Job Results:
Ligand
Structure
Job ID
b448ce30ce2c1c80e042f47c74d47daa
Job name
NA
Time
2026-01-10 22:40:02
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 21 | Hypoxia-inducible factor 2 alpha (HIF-2A) | 5TBM | 7.07 | |
Target general information Gen name EPAS1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms bHLHe73; PASD2; PAS domain-containing protein 2; Member of PAS protein 2; MOP2; Hypoxia-inducible factor 2-alpha; HLF; HIF2A; HIF2-alpha; HIF-2-alpha; HIF-1-alpha-like factor; Endothelial PAS domain-c Protein family NA Biochemical class NA Function Heterodimerizes with ARNT; heterodimer binds to core DNA sequence 5'-TACGTG-3' within the hypoxia response element (HRE) of target gene promoters. Regulates the vascular endothelial growth factor (VEGF) expression and seems to be implicated in the development of blood vessels and the tubular system of lung. May also play a role in the formation of the endothelium that gives rise to the blood brain barrier. Potent activator of the Tie-2 tyrosine kinase expression. Activation requires recruitment of transcriptional coactivators such as CREBBP and probably EP300. Interaction with redox regulatory protein APEX seems to activate CTAD. Transcription factor involved in the induction of oxygen regulated genes. Related diseases Erythrocytosis, familial, 4 (ECYT4) [MIM:611783]: An autosomal dominant disorder characterized by elevated serum hemoglobin and hematocrit, and normal platelet and leukocyte counts. {ECO:0000269|PubMed:18184961, ECO:0000269|PubMed:18378852, ECO:0000269|PubMed:19208626, ECO:0000269|PubMed:22367913}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB15463; DB12255 Interacts with P27540; Q96RK4; O00327-8; Q8WYA1-3; Q9GZT9; P60228; O60573; P09467; P61244; Q9BWF3-1; P08047; Q9Y2K6; P40818 EC number NA Uniprot keywords 3D-structure; Activator; Angiogenesis; Congenital erythrocytosis; Developmental protein; Differentiation; Disease variant; DNA-binding; Host-virus interaction; Hydroxylation; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Transcription; Transcription regulation; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 12249.8 Length 106 Aromaticity 0.11 Instability index 40.77 Isoelectric point 5.25 Charge (pH=7) -5.82 2D Binding mode Binding energy (Kcal/mol) -9.65 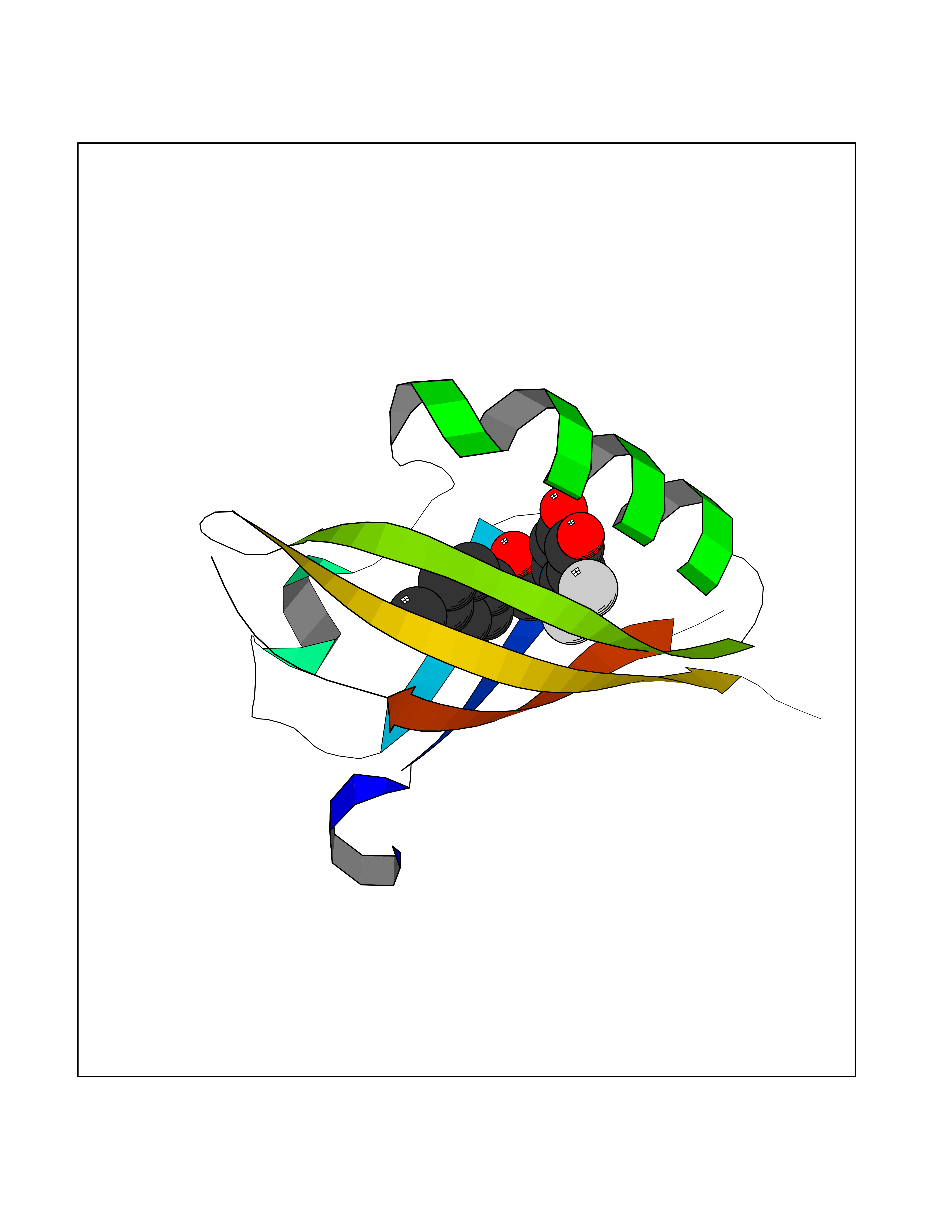 Molscript Map 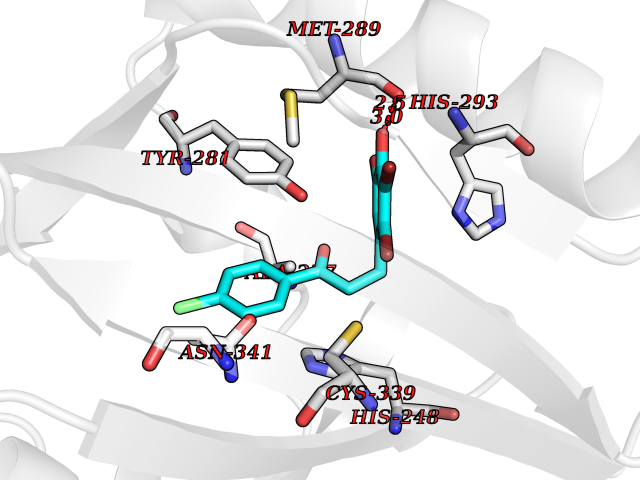 Pymol Map 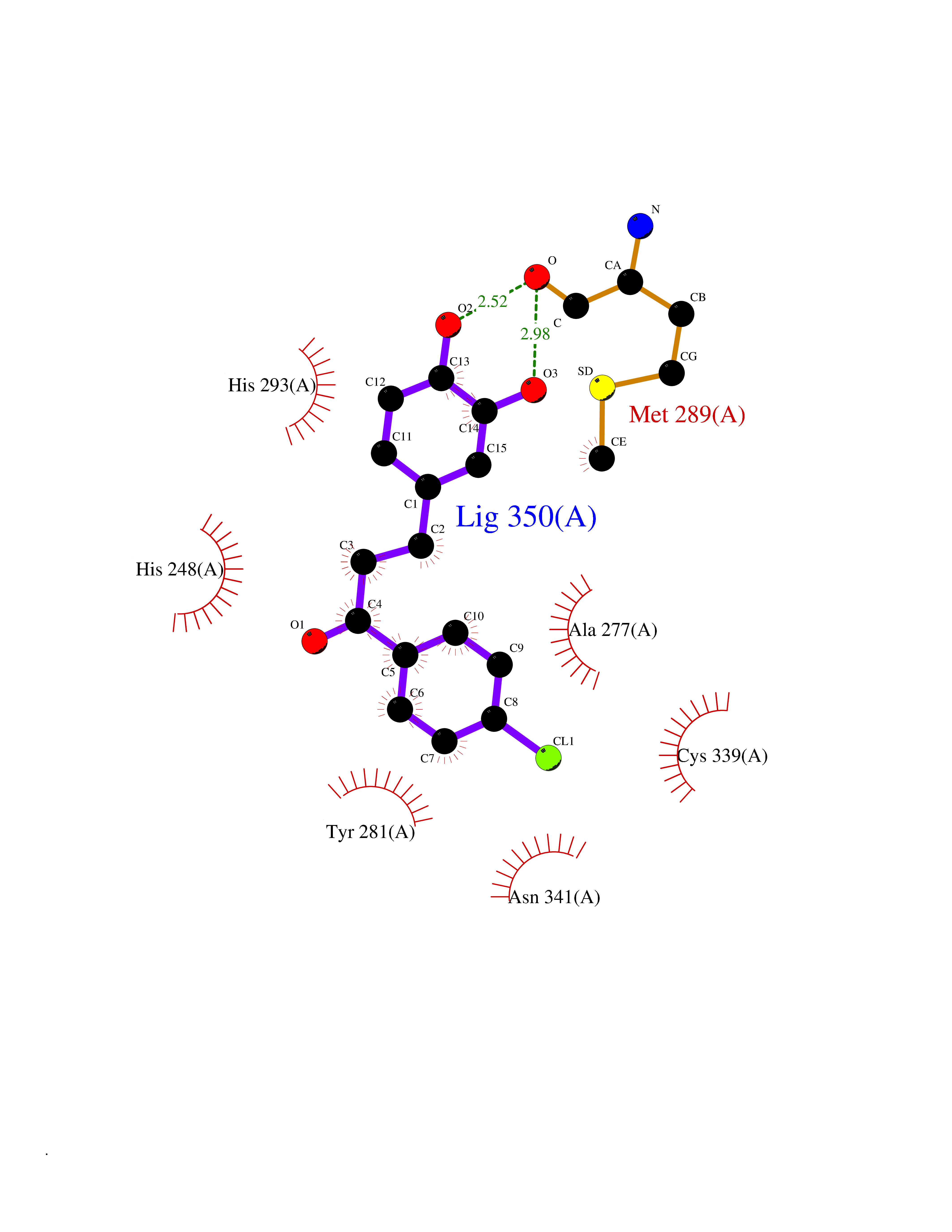 Ligplot Map 3D Binding mode Sequence LDSKTFLSEHSMDMKFTYCDDRITELIGYHPEELLGRSAYEFYHALDSENMTKSHQNLCTKGQVVSGQYRMLAKHGGYVWLETQGTVIYNPPQCIMCVNYVLSEIE Hydrogen bonds contact Hydrophobic contact | ||||
| 22 | Wnt-7a protein (WNT7A) | 4UZQ | 7.07 | |
Target general information Gen name WNT7A Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Protein Wnt-7a Protein family Wnt family Biochemical class NA Function Plays an important role in embryonic development, including dorsal versus ventral patterning during limb development, skeleton development and urogenital tract development. Required for central nervous system (CNS) angiogenesis and blood-brain barrier regulation. Required for normal, sexually dimorphic development of the Mullerian ducts, and for normal fertility in both sexes. Required for normal neural stem cell proliferation in the hippocampus dentate gyrus. Required for normal progress through the cell cycle in neural progenitor cells, for self-renewal of neural stem cells, and for normal neuronal differentiation and maturation. Promotes formation of synapses via its interaction with FZD5. Ligand for members of the frizzled family of seven transmembrane receptors that functions in the canonical Wnt/beta-catenin signaling pathway. Related diseases Limb pelvis hypoplasia aplasia syndrome (LPHAS) [MIM:276820]: A syndrome of severe deficiency of the extremities due to hypo- or aplasia of one or more long bones of one or more limbs. Pelvic manifestations include hip dislocation, hypoplastic iliac bone and aplastic pubic bones. Thoracic deformity, unusual facies and genitourinary anomalies can be present. {ECO:0000269|PubMed:16826533, ECO:0000269|PubMed:17431918, ECO:0000269|PubMed:20949531, ECO:0000269|PubMed:21271649, ECO:0000269|PubMed:27638328}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Fuhrmann syndrome (FUHRS) [MIM:228930]: Distinct limb-malformation disorder characterized also by various degrees of limb aplasia/hypoplasia and joint dysplasia. {ECO:0000269|PubMed:16826533}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P55212; P22607; P06396; P13473-2; Q9UMX0; Q9Y5W5; Q5T9L3; Q9Z0J1 EC number NA Uniprot keywords 3D-structure; Developmental protein; Disease variant; Disulfide bond; Extracellular matrix; Glycoprotein; Lipoprotein; Proteomics identification; Reference proteome; Secreted; Signal; Wnt signaling pathway Protein physicochemical properties Chain ID A,B Molecular weight (Da) 40475.5 Length 356 Aromaticity 0.1 Instability index 50.49 Isoelectric point 7.67 Charge (pH=7) 1.62 2D Binding mode Binding energy (Kcal/mol) -9.64 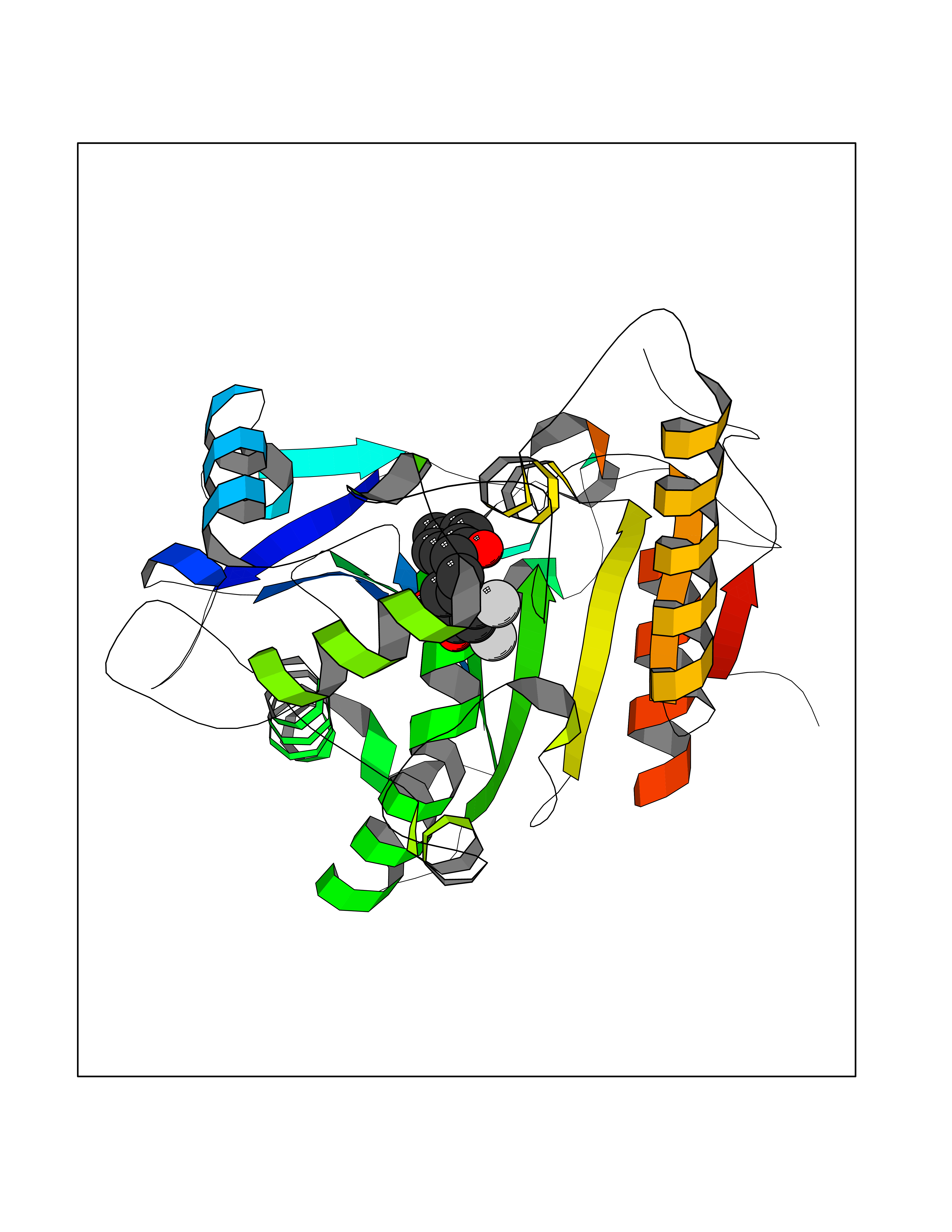 Molscript Map 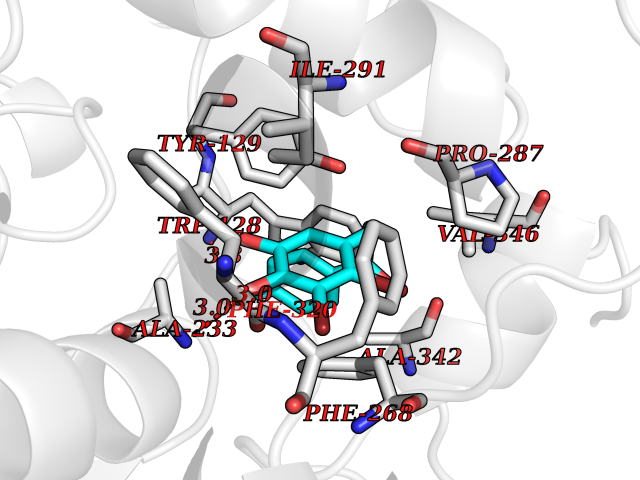 Pymol Map 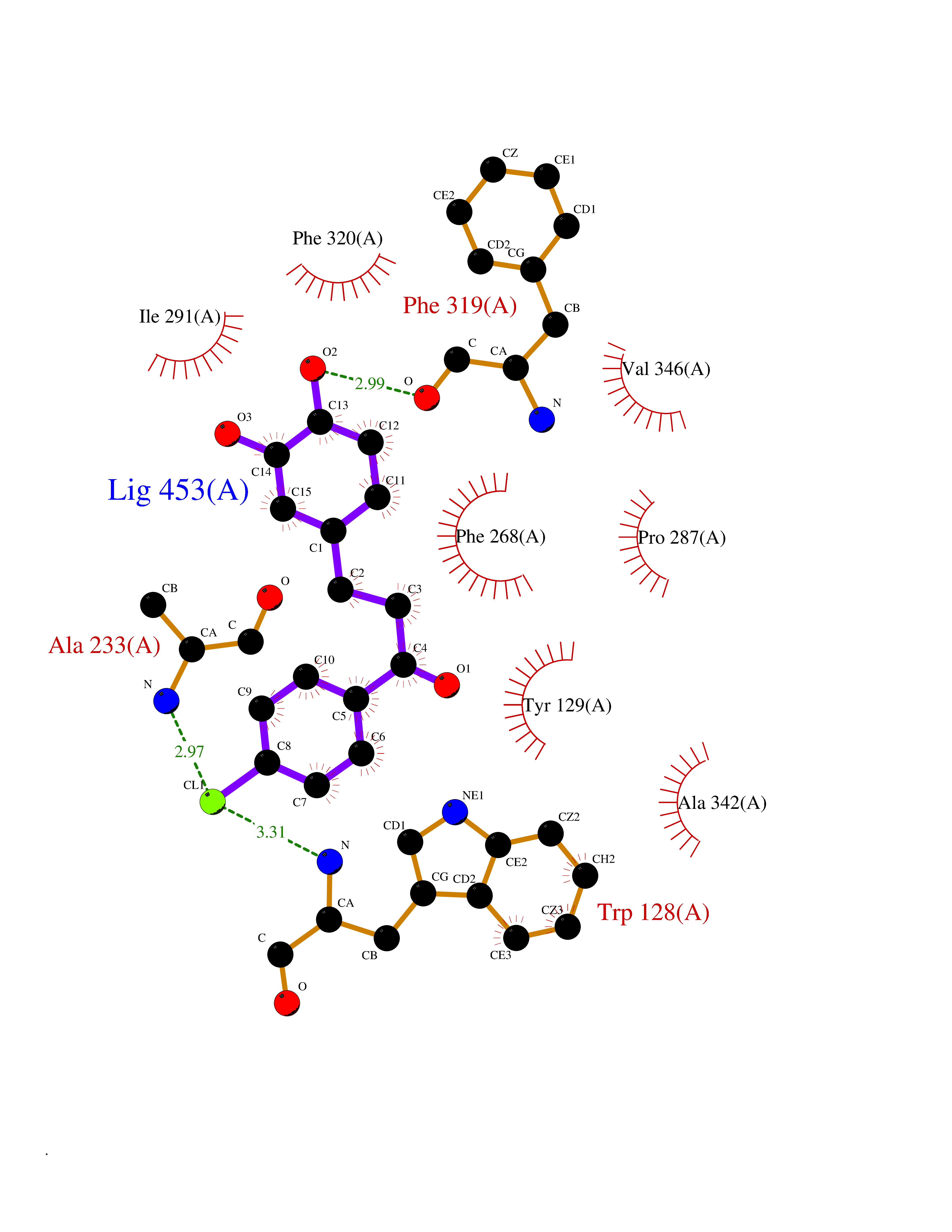 Ligplot Map 3D Binding mode Sequence EDLRLHLLLNTSVTCNDGSPAGYYLKESRGSRRWLLFLEGGWYCFNRENCDSRYDTMRRLMSSRDWPRTRTGTGILSSQPEENPYWWNANMVFIPYCSSDVWSGASSKSEKNEYAFMGALIIQEVVRELLGRGLSGAKVLLLAGSAAGGTGVLLNVDRVAEQLEKLGYPAIQVRGLADSGWFLDNKQYRHTDCVDTITCAPTEAIRRGIRYWNGVVPERCRRQFQEGEEWNCFFGYKVYPTLRSPVFVVQWLFDEAQLTVDNVHLTGQPVQEGLRLYIQNLGRELRHTLKDVPASFAPACLSHEIIIRSHWTDVQVKGTSLPRALHCWDRSLHKGCPVHLVDSCPWPHCNPSCPTS Hydrogen bonds contact Hydrophobic contact | ||||
| 23 | Bacterial Oxoacyl-[acyl-carrier-protein] synthase II (Bact fabF) | 2GFX | 7.06 | |
Target general information Gen name Bact fabF Organism Escherichia coli (strain K12) Uniprot ID TTD ID Synonyms KASB; KAS II; KAS 2; FabF; Condensing enzyme FabF; Beta-ketoacyl-acyl carrier protein synthase B; Beta-ketoacyl-ACP synthase II; Beta-ketoacyl-ACP synthase 2; 3-oxoacyl-[acyl-carrier-protein] synthase Protein family Thiolase-like superfamily, Beta-ketoacyl-ACP synthases family Biochemical class Acyltransferase Function Catalyzes the condensation reaction of fatty acid synthesis by the addition to an acyl acceptor of two carbons from malonyl-ACP. Has a preference for short chain acid substrates and may function to supply the octanoic substrates for lipoic acid biosynthesis. Related diseases Pigmentary disorder, reticulate, with systemic manifestations, X-linked (PDR) [MIM:301220]: An X-linked recessive disorder characterized by recurrent infections and sterile inflammation in various organs. Diffuse skin hyperpigmentation with a distinctive reticulate pattern is universally evident by early childhood. This is later followed in many patients by hypohidrosis, corneal inflammation and scarring, enterocolitis that resembles inflammatory bowel disease, and recurrent urethral strictures. Melanin and amyloid deposition is present in the dermis. Affected males also have a characteristic facies with frontally upswept hair and flared eyebrows. Female carriers have only restricted pigmentary changes along Blaschko's lines. {ECO:0000269|PubMed:27019227}. The disease is caused by variants affecting the gene represented in this entry. XLPDR is caused by a recurrent intronic mutation that results in missplicing and reduced POLA1 expression. This leads to a decrease in cytosolic RNA:DNA hybrids and constitutive activation of type I interferon responses, but has no effect on cell replication. {ECO:0000269|PubMed:27019227}.; DISEASE: Van Esch-O'Driscoll syndrome (VEODS) [MIM:301030]: An X-linked recessive syndrome characterized by different degrees of intellectual disability, moderate to severe short stature, microcephaly, hypogonadism, and variable congenital malformations. {ECO:0000269|PubMed:31006512}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08366; DB01034; DB03017; DB08407 Interacts with P0A6Y8 EC number EC 2.3.1.179 Uniprot keywords 3D-structure; Acyltransferase; Direct protein sequencing; Fatty acid biosynthesis; Fatty acid metabolism; Lipid biosynthesis; Lipid metabolism; Reference proteome; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 42852 Length 411 Aromaticity 0.06 Instability index 31.41 Isoelectric point 5.72 Charge (pH=7) -7.34 2D Binding mode Binding energy (Kcal/mol) -9.63 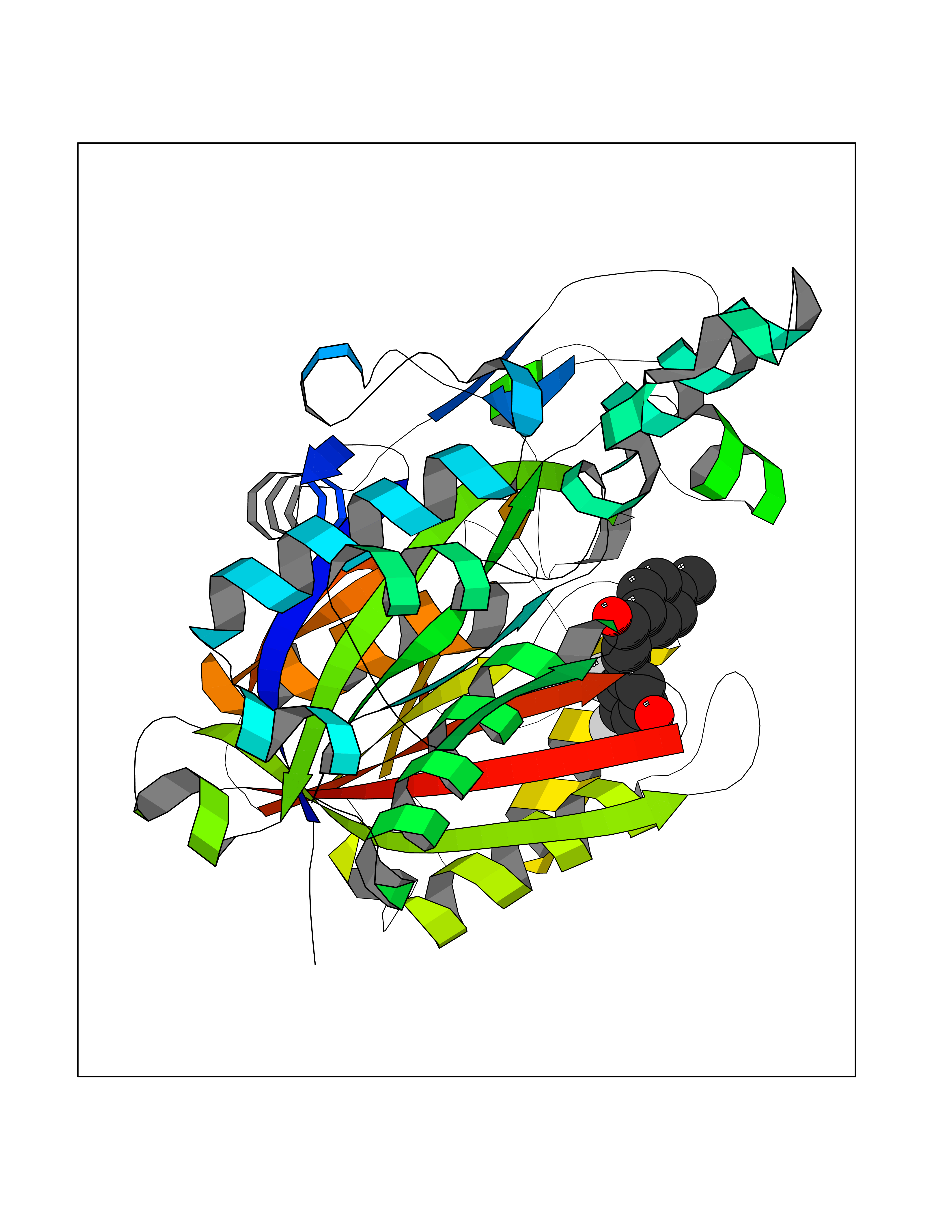 Molscript Map 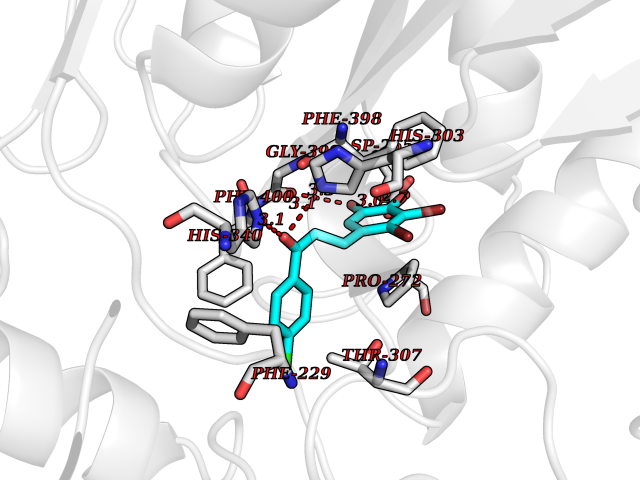 Pymol Map 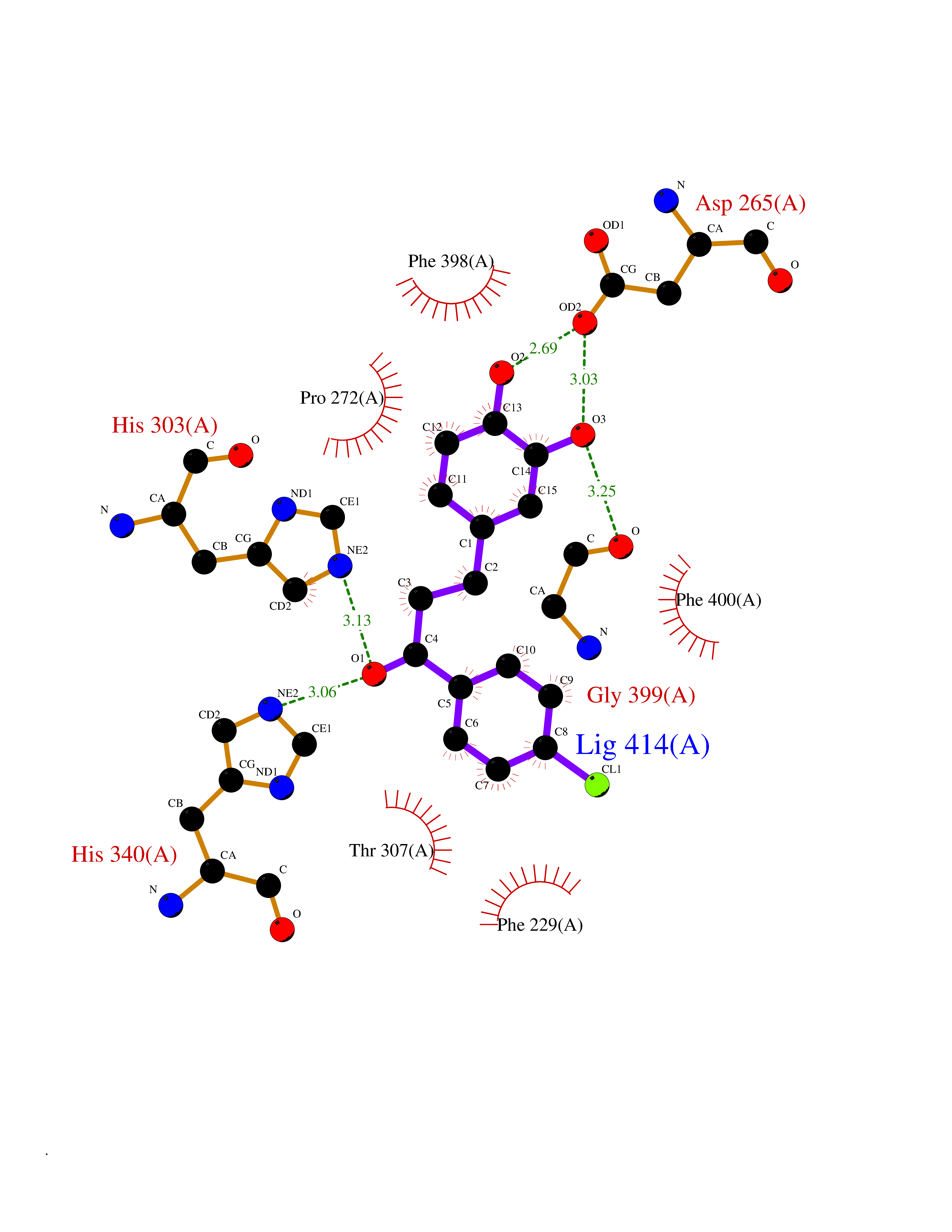 Ligplot Map 3D Binding mode Sequence KRRVVVTGLGMLSPVGNTVESTWKALLAGQSGISLIDHFDTSAYATKFAGLVKDFNCEDIISRKEQRKMDAFIQYGIVAGVQAMQDSGLEITEENATRIGAAIGSGIGGLGLIEENHTSLMNGGPRKISPFFVPSTIVNMVAGHLTIMYGLRGPSISIATAQTSGVHNIGHAARIIAYGDADVMVAGGAEKASTPLGVGGFGAARALSTRNDNPQAASRPWDKERDGFVLGDGAGMLVLEEYEHAKKRGAKIYAELVGFGMSSDAYHMTSPPENGAGAALAMANALRDAGIEASQIGYVNAHGTSTPAGDKAEAQAVKTIFGEAASRVLVSSTKSMTGHLLGAAGAVESIYSILALRDQAVPPTINLDNPDEGCDLDFVPHEARQVSGMEYTLCNSFGFGGTNGSLIFKKI Hydrogen bonds contact Hydrophobic contact | ||||
