Job Results:
Ligand
Structure
Job ID
7a36b9b0fd299388007ea8e58dfe56c6
Job name
NA
Time
2025-09-26 17:47:53
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 81 | Notch-1 receptor (NOTCH1) | 5L0R | 6.33 | |
Target general information Gen name NOTCH1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms hN1; Translocationassociated notch protein TAN1; Translocation-associated notch protein TAN-1; TAN1; Notch 1 intracellular domain; Notch 1; Neurogenic locus notch homolog protein 1; NICD Protein family NOTCH family Biochemical class Notch protein Function Upon ligand activation through the released notch intracellular domain (NICD) it forms a transcriptional activator complex with RBPJ/RBPSUH and activates genes of the enhancer of split locus. Affects the implementation of differentiation, proliferation and apoptotic programs. Involved in angiogenesis; negatively regulates endothelial cell proliferation and migration and angiogenic sprouting. Involved in the maturation of both CD4(+) and CD8(+) cells in the thymus. Important for follicular differentiation and possibly cell fate selection within the follicle. During cerebellar development, functions as a receptor for neuronal DNER and is involved in the differentiation of Bergmann glia. Represses neuronal and myogenic differentiation. May play an essential role in postimplantation development, probably in some aspect of cell specification and/or differentiation. May be involved in mesoderm development, somite formation and neurogenesis. May enhance HIF1A function by sequestering HIF1AN away from HIF1A. Required for the THBS4 function in regulating protective astrogenesis from the subventricular zone (SVZ) niche after injury. Involved in determination of left/right symmetry by modulating the balance between motile and immotile (sensory) cilia at the left-right organiser (LRO). Functions as a receptor for membrane-bound ligands Jagged-1 (JAG1), Jagged-2 (JAG2) and Delta-1 (DLL1) to regulate cell-fate determination. Related diseases Aortic valve disease 1 (AOVD1) [MIM:109730]: A common defect in the aortic valve in which two rather than three leaflets are present. It is often associated with aortic valve calcification, stenosis and insufficiency. In extreme cases, the blood flow may be so restricted that the left ventricle fails to grow, resulting in hypoplastic left heart syndrome. {ECO:0000269|PubMed:16025100}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Adams-Oliver syndrome 5 (AOS5) [MIM:616028]: A form of Adams-Oliver syndrome, a disorder characterized by the congenital absence of skin (aplasia cutis congenita) in combination with transverse limb defects. Aplasia cutis congenita can be located anywhere on the body, but in the vast majority of the cases, it is present on the posterior parietal region where it is often associated with an underlying defect of the parietal bones. Limb abnormalities are typically limb truncation defects affecting the distal phalanges or entire digits (true ectrodactyly). Only rarely, metatarsals/metacarpals or more proximal limb structures are also affected. Apart from transverse limb defects, syndactyly, most commonly of second and third toes, can also be observed. The clinical features are highly variable and can also include cardiovascular malformations, brain abnormalities and vascular defects such as cutis marmorata and dilated scalp veins. {ECO:0000269|PubMed:25132448}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q13315; Q969H0; Q16665; P78504; O60341; Q8N423; Q92585; P19838; P46531; Q13153; Q13526; Q06330; Q06330-6; Q13573; P98170; Q8IZL2; Q96JK9 EC number NA Uniprot keywords 3D-structure; Activator; Angiogenesis; ANK repeat; Calcium; Cell membrane; Developmental protein; Differentiation; Direct protein sequencing; Disease variant; Disulfide bond; EGF-like domain; Endosome; Glycoprotein; Hydroxylation; Isopeptide bond; Membrane; Metal-binding; Notch signaling pathway; Nucleus; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Repeat; Signal; Transcription; Transcription regulation; Transmembrane; Transmembrane helix; Ubl conjugation Protein physicochemical properties Chain ID A,B Molecular weight (Da) 46121.2 Length 394 Aromaticity 0.13 Instability index 39.95 Isoelectric point 7.22 Charge (pH=7) 0.63 2D Binding mode Binding energy (Kcal/mol) -8.63  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence KWKVFIDQINRSLENYEPCSSQNCSCYHGVIEEDLTPFRGGISRKMMAEVVRRKLGTHYQITKNRLYRENDCMFPSRCSGVEHFILEVIGRLPDMEMVINVRDYPQVPKWMEPAIPVFSFSKTSEYHDIMYPAWTFWEGGPAVWPIYPTGLGRWDLFREDLVRSAAQWPWKKKNSTAYFRGSRTSPERDPLILLSRKNPKLVDAEYTKNQAWKSMKDTLGKPAAKDVHLVDHCKYKYLFNFRGVAASFRFKHLFLCGSLVFHVGDEWLEFFYPQLKPWVHYIPVKTDLSNVQELLQFVKANDDVAQEIAERGSQFIRNHLQMDDITCYWENLLSEYSKFLSYNVTRRKGYDQIIPVNECVSNPCQNDATCLDQIGEFQCICMPGYEGVHCEVNT Hydrogen bonds contact Hydrophobic contact | ||||
| 82 | Receptor-type protein-tyrosine phosphatase zeta (PTPRZ1) | 5H08 | 6.33 | |
Target general information Gen name PTPRZ1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Receptor protein tyrosine phosphatase zeta; R-PTP-zeta; PTPRZ1 Protein family Protein-tyrosine phosphatase family, Receptor class 5 subfamily Biochemical class Phosphoric monoester hydrolase Function Protein tyrosine phosphatase that negatively regulates oligodendrocyte precursor proliferation in the embryonic spinal cord. Required for normal differentiation of the precursor cells into mature, fully myelinating oligodendrocytes. May play a role in protecting oligondendrocytes against apoptosis. May play a role in the establishment of contextual memory, probably via the dephosphorylation of proteins that are part of important signaling cascades. Related diseases Optic atrophy 1 (OPA1) [MIM:165500]: A condition that features progressive visual loss in association with optic atrophy. Atrophy of the optic disk indicates a deficiency in the number of nerve fibers which arise in the retina and converge to form the optic disk, optic nerve, optic chiasm and optic tracts. OPA1 is characterized by an insidious onset of visual impairment in early childhood with moderate to severe loss of visual acuity, temporal optic disk pallor, color vision deficits, and centrocecal scotoma of variable density. {ECO:0000269|PubMed:11017079, ECO:0000269|PubMed:11017080, ECO:0000269|PubMed:11440988, ECO:0000269|PubMed:11440989, ECO:0000269|PubMed:11810270, ECO:0000269|PubMed:12036970, ECO:0000269|PubMed:12566046, ECO:0000269|PubMed:14961560, ECO:0000269|PubMed:15948788, ECO:0000269|PubMed:16513463, ECO:0000269|PubMed:16617242, ECO:0000269|PubMed:18204809, ECO:0000269|PubMed:18360822, ECO:0000269|PubMed:19319978, ECO:0000269|PubMed:19325939, ECO:0000269|PubMed:19969356, ECO:0000269|PubMed:20185555, ECO:0000269|PubMed:22382025, ECO:0000269|PubMed:22857269, ECO:0000269|PubMed:23401657}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Dominant optic atrophy plus syndrome (DOA+) [MIM:125250]: A neurologic disorder characterized most commonly by an insidious onset of visual loss and sensorineural hearing loss in childhood with variable presentation of other clinical manifestations including progressive external ophthalmoplegia, muscle cramps, hyperreflexia, and ataxia. There appears to be a wide range of intermediate phenotypes. {ECO:0000269|PubMed:15531309, ECO:0000269|PubMed:16240368, ECO:0000269|PubMed:18065439, ECO:0000269|PubMed:18158317, ECO:0000269|PubMed:18195150, ECO:0000269|PubMed:20185555, ECO:0000269|PubMed:21112924, ECO:0000269|PubMed:23387428}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Behr syndrome (BEHRS) [MIM:210000]: An autosomal recessive syndrome characterized by optic atrophy beginning in early childhood associated with ataxia, pyramidal signs, spasticity, intellectual disability, and posterior column sensory loss. The ataxia, spasticity, and muscle contractures, mainly of