Job Results:
Ligand
Structure
Job ID
57b30c77c1e7b273b1e911481965e4cd
Job name
NA
Time
2025-10-13 17:21:20
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 61 | Protein kinase G1 (PRKG1) | 6BG2 | 6.58 | |
Target general information Gen name PRKG1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms cGMP-dependent protein kinase I; cGMP-dependent protein kinase 1; cGKI; cGK1; cGK 1; PRKGR1B; PRKGR1A; PRKG1B Protein family Protein kinase superfamily, AGC Ser/Thr protein kinase family, cGMP subfamily Biochemical class Kinase Function GMP binding activates PRKG1, which phosphorylates serines and threonines on many cellular proteins. Numerous protein targets for PRKG1 phosphorylation are implicated in modulating cellular calcium, but the contribution of each of these targets may vary substantially among cell types. Proteins that are phosphorylated by PRKG1 regulate platelet activation and adhesion, smooth muscle contraction, cardiac function, gene expression, feedback of the NO-signaling pathway, and other processes involved in several aspects of the CNS like axon guidance, hippocampal and cerebellar learning, circadian rhythm and nociception. Smooth muscle relaxation is mediated through lowering of intracellular free calcium, by desensitization of contractile proteins to calcium, and by decrease in the contractile state of smooth muscle or in platelet activation. Regulates intracellular calcium levels via several pathways: phosphorylates MRVI1/IRAG and inhibits IP3-induced Ca(2+) release from intracellular stores, phosphorylation of KCNMA1 (BKCa) channels decreases intracellular Ca(2+) levels, which leads to increased opening of this channel. PRKG1 phosphorylates the canonical transient receptor potential channel (TRPC) family which inactivates the associated inward calcium current. Another mode of action of NO/cGMP/PKGI signaling involves PKGI-mediated inactivation of the Ras homolog gene family member A (RhoA). Phosphorylation of RHOA by PRKG1 blocks the action of this protein in myriad processes: regulation of RHOA translocation; decreasing contraction; controlling vesicle trafficking, reduction of myosin light chain phosphorylation resulting in vasorelaxation. Activation of PRKG1 by NO signaling alters also gene expression in a number of tissues. In smooth muscle cells, increased cGMP and PRKG1 activity influence expression of smooth muscle-specific contractile proteins, levels of proteins in the NO/cGMP signaling pathway, down-regulation of the matrix proteins osteopontin and thrombospondin-1 to limit smooth muscle cell migration and phenotype. Regulates vasodilator-stimulated phosphoprotein (VASP) functions in platelets and smooth muscle. Serine/threonine protein kinase that acts as key mediator of the nitric oxide (NO)/cGMP signaling pathway. Related diseases Aortic aneurysm, familial thoracic 8 (AAT8) [MIM:615436]: A disease characterized by permanent dilation of the thoracic aorta usually due to degenerative changes in the aortic wall. It is primarily associated with a characteristic histologic appearance known as 'medial necrosis' or 'Erdheim cystic medial necrosis' in which there is degeneration and fragmentation of elastic fibers, loss of smooth muscle cells, and an accumulation of basophilic ground substance. {ECO:0000269|PubMed:23910461}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q13873; Q86V42; P25791; O76074; O15015; O94989; P25791-3; Q6FHY5; Q13976-2; Q9NYW8 EC number EC 2.7.11.12 Uniprot keywords 3D-structure; Acetylation; Allosteric enzyme; Alternative splicing; Aortic aneurysm; ATP-binding; cGMP; cGMP-binding; Coiled coil; Cytoplasm; Disease variant; Disulfide bond; Kinase; Nucleotide-binding; Phosphoprotein; Proteomics identification; Reference proteome; Serine/threonine-protein kinase; Transferase Protein physicochemical properties Chain ID C Molecular weight (Da) 38032 Length 335 Aromaticity 0.12 Instability index 38.26 Isoelectric point 6.23 Charge (pH=7) -2.54 2D Binding mode Binding energy (Kcal/mol) -8.98 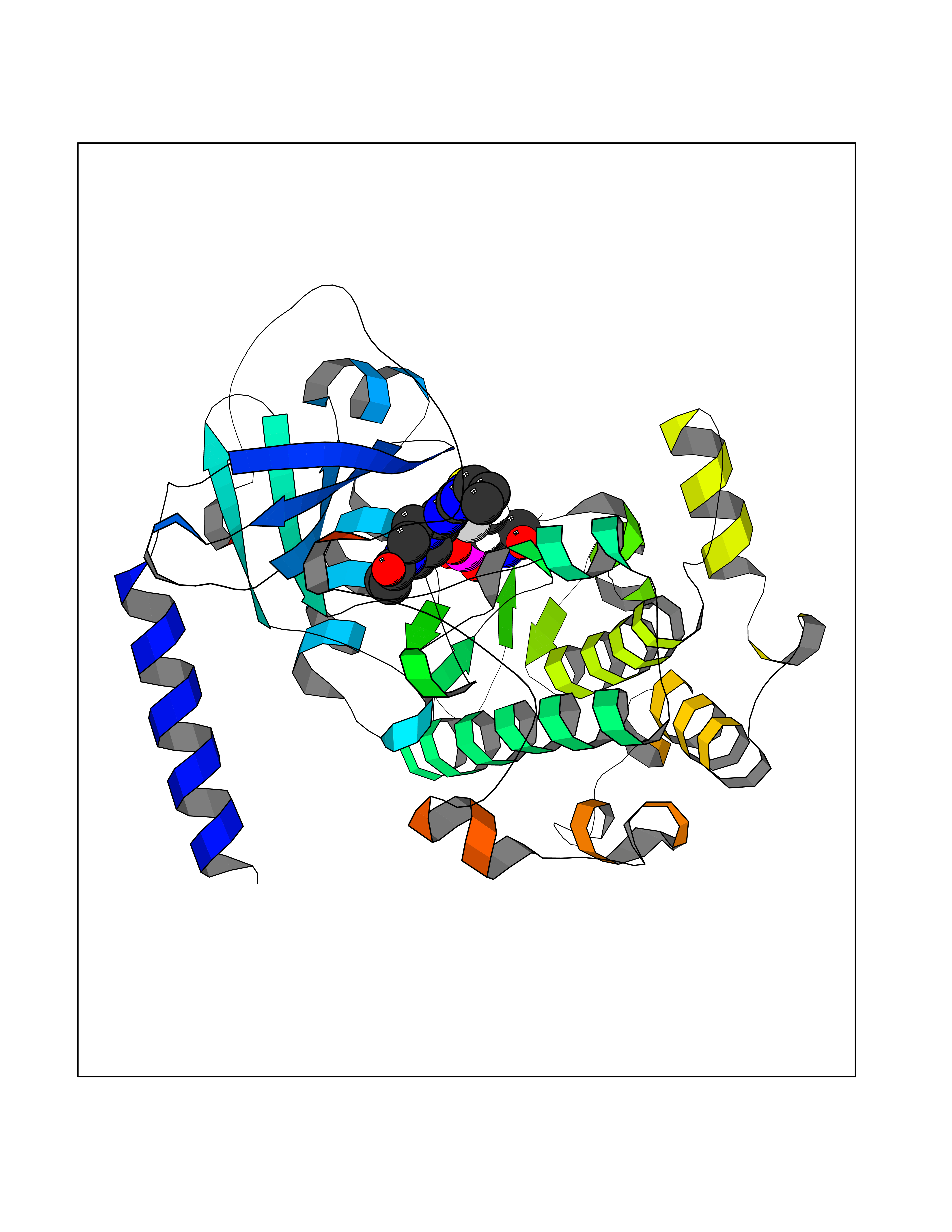 Molscript Map 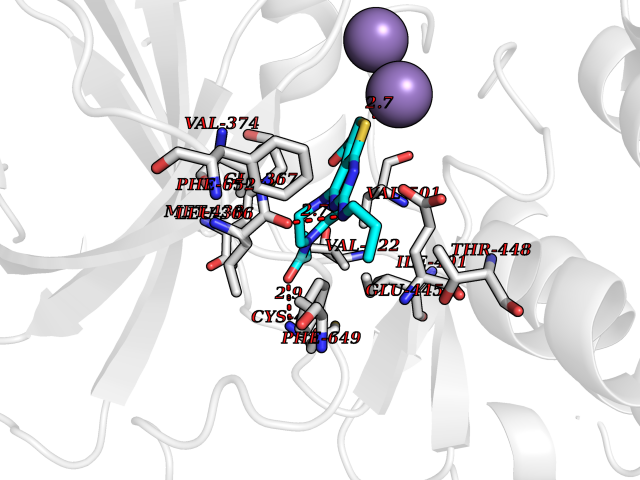 Pymol Map 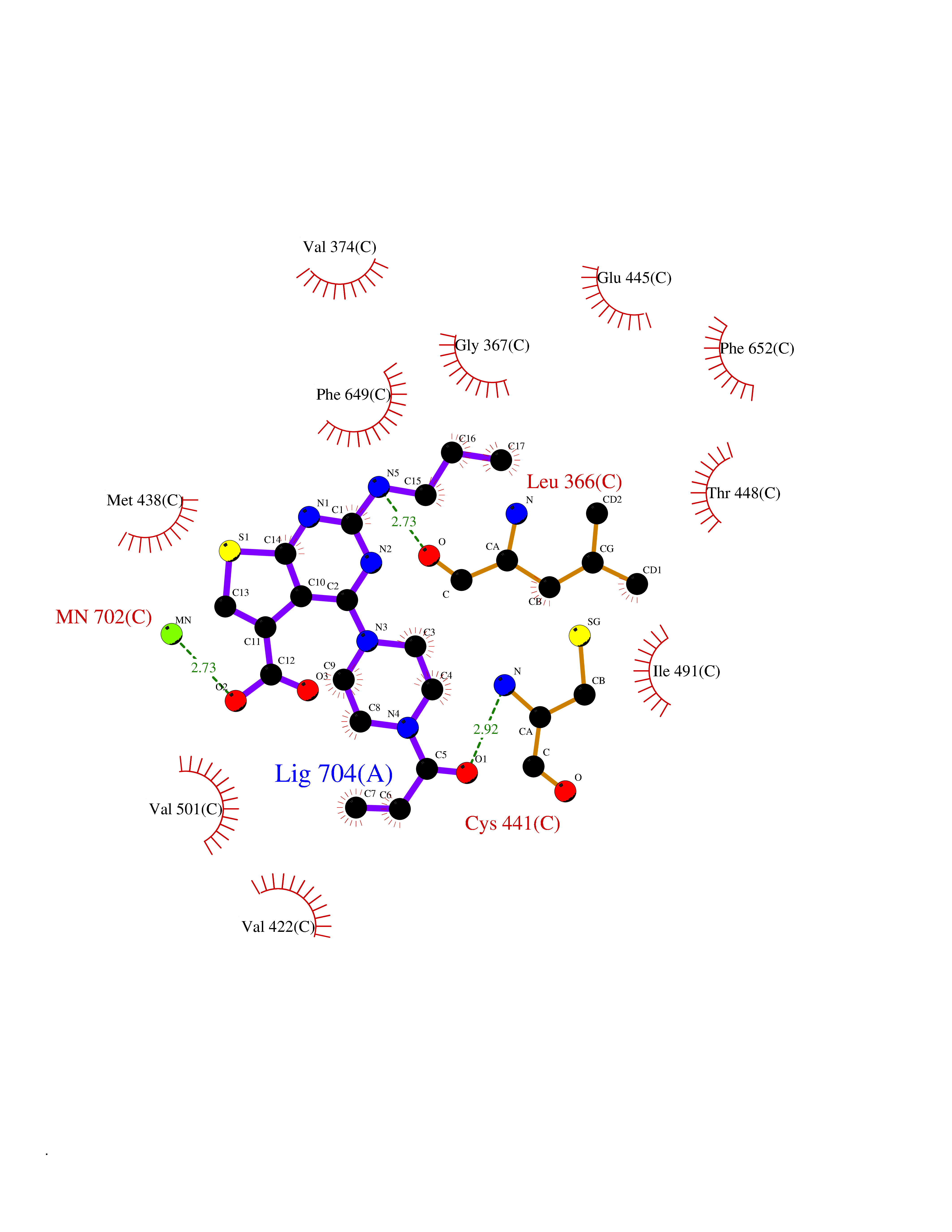 Ligplot Map 3D Binding mode Sequence EDAEAKAKYEAEAAFFANLKLSDFNIIDTLGVGGFGRVELVQLKSEESKTFAMKILKKRHIVDTRQQEHIRSEKQIMQGAHSDFIVRLYRTFKDSKYLYMLMEACLGGELWTILRDRGSFEDSTTRFYTACVVEAFAYLHSKGIIYRDLKPENLILDHRGYAKLVDFGFAKKIGFGKKTWXFCGTPEYVAPEIILNKGHDISADYWSLGILMYELLTGSPPFSGPDPMKTYNIILRGIDMIEFPKKIAKNAANLIKKLCRDNPSERLGNLKNGVKDIQKHKWFEGFNWEGLRKGTLTPPIIPSVASPTDTSNFDSFPEDNDEPPPDDNSGWDIDF Hydrogen bonds contact Hydrophobic contact | ||||
| 62 | Transferrin (TF) | 1RYO | 6.57 | |
Target general information Gen name TF Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Siderophilin; Serotransferrin; PRO1400; Beta-1 metal-binding globulin Protein family Transferrin family Biochemical class Transferrin Function It is responsible for the transport of iron from sites of absorption and heme degradation to those of storage and utilization. Serum transferrin may also have a further role in stimulating cell proliferation. Transferrins are iron binding transport proteins which can bind two Fe(3+) ions in association with the binding of an anion, usually bicarbonate. Related diseases Atransferrinemia (ATRAF) [MIM:209300]: A rare autosomal recessive disorder characterized by abnormal synthesis of transferrin leading to iron overload and microcytic hypochromic anemia. {ECO:0000269|PubMed:11110675, ECO:0000269|PubMed:15466165}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01370; DB14517; DB14518; DB01294; DB14526; DB14527; DB11136; DB14528; DB14529; DB14530; DB00515; DB09130; DB11397; DB13949; DB14490; DB14491; DB14488; DB14501; DB14489; DB13257; DB06215; DB06784; DB05260; DB01592; DB00893; DB00677; DB06757; DB11182; DB14520; DB01593; DB14487; DB14533; DB14548 Interacts with O43315; O00501; Q7Z7G2; Q9GZR5; Q9Y282; Q96KR6; P01350; P08034; Q8NBJ4; O15529; Q8TED1; Q7Z5P4; A8MZ59; O15173; Q96TC7; Q3KNW5; Q9BXS9-3; Q99523; O43278-2; Q8N9I0; P02786; Q4KMG9; Q9K0U9; Q09057; Q9K0V0; P02786 EC number NA Uniprot keywords 3D-structure; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; Ion transport; Iron; Iron transport; Metal-binding; Methylation; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Secreted; Signal; Transport Protein physicochemical properties Chain ID A Molecular weight (Da) 35854.5 Length 324 Aromaticity 0.1 Instability index 34.17 Isoelectric point 7.58 Charge (pH=7) 1.42 2D Binding mode Binding energy (Kcal/mol) -8.96 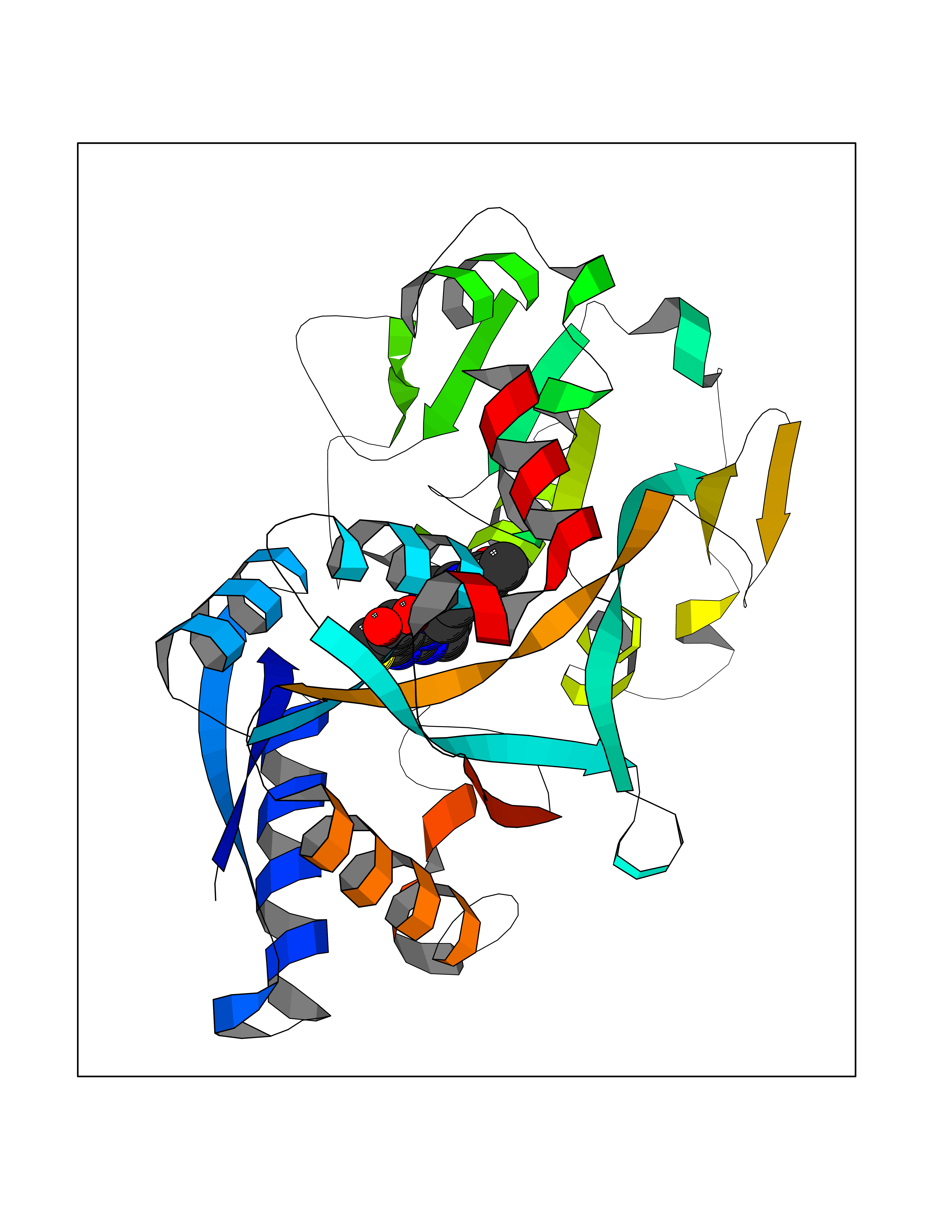 Molscript Map 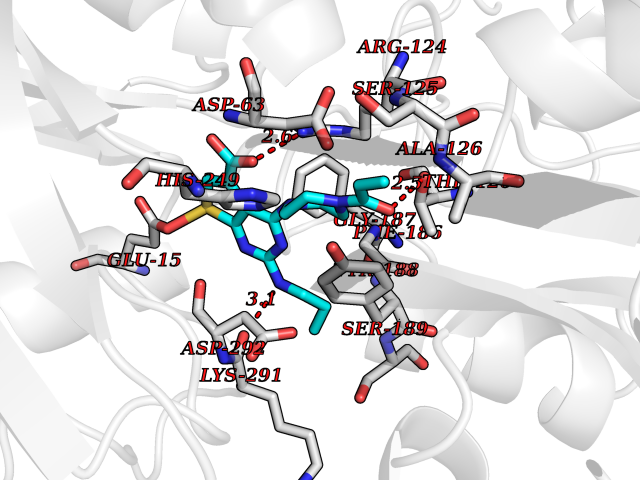 Pymol Map 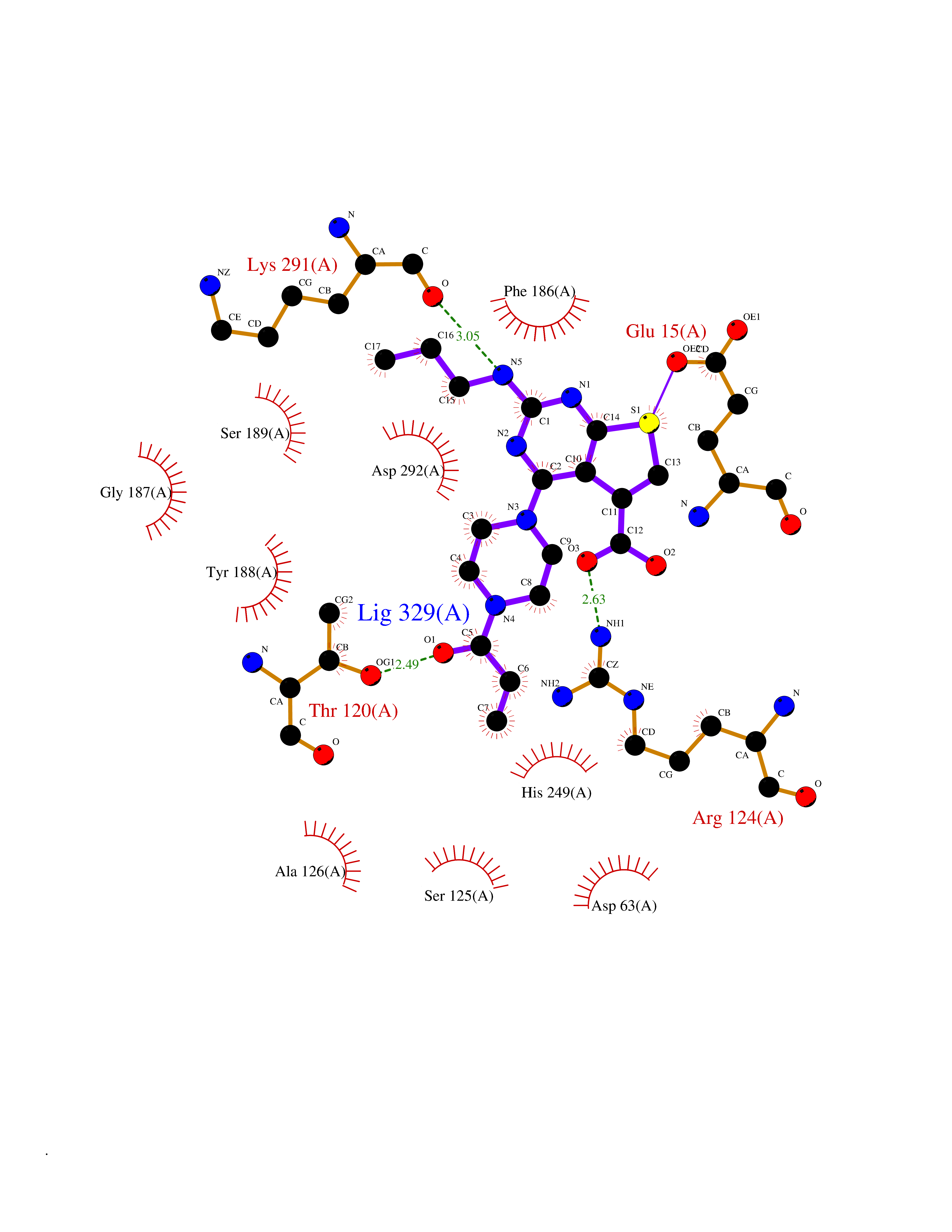 Ligplot Map 3D Binding mode Sequence KTVRWCAVSEHEATKCQSFRDHMKSVIPSDGPSVACVKKASYLDCIRAIAANEADAVTLDAGLVYDAYLAPNNLKPVVAEFYGSKEDPQTFYYAVAVVKKDSGFQMNQLRGKKSCHTGLGRSAGWNIPIGLLYCDLPEPRKPLEKAVANFFSGSCAPCADGTDFPQLCQLCPGCGCSTLNQYFGYSGAFKCLKDGAGDVAFVKHSTIFENLANKADRDQYELLCLDNTRKPVDEYKDCHLAQVPSHTVVARSMGGKEDLIWELLNQAQEHFGKDKSKEFQLFSSPHGKDLLFKDSAHGFLKVPPRMDAKMYLGYEYVTAIRNLR Hydrogen bonds contact Hydrophobic contact | ||||
| 63 | UDP-N-acetylglucosamine 1-carboxyvinyltransferase | 1C3R | 6.57 | |
Target general information Gen name murA Organism Aquifex aeolicus (strain VF5) Uniprot ID TTD ID NA Synonyms aq_1023 Protein family Histone deacetylase family Biochemical class Lyase Function UDP-N-acetylglucosamine 1-carboxyvinyltransferase activity. Related diseases Atrial fibrillation, familial, 14 (ATFB14) [MIM:615378]: A familial form of atrial fibrillation, a common sustained cardiac rhythm disturbance. Atrial fibrillation is characterized by disorganized atrial electrical activity and ineffective atrial contraction promoting blood stasis in the atria and reduces ventricular filling. It can result in palpitations, syncope, thromboembolic stroke, and congestive heart failure. {ECO:0000269|PubMed:19808477}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Genetic variations in SCN2B may be involved in Brugada syndrome (PubMed:23559163). This tachyarrhythmia is characterized by right bundle branch block and ST segment elevation on an electrocardiogram (ECG). It can cause the ventricles to beat so fast that the blood is prevented from circulating efficiently in the body. When this situation occurs, the individual will faint and may die in a few minutes if the heart is not reset. {ECO:0000269|PubMed:23559163}. Drugs (DrugBank ID) DB04297; DB02546 Interacts with NA EC number NA Uniprot keywords 3D-structure; Acetoin catabolism; Metal-binding; Reference proteome; Zinc Protein physicochemical properties Chain ID A,B Molecular weight (Da) 42338.9 Length 372 Aromaticity 0.12 Instability index 35.74 Isoelectric point 5.77 Charge (pH=7) -6.5 2D Binding mode Binding energy (Kcal/mol) -8.96 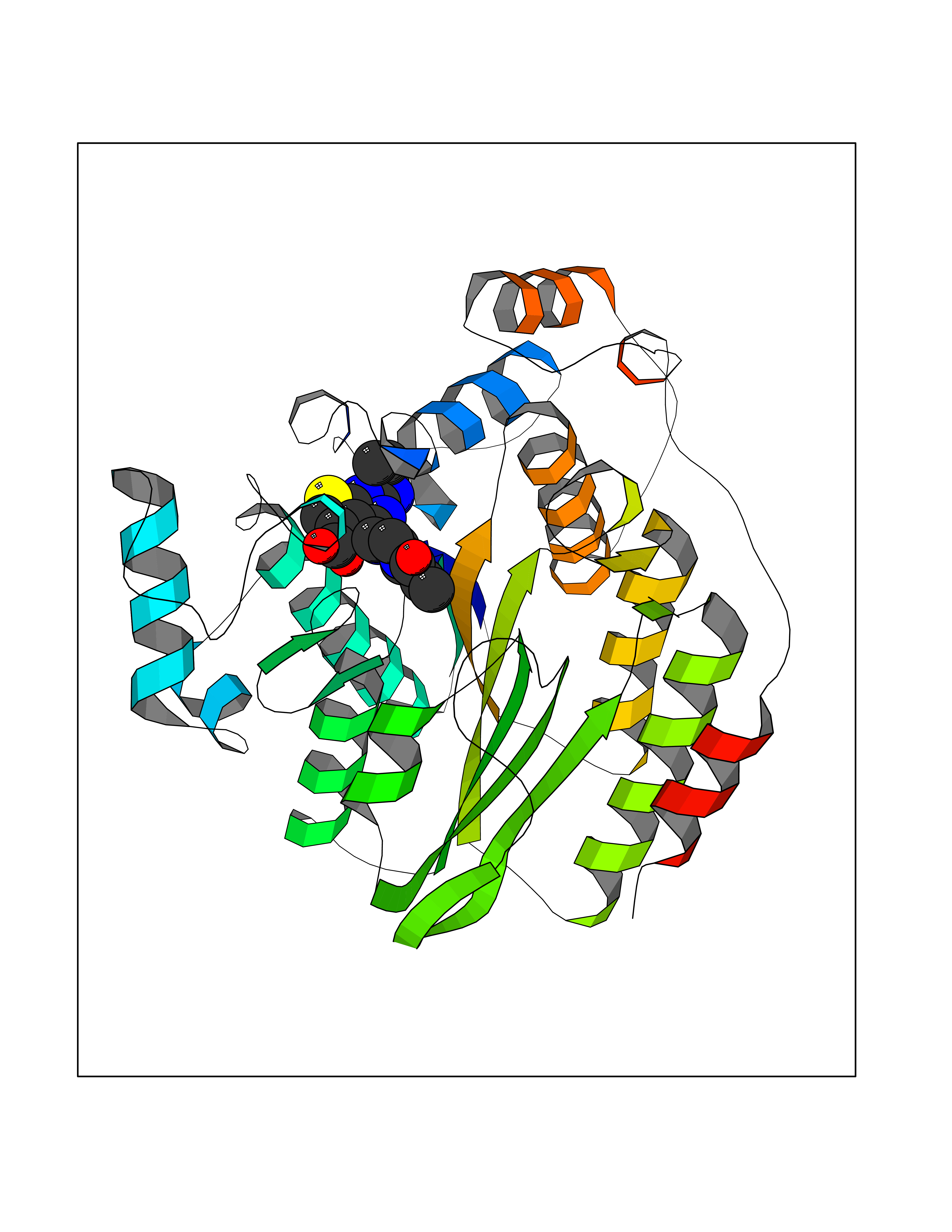 Molscript Map 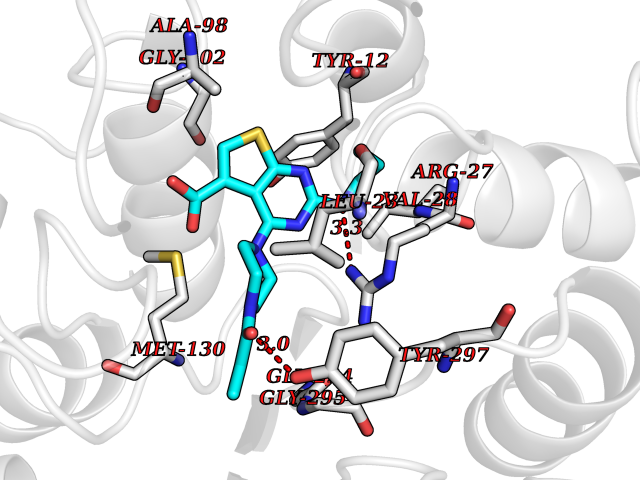 Pymol Map 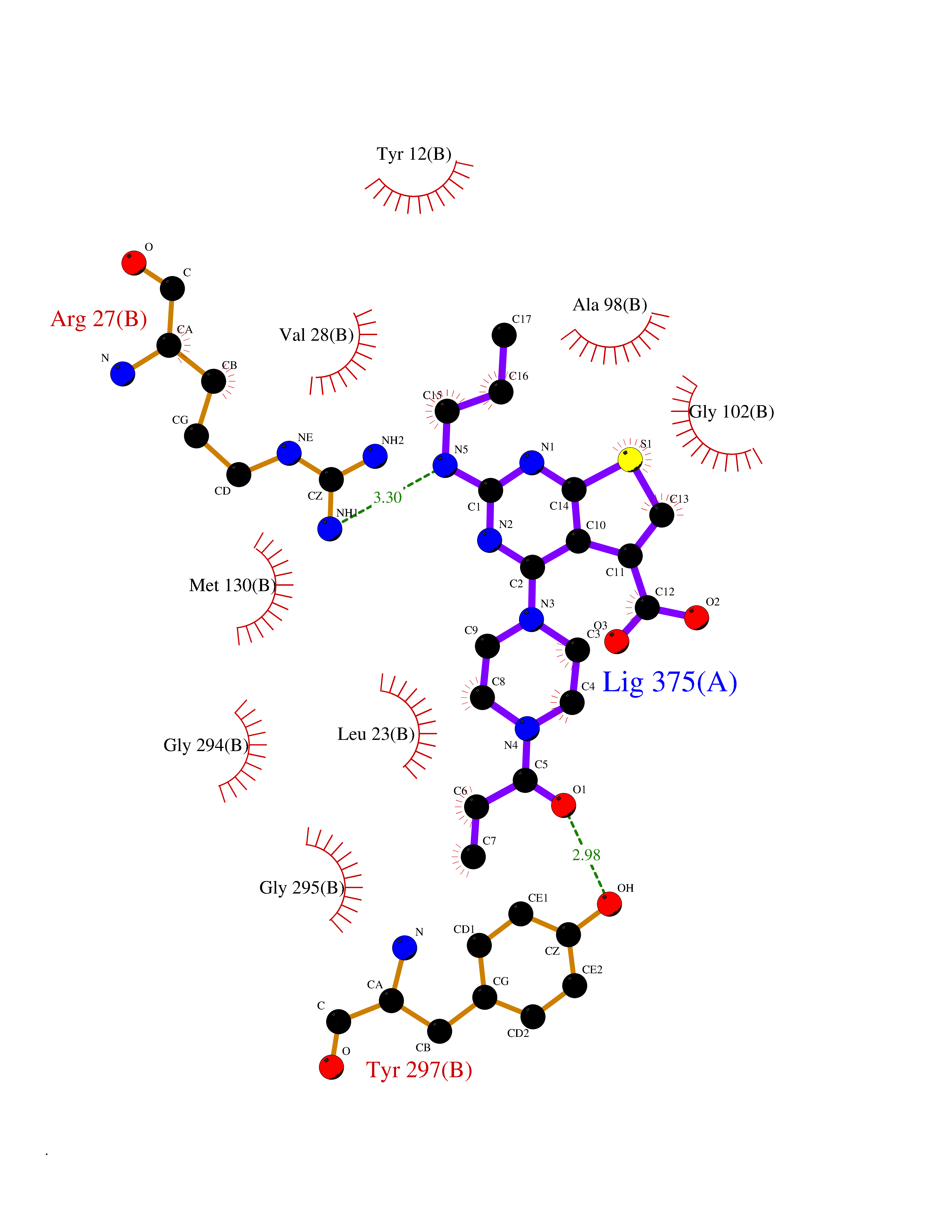 Ligplot Map 3D Binding mode Sequence KKVKLIGTLDYGKYRYPKNHPLKIPRVSLLLRFKDAMNLIDEKELIKSRPATKEELLLFHTEDYINTLMEAERSQSVPKGAREKYNIGGYENPVSYAMFTGSSLATGSTVQAIEEFLKGNVAFNPAGGMHHAFKSRANGFCYINNPAVGIEYLRKKGFKRILYIDLDAHHCDGVQEAFYDTDQVFVLSLHQSPEYAFPFEKGFLEEIGEGKGKGYNLNIPLPKGLNDNEFLFALEKSLEIVKEVFEPEVYLLQLGTDPLLEDYLSKFNLSNVAFLKAFNIVREVFGEGVYLGGGGYHPYALARAWTLIWCELSGREVPEKLNNKAKELLKSIDFEEFDDEVDRSYMLETLKDPWRGGEVRKEVKDTLEKAKA Hydrogen bonds contact Hydrophobic contact | ||||