| 24 | Retinoic acid receptor alpha (RARA) | 3KMR | 7.05 | |
Target general information Gen name RARA Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms RAR-alpha; RAR alpha; Nuclear receptor subfamily 1 group B member 1; NR1B1 Protein family Nuclear hormone receptor family, NR1 subfamily Biochemical class Nuclear hormone receptor Function Retinoic acid receptors bind as heterodimers to their target response elements in response to their ligands, all-trans or 9-cis retinoic acid, and regulate gene expression in various biological processes. The RXR/RAR heterodimers bind to the retinoic acid response elements (RARE) composed of tandem 5'-AGGTCA-3' sites known as DR1-DR5. In the absence of ligand, the RXR-RAR heterodimers associate with a multiprotein complex containing transcription corepressors that induce histone acetylation, chromatin condensation and transcriptional suppression. On ligand binding, the corepressors dissociate from the receptors and associate with the coactivators leading to transcriptional activation. RARA plays an essential role in the regulation of retinoic acid-induced germ cell development during spermatogenesis. Has a role in the survival of early spermatocytes at the beginning prophase of meiosis. In Sertoli cells, may promote the survival and development of early meiotic prophase spermatocytes. In concert with RARG, required for skeletal growth, matrix homeostasis and growth plate function. Receptor for retinoic acid. Related diseases Chromosomal aberrations involving RARA are commonly found in acute promyelocytic leukemia. Translocation t(11;17)(q32;q21) with ZBTB16/PLZF; translocation t(15;17)(q21;q21) with PML; translocation t(5;17)(q32;q11) with NPM. The PML-RARA oncoprotein requires both the PML ring structure and coiled-coil domain for both interaction with UBE2I, nuclear microspeckle location and sumoylation. In addition, the coiled-coil domain functions in blocking RA-mediated transactivation and cell differentiation. {ECO:0000269|PubMed:12691149, ECO:0000269|PubMed:8302850, ECO:0000269|PubMed:8562957}. Drugs (DrugBank ID) DB00459; DB00210; DB00523; DB00926; DB00982; DB05785; DB04942; DB00799; DB00755; DB12808 Interacts with O43707-1; O15296; Q15699; Q96RK4; O95273; P51946; Q15910; P50148; Q9UKP3; Q96EZ8; Q15648; Q71SY5; Q15788; Q9Y6Q9; O75376; Q9Y618; Q16236; P13056-2; P48552; Q9UPP1-2; Q9H8W4; P37231; P78527; P19793; P28702; P28702-3; P48443; Q96EB6; P63165; Q8WW24; Q2M1K9; Q91XC0; P59598; Q14457; P48552; Q96CV9; P28702; P48443; Q8WW24 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Chromosomal rearrangement; Cytoplasm; DNA-binding; Isopeptide bond; Metal-binding; Nucleus; Phosphoprotein; Proteomics identification; Proto-oncogene; Receptor; Reference proteome; Transcription; Transcription regulation; Ubl conjugation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A Molecular weight (Da) 27724.1 Length 244 Aromaticity 0.05 Instability index 50.8 Isoelectric point 5.82 Charge (pH=7) -3.61 2D Binding mode Binding energy (Kcal/mol) -9.61  Molscript Map 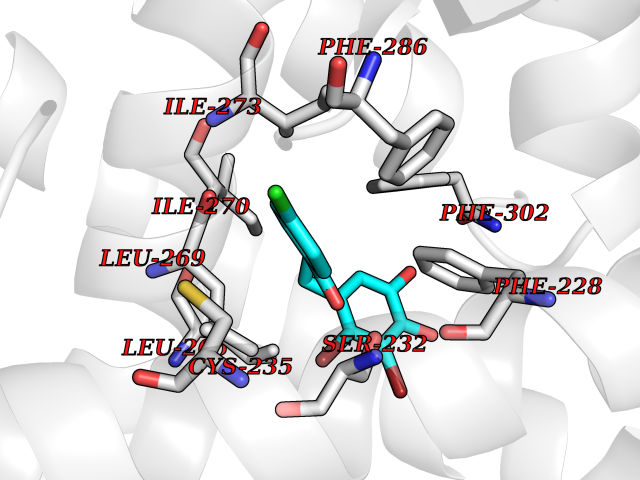 Pymol Map 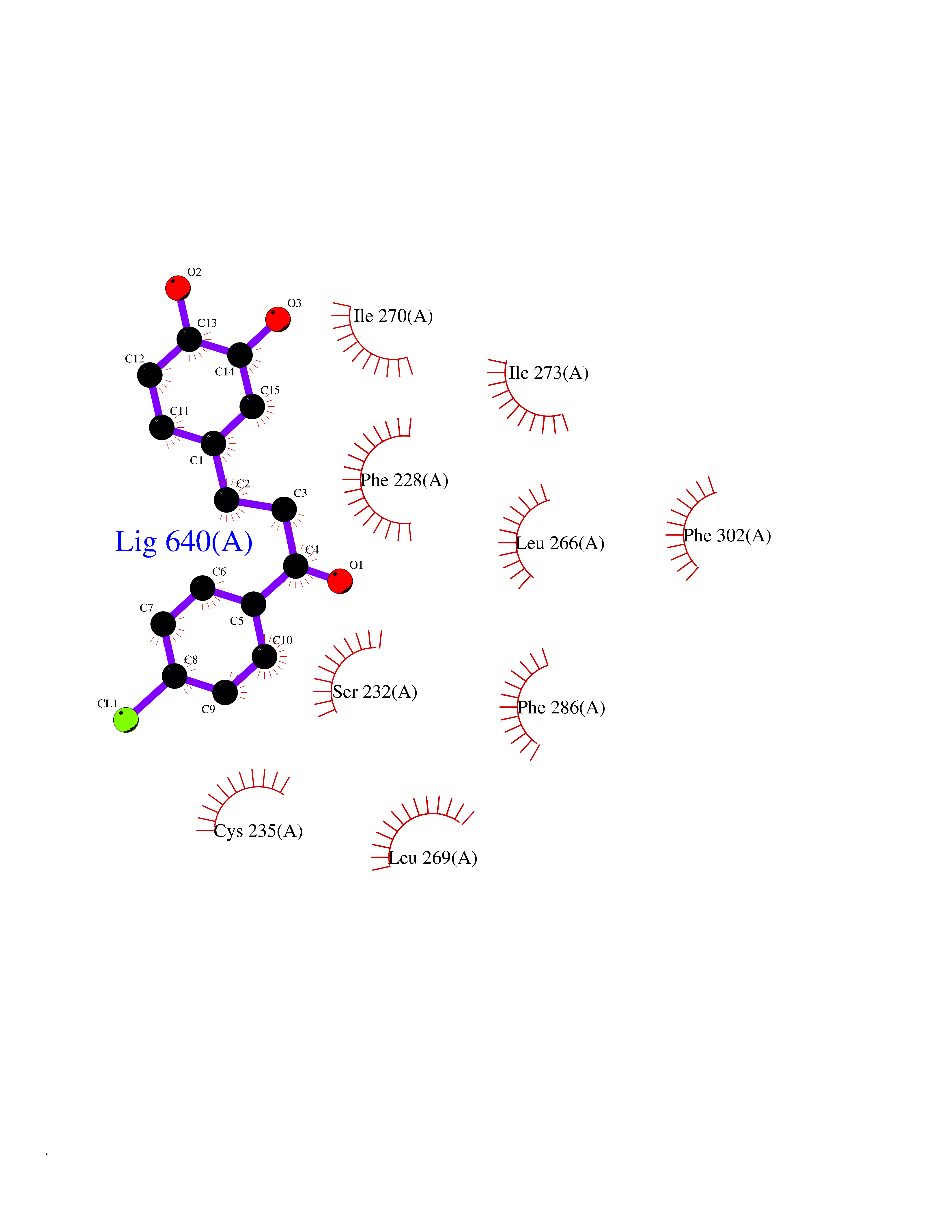 Ligplot Map 3D Binding mode Sequence PEVGELIEKVRKAHQETFPALCQLGKYTTNNSSEQRVSLDIDLWDKFSELSTKCIIKTVEFAKQLPGFTTLTIADQITLLKAACLDILILRICTRYTPEQDTMTFSDGLTLNRTQMHNAGFGPLTDLVFAFANQLLPLEMDDAETGLLSAICLICGDRQDLEQPDRVDMLQEPLLEALKVYVRKRRPSRPHMFPKMLMKITDLRSISAKGAERVITLKMEIPGSMPPLIQEMLEHKILHRLLQE Hydrogen bonds contact Hydrophobic contact | ||||
| 25 | Retinoic acid receptor gamma (RARG) | 1FCY | 7.05 | |
Target general information Gen name RARG Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms RAR-gamma; Nuclear receptor subfamily 1 group B member 3; NR1B3 Protein family Nuclear hormone receptor family, NR1 subfamily Biochemical class Nuclear hormone receptor Function Receptor for retinoic acid. Retinoic acid receptors bind as heterodimers to their target response elements in response to their ligands, all-trans or 9-cis retinoic acid, and regulate gene expression in various biological processes. The RAR/RXR heterodimers bind to the retinoic acid response elements (RARE) composed of tandem 5'-AGGTCA-3' sites known as DR1-DR5. In the absence of ligand, acts mainly as an activator of gene expression due to weak binding to corepressors. Required for limb bud development. In concert with RARA or RARB, required for skeletal growth, matrix homeostasis and growth plate function (By similarity). Related diseases Cystic fibrosis (CF) [MIM:219700]: A common generalized disorder of the exocrine glands which impairs clearance of secretions in a variety of organs. It is characterized by the triad of chronic bronchopulmonary disease (with recurrent respiratory infections), pancreatic insufficiency (which leads to malabsorption and growth retardation) and elevated sweat electrolytes. It is the most common genetic disease in Caucasians, with a prevalence of about 1 in 2'000 live births. Inheritance is autosomal recessive. {ECO:0000269|PubMed:10094564, ECO:0000269|PubMed:10869121, ECO:0000269|PubMed:10923036, ECO:0000269|PubMed:11242048, ECO:0000269|PubMed:12167682, ECO:0000269|PubMed:12394343, ECO:0000269|PubMed:12529365, ECO:0000269|PubMed:1284466, ECO:0000269|PubMed:1284468, ECO:0000269|PubMed:1284529, ECO:0000269|PubMed:1284530, ECO:0000269|PubMed:1284548, ECO:0000269|PubMed:1379210, ECO:0000269|PubMed:15528182, ECO:0000269|PubMed:15716351, ECO:0000269|PubMed:16822950, ECO:0000269|PubMed:1695717, ECO:0000269|PubMed:1699669, ECO:0000269|PubMed:17098864, ECO:0000269|PubMed:1710600, ECO:0000269|PubMed:1712898, ECO:0000269|PubMed:17182731, ECO:0000269|PubMed:20008117, ECO:0000269|PubMed:20150177, ECO:0000269|PubMed:20691141, ECO:0000269|PubMed:21884936, ECO:0000269|PubMed:2236053, ECO:0000269|PubMed:23818989, ECO:0000269|PubMed:25330774, ECO:0000269|PubMed:26846474, ECO:0000269|PubMed:27241308, ECO:0000269|PubMed:28001373, ECO:0000269|PubMed:28067262, ECO:0000269|PubMed:28087700, ECO:0000269|PubMed:32026723, ECO:0000269|PubMed:33572515, ECO:0000269|PubMed:7504969, ECO:0000269|PubMed:7505694, ECO:0000269|PubMed:7505767, ECO:0000269|PubMed:7508414, ECO:0000269|PubMed:7513296, ECO:0000269|PubMed:7517264, ECO:0000269|PubMed:7520022, ECO:0000269|PubMed:7522211, ECO:0000269|PubMed:7524909, ECO:0000269|PubMed:7524913, ECO:0000269|PubMed:7525450, ECO:0000269|PubMed:7537150, ECO:0000269|PubMed:7541273, ECO:0000269|PubMed:7541510, ECO:0000269|PubMed:7543567, ECO:0000269|PubMed:7544319, ECO:0000269|PubMed:7581407, ECO:0000269|PubMed:7606851, ECO:0000269|PubMed:7680525, ECO:0000269|PubMed:7683628, ECO:0000269|PubMed:7683954, ECO:0000269|PubMed:8081395, ECO:0000269|PubMed:8406518, ECO:0000269|PubMed:8522333, ECO:0000269|PubMed:8723693, ECO:0000269|PubMed:8723695, ECO:0000269|PubMed:8800923, ECO:0000269|PubMed:8829633, ECO:0000269|PubMed:8910473, ECO:0000269|PubMed:8956039, ECO:0000269|PubMed:9101301, ECO:0000269|PubMed:9222768, ECO:0000269|PubMed:9375855, ECO:0000269|PubMed:9401006, ECO:0000269|PubMed:9443874, ECO:0000269|PubMed:9452048, ECO:0000269|PubMed:9452054, ECO:0000269|PubMed:9452073, ECO:0000269|PubMed:9482579, ECO:0000269|PubMed:9507391, ECO:0000269|PubMed:9521595, ECO:0000269|PubMed:9554753, ECO:0000269|PubMed:9736778, ECO:0000269|PubMed:9804160, ECO:0000269|PubMed:9921909}. The disease is caused by variants affecting the gene represented in this entry. There is some evidence that the functional defect caused by the most common variant Phe-508 DEL can be corrected by the binding to the snake phospholipase A2 crotoxin basic subunit CB. This toxin both disrupts the Phe-508 DEL-cytokeratin 8 complex, allowing for the escape from degradation, and increases the chloride channel current (PubMed:27241308). {ECO:0000269|PubMed:27241308}.; DISEASE: Congenital bilateral absence of the vas deferens (CBAVD) [MIM:277180]: An autosomal recessive disease characterized by vas deferens aplasia resulting in azoospermia and male infertility. CBAVD may occur in isolation or as a manifestation of cystic fibrosis. {ECO:0000269|PubMed:10066035, ECO:0000269|PubMed:10651488, ECO:0000269|PubMed:17329263, ECO:0000269|PubMed:7529962, ECO:0000269|PubMed:7539342, ECO:0000269|PubMed:9067761, ECO:0000269|PubMed:9736778, ECO:0000269|Ref.117}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07294; DB07031; DB00459; DB00210; DB00523; DB02466; DB03466; DB02741; DB03279; DB00926; DB00982; DB05785; DB05467; DB02258; DB00799; DB00755; DB12808 Interacts with Q96RK4; P13349; P31321; P28702; P48443; O60504-2 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; DNA-binding; Isopeptide bond; Metal-binding; Methylation; Nucleus; Proteomics identification; Receptor; Reference proteome; Transcription; Transcription regulation; Ubl conjugation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A Molecular weight (Da) 26574.9 Length 236 Aromaticity 0.06 Instability index 49.98 Isoelectric point 5.76 Charge (pH=7) -2.95 2D Binding mode Binding energy (Kcal/mol) -9.62 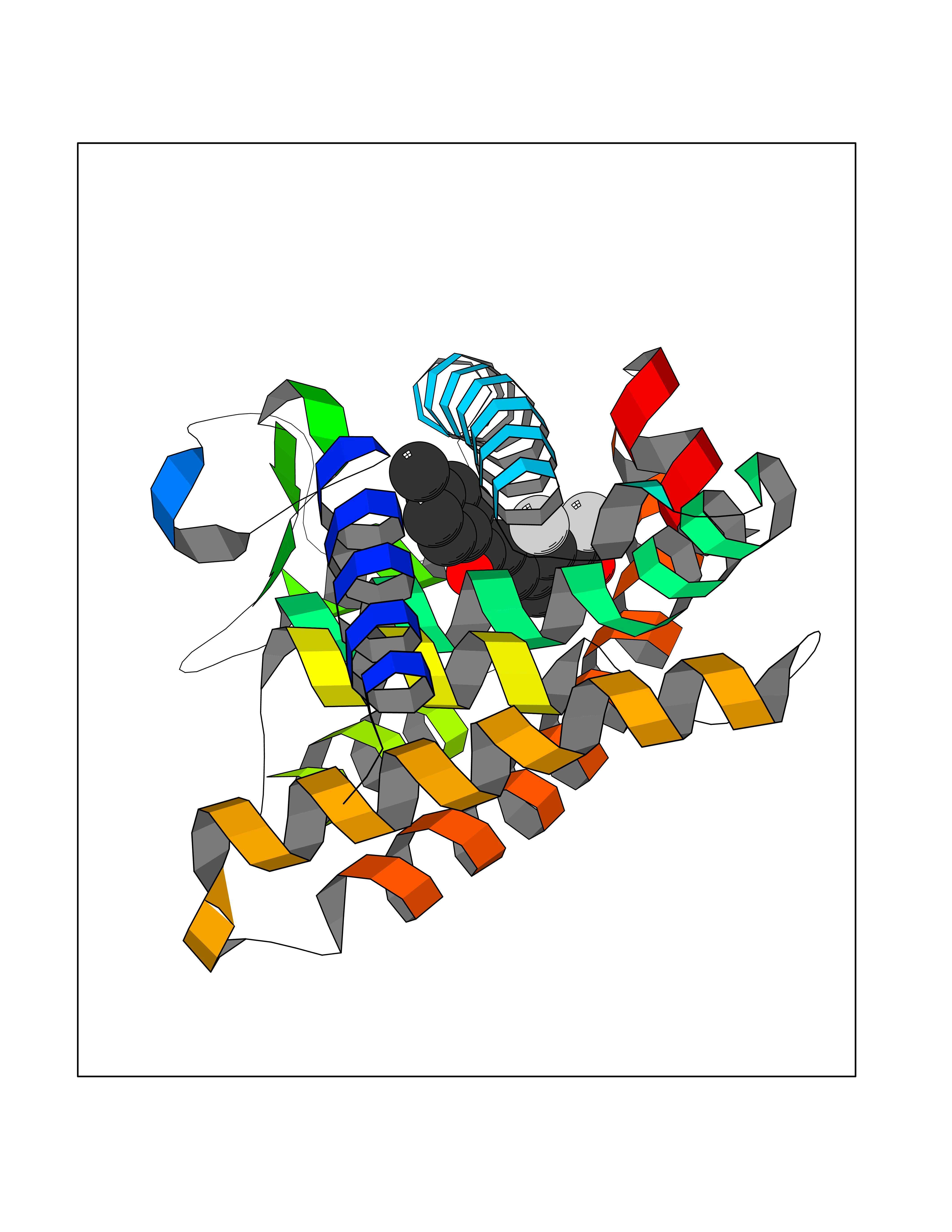 Molscript Map 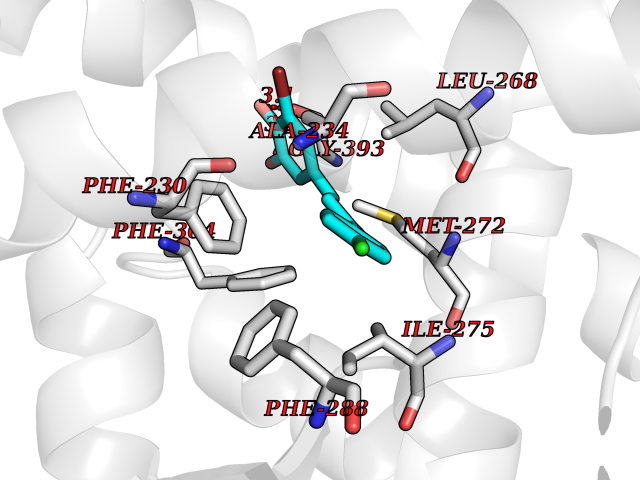 Pymol Map 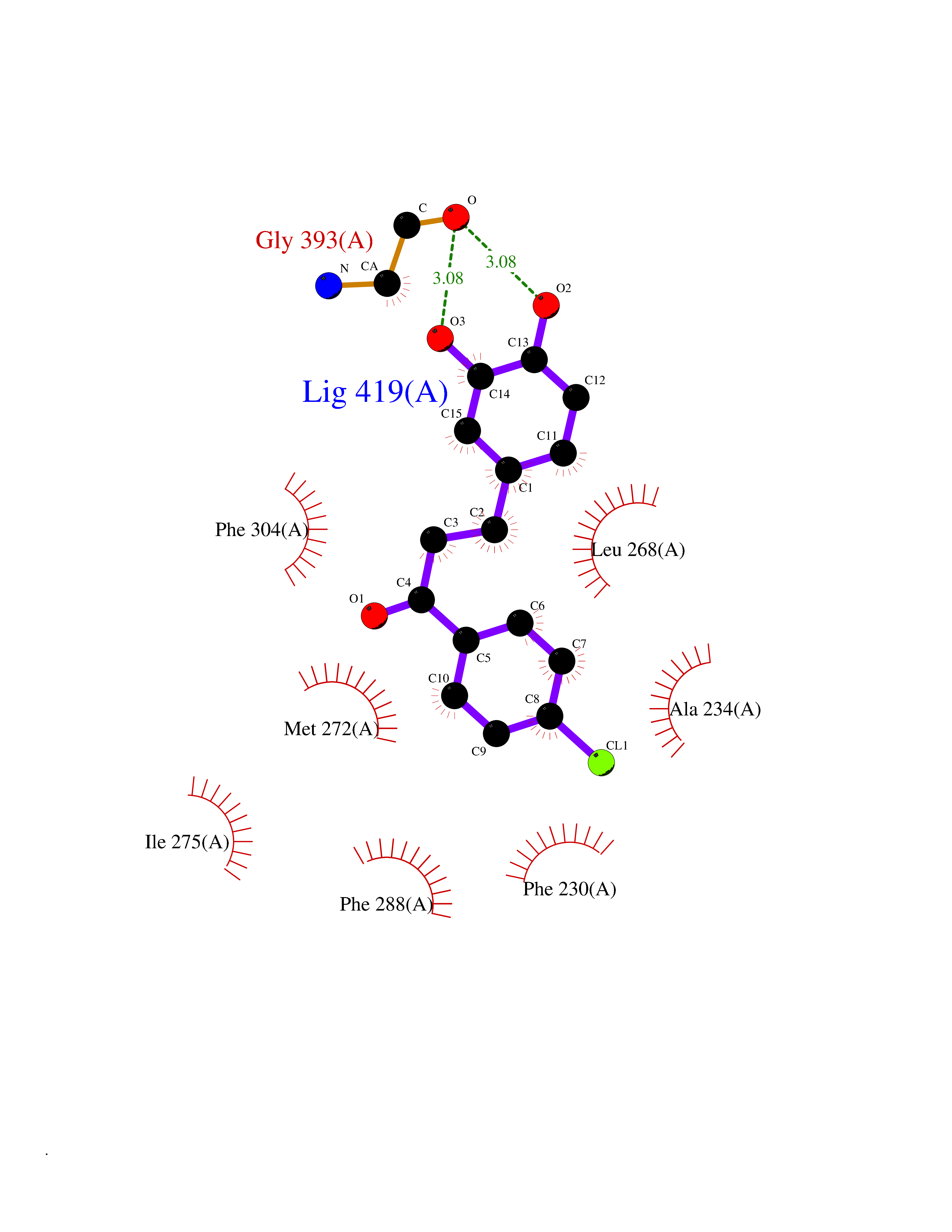 Ligplot Map 3D Binding mode Sequence ASPQLEELITKVSKAHQETFPSLCQLGKYTTNSSADHRVQLDLGLWDKFSELATKCIIKIVEFAKRLPGFTGLSIADQITLLKAACLDILMLRICTRYTPEQDTMTFSDGLTLNRTQMHNAGFGPLTDLVFAFAGQLLPLEMDDTETGLLSAICLICGDRMDLEEPEKVDKLQEPLLEALRLYARRRRPSQPYMFPRMLMKITDLRGISTKGAERAITLKMEIPGPMPPLIREMLE Hydrogen bonds contact Hydrophobic contact | ||||
