Job Results:
Ligand
Structure
Job ID
7a36b9b0fd299388007ea8e58dfe56c6
Job name
NA
Time
2025-09-26 17:47:53
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 61 | Squalene monooxygenase (SQLE) | 6C6N | 6.38 | |
Target general information Gen name SQLE Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Squalene epoxidase; SQLE; SE; Oxidosqaulene cyclase Protein family Squalene monooxygenase family Biochemical class Paired donor oxygen oxidoreductase Function Catalyzes the first oxygenation step in sterol biosynthesis and is suggested to be one of the rate-limiting enzymes in this pathway. Related diseases Developmental and epileptic encephalopathy 24 (DEE24) [MIM:615871]: A disease characterized by early-onset seizures, intellectual disability of varying degrees, and behavioral disturbances or autistic features in most individuals. {ECO:0000269|PubMed:24747641, ECO:0000269|PubMed:27864847, ECO:0000269|PubMed:30351409}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Generalized epilepsy with febrile seizures plus 10 (GEFSP10) [MIM:618482]: An autosomal dominant neurologic disorder with incomplete penetrance, characterized by variable types of seizures including absence, tonic-clonic, febrile, focal, and eyelid myoclonia. Some patients have normal neurologic development. Others have mild-to-moderate intellectual disability or autism spectrum disorder. {ECO:0000269|PubMed:29936235, ECO:0000269|PubMed:30351409}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01091; DB08846; DB00735; DB00857 Interacts with Q96BA8; Q9H6H4; Q9NUH8 EC number EC 1.14.14.17 Uniprot keywords 3D-structure; Endoplasmic reticulum; FAD; Flavoprotein; Lipid metabolism; Membrane; Microsome; Oxidoreductase; Proteomics identification; Reference proteome; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 50003.4 Length 450 Aromaticity 0.1 Instability index 37.4 Isoelectric point 7.8 Charge (pH=7) 1.9 2D Binding mode Binding energy (Kcal/mol) -8.7 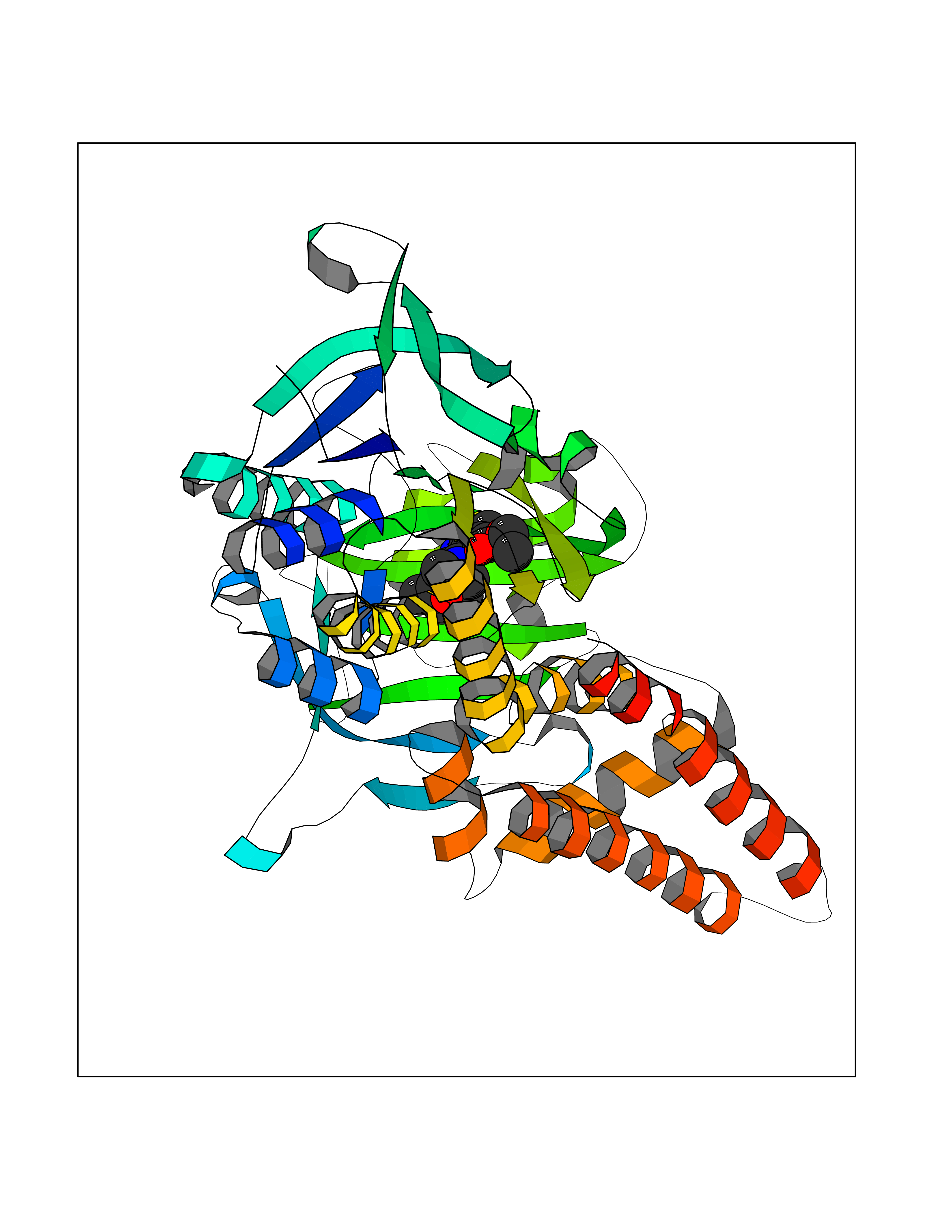 Molscript Map 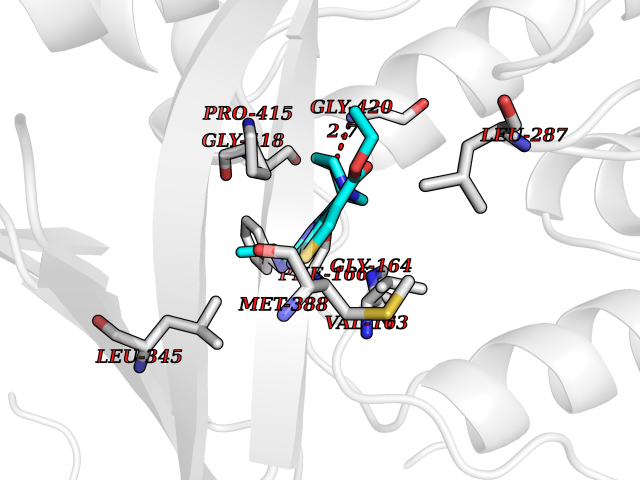 Pymol Map 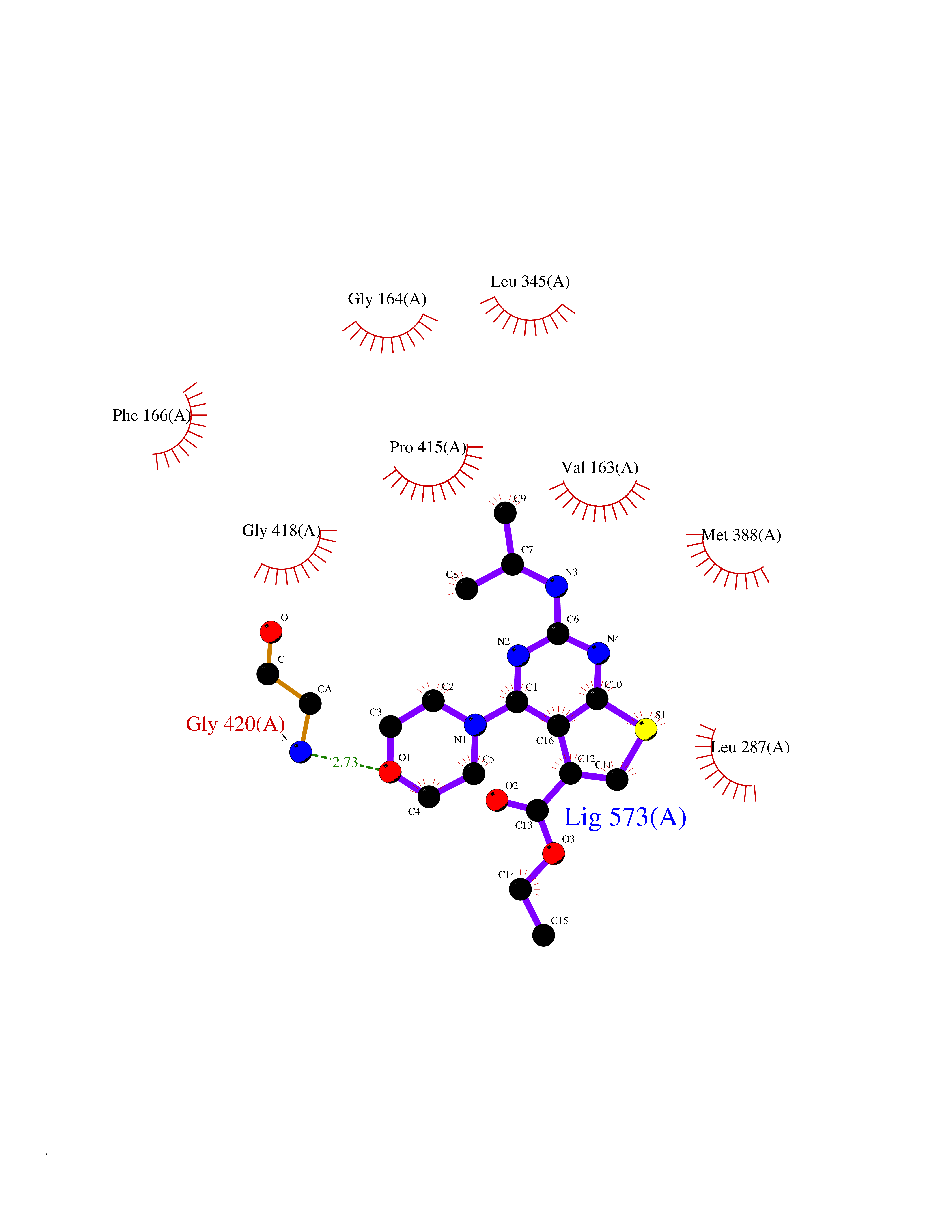 Ligplot Map 3D Binding mode Sequence NDPEVIIVGAGVLGSALAAVLSRDGRKVTVIERDLKEPDRIVGEFLQPGGYHVLKDLGLGDTVEGLDAQVVNGYMIHDQESKSEVQIPYPLSENNQVQSGRAFHHGRFIMSLRKAAMAEPNAKFIEGVVLQLLEEDDVVMGVQYKDKETGDIKELHAPLTVVADGLFSKFRKSLVSNKVSVSSHFVGFLMKNAPQFKANHAELILANPSPVLIYQISSSETRVLVDIRGEMPRNLREYMVEKIYPQIPDHLKEPFLEATDNSHLRSMPASFLPPSSVKKRGVLLLGDAYNMRHPLTGGGMTVAFKDIKLWRKLLKGIPDLYDDAAIFEAKKSFYWARKTSHSFVVNILAQALYELFSATDDSLHQLRKACFLYFKLGGECVAGPVGLLSVLSPNPLVLIGHFFAVAIYAVYFCFKSEPWITKPRALLSSGAVLYKACSVIFPLIYSEMKY Hydrogen bonds contact Hydrophobic contact | ||||
| 62 | Glycine oxidase | 1RYI | 6.37 | |
Target general information Gen name thiO Organism Bacillus subtilis (strain 168) Uniprot ID TTD ID NA Synonyms goxB;yjbR;BSU11670 Protein family DAO family, ThiO subfamily Biochemical class Oxidoreductase Function FAD binding.Glycine oxidase activity.Oxidoreductase activity. Related diseases Hypervalinemia and hyperleucine-isoleucinemia (HVLI) [MIM:618850]: An autosomal recessive metabolic disorder characterized by highly elevated plasma concentrations of valine and leucine/isoleucine. Affected individuals suffer from headache and mild memory impairment. {ECO:0000269|PubMed:25653144}. The disease is caused by variants affecting the gene represented in this entry. A patient with hypervalinemia and hyperleucine-isoleucinemia was identified as compound heterozygote for Gln-170 (inherited from his father) and Lys-264 (inherited from his mother), both variants reduced the catalytic activity of the enzyme. After treatment with vitamin B6, a precursor of pyridoxal 5'-phosphate, a BCAT2 cofactor, the blood levels of branched chain amino acids, especially valine, were decreased and brain lesions were improved. {ECO:0000269|PubMed:25653144}. Drugs (DrugBank ID) DB02713; DB03147 Interacts with NA EC number 1.4.3.19 Uniprot keywords 3D-structure; Cytoplasm; FAD; Flavoprotein; Herbicide resistance; Oxidoreductase; Reference proteome; Thiamine biosynthesis Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 40395.8 Length 364 Aromaticity 0.1 Instability index 36.07 Isoelectric point 6.02 Charge (pH=7) -6.35 2D Binding mode Binding energy (Kcal/mol) -8.69 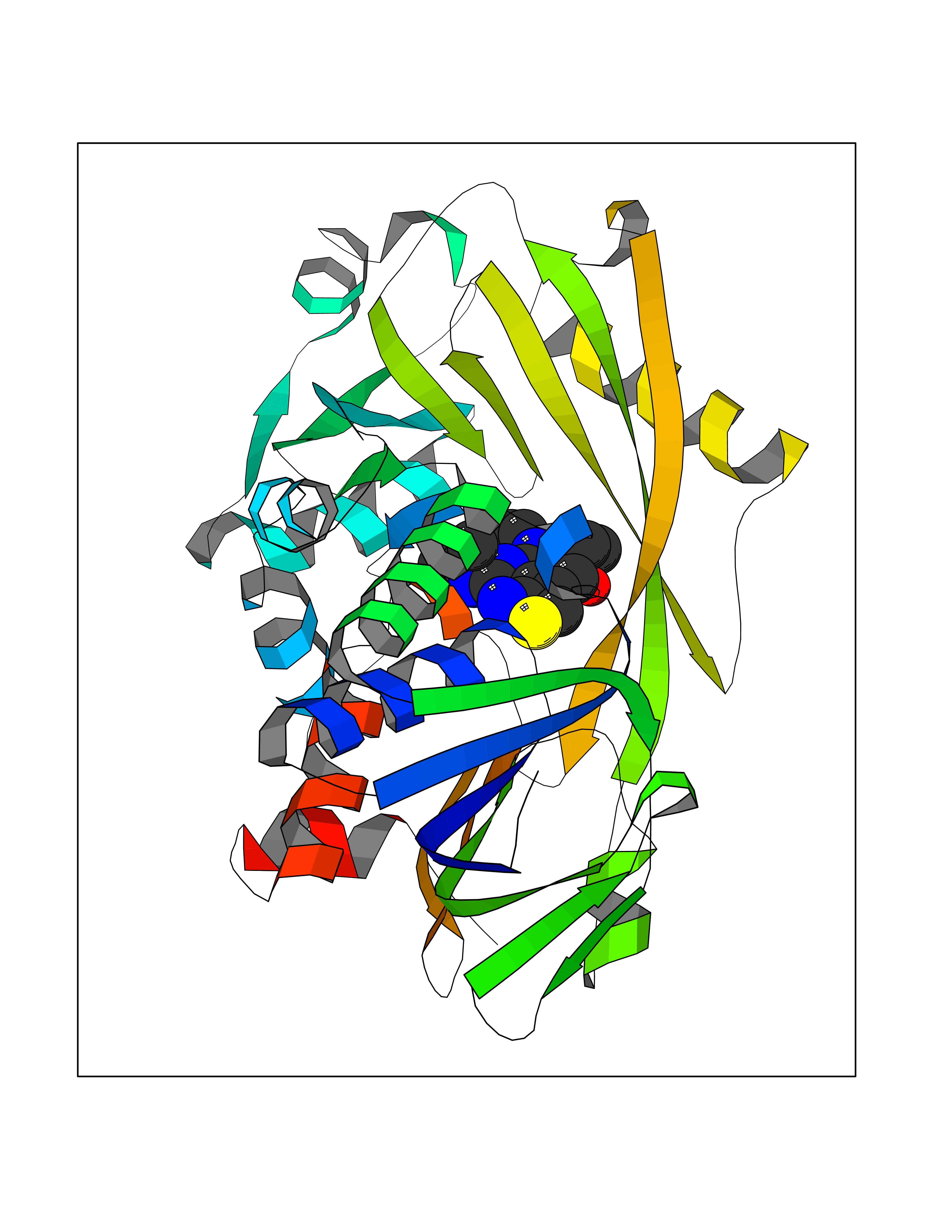 Molscript Map 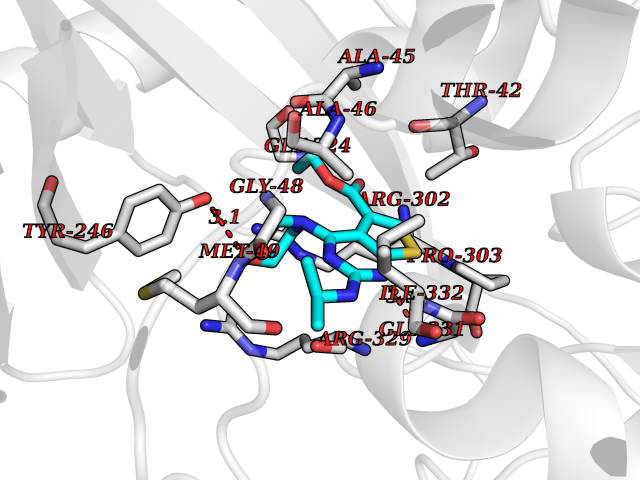 Pymol Map 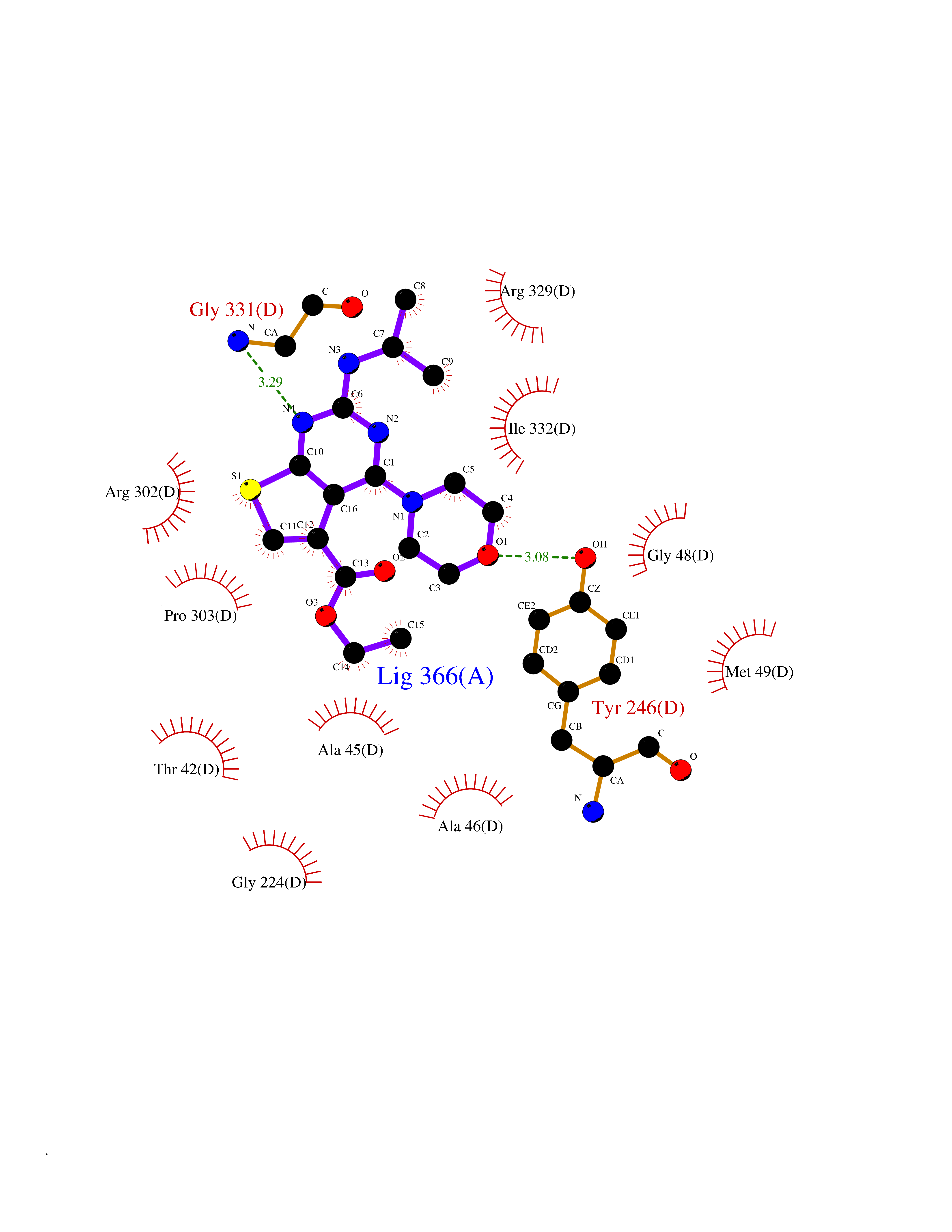 Ligplot Map 3D Binding mode Sequence MKRHYEAVVIGGGIIGSAIAYYLAKENKNTALFESGTMGGRTTSAAAGMLGAHAECEERDAFFDFAMHSQRLYKGLGEELYALSGVDIRQHNGGMFKLAFSEEDVLQLRQMDDLDSVSWYSKEEVLEKEPYASGDIFGASFIQDDVHVEPYFVCKAYVKAAKMLGAEIFEHTPVLHVERDGEALFIKTPSGDVWANHVVVASGVWSGMFFKQLGLNNAFLPVKGECLSVWNDDIPLTKTLYHDHCYIVPRKSGRLVVGATMKPGDWSETPDLGGLESVMKKAKTMLPAIQNMKVDRFWAGLRPGTKDGKPYIGRHPEDSRILFAAGHFRNGILLAPATGALISDLIMNKEVNQDWLHAFRIDRK Hydrogen bonds contact Hydrophobic contact | ||||
| 63 | "Periplasmic trehalase (EC 3.2.1.28) (Alpha,alpha-trehalase) (Alpha,alpha-trehalose glucohydrolase) (Tre37A)" | 2JG0 | 6.37 | |
Target general information Gen name treA Organism Escherichia coli (strain K12) Uniprot ID TTD ID NA Synonyms JW1186;osmA;b1197 Protein family Glycosyl hydrolase 37 family Biochemical class NA Function Provides the cells with the ability to utilize trehalose at high osmolarity by splitting it into glucose molecules that can subsequently be taken up by the phosphotransferase-mediated uptake system. Related diseases SRC kinase activity has been shown to be increased in several tumor tissues and tumor cell lines such as colon carcinoma cells. {ECO:0000269|PubMed:2498394, ECO:0000269|PubMed:3093483}.; DISEASE: Thrombocytopenia 6 (THC6) [MIM:616937]: A form of thrombocytopenia, a hematologic disorder defined by a decrease in the number of platelets in circulating blood, resulting in the potential for increased bleeding and decreased ability for clotting. THC6 is an autosomal dominant form. Affected individuals may also have bone abnormalities and an increased risk for myelofibrosis. {ECO:0000269|PubMed:26936507}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number 3.2.1.28 Uniprot keywords 3D-structure; Direct protein sequencing; Glycosidase; Hydrolase; Periplasm; Reference proteome; Signal Protein physicochemical properties Chain ID A Molecular weight (Da) 57508.9 Length 507 Aromaticity 0.11 Instability index 48.32 Isoelectric point 5.48 Charge (pH=7) -10.13 2D Binding mode Binding energy (Kcal/mol) -8.69 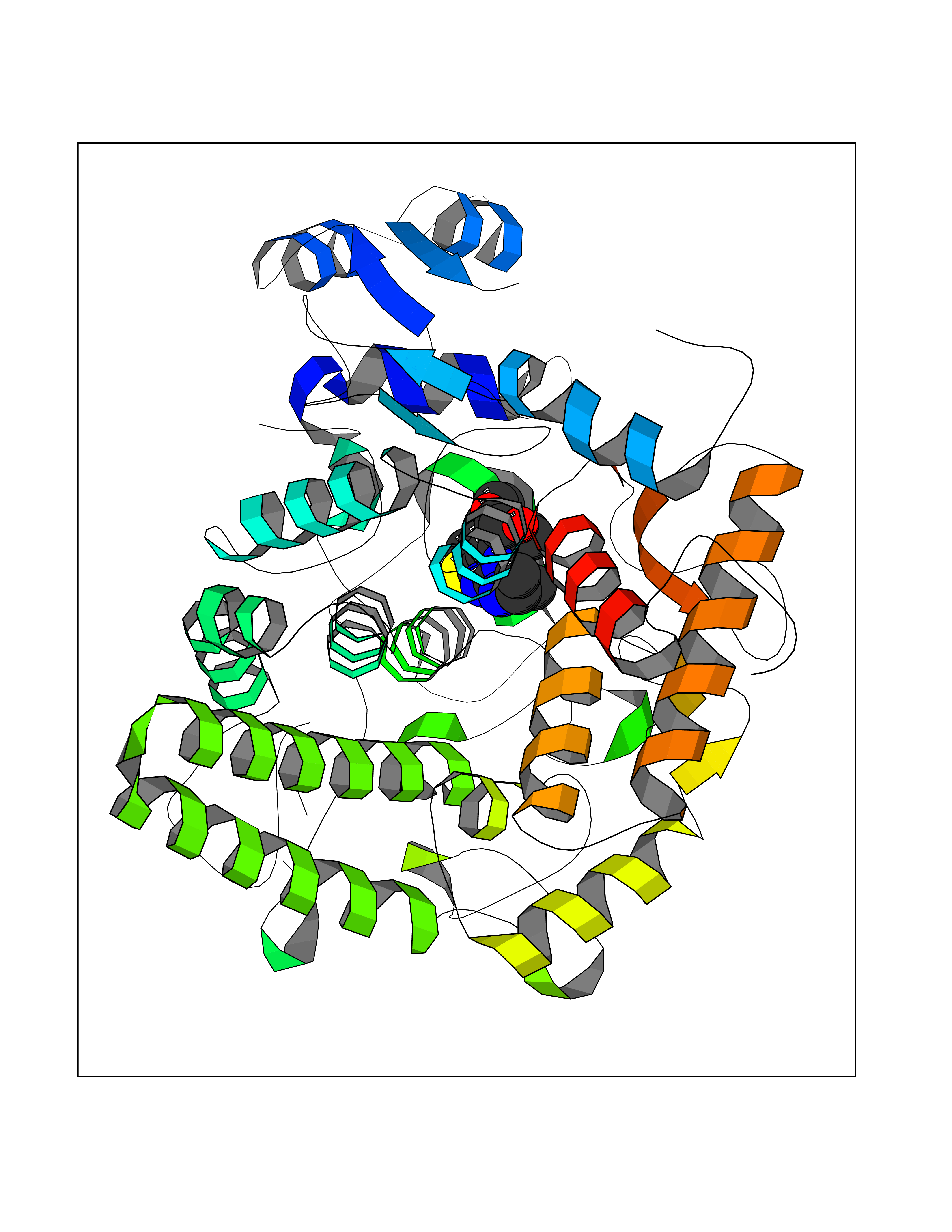 Molscript Map 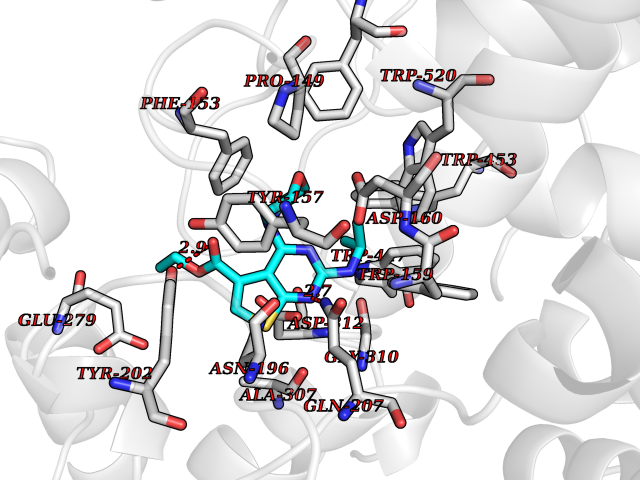 Pymol Map 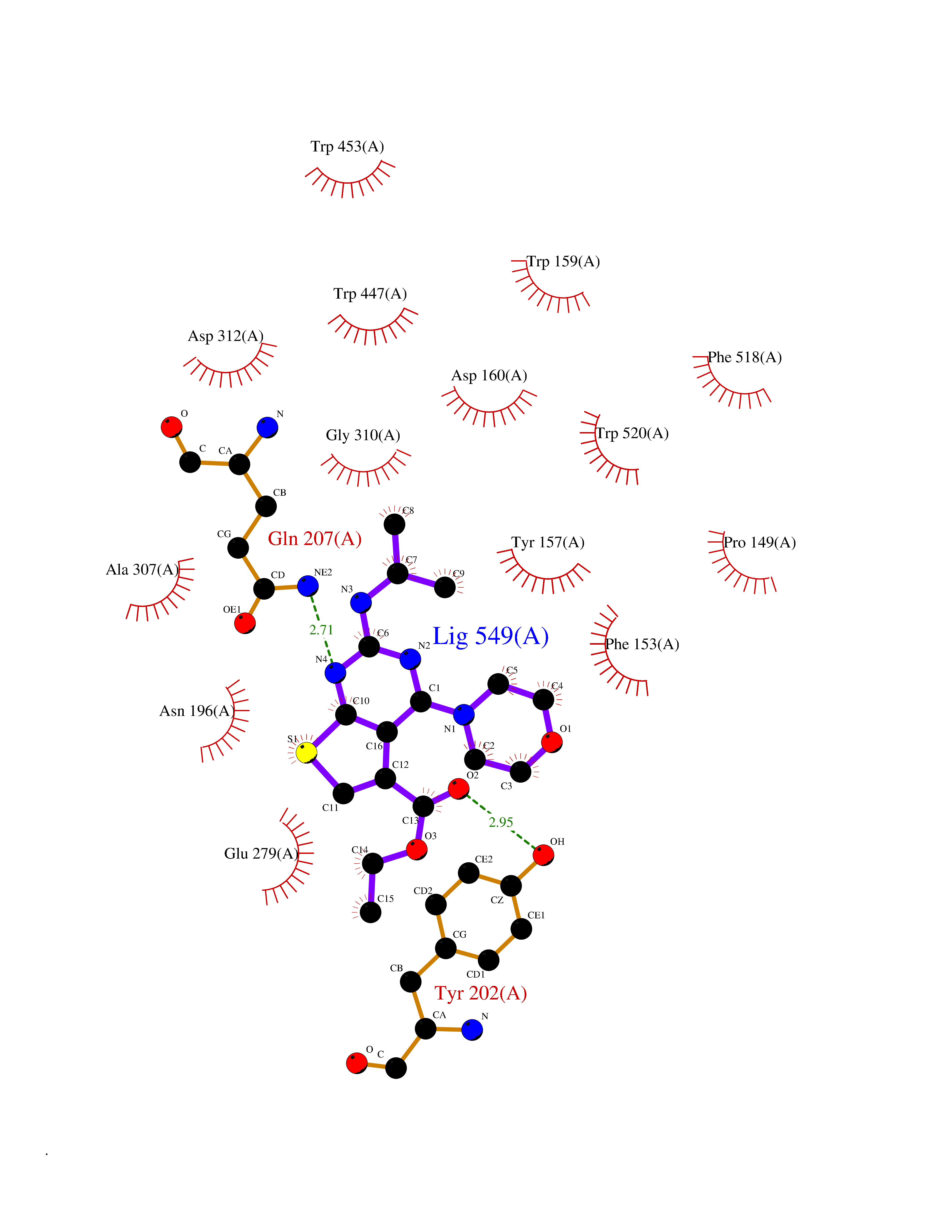 Ligplot Map 3D Binding mode Sequence PQPPDILLGPLFNDVQNAKLFPDQKTFADAVPNSDPLMILADYRMQQNQSGFDLRHFVNVNFTLPKYVPPEGQSLREHIDGLWPVLTRSTENTEKWDSLLPLPEPYVVPGGRFREVYYWDSYFTMLGLAESGHWDKVADMVANFAHEIDTYGHIPNGNRSYYLSRSQPPFFALMVELLAQHEGDAALKQYLPQMQKEYAYWMDGVENLQAGQQEKRVVKLQDGTLLNRYWDDRDTPRPESWVEDIATAKSNPNRPATEIYRDLRSAAASGWDFSSRWMDNPQQLNTLRTTSIVPVDLNSLMFKMEKILARASKAAGDNAMANQYETLANARQKGIEKYLWNDQQGWYADYDLKSHKVRNQLTAAALFPLYVNAAAKDRANKMATATKTHLLQPGGLNTTSVKSGQQWDAPNGWAPLQWVATEGLQNYGQKEVAMDISWHFLTNVQHTYDREKKLVEKYDVSTTGTGGGGGEYPLQDGFGWTNGVTLKMLDLICPKEQPCDNVPATRP Hydrogen bonds contact Hydrophobic contact | ||||
| 64 | Haemophilus influenzae NadR protein (Hae-influ nadR) | 1LW7 | 6.37 | |
