Job Results:
Ligand
Structure
Job ID
7a36b9b0fd299388007ea8e58dfe56c6
Job name
NA
Time
2025-09-26 17:47:53
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 1 | Folate receptor beta (FOLR2) | 4KN0 | 6.72 | |
Target general information Gen name FOLR2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Placental folate-binding protein; Folate receptor, fetal/placental; Folate receptor type-beta; Folate receptor 2; FR-beta; FOLR2 Protein family Folate receptor family Biochemical class Folate receptor Function Binds to folate and reduced folic acid derivatives and mediates delivery of 5-methyltetrahydrofolate and folate analogs into the interior of cells. Has high affinity for folate and folic acid analogs at neutral pH. Exposure to slightly acidic pH after receptor endocytosis triggers a conformation change that strongly reduces its affinity for folates and mediates their release. Related diseases Acute hepatic porphyria (AHEPP) [MIM:612740]: A form of porphyria. Porphyrias are inherited defects in the biosynthesis of heme, resulting in the accumulation and increased excretion of porphyrins or porphyrin precursors. They are classified as erythropoietic or hepatic, depending on whether the enzyme deficiency occurs in red blood cells or in the liver. AHP is characterized by attacks of gastrointestinal disturbances, abdominal colic, paralyses and peripheral neuropathy. Most attacks are precipitated by drugs, alcohol, caloric deprivation, infections, or endocrine factors. {ECO:0000269|PubMed:10706561, ECO:0000269|PubMed:1309003, ECO:0000269|PubMed:1569184, ECO:0000269|PubMed:17236137, ECO:0000269|PubMed:2063868}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00158; DB00563; DB05168 Interacts with NA EC number NA Uniprot keywords 3D-structure; Cell membrane; Direct protein sequencing; Disulfide bond; Folate-binding; Glycoprotein; GPI-anchor; Lipoprotein; Membrane; Proteomics identification; Receptor; Reference proteome; Secreted; Signal; Transport Protein physicochemical properties Chain ID A Molecular weight (Da) 23841.6 Length 205 Aromaticity 0.12 Instability index 56.78 Isoelectric point 7.92 Charge (pH=7) 2.58 2D Binding mode Binding energy (Kcal/mol) -9.17 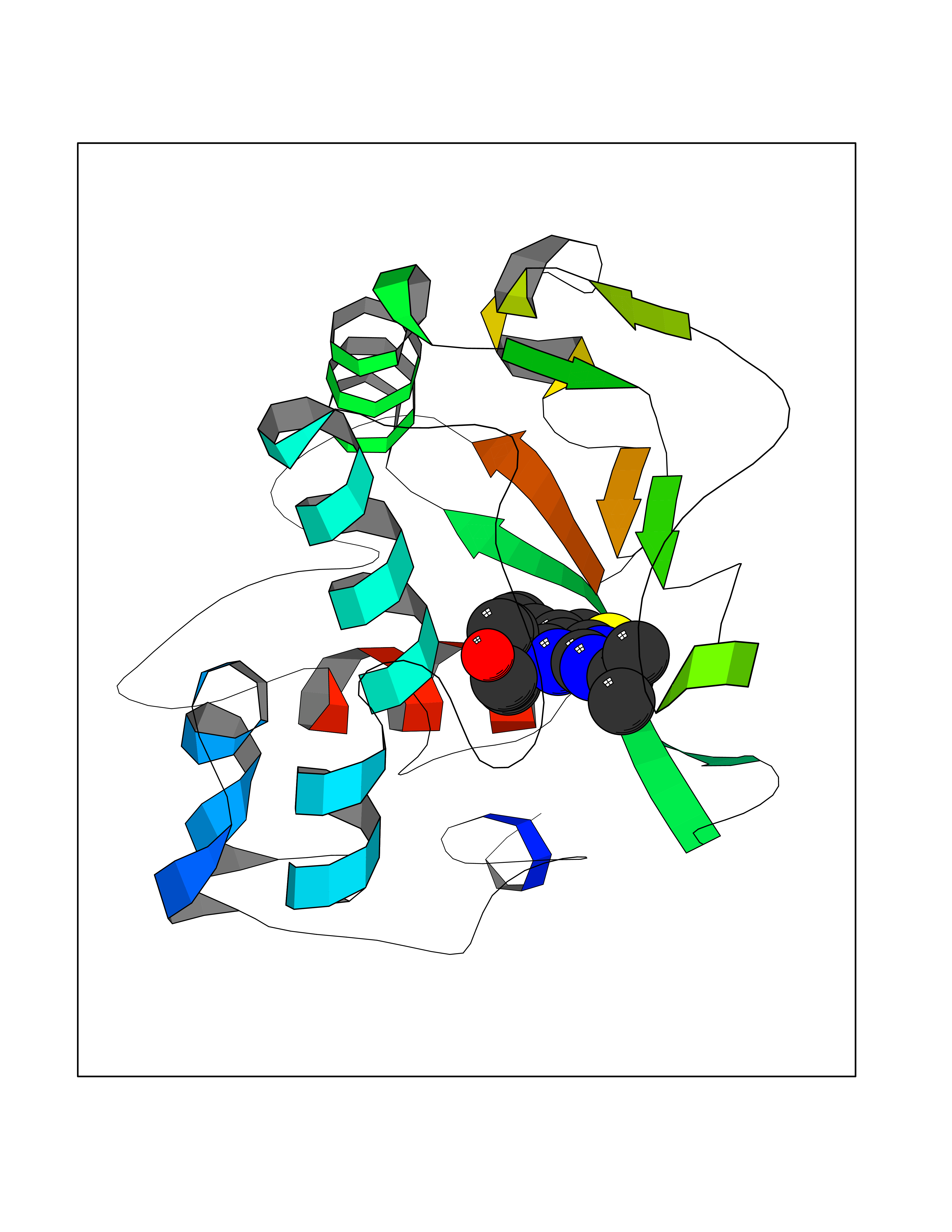 Molscript Map 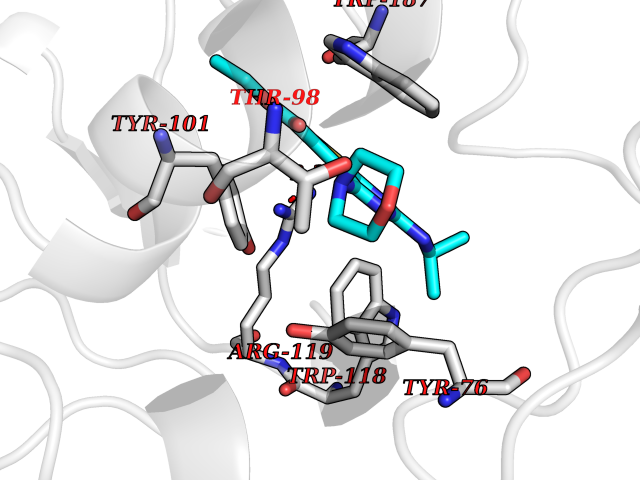 Pymol Map 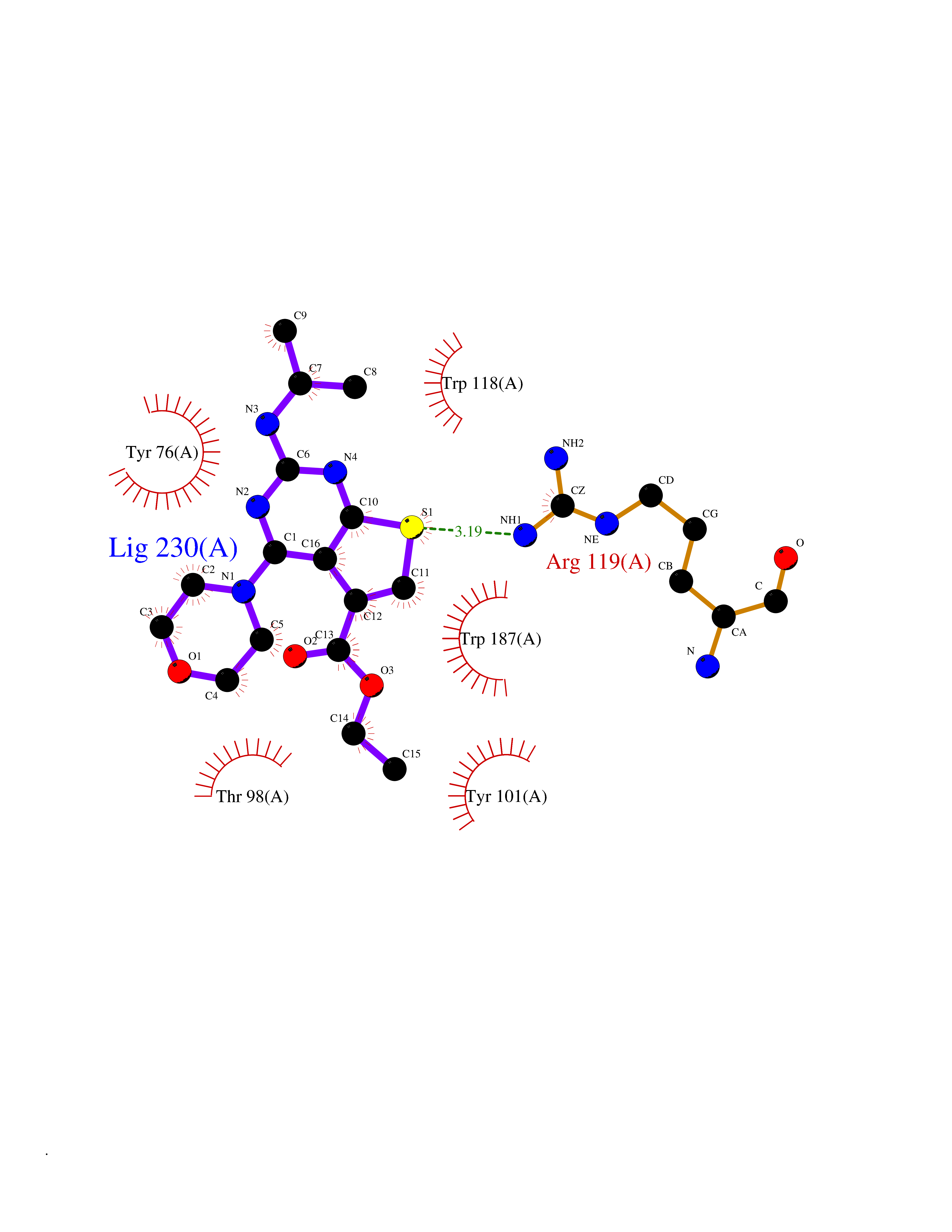 Ligplot Map 3D Binding mode Sequence RTDLLNVCMDAKHHKTKPGPEDKLHDQCSPWKKNACCTASTSQELHKDTSRLYNFNWDHCGKMEPACKRHFIQDTCLYECSPNLGPWIQQVNQSWRKERFLDVPLCKEDCQRWWEDCHTSHTCKSNWHRGWDWTSGVNKCPAGALCRTFESYFPTPAALCEGLWSHSYKVSNYSRGSGRCIQMWFDSAQGNPNEEVARFYAAAMH Hydrogen bonds contact Hydrophobic contact | ||||
| 2 | Plasmodium Dihydroorotate dehydrogenase (Malaria DHOdehase) | 1TV5 | 6.72 | |
Target general information Gen name Malaria DHOdehase Organism Plasmodium falciparum (isolate 3D7) Uniprot ID TTD ID Synonyms PFF0160c; Mitochondrially bound dihydroorotate-ubiqui oxidoreductase; Dihydroorotate oxidase of Plasmodium falciparum; Dihydroorotate dehydrogenase of Plasmodium falciparum; DHOdehase of Plasmodium fa Protein family Dihydroorotate dehydrogenase family, Type 2 subfamily Biochemical class CH-CH donor oxidoreductase Function Catalyzes the conversion of dihydroorotate to orotate with quinone as electron acceptor. Related diseases Combined oxidative phosphorylation deficiency 33 (COXPD33) [MIM:617713]: An autosomal recessive disorder caused by multiple mitochondrial respiratory chain defects and impaired mitochondrial energy metabolism. Clinical manifestations are highly variable. Affected infants present with cardiomyopathy accompanied by multisystemic features involving liver, kidney, and brain. Death in infancy is observed in some patients. Children and adults present with myopathy and progressive external ophthalmoplegia. {ECO:0000269|PubMed:28942965}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01117 Interacts with NA EC number EC 1.3.5.2 Uniprot keywords 3D-structure; Flavoprotein; FMN; Membrane; Mitochondrion; Mitochondrion inner membrane; Oxidoreductase; Pyrimidine biosynthesis; Reference proteome; Transit peptide; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 41846.8 Length 371 Aromaticity 0.1 Instability index 37.25 Isoelectric point 8.21 Charge (pH=7) 3.13 2D Binding mode Binding energy (Kcal/mol) -9.16 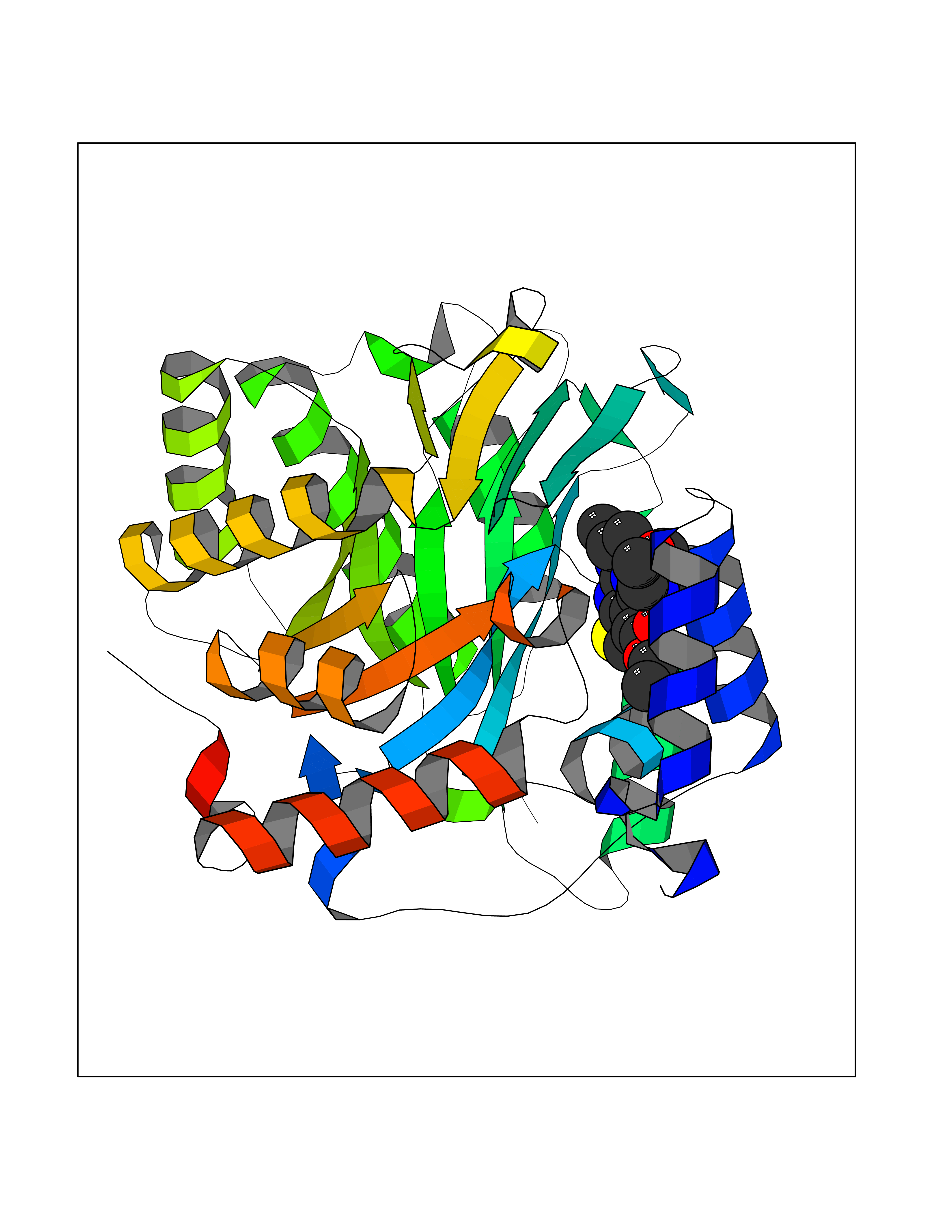 Molscript Map 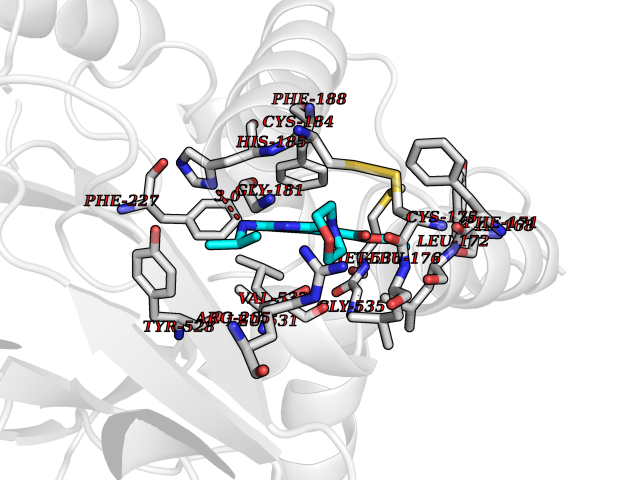 Pymol Map 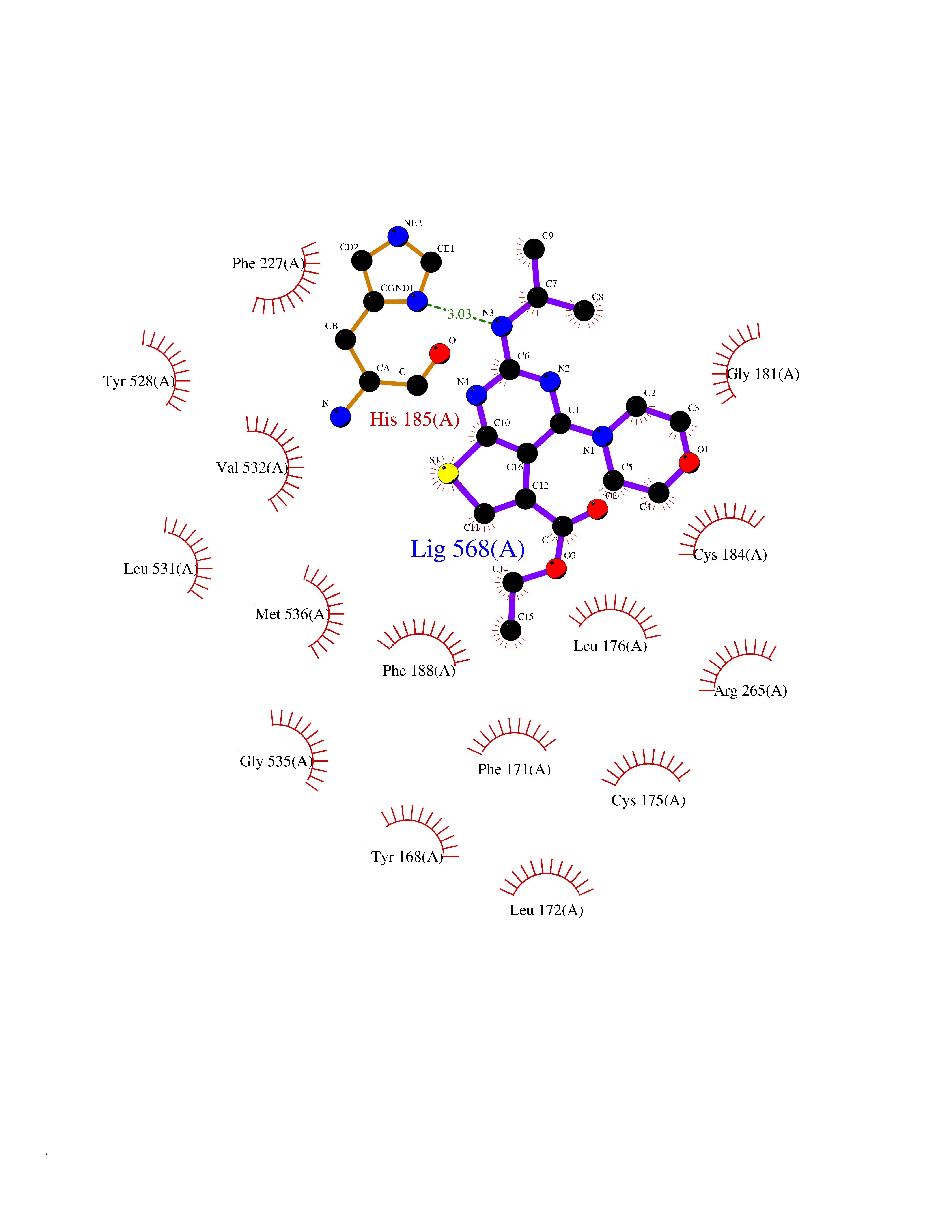 Ligplot Map 3D Binding mode Sequence FESYNPEFFLYDIFLKFCLKYIDGEICHDLFLLLGKYNILPYDTSNDSIYACTNIKHLDFINPFGVAAGFDKNGVCIDSILKLGFSFIEIGTITPRGQTGNAKPRIFRDVESRSIINSCGFNNMGCDKVTENLILFRKRQEEDKLLSKHIVGVSIGKNKDTVNIVDDLKYCINKIGRYADYIAINVSSPNTPGLRDNQEAGKLKNIILSVKEEIDNLEFLWFNTTKKKPLVFVKLAPDLNQEQKKEIADVLLETNIDGMIISNTTTQINDIKSFENKKGGVSGAKLKDISTKFICEMYNYTNKQIPIIASGGIFSGLDALEKIEAGASVCQLYSCLVFNGMKSAVQIKRELNHLLYQRGYYNLKEAIGRKH Hydrogen bonds contact Hydrophobic contact | ||||
| 3 | Bacterial Cystathionine gamma-synthase (Bact metB) | 1CS1 | 6.72 | |
Target general information Gen name Bact metB Organism Escherichia coli (strain K12) Uniprot ID TTD ID Synonyms metB; O-succinylhomoserine (Thiol)-lyase; CGS Protein family Trans-sulfuration enzymes family Biochemical class Alkyl aryl transferase Function Catalyzes the formation of L-cystathionine from O- succinyl-L-homoserine (OSHS) and L-cysteine, via a gamma- replacement reaction. In the absence of thiol, catalyzes gamma- elimination to form 2-oxobutanoate, succinate and ammonia. Related diseases Congenital disorder of glycosylation 2A (CDG2A) [MIM:212066]: A multisystem disorder caused by a defect in glycoprotein biosynthesis and characterized by under-glycosylated serum glycoproteins. Congenital disorders of glycosylation result in a wide variety of clinical features, such as defects in the nervous system development, psychomotor retardation, dysmorphic features, hypotonia, coagulation disorders, and immunodeficiency. The broad spectrum of features reflects the critical role of N-glycoproteins during embryonic development, differentiation, and maintenance of cell functions. {ECO:0000269|PubMed:11228641, ECO:0000269|PubMed:8808595}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB04083 Interacts with NA EC number EC 2.5.1.48 Uniprot keywords 3D-structure; Amino-acid biosynthesis; Cytoplasm; Direct protein sequencing; Methionine biosynthesis; Pyridoxal phosphate; Reference proteome; Transferase Protein physicochemical properties Chain ID A,C Molecular weight (Da) 82360.8 Length 767 Aromaticity 0.06 Instability index 34.34 Isoelectric point 5.9 Charge (pH=7) -12.3 2D Binding mode Binding energy (Kcal/mol) -9.17 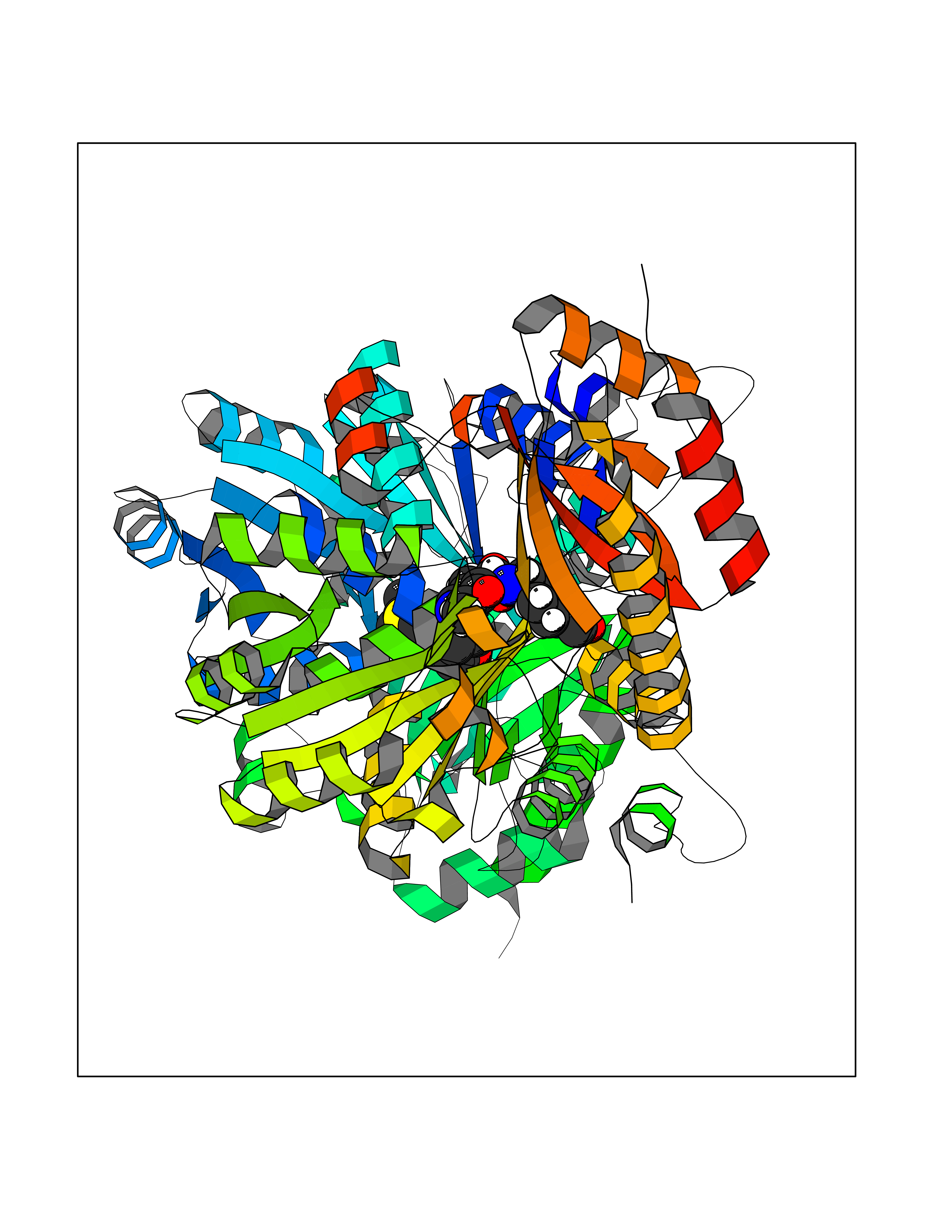 Molscript Map 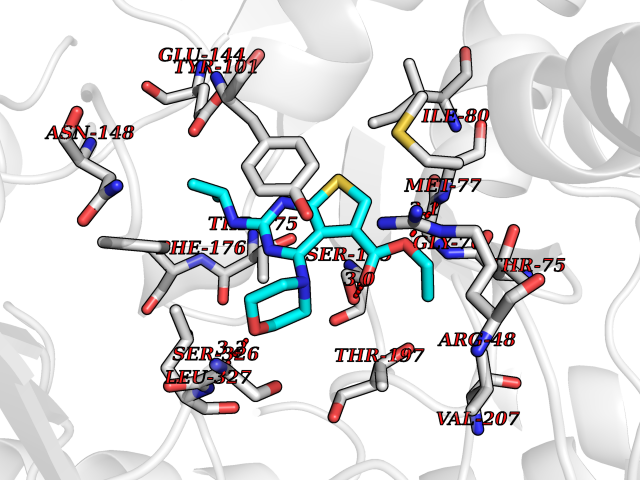 Pymol Map 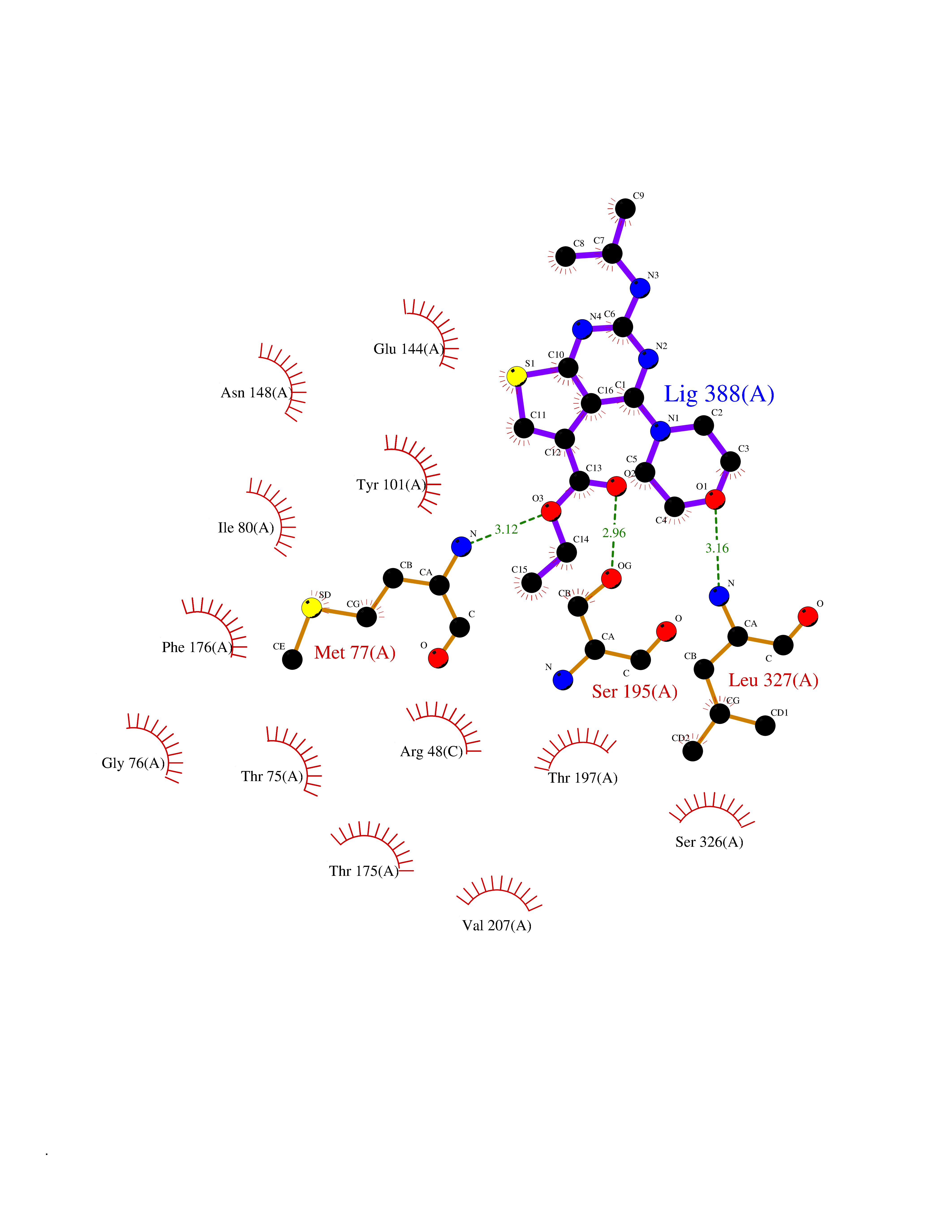 Ligplot Map 3D Binding mode Sequence RKQATIAVRSGLNDDEQYGCVVPPIHLSSTYNFTGFNEPRAHDYSRRGNPTRDVVQRALAELEGGAGAVLTNTGMSAIHLVTTVFLKPGDLLVAPHDCYGGSYRLFDSLAKRGCYRVLFVDQGDEQALRAALAEKPKLVLVESPSNPLLRVVDIAKICHLAREVGAVSVVDNTFLSPALQNPLALGADLVLHSCTYLNGHSDVVAGVVIAKDPDVVTELAWWANNIGVTGGAFDSYLLLRGLRTLVPRMELAQRNAQAIVKYLQTQPLVKKLYHPSLPENQGHEIAARQQKGFGAMLSFELDGDEQTLRRFLGGLSLFTLAESLGGVESLISHAATMTHAGMAPEARAAAGISETLLRISTGIEDGEDLIADLENGFRAANKGRKQATIAVRSGLNDDEQYGCVVPPIHLSSTYNFTGFNEPRAHDYSRRGNPTRDVVQRALAELEGGAGAVLTNTGMSAIHLVTTVFLKPGDLLVAPHDCYGGSYRLFDSLAKRGCYRVLFVDQGDEQALRAALAEKPKLVLVESPSNPLLRVVDIAKICHLAREVGAVSVVDNTFLSPALQNPLALGADLVLHSCTXYLNGHSDVVAGVVIAKDPDVVTELAWWANNIGVTGGAFDSYLLLRGLRTLVPRMELAQRNAQAIVKYLQTQPLVKKLYHPSLPENQGHEIAARQQKGFGAMLSFELDGDEQTLRRFLGGLSLFTLAESLGGVESLISHAATMTHAGMAPEARAAAGISETLLRISTGIEDGEDLIADLENGFRAANKG Hydrogen bonds contact Hydrophobic contact | ||||
| 4 | Mutated Histone H3.3 (H3F3A) | 4GUS | 6.71 | |