the hip adductors, hamstrings, and soleus, are progressive and become more prominent in the second decade. {ECO:0000269|PubMed:21636302, ECO:0000269|PubMed:25012220, ECO:0000269|PubMed:25146916}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Mitochondrial DNA depletion syndrome 14, cardioencephalomyopathic type (MTDPS14) [MIM:616896]: An autosomal recessive mitochondrial disorder characterized by lethal infantile encephalopathy, hypertrophic cardiomyopathy and optic atrophy. Skeletal muscle biopsies show significant mtDNA depletion and abnormal mitochondria. {ECO:0000269|PubMed:26561570}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q9UM73; Q12860 EC number EC 3.1.3.48 Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disulfide bond; Glycoprotein; Hydrolase; Membrane; Phosphoprotein; Protein phosphatase; Proteoglycan; Proteomics identification; Reference proteome; Repeat; Secreted; Signal; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 32208.1 Length 282 Aromaticity 0.11 Instability index 38.98 Isoelectric point 7.35 Charge (pH=7) 0.89 2D Binding mode Binding energy (Kcal/mol) -8.63 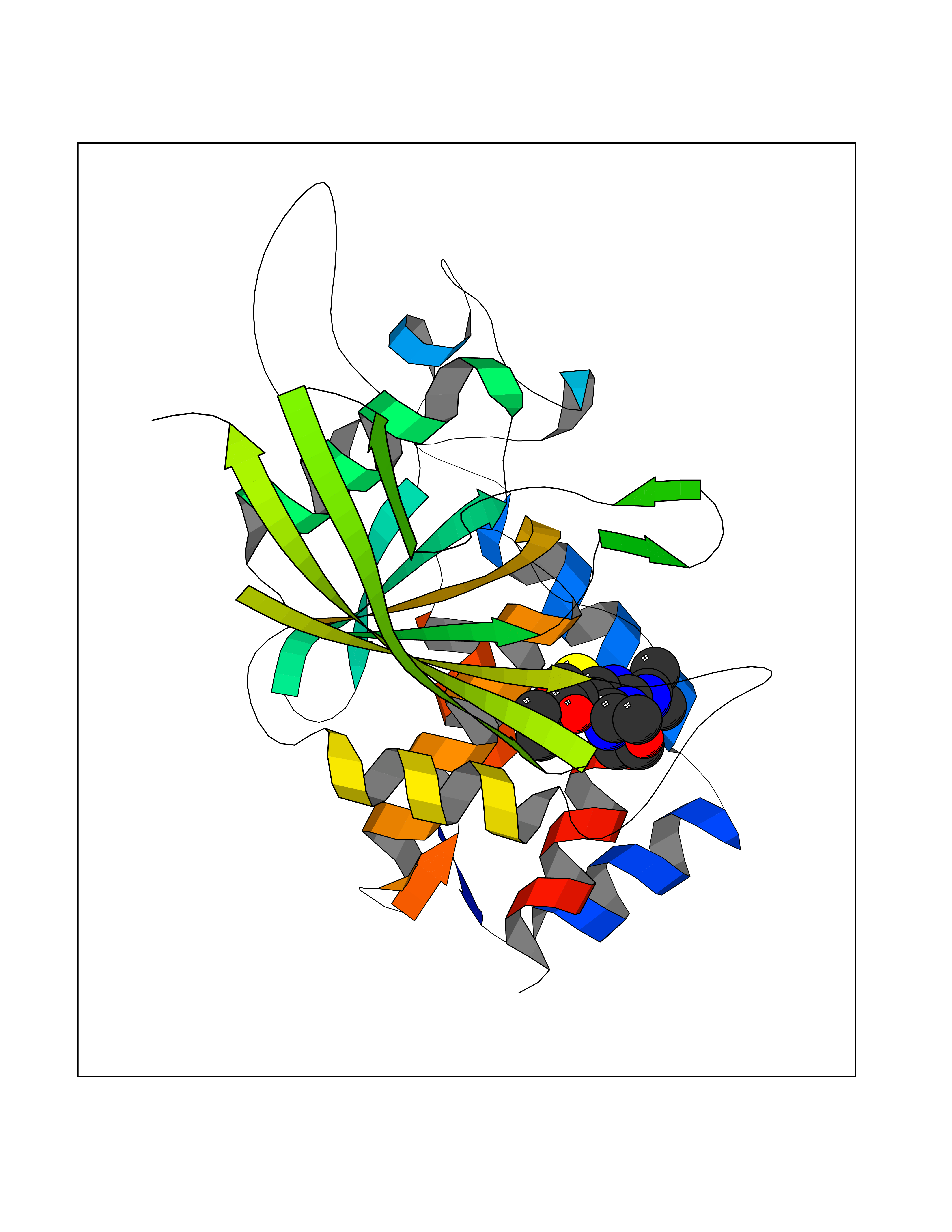 Molscript Map 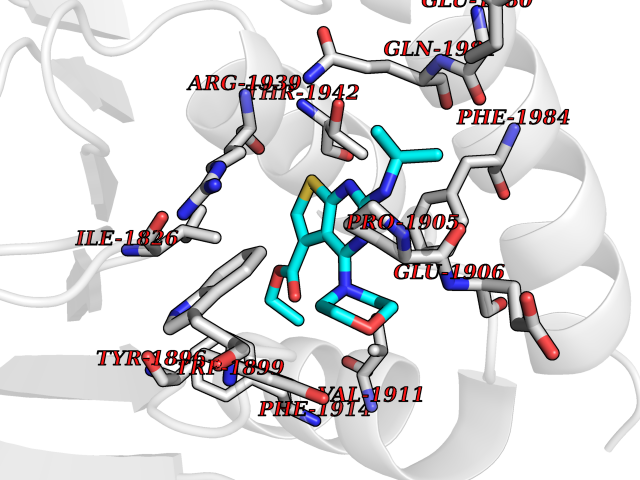 Pymol Map 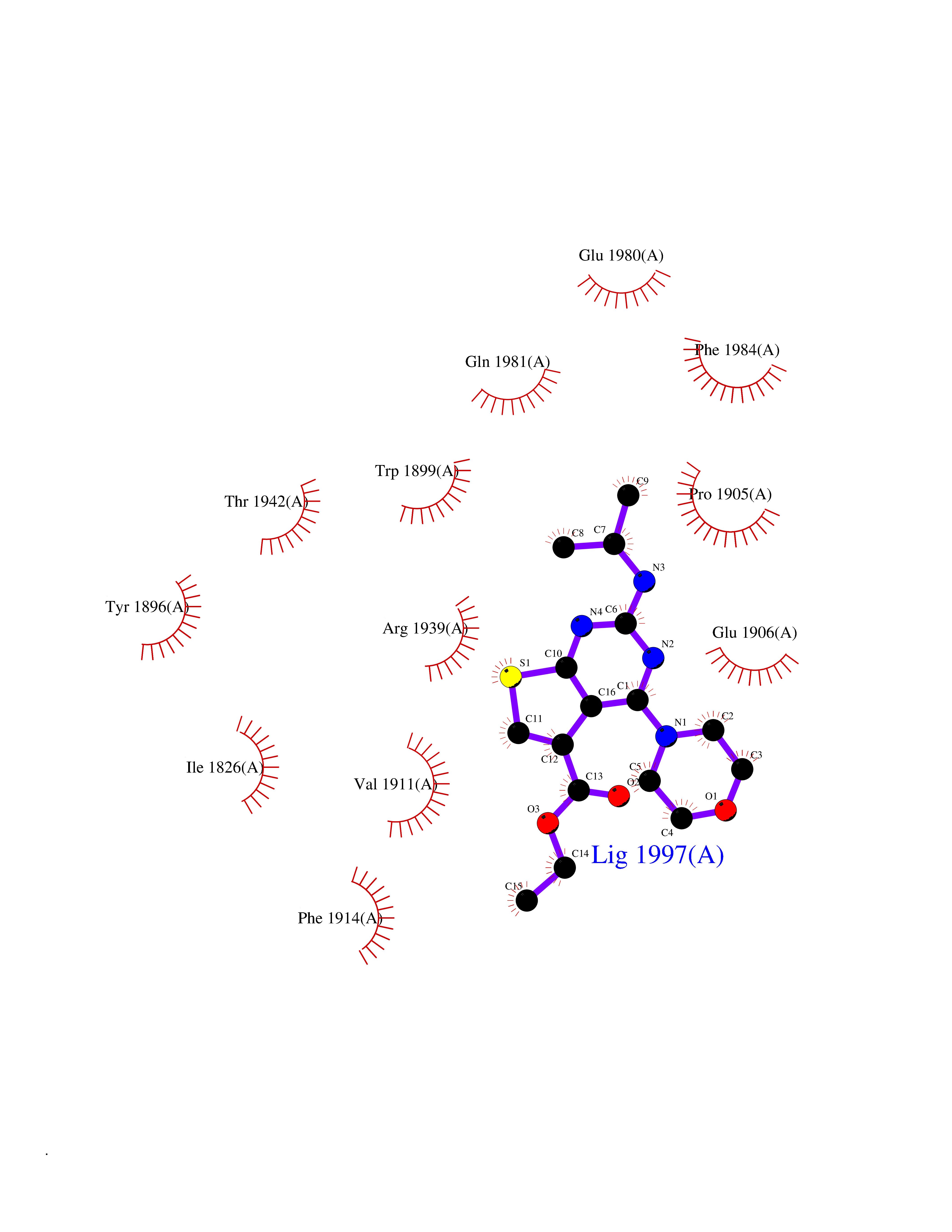 Ligplot Map 3D Binding mode Sequence GPAIPIKHFPKHVADLHASSGFTEEFEEVQSCTVDLGITADSSNHPDNKHKNRYINIVAYDHSRVKLAQLAEKDGKLTDYINANYVDGYNRPKAYIAAQGPLKSTAEDFWRMIWEHNVEVIVMITNLVEKGRRKCDQYWPADGSEEYGNFLVTQKSVQVLAYYTVRNFTLRNTKIRVVTQYHYTQWPDMGVPEYSLPVLTFVRKAAYAKRHAVGPVVVHCSAGVGRTGTYIVLDSMLQQIQHEGTVNIFGFLKHIRSQRNYLVQTEEQYVFIHDTLVEAILS Hydrogen bonds contact Hydrophobic contact | ||||
| 83 | Ornithine decarboxylase antizyme 1 | 4ZGY | 6.32 | |
Target general information Gen name OAZ1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms OAZ Protein family ODC antizyme family Biochemical class Lyase / lyase inhibitor Function Ornithine decarboxylase inhibitor activity. Related diseases LIMK1 is located in the Williams-Beuren syndrome (WBS) critical region. WBS results from a hemizygous deletion of several genes on chromosome 7q11.23, thought to arise as a consequence of unequal crossing over between highly homologous low-copy repeat sequences flanking the deleted region. Drugs (DrugBank ID) DB00129 Interacts with P30556 EC number NA Uniprot keywords 3D-structure; Polyamine biosynthesis; Proteomics identification; Reference proteome; Ribosomal frameshifting; Transport Protein physicochemical properties Chain ID B Molecular weight (Da) 55126.7 Length 492 Aromaticity 0.11 Instability index 46.89 Isoelectric point 6.19 Charge (pH=7) -3.86 2D Binding mode Binding energy (Kcal/mol) -8.61 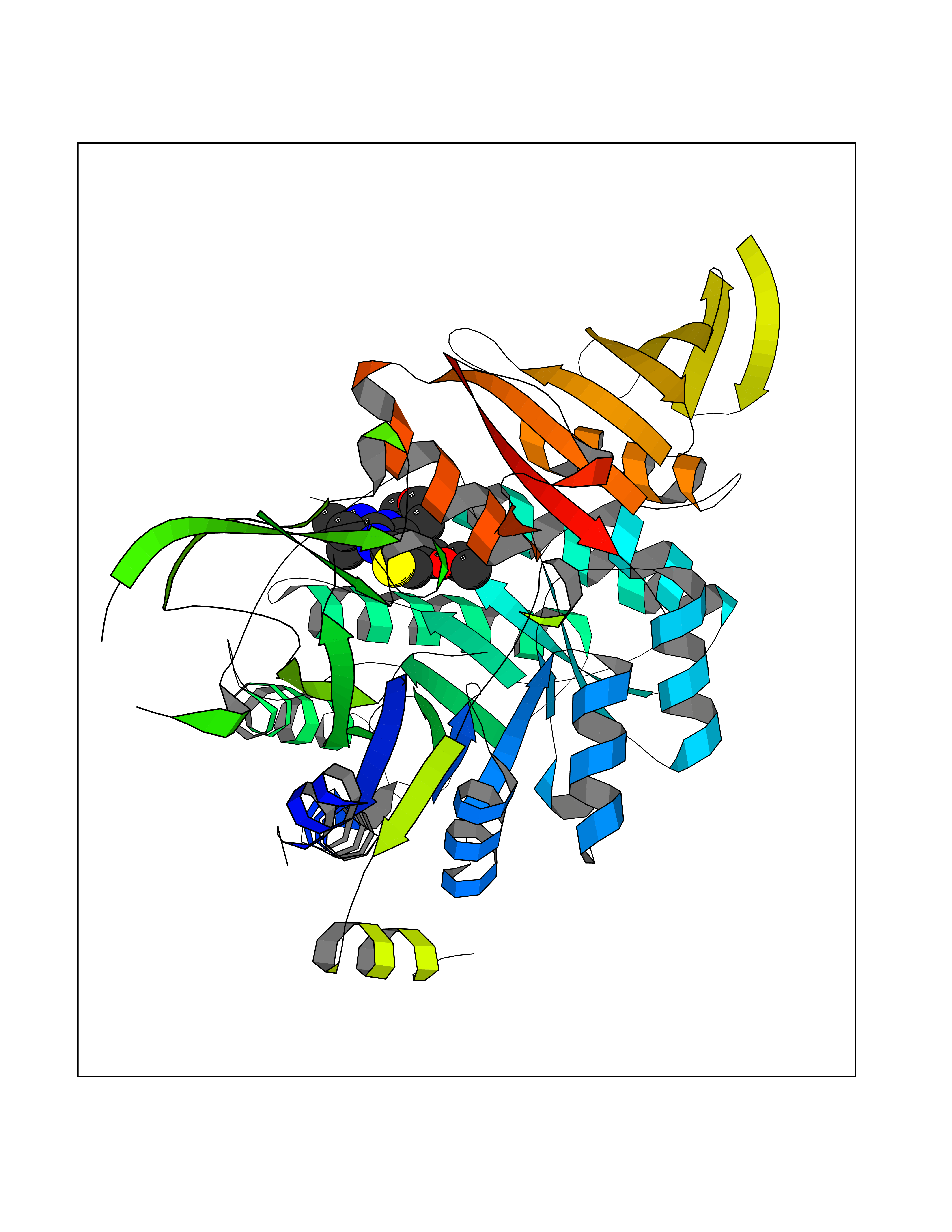 Molscript Map 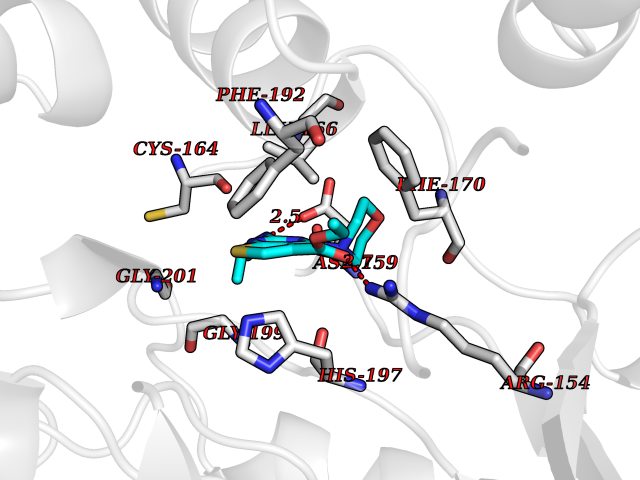 Pymol Map 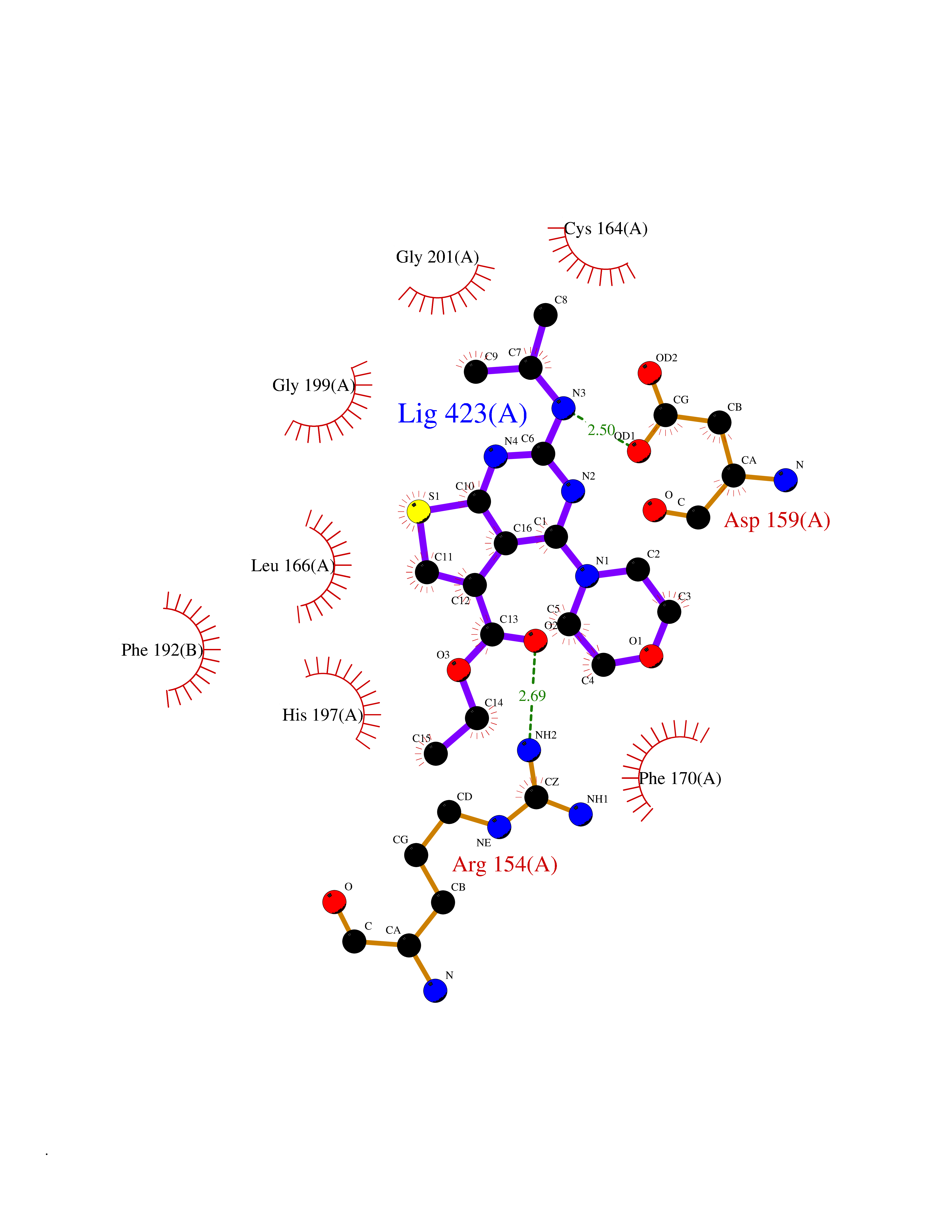 Ligplot Map 3D Binding mode Sequence TAKDILDQKINEKDAFYVADLGDILKKHLRWLKALPRVTPFYAVKCNDSKAIVKTLAATGTGFDCASKTEIQLVQSLGVPPERIIYANPCKQVSQIKYAANNGVQMMTFDSEVELMKVARAHPKAKLVLRIATDDSCRLSVKFGATLRTSRLLLERAKELNIDVVGVSFHVGSGCTDPETFVQAISDARCVFDMGAEVGFSMYLLDIGGGFPGSEDVKLKFEEITGVINPALDKYFPSDSGVRIIAEPGRYYVASAFTLAVNIIAMYYVNDGVYGSFNCILYDHAHVKPLLQKRPEKYYSSSIWGPTCDGLDRIVERCDLPEMHVGDWMLFENMGAYTVASTFNGFQRPTIYYVMSGPAWQLMQQFQFYSDDRLNVTEELTSNDKTRILNVQSRLTDAKRINWRTVLSGGSLYIEIPGGALPEGSKDSFAVLLEFAEEQLRADHVFICFHKNREDRAALLRTFSFLGFEIVRPGHPLVPKRPDACFMAYTFE Hydrogen bonds contact Hydrophobic contact | ||||
| 84 | Gastrin (GAST) | 5WRJ | 6.32 | |
Target general information Gen name GAST Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Gastrin6; GAST; G52; G34; G14 Protein family Gastrin/cholecystokinin family Biochemical class Gastrin cholecystokinin Function Gastrin stimulates the stomach mucosa to produce and secrete hydrochloric acid and the pancreas to secrete its digestive enzymes. It also stimulates smooth muscle contraction and increases blood circulation and water secretion in the stomach and intestine. Related diseases Orotic aciduria 1 (ORAC1) [MIM:258900]: A disorder of pyrimidine metabolism resulting in megaloblastic anemia and orotic acid crystalluria that is frequently associated with some degree of physical and intellectual disability. A minority of cases have additional features, particularly congenital malformations and immune deficiencies. {ECO:0000269|PubMed:9042911}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB12532 Interacts with Q13520; O43315; Q12797-6; Q9BXK5; Q8N5K1; Q96BA8; P00387; Q9Y282; Q5JX71; Q8NBJ4; Q8TDT2; P43628; O76011; Q6ZUX7; Q15546; P15941-11; Q13113; P60201-2; Q14973; P02787; Q4KMG9 EC number NA Uniprot keywords 3D-structure; Amidation; Cleavage on pair of basic residues; Direct protein sequencing; Hormone; Phosphoprotein; Proteomics identification; Pyrrolidone carboxylic acid; Reference proteome; Secreted; Signal; Sulfation Protein physicochemical properties Chain ID A,F Molecular weight (Da) 31827.9 Length 280 Aromaticity 0.08 Instability index 41.06 Isoelectric point 8.84 Charge (pH=7) 5.56 2D Binding mode Binding energy (Kcal/mol) -8.62 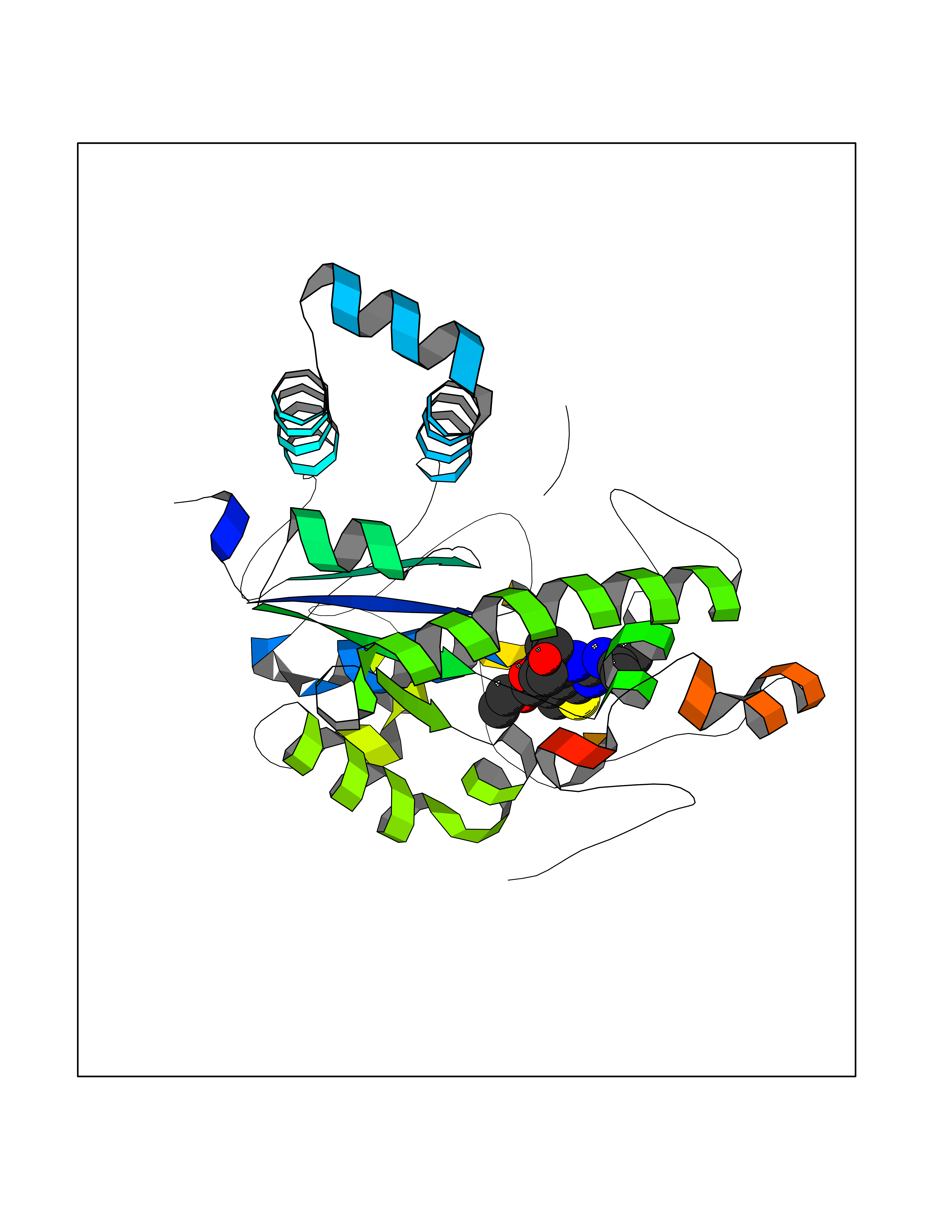 Molscript Map 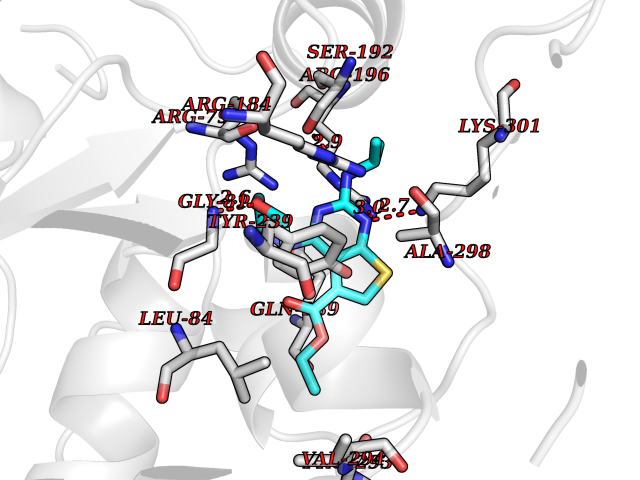 Pymol Map 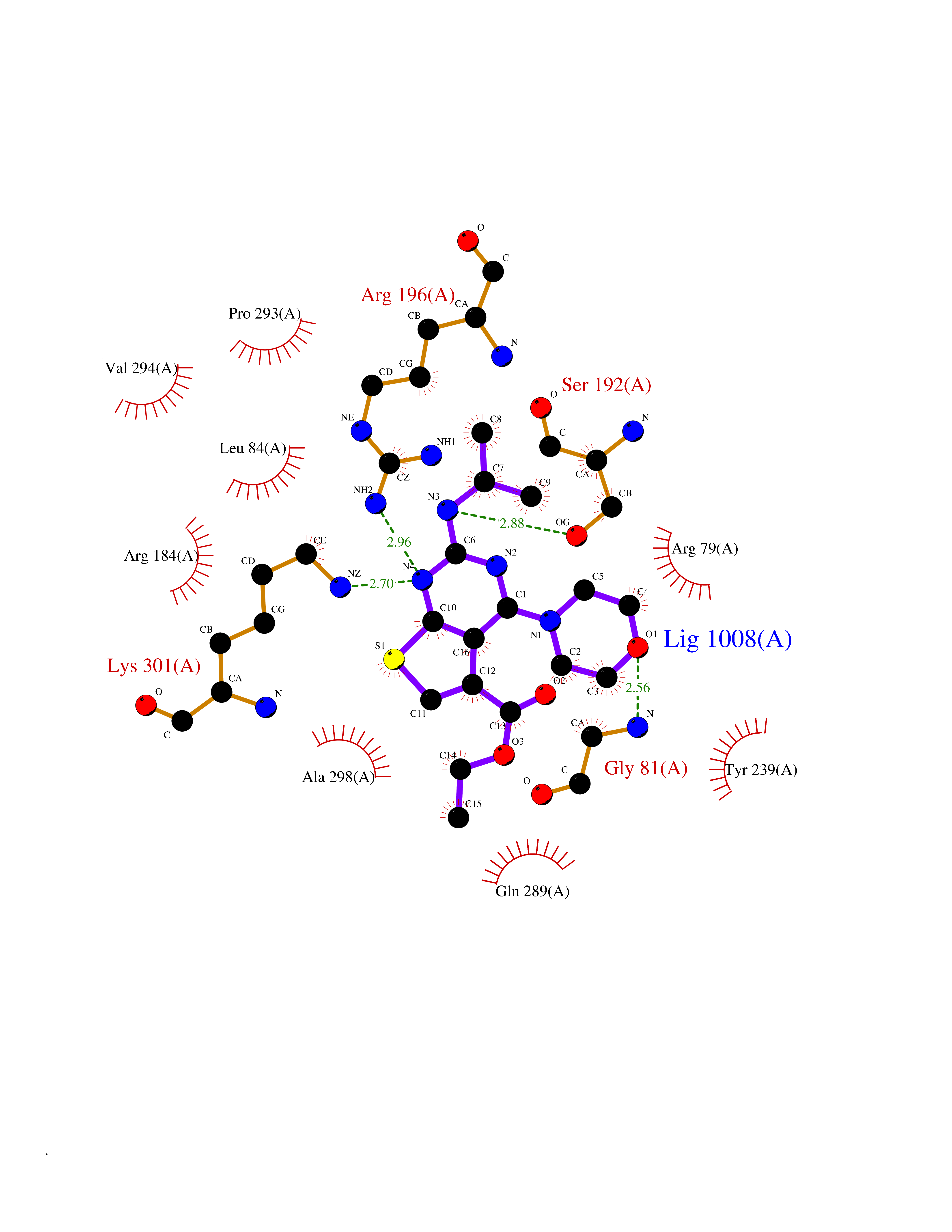 Ligplot Map 3D Binding mode Sequence AYHKDMPLIFIGGVPRSGTTLMRAMLDAHPDIRCGEETRVIPRILALKQMWSRSSKEKIRLDEAGVTDEVLDSAMQAFLLEIIVKHGEPAPYLCNKDPFALKSLTYLSRLFPNAKFLLMVRDGRASVHSMISRKVTIAGFDLNSYRDCLTKWNRAIETMYNQCMEVGYKKCMLVHYEQLVLHPERWMRTLLKFLQIPWNHSVLHHEEMIGKAGGVSLSKVERSTDQVIKPVNVGALSKWVGKIPPDVLQDMAVIAPMLAKLGYDPYANPPNYGKPEEEAY Hydrogen bonds contact Hydrophobic contact | ||||
| 85 | Melatonin receptor type 1B (MTNR1B) | 6ME9 | 6.32 | |
Target general information Gen name MTNR1B Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Mel1b receptor; Mel1b melatonin receptor; Mel-1B-R Protein family G-protein coupled receptor 1 family Biochemical class GPCR rhodopsin Function Likely to mediate the reproductive and circadian actions of melatonin. The activity of this receptor is mediated by pertussis toxin sensitive G proteins that inhibit adenylate cyclase activity. High affinity receptor for melatonin. Related diseases Insulin-like growth factor 1 resistance (IGF1RES) [MIM:270450]: A disorder characterized by intrauterine growth retardation, poor postnatal growth and increased plasma IGF1 levels. {ECO:0000269|PubMed:14657428, ECO:0000269|PubMed:15928254, ECO:0000269|PubMed:25040157}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB06594; DB01065; DB00980; DB02709; DB09071; DB15133 Interacts with P28335; P48039; O76081; Q14669 EC number NA Uniprot keywords 3D-structure; Cell membrane; Disulfide bond; G-protein coupled receptor; Glycoprotein; Membrane; Receptor; Reference proteome; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 50184.9 Length 448 Aromaticity 0.11 Instability index 37.2 Isoelectric point 5.72 Charge (pH=7) -5.68 2D Binding mode Binding energy (Kcal/mol) -8.62 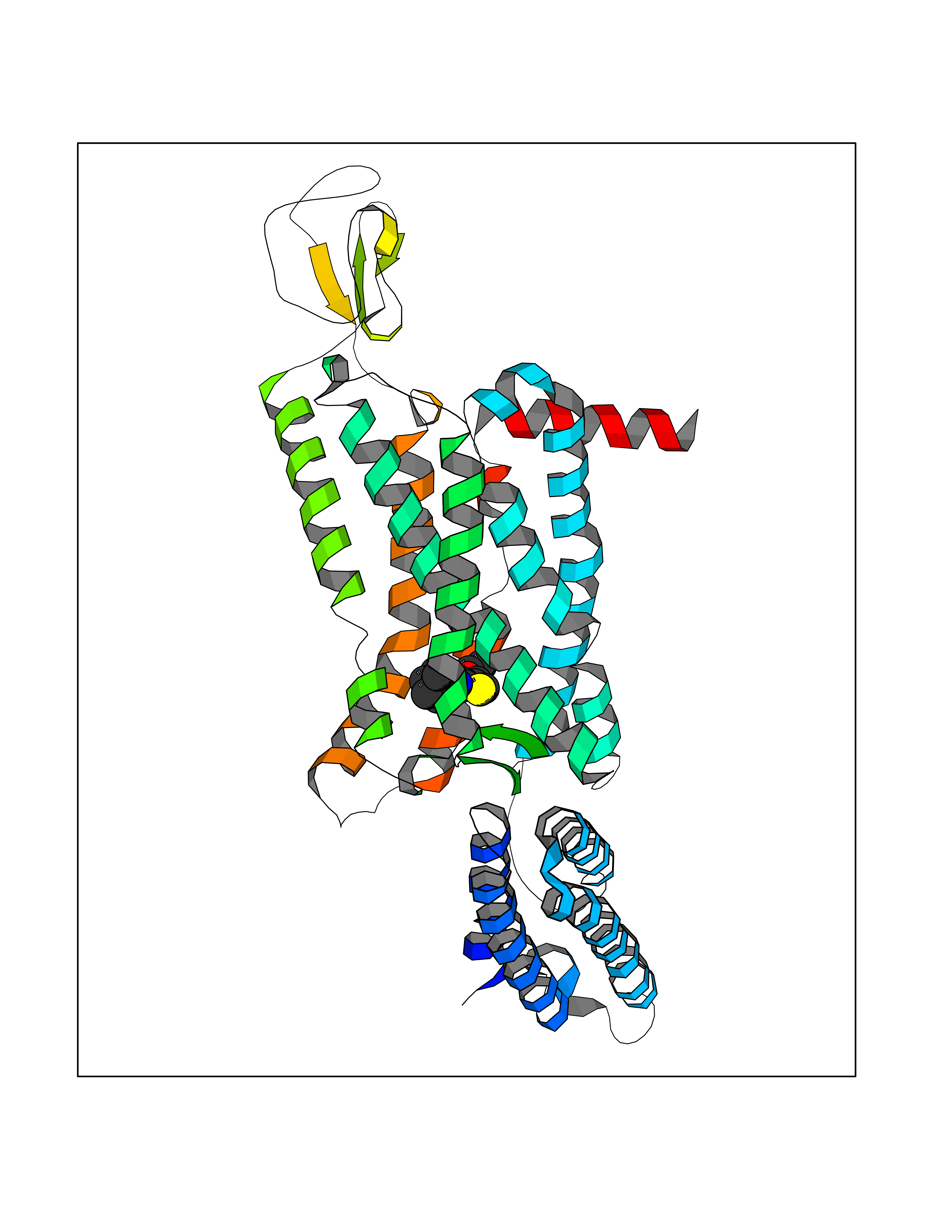 Molscript Map 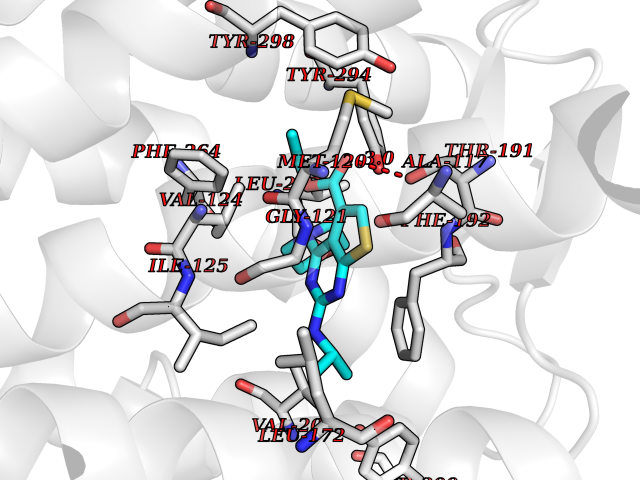 Pymol Map 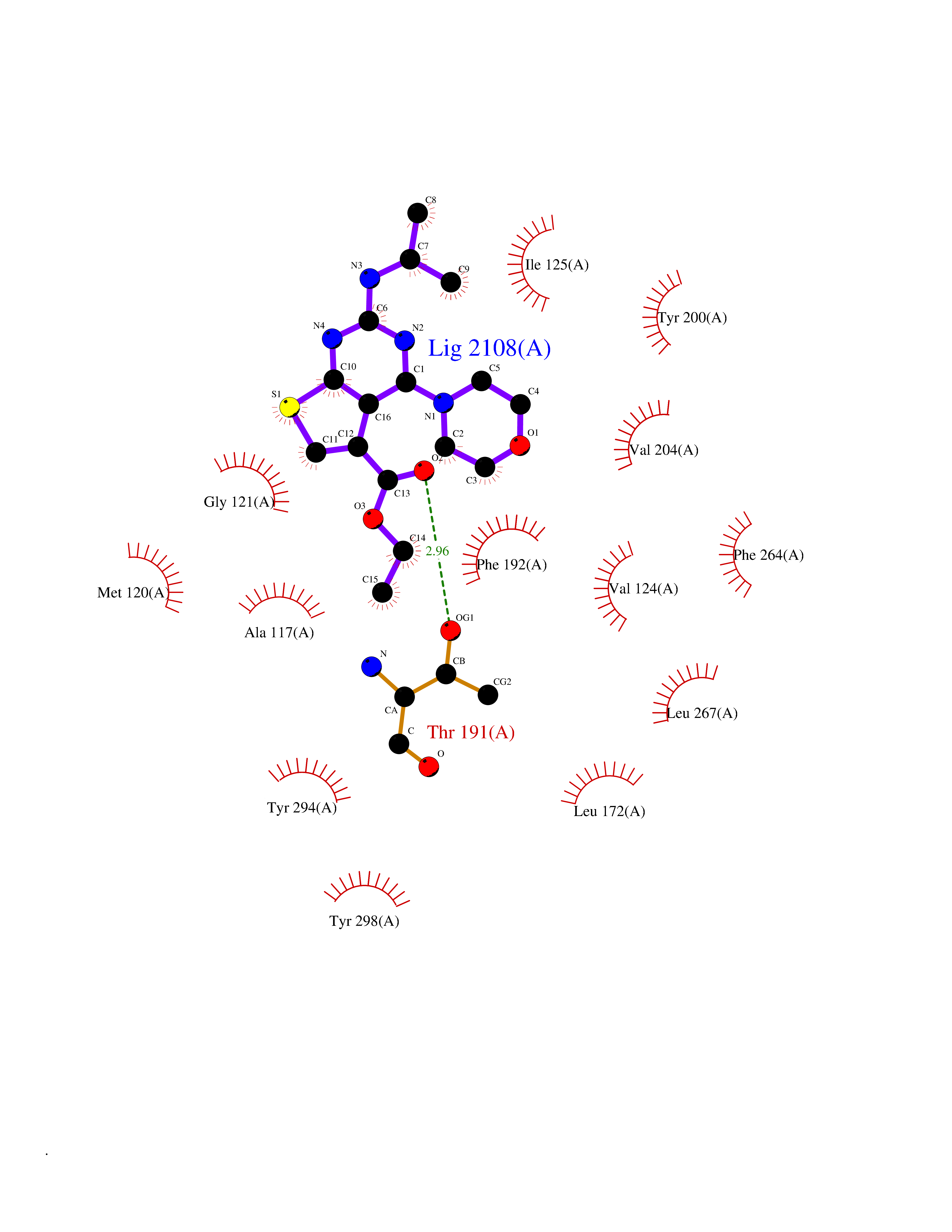 Ligplot Map 3D Binding mode Sequence ADLEDNWETLNDNLKVIEKADNAAQVKDALTKMRAAALDAQKATPPKLEDKSPDSPEMKDFRHGFDILVGQIDDALKLANEGKVKEAQAAAEQLKTTRNAYIQKYLGDGARPSWVAPALSAVLIVTTAVDVVGNLLVILSVLRNRKLRNAGNLFLVSLALANLVVAFYPYPLILVAIFYDGWAFGEEHCKASAFVMGLSVIGSVWNITAIAIDRYLYICHSMAYHRIYRRWHTPLHICLIWLLTVVALLPNFFVGSLEYDPRIYSCTFIQTASTQYTAAVVVIHFLLPIAVVSFCYLRIWVLVLQARMKKYTCTVCGYIYNPEDGDPDNGVNPGTDFKDIPDDWVCPLCGVGKDQFEEVECLKPSDLRSFLTMFVVFVIFAICFAPLNCIGLAVAINPQEMAPQIPEGLFVTSYLLAYFNSCLNPIVYGLLDQNFRREYKRILLALWN Hydrogen bonds contact Hydrophobic contact | ||||
| 86 | Thymidine phosphorylase (TYMP) | 2J0F | 6.32 | |