Target general information Gen name Hae-influ nadR Organism Haemophilus influenzae (strain ATCC 51907 / DSM 11121 / KW20 / Rd) Uniprot ID TTD ID Synonyms nadR; Transcriptional regulator nadR Protein family Bacterial NMN adenylyltransferase family; Bacterial RNK family Biochemical class Nicotinamide ribonucleoside uptake permease Function This enzyme has twoactivities: nicotinamide mononucleotide (NMN) adenylyltransferase and ribosylnicotinamide (RN) kinase. The RN kinase activity catalyzes the phosphorylation of RN to form nicotinamide ribonucleotide. The NMN adenylyltransferase activity catalyzes the transfer of the AMP moiety of ATP to nicotinamide ribonucleotide to form NAD(+). Related diseases Involved in the epigenetic regulation of ESR1 expression in breast cancer in a TFAP2C, IFI16 and HDAC4/5/6-dependent manner. {ECO:0000269|PubMed:24413532}. Drugs (DrugBank ID) NA Interacts with NA EC number NA Uniprot keywords 3D-structure; Alternative initiation; ATP-binding; Cell membrane; Cytoplasm; Kinase; Membrane; Multifunctional enzyme; NAD; Nucleotide-binding; Pyridine nucleotide biosynthesis; Reference proteome; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 39581.5 Length 344 Aromaticity 0.14 Instability index 41.39 Isoelectric point 6.94 Charge (pH=7) -0.18 2D Binding mode Binding energy (Kcal/mol) -8.69  Molscript Map 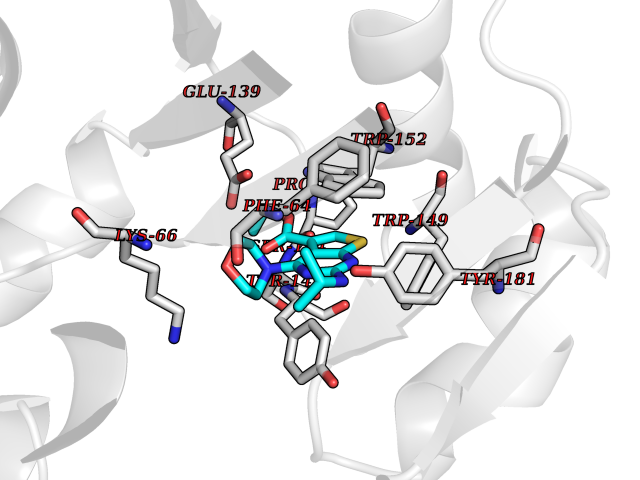 Pymol Map 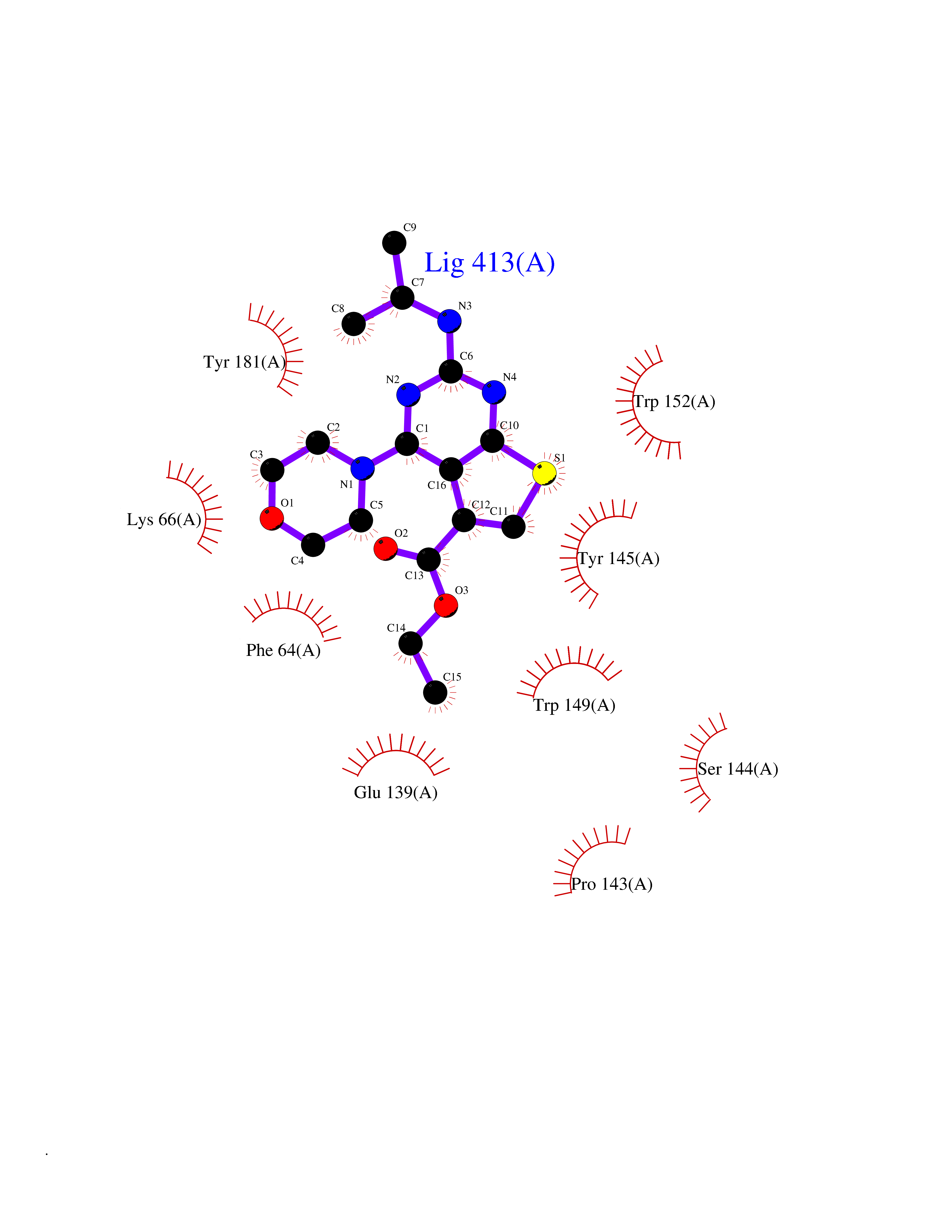 Ligplot Map 3D Binding mode Sequence EKKVGVIFGKFYPVHTGHINXIYEAFSKVDELHVIVCSDTVRDLKLFYDSKXKRXPTVQDRLRWXQQIFKYQKNQIFIHHLVEDGIPSYPNGWQSWSEAVKTLFHEKHFEPSIVFSSEPQDKAPYEKYLGLEVSLVDPDRTFFNVSATKIRTTPFQYWKFIPKEARPFFAKTVAILGGESSGKSVLVNKLAAVFNTTSAWEYGREFVFEKLGGDEQAMQYSDYPQXALGHQRYIDYAVRHSHKIAFIDTDFITTQAFCIQYEGKAHPFLDSXIKEYPFDVTILLKNNTEQKQRQQFQQLLKKLLDKYKVPYIEIESPSYLDRYNQVKAVIEKVLNEEEISELQN Hydrogen bonds contact Hydrophobic contact | ||||
| 65 | Wnt-7a protein (WNT7A) | 4UZQ | 6.37 | |
Target general information Gen name WNT7A Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Protein Wnt-7a Protein family Wnt family Biochemical class NA Function Plays an important role in embryonic development, including dorsal versus ventral patterning during limb development, skeleton development and urogenital tract development. Required for central nervous system (CNS) angiogenesis and blood-brain barrier regulation. Required for normal, sexually dimorphic development of the Mullerian ducts, and for normal fertility in both sexes. Required for normal neural stem cell proliferation in the hippocampus dentate gyrus. Required for normal progress through the cell cycle in neural progenitor cells, for self-renewal of neural stem cells, and for normal neuronal differentiation and maturation. Promotes formation of synapses via its interaction with FZD5. Ligand for members of the frizzled family of seven transmembrane receptors that functions in the canonical Wnt/beta-catenin signaling pathway. Related diseases Limb pelvis hypoplasia aplasia syndrome (LPHAS) [MIM:276820]: A syndrome of severe deficiency of the extremities due to hypo- or aplasia of one or more long bones of one or more limbs. Pelvic manifestations include hip dislocation, hypoplastic iliac bone and aplastic pubic bones. Thoracic deformity, unusual facies and genitourinary anomalies can be present. {ECO:0000269|PubMed:16826533, ECO:0000269|PubMed:17431918, ECO:0000269|PubMed:20949531, ECO:0000269|PubMed:21271649, ECO:0000269|PubMed:27638328}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Fuhrmann syndrome (FUHRS) [MIM:228930]: Distinct limb-malformation disorder characterized also by various degrees of limb aplasia/hypoplasia and joint dysplasia. {ECO:0000269|PubMed:16826533}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P55212; P22607; P06396; P13473-2; Q9UMX0; Q9Y5W5; Q5T9L3; Q9Z0J1 EC number NA Uniprot keywords 3D-structure; Developmental protein; Disease variant; Disulfide bond; Extracellular matrix; Glycoprotein; Lipoprotein; Proteomics identification; Reference proteome; Secreted; Signal; Wnt signaling pathway Protein physicochemical properties Chain ID A,B Molecular weight (Da) 40475.5 Length 356 Aromaticity 0.1 Instability index 50.49 Isoelectric point 7.67 Charge (pH=7) 1.62 2D Binding mode Binding energy (Kcal/mol) -8.68  Molscript Map 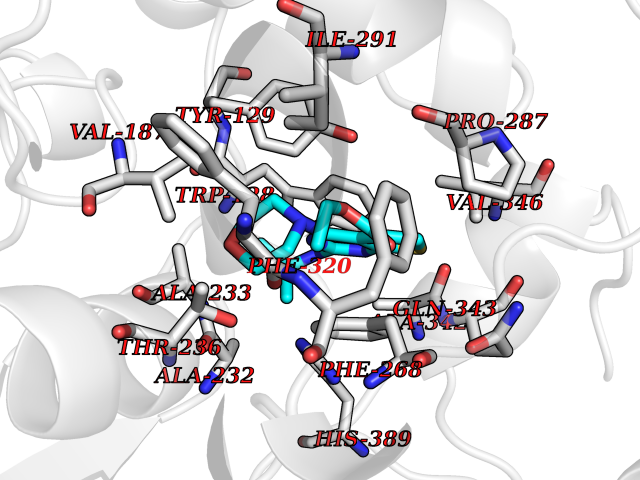 Pymol Map 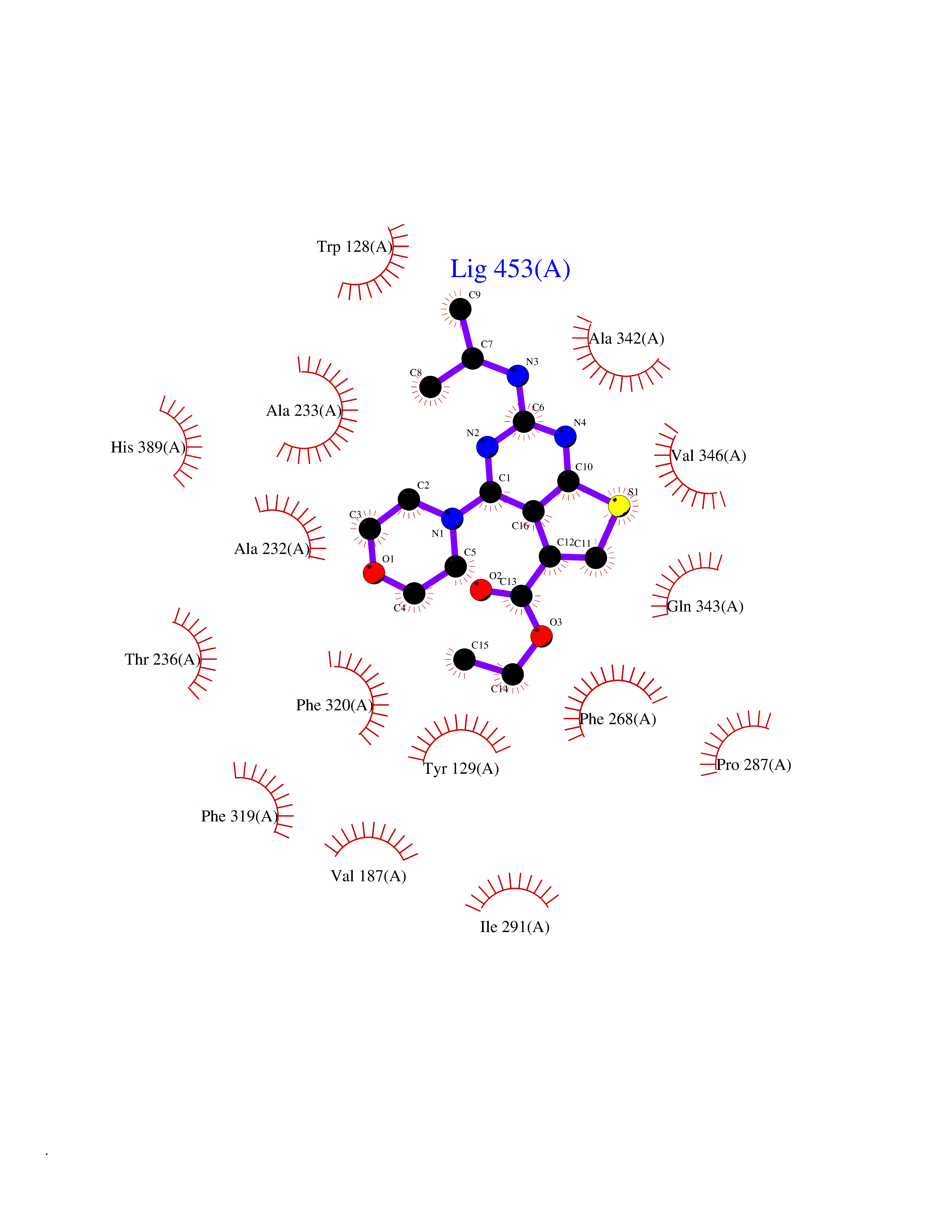 Ligplot Map 3D Binding mode Sequence EDLRLHLLLNTSVTCNDGSPAGYYLKESRGSRRWLLFLEGGWYCFNRENCDSRYDTMRRLMSSRDWPRTRTGTGILSSQPEENPYWWNANMVFIPYCSSDVWSGASSKSEKNEYAFMGALIIQEVVRELLGRGLSGAKVLLLAGSAAGGTGVLLNVDRVAEQLEKLGYPAIQVRGLADSGWFLDNKQYRHTDCVDTITCAPTEAIRRGIRYWNGVVPERCRRQFQEGEEWNCFFGYKVYPTLRSPVFVVQWLFDEAQLTVDNVHLTGQPVQEGLRLYIQNLGRELRHTLKDVPASFAPACLSHEIIIRSHWTDVQVKGTSLPRALHCWDRSLHKGCPVHLVDSCPWPHCNPSCPTS Hydrogen bonds contact Hydrophobic contact | ||||
| 66 | Retinoic acid receptor alpha (RARA) | 3KMR | 6.36 | |
Target general information Gen name RARA Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms RAR-alpha; RAR alpha; Nuclear receptor subfamily 1 group B member 1; NR1B1 Protein family Nuclear hormone receptor family, NR1 subfamily Biochemical class Nuclear hormone receptor Function Retinoic acid receptors bind as heterodimers to their target response elements in response to their ligands, all-trans or 9-cis retinoic acid, and regulate gene expression in various biological processes. The RXR/RAR heterodimers bind to the retinoic acid response elements (RARE) composed of tandem 5'-AGGTCA-3' sites known as DR1-DR5. In the absence of ligand, the RXR-RAR heterodimers associate with a multiprotein complex containing transcription corepressors that induce histone acetylation, chromatin condensation and transcriptional suppression. On ligand binding, the corepressors dissociate from the receptors and associate with the coactivators leading to transcriptional activation. RARA plays an essential role in the regulation of retinoic acid-induced germ cell development during spermatogenesis. Has a role in the survival of early spermatocytes at the beginning prophase of meiosis. In Sertoli cells, may promote the survival and development of early meiotic prophase spermatocytes. In concert with RARG, required for skeletal growth, matrix homeostasis and growth plate function. Receptor for retinoic acid. Related diseases Chromosomal aberrations involving RARA are commonly found in acute promyelocytic leukemia. Translocation t(11;17)(q32;q21) with ZBTB16/PLZF; translocation t(15;17)(q21;q21) with PML; translocation t(5;17)(q32;q11) with NPM. The PML-RARA oncoprotein requires both the PML ring structure and coiled-coil domain for both interaction with UBE2I, nuclear microspeckle location and sumoylation. In addition, the coiled-coil domain functions in blocking RA-mediated transactivation and cell differentiation. {ECO:0000269|PubMed:12691149, ECO:0000269|PubMed:8302850, ECO:0000269|PubMed:8562957}. Drugs (DrugBank ID) DB00459; DB00210; DB00523; DB00926; DB00982; DB05785; DB04942; DB00799; DB00755; DB12808 Interacts with O43707-1; O15296; Q15699; Q96RK4; O95273; P51946; Q15910; P50148; Q9UKP3; Q96EZ8; Q15648; Q71SY5; Q15788; Q9Y6Q9; O75376; Q9Y618; Q16236; P13056-2; P48552; Q9UPP1-2; Q9H8W4; P37231; P78527; P19793; P28702; P28702-3; P48443; Q96EB6; P63165; Q8WW24; Q2M1K9; Q91XC0; P59598; Q14457; P48552; Q96CV9; P28702; P48443; Q8WW24 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Chromosomal rearrangement; Cytoplasm; DNA-binding; Isopeptide bond; Metal-binding; Nucleus; Phosphoprotein; Proteomics identification; Proto-oncogene; Receptor; Reference proteome; Transcription; Transcription regulation; Ubl conjugation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A Molecular weight (Da) 27724.1 Length 244 Aromaticity 0.05 Instability index 50.8 Isoelectric point 5.82 Charge (pH=7) -3.61 2D Binding mode Binding energy (Kcal/mol) -8.67 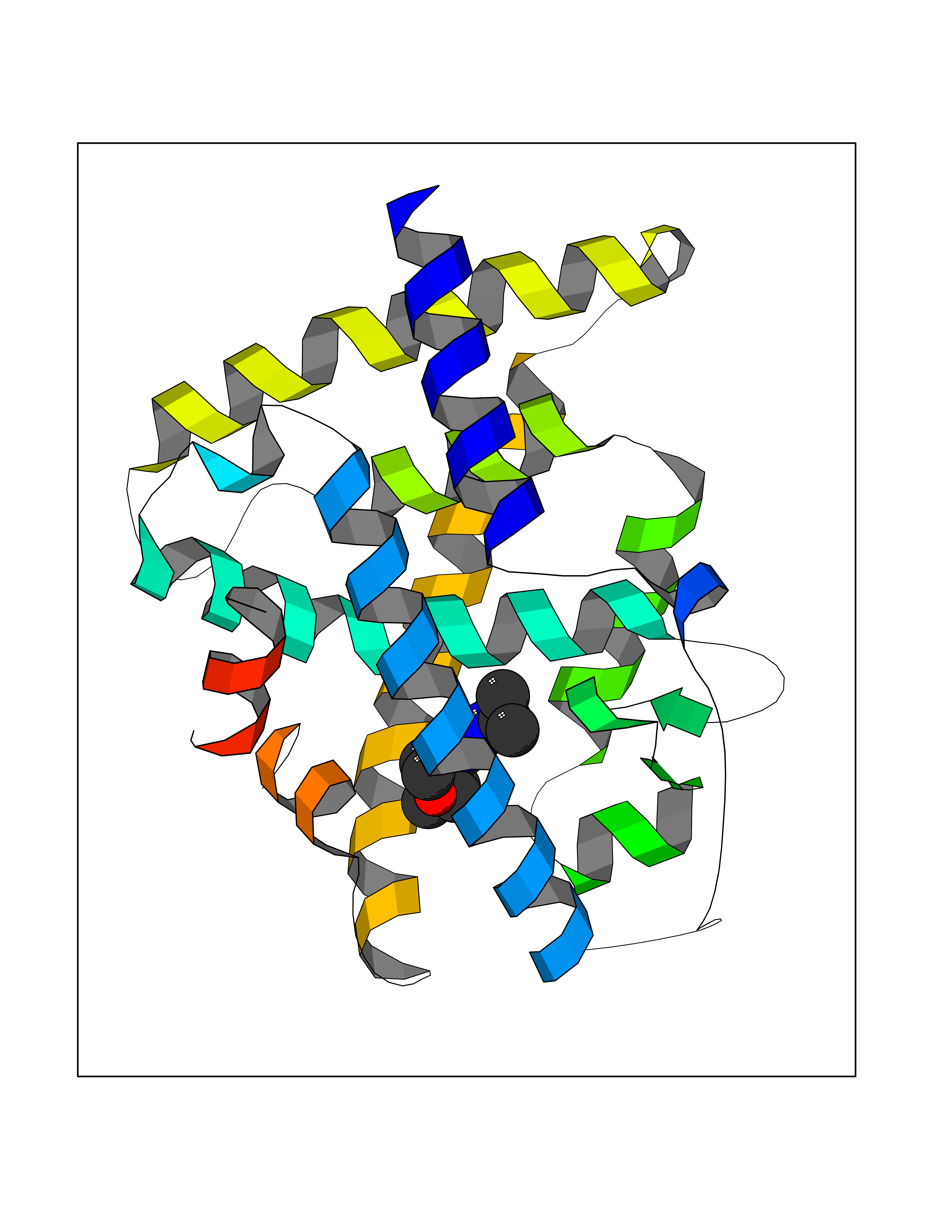 Molscript Map 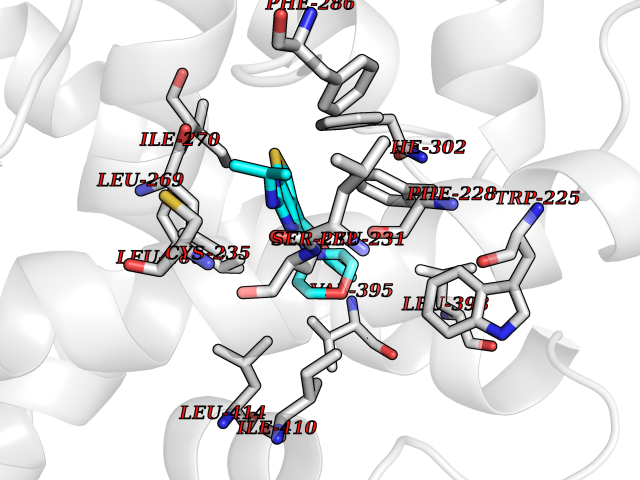 Pymol Map 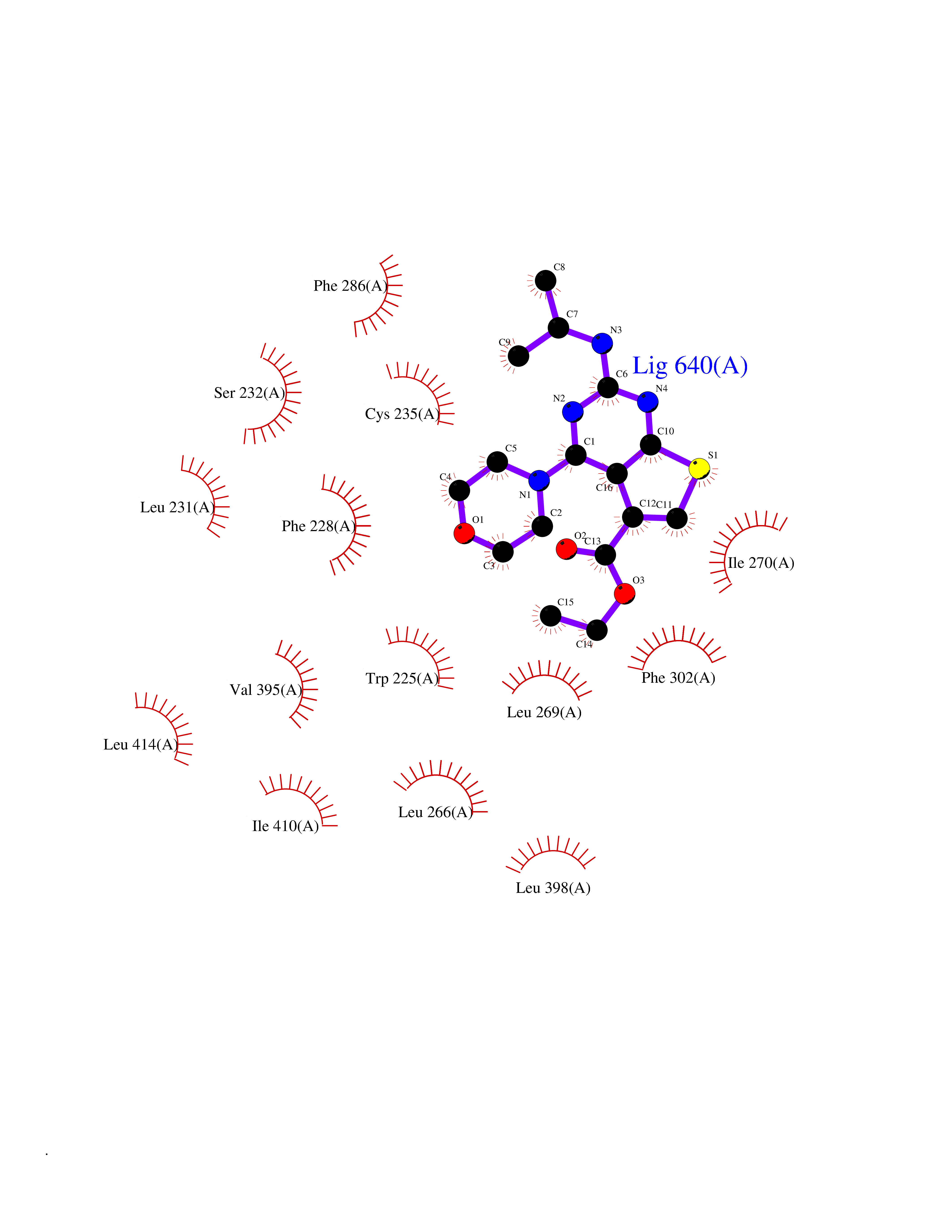 Ligplot Map 3D Binding mode Sequence PEVGELIEKVRKAHQETFPALCQLGKYTTNNSSEQRVSLDIDLWDKFSELSTKCIIKTVEFAKQLPGFTTLTIADQITLLKAACLDILILRICTRYTPEQDTMTFSDGLTLNRTQMHNAGFGPLTDLVFAFANQLLPLEMDDAETGLLSAICLICGDRQDLEQPDRVDMLQEPLLEALKVYVRKRRPSRPHMFPKMLMKITDLRSISAKGAERVITLKMEIPGSMPPLIQEMLEHKILHRLLQE Hydrogen bonds contact Hydrophobic contact | ||||
| 67 | Transferrin (TF) | 1RYO | 6.36 | |