Target general information Gen name H3F3A Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms PP781; Histone H3.3; H3F3; H3.3B; H3.3A Protein family Histone H3 family Biochemical class NA Function Variant histone H3 which replaces conventional H3 in a wide range of nucleosomes in active genes. Constitutes the predominant form of histone H3 in non-dividing cells and is incorporated into chromatin independently of DNA synthesis. Deposited at sites of nucleosomal displacement throughout transcribed genes, suggesting that it represents an epigenetic imprint of transcriptionally active chromatin. Nucleosomes wrap and compact DNA into chromatin, limiting DNA accessibility to the cellular machineries which require DNA as a template. Histones thereby play a central role in transcription regulation, DNA repair, DNA replication and chromosomal stability. DNA accessibility is regulated via a complex set of post-translational modifications of histones, also called histone code, and nucleosome remodeling. Related diseases Glioma (GLM) [MIM:137800]: Gliomas are benign or malignant central nervous system neoplasms derived from glial cells. They comprise astrocytomas and glioblastoma multiforme that are derived from astrocytes, oligodendrogliomas derived from oligodendrocytes and ependymomas derived from ependymocytes. {ECO:0000269|PubMed:22286061, ECO:0000269|PubMed:22286216, ECO:0000269|PubMed:23539269}. The gene represented in this entry is involved in disease pathogenesis. H3F3A mutations affecting residues involved in post-translational modifications of histone H3.3 are recurrent in malignant, aggressive gliomas including glioblastoma multiforme (GBM) and diffuse intrinsic pontine glioma (DIPG) (PubMed:22286061, PubMed:22286216). The mechanism through which mutations lead to tumorigenesis involves altered histones methylation, impaired regulation of Polycomb repressive complex 2 (PRC2) activity, and aberrant epigenetic regulation of gene expression (PubMed:23539183, PubMed:23539269, PubMed:23603901). {ECO:0000269|PubMed:22286061, ECO:0000269|PubMed:22286216, ECO:0000269|PubMed:23539183, ECO:0000269|PubMed:23539269, ECO:0000269|PubMed:23603901}.; DISEASE: Bryant-Li-Bhoj neurodevelopmental syndrome 1 (BRYLIB1) [MIM:619720]: An autosomal dominant disorder predominantly characterized by global developmental delay, impaired intellectual development, poor or absent speech, and delayed motor milestones. Clinical manifestations are highly variable, including abnormal head shape, dysmorphic facial features, oculomotor abnormalities, feeding problems, and non-specific brain imaging abnormalities. Additional features may include hearing loss, seizures, short stature, and mild skeletal defects. {ECO:0000269|PubMed:33268356, ECO:0000269|PubMed:34876591}. The disease is caused by variants affecting the gene represented in this entry. BRYLIB1 is caused by variants in H3-3A. {ECO:0000269|PubMed:33268356, ECO:0000269|PubMed:34876591}.; DISEASE: Bryant-Li-Bhoj neurodevelopmental syndrome 2 (BRYLIB2) [MIM:619721]: An autosomal dominant disorder predominantly characterized by global developmental delay, impaired intellectual development, poor or absent speech, and delayed motor milestones. Clinical manifestations are highly variable, including abnormal head shape, dysmorphic facial features, oculomotor abnormalities, feeding problems, and non-specific brain imaging abnormalities. Additional features may include hearing loss, seizures, short stature, and mild skeletal defects. {ECO:0000269|PubMed:33268356, ECO:0000269|PubMed:34876591}. The disease is caused by variants affecting the gene represented in this entry. BRYLIB2 is caused by variants in H3-3B. {ECO:0000269|PubMed:33268356, ECO:0000269|PubMed:34876591}.; DISEASE: H3F3A and H3F3B mutations affecting residues involved in post-translational modifications of histone H3.3 are implicated in the pathogenesis of some bone and cartilage neoplasms. Mutations have been found with high prevalence in chondroblastoma and giant cell tumors of bone, and with low frequency in osteosarcoma, conventional chondrosarcoma and clear cell chondrosarcoma. Chondroblastoma samples frequently carry a H3F3B mutation affecting residue Lys-37 (H3K36), although H3F3A is mutated in some cases. Most giant cell tumors of bone harbor H3F3A mutations affecting residue Gly-35 (H3G34). {ECO:0000269|PubMed:24162739}. Drugs (DrugBank ID) NA Interacts with Q9NVP2; P45973; Q13111; Q9UER7; Q9UER7-1; Q9Y6K1; P62805; P49321-2; Q8IZL8; Q5VWG9; Q9VK33; Q8R5C8 EC number NA Uniprot keywords 3D-structure; Acetylation; ADP-ribosylation; Chromosome; Citrullination; Direct protein sequencing; Disease variant; DNA-binding; Hydroxylation; Intellectual disability; Lipoprotein; Methylation; Nucleosome core; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Ubl conjugation Protein physicochemical properties Chain ID A,C Molecular weight (Da) 86148.9 Length 766 Aromaticity 0.1 Instability index 37.57 Isoelectric point 8.25 Charge (pH=7) 7.16 2D Binding mode Binding energy (Kcal/mol) -9.15 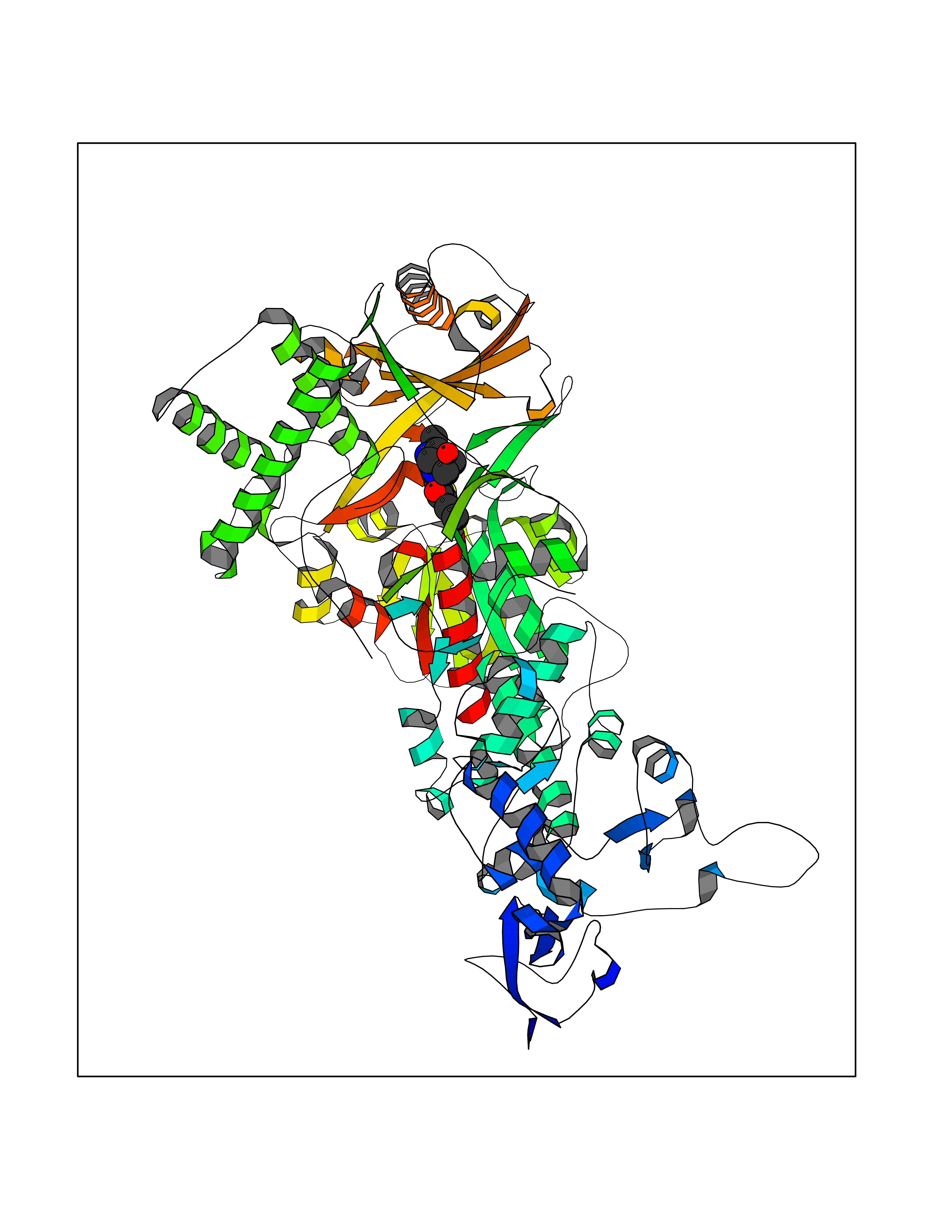 Molscript Map 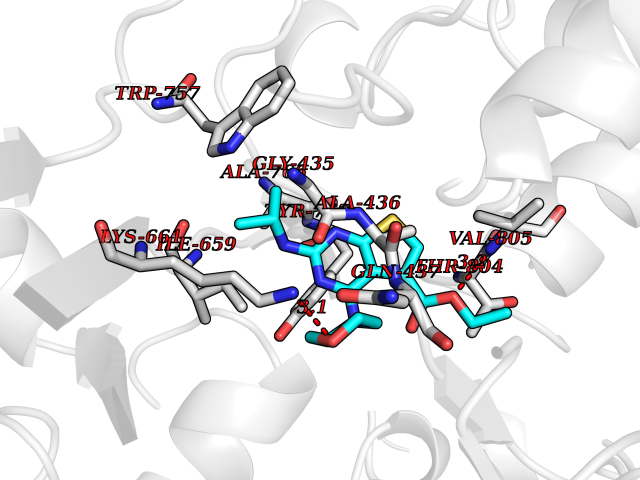 Pymol Map 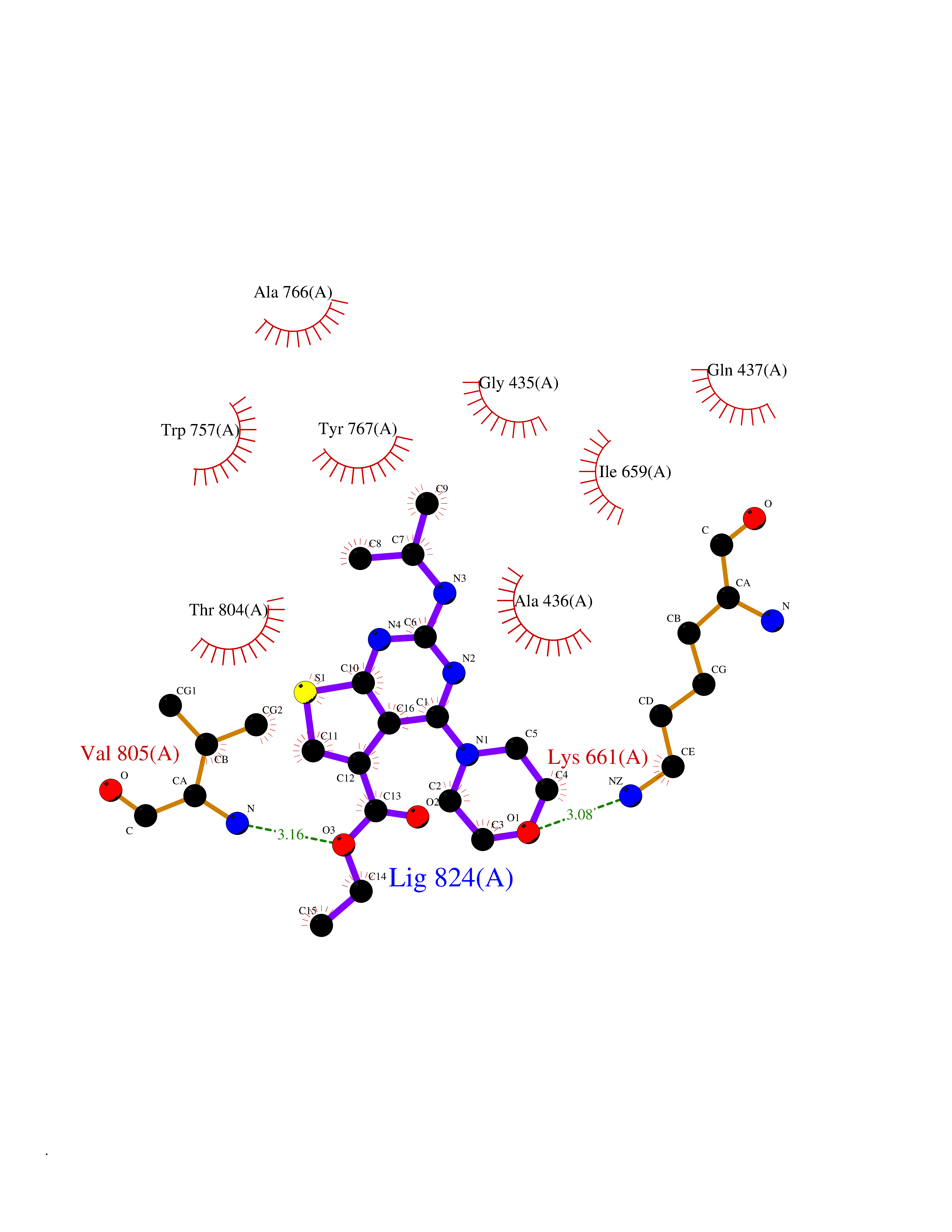 Ligplot Map 3D Binding mode Sequence GSRKCEKAGCTATCPVCFASASERCAKNGYTSRWYHLSCGEHFCNECFDHYYRSHKDGYDKYTTWKKIWTSNGKTEPSPKAFMADQQLPYWVQCTKPECRKWRQLTKEIQLTPQIAKTYRCGMKPNTAIKPETSDHCSLPEDLRVLEVSNHWWYSMLILPPLLKDSVAAPLLSAYYPDCVGMSPSCTGMNRYFQPFYQPNECGKALCVRPDVMELDELYEFPEYSRDPTMYLALRNLILALWYTNCKEALTPQKCIPHIIVRGLVRIRCVQEVERILYFMTRKGLINTGVLSVGADQYLLPKDYHNKSVIIIGAGPAGLAAARQLHNFGIKVTVLEAKDRIGGRVWDDKSFKGVTVGRGAQIVNGCINNPVALMCEQLGISMHKFGERCDLIQEGGRITDPTIDKRMDFHFNALLDVVSEWRKDKTQLQDVPLGEKIEEIYKAFIKESGIQFSELEGQVLQFHLSNLEYACGSNLHQVSARSWDHNEFFAQFAGDHTLLTPGYSVIIEKLAEGLDIQLKSPVQCIDYSGDEVQVTTTDGTGYSAQKVLVTVPLALLQKGAIQFNPPLSEKKMKAINSLGAGIIEKIALQFPYRFWDSKVQGADFFGHVPPSASKRGLFAVFYDMDPQKKHSVLMSVIAGEAVASVRTLDDKQVLQQCMATLRELFKEQEVPDPTKYFVTRWSTDPWIQMAYSFVKTGGSGEAYDIIAEDIQGTVFFAGEATNRHFPQTVTGAYLSGVREASKIAAFARTMQTARKSTGGKAPRKQL Hydrogen bonds contact Hydrophobic contact | ||||
| 5 | Gamma-aminobutyric acid type B receptor subunit 1 | 4MS4 | 6.67 | |
Target general information Gen name GABBR1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms GPRC3B;GPR51 Protein family G-protein coupled receptor 3 family, GABA-B receptor subfamily Biochemical class Signaling protein / antagonist Function G-protein coupled GABA receptor activity. Related diseases Neurodevelopmental disorder with poor language and loss of hand skills (NDPLHS) [MIM:617903]: An autosomal dominant disorder characterized by psychomotor developmental stagnation or regression. NDPLHS manifest in the first years of life as loss of purposeful hand movements, loss of language, and intellectual disability. {ECO:0000269|PubMed:26740508, ECO:0000269|PubMed:28856709, ECO:0000269|PubMed:29369404}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Developmental and epileptic encephalopathy 59 (DEE59) [MIM:617904]: A form of epileptic encephalopathy, a heterogeneous group of severe early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE59 is an autosomal dominant condition characterized by onset of refractory seizures in early infancy. {ECO:0000269|PubMed:28856709, ECO:0000269|PubMed:29100083, ECO:0000269|PubMed:29369404}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08891; DB08892; DB00181; DB00363; DB02530; DB05010; DB09072 Interacts with Q9UBS5; Q9UBS5-2; P46459; Q86UR5 EC number NA Uniprot keywords 3D-structure; Cell membrane; Coiled coil; Direct protein sequencing; Disease variant; Disulfide bond; Epilepsy; G-protein coupled receptor; Glycoprotein; Intellectual disability; Membrane; Phosphoprotein; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Signal; Synapse; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 46502.1 Length 408 Aromaticity 0.12 Instability index 50.05 Isoelectric point 5.78 Charge (pH=7) -5.62 2D Binding mode Binding energy (Kcal/mol) -9.1  Molscript Map 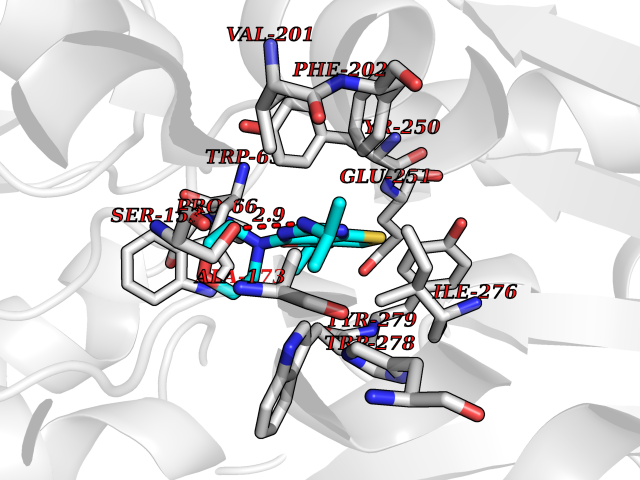 Pymol Map 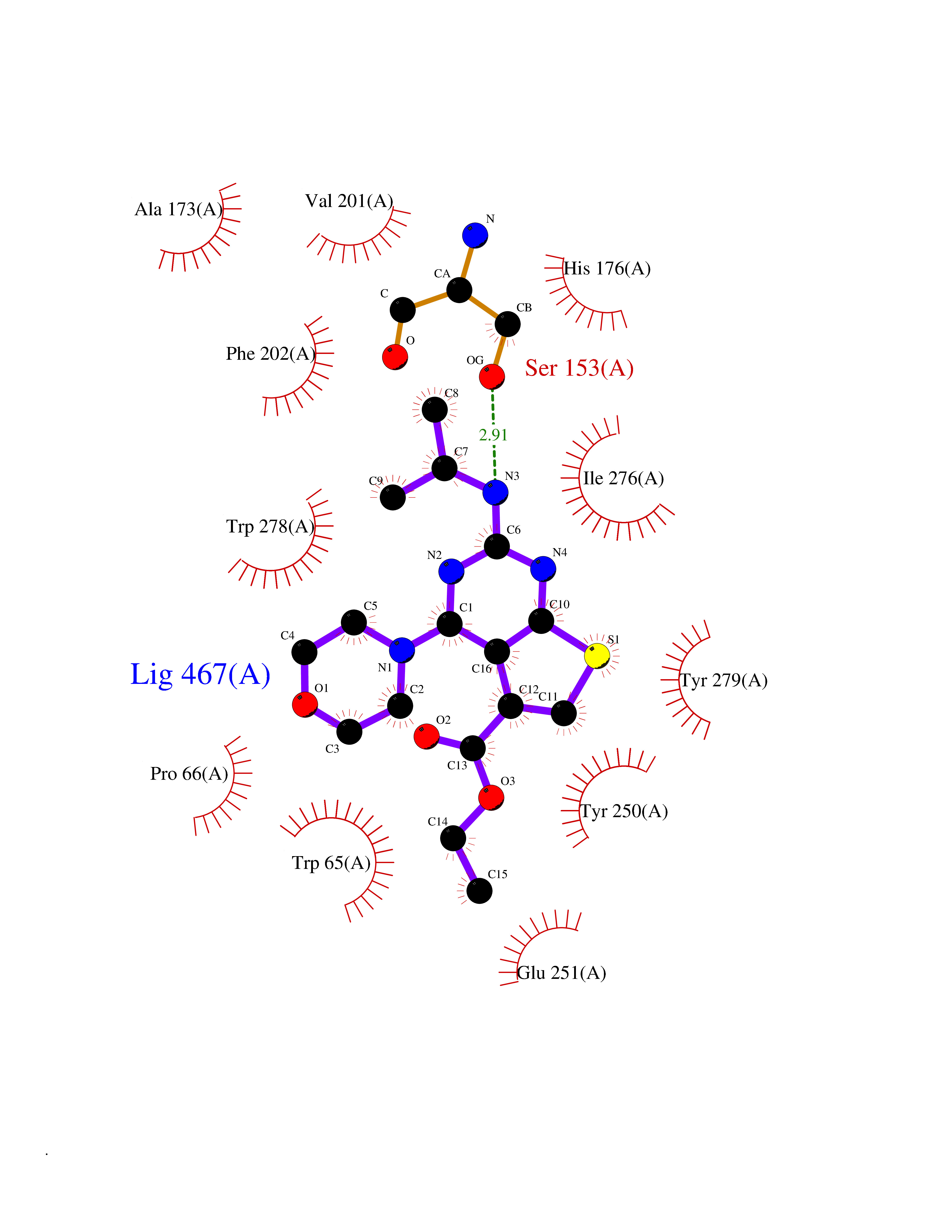 Ligplot Map 3D Binding mode Sequence RRAVYIGALFPMSGGWPGGQACQPAVEMALEDVNSRRDILPDYELKLIHHDSKCDPGQATKYLYELLYNDPIKIILMPGCSSVSTLVAEAARMWNLIVLSYGSSSPALSNRQRFPTFFRTHPSATLHNPTRVKLFEKWGWKKIATIQQTTEVFTSTLDDLEERVKEAGIEITFRQSFFSDPAVPVKNLKRQDARIIVGLFYETEARKVFCEVYKERLFGKKYVWFLIGWYADNWFKIYDPSINCTVDEMTEAVEGHITTEIVMLNPANTRSISNMTSQEFVEKLTKRLKRHPEETGGFQEAPLAYDAIWALALALNKTSRLEDFNYNNQTITDQIYRAMNSSSFEGVSGHVVFDASGSRMAWTLIEQLQGGSYKKIGYYDSTKDDLSWSKTDKWIGGSPPADDYKDDD Hydrogen bonds contact Hydrophobic contact | ||||