Target general information Gen name TYMP Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms TdRPase; TYMP; TP; Platelet-derived endothelial cell growth factor; PDECGF; PD-ECGF; Gliostatin Protein family Thymidine/pyrimidine-nucleoside phosphorylase family Biochemical class Pentosyltransferase Function Catalyzes the reversible phosphorolysis of thymidine. The produced molecules are then utilized as carbon and energy sources or in the rescue of pyrimidine bases for nucleotide synthesis. Related diseases Mitochondrial DNA depletion syndrome 1, MNGIE type (MTDPS1) [MIM:603041]: A multisystem disease associated with mitochondrial dysfunction. It is clinically characterized by onset between the second and fifth decades of life, ptosis, progressive external ophthalmoplegia, gastrointestinal dysmotility (often pseudoobstruction), diffuse leukoencephalopathy, cachexia, peripheral neuropathy, and myopathy. {ECO:0000269|PubMed:12177387, ECO:0000269|PubMed:9924029}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01101; DB00369; DB00322; DB00544; DB06433; DB09343; DB00432 Interacts with Q14696; Q9H0C1 EC number EC 2.4.2.4 Uniprot keywords 3D-structure; Alternative splicing; Angiogenesis; Chemotaxis; Developmental protein; Differentiation; Direct protein sequencing; Disease variant; Glycosyltransferase; Growth factor; Neuropathy; Phosphoprotein; Primary mitochondrial disease; Progressive external ophthalmoplegia; Proteomics identification; Reference proteome; Repeat; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 46508.2 Length 446 Aromaticity 0.03 Instability index 37.49 Isoelectric point 5.81 Charge (pH=7) -4.7 2D Binding mode Binding energy (Kcal/mol) -8.63 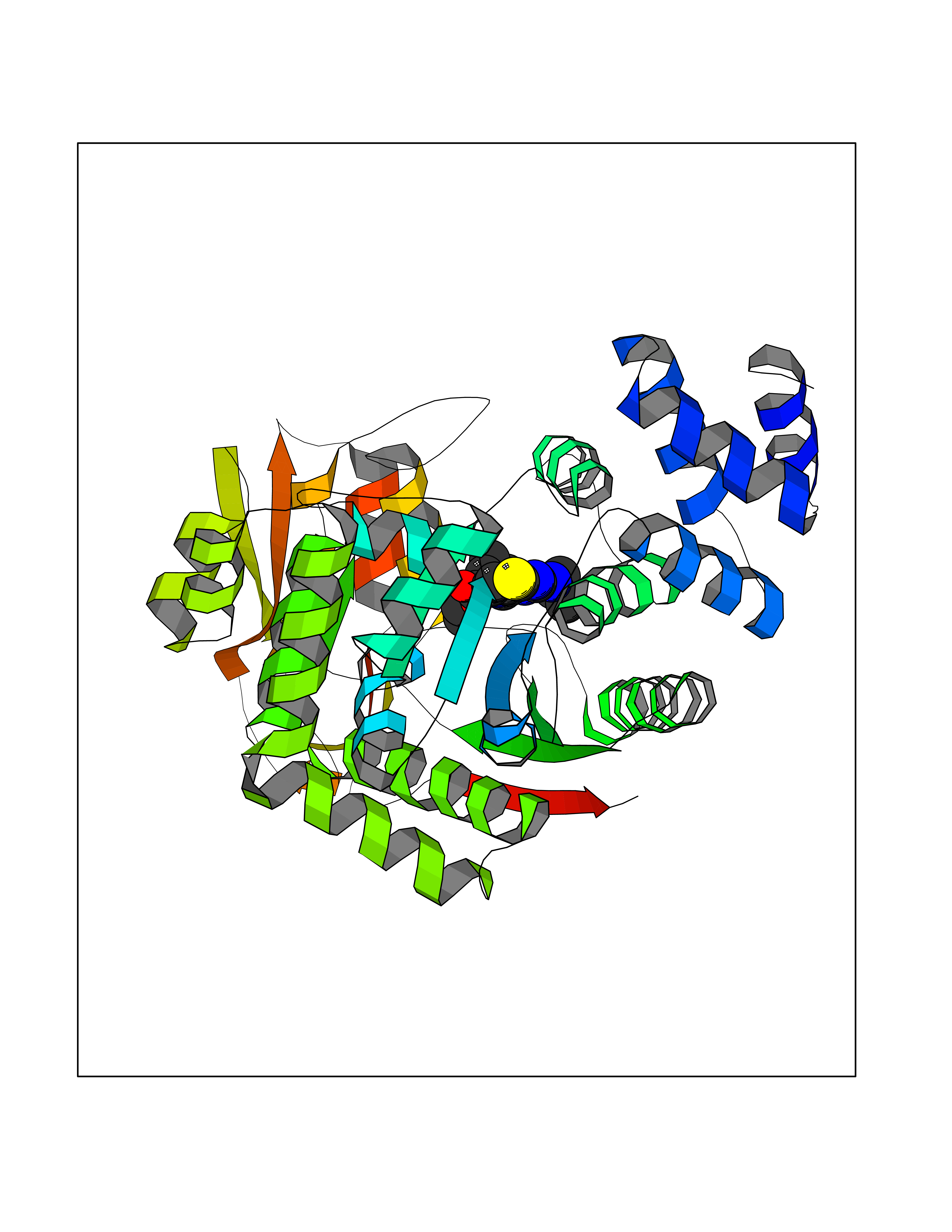 Molscript Map 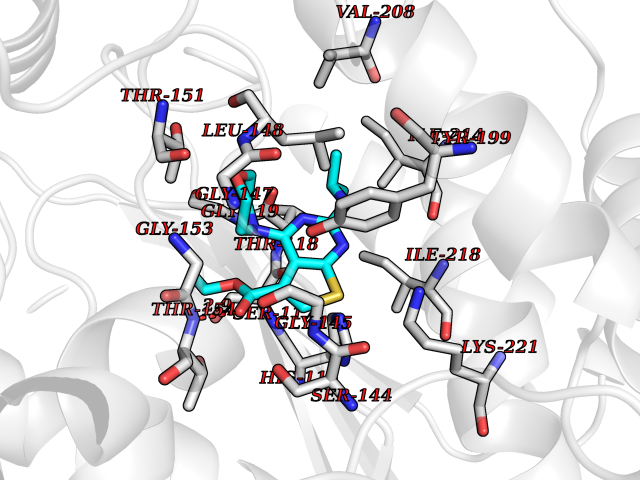 Pymol Map 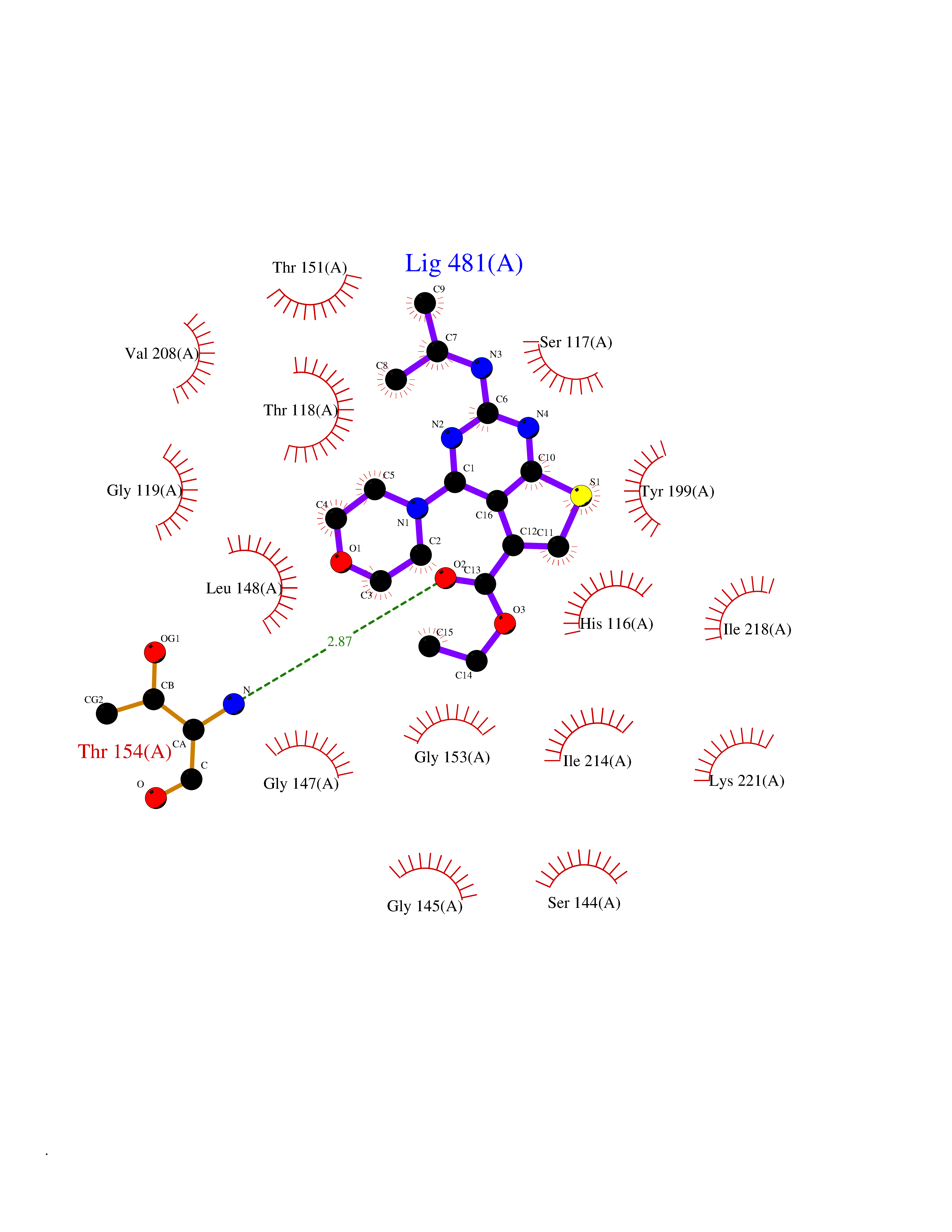 Ligplot Map 3D Binding mode Sequence KQLPELIRMKRDGGRLSEADIRGFVAAVVNGSAQGAQIGAMLMAIRLRGMDLEETSVLTQALAQSGQQLEWPEAWRQQLVDKHSTGGVGDKVSLVLAPALAACGCKVPMISGRGLGHTGGTLDKLESIPGFNVIQSPEQMQVLLDQAGCCIVGQSEQLVPADGILYAARDVTATVDSLPLITASILSKKLVEGLSALVVDVKFGAGAVFPNQEQARELAKTLVGVGASLGLRVAAALTAMDKPLGRCVGHALEVEEALLCMDGAGPPDLRDLVTTLGGALLWLSGHAGTQAQGAARVAAALDDGSALGRFERMLAAQGVDPGLARALCSGSPAERRQLLPRAREQEELLAPADGTVELVRALPLALVLHELGAGRSRAGEPLRLGVGAELLVDVGQRLRRGTPWLRVHRDGPALSGPQSRALQEALVLSDRAPFAAPLPFAELVLP Hydrogen bonds contact Hydrophobic contact | ||||
| 87 | Fungal Scytalone dehydratase (Fung SDH1) | 3STD | 6.32 | |
Target general information Gen name Fung SDH1 Organism Pyricularia oryzae (strain 70-15 / ATCC MYA-4617 / FGSC 8958) (Rice blast fungus) (Magnaporthe oryzae) Uniprot ID TTD ID Synonyms SDH1 Protein family Scytalone dehydratase family Biochemical class Alpha-carbonic anhydrase Function Catalyzes two steps in melanin biosynthesis. From scytalone they are two dehydration steps and one reduction step to yield melanin. Related diseases CODAS syndrome (CODASS) [MIM:600373]: A rare syndrome characterized by the combination of cerebral, ocular, dental, auricular, and skeletal features. These include developmental delay, craniofacial anomalies, cataracts, ptosis, median nasal groove, delayed tooth eruption, hearing loss, short stature, delayed epiphyseal ossification, metaphyseal hip dysplasia, and vertebral coronal clefts. {ECO:0000269|PubMed:25574826, ECO:0000269|PubMed:25808063}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number EC 4.2.1.94 Uniprot keywords 3D-structure; Calcium; Direct protein sequencing; Endosome; Lyase; Melanin biosynthesis; Metal-binding; Reference proteome Protein physicochemical properties Chain ID A Molecular weight (Da) 19102.4 Length 162 Aromaticity 0.14 Instability index 31.72 Isoelectric point 5.87 Charge (pH=7) -3.7 2D Binding mode Binding energy (Kcal/mol) -8.62 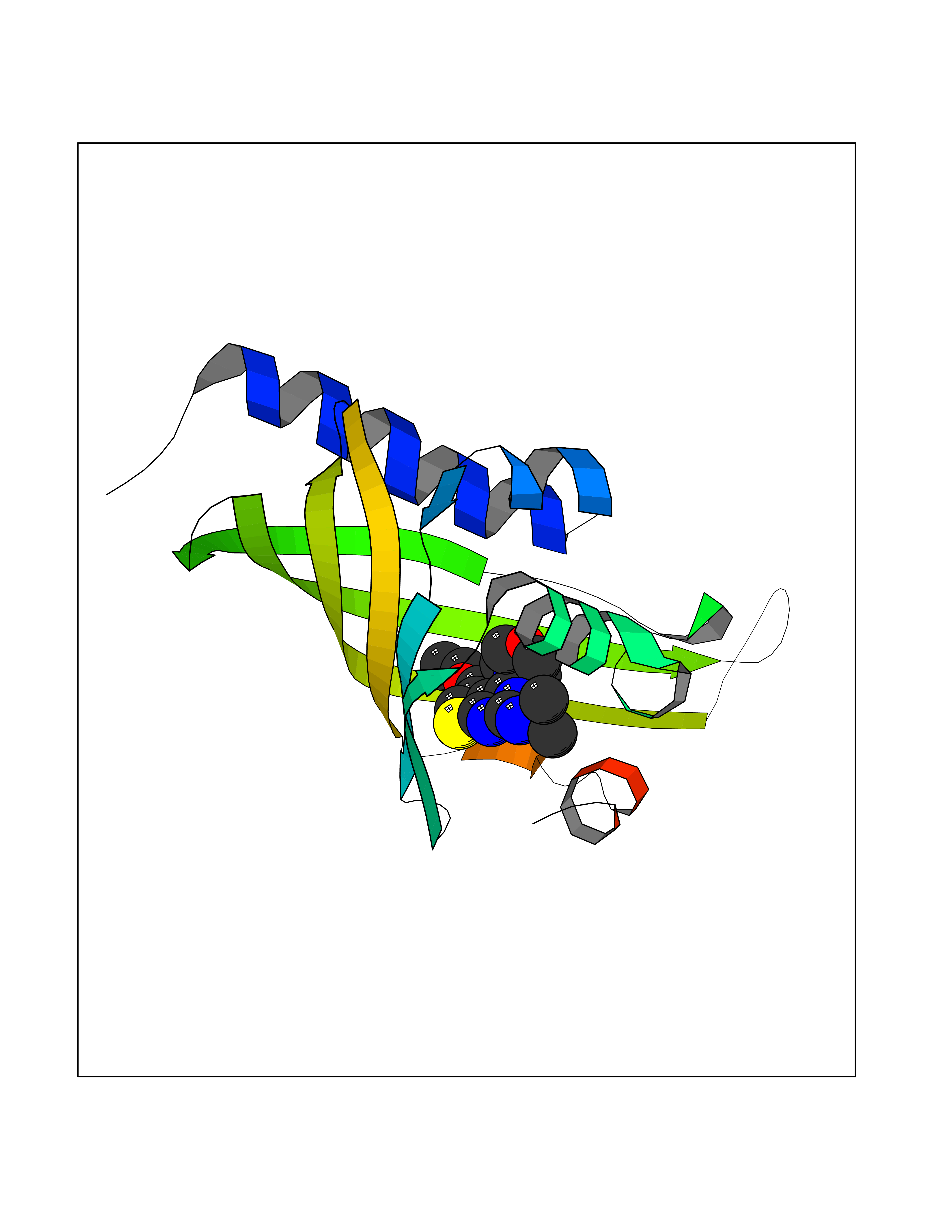 Molscript Map 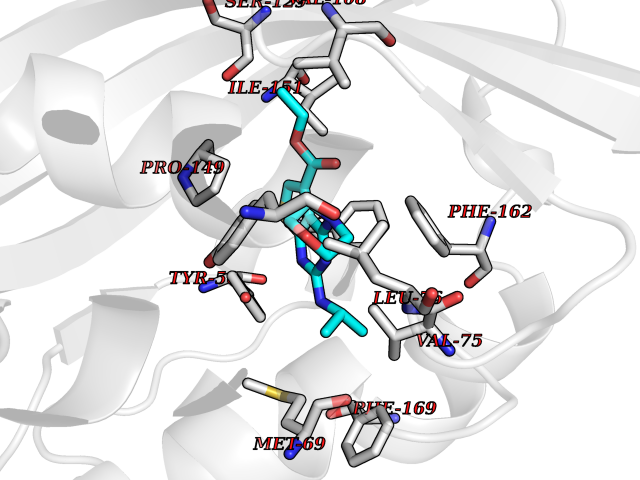 Pymol Map 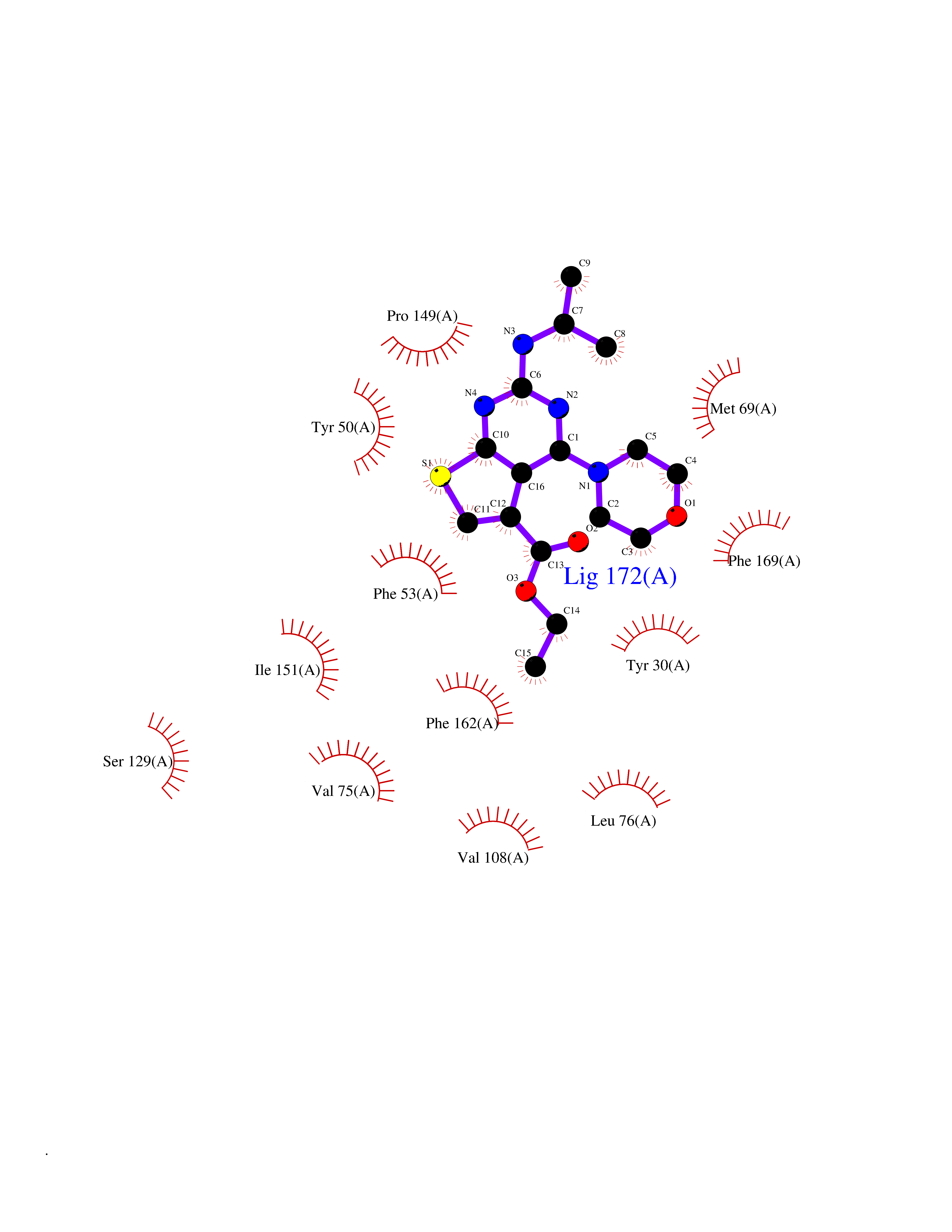 Ligplot Map 3D Binding mode Sequence GEITFSDYLGLMTCVYEWADSYDSKDWDRLRKVIAPTLRIDYRSFLDKLWEAMPAEEFVGMVSSKQVLGDPTLRTQHFIGGTRWEKVSEDEVIGYHQLRVPHQRYKDTTMKEVTMKGHAHSANLHWYKKIDGVWKFAGLKPDIRWGEFDFDRIFEDGRETFG Hydrogen bonds contact Hydrophobic contact | ||||
| 88 | Trypanosoma Trypanothione reductase (Trypano TPR) | 2WBA | 6.32 | |
Target general information Gen name Trypano TPR Organism Trypanosoma brucei brucei Uniprot ID TTD ID Synonyms TRYR; TPR; Parasite-specific trypanothione reductase; N(1),N(8)-bis(glutathionyl)spermidine reductase Protein family Class-I pyridine nucleotide-disulfide oxidoreductase family Biochemical class Sulfur donor oxidoreductase Function Trypanothione is the parasite analog of glutathione; this enzyme is the equivalent of glutathione reductase. Related diseases Immunodeficiency 57 with autoinflammation (IMD57) [MIM:618108]: An autosomal recessive primary immunodeficiency characterized by lymphopenia and recurrent viral, bacterial, and fungal infections. Patients exhibit early-onset inflammatory bowel disease involving the upper and lower gastrointestinal tract, and develop progressive polyarthritis. {ECO:0000269|PubMed:30026316}. The disease is caused by variants affecting the gene represented in this entry. RIPK1-deficient immune cells from IMD57 patients have impaired proinflammatory signaling leading to dysregulated cytokine secretion and are prone to necroptosis. {ECO:0000269|PubMed:30026316}.; DISEASE: Autoinflammation with episodic fever and lymphadenopathy (AIEFL) [MIM:618852]: An autosomal dominant immunologic disorder characterized by early onset of recurrent episodes of unexplained fever, lymphadenopathy, hepatosplenomegaly, and increased levels of inflammatory cytokines and chemokines in patient serum. {ECO:0000269|PubMed:31827280, ECO:0000269|PubMed:31827281}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number EC 1.8.1.12 Uniprot keywords 3D-structure; Cytoplasm; Disulfide bond; FAD; Flavoprotein; NADP; Oxidoreductase; Redox-active center Protein physicochemical properties Chain ID A,B Molecular weight (Da) 105578 Length 978 Aromaticity 0.08 Instability index 33.76 Isoelectric point 6.25 Charge (pH=7) -6.81 2D Binding mode Binding energy (Kcal/mol) -8.62 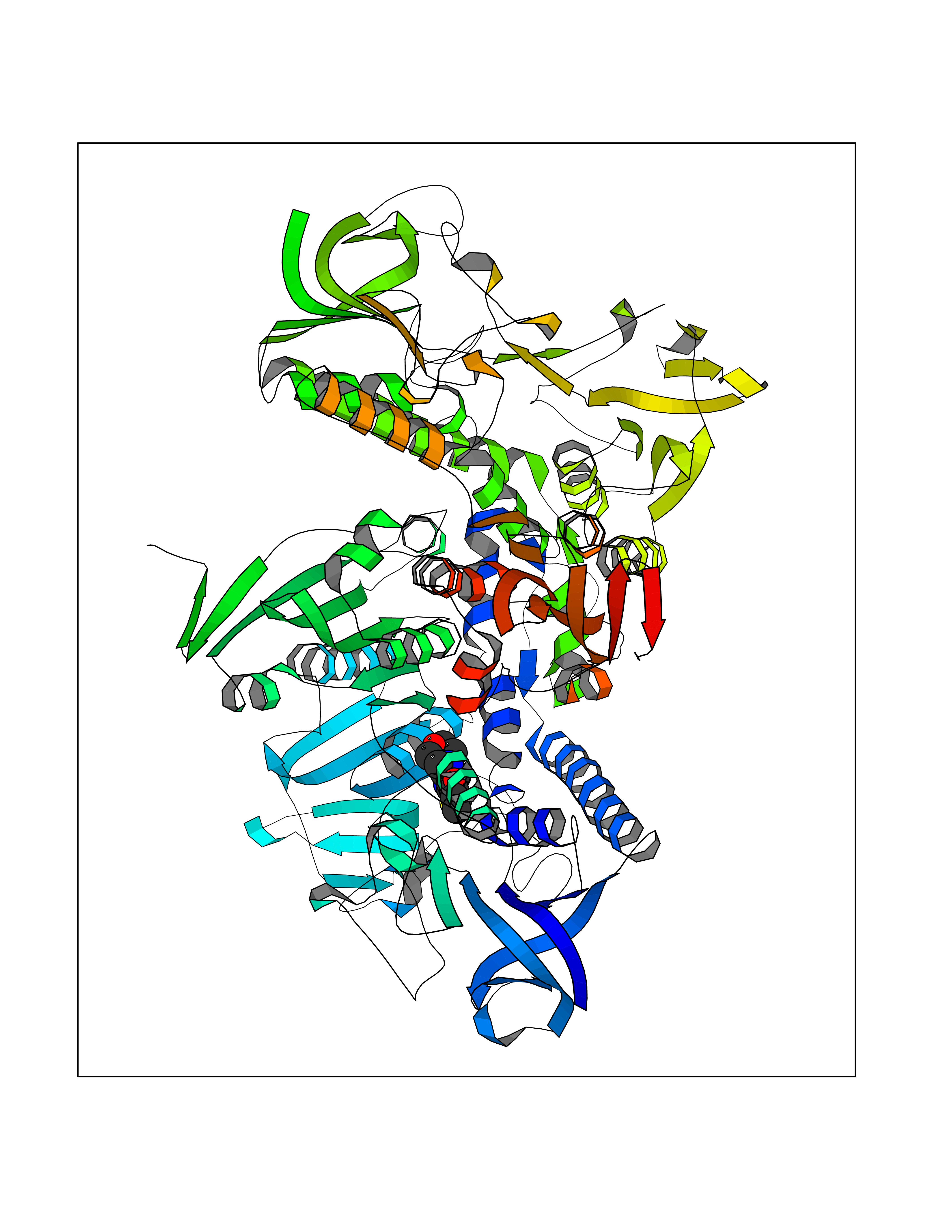 Molscript Map 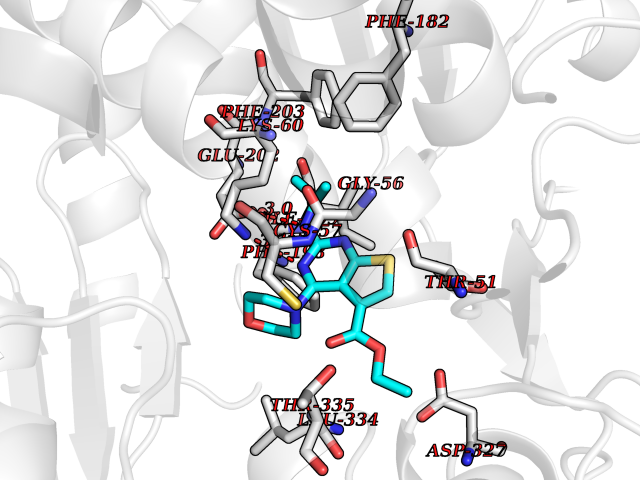 Pymol Map 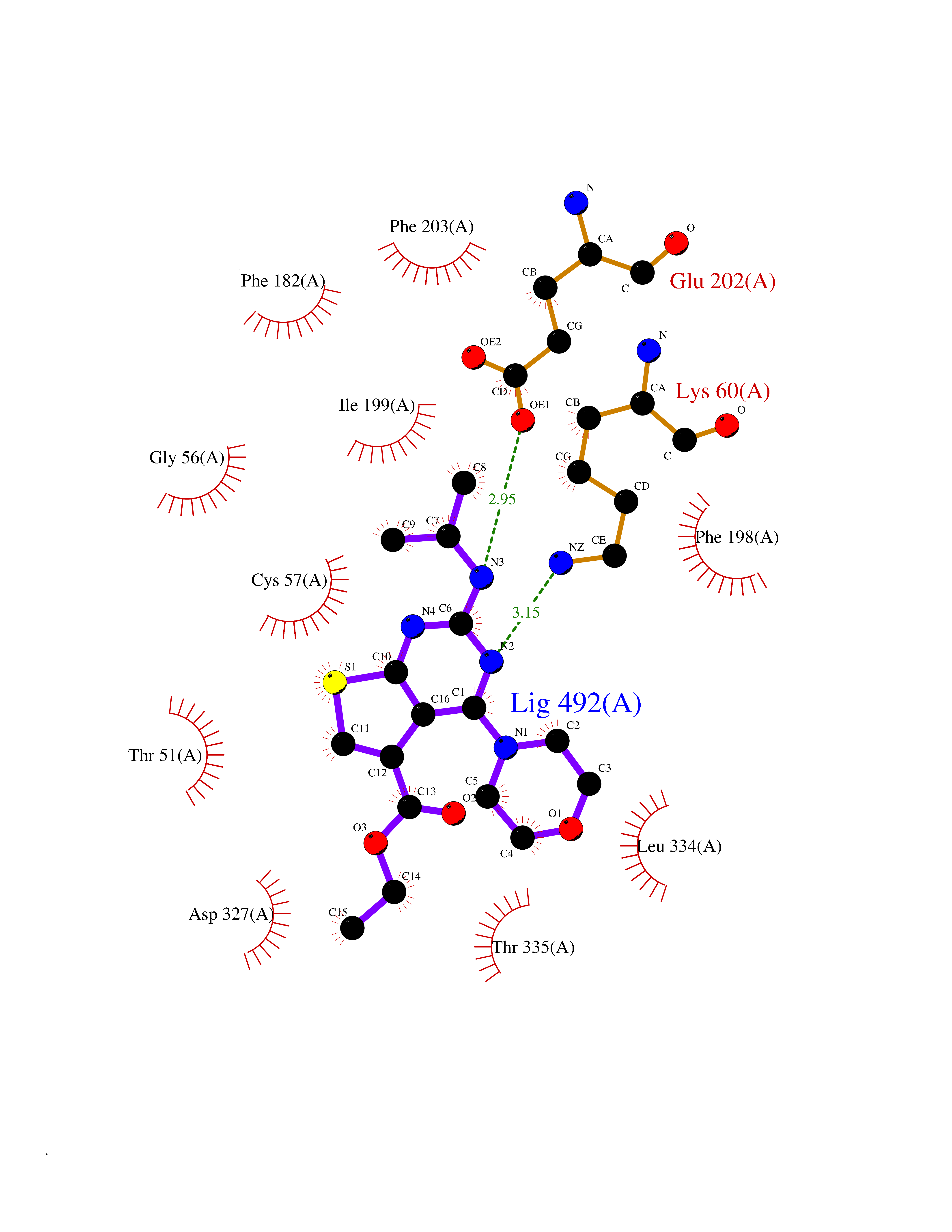 Ligplot Map 3D Binding mode Sequence SKAFDLVVIGAGSGGLEAGWNAATLYGKRVAVVDVQTSHGPPFYAALGGTCVNVGCVPKKLMVTGAQYMDHLRESAGFGWEFDGSSVKANWKKLIAAKNEAVLDINKSYEGMFNDTEGLDFFLGWGSLESKNVVVVRETADPKSAVKERLQADHILLATGSWPQMPAIPGIEHCISSNEAFYLPEPPRRVLTVGGGFISVEFAGIFNAYKPPGGKVTLCYRNNLILRGFDETIREEVTKQLTANGIEIMTNENPAKVSLNTDGSKHVTFESGKTLDVDVVMMAIGRIPRTNDLQLGNVGVKLTPKGGVQVDEFSRTNVPNIYAIGDITDRLMLTPVAINEGAALVDTVFGNKPRKTDHTRVASAVFSIPPIGTCGLIEEVAAKEFEKVAVYMSSFTPLMHNISGSKYKKFVAKIVTNHSDGTVLGVHLLGDGAPEIIQAVGVCLRLNAKISDFYNTIGVHPTSAEELCSMRTPSYYYVKGEKMEKLPDSSKAFDLVVIGAGSGGLEAGWNAATLYGKRVAVVDVQTSHGPPFYAALGGTCVNVGCVPKKLMVTGAQYMDHLRESAGFGWEFDGSSVKANWKKLIAAKNEAVLDINKSYEGMFNDTEGLDFFLGWGSLESKNVVVVRETADPKSAVKERLQADHILLATGSWPQMPAIPGIEHCISSNEAFYLPEPPRRVLTVGGGFISVEFAGIFNAYKPPGGKVTLCYRNNLILRGFDETIREEVTKQLTANGIEIMTNENPAKVSLNTDGSKHVTFESGKTLDVDVVMMAIGRIPRTNDLQLGNVGVKLTPKGGVQVDEFSRTNVPNIYAIGDITDRLMLTPVAINEGAALVDTVFGNKPRKTDHTRVASAVFSIPPIGTCGLIEEVAAKEFEKVAVYMSSFTPLMHNISGSKYKKFVAKIVTNHSDGTVLGVHLLGDGAPEIIQAVGVCLRLNAKISDFYNTIGVHPTSAEELCSMRTPSYYYVKGEKMEKLPDS Hydrogen bonds contact Hydrophobic contact | ||||
| 89 | Monoamine oxidase type B (MAO-B) | 2V5Z | 6.31 | |