Target general information Gen name TF Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Siderophilin; Serotransferrin; PRO1400; Beta-1 metal-binding globulin Protein family Transferrin family Biochemical class Transferrin Function It is responsible for the transport of iron from sites of absorption and heme degradation to those of storage and utilization. Serum transferrin may also have a further role in stimulating cell proliferation. Transferrins are iron binding transport proteins which can bind two Fe(3+) ions in association with the binding of an anion, usually bicarbonate. Related diseases Atransferrinemia (ATRAF) [MIM:209300]: A rare autosomal recessive disorder characterized by abnormal synthesis of transferrin leading to iron overload and microcytic hypochromic anemia. {ECO:0000269|PubMed:11110675, ECO:0000269|PubMed:15466165}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01370; DB14517; DB14518; DB01294; DB14526; DB14527; DB11136; DB14528; DB14529; DB14530; DB00515; DB09130; DB11397; DB13949; DB14490; DB14491; DB14488; DB14501; DB14489; DB13257; DB06215; DB06784; DB05260; DB01592; DB00893; DB00677; DB06757; DB11182; DB14520; DB01593; DB14487; DB14533; DB14548 Interacts with O43315; O00501; Q7Z7G2; Q9GZR5; Q9Y282; Q96KR6; P01350; P08034; Q8NBJ4; O15529; Q8TED1; Q7Z5P4; A8MZ59; O15173; Q96TC7; Q3KNW5; Q9BXS9-3; Q99523; O43278-2; Q8N9I0; P02786; Q4KMG9; Q9K0U9; Q09057; Q9K0V0; P02786 EC number NA Uniprot keywords 3D-structure; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; Ion transport; Iron; Iron transport; Metal-binding; Methylation; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Secreted; Signal; Transport Protein physicochemical properties Chain ID A Molecular weight (Da) 35854.5 Length 324 Aromaticity 0.1 Instability index 34.17 Isoelectric point 7.58 Charge (pH=7) 1.42 2D Binding mode Binding energy (Kcal/mol) -8.68 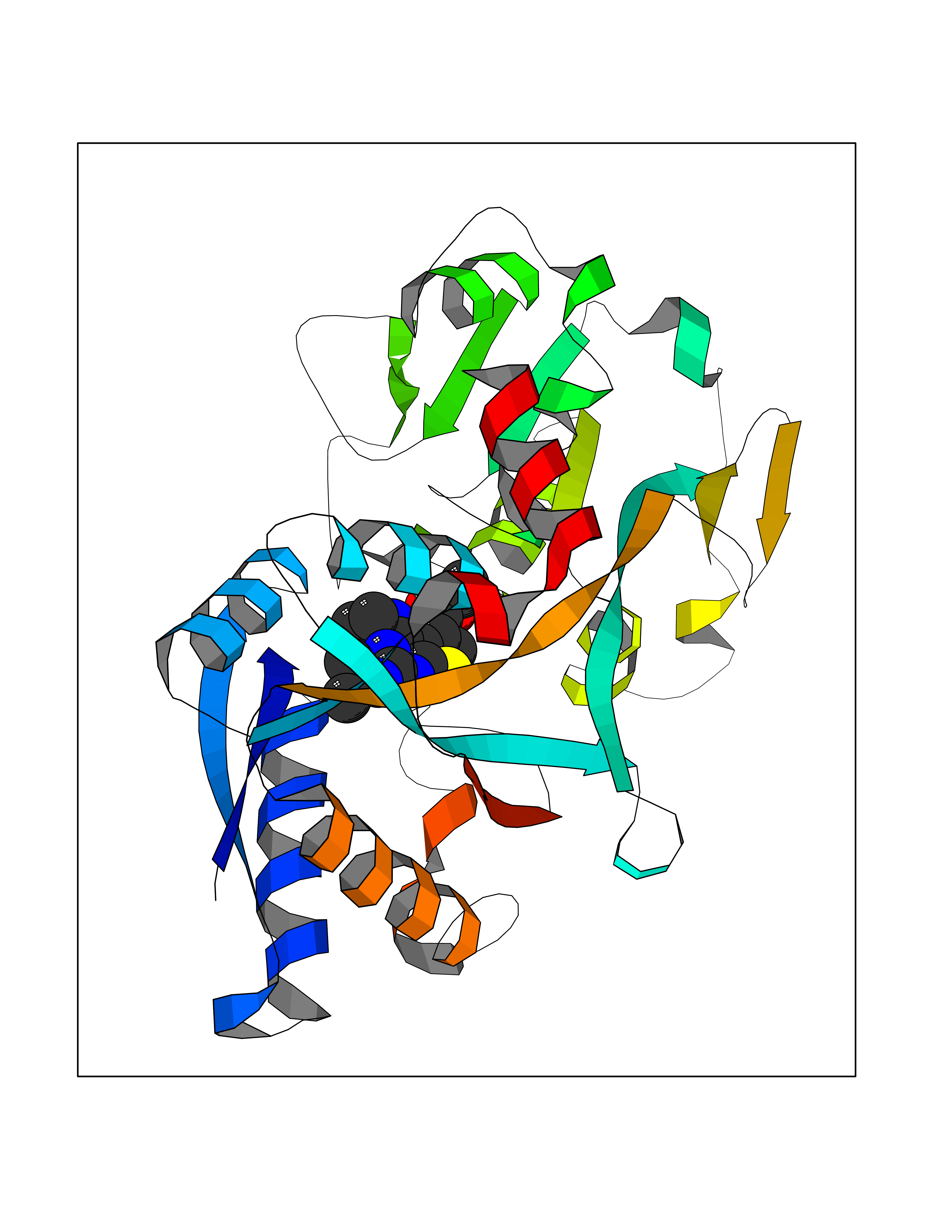 Molscript Map 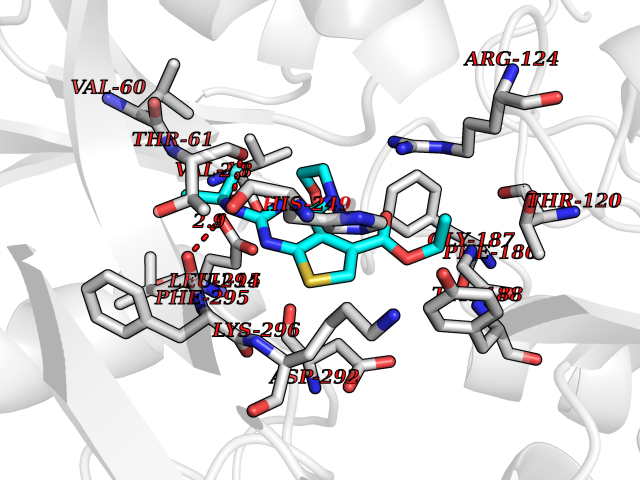 Pymol Map 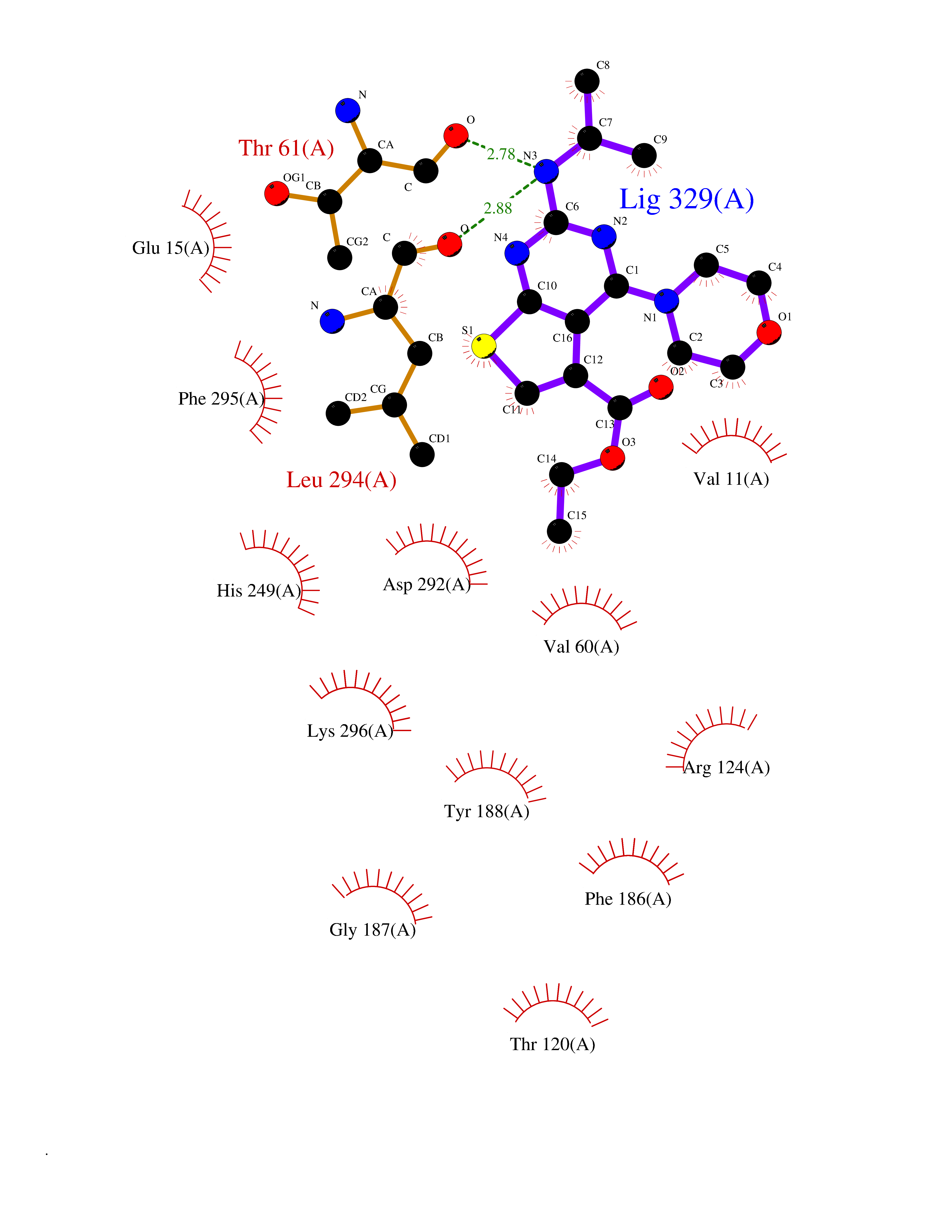 Ligplot Map 3D Binding mode Sequence KTVRWCAVSEHEATKCQSFRDHMKSVIPSDGPSVACVKKASYLDCIRAIAANEADAVTLDAGLVYDAYLAPNNLKPVVAEFYGSKEDPQTFYYAVAVVKKDSGFQMNQLRGKKSCHTGLGRSAGWNIPIGLLYCDLPEPRKPLEKAVANFFSGSCAPCADGTDFPQLCQLCPGCGCSTLNQYFGYSGAFKCLKDGAGDVAFVKHSTIFENLANKADRDQYELLCLDNTRKPVDEYKDCHLAQVPSHTVVARSMGGKEDLIWELLNQAQEHFGKDKSKEFQLFSSPHGKDLLFKDSAHGFLKVPPRMDAKMYLGYEYVTAIRNLR Hydrogen bonds contact Hydrophobic contact | ||||
| 68 | Macrophage colony-stimulating factor 1 receptor (CSF1R) | 2I1M | 6.36 | |
Target general information Gen name CSF1R Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Proto-oncogene c-Fms; M-CSF-R; FMS; CSF-1R; CSF-1-R; CSF-1 receptor; CD115 Protein family Protein kinase superfamily, Tyr protein kinase family, CSF-1/PDGF receptor subfamily Biochemical class Kinase Function Promotes the release of proinflammatory chemokines in response to IL34 and CSF1, and thereby plays an important role in innate immunity and in inflammatory processes. Plays an important role in the regulation of osteoclast proliferation and differentiation, the regulation of bone resorption, and is required for normal bone and tooth development. Required for normal male and female fertility, and for normal development of milk ducts and acinar structures in the mammary gland during pregnancy. Promotes reorganization of the actin cytoskeleton, regulates formation of membrane ruffles, cell adhesion and cell migration, and promotes cancer cell invasion. Activates several signaling pathways in response to ligand binding. Phosphorylates PIK3R1, PLCG2, GRB2, SLA2 and CBL. Activation of PLCG2 leads to the production of the cellular signaling molecules diacylglycerol and inositol 1,4,5-trisphosphate, that then lead to the activation of protein kinase C family members, especially PRKCD. Phosphorylation of PIK3R1, the regulatory subunit of phosphatidylinositol 3-kinase, leads to activation of the AKT1 signaling pathway. Activated CSF1R also mediates activation of the MAP kinases MAPK1/ERK2 and/or MAPK3/ERK1, and of the SRC family kinases SRC, FYN and YES1. Activated CSF1R transmits signals both via proteins that directly interact with phosphorylated tyrosine residues in its intracellular domain, or via adapter proteins, such as GRB2. Promotes activation of STAT family members STAT3, STAT5A and/or STAT5B. Promotes tyrosine phosphorylation of SHC1 and INPP5D/SHIP-1. Receptor signaling is down-regulated by protein phosphatases, such as INPP5D/SHIP-1, that dephosphorylate the receptor and its downstream effectors, and by rapid internalization of the activated receptor. Tyrosine-protein kinase that acts as cell-surface receptor for CSF1 and IL34 and plays an essential role in the regulation of survival, proliferation and differentiation of hematopoietic precursor cells, especially mononuclear phagocytes, such as macrophages and monocytes. Related diseases Aberrant expression of CSF1 or CSF1R can promote cancer cell proliferation, invasion and formation of metastases. Overexpression of CSF1 or CSF1R is observed in a significant percentage of breast, ovarian, prostate, and endometrial cancers.; DISEASE: Aberrant expression of CSF1 or CSF1R may play a role in inflammatory diseases, such as rheumatoid arthritis, glomerulonephritis, atherosclerosis, and allograft rejection.; DISEASE: Leukoencephalopathy, hereditary diffuse, with spheroids 1 (HDLS1) [MIM:221820]: An autosomal dominant adult-onset rapidly progressive neurodegenerative disorder characterized by variable behavioral, cognitive, and motor changes. Patients often die of dementia within 6 years of onset. Brain imaging shows patchy abnormalities in the cerebral white matter, predominantly affecting the frontal and parietal lobes. {ECO:0000269|PubMed:22197934, ECO:0000269|PubMed:23408870, ECO:0000269|PubMed:24336230, ECO:0000269|PubMed:24532199}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Brain abnormalities, neurodegeneration, and dysosteosclerosis (BANDDOS) [MIM:618476]: An autosomal recessive disease with variable manifestations. Main features are brain malformations with calcifying leukoencephalopathy, progressive neurodegeneration, and bone sclerotic features. The age at onset ranges from infancy to early adulthood. Neurologic features include loss of previous motor and language skills, cognitive impairment, spasticity, and focal seizures. Brain imaging shows periventricular white matter abnormalities and calcifications, large cisterna magna or Dandy-Walker malformation, and sometimes agenesis of the corpus callosum. {ECO:0000269|PubMed:30982608, ECO:0000269|PubMed:30982609}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07167; DB07202; DB12147; DB12010; DB00619; DB06080; DB12978; DB01268 Interacts with P09603; Q15375; P29323; Q6ZMJ4-1 EC number EC 2.7.10.1 Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Cell membrane; Disease variant; Disulfide bond; Glycoprotein; Immunity; Immunoglobulin domain; Inflammatory response; Innate immunity; Kinase; Membrane; Neurodegeneration; Nucleotide-binding; Phosphoprotein; Proteomics identification; Proto-oncogene; Receptor; Reference proteome; Repeat; Signal; Transferase; Transmembrane; Transmembrane helix; Tyrosine-protein kinase; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 35082.9 Length 311 Aromaticity 0.11 Instability index 44.6 Isoelectric point 8.13 Charge (pH=7) 2.42 2D Binding mode Binding energy (Kcal/mol) -8.68 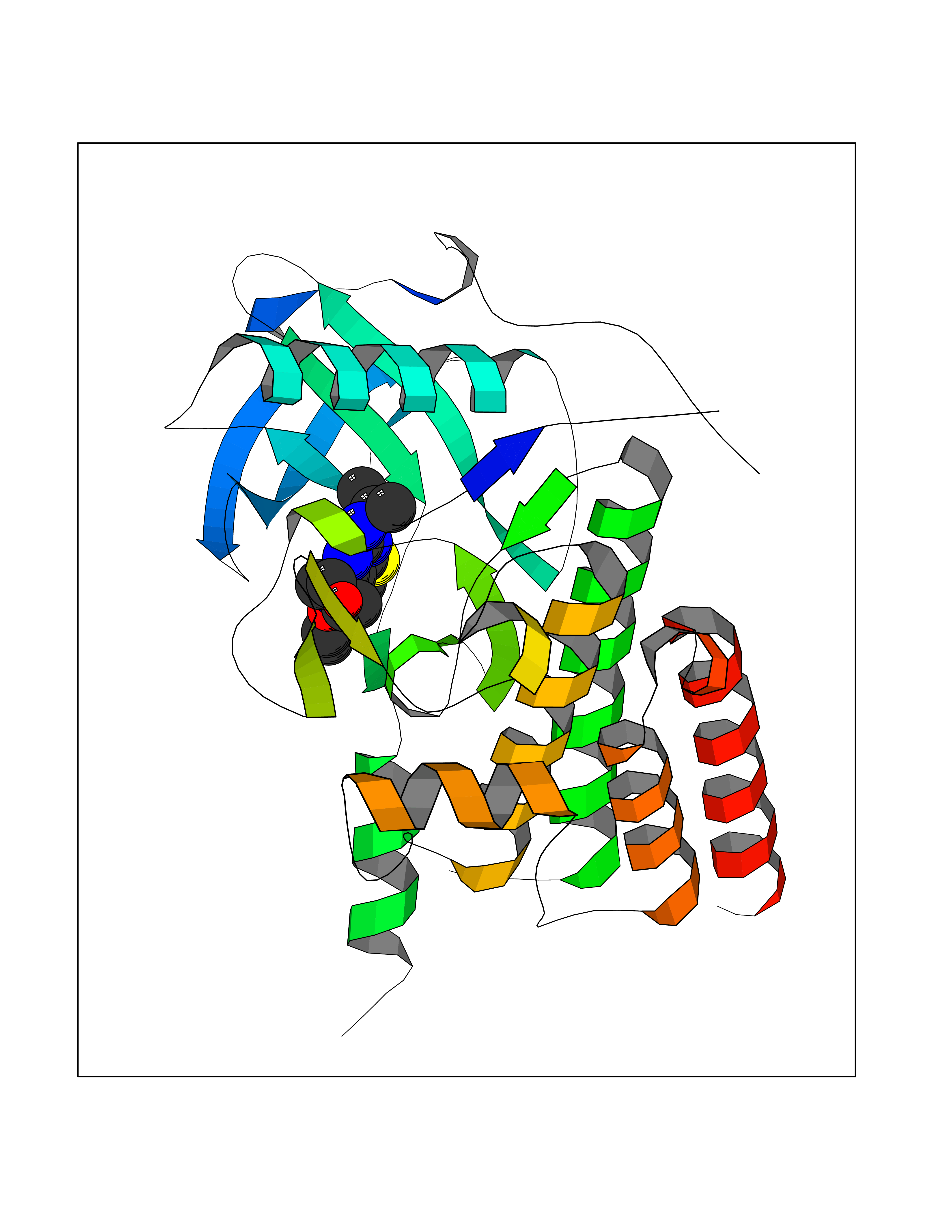 Molscript Map 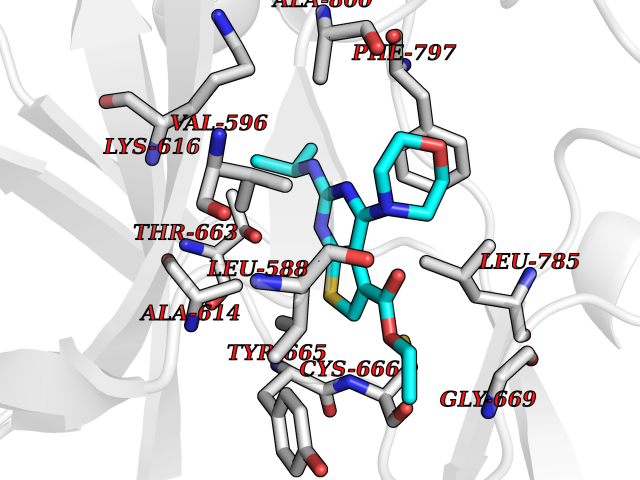 Pymol Map 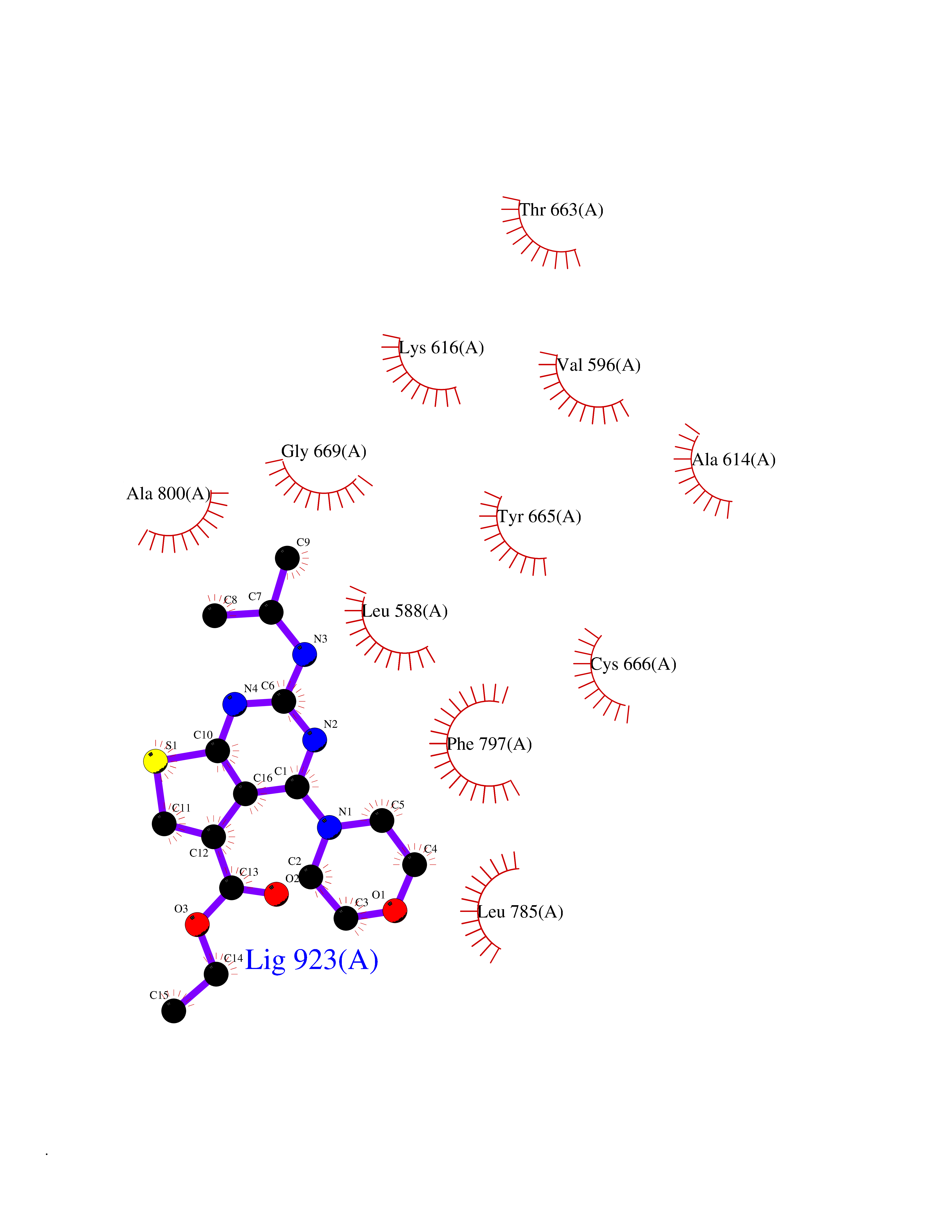 Ligplot Map 3D Binding mode Sequence QVRWKIIESYNSYTFIDPTQLPYNEKWEFPRNNLQFGKTLGAGAFGKVVEATAFGLGKEDAVLKVAVKMLKSTAHADEKEALMSELKIMSHLGQHENIVNLLGACTHGGPVLVITEYCCYGDLLNFLRRKSRVLETDSTASTRDLLHFSSQVAQGMAFLASKNCIHRDVAARNVLLTNGHVAKIGDFGLARDIMNDSNYIVKGNARLPVKWMAPESIFDCVYTVQSDVWSYGILLWEIFSLGLNPYPGILVNSKFYKLVKDGYQMAQPAFAPKNIYSIMQACWALEPTHRPTFQQICSFLQEQAQEDRRER Hydrogen bonds contact Hydrophobic contact | ||||
