Job Results:
Ligand
Structure
Job ID
5c976aa63f0cd6d4251a6978fa589574
Job name
NA
Time
2025-07-21 16:39:34
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 81 | Angiotensin-converting enzyme 2 (ACE2) | 1R42 | 5.30 | |
Target general information Gen name ACE2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Processed angiotensinconverting enzyme 2; Metalloprotease MPROT15; Angiotensinconverting enzyme homolog; Angiotensinconverting enzyme 2; ACErelated carboxypeptidase; ACEH; ACE2 Protein family Peptidase M2 family Biochemical class Peptidase Function Carboxypeptidase which converts angiotensin I to angiotensin 1-9, a peptide of unknown function, and angiotensin II to angiotensin 1-7, a vasodilator. Also able to hydrolyze apelin- 13 and dynorphin-13 with high efficiency. May be an important regulator of heart function. In case of human coronaviruses SARS and HCoV-NL63 infections, serve as functional receptor for the spike glycoprotein of both coronaviruses. Related diseases Cornelia de Lange syndrome 5 (CDLS5) [MIM:300882]: A form of Cornelia de Lange syndrome, a clinically heterogeneous developmental disorder associated with malformations affecting multiple systems. It is characterized by facial dysmorphisms, abnormal hands and feet, growth delay, cognitive retardation, hirsutism, gastroesophageal dysfunction and cardiac, ophthalmologic and genitourinary anomalies. {ECO:0000269|PubMed:22885700, ECO:0000269|PubMed:22889856}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB09019; DB00608; DB01611; DB15643; DB05203 Interacts with Q9BYF1; PRO_0000032457 [P01019]; PRO_0000032458 [P01019]; Q96CW1; PRO_0000000092 [P05067]; Q9H2X3; PRO_0000417390 [Q01523]; Q14416; P11021; P05556; O60341; PRO_0000006688 [P01042]; O14745; PRO_0000019524 [P30990]; Q5T2W1; P35247; Q9Y566; Q695T7; Q96L92; O15393; P08670; P52293; A0A6B9WHD3; A0A6G6A1M4; A0A6M3G9R1; P0DTC2; PRO_0000449647 [P0DTC2]; P59594; PRO_0000037209 [P59594]; Q5GDB5; Q6Q1S2 EC number EC 3.4.17.23 Uniprot keywords 3D-structure; Alternative splicing; Carboxypeptidase; Cell membrane; Cell projection; Chloride; Cytoplasm; Direct protein sequencing; Disulfide bond; Glycoprotein; Host cell receptor for virus entry; Host-virus interaction; Hydrolase; Isopeptide bond; Membrane; Metal-binding; Metalloprotease; Phosphoprotein; Protease; Proteomics identification; Receptor; Reference proteome; Secreted; Signal; Transmembrane; Transmembrane helix; Ubl conjugation; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 69085.2 Length 597 Aromaticity 0.13 Instability index 42.1 Isoelectric point 5.01 Charge (pH=7) -27.11 2D Binding mode Binding energy (Kcal/mol) -7.23  Molscript Map 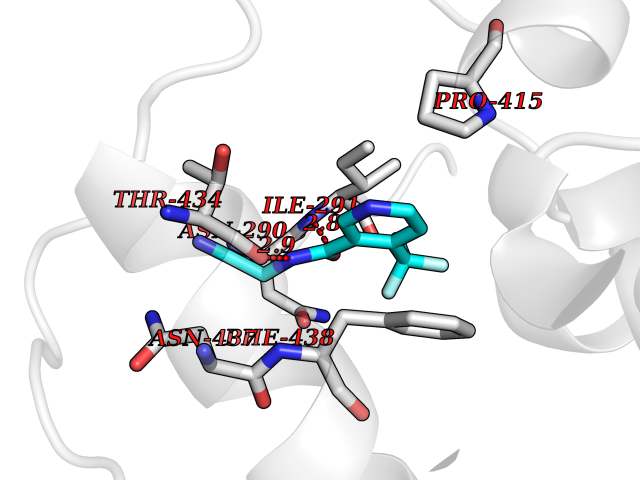 Pymol Map 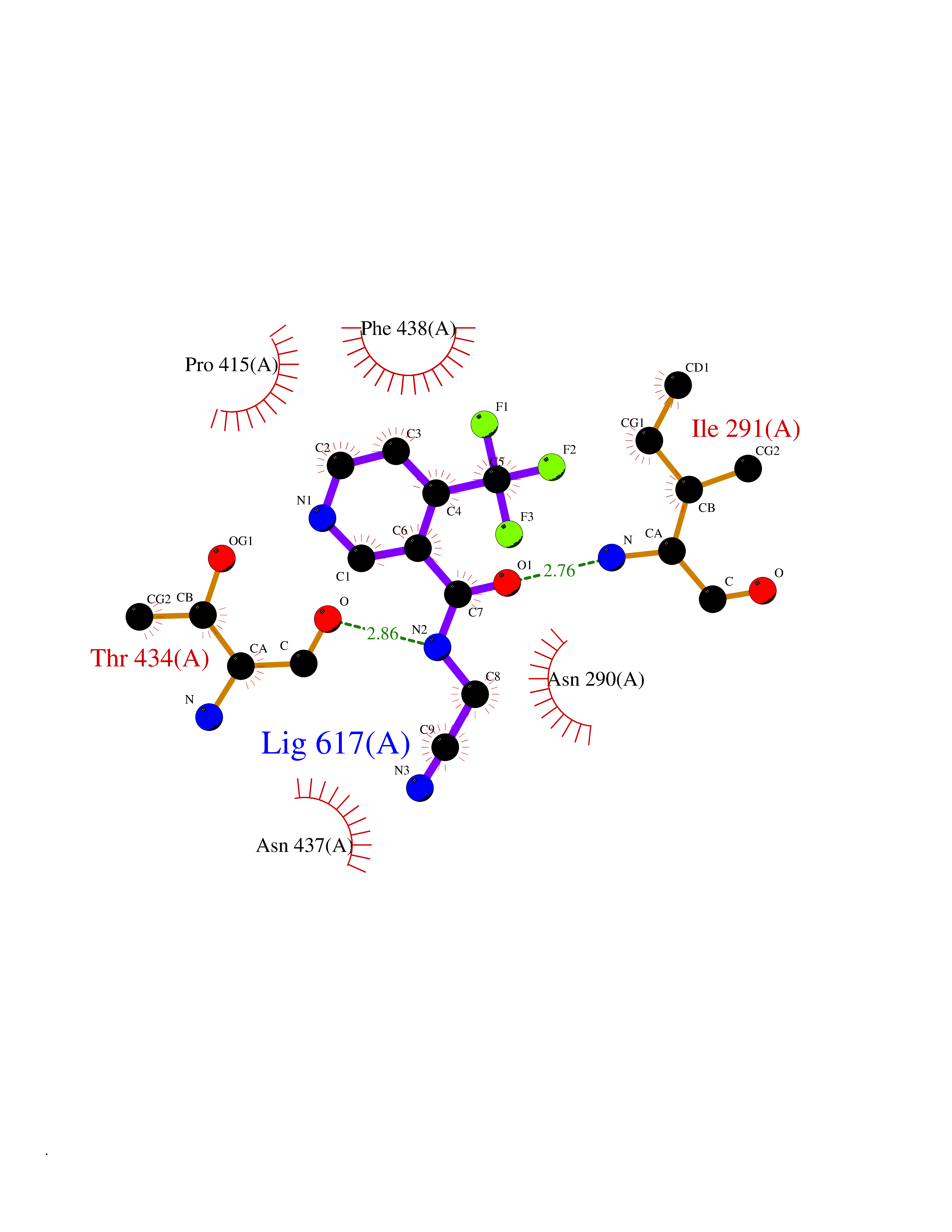 Ligplot Map 3D Binding mode Sequence STIEEQAKTFLDKFNHEAEDLFYQSSLASWNYNTNITEENVQNMNNAGDKWSAFLKEQSTLAQMYPLQEIQNLTVKLQLQALQQNGSSVLSEDKSKRLNTILNTMSTIYSTGKVCNPDNPQECLLLEPGLNEIMANSLDYNERLWAWESWRSEVGKQLRPLYEEYVVLKNEMARANHYEDYGDYWRGDYEVNGVDGYDYSRGQLIEDVEHTFEEIKPLYEHLHAYVRAKLMNAYPSYISPIGCLPAHLLGDMWGRFWTNLYSLTVPFGQKPNIDVTDAMVDQAWDAQRIFKEAEKFFVSVGLPNMTQGFWENSMLTDPGNVQKAVCHPTAWDLGKGDFRILMCTKVTMDDFLTAHHEMGHIQYDMAYAAQPFLLRNGANEGFHEAVGEIMSLSAATPKHLKSIGLLSPDFQEDNETEINFLLKQALTIVGTLPFTYMLEKWRWMVFKGEIPKDQWMKKWWEMKREIVGVVEPVPHDETYCDPASLFHVSNDYSFIRYYTRTLYQFQFQEALCQAAKHEGPLHKCDISNSTEAGQKLFNMLRLGKSEPWTLALENVVGAKNMNVRPLLNYFEPLFTWLKDQNKNSFVGWSTDWSPYAD Hydrogen bonds contact Hydrophobic contact | ||||
| 82 | PRKR-like endoplasmic reticulum kinase (PERK) | 4G31 | 5.30 | |
Target general information Gen name EIF2AK3 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms PEK Protein family Protein kinase superfamily, Ser/Thr protein kinase family, GCN2 subfamily Biochemical class Kinase Function Converts phosphorylated eIF-2-alpha/EIF2S1 either in a global protein synthesis inhibitor, leading to a reduced overall utilization of amino acids, or to a translation initiation activator of specific mRNAs, such as the transcriptional activator ATF4, and hence allowing ATF4-mediated reprogramming of amino acid biosynthetic gene expression to alleviate nutrient depletion. Serves as a critical effector of unfolded protein response (UPR)-induced G1 growth arrest due to the loss of cyclin-D1 (CCND1). Involved in control of mitochondrial morphology and function. Metabolic-stress sensing protein kinase that phosphorylates the alpha subunit of eukaryotic translation initiation factor 2 (eIF-2-alpha/EIF2S1) on 'Ser-52' during the unfolded protein response (UPR) and in response to low amino acid availability. Related diseases Wolcott-Rallison syndrome (WRS) [MIM:226980]: A rare autosomal recessive disorder, characterized by permanent neonatal or early infancy insulin-dependent diabetes and, at a later age, epiphyseal dysplasia, osteoporosis, growth retardation and other multisystem manifestations, such as hepatic and renal dysfunctions, intellectual disability and cardiovascular abnormalities. {ECO:0000269|PubMed:10932183, ECO:0000269|PubMed:12086964, ECO:0000269|PubMed:12960215, ECO:0000269|PubMed:16813601, ECO:0000269|PubMed:24168455, ECO:0000269|PubMed:24194294, ECO:0000269|PubMed:27145240, ECO:0000269|PubMed:28220546, ECO:0000269|PubMed:30906465, ECO:0000269|PubMed:30922274, ECO:0000269|PubMed:32216767, ECO:0000269|PubMed:34123975}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q9NZJ5; P11021 EC number EC 2.7.11.1 Uniprot keywords 3D-structure; ADP-ribosylation; ATP-binding; Diabetes mellitus; Disease variant; Endoplasmic reticulum; Glycoprotein; Kinase; Membrane; Nucleotide-binding; Phosphoprotein; Proteomics identification; Reference proteome; Serine/threonine-protein kinase; Signal; Stress response; Transferase; Translation regulation; Transmembrane; Transmembrane helix; Tyrosine-protein kinase; Unfolded protein response Protein physicochemical properties Chain ID A Molecular weight (Da) 29033.5 Length 248 Aromaticity 0.1 Instability index 43.71 Isoelectric point 7.75 Charge (pH=7) 1.27 2D Binding mode Binding energy (Kcal/mol) -7.23 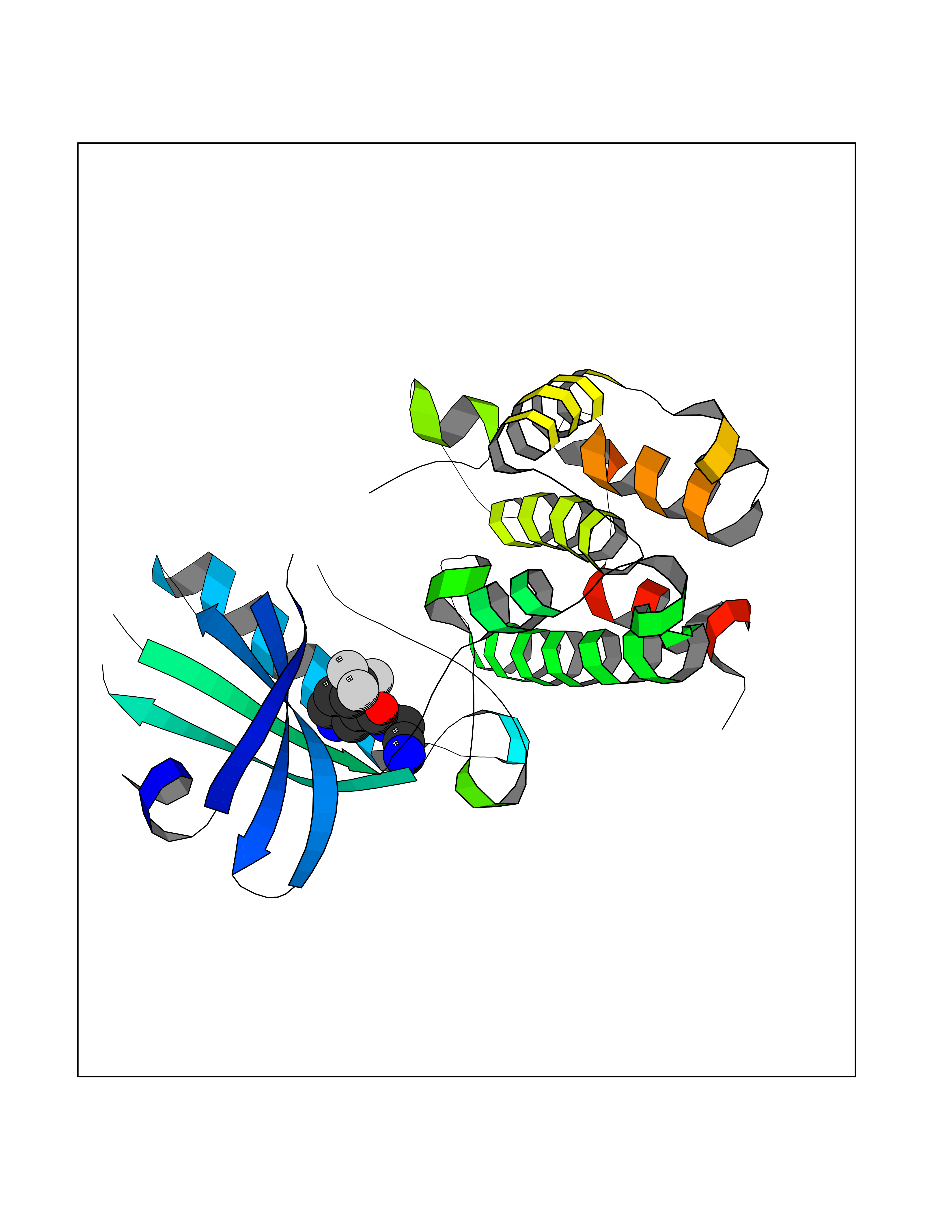 Molscript Map 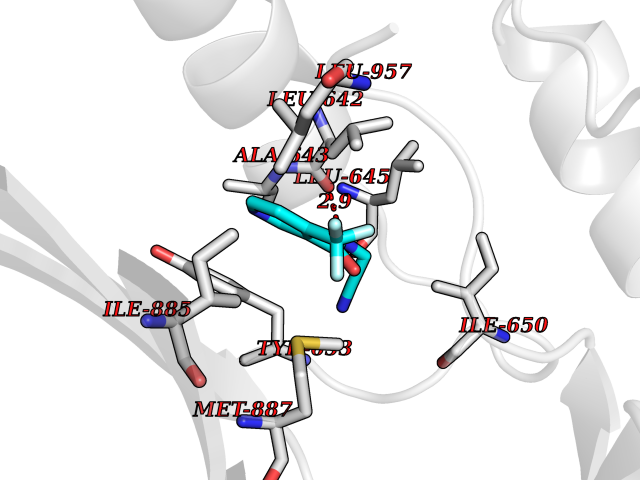 Pymol Map 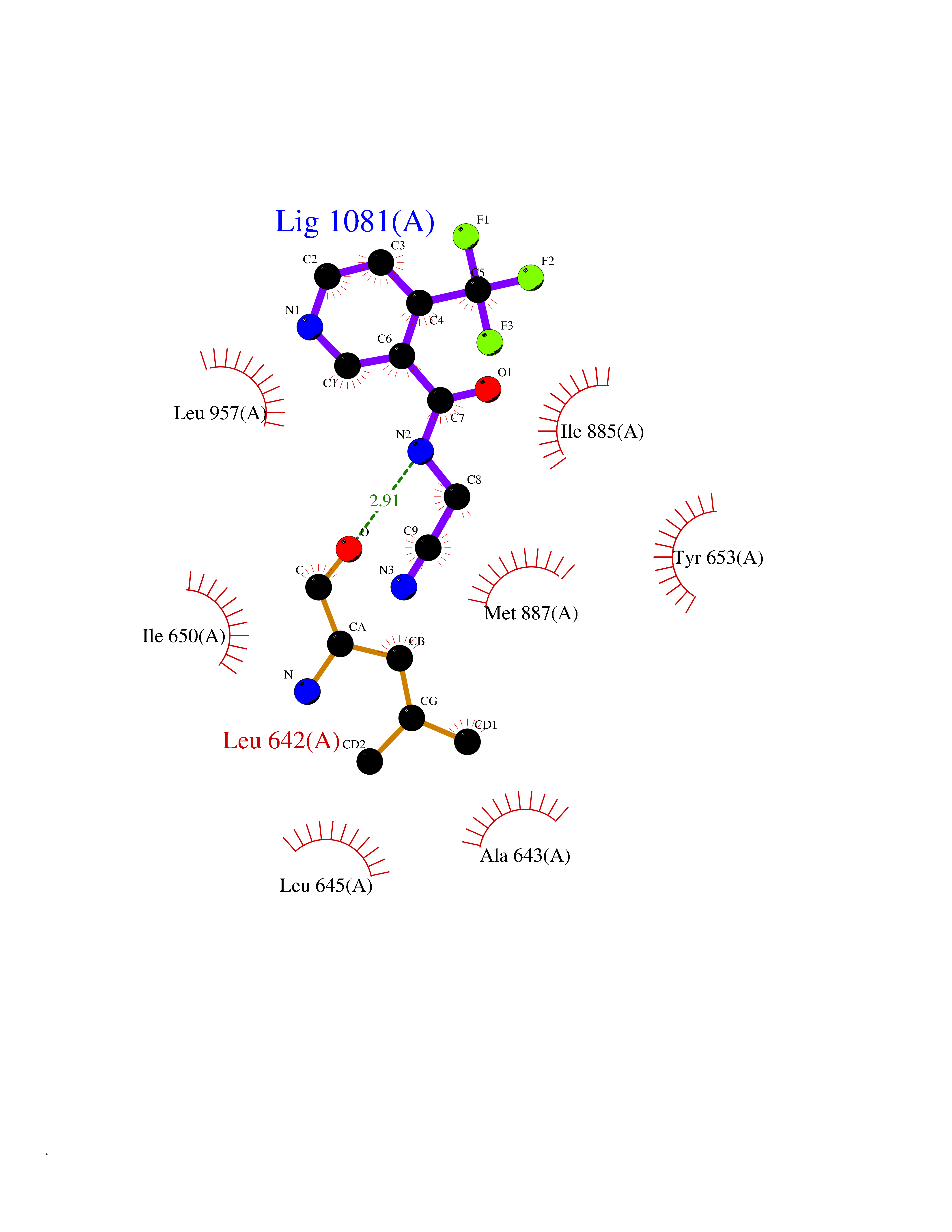 Ligplot Map 3D Binding mode Sequence GRYLTDFEPIQCLGRGGVVFEAKNKVDDCNYAIKRIRLPNRELAREKVMREVKALAKLEHPGIVRYFNAWLEKNKVYLYIQMQLCRKENLKDWMNGRCTIEERERSVCLHIFLQIAEAVEFLHSKGLMHRDLKPSNIFFTMDDVVKVGDFGLVGTKLYMSPEQIHGNSYSHKVDIFSLGLILFELLYPFSTQMERVRTLTDVRNLKFPPLFTQKYPCEYVMVQDMLSPSPMERPEAINIIENAVFEDL Hydrogen bonds contact Hydrophobic contact | ||||
| 83 | Tracheobronchial mucin 5A (MUC5AC) | 5AJP | 5.30 | |
Target general information Gen name MUC5AC Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Tracheobronchial mucin; TBM; Mucin5AC; Mucin5 subtype AC, tracheobronchial; Mucin-5AC; Mucin-5 subtype AC, tracheobronchial; Major airway glycoprotein; MUC5; MUC-5AC; Lewis B blood group antigen; LeB; Protein family NA Biochemical class NA Function Gel-forming glycoprotein of gastric and respiratoy tract epithelia that protects the mucosa from infection and chemical damage by binding to inhaled microrganisms and particles that are subsequently removed by the mucocilary system. Related diseases Orotic aciduria 1 (ORAC1) [MIM:258900]: A disorder of pyrimidine metabolism resulting in megaloblastic anemia and orotic acid crystalluria that is frequently associated with some degree of physical and intellectual disability. A minority of cases have additional features, particularly congenital malformations and immune deficiencies. {ECO:0000269|PubMed:9042911}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number NA Uniprot keywords 3D-structure; Copper; Direct protein sequencing; Disulfide bond; Glycoprotein; Metal-binding; Proteomics identification; Reference proteome; Repeat; Secreted; Signal Protein physicochemical properties Chain ID A,B Molecular weight (Da) 57738 Length 509 Aromaticity 0.09 Instability index 47.24 Isoelectric point 8.35 Charge (pH=7) 4.76 2D Binding mode Binding energy (Kcal/mol) -7.22 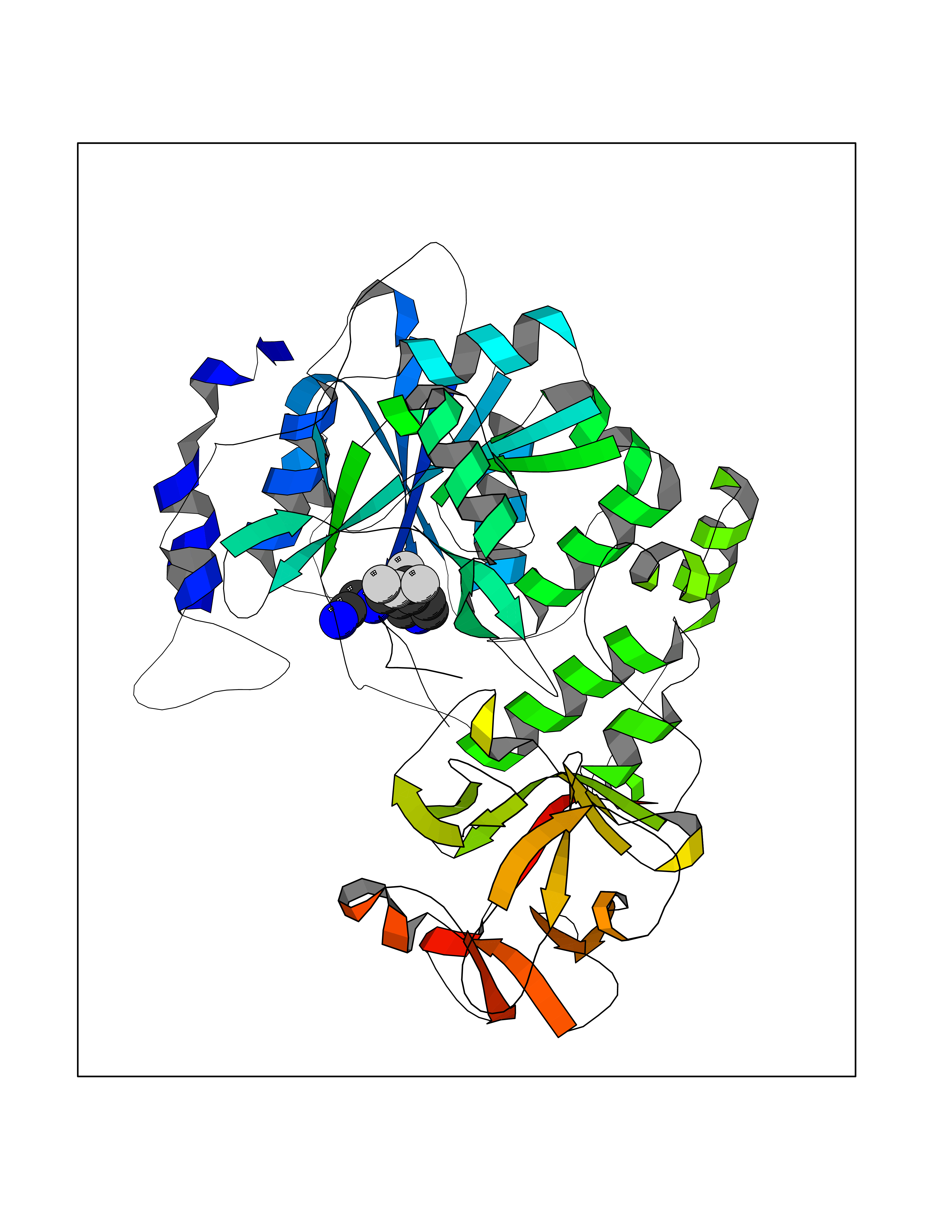 Molscript Map 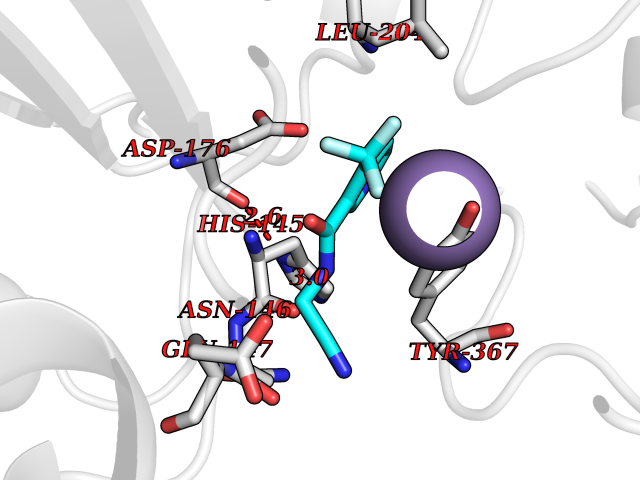 Pymol Map 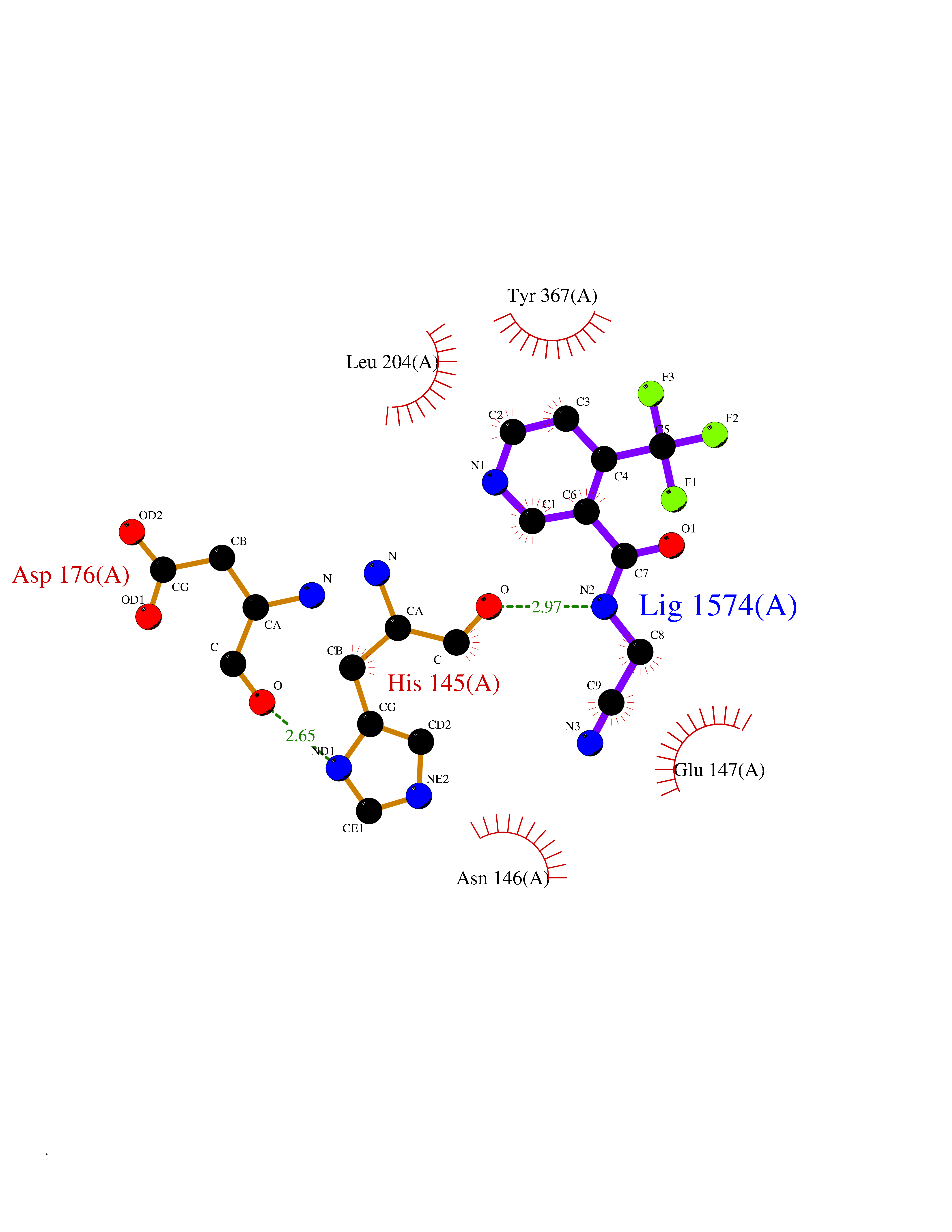 Ligplot Map 3D Binding mode Sequence KVRWPDFNQEAYVGGTMVRSGQDPYARNKFNQVESDKLRMDRAIPDTRHDQCQRKQWRVDLPATSVVITFHNEARSALLRTVVSVLKKSPPHLIKEIILVDDYSNDPEDGALLGKIEKVRVLRNDRREGLMRSRVRGADAAQAKVLTFLDSHCECNEHWLEPLLERVAEDRTRVVSPIIDVINMDNFQYVGASADLKGGFDWNLVFKWDYMTPEQRRSRQGNPVAPIKTPMIAGGLFVMDKFYFEELGKYDMMMDVWGGENLEISFRVWQCGGSLEIIPCSRVGHVFRKQHPYTFPGGSGTVFARNTRRAAEVWMDEYKNFYYAAVPSARNVPYGNIQSRLELRKKLSCKPFKWYLENVYPELRVPDHQDIAFGALQQGTNCLDTLGHFADGVVGVYECHNAGGNQEWALTKEKSVKHMDLCLTVVDRAPGSLIKLQGCREDDSRQKWEQIEGNSKLRHVGSNLCLDSRTAKSGGLSVEVCGPALSQQWKFTLNLTTPSPVPTTSTTSA Hydrogen bonds contact Hydrophobic contact | ||||
| 84 | Monoglyceride lipase (MAGL) | 3PE6 | 5.30 | |
