Job Results:
Ligand
Structure
Job ID
57b30c77c1e7b273b1e911481965e4cd
Job name
NA
Time
2025-10-13 17:21:20
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 21 | Pyruvate dehydrogenase [ubiquinone] | 3EYA | 6.78 | |
Target general information Gen name poxB Organism Escherichia coli (strain K12) Uniprot ID TTD ID NA Synonyms b0871;JW0855 Protein family TPP enzyme family Biochemical class Oxidoreductase Function Flavin adenine dinucleotide binding.Identical protein binding.Lipid binding.Magnesium ion binding.Pyruvate dehydrogenase (quinone) activity.Thiamine pyrophosphate binding. Related diseases Glycogen storage disease 6 (GSD6) [MIM:232700]: A metabolic disorder characterized by mild to moderate hypoglycemia, mild ketosis, growth retardation, and prominent hepatomegaly. Heart and skeletal muscle are not affected. {ECO:0000269|PubMed:9529348}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P07003 EC number 1.2.5.1 Uniprot keywords 3D-structure; Cell inner membrane; Cell membrane; Direct protein sequencing; FAD; Flavoprotein; Lipid-binding; Magnesium; Membrane; Metal-binding; Nucleotide-binding; Oxidoreductase; Pyruvate; Reference proteome; Thiamine pyrophosphate; Ubiquinone Protein physicochemical properties Chain ID A,B,C,D,E,F,G,H,I,J,K,L Molecular weight (Da) 113027 Length 1046 Aromaticity 0.07 Instability index 35.99 Isoelectric point 5.75 Charge (pH=7) -24.38 2D Binding mode Binding energy (Kcal/mol) -9.25  Molscript Map 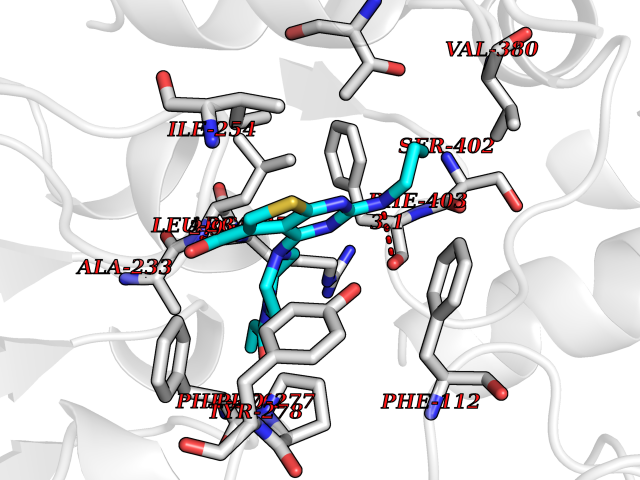 Pymol Map 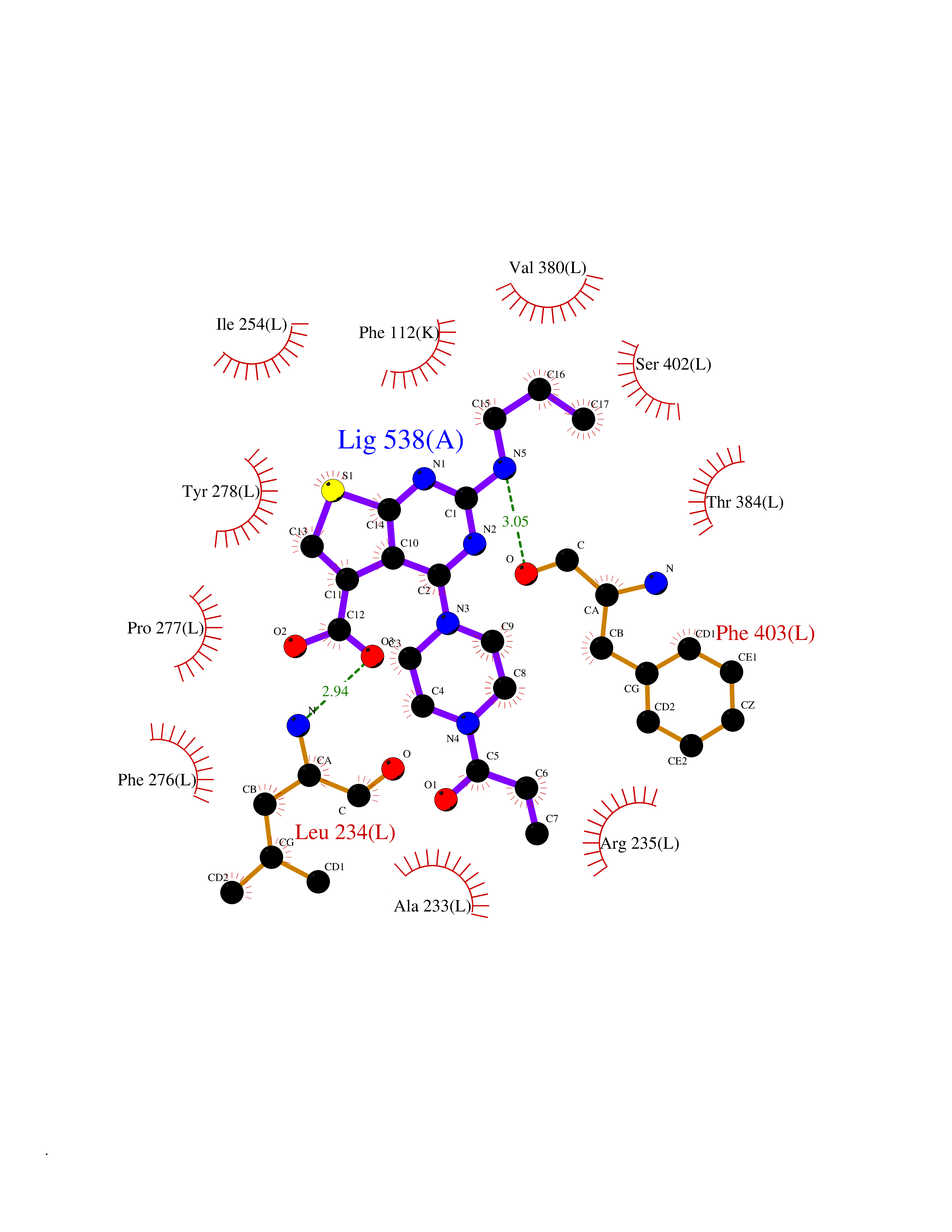 Ligplot Map 3D Binding mode Sequence MKQTVAAYIAKTLESAGVKRIWGVTGDSLNGLSDSLNRMGTIEWMSTRHEEVAAFAAGAEAQLSGELAVCAGSCGPGNLHLINGLFDCHRNHVPVLAIAAHIPSSEIGSGYFQETHPQELFRECSHYCELVSSPEQIPQVLAIAMRKAVLNRGVSVVVLPGDVALKPAPEGATMHWYHAPQPVVTPEEEELRKLAQLLRYSSNIALMCGSGCAGAHKELVEFAGKIKAPIVHALRGKEHVEYDNPYDVGMTGLIGFSSGFHTMMNADTLVLLGTQFPYRAFYPTDAKIIQIDINPASIGAHSKVDMALVGDIKSTLRALLPLVEEKADRKFLDKALEDYRDARKGLDDLAKPSEKAIHPQYLAQQISHFAADDAIFTCDVGTPTVWAARYLKMNGKRRLLGSFNHGSMANAMPQALGAQATEPERQVVAMCGDGGFSMLMGDFLSVVQMKLPVKIVVFNNSVLGFDGTELHDTNFARIAEACGITGIRVEKASEVDEALQRAFSIDGPVLVDVVVAKEELAIPMKQTVAAYIAKTLESAGVKRIWGVTGDSLNGLSDSLNRMGTIEWMSTRHEEVAAFAAGAEAQLSGELAVCAGSCGPGNLHLINGLFDCHRNHVPVLAIAAHIPSSEIGSGYFQETHPQELFRECSHYCELVSSPEQIPQVLAIAMRKAVLNRGVSVVVLPGDVALKPAPEGATMHWYHAPQPVVTPEEEELRKLAQLLRYSSNIALMCGSGCAGAHKELVEFAGKIKAPIVHALRGKEHVEYDNPYDVGMTGLIGFSSGFHTMMNADTLVLLGTQFPYRAFYPTDAKIIQIDINPASIGAHSKVDMALVGDIKSTLRALLPLVEEKADRKFLDKALEDYRDARKGLDDLAKPSEKAIHPQYLAQQISHFAADDAIFTCDVGTPTVWAARYLKMNGKRRLLGSFNHGSMANAMPQALGAQATEPERQVVAMCGDGGFSMLMGDFLSVVQMKLPVKIVVFNNSVLGFVGTELHDTNFARIAEACGITGIRVEKASEVDEALQRAFSIDGPVLVDVVVAKEELAIP Hydrogen bonds contact Hydrophobic contact | ||||
| 22 | Melatonin receptor type 1B (MTNR1B) | 6ME9 | 6.78 | |
Target general information Gen name MTNR1B Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Mel1b receptor; Mel1b melatonin receptor; Mel-1B-R Protein family G-protein coupled receptor 1 family Biochemical class GPCR rhodopsin Function Likely to mediate the reproductive and circadian actions of melatonin. The activity of this receptor is mediated by pertussis toxin sensitive G proteins that inhibit adenylate cyclase activity. High affinity receptor for melatonin. Related diseases Insulin-like growth factor 1 resistance (IGF1RES) [MIM:270450]: A disorder characterized by intrauterine growth retardation, poor postnatal growth and increased plasma IGF1 levels. {ECO:0000269|PubMed:14657428, ECO:0000269|PubMed:15928254, ECO:0000269|PubMed:25040157}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB06594; DB01065; DB00980; DB02709; DB09071; DB15133 Interacts with P28335; P48039; O76081; Q14669 EC number NA Uniprot keywords 3D-structure; Cell membrane; Disulfide bond; G-protein coupled receptor; Glycoprotein; Membrane; Receptor; Reference proteome; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 50184.9 Length 448 Aromaticity 0.11 Instability index 37.2 Isoelectric point 5.72 Charge (pH=7) -5.68 2D Binding mode Binding energy (Kcal/mol) -9.25  Molscript Map 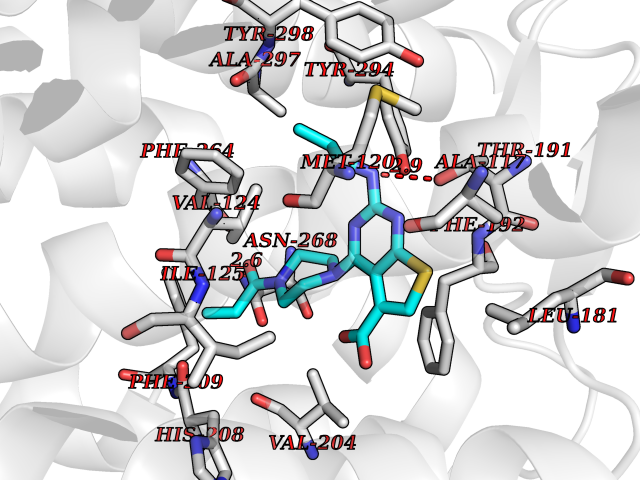 Pymol Map 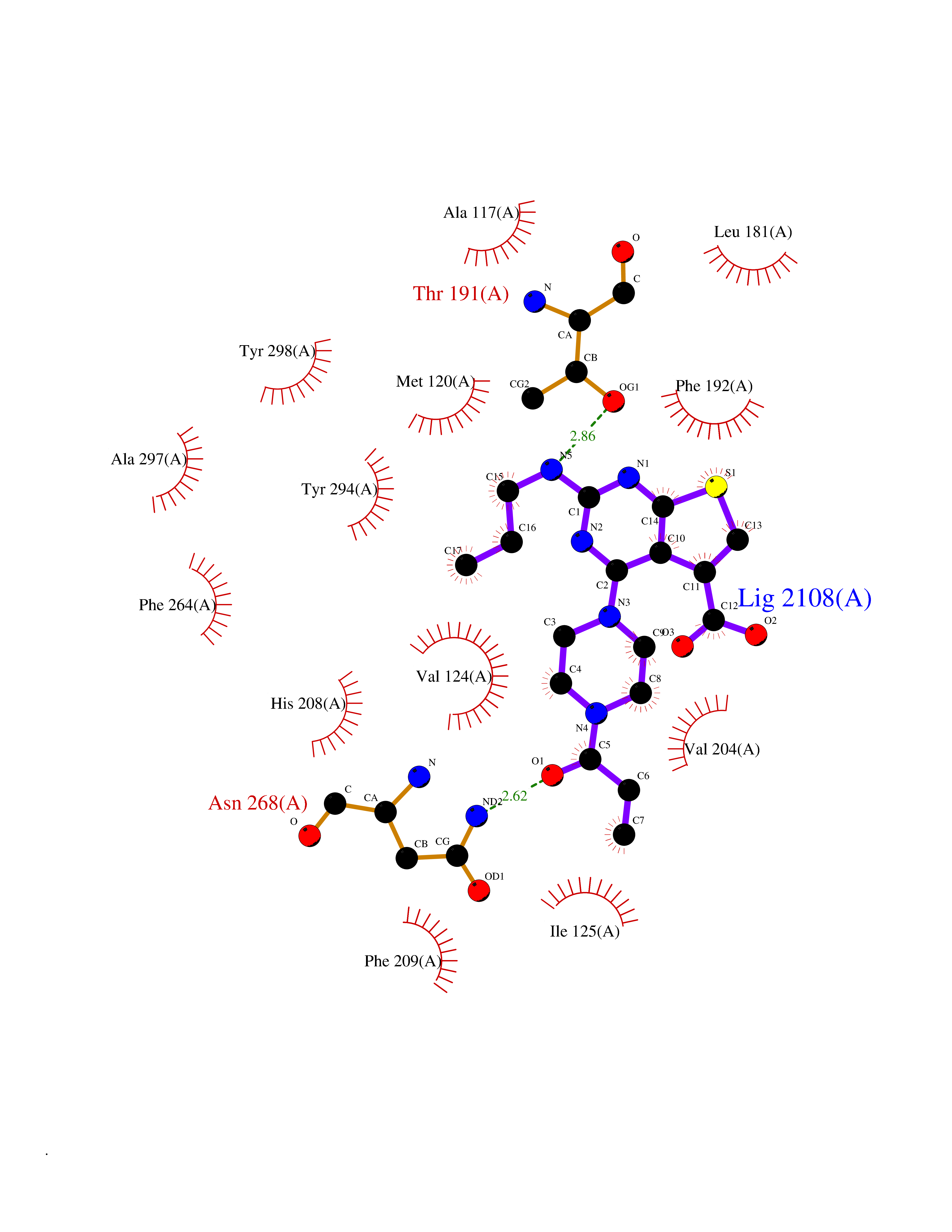 Ligplot Map 3D Binding mode Sequence ADLEDNWETLNDNLKVIEKADNAAQVKDALTKMRAAALDAQKATPPKLEDKSPDSPEMKDFRHGFDILVGQIDDALKLANEGKVKEAQAAAEQLKTTRNAYIQKYLGDGARPSWVAPALSAVLIVTTAVDVVGNLLVILSVLRNRKLRNAGNLFLVSLALANLVVAFYPYPLILVAIFYDGWAFGEEHCKASAFVMGLSVIGSVWNITAIAIDRYLYICHSMAYHRIYRRWHTPLHICLIWLLTVVALLPNFFVGSLEYDPRIYSCTFIQTASTQYTAAVVVIHFLLPIAVVSFCYLRIWVLVLQARMKKYTCTVCGYIYNPEDGDPDNGVNPGTDFKDIPDDWVCPLCGVGKDQFEEVECLKPSDLRSFLTMFVVFVIFAICFAPLNCIGLAVAINPQEMAPQIPEGLFVTSYLLAYFNSCLNPIVYGLLDQNFRREYKRILLALWN Hydrogen bonds contact Hydrophobic contact | ||||
| 23 | Aldo-keto reductase family 1 member C3 | 1S1P | 6.77 | |
Target general information Gen name AKR1C3 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms PGFS;DDH1;HSD17B5;KIAA0119 Protein family Aldo/keto reductase family Biochemical class Oxidoreductase Function 15-hydroxyprostaglandin-D dehydrogenase (NADP+) activity.Alditol:NADP+ 1-oxidoreductase activity.Aldo-keto reductase (NADP) activity.Androsterone dehydrogenase activity.Delta4-3-oxosteroid 5beta-reductase activity.Dihydrotestosterone 17-beta-dehydrogenase activity.Geranylgeranyl reductase activity.Indanol dehydrogenase activity.Ketoreductase activity.Ketosteroid monooxygenase activity.NADP-retinol dehydrogenase activity.Oxidoreductase activity, acting on NAD(P)H, quinone or similar compound as acceptor.Phenanthrene 9,10-monooxygenase activity.Prostaglandin D2 11-ketoreductase activity.Prostaglandin-F synthase activity.Prostaglandin H2 endoperoxidase reductase activity.Retinal dehydrogenase activity.Retinol dehydrogenase activity.Testosterone 17-beta-dehydrogenase (NADP+) activity.Testosterone dehydrogenase (NAD+) activity.Trans-1,2-dihydrobenzene-1,2-diol dehydrogenase activity. Related diseases Neurodevelopmental disorder with language impairment and behavioral abnormalities (NEDLIB) [MIM:618917]: A neurodevelopmental disorder characterized by global developmental delay, impaired intellectual development, poor or absent speech, and behavioral abnormalities, such as autism spectrum disorder, repetitive behaviors, and hyperactivity. Some patients develop seizures and manifest developmental regression. {ECO:0000269|PubMed:31300657}. The disease is caused by variants affecting the gene represented in this entry. The genetic variation producing the missense variant p.Q607E, associated with NEDLIB, is predicted to deeply affect RNA editing. In a physiological context, the adenosine (A) residue of the original glutamine (Q) codon CAG is post-transcriptionaly edited to inosine (I) by ADAR2, leading to a codon recognized by the ribosome as arginine (R). The glutamate (E) codon GAG, resulting from the genetic variation, is predicted to be edited 90% less than the normal CAG codon. If edited, the codon GIG would be translated as p.Q607G. {ECO:0000305|PubMed:31300657}. Drugs (DrugBank ID) DB07700; DB01561; DB01536; DB00997; DB01039; DB02266; DB13751; DB00328; DB06077; DB00959; DB00157; DB03461; DB09074; DB00776; DB02056; DB01698; DB02901 Interacts with P17516 EC number 1.1.1.-; 1.1.1.188; 1.1.1.210; 1.1.1.239; 1.1.1.357; 1.1.1.53; 1.1.1.62; 1.1.1.64 Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; Lipid metabolism; NAD; NADP; Oxidoreductase; Proteomics identification; Reference proteome Protein physicochemical properties Chain ID A Molecular weight (Da) 35846.8 Length 315 Aromaticity 0.09 Instability index 47.59 Isoelectric point 8.54 Charge (pH=7) 4.51 2D Binding mode Binding energy (Kcal/mol) -9.23  Molscript Map  Pymol Map 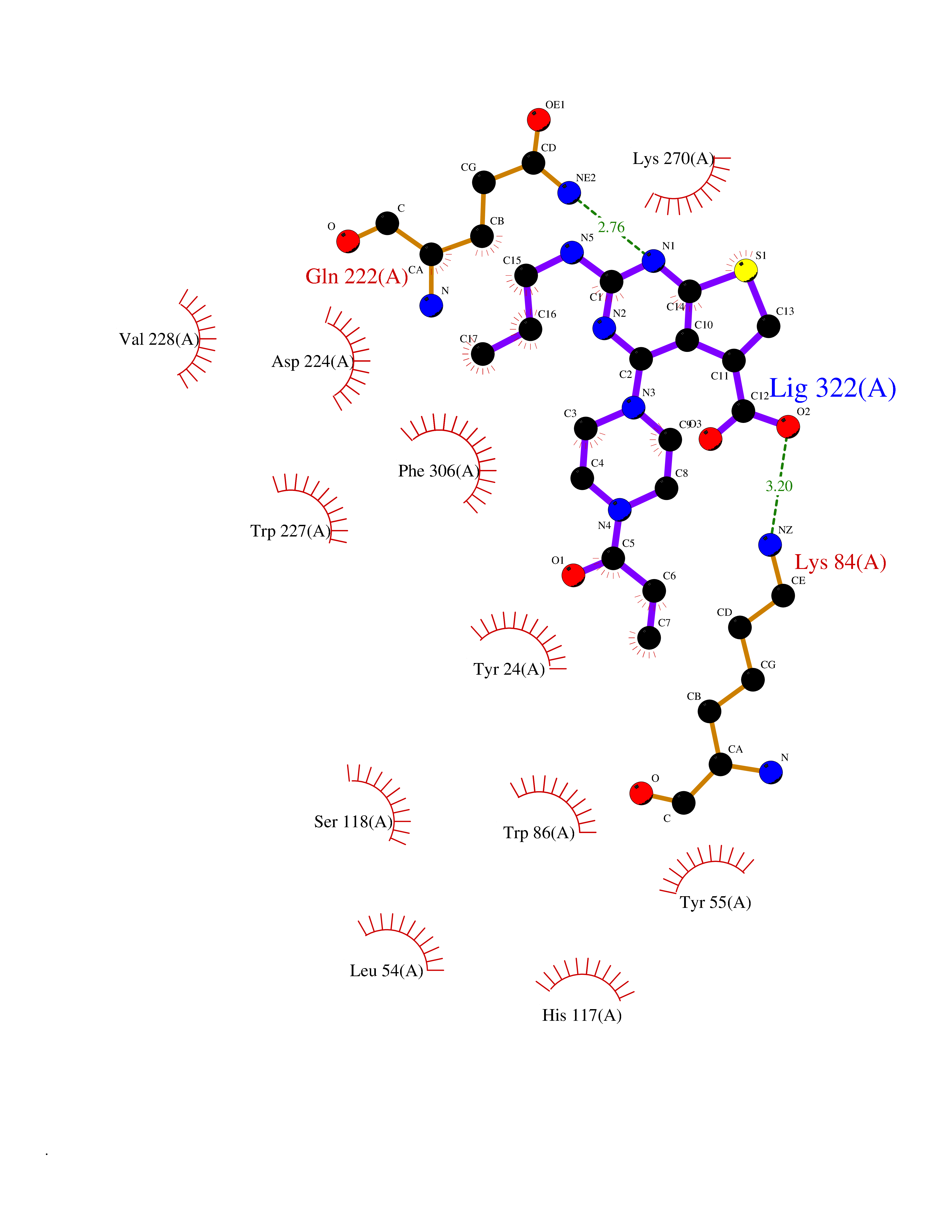 Ligplot Map 3D Binding mode Sequence QCVKLNDGHFMPVLGFGTYAPPEVPRSKALEVTKLAIEAGFRHIDSAHLYNNEEQVGLAIRSKIADGSVKREDIFYTSKLWSTFHRPELVRPALENSLKKAQLDYVDLYLIHSPMSLKPGEELSPTDENGKVIFDIVDLCTTWEAMEKCKDAGLAKSIGVSNFNRRQLEMILNKPGLKYKPVCNQVECHPYFNRSKLLDFCKSKDIVLVAYSALGSQRDKRWVDPNSPVLLEDPVLCALAKKHKRTPALIALRYQLQRGVVVLAKSYNEQRIRQNVQVFEFQLTAEDMKAIDGLDRNLHYFNSDSFASHPNYPYS Hydrogen bonds contact Hydrophobic contact | ||||
| 24 | Monomeric sarcosine oxidase | 2GF3 | 6.77 | |