| 26 | Retinoic acid receptor beta (RARB) | 4DM6 | 7.04 | |
Target general information Gen name RARB Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms RAR-epsilon; RAR-beta; Nuclear receptor subfamily 1 group B member 2; NR1B2; HBV-activated protein; HAP Protein family Nuclear hormone receptor family, NR1 subfamily Biochemical class Nuclear hormone receptor Function Retinoic acid receptors bind as heterodimers to their target response elements in response to their ligands, all-trans or 9-cis retinoic acid, and regulate gene expression in various biological processes. The RXR/RAR heterodimers bind to the retinoic acid response elements (RARE) composed of tandem 5'-AGGTCA-3' sites known as DR1-DR5. In the absence or presence of hormone ligand, acts mainly as an activator of gene expression due to weak binding to corepressors. In concert with RARG, required for skeletal growth, matrix homeostasis and growth plate function. Receptor for retinoic acid. Related diseases Microphthalmia, syndromic, 12 (MCOPS12) [MIM:615524]: A form of microphthalmia, a disorder of eye formation, ranging from small size of a single eye to complete bilateral absence of ocular tissues (anophthalmia). In many cases, microphthalmia/anophthalmia occurs in association with syndromes that include non-ocular abnormalities. MCOPS12 patients manifest variable features, including diaphragmatic hernia, pulmonary hypoplasia, and cardiac abnormalities. {ECO:0000269|PubMed:24075189, ECO:0000269|PubMed:27120018}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00459; DB00210; DB00523; DB02877; DB00926; DB05785; DB04942; DB00799; DB00755; DB12808 Interacts with O95273; P50222; Q9UBK2; P62195; P28702; P28702-3; P48443; P03255 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; Disease variant; DNA-binding; Metal-binding; Microphthalmia; Nucleus; Phosphoprotein; Proto-oncogene; Receptor; Reference proteome; Transcription; Transcription regulation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A,B Molecular weight (Da) 25904.1 Length 229 Aromaticity 0.06 Instability index 44.34 Isoelectric point 7.55 Charge (pH=7) 0.73 2D Binding mode Binding energy (Kcal/mol) -9.6  Molscript Map 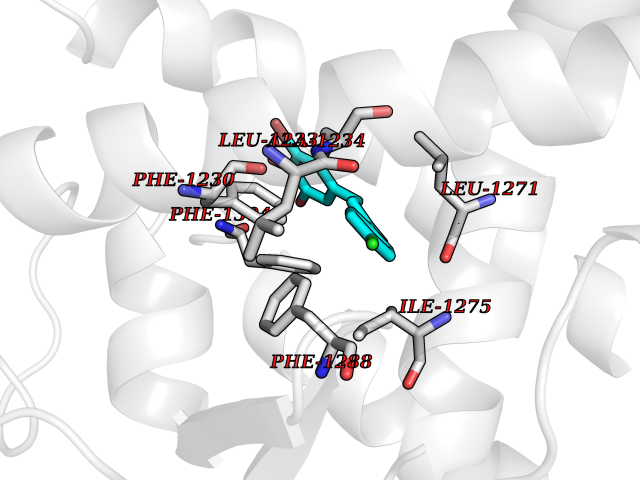 Pymol Map 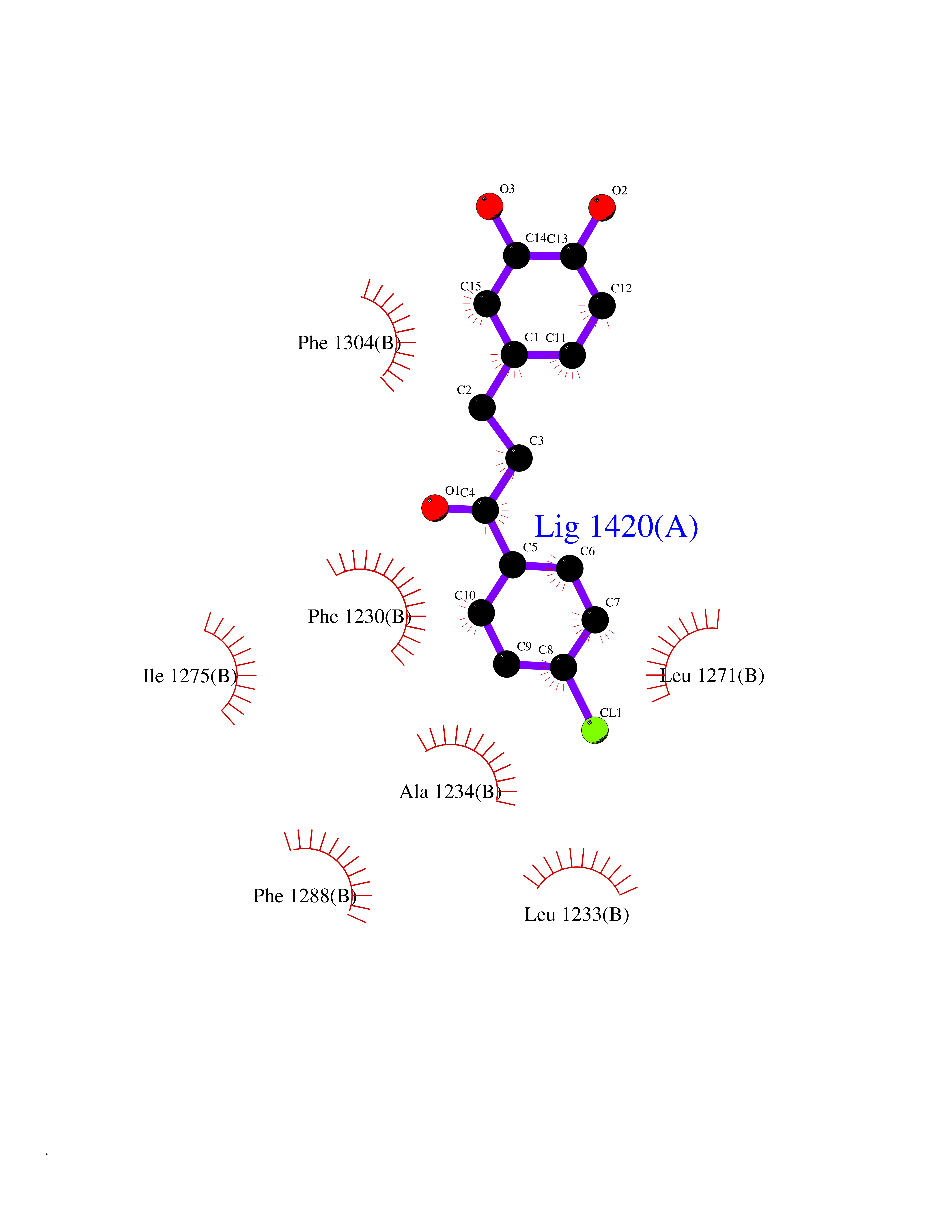 Ligplot Map 3D Binding mode Sequence TEKIRKAHQETFPSLCQLGKYTTNSSADHRVRLDLGLWDKFSELATKCIIKIVEFAKRLPGFTGLTIADQITLLKAACLDILILRICTRYTPEQDTMTFSDGLTLNRTQMHNAGFGPLTDLVFTFANQLLPLEMDDTETGLLSAICLICGDRQDLEEPTKVDKLQEPLLEALKIYIRKRRPSKPHMFPKILMKITDLRSISAKGAERVITLKMEIPGSMPPLIQEMLEN Hydrogen bonds contact Hydrophobic contact | ||||
| 27 | Sphingosine kinase 1 (SPHK1) | 3VZB | 7.04 | |
Target general information Gen name SPHK1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms SPK 1; SPK; SPHK1; SK 1; Acetyltransferase SPHK1 Protein family NA Biochemical class Kinase Function Acts on D-erythro-sphingosine and to a lesser extent sphinganine, but not other lipids, such as D,L-threo-dihydrosphingosine, N,N-dimethylsphingosine, diacylglycerol, ceramide, or phosphatidylinositol. In contrast to proapoptotic SPHK2, has a negative effect on intracellular ceramide levels, enhances cell growth and inhibits apoptosis. Involved in the regulation of inflammatory response and neuroinflammation. Via the product sphingosine 1-phosphate, stimulates TRAF2 E3 ubiquitin ligase activity, and promotes activation of NF-kappa-B in response to TNF signaling leading to IL17 secretion. In response to TNF and in parallel to NF-kappa-B activation, negatively regulates RANTES inducion through p38 MAPK signaling pathway. Involved in endocytic membrane trafficking induced by sphingosine, recruited to dilate endosomes, also plays a role on later stages of endosomal maturation and membrane fusion independently of its kinase activity. In Purkinje cells, seems to be also involved in the regulation of autophagosome-lysosome fusion upon VEGFA. Catalyzes the phosphorylation of sphingosine to form sphingosine 1-phosphate (SPP), a lipid mediator with both intra- and extracellular functions. Related diseases Intellectual developmental disorder, X-linked, syndromic, Claes-Jensen type (MRXSCJ) [MIM:300534]: A disorder characterized by significantly below average general intellectual functioning associated with impairments in adaptive behavior and manifested during the developmental period. MRXSCJ patients manifest intellectual disability associated with variable features such as slowly progressive spastic paraplegia, seizures, facial dysmorphism. {ECO:0000269|PubMed:15586325, ECO:0000269|PubMed:16538222, ECO:0000269|PubMed:16541399, ECO:0000269|PubMed:17320160, ECO:0000269|PubMed:17468742, ECO:0000269|PubMed:23356856, ECO:0000269|PubMed:25666439}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08868 Interacts with P07858; P68104; Q14192; Q2M3C7; Q9Y4K3; P13473-2; Q9Y371 EC number EC 2.7.1.91 Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Calmodulin-binding; Cell membrane; Coated pit; Cytoplasm; Endosome; Kinase; Lipid metabolism; Membrane; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Synapse; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 39813 Length 360 Aromaticity 0.08 Instability index 43.79 Isoelectric point 7.34 Charge (pH=7) 0.84 2D Binding mode Binding energy (Kcal/mol) -9.6  Molscript Map 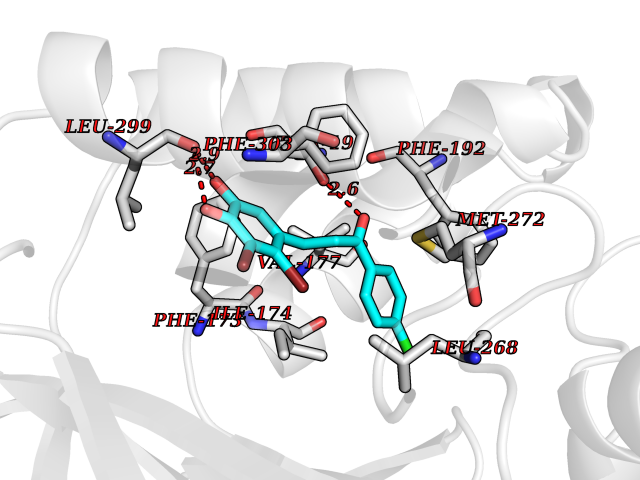 Pymol Map 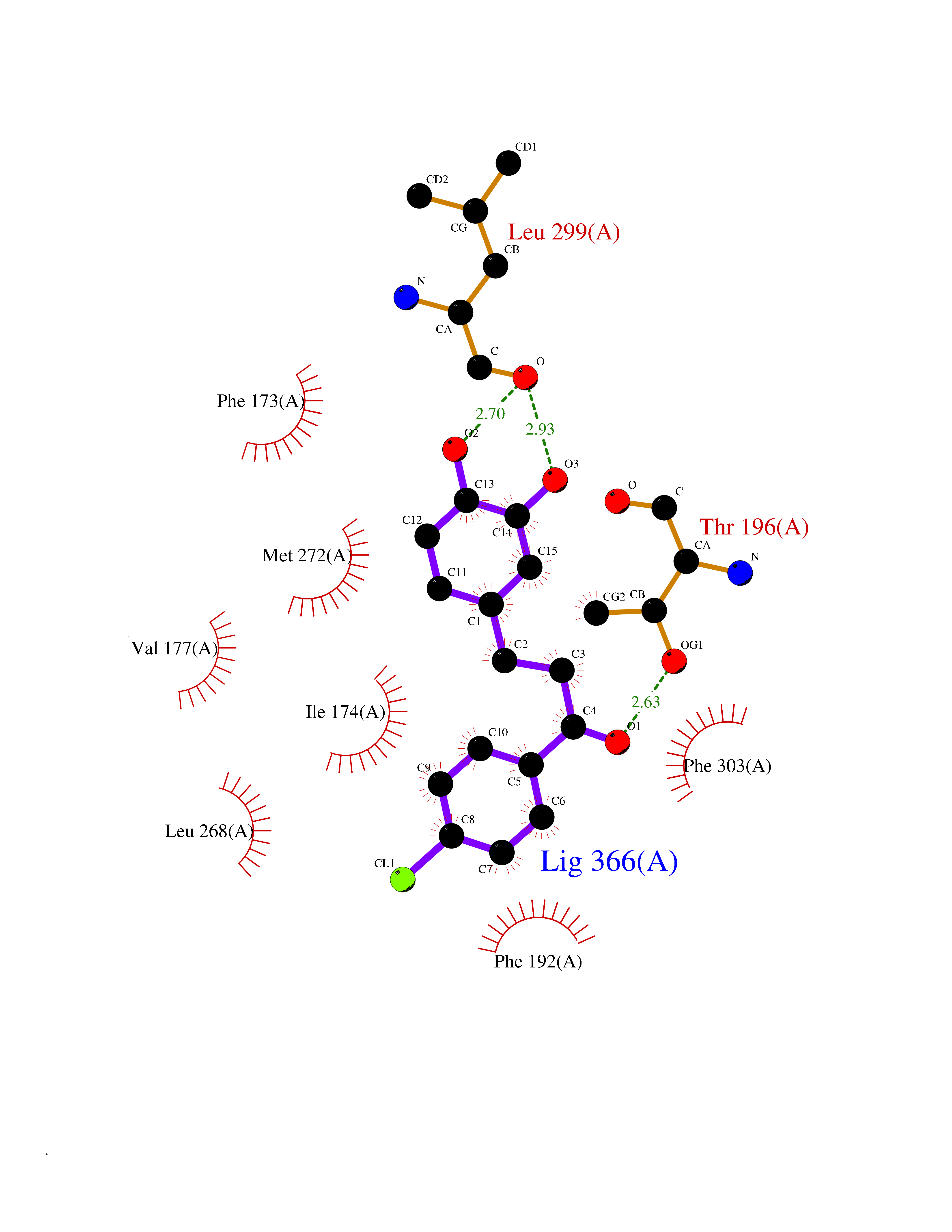 Ligplot Map 3D Binding mode Sequence AMGSGVLPRPCRVLVLLNPRGGKGKALQLFRSHVQPLLAEAEISFTLMLTERRNHARELVRSEELGRWDALVVMSGDGLMHEVVNGLMERPDWETAIQKPLCSLPAGSGNALAASLNHYAGYEQVTNEDLLTNCTLLLCRRLLSPMNLLSLHTASGLRLFSVLSLAWGFIADVDLESEKYRRLGEMRFTLGTFLRLAALRTYRGRLAYLPVGRVGSKTPASPVVVQQGPVDAHLVPLEEPVPSHWTVVPDEDFVLVLALLHSHLGSEMFAAPMGRCAAGVMHLFYVRAGVSRAMLLRLFLAMEKGRHMEYECPYLVYVPVVAFRLEPKDGKGVFAVDGELMVSEAVQGQVHPNYFWMVSG Hydrogen bonds contact Hydrophobic contact | ||||
| 28 | Janus kinase 2 (JAK-2) | 3UGC | 7.03 | |
Target general information Gen name JAK2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Tyrosine-protein kinase JAK2 Protein family Protein kinase superfamily, Tyr protein kinase family, JAK subfamily Biochemical class Kinase Function Mediates essential signaling events in both innate and adaptive immunity. In the cytoplasm, plays a pivotal role in signal transduction via its association with type I receptors such as growth hormone (GHR), prolactin (PRLR), leptin (LEPR), erythropoietin (EPOR), thrombopoietin (THPO); or type II receptors including IFN-alpha, IFN-beta, IFN-gamma and multiple interleukins. Following ligand-binding to cell surface receptors, phosphorylates specific tyrosine residues on the cytoplasmic tails of the receptor, creating docking sites for STATs proteins. Subsequently, phosphorylates the STATs proteins once they are recruited to the receptor. Phosphorylated STATs then form homodimer or heterodimers and translocate to the nucleus to activate gene transcription. For example, cell stimulation with erythropoietin (EPO) during erythropoiesis leads to JAK2 autophosphorylation, activation, and its association with erythropoietin receptor (EPOR) that becomes phosphorylated in its cytoplasmic domain. Then, STAT5 (STAT5A or STAT5B) is recruited, phosphorylated and activated by JAK2. Once activated, dimerized STAT5 translocates into the nucleus and promotes the transcription of several essential genes involved in the modulation of erythropoiesis. Part of a signaling cascade that is activated by increased cellular retinol and that leads to