| 64 | Smoothened homolog (SMO) | 4JKV | 6.57 | |
Target general information Gen name SMO Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Smo-D473H; SMOH; Protein Gx Protein family G-protein coupled receptor Fz/Smo family Biochemical class GPCR frizzled Function Binding of sonic hedgehog (SHH) to its receptor patched is thought to prevent normal inhibition by patched of smoothened (SMO). Required for the accumulation of KIF7, GLI2 and GLI3 in the cilia. Interacts with DLG5 at the ciliary base to induce the accumulation of KIF7 and GLI2 at the ciliary tip for GLI2 activation. G protein-coupled receptor that probably associates with the patched protein (PTCH) to transduce the hedgehog's proteins signal. Related diseases Curry-Jones syndrome (CRJS) [MIM:601707]: A multisystem disorder characterized by patchy skin lesions, polysyndactyly, diverse cerebral malformations, unicoronal craniosynostosis, iris colobomas, microphthalmia, and intestinal malrotation with myofibromas or hamartomas. {ECO:0000269|PubMed:24859340, ECO:0000269|PubMed:27236920}. The disease is caused by variants affecting the gene represented in this entry. 8 individuals have been identified with the disease-causing mutation Phe-412 and all were mosaic. The mutation could not be reliably detected in blood, greatest success rates were obtained with affected tissues obtained by invasive procedures. It is thought that the mutation has arisen postzygotically early during embryonic development (PubMed:27236920). This mutation has also been identified in ameloblastoma, medulloblastoma, meningioma, and basal cell carcinoma, and has been reported as the oncogenic driver in some of these tumors (PubMed:24859340). {ECO:0000269|PubMed:24859340, ECO:0000269|PubMed:27236920}. Drugs (DrugBank ID) DB01047; DB11978; DB06786; DB09143; DB08828 Interacts with NA EC number NA Uniprot keywords 3D-structure; Cell membrane; Cell projection; Developmental protein; Disease variant; Disulfide bond; G-protein coupled receptor; Glycoprotein; Membrane; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Signal; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A,B Molecular weight (Da) 37420.1 Length 333 Aromaticity 0.16 Instability index 25.17 Isoelectric point 6.65 Charge (pH=7) -0.76 2D Binding mode Binding energy (Kcal/mol) -8.96 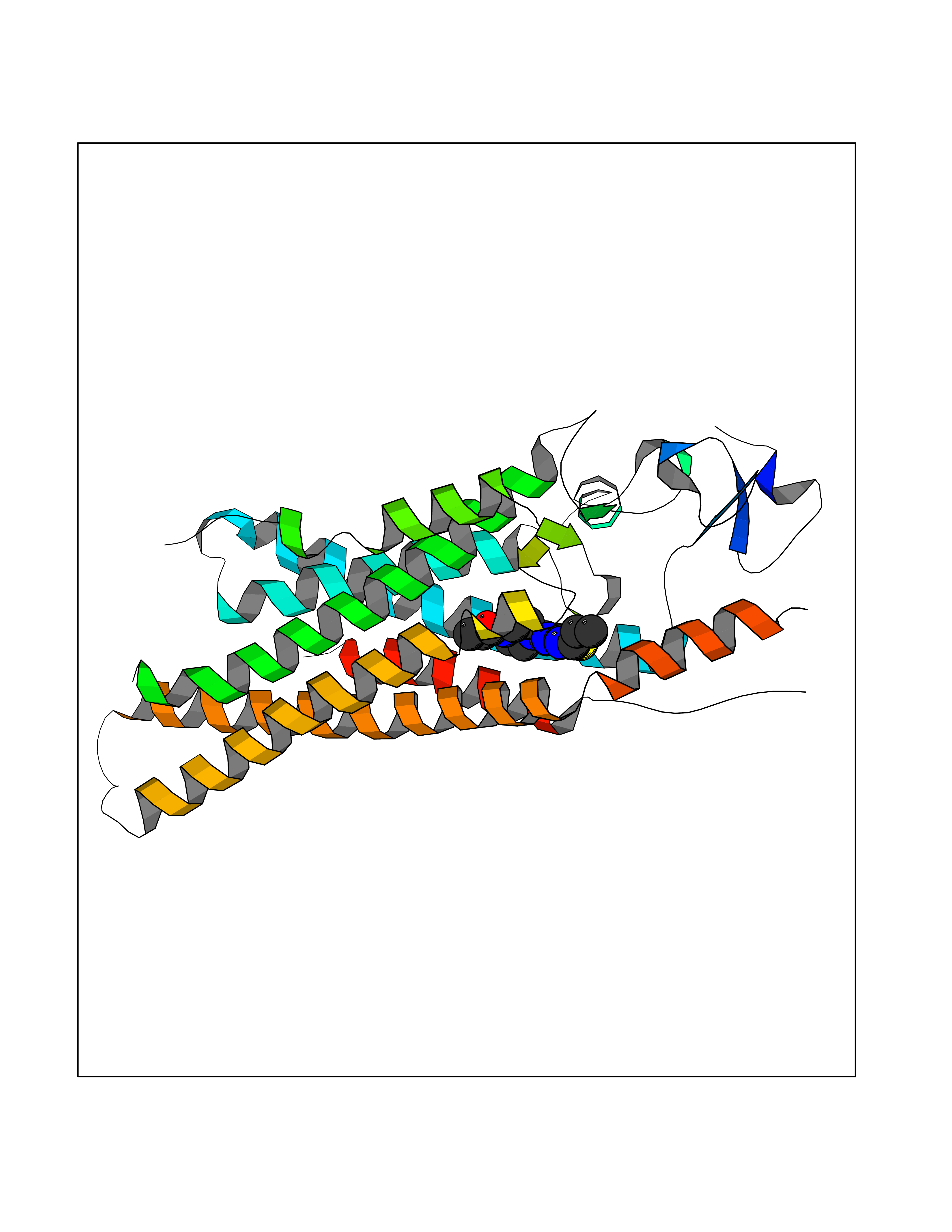 Molscript Map 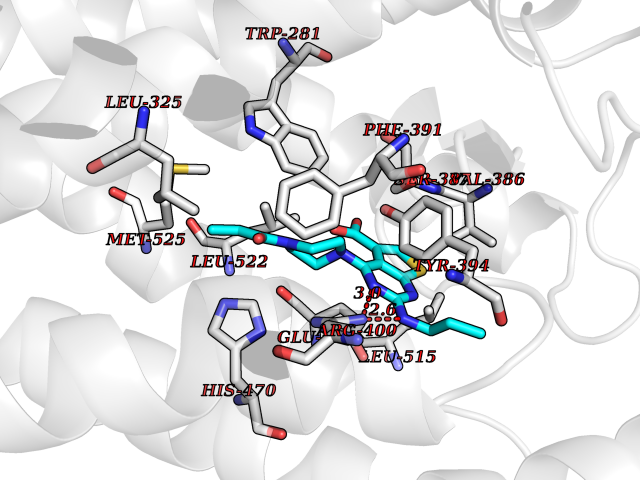 Pymol Map 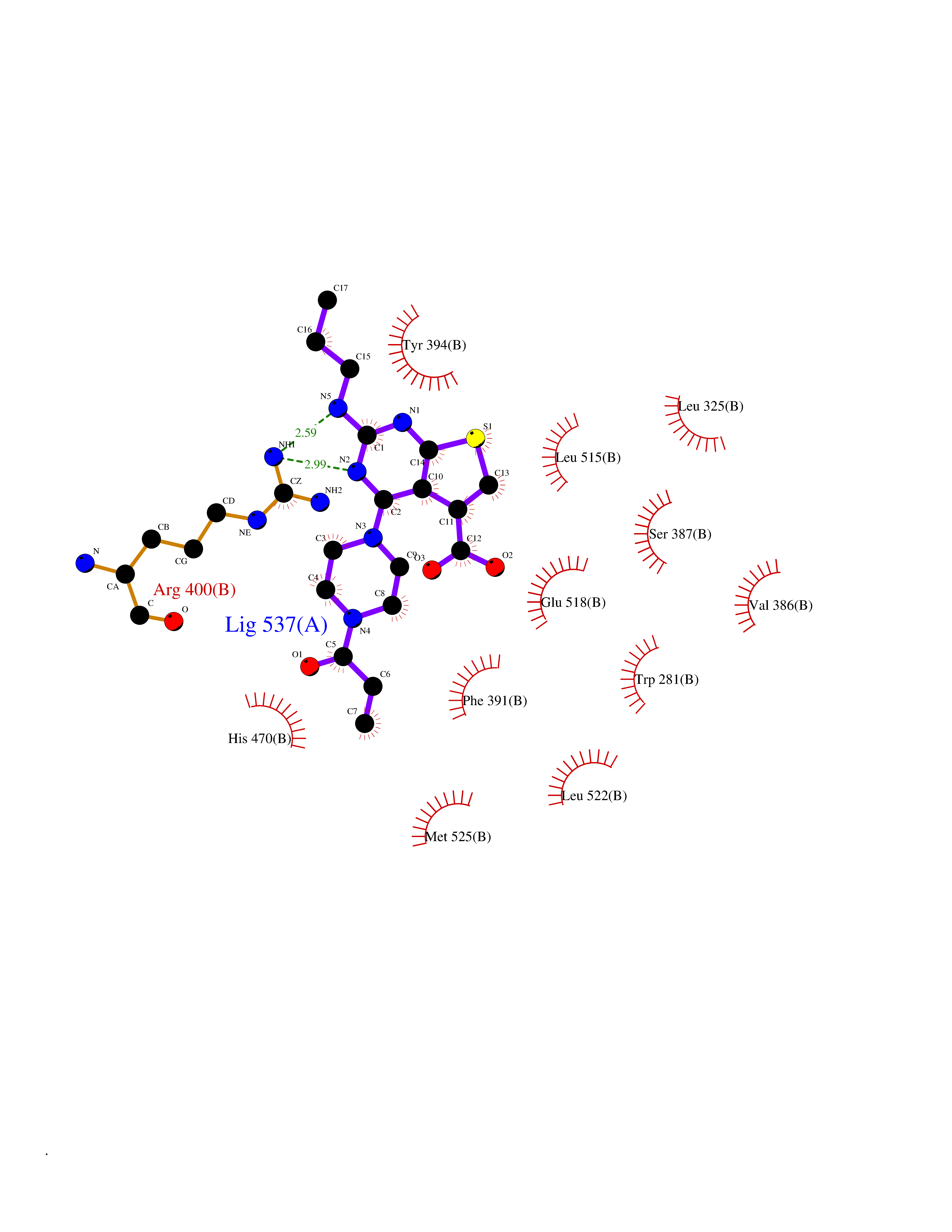 Ligplot Map 3D Binding mode Sequence AYIQKYLSGQCEVPLVRTDNPKSWYEDVEGCGIQCQNPLFTEAEHQDMHSYIAAFGAVTGLCTLFTLATFVADWRNSNRYPAVILFYVNACFFVGSIGWLAQFMDGARREIVCRADGTMRLGEPTSNETLSCVIIFVIVYYALMAGVVWFVVLTYAWHTSFKALGKTSYFHLLTWSLPFVLTVAILAVAQVDGDSVSGICFVGYKNYRYRAGFVLAPIGLVLIVGGYFLIRGVMTLFSIKSNHPGLLSEKAASKINETMLRLGIFGFLAFGFVLITFSCHFYDFFNQAEWERSFRDYVLCQANDCEIKNRPSLLVEKINLFAMFGTGIAMSTW Hydrogen bonds contact Hydrophobic contact | ||||
| 65 | Tankyrase-2 (TNKS-2) | 3U9H | 6.57 | |
Target general information Gen name TNKS2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Tankyrase-related protein; Tankyrase-like protein; Tankyrase II; TRF1-interacting ankyrin-related ADP-ribose polymerase 2; TNKL; TANK2; Protein poly-ADP-ribosyltransferase tankyrase-2; Poly [ADP-ribos Protein family ARTD/PARP family Biochemical class Glycosyltransferases Function Acts as an activator of the Wnt signaling pathway by mediating poly-ADP-ribosylation of AXIN1 and AXIN2, 2 key components of the beta-catenin destruction complex: poly-ADP-ribosylated target proteins are recognized by RNF146, which mediates their ubiquitination and subsequent degradation. Also mediates poly-ADP-ribosylation of BLZF1 and CASC3, followed by recruitment of RNF146 and subsequent ubiquitination. Mediates poly-ADP-ribosylation of TERF1, thereby contributing to the regulation of telomere length. Stimulates 26S proteasome activity. Poly-ADP-ribosyltransferase involved in various processes such as Wnt signaling pathway, telomere length and vesicle trafficking. Related diseases Intellectual developmental disorder with macrocephaly, seizures, and speech delay (IDDMSSD) [MIM:618158]: An autosomal dominant neurodevelopmental disorder characterized by impaired intellectual development, poor speech, postnatal macrocephaly, and seizures. {ECO:0000269|PubMed:30290153}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with O15084; Q7Z6K5-1; O15169; Q9NWV8; P11274; Q13698; Q9NRI5; Q6V0I7; Q9NWT6; P14652; Q9UIQ6; Q14980; Q9BZL4; Q92698; P78314; O43815; P54274; Q9C0C2; Q9UHP3; Q06649 EC number EC 2.4.2.30 Uniprot keywords 3D-structure; ADP-ribosylation; ANK repeat; Chromosome; Cytoplasm; Glycosyltransferase; Golgi apparatus; Hydroxylation; Membrane; Metal-binding; NAD; Nucleotidyltransferase; Nucleus; Proteomics identification; Reference proteome; Repeat; Telomere; Transferase; Ubl conjugation; Wnt signaling pathway; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 23695.5 Length 208 Aromaticity 0.11 Instability index 47.61 Isoelectric point 8.28 Charge (pH=7) 2.88 2D Binding mode Binding energy (Kcal/mol) -8.96 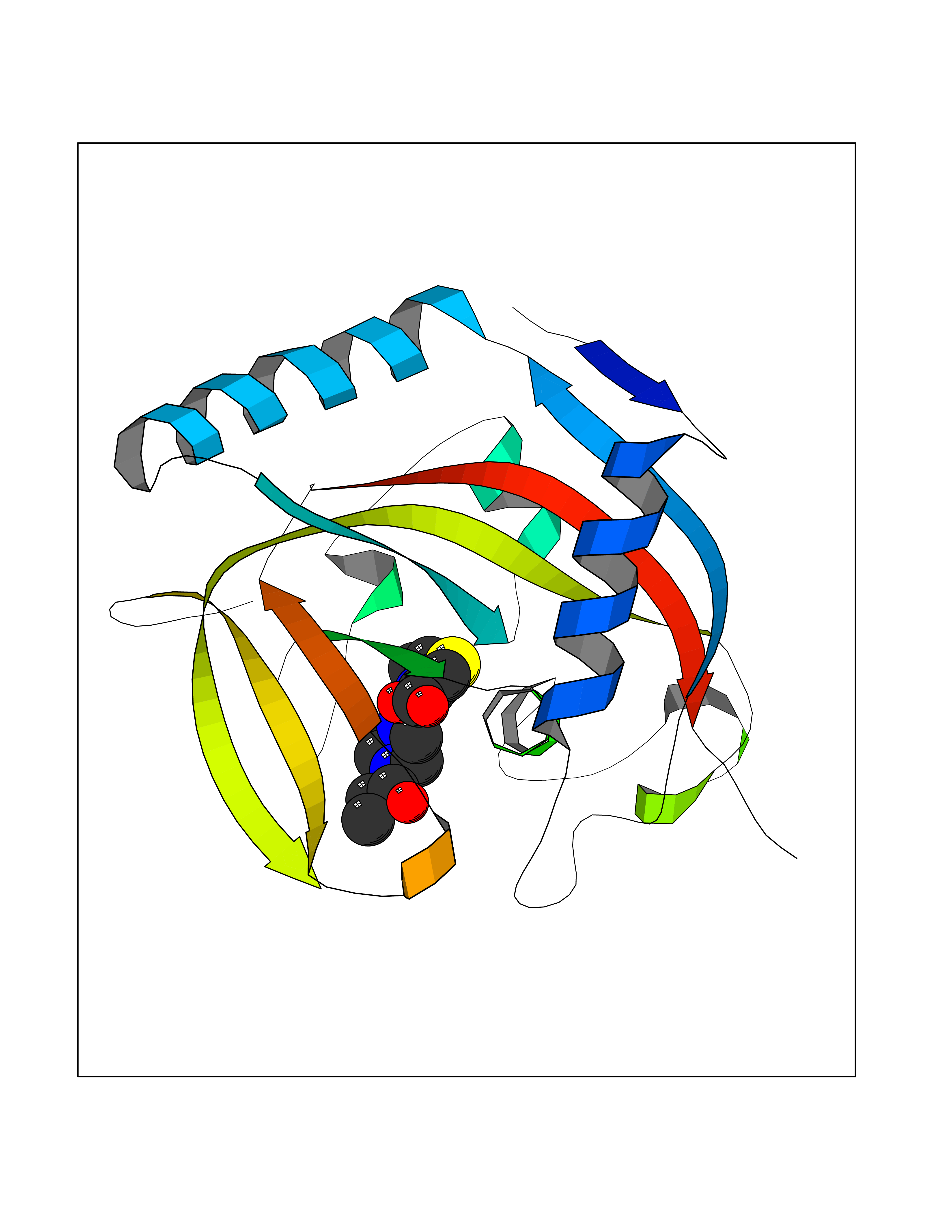 Molscript Map 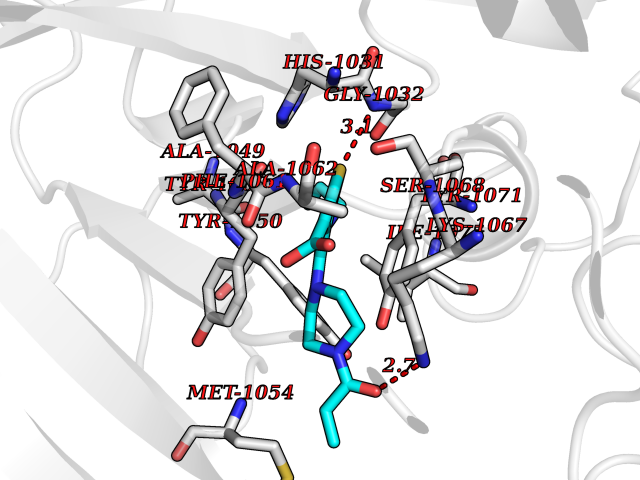 Pymol Map 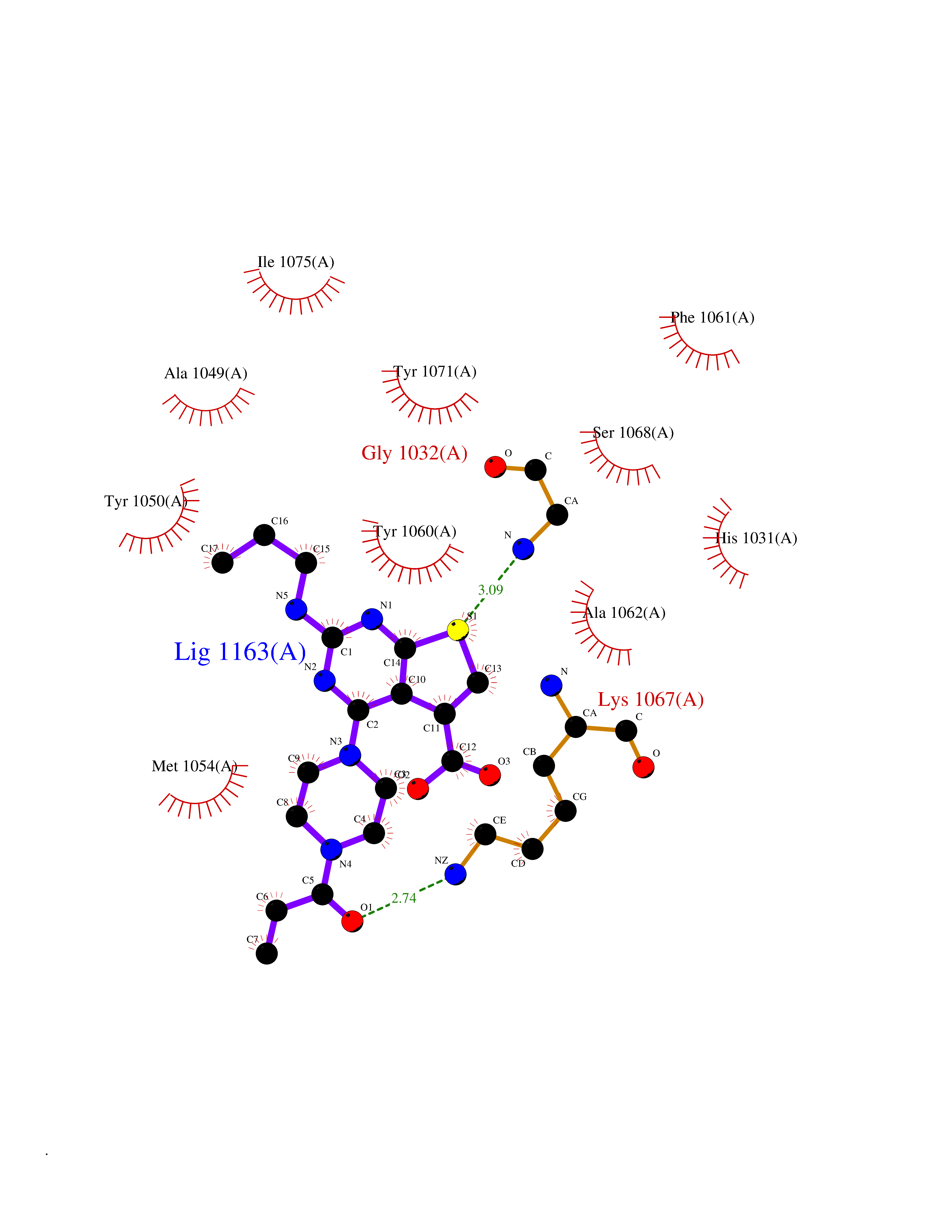 Ligplot Map 3D Binding mode Sequence GTILIDLSPDDKEFQSVEEEMQSTVREHRDGGHAGGIFNRYNILKIQKVCNKKLWERYTHRRKEVSEENHNHANERMLFHGSPFVNAIIHKGFDERHAYIGGMFGAGIYFAENSSKSNQYVYGIGGGTGCPVHKDRSCYICHRQLLFCRVTLGKSFLQFSAMAHSPPGHHSVTGRPSVNGLALAEYVIYRGEQAYPEYLITYQIMRPE Hydrogen bonds contact Hydrophobic contact | ||||
| 66 | Squalene synthetase (FDFT1) | 3WCM | 6.57 | |
Target general information Gen name FDFT1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Squalene synthase; SS; SQS; Farnesyl-diphosphate farnesyltransferase; FPP:FPP farnesyltransferase Protein family Phytoene/squalene synthase family Biochemical class Alkyl aryl transferase Function Participates in the isoprenoid biosynthetic pathway, catalyzing a two-step reaction in which two identical molecules of farnesyl pyrophosphate (FPP) are converted into squalene, with the consumption of NADPH. Related diseases Squalene synthase deficiency (SQSD) [MIM:618156]: An autosomal recessive disorder characterized by profound developmental delay, brain abnormalities, 2/3 syndactyly of the toes, facial dysmorphisms, low total and LDL-cholesterol, and abnormal urine organic acids. {ECO:0000269|PubMed:29909962}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB05317 Interacts with Q13520; Q3SXY8; P04233-2; P11912; O75503; O43889-2; Q9GZR5; Q5JX71; P48165; Q8TDT2; Q8N5M9; Q6IBW4-4; Q96RD7; Q14973; Q9NQQ7-3; Q96MV1; Q9Y320 EC number EC 2.5.1.21 Uniprot keywords 3D-structure; Alternative splicing; Cholesterol biosynthesis; Cholesterol metabolism; Endoplasmic reticulum; Lipid biosynthesis; Lipid metabolism; Magnesium; Membrane; Metal-binding; Multifunctional enzyme; NAD; NADP; Proteomics identification; Reference proteome; Steroid biosynthesis; Steroid metabolism; Sterol biosynthesis; Sterol metabolism; Transferase; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 37860 Length 329 Aromaticity 0.1 Instability index 40.19 Isoelectric point 5.47 Charge (pH=7) -7.65 2D Binding mode Binding energy (Kcal/mol) -8.96 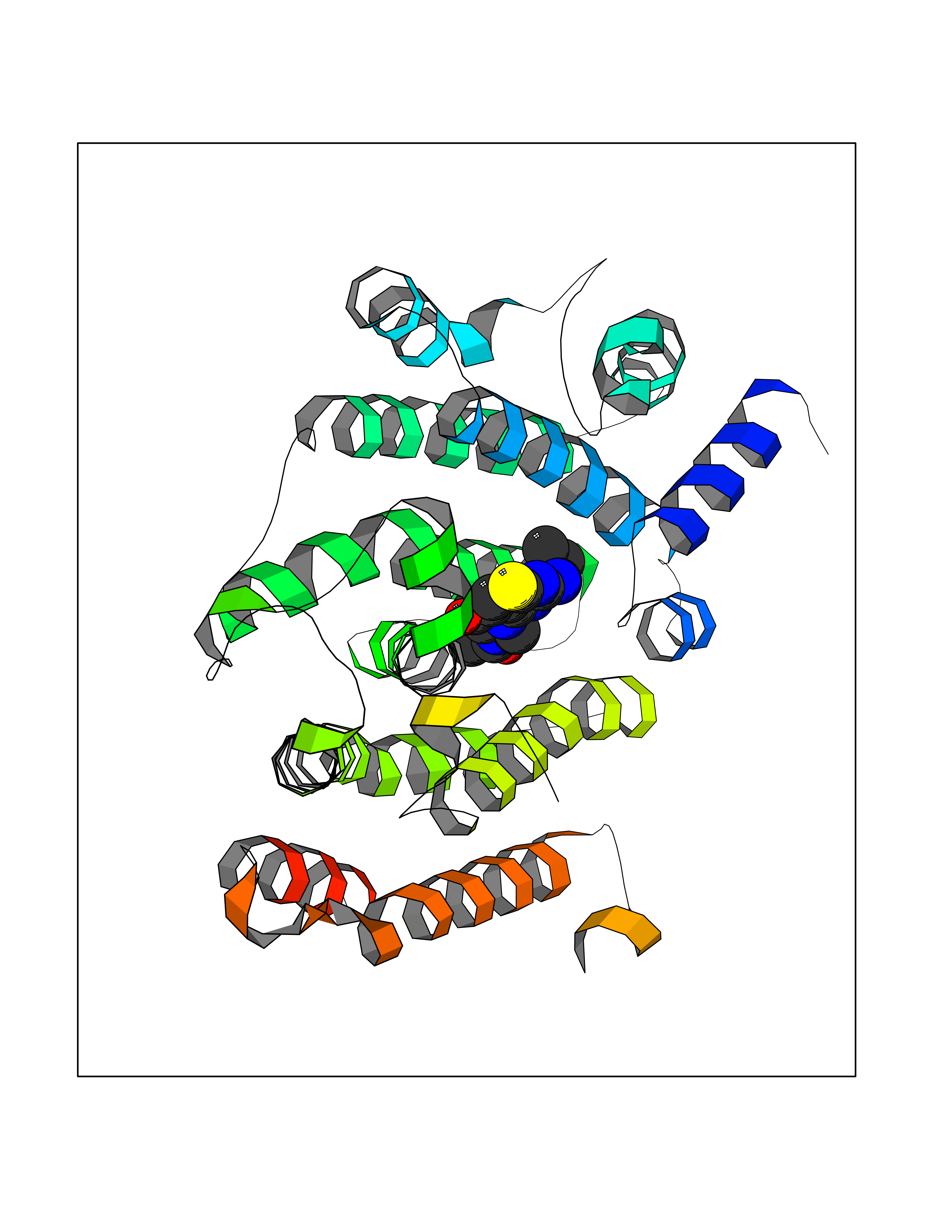 Molscript Map 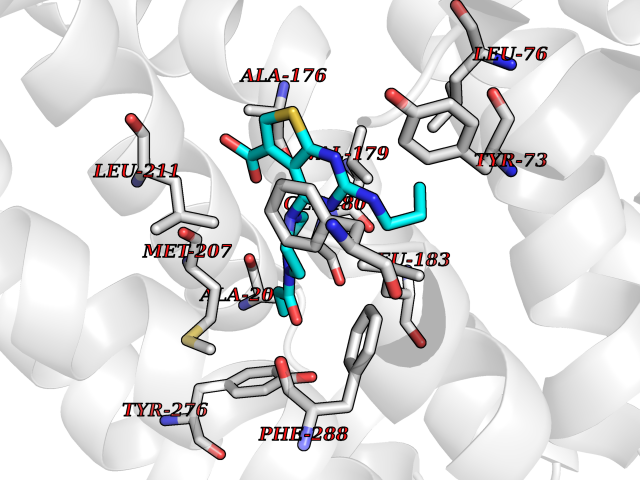 Pymol Map 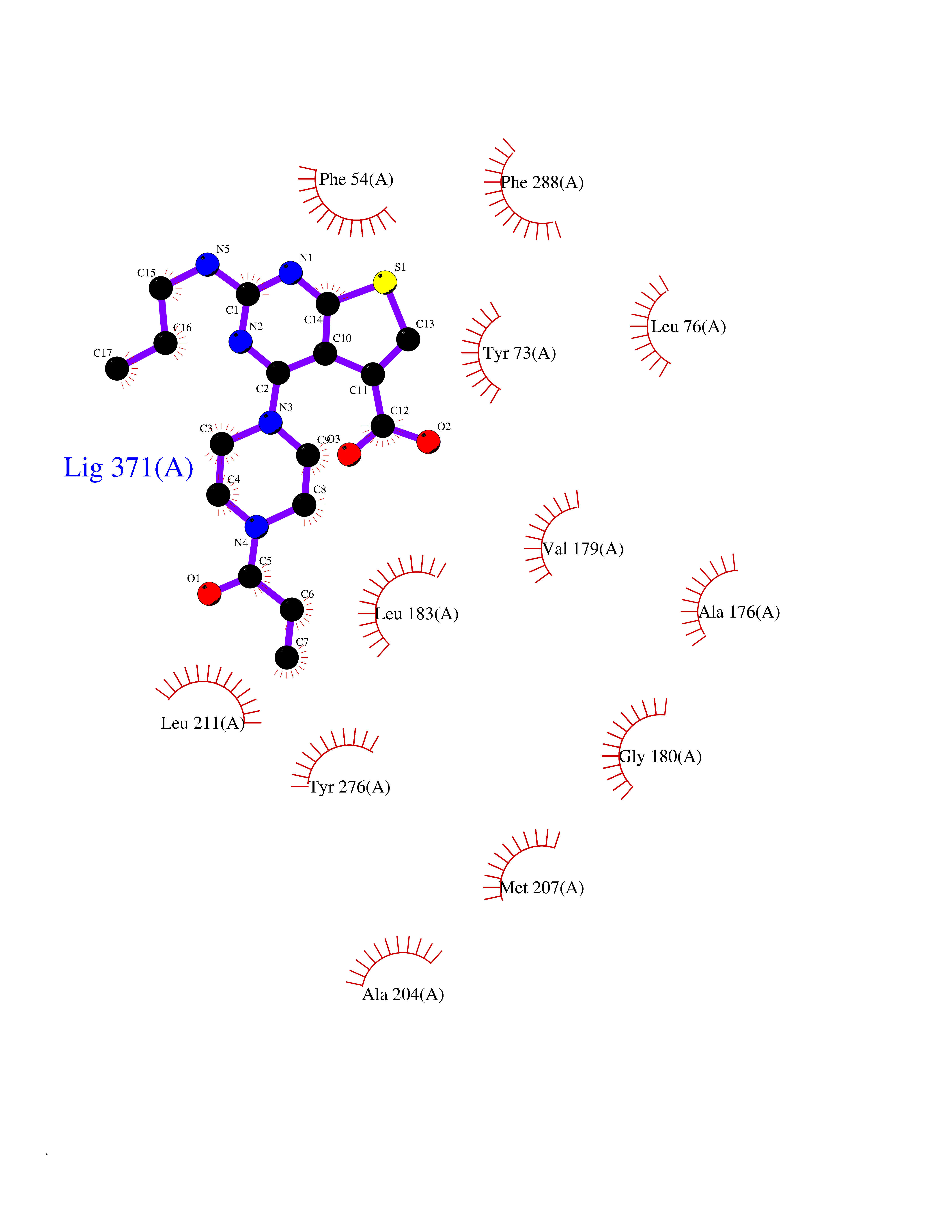 Ligplot Map 3D Binding mode Sequence LSSSLKTCYKYLNQTSRSFAAVIQALDGEMRNAVCIFYLVLRALDTLEDDMTISVEKKVPLLHNFHSFLYQPDWRFMESKEKDRQVLEDFPTISLEFRNLAEKYQTVIADICRRMGIGMAEFLDKHVTSEQEWDKYCHYVAGLVGIGLSRLFSASEFEDPLVGEDTERANSMGLFLQKTNIIRDYLEDQQGGREFWPQEVWSRYVKKLGDFALPENIDLAVQCLNELITNALHHIPDVITYLSRLRNQSVFNFCAIPQVMAIATLAACYNNQQVFKGAVLIVTLMMDATNMPAVKAIIYQYMEEIYHRIPDSNPSSSKTRQIISTIRTQ Hydrogen bonds contact Hydrophobic contact | ||||