| 6 | Dimethylglycine oxidase | 1PJ5 | 6.63 | |
Target general information Gen name dmg Organism Arthrobacter globiformis Uniprot ID TTD ID NA Synonyms NA Protein family GcvT family Biochemical class Oxidoreductase Function Dimethylglycine oxidase activity.Nucleotide binding. Related diseases Curry-Jones syndrome (CRJS) [MIM:601707]: A multisystem disorder characterized by patchy skin lesions, polysyndactyly, diverse cerebral malformations, unicoronal craniosynostosis, iris colobomas, microphthalmia, and intestinal malrotation with myofibromas or hamartomas. {ECO:0000269|PubMed:24859340, ECO:0000269|PubMed:27236920}. The disease is caused by variants affecting the gene represented in this entry. 8 individuals have been identified with the disease-causing mutation Phe-412 and all were mosaic. The mutation could not be reliably detected in blood, greatest success rates were obtained with affected tissues obtained by invasive procedures. It is thought that the mutation has arisen postzygotically early during embryonic development (PubMed:27236920). This mutation has also been identified in ameloblastoma, medulloblastoma, meningioma, and basal cell carcinoma, and has been reported as the oncogenic driver in some of these tumors (PubMed:24859340). {ECO:0000269|PubMed:24859340, ECO:0000269|PubMed:27236920}. Drugs (DrugBank ID) DB03256; DB03147 Interacts with NA EC number 1.5.3.10 Uniprot keywords 3D-structure; Direct protein sequencing; FAD; Flavoprotein; Nucleotide-binding; Oxidoreductase Protein physicochemical properties Chain ID A Molecular weight (Da) 45912.2 Length 427 Aromaticity 0.07 Instability index 43.46 Isoelectric point 4.83 Charge (pH=7) -20.69 2D Binding mode Binding energy (Kcal/mol) -9.04 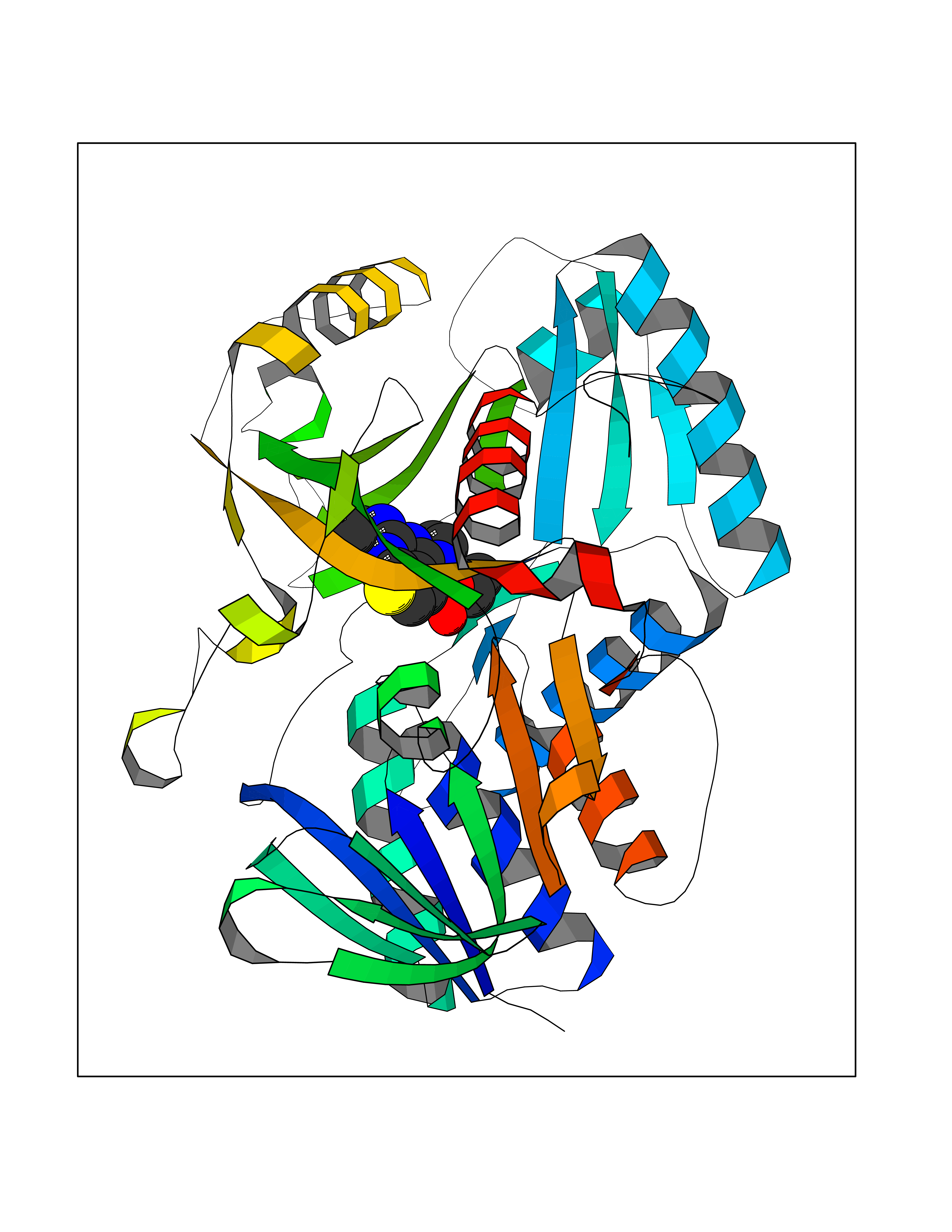 Molscript Map 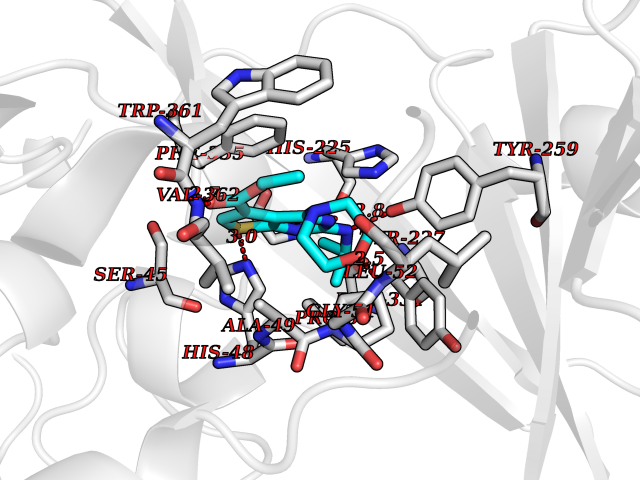 Pymol Map 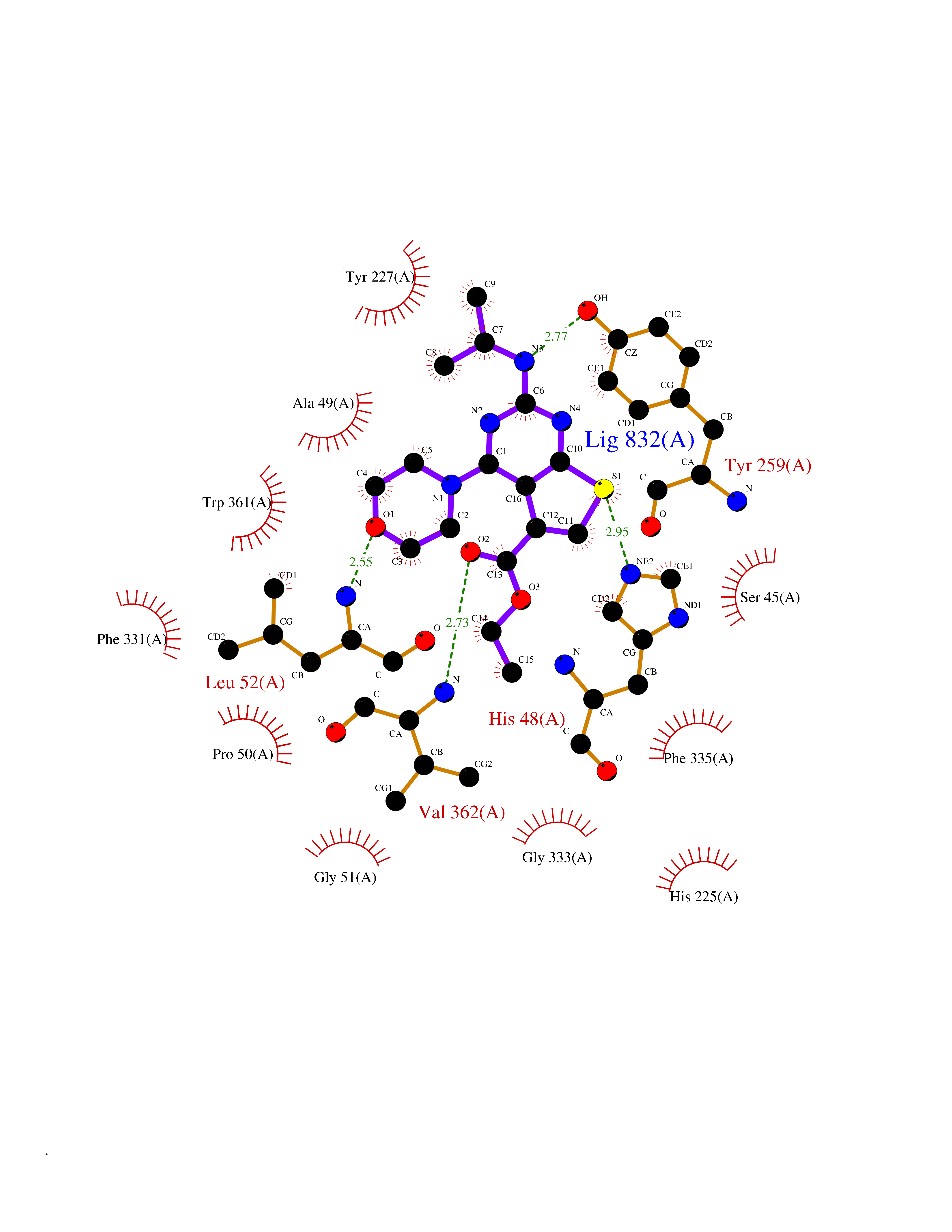 Ligplot Map 3D Binding mode Sequence TPRIVIIGAGIVGTNLADELVTRGWNNITVLDQGPLNMPGGSTSHAPGLVFQTNPSKTMASFAKYTVEKLLSLTEDGVSCFNQVGGLEVATTETRLADLKRKLGYAAAWGIEGRLLSPAECQELYPLLDGENILGGLHVPSDGLASAARAVQLLIKRTESAGVTYRGSTTVTGIEQSGGRVTGVQTADGVIPADIVVSCAGFWGAKIGAMIGMAVPLLPLAHQYVKTTPVPAQQGRNDQPNGARLPILRHQDQDLYYREHGDRYGIGSYAHRPMPVDVDTLGAYAPETVSEHHMPSRLDFTLEDFLPAWEATKQLLPALADSEIEDGFNGIFSFTPDGGPLLGESKELDGFYVAEAVWVTHSAGVAKAMAELLTTGRSETDLGECDITRFEDVQLTPEYVSETSQQNFVEIYDVLHPLQPRLSPRNL Hydrogen bonds contact Hydrophobic contact | ||||
| 7 | Aldehyde oxidoreductase | 4USA | 6.62 | |
Target general information Gen name mop Organism Megalodesulfovibrio gigas (Desulfovibrio gigas) Uniprot ID TTD ID NA Synonyms NA Protein family Xanthine dehydrogenase family Biochemical class Oxidoreductase Function 2 iron, 2 sulfur cluster binding.Aldehyde dehydrogenase (FAD-independent) activity.Electron carrier activity.Metal ion binding. Related diseases LTC4 synthase deficiency is associated with a neurometabolic developmental disorder characterized by muscular hypotonia, psychomotor retardation, failure to thrive, and microcephaly. {ECO:0000269|PubMed:10896305, ECO:0000269|PubMed:9820300}. Drugs (DrugBank ID) DB02137 Interacts with NA EC number 1.2.99.7 Uniprot keywords 2Fe-2S; 3D-structure; FAD; Flavoprotein; Iron; Iron-sulfur; Metal-binding; Molybdenum; NAD; Oxidoreductase Protein physicochemical properties Chain ID A Molecular weight (Da) 96930.4 Length 907 Aromaticity 0.07 Instability index 29.17 Isoelectric point 5.69 Charge (pH=7) -17.56 2D Binding mode Binding energy (Kcal/mol) -9.03 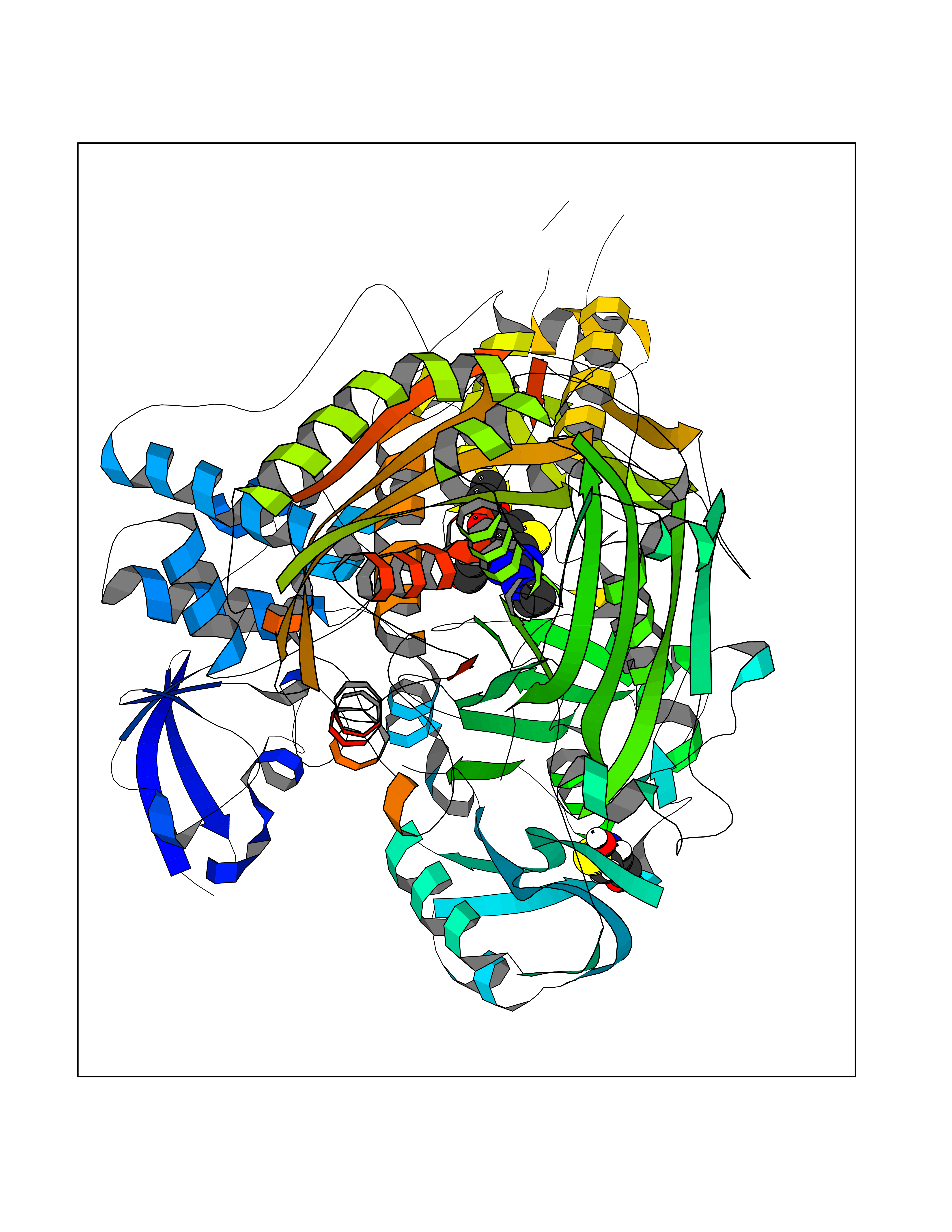 Molscript Map 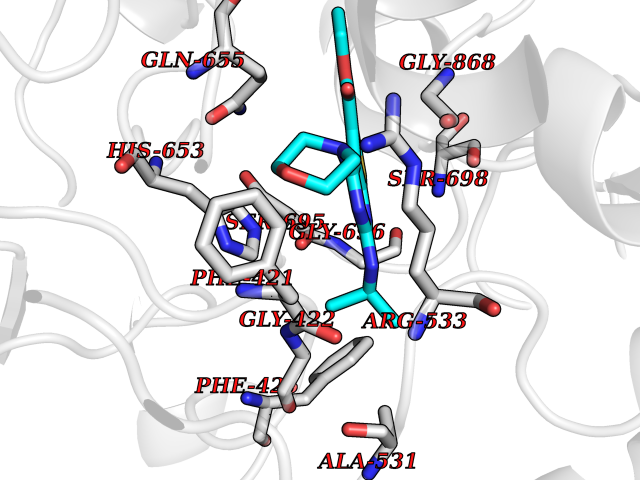 Pymol Map 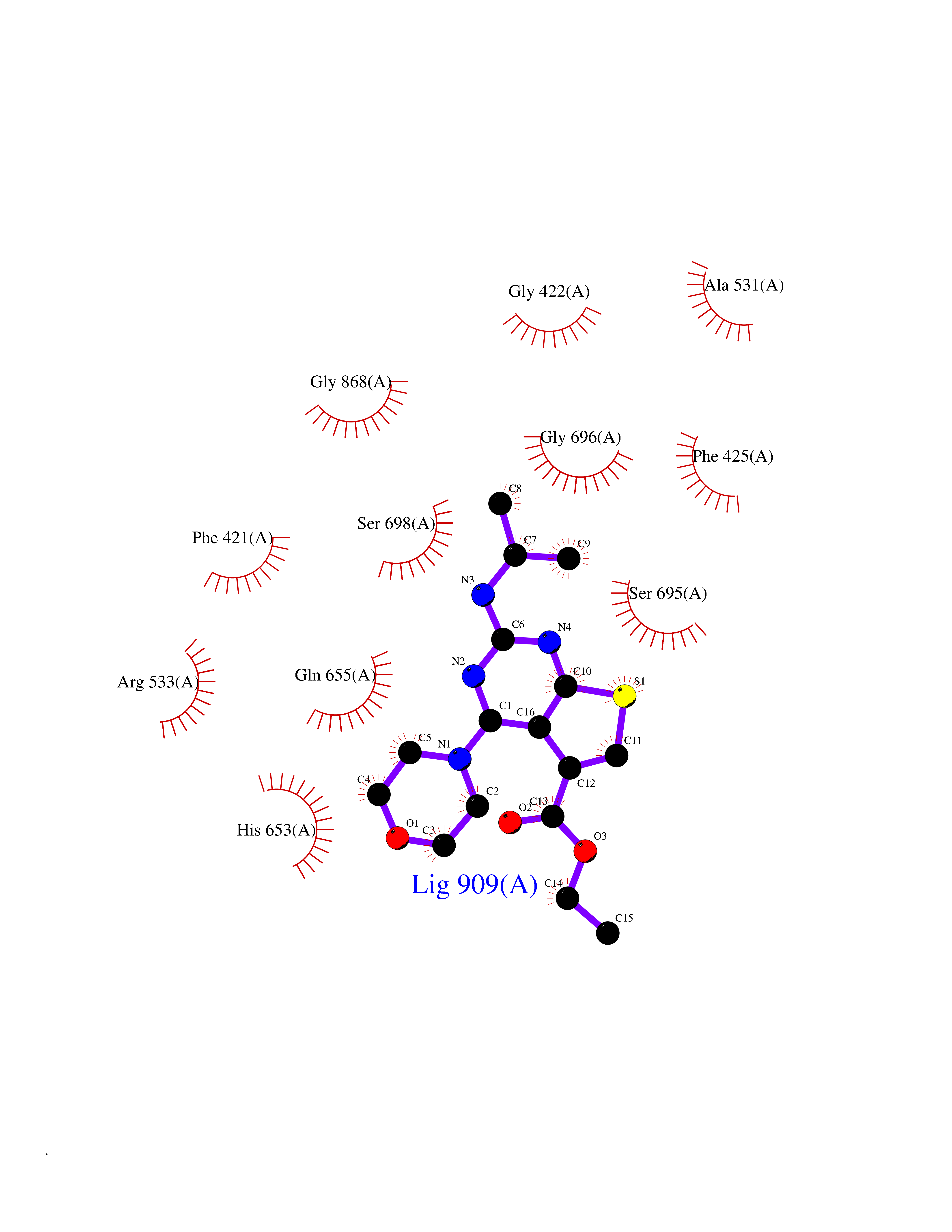 Ligplot Map 3D Binding mode Sequence MIQKVITVNGIEQNLFVDAEALLSDVLRQQLGLTGVKVGCEQGQCGACSVILDGKVVRACVTKMKRVADGAQITTIEGVGQPENLHPLQKAWVLHGGAQCGFCSPGFIVSAKGLLDTNADPSREDVRDWFQKHRNACRCTGYKPLVDAVMDAAAVINGKKPETDLEFKMPADGRIWGSKYPRPTAVAKVTGTLDYGADLGLKMPAGTLHLAMVQAKVSHANIKGIDTSEALTMPGVHSVITHKDVKGKNRITGLITFPTNKGDGWDRPILXDEKVFQYGDCIALVCADSEANARAAAEKVKVDLEELPAYMSGPAAAAEDAIEIHPGTPNVYFEQPIVKGEDTGPIFASADVTVEGDFYVGRQPHMPIEPDVAFAYMGDDGKCYIHSKSIGVHLHLYMIAPGVGLEPDQLVLVANPMGGTFGYKFSPTSEALVAVAAMATGRPVHLRYNYQQQQQYTGKRSPWEMNVKFAAKKDGTLLAMESDWLVDHGPYSEFGDLLTLRGAQFIGAGYNIPNIRGLGRTVATNHVWGSAFRGYGAPQSMFASECLMDMLAEKLGMDPLELRYKNAYRPGDTNPTGQEPEVFSLPDMIDQLRPKYQAALEKAQKESTATHKKGVGISIGVYGSGLDGPDASEAWAELNADGTITVHTAWEDHGQGADIGCVGTAHEALRPMGVAPEKIKFTWPNTATTPNSGPSGGSRQQVMTGNAIRVACENLLKACEKPGGGYYTYDELKAADKPTKITGNWTASGATHCDAVTGLGKPFVVYMYGVFMAEVTVDVATGQTTVDGMTLMADLGSLCNQLATDGQIYGGLAQGIGLALSEDFEDIKKHATLVGAGFPFIKQIPDKLDIVYVNHPRPDGPFGASGVGELPLTSPHAAIINAIKSATGVRIYRLPAYPEKVLEALKA Hydrogen bonds contact Hydrophobic contact | ||||
| 8 | Thyroid hormone receptor alpha (THRA) | 3ILZ | 6.61 | |
Target general information Gen name THRA Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms V-erbA-related protein 7; THRA2; THRA1; Nuclear receptor subfamily 1 group A member 1; NR1A1; ERBA1; EAR7; EAR-7; C-erbA-alpha; C-erbA-1 Protein family Nuclear hormone receptor family, NR1 subfamily Biochemical class Nuclear hormone receptor Function High affinity receptor for thyroid hormones, including triiodothyronine and thyroxine. Isoform Alpha-1: Nuclear hormone receptor that can act as a repressor or activator of transcription. Related diseases Hypothyroidism, congenital, non-goitrous, 6 (CHNG6) [MIM:614450]: A disease characterized by growth retardation, developmental retardation, skeletal dysplasia, borderline low thyroxine levels and high triiodothyronine levels. There is differential sensitivity to thyroid hormone action, with retention of hormone responsiveness in the hypothalamic pituitary axis and liver but skeletal, gastrointestinal, and myocardial resistance. {ECO:0000269|PubMed:22168587, ECO:0000269|PubMed:24969835, ECO:0000269|PubMed:25670821, ECO:0000269|PubMed:26037512}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01118; DB00509; DB04855; DB05035; DB03176; DB00451; DB00279; DB01583; DB05235; DB09100 Interacts with Q9Y2J4; Q9Y2J4-4; O95971; Q8TAP6; Q96JM7; Q15648; Q6FHY5; P31321; Q96A49; O75410-7; Q9JLI4 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Congenital hypothyroidism; Cytoplasm; Disease variant; DNA-binding; Metal-binding; Nucleus; Proteomics identification; Receptor; Reference proteome; Transcription; Transcription regulation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A Molecular weight (Da) 29910.1 Length 267 Aromaticity 0.07 Instability index 52.75 Isoelectric point 5.31 Charge (pH=7) -11.32 2D Binding mode Binding energy (Kcal/mol) -9.02 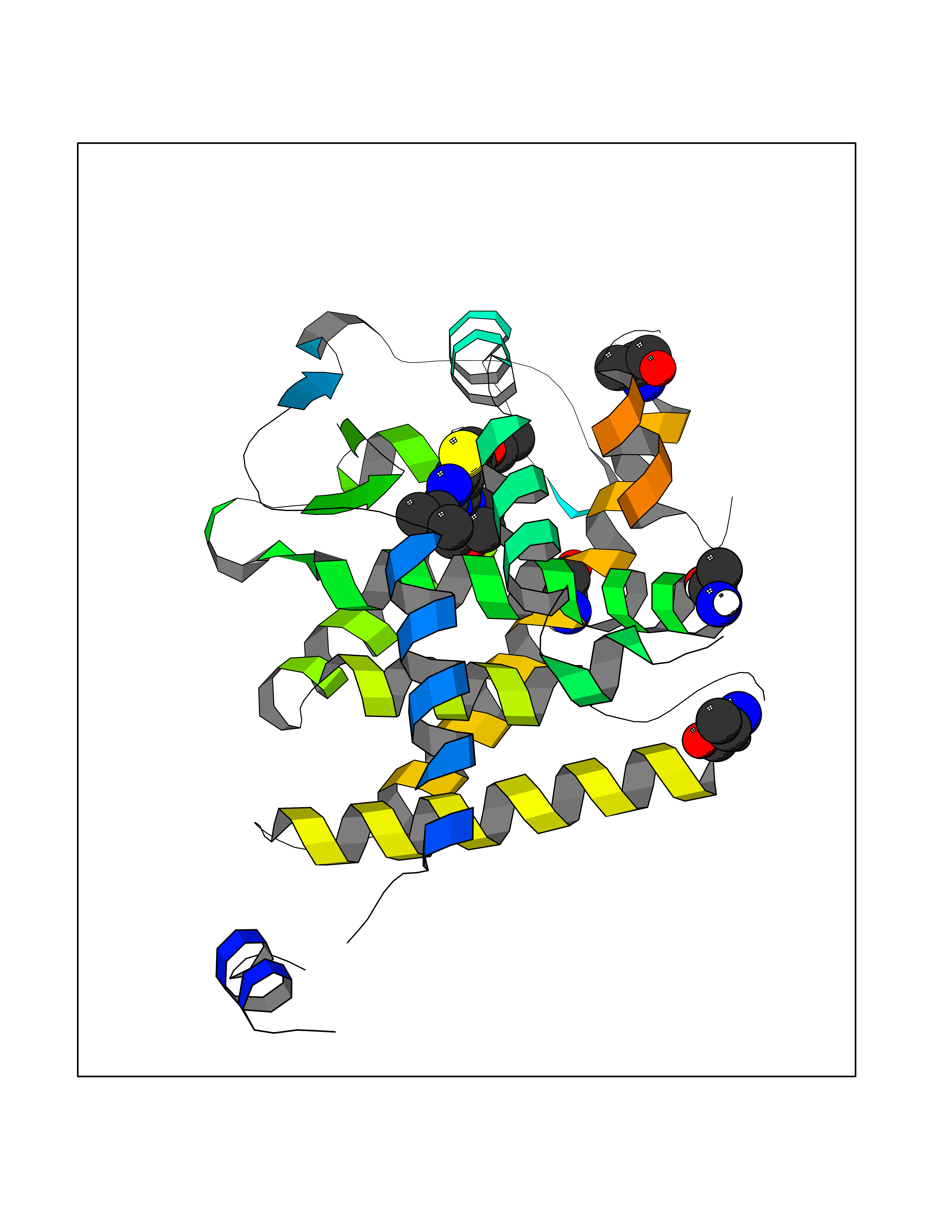 Molscript Map 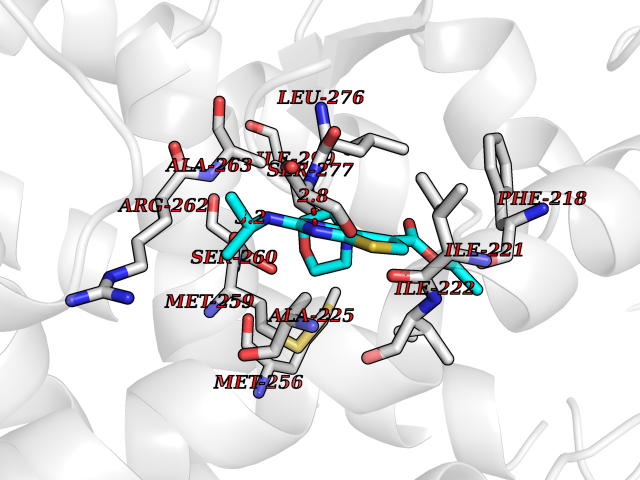 Pymol Map 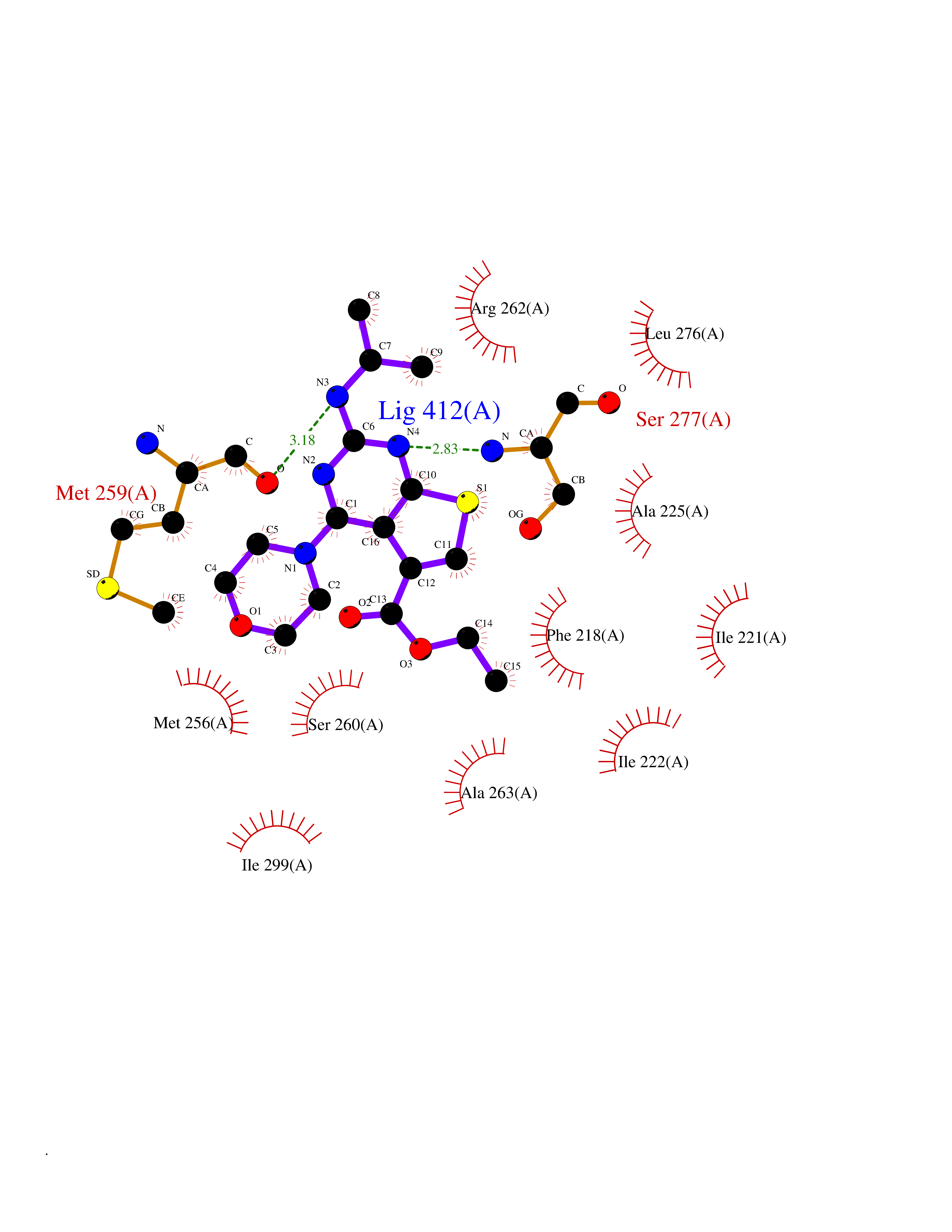 Ligplot Map 3D Binding mode Sequence GSHMEEMIRSLQQRPEPTPEEWDLIHIATEAHRSTNAQGSHWKQRRKFLPDDIGQSPIVSMPDGDKVDLEAFSEFTKIITPAITRVVDFAKKLPMFSELPXEDQIILLKGCCMEIMSLRAAVRYDPESDTLTLSGEMAVKREQLKNGGLGVVSDAIFELGKSLSAFNLDDTEVALLQAVLLMSTDRSGLLXVDKIEKSQEAYLLAFEHYVNHRKHNIPHFWPKLLMKVTDLRMIGAXHASRFLHMKVEXPTELFPPLFLEVFEDQEV Hydrogen bonds contact Hydrophobic contact | ||||