Target general information Gen name MGLL Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Monoacylglycerol lipase; MGL; Lysophospholipaselike; Lysophospholipase-like; Lysophospholipase homolog; HUK5; HU-K5 Protein family AB hydrolase superfamily, Monoacylglycerol lipase family Biochemical class Carboxylic ester hydrolase Function Hydrolyzes the endocannabinoid 2-arachidonoylglycerol, and thereby contributes to the regulation of endocannabinoid signaling, nociperception and perception of pain. Regulates the levels of fatty acids that serve as signaling molecules and promote cancer cell migration, invasion and tumor growth. Converts monoacylglycerides to free fatty acids and glycerol. Related diseases Systemic lupus erythematosus 9 (SLEB9) [MIM:610927]: A chronic, relapsing, inflammatory, and often febrile multisystemic disorder of connective tissue, characterized principally by involvement of the skin, joints, kidneys and serosal membranes. It is of unknown etiology, but is thought to represent a failure of the regulatory mechanisms of the autoimmune system. The disease is marked by a wide range of system dysfunctions, an elevated erythrocyte sedimentation rate, and the formation of LE cells in the blood or bone marrow. {ECO:0000269|PubMed:17360460}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Immunodeficiency, common variable, 7 (CVID7) [MIM:614699]: A primary immunodeficiency characterized by antibody deficiency, hypogammaglobulinemia, recurrent bacterial infections and an inability to mount an antibody response to antigen. The defect results from a failure of B-cell differentiation and impaired secretion of immunoglobulins; the numbers of circulating B-cells is usually in the normal range, but can be low. {ECO:0000269|PubMed:22035880}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P07550; P37235 EC number EC 3.1.1.23 Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; Fatty acid biosynthesis; Fatty acid metabolism; Hydrolase; Lipid biosynthesis; Lipid degradation; Lipid metabolism; Membrane; Nitration; Phosphoprotein; Proteomics identification; Reference proteome; Serine esterase Protein physicochemical properties Chain ID A Molecular weight (Da) 31808.4 Length 289 Aromaticity 0.08 Instability index 29.7 Isoelectric point 6.73 Charge (pH=7) -0.91 2D Binding mode Binding energy (Kcal/mol) -7.23 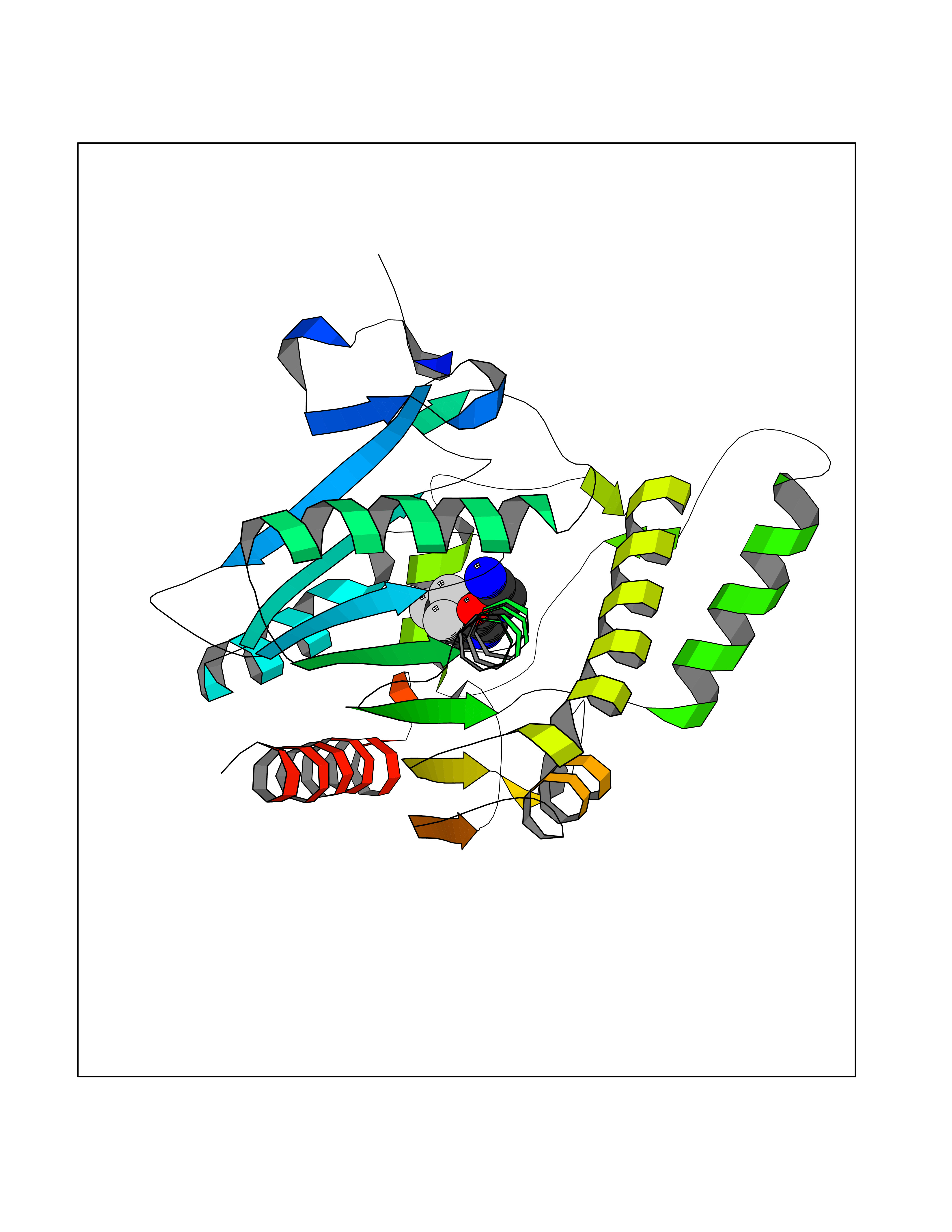 Molscript Map 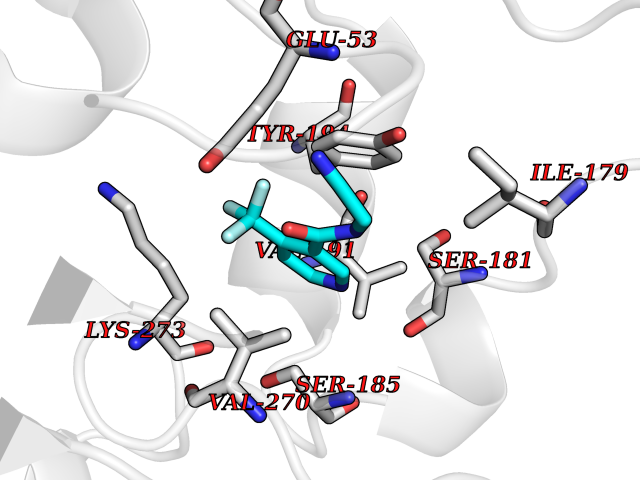 Pymol Map  Ligplot Map 3D Binding mode Sequence PRRTPQSIPYQDLPHLVNADGQYLFCRYWAPTGTPKALIFVSHGAGEHSGRYEELARMLMGLDLLVFAHDHVGHGQSEGERMVVSDFHVFVRDVLQHVDSMQKDYPGLPVFLLGHSMGGAIAILTAAERPGHFAGMVLISPLVLANPESATTFKVLAAKVLNSVLPNLSSGPIDSSVLSRNKTEVDIYNSDPLICRAGLKVCFGIQLLNAVSRVERALPKLTVPFLLLQGSADRLCDSKGAYLLMELAKSQDKTLKIYEGAYHVLHKELPEVTNSVFHEINMWVSQRTA Hydrogen bonds contact Hydrophobic contact | ||||
| 85 | GTPase HRas (HRAS) | 7L0F | 5.30 | |
Target general information Gen name HRAS Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms p21ras; cHras; c-H-ras; Transforming protein p21; HaRas; Ha-Ras; H-Ras-1; GTPase HRas, Nterminally processed Protein family Small GTPase superfamily, Ras family Biochemical class Small GTPase Function Ras proteins bind GDP/GTP and possess intrinsic GTPase activity. Involved in the activation of Ras protein signal transduction. Related diseases Costello syndrome (CSTLO) [MIM:218040]: A rare condition characterized by prenatally increased growth, postnatal growth deficiency, intellectual disability, distinctive facial appearance, cardiovascular abnormalities (typically pulmonic stenosis, hypertrophic cardiomyopathy and/or atrial tachycardia), tumor predisposition, skin and musculoskeletal abnormalities. {ECO:0000269|PubMed:16170316, ECO:0000269|PubMed:16329078, ECO:0000269|PubMed:16443854, ECO:0000269|PubMed:17054105, ECO:0000269|PubMed:18039947, ECO:0000269|PubMed:18247425, ECO:0000269|PubMed:19995790}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Congenital myopathy with excess of muscle spindles (CMEMS) [MIM:218040]: Variant of Costello syndrome. {ECO:0000269|PubMed:17412879}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Thyroid cancer, non-medullary, 2 (NMTC2) [MIM:188470]: A form of non-medullary thyroid cancer (NMTC), a cancer characterized by tumors originating from the thyroid follicular cells. NMTCs represent approximately 95% of all cases of thyroid cancer and are classified into papillary, follicular, Hurthle cell, and anaplastic neoplasms. {ECO:0000269|PubMed:12727991}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Mutations which change positions 12, 13 or 61 activate the potential of HRAS to transform cultured cells and are implicated in a variety of human tumors. {ECO:0000269|PubMed:3670300}.; DISEASE: Bladder cancer (BLC) [MIM:109800]: A malignancy originating in tissues of the urinary bladder. It often presents with multiple tumors appearing at different times and at different sites in the bladder. Most bladder cancers are transitional cell carcinomas that begin in cells that normally make up the inner lining of the bladder. Other types of bladder cancer include squamous cell carcinoma (cancer that begins in thin, flat cells) and adenocarcinoma (cancer that begins in cells that make and release mucus and other fluids). Bladder cancer is a complex disorder with both genetic and environmental influences. {ECO:0000269|PubMed:6298635, ECO:0000269|PubMed:6844927}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Schimmelpenning-Feuerstein-Mims syndrome (SFM) [MIM:163200]: A disease characterized by sebaceous nevi, often on the face, associated with variable ipsilateral abnormalities of the central nervous system, ocular anomalies, and skeletal defects. Many oral manifestations have been reported, not only including hypoplastic and malformed teeth, and mucosal papillomatosis, but also ankyloglossia, hemihyperplastic tongue, intraoral nevus, giant cell granuloma, ameloblastoma, bone cysts, follicular cysts, oligodontia, and odontodysplasia. Sebaceous nevi follow the lines of Blaschko and these can continue as linear intraoral lesions, as in mucosal papillomatosis. {ECO:0000269|PubMed:22683711}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB04315; DB04137; DB02210; DB08751; DB03226; DB15588 Interacts with Q99996-3; P53677-2; P10398; Q9NXL2-1; Q9UII2; Q9H7T9; Q00994; Q9H2G9; P15056; Q7Z569; Q5PSV4; Q9ULD4-2; Q96LL4; Q96HB5; Q49A88-3; Q96GN5-2; P24941; O95674; Q9H3R5; Q9Y4F5-3; Q86XR8; Q494V2-2; Q8WUX9; Q14117; Q9Y6W6; O14641; A0AVK6; Q8NB25; Q8IZU1; O94868-3; P15407; P15408; P52655; Q96CS2; Q9BT25; Q8IV36; O43248; Q53GQ0; P10809; Q8NDH6-2; Q8IY31-2; Q8NA54; Q13352; P28290-2; Q9BVG8-5; Q2M2Z5; Q6P597; P57682; Q9UH77; P08727; Q14525; Q14847-2; Q96LR2; P27338; Q99558; Q96EZ8; Q8TAC0; Q5JXC2; Q8NEH6; Q9Y605; Q96HT8; Q9GZM8; P21359; Q8N5V2; Q6PHZ7; Q9BZ95-3; A5D8V7; O43482; Q9BR81; O15534; Q9BUL5; O00329; O00329-2; Q9UPR0; Q96I34; Q15435-3; P04049; P11233; Q15311; Q12967; Q9NS23-2; Q9NS23-4; Q8WWW0; Q8TBY0; Q9P2K3-2; Q9NZL6; O15211; Q8IXN7; Q13671; Q13671-1; Q8WVD3; Q9BY12-3; Q13435; Q12824; Q13573; Q07889; Q86W54-2; Q92783-2; O75886; Q13586; Q8N4C7; O75528; P54274-2; Q9BXU0; Q5T0J7-2; Q5T1C6; Q8IUR5-4; P36406; Q86WT6-2; Q99598; Q6PF05; Q9UGJ1-2; Q9Y5Z9; P22415; Q495M9; Q9H270; Q8NEZ2; P19544-6; O43829; Q9C0F3; Q7Z637; Q86V28; P42337; Q9Z0S9; Q9EQZ6; P27671; Q5EBH1; Q5EBH1-1; P52306-5 EC number NA Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Cell membrane; Cytoplasm; Direct protein sequencing; Disease variant; Glycoprotein; Golgi apparatus; GTP-binding; Hydrolase; Isopeptide bond; Lipoprotein; Membrane; Methylation; Nucleotide-binding; Nucleus; Palmitate; Prenylation; Proteomics identification; Proto-oncogene; Reference proteome; S-nitrosylation; Ubl conjugation Protein physicochemical properties Chain ID E,F Molecular weight (Da) 28737.2 Length 259 Aromaticity 0.1 Instability index 30.69 Isoelectric point 5.64 Charge (pH=7) -4.15 2D Binding mode Binding energy (Kcal/mol) -7.24  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence MTEYKLVVVGAGGVGKSALTIQLIQNHFVDEYDPTIEDSYRKQVVIDGETCLLDILDTAGQEEYSAMRDQYMRTGEGFLCVFAINNTKSFEDIHQYREQIKRVKDSDDVPMVLVGNKCDLAARTVESRQAQDLARSYGIPYIETSAKTRQGVEDAFYTLVREIRQHSVPTKLEVVAATPTSLLISWDAPAVTVFFYIIAYGETGHGVGAFQAFRVPGSKSTATISGLKPGVDYTITVYARGYSKQGPYKPSPISINYRT Hydrogen bonds contact Hydrophobic contact | ||||
| 86 | Bacterial Threonine deaminase (Bact ilvA) | 1TDJ | 5.30 | |