| 67 | Fatty acid-binding protein, intestinal | 3AKM | 6.56 | |
Target general information Gen name FABP2 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms FABPI Protein family Calycin superfamily, Fatty-acid binding protein (FABP) family Biochemical class Transport protein Function Fatty acid binding.Transporter activity. Related diseases Usher syndrome 3B (USH3B) [MIM:614504]: A syndrome characterized by progressive vision and hearing loss during early childhood. Some patients have the so-called 'Charles Bonnet syndrome,' involving decreased visual acuity and vivid visual hallucinations. USH is a genetically heterogeneous condition characterized by the association of retinitis pigmentosa with sensorineural deafness. Age at onset and differences in auditory and vestibular function distinguish Usher syndrome type 1 (USH1), Usher syndrome type 2 (USH2) and Usher syndrome type 3 (USH3). USH3 is characterized by postlingual, progressive hearing loss, variable vestibular dysfunction, and onset of retinitis pigmentosa symptoms, including nyctalopia, constriction of the visual fields, and loss of central visual acuity, usually by the second decade of life. {ECO:0000269|PubMed:22279524}. The disease may be caused by variants affecting the gene represented in this entry.; DISEASE: Charcot-Marie-Tooth disease, axonal, 2W (CMT2W) [MIM:616625]: An autosomal dominant, axonal form of Charcot-Marie-Tooth disease, a disorder of the peripheral nervous system, characterized by progressive weakness and atrophy, initially of the peroneal muscles and later of the distal muscles of the arms. Charcot-Marie-Tooth disease is classified in two main groups on the basis of electrophysiologic properties and histopathology: primary peripheral demyelinating neuropathies (designated CMT1 when they are dominantly inherited) and primary peripheral axonal neuropathies (CMT2). Neuropathies of the CMT2 group are characterized by signs of axonal degeneration in the absence of obvious myelin alterations, normal or slightly reduced nerve conduction velocities, and progressive distal muscle weakness and atrophy. CMT2W patients manifest a peripheral neuropathy mainly affecting the lower limbs and resulting in gait difficulties and distal sensory impairment. Most patients also have upper limb involvement. {ECO:0000269|PubMed:22930593, ECO:0000269|PubMed:26072516, ECO:0000269|PubMed:29235198}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB04557; DB09213; DB00783; DB13952; DB13953; DB13954; DB13955; DB13956; DB01050; DB08231; DB03796; DB01138 Interacts with O95994; Q9NYB0 EC number NA Uniprot keywords 3D-structure; Acetylation; Cytoplasm; Lipid-binding; Proteomics identification; Reference proteome; Transport Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 15075.9 Length 131 Aromaticity 0.11 Instability index 32.01 Isoelectric point 6.88 Charge (pH=7) -0.09 2D Binding mode Binding energy (Kcal/mol) -8.95 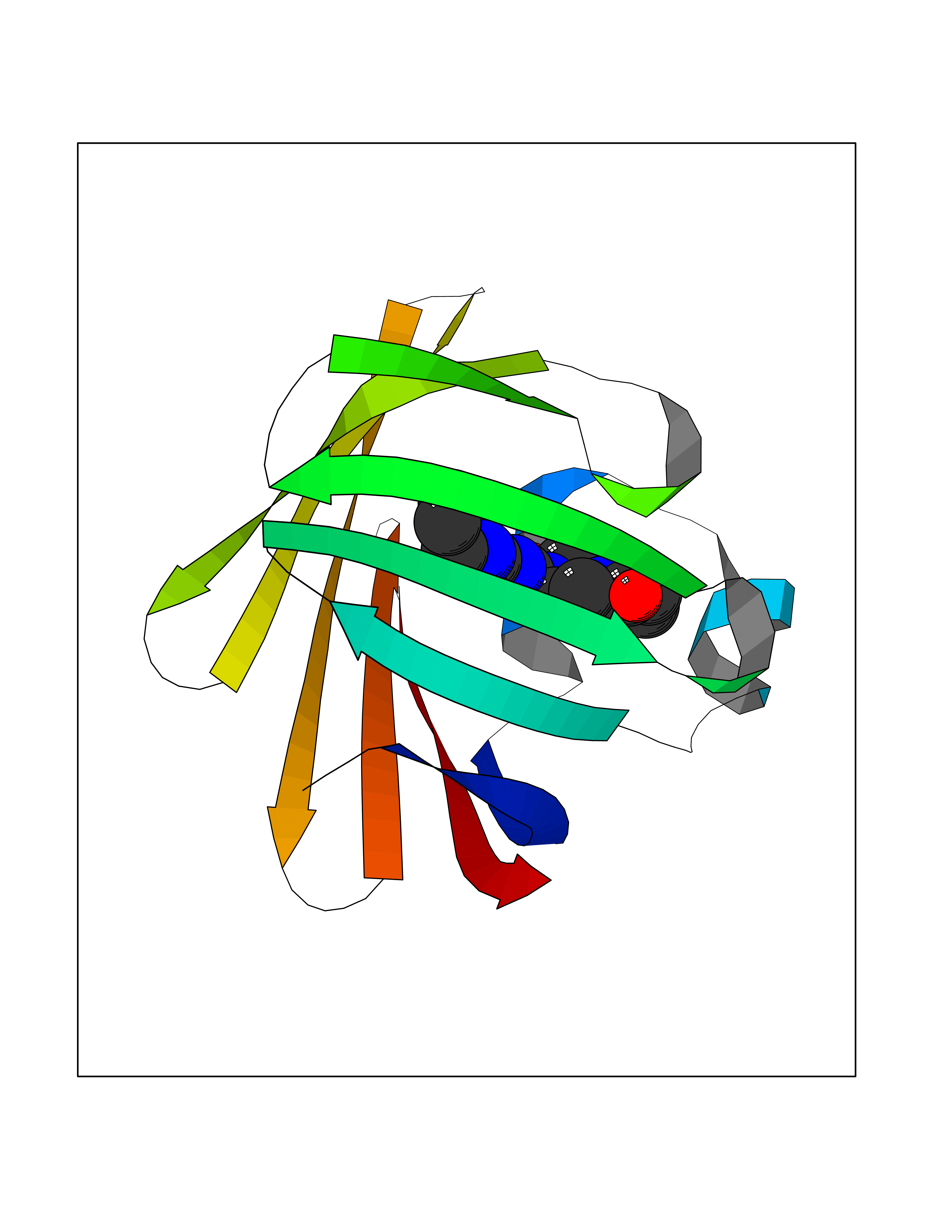 Molscript Map 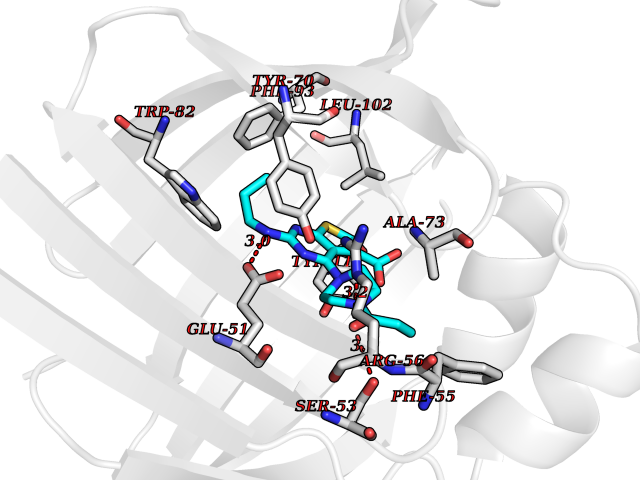 Pymol Map 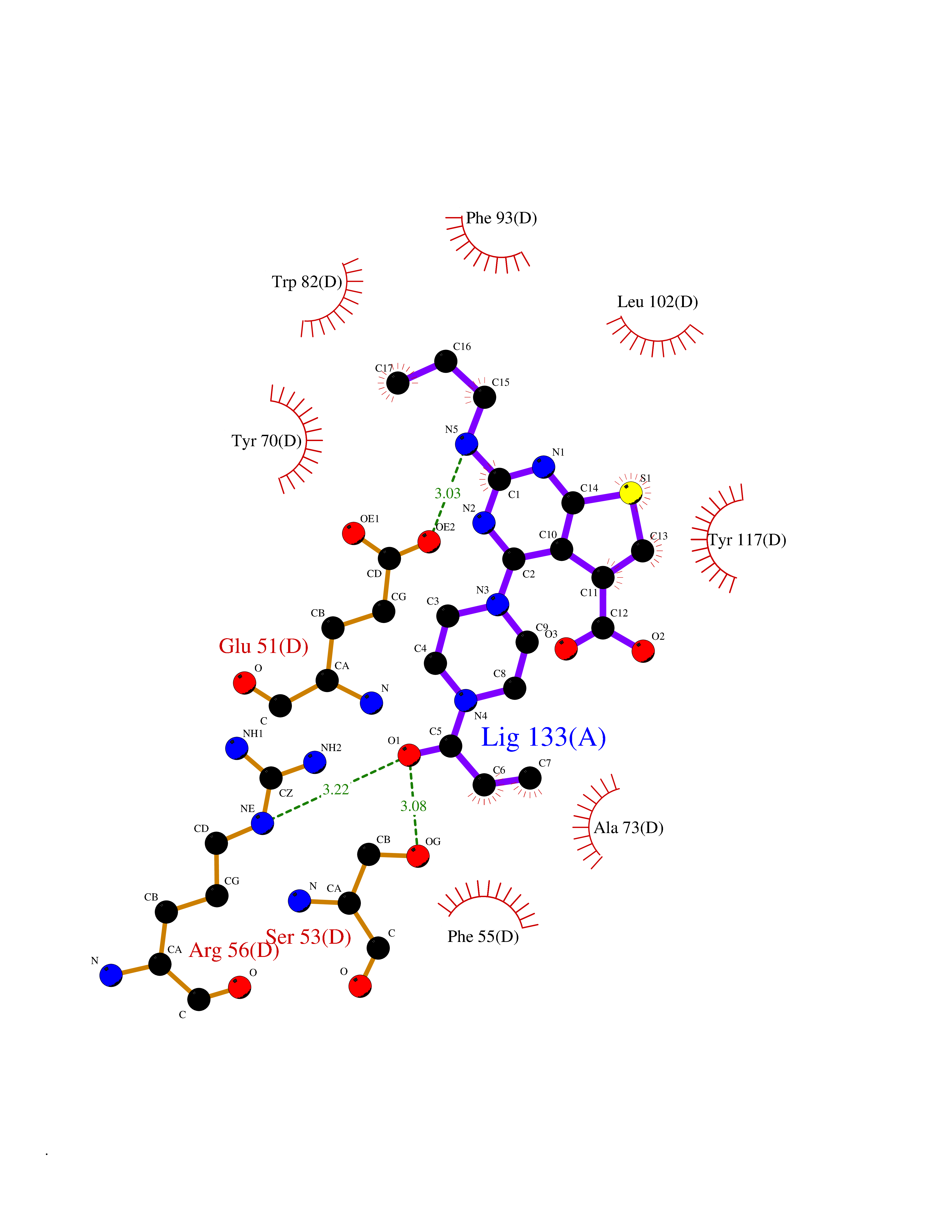 Ligplot Map 3D Binding mode Sequence AFDSTWKVDRSENYDKFMEKMGVNIVKRKLAAHDNLKLTITQEGNKFTVKESSAFRNIEVVFELGVTFNYNLADGTELRGTWSLEGNKLIGKFKRTDNGNELNTVREIIGDELVQTYVYEGVEAKRIFKKD Hydrogen bonds contact Hydrophobic contact | ||||
| 68 | Cytoplasmic aspartate aminotransferase (GOT1) | 3II0 | 6.56 | |
Target general information Gen name GOT1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Glutamate oxaloacetate transaminase-1; GOT1 Protein family Class-I pyridoxal-phosphate-dependent aminotransferase family Biochemical class Transaminase Function Biosynthesis of L-glutamate from L-aspartate or L- cysteine. Important regulator of levels of glutamate, the major excitatory neurotransmitter of the vertebrate central nervous system. Acts as a scavenger of glutamate in brain neuroprotection. The aspartate aminotransferase activity is involved in hepatic glucose synthesis during development and in adipocyte glyceroneogenesis. Using L-cysteine as substrate, regulates levels of mercaptopyruvate, an important source of hydrogen sulfide. Mercaptopyruvate is converted into H(2)S via the action of 3- mercaptopyruvate sulfurtransferase (3MST). Hydrogen sulfide is an important synaptic modulator and neuroprotectant in the brain. Related diseases Multiple fibroadenomas of the breast (MFAB) [MIM:615554]: A benign breast disease marked by lobuloalveolar growth with abnormally high proliferation of the epithelium, and characterized by the presence of more than 3 fibroadenomas in one breast. Fibroadenomas are adenomas containing fibrous tissue. {ECO:0000269|PubMed:18779591}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Hyperprolactinemia (HPRL) [MIM:615555]: A disorder characterized by increased levels of prolactin in the blood not associated with gestation or the puerperium. HPRL may result in infertility, hypogonadism, and galactorrhea. {ECO:0000269|PubMed:24195502}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00210; DB00128; DB09130; DB00151; DB00142; DB04299; DB00114 Interacts with NA EC number EC 2.6.1.1 Uniprot keywords 3D-structure; Alternative splicing; Amino-acid biosynthesis; Aminotransferase; Cytoplasm; Direct protein sequencing; Phosphoprotein; Proteomics identification; Pyridoxal phosphate; Reference proteome; Transferase Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 51732.3 Length 462 Aromaticity 0.1 Instability index 31.83 Isoelectric point 7.48 Charge (pH=7) 0.86 2D Binding mode Binding energy (Kcal/mol) -8.95 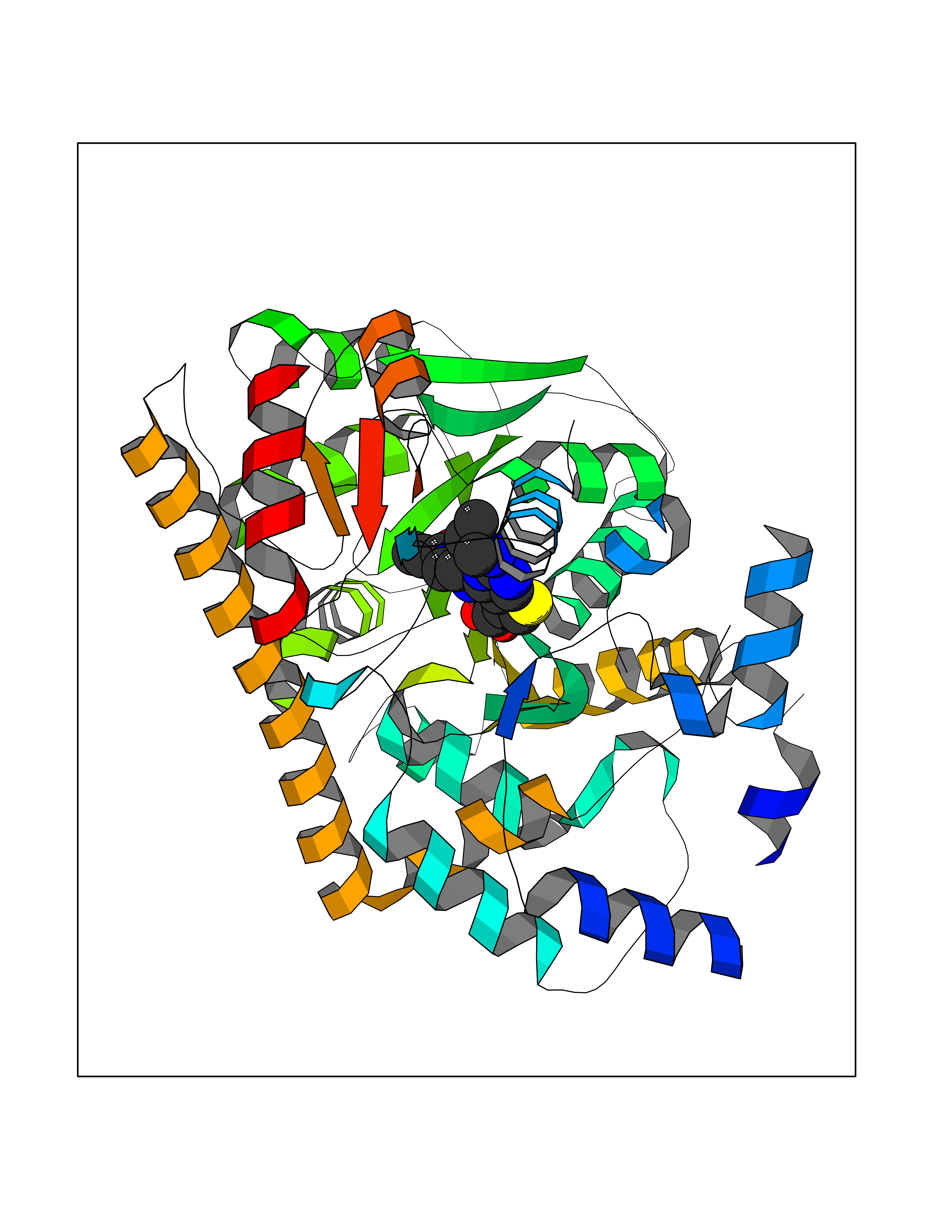 Molscript Map 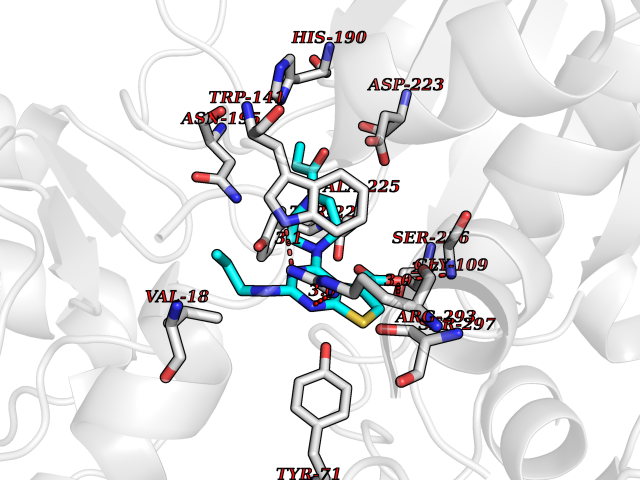 Pymol Map 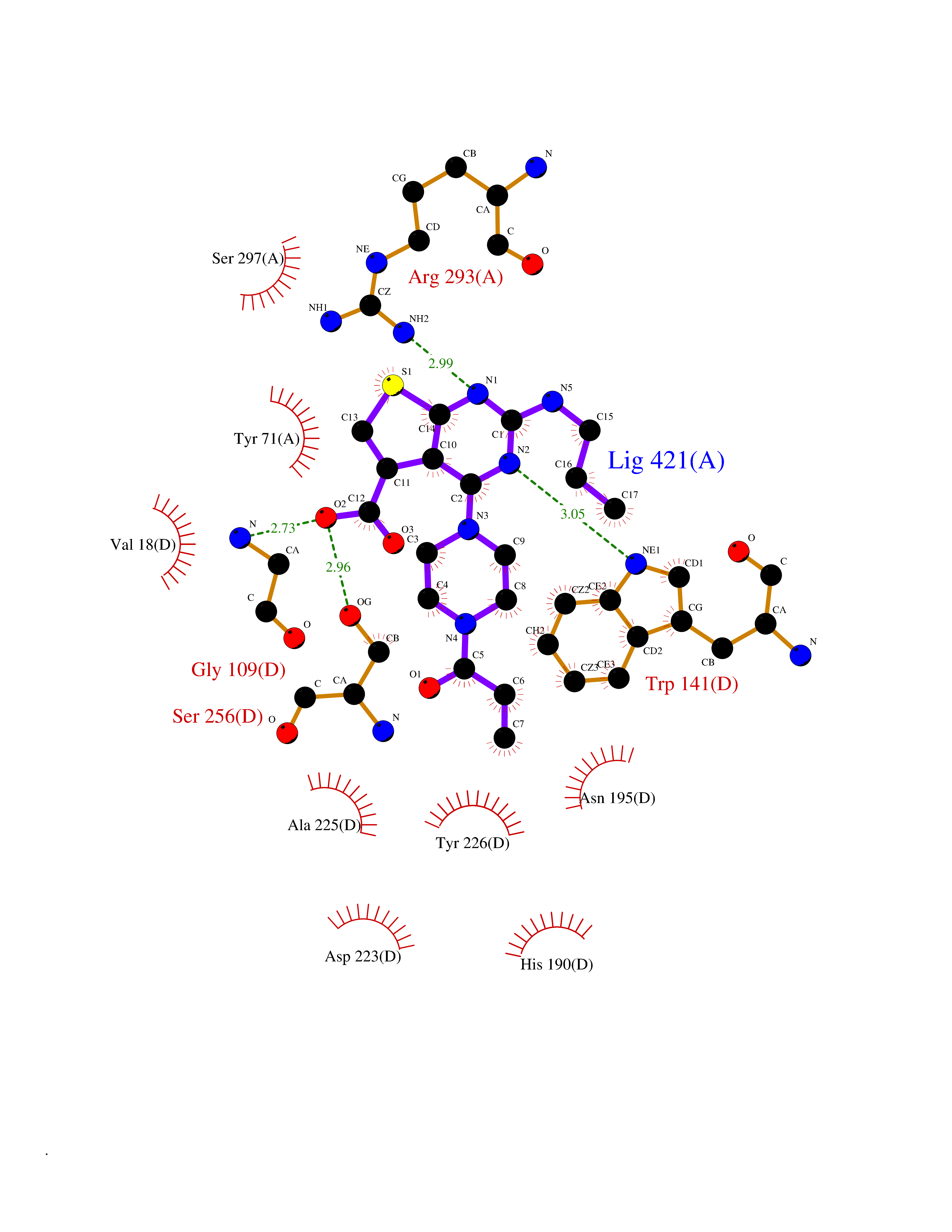 Ligplot Map 3D Binding mode Sequence PVLVFKLTVLPVVKKVEQKIANDNSLNHEYLPILGLAEFRVQSLGGTGALRIGADEKIVRITWSNPMQPVLVFKLTADFREDPDPRKVNLGVGAYRTDDCHPWVLPVVKKVEQKIANDNSLNHEYLPILGLAEFRSCASRLALGDDSPALKEKRVGGVQSLGGTGALRIGADFLARWYNGTNNKNTPVYVSSPTWENHNAVFSAAGFKDIRSYRYWDAEKRGLDLQGFLNDLENAPEFSIVVLHACAHNPTGIDPTPEQWKQIASVMKHRFLFPFFDSAYQGFASGNLERDAWAIRYFVSEGFEFFCAQSFSKNFGLYNERVGNLTVVGKEPESILQVLSQMEKIVRITWSNPPAQGARIVASTLSNPELFEEWTGNVKTMADRILTMRSELRARLEALKTPGTWNHITDQIGMFSFTGLNPKQVEYLVNEKHIYLLPSGRINVSGLTTKNLDYVATSIHEA Hydrogen bonds contact Hydrophobic contact | ||||
| 69 | Staphylococcus Enoyl ACP reductase (Stap-coc fabI) | 6YUR | 6.56 | |
Target general information Gen name Stap-coc fabI Organism Staphylococcus aureus (strain NCTC 8325 / PS 47) Uniprot ID TTD ID Synonyms NADPHdependent enoylACP reductase; Enoyl[acylcarrierprotein] reductase [NADPH] FabI Protein family Short-chain dehydrogenases/reductases (SDR) family, FabI subfamily Biochemical class Short-chain dehydrogenases reductase Function Catalyzes the reduction of a carbon-carbon double bond in an enoyl moiety that is covalently linked to an acyl carrier protein (ACP). It has a preference for a long chain (C12) substrate compared to the shorter (C4) acyl group. Involved in the elongation cycle of fatty acid which are used in the lipid metabolism. Related diseases Amyloidosis, hereditary systemic 4, Finnish type (AMYLD4) [MIM:105120]: A form of hereditary systemic amyloidosis, a disorder characterized by amyloid deposition in multiple tissues resulting in a wide clinical spectrum. AMYLD4 is due to gelsolin amyloid deposition and is typically characterized by cranial neuropathy and lattice corneal dystrophy. Most patients have modest involvement of internal organs, but severe systemic disease can develop in some individuals causing peripheral polyneuropathy, amyloid cardiomyopathy, and nephrotic syndrome leading to renal failure. AMYLD4 is usually inherited in an autosomal dominant pattern. However, homozygotes with a more severe phenotype have also been reported. {ECO:0000269|PubMed:1338910, ECO:0000269|PubMed:19666512, ECO:0000269|PubMed:2176481}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number NA Uniprot keywords 3D-structure; Fatty acid biosynthesis; Fatty acid metabolism; Lipid biosynthesis; Lipid metabolism; NAD; NADP; Oxidoreductase; Reference proteome Protein physicochemical properties Chain ID A,C Molecular weight (Da) 55674.6 Length 510 Aromaticity 0.07 Instability index 33.86 Isoelectric point 5.64 Charge (pH=7) -9.3 2D Binding mode Binding energy (Kcal/mol) -8.95  Molscript Map 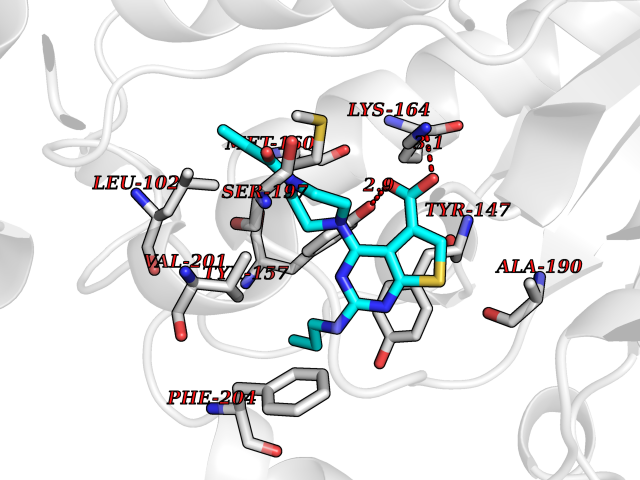 Pymol Map 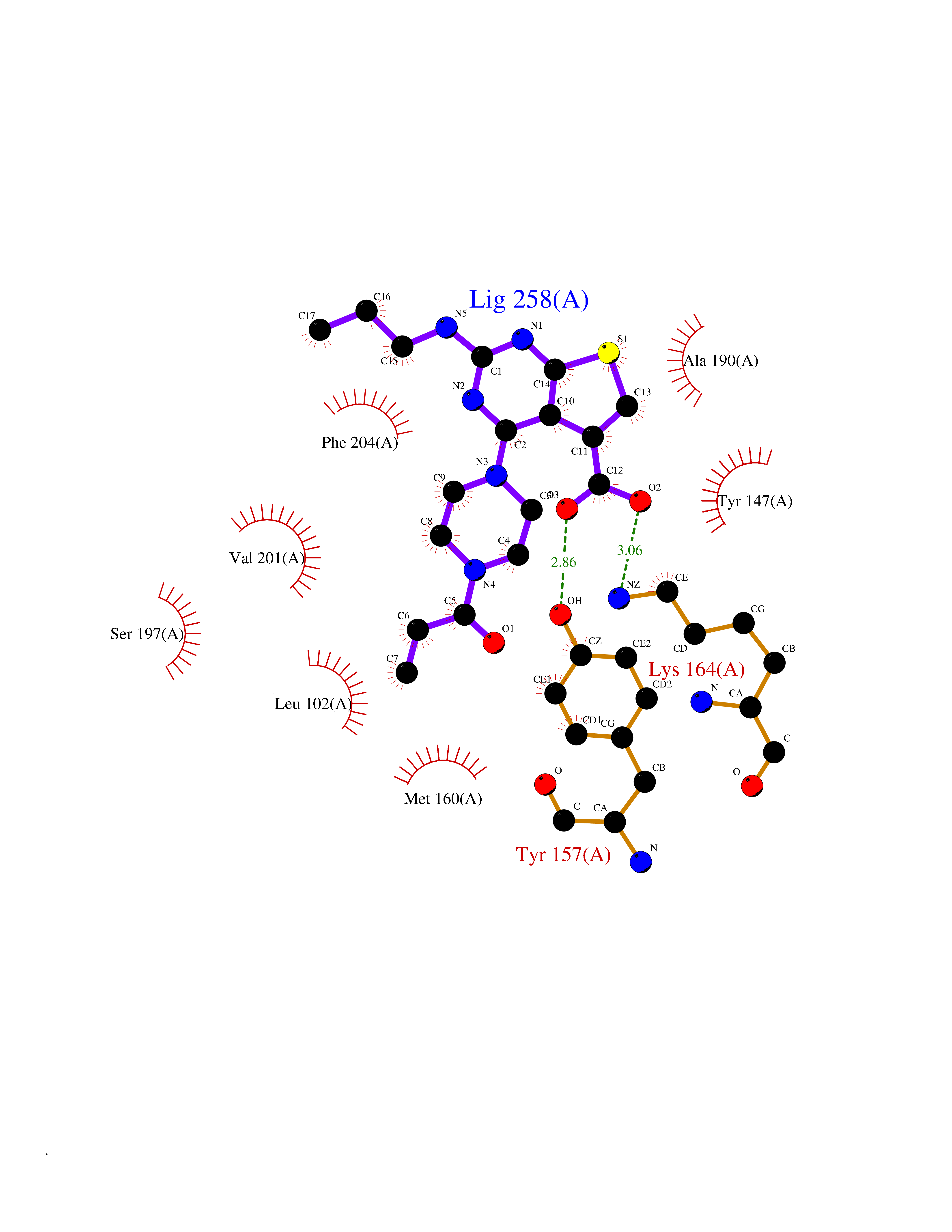 Ligplot Map 3D Binding mode Sequence VNLENKTYVIMGIANKRSIAFGVAKVLDQLGAKLVFTYRKERSRKELEKLLEQLNQPEAHLYQIDVQSDEEVINGFEQIGKDVGNIDGVYHSIAFANMEDLRGRFSETSREGFLLAQDISSYSLTIVAHEAKKLMPEGGSIVATTYLGGEFAVQNYNVMGVAKASLEANVKYLALDLGPDNIRVNAISAGPIRTLSAKGVGGFNTILKEIEERAPLKRNVDQVEVGKTAAYLLSDLSSGVTGENIHVDSGFHAIKVNLENKTYVIMGIANKRSIAFGVAKVLDQLGAKLVFTYRKERSRKELEKLLEQLNQPEAHLYQIDVQSDEEVINGFEQIGKDVGNIDGVYHSIAFANMEDLRGRFSETSREGFLLAQDISSYSLTIVAHEAKKLMPEGGSIVATTYLGGEFAVQNYNVMGVAKASLEANVKYLALDLGPDNIRVNAISAGPIRTLSAKGVGGFNTILKEIEERAPLKRNVDQVEVGKTAAYLLSDLSSGVTGENIHVDSGFHAIK Hydrogen bonds contact Hydrophobic contact | ||||
