Job Results:
Ligand
Structure
Job ID
7a36b9b0fd299388007ea8e58dfe56c6
Job name
NA
Time
2025-09-26 17:47:53
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 21 | Vitamin K epoxide reductase complex 1 (VKORC1) | 6WV3 | 6.50 | |
Target general information Gen name VKORC1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Vitamin K1 2,3-epoxide reductase subunit 1; VKORC1; VKOR; UNQ308/PRO351; MSTP576; MSTP134 Protein family VKOR family Biochemical class Short-chain dehydrogenases reductase Function Involved invitamin K metabolism. Catalytic subunit of the vitamin K epoxide reductase (VKOR) complex which reduces inactive vitamin K 2,3-epoxide to active vitamin K. Vitamin K is required for the gamma-carboxylation of various proteins, including clotting factors, and is required for normal blood coagulation, but also for normal bone development. Related diseases Combined deficiency of vitamin K-dependent clotting factors 2 (VKCFD2) [MIM:607473]: VKCFD leads to a bleeding tendency that is usually reversed by oral administration of vitamin K. {ECO:0000269|PubMed:14765194, ECO:0000269|PubMed:16270630}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Coumarin resistance (CMRES) [MIM:122700]: A condition characterized by partial or complete resistance to warfarin or other 4-hydroxycoumarin derivatives. These drugs are used as anti-coagulants for the prevention of thromboembolic diseases in subjects with deep vein thrombosis, atrial fibrillation, or mechanical heart valve replacement. {ECO:0000269|PubMed:14765194, ECO:0000269|PubMed:20946155}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01418; DB00266; DB09332; DB00170; DB00498; DB00946; DB01022; DB00682 Interacts with Q13323; Q7Z7G2; Q96BA8; Q9Y282; Q5JX71; Q96KR6; Q5T7V8; Q8TDT2; Q9NQG1; P15941-11; Q96TC7; Q9NR31; A0A0S2Z4U3; Q8TBB6; O15393-2; Q19QW4 EC number EC 1.17.4.4 Uniprot keywords 3D-structure; Alternative splicing; Disease variant; Disulfide bond; Endoplasmic reticulum; Membrane; Oxidoreductase; Proteomics identification; Quinone; Redox-active center; Reference proteome; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 42656.4 Length 381 Aromaticity 0.1 Instability index 32.12 Isoelectric point 7.73 Charge (pH=7) 1.93 2D Binding mode Binding energy (Kcal/mol) -8.86  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence KGEELFTGVVPILVELDGDVNGHKFSVRGEGEGDATNGKLTLKFICTTGKLPVPWPTLVTTLXVQCFSRYPDHMKRHDFFKSAMPEGYVQERTISFKDDGTYKTRAEVKFEGDTLVNRIELKGIDFKEDGNILGHKLEYNSTWGSPGWVRLALCLTGLVLSLYALHVKAARARDRDYRALCDVGTAISCSRVFSSRWGRGFGLVEHVLGQDSILNQSNSIFGCIFYTLQLLLGCLRTRWASVLMLLSSLVSLAGSVYLAWILFFVLYDFCIVCITTYAINVSLMWLSFRKVQENSHNVYITADKQKNGIKANFKIRHNVEDGSVQLADHYQQNTPIGDGPVLLPDNHYLSTQSVLSKDPNEKRDHMVLLEFVTAAGITHHH Hydrogen bonds contact Hydrophobic contact | ||||
| 22 | Protein arginine methyltransferase 5 (PRMT5) | 7MXC | 6.50 | |
Target general information Gen name PRMT5 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Shk1 kinase-binding protein 1 homolog; SKB1Hs; SKB1 homolog; SKB1; Protein arginine N-methyltransferase 5; Jak-binding protein 1; JBP1; IBP72; Histone-arginine N-methyltransferase PRMT5; HRMT1L5; 72 k Protein family Class I-like SAM-binding methyltransferase superfamily, Protein arginine N-methyltransferase family Biochemical class Methyltransferase Function Specifically mediates the symmetrical dimethylation of arginine residues in the small nuclear ribonucleoproteins Sm D1 (SNRPD1) and Sm D3 (SNRPD3); such methylation being required for the assembly and biogenesis of snRNP core particles. Methylates SUPT5H and may regulate its transcriptional elongation properties. Mono- and dimethylates arginine residues of myelin basic protein (MBP) in vitro. May play a role in cytokine-activated transduction pathways. Negatively regulates cyclin E1 promoter activity and cellular proliferation. Methylates histone H2A and H4 'Arg-3' during germ cell development. Methylates histone H3 'Arg-8', which may repress transcription. Methylates the Piwi proteins (PIWIL1, PIWIL2 and PIWIL4), methylation of Piwi proteins being required for the interaction with Tudor domain-containing proteins and subsequent localization to the meiotic nuage. Methylates RPS10. Attenuates EGF signaling through the MAPK1/MAPK3 pathway acting at 2 levels. First, monomethylates EGFR; this enhances EGFR 'Tyr-1197' phosphorylation and PTPN6 recruitment, eventually leading to reduced SOS1 phosphorylation. Second, methylates RAF1 and probably BRAF, hence destabilizing these 2 signaling proteins and reducing their catalytic activity. Required for induction of E-selectin and VCAM-1, on the endothelial cells surface at sites of inflammation. Methylates HOXA9. Methylates and regulates SRGAP2 which is involved in cell migration and differentiation. Acts as a transcriptional corepressor in CRY1-mediated repression of the core circadian component PER1 by regulating the H4R3 dimethylation at the PER1 promoter. Methylates GM130/GOLGA2, regulating Golgi ribbon formation. Methylates H4R3 in genes involved in glioblastomagenesis in a CHTOP- and/or TET1-dependent manner. Symmetrically methylates POLR2A, a modification that allows the recruitment to POLR2A of proteins including SMN1/SMN2 and SETX. This is required for resolving RNA-DNA hybrids created by RNA polymerase II, that form R-loop in transcription terminal regions, an important step in proper transcription termination. Along with LYAR, binds the promoter of gamma-globin HBG1/HBG2 and represses its expression. Symmetrically methylates NCL. Methylates TP53; methylation might possibly affect TP53 target gene specificity. Arginine methyltransferase that can both catalyze the formation of omega-N monomethylarginine (MMA) and symmetrical dimethylarginine (sDMA), with a preference for the formation of MMA. Related diseases Epilepsy, nocturnal frontal lobe, 3 (ENFL3) [MIM:605375]: An autosomal dominant focal epilepsy characterized by nocturnal seizures with hyperkinetic automatisms and poorly organized stereotyped movements. {ECO:0000269|PubMed:11062464, ECO:0000269|PubMed:11104662}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P01019; Q9NX04; Q8WUW1; Q08289; P78371; Q16543; Q8N8U2; P54105; P21964-2; Q9NQ92; Q16526; Q9Y6K1; Q01094; Q08426; P38919; Q14241; O15197-2; Q6ZV65; P01100; O95995; P62993; Q8TE85; Q9BX10; P62805; P31269; Q00613; Q63ZY3; P03952; Q8TBB1; P06858; Q86UQ8-1; Q96HA8; Q8WVJ2; P24928; O14744; Q86U06; Q9BRS2; O75044; Q96RU7; P31930; P40337-2; Q9BQA1; P63104; Q96E35; Q91XC0; P03418; Q6ZV65-2 EC number EC 2.1.1.320 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Biological rhythms; Chromatin regulator; Chromosome; Cytoplasm; Direct protein sequencing; Golgi apparatus; Methyltransferase; Nucleus; Proteomics identification; Reference proteome; Repressor; S-adenosyl-L-methionine; Transcription; Transcription regulation; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 71188.3 Length 621 Aromaticity 0.11 Instability index 44.6 Isoelectric point 5.95 Charge (pH=7) -9.68 2D Binding mode Binding energy (Kcal/mol) -8.86 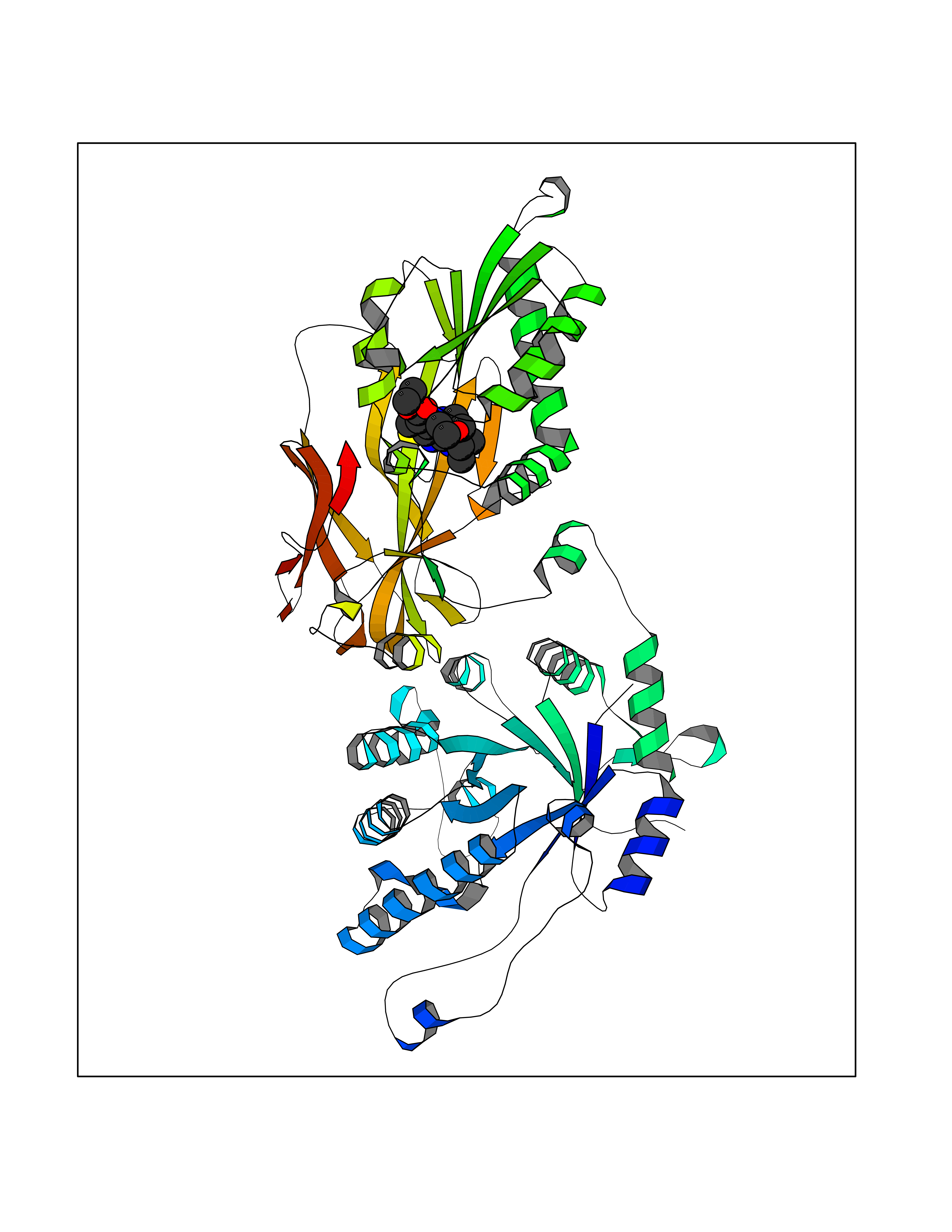 Molscript Map 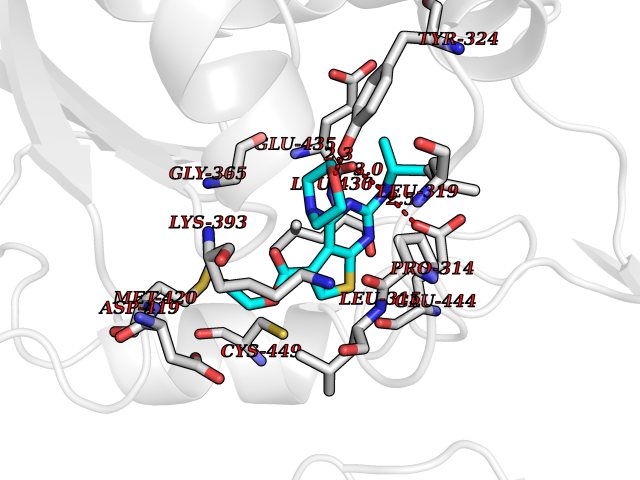 Pymol Map 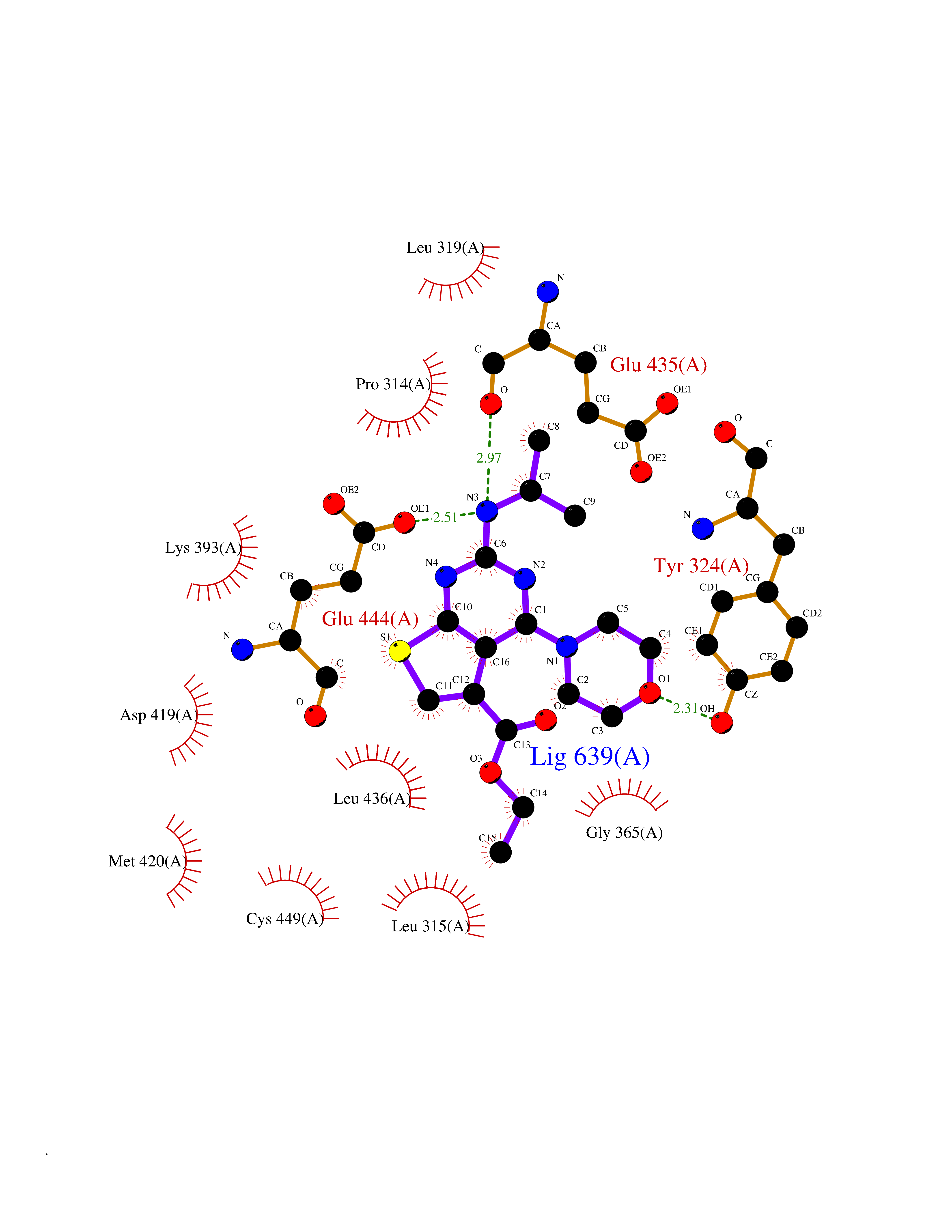 Ligplot Map 3D Binding mode Sequence RVSSGRDLNCVPEIADTLGAVAKQGFDFLCMPVFHPRFKREFIQEPAKNRPGPQTRSDLLLSGRDWNTLIVGKLSPWIRPDSKVEKIRRNSEAAMLQELNFGAYLGLPAFLLPLNQEDNTNLARVLTNHIHTGHHSSMFWMRVPLVAPEDLRDDIIENAPTTHTEEYSGEEKTWMWWHNFRTLCDYSKRIAVALEIGADLPSNHVIDRWLGEPIKAAILPTSIFLTNKKGFPVLSKMHQRLIFRLLKLEVQFIITGTNHHSCSYLQYLEYLSQNRPPPNAYELFAKGYEDYLQSPLQPLMDNLESQTYEVFEKDPIKYSQYQQAIYKCLLDRVPEEEKDTNVQVLMVLGAGRGPLVNASLRAAKQADRRIKLYAVEKNPNAVVTLENWQFEEWGSQVTVVSSDMREWVAPEKADIIVSELLGSFADNELSPECLDGAQHFLKDDGVSIPGEYTSFLAPISSSKLYNEVRACREKDRDPEAQFEMPYVVRLHNFHQLSAPQPCFTFSHPNRDPMIDNNRYCTLEFPVEVNTVLHGFAGYFETVLYQDITLSIRPETHSPGMFSWFPILFPIKQPITVREGQTICVRFWRCSNSKKVWYEWAVTAPVCSAIHNPTGRSYTIGL Hydrogen bonds contact Hydrophobic contact | ||||
