Job Results:
Ligand
Structure
Job ID
5c976aa63f0cd6d4251a6978fa589574
Job name
NA
Time
2025-07-21 16:39:34
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 1 | Cyclopropane mycolic acid synthase MmaA2 | 1TPY | 5.58 | |
Target general information Gen name mmaA2 Organism Mycobacterium tuberculosis (strain ATCC 25618 / H37Rv) Uniprot ID TTD ID NA Synonyms Rv0644c;mma2 Protein family CFA/CMAS family Biochemical class Transferase Function Cyclopropane-fatty-acyl-phospholipid synthase activity.Methyltransferase activity. Related diseases Oocyte/zygote/embryo maturation arrest 16 (OZEMA16) [MIM:617234]: A rare cause of female primary infertility. In affected women, ovulation and fertilization proceed normally but embryos are arrested at early stages of development. Inheritance is autosomal recessive. {ECO:0000269|PubMed:27545678}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01718; DB01752 Interacts with NA EC number 2.1.1.79 Uniprot keywords 3D-structure; Acetylation; Lipid biosynthesis; Lipid metabolism; Methyltransferase; Reference proteome; S-adenosyl-L-methionine; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 32493.6 Length 285 Aromaticity 0.1 Instability index 43.61 Isoelectric point 5.53 Charge (pH=7) -10.17 2D Binding mode Binding energy (Kcal/mol) -7.61 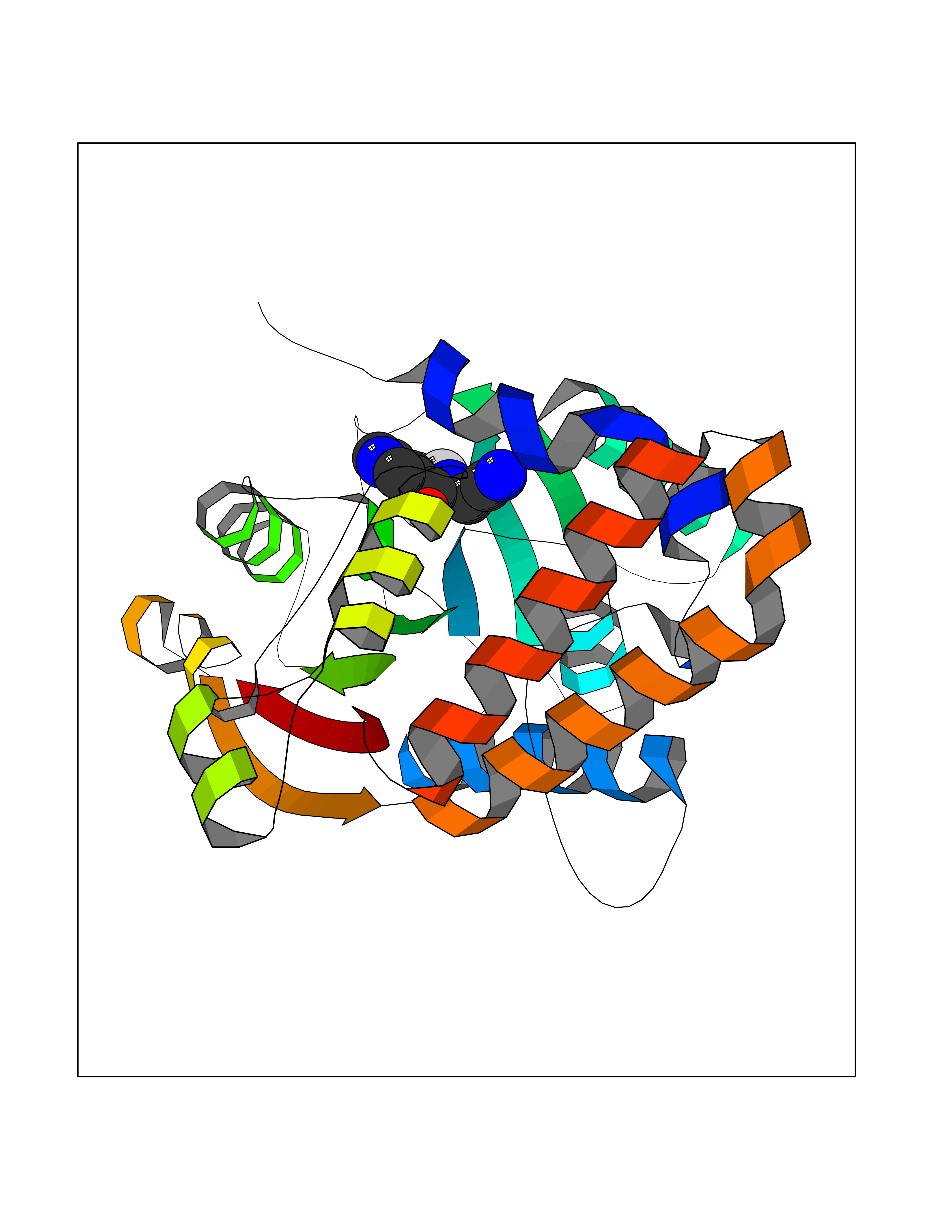 Molscript Map 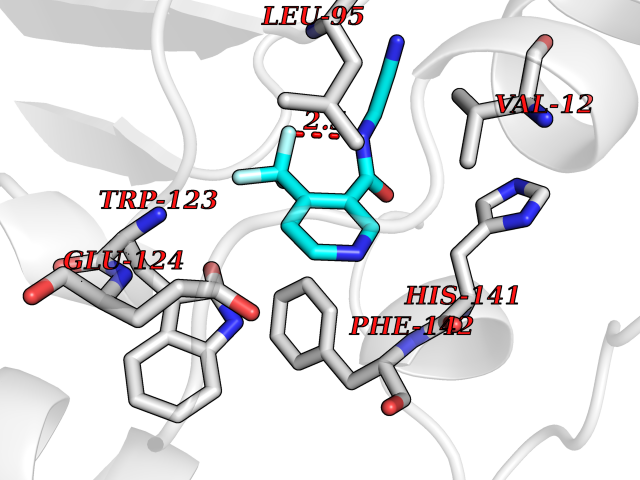 Pymol Map 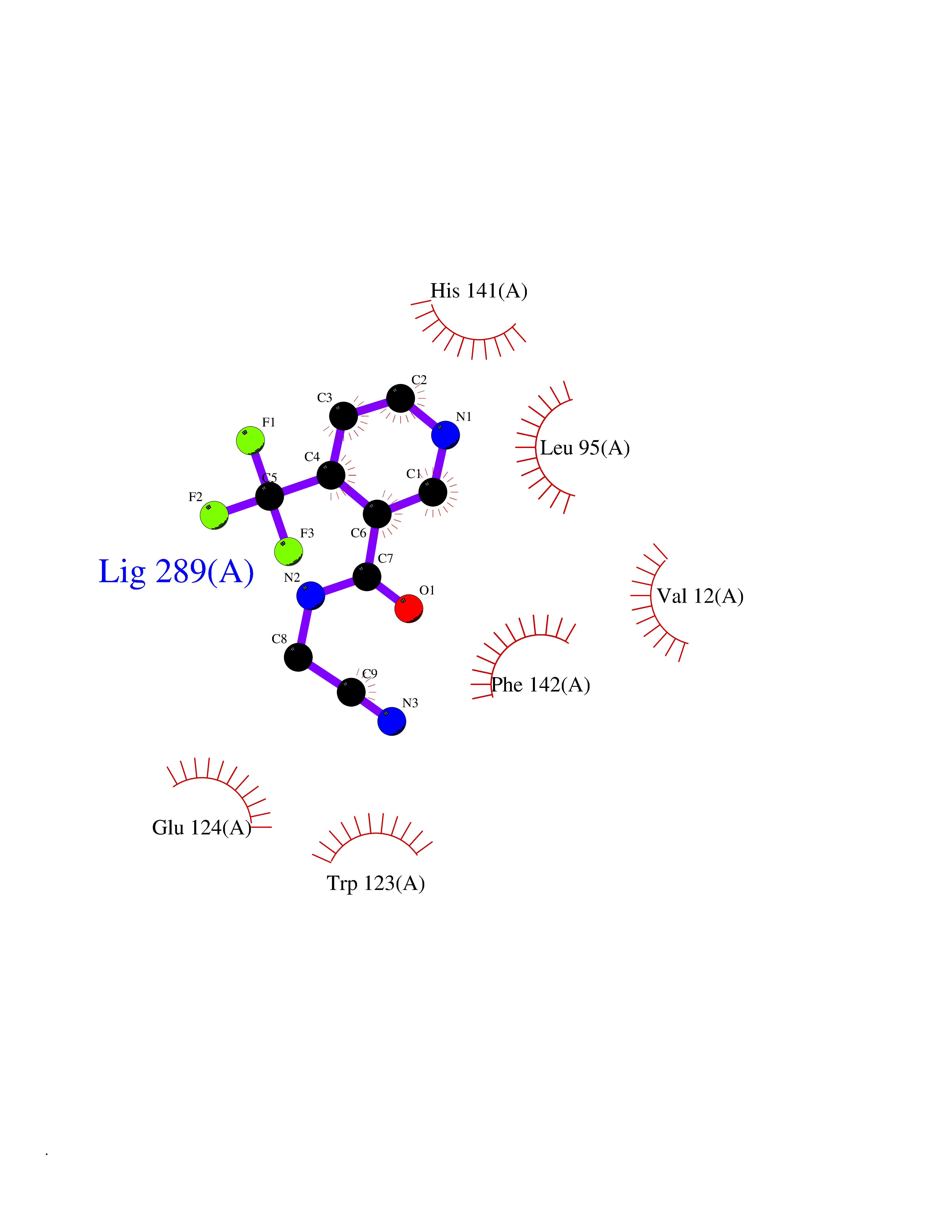 Ligplot Map 3D Binding mode Sequence NDLTPHFEDVQAHYDLSDDFFRLFLDPTQTYSCAHFEREDMTLEEAQIAKIDLALGKLGLQPGMTLLDIGCGWGATMRRAIAQYDVNVVGLTLSKNQAAHVQKSFDEMDTPRDRRVLLAGWEQFNEPVDRIVSIGAFEHFGHDRHADFFARAHKILPPDGVLLLHTITGLTRQQMVDHGLPLTLWLARFLKFIATEIFPGGQPPTIEMVEEQSAKTGFTLTRRQSLQPHYARTLDLWAEALQEHKSEAIAIQSEEVYERYMKYLTGCAKLFRVGYIDVNQFTLAK Hydrogen bonds contact Hydrophobic contact | ||||
| 2 | Acetylcholine receptor subunit alpha | 4ZJS | 5.55 | |
Target general information Gen name CHRNA1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms ACHRA;CHNRA Protein family Ligand-gated ion channel (TC 1.A.9) family, Acetylcholine receptor (TC 1.A.9.1) subfamily, Alpha-1/CHRNA1 sub-subfamily Biochemical class Immune system Function Acetylcholine binding.Acetylcholine-gated cation-selective channel activity.Acetylcholine receptor activity.Ion channel activity.Ligand-gated ion channel activity. Related diseases Multiple pterygium syndrome, lethal type (LMPS) [MIM:253290]: Multiple pterygia are found infrequently in children with arthrogryposis and in fetuses with fetal akinesia syndrome. In lethal multiple pterygium syndrome there is intrauterine growth retardation, multiple pterygia, and flexion contractures causing severe arthrogryposis and fetal akinesia. Subcutaneous edema can be severe, causing fetal hydrops with cystic hygroma and lung hypoplasia. Oligohydramnios and facial anomalies are frequent. {ECO:0000269|PubMed:18252226}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: The alpha subunit is the main focus for antibody binding in myasthenia gravis. Myasthenia gravis is characterized by sporadic muscular fatigability and weakness, occurring chiefly in muscles innervated by cranial nerves, and characteristically improved by cholinesterase-inhibiting drugs.; DISEASE: Myasthenic syndrome, congenital, 1A, slow-channel (CMS1A) [MIM:601462]: A common congenital myasthenic syndrome. Congenital myasthenic syndromes are characterized by muscle weakness affecting the axial and limb muscles (with hypotonia in early-onset forms), the ocular muscles (leading to ptosis and ophthalmoplegia), and the facial and bulbar musculature (affecting sucking and swallowing, and leading to dysphonia). The symptoms fluctuate and worsen with physical effort. CMS1A is a slow-channel myasthenic syndrome. It is caused by kinetic abnormalities of the AChR, resulting in prolonged AChR channel opening episodes, prolonged endplate currents, and depolarization block. This is associated with calcium overload, which may contribute to subsequent degeneration of the endplate and postsynaptic membrane. {ECO:0000269|PubMed:16685696, ECO:0000269|PubMed:7619526, ECO:0000269|PubMed:8872460, ECO:0000269|PubMed:9158151, ECO:0000269|PubMed:9221765}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Myasthenic syndrome, congenital, 1B, fast-channel (CMS1B) [MIM:608930]: A form of congenital myasthenic syndrome, a group of disorders characterized by failure of neuromuscular transmission, including pre-synaptic, synaptic, and post-synaptic disorders that are not of autoimmune origin. Clinical features are easy fatigability and muscle weakness affecting the axial and limb muscles (with hypotonia in early-onset forms), the ocular muscles (leading to ptosis and ophthalmoplegia), and the facial and bulbar musculature (affecting sucking and swallowing, and leading to dysphonia). The symptoms fluctuate and worsen with physical effort. CMS1B is a fast-channel myasthenic syndrome. It is caused by kinetic abnormalities of the AChR, resulting in brief opening and activity of the channel, with a rapid decay in endplate current, failure to achieve threshold depolarization of the endplate and consequent failure to fire an action potential. {ECO:0000269|PubMed:10195214, ECO:0000269|PubMed:12588888, ECO:0000269|PubMed:15079006}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08838; DB00565; DB00555 Interacts with NA EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Congenital myasthenic syndrome; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; Ion channel; Ion transport; Ligand-gated ion channel; Membrane; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Signal; Synapse; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A,B,C,D,E Molecular weight (Da) 46717.8 Length 411 Aromaticity 0.11 Instability index 38.02 Isoelectric point 4.77 Charge (pH=7) -22.31 2D Binding mode Binding energy (Kcal/mol) -7.57 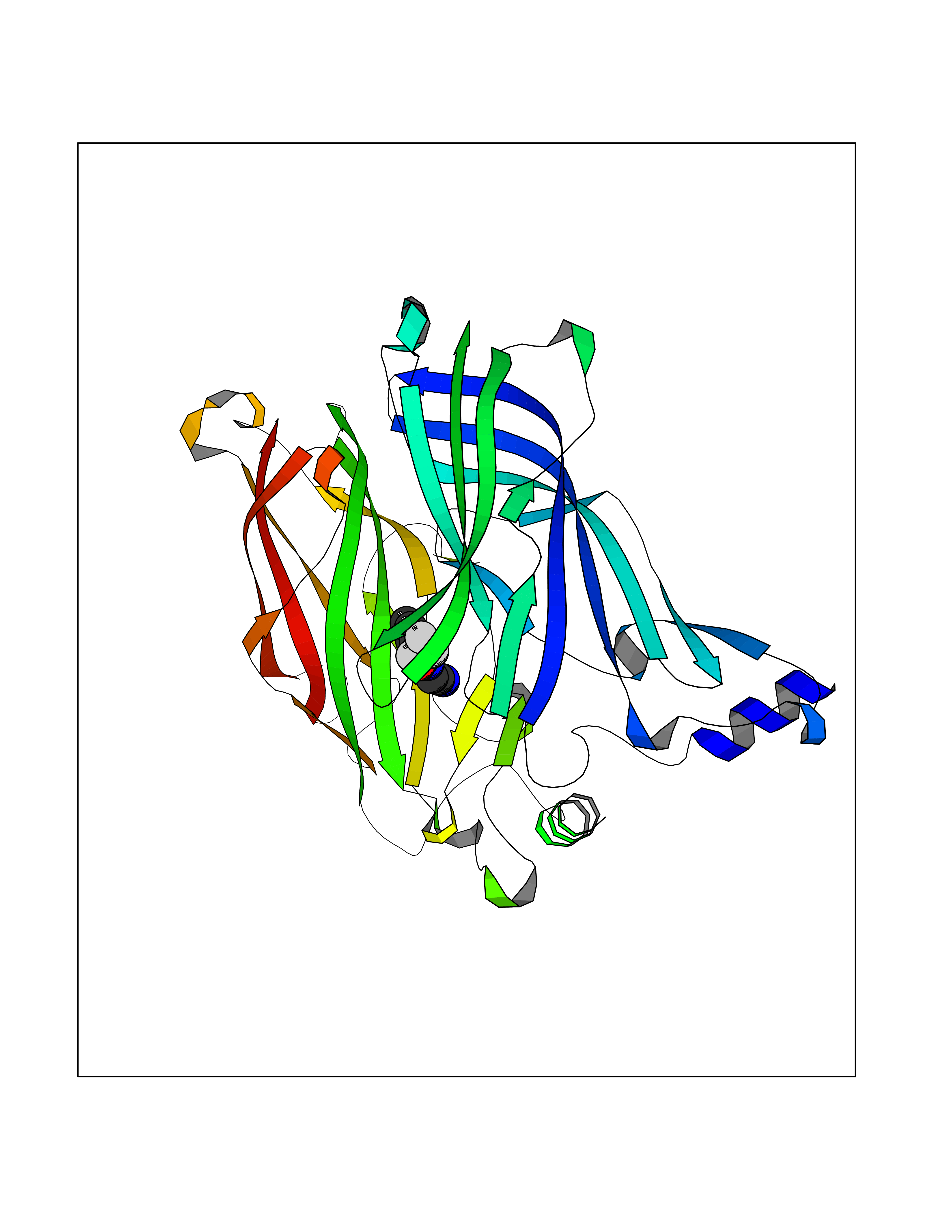 Molscript Map 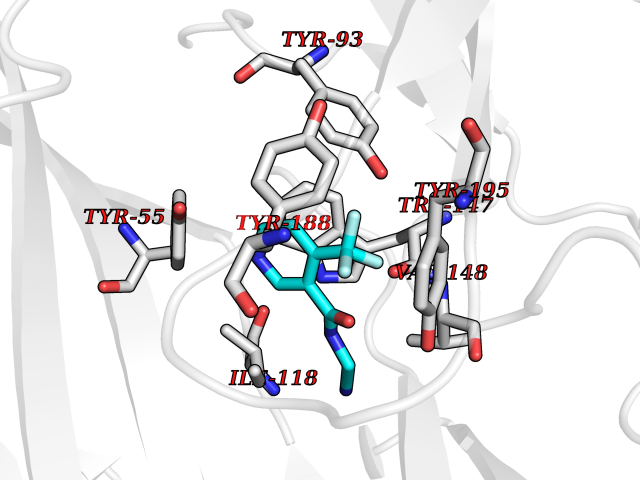 Pymol Map 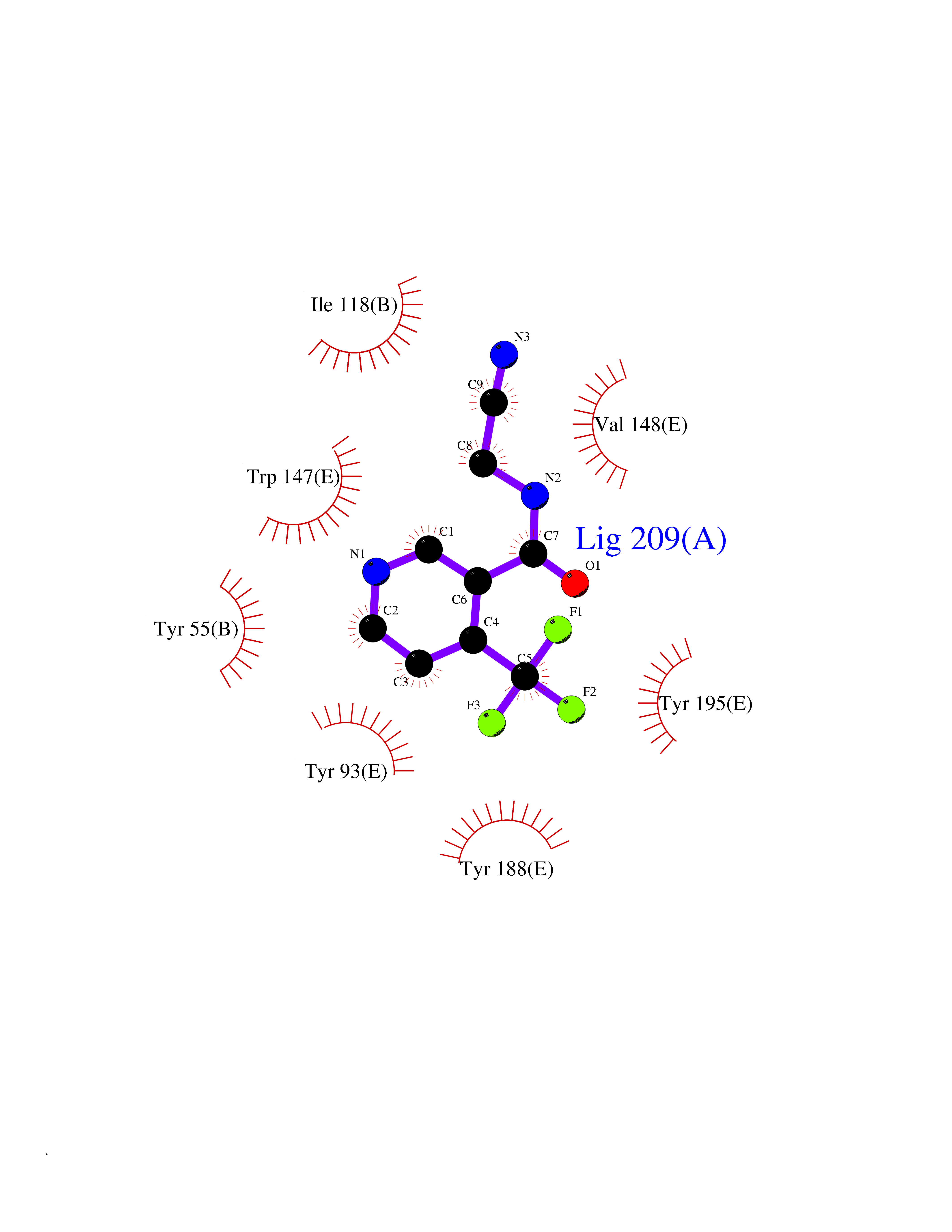 Ligplot Map 3D Binding mode Sequence EHETRLVAKLFKDYSSVVRPVEDHRQVVEVTLGFTLQDIVKADSSTNEVDLVYYEQQRWVDYNLKWNPDDYGGVKKIHIPAADIWTPDITAYSSTRPVQVLSPQIAVVTHDGSVMFIPAQRLSFMCDPTGVDSEEGATCAVKFGSWVYSGFEIDLKTDTDQVDLSSYYASSKYEILSATQTRQVQHYSCCPEPYIDVNLVVKFREEHETRLVAKLFKDYSSVVRPVEDHRQVVEVTLGFTLQDIVKADSSTNEVDLVYYEQQRWVDYNLKWNPDDYGGVKKIHIPAADIWTPDITAYSSTRPVQVLSPQIAVVTHDGSVMFIPAQRLSFMCDPTGVDSEEGATCAVKFGSWVYSGFEIDLKTDTDQVDLSSYYASSKYEILSATQTRQVQHYSCCPEPYIDVNLVVKFRER Hydrogen bonds contact Hydrophobic contact | ||||
| 3 | "Periplasmic trehalase (EC 3.2.1.28) (Alpha,alpha-trehalase) (Alpha,alpha-trehalose glucohydrolase) (Tre37A)" | 2JG0 | 5.54 | |
Target general information Gen name treA Organism Escherichia coli (strain K12) Uniprot ID TTD ID NA Synonyms JW1186;osmA;b1197 Protein family Glycosyl hydrolase 37 family Biochemical class NA Function Provides the cells with the ability to utilize trehalose at high osmolarity by splitting it into glucose molecules that can subsequently be taken up by the phosphotransferase-mediated uptake system. Related diseases SRC kinase activity has been shown to be increased in several tumor tissues and tumor cell lines such as colon carcinoma cells. {ECO:0000269|PubMed:2498394, ECO:0000269|PubMed:3093483}.; DISEASE: Thrombocytopenia 6 (THC6) [MIM:616937]: A form of thrombocytopenia, a hematologic disorder defined by a decrease in the number of platelets in circulating blood, resulting in the potential for increased bleeding and decreased ability for clotting. THC6 is an autosomal dominant form. Affected individuals may also have bone abnormalities and an increased risk for myelofibrosis. {ECO:0000269|PubMed:26936507}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number 3.2.1.28 Uniprot keywords 3D-structure; Direct protein sequencing; Glycosidase; Hydrolase; Periplasm; Reference proteome; Signal Protein physicochemical properties Chain ID A Molecular weight (Da) 57508.9 Length 507 Aromaticity 0.11 Instability index 48.32 Isoelectric point 5.48 Charge (pH=7) -10.13 2D Binding mode Binding energy (Kcal/mol) -7.55 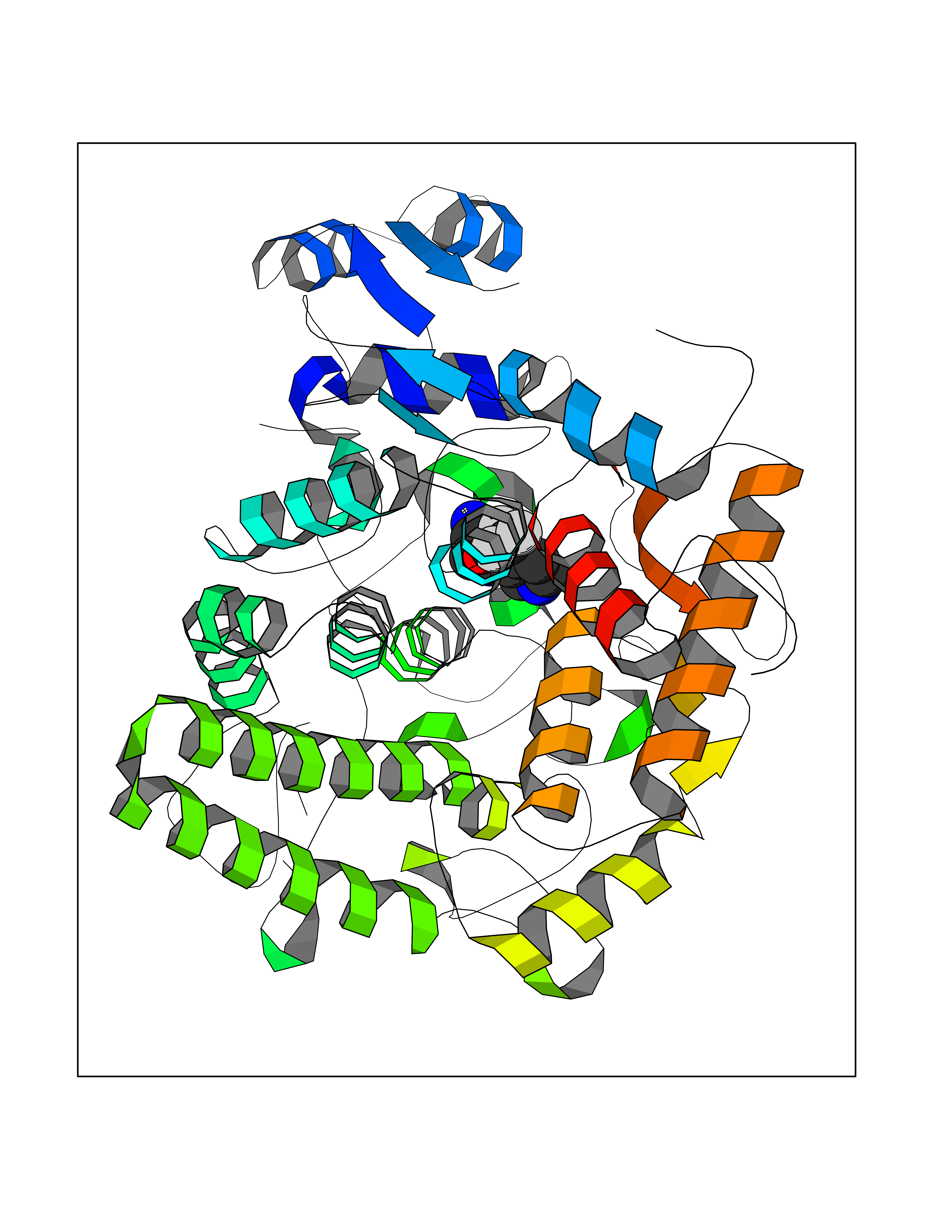 Molscript Map 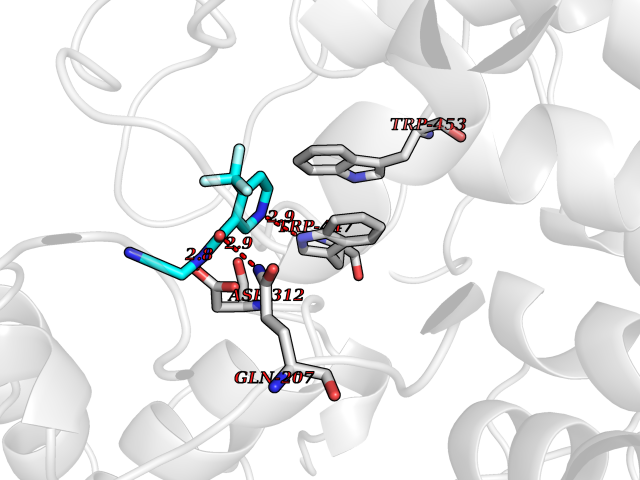 Pymol Map 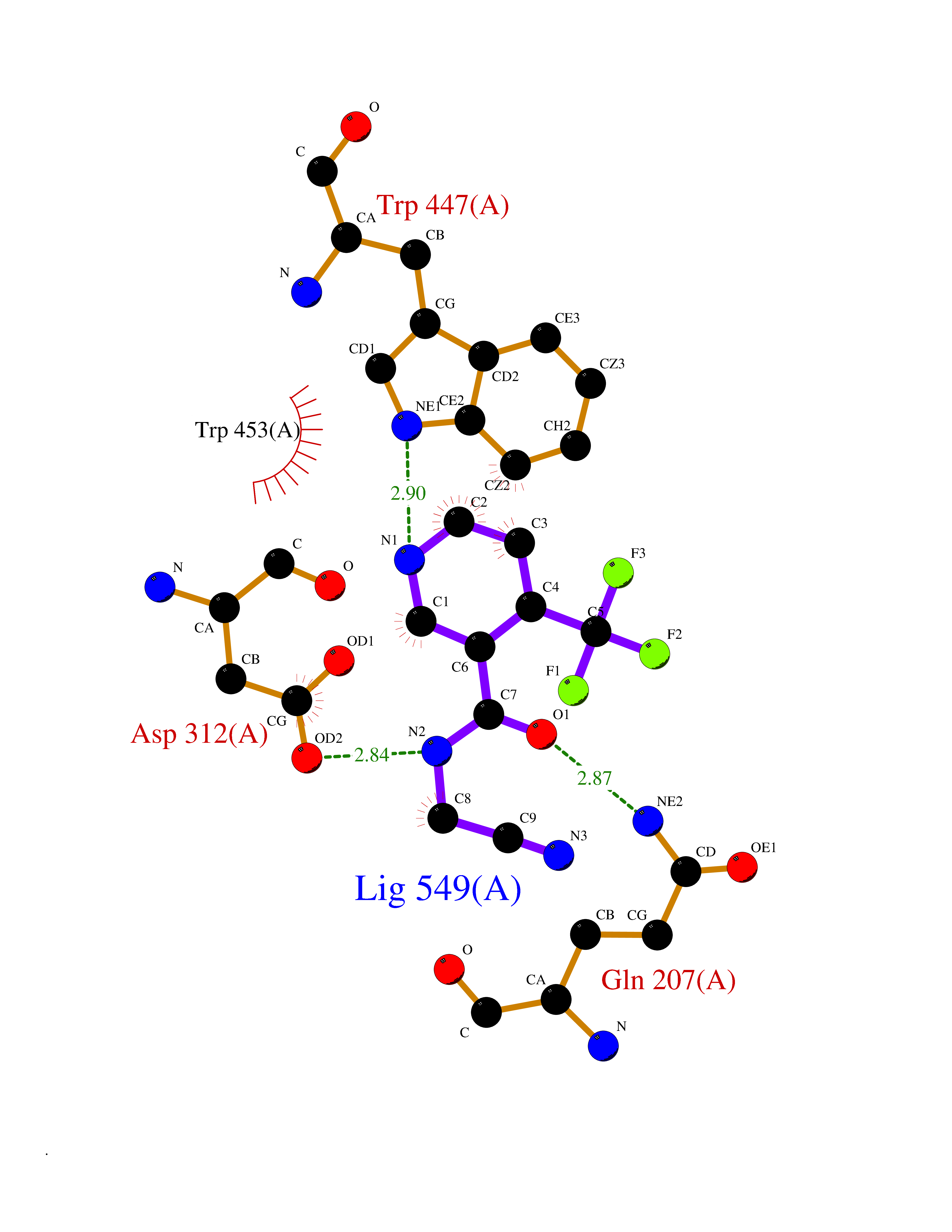 Ligplot Map 3D Binding mode Sequence PQPPDILLGPLFNDVQNAKLFPDQKTFADAVPNSDPLMILADYRMQQNQSGFDLRHFVNVNFTLPKYVPPEGQSLREHIDGLWPVLTRSTENTEKWDSLLPLPEPYVVPGGRFREVYYWDSYFTMLGLAESGHWDKVADMVANFAHEIDTYGHIPNGNRSYYLSRSQPPFFALMVELLAQHEGDAALKQYLPQMQKEYAYWMDGVENLQAGQQEKRVVKLQDGTLLNRYWDDRDTPRPESWVEDIATAKSNPNRPATEIYRDLRSAAASGWDFSSRWMDNPQQLNTLRTTSIVPVDLNSLMFKMEKILARASKAAGDNAMANQYETLANARQKGIEKYLWNDQQGWYADYDLKSHKVRNQLTAAALFPLYVNAAAKDRANKMATATKTHLLQPGGLNTTSVKSGQQWDAPNGWAPLQWVATEGLQNYGQKEVAMDISWHFLTNVQHTYDREKKLVEKYDVSTTGTGGGGGEYPLQDGFGWTNGVTLKMLDLICPKEQPCDNVPATRP Hydrogen bonds contact Hydrophobic contact | ||||
