Job Results:
Ligand
Structure
Job ID
13aa62e81038093cc0a5aa14a89b9a62
Job name
NA
Time
2026-01-10 22:45:10
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 41 | Plasma retinol-binding protein (RBP4) | 5NU7 | 7.22 | |
Target general information Gen name RBP4 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Retinol-binding protein 4; RBP4; RBP; Plasma retinol-binding protein(1-176); PRBP Protein family Calycin superfamily, Lipocalin family Biochemical class Calycin family Function Delivers retinol from the liver stores to the peripheral tissues. In plasma, the RBP-retinol complex interacts with transthyretin, this prevents its loss by filtration through the kidney glomeruli. Related diseases Retinal dystrophy, iris coloboma, and comedogenic acne syndrome (RDCCAS) [MIM:615147]: A disease characterized by retinal degeneration, ocular colobomas involving both the anterior and posterior segment, impaired night vision and loss of visual acuity. Additional characteristic features include developmental abnormalities and severe acne. {ECO:0000269|PubMed:10232633, ECO:0000269|PubMed:23189188, ECO:0000269|PubMed:9888420}. The disease is caused by variants affecting the gene represented in this entry. Loss of functional RBP4 protein results in serum retinol deficiency. Lack of normal levels of retinol impairs the visual cycle leading to night blindness at early stages; prolonged deficiency may lead to retinal degeneration. Additionally, retinol deficiency may result in dry skin, increased susceptibility to infection and acne (PubMed:23189188). {ECO:0000269|PubMed:23189188}.; DISEASE: Microphthalmia/Coloboma 10 (MCOPCB10) [MIM:616428]: A disorder of eye formation, ranging from small size of a single eye to complete bilateral absence of ocular tissues. Ocular abnormalities like opacities of the cornea and lens, scaring of the retina and choroid, and other abnormalities may also be present. Ocular colobomas are a set of malformations resulting from abnormal morphogenesis of the optic cup and stalk, and the fusion of the fetal fissure (optic fissure). {ECO:0000269|PubMed:25910211}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB06985; DB06755; DB05076; DB03917; DB00755; DB00162 Interacts with Q9UBX0; P02766; O55245 EC number NA Uniprot keywords 3D-structure; Direct protein sequencing; Disease variant; Disulfide bond; Methylation; Microphthalmia; Proteomics identification; Reference proteome; Retinol-binding; Secreted; Sensory transduction; Signal; Transport; Vision; Vitamin A Protein physicochemical properties Chain ID A Molecular weight (Da) 20030.2 Length 174 Aromaticity 0.13 Instability index 28.54 Isoelectric point 5.24 Charge (pH=7) -4.02 2D Binding mode Binding energy (Kcal/mol) -9.85  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence ERDCRVSSFRVKENFDKARFSGTWYAMAKKDPEGLFLQDNIVAEFSVDETGQMSATAKGRVRLLNNWDVCADMVGTFTDTEDPAKFKMKYWGVASFLQKGNDDHWIVDTDYDTYAVQYSCRLLNLDGTCADSYSFVFSRDPNGLPPEAQKIVRQRQEELCLARQYRLIVHNGYC Hydrogen bonds contact Hydrophobic contact | ||||
| 42 | Alpha-tubulin N-acetyltransferase 1 (ATAT1) | 4B5O | 7.22 | |
Target general information Gen name ATAT1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms MEC17; C6orf134; Alpha-TAT; Acetyltransferase mec-17 homolog Protein family Acetyltransferase ATAT1 family Biochemical class Acetyltransferase ATAT1 family Function Specifically acetylates 'Lys-40' in alpha-tubulin on the lumenal side of microtubules. Promotes microtubule destabilization and accelerates microtubule dynamics; this activity may be independent of acetylation activity. Acetylates alpha-tubulin with a slow enzymatic rate, due to a catalytic site that is not optimized for acetyl transfer. Enters the microtubule through each end and diffuses quickly throughout the lumen of microtubules. Acetylates only long/old microtubules because of its slow acetylation rate since it does not have time to act on dynamically unstable microtubules before the enzyme is released. Required for normal sperm flagellar function. Promotes directional cell locomotion and chemotaxis, through AP2A2-dependent acetylation of alpha-tubulin at clathrin-coated pits that are concentrated at the leading edge of migrating cells. May facilitate primary cilium assembly. Related diseases Anterior segment dysgenesis 6 (ASGD6) [MIM:617315]: A form of anterior segment dysgenesis, a group of defects affecting anterior structures of the eye including cornea, iris, lens, trabecular meshwork, and Schlemm canal. Anterior segment dysgeneses result from abnormal migration or differentiation of the neural crest derived mesenchymal cells that give rise to components of the anterior chamber during eye development. Different anterior segment anomalies may exist alone or in combination, including iris hypoplasia, enlarged or reduced corneal diameter, corneal vascularization and opacity, posterior embryotoxon, corectopia, polycoria, abnormal iridocorneal angle, ectopia lentis, and anterior synechiae between the iris and posterior corneal surface. Clinical conditions falling within the phenotypic spectrum of anterior segment dysgeneses include aniridia, Axenfeld anomaly, Reiger anomaly/syndrome, Peters anomaly, and iridogoniodysgenesis. ASGD6 patients predominantly manifest Peters anomaly. Peters anomaly consists of corneal leukoma, defects in the posterior structures of the cornea such as absence of the posterior corneal stroma and Descemet membrane, and a variable degree of iridocorneal and/or keratolenticular adhesions. Over 50% of patients develop glaucoma in childhood. {ECO:0000269|PubMed:11403040}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Glaucoma 3, primary congenital, A (GLC3A) [MIM:231300]: An autosomal recessive form of primary congenital glaucoma (PCG). PCG is characterized by marked increase of intraocular pressure at birth or early childhood, large ocular globes (buphthalmos) and corneal edema. It results from developmental defects of the trabecular meshwork and anterior chamber angle of the eye that prevent adequate drainage of aqueous humor. {ECO:0000269|PubMed:10227395, ECO:0000269|PubMed:10655546, ECO:0000269|PubMed:11184479, ECO:0000269|PubMed:11527932, ECO:0000269|PubMed:11774072, ECO:0000269|PubMed:11980847, ECO:0000269|PubMed:12036985, ECO:0000269|PubMed:12525557, ECO:0000269|PubMed:14635112, ECO:0000269|PubMed:14640114, ECO:0000269|PubMed:15255109, ECO:0000269|PubMed:15342693, ECO:0000269|PubMed:15475877, ECO:0000269|PubMed:16490498, ECO:0000269|PubMed:16688110, ECO:0000269|PubMed:16735994, ECO:0000269|PubMed:16862072, ECO:0000269|PubMed:18470941, ECO:0000269|PubMed:9463332, ECO:0000269|PubMed:9497261}. The disease is caused by variants affecting distinct genetic loci, including the gene represented in this entry.; DISEASE: Glaucoma 1, open angle, A (GLC1A) [MIM:137750]: A form of primary open angle glaucoma (POAG). POAG is characterized by a specific pattern of optic nerve and visual field defects. The angle of the anterior chamber of the eye is open, and usually the intraocular pressure is increased. However, glaucoma can occur at any intraocular pressure. The disease is generally asymptomatic until the late stages, by which time significant and irreversible optic nerve damage has already taken place. {ECO:0000269|PubMed:11774072}. The gene represented in this entry acts as a disease modifier. Digenic mutations in CYP1B1 and MYOC have been found in a family