| 23 | Xanthine dehydrogenase/oxidase (XDH) | 2E1Q | 6.48 | |
Target general information Gen name XDH Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Xanthine oxidase; Xanthine dehydrogenase; XDHA Protein family Xanthine dehydrogenase family Biochemical class CH/CH(2) oxidoreductase Function Catalyzes the oxidation of hypoxanthine to xanthine. Catalyzes the oxidation of xanthine to uric acid. Contributes to the generation of reactive oxygen species. Has also low oxidase activity towards aldehydes (in vitro). Key enzyme in purine degradation. Related diseases Xanthinuria 1 (XAN1) [MIM:278300]: A disorder characterized by excretion of very large amounts of xanthine in the urine and a tendency to form xanthine stones. Uric acid is strikingly diminished in serum and urine. XAN1 is due to isolated xanthine dehydrogenase deficiency. Patients can metabolize allopurinol. {ECO:0000269|PubMed:10844591, ECO:0000269|PubMed:11379872, ECO:0000269|PubMed:14551354, ECO:0000269|PubMed:9153281}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00640; DB00041; DB00437; DB00993; DB00958; DB01136; DB00856; DB00515; DB00746; DB03328; DB00997; DB03516; DB12466; DB04854; DB03147; DB04335; DB01020; DB00583; DB00170; DB01033; DB00157; DB03841; DB00336; DB01250; DB05262; DB06478; DB01168; DB00339; DB00127; DB01685; DB00831 Interacts with Q9Y3R0-3 EC number NA Uniprot keywords 2Fe-2S; 3D-structure; Cytoplasm; Disease variant; Disulfide bond; FAD; Flavoprotein; Iron; Iron-sulfur; Metal-binding; Molybdenum; NAD; Oxidoreductase; Peroxisome; Proteomics identification; Reference proteome; Secreted Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 143697 Length 1307 Aromaticity 0.08 Instability index 37.9 Isoelectric point 8.01 Charge (pH=7) 7.07 2D Binding mode Binding energy (Kcal/mol) -8.84  Molscript Map 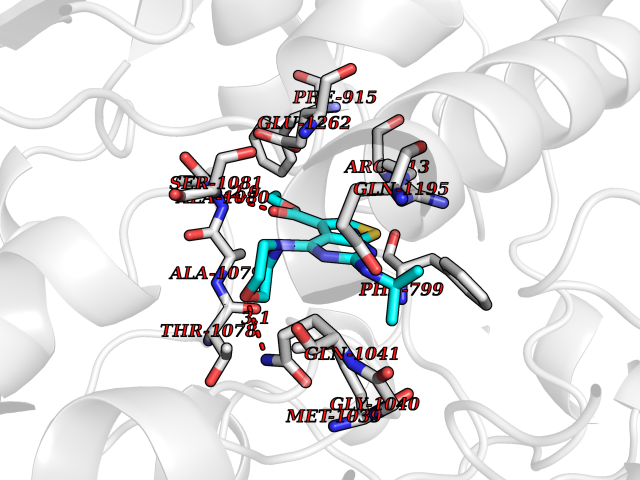 Pymol Map 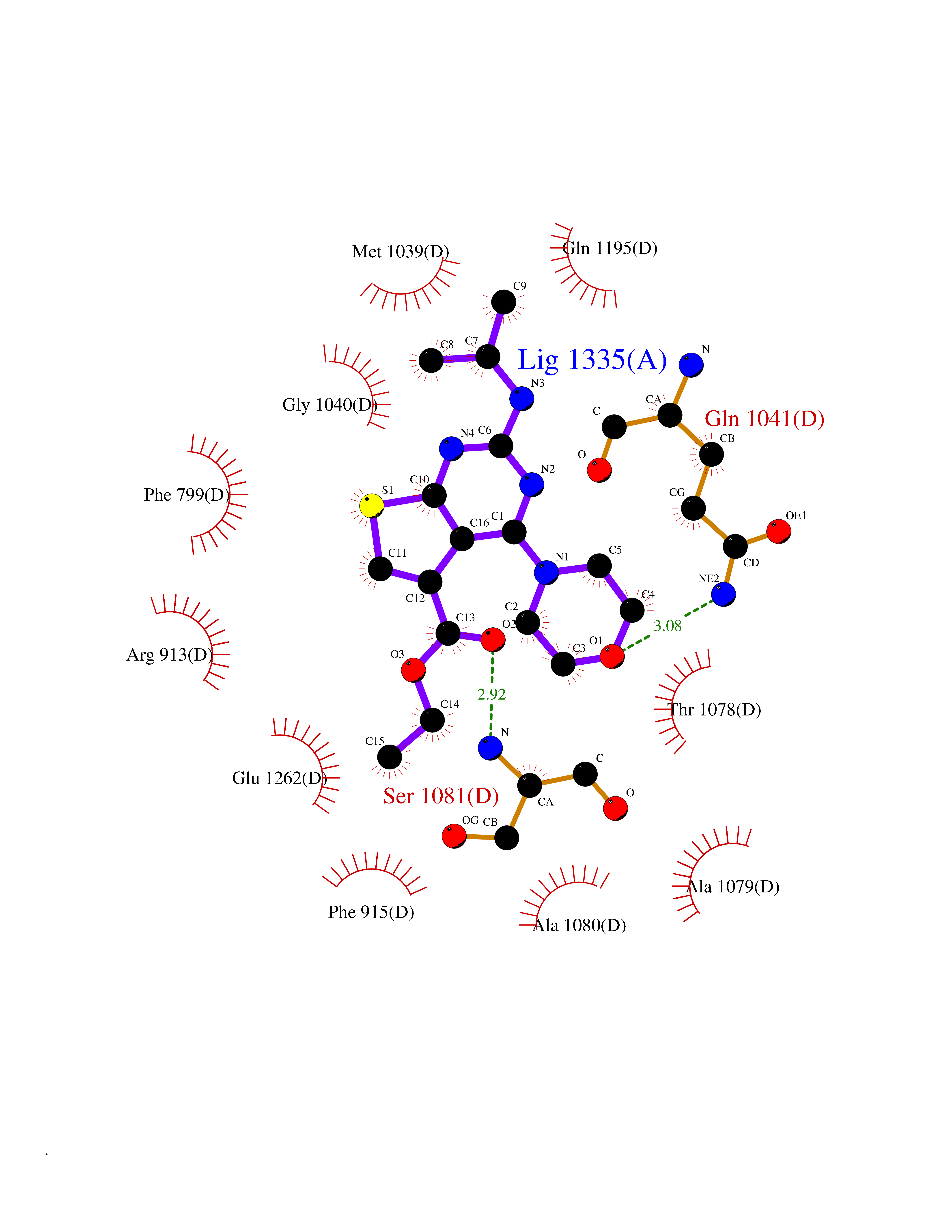 Ligplot Map 3D Binding mode Sequence ADKLVFFVNGRKVVEKNADPETTLLAYLRRKLGLSGTKLGCGEGGCGACTVMLSKYDRLQNKIVHFSANACLAPICSLHHVAVTTVEGIGSTKTRLHPVQERIAKSHGSQCGFCTPGIVMSMYTLLRNQPEPTMEEIENAFQGNLCRCTGYRPILQGFRTFARDGSPSLFKPEEFTPLDPTQEPIFPPELLRLKDTPRKQLRFEGERVTWIQASTLKELLDLKAQHPDAKLVVGNTEIGIEMKFKNMLFPMIVCPAWIPELNSVEHGPDGISFGAACPLSIVEKTLVDAVAKLPAQKTEVFRGVLEQLRWFAGKQVKSVASVGGNIITASPISDLNPVFMASGAKLTLVSRGTRRTVQMDHTFFPGYRKTLLSPEEILLSIEIPYSREGEYFSAFKQASRREDDIAKVTSGMRVLFKPGTTEVQELALCYGGMANRTISALKTTQRQLSKLWKEELLQDVCAGLAEELHLPPDAPGGMVDFRCTLTLSFFFKFYLTVLQKLGQENLEDKCGKLDPTFASATLLFQKDPPADVQLFQEVPKGQSEEDMVGRPLPHLAADMQASGEAVYCDDIPRYENELSLRLVTSTRAHAKIKSIDTSEAKKVPGFVCFISADDVPGSNITGICNDETVFAKDKVTCVGHIIGAVVADTPEHTQRAAQGVKITYEELPAIITIEDAIKNNSFYGPELKIEKGDLKKGFSEADNVVSGEIYIGGQEHFYLETHCTIAVPKGEAGEMELFVSTQNTMKTQSFVAKMLGVPANRIVVRVKRMGGGFGGKVTRSTVVSTAVALAAYKTGRPVRCMLDRDEDMLITGGRHPFLARYKVGFMKTGTVVALEVDHFSNVGNTQDLSQSIMERALFHMDNCYKIPNIRGTGRLCKTNLPSNTAFRGFGGPQGMLIAECWMSEVAVTCGMPAEEVRRKNLYKEGDLTHFNQKLEGFTLPRCWEECLASSQYHARKSEVDKFNKENCWKKRGLCIIPTKFGISFTVPFLNQAGALLHVYTDGSVLLTHGGTEMGQGLHTKMVQVASRALKIPTSKIYISETSTNTVPNTSPTAASVSADLNGQAVYAACQTILKRLEPYKKKNPSGSWEDWVTAAYMDTVSLSATGFYRTPNLGYSFETNSGNPFHYFSYGVACSEVEIDCLTGDHKNLRTDIVMDVGSSLNPAIDIGQVEGAFVQGLGLFTLEELHYSPEGSLHTRGPSTYKIPAFGSIPIEFRVSLLRDCPNKKAIYASKAVGEPPLFLAASIFFAIKDAIRAARAQHTGNNVKELFRLDSPATPEKIRNACVDKFTTLCVTGVPENCKPWSVRV Hydrogen bonds contact Hydrophobic contact | ||||
| 24 | Aldo-keto reductase family 1 member C3 | 1S1P | 6.48 | |
Target general information Gen name AKR1C3 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms PGFS;DDH1;HSD17B5;KIAA0119 Protein family Aldo/keto reductase family Biochemical class Oxidoreductase Function 15-hydroxyprostaglandin-D dehydrogenase (NADP+) activity.Alditol:NADP+ 1-oxidoreductase activity.Aldo-keto reductase (NADP) activity.Androsterone dehydrogenase activity.Delta4-3-oxosteroid 5beta-reductase activity.Dihydrotestosterone 17-beta-dehydrogenase activity.Geranylgeranyl reductase activity.Indanol dehydrogenase activity.Ketoreductase activity.Ketosteroid monooxygenase activity.NADP-retinol dehydrogenase activity.Oxidoreductase activity, acting on NAD(P)H, quinone or similar compound as acceptor.Phenanthrene 9,10-monooxygenase activity.Prostaglandin D2 11-ketoreductase activity.Prostaglandin-F synthase activity.Prostaglandin H2 endoperoxidase reductase activity.Retinal dehydrogenase activity.Retinol dehydrogenase activity.Testosterone 17-beta-dehydrogenase (NADP+) activity.Testosterone dehydrogenase (NAD+) activity.Trans-1,2-dihydrobenzene-1,2-diol dehydrogenase activity. Related diseases Neurodevelopmental disorder with language impairment and behavioral abnormalities (NEDLIB) [MIM:618917]: A neurodevelopmental disorder characterized by global developmental delay, impaired intellectual development, poor or absent speech, and behavioral abnormalities, such as autism spectrum disorder, repetitive behaviors, and hyperactivity. Some patients develop seizures and manifest developmental regression. {ECO:0000269|PubMed:31300657}. The disease is caused by variants affecting the gene represented in this entry. The genetic variation producing the missense variant p.Q607E, associated with NEDLIB, is predicted to deeply affect RNA editing. In a physiological context, the adenosine (A) residue of the original glutamine (Q) codon CAG is post-transcriptionaly edited to inosine (I) by ADAR2, leading to a codon recognized by the ribosome as arginine (R). The glutamate (E) codon GAG, resulting from the genetic variation, is predicted to be edited 90% less than the normal CAG codon. If edited, the codon GIG would be translated as p.Q607G. {ECO:0000305|PubMed:31300657}. Drugs (DrugBank ID) DB07700; DB01561; DB01536; DB00997; DB01039; DB02266; DB13751; DB00328; DB06077; DB00959; DB00157; DB03461; DB09074; DB00776; DB02056; DB01698; DB02901 Interacts with P17516 EC number 1.1.1.-; 1.1.1.188; 1.1.1.210; 1.1.1.239; 1.1.1.357; 1.1.1.53; 1.1.1.62; 1.1.1.64 Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; Lipid metabolism; NAD; NADP; Oxidoreductase; Proteomics identification; Reference proteome Protein physicochemical properties Chain ID A Molecular weight (Da) 35846.8 Length 315 Aromaticity 0.09 Instability index 47.59 Isoelectric point 8.54 Charge (pH=7) 4.51 2D Binding mode Binding energy (Kcal/mol) -8.83  Molscript Map 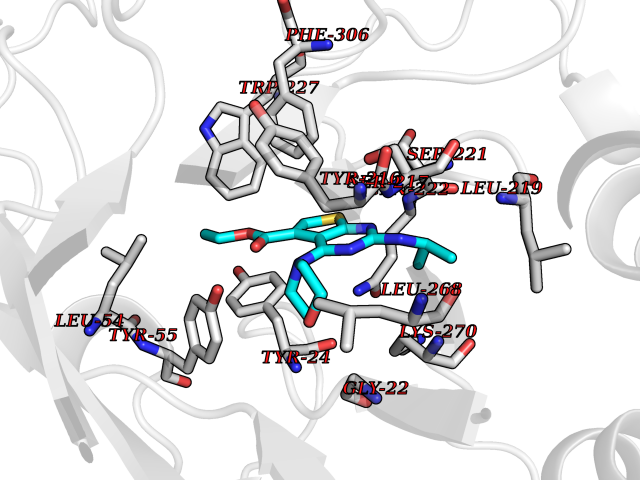 Pymol Map 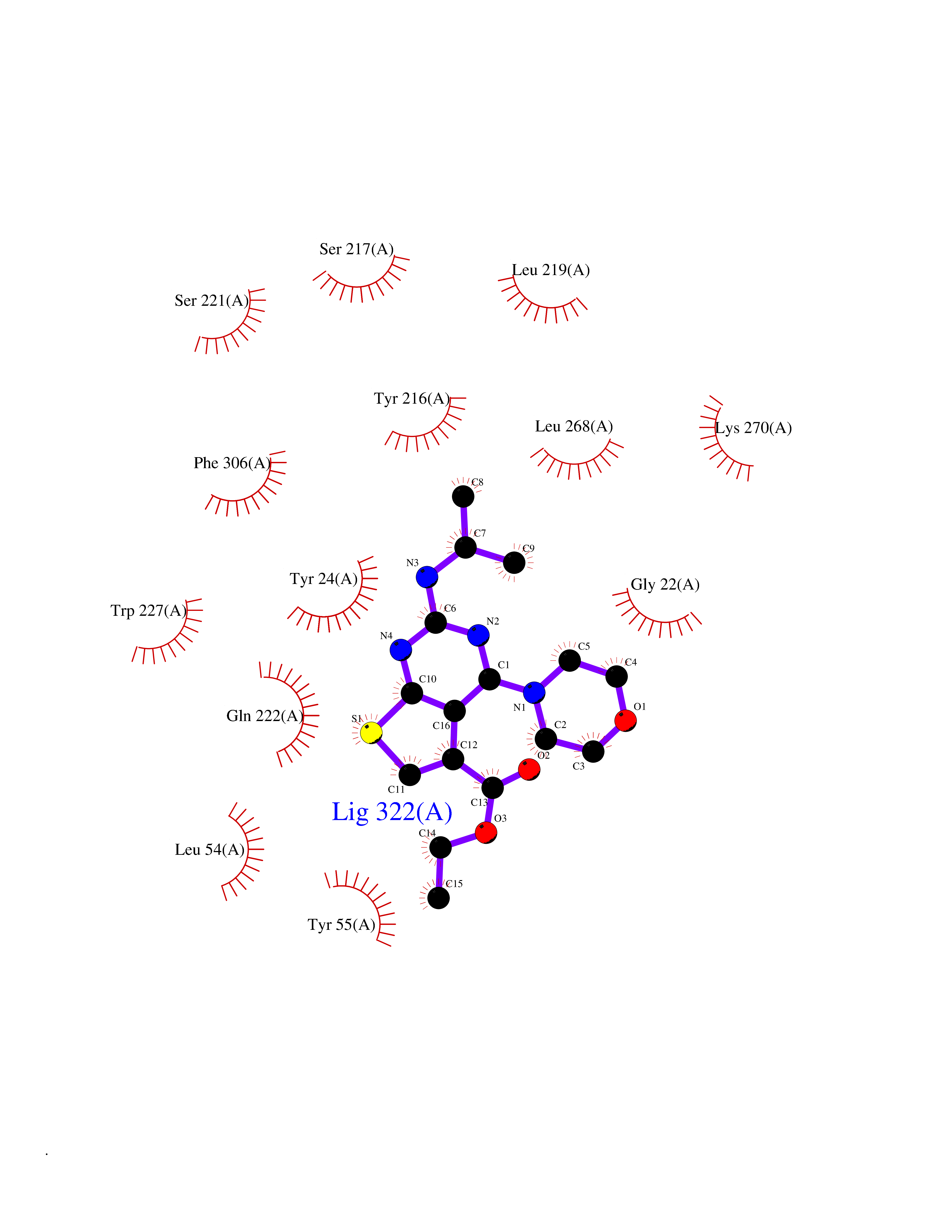 Ligplot Map 3D Binding mode Sequence QCVKLNDGHFMPVLGFGTYAPPEVPRSKALEVTKLAIEAGFRHIDSAHLYNNEEQVGLAIRSKIADGSVKREDIFYTSKLWSTFHRPELVRPALENSLKKAQLDYVDLYLIHSPMSLKPGEELSPTDENGKVIFDIVDLCTTWEAMEKCKDAGLAKSIGVSNFNRRQLEMILNKPGLKYKPVCNQVECHPYFNRSKLLDFCKSKDIVLVAYSALGSQRDKRWVDPNSPVLLEDPVLCALAKKHKRTPALIALRYQLQRGVVVLAKSYNEQRIRQNVQVFEFQLTAEDMKAIDGLDRNLHYFNSDSFASHPNYPYS Hydrogen bonds contact Hydrophobic contact | ||||
| 25 | Melatonin receptor type 1A (MTNR1A) | 7DB6 | 6.48 | |