| 4 | Dopamine beta-hydroxylase | 4ZEL | 5.53 | |
Target general information Gen name DBH Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Copper type II ascorbate-dependent monooxygenase family Biochemical class Oxidoreductase Function Catalytic activity.Copper ion binding.Dopamine beta-monooxygenase activity.L-ascorbic acid binding. Related diseases Orthostatic hypotension 1 (ORTHYP1) [MIM:223360]: A form of orthostatic hypotension due to congenital dopamine beta-hydroxylase deficiency. Orthostatic hypotension, also known as postural hypotension, is a finding defined as a 20-mm Hg decrease in systolic pressure or a 10-mm Hg decrease in diastolic pressure occurring 3 minutes after a person has risen from supine to standing. Symptoms include dizziness, blurred vision, and sometimes syncope. ORTHYP1 is an autosomal recessive condition apparent from infancy or early childhood and characterized by low plasma and urinary levels of norepinephrine and epinephrine, and episodic hypoglycemia. {ECO:0000269|PubMed:11857564}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00126; DB06774; DB09130; DB05394; DB00822; DB00988; DB00968; DB00550 Interacts with P00352; P63010-2; Q04656; Q8WUW1; Q9UNS2; Q71DI3; P61978; Q9Y2M5; Q92876; P08727; Q14693; P0DPK4; Q6GQQ9-2; P27986-2; Q9ULX5; Q96D59; Q8N6K7-2; Q9GZS3; Q8IUW3; Q86WT6-2 EC number 1.14.17.1 Uniprot keywords 3D-structure; Catecholamine biosynthesis; Copper; Cytoplasmic vesicle; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; Membrane; Metal-binding; Monooxygenase; Oxidoreductase; Proteomics identification; Reference proteome; Secreted; Signal-anchor; Transmembrane; Transmembrane helix; Vitamin C Protein physicochemical properties Chain ID A,B Molecular weight (Da) 123694 Length 1094 Aromaticity 0.1 Instability index 51.85 Isoelectric point 5.84 Charge (pH=7) -24.5 2D Binding mode Binding energy (Kcal/mol) -7.54  Molscript Map 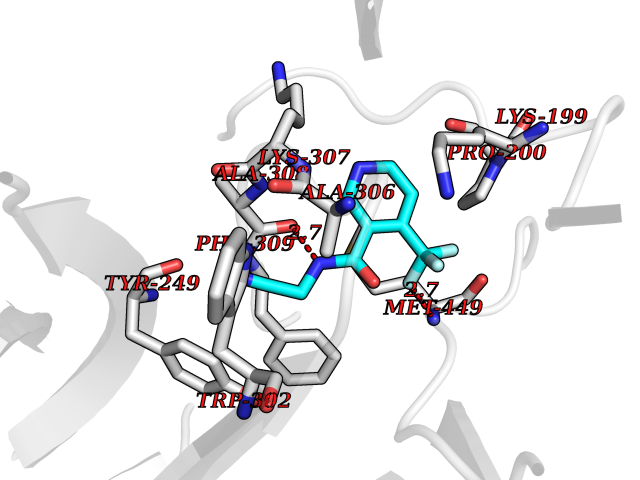 Pymol Map 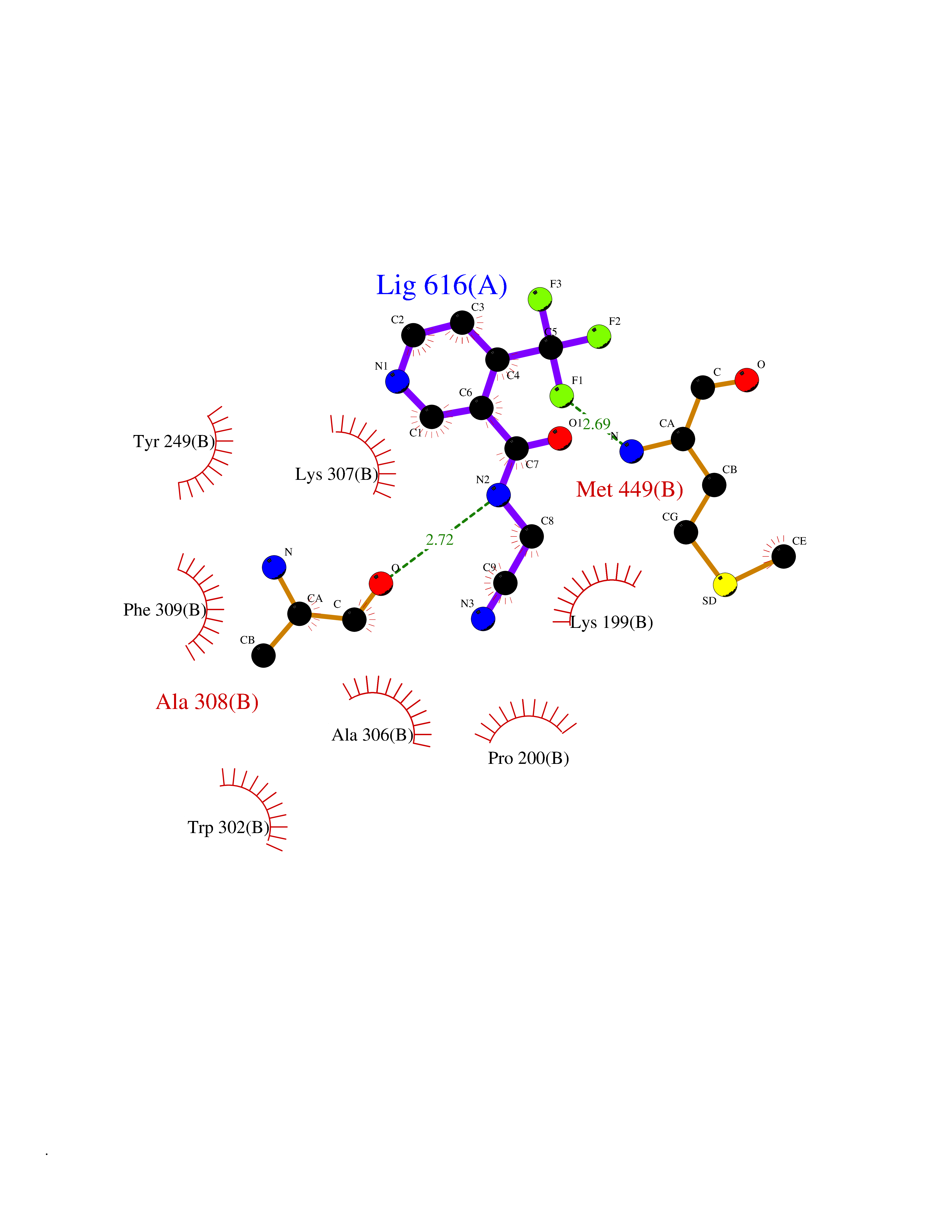 Ligplot Map 3D Binding mode Sequence PLPYHIPLDPEGSLELSWNVSYTQEAIHFQLLVRRLKAGVLFGMSDRGELENADLVVLWTDGDAYFADAWSDQKGQIHLDPQQDYQLLQVQRTPEGLTLLFKRPFGTCDPKDYLIEDGTVHLVYGILEEPFRSLEAINGSGLQMGLQRVQLLKPNIPEPELPSDACTMEVQAPNIQIPSQETTYWCYIKELPKGFSRHHIIKYEPIVTKGNEALVHHMEVFQCAPEMDSVPHFSGPCDSKMKPDRLNYCRHVLAAWALGAKAFYYPEEAGLAFGGPGSSRYLRLEVHYHNPLVIEGRNDSSGIRLYYTAKLRRFNAGIMELGLVYTPVMAIPPRETAFILTGYCTDKCTQLALPPSGIHIFASQLHTHLTGRKVVTVLVRDGREWEIVNQDNHYSPHFQEIRMLKKVVSVHPGDVLITSCTYNTEDRELATVGGFGILEEMCVNYVHYYPQTQLELCKSAVDAGFLQKYFHLINRFNNEDVCTCPQASVSQQFTSVPWNSFNRDVLKALYSFAPISMHCNKSSAVRFQGEWNLQPLPKVISTLEEPTVVSPLPYHIPLDPEGSLELSWNVSYTQEAIHFQLLVRRLKAGVLFGMSDRGELENADLVVLAYFADAWSDQKGQIHLDPQQDYQLLQVQRTPEGLTLLFKRPFGTCDPKDYLIEDGTVHLVYGILEEPFRSLEAINGSGLQMGLQRVQLLKPNIPEPELPSDACTMEVQAPNIQIPSQETTYWCYIKELPKGFSRHHIIKYEPIVTKGNEALVHHMEVFQCAPEVPHFSGPCDSKMLNYCRHVLAAWALGAKAFYYPEEAGLAFGGPGSSRYLRLEVHYHNPLVIEGRNDSSGIRLYYTAKLRRFNAGIMELGLVYTPVMAIPPRETAFILTGYCTDKCTQLALPPSGIHIFASQLHTHLTGRKVVTVLVRDGREWEIVNQDNHYSPHFQEIRMLKKVVSVHPGDVLITSCTYNTEDRELATVGGFGILEEMCVNYVHYYPQTQLELCKSAVDAGFLQKYFHLINRFNNEDVCTCPQASVSQQFTSVPWNSFNRDVLKALYSFAPISMHCNKSSAVRFQGEWNLQPLPKVISTLEEPTPQCVVSIGG Hydrogen bonds contact Hydrophobic contact | ||||
| 5 | Programmed cell death 1 ligand 1 (PD-L1) | 5J89 | 5.53 | |
Target general information Gen name CD274 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms hPD-L1; Programmed death ligand 1; PDL1; PDCD1LG1; PDCD1L1; PDCD1 ligand 1; B7H1; B7-H1; B7 homolog 1 Protein family Immunoglobulin superfamily, BTN/MOG family Biochemical class Immunoglobulin Function As a ligand for the inhibitory receptor PDCD1/PD-1, modulates the activation threshold of T-cells and limits T-cell effector response. Through a yet unknown activating receptor, may costimulate T-cell subsets that predominantly produce interleukin-10 (IL10). Plays a critical role in induction and maintenance of immune tolerance to self. Related diseases Truncation of the 3'-untranslated (3'-UTR) region of CD274 transcripts leads to elevated expression of CD274 in multiple cancers including T-cell leukemia, diffuse large B-cell lymphoma and stomach adenocarcinoma (PubMed:27281199). Disruption of 3'-UTR region is caused by structural variants that stabilize CD274 transcripts, leading to overexpression (PubMed:27281199). Increased expression in tumors promotes immune evasion and tumor cell growth by allowing malignant cells to escape destruction by the immune system (PubMed:27281199). {ECO:0000269|PubMed:27281199}. Drugs (DrugBank ID) DB15773; DB11595; DB15771; DB11945; DB15772; DB14776; DB15770; DB11714; DB15769; DB09035; DB09037; DB00203; DB00313 Interacts with P33681; Q8IZR5; Q9NX76; Q15116; Q15116 EC number NA Uniprot keywords 3D-structure; Adaptive immunity; Alternative splicing; Cell membrane; Disulfide bond; Endosome; Glycoprotein; Immunity; Immunoglobulin domain; Membrane; Nucleus; Proteomics identification; Receptor; Reference proteome; Repeat; Secreted; Signal; Transmembrane; Transmembrane helix; Ubl conjugation Protein physicochemical properties Chain ID C,D Molecular weight (Da) 28335.2 Length 249 Aromaticity 0.1 Instability index 35.39 Isoelectric point 6.15 Charge (pH=7) -3.43 2D Binding mode Binding energy (Kcal/mol) -7.54 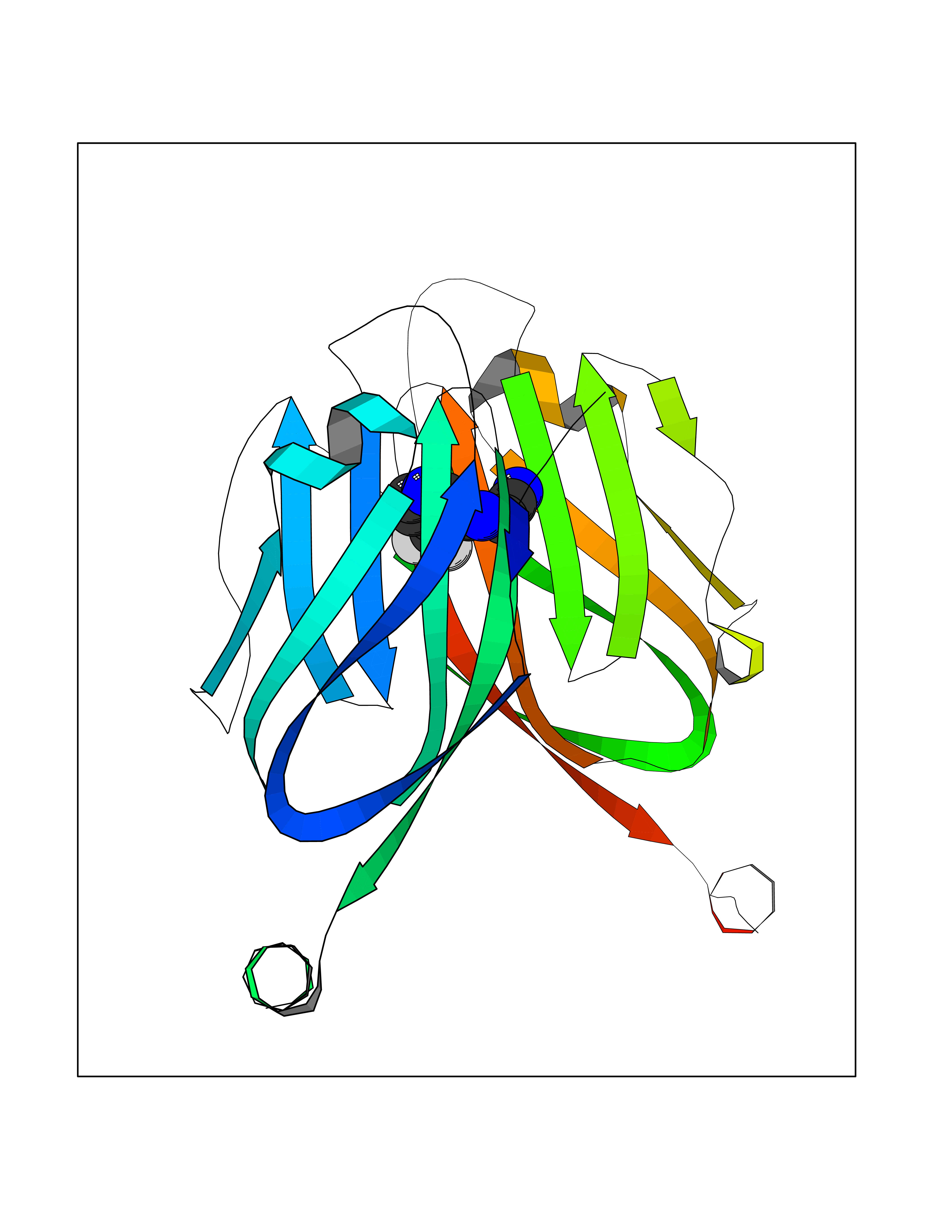 Molscript Map  Pymol Map 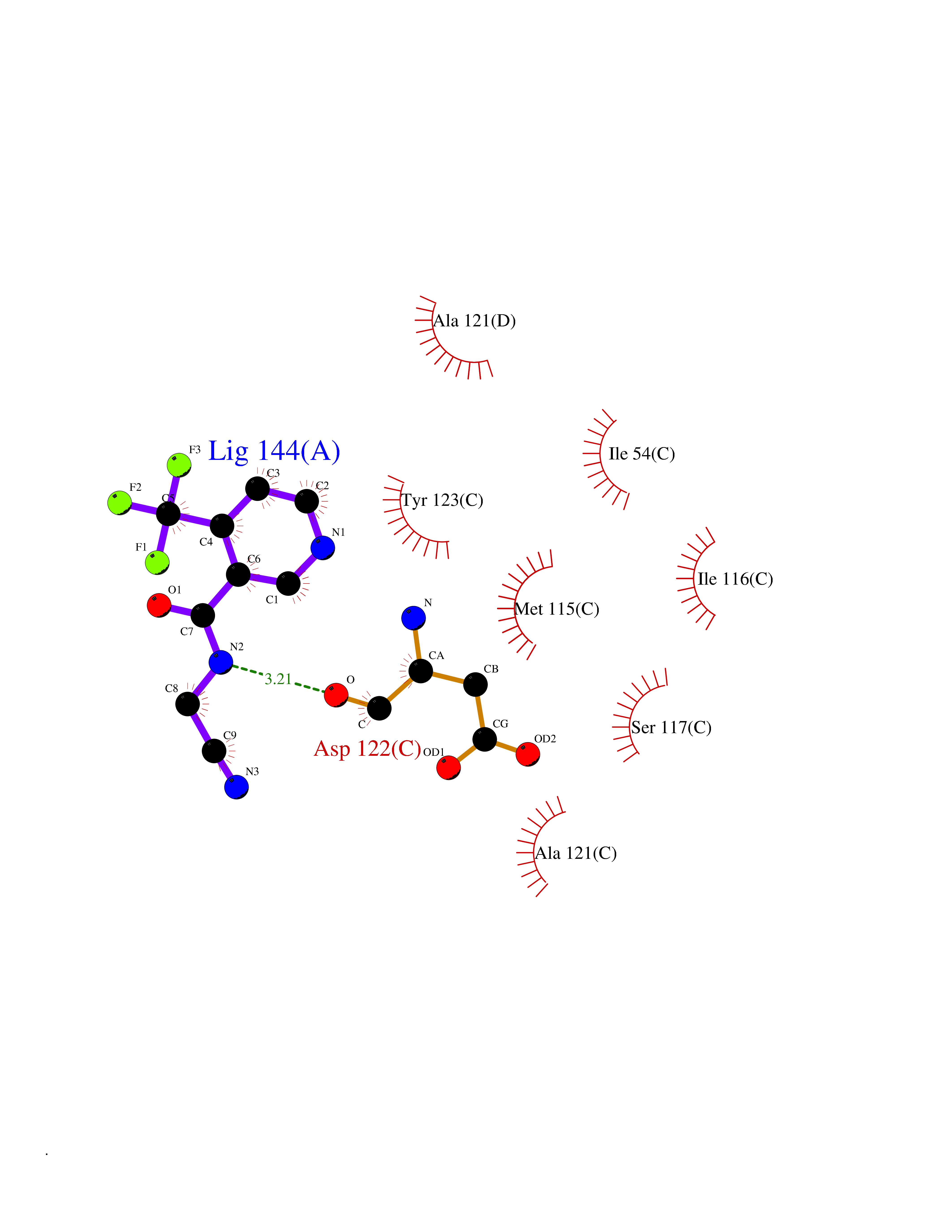 Ligplot Map 3D Binding mode Sequence AFTVTVPKDLYVVEYGSNMTIECKFPVEKQLDLAALIVYWEMEDKNIIQFVHGEEDLKVQHSSYRQRARLLKDQLSLGNAALQITDVKLQDAGVYRCMISYGGADYKRITVKVNAPYAAALEHHHAFTVTVPKDLYVVEYGSNMTIECKFPVEKQLDLAALIVYWEMEDKNIIQFVHGEEDLKVQHSSYRQRARLLKDQLSLGNAALQITDVKLQDAGVYRCMISYGGADYKRITVKVNAPYAAALEHH Hydrogen bonds contact Hydrophobic contact | ||||
| 6 | Spermine synthase | 3C6K | 5.51 | |
Target general information Gen name SMS Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Spermidine/spermine synthase family Biochemical class Transferase Function Spermine synthase activity. Related diseases Intellectual developmental disorder, X-linked, syndromic, Snyder-Robinson type (MRXSSR) [MIM:309583]: An X-linked intellectual disability syndrome characterized by a collection of clinical features including facial asymmetry, marfanoid habitus, hypertonia, osteoporosis and unsteady gait. {ECO:0000269|PubMed:14508504, ECO:0000269|PubMed:18550699, ECO:0000269|PubMed:19206178, ECO:0000269|PubMed:22612257, ECO:0000269|PubMed:23696453, ECO:0000269|PubMed:23897707}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00127 Interacts with NA EC number 2.5.1.22 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Direct protein sequencing; Intellectual disability; Phosphoprotein; Polyamine biosynthesis; Proteomics identification; Reference proteome; Transferase Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 27177.1 Length 238 Aromaticity 0.11 Instability index 41.31 Isoelectric point 4.82 Charge (pH=7) -9.19 2D Binding mode Binding energy (Kcal/mol) -7.52 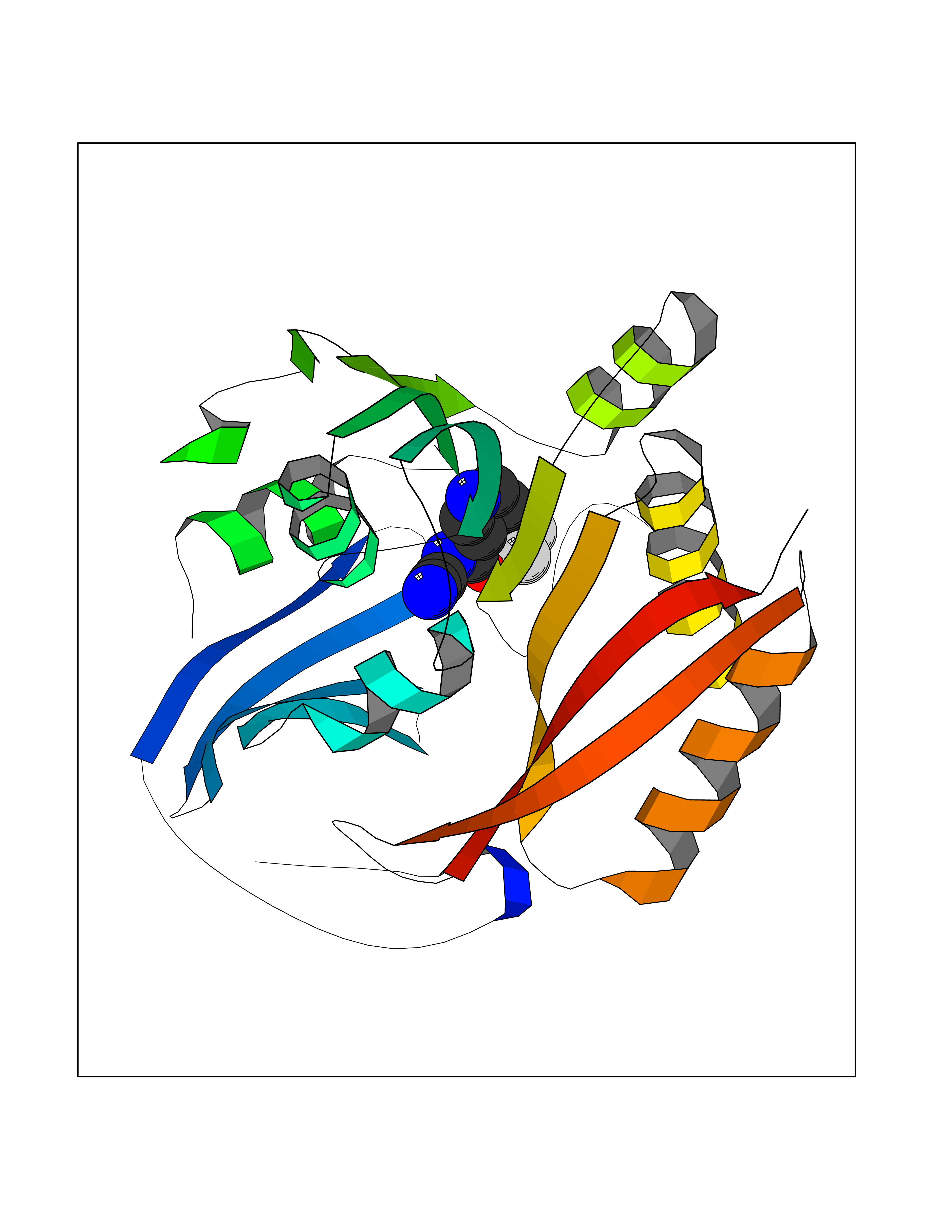 Molscript Map 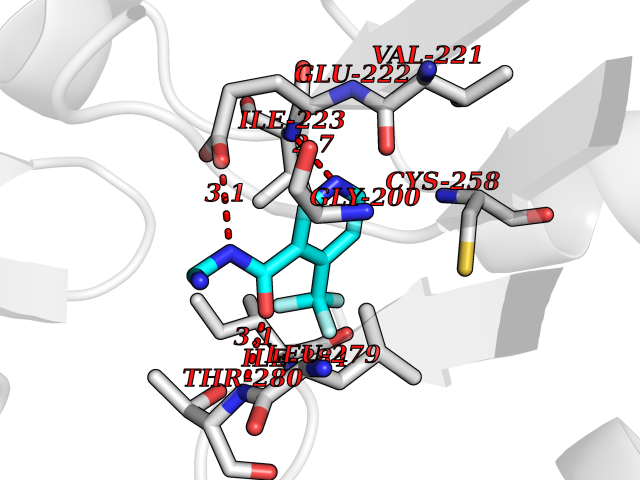 Pymol Map 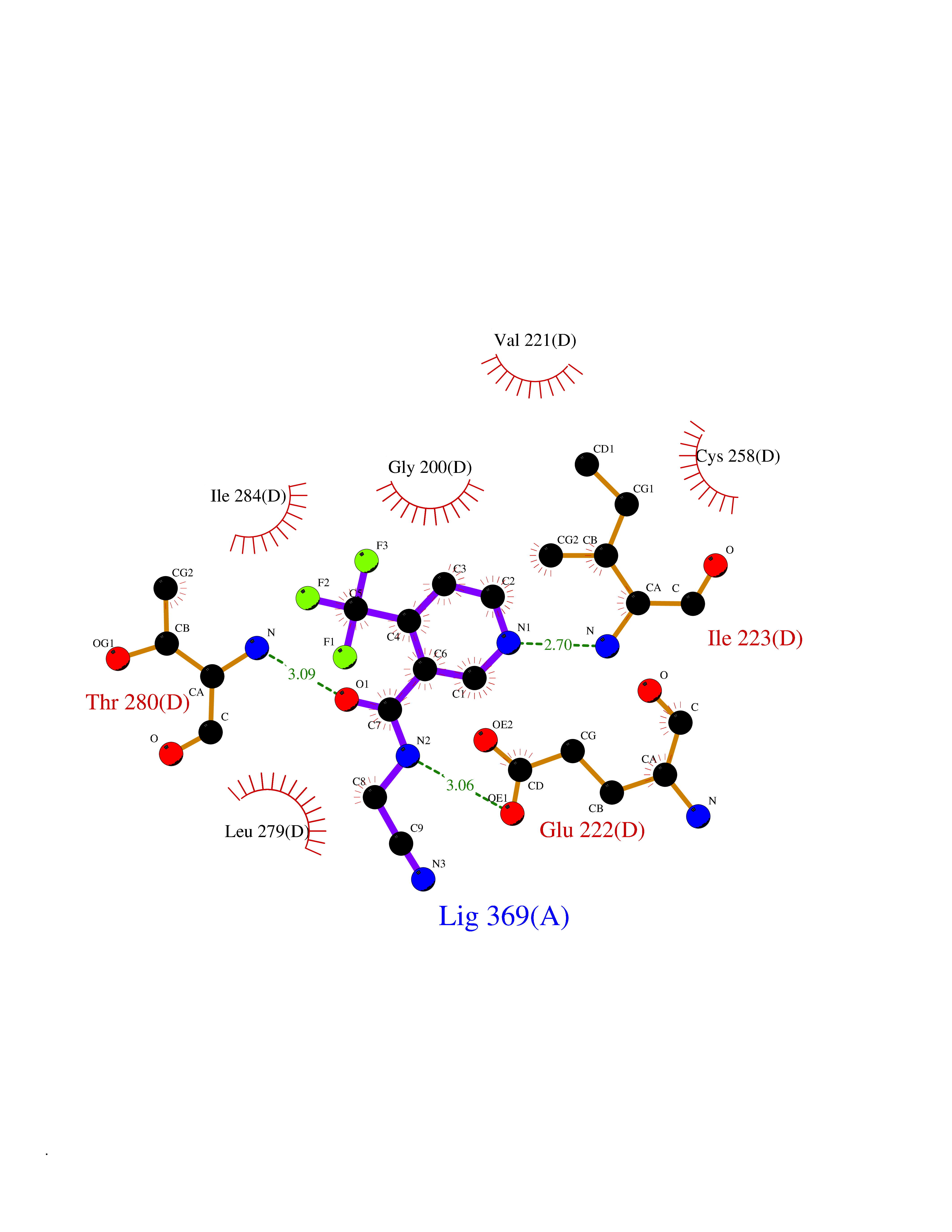 Ligplot Map 3D Binding mode Sequence RYWPTADGRLVEYDIDEVVYDEDSPYQNIKILHSKQFGNILILSGDVNLAESDLAYTRAIMGSGKEDYTGKDVLILGGGDGGILCEIVKLKPKMVTMVEIDQMVIDGCKKYMRKDVLDNLKGDCYQVLIEDCIPVLKRYAKEGREFDYVINDLTAVPISTSPSTWEFLRLILDLSMKVLKQDGKYFTQGNCVNLTEALSLYEEQLGRLYCPVEFSKEIVCVPSYLELWVFYTVWKKAK Hydrogen bonds contact Hydrophobic contact | ||||
| 7 | Geranyltranstransferase (FDPS) | 4KPJ | 5.51 | |
