Job Results:
Ligand
Structure
Job ID
5c976aa63f0cd6d4251a6978fa589574
Job name
NA
Time
2025-07-21 16:39:34
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 61 | Pyruvate synthase | 2C42 | 5.32 | |
Target general information Gen name por Organism Desulfocurvibacter africanus (Desulfovibrio africanus) Uniprot ID TTD ID NA Synonyms NA Protein family Pyruvate:ferredoxin/flavodoxin oxidoreductase family Biochemical class Oxidoreductase Function 4 iron, 4 sulfur cluster binding.Iron ion binding.Pyruvate synthase activity.Thiamine pyrophosphate binding. Related diseases Intellectual developmental disorder, autosomal dominant 62 (MRD62) [MIM:618793]: An autosomal dominant form of intellectual disability, a disorder characterized by significantly below average general intellectual functioning associated with impairments in adaptive behavior and manifested during the developmental period. MRD62 is characterized by mild to moderately impaired intellectual development. {ECO:0000269|PubMed:27479843, ECO:0000269|PubMed:29460436}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02410; DB01987; DB00507 Interacts with NA EC number 1.2.7.1 Uniprot keywords 3D-structure; 4Fe-4S; Calcium; Cytoplasm; Direct protein sequencing; Disulfide bond; Electron transport; Iron; Iron-sulfur; Magnesium; Metal-binding; Oxidoreductase; Pyruvate; Thiamine pyrophosphate; Transport Protein physicochemical properties Chain ID A,B Molecular weight (Da) 115569 Length 1065 Aromaticity 0.09 Instability index 31.51 Isoelectric point 6.32 Charge (pH=7) -5.62 2D Binding mode Binding energy (Kcal/mol) -7.26  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence GKKMMTTDGNTATAHVAYAMSEVAAIYPITPSSTMGEEADDWAAQGRKNIFGQTLTIREMQSEAGAAGAVHGALAAGALTTTFTASQGLLLMIPNMYKISGELLPGVFHVTARAIAAHALSIFGDHQDIYAARQTGFAMLASSSVQEAHDMALVAHLAAIESNVPFMHFFDGFRTSHEIQKIEVLDYADMASLVNQKALAEFRAKSPGIVAEYMQKVASLTGRSYKLFDYVGAPDAERVIVSMGSSCETIEEVINHLAAKGEKIGLIKVRLYRPFVSEAFFAALPASAKVITVLDRTKEPGAPGDPLYLDVCSAFVERGEAMPKILAGRYGLGSKEFSPAMVKSVYDNMSGAKKNHFTVGIEDDVTGTSLPVDNAFADTTPKGTIQCQFWGLGADGTVGANKQAIKIIGDNTDLFAQGYFSYDSKKSGGITISHLRFGEKPIQSTYLVNRADYVACHNPAYVGIYDILEGIKDGGTFVLNSPWSSLEDMDKHLPSGIKRTIANKKLKFYNIDAVKIATDVGLGGRINMIMQTAFFKLAGVLPFEKAVDLLKKSIHKAYGKKGEKIVKMNTDAVDQAVTSLQEFKYPDSWKDAPAETKAEPMTNEFFKNVVKPILTQQGDKLPVSAFEADGRFPLGTSQFEKRGVAINVPQWVPENCIQCNQCAFVCPHSAILPVLAKEEELVGAPANFTALEAKGKELKGYKFRIQINTLDCMGCGNCADICPPKEKALVMQPLDTQRDAQVPNLEYAARIPVKSEVLPRDSLKGSQFQEPLMEFSGACSGCGETPYVRVITQLFGERMFIANATGCSSIWGASAPSMPYKTNRLGQGPAWGNSLFEDAAEYGFGMSVWIFGGDGWAYDIGYGGLDHVLASGEDVNVFVMDTEVYSNTGGQSSKATPTGAVAKFAAAGKRTGKKDLARMVMTYGYVYVATVSMGYSKQQFLKVLKEAESFPGPSLVIAYATCINQGLRKGMGKSQDVMNTAVKSGYWPLFRYDPRLAAQGKNPFQLDSKAPDGSVEEFLMAQNRFAVLDRSFPEDAKRLRAQVAHELDVRFKELEHMAATNIFES Hydrogen bonds contact Hydrophobic contact | ||||
| 62 | Leucine carboxyl methyltransferase 1 | 3IEI | 5.32 | |
Target general information Gen name LCMT1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms CGI-68;LCMT Protein family Methyltransferase superfamily, LCMT family Biochemical class Transferase Function Protein C-terminal carboxyl O-methyltransferase activity.Protein C-terminal leucine carboxyl O-methyltransferase activity.S-adenosylmethionine-dependent methyltransferase activity. Related diseases Neurodevelopmental disorder, mitochondrial, with abnormal movements and lactic acidosis, with or without seizures (NEMMLAS) [MIM:617710]: An autosomal recessive, mitochondrial disorder with a broad phenotypic spectrum ranging from severe neonatal lactic acidosis, encephalomyopathy and early death to an attenuated course with milder manifestations. Clinical features include delayed psychomotor development, intellectual disability, hypotonia, dystonia, ataxia, and spasticity. Severe combined respiratory chain deficiency may be found in severely affected individuals. {ECO:0000269|PubMed:28236339, ECO:0000269|PubMed:28650581, ECO:0000269|PubMed:28905505, ECO:0000269|PubMed:30920170, ECO:0000269|PubMed:35074316}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Parkinsonism-dystonia 3, childhood-onset (PKDYS3) [MIM:619738]: An autosomal recessive neurodegenerative disorder with onset in infancy or early childhood. Affected individuals present with progressive movement abnormalities, including parkinsonism with tremor, dystonia, myoclonus ataxia, and hyperkinetic movements such as ballismus. The parkinsonism features may be responsive to treatment with levodopa, although many patients develop levodopa-induced dyskinesia. Some patients may have mild cognitive impairment or psychiatric disturbances. {ECO:0000269|PubMed:29120065, ECO:0000269|PubMed:31970218, ECO:0000269|PubMed:34890876}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00149 Interacts with P51116 EC number 2.1.1.233 Uniprot keywords 3D-structure; Alternative splicing; Methyltransferase; Proteomics identification; Reference proteome; S-adenosyl-L-methionine; Transferase Protein physicochemical properties Chain ID A,B,C,D,E,F,G,H Molecular weight (Da) 35803 Length 310 Aromaticity 0.08 Instability index 42.77 Isoelectric point 6.13 Charge (pH=7) -3.58 2D Binding mode Binding energy (Kcal/mol) -7.26  Molscript Map 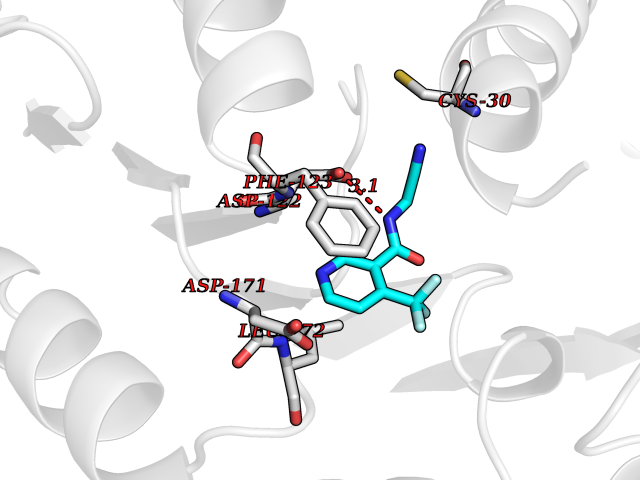 Pymol Map 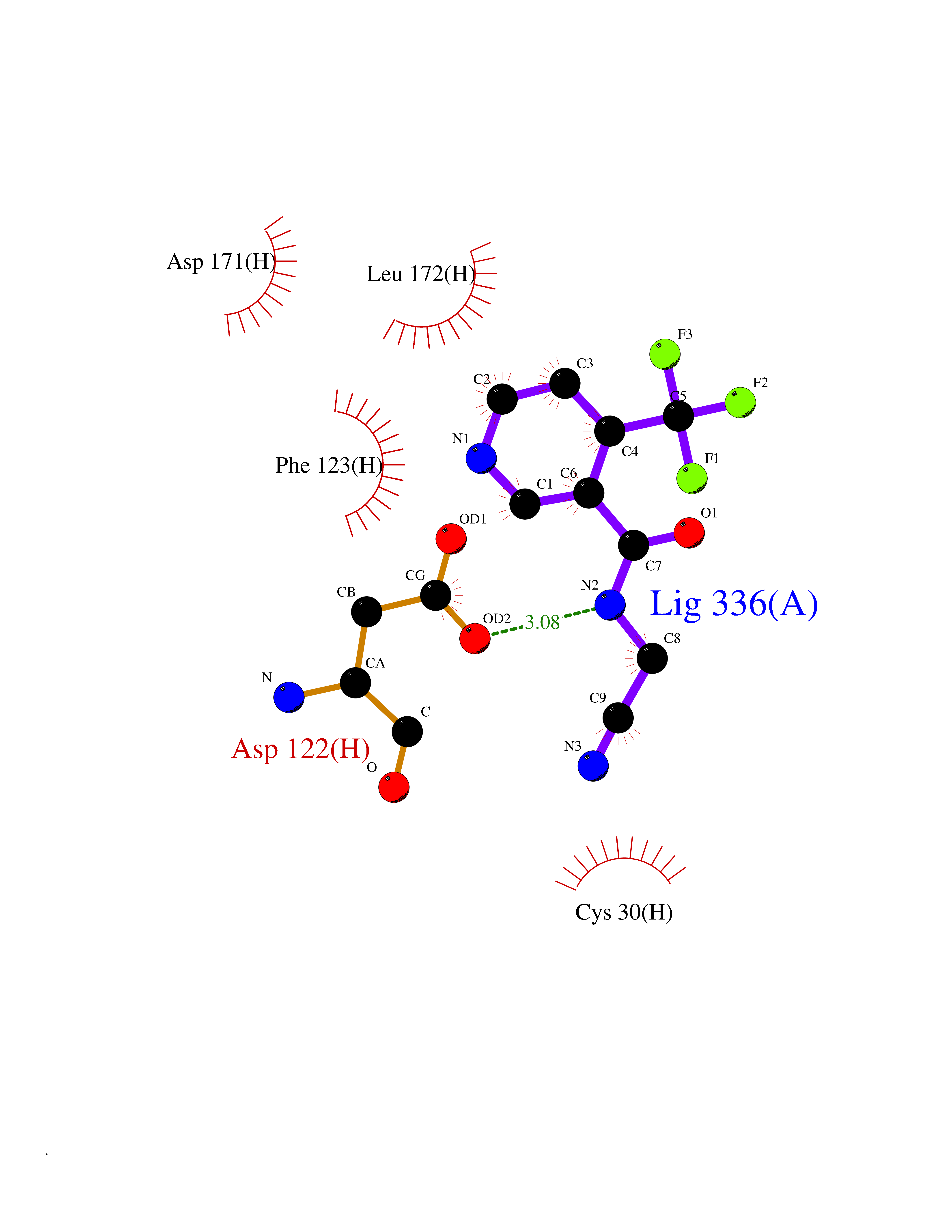 Ligplot Map 3D Binding mode Sequence GVRGTCEDASLCKRFAVSIGYWHDPYIQHFVRLSKERKAPEINRGYFARVHGVSQLIKAFLRKTECHCQIVNLGAGMDTTFWRLKDEDLLSSKYFEVDFPMIVTRKLHSIKCKPPLSSPILELHSEDTLQMDGHILDSKRYAVIGADLRDLSELEEKLKKCNMNTQLPTLLIAECVLVYMTPEQSANLLKWAANSFERAMFINYEQVNMGDRFGQIMIENLRRRQCDLAGVETCKSLESQKERLLSNGWETASAVDMMELYNRLPRAEVSRIESLEFLDEMELLEQLMRHYCLCWATKGGNELGLKEITY Hydrogen bonds contact Hydrophobic contact | ||||
| 63 | Plasmodium Dihydroorotate dehydrogenase (Malaria DHOdehase) | 1TV5 | 5.32 | |