| 70 | Calcitonin gene-related peptide receptor (CGRPR) | 3N7S | 6.56 | |
Target general information Gen name CALCRL Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Calcitonin receptor-like receptor; Calcitonin gene-related peptide type 1 receptor; CGRPR; CGRP type 1 receptor Protein family G-protein coupled receptor 2 family Biochemical class GPCR secretin Function Receptor for calcitonin-gene-related peptide (CGRP) together with RAMP1 and receptor for adrenomedullin together with RAMP3 (By similarity). Receptor for adrenomedullin together with RAMP2. The activity of this receptor is mediated by G proteins which activate adenylyl cyclase. Related diseases Lymphatic malformation 8 (LMPHM8) [MIM:618773]: A form of primary lymphedema, a disease characterized by swelling of body parts due to developmental anomalies and functional defects of the lymphatic system. Adult patients with lymphedema may suffer from recurrent local infections. Impaired lymphatic drainage in the fetus can develop into hydrops fetalis, a severe condition characterized by excessive fluid accumulation in more than two fetal extra-vascular compartments and body cavities, placental enlargement and edema, pericardial or pleural effusion, or ascites. LMPHM8 is an autosomal recessive form characterized by onset in utero and fetal death due to non-immune hydrops fetalis. {ECO:0000269|PubMed:30115739}. The disease may be caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB16098; DB14039; DB04869; DB12457; DB12228; DB15328; DB15688 Interacts with P06881; O60894; O60895 EC number NA Uniprot keywords 3D-structure; Cell membrane; Direct protein sequencing; Disease variant; Disulfide bond; G-protein coupled receptor; Glycoprotein; Membrane; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Signal; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID D,A,C Molecular weight (Da) 31335.1 Length 269 Aromaticity 0.14 Instability index 29.06 Isoelectric point 5.22 Charge (pH=7) -7.93 2D Binding mode Binding energy (Kcal/mol) -8.95  Molscript Map 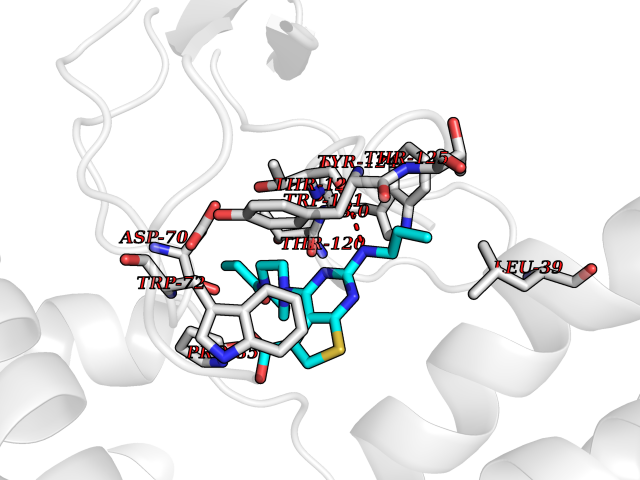 Pymol Map 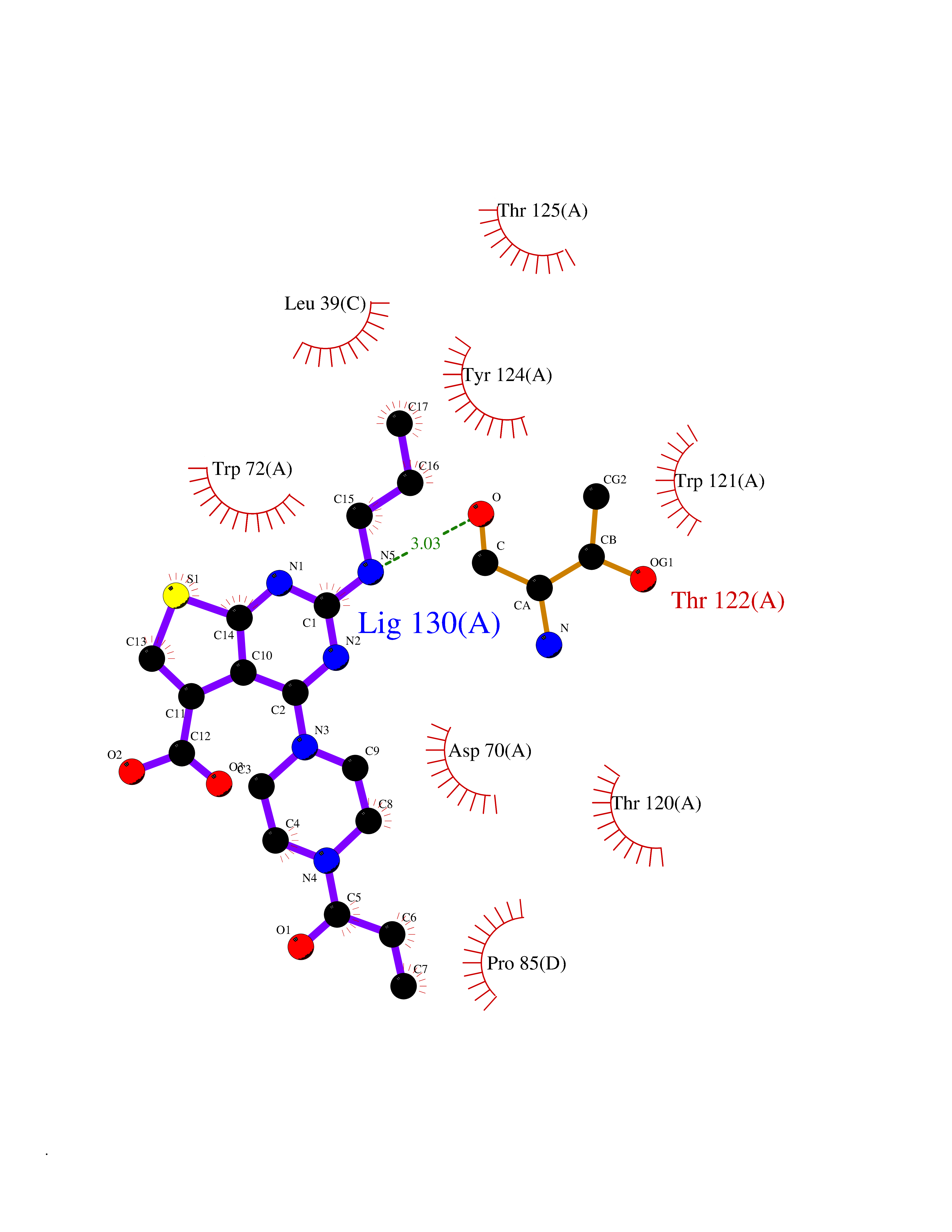 Ligplot Map 3D Binding mode Sequence IQLGVTRNKIMTAQYECYQKIMQDPIEGVYCNRTWDGWLCWNDVAAGTESMQLCPDYFQDFDPSEKVTKICDQDGNWFRHPASNRTWTNYTQCNACQEANYGALLRELCLTQFQVDMEAVGETLWCDWGRTIRSYRELADCTWHMAEKLGCFWPNAEVDRFFLAVHGRYFRSCPISGRAVRDPPGCQEANYGALLRELCLTQFQVDMEAVGETLWCDWGRTIRSYRELADCTWHMAEKLGCFWPNAEVDRFFLAVHGRYFRSCPISGRA Hydrogen bonds contact Hydrophobic contact | ||||
| 71 | N-acetylmannosamine kinase (GNE) | 4ZHT | 6.56 | |
Target general information Gen name GNE Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms UDPGlcNAc2epimerase/ManAc kinase; GNE; Bifunctional UDPNacetylglucosamine 2epimerase/Nacetylmannosamine kinase Protein family UDP-N-acetylglucosamine 2-epimerase family; ROK (NagC/XylR) family Biochemical class Kinase Function Regulates and initiates biosynthesis of N- acetylneuraminic acid (NeuAc), a precursor of sialic acids. Plays an essential role in early development. Required for normal sialylation in hematopoietic cells. Sialylation is implicated in cell adhesion, signal transduction, tumorigenicity and metastatic behavior of malignant cells. {ECO:0000250, ECO:0000269|PubMed:10334995}. Related diseases Sialuria (SIALURIA) [MIM:269921]: In sialuria, free sialic acid accumulates in the cytoplasm and gram quantities of neuraminic acid are secreted in the urine. The metabolic defect involves lack of feedback inhibition of UDP-GlcNAc 2-epimerase by CMP-Neu5Ac, resulting in constitutive overproduction of free Neu5Ac. Clinical features include variable degrees of developmental delay, coarse facial features and hepatomegaly. Sialuria inheritance is autosomal dominant. {ECO:0000269|PubMed:10330343, ECO:0000269|PubMed:10356312, ECO:0000269|PubMed:11326336, ECO:0000269|PubMed:2808337}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Nonaka myopathy (NM) [MIM:605820]: An autosomal recessive myopathy characterized by early adult onset and progressive distal muscle weakness that preferentially affects the anterior tibial muscles, usually sparing the quadriceps femoris. Some individuals may have involvement of the upper limbs or proximal muscles. Muscle biopsy reveals presence of rimmed vacuoles. {ECO:0000269|PubMed:11528398, ECO:0000269|PubMed:11916006, ECO:0000269|PubMed:12177386, ECO:0000269|PubMed:12325084, ECO:0000269|PubMed:12409274, ECO:0000269|PubMed:12473753, ECO:0000269|PubMed:12473769, ECO:0000269|PubMed:12473780, ECO:0000269|PubMed:12497639, ECO:0000269|PubMed:12811782, ECO:0000269|PubMed:12913203, ECO:0000269|PubMed:14707127, ECO:0000269|PubMed:15146476, ECO:0000269|PubMed:16503651}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Thrombocytopenia 12 with or without myopathy (THC12) [MIM:620757]: A form of thrombocytopenia, a hematologic disorder defined by a decrease in the number of platelets in circulating blood, resulting in the potential for increased bleeding and decreased ability for clotting. THC12 is an autosomal recessive form manifesting from infancy or early childhood with bleeding episodes. Clinical features include petechiae, easy bruising, epistaxis, hematomas, menorrhagia, and increased bleeding after trauma or surgery. Rare patients may have thrombocytopenia without bleeding. Some affected individuals have myopathic features, usually apparent in the second or third decades of life. {ECO:0000269|PubMed:25257349, ECO:0000269|PubMed:30171045, ECO:0000269|PubMed:33198675, ECO:0000269|PubMed:34788986, ECO:0000269|PubMed:34858435, ECO:0000269|PubMed:35052006, ECO:0000269|PubMed:38237079}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P12814; Q6UY14-3; Q969Y2; Q15323; P60370; P60409; P60410; P60411; Q9BQ66; P26371; Q9BYQ4; Q7Z3S9; O43597; Q6UY14-3; Q6P5X5; P27918; A8MQ03; Q16610; Q9UHF1; P28799; P49639; Q5T749; Q15323; O76011; Q6A162; P78385; P78386; O43790; Q07627; Q8IUG1; P60409; P60410; Q8IUC1; P60328; Q52LG2; Q3SY46; Q9BYP8; Q3LHN2; Q3SYF9; Q9BYR8; Q9BYR6; Q9BYQ7; Q9BYQ6; Q9BYR3; P26371; Q3LI64; Q3LI66; Q3LI67; Q9BYQ4; Q9BYQ3; Q9BYQ0; Q99750; Q8IV28; P0DPK4; O15496; O43609; O43610; P14373; Q8IWZ5; Q15654; O14817; Q2TAL6; Q9BRX9; O76024; Q9NZC7-5 EC number NA Uniprot keywords 3D-structure; Allosteric enzyme; Alternative splicing; ATP-binding; Cytoplasm; Disease variant; Hydrolase; Kinase; Metal-binding; Multifunctional enzyme; Nucleotide-binding; Phosphoprotein; Proteomics identification; Reference proteome; Transferase; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 41589.2 Length 384 Aromaticity 0.07 Instability index 35.07 Isoelectric point 7.05 Charge (pH=7) 0.19 2D Binding mode Binding energy (Kcal/mol) -8.95  Molscript Map 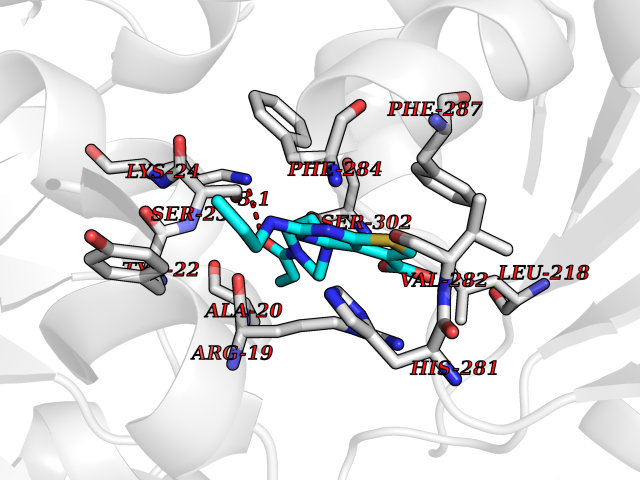 Pymol Map 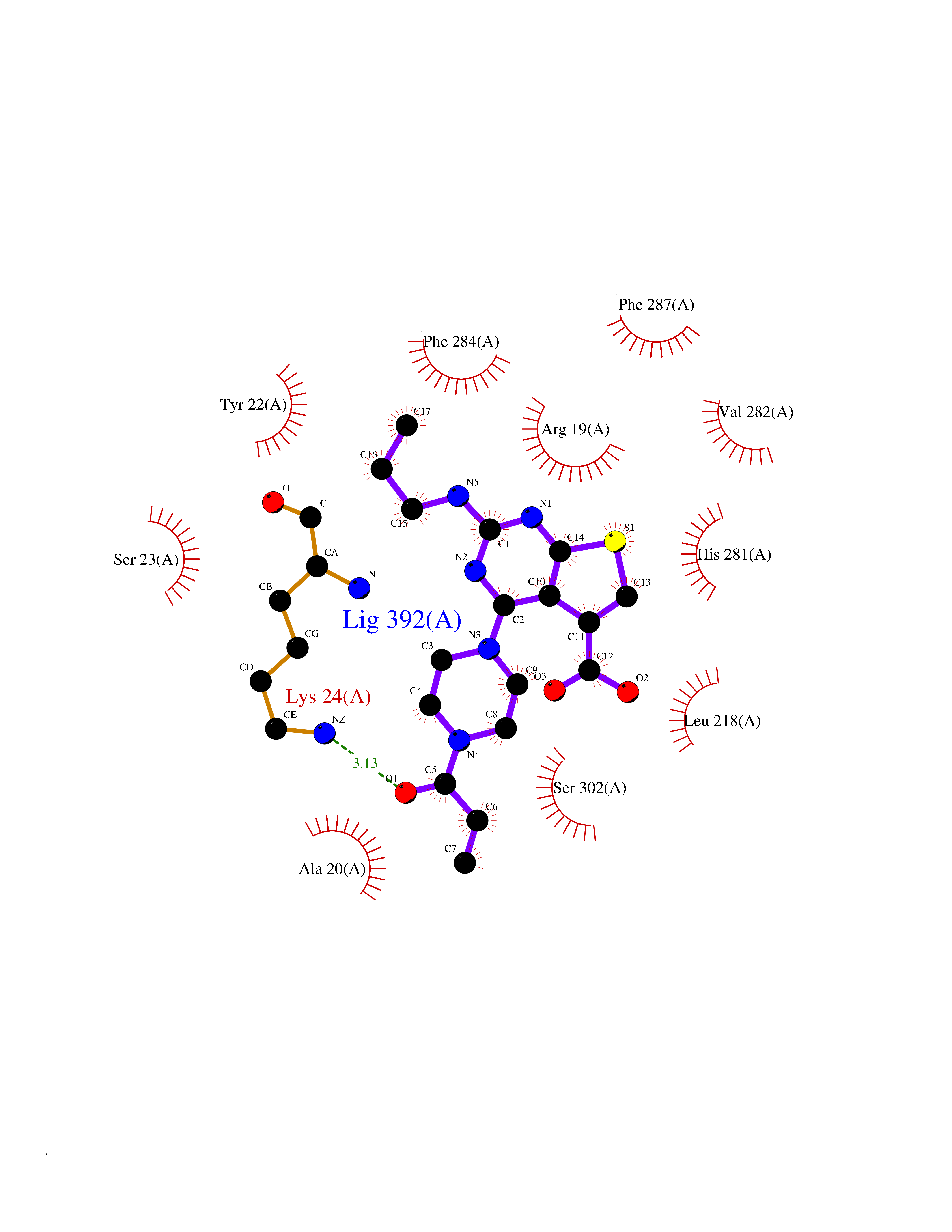 Ligplot Map 3D Binding mode Sequence NRKLRVCVATCNRADYSKLAPIXFGIKTEPEFFELDVVVLGSHLIDDYGNTYRXIEQDDFDINTRLHTIVRGEDEAAXVESVGLALVKLPDVLNRLKPDIXIVHGDRFDALALATSAALXNIRILHIEGGEVSGTIDDSIRHAITKLAHYHVCCTRSAEQHLISXCEDHDRILLAGCPSYDKLLSAKNKDYXSIIRXWLGDDVKSKDYIVALQHPVTTDIKHSIKXFELTLDALISFNKRTLVLFPNIDAGSKEXVRVXRKKGIEHHPNFRAVKHVPFDQFIQLVAHAGCXIGNSSCGVREVGAFGTPVINLGTRQIGRETGENVLHVRDADTQDKILQALHLQFGKQYPCSKIYGDGNAVPRILKFLKSIDLQEPLQKKFCFP Hydrogen bonds contact Hydrophobic contact | ||||
| 72 | Plasma retinol-binding protein (RBP4) | 5NU7 | 6.56 | |
