Job Results:
Ligand
Structure
Job ID
b5aa43c6924a21fc75a6162a600078fc
Job name
NA
Time
2026-03-04 17:25:26
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 1 | Monoamine oxidase type B (MAO-B) | 2V5Z | 8.77 | |
Target general information Gen name MAOB Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms MAO-B; Amine oxidase [flavin-containing] B Protein family Flavin monoamine oxidase family Biochemical class CH-NH(2) donor oxidoreductase Function Catalyzes the oxidative deamination of biogenic and xenobiotic amines and has important functions in the metabolism of neuroactive and vasoactive amines in the central nervous system and peripheral tissues. MAOB preferentially degrades benzylamine and phenylethylamine. Related diseases Microvascular complications of diabetes 5 (MVCD5) [MIM:612633]: Pathological conditions that develop in numerous tissues and organs as a consequence of diabetes mellitus. They include diabetic retinopathy, diabetic nephropathy leading to end-stage renal disease, and diabetic neuropathy. Diabetic retinopathy remains the major cause of new-onset blindness among diabetic adults. It is characterized by vascular permeability and increased tissue ischemia and angiogenesis. Disease susceptibility is associated with variants affecting the gene represented in this entry. Homozygosity for the Leu-55 allele is strongly associated with the development of retinal disease in diabetic patients. Drugs (DrugBank ID) DB08176; DB02211; DB08516; DB08480; DB01472; DB04307; DB07512; DB07513; DB00915; DB00182; DB06698; DB04889; DB00215; DB09130; DB04147; DB00988; DB01363; DB00668; DB01175; DB02509; DB03147; DB14914; DB00614; DB04818; DB02095; DB01247; DB00601; DB01577; DB01442; DB01171; DB08082; DB02643; DB04677; DB03894; DB08804; DB04820; DB00184; DB04821; DB12612; DB01626; DB00780; DB00191; DB00388; DB01132; DB00721; DB01168; DB01367; DB09363; DB06654; DB01037; DB01104; DB14569; DB09042; DB00752; DB16446; DB09185; DB04832; DB00909 Interacts with P55212; P28329-3; Q8NI60; Q5RI15; Q92915-2; P22607; Q53GS7; P06396; P01112; O14901; P13473-2; P21397; Q9BVL2; O75400-2; P62826; Q6NTF9-3; Q9Y371; Q7Z699; Q9UMX0; Q9Y649 EC number EC 1.4.3.4 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Direct protein sequencing; FAD; Flavoprotein; Membrane; Mitochondrion; Mitochondrion outer membrane; Oxidoreductase; Proteomics identification; Reference proteome; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A,B Molecular weight (Da) 56019.9 Length 494 Aromaticity 0.09 Instability index 34.81 Isoelectric point 6.51 Charge (pH=7) -2.2 2D Binding mode Binding energy (Kcal/mol) -11.96  Molscript Map 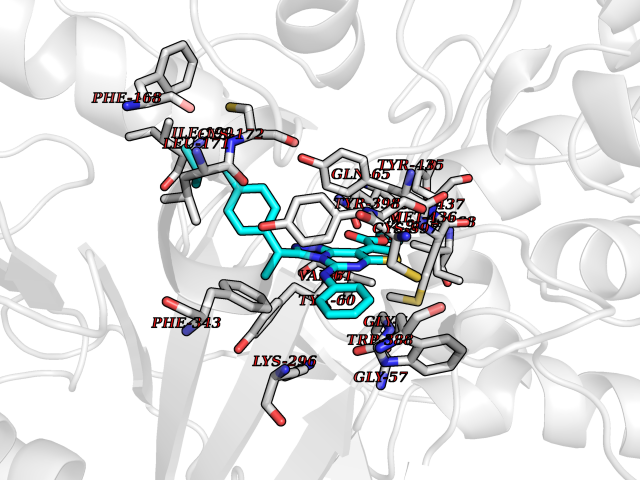 Pymol Map 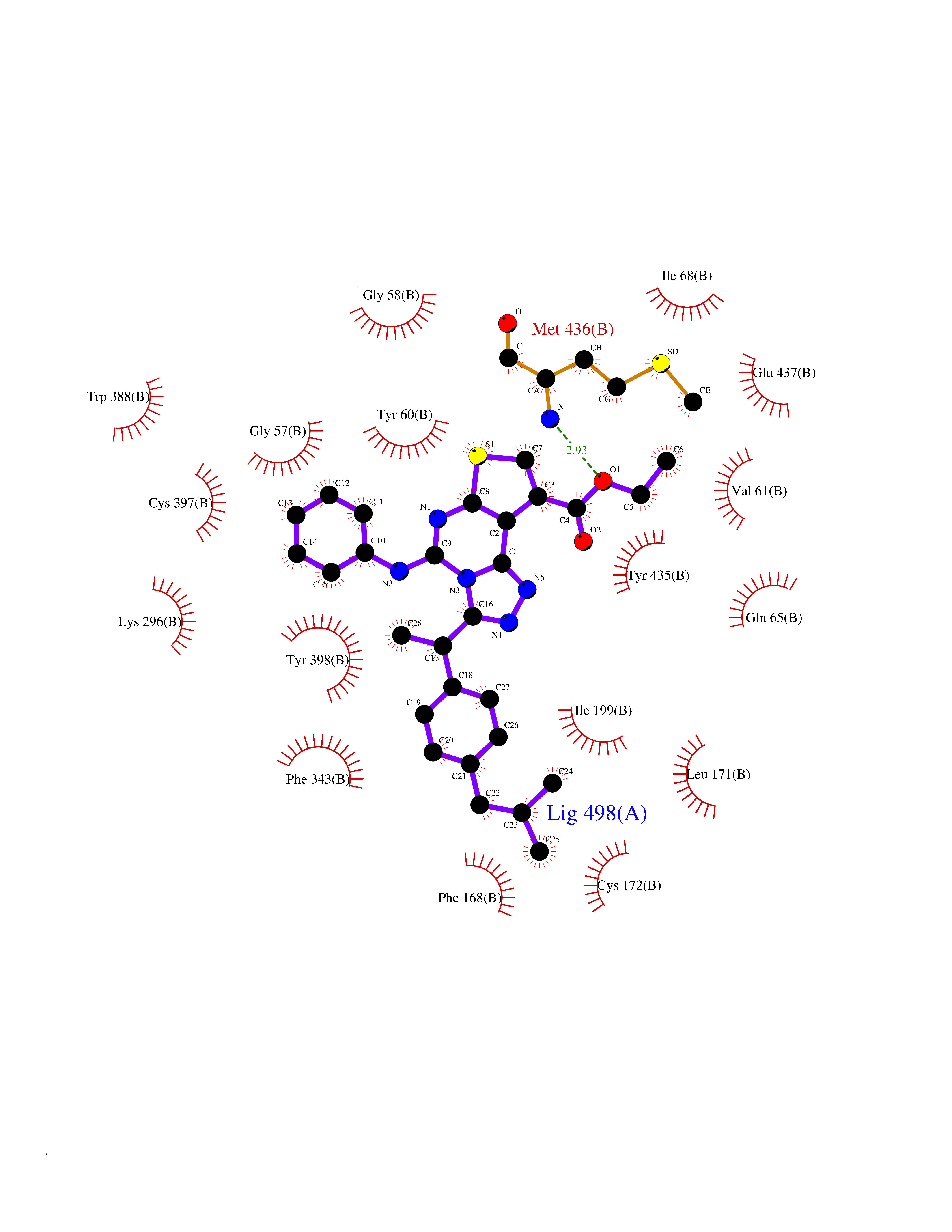 Ligplot Map 3D Binding mode Sequence NKCDVVVVGGGISGMAAAKLLHDSGLNVVVLEARDRVGGRTYTLRNQKVKYVDLGGSYVGPTQNRILRLAKELGLETYKVNEVERLIHHVKGKSYPFRGPFPPVWNPITYLDHNNFWRTMDDMGREIPSDAPWKAPLAEEWDNMTMKELLDKLCWTESAKQLATLFVNLCVTAETHEVSALWFLWYVKQCGGTTRIISTTNGGQERKFVGGSGQVSERIMDLLGDRVKLERPVIYIDQTRENVLVETLNHEMYEAKYVISAIPPTLGMKIHFNPPLPMMRNQMITRVPLGSVIKCIVYYKEPFWRKKDYCGTMIIDGEEAPVAYTLDDTKPEGNYAAIMGFILAHKARKLARLTKEERLKKLCELYAKVLGSLEALEPVHYEEKNWCEEQYSGGCYTTYFPPGILTQYGRVLRQPVDRIYFAGTETATHWSGYMEGAVEAGERAAREILHAMGKIPEDEIWQSEPESVDVPAQPITTTFLERHLPSVPGLLRLI Hydrogen bonds contact Hydrophobic contact | ||||
| 2 | Phenylethanolamine N-methyltransferase (PNMT) | 2G72 | 8.77 | |
Target general information Gen name PNMT Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms PNMTase; PENT; Noradrenaline N-methyltransferase Protein family Class I-like SAM-binding methyltransferase superfamily, NNMT/PNMT/TEMT family Biochemical class NA Function Converts noradrenaline to adrenaline. Related diseases A chromosomal aberration involving TRIM24/TIF1 is found in papillary thyroid carcinomas (PTCs). Translocation t(7;10)(q32;q11) with RET. The translocation generates the TRIM24/RET (PTC6) oncogene. {ECO:0000269|PubMed:10439047}. Drugs (DrugBank ID) DB08129; DB08128; DB07739; DB07798; DB07747; DB03468; DB08550; DB03824; DB04273; DB07906; DB07597; DB09571; DB00968; DB08631; DB01752; DB08654 Interacts with Q9P2G9-2; Q8TBB1 EC number EC 2.1.1.28 Uniprot keywords 3D-structure; Catecholamine biosynthesis; Direct protein sequencing; Methyltransferase; Phosphoprotein; Proteomics identification; Reference proteome; S-adenosyl-L-methionine; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 29198.9 Length 264 Aromaticity 0.09 Instability index 54.33 Isoelectric point 5.91 Charge (pH=7) -3.69 2D Binding mode Binding energy (Kcal/mol) -11.96 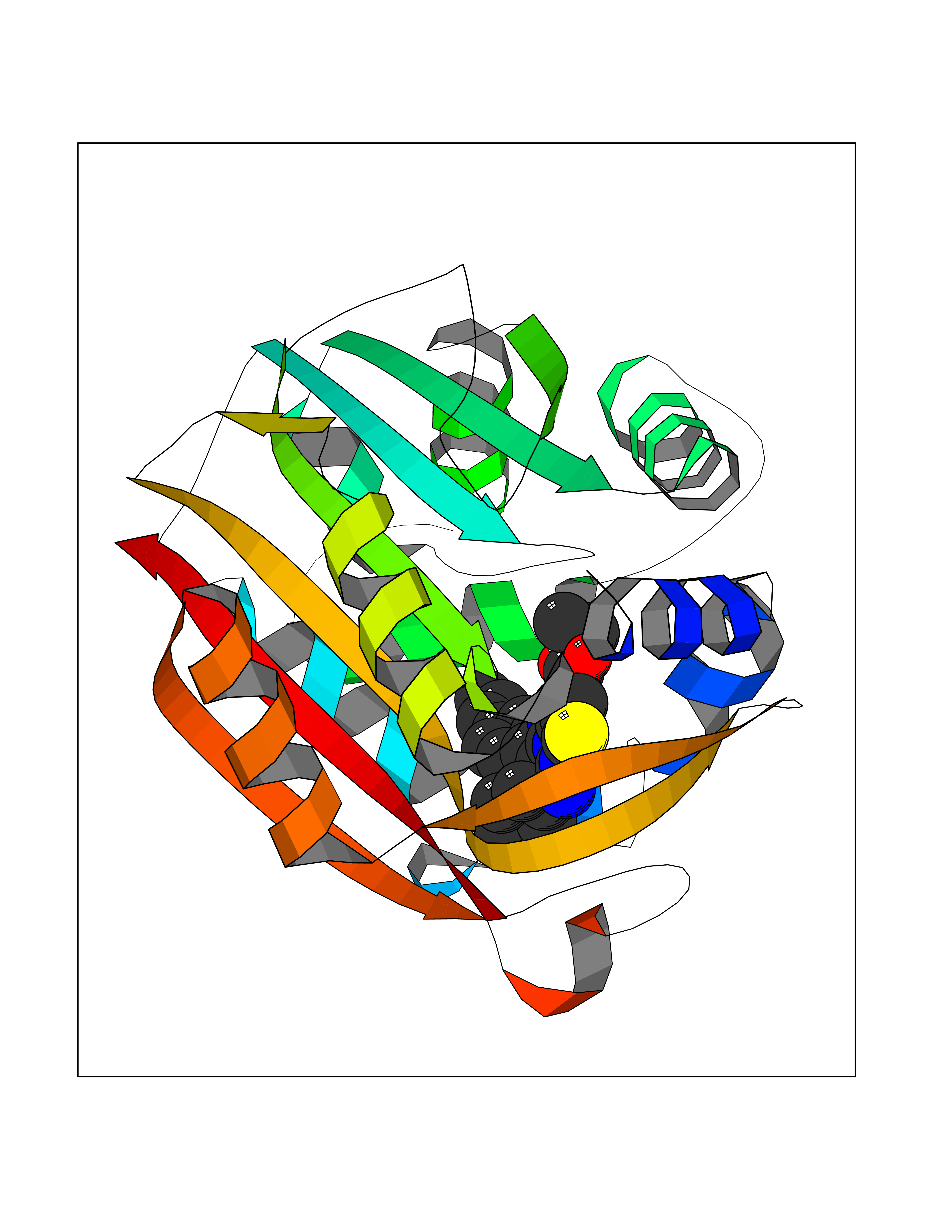 Molscript Map 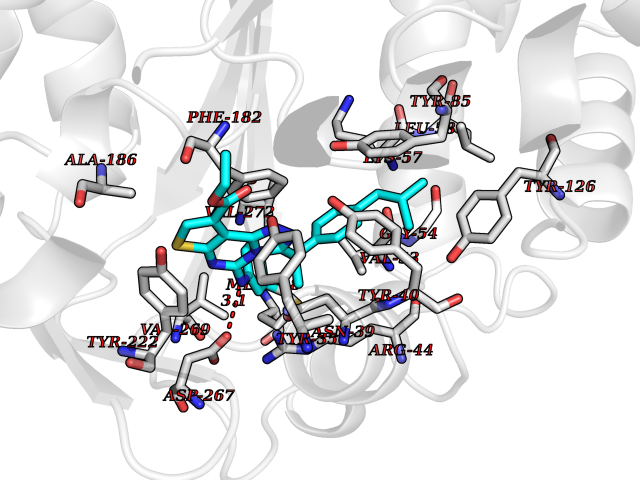 Pymol Map 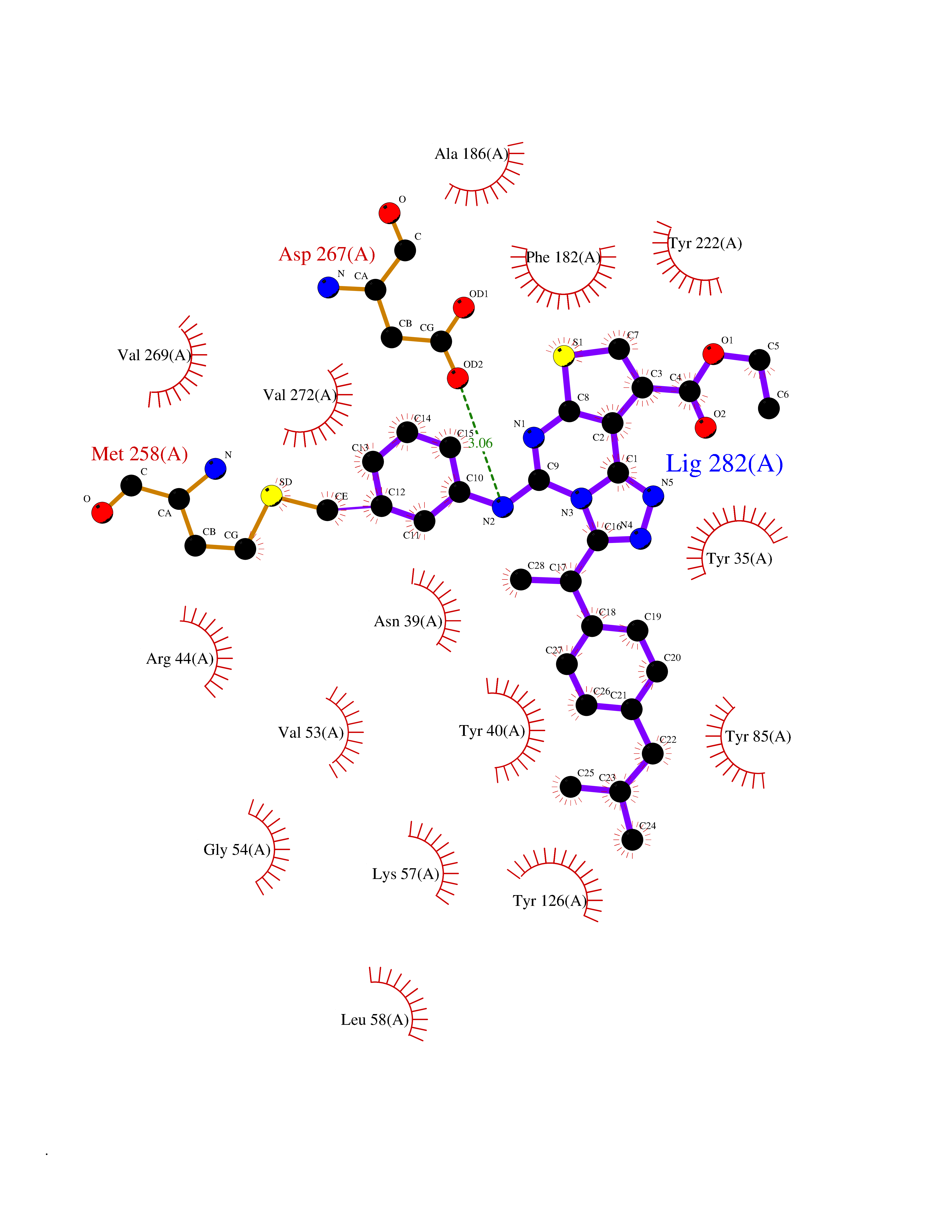 Ligplot Map 3D Binding mode Sequence APGQAAVASAYQRFEPRAYLRNNYAPPRGDLCNPNGVGPWKLRCLAQTFATGEVSGRTLIDIGSGPTVYQLLSACSHFEDITMTDFLEVNRQELGRWLQEEPGAFNWSMYSQHACLIEGKGECWQDKERQLRARVKRVLPIDVHQPQPLGAGSPAPLPADALVSAFCLEAVSPDLASFQRALDHITTLLRPGGHLLLIGALEESWYLAGEARLTVVPVSEEEVREALVRSGYKVRDLRTYIMPAHLQTGVDDVKGVFFAWAQKV Hydrogen bonds contact Hydrophobic contact | ||||