Target general information Gen name Malaria DHOdehase Organism Plasmodium falciparum (isolate 3D7) Uniprot ID TTD ID Synonyms PFF0160c; Mitochondrially bound dihydroorotate-ubiqui oxidoreductase; Dihydroorotate oxidase of Plasmodium falciparum; Dihydroorotate dehydrogenase of Plasmodium falciparum; DHOdehase of Plasmodium fa Protein family Dihydroorotate dehydrogenase family, Type 2 subfamily Biochemical class CH-CH donor oxidoreductase Function Catalyzes the conversion of dihydroorotate to orotate with quinone as electron acceptor. Related diseases Combined oxidative phosphorylation deficiency 33 (COXPD33) [MIM:617713]: An autosomal recessive disorder caused by multiple mitochondrial respiratory chain defects and impaired mitochondrial energy metabolism. Clinical manifestations are highly variable. Affected infants present with cardiomyopathy accompanied by multisystemic features involving liver, kidney, and brain. Death in infancy is observed in some patients. Children and adults present with myopathy and progressive external ophthalmoplegia. {ECO:0000269|PubMed:28942965}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01117 Interacts with NA EC number EC 1.3.5.2 Uniprot keywords 3D-structure; Flavoprotein; FMN; Membrane; Mitochondrion; Mitochondrion inner membrane; Oxidoreductase; Pyrimidine biosynthesis; Reference proteome; Transit peptide; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 41846.8 Length 371 Aromaticity 0.1 Instability index 37.25 Isoelectric point 8.21 Charge (pH=7) 3.13 2D Binding mode Binding energy (Kcal/mol) -7.25  Molscript Map 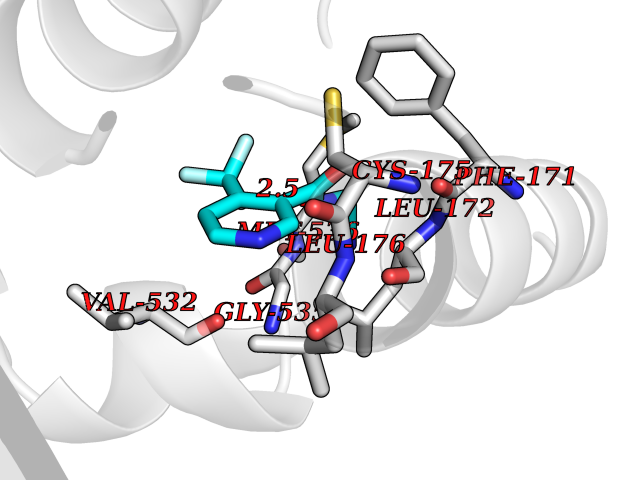 Pymol Map 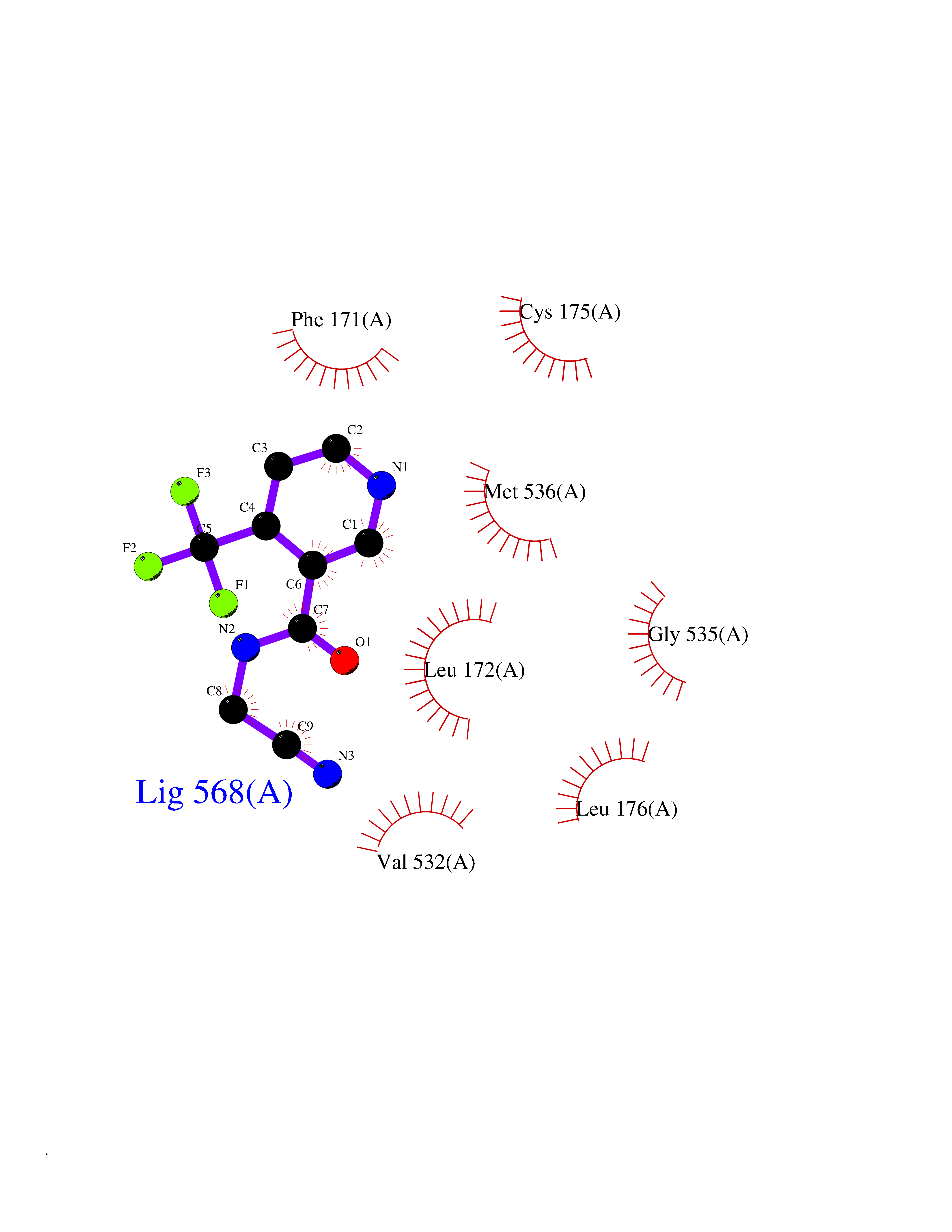 Ligplot Map 3D Binding mode Sequence FESYNPEFFLYDIFLKFCLKYIDGEICHDLFLLLGKYNILPYDTSNDSIYACTNIKHLDFINPFGVAAGFDKNGVCIDSILKLGFSFIEIGTITPRGQTGNAKPRIFRDVESRSIINSCGFNNMGCDKVTENLILFRKRQEEDKLLSKHIVGVSIGKNKDTVNIVDDLKYCINKIGRYADYIAINVSSPNTPGLRDNQEAGKLKNIILSVKEEIDNLEFLWFNTTKKKPLVFVKLAPDLNQEQKKEIADVLLETNIDGMIISNTTTQINDIKSFENKKGGVSGAKLKDISTKFICEMYNYTNKQIPIIASGGIFSGLDALEKIEAGASVCQLYSCLVFNGMKSAVQIKRELNHLLYQRGYYNLKEAIGRKH Hydrogen bonds contact Hydrophobic contact | ||||
| 64 | Bacterial Cystathionine beta-lyase (Bact metC) | 4ITX | 5.32 | |
Target general information Gen name Bact metC Organism Escherichia coli (strain K12) Uniprot ID TTD ID Synonyms Cysteine-S-conjugate beta-lyase MetC; Cysteine lyase MetC; Cysteine desulfhydrase MetC; Cystathionine beta-lyase MetC; CL; CBL; Beta-cystathionase MetC; Bacterial CD Protein family Trans-sulfuration enzymes family Biochemical class Carbon-sulfur lyases Function Primarily catalyzes the cleavage of cystathionine to homocysteine, pyruvate and ammonia during methionine biosynthesis. Also exhibits cysteine desulfhydrase activity, producing sulfide from cysteine. In addition, under certain growth conditions, exhibits significant alanine racemase coactivity. Related diseases Coronary artery disease, autosomal dominant, 2 (ADCAD2) [MIM:610947]: A common heart disease characterized by reduced or absent blood flow in one or more of the arteries that encircle and supply the heart. Its most important complication is acute myocardial infarction. {ECO:0000269|PubMed:17332414, ECO:0000269|PubMed:23703864}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Tooth agenesis, selective, 7 (STHAG7) [MIM:616724]: An autosomal dominant form of selective tooth agenesis, a common anomaly characterized by the congenital absence of one or more teeth. Selective tooth agenesis without associated systemic disorders has sometimes been divided into 2 types: oligodontia, defined as agenesis of 6 or more permanent teeth, and hypodontia, defined as agenesis of less than 6 teeth. The number in both cases does not include absence of third molars (wisdom teeth). {ECO:0000269|PubMed:26387593}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number EC 4.4.1.13 Uniprot keywords 3D-structure; Amino-acid biosynthesis; Cytoplasm; Direct protein sequencing; Lyase; Methionine biosynthesis; Pyridoxal phosphate; Reference proteome Protein physicochemical properties Chain ID A Molecular weight (Da) 42756.3 Length 391 Aromaticity 0.08 Instability index 27.11 Isoelectric point 6.01 Charge (pH=7) -6.11 2D Binding mode Binding energy (Kcal/mol) -7.26  Molscript Map 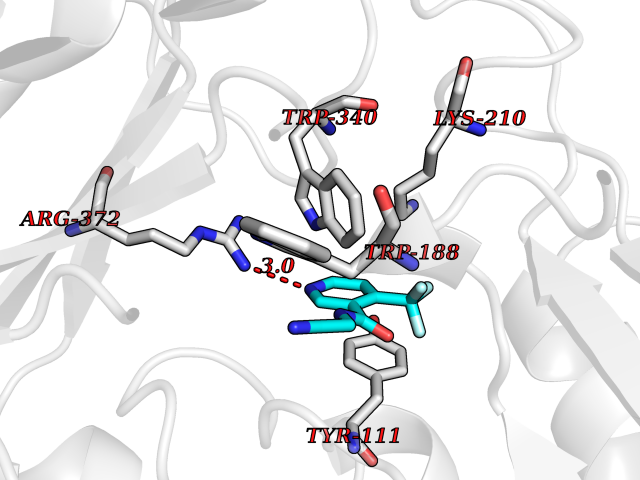 Pymol Map 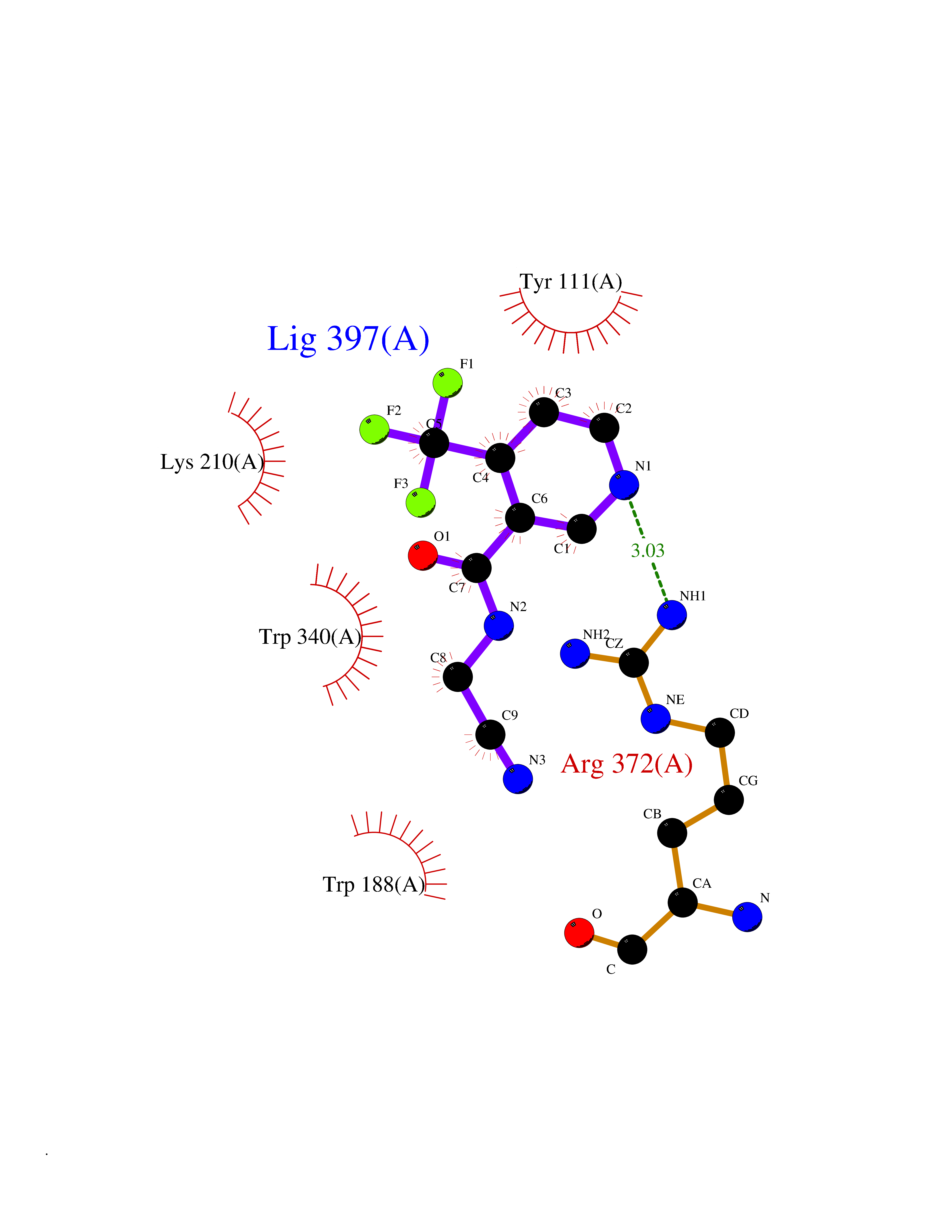 Ligplot Map 3D Binding mode Sequence KLDTQLVNAGRSKKYTLGAVNSVIQRASSLVFDSVEAKKHATRNRANGELFYGRRGTLTHFSLQQAMCELEGGAGCVLFPCGAAAVANSILAFIEQGDHVLMTNTAYESSQDFCSKILSKLGVTTSWFDPLIGADIVKHLQPNTKIVFLESPGSITMEVHDVPAIVAAVRSVVPDAIIMIDNTWAAGVLFKALDFGIDVSIQAATKYLVGHSDAMIGTAVCNARCWEQLRENAYLMGQMVDADTAYITSRGLRTLGVRLRQHHESSLKVAEWLAEHPQVARVNHPALPGSKGHEFWKRDFTGSSGLFSFVLKKKLNNEELANYLDNFSLFSMAYSWGGYESLILANQPEHIAAIRPQGEIDFSGTLIRLHIGLEDVDDLIADLDAGFARIV Hydrogen bonds contact Hydrophobic contact | ||||
| 65 | Phenylethanolamine N-methyltransferase (PNMT) | 2G72 | 5.32 | |
Target general information Gen name PNMT Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms PNMTase; PENT; Noradrenaline N-methyltransferase Protein family Class I-like SAM-binding methyltransferase superfamily, NNMT/PNMT/TEMT family Biochemical class NA Function Converts noradrenaline to adrenaline. Related diseases A chromosomal aberration involving TRIM24/TIF1 is found in papillary thyroid carcinomas (PTCs). Translocation t(7;10)(q32;q11) with RET. The translocation generates the TRIM24/RET (PTC6) oncogene. {ECO:0000269|PubMed:10439047}. Drugs (DrugBank ID) DB08129; DB08128; DB07739; DB07798; DB07747; DB03468; DB08550; DB03824; DB04273; DB07906; DB07597; DB09571; DB00968; DB08631; DB01752; DB08654 Interacts with Q9P2G9-2; Q8TBB1 EC number EC 2.1.1.28 Uniprot keywords 3D-structure; Catecholamine biosynthesis; Direct protein sequencing; Methyltransferase; Phosphoprotein; Proteomics identification; Reference proteome; S-adenosyl-L-methionine; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 29198.9 Length 264 Aromaticity 0.09 Instability index 54.33 Isoelectric point 5.91 Charge (pH=7) -3.69 2D Binding mode Binding energy (Kcal/mol) -7.26  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence APGQAAVASAYQRFEPRAYLRNNYAPPRGDLCNPNGVGPWKLRCLAQTFATGEVSGRTLIDIGSGPTVYQLLSACSHFEDITMTDFLEVNRQELGRWLQEEPGAFNWSMYSQHACLIEGKGECWQDKERQLRARVKRVLPIDVHQPQPLGAGSPAPLPADALVSAFCLEAVSPDLASFQRALDHITTLLRPGGHLLLIGALEESWYLAGEARLTVVPVSEEEVREALVRSGYKVRDLRTYIMPAHLQTGVDDVKGVFFAWAQKV Hydrogen bonds contact Hydrophobic contact | ||||
| 66 | Corynebacterium Pup-protein ligase (Cory pafA) | 4BJR | 5.32 | |
Target general information Gen name Cory pafA Organism Corynebacterium glutamicum (strain ATCC 13032 / DSM 20300 / JCM 1318 / BCRC 11384 / CCUG 27702 / LMG 3730 / NBRC 12168 / NCIMB 10025 / NRRL B-2784 / 534) Uniprot ID TTD ID Synonyms Pup-conjugating enzyme; Pup--protein ligase; Proteasome accessory factor A Protein family Pup ligase/Pup deamidase family, Pup-conjugating enzyme subfamily Biochemical class NA Function Catalyzes the covalent attachment of the prokaryotic ubiquitin-like protein modifier Pup to the proteasomal substrate proteins, thereby targeting them for proteasomal degradation. This tagging system is termed pupylation. The ligation reaction involves the side-chain carboxylate of the C-terminal glutamate of Pup and the side-chain amino group of a substrate lysine. Related diseases Anemia, non-spherocytic hemolytic, due to G6PD deficiency (NSHA) [MIM:300908]: A disease characterized by G6PD deficiency, acute hemolytic anemia, fatigue, back pain, and jaundice. In most patients, the disease is triggered by an exogenous agent, such as some drugs, food, or infection. Increased unconjugated bilirubin, lactate dehydrogenase, and