Target general information Gen name RBP4 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Retinol-binding protein 4; RBP4; RBP; Plasma retinol-binding protein(1-176); PRBP Protein family Calycin superfamily, Lipocalin family Biochemical class Calycin family Function Delivers retinol from the liver stores to the peripheral tissues. In plasma, the RBP-retinol complex interacts with transthyretin, this prevents its loss by filtration through the kidney glomeruli. Related diseases Retinal dystrophy, iris coloboma, and comedogenic acne syndrome (RDCCAS) [MIM:615147]: A disease characterized by retinal degeneration, ocular colobomas involving both the anterior and posterior segment, impaired night vision and loss of visual acuity. Additional characteristic features include developmental abnormalities and severe acne. {ECO:0000269|PubMed:10232633, ECO:0000269|PubMed:23189188, ECO:0000269|PubMed:9888420}. The disease is caused by variants affecting the gene represented in this entry. Loss of functional RBP4 protein results in serum retinol deficiency. Lack of normal levels of retinol impairs the visual cycle leading to night blindness at early stages; prolonged deficiency may lead to retinal degeneration. Additionally, retinol deficiency may result in dry skin, increased susceptibility to infection and acne (PubMed:23189188). {ECO:0000269|PubMed:23189188}.; DISEASE: Microphthalmia/Coloboma 10 (MCOPCB10) [MIM:616428]: A disorder of eye formation, ranging from small size of a single eye to complete bilateral absence of ocular tissues. Ocular abnormalities like opacities of the cornea and lens, scaring of the retina and choroid, and other abnormalities may also be present. Ocular colobomas are a set of malformations resulting from abnormal morphogenesis of the optic cup and stalk, and the fusion of the fetal fissure (optic fissure). {ECO:0000269|PubMed:25910211}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB06985; DB06755; DB05076; DB03917; DB00755; DB00162 Interacts with Q9UBX0; P02766; O55245 EC number NA Uniprot keywords 3D-structure; Direct protein sequencing; Disease variant; Disulfide bond; Methylation; Microphthalmia; Proteomics identification; Reference proteome; Retinol-binding; Secreted; Sensory transduction; Signal; Transport; Vision; Vitamin A Protein physicochemical properties Chain ID A Molecular weight (Da) 20030.2 Length 174 Aromaticity 0.13 Instability index 28.54 Isoelectric point 5.24 Charge (pH=7) -4.02 2D Binding mode Binding energy (Kcal/mol) -8.95 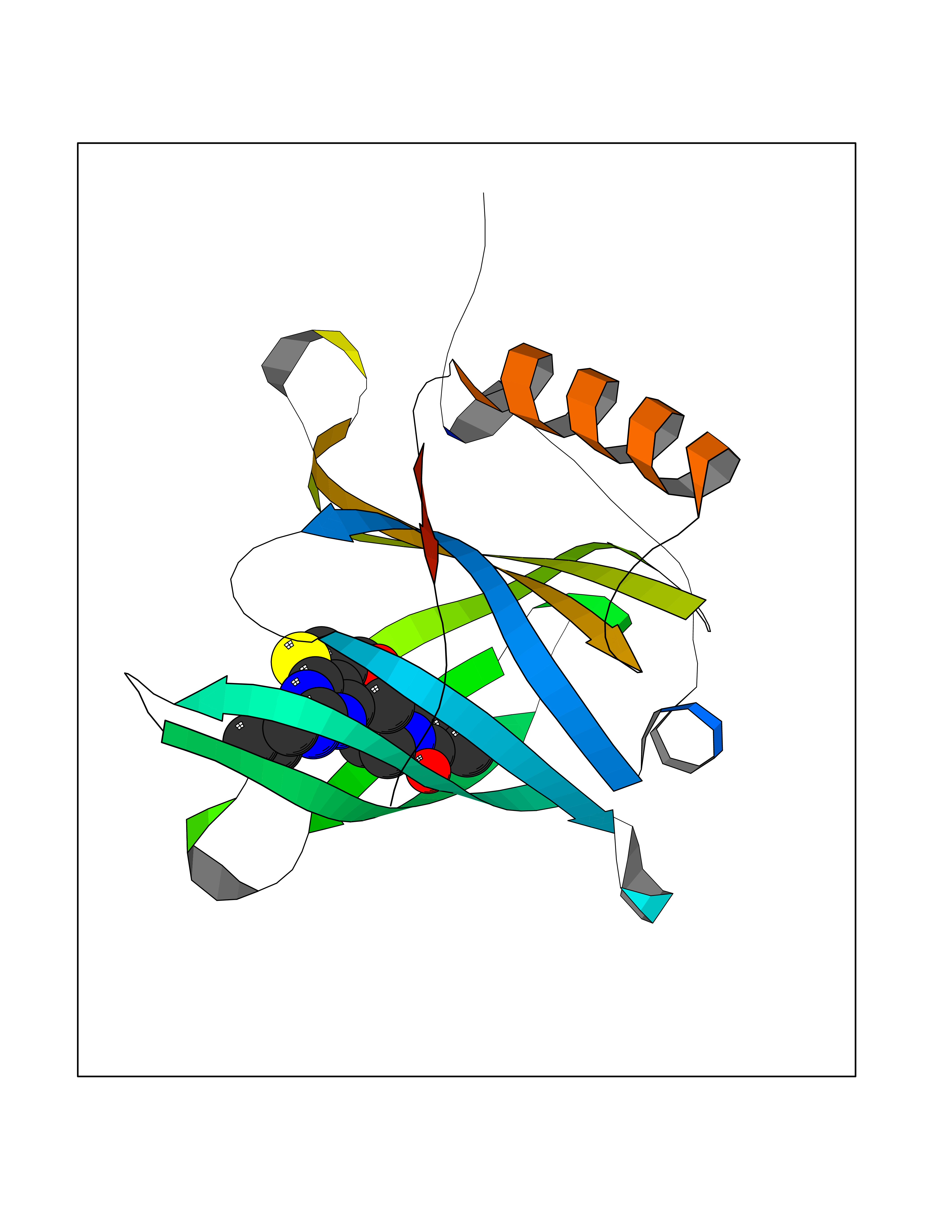 Molscript Map 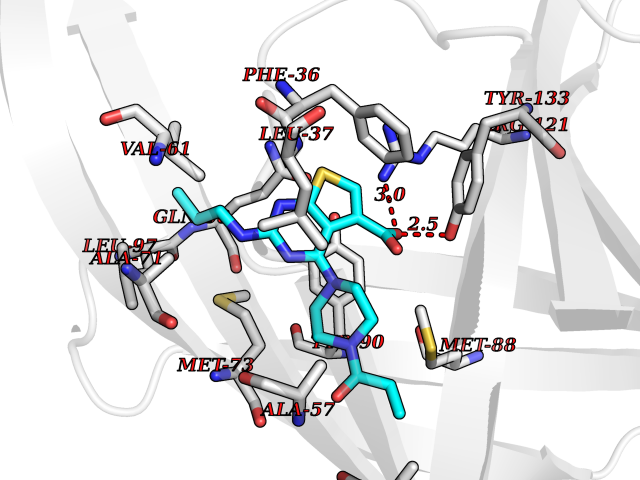 Pymol Map  Ligplot Map 3D Binding mode Sequence ERDCRVSSFRVKENFDKARFSGTWYAMAKKDPEGLFLQDNIVAEFSVDETGQMSATAKGRVRLLNNWDVCADMVGTFTDTEDPAKFKMKYWGVASFLQKGNDDHWIVDTDYDTYAVQYSCRLLNLDGTCADSYSFVFSRDPNGLPPEAQKIVRQRQEELCLARQYRLIVHNGYC Hydrogen bonds contact Hydrophobic contact | ||||
| 73 | Beta-ketoacyl-ACP synthase (OXSM) | 2IWZ | 6.56 | |
Target general information Gen name OXSM Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms 3-oxoacyl-[acyl-carrier-protein] synthase, mitochondrial Protein family Thiolase-like superfamily, Beta-ketoacyl-ACP synthases family Biochemical class Acyltransferase Function May play a role in the biosynthesis of lipoic acid as well as longer chain fatty acids required for optimal mitochondrial function. Related diseases Lethal congenital contracture syndrome 2 (LCCS2) [MIM:607598]: A form of lethal congenital contracture syndrome, an autosomal recessive disorder characterized by degeneration of anterior horn neurons, extreme skeletal muscle atrophy, and congenital non-progressive joint contractures (arthrogryposis). The contractures can involve the upper or lower limbs and/or the vertebral column, leading to various degrees of flexion or extension limitations evident at birth. LCCS2 patients manifest craniofacial/ocular findings, lack of hydrops, multiple pterygia, and fractures, as well as a normal duration of pregnancy and a unique feature of a markedly distended urinary bladder (neurogenic bladder defect). The phenotype suggests a spinal cord neuropathic etiology. {ECO:0000269|PubMed:17701904}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Erythroleukemia, familial (FERLK) [MIM:133180]: An autosomal dominant myeloproliferative disorder characterized by neoplastic proliferation of erythroblastic and myeloblastic elements with atypical erythroblasts and myeloblasts in the peripheral blood. Disease penetrance is incomplete. {ECO:0000269|PubMed:27416908}. Disease susceptibility may be associated with variants affecting the gene represented in this entry.; DISEASE: Visceral neuropathy, familial, 1, autosomal recessive (VSCN1) [MIM:243180]: An autosomal recessive disorder characterized by intestinal dysmotility due to aganglionosis (Hirschsprung disease), hypoganglionosis, and/or chronic intestinal pseudoobstruction. Additional variable features are progressive peripheral neuropathy, arthrogryposis, hypoplasia or aplasia of the olfactory bulb and of the external auditory canals, microtia or anotia, and facial dysmorphism. Some patients present structural cardiac anomalies and arthrogryposis with multiple pterygia. {ECO:0000269|PubMed:33497358}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number EC 2.3.1.41 Uniprot keywords 3D-structure; Acetylation; Acyltransferase; Alternative splicing; Fatty acid biosynthesis; Fatty acid metabolism; Lipid biosynthesis; Lipid metabolism; Mitochondrion; Proteomics identification; Reference proteome; Transferase; Transit peptide Protein physicochemical properties Chain ID A,B Molecular weight (Da) 89949.3 Length 852 Aromaticity 0.07 Instability index 35.84 Isoelectric point 6.13 Charge (pH=7) -10.02 2D Binding mode Binding energy (Kcal/mol) -8.95  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence IEGRSRLHRRVVITGIGLVTPLGVGTHLVWDRLIGGESGIVSLVGEEYKSIPCSVAAYVPRGSDEGQFNEQNFVSKSDIKSMSSPTIMAIGAAELAMKDSGWHPQSEADQVATGVAIGMGMIPLEVVSETALNFQTKGYNKVSPFFVPKILVNMAAGQVSIRYKLKGPNHAVSTACTTGAHAVGDSFRFIAHGDADVMVAGGTDSCISPLSLAGFSRARALSTNSDPKLACRPFHPKRDGFVMGEGAAVLVLEEYEHAVQRRARIYAEVLGYGLSGDAGHITAPDPEGEGALRCMAAALKDAGVQPEEISYINAHATSTPLGDAAENKAIKHLFKDHAYALAVSSTKGATGHLLGAAGAVEAAFTTLACYYQKLPPTLNLDCSEPEFDLNYVPLKAQEWKTEKRFIGLTNSFGFGGTNATLCIAGLIEGRSRLHRRVVITGIGLVTPLGVGTHLVWDRLIGGESGIVSLVGEEYKSIPCSVAAYVPRGSDEGQFNEQNFVSKSDIKSMSSPTIMAIGAAELAMKDSGWHPQSEADQVATGVAIGMGMIPLEVVSETALNFQTKGYNKVSPFFVPKILVNMAAGQVSIRYKLKGPNHAVSTACTTGAHAVGDSFRFIAHGDADVMVAGGTDSCISPLSLAGFSRARALSTNSDPKLACRPFHPKRDGFVMGEGAAVLVLEEYEHAVQRRARIYAEVLGYGLSGDAGHITAPDPEGEGALRCMAAALKDAGVQPEEISYINAHATSTPLGDAAENKAIKHLFKDHAYALAVSSTKGATGHLLGAAGAVEAAFTTLACYYQKLPPTLNLDCSEPEFDLNYVPLKAQEWKTEKRFIGLTNSFGFGGTNATLCIAGL Hydrogen bonds contact Hydrophobic contact | ||||
| 74 | Quinone-dependent D-lactate dehydrogenase | 1F0X | 6.55 | |
Target general information Gen name dld Organism Escherichia coli (strain K12) Uniprot ID TTD ID NA Synonyms JW2121;b2133 Protein family Quinone-dependent D-lactate dehydrogenase family Biochemical class Oxidoreductase Function (R)-2-hydroxyglutarate dehydrogenase activity.D-lactate dehydrogenase (cytochrome) activity.Electron carrier activity.FAD binding.Flavin adenine dinucleotide binding.NAD binding.Oxidoreductase activity, acting on the CH-OH group of donors, quinone or similar compound as acceptor.Quinone binding. Related diseases Myeloperoxidase deficiency (MPOD) [MIM:254600]: A disorder characterized by decreased myeloperoxidase activity in neutrophils and monocytes that results in disseminated candidiasis. {ECO:0000269|PubMed:37198333, ECO:0000269|PubMed:7904599, ECO:0000269|PubMed:8142659, ECO:0000269|PubMed:8621627, ECO:0000269|PubMed:9354683, ECO:0000269|PubMed:9637725}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03147; DB00756 Interacts with NA EC number 1.1.5.12 Uniprot keywords 3D-structure; Cell inner membrane; Cell membrane; Direct protein sequencing; FAD; Flavoprotein; Membrane; Oxidoreductase; Quinone; Reference proteome Protein physicochemical properties Chain ID A,B Molecular weight (Da) 56475.2 Length 502 Aromaticity 0.09 Instability index 32.5 Isoelectric point 5.97 Charge (pH=7) -10.38 2D Binding mode Binding energy (Kcal/mol) -8.94  Molscript Map 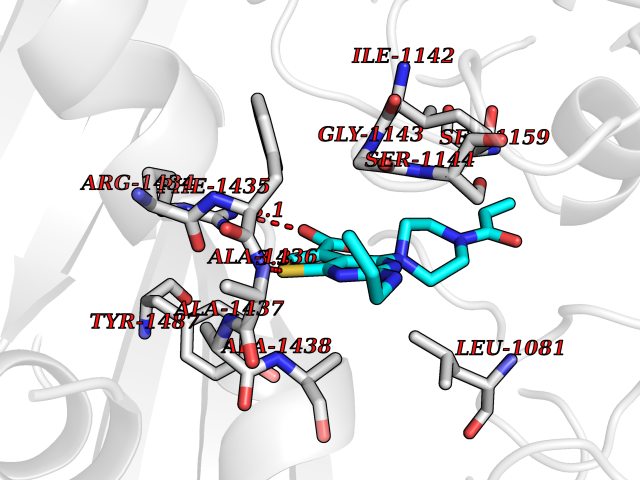 Pymol Map 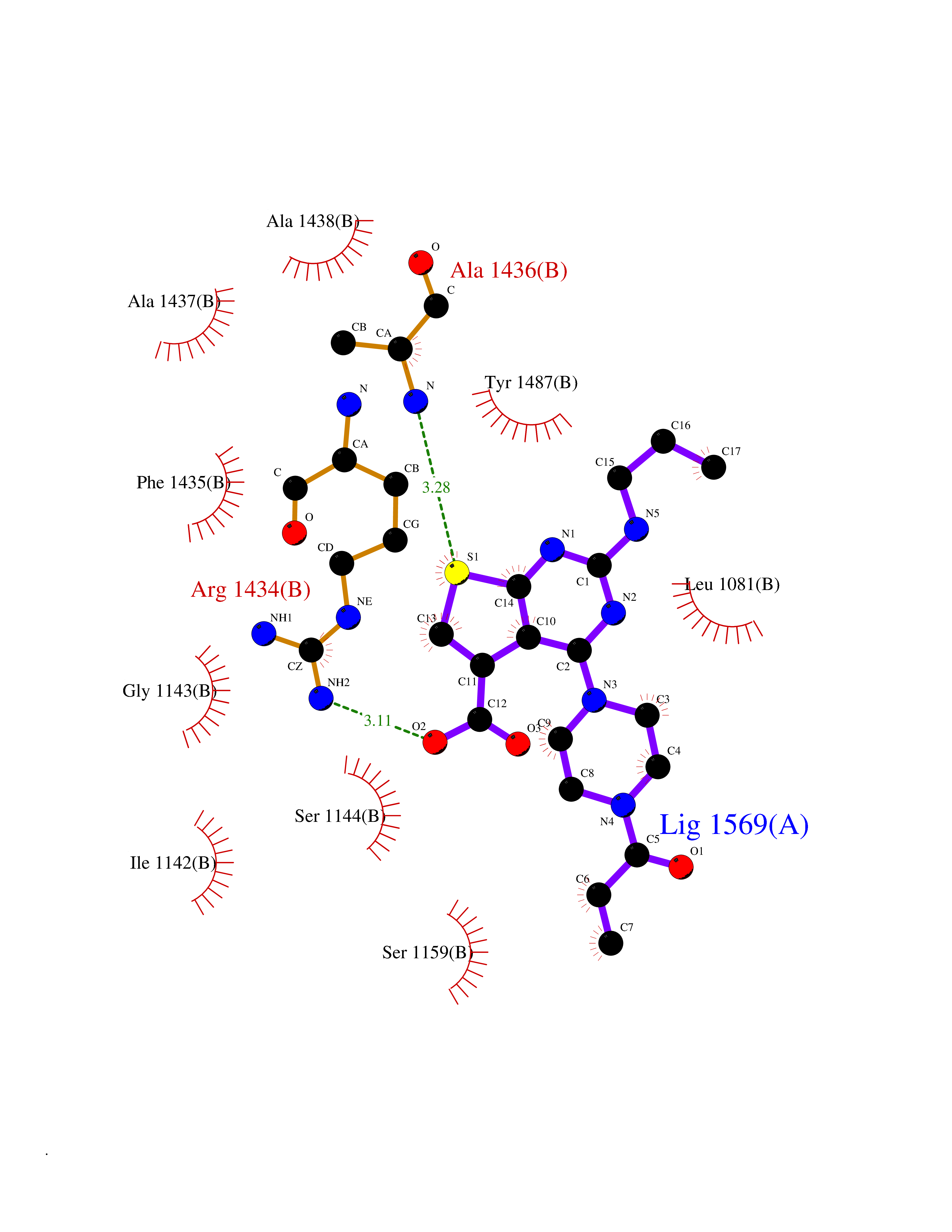 Ligplot Map 3D Binding mode Sequence NKAFLNELARLVGSSHLLTDPAKTARYRKGFRSGQGDALAVVFPGSLLELWRVLKACVTADKIILMQAANTGLTEGSTPNGNDYDRDVVIISTLRLDKLHVLGKGEQVLAYPGTTLYSLEKALKPLGREPHSVIGSSCIGASVIGGICNNSGGSLVQRGPAYTEMSLFARINEDGKLTLVNHLGIDLGETPEQILSKLDDDRIKDDDVRHDGRHAHDYDYVHRVRDIEADTPARYNADPDRLFESSGCAGKLAVFAVRLDTFEAEKNQQVFYIGTNQPEVLTEIRRHILANFENLPVAGEYMHRDIYDIAELPPRMKNWRDKYEHHLLLKMAGDGVGEAKSWLVDYFKQAEGDFFVCTPEEGSKAFLHRFAAAGAAIRYQAVHSDEVEDILALDIALRRNDTEWYEHLPPEIDSQLVHKLYYGHFMCYVFHQDYIVKKGVDVHALKEQMLELLQQRGAQYPAEHNVGHLYKAPETLQKFYRENDPTNSMNPGIGKTSKRKNW Hydrogen bonds contact Hydrophobic contact | ||||
| 75 | Hemoglobin subunit alpha | 1IRD | 6.55 | |
