Job Results:
Ligand
Structure
Job ID
b448ce30ce2c1c80e042f47c74d47daa
Job name
NA
Time
2026-01-10 22:40:02
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 61 | SET and MYND domain-containing protein 2 (SMYD2) | 5ARF | 6.94 | |
Target general information Gen name SMYD2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Nlysine methyltransferase SMYD2; N-lysine methyltransferase SMYD2; Lysine Nmethyltransferase 3C; Lysine N-methyltransferase 3C; KMT3C; Histone methyltransferase SMYD2; HSKMB; HSKM-B Protein family Class V-like SAM-binding methyltransferase superfamily Biochemical class Methyltransferase Function Specifically methylates histone H3 'Lys-4' (H3K4me) and dimethylates histone H3 'Lys-36' (H3K36me2). Shows even higher methyltransferase activity on p53/TP53. Monomethylates 'Lys-370' of p53/TP53, leading to decreased DNA-binding activity and subsequent transcriptional regulation activity of p53/TP53. Monomethylates RB1 at 'Lys-860'. Protein-lysine N-methyltransferase that methylates both histones and non-histone proteins, including p53/TP53 and RB1. Related diseases Pulmonary hypertension, primary, 4 (PPH4) [MIM:615344]: A rare disorder characterized by plexiform lesions of proliferating endothelial cells in pulmonary arterioles. The lesions lead to elevated pulmonary arterial pression, right ventricular failure, and death. The disease can occur from infancy throughout life and it has a mean age at onset of 36 years. Penetrance is reduced. Although familial pulmonary hypertension is rare, cases secondary to known etiologies are more common and include those associated with the appetite-suppressant drugs. {ECO:0000269|PubMed:23883380}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Defects in this gene may cause developmental delay with sleep apnea (DDSA). A disorder characterized by developmental neurologic, skeletal and respiratory anomalies including microcephaly, arthrogryposis, scoliosis, cleft palate, facial dysmorphology, bilateral talipes, feeding difficulties and central and/or obstructive sleep apnea. Malformations are detected as early as 21 weeks post gestation. Severely affected patients require ongoing treatment with nocturnal O2 or pressure-controlled ventilation. The disease is associated with recurrent de novo gain of function variants. {ECO:0000269|PubMed:36195757}. Drugs (DrugBank ID) NA Interacts with P20290-2; Q96K17; Q9UPZ9; Q9Y5W9; P04637 EC number EC 2.1.1.- Uniprot keywords 3D-structure; Alternative splicing; Chromatin regulator; Cytoplasm; Metal-binding; Methyltransferase; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; S-adenosyl-L-methionine; Transcription; Transcription regulation; Transferase; Zinc; Zinc-finger Protein physicochemical properties Chain ID A Molecular weight (Da) 48789.6 Length 425 Aromaticity 0.1 Instability index 51.93 Isoelectric point 6.35 Charge (pH=7) -4.02 2D Binding mode Binding energy (Kcal/mol) -9.47 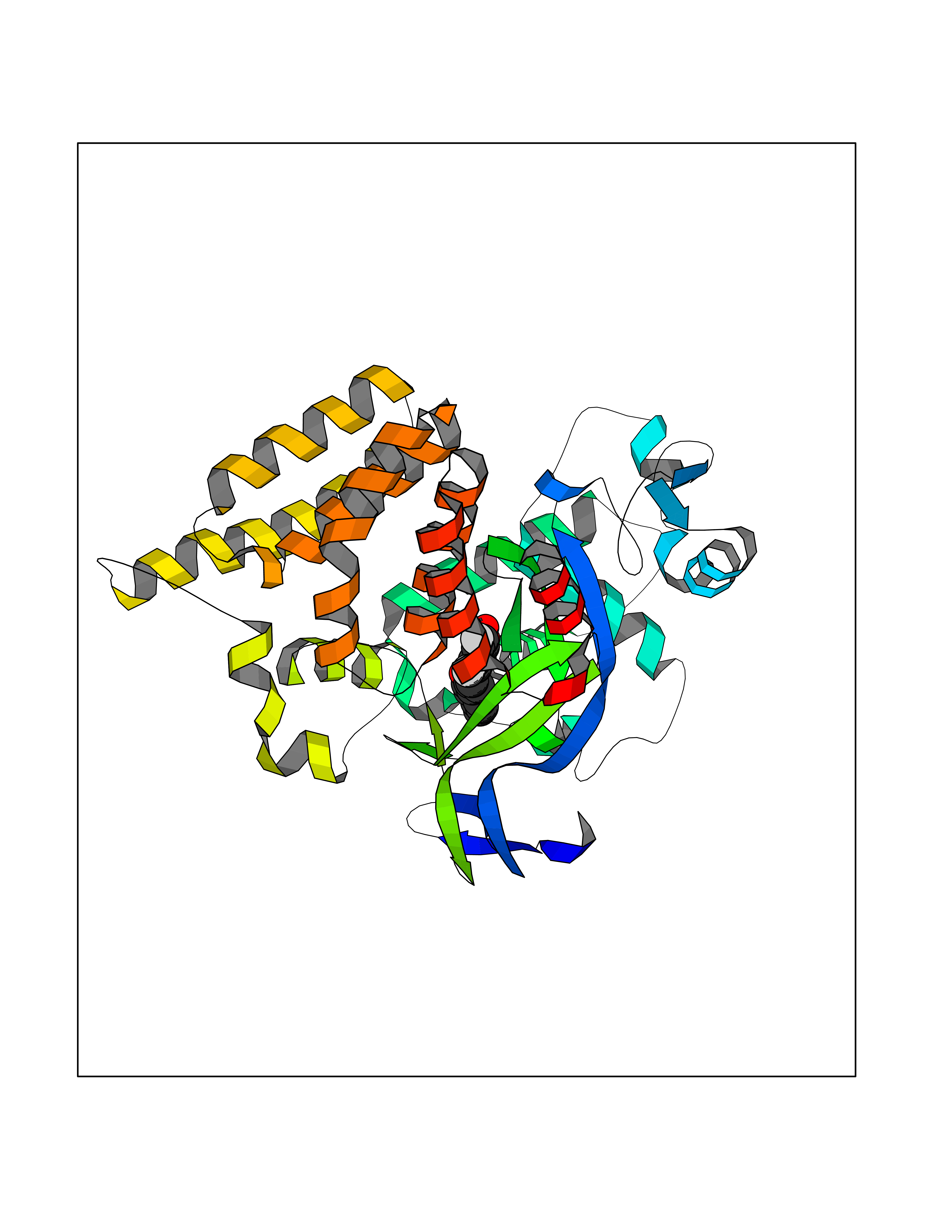 Molscript Map 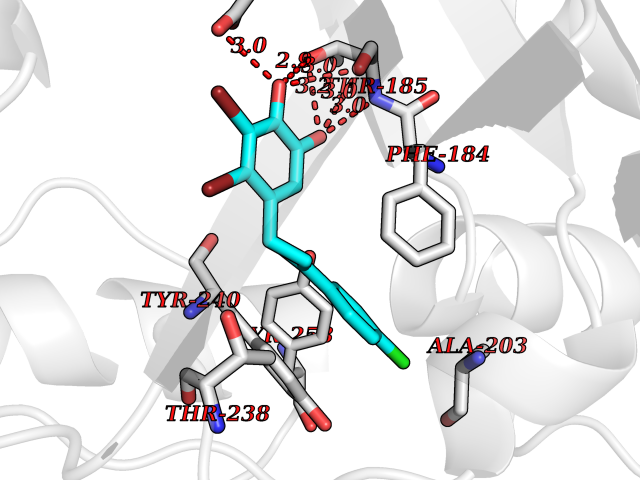 Pymol Map 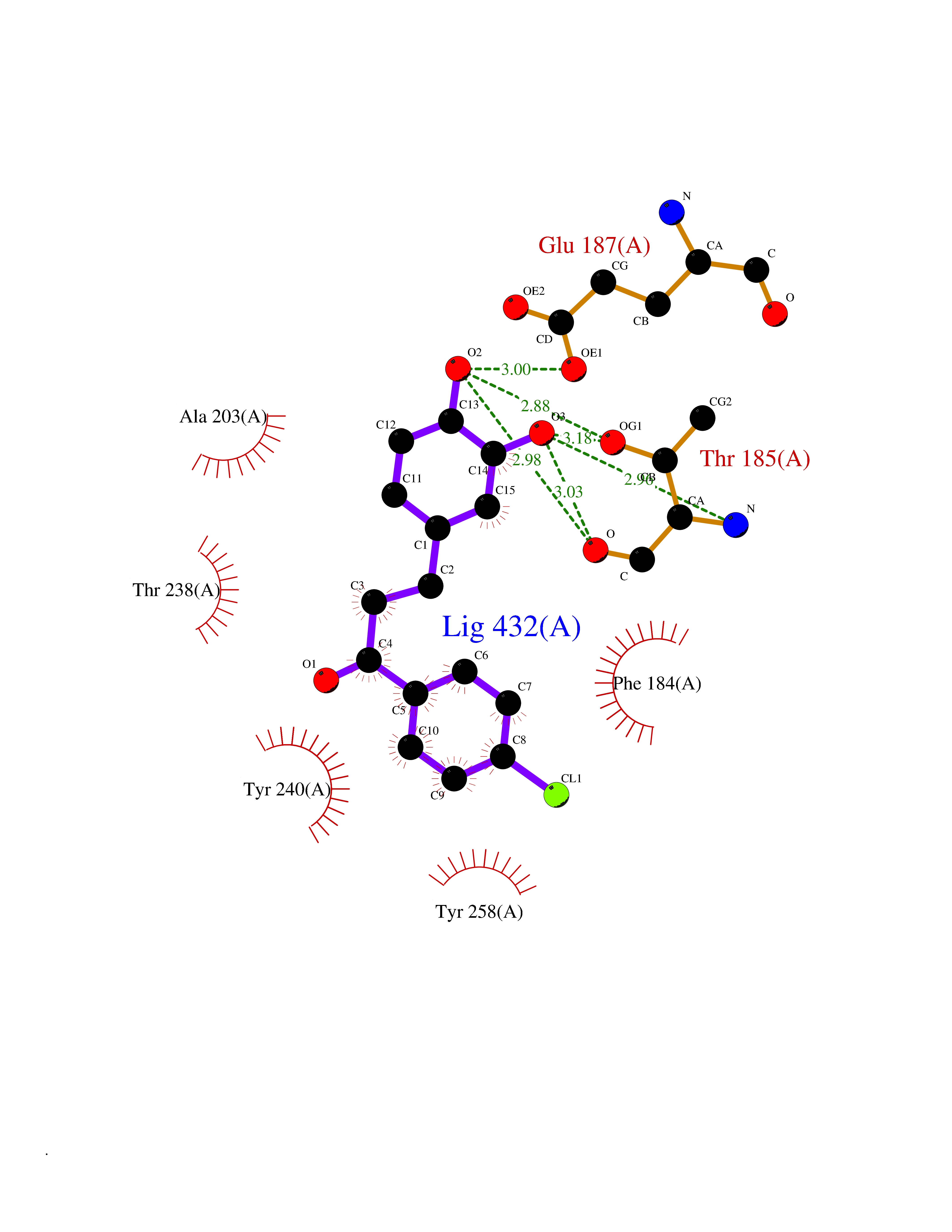 Ligplot Map 3D Binding mode Sequence LGGLERFCSPGKGRGLRALQPFQVGDLLFSCPAYAYVLTVNERGNHCEYCFTRKEGLSKCGRCKQAFYCNVECQKEDWPMHKLECSPMVVFGENWNPSETVRLTARILAKQKIHPERTPSEKLLAVKEFESHLDKLDNEKKDLIQSDIAALHHFYSKHLGFPDNDSLVVLFAQVNCNGFTIEDEELSHLGSAIFPDVALMNHSCCPNVIVTYKGTLAEVRAVQEIKPGEEVFTSYIDLLYPTEDRNDRLRDSYFFTCECQECTTKDKDKAKVEIRKLSDPPKAEAIRDMVRYARNVIEEFRRAKHYKSPSELLEICELSQEKMSSVFEDSNVYMLHMMYQAMGVCLYMQDWEGALQYGQKIIKPYSKHYPLYSLNVASMWLKLGRLYMGLEHKAAGEKALKKAIAIMEVAHGKDHPYISEIKQEI Hydrogen bonds contact Hydrophobic contact | ||||
| 62 | Corynebacterium Pup-protein ligase (Cory pafA) | 4BJR | 6.94 | |
Target general information Gen name Cory pafA Organism Corynebacterium glutamicum (strain ATCC 13032 / DSM 20300 / JCM 1318 / BCRC 11384 / CCUG 27702 / LMG 3730 / NBRC 12168 / NCIMB 10025 / NRRL B-2784 / 534) Uniprot ID TTD ID Synonyms Pup-conjugating enzyme; Pup--protein ligase; Proteasome accessory factor A Protein family Pup ligase/Pup deamidase family, Pup-conjugating enzyme subfamily Biochemical class NA Function Catalyzes the covalent attachment of the prokaryotic ubiquitin-like protein modifier Pup to the proteasomal substrate proteins, thereby targeting them for proteasomal degradation. This tagging system is termed pupylation. The ligation reaction involves the side-chain carboxylate of the C-terminal glutamate of Pup and the side-chain amino group of a substrate lysine. Related diseases Anemia, non-spherocytic hemolytic, due to G6PD deficiency (NSHA) [MIM:300908]: A disease characterized by G6PD deficiency, acute hemolytic anemia, fatigue, back pain, and jaundice. In most patients, the disease is triggered by an exogenous agent, such as some drugs, food, or infection. Increased unconjugated bilirubin, lactate dehydrogenase, and reticulocytosis are markers of the disorder. Although G6PD deficiency can be life-threatening, most patients are asymptomatic throughout their life. {ECO:0000269|PubMed:12524354, ECO:0000269|PubMed:1303180, ECO:0000269|PubMed:1303182, ECO:0000269|PubMed:1536798, ECO:0000269|PubMed:1611091, ECO:0000269|PubMed:1889820, ECO:0000269|PubMed:1945893, ECO:0000269|PubMed:20007901, ECO:0000269|PubMed:26479991, ECO:0000269|PubMed:2836867, ECO:0000269|PubMed:2912069, ECO:0000269|PubMed:30988594, ECO:0000269|PubMed:38066190, ECO:0000269|PubMed:7858267, ECO:0000269|PubMed:7959695, ECO:0000269|PubMed:8193373, ECO:0000269|PubMed:8490627, ECO:0000269|PubMed:8533762, ECO:0000269|PubMed:8733135, ECO:0000269|PubMed:9452072}. The disease is caused by variants affecting the gene represented in this entry. Deficiency of G6PD is associated with hemolytic anemia in two different situations. First, in areas in which malaria has been endemic, G6PD-deficiency alleles have reached high frequencies (1% to 50%) and deficient individuals, though essentially asymptomatic in the steady state, have a high risk of acute hemolytic attacks. Secondly, sporadic cases of G6PD deficiency occur at a very low frequencies, and they usually present a more severe phenotype. Several types of NSHA are recognized. Class-I variants are associated with severe NSHA; class-II have an activity <10% of normal; class-III have an activity of 10% to 60% of normal; class-IV have near normal activity. Drugs (DrugBank ID) NA Interacts with NA EC number NA Uniprot keywords 3D-structure; ATP-binding; Ligase; Magnesium; Metal-binding; Nucleotide-binding; Reference proteome; Ubl conjugation pathway Protein physicochemical properties Chain ID A,B Molecular weight (Da) 110572 Length 994 Aromaticity 0.07 Instability index 42.33 Isoelectric point 5.26 Charge (pH=7) -34.19 2D Binding mode Binding energy (Kcal/mol) -9.46  Molscript Map 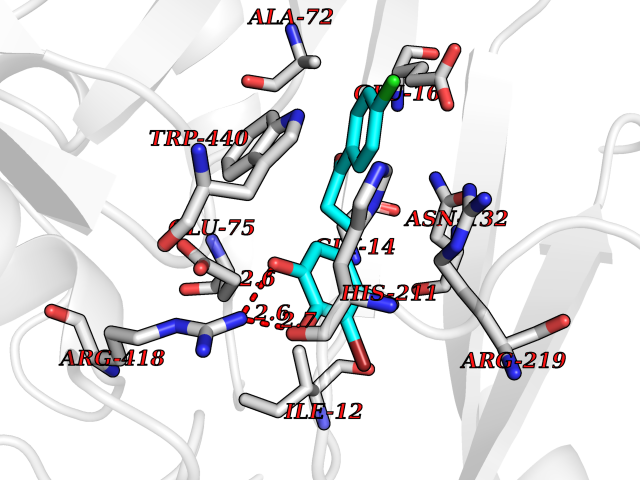 Pymol Map 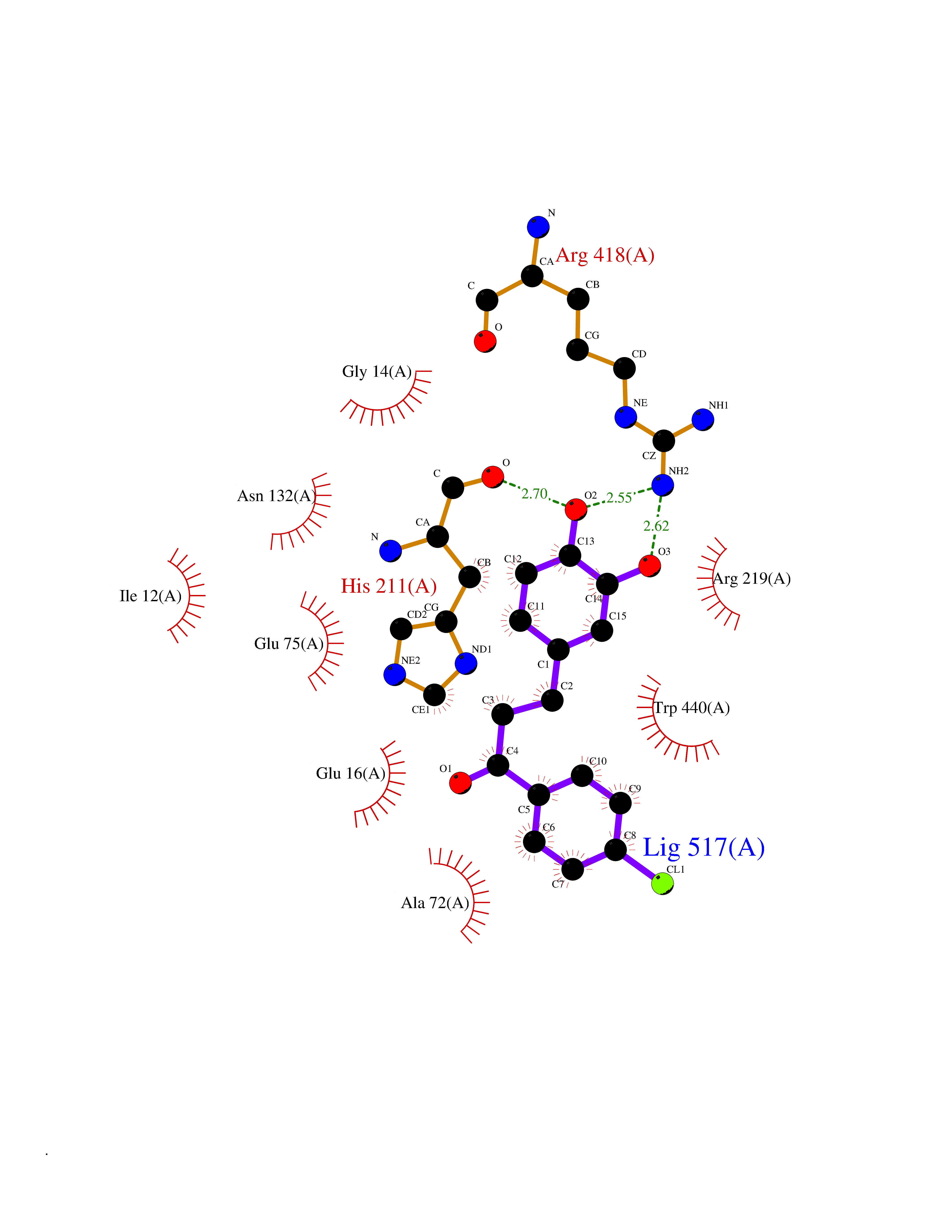 Ligplot Map 3D Binding mode Sequence TVESALTRRIMGIETEYGLTFVDGDSKKLRPDEIARRMFRPIVEKYSSSNIFIPNGSRLYLNVGSHPEYATAECDNLTQLINFEKAGDVIADRMAVDAEESLAKEDIAGQVYLFKNNVDSVGNSYGCHENYLVGRSMPLKALGKRLMPFLITRQLICGAGRIHHPNPSFPLGYCISQRSDHVWEGVSSASRPIINTRDEPHADSHSYRRLHVIVGDANMAEPSIALKVGSTLLVLEMIEADFGLPSLELANDIASIREISRDATGSTLLSLKDGTTMTALQIQQVVFEHASKWLEQRPEPEFSGTSNTEMARVLDLWGRMLKAIESGDFSEVDTEIDWVIKKKLIDRFIQRGNLGLDDPKLAQVDLTYHDIRPGRGLFSVLQSRGMIKRWTTDEAILAAVDTAPDTTRAHLRGRILKAADTLGVPVTVDWMRHKVNRPEPQSVELGDPFSAVNSEVDQLIEYMTVHAGSASGTSLLDEIDGLLENNAEEFVRSYVQKGGETVESALTRRIMGIETEYGLTFVDGDSKKLRPDEIARRMFRPIVEKYSSSNIFIPNGSRLYLNVGSHPEYATAECDNLTQLINFEKAGDVIADRMAVDAEESLAKEDIAGQVYLFKNNVDSVGNSYGCHENYLVGRSMPLKALGKRLMPFLITRQLICGAGRIHHPNPSFPLGYCISQRSDHVWEGVSSASRPIINTRDEPHADSHSYRRLHVIVGDANMAEPSIALKVGSTLLVLEMIEADFGLPSLELANDIASIREISRDATGSTLLSLKDGTTMTALQIQQVVFEHASKWLEQRPEPEFSGTSNTEMARVLDLWGRMLKAIESGDFSEVDTEIDWVIKKKLIDRFIQRGNLGLDDPKLAQVDLTYHDIRPGRGLFSVLQSRGMIKRWTTDEAILAAVDTAPDTTRAHLRGRILKAADTLGVPVTVDWMRHKVNRPEPQSVELGDPFSAVNSEVDQLIEYMTVHASLLDEIDGLLENNAEEFVRSYVQKGGE Hydrogen bonds contact Hydrophobic contact | ||||
| 63 | Purine nucleoside phosphorylase (PNP) | 4EAR | 6.93 | |