Target general information Gen name MAOB Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms MAO-B; Amine oxidase [flavin-containing] B Protein family Flavin monoamine oxidase family Biochemical class CH-NH(2) donor oxidoreductase Function Catalyzes the oxidative deamination of biogenic and xenobiotic amines and has important functions in the metabolism of neuroactive and vasoactive amines in the central nervous system and peripheral tissues. MAOB preferentially degrades benzylamine and phenylethylamine. Related diseases Microvascular complications of diabetes 5 (MVCD5) [MIM:612633]: Pathological conditions that develop in numerous tissues and organs as a consequence of diabetes mellitus. They include diabetic retinopathy, diabetic nephropathy leading to end-stage renal disease, and diabetic neuropathy. Diabetic retinopathy remains the major cause of new-onset blindness among diabetic adults. It is characterized by vascular permeability and increased tissue ischemia and angiogenesis. Disease susceptibility is associated with variants affecting the gene represented in this entry. Homozygosity for the Leu-55 allele is strongly associated with the development of retinal disease in diabetic patients. Drugs (DrugBank ID) DB08176; DB02211; DB08516; DB08480; DB01472; DB04307; DB07512; DB07513; DB00915; DB00182; DB06698; DB04889; DB00215; DB09130; DB04147; DB00988; DB01363; DB00668; DB01175; DB02509; DB03147; DB14914; DB00614; DB04818; DB02095; DB01247; DB00601; DB01577; DB01442; DB01171; DB08082; DB02643; DB04677; DB03894; DB08804; DB04820; DB00184; DB04821; DB12612; DB01626; DB00780; DB00191; DB00388; DB01132; DB00721; DB01168; DB01367; DB09363; DB06654; DB01037; DB01104; DB14569; DB09042; DB00752; DB16446; DB09185; DB04832; DB00909 Interacts with P55212; P28329-3; Q8NI60; Q5RI15; Q92915-2; P22607; Q53GS7; P06396; P01112; O14901; P13473-2; P21397; Q9BVL2; O75400-2; P62826; Q6NTF9-3; Q9Y371; Q7Z699; Q9UMX0; Q9Y649 EC number EC 1.4.3.4 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Direct protein sequencing; FAD; Flavoprotein; Membrane; Mitochondrion; Mitochondrion outer membrane; Oxidoreductase; Proteomics identification; Reference proteome; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A,B Molecular weight (Da) 56019.9 Length 494 Aromaticity 0.09 Instability index 34.81 Isoelectric point 6.51 Charge (pH=7) -2.2 2D Binding mode Binding energy (Kcal/mol) -8.6  Molscript Map 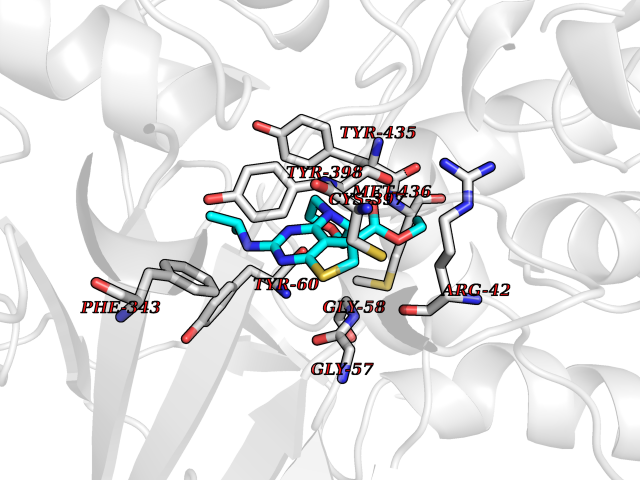 Pymol Map 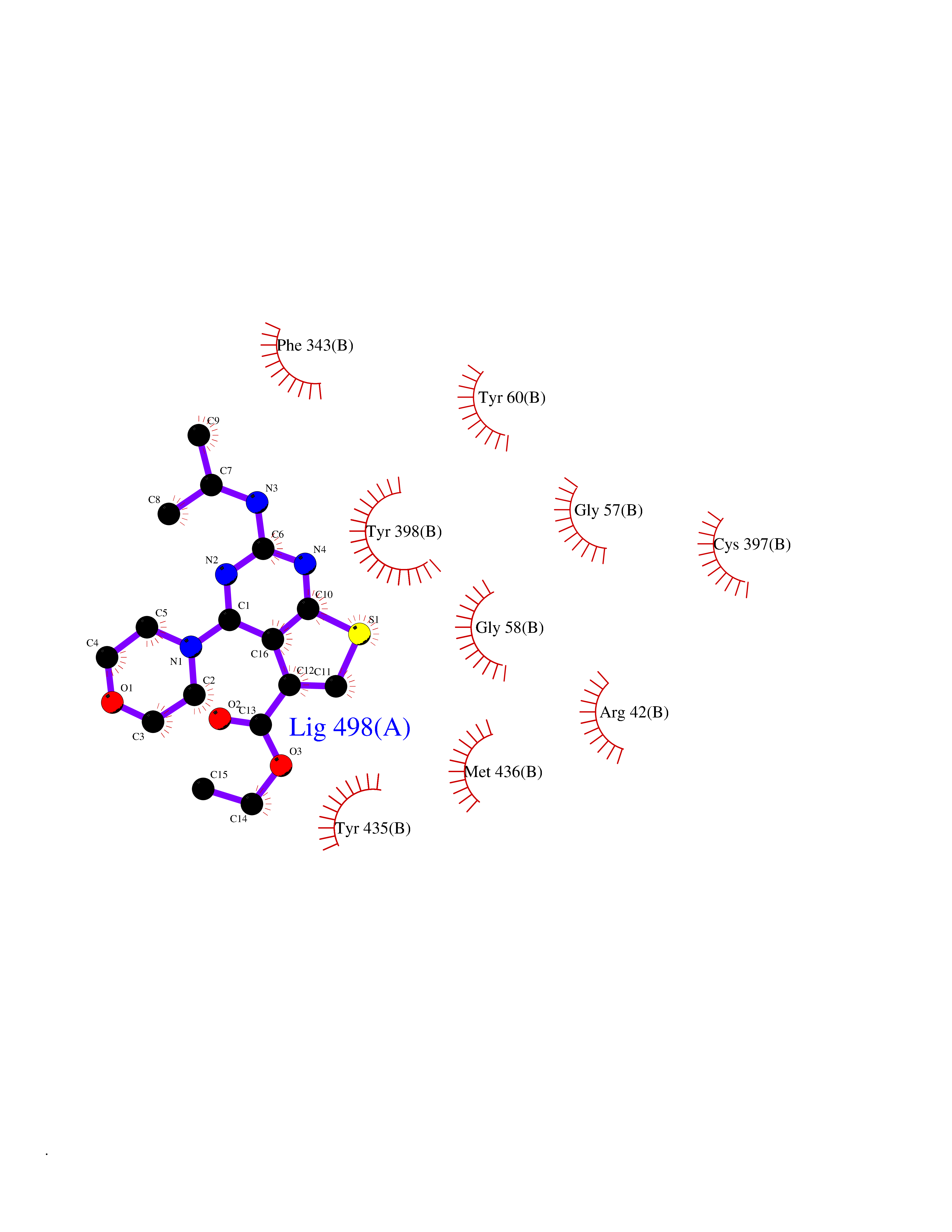 Ligplot Map 3D Binding mode Sequence NKCDVVVVGGGISGMAAAKLLHDSGLNVVVLEARDRVGGRTYTLRNQKVKYVDLGGSYVGPTQNRILRLAKELGLETYKVNEVERLIHHVKGKSYPFRGPFPPVWNPITYLDHNNFWRTMDDMGREIPSDAPWKAPLAEEWDNMTMKELLDKLCWTESAKQLATLFVNLCVTAETHEVSALWFLWYVKQCGGTTRIISTTNGGQERKFVGGSGQVSERIMDLLGDRVKLERPVIYIDQTRENVLVETLNHEMYEAKYVISAIPPTLGMKIHFNPPLPMMRNQMITRVPLGSVIKCIVYYKEPFWRKKDYCGTMIIDGEEAPVAYTLDDTKPEGNYAAIMGFILAHKARKLARLTKEERLKKLCELYAKVLGSLEALEPVHYEEKNWCEEQYSGGCYTTYFPPGILTQYGRVLRQPVDRIYFAGTETATHWSGYMEGAVEAGERAAREILHAMGKIPEDEIWQSEPESVDVPAQPITTTFLERHLPSVPGLLRLI Hydrogen bonds contact Hydrophobic contact | ||||
| 90 | Guanidinoacetate N-methyltransferase | 3ORH | 6.31 | |
Target general information Gen name GAMT Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Class I-like SAM-binding methyltransferase superfamily, RMT2 methyltransferase family Biochemical class Transferase Function Guanidinoacetate N-methyltransferase activity.Methyltransferase activity. Related diseases Cerebral creatine deficiency syndrome 2 (CCDS2) [MIM:612736]: An autosomal recessive disorder characterized by developmental delay and regression, intellectual disability, severe disturbance of expressive and cognitive speech, intractable seizures, movement disturbances, severe depletion of creatine and phosphocreatine in the brain, and accumulation of guanidinoacetic acid in brain and body fluids. {ECO:0000269|PubMed:12468279, ECO:0000269|PubMed:15108290, ECO:0000269|PubMed:15651030, ECO:0000269|PubMed:16293431, ECO:0000269|PubMed:16855203, ECO:0000269|PubMed:17101918, ECO:0000269|PubMed:17466557, ECO:0000269|PubMed:19388150, ECO:0000269|PubMed:23660394, ECO:0000269|PubMed:24415674, ECO:0000269|PubMed:8651275}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00148; DB02751; DB00536; DB13191; DB01752 Interacts with O95363; Q969Q5; Q9HCM9-2 EC number 2.1.1.2 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Disease variant; Methyltransferase; Proteomics identification; Reference proteome; S-adenosyl-L-methionine; Transferase Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 24656 Length 219 Aromaticity 0.11 Instability index 46.5 Isoelectric point 5.91 Charge (pH=7) -4.34 2D Binding mode Binding energy (Kcal/mol) -8.6  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence PAWGAAPAAYDAADTHLRILGKPVMERWETPYMHALAAAASSKGGRVLEVGFGMAIAASKVQEAPIDEHWIIECNDGVFQRLRDWAPRQTHKVIPLKGLWEDVAPTLPDGHFDGILYDTYPLSEETWHTHQFNFIKNHAFRLLKPGGVLTYCNLTSWGELMKSKYSDITIMFEETQVPALLEAGFRRENIRTEVMALVPPADCRYYAFPQMITPLVTKG Hydrogen bonds contact Hydrophobic contact | ||||
| 91 | NAD-dependent deacetylase sirtuin-5 (SIRT5) | 3RIY | 6.31 | |
Target general information Gen name SIRT5 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms SIR2L5; SIR2-like protein 5; Regulatory protein SIR2 homolog 5 Protein family Sirtuin family, Class III subfamily Biochemical class Sirtuin family. Class III subfamily Function NAD-dependent lysine demalonylase, desuccinylase and deglutarylase that specifically removes malonyl, succinyl and glutaryl groups on target proteins (PubMed:21908771, PubMed:22076378, PubMed:24703693, PubMed:29180469). Activates CPS1 and contributes to the regulation of blood ammonia levels during prolonged fasting: acts by mediating desuccinylation and deglutarylation of CPS1, thereby increasing CPS1 activity in response to elevated NAD levels during fasting (PubMed:22076378, PubMed:24703693). Activates SOD1 by mediating its desuccinylation, leading to reduced reactive oxygen species (PubMed:24140062). Activates SHMT2 by mediating its desuccinylation (PubMed:29180469). Modulates ketogenesis through the desuccinylation and activation of HMGCS2. Has weak NAD-dependent protein deacetylase activity; however this activity may not be physiologically relevant in vivo. Can deacetylate cytochrome c (CYCS) and a number of other proteins in vitro such as UOX. Related diseases Immunodeficiency 98 with autoinflammation, X-linked (IMD98) [MIM:301078]: An X-linked disorder characterized by onset of recurrent infections associated with lymphoproliferation and autoinflammation in the first decade of life. Mostly males are affected; carrier females may have mild symptoms. Features include mouth ulcers, fever, poor early growth, hepatosplenomegaly, lymphadenopathy, polyarthritis, and non-infectious enteritis. {ECO:0000269|PubMed:33512449, ECO:0000269|PubMed:34981838}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03478; DB02059; DB15493; DB02701; DB04786 Interacts with NA EC number EC 3.5.1.- Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; Metal-binding; Mitochondrion; NAD; Nucleus; Proteomics identification; Reference proteome; Transferase; Transit peptide; Zinc Protein physicochemical properties Chain ID A,B Molecular weight (Da) 28525.2 Length 262 Aromaticity 0.08 Instability index 43.37 Isoelectric point 8.39 Charge (pH=7) 3.54 2D Binding mode Binding energy (Kcal/mol) -8.61  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence ADFRKFFAKAKHIVIISGAGVSAESGVPTFRGAGGYWRKWQAQDLATPLAFAHNPSRVWEFYHYRREVMGSKEPNAGHRAIAECETRLGKQGRRVVVITQNIDELHRKAGTKNLLEIHGSLFKTRCTSCGVVAENYKSPICPALSGKGAPEPGTQDASIPVEKLPRCEEAGCGGLLRPHVVWFGENLDPAILEEVDRELAHCDLCLVVGTSSVVYPAAMFAPQVAARGVPVAEFNTETTPATNRFRFHFQGPCGTTLPEALA Hydrogen bonds contact Hydrophobic contact | ||||
| 92 | SEC14-like protein 3 | 4UYB | 6.31 | |