Target general information Gen name Bact ilvA Organism Escherichia coli (strain K12) Uniprot ID TTD ID Synonyms ilvA; Putative threonine dehydratase Protein family Serine/threonine dehydratase family Biochemical class Carbon-nitrogen lyases Function Catalyzes the anaerobic formation of alpha-ketobutyrate and ammonia from threonine in a two-step reaction. The first step involved a dehydration of threonine and a production of enamine intermediates (aminocrotonate), which tautomerizes to its imine form(iminobutyrate). Both intermediates are unstable and short- lived. The second step is the nonenzymatic hydrolysis of the enamine/imine intermediates to form 2-ketobutyrate and free ammonia. In the low water environment of the cell, the second step is accelerated by RidA. Related diseases Familial male precocious puberty (FMPP) [MIM:176410]: In FMPP the receptor is constitutively activated. {ECO:0000269|PubMed:11134146, ECO:0000269|PubMed:11391350, ECO:0000269|PubMed:7629248, ECO:0000269|PubMed:7692306, ECO:0000269|PubMed:7714085, ECO:0000269|PubMed:7757065, ECO:0000269|PubMed:8281137, ECO:0000269|PubMed:8829636, ECO:0000269|PubMed:8929952, ECO:0000269|PubMed:9467560, ECO:0000269|PubMed:9661624}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Luteinizing hormone resistance (LHR) [MIM:238320]: An autosomal recessive disorder characterized by unresponsiveness to luteinizing hormone, defective sexual development in males, and defective follicular development and ovulation, amenorrhea and infertility in females. Two forms of the disorder have been defined in males. Type 1 is a severe form characterized by complete 46,XY male pseudohermaphroditism, low testosterone and high luteinizing hormone levels, total lack of responsiveness to luteinizing and chorionic gonadotropin hormones, lack of breast development, and absent development of secondary male sex characteristics. Type 2, a milder form, displays a broader range of phenotypic expression ranging from micropenis to severe hypospadias. {ECO:0000269|PubMed:12050206, ECO:0000269|PubMed:15372531, ECO:0000269|PubMed:15472221, ECO:0000269|PubMed:19551906, ECO:0000269|PubMed:7719343, ECO:0000269|PubMed:8559204, ECO:0000269|PubMed:9215288, ECO:0000269|PubMed:9514160, ECO:0000269|PubMed:9626144, ECO:0000269|PubMed:9626653}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number EC 4.3.1.19 Uniprot keywords 3D-structure; Allosteric enzyme; Amino-acid biosynthesis; Branched-chain amino acid biosynthesis; Isoleucine biosynthesis; Lyase; Pyridoxal phosphate; Reference proteome; Repeat Protein physicochemical properties Chain ID A Molecular weight (Da) 53966.2 Length 494 Aromaticity 0.08 Instability index 41.16 Isoelectric point 5.88 Charge (pH=7) -8.91 2D Binding mode Binding energy (Kcal/mol) -7.22  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence QPLSGAPEGAEYLRAVLRAPVYEAAQVTPLQKMEKLSSRLDNVILVKREDRQPVHSFKLRGAYAMMAGLTEEQKAHGVITASAGNHAQGVAFSSARLGVKALIVMPTATADIKVDAVRGFGGEVLLHGANFDEAKAKAIELSQQQGFTWVPPFDHPMVIAGQGTLALELLQQDAHLDRVFVPVGGGGLAAGVAVLIKQLMPQIKVIAVEAEDSACLKAALDAGHPVDLPRVGLFAEGVAVKRIGDETFRLCQEYLDDIITVDSDAICAAMKDLFEDVRAVAEPSGALALAGMKKYIALHNIRGERLAHILSGANVNFHGLRYVSERCELGEQREALLAVTIPEEKGSFLKFCQLLGGRSVTEFNYRFADAKNACIFVGVRLSRGLEERKEILQMLNDGGYSVVDLSDDEMAKLHVRYMVGGRPSHPLQERLYSFEFPESPGALLRFLNTLGTYWNISLFHYRSHGTDYGRVLAAFEYDCHDETNNPAFRFFLAG Hydrogen bonds contact Hydrophobic contact | ||||
| 87 | Cytochrome P450 1A2 | 2HI4 | 5.29 | |
Target general information Gen name CYP1A2 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Cytochrome P450 family Biochemical class Oxidoreductase Function Aromatase activity.Caffeine oxidase activity.Demethylase activity.Electron carrier activity.Enzyme binding.Heme binding.Iron ion binding.Monooxygenase activity.Oxidoreductase activity.Oxidoreductase activity, acting on paired donors, with incorporation or reduction of molecular oxygen, reduced flavin or flavoprotein as one donor, and incorporation of one atom of oxygen.Oxygen binding. Related diseases Myeloperoxidase deficiency (MPOD) [MIM:254600]: A disorder characterized by decreased myeloperoxidase activity in neutrophils and monocytes that results in disseminated candidiasis. {ECO:0000269|PubMed:37198333, ECO:0000269|PubMed:7904599, ECO:0000269|PubMed:8142659, ECO:0000269|PubMed:8621627, ECO:0000269|PubMed:9354683, ECO:0000269|PubMed:9637725}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08496; DB01667; DB14132; DB04356; DB02489; DB11932; DB12001; DB05812; DB13573; DB01418; DB00316; DB15568; DB06594; DB00518; DB05396; DB00969; DB07453; DB01424; DB01223; DB01118; DB00321; DB00261; DB01217; DB01435; DB06605; DB05676; DB06413; DB06216; DB01072; DB15011; DB06442; DB06626; DB00993; DB00972; DB13203; DB05015; DB16703; DB06769; DB01086; DB06770; DB06771; DB06732; DB00195; DB04889; DB11967; DB13975; DB00188; DB12151; DB01558; DB14018; DB13812; DB00201; DB09061; DB14737; DB11791; DB06774; DB00564; DB06016; DB01136; DB12814; DB00477; DB00356; DB01166; DB00501; DB01012; DB00568; DB00827; DB00537; DB00215; DB12499; DB14025; DB00349; DB01242; DB00575; DB00758; DB00363; DB00286; DB11672; DB14635; DB00924; DB08912; DB00851; DB06292; DB01254; DB01609; DB01151; DB16650; DB12161; DB01191; DB00633; DB11994; DB00586; DB11511; DB12945; DB00280; DB01184; DB09167; DB05928; DB01142; DB09273; DB00470; DB00476; DB00625; DB15444; DB06210; DB13874; DB11718; DB00467; DB11404; DB00530; DB00783; DB13952; DB13953; DB13954; DB13955; DB13956; DB00655; DB04574; DB13592; DB00330; DB00898; DB00977; DB00773; DB01628; DB00927; DB04854; DB01482; DB00574; DB12265; DB15669; DB01195; DB08972; DB04841; DB00544; DB00472; DB00499; DB00176; DB01320; DB00998; DB14029; DB06160; DB01044; DB01241; DB01155; DB01645; DB01381; DB00986; DB00365; DB00400; DB05708; DB00629; DB00502; DB01094; DB14999; DB04076; DB11737; DB00619; DB00458; DB11564; DB01306; DB09456; DB09564; DB01307; DB00047; DB01309; DB00030; DB00046; DB11567; DB00071; DB11568; DB05258; DB00034; DB00105; DB15131; DB00011; DB00018; DB00069; DB00060; DB00068; DB00033; DB00951; DB11757; DB09570; DB01026; DB01097; DB16217; DB09078; DB01002; DB05667; DB00281; DB12406; DB09198; DB04948; DB00978; DB06448; DB16220; DB01601; DB00455; DB04871; DB06077; DB01283; DB00772; DB00934; DB06234; DB14009; DB00784; DB01065; DB00170; DB00454; DB00532; DB00333; DB00763; DB00553; DB01028; DB09241; DB01233; DB00379; DB06148; DB01388; DB06595; DB00370; DB16236; DB00745; DB11763; DB00218; DB06510; DB14011; DB00461; DB00607; DB00779; DB00788; DB06600; DB00238; DB06803; DB00184; DB01115; DB11793; DB00435; DB05115; DB00717; DB01059; DB00540; DB05990; DB01165; DB00334; DB16267; DB00338; DB00904; DB11632; DB11443; DB01173; DB11837; DB09330; DB01303; DB11697; DB00377; DB00715; DB06589; DB11774; DB00487; DB00008; DB00022; DB09122; DB13634; DB00806; DB11198; DB08883; DB00850; DB03783; DB01174; DB00388; DB00252; DB11450; DB01100; DB13823; DB04951; DB17472; DB11642; DB08910; DB15822; DB01058; DB01087; DB00794; DB00420; DB09288; DB01182; DB06479; DB00818; DB00571; DB13449; DB11892; DB04216; DB00908; DB00468; DB01129; DB00980; DB09290; DB00863; DB01367; DB00409; DB02709; DB13174; DB01045; DB11753; DB00740; DB14924; DB00503; DB00533; DB01656; DB15119; DB00268; DB00296; DB00412; DB00817; DB12332; DB13772; DB06654; DB11491; DB00418; DB01037; DB11689; DB06290; DB13261; DB15093; DB00052; DB00398; DB01208; DB09118; DB00428; DB06820; DB00382; DB00675; DB06083; DB09071; DB05488; DB09256; DB01079; DB01405; DB00857; DB08880; DB11712; DB01412; DB00277; DB00730; DB01623; DB00208; DB06137; DB00697; DB01056; DB06264; DB00752; DB00384; DB12245; DB00831; DB15442; DB00440; DB00685; DB08867; DB14989; DB13609; DB06235; DB00313; DB08881; DB00661; DB09185; DB12026; DB00682; DB02134; DB00549; DB00744; DB00315; DB00425; DB09225; DB09120 Interacts with O95870 EC number 1.14.14.1; 4.2.1.152 Uniprot keywords 3D-structure; Direct protein sequencing; Endoplasmic reticulum; Fatty acid metabolism; Glycoprotein; Heme; Iron; Lipid metabolism; Lyase; Membrane; Metal-binding; Microsome; Monooxygenase; Oxidoreductase; Proteomics identification; Reference proteome; Steroid metabolism; Sterol metabolism Protein physicochemical properties Chain ID A Molecular weight (Da) 54475 Length 480 Aromaticity 0.1 Instability index 40.43 Isoelectric point 9.16 Charge (pH=7) 9.89 2D Binding mode Binding energy (Kcal/mol) -7.21  Molscript Map 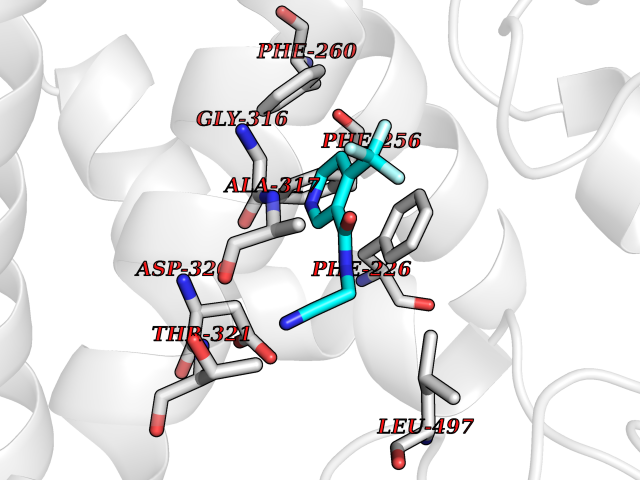 Pymol Map 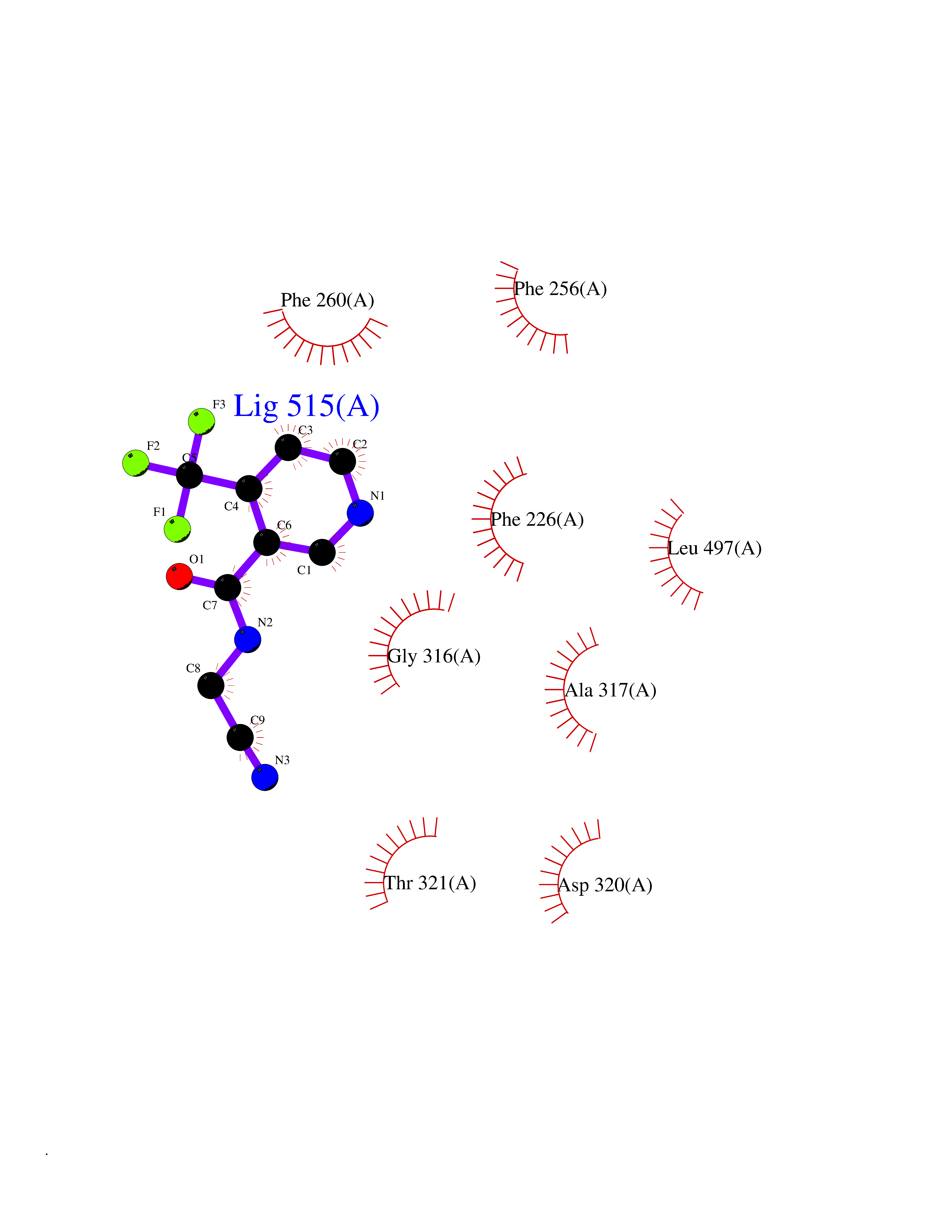 Ligplot Map 3D Binding mode Sequence RVPKGLKSPPEPWGWPLLGHVLTLGKNPHLALSRMSQRYGDVLQIRIGSTPVLVLSRLDTIRQALVRQGDDFKGRPDLYTSTLITDGQSLTFSTDSGPVWAARRRLAQNALNTFSIASDPASSSSCYLEEHVSKEAKALISRLQELMAGPGHFDPYNQVVVSVANVIGAMCFGQHFPESSDEMLSLVKNTHEFVETASSGNPLDFFPILRYLPNPALQRFKAFNQRFLWFLQKTVQEHYQDFDKNSVRDITGALFKHSKKGPRASGNLIPQEKIVNLVNDIFGAGFDTVTTAISWSLMYLVTKPEIQRKIQKELDTVIGRERRPRLSDRPQLPYLEAFILETFRHSSFLPFTIPHSTTRDTTLNGFYIPKKCCVFVNQWQVNHDPELWEDPSEFRPERFLTADGTAINKPLSEKMMLFGMGKRRCIGEVLAKWEIFLFLAILLQQLEFSVPPGVKVDLTPIYGLTMKHARCEHVQARRFS Hydrogen bonds contact Hydrophobic contact | ||||
| 88 | Histamine N-methyltransferase (HNMT) | 2AOT | 5.29 | |