Target general information Gen name soxA Organism Bacillus sp. (strain B-0618) Uniprot ID TTD ID NA Synonyms sox Protein family MSOX/MTOX family, MSOX subfamily Biochemical class Oxidoreductase Function Sarcosine oxidase activity. Related diseases Defects in PPARG can lead to type 2 insulin-resistant diabetes and hyptertension. PPARG mutations may be associated with colon cancer. {ECO:0000269|PubMed:10394368}.; DISEASE: Obesity (OBESITY) [MIM:601665]: A condition characterized by an increase of body weight beyond the limitation of skeletal and physical requirements, as the result of excessive accumulation of body fat. {ECO:0000269|PubMed:9753710}. Disease susceptibility may be associated with variants affecting the gene represented in this entry.; DISEASE: Lipodystrophy, familial partial, 3 (FPLD3) [MIM:604367]: A form of lipodystrophy characterized by marked loss of subcutaneous fat from the extremities. Facial adipose tissue may be increased, decreased or normal. Affected individuals show an increased preponderance of insulin resistance, diabetes mellitus and dyslipidemia. {ECO:0000269|PubMed:11788685, ECO:0000269|PubMed:12453919}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Glioma 1 (GLM1) [MIM:137800]: Gliomas are benign or malignant central nervous system neoplasms derived from glial cells. They comprise astrocytomas and glioblastoma multiforme that are derived from astrocytes, oligodendrogliomas derived from oligodendrocytes and ependymomas derived from ependymocytes. {ECO:0000269|PubMed:10851250}. Disease susceptibility may be associated with variants affecting the gene represented in this entry. Polymorphic PPARG alleles have been found to be significantly over-represented among a cohort of American patients with sporadic glioblastoma multiforme suggesting a possible contribution to disease susceptibility. Drugs (DrugBank ID) DB03098; DB01918; DB03517; DB03147; DB03366; DB02083; DB02543 Interacts with NA EC number 1.5.3.1 Uniprot keywords 3D-structure; Cytoplasm; Direct protein sequencing; FAD; Flavoprotein; Oxidoreductase Protein physicochemical properties Chain ID A,B Molecular weight (Da) 42606.4 Length 385 Aromaticity 0.1 Instability index 26.97 Isoelectric point 5.27 Charge (pH=7) -17.18 2D Binding mode Binding energy (Kcal/mol) -9.24  Molscript Map 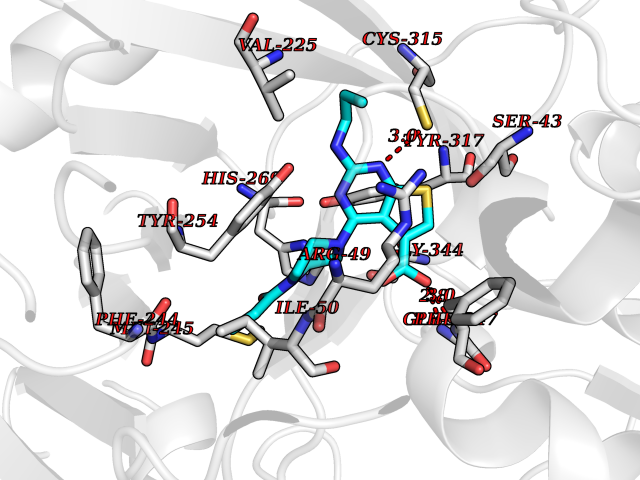 Pymol Map 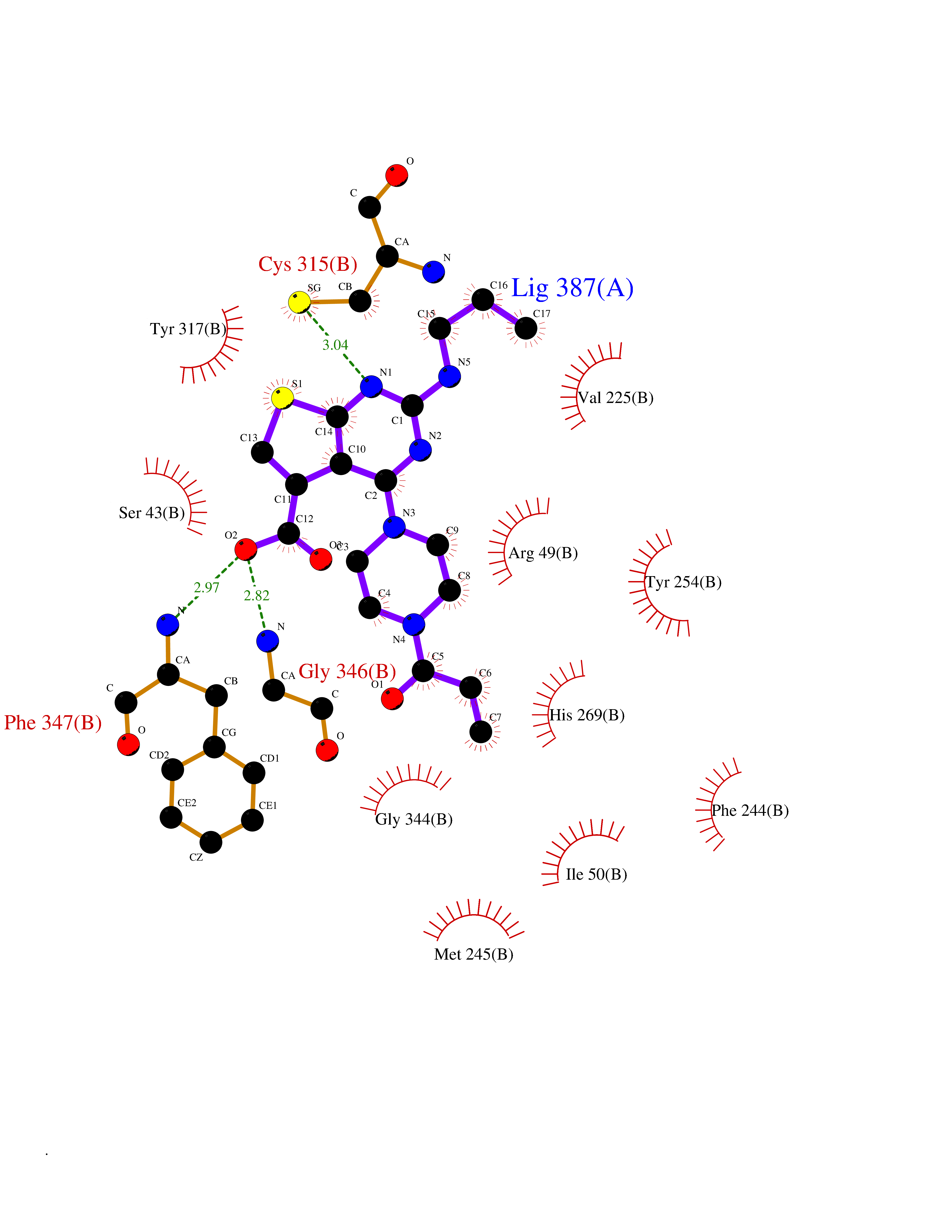 Ligplot Map 3D Binding mode Sequence STHFDVIVVGAGSMGMAAGYQLAKQGVKTLLVDAFDPPHTNGSHHGDTRIIRHAYGEGREYVPLALRSQELWYELEKETHHKIFTKTGVLVFGPKGESAFVAETMEAAKEHSLTVDLLEGDEINKRWPGITVPENYNAIFEPNSGVLFSENCIRAYRELAEARGAKVLTHTRVEDFDISPDSVKIETANGSYTADKLIVSMGAWNSKLLSKLNLDIPLQPYRQVVGFFESDESKYSNDIDFPGFMVEVPNGIYYGFPSFGGCGLKLGYHTFGQKIDPDTINREFGVYPEDESNLRAFLEEYMPGANGELKRGAVCMYTKTLDEHFIIDLHPEHSNVVIAAGFSGHGFKFSSGVGEVLSQLALTGKTEHDISIFSINRPALKESLQ Hydrogen bonds contact Hydrophobic contact | ||||
| 25 | Opioid receptor sigma 1 (OPRS1) | 5HK1 | 6.77 | |
Target general information Gen name SIGMAR1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms hSigmaR1; Sigma1R; Sigma1-receptor; Sigma non-opioid intracellular receptor 1; Sigma 1-type opioid receptor; SRBP; SR31747-binding protein; SR31747 binding protein 1; SR-BP; SIG-1R; Opioid receptor, s Protein family ERG2 family Biochemical class GPCR rhodopsin Function Involved in the regulation of different receptors it plays a role in BDNF signaling and EGF signaling. Also regulates ion channels like the potassium channel and could modulate neurotransmitter release. Plays a role in calcium signaling through modulation together with ANK2 of the ITP3R-dependent calcium efflux at the endoplasmic reticulum. Plays a role in several other cell functions including proliferation, survival and death. Originally identified for its ability to bind various psychoactive drugs it is involved in learning processes, memory and mood alteration. Necessary for proper mitochondrial axonal transport in motor neurons, in particular the retrograde movement of mitochondria. Plays a role in protecting cells against oxidative stress-induced cell death via its interaction with RNF112. Functions in lipid transport from the endoplasmic reticulum and is involved in a wide array of cellular functions probably through regulation of the biogenesis of lipid microdomains at the plasma membrane. Related diseases Amyotrophic lateral sclerosis 16, juvenile (ALS16) [MIM:614373]: A neurodegenerative disorder affecting upper motor neurons in the brain and lower motor neurons in the brain stem and spinal cord, resulting in fatal paralysis. Sensory abnormalities are absent. The pathologic hallmarks of the disease include pallor of the corticospinal tract due to loss of motor neurons, presence of ubiquitin-positive inclusions within surviving motor neurons, and deposition of pathologic aggregates. The etiology of amyotrophic lateral sclerosis is likely to be multifactorial, involving both genetic and environmental factors. The disease is inherited in 5-10% of the cases. {ECO:0000269|PubMed:21842496}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Neuronopathy, distal hereditary motor, autosomal recessive 2 (HMNR2) [MIM:605726]: A form of distal hereditary motor neuronopathy, a heterogeneous group of neuromuscular disorders caused by selective degeneration of motor neurons in the anterior horn of the spinal cord, without sensory deficit in the posterior horn. The overall clinical picture consists of a classical distal muscular atrophy syndrome in the legs without clinical sensory loss. The disease starts with weakness and wasting of distal muscles of the anterior tibial and peroneal compartments of the legs. Later on, weakness and atrophy may expand to the proximal muscles of the lower limbs and/or to the distal upper limbs. HMNR2 is characterized by onset of distal muscle weakness and wasting affecting the lower and upper limbs in the first decade. {ECO:0000269|PubMed:26078401, ECO:0000269|PubMed:27629094}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00321; DB09014; DB00907; DB00514; DB01488; DB00574; DB00502; DB00956; DB00704; DB00540; DB06174; DB00652; DB11186; DB03575; DB05316; DB01708; DB00409; DB01104 Interacts with Q92847-1; Q99720-1; O00213-2; P17612; P50454; P37173 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Amyotrophic lateral sclerosis; Cell junction; Cell membrane; Cell projection; Cytoplasmic vesicle; Direct protein sequencing; Disease variant; Endoplasmic reticulum; Lipid droplet; Lipid transport; Membrane; Neurodegeneration; Neuropathy; Nucleus; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Synapse; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A,B,C Molecular weight (Da) 20805.3 Length 185 Aromaticity 0.14 Instability index 31.72 Isoelectric point 5.44 Charge (pH=7) -6.63 2D Binding mode Binding energy (Kcal/mol) -9.23 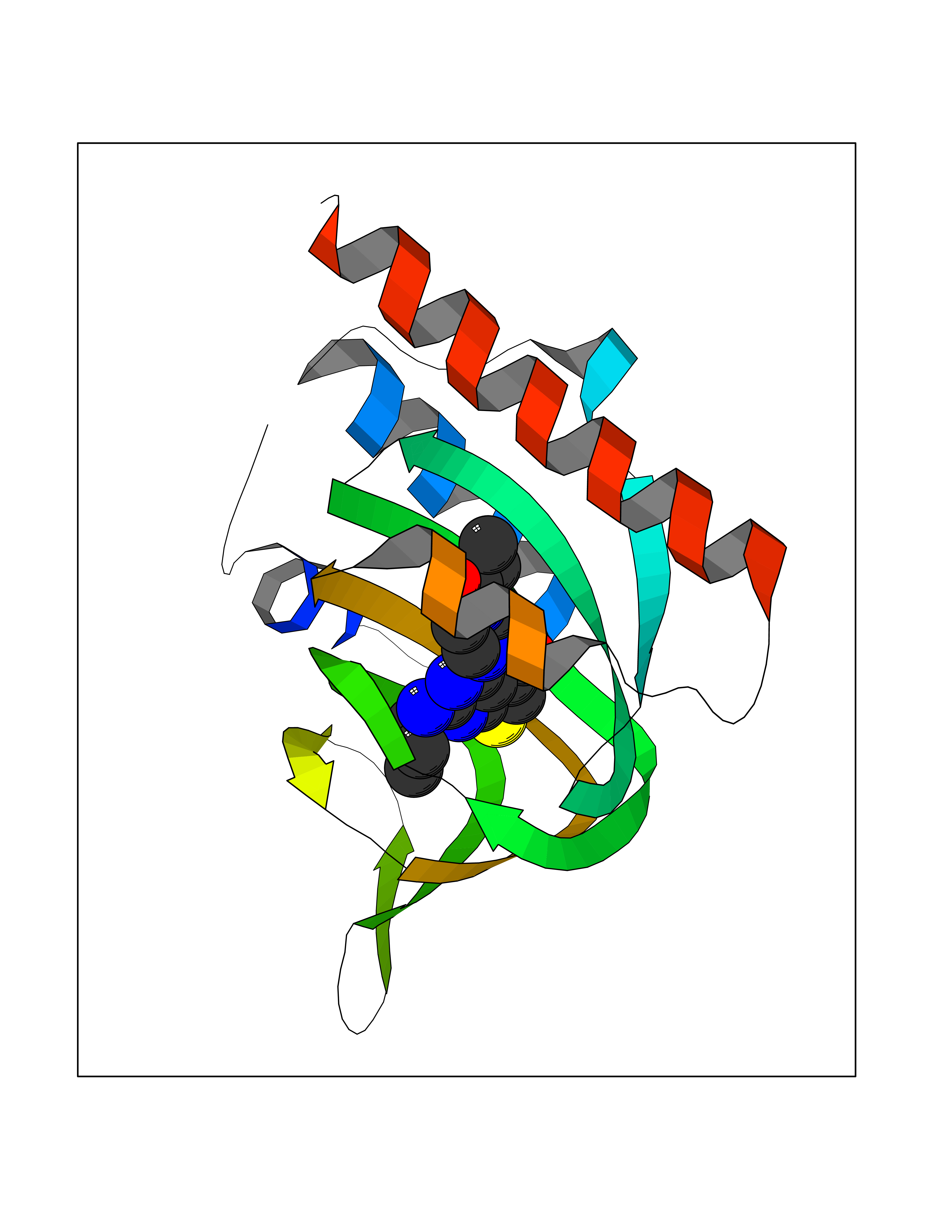 Molscript Map 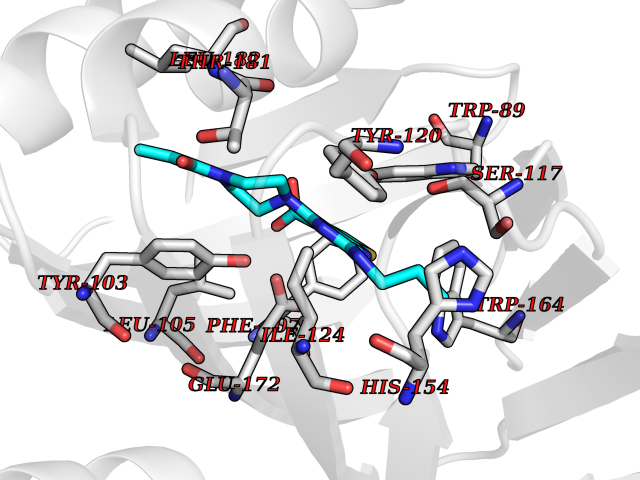 Pymol Map 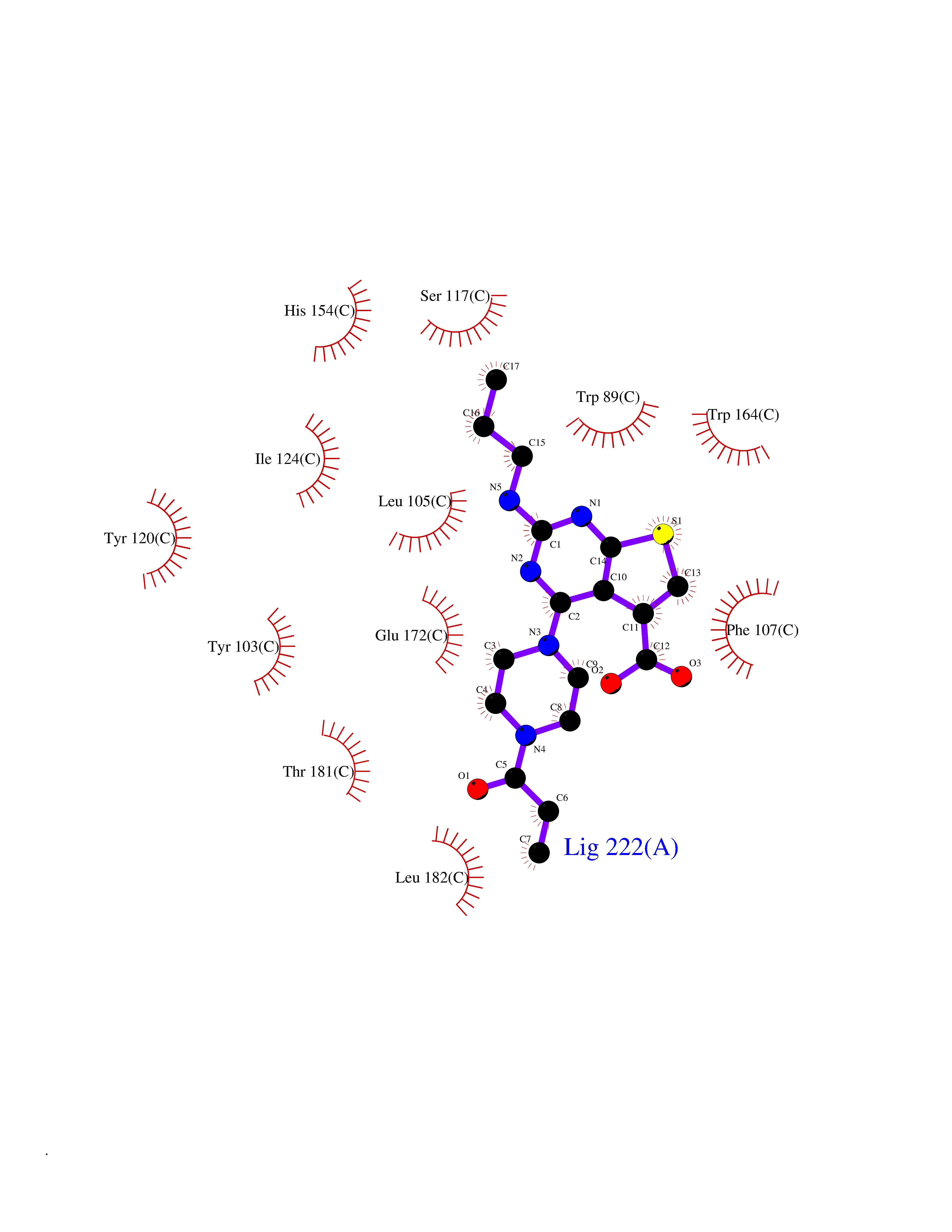 Ligplot Map 3D Binding mode Sequence VFQREEIAQLARQYAGLDHELAFSRLIVELRRLHPGHVLPDEELQWVFVNAGGWMGAMCLLHASLSEYVLLFGTALGSRGHSGRYWAEISDTIISGTFHQWREGTTKSEVFYPGETVVHGPGEATAVEWGPNTWMVEYGRGVIPSTLAFALADTVFSTQDFLTLFYTLRSYARGLRLELTTYLFG Hydrogen bonds contact Hydrophobic contact | ||||
| 26 | Cytochrome c oxidase subunit 2 | 3VRJ | 6.75 | |
Target general information Gen name MT-CO2 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms MTCO2;COXII;COII;COX2 Protein family Cytochrome c oxidase subunit 2 family Biochemical class Immune system Function Copper ion binding.Cytochrome-c oxidase activity. Related diseases Mitochondrial complex IV deficiency (MT-C4D) [MIM:220110]: A disorder of the mitochondrial respiratory chain with heterogeneous clinical manifestations, ranging from isolated myopathy to severe multisystem disease affecting several tissues and organs. Features include hypertrophic cardiomyopathy, hepatomegaly and liver dysfunction, hypotonia, muscle weakness, exercise intolerance, developmental delay, delayed motor development and intellectual disability. Some affected individuals manifest a fatal hypertrophic cardiomyopathy resulting in neonatal death. A subset of patients manifest Leigh syndrome. {ECO:0000269|PubMed:10486321}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02659; DB04464; DB05412 Interacts with Q9NZ94-2; P49281-3 EC number 7.1.1.9 Uniprot keywords 3D-structure; Copper; Disease variant; Electron transport; Magnesium; Membrane; Metal-binding; Mitochondrion; Mitochondrion inner membrane; Primary mitochondrial disease; Proteomics identification; Reference proteome; Respiratory chain; Translocase; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID C Molecular weight (Da) 21687.9 Length 189 Aromaticity 0.11 Instability index 38 Isoelectric point 5.68 Charge (pH=7) -3.26 2D Binding mode Binding energy (Kcal/mol) -9.2 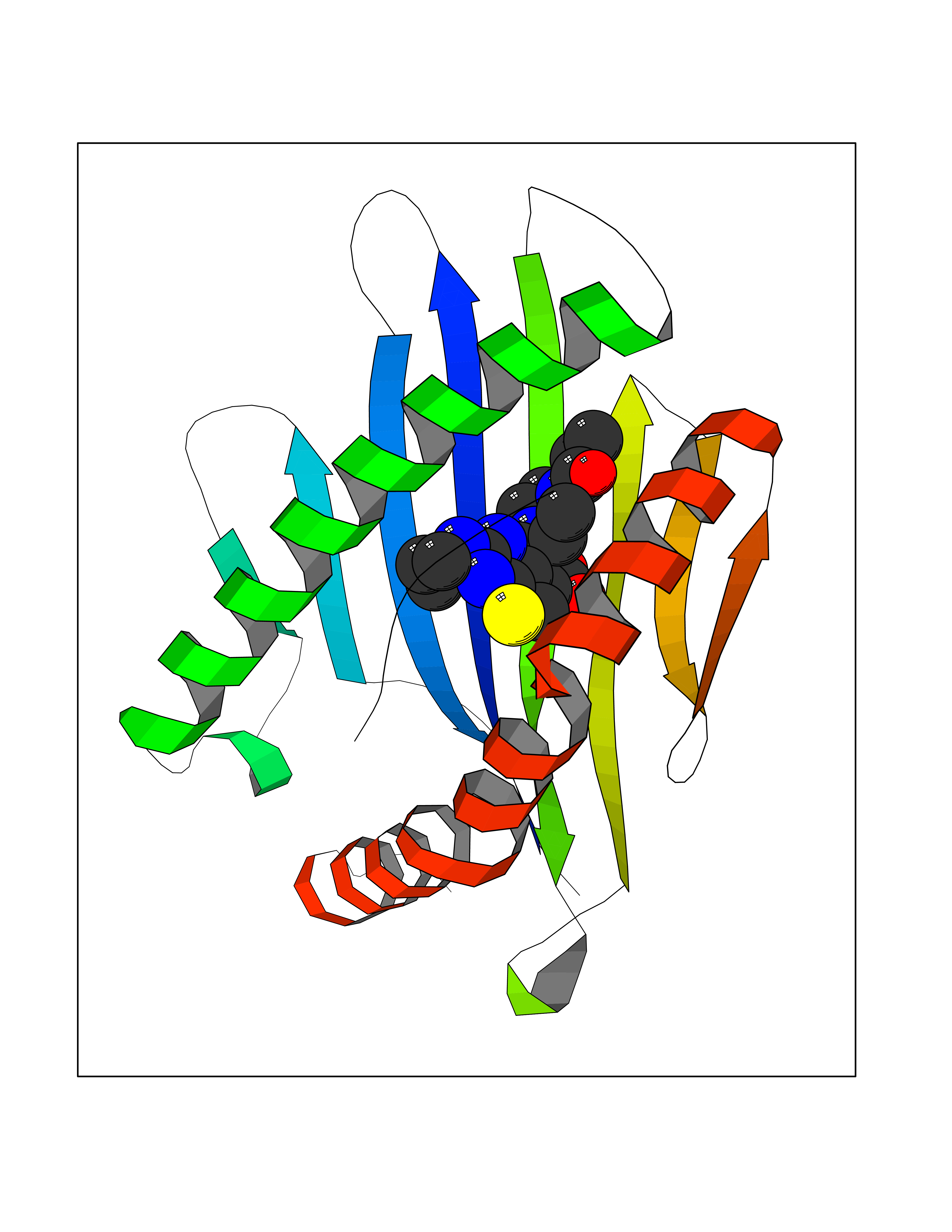 Molscript Map 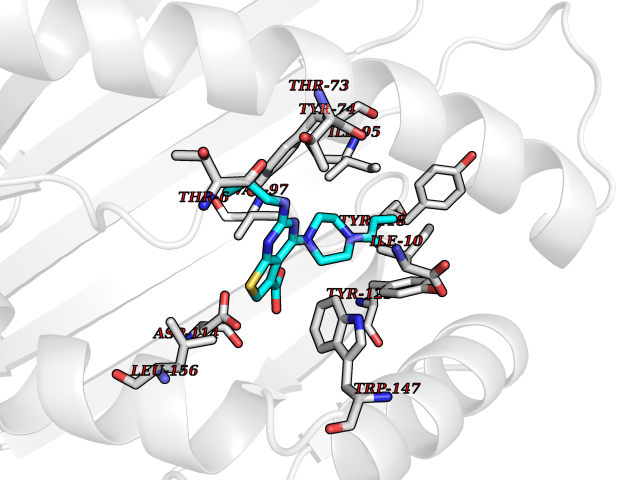 Pymol Map  Ligplot Map 3D Binding mode Sequence SHSMRYFYTAMSRPGRGEPRFIAVGYVDDTQFVRFDSDAASPRMAPRAPWIEQEGPEYWDGETRNMKASAQTYRENLRIALRYYNQSEAGSHIIQVMYGCDVGPDGRLLRGHDQSAYDGKDYIALNEDLSSWTAADTAAQITQRKWEAARVAEQLRAYLEGLCVEWLRRYLENGKETLQLTTKLTNTNI Hydrogen bonds contact Hydrophobic contact | ||||