segregating both primary adult-onset and juvenile forms of open angle glaucoma (PubMed:11774072). All affected family members with mutations in both MYOC and CYP1B1 had juvenile glaucoma, whereas those with only the MYOC mutation had the adult-onset form (PubMed:11774072). {ECO:0000269|PubMed:11774072}. Drugs (DrugBank ID) NA Interacts with P62258 EC number EC 2.3.1.108 Uniprot keywords 3D-structure; Acetylation; Acyltransferase; Alternative splicing; Cell junction; Cell projection; Coated pit; Cytoplasm; Cytoskeleton; Membrane; Methylation; Phosphoprotein; Proteomics identification; Reference proteome; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 22197.2 Length 192 Aromaticity 0.09 Instability index 47 Isoelectric point 8.69 Charge (pH=7) 2.35 2D Binding mode Binding energy (Kcal/mol) -9.85 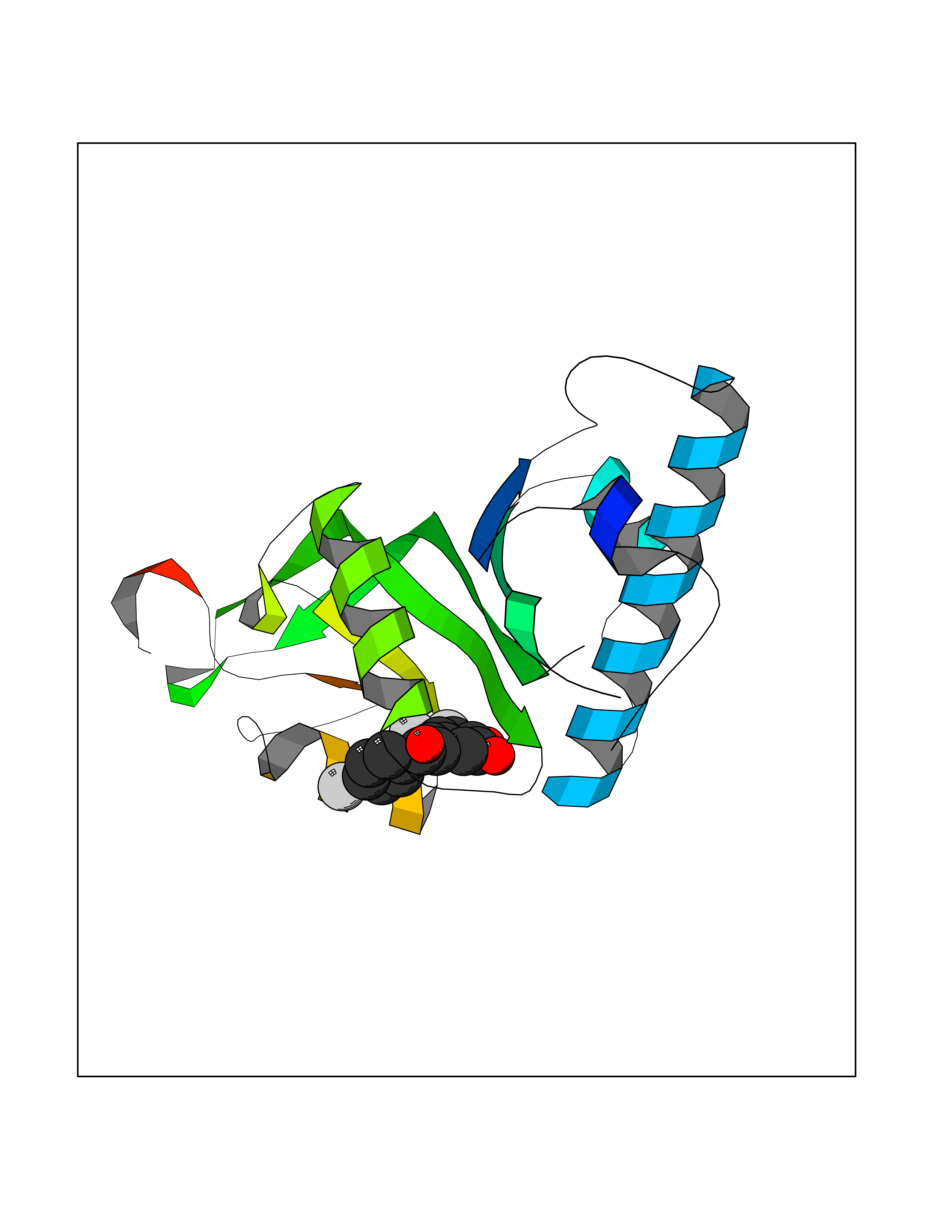 Molscript Map 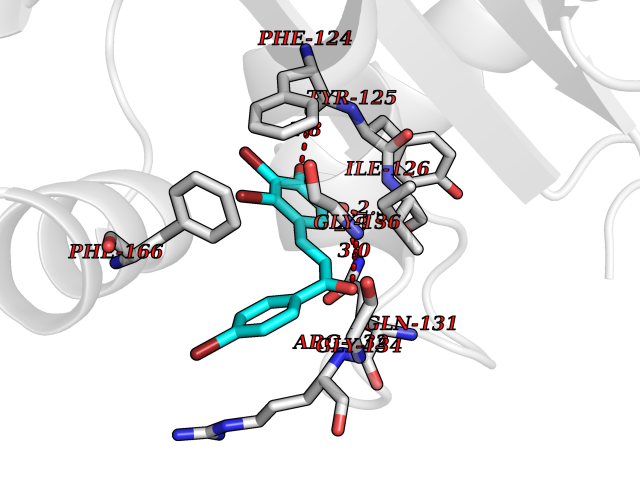 Pymol Map 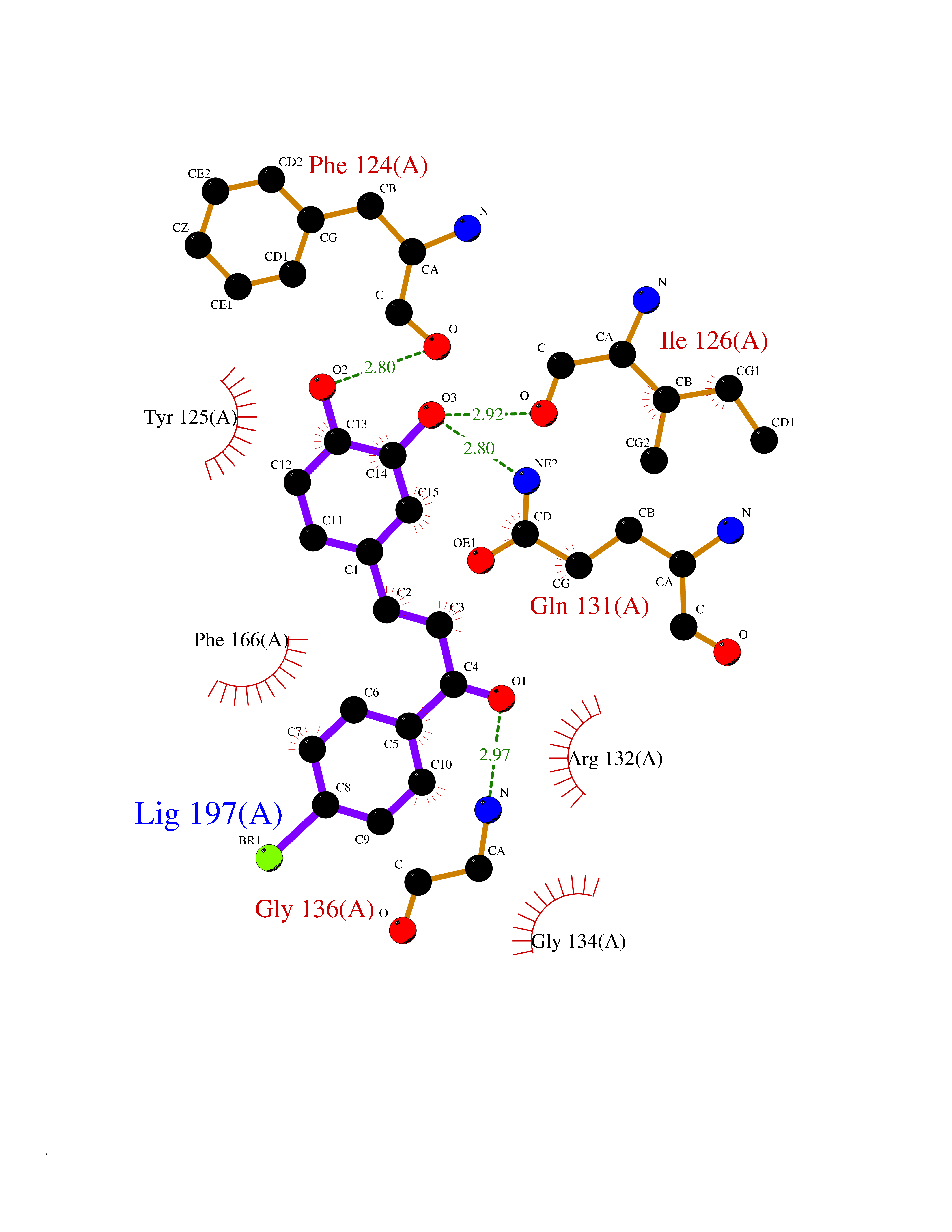 Ligplot Map 3D Binding mode Sequence SMEFPFDVDALFPERITVLDQHLRPPARRPGTTTPARVDLQQQIMTIIDELGKASAKAQNLSAPITSASRMQSNRHVVYILKDSSARPAIIGFIKVGYKKLFVLDDREAHNEVEPLCILDFYIHESVQRHGHGRELFQYMLQKERVEPHQLAIDRPSQKLLKFLNKHYNLETTVPQVNNFVIFEGFFAHQHR Hydrogen bonds contact Hydrophobic contact | ||||
| 43 | Catechol-O-methyl-transferase (COMT) | 3BWY | 7.21 | |
Target general information Gen name COMT Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms S-COMT; MB-COMT; Catechol-O-methyltransferase; COMT Protein family Class I-like SAM-binding methyltransferase superfamily, Cation-dependent O-methyltransferase family Biochemical class Methyltransferase Function Catalyzes the O-methylation, and thereby the inactivation, of catecholamine neurotransmitters and catechol hormones. Also shortens the biological half-lives of certain neuroactive drugs, like L-DOPA, alpha-methyl DOPA and isoproterenol. Related diseases Schizophrenia (SCZD) [MIM:181500]: A complex, multifactorial psychotic disorder or group of disorders characterized by disturbances in the form and content of thought (e.g. delusions, hallucinations), in mood (e.g. inappropriate affect), in sense of self and relationship to the external world (e.g. loss of ego boundaries, withdrawal), and in behavior (e.g bizarre or apparently purposeless behavior). Although it affects emotions, it is distinguished from mood disorders in which such disturbances are primary. Similarly, there may be mild impairment of cognitive function, and it is distinguished from the dementias in which disturbed cognitive function is considered primary. Some patients manifest schizophrenic as well as bipolar disorder symptoms and are often given the diagnosis of schizoaffective disorder. {ECO:0000269|PubMed:15645182}. Disease susceptibility may be associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07462; DB02342; DB02105; DB08049; DB00118; DB00714; DB03336; DB00286; DB00255; DB00841; DB00988; DB15488; DB00494; DB00668; DB00783; DB00977; DB01064; DB00968; DB01141; DB03907; DB04820; DB06152; DB11632; DB00252; DB01420; DB00323 Interacts with Q6P5T0; P30518; Q8NFU1; Q8NHW4; P34972; Q96BA8; P50402; Q5JX71; O14843; O00258; P08034; O75712; Q9NTQ9; O95377; Q8TDT2; Q8N6U8; O15529; P31937; Q9H2F3; O95279; Q5SR56; A6NDP7; Q0D2K0; Q7RTS5; Q9UHJ9-5; Q8IY26; Q9H6H4; Q6NTF9-3; O75783; Q99500; Q9Y6D0; Q3KNW5; O60669; P22732; Q96G79; Q5T1Q4; Q9NY26; Q9NP94; Q6P1K1; P30825; Q9UHI5; B2RUZ4; Q9UPZ6; Q96MV1; Q9NV29; A0PK00; Q9NUH8; Q9P0S9; Q14656; Q6UW68; Q9H0R3; O95807; P34981; Q15645; Q15836; O95183; O76024; P30260; Q9H816; Q92997; P29323-3; P22607; P06396; Q15323; Q6A162; P26371; O15116; P20645; O14744; Q5T160; Q9UJD0; Q2MKA7; Q8N488; O75880; Q14141; Q9UNE7; Q15645; Q9NYH9; Q8NA23-2 EC number EC 2.1.1.6 Uniprot keywords 3D-structure; Alternative initiation; Catecholamine metabolism; Cell membrane; Cytoplasm; Direct protein sequencing; Lipid metabolism; Magnesium; Membrane; Metal-binding; Methyltransferase; Neurotransmitter degradation; Phosphoprotein; Proteomics identification; Reference proteome; S-adenosyl-L-methionine; Schizophrenia; Signal-anchor; Transferase; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 23851.2 Length 214 Aromaticity 0.07 Instability index 25.99 Isoelectric point 5.25 Charge (pH=7) -7.75 2D Binding mode Binding energy (Kcal/mol) -9.83 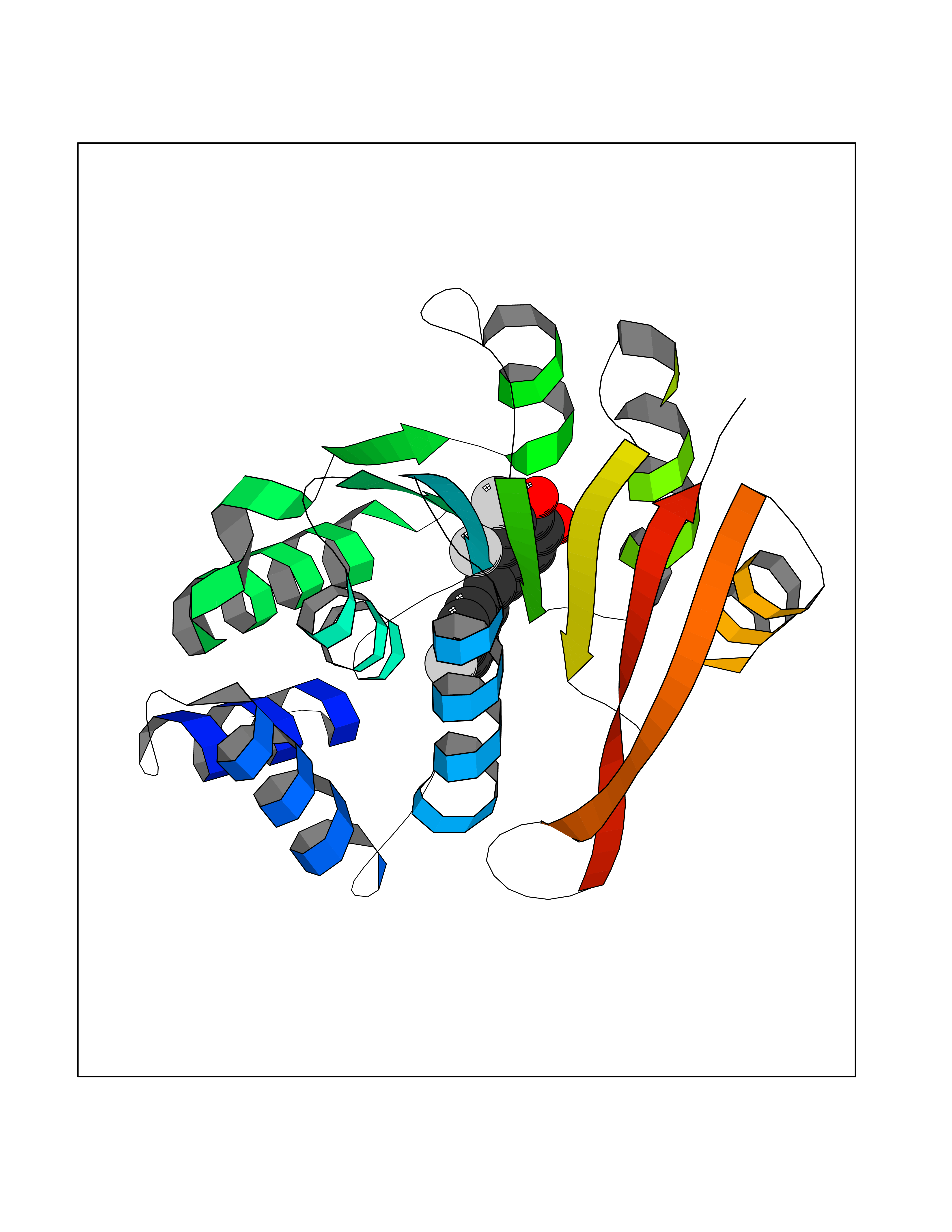 Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence GDTKEQRILNHVLQHAEPGNAQSVLEAIDTYCEQKEWAMNVGDKKGKIVDAVIQEHQPSVLLELGAYCGYSAVRMARLLSPGARLITIEINPDCAAITQRMVDFAGMKDKVTLVVGASQDIIPQLKKKYDVDTLDMVFLDHWKDRYLPDTLLLEECGLLRKGTVLLADNVICPGAPDFLAHVRGSSCFECTHYQSFLEYREVVDGLEKAIYKGP Hydrogen bonds contact Hydrophobic contact | ||||
| 44 | Toll-like receptor 1 (TLR1) | 2Z7X | 7.21 | |
Target general information Gen name TLR1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Toll/interleukin-1 receptor-like protein; TIL; KIAA0012; CD281 Protein family Toll-like receptor family Biochemical class Toll-like receptor Function Specifically recognizes diacylated and triacylated lipopeptides. Cooperates with TLR2 to mediate the innate immune response to bacterial lipoproteins or lipopeptides. Forms the activation cluster TLR2:TLR1:CD14 in response to triacylated lipopeptides, this cluster triggers signaling from the cell surface and subsequently is targeted to the Golgi in a lipid-raft dependent pathway. Acts via MYD88 and TRAF6, leading to NF-kappa-B activation, cytokine secretion and the inflammatory response. Participates in the innate immune response to microbial agents. Related diseases Hao-Fountain syndrome (HAFOUS) [MIM:616863]: An autosomal dominant neurodevelopmental disorder characterized by global developmental delay, varying degrees of intellectual disability, autism spectrum disorder, poor or absent speech, and mild facial dysmorphism. Most patients develop seizures. Additional variable features include hypotonia, hypogonadism in males, and ocular anomalies. {ECO:0000269|PubMed:26365382, ECO:0000269|PubMed:30679821}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q15399; Q9BXR5; O60603; Q9Y2C9 EC number NA Uniprot keywords 3D-structure; Cell membrane; Cytoplasmic vesicle; Direct protein sequencing; Disulfide bond; Glycoprotein; Golgi apparatus; Immunity; Inflammatory response; Innate immunity; Leucine-rich repeat; Membrane; NAD; Proteomics identification; Receptor; Reference proteome; Repeat; Signal; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A,C Molecular weight (Da) 62767.4 Length 555 Aromaticity 0.07 Instability index 38.79 Isoelectric point 6.85 Charge (pH=7) -0.55 2D Binding mode Binding energy (Kcal/mol) -9.84  Molscript Map  Pymol Map 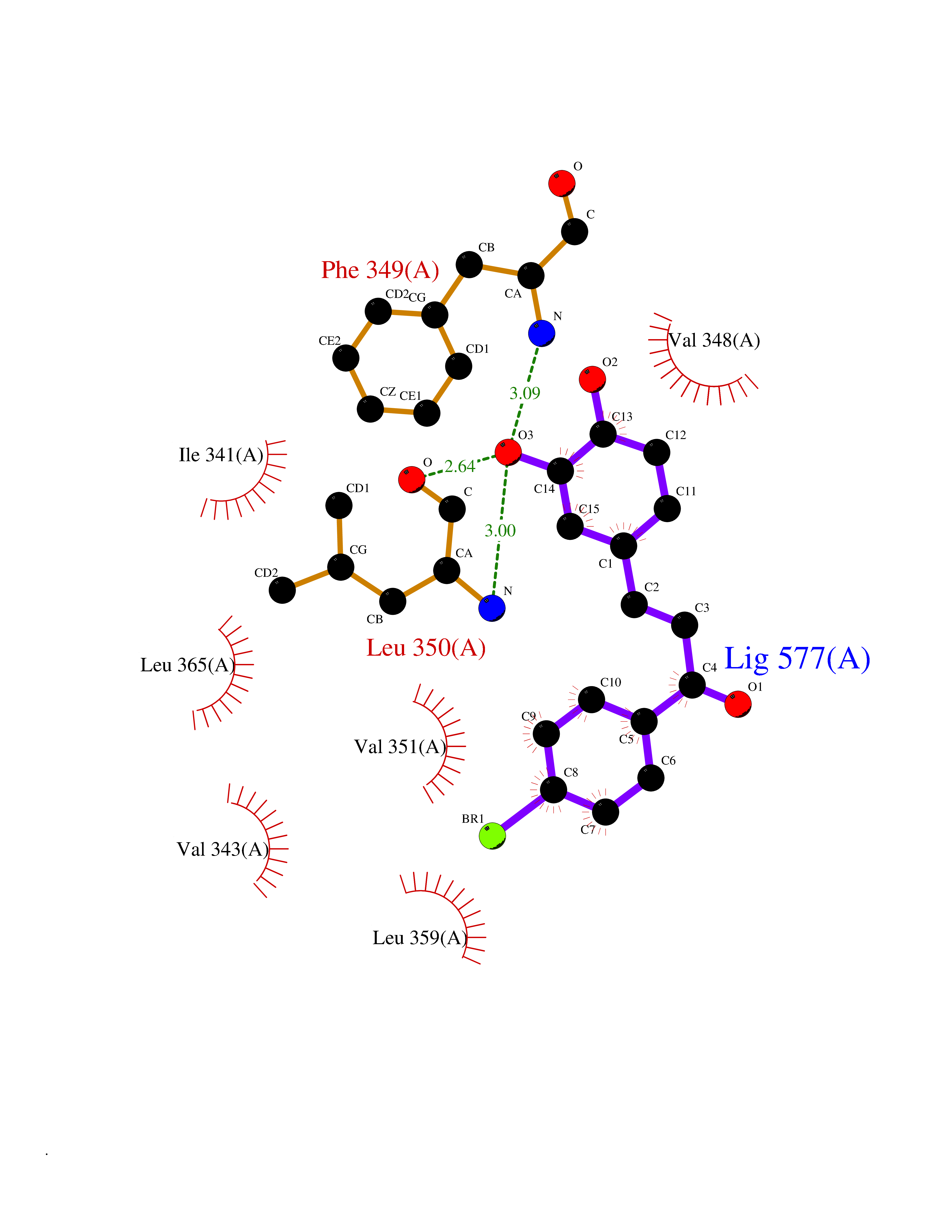 Ligplot Map 3D Binding mode Sequence SLSCDRNGICKGSSGSLNSIPSGLTEAVKSLDLSNNRITYISNSDLQRCVNLQALVLTSNGINTIEEDSFSSLGSLEHLDLSYNYLSNLSSSWFKPLSSLTFLNLLGNPYKTLGETSLFSHLTKLQILRVGNMDTFTKIQRKDFAGLTFLEELEIDASDLQSYEPKSLKSIQNVSHLILHMKQHILLLEIFVDVTSSVECLELRDTDLDTFHFSELSTGETNSLIKKFTFRNVKITDESLFQVMKLLNQISGLLELEFDDCTLNGVGNFRASDNDRVIDPGKVETLTIRRLHIPRFYLFYDLSTLYSLTERVKRITVENSKVFLVPCLLSQHLKSLEYLDLSENLMVEEYLKNSACEDAWPSLQTLILRQNHLASLEKTGETLLTLKNLTNIDISKNSFHSMPETCQWPEKMKYLNLSSTRIHSVTGCIPKTLEILDVSNNNLNLFSLNLPQLKELYISRNKLMTLPDASLLPMLLVLKISRNQLKSVPDGIFDRLTSLQKIWLHTNPWDCSCPRIDYLSRWLNKNSQKEQGSAKCSGSGKPVRSIICPXSKKKK Hydrogen bonds contact Hydrophobic contact | ||||
| 45 | Toll-like receptor 2 (TLR2) | 2Z7X | 7.21 | |