Target general information Gen name PNP Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms PNP; Inosine phosphorylase Protein family PNP/MTAP phosphorylase family Biochemical class Pentosyltransferase Function The purine nucleoside phosphorylases catalyze the phosphorolytic breakdown of the N-glycosidic bond in the beta- (deoxy)ribonucleoside molecules, with the formation of the corresponding free purine bases and pentose-1-phosphate. Related diseases Purine nucleoside phosphorylase deficiency (PNPD) [MIM:613179]: A disorder that interrupts both the catabolism of inosine into hypoxanthine and guanosine into guanine, and leads to the accumulation of guanosine, inosine, and their deoxified by-products. The main clinical presentation is recurrent infections due to severe T-cell immunodeficiency. Some patients also have neurologic impairment. {ECO:0000269|PubMed:1384322, ECO:0000269|PubMed:3029074, ECO:0000269|PubMed:8931706}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03881; DB03551; DB02222; DB02391; DB03609; DB01667; DB04260; DB02796; DB04753; DB00640; DB00242; DB00900; DB06185; DB02377; DB02857; DB04754; DB04757; DB04076; DB02230; DB04335; DB02568; DB03101 Interacts with P05067; Q9UQM7; O14576-2; P06241; P14136; Q92993-2; Q9BXM7; P00491; P17612; P63000; Q92673; Q15583 EC number EC 2.4.2.1 Uniprot keywords 3D-structure; Acetylation; Cytoplasm; Direct protein sequencing; Disease variant; Glycosyltransferase; Phosphoprotein; Proteomics identification; Purine salvage; Reference proteome; Transferase Protein physicochemical properties Chain ID A,B,C Molecular weight (Da) 31849.2 Length 288 Aromaticity 0.1 Instability index 34.77 Isoelectric point 6.42 Charge (pH=7) -1.63 2D Binding mode Binding energy (Kcal/mol) -9.45  Molscript Map 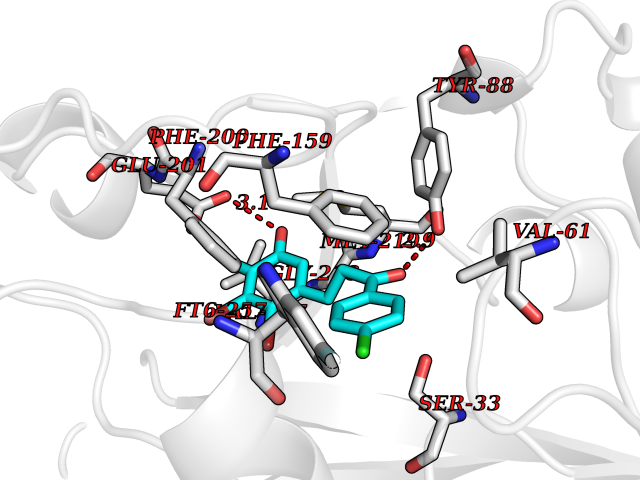 Pymol Map 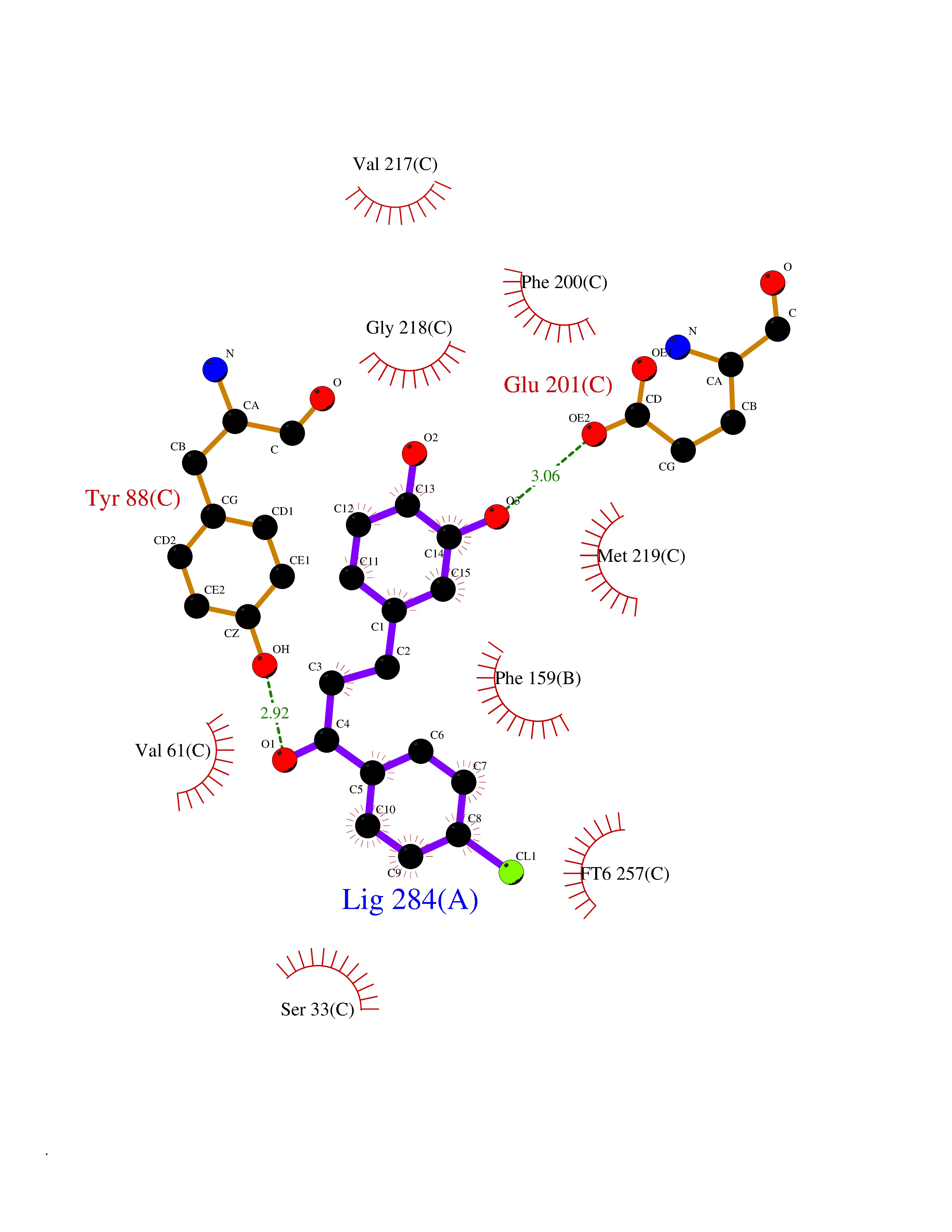 Ligplot Map 3D Binding mode Sequence GYTYEDYKNTAEYLLSHTKHRPQVAIICGSGLGGLTDKLTQAQIFDYSEIPNFPRSTVPGHAGRLVFGFLNGRACVMMQGRFHMYEGYPLYKVTFPVRVFHLLGVDTLVVTNAAGGLNPKFEVGDIMLIRDHINLPGFSGQNPLRGPNDERFGDRFPAMSDAYDRTMRQRALSTYKQMGEQRELQEGTYVMVAGPSFETVAECRVLQKLGADAVGMSTVPEVIVARHCGLRVFGFSLITNKVIMDYESLEKANXEEVLAAGKQAAQKLEQFVSILMASIDRFPAMSDA Hydrogen bonds contact Hydrophobic contact | ||||
| 64 | Ribonucleoside-diphosphate reductase large subunit | 2WGH | 6.93 | |
Target general information Gen name RRM1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms RR1 Protein family Ribonucleoside diphosphate reductase large chain family Biochemical class Oxidoreductase Function ATP binding.Identical protein binding.Ribonucleoside-diphosphate reductase activity, thioredoxin disulfide as acceptor. Related diseases Progressive external ophthalmoplegia with mitochondrial DNA deletions, autosomal recessive 6 (PEOB6) [MIM:620647]: A form of progressive external ophthalmoplegia, a mitochondrial myopathy characterized by progressive paralysis of the levator palpebrae, orbicularis oculi, and extraocular muscles. Ragged red fibers are seen on muscle biopsy. {ECO:0000269|PubMed:35617047}. The disease may be caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00242; DB00631; DB01073; DB05420; DB00441; DB01005; DB05003; DB01280; DB06433 Interacts with P23921; P31350; Q8N720 EC number 1.17.4.1 Uniprot keywords 3D-structure; Acetylation; Allosteric enzyme; ATP-binding; Cytoplasm; Deoxyribonucleotide synthesis; Disulfide bond; Nucleotide-binding; Oxidoreductase; Phosphoprotein; Primary mitochondrial disease; Progressive external ophthalmoplegia; Proteomics identification; Reference proteome Protein physicochemical properties Chain ID A,B Molecular weight (Da) 147968 Length 1301 Aromaticity 0.1 Instability index 43.58 Isoelectric point 7.14 Charge (pH=7) 0.86 2D Binding mode Binding energy (Kcal/mol) -9.46 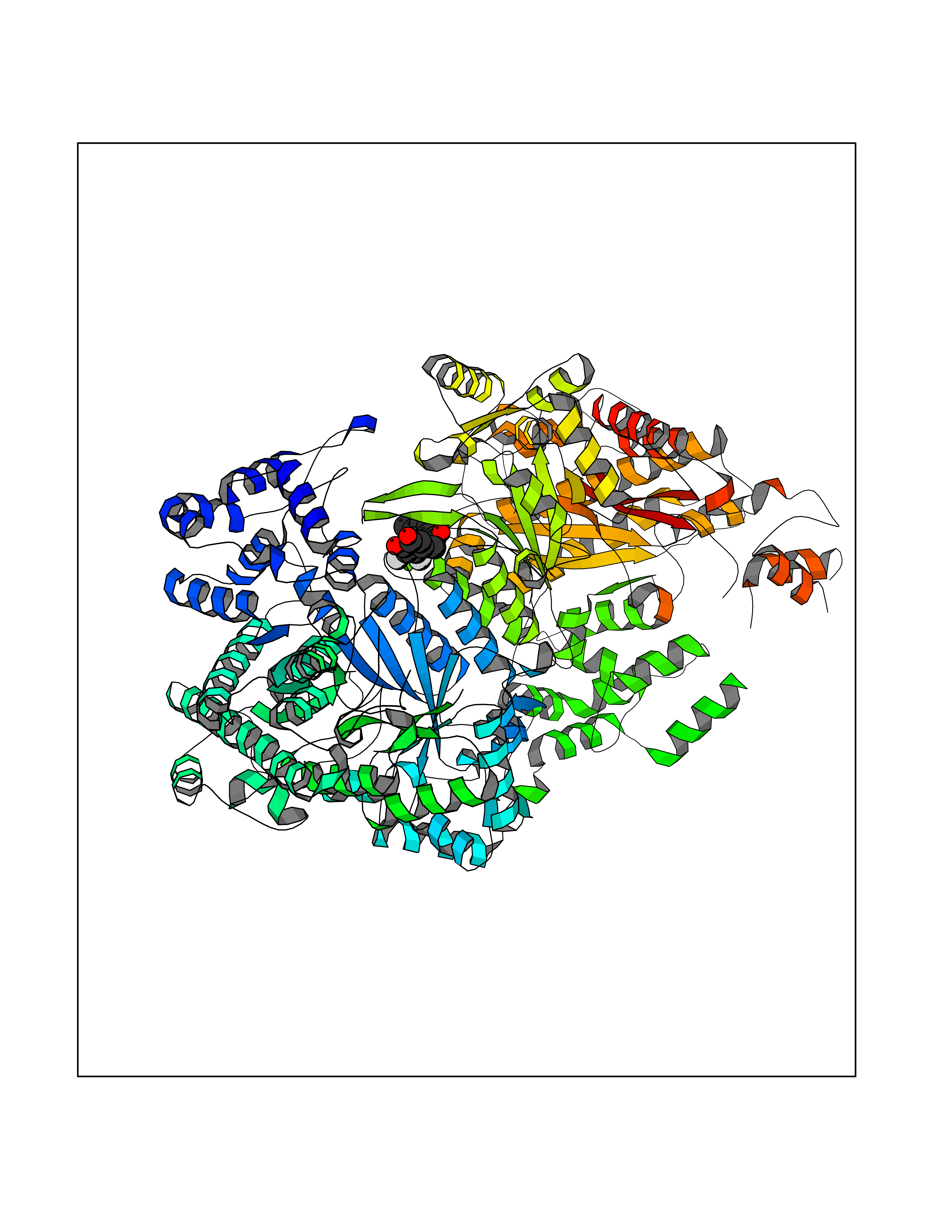 Molscript Map 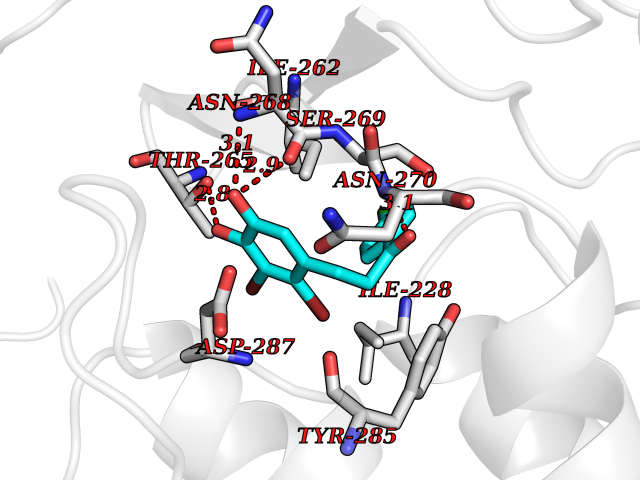 Pymol Map 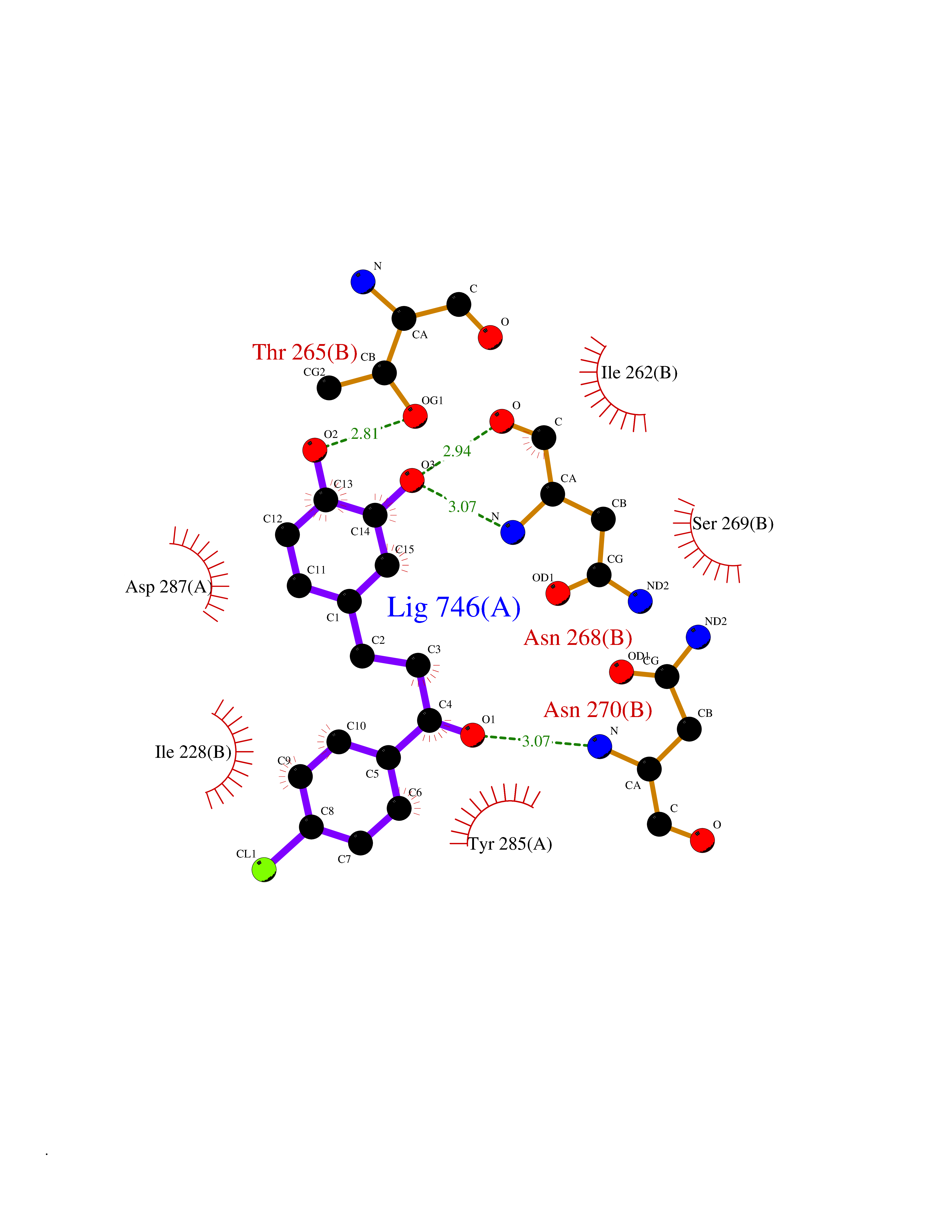 Ligplot Map 3D Binding mode Sequence LAARIAVSNLHKETKKVFSDVMEDLYNYINPHNGKHSPMVAKSTLDIVLANKDRLNSAIIYDRDFSYNYFGFKTLERSYLLKINGKVAERPQHMLMRVSVGIHKEDIDAAIETYNLLSERWFTHASPTLFNAGTNRPQLSSCFLLSMKDDSIEGIYDTLKQCALISKSAGGIGVAVSCIRATGSYIAGTNGNSNGLVPMLRVYNNTARYVDQGGNKRPGAFAIYLEPWHLDIFEFLDLKKNTGKEEQRARDLFFALWIPDLFMKRVETNQDWSLMCPNECPGLDEVWGEEFEKLYASYEKQGRVRKVVKAQQLWYAIIESQTETGTPYMLYKDSCNRKSNQQNLGTIKCSNLCTEIVEYTSKDEVAVCNLASLALNMYVTSEHTYDFKKLAEVTKVVVRNLNKIIDINYYPVPEACLSNKRHRPIGIGVQGLADAFILMRYPFESAEAQLLNKQIFETIYYGALEASCDLAKEQGPYETYEGSPVSKGILQYDMWNVTPTDLWDWKVLKEKIAKYGIRNSLLIAPMPTASTAQILGNNESIEPYTSNIYQIVNPHLLKDLTERGLGSIQSIPEIPDDLKQLYKTVWEISQKTVLKMAAERGAFIDQSQSLNIHIAEPNYGKLTSMHFYGWKQGLKTGMYYLRTRAARIAVSNLHKETKKVFSDVMEDLYNYINPHNGKHSPMVAKSTLDIVLANKDRLNSAIIYDRDFSYNYFGFKTLERSYLLKINGKVAERPQHMLMRVSVGIHKEDIDAAIETYNLLSERWFTHASPTLFNAGTNRPQLSSCFLLSMKDDSIEGIYDTLKQCALISKSAGGIGVAVSCIRATGSYIAGTNGNSNGLVPMLRVYNNTARYVDQGGNKRPGAFAIYLEPWHLDIFEFLDLKKNTGKEEQRARDLFFALWIPDLFMKRVETNQDWSLMCPNECPGLDEVWGEEFEKLYASYEKQGRVRKVVKAQQLWYAIIESQTETGTPYMLYKDSCNRKSNQQNLGTIKCSNLCTEIVEYTSKDEVAVCNLASLALNMYVTSEHTYDFKKLAEVTKVVVRNLNKIIDINYYPVPEACLSNKRHRPIGIGVQGLADAFILMRYPFESAEAQLLNKQIFETIYYGALEASCDLAKEQGPYETYEGSPVSKGILQYDMWNVTPTDLWDWKVLKEKIAKYGIRNSLLIAPMPTASTAQILGNNESIEPYTSNIYQIVNPHLLKDLTERGLWEEMKNQIIACNGSIQSIPEIPDDLKQLYKTVWEISQKTVLKMAAERGAFIDQSQSLNIHIAEPNYGKLTSMHFYGWKQGLKTGMYYLRTRAH Hydrogen bonds contact Hydrophobic contact | ||||
| 65 | Dihydroorotate dehydrogenase (DHODH) | 4OQV | 6.93 | |
Target general information Gen name DHODH Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Dihydroorotate oxidase; Dihydroorotate dehydrogenase (quinone), mitochondrial; DHOdehase; DHODH Protein family Dihydroorotate dehydrogenase family, Type 2 subfamily Biochemical class CH-CH donor oxidoreductase Function Catalyzes the conversion of dihydroorotate to orotate with quinone as electron acceptor. Related diseases Postaxial acrofacial dysostosis (POADS) [MIM:263750]: POADS is characterized by severe micrognathia, cleft lip and/or palate, hypoplasia or aplasia of the posterior elements of the limbs, coloboma of the eyelids and supernumerary nipples. POADS is a very rare disorder: only 2 multiplex families, each consisting of 2 affected siblings born to unaffected, nonconsanguineous parents, have been described among a total of around 30 reported cases. {ECO:0000269|PubMed:19915526}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07559; DB07561; DB08172; DB08169; DB07443; DB07978; DB07975; DB04281; DB08249; DB07977; DB07976; DB04583; DB08008; DB01117; DB03523; DB03480; DB02613; DB04147; DB03247; DB01097; DB06481; DB08006; DB02262; DB05125; DB08880; DB07646 Interacts with Q6ZMZ0; P49638 EC number EC 1.3.5.2 Uniprot keywords 3D-structure; Disease variant; Flavoprotein; FMN; Membrane; Mitochondrion; Mitochondrion inner membrane; Oxidoreductase; Proteomics identification; Pyrimidine biosynthesis; Reference proteome; Transit peptide; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 38341.4 Length 353 Aromaticity 0.05 Instability index 39.27 Isoelectric point 9.28 Charge (pH=7) 5.52 2D Binding mode Binding energy (Kcal/mol) -9.46 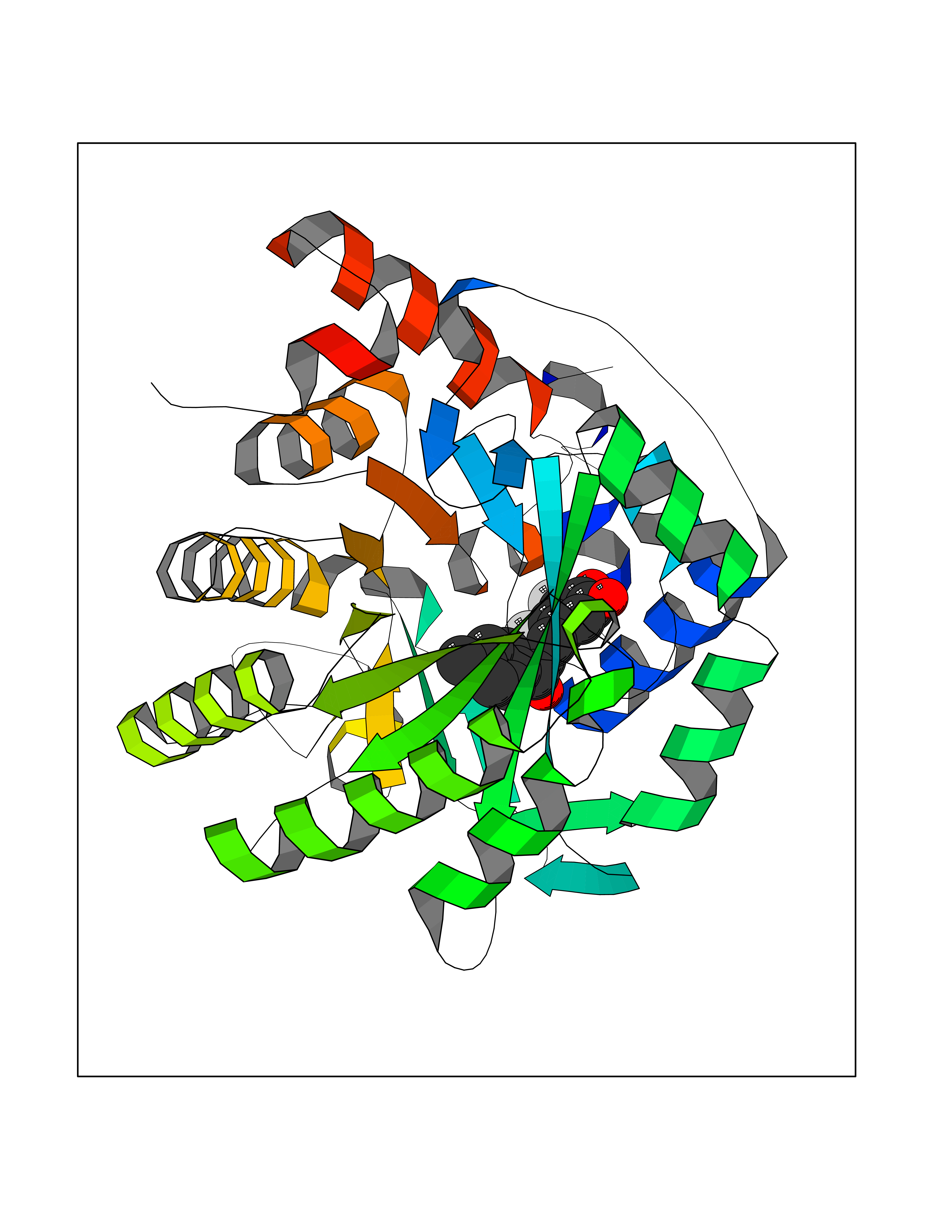 Molscript Map 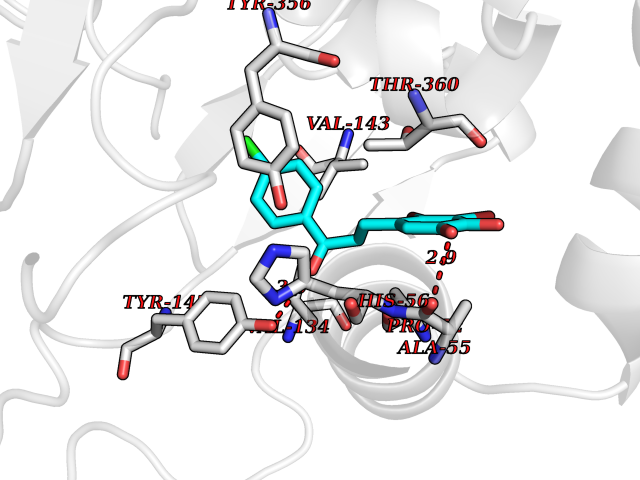 Pymol Map 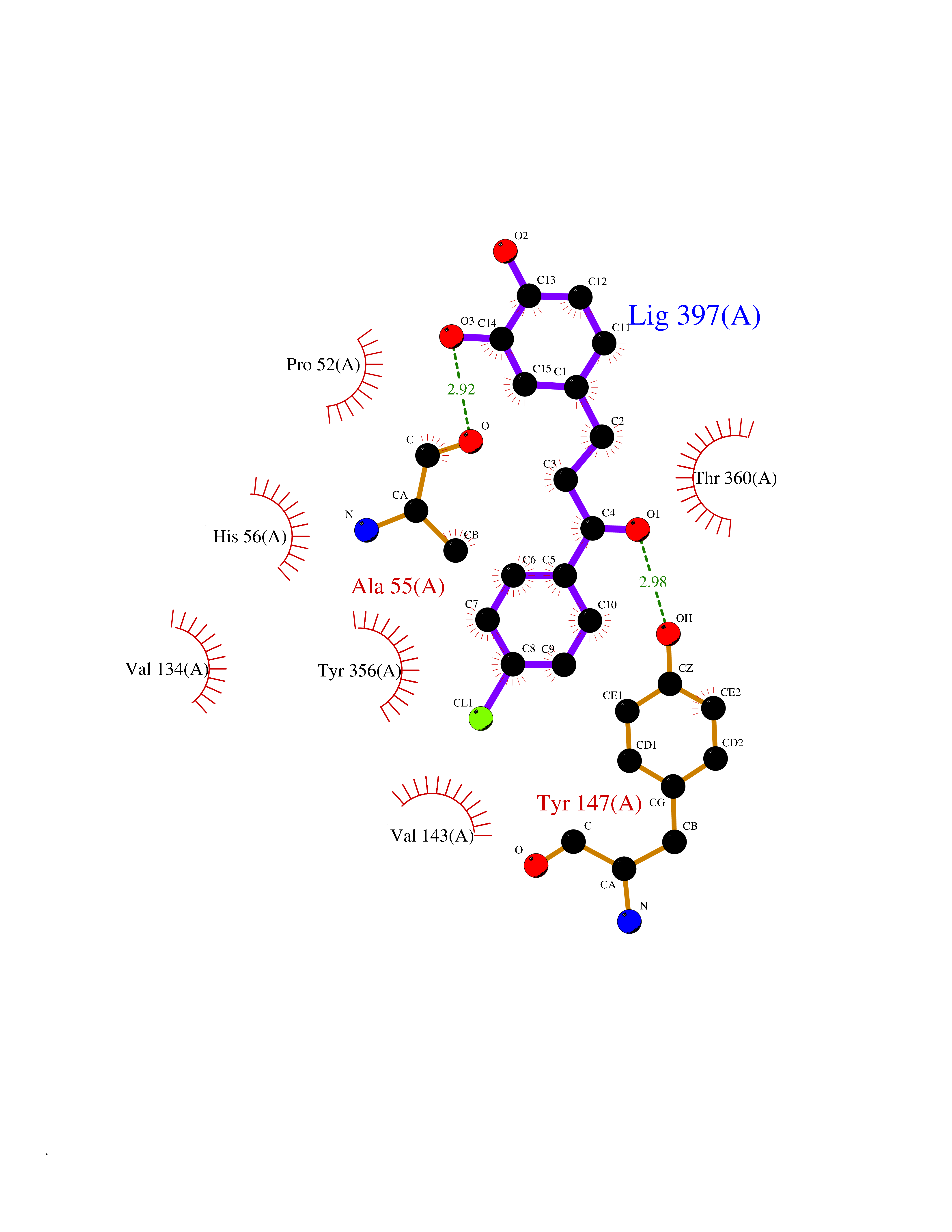 Ligplot Map 3D Binding mode Sequence DERFYAEHLMPTLQGLLDPESAHRLAVRFTSLGLLPRARFQDSDMLEVRVLGHKFRNPVGIAAGFDKHGEAVDGLYKMGFGFVEIGSVTPKPQEGNPRPRVFRLPEDQAVINRYGFNSHGLSVVEHRLRARQQKQAKLTEDGLPLGVNLGKNKTSVDAAEDYAEGVRVLGPLADYLVVNVSSPGKAELRRLLTKVLQERDGLRRVHRPAVLVKIAPDLTSQDKEDIASVVKELGIDGLIVTNTTVSRPAGLQGALRSETGGLSGKPLRDLSTQTIREMYALTQGRVPIIGVGGVSSGQDALEKIRAGASLVQLYTALTFWGPPVVGKVKRELEALLKEQGFGGVTDAIGADHR Hydrogen bonds contact Hydrophobic contact | ||||
| 66 | Glycolipid transfer protein | 3RZN | 6.93 | |
Target general information Gen name GLTP Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family GLTP family Biochemical class Lipid transport Function Glycolipid binding.Glycolipid transporter activity.Identical protein binding.Intermembrane lipid transfer activity.Lipid binding. Related diseases Brugada syndrome 7 (BRGDA7) [MIM:613120]: A tachyarrhythmia characterized by right bundle branch block and ST segment elevation on an electrocardiogram (ECG). It can cause the ventricles to beat so fast that the blood is prevented from circulating efficiently in the body. When this situation occurs, the individual will faint and may die in a few minutes if the heart is not reset. {ECO:0000269|PubMed:20031595}. The gene represented in this entry may be involved in disease pathogenesis.; DISEASE: Atrial fibrillation, familial, 16 (ATFB16) [MIM:613120]: A familial form of atrial fibrillation, a common sustained cardiac rhythm disturbance. Atrial fibrillation is characterized by disorganized atrial electrical activity and ineffective atrial contraction promoting blood stasis in the atria and reduces ventricular filling. It can result in palpitations, syncope, thromboembolic stroke, and congestive heart failure. {ECO:0000269|PubMed:20558140, ECO:0000269|PubMed:21051419}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03600; DB04465; DB03017; DB03203 Interacts with Q96DZ9; Q9NZD2 EC number NA Uniprot keywords 3D-structure; Acetylation; Cytoplasm; Lipid transport; Proteomics identification; Reference proteome; Repeat; Transport Protein physicochemical properties Chain ID A Molecular weight (Da) 23534.1 Length 206 Aromaticity 0.11 Instability index 36.45 Isoelectric point 7.08 Charge (pH=7) 0.1 2D Binding mode Binding energy (Kcal/mol) -9.45 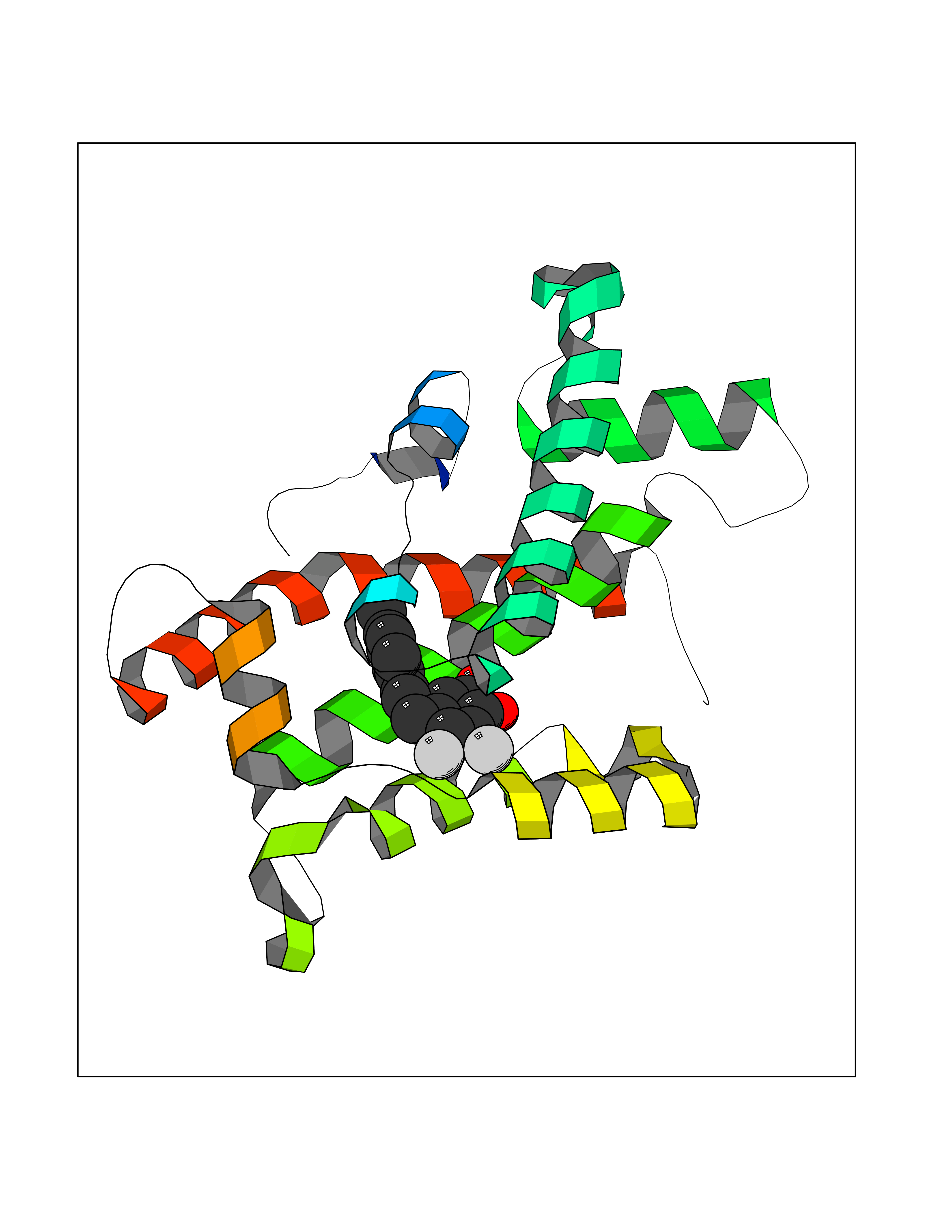 Molscript Map 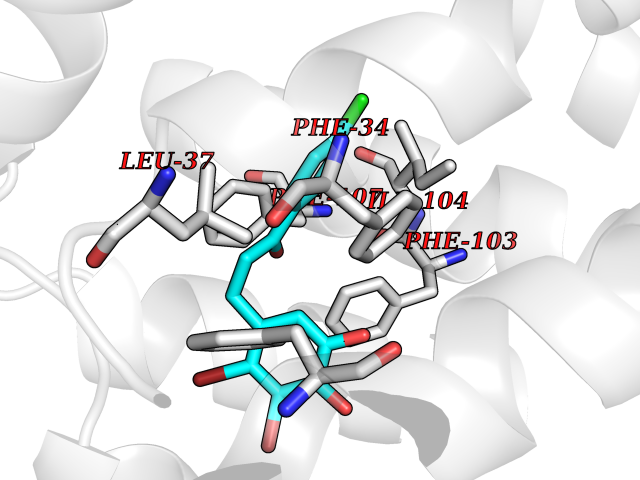 Pymol Map 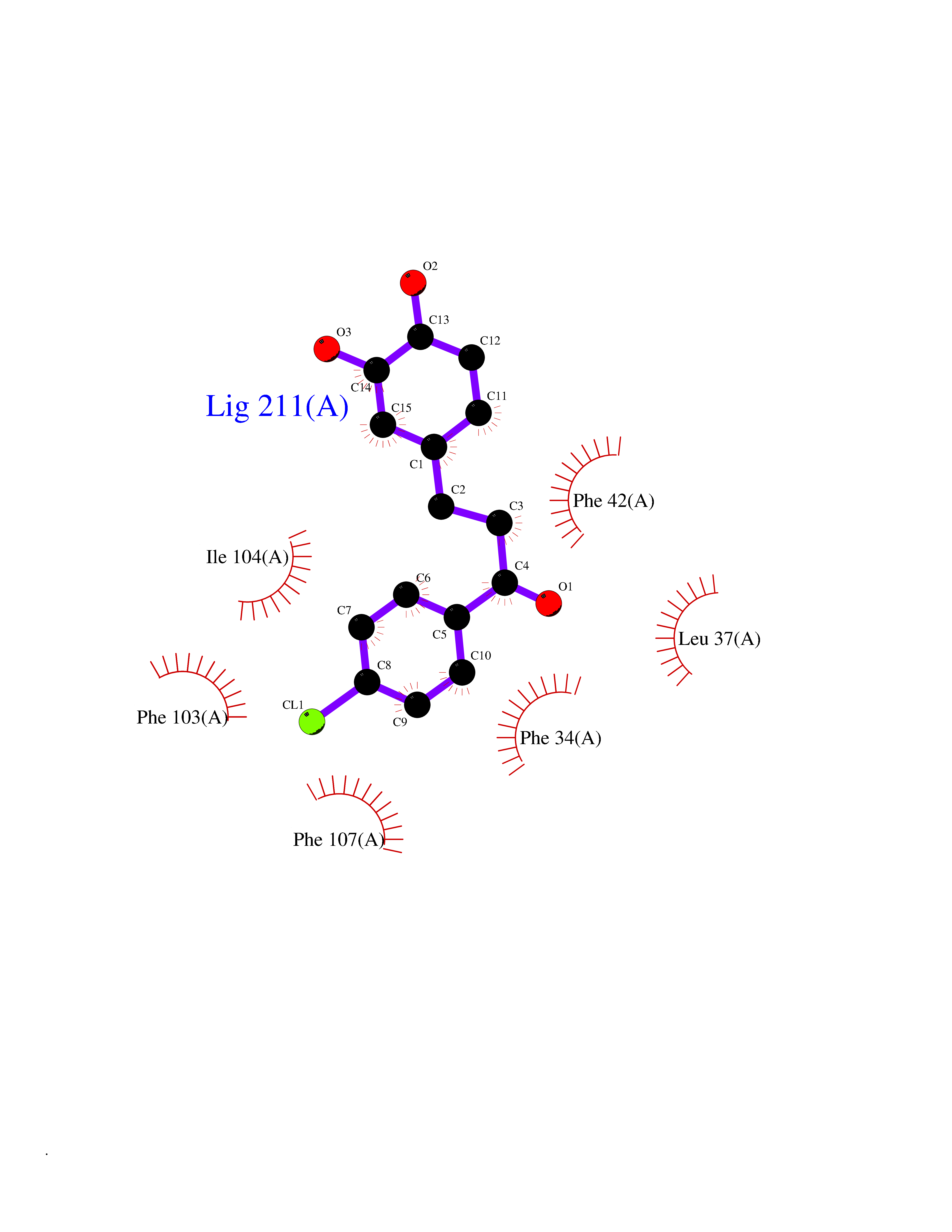 Ligplot Map 3D Binding mode Sequence LAEHLLKPLPADKQIETGPFLEAVSHLPPFFDCLGSPVFTPIKADISGNITKIKAVYDTNPAKFRTLQNILEVEKEMYGAEWPKVGATLALMWLKRGLRFIQVFLQSICDGERDENHPNLIRVNATKAYEMALKKYHGWIVQKIFQAALYAAPYKSDFLKALSKGQNVTEEECLEKIRLFLVNYTATIDVIYEMYTQMNAELNYKV Hydrogen bonds contact Hydrophobic contact | ||||
| 67 | cAMP-dependent protein kinase A type I (PRKAR1A) | 5KJZ | 6.93 | |