| 27 | Adrenergic receptor alpha-2A (ADRA2A) | 7EJ8 | 6.75 | |
Target general information Gen name ADRA2A Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Alpha-2AAR; Alpha-2A adrenoreceptor; Alpha-2A adrenoceptor; Alpha-2A adrenergic receptor; Alpha-2 adrenergic receptor subtype C10; ADRAR; ADRA2R Protein family G-protein coupled receptor 1 family, Adrenergic receptor subfamily, ADRA2A sub-subfamily Biochemical class GPCR rhodopsin Function The rank order of potency for agonists of this receptor is oxymetazoline > clonidine > epinephrine > norepinephrine > phenylephrine > dopamine > p-synephrine > p-tyramine > serotonin = p-octopamine. For antagonists, the rank order is yohimbine > phentolamine = mianserine > chlorpromazine = spiperone = prazosin > propanolol > alprenolol = pindolol. Alpha-2 adrenergic receptors mediate the catecholamine-induced inhibition of adenylate cyclase through the action of G proteins. Related diseases Lipodystrophy, familial partial, 8 (FPLD8) [MIM:620679]: An autosomal dominant form of partial lipodystrophy, a disorder characterized by abnormal subcutaneous fat distribution. FPLD8 patients show selective loss of subcutaneous adipose tissue from the limbs, beginning around 13 to 15 years of age, and abnormal accumulation of subcutaneous adipose tissue in the dorsal neck and face, as well as in the posterior thoracic and abdominal regions. The disorder is associated with metabolic abnormalities, including diabetes mellitus and hyperlipidemia. {ECO:0000269|PubMed:27376152}. The disease may be caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01472; DB00321; DB00543; DB00182; DB00714; DB00964; DB09229; DB01238; DB14185; DB06216; DB00865; DB00217; DB00484; DB01200; DB00248; DB01136; DB04846; DB00477; DB09202; DB00575; DB00363; DB01151; DB00633; DB01576; DB11273; DB13345; DB00320; DB00449; DB11278; DB09167; DB04855; DB06262; DB01363; DB05492; DB00751; DB00668; DB01049; DB00696; DB01175; DB06678; DB09194; DB00800; DB06623; DB00629; DB01018; DB00502; DB11577; DB00555; DB06707; DB00589; DB04948; DB09195; DB00408; DB08815; DB00934; DB01365; DB01577; DB01403; DB00968; DB06148; DB00370; DB09205; DB09242; DB06711; DB01149; DB00368; DB00540; DB06229; DB00935; DB01267; DB00715; DB01186; DB01608; DB00925; DB00692; DB00397; DB09286; DB09244; DB06153; DB00413; DB00457; DB00433; DB01069; DB00852; DB01224; DB11124; DB11738; DB00268; DB09304; DB06764; DB13025; DB00697; DB00797; DB00193; DB00656; DB00726; DB11477; DB06694; DB01392; DB00246; DB01624 Interacts with NA EC number NA Uniprot keywords 3D-structure; Cell membrane; Direct protein sequencing; Disulfide bond; G-protein coupled receptor; Glycoprotein; Lipoprotein; Membrane; Methylation; Palmitate; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID R Molecular weight (Da) 30303.9 Length 263 Aromaticity 0.16 Instability index 35.08 Isoelectric point 9.66 Charge (pH=7) 16.82 2D Binding mode Binding energy (Kcal/mol) -9.21  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence YSLQVTLTLVCLAGLLMLLTVFGNVLVIIAVFTSRALKAPQNLFLVSLASADILVATLVIPFSLANEVMGYWYFGKAWCEIYLALDVLFCTSSIVHLCAISLDRYWSITQAIEYNLKRTPRRIKAIIITVWVISAVISFPPRCEINDQKWYVISSCIGSFFAPCLIMILVYVRIYQIAKRRTRRGRQNREKRFTFVLAVVIGVFVVCWFPFFFTYTLTAVGCSVPRTLFKFFFWFGYCNSSLNPVIYTIFNHDFRRAFKKILC Hydrogen bonds contact Hydrophobic contact | ||||
| 28 | Glycine oxidase | 1RYI | 6.74 | |
Target general information Gen name thiO Organism Bacillus subtilis (strain 168) Uniprot ID TTD ID NA Synonyms goxB;yjbR;BSU11670 Protein family DAO family, ThiO subfamily Biochemical class Oxidoreductase Function FAD binding.Glycine oxidase activity.Oxidoreductase activity. Related diseases Hypervalinemia and hyperleucine-isoleucinemia (HVLI) [MIM:618850]: An autosomal recessive metabolic disorder characterized by highly elevated plasma concentrations of valine and leucine/isoleucine. Affected individuals suffer from headache and mild memory impairment. {ECO:0000269|PubMed:25653144}. The disease is caused by variants affecting the gene represented in this entry. A patient with hypervalinemia and hyperleucine-isoleucinemia was identified as compound heterozygote for Gln-170 (inherited from his father) and Lys-264 (inherited from his mother), both variants reduced the catalytic activity of the enzyme. After treatment with vitamin B6, a precursor of pyridoxal 5'-phosphate, a BCAT2 cofactor, the blood levels of branched chain amino acids, especially valine, were decreased and brain lesions were improved. {ECO:0000269|PubMed:25653144}. Drugs (DrugBank ID) DB02713; DB03147 Interacts with NA EC number 1.4.3.19 Uniprot keywords 3D-structure; Cytoplasm; FAD; Flavoprotein; Herbicide resistance; Oxidoreductase; Reference proteome; Thiamine biosynthesis Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 40395.8 Length 364 Aromaticity 0.1 Instability index 36.07 Isoelectric point 6.02 Charge (pH=7) -6.35 2D Binding mode Binding energy (Kcal/mol) -9.2 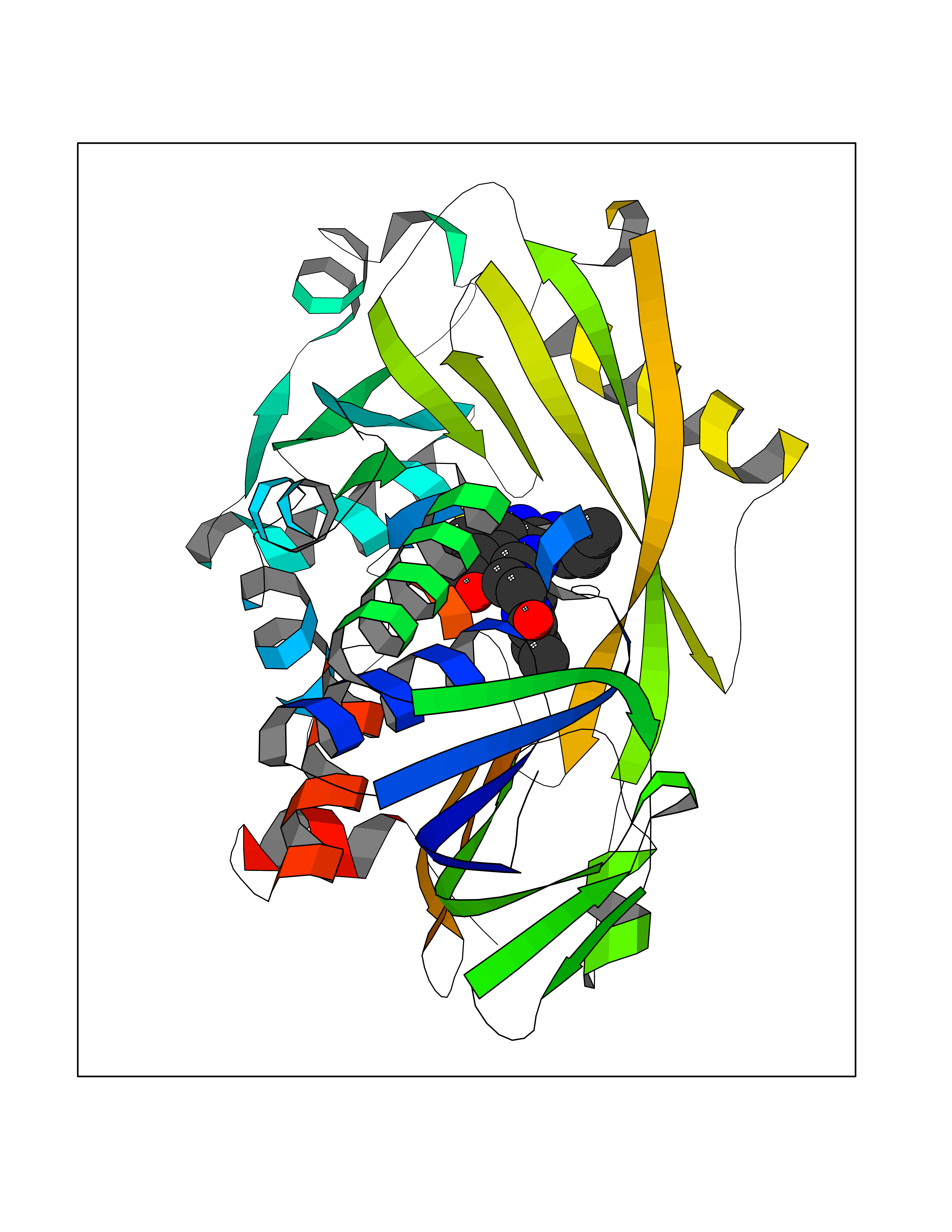 Molscript Map 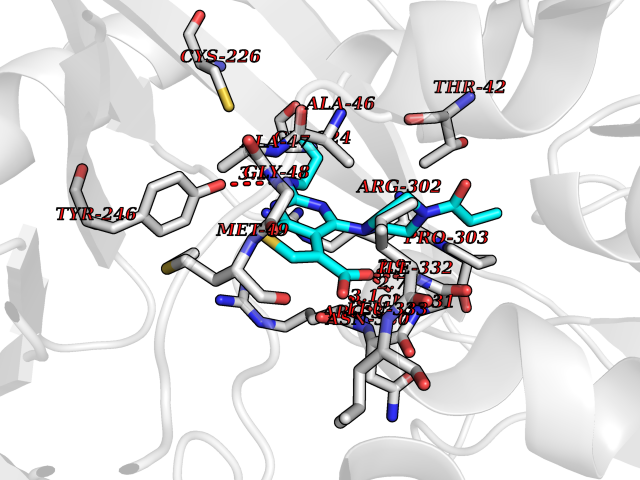 Pymol Map 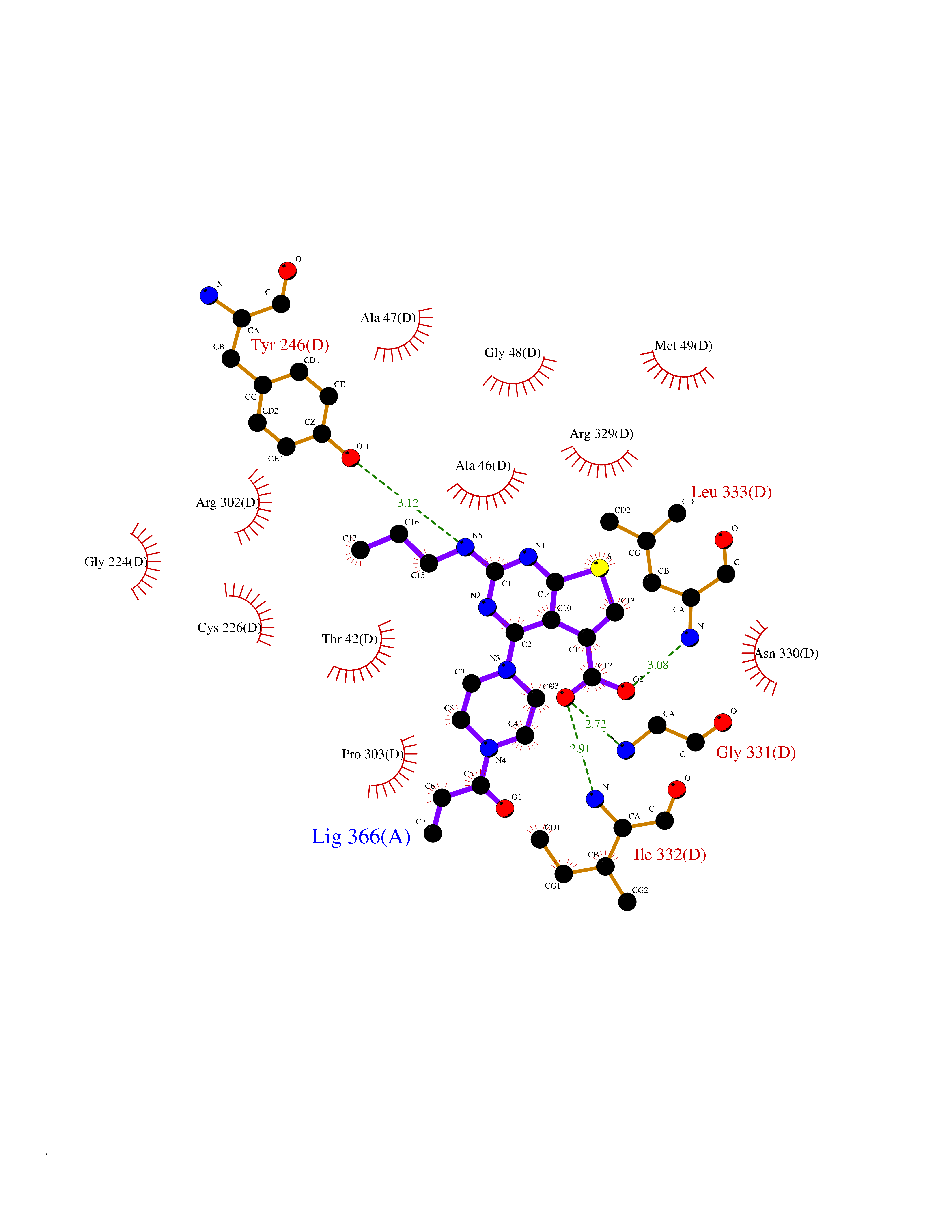 Ligplot Map 3D Binding mode Sequence MKRHYEAVVIGGGIIGSAIAYYLAKENKNTALFESGTMGGRTTSAAAGMLGAHAECEERDAFFDFAMHSQRLYKGLGEELYALSGVDIRQHNGGMFKLAFSEEDVLQLRQMDDLDSVSWYSKEEVLEKEPYASGDIFGASFIQDDVHVEPYFVCKAYVKAAKMLGAEIFEHTPVLHVERDGEALFIKTPSGDVWANHVVVASGVWSGMFFKQLGLNNAFLPVKGECLSVWNDDIPLTKTLYHDHCYIVPRKSGRLVVGATMKPGDWSETPDLGGLESVMKKAKTMLPAIQNMKVDRFWAGLRPGTKDGKPYIGRHPEDSRILFAAGHFRNGILLAPATGALISDLIMNKEVNQDWLHAFRIDRK Hydrogen bonds contact Hydrophobic contact | ||||
| 29 | Monoamine oxidase type B (MAO-B) | 2V5Z | 6.74 | |
Target general information Gen name MAOB Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms MAO-B; Amine oxidase [flavin-containing] B Protein family Flavin monoamine oxidase family Biochemical class CH-NH(2) donor oxidoreductase Function Catalyzes the oxidative deamination of biogenic and xenobiotic amines and has important functions in the metabolism of neuroactive and vasoactive amines in the central nervous system and peripheral tissues. MAOB preferentially degrades benzylamine and phenylethylamine. Related diseases Microvascular complications of diabetes 5 (MVCD5) [MIM:612633]: Pathological conditions that develop in numerous tissues and organs as a consequence of diabetes mellitus. They include diabetic retinopathy, diabetic nephropathy leading to end-stage renal disease, and diabetic neuropathy. Diabetic retinopathy remains the major cause of new-onset blindness among diabetic adults. It is characterized by vascular permeability and increased tissue ischemia and angiogenesis. Disease susceptibility is associated with variants affecting the gene represented in this entry. Homozygosity for the Leu-55 allele is strongly associated with the development of retinal disease in diabetic patients. Drugs (DrugBank ID) DB08176; DB02211; DB08516; DB08480; DB01472; DB04307; DB07512; DB07513; DB00915; DB00182; DB06698; DB04889; DB00215; DB09130; DB04147; DB00988; DB01363; DB00668; DB01175; DB02509; DB03147; DB14914; DB00614; DB04818; DB02095; DB01247; DB00601; DB01577; DB01442; DB01171; DB08082; DB02643; DB04677; DB03894; DB08804; DB04820; DB00184; DB04821; DB12612; DB01626; DB00780; DB00191; DB00388; DB01132; DB00721; DB01168; DB01367; DB09363; DB06654; DB01037; DB01104; DB14569; DB09042; DB00752; DB16446; DB09185; DB04832; DB00909 Interacts with P55212; P28329-3; Q8NI60; Q5RI15; Q92915-2; P22607; Q53GS7; P06396; P01112; O14901; P13473-2; P21397; Q9BVL2; O75400-2; P62826; Q6NTF9-3; Q9Y371; Q7Z699; Q9UMX0; Q9Y649 EC number EC 1.4.3.4 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Direct protein sequencing; FAD; Flavoprotein; Membrane; Mitochondrion; Mitochondrion outer membrane; Oxidoreductase; Proteomics identification; Reference proteome; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A,B Molecular weight (Da) 56019.9 Length 494 Aromaticity 0.09 Instability index 34.81 Isoelectric point 6.51 Charge (pH=7) -2.2 2D Binding mode Binding energy (Kcal/mol) -9.2 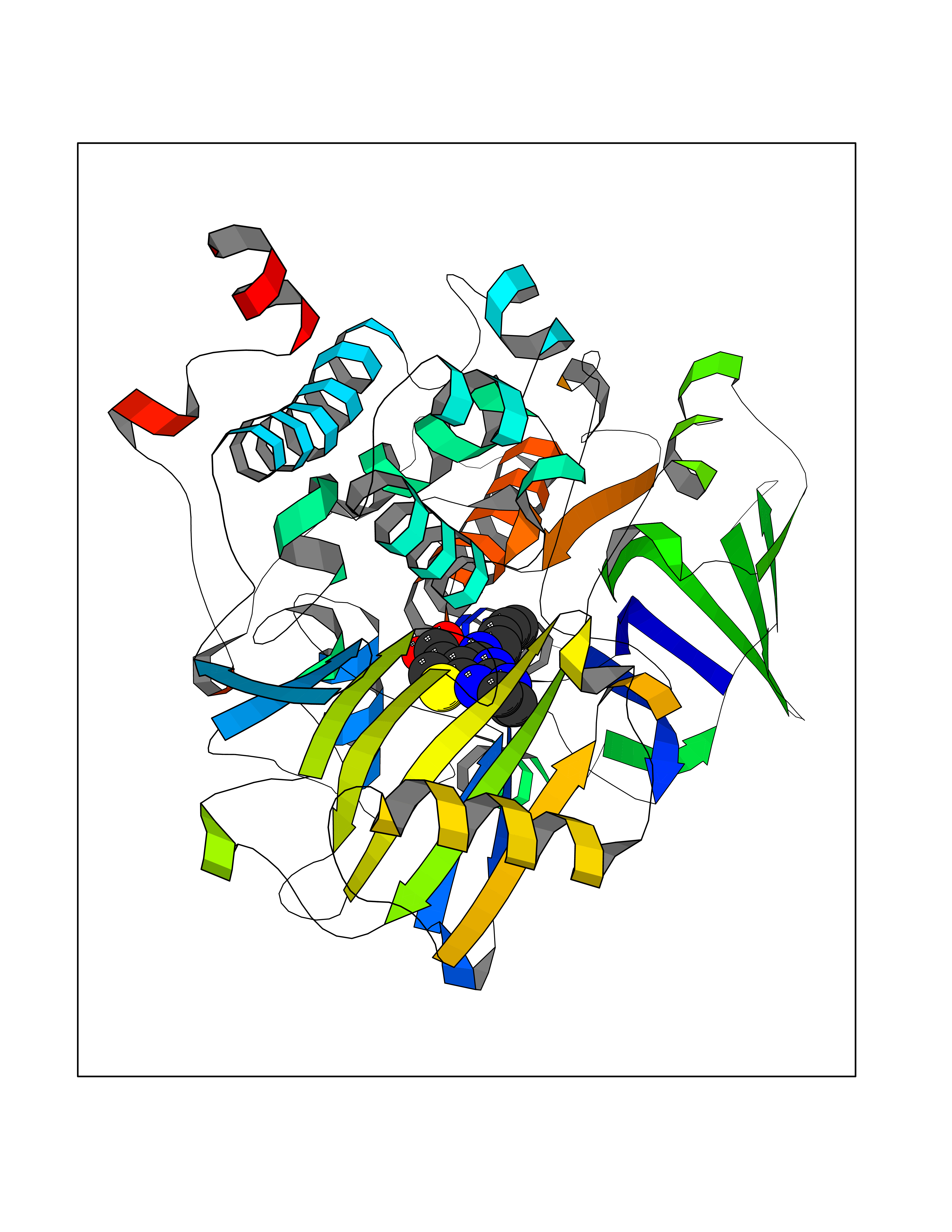 Molscript Map 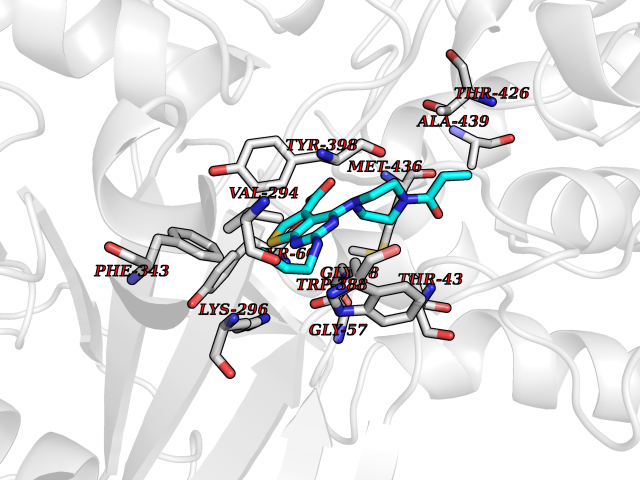 Pymol Map 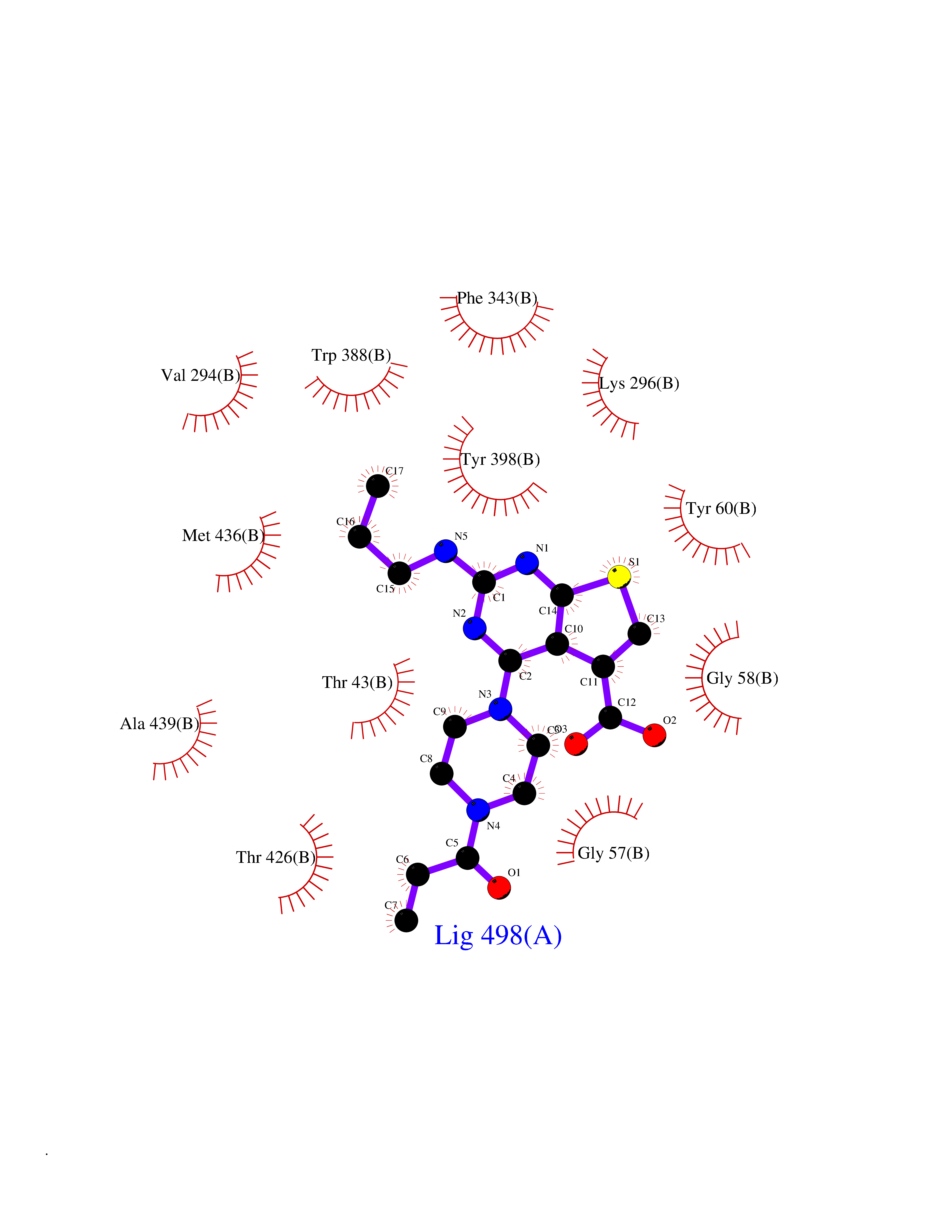 Ligplot Map 3D Binding mode Sequence NKCDVVVVGGGISGMAAAKLLHDSGLNVVVLEARDRVGGRTYTLRNQKVKYVDLGGSYVGPTQNRILRLAKELGLETYKVNEVERLIHHVKGKSYPFRGPFPPVWNPITYLDHNNFWRTMDDMGREIPSDAPWKAPLAEEWDNMTMKELLDKLCWTESAKQLATLFVNLCVTAETHEVSALWFLWYVKQCGGTTRIISTTNGGQERKFVGGSGQVSERIMDLLGDRVKLERPVIYIDQTRENVLVETLNHEMYEAKYVISAIPPTLGMKIHFNPPLPMMRNQMITRVPLGSVIKCIVYYKEPFWRKKDYCGTMIIDGEEAPVAYTLDDTKPEGNYAAIMGFILAHKARKLARLTKEERLKKLCELYAKVLGSLEALEPVHYEEKNWCEEQYSGGCYTTYFPPGILTQYGRVLRQPVDRIYFAGTETATHWSGYMEGAVEAGERAAREILHAMGKIPEDEIWQSEPESVDVPAQPITTTFLERHLPSVPGLLRLI Hydrogen bonds contact Hydrophobic contact | ||||