reticulocytosis are markers of the disorder. Although G6PD deficiency can be life-threatening, most patients are asymptomatic throughout their life. {ECO:0000269|PubMed:12524354, ECO:0000269|PubMed:1303180, ECO:0000269|PubMed:1303182, ECO:0000269|PubMed:1536798, ECO:0000269|PubMed:1611091, ECO:0000269|PubMed:1889820, ECO:0000269|PubMed:1945893, ECO:0000269|PubMed:20007901, ECO:0000269|PubMed:26479991, ECO:0000269|PubMed:2836867, ECO:0000269|PubMed:2912069, ECO:0000269|PubMed:30988594, ECO:0000269|PubMed:38066190, ECO:0000269|PubMed:7858267, ECO:0000269|PubMed:7959695, ECO:0000269|PubMed:8193373, ECO:0000269|PubMed:8490627, ECO:0000269|PubMed:8533762, ECO:0000269|PubMed:8733135, ECO:0000269|PubMed:9452072}. The disease is caused by variants affecting the gene represented in this entry. Deficiency of G6PD is associated with hemolytic anemia in two different situations. First, in areas in which malaria has been endemic, G6PD-deficiency alleles have reached high frequencies (1% to 50%) and deficient individuals, though essentially asymptomatic in the steady state, have a high risk of acute hemolytic attacks. Secondly, sporadic cases of G6PD deficiency occur at a very low frequencies, and they usually present a more severe phenotype. Several types of NSHA are recognized. Class-I variants are associated with severe NSHA; class-II have an activity <10% of normal; class-III have an activity of 10% to 60% of normal; class-IV have near normal activity. Drugs (DrugBank ID) NA Interacts with NA EC number NA Uniprot keywords 3D-structure; ATP-binding; Ligase; Magnesium; Metal-binding; Nucleotide-binding; Reference proteome; Ubl conjugation pathway Protein physicochemical properties Chain ID A,B Molecular weight (Da) 110572 Length 994 Aromaticity 0.07 Instability index 42.33 Isoelectric point 5.26 Charge (pH=7) -34.19 2D Binding mode Binding energy (Kcal/mol) -7.26  Molscript Map  Pymol Map 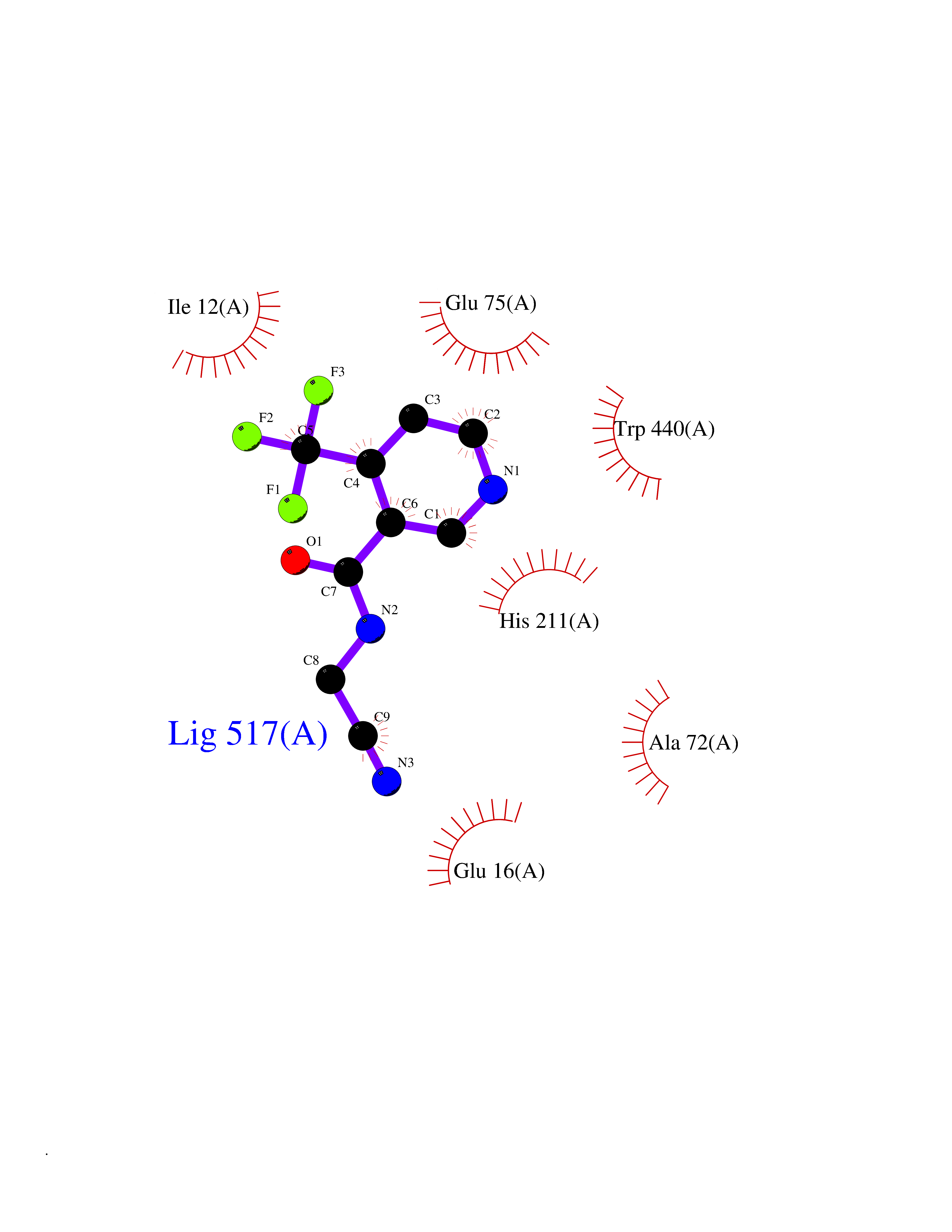 Ligplot Map 3D Binding mode Sequence TVESALTRRIMGIETEYGLTFVDGDSKKLRPDEIARRMFRPIVEKYSSSNIFIPNGSRLYLNVGSHPEYATAECDNLTQLINFEKAGDVIADRMAVDAEESLAKEDIAGQVYLFKNNVDSVGNSYGCHENYLVGRSMPLKALGKRLMPFLITRQLICGAGRIHHPNPSFPLGYCISQRSDHVWEGVSSASRPIINTRDEPHADSHSYRRLHVIVGDANMAEPSIALKVGSTLLVLEMIEADFGLPSLELANDIASIREISRDATGSTLLSLKDGTTMTALQIQQVVFEHASKWLEQRPEPEFSGTSNTEMARVLDLWGRMLKAIESGDFSEVDTEIDWVIKKKLIDRFIQRGNLGLDDPKLAQVDLTYHDIRPGRGLFSVLQSRGMIKRWTTDEAILAAVDTAPDTTRAHLRGRILKAADTLGVPVTVDWMRHKVNRPEPQSVELGDPFSAVNSEVDQLIEYMTVHAGSASGTSLLDEIDGLLENNAEEFVRSYVQKGGETVESALTRRIMGIETEYGLTFVDGDSKKLRPDEIARRMFRPIVEKYSSSNIFIPNGSRLYLNVGSHPEYATAECDNLTQLINFEKAGDVIADRMAVDAEESLAKEDIAGQVYLFKNNVDSVGNSYGCHENYLVGRSMPLKALGKRLMPFLITRQLICGAGRIHHPNPSFPLGYCISQRSDHVWEGVSSASRPIINTRDEPHADSHSYRRLHVIVGDANMAEPSIALKVGSTLLVLEMIEADFGLPSLELANDIASIREISRDATGSTLLSLKDGTTMTALQIQQVVFEHASKWLEQRPEPEFSGTSNTEMARVLDLWGRMLKAIESGDFSEVDTEIDWVIKKKLIDRFIQRGNLGLDDPKLAQVDLTYHDIRPGRGLFSVLQSRGMIKRWTTDEAILAAVDTAPDTTRAHLRGRILKAADTLGVPVTVDWMRHKVNRPEPQSVELGDPFSAVNSEVDQLIEYMTVHASLLDEIDGLLENNAEEFVRSYVQKGGE Hydrogen bonds contact Hydrophobic contact | ||||
| 67 | Neuronal acetylcholine receptor alpha-3/beta-4 (CHRNA3/B4) | 6PV7 | 5.32 | |
Target general information Gen name CHRNA3-CHRNB4 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Neuronal acetylcholine receptor Protein family Ligand-gated ion channel (TC 1.A.9) family, Acetylcholine receptor (TC 1.A.9.1) subfamily, Alpha-3/CHRNA3 sub-subfamily Biochemical class Neurotransmitter receptor Function A type of nicotinic acetylcholine receptor, consisting of 3 and 4 subunits. Related diseases Bladder dysfunction, autonomic, with impaired pupillary reflex and secondary CAKUT (BAIPRCK) [MIM:191800]: An autosomal recessive disease characterized by impaired innervation and autonomic dysfunction of the urinary bladder, hydronephrosis, vesicoureteral reflux, small kidneys, recurrent urinary tract infections, and progressive renal insufficiency. Additional autonomic features are impaired pupillary reflex and orthostatic hypotension. The disease manifests in utero or early childhood. {ECO:0000269|PubMed:31708116}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00915; DB01156; DB00237; DB00565; DB09028; DB00514; DB07720; DB00898; DB00472; DB05710; DB01227; DB00848; DB00333; DB00184; DB01090; DB00202; DB01273 Interacts with NA EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disease variant; Disulfide bond; Endoplasmic reticulum; Glycoprotein; Golgi apparatus; Ion channel; Ion transport; Ligand-gated ion channel; Membrane; Phosphoprotein; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Signal; Synapse; Transmembrane; Transmembrane helix; Transport; Ubl conjugation Protein physicochemical properties Chain ID A,B Molecular weight (Da) 89661.2 Length 775 Aromaticity 0.12 Instability index 35.11 Isoelectric point 6.07 Charge (pH=7) -5.67 2D Binding mode Binding energy (Kcal/mol) -7.25 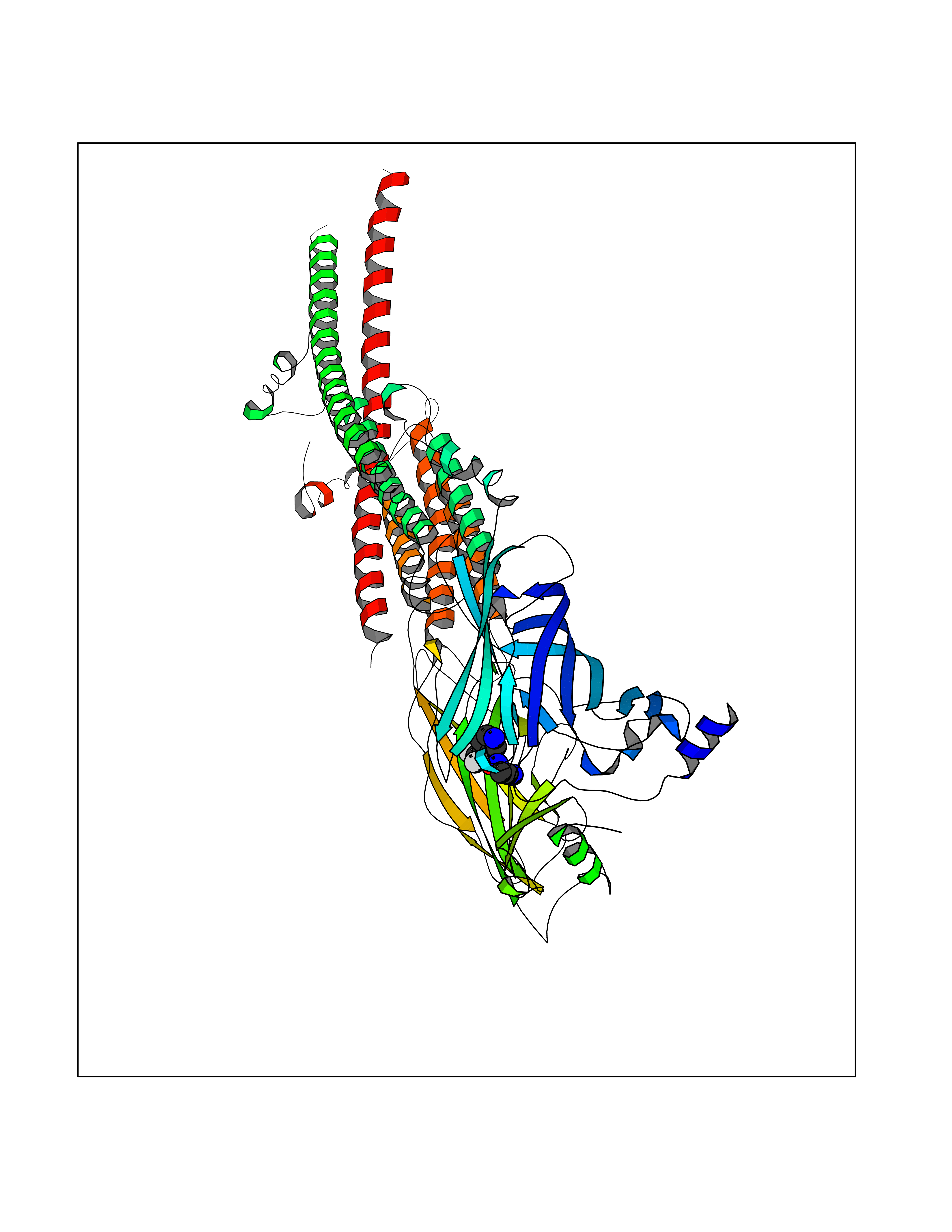 Molscript Map  Pymol Map 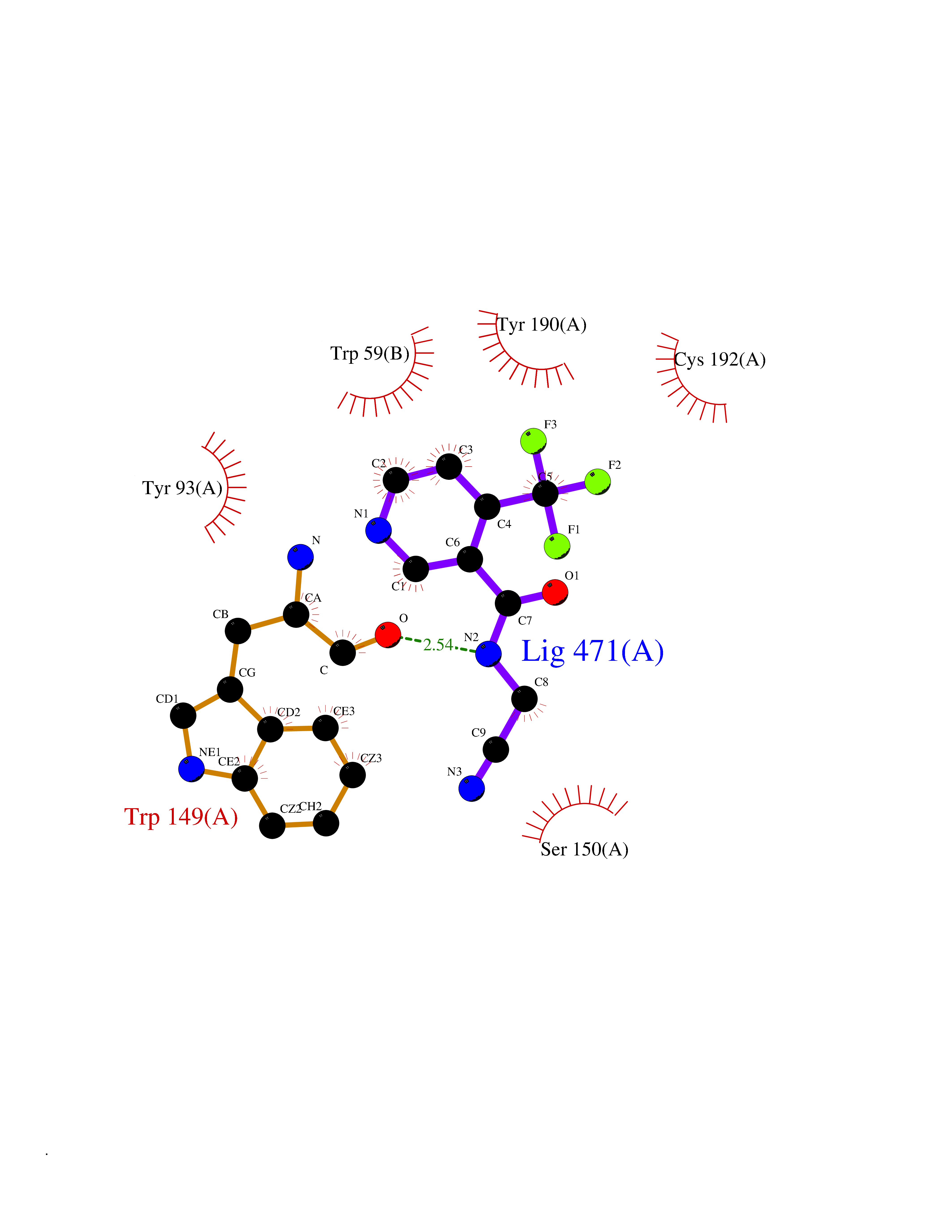 Ligplot Map 3D Binding mode Sequence SEAEHRLFERLFEDYNEIIRPVANVSDPVIIHFEVSMSQLVKVDEVNQIMETNLWLKQIWNDYKLKWNPSDYGGAEFMRVPAQKIWKPDIVLYNNAVGDFQVDDKTKALLKYTGEVTWIPPAIFKSSCKIDVTYFPFDYQNCTMKFGSWSYDKAKIDLVLIGSSMNLKDYWESGEWAIIKAPGYKHDIKYNCCEEIYPDITYSLYIRRLPLFYTINLIIPCLLISFLTVLVFYLPSDCGEKVTLCISVLLSLTVFLLVITETIPSTSLVIPLIGEYLLFTMIFVTLSIVITVFVLNVHYRTPTTHTMPSWVKTVFLNLLPRVMFMTRIKEAIQSVKYIAENMKAQNEAKEIQDDWKYVAMVIDRIFLWVFTLVCILGTAGLFLQPLMRVANAEEKLMDDLLNKTRYNNLIRPATSSSQLISIKLQLSLAQLISVNEREQIMTTNVWLKQEWTDYRLTWNSSRYEGVNILRIPAKRIWLPDIVLYNNADGTYEVSVYTNLIVRSNGSVLWLPPAIYKSACKIEVKYFPFDQQNCTLKFRSWTYDHTEIDMVLMTPTASMDDFTPSGEWDIVALPGRRTVNPQDPSYVDVTYDFIIKRKPLFYTINLIIPCVLTTLLAILVFYLPSDCGEKMTLCISVLLALTFFLLLISKIVPPTSLDVPLIGKYLMFTMVLVTFSIVTSVCVLNVHHRSPSTHTMAPWVKRCFLHKLPTFLFMKRRQDVQEALEGVSFIAQHMKNDDEDQSVVEDWKYVAMVVDRLFLWVFMFVCVLGTVGLFLP Hydrogen bonds contact Hydrophobic contact | ||||
| 68 | Histone-lysine N-methyltransferase (HLNM) | 3QOW | 5.32 | |