Target general information Gen name FDPS Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms KIAA1293; Geranylgeranyl pyrophosphate synthase; Geranylgeranyl diphosphate synthase; GGPS1; GGPPSase; GGPP synthase; Farnesyltranstransferase; Farnesyl pyrophosphate synthase; Farnesyl diphosphate sy Protein family FPP/GGPP synthase family Biochemical class Alkyl aryl transferase Function FPP also serves as substrate for protein farnesylation and geranylgeranylation. Catalyzes the sequential condensation of isopentenyl pyrophosphate with the allylic pyrophosphates, dimethylallyl pyrophosphate, and then with the resultant geranylpyrophosphate to the ultimate product farnesyl pyrophosphate. Key enzyme in isoprenoid biosynthesis which catalyzes the formation of farnesyl diphosphate (FPP), a precursor for several classes of essential metabolites including sterols, dolichols, carotenoids, and ubiquinones. Related diseases Porokeratosis 9, multiple types (POROK9) [MIM:616631]: A form of porokeratosis, a disorder of faulty keratinization characterized by one or more atrophic patches surrounded by a distinctive hyperkeratotic ridgelike border called the cornoid lamella. The keratotic lesions can progress to overt cutaneous neoplasms, typically squamous cell carcinomas. Multiple clinical variants of porokeratosis are recognized, including porokeratosis of Mibelli, linear porokeratosis, disseminated superficial actinic porokeratosis, palmoplantar porokeratosis, and punctate porokeratosis. Different clinical presentations can be observed among members of the same family. Individuals expressing more than one variant have also been reported. {ECO:0000269|PubMed:26202976}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00630; DB01785; DB07780; DB02552; DB07841; DB00710; DB06255; DB04714; DB02508; DB06548; DB00282; DB00884; DB00399 Interacts with O95870; P54253; Q6ZMZ0; Q9BRI3; Q8WWF3 EC number EC 2.5.1.10 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Cholesterol biosynthesis; Cholesterol metabolism; Cytoplasm; Disease variant; Host-virus interaction; Hydroxylation; Isoprene biosynthesis; Lipid biosynthesis; Lipid metabolism; Magnesium; Metal-binding; Proteomics identification; Reference proteome; Steroid biosynthesis; Steroid metabolism; Sterol biosynthesis; Sterol metabolism; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 38705 Length 338 Aromaticity 0.12 Instability index 44.66 Isoelectric point 5.1 Charge (pH=7) -9.92 2D Binding mode Binding energy (Kcal/mol) -7.52 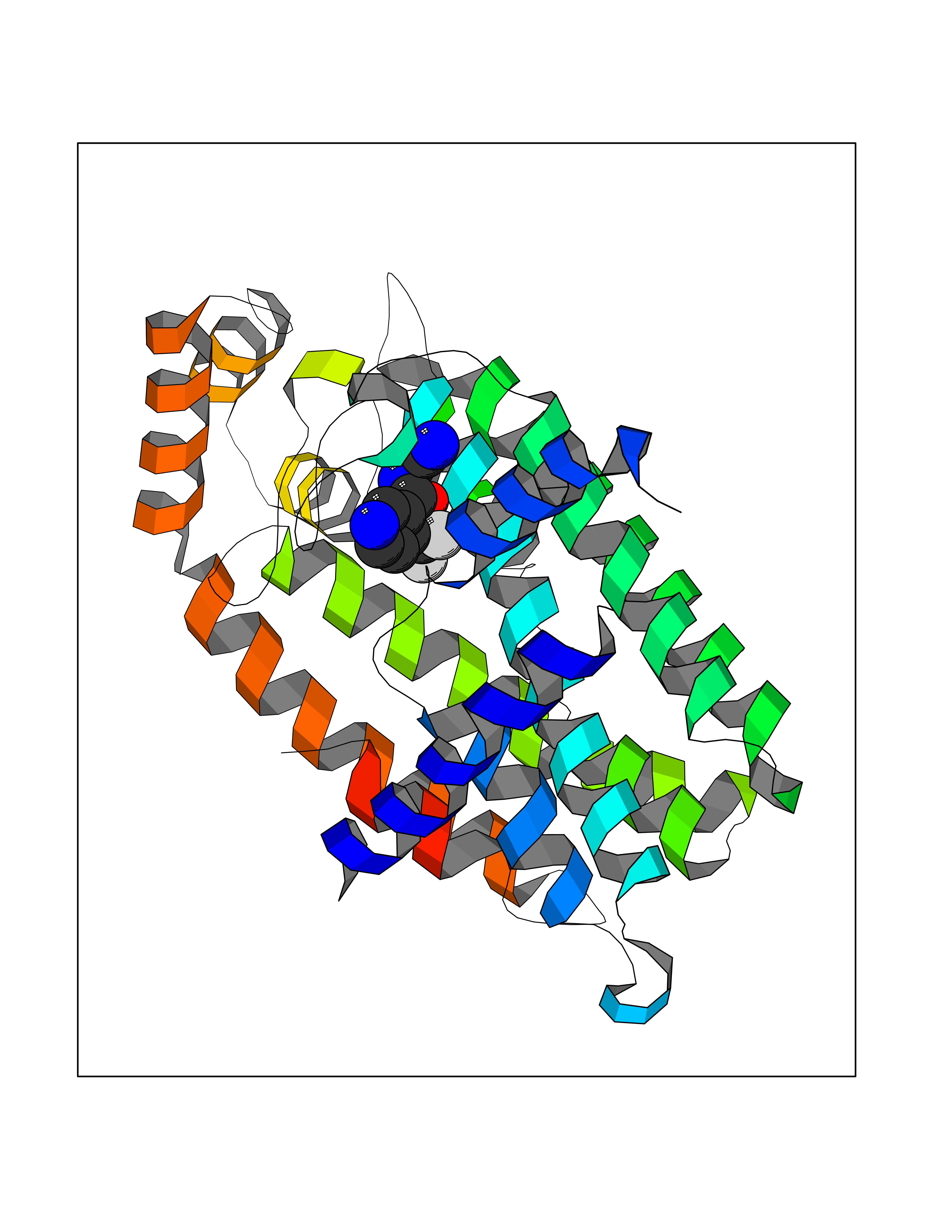 Molscript Map 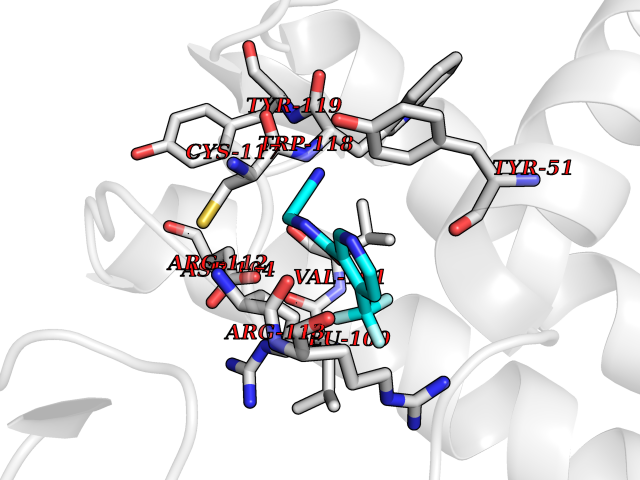 Pymol Map 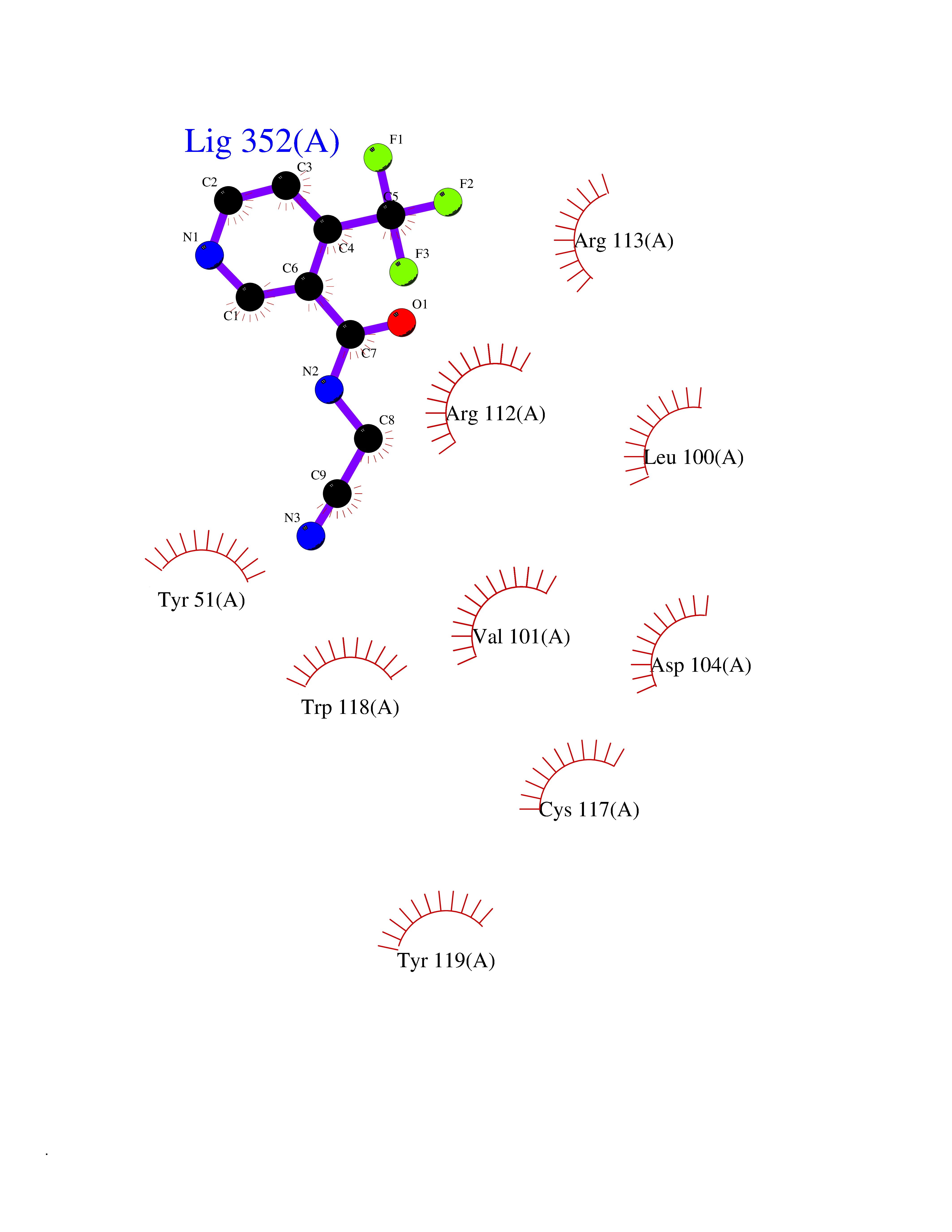 Ligplot Map 3D Binding mode Sequence VYAQEKQDFVQHFSQIVRVLTEHPEIGDAIARLKEVLEYNAIGGKYNRGLTVVVAFRELVEPRKQDADSLQRAWTVGWCVELLQAFFLVADDIMDSSLTRRGQICWYQKPGVGLDAINDANLLEACIYRLLKLYCREQPYYLNLIELFLQSSYQTEIGQTLDLLTAPQGNVDLVRFTEKRYKSIVKYKTAFASFYLPIAAAMYMAGIDGEKEHANAKKILLEMGEFFQIQDDYLDLFGDPSVTGKIGTDIQDNKCSWLVVQCLQRATPEQYQILKENYGQKEAEKVARVKALYEELDLPAVFLQYEEDSYSHIMALIEQYAAPLPPAVFLGLARKIYK Hydrogen bonds contact Hydrophobic contact | ||||
| 8 | Mycobacterium Cytochrome p450 121 (MycB cyp121) | 5EDT | 5.50 | |
Target general information Gen name MycB cyp121 Organism Mycobacterium bovis (strain ATCC BAA-935 / AF2122/97) Uniprot ID TTD ID Synonyms Cytochrome P450 MT2; Cytochrome P450 121 Protein family Cytochrome P450 family Biochemical class Paired donor oxygen oxidoreductase Function Catalyzes the formation of an intramolecular C-C bond between two tyrosyl carbon atoms of Cyy. Related diseases Long QT syndrome 1 (LQT1) [MIM:192500]: A heart disorder characterized by a prolonged QT interval on the ECG and polymorphic ventricular arrhythmias. They cause syncope and sudden death in response to exercise or emotional stress, and can present with a sentinel event of sudden cardiac death in infancy. {ECO:0000269|PubMed:10024302, ECO:0000269|PubMed:10220144, ECO:0000269|PubMed:10220146, ECO:0000269|PubMed:10367071, ECO:0000269|PubMed:10409658, ECO:0000269|PubMed:10482963, ECO:0000269|PubMed:10728423, ECO:0000269|PubMed:10973849, ECO:0000269|PubMed:11799244, ECO:0000269|PubMed:12442276, ECO:0000269|PubMed:15840476, ECO:0000269|PubMed:16414944, ECO:0000269|PubMed:16556865, ECO:0000269|PubMed:16922724, ECO:0000269|PubMed:18165683, ECO:0000269|PubMed:18400097, ECO:0000269|PubMed:19540844, ECO:0000269|PubMed:19716085, ECO:0000269|PubMed:19808498, ECO:0000269|PubMed:21241800, ECO:0000269|PubMed:24184248, ECO:0000269|PubMed:24269949, ECO:0000269|PubMed:24713462, ECO:0000269|PubMed:25037568, ECO:0000269|PubMed:25139741, ECO:0000269|PubMed:25705178, ECO:0000269|PubMed:28096388, ECO:0000269|PubMed:34398675, ECO:0000269|PubMed:8528244, ECO:0000269|PubMed:8818942, ECO:0000269|PubMed:8872472, ECO:0000269|PubMed:9024139, ECO:0000269|PubMed:9272155, ECO:0000269|PubMed:9302275, ECO:0000269|PubMed:9312006, ECO:0000269|PubMed:9323054, ECO:0000269|PubMed:9386136, ECO:0000269|PubMed:9482580, ECO:0000269|PubMed:9570196, ECO:0000269|PubMed:9641694, ECO:0000269|PubMed:9693036, ECO:0000269|PubMed:9702906, ECO:0000269|PubMed:9799083, ECO:0000269|PubMed:9927399, ECO:0000269|Ref.36}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Jervell and Lange-Nielsen syndrome 1 (JLNS1) [MIM:220400]: An autosomal recessive disorder characterized by congenital deafness, prolongation of the QT interval, syncopal attacks due to ventricular arrhythmias, and a high risk of sudden death. {ECO:0000269|PubMed:10090886, ECO:0000269|PubMed:10728423, ECO:0000269|PubMed:18400097, ECO:0000269|PubMed:18441444, ECO:0000269|PubMed:25705178, ECO:0000269|PubMed:9781056}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Atrial fibrillation, familial, 3 (ATFB3) [MIM:607554]: An autosomal dominant form of atrial fibrillation, a common sustained cardiac rhythm disturbance. Atrial fibrillation is characterized by disorganized atrial electrical activity and ineffective atrial contraction promoting blood stasis in the atria and reduces ventricular filling. It can result in palpitations, syncope, thromboembolic stroke, and congestive heart failure. {ECO:0000269|PubMed:12522251}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Short QT syndrome 2 (SQT2) [MIM:609621]: An autosomal dominant form of short QT syndrome, a heart disorder characterized by idiopathic persistently and uniformly short QT interval on ECG in the absence of structural heart disease in affected individuals. It can cause syncope and sudden death. {ECO:0000269|PubMed:15159330}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Type 2 diabetes mellitus (T2D) [MIM:125853]: A multifactorial disorder of glucose homeostasis caused by a lack of sensitivity to insulin. Affected individuals usually have an obese body habitus and manifestations of a metabolic syndrome characterized by diabetes, insulin resistance, hypertension and hypertriglyceridemia. The disease results in long-term complications that affect the eyes, kidneys, nerves, and blood vessels. {ECO:0000269|PubMed:18711366, ECO:0000269|PubMed:18711367, ECO:0000269|PubMed:24390345}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number EC 1.14.-.- Uniprot keywords 3D-structure; Cytoplasm; Heme; Iron; Metal-binding; Monooxygenase; Oxidoreductase; Reference proteome Protein physicochemical properties Chain ID A Molecular weight (Da) 43023.2 Length 394 Aromaticity 0.06 Instability index 39.15 Isoelectric point 6.23 Charge (pH=7) -2.55 2D Binding mode Binding energy (Kcal/mol) -7.5 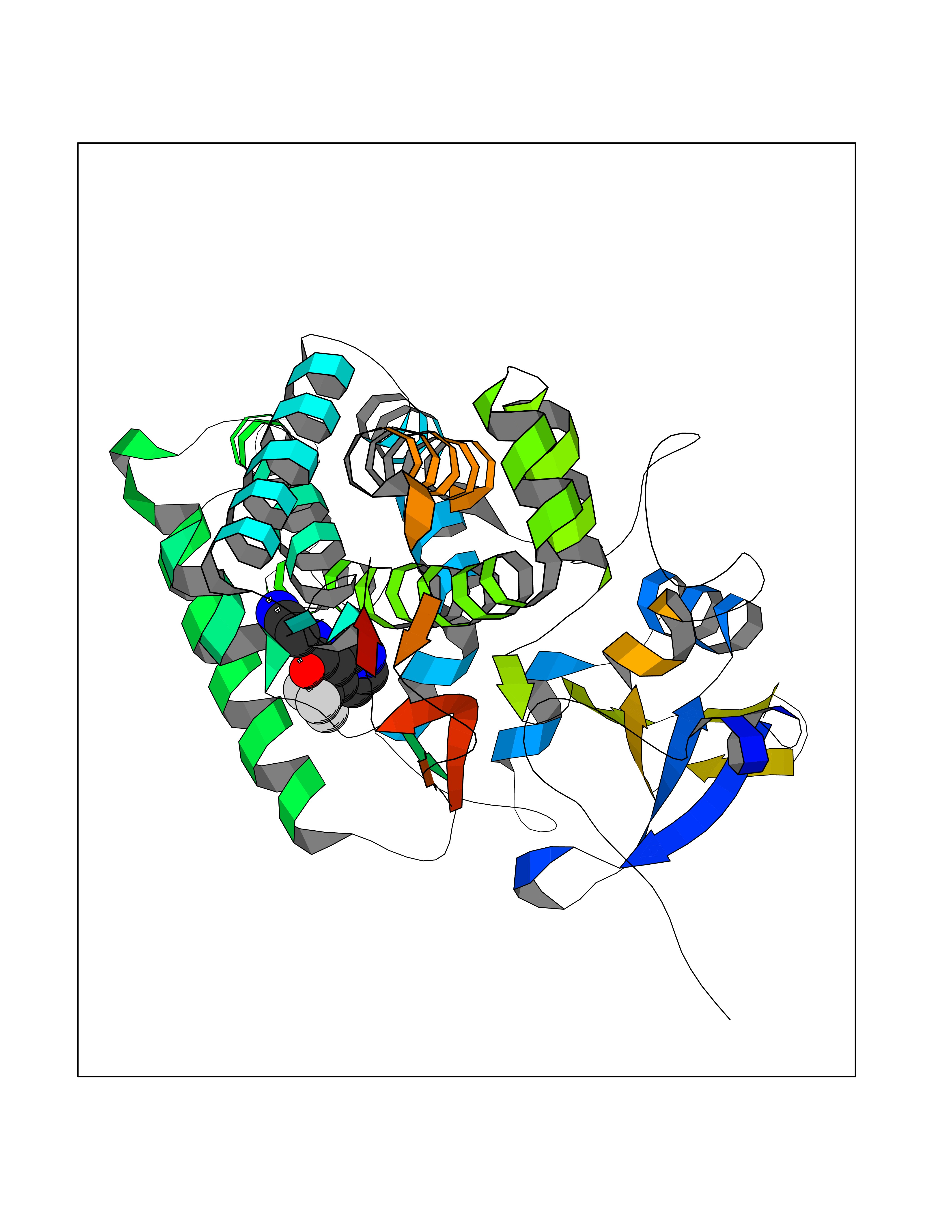 Molscript Map 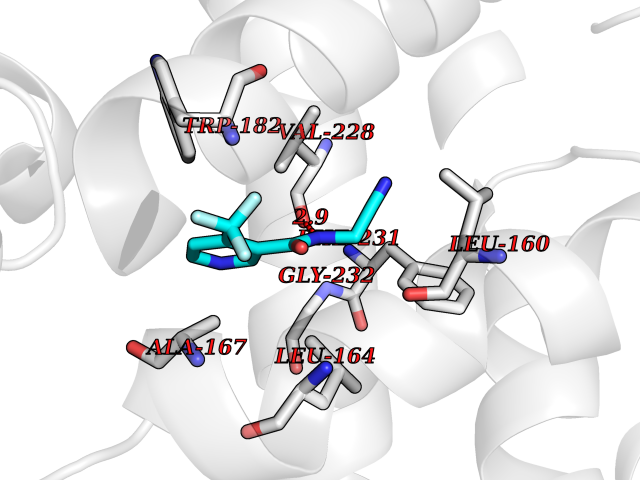 Pymol Map 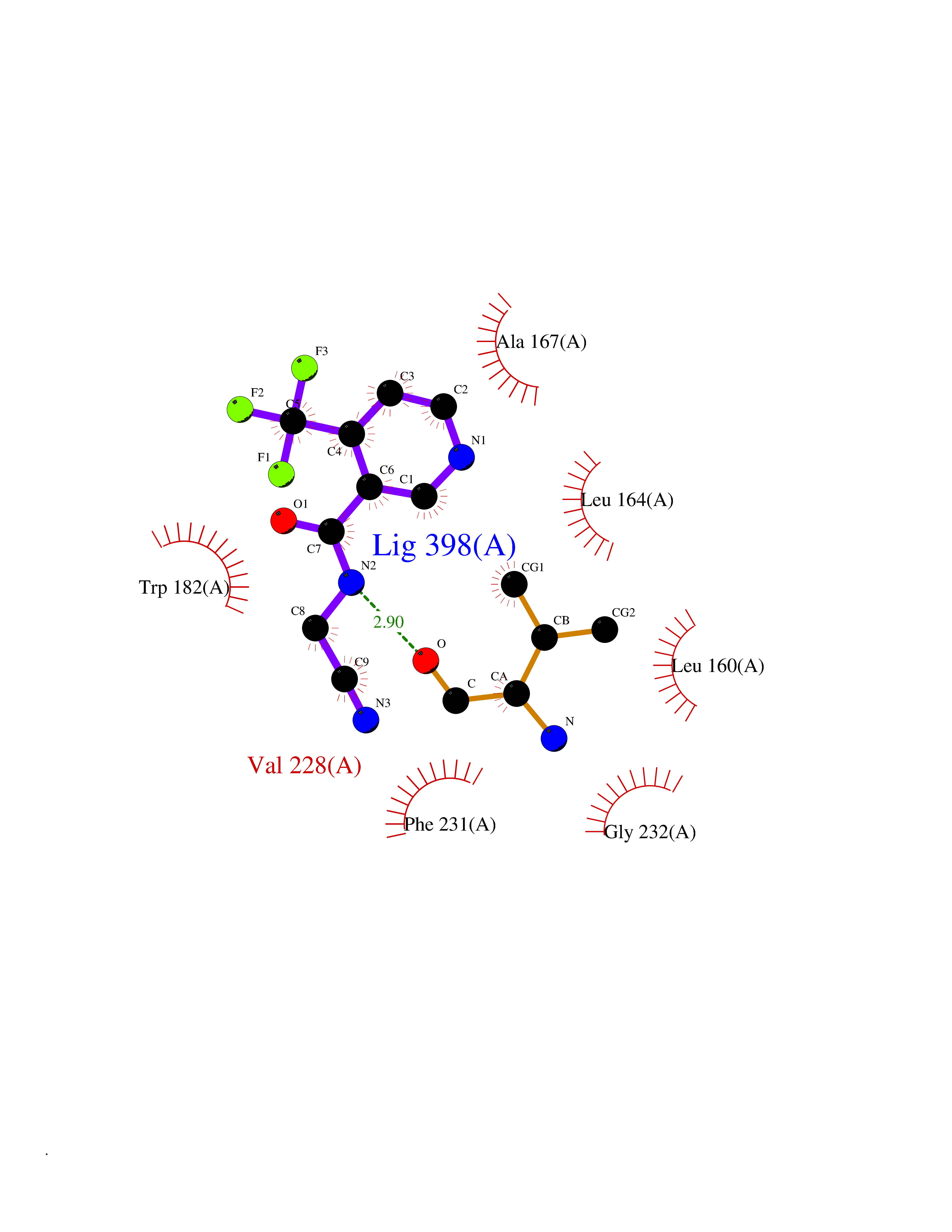 Ligplot Map 3D Binding mode Sequence ATVLLEVPFSARGDRIPDAVAELRTREPIRKVRTITGAEAWLVSSYALCTQVLEDRRFSMKETAAAGAPRLNALTVPPEVVNNMGNIADAGLRKAVMKAITPKAPGLEQFLRDTANSLLDNLITEGAPADLRNDFADPLATALHCKVLGIPQEDGPKLFRSLSIAFMSSADPIPAAKINWDRDIEYMAGILENPNITTGLMGELSRLRKDPAYSHVSDELFATIGVTFFGAGVISTGSFLTTALISLIQRPQLRNLLHEKPELIPAGVEELLRINLSFADGLPRLATADIQVGDVLVRKGELVLVLLEGANFDPEHFPNPGSIELDRPNPTSHLAFGRGQHFCPGSALGRRHAQIGIEALLKKMPGVDLAVPIDQLVWRTRFQRRIPERLPVLW Hydrogen bonds contact Hydrophobic contact | ||||
| 9 | Serine--pyruvate aminotransferase | 5HHY | 5.49 | |