Target general information Gen name HBA1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms HBA2 Protein family Globin family Biochemical class Oxygen storage / transport Function Heme binding.Iron ion binding.Oxygen binding.Oxygen transporter activity. Related diseases Heinz body anemias (HEIBAN) [MIM:140700]: Form of non-spherocytic hemolytic anemia of Dacie type 1. After splenectomy, which has little benefit, basophilic inclusions called Heinz bodies are demonstrable in the erythrocytes. Before splenectomy, diffuse or punctate basophilia may be evident. Most of these cases are probably instances of hemoglobinopathy. The hemoglobin demonstrates heat lability. Heinz bodies are observed also with the Ivemark syndrome (asplenia with cardiovascular anomalies) and with glutathione peroxidase deficiency. {ECO:0000269|PubMed:2833478}. The disease may be caused by variants affecting the gene represented in this entry.; DISEASE: Alpha-thalassemia (A-THAL) [MIM:604131]: A form of thalassemia. Thalassemias are common monogenic diseases occurring mostly in Mediterranean and Southeast Asian populations. The hallmark of alpha-thalassemia is an imbalance in globin-chain production in the adult HbA molecule. The level of alpha chain production can range from none to very nearly normal levels. Deletion of both copies of each of the two alpha-globin genes causes alpha(0)-thalassemia, also known as homozygous alpha thalassemia. Due to the complete absence of alpha chains, the predominant fetal hemoglobin is a tetramer of gamma-chains (Bart hemoglobin) that has essentially no oxygen carrying capacity. This causes oxygen starvation in the fetal tissues leading to prenatal lethality or early neonatal death. The loss of two alpha genes results in mild alpha-thalassemia, also known as heterozygous alpha-thalassemia. Affected individuals have small red cells and a mild anemia (microcytosis). If three of the four alpha-globin genes are functional, individuals are completely asymptomatic. Some rare forms of alpha-thalassemia are due to point mutations (non-deletional alpha-thalassemia). The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Alpha(0)-thalassemia is associated with non-immune hydrops fetalis, a generalized edema of the fetus with fluid accumulation in the body cavities due to non-immune causes. Non-immune hydrops fetalis is not a diagnosis in itself but a symptom, a feature of many genetic disorders, and the end-stage of a wide variety of disorders.; DISEASE: Hemoglobin H disease (HBH) [MIM:613978]: A form of alpha-thalassemia due to the loss of three alpha genes. This results in high levels of a tetramer of four beta chains (hemoglobin H), causing a severe and life-threatening anemia. Untreated, most patients die in childhood or early adolescence. {ECO:0000269|PubMed:10569720}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08262; DB07427; DB08077; DB07428; DB02126; DB09130; DB08486; DB15617; DB09147; DB13995; DB14490; DB14491; DB14488; DB14501; DB14489; DB13257; DB01592; DB00893; DB09112; DB09140; DB06154; DB07645; DB09517; DB08632; DB14975; DB01593; DB14487; DB14533; DB14548 Interacts with Q9NZD4; Q2TAC2; P00387; P68871; P02042; P02100; P69892; P09105; Q15323; O76011; Q6A162; P29474; P0DPK4 EC number NA Uniprot keywords 3D-structure; Acetylation; Direct protein sequencing; Disease variant; Glycation; Glycoprotein; Heme; Hereditary hemolytic anemia; Iron; Metal-binding; Oxygen transport; Phosphoprotein; Proteomics identification; Reference proteome; Transport Protein physicochemical properties Chain ID A Molecular weight (Da) 30975.2 Length 287 Aromaticity 0.08 Instability index 6.57 Isoelectric point 7.92 Charge (pH=7) 2.38 2D Binding mode Binding energy (Kcal/mol) -8.93 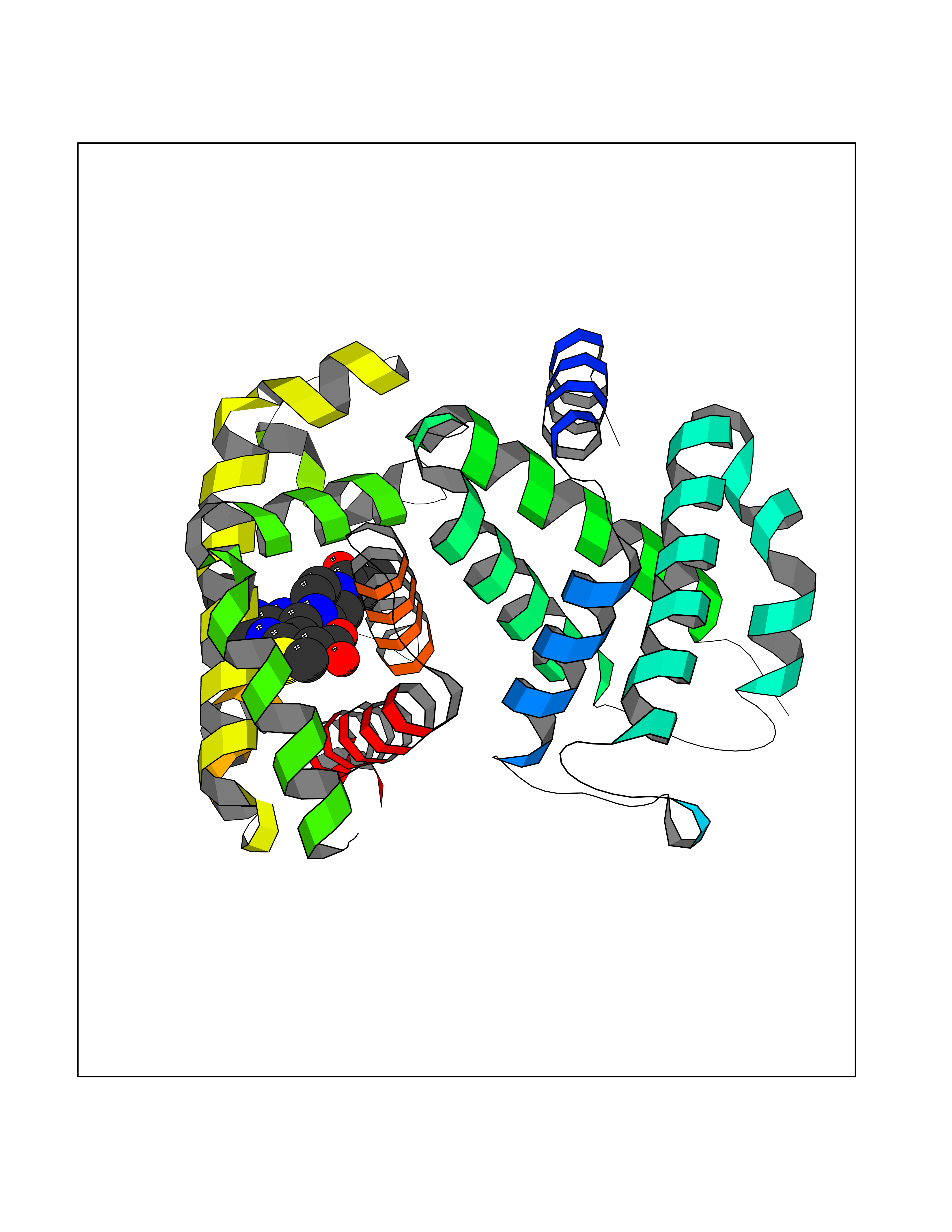 Molscript Map 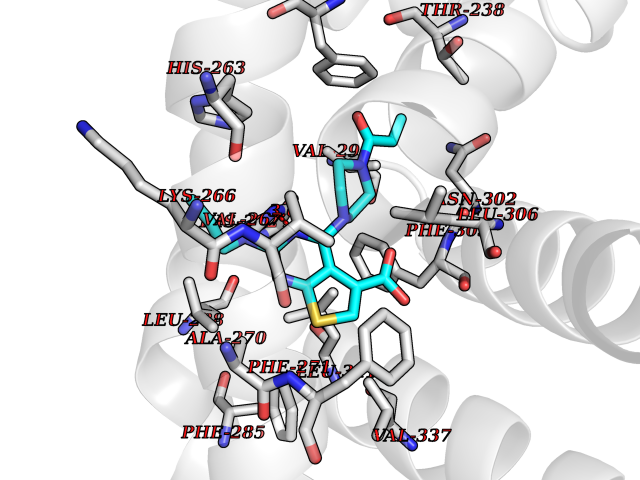 Pymol Map 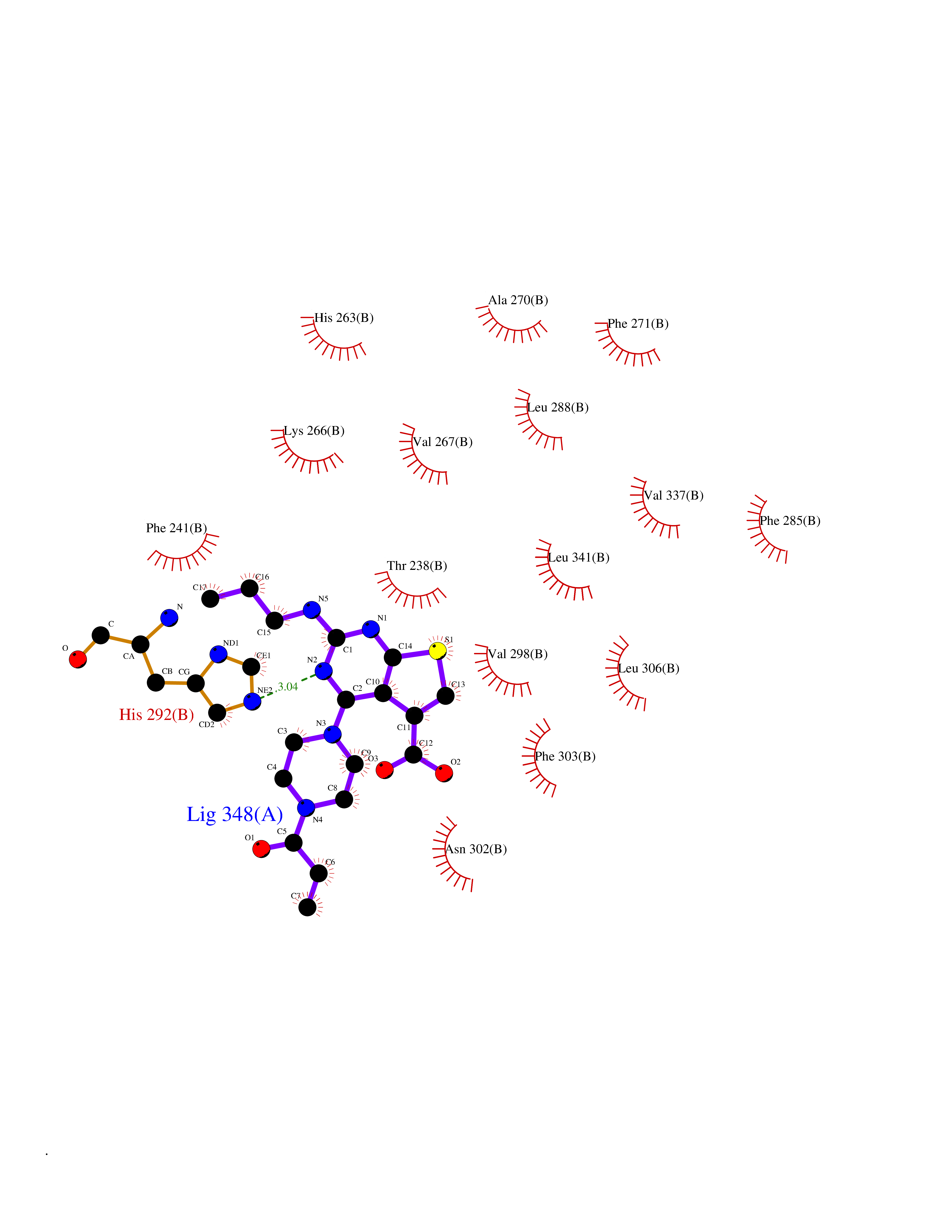 Ligplot Map 3D Binding mode Sequence VLSPADKTNVKAAWGKVGAHAGEYGAEALERMFLSFPTTKTYFPHFDLSHGSAQVKGHGKKVADALTNAVAHVDDMPNALSALSDLHAHKLRVDPVNFKLLSHCLLVTLAAHLPAEFTPAVHASLDKFLASVSTVLTSKYRVHLTPEEKSAVTALWGKVNVDEVGGEALGRLLVVYPWTQRFFESFGDLSTPDAVMGNPKVKAHGKKVLGAFSDGLAHLDNLKGTFATLSELHCDKLHVDPENFRLLGNVLVCVLAHHFGKEFTPPVQAAYQKVVAGVANALAHKYH Hydrogen bonds contact Hydrophobic contact | ||||
| 76 | Protein kinase C epsilon (PRKCE) | 5LIH | 6.55 | |
Target general information Gen name PRKCE Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Protein kinase C epsilon type; PKCE; PKC epsilon; NPKC-epsilon Protein family Protein kinase superfamily, AGC Ser/Thr protein kinase family, PKC subfamily Biochemical class Kinase Function Mediates cell adhesion to the extracellular matrix via integrin-dependent signaling, by mediating angiotensin-2-induced activation of integrin beta-1 (ITGB1) in cardiac fibroblasts. Phosphorylates MARCKS, which phosphorylates and activates PTK2/FAK, leading to the spread of cardiomyocytes. Involved in the control of the directional transport of ITGB1 in mesenchymal cells by phosphorylating vimentin (VIM), an intermediate filament (IF) protein. In epithelial cells, associates with and phosphorylates keratin-8 (KRT8), which induces targeting of desmoplakin at desmosomes and regulates cell-cell contact. Phosphorylates IQGAP1, which binds to CDC42, mediating epithelial cell-cell detachment prior to migration. In HeLa cells, contributes to hepatocyte growth factor (HGF)-induced cell migration, and in human corneal epithelial cells, plays a critical role in wound healing after activation by HGF. During cytokinesis, forms a complex with YWHAB, which is crucial for daughter cell separation, and facilitates abscission by a mechanism which may implicate the regulation of RHOA. In cardiac myocytes, regulates myofilament function and excitation coupling at the Z-lines, where it is indirectly associated with F-actin via interaction with COPB1. During endothelin-induced cardiomyocyte hypertrophy, mediates activation of PTK2/FAK, which is critical for cardiomyocyte survival and regulation of sarcomere length. Plays a role in the pathogenesis of dilated cardiomyopathy via persistent phosphorylation of troponin I (TNNI3). Involved in nerve growth factor (NFG)-induced neurite outgrowth and neuron morphological change independently of its kinase activity, by inhibition of RHOA pathway, activation of CDC42 and cytoskeletal rearrangement. May be involved in presynaptic facilitation by mediating phorbol ester-induced synaptic potentiation. Phosphorylates gamma-aminobutyric acid receptor subunit gamma-2 (GABRG2), which reduces the response of GABA receptors to ethanol and benzodiazepines and may mediate acute tolerance to the intoxicating effects of ethanol. Upon PMA treatment, phosphorylates the capsaicin- and heat-activated cation channel TRPV1, which is required for bradykinin-induced sensitization of the heat response in nociceptive neurons. Is able to form a complex with PDLIM5 and N-type calcium channel, and may enhance channel activities and potentiates fast synaptic transmission by phosphorylating the pore-forming alpha subunit CACNA1B (CaV2. 2). In prostate cancer cells, interacts with and phosphorylates STAT3, which increases DNA-binding and transcriptional activity of STAT3 and seems to be essential for prostate cancer cell invasion. Downstream of TLR4, plays an important role in the lipopolysaccharide (LPS)-induced immune response by phosphorylating and activating TICAM2/TRAM, which in turn activates the transcription factor IRF3 and subsequent cytokines production. In differentiating erythroid progenitors, is regulated by EPO and controls the protection against the TNFSF10/TRAIL-mediated apoptosis, via BCL2. May be involved in the regulation of the insulin-induced phosphorylation and activation of AKT1. Calcium-independent, phospholipid- and diacylglycerol (DAG)-dependent serine/threonine-protein kinase that plays essential roles in the regulation of multiple cellular processes linked to cytoskeletal proteins, such as cell adhesion, motility, migration and cell cycle, functions in neuron growth and ion channel regulation, and is involved in immune response, cancer cell invasion and regulation of apoptosis. Related diseases Hyperinsulinemic hypoglycemia, familial, 2 (HHF2) [MIM:601820]: A form of hyperinsulinemic hypoglycemia, a clinically and genetically heterogeneous disorder characterized by inappropriate insulin secretion from the pancreatic beta-cells in the presence of low blood glucose levels. HHF2 is a common cause of persistent hypoglycemia in infancy. Unless early and aggressive intervention is undertaken, brain damage from recurrent episodes of hypoglycemia may occur. HHF2 inheritance can be autosomal dominant or autosomal recessive. {ECO:0000269|PubMed:10204114, ECO:0000269|PubMed:12364426, ECO:0000269|PubMed:15562009, ECO:0000269|PubMed:15579781, ECO:0000269|PubMed:15807877, ECO:0000269|PubMed:15998776, ECO:0000269|PubMed:16332676, ECO:0000269|PubMed:16357843, ECO:0000269|PubMed:18596924, ECO:0000269|PubMed:19357197, ECO:0000269|PubMed:7847376, ECO:0000269|PubMed:8923010}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Diabetes