Target general information Gen name SEC14L3 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms TAP2 Protein family NA Biochemical class Transport protein Function Lipid binding.Transporter activity. Related diseases Chondrodysplasia with platyspondyly, distinctive brachydactyly, hydrocephaly, and microphthalmia (CDP-PBHM) [MIM:300863]: A disease characterized by chondrodysplasia, severe platyspondyly, hydrocephaly, and facial features with microphthalmia. Bone abnormalities include a distinctive metaphyseal cupping of the metacarpals, metatarsals, and phalanges. Affected females show a milder phenotype with small stature, sometimes associated with body asymmetry and mild intellectual disability. {ECO:0000269|PubMed:20181727}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB14003; DB14001; DB14002; DB11635; DB11251; DB00163 Interacts with NA EC number NA Uniprot keywords 3D-structure; Alternative splicing; Lipid-binding; Proteomics identification; Reference proteome; Transport Protein physicochemical properties Chain ID A Molecular weight (Da) 46148.7 Length 401 Aromaticity 0.1 Instability index 45.19 Isoelectric point 5.79 Charge (pH=7) -5.94 2D Binding mode Binding energy (Kcal/mol) -8.61  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence SMSGRVGDLSPKQAETLAKFRENVQDVLPALPNPDDYFLLRWLRARNFDLQKSEALLRKYMEFRKTMDIDHILDWQPPEVIQKYMPGGLCGYDRDGCPVWYDIIGPLDPKGLLFSVTKQDLLKTKMRDCERILHECDLQTERLGKKIETIVMIFDCEGLGLKHFWKPLVEVYQEFFGLLEENYPETLKFMLIVKATKLFPVGYNLMKPFLSEDTRRKIIVLGNNWKEGLLKLISPEELPAQFGGTLTDPDGNPKCLTKINYGGEIPKSMYVRDQVKTQYEHSVQINRGSSHQVEYEILFPGCVLRWQFSSDGADIGFGVFLKTKMGERQRAGEMTEVLPSQRYNAHMVPEDGNLTCSEAGVYVLRFDNTYSFVHAKKVSFTVEVLLPDEGMQKYDKELTPV Hydrogen bonds contact Hydrophobic contact | ||||
| 93 | Beta-arrestin-1 (ARRB1) | 6TKO | 6.31 | |
Target general information Gen name ARRB1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Non-visual arrestin-2; Betaarrestin1; Arrestin beta1; Arrestin beta-1; ARR1 Protein family Arrestin family Biochemical class Arrestin protein Function During homologous desensitization, beta-arrestins bind to the GPRK-phosphorylated receptor and sterically preclude its coupling to the cognate G-protein; the binding appears to require additional receptor determinants exposed only in the active receptor conformation. The beta-arrestins target many receptors for internalization by acting as endocytic adapters (CLASPs, clathrin-associated sorting proteins) and recruiting the GPRCs to the adapter protein 2 complex 2 (AP-2) in clathrin-coated pits (CCPs). However, the extent of beta-arrestin involvement appears to vary significantly depending on the receptor, agonist and cell type. Internalized arrestin-receptor complexes traffic to intracellular endosomes, where they remain uncoupled from G-proteins. Two different modes of arrestin-mediated internalization occur. Class A receptors, like ADRB2, OPRM1, ENDRA, D1AR and ADRA1B dissociate from beta-arrestin at or near the plasma membrane and undergo rapid recycling. Class B receptors, like AVPR2, AGTR1, NTSR1, TRHR and TACR1 internalize as a complex with arrestin and traffic with it to endosomal vesicles, presumably as desensitized receptors, for extended periods of time. Receptor resensitization then requires that receptor-bound arrestin is removed so that the receptor can be dephosphorylated and returned to the plasma membrane. Involved in internalization of P2RY4 and UTP-stimulated internalization of P2RY2. Involved in phosphorylation-dependent internalization of OPRD1 ands subsequent recycling. Involved in the degradation of cAMP by recruiting cAMP phosphodiesterases to ligand-activated receptors. Beta-arrestins function as multivalent adapter proteins that can switch the GPCR from a G-protein signaling mode that transmits short-lived signals from the plasma membrane via small molecule second messengers and ion channels to a beta-arrestin signaling mode that transmits a distinct set of signals that are initiated as the receptor internalizes and transits the intracellular compartment. Acts as signaling scaffold for MAPK pathways such as MAPK1/3 (ERK1/2). ERK1/2 activated by the beta-arrestin scaffold is largely excluded from the nucleus and confined to cytoplasmic locations such as endocytic vesicles, also called beta-arrestin signalosomes. Recruits c-Src/SRC to ADRB2 resulting in ERK activation. GPCRs for which the beta-arrestin-mediated signaling relies on both ARRB1 and ARRB2 (codependent regulation) include ADRB2, F2RL1 and PTH1R. For some GPCRs the beta-arrestin-mediated signaling relies on either ARRB1 or ARRB2 and is inhibited by the other respective beta-arrestin form (reciprocal regulation). Inhibits ERK1/2 signaling in AGTR1- and AVPR2-mediated activation (reciprocal regulation). Is required for SP-stimulated endocytosis of NK1R and recruits c-Src/SRC to internalized NK1R resulting in ERK1/2 activation, which is required for the antiapoptotic effects of SP. Is involved in proteinase-activated F2RL1-mediated ERK activity. Acts as signaling scaffold for the AKT1 pathway. Is involved in alpha-thrombin-stimulated AKT1 signaling. Is involved in IGF1-stimulated AKT1 signaling leading to increased protection from apoptosis. Involved in activation of the p38 MAPK signaling pathway and in actin bundle formation. Involved in F2RL1-mediated cytoskeletal rearrangement and chemotaxis. Involved in AGTR1-mediated stress fiber formation by acting together with GNAQ to activate RHOA. Appears to function as signaling scaffold involved in regulation of MIP-1-beta-stimulated CCR5-dependent chemotaxis. Involved in attenuation of NF-kappa-B-dependent transcription in response to GPCR or cytokine stimulation by interacting with and stabilizing CHUK. May serve as nuclear messenger for GPCRs. Involved in OPRD1-stimulated transcriptional regulation by translocating to CDKN1B and FOS promoter regions and recruiting EP300 resulting in acetylation of histone H4. Involved in regulation of LEF1 transcriptional activity via interaction with DVL1 and/or DVL2 Also involved in regulation of receptors other than GPCRs. Involved in Toll-like receptor and IL-1 receptor signaling through the interaction with TRAF6 which prevents TRAF6 autoubiquitination and oligomerization required for activation of NF-kappa-B and JUN. Binds phosphoinositides. Binds inositolhexakisphosphate (InsP6). Involved in IL8-mediated granule release in neutrophils. Required for atypical chemokine receptor ACKR2-induced RAC1-LIMK1-PAK1-dependent phosphorylation of cofilin (CFL1) and for the up-regulation of ACKR2 from endosomal compartment to cell membrane, increasing its efficiency in chemokine uptake and degradation. Involved in the internalization of the atypical chemokine receptor ACKR3. Negatively regulates the NOTCH signaling pathway by mediating the ubiquitination and degradation of NOTCH1 by ITCH. Participates to the recruitment of the ubiquitin-protein ligase to the receptor. Functions in regulating agonist-mediated G-protein coupled receptor (GPCR) signaling by mediating both receptor desensitization and resensitization processes. Related diseases Intellectual developmental disorder with dysmorphic facies and ptosis (IDDDFP) [MIM:617333]: An autosomal dominant neurodevelopmental disorder characterized by delayed psychomotor development, intellectual disability, delayed language, and facial dysmorphisms, most notably ptosis. Additional features may include poor growth, hypotonia, and seizures. {ECO:0000269|PubMed:27939639, ECO:0000269|PubMed:27939640}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P63010-2; O15169; P0DP25; P20963; P25101; P50148; Q5JWF2; Q14749; P06396; Q16665; P11142; Q99683; P53779; P45984; Q00987; P19338; Q14978; P14618; P14859-6; P35813; O75688; Q13523; P06702; P12931; Q15208; Q13428; P04637; P27348; P25490; O43298; O95218; Q7DB77 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Cell projection; Coated pit; Cytoplasm; Cytoplasmic vesicle; Membrane; Nucleus; Phosphoprotein; Protein transport; Proteomics identification; Reference proteome; Signal transduction inhibitor; Transcription; Transcription regulation; Transport; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 32455.6 Length 293 Aromaticity 0.12 Instability index 28.99 Isoelectric point 9.12 Charge (pH=7) 9.86 2D Binding mode Binding energy (Kcal/mol) -8.61  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence AGCSLLMALVVLLIVAGNVLVIAAIGRTQRLQTLTNLFITSLACADLVVGLLVVPFGATLVCRGTWLWGSFLCELWTSLDVLCVTASIWTLCVIAIDRYLAITSPFRYQSLMTRARAKVIICTVWAISALVSFLPIMMHWWRDEDPQALKCYQDPGCCDFVTNRAYAIASSIISFYIPLLIMIFVYLRVYREAKEQIRKIDVMAMREHKALKTLGIIMGVFTLCWLPFFLVNIVNVFNRDLVPKWLFVAFNWLGYANSAMNPIIYCRSPDFRKAFKRLLAEXAXXAXXXLAKD Hydrogen bonds contact Hydrophobic contact | ||||