| 69 | Cystathionine gamma-lyase (CTH) | 3COG | 6.36 | |
Target general information Gen name CTH Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Gamma-cystathionase; Cysteine-protein sulfhydrase Protein family Trans-sulfuration enzymes family Biochemical class NA Function Catalyzes the last step in the trans-sulfuration pathway from methionine to cysteine. Has broad substrate specificity. Converts cystathionine to cysteine, ammonia and 2-oxobutanoate. Converts two cysteine molecules to lanthionine and hydrogen sulfide. Can also accept homocysteine as substrate. Specificity depends on the levels of the endogenous substrates. Generates the endogenous signaling molecule hydrogen sulfide (H2S), and so contributes to the regulation of blood pressure. Acts as a cysteine-protein sulfhydrase by mediating sulfhydration of target proteins: sulfhydration consists of converting -SH groups into -SSH on specific cysteine residues of target proteins such as GAPDH, PTPN1 and NF-kappa-B subunit RELA, thereby regulating their function. Related diseases Cystathioninuria (CSTNU) [MIM:219500]: Autosomal recessive phenotype characterized by abnormal accumulation of plasma cystathionine, leading to increased urinary excretion. {ECO:0000269|PubMed:12574942, ECO:0000269|PubMed:18476726}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02328; DB03928; DB00151; DB04217; DB00114 Interacts with P32929; Q96NT3; Q96NT3-2; Q96HA8; Q6P9E2 EC number EC 4.4.1.1 Uniprot keywords 3D-structure; Alternative splicing; Amino-acid biosynthesis; Calmodulin-binding; Cysteine biosynthesis; Cytoplasm; Disease variant; Lipid metabolism; Lyase; Proteomics identification; Pyridoxal phosphate; Reference proteome Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 86026 Length 782 Aromaticity 0.08 Instability index 32.4 Isoelectric point 6.27 Charge (pH=7) -9.46 2D Binding mode Binding energy (Kcal/mol) -8.67 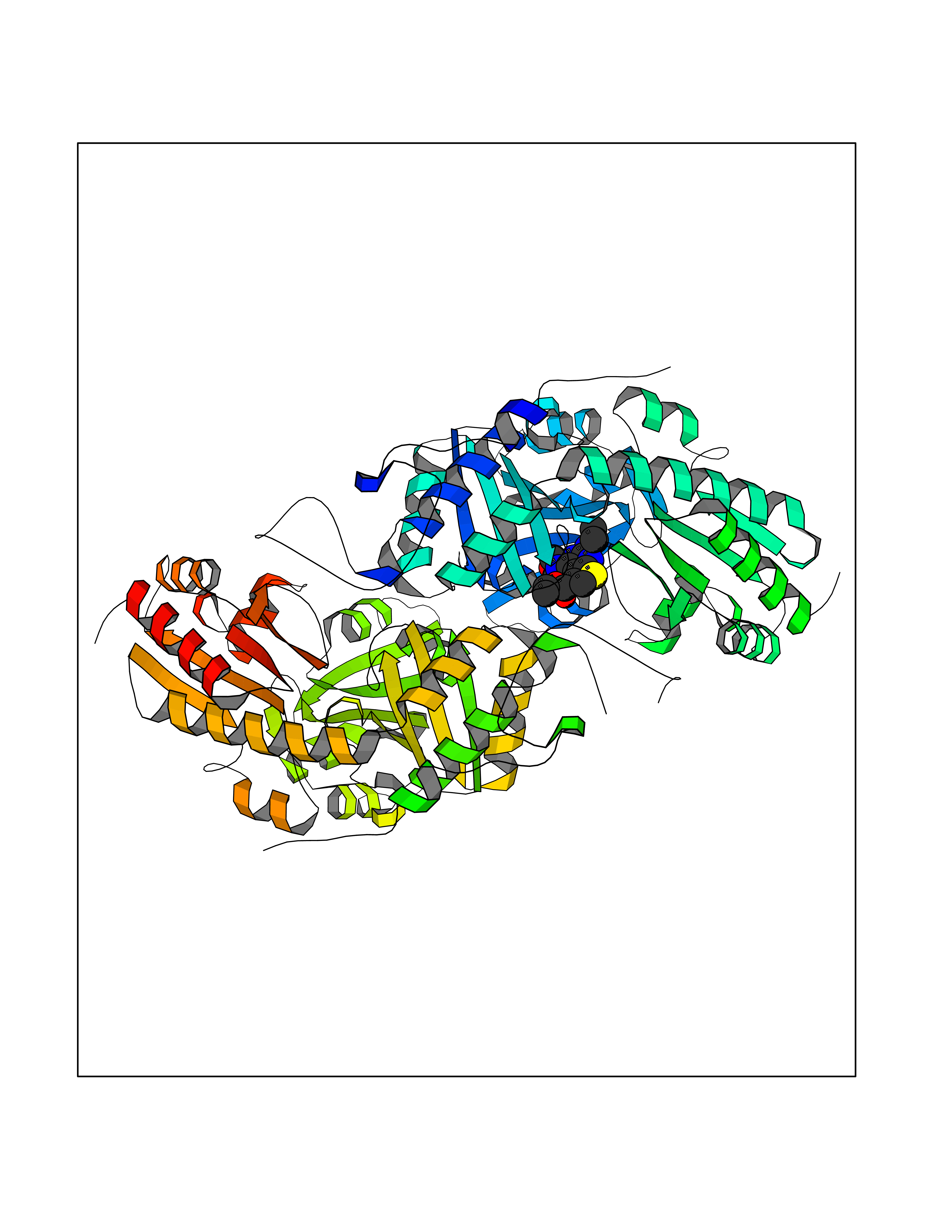 Molscript Map 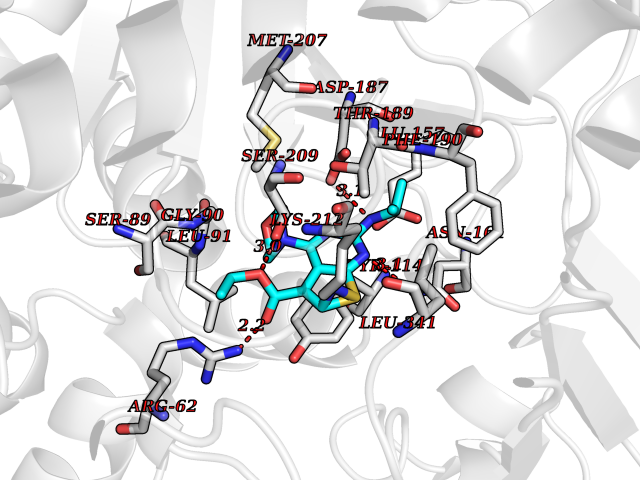 Pymol Map 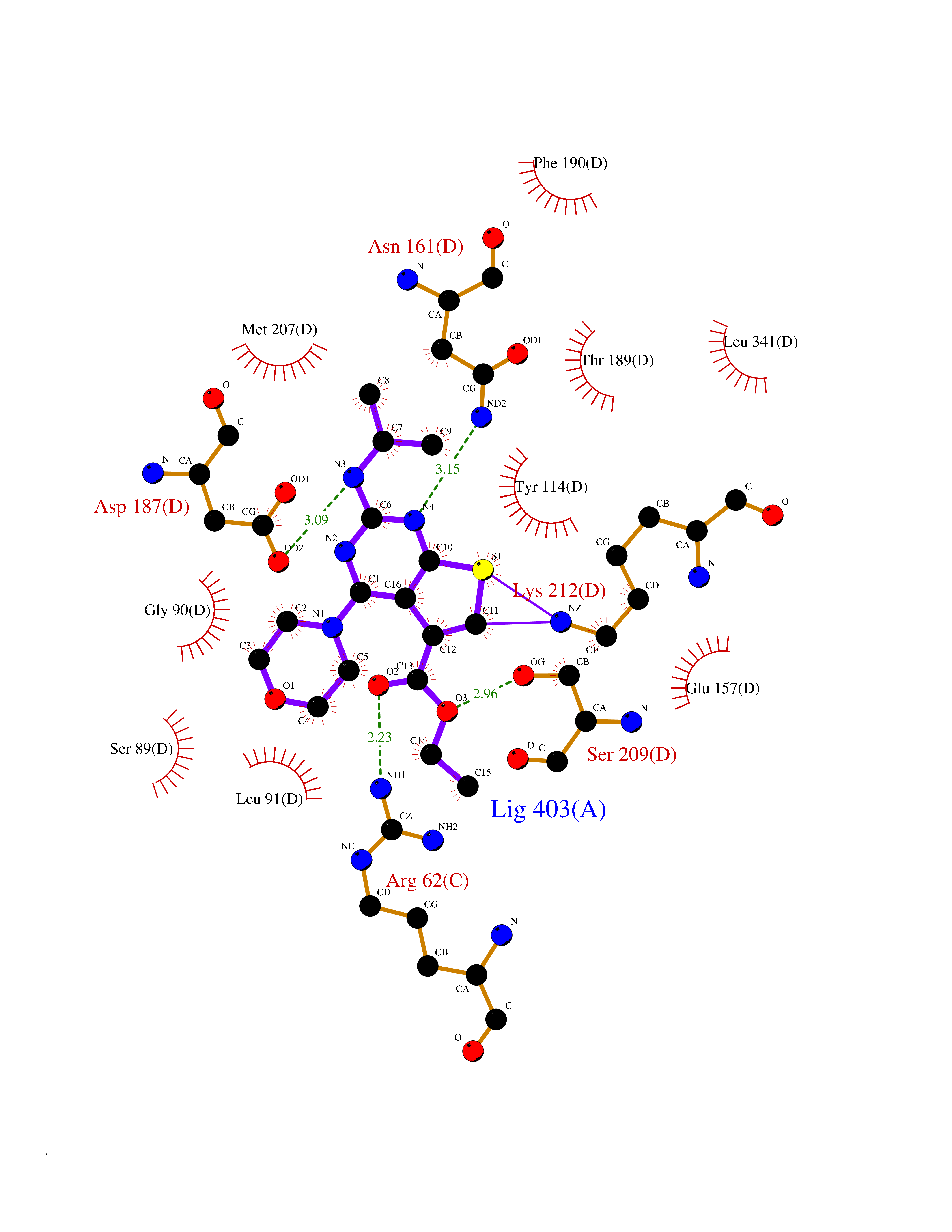 Ligplot Map 3D Binding mode Sequence GFLPHFQHFATQAIHVGQDPEQWTSRAVVPPISLSTTFKQGAPGQHSGFEYSRSGNPTRNCLEKAVAALDGAKYCLAFASGLAATVTITHLLKAGDQIICMDDVYGGTNRYFRQVASEFGLKISFVDCSKIKLLEAAITPETKLVWIETPTNPTQKVIDIEGCAHIVHKHGDIILVVDNTFMSPYFQRPLALGADISMYSATKYMNGHSDVVMGLVSVNCESLHNRLRFLQNSLGAVPSPIDCYLCNRGLKTLHVRMEKHFKNGMAVAQFLESNPWVEKVIYPGLPSHPQHELVKRQCTGCTGMVTFYIKGTLQHAEIFLKNLKLFTLAESLGGFESLAELPAIMTHASVLKNDRDVLGISDTLIRLSVGLEDEEDLLEDLDQALKAAHPPSGFLPHFQHFATQAIHVGQDPEQWTSRAVVPPISLSTTFKQGAPGQGFEYSRSGNPTRNCLEKAVAALDGAKYCLAFASGLAATVTITHLLKAGDQIICMDDVYGGTNRYFRQVASEFGLKISFVDCSKIKLLEAAITPETKLVWIETPTNPTQKVIDIEGCAHIVHKHGDIILVVDNTFMSPYFQRPLALGADISMYSATKYMNGHSDVVMGLVSVNCESLHNRLRFLQNSLGAVPSPIDCYLCNRGLKTLHVRMEKHFKNGMAVAQFLESNPWVEKVIYPGLPSHPQHELVKRQCTGCTGMVTFYIKGTLQHAEIFLKNLKLFTLAESLGGFESLAELPAIMTHASVLKNDRDVLGISDTLIRLSVGLEDEEDLLEDLDQALKAAHPPS Hydrogen bonds contact Hydrophobic contact | ||||
| 70 | Oxalosuccinate decarboxylase (IDH1) | 6ADG | 6.36 | |
Target general information Gen name IDH1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms PICD; NADP(+)-specific ICDH; Isocitrate dehydrogenase [NADP] cytoplasmic; IDP; IDH; Cytosolic NADP-isocitrate dehydrogenase Protein family Isocitrate and isopropylmalate dehydrogenases family Biochemical class Short-chain dehydrogenases reductase Function Catalyses the NADPH-dependent reduction of alpha-ketoglutarate to R(-)-2-hydroxyglutarate (2HG). Related diseases Glioma (GLM) [MIM:137800]: Gliomas are benign or malignant central nervous system neoplasms derived from glial cells. They comprise astrocytomas and glioblastoma multiforme that are derived from astrocytes, oligodendrogliomas derived from oligodendrocytes and ependymomas derived from ependymocytes. {ECO:0000269|PubMed:19117336, ECO:0000269|PubMed:19935646}. The gene represented in this entry is involved in disease pathogenesis. Mutations affecting Arg-132 are tissue-specific, and suggest that this residue plays a unique role in the development of high-grade gliomas. Mutations of Arg-132 to Cys, His, Leu or Ser abolish magnesium binding and abolish the conversion of isocitrate to alpha-ketoglutarate. Instead, alpha-ketoglutarate is converted to R(-)-2-hydroxyglutarate. Elevated levels of R(-)-2-hydroxyglutarate are correlated with an elevated risk of malignant brain tumors. {ECO:0000269|PubMed:19935646}.; DISEASE: Genetic variations are associated with cartilaginous tumors such as enchondroma or chondrosarcoma. Mutations of Arg-132 to Cys, Gly or His abolish the conversion of isocitrate to alpha-ketoglutarate. Instead, alpha-ketoglutarate is converted to R(-)-2-hydroxyglutarate. {ECO:0000269|PubMed:26161668}. Drugs (DrugBank ID) DB09374; DB01727; DB14568; DB03461; DB16267 Interacts with P0DP23; P27797; P36957; O75874; Q8TDX7; P16284; P17612; P50454; P37173; Q05086-3 EC number EC 1.1.1.42 Uniprot keywords 3D-structure; Acetylation; Cytoplasm; Direct protein sequencing; Glyoxylate bypass; Magnesium; Manganese; Metal-binding; NADP; Oxidoreductase; Peroxisome; Phosphoprotein; Proteomics identification; Reference proteome; Tricarboxylic acid cycle Protein physicochemical properties Chain ID A,B Molecular weight (Da) 92711.7 Length 823 Aromaticity 0.1 Instability index 26.74 Isoelectric point 6.42 Charge (pH=7) -4.48 2D Binding mode Binding energy (Kcal/mol) -8.68 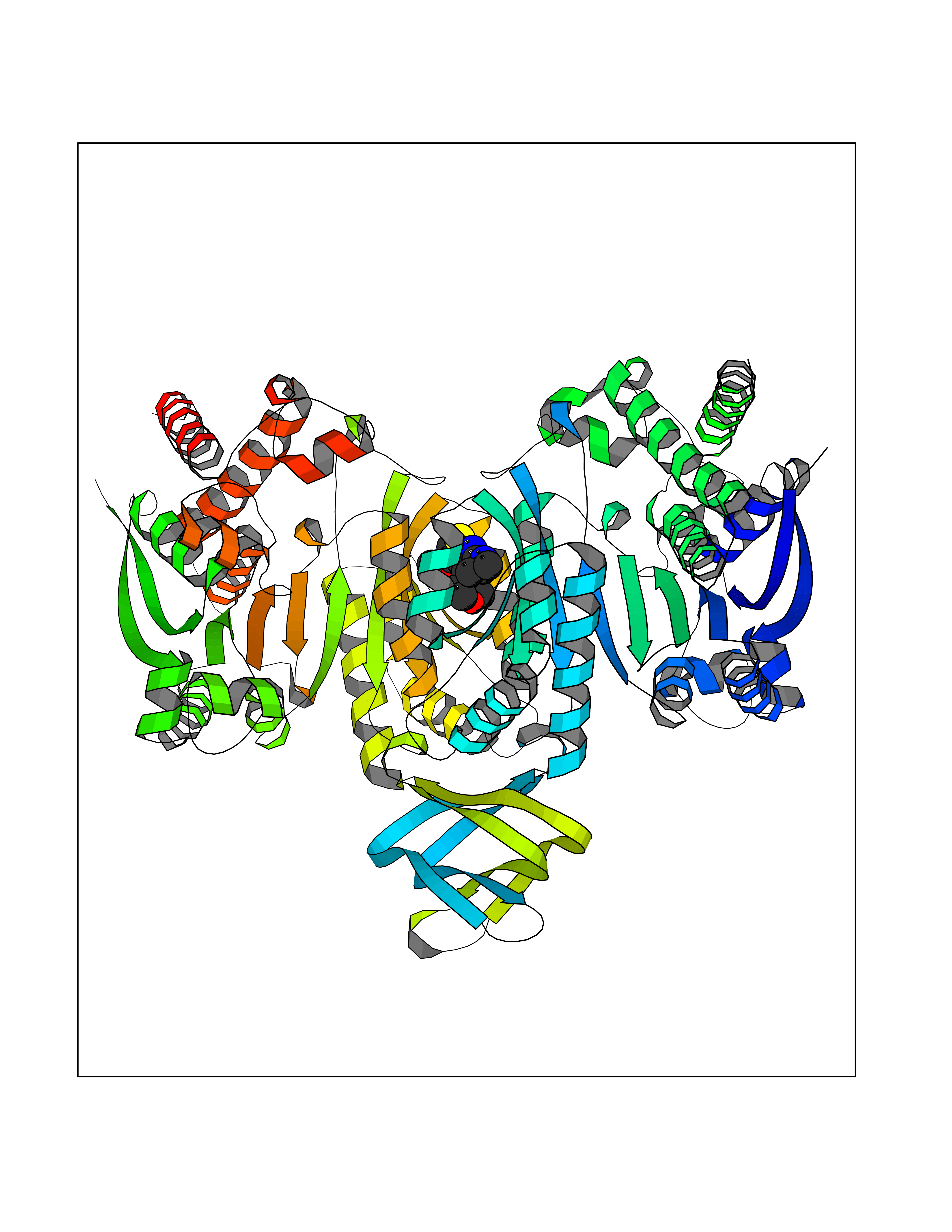 Molscript Map 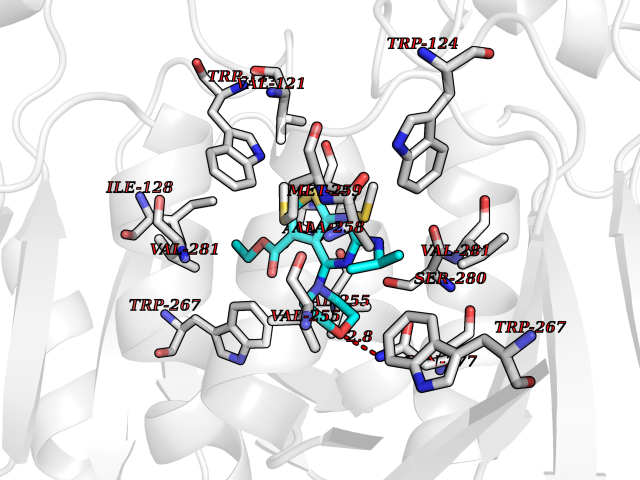 Pymol Map 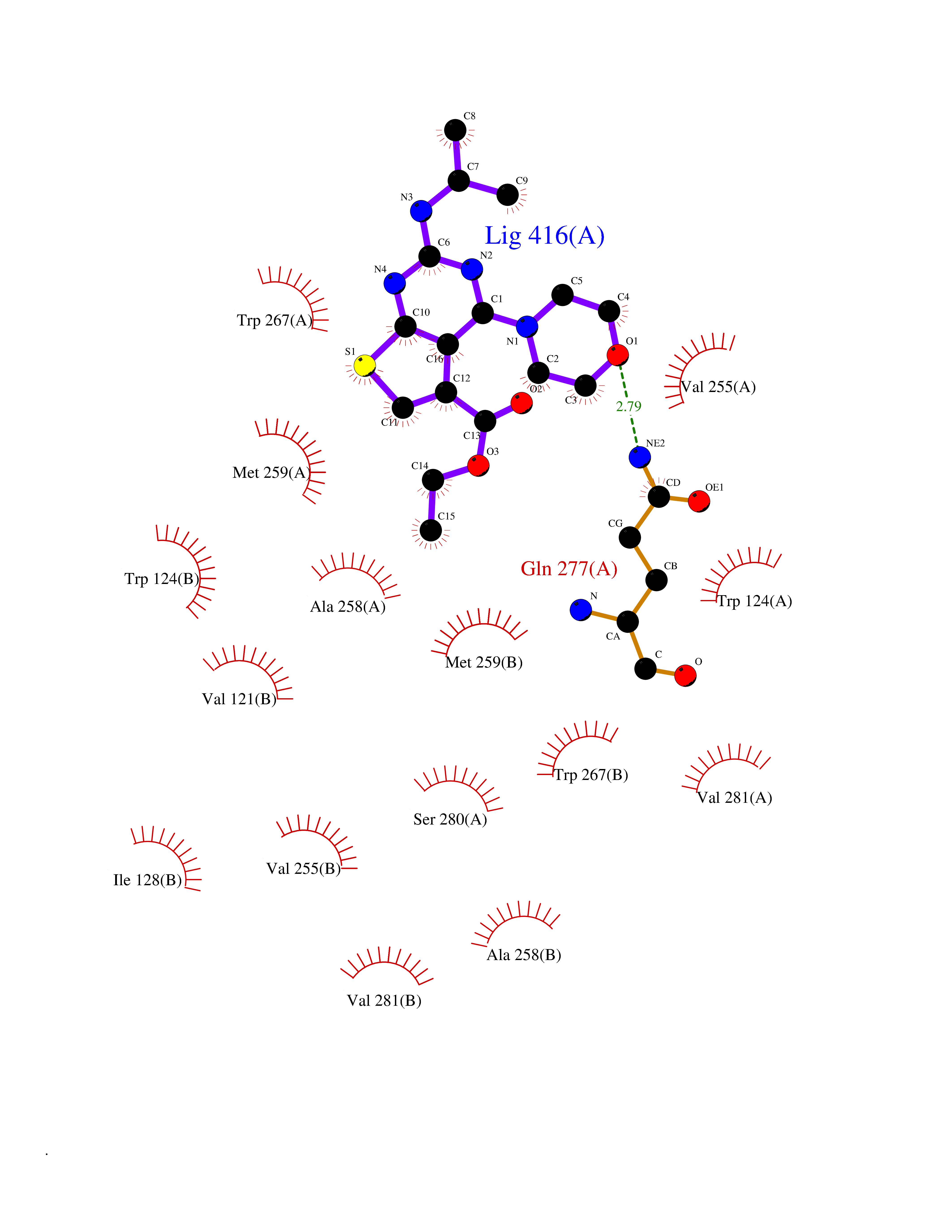 Ligplot Map 3D Binding mode Sequence KKISGGSVVEMQGDEMTRIIWELIKEKLIFPYVELDLHSYDLGIENRDATNDQVTKDAAEAIKKHNVGVKCATITPDEKRVEEFKLKQMWKSPNGTIRNILGGTVFREAIICKNIPRLVSGWVKPIIIGHHAYGDQYRATDFVVPGPGKVEITYTPSDGTQKVTYLVHNFEEGGGVAMGMYNQDKSIEDFAHSSFQMALSKGWPLYLSTKNTILKKYDGRFKDIFQEIYDKQYKSQFEAQKIWYEHRLIDDMVAQAMKSEGGFIWACKNYDGDVQSDSVAQGYGSLGMMTSVLVCPDGKTVEAEAAHGTVTRHYRMYQKGQETSTNPIASIFAWTRGLAHRAKLDNNKELAFFANALEEVSIETIEAGFMTKDLAACIKGLPNVQRSDYLNTFEFMDKLGENLKIKLAQAKLKKISGGSVVEMQGDEMTRIIWELIKEKLIFPYVELDLHSYDLGIENRDATNDQVTKDAAEAIKKHNVGVKCATITPDEKRVEEFKLKQMWKSPNGTIRNILGGTVFREAIICKNIPRLVSGWVKPIIIGHHAYGDQYRATDFVVPGPGKVEITYTPSDGTQKVTYLVHNFEEGGGVAMGMYNQDKSIEDFAHSSFQMALSKGWPLYLSTKNTILKKYDGRFKDIFQEIYDKQYKSQFEAQKIWYEHRLIDDMVAQAMKSEGGFIWACKNYDGDVQSDSVAQGYGSLGMMTSVLVCPDGKTVEAEAAHGTVTRHYRMYQKGQETSTNPIASIFAWTRGLAHRAKLDNNKELAFFANALEEVSIETIEAGFMTKDLAACIKGLPNVQRSDYLNTFEFMDKLGENLKIKLAQAK Hydrogen bonds contact Hydrophobic contact | ||||
| 71 | Mutated oxalosuccinate decarboxylase (mIDH1) | 6ADG | 6.36 | |
Target general information Gen name IDH1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms PICD (mutated); Oxalosuccinate decarboxylase (mutated); NADP(+)-specific ICDH (mutated); Isocitrate dehydrogenase [NADP] cytoplasmic (mutated); IDP (mutated); IDH (mutated); Cytosolic NADP-isocitrate Protein family Isocitrate and isopropylmalate dehydrogenases family Biochemical class Short-chain dehydrogenases reductase Function Catalyses the NADPH-dependent reduction of alpha-ketoglutarate to R(-)-2-hydroxyglutarate (2HG). Related diseases Glioma (GLM) [MIM:137800]: Gliomas are benign or malignant central nervous system neoplasms derived from glial cells. They comprise astrocytomas and glioblastoma multiforme that are derived from astrocytes, oligodendrogliomas derived from oligodendrocytes and ependymomas derived from ependymocytes. {ECO:0000269|PubMed:19117336, ECO:0000269|PubMed:19935646}. The gene represented in this entry is involved in disease pathogenesis. Mutations affecting Arg-132 are tissue-specific, and suggest that this residue plays a unique role in the development of high-grade gliomas. Mutations of Arg-132 to Cys, His, Leu or Ser abolish magnesium binding and abolish the conversion of isocitrate to alpha-ketoglutarate. Instead, alpha-ketoglutarate is converted to R(-)-2-hydroxyglutarate. Elevated levels of R(-)-2-hydroxyglutarate are correlated with an elevated risk of malignant brain