Target general information Gen name DOT1L Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Lysine N-methyltransferase 4; KMT4; KIAA1814; Histone-lysine N-methyltransferase, H3 lysine-79 specific; Histone H3-K79 methyltransferase; H3-K79-HMTase; DOT1-like protein Protein family Class I-like SAM-binding methyltransferase superfamily, DOT1 family Biochemical class Methyltransferase Function Histone methyltransferase. Methylates 'Lys-79' of histone H3. Nucleosomes are preferred as substrate compared to free histones. Binds to DNA. Related diseases Defects in DOTL1 are associated with an autosomal dominant form of global developmental delay and intellectual disability, with or without one or more major congenital anomalies (PubMed:37827158). The patient phenotypes are characterized by central nervous system (CNS) dysfunction, such as mild motor delay and significant speech and language delay, and a range of congenital anomalies, including brain structural anomalies, cardiac defects, varied urogenital features and growth restriction (PubMed:37827158). Variants may cause a gain-of-function effect leading to an increase in cellular H3K79 methylation levels (PubMed:37827158). {ECO:0000269|PubMed:37827158}. Drugs (DrugBank ID) NA Interacts with Q03111; P42568 EC number EC 2.1.1.43 Uniprot keywords 3D-structure; Alternative splicing; Chromatin regulator; Disease variant; DNA-binding; Methyltransferase; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; S-adenosyl-L-methionine; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 37367.1 Length 322 Aromaticity 0.11 Instability index 32.71 Isoelectric point 6.03 Charge (pH=7) -5.25 2D Binding mode Binding energy (Kcal/mol) -7.26  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence KLELRLKSPVGAEPAVYPWPLPVYDKHHDAAHEIIETIRWVCEEIPDLKLAMENYLIDYDTKSFESMQRLCDKYNRAIDSIHQLWKGTTQPMKLNTRPSTGLLRHILQQVYNHSVTDPEKLNNYEPFSPEVYGETSFDLVAQMIDEIKMTDDDLFVDLGSGVGQVVLQVAAATNCKHHYGVEKADIPAKYAETMDREFRKWMKWYGKKHAEYTLERGDFLSEEWRERIANTSVIFVNNFAFGPEVDHQLKERFANMKEGGRIVSSKPFAPLNFRINSRNLSDIGTIMRVVELSPLKWTGKPVSYYLHTIDRTILENYFSSLK Hydrogen bonds contact Hydrophobic contact | ||||
| 69 | Deoxycytidine kinase (DCK) | 1P5Z | 5.31 | |
Target general information Gen name DCK Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms dCK Protein family DCK/DGK family Biochemical class Kinase Function Required for the phosphorylation of the deoxyribonucleosides deoxycytidine (dC), deoxyguanosine (dG) and deoxyadenosine (dA). Has broad substrate specificity, and does not display selectivity based on the chirality of the substrate. It is also an essential enzyme for the phosphorylation of numerous nucleoside analogs widely employed as antiviral and chemotherapeutic agents. Related diseases Microvascular complications of diabetes 5 (MVCD5) [MIM:612633]: Pathological conditions that develop in numerous tissues and organs as a consequence of diabetes mellitus. They include diabetic retinopathy, diabetic nephropathy leading to end-stage renal disease, and diabetic neuropathy. Diabetic retinopathy remains the major cause of new-onset blindness among diabetic adults. It is characterized by vascular permeability and increased tissue ischemia and angiogenesis. Disease susceptibility is associated with variants affecting the gene represented in this entry. Homozygosity for the Leu-55 allele is strongly associated with the development of retinal disease in diabetic patients. Drugs (DrugBank ID) DB02594; DB00242; DB00631; DB00987; DB01262; DB05494; DB00879; DB01073; DB00441; DB00709; DB01280; DB00642; DB04961; DB00943 Interacts with Q16854 EC number EC 2.7.1.74 Uniprot keywords 3D-structure; ATP-binding; Direct protein sequencing; Kinase; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Transferase Protein physicochemical properties Chain ID B Molecular weight (Da) 27128.5 Length 229 Aromaticity 0.13 Instability index 52.5 Isoelectric point 5.26 Charge (pH=7) -7.8 2D Binding mode Binding energy (Kcal/mol) -7.24  Molscript Map 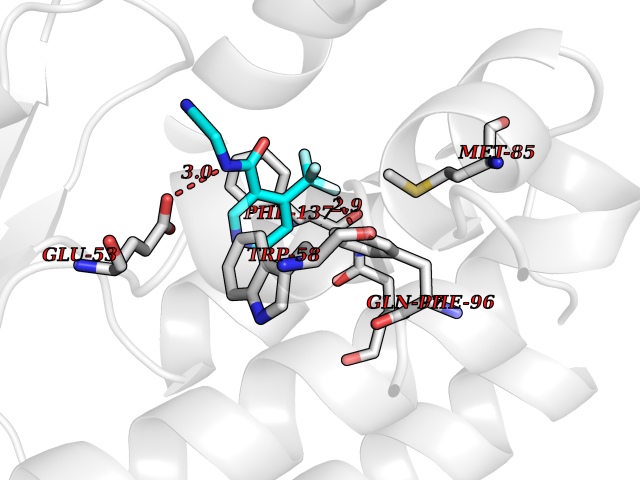 Pymol Map 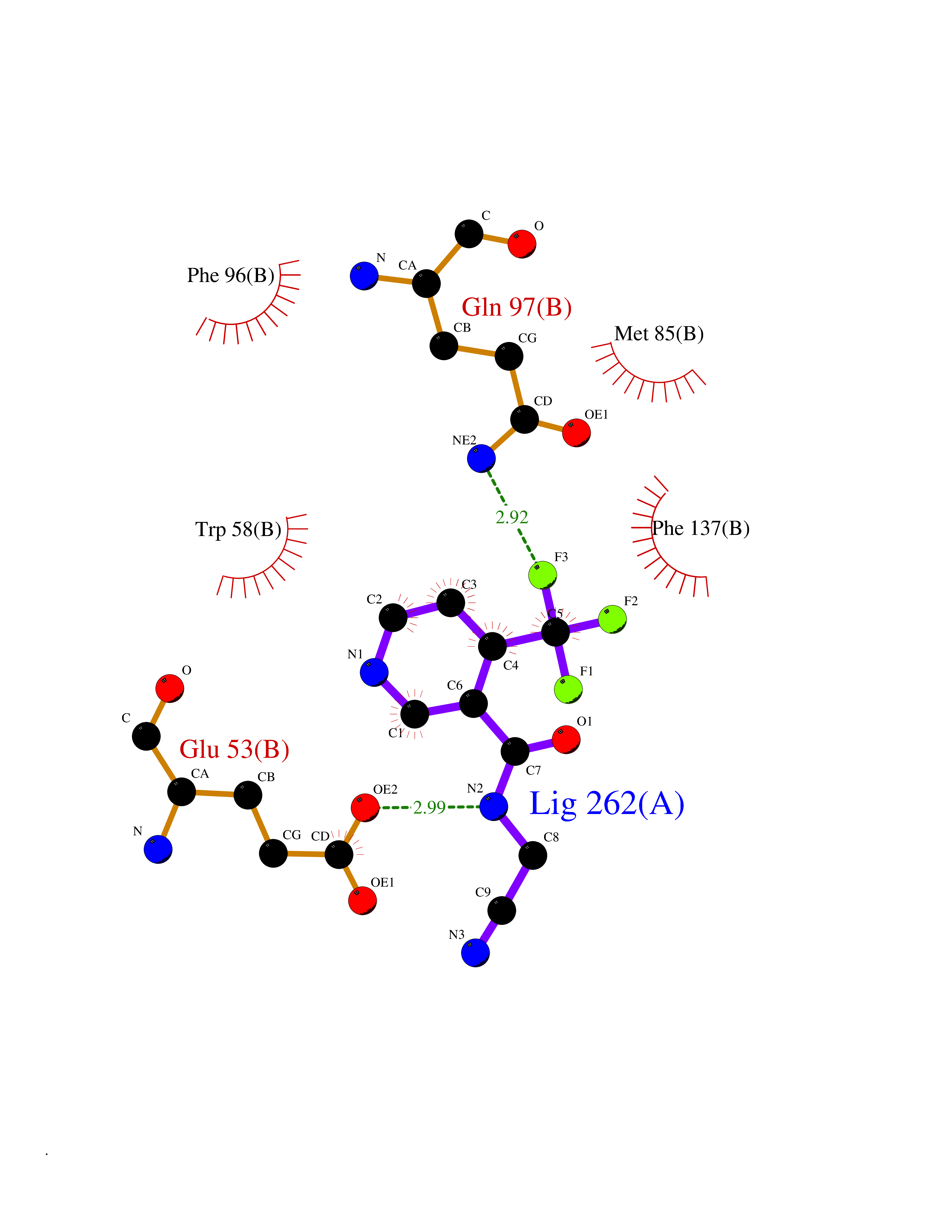 Ligplot Map 3D Binding mode Sequence RIKKISIEGNIAAGKSTFVNILKQLCEDWEVVPEPVARWCNVQSTNGGNVLQMMYEKPERWSFTFQTYACLSRIRAQLASLNGKLKDAEKPVLFFERSVYSDRYIFASNLYESECMNETEWTIYQDWHDWMNNQFGQSLELDGIIYLQATPETCLHRIYLRGRNEEQGIPLEYLEKLHYKHESWLLHRTLKTNFDYLQEVPILTLDVNEDFKDKYESLVEKVKEFLSTL Hydrogen bonds contact Hydrophobic contact | ||||
| 70 | Glutamate receptor ionotropic kainate 1 (GRIK1) | 3FV2 | 5.31 | |
Target general information Gen name GRIK1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Glutamate receptor 5; GluR5 kainate receptor; GluR5; GluR-5; GRIK1; Excitatory amino acid receptor 3; EAA3 Protein family Glutamate-gated ion channel (TC 1.A.10.1) family, GRIK1 subfamily Biochemical class Glutamate-gated ion channel Function Ionotropic glutamate receptor. L-glutamate acts as an excitatory neurotransmitter at many synapses in the central nervous system. Binding of the excitatory neurotransmitter L- glutamate induces a conformation change, leading tothe opening of the cation channel, and thereby converts the chemical signal to an electrical impulse. The receptor then desensitizes rapidly and enters a transient inactive state, characterized by the presence of bound agonist. May be involved in the transmission of light information from the retina to the hypothalamus. Related diseases Defects in PPARG can lead to type 2 insulin-resistant diabetes and hyptertension. PPARG mutations may be associated with colon cancer. {ECO:0000269|PubMed:10394368}.; DISEASE: Obesity (OBESITY) [MIM:601665]: A condition characterized by an increase of body weight beyond the limitation of skeletal and physical requirements, as the result of excessive accumulation of body fat. {ECO:0000269|PubMed:9753710}. Disease susceptibility may be associated with variants affecting the gene represented in this entry.; DISEASE: Lipodystrophy, familial partial, 3 (FPLD3) [MIM:604367]: A form of lipodystrophy characterized by marked loss of subcutaneous fat from the extremities. Facial adipose tissue may be increased, decreased or normal. Affected individuals show an increased preponderance of insulin resistance, diabetes mellitus and dyslipidemia. {ECO:0000269|PubMed:11788685, ECO:0000269|PubMed:12453919}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Glioma 1 (GLM1) [MIM:137800]: Gliomas are benign or malignant central nervous system neoplasms derived from glial cells. They comprise astrocytomas and glioblastoma multiforme that are derived from astrocytes, oligodendrogliomas derived from oligodendrocytes and ependymomas derived from ependymocytes. {ECO:0000269|PubMed:10851250}. Disease susceptibility may be associated with variants affecting the gene represented in this entry. Polymorphic PPARG alleles have been found to be significantly over-represented among a cohort of American patients with sporadic glioblastoma multiforme suggesting a possible contribution to disease susceptibility. Drugs (DrugBank ID) DB00237; DB00142; DB06354; DB00273 Interacts with NA EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disulfide bond; Glycoprotein; Ion channel; Ion transport; Ligand-gated ion channel; Membrane; Phosphoprotein; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; RNA editing; Signal; Synapse; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A,B Molecular weight (Da) 29057.1 Length 256 Aromaticity 0.1 Instability index 24.38 Isoelectric point 8.3 Charge (pH=7) 1.97 2D Binding mode Binding energy (Kcal/mol) -7.24 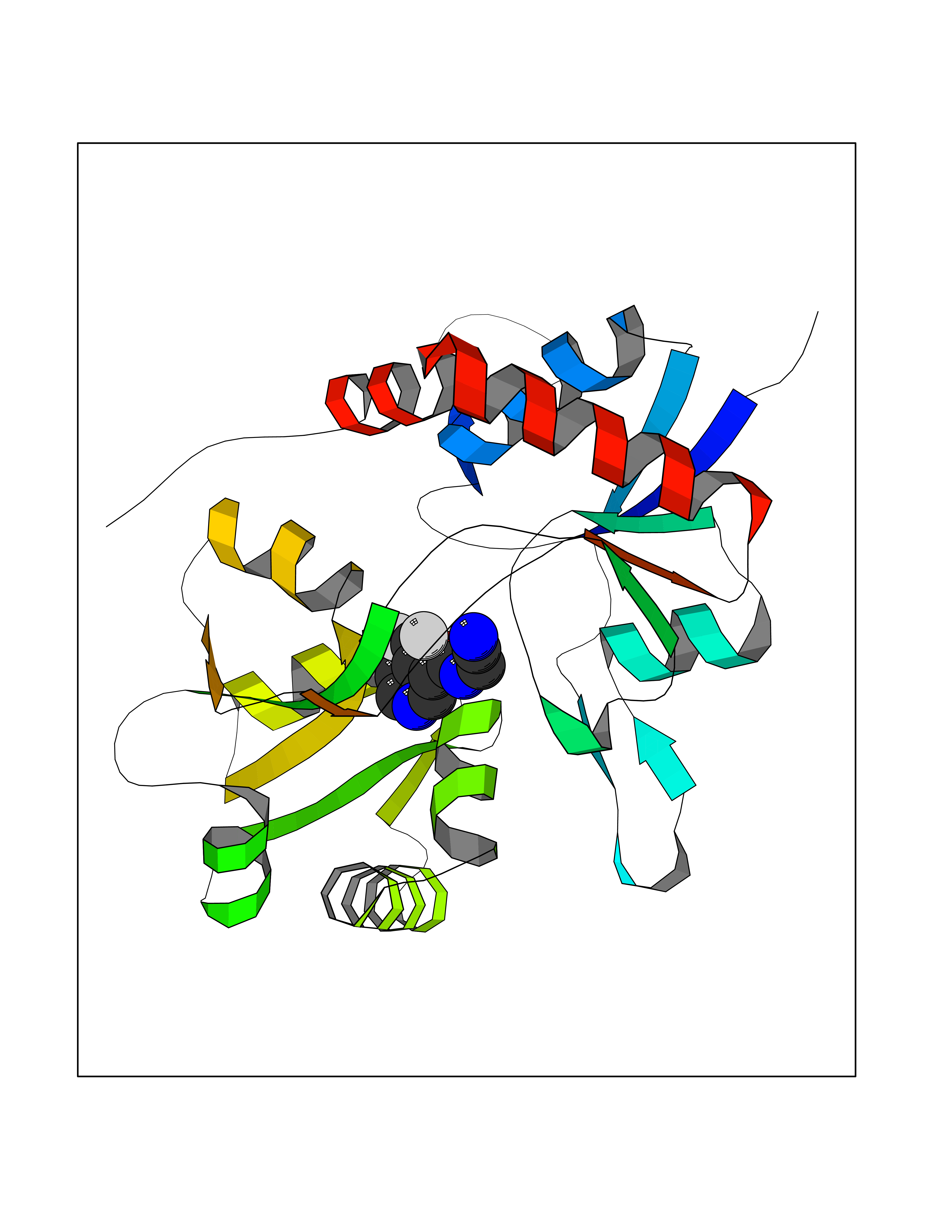 Molscript Map 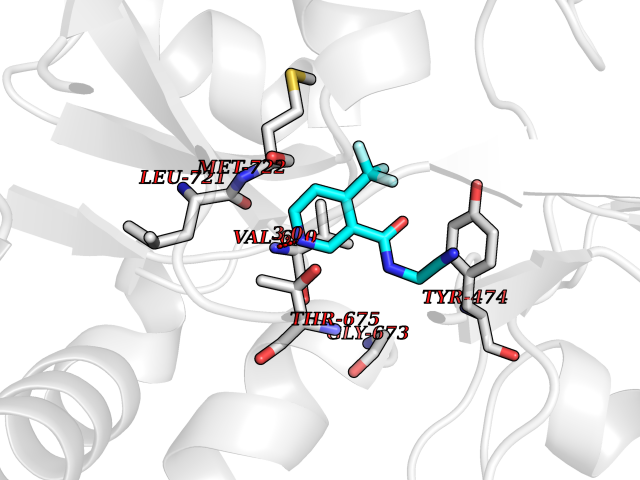 Pymol Map 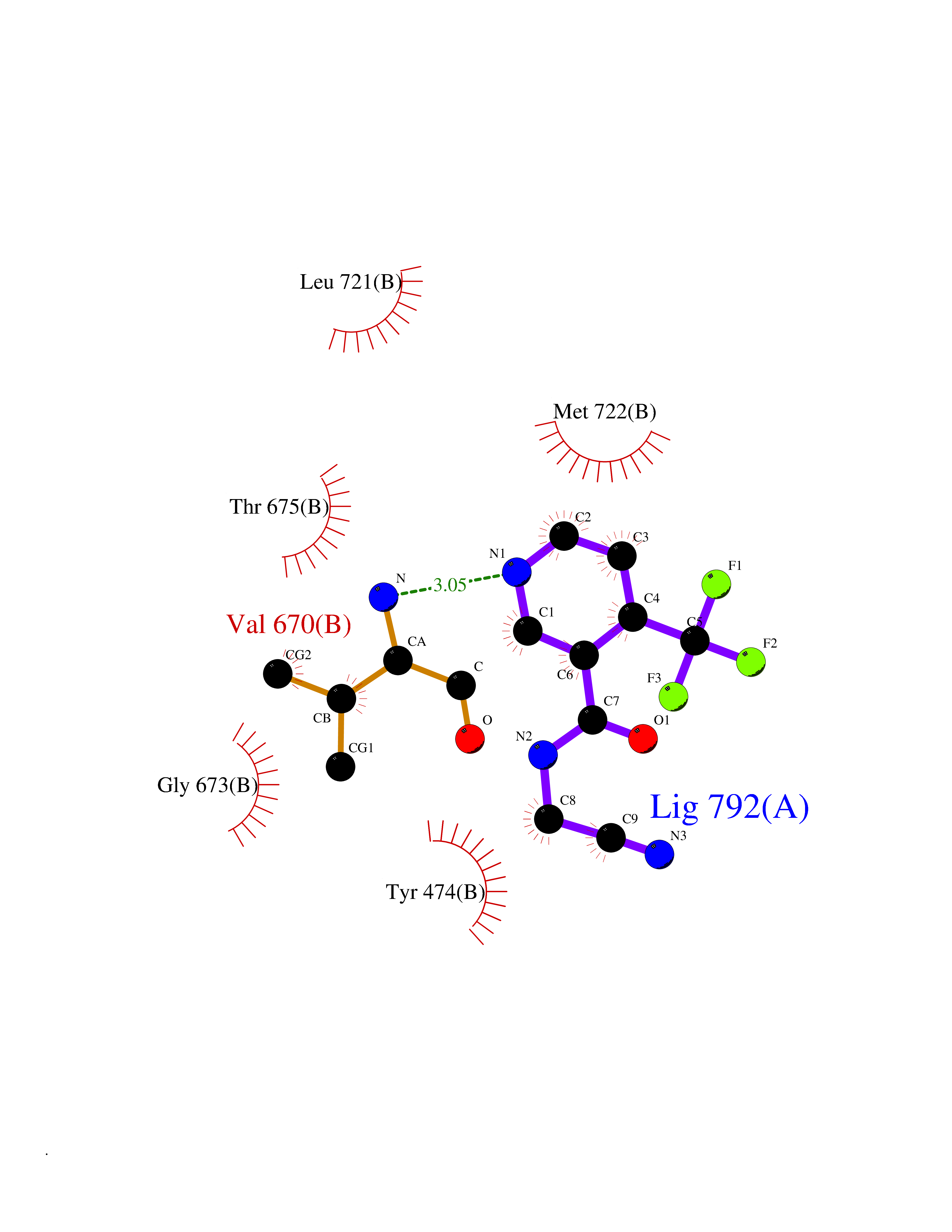 Ligplot Map 3D Binding mode Sequence ANRTLIVTTILEEPYVMYRKSDKPLYGNDRFEGYCLDLLKELSNILGFIYDVKLVPDGKYGAQNDKGEWNGMVKELIDHRADLAVAPLTITYVREKVIDFSKPFMTLGISILYRKGTPIDSADDLAKQTKIEYGAVRDGSTMTFFKKSKISTYEKMWAFMSSRQQTALVRNSDEGIQRVLTTDYALLMESTSIEYVTQRNCNLTQIGGLIDSKGYGVGTPIGSPYRDKITIAILQLQEEGKLHMMKEKWWRGNGCP Hydrogen bonds contact Hydrophobic contact | ||||