the activation of STAT5 (STAT5A or STAT5B). In addition, JAK2 mediates angiotensin-2-induced ARHGEF1 phosphorylation. Plays a role in cell cycle by phosphorylating CDKN1B. Cooperates with TEC through reciprocal phosphorylation to mediate cytokine-driven activation of FOS transcription. In the nucleus, plays a key role in chromatin by specifically mediating phosphorylation of 'Tyr-41' of histone H3 (H3Y41ph), a specific tag that promotes exclusion of CBX5 (HP1 alpha) from chromatin. Non-receptor tyrosine kinase involved in various processes such as cell growth, development, differentiation or histone modifications. Related diseases Chromosomal aberrations involving JAK2 are found in both chronic and acute forms of eosinophilic, lymphoblastic and myeloid leukemia. Translocation t(8;9)(p22;p24) with PCM1 links the protein kinase domain of JAK2 to the major portion of PCM1. Translocation t(9;12)(p24;p13) with ETV6.; DISEASE: Budd-Chiari syndrome (BDCHS) [MIM:600880]: A syndrome caused by obstruction of hepatic venous outflow involving either the hepatic veins or the terminal segment of the inferior vena cava. Obstructions are generally caused by thrombosis and lead to hepatic congestion and ischemic necrosis. Clinical manifestations observed in the majority of patients include hepatomegaly, right upper quadrant pain and abdominal ascites. Budd-Chiari syndrome is associated with a combination of disease states including primary myeloproliferative syndromes and thrombophilia due to factor V Leiden, protein C deficiency and antithrombin III deficiency. Budd-Chiari syndrome is a rare but typical complication in patients with polycythemia vera. {ECO:0000269|PubMed:16707754}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Polycythemia vera (PV) [MIM:263300]: A myeloproliferative disorder characterized by abnormal proliferation of all hematopoietic bone marrow elements, erythroid hyperplasia, an absolute increase in total blood volume, but also by myeloid leukocytosis, thrombocytosis and splenomegaly. {ECO:0000269|PubMed:15781101, ECO:0000269|PubMed:15793561, ECO:0000269|PubMed:15858187, ECO:0000269|PubMed:16603627, ECO:0000269|PubMed:25644777}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Thrombocythemia 3 (THCYT3) [MIM:614521]: A myeloproliferative disorder characterized by excessive platelet production, resulting in increased numbers of circulating platelets. It can be associated with spontaneous hemorrhages and thrombotic episodes. {ECO:0000269|PubMed:16325696, ECO:0000269|PubMed:22397670}. The disease may be caused by variants affecting the gene represented in this entry.; DISEASE: Myelofibrosis (MYELOF) [MIM:254450]: A disorder characterized by replacement of the bone marrow by fibrous tissue, occurring in association with a myeloproliferative disorder. Clinical manifestations may include anemia, pallor, splenomegaly, hypermetabolic state, petechiae, ecchymosis, bleeding, lymphadenopathy, hepatomegaly, portal hypertension. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Leukemia, acute myelogenous (AML) [MIM:601626]: A subtype of acute leukemia, a cancer of the white blood cells. AML is a malignant disease of bone marrow characterized by maturational arrest of hematopoietic precursors at an early stage of development. Clonal expansion of myeloid blasts occurs in bone marrow, blood, and other tissue. Myelogenous leukemias develop from changes in cells that normally produce neutrophils, basophils, eosinophils and monocytes. {ECO:0000269|PubMed:16247455}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB04716; DB07162; DB08067; DB07161; DB14973; DB11817; DB11986; DB12500; DB12010; DB11763; DB11697; DB15822; DB08877; DB08895; DB05243; DB15035 Interacts with P32927; Q01344; P23458; O60674; P40238; P16333; P18031; O75116; P29597; Q9JHI9 EC number EC 2.7.10.2 Uniprot keywords 3D-structure; Adaptive immunity; ATP-binding; Chromatin regulator; Chromosomal rearrangement; Cytoplasm; Disease variant; Immunity; Innate immunity; Kinase; Magnesium; Membrane; Metal-binding; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Proto-oncogene; Reference proteome; Repeat; SH2 domain; Transferase; Tyrosine-protein kinase; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 32174.5 Length 274 Aromaticity 0.1 Instability index 50.94 Isoelectric point 7.78 Charge (pH=7) 1.46 2D Binding mode Binding energy (Kcal/mol) -9.59  Molscript Map 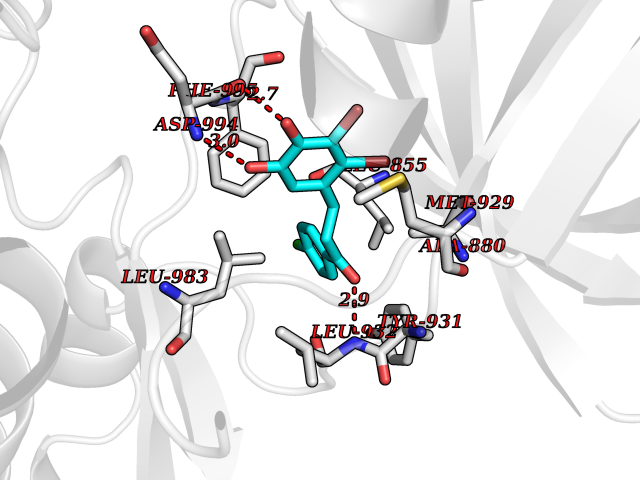 Pymol Map 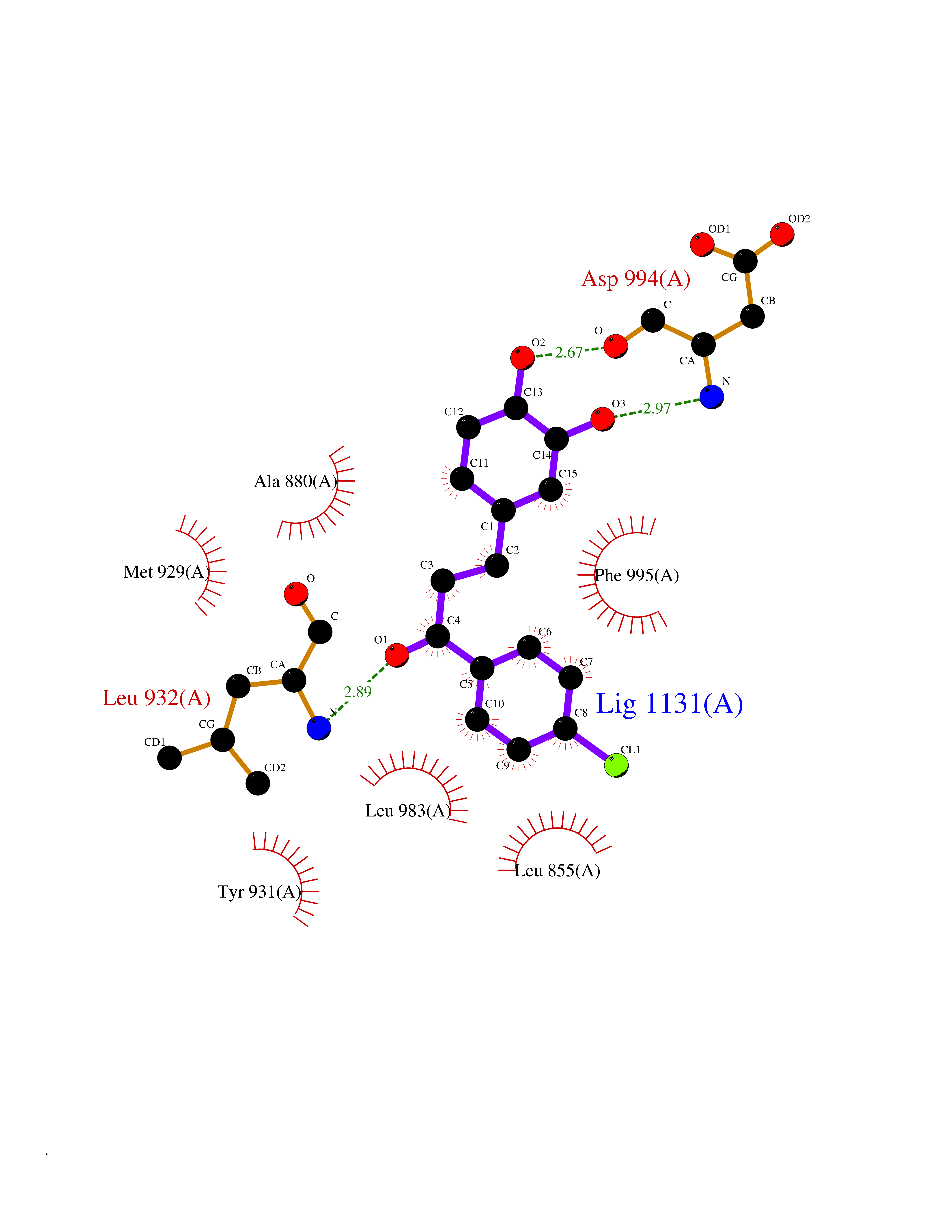 Ligplot Map 3D Binding mode Sequence QFEERHLKFLQQLGKGNFGSVEMCRYDPLQDNTGEVVAVKKLQHSTEEHLRDFEREIEILKSLQHDNIVKYKGVCYSAGRRNLKLIMEYLPYGSLRDYLQKHKERIDHIKLLQYTSQICKGMEYLGTKRYIHRDLATRNILVENENRVKIGDFGLTKPGESPIFWYAPESLTESKFSVASDVWSFGVVLYELFTYIEKSKSPPAEFMRMIGNDKQGQMIVFHLIELLKNNGRLPRPDGCPDEIYMIMTECWNNNVNQRPSFRDLALRVDQIRDN Hydrogen bonds contact Hydrophobic contact | ||||
| 29 | Protein cereblon (CRBN) | 5FQD | 7.03 | |
Target general information Gen name CRBN Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Protein cereblon Protein family CRBN family Biochemical class NA Function Substrate recognition component of a DCX (DDB1-CUL4-X-box) E3 protein ligase complex that mediates the ubiquitination and subsequent proteasomal degradation of target proteins, such as MEIS2. Normal degradation of key regulatory proteins is required for normal limb outgrowth and expression of the fibroblast growth factor FGF8. May play a role in memory and learning by regulating the assembly and neuronal surface expression of large-conductance calcium-activated potassium channels in brain regions involved in memory and learning via its interaction with KCNT1. Binding of pomalidomide and other thalidomide-related drugs changes the substrate specificity of the human protein, leading to decreased degradation of MEIS2 and other target proteins and increased degradation of MYC, IRF4, IKZF1 and IKZF3. Related diseases Intellectual developmental disorder, autosomal recessive 2 (MRT2) [MIM:607417]: A disorder characterized by significantly below average general intellectual functioning associated with impairments in adaptive behavior and manifested during the developmental period. MRT2 patients display mild intellectual disability with a standard IQ ranged from 50 to 70. IQ scores are lower in males than females. Developmental milestones are mildly delayed. There are no dysmorphic or autistic features. {ECO:0000269|PubMed:15557513, ECO:0000269|PubMed:28143899}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00480; DB08910; DB01041 Interacts with Q96A83-2; P48729; Q16531; O14901; Q8IVT2; Q9P286; A0A6Q8PF08; Q93062; Q16531; Q13422-7 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; Disease variant; Intellectual disability; Membrane; Metal-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Ubl conjugation; Ubl conjugation pathway; Zinc Protein physicochemical properties Chain ID B,E Molecular weight (Da) 38245.7 Length 337 Aromaticity 0.08 Instability index 40.62 Isoelectric point 5.7 Charge (pH=7) -6.53 2D Binding mode Binding energy (Kcal/mol) -9.58 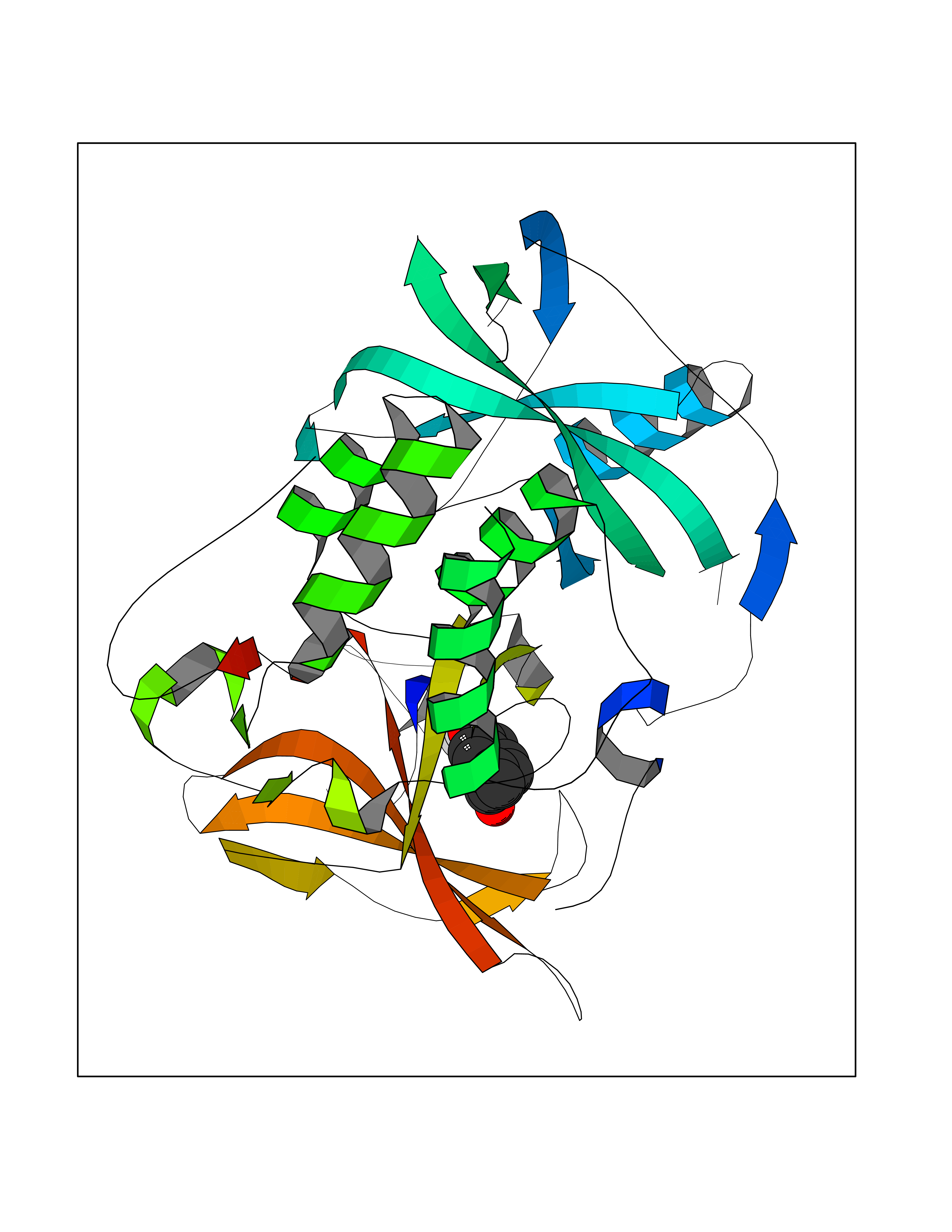 Molscript Map 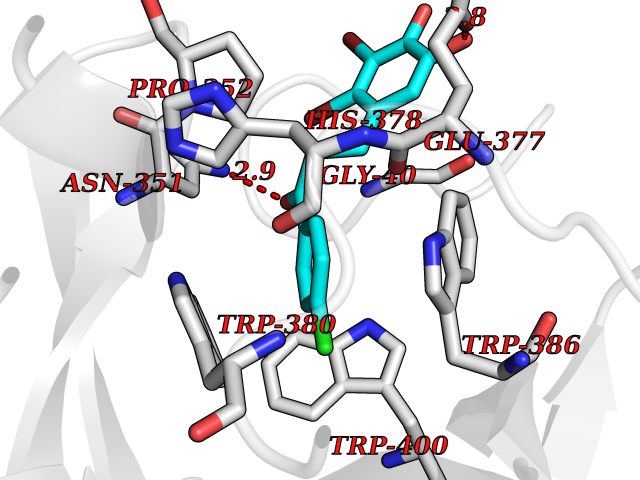 Pymol Map 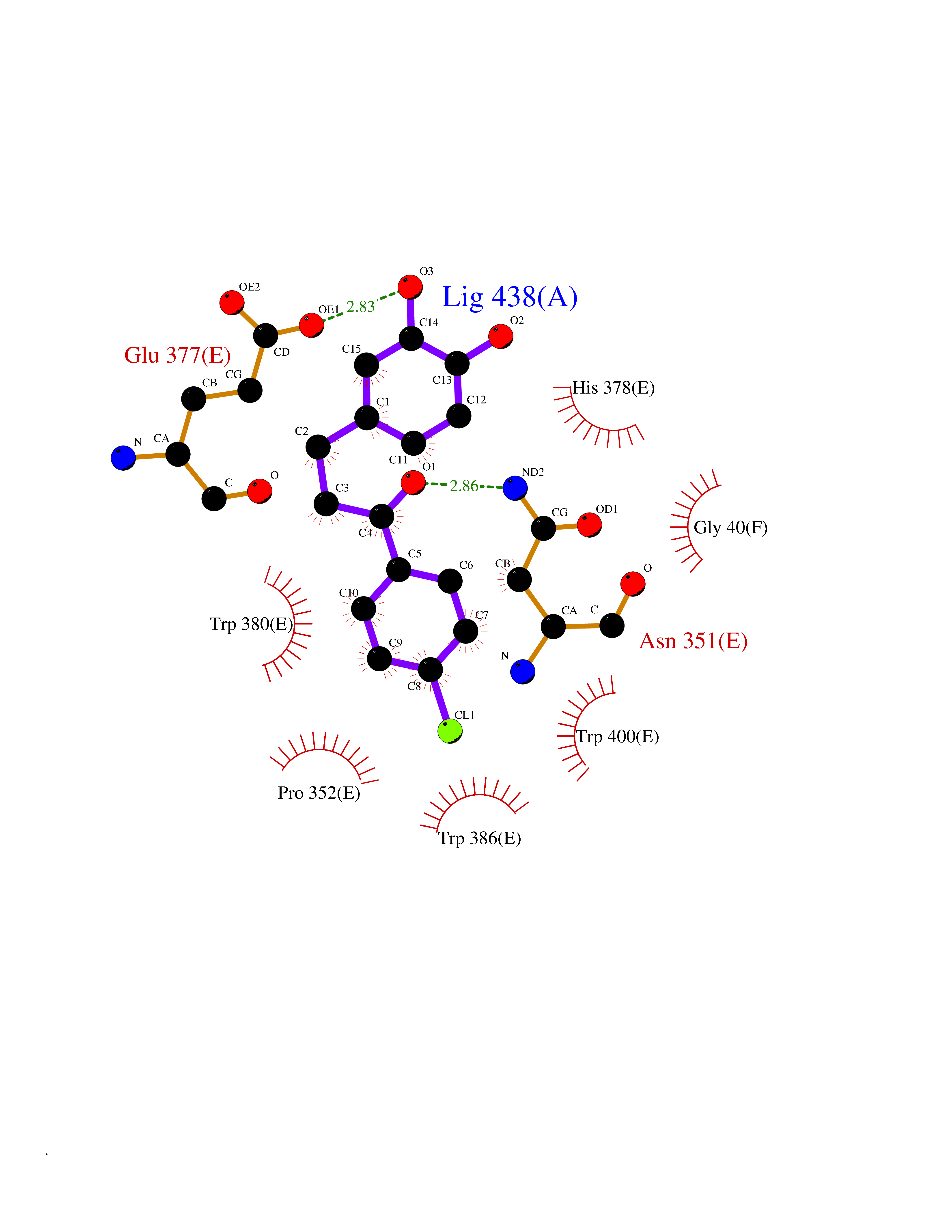 Ligplot Map 3D Binding mode Sequence EFIVGGKYKLNITNGEEVAVINFDTSLPTSHTYLGADMEEFHGRTLHDDDSCQVIPVLPQVMMILIPGQTLPLQLFHPQEVSMVRNLIQKDRTFAVLAYSNVQEREAQFGTTAEIYAYREEIVKVKAIGRQRFKVLEQQAKVQILPECVLAETLMDRIKKQLREWDENLKDDSLPSNPIDFSYRVAACLPIDDVLRIQLLKIGSAIQRLRCELDIMNKCTSLCCKQCQETEITTKNEIFSLSLCGPMAAYVNPHGYVHETLTVYKACNLNLIGRPSTEHSWFPGYAWTVAQCKICASHIGWKFTATKKDMSPQKFWGLTRSALLPTIPDTEDEISPD Hydrogen bonds contact Hydrophobic contact | ||||
| 30 | BUB1 mitotic checkpoint serine/threonine kinase (BUB1) | 6F7B | 7.03 | |