Target general information Gen name TLR2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Toll/interleukin-1 receptor-like protein 4; TIL4; CD282 Protein family Toll-like receptor family Biochemical class Toll-like receptor Function Cooperates with TLR1 or TLR6 to mediate the innate immune response to bacterial lipoproteins or lipopeptides. Acts via MYD88 and TRAF6, leading to NF-kappa-B activation, cytokine secretion and the inflammatory response. May also activate immune cells and promote apoptosis in response to the lipid moiety of lipoproteins. Recognizes mycoplasmal macrophage-activating lipopeptide-2kD (MALP-2), soluble tuberculosis factor (STF), phenol-soluble modulin (PSM) and B. burgdorferi outer surface protein A lipoprotein (OspA-L) cooperatively with TLR6. Stimulation of monocytes in vitro with M. tuberculosis PstS1 induces p38 MAPK and ERK1/2 activation primarily via this receptor, but also partially via TLR4. MAPK activation in response to bacterial peptidoglycan also occurs via this receptor. Acts as a receptor for M. tuberculosis lipoproteins LprA, LprG, LpqH and PstS1, some lipoproteins are dependent on other coreceptors (TLR1, CD14 and/or CD36); the lipoproteins act as agonists to modulate antigen presenting cell functions in response to the pathogen. M. tuberculosis HSP70 (dnaK) but not HSP65 (groEL-2) acts via this protein to stimulate NF-kappa-B expression. Recognizes M. tuberculosis major T-antigen EsxA (ESAT-6) which inhibits downstream MYD88-dependent signaling (shown in mouse). Forms activation clusters composed of several receptors depending on the ligand, these clusters trigger signaling from the cell surface and subsequently are targeted to the Golgi in a lipid-raft dependent pathway. Forms the cluster TLR2:TLR6:CD14:CD36 in response to diacylated lipopeptides and TLR2:TLR1:CD14 in response to triacylated lipopeptides. Required for normal uptake of M. tuberculosis, a process that is inhibited by M. tuberculosis LppM. Cooperates with LY96 to mediate the innate immune response to bacterial lipoproteins and other microbial cell wall components. Related diseases Squalene synthase deficiency (SQSD) [MIM:618156]: An autosomal recessive disorder characterized by profound developmental delay, brain abnormalities, 2/3 syndactyly of the toes, facial dysmorphisms, low total and LDL-cholesterol, and abnormal urine organic acids. {ECO:0000269|PubMed:29909962}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00210; DB05475; DB00045; DB16474; DB03963; DB11601 Interacts with P61073; P00533; Q99836; Q15399; Q9BXR5; O60603; Q9Y2C9; Q96DA0 EC number NA Uniprot keywords 3D-structure; Cytoplasmic vesicle; Disulfide bond; Glycoprotein; Immunity; Inflammatory response; Innate immunity; Isopeptide bond; Leucine-rich repeat; Membrane; NAD; Proteomics identification; Receptor; Reference proteome; Repeat; Signal; Transmembrane; Transmembrane helix; Ubl conjugation Protein physicochemical properties Chain ID A,C Molecular weight (Da) 62767.4 Length 555 Aromaticity 0.07 Instability index 38.79 Isoelectric point 6.85 Charge (pH=7) -0.55 2D Binding mode Binding energy (Kcal/mol) -9.83  Molscript Map 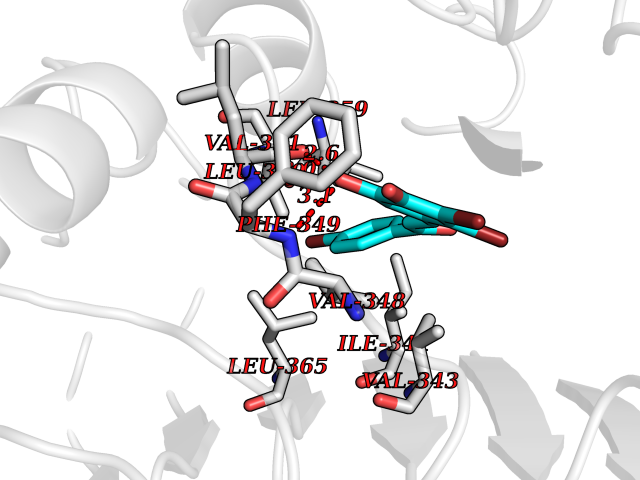 Pymol Map 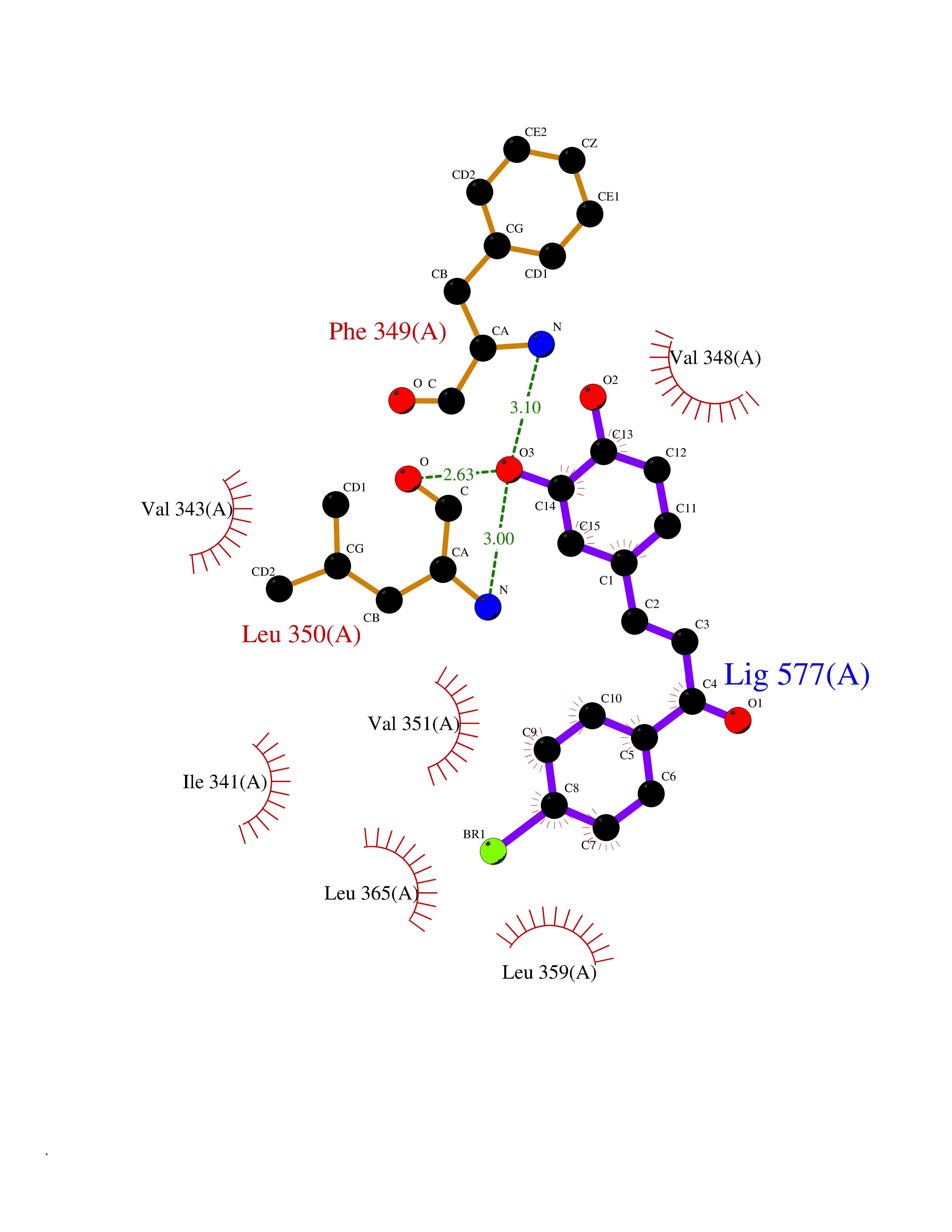 Ligplot Map 3D Binding mode Sequence SLSCDRNGICKGSSGSLNSIPSGLTEAVKSLDLSNNRITYISNSDLQRCVNLQALVLTSNGINTIEEDSFSSLGSLEHLDLSYNYLSNLSSSWFKPLSSLTFLNLLGNPYKTLGETSLFSHLTKLQILRVGNMDTFTKIQRKDFAGLTFLEELEIDASDLQSYEPKSLKSIQNVSHLILHMKQHILLLEIFVDVTSSVECLELRDTDLDTFHFSELSTGETNSLIKKFTFRNVKITDESLFQVMKLLNQISGLLELEFDDCTLNGVGNFRASDNDRVIDPGKVETLTIRRLHIPRFYLFYDLSTLYSLTERVKRITVENSKVFLVPCLLSQHLKSLEYLDLSENLMVEEYLKNSACEDAWPSLQTLILRQNHLASLEKTGETLLTLKNLTNIDISKNSFHSMPETCQWPEKMKYLNLSSTRIHSVTGCIPKTLEILDVSNNNLNLFSLNLPQLKELYISRNKLMTLPDASLLPMLLVLKISRNQLKSVPDGIFDRLTSLQKIWLHTNPWDCSCPRIDYLSRWLNKNSQKEQGSAKCSGSGKPVRSIICPXSKKKK Hydrogen bonds contact Hydrophobic contact | ||||
| 46 | Serine--pyruvate aminotransferase | 5HHY | 7.20 | |
Target general information Gen name AGXT Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms AGT1;SPAT Protein family Class-V pyridoxal-phosphate-dependent aminotransferase family Biochemical class Transferase Function Alanine-glyoxylate transaminase activity.Amino acid binding.Identical protein binding.Protein homodimerization activity.Protein self-association.Pyridoxal phosphate binding.Receptor binding.Serine-pyruvate transaminase activity.Transaminase activity. Related diseases Hyperoxaluria primary 1 (HP1) [MIM:259900]: An inborn error of glyoxylate metabolism characterized by increased excretion of oxalate and glycolate, and progressive tissue accumulation of insoluble calcium oxalate. Affected individuals are at risk for nephrolithiasis, nephrocalcinosis and early onset end-stage renal disease. {ECO:0000269|PubMed:10394939, ECO:0000269|PubMed:10453743, ECO:0000269|PubMed:10541294, ECO:0000269|PubMed:10862087, ECO:0000269|PubMed:10960483, ECO:0000269|PubMed:12559847, ECO:0000269|PubMed:12777626, ECO:0000269|PubMed:1301173, ECO:0000269|PubMed:1349575, ECO:0000269|PubMed:15253729, ECO:0000269|PubMed:15849466, ECO:0000269|PubMed:15961946, ECO:0000269|PubMed:15963748, ECO:0000269|PubMed:16971151, ECO:0000269|PubMed:1703535, ECO:0000269|PubMed:17495019, ECO:0000269|PubMed:2039493, ECO:0000269|PubMed:23229545, ECO:0000269|PubMed:24055001, ECO:0000269|PubMed:24934730, ECO:0000269|PubMed:26149463, ECO:0000269|PubMed:8101040, ECO:0000269|PubMed:9192270, ECO:0000269|PubMed:9604803}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08060; DB00160; DB02079; DB00145; DB04083; DB00114; DB00133 Interacts with Q5BKX5-3; P21549; P0C7T5; Q9NR55; Q9H5F2; Q9UKJ5; A8MQ03; O43281-2; A1KXE4-2; Q5TD97; P49356; P53539; P49639; Q15323; O76011; O76014; Q6A162; Q07627; P60328; Q9BYR8; Q3LI67; Q8IUC2; Q9BYQ4; O60711; Q99750; Q5VZ52; P0DPK4; O43482; P50542-1; O15496; Q6ZR37; Q9NQX0; Q8HWS3; Q5W111-2; Q8WVR3; Q6DKK2 EC number 2.6.1.44; 2.6.1.51 Uniprot keywords 3D-structure; Acetylation; Aminotransferase; Disease variant; Peroxisome; Phosphoprotein; Proteomics identification; Pyridoxal phosphate; Reference proteome; Transferase Protein physicochemical properties Chain ID A,B Molecular weight (Da) 84537 Length 771 Aromaticity 0.07 Instability index 36.81 Isoelectric point 8.42 Charge (pH=7) 6.81 2D Binding mode Binding energy (Kcal/mol) -9.81  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence LLVTPPKALLKPLSIPNQLLLGPGPSNLPPRIMAAGGLQMIGSMSKDMYQIMDEIKEGIQYVFQTRNPLTLVISGSGHCALEAALVNVLEPGDSFLVGANGIWGQRAVDIGERIGARVHPMTKDPGGHYTLQEVEEGLAQHKPVLLFLTHGESSTGVLQPLDGFGELCHRYKCLLLVDSVASLGGTPLYMDRQGIDILYSGSQKALNAPPGTSLISFSDKAKKKMYSRKTKPFSFYLDIKWLANFWGCDDQPRMYHHTIPVISLYSLRESLALIAEQGLENSWRQHREAAAYLHGRLQALGLQLFVKDPALRLPTVTTVAVPAGYDWRDIVSYVIDHFDIEIMGGLGPSTGKVLRIGLLGCNATRENVDRVTEALRAALQHCPKKLLVTPPKALLKPLSIPNQLLLGPGPSNLPPRIMAAGGLQMIGSMSKDMYQIMDEIKEGIQYVFQTRNPLTLVISGSGHCALEAALVNVLEPGDSFLVGANGIWGQRAVDIGERIGARVHPMTKDPGGHYTLQEVEEGLAQHKPVLLFLTHGESSTGVLQPLDGFGELCHRYKCLLLVDSVASLGGTPLYMDRQGIDILYSGSQKALNAPPGTSLISFSDKAKKKMYSRKTKPFSFYLDIKWLANFWGCDDQPRMYHHTIPVISLYSLRESLALIAEQGLENSWRQHREAAAYLHGRLQALGLQLFVKDPALRLPTVTTVAVPAGYDWRDIVSYVIDHFDIEIMGGLGPSTGKVLRIGLLGCNATRENVDRVTEALRAALQHCPKKK Hydrogen bonds contact Hydrophobic contact | ||||
| 47 | Xanthine dehydrogenase/oxidase (XDH) | 2E1Q | 7.20 | |