mellitus, permanent neonatal, 2 (PNDM2) [MIM:618856]: A form of permanent neonatal diabetes mellitus, a type of diabetes characterized by onset of persistent hyperglycemia within the first six months of life. Initial clinical manifestations include intrauterine growth retardation, hyperglycemia, glycosuria, osmotic polyuria, severe dehydration, and failure to thrive. Some PNDM2 patients may also have developmental delay, muscle weakness, epilepsy and dysmorphic features. PNDM2 transmission pattern is consistent with autosomal dominant inheritance. {ECO:0000269|PubMed:15115830, ECO:0000269|PubMed:15292329, ECO:0000269|PubMed:15448106, ECO:0000269|PubMed:15448107, ECO:0000269|PubMed:15580558, ECO:0000269|PubMed:15583126, ECO:0000269|PubMed:16609879, ECO:0000269|PubMed:16731833, ECO:0000269|PubMed:17213273, ECO:0000269|PubMed:17652641, ECO:0000269|PubMed:17855752, ECO:0000269|PubMed:20022885, ECO:0000269|PubMed:28842488}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Transient neonatal diabetes mellitus 3 (TNDM3) [MIM:610582]: Neonatal diabetes mellitus, defined as insulin-requiring hyperglycemia within the first month of life, is a rare entity. In about half of the neonates, diabetes is transient and resolves at a median age of 3 months, whereas the rest have a permanent form of diabetes. In a significant number of patients with transient neonatal diabetes mellitus, diabetes type 2 appears later in life. The onset and severity of TNDM3 is variable with childhood-onset diabetes, gestational diabetes or adult-onset diabetes described. {ECO:0000269|PubMed:15718250, ECO:0000269|PubMed:15784703}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Defects in KCNJ11 may contribute to non-insulin-dependent diabetes mellitus (NIDDM), also known as diabetes mellitus type 2.; DISEASE: Maturity-onset diabetes of the young 13 (MODY13) [MIM:616329]: A form of diabetes that is characterized by an autosomal dominant mode of inheritance, onset in childhood or early adulthood (usually before 25 years of age), a primary defect in insulin secretion and frequent insulin-independence at the beginning of the disease. {ECO:0000269|PubMed:22701567}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB09096; DB11752; DB04209; DB12010; DB06064; DB02010; DB00675 Interacts with P05067; P15056; P08238; C6GKH1; Q9BPZ7; P17252; Q15349; P31947; P37840; P62258; P63104 EC number EC 2.7.11.13 Uniprot keywords 3D-structure; ATP-binding; Cell adhesion; Cell cycle; Cell division; Cell membrane; Cytoplasm; Cytoskeleton; Immunity; Kinase; Membrane; Metal-binding; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Serine/threonine-protein kinase; Transferase; Zinc; Zinc-finger Protein physicochemical properties Chain ID A,F Molecular weight (Da) 39129.4 Length 339 Aromaticity 0.12 Instability index 42.21 Isoelectric point 6.17 Charge (pH=7) -3.73 2D Binding mode Binding energy (Kcal/mol) -8.94 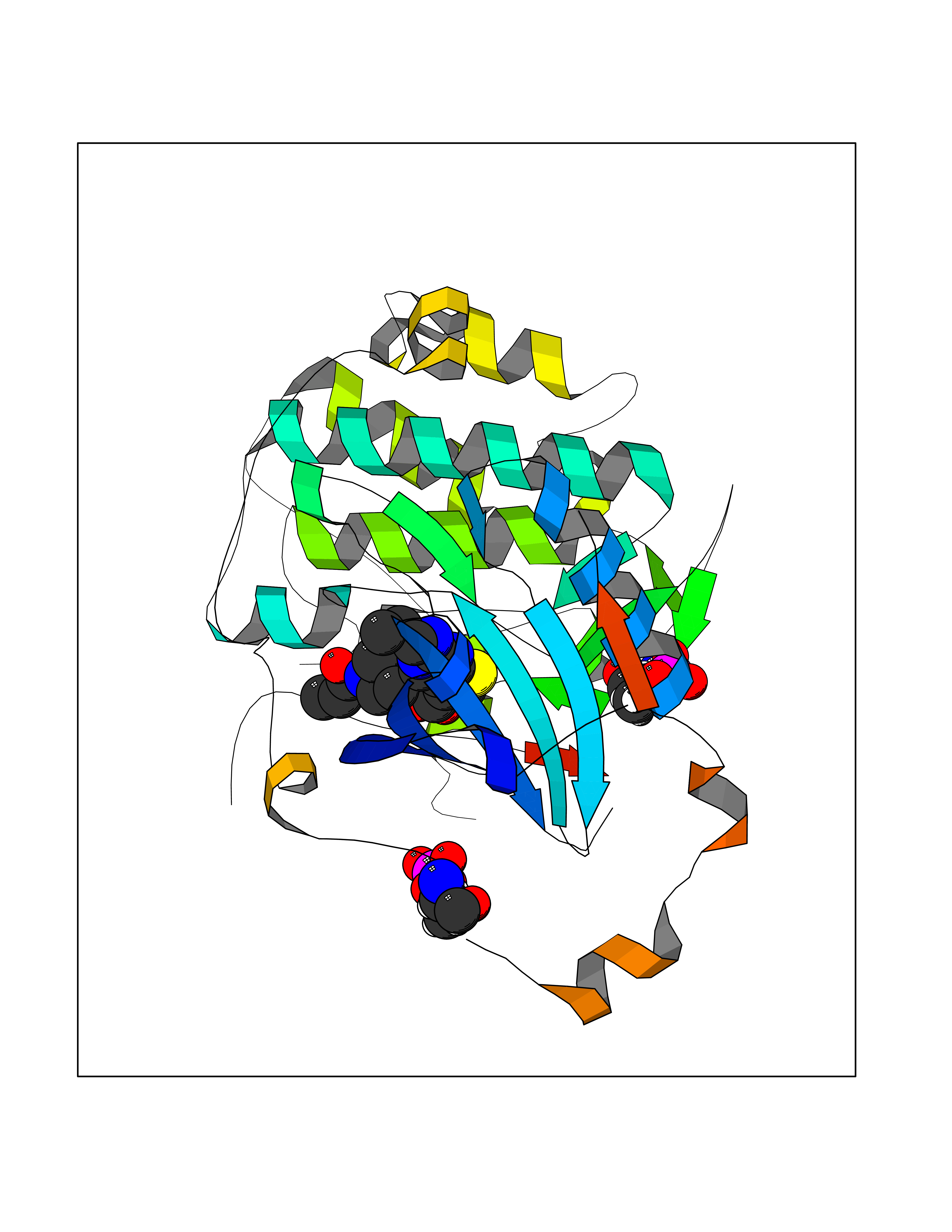 Molscript Map 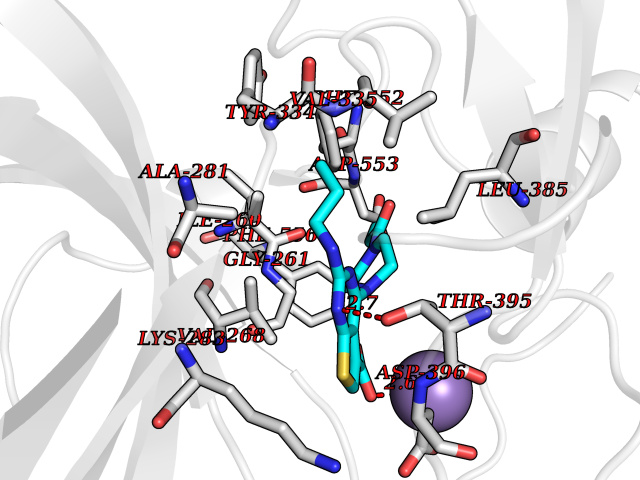 Pymol Map 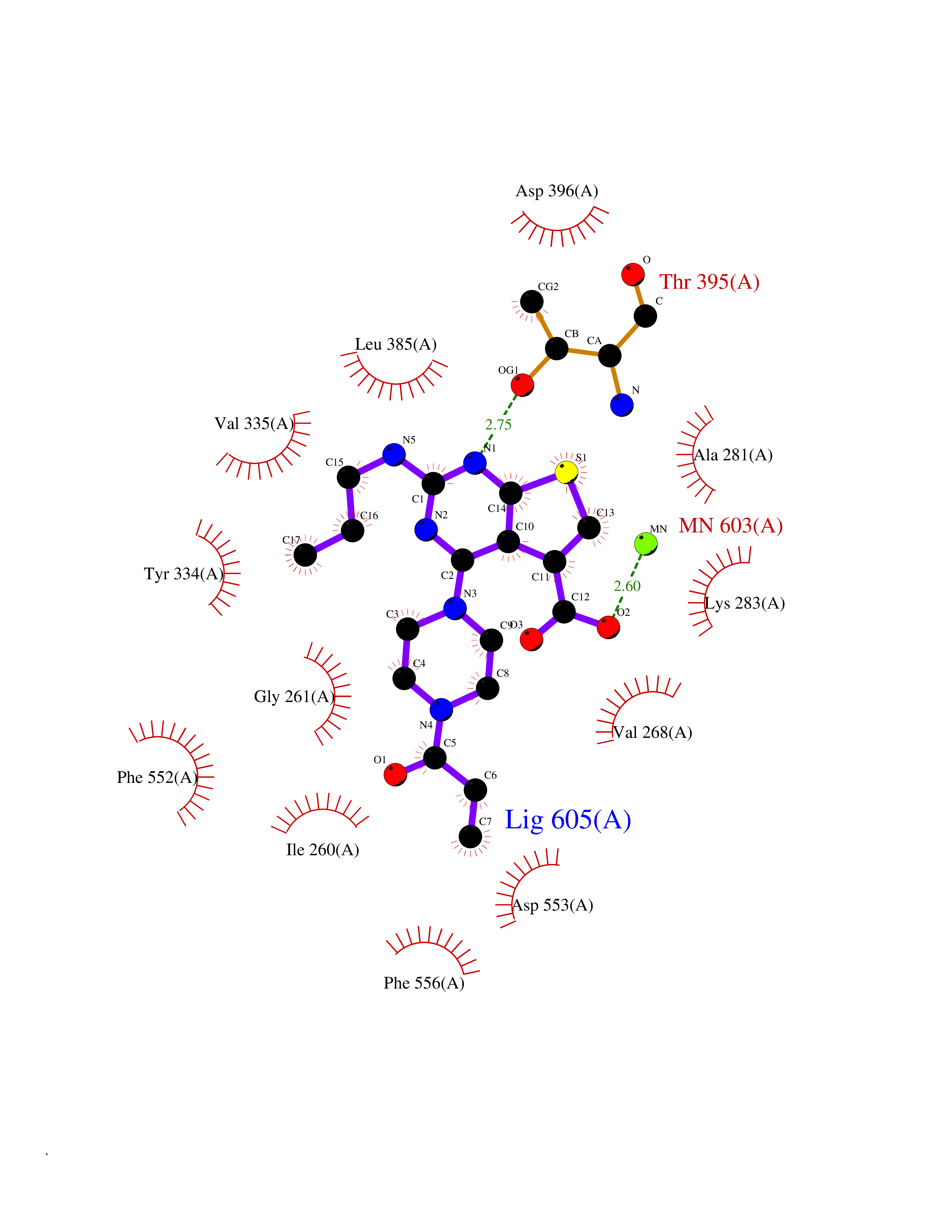 Ligplot Map 3D Binding mode Sequence SLGLQDFDLLRVIGRGSYAKVLLVRLKKTDRIYAMKVVKKELVWVQTEKHVFEQASNHPFLVGLHSCFQTESRLFFVIEYVNGGDLMFHMQRQRKLPEEHARFYSAEISLALNYLHERGIIYRDLKLDNVLLDSEGHIKLTDYGMCKEGLRPGDTTSXFCGTPNYIAPEILRGEDYGFSVDWWALGVLMFEMMAGRSPFDIQNTEDYLFQVILEKQIRIPRSLSVKAASVLKSFLNKDPKERLGCHPQTGFADIQGHPFFRNVDWDMMEQKQVVPPFKPNISGEFGLDNFDSQFTNEPVQLXPDDDDIVRKIDQSEFEGFEYINPLRMRPFKRQGSVRR Hydrogen bonds contact Hydrophobic contact | ||||
| 77 | Guanylate cyclase soluble beta-1 (GUCY1B1) | 7D9R | 6.55 | |
Target general information Gen name GUCY1B1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Soluble guanylate cyclase small subunit; Guanylate cyclase soluble subunit beta-3; Guanylate cyclase soluble subunit beta-1; GUCY1B3; GUCSB3; GUC1B3; GCS-beta-3; GCS-beta-1 Protein family Adenylyl cyclase class-4/guanylyl cyclase family Biochemical class Phosphorus-oxygen lyase Function Mediates responses to nitric oxide (NO) by catalyzing the biosynthesis of the signaling molecule cGMP. Related diseases Neurodevelopmental disorder with seizures, hypotonia, and brain imaging abnormalities (NEDSHBA) [MIM:618922]: An autosomal recessive neurodevelopmental disorder characterized by global developmental delay, hypotonia, severe to profound intellectual disability, early-onset epilepsy, and microcephaly. Neuroimaging shows cerebral atrophy, thin corpus callosum and hypomyelination in a majority of cases. Death in childhood may occur. {ECO:0000269|PubMed:27435318, ECO:0000269|PubMed:28097321, ECO:0000269|PubMed:32286009, ECO:0000269|PubMed:33476302, ECO:0000269|PubMed:33500274}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB09401; DB15456 Interacts with Q02108; Q02108-1 EC number EC 4.6.1.2 Uniprot keywords 3D-structure; Alternative splicing; cGMP biosynthesis; Cytoplasm; Direct protein sequencing; GTP-binding; Heme; Iron; Lyase; Metal-binding; Nucleotide-binding; Proteomics identification; Reference proteome Protein physicochemical properties Chain ID A,B Molecular weight (Da) 121177 Length 1071 Aromaticity 0.09 Instability index 42.35 Isoelectric point 6.18 Charge (pH=7) -11.86 2D Binding mode Binding energy (Kcal/mol) -8.94  Molscript Map 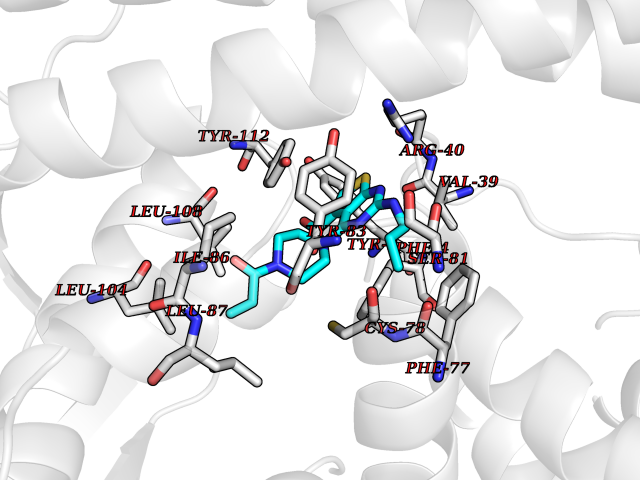 Pymol Map 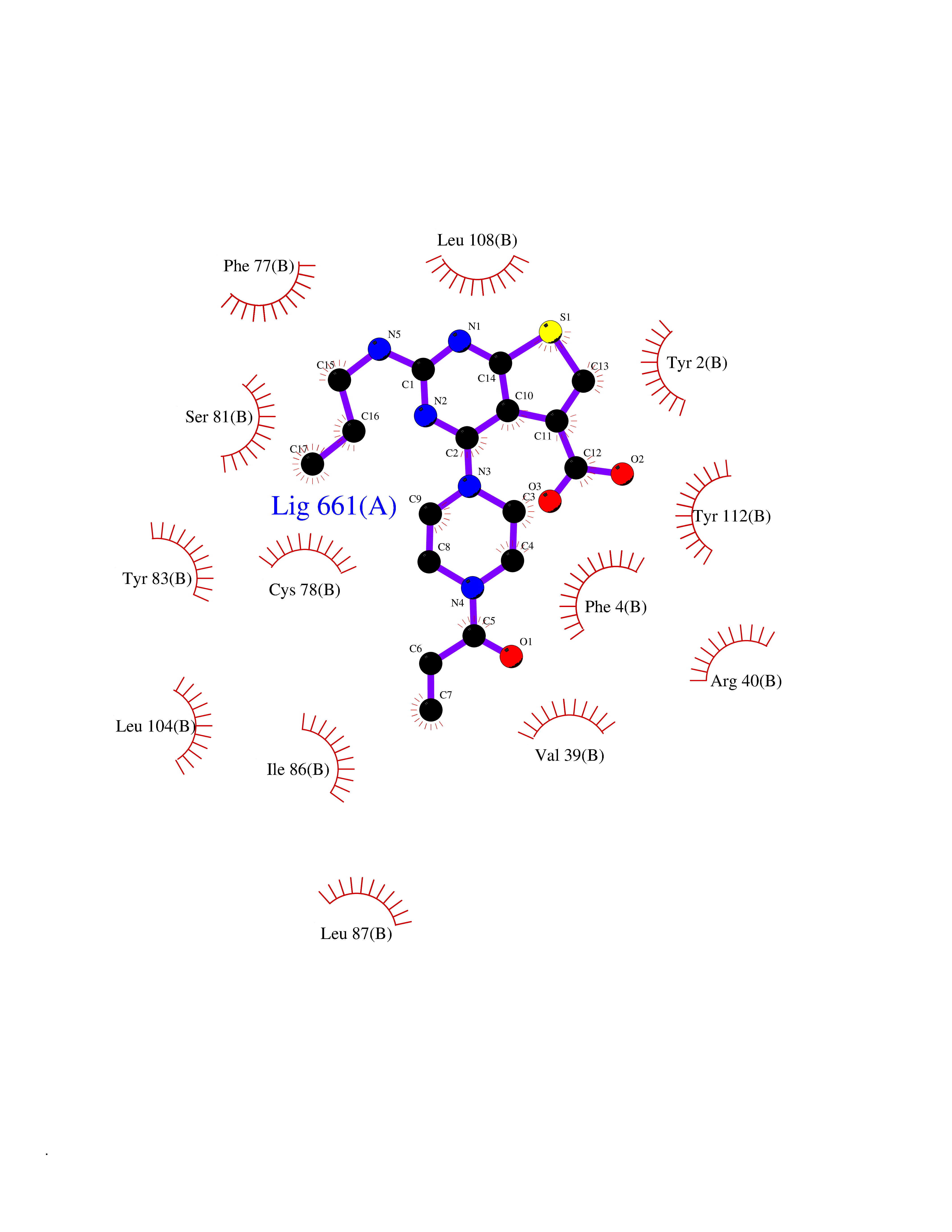 Ligplot Map 3D Binding mode Sequence SRVYLHTLAESICKLIFPEFERLNVALQRTLAKHKFEKTIAEQAVAAGVPVEVIKESLGEEVFKICYEEDENILGVVGGTLKDFLNSFSTLLKQASILCLLHVYYFTTSLILPGIIKAAAHVLYETEVEVSLYLLYSVHSLVIPTSLFCKTFPFHFMFDKDMTILQFGNGIRRLMNRRDKPNFEEYFEILTPKINQTFSGIMTMLNMQFVVRVRRVMDLKGQMIYIVESSAILFLGSPCVLYLSDIPIHNALRDVVLIGEQARAQDGLKKRLGKLKATLEQAHQALEEEKKKTVDLLCSIFPCEVAQQLWQGQVVQAKKFSNVTMLFSDIVGFTAICSQCSPLQVITMLNALYTRFDQQCGELDVYKVETIGDAYCVAGGLHKESDTHAVQIALMAVKMMELSDEVMSPHGEPIKMRIGLHSGSVFAGVVGVKMPRYCLFGNNVTLANKFESCSVPRKINVSPTTYRLLKDCPGFVFTPRSREELPPNFPSEIPGICHFLDAMYGFVNHALELLVIRNYGPEVWEDIKKEAQLDEEGQFLVRIIYDDSKTYDLVAAASKVLNLNAGEILQMFGKMFFVFCQESGYDTILRVLGSNVREFLQNLDALHDHLATIYPGMRAPSFRCTDAEKGKGLILHYYSEREGLQDIVIGIIKTVAQQIHGTEIDMKVIQQRNEECDHTQFLIEEKEESRISPYTFCKAFPFHIIFDRDLVVTQCGNAIYRVLPQLQPGNCSLLSVFSLVRPHIDISFHGILSHINTVFVLRSKEGLLDSCLRLKGQMIYLPEADSILFLCSPSVMNLDDLTRRGLYLSDIPLHDATRDLVLLGEQFREEYKLTQELEILTDRLQLTLRALEDEKKKTDTLLYSVLPPSVANELRHKRPVPAKRYDNVTILFSGIVGFNAFCSKHAGAMKIVNLLNDLYTRFDTLTDSRKNPFVYKVETVGDKYMTVSGLPEPCIHHARSICHLALDMMEIAGQVQVDGESVQITIGIHTGEVVTGVIGQRMPRYCLFGNTVNLTSRTETTGEKGKINVSEYTYRCLMSPENSDPQFHLEHRGPVSMKGKKEPMQVWFLSR Hydrogen bonds contact Hydrophobic contact | ||||
| 78 | C-C chemokine receptor type 9 (CCR9) | 5LWE | 6.55 | |
Target general information Gen name CCR9 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Gprotein coupled receptor 28; GPR96; GPR28; GPR-9-6; G-protein coupled receptor 28; CDw199; CCR-9; CCCKR9; CC-CKR-9; CC chemokine receptor type 9; CC CKR9; C-C CKR-9 Protein family G-protein coupled receptor 1 family Biochemical class GPCR rhodopsin Function Subsequently transduces a signal by increasing the intracellular calcium ions level. Receptor for chemokine SCYA25/TECK. Related diseases Split-foot malformation with mesoaxial polydactyly (SFMMP) [MIM:616890]: An autosomal recessive disorder characterized by a split-foot defect, mesoaxial polydactyly, nail abnormalities of the hands, and sensorineural hearing loss. {ECO:0000269|PubMed:26755636, ECO:0000269|PubMed:32266845}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Myopathy, centronuclear, 6, with fiber-type disproportion (CNM6) [MIM:617760]: A form of centronuclear myopathy, a congenital muscle disorder characterized by progressive muscular weakness and wasting involving mainly limb girdle, trunk, and neck muscles. It may also affect distal muscles. Weakness may be present during childhood or adolescence or may not become evident until the third decade of life. Ptosis is a frequent clinical feature. The most prominent histopathologic features include high frequency of centrally located nuclei in muscle fibers not secondary to regeneration, radial arrangement of sarcoplasmic strands around the central nuclei, and predominance and hypotrophy of type 1 fibers. CNM6 is an autosomal recessive, slowly progressive form with onset in infancy or early childhood. {ECO:0000269|PubMed:27816943, ECO:0000269|PubMed:30237576}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB15250 Interacts with NA EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disulfide bond; G-protein coupled receptor; Glycoprotein; Host-virus interaction; Membrane; Receptor; Reference proteome; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 31137.4 Length 275 Aromaticity 0.13 Instability index 25.98 Isoelectric point 9.31 Charge (pH=7) 15.22 2D Binding mode Binding energy (Kcal/mol) -8.94  Molscript Map 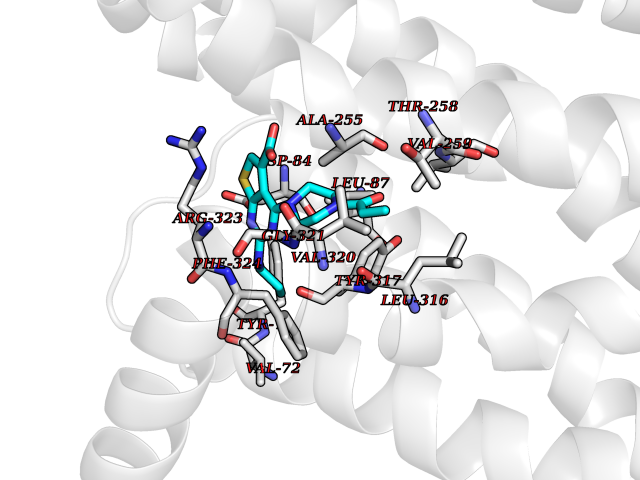 Pymol Map 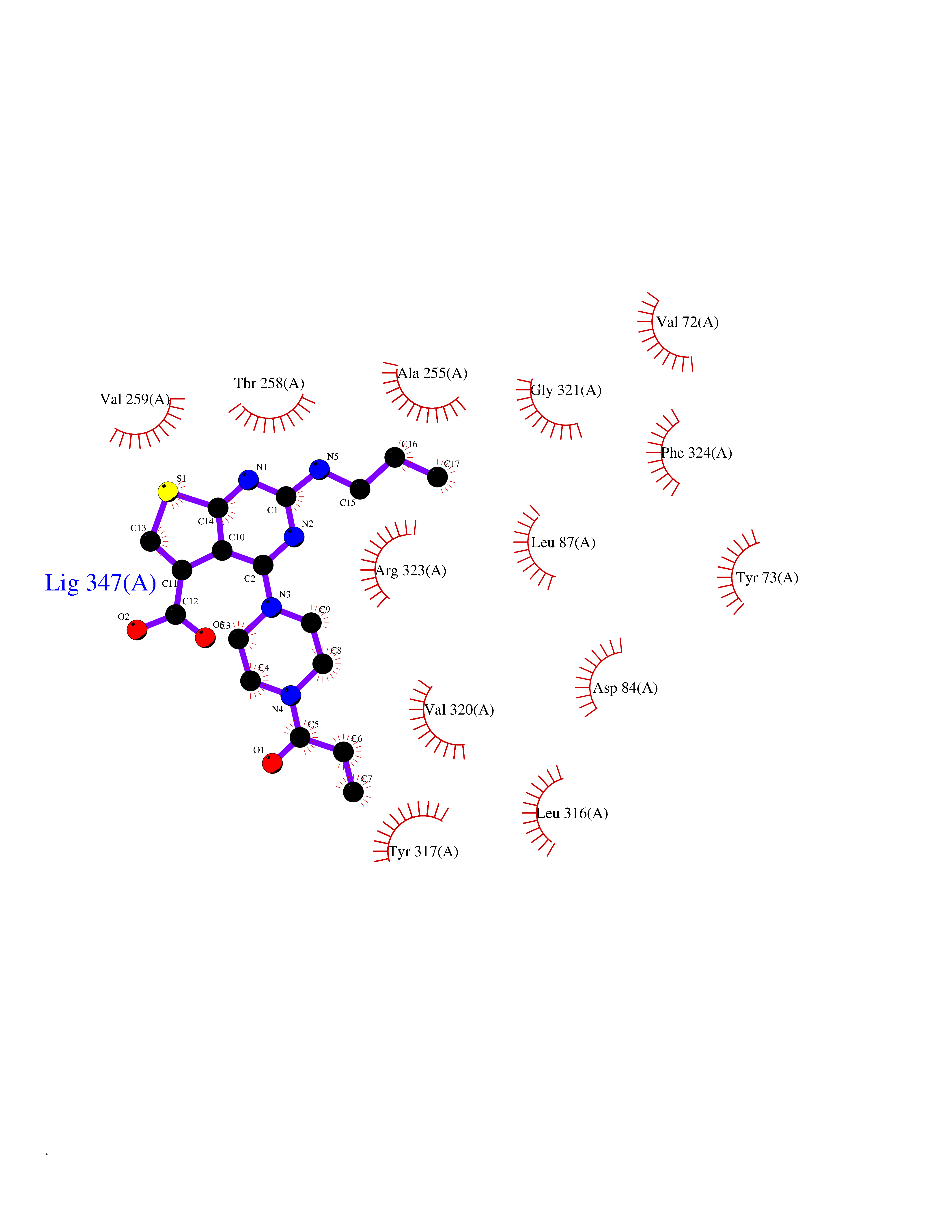 Ligplot Map 3D Binding mode Sequence RQFASHFLPPLYWLVFIVGALGNSLVILVYWYCARAKTATDMFLLNLAIADLLFLVTLPFWAIATFMCKVVNSMYKMNFYSCVLLIMCICVDRYIAIAQAMRAHTWREKRLLYSKMVCFTIWVLAAALCIPEILYCTTKLKSAVLALKVILGFFLPFVVMACCYTIIIHTLIQAKKSSKHKALKATITVLTVFVLSQFPYNCILLVQTIDAYAMFISNCAVSTAIDICFQVTQAIAFFHSCLNPVLYVFVGERFRRDLVKTLKNLGAISQAAAHH Hydrogen bonds contact Hydrophobic contact | ||||