Target general information Gen name BUB1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms hBUB1; Mitotic checkpoint serine/threonine-protein kinase BUB1; BUB1L; BUB1A Protein family Protein kinase superfamily, Ser/Thr protein kinase family, BUB1 subfamily Biochemical class Kinase Function Has a key role in the assembly of checkpoint proteins at the kinetochore, being required for the subsequent localization of CENPF, BUB1B, CENPE and MAD2L1. Required for the kinetochore localization of PLK1. Required for centromeric enrichment of AUKRB in prometaphase. Plays an important role in defining SGO1 localization and thereby affects sister chromatid cohesion. Acts as a substrate for anaphase-promoting complex or cyclosome (APC/C) in complex with its activator CDH1 (APC/C-Cdh1). Necessary for ensuring proper chromosome segregation and binding to BUB3 is essential for this function. Can regulate chromosome segregation in a kinetochore-independent manner. Can phosphorylate BUB3. The BUB1-BUB3 complex plays a role in the inhibition of APC/C when spindle-assembly checkpoint is activated and inhibits the ubiquitin ligase activity of APC/C by phosphorylating its activator CDC20. This complex can also phosphorylate MAD1L1. Kinase activity is essential for inhibition of APC/CCDC20 and for chromosome alignment but does not play a major role in the spindle-assembly checkpoint activity. Mediates cell death in response to chromosome missegregation and acts to suppress spontaneous tumorigenesis. Serine/threonine-protein kinase that performs 2 crucial functions during mitosis: it is essential for spindle-assembly checkpoint signaling and for correct chromosome alignment. Related diseases Microcephaly 30, primary, autosomal recessive (MCPH30) [MIM:620183]: A form of microcephaly, a disease defined as a head circumference more than 3 standard deviations below the age, sex and ethnically matched mean. Brain weight is markedly reduced and the cerebral cortex is disproportionately small. MCPH30 is characterized by small head, poor overall growth, and global developmental delay with variably impaired intellectual development. Affected individuals may also have variable congenital anomalies, including atrial septal defect, dysmorphic facial features, tracheal stenosis, and anomalies of the skin and teeth. {ECO:0000269|PubMed:35044816}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with O95376; O60566; O43684; P46108; Q8NG31; Q8NG31-2; Q9GZQ8; P03070 EC number EC 2.7.11.1 Uniprot keywords 3D-structure; Alternative splicing; Apoptosis; ATP-binding; Cell cycle; Cell division; Centromere; Chromosome; Chromosome partition; Host-virus interaction; Intellectual disability; Kinase; Kinetochore; Mitosis; Nucleotide-binding; Nucleus; Phosphoprotein; Primary microcephaly; Proteomics identification; Reference proteome; Serine/threonine-protein kinase; Transferase; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 39564.8 Length 343 Aromaticity 0.12 Instability index 31.17 Isoelectric point 8.49 Charge (pH=7) 4.7 2D Binding mode Binding energy (Kcal/mol) -9.59 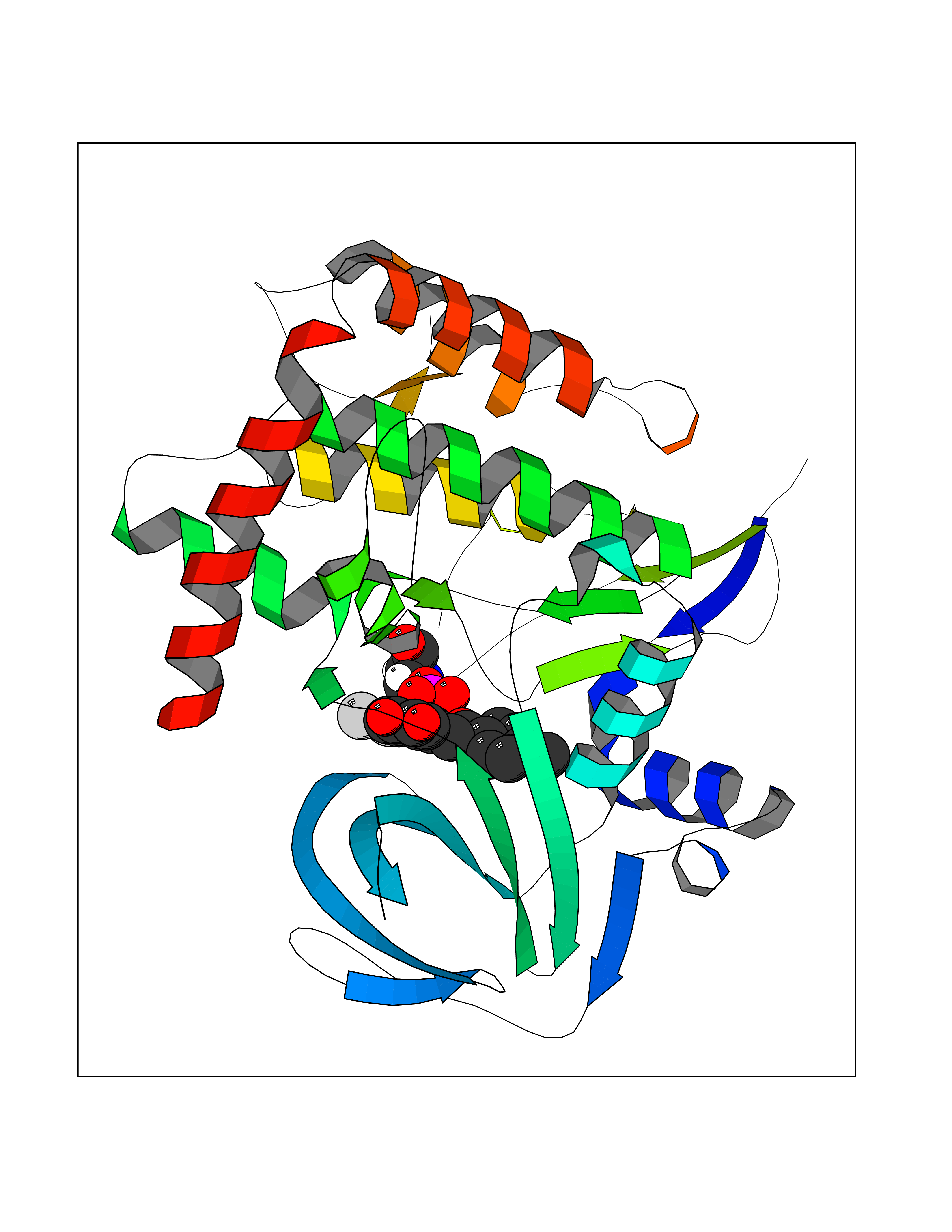 Molscript Map 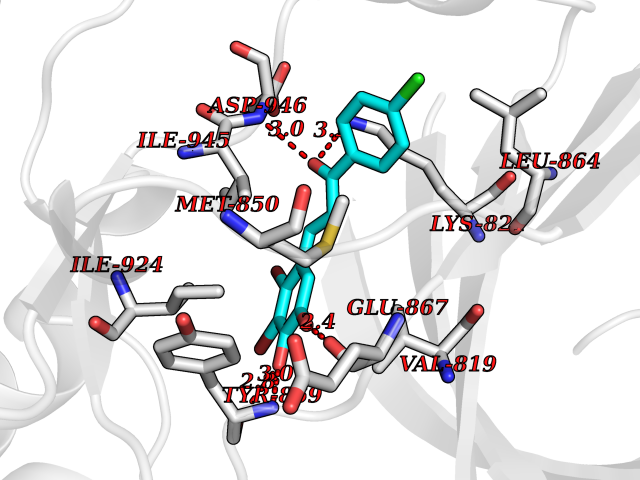 Pymol Map 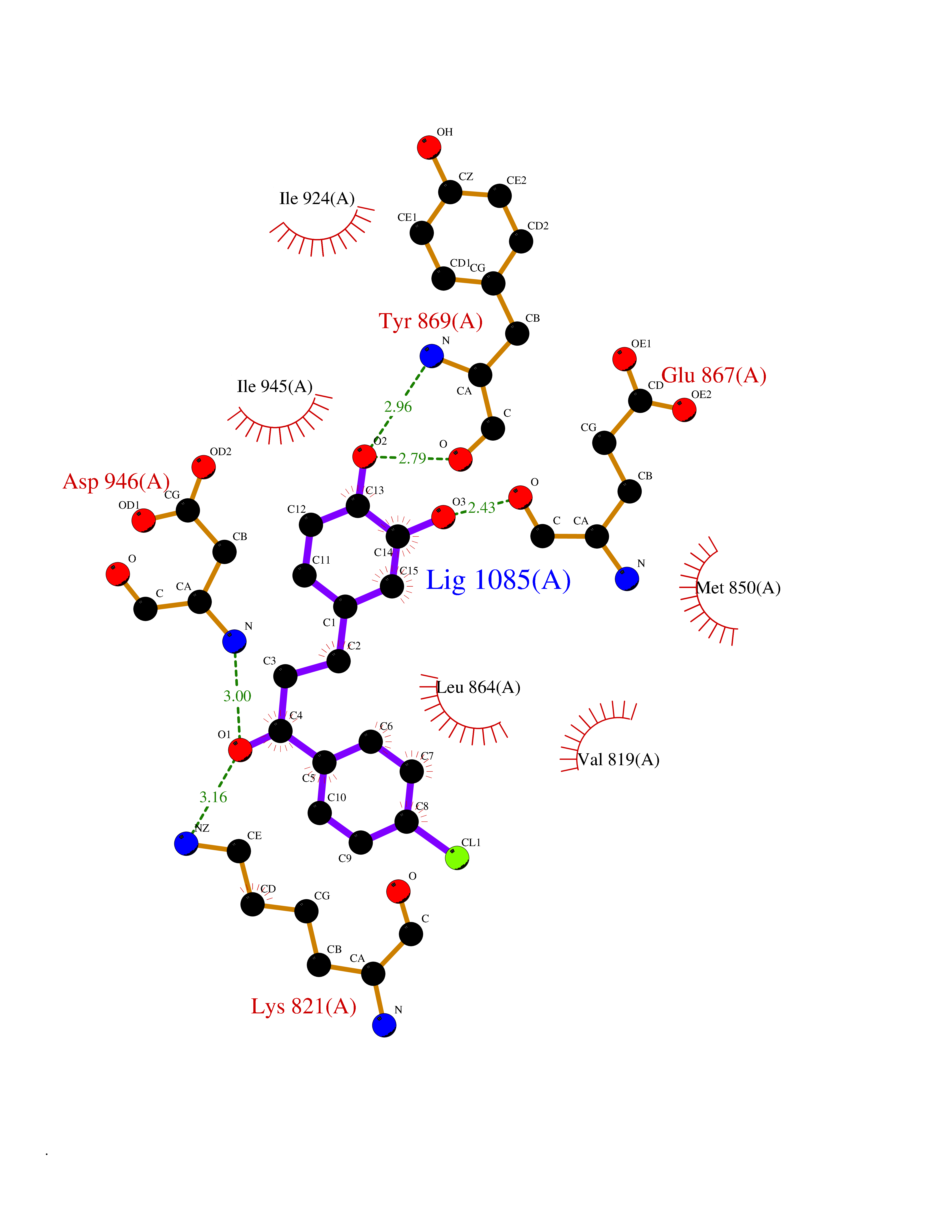 Ligplot Map 3D Binding mode Sequence APNFIVGNPWDDKLIFKLLSGLSKPVSSYPNTFEWQCKLPAIKPKTEFQLGSKLVYVHHLLGEGAFAQVYEATQKNKQKFVLKVQKPANPWEFYIGTQLMERLKPSMQHMFMKFYSAHLFQNGSVLVGELYSYGTLLNAINLYKNTPEKVMPQGLVISFAMRMLYMIEQVHDCEIIHGDIKPDNFILGNGFLEQDDEDDLSAGLALIDLGQSIDMKLFPKGTIFTAKCETXGFQCVEMLSNKPWNYQIDYFGVAATVYCMLFGTYMKVKNEECKPEGLFRRLPHLDMWNEFFHVMLNIPDCHHLPSLDLLRQKLKKVFQQHYTNKIRALRNRLIVLLLECKRS Hydrogen bonds contact Hydrophobic contact | ||||
| 31 | WNK lysine-deficient protein kinase 3 (WNK3) | 5O2B | 7.03 | |
Target general information Gen name WNK3 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Serine/threonine-protein kinase WNK3; Protein kinase with no lysine 3; Protein kinase lysine-deficient 3; KIAA1566 Protein family Protein kinase superfamily, Ser/Thr protein kinase family, WNK subfamily Biochemical class NA Function Serine/threonine kinase which plays an important role in the regulation of electrolyte homeostasis, cell signaling, survival and proliferation. Acts as an activator and inhibitor of sodium-coupled chloride cotransporters and potassium-coupled chloride cotransporters respectively. Phosphorylates WNK4. Regulates the phosphorylation of SLC12A1 and SLC12A2. Increases Ca(2+) influx mediated by TRPV5 and TRPV6 by enhancing their membrane expression level via a kinase-dependent pathway. Inhibits the activity of KCNJ1 by decreasing its expression at the cell membrane in a non-catalytic manner. Related diseases Prieto syndrome (PRS) [MIM:309610]: An X-linked recessive disorder characterized by impaired intellectual development, developmental delay, autism spectrum disorder, variable epilepsy, craniofacial dysmorphism, and structural brain abnormalities including polymicrogyria and cerebral atrophy. {ECO:0000269|PubMed:35678782}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P42574; P52954; Q04864-2 EC number EC 2.7.11.1 Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Cytoplasm; Disease variant; Intellectual disability; Kinase; Nucleotide-binding; Phosphoprotein; Proteomics identification; Reference proteome; Serine/threonine-protein kinase; Transferase; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 30818.4 Length 269 Aromaticity 0.1 Instability index 41.03 Isoelectric point 6.27 Charge (pH=7) -2.07 2D Binding mode Binding energy (Kcal/mol) -9.59 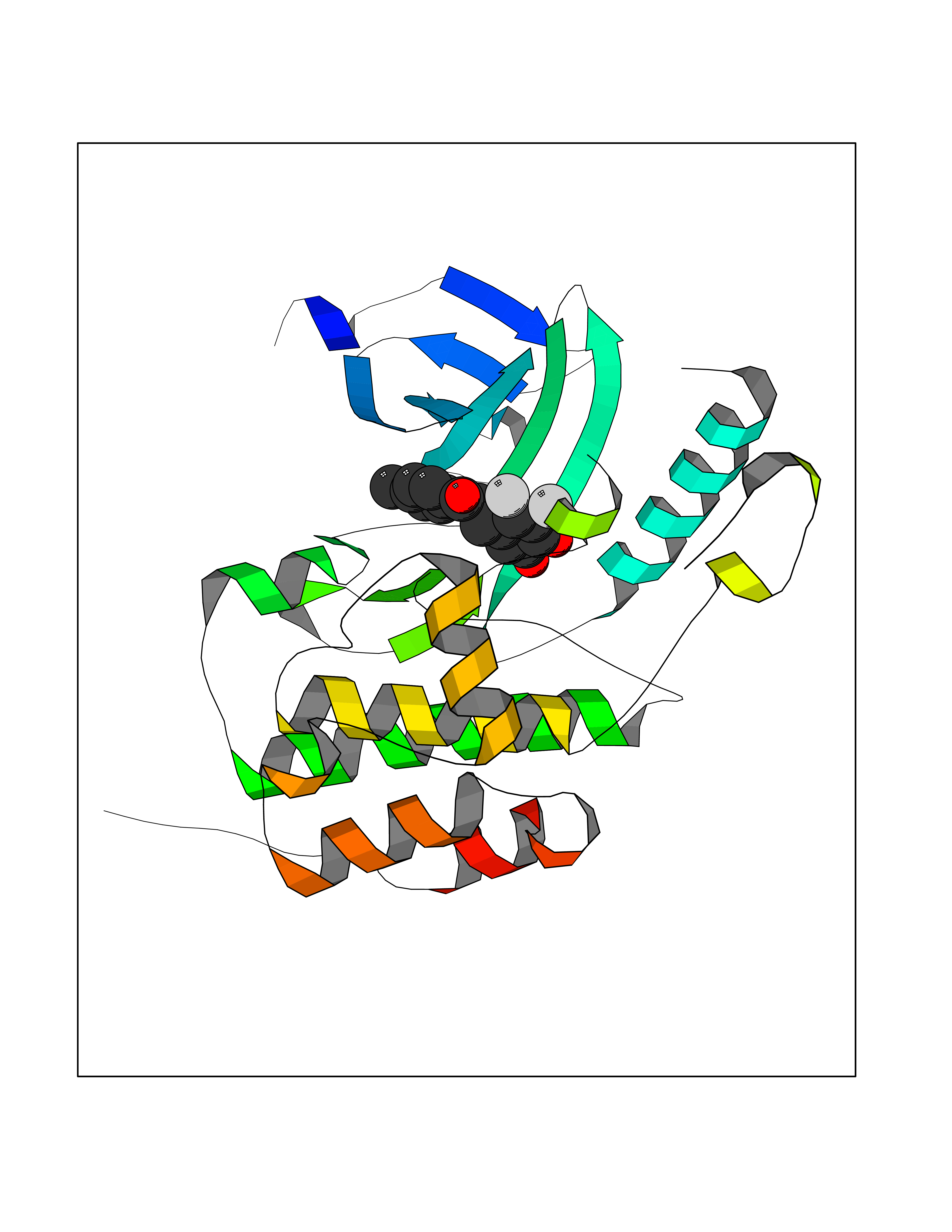 Molscript Map 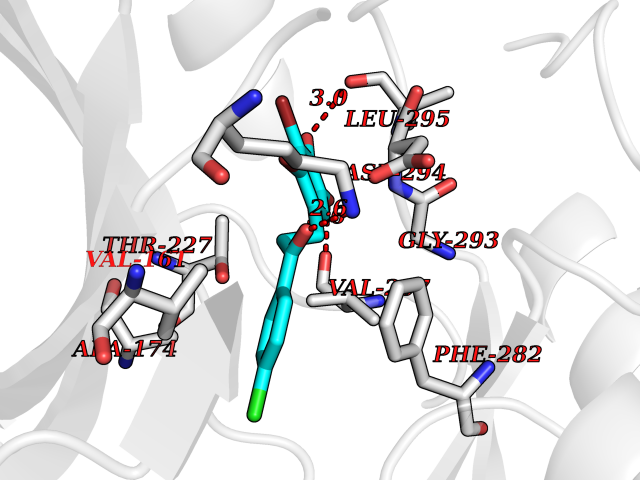 Pymol Map 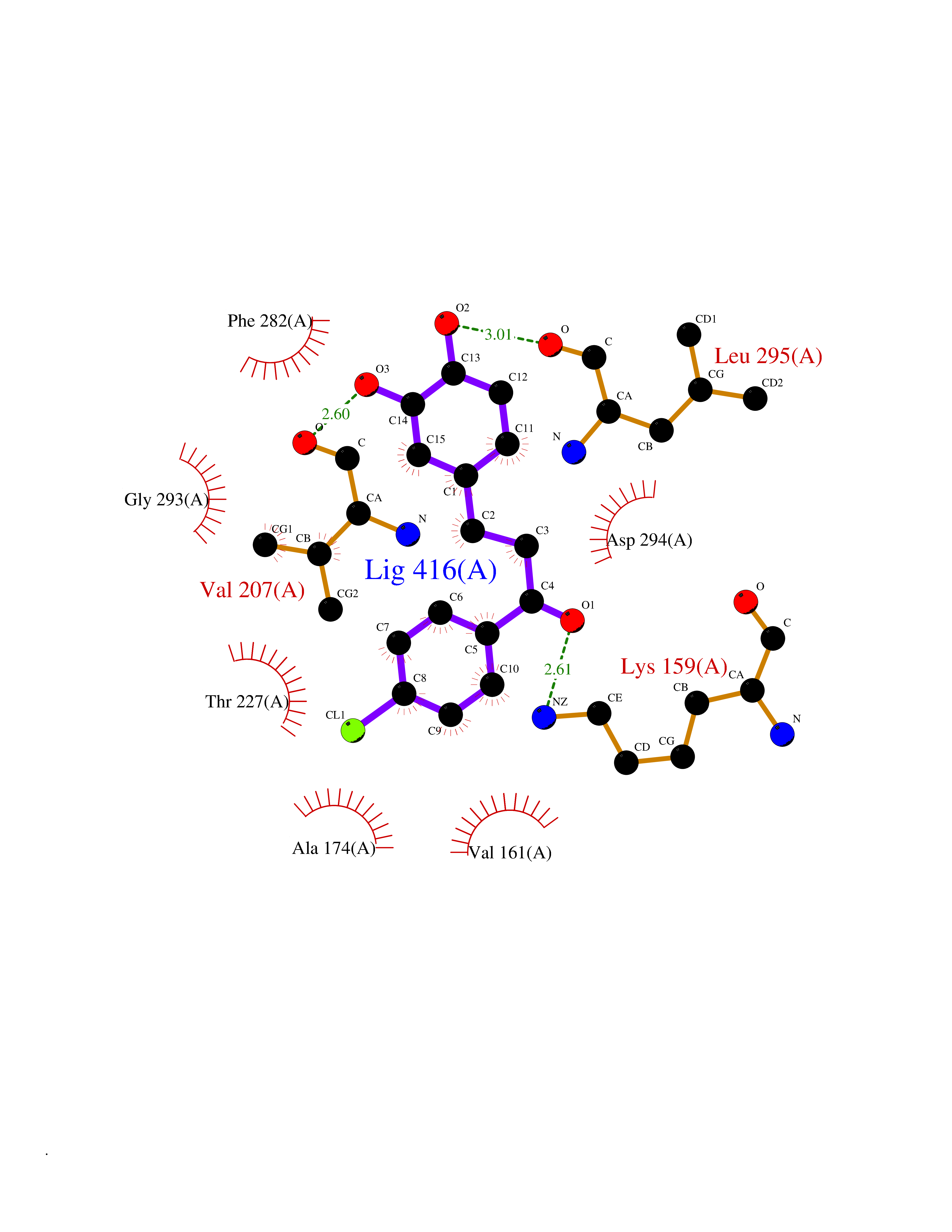 Ligplot Map 3D Binding mode Sequence MEAEMKAVATSPSGRFLKFDIELGRGAFKTVYKGLDTETWVEVAWCELQLTKAEQQRFKEEAEMLKGLQHPNIVRFYDSWESIKCIVLVTELMTSGTLKTYLKRFKVMKPKVLRSWCRQILKGLQFLHTRTPPIIHRDLKCDNIFITGPTGSVKIGDLGLATLMIGTPEFMAPEMYEEHYDESVDVYAFGMCMLEMATSEYPYSECQNAAQIYRKVTSGIKPASFNKVTDPEVKEIIEGCIRQNKSERLSIRDLLNHAFFAEDTGLRVE Hydrogen bonds contact Hydrophobic contact | ||||
| 32 | Neuronal acetylcholine receptor beta-2 (CHRNB2) | 6CNJ | 7.03 | |
Target general information Gen name CHRNB2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Nicotinic acetylcholine receptor beta2; Nicotinic acetylcholine receptor beta 2-subunit protein; CHRNB2; Beta-2 nAChR; Alpha-4/beta-2 nicotinic receptor Protein family Ligand-gated ion channel (TC 1.A.9) family, Acetylcholine receptor (TC 1.A.9.1) subfamily, Beta-2/CHRNB2 sub-subfamily Biochemical class Neurotransmitter receptor Function After binding acetylcholine, the AChR responds by an extensive change in conformation that affects all subunits and leads to opening of an ion-conducting channel across the plasma membrane permeable to sodiun ions. Related diseases Epilepsy, nocturnal frontal lobe, 3 (ENFL3) [MIM:605375]: An autosomal dominant focal epilepsy characterized by nocturnal seizures with hyperkinetic automatisms and poorly organized stereotyped movements. {ECO:0000269|PubMed:11062464, ECO:0000269|PubMed:11104662}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00572; DB00237; DB00565; DB09028; DB01245; DB00514; DB07720; DB00898; DB00753; DB00657; DB00333; DB00184; DB00981; DB05458; DB05855; DB05740; DB00747; DB00202; DB01273 Interacts with P43681-1; P30532 EC number NA Uniprot keywords 3D-structure; Cell membrane; Disease variant; Disulfide bond; Epilepsy; Glycoprotein; Ion channel; Ion transport; Ligand-gated ion channel; Membrane; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Signal; Synapse; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A,B Molecular weight (Da) 84601.2 Length 728 Aromaticity 0.13 Instability index 39.72 Isoelectric point 5.86 Charge (pH=7) -9.84 2D Binding mode Binding energy (Kcal/mol) -9.59 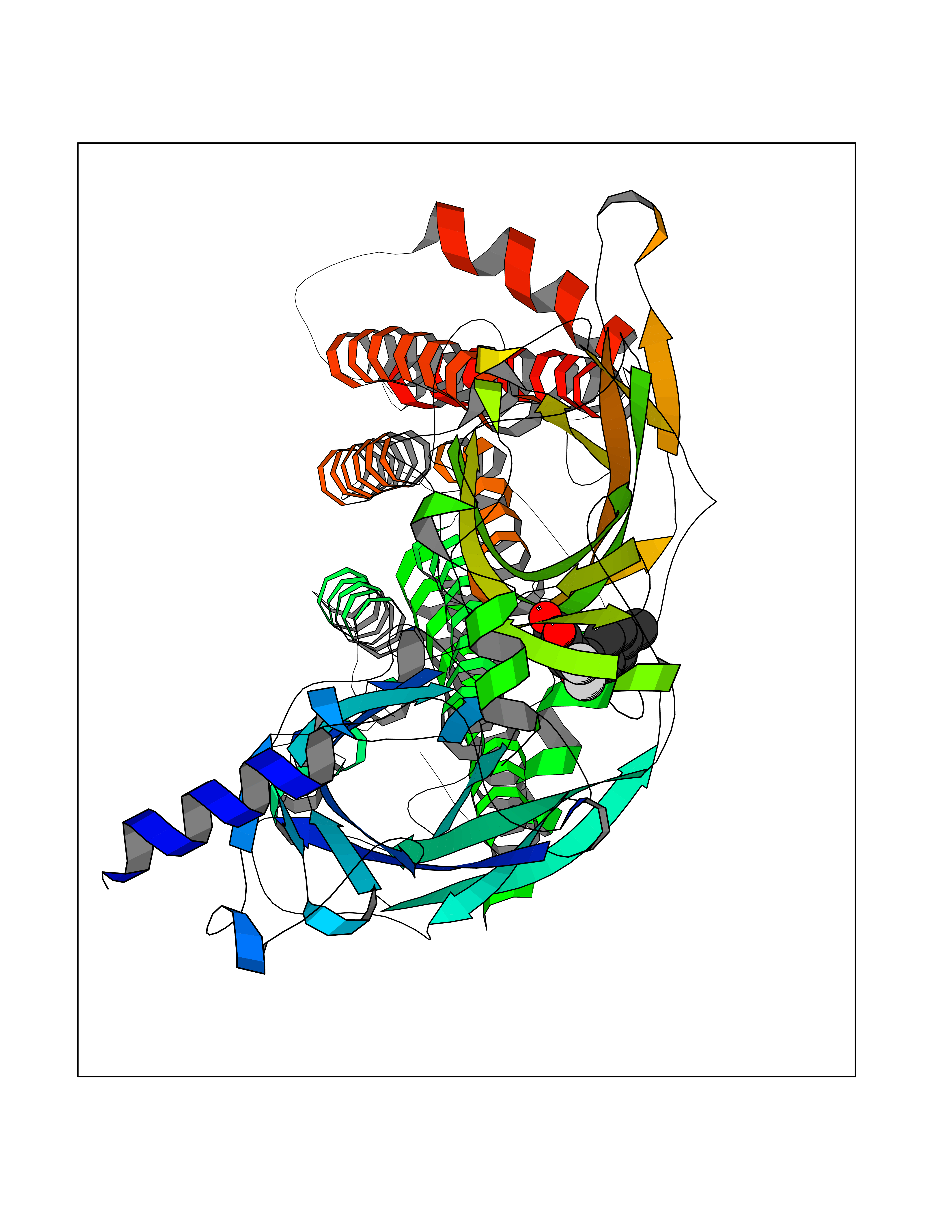 Molscript Map 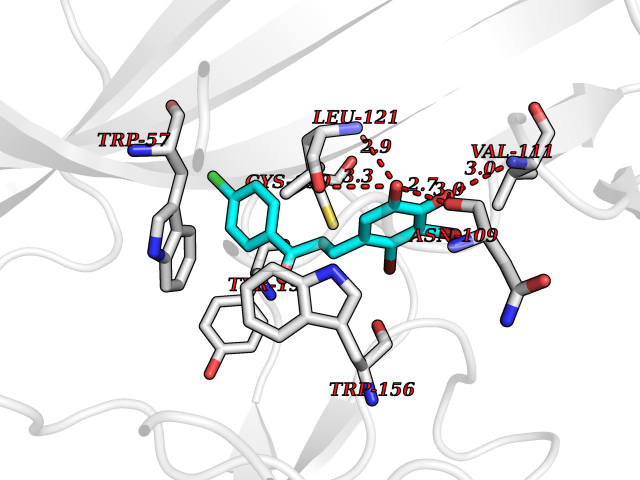 Pymol Map 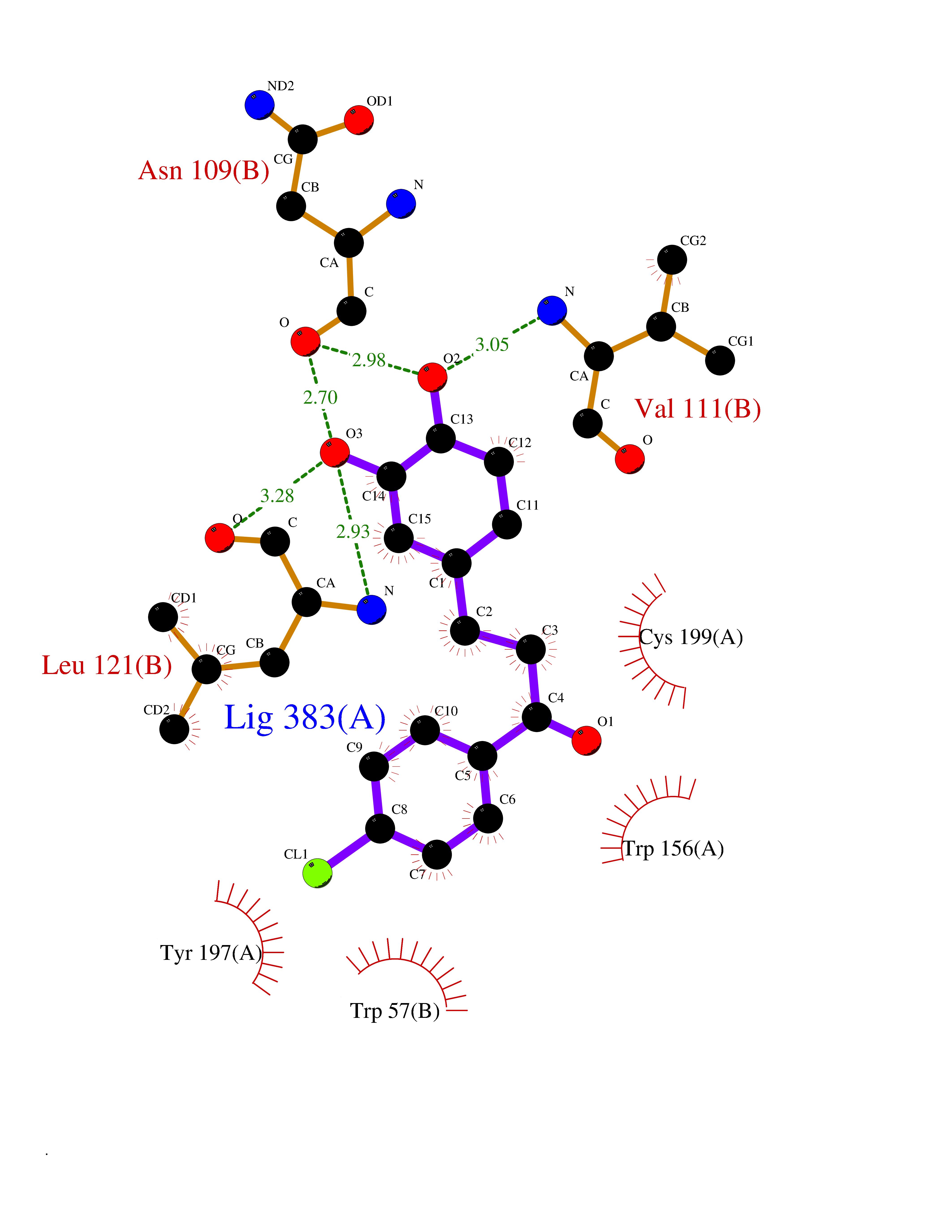 Ligplot Map 3D Binding mode Sequence ETRAHAEERLLKKLFSGYNKWSRPVANISDVVLVRFGLSIAQLIDVDEKNQMMTTNVWVKQEWHDYKLRWDPADYENVTSIRIPSELIWRPDIVLYNNADGDFAVTHLTKAHLFHDGRVQWTPPAIYKSSCSIDVTFFPFDQQNCTMKFGSWTYDKAKIDLVNMHSRVDQLDFWESGEWVIVDAVGTYNTRKYECCAEIYPDITYAFVIRRLPLFYTINLIIPCLLISCLTVLVFYLPSECGEKITLCISVLLSLTVFLLLITEIIPSTSLVIPLIGEYLLFTMIFVTLSIVITVFVLNVHHRSPRTHTMPTWVRRVFLDIVPRLLLMKRFERSVKEDWKYVAMVIDRIFLWMFIIVCLLGTVGLFLPPWDTEERLVEHLLDPSRYNKLIRPATNGSELVTVQLMVSLAQLISVHEREQIMTTNVWLTQEWEDYRLTWKPEEFDNMKKVRLPSKHIWLPDVVLYNNADGMYEVSFYSNAVVSYDGSIFWLPPAIYKSACKIEVKHFPFDQQNCTMKFRSWTYDRTEIDLVLKSEVASLDDFTPSGEWDIVALPGRRNENPDDSTYVDITYDFIIRRKPLFYTINLIIPCVLITSLAILVFYLPSDCGEKMTLCISVLLALTVFLLLISKIVPPTSLDVPLVGKYLMFTMVLVTFSIVTSVCVLNVHHRSPTTHTMAPWVKVVFLEKLPALLFMQQSVSEDWKYVAMVIDRLFLWIFVFVCVFGTIGMF Hydrogen bonds contact Hydrophobic contact | ||||
| 33 | Histidine decarboxylase (HDC) | 4E1O | 7.02 | |