Target general information Gen name AGXT Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms AGT1;SPAT Protein family Class-V pyridoxal-phosphate-dependent aminotransferase family Biochemical class Transferase Function Alanine-glyoxylate transaminase activity.Amino acid binding.Identical protein binding.Protein homodimerization activity.Protein self-association.Pyridoxal phosphate binding.Receptor binding.Serine-pyruvate transaminase activity.Transaminase activity. Related diseases Hyperoxaluria primary 1 (HP1) [MIM:259900]: An inborn error of glyoxylate metabolism characterized by increased excretion of oxalate and glycolate, and progressive tissue accumulation of insoluble calcium oxalate. Affected individuals are at risk for nephrolithiasis, nephrocalcinosis and early onset end-stage renal disease. {ECO:0000269|PubMed:10394939, ECO:0000269|PubMed:10453743, ECO:0000269|PubMed:10541294, ECO:0000269|PubMed:10862087, ECO:0000269|PubMed:10960483, ECO:0000269|PubMed:12559847, ECO:0000269|PubMed:12777626, ECO:0000269|PubMed:1301173, ECO:0000269|PubMed:1349575, ECO:0000269|PubMed:15253729, ECO:0000269|PubMed:15849466, ECO:0000269|PubMed:15961946, ECO:0000269|PubMed:15963748, ECO:0000269|PubMed:16971151, ECO:0000269|PubMed:1703535, ECO:0000269|PubMed:17495019, ECO:0000269|PubMed:2039493, ECO:0000269|PubMed:23229545, ECO:0000269|PubMed:24055001, ECO:0000269|PubMed:24934730, ECO:0000269|PubMed:26149463, ECO:0000269|PubMed:8101040, ECO:0000269|PubMed:9192270, ECO:0000269|PubMed:9604803}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08060; DB00160; DB02079; DB00145; DB04083; DB00114; DB00133 Interacts with Q5BKX5-3; P21549; P0C7T5; Q9NR55; Q9H5F2; Q9UKJ5; A8MQ03; O43281-2; A1KXE4-2; Q5TD97; P49356; P53539; P49639; Q15323; O76011; O76014; Q6A162; Q07627; P60328; Q9BYR8; Q3LI67; Q8IUC2; Q9BYQ4; O60711; Q99750; Q5VZ52; P0DPK4; O43482; P50542-1; O15496; Q6ZR37; Q9NQX0; Q8HWS3; Q5W111-2; Q8WVR3; Q6DKK2 EC number 2.6.1.44; 2.6.1.51 Uniprot keywords 3D-structure; Acetylation; Aminotransferase; Disease variant; Peroxisome; Phosphoprotein; Proteomics identification; Pyridoxal phosphate; Reference proteome; Transferase Protein physicochemical properties Chain ID A,B Molecular weight (Da) 84537 Length 771 Aromaticity 0.07 Instability index 36.81 Isoelectric point 8.42 Charge (pH=7) 6.81 2D Binding mode Binding energy (Kcal/mol) -7.49  Molscript Map  Pymol Map 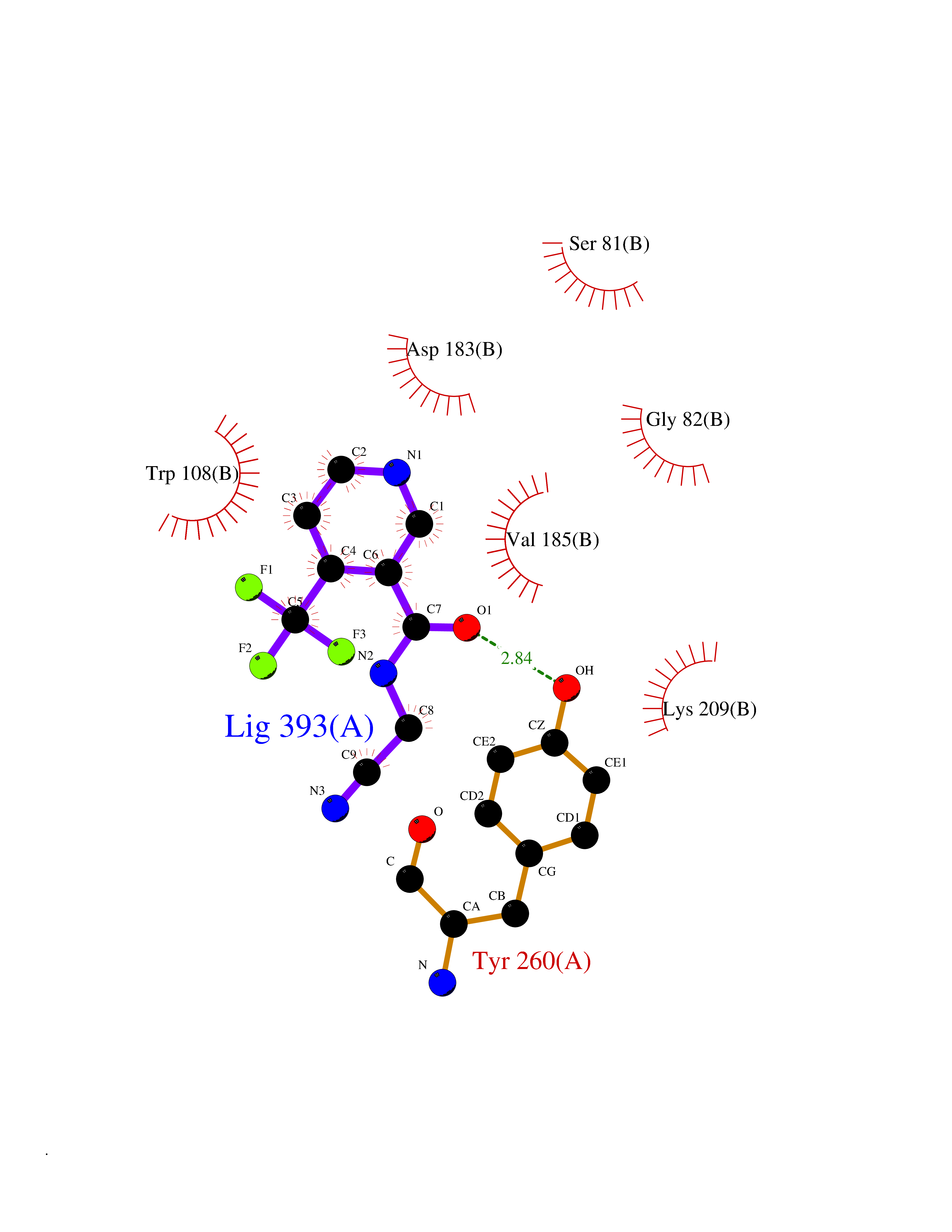 Ligplot Map 3D Binding mode Sequence LLVTPPKALLKPLSIPNQLLLGPGPSNLPPRIMAAGGLQMIGSMSKDMYQIMDEIKEGIQYVFQTRNPLTLVISGSGHCALEAALVNVLEPGDSFLVGANGIWGQRAVDIGERIGARVHPMTKDPGGHYTLQEVEEGLAQHKPVLLFLTHGESSTGVLQPLDGFGELCHRYKCLLLVDSVASLGGTPLYMDRQGIDILYSGSQKALNAPPGTSLISFSDKAKKKMYSRKTKPFSFYLDIKWLANFWGCDDQPRMYHHTIPVISLYSLRESLALIAEQGLENSWRQHREAAAYLHGRLQALGLQLFVKDPALRLPTVTTVAVPAGYDWRDIVSYVIDHFDIEIMGGLGPSTGKVLRIGLLGCNATRENVDRVTEALRAALQHCPKKLLVTPPKALLKPLSIPNQLLLGPGPSNLPPRIMAAGGLQMIGSMSKDMYQIMDEIKEGIQYVFQTRNPLTLVISGSGHCALEAALVNVLEPGDSFLVGANGIWGQRAVDIGERIGARVHPMTKDPGGHYTLQEVEEGLAQHKPVLLFLTHGESSTGVLQPLDGFGELCHRYKCLLLVDSVASLGGTPLYMDRQGIDILYSGSQKALNAPPGTSLISFSDKAKKKMYSRKTKPFSFYLDIKWLANFWGCDDQPRMYHHTIPVISLYSLRESLALIAEQGLENSWRQHREAAAYLHGRLQALGLQLFVKDPALRLPTVTTVAVPAGYDWRDIVSYVIDHFDIEIMGGLGPSTGKVLRIGLLGCNATRENVDRVTEALRAALQHCPKKK Hydrogen bonds contact Hydrophobic contact | ||||
| 10 | Dopamine beta-hydroxylase | 4ZEL | 5.48 | |
Target general information Gen name DBH Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Copper type II ascorbate-dependent monooxygenase family Biochemical class Oxidoreductase Function Catalytic activity.Copper ion binding.Dopamine beta-monooxygenase activity.L-ascorbic acid binding. Related diseases Orthostatic hypotension 1 (ORTHYP1) [MIM:223360]: A form of orthostatic hypotension due to congenital dopamine beta-hydroxylase deficiency. Orthostatic hypotension, also known as postural hypotension, is a finding defined as a 20-mm Hg decrease in systolic pressure or a 10-mm Hg decrease in diastolic pressure occurring 3 minutes after a person has risen from supine to standing. Symptoms include dizziness, blurred vision, and sometimes syncope. ORTHYP1 is an autosomal recessive condition apparent from infancy or early childhood and characterized by low plasma and urinary levels of norepinephrine and epinephrine, and episodic hypoglycemia. {ECO:0000269|PubMed:11857564}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00126; DB06774; DB09130; DB05394; DB00822; DB00988; DB00968; DB00550 Interacts with P00352; P63010-2; Q04656; Q8WUW1; Q9UNS2; Q71DI3; P61978; Q9Y2M5; Q92876; P08727; Q14693; P0DPK4; Q6GQQ9-2; P27986-2; Q9ULX5; Q96D59; Q8N6K7-2; Q9GZS3; Q8IUW3; Q86WT6-2 EC number 1.14.17.1 Uniprot keywords 3D-structure; Catecholamine biosynthesis; Copper; Cytoplasmic vesicle; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; Membrane; Metal-binding; Monooxygenase; Oxidoreductase; Proteomics identification; Reference proteome; Secreted; Signal-anchor; Transmembrane; Transmembrane helix; Vitamin C Protein physicochemical properties Chain ID A,B Molecular weight (Da) 123694 Length 1094 Aromaticity 0.1 Instability index 51.85 Isoelectric point 5.84 Charge (pH=7) -24.5 2D Binding mode Binding energy (Kcal/mol) -7.47 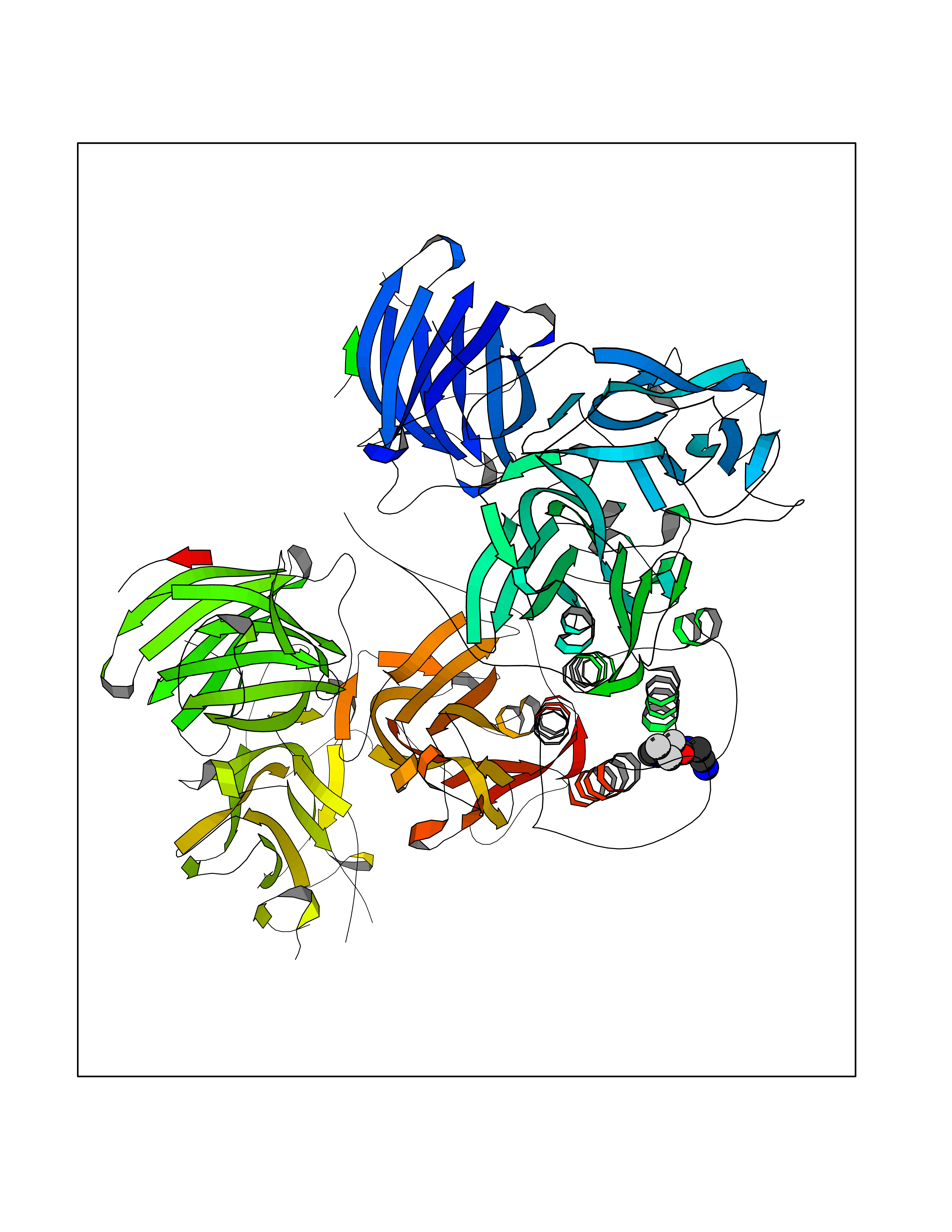 Molscript Map 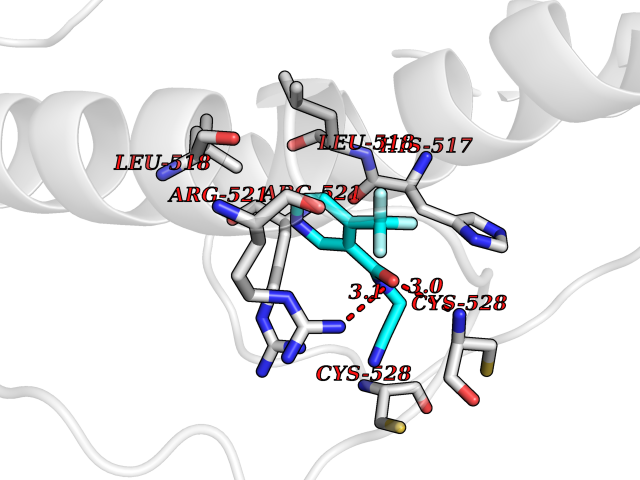 Pymol Map 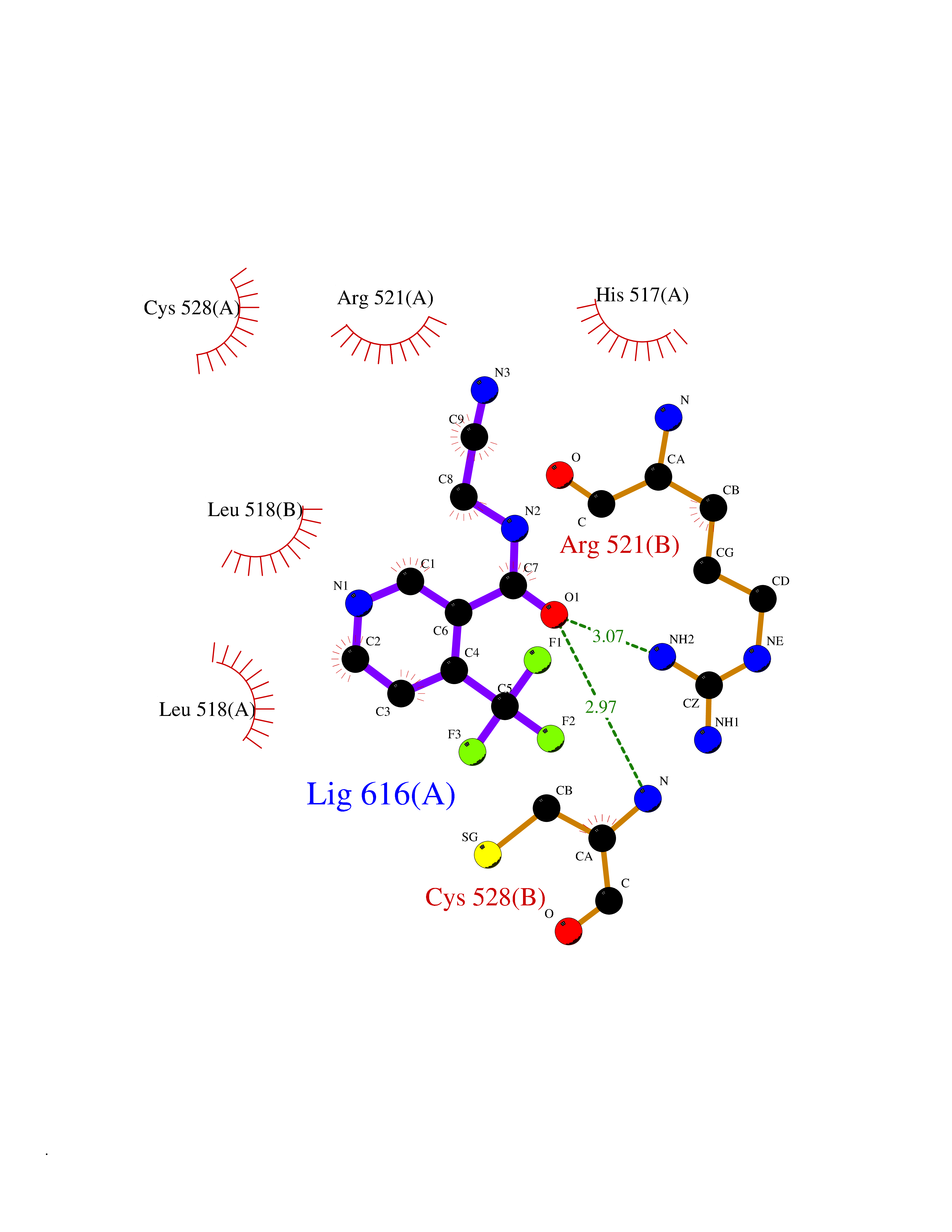 Ligplot Map 3D Binding mode Sequence PLPYHIPLDPEGSLELSWNVSYTQEAIHFQLLVRRLKAGVLFGMSDRGELENADLVVLWTDGDAYFADAWSDQKGQIHLDPQQDYQLLQVQRTPEGLTLLFKRPFGTCDPKDYLIEDGTVHLVYGILEEPFRSLEAINGSGLQMGLQRVQLLKPNIPEPELPSDACTMEVQAPNIQIPSQETTYWCYIKELPKGFSRHHIIKYEPIVTKGNEALVHHMEVFQCAPEMDSVPHFSGPCDSKMKPDRLNYCRHVLAAWALGAKAFYYPEEAGLAFGGPGSSRYLRLEVHYHNPLVIEGRNDSSGIRLYYTAKLRRFNAGIMELGLVYTPVMAIPPRETAFILTGYCTDKCTQLALPPSGIHIFASQLHTHLTGRKVVTVLVRDGREWEIVNQDNHYSPHFQEIRMLKKVVSVHPGDVLITSCTYNTEDRELATVGGFGILEEMCVNYVHYYPQTQLELCKSAVDAGFLQKYFHLINRFNNEDVCTCPQASVSQQFTSVPWNSFNRDVLKALYSFAPISMHCNKSSAVRFQGEWNLQPLPKVISTLEEPTVVSPLPYHIPLDPEGSLELSWNVSYTQEAIHFQLLVRRLKAGVLFGMSDRGELENADLVVLAYFADAWSDQKGQIHLDPQQDYQLLQVQRTPEGLTLLFKRPFGTCDPKDYLIEDGTVHLVYGILEEPFRSLEAINGSGLQMGLQRVQLLKPNIPEPELPSDACTMEVQAPNIQIPSQETTYWCYIKELPKGFSRHHIIKYEPIVTKGNEALVHHMEVFQCAPEVPHFSGPCDSKMLNYCRHVLAAWALGAKAFYYPEEAGLAFGGPGSSRYLRLEVHYHNPLVIEGRNDSSGIRLYYTAKLRRFNAGIMELGLVYTPVMAIPPRETAFILTGYCTDKCTQLALPPSGIHIFASQLHTHLTGRKVVTVLVRDGREWEIVNQDNHYSPHFQEIRMLKKVVSVHPGDVLITSCTYNTEDRELATVGGFGILEEMCVNYVHYYPQTQLELCKSAVDAGFLQKYFHLINRFNNEDVCTCPQASVSQQFTSVPWNSFNRDVLKALYSFAPISMHCNKSSAVRFQGEWNLQPLPKVISTLEEPTPQCVVSIGG Hydrogen bonds contact Hydrophobic contact | ||||
| 11 | 4-hydroxyphenylpyruvate dioxygenase | 3ISQ | 5.48 | |
Target general information Gen name HPD Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms PPD Protein family 4HPPD family Biochemical class Oxidoreductase Function 4-hydroxyphenylpyruvate dioxygenase activity.Metal ion binding. Related diseases Tyrosinemia 3 (TYRSN3) [MIM:276710]: An inborn error of metabolism characterized by elevations of tyrosine in the blood and urine, seizures and mild intellectual disability. {ECO:0000269|PubMed:10942115, ECO:0000269|PubMed:11073718}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Hawkinsinuria (HWKS) [MIM:140350]: An inborn error of tyrosine metabolism characterized by failure to thrive, persistent metabolic acidosis, fine and sparse hair, and excretion of the unusual cyclic amino acid metabolite, hawkinsin, in the urine. {ECO:0000269|PubMed:11073718}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02850; DB00348 Interacts with NA EC number 1.13.11.27 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Cytoplasm; Dioxygenase; Disease variant; Endoplasmic reticulum; Golgi apparatus; Intellectual disability; Iron; Membrane; Metal-binding; Oxidoreductase; Phenylalanine catabolism; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Tyrosine catabolism Protein physicochemical properties Chain ID A Molecular weight (Da) 43164.8 Length 376 Aromaticity 0.11 Instability index 32.38 Isoelectric point 6.73 Charge (pH=7) -1.04 2D Binding mode Binding energy (Kcal/mol) -7.48  Molscript Map 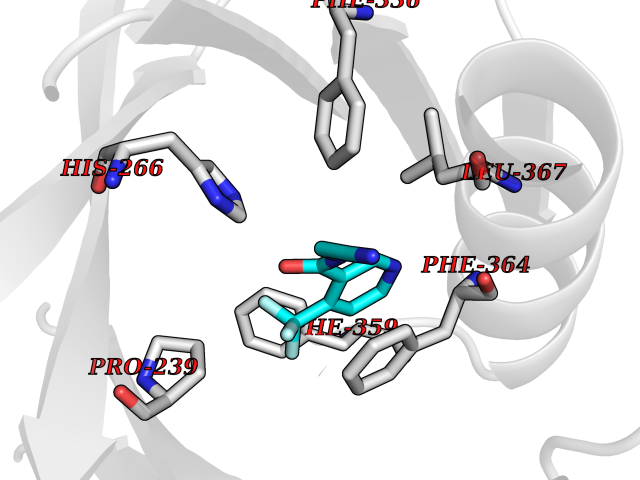 Pymol Map 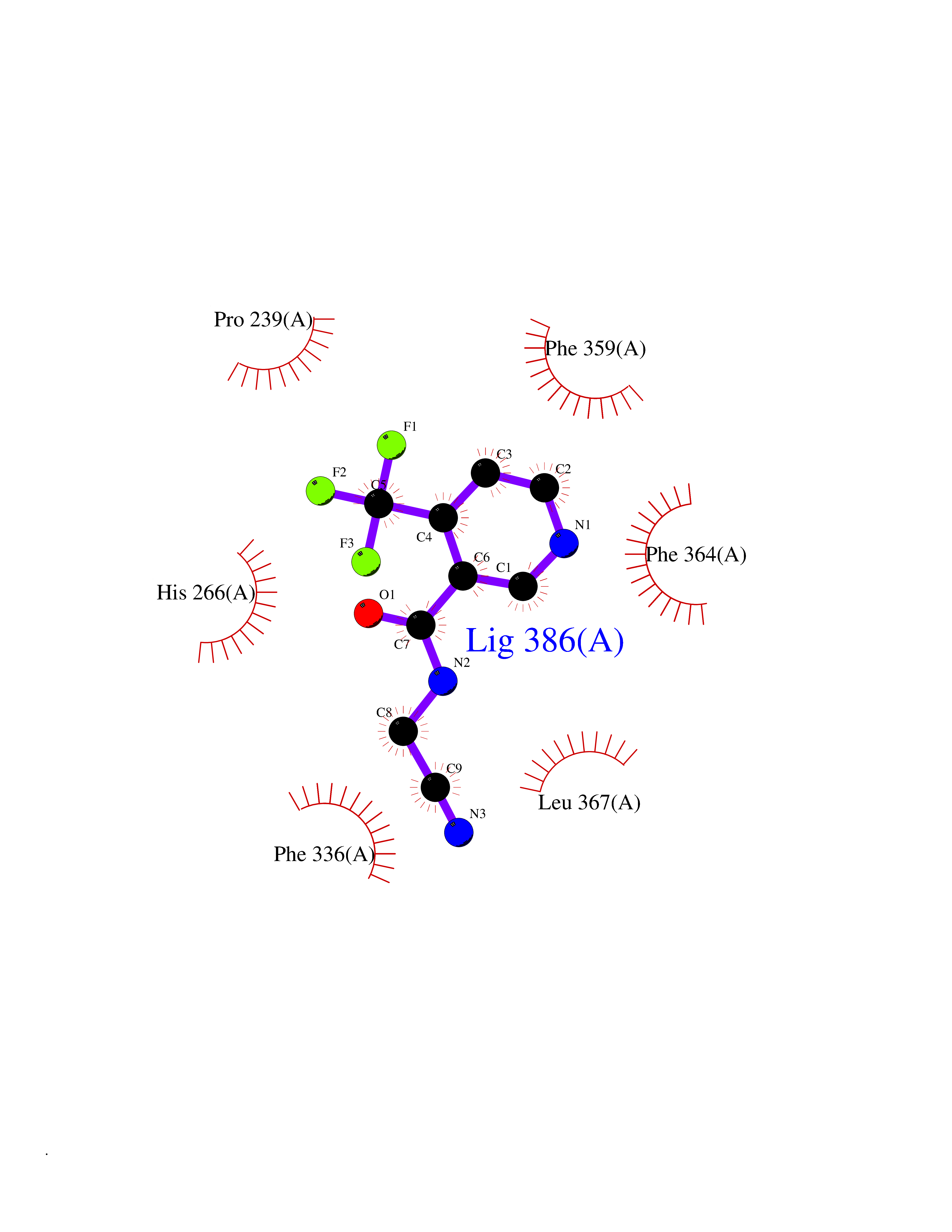 Ligplot Map 3D Binding mode Sequence AKPERGRFLHFHSVTFWVGNAKQAASFYCSKMGFEPLAYRGLETGSREVVSHVIKQGKIVFVLSSALNPWNKEMGDHLVKHGDGVKDIAFEVEDCDYIVQKARERGAKIMREPWVEQDKFGKVKFAVLQTYGDTTHTLVEKMNYIGQFLPGYEAPAFMDPLLPKLPKCSLEMIDHIVGNQPDQEMVSASEWYLKNLQFHRFWSVDDTQVHTEYSSLRSIVVANYEESIKMPINEPAPGKKKSQIQEYVDYNGGAGVQHIALKTEDIITAIRHLRERGLEFLSVPSTYYKQLREKLKTAKIKVKENIDALEELKILVDYDEKGYLLQIFTKPVQDRPTLFLEVIQRHNHQGFGAGNFNSLFKAFEEEQNLRGNLTNM Hydrogen bonds contact Hydrophobic contact | ||||
| 12 | Leukotriene A-4 hydrolase (LTA4H) | 3U9W | 5.47 | |