| 3 | LIM domain kinase-2 (LIMK-2) | 7QHG | 8.72 | |
Target general information Gen name LIMK2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms LIMK-2; LIM domain kinase 2 Protein family Protein kinase superfamily, TKL Ser/Thr protein kinase family Biochemical class Kinase Function Displays serine/threonine-specific phosphorylation of myelin basic protein and histone (MBP) in vitro. Related diseases Intellectual developmental disorder, autosomal recessive 80, with variant lissencephaly (MRT80) [MIM:620653]: An autosomal recessive disorder characterized by global developmental delay, mildly to moderately impaired intellectual development, attention deficit-hyperactivity disorder, hypotonia, seizure, poor social skills, and autistic traits. Brain imaging shows fronto-temporal lissencephaly and pachygyria. {ECO:0000269|PubMed:37880421}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB11718; DB12010 Interacts with Q16543; P08238; Q96C90; P62258 EC number EC 2.7.11.1 Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Cytoplasm; Cytoskeleton; Kinase; LIM domain; Metal-binding; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Serine/threonine-protein kinase; Transferase; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 32109.2 Length 283 Aromaticity 0.1 Instability index 27.28 Isoelectric point 6.01 Charge (pH=7) -3.93 2D Binding mode Binding energy (Kcal/mol) -11.89 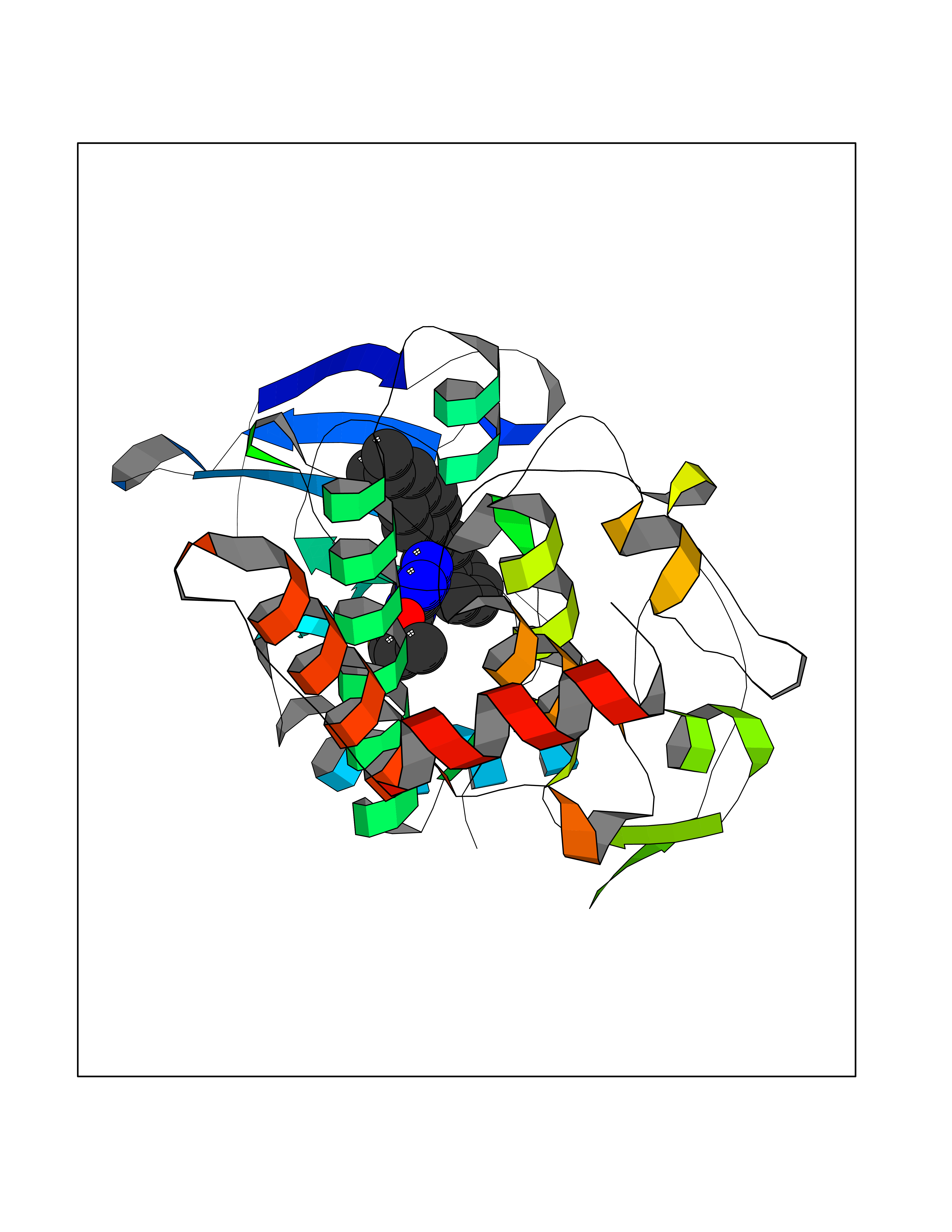 Molscript Map 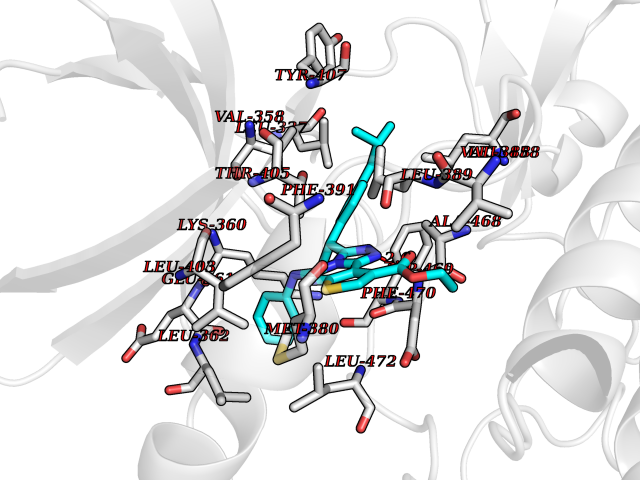 Pymol Map 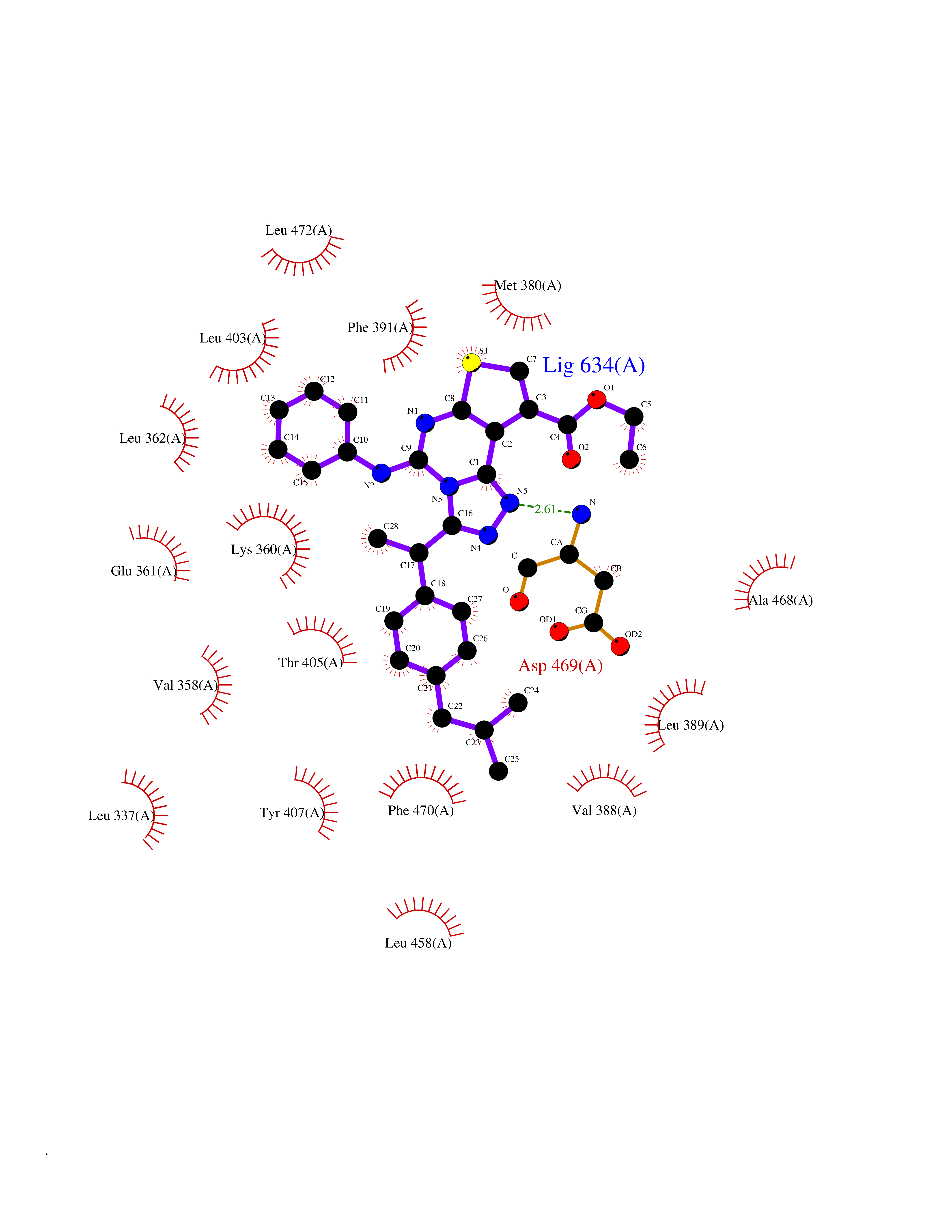 Ligplot Map 3D Binding mode Sequence MDLIHGEVLGKGFFGQAIKVTHKATGKVMVMKELIRCDEETQKTFLTEVKVMRSLDHPNVLKFIGVLYKDKKLNLLTEYIEGGTLKDFLRSMDPFPWQQKVRFAKGIASGMAYLHSMCIIHRDLNSHNCLIKLDKTVVVADFGLSRLIVDRKKRYTVVGNPYWMAPEMLNGKSYDETVDIFSFGIVLCEIIGQVYADPDCLPRTLDFGLNVKLFWEKFVPTDCPPAFFPLAAICCRLEPESRPAFSKLEDSFEALSLYLGELGIPLPAELEELDHTVSMQYGL Hydrogen bonds contact Hydrophobic contact | ||||
| 4 | Vitamin K epoxide reductase complex 1 (VKORC1) | 6WV3 | 8.68 | |
Target general information Gen name VKORC1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Vitamin K1 2,3-epoxide reductase subunit 1; VKORC1; VKOR; UNQ308/PRO351; MSTP576; MSTP134 Protein family VKOR family Biochemical class Short-chain dehydrogenases reductase Function Involved invitamin K metabolism. Catalytic subunit of the vitamin K epoxide reductase (VKOR) complex which reduces inactive vitamin K 2,3-epoxide to active vitamin K. Vitamin K is required for the gamma-carboxylation of various proteins, including clotting factors, and is required for normal blood coagulation, but also for normal bone development. Related diseases Combined deficiency of vitamin K-dependent clotting factors 2 (VKCFD2) [MIM:607473]: VKCFD leads to a bleeding tendency that is usually reversed by oral administration of vitamin K. {ECO:0000269|PubMed:14765194, ECO:0000269|PubMed:16270630}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Coumarin resistance (CMRES) [MIM:122700]: A condition characterized by partial or complete resistance to warfarin or other 4-hydroxycoumarin derivatives. These drugs are used as anti-coagulants for the prevention of thromboembolic diseases in subjects with deep vein thrombosis, atrial fibrillation, or mechanical heart valve replacement. {ECO:0000269|PubMed:14765194, ECO:0000269|PubMed:20946155}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01418; DB00266; DB09332; DB00170; DB00498; DB00946; DB01022; DB00682 Interacts with Q13323; Q7Z7G2; Q96BA8; Q9Y282; Q5JX71; Q96KR6; Q5T7V8; Q8TDT2; Q9NQG1; P15941-11; Q96TC7; Q9NR31; A0A0S2Z4U3; Q8TBB6; O15393-2; Q19QW4 EC number EC 1.17.4.4 Uniprot keywords 3D-structure; Alternative splicing; Disease variant; Disulfide bond; Endoplasmic reticulum; Membrane; Oxidoreductase; Proteomics identification; Quinone; Redox-active center; Reference proteome; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 42656.4 Length 381 Aromaticity 0.1 Instability index 32.12 Isoelectric point 7.73 Charge (pH=7) 1.93 2D Binding mode Binding energy (Kcal/mol) -11.84 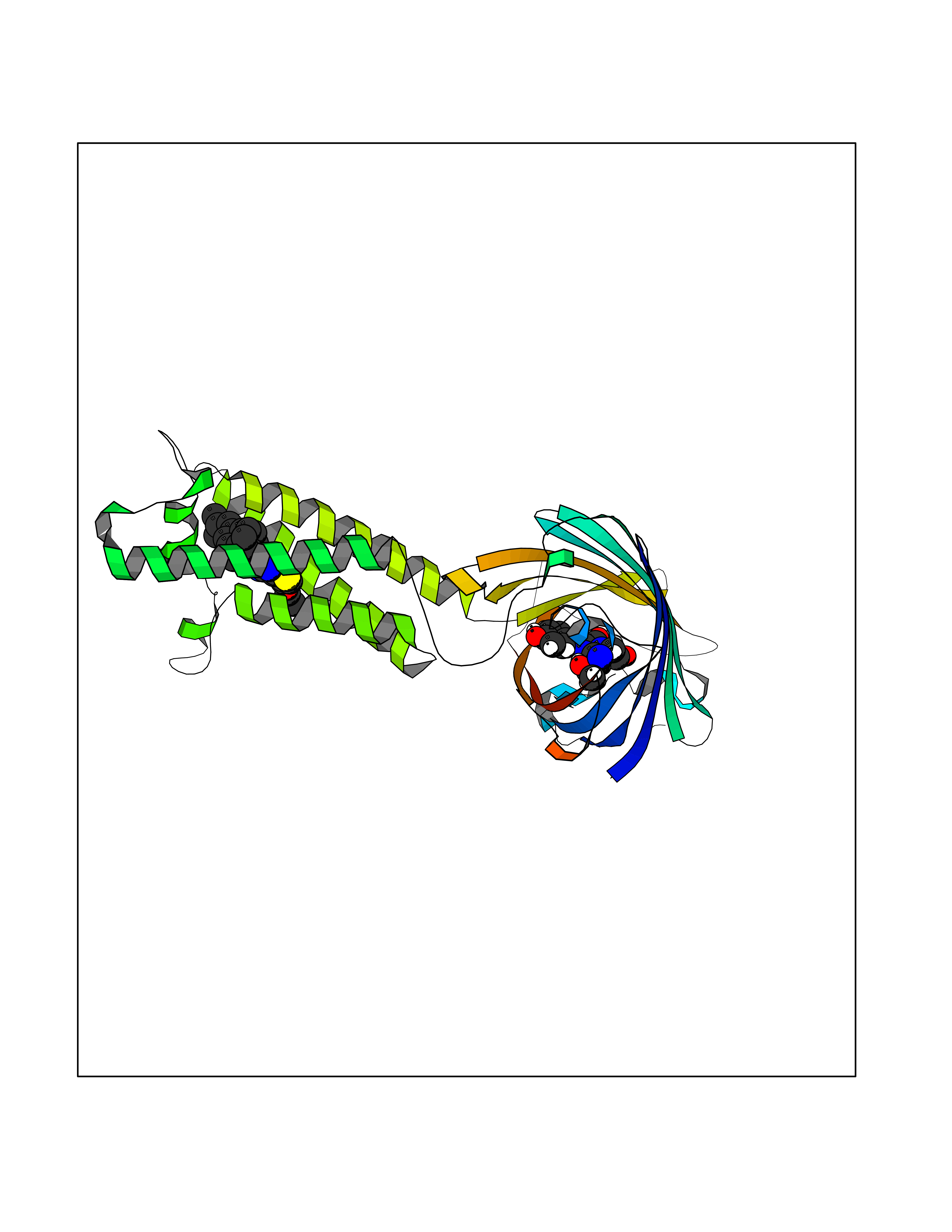 Molscript Map 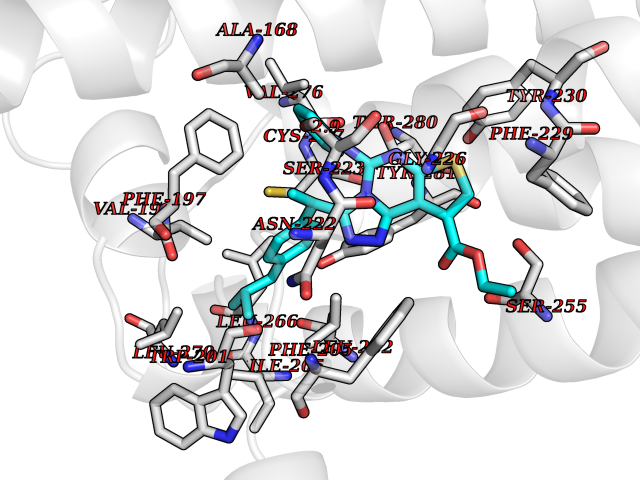 Pymol Map 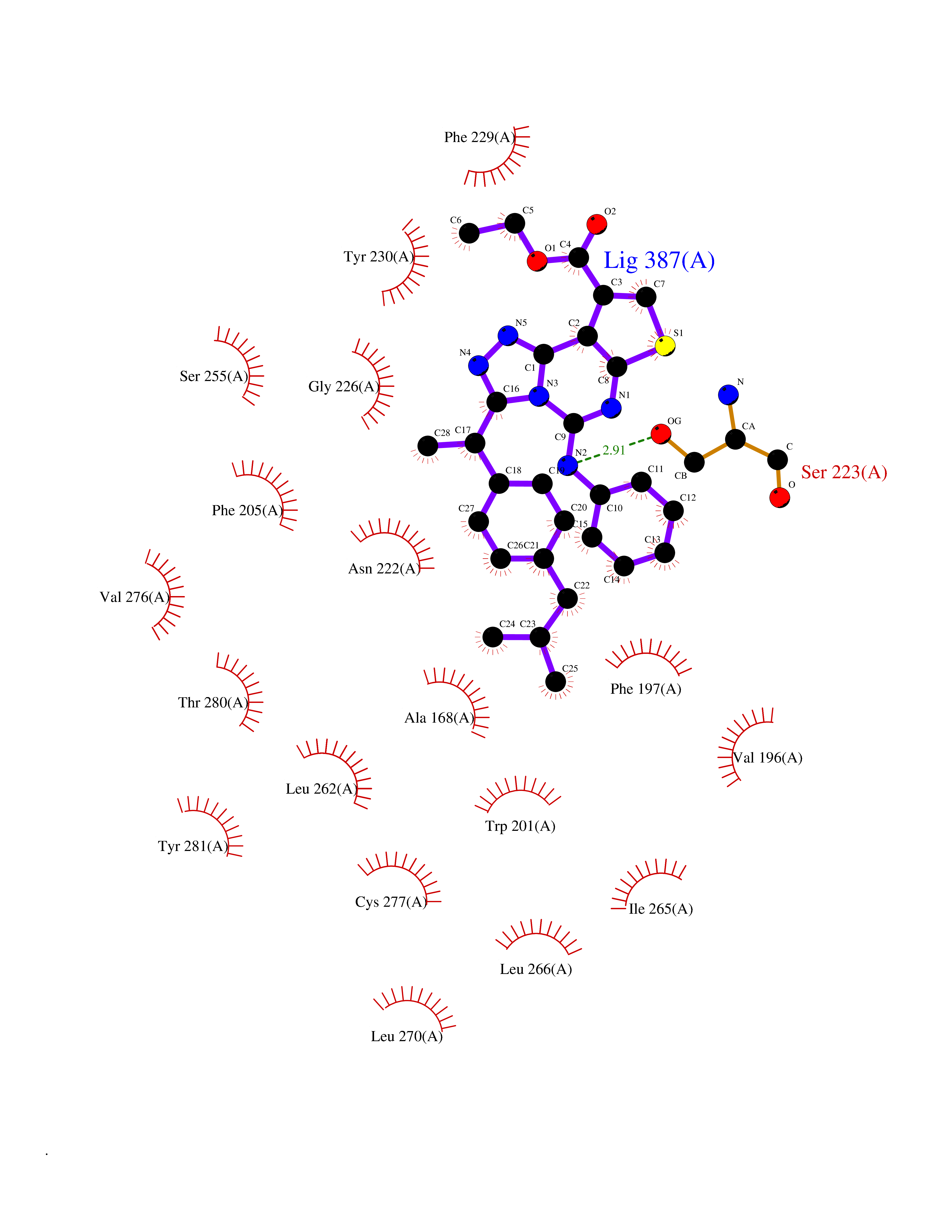 Ligplot Map 3D Binding mode Sequence KGEELFTGVVPILVELDGDVNGHKFSVRGEGEGDATNGKLTLKFICTTGKLPVPWPTLVTTLXVQCFSRYPDHMKRHDFFKSAMPEGYVQERTISFKDDGTYKTRAEVKFEGDTLVNRIELKGIDFKEDGNILGHKLEYNSTWGSPGWVRLALCLTGLVLSLYALHVKAARARDRDYRALCDVGTAISCSRVFSSRWGRGFGLVEHVLGQDSILNQSNSIFGCIFYTLQLLLGCLRTRWASVLMLLSSLVSLAGSVYLAWILFFVLYDFCIVCITTYAINVSLMWLSFRKVQENSHNVYITADKQKNGIKANFKIRHNVEDGSVQLADHYQQNTPIGDGPVLLPDNHYLSTQSVLSKDPNEKRDHMVLLEFVTAAGITHHH Hydrogen bonds contact Hydrophobic contact | ||||