Target general information Gen name XDH Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Xanthine oxidase; Xanthine dehydrogenase; XDHA Protein family Xanthine dehydrogenase family Biochemical class CH/CH(2) oxidoreductase Function Catalyzes the oxidation of hypoxanthine to xanthine. Catalyzes the oxidation of xanthine to uric acid. Contributes to the generation of reactive oxygen species. Has also low oxidase activity towards aldehydes (in vitro). Key enzyme in purine degradation. Related diseases Xanthinuria 1 (XAN1) [MIM:278300]: A disorder characterized by excretion of very large amounts of xanthine in the urine and a tendency to form xanthine stones. Uric acid is strikingly diminished in serum and urine. XAN1 is due to isolated xanthine dehydrogenase deficiency. Patients can metabolize allopurinol. {ECO:0000269|PubMed:10844591, ECO:0000269|PubMed:11379872, ECO:0000269|PubMed:14551354, ECO:0000269|PubMed:9153281}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00640; DB00041; DB00437; DB00993; DB00958; DB01136; DB00856; DB00515; DB00746; DB03328; DB00997; DB03516; DB12466; DB04854; DB03147; DB04335; DB01020; DB00583; DB00170; DB01033; DB00157; DB03841; DB00336; DB01250; DB05262; DB06478; DB01168; DB00339; DB00127; DB01685; DB00831 Interacts with Q9Y3R0-3 EC number NA Uniprot keywords 2Fe-2S; 3D-structure; Cytoplasm; Disease variant; Disulfide bond; FAD; Flavoprotein; Iron; Iron-sulfur; Metal-binding; Molybdenum; NAD; Oxidoreductase; Peroxisome; Proteomics identification; Reference proteome; Secreted Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 143697 Length 1307 Aromaticity 0.08 Instability index 37.9 Isoelectric point 8.01 Charge (pH=7) 7.07 2D Binding mode Binding energy (Kcal/mol) -9.83  Molscript Map 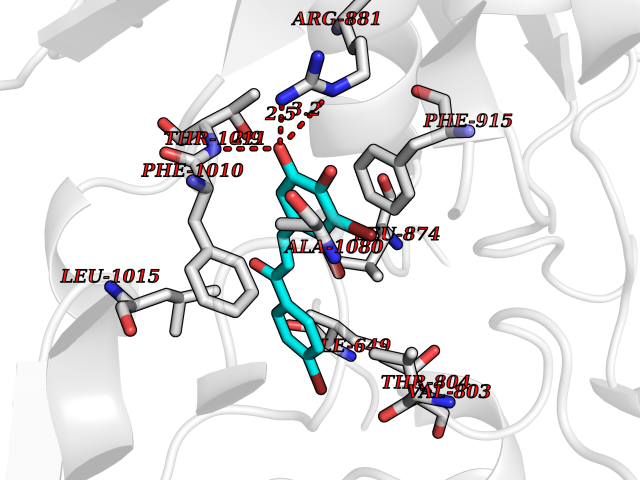 Pymol Map 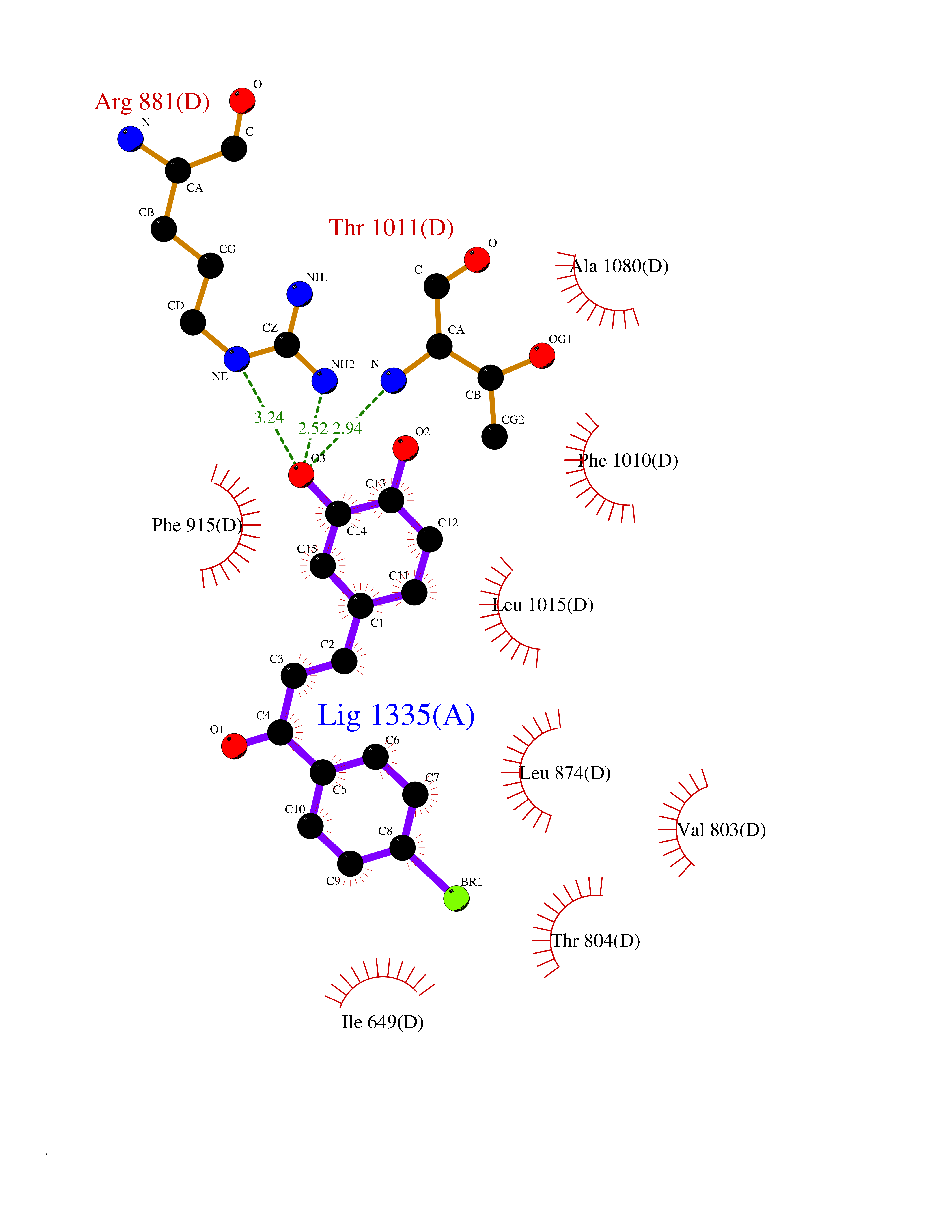 Ligplot Map 3D Binding mode Sequence ADKLVFFVNGRKVVEKNADPETTLLAYLRRKLGLSGTKLGCGEGGCGACTVMLSKYDRLQNKIVHFSANACLAPICSLHHVAVTTVEGIGSTKTRLHPVQERIAKSHGSQCGFCTPGIVMSMYTLLRNQPEPTMEEIENAFQGNLCRCTGYRPILQGFRTFARDGSPSLFKPEEFTPLDPTQEPIFPPELLRLKDTPRKQLRFEGERVTWIQASTLKELLDLKAQHPDAKLVVGNTEIGIEMKFKNMLFPMIVCPAWIPELNSVEHGPDGISFGAACPLSIVEKTLVDAVAKLPAQKTEVFRGVLEQLRWFAGKQVKSVASVGGNIITASPISDLNPVFMASGAKLTLVSRGTRRTVQMDHTFFPGYRKTLLSPEEILLSIEIPYSREGEYFSAFKQASRREDDIAKVTSGMRVLFKPGTTEVQELALCYGGMANRTISALKTTQRQLSKLWKEELLQDVCAGLAEELHLPPDAPGGMVDFRCTLTLSFFFKFYLTVLQKLGQENLEDKCGKLDPTFASATLLFQKDPPADVQLFQEVPKGQSEEDMVGRPLPHLAADMQASGEAVYCDDIPRYENELSLRLVTSTRAHAKIKSIDTSEAKKVPGFVCFISADDVPGSNITGICNDETVFAKDKVTCVGHIIGAVVADTPEHTQRAAQGVKITYEELPAIITIEDAIKNNSFYGPELKIEKGDLKKGFSEADNVVSGEIYIGGQEHFYLETHCTIAVPKGEAGEMELFVSTQNTMKTQSFVAKMLGVPANRIVVRVKRMGGGFGGKVTRSTVVSTAVALAAYKTGRPVRCMLDRDEDMLITGGRHPFLARYKVGFMKTGTVVALEVDHFSNVGNTQDLSQSIMERALFHMDNCYKIPNIRGTGRLCKTNLPSNTAFRGFGGPQGMLIAECWMSEVAVTCGMPAEEVRRKNLYKEGDLTHFNQKLEGFTLPRCWEECLASSQYHARKSEVDKFNKENCWKKRGLCIIPTKFGISFTVPFLNQAGALLHVYTDGSVLLTHGGTEMGQGLHTKMVQVASRALKIPTSKIYISETSTNTVPNTSPTAASVSADLNGQAVYAACQTILKRLEPYKKKNPSGSWEDWVTAAYMDTVSLSATGFYRTPNLGYSFETNSGNPFHYFSYGVACSEVEIDCLTGDHKNLRTDIVMDVGSSLNPAIDIGQVEGAFVQGLGLFTLEELHYSPEGSLHTRGPSTYKIPAFGSIPIEFRVSLLRDCPNKKAIYASKAVGEPPLFLAASIFFAIKDAIRAARAQHTGNNVKELFRLDSPATPEKIRNACVDKFTTLCVTGVPENCKPWSVRV Hydrogen bonds contact Hydrophobic contact | ||||
| 48 | Glutathione S-transferase kappa 1 | 3RPN | 7.20 | |
Target general information Gen name GSTK1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms HDCMD47P Protein family GST superfamily, Kappa family Biochemical class Transferase / transferase inhibitor Function Glutathione peroxidase activity.Glutathione transferase activity.Protein disulfide oxidoreductase activity.Receptor binding. Related diseases Dyskinesia, limb and orofacial, infantile-onset (IOLOD) [MIM:616921]: An autosomal recessive, early-onset hyperkinetic movement disorder characterized by axial hypotonia, dyskinesia of the limbs and trunk, orofacial dyskinesia, drooling, and dysarthria. The severity of the hyperkinesis is variable. {ECO:0000269|PubMed:27058446}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Striatal degeneration, autosomal dominant 2 (ADSD2) [MIM:616922]: An autosomal dominant disorder characterized by striatal degeneration and dysfunction of basal ganglia, resulting in hyperkinesis. {ECO:0000269|PubMed:27058447}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00143; DB04700 Interacts with O95273; Q8IZU0; Q60994; Q7Z3Y8 EC number 2.5.1.18 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Peroxisome; Proteomics identification; Reference proteome; Transferase Protein physicochemical properties Chain ID A,B,C,D,E,F Molecular weight (Da) 24811.7 Length 220 Aromaticity 0.08 Instability index 47.17 Isoelectric point 7.96 Charge (pH=7) 1.39 2D Binding mode Binding energy (Kcal/mol) -9.82  Molscript Map 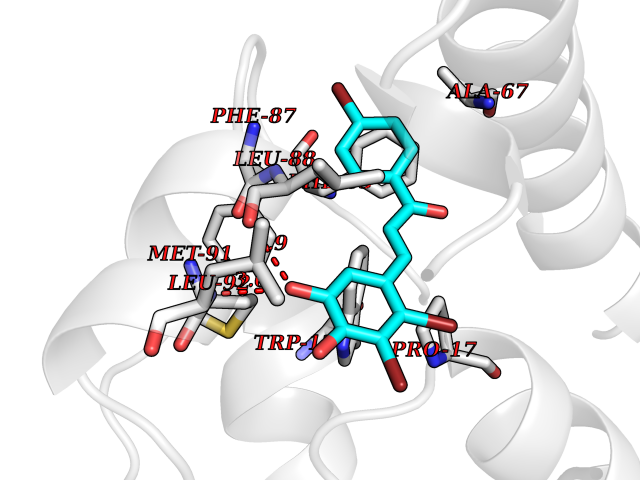 Pymol Map 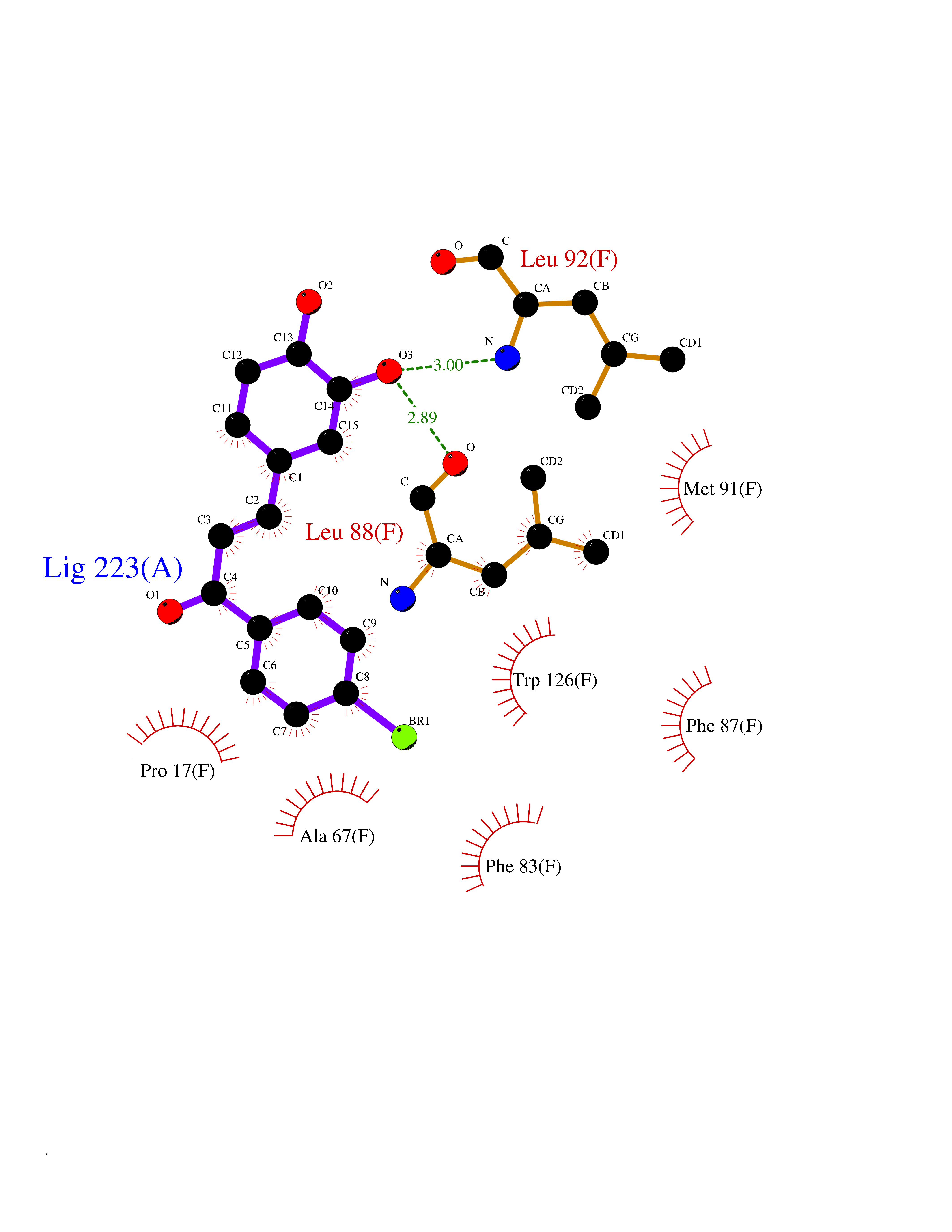 Ligplot Map 3D Binding mode Sequence GPLPRTVELFYDVLSPYSWLGFEILCRYQNIWNINLQLRPSLITGIMKDSGNKPPGLLPRKGLYMANDLKLLRHHLQIPIHFPKDFLSVMLEKGSLSAMRFLTAVNLEHPEMLEKASRELWMRVWSRNEDITEPQSILAAAEKAGMSAEQAQGLLEKIATPKVKNQLKETTEAACRYGAFGLPITVAHVDGQTHMLFGSDRMELLAHLLGEKWMGPIPPA Hydrogen bonds contact Hydrophobic contact | ||||
| 49 | "Periplasmic trehalase (EC 3.2.1.28) (Alpha,alpha-trehalase) (Alpha,alpha-trehalose glucohydrolase) (Tre37A)" | 2JG0 | 7.19 | |
Target general information Gen name treA Organism Escherichia coli (strain K12) Uniprot ID TTD ID NA Synonyms JW1186;osmA;b1197 Protein family Glycosyl hydrolase 37 family Biochemical class NA Function Provides the cells with the ability to utilize trehalose at high osmolarity by splitting it into glucose molecules that can subsequently be taken up by the phosphotransferase-mediated uptake system. Related diseases SRC kinase activity has been shown to be increased in several tumor tissues and tumor cell lines such as colon carcinoma cells. {ECO:0000269|PubMed:2498394, ECO:0000269|PubMed:3093483}.; DISEASE: Thrombocytopenia 6 (THC6) [MIM:616937]: A form of thrombocytopenia, a hematologic disorder defined by a decrease in the number of platelets in circulating blood, resulting in the potential for increased bleeding and decreased ability for clotting. THC6 is an autosomal dominant form. Affected individuals may also have bone abnormalities and an increased risk for myelofibrosis. {ECO:0000269|PubMed:26936507}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number 3.2.1.28 Uniprot keywords 3D-structure; Direct protein sequencing; Glycosidase; Hydrolase; Periplasm; Reference proteome; Signal Protein physicochemical properties Chain ID A Molecular weight (Da) 57508.9 Length 507 Aromaticity 0.11 Instability index 48.32 Isoelectric point 5.48 Charge (pH=7) -10.13 2D Binding mode Binding energy (Kcal/mol) -9.81  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence PQPPDILLGPLFNDVQNAKLFPDQKTFADAVPNSDPLMILADYRMQQNQSGFDLRHFVNVNFTLPKYVPPEGQSLREHIDGLWPVLTRSTENTEKWDSLLPLPEPYVVPGGRFREVYYWDSYFTMLGLAESGHWDKVADMVANFAHEIDTYGHIPNGNRSYYLSRSQPPFFALMVELLAQHEGDAALKQYLPQMQKEYAYWMDGVENLQAGQQEKRVVKLQDGTLLNRYWDDRDTPRPESWVEDIATAKSNPNRPATEIYRDLRSAAASGWDFSSRWMDNPQQLNTLRTTSIVPVDLNSLMFKMEKILARASKAAGDNAMANQYETLANARQKGIEKYLWNDQQGWYADYDLKSHKVRNQLTAAALFPLYVNAAAKDRANKMATATKTHLLQPGGLNTTSVKSGQQWDAPNGWAPLQWVATEGLQNYGQKEVAMDISWHFLTNVQHTYDREKKLVEKYDVSTTGTGGGGGEYPLQDGFGWTNGVTLKMLDLICPKEQPCDNVPATRP Hydrogen bonds contact Hydrophobic contact | ||||
| 50 | Thiopurine S-methyltransferase | 2BZG | 7.19 | |