Target general information Gen name PRKAR1A Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms cAMP-dependent protein kinase type I-alpha regulatory subunit; Tissue-specific extinguisher 1; TSE1; PRKAR1; PKR1 Protein family CAMP-dependent kinase regulatory chain family Biochemical class Kinase Function Regulatory subunit of the cAMP-dependent protein kinases involved in cAMP signaling in cells. Related diseases Carney complex 1 (CNC1) [MIM:160980]: CNC is a multiple neoplasia syndrome characterized by spotty skin pigmentation, cardiac and other myxomas, endocrine tumors, and psammomatous melanotic schwannomas. {ECO:0000269|PubMed:15371594, ECO:0000269|PubMed:18241045, ECO:0000269|PubMed:22785148, ECO:0000269|PubMed:23323113, ECO:0000269|PubMed:26405036}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Intracardiac myxoma (INTMYX) [MIM:255960]: Inheritance is autosomal recessive. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Primary pigmented nodular adrenocortical disease 1 (PPNAD1) [MIM:610489]: A rare bilateral adrenal defect causing ACTH-independent Cushing syndrome. Macroscopic appearance of the adrenals is characteristic with small pigmented micronodules observed in the cortex. Clinical manifestations of Cushing syndrome include facial and truncal obesity, abdominal striae, muscular weakness, osteoporosis, arterial hypertension, diabetes. PPNAD1 is most often diagnosed in patients with Carney complex, a multiple neoplasia syndrome. However it can also be observed in patients without other manifestations. {ECO:0000269|PubMed:12213893}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Acrodysostosis 1, with or without hormone resistance (ACRDYS1) [MIM:101800]: A form of skeletal dysplasia characterized by short stature, severe brachydactyly, facial dysostosis, and nasal hypoplasia. Affected individuals often have advanced bone age and obesity. Laboratory studies show resistance to multiple hormones, including parathyroid, thyrotropin, calcitonin, growth hormone-releasing hormone, and gonadotropin. However, not all patients show endocrine abnormalities. {ECO:0000269|PubMed:21651393, ECO:0000269|PubMed:22464250, ECO:0000269|PubMed:22464252, ECO:0000269|PubMed:22723333, ECO:0000269|PubMed:23043190, ECO:0000269|PubMed:23425300, ECO:0000269|PubMed:26405036}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01790; DB02527; DB02315; DB05798 Interacts with Q9GZX7; P24588; O43687-2; Q9BSF0; Q9H6J7-2; Q86Y01; P0C7A2-2; Q9H0R8; Q9H8W4; P17612; P31321; P51817; P35250; Q86UC2; Q01105; Q8N0X7; O96006; P03259-2 EC number NA Uniprot keywords 3D-structure; Acetylation; Alternative splicing; cAMP; cAMP-binding; Cell membrane; Cushing syndrome; Direct protein sequencing; Disease variant; Disulfide bond; Membrane; Nucleotide-binding; Phosphoprotein; Proteomics identification; Reference proteome; Repeat Protein physicochemical properties Chain ID A Molecular weight (Da) 17007.4 Length 149 Aromaticity 0.08 Instability index 49.36 Isoelectric point 6.36 Charge (pH=7) -0.52 2D Binding mode Binding energy (Kcal/mol) -9.45 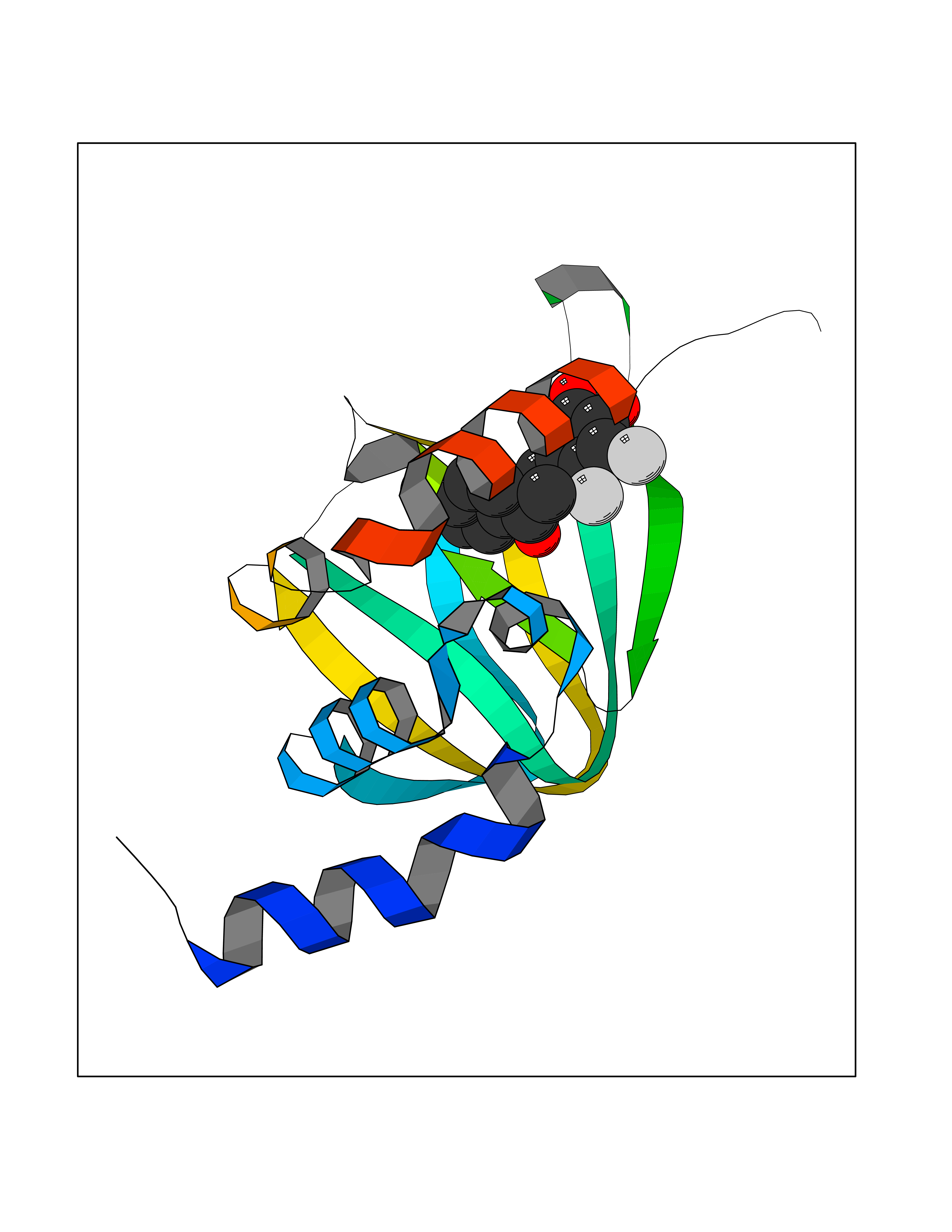 Molscript Map 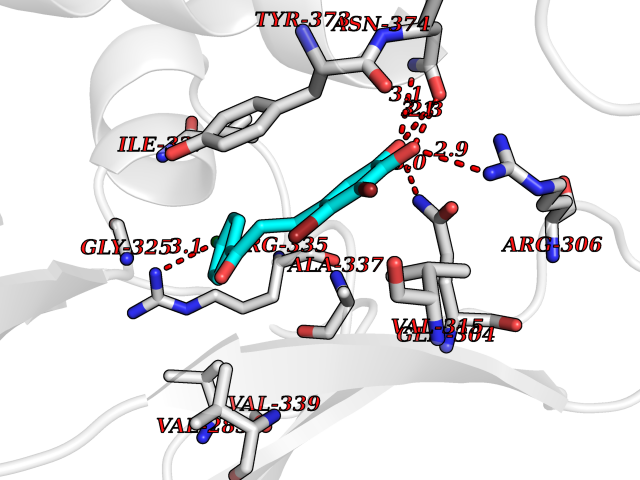 Pymol Map 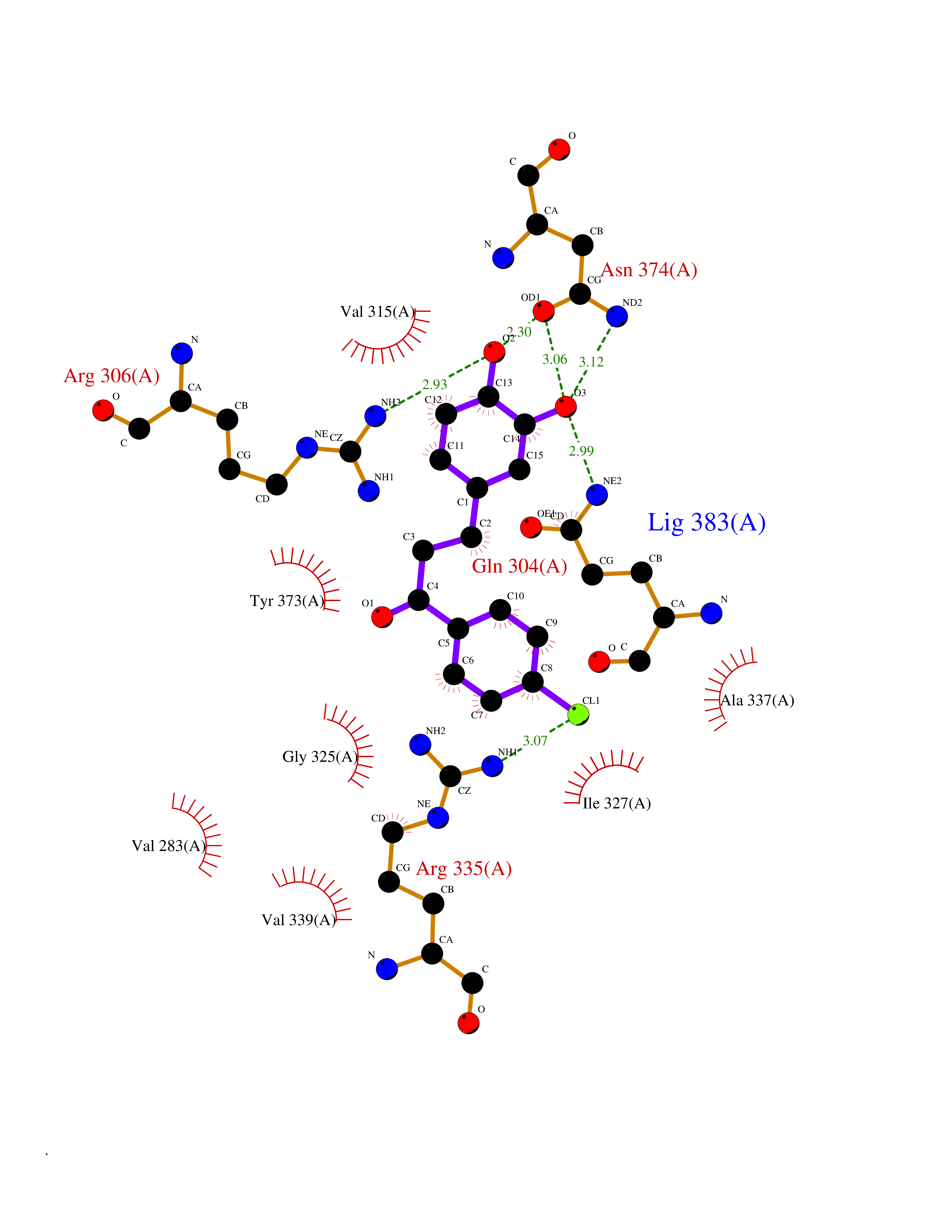 Ligplot Map 3D Binding mode Sequence SILMGSTLRKRKMYEEFLSKVSILESLDKWERLTVADALEPVQFEDGQKIVVQGEPGDEFFIILEGSAAVLQRRSENEEFVEVRRLGPSDYFGEIALLMNRPRTATVVARGPLKCVKLDRPRFERVLGPCSDILKRNIQQYNSFVSLSV Hydrogen bonds contact Hydrophobic contact | ||||
| 68 | Histone-lysine N-methyltransferase SMYD3 (SMYD3) | 6P7Z | 6.93 | |
Target general information Gen name SMYD3 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms SET and MYND domain-containing protein 3; Zinc finger MYND domain-containing protein 1 Protein family Class V-like SAM-binding methyltransferase superfamily, Histone-lysine methyltransferase family Biochemical class NA Function Histone methyltransferase. Specifically methylates 'Lys-4' of histone H3, inducing di- and tri-methylation, but not monomethylation . Also methylates 'Lys-5' of histone H4. Plays an important role in transcriptional activation as a member of an RNA polymerase complex. Binds DNA containing 5'-CCCTCC-3' or 5'-GAGGGG-3' sequences. Related diseases Leukemia, juvenile myelomonocytic (JMML) [MIM:607785]: An aggressive pediatric myelodysplastic syndrome/myeloproliferative disorder characterized by malignant transformation in the hematopoietic stem cell compartment with proliferation of differentiated progeny. Patients have splenomegaly, enlarged lymph nodes, rashes, and hemorrhages. {ECO:0000269|PubMed:17332249}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Noonan syndrome 6 (NS6) [MIM:613224]: A form of Noonan syndrome, a disease characterized by short stature, facial dysmorphic features such as hypertelorism, a downward eyeslant and low-set posteriorly rotated ears, and a high incidence of congenital heart defects and hypertrophic cardiomyopathy. Other features can include a short neck with webbing or redundancy of skin, deafness, motor delay, variable intellectual deficits, multiple skeletal defects, cryptorchidism, and bleeding diathesis. Individuals with Noonan syndrome are at risk of juvenile myelomonocytic leukemia, a myeloproliferative disorder characterized by excessive production of myelomonocytic cells. {ECO:0000269|PubMed:19966803}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: RAS-associated autoimmune leukoproliferative disorder (RALD) [MIM:614470]: A disorder of apoptosis, characterized by chronic accumulation of non-malignant lymphocytes, defective lymphocyte apoptosis, and an increased risk for the development of hematologic malignancies. {ECO:0000269|PubMed:17517660}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Melanocytic nevus syndrome, congenital (CMNS) [MIM:137550]: A syndrome characterized by congenital pigmentary skin lesions which can occur at any site and can cover most of the body surface. These lesions may or may not be hairy. Congenital melanocytic nevi are associated with neuromelanosis (the presence of melanin-producing cells within the brain parenchyma or leptomeninges). Less commonly they are associated with malignant melanoma in childhood, both in the skin and the central nervous system. CMNS patients also tend to have a characteristic facial appearance, including wide or prominent forehead, periorbital fullness, small short nose with narrow nasal bridge, round face, full cheeks, prominent premaxilla, and everted lower lip. {ECO:0000269|PubMed:18633438, ECO:0000269|PubMed:23392294}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Melanosis, neurocutaneous (NCMS) [MIM:249400]: A rare congenital disease characterized by the presence of giant or multiple melanocytic nevi on the skin, foci of melanin-producing cells within the brain parenchyma, and infiltration of leptomeninges by abnormal melanin deposits. Neurologic abnormalities include seizures, hydrocephalus, arachnoid cysts, tumors, and syringomyelia. Some patients may develop malignant melanoma. {ECO:0000269|PubMed:23392294}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Keratinocytic non-epidermolytic nevus (KNEN) [MIM:162900]: Epidermal nevi of the common, non-organoid and non-epidermolytic type are benign skin lesions and may vary in their extent from a single (usually linear) lesion to widespread and systematized involvement. They may be present at birth or develop early during childhood. {ECO:0000269|PubMed:22499344}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Thyroid cancer, non-medullary, 2 (NMTC2) [MIM:188470]: A form of non-medullary thyroid cancer (NMTC), a cancer characterized by tumors originating from the thyroid follicular cells. NMTCs represent approximately 95% of all cases of thyroid cancer and are classified into papillary, follicular, Hurthle cell, and anaplastic neoplasms. {ECO:0000269|PubMed:12727991}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q9H0L4; Q9H0I2; Q13064; Q7Z3B4; Q16512; Q92529; Q15915; Q9Y2U5 EC number EC 2.1.1.354 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Chromatin regulator; Cytoplasm; Metal-binding; Methyltransferase; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; S-adenosyl-L-methionine; Transferase; Zinc; Zinc-finger Protein physicochemical properties Chain ID A Molecular weight (Da) 48694.1 Length 425 Aromaticity 0.08 Instability index 44.91 Isoelectric point 6.86 Charge (pH=7) -0.4 2D Binding mode Binding energy (Kcal/mol) -9.46 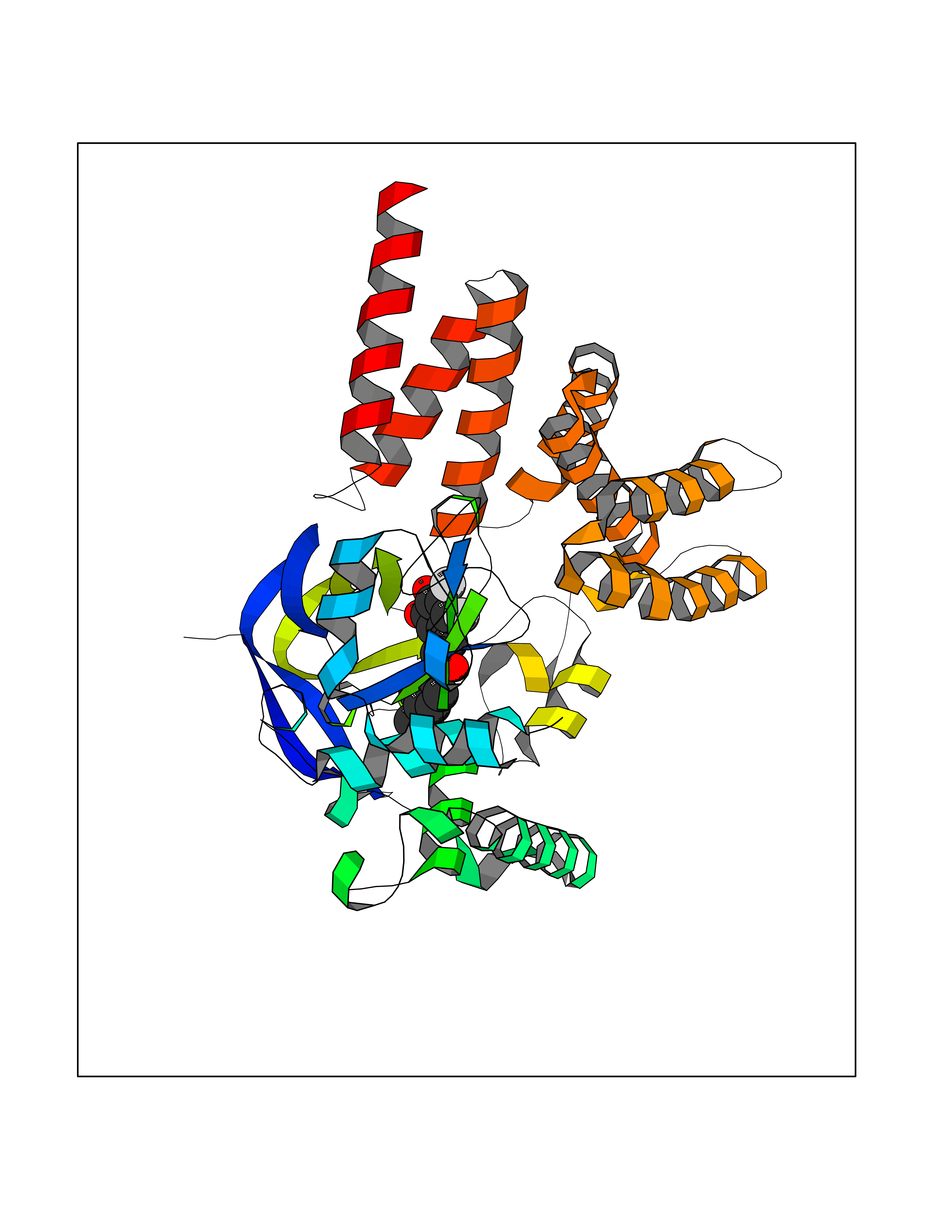 Molscript Map 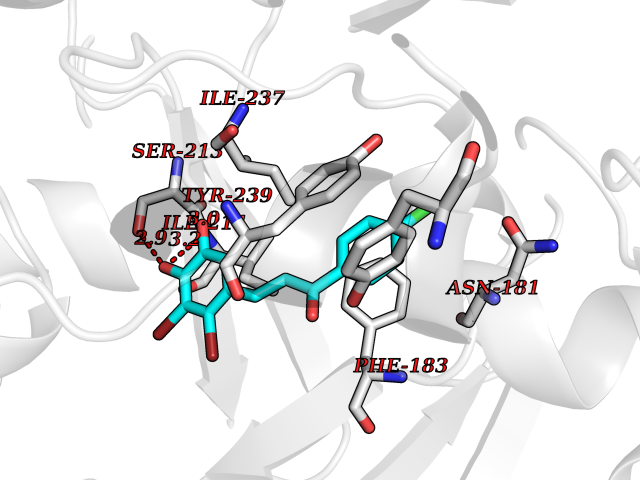 Pymol Map 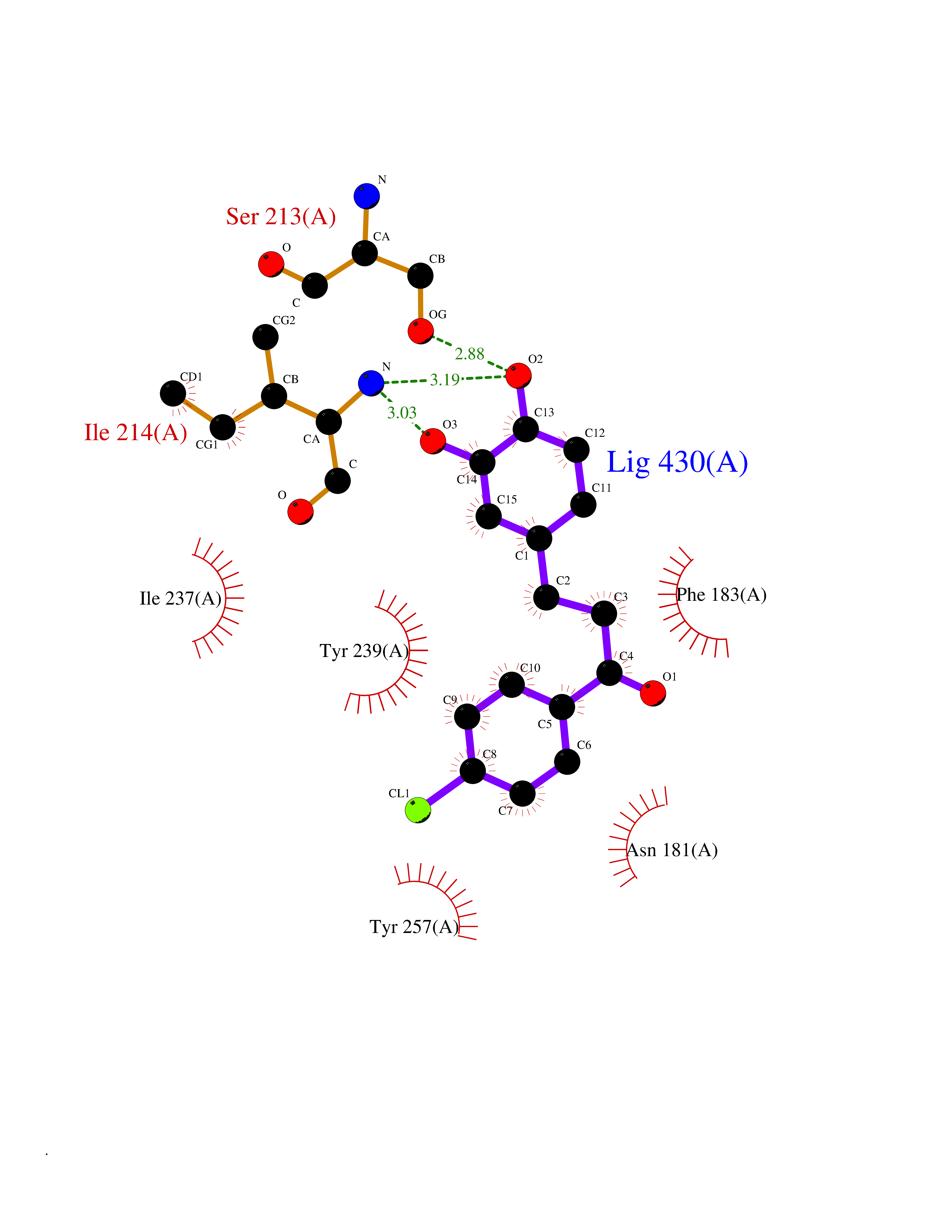 Ligplot Map 3D Binding mode Sequence PLKVEKFATANRGNGLRAVTPLRPGELLFRSDPLAYTVCKGSRGVVCDRCLLGKEKLMRCSQCRVAKYCSAKCQKKAWPDHKRECKCLKSCPRYPPDSVRLLGRVVFKLMDGAPSESEKLYSFYDLESNINKLTEDKKEGLRQLVMTFQHFMREEIQDASQLPPAFDLFEAFAKVICNSFTICNAEMQEVGVGLYPSISLLNHSCDPNCSIVFNGPHLLLRAVRDIEVGEELTICYLDMLMTSEERRKQLRDQYCFECDCFRCQTQDKDADMLTGDEQVWKEVQESLKKIEELKAHWKWEQVLAMCQAIISSNSERLPDINIYQLKVLDCAMDACINLGLLEEALFYGTRTMEPYRIFFPGSHPVRGVQVMKVGKLQLHQGMFPQAMKNLRLAFDIMRVTHGREHSLIEDLILLLEECDANIRAS Hydrogen bonds contact Hydrophobic contact | ||||