| 30 | T-cell receptor beta constant 1 (TRBC1) | 4LCC | 6.74 | |
Target general information Gen name TRBC1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms TCRBC1; BV05S1J2.2 Protein family NA Biochemical class NA Function Alpha-beta T cell receptors are antigen specific receptors which are essential to the immune response and are present on the cell surface of T lymphocytes. Recognize peptide-major histocompatibility (MH) (pMH) complexes that are displayed by antigen presenting cells (APC), a prerequisite for efficient T cell adaptive immunity against pathogens. Binding of alpha-beta TR to pMH complex initiates TR-CD3 clustering on the cell surface and intracellular activation of LCK that phosphorylates the ITAM motifs of CD3G, CD3D, CD3E and CD247 enabling the recruitment of ZAP70. In turn, ZAP70 phosphorylates LAT, which recruits numerous signaling molecules to form the LAT signalosome. The LAT signalosome propagates signal branching to three major signaling pathways, the calcium, the mitogen-activated protein kinase (MAPK) kinase and the nuclear factor NF-kappa-B (NF-kB) pathways, leading to the mobilization of transcription factors that are critical for gene expression and essential for T cell growth and differentiation. The T cell repertoire is generated in the thymus, by V-(D)-J rearrangement. This repertoire is then shaped by intrathymic selection events to generate a peripheral T cell pool of self-MH restricted, non-autoaggressive T cells. Post-thymic interaction of alpha-beta TR with the pMH complexes shapes TR structural and functional avidity. Constant region of T cell receptor (TR) alpha chain. Related diseases Immunodeficiency 57 with autoinflammation (IMD57) [MIM:618108]: An autosomal recessive primary immunodeficiency characterized by lymphopenia and recurrent viral, bacterial, and fungal infections. Patients exhibit early-onset inflammatory bowel disease involving the upper and lower gastrointestinal tract, and develop progressive polyarthritis. {ECO:0000269|PubMed:30026316}. The disease is caused by variants affecting the gene represented in this entry. RIPK1-deficient immune cells from IMD57 patients have impaired proinflammatory signaling leading to dysregulated cytokine secretion and are prone to necroptosis. {ECO:0000269|PubMed:30026316}.; DISEASE: Autoinflammation with episodic fever and lymphadenopathy (AIEFL) [MIM:618852]: An autosomal dominant immunologic disorder characterized by early onset of recurrent episodes of unexplained fever, lymphadenopathy, hepatosplenomegaly, and increased levels of inflammatory cytokines and chemokines in patient serum. {ECO:0000269|PubMed:31827280, ECO:0000269|PubMed:31827281}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02740 Interacts with NA EC number NA Uniprot keywords 3D-structure; Adaptive immunity; Cell membrane; Disulfide bond; Glycoprotein; Immunity; Immunoglobulin domain; Membrane; Proteomics identification; Receptor; Reference proteome; T cell receptor; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID C,B,A Molecular weight (Da) 84668.2 Length 736 Aromaticity 0.13 Instability index 39.96 Isoelectric point 5.8 Charge (pH=7) -14.36 2D Binding mode Binding energy (Kcal/mol) -9.2 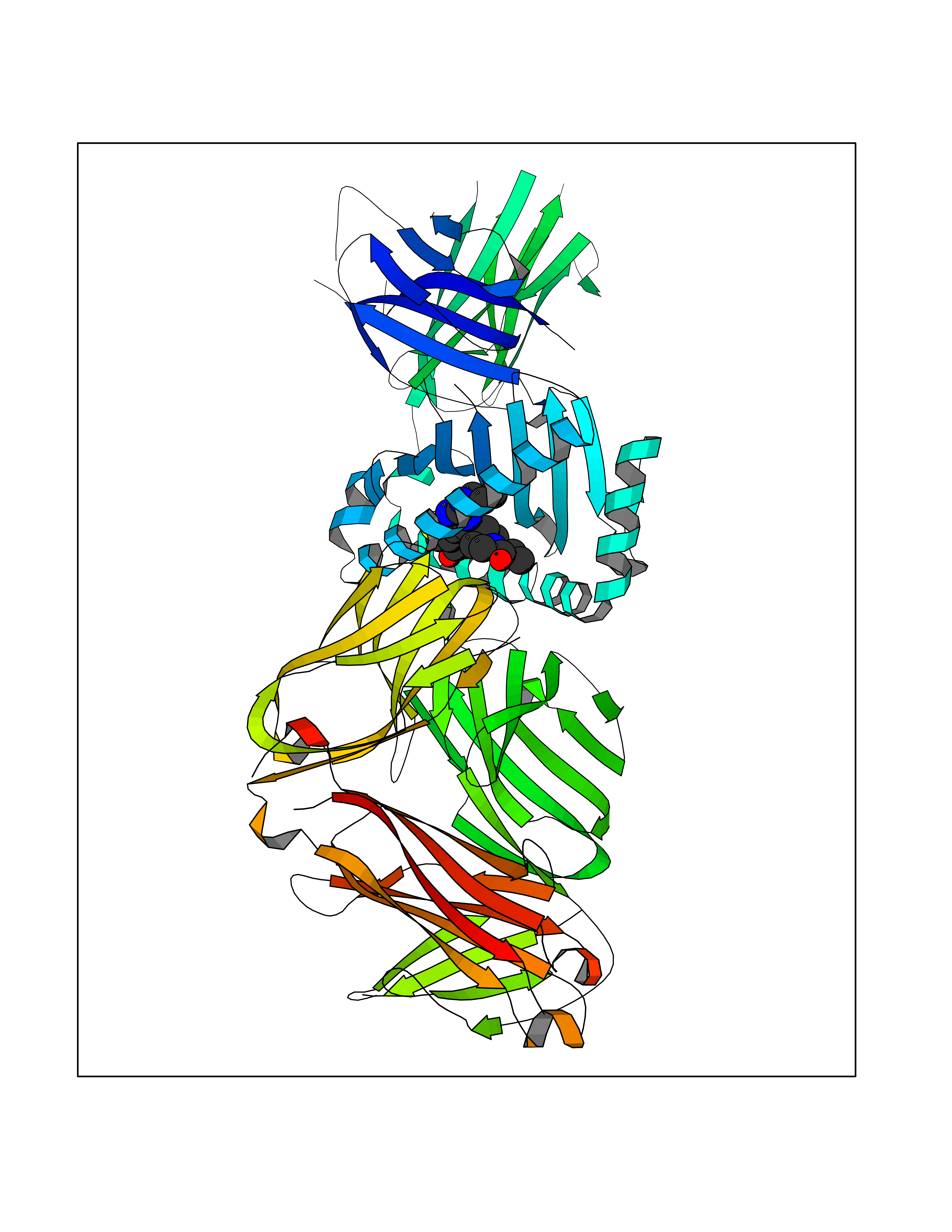 Molscript Map 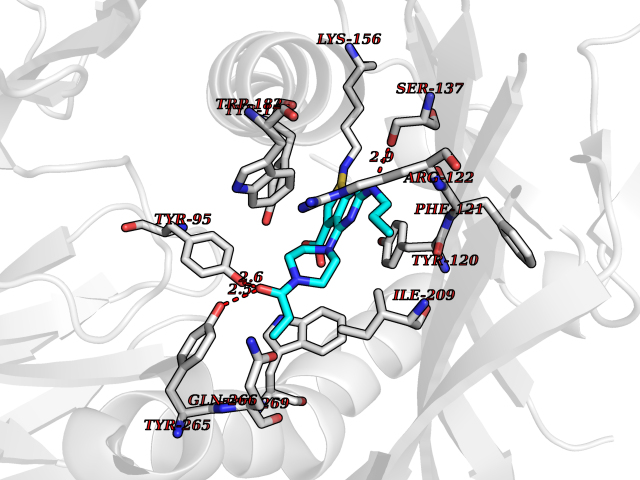 Pymol Map 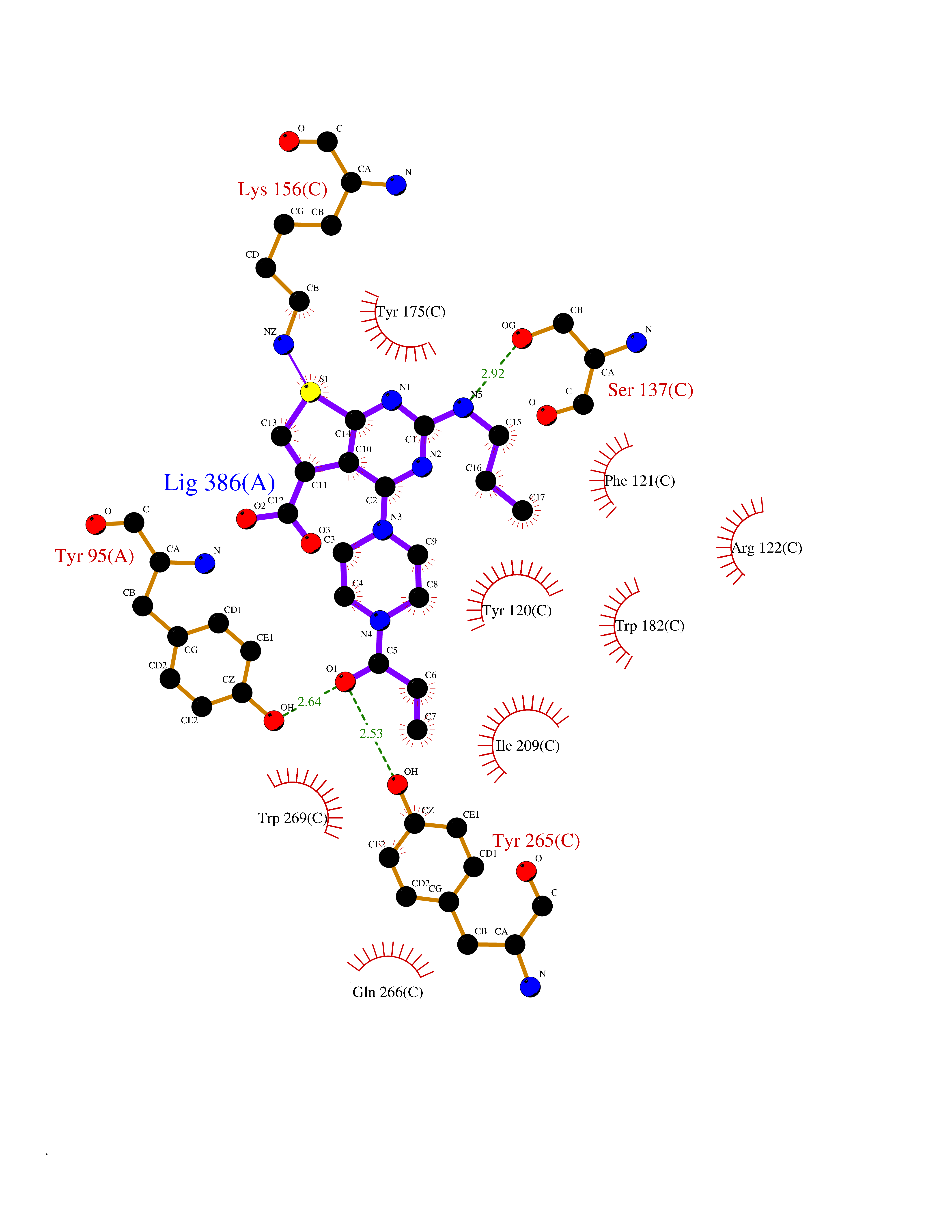 Ligplot Map 3D Binding mode Sequence IQRPPKIQVYSRHPPNYLNCYVYGFHPPQIEIDLLKIKSEQSDLSFSKDWSFYLLSHATPNSKDQYSCRVKHVTLEQPRIVKWDRTHSLRYFRLGISEIPEFISAGYVDSHPITMYNSVSQLKEPRALWMEENLAPDHWERYTQLLRGWQQMFKVELKQLQHHYNHSGFHTYQRMIGCELLEDGSITGFLQYAYDGQDFLIFNKDTLSWMAMDNVADIIRRVWEANQHELLYQKNWLEEECIAWLKRFLEYGKDALQRTEPPKVRVNHKTTLYCRAYGFYPPEISINWMKNGEEIFQDTDYGGILPSGDGTYQTWVSVELGDIYSCHVEHGGVHMVLQGFQQNIDQPTEMTATEGAIVQINCTYQTSGFNGLFWYQQHAGEAPTFLSYNVLDGLEEKGRFSSFLSRSKGYSYLLLKELQMKDSASYLCAVKDSNYQLIWGAGTKLIIKPNIQNPDPAVYQLRDSKSSDKSVCLFTDFDKDSDVYITDKKSNSAVAWSNAGVTQTPKFQVLKTGQSMTLQCAQDMNHNSMYWYRQDPGMGLRLIYYSASEGTTDKGEVPNGYNVSRLNKREFSLRLESAAPSQTSVYFCASSVWTGEGSGELFFGEGSRLTVLEDLKNVFPPEVAVFEPSEAEISHTQKATLVCLATGFYPDHVELSWWVNGKEVHSGVCTDPQPLKEQPALNDSRYALSSRLRVSATFWQNPRNHFRCQVQFYGLSENDEWKPVTQIVSAEAWGRA Hydrogen bonds contact Hydrophobic contact | ||||
| 31 | Monoamine oxidase type B (MAO-B) | 2V5Z | 6.73 | |
Target general information Gen name MAOB Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms MAO-B; Amine oxidase [flavin-containing] B Protein family Flavin monoamine oxidase family Biochemical class CH-NH(2) donor oxidoreductase Function Catalyzes the oxidative deamination of biogenic and xenobiotic amines and has important functions in the metabolism of neuroactive and vasoactive amines in the central nervous system and peripheral tissues. MAOB preferentially degrades benzylamine and phenylethylamine. Related diseases Microvascular complications of diabetes 5 (MVCD5) [MIM:612633]: Pathological conditions that develop in numerous tissues and organs as a consequence of diabetes mellitus. They include diabetic retinopathy, diabetic nephropathy leading to end-stage renal disease, and diabetic neuropathy. Diabetic retinopathy remains the major cause of new-onset blindness among diabetic adults. It is characterized by vascular permeability and increased tissue ischemia and angiogenesis. Disease susceptibility is associated with variants affecting the gene represented in this entry. Homozygosity for the Leu-55 allele is strongly associated with the development of retinal disease in diabetic patients. Drugs (DrugBank ID) DB08176; DB02211; DB08516; DB08480; DB01472; DB04307; DB07512; DB07513; DB00915; DB00182; DB06698; DB04889; DB00215; DB09130; DB04147; DB00988; DB01363; DB00668; DB01175; DB02509; DB03147; DB14914; DB00614; DB04818; DB02095; DB01247; DB00601; DB01577; DB01442; DB01171; DB08082; DB02643; DB04677; DB03894; DB08804; DB04820; DB00184; DB04821; DB12612; DB01626; DB00780; DB00191; DB00388; DB01132; DB00721; DB01168; DB01367; DB09363; DB06654; DB01037; DB01104; DB14569; DB09042; DB00752; DB16446; DB09185; DB04832; DB00909 Interacts with P55212; P28329-3; Q8NI60; Q5RI15; Q92915-2; P22607; Q53GS7; P06396; P01112; O14901; P13473-2; P21397; Q9BVL2; O75400-2; P62826; Q6NTF9-3; Q9Y371; Q7Z699; Q9UMX0; Q9Y649 EC number EC 1.4.3.4 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Direct protein sequencing; FAD; Flavoprotein; Membrane; Mitochondrion; Mitochondrion outer membrane; Oxidoreductase; Proteomics identification; Reference proteome; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A,B Molecular weight (Da) 56019.9 Length 494 Aromaticity 0.09 Instability index 34.81 Isoelectric point 6.51 Charge (pH=7) -2.2 2D Binding mode Binding energy (Kcal/mol) -9.18 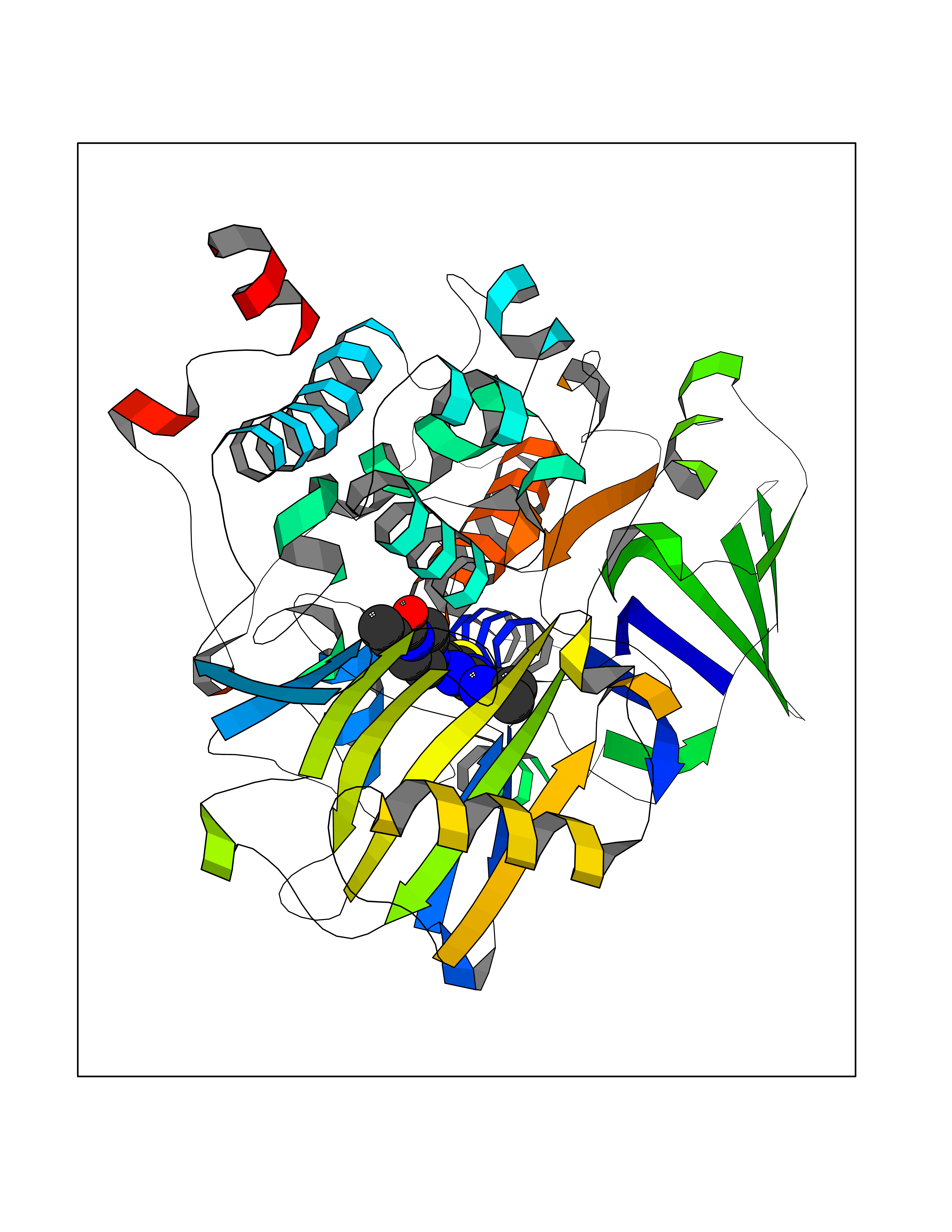 Molscript Map 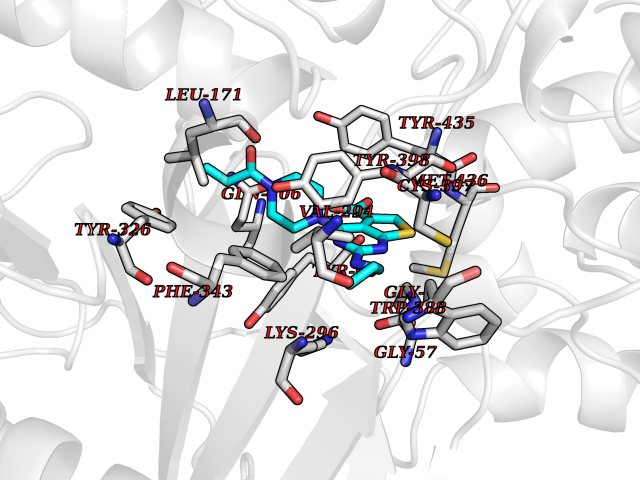 Pymol Map 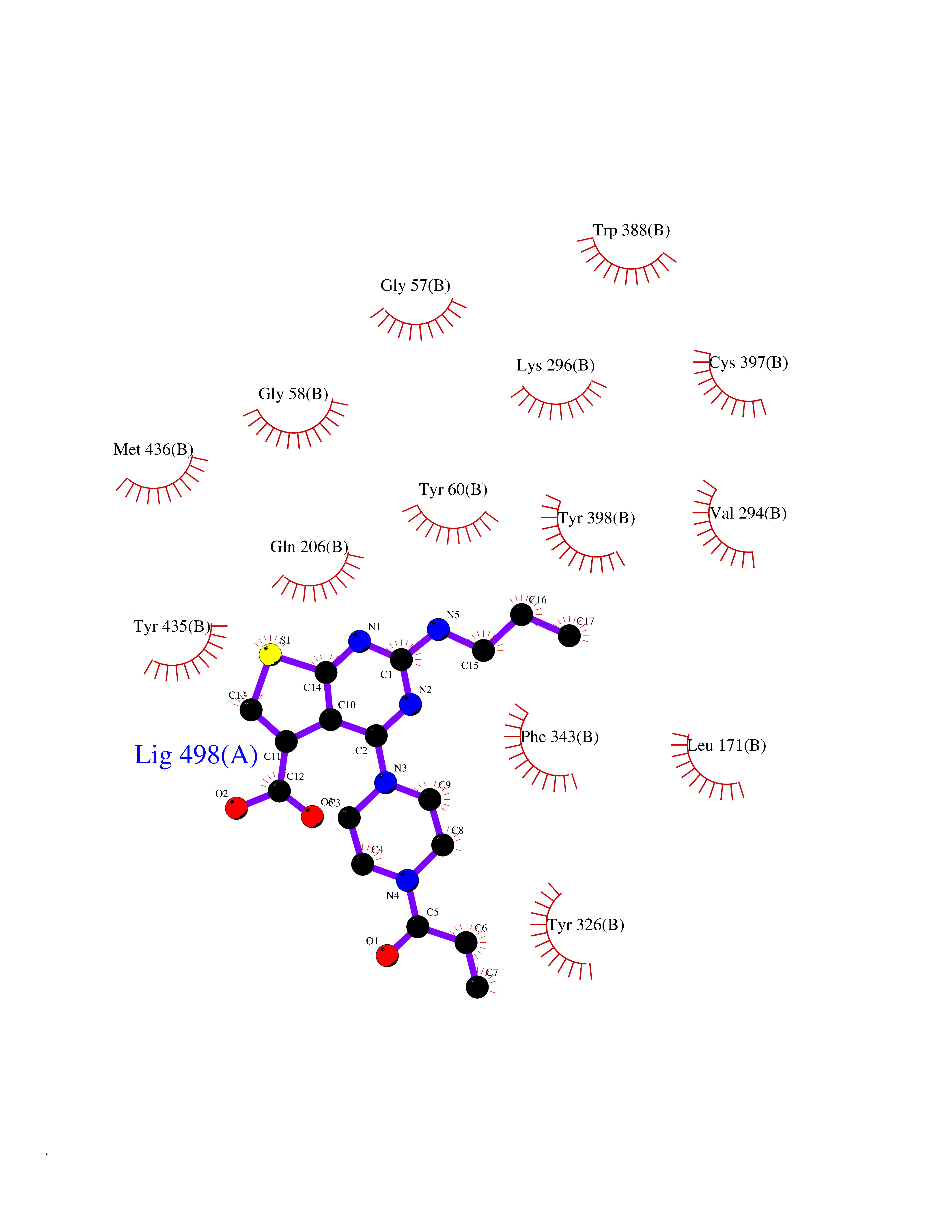 Ligplot Map 3D Binding mode Sequence NKCDVVVVGGGISGMAAAKLLHDSGLNVVVLEARDRVGGRTYTLRNQKVKYVDLGGSYVGPTQNRILRLAKELGLETYKVNEVERLIHHVKGKSYPFRGPFPPVWNPITYLDHNNFWRTMDDMGREIPSDAPWKAPLAEEWDNMTMKELLDKLCWTESAKQLATLFVNLCVTAETHEVSALWFLWYVKQCGGTTRIISTTNGGQERKFVGGSGQVSERIMDLLGDRVKLERPVIYIDQTRENVLVETLNHEMYEAKYVISAIPPTLGMKIHFNPPLPMMRNQMITRVPLGSVIKCIVYYKEPFWRKKDYCGTMIIDGEEAPVAYTLDDTKPEGNYAAIMGFILAHKARKLARLTKEERLKKLCELYAKVLGSLEALEPVHYEEKNWCEEQYSGGCYTTYFPPGILTQYGRVLRQPVDRIYFAGTETATHWSGYMEGAVEAGERAAREILHAMGKIPEDEIWQSEPESVDVPAQPITTTFLERHLPSVPGLLRLI Hydrogen bonds contact Hydrophobic contact | ||||
| 32 | Protein arginine methyltransferase 5 (PRMT5) | 7MXC | 6.73 | |