tumors. {ECO:0000269|PubMed:19935646}.; DISEASE: Genetic variations are associated with cartilaginous tumors such as enchondroma or chondrosarcoma. Mutations of Arg-132 to Cys, Gly or His abolish the conversion of isocitrate to alpha-ketoglutarate. Instead, alpha-ketoglutarate is converted to R(-)-2-hydroxyglutarate. {ECO:0000269|PubMed:26161668}. Drugs (DrugBank ID) DB09374; DB01727; DB14568; DB03461; DB16267 Interacts with P0DP23; P27797; P36957; O75874; Q8TDX7; P16284; P17612; P50454; P37173; Q05086-3 EC number EC 1.1.1.42 Uniprot keywords 3D-structure; Acetylation; Cytoplasm; Direct protein sequencing; Glyoxylate bypass; Magnesium; Manganese; Metal-binding; NADP; Oxidoreductase; Peroxisome; Phosphoprotein; Proteomics identification; Reference proteome; Tricarboxylic acid cycle Protein physicochemical properties Chain ID A,B Molecular weight (Da) 92711.7 Length 823 Aromaticity 0.1 Instability index 26.74 Isoelectric point 6.42 Charge (pH=7) -4.48 2D Binding mode Binding energy (Kcal/mol) -8.68 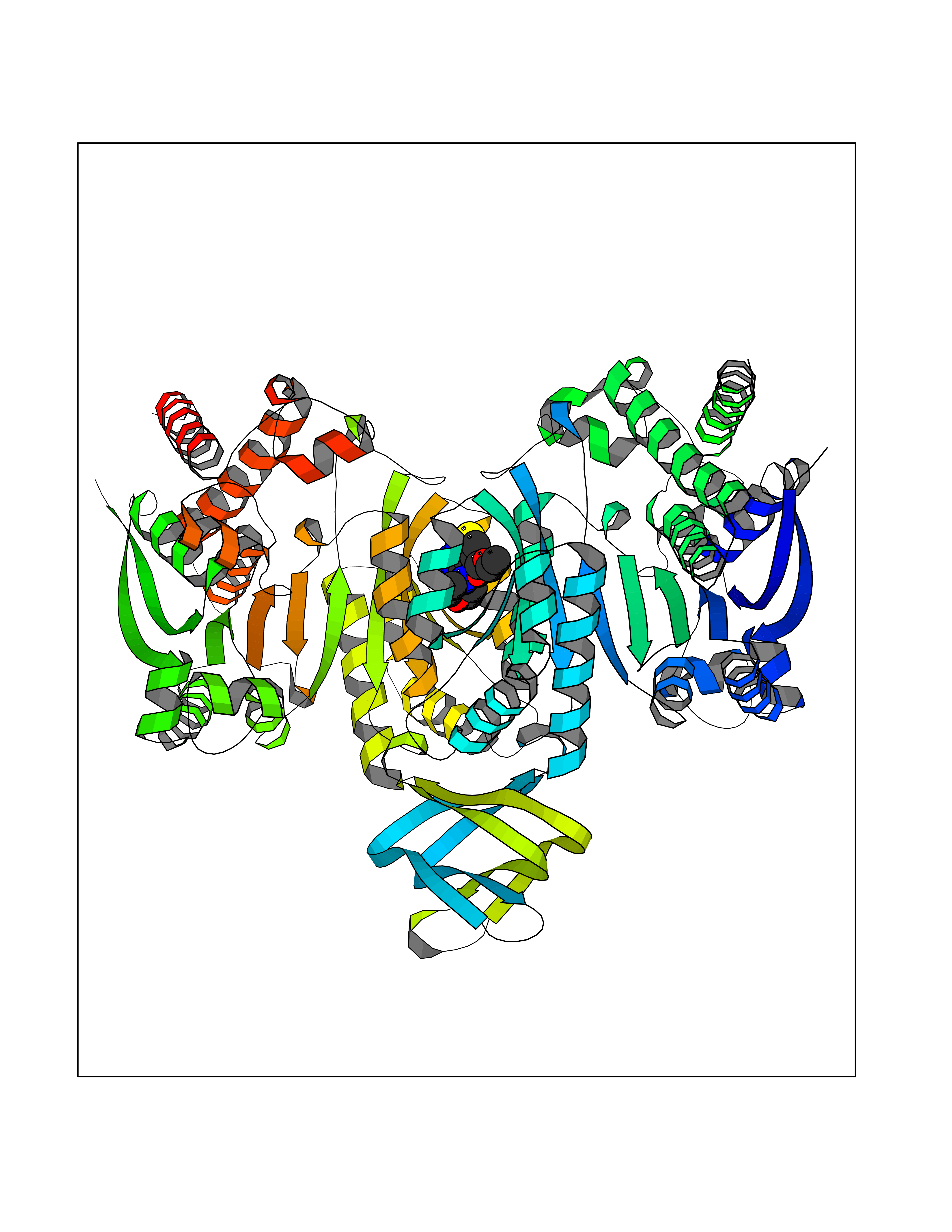 Molscript Map 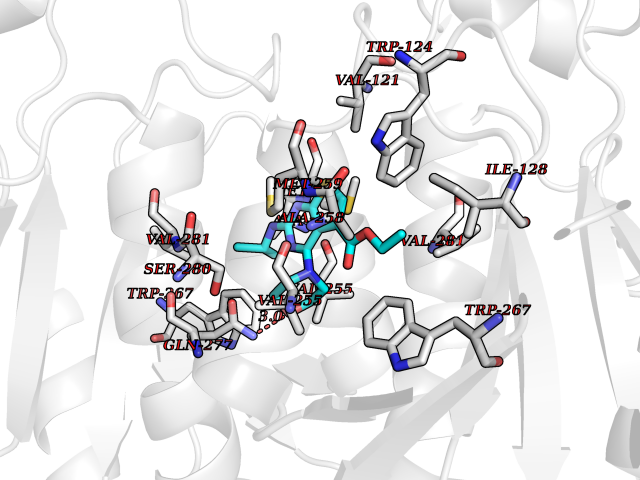 Pymol Map 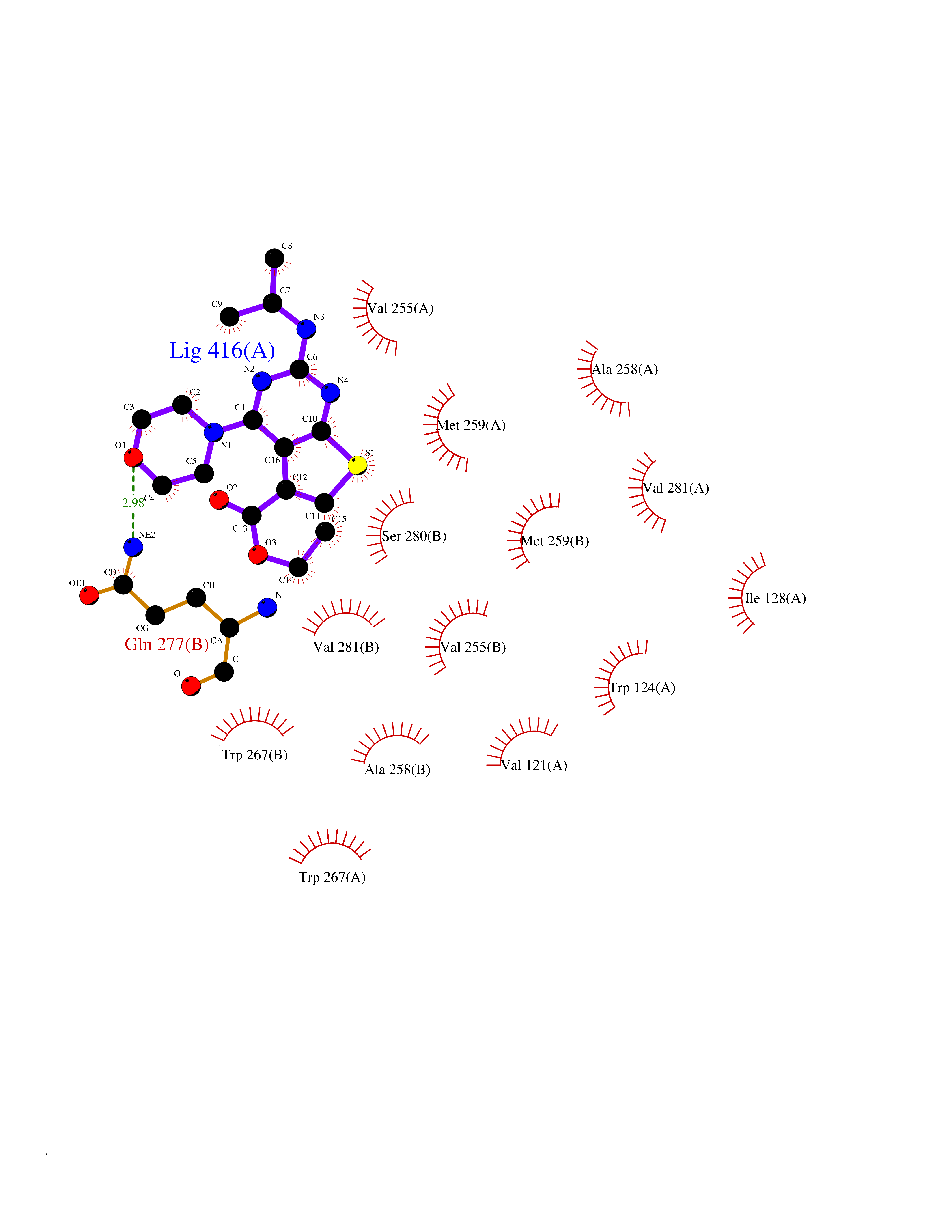 Ligplot Map 3D Binding mode Sequence KKISGGSVVEMQGDEMTRIIWELIKEKLIFPYVELDLHSYDLGIENRDATNDQVTKDAAEAIKKHNVGVKCATITPDEKRVEEFKLKQMWKSPNGTIRNILGGTVFREAIICKNIPRLVSGWVKPIIIGHHAYGDQYRATDFVVPGPGKVEITYTPSDGTQKVTYLVHNFEEGGGVAMGMYNQDKSIEDFAHSSFQMALSKGWPLYLSTKNTILKKYDGRFKDIFQEIYDKQYKSQFEAQKIWYEHRLIDDMVAQAMKSEGGFIWACKNYDGDVQSDSVAQGYGSLGMMTSVLVCPDGKTVEAEAAHGTVTRHYRMYQKGQETSTNPIASIFAWTRGLAHRAKLDNNKELAFFANALEEVSIETIEAGFMTKDLAACIKGLPNVQRSDYLNTFEFMDKLGENLKIKLAQAKLKKISGGSVVEMQGDEMTRIIWELIKEKLIFPYVELDLHSYDLGIENRDATNDQVTKDAAEAIKKHNVGVKCATITPDEKRVEEFKLKQMWKSPNGTIRNILGGTVFREAIICKNIPRLVSGWVKPIIIGHHAYGDQYRATDFVVPGPGKVEITYTPSDGTQKVTYLVHNFEEGGGVAMGMYNQDKSIEDFAHSSFQMALSKGWPLYLSTKNTILKKYDGRFKDIFQEIYDKQYKSQFEAQKIWYEHRLIDDMVAQAMKSEGGFIWACKNYDGDVQSDSVAQGYGSLGMMTSVLVCPDGKTVEAEAAHGTVTRHYRMYQKGQETSTNPIASIFAWTRGLAHRAKLDNNKELAFFANALEEVSIETIEAGFMTKDLAACIKGLPNVQRSDYLNTFEFMDKLGENLKIKLAQAK Hydrogen bonds contact Hydrophobic contact | ||||
| 72 | Heat shock protein 90 beta (HSP90B) | 7Z38 | 6.36 | |
Target general information Gen name HSP90AB1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Heat shock 84 kDa; HSP84; HSP 84 Protein family Heat shock protein 90 family Biochemical class Heat shock protein Function Molecular chaperone that promotes the maturation, structural maintenance and proper regulation of specific target proteins involved for instance in cell cycle control and signal transduction. Undergoes a functional cycle that is linked to its ATPase activity. This cycle probably induces conformational changes in the client proteins, thereby causing their activation. Interacts dynamically with various co-chaperones that modulate its substrate recognition, ATPase cycle and chaperone function (PubMed:16478993, PubMed:19696785). Engages with a range of client protein classes via its interaction with various co-chaperone proteins or complexes, that act as adapters, simultaneously able to interact with the specific client and the central chaperone itself. Recruitment of ATP and co-chaperone followed by client protein forms a functional chaperone. After the completion of the chaperoning process, properly folded client protein and co-chaperone leave HSP90 in an ADP-bound partially open conformation and finally, ADP is released from HSP90 which acquires an open conformation for the next cycle (PubMed:27295069, PubMed:26991466). Apart from its chaperone activity, it also plays a role in the regulation of the transcription machinery. HSP90 and its co-chaperones modulate transcription at least at three different levels. In the first place, they alter the steady-state levels of certain transcription factors in response to various physiological cues. Second, they modulate the activity of certain epigenetic modifiers, such as histone deacetylases or DNA methyl transferases, and thereby respond to the change in the environment. Third, they participate in the eviction of histones from the promoter region of certain genes and thereby turn on gene expression (PubMed:25973397). Antagonizes STUB1-mediated inhibition of TGF-beta signaling via inhibition of STUB1-mediated SMAD3 ubiquitination and degradation (PubMed:24613385). Promotes cell differentiation by chaperoning BIRC2 and thereby protecting from auto-ubiquitination and degradation by the proteasomal machinery (PubMed:18239673). Main chaperone that is involved in the phosphorylation/activation of the STAT1 by chaperoning both JAK2 and PRKCE under heat shock and in turn, activates its own transcription (PubMed:20353823). Related diseases Galactosemia 3 (GALAC3) [MIM:230350]: A form of galactosemia, an inborn error of galactose metabolism typically manifesting in the neonatal period, after ingestion of galactose, with jaundice, hepatosplenomegaly, hepatocellular insufficiency, food intolerance, hypoglycemia, renal tubular dysfunction, muscle hypotonia, sepsis and cataract. GALAC3 is an autosomal recessive form caused by galactose epimerase deficiency. It can manifest as benign, peripheral form with mild symptoms and enzymatic deficiency in circulating blood cells only. A second form, known as generalized epimerase deficiency, is characterized by undetectable levels of enzyme activity in all tissues and severe clinical features, including restricted growth and intellectual disability. {ECO:0000269|PubMed:11279193, ECO:0000269|PubMed:11903335, ECO:0000269|PubMed:15639193, ECO:0000269|PubMed:16301867, ECO:0000269|PubMed:16302980, ECO:0000269|PubMed:9326324, ECO:0000269|PubMed:9538513, ECO:0000269|PubMed:9973283}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Thrombocytopenia 13, syndromic (THC13) [MIM:620776]: An autosomal recessive form of thrombocytopenia, a hematologic disorder defined by a decrease in the number of platelets in circulating blood, resulting in the potential for increased bleeding and decreased ability for clotting. THC13 patients have enlarged, gray platelets with defective function. Some affected individuals have leukopenia or anemia and pancytopenia. Additional variable features include mitral valve malformations, pyloric stenosis, and impaired intellectual development. {ECO:0000269|PubMed:30247636, ECO:0000269|PubMed:33510604, ECO:0000269|PubMed:34159722, ECO:0000269|PubMed:36395340}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08293; DB08153; DB08292; DB08346; DB08465; DB08045; DB07877; DB02754; DB07594; DB02424; DB08464; DB09221; DB03758; DB06070; DB05134 Interacts with P36896; Q9UL18; O95433; O00170; P31749; P31751; Q9UM73; Q16671; Q01432; P10398; Q96GD4; P30530; P51451; P51813; P15056; Q06187; Q9UQM7; Q13555; Q16543; Q7L3B6; Q15131; Q9UQ88; O94921; Q96Q40; Q00526; P11802; Q00534; P50613; P50750; O14757; Q9UHD1; O15111; Q9UPZ9; P49761; P48729; P49674; P68400; Q13616; Q13617; Q13618; Q13619; Q13620; Q16832; Q9Y463; Q9NR20; P00533; Q9BQI3; P29317; O15197; P04626; P21860; Q15303; Q96A26; Q9UKC9; O75426; Q9UKT8; P16591; P11362; P22607; P09769; Q02790; Q13451; Q14318; P35916; P06241; P43250; Q8WTQ7; P49840; Q8TF76; P08631; Q8NE63; P08238; Q14164; Q9Y6K9; Q13418; P14616; Q08881; Q2WGJ6; P06239; P53671; P07948; Q8N7X4; Q13163; O14733; Q99558; P41279; P80192; Q92918; Q8TD08; P31152; P10636-8; P42679; P00540; O15146; Q9H1R3; Q86YV6; Q8WXR4; Q8NG66; Q86SG6; Q8TD19; O75469; P04629; Q8N165; Q9BXM7; Q9P215; P53041; Q13131; P22694; Q02156; P05129; Q05513; Q15139; O60260; P51817; P11801; Q96QS6; Q15185; Q13882; P04049; P49758; Q01974; Q9H6T3; P62913; Q15418; P51812; Q9HBY8; P31948; Q15831; Q86UX6; Q15208; Q9UNE7; Q6J9G0; Q9Y2Z0-2; Q9UHD2; Q96S53; Q07912; Q9BXA7; Q96PF2; Q9BXA6; O95801; P29597; Q06418; Q8IWX7; P07947 EC number NA Uniprot keywords 3D-structure; Acetylation; ATP-binding; Cell membrane; Chaperone; Cytoplasm; Direct protein