| 71 | Histone-lysine N-methyltransferase KMT5B (KMT5B) | 3S8P | 5.31 | |
Target general information Gen name KMT5B Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Lysine N-methyltransferase 5B; Lysine-specific methyltransferase 5B; Suppressor of variegation 4-20 homolog 1; Su(var)4-20 homolog 1; Suv4-20h1; [histone H4]-N-methyl-L-lysine20 N-methyltransferase KM Protein family Class V-like SAM-binding methyltransferase superfamily, Histone-lysine methyltransferase family, Suvar4-20 subfamily Biochemical class NA Function Histone methyltransferase that specifically methylates monomethylated 'Lys-20' (H4K20me1) and dimethylated 'Lys-20' (H4K20me2) of histone H4 to produce respectively dimethylated 'Lys-20' (H4K20me2) and trimethylated 'Lys-20' (H4K20me3) and thus regulates transcription and maintenance of genome integrity. In vitro also methylates unmodified 'Lys-20' (H4K20me0) of histone H4 and nucleosomes. H4 'Lys-20' trimethylation represents a specific tag for epigenetic transcriptional repression. Mainly functions in pericentric heterochromatin regions, thereby playing a central role in the establishment of constitutive heterochromatin in these regions. KMT5B is targeted to histone H3 via its interaction with RB1 family proteins (RB1, RBL1 and RBL2) (By similarity). Plays a role in myogenesis by regulating the expression of target genes, such as EID3. Facilitates TP53BP1 foci formation upon DNA damage and proficient non-homologous end-joining (NHEJ)-directed DNA repair by catalyzing the di- and trimethylation of 'Lys-20' of histone H4. May play a role in class switch reconbination by catalyzing the di- and trimethylation of 'Lys-20' of histone H4 (By similarity). Related diseases Intellectual developmental disorder, autosomal dominant 51 (MRD51) [MIM:617788]: A disorder characterized by significantly below average general intellectual functioning associated with impairments in adaptive behavior and manifested during the developmental period. {ECO:0000269|PubMed:28191889, ECO:0000269|PubMed:29276005}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q9H2G4; Q61026 EC number EC 2.1.1.361 Uniprot keywords 3D-structure; Alternative splicing; Chromatin regulator; Chromosome; Disease variant; Intellectual disability; Isopeptide bond; Metal-binding; Methyltransferase; Myogenesis; Nucleus; Proteomics identification; Reference proteome; Repressor; S-adenosyl-L-methionine; Transcription; Transcription regulation; Transferase; Ubl conjugation; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 26230.2 Length 233 Aromaticity 0.11 Instability index 42.29 Isoelectric point 5.64 Charge (pH=7) -6.07 2D Binding mode Binding energy (Kcal/mol) -7.24  Molscript Map  Pymol Map 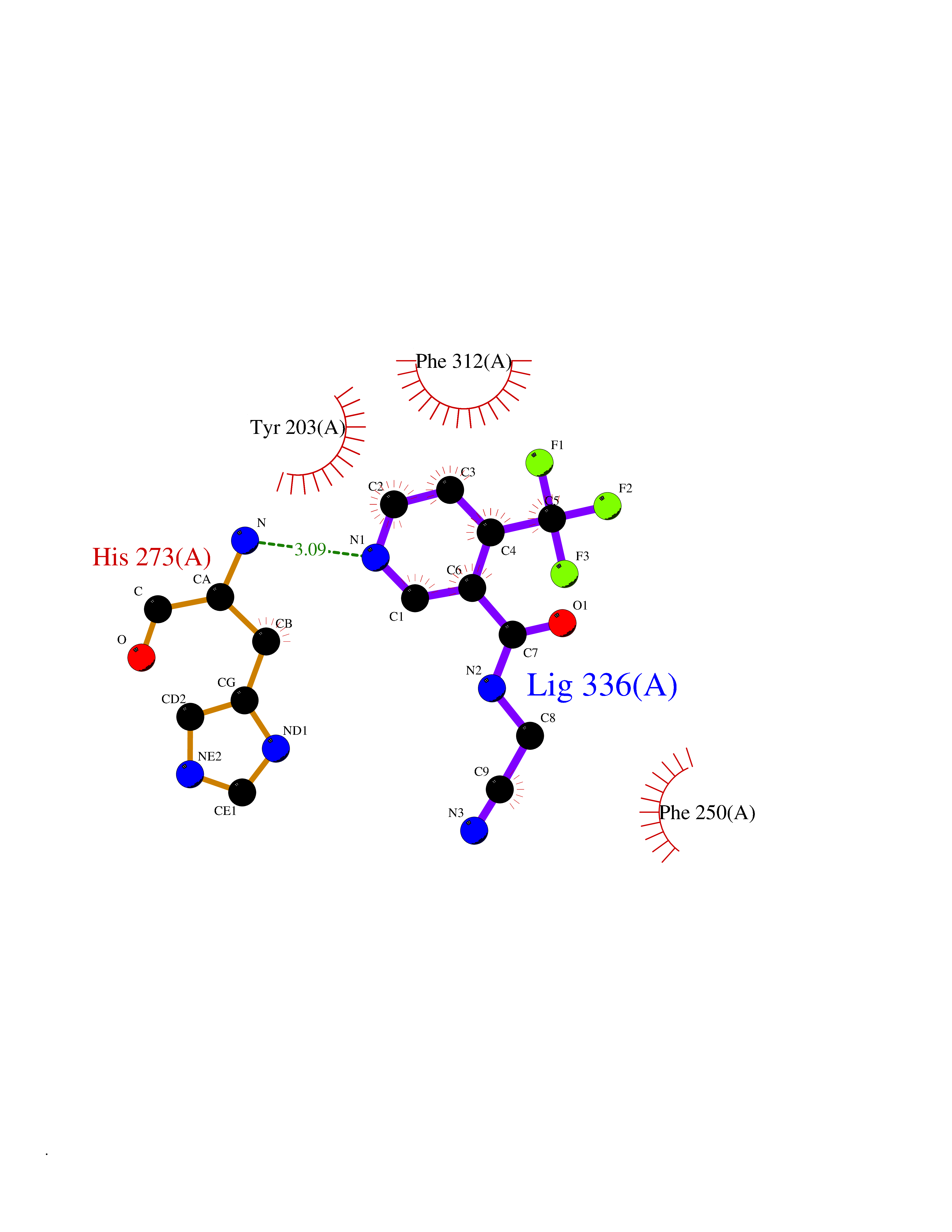 Ligplot Map 3D Binding mode Sequence XSAKELCENDDLATSLVLDPYLGFQTHKXNTRQEELKEVIERFKKDEHLEKAFKCLTSGEWARHYFLNKNKXQEKLFKEHVFIYLRXFATDSGFEILPCNRYSSEQNGAKIVATKEWKRNDKIELLVGCIAELSEIEENXLLRHGENDFSVXYSTRKNCAQLWLGPAAFINHDCRPNCKFVSTGRDTACVKALRDIEPGEEISCYYGDGFFGENNEFCECYTCERRGTGAFKS Hydrogen bonds contact Hydrophobic contact | ||||
| 72 | SET and MYND domain-containing protein 2 (SMYD2) | 5ARF | 5.31 | |
Target general information Gen name SMYD2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Nlysine methyltransferase SMYD2; N-lysine methyltransferase SMYD2; Lysine Nmethyltransferase 3C; Lysine N-methyltransferase 3C; KMT3C; Histone methyltransferase SMYD2; HSKMB; HSKM-B Protein family Class V-like SAM-binding methyltransferase superfamily Biochemical class Methyltransferase Function Specifically methylates histone H3 'Lys-4' (H3K4me) and dimethylates histone H3 'Lys-36' (H3K36me2). Shows even higher methyltransferase activity on p53/TP53. Monomethylates 'Lys-370' of p53/TP53, leading to decreased DNA-binding activity and subsequent transcriptional regulation activity of p53/TP53. Monomethylates RB1 at 'Lys-860'. Protein-lysine N-methyltransferase that methylates both histones and non-histone proteins, including p53/TP53 and RB1. Related diseases Pulmonary hypertension, primary, 4 (PPH4) [MIM:615344]: A rare disorder characterized by plexiform lesions of proliferating endothelial cells in pulmonary arterioles. The lesions lead to elevated pulmonary arterial pression, right ventricular failure, and death. The disease can occur from infancy throughout life and it has a mean age at onset of 36 years. Penetrance is reduced. Although familial pulmonary hypertension is rare, cases secondary to known etiologies are more common and include those associated with the appetite-suppressant drugs. {ECO:0000269|PubMed:23883380}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Defects in this gene may cause developmental delay with sleep apnea (DDSA). A disorder characterized by developmental neurologic, skeletal and respiratory anomalies including microcephaly, arthrogryposis, scoliosis, cleft palate, facial dysmorphology, bilateral talipes, feeding difficulties and central and/or obstructive sleep apnea. Malformations are detected as early as 21 weeks post gestation. Severely affected patients require ongoing treatment with nocturnal O2 or pressure-controlled ventilation. The disease is associated with recurrent de novo gain of function variants. {ECO:0000269|PubMed:36195757}. Drugs (DrugBank ID) NA Interacts with P20290-2; Q96K17; Q9UPZ9; Q9Y5W9; P04637 EC number EC 2.1.1.- Uniprot keywords 3D-structure; Alternative splicing; Chromatin regulator; Cytoplasm; Metal-binding; Methyltransferase; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; S-adenosyl-L-methionine; Transcription; Transcription regulation; Transferase; Zinc; Zinc-finger Protein physicochemical properties Chain ID A Molecular weight (Da) 48789.6 Length 425 Aromaticity 0.1 Instability index 51.93 Isoelectric point 6.35 Charge (pH=7) -4.02 2D Binding mode Binding energy (Kcal/mol) -7.24 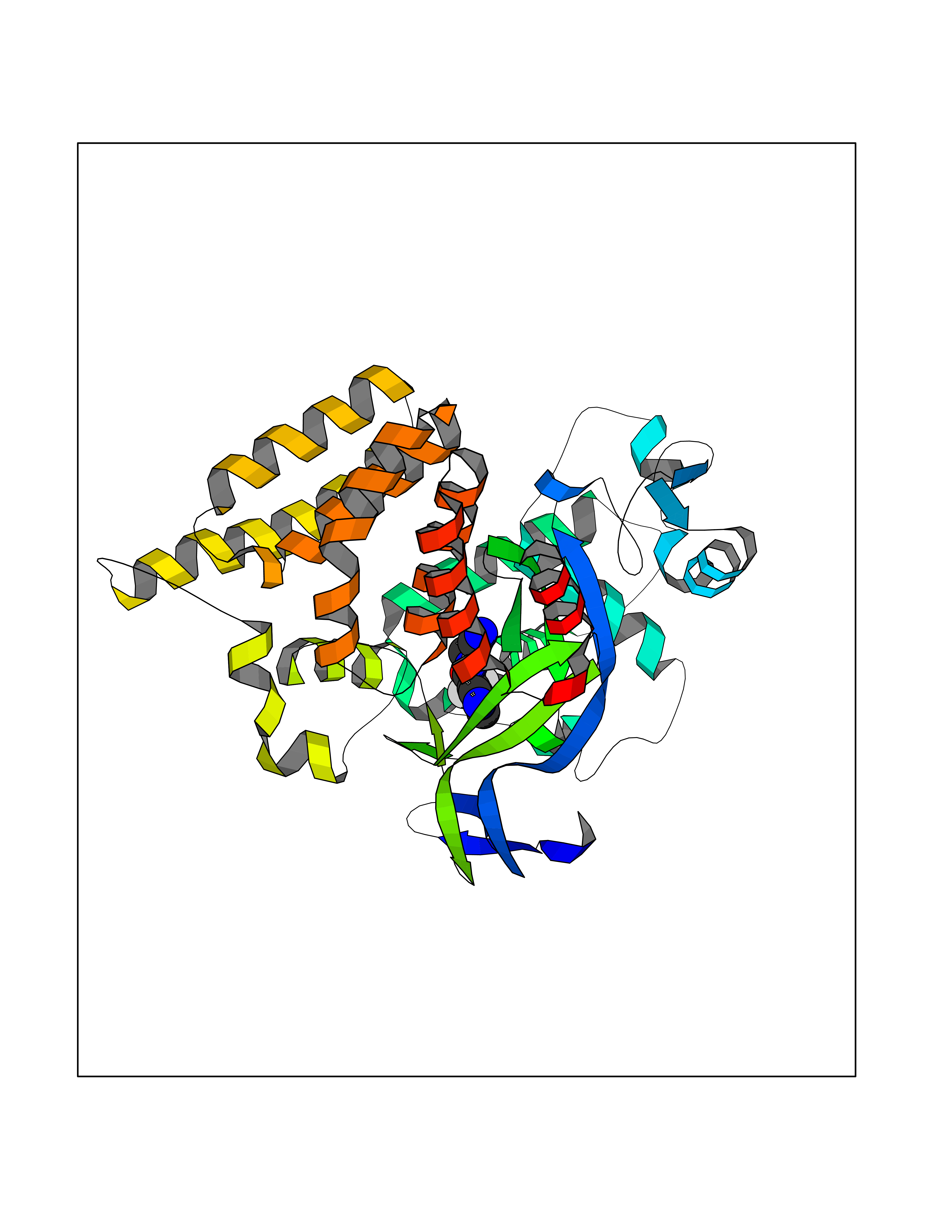 Molscript Map 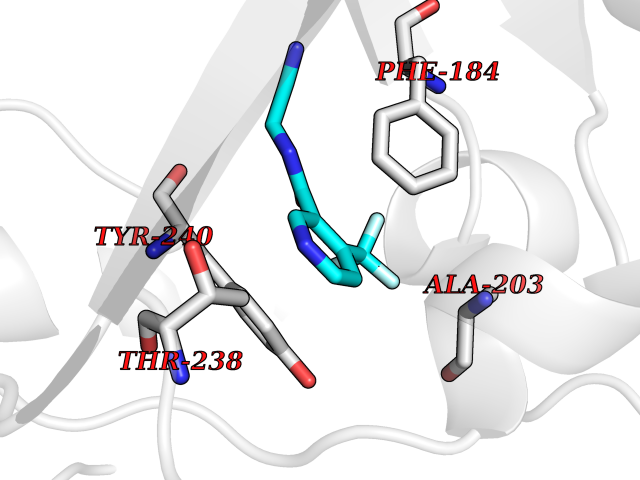 Pymol Map 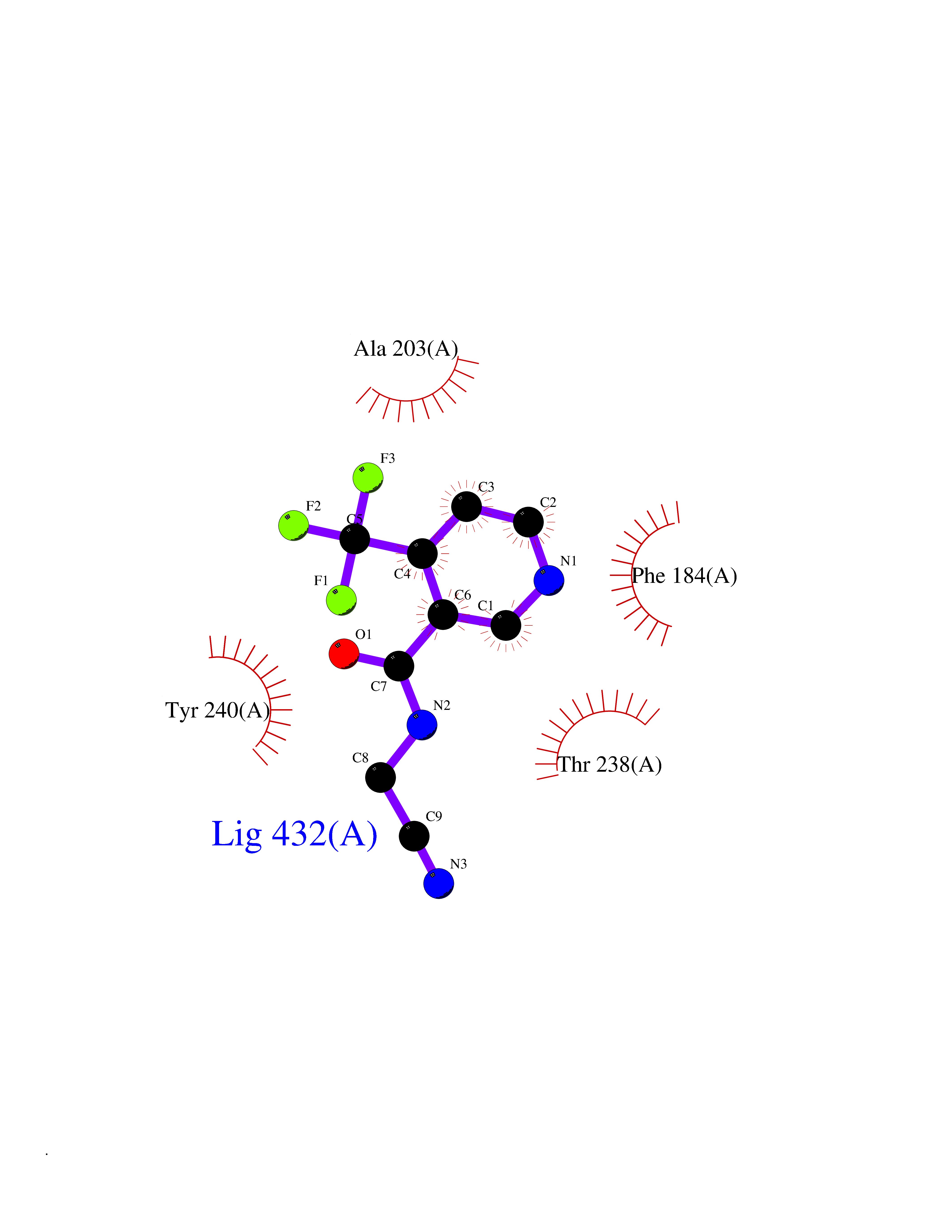 Ligplot Map 3D Binding mode Sequence LGGLERFCSPGKGRGLRALQPFQVGDLLFSCPAYAYVLTVNERGNHCEYCFTRKEGLSKCGRCKQAFYCNVECQKEDWPMHKLECSPMVVFGENWNPSETVRLTARILAKQKIHPERTPSEKLLAVKEFESHLDKLDNEKKDLIQSDIAALHHFYSKHLGFPDNDSLVVLFAQVNCNGFTIEDEELSHLGSAIFPDVALMNHSCCPNVIVTYKGTLAEVRAVQEIKPGEEVFTSYIDLLYPTEDRNDRLRDSYFFTCECQECTTKDKDKAKVEIRKLSDPPKAEAIRDMVRYARNVIEEFRRAKHYKSPSELLEICELSQEKMSSVFEDSNVYMLHMMYQAMGVCLYMQDWEGALQYGQKIIKPYSKHYPLYSLNVASMWLKLGRLYMGLEHKAAGEKALKKAIAIMEVAHGKDHPYISEIKQEI Hydrogen bonds contact Hydrophobic contact | ||||