Target general information Gen name HDC Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Human histidine decarboxylase Protein family Group II decarboxylase family Biochemical class Carbon-carbon lyase Function Catalyzes the biosynthesis of histamine from histidine. Related diseases Corticosterone methyloxidase 1 deficiency (CMO-1 deficiency) [MIM:203400]: Autosomal recessive disorder of aldosterone biosynthesis. There are two biochemically different forms of selective aldosterone deficiency be termed corticosterone methyloxidase (CMO) deficiency type 1 and type 2. In CMO-1 deficiency, aldosterone is undetectable in plasma, while its immediate precursor, 18-hydroxycorticosterone, is low or normal. {ECO:0000269|PubMed:11238478, ECO:0000269|PubMed:8439335, ECO:0000269|PubMed:9177280}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Corticosterone methyloxidase 2 deficiency (CMO-2 deficiency) [MIM:610600]: Autosomal recessive disorder of aldosterone biosynthesis. In CMO-2 deficiency, aldosterone can be low or normal, but at the expense of increased secretion of 18-hydroxycorticosterone. Consequently, patients have a greatly increased ratio of 18-hydroxycorticosterone to aldosterone and a low ratio of corticosterone to 18-hydroxycorticosterone in serum. {ECO:0000269|PubMed:12788848, ECO:0000269|PubMed:1346492, ECO:0000269|PubMed:1594605, ECO:0000269|PubMed:9625333, ECO:0000269|PubMed:9814506}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00117; DB00114 Interacts with Q86UW9 EC number EC 4.1.1.22 Uniprot keywords 3D-structure; Alternative splicing; Catecholamine biosynthesis; Decarboxylase; Lyase; Proteomics identification; Pyridoxal phosphate; Reference proteome Protein physicochemical properties Chain ID A,B,C,D,E,F Molecular weight (Da) 107706 Length 956 Aromaticity 0.1 Instability index 55.17 Isoelectric point 6.23 Charge (pH=7) -9.63 2D Binding mode Binding energy (Kcal/mol) -9.57  Molscript Map 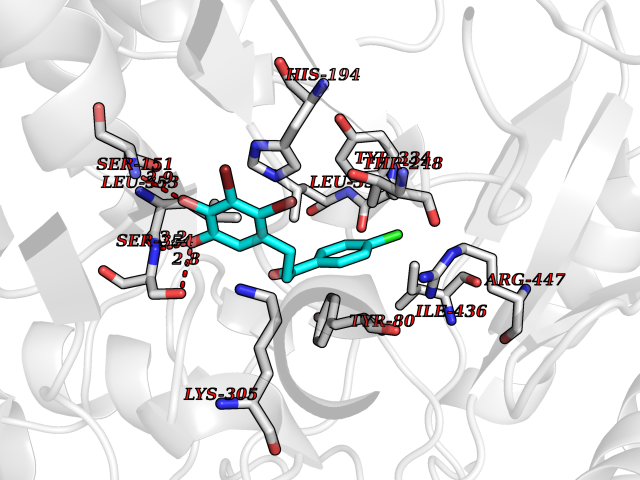 Pymol Map 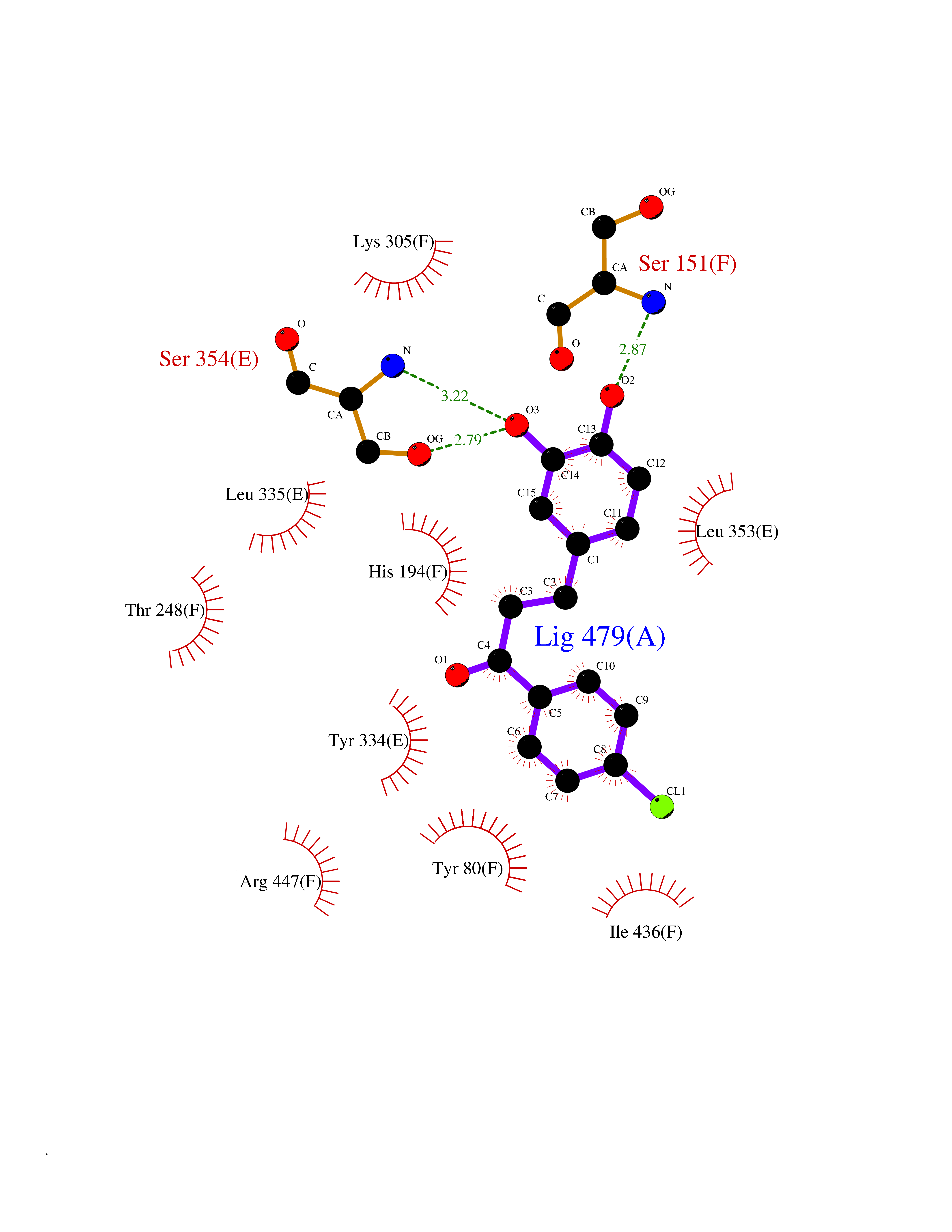 Ligplot Map 3D Binding mode Sequence GSMEPEEYRERGREMVDYICQYLSTVRERRVTPDVQPGYLRAQLPESAPEDPDSWDSIFGDIERIIMPGVVHWQSPHMHAYYPALTSWPSLLGDMLADAINCLGFTWASSPACTELEMNVMDWLAKMLGLPEHFLHHHPSSQGGGVLQSTVSESTLIALLAARKNKILEMKTSEPDADESSLNARLVAYASDQAHSSVEKAGLISLVKMKFLPVDDNFSLRGEALQKAIEEDKQRGLVPVFVCATLGTTGVCAFDXLSELGPICAREGLWLHIDAAYAGTAFLCPEFRGFLKGIEYADSFTFNPSKWMMVHFDCTGFWVKDKYKLQQTFSVNPIYLRHANSGVATDFMHWQIPLSRRFRSVKLWFVIRSFGVKNLQAHVRHGTEMAKYFESLVRNDPSFEIPAKRHLGLVVFRLKGPNSLTENVLKEIAKAGRLFLIPATIQDKLIIRFTVTSQFTTRDDILRDWNLIRDAATLILSQGSMEPEEYRERGREMVDYICQYLSTVRERRVTPDVQPGYLRAQLPESAPEDPDSWDSIFGDIERIIMPGVVHWQSPHMHAYYPALTSWPSLLGDMLADAINCLGFTWASSPACTELEMNVMDWLAKMLGLPEHFLHHHPSSQGGGVLQSTVSESTLIALLAARKNKILEMKTSEPDADESSLNARLVAYASDQAHSSVEKAGLISLVKMKFLPVDDNFSLRGEALQKAIEEDKQRGLVPVFVCATLGTTGVCAFDXLSELGPICAREGLWLHIDAAYAGTAFLCPEFRGFLKGIEYADSFTFNPSKWMMVHFDCTGFWVKDKYKLQQTFSVNPIYLRHANSGVATDFMHWQIPLSRRFRSVKLWFVIRSFGVKNLQAHVRHGTEMAKYFESLVRNDPSFEIPAKRHLGLVVFRLKGPNSLTENVLKEIAKAGRLFLIPATIQDKLIIRFTVTSQFTTRDDILRDWNLIRDAATLILSQ Hydrogen bonds contact Hydrophobic contact | ||||
| 34 | Monoamine oxidase type B (MAO-B) | 2V5Z | 7.02 | |
Target general information Gen name MAOB Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms MAO-B; Amine oxidase [flavin-containing] B Protein family Flavin monoamine oxidase family Biochemical class CH-NH(2) donor oxidoreductase Function Catalyzes the oxidative deamination of biogenic and xenobiotic amines and has important functions in the metabolism of neuroactive and vasoactive amines in the central nervous system and peripheral tissues. MAOB preferentially degrades benzylamine and phenylethylamine. Related diseases Microvascular complications of diabetes 5 (MVCD5) [MIM:612633]: Pathological conditions that develop in numerous tissues and organs as a consequence of diabetes mellitus. They include diabetic retinopathy, diabetic nephropathy leading to end-stage renal disease, and diabetic neuropathy. Diabetic retinopathy remains the major cause of new-onset blindness among diabetic adults. It is characterized by vascular permeability and increased tissue ischemia and angiogenesis. Disease susceptibility is associated with variants affecting the gene represented in this entry. Homozygosity for the Leu-55 allele is strongly associated with the development of retinal disease in diabetic patients. Drugs (DrugBank ID) DB08176; DB02211; DB08516; DB08480; DB01472; DB04307; DB07512; DB07513; DB00915; DB00182; DB06698; DB04889; DB00215; DB09130; DB04147; DB00988; DB01363; DB00668; DB01175; DB02509; DB03147; DB14914; DB00614; DB04818; DB02095; DB01247; DB00601; DB01577; DB01442; DB01171; DB08082; DB02643; DB04677; DB03894; DB08804; DB04820; DB00184; DB04821; DB12612; DB01626; DB00780; DB00191; DB00388; DB01132; DB00721; DB01168; DB01367; DB09363; DB06654; DB01037; DB01104; DB14569; DB09042; DB00752; DB16446; DB09185; DB04832; DB00909 Interacts with P55212; P28329-3; Q8NI60; Q5RI15; Q92915-2; P22607; Q53GS7; P06396; P01112; O14901; P13473-2; P21397; Q9BVL2; O75400-2; P62826; Q6NTF9-3; Q9Y371; Q7Z699; Q9UMX0; Q9Y649 EC number EC 1.4.3.4 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Direct protein sequencing; FAD; Flavoprotein; Membrane; Mitochondrion; Mitochondrion outer membrane; Oxidoreductase; Proteomics identification; Reference proteome; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A,B Molecular weight (Da) 56019.9 Length 494 Aromaticity 0.09 Instability index 34.81 Isoelectric point 6.51 Charge (pH=7) -2.2 2D Binding mode Binding energy (Kcal/mol) -9.58  Molscript Map 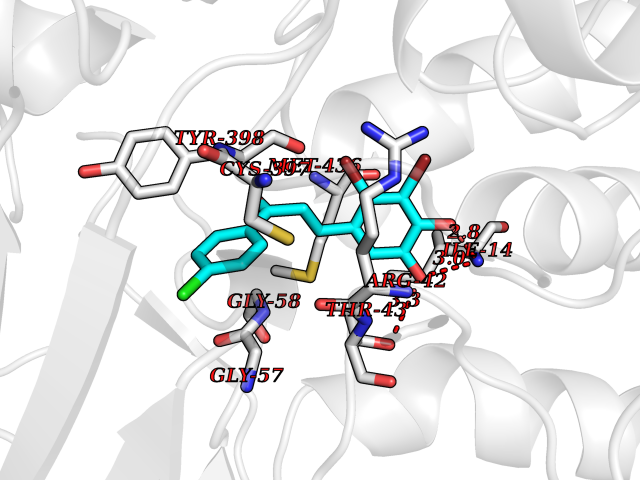 Pymol Map 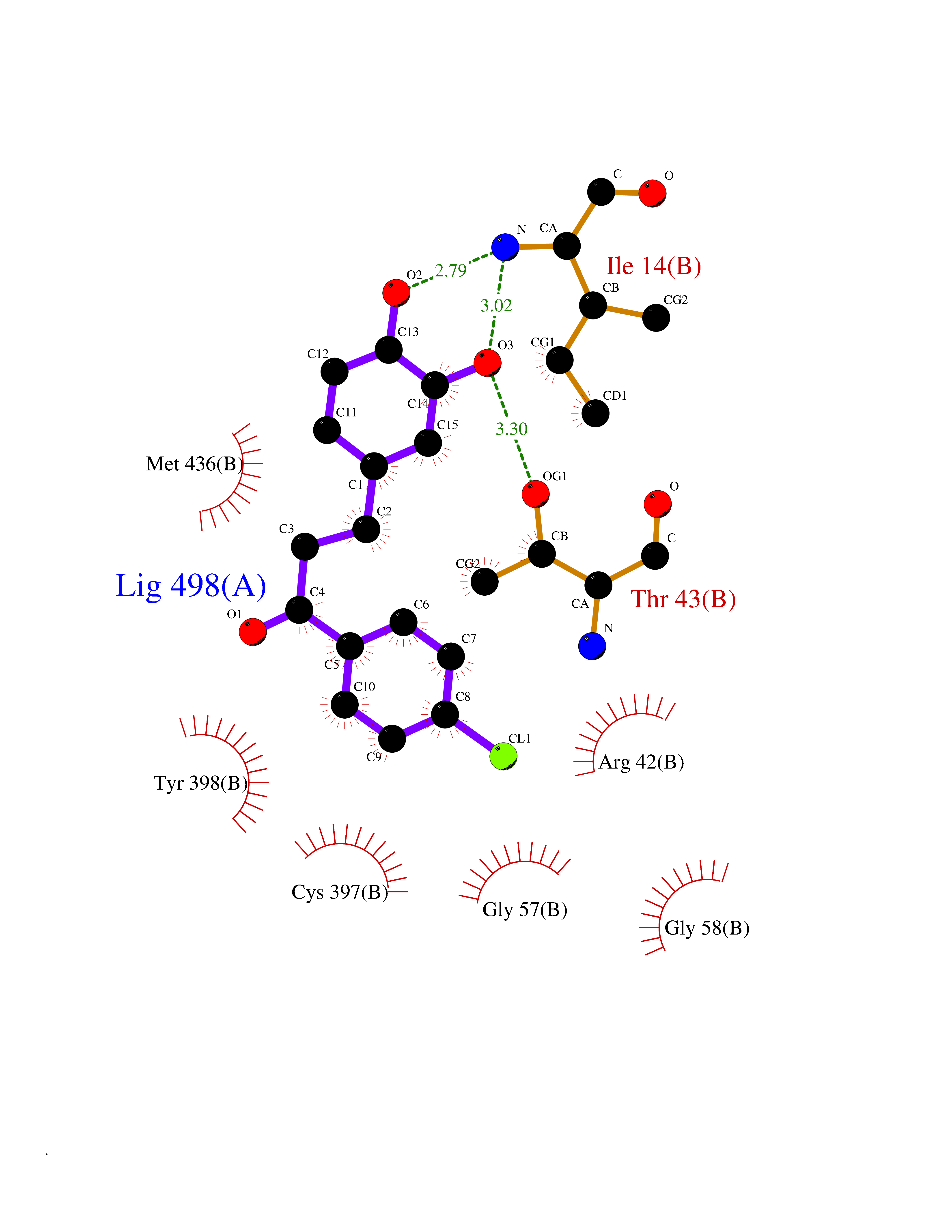 Ligplot Map 3D Binding mode Sequence NKCDVVVVGGGISGMAAAKLLHDSGLNVVVLEARDRVGGRTYTLRNQKVKYVDLGGSYVGPTQNRILRLAKELGLETYKVNEVERLIHHVKGKSYPFRGPFPPVWNPITYLDHNNFWRTMDDMGREIPSDAPWKAPLAEEWDNMTMKELLDKLCWTESAKQLATLFVNLCVTAETHEVSALWFLWYVKQCGGTTRIISTTNGGQERKFVGGSGQVSERIMDLLGDRVKLERPVIYIDQTRENVLVETLNHEMYEAKYVISAIPPTLGMKIHFNPPLPMMRNQMITRVPLGSVIKCIVYYKEPFWRKKDYCGTMIIDGEEAPVAYTLDDTKPEGNYAAIMGFILAHKARKLARLTKEERLKKLCELYAKVLGSLEALEPVHYEEKNWCEEQYSGGCYTTYFPPGILTQYGRVLRQPVDRIYFAGTETATHWSGYMEGAVEAGERAAREILHAMGKIPEDEIWQSEPESVDVPAQPITTTFLERHLPSVPGLLRLI Hydrogen bonds contact Hydrophobic contact | ||||
| 35 | Penicillin acylase | 2PVA | 7.01 | |
Target general information Gen name N/A Organism Lysinibacillus sphaericus (Bacillus sphaericus) Uniprot ID TTD ID NA Synonyms NA Protein family Peptidase C59 family Biochemical class Hydrolase Function Penicillin amidase activity. Related diseases Usher syndrome 3B (USH3B) [MIM:614504]: A syndrome characterized by progressive vision and hearing loss during early childhood. Some patients have the so-called 'Charles Bonnet syndrome,' involving decreased visual acuity and vivid visual hallucinations. USH is a genetically heterogeneous condition characterized by the association of retinitis pigmentosa with sensorineural deafness. Age at onset and differences in auditory and vestibular function distinguish Usher syndrome type 1 (USH1), Usher syndrome type 2 (USH2) and Usher syndrome type 3 (USH3). USH3 is characterized by postlingual, progressive hearing loss, variable vestibular dysfunction, and onset of retinitis pigmentosa symptoms, including nyctalopia, constriction of the visual fields, and loss of central visual acuity, usually by the second decade of life. {ECO:0000269|PubMed:22279524}. The disease may be caused by variants affecting the gene represented in this entry.; DISEASE: Charcot-Marie-Tooth disease, axonal, 2W (CMT2W) [MIM:616625]: An autosomal dominant, axonal form of Charcot-Marie-Tooth disease, a disorder of the peripheral nervous system, characterized by progressive weakness and atrophy, initially of the peroneal muscles and later of the distal muscles of the arms. Charcot-Marie-Tooth disease is classified in two main groups on the basis of electrophysiologic properties and histopathology: primary peripheral demyelinating neuropathies (designated CMT1 when they are dominantly inherited) and primary peripheral axonal neuropathies (CMT2). Neuropathies of the CMT2 group are characterized by signs of axonal degeneration in the absence of obvious myelin alterations, normal or slightly reduced nerve conduction velocities, and progressive distal muscle weakness and atrophy. CMT2W patients manifest a peripheral neuropathy mainly affecting the lower limbs and resulting in gait difficulties and distal sensory impairment. Most patients also have upper limb involvement. {ECO:0000269|PubMed:22930593, ECO:0000269|PubMed:26072516, ECO:0000269|PubMed:29235198}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01822; DB03661; DB00417 Interacts with NA EC number 3.5.1.11 Uniprot keywords 3D-structure; Antibiotic resistance; Direct protein sequencing; Hydrolase; Zymogen Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 32972.1 Length 295 Aromaticity 0.11 Instability index 30.94 Isoelectric point 5.65 Charge (pH=7) -4.18 2D Binding mode Binding energy (Kcal/mol) -9.56 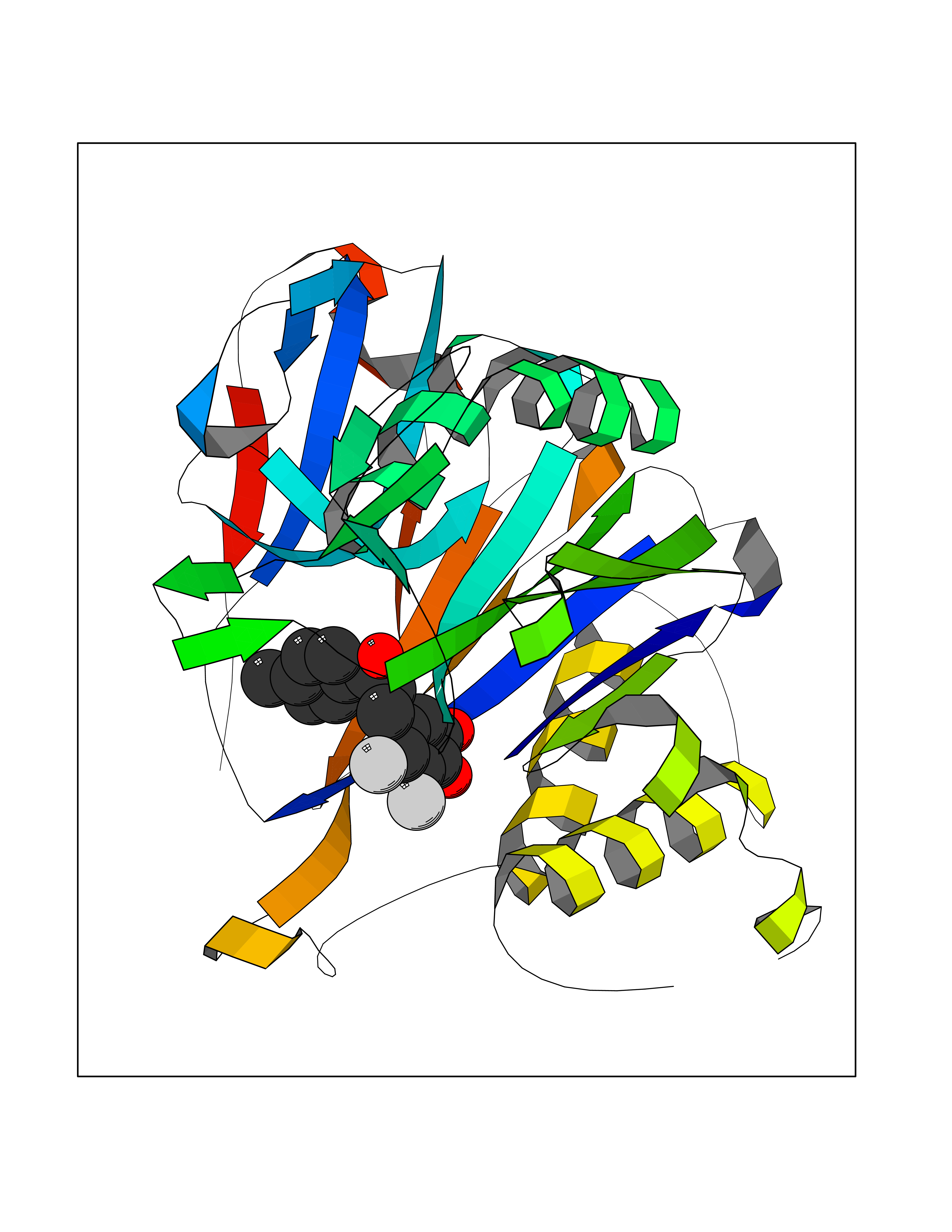 Molscript Map 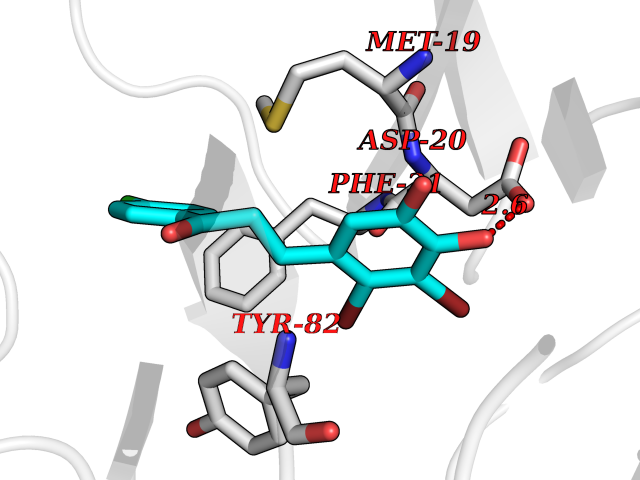 Pymol Map 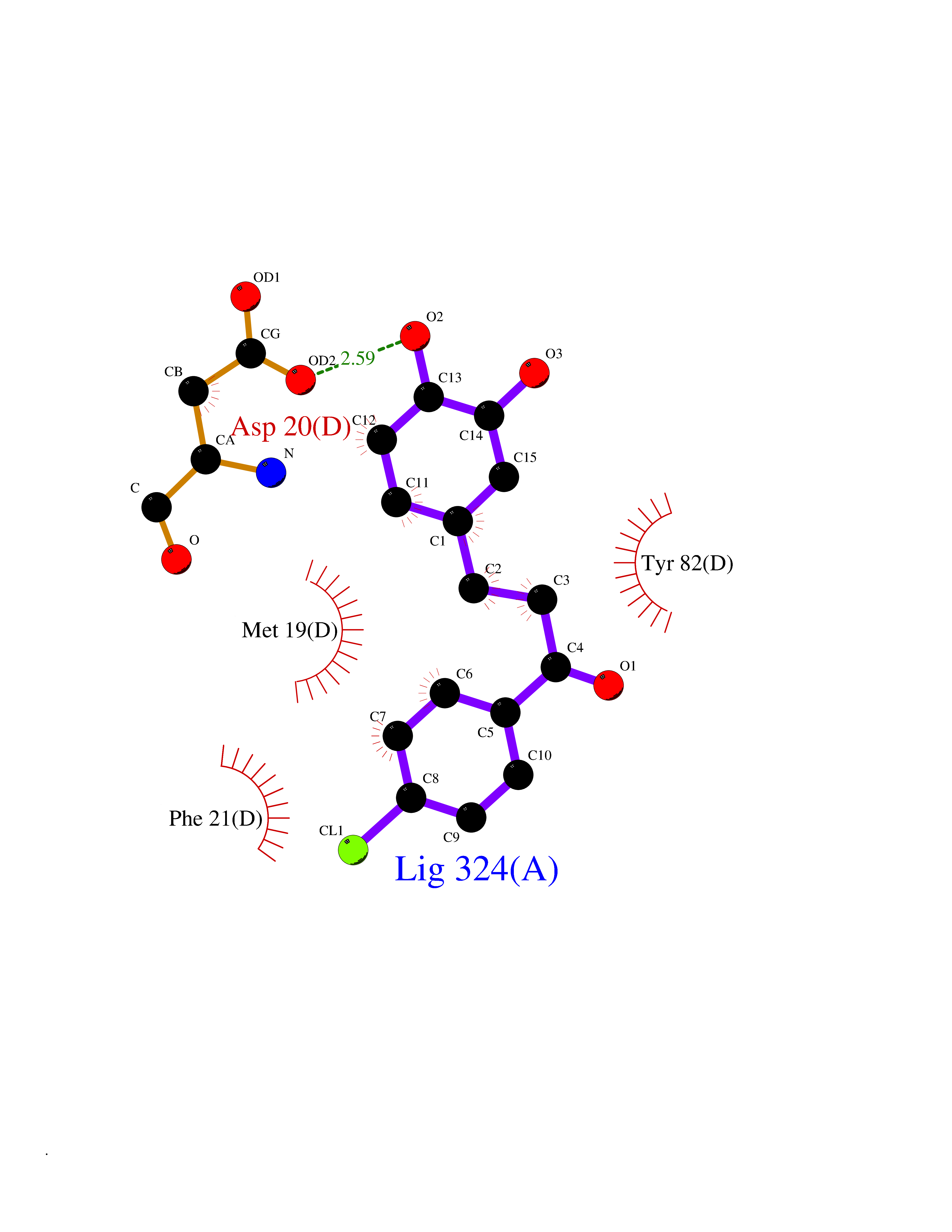 Ligplot Map 3D Binding mode Sequence SSLSIRTTDDKSLFARTMDFTMEPDSKVIIVPRNYGIRLLEKENVVINNSYAFVGMGSTDITSPVLYDGVNEKGLMGAMLYYATFATYADEPKKGTTGINPVYVISQVLGNCVTVDDVIEKLTSYTLLNEANIILGFAPPLHYTFTDASGESIVIEPDKTGITIHRKTIGVMTNSPGYEWHQTNLRAYIGVLPGDFTPSARFLRVAYWKKYTEKAKNETEGVTNLFHILSSVNIPKGVVLTNEGKTDYTIYTSAMCAQSKNYYFKLYDNSRISAVSLMAENLNSQDLITFEWDRK Hydrogen bonds contact Hydrophobic contact | ||||