Target general information Gen name MTNR1A Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Mel1a receptor; Mel1AR; Mel-1A-R Protein family G-protein coupled receptor 1 family Biochemical class GPCR rhodopsin Function Likely to mediate the reproductive and circadian actions of melatonin. The activity of this receptor is mediated by pertussis toxin sensitive G proteins that inhibit adenylate cyclase activity. High affinity receptor for melatonin. Related diseases Spermatogenic failure 5 (SPGF5) [MIM:243060]: An infertility disorder caused by spermatogenesis defects. Semen from affected men show close to 100% morphologically abnormal multiflagellar spermatozoa with low motility, oversized irregular heads, and abnormal midpiece and acrosome. {ECO:0000269|PubMed:17435757, ECO:0000269|PubMed:21733974}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB06594; DB01065; DB00980; DB02709; DB09071 Interacts with P27797; A8MQ03; Q8IUG1; P49286; O76081; P57088 EC number NA Uniprot keywords 3D-structure; Cell membrane; Disulfide bond; G-protein coupled receptor; Glycoprotein; Membrane; Receptor; Reference proteome; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID D Molecular weight (Da) 31301 Length 276 Aromaticity 0.15 Instability index 37.33 Isoelectric point 9.22 Charge (pH=7) 9.92 2D Binding mode Binding energy (Kcal/mol) -8.83 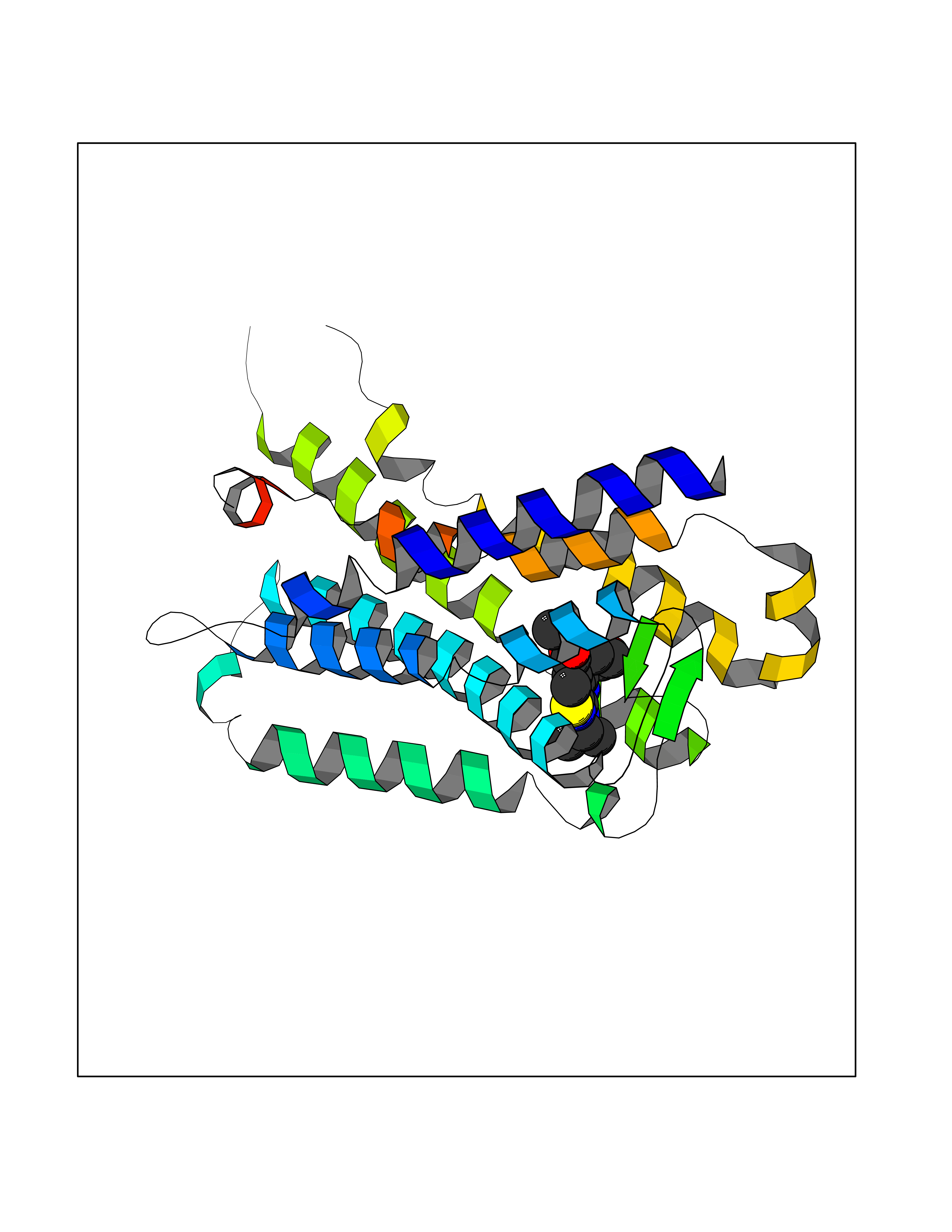 Molscript Map 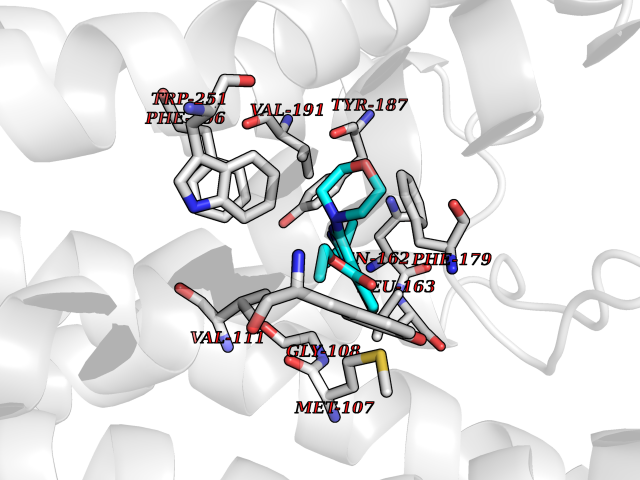 Pymol Map 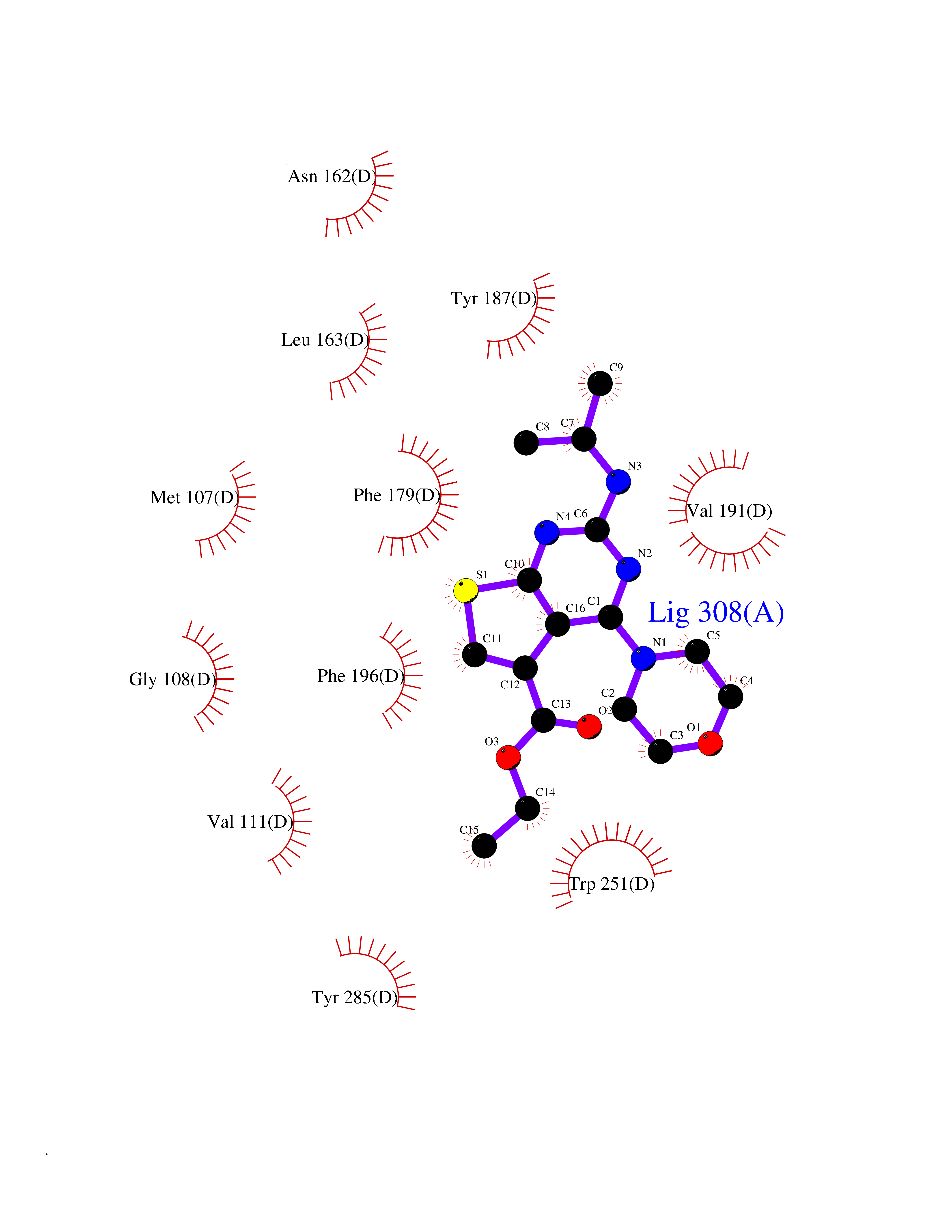 Ligplot Map 3D Binding mode Sequence RPSWLASALACVLIFTIVVDILGNLLVILSVYRNKKLRNAGNIFVVSLAVADLVVAIYPYPLVLMSIFNNGWNLGYLHCQVSGFLMGLSVIGSIFNITGIAINRYCYICHSLKYDKLYSSKNSLCYVLLIWLLTLAAVLPNLRAGTLQYDPRIYSCTFAQSVSSAYTIAVVVFHFLVPMIIVIFCYLRIWILVLQVRQRVPQDFRNFVTMFVVFVLFAICWAPLNFIGLAVASDPASMVPRIPEWLFVASYYMAYFNSCLNAIIYGLLNQNFRKEY Hydrogen bonds contact Hydrophobic contact | ||||
| 26 | Glucarate dehydratase | 1EC8 | 6.47 | |
Target general information Gen name gudD Organism Escherichia coli (strain K12) Uniprot ID TTD ID NA Synonyms b2787;ygcX;JW2758 Protein family Mandelate racemase/muconate lactonizing enzyme family, GlucD subfamily Biochemical class Lyase Function Glucarate dehydratase activity.Magnesium ion binding. Related diseases Pigmentary disorder, reticulate, with systemic manifestations, X-linked (PDR) [MIM:301220]: An X-linked recessive disorder characterized by recurrent infections and sterile inflammation in various organs. Diffuse skin hyperpigmentation with a distinctive reticulate pattern is universally evident by early childhood. This is later followed in many patients by hypohidrosis, corneal inflammation and scarring, enterocolitis that resembles inflammatory bowel disease, and recurrent urethral strictures. Melanin and amyloid deposition is present in the dermis. Affected males also have a characteristic facies with frontally upswept hair and flared eyebrows. Female carriers have only restricted pigmentary changes along Blaschko's lines. {ECO:0000269|PubMed:27019227}. The disease is caused by variants affecting the gene represented in this entry. XLPDR is caused by a recurrent intronic mutation that results in missplicing and reduced POLA1 expression. This leads to a decrease in cytosolic RNA:DNA hybrids and constitutive activation of type I interferon responses, but has no effect on cell replication. {ECO:0000269|PubMed:27019227}.; DISEASE: Van Esch-O'Driscoll syndrome (VEODS) [MIM:301030]: An X-linked recessive syndrome characterized by different degrees of intellectual disability, moderate to severe short stature, microcephaly, hypogonadism, and variable congenital malformations. {ECO:0000269|PubMed:31006512}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03237; DB03212; DB03603; DB03734 Interacts with NA EC number 4.2.1.40 Uniprot keywords 3D-structure; Direct protein sequencing; Lyase; Magnesium; Metal-binding; Reference proteome Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 48707 Length 442 Aromaticity 0.08 Instability index 26.98 Isoelectric point 5.69 Charge (pH=7) -10.89 2D Binding mode Binding energy (Kcal/mol) -8.83 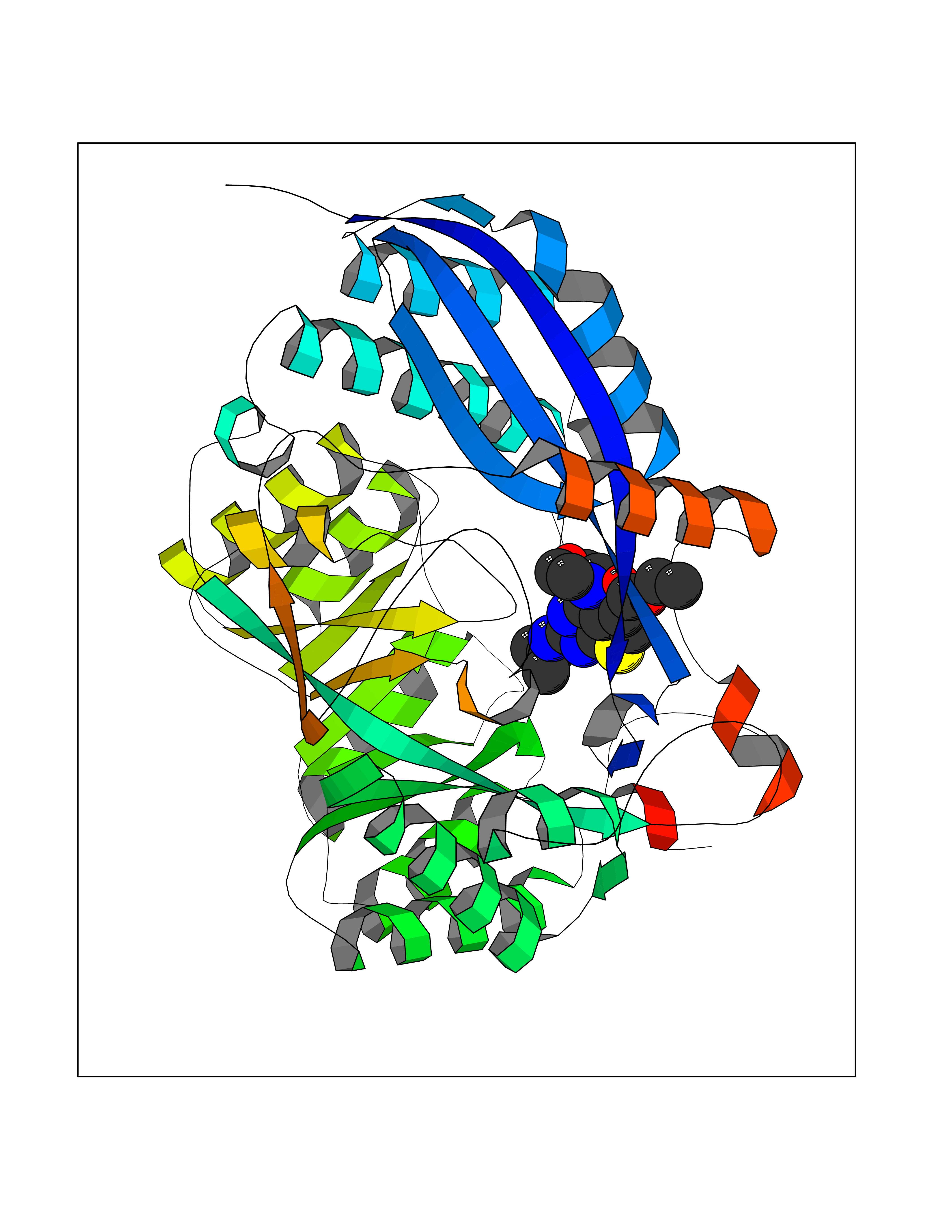 Molscript Map 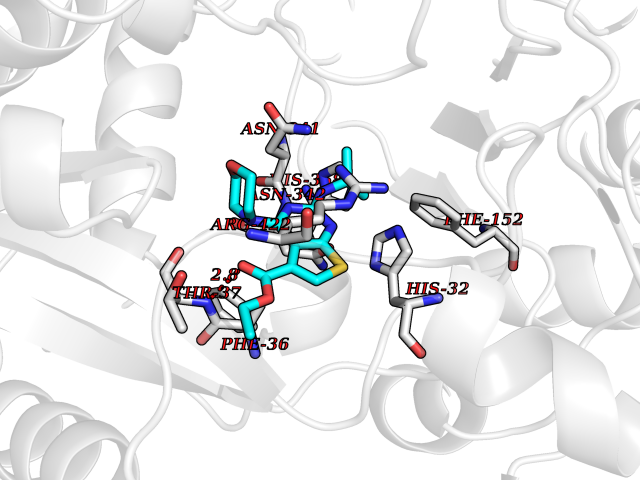 Pymol Map 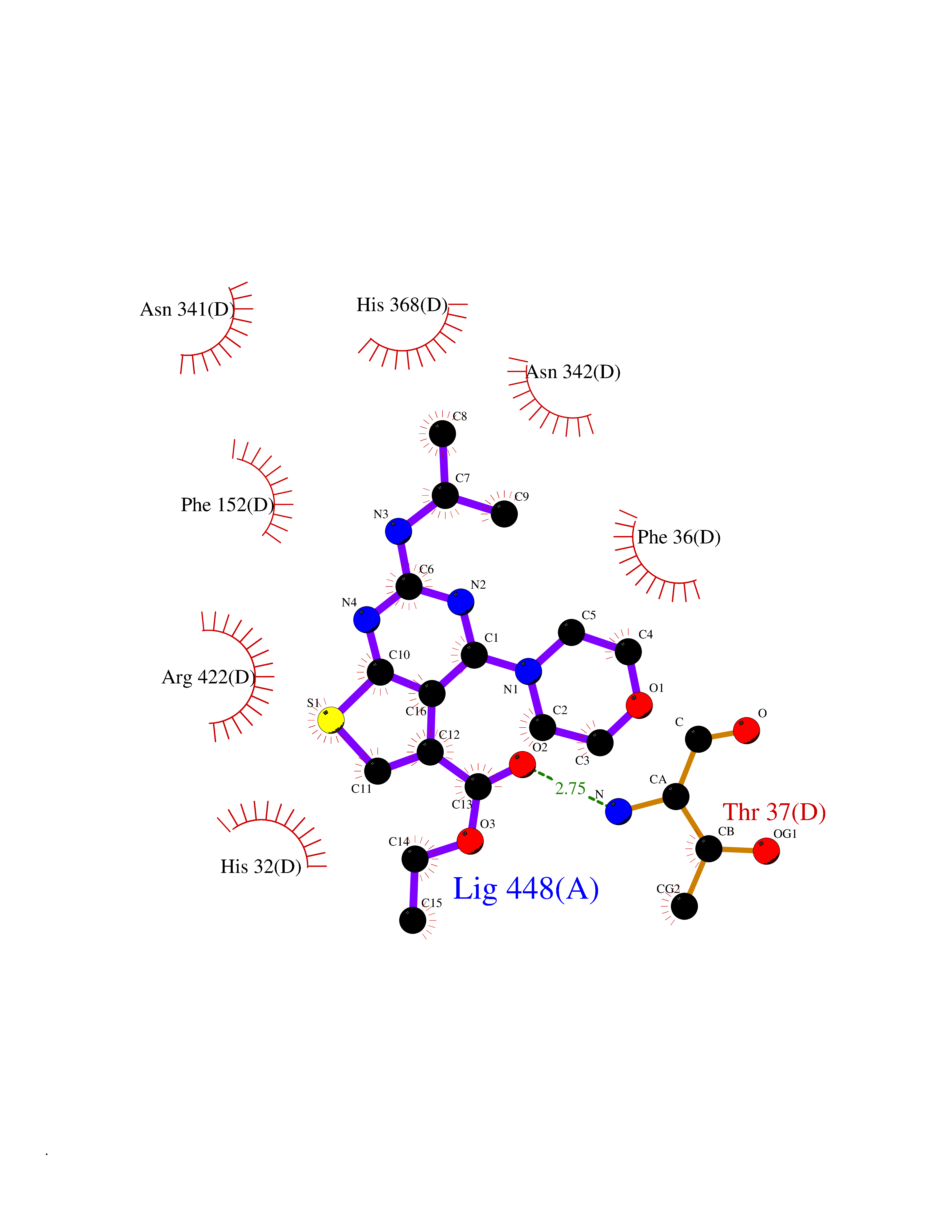 Ligplot Map 3D Binding mode Sequence FTTPVVTEMQVIPVAGHDSMLMNLSGAHAPFFTRNIVIIKDNSGHTGVGEIPGGEKIRKTLEDAIPLVVGKTLGEYKNVLTLVRNTFADRDAGGRGLQTFDLRTTIHVVTGIEAAMLDLLGQHLGVNVASLLGDGQQRSEVEMLGYLFFVGNRKATPLPYQSQPDDSCDWYRLRHEEAMTPDAVVRLAEAAYEKYGFNDFKLKGGVLAGEEEAESIVALAQRFPQARITLDPNGAWSLNEAIKIGKYLKGSLAYAEDPCGAEQGFSGREVMAEFRRATGLPTATNMIATDWRQMGHTLSLQSVDIPLADPHFWTMQGSVRVAQMCHEFGLTWGSHSNNHFDISLAMFTHVAAAAPGKITAIDTHWIWQEGNQRLTKEPFEIKGGLVQVPEKPGLGVEIDMDQVMKAHELYQKHGLGARDDAMGMQYLIPGWTFDNKRPCMVR Hydrogen bonds contact Hydrophobic contact | ||||
| 27 | Thiopurine S-methyltransferase | 2BZG | 6.47 | |
Target general information Gen name TPMT Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Class I-like SAM-binding methyltransferase superfamily, TPMT family Biochemical class Transferase Function Thiopurine S-methyltransferase activity. Related diseases Cerebral creatine deficiency syndrome 3 (CCDS3) [MIM:612718]: An autosomal recessive disorder characterized by developmental delay/regression, intellectual disability, severe disturbance of expressive and cognitive speech, and severe depletion of creatine/phosphocreatine in the brain. Most patients develop a myopathy characterized by muscle weakness and atrophy later in life. {ECO:0000269|PubMed:11555793, ECO:0000269|PubMed:20682460, ECO:0000269|PubMed:22386973, ECO:0000269|PubMed:23660394, ECO:0000269|PubMed:23770102, ECO:0000269|PubMed:26490222, ECO:0000269|PubMed:27233232}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Fanconi renotubular syndrome 1 (FRTS1) [MIM:134600]: A form of Fanconi renotubular syndrome, a disease due to a generalized dysfunction of the proximal kidney tubule resulting in decreased solute and water reabsorption. Patients have polydipsia and polyuria with phosphaturia, glycosuria and aminoaciduria. They may develop hypophosphatemic rickets or osteomalacia, acidosis and a tendency toward dehydration. Some eventually develop renal insufficiency. FRTS1 inheritance is autosomal dominant. {ECO:0000269|PubMed:29654216}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00993; DB00436; DB01327; DB01033; DB01250; DB01021 Interacts with Q8TAP4-4; Q15047-2; P61981 EC number 2.1.1.67 Uniprot keywords 3D-structure; Acetylation; Cytoplasm; Direct protein sequencing; Methyltransferase; Phosphoprotein; Proteomics identification; Reference proteome; S-adenosyl-L-methionine; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 25971.5 Length 229 Aromaticity 0.12 Instability index 32.58 Isoelectric point 6.74 Charge (pH=7) -0.6 2D Binding mode Binding energy (Kcal/mol) -8.83 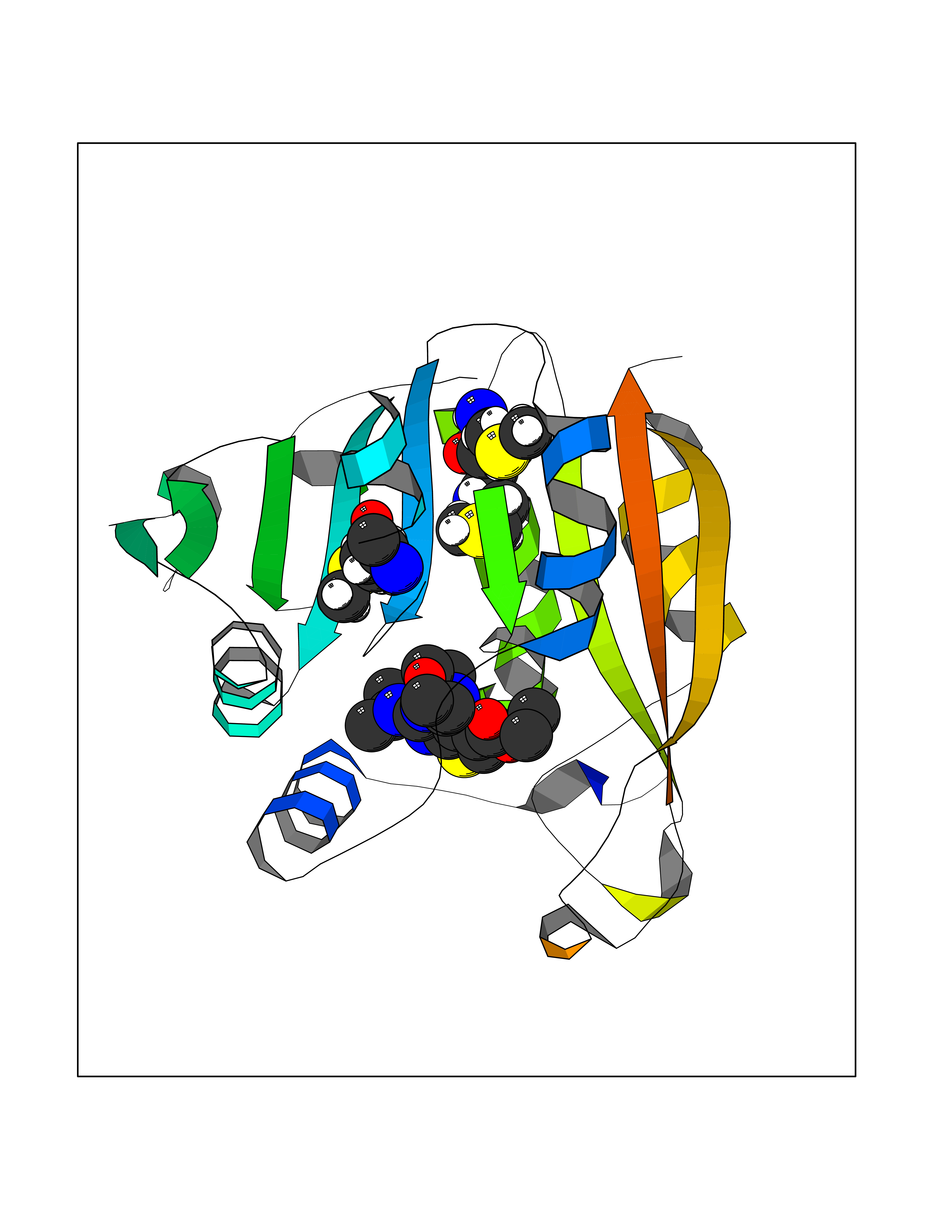 Molscript Map 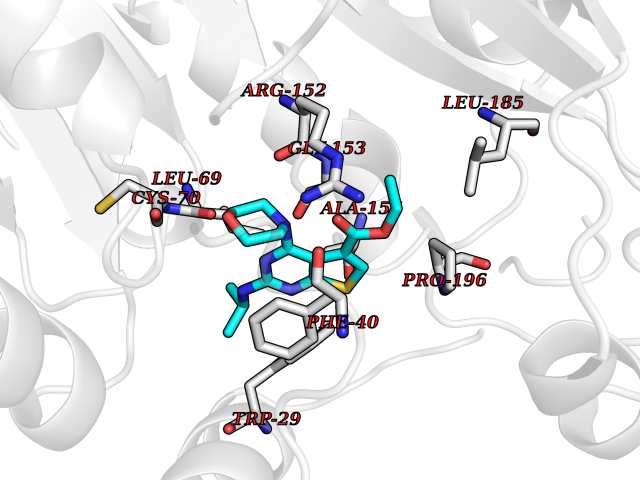 Pymol Map 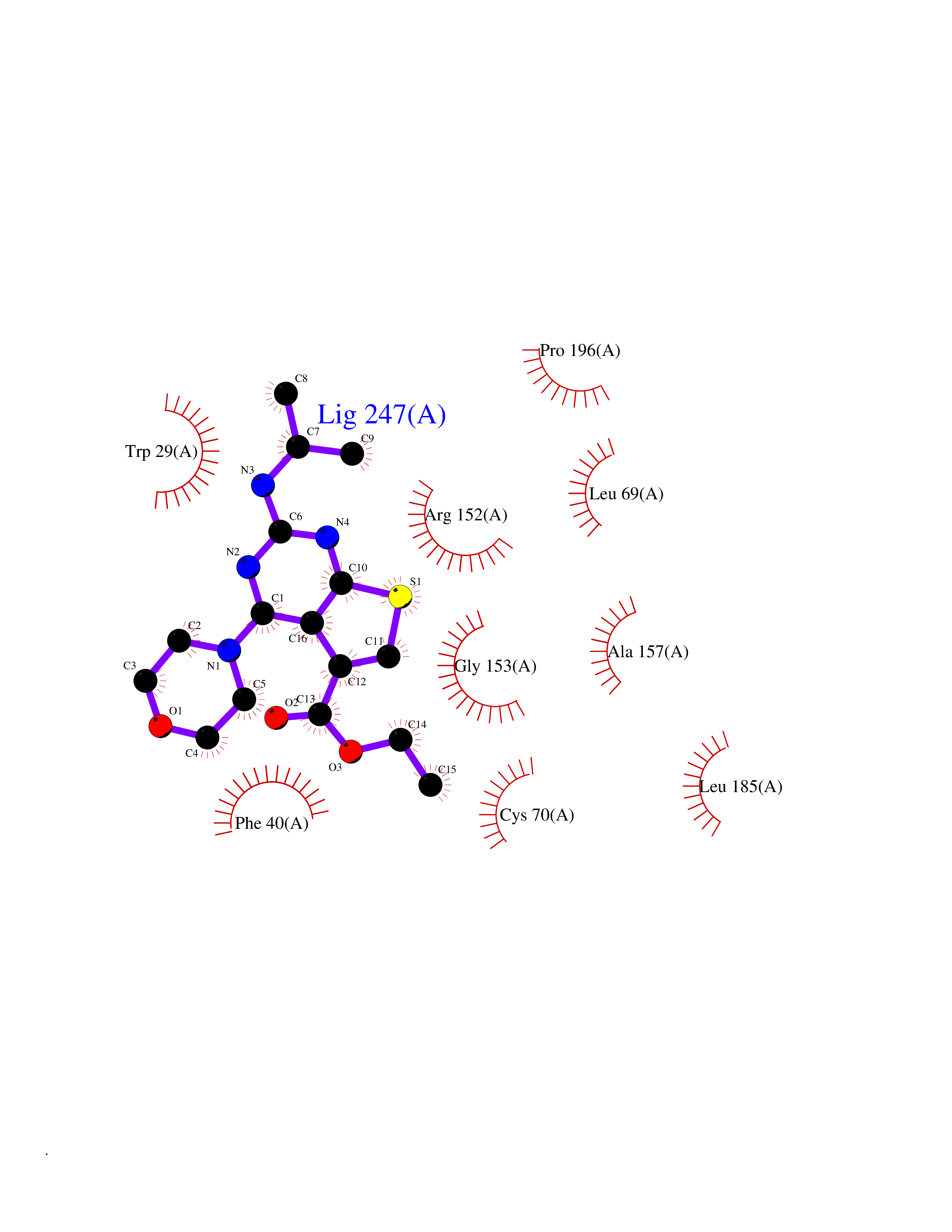 Ligplot Map 3D Binding mode Sequence EVQKNQVLTLEEWQDKWVNGKTAFHQEQGHQLLKKHLDTFLKGKSGLRVFFPLCGKAVEXKWFADRGHSVVGVEISELGIQEFFTEQNLSYSEEPITEIPGTKVFKSSSGNISLYCCSIFDLPRTNIGKFDXIWDRGALVAINPGDRKCYADTXFSLLGKKFQYLLCVLSYDPTKHPGPPFYVPHAEIERLFGKICNIRCLEKVDAFEERHKSWGIDCLFEKLYLLTEK Hydrogen bonds contact Hydrophobic contact | ||||
| 28 | GTPase HRas (HRAS) | 7L0F | 6.46 | |