| 69 | Programmed cell death 1 ligand 1 (PD-L1) | 5J89 | 6.92 | |
Target general information Gen name CD274 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms hPD-L1; Programmed death ligand 1; PDL1; PDCD1LG1; PDCD1L1; PDCD1 ligand 1; B7H1; B7-H1; B7 homolog 1 Protein family Immunoglobulin superfamily, BTN/MOG family Biochemical class Immunoglobulin Function As a ligand for the inhibitory receptor PDCD1/PD-1, modulates the activation threshold of T-cells and limits T-cell effector response. Through a yet unknown activating receptor, may costimulate T-cell subsets that predominantly produce interleukin-10 (IL10). Plays a critical role in induction and maintenance of immune tolerance to self. Related diseases Truncation of the 3'-untranslated (3'-UTR) region of CD274 transcripts leads to elevated expression of CD274 in multiple cancers including T-cell leukemia, diffuse large B-cell lymphoma and stomach adenocarcinoma (PubMed:27281199). Disruption of 3'-UTR region is caused by structural variants that stabilize CD274 transcripts, leading to overexpression (PubMed:27281199). Increased expression in tumors promotes immune evasion and tumor cell growth by allowing malignant cells to escape destruction by the immune system (PubMed:27281199). {ECO:0000269|PubMed:27281199}. Drugs (DrugBank ID) DB15773; DB11595; DB15771; DB11945; DB15772; DB14776; DB15770; DB11714; DB15769; DB09035; DB09037; DB00203; DB00313 Interacts with P33681; Q8IZR5; Q9NX76; Q15116; Q15116 EC number NA Uniprot keywords 3D-structure; Adaptive immunity; Alternative splicing; Cell membrane; Disulfide bond; Endosome; Glycoprotein; Immunity; Immunoglobulin domain; Membrane; Nucleus; Proteomics identification; Receptor; Reference proteome; Repeat; Secreted; Signal; Transmembrane; Transmembrane helix; Ubl conjugation Protein physicochemical properties Chain ID C,D Molecular weight (Da) 28335.2 Length 249 Aromaticity 0.1 Instability index 35.39 Isoelectric point 6.15 Charge (pH=7) -3.43 2D Binding mode Binding energy (Kcal/mol) -9.44  Molscript Map 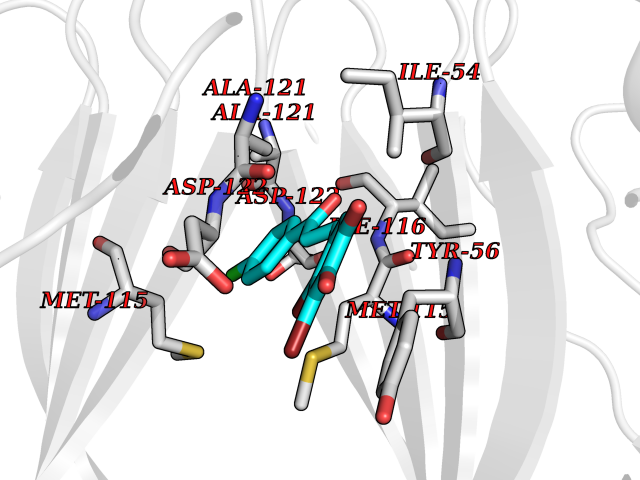 Pymol Map 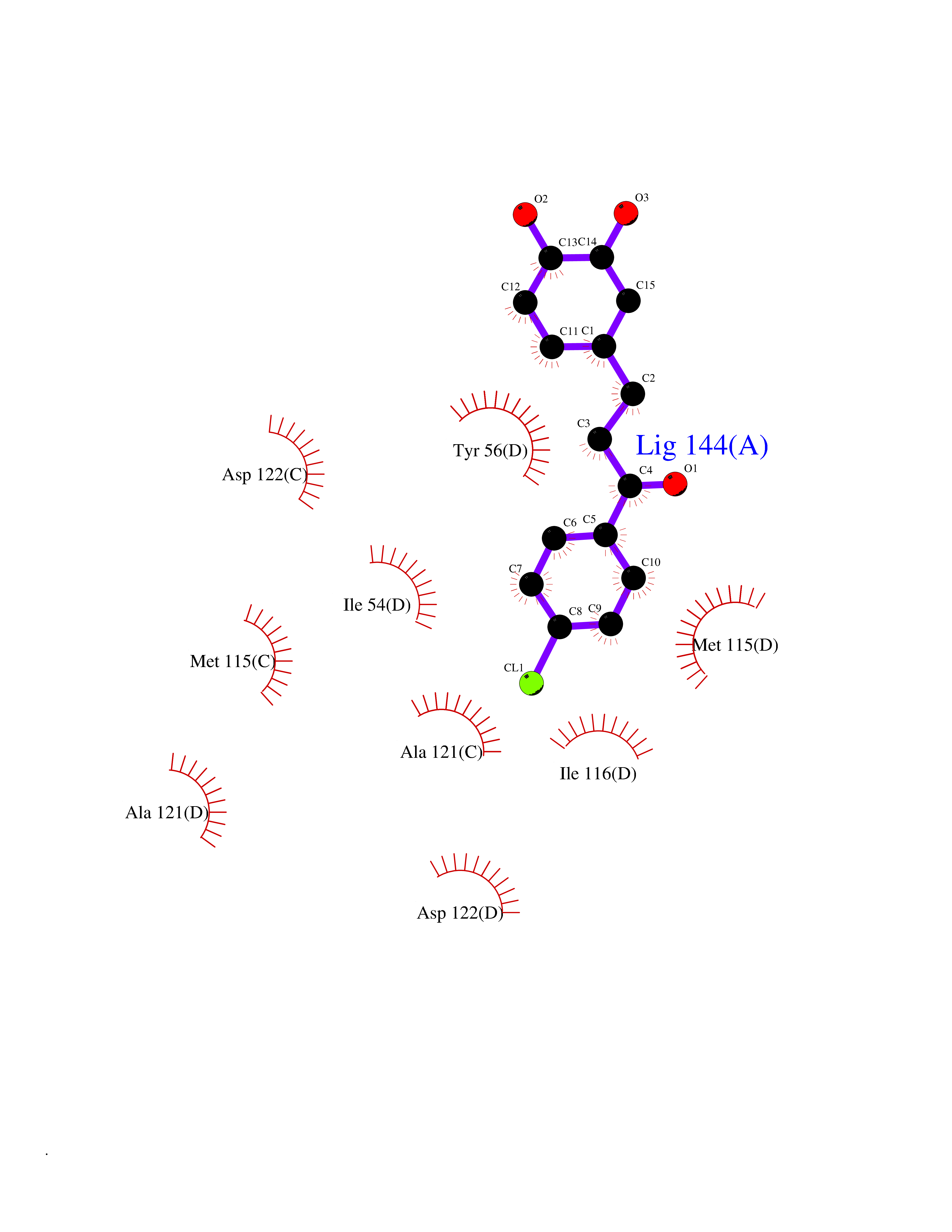 Ligplot Map 3D Binding mode Sequence AFTVTVPKDLYVVEYGSNMTIECKFPVEKQLDLAALIVYWEMEDKNIIQFVHGEEDLKVQHSSYRQRARLLKDQLSLGNAALQITDVKLQDAGVYRCMISYGGADYKRITVKVNAPYAAALEHHHAFTVTVPKDLYVVEYGSNMTIECKFPVEKQLDLAALIVYWEMEDKNIIQFVHGEEDLKVQHSSYRQRARLLKDQLSLGNAALQITDVKLQDAGVYRCMISYGGADYKRITVKVNAPYAAALEHH Hydrogen bonds contact Hydrophobic contact | ||||
| 70 | Matrix metalloproteinase-9 (MMP-9) | 4XCT | 6.92 | |
Target general information Gen name MMP9 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Matrix metalloproteinase 9; GELB; CLG4B; 92 kDa type IV collagenase; 92 kDa gelatinase Protein family Peptidase M10A family Biochemical class Peptidase Function Could play a role in bone osteoclastic resorption. Cleaves KiSS1 at a Gly-|-Leu bond. Cleaves type IV and type V collagen into large C-terminal three quarter fragments and shorter N-terminal one quarter fragments. Degrades fibronectin but not laminin or Pz-peptide. May play an essential role in local proteolysis of the extracellular matrix and in leukocyte migration. Related diseases Intervertebral disc disease (IDD) [MIM:603932]: A common musculo-skeletal disorder caused by degeneration of intervertebral disks of the lumbar spine. It results in low-back pain and unilateral leg pain. {ECO:0000269|PubMed:18455130}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Metaphyseal anadysplasia 2 (MANDP2) [MIM:613073]: A bone development disorder characterized by skeletal anomalies that resolve spontaneously with age. Clinical characteristics are evident from the first months of life and include slight shortness of stature and a mild varus deformity of the legs. Patients attain a normal stature in adolescence and show improvement or complete resolution of varus deformity of the legs and rhizomelic micromelia. {ECO:0000269|PubMed:19615667}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07246; DB07285; DB01949; DB03683; DB07117; DB01197; DB06423; DB00143; DB00786; DB01017; DB05387; DB12843; DB05495; DB01593; DB14487; DB14533; DB14548 Interacts with Q16819; Q16820; P14780; Q8IX30; P13611; Q9ZFS6 EC number EC 3.4.24.35 Uniprot keywords 3D-structure; Calcium; Collagen degradation; Direct protein sequencing; Disulfide bond; Extracellular matrix; Glycoprotein; Hydrolase; Metal-binding; Metalloprotease; Protease; Proteomics identification; Reference proteome; Repeat; Secreted; Signal; Zinc; Zymogen Protein physicochemical properties Chain ID A Molecular weight (Da) 17550.3 Length 157 Aromaticity 0.14 Instability index 27.98 Isoelectric point 5.12 Charge (pH=7) -11.26 2D Binding mode Binding energy (Kcal/mol) -9.44  Molscript Map 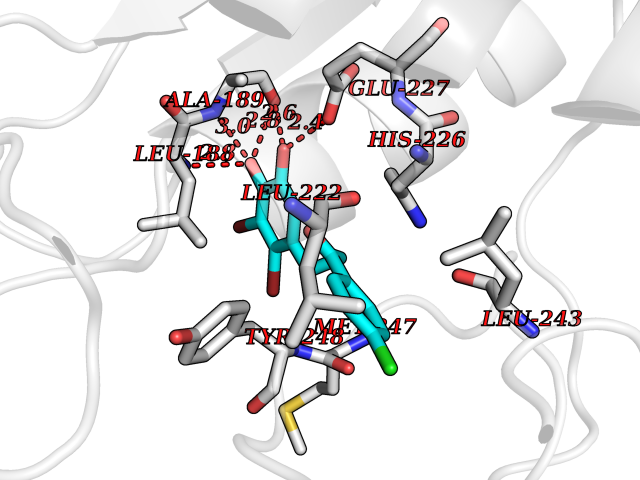 Pymol Map 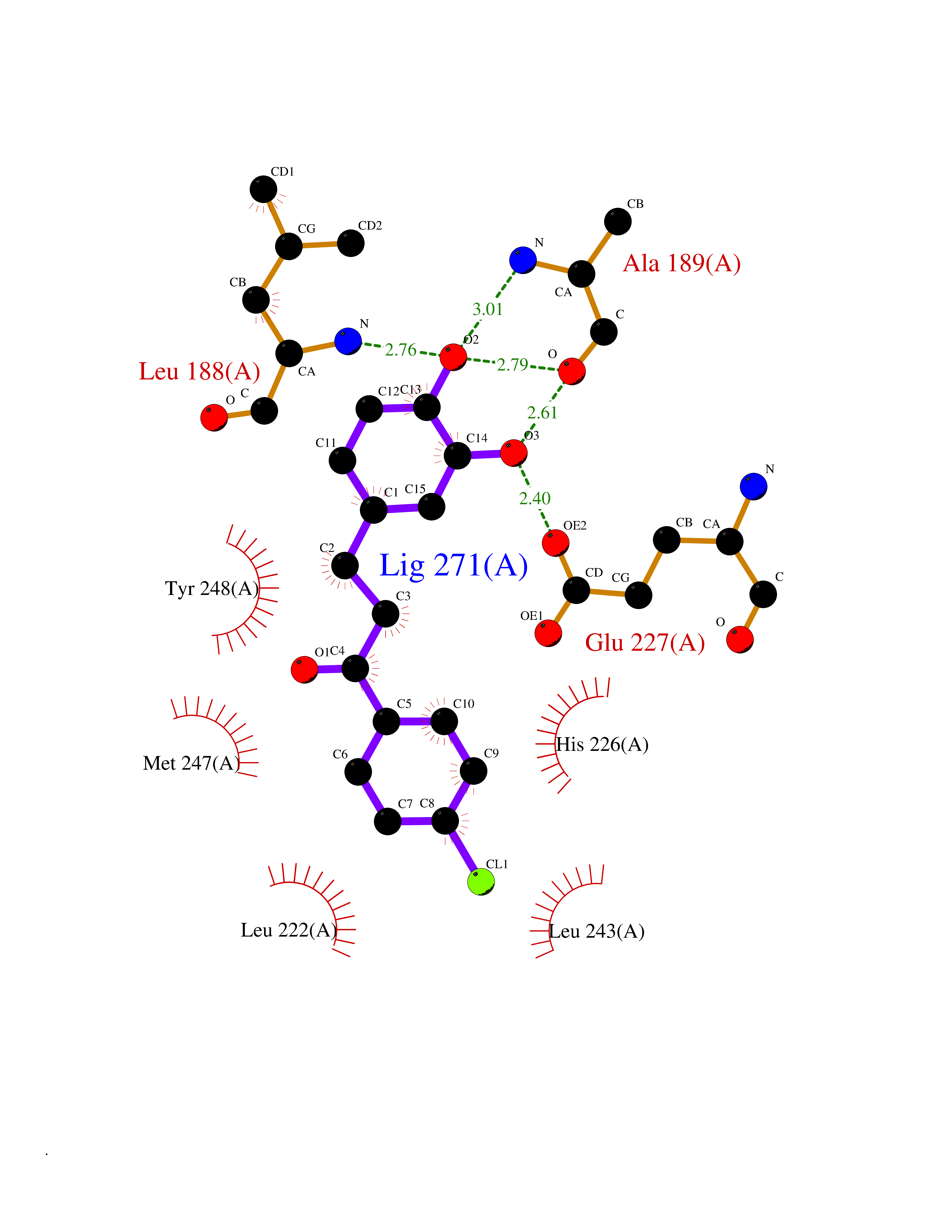 Ligplot Map 3D Binding mode Sequence DLKWHHHNITYWIQNYSEDLPRAVIDDAFARAFALWSAVTPLTFTRVYSRDADIVIQFGVAEHGDGYPFDGKDGLLAHAFPPGPGIQGDAHFDDDELWSLGKGVGYSLFLVAAHEFGHALGLDHSSVPEALMYPMYRFTEGPPLHKDDVNGIRHLYG Hydrogen bonds contact Hydrophobic contact | ||||
| 71 | Vitamin D3 receptor (VDR) | 3B0T | 6.92 | |
Target general information Gen name VDR Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Vitamin D(3) receptor; Nuclear vitamin D receptor; Nuclear receptor subfamily 1 group I member 1; NR1I1; 1,25-dihydroxyvitamin D3 receptor Protein family Nuclear hormone receptor family, NR1 subfamily Biochemical class Nuclear hormone receptor Function Enters the nucleus upon vitamin D3 binding where it forms heterodimers with the retinoid X receptor/RXR. The VDR-RXR heterodimers bind to specific response elements on DNA and activate the transcription of vitamin D3-responsive target genes. Plays a central role in calcium homeostasis. Nuclear receptor for calcitriol, the active form of vitamin D3 which mediates the action of this vitamin on cells. Related diseases Rickets vitamin D-dependent 2A (VDDR2A) [MIM:277440]: A disorder of vitamin D metabolism resulting in severe rickets, hypocalcemia and secondary hyperparathyroidism. Most patients have total alopecia in addition to rickets. {ECO:0000269|PubMed:1652893, ECO:0000269|PubMed:17970811, ECO:0000269|PubMed:2177843, ECO:0000269|PubMed:2849209, ECO:0000269|PubMed:28698609, ECO:0000269|PubMed:7828346, ECO:0000269|PubMed:8106618, ECO:0000269|PubMed:8381803, ECO:0000269|PubMed:8392085, ECO:0000269|PubMed:8675579, ECO:0000269|PubMed:8961271, ECO:0000269|PubMed:9005998}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07530; DB08742; DB01436; DB04891; DB00146; DB02300; DB00136; DB00169; DB04540; DB05024; DB11672; DB14635; DB01070; DB06410; DB05295; DB06194; DB00153; DB04796; DB03451; DB00910; DB04258; DB11094 Interacts with P35222; Q09472; Q15648; P50222; Q15788; P26045; P19793; Q13573; Q13501; P04637; Q15645; Q9JLI4; P28700; X5D778; Q96HA8; Q01804; Q96S38; P48443 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; Disease variant; DNA-binding; Metal-binding; Nucleus; Proteomics identification; Receptor; Reference proteome; Transcription; Transcription regulation; Ubl conjugation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A Molecular weight (Da) 28781 Length 254 Aromaticity 0.07 Instability index 47.69 Isoelectric point 6.15 Charge (pH=7) -3.44 2D Binding mode Binding energy (Kcal/mol) -9.44  Molscript Map 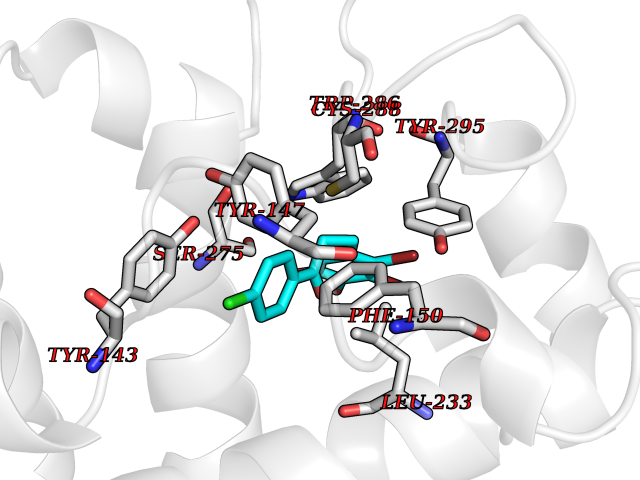 Pymol Map 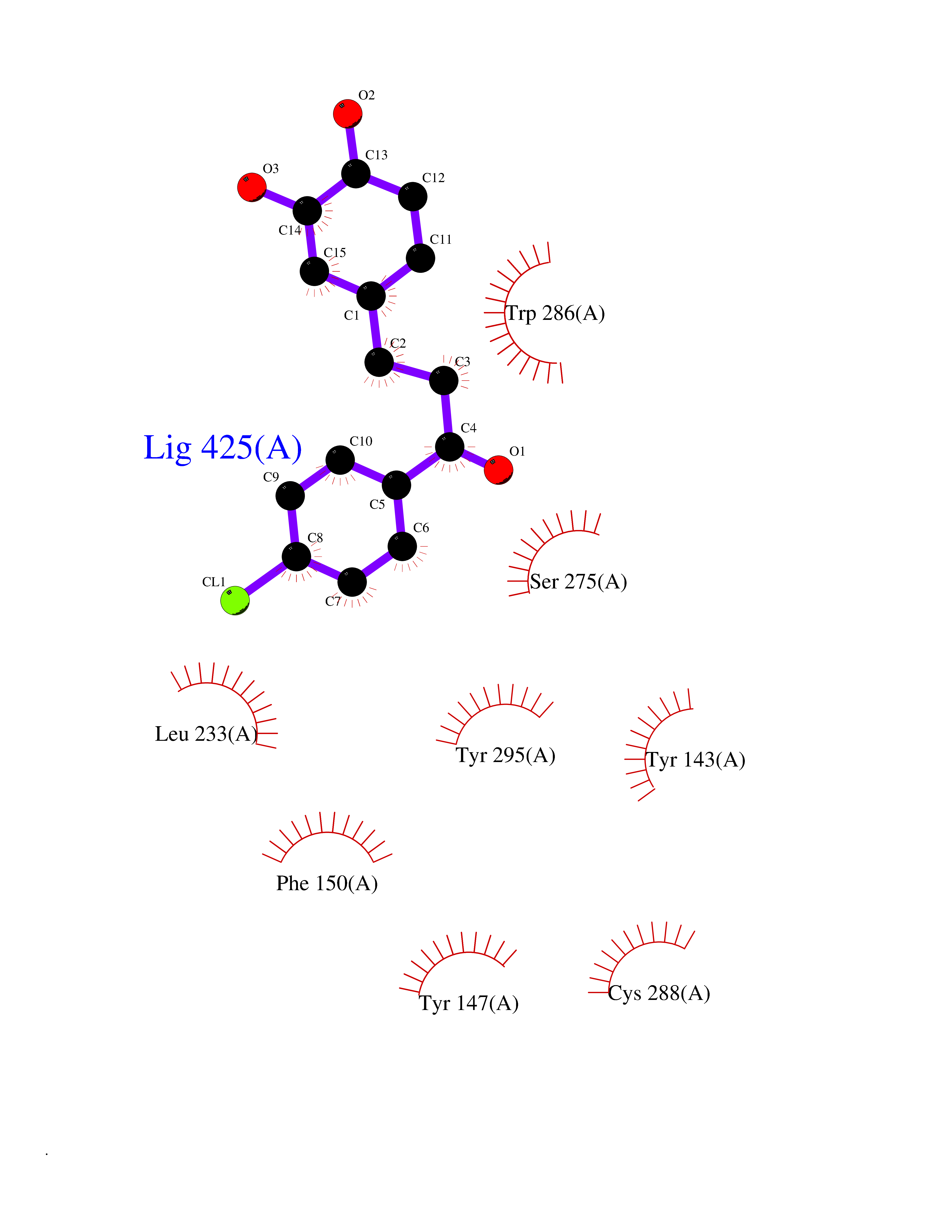 Ligplot Map 3D Binding mode Sequence ALRPKLSEEQQRIIAILLDAHHKTYDPTYSDFCQFRPPVRVNDGGGSVTLELSQLSMLPHLADLVSYSIQKVIGFAKMIPGFRDLTSEDQIVLLKSSAIEVIMLRSNESFTMDDMSWTCGNQDYKYRVSDVTKAGHSLELIEPLIKFQVGLKKLNLHEEEHVLLMAICIVSPDRPGVQDAALIEAIQDRLSNTLQTYIRCRHPPPGSHLLYAKMIQKLADLRSLNEEHSKQYRCLSFQPECSMKLTPLVLEVFG Hydrogen bonds contact Hydrophobic contact | ||||
| 72 | Androgen receptor (AR) | 2AM9 | 6.92 | |