Target general information Gen name TPMT Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Class I-like SAM-binding methyltransferase superfamily, TPMT family Biochemical class Transferase Function Thiopurine S-methyltransferase activity. Related diseases Cerebral creatine deficiency syndrome 3 (CCDS3) [MIM:612718]: An autosomal recessive disorder characterized by developmental delay/regression, intellectual disability, severe disturbance of expressive and cognitive speech, and severe depletion of creatine/phosphocreatine in the brain. Most patients develop a myopathy characterized by muscle weakness and atrophy later in life. {ECO:0000269|PubMed:11555793, ECO:0000269|PubMed:20682460, ECO:0000269|PubMed:22386973, ECO:0000269|PubMed:23660394, ECO:0000269|PubMed:23770102, ECO:0000269|PubMed:26490222, ECO:0000269|PubMed:27233232}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Fanconi renotubular syndrome 1 (FRTS1) [MIM:134600]: A form of Fanconi renotubular syndrome, a disease due to a generalized dysfunction of the proximal kidney tubule resulting in decreased solute and water reabsorption. Patients have polydipsia and polyuria with phosphaturia, glycosuria and aminoaciduria. They may develop hypophosphatemic rickets or osteomalacia, acidosis and a tendency toward dehydration. Some eventually develop renal insufficiency. FRTS1 inheritance is autosomal dominant. {ECO:0000269|PubMed:29654216}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00993; DB00436; DB01327; DB01033; DB01250; DB01021 Interacts with Q8TAP4-4; Q15047-2; P61981 EC number 2.1.1.67 Uniprot keywords 3D-structure; Acetylation; Cytoplasm; Direct protein sequencing; Methyltransferase; Phosphoprotein; Proteomics identification; Reference proteome; S-adenosyl-L-methionine; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 25971.5 Length 229 Aromaticity 0.12 Instability index 32.58 Isoelectric point 6.74 Charge (pH=7) -0.6 2D Binding mode Binding energy (Kcal/mol) -9.8 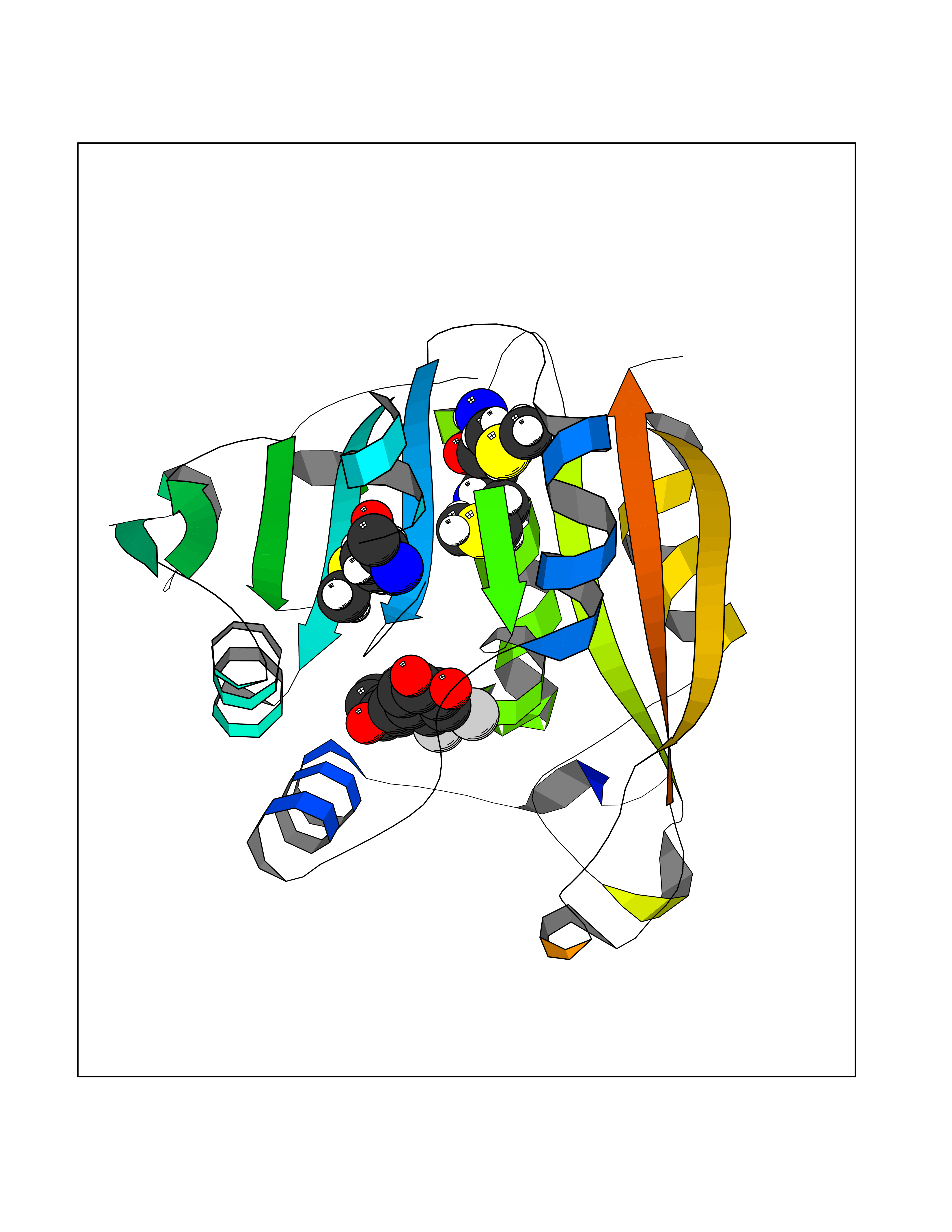 Molscript Map 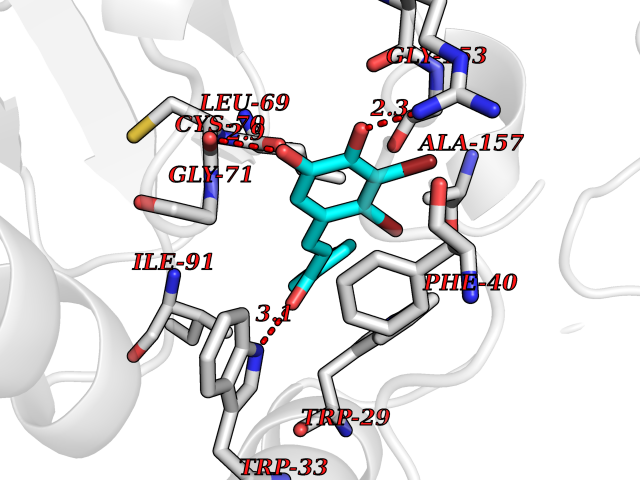 Pymol Map 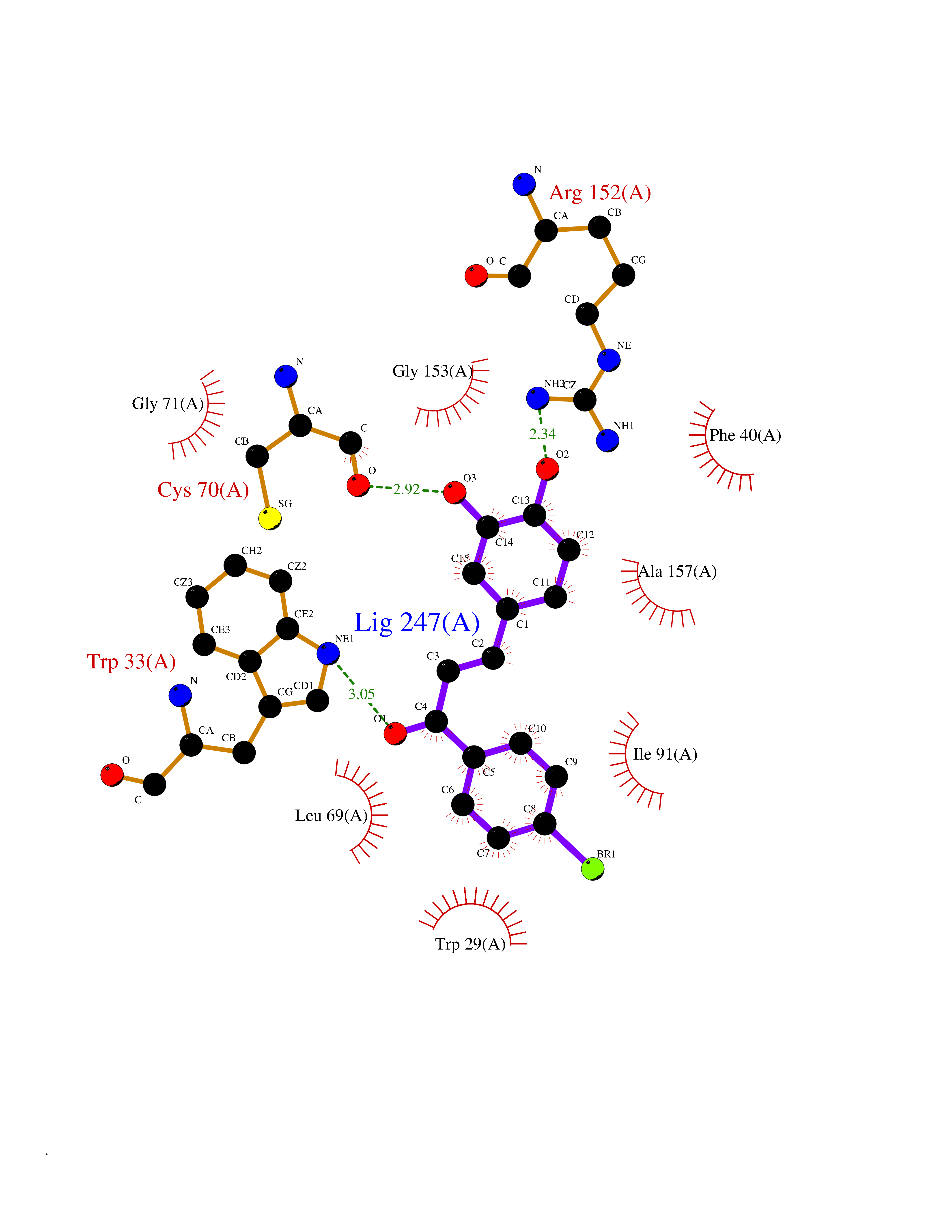 Ligplot Map 3D Binding mode Sequence EVQKNQVLTLEEWQDKWVNGKTAFHQEQGHQLLKKHLDTFLKGKSGLRVFFPLCGKAVEXKWFADRGHSVVGVEISELGIQEFFTEQNLSYSEEPITEIPGTKVFKSSSGNISLYCCSIFDLPRTNIGKFDXIWDRGALVAINPGDRKCYADTXFSLLGKKFQYLLCVLSYDPTKHPGPPFYVPHAEIERLFGKICNIRCLEKVDAFEERHKSWGIDCLFEKLYLLTEK Hydrogen bonds contact Hydrophobic contact | ||||
| 51 | Dihydroorotate dehydrogenase (quinone), mitochondrial | 4CQ8 | 7.19 | |
Target general information Gen name PFF0160c Organism Plasmodium falciparum (isolate 3D7) Uniprot ID TTD ID NA Synonyms NA Protein family Dihydroorotate dehydrogenase family, Type 2 subfamily Biochemical class Oxidoreductase Function Dihydroorotate dehydrogenase activity. Related diseases Combined oxidative phosphorylation deficiency 33 (COXPD33) [MIM:617713]: An autosomal recessive disorder caused by multiple mitochondrial respiratory chain defects and impaired mitochondrial energy metabolism. Clinical manifestations are highly variable. Affected infants present with cardiomyopathy accompanied by multisystemic features involving liver, kidney, and brain. Death in infancy is observed in some patients. Children and adults present with myopathy and progressive external ophthalmoplegia. {ECO:0000269|PubMed:28942965}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01117 Interacts with NA EC number 1.3.5.2 Uniprot keywords 3D-structure; Flavoprotein; FMN; Membrane; Mitochondrion; Mitochondrion inner membrane; Oxidoreductase; Pyrimidine biosynthesis; Reference proteome; Transit peptide; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A,B Molecular weight (Da) 42573.5 Length 378 Aromaticity 0.1 Instability index 36.63 Isoelectric point 8.21 Charge (pH=7) 3.17 2D Binding mode Binding energy (Kcal/mol) -9.81 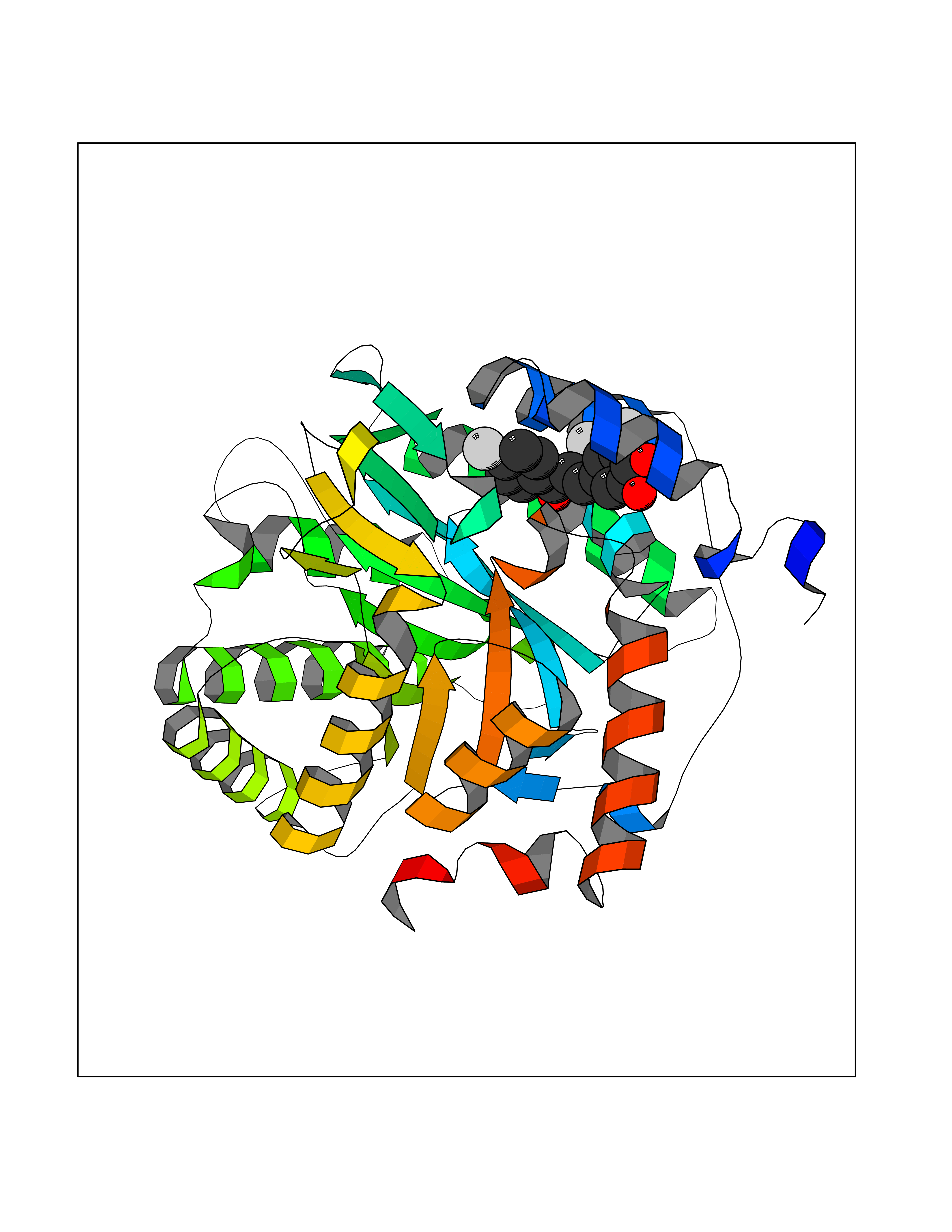 Molscript Map 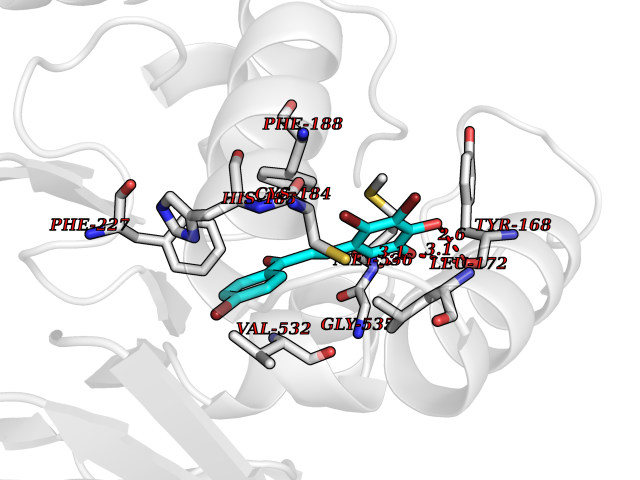 Pymol Map 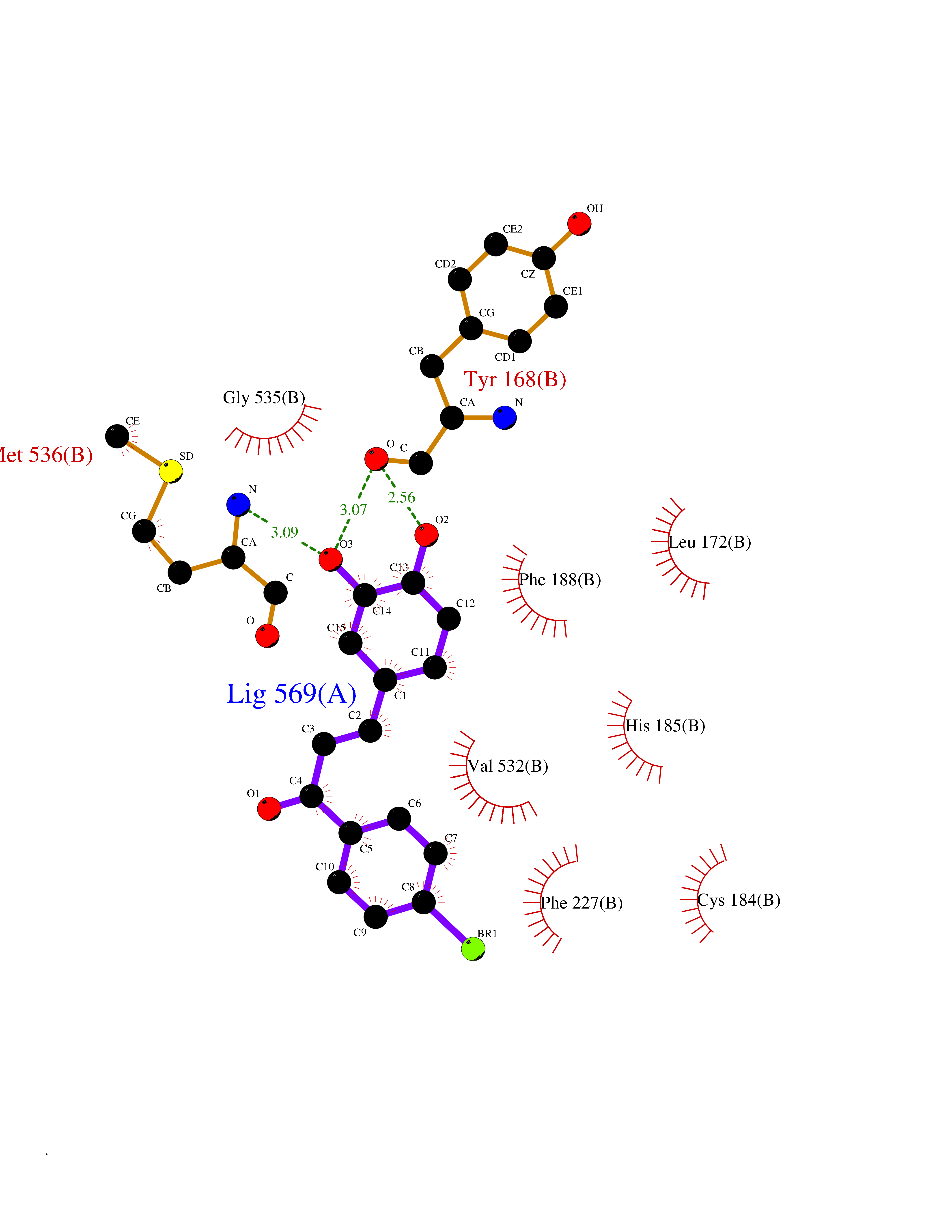 Ligplot Map 3D Binding mode Sequence ADPFESYNPEFFLYDIFLKFCLKYIDGEICHDLFLLLGKYNILPYDTSNDSIYACTNIKHLDFINPFGVAAGFDKNGVCIDSILKLGFSFIEIGTITPRGQTGNAKPRIFRDVESRSIINSCGFNNMGCDKVTENLILFRKRQEEDKLLSKHIVGVSIGKNKDTVNIVDDLKYCINKIGRYADYIAINVSSPNTPGLRDNQEAGKLKNIILSVKEEIDNLEKNNFLWFNTTKKKPLVFVKLAPDLNQEQKKEIADVLLETNIDGMIISNTTTQINDIKSFENKKGGVSGAKLKDISTKFICEMYNYTNKQIPIIASGGIFSGLDALEKIEAGASVCQLYSCLVFNGMKSAVQIKRELNHLLYQRGYYNLKEAIGRKHS Hydrogen bonds contact Hydrophobic contact | ||||
| 52 | WNK lysine-deficient protein kinase 3 (WNK3) | 5O2B | 7.19 | |
Target general information Gen name WNK3 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Serine/threonine-protein kinase WNK3; Protein kinase with no lysine 3; Protein kinase lysine-deficient 3; KIAA1566 Protein family Protein kinase superfamily, Ser/Thr protein kinase family, WNK subfamily Biochemical class NA Function Serine/threonine kinase which plays an important role in the regulation of electrolyte homeostasis, cell signaling, survival and proliferation. Acts as an activator and inhibitor of sodium-coupled chloride cotransporters and potassium-coupled chloride cotransporters respectively. Phosphorylates WNK4. Regulates the phosphorylation of SLC12A1 and SLC12A2. Increases Ca(2+) influx mediated by TRPV5 and TRPV6 by enhancing their membrane expression level via a kinase-dependent pathway. Inhibits the activity of KCNJ1 by decreasing its expression at the cell membrane in a non-catalytic manner. Related diseases Prieto syndrome (PRS) [MIM:309610]: An X-linked recessive disorder characterized by impaired intellectual development, developmental delay, autism spectrum disorder, variable epilepsy, craniofacial dysmorphism, and structural brain abnormalities including polymicrogyria and cerebral atrophy. {ECO:0000269|PubMed:35678782}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P42574; P52954; Q04864-2 EC number EC 2.7.11.1 Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Cytoplasm; Disease variant; Intellectual disability; Kinase; Nucleotide-binding; Phosphoprotein; Proteomics identification; Reference proteome; Serine/threonine-protein kinase; Transferase; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 30818.4 Length 269 Aromaticity 0.1 Instability index 41.03 Isoelectric point 6.27 Charge (pH=7) -2.07 2D Binding mode Binding energy (Kcal/mol) -9.8 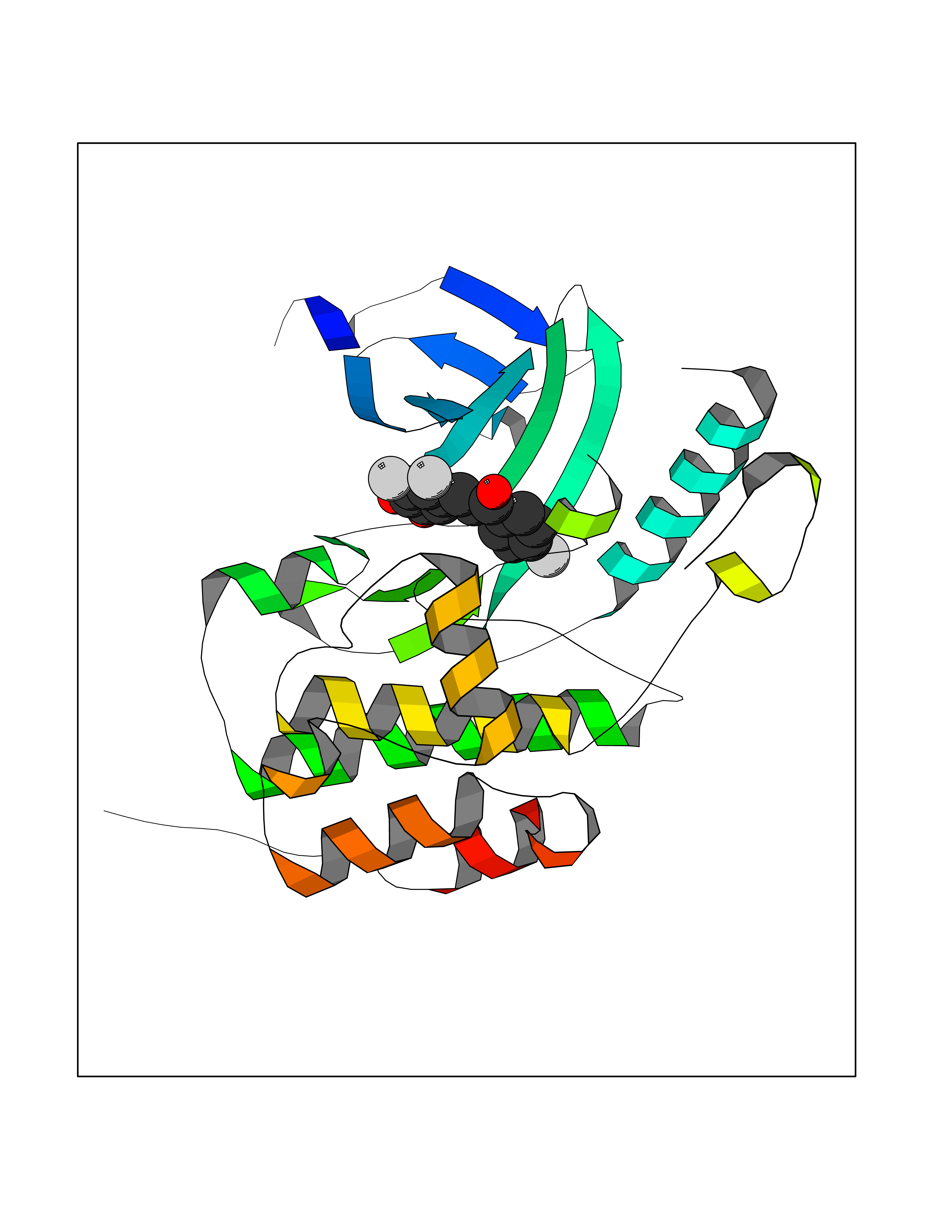 Molscript Map 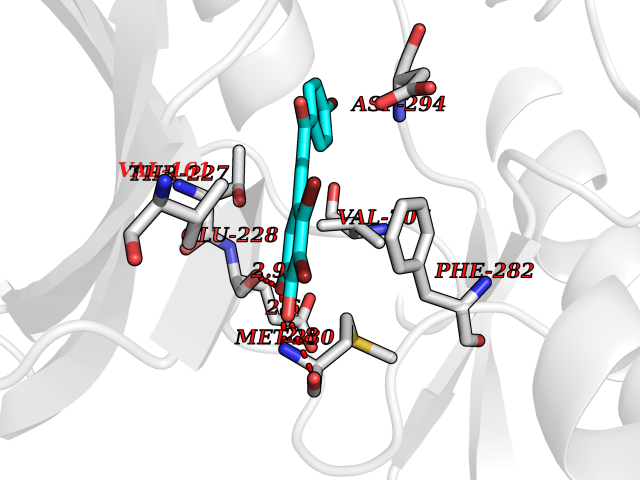 Pymol Map 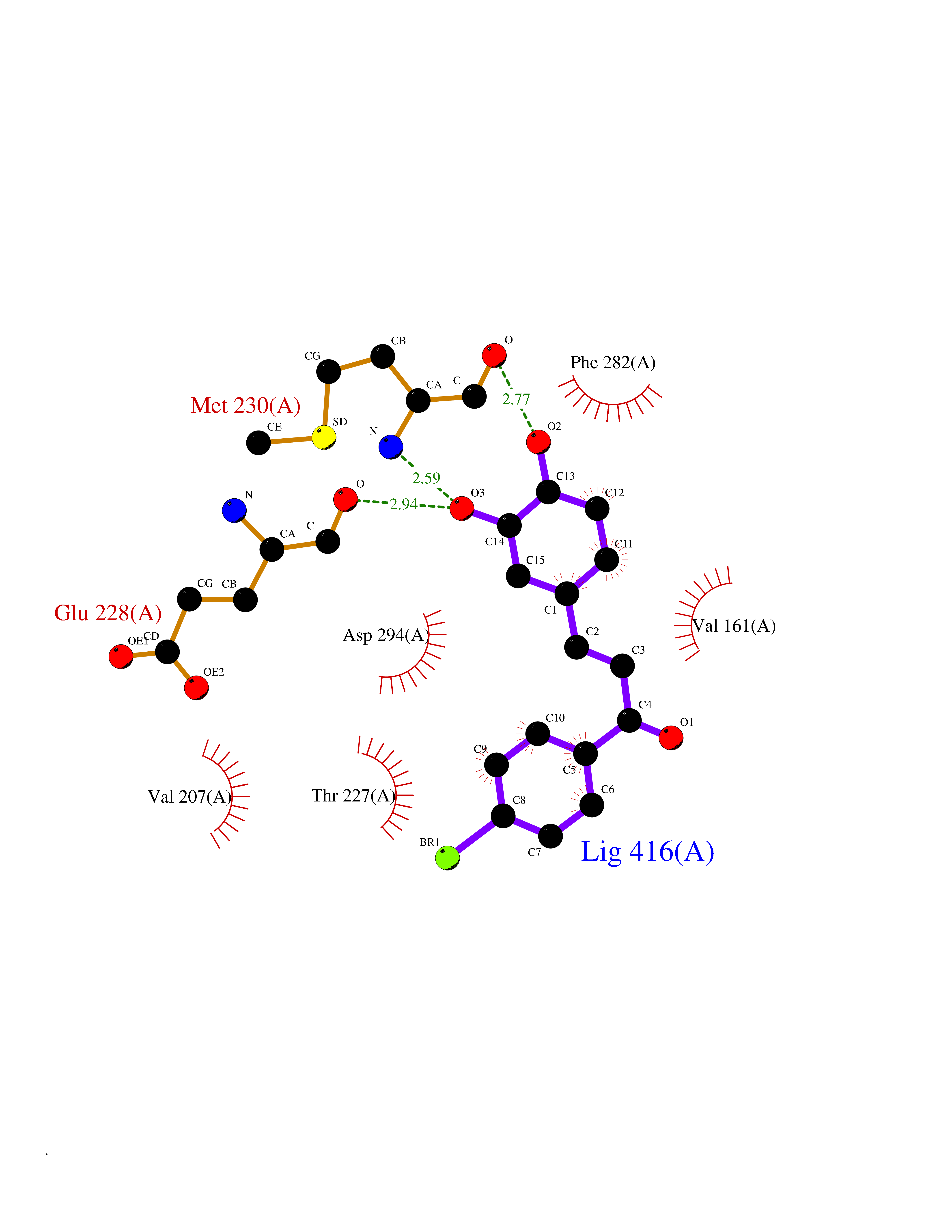 Ligplot Map 3D Binding mode Sequence MEAEMKAVATSPSGRFLKFDIELGRGAFKTVYKGLDTETWVEVAWCELQLTKAEQQRFKEEAEMLKGLQHPNIVRFYDSWESIKCIVLVTELMTSGTLKTYLKRFKVMKPKVLRSWCRQILKGLQFLHTRTPPIIHRDLKCDNIFITGPTGSVKIGDLGLATLMIGTPEFMAPEMYEEHYDESVDVYAFGMCMLEMATSEYPYSECQNAAQIYRKVTSGIKPASFNKVTDPEVKEIIEGCIRQNKSERLSIRDLLNHAFFAEDTGLRVE Hydrogen bonds contact Hydrophobic contact | ||||
| 53 | Bacterial NADH-dependent enoyl-ACP reductase (Bact fabI) | 1MFP | 7.19 | |