Target general information Gen name HRAS Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms p21ras; cHras; c-H-ras; Transforming protein p21; HaRas; Ha-Ras; H-Ras-1; GTPase HRas, Nterminally processed Protein family Small GTPase superfamily, Ras family Biochemical class Small GTPase Function Ras proteins bind GDP/GTP and possess intrinsic GTPase activity. Involved in the activation of Ras protein signal transduction. Related diseases Costello syndrome (CSTLO) [MIM:218040]: A rare condition characterized by prenatally increased growth, postnatal growth deficiency, intellectual disability, distinctive facial appearance, cardiovascular abnormalities (typically pulmonic stenosis, hypertrophic cardiomyopathy and/or atrial tachycardia), tumor predisposition, skin and musculoskeletal abnormalities. {ECO:0000269|PubMed:16170316, ECO:0000269|PubMed:16329078, ECO:0000269|PubMed:16443854, ECO:0000269|PubMed:17054105, ECO:0000269|PubMed:18039947, ECO:0000269|PubMed:18247425, ECO:0000269|PubMed:19995790}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Congenital myopathy with excess of muscle spindles (CMEMS) [MIM:218040]: Variant of Costello syndrome. {ECO:0000269|PubMed:17412879}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Thyroid cancer, non-medullary, 2 (NMTC2) [MIM:188470]: A form of non-medullary thyroid cancer (NMTC), a cancer characterized by tumors originating from the thyroid follicular cells. NMTCs represent approximately 95% of all cases of thyroid cancer and are classified into papillary, follicular, Hurthle cell, and anaplastic neoplasms. {ECO:0000269|PubMed:12727991}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Mutations which change positions 12, 13 or 61 activate the potential of HRAS to transform cultured cells and are implicated in a variety of human tumors. {ECO:0000269|PubMed:3670300}.; DISEASE: Bladder cancer (BLC) [MIM:109800]: A malignancy originating in tissues of the urinary bladder. It often presents with multiple tumors appearing at different times and at different sites in the bladder. Most bladder cancers are transitional cell carcinomas that begin in cells that normally make up the inner lining of the bladder. Other types of bladder cancer include squamous cell carcinoma (cancer that begins in thin, flat cells) and adenocarcinoma (cancer that begins in cells that make and release mucus and other fluids). Bladder cancer is a complex disorder with both genetic and environmental influences. {ECO:0000269|PubMed:6298635, ECO:0000269|PubMed:6844927}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Schimmelpenning-Feuerstein-Mims syndrome (SFM) [MIM:163200]: A disease characterized by sebaceous nevi, often on the face, associated with variable ipsilateral abnormalities of the central nervous system, ocular anomalies, and skeletal defects. Many oral manifestations have been reported, not only including hypoplastic and malformed teeth, and mucosal papillomatosis, but also ankyloglossia, hemihyperplastic tongue, intraoral nevus, giant cell granuloma, ameloblastoma, bone cysts, follicular cysts, oligodontia, and odontodysplasia. Sebaceous nevi follow the lines of Blaschko and these can continue as linear intraoral lesions, as in mucosal papillomatosis. {ECO:0000269|PubMed:22683711}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB04315; DB04137; DB02210; DB08751; DB03226; DB15588 Interacts with Q99996-3; P53677-2; P10398; Q9NXL2-1; Q9UII2; Q9H7T9; Q00994; Q9H2G9; P15056; Q7Z569; Q5PSV4; Q9ULD4-2; Q96LL4; Q96HB5; Q49A88-3; Q96GN5-2; P24941; O95674; Q9H3R5; Q9Y4F5-3; Q86XR8; Q494V2-2; Q8WUX9; Q14117; Q9Y6W6; O14641; A0AVK6; Q8NB25; Q8IZU1; O94868-3; P15407; P15408; P52655; Q96CS2; Q9BT25; Q8IV36; O43248; Q53GQ0; P10809; Q8NDH6-2; Q8IY31-2; Q8NA54; Q13352; P28290-2; Q9BVG8-5; Q2M2Z5; Q6P597; P57682; Q9UH77; P08727; Q14525; Q14847-2; Q96LR2; P27338; Q99558; Q96EZ8; Q8TAC0; Q5JXC2; Q8NEH6; Q9Y605; Q96HT8; Q9GZM8; P21359; Q8N5V2; Q6PHZ7; Q9BZ95-3; A5D8V7; O43482; Q9BR81; O15534; Q9BUL5; O00329; O00329-2; Q9UPR0; Q96I34; Q15435-3; P04049; P11233; Q15311; Q12967; Q9NS23-2; Q9NS23-4; Q8WWW0; Q8TBY0; Q9P2K3-2; Q9NZL6; O15211; Q8IXN7; Q13671; Q13671-1; Q8WVD3; Q9BY12-3; Q13435; Q12824; Q13573; Q07889; Q86W54-2; Q92783-2; O75886; Q13586; Q8N4C7; O75528; P54274-2; Q9BXU0; Q5T0J7-2; Q5T1C6; Q8IUR5-4; P36406; Q86WT6-2; Q99598; Q6PF05; Q9UGJ1-2; Q9Y5Z9; P22415; Q495M9; Q9H270; Q8NEZ2; P19544-6; O43829; Q9C0F3; Q7Z637; Q86V28; P42337; Q9Z0S9; Q9EQZ6; P27671; Q5EBH1; Q5EBH1-1; P52306-5 EC number NA Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Cell membrane; Cytoplasm; Direct protein sequencing; Disease variant; Glycoprotein; Golgi apparatus; GTP-binding; Hydrolase; Isopeptide bond; Lipoprotein; Membrane; Methylation; Nucleotide-binding; Nucleus; Palmitate; Prenylation; Proteomics identification; Proto-oncogene; Reference proteome; S-nitrosylation; Ubl conjugation Protein physicochemical properties Chain ID E,F Molecular weight (Da) 28737.2 Length 259 Aromaticity 0.1 Instability index 30.69 Isoelectric point 5.64 Charge (pH=7) -4.15 2D Binding mode Binding energy (Kcal/mol) -8.81  Molscript Map  Pymol Map 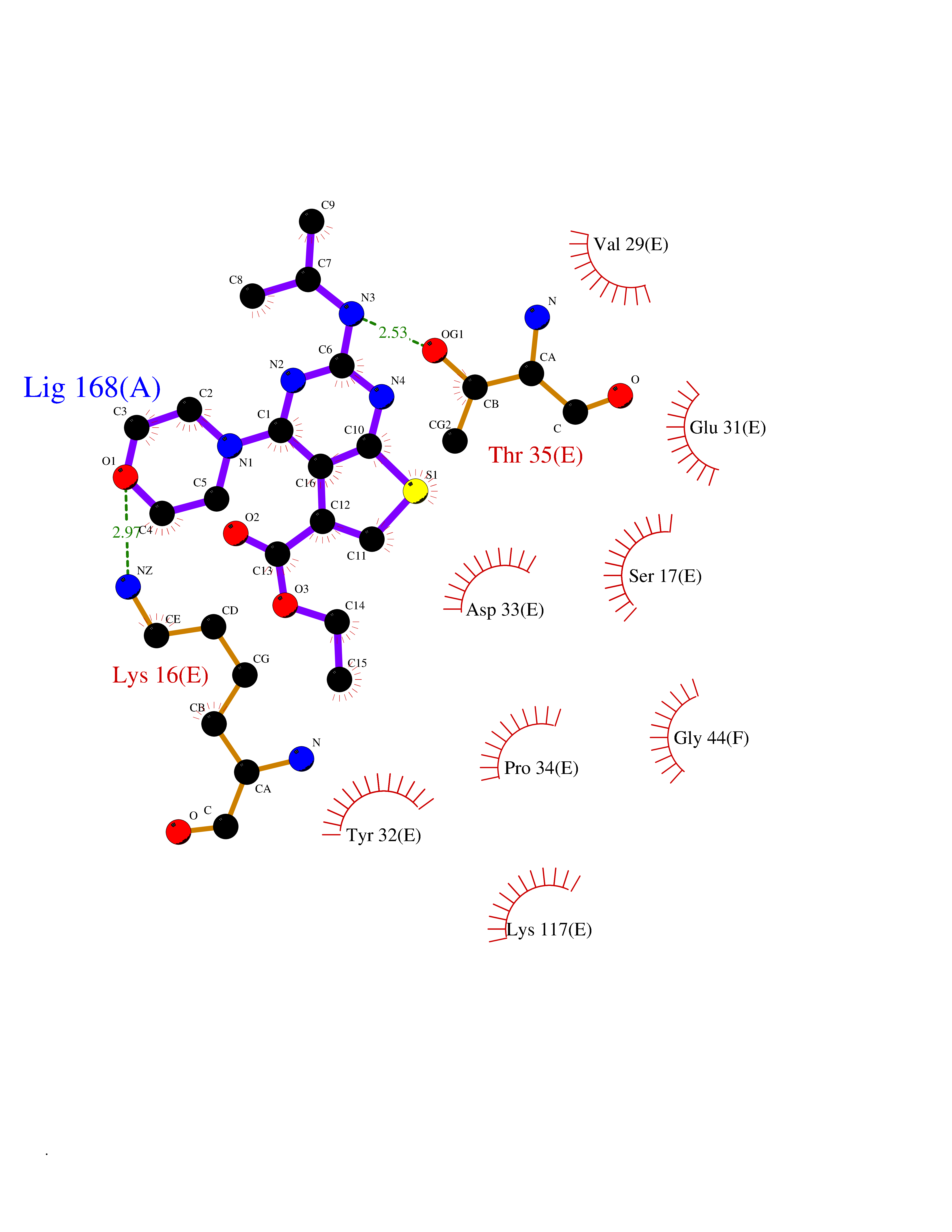 Ligplot Map 3D Binding mode Sequence MTEYKLVVVGAGGVGKSALTIQLIQNHFVDEYDPTIEDSYRKQVVIDGETCLLDILDTAGQEEYSAMRDQYMRTGEGFLCVFAINNTKSFEDIHQYREQIKRVKDSDDVPMVLVGNKCDLAARTVESRQAQDLARSYGIPYIETSAKTRQGVEDAFYTLVREIRQHSVPTKLEVVAATPTSLLISWDAPAVTVFFYIIAYGETGHGVGAFQAFRVPGSKSTATISGLKPGVDYTITVYARGYSKQGPYKPSPISINYRT Hydrogen bonds contact Hydrophobic contact | ||||
| 29 | Folate receptor alpha (FOLR1) | 4LRH | 6.46 | |
Target general information Gen name FOLR1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Ovarian tumorassociated antigen MOv18; KB cells FBP; Folate receptor, adult; Folate receptor 1; FRalpha; FOLR1; Adult folatebinding protein Protein family Folate receptor family Biochemical class Folate receptor Function Binds to folate and reduced folic acid derivatives and mediates delivery of 5-methyltetrahydrofolate and folate analogs into the interior of cells. Has high affinity for folate and folic acid analogs at neutral pH. Exposure to slightly acidic pHafter receptor endocytosis triggers a conformation change that strongly reduces its affinity for folates and mediates their release. Required for normal embryonic development and normal cell proliferation. Related diseases Neurodegeneration due to cerebral folate transport deficiency (NCFTD) [MIM:613068]: An autosomal recessive neurodegenerative disorder resulting from brain-specific folate deficiency early in life. Onset is apparent in late infancy with severe developmental regression, movement disturbances, epilepsy and leukodystrophy. {ECO:0000269|PubMed:19732866}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB05595; DB00158; DB00563; DB12489; DB15413; DB05168 Interacts with Q8N357 EC number NA Uniprot keywords 3D-structure; Cell membrane; Cytoplasmic vesicle; Direct protein sequencing; Disulfide bond; Endosome; Folate-binding; Glycoprotein; GPI-anchor; Lipoprotein; Membrane; Neurodegeneration; Proteomics identification; Receptor; Reference proteome; Secreted; Signal; Transport Protein physicochemical properties Chain ID A Molecular weight (Da) 24216 Length 207 Aromaticity 0.13 Instability index 49.36 Isoelectric point 8.14 Charge (pH=7) 3.41 2D Binding mode Binding energy (Kcal/mol) -8.81 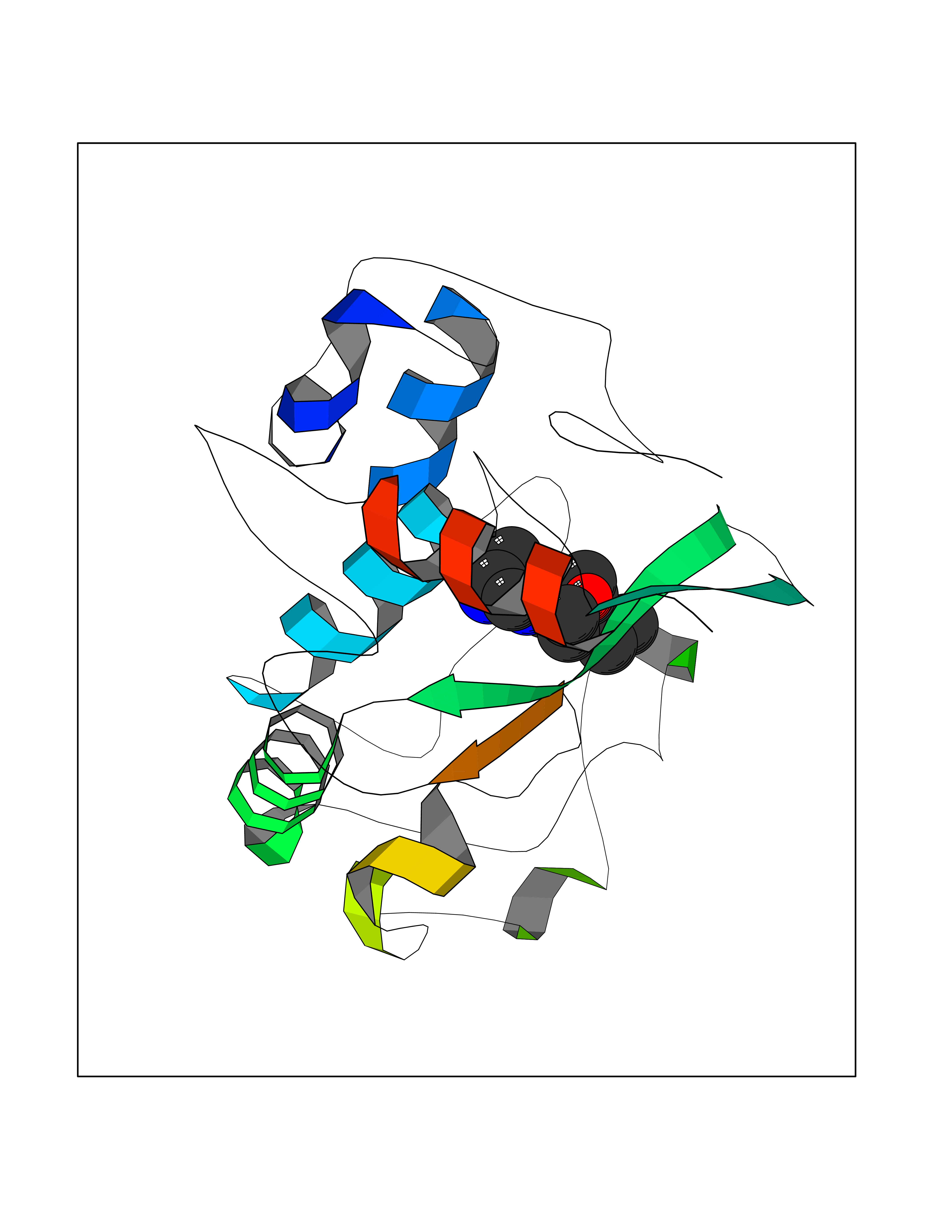 Molscript Map 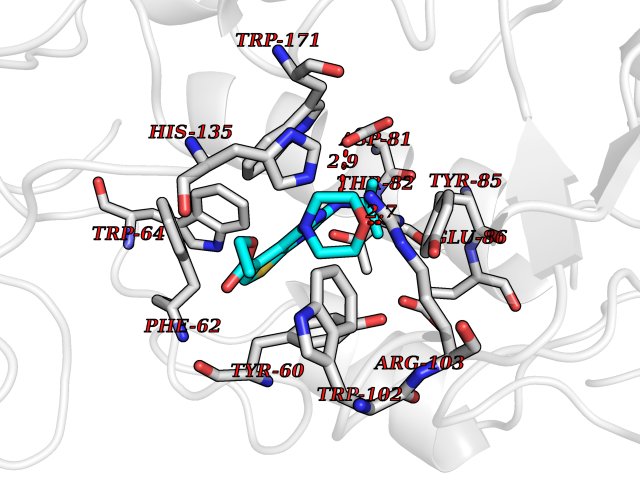 Pymol Map 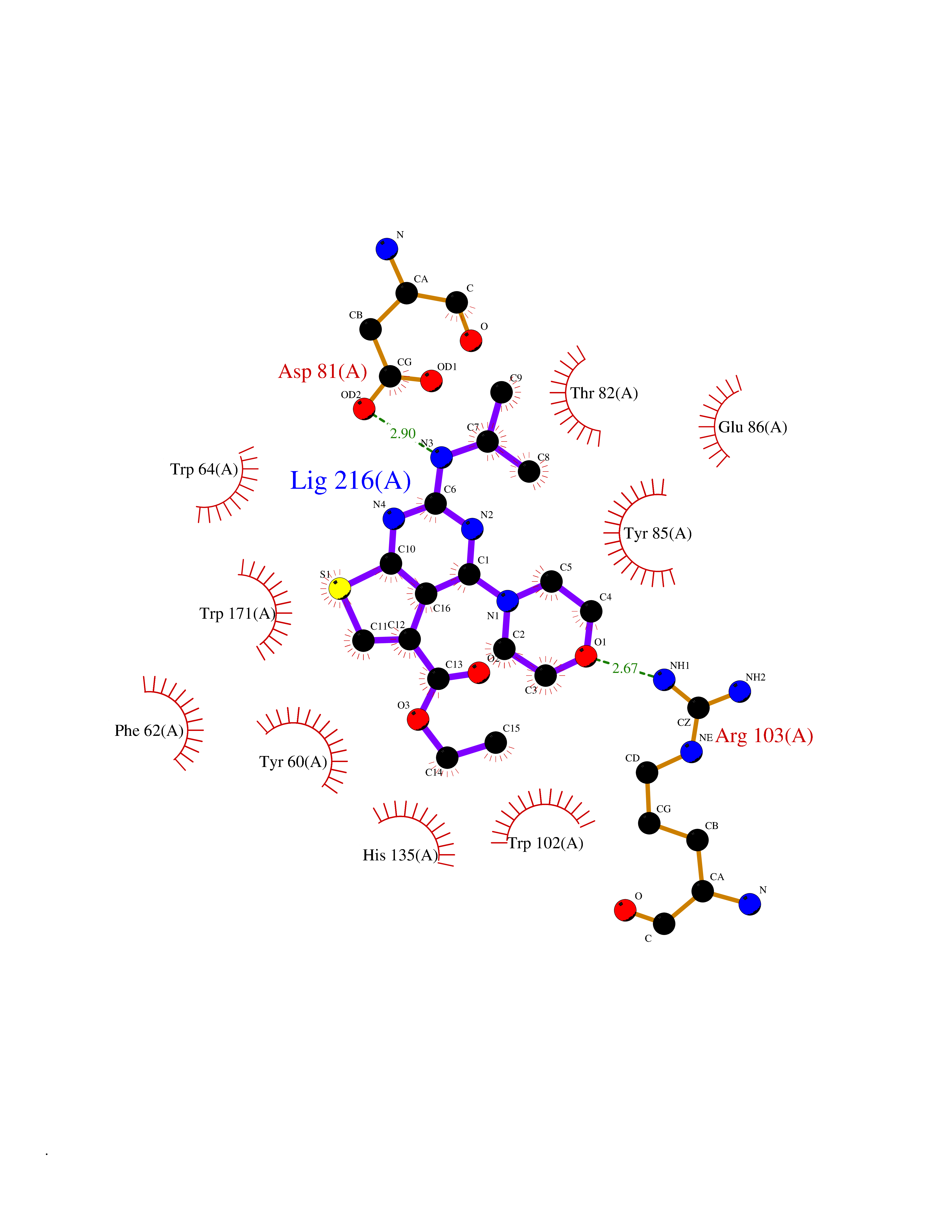 Ligplot Map 3D Binding mode Sequence RTELLNVCMNAKHHKEKPGPEDKLHEQCRPWRKNACCSTNTSQEAHKDVSYLYRFNWNHCGEMAPACKRHFIQDTCLYECSPNLGPWIQQVDQSWRKERVLNVPLCKEDCEQWWEDCRTSYTCKSNWHKGWNWTSGFNKCAVGAACQPFHFYFPTPTVLCNEIWTHSYKVSNYSRGSGRCIQMWFDPAQGNPNEEVARFYAAAMSGT Hydrogen bonds contact Hydrophobic contact | ||||