Target general information Gen name LTA4H Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Leukotriene A4 hydrolase; Leukotriene A(4)Leukotriene A-4 hydrolase hydrolase; Leukotriene A(4) hydrolase; LTA4; LTA-H; LTA-4hydrolase; LTA-4 hydrolase Protein family Peptidase M1 family Biochemical class Ether bond hydrolase Function Has also aminopeptidase activity. Epoxide hydrolase that catalyzes the final step in the biosynthesis of the proinflammatory mediator leukotriene B4. Related diseases Pigmentary disorder, reticulate, with systemic manifestations, X-linked (PDR) [MIM:301220]: An X-linked recessive disorder characterized by recurrent infections and sterile inflammation in various organs. Diffuse skin hyperpigmentation with a distinctive reticulate pattern is universally evident by early childhood. This is later followed in many patients by hypohidrosis, corneal inflammation and scarring, enterocolitis that resembles inflammatory bowel disease, and recurrent urethral strictures. Melanin and amyloid deposition is present in the dermis. Affected males also have a characteristic facies with frontally upswept hair and flared eyebrows. Female carriers have only restricted pigmentary changes along Blaschko's lines. {ECO:0000269|PubMed:27019227}. The disease is caused by variants affecting the gene represented in this entry. XLPDR is caused by a recurrent intronic mutation that results in missplicing and reduced POLA1 expression. This leads to a decrease in cytosolic RNA:DNA hybrids and constitutive activation of type I interferon responses, but has no effect on cell replication. {ECO:0000269|PubMed:27019227}.; DISEASE: Van Esch-O'Driscoll syndrome (VEODS) [MIM:301030]: An X-linked recessive syndrome characterized by different degrees of intellectual disability, moderate to severe short stature, microcephaly, hypogonadism, and variable congenital malformations. {ECO:0000269|PubMed:31006512}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07102; DB06917; DB07258; DB07094; DB07259; DB02352; DB07292; DB07104; DB06828; DB08466; DB01197; DB05177; DB03366; DB08040; DB06851; DB02062; DB07099; DB07260; DB07196; DB11781; DB03424; DB07237 Interacts with Q9BSI4 EC number EC 3.3.2.6 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Cytoplasm; Direct protein sequencing; Hydrolase; Leukotriene biosynthesis; Lipid metabolism; Metal-binding; Metalloprotease; Phosphoprotein; Protease; Proteomics identification; Reference proteome; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 68927 Length 608 Aromaticity 0.1 Instability index 38.84 Isoelectric point 5.87 Charge (pH=7) -9.86 2D Binding mode Binding energy (Kcal/mol) -7.46  Molscript Map 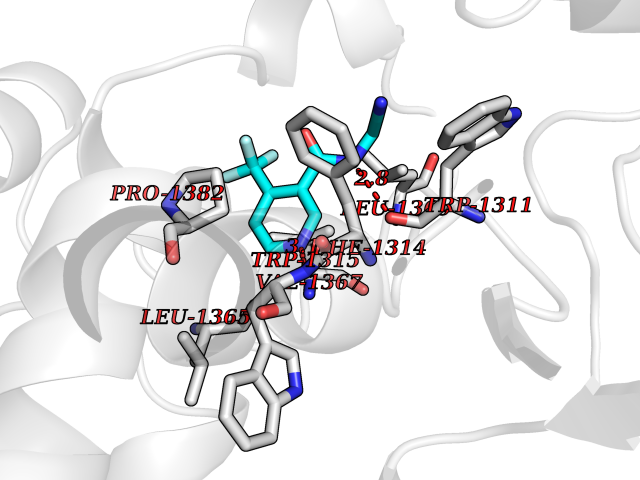 Pymol Map 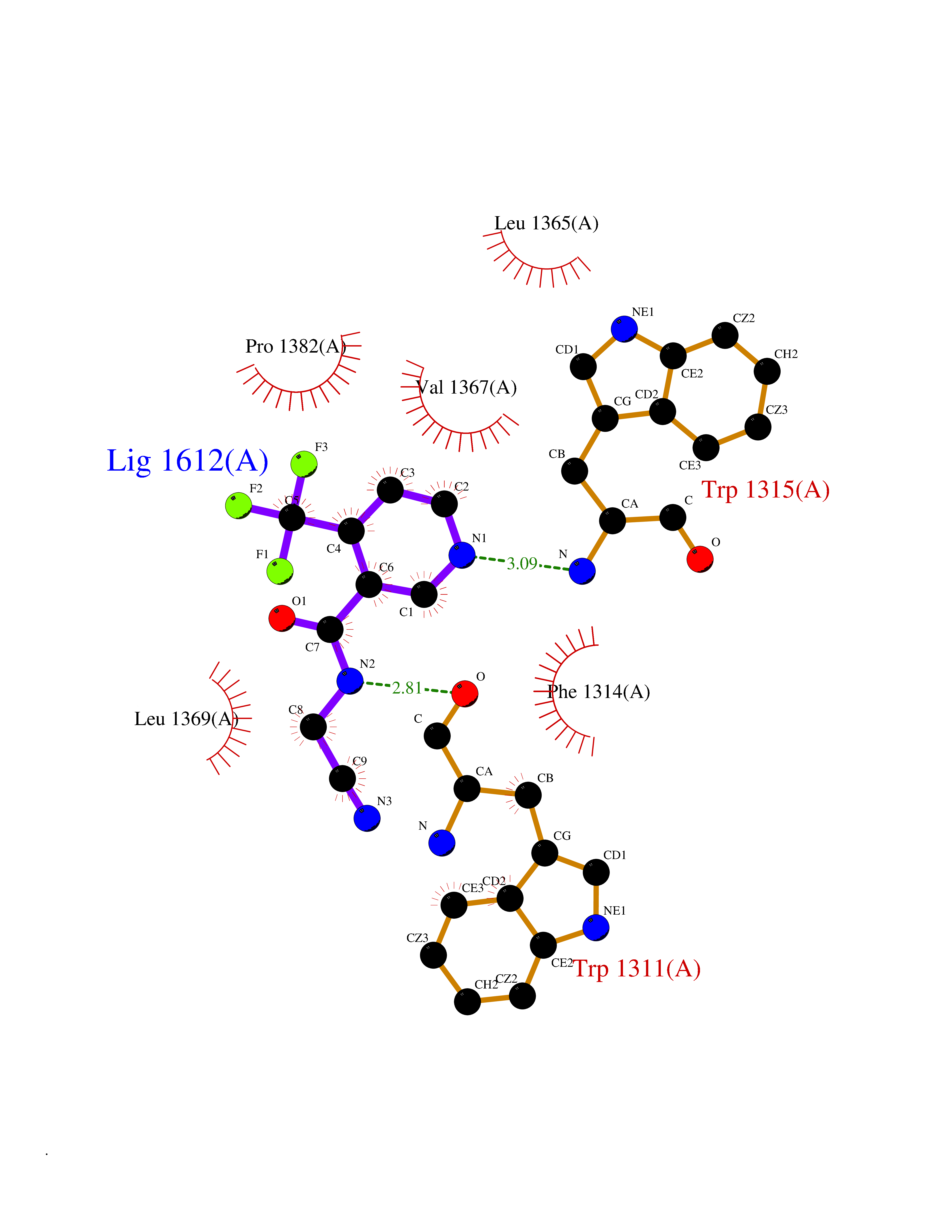 Ligplot Map 3D Binding mode Sequence IVDTCSLASPASVCRTKHLHLRCSVDFTRRTLTGTAALTVQSQEDNLRSLVLDTKDLTIEKVVINGQEVKYALGERQSYKGSPMEISLPIALSKNQEIVIEISFETSPKSSALQWLTPEQTSGKEHPYLFSQCQAIHCRAILPCQDTPSVKLTYTAEVSVPKELVALMSAIRDGETPDPEDPSRKIYKFIQKVPIPCYLIALVVGALESRQIGPRTLVWSEKEQVEKSAYEFSETESMLKIAEDLGGPYVWGQYDLLVLPPSFPYGGMENPCLTFVTPTLLAGDKSLSNVIAHEISHSWTGNLVTNKTWDHFWLNEGHTVYLERHICGRLFGEKFRHFNALGGWGELQNSVKTFGETHPFTKLVVDLTDIDPDVAYSSVPYEKGFALLFYLEQLLGGPEIFLGFLKAYVEKFSYKSITTDDWKDFLYSYFKDKVDVLNQVDWNAWLYSPGLPPIKPNYDMTLTNACIALSQRWITAKEDDLNSFNATDLKDLSSHQLNEFLAQTLQRAPLPLGHIKRMQEVYNFNAINNSEIRFRWLRLCIQSKWEDAIPLALKMATEQGRMKFTRPLFKDLAAFDKSHDQAVRTYQEHKASMHPVTAMLVGKDLKVD Hydrogen bonds contact Hydrophobic contact | ||||
| 13 | Metabotropic glutamate receptor 3 (mGluR3) | 4XAR | 5.47 | |
Target general information Gen name GRM3 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms mGLUR3; Group III metabotropic glutamate receptor; GPRC1C Protein family G-protein coupled receptor 3 family Biochemical class GPCR glutamate Function Ligand binding causes a conformation change that triggers signaling via guanine nucleotide-binding proteins (G proteins) and modulates the activity of down-stream effectors. Signaling inhibits adenylate cyclase activity. G-protein coupled receptor for glutamate. Related diseases Paramyotonia congenita (PMC) [MIM:168300]: An autosomal dominant channelopathy characterized by myotonia, increased by exposure to cold, intermittent flaccid paresis, not necessarily dependent on cold or myotonia, lability of serum potassium, non-progressive nature and lack of atrophy or hypertrophy of muscles. In some patients, myotonia is not increased by cold exposure (paramyotonia without cold paralysis). Patients may have a combination phenotype of PMC and HYPP. {ECO:0000269|PubMed:10369308, ECO:0000269|PubMed:10727489, ECO:0000269|PubMed:1310898, ECO:0000269|PubMed:1316765, ECO:0000269|PubMed:1338909, ECO:0000269|PubMed:15318338, ECO:0000269|PubMed:15790667, ECO:0000269|PubMed:16786525, ECO:0000269|PubMed:18166706, ECO:0000269|PubMed:18690054, ECO:0000269|PubMed:19077043, ECO:0000269|PubMed:20076800, ECO:0000269|PubMed:8242056, ECO:0000269|PubMed:8308722, ECO:0000269|PubMed:8388676, ECO:0000269|PubMed:8580427}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Periodic paralysis hypokalemic 2 (HOKPP2) [MIM:613345]: An autosomal dominant disorder manifested by episodic flaccid generalized muscle weakness associated with falls of serum potassium levels. {ECO:0000269|PubMed:10599760, ECO:0000269|PubMed:10851391, ECO:0000269|PubMed:10944223, ECO:0000269|PubMed:11558801, ECO:0000269|PubMed:11591859, ECO:0000269|PubMed:16890191, ECO:0000269|PubMed:17898326, ECO:0000269|PubMed:18162704, ECO:0000269|PubMed:19118277, ECO:0000269|PubMed:20522878, ECO:0000269|PubMed:21043388, ECO:0000269|PubMed:24549961}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Periodic paralysis hyperkalemic (HYPP) [MIM:170500]: An autosomal dominant channelopathy characterized by episodic flaccid generalized muscle weakness associated with high levels of serum potassium. Concurrence of myotonia is found in HYPP patients. {ECO:0000269|PubMed:1659668, ECO:0000269|PubMed:1659948, ECO:0000269|PubMed:20076800}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Periodic paralysis normokalemic (NKPP) [MIM:170500]: A disorder closely related to hyperkalemic periodic paralysis, but marked by a lack of alterations in potassium levels during attacks of muscle weakness. {ECO:0000269|PubMed:15596759, ECO:0000269|PubMed:18046642, ECO:0000269|PubMed:20522878}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Myotonia SCN4A-related (MYOSCN4A) [MIM:608390]: A phenotypically highly variable myotonia aggravated by potassium loading, and sometimes by cold. Myotonia is characterized by sustained muscle tensing that prevents muscles from relaxing normally. It causes muscle stiffness that can interfere with movement. In some people the stiffness is very mild, while in other cases it may be severe enough to interfere with walking, running, and other activities of daily life. Myotonia SCN4A-related includes myotonia permanens and myotonia fluctuans. In myotonia permanens, the myotonia is generalized and there is a hypertrophy of the muscle, particularly in the neck and the shoulder. Attacks of severe muscle stiffness of the thoracic muscles may be life threatening due to impaired ventilation. In myotonia fluctuans, the muscle stiffness may fluctuate from day to day, provoked by exercise. {ECO:0000269|PubMed:10218481, ECO:0000269|PubMed:16786525, ECO:0000269|PubMed:16832098, ECO:0000269|PubMed:17212350, ECO:0000269|PubMed:17998485, ECO:0000269|PubMed:18203179, ECO:0000269|PubMed:18337100, ECO:0000269|PubMed:19015483, ECO:0000269|PubMed:19347921, ECO:0000269|PubMed:20076800, ECO:0000269|PubMed:27653901, ECO:0000269|PubMed:8058156, ECO:0000269|PubMed:9392583}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Myasthenic syndrome, congenital, 16 (CMS16) [MIM:614198]: A form of congenital myasthenic syndrome, a group of disorders characterized by failure of neuromuscular transmission, including pre-synaptic, synaptic, and post-synaptic disorders that are not of autoimmune origin. Clinical features are easy fatigability and muscle weakness. CMS16 is characterized by fatigable generalized weakness and recurrent attacks of respiratory and bulbar paralysis since birth. The fatigable weakness involves lid-elevator, external ocular, facial, limb and truncal muscles and an decremental response of the compound muscle action potential on repetitive stimulation. {ECO:0000269|PubMed:12766226, ECO:0000269|PubMed:25707578, ECO:0000269|PubMed:26659129}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Congenital myopathy 22A, classic (CMYO22A) [MIM:620351]: A form of congenital myopathy, a clinically and genetically heterogeneous group of muscle disorders characterized by hypotonia and muscle