Target general information Gen name AR Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Testosterone receptor; Nuclear receptor subfamily 3 group C member 4; NR3C4; Dihydrotestosterone receptor; DHTR Protein family Nuclear hormone receptor family, NR3 subfamily Biochemical class Nuclear hormone receptor Function Transcription factor activity is modulated by bound coactivator and corepressor proteins like ZBTB7A that recruits NCOR1 and NCOR2 to the androgen response elements/ARE on target genes, negatively regulating androgen receptor signaling and androgen-induced cell proliferation. Transcription activation is also down-regulated by NR0B2. Activated, but not phosphorylated, by HIPK3 and ZIPK/DAPK3. Steroid hormone receptors are ligand-activated transcription factors that regulate eukaryotic gene expression and affect cellular proliferation and differentiation in target tissues. Related diseases Androgen insensitivity syndrome (AIS) [MIM:300068]: An X-linked recessive form of pseudohermaphroditism due end-organ resistance to androgen. Affected males have female external genitalia, female breast development, blind vagina, absent uterus and female adnexa, and abdominal or inguinal testes, despite a normal 46,XY karyotype. {ECO:0000269|PubMed:10022458, ECO:0000269|PubMed:10221692, ECO:0000269|PubMed:10221770, ECO:0000269|PubMed:10404311, ECO:0000269|PubMed:10458483, ECO:0000269|PubMed:10571951, ECO:0000269|PubMed:10590024, ECO:0000269|PubMed:10690872, ECO:0000269|PubMed:11587068, ECO:0000269|PubMed:11744994, ECO:0000269|PubMed:1307250, ECO:0000269|PubMed:1316540, ECO:0000269|PubMed:1426313, ECO:0000269|PubMed:1430233, ECO:0000269|PubMed:1464650, ECO:0000269|PubMed:14756668, ECO:0000269|PubMed:1480178, ECO:0000269|PubMed:1487249, ECO:0000269|PubMed:1569163, ECO:0000269|PubMed:1609793, ECO:0000269|PubMed:16129672, ECO:0000269|PubMed:16595706, ECO:0000269|PubMed:1775137, ECO:0000269|PubMed:1999491, ECO:0000269|PubMed:2082179, ECO:0000269|PubMed:2594783, ECO:0000269|PubMed:7537149, ECO:0000269|PubMed:7581399, ECO:0000269|PubMed:7633398, ECO:0000269|PubMed:7641413, ECO:0000269|PubMed:7671849, ECO:0000269|PubMed:7929841, ECO:0000269|PubMed:7962294, ECO:0000269|PubMed:7970939, ECO:0000269|PubMed:7981687, ECO:0000269|PubMed:7981689, ECO:0000269|PubMed:7993455, ECO:0000269|PubMed:8040309, ECO:0000269|PubMed:8096390, ECO:0000269|PubMed:8103398, ECO:0000269|PubMed:8162033, ECO:0000269|PubMed:8224266, ECO:0000269|PubMed:8281140, ECO:0000269|PubMed:8325950, ECO:0000269|PubMed:8339746, ECO:0000269|PubMed:8413310, ECO:0000269|PubMed:8446106, ECO:0000269|PubMed:8626869, ECO:0000269|PubMed:8647313, ECO:0000269|PubMed:8683794, ECO:0000269|PubMed:8723113, ECO:0000269|PubMed:8768864, ECO:0000269|PubMed:8809734, ECO:0000269|PubMed:8830623, ECO:0000269|PubMed:8918984, ECO:0000269|PubMed:8990010, ECO:0000269|PubMed:9001799, ECO:0000269|PubMed:9007482, ECO:0000269|PubMed:9039340, ECO:0000269|PubMed:9106550, ECO:0000269|PubMed:9160185, ECO:0000269|PubMed:9252933, ECO:0000269|PubMed:9255042, ECO:0000269|PubMed:9302173, ECO:0000269|PubMed:9328206, ECO:0000269|PubMed:9544375, ECO:0000269|PubMed:9554754, ECO:0000269|PubMed:9610419, ECO:0000269|PubMed:9627582, ECO:0000269|PubMed:9698822, ECO:0000269|PubMed:9851768, ECO:0000269|PubMed:9856504, ECO:0000269|Ref.116, ECO:0000269|Ref.182}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Spinal and bulbar muscular atrophy X-linked 1 (SMAX1) [MIM:313200]: An X-linked recessive form of spinal muscular atrophy. Spinal muscular atrophy refers to a group of neuromuscular disorders characterized by degeneration of the anterior horn cells of the spinal cord, leading to symmetrical muscle weakness and atrophy. SMAX1 occurs only in men. Age at onset is usually in the third to fifth decade of life, but earlier involvement has been reported. It is characterized by slowly progressive limb and bulbar muscle weakness with fasciculations, muscle atrophy, and gynecomastia. The disorder is clinically similar to classic forms of autosomal spinal muscular atrophy. {ECO:0000269|PubMed:15851746}. The disease is caused by variants affecting the gene represented in this entry. Caused by trinucleotide CAG repeat expansion. In SMAX1 patients the number of Gln ranges from 38 to 62. Longer expansions result in earlier onset and more severe clinical manifestations of the disease.; DISEASE: Prostate cancer, hereditary, X-linked 3 (HPCX3) [MIM:301120]: A condition associated with familial predisposition to cancer of the prostate. Most prostate cancers are adenocarcinomas that develop in the acini of the prostatic ducts. Other rare histopathologic types of prostate cancer that occur in approximately 5% of patients include small cell carcinoma, mucinous carcinoma, prostatic ductal carcinoma, transitional cell carcinoma, squamous cell carcinoma, basal cell carcinoma, adenoid cystic carcinoma (basaloid), signet-ring cell carcinoma and neuroendocrine carcinoma. {ECO:0000269|PubMed:8530589}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Defects in AR may play a role in metastatic prostate cancer. The mutated receptor stimulates prostate growth and metastases development despite of androgen ablation. This treatment can reduce primary and metastatic lesions probably by inducing apoptosis of tumor cells when they express the wild-type receptor. {ECO:0000269|PubMed:10363963, ECO:0000269|PubMed:10569618, ECO:0000269|PubMed:1562539, ECO:0000269|PubMed:16129672, ECO:0000269|PubMed:17311914, ECO:0000269|PubMed:2260966, ECO:0000269|PubMed:25091737, ECO:0000269|PubMed:8187068, ECO:0000269|PubMed:8274409, ECO:0000269|PubMed:8827083}.; DISEASE: Androgen insensitivity, partial (PAIS) [MIM:312300]: A disorder that is characterized by hypospadias, hypogonadism, gynecomastia, genital ambiguity, normal XY karyotype, and a pedigree pattern consistent with X-linked recessive inheritance. Some patients present azoospermia or severe oligospermia without other clinical manifestations. {ECO:0000269|PubMed:10022458, ECO:0000269|PubMed:10221692, ECO:0000269|PubMed:10470409, ECO:0000269|PubMed:10502786, ECO:0000269|PubMed:10543676, ECO:0000269|PubMed:11587068, ECO:0000269|PubMed:1303262, ECO:0000269|PubMed:1307250, ECO:0000269|PubMed:1316540, ECO:0000269|PubMed:1424203, ECO:0000269|PubMed:1430233, ECO:0000269|PubMed:14756668, ECO:0000269|PubMed:2010552, ECO:0000269|PubMed:7581399, ECO:0000269|PubMed:7649358, ECO:0000269|PubMed:7671849, ECO:0000269|PubMed:7909256, ECO:0000269|PubMed:7910529, ECO:0000269|PubMed:7929841, ECO:0000269|PubMed:7970939, ECO:0000269|PubMed:7981687, ECO:0000269|PubMed:8033918, ECO:0000269|PubMed:8097257, ECO:0000269|PubMed:8126121, ECO:0000269|PubMed:8205256, ECO:0000269|PubMed:8281139, ECO:0000269|PubMed:8325932, ECO:0000269|PubMed:8325950, ECO:0000269|PubMed:8446106, ECO:0000269|PubMed:8550758, ECO:0000269|PubMed:8809734, ECO:0000269|PubMed:8823308, ECO:0000269|PubMed:8824883, ECO:0000269|PubMed:9039340, ECO:0000269|PubMed:9196614, ECO:0000269|PubMed:9302173, ECO:0000269|PubMed:9329414, ECO:0000269|PubMed:9543136, ECO:0000269|PubMed:9607727, ECO:0000269|PubMed:9768671, ECO:0000269|PubMed:9856504, ECO:0000269|Ref.124}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Hypospadias 1, X-linked (HYSP1) [MIM:300633]: A common malformation in which the urethra opens on the ventral side of the penis, due to developmental arrest of urethral fusion. The opening can be located glandular, penile, or even more posterior in the scrotum or perineum. Hypospadias is a feature of several syndromic disorders, including the androgen insensitivity syndrome and Opitz syndrome. {ECO:0000269|PubMed:8097257}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07422; DB07039; DB04709; DB07717; DB07454; DB02932; DB08035; DB01481; DB08088; DB08461; DB08087; DB07421; DB01063; DB07423; DB11901; DB01128; DB07286; DB01541; DB14639; DB01564; DB12499; DB04839; DB01406; DB12941; DB09123; DB00255; DB06133; DB01395; DB00858; DB15488; DB11219; DB08899; DB13155; DB00655; DB09086; DB02266; DB01185; DB00623; DB00499; DB11619; DB11064; DB01026; DB15647; DB00367; DB08089; DB05234; DB13934; DB11425; DB06710; DB02998; DB11429; DB00648; DB08804; DB00984; DB00665; DB06713; DB00717; DB09371; DB00957; DB09389; DB00621; DB01428; DB06412; DB01608; DB11447; DB01708; DB00396; DB07419; DB07769; DB14583; DB00421; DB02901; DB13951; DB06718; DB00675; DB00624; DB13943; DB13944; DB01420; DB13946; DB06870; DB08604; DB08867 Interacts with P00519; Q9UBL3; P51451; Q8WV28; O60885-1; P78543; Q14790; P24385; Q92793; O14595; P35222; Q9UER7; P20711; P11308; P07332; P09769; Q02790; P55317; O75593; Q14451; P06396; P56524; Q16665; Q16666; O15357; Q15652; O95251; Q9BY66; Q9BY66-3; Q03164; O14686; P06239; P07948; P20794; P42679; Q00987; Q15596; Q14686; O96028; Q99497; P27986; O00459; Q92569; P19174; P16885; Q06830; P78527; Q06124; P20936; Q9UBS8; Q9Y252; O14796; Q9NP31; P29353; Q6S5L8; Q5VZ18; Q15797; O14544; P12931; Q9ULZ2; P63165; Q9HBL0; P07947; Q9R1E0; Q06986 EC number NA Uniprot keywords 3D-structure; Activator; Alternative splicing; Cytoplasm; Disease variant; DNA-binding; Isopeptide bond; Lipid-binding; Lipoprotein; Metal-binding; Neurodegeneration; Nucleus; Palmitate; Phosphoprotein; Proteomics identification; Pseudohermaphroditism; Receptor; Reference proteome; Steroid-binding; Transcription; Transcription regulation; Triplet repeat expansion; Ubl conjugation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A Molecular weight (Da) 29137.9 Length 250 Aromaticity 0.11 Instability index 42.11 Isoelectric point 8.94 Charge (pH=7) 5.43 2D Binding mode Binding energy (Kcal/mol) -9.44  Molscript Map  Pymol Map 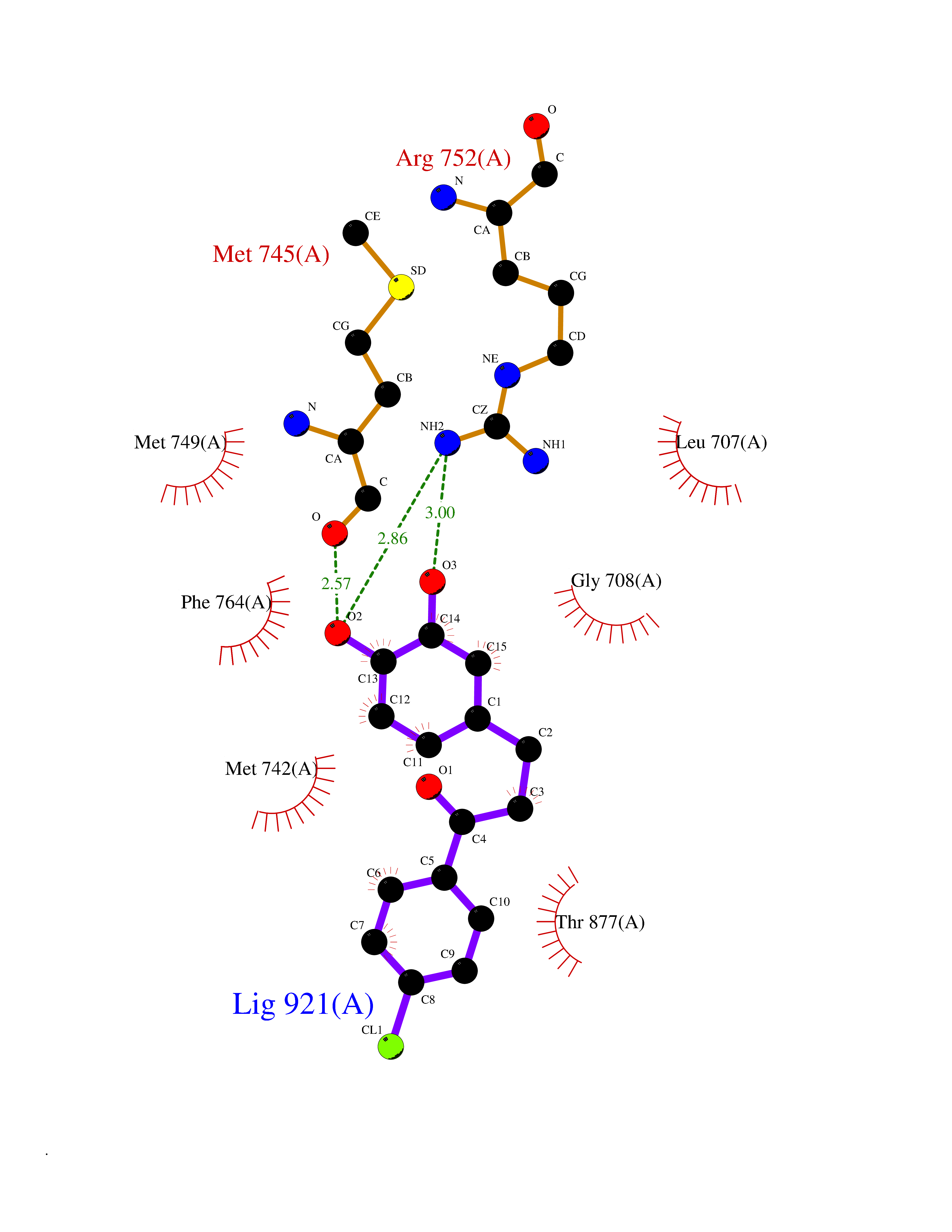 Ligplot Map 3D Binding mode Sequence QPIFLNVLEAIEPGVVCAGHDNNQPDSFAALLSSLNELGERQLVHVVKWAKALPGFRNLHVDDQMAVIQYSWMGLMVFAMGWRSFTNVNSRMLYFAPDLVFNEYRMHKSRMYSQCVRMRHLSQEFGWLQITPQEFLCMKALLLFSIIPVDGLKNQKFFDELRMNYIKELDRIIACKRKNPTSCSRRFYQLTKLLDSVQPIARELHQFTFDLLIKSHMVSVDFPEMMAEIISVQVPKILSGKVKPIYFHTQ Hydrogen bonds contact Hydrophobic contact | ||||