| 30 | Monoamine oxidase type B (MAO-B) | 2V5Z | 6.44 | |
Target general information Gen name MAOB Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms MAO-B; Amine oxidase [flavin-containing] B Protein family Flavin monoamine oxidase family Biochemical class CH-NH(2) donor oxidoreductase Function Catalyzes the oxidative deamination of biogenic and xenobiotic amines and has important functions in the metabolism of neuroactive and vasoactive amines in the central nervous system and peripheral tissues. MAOB preferentially degrades benzylamine and phenylethylamine. Related diseases Microvascular complications of diabetes 5 (MVCD5) [MIM:612633]: Pathological conditions that develop in numerous tissues and organs as a consequence of diabetes mellitus. They include diabetic retinopathy, diabetic nephropathy leading to end-stage renal disease, and diabetic neuropathy. Diabetic retinopathy remains the major cause of new-onset blindness among diabetic adults. It is characterized by vascular permeability and increased tissue ischemia and angiogenesis. Disease susceptibility is associated with variants affecting the gene represented in this entry. Homozygosity for the Leu-55 allele is strongly associated with the development of retinal disease in diabetic patients. Drugs (DrugBank ID) DB08176; DB02211; DB08516; DB08480; DB01472; DB04307; DB07512; DB07513; DB00915; DB00182; DB06698; DB04889; DB00215; DB09130; DB04147; DB00988; DB01363; DB00668; DB01175; DB02509; DB03147; DB14914; DB00614; DB04818; DB02095; DB01247; DB00601; DB01577; DB01442; DB01171; DB08082; DB02643; DB04677; DB03894; DB08804; DB04820; DB00184; DB04821; DB12612; DB01626; DB00780; DB00191; DB00388; DB01132; DB00721; DB01168; DB01367; DB09363; DB06654; DB01037; DB01104; DB14569; DB09042; DB00752; DB16446; DB09185; DB04832; DB00909 Interacts with P55212; P28329-3; Q8NI60; Q5RI15; Q92915-2; P22607; Q53GS7; P06396; P01112; O14901; P13473-2; P21397; Q9BVL2; O75400-2; P62826; Q6NTF9-3; Q9Y371; Q7Z699; Q9UMX0; Q9Y649 EC number EC 1.4.3.4 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Direct protein sequencing; FAD; Flavoprotein; Membrane; Mitochondrion; Mitochondrion outer membrane; Oxidoreductase; Proteomics identification; Reference proteome; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A,B Molecular weight (Da) 56019.9 Length 494 Aromaticity 0.09 Instability index 34.81 Isoelectric point 6.51 Charge (pH=7) -2.2 2D Binding mode Binding energy (Kcal/mol) -8.78  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence NKCDVVVVGGGISGMAAAKLLHDSGLNVVVLEARDRVGGRTYTLRNQKVKYVDLGGSYVGPTQNRILRLAKELGLETYKVNEVERLIHHVKGKSYPFRGPFPPVWNPITYLDHNNFWRTMDDMGREIPSDAPWKAPLAEEWDNMTMKELLDKLCWTESAKQLATLFVNLCVTAETHEVSALWFLWYVKQCGGTTRIISTTNGGQERKFVGGSGQVSERIMDLLGDRVKLERPVIYIDQTRENVLVETLNHEMYEAKYVISAIPPTLGMKIHFNPPLPMMRNQMITRVPLGSVIKCIVYYKEPFWRKKDYCGTMIIDGEEAPVAYTLDDTKPEGNYAAIMGFILAHKARKLARLTKEERLKKLCELYAKVLGSLEALEPVHYEEKNWCEEQYSGGCYTTYFPPGILTQYGRVLRQPVDRIYFAGTETATHWSGYMEGAVEAGERAAREILHAMGKIPEDEIWQSEPESVDVPAQPITTTFLERHLPSVPGLLRLI Hydrogen bonds contact Hydrophobic contact | ||||
| 31 | Gamma-aminobutyric acid receptor subunit beta-3 | 4COF | 6.44 | |
Target general information Gen name GABRB3 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Ligand-gated ion channel (TC 1.A.9) family, Gamma-aminobutyric acid receptor (TC 1.A.9.5) subfamily, GABRB3 sub-subfamily Biochemical class Transport protein Function GABA-A receptor activity.GABA-gated chloride ion channel activity.Identical protein binding. Related diseases Epilepsy, childhood absence 5 (ECA5) [MIM:612269]: A subtype of idiopathic generalized epilepsy characterized by an onset at age 6-7 years, frequent absence seizures (several per day) and bilateral, synchronous, symmetric 3-Hz spike waves on EEG. Tonic-clonic seizures often develop in adolescence. Absence seizures may either remit or persist into adulthood. {ECO:0000269|PubMed:18514161, ECO:0000269|PubMed:22303015}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Developmental and epileptic encephalopathy 43 (DEE43) [MIM:617113]: A form of epileptic encephalopathy, a heterogeneous group of severe early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE43 inheritance is autosomal dominant. {ECO:0000269|PubMed:23934111, ECO:0000269|PubMed:25356899, ECO:0000269|PubMed:26950270, ECO:0000269|PubMed:26993267, ECO:0000269|PubMed:27476654, ECO:0000269|PubMed:27864847}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB12537; DB00546; DB00404; DB00543; DB11901; DB14719; DB11859; DB01558; DB09017; DB00237; DB00241; DB01489; DB00475; DB14715; DB01594; DB00349; DB01068; DB00628; DB01559; DB01553; DB01511; DB01189; DB00829; DB13837; DB00228; DB01215; DB00402; DB00898; DB00189; DB01545; DB09166; DB00292; DB01567; DB01205; DB01544; DB00690; DB06716; DB05087; DB01437; DB00801; DB01159; DB00753; DB01587; DB00555; DB00431; DB13643; DB00186; DB13872; DB13437; DB00603; DB01043; DB00371; DB00463; DB01028; DB01107; DB15489; DB00683; DB12458; DB01595; DB14028; DB00842; DB14672; DB00312; DB00252; DB13335; DB00592; DB01708; DB01588; DB00794; DB00818; DB01589; DB12404; DB01236; DB09118; DB00306; DB01956; DB00231; DB11582; DB00897; DB15490 Interacts with P28472 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Chloride; Chloride channel; Cytoplasmic vesicle; Direct protein sequencing; Disease variant; Disulfide bond; Epilepsy; Glycoprotein; Ion channel; Ion transport; Ligand-gated ion channel; Membrane; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Signal; Synapse; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A,B,C,D,E Molecular weight (Da) 76970.6 Length 663 Aromaticity 0.15 Instability index 39.4 Isoelectric point 8.37 Charge (pH=7) 4.12 2D Binding mode Binding energy (Kcal/mol) -8.78  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence SFVKETVDKLLKGYDIRLRPDFGGPPVCVGMNIDIASIDMVSEVNMDYTLTMYFQQYWRDKRLAYSGIPLNLTLDNRVADQLWVPDTYFLNDKKSFVHGVTVKNRMIRLHPDGTVLYGLRITTTAACMMDLRRYPLDEQNCTLEIESYGYTTDDIEFYWRGGDKAVTGVERIELPQFSIVEHRLVSRNVVFATGAYPRLSLSFRLKRNIGYFILQTYMPSILITILSWVSFWINYDASAARVALGITTVLTMTTINTHLRETLPKIPYVKAIDMYLMGCFVFVFLALLEYAFVNYIFFSQPARAAAIDRWSRIVFPFTFSLFNLVYWLYYVNSFVKETVDKLLKGYDIRLRPDFGGPPVCVGMNIDIASIDMVSEVNMDYTLTMYFQQYWRDKRLAYSGIPLNLTLDNRVADQLWVPDTYFLNDKKSFVHGVTVKNRMIRLHPDGTVLYGLRITTTAACMMDLRRYPLDEQNCTLEIESYGYTTDDIEFYWRGGDKAVTGVERIELPQFSIVEHRLVSRNVVFATGAYPRLSLSFRLKRNIGYFILQTYMPSILITILSWVSFWINYDASAARVALGITTVLTMTTINTHLRETLPKIPYVKAIDMYLMGCFVFVFLALLEYAFVNYIFFSQPARAAAIDRWSRIVFPFTFSLFNLVYWLYYV Hydrogen bonds contact Hydrophobic contact | ||||
| 32 | Ferredoxin reductase | 2GQW | 6.44 | |