sequencing; Glycoprotein; Membrane; Methylation; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; S-nitrosylation; Secreted; Stress response; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 73399.1 Length 636 Aromaticity 0.08 Instability index 37.9 Isoelectric point 5.82 Charge (pH=7) -9.12 2D Binding mode Binding energy (Kcal/mol) -8.67 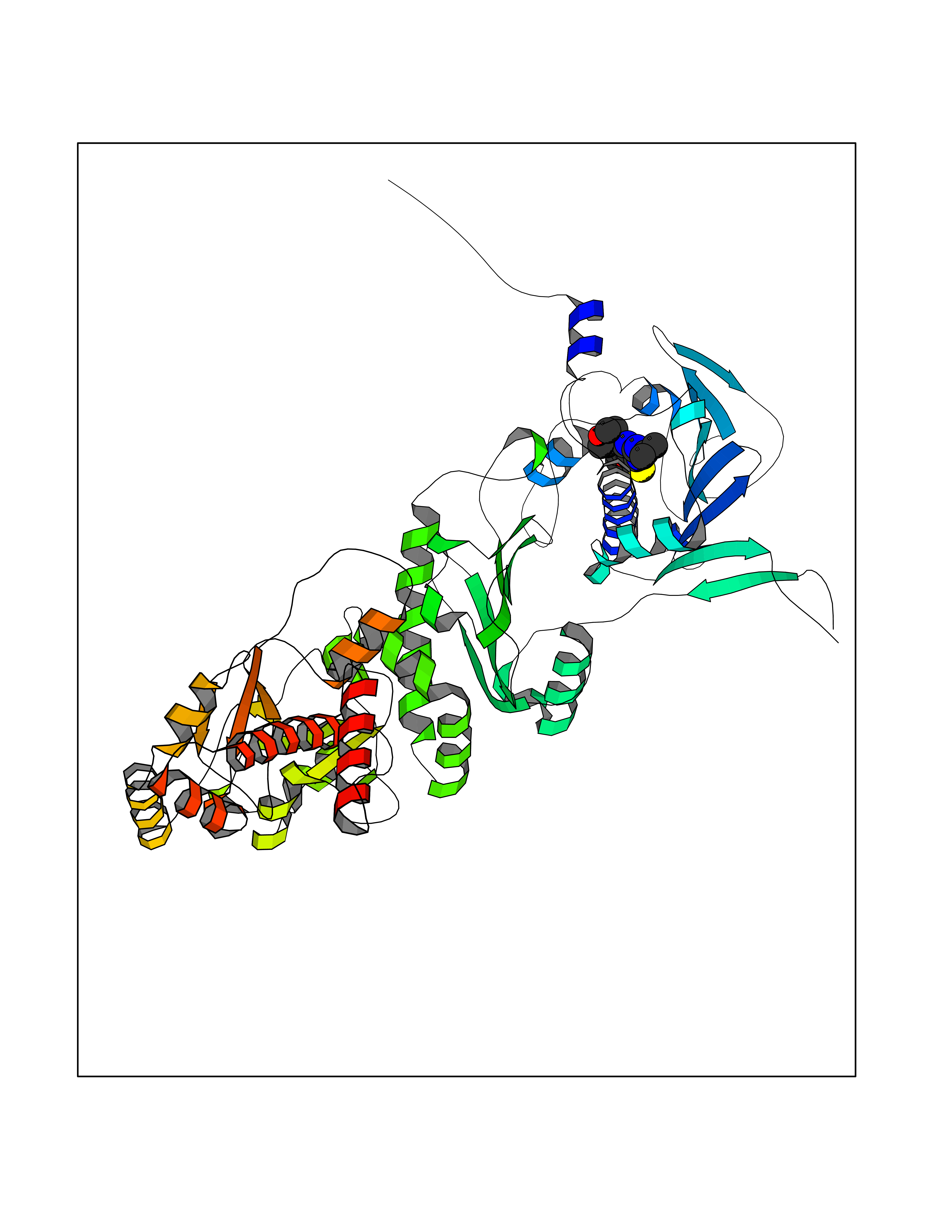 Molscript Map 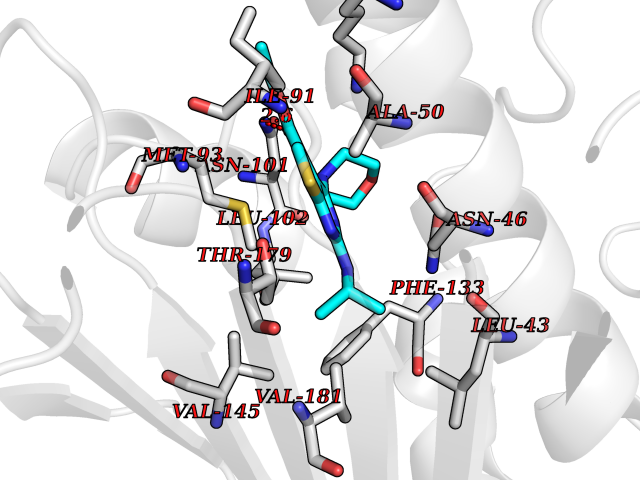 Pymol Map 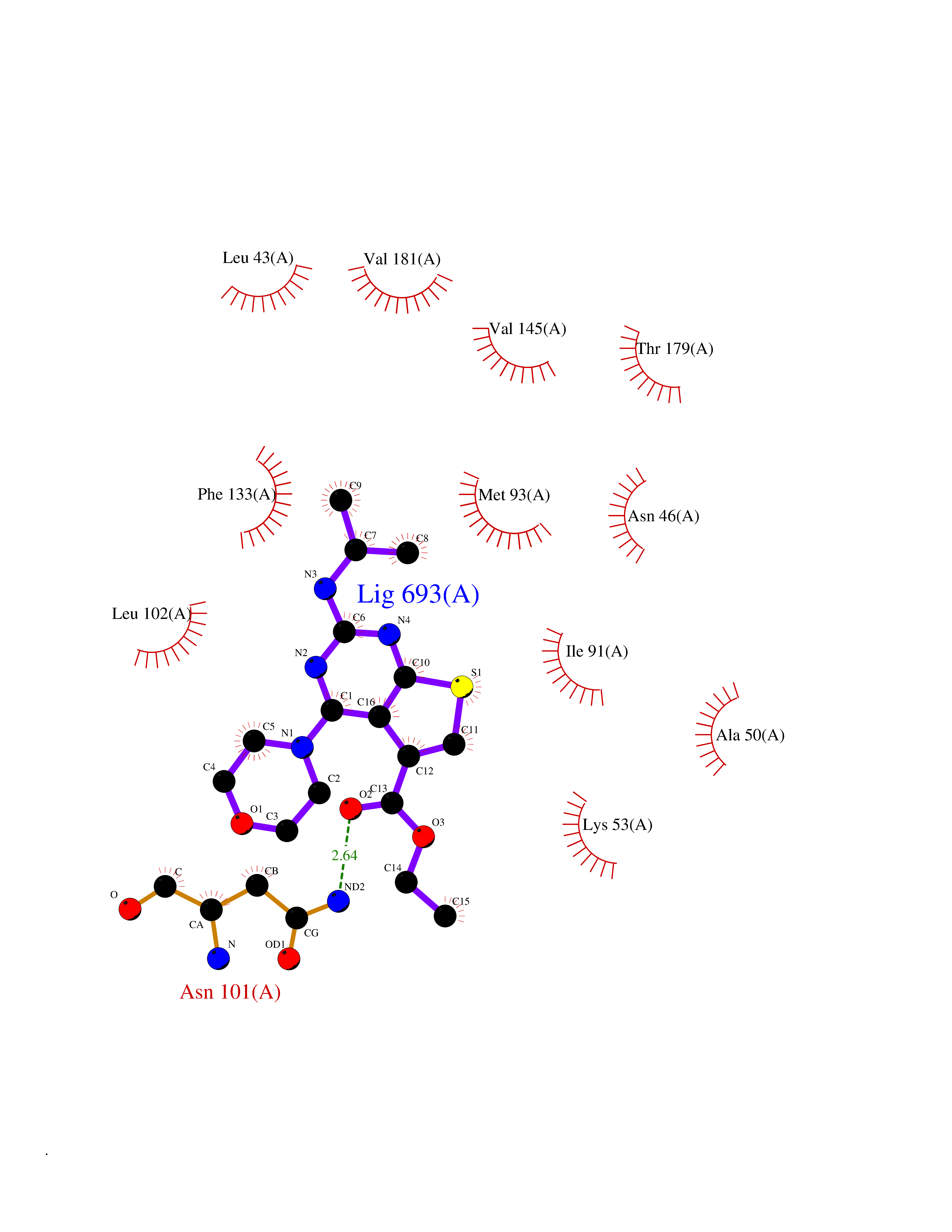 Ligplot Map 3D Binding mode Sequence EEVETFAFQAEIAQLMSLIINTFYSNKEIFLRELISNASDALDKIRYESLTDPSKLDSGKELKIDIIPNPQERTLTLVDTGIGMTKADLINNLGTIAKSGTKAFMEALQAGADISMIGQFGVGFYSAYLVAEKVVVITKHNDDEQYAWESSAGGSFTVRADHGEPIGRGTKVILHLKEDQTEYLEERRVKEVVKKHSQFIGYPITLYLEKEREKKKIKEKYIDQEELNKTKPIWTRNPDDITQEEYGEFYKSLTNDWEDHLAVKHFSVEGQLEFRALLFIPRRAPFDLFENKKKKNNIKLYVRRVFIMDSCDELIPEYLNFIRGVVDSEDLPLNISREMLQQSKILKVIRKNIVKKCLELFSELAEDKENYKKFYEAFSKNLKLGIHEDSTNRRRLSELLRYHTSQSGDEMTSLSEYVSRMKETQKSIYYITGESKEQVANSAFVERVRKRGFEVVYMTEPIDEYCVQQLKEFDGKSLVSVTKEGLELPEDEEEKKKMEESKAKFENLCKLMKEILDKKVEKVTISNRLVSSPCCIVTSTYGWTANMERIMKAQALRDNSTMGYMMAKKHLEINPDHPIVETLRQKAEADKNDKAVKDLVVLLFETALLSSGFSLEDPQTHSNRIYRMIKLGLGID Hydrogen bonds contact Hydrophobic contact | ||||
| 73 | Quinone-dependent D-lactate dehydrogenase | 1F0X | 6.35 | |
Target general information Gen name dld Organism Escherichia coli (strain K12) Uniprot ID TTD ID NA Synonyms JW2121;b2133 Protein family Quinone-dependent D-lactate dehydrogenase family Biochemical class Oxidoreductase Function (R)-2-hydroxyglutarate dehydrogenase activity.D-lactate dehydrogenase (cytochrome) activity.Electron carrier activity.FAD binding.Flavin adenine dinucleotide binding.NAD binding.Oxidoreductase activity, acting on the CH-OH group of donors, quinone or similar compound as acceptor.Quinone binding. Related diseases Myeloperoxidase deficiency (MPOD) [MIM:254600]: A disorder characterized by decreased myeloperoxidase activity in neutrophils and monocytes that results in disseminated candidiasis. {ECO:0000269|PubMed:37198333, ECO:0000269|PubMed:7904599, ECO:0000269|PubMed:8142659, ECO:0000269|PubMed:8621627, ECO:0000269|PubMed:9354683, ECO:0000269|PubMed:9637725}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03147; DB00756 Interacts with NA EC number 1.1.5.12 Uniprot keywords 3D-structure; Cell inner membrane; Cell membrane; Direct protein sequencing; FAD; Flavoprotein; Membrane; Oxidoreductase; Quinone; Reference proteome Protein physicochemical properties Chain ID A,B Molecular weight (Da) 56475.2 Length 502 Aromaticity 0.09 Instability index 32.5 Isoelectric point 5.97 Charge (pH=7) -10.38 2D Binding mode Binding energy (Kcal/mol) -8.66  Molscript Map 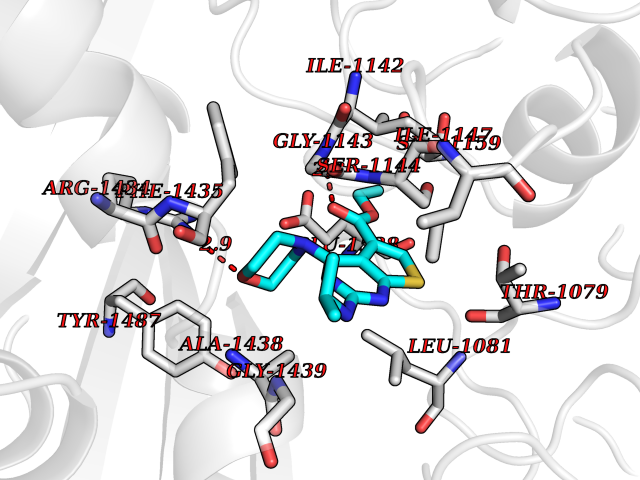 Pymol Map 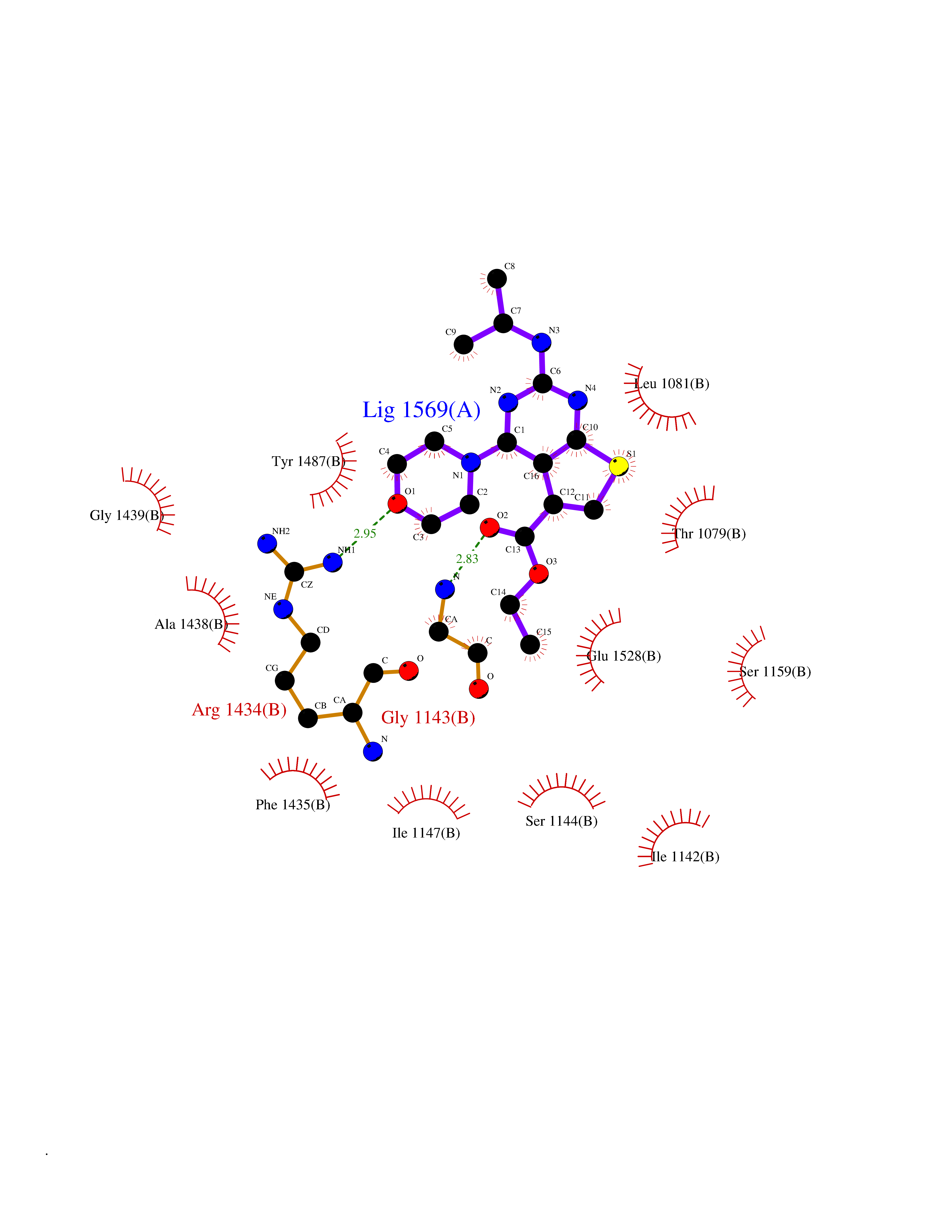 Ligplot Map 3D Binding mode Sequence NKAFLNELARLVGSSHLLTDPAKTARYRKGFRSGQGDALAVVFPGSLLELWRVLKACVTADKIILMQAANTGLTEGSTPNGNDYDRDVVIISTLRLDKLHVLGKGEQVLAYPGTTLYSLEKALKPLGREPHSVIGSSCIGASVIGGICNNSGGSLVQRGPAYTEMSLFARINEDGKLTLVNHLGIDLGETPEQILSKLDDDRIKDDDVRHDGRHAHDYDYVHRVRDIEADTPARYNADPDRLFESSGCAGKLAVFAVRLDTFEAEKNQQVFYIGTNQPEVLTEIRRHILANFENLPVAGEYMHRDIYDIAELPPRMKNWRDKYEHHLLLKMAGDGVGEAKSWLVDYFKQAEGDFFVCTPEEGSKAFLHRFAAAGAAIRYQAVHSDEVEDILALDIALRRNDTEWYEHLPPEIDSQLVHKLYYGHFMCYVFHQDYIVKKGVDVHALKEQMLELLQQRGAQYPAEHNVGHLYKAPETLQKFYRENDPTNSMNPGIGKTSKRKNW Hydrogen bonds contact Hydrophobic contact | ||||
| 74 | Hydroxymethylglutaryl-CoA synthase 2 (HMGCS2) | 2WYA | 6.35 | |
Target general information Gen name HMGCS2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms HMGCS2; HMG-CoAsynthase; 3-hydroxy-3-methylglutaryl coenzyme A synthase 2 Protein family Thiolase-like superfamily, HMG-CoA synthase family Biochemical class Acyltransferase Function This enzyme condenses acetyl-CoA with acetoacetyl-CoA to form HMG-CoA, which is the substrate for HMG-CoA reductase. Related diseases 3-hydroxy-3-methylglutaryl-CoA synthase-2 deficiency (HMGCS2D) [MIM:605911]: A metabolic disorder characterized by severe hypoketotic hypoglycemia, encephalopathy, and hepatomegaly. {ECO:0000269|PubMed:11228257, ECO:0000269|PubMed:11479731, ECO:0000269|PubMed:12647205, ECO:0000269|PubMed:16601895, ECO:0000269|PubMed:23751782, ECO:0000269|PubMed:25511235, ECO:0000269|PubMed:29597274}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number EC 2.3.3.10 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Cholesterol biosynthesis; Cholesterol metabolism; Disease variant; Lipid biosynthesis; Lipid metabolism; Mitochondrion; Phosphoprotein; Proteomics identification; Reference proteome; Steroid biosynthesis; Steroid metabolism; Sterol biosynthesis; Sterol metabolism; Transferase; Transit peptide Protein physicochemical properties Chain ID A,D Molecular weight (Da) 102499 Length 920 Aromaticity 0.1 Instability index 33.48 Isoelectric point 6.72 Charge (pH=7) -1.41 2D Binding mode Binding energy (Kcal/mol) -8.66 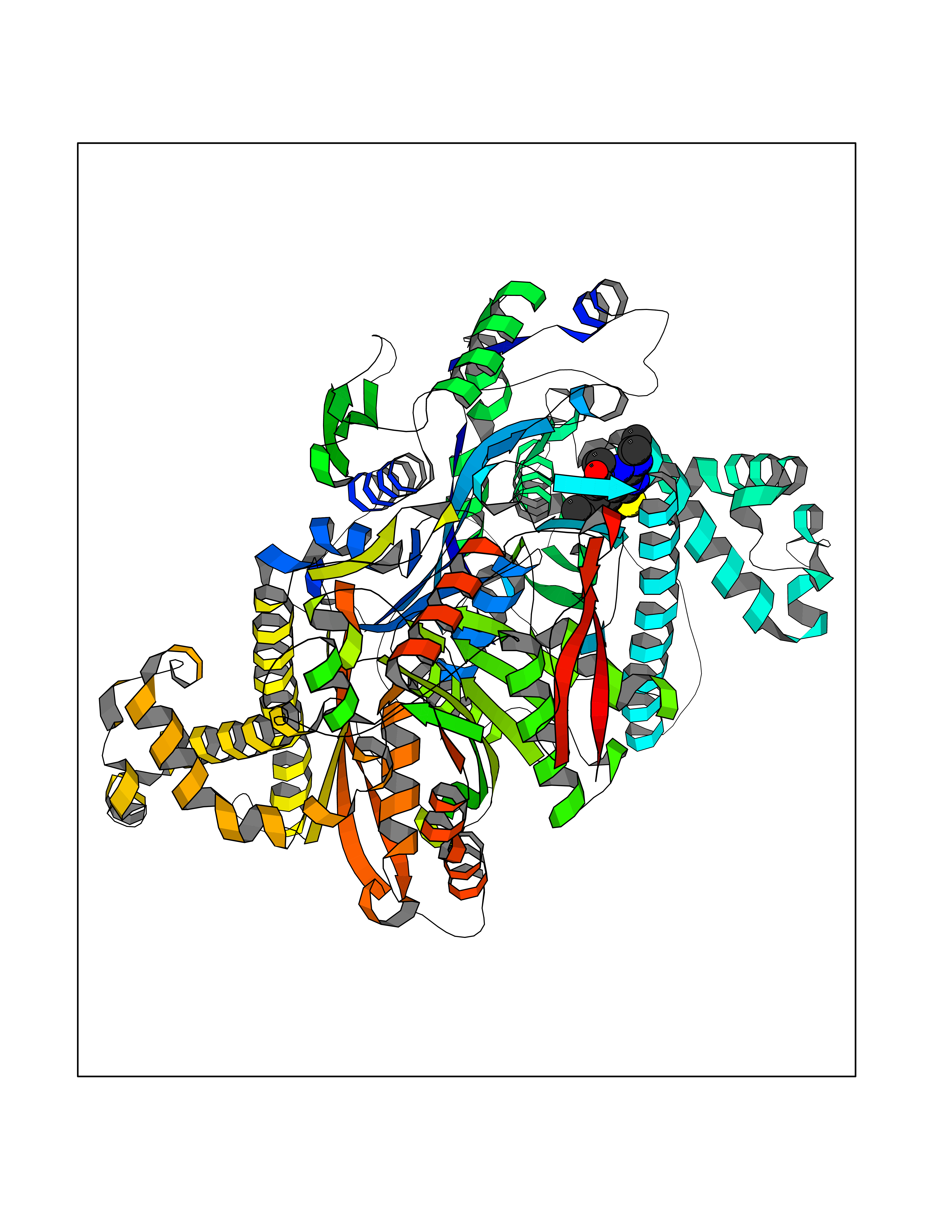 Molscript Map 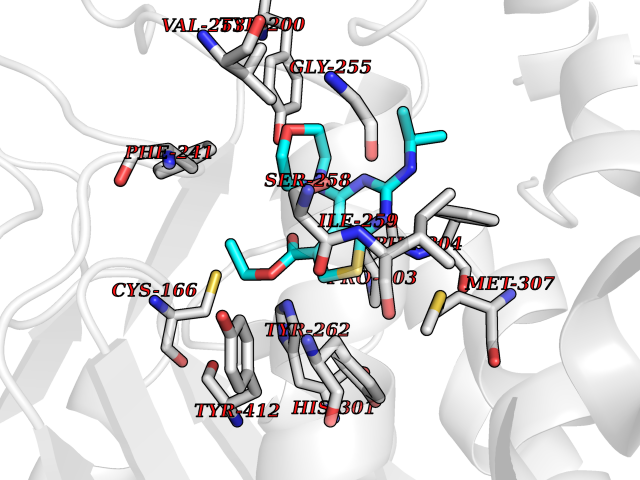 Pymol Map 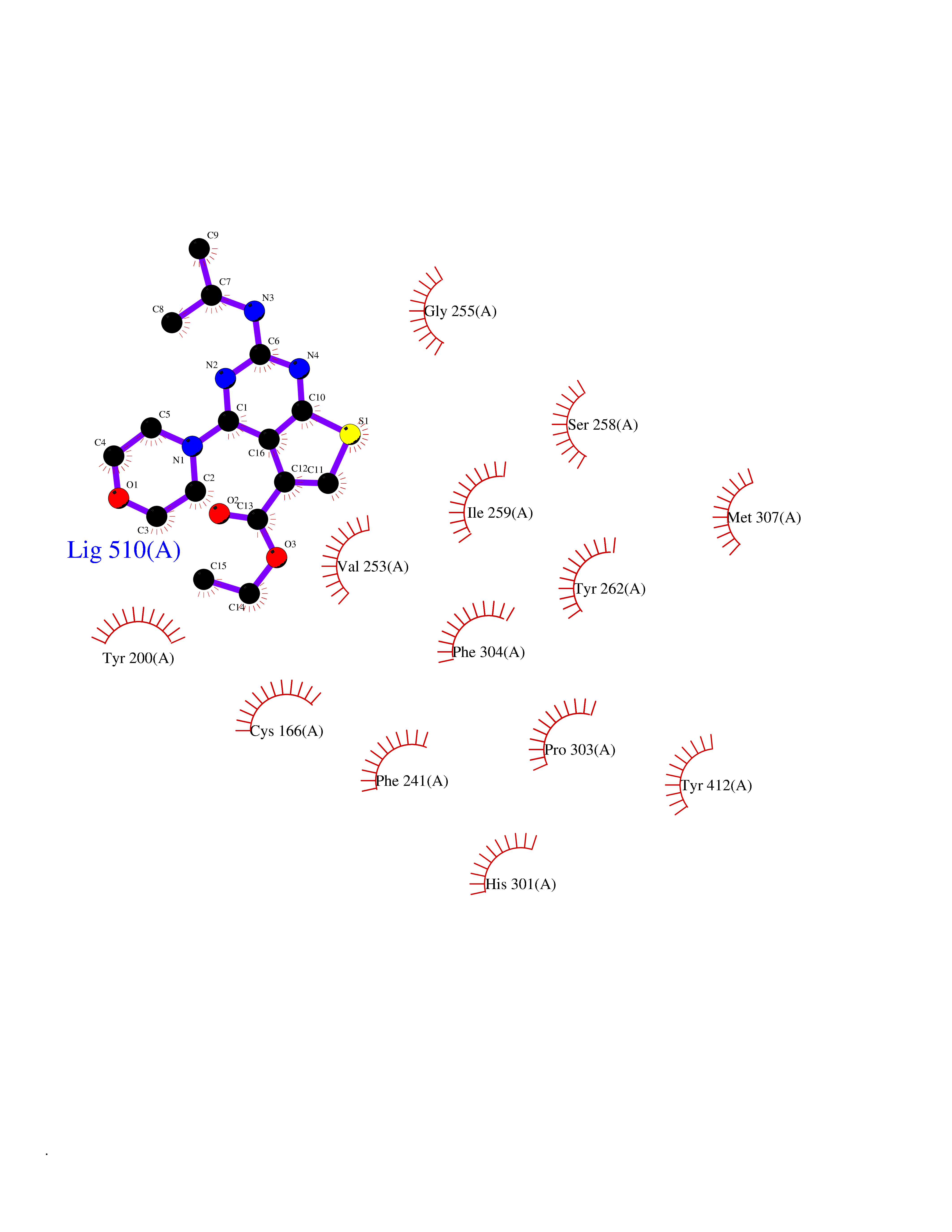 Ligplot Map 3D Binding mode Sequence SMPKDVGILALEVYFPAQYVDQTDLEKYNNVEAGKYTVGLGQTRMGFCSVQEDINSLCLTVVQRLMERIQLPWDSVGRLEVGTETIIDKSKAVKTVLMELFQDSGNTDIEGIDTTNACYGGTASLFNAANWMESSSWDGRYAMVVCGDIAVYPSGNARPTGGAGAVAMLIGPKAPLALERGLRGTHMENVYDFYKPNLASEYPIVDGKLSIQCYLRALDRCYTSYRKKIQNQWKQAGSDRPFTLDDLQYMIFHTPFCKMVQKSLARLMFNDFLSASSDTQTSLYKGLEAFGGLKLEDTYTNKDLDKALLKASQDMFDKKTKASLYLSTHNGNMYTSSLYGCLASLLSHHSAQELAGSRIGAFSYGSGLAASFFSFRVSQDAAPGSPLDKLVSSTSDLPKRLASRKCVSPEEFTEIMNQREQFYHKVNFSPPGDTNSLFPGTWYLERVDEQHRRKYARRPVSMPKDVGILALEVYFPAQYVDQTDLEKYNNVEAGKYTVGLGQTRMGFCSVQEDINSLCLTVVQRLMERIQLPWDSVGRLEVGTETIIDKSKAVKTVLMELFQDSGNTDIEGIDTTNACYGGTASLFNAANWMESSSWDGRYAMVVCGDIAVYPSGNARPTGGAGAVAMLIGPKAPLALERGLRGTHMENVYDFYKPNLASEYPIVDGKLSIQCYLRALDRCYTSYRKKIQNQWKQAGSDRPFTLDDLQYMIFHTPFCKMVQKSLARLMFNDFLSASSDTQTSLYKGLEAFGGLKLEDTYTNKDLDKALLKASQDMFDKKTKASLYLSTHNGNMYTSSLYGCLASLLSHHSAQELAGSRIGAFSYGSGLAASFFSFRVSQDAAPGSPLDKLVSSTSDLPKRLASRKCVSPEEFTEIMNQREQFYHKVNFSPPGDTNSLFPGTWYLERVDEQHRRKYARRPV Hydrogen bonds contact Hydrophobic contact | ||||
| 75 | Beta-ketoacyl-ACP synthase (OXSM) | 2IWZ | 6.35 | |
Target general information Gen name OXSM Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms 3-oxoacyl-[acyl-carrier-protein] synthase, mitochondrial Protein family Thiolase-like superfamily, Beta-ketoacyl-ACP synthases family Biochemical class Acyltransferase Function May play a role in the biosynthesis of lipoic acid as well as longer chain fatty acids required for optimal mitochondrial function. Related diseases Lethal congenital contracture syndrome 2 (LCCS2) [MIM:607598]: A form of lethal congenital contracture syndrome, an autosomal recessive disorder characterized by degeneration of anterior horn neurons, extreme skeletal muscle atrophy, and congenital non-progressive joint contractures (arthrogryposis). The contractures can involve the upper or lower limbs and/or the vertebral column, leading to various degrees of flexion or extension limitations evident at birth. LCCS2 patients manifest craniofacial/ocular findings, lack of hydrops, multiple pterygia, and fractures, as well as a normal duration of pregnancy and a unique feature of a markedly distended urinary bladder (neurogenic bladder defect). The phenotype suggests a spinal cord neuropathic etiology. {ECO:0000269|PubMed:17701904}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Erythroleukemia, familial (FERLK) [MIM:133180]: An autosomal dominant myeloproliferative disorder characterized by neoplastic proliferation of erythroblastic and myeloblastic elements with atypical erythroblasts and myeloblasts in the peripheral blood. Disease penetrance is incomplete. {ECO:0000269|PubMed:27416908}. Disease susceptibility may be associated with variants affecting the gene represented in this entry.; DISEASE: Visceral neuropathy, familial, 1, autosomal recessive (VSCN1) [MIM:243180]: An autosomal recessive disorder characterized