| 9 | Monomeric sarcosine oxidase | 2GF3 | 6.61 | |
Target general information Gen name soxA Organism Bacillus sp. (strain B-0618) Uniprot ID TTD ID NA Synonyms sox Protein family MSOX/MTOX family, MSOX subfamily Biochemical class Oxidoreductase Function Sarcosine oxidase activity. Related diseases Defects in PPARG can lead to type 2 insulin-resistant diabetes and hyptertension. PPARG mutations may be associated with colon cancer. {ECO:0000269|PubMed:10394368}.; DISEASE: Obesity (OBESITY) [MIM:601665]: A condition characterized by an increase of body weight beyond the limitation of skeletal and physical requirements, as the result of excessive accumulation of body fat. {ECO:0000269|PubMed:9753710}. Disease susceptibility may be associated with variants affecting the gene represented in this entry.; DISEASE: Lipodystrophy, familial partial, 3 (FPLD3) [MIM:604367]: A form of lipodystrophy characterized by marked loss of subcutaneous fat from the extremities. Facial adipose tissue may be increased, decreased or normal. Affected individuals show an increased preponderance of insulin resistance, diabetes mellitus and dyslipidemia. {ECO:0000269|PubMed:11788685, ECO:0000269|PubMed:12453919}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Glioma 1 (GLM1) [MIM:137800]: Gliomas are benign or malignant central nervous system neoplasms derived from glial cells. They comprise astrocytomas and glioblastoma multiforme that are derived from astrocytes, oligodendrogliomas derived from oligodendrocytes and ependymomas derived from ependymocytes. {ECO:0000269|PubMed:10851250}. Disease susceptibility may be associated with variants affecting the gene represented in this entry. Polymorphic PPARG alleles have been found to be significantly over-represented among a cohort of American patients with sporadic glioblastoma multiforme suggesting a possible contribution to disease susceptibility. Drugs (DrugBank ID) DB03098; DB01918; DB03517; DB03147; DB03366; DB02083; DB02543 Interacts with NA EC number 1.5.3.1 Uniprot keywords 3D-structure; Cytoplasm; Direct protein sequencing; FAD; Flavoprotein; Oxidoreductase Protein physicochemical properties Chain ID A,B Molecular weight (Da) 42606.4 Length 385 Aromaticity 0.1 Instability index 26.97 Isoelectric point 5.27 Charge (pH=7) -17.18 2D Binding mode Binding energy (Kcal/mol) -9.02  Molscript Map  Pymol Map 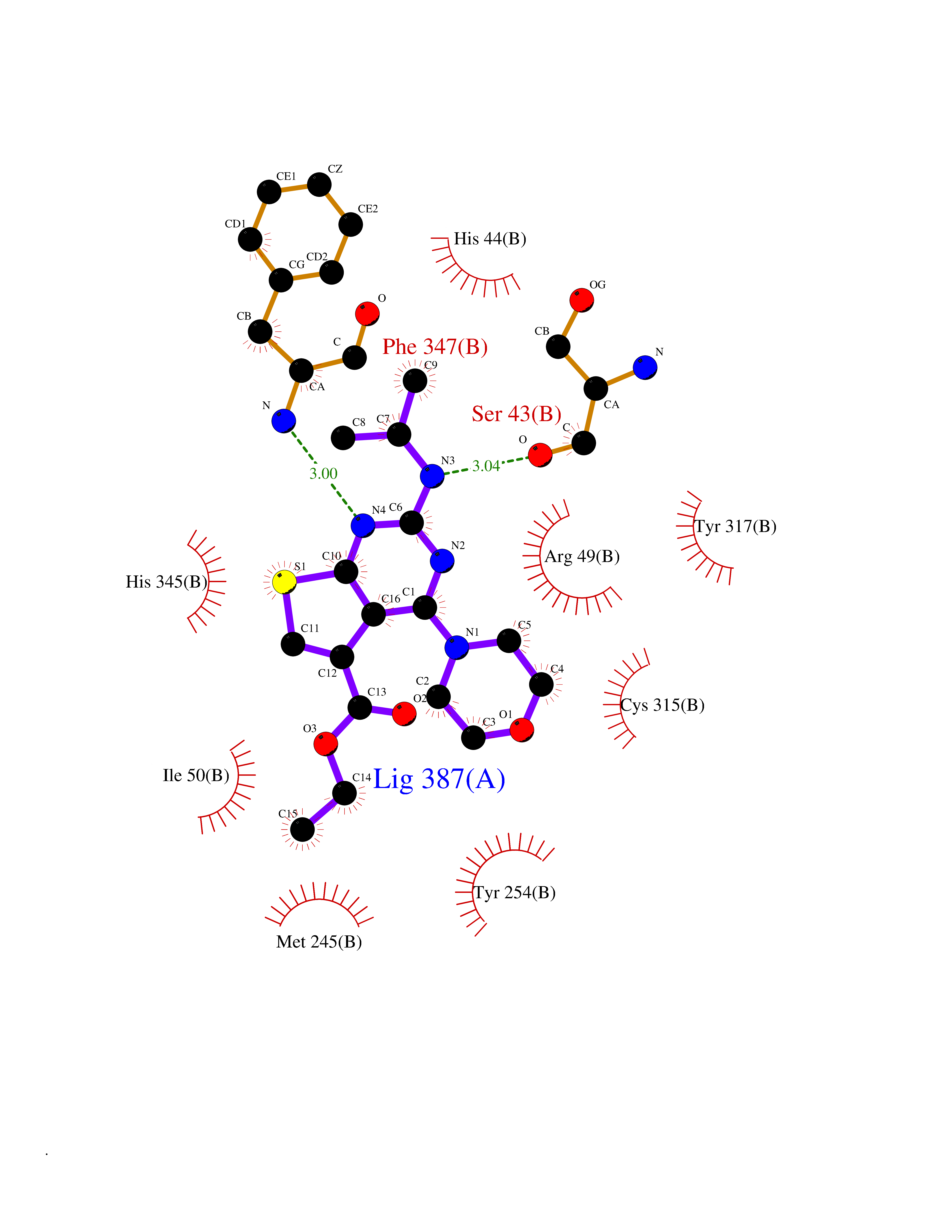 Ligplot Map 3D Binding mode Sequence STHFDVIVVGAGSMGMAAGYQLAKQGVKTLLVDAFDPPHTNGSHHGDTRIIRHAYGEGREYVPLALRSQELWYELEKETHHKIFTKTGVLVFGPKGESAFVAETMEAAKEHSLTVDLLEGDEINKRWPGITVPENYNAIFEPNSGVLFSENCIRAYRELAEARGAKVLTHTRVEDFDISPDSVKIETANGSYTADKLIVSMGAWNSKLLSKLNLDIPLQPYRQVVGFFESDESKYSNDIDFPGFMVEVPNGIYYGFPSFGGCGLKLGYHTFGQKIDPDTINREFGVYPEDESNLRAFLEEYMPGANGELKRGAVCMYTKTLDEHFIIDLHPEHSNVVIAAGFSGHGFKFSSGVGEVLSQLALTGKTEHDISIFSINRPALKESLQ Hydrogen bonds contact Hydrophobic contact | ||||
| 10 | Cytochrome c oxidase subunit 2 | 3VRJ | 6.58 | |
Target general information Gen name MT-CO2 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms MTCO2;COXII;COII;COX2 Protein family Cytochrome c oxidase subunit 2 family Biochemical class Immune system Function Copper ion binding.Cytochrome-c oxidase activity. Related diseases Mitochondrial complex IV deficiency (MT-C4D) [MIM:220110]: A disorder of the mitochondrial respiratory chain with heterogeneous clinical manifestations, ranging from isolated myopathy to severe multisystem disease affecting several tissues and organs. Features include hypertrophic cardiomyopathy, hepatomegaly and liver dysfunction, hypotonia, muscle weakness, exercise intolerance, developmental delay, delayed motor development and intellectual disability. Some affected individuals manifest a fatal hypertrophic cardiomyopathy resulting in neonatal death. A subset of patients manifest Leigh syndrome. {ECO:0000269|PubMed:10486321}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02659; DB04464; DB05412 Interacts with Q9NZ94-2; P49281-3 EC number 7.1.1.9 Uniprot keywords 3D-structure; Copper; Disease variant; Electron transport; Magnesium; Membrane; Metal-binding; Mitochondrion; Mitochondrion inner membrane; Primary mitochondrial disease; Proteomics identification; Reference proteome; Respiratory chain; Translocase; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID C Molecular weight (Da) 21687.9 Length 189 Aromaticity 0.11 Instability index 38 Isoelectric point 5.68 Charge (pH=7) -3.26 2D Binding mode Binding energy (Kcal/mol) -8.97  Molscript Map 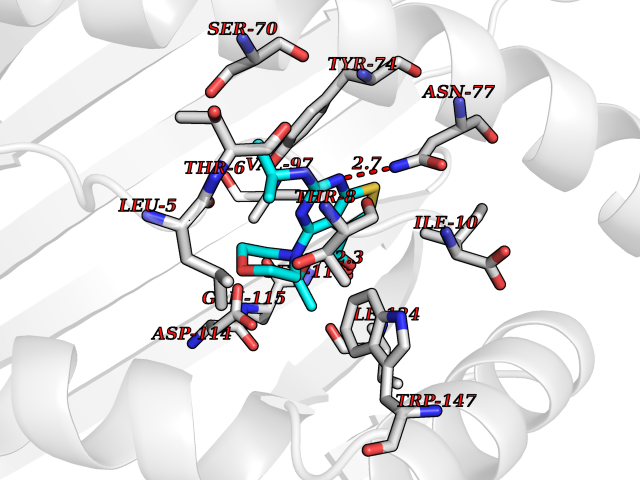 Pymol Map 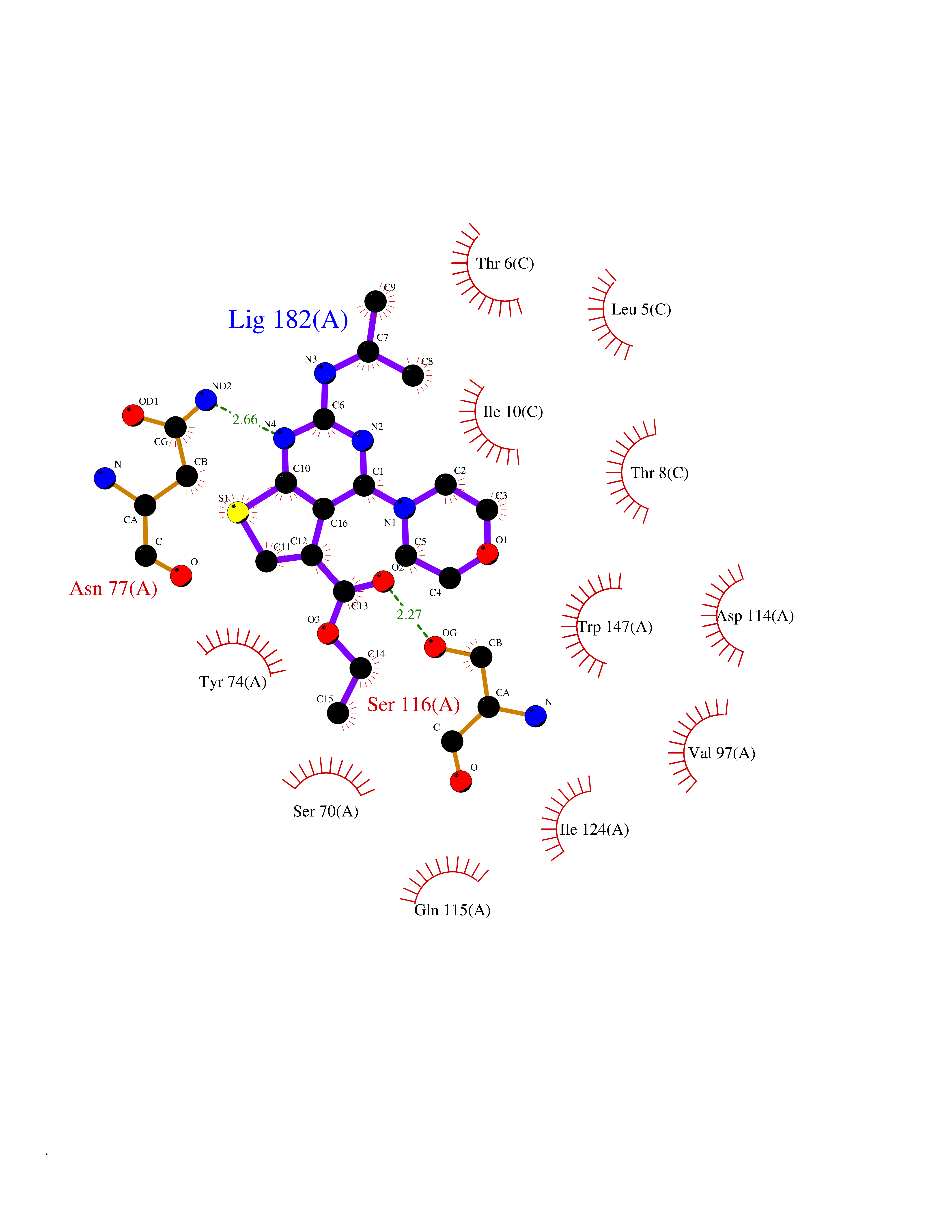 Ligplot Map 3D Binding mode Sequence SHSMRYFYTAMSRPGRGEPRFIAVGYVDDTQFVRFDSDAASPRMAPRAPWIEQEGPEYWDGETRNMKASAQTYRENLRIALRYYNQSEAGSHIIQVMYGCDVGPDGRLLRGHDQSAYDGKDYIALNEDLSSWTAADTAAQITQRKWEAARVAEQLRAYLEGLCVEWLRRYLENGKETLQLTTKLTNTNI Hydrogen bonds contact Hydrophobic contact | ||||
| 11 | Cytoplasmic aspartate aminotransferase (GOT1) | 3II0 | 6.55 | |
Target general information Gen name GOT1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Glutamate oxaloacetate transaminase-1; GOT1 Protein family Class-I pyridoxal-phosphate-dependent aminotransferase family Biochemical class Transaminase Function Biosynthesis of L-glutamate from L-aspartate or L- cysteine. Important regulator of levels of glutamate, the major excitatory neurotransmitter of the vertebrate central nervous system. Acts as a scavenger of glutamate in brain neuroprotection. The aspartate aminotransferase activity is involved in hepatic glucose synthesis during development and in adipocyte glyceroneogenesis. Using L-cysteine as substrate, regulates levels of mercaptopyruvate, an important source of hydrogen sulfide. Mercaptopyruvate is converted into H(2)S via the action of 3- mercaptopyruvate sulfurtransferase (3MST). Hydrogen sulfide is an important synaptic modulator and neuroprotectant in the brain. Related diseases Multiple fibroadenomas of the breast (MFAB) [MIM:615554]: A benign breast disease marked by lobuloalveolar growth with abnormally high proliferation of the epithelium, and characterized by the presence of more than 3 fibroadenomas in one breast. Fibroadenomas are adenomas containing fibrous tissue. {ECO:0000269|PubMed:18779591}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Hyperprolactinemia (HPRL) [MIM:615555]: A disorder characterized by increased levels of prolactin in the blood not associated with gestation or the puerperium. HPRL may result in infertility, hypogonadism, and galactorrhea. {ECO:0000269|PubMed:24195502}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00210; DB00128; DB09130; DB00151; DB00142; DB04299; DB00114 Interacts with NA EC number EC 2.6.1.1 Uniprot keywords 3D-structure; Alternative splicing; Amino-acid biosynthesis; Aminotransferase; Cytoplasm; Direct protein sequencing; Phosphoprotein; Proteomics identification; Pyridoxal phosphate; Reference proteome; Transferase Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 51732.3 Length 462 Aromaticity 0.1 Instability index 31.83 Isoelectric point 7.48 Charge (pH=7) 0.86 2D Binding mode Binding energy (Kcal/mol) -8.93 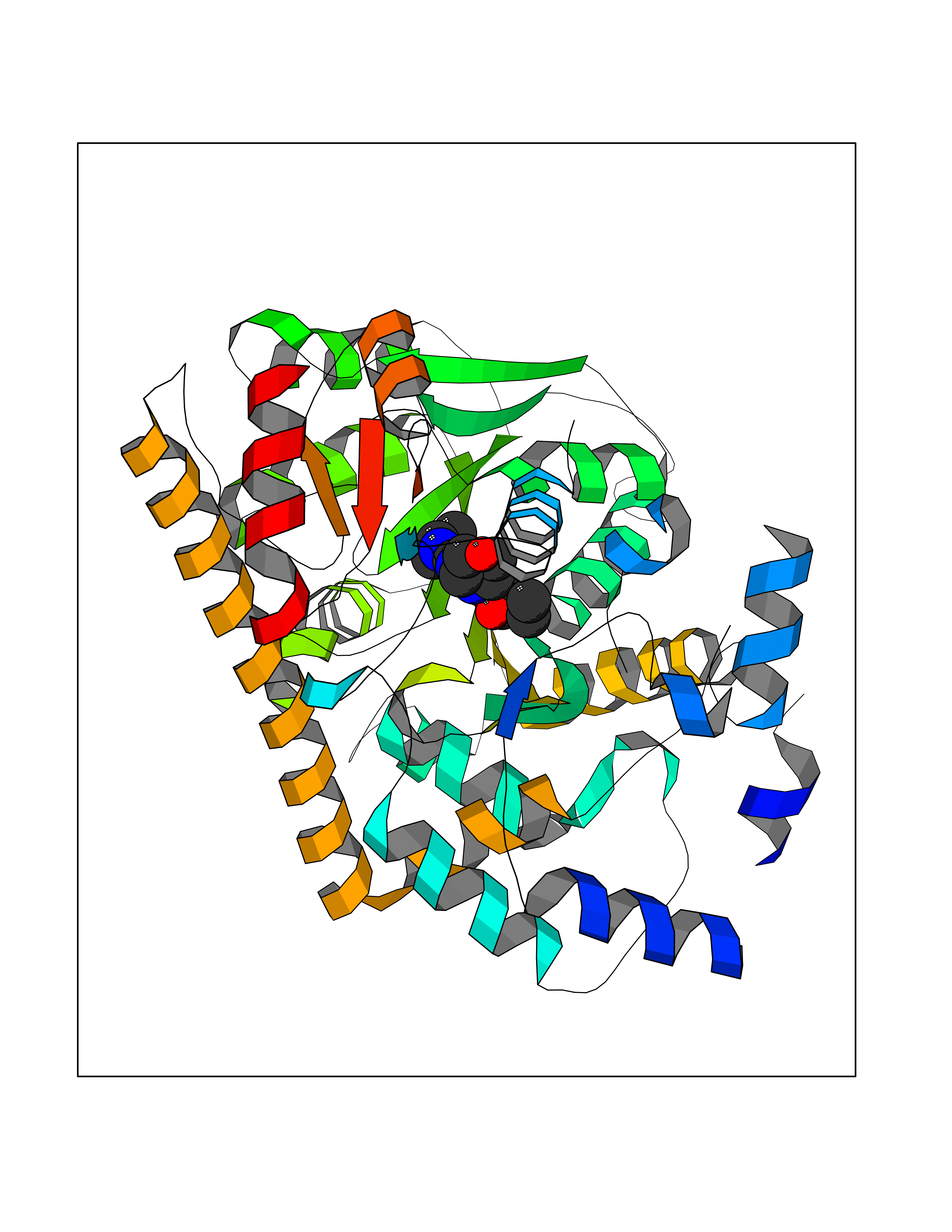 Molscript Map 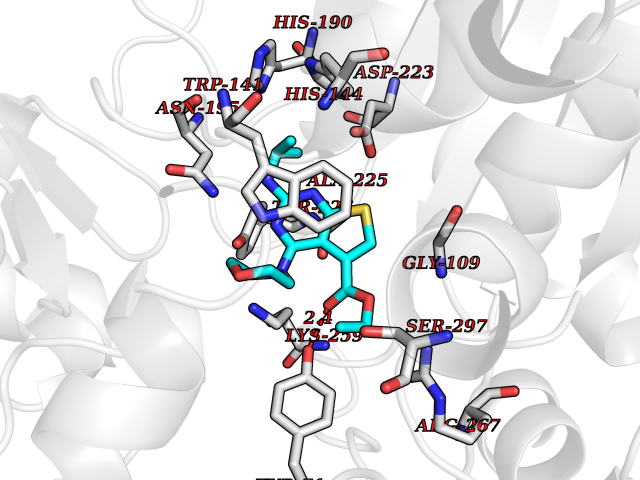 Pymol Map 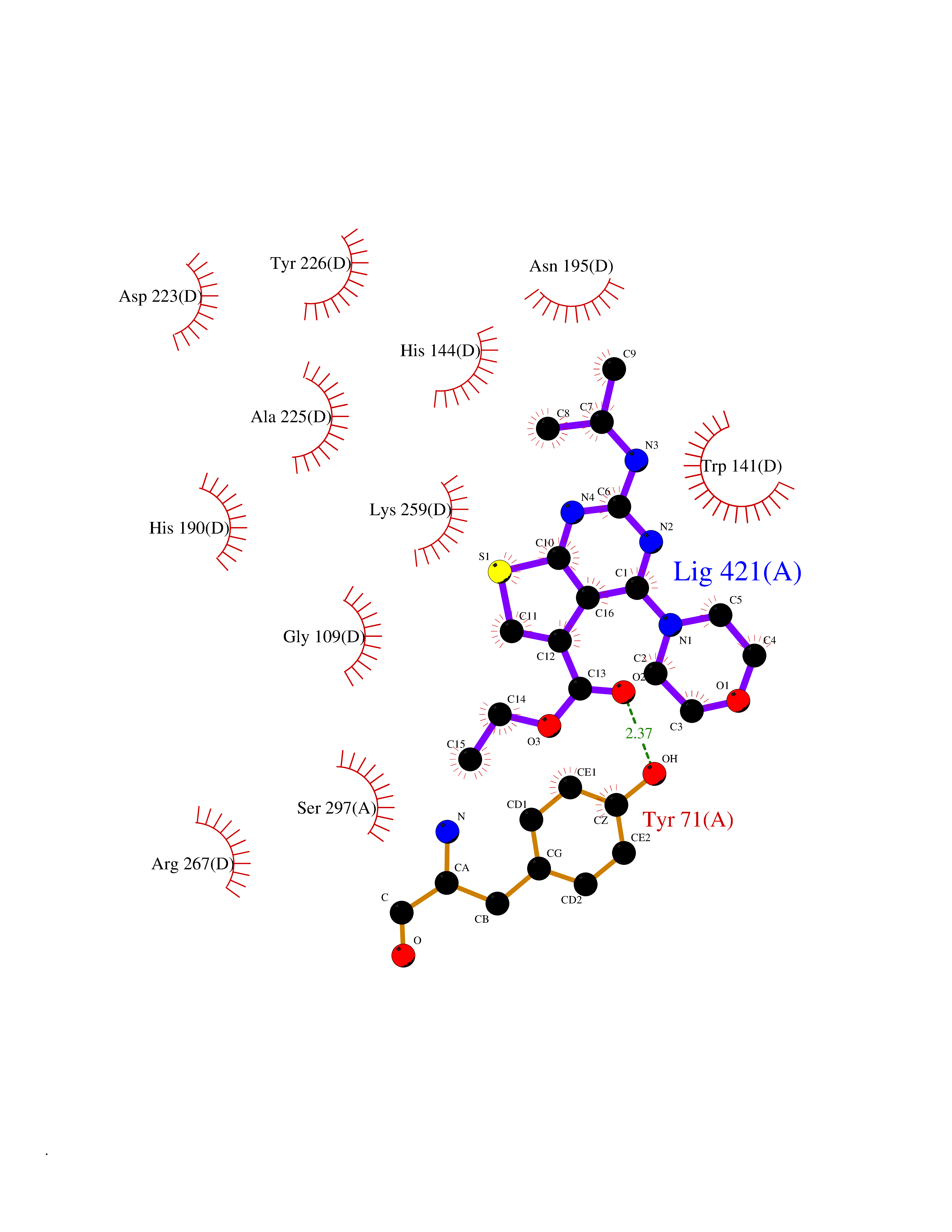 Ligplot Map 3D Binding mode Sequence PVLVFKLTVLPVVKKVEQKIANDNSLNHEYLPILGLAEFRVQSLGGTGALRIGADEKIVRITWSNPMQPVLVFKLTADFREDPDPRKVNLGVGAYRTDDCHPWVLPVVKKVEQKIANDNSLNHEYLPILGLAEFRSCASRLALGDDSPALKEKRVGGVQSLGGTGALRIGADFLARWYNGTNNKNTPVYVSSPTWENHNAVFSAAGFKDIRSYRYWDAEKRGLDLQGFLNDLENAPEFSIVVLHACAHNPTGIDPTPEQWKQIASVMKHRFLFPFFDSAYQGFASGNLERDAWAIRYFVSEGFEFFCAQSFSKNFGLYNERVGNLTVVGKEPESILQVLSQMEKIVRITWSNPPAQGARIVASTLSNPELFEEWTGNVKTMADRILTMRSELRARLEALKTPGTWNHITDQIGMFSFTGLNPKQVEYLVNEKHIYLLPSGRINVSGLTTKNLDYVATSIHEA Hydrogen bonds contact Hydrophobic contact | ||||