| 94 | BMP-2-inducible protein kinase (BMP2K) | 4W9X | 6.31 | |
Target general information Gen name BMP2K Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms HRIHFB2017; BIKe Protein family Protein kinase superfamily, Ser/Thr protein kinase family Biochemical class Kinase Function May be involved in osteoblast differentiation. Related diseases Glioma (GLM) [MIM:137800]: Gliomas are benign or malignant central nervous system neoplasms derived from glial cells. They comprise astrocytomas and glioblastoma multiforme that are derived from astrocytes, oligodendrogliomas derived from oligodendrocytes and ependymomas derived from ependymocytes. {ECO:0000269|PubMed:22286061, ECO:0000269|PubMed:22286216, ECO:0000269|PubMed:23539269}. The gene represented in this entry is involved in disease pathogenesis. H3F3A mutations affecting residues involved in post-translational modifications of histone H3.3 are recurrent in malignant, aggressive gliomas including glioblastoma multiforme (GBM) and diffuse intrinsic pontine glioma (DIPG) (PubMed:22286061, PubMed:22286216). The mechanism through which mutations lead to tumorigenesis involves altered histones methylation, impaired regulation of Polycomb repressive complex 2 (PRC2) activity, and aberrant epigenetic regulation of gene expression (PubMed:23539183, PubMed:23539269, PubMed:23603901). {ECO:0000269|PubMed:22286061, ECO:0000269|PubMed:22286216, ECO:0000269|PubMed:23539183, ECO:0000269|PubMed:23539269, ECO:0000269|PubMed:23603901}.; DISEASE: Bryant-Li-Bhoj neurodevelopmental syndrome 1 (BRYLIB1) [MIM:619720]: An autosomal dominant disorder predominantly characterized by global developmental delay, impaired intellectual development, poor or absent speech, and delayed motor milestones. Clinical manifestations are highly variable, including abnormal head shape, dysmorphic facial features, oculomotor abnormalities, feeding problems, and non-specific brain imaging abnormalities. Additional features may include hearing loss, seizures, short stature, and mild skeletal defects. {ECO:0000269|PubMed:33268356, ECO:0000269|PubMed:34876591}. The disease is caused by variants affecting the gene represented in this entry. BRYLIB1 is caused by variants in H3-3A. {ECO:0000269|PubMed:33268356, ECO:0000269|PubMed:34876591}.; DISEASE: Bryant-Li-Bhoj neurodevelopmental syndrome 2 (BRYLIB2) [MIM:619721]: An autosomal dominant disorder predominantly characterized by global developmental delay, impaired intellectual development, poor or absent speech, and delayed motor milestones. Clinical manifestations are highly variable, including abnormal head shape, dysmorphic facial features, oculomotor abnormalities, feeding problems, and non-specific brain imaging abnormalities. Additional features may include hearing loss, seizures, short stature, and mild skeletal defects. {ECO:0000269|PubMed:33268356, ECO:0000269|PubMed:34876591}. The disease is caused by variants affecting the gene represented in this entry. BRYLIB2 is caused by variants in H3-3B. {ECO:0000269|PubMed:33268356, ECO:0000269|PubMed:34876591}.; DISEASE: H3F3A and H3F3B mutations affecting residues involved in post-translational modifications of histone H3.3 are implicated in the pathogenesis of some bone and cartilage neoplasms. Mutations have been found with high prevalence in chondroblastoma and giant cell tumors of bone, and with low frequency in osteosarcoma, conventional chondrosarcoma and clear cell chondrosarcoma. Chondroblastoma samples frequently carry a H3F3B mutation affecting residue Lys-37 (H3K36), although H3F3A is mutated in some cases. Most giant cell tumors of bone harbor H3F3A mutations affecting residue Gly-35 (H3G34). {ECO:0000269|PubMed:24162739}. Drugs (DrugBank ID) DB12010 Interacts with Q14677; O95630 EC number EC 2.7.11.1 Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Kinase; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Serine/threonine-protein kinase; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 33719.6 Length 300 Aromaticity 0.09 Instability index 38.09 Isoelectric point 6.42 Charge (pH=7) -1.74 2D Binding mode Binding energy (Kcal/mol) -8.61  Molscript Map 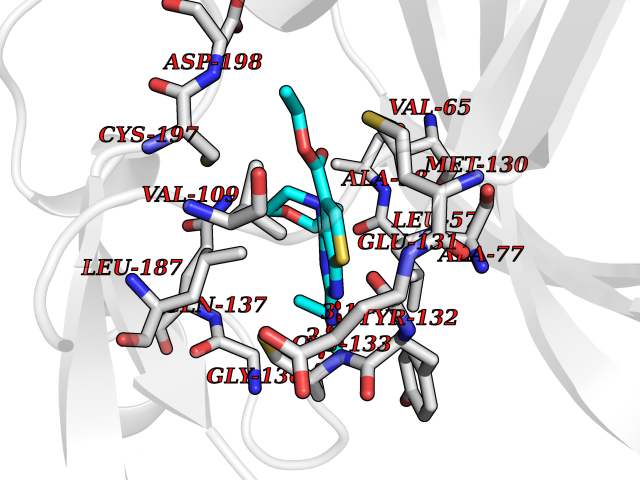 Pymol Map 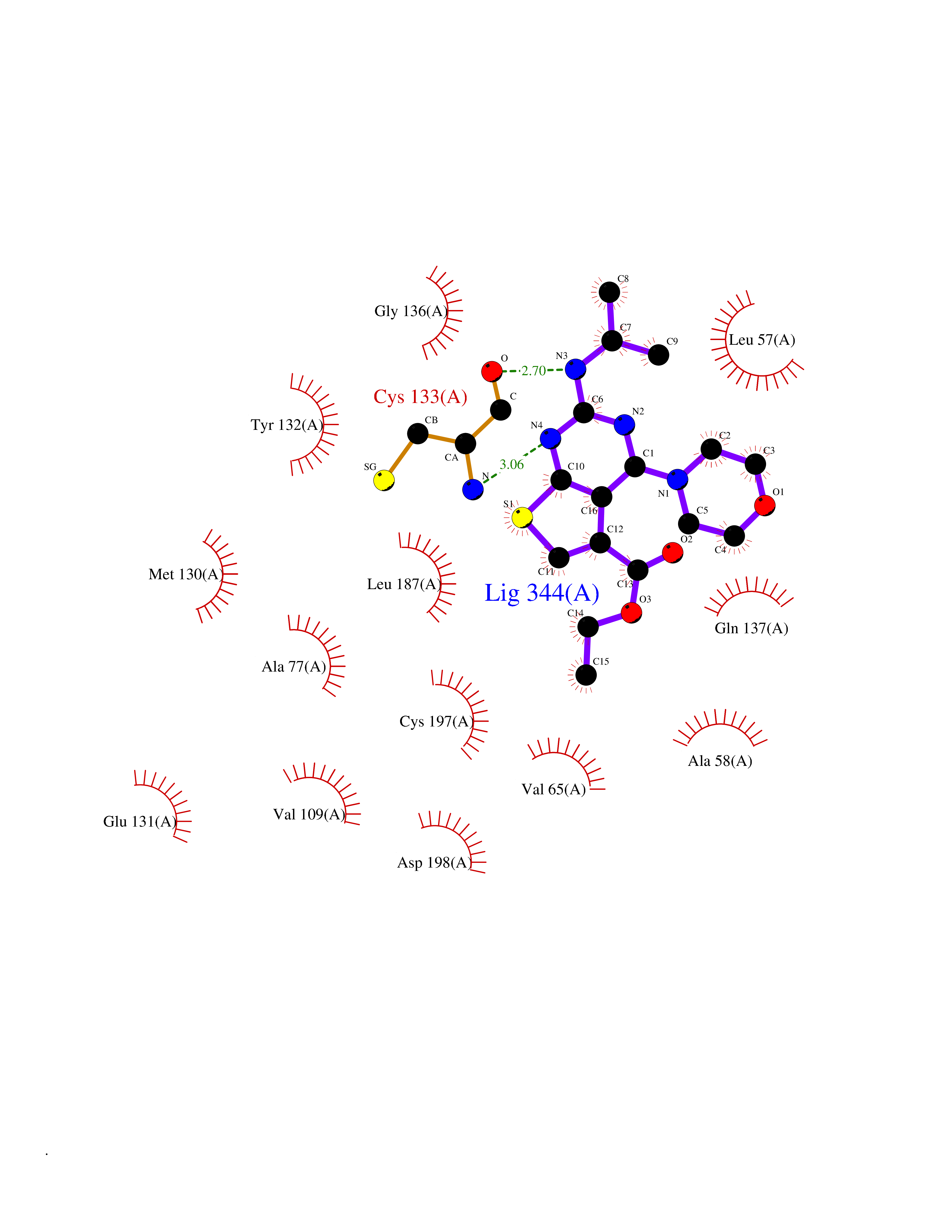 Ligplot Map 3D Binding mode Sequence VGVRVFAVGRHQVTLEESLAEGGFSTVFLVRTHGGIRCALKRMYVNNMPDLNVCKREITIMKELSGHKNIVGYLDCAVNSISWEVLILMEYCRAGQVVNQMNKKQTGFTEPEVLQIFCDTCEAVARLHQCKTPIIHRDLKVENILLNDGGNYVLCDFGSATNKFLNPQKDGVNVVEEEIKKYTTLSYRAPEMINLYGGKPITTKADIWALGCLLYKLCFFTLPFGESQVAICDGNFTIPDNSRYSRNIHCLIRFMLEPDPEHRPDIFQVSYFAFKFAAADCPVSNINNSSIPSALPEPMT Hydrogen bonds contact Hydrophobic contact | ||||
| 95 | L-aspartate oxidase | 1KNR | 6.30 | |
Target general information Gen name nadB Organism Escherichia coli (strain K12) Uniprot ID TTD ID NA Synonyms JW2558;nicB;b2574 Protein family FAD-dependent oxidoreductase 2 family, NadB subfamily Biochemical class Oxidoreductase Function Flavin adenine dinucleotide binding.L-aspartate:fumarate oxidoreductase activity.L-aspartate oxidase activity. Related diseases Thyroid hormone resistance, generalized, autosomal dominant (GRTHD) [MIM:188570]: An autosomal dominant disease characterized by high levels of circulating thyroid hormones (T3-T4), goiter, abnormal mental functions, increased susceptibility to infections, abnormal growth and bone maturation, tachycardia and deafness. Affected individuals may also have attention deficit-hyperactivity disorders (ADHD) and language difficulties. Patients have normal or slightly elevated thyroid stimulating hormone (TSH). {ECO:0000269|PubMed:10660344, ECO:0000269|PubMed:12511610, ECO:0000269|PubMed:12554782, ECO:0000269|PubMed:1314846, ECO:0000269|PubMed:1324420, ECO:0000269|PubMed:1563081, ECO:0000269|PubMed:1587388, ECO:0000269|PubMed:1619012, ECO:0000269|PubMed:1661299, ECO:0000269|PubMed:16804041, ECO:0000269|PubMed:1846005, ECO:0000269|PubMed:19268523, ECO:0000269|PubMed:2153155, ECO:0000269|PubMed:2510172, ECO:0000269|PubMed:7833659, ECO:0000269|PubMed:8175986, ECO:0000269|PubMed:8514853, ECO:0000269|PubMed:8664910, ECO:0000269|PubMed:8889584}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Thyroid hormone resistance, generalized, autosomal recessive (GRTHR) [MIM:274300]: An autosomal recessive disorder characterized by goiter, clinical euthyroidism, end-organ unresponsiveness to thyroid hormone, abnormal growth and bone maturation, and deafness. Patients also have high levels of circulating thyroid hormones, with elevated thyroid stimulating hormone. {ECO:0000269|PubMed:1653889}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Selective pituitary thyroid hormone resistance (PRTH) [MIM:145650]: Variant form of thyroid hormone resistance and is characterized by clinical hyperthyroidism, with elevated free thyroid hormones, but inappropriately normal serum TSH. Unlike GRTH, where the syndrome usually segregates with a dominant allele, the mode of inheritance in PRTH has not been established. {ECO:0000269|PubMed:7528740, ECO:0000269|PubMed:8381821}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03147 Interacts with NA EC number 1.4.3.16; 1.5.99.- Uniprot keywords 3D-structure; Cytoplasm; Direct protein sequencing; FAD; Flavoprotein; Nucleotide-binding; Oxidoreductase; Pyridine nucleotide biosynthesis; Reference proteome Protein physicochemical properties Chain ID A Molecular weight (Da) 58993.2 Length 529 Aromaticity 0.07 Instability index 36.96 Isoelectric point 5.76 Charge (pH=7) -15.84 2D Binding mode Binding energy (Kcal/mol) -8.59 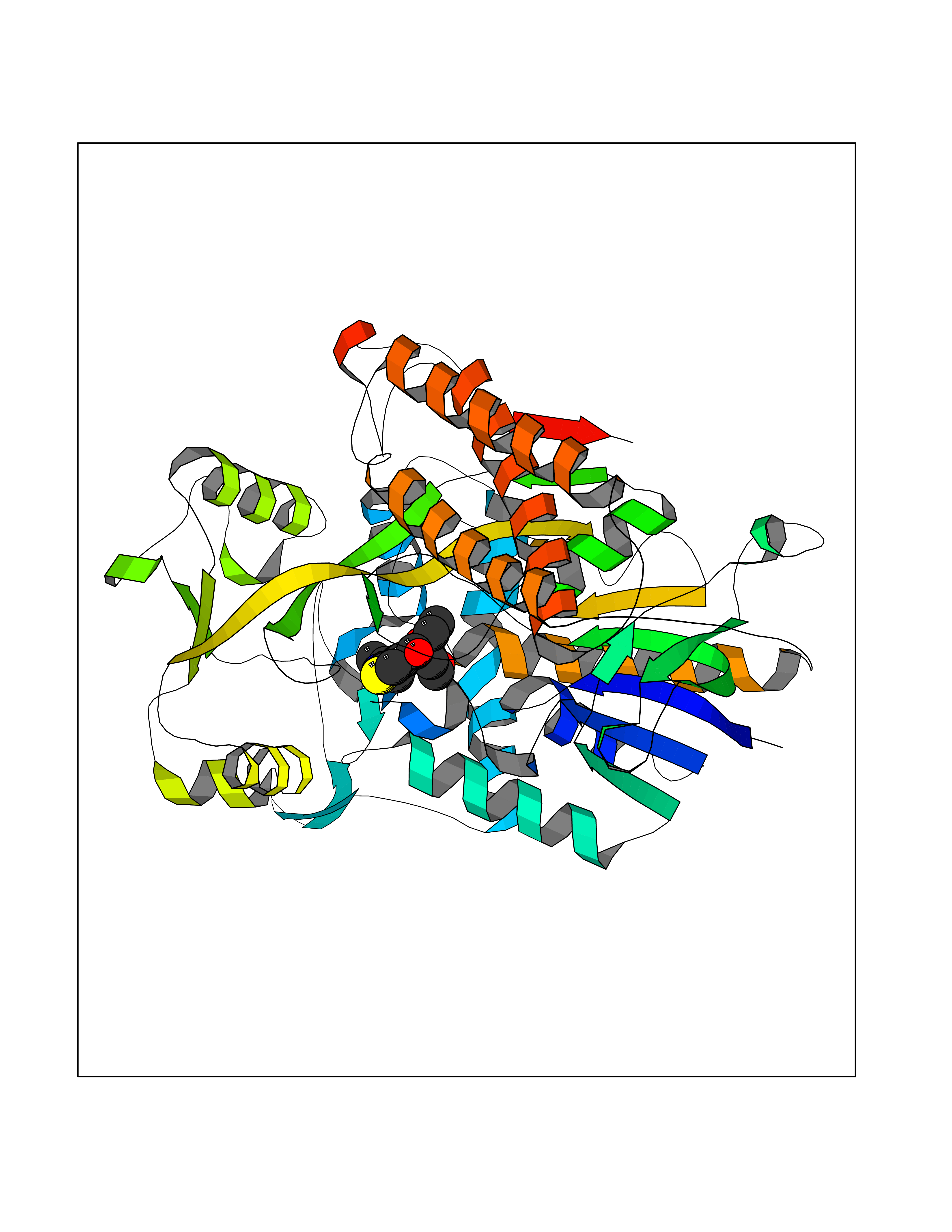 Molscript Map 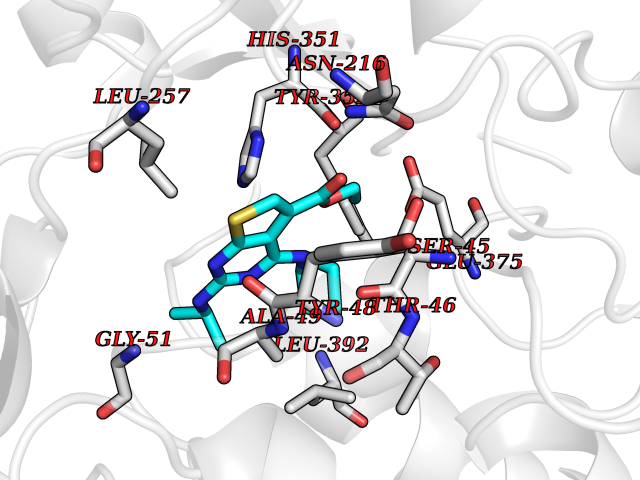 Pymol Map 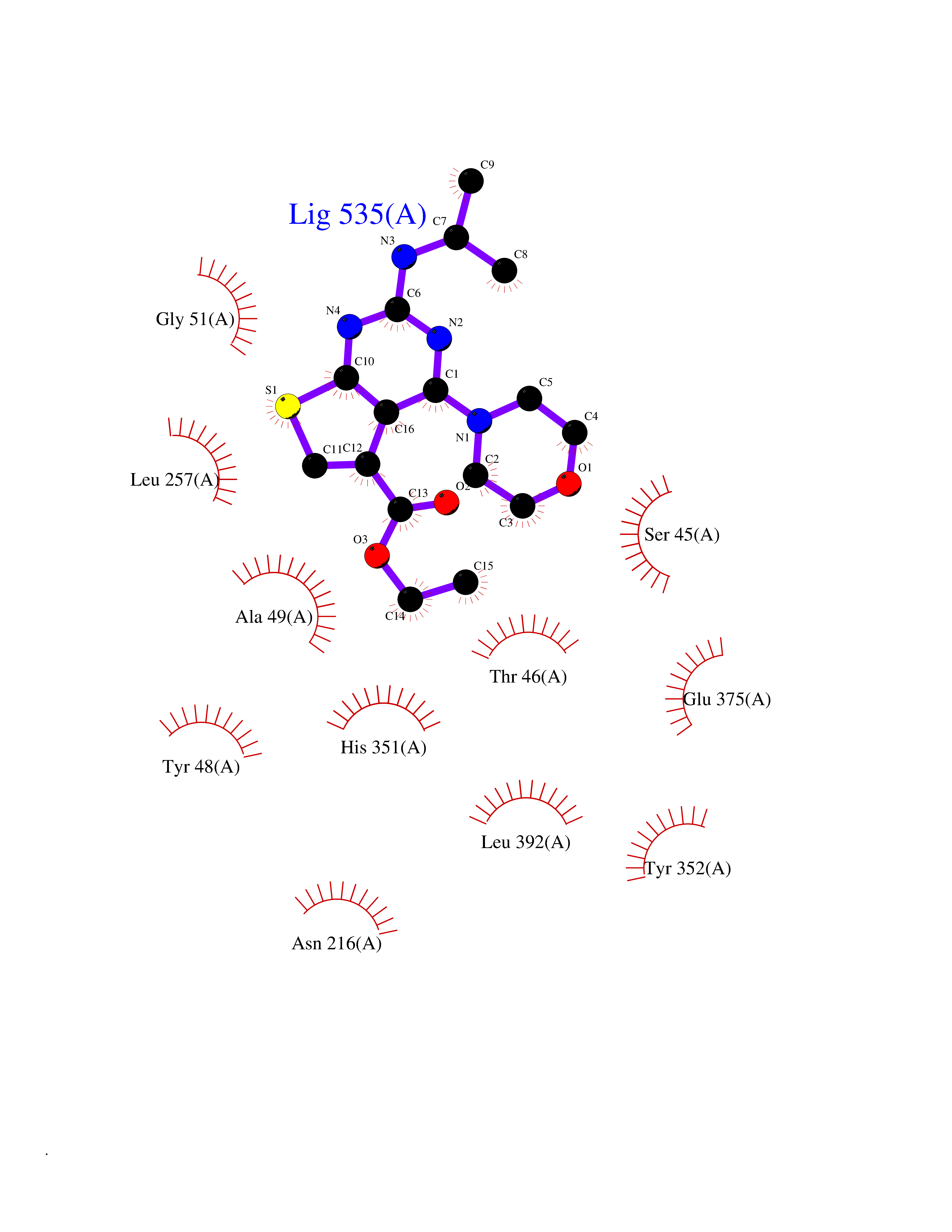 Ligplot Map 3D Binding mode Sequence PEHSCDVLIIGSGAAGLSLALRLADQHQVIVLSKGPVTEGSTFYAQGGIAAVFDETDSIDSHVEDTLIAGAGICDRHAVEFVASNARSCVQWLIDQGVLFDTHIQPNGEESYHLTREGGHSHRRILHAADATGREVETTLVSKALNHPNIRVLERTNAVDLIVSDKIGLPGTRRVVGAWVWNRNKETVETCHAKAVVLATGGASKVYQYTTNPDISSGDGIAMAWRAGCRVANLEFNQFHPTALYHPQARNFLLTEALRGEGAYLKRPDGTRFMPDFDERGELAPRDIVARAIDHEMKRLGADCMFLDISHKPADFIRQHFPMIYEKLLGLGIDLTQEPVPIVPAAHYTCGGVMVDDHGRTDVEGLYAIGEVSYTGLHGANLMASNSLLECLVYGWSAAEDITRRMPYAHDISTLPPWDESRVENPDERVVIQHNWHELRLFMWDYVGIVRTTKRLERALRRITMLQQEIDEYYAHFRVSNNLLELRNLVQVAELIVRCAMMRKESRGLHFTLDYPELLTHSGPSILSP Hydrogen bonds contact Hydrophobic contact | ||||