Target general information Gen name PRMT5 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Shk1 kinase-binding protein 1 homolog; SKB1Hs; SKB1 homolog; SKB1; Protein arginine N-methyltransferase 5; Jak-binding protein 1; JBP1; IBP72; Histone-arginine N-methyltransferase PRMT5; HRMT1L5; 72 k Protein family Class I-like SAM-binding methyltransferase superfamily, Protein arginine N-methyltransferase family Biochemical class Methyltransferase Function Specifically mediates the symmetrical dimethylation of arginine residues in the small nuclear ribonucleoproteins Sm D1 (SNRPD1) and Sm D3 (SNRPD3); such methylation being required for the assembly and biogenesis of snRNP core particles. Methylates SUPT5H and may regulate its transcriptional elongation properties. Mono- and dimethylates arginine residues of myelin basic protein (MBP) in vitro. May play a role in cytokine-activated transduction pathways. Negatively regulates cyclin E1 promoter activity and cellular proliferation. Methylates histone H2A and H4 'Arg-3' during germ cell development. Methylates histone H3 'Arg-8', which may repress transcription. Methylates the Piwi proteins (PIWIL1, PIWIL2 and PIWIL4), methylation of Piwi proteins being required for the interaction with Tudor domain-containing proteins and subsequent localization to the meiotic nuage. Methylates RPS10. Attenuates EGF signaling through the MAPK1/MAPK3 pathway acting at 2 levels. First, monomethylates EGFR; this enhances EGFR 'Tyr-1197' phosphorylation and PTPN6 recruitment, eventually leading to reduced SOS1 phosphorylation. Second, methylates RAF1 and probably BRAF, hence destabilizing these 2 signaling proteins and reducing their catalytic activity. Required for induction of E-selectin and VCAM-1, on the endothelial cells surface at sites of inflammation. Methylates HOXA9. Methylates and regulates SRGAP2 which is involved in cell migration and differentiation. Acts as a transcriptional corepressor in CRY1-mediated repression of the core circadian component PER1 by regulating the H4R3 dimethylation at the PER1 promoter. Methylates GM130/GOLGA2, regulating Golgi ribbon formation. Methylates H4R3 in genes involved in glioblastomagenesis in a CHTOP- and/or TET1-dependent manner. Symmetrically methylates POLR2A, a modification that allows the recruitment to POLR2A of proteins including SMN1/SMN2 and SETX. This is required for resolving RNA-DNA hybrids created by RNA polymerase II, that form R-loop in transcription terminal regions, an important step in proper transcription termination. Along with LYAR, binds the promoter of gamma-globin HBG1/HBG2 and represses its expression. Symmetrically methylates NCL. Methylates TP53; methylation might possibly affect TP53 target gene specificity. Arginine methyltransferase that can both catalyze the formation of omega-N monomethylarginine (MMA) and symmetrical dimethylarginine (sDMA), with a preference for the formation of MMA. Related diseases Epilepsy, nocturnal frontal lobe, 3 (ENFL3) [MIM:605375]: An autosomal dominant focal epilepsy characterized by nocturnal seizures with hyperkinetic automatisms and poorly organized stereotyped movements. {ECO:0000269|PubMed:11062464, ECO:0000269|PubMed:11104662}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P01019; Q9NX04; Q8WUW1; Q08289; P78371; Q16543; Q8N8U2; P54105; P21964-2; Q9NQ92; Q16526; Q9Y6K1; Q01094; Q08426; P38919; Q14241; O15197-2; Q6ZV65; P01100; O95995; P62993; Q8TE85; Q9BX10; P62805; P31269; Q00613; Q63ZY3; P03952; Q8TBB1; P06858; Q86UQ8-1; Q96HA8; Q8WVJ2; P24928; O14744; Q86U06; Q9BRS2; O75044; Q96RU7; P31930; P40337-2; Q9BQA1; P63104; Q96E35; Q91XC0; P03418; Q6ZV65-2 EC number EC 2.1.1.320 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Biological rhythms; Chromatin regulator; Chromosome; Cytoplasm; Direct protein sequencing; Golgi apparatus; Methyltransferase; Nucleus; Proteomics identification; Reference proteome; Repressor; S-adenosyl-L-methionine; Transcription; Transcription regulation; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 71188.3 Length 621 Aromaticity 0.11 Instability index 44.6 Isoelectric point 5.95 Charge (pH=7) -9.68 2D Binding mode Binding energy (Kcal/mol) -9.18 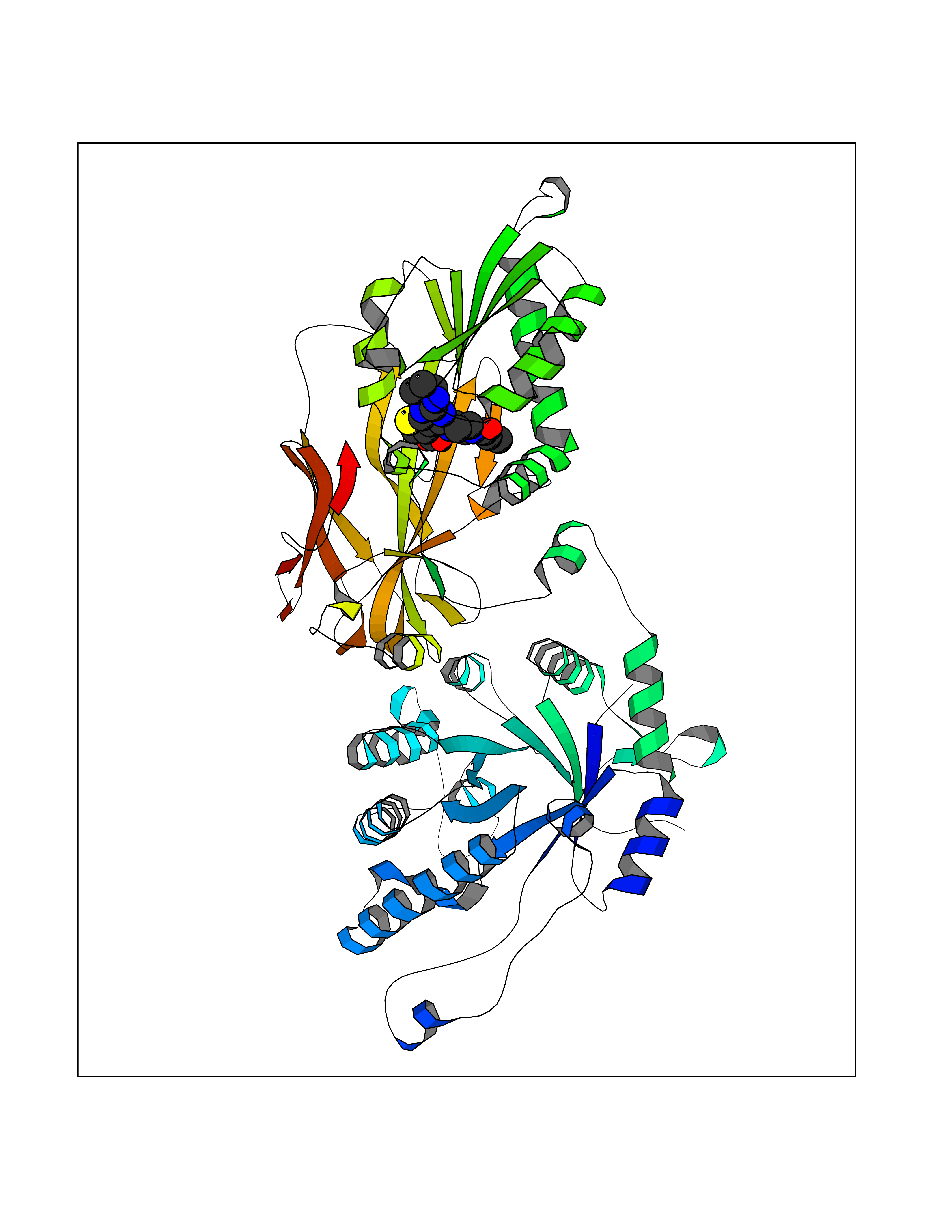 Molscript Map 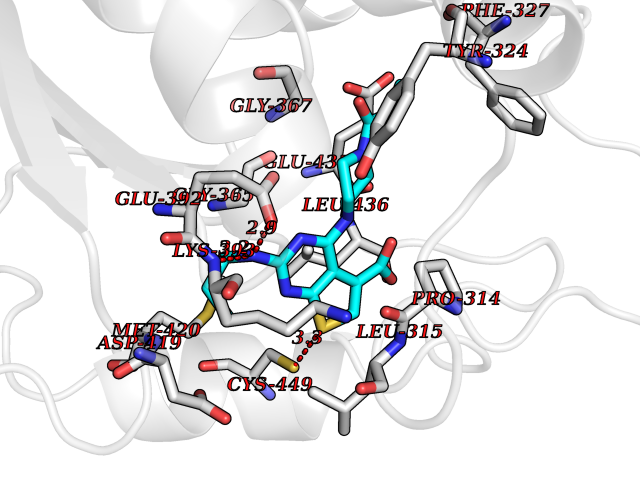 Pymol Map 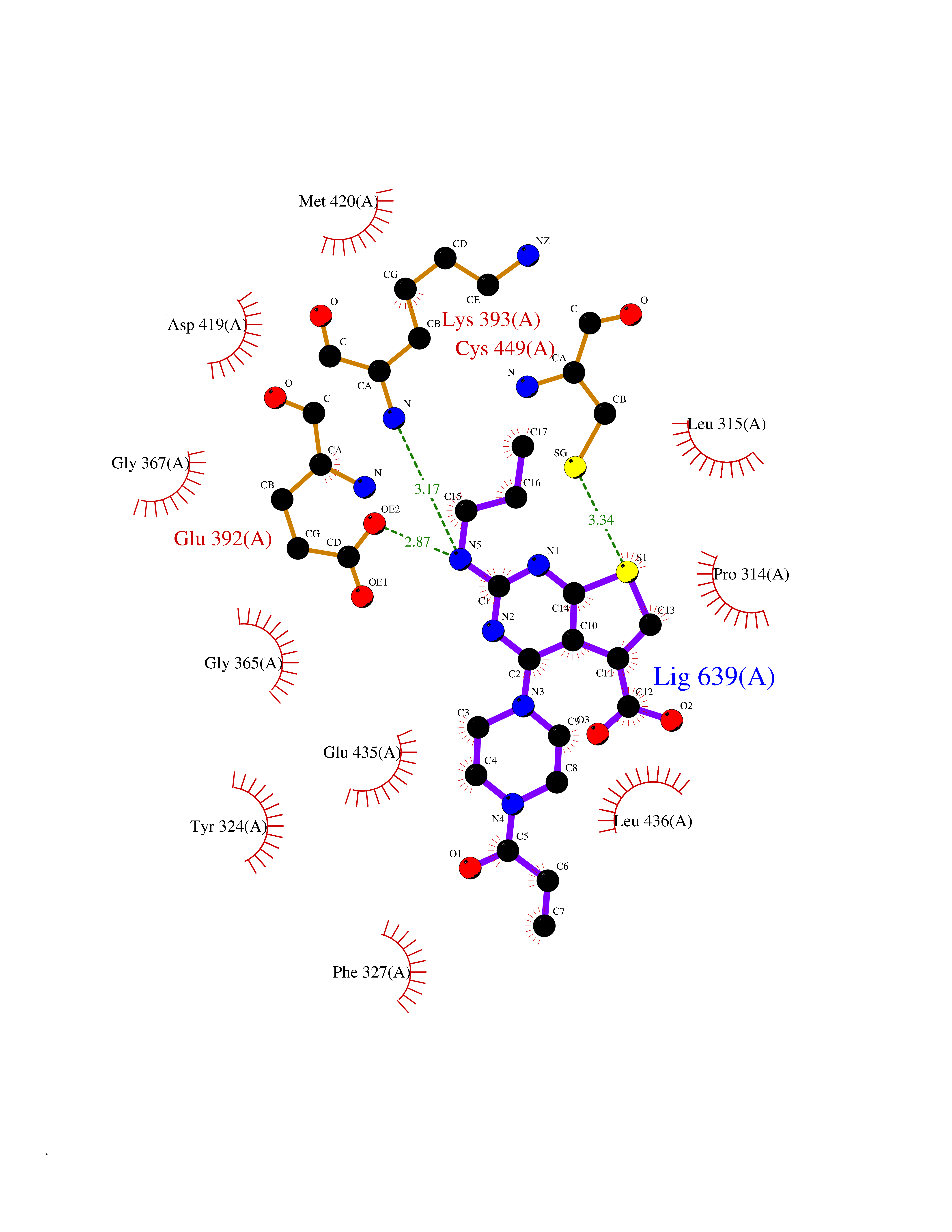 Ligplot Map 3D Binding mode Sequence RVSSGRDLNCVPEIADTLGAVAKQGFDFLCMPVFHPRFKREFIQEPAKNRPGPQTRSDLLLSGRDWNTLIVGKLSPWIRPDSKVEKIRRNSEAAMLQELNFGAYLGLPAFLLPLNQEDNTNLARVLTNHIHTGHHSSMFWMRVPLVAPEDLRDDIIENAPTTHTEEYSGEEKTWMWWHNFRTLCDYSKRIAVALEIGADLPSNHVIDRWLGEPIKAAILPTSIFLTNKKGFPVLSKMHQRLIFRLLKLEVQFIITGTNHHSCSYLQYLEYLSQNRPPPNAYELFAKGYEDYLQSPLQPLMDNLESQTYEVFEKDPIKYSQYQQAIYKCLLDRVPEEEKDTNVQVLMVLGAGRGPLVNASLRAAKQADRRIKLYAVEKNPNAVVTLENWQFEEWGSQVTVVSSDMREWVAPEKADIIVSELLGSFADNELSPECLDGAQHFLKDDGVSIPGEYTSFLAPISSSKLYNEVRACREKDRDPEAQFEMPYVVRLHNFHQLSAPQPCFTFSHPNRDPMIDNNRYCTLEFPVEVNTVLHGFAGYFETVLYQDITLSIRPETHSPGMFSWFPILFPIKQPITVREGQTICVRFWRCSNSKKVWYEWAVTAPVCSAIHNPTGRSYTIGL Hydrogen bonds contact Hydrophobic contact | ||||
| 33 | Metabotropic glutamate receptor 5 (mGluR5) | 4OO9 | 6.72 | |
Target general information Gen name GRM5 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms MGLUR5; GPRC1E Protein family G-protein coupled receptor 3 family Biochemical class GPCR glutamate Function G-protein coupled receptor for glutamate. Ligand binding causes a conformation change that triggers signaling via guanine nucleotide-binding proteins (G proteins) and modulates the activity of down-stream effectors. Signaling activates a phosphatidylinositol-calcium second messenger system and generates a calcium-activated chloride current. Plays an important role in the regulation of synaptic plasticity and the modulation of the neural network activity. Related diseases Charcot-Marie-Tooth disease, axonal, 2D (CMT2D) [MIM:601472]: A dominant axonal form of Charcot-Marie-Tooth disease, a disorder of the peripheral nervous system, characterized by progressive weakness and atrophy, initially of the peroneal muscles and later of the distal muscles of the arms. Charcot-Marie-Tooth disease is classified in two main groups on the basis of electrophysiologic properties and histopathology: primary peripheral demyelinating neuropathies (designated CMT1 when they are dominantly inherited) and primary peripheral axonal neuropathies (CMT2). Neuropathies of the CMT2 group are characterized by signs of axonal degeneration in the absence of obvious myelin alterations, normal or slightly reduced nerve conduction velocities, and progressive distal muscle weakness and atrophy. {ECO:0000269|PubMed:12690580, ECO:0000269|PubMed:17035524, ECO:0000269|PubMed:17101916, ECO:0000269|PubMed:17663003, ECO:0000269|PubMed:20169446, ECO:0000269|PubMed:24604904, ECO:0000269|PubMed:25168514, ECO:0000269|PubMed:26244500, ECO:0000269|PubMed:26503042, ECO:0000269|PubMed:31173493}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Neuronopathy, distal hereditary motor, autosomal dominant 5 (HMND5) [MIM:600794]: A form of distal hereditary motor neuronopathy, a heterogeneous group of neuromuscular disorders caused by selective degeneration of motor neurons in the anterior horn of the spinal cord, without sensory deficit in the posterior horn. The overall clinical picture consists of a classical distal muscular atrophy syndrome in the legs without clinical sensory loss. The disease starts with weakness and wasting of distal muscles of the anterior tibial and peroneal compartments of the legs. Later on, weakness and atrophy may expand to the proximal muscles of the lower limbs and/or to the distal upper limbs. {ECO:0000269|PubMed:12690580, ECO:0000269|PubMed:17035524, ECO:0000269|PubMed:23279345, ECO:0000269|PubMed:24627108, ECO:0000269|PubMed:26503042}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Spinal muscular atrophy, infantile, James type (SMAJI) [MIM:619042]: An autosomal dominant form of spinal muscular atrophy, a group of neuromuscular disorders characterized by degeneration of the anterior horn cells of the spinal cord, leading to symmetrical muscle weakness and atrophy. SMAJI is a severe disease characterized by hypotonia manifesting in the first weeks or months of life, delayed motor development, motor regression, and muscle weakness and atrophy primarily affecting distal muscles. Additional variable features include feeding difficulties, poor overall growth, foot deformities, kyphosis, hyperlordosis, scoliosis, vocal cord dysfunction, and respiratory insufficiency. {ECO:0000269|PubMed:32181591}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00659; DB05070; DB12733; DB06201 Interacts with P41594; Q7Z6G3 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disulfide bond; G-protein coupled receptor; Glycoprotein; Membrane; Methylation; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Signal; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 27065.4 Length 247 Aromaticity 0.13 Instability index 42.92 Isoelectric point 9.24 Charge (pH=7) 11.34 2D Binding mode Binding energy (Kcal/mol) -9.16 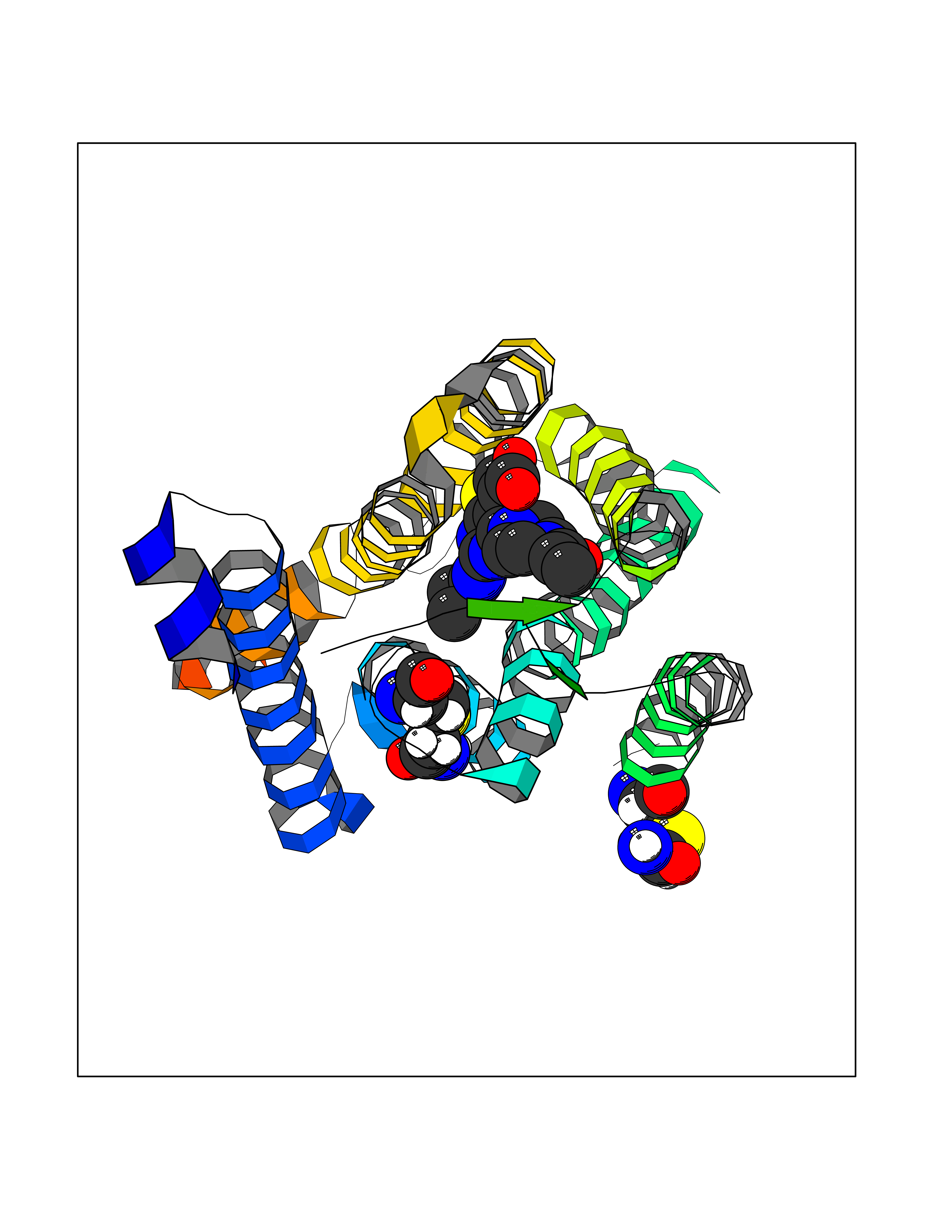 Molscript Map 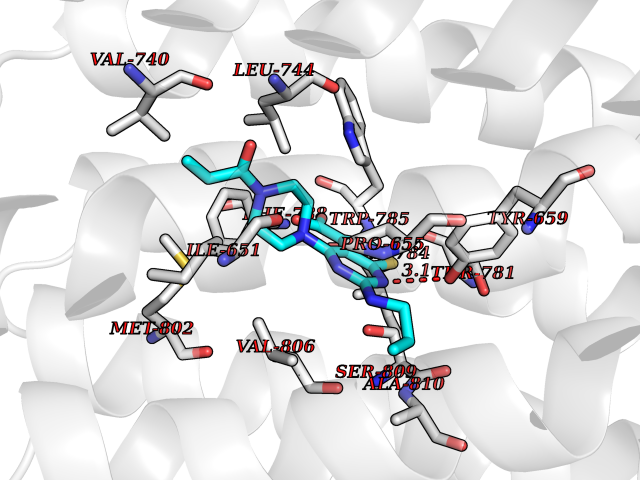 Pymol Map 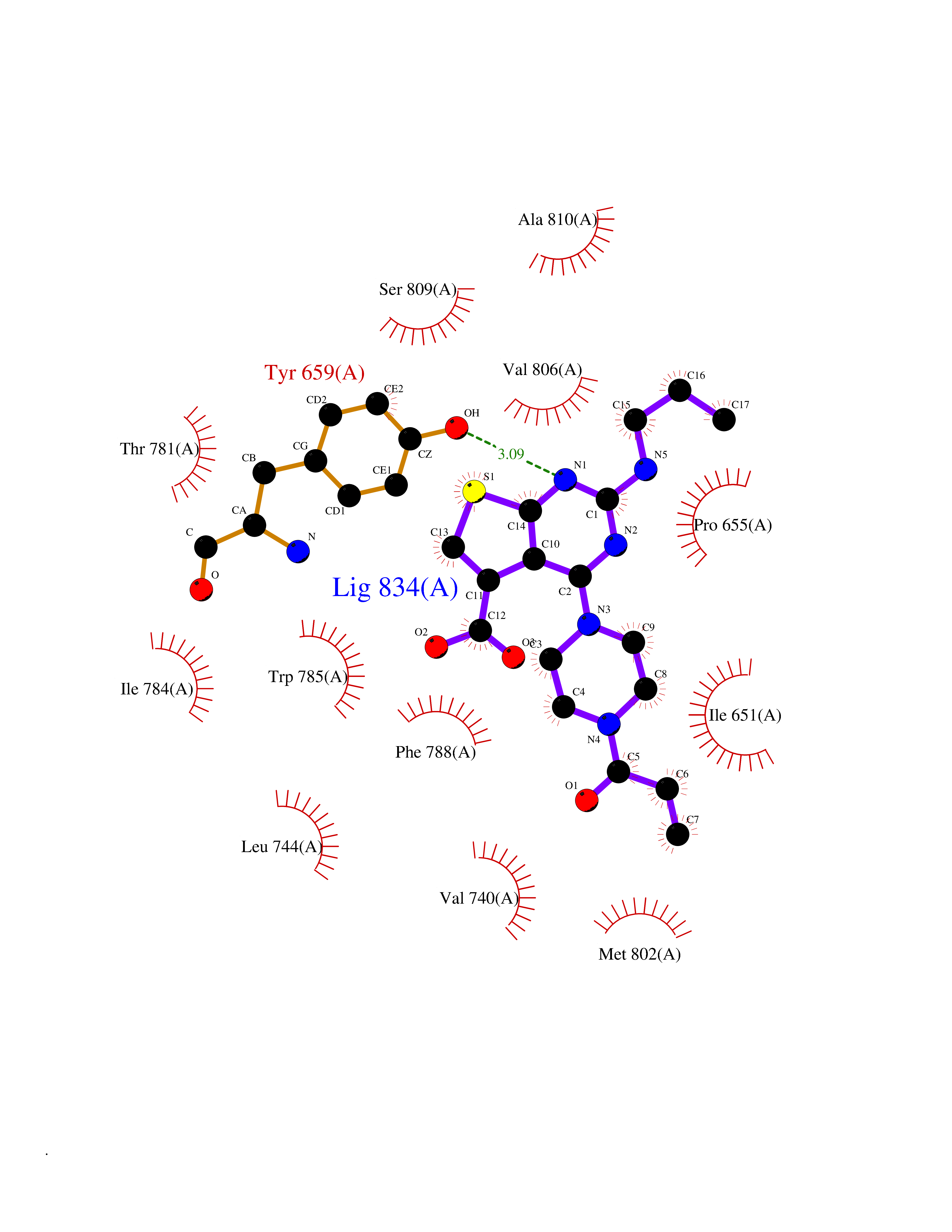 Ligplot Map 3D Binding mode Sequence SPVQYLRWGDPAPIAAVVFACLGLLATLFVTVVFIIYRDTPVVKSSSRELCYIILAGICLGYLCTFXLIAKPKQIYCYLQRIGIGLSPAMSYSALVTKTYRAARILAMSKKSAXAQLVIAFILICIQLGIIVALFIMEPPDIMVYLICNTTNLGVVAPLGYNGLLILACTFYAFKTRNVPANFNEAKYIAFTMYTTCIIWLAFVPIYFGSNYKIITMCFSVSLSATVALGCMFVPKVYIILAKPERN Hydrogen bonds contact Hydrophobic contact | ||||