weakness apparent at birth, and specific pathological features on muscle biopsy. CMYO22A is an autosomal recessive form characterized by fetal hypokinesia, polyhydramnios, and severe neonatal hypotonia associated with respiratory insufficiency. Affected individuals who survive the neonatal period have delayed motor development, difficulty walking, proximal muscle weakness of the upper and lower limbs, facial and neck muscle weakness, easy fatigability, and mild limb contractures or foot deformities. {ECO:0000269|PubMed:26700687, ECO:0000269|PubMed:28262468, ECO:0000269|PubMed:36090556}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Congenital myopathy 22B, severe fetal (CMYO22B) [MIM:620369]: A severe congenital myopathy, a clinically and genetically heterogeneous group of muscle disorders characterized by hypotonia and muscle weakness apparent at birth, and specific pathological features on muscle biopsy. CMYO22B is an autosomal recessive form characterized by onset in utero. Affected individuals show fetal akinesia, and develop fetal hydrops with pulmonary hypoplasia, severe joint contractures, and generalized muscle hypoplasia. Death occurs in utero or soon after birth. {ECO:0000269|PubMed:26700687}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB05096 Interacts with NA EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disulfide bond; G-protein coupled receptor; Glycoprotein; Membrane; Proteomics identification; Receptor; Reference proteome; Signal; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 50355.5 Length 445 Aromaticity 0.11 Instability index 38.26 Isoelectric point 6.52 Charge (pH=7) -1.53 2D Binding mode Binding energy (Kcal/mol) -7.46  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence RREIKIEGDLVLGGLFPINEKGTGTEECGRINEDRGIQRLEAMLFAIDEINKDDYLLPGVKLGVHILDTCSRDTYALEQSLEFVRASLLLIAGVIGGSYSSVSIQVANLLRLFQIPQISYASTSAKLSDKSRYDYFARTVPPDFYQAKAMAEILRFFNWTYVSTVASEGDYGETGIEAFEQEARLRNISIATAEKVGRSNIRKSYDSVIRELLQKPNARVVVLFMRSDDSRELIAAASRANASFTWVASDGWGAQESIIKGSEHVAYGAITLELASQPVRQFDRYFQSLNPYNNHRNPWFRDFWEQKFQCSLRVCDKHLAIDSSNYEQESKIMFVVNAVYAMAHALHKMQRTLCPNTTKLCDAMKILDGKKLYKDYLLKINFTAPDADSIVKFDTFGDGMGRYNVFNFQNVGGKYSYLKVGHWAETLSLDVNSIHWSRNSVPTSE Hydrogen bonds contact Hydrophobic contact | ||||
| 14 | Neprilysin | 1R1H | 5.45 | |
Target general information Gen name MME Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms EPN Protein family Peptidase M13 family Biochemical class Hydrolase Function Endopeptidase activity.Exopeptidase activity.Metalloendopeptidase activity.Metallopeptidase activity.Peptide binding.Zinc ion binding. Related diseases Charcot-Marie-Tooth disease, axonal, 2T (CMT2T) [MIM:617017]: An axonal form of Charcot-Marie-Tooth disease, a disorder of the peripheral nervous system, characterized by progressive weakness and atrophy, initially of the peroneal muscles and later of the distal muscles of the arms. Charcot-Marie-Tooth disease is classified in two main groups on the basis of electrophysiologic properties and histopathology: primary peripheral demyelinating neuropathies (designated CMT1 when they are dominantly inherited) and primary peripheral axonal neuropathies (CMT2). Neuropathies of the CMT2 group are characterized by signs of axonal degeneration in the absence of obvious myelin alterations, normal or slightly reduced nerve conduction velocities, and progressive distal muscle weakness and atrophy. {ECO:0000269|PubMed:26991897, ECO:0000269|PubMed:27588448}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Spinocerebellar ataxia 43 (SCA43) [MIM:617018]: A form of spinocerebellar ataxia, a clinically and genetically heterogeneous group of cerebellar disorders. Patients show progressive incoordination of gait and often poor coordination of hands, speech and eye movements, due to degeneration of the cerebellum with variable involvement of the brainstem and spinal cord. SCA43 is a slowly progressive, autosomal dominant form. {ECO:0000269|PubMed:27583304}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08575; DB02597; DB00616; DB11623; DB05796; DB06655; DB02558; DB02062; DB00886; DB02557; DB09292; DB13928; DB08626 Interacts with P05067; P21926; Q06787-7; P08107; P04792 EC number 3.4.24.11 Uniprot keywords 3D-structure; Cell membrane; Charcot-Marie-Tooth disease; Disease variant; Disulfide bond; Glycoprotein; Hydrolase; Lipoprotein; Membrane; Metal-binding; Metalloprotease; Myristate; Neurodegeneration; Neuropathy; Phosphoprotein; Protease; Proteomics identification; Reference proteome; Signal-anchor; Spinocerebellar ataxia; Transmembrane; Transmembrane helix; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 79435.8 Length 696 Aromaticity 0.11 Instability index 37.5 Isoelectric point 5.53 Charge (pH=7) -11.46 2D Binding mode Binding energy (Kcal/mol) -7.43  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence GICKSSDCIKSAARLIQNMDATTEPCTDFFKYACGGWLKRNVIPETSSRYGNFDILRDELEVVLKDVLQEPKTEDIVAVQKAKALYRSCINESAIDSRGGEPLLKLLPDIYGWPVATENWEQKYGASWTAEKAIAQLNSKYGKKVLINLFVGTDDKNSVNHVIHIDQPRLGLPSRDYYECTGIYKEACTAYVDFMISVARLIRQEERLPIDENQLALEMNKVMELEKEIANATAKPEDRNDPMLLYNKMTLAQIQNNFSLEINGKPFSWLNFTNEIMSTVNISITNEEDVVVYAPEYLTKLKPILTKYSARDLQNLMSWRFIMDLVSSLSRTYKESRNAFRKALYGTTSETATWRRCANYVNGNMENAVGRLYVEAAFAGESKHVVEDLIAQIREVFIQTLDDLTWMDAETKKRAEEKALAIKERIGYPDDIVSNDNKLNNEYLELNYKEDEYFENIIQNLKFSQSKQLKKLREKVDKDEWISGAAVVNAFYSSGRNQIVFPAGILQPPFFSAQQSNSLNYGGIGMVIGHEITHGFDDNGRNFNKDGDLVDWWTQQSASNFKEQSQCMVYQYGNFSWDLAGGQHLNGINTLGENIADNGGLGQAYRAYQNYIKKNGEEKLLPGLDLNHKQLFFLNFAQVWCGTYRPEYAVNSIKTDVHSPGNFRIIGTLQNSAEFSEAFHCRKNSYMNPEKKCRVW Hydrogen bonds contact Hydrophobic contact | ||||
| 15 | Cystine/glutamate transporter (SLC7A11) | 7EPZ | 5.45 | |
Target general information Gen name SLC7A11 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms XCT; X(c)-cystine transporter; X(c)- plasma membrane cystine transporter; Solute carrier family 7 member 11; Calcium channel blocker resistance protein CCBR1; Amino acid transport system xc-; Amino ac Protein family Amino acid-polyamine-organocation (APC) superfamily, L-type amino acid transporter (LAT) (TC 2.A.3.8) family Biochemical class Amino acid-polyamine-organocation Function Sodium-independent, high-affinity exchange of anionic amino acids with high specificity for anionic form of cystine and glutamate. Related diseases Lipodystrophy, familial partial, 8 (FPLD8) [MIM:620679]: An autosomal dominant form of partial lipodystrophy, a disorder characterized by abnormal subcutaneous fat distribution. FPLD8 patients show selective loss of subcutaneous adipose tissue from the limbs, beginning around 13 to 15 years of age, and abnormal accumulation of subcutaneous adipose tissue in the dorsal neck and face, as well as in the posterior thoracic and abdominal regions. The disorder is associated with metabolic abnormalities, including diabetes mellitus and hyperlipidemia. {ECO:0000269|PubMed:27376152}. The disease may be caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB06151; DB05540; DB00138; DB00142; DB00740; DB01098; DB00795; DB08833; DB04348; DB08834; DB11590 Interacts with P16070; Q07627; Q8IUG1; P08195; P08195-4 EC number NA Uniprot keywords 3D-structure; Amino-acid transport; Cell membrane; Cell projection; Disulfide bond; Glycoprotein; Membrane; Phosphoprotein; Proteomics identification; Reference proteome; Transmembrane; Transmembrane helix; Transport; Ubl conjugation Protein physicochemical properties Chain ID B Molecular weight (Da) 50166.3 Length 453 Aromaticity 0.13 Instability index 35.95 Isoelectric point 8.86 Charge (pH=7) 5.88 2D Binding mode Binding energy (Kcal/mol) -7.44  Molscript Map  Pymol Map 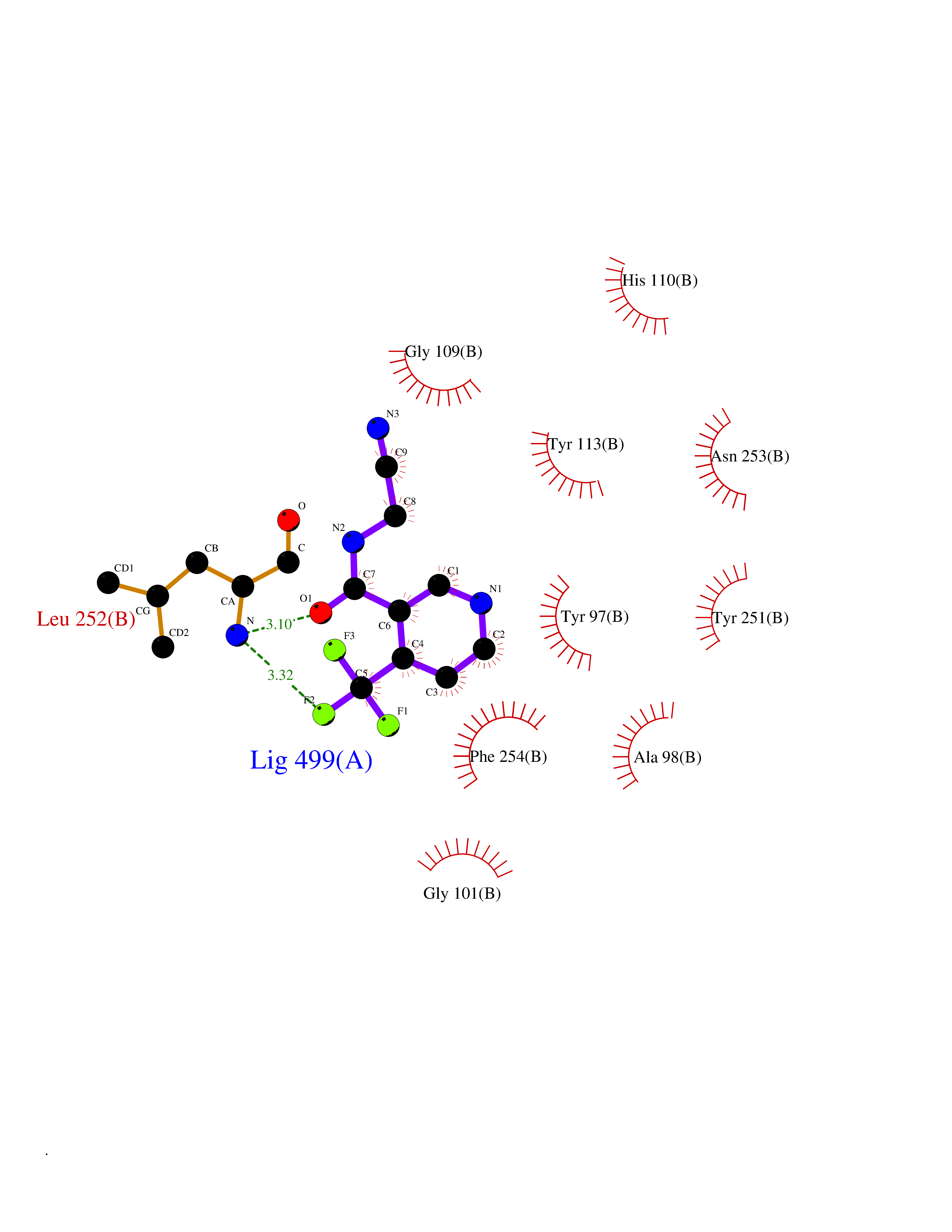 Ligplot Map 3D Binding mode Sequence TLLRGVSIIIGTIIGAGIFISPKGVLQNTGSVGMSLTIWTVCGVLSLFGALSYAELGTTIKKSGGHYTYILEVFGPLPAFVRVWVELLIIRPAATAVISLAFGRYILEPFFIQCEIPELAIKLITAVGITVVMVLNSMSVSWSARIQIFLTFCKLTAILIIIVPGVMQLIKGQTQNFKDAFSGRDSSITRLPLAFYYGMYAYAGWFYLNFVTEEVENPEKTIPLAICISMAIVTIGYVLTNVAYFTTINAEELLLSNAVAVTFSERLLGNFSLAVPIFVALSCFGSMNGGVFAVSRLFYVASREGHLPEILSMIHVRKHTPLPAVIVLHPLTMIMLFSGDLDSLLNFLSFARWLFIGLAVAGLIYLRYKCPDMHRPFKVPLFIPALFSFTCLFMVALSLYSDPFSTGIGFVITLTGVPAYYLFIIWDKKPRWFRIMSEKITRTLQIILEVVPE Hydrogen bonds contact Hydrophobic contact | ||||
| 16 | Bacterial Oxoacyl-[acyl-carrier-protein] synthase II (Bact fabF) | 2GFX | 5.45 | |