Target general information Gen name HNMT Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Histamine-N-methyltransferase; HNMT; HMT Protein family Class I-like SAM-binding methyltransferase superfamily, HNMT family Biochemical class Methyltransferase Function Inactivates histamine by N-methylation. Plays an important role in degrading histamine and in regulating the airway response to histamine. Related diseases Intellectual developmental disorder, autosomal recessive 51 (MRT51) [MIM:616739]: A disorder characterized by significantly below average general intellectual functioning associated with impairments in adaptive behavior and manifested during the developmental period. {ECO:0000269|PubMed:26206890}. The disease is caused by variants affecting distinct genetic loci, including the gene represented in this entry. Drugs (DrugBank ID) DB00613; DB13875; DB05381; DB04655; DB01103; DB01752; DB07106 Interacts with NA EC number EC 2.1.1.8 Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; Disease variant; Intellectual disability; Methyltransferase; Proteomics identification; Reference proteome; S-adenosyl-L-methionine; Transferase Protein physicochemical properties Chain ID A,B Molecular weight (Da) 32712 Length 288 Aromaticity 0.1 Instability index 36.38 Isoelectric point 5.18 Charge (pH=7) -9.97 2D Binding mode Binding energy (Kcal/mol) -7.21 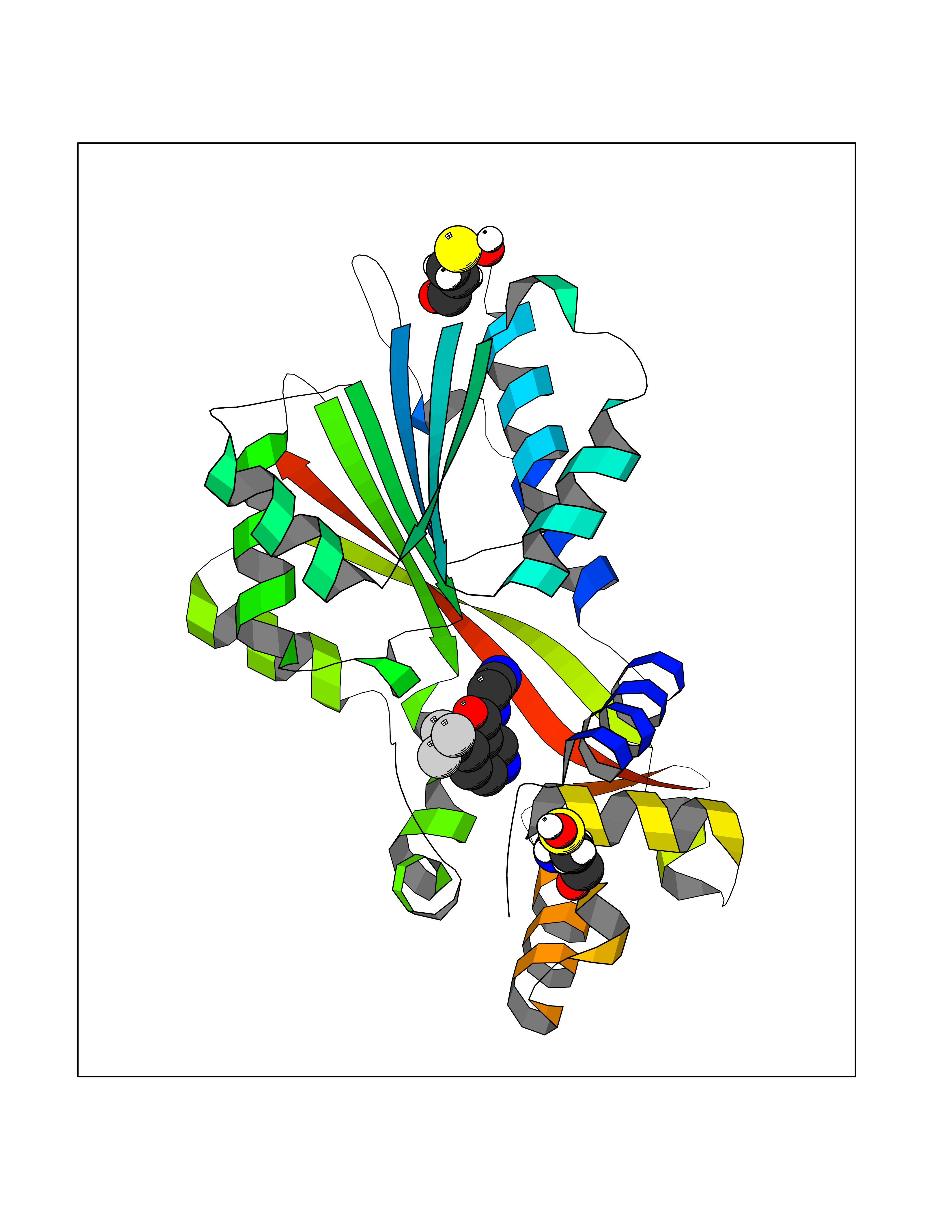 Molscript Map 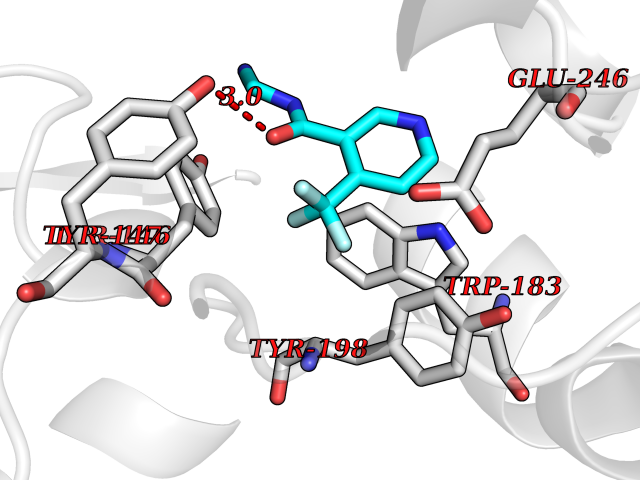 Pymol Map  Ligplot Map 3D Binding mode Sequence MRSLFSDHGKYVESFRRFLNHSTEHQCMQEFMDKKLPGIIGRIGDTKSEIKILSIGGGAGEIDLQILSKVQAQYPGVXINNEVVEPSAEQIAKYKELVAKTSNLENVKFAWHKETSSEYQSRMLEKKELQKWDFIHMIQMLYYVKDIPATLKFFHSLLGTNAKMLIIVVSGSSGWDKLWKKYGSRFPQDDLCQYITSDDLTQMLDNLGLKYECYDLLSTMDISDCFIDGNENGDLLWDFLTETXNFNATAPPDLRAELGKDLQEPEFSAKKEGKVLFNNTLSFIVIEA Hydrogen bonds contact Hydrophobic contact | ||||
| 89 | Opioid receptor sigma 1 (OPRS1) | 5HK1 | 5.29 | |
Target general information Gen name SIGMAR1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms hSigmaR1; Sigma1R; Sigma1-receptor; Sigma non-opioid intracellular receptor 1; Sigma 1-type opioid receptor; SRBP; SR31747-binding protein; SR31747 binding protein 1; SR-BP; SIG-1R; Opioid receptor, s Protein family ERG2 family Biochemical class GPCR rhodopsin Function Involved in the regulation of different receptors it plays a role in BDNF signaling and EGF signaling. Also regulates ion channels like the potassium channel and could modulate neurotransmitter release. Plays a role in calcium signaling through modulation together with ANK2 of the ITP3R-dependent calcium efflux at the endoplasmic reticulum. Plays a role in several other cell functions including proliferation, survival and death. Originally identified for its ability to bind various psychoactive drugs it is involved in learning processes, memory and mood alteration. Necessary for proper mitochondrial axonal transport in motor neurons, in particular the retrograde movement of mitochondria. Plays a role in protecting cells against oxidative stress-induced cell death via its interaction with RNF112. Functions in lipid transport from the endoplasmic reticulum and is involved in a wide array of cellular functions probably through regulation of the biogenesis of lipid microdomains at the plasma membrane. Related diseases Amyotrophic lateral sclerosis 16, juvenile (ALS16) [MIM:614373]: A neurodegenerative disorder affecting upper motor neurons in the brain and lower motor neurons in the brain stem and spinal cord, resulting in fatal paralysis. Sensory abnormalities are absent. The pathologic hallmarks of the disease include pallor of the corticospinal tract due to loss of motor neurons, presence of ubiquitin-positive inclusions within surviving motor neurons, and deposition of pathologic aggregates. The etiology of amyotrophic lateral sclerosis is likely to be multifactorial, involving both genetic and environmental factors. The disease is inherited in 5-10% of the cases. {ECO:0000269|PubMed:21842496}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Neuronopathy, distal hereditary motor, autosomal recessive 2 (HMNR2) [MIM:605726]: A form of distal hereditary motor neuronopathy, a heterogeneous group of neuromuscular disorders caused by selective degeneration of motor neurons in the anterior horn of the spinal cord, without sensory deficit in the posterior horn. The overall clinical picture consists of a classical distal muscular atrophy syndrome in the legs without clinical sensory loss. The disease starts with weakness and wasting of distal muscles of the anterior tibial and peroneal compartments of the legs. Later on, weakness and atrophy may expand to the proximal muscles of the lower limbs and/or to the distal upper limbs. HMNR2 is characterized by onset of distal muscle weakness and wasting affecting the lower and upper limbs in the first decade. {ECO:0000269|PubMed:26078401, ECO:0000269|PubMed:27629094}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00321; DB09014; DB00907; DB00514; DB01488; DB00574; DB00502; DB00956; DB00704; DB00540; DB06174; DB00652; DB11186; DB03575; DB05316; DB01708; DB00409; DB01104 Interacts with Q92847-1; Q99720-1; O00213-2; P17612; P50454; P37173 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Amyotrophic lateral sclerosis; Cell junction; Cell membrane; Cell projection; Cytoplasmic vesicle; Direct protein sequencing; Disease variant; Endoplasmic reticulum; Lipid droplet; Lipid transport; Membrane; Neurodegeneration; Neuropathy; Nucleus; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Synapse; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A,B,C Molecular weight (Da) 20805.3 Length 185 Aromaticity 0.14 Instability index 31.72 Isoelectric point 5.44 Charge (pH=7) -6.63 2D Binding mode Binding energy (Kcal/mol) -7.21  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence VFQREEIAQLARQYAGLDHELAFSRLIVELRRLHPGHVLPDEELQWVFVNAGGWMGAMCLLHASLSEYVLLFGTALGSRGHSGRYWAEISDTIISGTFHQWREGTTKSEVFYPGETVVHGPGEATAVEWGPNTWMVEYGRGVIPSTLAFALADTVFSTQDFLTLFYTLRSYARGLRLELTTYLFG Hydrogen bonds contact Hydrophobic contact | ||||
| 90 | Serine/threonine-protein kinase cot (COT) | 4Y85 | 5.29 | |
Target general information Gen name MAP3K8 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Tumor progression locus 2; TPL-2; Proto-oncogene c-Cot; Mitogen-activated protein kinase kinase kinase 8; ESTF; Cancer Osaka thyroid oncogene; COT Protein family Protein kinase superfamily, STE Ser/Thr protein kinase family, MAP kinase kinase kinase subfamily Biochemical class NA Function Required for lipopolysaccharide (LPS)-induced, TLR4-mediated activation of the MAPK/ERK pathway in macrophages, thus being critical for production of the proinflammatory cytokine TNF-alpha (TNF) during immune responses. Involved in the regulation of T-helper cell differentiation and IFNG expression in T-cells. Involved in mediating host resistance to bacterial infection through negative regulation of type I interferon (IFN) production. In vitro, activates MAPK/ERK pathway in response to IL1 in an IRAK1-independent manner, leading to up-regulation of IL8 and CCL4. Transduces CD40 and TNFRSF1A signals that activate ERK in B-cells and macrophages, and thus may play a role in the regulation of immunoglobulin production. May also play a role in the transduction of TNF signals that activate JNK and NF-kappa-B in some cell types. In adipocytes, activates MAPK/ERK pathway in an IKBKB-dependent manner in response to IL1B and TNF, but not insulin, leading to induction of lipolysis. Plays a role in the cell cycle. Isoform 1 shows some transforming activity, although it is much weaker than that of the activated oncogenic variant. Related diseases Hyperinsulinemic hypoglycemia, familial, 2 (HHF2) [MIM:601820]: A form of hyperinsulinemic hypoglycemia, a clinically and genetically heterogeneous disorder characterized by inappropriate insulin secretion from the pancreatic beta-cells in the presence of low blood glucose levels. HHF2 is a common cause of persistent hypoglycemia in infancy. Unless early and aggressive intervention is undertaken, brain damage from recurrent episodes of hypoglycemia may occur. HHF2 inheritance can be autosomal dominant or autosomal recessive. {ECO:0000269|PubMed:10204114, ECO:0000269|PubMed:12364426, ECO:0000269|PubMed:15562009, ECO:0000269|PubMed:15579781, ECO:0000269|PubMed:15807877, ECO:0000269|PubMed:15998776, ECO:0000269|PubMed:16332676, ECO:0000269|PubMed:16357843, ECO:0000269|PubMed:18596924, ECO:0000269|PubMed:19357197, ECO:0000269|PubMed:7847376, ECO:0000269|PubMed:8923010}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Diabetes mellitus, permanent neonatal, 2 (PNDM2) [MIM:618856]: A form of permanent neonatal diabetes mellitus, a type of diabetes characterized by onset of persistent hyperglycemia within the first six months of life. Initial clinical manifestations include intrauterine growth retardation, hyperglycemia, glycosuria, osmotic polyuria, severe dehydration, and failure to thrive. Some PNDM2 patients may also have developmental delay, muscle weakness, epilepsy and dysmorphic features. PNDM2 transmission pattern is consistent with autosomal dominant inheritance. {ECO:0000269|PubMed:15115830, ECO:0000269|PubMed:15292329, ECO:0000269|PubMed:15448106, ECO:0000269|PubMed:15448107, ECO:0000269|PubMed:15580558, ECO:0000269|PubMed:15583126, ECO:0000269|PubMed:16609879, ECO:0000269|PubMed:16731833, ECO:0000269|PubMed:17213273, ECO:0000269|PubMed:17652641, ECO:0000269|PubMed:17855752, ECO:0000269|PubMed:20022885, ECO:0000269|PubMed:28842488}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Transient neonatal diabetes mellitus 3 (TNDM3) [MIM:610582]: Neonatal diabetes mellitus, defined as insulin-requiring hyperglycemia within the first month