Target general information Gen name Bact fabI Organism Escherichia coli (strain K12) Uniprot ID TTD ID Synonyms Enoyl[acylcarrierprotein] reductase [NADH] FabI; ENR of Mycobacterium tuberculosis Protein family Short-chain dehydrogenases/reductases (SDR) family, FabI subfamily Biochemical class Short-chain dehydrogenases reductase Function Catalyzes the reduction of a carbon-carbon double bond in an enoyl moiety that is covalently linked to an acyl carrier protein (ACP). Involved in the elongation cycle of fatty acid which are used in the lipid metabolism and in the biotin biosynthesis. Related diseases Pigmentary disorder, reticulate, with systemic manifestations, X-linked (PDR) [MIM:301220]: An X-linked recessive disorder characterized by recurrent infections and sterile inflammation in various organs. Diffuse skin hyperpigmentation with a distinctive reticulate pattern is universally evident by early childhood. This is later followed in many patients by hypohidrosis, corneal inflammation and scarring, enterocolitis that resembles inflammatory bowel disease, and recurrent urethral strictures. Melanin and amyloid deposition is present in the dermis. Affected males also have a characteristic facies with frontally upswept hair and flared eyebrows. Female carriers have only restricted pigmentary changes along Blaschko's lines. {ECO:0000269|PubMed:27019227}. The disease is caused by variants affecting the gene represented in this entry. XLPDR is caused by a recurrent intronic mutation that results in missplicing and reduced POLA1 expression. This leads to a decrease in cytosolic RNA:DNA hybrids and constitutive activation of type I interferon responses, but has no effect on cell replication. {ECO:0000269|PubMed:27019227}.; DISEASE: Van Esch-O'Driscoll syndrome (VEODS) [MIM:301030]: An X-linked recessive syndrome characterized by different degrees of intellectual disability, moderate to severe short stature, microcephaly, hypogonadism, and variable congenital malformations. {ECO:0000269|PubMed:31006512}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB04030; DB08265; DB01865; DB03534; DB03030; DB08605; DB02379; DB01691; DB08604 Interacts with P0AF90 EC number NA Uniprot keywords 3D-structure; Antibiotic resistance; Direct protein sequencing; Fatty acid biosynthesis; Fatty acid metabolism; Lipid biosynthesis; Lipid metabolism; NAD; Oxidoreductase; Reference proteome Protein physicochemical properties Chain ID A Molecular weight (Da) 27362 Length 258 Aromaticity 0.08 Instability index 21.64 Isoelectric point 5.57 Charge (pH=7) -4.81 2D Binding mode Binding energy (Kcal/mol) -9.8  Molscript Map 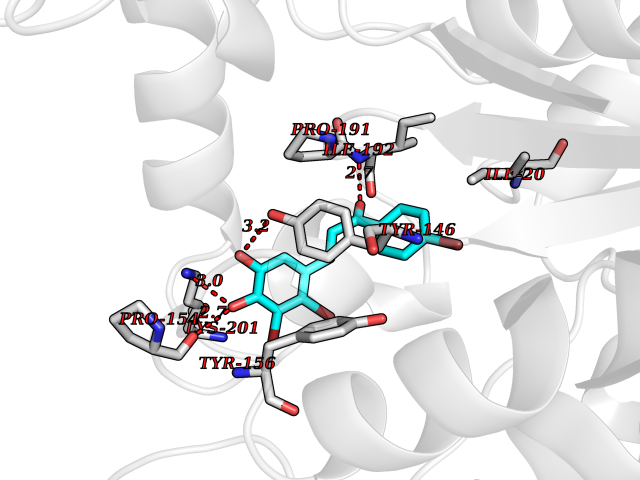 Pymol Map 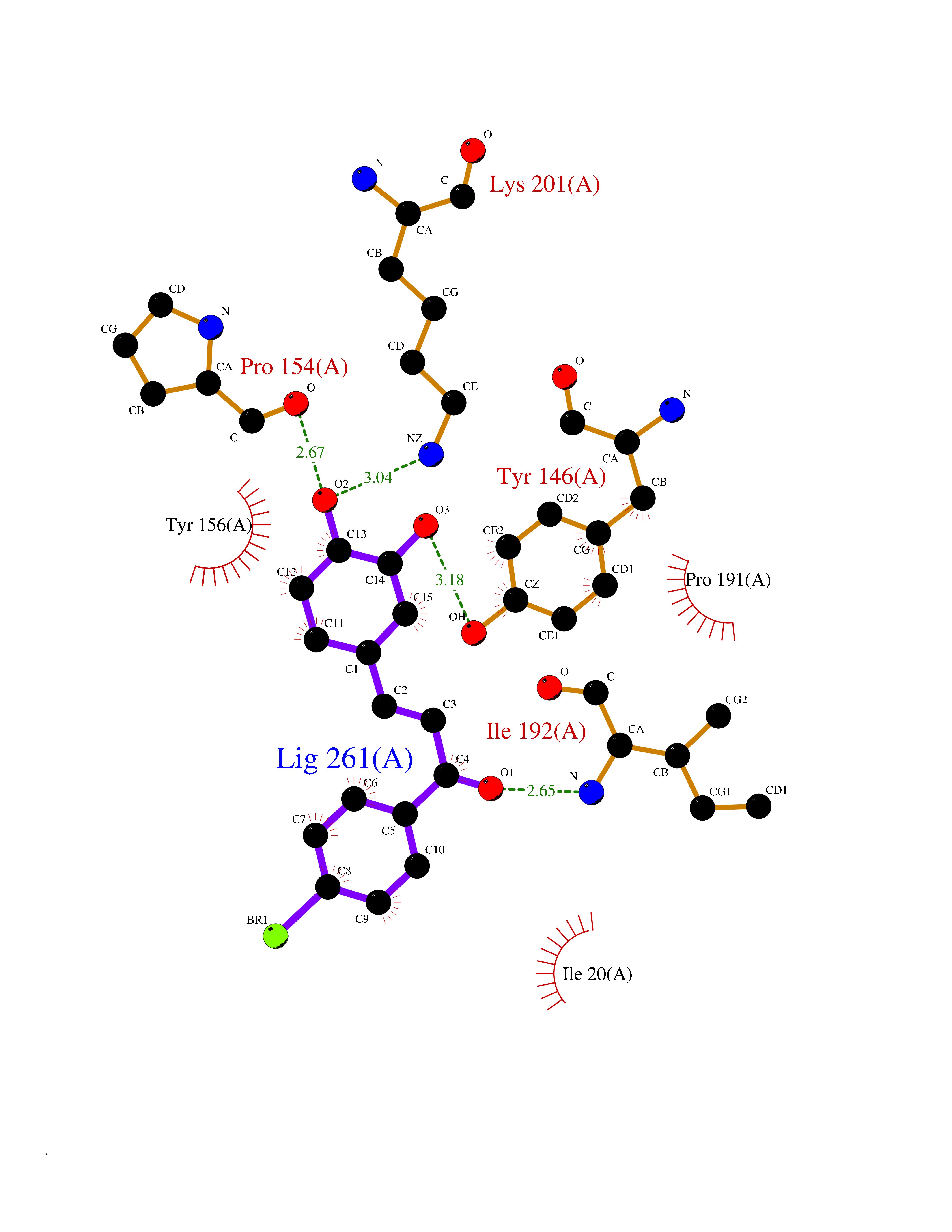 Ligplot Map 3D Binding mode Sequence GFLSGKRILVTGVASKLSIAYGIAQAMHREGAELAFTYQNDKLKGRVEEFAAQLGSDIVLQCDVAEDASIDTMFAELGKVWPKFDGFVHSIGFAPGDQLDGDYVNAVTREGFKIAHDISSYSFVAMAKACRSMLNPGSALLTLSYLGAERAIPNYNVMGLAKASLEANVRYMANAMGPEGVRVNAISAGPIRTLAASGIKDFRKMLAHCEAVTPIRRTVTIEDVGNSAAFLCSDLSAGISGEVVHVDGGFSIAAMNEL Hydrogen bonds contact Hydrophobic contact | ||||
| 54 | Multidrug efflux pump subunit AcrB | 5ENQ | 7.18 | |
Target general information Gen name acrB Organism Escherichia coli (strain K12) Uniprot ID TTD ID NA Synonyms b0462;acrE;JW0451 Protein family Resistance-nodulation-cell division (RND) (TC 2.A.6) family Biochemical class Transport protein Function Drug:proton antiporter activity.Drug transmembrane transporter activity.Identical protein binding. Related diseases Pyruvate kinase hyperactivity (PKHYP) [MIM:102900]: Autosomal dominant phenotype characterized by increase of red blood cell ATP. {ECO:0000269|PubMed:9090535}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Pyruvate kinase deficiency of red cells (PKRD) [MIM:266200]: A frequent cause of hereditary non-spherocytic hemolytic anemia. Clinically, pyruvate kinase-deficient patients suffer from a highly variable degree of chronic hemolysis, ranging from severe neonatal jaundice and fatal anemia at birth, severe transfusion-dependent chronic hemolysis, moderate hemolysis with exacerbation during infection, to a fully compensated hemolysis without apparent anemia. {ECO:0000269|PubMed:10087985, ECO:0000269|PubMed:10772876, ECO:0000269|PubMed:11328279, ECO:0000269|PubMed:11960989, ECO:0000269|PubMed:1536957, ECO:0000269|PubMed:1896471, ECO:0000269|PubMed:19085939, ECO:0000269|PubMed:2018831, ECO:0000269|PubMed:21794208, ECO:0000269|PubMed:7706479, ECO:0000269|PubMed:8161798, ECO:0000269|PubMed:8180378, ECO:0000269|PubMed:8476433, ECO:0000269|PubMed:8481523, ECO:0000269|PubMed:8483951, ECO:0000269|PubMed:8664896, ECO:0000269|PubMed:8807089, ECO:0000269|PubMed:9075576, ECO:0000269|PubMed:9482576, ECO:0000269|PubMed:9827908, ECO:0000269|PubMed:9886305, ECO:0000269|Ref.24}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03619; DB04209; DB03825 Interacts with P31224; P0AAW9; P0ADZ7 EC number NA Uniprot keywords 3D-structure; Cell inner membrane; Cell membrane; Membrane; Reference proteome; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A,B,C Molecular weight (Da) 60242.4 Length 553 Aromaticity 0.08 Instability index 29.1 Isoelectric point 4.76 Charge (pH=7) -18.86 2D Binding mode Binding energy (Kcal/mol) -9.79 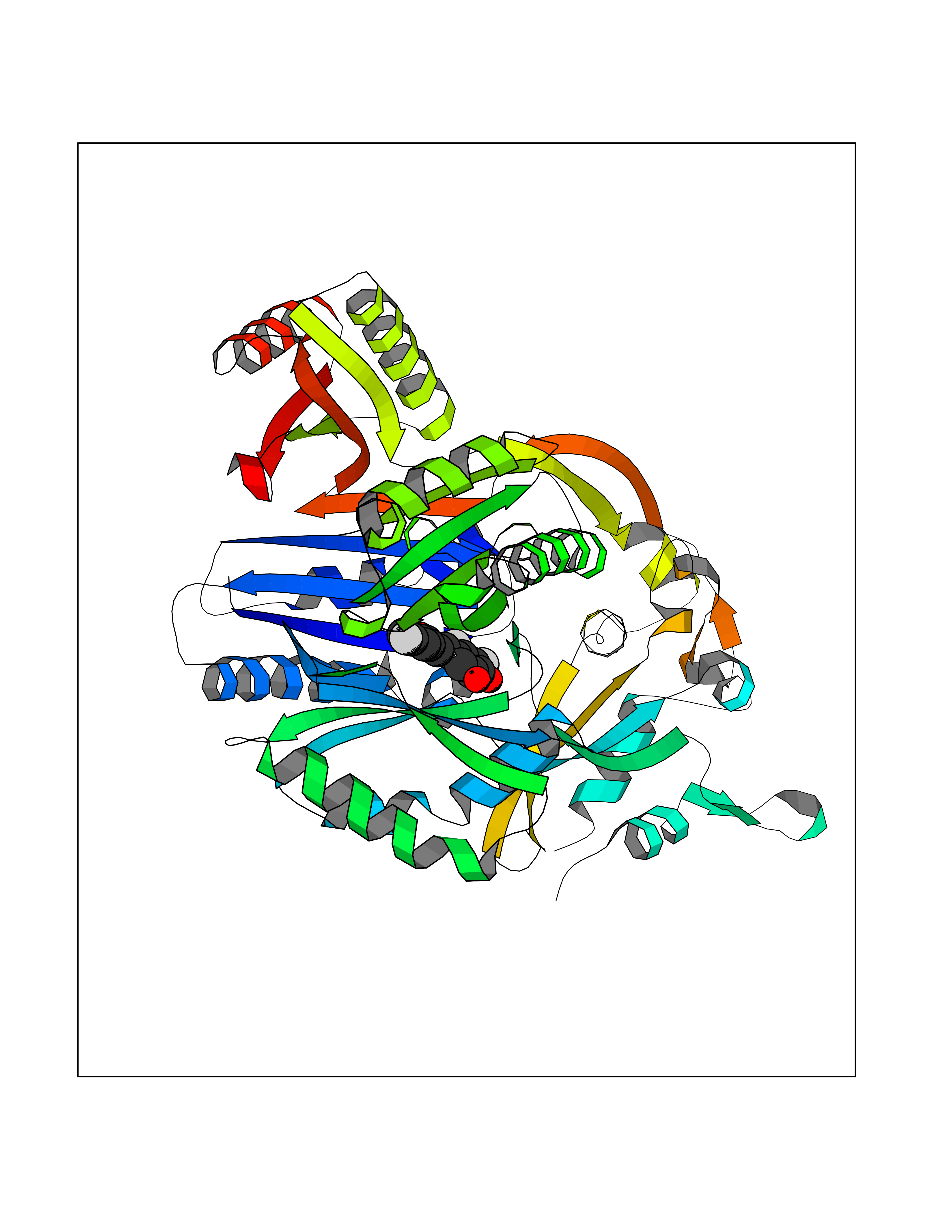 Molscript Map 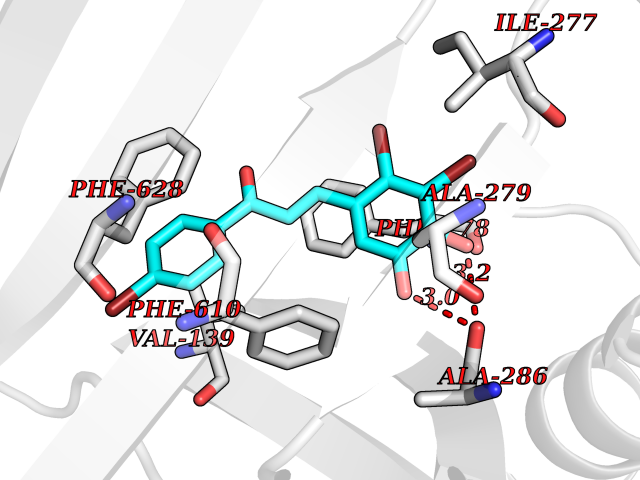 Pymol Map 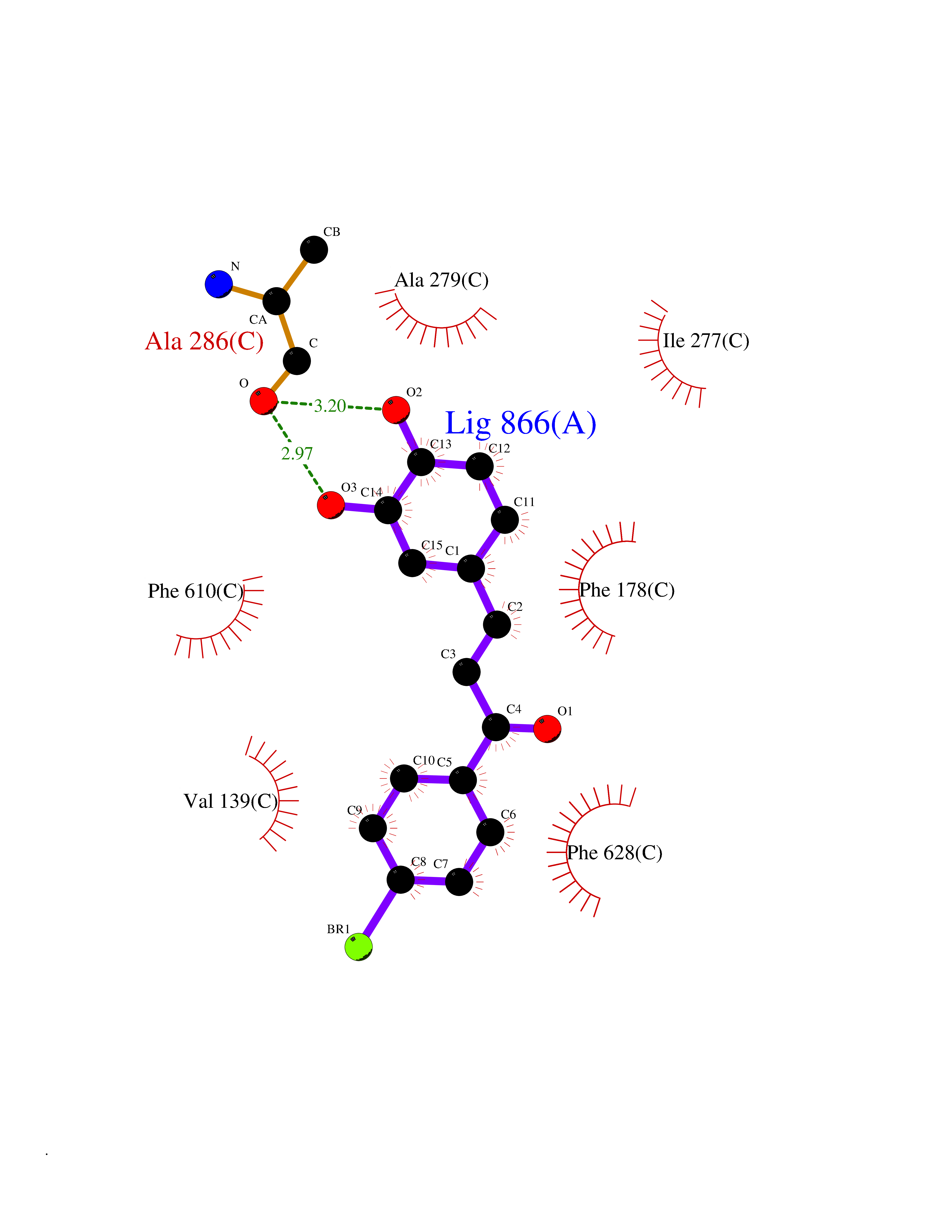 Ligplot Map 3D Binding mode Sequence APPAVTISASYPGADAKTVQDTVTQVIEQNMNGIDNLMYMSSNSDSTGTVQITLTFESGTDADIAQVQVQNKLQLAMPLLPQEVQQQGVSVEKSSSSFLMVVGVINTDGTMTQEDISDYVAANMKDAISRTSGVGDVQLFGSQYAMRIWMNPNELNKFQLTPVDVITAIKAQNATRLTSTEEFGKILLKVNQDGSRVLLRDVAKIELGGENYDIIAEFNGQPASGLGIKLATGANALDTAAAIRAELAKMEPFFPSGLKIVYPYDTGVFMTMVQLPAGATQERTQKVLNEVTHYYLTKEKNNVESVFAVNGFGFAGRGQNTGIAFVSLKDWADRPGEENKVEAITMRATRAFSQIKDAMVFAFNLATGFDFELIDQAGLGHEKLTQARNQLLAEAAKHPDMLTSVRPNGLEDTPQFKIDIDQEKAQALGVSINDINTTLGAAWGGSYVNDFIDRGRVKKVYVMSEAKYRMLPDDIGDWYVRAADGQMVPFSAFSSSRWEYGSPRLERYNGLPSMEILGQAAPGKSTGEAMELMEQLASKLPTGVGYDWTGMSY Hydrogen bonds contact Hydrophobic contact | ||||
| 55 | Nitric-oxide synthase inducible (NOS2) | 3E7G | 7.18 | |
Target general information Gen name NOS2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms iNOS; Peptidyl-cysteine S-nitrosylase NOS2; Nitric oxide synthase, inducible; NOS2A; NOS type II; Inducible NOS; Inducible NO synthase; Hepatocyte NOS; HEP-NOS Protein family NOS family Biochemical class Paired donor oxygen oxidoreductase Function Produces nitric oxide (NO) which is a messenger molecule with diverse functions throughout the body. In macrophages, NO mediates tumoricidal and bactericidal actions. Also has nitrosylase activity and mediates cysteine S-nitrosylation of cytoplasmic target proteins such PTGS2/COX2 (By similarity). As component of the iNOS-S100A8/9 transnitrosylase complex involved in the selective inflammatory stimulus-dependent S-nitrosylation of GAPDH on 'Cys-247' implicated in regulation of the GAIT complex activity and probably multiple targets including ANXA5, EZR, MSN and VIM. Involved in inflammation, enhances the synthesis of proinflammatory mediators such as IL6 and IL8. Related diseases Cerebellar ataxia, impaired intellectual development, and dysequilibrium syndrome 3 (CAMRQ3) [MIM:613227]: An autosomal recessive, congenital cerebellar ataxia associated with dysarthia, quadrupedal gait and intellectual disability. {ECO:0000269|PubMed:19461874}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07003; DB07007; DB07011; DB07405; DB08750; DB01997; DB07029; DB07008; DB08214; DB07002; DB01835; DB06879; DB04534; DB03100; DB02207; DB00125; DB00155; DB01234; DB14649; DB11327; DB00997; DB07306; DB07388; DB05252; DB01381; DB03366; DB05214; DB04400; DB09237; DB00244; DB01110; DB01017; DB03144; DB01686; DB03449; DB06916; DB07318; DB07389; DB02044; DB02644; DB05383; DB02234; DB03953; DB02462; DB08814 Interacts with P04406 EC number EC 1.14.13.39 Uniprot keywords 3D-structure; Alternative splicing; Calcium; Calmodulin-binding; Cytoplasm; FAD; Flavoprotein; FMN; Heme; Iron; Metal-binding; NADP; Oxidoreductase; Phosphoprotein; Proteomics identification; Reference proteome; Ubl conjugation; Zinc Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 48633 Length 421 Aromaticity 0.12 Instability index 46.5 Isoelectric point 6.75 Charge (pH=7) -1.04 2D Binding mode Binding energy (Kcal/mol) -9.79 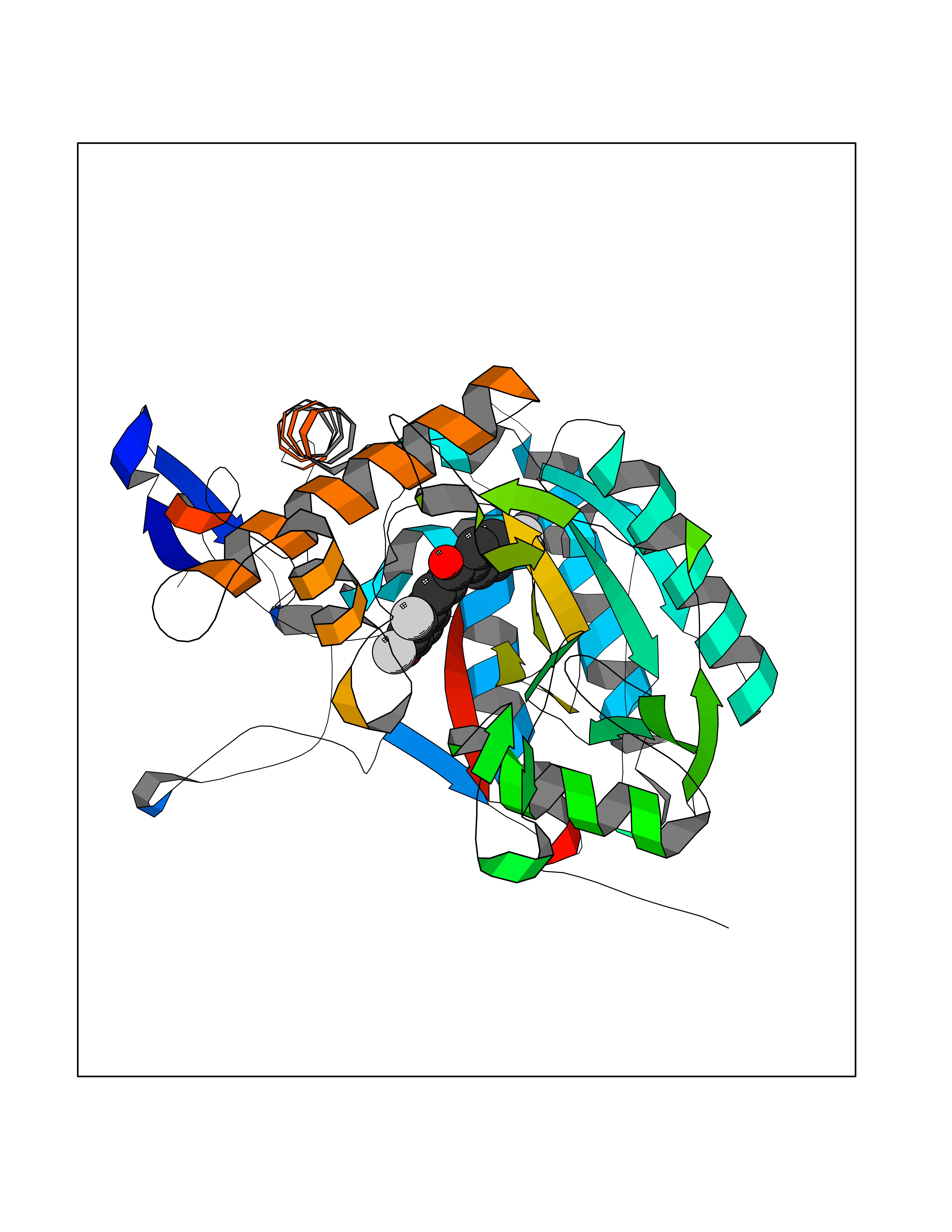 Molscript Map 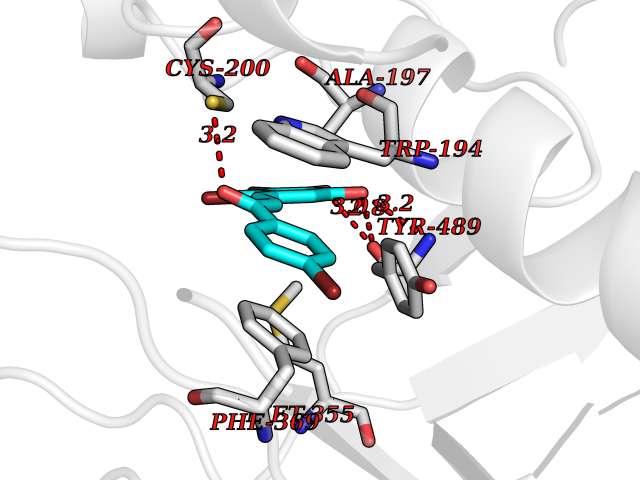 Pymol Map 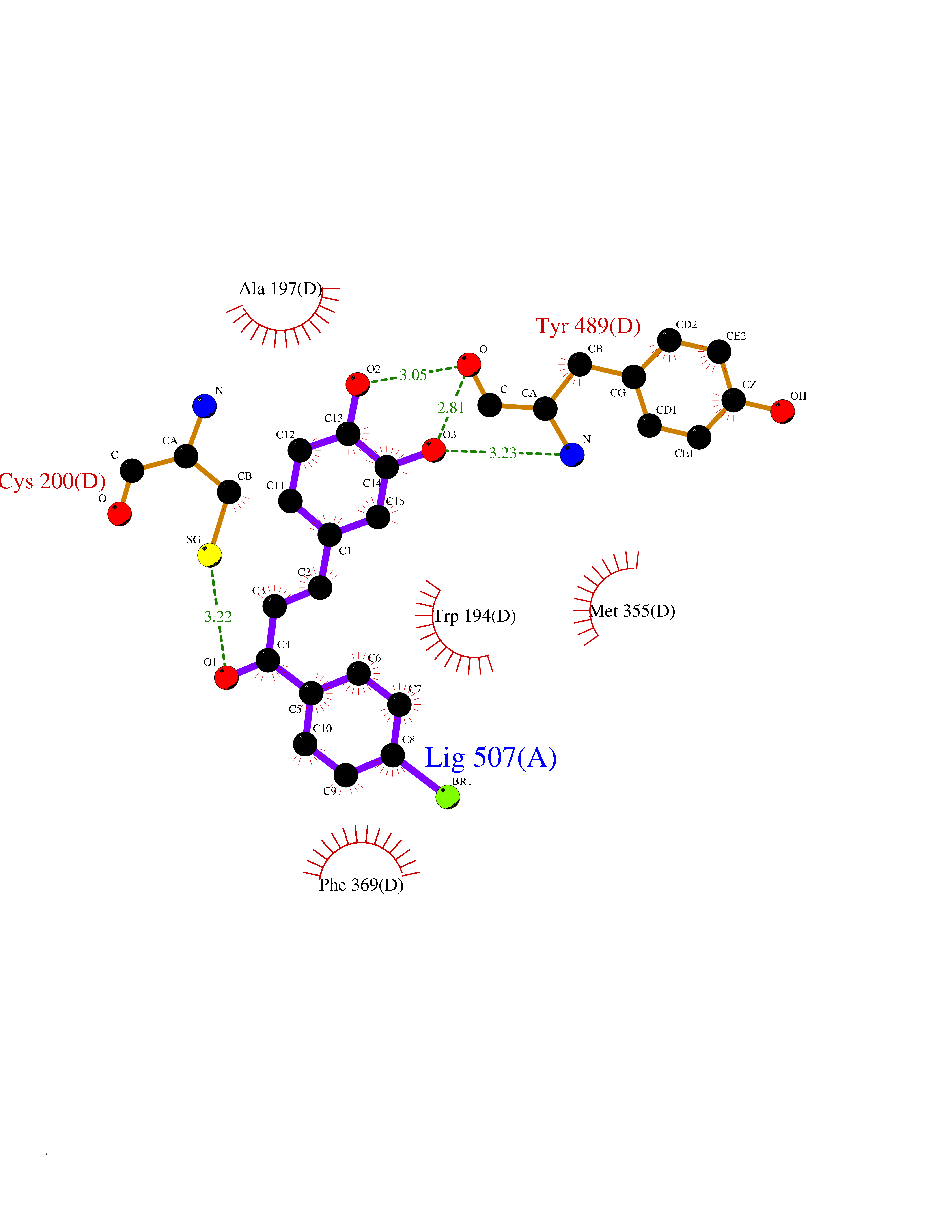 Ligplot Map 3D Binding mode Sequence RHVRIKNWGSGMTFQDTLHHKAKGILTCRSKSCLGSIMTPKSLTRGPRDKPTPPDELLPQAIEFVNQYYGSFKEAKIEEHLARVEAVTKEIETTGTYQLTGDELIFATKQAWRNAPRCIGRIQWSNLQVFDARSCSTAREMFEHICRHVRYSTNNGNIRSAITVFPQRSDGKHDFRVWNAQLIRYAGYQMPDGSIRGDPANVEFTQLCIDLGWKPKYGRFDVVPLVLQANGRDPELFEIPPDLVLEVAMEHPKYEWFRELELKWYALPAVANMLLEVGGLEFPGCPFNGWYMGTEIGVRDFCDVQRYNILEEVGRRMGLETHKLASLWKDQAVVEINIAVLHSFQKQNVTIMDHHSAAESFMKYMQNEYRSRGGCPADWIWLVPPMSGSITPVFHQEMLNYVLSPFYYYQVEAWKTHVWQD Hydrogen bonds contact Hydrophobic contact | ||||