| 5 | Monoamine oxidase type A (MAO-A) | 2Z5Y | 8.67 | |
Target general information Gen name MAOA Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Monoamine oxidase A; Amine oxidase [flavin-containing] A Protein family Flavin monoamine oxidase family Biochemical class CH-NH(2) donor oxidoreductase Function MAOA preferentially oxidizes biogenic amines such as 5-hydroxytryptamine (5-HT), norepinephrine and epinephrine. Catalyzes the oxidative deamination of biogenic and xenobiotic amines and has important functions in the metabolism of neuroactive and vasoactive amines in the central nervous system and peripheral tissues. Related diseases Brunner syndrome (BRNRS) [MIM:300615]: A form of X-linked non-dysmorphic mild intellectual disability. Male patients are affected by borderline intellectual deficit and exhibit abnormal behavior, including disturbed regulation of impulsive aggression. Obligate female carriers have normal intelligence and behavior. {ECO:0000269|PubMed:8211186}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01472; DB00918; DB00182; DB06698; DB04889; DB13876; DB01445; DB06774; DB00215; DB04017; DB09130; DB05205; DB07641; DB00988; DB01363; DB00668; DB12329; DB01175; DB03147; DB14914; DB00614; DB01381; DB07919; DB04818; DB01247; DB00601; DB01577; DB00805; DB01442; DB01171; DB08804; DB00952; DB04820; DB00184; DB04821; DB06412; DB01626; DB00780; DB00191; DB00388; DB00397; DB09244; DB04850; DB00721; DB01168; DB00571; DB00852; DB09363; DB00140; DB00953; DB06654; DB01037; DB01104; DB00669; DB14569; DB09042; DB00624; DB13943; DB13944; DB13946; DB09245; DB00752; DB15328; DB09185; DB04832; DB00315; DB00909 Interacts with P27338 EC number EC 1.4.3.4 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Catecholamine metabolism; Direct protein sequencing; Disease variant; FAD; Flavoprotein; Intellectual disability; Membrane; Mitochondrion; Mitochondrion outer membrane; Neurotransmitter degradation; Oxidoreductase; Phosphoprotein; Proteomics identification; Reference proteome; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 58195.3 Length 513 Aromaticity 0.11 Instability index 34.97 Isoelectric point 7.98 Charge (pH=7) 2.87 2D Binding mode Binding energy (Kcal/mol) -11.82 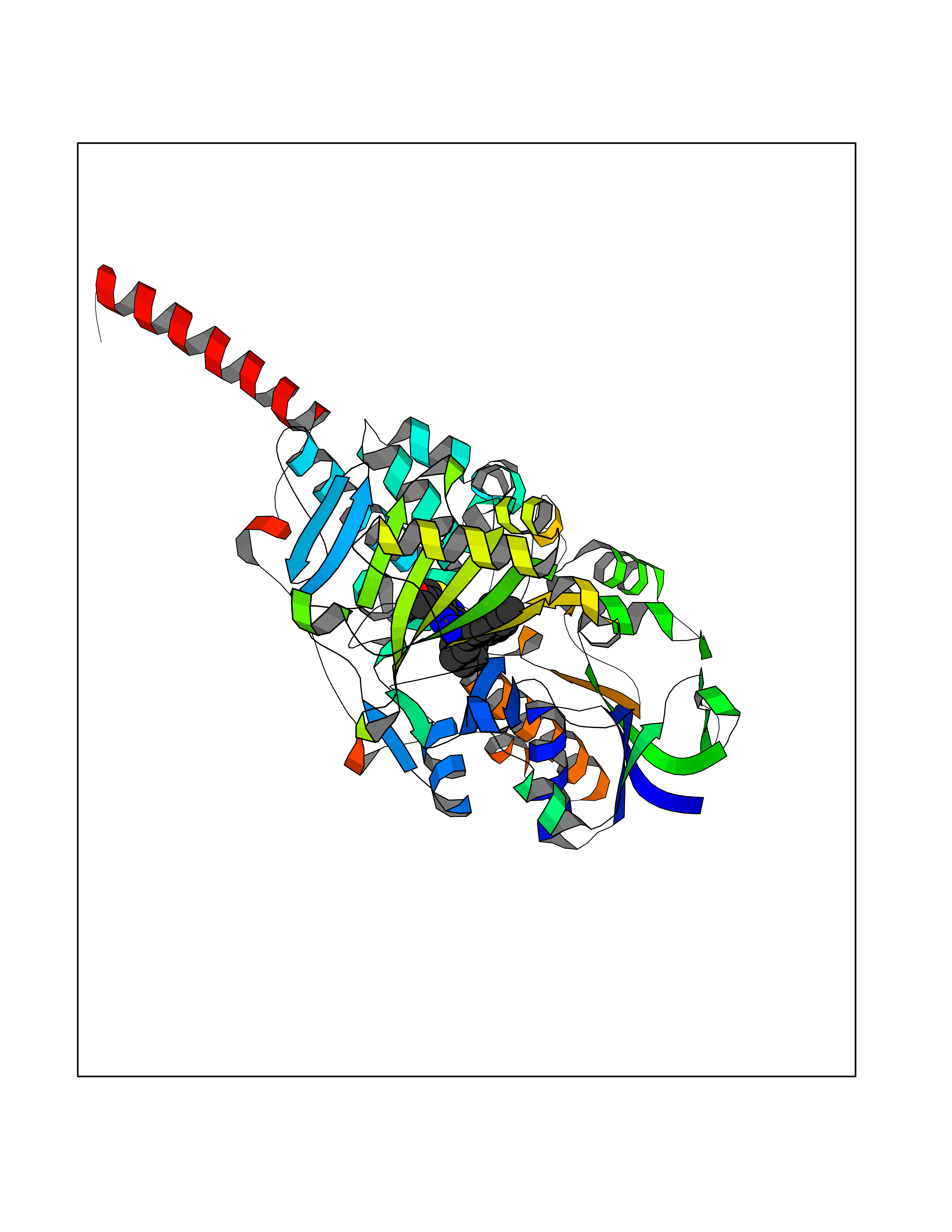 Molscript Map 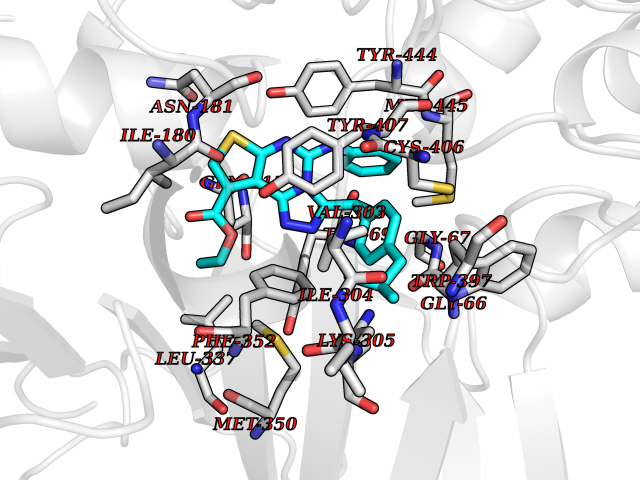 Pymol Map 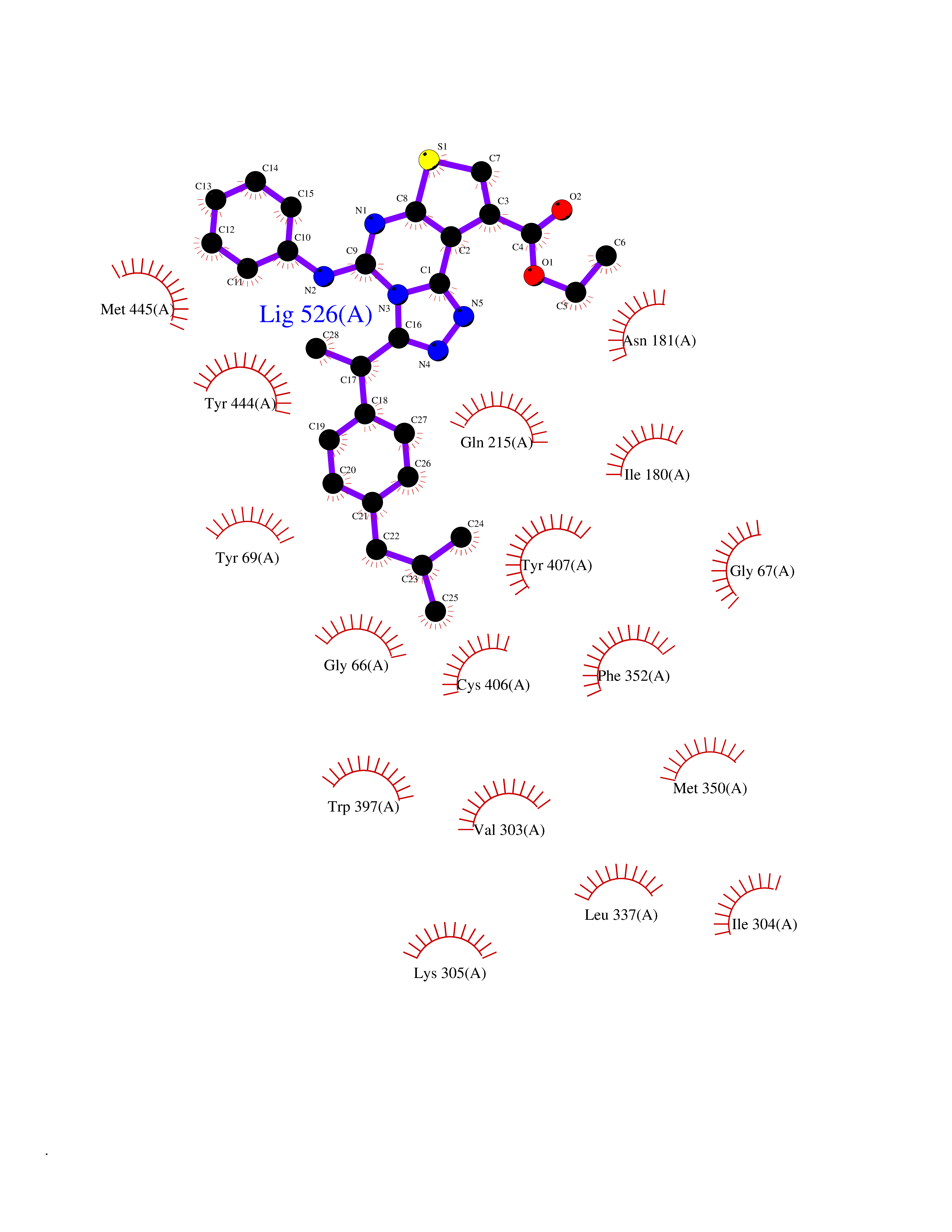 Ligplot Map 3D Binding mode Sequence HMFDVVVIGGGISGLSAAKLLTEYGVSVLVLEARDRVGGRTYTIRNEHVDYVDVGGAYVGPTQNRILRLSKELGIETYKVNVSERLVQYVKGKTYPFRAAFPPVWNPIAYLDYNNLWRTIDNMGKEIPTDAPWEAQHADKWDKMTMKELIDKICWTKTARRFAYLFVNINVTSEPHEVSALWFLWYVKQCGGTTRIFSVTNGGQERKFVGGSGQVSERIMDLLGDQVKLNHPVTHVDQSSDNIIIETLNHEHYECKYVINAIPPTLTAKIHFRPELPAERNQLIQRLPMGAVIKCMMYYKEAFWKKKDYCGCMIIEDEDAPISITLDDTKPDGSLPAIMGFILARKADRLAKLHKEIRKKKICELYAKVLGSQEALHPVHYEEKNWCEEQYSGGCYTAYFPPGIMTQYGRVIRQPVGRIFFAGTETATKWSGYMEGAVEAGERAAREVLNGLGKVTEKDIWVQEPESKDVPAVEITHTFWERNLPSVSGLLKIIGFSTSVTALGFVLYKYKLL Hydrogen bonds contact Hydrophobic contact | ||||
| 6 | Oxysterols receptor LXR-alpha (NR1H3) | 3IPQ | 8.66 | |
Target general information Gen name NR1H3 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Nuclear receptor subfamily 1 group H member 3; Nuclear receptor LXRalpha; Nuclear orphan receptor LXR-alpha; Liver X receptor alpha; LXRalpha; LXRA Protein family Nuclear hormone receptor family, NR1 subfamily Biochemical class Nuclear hormone receptor Function Interaction with retinoic acid receptor (RXR) shifts RXR from its role as a silent DNA-binding partner to an active ligand-binding subunit in mediating retinoid responses through target genes defined by LXRES. LXRES are DR4-type response elements characterized by direct repeats of two similar hexanuclotide half-sites spaced by four nucleotides. Plays an important role in the regulation of cholesterol homeostasis, regulating cholesterol uptake through MYLIP-dependent ubiquitination of LDLR, VLDLR and LRP8. Interplays functionally with RORA for the regulation of genes involved in liver metabolism. Nuclear receptor that exhibits a ligand-dependent transcriptional activation activity. Related diseases Okur-Chung neurodevelopmental syndrome (OCNDS) [MIM:617062]: An autosomal dominant neurodevelopmental disorder characterized by developmental delay, intellectual disability, behavioral problems, hypotonia, speech problems, microcephaly, pachygyria and variable dysmorphic features. {ECO:0000269|PubMed:27048600}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08175; DB08063; DB11994; DB07929; DB13174; DB07080 Interacts with O60869; O60341; Q99750; Q15788; O75376; Q07869; Q07869-1; Q03181; P37231; P19793; P28702; P48443; O43463; P42858; Q99750; O95817; G5E9A7; O95872; P02545; Q99750; P28702; P28702-3; P48443; Q7Z699 EC number NA Uniprot