| 79 | Melatonin receptor type 1A (MTNR1A) | 7DB6 | 6.55 | |
Target general information Gen name MTNR1A Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Mel1a receptor; Mel1AR; Mel-1A-R Protein family G-protein coupled receptor 1 family Biochemical class GPCR rhodopsin Function Likely to mediate the reproductive and circadian actions of melatonin. The activity of this receptor is mediated by pertussis toxin sensitive G proteins that inhibit adenylate cyclase activity. High affinity receptor for melatonin. Related diseases Spermatogenic failure 5 (SPGF5) [MIM:243060]: An infertility disorder caused by spermatogenesis defects. Semen from affected men show close to 100% morphologically abnormal multiflagellar spermatozoa with low motility, oversized irregular heads, and abnormal midpiece and acrosome. {ECO:0000269|PubMed:17435757, ECO:0000269|PubMed:21733974}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB06594; DB01065; DB00980; DB02709; DB09071 Interacts with P27797; A8MQ03; Q8IUG1; P49286; O76081; P57088 EC number NA Uniprot keywords 3D-structure; Cell membrane; Disulfide bond; G-protein coupled receptor; Glycoprotein; Membrane; Receptor; Reference proteome; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID D Molecular weight (Da) 31301 Length 276 Aromaticity 0.15 Instability index 37.33 Isoelectric point 9.22 Charge (pH=7) 9.92 2D Binding mode Binding energy (Kcal/mol) -8.94 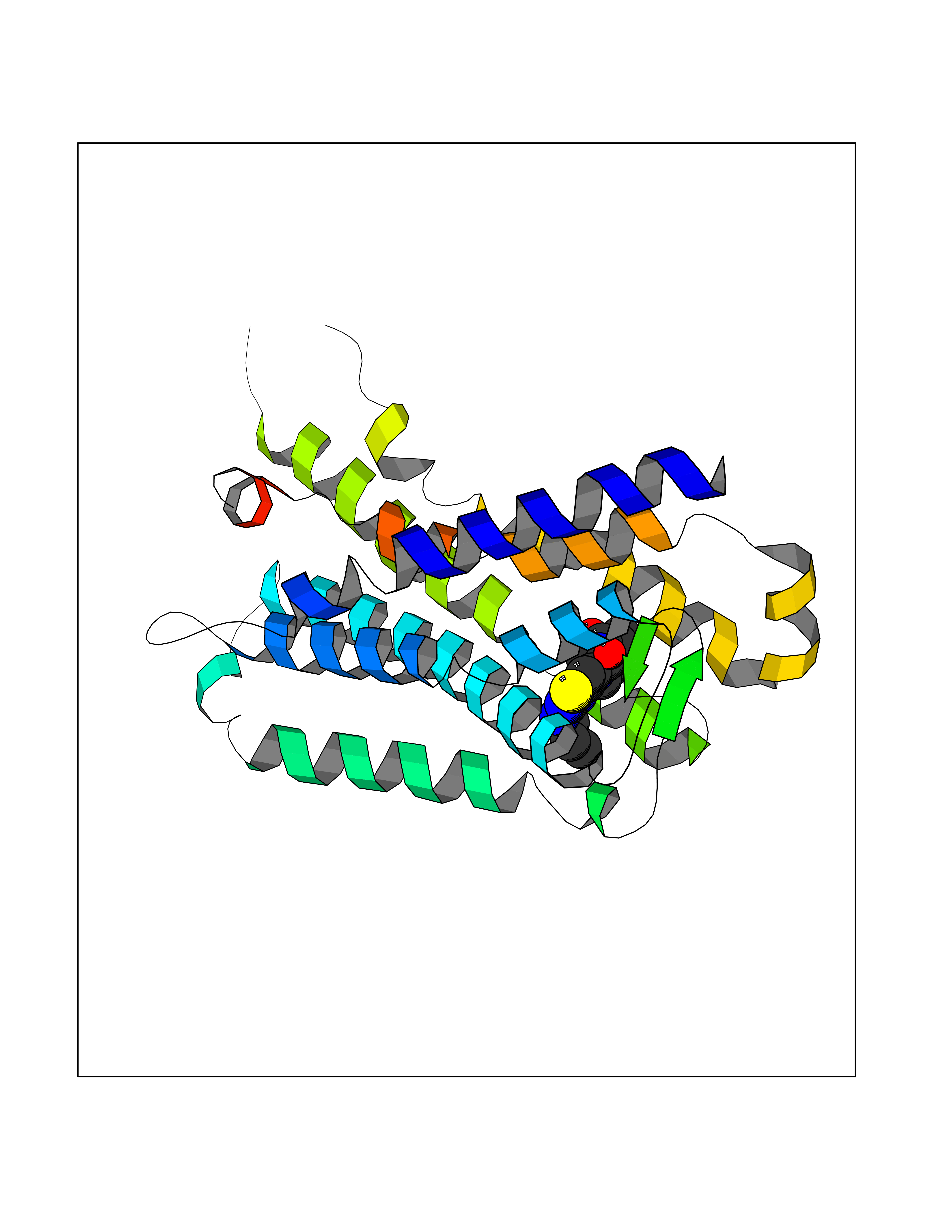 Molscript Map 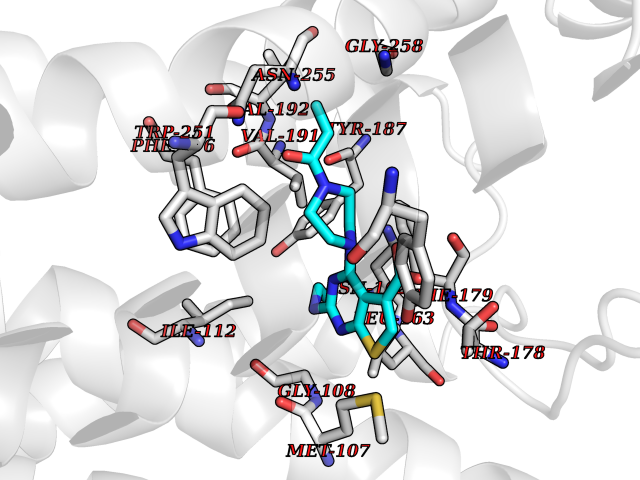 Pymol Map 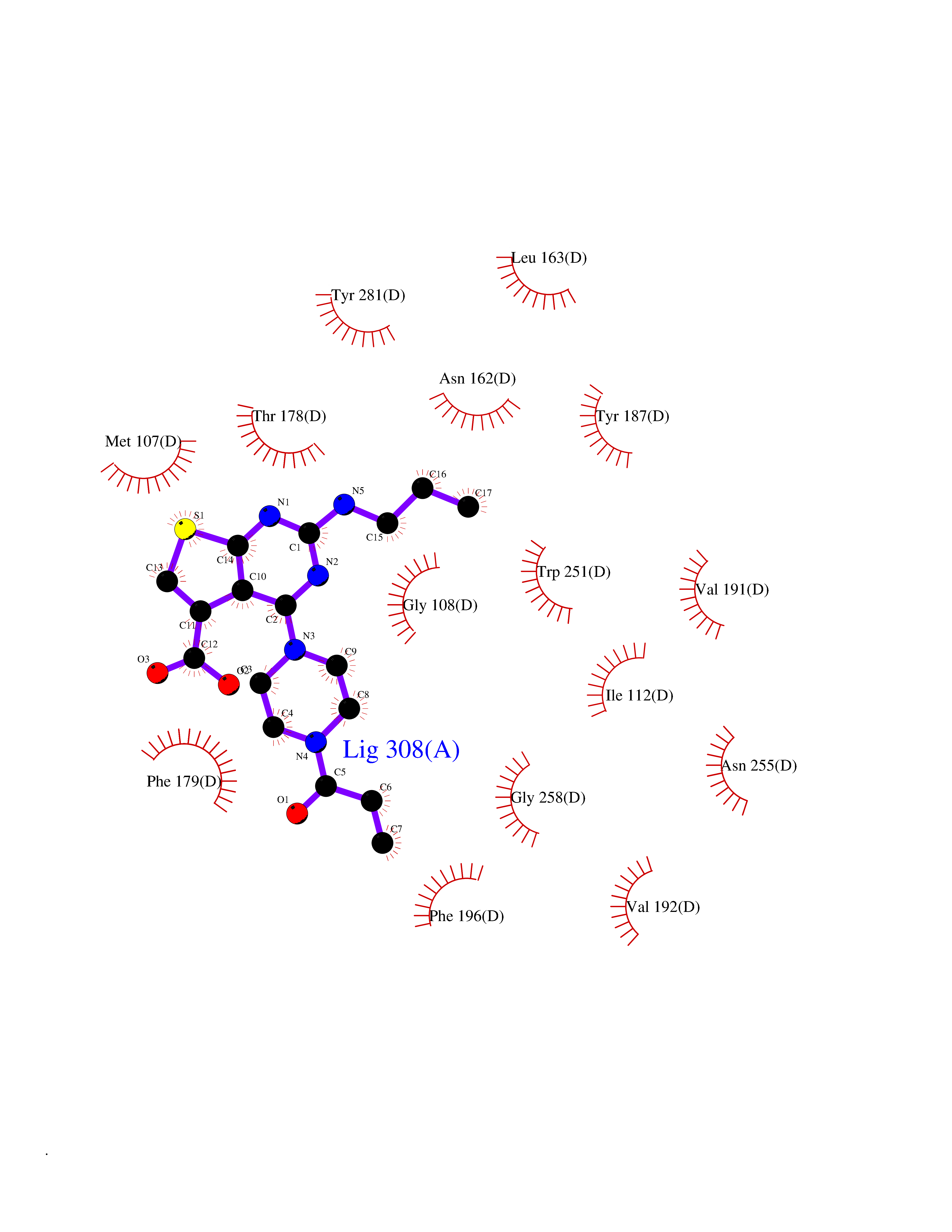 Ligplot Map 3D Binding mode Sequence RPSWLASALACVLIFTIVVDILGNLLVILSVYRNKKLRNAGNIFVVSLAVADLVVAIYPYPLVLMSIFNNGWNLGYLHCQVSGFLMGLSVIGSIFNITGIAINRYCYICHSLKYDKLYSSKNSLCYVLLIWLLTLAAVLPNLRAGTLQYDPRIYSCTFAQSVSSAYTIAVVVFHFLVPMIIVIFCYLRIWILVLQVRQRVPQDFRNFVTMFVVFVLFAICWAPLNFIGLAVASDPASMVPRIPEWLFVASYYMAYFNSCLNAIIYGLLNQNFRKEY Hydrogen bonds contact Hydrophobic contact | ||||
| 80 | Retinoic acid receptor gamma (RARG) | 1FCY | 6.54 | |
Target general information Gen name RARG Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms RAR-gamma; Nuclear receptor subfamily 1 group B member 3; NR1B3 Protein family Nuclear hormone receptor family, NR1 subfamily Biochemical class Nuclear hormone receptor Function Receptor for retinoic acid. Retinoic acid receptors bind as heterodimers to their target response elements in response to their ligands, all-trans or 9-cis retinoic acid, and regulate gene expression in various biological processes. The RAR/RXR heterodimers bind to the retinoic acid response elements (RARE) composed of tandem 5'-AGGTCA-3' sites known as DR1-DR5. In the absence of ligand, acts mainly as an activator of gene expression due to weak binding to corepressors. Required for limb bud development. In concert with RARA or RARB, required for skeletal growth, matrix homeostasis and growth plate function (By similarity). Related diseases Cystic fibrosis (CF) [MIM:219700]: A common generalized disorder of the exocrine glands which impairs clearance of secretions in a variety of organs. It is characterized by the triad of chronic bronchopulmonary disease (with recurrent respiratory infections), pancreatic insufficiency (which leads to malabsorption and growth retardation) and elevated sweat electrolytes. It is the most common genetic disease in Caucasians, with a prevalence of about 1 in 2'000 live births. Inheritance is autosomal recessive. {ECO:0000269|PubMed:10094564, ECO:0000269|PubMed:10869121, ECO:0000269|PubMed:10923036, ECO:0000269|PubMed:11242048, ECO:0000269|PubMed:12167682, ECO:0000269|PubMed:12394343, ECO:0000269|PubMed:12529365, ECO:0000269|PubMed:1284466, ECO:0000269|PubMed:1284468, ECO:0000269|PubMed:1284529, ECO:0000269|PubMed:1284530, ECO:0000269|PubMed:1284548, ECO:0000269|PubMed:1379210, ECO:0000269|PubMed:15528182, ECO:0000269|PubMed:15716351, ECO:0000269|PubMed:16822950, ECO:0000269|PubMed:1695717, ECO:0000269|PubMed:1699669, ECO:0000269|PubMed:17098864, ECO:0000269|PubMed:1710600, ECO:0000269|PubMed:1712898, ECO:0000269|PubMed:17182731, ECO:0000269|PubMed:20008117, ECO:0000269|PubMed:20150177, ECO:0000269|PubMed:20691141, ECO:0000269|PubMed:21884936, ECO:0000269|PubMed:2236053, ECO:0000269|PubMed:23818989, ECO:0000269|PubMed:25330774, ECO:0000269|PubMed:26846474, ECO:0000269|PubMed:27241308, ECO:0000269|PubMed:28001373, ECO:0000269|PubMed:28067262, ECO:0000269|PubMed:28087700, ECO:0000269|PubMed:32026723, ECO:0000269|PubMed:33572515, ECO:0000269|PubMed:7504969, ECO:0000269|PubMed:7505694, ECO:0000269|PubMed:7505767, ECO:0000269|PubMed:7508414, ECO:0000269|PubMed:7513296, ECO:0000269|PubMed:7517264, ECO:0000269|PubMed:7520022, ECO:0000269|PubMed:7522211, ECO:0000269|PubMed:7524909, ECO:0000269|PubMed:7524913, ECO:0000269|PubMed:7525450, ECO:0000269|PubMed:7537150, ECO:0000269|PubMed:7541273, ECO:0000269|PubMed:7541510, ECO:0000269|PubMed:7543567, ECO:0000269|PubMed:7544319, ECO:0000269|PubMed:7581407, ECO:0000269|PubMed:7606851, ECO:0000269|PubMed:7680525, ECO:0000269|PubMed:7683628, ECO:0000269|PubMed:7683954, ECO:0000269|PubMed:8081395, ECO:0000269|PubMed:8406518, ECO:0000269|PubMed:8522333, ECO:0000269|PubMed:8723693, ECO:0000269|PubMed:8723695, ECO:0000269|PubMed:8800923, ECO:0000269|PubMed:8829633, ECO:0000269|PubMed:8910473, ECO:0000269|PubMed:8956039, ECO:0000269|PubMed:9101301, ECO:0000269|PubMed:9222768, ECO:0000269|PubMed:9375855, ECO:0000269|PubMed:9401006, ECO:0000269|PubMed:9443874, ECO:0000269|PubMed:9452048, ECO:0000269|PubMed:9452054, ECO:0000269|PubMed:9452073, ECO:0000269|PubMed:9482579, ECO:0000269|PubMed:9507391, ECO:0000269|PubMed:9521595, ECO:0000269|PubMed:9554753, ECO:0000269|PubMed:9736778, ECO:0000269|PubMed:9804160, ECO:0000269|PubMed:9921909}. The disease is caused by variants affecting the gene represented in this entry. There is some evidence that the functional defect caused by the most common variant Phe-508 DEL can be corrected by the binding to the snake phospholipase A2 crotoxin basic subunit CB. This toxin both disrupts the Phe-508 DEL-cytokeratin 8 complex, allowing for the escape from degradation, and increases the chloride channel current (PubMed:27241308). {ECO:0000269|PubMed:27241308}.; DISEASE: Congenital bilateral absence of the vas deferens (CBAVD) [MIM:277180]: An autosomal recessive disease characterized by vas deferens aplasia resulting in azoospermia and male infertility. CBAVD may occur in isolation or as a manifestation of cystic fibrosis. {ECO:0000269|PubMed:10066035, ECO:0000269|PubMed:10651488, ECO:0000269|PubMed:17329263, ECO:0000269|PubMed:7529962, ECO:0000269|PubMed:7539342, ECO:0000269|PubMed:9067761, ECO:0000269|PubMed:9736778, ECO:0000269|Ref.117}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07294; DB07031; DB00459; DB00210; DB00523; DB02466; DB03466; DB02741; DB03279; DB00926; DB00982; DB05785; DB05467; DB02258; DB00799; DB00755; DB12808 Interacts with Q96RK4; P13349; P31321; P28702; P48443; O60504-2 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; DNA-binding; Isopeptide bond; Metal-binding; Methylation; Nucleus; Proteomics identification; Receptor; Reference proteome; Transcription; Transcription regulation; Ubl conjugation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A Molecular weight (Da) 26574.9 Length 236 Aromaticity 0.06 Instability index 49.98 Isoelectric point 5.76 Charge (pH=7) -2.95 2D Binding mode Binding energy (Kcal/mol) -8.92 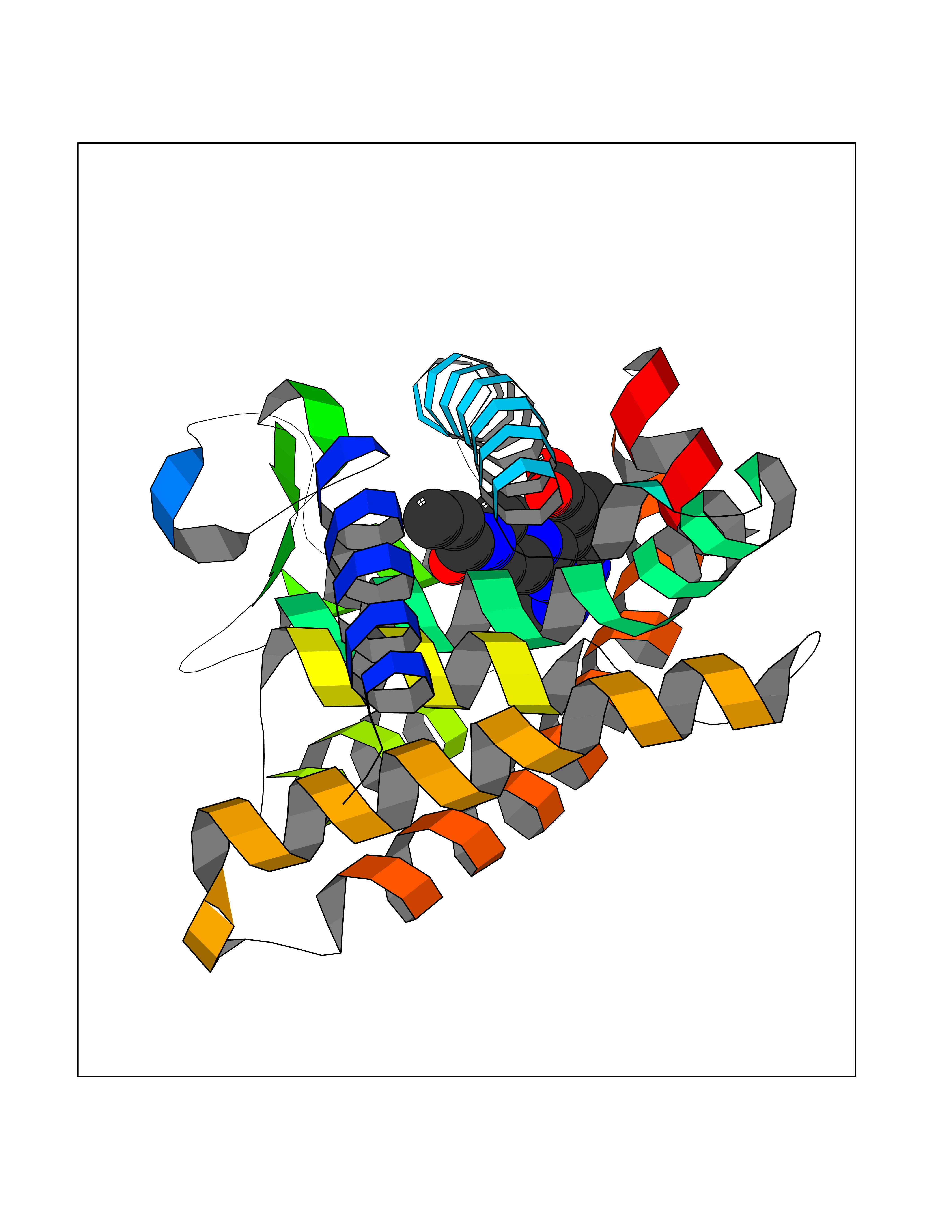 Molscript Map 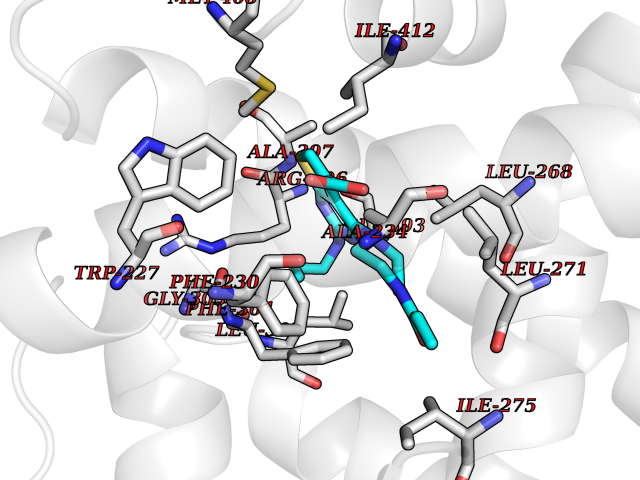 Pymol Map 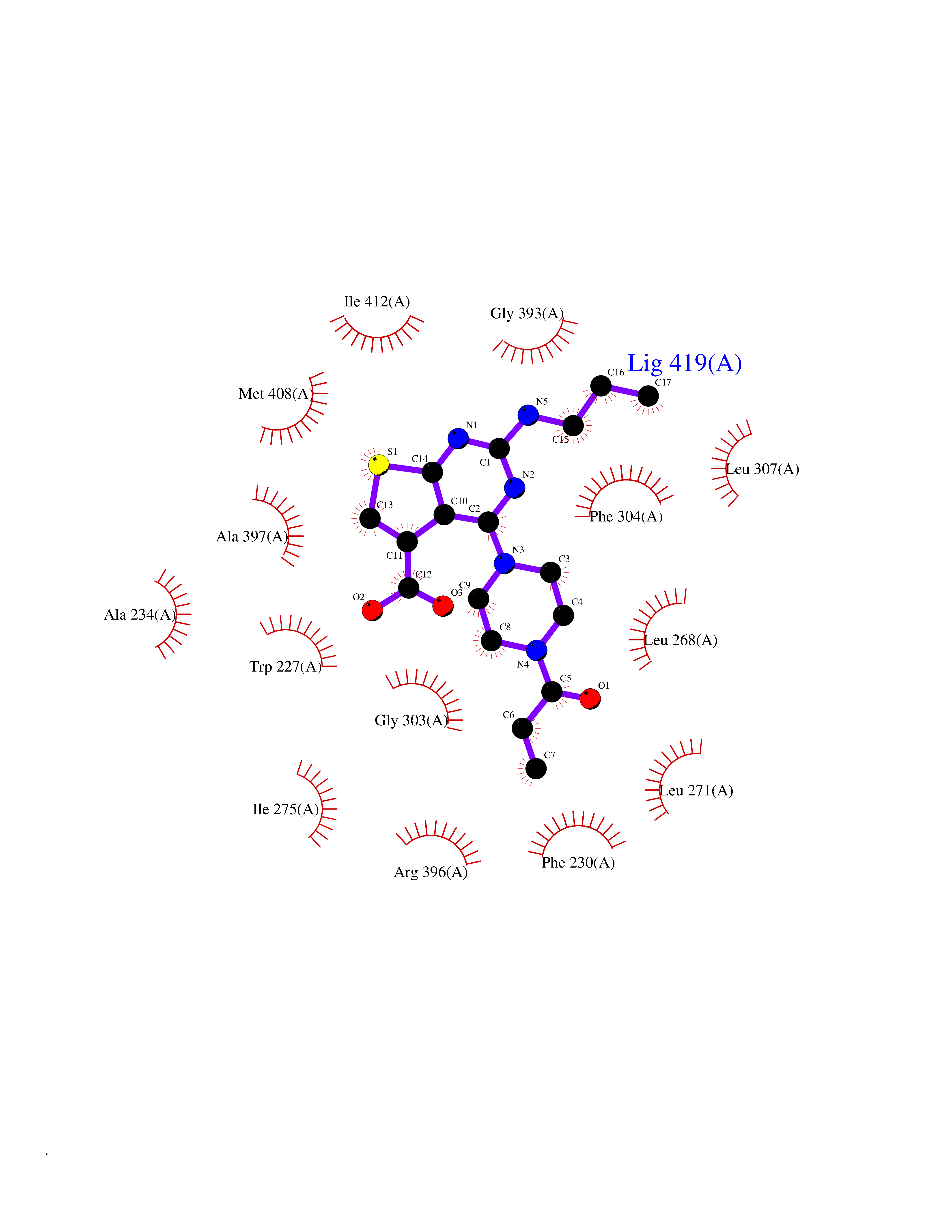 Ligplot Map 3D Binding mode Sequence ASPQLEELITKVSKAHQETFPSLCQLGKYTTNSSADHRVQLDLGLWDKFSELATKCIIKIVEFAKRLPGFTGLSIADQITLLKAACLDILMLRICTRYTPEQDTMTFSDGLTLNRTQMHNAGFGPLTDLVFAFAGQLLPLEMDDTETGLLSAICLICGDRMDLEEPEKVDKLQEPLLEALRLYARRRRPSQPYMFPRMLMKITDLRGISTKGAERAITLKMEIPGPMPPLIREMLE Hydrogen bonds contact Hydrophobic contact | ||||