of life, is a rare entity. In about half of the neonates, diabetes is transient and resolves at a median age of 3 months, whereas the rest have a permanent form of diabetes. In a significant number of patients with transient neonatal diabetes mellitus, diabetes type 2 appears later in life. The onset and severity of TNDM3 is variable with childhood-onset diabetes, gestational diabetes or adult-onset diabetes described. {ECO:0000269|PubMed:15718250, ECO:0000269|PubMed:15784703}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Defects in KCNJ11 may contribute to non-insulin-dependent diabetes mellitus (NIDDM), also known as diabetes mellitus type 2.; DISEASE: Maturity-onset diabetes of the young 13 (MODY13) [MIM:616329]: A form of diabetes that is characterized by an autosomal dominant mode of inheritance, onset in childhood or early adulthood (usually before 25 years of age), a primary defect in insulin secretion and frequent insulin-independence at the beginning of the disease. {ECO:0000269|PubMed:22701567}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P08238; P19838; Q00653; Q13526; Q8NFZ5 EC number EC 2.7.11.25 Uniprot keywords 3D-structure; Alternative initiation; ATP-binding; Cell cycle; Cytoplasm; Immunity; Kinase; Magnesium; Metal-binding; Nucleotide-binding; Phosphoprotein; Proteomics identification; Proto-oncogene; Reference proteome; Serine/threonine-protein kinase; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 34773.8 Length 307 Aromaticity 0.09 Instability index 39.24 Isoelectric point 6.68 Charge (pH=7) -1.2 2D Binding mode Binding energy (Kcal/mol) -7.21 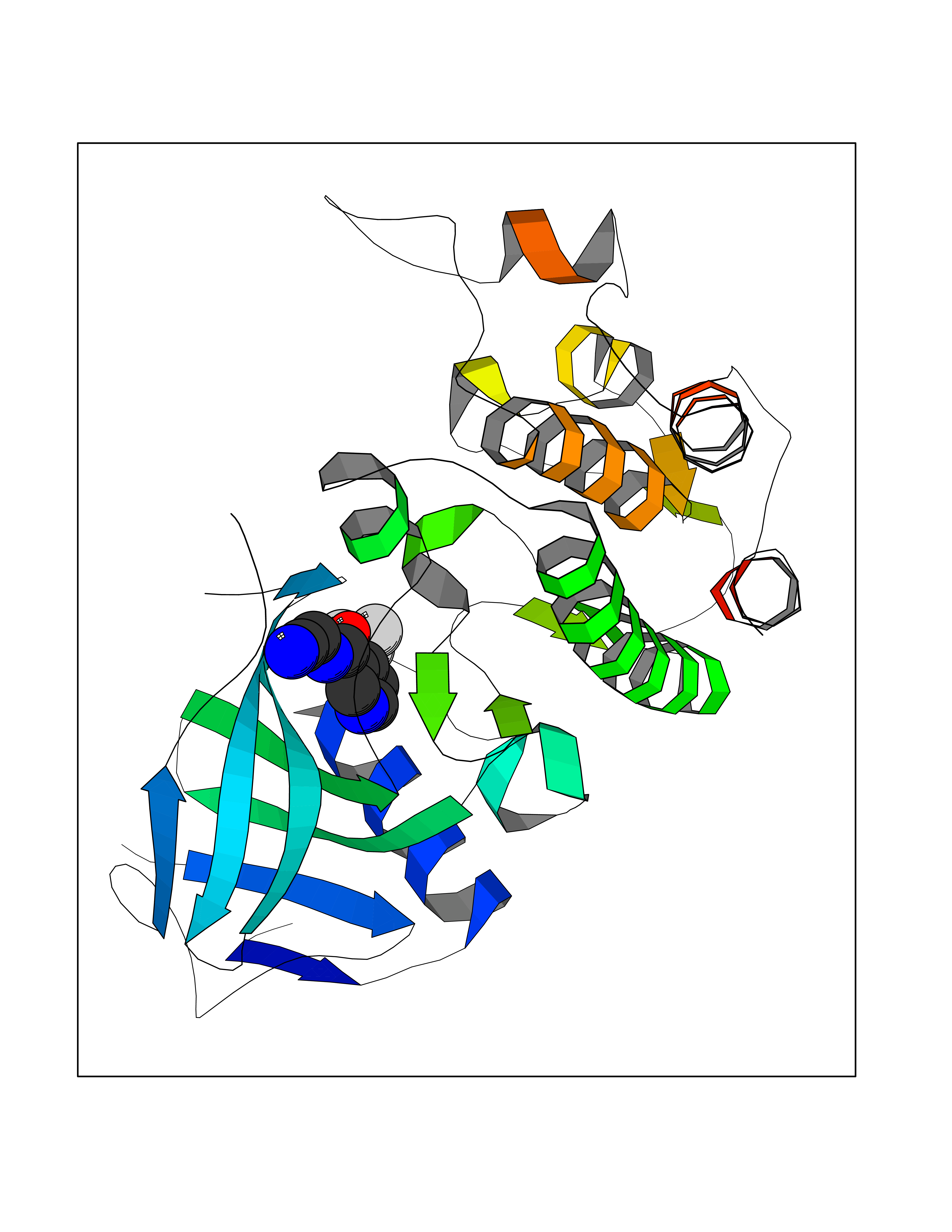 Molscript Map 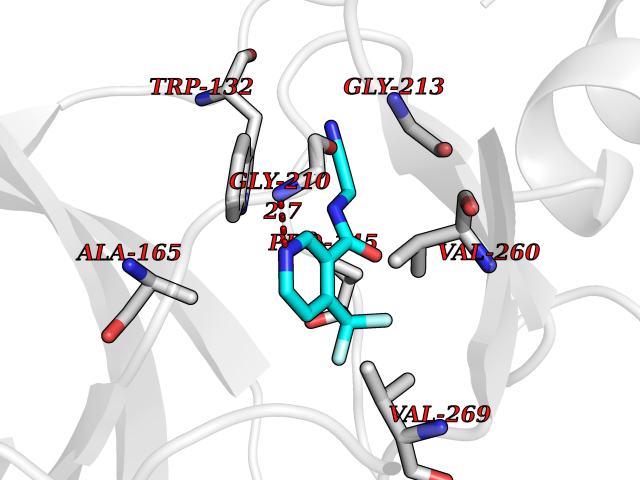 Pymol Map 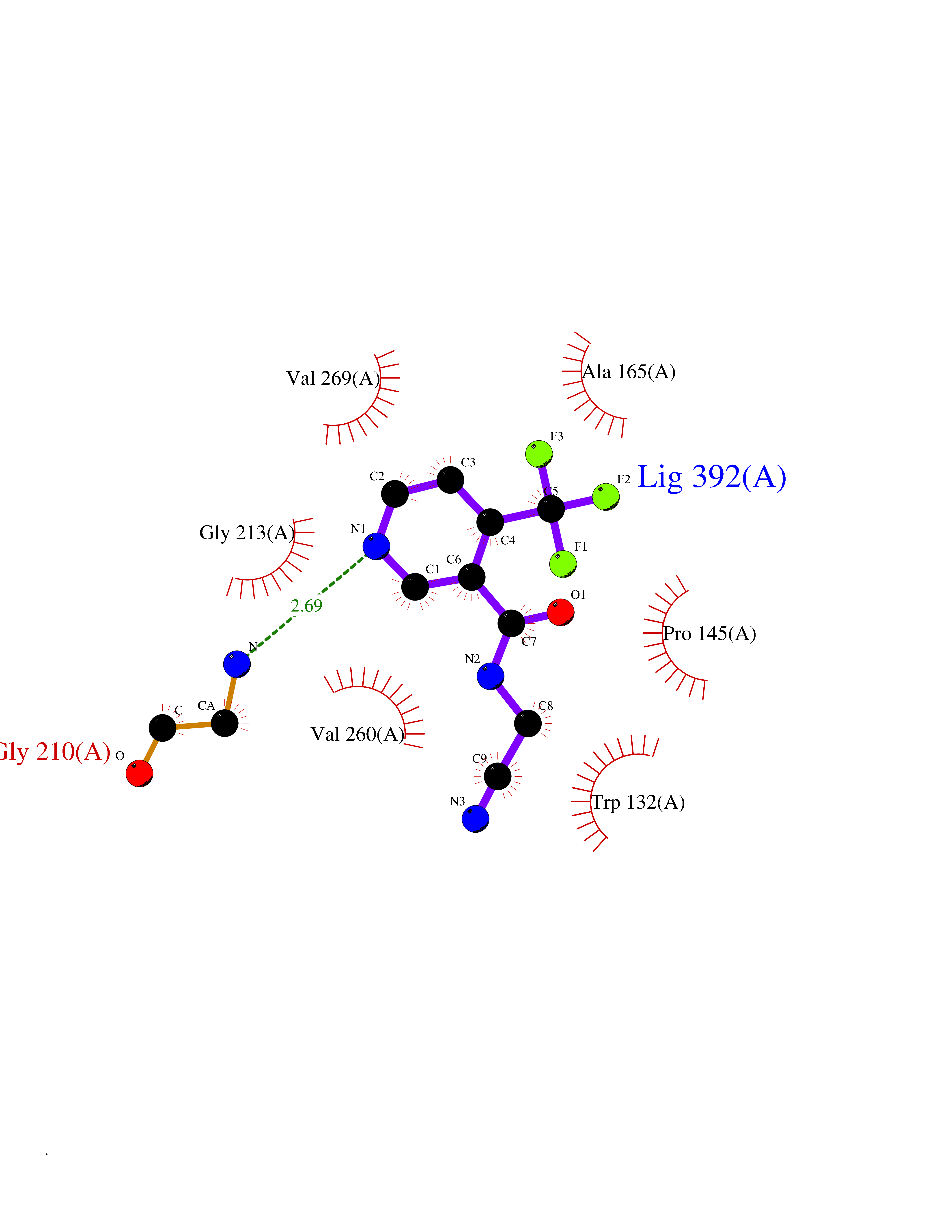 Ligplot Map 3D Binding mode Sequence LSSVRYGTVEDLLAFANHISNTPQESGILLNMVITPQNGRYQIDSDVLLIPWKLTYRNIFIPRGAFGKVYLAQDIKTKKRMACKLIPVDQFKPSDVEIQACFRHENIAELYGAVLWGETVHLFMEAGEGGSVLEKLESCGPMREFEIIWVTKHVLKGLDFLHSKKVIHHDIKPSNIVFMSTKAVLVDFGLSVQMTEDVYFPKDLRGTEIYMSPEVILCRGHSTKADIYSLGATLIHMQTGTPPWVKRYPRSAYPSYLYIIHKQAPPLEDIADDCSPGMRELIEASLERNPNHRPRAADLLKHEALNP Hydrogen bonds contact Hydrophobic contact | ||||
| 91 | Histamine H3 receptor (H3R) | 7F61 | 5.29 | |
Target general information Gen name HRH3 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Histamine receptor 3; HH3R; GPCR97; G-protein coupled receptor 97; G protein-coupled receptor 97 Protein family G-protein coupled receptor 1 family Biochemical class GPCR rhodopsin Function Signals through the inhibition of adenylate cyclase and displays high constitutive activity (spontaneous activity in the absence of agonist). Agonist stimulation of isoform 3 neither modified adenylate cyclase activity nor induced intracellular calcium mobilization. The H3 subclass of histamine receptors could mediate the histamine signals in CNS and peripheral nervous system. Related diseases Immunodeficiency 48 (IMD48) [MIM:269840]: A form of severe immunodeficiency characterized by a selective absence of CD8+ T-cells. {ECO:0000269|PubMed:11123350, ECO:0000269|PubMed:11412303, ECO:0000269|PubMed:18509675, ECO:0000269|PubMed:8124727, ECO:0000269|PubMed:8202713}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Autoimmune disease, multisystem, infantile-onset, 2 (ADMIO2) [MIM:617006]: An autosomal recessive, autoimmune disorder characterized by systemic manifestations including blistering skin disease, uncontrollable bullous pemphigoid, inflammatory colitis, autoimmune hypothyroidism, proteinuria and nephrotic syndrome. {ECO:0000269|PubMed:26783323}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01238; DB06698; DB05381; DB17087; DB05080; DB00768; DB11642 Interacts with NA EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disulfide bond; G-protein coupled receptor; Glycoprotein; Membrane; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 34321.1 Length 301 Aromaticity 0.16 Instability index 32.22 Isoelectric point 9.63 Charge (pH=7) 15.11 2D Binding mode Binding energy (Kcal/mol) -7.21 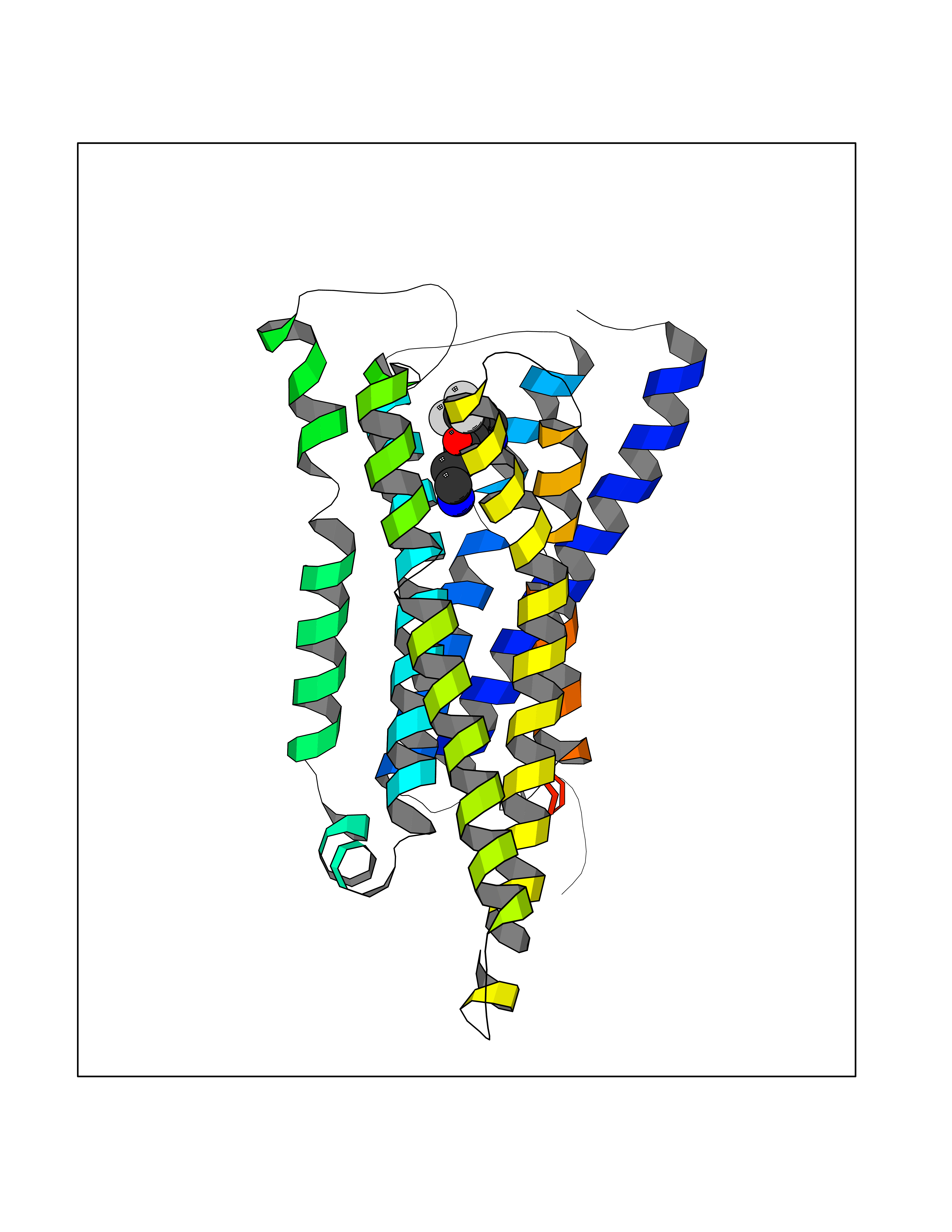 Molscript Map 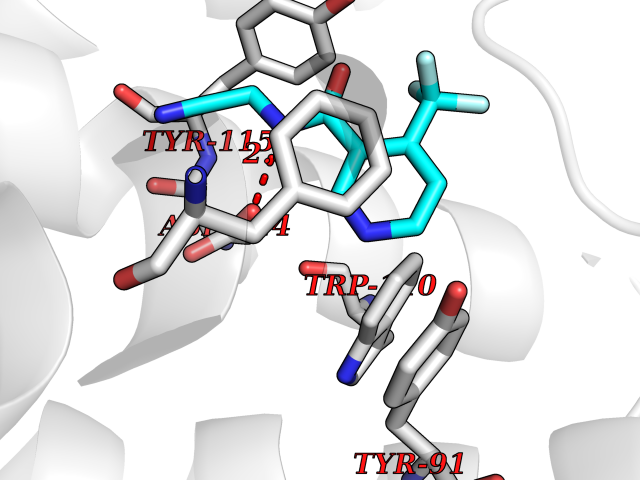 Pymol Map 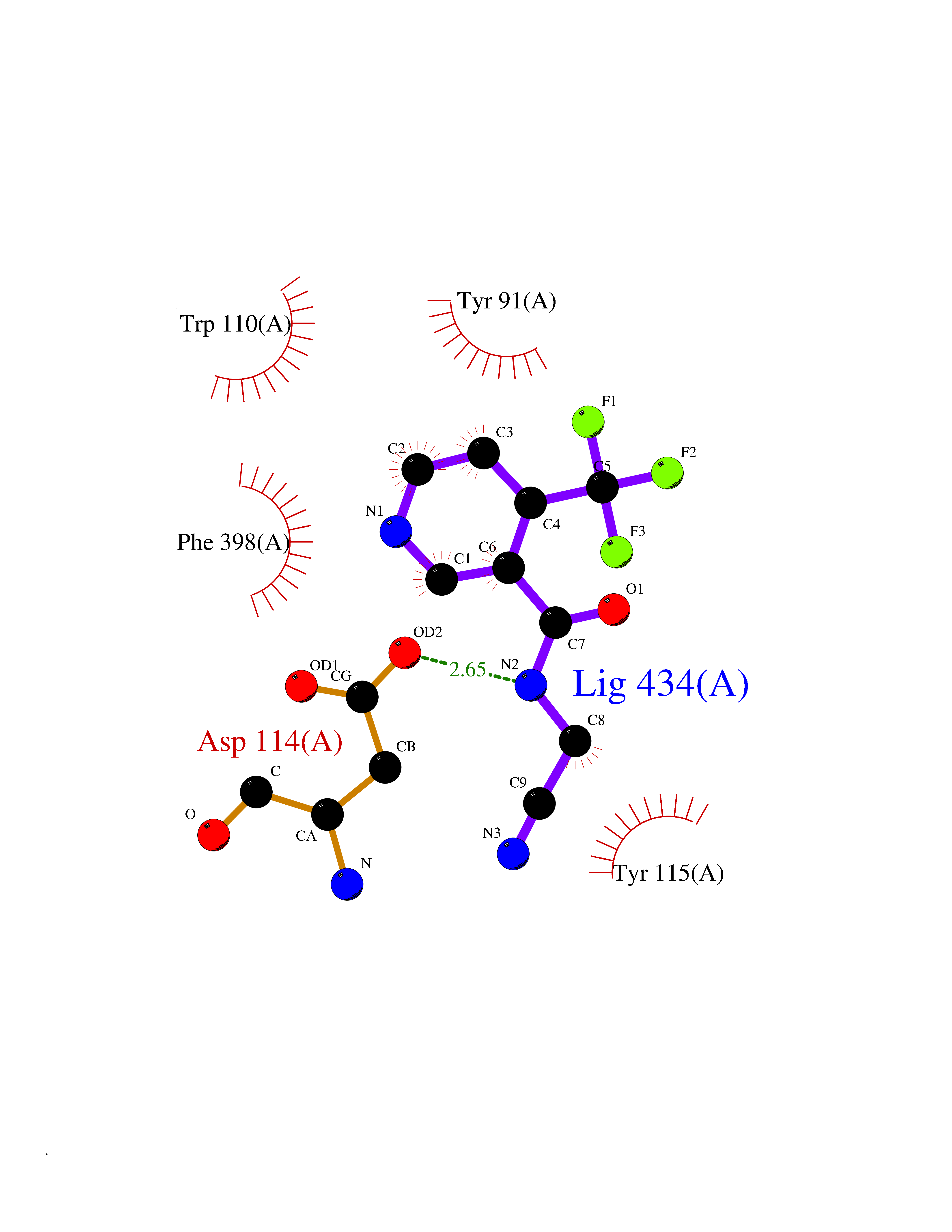 Ligplot Map 3D Binding mode Sequence RGFSAAWTAVLAALMALLIVATVLGNALVMLAFVADSSLRTQNNFFLLNLAISDFLVGAFCIPLYVPYVLTGRWTFGRGLCKLWLVVDYLLCTSKAFNIVLISYDRFLSVTRAVSYRAQQGDTRRAVRKMLLVWVLAFLLYGPAILSWEYLSGGSSIPEGHCYAEFFYNWYFLITASTLEFFTPFLSVTFFNLSIYLNIQRRTRLRLDGAREAAGRFRLSRDRKVAKSLAVIVSIFGLCWAPYTLLMIIRAACHGHCVPDYWYETSFWLLWANSAVNPVLYPLCHHSFRRAFTKLLCPQKL Hydrogen bonds contact Hydrophobic contact | ||||
| 92 | Caspase-7 (CASP7) | 1SHJ | 5.29 | |
Target general information Gen name CASP7 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms MCH3; ICE-like apoptotic protease 3; ICE-LAP3; CMH-1; CASP-7; Apoptotic protease Mch-3 Protein family Peptidase C14A family Biochemical class Peptidase Function Cleaves and activates sterol regulatory element binding proteins (SREBPs). Proteolytically cleaves poly(ADP-ribose) polymerase (PARP) at a '216-Asp-|-Gly-217' bond. Overexpression promotes programmed cell death. Involved in the activation cascade of caspases responsible for apoptosis execution. Related diseases Pregnancy loss, recurrent, 3 (RPRGL3) [MIM:614391]: A common complication of pregnancy, resulting in spontaneous abortion before the fetus has reached viability. The term includes all miscarriages from the time of conception until 24 weeks of gestation. Recurrent pregnancy loss is defined as 3 or more consecutive spontaneous abortions. {ECO:0000269|PubMed:17339269}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB05408; DB03384; DB06255 Interacts with Q13490; P83105; P42858; Q8N4N3-2; P43364; Q16236; Q9GZT8; Q13177; P27986-2; P21673; Q86WV1-2; P17405; P98170 EC number EC 3.4.22.60 Uniprot keywords 3D-structure; Acetylation; Allosteric enzyme; Alternative splicing; Apoptosis; Cytoplasm; Hydrolase; Nucleus; Phosphoprotein; Protease; Proteomics identification; Reference proteome; RNA-binding; Secreted; Thiol protease; Ubl conjugation; Zymogen Protein physicochemical properties Chain ID A,B Molecular weight (Da) 47441.5 Length 417 Aromaticity 0.11 Instability index 20.98 Isoelectric point 8.38 Charge (pH=7) 6.12 2D Binding mode Binding energy (Kcal/mol) -7.21 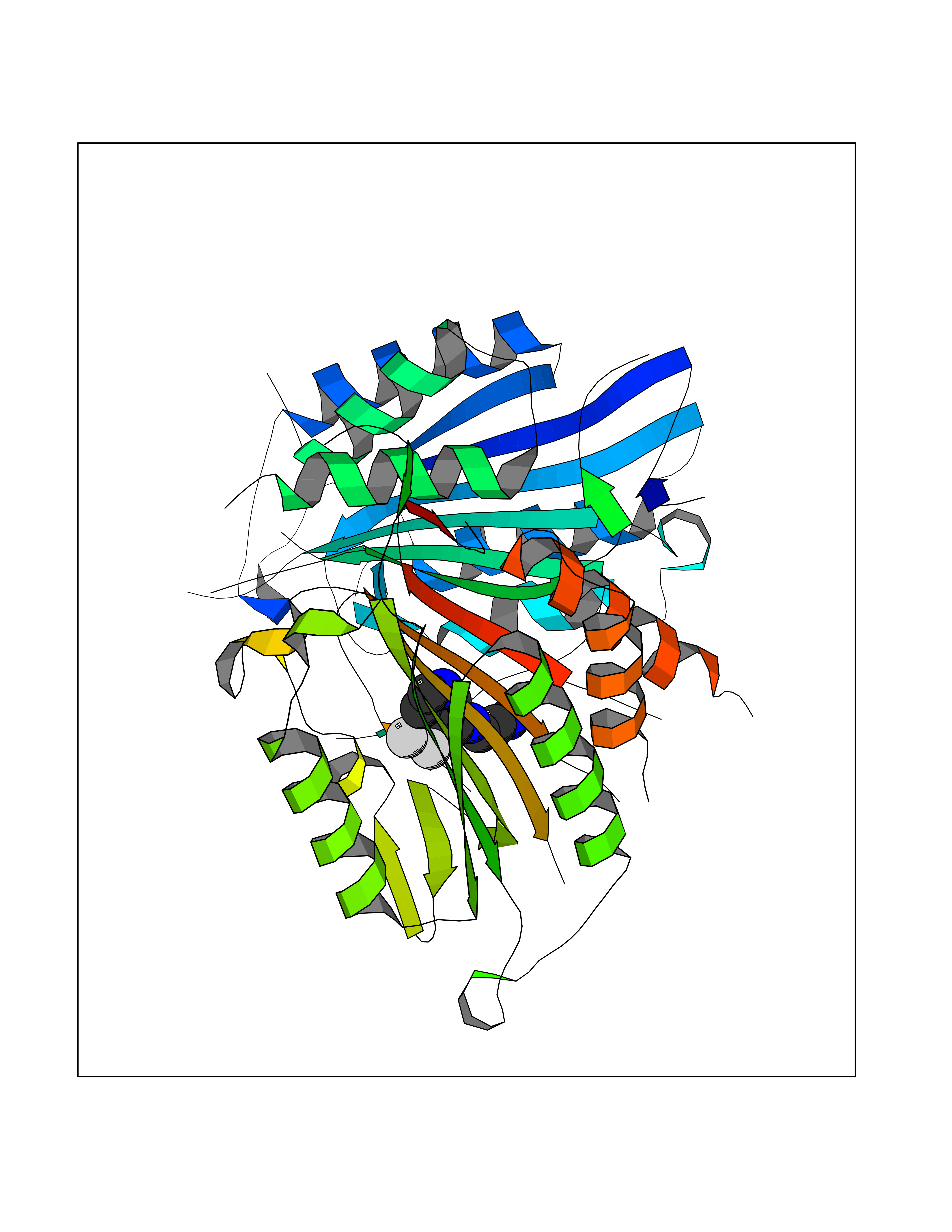 Molscript Map  Pymol Map 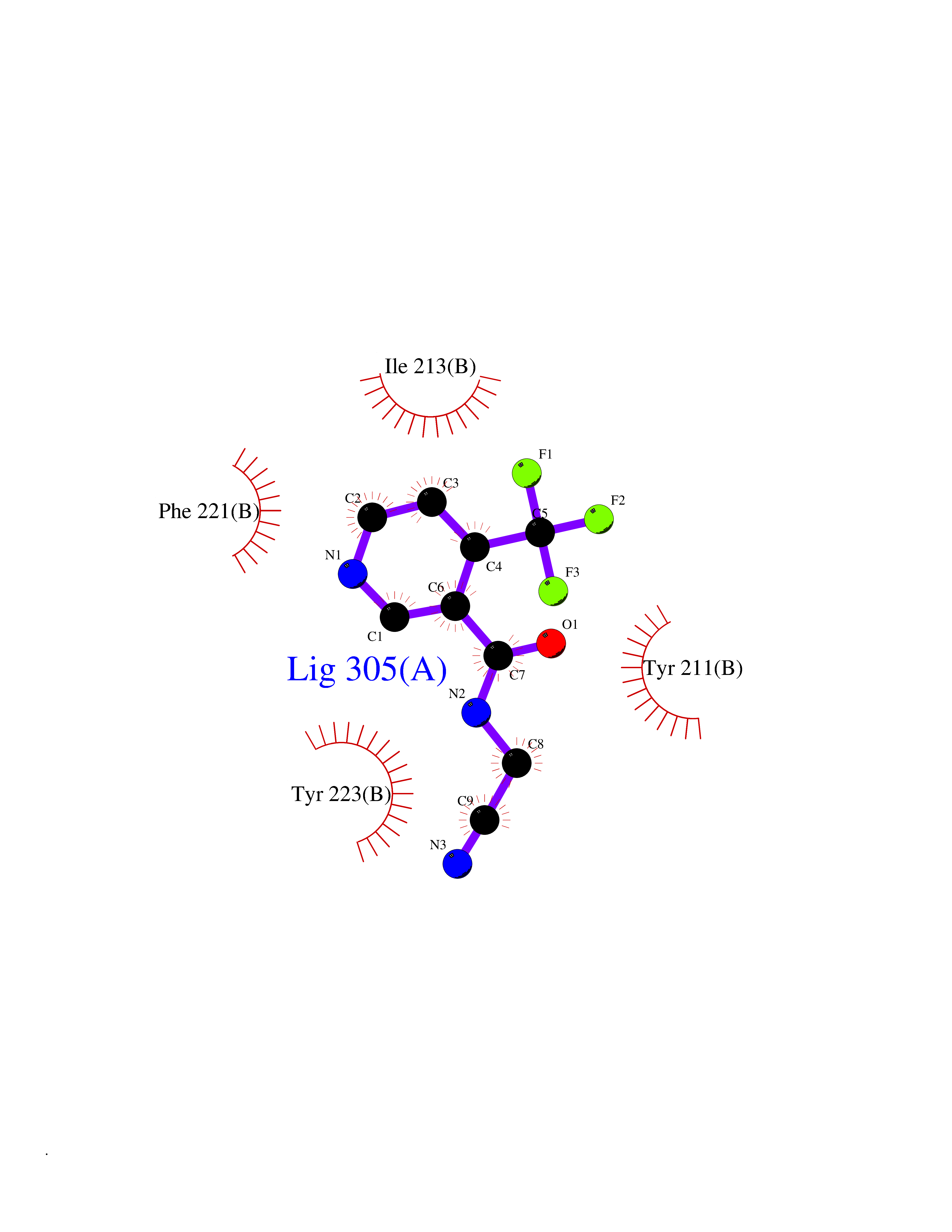 Ligplot Map 3D Binding mode Sequence TYQYNMNFEKLGKCIIINNKNFDKVTGMGVRNGTDKDAEALFKCFRSLGFDVIVYNDCSCAKMQDLLKKASEEDHTNAACFACILLSHGEENVIYGKDGVTPIKDLTAHFRGARCKTLLEKPKLFFIQACRGTEPRYKIPVEADFLFAYSTVRGSWFVQALCSILEEHGKDLEIMQILTRVNDRVARHFKKQIPCVVSMLTKELYFSQVPTYQYNMNFEKLGKCIIINNKNFDKVTGMGVRNGTDKDAEALFKCFRSLGFDVIVYNDCSCAKMQDLLKKASEEDHTNAACFACILLSHGEENVIYGKDGVTPIKDLTAHFRGARCKTLLEKPKLFFIQACRGPRYKIPVEADFLFAYSTVPGSWFVQALCSILEEHGKDLEIMQILTRVNDRVARHFESKQIPCVVSMLTKELYFSQ Hydrogen bonds contact Hydrophobic contact | ||||
| 93 | Cytochrome P450 1B1 (CYP1B1) | 3PM0 | 5.29 | |
Target general information Gen name CYP1B1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms CYPIB1 Protein family Cytochrome P450 family Biochemical class Paired donor oxygen oxidoreductase Function In liver microsomes, this enzyme is involved in an NADPH-dependent electron transport pathway. It oxidizes a variety of structurally unrelated compounds, including steroids, fatty acids, retinoid and xenobiotics. Preferentially oxidizes 17beta-estradiol to the carcinogenic 4-hydroxy derivative, and a variety of procarcinogenic compounds to their activated forms, including polycyclic aromatic hydrocarbons. Promotes angiogenesis by removing cellular oxygenation products, thereby decreasing oxidative stress, release of antiangiogenic factor THBS2, then allowing endothelial cells migration, cell adhesion and capillary morphogenesis. These changes are concommitant with the endothelial nitric oxide synthase activity and nitric oxide synthesis. Plays an important role in the regulation of perivascular cell proliferation, migration, and survival through modulation of the intracellular oxidative state and NF-kappa-B expression and/or activity, during angiogenesis. Contributes to oxidative homeostasis and ultrastructural organization and function of trabecular meshwork tissue through modulation of POSTN expression. Cytochromes P450 are a group of heme-thiolate monooxygenases. Related diseases Anterior segment dysgenesis 6 (ASGD6) [MIM:617315]: A form of anterior segment dysgenesis, a group of defects affecting anterior structures of the eye including cornea, iris, lens, trabecular meshwork, and Schlemm canal. Anterior segment dysgeneses result from abnormal migration or differentiation of the neural crest derived mesenchymal cells that give rise to components of the anterior chamber during eye development. Different anterior segment anomalies may exist alone or in combination, including iris hypoplasia, enlarged or reduced corneal diameter, corneal vascularization and opacity, posterior embryotoxon, corectopia, polycoria, abnormal iridocorneal angle, ectopia lentis, and anterior