keywords 3D-structure; Activator; Alternative splicing; Cytoplasm; DNA-binding; Metal-binding; Nucleus; Proteomics identification; Receptor; Reference proteome; Transcription; Transcription regulation; Ubl conjugation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A Molecular weight (Da) 25389.9 Length 220 Aromaticity 0.09 Instability index 46.42 Isoelectric point 5.51 Charge (pH=7) -6.58 2D Binding mode Binding energy (Kcal/mol) -11.81 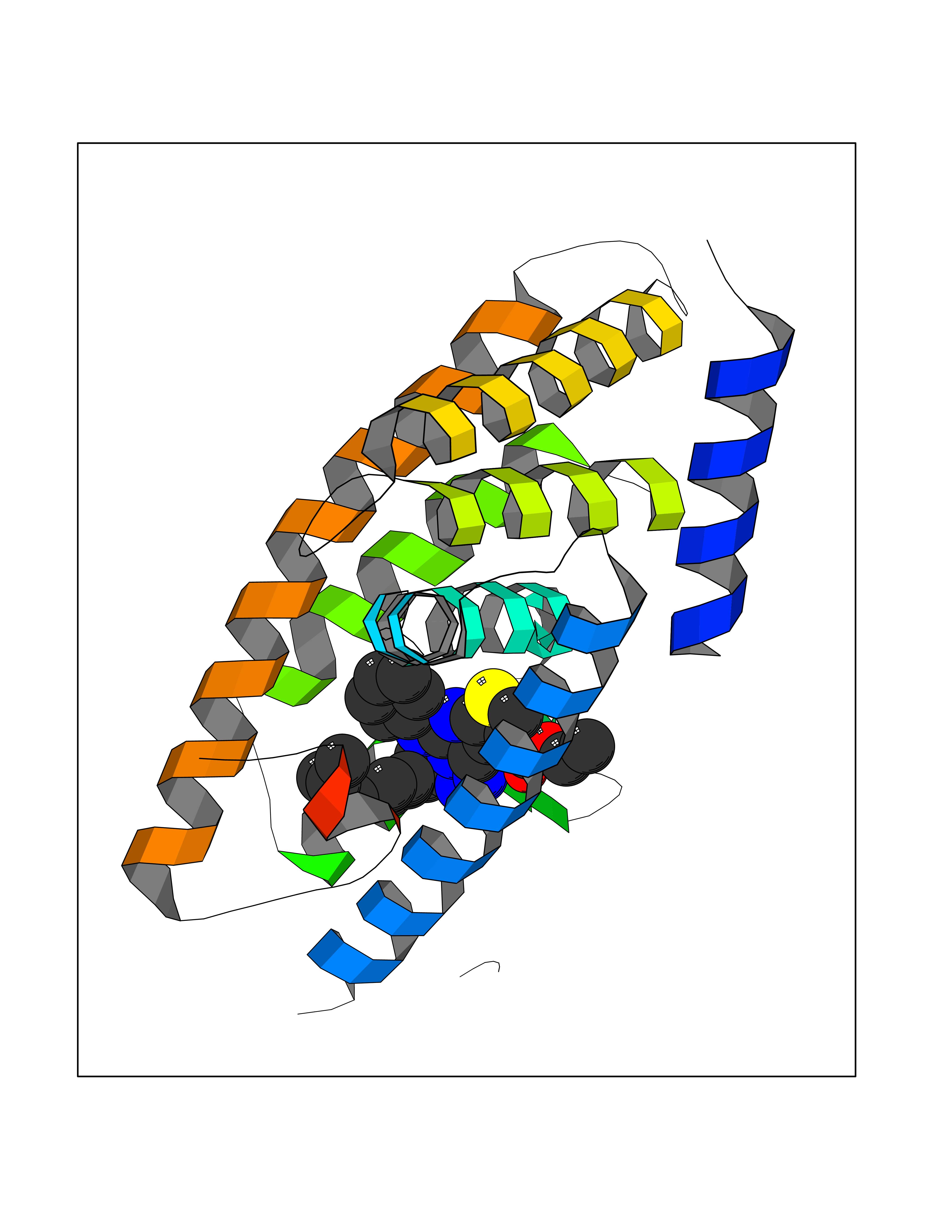 Molscript Map 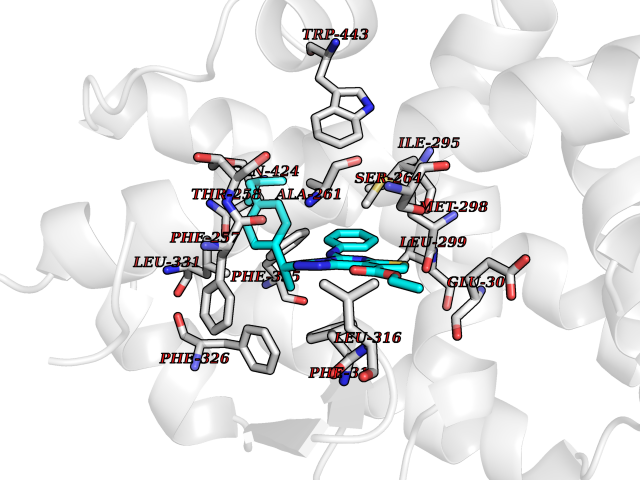 Pymol Map 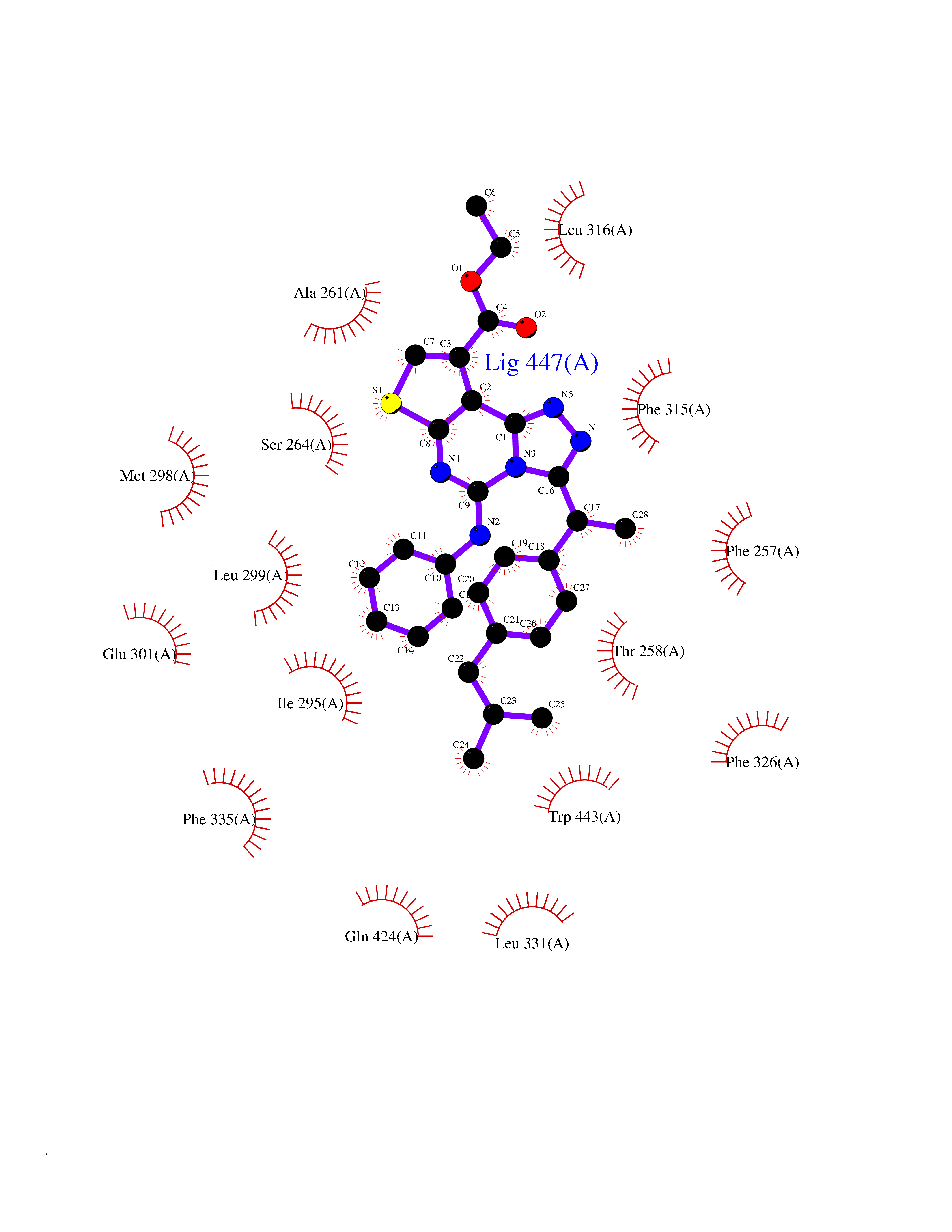 Ligplot Map 3D Binding mode Sequence QLSPEQLGMIEKLVAAQQTPWPEARQQRFAHFTELAIVSVQEIVDFAKQLPGFLQLSREDQIALLKTSAIEVMLLETSRRYNPGSESITFLKDFSYNREDFAKAGLQVEFINPIFEFSRAMNELQLNDAEFALLIAISIFSADRPNVQDQLQVERLQHTYVEALHAYVSIHHPHDRLMFPRMLMKLVSLRTLSSVHSEQVFALRLQDKKLPPLLSEIWDV Hydrogen bonds contact Hydrophobic contact | ||||
| 7 | Monoamine oxidase type B (MAO-B) | 2V5Z | 8.64 | |
Target general information Gen name MAOB Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms MAO-B; Amine oxidase [flavin-containing] B Protein family Flavin monoamine oxidase family Biochemical class CH-NH(2) donor oxidoreductase Function Catalyzes the oxidative deamination of biogenic and xenobiotic amines and has important functions in the metabolism of neuroactive and vasoactive amines in the central nervous system and peripheral tissues. MAOB preferentially degrades benzylamine and phenylethylamine. Related diseases Microvascular complications of diabetes 5 (MVCD5) [MIM:612633]: Pathological conditions that develop in numerous tissues and organs as a consequence of diabetes mellitus. They include diabetic retinopathy, diabetic nephropathy leading to end-stage renal disease, and diabetic neuropathy. Diabetic retinopathy remains the major cause of new-onset blindness among diabetic adults. It is characterized by vascular permeability and increased tissue ischemia and angiogenesis. Disease susceptibility is associated with variants affecting the gene represented in this entry. Homozygosity for the Leu-55 allele is strongly associated with the development of retinal disease in diabetic patients. Drugs (DrugBank ID) DB08176; DB02211; DB08516; DB08480; DB01472; DB04307; DB07512; DB07513; DB00915; DB00182; DB06698; DB04889; DB00215; DB09130; DB04147; DB00988; DB01363; DB00668; DB01175; DB02509; DB03147; DB14914; DB00614; DB04818; DB02095; DB01247; DB00601; DB01577; DB01442; DB01171; DB08082; DB02643; DB04677; DB03894; DB08804; DB04820; DB00184; DB04821; DB12612; DB01626; DB00780; DB00191; DB00388; DB01132; DB00721; DB01168; DB01367; DB09363; DB06654; DB01037; DB01104; DB14569; DB09042; DB00752; DB16446; DB09185; DB04832; DB00909 Interacts with P55212; P28329-3; Q8NI60; Q5RI15; Q92915-2; P22607; Q53GS7; P06396; P01112; O14901; P13473-2; P21397; Q9BVL2; O75400-2; P62826; Q6NTF9-3; Q9Y371; Q7Z699; Q9UMX0; Q9Y649 EC number EC 1.4.3.4 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Direct protein sequencing; FAD; Flavoprotein; Membrane; Mitochondrion; Mitochondrion outer membrane; Oxidoreductase; Proteomics identification; Reference proteome; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A,B Molecular weight (Da) 56019.9 Length 494 Aromaticity 0.09 Instability index 34.81 Isoelectric point 6.51 Charge (pH=7) -2.2 2D Binding mode Binding energy (Kcal/mol) -11.79 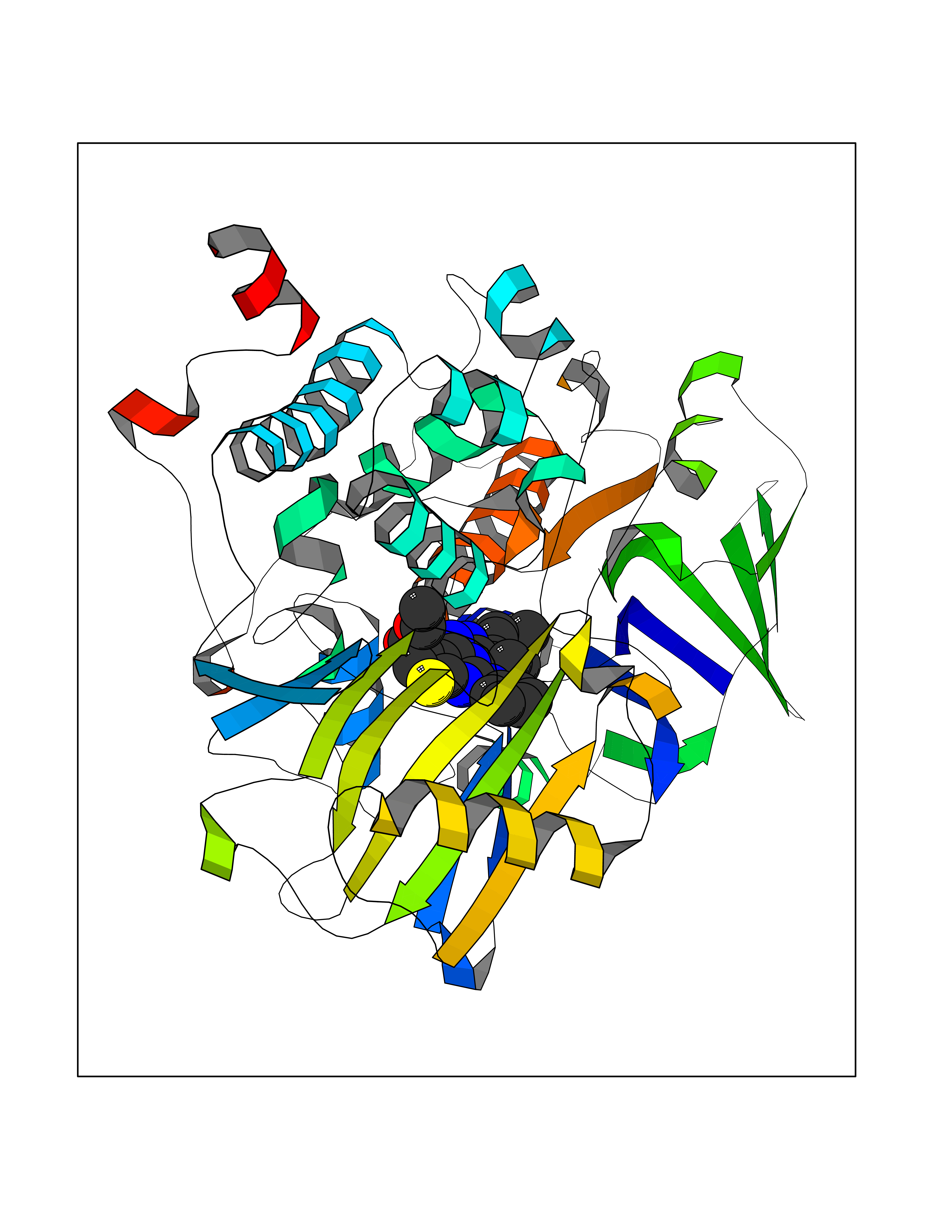 Molscript Map 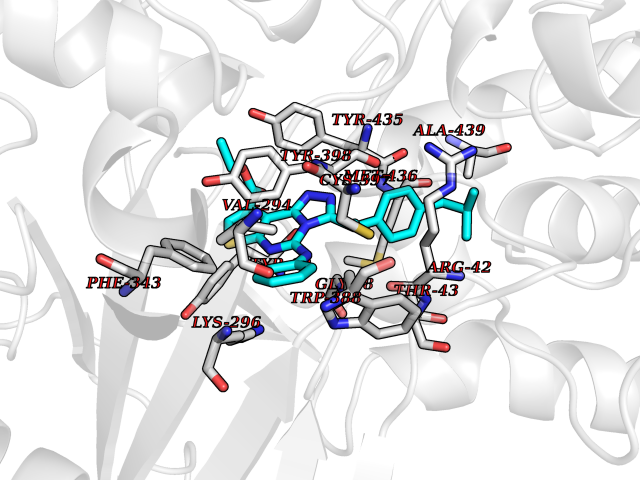 Pymol Map 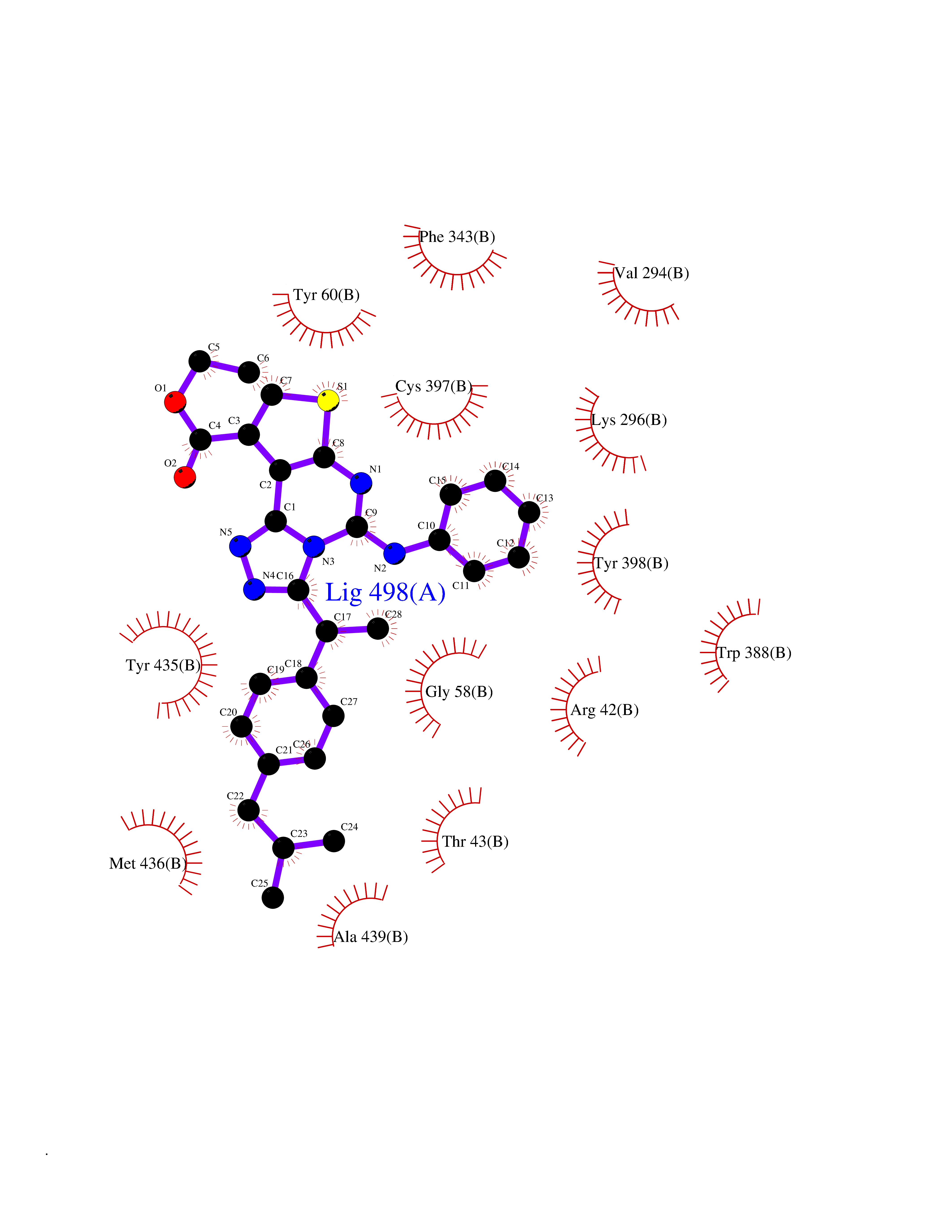 Ligplot Map 3D Binding mode Sequence NKCDVVVVGGGISGMAAAKLLHDSGLNVVVLEARDRVGGRTYTLRNQKVKYVDLGGSYVGPTQNRILRLAKELGLETYKVNEVERLIHHVKGKSYPFRGPFPPVWNPITYLDHNNFWRTMDDMGREIPSDAPWKAPLAEEWDNMTMKELLDKLCWTESAKQLATLFVNLCVTAETHEVSALWFLWYVKQCGGTTRIISTTNGGQERKFVGGSGQVSERIMDLLGDRVKLERPVIYIDQTRENVLVETLNHEMYEAKYVISAIPPTLGMKIHFNPPLPMMRNQMITRVPLGSVIKCIVYYKEPFWRKKDYCGTMIIDGEEAPVAYTLDDTKPEGNYAAIMGFILAHKARKLARLTKEERLKKLCELYAKVLGSLEALEPVHYEEKNWCEEQYSGGCYTTYFPPGILTQYGRVLRQPVDRIYFAGTETATHWSGYMEGAVEAGERAAREILHAMGKIPEDEIWQSEPESVDVPAQPITTTFLERHLPSVPGLLRLI Hydrogen bonds contact Hydrophobic contact | ||||
| 8 | Mutated Histone H3.3 (H3F3A) | 4GUS | 8.64 | |