| 73 | Monoglyceride lipase (MAGL) | 3PE6 | 6.92 | |
Target general information Gen name MGLL Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Monoacylglycerol lipase; MGL; Lysophospholipaselike; Lysophospholipase-like; Lysophospholipase homolog; HUK5; HU-K5 Protein family AB hydrolase superfamily, Monoacylglycerol lipase family Biochemical class Carboxylic ester hydrolase Function Hydrolyzes the endocannabinoid 2-arachidonoylglycerol, and thereby contributes to the regulation of endocannabinoid signaling, nociperception and perception of pain. Regulates the levels of fatty acids that serve as signaling molecules and promote cancer cell migration, invasion and tumor growth. Converts monoacylglycerides to free fatty acids and glycerol. Related diseases Systemic lupus erythematosus 9 (SLEB9) [MIM:610927]: A chronic, relapsing, inflammatory, and often febrile multisystemic disorder of connective tissue, characterized principally by involvement of the skin, joints, kidneys and serosal membranes. It is of unknown etiology, but is thought to represent a failure of the regulatory mechanisms of the autoimmune system. The disease is marked by a wide range of system dysfunctions, an elevated erythrocyte sedimentation rate, and the formation of LE cells in the blood or bone marrow. {ECO:0000269|PubMed:17360460}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Immunodeficiency, common variable, 7 (CVID7) [MIM:614699]: A primary immunodeficiency characterized by antibody deficiency, hypogammaglobulinemia, recurrent bacterial infections and an inability to mount an antibody response to antigen. The defect results from a failure of B-cell differentiation and impaired secretion of immunoglobulins; the numbers of circulating B-cells is usually in the normal range, but can be low. {ECO:0000269|PubMed:22035880}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P07550; P37235 EC number EC 3.1.1.23 Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; Fatty acid biosynthesis; Fatty acid metabolism; Hydrolase; Lipid biosynthesis; Lipid degradation; Lipid metabolism; Membrane; Nitration; Phosphoprotein; Proteomics identification; Reference proteome; Serine esterase Protein physicochemical properties Chain ID A Molecular weight (Da) 31808.4 Length 289 Aromaticity 0.08 Instability index 29.7 Isoelectric point 6.73 Charge (pH=7) -0.91 2D Binding mode Binding energy (Kcal/mol) -9.44  Molscript Map 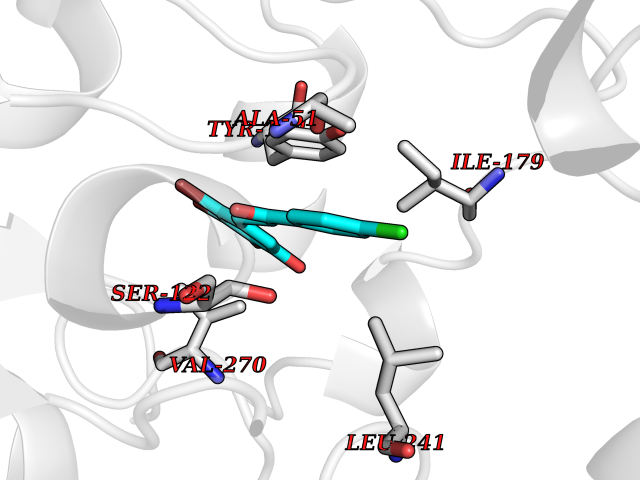 Pymol Map 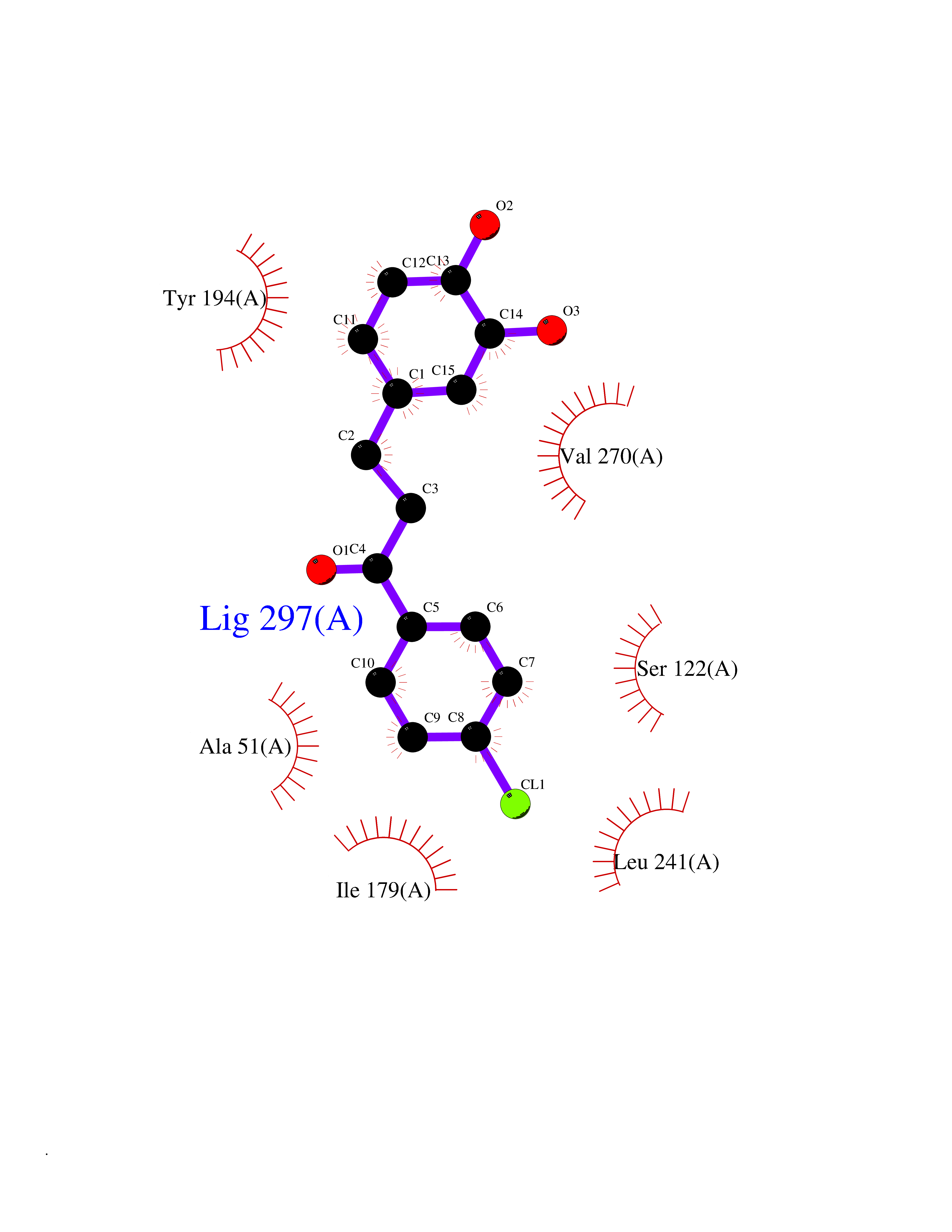 Ligplot Map 3D Binding mode Sequence PRRTPQSIPYQDLPHLVNADGQYLFCRYWAPTGTPKALIFVSHGAGEHSGRYEELARMLMGLDLLVFAHDHVGHGQSEGERMVVSDFHVFVRDVLQHVDSMQKDYPGLPVFLLGHSMGGAIAILTAAERPGHFAGMVLISPLVLANPESATTFKVLAAKVLNSVLPNLSSGPIDSSVLSRNKTEVDIYNSDPLICRAGLKVCFGIQLLNAVSRVERALPKLTVPFLLLQGSADRLCDSKGAYLLMELAKSQDKTLKIYEGAYHVLHKELPEVTNSVFHEINMWVSQRTA Hydrogen bonds contact Hydrophobic contact | ||||
| 74 | Plasmodium Hypoxanthine-guanine phosphoribosyltransferase (Malaria LACZ) | 3OZF | 6.92 | |
Target general information Gen name Malaria LACZ Organism Plasmodium falciparum (isolate FCR-3 / Gambia) Uniprot ID TTD ID Synonyms LACZ of Plasmodium falciparum (isolate FCR-3 / Gambia); Hypoxanthine phosphoribosyltransferase; HPRT; HGPRTase; HGPRT of Plasmodium falciparum (isolate FCR-3 / Gambia); Guanine phosphoribosyltransfera Protein family Purine/pyrimidine phosphoribosyltransferase family Biochemical class Pentosyltransferase Function Converts guanine to guanosine monophosphate, and hypoxanthine to inosine monophosphate. Transfers the 5- phosphoribosyl group from 5-phosphoribosylpyrophosphate onto the purine. Plays a central role in the generation of purine nucleotides through the purine salvage pathway. Related diseases Intellectual developmental disorder with dysmorphic facies and ptosis (IDDDFP) [MIM:617333]: An autosomal dominant neurodevelopmental disorder characterized by delayed psychomotor development, intellectual disability, delayed language, and facial dysmorphisms, most notably ptosis. Additional features may include poor growth, hypotonia, and seizures. {ECO:0000269|PubMed:27939639, ECO:0000269|PubMed:27939640}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02075; DB11638 Interacts with NA EC number NA Uniprot keywords 3D-structure; Cytoplasm; Glycosyltransferase; Magnesium; Metal-binding; Nucleotide-binding; Purine salvage; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 26581.3 Length 233 Aromaticity 0.12 Instability index 29.77 Isoelectric point 7.92 Charge (pH=7) 1.69 2D Binding mode Binding energy (Kcal/mol) -9.44  Molscript Map 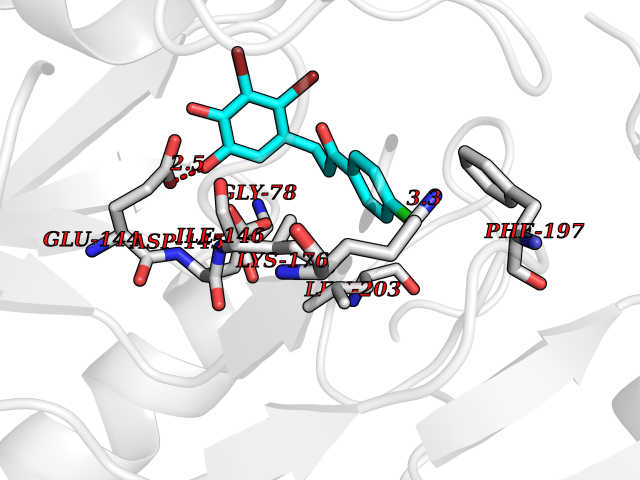 Pymol Map 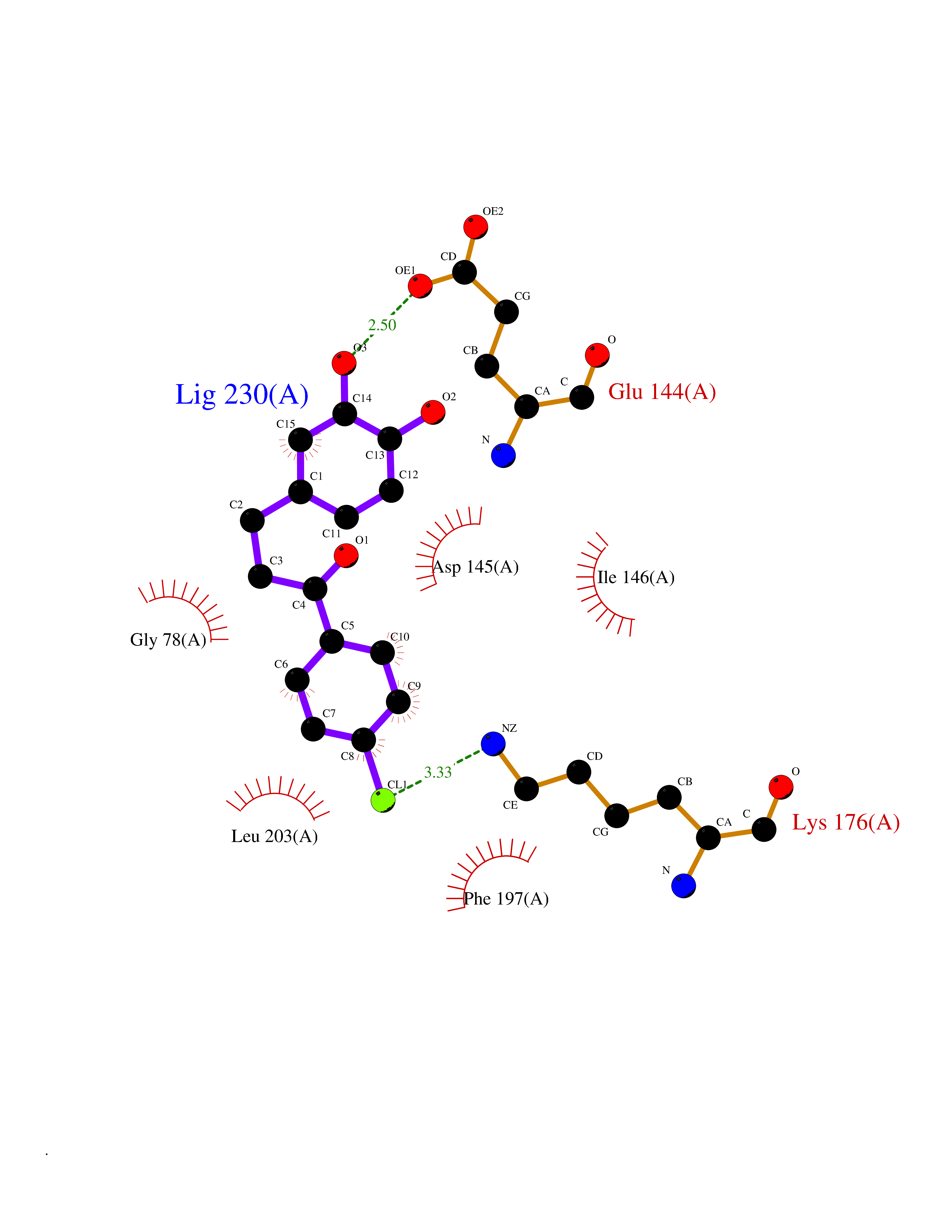 Ligplot Map 3D Binding mode Sequence PRGSHMPIPNNPGAGENAFDPVFVNDDDGYDLDSFMIPAHYKKYLTKVLVPNGVIKNRIEKLAYDIKKVYNNEEFHILCLLKGSRGFFTALLKHLSRIHNYSAVETSKPLFGEHYVRVKSYCNDQSTGTLEIVSEDLSCLKGKHVLIVEDIIDTGKTLVKFCEYLKKFEIKTVAIACLFIKRTPLWNGFKADFVGFSIPDHFVVGYSLDYNEIFRDLDHCCLVNDEGKKKYKA Hydrogen bonds contact Hydrophobic contact | ||||
| 75 | Neuronal acetylcholine receptor alpha-4 (CHRNA4) | 6CNJ | 6.92 | |
Target general information Gen name CHRNA4 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Nicotinic acetylcholine receptor alpha4; CHRNA4; Alpha-4 nAChR Protein family Ligand-gated ion channel (TC 1.A.9) family, Acetylcholine receptor (TC 1.A.9.1) subfamily, Alpha-4/CHRNA4 sub-subfamily Biochemical class Neurotransmitter receptor Function After binding acetylcholine, the AChR responds by an extensive change in conformation that affects all subunits and leads to opening of an ion-conducting channel across the plasmamembrane permeable to sodium ions. Related diseases Epilepsy, nocturnal frontal lobe, 1 (ENFL1) [MIM:600513]: An autosomal dominant focal epilepsy characterized by nocturnal seizures with hyperkinetic automatisms and poorly organized stereotyped movements. {ECO:0000269|PubMed:10563623, ECO:0000269|PubMed:14623738, ECO:0000269|PubMed:7550350}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00915; DB01351; DB01352; DB00572; DB01483; DB00237; DB00241; DB01353; DB00564; DB00565; DB09028; DB01245; DB00514; DB01496; DB07720; DB00783; DB13952; DB13953; DB13954; DB13955; DB13956; DB00898; DB01354; DB01355; DB00753; DB00657; DB00333; DB00463; DB00849; DB00184; DB00312; DB01174; DB00981; DB05458; DB00794; DB05740; DB00747; DB00418; DB00202; DB00306; DB00599; DB01273 Interacts with Q6UY14-3; P05067; P83916; Q6UXH1-1; Q6UXH1-3; P20042; Q9NZR2; Q92673; P17787 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disease variant; Disulfide bond; Epilepsy; Glycoprotein; Ion channel; Ion transport; Ligand-gated ion channel; Lipoprotein; Membrane; Palmitate; Phosphoprotein; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Signal; Synapse; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A,B Molecular weight (Da) 84601.2 Length 728 Aromaticity 0.13 Instability index 39.72 Isoelectric point 5.86 Charge (pH=7) -9.84 2D Binding mode Binding energy (Kcal/mol) -9.43  Molscript Map 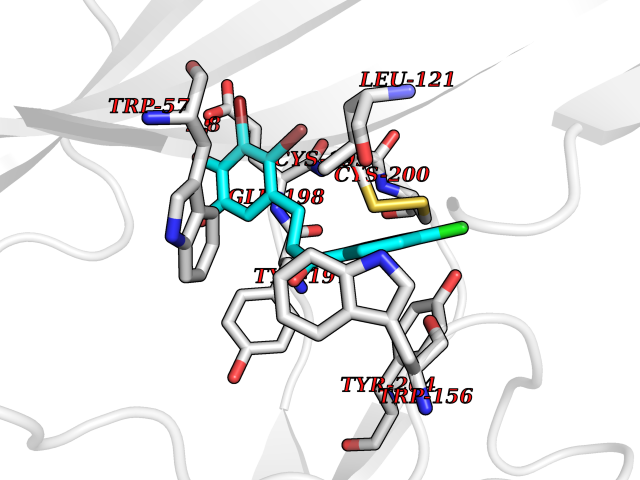 Pymol Map 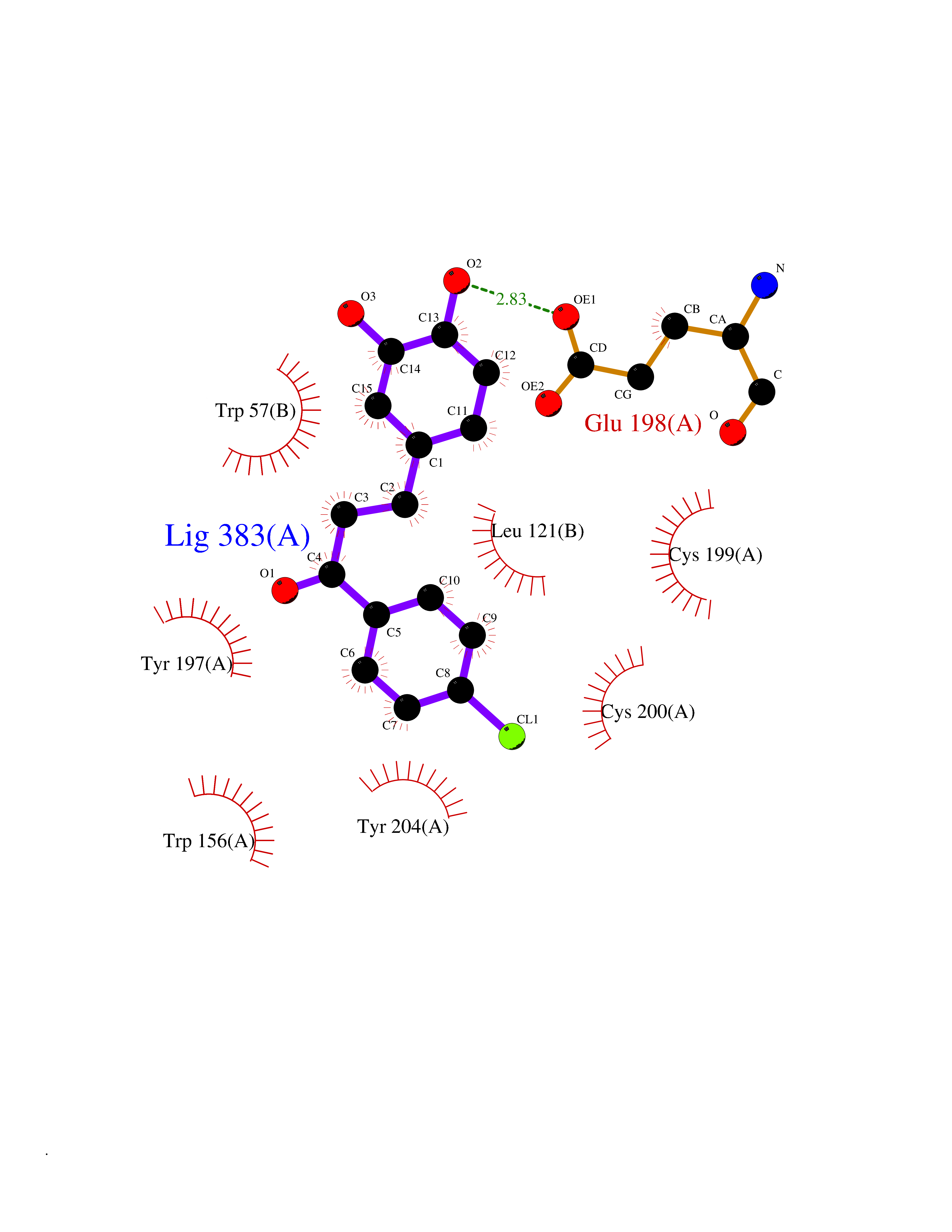 Ligplot Map 3D Binding mode Sequence ETRAHAEERLLKKLFSGYNKWSRPVANISDVVLVRFGLSIAQLIDVDEKNQMMTTNVWVKQEWHDYKLRWDPADYENVTSIRIPSELIWRPDIVLYNNADGDFAVTHLTKAHLFHDGRVQWTPPAIYKSSCSIDVTFFPFDQQNCTMKFGSWTYDKAKIDLVNMHSRVDQLDFWESGEWVIVDAVGTYNTRKYECCAEIYPDITYAFVIRRLPLFYTINLIIPCLLISCLTVLVFYLPSECGEKITLCISVLLSLTVFLLLITEIIPSTSLVIPLIGEYLLFTMIFVTLSIVITVFVLNVHHRSPRTHTMPTWVRRVFLDIVPRLLLMKRFERSVKEDWKYVAMVIDRIFLWMFIIVCLLGTVGLFLPPWDTEERLVEHLLDPSRYNKLIRPATNGSELVTVQLMVSLAQLISVHEREQIMTTNVWLTQEWEDYRLTWKPEEFDNMKKVRLPSKHIWLPDVVLYNNADGMYEVSFYSNAVVSYDGSIFWLPPAIYKSACKIEVKHFPFDQQNCTMKFRSWTYDRTEIDLVLKSEVASLDDFTPSGEWDIVALPGRRNENPDDSTYVDITYDFIIRRKPLFYTINLIIPCVLITSLAILVFYLPSDCGEKMTLCISVLLALTVFLLLISKIVPPTSLDVPLVGKYLMFTMVLVTFSIVTSVCVLNVHHRSPTTHTMAPWVKVVFLEKLPALLFMQQSVSEDWKYVAMVIDRLFLWIFVFVCVFGTIGMF Hydrogen bonds contact Hydrophobic contact | ||||
| 76 | Lysine N-methyltransferase 3A (SETD2) | 7LZD | 6.92 | |