by intestinal dysmotility due to aganglionosis (Hirschsprung disease), hypoganglionosis, and/or chronic intestinal pseudoobstruction. Additional variable features are progressive peripheral neuropathy, arthrogryposis, hypoplasia or aplasia of the olfactory bulb and of the external auditory canals, microtia or anotia, and facial dysmorphism. Some patients present structural cardiac anomalies and arthrogryposis with multiple pterygia. {ECO:0000269|PubMed:33497358}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number EC 2.3.1.41 Uniprot keywords 3D-structure; Acetylation; Acyltransferase; Alternative splicing; Fatty acid biosynthesis; Fatty acid metabolism; Lipid biosynthesis; Lipid metabolism; Mitochondrion; Proteomics identification; Reference proteome; Transferase; Transit peptide Protein physicochemical properties Chain ID A,B Molecular weight (Da) 89949.3 Length 852 Aromaticity 0.07 Instability index 35.84 Isoelectric point 6.13 Charge (pH=7) -10.02 2D Binding mode Binding energy (Kcal/mol) -8.67 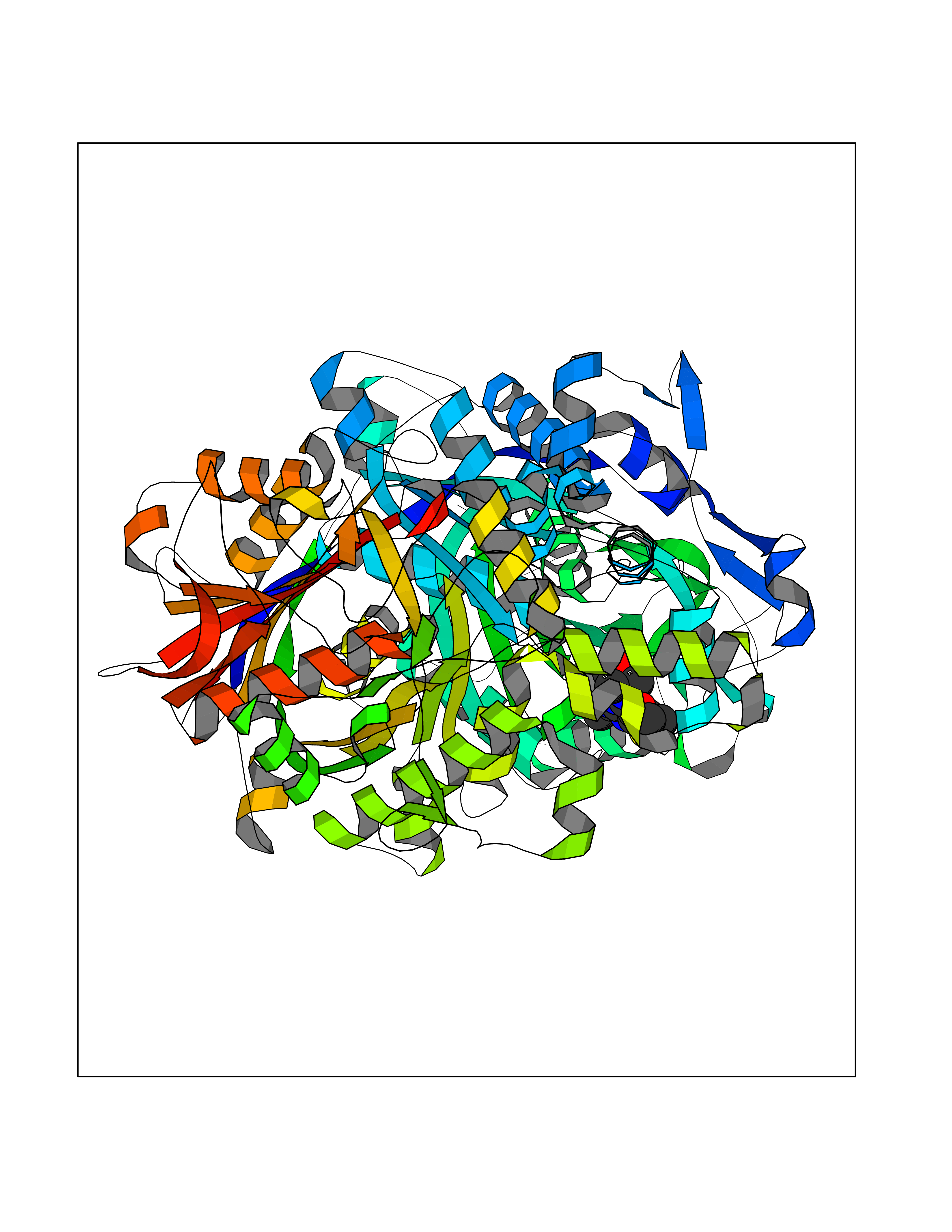 Molscript Map 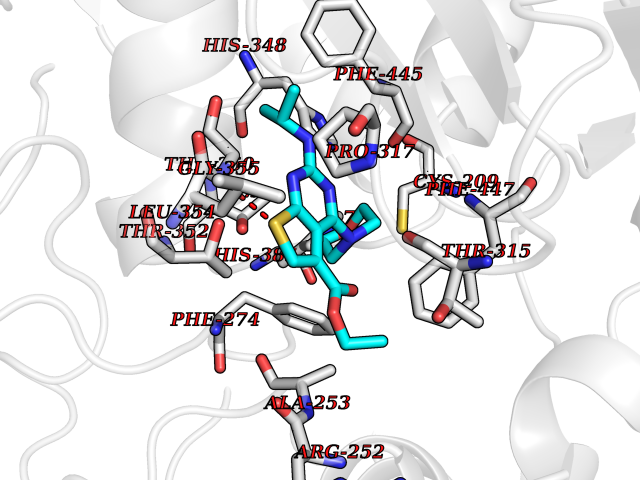 Pymol Map 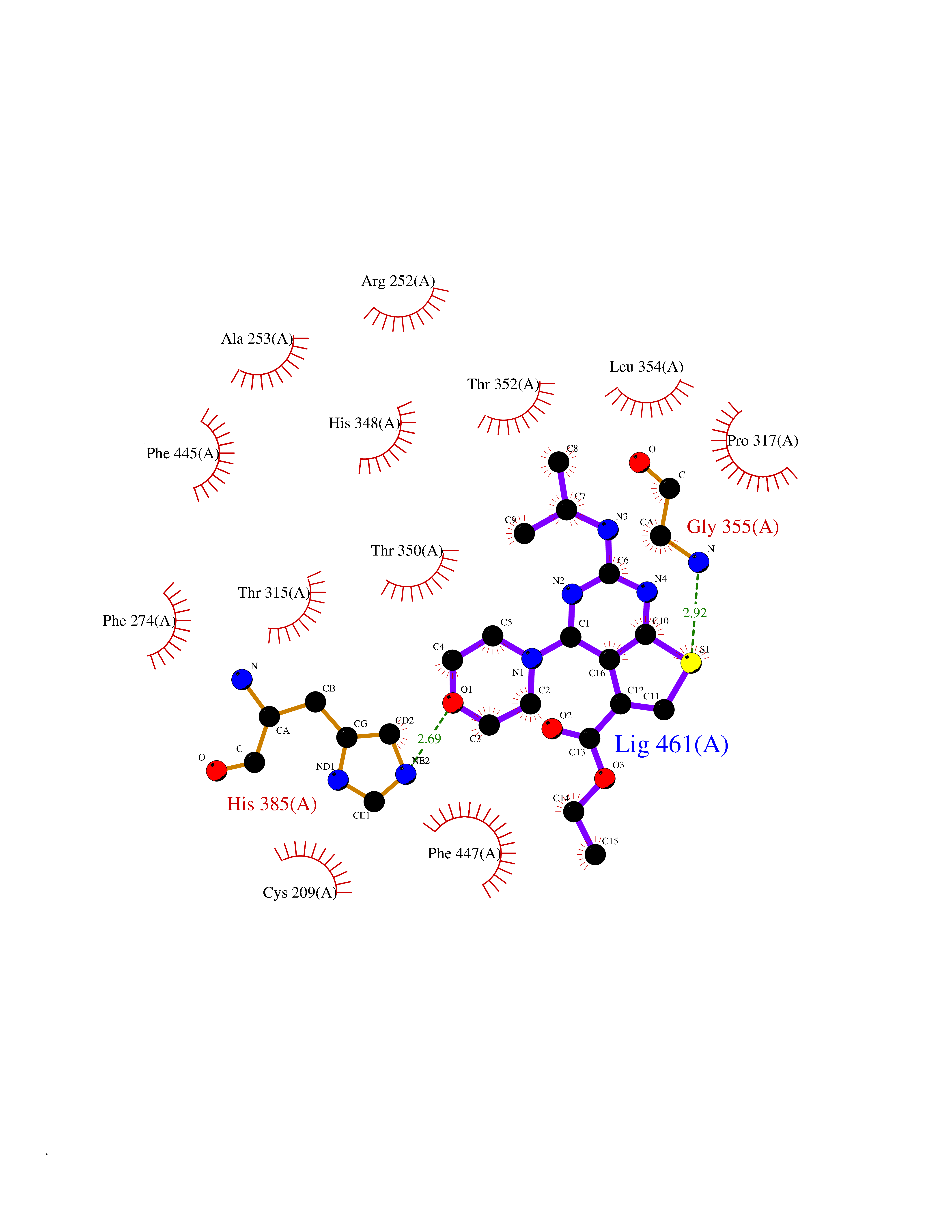 Ligplot Map 3D Binding mode Sequence IEGRSRLHRRVVITGIGLVTPLGVGTHLVWDRLIGGESGIVSLVGEEYKSIPCSVAAYVPRGSDEGQFNEQNFVSKSDIKSMSSPTIMAIGAAELAMKDSGWHPQSEADQVATGVAIGMGMIPLEVVSETALNFQTKGYNKVSPFFVPKILVNMAAGQVSIRYKLKGPNHAVSTACTTGAHAVGDSFRFIAHGDADVMVAGGTDSCISPLSLAGFSRARALSTNSDPKLACRPFHPKRDGFVMGEGAAVLVLEEYEHAVQRRARIYAEVLGYGLSGDAGHITAPDPEGEGALRCMAAALKDAGVQPEEISYINAHATSTPLGDAAENKAIKHLFKDHAYALAVSSTKGATGHLLGAAGAVEAAFTTLACYYQKLPPTLNLDCSEPEFDLNYVPLKAQEWKTEKRFIGLTNSFGFGGTNATLCIAGLIEGRSRLHRRVVITGIGLVTPLGVGTHLVWDRLIGGESGIVSLVGEEYKSIPCSVAAYVPRGSDEGQFNEQNFVSKSDIKSMSSPTIMAIGAAELAMKDSGWHPQSEADQVATGVAIGMGMIPLEVVSETALNFQTKGYNKVSPFFVPKILVNMAAGQVSIRYKLKGPNHAVSTACTTGAHAVGDSFRFIAHGDADVMVAGGTDSCISPLSLAGFSRARALSTNSDPKLACRPFHPKRDGFVMGEGAAVLVLEEYEHAVQRRARIYAEVLGYGLSGDAGHITAPDPEGEGALRCMAAALKDAGVQPEEISYINAHATSTPLGDAAENKAIKHLFKDHAYALAVSSTKGATGHLLGAAGAVEAAFTTLACYYQKLPPTLNLDCSEPEFDLNYVPLKAQEWKTEKRFIGLTNSFGFGGTNATLCIAGL Hydrogen bonds contact Hydrophobic contact | ||||
| 76 | Smoothened homolog (SMO) | 4JKV | 6.34 | |
Target general information Gen name SMO Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Smo-D473H; SMOH; Protein Gx Protein family G-protein coupled receptor Fz/Smo family Biochemical class GPCR frizzled Function Binding of sonic hedgehog (SHH) to its receptor patched is thought to prevent normal inhibition by patched of smoothened (SMO). Required for the accumulation of KIF7, GLI2 and GLI3 in the cilia. Interacts with DLG5 at the ciliary base to induce the accumulation of KIF7 and GLI2 at the ciliary tip for GLI2 activation. G protein-coupled receptor that probably associates with the patched protein (PTCH) to transduce the hedgehog's proteins signal. Related diseases Curry-Jones syndrome (CRJS) [MIM:601707]: A multisystem disorder characterized by patchy skin lesions, polysyndactyly, diverse cerebral malformations, unicoronal craniosynostosis, iris colobomas, microphthalmia, and intestinal malrotation with myofibromas or hamartomas. {ECO:0000269|PubMed:24859340, ECO:0000269|PubMed:27236920}. The disease is caused by variants affecting the gene represented in this entry. 8 individuals have been identified with the disease-causing mutation Phe-412 and all were mosaic. The mutation could not be reliably detected in blood, greatest success rates were obtained with affected tissues obtained by invasive procedures. It is thought that the mutation has arisen postzygotically early during embryonic development (PubMed:27236920). This mutation has also been identified in ameloblastoma, medulloblastoma, meningioma, and basal cell carcinoma, and has been reported as the oncogenic driver in some of these tumors (PubMed:24859340). {ECO:0000269|PubMed:24859340, ECO:0000269|PubMed:27236920}. Drugs (DrugBank ID) DB01047; DB11978; DB06786; DB09143; DB08828 Interacts with NA EC number NA Uniprot keywords 3D-structure; Cell membrane; Cell projection; Developmental protein; Disease variant; Disulfide bond; G-protein coupled receptor; Glycoprotein; Membrane; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Signal; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A,B Molecular weight (Da) 37420.1 Length 333 Aromaticity 0.16 Instability index 25.17 Isoelectric point 6.65 Charge (pH=7) -0.76 2D Binding mode Binding energy (Kcal/mol) -8.65 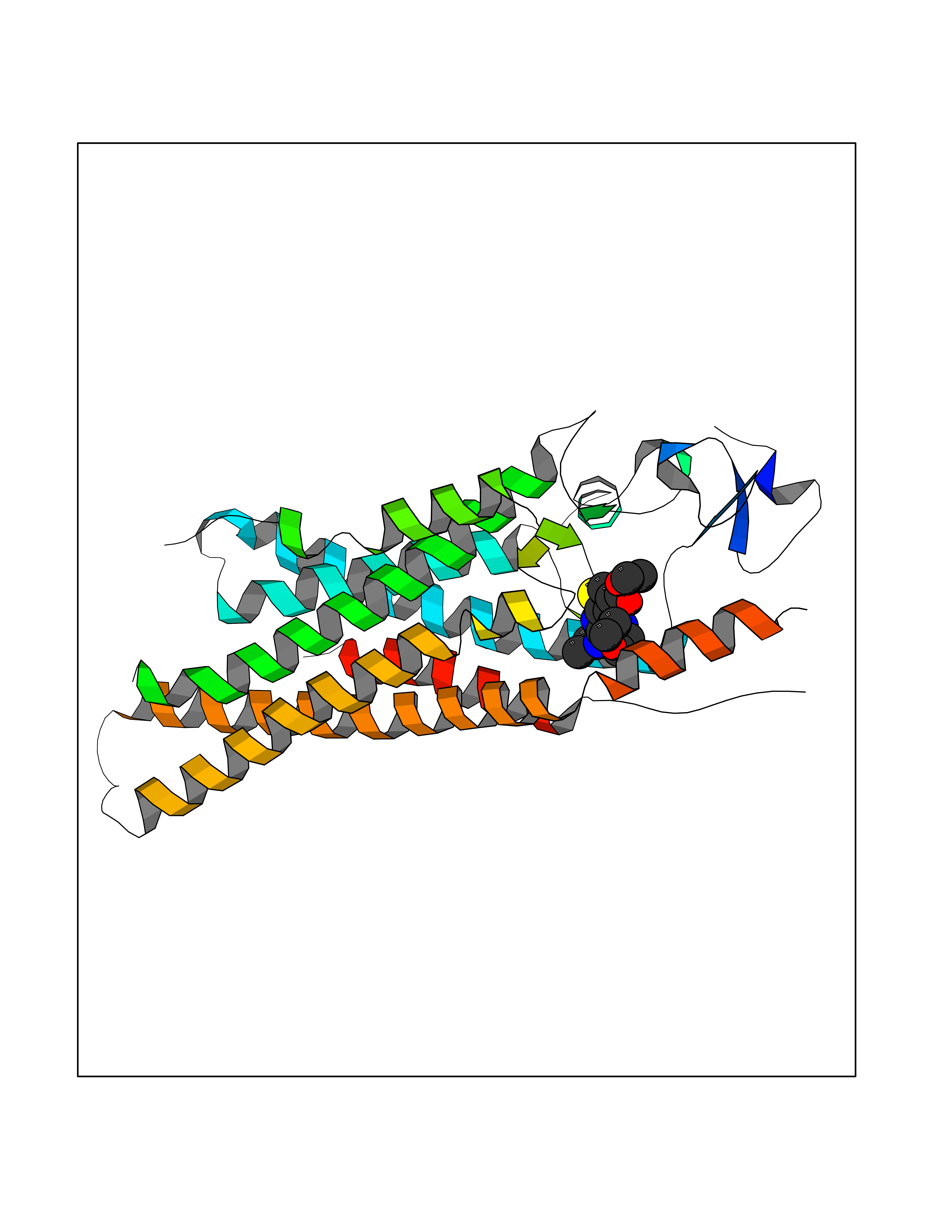 Molscript Map 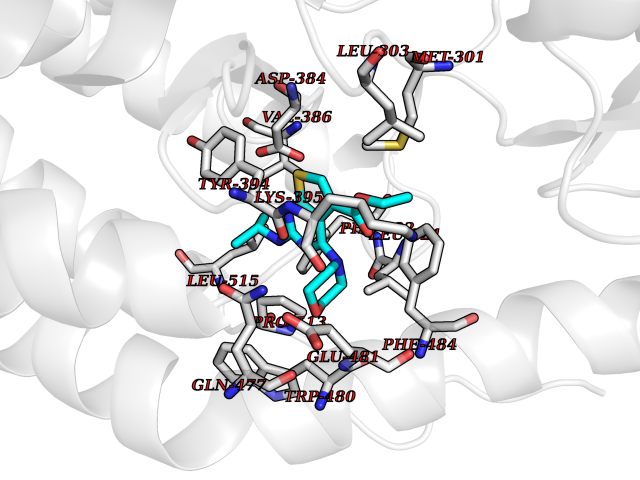 Pymol Map 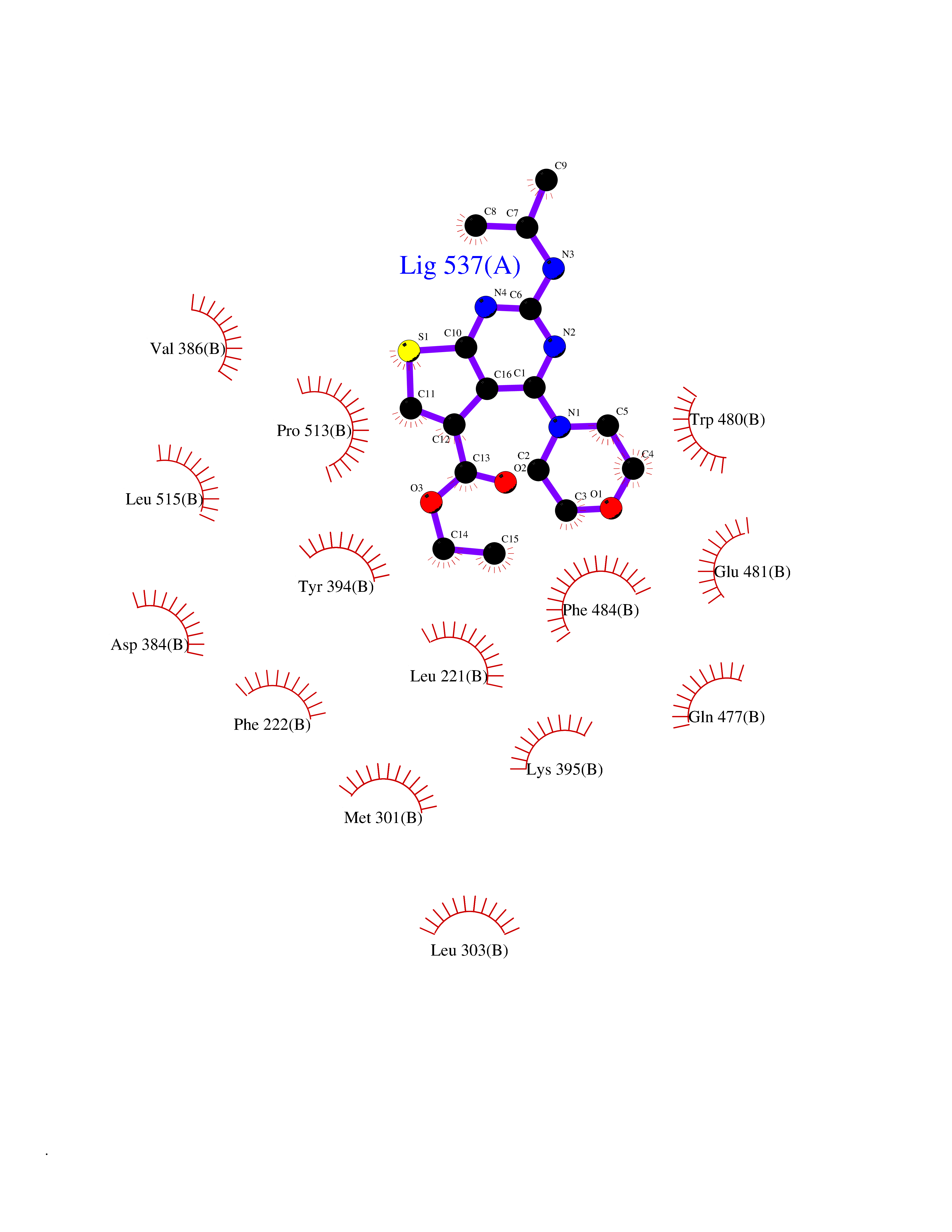 Ligplot Map 3D Binding mode Sequence AYIQKYLSGQCEVPLVRTDNPKSWYEDVEGCGIQCQNPLFTEAEHQDMHSYIAAFGAVTGLCTLFTLATFVADWRNSNRYPAVILFYVNACFFVGSIGWLAQFMDGARREIVCRADGTMRLGEPTSNETLSCVIIFVIVYYALMAGVVWFVVLTYAWHTSFKALGKTSYFHLLTWSLPFVLTVAILAVAQVDGDSVSGICFVGYKNYRYRAGFVLAPIGLVLIVGGYFLIRGVMTLFSIKSNHPGLLSEKAASKINETMLRLGIFGFLAFGFVLITFSCHFYDFFNQAEWERSFRDYVLCQANDCEIKNRPSLLVEKINLFAMFGTGIAMSTW Hydrogen bonds contact Hydrophobic contact | ||||
| 77 | Sphingosine-1-phosphate receptor 1 (S1PR1) | 7EW0 | 6.34 | |
Target general information Gen name S1PR1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Sphingosine 1-phosphate receptor Edg-1; S1P1; S1P receptor Edg-1; S1P receptor 1; Endothelial differentiation G-protein coupled receptor 1; CHEDG1; CD363 Protein family G-protein coupled receptor 1 family Biochemical class GPCR rhodopsin Function Signaling leads to the activation of RAC1, SRC, PTK2/FAK1 and MAP kinases. Plays an important role in cell migration, probably via its role in the reorganization of the actin cytoskeleton and the formation of lamellipodia in response to stimuli that increase the activity of the sphingosine kinase SPHK1. Required for normal chemotaxis toward sphingosine 1-phosphate. Required for normal embryonic heart development and normal cardiac morphogenesis. Plays an important role in the regulation of sprouting angiogenesis and vascular maturation. Inhibits sprouting angiogenesis to prevent excessive sprouting during blood vessel development. Required for normal egress of mature T-cells from the thymus into the blood stream and into peripheral lymphoid organs. Plays a role in the migration of osteoclast precursor cells, the regulation of bone mineralization and bone homeostasis. Plays a role in responses to oxidized 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphocholine by pulmonary endothelial cells and in the protection against ventilator-induced lung injury. G-protein coupled receptor for the bioactive lysosphingolipid sphingosine 1-phosphate (S1P) that seems to be coupled to the G(i) subclass of heteromeric G proteins. Related diseases Wolcott-Rallison syndrome (WRS) [MIM:226980]: A rare autosomal recessive disorder, characterized by permanent neonatal or early infancy insulin-dependent diabetes and, at a later age, epiphyseal dysplasia, osteoporosis, growth retardation and other multisystem manifestations, such as hepatic and renal dysfunctions, intellectual disability and cardiovascular abnormalities. {ECO:0000269|PubMed:10932183, ECO:0000269|PubMed:12086964, ECO:0000269|PubMed:12960215, ECO:0000269|PubMed:16813601, ECO:0000269|PubMed:24168455, ECO:0000269|PubMed:24194294, ECO:0000269|PubMed:27145240, ECO:0000269|PubMed:28220546, ECO:0000269|PubMed:30906465, ECO:0000269|PubMed:30922274, ECO:0000269|PubMed:32216767, ECO:0000269|PubMed:34123975}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB14766; DB08868; DB12612; DB12016; DB12371 Interacts with Q07108 EC number NA Uniprot keywords 3D-structure; Acetylation; Angiogenesis; Cell membrane; Chemotaxis; Disulfide bond; Endosome; G-protein coupled receptor; Glycoprotein; Lipoprotein; Membrane; Palmitate; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID D Molecular weight (Da) 32418.9 Length 284 Aromaticity 0.13 Instability index 39.5 Isoelectric point 9.71 Charge (pH=7) 19.09 2D Binding mode Binding energy (Kcal/mol) -8.64 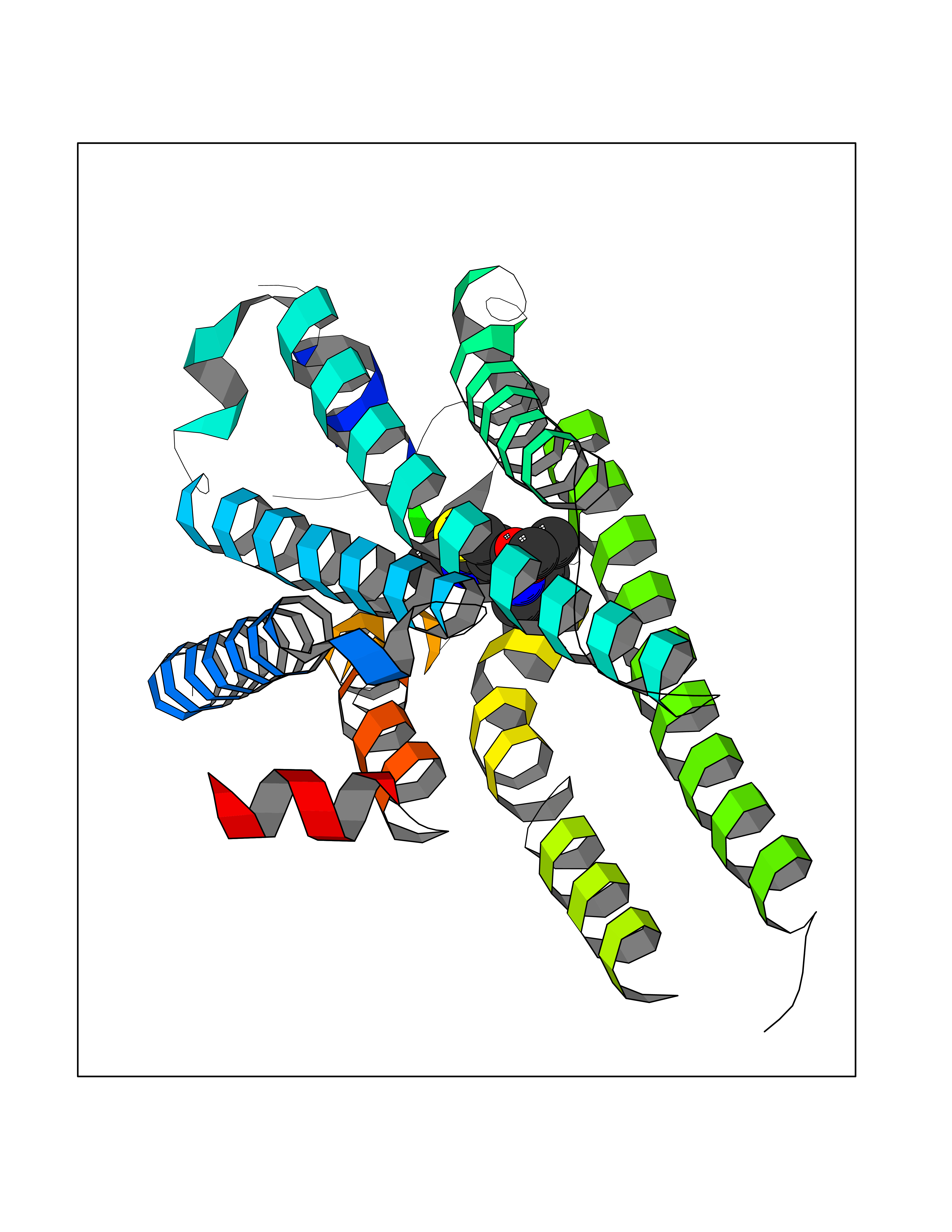 Molscript Map 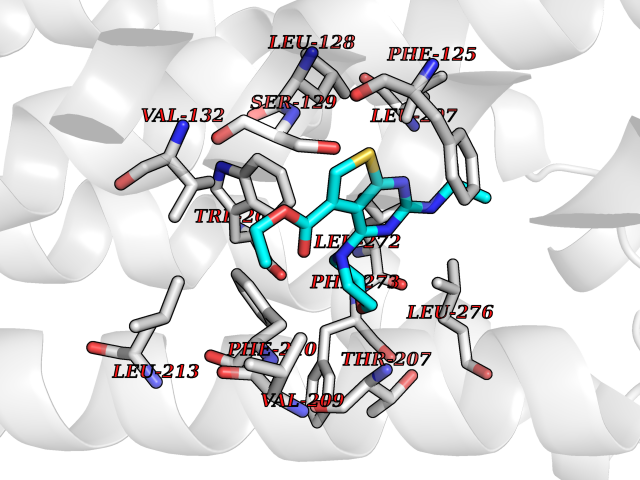 Pymol Map 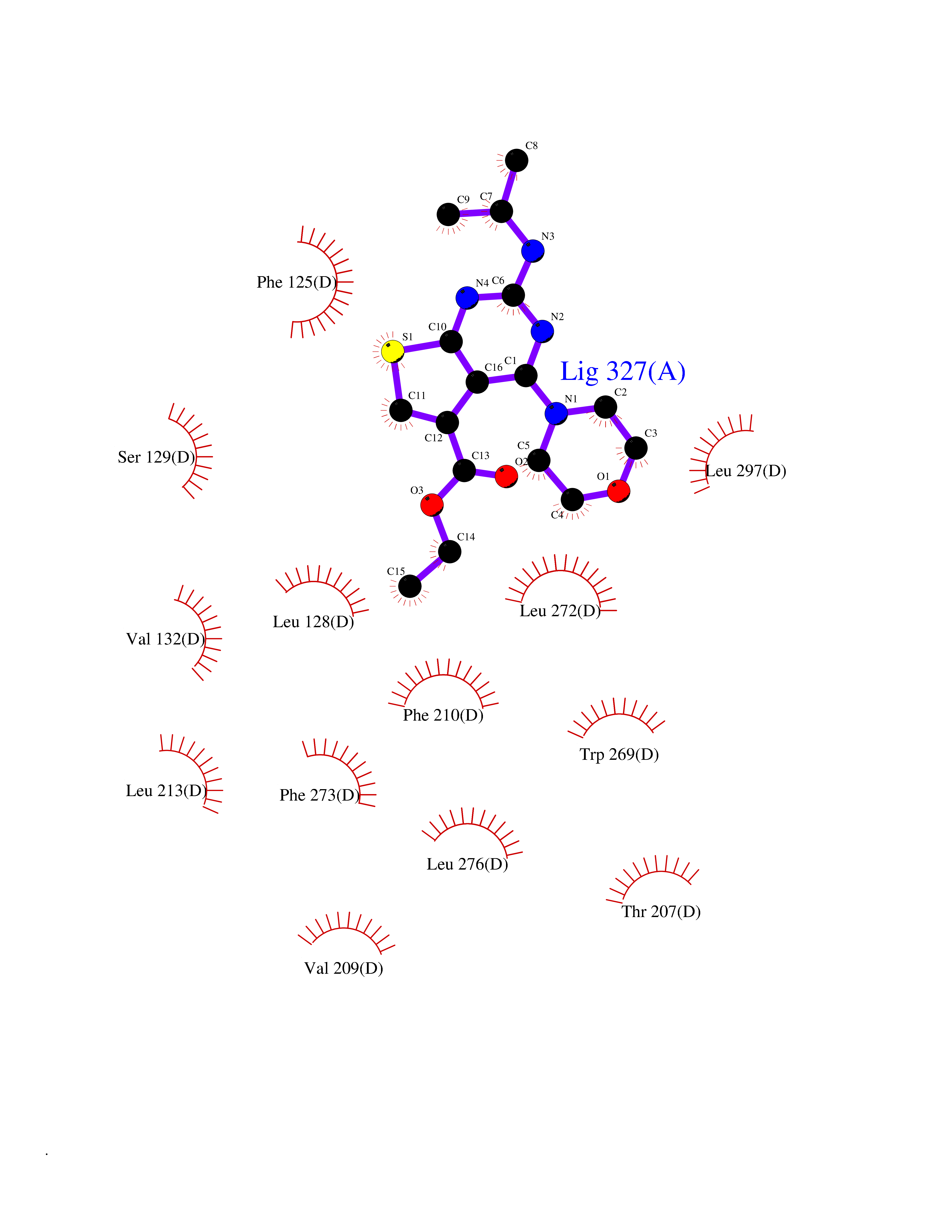 Ligplot Map 3D Binding mode Sequence YDIIVRHYNYTGKLTSVVFILICCFIILENIFVLLTIWKTKKFHRPMYYFIGNLALSDLLAGVAYTANLLLSGATTYKLTPAQWFLREGSMFVALSASVFSLLAIAIERYITMLKMKLHNGSNNFRLFLLISACWVISLILGGLPIMGWNCISALSSCSTVLPLYHKHYILFCTTVFTLLLLSIVILYCRIYSLVRTRSRRLTFRKSEKSLALLKTVIIVLSVFIACWAPLFILLLLDVGCKVKTCDILFRAEYFLVLAVLNSGTNPIIYTLTNKEMRRAFIRI Hydrogen bonds contact Hydrophobic contact | ||||