Target general information Gen name H3F3A Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms PP781; Histone H3.3; H3F3; H3.3B; H3.3A Protein family Histone H3 family Biochemical class NA Function Variant histone H3 which replaces conventional H3 in a wide range of nucleosomes in active genes. Constitutes the predominant form of histone H3 in non-dividing cells and is incorporated into chromatin independently of DNA synthesis. Deposited at sites of nucleosomal displacement throughout transcribed genes, suggesting that it represents an epigenetic imprint of transcriptionally active chromatin. Nucleosomes wrap and compact DNA into chromatin, limiting DNA accessibility to the cellular machineries which require DNA as a template. Histones thereby play a central role in transcription regulation, DNA repair, DNA replication and chromosomal stability. DNA accessibility is regulated via a complex set of post-translational modifications of histones, also called histone code, and nucleosome remodeling. Related diseases Glioma (GLM) [MIM:137800]: Gliomas are benign or malignant central nervous system neoplasms derived from glial cells. They comprise astrocytomas and glioblastoma multiforme that are derived from astrocytes, oligodendrogliomas derived from oligodendrocytes and ependymomas derived from ependymocytes. {ECO:0000269|PubMed:22286061, ECO:0000269|PubMed:22286216, ECO:0000269|PubMed:23539269}. The gene represented in this entry is involved in disease pathogenesis. H3F3A mutations affecting residues involved in post-translational modifications of histone H3.3 are recurrent in malignant, aggressive gliomas including glioblastoma multiforme (GBM) and diffuse intrinsic pontine glioma (DIPG) (PubMed:22286061, PubMed:22286216). The mechanism through which mutations lead to tumorigenesis involves altered histones methylation, impaired regulation of Polycomb repressive complex 2 (PRC2) activity, and aberrant epigenetic regulation of gene expression (PubMed:23539183, PubMed:23539269, PubMed:23603901). {ECO:0000269|PubMed:22286061, ECO:0000269|PubMed:22286216, ECO:0000269|PubMed:23539183, ECO:0000269|PubMed:23539269, ECO:0000269|PubMed:23603901}.; DISEASE: Bryant-Li-Bhoj neurodevelopmental syndrome 1 (BRYLIB1) [MIM:619720]: An autosomal dominant disorder predominantly characterized by global developmental delay, impaired intellectual development, poor or absent speech, and delayed motor milestones. Clinical manifestations are highly variable, including abnormal head shape, dysmorphic facial features, oculomotor abnormalities, feeding problems, and non-specific brain imaging abnormalities. Additional features may include hearing loss, seizures, short stature, and mild skeletal defects. {ECO:0000269|PubMed:33268356, ECO:0000269|PubMed:34876591}. The disease is caused by variants affecting the gene represented in this entry. BRYLIB1 is caused by variants in H3-3A. {ECO:0000269|PubMed:33268356, ECO:0000269|PubMed:34876591}.; DISEASE: Bryant-Li-Bhoj neurodevelopmental syndrome 2 (BRYLIB2) [MIM:619721]: An autosomal dominant disorder predominantly characterized by global developmental delay, impaired intellectual development, poor or absent speech, and delayed motor milestones. Clinical manifestations are highly variable, including abnormal head shape, dysmorphic facial features, oculomotor abnormalities, feeding problems, and non-specific brain imaging abnormalities. Additional features may include hearing loss, seizures, short stature, and mild skeletal defects. {ECO:0000269|PubMed:33268356, ECO:0000269|PubMed:34876591}. The disease is caused by variants affecting the gene represented in this entry. BRYLIB2 is caused by variants in H3-3B. {ECO:0000269|PubMed:33268356, ECO:0000269|PubMed:34876591}.; DISEASE: H3F3A and H3F3B mutations affecting residues involved in post-translational modifications of histone H3.3 are implicated in the pathogenesis of some bone and cartilage neoplasms. Mutations have been found with high prevalence in chondroblastoma and giant cell tumors of bone, and with low frequency in osteosarcoma, conventional chondrosarcoma and clear cell chondrosarcoma. Chondroblastoma samples frequently carry a H3F3B mutation affecting residue Lys-37 (H3K36), although H3F3A is mutated in some cases. Most giant cell tumors of bone harbor H3F3A mutations affecting residue Gly-35 (H3G34). {ECO:0000269|PubMed:24162739}. Drugs (DrugBank ID) NA Interacts with Q9NVP2; P45973; Q13111; Q9UER7; Q9UER7-1; Q9Y6K1; P62805; P49321-2; Q8IZL8; Q5VWG9; Q9VK33; Q8R5C8 EC number NA Uniprot keywords 3D-structure; Acetylation; ADP-ribosylation; Chromosome; Citrullination; Direct protein sequencing; Disease variant; DNA-binding; Hydroxylation; Intellectual disability; Lipoprotein; Methylation; Nucleosome core; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Ubl conjugation Protein physicochemical properties Chain ID A,C Molecular weight (Da) 86148.9 Length 766 Aromaticity 0.1 Instability index 37.57 Isoelectric point 8.25 Charge (pH=7) 7.16 2D Binding mode Binding energy (Kcal/mol) -11.79 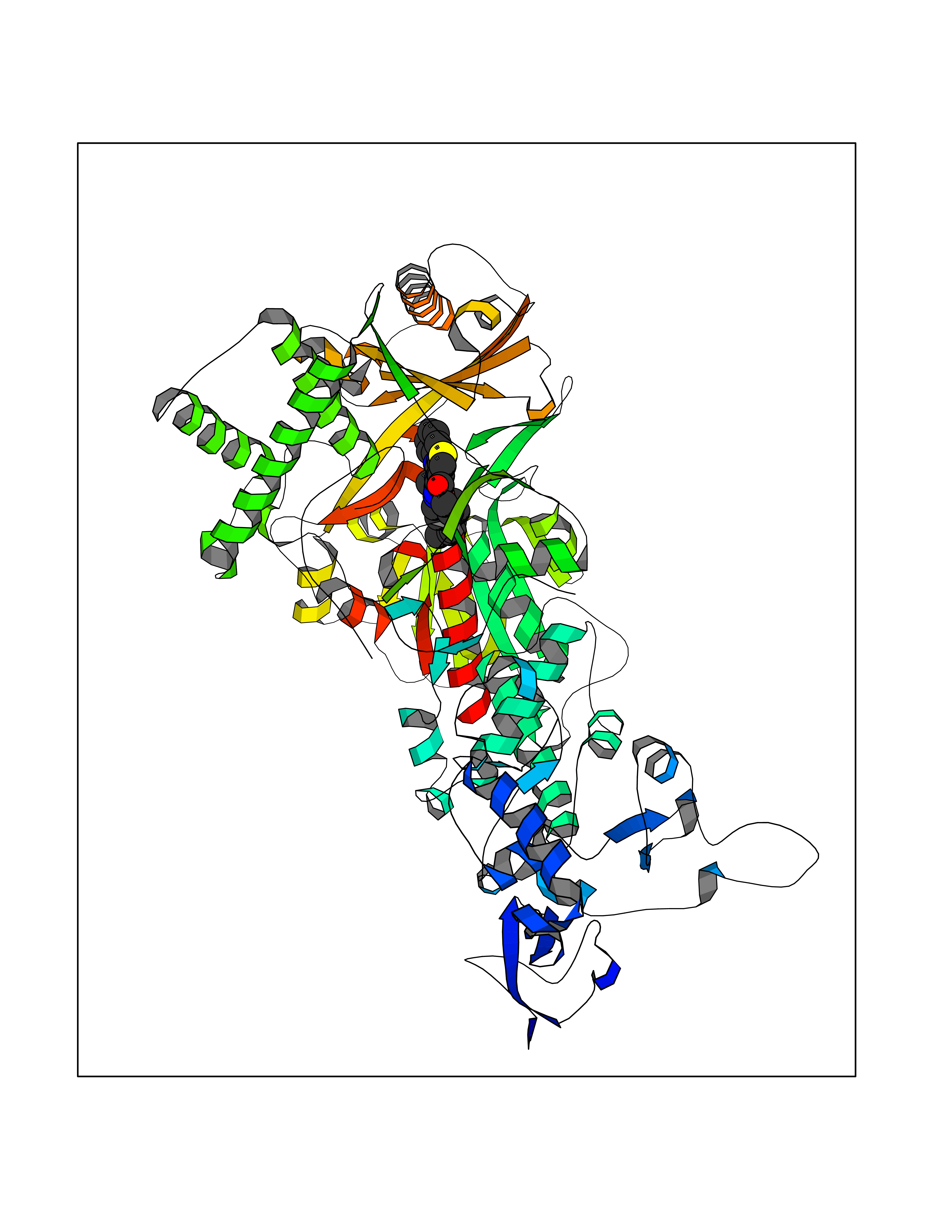 Molscript Map 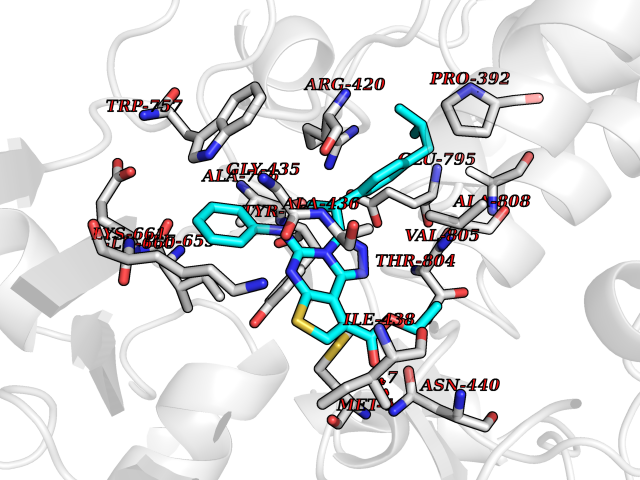 Pymol Map 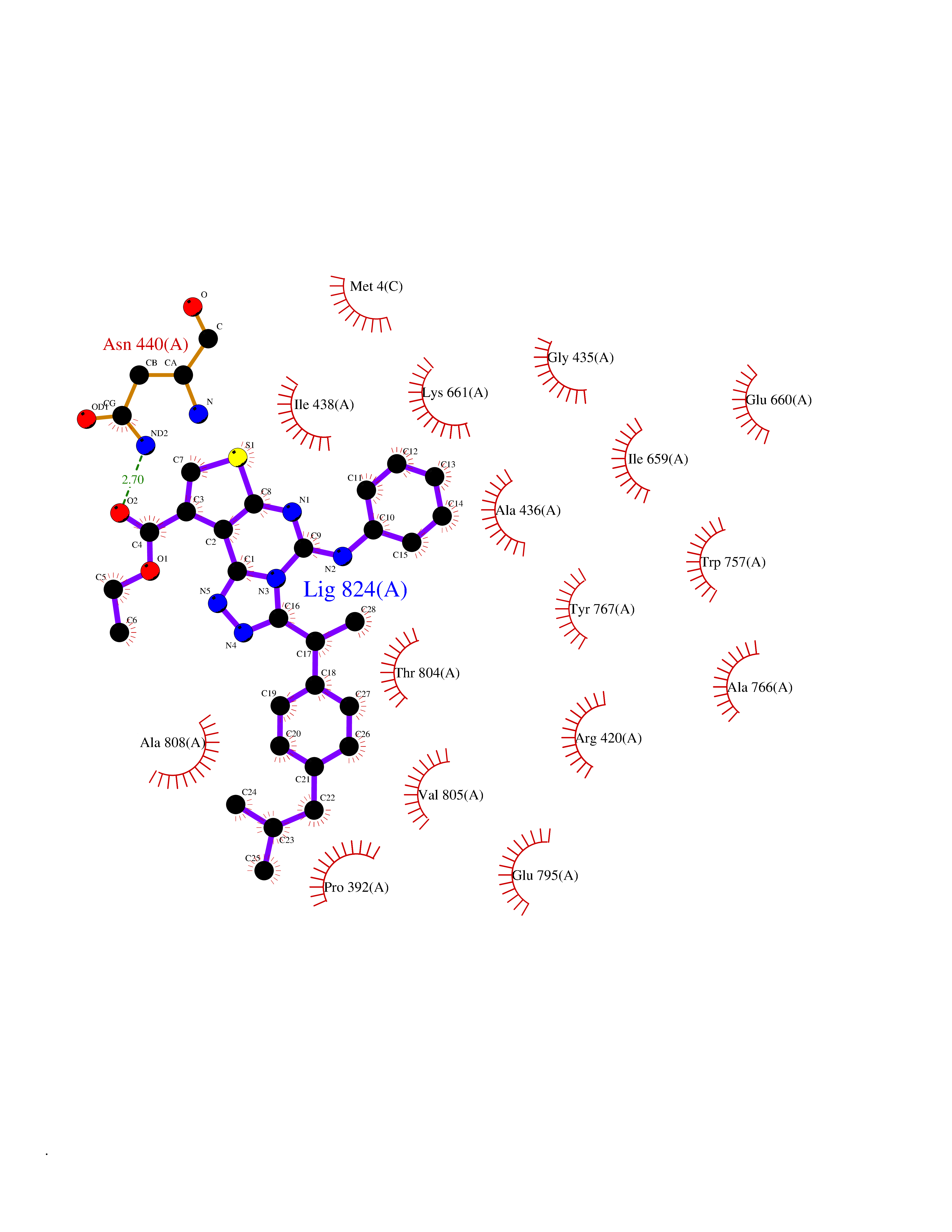 Ligplot Map 3D Binding mode Sequence GSRKCEKAGCTATCPVCFASASERCAKNGYTSRWYHLSCGEHFCNECFDHYYRSHKDGYDKYTTWKKIWTSNGKTEPSPKAFMADQQLPYWVQCTKPECRKWRQLTKEIQLTPQIAKTYRCGMKPNTAIKPETSDHCSLPEDLRVLEVSNHWWYSMLILPPLLKDSVAAPLLSAYYPDCVGMSPSCTGMNRYFQPFYQPNECGKALCVRPDVMELDELYEFPEYSRDPTMYLALRNLILALWYTNCKEALTPQKCIPHIIVRGLVRIRCVQEVERILYFMTRKGLINTGVLSVGADQYLLPKDYHNKSVIIIGAGPAGLAAARQLHNFGIKVTVLEAKDRIGGRVWDDKSFKGVTVGRGAQIVNGCINNPVALMCEQLGISMHKFGERCDLIQEGGRITDPTIDKRMDFHFNALLDVVSEWRKDKTQLQDVPLGEKIEEIYKAFIKESGIQFSELEGQVLQFHLSNLEYACGSNLHQVSARSWDHNEFFAQFAGDHTLLTPGYSVIIEKLAEGLDIQLKSPVQCIDYSGDEVQVTTTDGTGYSAQKVLVTVPLALLQKGAIQFNPPLSEKKMKAINSLGAGIIEKIALQFPYRFWDSKVQGADFFGHVPPSASKRGLFAVFYDMDPQKKHSVLMSVIAGEAVASVRTLDDKQVLQQCMATLRELFKEQEVPDPTKYFVTRWSTDPWIQMAYSFVKTGGSGEAYDIIAEDIQGTVFFAGEATNRHFPQTVTGAYLSGVREASKIAAFARTMQTARKSTGGKAPRKQL Hydrogen bonds contact Hydrophobic contact | ||||