| 56 | Tankyrase-2 (TNKS-2) | 3U9H | 7.18 | |
Target general information Gen name TNKS2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Tankyrase-related protein; Tankyrase-like protein; Tankyrase II; TRF1-interacting ankyrin-related ADP-ribose polymerase 2; TNKL; TANK2; Protein poly-ADP-ribosyltransferase tankyrase-2; Poly [ADP-ribos Protein family ARTD/PARP family Biochemical class Glycosyltransferases Function Acts as an activator of the Wnt signaling pathway by mediating poly-ADP-ribosylation of AXIN1 and AXIN2, 2 key components of the beta-catenin destruction complex: poly-ADP-ribosylated target proteins are recognized by RNF146, which mediates their ubiquitination and subsequent degradation. Also mediates poly-ADP-ribosylation of BLZF1 and CASC3, followed by recruitment of RNF146 and subsequent ubiquitination. Mediates poly-ADP-ribosylation of TERF1, thereby contributing to the regulation of telomere length. Stimulates 26S proteasome activity. Poly-ADP-ribosyltransferase involved in various processes such as Wnt signaling pathway, telomere length and vesicle trafficking. Related diseases Intellectual developmental disorder with macrocephaly, seizures, and speech delay (IDDMSSD) [MIM:618158]: An autosomal dominant neurodevelopmental disorder characterized by impaired intellectual development, poor speech, postnatal macrocephaly, and seizures. {ECO:0000269|PubMed:30290153}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with O15084; Q7Z6K5-1; O15169; Q9NWV8; P11274; Q13698; Q9NRI5; Q6V0I7; Q9NWT6; P14652; Q9UIQ6; Q14980; Q9BZL4; Q92698; P78314; O43815; P54274; Q9C0C2; Q9UHP3; Q06649 EC number EC 2.4.2.30 Uniprot keywords 3D-structure; ADP-ribosylation; ANK repeat; Chromosome; Cytoplasm; Glycosyltransferase; Golgi apparatus; Hydroxylation; Membrane; Metal-binding; NAD; Nucleotidyltransferase; Nucleus; Proteomics identification; Reference proteome; Repeat; Telomere; Transferase; Ubl conjugation; Wnt signaling pathway; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 23695.5 Length 208 Aromaticity 0.11 Instability index 47.61 Isoelectric point 8.28 Charge (pH=7) 2.88 2D Binding mode Binding energy (Kcal/mol) -9.8 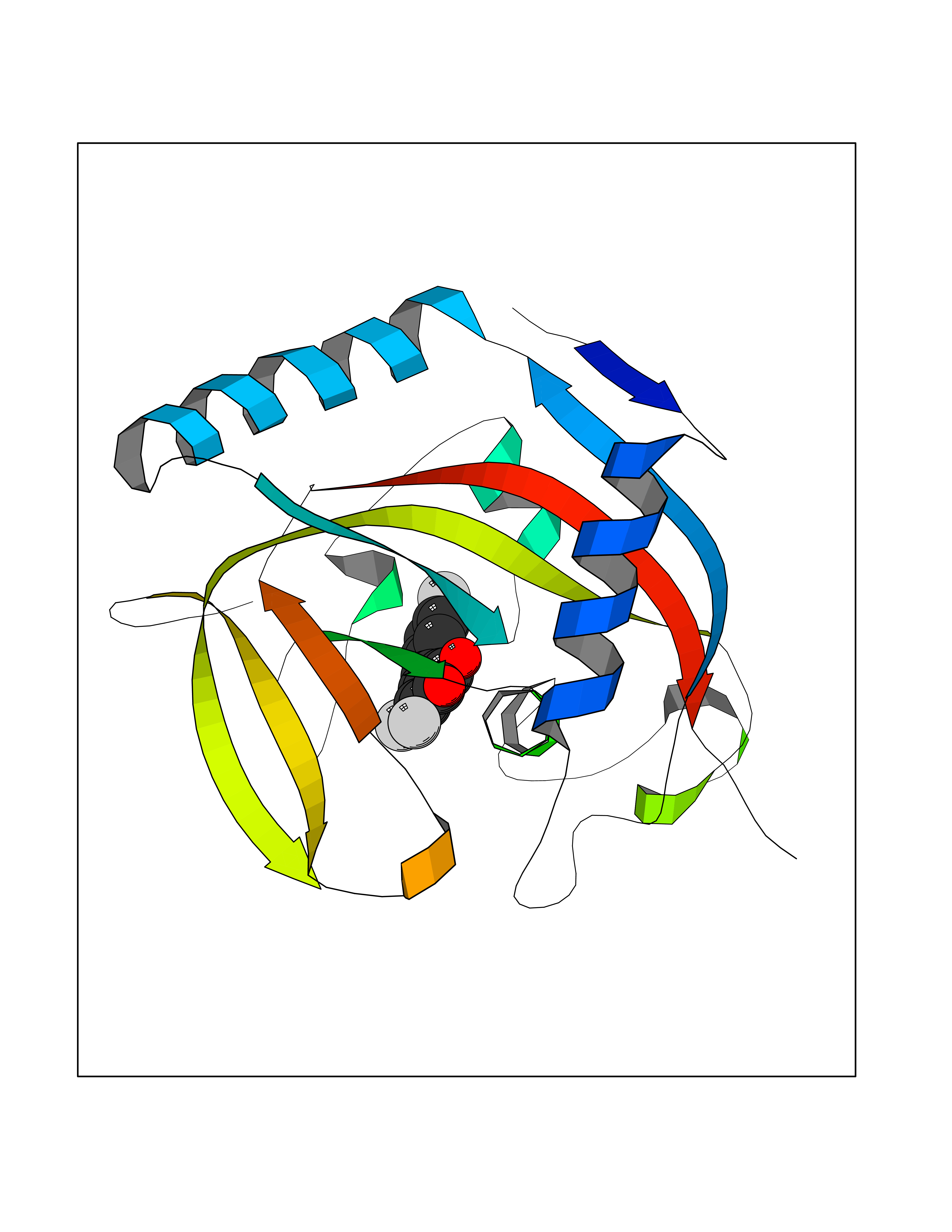 Molscript Map 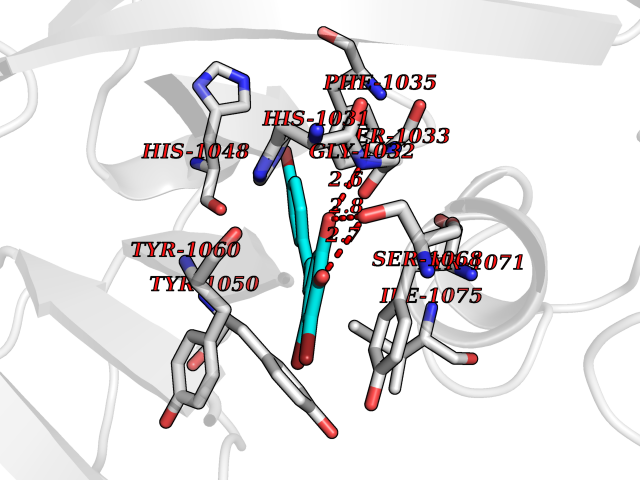 Pymol Map 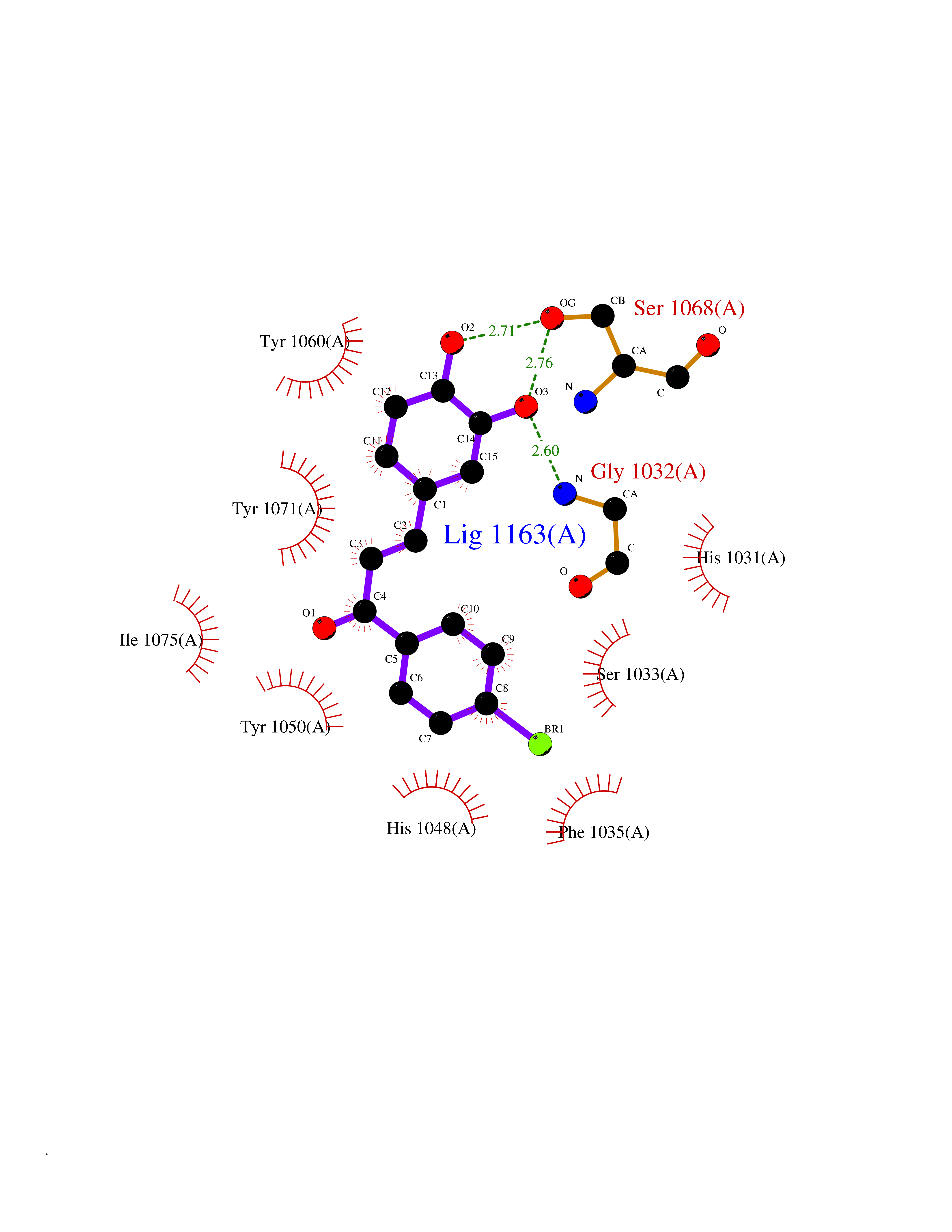 Ligplot Map 3D Binding mode Sequence GTILIDLSPDDKEFQSVEEEMQSTVREHRDGGHAGGIFNRYNILKIQKVCNKKLWERYTHRRKEVSEENHNHANERMLFHGSPFVNAIIHKGFDERHAYIGGMFGAGIYFAENSSKSNQYVYGIGGGTGCPVHKDRSCYICHRQLLFCRVTLGKSFLQFSAMAHSPPGHHSVTGRPSVNGLALAEYVIYRGEQAYPEYLITYQIMRPE Hydrogen bonds contact Hydrophobic contact | ||||
| 57 | Receptor-interacting protein 1 (RIPK1) | 5TX5 | 7.18 | |
Target general information Gen name RIPK1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Receptor-interacting serine/threonine-protein kinase 1; RIP1; RIP-1; RIP; Cell death protein RIP Protein family Protein kinase superfamily, TKL Ser/Thr protein kinase family Biochemical class Kinase Function Upon activation of TNFR1 by the TNF-alpha family cytokines, TRADD and TRAF2 are recruited to the receptor. Phosphorylates DAB2IP at 'Ser-728' in a TNF-alpha-dependent manner, and thereby activates the MAP3K5-JNK apoptotic cascade. Ubiquitination by TRAF2 via 'Lys-63'-link chains acts as a critical enhancer of communication with downstream signal transducers in the mitogen-activated protein kinase pathway and the NF-kappa-B pathway, which in turn mediate downstream events including the activation of genes encoding inflammatory molecules. Polyubiquitinated protein binds to IKBKG/NEMO, the regulatory subunit of the IKK complex, a critical event for NF-kappa-B activation. Interaction with other cellular RHIM-containing adapters initiates gene activation and cell death. RIPK1 and RIPK3 association, in particular, forms a necrosis-inducing complex. Serine-threonine kinase which transduces inflammatory and cell-death signals (programmed necrosis) following death receptors ligation, activation of pathogen recognition receptors (PRRs), and DNA damage. Related diseases Immunodeficiency 57 with autoinflammation (IMD57) [MIM:618108]: An autosomal recessive primary immunodeficiency characterized by lymphopenia and recurrent viral, bacterial, and fungal infections. Patients exhibit early-onset inflammatory bowel disease involving the upper and lower gastrointestinal tract, and develop progressive polyarthritis. {ECO:0000269|PubMed:30026316}. The disease is caused by variants affecting the gene represented in this entry. RIPK1-deficient immune cells from IMD57 patients have impaired proinflammatory signaling leading to dysregulated cytokine secretion and are prone to necroptosis. {ECO:0000269|PubMed:30026316}.; DISEASE: Autoinflammation with episodic fever and lymphadenopathy (AIEFL) [MIM:618852]: An autosomal dominant immunologic disorder characterized by early onset of recurrent episodes of unexplained fever, lymphadenopathy, hepatosplenomegaly, and increased levels of inflammatory cytokines and chemokines in patient serum. {ECO:0000269|PubMed:31827280, ECO:0000269|PubMed:31827281}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB12010 Interacts with P04083; Q13490; Q13489; Q92851; Q14790; Q8IVM0; P48729; Q13158; Q9Y6K9; Q96AB6; Q9ULZ3; Q13546; Q9Y572; P19438; Q13077; Q12933; Q13114; Q13107; B7UI21; PRO_0000449629 [P0DTD1]; U5TQE9 EC number EC 2.7.11.1 Uniprot keywords 3D-structure; Alternative splicing; Apoptosis; ATP-binding; Cell membrane; Cytoplasm; Disease variant; Glycoprotein; Host-virus interaction; Inflammatory response; Isopeptide bond; Kinase; Membrane; Necrosis; Nucleotide-binding; Phosphoprotein; Proteomics identification; Reference proteome; Serine/threonine-protein kinase; Transferase; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 29554.2 Length 259 Aromaticity 0.08 Instability index 48.26 Isoelectric point 6.29 Charge (pH=7) -2.52 2D Binding mode Binding energy (Kcal/mol) -9.8 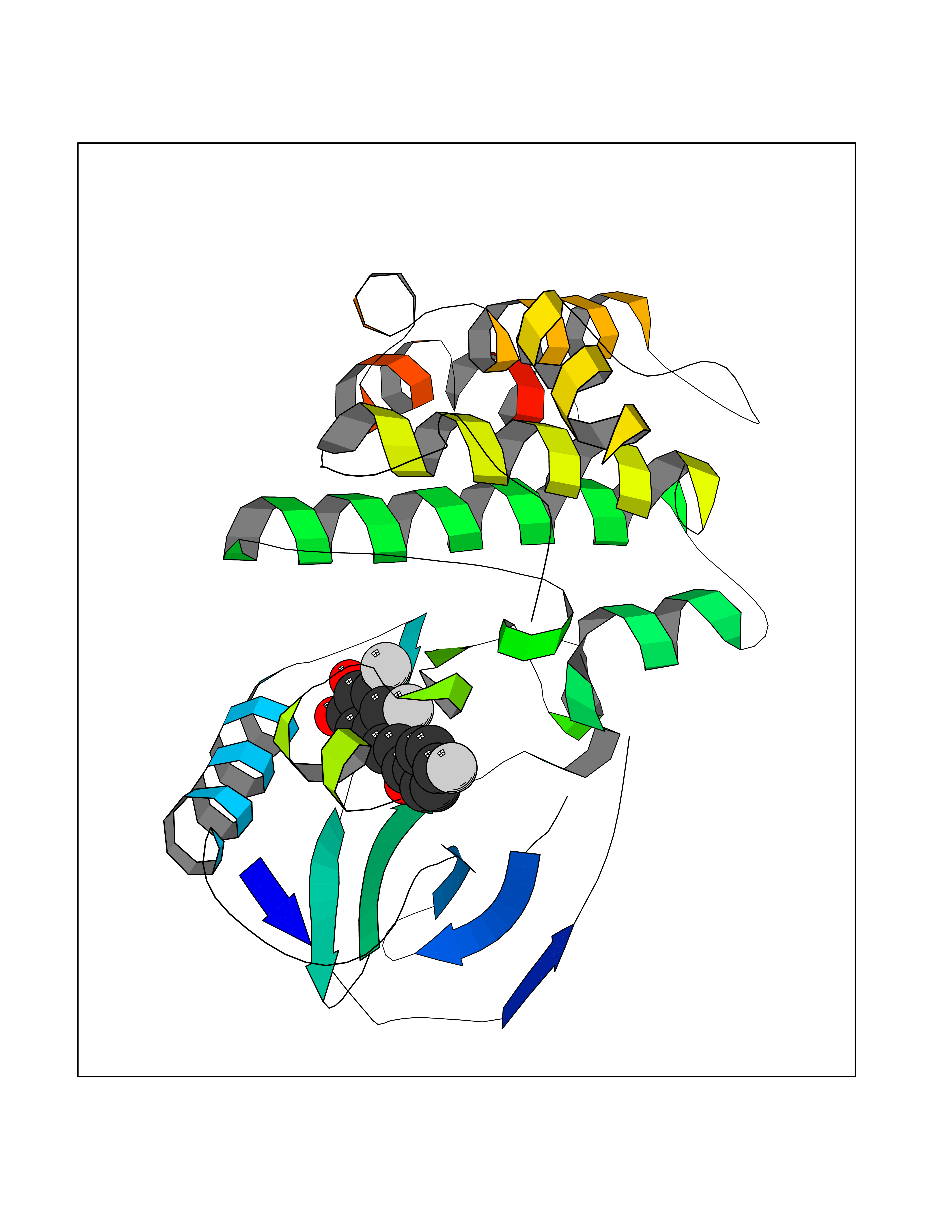 Molscript Map 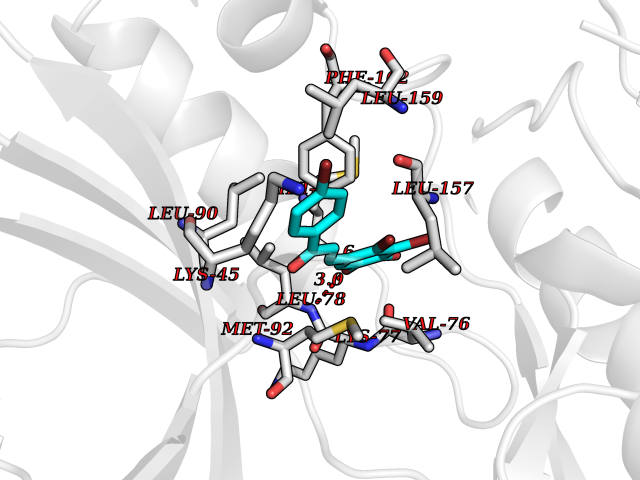 Pymol Map 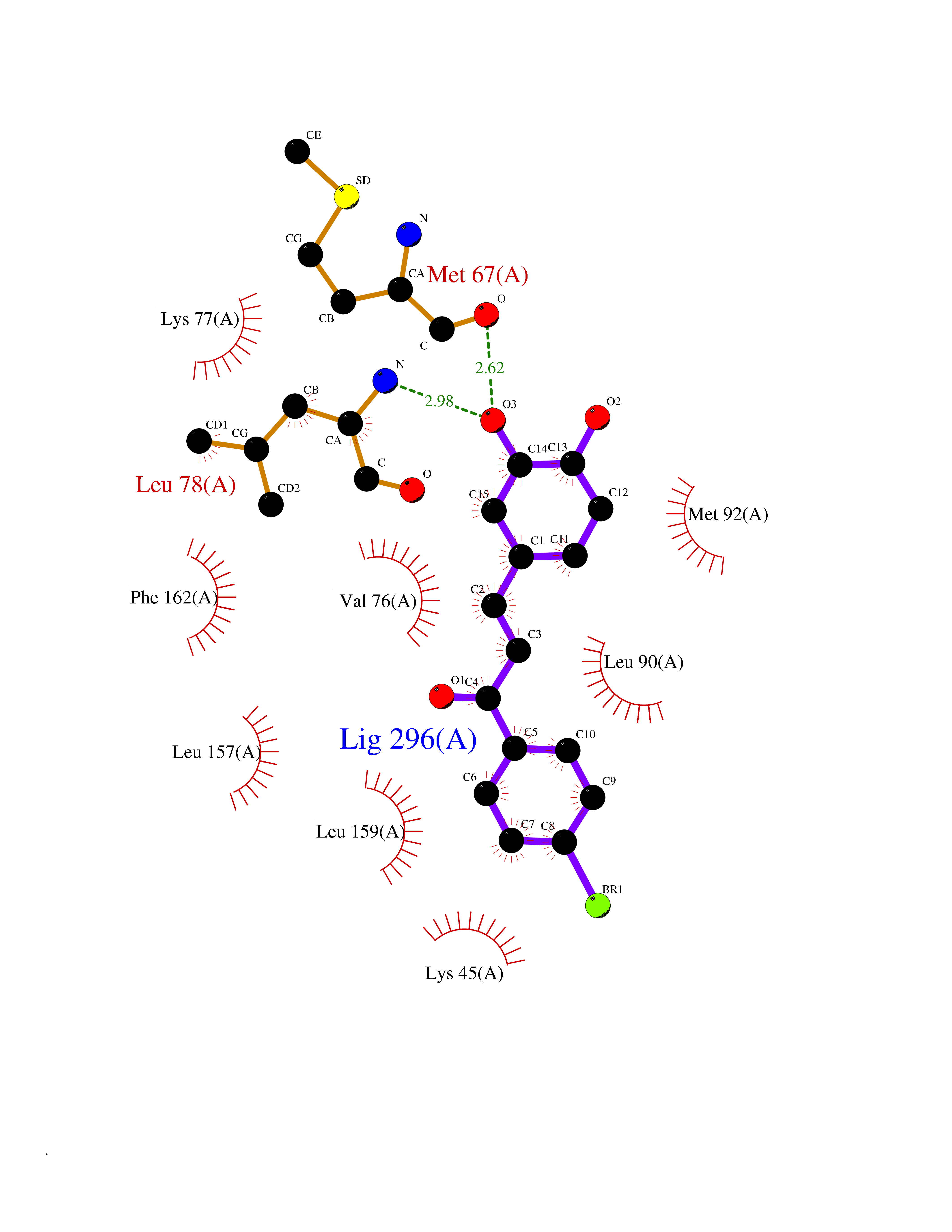 Ligplot Map 3D Binding mode Sequence IKMKSSDFLESAELDSGGKVSLAFHRTQGLMIMKTVYKGPNCIEHNEALLEEAKMMNRLRHSRVVKLLGVIIEEGKYSLVMEYMEKGNLMHVLKAEMSTPLSVKGRIILEIIEGMAYLHGKGVIHKDLKPENILVDNDFHIKIADLGLASFKMWSKLNGTLYYMAPEHLNDVNAKPTEKSDVYSFAVVLWAIFANKEPYQQLIMAIKSGNRPDVDDITEYCPREIISLMKLCWEANPEARPTFPGIEEKFRPFYLSQLE Hydrogen bonds contact Hydrophobic contact | ||||
| 58 | Oxysterols receptor LXR-alpha (NR1H3) | 3IPQ | 7.17 | |