| 96 | Dihydroorotate dehydrogenase (quinone), mitochondrial | 4CQ8 | 6.30 | |
Target general information Gen name PFF0160c Organism Plasmodium falciparum (isolate 3D7) Uniprot ID TTD ID NA Synonyms NA Protein family Dihydroorotate dehydrogenase family, Type 2 subfamily Biochemical class Oxidoreductase Function Dihydroorotate dehydrogenase activity. Related diseases Combined oxidative phosphorylation deficiency 33 (COXPD33) [MIM:617713]: An autosomal recessive disorder caused by multiple mitochondrial respiratory chain defects and impaired mitochondrial energy metabolism. Clinical manifestations are highly variable. Affected infants present with cardiomyopathy accompanied by multisystemic features involving liver, kidney, and brain. Death in infancy is observed in some patients. Children and adults present with myopathy and progressive external ophthalmoplegia. {ECO:0000269|PubMed:28942965}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01117 Interacts with NA EC number 1.3.5.2 Uniprot keywords 3D-structure; Flavoprotein; FMN; Membrane; Mitochondrion; Mitochondrion inner membrane; Oxidoreductase; Pyrimidine biosynthesis; Reference proteome; Transit peptide; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A,B Molecular weight (Da) 42573.5 Length 378 Aromaticity 0.1 Instability index 36.63 Isoelectric point 8.21 Charge (pH=7) 3.17 2D Binding mode Binding energy (Kcal/mol) -8.6 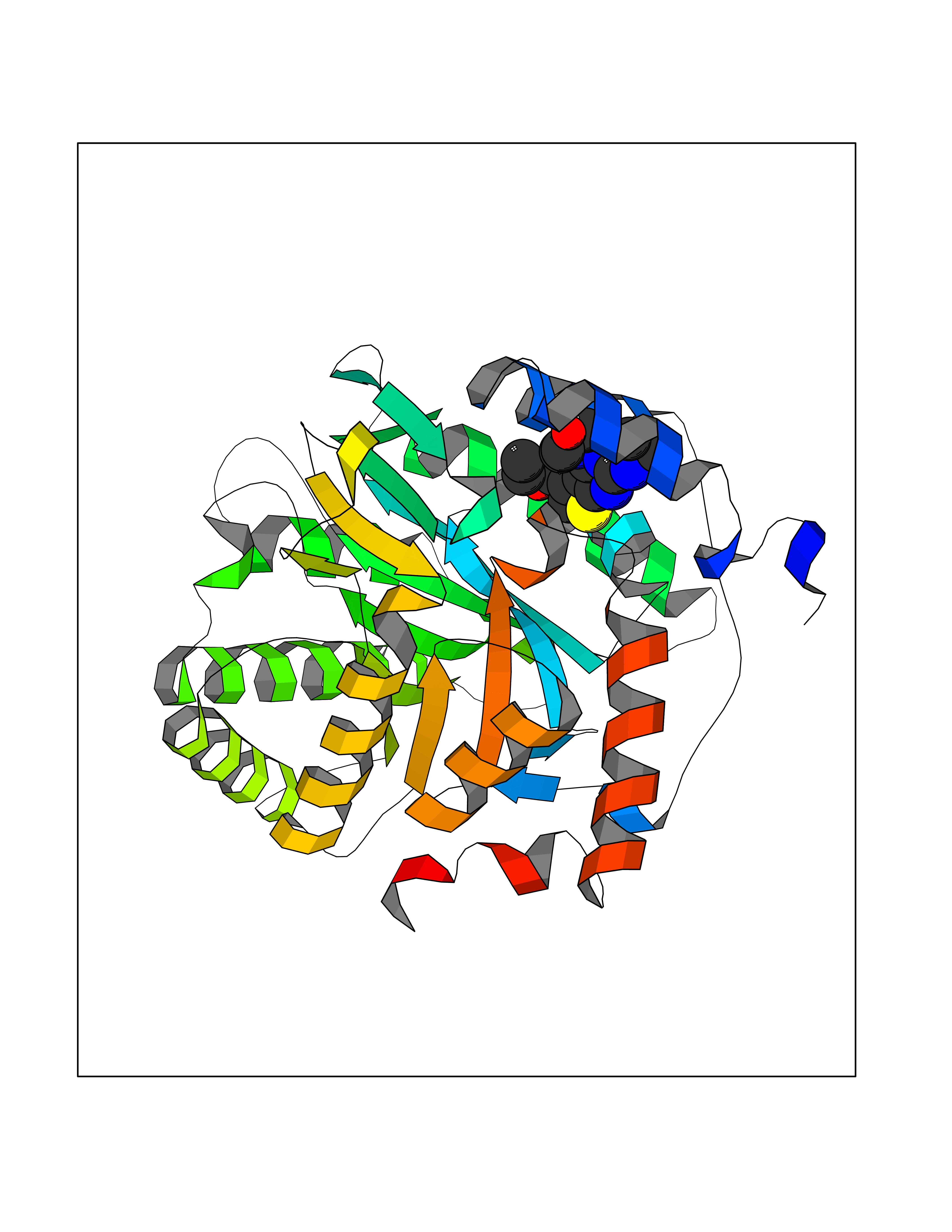 Molscript Map 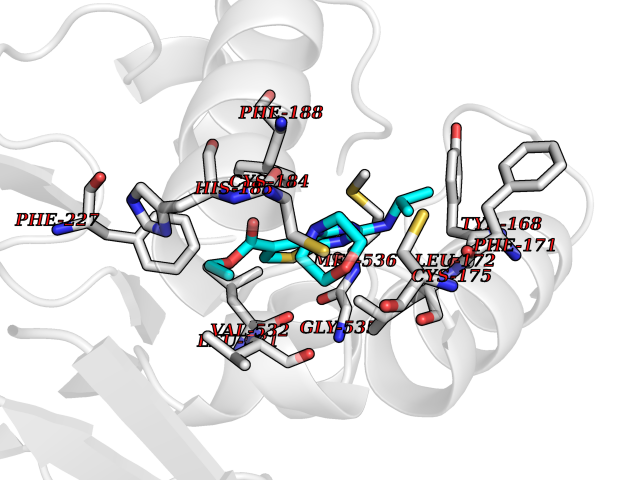 Pymol Map 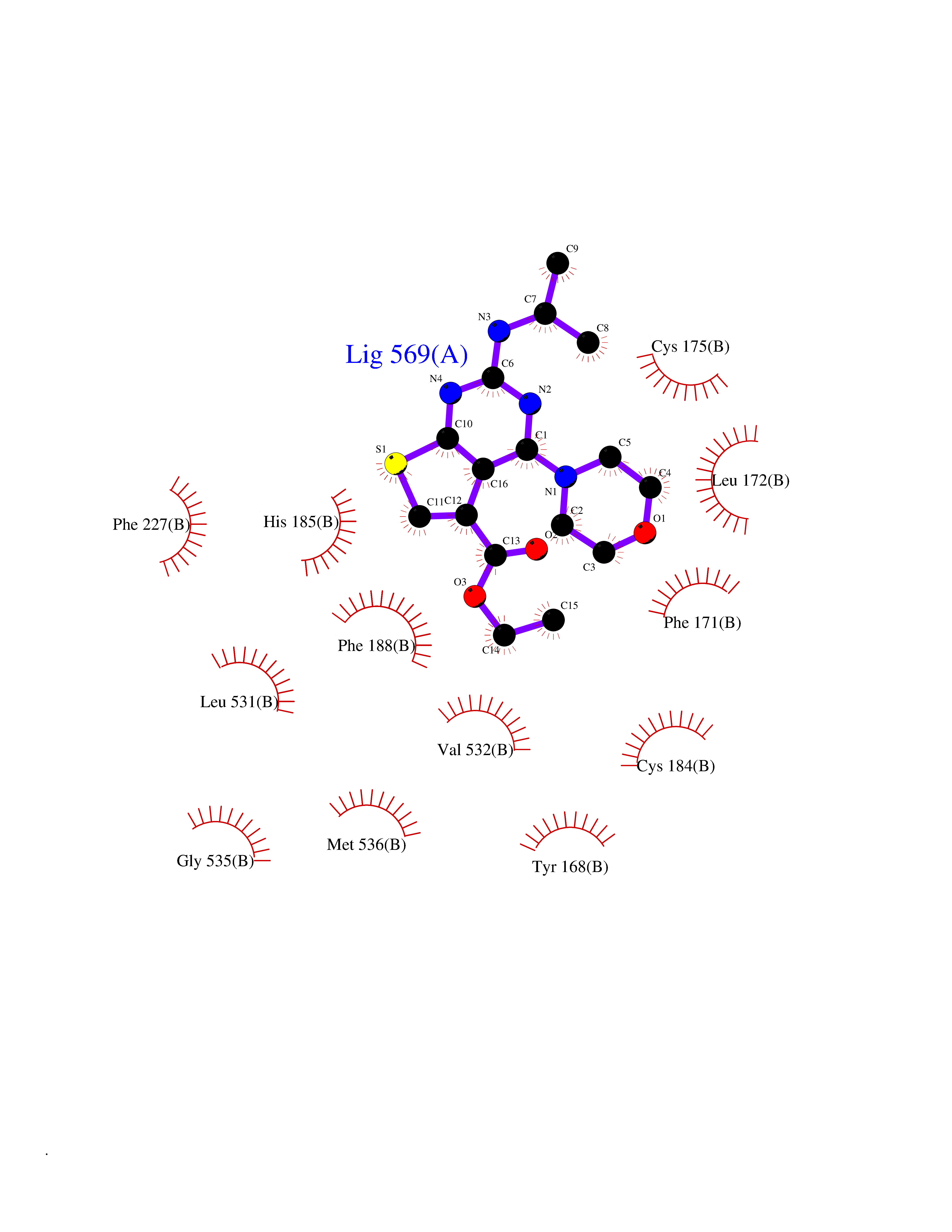 Ligplot Map 3D Binding mode Sequence ADPFESYNPEFFLYDIFLKFCLKYIDGEICHDLFLLLGKYNILPYDTSNDSIYACTNIKHLDFINPFGVAAGFDKNGVCIDSILKLGFSFIEIGTITPRGQTGNAKPRIFRDVESRSIINSCGFNNMGCDKVTENLILFRKRQEEDKLLSKHIVGVSIGKNKDTVNIVDDLKYCINKIGRYADYIAINVSSPNTPGLRDNQEAGKLKNIILSVKEEIDNLEKNNFLWFNTTKKKPLVFVKLAPDLNQEQKKEIADVLLETNIDGMIISNTTTQINDIKSFENKKGGVSGAKLKDISTKFICEMYNYTNKQIPIIASGGIFSGLDALEKIEAGASVCQLYSCLVFNGMKSAVQIKRELNHLLYQRGYYNLKEAIGRKHS Hydrogen bonds contact Hydrophobic contact | ||||
| 97 | ADP-ribosyl cyclase/cyclic ADP-ribose hydrolase 2 | 1ISI | 6.30 | |
Target general information Gen name BST1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family ADP-ribosyl cyclase family Biochemical class Hydrolase Function ADP-ribosyl cyclase activity.Cyclic ADP-ribose hydrolase.NAD(P)+ nucleosidase activity.NAD+ nucleosidase activity.NAD+ nucleotidase, cyclic ADP-ribose generating.Phosphorus-oxygen lyase activity.Transferase activity. Related diseases Lymphoproliferative syndrome 1 (LPFS1) [MIM:613011]: A rare immunodeficiency characterized by extreme susceptibility to infection with Epstein-Barr virus (EBV). Inadequate immune response to EBV can have a fatal outcome. Clinical features include splenomegaly, lymphadenopathy, anemia, thrombocytopenia, pancytopenia, recurrent infections. There is an increased risk for lymphoma. {ECO:0000269|PubMed:19425169}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02930; DB02483; DB03732; DB02701; DB03227 Interacts with NA EC number 3.2.2.6 Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Direct protein sequencing; Disulfide bond; Glycoprotein; GPI-anchor; Hydrolase; Lipoprotein; Membrane; NAD; Proteomics identification; Reference proteome; Signal; Transferase Protein physicochemical properties Chain ID A,B Molecular weight (Da) 57100.1 Length 500 Aromaticity 0.1 Instability index 39.83 Isoelectric point 5.83 Charge (pH=7) -9.14 2D Binding mode Binding energy (Kcal/mol) -8.59 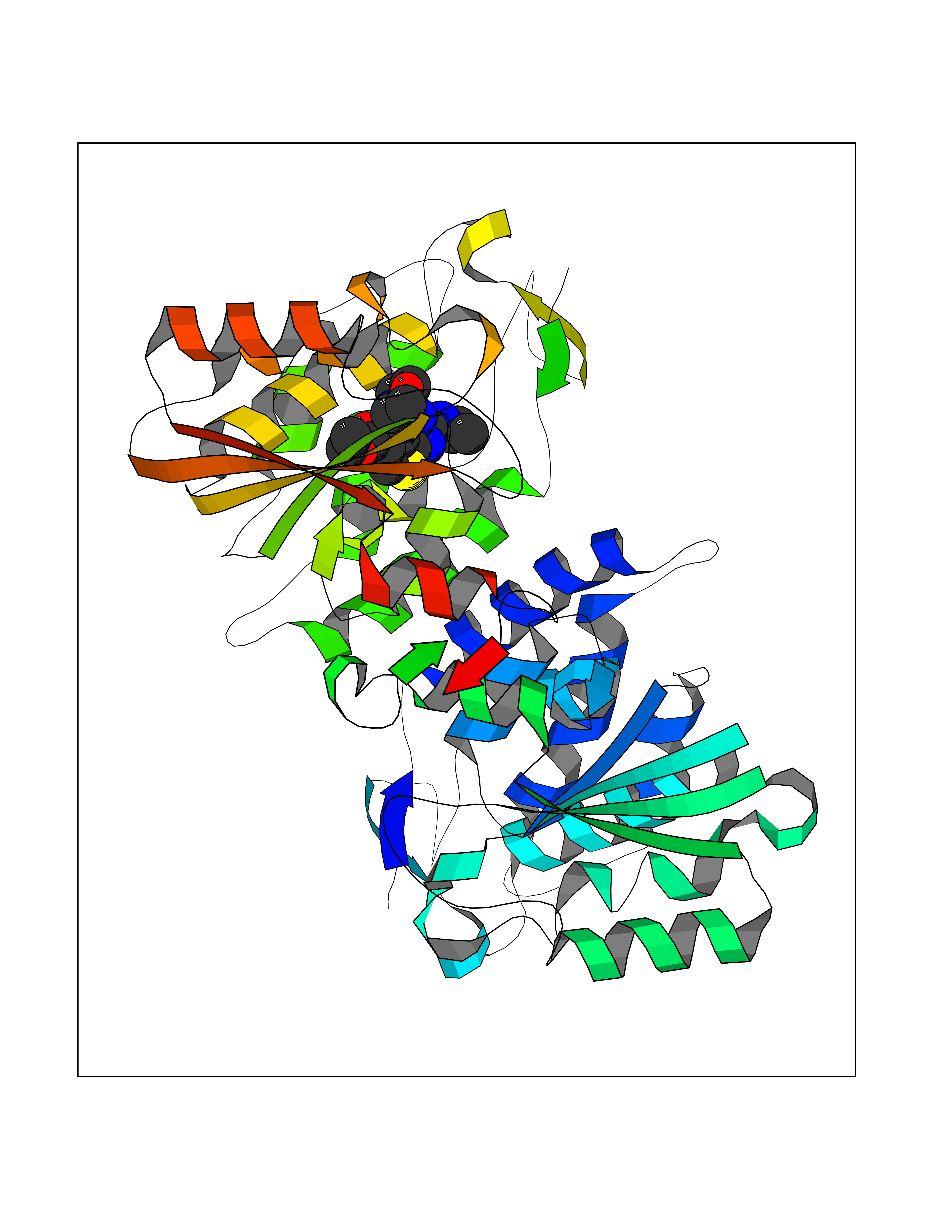 Molscript Map 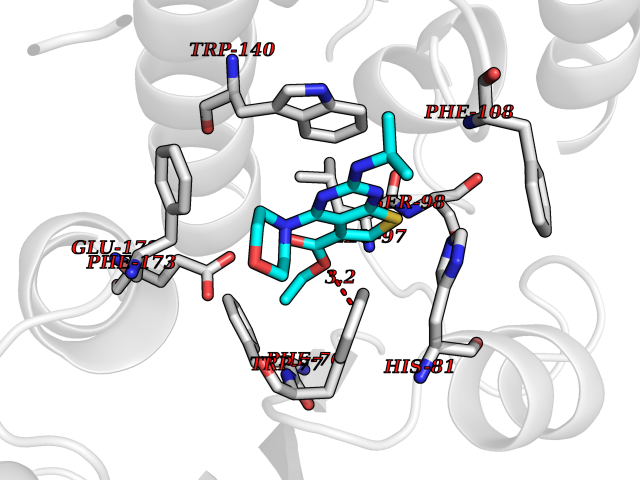 Pymol Map 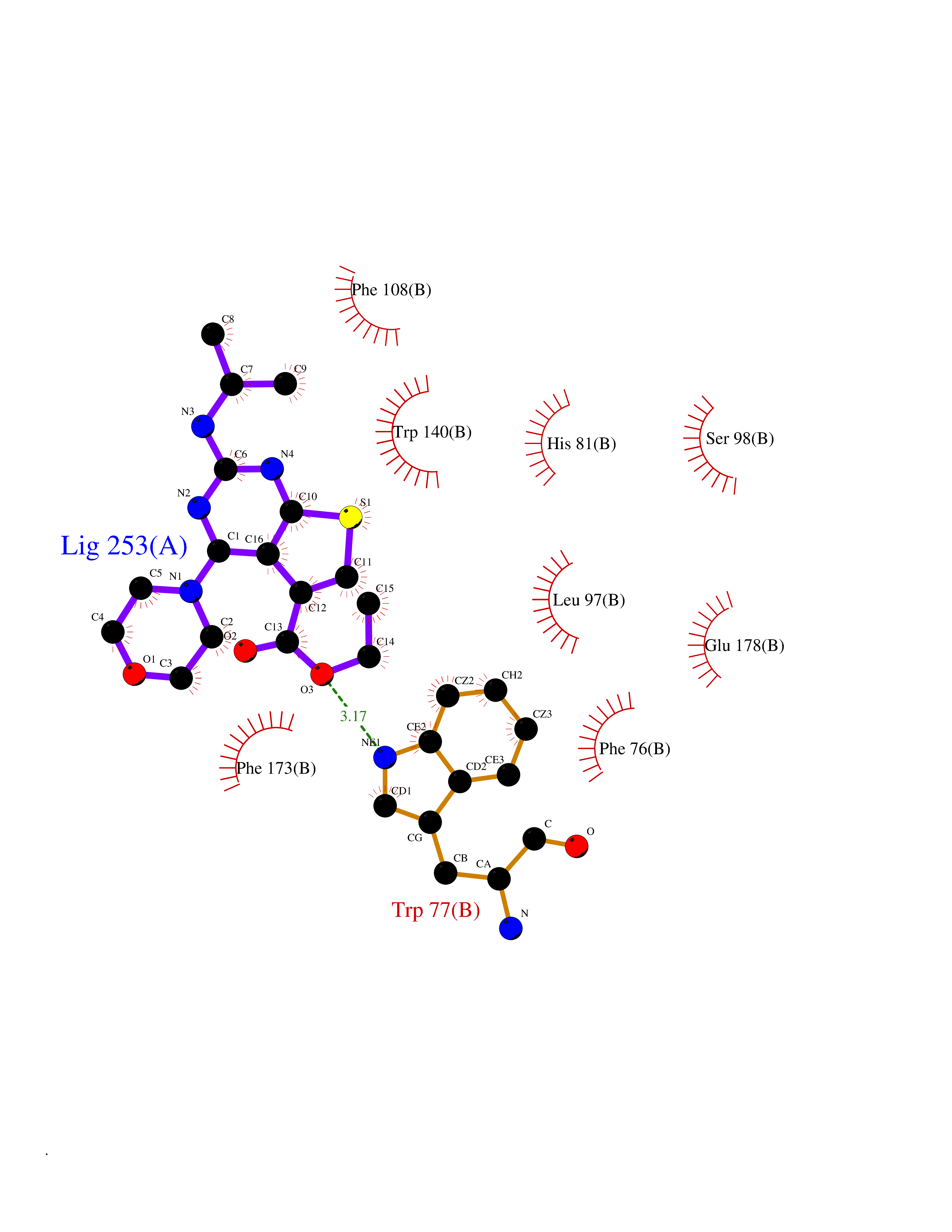 Ligplot Map 3D Binding mode Sequence WRAEGTSAHLRDIFLGRCAEYRALLSPEQRNKDCTAIWEAFKVALDKDPCSVLPSDYDLFITLSRHSIPRDKSLFWENSHLLVNSFADNTRRFMPLSDVLYGRVADFLSWCRQKADSGLDYQSCPTSEDCENNPVDSFWKRASIQYSKDSSGVIHVMLNGSEPTGAYPIKGFFADYEIPNLQKEKITRIEIWVMHEIGGPNVESCGEGSMKVLEKRLKDMGFQYSCINDYRPVKLLQCVDHSTHPDCALKWRAEGTSAHLRDIFLGRCAEYRALLSPEQRNKDCTAIWEAFKVALDKDPCSVLPSDYDLFITLSRHSIPRDKSLFWENSHLLVNSFADNTRRFMPLSDVLYGRVADFLSWCRQKADSGLDYQSCPTSEDCENNPVDSFWKRASIQYSKDSSGVIHVMLNGSEPTGAYPIKGFFADYEIPNLQKEKITRIEIWVMHEIGGPNVESCGEGSMKVLEKRLKDMGFQYSCINDYRPVKLLQCVDHSTHPDCALK Hydrogen bonds contact Hydrophobic contact | ||||
| 98 | Opioid receptor sigma 1 (OPRS1) | 6DJZ | 6.30 | |
Target general information Gen name SIGMAR1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms hSigmaR1; Sigma1R; Sigma1-receptor; Sigma non-opioid intracellular receptor 1; Sigma 1-type opioid receptor; SRBP; SR31747-binding protein; SR31747 binding protein 1; SR-BP; SIG-1R; Opioid receptor, s Protein family ERG2 family Biochemical class GPCR rhodopsin Function Involved in the regulation of different receptors it plays a role in BDNF signaling and EGF signaling. Also regulates ion channels like the potassium channel and could modulate neurotransmitter release. Plays a role in calcium signaling through modulation together with ANK2 of the ITP3R-dependent calcium efflux at the endoplasmic reticulum. Plays a role in several other cell functions including proliferation, survival and death. Originally identified for its ability to bind various psychoactive drugs it is involved in learning processes, memory and mood alteration. Necessary for proper mitochondrial axonal transport in motor neurons, in particular the retrograde movement of mitochondria. Plays a role in protecting cells against oxidative stress-induced cell death via its interaction with RNF112. Functions in lipid transport from the endoplasmic reticulum and is involved in a wide array of cellular functions probably through regulation of the biogenesis of lipid microdomains at the plasma membrane. Related diseases Amyotrophic lateral sclerosis 16, juvenile (ALS16) [MIM:614373]: A neurodegenerative disorder affecting upper motor neurons in the brain and lower motor neurons in the brain stem and spinal cord, resulting in fatal paralysis. Sensory abnormalities are absent. The pathologic hallmarks of the disease include pallor of the corticospinal tract due to loss of motor neurons, presence of ubiquitin-positive inclusions within surviving motor neurons, and deposition of pathologic aggregates. The etiology of amyotrophic lateral sclerosis is likely to be multifactorial, involving both genetic and environmental factors. The disease is inherited in 5-10% of the cases. {ECO:0000269|PubMed:21842496}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Neuronopathy, distal hereditary motor, autosomal recessive 2 (HMNR2) [MIM:605726]: A form of distal hereditary motor neuronopathy, a heterogeneous group of neuromuscular disorders caused by selective degeneration of motor neurons in the anterior horn of the spinal cord, without sensory deficit in the posterior horn. The overall clinical picture consists of a classical distal muscular atrophy syndrome in the legs without clinical sensory loss. The disease starts with weakness and wasting of distal muscles of the anterior tibial and peroneal compartments of the legs. Later on, weakness and atrophy may expand to the proximal muscles of the lower limbs and/or to the distal upper limbs. HMNR2 is characterized by onset of distal muscle weakness and wasting affecting the