| 9 | SEC14-like protein 2 | 4OMJ | 8.63 | |
Target general information Gen name SEC14L2 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms KIAA1658;C22orf6;KIAA1186 Protein family NA Biochemical class Transport protein Function Phospholipid binding.Transporter activity.Vitamin E binding. Related diseases Neurodevelopmental disorder with poor language and loss of hand skills (NDPLHS) [MIM:617903]: An autosomal dominant disorder characterized by psychomotor developmental stagnation or regression. NDPLHS manifest in the first years of life as loss of purposeful hand movements, loss of language, and intellectual disability. {ECO:0000269|PubMed:26740508, ECO:0000269|PubMed:28856709, ECO:0000269|PubMed:29369404}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Developmental and epileptic encephalopathy 59 (DEE59) [MIM:617904]: A form of epileptic encephalopathy, a heterogeneous group of severe early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE59 is an autosomal dominant condition characterized by onset of refractory seizures in early infancy. {ECO:0000269|PubMed:28856709, ECO:0000269|PubMed:29100083, ECO:0000269|PubMed:29369404}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB14003; DB14001; DB14002; DB11635; DB11251; DB00163 Interacts with NA EC number NA Uniprot keywords 3D-structure; Activator; Alternative splicing; Cytoplasm; Lipid-binding; Nucleus; Proteomics identification; Reference proteome; Transcription; Transcription regulation; Transport Protein physicochemical properties Chain ID A,B Molecular weight (Da) 31533.3 Length 274 Aromaticity 0.1 Instability index 49.26 Isoelectric point 8.34 Charge (pH=7) 2.81 2D Binding mode Binding energy (Kcal/mol) -11.77 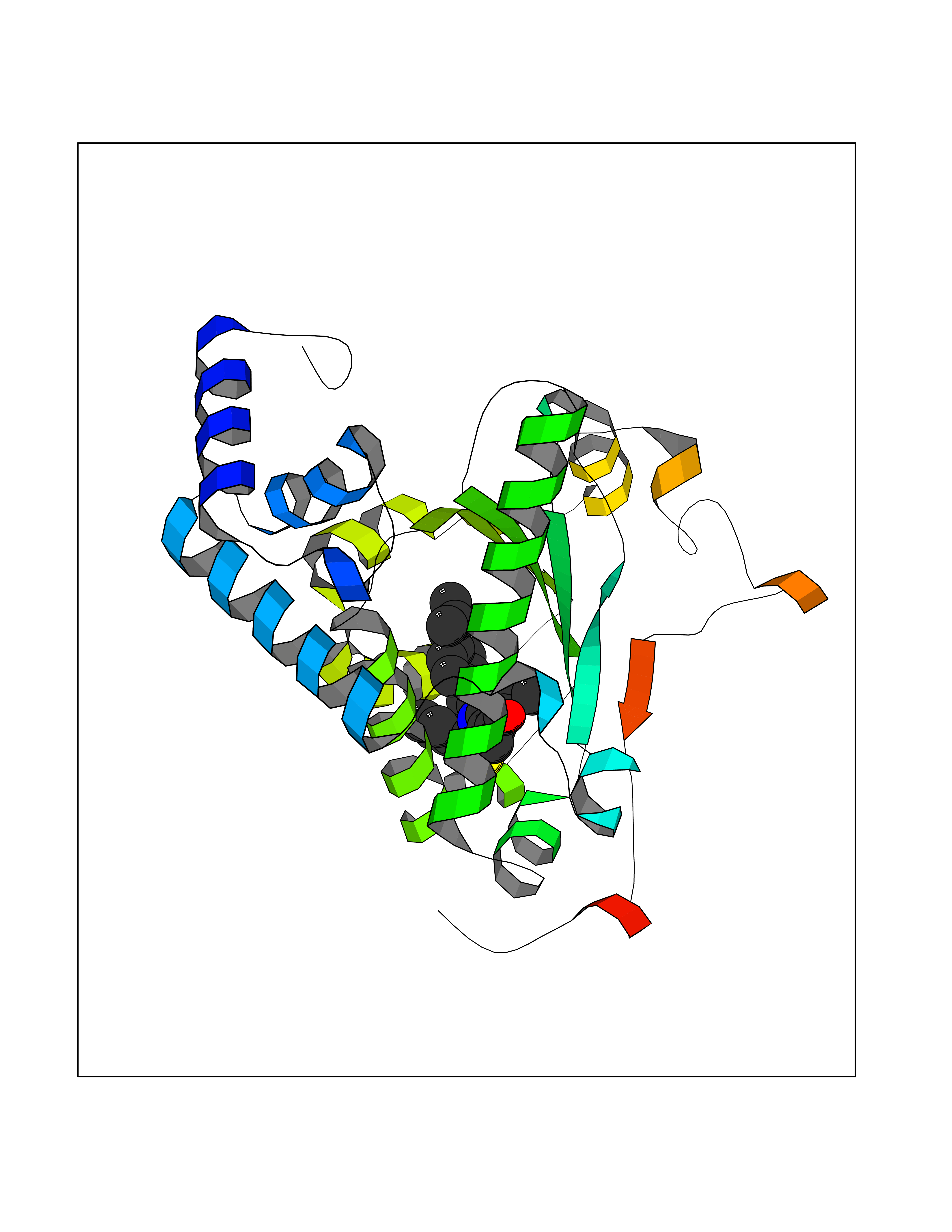 Molscript Map 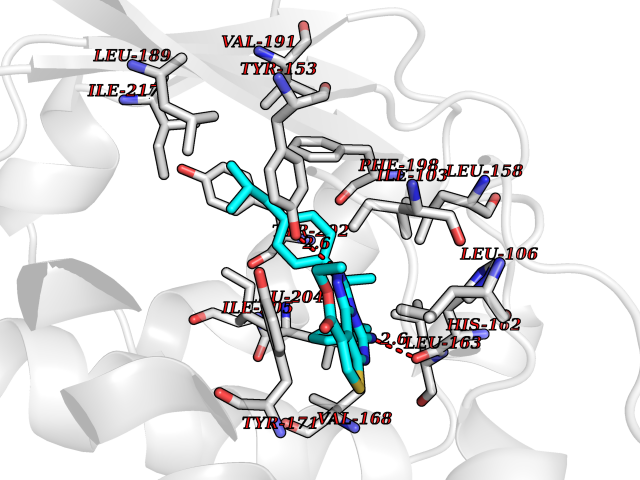 Pymol Map 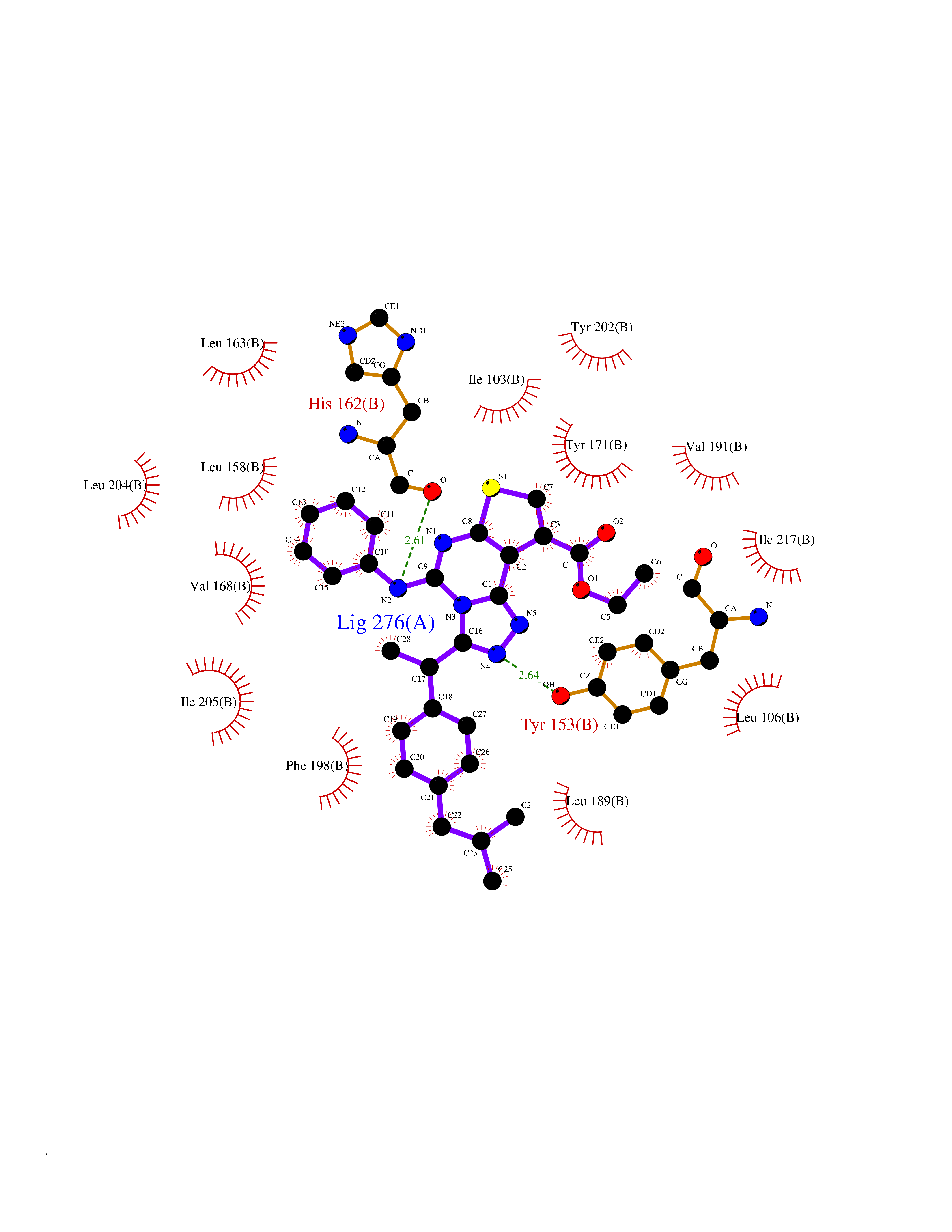 Ligplot Map 3D Binding mode Sequence MSGRVGDLSPRQKEALAKFRENVQDVLPALPNPDDYFLLRWLRARSFDLQKSEAMLRKHVEFRKQKDIDNIISWQPPEVIQQYLSGGMCGYDLDGCPVWYDIIGPLDAKGLLFSASKQDLLRTKMRECELLLQECAHQTTKLGRKVETITIIYDCEGLGLKHLWKPAVEAYGEFLCMFEENYPETLKRLFVVKAPKLFPVAYNLIKPFLSEDTRKKIMVLGANWKEVLLKHISPDQVPVEYGGTMTDPDGNPKCKSKINYGGDIPRKYYVRDQV Hydrogen bonds contact Hydrophobic contact | ||||
| 10 | Melatonin receptor type 1A (MTNR1A) | 7DB6 | 8.59 | |
Target general information Gen name MTNR1A Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Mel1a receptor; Mel1AR; Mel-1A-R Protein family G-protein coupled receptor 1 family Biochemical class GPCR rhodopsin Function Likely to mediate the reproductive and circadian actions of melatonin. The activity of this receptor is mediated by pertussis toxin sensitive G proteins that inhibit adenylate cyclase activity. High affinity receptor for melatonin. Related diseases Spermatogenic failure 5 (SPGF5) [MIM:243060]: An infertility disorder caused by spermatogenesis defects. Semen from affected men show close to 100% morphologically abnormal multiflagellar spermatozoa with low motility, oversized irregular heads, and abnormal midpiece and acrosome. {ECO:0000269|PubMed:17435757, ECO:0000269|PubMed:21733974}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB06594; DB01065; DB00980; DB02709; DB09071 Interacts with P27797; A8MQ03; Q8IUG1; P49286; O76081; P57088 EC number NA Uniprot keywords 3D-structure; Cell membrane; Disulfide bond; G-protein coupled receptor; Glycoprotein; Membrane; Receptor; Reference proteome; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID D Molecular weight (Da) 31301 Length 276 Aromaticity 0.15 Instability index 37.33 Isoelectric point 9.22 Charge (pH=7) 9.92 2D Binding mode Binding energy (Kcal/mol) -11.72  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence RPSWLASALACVLIFTIVVDILGNLLVILSVYRNKKLRNAGNIFVVSLAVADLVVAIYPYPLVLMSIFNNGWNLGYLHCQVSGFLMGLSVIGSIFNITGIAINRYCYICHSLKYDKLYSSKNSLCYVLLIWLLTLAAVLPNLRAGTLQYDPRIYSCTFAQSVSSAYTIAVVVFHFLVPMIIVIFCYLRIWILVLQVRQRVPQDFRNFVTMFVVFVLFAICWAPLNFIGLAVASDPASMVPRIPEWLFVASYYMAYFNSCLNAIIYGLLNQNFRKEY Hydrogen bonds contact Hydrophobic contact | ||||
| 11 | Retinoic acid receptor beta (RARB) | 4DM6 | 8.58 | |