| 12 | Metabotropic glutamate receptor 3 (mGluR3) | 4XAR | 6.55 | |
Target general information Gen name GRM3 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms mGLUR3; Group III metabotropic glutamate receptor; GPRC1C Protein family G-protein coupled receptor 3 family Biochemical class GPCR glutamate Function Ligand binding causes a conformation change that triggers signaling via guanine nucleotide-binding proteins (G proteins) and modulates the activity of down-stream effectors. Signaling inhibits adenylate cyclase activity. G-protein coupled receptor for glutamate. Related diseases Paramyotonia congenita (PMC) [MIM:168300]: An autosomal dominant channelopathy characterized by myotonia, increased by exposure to cold, intermittent flaccid paresis, not necessarily dependent on cold or myotonia, lability of serum potassium, non-progressive nature and lack of atrophy or hypertrophy of muscles. In some patients, myotonia is not increased by cold exposure (paramyotonia without cold paralysis). Patients may have a combination phenotype of PMC and HYPP. {ECO:0000269|PubMed:10369308, ECO:0000269|PubMed:10727489, ECO:0000269|PubMed:1310898, ECO:0000269|PubMed:1316765, ECO:0000269|PubMed:1338909, ECO:0000269|PubMed:15318338, ECO:0000269|PubMed:15790667, ECO:0000269|PubMed:16786525, ECO:0000269|PubMed:18166706, ECO:0000269|PubMed:18690054, ECO:0000269|PubMed:19077043, ECO:0000269|PubMed:20076800, ECO:0000269|PubMed:8242056, ECO:0000269|PubMed:8308722, ECO:0000269|PubMed:8388676, ECO:0000269|PubMed:8580427}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Periodic paralysis hypokalemic 2 (HOKPP2) [MIM:613345]: An autosomal dominant disorder manifested by episodic flaccid generalized muscle weakness associated with falls of serum potassium levels. {ECO:0000269|PubMed:10599760, ECO:0000269|PubMed:10851391, ECO:0000269|PubMed:10944223, ECO:0000269|PubMed:11558801, ECO:0000269|PubMed:11591859, ECO:0000269|PubMed:16890191, ECO:0000269|PubMed:17898326, ECO:0000269|PubMed:18162704, ECO:0000269|PubMed:19118277, ECO:0000269|PubMed:20522878, ECO:0000269|PubMed:21043388, ECO:0000269|PubMed:24549961}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Periodic paralysis hyperkalemic (HYPP) [MIM:170500]: An autosomal dominant channelopathy characterized by episodic flaccid generalized muscle weakness associated with high levels of serum potassium. Concurrence of myotonia is found in HYPP patients. {ECO:0000269|PubMed:1659668, ECO:0000269|PubMed:1659948, ECO:0000269|PubMed:20076800}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Periodic paralysis normokalemic (NKPP) [MIM:170500]: A disorder closely related to hyperkalemic periodic paralysis, but marked by a lack of alterations in potassium levels during attacks of muscle weakness. {ECO:0000269|PubMed:15596759, ECO:0000269|PubMed:18046642, ECO:0000269|PubMed:20522878}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Myotonia SCN4A-related (MYOSCN4A) [MIM:608390]: A phenotypically highly variable myotonia aggravated by potassium loading, and sometimes by cold. Myotonia is characterized by sustained muscle tensing that prevents muscles from relaxing normally. It causes muscle stiffness that can interfere with movement. In some people the stiffness is very mild, while in other cases it may be severe enough to interfere with walking, running, and other activities of daily life. Myotonia SCN4A-related includes myotonia permanens and myotonia fluctuans. In myotonia permanens, the myotonia is generalized and there is a hypertrophy of the muscle, particularly in the neck and the shoulder. Attacks of severe muscle stiffness of the thoracic muscles may be life threatening due to impaired ventilation. In myotonia fluctuans, the muscle stiffness may fluctuate from day to day, provoked by exercise. {ECO:0000269|PubMed:10218481, ECO:0000269|PubMed:16786525, ECO:0000269|PubMed:16832098, ECO:0000269|PubMed:17212350, ECO:0000269|PubMed:17998485, ECO:0000269|PubMed:18203179, ECO:0000269|PubMed:18337100, ECO:0000269|PubMed:19015483, ECO:0000269|PubMed:19347921, ECO:0000269|PubMed:20076800, ECO:0000269|PubMed:27653901, ECO:0000269|PubMed:8058156, ECO:0000269|PubMed:9392583}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Myasthenic syndrome, congenital, 16 (CMS16) [MIM:614198]: A form of congenital myasthenic syndrome, a group of disorders characterized by failure of neuromuscular transmission, including pre-synaptic, synaptic, and post-synaptic disorders that are not of autoimmune origin. Clinical features are easy fatigability and muscle weakness. CMS16 is characterized by fatigable generalized weakness and recurrent attacks of respiratory and bulbar paralysis since birth. The fatigable weakness involves lid-elevator, external ocular, facial, limb and truncal muscles and an decremental response of the compound muscle action potential on repetitive stimulation. {ECO:0000269|PubMed:12766226, ECO:0000269|PubMed:25707578, ECO:0000269|PubMed:26659129}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Congenital myopathy 22A, classic (CMYO22A) [MIM:620351]: A form of congenital myopathy, a clinically and genetically heterogeneous group of muscle disorders characterized by hypotonia and muscle weakness apparent at birth, and specific pathological features on muscle biopsy. CMYO22A is an autosomal recessive form characterized by fetal hypokinesia, polyhydramnios, and severe neonatal hypotonia associated with respiratory insufficiency. Affected individuals who survive the neonatal period have delayed motor development, difficulty walking, proximal muscle weakness of the upper and lower limbs, facial and neck muscle weakness, easy fatigability, and mild limb contractures or foot deformities. {ECO:0000269|PubMed:26700687, ECO:0000269|PubMed:28262468, ECO:0000269|PubMed:36090556}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Congenital myopathy 22B, severe fetal (CMYO22B) [MIM:620369]: A severe congenital myopathy, a clinically and genetically heterogeneous group of muscle disorders characterized by hypotonia and muscle weakness apparent at birth, and specific pathological features on muscle biopsy. CMYO22B is an autosomal recessive form characterized by onset in utero. Affected individuals show fetal akinesia, and develop fetal hydrops with pulmonary hypoplasia, severe joint contractures, and generalized muscle hypoplasia. Death occurs in utero or soon after birth. {ECO:0000269|PubMed:26700687}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB05096 Interacts with NA EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disulfide bond; G-protein coupled receptor; Glycoprotein; Membrane; Proteomics identification; Receptor; Reference proteome; Signal; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 50355.5 Length 445 Aromaticity 0.11 Instability index 38.26 Isoelectric point 6.52 Charge (pH=7) -1.53 2D Binding mode Binding energy (Kcal/mol) -8.94  Molscript Map 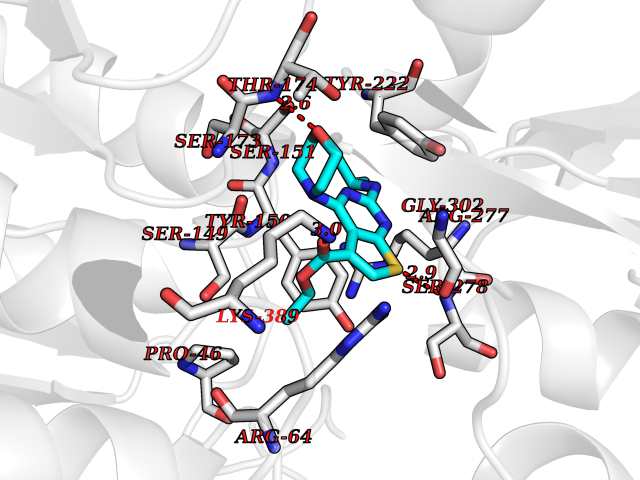 Pymol Map 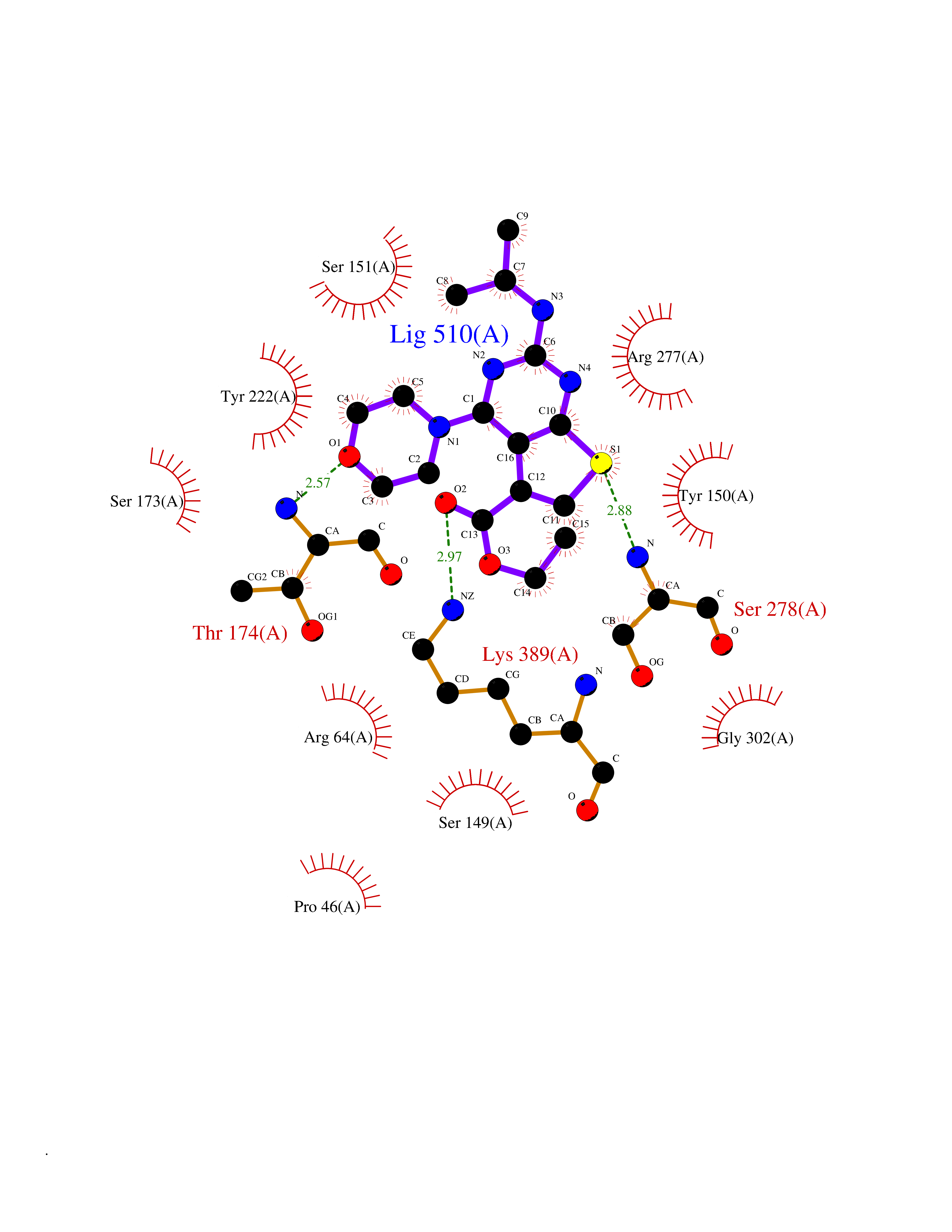 Ligplot Map 3D Binding mode Sequence RREIKIEGDLVLGGLFPINEKGTGTEECGRINEDRGIQRLEAMLFAIDEINKDDYLLPGVKLGVHILDTCSRDTYALEQSLEFVRASLLLIAGVIGGSYSSVSIQVANLLRLFQIPQISYASTSAKLSDKSRYDYFARTVPPDFYQAKAMAEILRFFNWTYVSTVASEGDYGETGIEAFEQEARLRNISIATAEKVGRSNIRKSYDSVIRELLQKPNARVVVLFMRSDDSRELIAAASRANASFTWVASDGWGAQESIIKGSEHVAYGAITLELASQPVRQFDRYFQSLNPYNNHRNPWFRDFWEQKFQCSLRVCDKHLAIDSSNYEQESKIMFVVNAVYAMAHALHKMQRTLCPNTTKLCDAMKILDGKKLYKDYLLKINFTAPDADSIVKFDTFGDGMGRYNVFNFQNVGGKYSYLKVGHWAETLSLDVNSIHWSRNSVPTSE Hydrogen bonds contact Hydrophobic contact | ||||