Target general information Gen name bphA4 Organism Pseudomonas sp. (strain KKS102) Uniprot ID TTD ID NA Synonyms NA Protein family NA Biochemical class Oxidoreductase Function Flavin adenine dinucleotide binding.Oxidoreductase activity. Related diseases LTC4 synthase deficiency is associated with a neurometabolic developmental disorder characterized by muscular hypotonia, psychomotor retardation, failure to thrive, and microcephaly. {ECO:0000269|PubMed:10896305, ECO:0000269|PubMed:9820300}. Drugs (DrugBank ID) DB03147 Interacts with NA EC number NA Uniprot keywords 3D-structure; FAD; Flavoprotein; Nucleotide-binding Protein physicochemical properties Chain ID A Molecular weight (Da) 42484 Length 401 Aromaticity 0.05 Instability index 32.45 Isoelectric point 6.13 Charge (pH=7) -2.69 2D Binding mode Binding energy (Kcal/mol) -8.78  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence LKAPVVVLGAGLASVSFVAELRQAGYQGLITVVGDEAERPYDRPPLSKDFMAHGDAEKIRLDCKRAPEVEWLLGVTAQSFDPQAHTVALSDGRTLPYGTLVLATGAAPRALPTLQGATMPVHTLRTLEDARRIQAGLRPQSRLLIVGGGVIGLELAATARTAGVHVSLVETQPRLMSRAAPATLADFVARYHAAQGVDLRFERSVTGSVDGVVLLDDGTRIAADMVVVGIGVLANDALARAAGLACDDGIFVDAYGRTTCPDVYALGDVTRQRNPLSGRFERIETWSNAQNQGIAVARHLVDPTAPGYAELPWYWSDQGALRIQVAGLASGDEEIVRGEVSLDAPKFTLIELQKGRIVGATCVNNARDFAPLRRLLAVGAKPDRAALADPATDLRKLAAAV Hydrogen bonds contact Hydrophobic contact | ||||
| 33 | Tracheobronchial mucin 5A (MUC5AC) | 5AJP | 6.44 | |
Target general information Gen name MUC5AC Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Tracheobronchial mucin; TBM; Mucin5AC; Mucin5 subtype AC, tracheobronchial; Mucin-5AC; Mucin-5 subtype AC, tracheobronchial; Major airway glycoprotein; MUC5; MUC-5AC; Lewis B blood group antigen; LeB; Protein family NA Biochemical class NA Function Gel-forming glycoprotein of gastric and respiratoy tract epithelia that protects the mucosa from infection and chemical damage by binding to inhaled microrganisms and particles that are subsequently removed by the mucocilary system. Related diseases Orotic aciduria 1 (ORAC1) [MIM:258900]: A disorder of pyrimidine metabolism resulting in megaloblastic anemia and orotic acid crystalluria that is frequently associated with some degree of physical and intellectual disability. A minority of cases have additional features, particularly congenital malformations and immune deficiencies. {ECO:0000269|PubMed:9042911}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number NA Uniprot keywords 3D-structure; Copper; Direct protein sequencing; Disulfide bond; Glycoprotein; Metal-binding; Proteomics identification; Reference proteome; Repeat; Secreted; Signal Protein physicochemical properties Chain ID A,B Molecular weight (Da) 57738 Length 509 Aromaticity 0.09 Instability index 47.24 Isoelectric point 8.35 Charge (pH=7) 4.76 2D Binding mode Binding energy (Kcal/mol) -8.79  Molscript Map 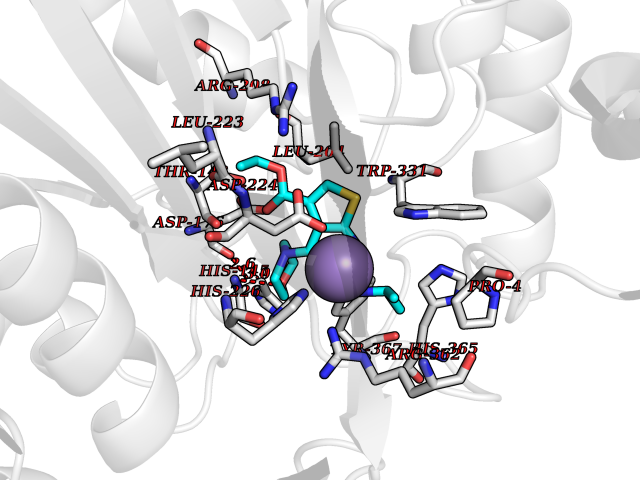 Pymol Map 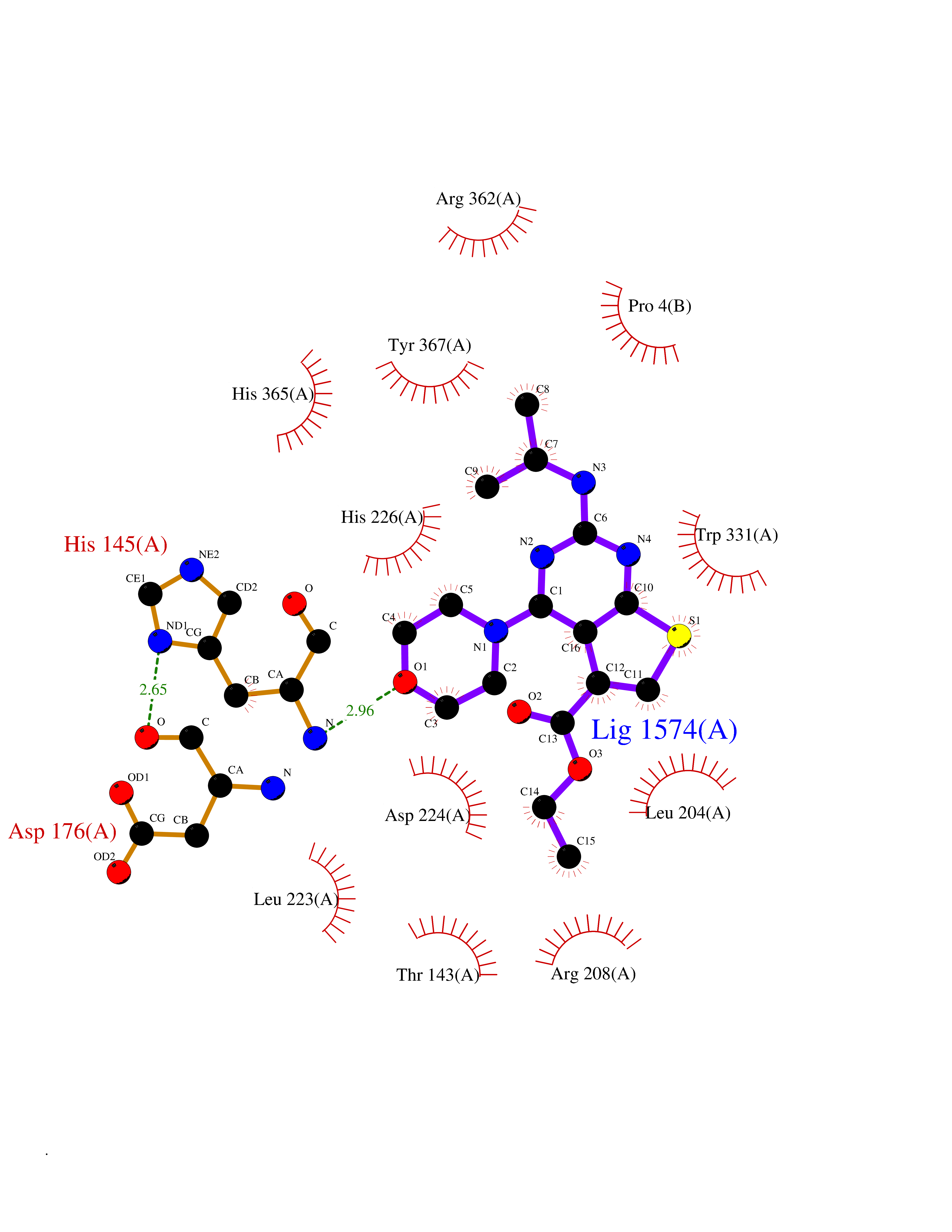 Ligplot Map 3D Binding mode Sequence KVRWPDFNQEAYVGGTMVRSGQDPYARNKFNQVESDKLRMDRAIPDTRHDQCQRKQWRVDLPATSVVITFHNEARSALLRTVVSVLKKSPPHLIKEIILVDDYSNDPEDGALLGKIEKVRVLRNDRREGLMRSRVRGADAAQAKVLTFLDSHCECNEHWLEPLLERVAEDRTRVVSPIIDVINMDNFQYVGASADLKGGFDWNLVFKWDYMTPEQRRSRQGNPVAPIKTPMIAGGLFVMDKFYFEELGKYDMMMDVWGGENLEISFRVWQCGGSLEIIPCSRVGHVFRKQHPYTFPGGSGTVFARNTRRAAEVWMDEYKNFYYAAVPSARNVPYGNIQSRLELRKKLSCKPFKWYLENVYPELRVPDHQDIAFGALQQGTNCLDTLGHFADGVVGVYECHNAGGNQEWALTKEKSVKHMDLCLTVVDRAPGSLIKLQGCREDDSRQKWEQIEGNSKLRHVGSNLCLDSRTAKSGGLSVEVCGPALSQQWKFTLNLTTPSPVPTTSTTSA Hydrogen bonds contact Hydrophobic contact | ||||
| 34 | Sodium/glucose cotransporter 2 (SGLT2) | 7VSI | 6.44 | |
Target general information Gen name SLC5A2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Solute carrier family 5 member 2; Na(+)/glucose cotransporter 2; Low affinity sodium-glucose cotransporter Protein family Sodium:solute symporter (SSF) (TC 2.A.21) family Biochemical class Solute:sodium symporter Function Has a Na(+) to glucose coupling ratio of 1:1. Sodium-dependent glucose transporter. Related diseases Renal glucosuria (GLYS) [MIM:233100]: A disorder characterized by persistent isolated glucosuria, normal fasting serum glucose concentration, decreased renal tubular resorption of glucose from the urine, and absence of any other signs of tubular dysfunction. {ECO:0000269|PubMed:14614622}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB12236; DB08907; DB01914; DB06292; DB09038; DB11827; DB12713 Interacts with O14556; Q13113 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disease variant; Disulfide bond; Glycoprotein; Ion transport; Membrane; Metal-binding; Proteomics identification; Reference proteome; Sodium; Sodium transport; Sugar transport; Symport; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A Molecular weight (Da) 63858.9 Length 586 Aromaticity 0.12 Instability index 39.46 Isoelectric point 8.62 Charge (pH=7) 7.41 2D Binding mode Binding energy (Kcal/mol) -8.78 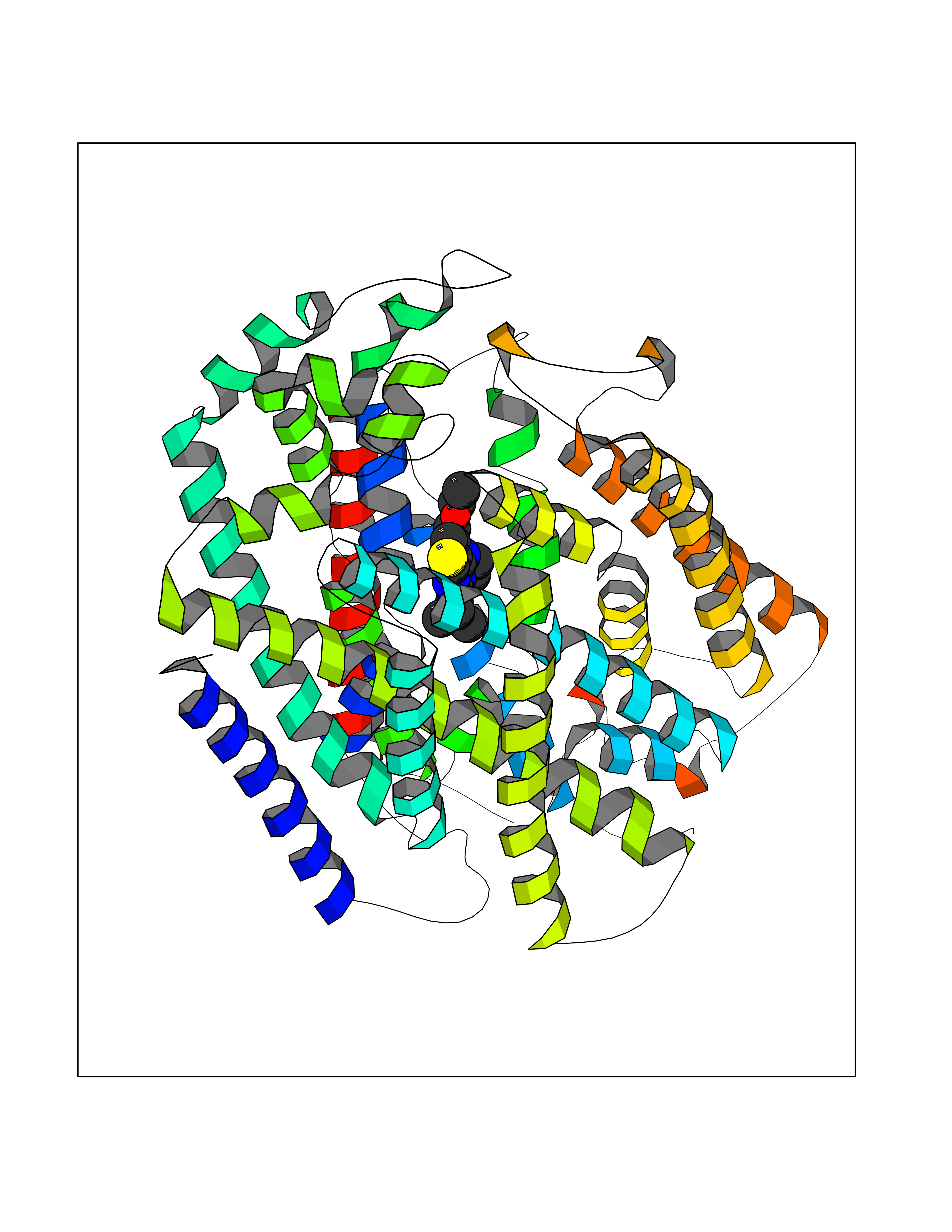 Molscript Map 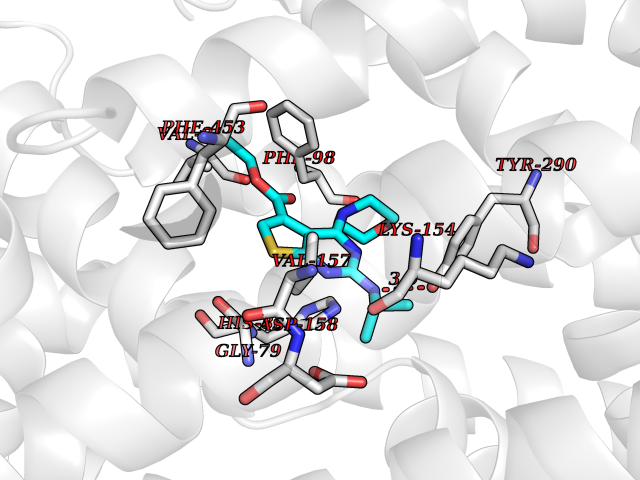 Pymol Map 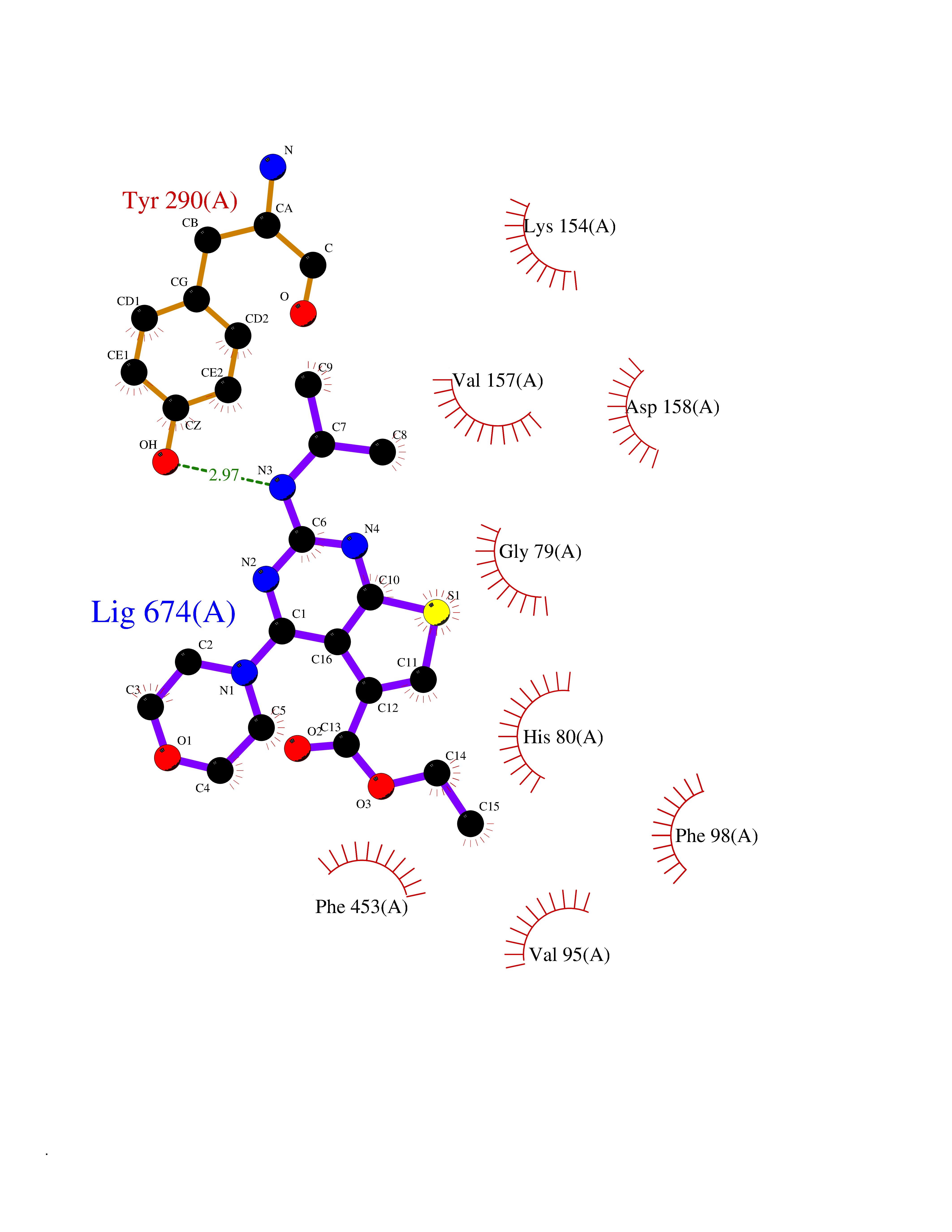 Ligplot Map 3D Binding mode Sequence DNPADILVIAAYFLLVIGVGLWSMCRTNRGTVGGYFLAGRSMVWWPVGASLFASNIGSGHFVGLAGTGAASGLAVAGFEWNALFVVLLLGWLFAPVYLTAGVITMPQYLRKRFGGRRIRLYLSVLSLFLYIFTKISVDMFSGAVFIQQALGWNIYASVIALLGITMIYTVTGGLAALMYTDTVQTFVILGGACILMGYAFHEVGGYSGLFDKYLGAATSLTVSEDPAVGNISSFCYRPRPDSYHLLRHPVTGDLPWPALLLGLTIVSGWYWCSDQVIVQRCLAGKSLTHIKAGCILCGYLKLTPMFLMVMPGMISRILYPDEVACVVPEVCRRVCGTEVGCSNIAYPRLVVKLMPNGLRGLMLAVMLAALMSSLASIFNSSSTLFTMDIYTRLRPRAGDRELLLVGRLWVVFIVVVSVAWLPVVQAAQGGQLFDYIQAVSSYLAPPVSAVFVLALFVPRVNEQGAFWGLIGGLLMGLARLIPEFSFGSGSCVQPSACPAFLCGVHYLYFAIVLFFCSGLLTLTVSLCTAPIPRKHLHRLVFSLRHSKEEREDLDEDISEDPSWARVVNLNALLMMAVAVFLWGFYA Hydrogen bonds contact Hydrophobic contact | ||||
| 35 | Plasmodium Adenylosuccinate synthetase (Malaria Adss) | 1P9B | 6.44 | |
Target general information Gen name Malaria Adss Organism Plasmodium falciparum Uniprot ID TTD ID Synonyms IMP--aspartate ligase; Adenylosuccinate synthase; AdSS; AMPSase Protein family Adenylosuccinate synthetase family Biochemical class Carbon-nitrogen ligase Function Plays an important role in the salvage pathway for purine nucleotide biosynthesis. Catalyzes the first committed step in the biosynthesis of AMP from IMP. Related diseases Hypertension and brachydactyly syndrome (HTNB) [MIM:112410]: A syndrome characterized by brachydactyly type E, severe salt-independent but age-dependent hypertension, an increased fibroblast growth rate, neurovascular contact at the rostral-ventrolateral medulla, and altered baroreflex blood pressure regulation. It results in death from stroke before age 50 years when untreated. Brachydactyly type E is characterized by shortening of the fingers mainly in the metacarpals and metatarsals. {ECO:0000269|PubMed:25961942}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03510; DB04315; DB02109 Interacts with NA EC number EC 6.3.4.4 Uniprot keywords 3D-structure; Cytoplasm; GTP-binding; Ligase; Magnesium; Metal-binding; Nucleotide-binding; Purine biosynthesis Protein physicochemical properties Chain ID A Molecular weight (Da) 47877.9 Length 424 Aromaticity 0.09 Instability index 31.72 Isoelectric point 7.63 Charge (pH=7) 1.58 2D Binding mode Binding energy (Kcal/mol) -8.78  Molscript Map 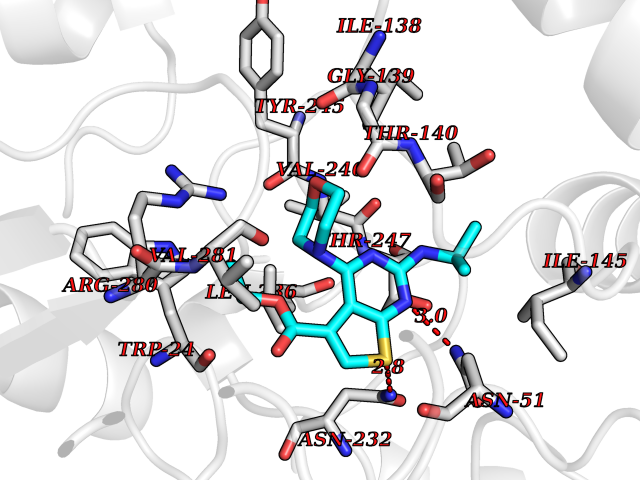 Pymol Map 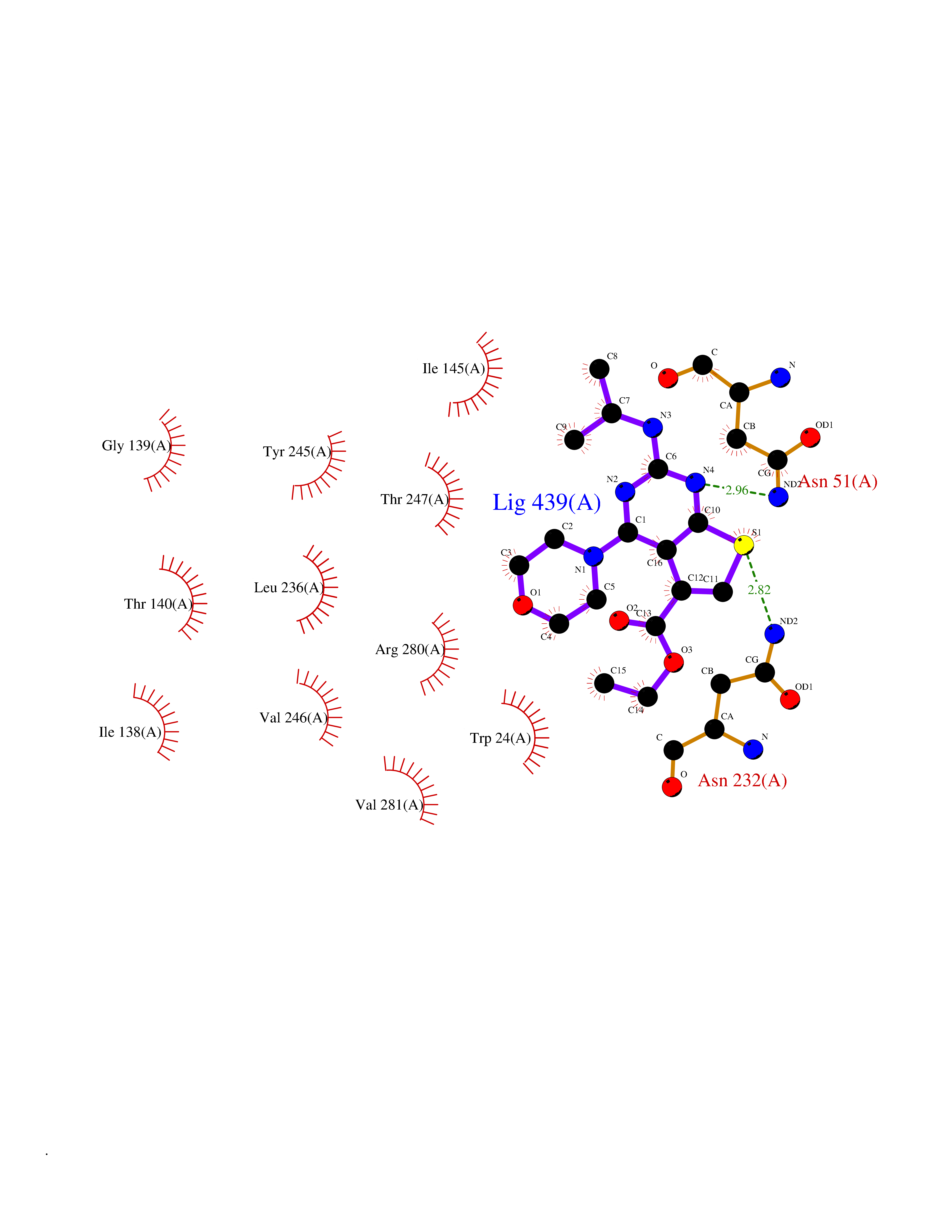 Ligplot Map 3D Binding mode Sequence GNVVAILGAQWGDEGKGKIIDMLSEYSDITCRFNGGANAGHTISVNDKKYALHLLPCGVLYDNNISVLGNGMVIHVKSLMEEIESVGGKLLDRLYLSNKAHILFDIHQIIDSIQETKKLKEGKQIGTTKRGIGPCYSTKASRIGIRLGTLKNFENFKNMYSKLIDHLMDLYNITEYDKEKELNLFYNYHIKLRDRIVDVISFMNTNLENNKKVLIEGANAAMLDIDFGTYPYVTSSCTTVGGVFSGLGIHHKKLNLVVGVVKSYLTRVGCGPFLTELNNDVGQYLREKGHEYGTTTKRPRRCGWLDIPMLLYVKCINSIDMINLTKLDVLSGLEEILLCVNFKNKKTGELLEKGCYPVEEEISEEYEPVYEKFSGWKEDISTCNEFDELPENAKKYILAIEKYLKTPIVWIGVGPNRKNMIVKK Hydrogen bonds contact Hydrophobic contact | ||||
| 36 | Thyroid hormone receptor beta (THRB) | 1N46 | 6.43 | |