Target general information Gen name RARB Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms RAR-epsilon; RAR-beta; Nuclear receptor subfamily 1 group B member 2; NR1B2; HBV-activated protein; HAP Protein family Nuclear hormone receptor family, NR1 subfamily Biochemical class Nuclear hormone receptor Function Retinoic acid receptors bind as heterodimers to their target response elements in response to their ligands, all-trans or 9-cis retinoic acid, and regulate gene expression in various biological processes. The RXR/RAR heterodimers bind to the retinoic acid response elements (RARE) composed of tandem 5'-AGGTCA-3' sites known as DR1-DR5. In the absence or presence of hormone ligand, acts mainly as an activator of gene expression due to weak binding to corepressors. In concert with RARG, required for skeletal growth, matrix homeostasis and growth plate function. Receptor for retinoic acid. Related diseases Microphthalmia, syndromic, 12 (MCOPS12) [MIM:615524]: A form of microphthalmia, a disorder of eye formation, ranging from small size of a single eye to complete bilateral absence of ocular tissues (anophthalmia). In many cases, microphthalmia/anophthalmia occurs in association with syndromes that include non-ocular abnormalities. MCOPS12 patients manifest variable features, including diaphragmatic hernia, pulmonary hypoplasia, and cardiac abnormalities. {ECO:0000269|PubMed:24075189, ECO:0000269|PubMed:27120018}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00459; DB00210; DB00523; DB02877; DB00926; DB05785; DB04942; DB00799; DB00755; DB12808 Interacts with O95273; P50222; Q9UBK2; P62195; P28702; P28702-3; P48443; P03255 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; Disease variant; DNA-binding; Metal-binding; Microphthalmia; Nucleus; Phosphoprotein; Proto-oncogene; Receptor; Reference proteome; Transcription; Transcription regulation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A,B Molecular weight (Da) 25904.1 Length 229 Aromaticity 0.06 Instability index 44.34 Isoelectric point 7.55 Charge (pH=7) 0.73 2D Binding mode Binding energy (Kcal/mol) -11.7  Molscript Map  Pymol Map 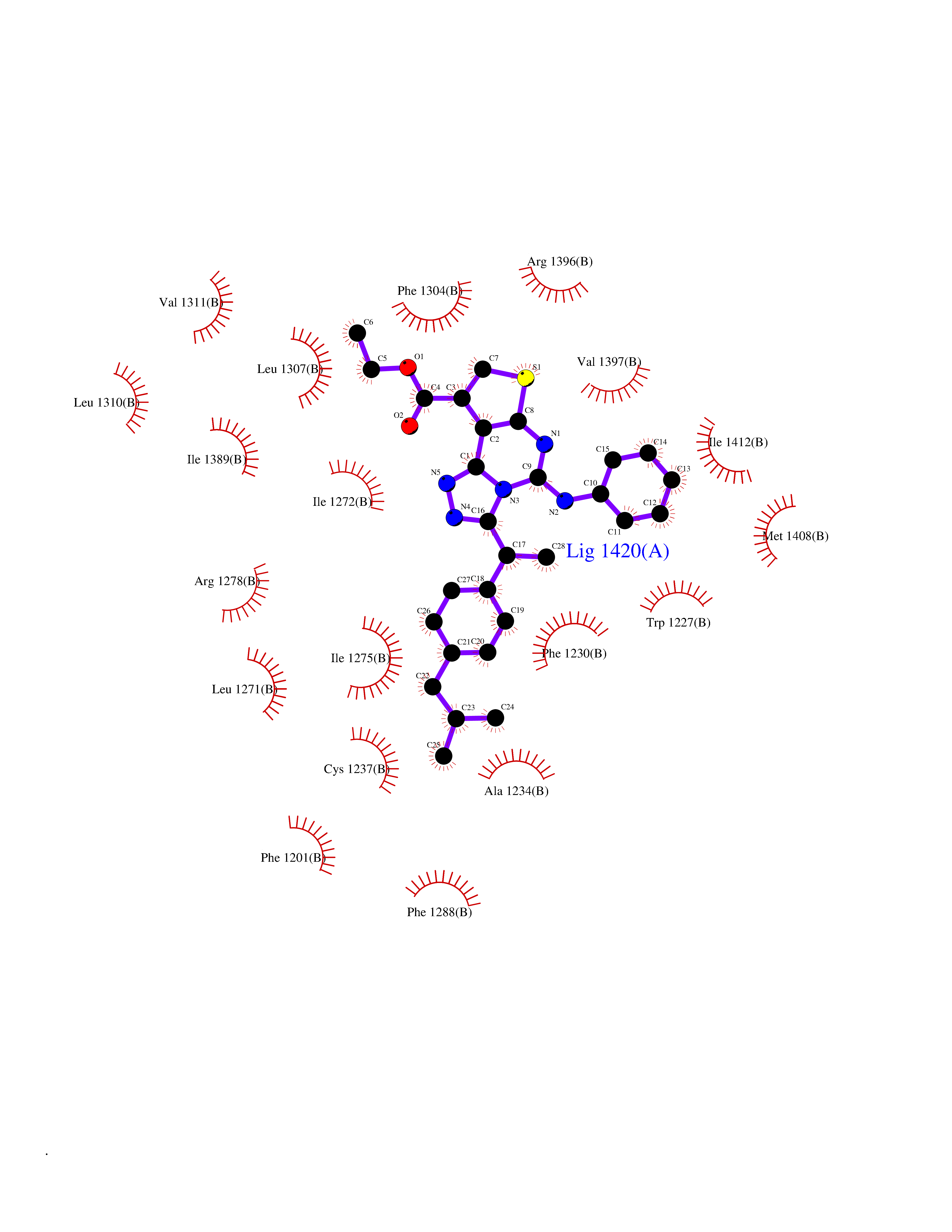 Ligplot Map 3D Binding mode Sequence TEKIRKAHQETFPSLCQLGKYTTNSSADHRVRLDLGLWDKFSELATKCIIKIVEFAKRLPGFTGLTIADQITLLKAACLDILILRICTRYTPEQDTMTFSDGLTLNRTQMHNAGFGPLTDLVFTFANQLLPLEMDDTETGLLSAICLICGDRQDLEEPTKVDKLQEPLLEALKIYIRKRRPSKPHMFPKILMKITDLRSISAKGAERVITLKMEIPGSMPPLIQEMLEN Hydrogen bonds contact Hydrophobic contact | ||||
| 12 | Lysine N-methyltransferase 3A (SETD2) | 7LZD | 8.58 | |
Target general information Gen name SETD2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms p231HBP; hSET2; SET2; SET domain-containing protein 2; Protein-lysine N-methyltransferase SETD2; KMT3A; KIAA1732; Huntingtin-interacting protein B; Huntingtin-interacting protein 1; Huntingtin yeast p Protein family Class V-like SAM-binding methyltransferase superfamily, Histone-lysine methyltransferase family, SET2 subfamily Biochemical class Methyltransferase Function Represents the main enzyme generating H3K36me3, a specific tag for epigenetic transcriptional activation. Plays a role in chromatin structure modulation during elongation by coordinating recruitment of the FACT complex and by interacting with hyperphosphorylated POLR2A. Acts as a key regulator of DNA mismatch repair in G1 and early S phase by generating H3K36me3, a mark required to recruit MSH6 subunit of the MutS alpha complex: early recruitment of the MutS alpha complex to chromatin to be replicated allows a quick identification of mismatch DNA to initiate the mismatch repair reaction. Required for DNA double-strand break repair in response to DNA damage: acts by mediating formation of H3K36me3, promoting recruitment of RAD51 and DNA repair via homologous recombination (HR). Acts as a tumor suppressor. H3K36me3 also plays an essential role in the maintenance of a heterochromatic state, by recruiting DNA methyltransferase DNMT3A. H3K36me3 is also enhanced in intron-containing genes, suggesting that SETD2 recruitment is enhanced by splicing and that splicing is coupled to recruitment of elongating RNA polymerase. Required during angiogenesis. Required for endoderm development by promoting embryonic stem cell differentiation toward endoderm: acts by mediating formation of H3K36me3 in distal promoter regions of FGFR3, leading to regulate transcription initiation of FGFR3. In addition to histones, also mediates methylation of other proteins, such as tubulins and STAT1. Trimethylates 'Lys-40' of alpha-tubulins such as TUBA1B (alpha-TubK40me3); alpha-TubK40me3 is required for normal mitosis and cytokinesis and may be a specific tag in cytoskeletal remodeling. Involved in interferon-alpha-induced antiviral defense by mediating both monomethylation of STAT1 at 'Lys-525' and catalyzing H3K36me3 on promoters of some interferon-stimulated genes (ISGs) to activate gene transcription. Histone methyltransferase