| 34 | Receptor-type protein-tyrosine phosphatase zeta (PTPRZ1) | 5H08 | 6.72 | |
Target general information Gen name PTPRZ1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Receptor protein tyrosine phosphatase zeta; R-PTP-zeta; PTPRZ1 Protein family Protein-tyrosine phosphatase family, Receptor class 5 subfamily Biochemical class Phosphoric monoester hydrolase Function Protein tyrosine phosphatase that negatively regulates oligodendrocyte precursor proliferation in the embryonic spinal cord. Required for normal differentiation of the precursor cells into mature, fully myelinating oligodendrocytes. May play a role in protecting oligondendrocytes against apoptosis. May play a role in the establishment of contextual memory, probably via the dephosphorylation of proteins that are part of important signaling cascades. Related diseases Optic atrophy 1 (OPA1) [MIM:165500]: A condition that features progressive visual loss in association with optic atrophy. Atrophy of the optic disk indicates a deficiency in the number of nerve fibers which arise in the retina and converge to form the optic disk, optic nerve, optic chiasm and optic tracts. OPA1 is characterized by an insidious onset of visual impairment in early childhood with moderate to severe loss of visual acuity, temporal optic disk pallor, color vision deficits, and centrocecal scotoma of variable density. {ECO:0000269|PubMed:11017079, ECO:0000269|PubMed:11017080, ECO:0000269|PubMed:11440988, ECO:0000269|PubMed:11440989, ECO:0000269|PubMed:11810270, ECO:0000269|PubMed:12036970, ECO:0000269|PubMed:12566046, ECO:0000269|PubMed:14961560, ECO:0000269|PubMed:15948788, ECO:0000269|PubMed:16513463, ECO:0000269|PubMed:16617242, ECO:0000269|PubMed:18204809, ECO:0000269|PubMed:18360822, ECO:0000269|PubMed:19319978, ECO:0000269|PubMed:19325939, ECO:0000269|PubMed:19969356, ECO:0000269|PubMed:20185555, ECO:0000269|PubMed:22382025, ECO:0000269|PubMed:22857269, ECO:0000269|PubMed:23401657}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Dominant optic atrophy plus syndrome (DOA+) [MIM:125250]: A neurologic disorder characterized most commonly by an insidious onset of visual loss and sensorineural hearing loss in childhood with variable presentation of other clinical manifestations including progressive external ophthalmoplegia, muscle cramps, hyperreflexia, and ataxia. There appears to be a wide range of intermediate phenotypes. {ECO:0000269|PubMed:15531309, ECO:0000269|PubMed:16240368, ECO:0000269|PubMed:18065439, ECO:0000269|PubMed:18158317, ECO:0000269|PubMed:18195150, ECO:0000269|PubMed:20185555, ECO:0000269|PubMed:21112924, ECO:0000269|PubMed:23387428}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Behr syndrome (BEHRS) [MIM:210000]: An autosomal recessive syndrome characterized by optic atrophy beginning in early childhood associated with ataxia, pyramidal signs, spasticity, intellectual disability, and posterior column sensory loss. The ataxia, spasticity, and muscle contractures, mainly of the hip adductors, hamstrings, and soleus, are progressive and become more prominent in the second decade. {ECO:0000269|PubMed:21636302, ECO:0000269|PubMed:25012220, ECO:0000269|PubMed:25146916}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Mitochondrial DNA depletion syndrome 14, cardioencephalomyopathic type (MTDPS14) [MIM:616896]: An autosomal recessive mitochondrial disorder characterized by lethal infantile encephalopathy, hypertrophic cardiomyopathy and optic atrophy. Skeletal muscle biopsies show significant mtDNA depletion and abnormal mitochondria. {ECO:0000269|PubMed:26561570}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q9UM73; Q12860 EC number EC 3.1.3.48 Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disulfide bond; Glycoprotein; Hydrolase; Membrane; Phosphoprotein; Protein phosphatase; Proteoglycan; Proteomics identification; Reference proteome; Repeat; Secreted; Signal; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 32208.1 Length 282 Aromaticity 0.11 Instability index 38.98 Isoelectric point 7.35 Charge (pH=7) 0.89 2D Binding mode Binding energy (Kcal/mol) -9.16 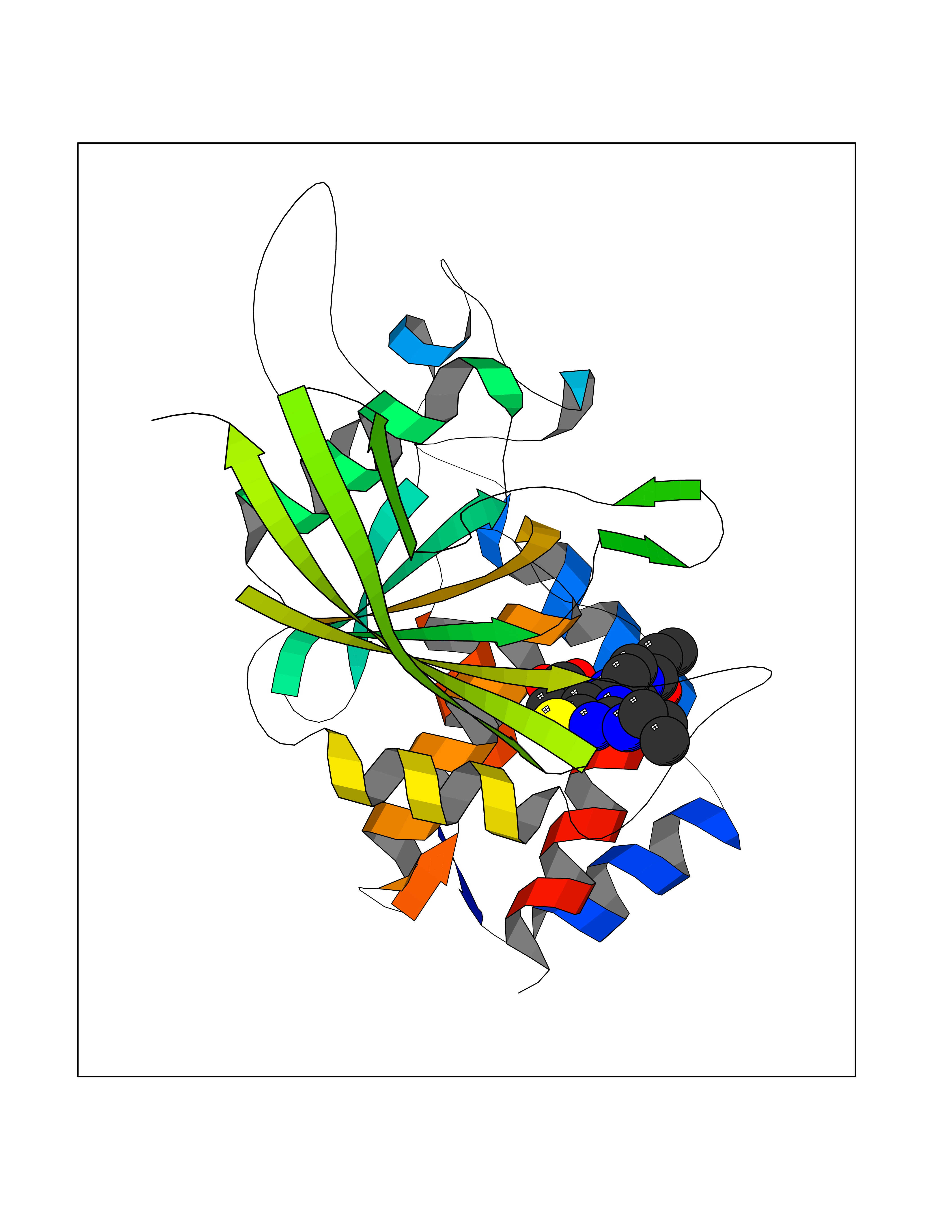 Molscript Map 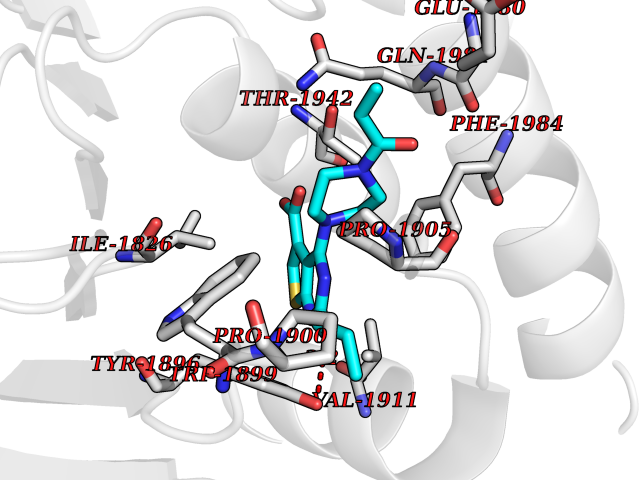 Pymol Map 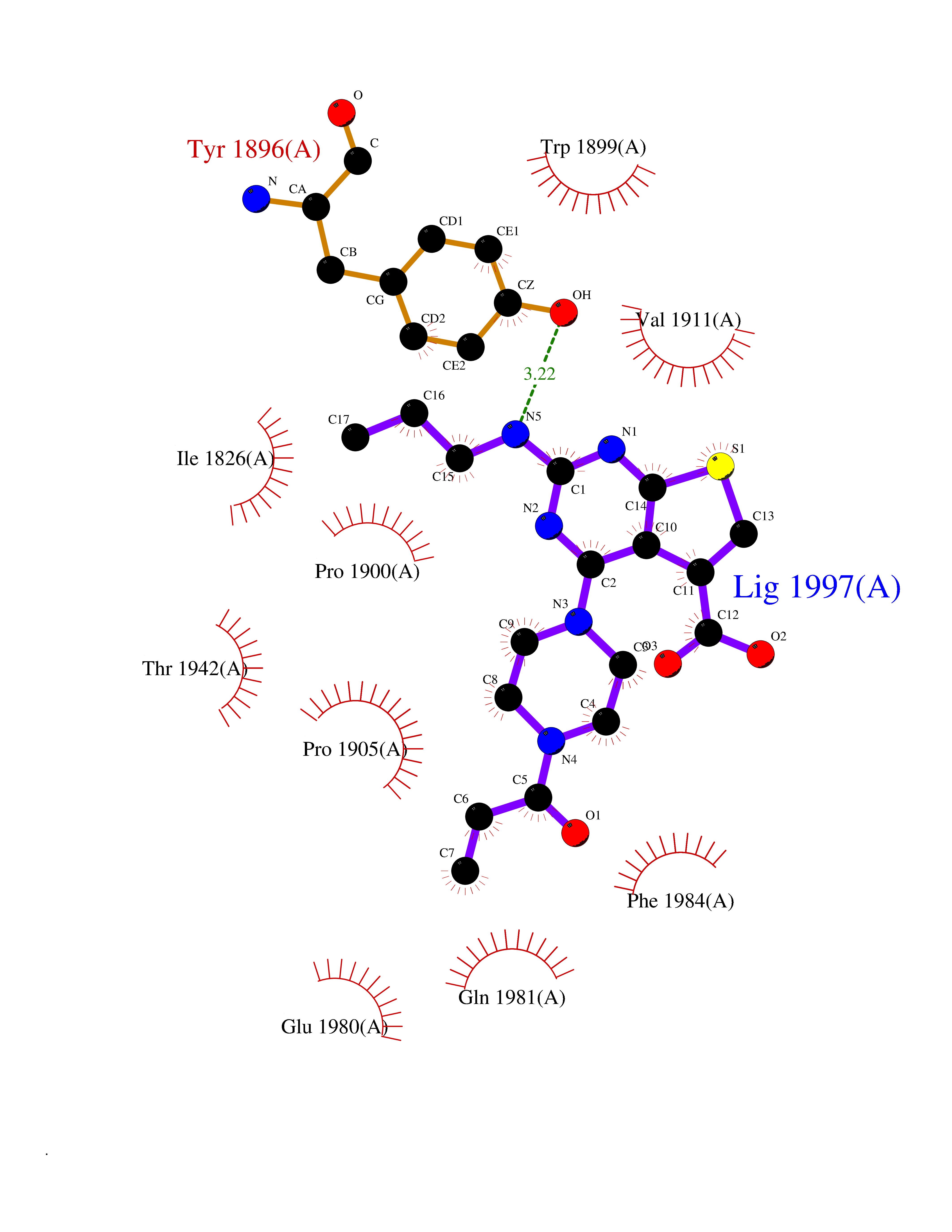 Ligplot Map 3D Binding mode Sequence GPAIPIKHFPKHVADLHASSGFTEEFEEVQSCTVDLGITADSSNHPDNKHKNRYINIVAYDHSRVKLAQLAEKDGKLTDYINANYVDGYNRPKAYIAAQGPLKSTAEDFWRMIWEHNVEVIVMITNLVEKGRRKCDQYWPADGSEEYGNFLVTQKSVQVLAYYTVRNFTLRNTKIRVVTQYHYTQWPDMGVPEYSLPVLTFVRKAAYAKRHAVGPVVVHCSAGVGRTGTYIVLDSMLQQIQHEGTVNIFGFLKHIRSQRNYLVQTEEQYVFIHDTLVEAILS Hydrogen bonds contact Hydrophobic contact | ||||
| 35 | LIM domain kinase-2 (LIMK-2) | 7QHG | 6.70 | |
Target general information Gen name LIMK2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms LIMK-2; LIM domain kinase 2 Protein family Protein kinase superfamily, TKL Ser/Thr protein kinase family Biochemical class Kinase Function Displays serine/threonine-specific phosphorylation of myelin basic protein and histone (MBP) in vitro. Related diseases Intellectual developmental disorder, autosomal recessive 80, with variant lissencephaly (MRT80) [MIM:620653]: An autosomal recessive disorder characterized by global developmental delay, mildly to moderately impaired intellectual development, attention deficit-hyperactivity disorder, hypotonia, seizure, poor social skills, and autistic traits. Brain imaging shows fronto-temporal lissencephaly and pachygyria. {ECO:0000269|PubMed:37880421}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB11718; DB12010 Interacts with Q16543; P08238; Q96C90; P62258 EC number EC 2.7.11.1 Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Cytoplasm; Cytoskeleton; Kinase; LIM domain; Metal-binding; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Serine/threonine-protein kinase; Transferase; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 32109.2 Length 283 Aromaticity 0.1 Instability index 27.28 Isoelectric point 6.01 Charge (pH=7) -3.93 2D Binding mode Binding energy (Kcal/mol) -9.14  Molscript Map 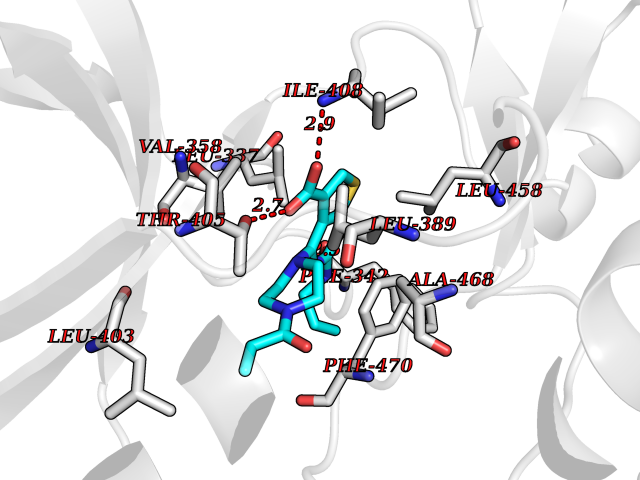 Pymol Map 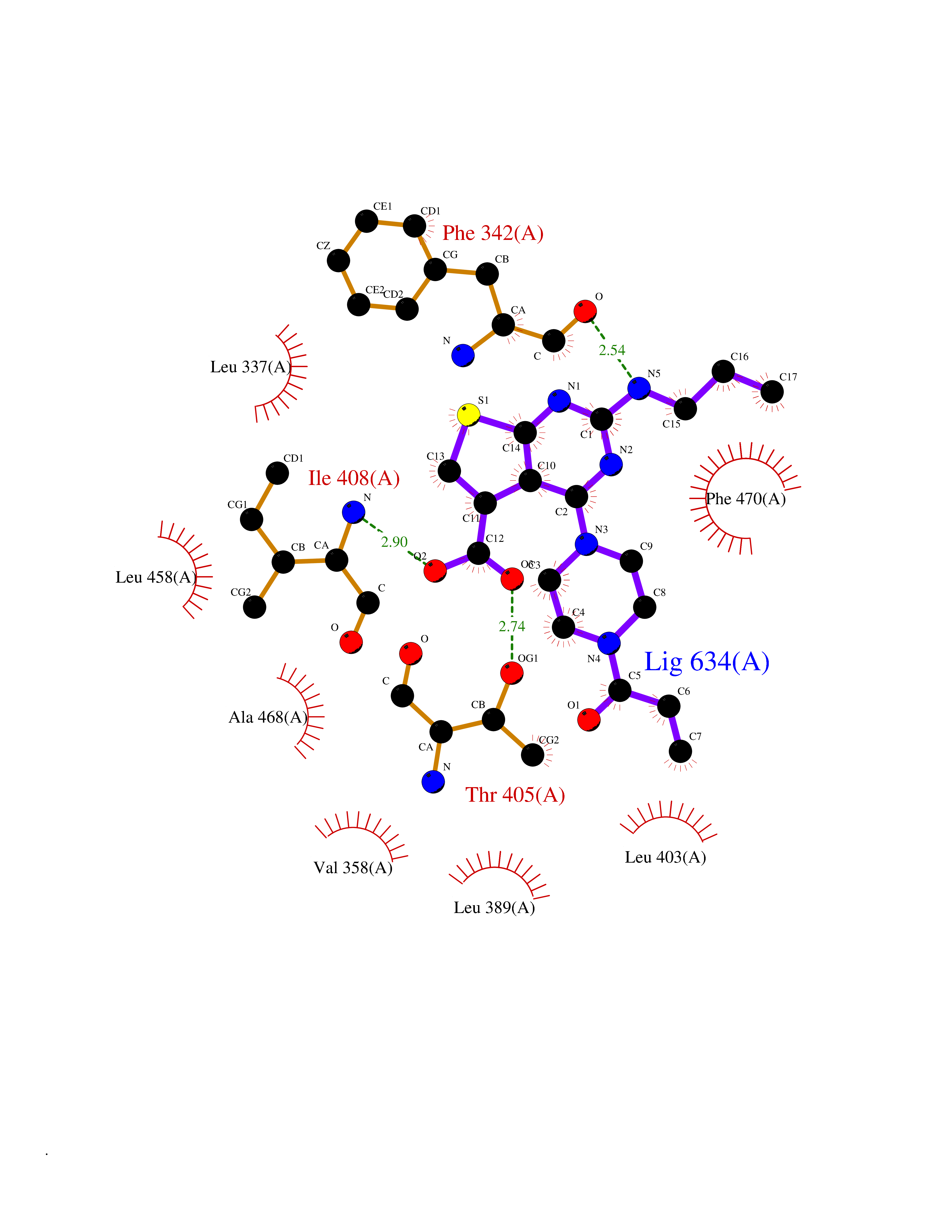 Ligplot Map 3D Binding mode Sequence MDLIHGEVLGKGFFGQAIKVTHKATGKVMVMKELIRCDEETQKTFLTEVKVMRSLDHPNVLKFIGVLYKDKKLNLLTEYIEGGTLKDFLRSMDPFPWQQKVRFAKGIASGMAYLHSMCIIHRDLNSHNCLIKLDKTVVVADFGLSRLIVDRKKRYTVVGNPYWMAPEMLNGKSYDETVDIFSFGIVLCEIIGQVYADPDCLPRTLDFGLNVKLFWEKFVPTDCPPAFFPLAAICCRLEPESRPAFSKLEDSFEALSLYLGELGIPLPAELEELDHTVSMQYGL Hydrogen bonds contact Hydrophobic contact | ||||
| 36 | Retinoic acid receptor alpha (RARA) | 3KMR | 6.69 | |
Target general information Gen name RARA Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms RAR-alpha; RAR alpha; Nuclear receptor subfamily 1 group B member 1; NR1B1 Protein family Nuclear hormone receptor family, NR1 subfamily Biochemical class Nuclear hormone receptor Function Retinoic acid receptors bind as heterodimers to their target response elements in response to their ligands, all-trans or 9-cis retinoic acid, and regulate gene expression in various biological processes. The RXR/RAR heterodimers bind to the retinoic acid response elements (RARE) composed of tandem 5'-AGGTCA-3' sites known as DR1-DR5. In the absence of ligand, the RXR-RAR heterodimers associate with a multiprotein complex containing transcription corepressors that induce histone acetylation, chromatin condensation and transcriptional suppression. On ligand binding, the corepressors dissociate from the receptors and associate with the coactivators leading to transcriptional activation. RARA plays an essential role in the regulation of retinoic acid-induced germ cell development during spermatogenesis. Has a role in the survival of early spermatocytes at the beginning prophase of meiosis. In Sertoli cells, may promote the survival and development of early meiotic prophase spermatocytes. In concert with RARG, required for skeletal growth, matrix homeostasis and growth plate function. Receptor for retinoic acid. Related diseases Chromosomal aberrations involving RARA are commonly found in acute promyelocytic leukemia. Translocation