| 73 | Bromodomain-containing protein 9 (BRD9) | 6V0X | 5.31 | |
Target general information Gen name BRD9 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Rhabdomyosarcoma antigen MU-RMS-40.8 Protein family NA Biochemical class Bromodomain Function Plays a role in chromatin remodeling and regulation of transcription. Acts as a chromatin reader that recognizes and binds acylated histones: binds histones that are acetylated and/or butyrylated. Component of SWI/SNF chromatin remodeling subcomplex GBAF that carries out key enzymatic activities, changing chromatin structure by altering DNA-histone contacts within a nucleosome in an ATP-dependent manner. Related diseases Major depressive disorder (MDD) [MIM:608516]: A common psychiatric disorder. It is a complex trait characterized by one or more major depressive episodes without a history of manic, mixed, or hypomanic episodes. A major depressive episode is characterized by at least 2 weeks during which there is a new onset or clear worsening of either depressed mood or loss of interest or pleasure in nearly all activities. Four additional symptoms must also be present including changes in appetite, weight, sleep, and psychomotor activity; decreased energy; feelings of worthlessness or guilt; difficulty thinking, concentrating, or making decisions; or recurrent thoughts of death or suicidal ideation, plans, or attempts. The episode must be accompanied by distress or impairment in social, occupational, or other important areas of functioning. {ECO:0000269|PubMed:15229186}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q7Z7H3; Q7Z7H3 EC number NA Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Bromodomain; Chromatin regulator; Isopeptide bond; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Transcription; Transcription regulation; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 11491.3 Length 99 Aromaticity 0.13 Instability index 13.58 Isoelectric point 9.24 Charge (pH=7) 3.8 2D Binding mode Binding energy (Kcal/mol) -7.24 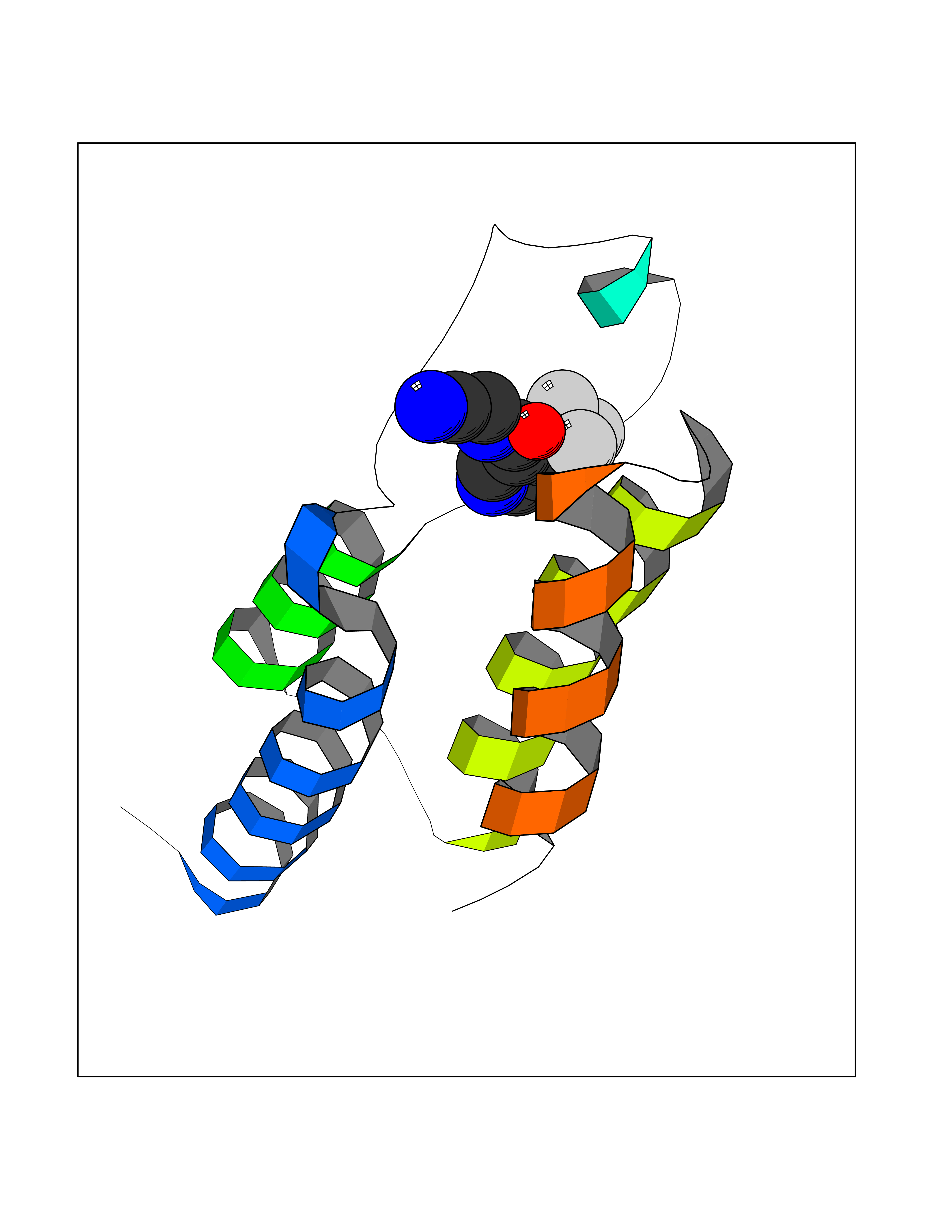 Molscript Map 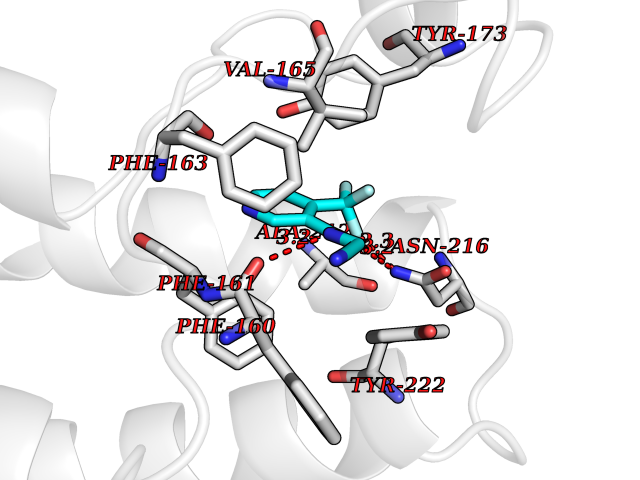 Pymol Map 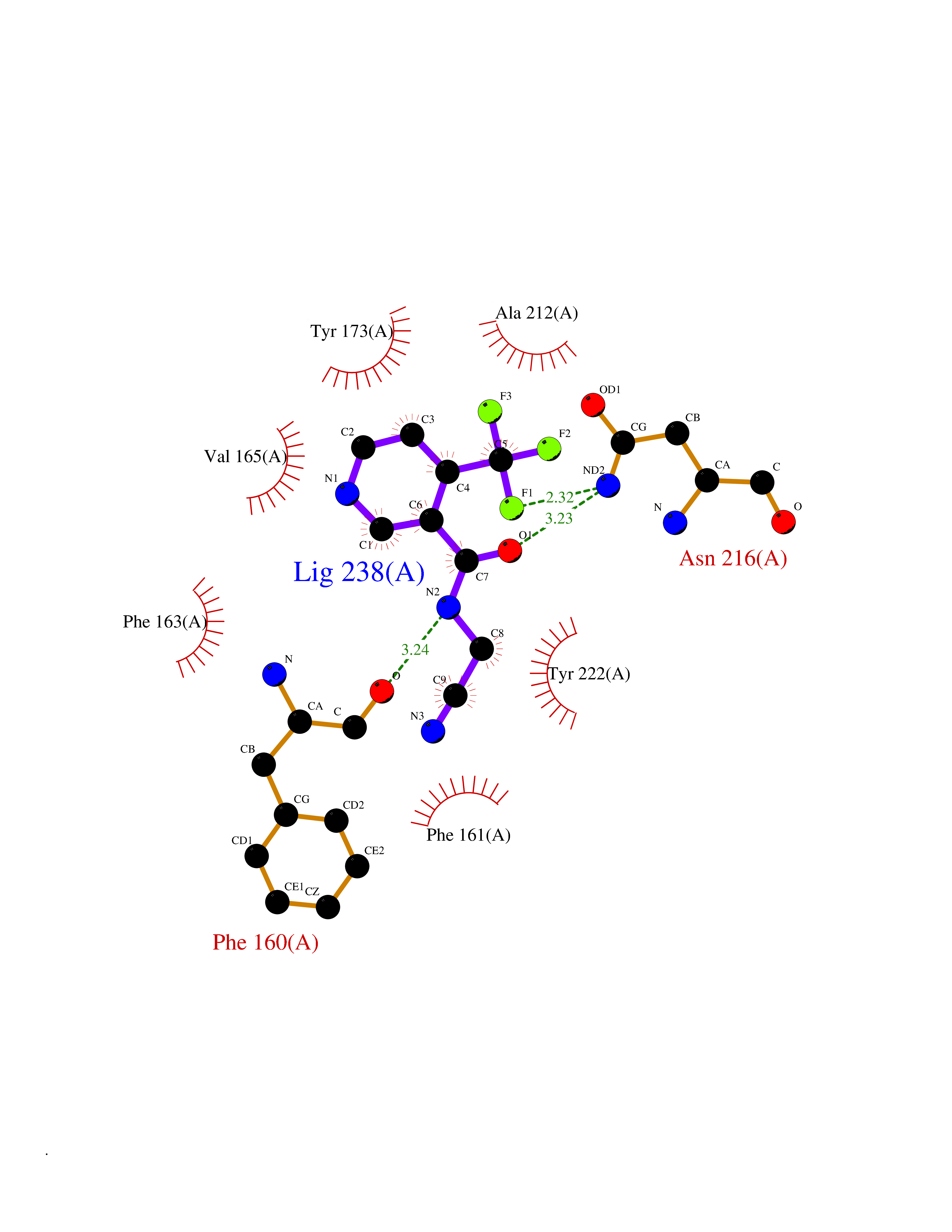 Ligplot Map 3D Binding mode Sequence STPIQQLLEHFLRQLQRKDPHGFFAFPVTDAIAPGYSMIIKHPMDFGTMKDKIVANEYKSVTEFKADFKLMCDNAMTYNRPDTVYYKLAKKILHAGFKM Hydrogen bonds contact Hydrophobic contact | ||||
| 74 | Schistosoma Purine nucleoside phosphorylase (sch PNP) | 3F8W | 5.31 | |
Target general information Gen name sch PNP Organism Schistosoma mansoni (Blood fluke) Uniprot ID TTD ID Synonyms sch Purine nucleoside phosphorylase; sch Inosine-guanosine phosphorylase Protein family PNP/MTAP phosphorylase family Biochemical class NA Function The purine nucleoside phosphorylases catalyze the phosphorolytic breakdown of the N-glycosidic bond in the beta-(deoxy)ribonucleoside molecules, with the formation of the corresponding free purine bases and pentose-1-phosphate. Related diseases Hemolytic anemia, non-spherocytic, due to glucose phosphate isomerase deficiency (HA-GPID) [MIM:613470]: A form of anemia in which there is no abnormal hemoglobin or spherocytosis. It is caused by glucose phosphate isomerase deficiency. {ECO:0000269|PubMed:28803808, ECO:0000269|PubMed:7989588, ECO:0000269|PubMed:8499925, ECO:0000269|PubMed:8822952, ECO:0000269|PubMed:8822954, ECO:0000269|PubMed:9446754, ECO:0000269|PubMed:9856489}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number EC 2.4.2.1 Uniprot keywords 3D-structure; Glycosyltransferase; Transferase Protein physicochemical properties Chain ID A,B Molecular weight (Da) 61140.6 Length 564 Aromaticity 0.06 Instability index 29.8 Isoelectric point 7.6 Charge (pH=7) 1.82 2D Binding mode Binding energy (Kcal/mol) -7.25  Molscript Map 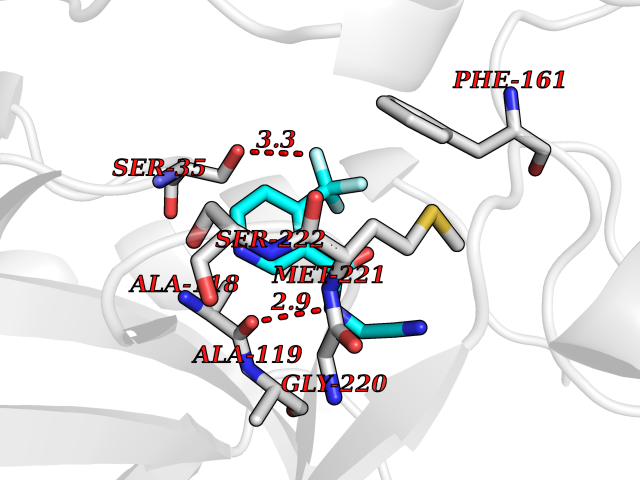 Pymol Map 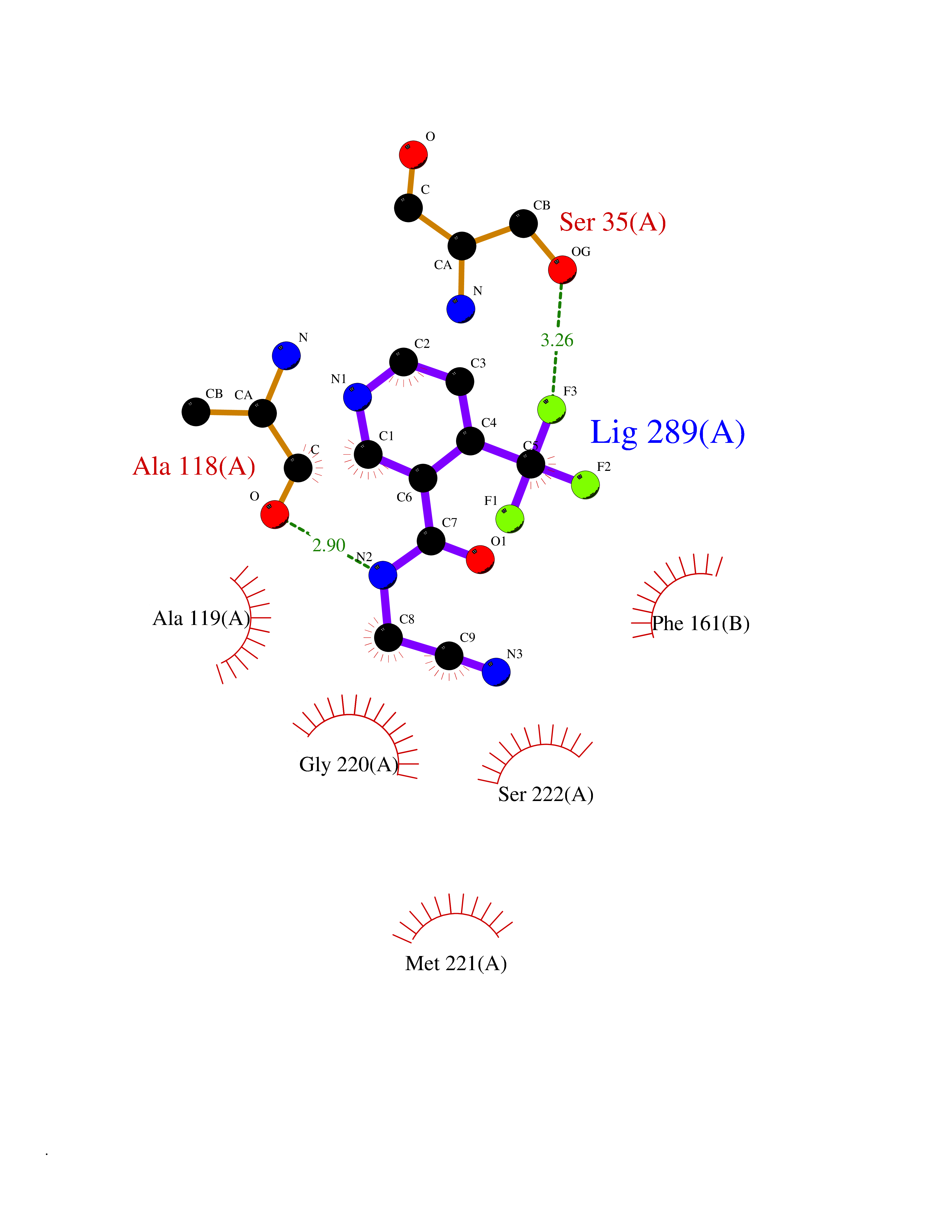 Ligplot Map 3D Binding mode Sequence SVTANIENVKKVAHHIQKLTSIVPEIGIICGSGLGKLADGVKDKITIPYTKIPNFPQTSHSGNLIFGTLSGRKVVVMQGRFHMYEGYSNDTVALPIRVMKLLGVKILMVSNAAGGLNRSLKLGDFVILKDHIYLPGLGLNNILVGPNQEAFGTRFPALSNAYDRDLRKLAVQVAEENGFGNLVHQGVYVMNGGPCYETPAECTMLLNMGCDVVGMSTIPEVVIARHCGIQVFAVSLVTNISVLDVESDLKPNHEEVLATGAQRAELMQSWFEKIIEKLPKSVTANIENVKKVAHHIQKLTSIVPEIGIICGSGLGKLADGVKDKITIPYTKIPNFPQTSVVGHSGNLIFGTLSGRKVVVMQGRFHMYEGYSNDTVALPIRVMKLLGVKILMVSNAAGGLNRSLKLGDFVILKDHIYLPGLGLNNILVGPNQEAFGTRFPALSNAYDRDLRKLAVQVAEENGFGNLVHQGVYVMNGGPCYETPAECTMLLNMGCDVVGMSTIPEVVIARHCGIQVFAVSLVTNISVLDVESDLKPNHEEVLATGAQRAELMQSWFEKIIEKLPKD Hydrogen bonds contact Hydrophobic contact | ||||
| 75 | Bacterial Fatty acid synthetase I (Bact inhA) | 4DTI | 5.31 | |
Target general information Gen name Bact inhA Organism Mycobacterium tuberculosis (strain ATCC 25618 / H37Rv) Uniprot ID TTD ID Synonyms Enoyl-[acyl-carrier-protein] reductase [NADH]; Bacterial InhA Protein family Short-chain dehydrogenases/reductases (SDR) family, FabI subfamily Biochemical class CH-CH donor oxidoreductase Function Involved in the resistance against the antituberculosis drugs isoniazid and ethionamide. Related diseases Immunodeficiency 35 (IMD35) [MIM:611521]: A primary immunodeficiency characterized by recurrent skin abscesses, pneumonia, and highly elevated serum IgE. {ECO:0000269|PubMed:17088085}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07155; DB07188; DB07192; DB07090; DB07222; DB07287; DB07178; DB00609; DB04289; DB00951; DB07123; DB05154; DB02990; DB08604 Interacts with NA EC number NA Uniprot keywords 3D-structure; Antibiotic resistance; Fatty acid biosynthesis; Fatty acid metabolism; Lipid biosynthesis; Lipid metabolism; NAD; Oxidoreductase; Phosphoprotein; Reference proteome Protein physicochemical properties Chain ID A Molecular weight (Da) 28279.2 Length 267 Aromaticity 0.06 Instability index 39.93 Isoelectric point 5.74 Charge (pH=7) -3.78 2D Binding mode Binding energy (Kcal/mol) -7.24  Molscript Map  Pymol Map 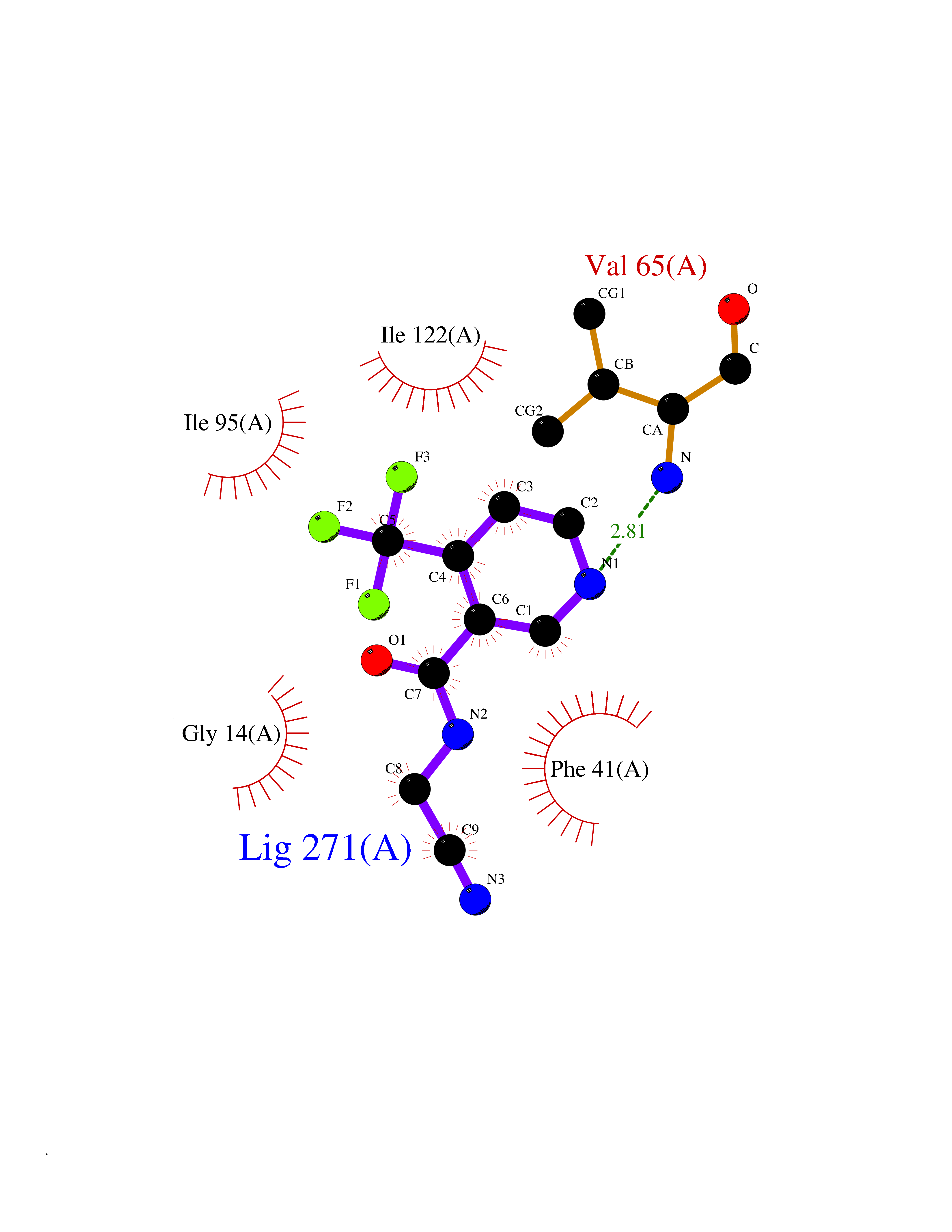 Ligplot Map 3D Binding mode Sequence GLLDGKRILVSGIITDSSIAFHIARVAQEQGAQLVLTGFDRLRLIQRITDRLPAKAPLLELDVQNEEHLASLAGRVTEAIGAGNKLDGVVHAIGFMPQTGMGINPFFDAPYADVSKGIHISAYSYASMAKALLPIMNPGGSIVGMDFDPSRAMPAYNWMTVAKSALESVNRFVAREAGKYGVRSNLVAAGPIRTLAMSAIVGGALGEEAGAQIQLLEEGWDQRAPIGWNMKDATPVAKTVCALLSDWLPATTGDIIYADGGAHTQLL Hydrogen bonds contact Hydrophobic contact | ||||
| 76 | UDP-glucose 4-epimerase (GALE) | 1EK6 | 5.31 | |