Target general information Gen name Bact fabF Organism Escherichia coli (strain K12) Uniprot ID TTD ID Synonyms KASB; KAS II; KAS 2; FabF; Condensing enzyme FabF; Beta-ketoacyl-acyl carrier protein synthase B; Beta-ketoacyl-ACP synthase II; Beta-ketoacyl-ACP synthase 2; 3-oxoacyl-[acyl-carrier-protein] synthase Protein family Thiolase-like superfamily, Beta-ketoacyl-ACP synthases family Biochemical class Acyltransferase Function Catalyzes the condensation reaction of fatty acid synthesis by the addition to an acyl acceptor of two carbons from malonyl-ACP. Has a preference for short chain acid substrates and may function to supply the octanoic substrates for lipoic acid biosynthesis. Related diseases Pigmentary disorder, reticulate, with systemic manifestations, X-linked (PDR) [MIM:301220]: An X-linked recessive disorder characterized by recurrent infections and sterile inflammation in various organs. Diffuse skin hyperpigmentation with a distinctive reticulate pattern is universally evident by early childhood. This is later followed in many patients by hypohidrosis, corneal inflammation and scarring, enterocolitis that resembles inflammatory bowel disease, and recurrent urethral strictures. Melanin and amyloid deposition is present in the dermis. Affected males also have a characteristic facies with frontally upswept hair and flared eyebrows. Female carriers have only restricted pigmentary changes along Blaschko's lines. {ECO:0000269|PubMed:27019227}. The disease is caused by variants affecting the gene represented in this entry. XLPDR is caused by a recurrent intronic mutation that results in missplicing and reduced POLA1 expression. This leads to a decrease in cytosolic RNA:DNA hybrids and constitutive activation of type I interferon responses, but has no effect on cell replication. {ECO:0000269|PubMed:27019227}.; DISEASE: Van Esch-O'Driscoll syndrome (VEODS) [MIM:301030]: An X-linked recessive syndrome characterized by different degrees of intellectual disability, moderate to severe short stature, microcephaly, hypogonadism, and variable congenital malformations. {ECO:0000269|PubMed:31006512}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08366; DB01034; DB03017; DB08407 Interacts with P0A6Y8 EC number EC 2.3.1.179 Uniprot keywords 3D-structure; Acyltransferase; Direct protein sequencing; Fatty acid biosynthesis; Fatty acid metabolism; Lipid biosynthesis; Lipid metabolism; Reference proteome; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 42852 Length 411 Aromaticity 0.06 Instability index 31.41 Isoelectric point 5.72 Charge (pH=7) -7.34 2D Binding mode Binding energy (Kcal/mol) -7.43  Molscript Map 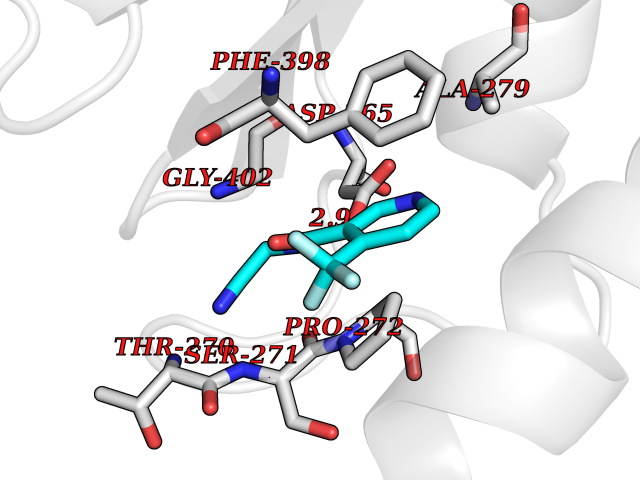 Pymol Map 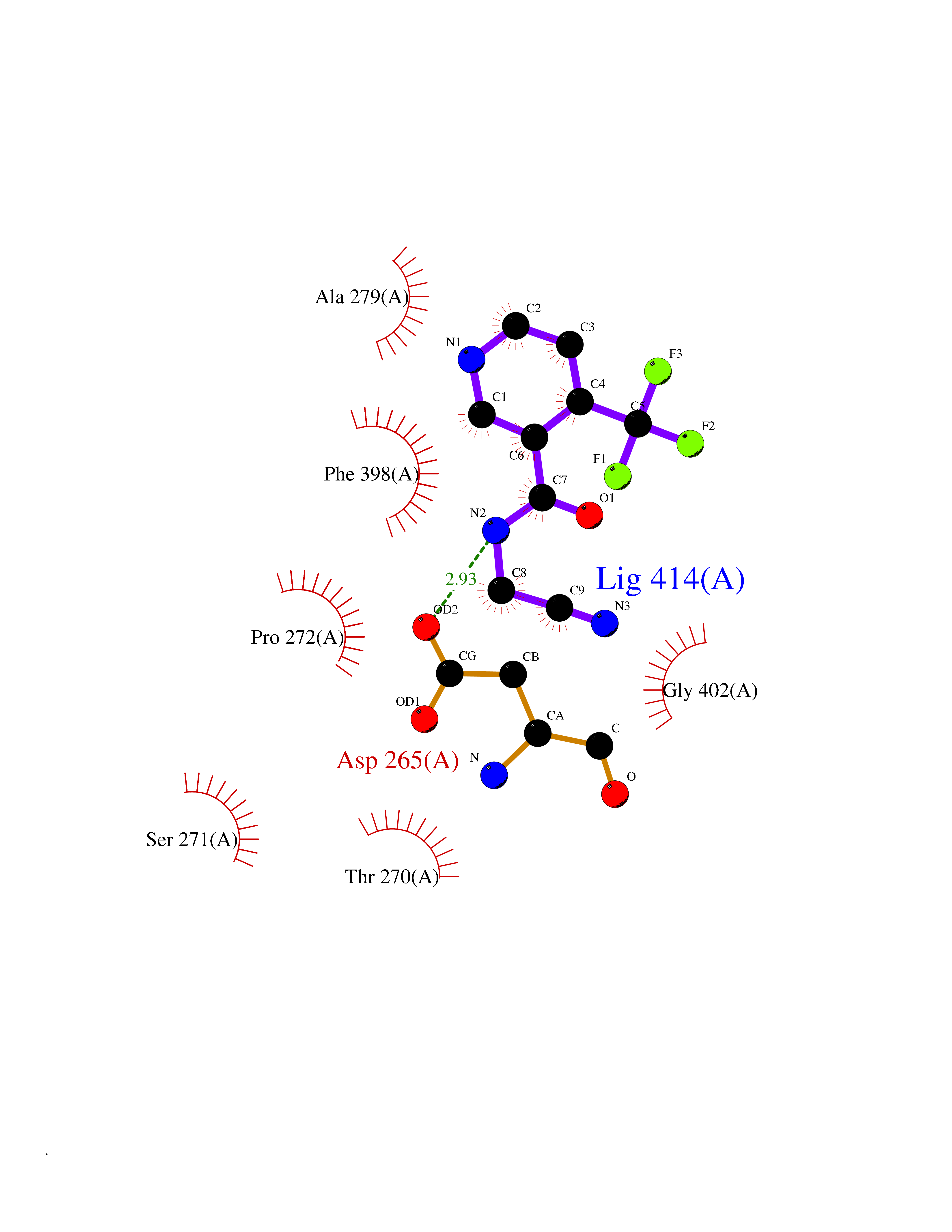 Ligplot Map 3D Binding mode Sequence KRRVVVTGLGMLSPVGNTVESTWKALLAGQSGISLIDHFDTSAYATKFAGLVKDFNCEDIISRKEQRKMDAFIQYGIVAGVQAMQDSGLEITEENATRIGAAIGSGIGGLGLIEENHTSLMNGGPRKISPFFVPSTIVNMVAGHLTIMYGLRGPSISIATAQTSGVHNIGHAARIIAYGDADVMVAGGAEKASTPLGVGGFGAARALSTRNDNPQAASRPWDKERDGFVLGDGAGMLVLEEYEHAKKRGAKIYAELVGFGMSSDAYHMTSPPENGAGAALAMANALRDAGIEASQIGYVNAHGTSTPAGDKAEAQAVKTIFGEAASRVLVSSTKSMTGHLLGAAGAVESIYSILALRDQAVPPTINLDNPDEGCDLDFVPHEARQVSGMEYTLCNSFGFGGTNGSLIFKKI Hydrogen bonds contact Hydrophobic contact | ||||
| 17 | Mutated Histone H3.3 (H3F3A) | 4GUS | 5.45 | |
Target general information Gen name H3F3A Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms PP781; Histone H3.3; H3F3; H3.3B; H3.3A Protein family Histone H3 family Biochemical class NA Function Variant histone H3 which replaces conventional H3 in a wide range of nucleosomes in active genes. Constitutes the predominant form of histone H3 in non-dividing cells and is incorporated into chromatin independently of DNA synthesis. Deposited at sites of nucleosomal displacement throughout transcribed genes, suggesting that it represents an epigenetic imprint of transcriptionally active chromatin. Nucleosomes wrap and compact DNA into chromatin, limiting DNA accessibility to the cellular machineries which require DNA as a template. Histones thereby play a central role in transcription regulation, DNA repair, DNA replication and chromosomal stability. DNA accessibility is regulated via a complex set of post-translational modifications of histones, also called histone code, and nucleosome remodeling. Related diseases Glioma (GLM) [MIM:137800]: Gliomas are benign or malignant central nervous system neoplasms derived from glial cells. They comprise astrocytomas and glioblastoma multiforme that are derived from astrocytes, oligodendrogliomas derived from oligodendrocytes and ependymomas derived from ependymocytes. {ECO:0000269|PubMed:22286061, ECO:0000269|PubMed:22286216, ECO:0000269|PubMed:23539269}. The gene represented in this entry is involved in disease pathogenesis. H3F3A mutations affecting residues involved in post-translational modifications of histone H3.3 are recurrent in malignant, aggressive gliomas including glioblastoma multiforme (GBM) and diffuse intrinsic pontine glioma (DIPG) (PubMed:22286061, PubMed:22286216). The mechanism through which mutations lead to tumorigenesis involves altered histones methylation, impaired regulation of Polycomb repressive complex 2 (PRC2) activity, and aberrant epigenetic regulation of gene expression (PubMed:23539183, PubMed:23539269, PubMed:23603901). {ECO:0000269|PubMed:22286061, ECO:0000269|PubMed:22286216, ECO:0000269|PubMed:23539183, ECO:0000269|PubMed:23539269, ECO:0000269|PubMed:23603901}.; DISEASE: Bryant-Li-Bhoj neurodevelopmental syndrome 1 (BRYLIB1) [MIM:619720]: An autosomal dominant disorder predominantly characterized by global developmental delay, impaired intellectual development, poor or absent speech, and delayed motor milestones. Clinical manifestations are highly variable, including abnormal head shape, dysmorphic facial features, oculomotor abnormalities, feeding problems, and non-specific brain imaging abnormalities. Additional features may include hearing loss, seizures, short stature, and mild skeletal defects. {ECO:0000269|PubMed:33268356, ECO:0000269|PubMed:34876591}. The disease is caused by variants affecting the gene represented in this entry. BRYLIB1 is caused by variants in H3-3A. {ECO:0000269|PubMed:33268356, ECO:0000269|PubMed:34876591}.; DISEASE: Bryant-Li-Bhoj neurodevelopmental syndrome 2 (BRYLIB2) [MIM:619721]: An autosomal dominant disorder predominantly characterized by global developmental delay, impaired intellectual development, poor or absent speech, and delayed motor milestones. Clinical manifestations are highly variable, including abnormal head shape, dysmorphic facial features, oculomotor abnormalities, feeding problems, and non-specific brain imaging abnormalities. Additional features may include hearing loss, seizures, short stature, and mild skeletal defects. {ECO:0000269|PubMed:33268356, ECO:0000269|PubMed:34876591}. The disease is caused by variants affecting the gene represented in this entry. BRYLIB2 is caused by variants in H3-3B. {ECO:0000269|PubMed:33268356, ECO:0000269|PubMed:34876591}.; DISEASE: H3F3A and H3F3B mutations affecting residues involved in post-translational modifications of histone H3.3 are implicated in the pathogenesis of some bone and cartilage neoplasms. Mutations have been found with high prevalence in chondroblastoma and giant cell tumors of bone, and with low frequency in osteosarcoma, conventional chondrosarcoma and clear cell chondrosarcoma. Chondroblastoma samples frequently carry a H3F3B mutation affecting residue Lys-37 (H3K36), although H3F3A is mutated in some cases. Most giant cell tumors of bone harbor H3F3A mutations affecting residue Gly-35 (H3G34). {ECO:0000269|PubMed:24162739}. Drugs (DrugBank ID) NA Interacts with Q9NVP2; P45973; Q13111; Q9UER7; Q9UER7-1; Q9Y6K1; P62805; P49321-2; Q8IZL8; Q5VWG9; Q9VK33; Q8R5C8 EC number NA Uniprot keywords 3D-structure; Acetylation; ADP-ribosylation; Chromosome; Citrullination; Direct protein sequencing; Disease variant; DNA-binding; Hydroxylation; Intellectual disability; Lipoprotein; Methylation; Nucleosome core; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Ubl conjugation Protein physicochemical properties Chain ID A,C Molecular weight (Da) 86148.9 Length 766 Aromaticity 0.1 Instability index 37.57 Isoelectric point 8.25 Charge (pH=7) 7.16 2D Binding mode Binding energy (Kcal/mol) -7.43  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence GSRKCEKAGCTATCPVCFASASERCAKNGYTSRWYHLSCGEHFCNECFDHYYRSHKDGYDKYTTWKKIWTSNGKTEPSPKAFMADQQLPYWVQCTKPECRKWRQLTKEIQLTPQIAKTYRCGMKPNTAIKPETSDHCSLPEDLRVLEVSNHWWYSMLILPPLLKDSVAAPLLSAYYPDCVGMSPSCTGMNRYFQPFYQPNECGKALCVRPDVMELDELYEFPEYSRDPTMYLALRNLILALWYTNCKEALTPQKCIPHIIVRGLVRIRCVQEVERILYFMTRKGLINTGVLSVGADQYLLPKDYHNKSVIIIGAGPAGLAAARQLHNFGIKVTVLEAKDRIGGRVWDDKSFKGVTVGRGAQIVNGCINNPVALMCEQLGISMHKFGERCDLIQEGGRITDPTIDKRMDFHFNALLDVVSEWRKDKTQLQDVPLGEKIEEIYKAFIKESGIQFSELEGQVLQFHLSNLEYACGSNLHQVSARSWDHNEFFAQFAGDHTLLTPGYSVIIEKLAEGLDIQLKSPVQCIDYSGDEVQVTTTDGTGYSAQKVLVTVPLALLQKGAIQFNPPLSEKKMKAINSLGAGIIEKIALQFPYRFWDSKVQGADFFGHVPPSASKRGLFAVFYDMDPQKKHSVLMSVIAGEAVASVRTLDDKQVLQQCMATLRELFKEQEVPDPTKYFVTRWSTDPWIQMAYSFVKTGGSGEAYDIIAEDIQGTVFFAGEATNRHFPQTVTGAYLSGVREASKIAAFARTMQTARKSTGGKAPRKQL Hydrogen bonds contact Hydrophobic contact | ||||