| 36 | Histone acetyltransferase p300 (EP300) | 5LKX | 7.01 | |
Target general information Gen name EP300 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms p300 HAT; Protein propionyltransferase p300; P300; Histone crotonyltransferase p300; Histone butyryltransferase p300; E1Aassociated protein p300; E1A-associated protein p300 Protein family NA Biochemical class Acyltransferase Function Acetylates all four core histones in nucleosomes. Histone acetylation gives an epigenetic tag for transcriptional activation. Mediates cAMP-gene regulation by binding specifically to phosphorylated CREB protein. Mediates acetylation of histone H3 at 'Lys-122' (H3K122ac), a modification that localizes at the surface of the histone octamer and stimulates transcription, possibly by promoting nucleosome instability. Mediates acetylation of histone H3 at 'Lys-27' (H3K27ac). Also functions as acetyltransferase for non-histone targets, such as ALX1, HDAC1, PRMT1 or SIRT2. Acetylates 'Lys-131' of ALX1 and acts as its coactivator. Acetylates SIRT2 and is proposed to indirectly increase the transcriptional activity of TP53 through acetylation and subsequent attenuation of SIRT2 deacetylase function. Acetylates HDAC1 leading to its inactivation and modulation of transcription. Acts as a TFAP2A-mediated transcriptional coactivator in presence of CITED2. Plays a role as a coactivator of NEUROD1-dependent transcription of the secretin and p21 genes and controls terminal differentiation of cells in the intestinal epithelium. Promotes cardiac myocyte enlargement. Can also mediate transcriptional repression. Acetylates FOXO1 and enhances its transcriptional activity. Acetylates BCL6 wich disrupts its ability to recruit histone deacetylases and hinders its transcriptional repressor activity. Participates in CLOCK or NPAS2-regulated rhythmic gene transcription; exhibits a circadian association with CLOCK or NPAS2, correlating with increase in PER1/2 mRNA and histone H3 acetylation on the PER1/2 promoter. Acetylates MTA1 at 'Lys-626' which is essential for its transcriptional coactivator activity. Acetylates XBP1 isoform 2; acetylation increases protein stability of XBP1 isoform 2 and enhances its transcriptional activity. Acetylates PCNA; acetylation promotes removal of chromatin-bound PCNA and its degradation during nucleotide excision repair (NER). Acetylates MEF2D. Acetylates and stabilizes ZBTB7B protein by antagonizing ubiquitin conjugation and degragation, this mechanism may be involved in CD4/CD8 lineage differentiation. In addition to protein acetyltransferase, can use different acyl-CoA substrates, such as (2E)-butenoyl-CoA (crotonyl-CoA), butanoyl-CoA (butyryl-CoA) or propanoyl-CoA (propionyl-CoA), and is able to mediate protein crotonylation, butyrylation or propionylation, respectively. Acts as a histone crotonyltransferase; crotonylation marks active promoters and enhancers and confers resistance to transcriptional repressors. Histone crotonyltransferase activity is dependent on the concentration of (2E)-butenoyl-CoA (crotonyl-CoA) substrate and such activity is weak when (E)-but-2-enoyl-CoA (crotonyl-CoA) concentration is low. Also acts as a histone butyryltransferase; butyrylation marks active promoters. Functions as a transcriptional coactivator for SMAD4 in the TGF-beta signaling pathway. Acetylates PCK1 and promotes PCK1 anaplerotic activity. Functions as histone acetyltransferase and regulates transcription via chromatin remodeling. Related diseases Defects in EP300 may play a role in epithelial cancer.; DISEASE: Chromosomal aberrations involving EP300 may be a cause of acute myeloid leukemias. Translocation t(8;22)(p11;q13) with KAT6A.; DISEASE: Rubinstein-Taybi syndrome 2 (RSTS2) [MIM:613684]: A disorder characterized by craniofacial abnormalities, postnatal growth deficiency, broad thumbs, broad big toes, intellectual disability and a propensity for development of malignancies. Some individuals with RSTS2 have less severe mental impairment, more severe microcephaly, and a greater degree of changes in facial bone structure than RSTS1 patients. {ECO:0000269|PubMed:15706485}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Menke-Hennekam syndrome 2 (MKHK2) [MIM:618333]: A form of Menke-Hennekam syndrome, a congenital autosomal dominant disease characterized by developmental delay, growth retardation, and craniofacial dysmorphism. Patients have intellectual disability of variable severity, speech delay, autistic behavior, short stature and microcephaly. Main facial characteristics include short palpebral fissures, telecanthi, depressed nasal ridge, short nose, anteverted nares, short columella and long philtrum. {ECO:0000269|PubMed:29460469}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q9NXW9; P27695; Q9UBL3; Q8WXX7; Q9NPI1; P24941; Q99967; P61201; P16220-1; P17844; Q01844; P35637; Q00403; Q16665; Q9H2X6; Q92831; P55209; O60934; P20265; Q96KQ4; Q8WUF5; Q13761; Q96EB6; Q13309; O95863; P42226; Q9UL17; P56279; P05549; P04637; Q13625; O15350; P11473; P67809; K4P3M7; P03122; P06422; P06790; Q61221; Q9QXM1; P04608; P03070; P03255; P03255-2; P03259 EC number EC 2.3.1.48 Uniprot keywords 3D-structure; Acetylation; Acyltransferase; Biological rhythms; Bromodomain; Cell cycle; Chromosomal rearrangement; Chromosome; Citrullination; Cytoplasm; Direct protein sequencing; Disease variant; Host-virus interaction; Intellectual disability; Isopeptide bond; Metal-binding; Methylation; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Transcription; Transcription regulation; Transferase; Ubl conjugation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A Molecular weight (Da) 64477.2 Length 554 Aromaticity 0.12 Instability index 45.78 Isoelectric point 7.01 Charge (pH=7) 0.05 2D Binding mode Binding energy (Kcal/mol) -9.56 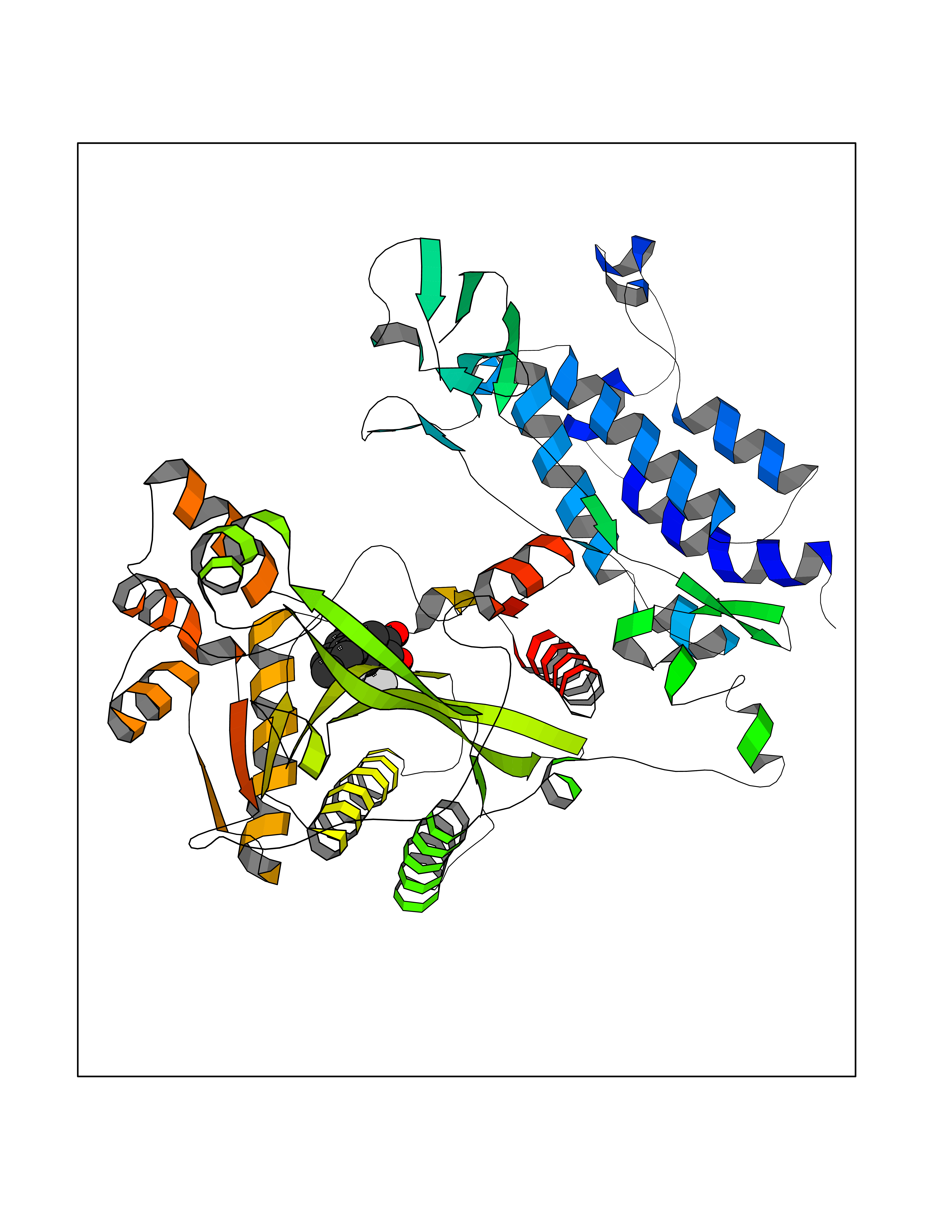 Molscript Map 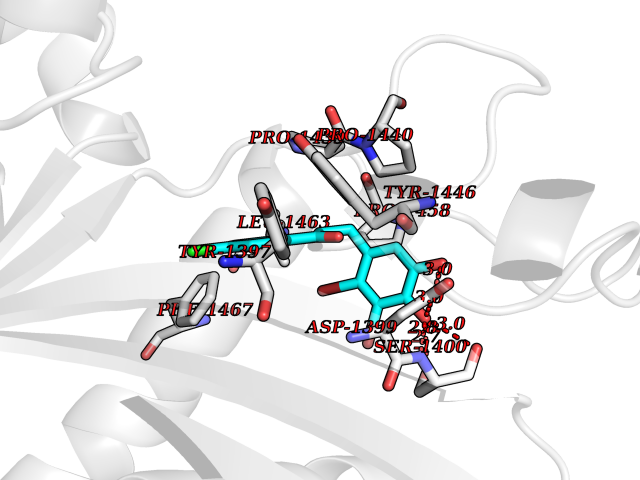 Pymol Map  Ligplot Map 3D Binding mode Sequence KKIFKPEELRQALMPTLEALYRQDPESLPFRQPVDPQLLGIPDYFDIVKSPMDLSTIKRKLDTGQYQEPWQYVDDIWLMFNNAWLYNRKTSRVYKYCSKLSEVFEQEIDPVMQSLGYCCGRKLEFSPQTLCCYGKQLCTIPRDATYYSYQNRYHFCEKCFNEIQGESVSLGQTTINKEQFSKRKNDTLDPELFVECTECGRKMHQICVLHHEIIWPAGFVCDGCLKKSARTRKENKFSAKRLPSTRLGTFLENRVNDFLRRQNHPESGEVTVRVVHASDKTVEVKPGMKARFVDSGEMAESFPYRTKALFAFEEIDGVDLCFFGMHVQEYGSDCPPPNQRRVYISYLDSVHFFRPKCLRTAVYHEILIGYLEYVKKLGYTTGHIWACPPSEGDDYIFHCHPPDQKIPKPKRLQEWFKKMLDKAVSERIVHDYKDIFKQATEDRLTSAKELPYFEGDFWPNVLEESIKESGGSGSQKLYATMEKHKEVFFVIRLIAGPAANSLPPIVDPDPLIPCDLMDGRDAFLTLARDKHLEFSSLRRAQWSTMCMLVELHTQ Hydrogen bonds contact Hydrophobic contact | ||||
| 37 | Toll-like receptor 1 (TLR1) | 2Z7X | 7.01 | |
Target general information Gen name TLR1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Toll/interleukin-1 receptor-like protein; TIL; KIAA0012; CD281 Protein family Toll-like receptor family Biochemical class Toll-like receptor Function Specifically recognizes diacylated and triacylated lipopeptides. Cooperates with TLR2 to mediate the innate immune response to bacterial lipoproteins or lipopeptides. Forms the activation cluster TLR2:TLR1:CD14 in response to triacylated lipopeptides, this cluster triggers signaling from the cell surface and subsequently is targeted to the Golgi in a lipid-raft dependent pathway. Acts via MYD88 and TRAF6, leading to NF-kappa-B activation, cytokine secretion and the inflammatory response. Participates in the innate immune response to microbial agents. Related diseases Hao-Fountain syndrome (HAFOUS) [MIM:616863]: An autosomal dominant neurodevelopmental disorder characterized by global developmental delay, varying degrees of intellectual disability, autism spectrum disorder, poor or absent speech, and mild facial dysmorphism. Most patients develop seizures. Additional variable features include hypotonia, hypogonadism in males, and ocular anomalies. {ECO:0000269|PubMed:26365382, ECO:0000269|PubMed:30679821}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q15399; Q9BXR5; O60603; Q9Y2C9 EC number NA Uniprot keywords 3D-structure; Cell membrane; Cytoplasmic vesicle; Direct protein sequencing; Disulfide bond; Glycoprotein; Golgi apparatus; Immunity; Inflammatory response; Innate immunity; Leucine-rich repeat; Membrane; NAD; Proteomics identification; Receptor; Reference proteome; Repeat; Signal; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A,C Molecular weight (Da) 62767.4 Length 555 Aromaticity 0.07 Instability index 38.79 Isoelectric point 6.85 Charge (pH=7) -0.55 2D Binding mode Binding energy (Kcal/mol) -9.56 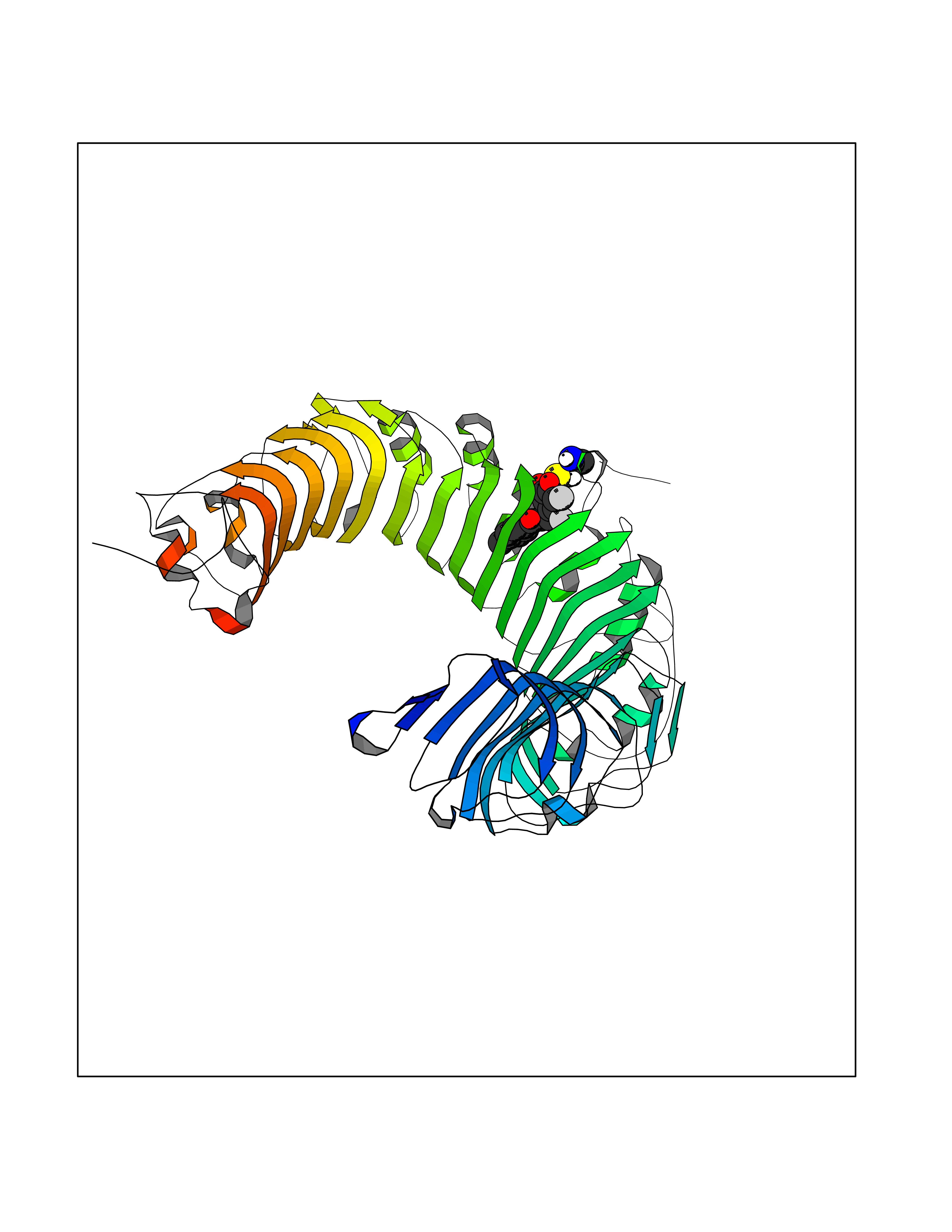 Molscript Map 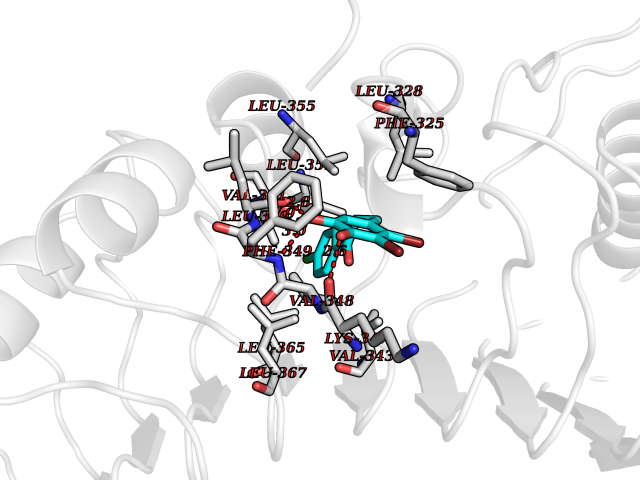 Pymol Map 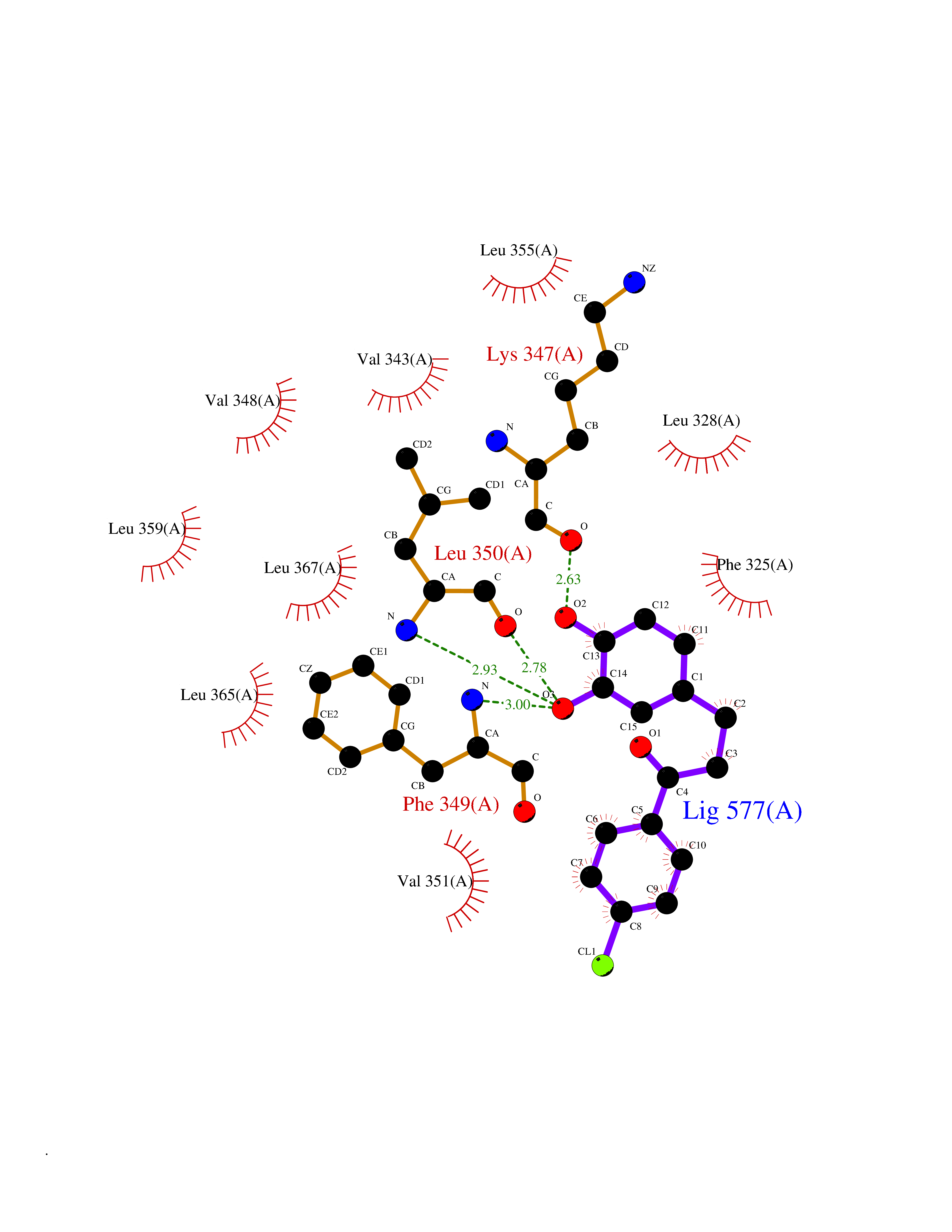 Ligplot Map 3D Binding mode Sequence SLSCDRNGICKGSSGSLNSIPSGLTEAVKSLDLSNNRITYISNSDLQRCVNLQALVLTSNGINTIEEDSFSSLGSLEHLDLSYNYLSNLSSSWFKPLSSLTFLNLLGNPYKTLGETSLFSHLTKLQILRVGNMDTFTKIQRKDFAGLTFLEELEIDASDLQSYEPKSLKSIQNVSHLILHMKQHILLLEIFVDVTSSVECLELRDTDLDTFHFSELSTGETNSLIKKFTFRNVKITDESLFQVMKLLNQISGLLELEFDDCTLNGVGNFRASDNDRVIDPGKVETLTIRRLHIPRFYLFYDLSTLYSLTERVKRITVENSKVFLVPCLLSQHLKSLEYLDLSENLMVEEYLKNSACEDAWPSLQTLILRQNHLASLEKTGETLLTLKNLTNIDISKNSFHSMPETCQWPEKMKYLNLSSTRIHSVTGCIPKTLEILDVSNNNLNLFSLNLPQLKELYISRNKLMTLPDASLLPMLLVLKISRNQLKSVPDGIFDRLTSLQKIWLHTNPWDCSCPRIDYLSRWLNKNSQKEQGSAKCSGSGKPVRSIICPXSKKKK Hydrogen bonds contact Hydrophobic contact | ||||
