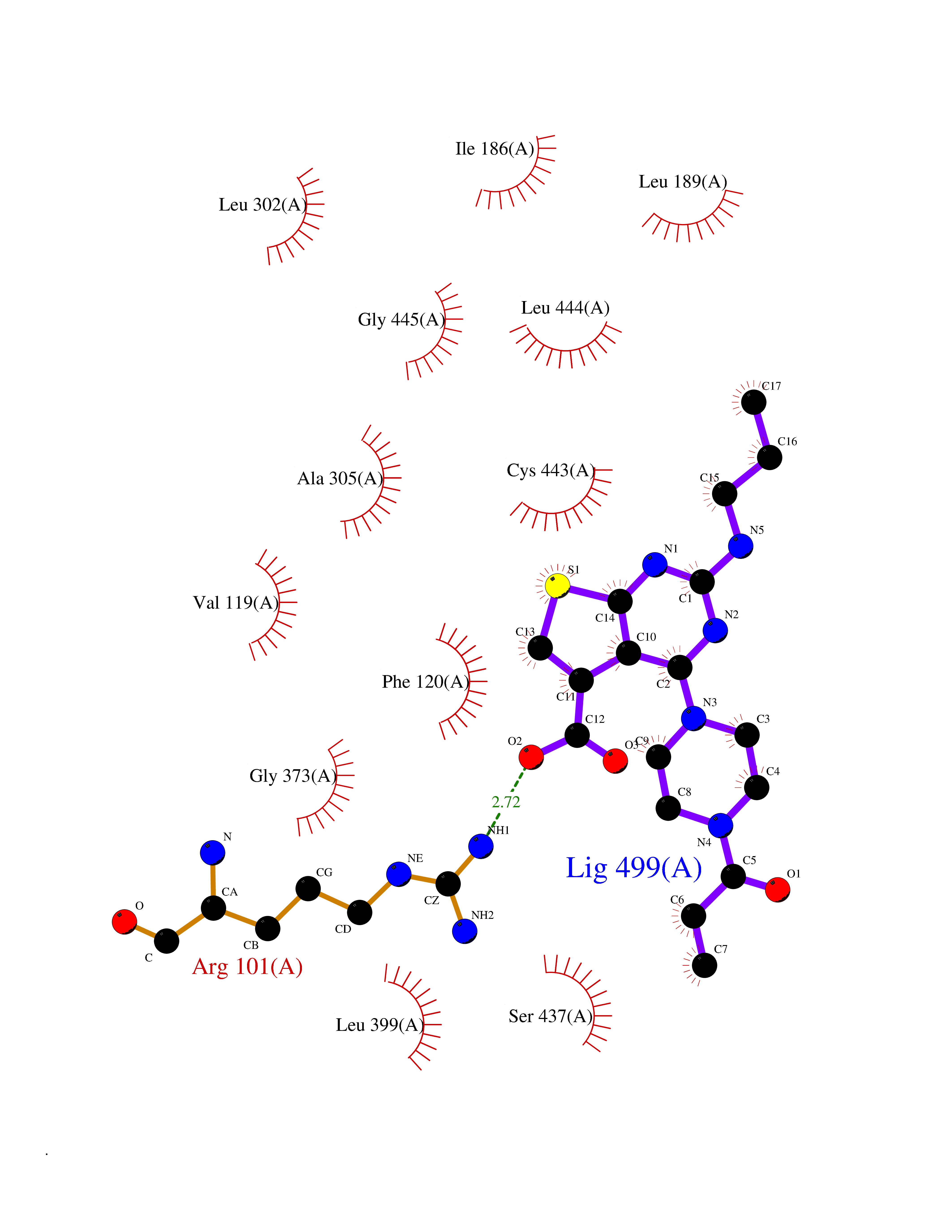 Ligplot Map 3D Binding mode Sequence GKLPPGPLPLPGLGNLLFQNTPYCFDQLRRRFGDVFSLQLAWTPVVVLNGLAAVREALVTHGEDTADRPPVPITQILGFGPRSQGVFLARYGPAWREQRRFSVSTLRNLGLGKKSLEQWVTEEAACLCAAFANHSGRPFRPNGLLDKAVSNVIASLTCGRRFEYDDPRFLRLLDLAQEGLKEESGFLREVLNAVPVLLHIPALAGKVLRFQKAFLTQLDELLTEHRMTWDPAQPPRDLTEAFLAEMEKAKGNPESSFNDENLRIVVADLFSAGMVTTSTTLAWGLLLMILHPDVQRRVQQEIDDVIGQVRRPEMGDQAHMPYTTAVIHEVQRFGDIVPLGVTHMTSRDIEVQGFRIPKGTTLITNLSSVLKDEAVWEKPFRFHPEHFLDAQGHFVKPEAFLPFSAGRRACLGEPLARMELFLFFTSLLQHFSFSVPTGQPRPSHHGVFAFLVSPSPYELCAVPR Hydrogen bonds contact Hydrophobic contact | ||||