| 18 | Orexin receptor type 2 (HCRTR2) | 4S0V | 5.43 | |
Target general information Gen name HCRTR2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Ox2r; Ox2-R; Ox-2-R; Orexin-2 receptor; Hypocretin receptor type 2; HFGANP Protein family G-protein coupled receptor 1 family Biochemical class GPCR rhodopsin Function Triggers an increase in cytoplasmic Ca(2+) levels in response to orexin-A binding. Nonselective, high-affinity receptor for both orexin-A and orexin-B neuropeptides. Related diseases Hyperchlorhidrosis, isolated (HYCHL) [MIM:143860]: An autosomal recessive disorder characterized by excessive sweating and increased sweat chloride levels. Affected individuals suffer from episodes of hyponatremic dehydration and report increased amounts of visible salt precipitates in sweat. {ECO:0000269|PubMed:21035102, ECO:0000269|PubMed:21184099, ECO:0000269|PubMed:26911677}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB15031; DB11951; DB09034 Interacts with P62937 EC number NA Uniprot keywords 3D-structure; Cell membrane; Disulfide bond; G-protein coupled receptor; Glycoprotein; Membrane; Receptor; Reference proteome; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 32524.7 Length 282 Aromaticity 0.14 Instability index 38.96 Isoelectric point 9.18 Charge (pH=7) 12.36 2D Binding mode Binding energy (Kcal/mol) -7.4  Molscript Map 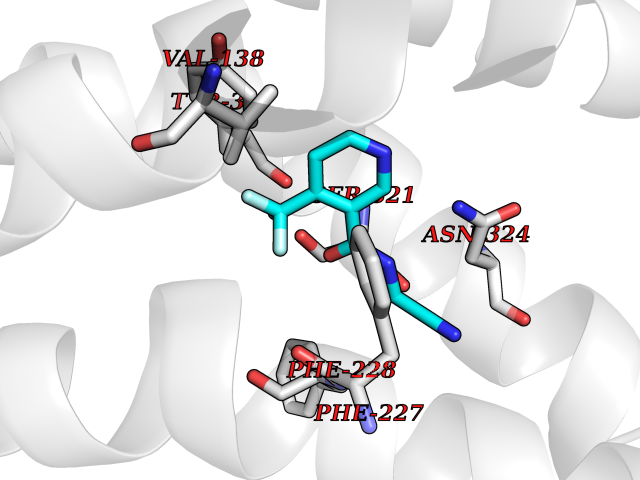 Pymol Map 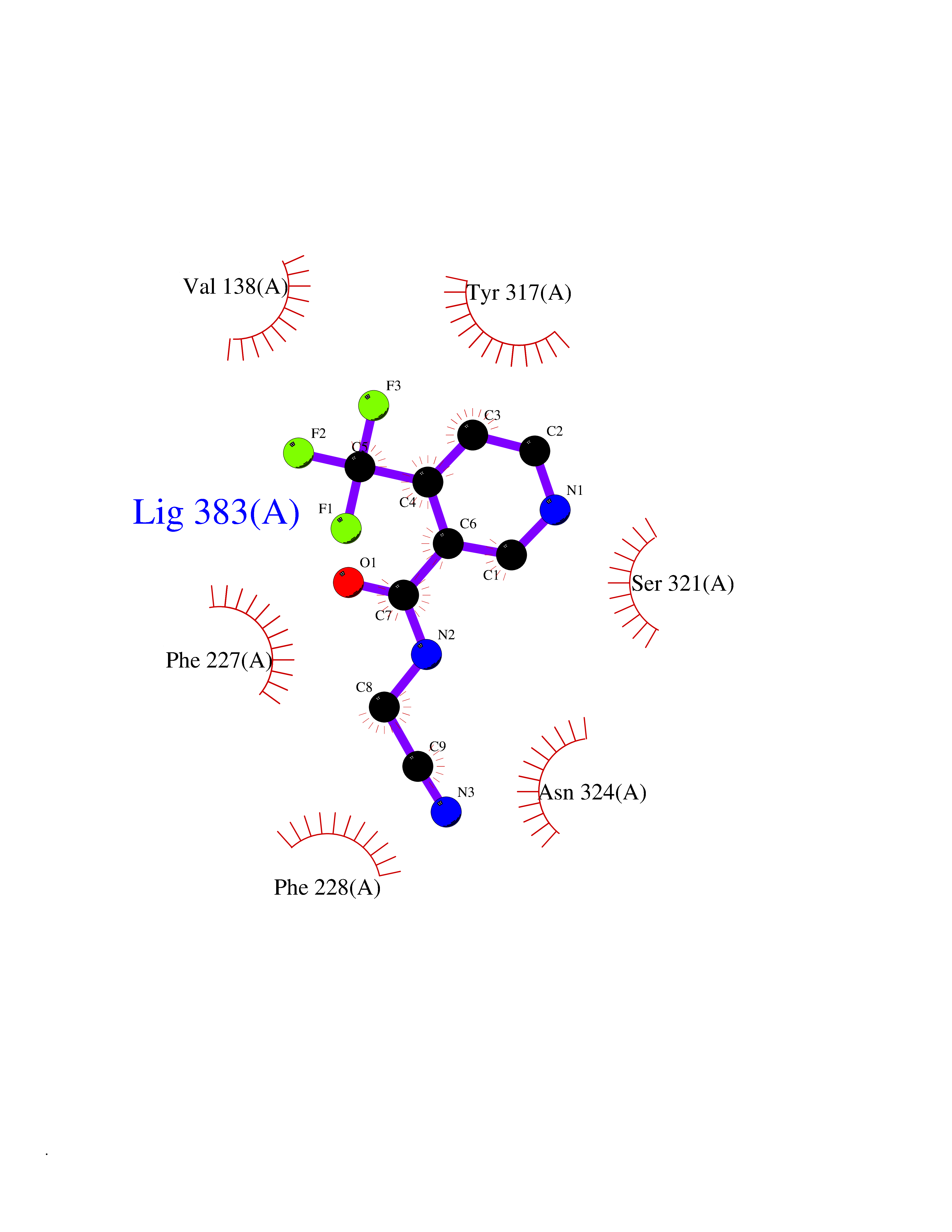 Ligplot Map 3D Binding mode Sequence PKEYEWVLIAGYIIVFVVALIGNVLVCVAVWKNHHMRTVTNYFIVNLSLADVLVTITCLPATLVVDITETWFFGQSLCKVIPYLQTVSVSVSVLTLSCIALDRWYAICHPSTAKRARNSIVIIWIVSCIIMIPQAIVMECSTVFKTTLFTVCDERWGGEIYPKMYHICFFLVTYMAPLCLMVLAYLQIFRKLWCRQKQIRARRKTARMLMVVLLVFAICYLPISILNVLKRVFGMFAHDRETVYAWFTFSHWLVYANSAANPIIYNFLSGKFREEFKAAFSC Hydrogen bonds contact Hydrophobic contact | ||||
| 19 | Tankyrase-2 (TNKS-2) | 3U9H | 5.43 | |
Target general information Gen name TNKS2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Tankyrase-related protein; Tankyrase-like protein; Tankyrase II; TRF1-interacting ankyrin-related ADP-ribose polymerase 2; TNKL; TANK2; Protein poly-ADP-ribosyltransferase tankyrase-2; Poly [ADP-ribos Protein family ARTD/PARP family Biochemical class Glycosyltransferases Function Acts as an activator of the Wnt signaling pathway by mediating poly-ADP-ribosylation of AXIN1 and AXIN2, 2 key components of the beta-catenin destruction complex: poly-ADP-ribosylated target proteins are recognized by RNF146, which mediates their ubiquitination and subsequent degradation. Also mediates poly-ADP-ribosylation of BLZF1 and CASC3, followed by recruitment of RNF146 and subsequent ubiquitination. Mediates poly-ADP-ribosylation of TERF1, thereby contributing to the regulation of telomere length. Stimulates 26S proteasome activity. Poly-ADP-ribosyltransferase involved in various processes such as Wnt signaling pathway, telomere length and vesicle trafficking. Related diseases Intellectual developmental disorder with macrocephaly, seizures, and speech delay (IDDMSSD) [MIM:618158]: An autosomal dominant neurodevelopmental disorder characterized by impaired intellectual development, poor speech, postnatal macrocephaly, and seizures. {ECO:0000269|PubMed:30290153}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with O15084; Q7Z6K5-1; O15169; Q9NWV8; P11274; Q13698; Q9NRI5; Q6V0I7; Q9NWT6; P14652; Q9UIQ6; Q14980; Q9BZL4; Q92698; P78314; O43815; P54274; Q9C0C2; Q9UHP3; Q06649 EC number EC 2.4.2.30 Uniprot keywords 3D-structure; ADP-ribosylation; ANK repeat; Chromosome; Cytoplasm; Glycosyltransferase; Golgi apparatus; Hydroxylation; Membrane; Metal-binding; NAD; Nucleotidyltransferase; Nucleus; Proteomics identification; Reference proteome; Repeat; Telomere; Transferase; Ubl conjugation; Wnt signaling pathway; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 23695.5 Length 208 Aromaticity 0.11 Instability index 47.61 Isoelectric point 8.28 Charge (pH=7) 2.88 2D Binding mode Binding energy (Kcal/mol) -7.41 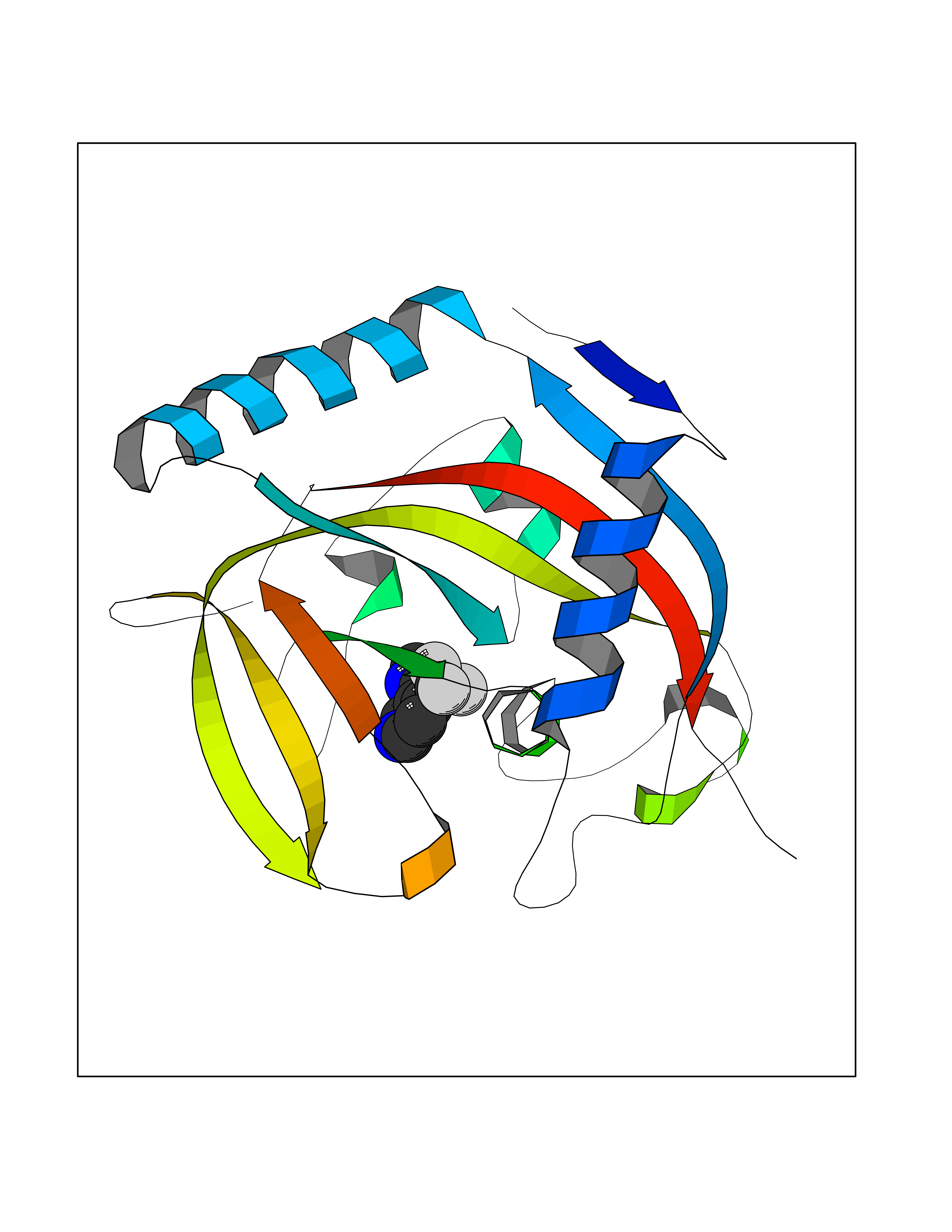 Molscript Map 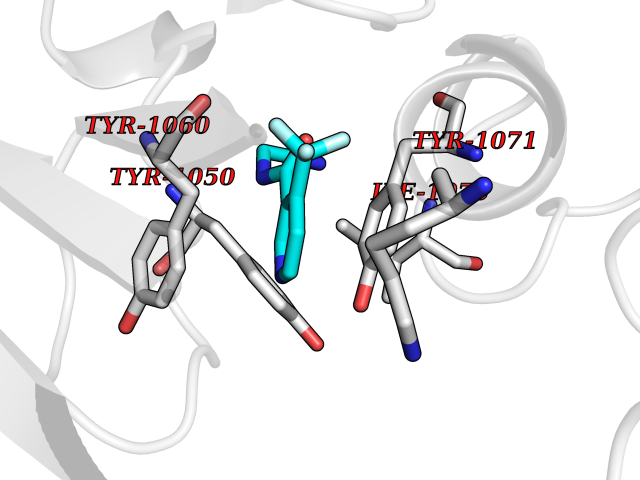 Pymol Map 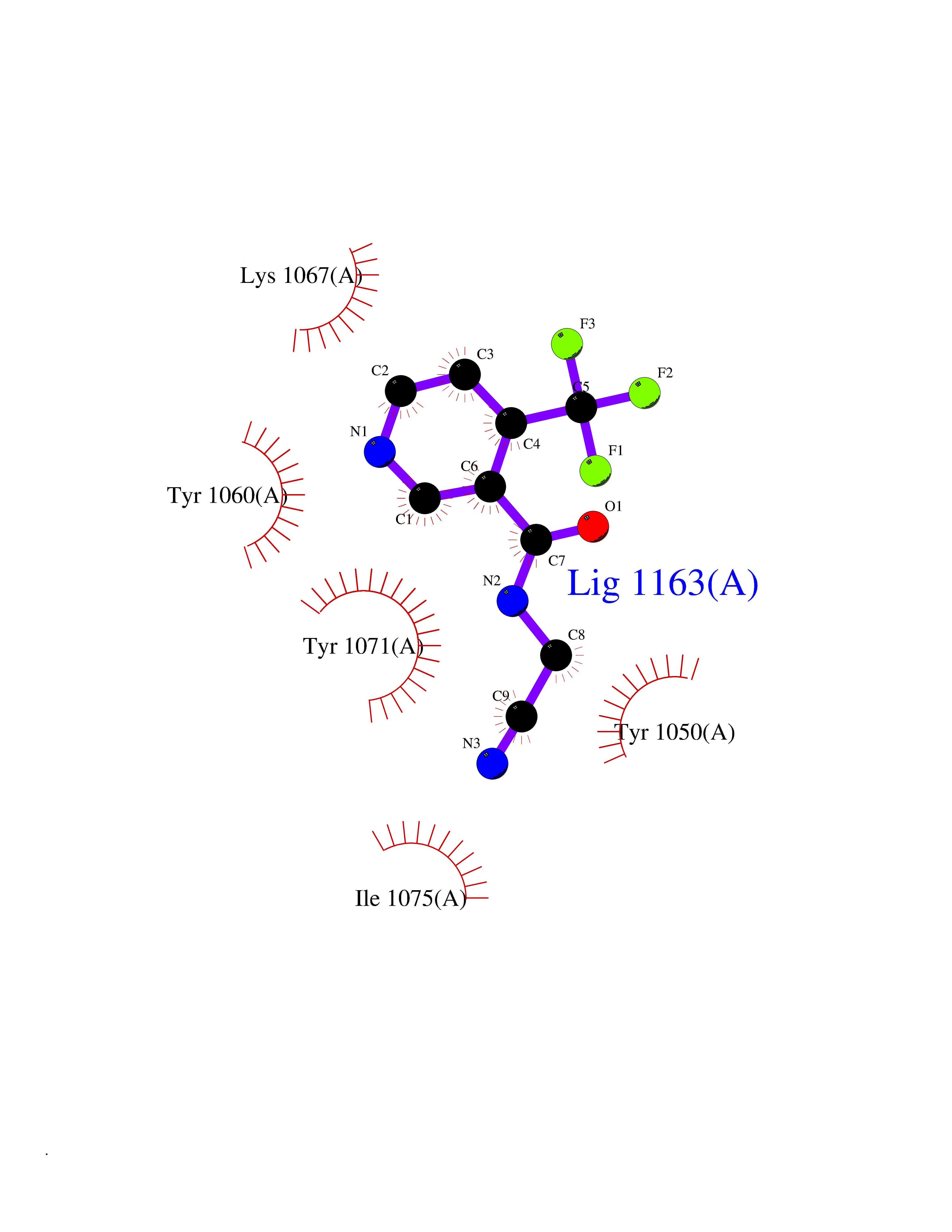 Ligplot Map 3D Binding mode Sequence GTILIDLSPDDKEFQSVEEEMQSTVREHRDGGHAGGIFNRYNILKIQKVCNKKLWERYTHRRKEVSEENHNHANERMLFHGSPFVNAIIHKGFDERHAYIGGMFGAGIYFAENSSKSNQYVYGIGGGTGCPVHKDRSCYICHRQLLFCRVTLGKSFLQFSAMAHSPPGHHSVTGRPSVNGLALAEYVIYRGEQAYPEYLITYQIMRPE Hydrogen bonds contact Hydrophobic contact | ||||
| 20 | TNF alpha converting enzyme (ADAM17) | 2FV5 | 5.43 | |
Target general information Gen name ADAM17 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms TNFalpha converting enzyme; TNF-alpha-converting enzyme; TNF-alpha converting enzyme; TNF-alpha convertase; TACE; Snake venom-like protease; Disintegrin and metalloproteinase domain-containing protein Protein family NA Biochemical class Peptidase Function Responsible for the proteolytical release of soluble JAM3 from endothelial cells surface. Responsible for the proteolytic release of several other cell-surface proteins, including p75 TNF-receptor, interleukin 1 receptor type II, p55 TNF-receptor, transforming growth factor-alpha, L-selectin, growth hormone receptor, MUC1 and the amyloid precursor protein. Acts as an activator of Notch pathway by mediating cleavage of Notch, generating the membrane-associated intermediate fragment called Notch extracellular truncation (NEXT). Plays a role in the proteolytic processing of ACE2. Plays a role in hemostasis through shedding of GP1BA, the platelet glycoprotein Ib alpha chain. Mediates the proteolytic cleavage of LAG3, leading to release the secreted form of LAG3. Cleaves the membrane-bound precursor of TNF-alpha to its mature soluble form. Related diseases Inflammatory skin and bowel disease, neonatal, 1 (NISBD1) [MIM:614328]: A disorder characterized by inflammatory features with neonatal onset, involving the skin, hair, and gut. The skin lesions involve perioral and perianal erythema, psoriasiform erythroderma, with flares of erythema, scaling, and widespread pustules. Gastrointestinal symptoms include malabsorptive diarrhea that is exacerbated by intercurrent gastrointestinal infections. The hair is short or broken, and the eyelashes and eyebrows are wiry and disorganized. {ECO:0000269|PubMed:22010916}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07189; DB07145; DB06943; DB07964; DB07079; DB07121; DB07147; DB07233 Interacts with Q12959; P05556; Q13257; Q80WQ6 EC number EC 3.4.24.86 Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Cleavage on pair of basic residues; Direct protein sequencing; Disulfide bond; Glycoprotein; Hydrolase; Membrane; Metal-binding; Metalloprotease; Notch signaling pathway; Phosphoprotein; Protease; Proteomics identification; Reference proteome; SH3-binding; Signal; Transmembrane; Transmembrane helix; Zinc; Zymogen Protein physicochemical properties Chain ID A Molecular weight (Da) 29343.5 Length 261 Aromaticity 0.1 Instability index 44.79 Isoelectric point 5.53 Charge (pH=7) -7.53 2D Binding mode Binding energy (Kcal/mol) -7.4  Molscript Map 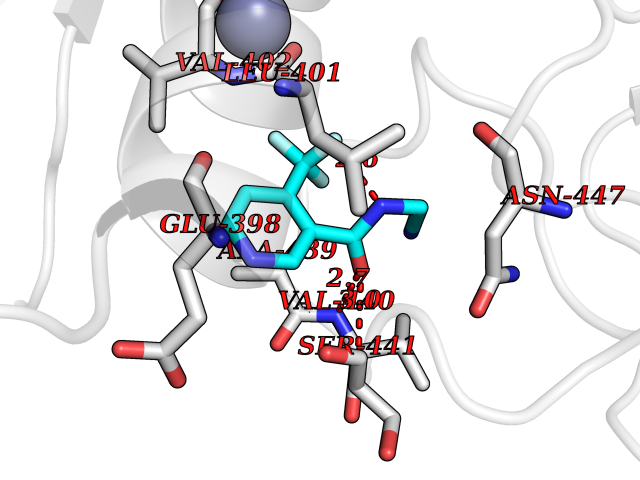 Pymol Map 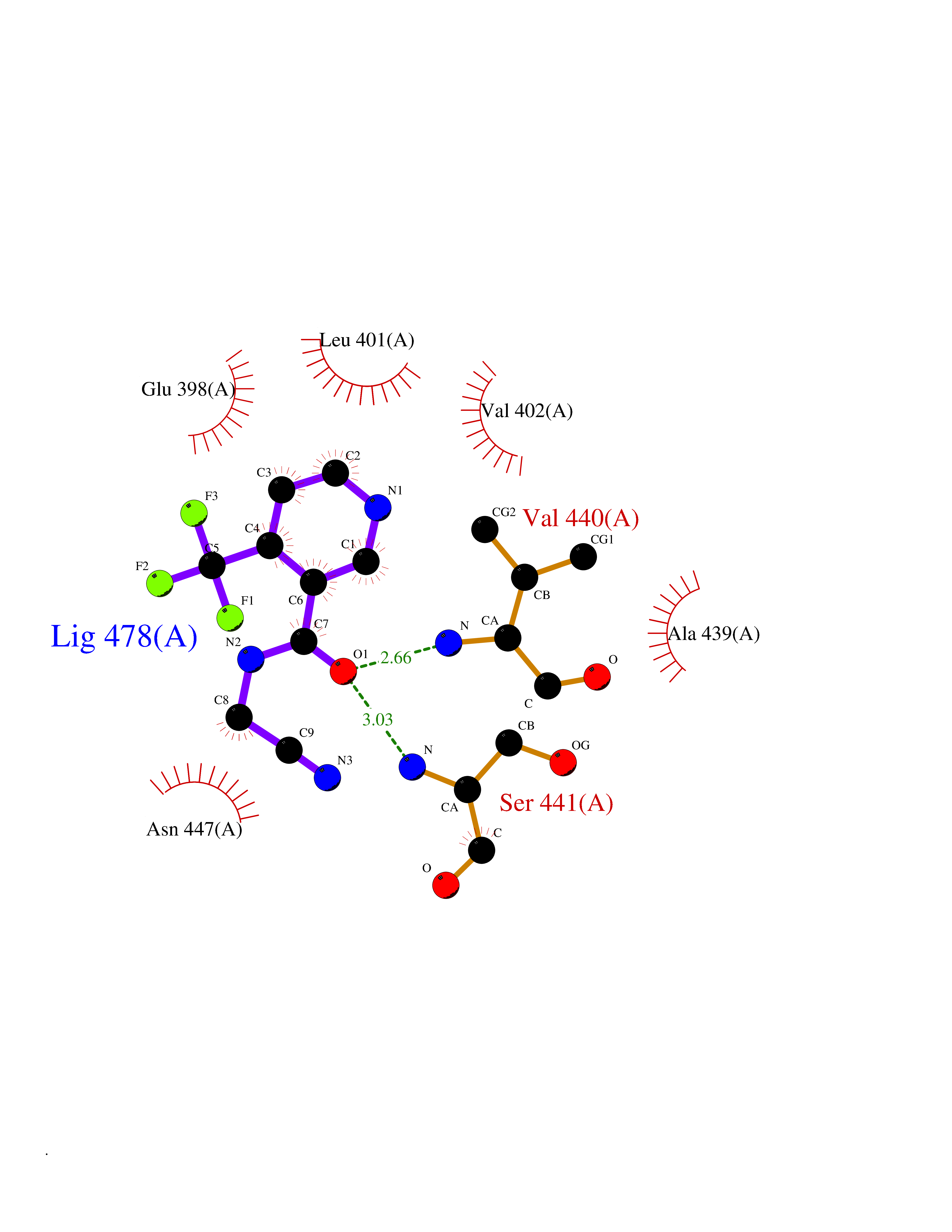 Ligplot Map 3D Binding mode Sequence ADPDPMKNTCKLLVVADHRFYRYMGRGEESTTTNYLIELIDRVDDIYRNTAWDNAGFKGYGIQIEQIRILKSPQEVKPGEKHYNMAKSYPNEEKDAWDVKMLLEQFSFDIAEEASKVCLAHLFTYQDFDMGTLGLAYGGSPRANSHGGVCPKAYYSPVGKKNIYLNSGLTSTKNYGKTILTKEADLVTTHELGHNFGAEHDPDGLAECAPNEDQGGKYVMYPIAVSGDHENNKMFSQCSKQSIYKTIESKAQECFQERSNA Hydrogen bonds contact Hydrophobic contact | ||||