| 38 | Histamine H3 receptor (H3R) | 7F61 | 7.01 | |
Target general information Gen name HRH3 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Histamine receptor 3; HH3R; GPCR97; G-protein coupled receptor 97; G protein-coupled receptor 97 Protein family G-protein coupled receptor 1 family Biochemical class GPCR rhodopsin Function Signals through the inhibition of adenylate cyclase and displays high constitutive activity (spontaneous activity in the absence of agonist). Agonist stimulation of isoform 3 neither modified adenylate cyclase activity nor induced intracellular calcium mobilization. The H3 subclass of histamine receptors could mediate the histamine signals in CNS and peripheral nervous system. Related diseases Immunodeficiency 48 (IMD48) [MIM:269840]: A form of severe immunodeficiency characterized by a selective absence of CD8+ T-cells. {ECO:0000269|PubMed:11123350, ECO:0000269|PubMed:11412303, ECO:0000269|PubMed:18509675, ECO:0000269|PubMed:8124727, ECO:0000269|PubMed:8202713}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Autoimmune disease, multisystem, infantile-onset, 2 (ADMIO2) [MIM:617006]: An autosomal recessive, autoimmune disorder characterized by systemic manifestations including blistering skin disease, uncontrollable bullous pemphigoid, inflammatory colitis, autoimmune hypothyroidism, proteinuria and nephrotic syndrome. {ECO:0000269|PubMed:26783323}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01238; DB06698; DB05381; DB17087; DB05080; DB00768; DB11642 Interacts with NA EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disulfide bond; G-protein coupled receptor; Glycoprotein; Membrane; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 34321.1 Length 301 Aromaticity 0.16 Instability index 32.22 Isoelectric point 9.63 Charge (pH=7) 15.11 2D Binding mode Binding energy (Kcal/mol) -9.56 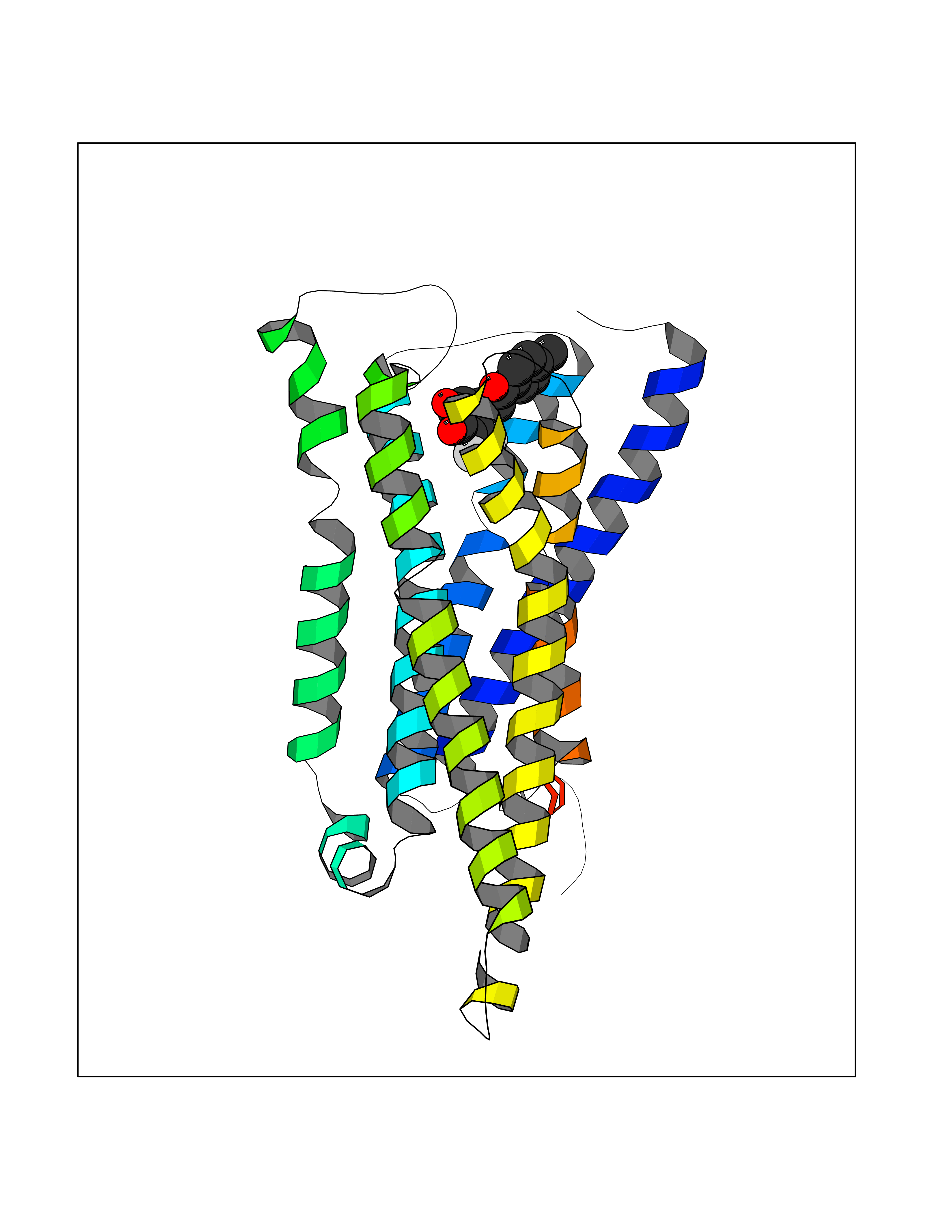 Molscript Map 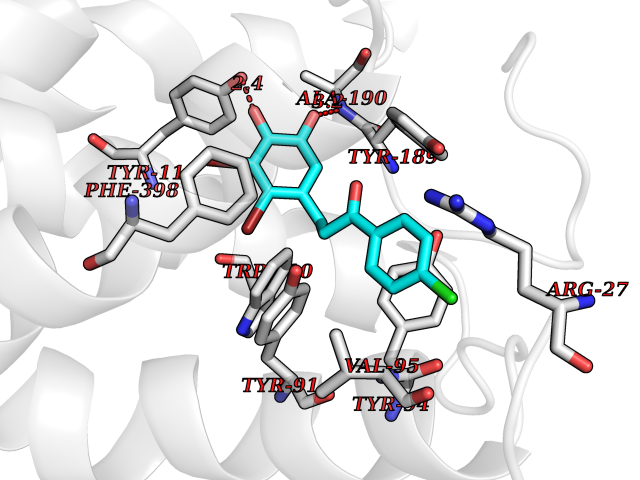 Pymol Map 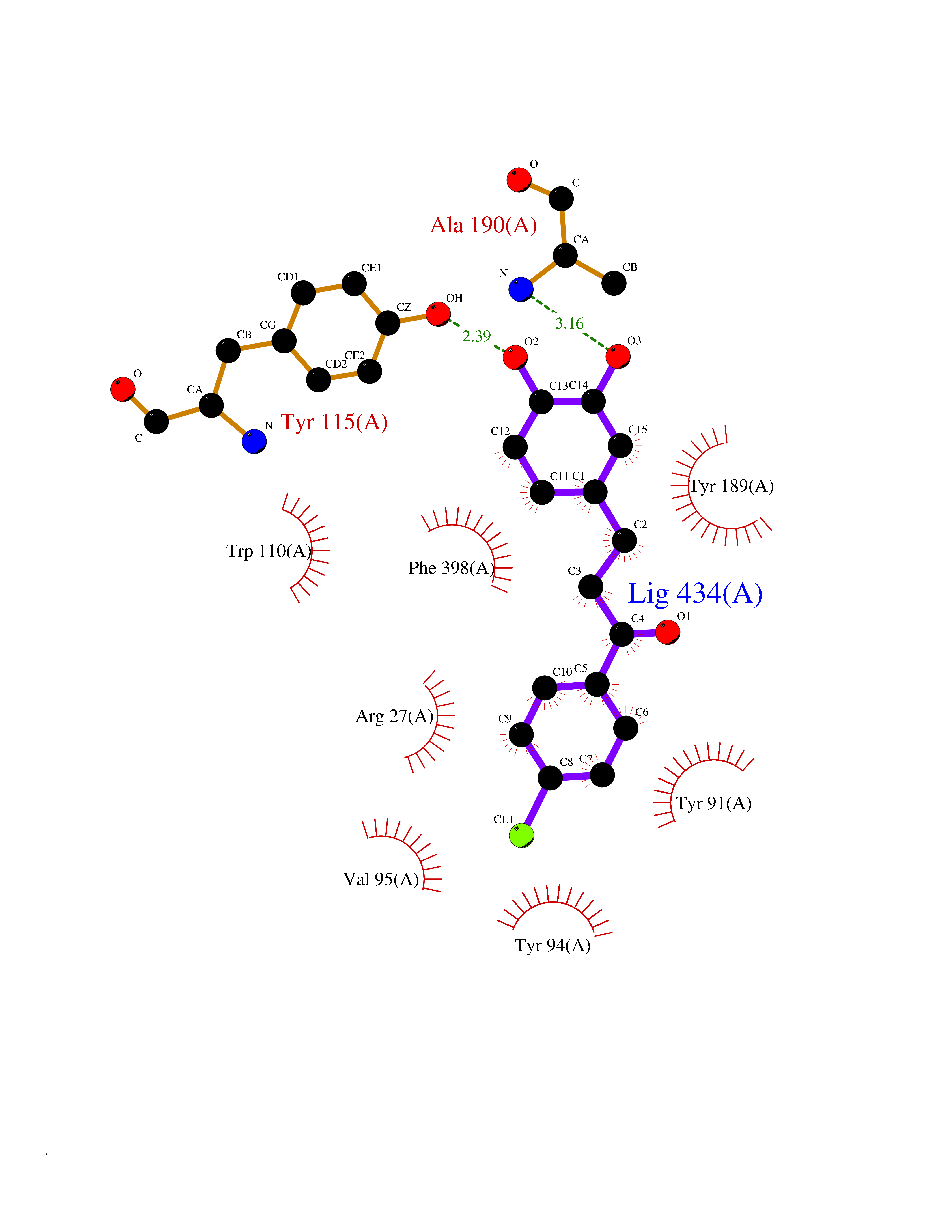 Ligplot Map 3D Binding mode Sequence RGFSAAWTAVLAALMALLIVATVLGNALVMLAFVADSSLRTQNNFFLLNLAISDFLVGAFCIPLYVPYVLTGRWTFGRGLCKLWLVVDYLLCTSKAFNIVLISYDRFLSVTRAVSYRAQQGDTRRAVRKMLLVWVLAFLLYGPAILSWEYLSGGSSIPEGHCYAEFFYNWYFLITASTLEFFTPFLSVTFFNLSIYLNIQRRTRLRLDGAREAAGRFRLSRDRKVAKSLAVIVSIFGLCWAPYTLLMIIRAACHGHCVPDYWYETSFWLLWANSAVNPVLYPLCHHSFRRAFTKLLCPQKL Hydrogen bonds contact Hydrophobic contact | ||||
| 39 | Glutamate receptor ionotropic kainate 1 (GRIK1) | 3FV2 | 7.00 | |
Target general information Gen name GRIK1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Glutamate receptor 5; GluR5 kainate receptor; GluR5; GluR-5; GRIK1; Excitatory amino acid receptor 3; EAA3 Protein family Glutamate-gated ion channel (TC 1.A.10.1) family, GRIK1 subfamily Biochemical class Glutamate-gated ion channel Function Ionotropic glutamate receptor. L-glutamate acts as an excitatory neurotransmitter at many synapses in the central nervous system. Binding of the excitatory neurotransmitter L- glutamate induces a conformation change, leading tothe opening of the cation channel, and thereby converts the chemical signal to an electrical impulse. The receptor then desensitizes rapidly and enters a transient inactive state, characterized by the presence of bound agonist. May be involved in the transmission of light information from the retina to the hypothalamus. Related diseases Defects in PPARG can lead to type 2 insulin-resistant diabetes and hyptertension. PPARG mutations may be associated with colon cancer. {ECO:0000269|PubMed:10394368}.; DISEASE: Obesity (OBESITY) [MIM:601665]: A condition characterized by an increase of body weight beyond the limitation of skeletal and physical requirements, as the result of excessive accumulation of body fat. {ECO:0000269|PubMed:9753710}. Disease susceptibility may be associated with variants affecting the gene represented in this entry.; DISEASE: Lipodystrophy, familial partial, 3 (FPLD3) [MIM:604367]: A form of lipodystrophy characterized by marked loss of subcutaneous fat from the extremities. Facial adipose tissue may be increased, decreased or normal. Affected individuals show an increased preponderance of insulin resistance, diabetes mellitus and dyslipidemia. {ECO:0000269|PubMed:11788685, ECO:0000269|PubMed:12453919}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Glioma 1 (GLM1) [MIM:137800]: Gliomas are benign or malignant central nervous system neoplasms derived from glial cells. They comprise astrocytomas and glioblastoma multiforme that are derived from astrocytes, oligodendrogliomas derived from oligodendrocytes and ependymomas derived from ependymocytes. {ECO:0000269|PubMed:10851250}. Disease susceptibility may be associated with variants affecting the gene represented in this entry. Polymorphic PPARG alleles have been found to be significantly over-represented among a cohort of American patients with sporadic glioblastoma multiforme suggesting a possible contribution to disease susceptibility. Drugs (DrugBank ID) DB00237; DB00142; DB06354; DB00273 Interacts with NA EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disulfide bond; Glycoprotein; Ion channel; Ion transport; Ligand-gated ion channel; Membrane; Phosphoprotein; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; RNA editing; Signal; Synapse; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A,B Molecular weight (Da) 29057.1 Length 256 Aromaticity 0.1 Instability index 24.38 Isoelectric point 8.3 Charge (pH=7) 1.97 2D Binding mode Binding energy (Kcal/mol) -9.54 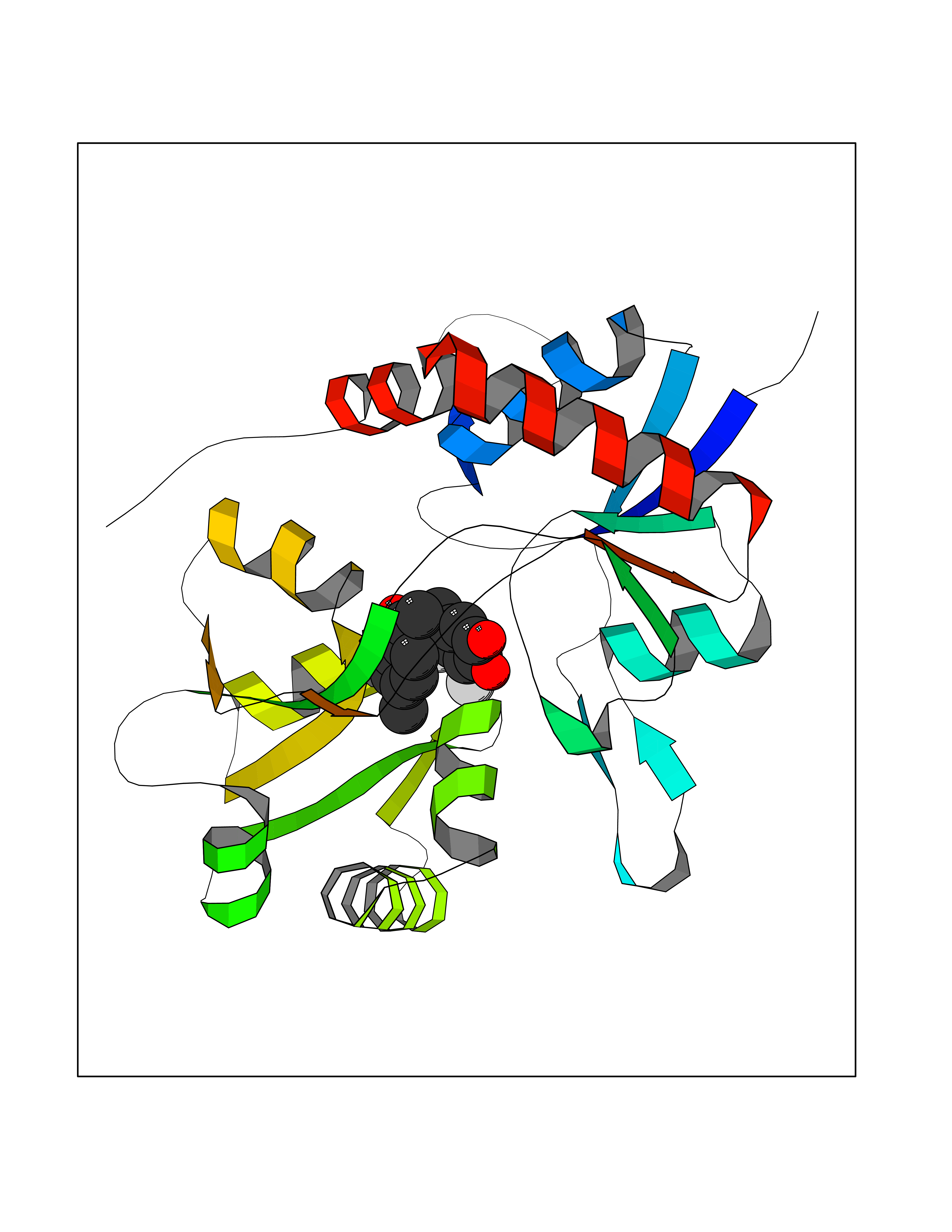 Molscript Map 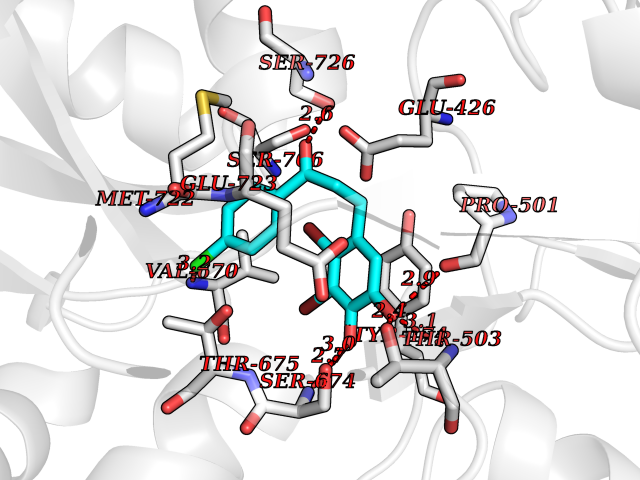 Pymol Map 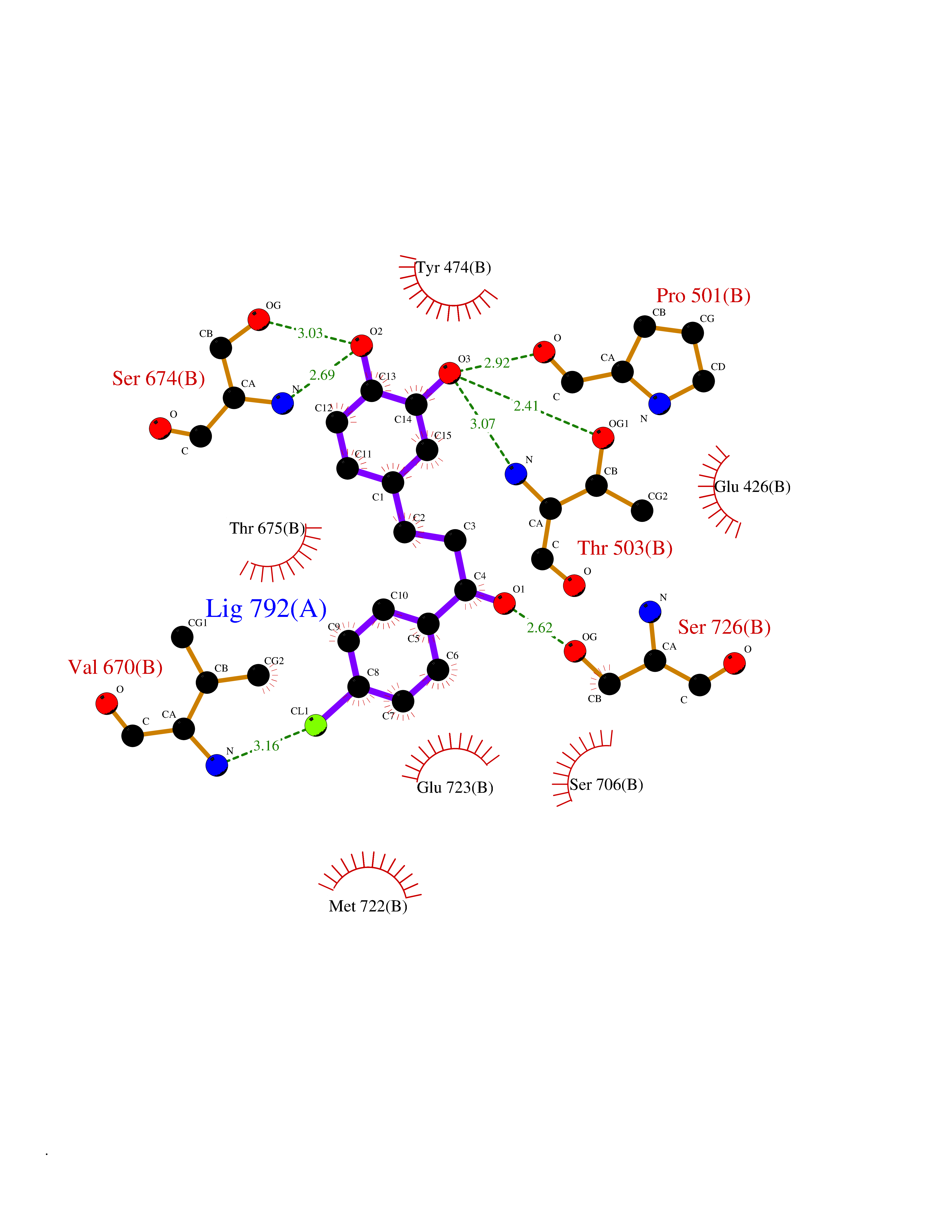 Ligplot Map 3D Binding mode Sequence ANRTLIVTTILEEPYVMYRKSDKPLYGNDRFEGYCLDLLKELSNILGFIYDVKLVPDGKYGAQNDKGEWNGMVKELIDHRADLAVAPLTITYVREKVIDFSKPFMTLGISILYRKGTPIDSADDLAKQTKIEYGAVRDGSTMTFFKKSKISTYEKMWAFMSSRQQTALVRNSDEGIQRVLTTDYALLMESTSIEYVTQRNCNLTQIGGLIDSKGYGVGTPIGSPYRDKITIAILQLQEEGKLHMMKEKWWRGNGCP Hydrogen bonds contact Hydrophobic contact | ||||
| 40 | SEC14-like protein 4 | 4TLG | 7.00 | |
Target general information Gen name SEC14L4 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms TAP3 Protein family NA Biochemical class Transport protein Function Lipid binding.Transporter activity. Related diseases Chondrodysplasia with platyspondyly, distinctive brachydactyly, hydrocephaly, and microphthalmia (CDP-PBHM) [MIM:300863]: A disease characterized by chondrodysplasia, severe platyspondyly, hydrocephaly, and facial features with microphthalmia. Bone abnormalities include a distinctive metaphyseal cupping of the metacarpals, metatarsals, and phalanges. Affected females show a milder phenotype with small stature, sometimes associated with body asymmetry and mild intellectual disability. {ECO:0000269|PubMed:20181727}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB14003; DB11635; DB11251; DB00163 Interacts with Q96LC9; O43186; P78358; Q9NYQ3; Q0VD86; Q15323; O76011; P50221; Q6FHY5; Q02548; P26367; Q9H8W4; Q04864; Q04864-2; Q9UHV2; P15884; P15884-3; Q96N21; Q9BYV2; Q8N6Y0; Q9H0C1 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Lipid-binding; Proteomics identification; Reference proteome; Transport Protein physicochemical properties Chain ID A,B Molecular weight (Da) 23947.6 Length 210 Aromaticity 0.1 Instability index 50.84 Isoelectric point 5.55 Charge (pH=7) -3.11 2D Binding mode Binding energy (Kcal/mol) -9.55  Molscript Map 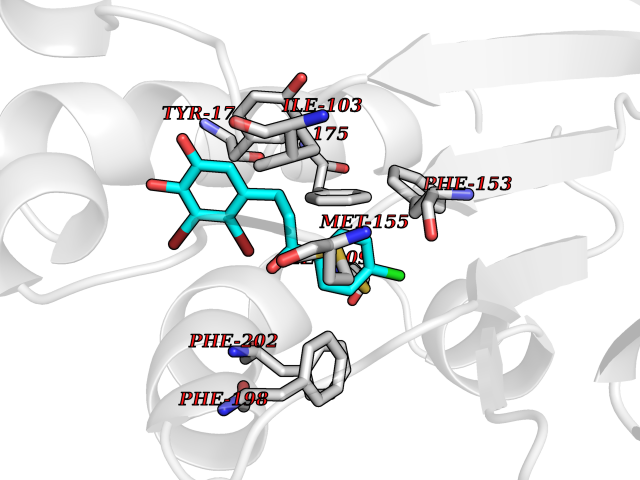 Pymol Map 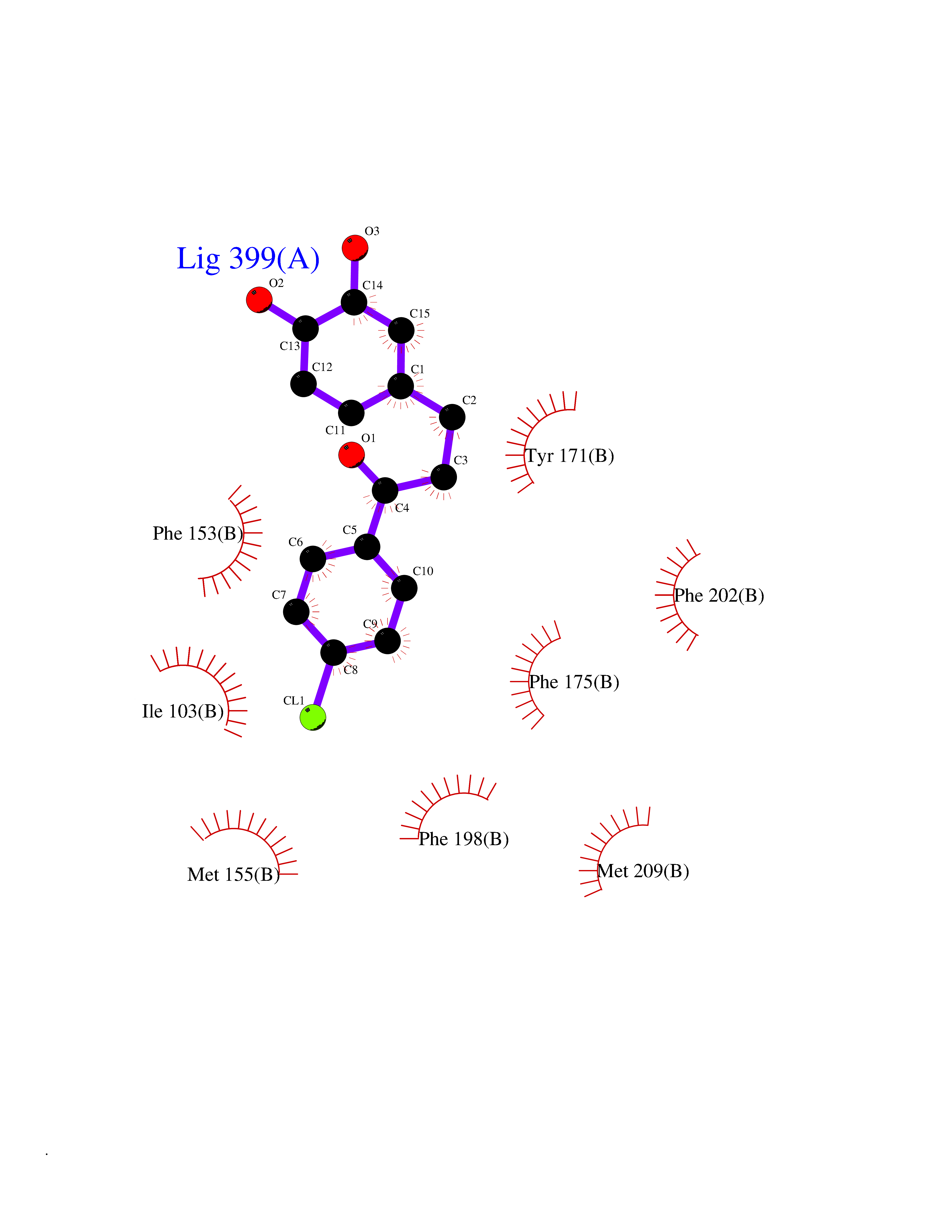 Ligplot Map 3D Binding mode Sequence VTWQPPEVIQLYDSGGLCGYDYEGCPVYFNIIGSLDPKGLLLSASKQDMIRKRIKVCELLLHECELQTQKLGRKIEMALMVFDMEGLSLKHLWKPAVEVYQQFFSILEANYPETLKNLIVIRAPKLFPVAFNLVKSFMSEETRRKIVILGDNWKQELTKFISPDQLPVEFGGTMTDPDGNPKCLTKINYGGEVPKSYYPDKASEETLQSM Hydrogen bonds contact Hydrophobic contact | ||||