Target general information Gen name THRB Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms c-erbA-beta; c-erbA-2; THR1; Nuclear receptor subfamily 1 group A member 2; NR1A2; ERBA2 Protein family Nuclear hormone receptor family, NR1 subfamily Biochemical class Nuclear hormone receptor Function High affinity receptor for thyroid hormones, including triiodothyronine and thyroxine. Nuclear hormone receptor that can act as a repressor or activator of transcription. Related diseases Thyroid hormone resistance, generalized, autosomal dominant (GRTHD) [MIM:188570]: An autosomal dominant disease characterized by high levels of circulating thyroid hormones (T3-T4), goiter, abnormal mental functions, increased susceptibility to infections, abnormal growth and bone maturation, tachycardia and deafness. Affected individuals may also have attention deficit-hyperactivity disorders (ADHD) and language difficulties. Patients have normal or slightly elevated thyroid stimulating hormone (TSH). {ECO:0000269|PubMed:10660344, ECO:0000269|PubMed:12511610, ECO:0000269|PubMed:12554782, ECO:0000269|PubMed:1314846, ECO:0000269|PubMed:1324420, ECO:0000269|PubMed:1563081, ECO:0000269|PubMed:1587388, ECO:0000269|PubMed:1619012, ECO:0000269|PubMed:1661299, ECO:0000269|PubMed:16804041, ECO:0000269|PubMed:1846005, ECO:0000269|PubMed:19268523, ECO:0000269|PubMed:2153155, ECO:0000269|PubMed:2510172, ECO:0000269|PubMed:7833659, ECO:0000269|PubMed:8175986, ECO:0000269|PubMed:8514853, ECO:0000269|PubMed:8664910, ECO:0000269|PubMed:8889584}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Thyroid hormone resistance, generalized, autosomal recessive (GRTHR) [MIM:274300]: An autosomal recessive disorder characterized by goiter, clinical euthyroidism, end-organ unresponsiveness to thyroid hormone, abnormal growth and bone maturation, and deafness. Patients also have high levels of circulating thyroid hormones, with elevated thyroid stimulating hormone. {ECO:0000269|PubMed:1653889}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Selective pituitary thyroid hormone resistance (PRTH) [MIM:145650]: Variant form of thyroid hormone resistance and is characterized by clinical hyperthyroidism, with elevated free thyroid hormones, but inappropriately normal serum TSH. Unlike GRTH, where the syndrome usually segregates with a dominant allele, the mode of inheritance in PRTH has not been established. {ECO:0000269|PubMed:7528740, ECO:0000269|PubMed:8381821}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08085; DB03181; DB02106; DB01118; DB00509; DB05035; DB03788; DB03176; DB00451; DB00279; DB01583; DB05192; DB07425; DB09100; DB03604 Interacts with Q60974; Q9Y618 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Deafness; Disease variant; DNA-binding; Metal-binding; Nucleus; Proteomics identification; Receptor; Reference proteome; Transcription; Transcription regulation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A,B Molecular weight (Da) 27235.4 Length 239 Aromaticity 0.09 Instability index 43.29 Isoelectric point 5.42 Charge (pH=7) -8.55 2D Binding mode Binding energy (Kcal/mol) -8.77 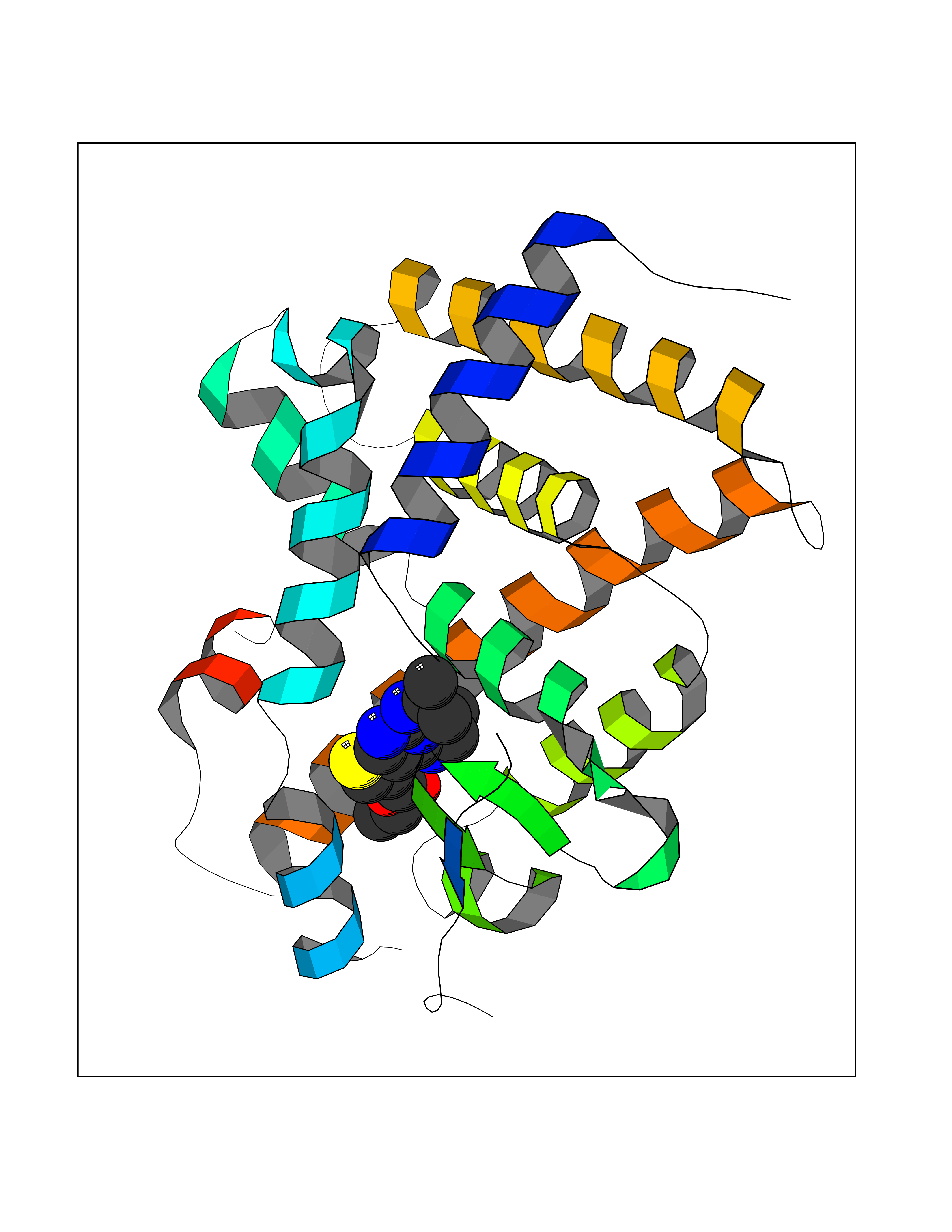 Molscript Map 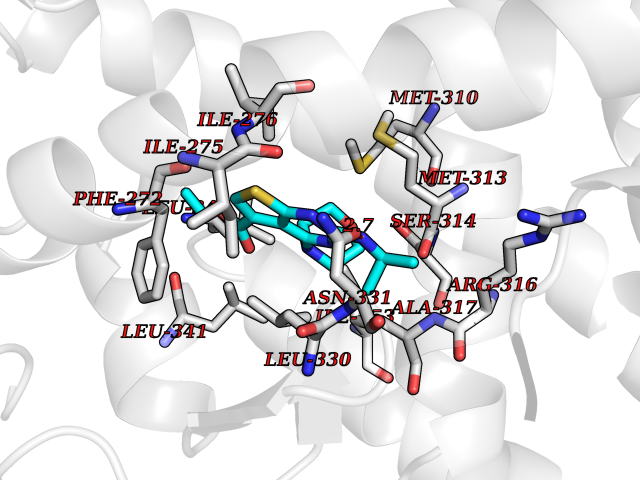 Pymol Map  Ligplot Map 3D Binding mode Sequence KPEPTDEEWELIKTVTEAHVATNAQWKQKRKFLPEDIGQAKVDLEAFSHFTKIITPAITRVVDFAKKLPMFCELPCEDQIILLKGCCMEIMSLRAAVRYDPESETLTLNGEMAVTRGQLKNGGLGVVSDAIFDLGMSLSSFNLDDTEVALLQAVLLMSSDRPGLACVERIEKYQDSFLLAFEHYINYRKHHVTHFWPKLLMKVTDLRMIGACHASRFLHMKVECPTELFPPLFLEVFED Hydrogen bonds contact Hydrophobic contact | ||||
| 37 | Choline O-acetyltransferase | 2FY3 | 6.43 | |
Target general information Gen name CHAT Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Carnitine/choline acetyltransferase family Biochemical class Transferase Function Choline O-acetyltransferase activity. Related diseases Myasthenic syndrome, congenital, 6, presynaptic (CMS6) [MIM:254210]: A form of congenital myasthenic syndrome, a group of disorders characterized by failure of neuromuscular transmission, including pre-synaptic, synaptic, and post-synaptic disorders that are not of autoimmune origin. Clinical features are easy fatigability and muscle weakness affecting the axial and limb muscles (with hypotonia in early-onset forms), the ocular muscles (leading to ptosis and ophthalmoplegia), and the facial and bulbar musculature (affecting sucking and swallowing, and leading to dysphonia). The symptoms fluctuate and worsen with physical effort. CMS6 affected individuals have myasthenic symptoms since birth or early infancy, negative tests for anti-AChR antibodies, and abrupt episodic crises with increased weakness, bulbar paralysis, and apnea precipitated by undue exertion, fever, or excitement. CMS6 inheritance is autosomal recessive. {ECO:0000269|PubMed:11172068, ECO:0000269|PubMed:12756141}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00122; DB14006; DB00184 Interacts with Q6H8Q1-8; Q8N302-2; Q9NXL2-1; Q6XD76; Q9UII2; Q8TBE0; Q9UQB8-6; Q9ULD4-2; Q9NSI6-4; Q6P5X5; Q96LL4; P20807-4; O00257-3; Q6ZP82-1; O95674; Q9H3R5; Q8WUX9; Q9H2A9; Q3SX64; Q92782-2; Q14117; O14641; Q658K8; Q6UXG2-3; O00472; Q6NXG1; Q15910-2; Q8IZU1; P15407; P55318; Q06547-3; P23769-2; P23771; Q15486; Q8IV36; Q4VB01; Q53GQ0; P10809; P41134; Q9NZH6; Q8NA54; Q86U28; P17275; Q8N5Z5; Q6P597; P08727; Q14525; Q8IUC2; Q6IAA8; Q14847-2; P27338; Q9GZQ8; Q53S70; Q5JXC2; A0A0A0MR05; Q8NEH6; Q8TCY5; Q6IN84-2; Q96H12; P01106; P41271-2; P14598; Q9GZM8; Q5BJF6-2; Q9H8K7; Q9NR21-5; Q5VU43-8; Q13956; Q5SXH7-1; Q96T60; Q96I34; Q86UA1; Q15311; Q8TBY0; Q04206; P47804-3; Q9H0X6; P62899; Q66K80; Q9BY12-3; Q86SQ7-2; Q7Z6I5; Q496A3; Q7Z698; Q9C004; Q92783-2; Q8N4C7; O75528; Q15814; O15273; Q96A09; Q8WTV1; Q53NU3; Q71RG4-4; Q86WT6-2; Q9Y3Q8; Q99598; P49459; P11441; Q9H270; P19544-6; Q53FD0-2; Q3KNS6-3 EC number 2.3.1.6 Uniprot keywords 3D-structure; Acyltransferase; Alternative splicing; Congenital myasthenic syndrome; Direct protein sequencing; Disease variant; Neurotransmitter biosynthesis; Phosphoprotein; Proteomics identification; Reference proteome; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 66365.9 Length 595 Aromaticity 0.08 Instability index 53.36 Isoelectric point 8.16 Charge (pH=7) 4.64 2D Binding mode Binding energy (Kcal/mol) -8.77  Molscript Map 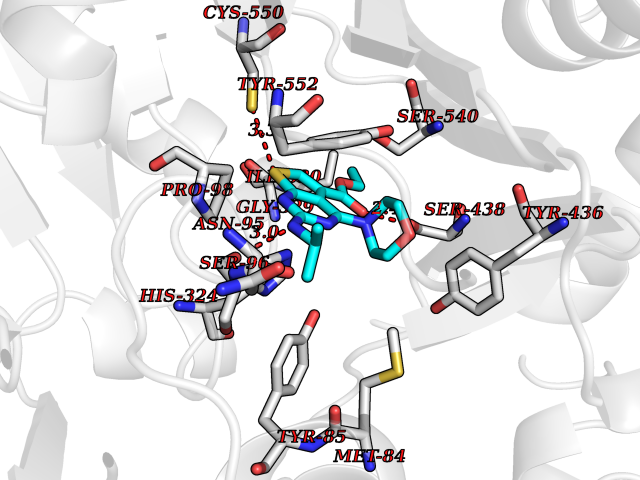 Pymol Map 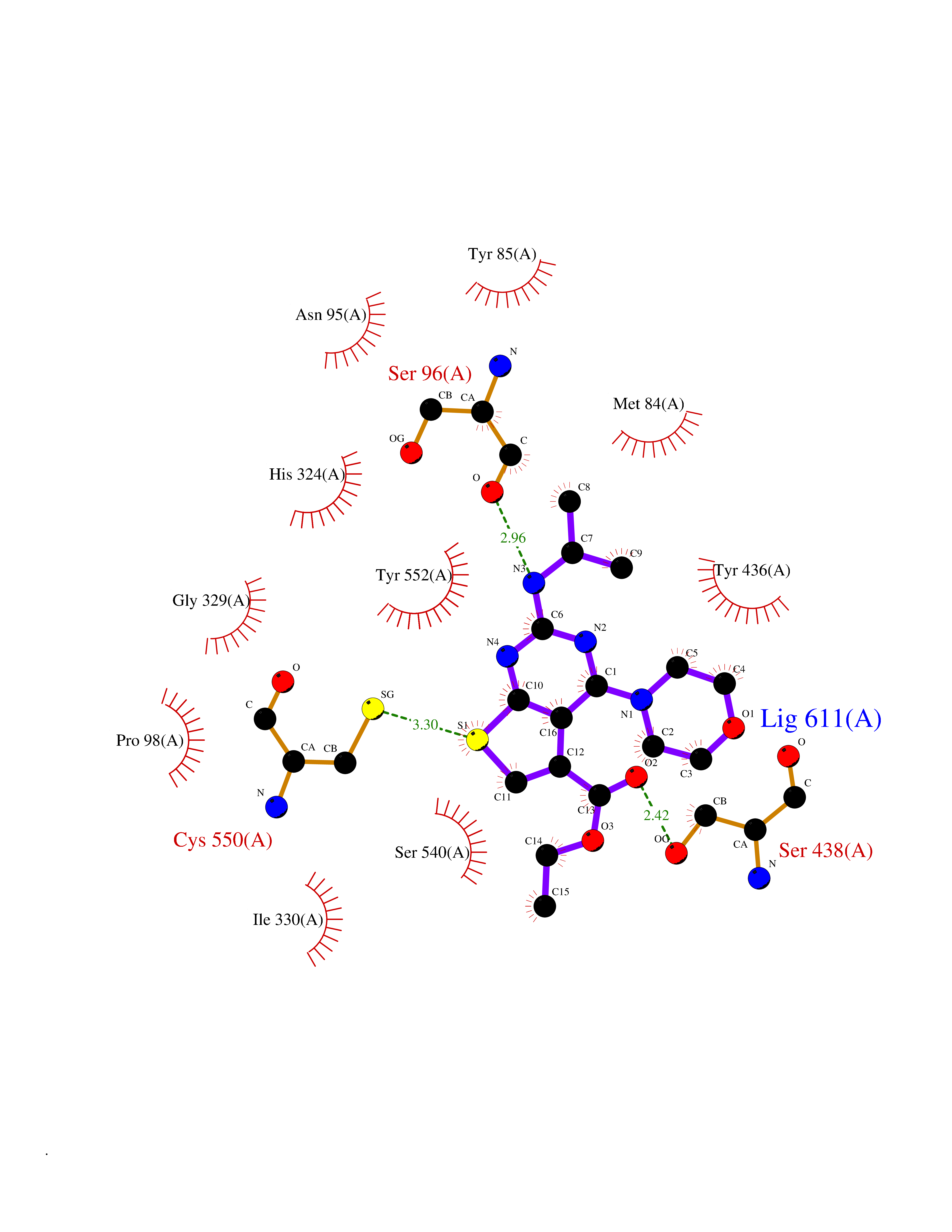 Ligplot Map 3D Binding mode Sequence SEESGLPKLPVPPLQQTLATYLQCMRHLVSEEQFRKSQAIVQQFGAPGGLGETLQQKLLERQEKTANWVSEYWLNDMYLNNRLALPVNSSPAVIFARQHFPGTDDQLRFAASLISGVLSYKALLDSHSIPTDCAKGQPLCMKQYYGLFSSYRLPGHTQDTLVAQNSSIMPEPEHVIVACCNQFFVLDVVINFRRLSEGDLFTQLRKIVKMASNAAARLPPIGLLTSDGRSEWAEARTVLVKDSTNRDSLDMIERCICLVCLDAPGGVELSDTHRALQLLHGGGYSKNGANRWYDKSLQFVVGRDGTCGVVCEHSPFDGIVLVQCTEHLLKHMTQPELVRSPMVPLPAPRRLRWKCSPEIQGHLASSAEKLQRIVKNLDFIVYKFDNYGKTFIKKQKCSPDAFIQVALQLAFYRLHRRLVPTYESASIRRFQEGRVDNIRSATPEALAFVRAVTDHKAAVPASEKLLLLKDAIRAQTAYTVMAITGMAIDNHLLALRELARAMCAALPEMFMDETYLMSNRFVLSTSQVPTTTEMFCCYGPVVPNGYGACYNPQPETILFCISSFHSCAATSSSKFAKAVEESLIDMRDLCSLLPP Hydrogen bonds contact Hydrophobic contact | ||||
| 38 | Dipeptidyl peptidase 8 (DPP-8) | 6EOP | 6.43 | |