lower and upper limbs in the first decade. {ECO:0000269|PubMed:26078401, ECO:0000269|PubMed:27629094}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00321; DB09014; DB00907; DB00514; DB01488; DB00574; DB00502; DB00956; DB00704; DB00540; DB06174; DB00652; DB11186; DB03575; DB05316; DB01708; DB00409; DB01104 Interacts with Q92847-1; Q99720-1; O00213-2; P17612; P50454; P37173 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Amyotrophic lateral sclerosis; Cell junction; Cell membrane; Cell projection; Cytoplasmic vesicle; Direct protein sequencing; Disease variant; Endoplasmic reticulum; Lipid droplet; Lipid transport; Membrane; Neurodegeneration; Neuropathy; Nucleus; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Synapse; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A Molecular weight (Da) 23901 Length 212 Aromaticity 0.14 Instability index 33.12 Isoelectric point 5.61 Charge (pH=7) -5.6 2D Binding mode Binding energy (Kcal/mol) -8.59 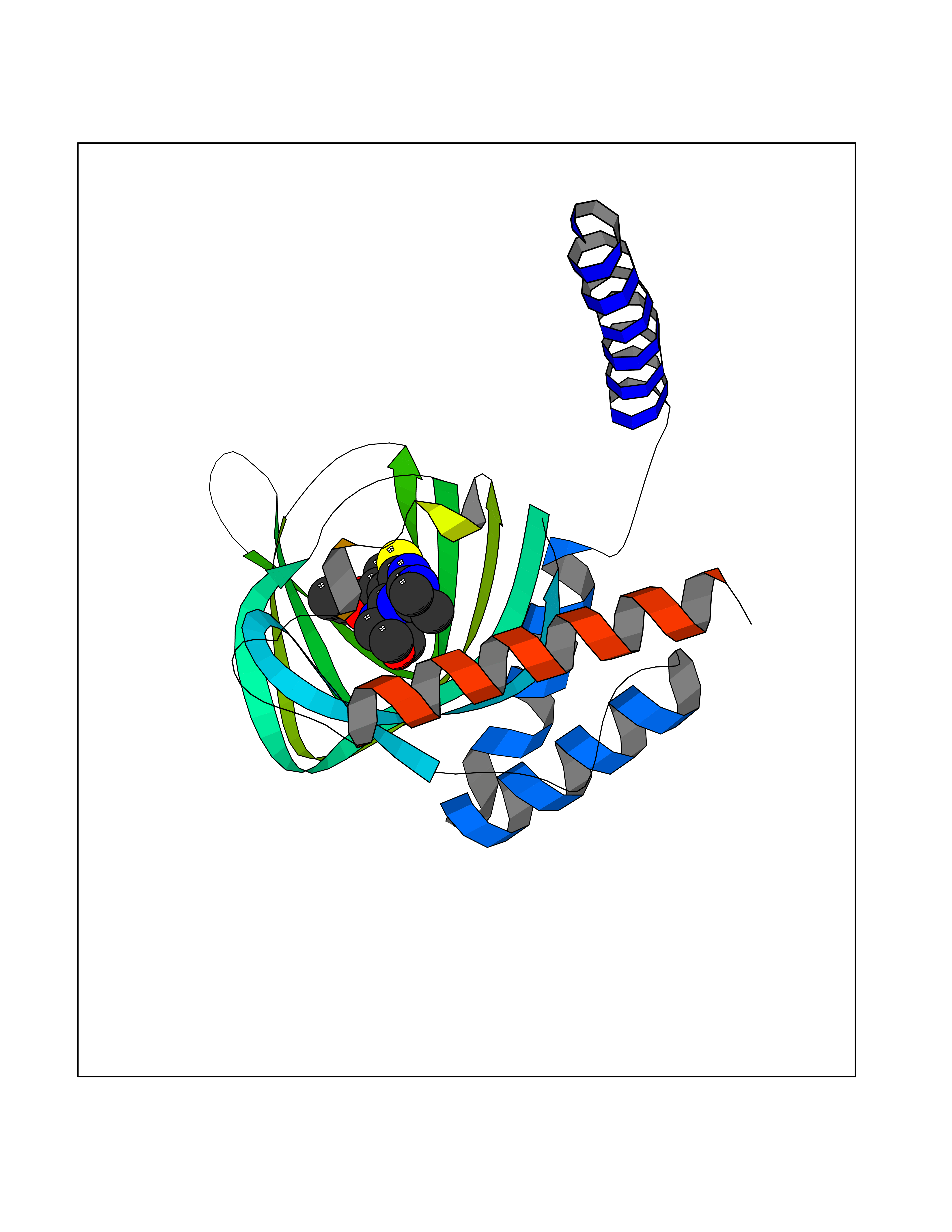 Molscript Map 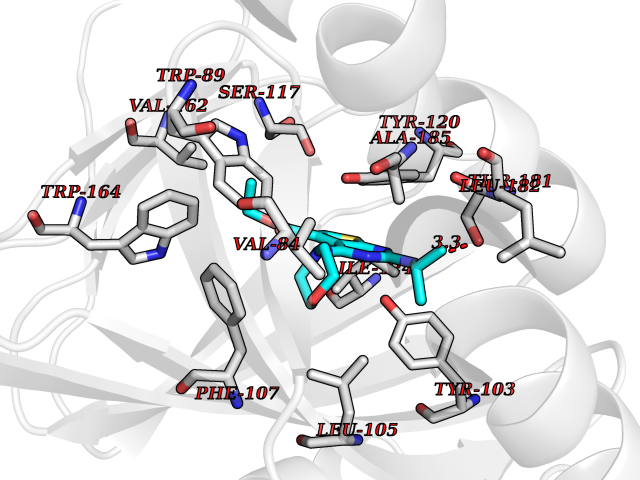 Pymol Map 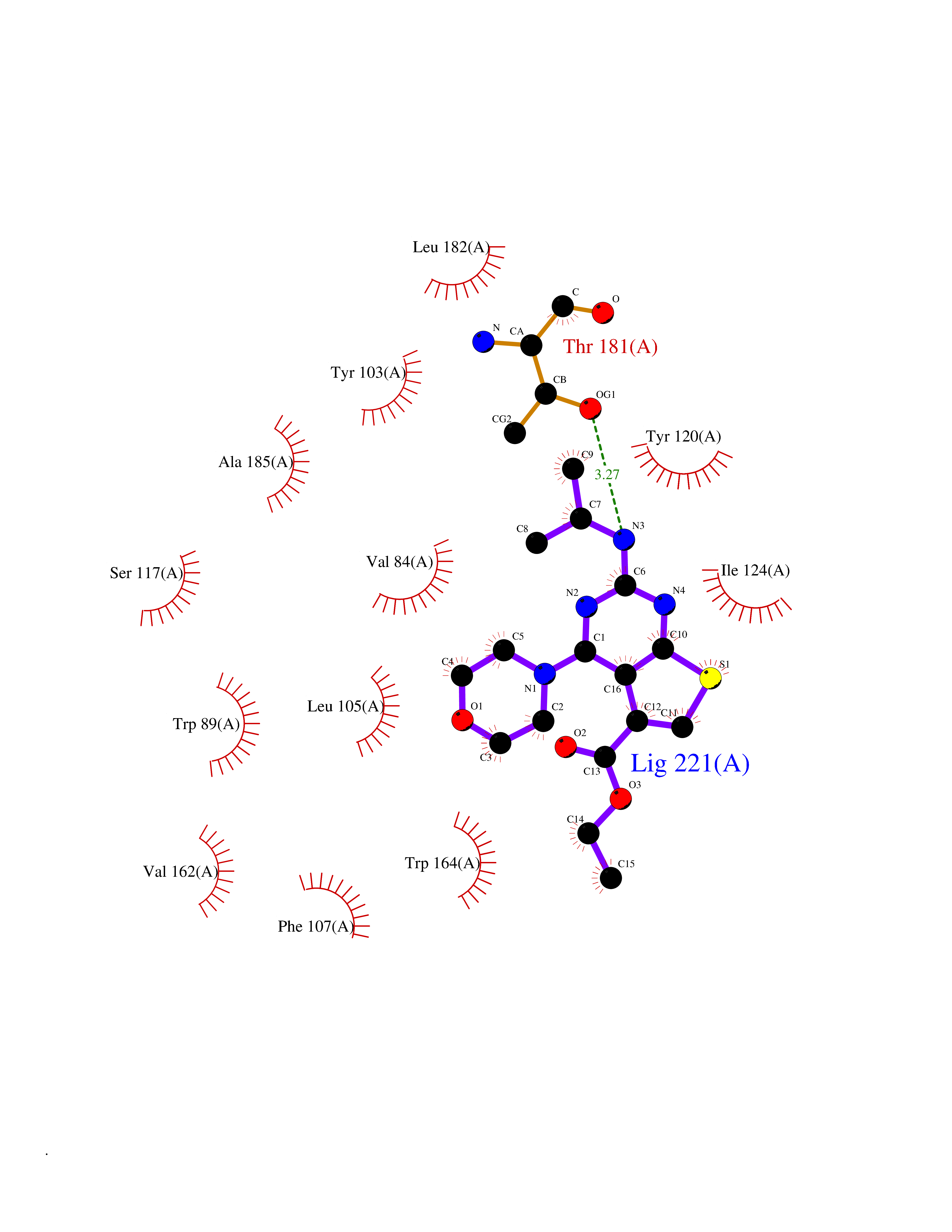 Ligplot Map 3D Binding mode Sequence RWAWAALLLAVAAVLTQVVWLWLGTQSFVFQREEIAQLARQYAGLDHELAFSRLIVELRRLHPGHVLPDEELQWVFVNAGGWMGAMCLLHASLSEYVLLFGTALGSRGHSGRYWAEISDTIISGTFHQWREGTTKSEVFYPGETVVHGPGEATAVEWGPNTWMVEYGRGVIPSTLAFALADTVFSTQDFLTLFYTLRSYARGLRLELTTYLF Hydrogen bonds contact Hydrophobic contact | ||||
| 99 | Metabotropic glutamate receptor 4 (mGluR4) | 7E9H | 6.30 | |
Target general information Gen name GRM4 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms mGluR4; Group III metabotropic glutamate receptor 4; GPRC1D Protein family G-protein coupled receptor 3 family Biochemical class GPCR glutamate Function Ligand binding causes a conformation change that triggers signaling via guanine nucleotide-binding proteins (G proteins) and modulates the activity of down-stream effectors. Signaling inhibits adenylate cyclase activity. G-protein coupled receptor for glutamate. Related diseases Immunodeficiency 57 with autoinflammation (IMD57) [MIM:618108]: An autosomal recessive primary immunodeficiency characterized by lymphopenia and recurrent viral, bacterial, and fungal infections. Patients exhibit early-onset inflammatory bowel disease involving the upper and lower gastrointestinal tract, and develop progressive polyarthritis. {ECO:0000269|PubMed:30026316}. The disease is caused by variants affecting the gene represented in this entry. RIPK1-deficient immune cells from IMD57 patients have impaired proinflammatory signaling leading to dysregulated cytokine secretion and are prone to necroptosis. {ECO:0000269|PubMed:30026316}.; DISEASE: Autoinflammation with episodic fever and lymphadenopathy (AIEFL) [MIM:618852]: An autosomal dominant immunologic disorder characterized by early onset of recurrent episodes of unexplained fever, lymphadenopathy, hepatosplenomegaly, and increased levels of inflammatory cytokines and chemokines in patient serum. {ECO:0000269|PubMed:31827280, ECO:0000269|PubMed:31827281}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00142 Interacts with NA EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disulfide bond; G-protein coupled receptor; Glycoprotein; Membrane; Proteomics identification; Receptor; Reference proteome; Signal; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID R Molecular weight (Da) 85773.3 Length 766 Aromaticity 0.1 Instability index 34.05 Isoelectric point 8.68 Charge (pH=7) 11.83 2D Binding mode Binding energy (Kcal/mol) -8.6  Molscript Map 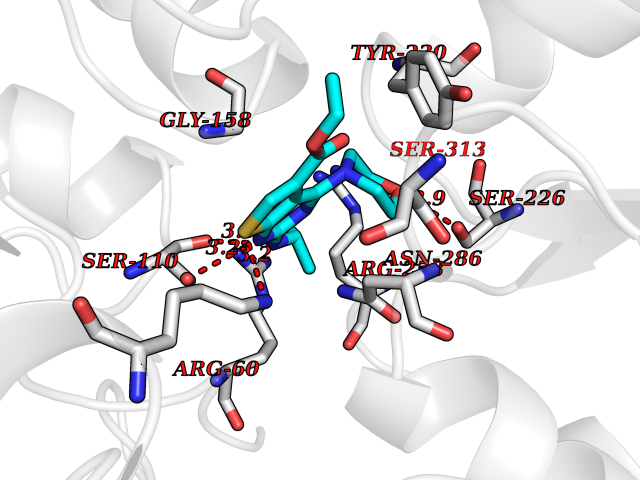 Pymol Map 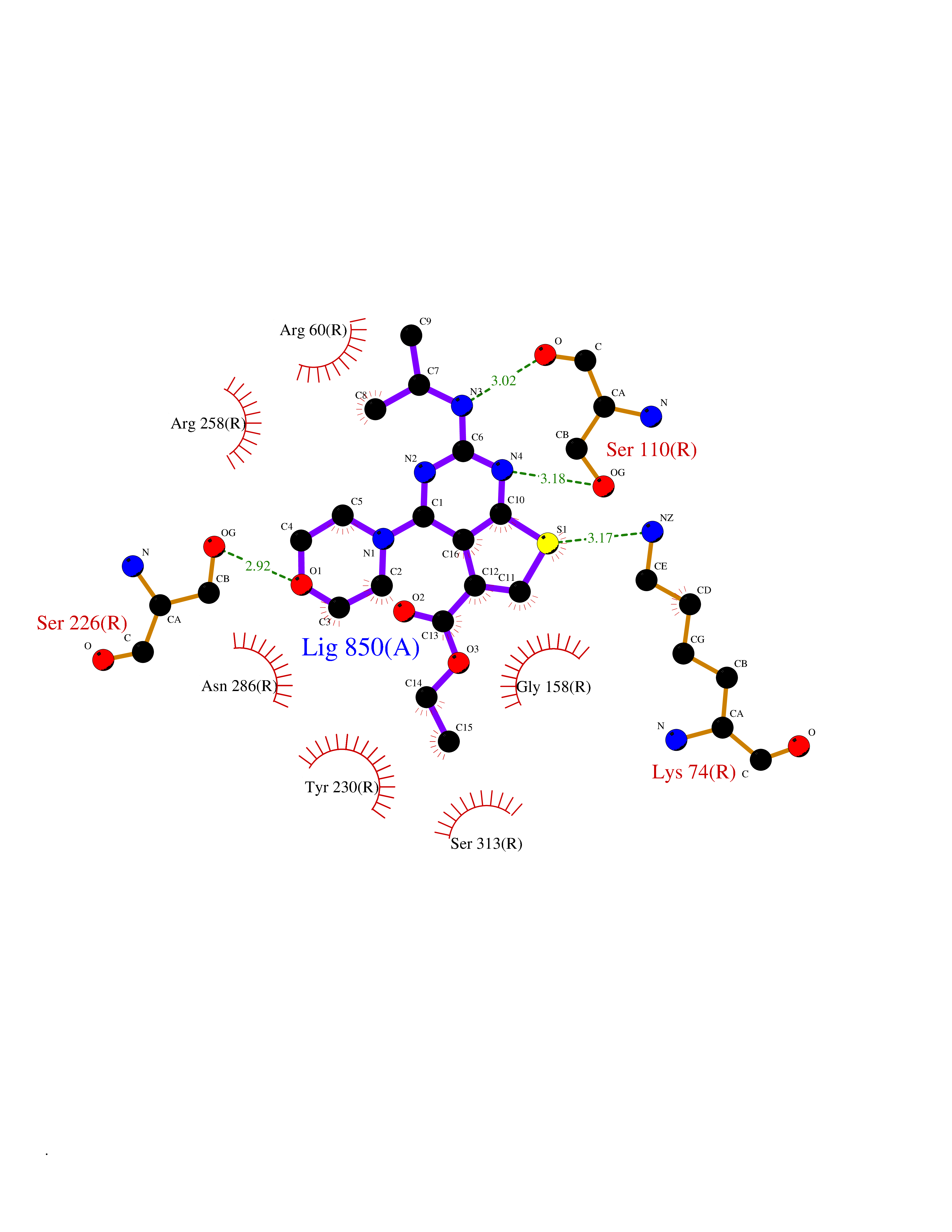 Ligplot Map 3D Binding mode Sequence NSIRIDGDITLGGLFPVHGRGSEGKPCGELKKEKGIHRLEAMLFALDRINNDPDLLPNITLGARILDTCSRDTHALEQSLTFVQALIERVVGVIGASGSSVSIMVANILRLFKIPQISYASTAPDLSDNSRYDFFSRVVPSDTYQAQAMVDIVRALKWNYVSTVASEGSYGESGVEAFIQKSREDGGVCIAQSVKIPREPKAGEFDKIIRRLLETSNARAVIIFANEDDIRRVLEAARRANQTGHFFWMGSDSWGSKIAPVLHLEEVAEGAVTILPKRMSVRGFDRYFSSRTLDNNRRNIWFAEFWEDNFHCKLVKKCTNRERIGQDSAYEQEGKVQFVIDAVYAMGHALHAMHRDLCPGRVGLCPRMDPVDGTQLLKYIRNVNFSGIAGNPVTFNENGDAPGRYDIYQYQLRAEYKVIGSWTDHLHLRIQQLPRSICSLPCQPGERKKTVKGMPCCWHCEPCTGYQYQVDRYTCKTCPYDMRPTENRTGCRPIPIIKLEWGSPWAVLPLFLAVVGIAATLFVVITFVRYNDTPIVKASGRELSYVLLAGIFLCYATTFLMIAEPDLGTCSLRRIFLGLGMSISYAALLTKTNRIYRIFEQGKRSVSAPRFISPASQLAITFSLISLQLLGICVWFVVDPSHSVVDFQDQRTLDPRFARGVLKCDISDLSLICLLGYSMLLMVTCTVYAIKTRGVPETFNEAKPIGFTMYTTCIVWLAFIPIFFGTSQSADKLYIQTTTLTVSVSLSASVSLGMLYMPKVYIILFH Hydrogen bonds contact Hydrophobic contact | ||||
| 100 | Progesterone receptor (PGR) | 1SQN | 6.29 | |
Target general information Gen name PGR Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms PR; Nuclear receptor subfamily 3 group C member 3; NR3C3 Protein family Nuclear hormone receptor family, NR3 subfamily Biochemical class Nuclear hormone receptor Function Depending on the isoform, progesterone receptor functions as transcriptional activator or repressor. The steroid hormones and their receptors are involved in the regulation of eukaryotic gene expression and affect cellular proliferation and differentiation in target tissues. Related diseases Butyrylcholinesterase deficiency (BCHED) [MIM:617936]: An autosomal recessive metabolic condition characterized by increased sensitivity to certain anesthetic drugs, including the muscle relaxants succinylcholine or mivacurium. BCHED results in slower hydrolysis of these drugs and, consequently, a prolonged neuromuscular block, leading to apnea. The duration of the prolonged apnea varies significantly depending on the extent of the enzyme deficiency. {ECO:0000269|PubMed:10404729, ECO:0000269|PubMed:11928765, ECO:0000269|PubMed:12881446, ECO:0000269|PubMed:1306123, ECO:0000269|PubMed:1349196, ECO:0000269|PubMed:1415224, ECO:0000269|PubMed:15563885, ECO:0000269|PubMed:15781196, ECO:0000269|PubMed:1611188, ECO:0000269|PubMed:16788378, ECO:0000269|PubMed:17700357, ECO:0000269|PubMed:18075469, ECO:0000269|PubMed:18300943, ECO:0000269|PubMed:25054547, ECO:0000269|PubMed:25264279, ECO:0000269|PubMed:2915989, ECO:0000269|PubMed:7634491, ECO:0000269|PubMed:8554068, ECO:0000269|PubMed:8680411, ECO:0000269|PubMed:9110359, ECO:0000269|PubMed:9191541, ECO:0000269|PubMed:9388484, ECO:0000269|PubMed:9543549, ECO:0000269|PubMed:9694584}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01431; DB06680; DB01406; DB12941; DB13857; DB00304; DB09123; DB01395; DB00378; DB11219; DB00823; DB00294; DB13867; DB08906; DB00588; DB06730; DB11619; DB11064; DB06789; DB00367; DB00431; DB09124; DB00603; DB00351; DB02998; DB00834; DB00648; DB00764; DB14512; DB06713; DB00717; DB00957; DB09389; DB01428; DB02746; DB00396; DB14583; DB00421; DB04787; DB05253; DB08867 Interacts with Q9H467; P03372; P06401; P40763; P03372 EC number NA Uniprot keywords 3D-structure; Alternative promoter usage; Alternative splicing; Cytoplasm; Direct protein sequencing; DNA-binding; Isopeptide bond; Lipid-binding; Lipoprotein; Membrane; Metal-binding; Mitochondrion; Mitochondrion outer membrane; Nucleus; Palmitate; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Steroid-binding; Transcription; Transcription regulation; Ubl conjugation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A,B Molecular weight (Da) 28853.6 Length 250 Aromaticity 0.09 Instability index 54.82 Isoelectric point 8.4 Charge (pH=7) 2.28 2D Binding mode Binding energy (Kcal/mol) -8.58 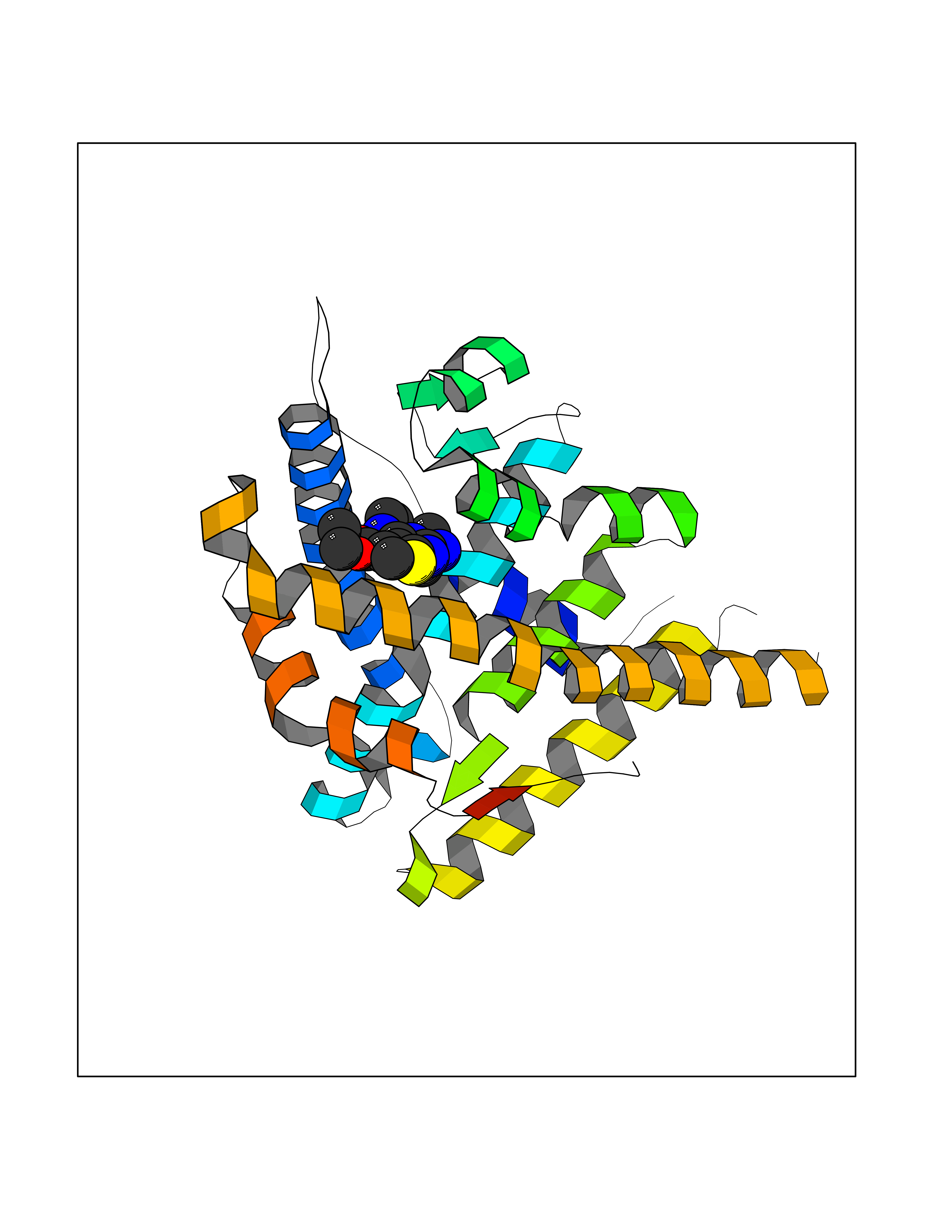 Molscript Map 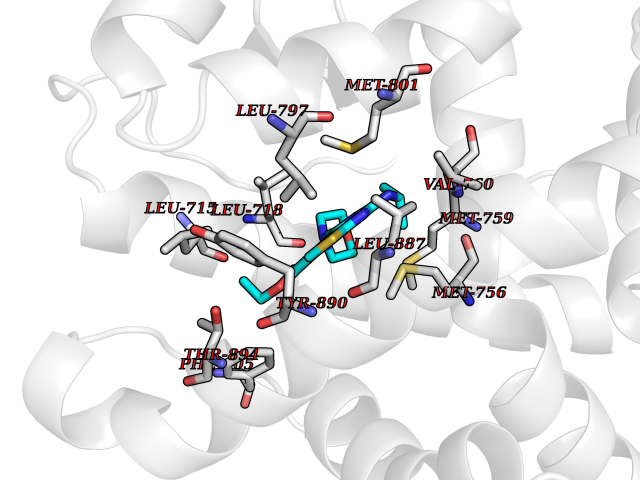 Pymol Map 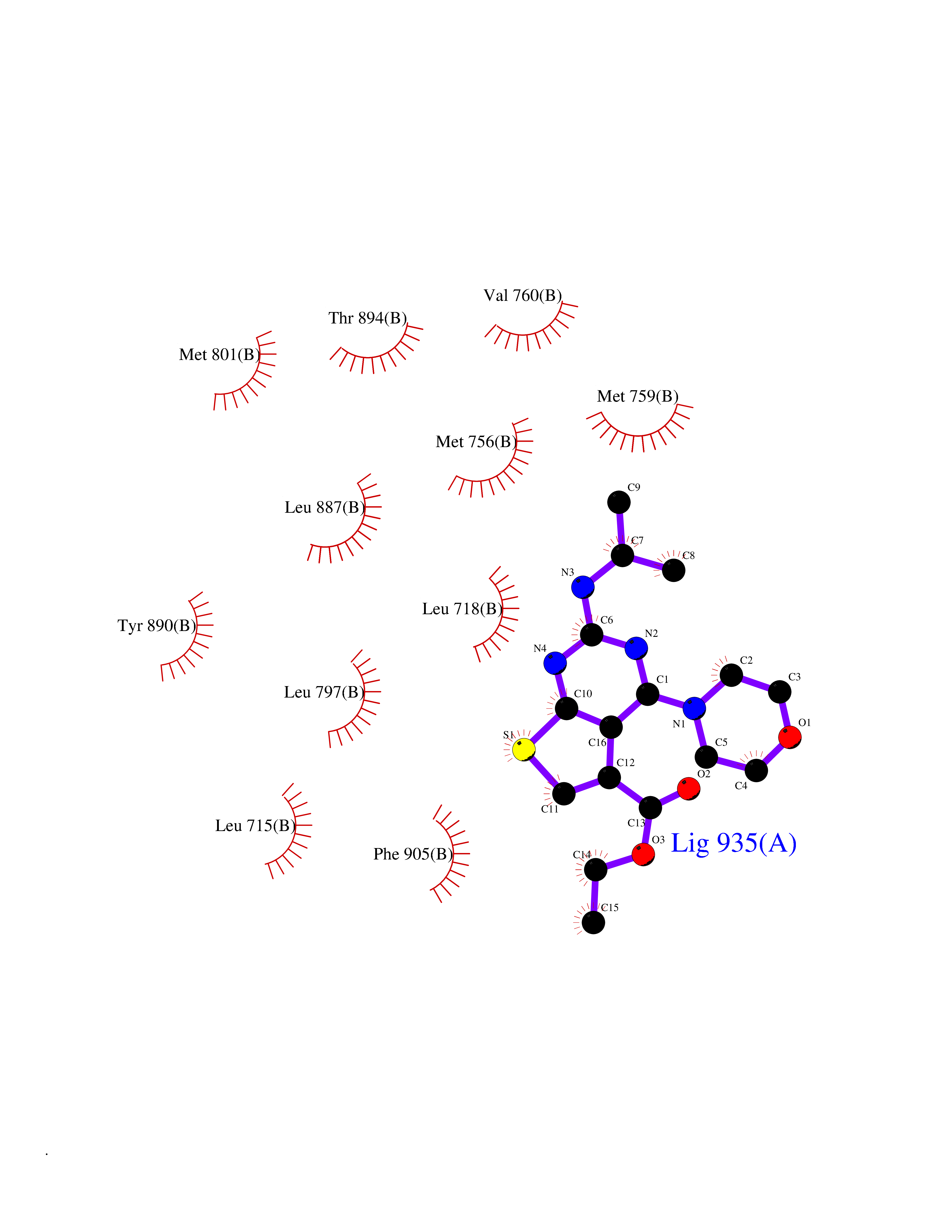 Ligplot Map 3D Binding mode Sequence LIPPLINLLMSIEPDVIYAGHDNTKPDTSSSLLTSLNQLGERQLLSVVKWSKSLPGFRNLHIDDQITLIQYSWMSLMVFGLGWRSYKHVSGQMLYFAPDLILNEQRMKESSFYSLCLTMWQIPQEFVKLQVSQEEFLCMKVLLLLNTIPLEGLRSQTQFEEMRSSYIRELIKAIGLRQGVVSSSQRFYQLTKLLDNLHDLVKQLHLYCLNTFIQSRALSVEFPEMMSEVIAAQLPKILAGMVKPLLFHKK Hydrogen bonds contact Hydrophobic contact | ||||