| 13 | Gamma-butyrobetaine dioxygenase | 4C5W | 6.53 | |
Target general information Gen name BBOX1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms BBH;BBOX Protein family Gamma-BBH/TMLD family Biochemical class Oxidoreductase Function Gamma-butyrobetaine dioxygenase activity.Identical protein binding.Iron ion binding.Zinc ion binding. Related diseases Neurodevelopmental disorder with poor language and loss of hand skills (NDPLHS) [MIM:617903]: An autosomal dominant disorder characterized by psychomotor developmental stagnation or regression. NDPLHS manifest in the first years of life as loss of purposeful hand movements, loss of language, and intellectual disability. {ECO:0000269|PubMed:26740508, ECO:0000269|PubMed:28856709, ECO:0000269|PubMed:29369404}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Developmental and epileptic encephalopathy 59 (DEE59) [MIM:617904]: A form of epileptic encephalopathy, a heterogeneous group of severe early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE59 is an autosomal dominant condition characterized by onset of refractory seizures in early infancy. {ECO:0000269|PubMed:28856709, ECO:0000269|PubMed:29100083, ECO:0000269|PubMed:29369404}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00126 Interacts with O75936; A0MZ66-7 EC number 1.14.11.1 Uniprot keywords 3D-structure; Carnitine biosynthesis; Cytoplasm; Dioxygenase; Iron; Metal-binding; Oxidoreductase; Phosphoprotein; Proteomics identification; Reference proteome; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 31642.5 Length 275 Aromaticity 0.12 Instability index 35.68 Isoelectric point 6.33 Charge (pH=7) -2.46 2D Binding mode Binding energy (Kcal/mol) -8.91  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence FPECQYWGSELQLPTLDFEDVLRYDEHAYKWLSTLKKVGIVRLTGASDKPGEVSKLGKRMGFLYLTFYGHTWQVQDKIDANNVAYTTGKLSFHTDYPALHHPPGVQLLHCIKQTVTGGDSEIVDGFNVCQKLKKNNPQAFQILSSTFVDFTDIGVDYCDFSVQSKHKIIELDDKGQVVRINFNNATRDTIFDVPVERVQPFYAALKEFVDLMNSKESKFTFKMNPGDVITFDNWRLLHGRRSYEAGTEISRHLEGAYADWDVVMSRLRILRQRVE Hydrogen bonds contact Hydrophobic contact | ||||
| 14 | Fumarate reductase flavoprotein subunit | 1KF6 | 6.53 | |
Target general information Gen name frdA Organism Escherichia coli (strain K12) Uniprot ID TTD ID NA Synonyms JW4115;b4154 Protein family FAD-dependent oxidoreductase 2 family, FRD/SDH subfamily Biochemical class Oxidoreductase Function Electron carrier activity.FAD binding.Fumarate reductase (menaquinone).Succinate dehydrogenase activity. Related diseases Glycogen storage disease 11 (GSD11) [MIM:612933]: A metabolic disorder that results in exertional myoglobinuria, pain, cramps and easy fatigue. {ECO:0000269|PubMed:2334430}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07490; DB07918; DB00730 Interacts with P0AC47; P0ACB4; P76111 EC number 1.3.5.1 Uniprot keywords 3D-structure; Cell inner membrane; Cell membrane; Direct protein sequencing; Electron transport; FAD; Flavoprotein; Membrane; Nucleotide-binding; Oxidoreductase; Reference proteome; Transport Protein physicochemical properties Chain ID A,M Molecular weight (Da) 90370.7 Length 820 Aromaticity 0.08 Instability index 28.88 Isoelectric point 5.86 Charge (pH=7) -16.21 2D Binding mode Binding energy (Kcal/mol) -8.91  Molscript Map 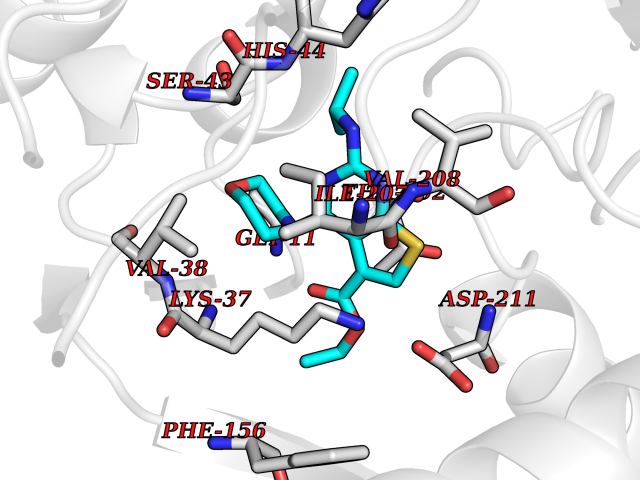 Pymol Map 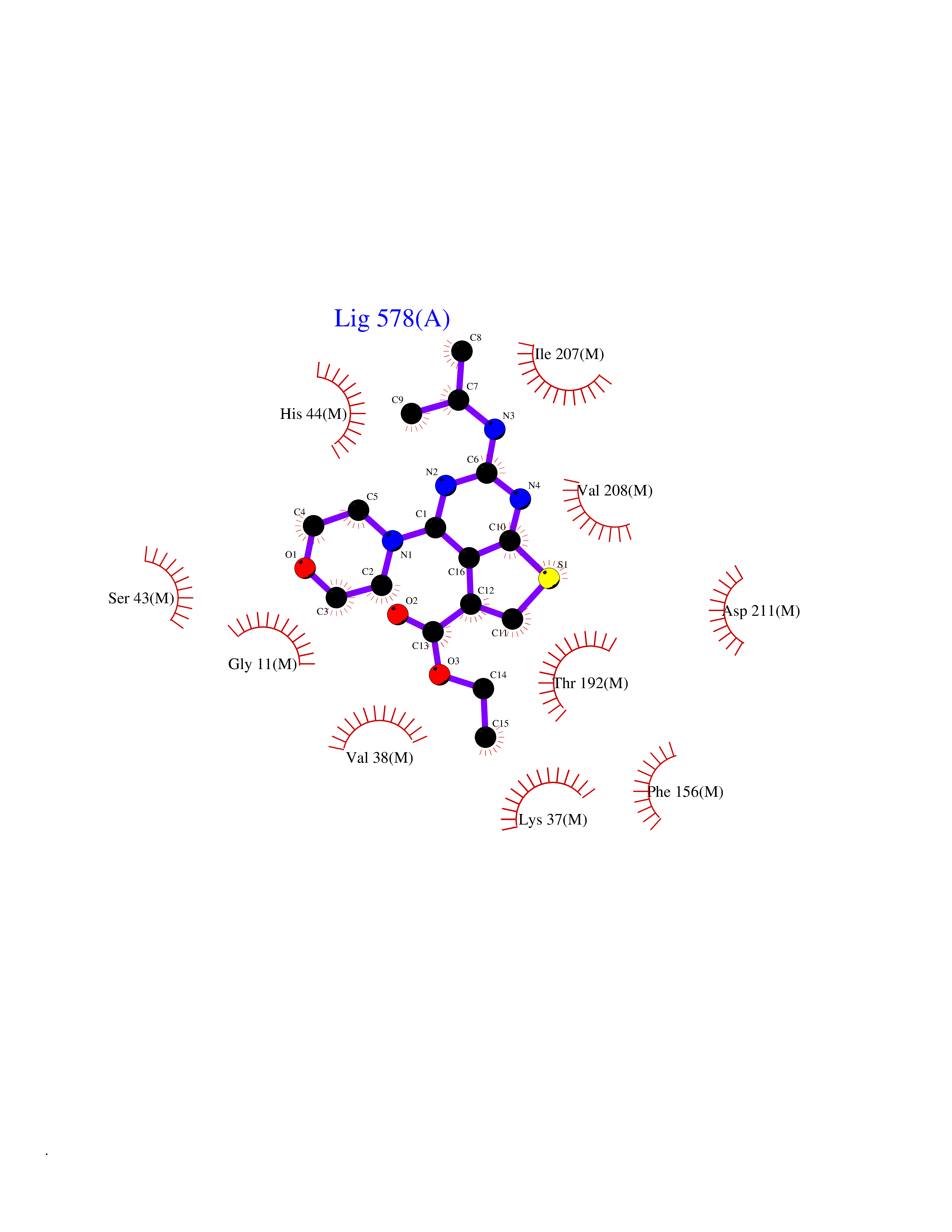 Ligplot Map 3D Binding mode Sequence MQTFQADLAIVGAGGAGLRAAIAAAQANPNAKIALISKVYPMRSHTVAAEGGSAAVAQDHDSFEYHFHDTVAGGDWLCEQDVVDYFVHHCPTEMTQLELWGCPWSRRPDGSVNVRRFGGMKIERTWFAADKTGFHMLHTLFQTSLQFPQIQRFDEHFVLDILVDDGHVRGLVAMNMMEGTLVQIRANAVVMATGGAGRVYRYNTNGGIVTGDGMGMALSHGVPLRDMEFVQYHPTGLPGSGILMTEGCRGEGGILVNKNGYRYLQDYGMGPETPLGEPKNKYMELGPRDKVSQAFWHEWRKGNTISTPRGDVVYLDLRHLGEKKLHERLPFICELAKAYVGVDPVKEPIPVRPTAHYTMGGIETDQNCETRIKGLFAVGECSSVGLHGANRLGSNSLAELVVFGRLAGEQATERAATAGNGNEAAIEAQAAGVEQRLKDLVNQDGGENWAKIRDEMGLAMEEGCGIYRTPELMQKTIDKLAELQERFKRVRITDTSSVFNTDLLYTIELGHGLNVAECMAHSAMARKESRGAHQRLDEGCTERDDVNFLKHTLAFRDADGTTRLEYSDVKITTLPPAAEMKNLKIEVVRYNPEVDTAPHSAFYEVPYDATTSLLDALGYIKDNLAPDLSYRWSCRMAICGSCGMMVNNVPKLACKTFLRDYTDGMKVEALANFPIERDLVVDMTHFIESLEAIKPYIIGNSRTADQGTNIQTPAQMAKYHQFSGCINCGLCYAACPQFGLNPEFIGPAAITLAHRYNEDSRDHGKKERMAQLNSQNGVWSCTFVGYCSEVCPKHVDPAAAIQQGKVESSKDFLIATLKPR Hydrogen bonds contact Hydrophobic contact | ||||
| 15 | Histidine decarboxylase (HDC) | 4E1O | 6.53 | |
Target general information Gen name HDC Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Human histidine decarboxylase Protein family Group II decarboxylase family Biochemical class Carbon-carbon lyase Function Catalyzes the biosynthesis of histamine from histidine. Related diseases Corticosterone methyloxidase 1 deficiency (CMO-1 deficiency) [MIM:203400]: Autosomal recessive disorder of aldosterone biosynthesis. There are two biochemically different forms of selective aldosterone deficiency be termed corticosterone methyloxidase (CMO) deficiency type 1 and type 2. In CMO-1 deficiency, aldosterone is undetectable in plasma, while its immediate precursor, 18-hydroxycorticosterone, is low or normal. {ECO:0000269|PubMed:11238478, ECO:0000269|PubMed:8439335, ECO:0000269|PubMed:9177280}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Corticosterone methyloxidase 2 deficiency (CMO-2 deficiency) [MIM:610600]: Autosomal recessive disorder of aldosterone biosynthesis. In CMO-2 deficiency, aldosterone can be low or normal, but at the expense of increased secretion of 18-hydroxycorticosterone. Consequently, patients have a greatly increased ratio of 18-hydroxycorticosterone to aldosterone and a low ratio of corticosterone to 18-hydroxycorticosterone in serum. {ECO:0000269|PubMed:12788848, ECO:0000269|PubMed:1346492, ECO:0000269|PubMed:1594605, ECO:0000269|PubMed:9625333, ECO:0000269|PubMed:9814506}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00117; DB00114 Interacts with Q86UW9 EC number EC 4.1.1.22 Uniprot keywords 3D-structure; Alternative splicing; Catecholamine biosynthesis; Decarboxylase; Lyase; Proteomics identification; Pyridoxal phosphate; Reference proteome Protein physicochemical properties Chain ID A,B,C,D,E,F Molecular weight (Da) 107706 Length 956 Aromaticity 0.1 Instability index 55.17 Isoelectric point 6.23 Charge (pH=7) -9.63 2D Binding mode Binding energy (Kcal/mol) -8.91  Molscript Map 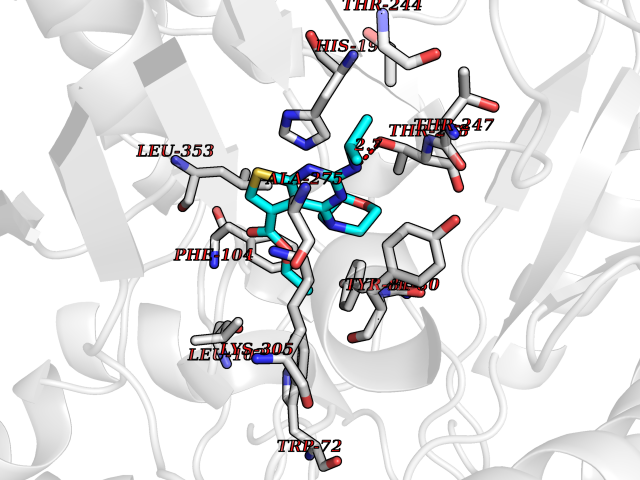 Pymol Map 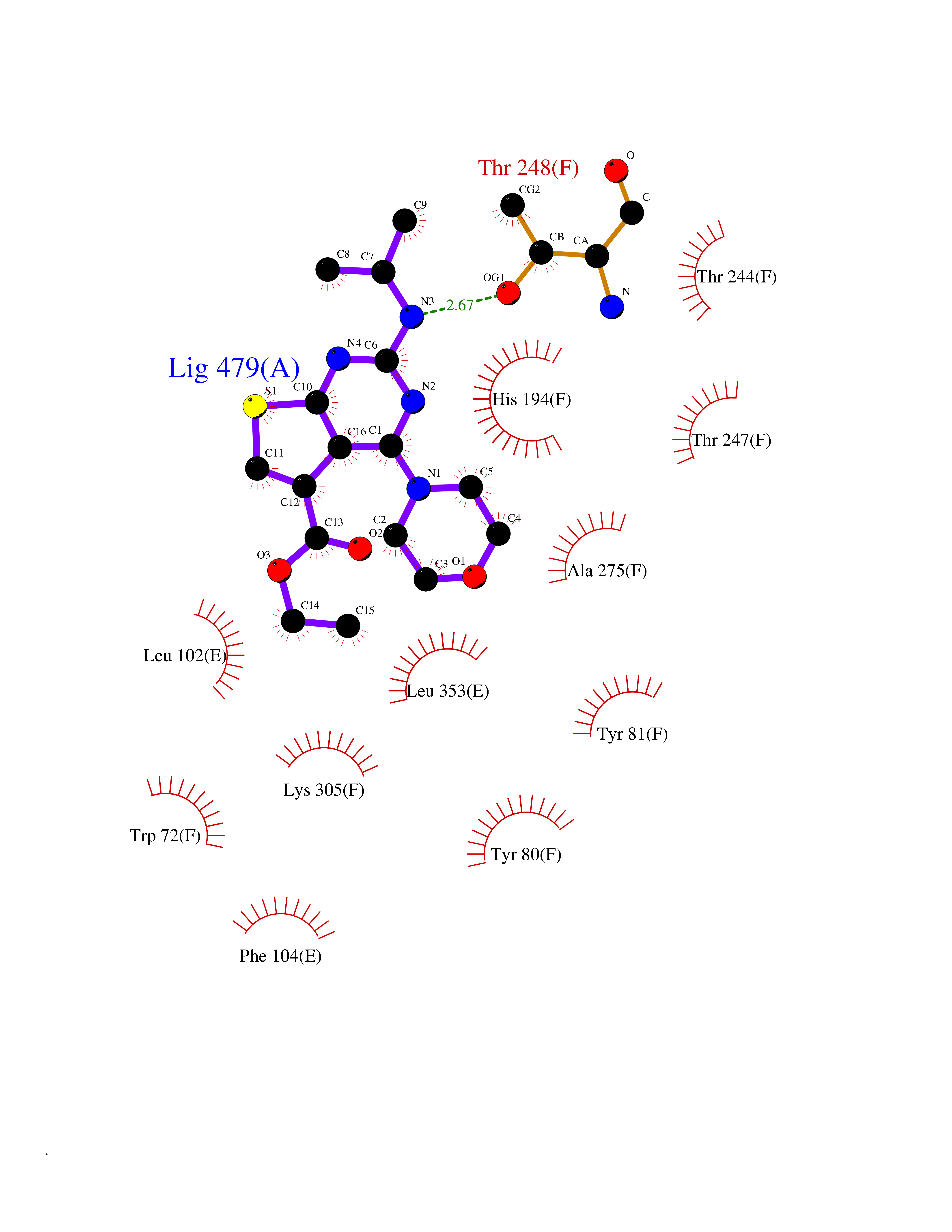 Ligplot Map 3D Binding mode Sequence GSMEPEEYRERGREMVDYICQYLSTVRERRVTPDVQPGYLRAQLPESAPEDPDSWDSIFGDIERIIMPGVVHWQSPHMHAYYPALTSWPSLLGDMLADAINCLGFTWASSPACTELEMNVMDWLAKMLGLPEHFLHHHPSSQGGGVLQSTVSESTLIALLAARKNKILEMKTSEPDADESSLNARLVAYASDQAHSSVEKAGLISLVKMKFLPVDDNFSLRGEALQKAIEEDKQRGLVPVFVCATLGTTGVCAFDXLSELGPICAREGLWLHIDAAYAGTAFLCPEFRGFLKGIEYADSFTFNPSKWMMVHFDCTGFWVKDKYKLQQTFSVNPIYLRHANSGVATDFMHWQIPLSRRFRSVKLWFVIRSFGVKNLQAHVRHGTEMAKYFESLVRNDPSFEIPAKRHLGLVVFRLKGPNSLTENVLKEIAKAGRLFLIPATIQDKLIIRFTVTSQFTTRDDILRDWNLIRDAATLILSQGSMEPEEYRERGREMVDYICQYLSTVRERRVTPDVQPGYLRAQLPESAPEDPDSWDSIFGDIERIIMPGVVHWQSPHMHAYYPALTSWPSLLGDMLADAINCLGFTWASSPACTELEMNVMDWLAKMLGLPEHFLHHHPSSQGGGVLQSTVSESTLIALLAARKNKILEMKTSEPDADESSLNARLVAYASDQAHSSVEKAGLISLVKMKFLPVDDNFSLRGEALQKAIEEDKQRGLVPVFVCATLGTTGVCAFDXLSELGPICAREGLWLHIDAAYAGTAFLCPEFRGFLKGIEYADSFTFNPSKWMMVHFDCTGFWVKDKYKLQQTFSVNPIYLRHANSGVATDFMHWQIPLSRRFRSVKLWFVIRSFGVKNLQAHVRHGTEMAKYFESLVRNDPSFEIPAKRHLGLVVFRLKGPNSLTENVLKEIAKAGRLFLIPATIQDKLIIRFTVTSQFTTRDDILRDWNLIRDAATLILSQ Hydrogen bonds contact Hydrophobic contact | ||||
| 16 | Serine--pyruvate aminotransferase | 5HHY | 6.53 | |