Target general information Gen name GALE Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms UDP-galactose4-epimerase; UDP-galactose4'-epimerase; UDP-galactose 4-epimerase; UDP-N-acetylglucosamine 4-epimerase; UDP-N-acetylgalactosamine 4-epimerase; UDP-GlcNAc 4-epimerase; UDP-GalNAc 4-epimera Protein family NAD(P)-dependent epimerase/dehydratase family Biochemical class Racemases and epimerase Function The reaction with UDP-Gal plays a critical role in the Leloir pathway of galactose catabolism in which galactose is converted to the glycolytic intermediate glucose 6-phosphate. It contributes to the catabolism of dietary galactose and enables the endogenous biosynthesis of both UDP-Gal and UDP-GalNAc when exogenous sources are limited. Both UDP-sugar interconversions are important in the synthesis of glycoproteins and glycolipids. Catalyzes two distinct but analogous reactions: the reversible epimerization of UDP-glucose to UDP-galactose and the reversible epimerization of UDP-N-acetylglucosamine to UDP-N-acetylgalactosamine. Related diseases Galactosemia 3 (GALAC3) [MIM:230350]: A form of galactosemia, an inborn error of galactose metabolism typically manifesting in the neonatal period, after ingestion of galactose, with jaundice, hepatosplenomegaly, hepatocellular insufficiency, food intolerance, hypoglycemia, renal tubular dysfunction, muscle hypotonia, sepsis and cataract. GALAC3 is an autosomal recessive form caused by galactose epimerase deficiency. It can manifest as benign, peripheral form with mild symptoms and enzymatic deficiency in circulating blood cells only. A second form, known as generalized epimerase deficiency, is characterized by undetectable levels of enzyme activity in all tissues and severe clinical features, including restricted growth and intellectual disability. {ECO:0000269|PubMed:11279193, ECO:0000269|PubMed:11903335, ECO:0000269|PubMed:15639193, ECO:0000269|PubMed:16301867, ECO:0000269|PubMed:16302980, ECO:0000269|PubMed:9326324, ECO:0000269|PubMed:9538513, ECO:0000269|PubMed:9973283}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Thrombocytopenia 13, syndromic (THC13) [MIM:620776]: An autosomal recessive form of thrombocytopenia, a hematologic disorder defined by a decrease in the number of platelets in circulating blood, resulting in the potential for increased bleeding and decreased ability for clotting. THC13 patients have enlarged, gray platelets with defective function. Some affected individuals have leukopenia or anemia and pancytopenia. Additional variable features include mitral valve malformations, pyloric stenosis, and impaired intellectual development. {ECO:0000269|PubMed:30247636, ECO:0000269|PubMed:33510604, ECO:0000269|PubMed:34159722, ECO:0000269|PubMed:36395340}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03501; DB03095; DB03041; DB01861; DB02196; DB03397 Interacts with Q14376 EC number EC 5.1.3.2 Uniprot keywords 3D-structure; Alternative splicing; Carbohydrate metabolism; Disease variant; Galactose metabolism; Isomerase; NAD; Proteomics identification; Reference proteome Protein physicochemical properties Chain ID A Molecular weight (Da) 38082 Length 346 Aromaticity 0.09 Instability index 33.02 Isoelectric point 6.26 Charge (pH=7) -2.83 2D Binding mode Binding energy (Kcal/mol) -7.25  Molscript Map 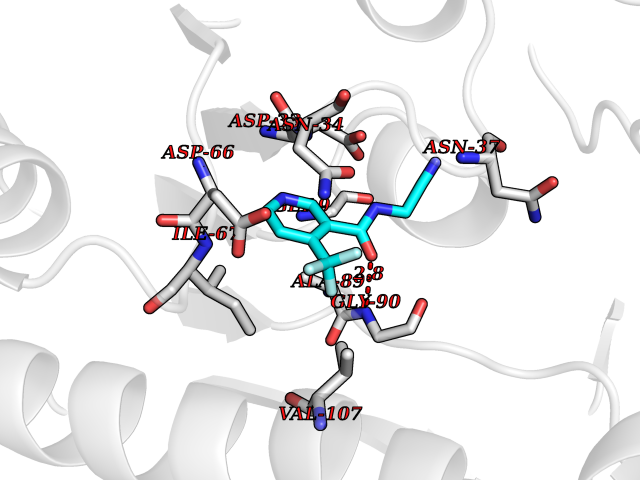 Pymol Map 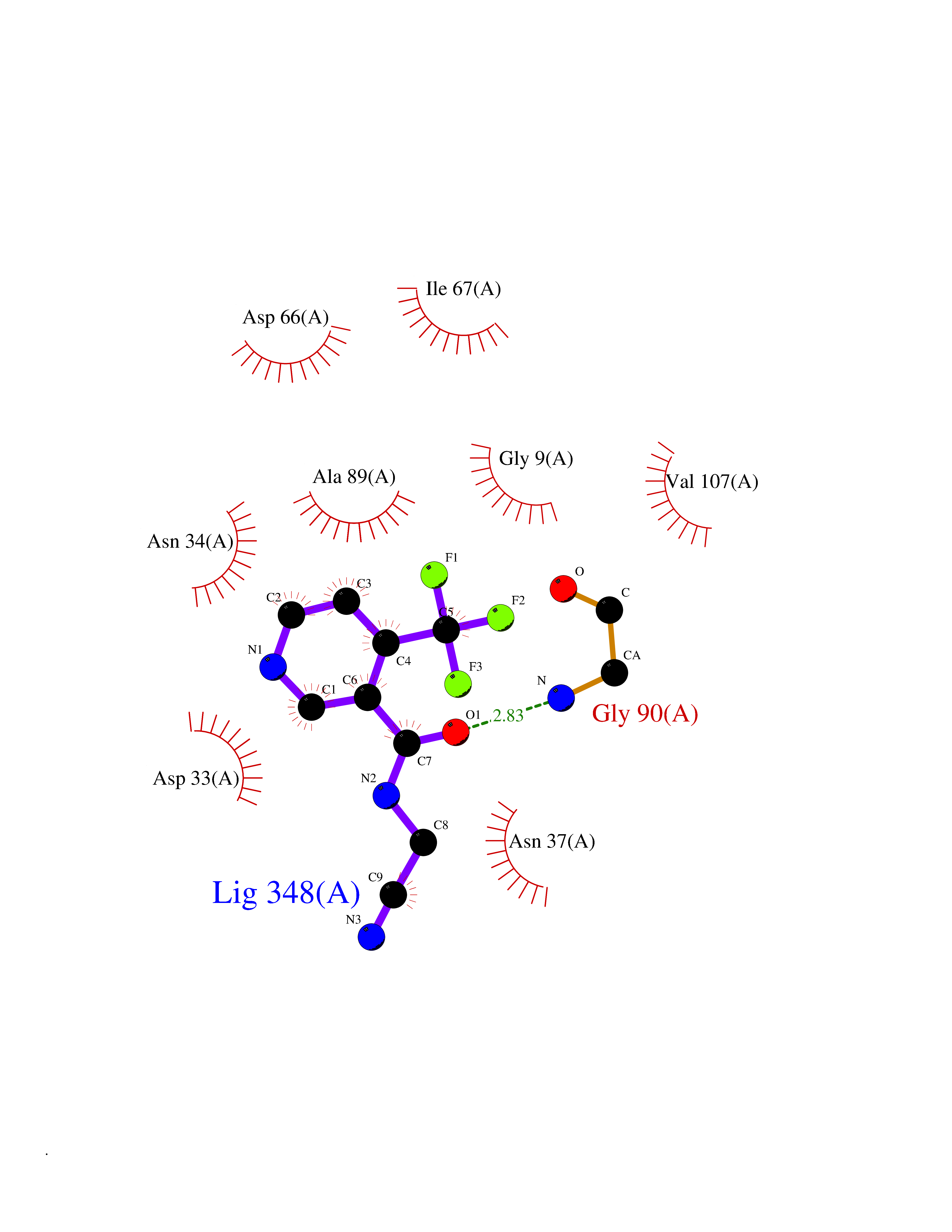 Ligplot Map 3D Binding mode Sequence MAEKVLVTGGAGYIGSHTVLELLEAGYLPVVIDNFHNAFRGGGSLPESLRRVQELTGRSVEFEEMDILDQGALQRLFKKYSFMAVIHFAGLKAVGESVQKPLDYYRVNLTGTIQLLEIMKAHGVKNLVFSSSATVYGNPQYLPLDEAHPTGGCTNPYGKSKFFIEEMIRDLCQADKTWNAVLLRYFNPTGAHASGCIGEDPQGIPNNLMPYVSQVAIGRREALNVFGNDYDTEDGTGVRDYIHVVDLAKGHIAALRKLKEQCGCRIYNLGTGTGYSVLQMVQAMEKASGKKIPYKVVARREGDVAACYANPSLAQEELGWTAALGLDRMCEDLWRWQKQNPSGFGT Hydrogen bonds contact Hydrophobic contact | ||||
| 77 | Human immunodeficiency virus Negative factor (HIV nef) | 6B72 | 5.30 | |
Target general information Gen name HIV nef Organism Human immunodeficiency virus type 1 group M subtype B (isolate ARV2/SF2) (HIV-1) Uniprot ID TTD ID Synonyms nef; Nef protein; F-protein; 3'ORF; 27 kDa protein Protein family Lentivirus primate group Nef protein family Biochemical class Lentivirus primate group Nef Function Extracellular Nef protein targets CD4(+) T-lymphocytes for apoptosis by interacting with CXCR4 surface receptors. Related diseases Neurodegeneration due to cerebral folate transport deficiency (NCFTD) [MIM:613068]: An autosomal recessive neurodegenerative disorder resulting from brain-specific folate deficiency early in life. Onset is apparent in late infancy with severe developmental regression, movement disturbances, epilepsy and leukodystrophy. {ECO:0000269|PubMed:19732866}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q14457; P01730 EC number NA Uniprot keywords 3D-structure; AIDS; Apoptosis; Early protein; Host cell membrane; Host Golgi apparatus; Host membrane; Host-virus interaction; Inhibition of host adaptive immune response by virus; Inhibition of host autophagy by virus; Inhibition of host MHC class I molecule presentation by virus; Inhibition of host MHC class II molecule presentation by virus; Lipoprotein; Membrane; Myristate; Phosphoprotein; Reference proteome; Secreted; SH3-binding; Viral immunoevasion; Virion; Virulence Protein physicochemical properties Chain ID A,B Molecular weight (Da) 29232.1 Length 245 Aromaticity 0.16 Instability index 50.97 Isoelectric point 5.71 Charge (pH=7) -7.32 2D Binding mode Binding energy (Kcal/mol) -7.23  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence ADCAWLEAQEEEEVGFPVRPQVPLRPMTYKAALDISHFLKEKGGLEGLIWSQRRQEILDLWIYHTQGYFPDWQNYTPGPGIRYPLTFGWCFKLVPVEEKEVLVWRFDSKLAFHHMARELHPEYYCAWLEAQEEEEVGFPVRPQVPLRPMTYKAALDISHFLKEKGGLEGLIWSQRRQEILDLWIYHTQGYFPDWQNYTPGPGIRYPLTFGWCFKLVPVEKEVLVWRFDSKLAFHHMARELHPEYY Hydrogen bonds contact Hydrophobic contact | ||||
| 78 | Purine nucleoside phosphorylase (PNP) | 4EAR | 5.30 | |