Target general information Gen name NR1H3 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Nuclear receptor subfamily 1 group H member 3; Nuclear receptor LXRalpha; Nuclear orphan receptor LXR-alpha; Liver X receptor alpha; LXRalpha; LXRA Protein family Nuclear hormone receptor family, NR1 subfamily Biochemical class Nuclear hormone receptor Function Interaction with retinoic acid receptor (RXR) shifts RXR from its role as a silent DNA-binding partner to an active ligand-binding subunit in mediating retinoid responses through target genes defined by LXRES. LXRES are DR4-type response elements characterized by direct repeats of two similar hexanuclotide half-sites spaced by four nucleotides. Plays an important role in the regulation of cholesterol homeostasis, regulating cholesterol uptake through MYLIP-dependent ubiquitination of LDLR, VLDLR and LRP8. Interplays functionally with RORA for the regulation of genes involved in liver metabolism. Nuclear receptor that exhibits a ligand-dependent transcriptional activation activity. Related diseases Okur-Chung neurodevelopmental syndrome (OCNDS) [MIM:617062]: An autosomal dominant neurodevelopmental disorder characterized by developmental delay, intellectual disability, behavioral problems, hypotonia, speech problems, microcephaly, pachygyria and variable dysmorphic features. {ECO:0000269|PubMed:27048600}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08175; DB08063; DB11994; DB07929; DB13174; DB07080 Interacts with O60869; O60341; Q99750; Q15788; O75376; Q07869; Q07869-1; Q03181; P37231; P19793; P28702; P48443; O43463; P42858; Q99750; O95817; G5E9A7; O95872; P02545; Q99750; P28702; P28702-3; P48443; Q7Z699 EC number NA Uniprot keywords 3D-structure; Activator; Alternative splicing; Cytoplasm; DNA-binding; Metal-binding; Nucleus; Proteomics identification; Receptor; Reference proteome; Transcription; Transcription regulation; Ubl conjugation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A Molecular weight (Da) 25389.9 Length 220 Aromaticity 0.09 Instability index 46.42 Isoelectric point 5.51 Charge (pH=7) -6.58 2D Binding mode Binding energy (Kcal/mol) -9.78  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence QLSPEQLGMIEKLVAAQQTPWPEARQQRFAHFTELAIVSVQEIVDFAKQLPGFLQLSREDQIALLKTSAIEVMLLETSRRYNPGSESITFLKDFSYNREDFAKAGLQVEFINPIFEFSRAMNELQLNDAEFALLIAISIFSADRPNVQDQLQVERLQHTYVEALHAYVSIHHPHDRLMFPRMLMKLVSLRTLSSVHSEQVFALRLQDKKLPPLLSEIWDV Hydrogen bonds contact Hydrophobic contact | ||||
| 59 | SEC14-like protein 2 | 4OMJ | 7.16 | |
Target general information Gen name SEC14L2 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms KIAA1658;C22orf6;KIAA1186 Protein family NA Biochemical class Transport protein Function Phospholipid binding.Transporter activity.Vitamin E binding. Related diseases Neurodevelopmental disorder with poor language and loss of hand skills (NDPLHS) [MIM:617903]: An autosomal dominant disorder characterized by psychomotor developmental stagnation or regression. NDPLHS manifest in the first years of life as loss of purposeful hand movements, loss of language, and intellectual disability. {ECO:0000269|PubMed:26740508, ECO:0000269|PubMed:28856709, ECO:0000269|PubMed:29369404}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Developmental and epileptic encephalopathy 59 (DEE59) [MIM:617904]: A form of epileptic encephalopathy, a heterogeneous group of severe early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE59 is an autosomal dominant condition characterized by onset of refractory seizures in early infancy. {ECO:0000269|PubMed:28856709, ECO:0000269|PubMed:29100083, ECO:0000269|PubMed:29369404}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB14003; DB14001; DB14002; DB11635; DB11251; DB00163 Interacts with NA EC number NA Uniprot keywords 3D-structure; Activator; Alternative splicing; Cytoplasm; Lipid-binding; Nucleus; Proteomics identification; Reference proteome; Transcription; Transcription regulation; Transport Protein physicochemical properties Chain ID A,B Molecular weight (Da) 31533.3 Length 274 Aromaticity 0.1 Instability index 49.26 Isoelectric point 8.34 Charge (pH=7) 2.81 2D Binding mode Binding energy (Kcal/mol) -9.77  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence MSGRVGDLSPRQKEALAKFRENVQDVLPALPNPDDYFLLRWLRARSFDLQKSEAMLRKHVEFRKQKDIDNIISWQPPEVIQQYLSGGMCGYDLDGCPVWYDIIGPLDAKGLLFSASKQDLLRTKMRECELLLQECAHQTTKLGRKVETITIIYDCEGLGLKHLWKPAVEAYGEFLCMFEENYPETLKRLFVVKAPKLFPVAYNLIKPFLSEDTRKKIMVLGANWKEVLLKHISPDQVPVEYGGTMTDPDGNPKCKSKINYGGDIPRKYYVRDQV Hydrogen bonds contact Hydrophobic contact | ||||
| 60 | Monoamine oxidase type A (MAO-A) | 2Z5Y | 7.16 | |
Target general information Gen name MAOA Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Monoamine oxidase A; Amine oxidase [flavin-containing] A Protein family Flavin monoamine oxidase family Biochemical class CH-NH(2) donor oxidoreductase Function MAOA preferentially oxidizes biogenic amines such as 5-hydroxytryptamine (5-HT), norepinephrine and epinephrine. Catalyzes the oxidative deamination of biogenic and xenobiotic amines and has important functions in the metabolism of neuroactive and vasoactive amines in the central nervous system and peripheral tissues. Related diseases Brunner syndrome (BRNRS) [MIM:300615]: A form of X-linked non-dysmorphic mild intellectual disability. Male patients are affected by borderline intellectual deficit and exhibit abnormal behavior, including disturbed regulation of impulsive aggression. Obligate female carriers have normal intelligence and behavior. {ECO:0000269|PubMed:8211186}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01472; DB00918; DB00182; DB06698; DB04889; DB13876; DB01445; DB06774; DB00215; DB04017; DB09130; DB05205; DB07641; DB00988; DB01363; DB00668; DB12329; DB01175; DB03147; DB14914; DB00614; DB01381; DB07919; DB04818; DB01247; DB00601; DB01577; DB00805; DB01442; DB01171; DB08804; DB00952; DB04820; DB00184; DB04821; DB06412; DB01626; DB00780; DB00191; DB00388; DB00397; DB09244; DB04850; DB00721; DB01168; DB00571; DB00852; DB09363; DB00140; DB00953; DB06654; DB01037; DB01104; DB00669; DB14569; DB09042; DB00624; DB13943; DB13944; DB13946; DB09245; DB00752; DB15328; DB09185; DB04832; DB00315; DB00909 Interacts with P27338 EC number EC 1.4.3.4 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Catecholamine metabolism; Direct protein sequencing; Disease variant; FAD; Flavoprotein; Intellectual disability; Membrane; Mitochondrion; Mitochondrion outer membrane; Neurotransmitter degradation; Oxidoreductase; Phosphoprotein; Proteomics identification; Reference proteome; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 58195.3 Length 513 Aromaticity 0.11 Instability index 34.97 Isoelectric point 7.98 Charge (pH=7) 2.87 2D Binding mode Binding energy (Kcal/mol) -9.77  Molscript Map  Pymol Map 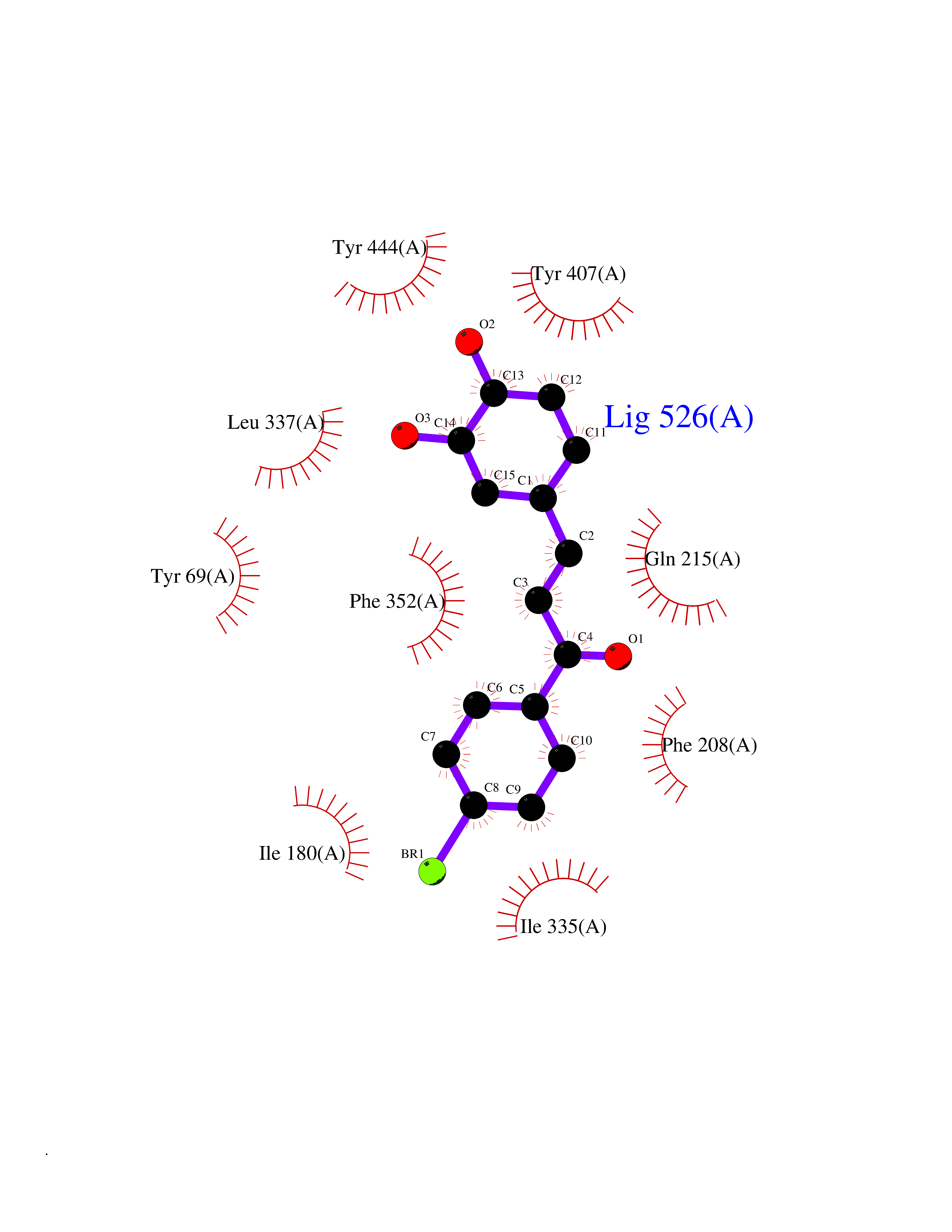 Ligplot Map 3D Binding mode Sequence HMFDVVVIGGGISGLSAAKLLTEYGVSVLVLEARDRVGGRTYTIRNEHVDYVDVGGAYVGPTQNRILRLSKELGIETYKVNVSERLVQYVKGKTYPFRAAFPPVWNPIAYLDYNNLWRTIDNMGKEIPTDAPWEAQHADKWDKMTMKELIDKICWTKTARRFAYLFVNINVTSEPHEVSALWFLWYVKQCGGTTRIFSVTNGGQERKFVGGSGQVSERIMDLLGDQVKLNHPVTHVDQSSDNIIIETLNHEHYECKYVINAIPPTLTAKIHFRPELPAERNQLIQRLPMGAVIKCMMYYKEAFWKKKDYCGCMIIEDEDAPISITLDDTKPDGSLPAIMGFILARKADRLAKLHKEIRKKKICELYAKVLGSQEALHPVHYEEKNWCEEQYSGGCYTAYFPPGIMTQYGRVIRQPVGRIFFAGTETATKWSGYMEGAVEAGERAAREVLNGLGKVTEKDIWVQEPESKDVPAVEITHTFWERNLPSVSGLLKIIGFSTSVTALGFVLYKYKLL Hydrogen bonds contact Hydrophobic contact | ||||