t(11;17)(q32;q21) with ZBTB16/PLZF; translocation t(15;17)(q21;q21) with PML; translocation t(5;17)(q32;q11) with NPM. The PML-RARA oncoprotein requires both the PML ring structure and coiled-coil domain for both interaction with UBE2I, nuclear microspeckle location and sumoylation. In addition, the coiled-coil domain functions in blocking RA-mediated transactivation and cell differentiation. {ECO:0000269|PubMed:12691149, ECO:0000269|PubMed:8302850, ECO:0000269|PubMed:8562957}. Drugs (DrugBank ID) DB00459; DB00210; DB00523; DB00926; DB00982; DB05785; DB04942; DB00799; DB00755; DB12808 Interacts with O43707-1; O15296; Q15699; Q96RK4; O95273; P51946; Q15910; P50148; Q9UKP3; Q96EZ8; Q15648; Q71SY5; Q15788; Q9Y6Q9; O75376; Q9Y618; Q16236; P13056-2; P48552; Q9UPP1-2; Q9H8W4; P37231; P78527; P19793; P28702; P28702-3; P48443; Q96EB6; P63165; Q8WW24; Q2M1K9; Q91XC0; P59598; Q14457; P48552; Q96CV9; P28702; P48443; Q8WW24 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Chromosomal rearrangement; Cytoplasm; DNA-binding; Isopeptide bond; Metal-binding; Nucleus; Phosphoprotein; Proteomics identification; Proto-oncogene; Receptor; Reference proteome; Transcription; Transcription regulation; Ubl conjugation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A Molecular weight (Da) 27724.1 Length 244 Aromaticity 0.05 Instability index 50.8 Isoelectric point 5.82 Charge (pH=7) -3.61 2D Binding mode Binding energy (Kcal/mol) -9.13 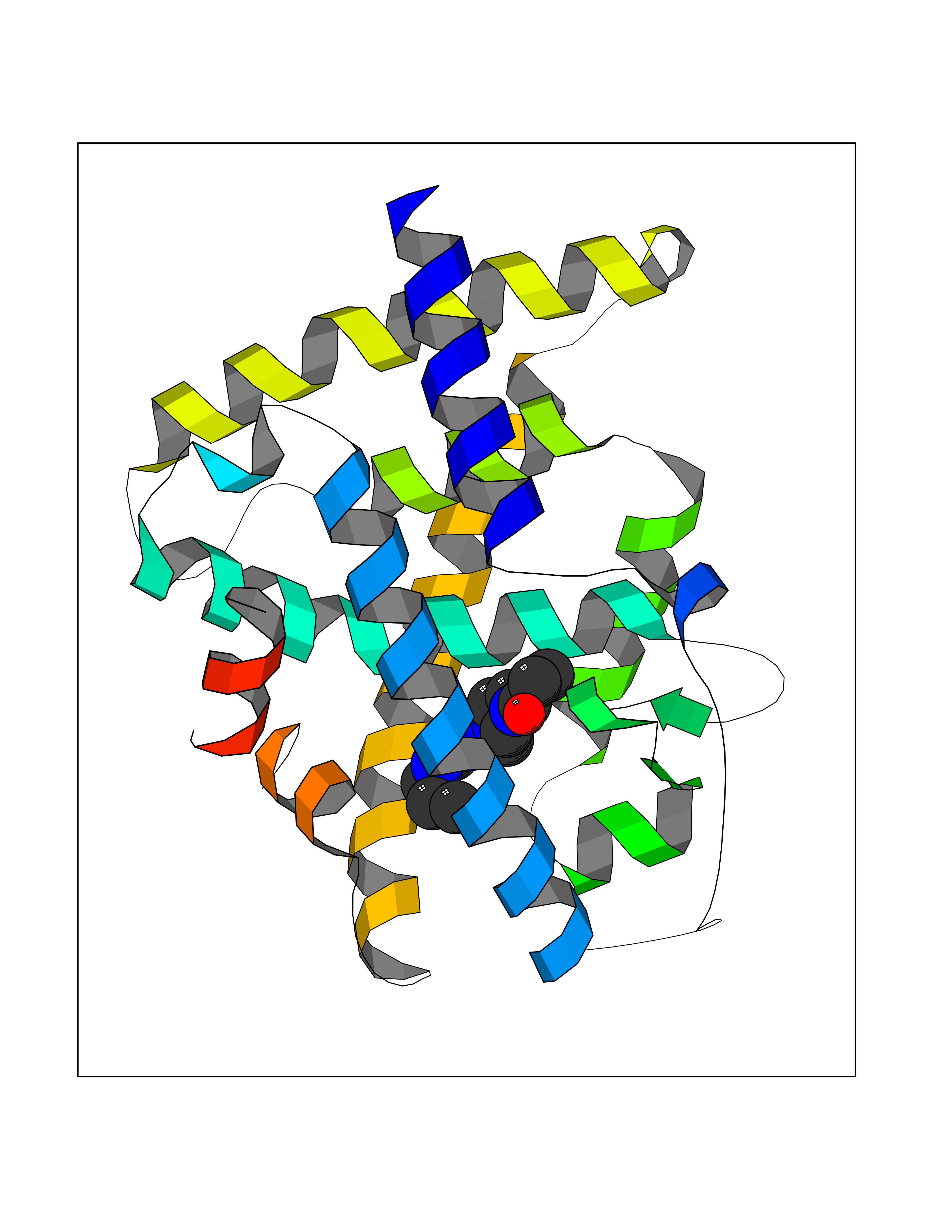 Molscript Map 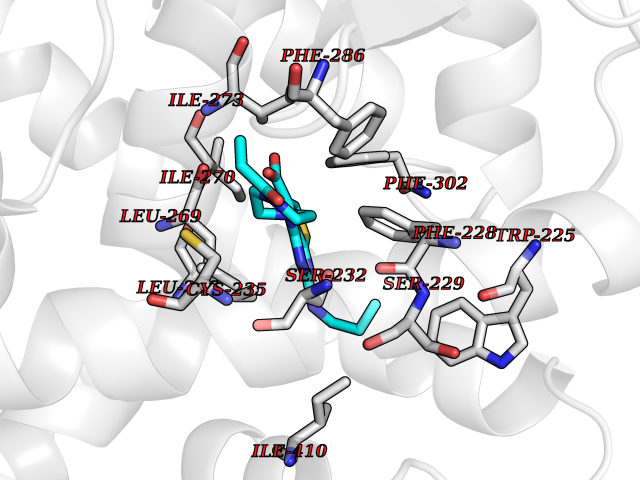 Pymol Map 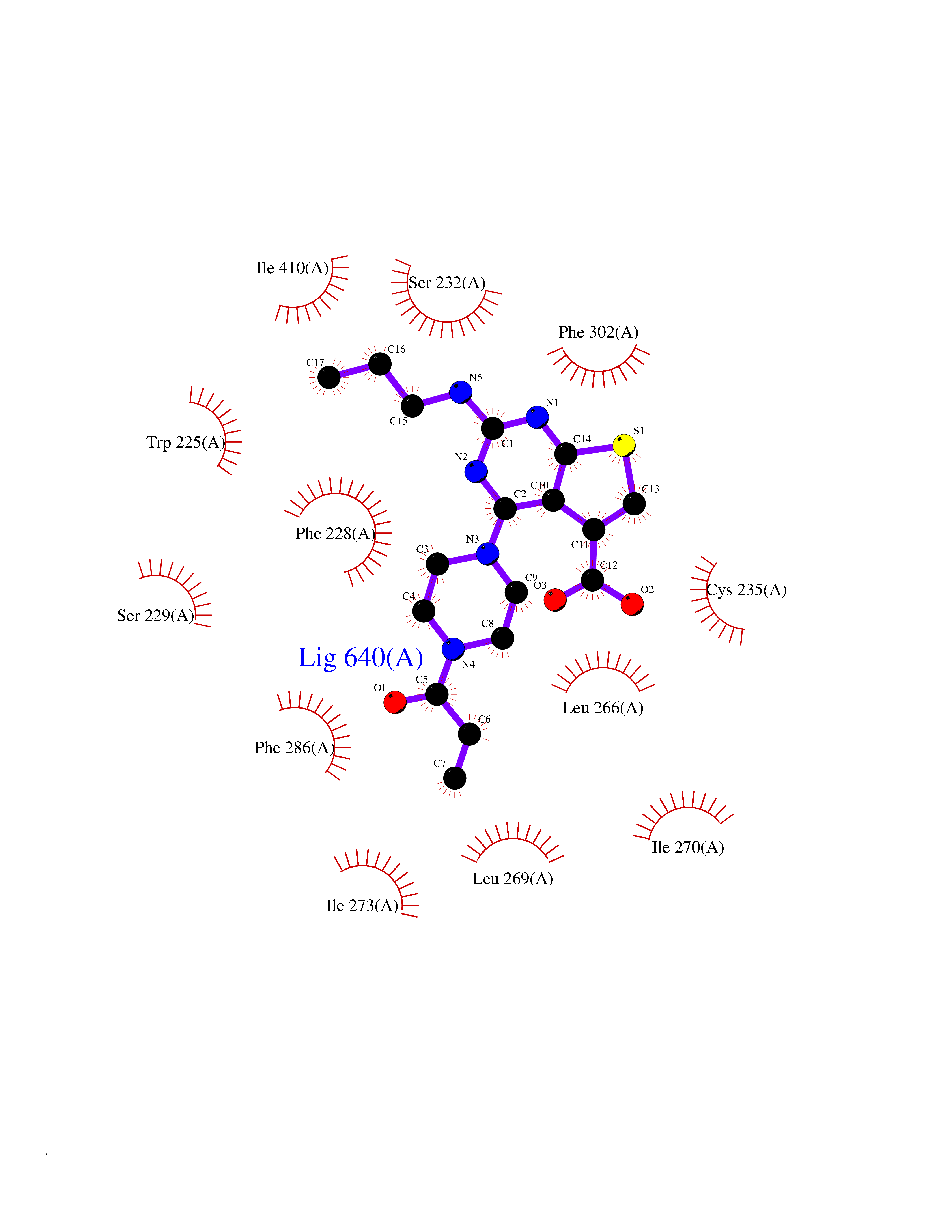 Ligplot Map 3D Binding mode Sequence PEVGELIEKVRKAHQETFPALCQLGKYTTNNSSEQRVSLDIDLWDKFSELSTKCIIKTVEFAKQLPGFTTLTIADQITLLKAACLDILILRICTRYTPEQDTMTFSDGLTLNRTQMHNAGFGPLTDLVFAFANQLLPLEMDDAETGLLSAICLICGDRQDLEQPDRVDMLQEPLLEALKVYVRKRRPSRPHMFPKMLMKITDLRSISAKGAERVITLKMEIPGSMPPLIQEMLEHKILHRLLQE Hydrogen bonds contact Hydrophobic contact | ||||
| 37 | Mutated Histone H3.3 (H3F3A) | 4GUS | 6.68 | |
Target general information Gen name H3F3A Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms PP781; Histone H3.3; H3F3; H3.3B; H3.3A Protein family Histone H3 family Biochemical class NA Function Variant histone H3 which replaces conventional H3 in a wide range of nucleosomes in active genes. Constitutes the predominant form of histone H3 in non-dividing cells and is incorporated into chromatin independently of DNA synthesis. Deposited at sites of nucleosomal displacement throughout transcribed genes, suggesting that it represents an epigenetic imprint of transcriptionally active chromatin. Nucleosomes wrap and compact DNA into chromatin, limiting DNA accessibility to the cellular machineries which require DNA as a template. Histones thereby play a central role in transcription regulation, DNA repair, DNA replication and chromosomal stability. DNA accessibility is regulated via a complex set of post-translational modifications of histones, also called histone code, and nucleosome remodeling. Related diseases Glioma (GLM) [MIM:137800]: Gliomas are benign or malignant central nervous system neoplasms derived from glial cells. They comprise astrocytomas and glioblastoma multiforme that are derived from astrocytes, oligodendrogliomas derived from oligodendrocytes and ependymomas derived from ependymocytes. {ECO:0000269|PubMed:22286061, ECO:0000269|PubMed:22286216, ECO:0000269|PubMed:23539269}. The gene represented in this entry is involved in disease pathogenesis. H3F3A mutations affecting residues involved in post-translational modifications of histone H3.3 are recurrent in malignant, aggressive gliomas including glioblastoma multiforme (GBM) and diffuse intrinsic pontine glioma (DIPG) (PubMed:22286061, PubMed:22286216). The mechanism through which mutations lead to tumorigenesis involves altered histones methylation, impaired regulation of Polycomb repressive complex 2 (PRC2) activity, and aberrant epigenetic regulation of gene expression (PubMed:23539183, PubMed:23539269, PubMed:23603901). {ECO:0000269|PubMed:22286061, ECO:0000269|PubMed:22286216, ECO:0000269|PubMed:23539183, ECO:0000269|PubMed:23539269, ECO:0000269|PubMed:23603901}.; DISEASE: Bryant-Li-Bhoj neurodevelopmental syndrome 1 (BRYLIB1) [MIM:619720]: An autosomal dominant disorder predominantly characterized by global developmental delay, impaired intellectual development, poor or absent speech, and delayed motor milestones. Clinical manifestations are highly variable, including abnormal head shape, dysmorphic facial features, oculomotor abnormalities, feeding problems, and non-specific brain imaging abnormalities. Additional features may include hearing loss, seizures, short stature, and mild skeletal defects. {ECO:0000269|PubMed:33268356, ECO:0000269|PubMed:34876591}. The disease is caused by variants affecting the gene represented in this entry. BRYLIB1 is caused by variants in H3-3A. {ECO:0000269|PubMed:33268356, ECO:0000269|PubMed:34876591}.; DISEASE: Bryant-Li-Bhoj neurodevelopmental syndrome 2 (BRYLIB2) [MIM:619721]: An autosomal dominant disorder predominantly characterized by global developmental delay, impaired intellectual development, poor or absent speech, and delayed motor milestones. Clinical manifestations are highly variable, including abnormal head shape, dysmorphic facial features, oculomotor abnormalities, feeding problems, and non-specific brain imaging abnormalities. Additional features may include hearing loss, seizures, short stature, and mild skeletal defects. {ECO:0000269|PubMed:33268356, ECO:0000269|PubMed:34876591}. The disease is caused by variants affecting the gene represented in this entry. BRYLIB2 is caused by variants in H3-3B. {ECO:0000269|PubMed:33268356, ECO:0000269|PubMed:34876591}.; DISEASE: H3F3A and H3F3B mutations affecting residues involved in post-translational modifications of histone H3.3 are implicated in the pathogenesis of some bone and cartilage neoplasms. Mutations have been found with high prevalence in chondroblastoma and giant cell tumors of bone, and with low frequency in osteosarcoma, conventional chondrosarcoma and clear cell chondrosarcoma. Chondroblastoma samples frequently carry a H3F3B mutation affecting residue Lys-37 (H3K36), although H3F3A is mutated in some cases. Most giant cell tumors of bone harbor H3F3A mutations affecting residue Gly-35 (H3G34). {ECO:0000269|PubMed:24162739}. Drugs (DrugBank ID) NA Interacts with Q9NVP2; P45973; Q13111; Q9UER7; Q9UER7-1; Q9Y6K1; P62805; P49321-2; Q8IZL8; Q5VWG9; Q9VK33; Q8R5C8 EC number NA Uniprot keywords 3D-structure; Acetylation; ADP-ribosylation; Chromosome; Citrullination; Direct protein sequencing; Disease variant; DNA-binding; Hydroxylation; Intellectual disability; Lipoprotein; Methylation; Nucleosome core; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Ubl conjugation Protein physicochemical properties Chain ID A,C Molecular weight (Da) 86148.9 Length 766 Aromaticity 0.1 Instability index 37.57 Isoelectric point 8.25 Charge (pH=7) 7.16 2D Binding mode Binding energy (Kcal/mol) -9.11 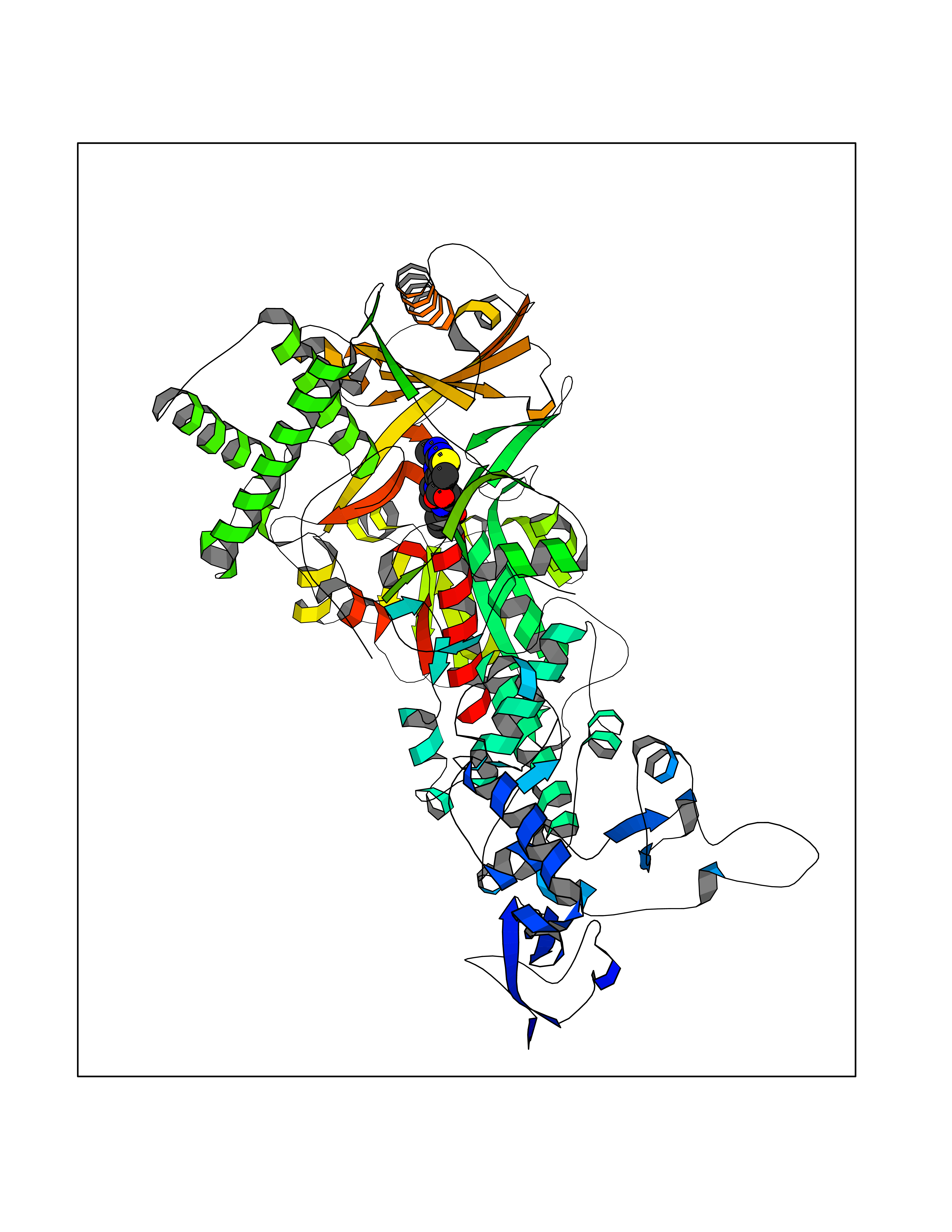 Molscript Map 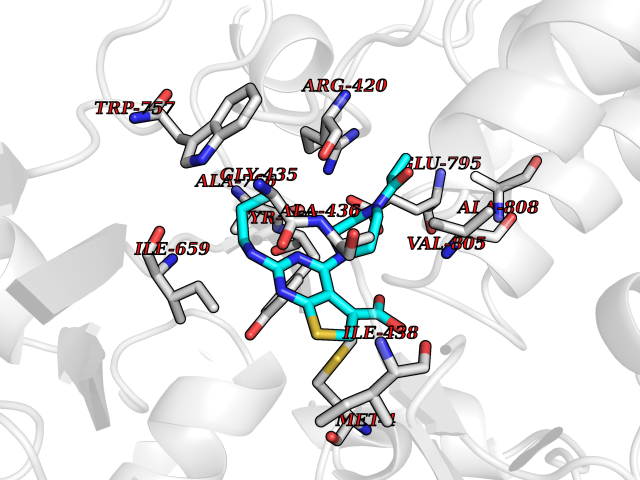 Pymol Map 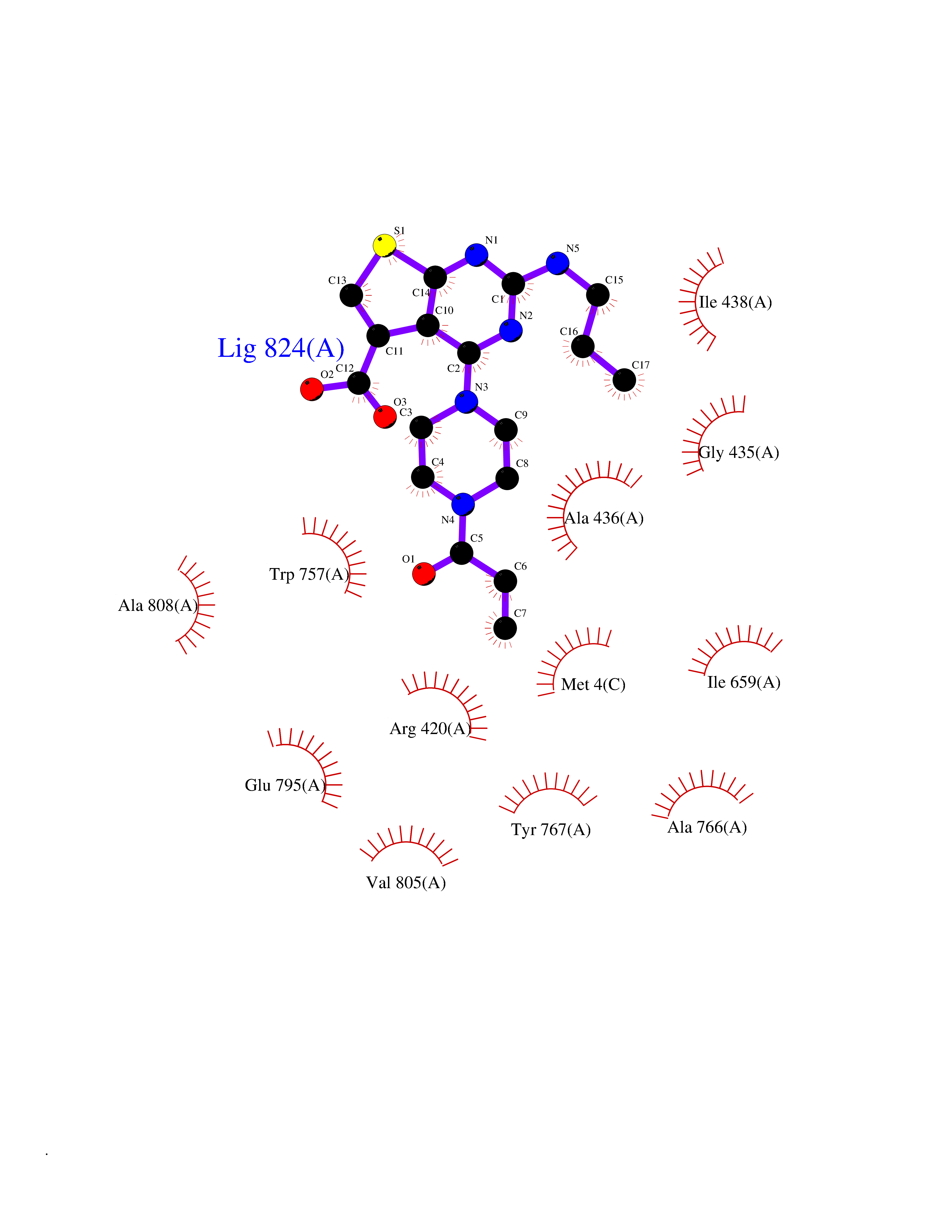 Ligplot Map 3D Binding mode Sequence GSRKCEKAGCTATCPVCFASASERCAKNGYTSRWYHLSCGEHFCNECFDHYYRSHKDGYDKYTTWKKIWTSNGKTEPSPKAFMADQQLPYWVQCTKPECRKWRQLTKEIQLTPQIAKTYRCGMKPNTAIKPETSDHCSLPEDLRVLEVSNHWWYSMLILPPLLKDSVAAPLLSAYYPDCVGMSPSCTGMNRYFQPFYQPNECGKALCVRPDVMELDELYEFPEYSRDPTMYLALRNLILALWYTNCKEALTPQKCIPHIIVRGLVRIRCVQEVERILYFMTRKGLINTGVLSVGADQYLLPKDYHNKSVIIIGAGPAGLAAARQLHNFGIKVTVLEAKDRIGGRVWDDKSFKGVTVGRGAQIVNGCINNPVALMCEQLGISMHKFGERCDLIQEGGRITDPTIDKRMDFHFNALLDVVSEWRKDKTQLQDVPLGEKIEEIYKAFIKESGIQFSELEGQVLQFHLSNLEYACGSNLHQVSARSWDHNEFFAQFAGDHTLLTPGYSVIIEKLAEGLDIQLKSPVQCIDYSGDEVQVTTTDGTGYSAQKVLVTVPLALLQKGAIQFNPPLSEKKMKAINSLGAGIIEKIALQFPYRFWDSKVQGADFFGHVPPSASKRGLFAVFYDMDPQKKHSVLMSVIAGEAVASVRTLDDKQVLQQCMATLRELFKEQEVPDPTKYFVTRWSTDPWIQMAYSFVKTGGSGEAYDIIAEDIQGTVFFAGEATNRHFPQTVTGAYLSGVREASKIAAFARTMQTARKSTGGKAPRKQL Hydrogen bonds contact Hydrophobic contact | ||||
| 38 | Cyclopropane mycolic acid synthase MmaA2 | 1TPY | 6.67 | |