| 78 | Glucose-dependent insulinotropic receptor (GPR119) | 7XZ6 | 6.34 | |
Target general information Gen name GPR119 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms GPR119; G-protein coupled receptor 119 Protein family G-protein coupled receptor 1 family Biochemical class GPCR rhodopsin Function Receptor for the endogenous fatty-acid ethanolamide oleoylethanolamide (OEA) and lysophosphatidylcholine (LPC). Functions as a glucose-dependent insulinotropic receptor. The activity of this receptor is mediated by G proteins which activate adenylate cyclase. Seems to act through a G(s) mediated pathway. Related diseases Developmental and epileptic encephalopathy 24 (DEE24) [MIM:615871]: A disease characterized by early-onset seizures, intellectual disability of varying degrees, and behavioral disturbances or autistic features in most individuals. {ECO:0000269|PubMed:24747641, ECO:0000269|PubMed:27864847, ECO:0000269|PubMed:30351409}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Generalized epilepsy with febrile seizures plus 10 (GEFSP10) [MIM:618482]: An autosomal dominant neurologic disorder with incomplete penetrance, characterized by variable types of seizures including absence, tonic-clonic, febrile, focal, and eyelid myoclonia. Some patients have normal neurologic development. Others have mild-to-moderate intellectual disability or autism spectrum disorder. {ECO:0000269|PubMed:29936235, ECO:0000269|PubMed:30351409}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB05166 Interacts with Q12797-6 EC number NA Uniprot keywords 3D-structure; Cell membrane; G-protein coupled receptor; Lipid-binding; Membrane; Receptor; Reference proteome; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID R Molecular weight (Da) 32134.1 Length 292 Aromaticity 0.12 Instability index 34.96 Isoelectric point 9.12 Charge (pH=7) 8.03 2D Binding mode Binding energy (Kcal/mol) -8.64  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence MESSFSFGVILAVLASLIIATNTLVAVAVLLLIHKNDGVSLCFTLNLAVADTLIGVAISGLLTDQLSSPSRPTQKTLCSLRMAFVTSSAAASVLTVMLITFDRYLAIKQPFRYLKIMSGFVAGACIAGLWLVSYLIGFLPLGIPMFQQTAYKGQCSFFAVFHPHFVLTLSCVGFFPAMLLFVFFYCDMLKIASMHSQQIRKMEHAGAMAGSDFKALRTVSVLIGSFALSWTPFLITGIVQVACQECHLYLVLERYLWLLGVGNSLLNPLIYAYWQKEVRLQLYHMALGVKKV Hydrogen bonds contact Hydrophobic contact | ||||
| 79 | Aspartate--tRNA ligase, cytoplasmic | 4J15 | 6.33 | |
Target general information Gen name DARS Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms PIG40;DARS Protein family Class-II aminoacyl-tRNA synthetase family, Type 2 subfamily Biochemical class Ligase Function Aminoacylase activity.Aspartate-tRNA ligase activity.ATP binding.RNA binding. Related diseases Hypomyelination with brainstem and spinal cord involvement and leg spasticity (HBSL) [MIM:615281]: An autosomal recessive leukoencephalopathy characterized by onset in the first year of life of severe spasticity, mainly affecting the lower limbs and resulting in an inability to achieve independent ambulation. Affected individuals show delayed motor development and nystagmus; some may have mild intellectual disability. Brain MRI shows hypomyelination and white matter lesions in the cerebrum, brainstem, cerebellum, and spinal cord. {ECO:0000269|PubMed:23643384}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00128 Interacts with Q13155; Q08050; P62993 EC number 6.1.1.12 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Aminoacyl-tRNA synthetase; ATP-binding; Cytoplasm; Disease variant; Ligase; Nucleotide-binding; Phosphoprotein; Protein biosynthesis; Proteomics identification; Reference proteome Protein physicochemical properties Chain ID A,B Molecular weight (Da) 49374.2 Length 430 Aromaticity 0.09 Instability index 42.82 Isoelectric point 6.11 Charge (pH=7) -5.14 2D Binding mode Binding energy (Kcal/mol) -8.63  Molscript Map 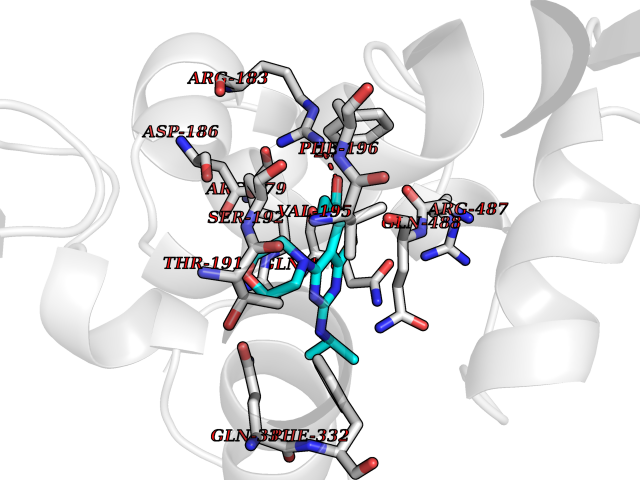 Pymol Map 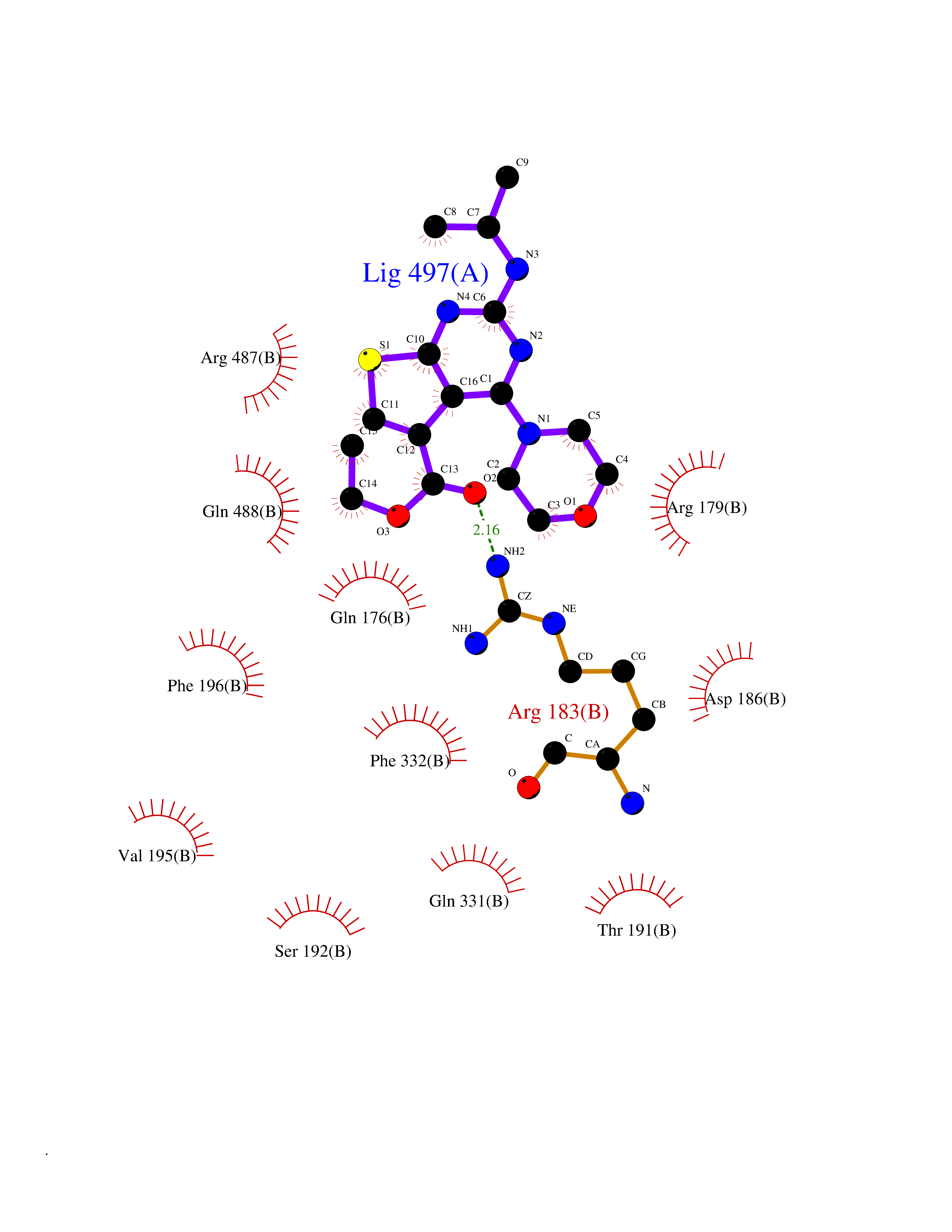 Ligplot Map 3D Binding mode Sequence AEDYAKERYGISSMIQSQEKPDRVLVRVRDLTIQKADEVVWVRARVHTSRAKGKQCFLVLRQQQFNVQALVAVGDHASKQMVKFAANINKESIVDVEGVVRKVNQKIGSCTQQDVELHVQKIYVISLAEPRLPLQLDDAVRPTVNQDTRLDNRVIDLRTSTSQAVFRLQSGICHLFRETLINKGFVEIQTPKISPQLYKQMCICADFEKVFSIGPVFLTEFVGLDIEMAFNYHYHEVMEEIADTMVQIFKGLQERFQTEIQTVNKQFPCEPFKFLEPTLRLEYCEALAMLREAGVEMGDEDDLSTPNEKLLGHLVKEKYDTDFYILDKYPLAVRPFYTMPDPRNPKQSNSYDMFMRGEEILSGAQRIHDPQLLTERALHHGIDLEKIKAYIDSFRFGAPPHAGGGIGLERVTMLFLGLHNVRQTSMFPRD Hydrogen bonds contact Hydrophobic contact | ||||
| 80 | Retinoic acid receptor RXR-beta (RXRB) | 5HJP | 6.33 | |
Target general information Gen name RXRB Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Retinoid X receptor beta; Nuclear receptor subfamily 2 group B member 2; NR2B2 Protein family Nuclear hormone receptor family, NR2 subfamily Biochemical class Nuclear hormone receptor Function Retinoic acid receptors bind as heterodimers to their target response elements in response to their ligands, all-trans or 9-cis retinoic acid, and regulate gene expression in various biological processes. The RAR/RXR heterodimers bind to the retinoic acid response elements (RARE). Receptor for retinoic acid. Related diseases Noonan syndrome 13 (NS13) [MIM:619087]: A form of Noonan syndrome, a disease characterized by short stature, facial dysmorphic features such as hypertelorism, a downward eyeslant and low-set posteriorly rotated ears, and a high incidence of congenital heart defects and hypertrophic cardiomyopathy. Other features can include a short neck with webbing or redundancy of skin, deafness, motor delay, variable intellectual deficits, multiple skeletal defects, cryptorchidism, and bleeding diathesis. Individuals with Noonan syndrome are at risk of juvenile myelomonocytic leukemia, a myeloproliferative disorder characterized by excessive production of myelomonocytic cells. NS13 inheritance is autosomal dominant. There is considerable variability in severity. {ECO:0000269|PubMed:32721402}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08175; DB00459; DB00210; DB00523; DB00307; DB01393; DB03756; DB00926; DB01941; DB07929; DB02746; DB00412; DB00799; DB07080; DB00755 Interacts with Q00975; Q9HB07; F1D8P7; Q13133; Q13133-3; Q96RI1-1; P04150; Q9NRD5; P37231; P10276; P10276-2; P10826-2; P13631; Q6IQ16; Q13137; Q96B26; Q08379; Q6A162; Q9UJV3-2; Q13133-3; Q96RI1-1; O43586; P10276; P10826-2; Q8IUQ4-2; O75528; Q12800; Q9UBB9; Q05BL1; P14373; O94972; Q96S82 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; DNA-binding; Metal-binding; Methylation; Nucleus; Proteomics identification; Receptor; Reference proteome; Transcription; Transcription regulation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A,C Molecular weight (Da) 28845.8 Length 251 Aromaticity 0.08 Instability index 54.86 Isoelectric point 6.74 Charge (pH=7) -0.6 2D Binding mode Binding energy (Kcal/mol) -8.64 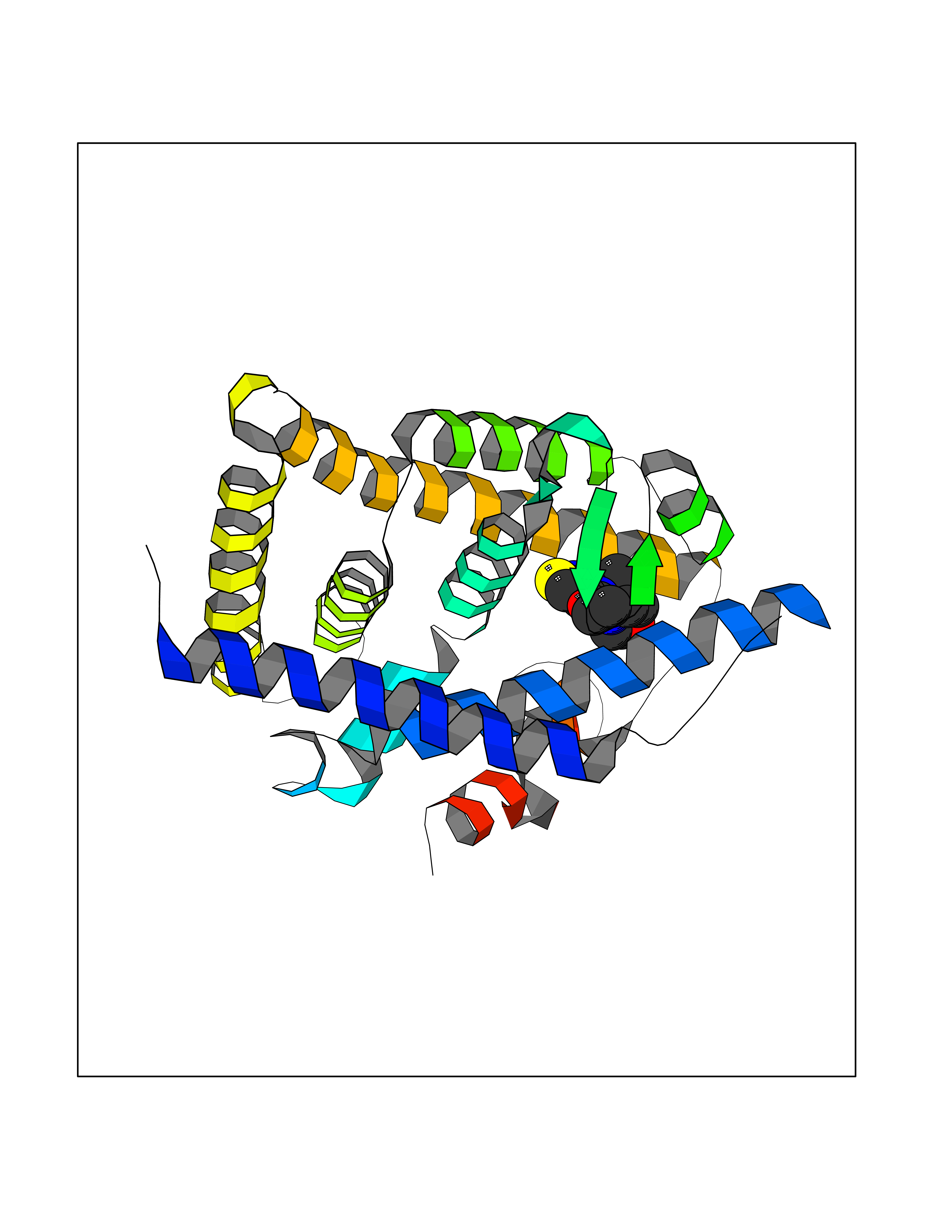 Molscript Map 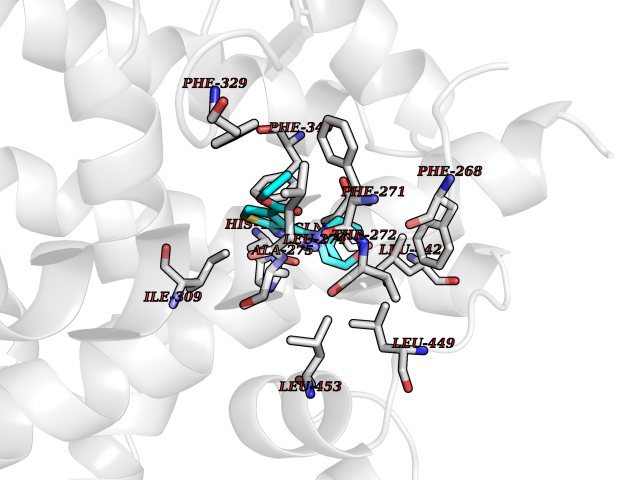 Pymol Map 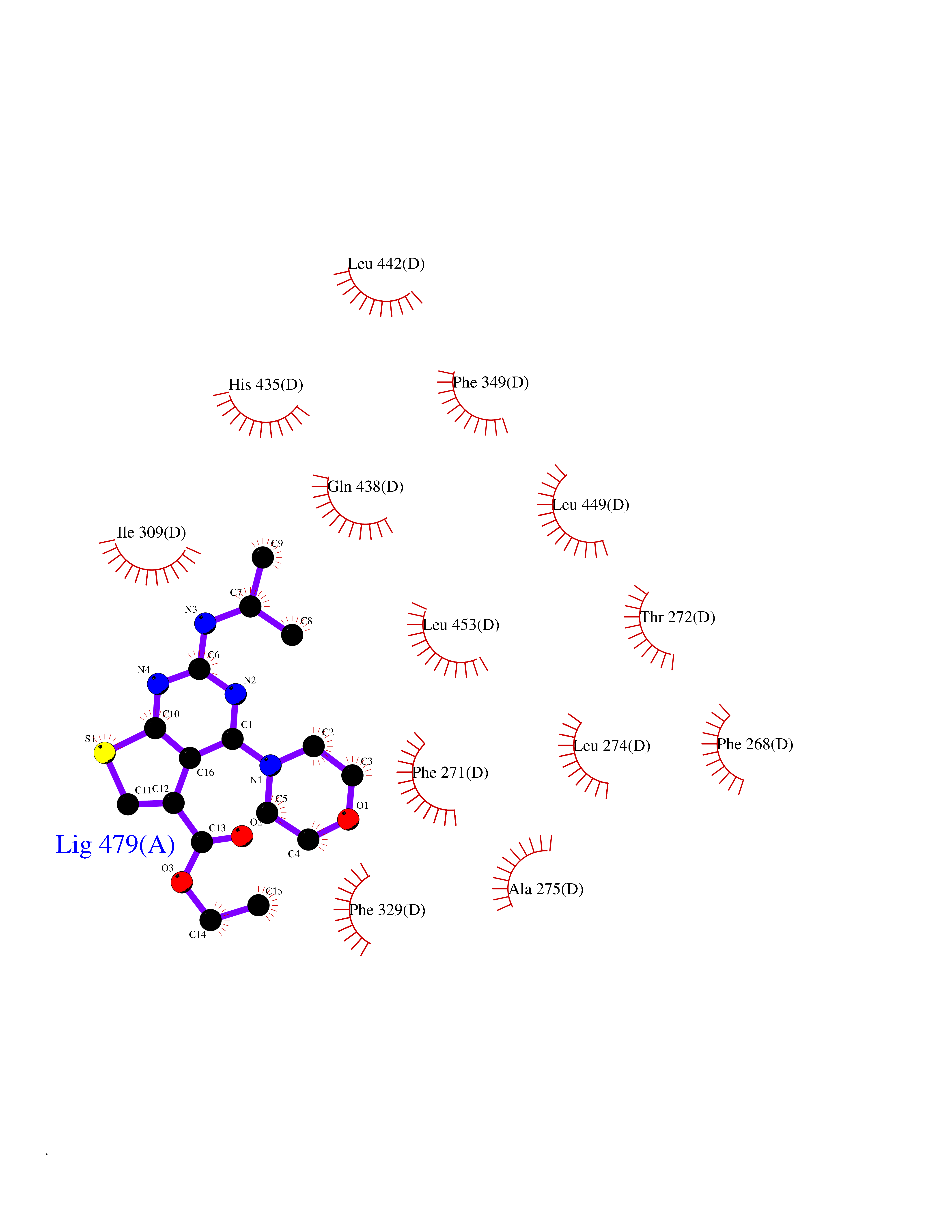 Ligplot Map 3D Binding mode Sequence QLTAAQELMIQQLVAAQLQCNKRSFSDQPKVTPWPSASQQRFAHFTELAIISVQEIVDFAKQVPGFLQLGREDQIALLKASTIEIMLLETARRYNHETECITFLKDFTYSKDDFHRAGLQVEFINPIFEFSRAMRRLGLDDAEYALLIAINIFSADRPNVQEPGRVEALQQPYVEALLSYTRIKRPQDQLRFPRMLMKLVSLRTLSSVHSEQVFALRLQDKKLPPLLSEIWDVHEGSGSGSHKILHRLLQD Hydrogen bonds contact Hydrophobic contact | ||||