Target general information Gen name SETD2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms p231HBP; hSET2; SET2; SET domain-containing protein 2; Protein-lysine N-methyltransferase SETD2; KMT3A; KIAA1732; Huntingtin-interacting protein B; Huntingtin-interacting protein 1; Huntingtin yeast p Protein family Class V-like SAM-binding methyltransferase superfamily, Histone-lysine methyltransferase family, SET2 subfamily Biochemical class Methyltransferase Function Represents the main enzyme generating H3K36me3, a specific tag for epigenetic transcriptional activation. Plays a role in chromatin structure modulation during elongation by coordinating recruitment of the FACT complex and by interacting with hyperphosphorylated POLR2A. Acts as a key regulator of DNA mismatch repair in G1 and early S phase by generating H3K36me3, a mark required to recruit MSH6 subunit of the MutS alpha complex: early recruitment of the MutS alpha complex to chromatin to be replicated allows a quick identification of mismatch DNA to initiate the mismatch repair reaction. Required for DNA double-strand break repair in response to DNA damage: acts by mediating formation of H3K36me3, promoting recruitment of RAD51 and DNA repair via homologous recombination (HR). Acts as a tumor suppressor. H3K36me3 also plays an essential role in the maintenance of a heterochromatic state, by recruiting DNA methyltransferase DNMT3A. H3K36me3 is also enhanced in intron-containing genes, suggesting that SETD2 recruitment is enhanced by splicing and that splicing is coupled to recruitment of elongating RNA polymerase. Required during angiogenesis. Required for endoderm development by promoting embryonic stem cell differentiation toward endoderm: acts by mediating formation of H3K36me3 in distal promoter regions of FGFR3, leading to regulate transcription initiation of FGFR3. In addition to histones, also mediates methylation of other proteins, such as tubulins and STAT1. Trimethylates 'Lys-40' of alpha-tubulins such as TUBA1B (alpha-TubK40me3); alpha-TubK40me3 is required for normal mitosis and cytokinesis and may be a specific tag in cytoskeletal remodeling. Involved in interferon-alpha-induced antiviral defense by mediating both monomethylation of STAT1 at 'Lys-525' and catalyzing H3K36me3 on promoters of some interferon-stimulated genes (ISGs) to activate gene transcription. Histone methyltransferase that specifically trimethylates 'Lys-36' of histone H3 (H3K36me3) using dimethylated 'Lys-36' (H3K36me2) as substrate. Related diseases Renal cell carcinoma (RCC) [MIM:144700]: Renal cell carcinoma is a heterogeneous group of sporadic or hereditary carcinoma derived from cells of the proximal renal tubular epithelium. It is subclassified into clear cell renal carcinoma (non-papillary carcinoma), papillary renal cell carcinoma, chromophobe renal cell carcinoma, collecting duct carcinoma with medullary carcinoma of the kidney, and unclassified renal cell carcinoma. Clear cell renal cell carcinoma is the most common subtype. {ECO:0000269|PubMed:20054297, ECO:0000269|PubMed:23622243, ECO:0000269|PubMed:23792563, ECO:0000269|PubMed:25728682}. The disease may be caused by variants affecting the gene represented in this entry. Defects of SETD2 are associated with loss of DNA methylation at non-promoter regions (PubMed:23792563). SETD2 defects lead to aberrant and reduced nucleosome compaction and chromatin association of key replication proteins, such as MCM7 and DNA polymerase delta, leading to hinder replication fork progression and prevent loading of RAD51 homologous recombination repair factor at DNA breaks (PubMed:25728682). {ECO:0000269|PubMed:23792563, ECO:0000269|PubMed:25728682}.; DISEASE: Luscan-Lumish syndrome (LLS) [MIM:616831]: An autosomal dominant syndrome with a variable phenotype. Clinical features include macrocephaly, distinctive facial appearance, postnatal overgrowth, various degrees of learning difficulties, autism spectrum disorder, and intellectual disability. {ECO:0000269|PubMed:23160955, ECO:0000269|PubMed:24852293, ECO:0000269|PubMed:26084711, ECO:0000269|PubMed:27317772}. The disease may be caused by variants affecting the gene represented in this entry.; DISEASE: Leukemia, acute lymphoblastic (ALL) [MIM:613065]: A subtype of acute leukemia, a cancer of the white blood cells. ALL is a malignant disease of bone marrow and the most common malignancy diagnosed in children. The malignant cells are lymphoid precursor cells (lymphoblasts) that are arrested in an early stage of development. The lymphoblasts replace the normal marrow elements, resulting in a marked decrease in the production of normal blood cells. Consequently, anemia, thrombocytopenia, and neutropenia occur to varying degrees. The lymphoblasts also proliferate in organs other than the marrow, particularly the liver, spleen, and lymphnodes. {ECO:0000269|PubMed:24509477, ECO:0000269|PubMed:24662245}. The disease may be caused by variants affecting distinct genetic loci, including the gene represented in this entry.; DISEASE: Leukemia, acute myelogenous (AML) [MIM:601626]: A subtype of acute leukemia, a cancer of the white blood cells. AML is a malignant disease of bone marrow characterized by maturational arrest of hematopoietic precursors at an early stage of development. Clonal expansion of myeloid blasts occurs in bone marrow, blood, and other tissue. Myelogenous leukemias develop from changes in cells that normally produce neutrophils, basophils, eosinophils and monocytes. {ECO:0000269|PubMed:16314571, ECO:0000269|PubMed:24509477}. The disease may be caused by variants affecting distinct genetic loci, including the gene represented in this entry.; DISEASE: Intellectual developmental disorder, autosomal dominant 70 (MRD70) [MIM:620157]: An autosomal dominant disorder characterized by mild global developmental delay, moderately impaired intellectual disability with speech difficulties, and behavioral abnormalities. {ECO:0000269|PubMed:32710489}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Rabin-Pappas syndrome (RAPAS) [MIM:620155]: An autosomal dominant neurodevelopmental disorder characterized by severely impaired global development, intellectual disability, microcephaly, facial dysmorphism, and variable congenital anomalies affecting the skeletal, genitourinary, cardiac, and other organ systems. {ECO:0000269|PubMed:32710489}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P42858; P84022 EC number EC 2.1.1.43 Uniprot keywords 3D-structure; Activator; Alternative splicing; Antiviral defense; Autism spectrum disorder; Chromatin regulator; Chromosome; Coiled coil; Developmental protein; Differentiation; Disease variant; DNA damage; DNA repair; Host-virus interaction; Immunity; Innate immunity; Intellectual disability; Isopeptide bond; Metal-binding; Methyltransferase; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; S-adenosyl-L-methionine; Transcription; Transcription regulation; Transferase; Tumor suppressor; Ubl conjugation; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 27416.8 Length 237 Aromaticity 0.11 Instability index 55.47 Isoelectric point 7.51 Charge (pH=7) 0.99 2D Binding mode Binding energy (Kcal/mol) -9.44 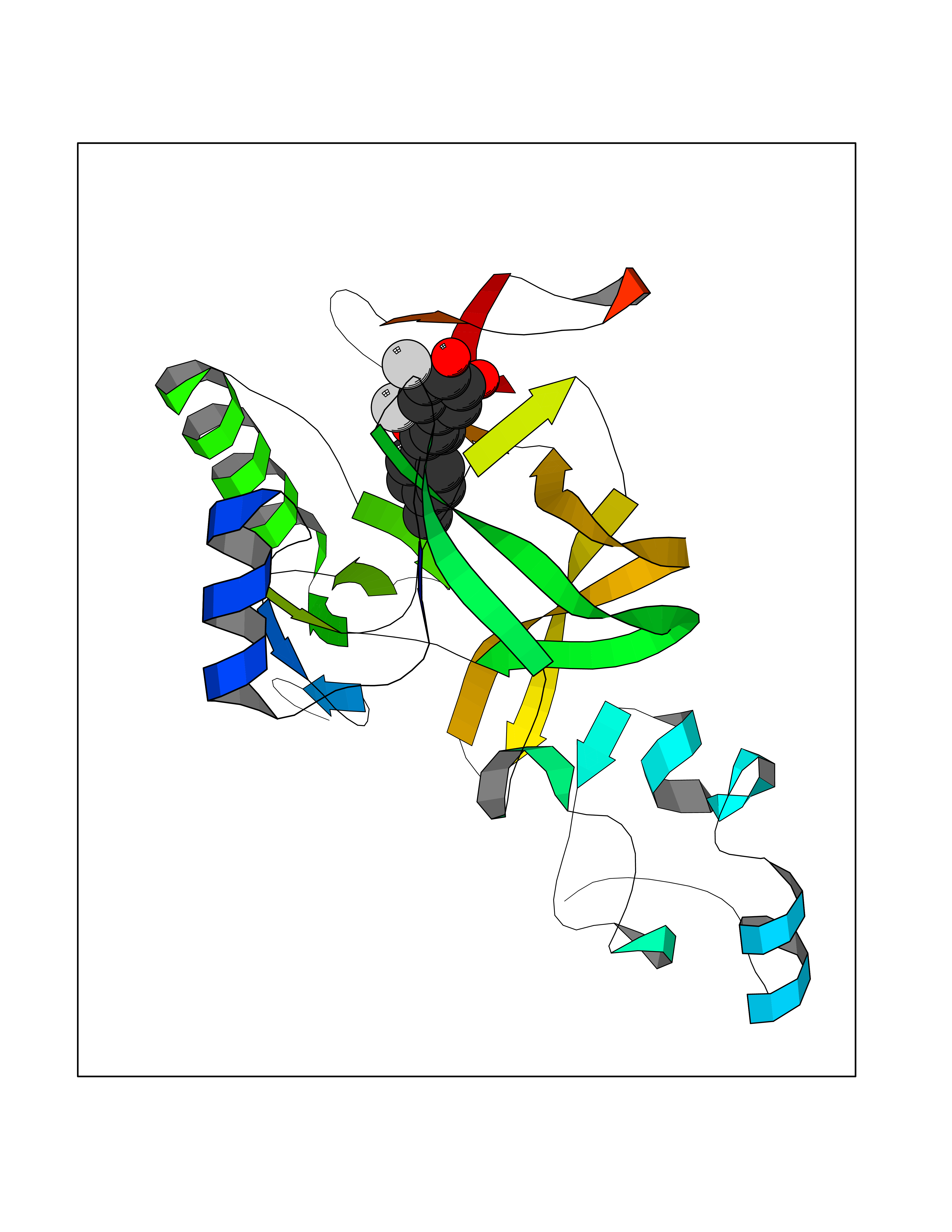 Molscript Map 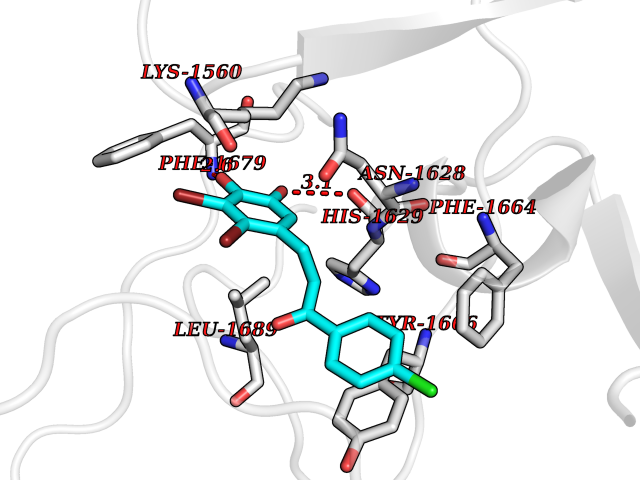 Pymol Map 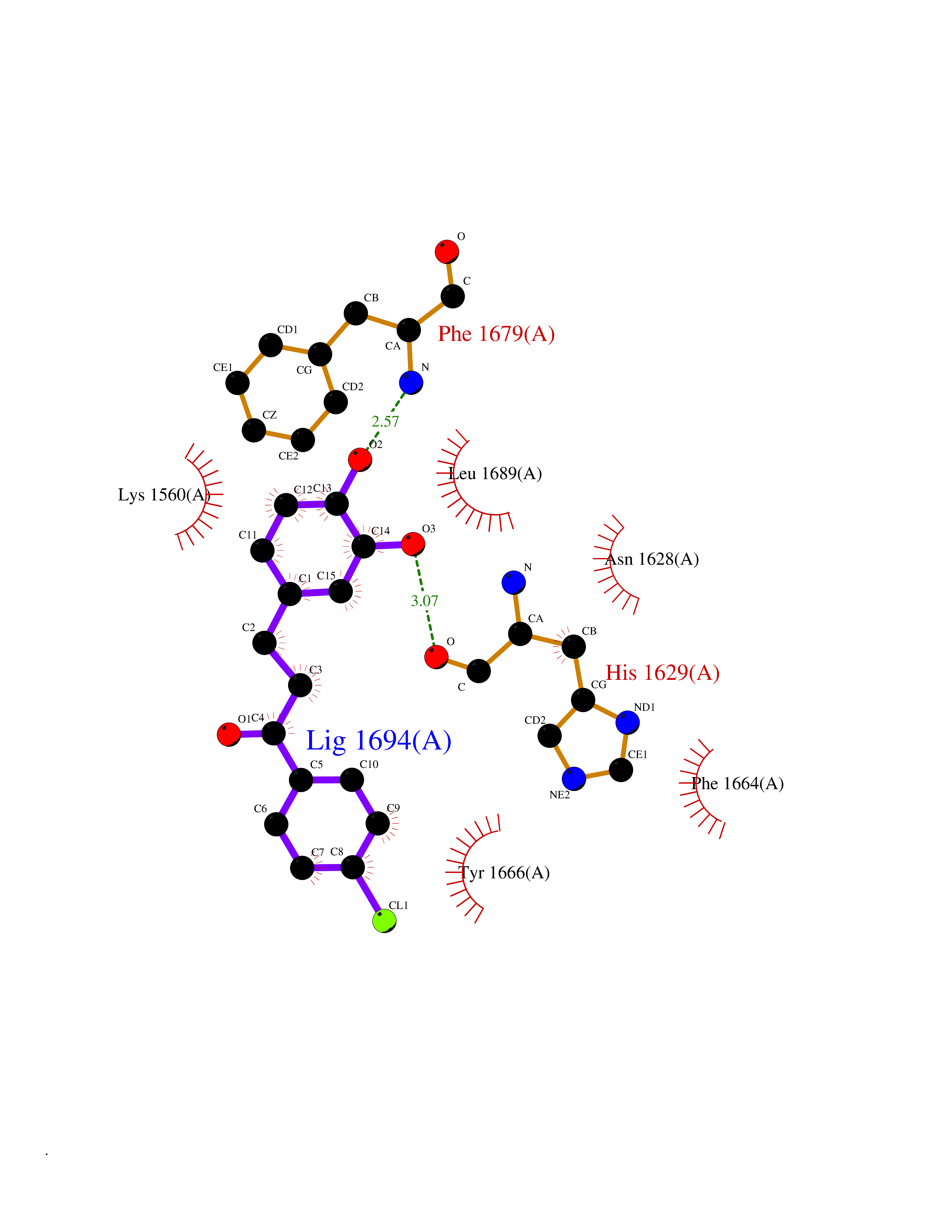 Ligplot Map 3D Binding mode Sequence GPSCVMDDFRDPQRWKECAKQGKMPCYFDLIEENVYLTERRMQCECTPLSKDERAQGEIACGEDCLNRLLMIECSSRCPNGDYCSNRRFQRKQHADVEVILTEKKGWGLRAAKDLPSNTFVLEYCGEVLDHKEFKARVKEYARNKNIHYYFMALKNDEIIDATQKGNCSRFMNHSCEPNCETQKWTVNGQLRVGFFTTKLVPSGSELTFDYQFQRYGKEAQKCFCGSANCRGYLGGE Hydrogen bonds contact Hydrophobic contact | ||||
| 77 | Zinc finger protein Helios (IKZF2) | 7LPS | 6.92 | |
Target general information Gen name IKZF2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Ikaros family zinc finger protein 2 Protein family Ikaros C2H2-type zinc-finger protein family Biochemical class NA Function Associates with Ikaros at centromeric heterochromatin. Related diseases Developmental and epileptic encephalopathy 25, with amelogenesis imperfecta (DEE25) [MIM:615905]: An autosomal recessive disease characterized by subclinical seizures appearing in the first days of life, evolving to severe epileptic disease. Affected individuals have profound or severe delayed development with lack of speech, and most patients do not acquire the ability to sit. Additional variable features include axial hypotonia, peripheral hypertonia, and abnormal involuntary movements such as dystonia and choreoathetosis. Dental abnormalities, including delayed eruption, hypodontia, tooth hypoplasia, yellow discoloration, thin enamel, and enamel chipping are observed in most patients. {ECO:0000269|PubMed:24995870, ECO:0000269|PubMed:26384929, ECO:0000269|PubMed:30054523}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P29972; P56545; P56545-3; Q17RB8; P09022; Q8N8B7-2 EC number NA Uniprot keywords 3D-structure; Acetylation; Activator; Alternative splicing; DNA-binding; Isopeptide bond; Metal-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Transcription; Transcription regulation; Ubl conjugation; Zinc; Zinc-finger Protein physicochemical properties Chain ID B,C Molecular weight (Da) 47006.6 Length 410 Aromaticity 0.09 Instability index 44.28 Isoelectric point 7.23 Charge (pH=7) 0.69 2D Binding mode Binding energy (Kcal/mol) -9.44 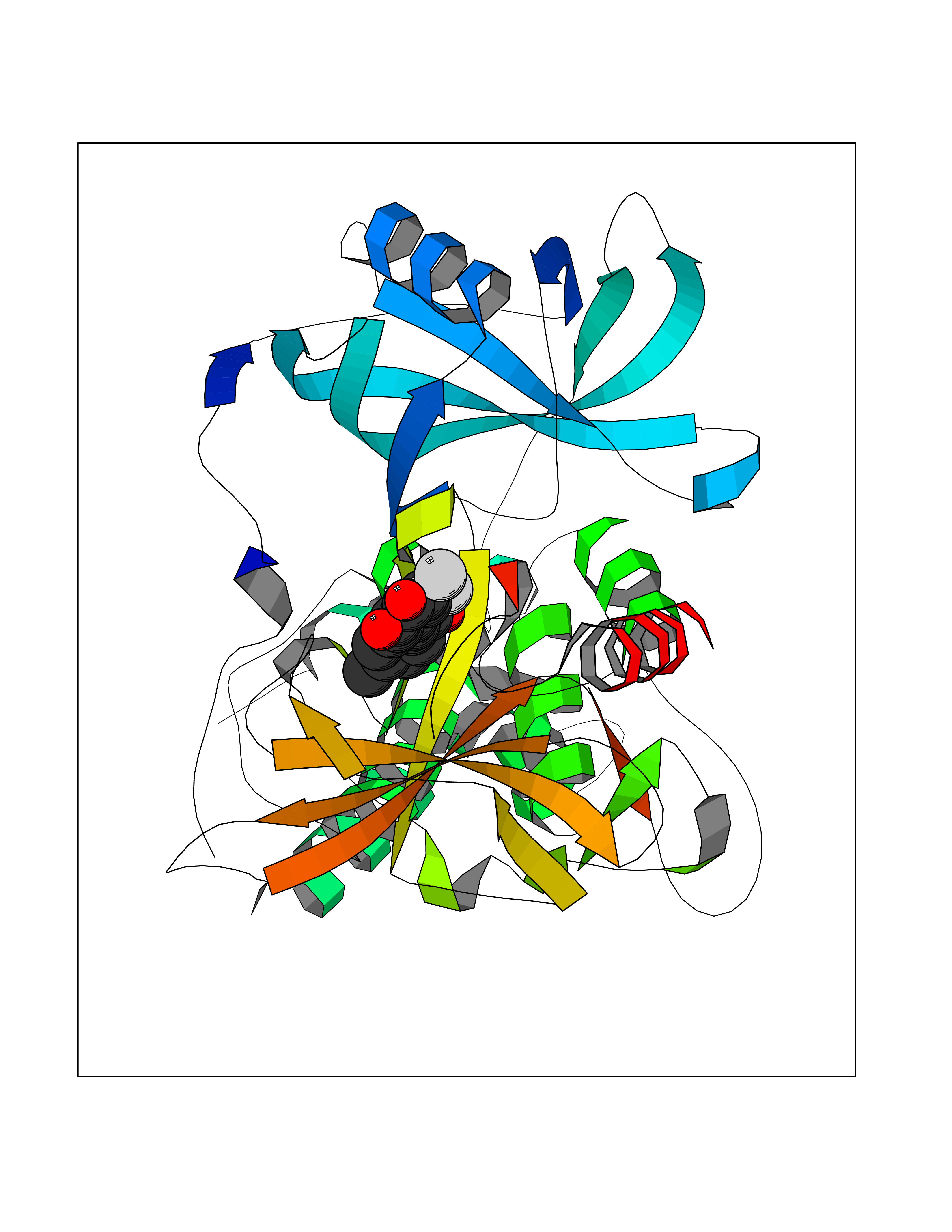 Molscript Map 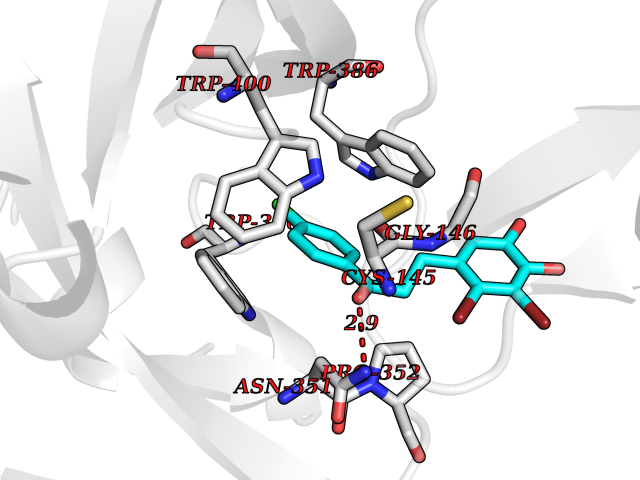 Pymol Map 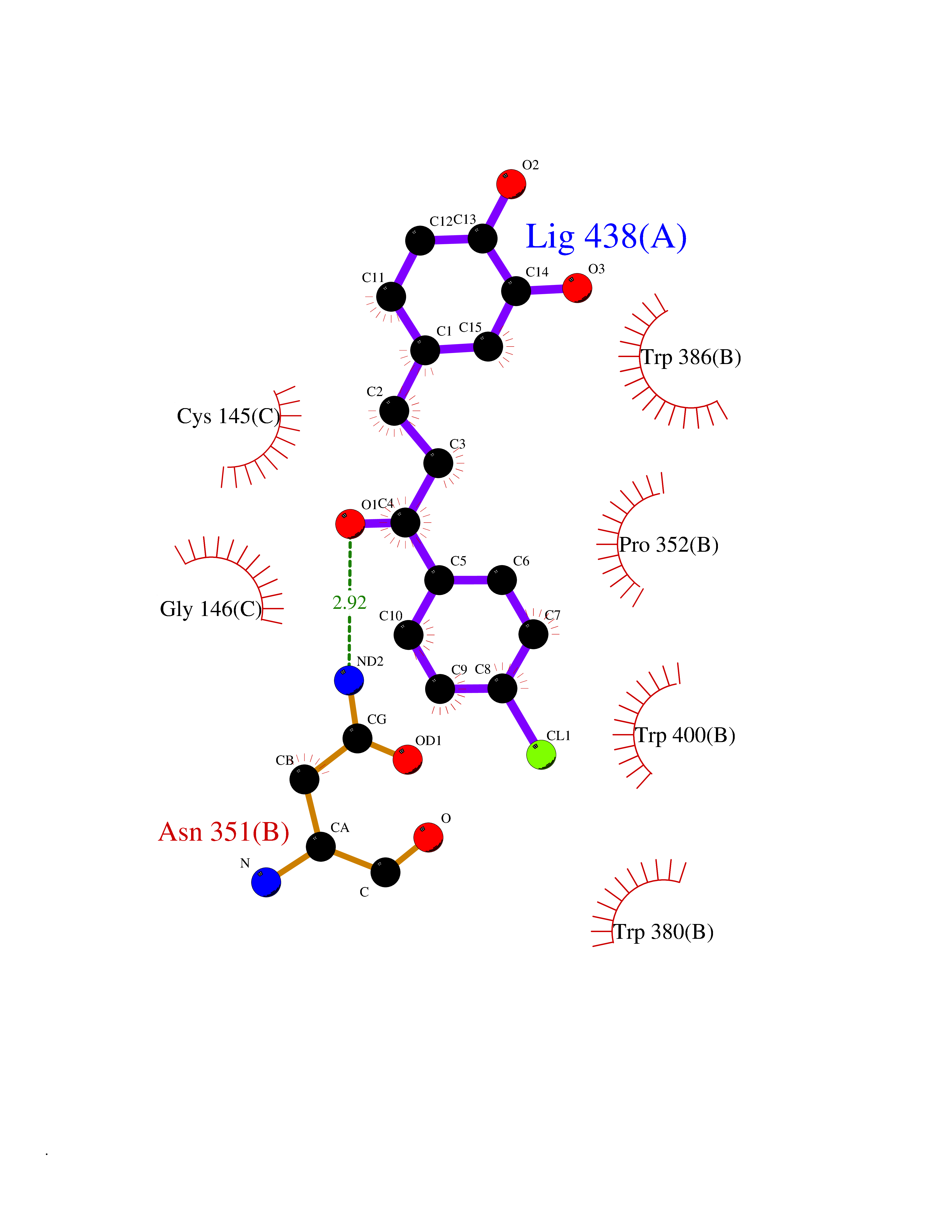 Ligplot Map 3D Binding mode Sequence INFDTSLPTSHTYLGADMEEFHGRTLHDDDSCQVIPVLPQVMMILIPGQTLPLQLFHPQEVSMVRNLIQKDRTFAVLAYSNVQEREAQFGTTAEIYAYREEQDFGIEIVKVKAIGRQRFKVLELRTQSDGIQQAKVQILPECVLPSTMSAVQLESLNKCQIFPCSYKWWQKYQKRKFHCANLTSWPRWLYSLYDAETLMDRIKKQLREWDENLKDDSLPSNPIDFSYRVAACLPIDDVLRIQLLKIGSAIQRLRCELDIMNKCTSLCCKQCQETEITTKNEIFSLSLCGPMAAYVNPHGYVHETLTVYKACNLNLIGRPSTEHSWFPGYAWTVAQCKICASHIGWKFTATKKDMSPQKFWGLTRSALLPTIPDTEDEISPDGERPFHCNQCGASFTQKGNLLRHIKLHSG Hydrogen bonds contact Hydrophobic contact | ||||