Target general information Gen name AGXT Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms AGT1;SPAT Protein family Class-V pyridoxal-phosphate-dependent aminotransferase family Biochemical class Transferase Function Alanine-glyoxylate transaminase activity.Amino acid binding.Identical protein binding.Protein homodimerization activity.Protein self-association.Pyridoxal phosphate binding.Receptor binding.Serine-pyruvate transaminase activity.Transaminase activity. Related diseases Hyperoxaluria primary 1 (HP1) [MIM:259900]: An inborn error of glyoxylate metabolism characterized by increased excretion of oxalate and glycolate, and progressive tissue accumulation of insoluble calcium oxalate. Affected individuals are at risk for nephrolithiasis, nephrocalcinosis and early onset end-stage renal disease. {ECO:0000269|PubMed:10394939, ECO:0000269|PubMed:10453743, ECO:0000269|PubMed:10541294, ECO:0000269|PubMed:10862087, ECO:0000269|PubMed:10960483, ECO:0000269|PubMed:12559847, ECO:0000269|PubMed:12777626, ECO:0000269|PubMed:1301173, ECO:0000269|PubMed:1349575, ECO:0000269|PubMed:15253729, ECO:0000269|PubMed:15849466, ECO:0000269|PubMed:15961946, ECO:0000269|PubMed:15963748, ECO:0000269|PubMed:16971151, ECO:0000269|PubMed:1703535, ECO:0000269|PubMed:17495019, ECO:0000269|PubMed:2039493, ECO:0000269|PubMed:23229545, ECO:0000269|PubMed:24055001, ECO:0000269|PubMed:24934730, ECO:0000269|PubMed:26149463, ECO:0000269|PubMed:8101040, ECO:0000269|PubMed:9192270, ECO:0000269|PubMed:9604803}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08060; DB00160; DB02079; DB00145; DB04083; DB00114; DB00133 Interacts with Q5BKX5-3; P21549; P0C7T5; Q9NR55; Q9H5F2; Q9UKJ5; A8MQ03; O43281-2; A1KXE4-2; Q5TD97; P49356; P53539; P49639; Q15323; O76011; O76014; Q6A162; Q07627; P60328; Q9BYR8; Q3LI67; Q8IUC2; Q9BYQ4; O60711; Q99750; Q5VZ52; P0DPK4; O43482; P50542-1; O15496; Q6ZR37; Q9NQX0; Q8HWS3; Q5W111-2; Q8WVR3; Q6DKK2 EC number 2.6.1.44; 2.6.1.51 Uniprot keywords 3D-structure; Acetylation; Aminotransferase; Disease variant; Peroxisome; Phosphoprotein; Proteomics identification; Pyridoxal phosphate; Reference proteome; Transferase Protein physicochemical properties Chain ID A,B Molecular weight (Da) 84537 Length 771 Aromaticity 0.07 Instability index 36.81 Isoelectric point 8.42 Charge (pH=7) 6.81 2D Binding mode Binding energy (Kcal/mol) -8.9  Molscript Map 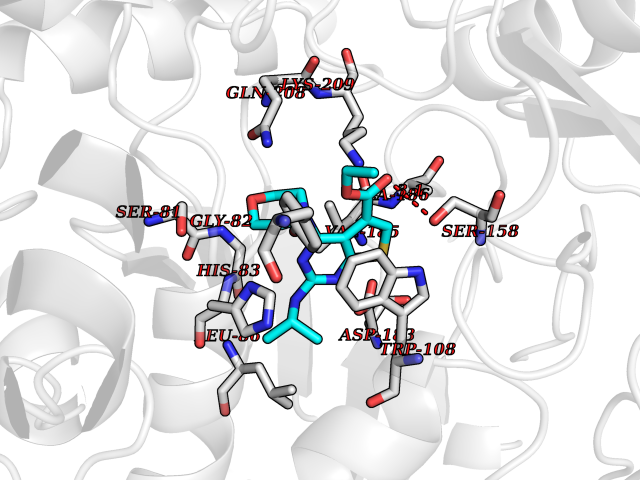 Pymol Map 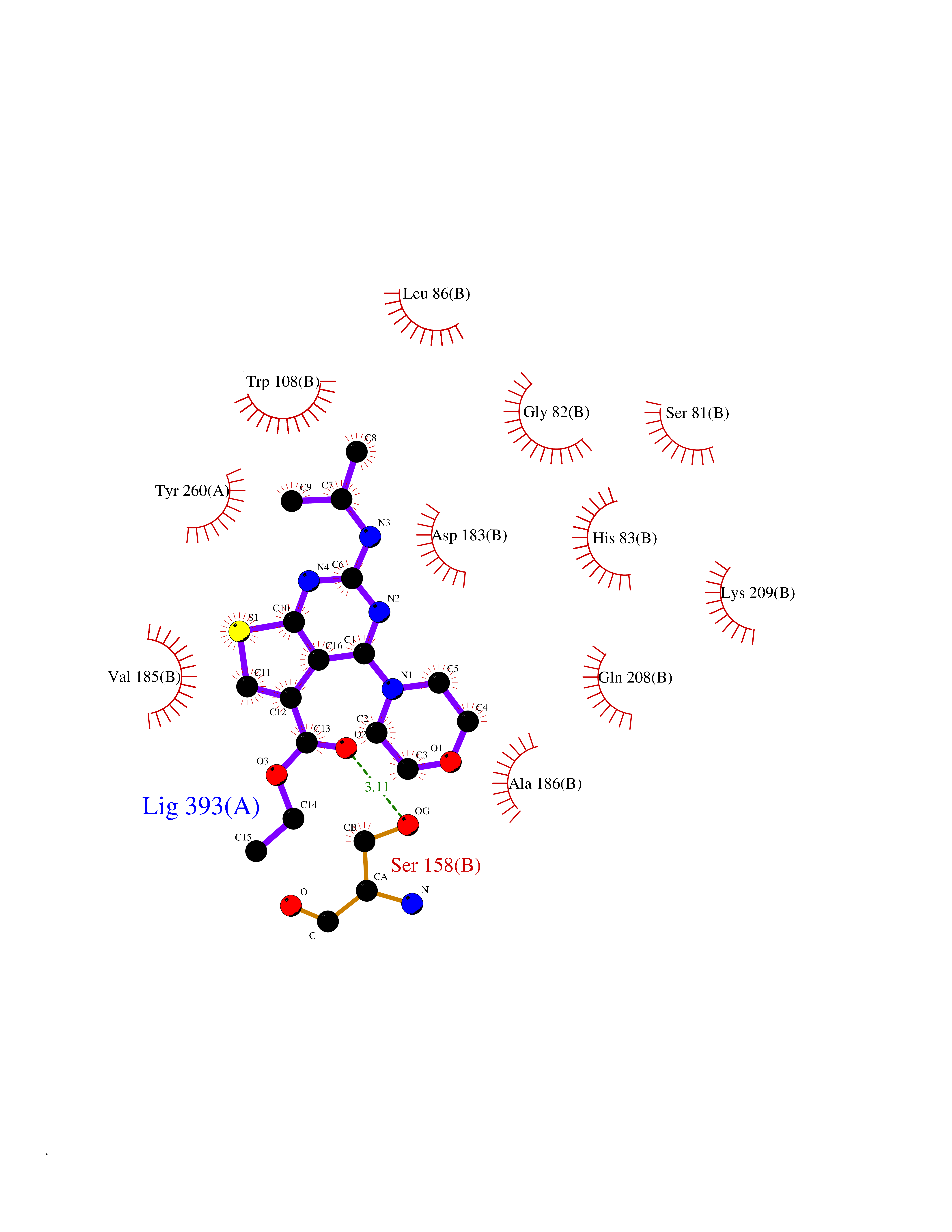 Ligplot Map 3D Binding mode Sequence LLVTPPKALLKPLSIPNQLLLGPGPSNLPPRIMAAGGLQMIGSMSKDMYQIMDEIKEGIQYVFQTRNPLTLVISGSGHCALEAALVNVLEPGDSFLVGANGIWGQRAVDIGERIGARVHPMTKDPGGHYTLQEVEEGLAQHKPVLLFLTHGESSTGVLQPLDGFGELCHRYKCLLLVDSVASLGGTPLYMDRQGIDILYSGSQKALNAPPGTSLISFSDKAKKKMYSRKTKPFSFYLDIKWLANFWGCDDQPRMYHHTIPVISLYSLRESLALIAEQGLENSWRQHREAAAYLHGRLQALGLQLFVKDPALRLPTVTTVAVPAGYDWRDIVSYVIDHFDIEIMGGLGPSTGKVLRIGLLGCNATRENVDRVTEALRAALQHCPKKLLVTPPKALLKPLSIPNQLLLGPGPSNLPPRIMAAGGLQMIGSMSKDMYQIMDEIKEGIQYVFQTRNPLTLVISGSGHCALEAALVNVLEPGDSFLVGANGIWGQRAVDIGERIGARVHPMTKDPGGHYTLQEVEEGLAQHKPVLLFLTHGESSTGVLQPLDGFGELCHRYKCLLLVDSVASLGGTPLYMDRQGIDILYSGSQKALNAPPGTSLISFSDKAKKKMYSRKTKPFSFYLDIKWLANFWGCDDQPRMYHHTIPVISLYSLRESLALIAEQGLENSWRQHREAAAYLHGRLQALGLQLFVKDPALRLPTVTTVAVPAGYDWRDIVSYVIDHFDIEIMGGLGPSTGKVLRIGLLGCNATRENVDRVTEALRAALQHCPKKK Hydrogen bonds contact Hydrophobic contact | ||||
| 17 | 4-cresol dehydrogenase [hydroxylating] flavoprotein subunit | 1WVF | 6.51 | |
Target general information Gen name pchF Organism Pseudomonas putida (Arthrobacter siderocapsulatus) Uniprot ID TTD ID NA Synonyms NA Protein family NA Biochemical class Oxidoreductase Function 4-cresol dehydrogenase (hydroxylating) activity.Flavin adenine dinucleotide binding.Oxidoreductase activity, acting on CH-OH group of donors. Related diseases Dihydrolipoamide dehydrogenase deficiency (DLDD) [MIM:246900]: An autosomal recessive metabolic disorder characterized biochemically by a combined deficiency of the branched-chain alpha-keto acid dehydrogenase complex (BCKDC), pyruvate dehydrogenase complex (PDC), and alpha-ketoglutarate dehydrogenase complex (KGDC). Clinically, affected individuals have lactic acidosis and neurologic deterioration due to sensitivity of the central nervous system to defects in oxidative metabolism. {ECO:0000269|PubMed:10448086, ECO:0000269|PubMed:11687750, ECO:0000269|PubMed:12925875, ECO:0000269|PubMed:15712224, ECO:0000269|PubMed:16442803, ECO:0000269|PubMed:16770810, ECO:0000269|PubMed:17404228, ECO:0000269|PubMed:20160912, ECO:0000269|PubMed:8506365, ECO:0000269|PubMed:8968745, ECO:0000269|PubMed:9540846, ECO:0000269|PubMed:9934985}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03147 Interacts with NA EC number 1.17.9.1 Uniprot keywords 3D-structure; Direct protein sequencing; FAD; Flavoprotein; Oxidoreductase; Plasmid Protein physicochemical properties Chain ID A Molecular weight (Da) 57240.8 Length 515 Aromaticity 0.1 Instability index 30.94 Isoelectric point 6.06 Charge (pH=7) -4.42 2D Binding mode Binding energy (Kcal/mol) -8.88  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence AVLPKGVTQGEFNKAVQKFRALLGDDNVLVESDQLVPYNKIMMPVENAAHAPSAAVTATTVEQVQGVVKICNEHKIPIWTISTGRNFGYGSAAPVQRGQVILDLKKMNKIIKIDPEMCYALVEPGVTFGQMYDYIQENNLPVMLSFSAPSAIAGPVGNTMDRGVGYTPYGEHFMMQCGMEVVLANGDVYRTGMGGVPGSNTWQIFKWGYGPTLDGMFTQANYGICTKMGFWLMPKPPVFKPFEVIFEDEADIVEIVDALRPLRMSNTIPNSVVIASTLWEAGSAHLTRAQYTTEPGHTPDSVIKQMQKDTGMGAWNLYAALYGTQEQVDVNWKIVTDVFKKLGKGRIVTQEEAGDTQPFKYRAQLMSGVPNLQEFGLYNWRGGGGSMWFAPVSEARGSECKKQAAMAKRVLHKYGLDYVAEFIVAPRDMHHVIDVLYDRTNPEETKRADACFNELLDEFEKEGYAVYRVNTRFQDRVAQSYGPVKRKLEHAIKRAVDPNNILAPGRSGIDLNNDF Hydrogen bonds contact Hydrophobic contact | ||||
| 18 | Monoamine oxidase type A (MAO-A) | 2Z5Y | 6.51 | |
Target general information Gen name MAOA Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Monoamine oxidase A; Amine oxidase [flavin-containing] A Protein family Flavin monoamine oxidase family Biochemical class CH-NH(2) donor oxidoreductase Function MAOA preferentially oxidizes biogenic amines such as 5-hydroxytryptamine (5-HT), norepinephrine and epinephrine. Catalyzes the oxidative deamination of biogenic and xenobiotic amines and has important functions in the metabolism of neuroactive and vasoactive amines in the central nervous system and peripheral tissues. Related diseases Brunner syndrome (BRNRS) [MIM:300615]: A form of X-linked non-dysmorphic mild intellectual disability. Male patients are affected by borderline intellectual deficit and exhibit abnormal behavior, including disturbed regulation of impulsive aggression. Obligate female carriers have normal intelligence and behavior. {ECO:0000269|PubMed:8211186}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01472; DB00918; DB00182; DB06698; DB04889; DB13876; DB01445; DB06774; DB00215; DB04017; DB09130; DB05205; DB07641; DB00988; DB01363; DB00668; DB12329; DB01175; DB03147; DB14914; DB00614; DB01381; DB07919; DB04818; DB01247; DB00601; DB01577; DB00805; DB01442; DB01171; DB08804; DB00952; DB04820; DB00184; DB04821; DB06412; DB01626; DB00780; DB00191; DB00388; DB00397; DB09244; DB04850; DB00721; DB01168; DB00571; DB00852; DB09363; DB00140; DB00953; DB06654; DB01037; DB01104; DB00669; DB14569; DB09042; DB00624; DB13943; DB13944; DB13946; DB09245; DB00752; DB15328; DB09185; DB04832; DB00315; DB00909 Interacts with P27338 EC number EC 1.4.3.4 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Catecholamine metabolism; Direct protein sequencing; Disease variant; FAD; Flavoprotein; Intellectual disability; Membrane; Mitochondrion; Mitochondrion outer membrane; Neurotransmitter degradation; Oxidoreductase; Phosphoprotein; Proteomics identification; Reference proteome; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 58195.3 Length 513 Aromaticity 0.11 Instability index 34.97 Isoelectric point 7.98 Charge (pH=7) 2.87 2D Binding mode Binding energy (Kcal/mol) -8.88  Molscript Map 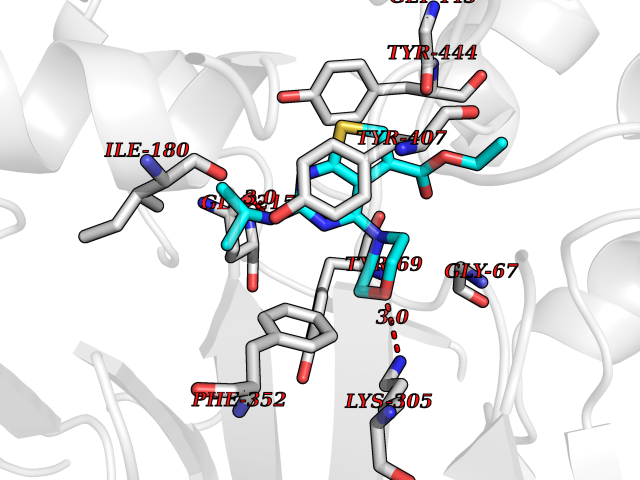 Pymol Map 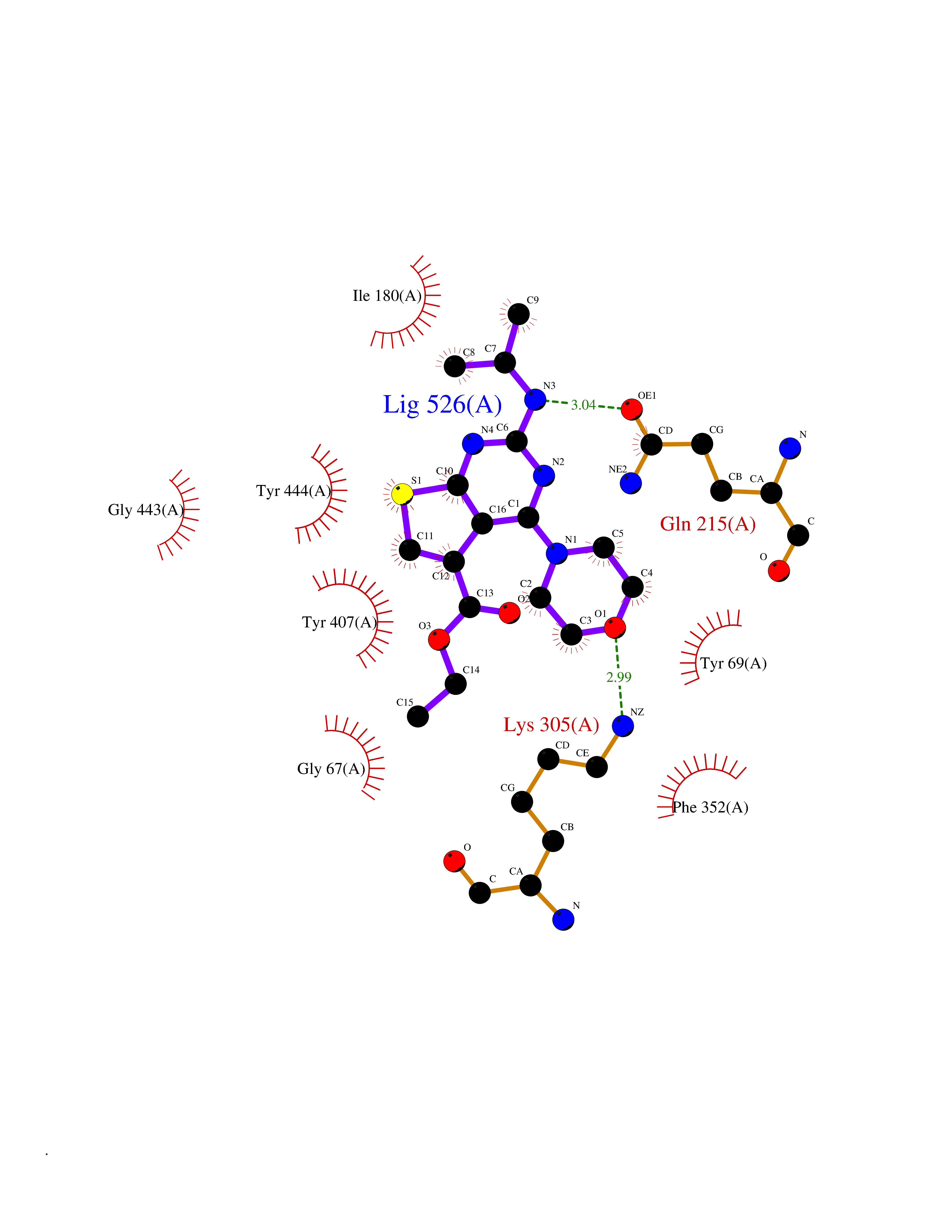 Ligplot Map 3D Binding mode Sequence HMFDVVVIGGGISGLSAAKLLTEYGVSVLVLEARDRVGGRTYTIRNEHVDYVDVGGAYVGPTQNRILRLSKELGIETYKVNVSERLVQYVKGKTYPFRAAFPPVWNPIAYLDYNNLWRTIDNMGKEIPTDAPWEAQHADKWDKMTMKELIDKICWTKTARRFAYLFVNINVTSEPHEVSALWFLWYVKQCGGTTRIFSVTNGGQERKFVGGSGQVSERIMDLLGDQVKLNHPVTHVDQSSDNIIIETLNHEHYECKYVINAIPPTLTAKIHFRPELPAERNQLIQRLPMGAVIKCMMYYKEAFWKKKDYCGCMIIEDEDAPISITLDDTKPDGSLPAIMGFILARKADRLAKLHKEIRKKKICELYAKVLGSQEALHPVHYEEKNWCEEQYSGGCYTAYFPPGIMTQYGRVIRQPVGRIFFAGTETATKWSGYMEGAVEAGERAAREVLNGLGKVTEKDIWVQEPESKDVPAVEITHTFWERNLPSVSGLLKIIGFSTSVTALGFVLYKYKLL Hydrogen bonds contact Hydrophobic contact | ||||