Target general information Gen name DPP8 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Prolyl dipeptidase DPP8; MSTP141; MSTP135; MSTP097; Dipeptidyl peptidase VIII; Dipeptidyl peptidase IV-related protein 1; DPRP1; DPRP-1; DPP VIII; DP8 Protein family Peptidase S9B family, DPPIV subfamily Biochemical class Peptidase Function Dipeptidyl peptidase that cleaves off N-terminal dipeptides from proteins having a Pro or Ala residue at position 2. Related diseases Orotic aciduria 1 (ORAC1) [MIM:258900]: A disorder of pyrimidine metabolism resulting in megaloblastic anemia and orotic acid crystalluria that is frequently associated with some degree of physical and intellectual disability. A minority of cases have additional features, particularly congenital malformations and immune deficiencies. {ECO:0000269|PubMed:9042911}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number EC 3.4.14.5 Uniprot keywords 3D-structure; Alternative splicing; Aminopeptidase; Apoptosis; Cytoplasm; Hydrolase; Protease; Proteomics identification; Reference proteome; Serine protease Protein physicochemical properties Chain ID A,D Molecular weight (Da) 97764.9 Length 849 Aromaticity 0.12 Instability index 47.71 Isoelectric point 5.69 Charge (pH=7) -21.66 2D Binding mode Binding energy (Kcal/mol) -8.77  Molscript Map 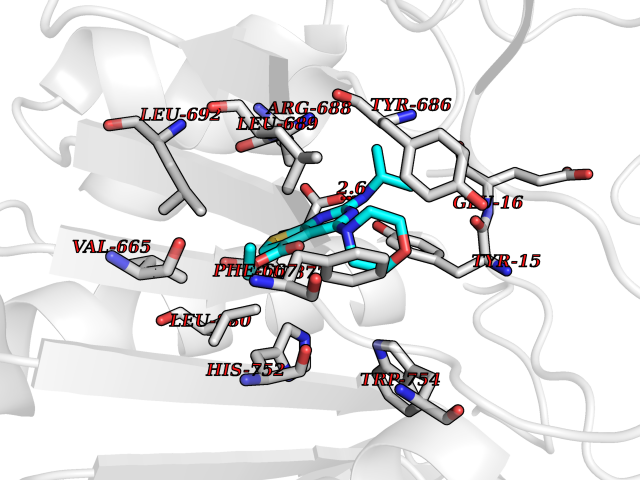 Pymol Map 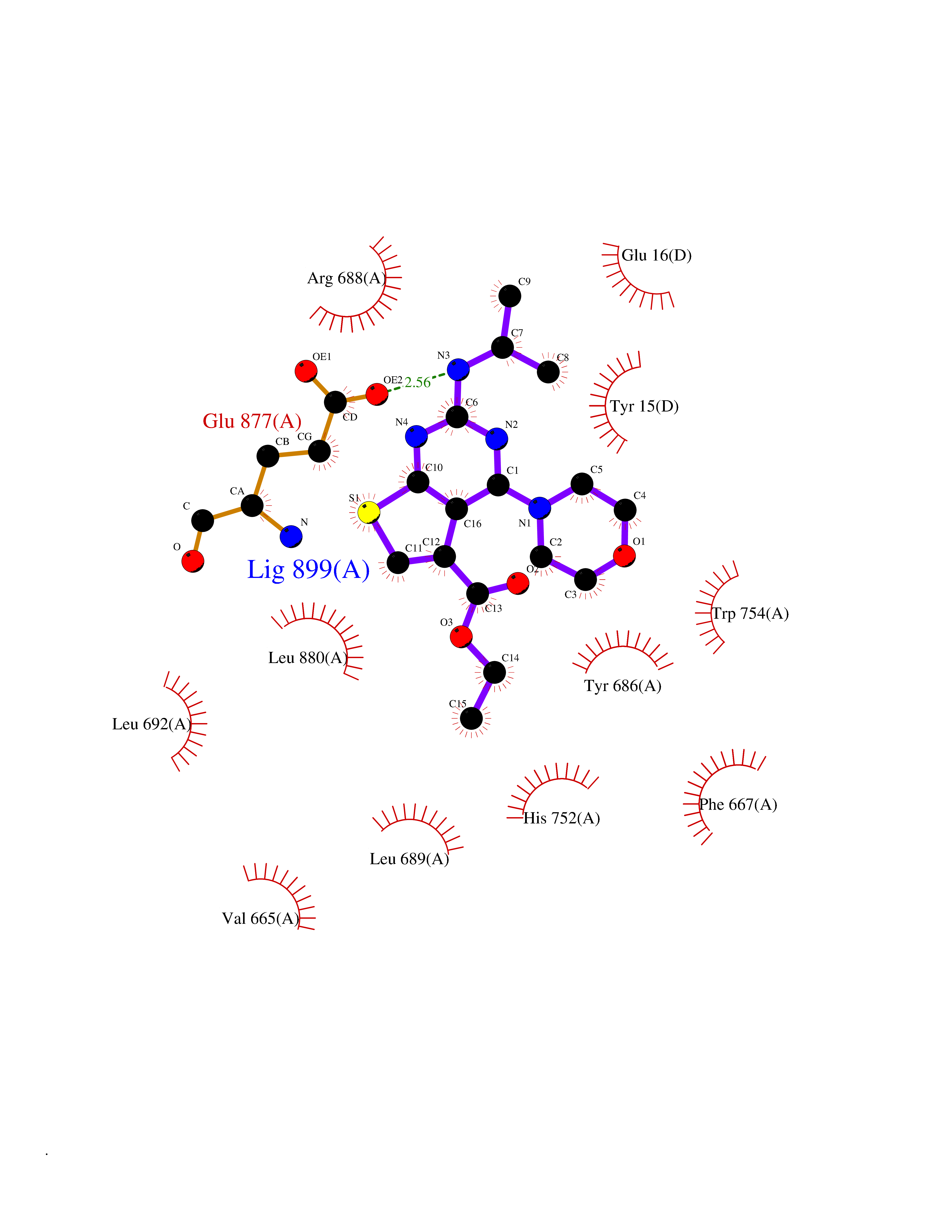 Ligplot Map 3D Binding mode Sequence LEPFYVERYSWSQLKKLLADTRKYHGYMMAKAPHDFMFVKRNDPDGPHSDRIYYLAMSNRENTLFYSEIPKTINRAAVLMLSWKPLLDLFQYSREEELLRERKRIGTVGIASYDYHQGSGTFLFQAGSGIYHVKDGGPQGFTQQPLRPNLVETSCPNIRMDPKLCPADPDWIAFIHSNDIWISNIVTREERRLTYVHNELANMEEDARSAGVATFVLQEEFDRYSGYWWCPKAETTPSGGKILRILYEENDESEVEIIHVTSPMLETRRADSFRYPKTGTANPKVTFKMSEIMIDAEGRIIDVIDKELIQPFEILFEGVEYIARAGWTPEGKYAWSILLDRSQTRLQIVLISPELFIPVEDDVMERQRLIESVPDSVTPLIIYEETTDIWINIHDIFHVFPQSHEEEIEFIFASECKTGFRHLYKITSILKESKYKRSSGGLPAPSDFKCPIKEEIAITSGEWEVLGRHGSNIQVDEVRRLVYFEGTKDSPLEHHLYVVSYVNPGEVTRLTDRGYSHSCCISQHCDFFISKYSNQKNPHCVSLYKLSSPEDDPTCKTKEFWATILDSAGPLPDYTPPEIFSFESTTGFTLYGMLYKPHDLQPGKKYPTVLFIYGGPQVQLVNNRFKGVKYFRLNTLASLGYVVVVIDNRGSXHRGLKFEGAFKYKMGQIEIDDQVEGLQYLASRYDFIDLDRVGIHGWSYGGYLSLMALMQRSDIFRVAIAGAPVTLWIFYDTGYTERYMGHPDQNEQGYYLGSVAMQAEKFPSEPNRLLLLHGFLDENVHFAHTSILLSFLVRAGKPYDLQIYPQERHSIRVPESGEHYELHLLHYLQENLGSRIAALKVSLRFLYEG Hydrogen bonds contact Hydrophobic contact | ||||
| 39 | Bacterial Botulinum toxin A (Bact botA) | 6XCF | 6.43 | |
Target general information Gen name Bact botA Organism Clostridium botulinum Uniprot ID TTD ID Synonyms botA Protein family Peptidase M27 family Biochemical class Peptidase Function Inhibits acetylcholine release. The botulinum toxin binds with high affinity to peripheral neuronal presynaptic membrane to the secretory vesicle protein SV2. It binds directly to the largest luminal loop of SV2A, SV2B and SV2C. It is then internalized by receptor-mediated endocytosis. The C-terminus of the heavy chain (H) is responsible for the adherence of the toxin to the cell surface while the N-terminus mediates transport of the light chain from the endocytic vesicle to the cytosol. After translocation, the light chain (L) hydrolyzes the 197-Gln-|-Arg- 198 bond in SNAP-25, thereby blocking neurotransmitter release. Inhibition of acetylcholine release results in flaccid paralysis, with frequent heart or respiratory failure. Related diseases Major depressive disorder (MDD) [MIM:608516]: A common psychiatric disorder. It is a complex trait characterized by one or more major depressive episodes without a history of manic, mixed, or hypomanic episodes. A major depressive episode is characterized by at least 2 weeks during which there is a new onset or clear worsening of either depressed mood or loss of interest or pleasure in nearly all activities. Four additional symptoms must also be present including changes in appetite, weight, sleep, and psychomotor activity; decreased energy; feelings of worthlessness or guilt; difficulty thinking, concentrating, or making decisions; or recurrent thoughts of death or suicidal ideation, plans, or attempts. The episode must be accompanied by distress or impairment in social, occupational, or other important areas of functioning. {ECO:0000269|PubMed:15229186}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q02563; Q496J9; Q9Z2I6 EC number NA Uniprot keywords 3D-structure; Cell wall; Direct protein sequencing; Disulfide bond; Host cell membrane; Host cytoplasm; Host cytoplasmic vesicle; Host membrane; Host synapse; Hydrolase; Lipid-binding; Membrane; Metal-binding; Metalloprotease; Neurotoxin; Pharmaceutical; Protease; Secreted; Toxin; Transmembrane; Transmembrane helix; Ubl conjugation; Virulence; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 47759.6 Length 417 Aromaticity 0.13 Instability index 21.7 Isoelectric point 6.36 Charge (pH=7) -1.96 2D Binding mode Binding energy (Kcal/mol) -8.77  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence MQFVNKQFNYKDPVNGVDIAYIKIPNVGQMQPVKAFKIHNKIWVIPERDTFTNPEEGDLNPPPPVSYYDSTYLSTDNEKDNYLKGVTKLFERIYSTDLGRMLLTSIVRGIPFWGGSTIDTELKVIDTNCINVIQPDGSYRSEELNLVIIGPSADIIQFECKSFGHEVLNLTRNGYGSTQYIRFSPDFTFGFEESLEVDTNPLLGAGKFATDPAVTLAHELIHAGHRLYGIAINPNRVFKVNTNAYYEMSGLEVSFEELRTFGGHDAKFIDSLQENEFRLYYYNKFKDIASTLNKAKSIVGTTASLQYMKNVFKEKYLLSEDTSGKFSVDKLKFDKLYKMLTEIYTEDNFVKFFKVLNRKTYLNFDKAVFKINIVPKVNYTIYDGFNLRNTNLAANFNGQNTEINNMNFTKLKNFTGL Hydrogen bonds contact Hydrophobic contact | ||||
| 40 | N-acylethanolamine-hydrolyzing acidamidase (NAAA) | 6DXX | 6.43 | |
Target general information Gen name NAAA Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Nacylsphingosine amidohydrolaselike; Nacylethanolaminehydrolyzing acid amidase subunit beta; NAAA; Acid ceramidaselike protein; ASAHlike protein Protein family Acid ceramidase family Biochemical class Carbon-nitrogen hydrolase Function Degrades bioactive fatty acid amides to their corresponding acids, with the following preference: N- palmitoylethanolamine > N-myristoylethanolamine > N- lauroylethanolamine = N-stearoylethanolamine > N- arachidonoylethanolamine > N-oleoylethanolamine. Also exhibits weak hydrolytic activity against the ceramides N- lauroylsphingosine and N-palmitoylsphingosine. Related diseases Hypertriglyceridemia, transient infantile (HTGTI) [MIM:614480]: An autosomal recessive disorder characterized by onset of moderate to severe transient hypertriglyceridemia in infancy that normalizes with age. The hypertriglyceridemia is associated with hepatomegaly, moderately elevated transaminases, persistent fatty liver, and the development of hepatic fibrosis. {ECO:0000269|PubMed:22226083, ECO:0000269|PubMed:24549054}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB09061; DB14009; DB14011 Interacts with NA EC number EC 3.5.1.- Uniprot keywords 3D-structure; Alternative splicing; Autocatalytic cleavage; Direct protein sequencing; Disulfide bond; Fatty acid metabolism; Glycoprotein; Hydrolase; Lipid degradation; Lipid metabolism; Lysosome; Membrane; Proteomics identification; Reference proteome; Signal; Zymogen Protein physicochemical properties Chain ID A,B Molecular weight (Da) 36877.8 Length 328 Aromaticity 0.11 Instability index 44.37 Isoelectric point 7.72 Charge (pH=7) 1.08 2D Binding mode Binding energy (Kcal/mol) -8.77 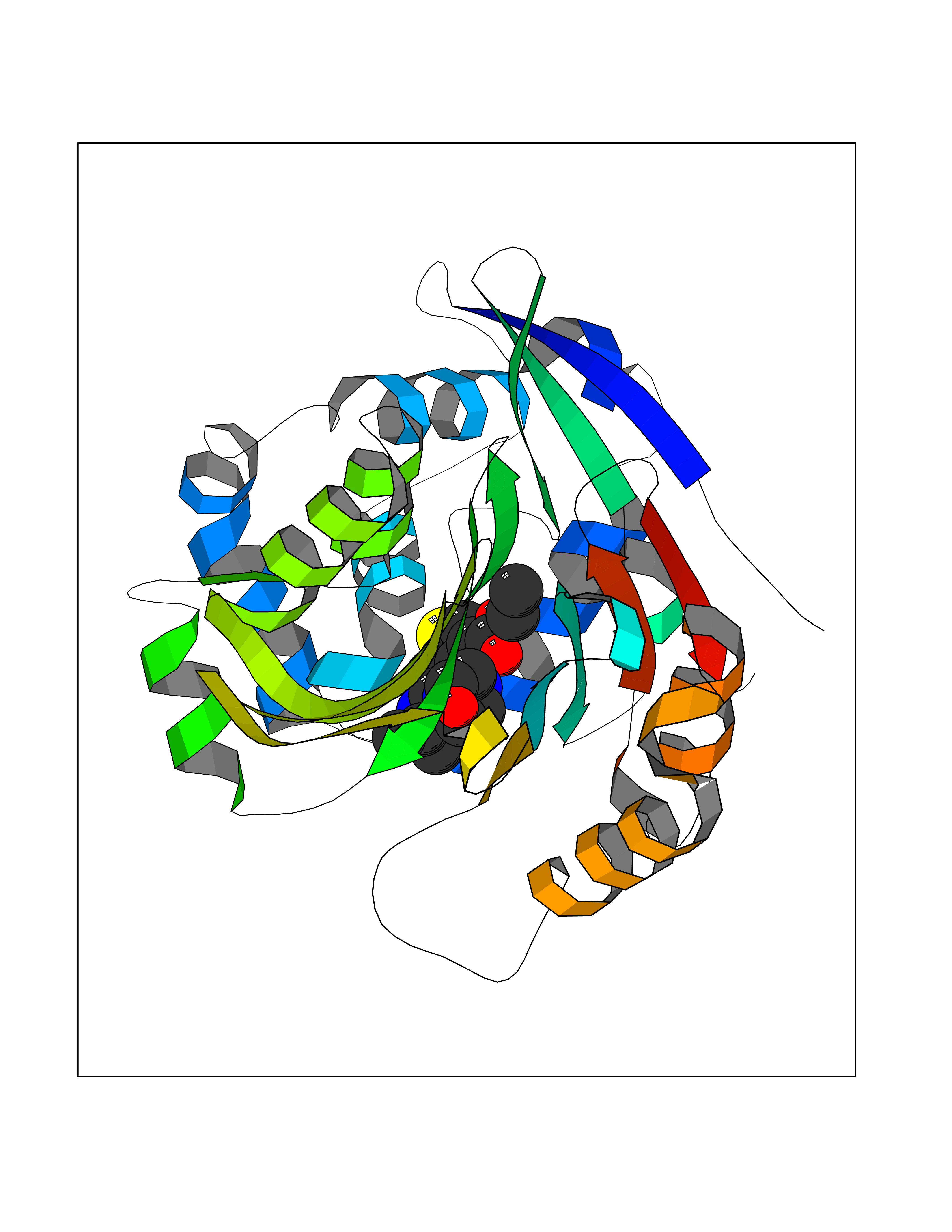 Molscript Map 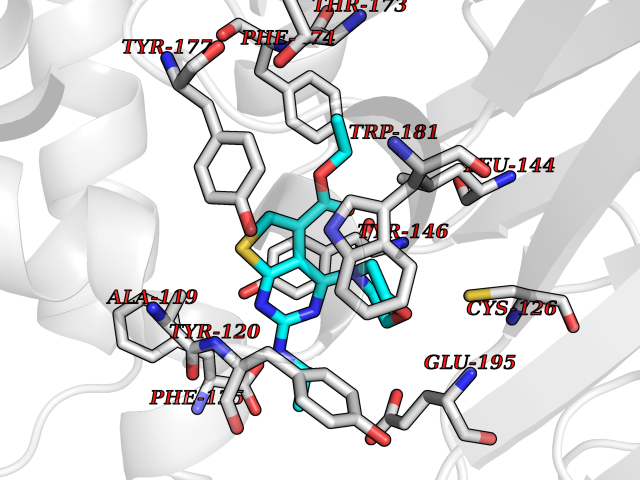 Pymol Map 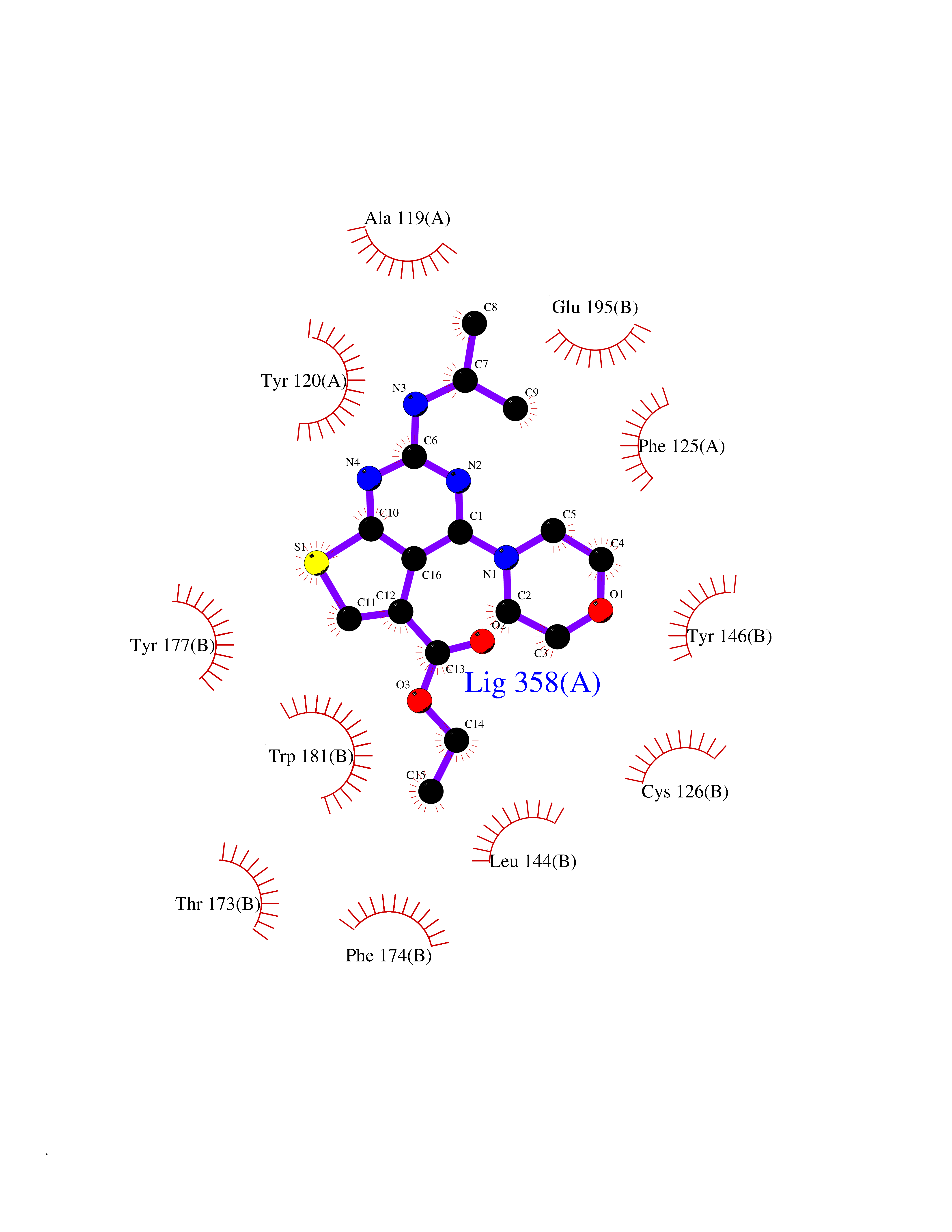 Ligplot Map 3D Binding mode Sequence SPPAAPRFNVSLDSVPELRWLPVLRHYDLDLVRAAMAQVIGDRVPKWVHVLIGKVVLELERFLPQPFTGEIRGMCDFMNLSLADCLLVNLAYESSVFCTSIVAQDSRGHIYHGRNLDYPFGNVLRKLTVDVQFLKNGQIAFTGTTFIGYVGLWTGQSPHKFTVSGDERDKGWWWENAIAALFRRHIPVSWLIRATLSESENFEAAVGKLAKTPLIADVYYIVGGTSPREGVVITRNRDGPADIWPLDPLNGAWFRVETNYDHWKPAPKEDDRRTSAIKALNATGQANLSLEALFQILSVVPVYNNFTIYTTVMSAGSPDKYMTRIRNP Hydrogen bonds contact Hydrophobic contact | ||||