Target general information Gen name mmaA2 Organism Mycobacterium tuberculosis (strain ATCC 25618 / H37Rv) Uniprot ID TTD ID NA Synonyms Rv0644c;mma2 Protein family CFA/CMAS family Biochemical class Transferase Function Cyclopropane-fatty-acyl-phospholipid synthase activity.Methyltransferase activity. Related diseases Oocyte/zygote/embryo maturation arrest 16 (OZEMA16) [MIM:617234]: A rare cause of female primary infertility. In affected women, ovulation and fertilization proceed normally but embryos are arrested at early stages of development. Inheritance is autosomal recessive. {ECO:0000269|PubMed:27545678}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01718; DB01752 Interacts with NA EC number 2.1.1.79 Uniprot keywords 3D-structure; Acetylation; Lipid biosynthesis; Lipid metabolism; Methyltransferase; Reference proteome; S-adenosyl-L-methionine; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 32493.6 Length 285 Aromaticity 0.1 Instability index 43.61 Isoelectric point 5.53 Charge (pH=7) -10.17 2D Binding mode Binding energy (Kcal/mol) -9.1  Molscript Map 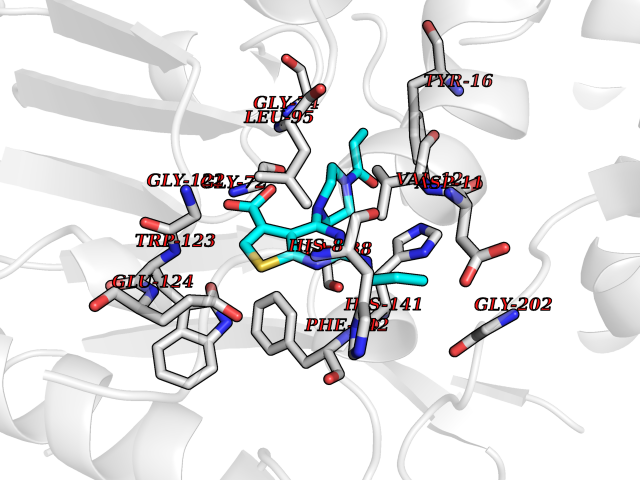 Pymol Map  Ligplot Map 3D Binding mode Sequence NDLTPHFEDVQAHYDLSDDFFRLFLDPTQTYSCAHFEREDMTLEEAQIAKIDLALGKLGLQPGMTLLDIGCGWGATMRRAIAQYDVNVVGLTLSKNQAAHVQKSFDEMDTPRDRRVLLAGWEQFNEPVDRIVSIGAFEHFGHDRHADFFARAHKILPPDGVLLLHTITGLTRQQMVDHGLPLTLWLARFLKFIATEIFPGGQPPTIEMVEEQSAKTGFTLTRRQSLQPHYARTLDLWAEALQEHKSEAIAIQSEEVYERYMKYLTGCAKLFRVGYIDVNQFTLAK Hydrogen bonds contact Hydrophobic contact | ||||
| 39 | Opioid receptor sigma 1 (OPRS1) | 6DJZ | 6.67 | |
Target general information Gen name SIGMAR1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms hSigmaR1; Sigma1R; Sigma1-receptor; Sigma non-opioid intracellular receptor 1; Sigma 1-type opioid receptor; SRBP; SR31747-binding protein; SR31747 binding protein 1; SR-BP; SIG-1R; Opioid receptor, s Protein family ERG2 family Biochemical class GPCR rhodopsin Function Involved in the regulation of different receptors it plays a role in BDNF signaling and EGF signaling. Also regulates ion channels like the potassium channel and could modulate neurotransmitter release. Plays a role in calcium signaling through modulation together with ANK2 of the ITP3R-dependent calcium efflux at the endoplasmic reticulum. Plays a role in several other cell functions including proliferation, survival and death. Originally identified for its ability to bind various psychoactive drugs it is involved in learning processes, memory and mood alteration. Necessary for proper mitochondrial axonal transport in motor neurons, in particular the retrograde movement of mitochondria. Plays a role in protecting cells against oxidative stress-induced cell death via its interaction with RNF112. Functions in lipid transport from the endoplasmic reticulum and is involved in a wide array of cellular functions probably through regulation of the biogenesis of lipid microdomains at the plasma membrane. Related diseases Amyotrophic lateral sclerosis 16, juvenile (ALS16) [MIM:614373]: A neurodegenerative disorder affecting upper motor neurons in the brain and lower motor neurons in the brain stem and spinal cord, resulting in fatal paralysis. Sensory abnormalities are absent. The pathologic hallmarks of the disease include pallor of the corticospinal tract due to loss of motor neurons, presence of ubiquitin-positive inclusions within surviving motor neurons, and deposition of pathologic aggregates. The etiology of amyotrophic lateral sclerosis is likely to be multifactorial, involving both genetic and environmental factors. The disease is inherited in 5-10% of the cases. {ECO:0000269|PubMed:21842496}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Neuronopathy, distal hereditary motor, autosomal recessive 2 (HMNR2) [MIM:605726]: A form of distal hereditary motor neuronopathy, a heterogeneous group of neuromuscular disorders caused by selective degeneration of motor neurons in the anterior horn of the spinal cord, without sensory deficit in the posterior horn. The overall clinical picture consists of a classical distal muscular atrophy syndrome in the legs without clinical sensory loss. The disease starts with weakness and wasting of distal muscles of the anterior tibial and peroneal compartments of the legs. Later on, weakness and atrophy may expand to the proximal muscles of the lower limbs and/or to the distal upper limbs. HMNR2 is characterized by onset of distal muscle weakness and wasting affecting the lower and upper limbs in the first decade. {ECO:0000269|PubMed:26078401, ECO:0000269|PubMed:27629094}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00321; DB09014; DB00907; DB00514; DB01488; DB00574; DB00502; DB00956; DB00704; DB00540; DB06174; DB00652; DB11186; DB03575; DB05316; DB01708; DB00409; DB01104 Interacts with Q92847-1; Q99720-1; O00213-2; P17612; P50454; P37173 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Amyotrophic lateral sclerosis; Cell junction; Cell membrane; Cell projection; Cytoplasmic vesicle; Direct protein sequencing; Disease variant; Endoplasmic reticulum; Lipid droplet; Lipid transport; Membrane; Neurodegeneration; Neuropathy; Nucleus; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Synapse; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A Molecular weight (Da) 23901 Length 212 Aromaticity 0.14 Instability index 33.12 Isoelectric point 5.61 Charge (pH=7) -5.6 2D Binding mode Binding energy (Kcal/mol) -9.1  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence RWAWAALLLAVAAVLTQVVWLWLGTQSFVFQREEIAQLARQYAGLDHELAFSRLIVELRRLHPGHVLPDEELQWVFVNAGGWMGAMCLLHASLSEYVLLFGTALGSRGHSGRYWAEISDTIISGTFHQWREGTTKSEVFYPGETVVHGPGEATAVEWGPNTWMVEYGRGVIPSTLAFALADTVFSTQDFLTLFYTLRSYARGLRLELTTYLF Hydrogen bonds contact Hydrophobic contact | ||||
| 40 | Bacterial NADH-dependent enoyl-ACP reductase (Bact fabI) | 1MFP | 6.67 | |
Target general information Gen name Bact fabI Organism Escherichia coli (strain K12) Uniprot ID TTD ID Synonyms Enoyl[acylcarrierprotein] reductase [NADH] FabI; ENR of Mycobacterium tuberculosis Protein family Short-chain dehydrogenases/reductases (SDR) family, FabI subfamily Biochemical class Short-chain dehydrogenases reductase Function Catalyzes the reduction of a carbon-carbon double bond in an enoyl moiety that is covalently linked to an acyl carrier protein (ACP). Involved in the elongation cycle of fatty acid which are used in the lipid metabolism and in the biotin biosynthesis. Related diseases Pigmentary disorder, reticulate, with systemic manifestations, X-linked (PDR) [MIM:301220]: An X-linked recessive disorder characterized by recurrent infections and sterile inflammation in various organs. Diffuse skin hyperpigmentation with a distinctive reticulate pattern is universally evident by early childhood. This is later followed in many patients by hypohidrosis, corneal inflammation and scarring, enterocolitis that resembles inflammatory bowel disease, and recurrent urethral strictures. Melanin and amyloid deposition is present in the dermis. Affected males also have a characteristic facies with frontally upswept hair and flared eyebrows. Female carriers have only restricted pigmentary changes along Blaschko's lines. {ECO:0000269|PubMed:27019227}. The disease is caused by variants affecting the gene represented in this entry. XLPDR is caused by a recurrent intronic mutation that results in missplicing and reduced POLA1 expression. This leads to a decrease in cytosolic RNA:DNA hybrids and constitutive activation of type I interferon responses, but has no effect on cell replication. {ECO:0000269|PubMed:27019227}.; DISEASE: Van Esch-O'Driscoll syndrome (VEODS) [MIM:301030]: An X-linked recessive syndrome characterized by different degrees of intellectual disability, moderate to severe short stature, microcephaly, hypogonadism, and variable congenital malformations. {ECO:0000269|PubMed:31006512}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB04030; DB08265; DB01865; DB03534; DB03030; DB08605; DB02379; DB01691; DB08604 Interacts with P0AF90 EC number NA Uniprot keywords 3D-structure; Antibiotic resistance; Direct protein sequencing; Fatty acid biosynthesis; Fatty acid metabolism; Lipid biosynthesis; Lipid metabolism; NAD; Oxidoreductase; Reference proteome Protein physicochemical properties Chain ID A Molecular weight (Da) 27362 Length 258 Aromaticity 0.08 Instability index 21.64 Isoelectric point 5.57 Charge (pH=7) -4.81 2D Binding mode Binding energy (Kcal/mol) -9.1 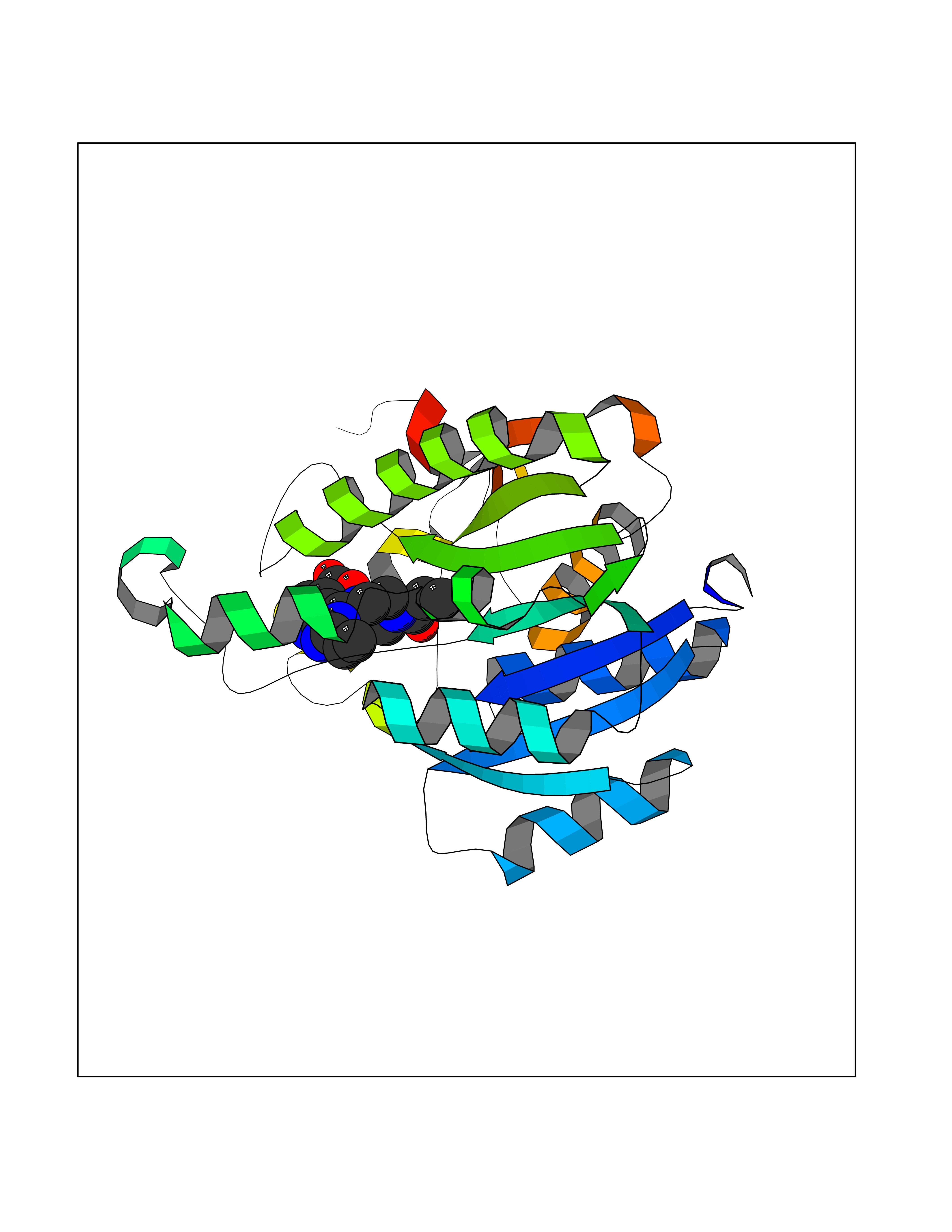 Molscript Map 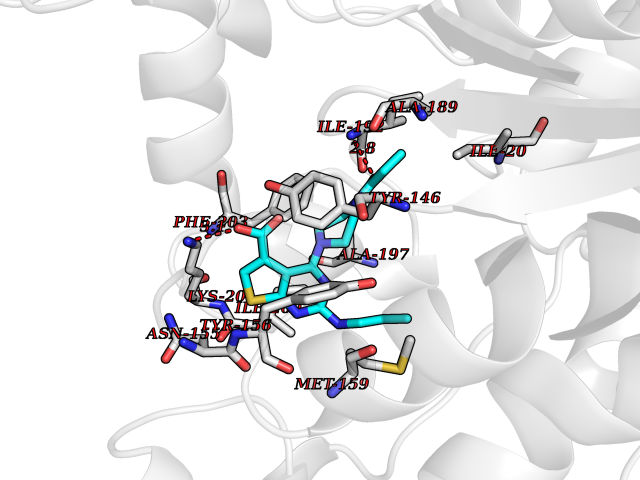 Pymol Map 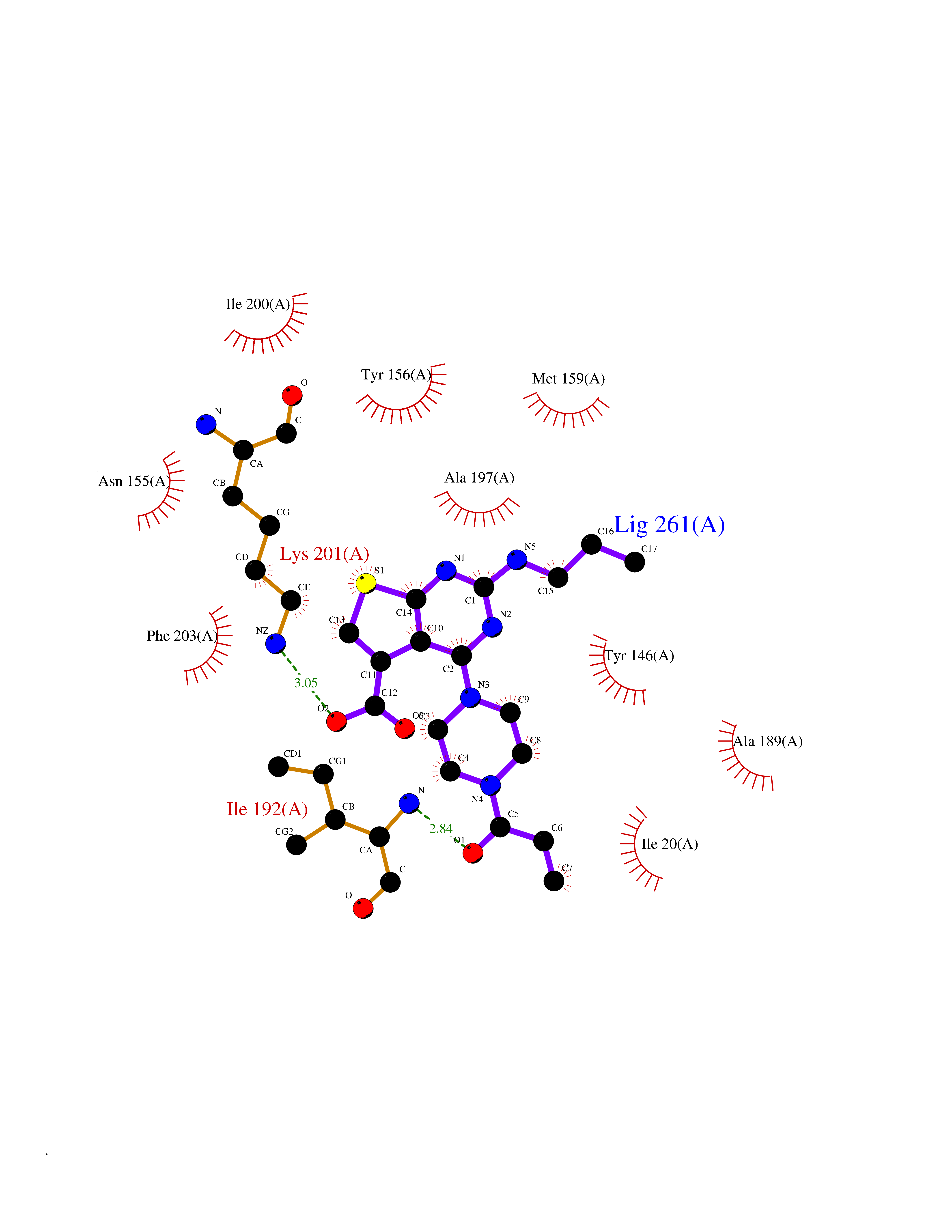 Ligplot Map 3D Binding mode Sequence GFLSGKRILVTGVASKLSIAYGIAQAMHREGAELAFTYQNDKLKGRVEEFAAQLGSDIVLQCDVAEDASIDTMFAELGKVWPKFDGFVHSIGFAPGDQLDGDYVNAVTREGFKIAHDISSYSFVAMAKACRSMLNPGSALLTLSYLGAERAIPNYNVMGLAKASLEANVRYMANAMGPEGVRVNAISAGPIRTLAASGIKDFRKMLAHCEAVTPIRRTVTIEDVGNSAAFLCSDLSAGISGEVVHVDGGFSIAAMNEL Hydrogen bonds contact Hydrophobic contact | ||||