| 19 | Lysine-specific histone demethylase 1B (KDM1B) | 4HSU | 6.51 | |
Target general information Gen name KDM1B Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Lysine-specific histone demethylase 2; LSD2; Flavin-containing amine oxidase domain-containing protein 1; C6orf193; AOF1 Protein family Flavin monoamine oxidase family Biochemical class CH-NH(2) donor oxidoreductase Function Required for de novo DNA methylation of a subset of imprinted genes during oogenesis. Acts by oxidizing the substrate by FAD to generate the corresponding imine that is subsequently hydrolyzed. Demethylates both mono- and di-methylated 'Lys-4' of histone H3. Has no effect on tri-methylated 'Lys-4', mono-, di- or tri-methylated 'Lys-9', mono-, di- or tri-methylated 'Lys-27', mono-, di- or tri-methylated 'Lys-36' of histone H3, or on mono-, di- or tri-methylated 'Lys-20' of histone H4. Histone demethylase that demethylates 'Lys-4' of histone H3, a specific tag for epigenetic transcriptional activation, thereby acting as a corepressor. Related diseases Angioedema, hereditary, 1 (HAE1) [MIM:106100]: An autosomal dominant disorder characterized by episodic local swelling involving subcutaneous or submucous tissue of the upper respiratory and gastrointestinal tracts, face, extremities, and genitalia. Hereditary angioedema due to C1 esterase inhibitor deficiency is comprised of two clinically indistinguishable forms. In hereditary angioedema type 1, serum levels of C1 esterase inhibitor are decreased, while in type 2, the levels are normal or elevated, but the protein is non-functional. {ECO:0000269|PubMed:12773530, ECO:0000269|PubMed:1363816, ECO:0000269|PubMed:1451784, ECO:0000269|PubMed:14635117, ECO:0000269|PubMed:16409206, ECO:0000269|PubMed:2118657, ECO:0000269|PubMed:2296585, ECO:0000269|PubMed:22994404, ECO:0000269|PubMed:2365061, ECO:0000269|PubMed:24456027, ECO:0000269|PubMed:3178731, ECO:0000269|PubMed:7814636, ECO:0000269|PubMed:7883978, ECO:0000269|PubMed:8172583, ECO:0000269|PubMed:8529136, ECO:0000269|PubMed:8755917, ECO:0000269|Ref.41}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q96L03 EC number EC 1.-.-.- Uniprot keywords 3D-structure; Alternative splicing; Chromatin regulator; Chromosome; Developmental protein; FAD; Flavoprotein; Metal-binding; Nucleus; Oxidoreductase; Phosphoprotein; Proteomics identification; Reference proteome; Repressor; Transcription; Transcription regulation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A,C Molecular weight (Da) 85795.5 Length 763 Aromaticity 0.1 Instability index 37.87 Isoelectric point 8.41 Charge (pH=7) 9.16 2D Binding mode Binding energy (Kcal/mol) -8.88 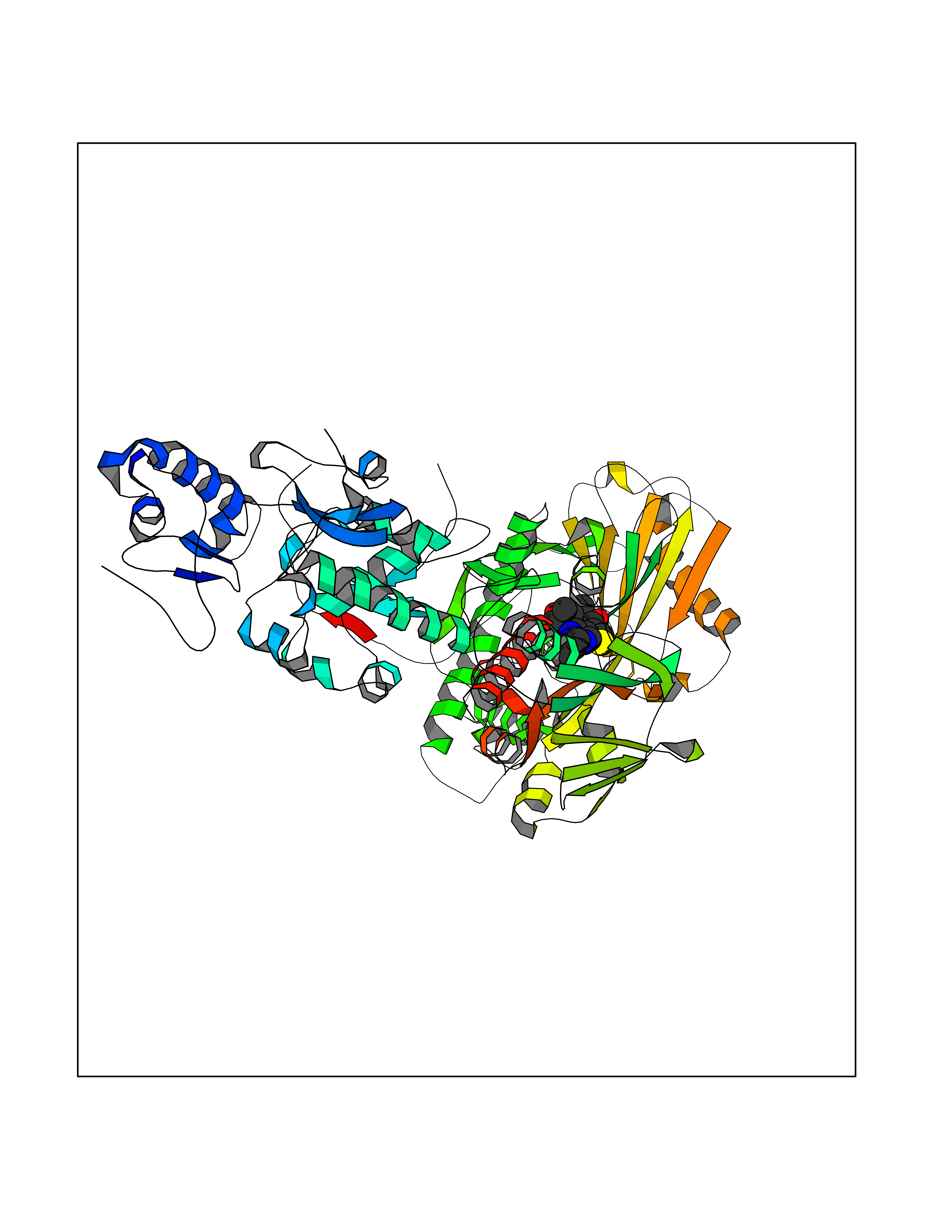 Molscript Map 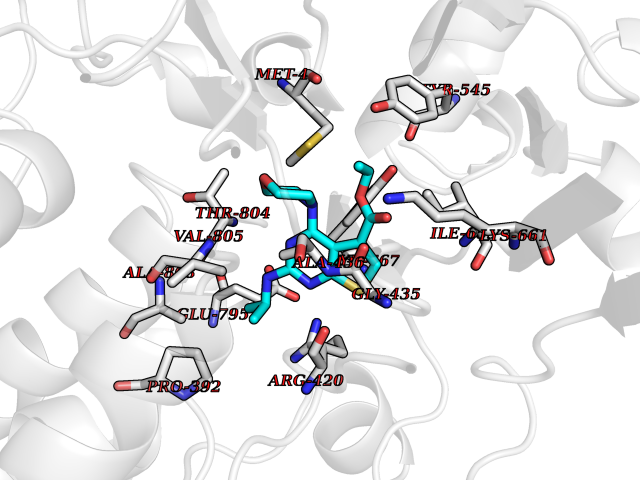 Pymol Map 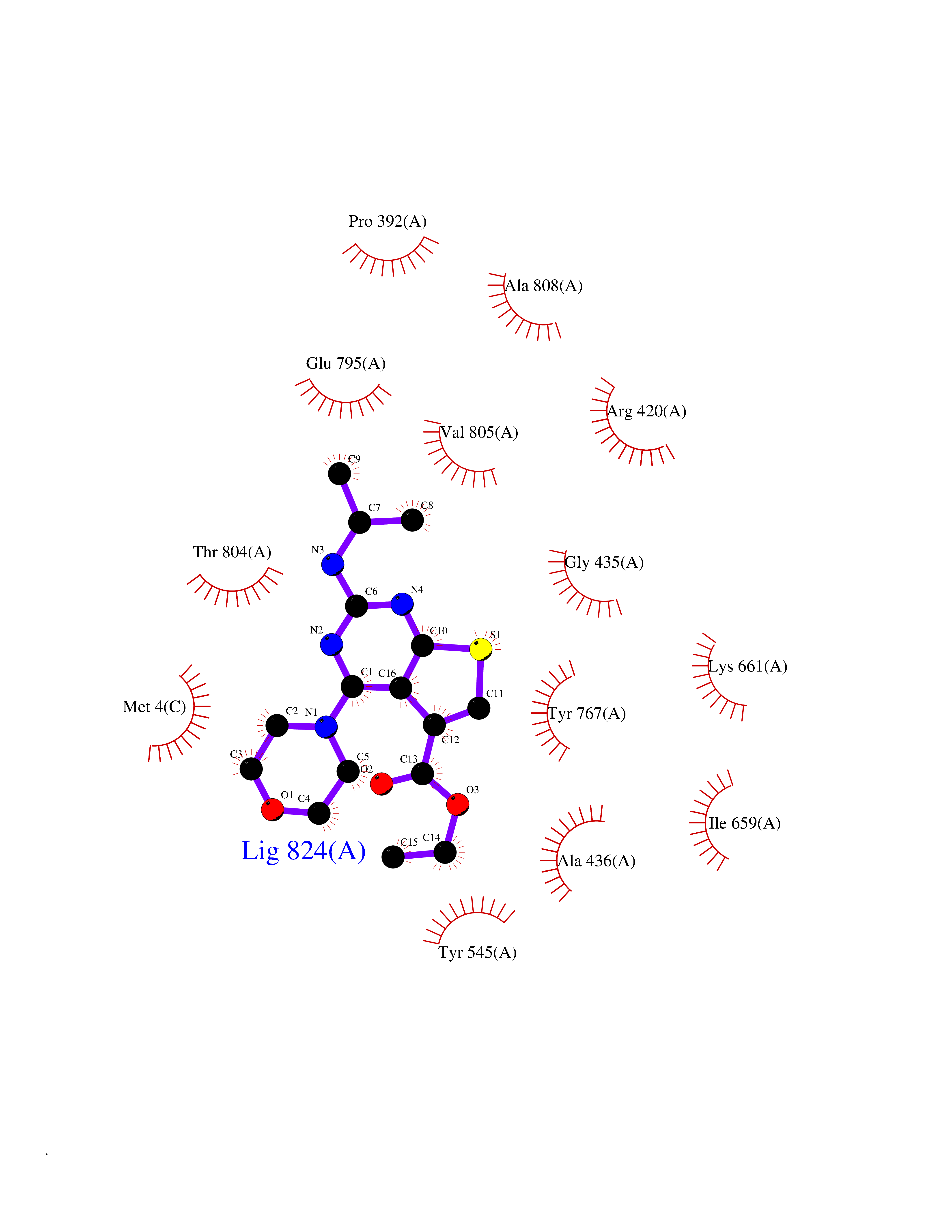 Ligplot Map 3D Binding mode Sequence GSRKCEKAGCTATCPVCFASASERCAKNGYTSRWYHLSCGEHFCNECFDHYYRSHKDGYDKYTTWKKIWTSNGKTEPSPKAFMADQQLPYWVQCTKPECRKWRQLTKEIQLTPQIAKTYRCGMKSDHCSLPEDLRVLEVSNHWWYSMLILPPLLKDSVAAPLLSAYYPDCVGMSPSCTGMNRYFQPFYQPNECGKALCVRPDVMELDELYEFPEYSRDPTMYLALRNLILALWYTNCKEALTPQKCIPHIIVRGLVRIRCVQEVERILYFMTRKGLINTGVLSVGADQYLLPKDYHNKSVIIIGAGPAGLAAARQLHNFGIKVTVLEAKDRIGGRVWDDKSFKGVTVGRGAQIVNGCINNPVALMCEQLGISMHKFGERCDLIQEGGRITDPTIDKRMDFHFNALLDVVSEWRKDKTQLQDVPLGEKIEEIYKAFIKESGIQFSELEGQVLQFHLSNLEYACGSNLHQVSARSWDHNEFFAQFAGDHTLLTPGYSVIIEKLAEGLDIQLKSPVQCIDYSGDEVQVTTTDGTGYSAQKVLVTVPLALLQKGAIQFNPPLSEKKMKAINSLGAGIIEKIALQFPYRFWDSKVQGADFFGHVPPSASKRGLFAVFYDMDPQKKHSVLMSVIAGEAVASVRTLDDKQVLQQCMATLRELFKEQEVPDPTKYFVTRWSTDPWIQMAYSFVKTGGSGEAYDIIAEDIQGTVFFAGEATNRHFPQTVTGAYLSGVREASKIAAFARTMQTARKSTGGKAPRKQLATKAAR Hydrogen bonds contact Hydrophobic contact | ||||
| 20 | Fumarate reductase flavoprotein subunit | 1Y0P | 6.50 | |
Target general information Gen name fccA Organism Shewanella frigidimarina Uniprot ID TTD ID NA Synonyms fcc3 Protein family FAD-dependent oxidoreductase 2 family, FRD/SDH subfamily Biochemical class Oxidoreductase Function Electron carrier activity.Fumarate reductase (menaquinone).Metal ion binding.Nucleic acid binding.Succinate dehydrogenase activity. Related diseases Pigmentary disorder, reticulate, with systemic manifestations, X-linked (PDR) [MIM:301220]: An X-linked recessive disorder characterized by recurrent infections and sterile inflammation in various organs. Diffuse skin hyperpigmentation with a distinctive reticulate pattern is universally evident by early childhood. This is later followed in many patients by hypohidrosis, corneal inflammation and scarring, enterocolitis that resembles inflammatory bowel disease, and recurrent urethral strictures. Melanin and amyloid deposition is present in the dermis. Affected males also have a characteristic facies with frontally upswept hair and flared eyebrows. Female carriers have only restricted pigmentary changes along Blaschko's lines. {ECO:0000269|PubMed:27019227}. The disease is caused by variants affecting the gene represented in this entry. XLPDR is caused by a recurrent intronic mutation that results in missplicing and reduced POLA1 expression. This leads to a decrease in cytosolic RNA:DNA hybrids and constitutive activation of type I interferon responses, but has no effect on cell replication. {ECO:0000269|PubMed:27019227}.; DISEASE: Van Esch-O'Driscoll syndrome (VEODS) [MIM:301030]: An X-linked recessive syndrome characterized by different degrees of intellectual disability, moderate to severe short stature, microcephaly, hypogonadism, and variable congenital malformations. {ECO:0000269|PubMed:31006512}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB04734; DB03147; DB01677; DB03343 Interacts with NA EC number 1.3.2.4 Uniprot keywords 3D-structure; Electron transport; FAD; Flavoprotein; Heme; Iron; Metal-binding; Oxidoreductase; Periplasm; Transport Protein physicochemical properties Chain ID A Molecular weight (Da) 60177.2 Length 568 Aromaticity 0.06 Instability index 27.7 Isoelectric point 6 Charge (pH=7) -8.64 2D Binding mode Binding energy (Kcal/mol) -8.87  Molscript Map 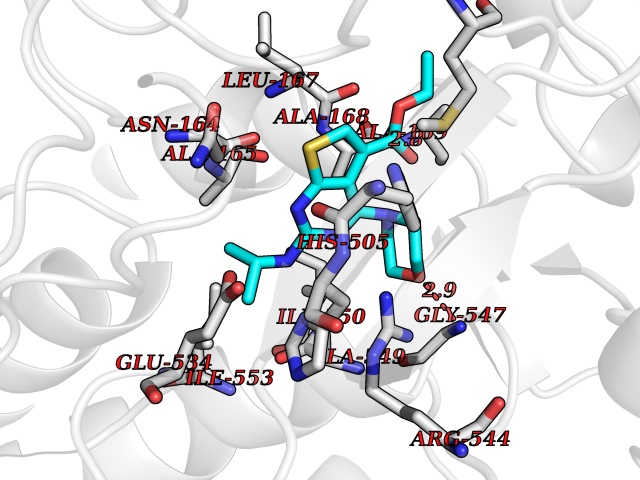 Pymol Map 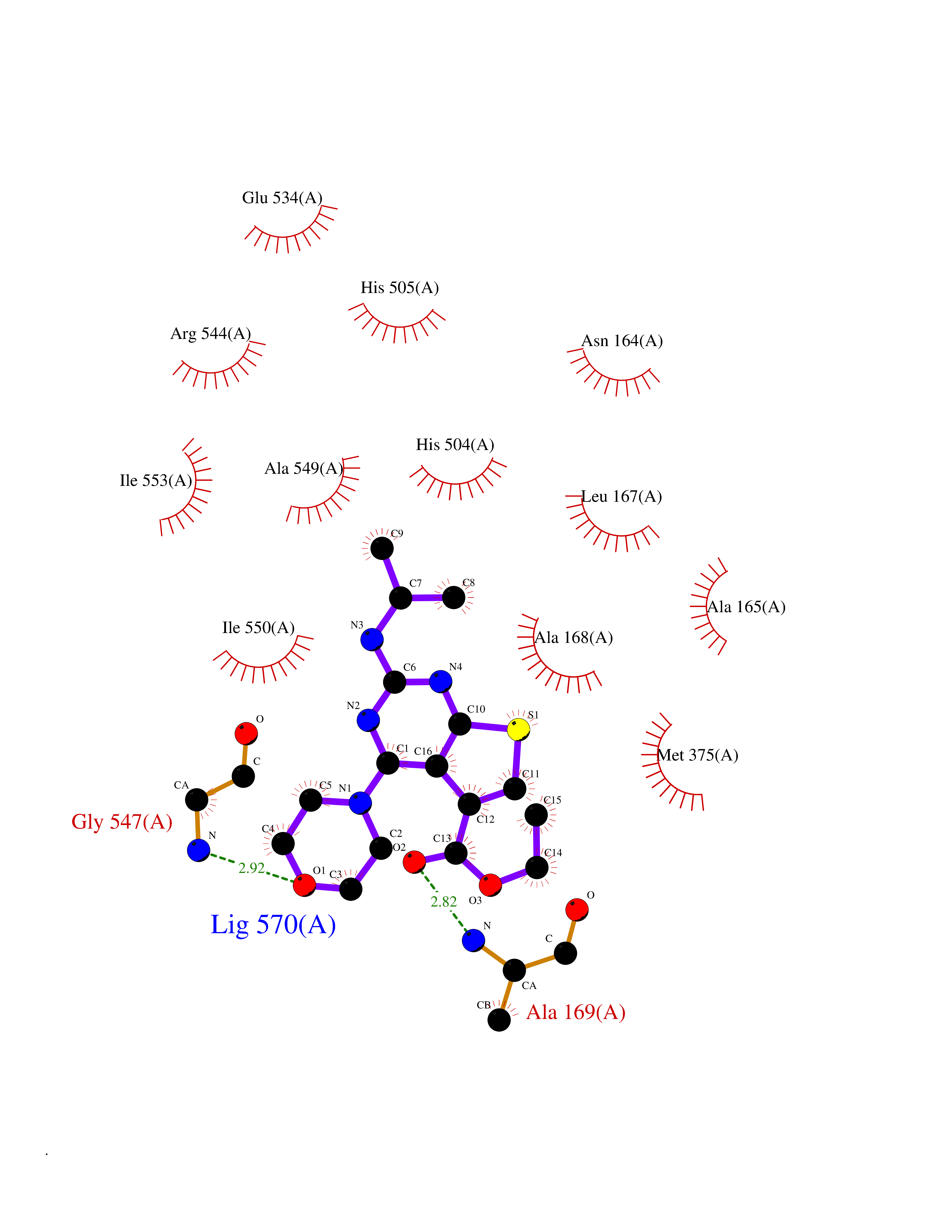 Ligplot Map 3D Binding mode Sequence ADNLAEFHVQNQECDSCHTPDGELSNDSLTYENTQCVSCHGTLAEVAETTKHEHYNAHASHFPGEVACTSCHSAHEKSMVYCDSCHSFDFNMPYAKKWLRDEPTIAELAKDKSERQAALASAPHDTVDVVVVGSGGAGFSAAISATDSGAKVILIEKEPVIGGNAKLAAGGMNAAWTDQQKAKKITDSPELMFEDTMKGGQNINDPALVKVLSSHSKDSVDWMTAMGADLTDVGMMGGASVNRAHRPTGGAGVGAHVVQVLYDNAVKRNIDLRMNTRGIEVLKDDKGTVKGILVKGMYKGYYWVKADAVILATGGFAKNNERVAKLDPSLKGFISTNQPGAVGDGLDVAENAGGALKDMQYIQAHPTLSVKGGVMVTEAVRGNGAILVNREGKRFVNEITTRDKASAAILAQTGKSAYLIFDDSVRKSLSKIDKYIGLGVAPTADSLVKLGKMEGIDGKALTETVARYNSLVSSGKDTDFERPNLPRALNEGNYYAIEVTPGVHHTMGGVMIDTKAEVMNAKKQVIPGLYGAGEVTGGVHGANRLGGNAISDIITFGRLAGEEAAKYS Hydrogen bonds contact Hydrophobic contact | ||||