| 78 | Calmodulin-dependent kinase II (CAMKK2) | 5YVC | 6.92 | |
Target general information Gen name CAMKK2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms KIAA0787; Calcium/calmodulin-dependent protein kinase kinase beta; Calcium/calmodulin-dependent protein kinase kinase 2; CaMKK beta; CaMKK 2; CaM-kinase kinase beta; CaM-kinase kinase 2; CaM-KK beta; Protein family Protein kinase superfamily, Ser/Thr protein kinase family Biochemical class Kinase Function Calcium/calmodulin-dependent protein kinase belonging to a proposed calcium-triggered signaling cascade involved in a number of cellular processes. Isoform 1, isoform 2 and isoform 3 phosphorylate CAMK1 and CAMK4. Isoform 3 phosphorylates CAMK1D. Isoform 4, isoform 5 and isoform 6 lacking part of the calmodulin-binding domain are inactive. Efficiently phosphorylates 5'-AMP-activated protein kinase (AMPK) trimer, including that consisting of PRKAA1, PRKAB1 and PRKAG1. This phosphorylation is stimulated in response to Ca(2+) signals (By similarity). Seems to be involved in hippocampal activation of CREB1 (By similarity). May play a role in neurite growth. Isoform 3 may promote neurite elongation, while isoform 1 may promoter neurite branching. Related diseases Squalene synthase deficiency (SQSD) [MIM:618156]: An autosomal recessive disorder characterized by profound developmental delay, brain abnormalities, 2/3 syndactyly of the toes, facial dysmorphisms, low total and LDL-cholesterol, and abnormal urine organic acids. {ECO:0000269|PubMed:29909962}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB12010 Interacts with NA EC number EC 2.7.11.17 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; ATP-binding; Calmodulin-binding; Cell projection; Cytoplasm; Kinase; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Serine/threonine-protein kinase; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 29603.8 Length 259 Aromaticity 0.09 Instability index 26.4 Isoelectric point 5.32 Charge (pH=7) -8.63 2D Binding mode Binding energy (Kcal/mol) -9.44 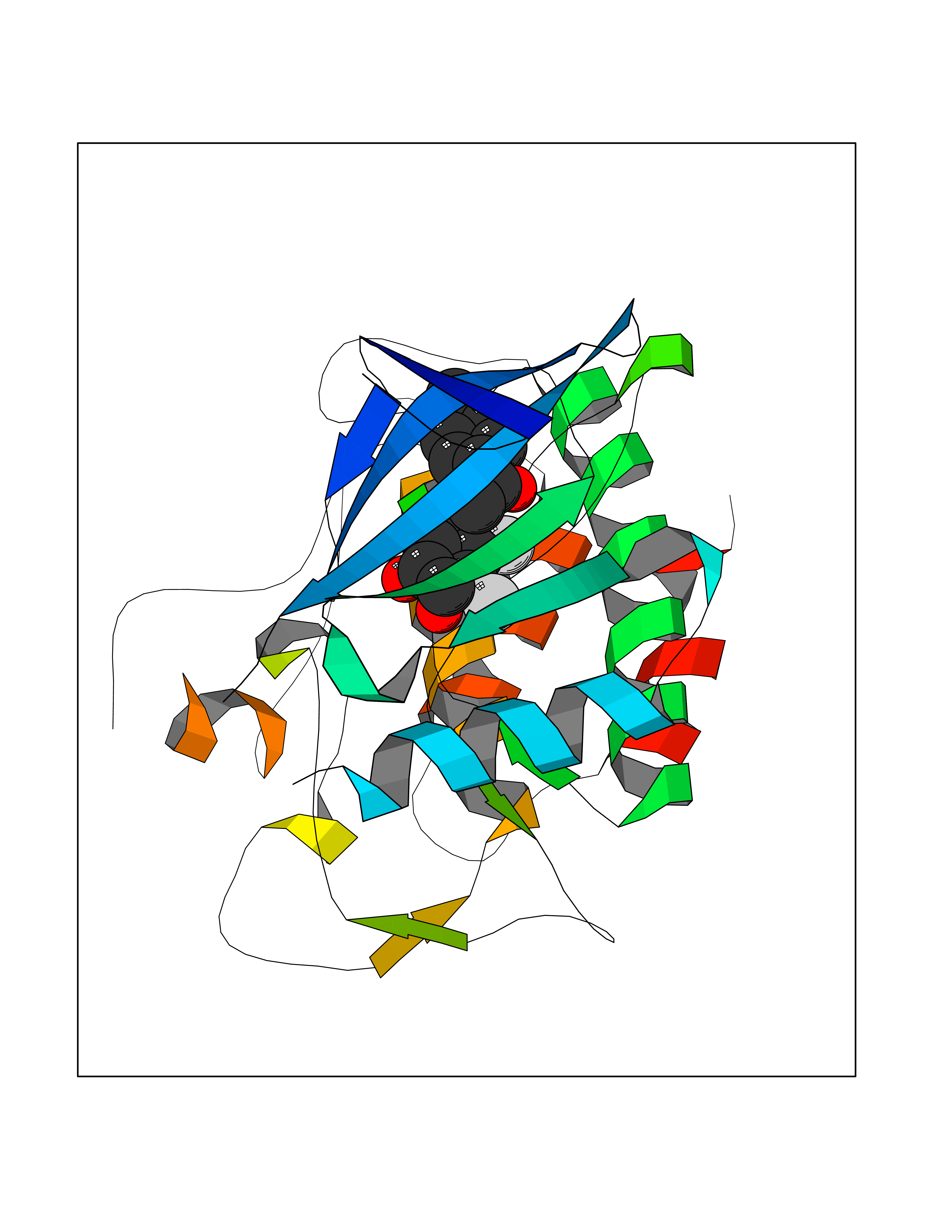 Molscript Map 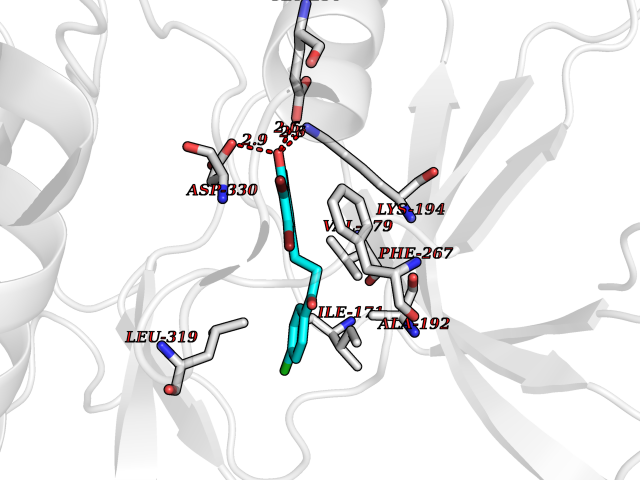 Pymol Map 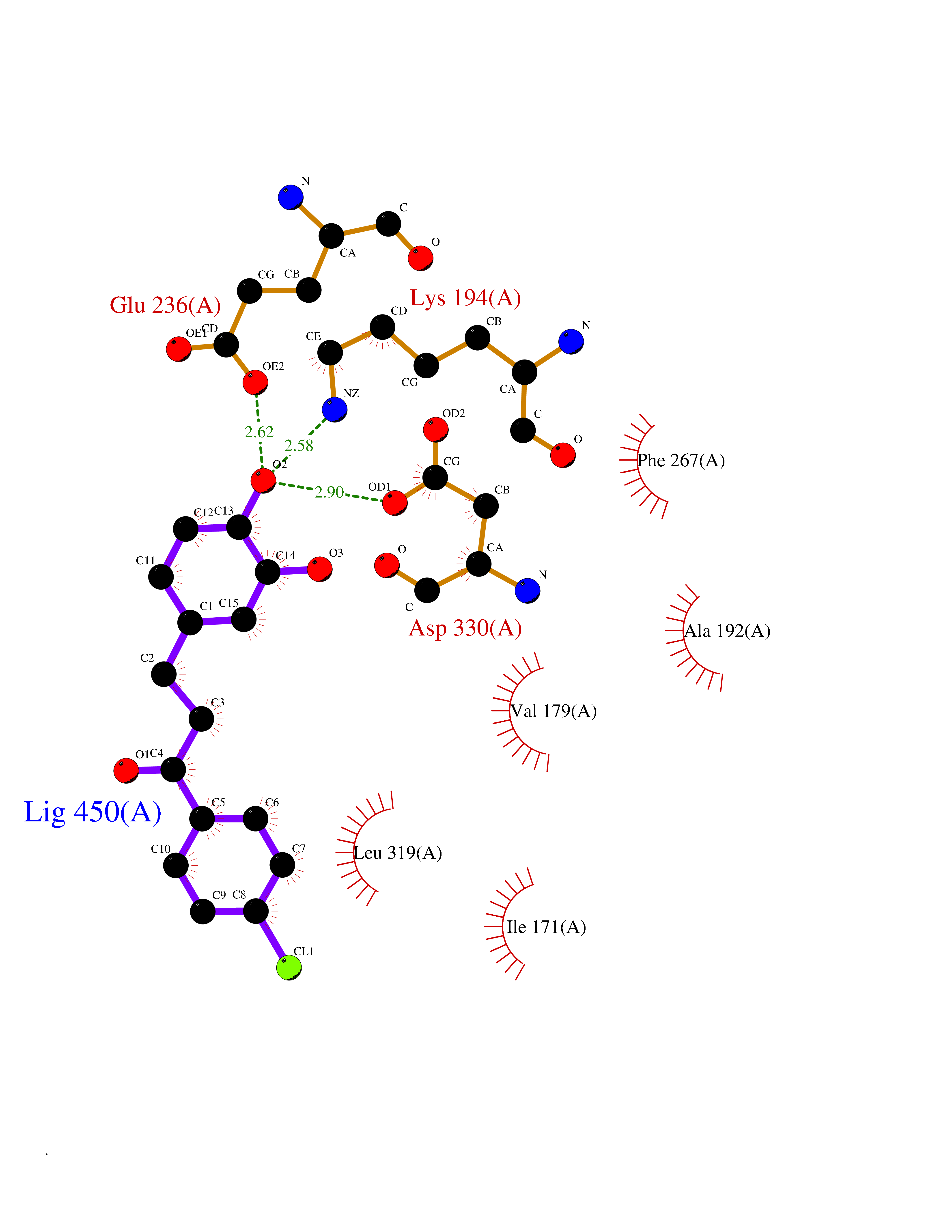 Ligplot Map 3D Binding mode Sequence VQLNQYTLKDEIGKGSYGVVKLAYNENDNTYYAMKVLSKKPIEQVYQEIAILKKLDHPNVVKLVEVLDDPNEDHLYMVFELVNQGPVMEVPTLKPLSEDQARFYFQDLIKGIEYLHYQKIIHRDIKPSNLLVGEDGHIKIADFGVSNEFKGSDALLSNTVGTPAFMAPESLSETRKIFSGKALDVWAMGVTLYCFVFGQCPFMDERIMLHSKIKSQALEFPDQPDIAEDLKDLITRMLDKNPESRIVVPEIKLHPWVTR Hydrogen bonds contact Hydrophobic contact | ||||
| 79 | SEC14-like protein 2 | 4OMJ | 6.91 | |
Target general information Gen name SEC14L2 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms KIAA1658;C22orf6;KIAA1186 Protein family NA Biochemical class Transport protein Function Phospholipid binding.Transporter activity.Vitamin E binding. Related diseases Neurodevelopmental disorder with poor language and loss of hand skills (NDPLHS) [MIM:617903]: An autosomal dominant disorder characterized by psychomotor developmental stagnation or regression. NDPLHS manifest in the first years of life as loss of purposeful hand movements, loss of language, and intellectual disability. {ECO:0000269|PubMed:26740508, ECO:0000269|PubMed:28856709, ECO:0000269|PubMed:29369404}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Developmental and epileptic encephalopathy 59 (DEE59) [MIM:617904]: A form of epileptic encephalopathy, a heterogeneous group of severe early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE59 is an autosomal dominant condition characterized by onset of refractory seizures in early infancy. {ECO:0000269|PubMed:28856709, ECO:0000269|PubMed:29100083, ECO:0000269|PubMed:29369404}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB14003; DB14001; DB14002; DB11635; DB11251; DB00163 Interacts with NA EC number NA Uniprot keywords 3D-structure; Activator; Alternative splicing; Cytoplasm; Lipid-binding; Nucleus; Proteomics identification; Reference proteome; Transcription; Transcription regulation; Transport Protein physicochemical properties Chain ID A,B Molecular weight (Da) 31533.3 Length 274 Aromaticity 0.1 Instability index 49.26 Isoelectric point 8.34 Charge (pH=7) 2.81 2D Binding mode Binding energy (Kcal/mol) -9.42 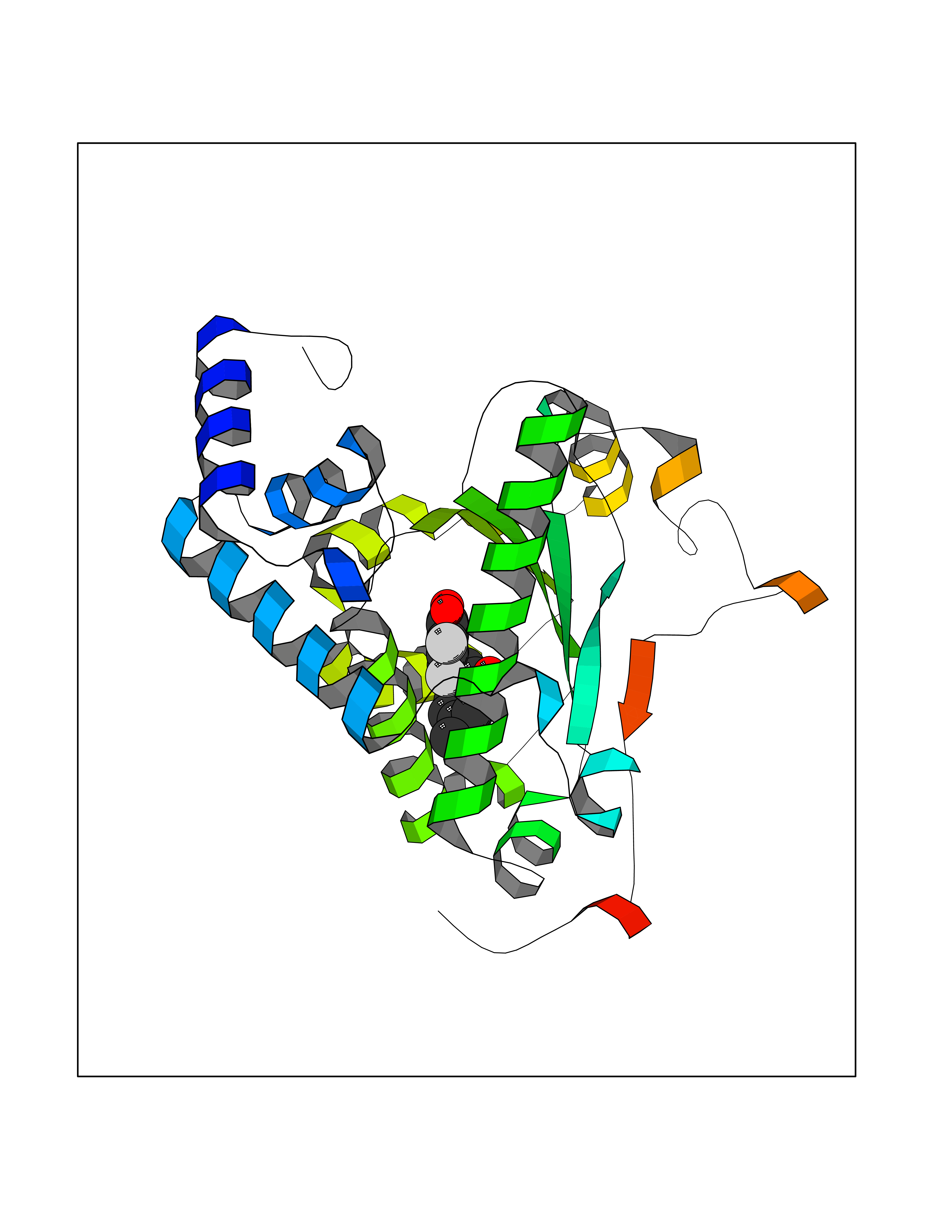 Molscript Map 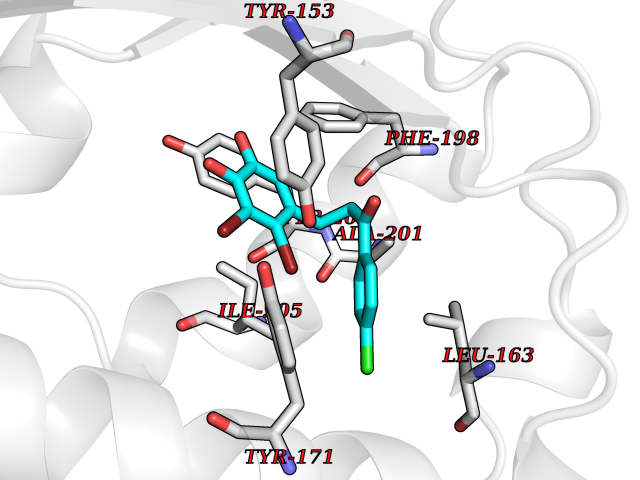 Pymol Map 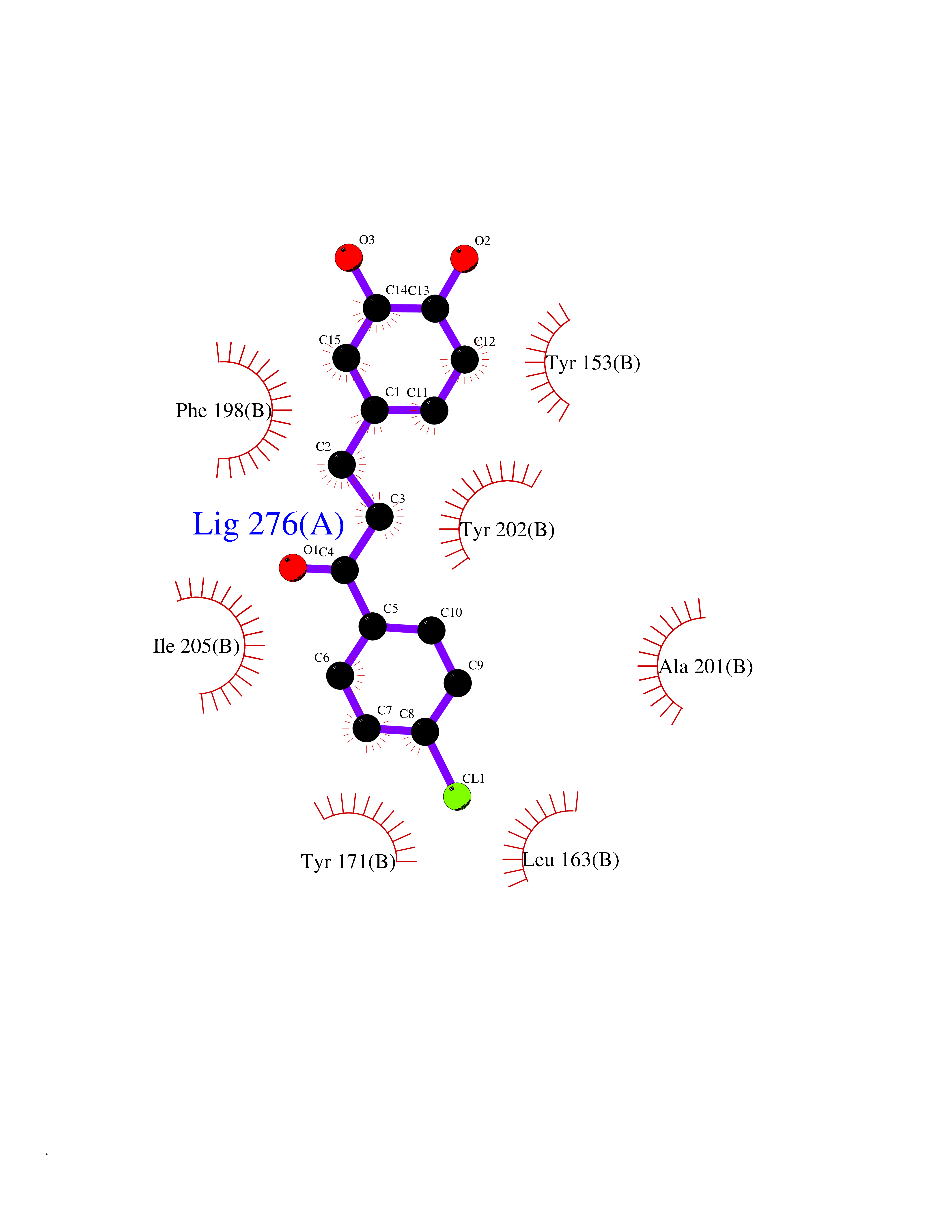 Ligplot Map 3D Binding mode Sequence MSGRVGDLSPRQKEALAKFRENVQDVLPALPNPDDYFLLRWLRARSFDLQKSEAMLRKHVEFRKQKDIDNIISWQPPEVIQQYLSGGMCGYDLDGCPVWYDIIGPLDAKGLLFSASKQDLLRTKMRECELLLQECAHQTTKLGRKVETITIIYDCEGLGLKHLWKPAVEAYGEFLCMFEENYPETLKRLFVVKAPKLFPVAYNLIKPFLSEDTRKKIMVLGANWKEVLLKHISPDQVPVEYGGTMTDPDGNPKCKSKINYGGDIPRKYYVRDQV Hydrogen bonds contact Hydrophobic contact | ||||
| 80 | Pyruvate kinase M2 (PKM) | 3GR4 | 6.91 | |
Target general information Gen name PKM Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms p58; Tumor M2-PK; Thyroid hormone-binding protein 1; THBP1; Pyruvate kinase muscle isozyme; Pyruvate kinase isozymes M1/M2; Pyruvate kinase PKM; Pyruvate kinase 2/3; PKM2; PK3; PK2; Opa-interacting pr Protein family Pyruvate kinase family Biochemical class Kinase Function Stimulates POU5F1-mediated transcriptional activation. Plays a general role in caspase independent cell death of tumor cells. The ratio between the highly active tetrameric form and nearly inactive dimeric form determines whether glucose carbons are channeled to biosynthetic processes or used for glycolytic ATP production. The transition between the 2 forms contributes to the control of glycolysis and is important for tumor cell proliferation and survival. Promotes in a STAT1-dependent manner, the expression of the immune checkpoint protein CD274 in ARNTL/BMAL1-deficient macrophages. Glycolytic enzyme that catalyzes the transfer of a phosphoryl group from phosphoenolpyruvate (PEP) to ADP, generating ATP. Related diseases Congenital sucrase-isomaltase deficiency (CSID) [MIM:222900]: Autosomal recessive intestinal disorder that is clinically characterized by fermentative diarrhea, abdominal pain, and cramps upon ingestion of sugar. The symptoms are the consequence of absent or drastically reduced enzymatic activities of sucrase and isomaltase. The prevalence of CSID is 0.02 % in individuals of European descent and appears to be much higher in Greenland, Alaskan, and Canadian native people. CSID arises due to post-translational perturbations in the intracellular transport, polarized sorting, aberrant processing, and defective function of SI. {ECO:0000269|PubMed:10903344, ECO:0000269|PubMed:11340066, ECO:0000269|PubMed:14724820, ECO:0000269|PubMed:16329100, ECO:0000269|PubMed:8609217}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07697; DB07692; DB02726; DB07628; DB00787; DB11638; DB09130; DB08951; DB01733; DB11263; DB00119 Interacts with P49407; P32121; Q96IK1-2; P35222; P53355; P22607; P42858; P04049; Q8N488; Q7Z699; Q9BSI4; Q9UMX0; Q9Y649; Q9WMX2; P35222; P53355; Q9H6Z9; P68431; Q16665; P27361 EC number EC 2.7.1.40 Uniprot keywords 3D-structure; Acetylation; Allosteric enzyme; Alternative splicing; ATP-binding; Cytoplasm; Direct protein sequencing; Glycolysis; Hydroxylation; Isopeptide bond; Kinase; Magnesium; Metal-binding; Methylation; Nucleotide-binding; Nucleus; Phosphoprotein; Potassium; Proteomics identification; Pyruvate; Reference proteome; S-nitrosylation; Transferase; Translation regulation; Ubl conjugation Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 112053 Length 1024 Aromaticity 0.05 Instability index 27.06 Isoelectric point 7.34 Charge (pH=7) 1.66 2D Binding mode Binding energy (Kcal/mol) -9.43 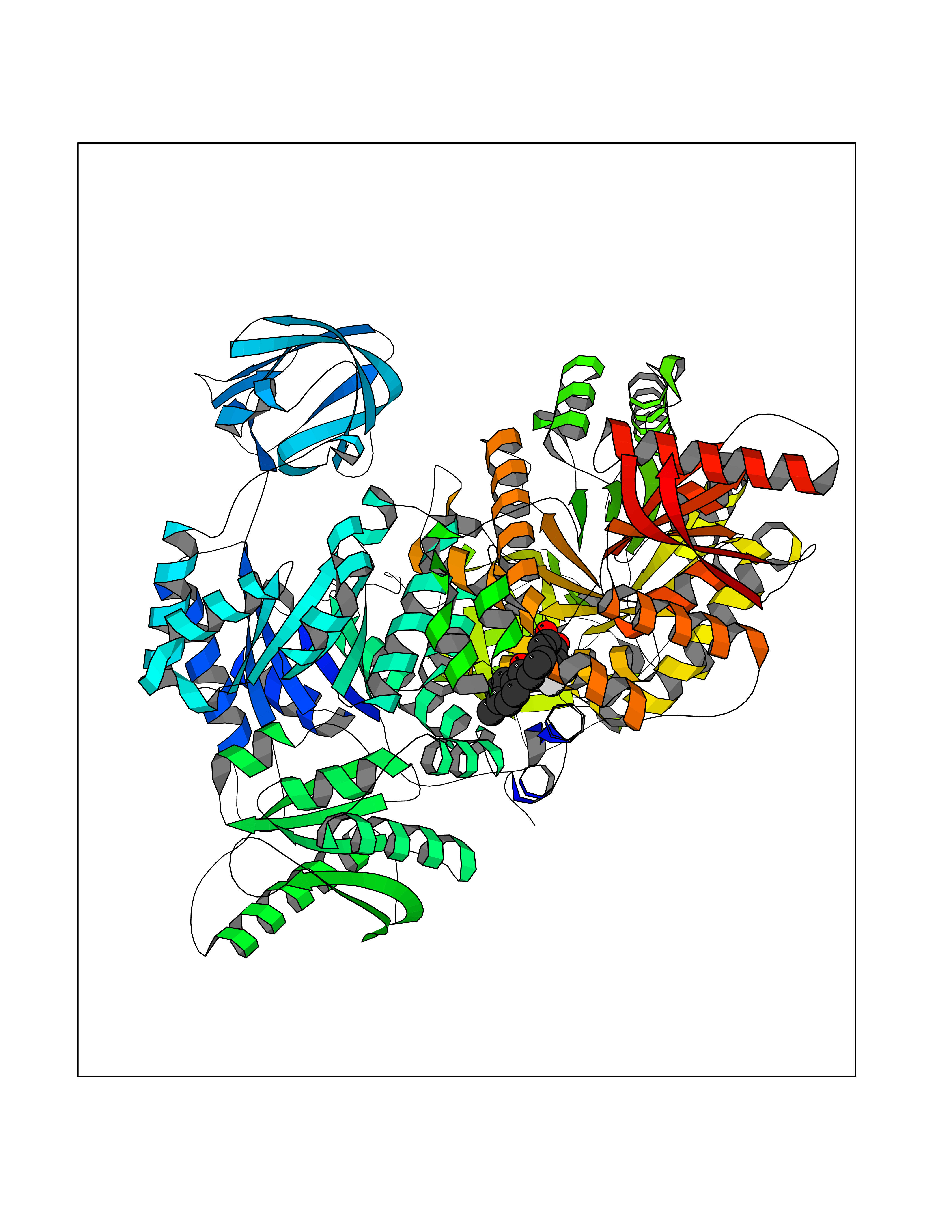 Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence IQTQQLHAAMADTFLEHMCRLDIDSPPITARNTGIICTIGPASRSVETLKEMIKSGMNVARLNFSHGTHEYHAETIKNVRTATESFASDPILYRPVAVALDTKGPEIRTGLIKGSGTAEVELKKGATLKITLDNAYMEKCDENILWLDYKNICKVVEVGSKIYVDDGLISLQVKQKGADFLVTEVENGGSLGSKKGVNLPGAAVDLPAVSEKDIQDLKFGVEQDVDMVFASFIRKASDVHEVRKVLGEKGKNIKIISKIENHEGVRRFDEILEASDGIMVARGDLGIEIPAEKVFLAQKMMIGRCNRAGKPVICATQMLESMIKKPRPTRAEGSDVANAVLDGADCIMLSGETAKGDYPLEAVRMQHLIAREAEAAIYHLQLFEELRRLAPITSDPTEATAVGAVEASFKCCSGAIIVLTKSGRSAHQVARYRPRAPIIAVTRNPQTARQAHLYRGIFPVLCKDPVQEAWAEDVDLRVNFAMNVGKARGFFKKGDVVIVLTGWRPGSGFTNTMRVVPVPIQTQQLHAAMADTFLEHMCRLDIDSPPITARNTGIICTIGPASRSVETLKEMIKSGMNVARLNFSHGTHEYHAETIKNVRTATESFASDPILYRPVAVALDTKGPEIRTGLIKEVEATLKITLDNAYMEKCDENILWLDYKNICKVVEVGSKIYVDDGLISLQVDFLVTEVENGGSLGSKKGVNLPGAAVDLPAVSEKDIQDLKFGVEQDVDMVFASFIRKASDVHEVRKVLGEKGKNIKIISKIENHEGVRRFDEILEASDGIMVARGDLGIEIPAEKVFLAQKMMIGRCNRAGKPVICATQMLESMIKKPRPTRAEGSDVANAVLDGADCIMLSGETAKGDYPLEAVRMQHLIAREAEAAIYHLQLFEELRRLAPITSDPTEATAVGAVEASFKCCSGAIIVLTKSGRSAHQVARYRPRAPIIAVTRNPQTARQAHLYRGIFPVLCKDPVQEAWAEDVDLRVNFAMNVGKARGFFKKGDVVIVLTGWRPGSGFTNTMRVVPVP Hydrogen bonds contact Hydrophobic contact | ||||