synechiae between the iris and posterior corneal surface. Clinical conditions falling within the phenotypic spectrum of anterior segment dysgeneses include aniridia, Axenfeld anomaly, Reiger anomaly/syndrome, Peters anomaly, and iridogoniodysgenesis. ASGD6 patients predominantly manifest Peters anomaly. Peters anomaly consists of corneal leukoma, defects in the posterior structures of the cornea such as absence of the posterior corneal stroma and Descemet membrane, and a variable degree of iridocorneal and/or keratolenticular adhesions. Over 50% of patients develop glaucoma in childhood. {ECO:0000269|PubMed:11403040}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Glaucoma 3, primary congenital, A (GLC3A) [MIM:231300]: An autosomal recessive form of primary congenital glaucoma (PCG). PCG is characterized by marked increase of intraocular pressure at birth or early childhood, large ocular globes (buphthalmos) and corneal edema. It results from developmental defects of the trabecular meshwork and anterior chamber angle of the eye that prevent adequate drainage of aqueous humor. {ECO:0000269|PubMed:10227395, ECO:0000269|PubMed:10655546, ECO:0000269|PubMed:11184479, ECO:0000269|PubMed:11527932, ECO:0000269|PubMed:11774072, ECO:0000269|PubMed:11980847, ECO:0000269|PubMed:12036985, ECO:0000269|PubMed:12525557, ECO:0000269|PubMed:14635112, ECO:0000269|PubMed:14640114, ECO:0000269|PubMed:15255109, ECO:0000269|PubMed:15342693, ECO:0000269|PubMed:15475877, ECO:0000269|PubMed:16490498, ECO:0000269|PubMed:16688110, ECO:0000269|PubMed:16735994, ECO:0000269|PubMed:16862072, ECO:0000269|PubMed:18470941, ECO:0000269|PubMed:9463332, ECO:0000269|PubMed:9497261}. The disease is caused by variants affecting distinct genetic loci, including the gene represented in this entry.; DISEASE: Glaucoma 1, open angle, A (GLC1A) [MIM:137750]: A form of primary open angle glaucoma (POAG). POAG is characterized by a specific pattern of optic nerve and visual field defects. The angle of the anterior chamber of the eye is open, and usually the intraocular pressure is increased. However, glaucoma can occur at any intraocular pressure. The disease is generally asymptomatic until the late stages, by which time significant and irreversible optic nerve damage has already taken place. {ECO:0000269|PubMed:11774072}. The gene represented in this entry acts as a disease modifier. Digenic mutations in CYP1B1 and MYOC have been found in a family segregating both primary adult-onset and juvenile forms of open angle glaucoma (PubMed:11774072). All affected family members with mutations in both MYOC and CYP1B1 had juvenile glaucoma, whereas those with only the MYOC mutation had the adult-onset form (PubMed:11774072). {ECO:0000269|PubMed:11774072}. Drugs (DrugBank ID) DB02342; DB00613; DB06732; DB00443; DB00121; DB01222; DB00201; DB09061; DB14737; DB01254; DB00694; DB01248; DB00997; DB00470; DB00530; DB00783; DB13952; DB13953; DB13954; DB13955; DB13956; DB00655; DB07776; DB00499; DB01645; DB01381; DB00741; DB01064; DB01026; DB00448; DB14009; DB01065; DB00170; DB00959; DB01204; DB14011; DB03467; DB00338; DB01229; DB14631; DB00635; DB01087; DB00396; DB00818; DB04216; DB02709; DB00675; DB00624; DB13946; DB00277; DB12245; DB11155 Interacts with Q02763 EC number EC 1.14.14.- Uniprot keywords 3D-structure; Disease variant; Endoplasmic reticulum; Fatty acid metabolism; Glaucoma; Heme; Iron; Lipid metabolism; Lyase; Membrane; Metal-binding; Microsome; Mitochondrion; Monooxygenase; Oxidoreductase; Peters anomaly; Proteomics identification; Reference proteome; Steroid metabolism Protein physicochemical properties Chain ID A Molecular weight (Da) 51875.9 Length 459 Aromaticity 0.1 Instability index 34.16 Isoelectric point 8.64 Charge (pH=7) 4.89 2D Binding mode Binding energy (Kcal/mol) -7.21  Molscript Map 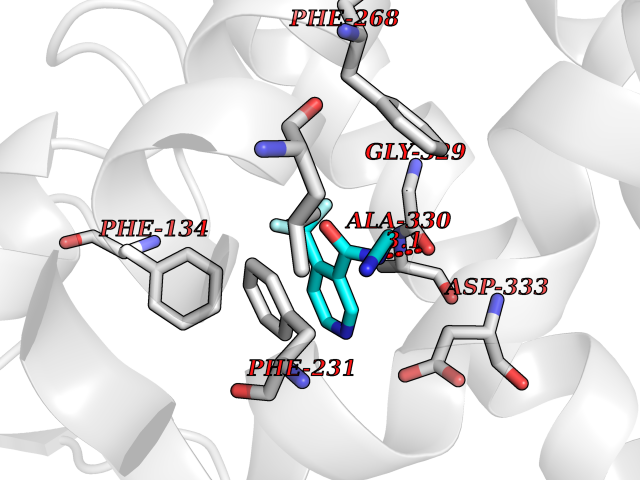 Pymol Map 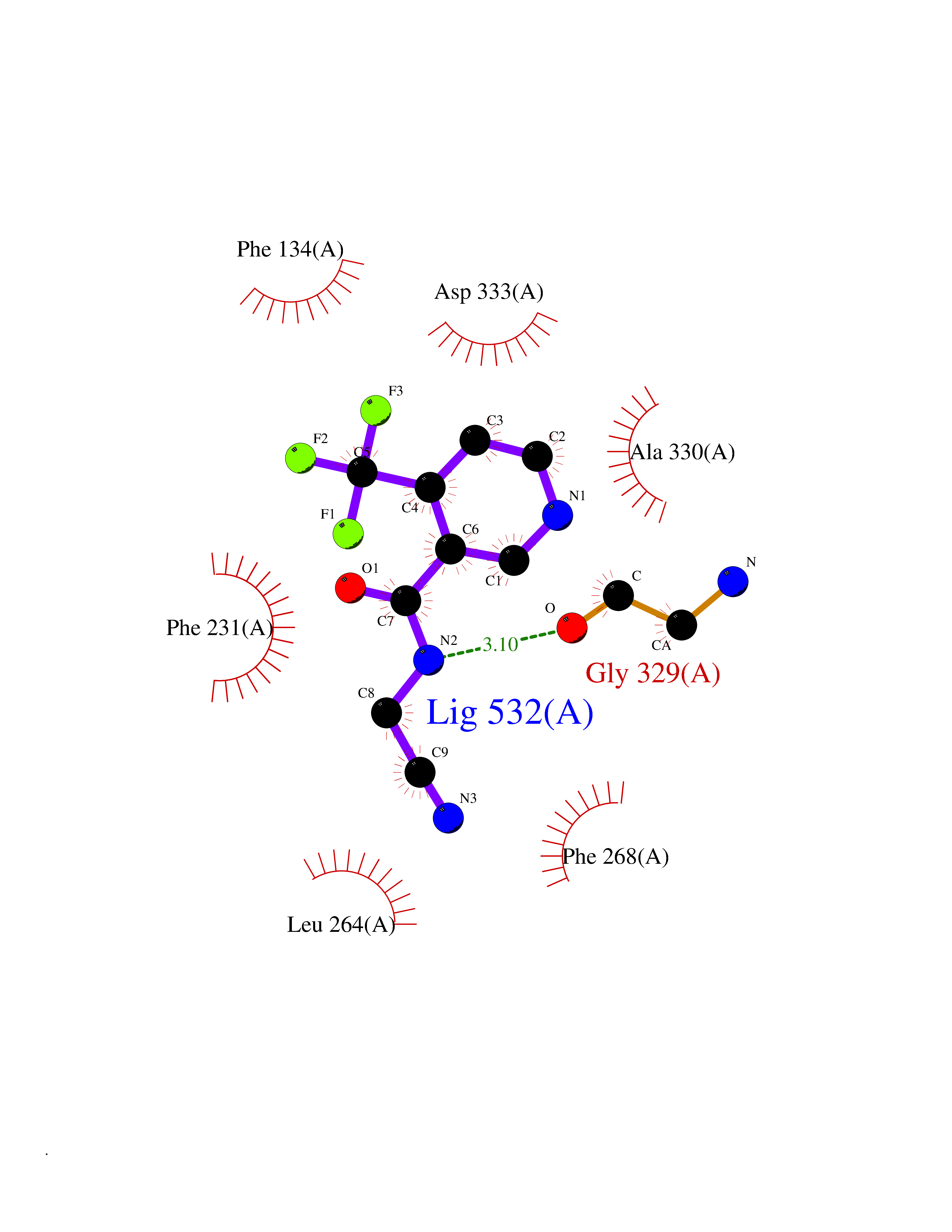 Ligplot Map 3D Binding mode Sequence QAAHLSFARLARRYGDVFQIRLGSCPIVVLNGERAIHQALVQQGSAFADRPSFASFRVVSGGRSMAFGHYSEHWKVQRRAAHSMMRNFFTRQPRSRQVLEGHVLSEARELVALLVRGSADGAFLDPRPLTVVAVANVMSAVCFGCRYSHDDPEFRELLSHNEEFGRTVGAGSLVDVMPWLQYFPNPVRTVFREFEQLNRNFSNFILDKFLRHCESLRPGAAPRDMMDAFILSAEKKAAGDGARLDLENVPATITDIFGASQDTLSTALQWLLLLFTRYPDVQTRVQAELDQVVGRDRLPCMGDQPNLPYVLAFLYEAMRFSSFVPVTIPHATTANTSVLGYHIPKDTVVFVNQWSVNHDPLKWPNPENFDPARFLDKDGLINKDLTSRVMIFSVGKRRCIGEELSKMQLFLFISILAHQCDFRANPNEPAKMNFSYGLTIKPKSFKVNVTLRESMELLD Hydrogen bonds contact Hydrophobic contact | ||||
| 94 | Alpha-tubulin N-acetyltransferase 1 (ATAT1) | 4B5O | 5.29 | |
Target general information Gen name ATAT1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms MEC17; C6orf134; Alpha-TAT; Acetyltransferase mec-17 homolog Protein family Acetyltransferase ATAT1 family Biochemical class Acetyltransferase ATAT1 family Function Specifically acetylates 'Lys-40' in alpha-tubulin on the lumenal side of microtubules. Promotes microtubule destabilization and accelerates microtubule dynamics; this activity may be independent of acetylation activity. Acetylates alpha-tubulin with a slow enzymatic rate, due to a catalytic site that is not optimized for acetyl transfer. Enters the microtubule through each end and diffuses quickly throughout the lumen of microtubules. Acetylates only long/old microtubules because of its slow acetylation rate since it does not have time to act on dynamically unstable microtubules before the enzyme is released. Required for normal sperm flagellar function. Promotes directional cell locomotion and chemotaxis, through AP2A2-dependent acetylation of alpha-tubulin at clathrin-coated pits that are concentrated at the leading edge of migrating cells. May facilitate primary cilium assembly. Related diseases Anterior segment dysgenesis 6 (ASGD6) [MIM:617315]: A form of anterior segment dysgenesis, a group of defects affecting anterior structures of the eye including cornea, iris, lens, trabecular meshwork, and Schlemm canal. Anterior segment dysgeneses result from abnormal migration or differentiation of the neural crest derived mesenchymal cells that give rise to components of the anterior chamber during eye development. Different anterior segment anomalies may exist alone or in combination, including iris hypoplasia, enlarged or reduced corneal diameter, corneal vascularization and opacity, posterior embryotoxon, corectopia, polycoria, abnormal iridocorneal angle, ectopia lentis, and anterior synechiae between the iris and posterior corneal surface. Clinical conditions falling within the phenotypic spectrum of anterior segment dysgeneses include aniridia, Axenfeld anomaly, Reiger anomaly/syndrome, Peters anomaly, and iridogoniodysgenesis. ASGD6 patients predominantly manifest Peters anomaly. Peters anomaly consists of corneal leukoma, defects in the posterior structures of the cornea such as absence of the posterior corneal stroma and Descemet membrane, and a variable degree of iridocorneal and/or keratolenticular adhesions. Over 50% of patients develop glaucoma in childhood. {ECO:0000269|PubMed:11403040}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Glaucoma 3, primary congenital, A (GLC3A) [MIM:231300]: An autosomal recessive form of primary congenital glaucoma (PCG). PCG is characterized by marked increase of intraocular pressure at birth or early childhood, large ocular globes (buphthalmos) and corneal edema. It results from developmental defects of the trabecular meshwork and anterior chamber angle of the eye that prevent adequate drainage of aqueous humor. {ECO:0000269|PubMed:10227395, ECO:0000269|PubMed:10655546, ECO:0000269|PubMed:11184479, ECO:0000269|PubMed:11527932, ECO:0000269|PubMed:11774072, ECO:0000269|PubMed:11980847, ECO:0000269|PubMed:12036985, ECO:0000269|PubMed:12525557, ECO:0000269|PubMed:14635112, ECO:0000269|PubMed:14640114, ECO:0000269|PubMed:15255109, ECO:0000269|PubMed:15342693, ECO:0000269|PubMed:15475877, ECO:0000269|PubMed:16490498, ECO:0000269|PubMed:16688110, ECO:0000269|PubMed:16735994, ECO:0000269|PubMed:16862072, ECO:0000269|PubMed:18470941, ECO:0000269|PubMed:9463332, ECO:0000269|PubMed:9497261}. The disease is caused by variants affecting distinct genetic loci, including the gene represented in this entry.; DISEASE: Glaucoma 1, open angle, A (GLC1A) [MIM:137750]: A form of primary open angle glaucoma (POAG). POAG is characterized by a specific pattern of optic nerve and visual field defects. The angle of the anterior chamber of the eye is open, and usually the intraocular pressure is increased. However, glaucoma can occur at any intraocular pressure. The disease is generally asymptomatic until the late stages, by which time significant and irreversible optic nerve damage has already taken place. {ECO:0000269|PubMed:11774072}. The gene represented in this entry acts as a disease modifier. Digenic mutations in CYP1B1 and MYOC have been found in a family segregating both primary adult-onset and juvenile forms of open angle glaucoma (PubMed:11774072). All affected family members with mutations in both MYOC and CYP1B1 had juvenile glaucoma, whereas those with only the MYOC mutation had the adult-onset form (PubMed:11774072). {ECO:0000269|PubMed:11774072}. Drugs (DrugBank ID) NA Interacts with P62258 EC number EC 2.3.1.108 Uniprot keywords 3D-structure; Acetylation; Acyltransferase; Alternative splicing; Cell junction; Cell projection; Coated pit; Cytoplasm; Cytoskeleton; Membrane; Methylation; Phosphoprotein; Proteomics identification; Reference proteome; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 22197.2 Length 192 Aromaticity 0.09 Instability index 47 Isoelectric point 8.69 Charge (pH=7) 2.35 2D Binding mode Binding energy (Kcal/mol) -7.21 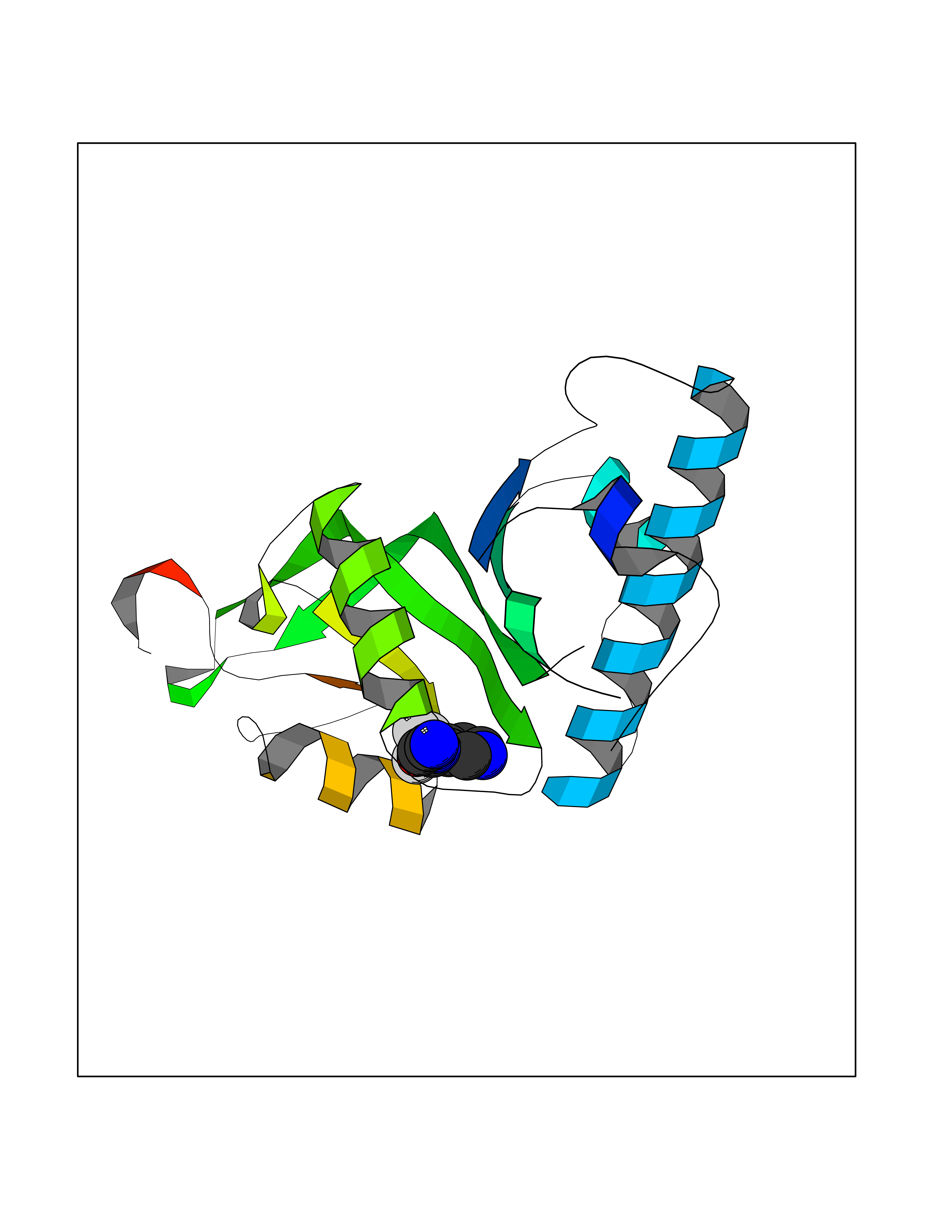 Molscript Map 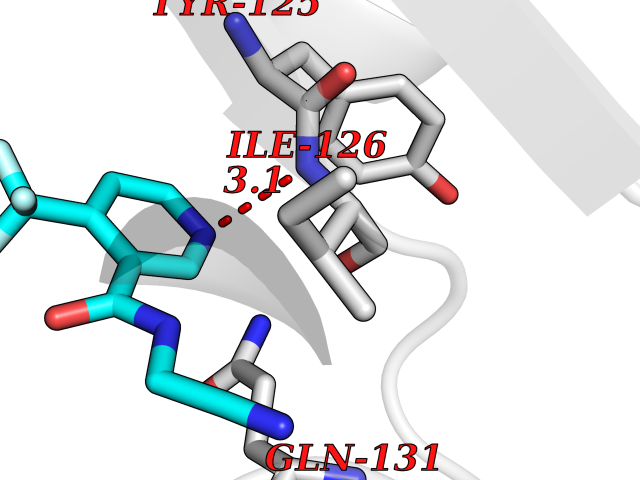 Pymol Map 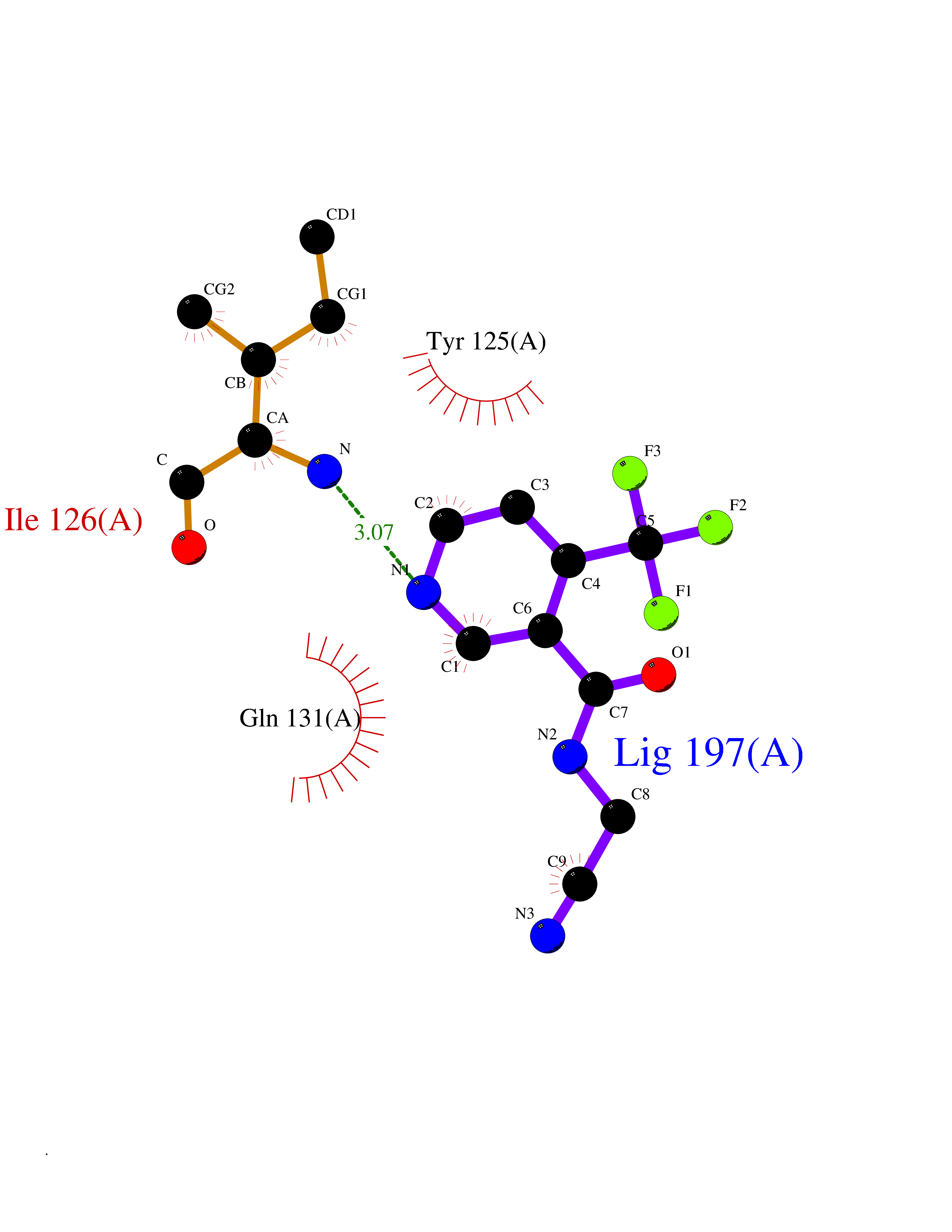 Ligplot Map 3D Binding mode Sequence SMEFPFDVDALFPERITVLDQHLRPPARRPGTTTPARVDLQQQIMTIIDELGKASAKAQNLSAPITSASRMQSNRHVVYILKDSSARPAIIGFIKVGYKKLFVLDDREAHNEVEPLCILDFYIHESVQRHGHGRELFQYMLQKERVEPHQLAIDRPSQKLLKFLNKHYNLETTVPQVNNFVIFEGFFAHQHR Hydrogen bonds contact Hydrophobic contact | ||||