Target general information Gen name PNP Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms PNP; Inosine phosphorylase Protein family PNP/MTAP phosphorylase family Biochemical class Pentosyltransferase Function The purine nucleoside phosphorylases catalyze the phosphorolytic breakdown of the N-glycosidic bond in the beta- (deoxy)ribonucleoside molecules, with the formation of the corresponding free purine bases and pentose-1-phosphate. Related diseases Purine nucleoside phosphorylase deficiency (PNPD) [MIM:613179]: A disorder that interrupts both the catabolism of inosine into hypoxanthine and guanosine into guanine, and leads to the accumulation of guanosine, inosine, and their deoxified by-products. The main clinical presentation is recurrent infections due to severe T-cell immunodeficiency. Some patients also have neurologic impairment. {ECO:0000269|PubMed:1384322, ECO:0000269|PubMed:3029074, ECO:0000269|PubMed:8931706}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03881; DB03551; DB02222; DB02391; DB03609; DB01667; DB04260; DB02796; DB04753; DB00640; DB00242; DB00900; DB06185; DB02377; DB02857; DB04754; DB04757; DB04076; DB02230; DB04335; DB02568; DB03101 Interacts with P05067; Q9UQM7; O14576-2; P06241; P14136; Q92993-2; Q9BXM7; P00491; P17612; P63000; Q92673; Q15583 EC number EC 2.4.2.1 Uniprot keywords 3D-structure; Acetylation; Cytoplasm; Direct protein sequencing; Disease variant; Glycosyltransferase; Phosphoprotein; Proteomics identification; Purine salvage; Reference proteome; Transferase Protein physicochemical properties Chain ID A,B,C Molecular weight (Da) 31849.2 Length 288 Aromaticity 0.1 Instability index 34.77 Isoelectric point 6.42 Charge (pH=7) -1.63 2D Binding mode Binding energy (Kcal/mol) -7.23  Molscript Map  Pymol Map 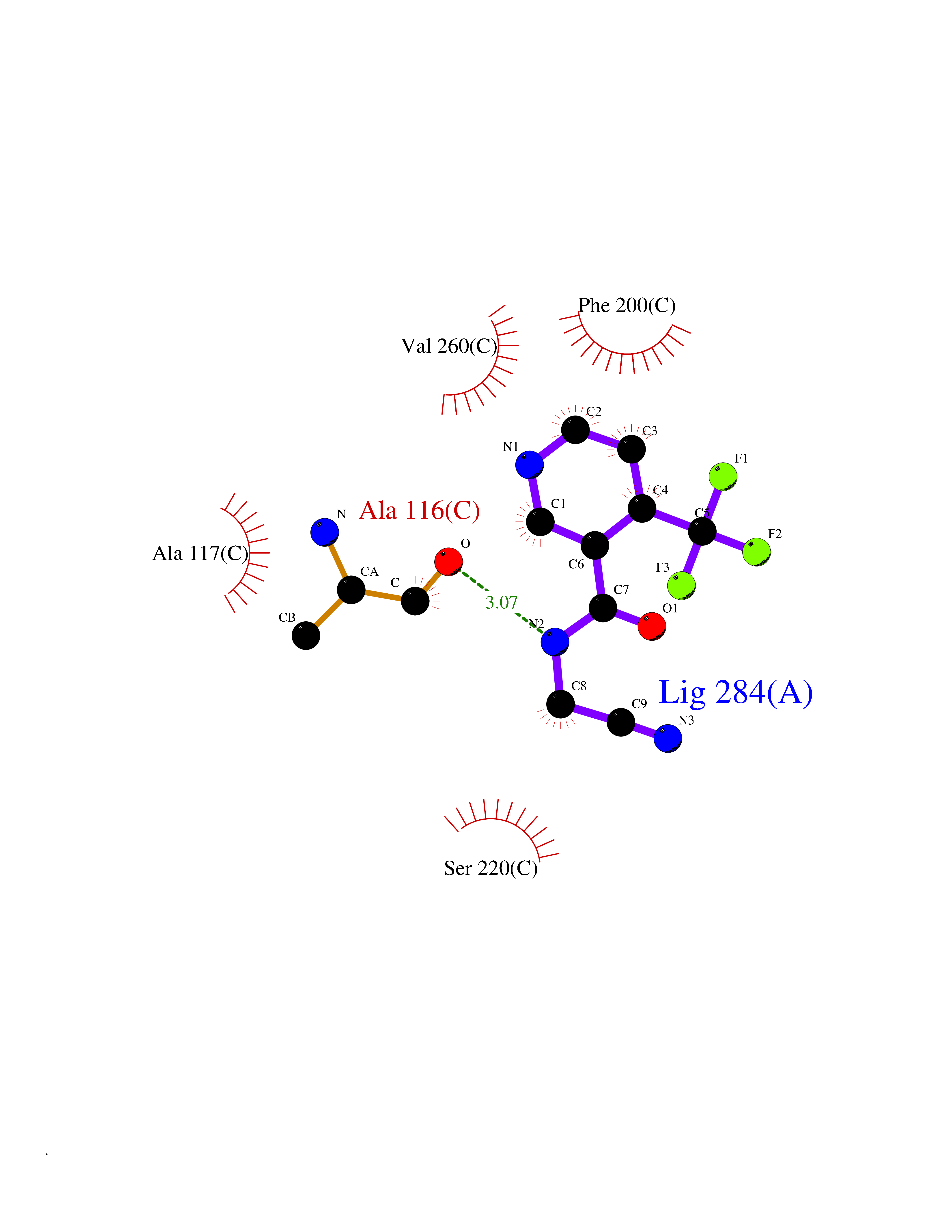 Ligplot Map 3D Binding mode Sequence GYTYEDYKNTAEYLLSHTKHRPQVAIICGSGLGGLTDKLTQAQIFDYSEIPNFPRSTVPGHAGRLVFGFLNGRACVMMQGRFHMYEGYPLYKVTFPVRVFHLLGVDTLVVTNAAGGLNPKFEVGDIMLIRDHINLPGFSGQNPLRGPNDERFGDRFPAMSDAYDRTMRQRALSTYKQMGEQRELQEGTYVMVAGPSFETVAECRVLQKLGADAVGMSTVPEVIVARHCGLRVFGFSLITNKVIMDYESLEKANXEEVLAAGKQAAQKLEQFVSILMASIDRFPAMSDA Hydrogen bonds contact Hydrophobic contact | ||||
| 79 | Catechol-O-methyl-transferase (COMT) | 3BWY | 5.30 | |
Target general information Gen name COMT Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms S-COMT; MB-COMT; Catechol-O-methyltransferase; COMT Protein family Class I-like SAM-binding methyltransferase superfamily, Cation-dependent O-methyltransferase family Biochemical class Methyltransferase Function Catalyzes the O-methylation, and thereby the inactivation, of catecholamine neurotransmitters and catechol hormones. Also shortens the biological half-lives of certain neuroactive drugs, like L-DOPA, alpha-methyl DOPA and isoproterenol. Related diseases Schizophrenia (SCZD) [MIM:181500]: A complex, multifactorial psychotic disorder or group of disorders characterized by disturbances in the form and content of thought (e.g. delusions, hallucinations), in mood (e.g. inappropriate affect), in sense of self and relationship to the external world (e.g. loss of ego boundaries, withdrawal), and in behavior (e.g bizarre or apparently purposeless behavior). Although it affects emotions, it is distinguished from mood disorders in which such disturbances are primary. Similarly, there may be mild impairment of cognitive function, and it is distinguished from the dementias in which disturbed cognitive function is considered primary. Some patients manifest schizophrenic as well as bipolar disorder symptoms and are often given the diagnosis of schizoaffective disorder. {ECO:0000269|PubMed:15645182}. Disease susceptibility may be associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07462; DB02342; DB02105; DB08049; DB00118; DB00714; DB03336; DB00286; DB00255; DB00841; DB00988; DB15488; DB00494; DB00668; DB00783; DB00977; DB01064; DB00968; DB01141; DB03907; DB04820; DB06152; DB11632; DB00252; DB01420; DB00323 Interacts with Q6P5T0; P30518; Q8NFU1; Q8NHW4; P34972; Q96BA8; P50402; Q5JX71; O14843; O00258; P08034; O75712; Q9NTQ9; O95377; Q8TDT2; Q8N6U8; O15529; P31937; Q9H2F3; O95279; Q5SR56; A6NDP7; Q0D2K0; Q7RTS5; Q9UHJ9-5; Q8IY26; Q9H6H4; Q6NTF9-3; O75783; Q99500; Q9Y6D0; Q3KNW5; O60669; P22732; Q96G79; Q5T1Q4; Q9NY26; Q9NP94; Q6P1K1; P30825; Q9UHI5; B2RUZ4; Q9UPZ6; Q96MV1; Q9NV29; A0PK00; Q9NUH8; Q9P0S9; Q14656; Q6UW68; Q9H0R3; O95807; P34981; Q15645; Q15836; O95183; O76024; P30260; Q9H816; Q92997; P29323-3; P22607; P06396; Q15323; Q6A162; P26371; O15116; P20645; O14744; Q5T160; Q9UJD0; Q2MKA7; Q8N488; O75880; Q14141; Q9UNE7; Q15645; Q9NYH9; Q8NA23-2 EC number EC 2.1.1.6 Uniprot keywords 3D-structure; Alternative initiation; Catecholamine metabolism; Cell membrane; Cytoplasm; Direct protein sequencing; Lipid metabolism; Magnesium; Membrane; Metal-binding; Methyltransferase; Neurotransmitter degradation; Phosphoprotein; Proteomics identification; Reference proteome; S-adenosyl-L-methionine; Schizophrenia; Signal-anchor; Transferase; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 23851.2 Length 214 Aromaticity 0.07 Instability index 25.99 Isoelectric point 5.25 Charge (pH=7) -7.75 2D Binding mode Binding energy (Kcal/mol) -7.23  Molscript Map  Pymol Map 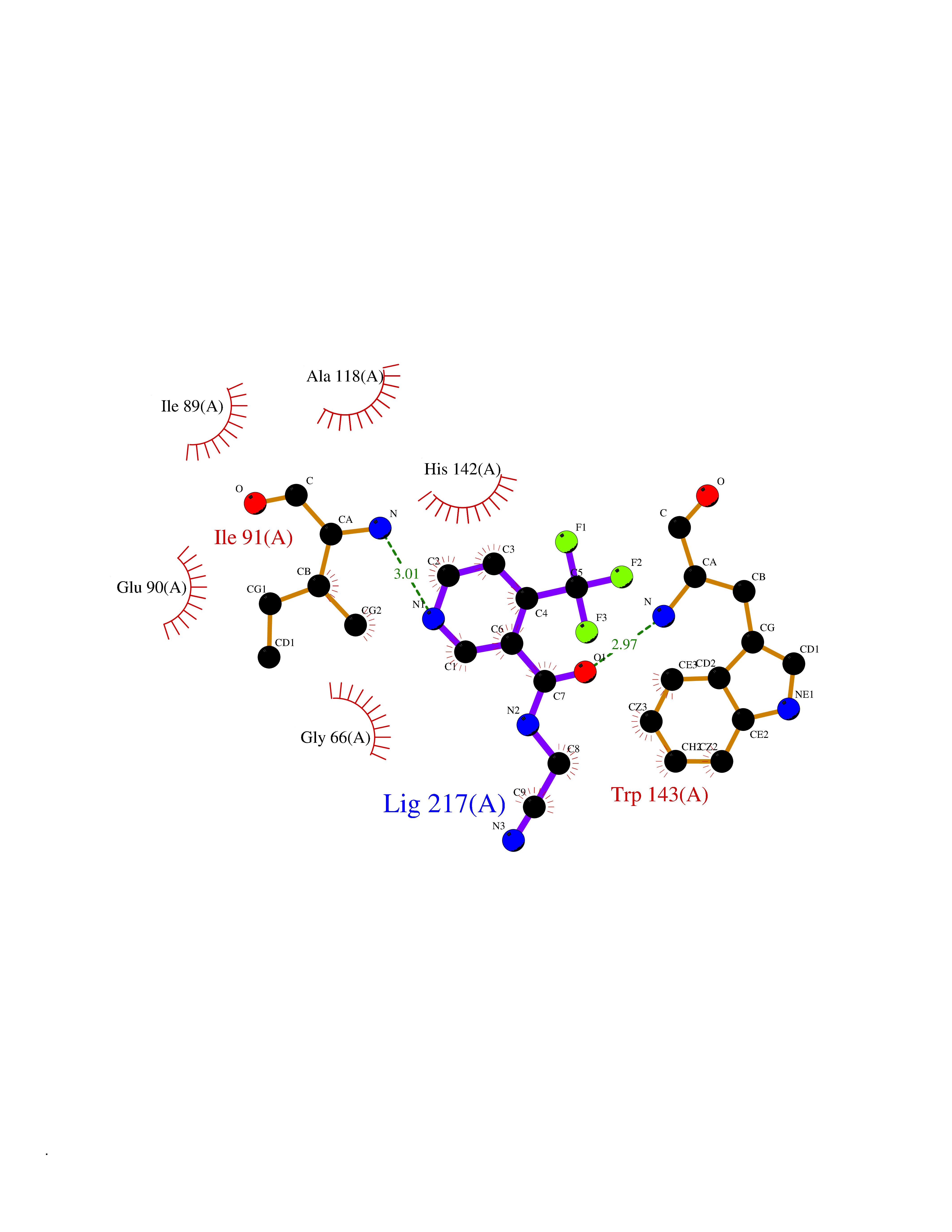 Ligplot Map 3D Binding mode Sequence GDTKEQRILNHVLQHAEPGNAQSVLEAIDTYCEQKEWAMNVGDKKGKIVDAVIQEHQPSVLLELGAYCGYSAVRMARLLSPGARLITIEINPDCAAITQRMVDFAGMKDKVTLVVGASQDIIPQLKKKYDVDTLDMVFLDHWKDRYLPDTLLLEECGLLRKGTVLLADNVICPGAPDFLAHVRGSSCFECTHYQSFLEYREVVDGLEKAIYKGP Hydrogen bonds contact Hydrophobic contact | ||||
| 80 | Adenosine kinase (ADK) | 1BX4 | 5.30 | |
Target general information Gen name ADK Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Adenosine 5'-phosphotransferase; AK Protein family Carbohydrate kinase PfkB family Biochemical class Kinase Function Serves as a potential regulator of concentrations of extracellular adenosine and intracellular adenine nucleotides. ATP dependent phosphorylation of adenosine and other related nucleoside analogs to monophosphate derivatives. Related diseases Hypermethioninemia due to adenosine kinase deficiency (HMAKD) [MIM:614300]: A metabolic disorder characterized by global developmental delay, early-onset seizures, mild dysmorphic features, and characteristic biochemical anomalies, including persistent hypermethioninemia with increased levels of S-adenosylmethionine and S-adenosylhomocysteine. Homocysteine levels are typically normal. {ECO:0000269|PubMed:21963049}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07280; DB07173; DB01048; DB00640; DB00131; DB00171; DB00811 Interacts with NA EC number EC 2.7.1.20 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; ATP-binding; Cytoplasm; Direct protein sequencing; Disease variant; Kinase; Magnesium; Metal-binding; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Purine salvage; Reference proteome; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 38383.4 Length 342 Aromaticity 0.09 Instability index 37.64 Isoelectric point 6.24 Charge (pH=7) -3.41 2D Binding mode Binding energy (Kcal/mol) -7.23  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence VRENILFGMGNPLLDISAVVDKDFLDKYSLKPNDQILAEDKHKELFDELVKKFKVEYHAGGSTQNSIKVAQWMIQQPHKAATFFGCIGIDKFGEILKRKAAEAHVDAHYYEQNEQPTGTCAACITGDNRSLIANLAAANCYKKEKHLDLEKNWMLVEKARVCYIAGFFLTVSPESVLKVAHHASENNRIFTLNLSAPFISQFYKESLMKVMPYVDILFGNETEAATFAREQGFETKDIKEIAKKTQALPKMNSKRQRIVIFTQGRDDTIMATESEVTAFAVLDQDQKEIIDTNGAGDAFVGGFLSQLVSDKPLTECIRAGHYAASIIIRRTGCTFPEKPDFH Hydrogen bonds contact Hydrophobic contact | ||||