| 95 | Aggrecanase (ADAMTS5) | 3HY7 | 5.29 | |
Target general information Gen name ADAMTS5 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Aggrecanase-2; ADMP-2; ADAMTS5; ADAMTS-5; ADAM-TS5; ADAM-TS 5; ADAM-TS 11; A disintegrin and metalloproteinase with thrombospondinmotifs 5 Protein family NA Biochemical class Peptidase Function Cleaves aggrecan, a cartilage proteoglycan, and may be involved in its turnover. May play an important role in the destruction of aggrecan in arthritic diseases. May play a role in proteolytic processing mostly during the peri-implantation period. Related diseases Mucopolysaccharidosis 1H (MPS1H) [MIM:607014]: A severe form of mucopolysaccharidosis type 1, a rare lysosomal storage disease characterized by progressive physical deterioration with urinary excretion of dermatan sulfate and heparan sulfate. Patients with MPS1H usually present, within the first year of life, a combination of hepatosplenomegaly, skeletal deformities, corneal clouding and severe intellectual disability. Obstructive airways disease, respiratory infection and cardiac complications usually result in death before 10 years of age. {ECO:0000269|PubMed:10466419, ECO:0000269|PubMed:10735634, ECO:0000269|PubMed:12559846, ECO:0000269|PubMed:1301941, ECO:0000269|PubMed:15300847, ECO:0000269|PubMed:19396826, ECO:0000269|PubMed:21394825, ECO:0000269|PubMed:24036510, ECO:0000269|PubMed:31194252, ECO:0000269|PubMed:7550232, ECO:0000269|PubMed:7550242, ECO:0000269|PubMed:7951228, ECO:0000269|PubMed:8019563, ECO:0000269|PubMed:8328452, ECO:0000269|PubMed:8401515, ECO:0000269|Ref.20}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Mucopolysaccharidosis 1H/S (MPS1H/S) [MIM:607015]: A form of mucopolysaccharidosis type 1, a rare lysosomal storage disease characterized by progressive physical deterioration with urinary excretion of dermatan sulfate and heparan sulfate. MPS1H/S represents an intermediate phenotype of the MPS1 clinical spectrum. It is characterized by relatively little neurological involvement, but most of the somatic symptoms described for severe MPS1 develop in the early to mid-teens, causing considerable loss of mobility. {ECO:0000269|PubMed:10466419, ECO:0000269|PubMed:10735634, ECO:0000269|PubMed:12559846, ECO:0000269|PubMed:15300847, ECO:0000269|PubMed:21394825, ECO:0000269|PubMed:7550232, ECO:0000269|PubMed:7550242, ECO:0000269|PubMed:8401515}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Mucopolysaccharidosis 1S (MPS1S) [MIM:607016]: A mild form of mucopolysaccharidosis type 1, a rare lysosomal storage disease characterized by progressive physical deterioration with urinary excretion of dermatan sulfate and heparan sulfate. Patients with MPS1S may have little or no neurological involvement, normal stature and life span, but present development of joints stiffness, mild hepatosplenomegaly, aortic valve disease and corneal clouding. {ECO:0000269|PubMed:12559846, ECO:0000269|PubMed:15300847, ECO:0000269|PubMed:19396826, ECO:0000269|PubMed:21394825, ECO:0000269|PubMed:25256405, ECO:0000269|PubMed:7550232, ECO:0000269|PubMed:7550242, ECO:0000269|PubMed:8213840}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB06837; DB03880; DB06945 Interacts with P13608 EC number EC 3.4.24.- Uniprot keywords 3D-structure; Cleavage on pair of basic residues; Disulfide bond; Extracellular matrix; Glycoprotein; Hydrolase; Metal-binding; Metalloprotease; Protease; Proteomics identification; Reference proteome; Repeat; Secreted; Signal; Zinc; Zymogen Protein physicochemical properties Chain ID A Molecular weight (Da) 23997.8 Length 217 Aromaticity 0.06 Instability index 46.68 Isoelectric point 5.82 Charge (pH=7) -8.36 2D Binding mode Binding energy (Kcal/mol) -7.22 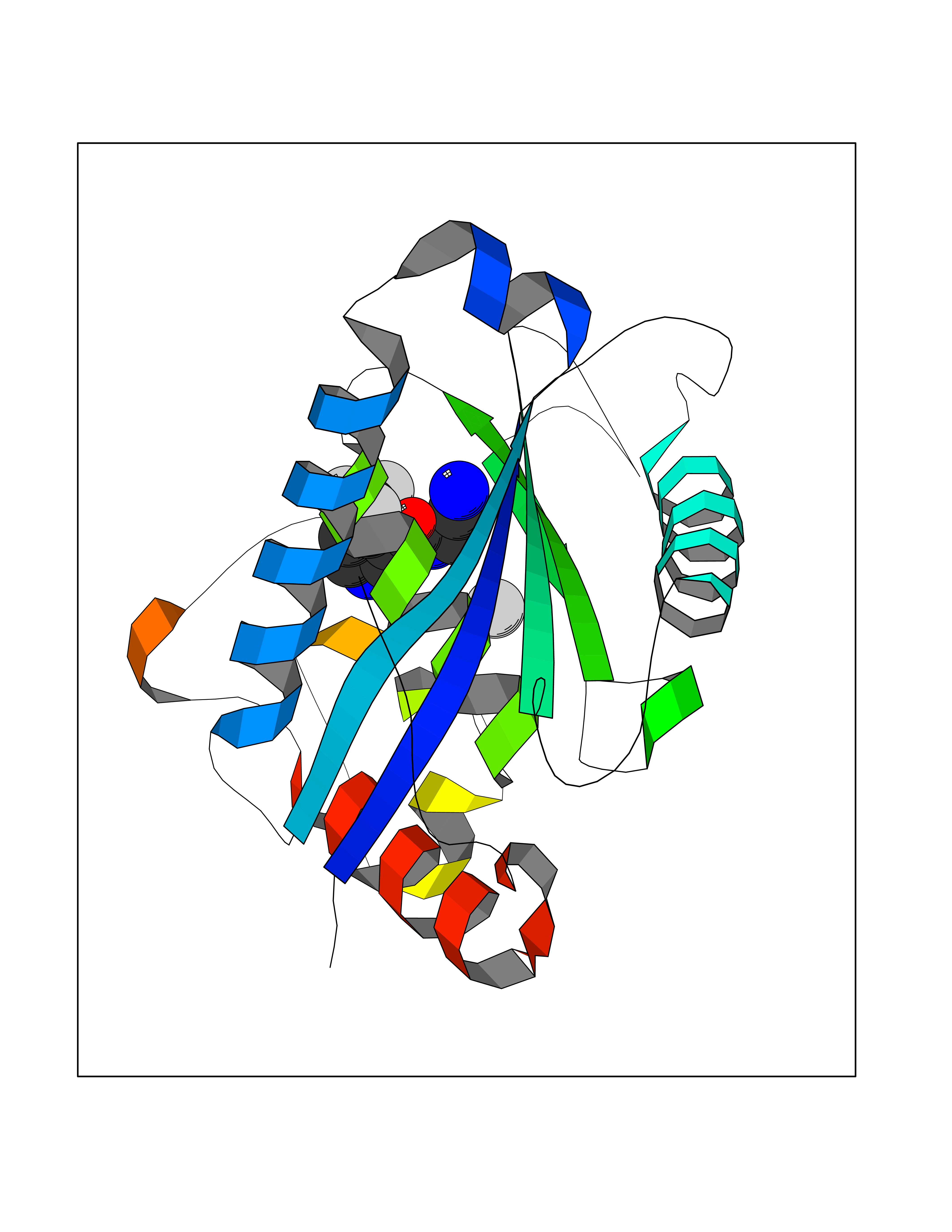 Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence SRARQVELLLVADASMARKYGRGLQHYLLTLASIANRLYSHASIENHIRLAVVKVVVLGDKDKSLEVSKNAATTLKNFCKWQHQHNQLGDDHEEHYDAAILFTREDLCGHHSCDTLGMADVGTICSPERSCAVIEDDGLHAAFTVAHEIGHLLGLSHDDSKFCEETFGSTEDKRLMSSILTSIDASKPWSKCTSATITEFLDDGHGNCLLDLPRKQI Hydrogen bonds contact Hydrophobic contact | ||||
| 96 | UDP-galactopyranose mutase | 1I8T | 5.28 | |
Target general information Gen name glf Organism Escherichia coli (strain K12) Uniprot ID TTD ID NA Synonyms b2036;JW2021;yefE Protein family UDP-galactopyranose/dTDP-fucopyranose mutase family Biochemical class Isomerase Function Flavin adenine dinucleotide binding.UDP-galactopyranose mutase activity. Related diseases Defects in PPARG can lead to type 2 insulin-resistant diabetes and hyptertension. PPARG mutations may be associated with colon cancer. {ECO:0000269|PubMed:10394368}.; DISEASE: Obesity (OBESITY) [MIM:601665]: A condition characterized by an increase of body weight beyond the limitation of skeletal and physical requirements, as the result of excessive accumulation of body fat. {ECO:0000269|PubMed:9753710}. Disease susceptibility may be associated with variants affecting the gene represented in this entry.; DISEASE: Lipodystrophy, familial partial, 3 (FPLD3) [MIM:604367]: A form of lipodystrophy characterized by marked loss of subcutaneous fat from the extremities. Facial adipose tissue may be increased, decreased or normal. Affected individuals show an increased preponderance of insulin resistance, diabetes mellitus and dyslipidemia. {ECO:0000269|PubMed:11788685, ECO:0000269|PubMed:12453919}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Glioma 1 (GLM1) [MIM:137800]: Gliomas are benign or malignant central nervous system neoplasms derived from glial cells. They comprise astrocytomas and glioblastoma multiforme that are derived from astrocytes, oligodendrogliomas derived from oligodendrocytes and ependymomas derived from ependymocytes. {ECO:0000269|PubMed:10851250}. Disease susceptibility may be associated with variants affecting the gene represented in this entry. Polymorphic PPARG alleles have been found to be significantly over-represented among a cohort of American patients with sporadic glioblastoma multiforme suggesting a possible contribution to disease susceptibility. Drugs (DrugBank ID) DB03147 Interacts with P11868 EC number 5.4.99.9 Uniprot keywords 3D-structure; Direct protein sequencing; FAD; Flavoprotein; Isomerase; Lipopolysaccharide biosynthesis; Reference proteome Protein physicochemical properties Chain ID A,B Molecular weight (Da) 42965.3 Length 367 Aromaticity 0.14 Instability index 32.48 Isoelectric point 6.62 Charge (pH=7) -1.52 2D Binding mode Binding energy (Kcal/mol) -7.2  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence MYDYIIVGSGLFGAVCANELKKLNKKVLVIEKRNHIGGNAYTEDCEGIQIHKYGAHIFHTNDKYIWDYVNDLVEFNRFTNSPLAIYKDKLFNLPFNMNTFHQMWGVKDPQEAQNIINAQKKKYGDKVPENLEEQAISLVGEDLYQALIKGYTEKQWGRSAKELPAFIIKRIPVRFTFDNNYFSDRYQGIPVGGYTKLIEKMLEGVDVKLGIDFLKDKDSLASKAHRIIYTGPIDQYFDYRFGALEYRSLKFETERHEFPNFQGNAVINFTDANVPYTRIIEHKHFDYVETKHTVVTKEYPLEWKVGDEPYYPVNDNKNMELFKKYRELASREDKVIFGGRLAEYKYYDMHQVISAALYQVKNIMSTD Hydrogen bonds contact Hydrophobic contact | ||||
| 97 | Estrogen sulfotransferase | 1G3M | 5.28 | |
Target general information Gen name SULT1E1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms STE Protein family Sulfotransferase 1 family Biochemical class Transferase Function Aryl sulfotransferase activity.Estrone sulfotransferase activity.Flavonol 3-sulfotransferase activity.Steroid binding.Steroid sulfotransferase activity.Sulfotransferase activity. Related diseases Neuromyotonia and axonal neuropathy, autosomal recessive (NMAN) [MIM:137200]: An autosomal recessive neurologic disorder characterized by onset in the first or second decade of a peripheral axonal neuropathy predominantly affecting motor more than sensory nerves. The axonal neuropathy is reminiscent of Charcot-Marie-Tooth disease type 2 and distal hereditary motor neuropathy. Individuals with NMAN also have delayed muscle relaxation and action myotonia associated with neuromyotonic discharges on needle EMG resulting from hyperexcitability of the peripheral nerves. {ECO:0000269|PubMed:16835243, ECO:0000269|PubMed:22961002, ECO:0000269|PubMed:28691797, ECO:0000269|PubMed:29787766, ECO:0000269|PubMed:31088288}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02902; DB03346; DB01812; DB00714; DB14635; DB01176; DB00977; DB09288; DB00675; DB09100 Interacts with O76083; O76083-2 EC number 2.8.2.4 Uniprot keywords 3D-structure; Cytoplasm; Lipid metabolism; Lipid-binding; Proteomics identification; Reference proteome; Steroid-binding; Transferase Protein physicochemical properties Chain ID A,B Molecular weight (Da) 34022.7 Length 285 Aromaticity 0.15 Instability index 32.76 Isoelectric point 6.33 Charge (pH=7) -2.75 2D Binding mode Binding energy (Kcal/mol) -7.2  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence SELDYYEKFEEVHGILMYKDFVKYWDNVEAFQARPDDLVIATYPKSGTTWVSEIVYMIYKEGDVEKCKEDVIFNRIPFLECRNGVKQLDEMNSPRIVKTHLPPELLPASFWEKDCKIIYLCRNAKDVAVSFYYFFLMVAGHPNPGSFPEFVEKFMQGQVPYGSWYKHVKSWWEKGKSPRVLFLFYEDLKEDIRKEVIKLIHFLERKPSEELVDRIIHHTSFQEMKNNPSTNYTTLPDEIMNQKLSPFMRKGITGDWKNHFTVALNEKFDKHYEQQMKESTLKFRT Hydrogen bonds contact Hydrophobic contact | ||||
| 98 | Histamine N-methyltransferase (HNMT) | 2AOT | 5.28 | |
Target general information Gen name HNMT Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Histamine-N-methyltransferase; HNMT; HMT Protein family Class I-like SAM-binding methyltransferase superfamily, HNMT family Biochemical class Methyltransferase Function Inactivates histamine by N-methylation. Plays an important role in degrading histamine and in regulating the airway response to histamine. Related diseases Intellectual developmental disorder, autosomal recessive 51 (MRT51) [MIM:616739]: A disorder characterized by significantly below average general intellectual functioning associated with impairments in adaptive behavior and manifested during the developmental period. {ECO:0000269|PubMed:26206890}. The disease is caused by variants affecting distinct genetic loci, including the gene represented in this entry. Drugs (DrugBank ID) DB00613; DB13875; DB05381; DB04655; DB01103; DB01752; DB07106 Interacts with NA EC number EC 2.1.1.8 Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; Disease variant; Intellectual disability; Methyltransferase; Proteomics identification; Reference proteome; S-adenosyl-L-methionine; Transferase Protein physicochemical properties Chain ID A,B Molecular weight (Da) 32712 Length 288 Aromaticity 0.1 Instability index 36.38 Isoelectric point 5.18 Charge (pH=7) -9.97 2D Binding mode Binding energy (Kcal/mol) -7.2  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence MRSLFSDHGKYVESFRRFLNHSTEHQCMQEFMDKKLPGIIGRIGDTKSEIKILSIGGGAGEIDLQILSKVQAQYPGVXINNEVVEPSAEQIAKYKELVAKTSNLENVKFAWHKETSSEYQSRMLEKKELQKWDFIHMIQMLYYVKDIPATLKFFHSLLGTNAKMLIIVVSGSSGWDKLWKKYGSRFPQDDLCQYITSDDLTQMLDNLGLKYECYDLLSTMDISDCFIDGNENGDLLWDFLTETXNFNATAPPDLRAELGKDLQEPEFSAKKEGKVLFNNTLSFIVIEA Hydrogen bonds contact Hydrophobic contact | ||||
| 99 | Dimethylglycine oxidase | 1PJ5 | 5.28 | |
Target general information Gen name dmg Organism Arthrobacter globiformis Uniprot ID TTD ID NA Synonyms NA Protein family GcvT family Biochemical class Oxidoreductase Function Dimethylglycine oxidase activity.Nucleotide binding. Related diseases Curry-Jones syndrome (CRJS) [MIM:601707]: A multisystem disorder characterized by patchy skin lesions, polysyndactyly, diverse cerebral malformations, unicoronal craniosynostosis, iris colobomas, microphthalmia, and intestinal malrotation with myofibromas or hamartomas. {ECO:0000269|PubMed:24859340, ECO:0000269|PubMed:27236920}. The disease is caused by variants affecting the gene represented in this entry. 8 individuals have been identified with the disease-causing mutation Phe-412 and all were mosaic. The mutation could not be reliably detected in blood, greatest success rates were obtained with affected tissues obtained by invasive procedures. It is thought that the mutation has arisen postzygotically early during embryonic development (PubMed:27236920). This mutation has also been identified in ameloblastoma, medulloblastoma, meningioma, and basal cell carcinoma, and has been reported as the oncogenic driver in some of these tumors (PubMed:24859340). {ECO:0000269|PubMed:24859340, ECO:0000269|PubMed:27236920}. Drugs (DrugBank ID) DB03256; DB03147 Interacts with NA EC number 1.5.3.10 Uniprot keywords 3D-structure; Direct protein sequencing; FAD; Flavoprotein; Nucleotide-binding; Oxidoreductase Protein physicochemical properties Chain ID A Molecular weight (Da) 45912.2 Length 427 Aromaticity 0.07 Instability index 43.46 Isoelectric point 4.83 Charge (pH=7) -20.69 2D Binding mode Binding energy (Kcal/mol) -7.2  Molscript Map 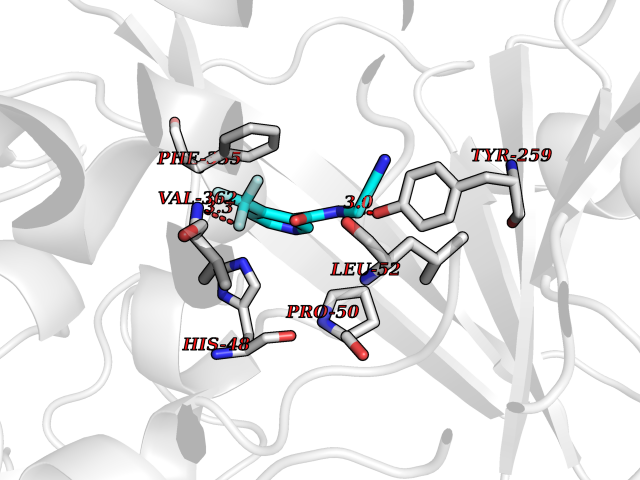 Pymol Map 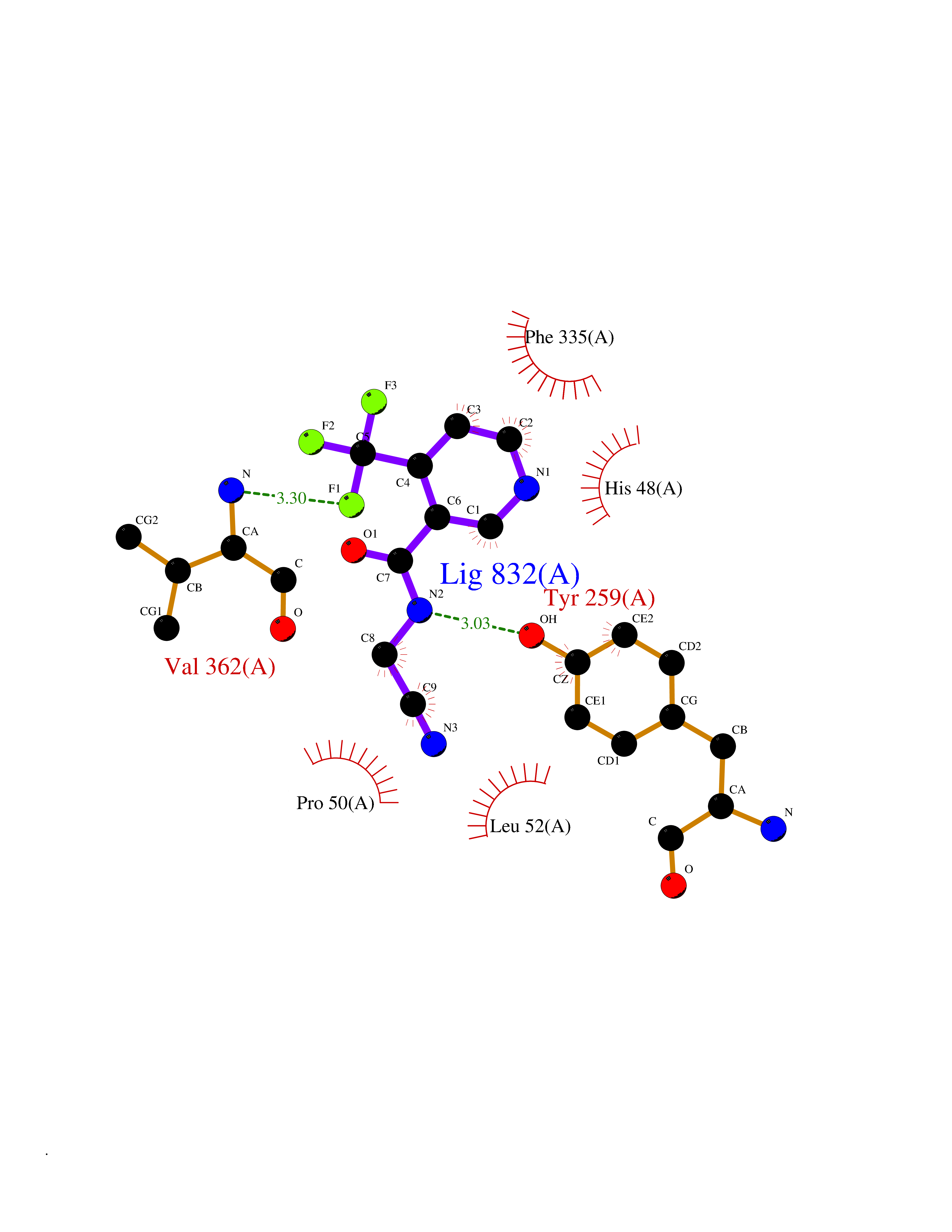 Ligplot Map 3D Binding mode Sequence TPRIVIIGAGIVGTNLADELVTRGWNNITVLDQGPLNMPGGSTSHAPGLVFQTNPSKTMASFAKYTVEKLLSLTEDGVSCFNQVGGLEVATTETRLADLKRKLGYAAAWGIEGRLLSPAECQELYPLLDGENILGGLHVPSDGLASAARAVQLLIKRTESAGVTYRGSTTVTGIEQSGGRVTGVQTADGVIPADIVVSCAGFWGAKIGAMIGMAVPLLPLAHQYVKTTPVPAQQGRNDQPNGARLPILRHQDQDLYYREHGDRYGIGSYAHRPMPVDVDTLGAYAPETVSEHHMPSRLDFTLEDFLPAWEATKQLLPALADSEIEDGFNGIFSFTPDGGPLLGESKELDGFYVAEAVWVTHSAGVAKAMAELLTTGRSETDLGECDITRFEDVQLTPEYVSETSQQNFVEIYDVLHPLQPRLSPRNL Hydrogen bonds contact Hydrophobic contact | ||||
| 100 | Dibasic-processing enzyme (Furin) | 7LCU | 5.28 | |
Target general information Gen name FURIN Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Paired basic amino acid residuecleaving enzyme; Paired basic amino acid residue-cleaving enzyme; PCSK3; PACE; FUR; Dibasicprocessing enzyme Protein family Peptidase S8 family, Furin subfamily Biochemical class Peptidase Function Mediates processing of TGFB1, an essential step in TGF-beta-1 activation. Ubiquitous endoprotease within constitutive secretory pathways capable of cleavage at the RX(K/R)R consensus motif. Related diseases Brachydactyly A2 (BDA2) [MIM:112600]: A form of brachydactyly. Brachydactyly defines a group of inherited malformations characterized by shortening of the digits due to abnormal development of the phalanges and/or the metacarpals. In brachydactyly type A2 shortening of the middle phalanges is confined to the index finger and the second toe, all other digits being more or less normal. Because of a rhomboid or triangular shape of the affected middle phalanx, the end of the second finger usually deviates radially. {ECO:0000269|PubMed:19327734, ECO:0000269|PubMed:21357617}. The gene represented in this entry is involved in disease pathogenesis. Duplications of a cis-regulatory element located approximately 110 kb downstream of BMP2 have been found in BDA2 families. They likely cause altered BMP2 expression with pathological consequences. {ECO:0000269|PubMed:19327734, ECO:0000269|PubMed:21357617}.; DISEASE: Short stature, facial dysmorphism, and skeletal anomalies with or without cardiac anomalies 1 (SSFSC1) [MIM:617877]: An autosomal dominant disorder characterized by short stature, facial dysmorphism, skeletal anomalies, and variable cardiac defects. Distinctive facial features include midface retrusion, short upturned nose, long philtrum, high-arched or cleft palate, and variable degrees of micrognathia and dental crowding. Skeletal anomalies include patterning defects of the axial skeleton, characterized by 11 pairs of ribs and brachydactyly of the fifth ray. Congenital heart defects are variably observed and appear to involve primarily the cardiac outflow tract. {ECO:0000269|PubMed:29198724}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03600 Interacts with P05067; P50281; Q9H239; O14793; K9N5Q8; P0DTC2; Q91QT1 EC number EC 3.4.21.75 Uniprot keywords 3D-structure; Autocatalytic cleavage; Calcium; Cell membrane; Cleavage on pair of basic residues; Disulfide bond; Endosome; Glycoprotein; Golgi apparatus; Heparin-binding; Host-virus interaction; Hydrolase; Membrane; Metal-binding; Phosphoprotein; Protease; Proteomics identification; Reference proteome; Repeat; Secreted; Serine protease; Signal; Transmembrane; Transmembrane helix; Zymogen Protein physicochemical properties Chain ID A Molecular weight (Da) 51029.8 Length 470 Aromaticity 0.08 Instability index 26.23 Isoelectric point 5.23 Charge (pH=7) -16.94 2D Binding mode Binding energy (Kcal/mol) -7.2 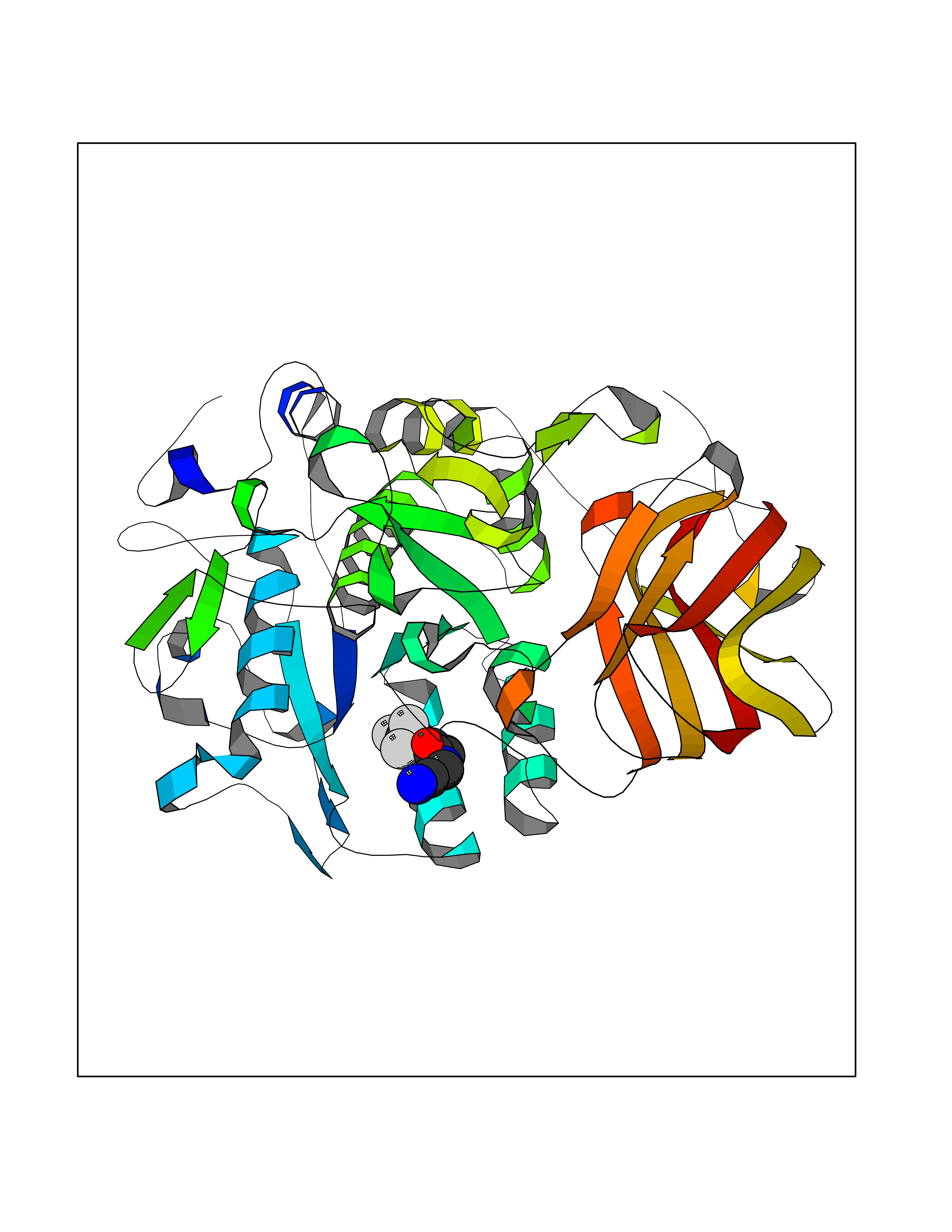 Molscript Map 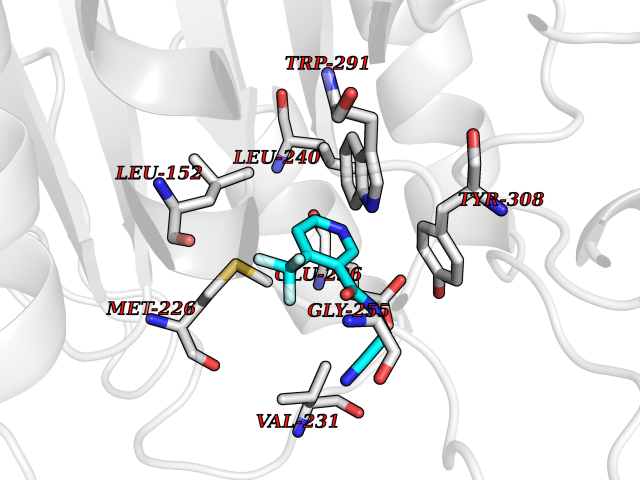 Pymol Map 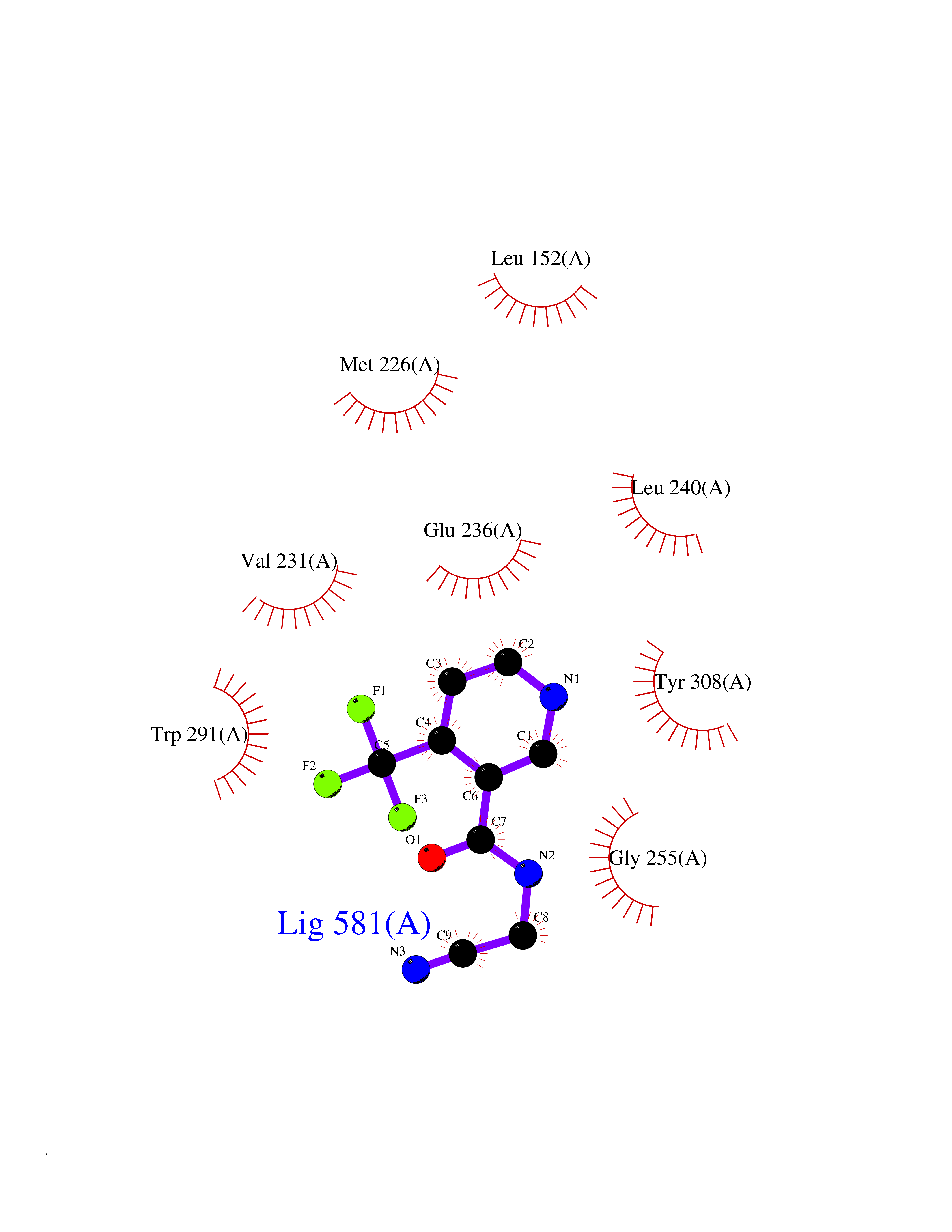 Ligplot Map 3D Binding mode Sequence YQEPTDPKFPQQWYLSGVTQRDLNVKAAWAQGYTGHGIVVSILDDGIEKNHPDLAGNYDPGASFDVNDQDPDPQPRYTQMNDNRHGTRCAGEVAAVANNGVCGVGVAYNARIGGVRMLDGEVTDAVEARSLGLNPNHIHIYSASWGPEDDGKTVDGPARLAEEAFFRGVSQGRGGLGSIFVWASGNGGREHDSCNCDGYTNSIYTLSISSATQFGNVPWYSEACSSTLATTYSSGNQNEKQIVTTDLRQKCTESHTGTSASAPLAAGIIALTLEANKDLTWRDMQHLVVQTSKPAHLNANDWATNGVGRKVSHSYGYGLLDAGAMVALAQDWTTVAPQRKCIIDILTEPKDIGKRLEVRKTVTACLGEPNHITRLEHAQARLTLSYNRRGDLAIHLVSPMGTRSTLLAARPHDYSADGFNDWAFMTTHSWDEDPSGEWVLEIENTSEANNYGTLTKFTLVLYGTAGENLY Hydrogen bonds contact Hydrophobic contact | ||||