that specifically trimethylates 'Lys-36' of histone H3 (H3K36me3) using dimethylated 'Lys-36' (H3K36me2) as substrate. Related diseases Renal cell carcinoma (RCC) [MIM:144700]: Renal cell carcinoma is a heterogeneous group of sporadic or hereditary carcinoma derived from cells of the proximal renal tubular epithelium. It is subclassified into clear cell renal carcinoma (non-papillary carcinoma), papillary renal cell carcinoma, chromophobe renal cell carcinoma, collecting duct carcinoma with medullary carcinoma of the kidney, and unclassified renal cell carcinoma. Clear cell renal cell carcinoma is the most common subtype. {ECO:0000269|PubMed:20054297, ECO:0000269|PubMed:23622243, ECO:0000269|PubMed:23792563, ECO:0000269|PubMed:25728682}. The disease may be caused by variants affecting the gene represented in this entry. Defects of SETD2 are associated with loss of DNA methylation at non-promoter regions (PubMed:23792563). SETD2 defects lead to aberrant and reduced nucleosome compaction and chromatin association of key replication proteins, such as MCM7 and DNA polymerase delta, leading to hinder replication fork progression and prevent loading of RAD51 homologous recombination repair factor at DNA breaks (PubMed:25728682). {ECO:0000269|PubMed:23792563, ECO:0000269|PubMed:25728682}.; DISEASE: Luscan-Lumish syndrome (LLS) [MIM:616831]: An autosomal dominant syndrome with a variable phenotype. Clinical features include macrocephaly, distinctive facial appearance, postnatal overgrowth, various degrees of learning difficulties, autism spectrum disorder, and intellectual disability. {ECO:0000269|PubMed:23160955, ECO:0000269|PubMed:24852293, ECO:0000269|PubMed:26084711, ECO:0000269|PubMed:27317772}. The disease may be caused by variants affecting the gene represented in this entry.; DISEASE: Leukemia, acute lymphoblastic (ALL) [MIM:613065]: A subtype of acute leukemia, a cancer of the white blood cells. ALL is a malignant disease of bone marrow and the most common malignancy diagnosed in children. The malignant cells are lymphoid precursor cells (lymphoblasts) that are arrested in an early stage of development. The lymphoblasts replace the normal marrow elements, resulting in a marked decrease in the production of normal blood cells. Consequently, anemia, thrombocytopenia, and neutropenia occur to varying degrees. The lymphoblasts also proliferate in organs other than the marrow, particularly the liver, spleen, and lymphnodes. {ECO:0000269|PubMed:24509477, ECO:0000269|PubMed:24662245}. The disease may be caused by variants affecting distinct genetic loci, including the gene represented in this entry.; DISEASE: Leukemia, acute myelogenous (AML) [MIM:601626]: A subtype of acute leukemia, a cancer of the white blood cells. AML is a malignant disease of bone marrow characterized by maturational arrest of hematopoietic precursors at an early stage of development. Clonal expansion of myeloid blasts occurs in bone marrow, blood, and other tissue. Myelogenous leukemias develop from changes in cells that normally produce neutrophils, basophils, eosinophils and monocytes. {ECO:0000269|PubMed:16314571, ECO:0000269|PubMed:24509477}. The disease may be caused by variants affecting distinct genetic loci, including the gene represented in this entry.; DISEASE: Intellectual developmental disorder, autosomal dominant 70 (MRD70) [MIM:620157]: An autosomal dominant disorder characterized by mild global developmental delay, moderately impaired intellectual disability with speech difficulties, and behavioral abnormalities. {ECO:0000269|PubMed:32710489}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Rabin-Pappas syndrome (RAPAS) [MIM:620155]: An autosomal dominant neurodevelopmental disorder characterized by severely impaired global development, intellectual disability, microcephaly, facial dysmorphism, and variable congenital anomalies affecting the skeletal, genitourinary, cardiac, and other organ systems. {ECO:0000269|PubMed:32710489}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P42858; P84022 EC number EC 2.1.1.43 Uniprot keywords 3D-structure; Activator; Alternative splicing; Antiviral defense; Autism spectrum disorder; Chromatin regulator; Chromosome; Coiled coil; Developmental protein; Differentiation; Disease variant; DNA damage; DNA repair; Host-virus interaction; Immunity; Innate immunity; Intellectual disability; Isopeptide bond; Metal-binding; Methyltransferase; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; S-adenosyl-L-methionine; Transcription; Transcription regulation; Transferase; Tumor suppressor; Ubl conjugation; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 27416.8 Length 237 Aromaticity 0.11 Instability index 55.47 Isoelectric point 7.51 Charge (pH=7) 0.99 2D Binding mode Binding energy (Kcal/mol) -11.7 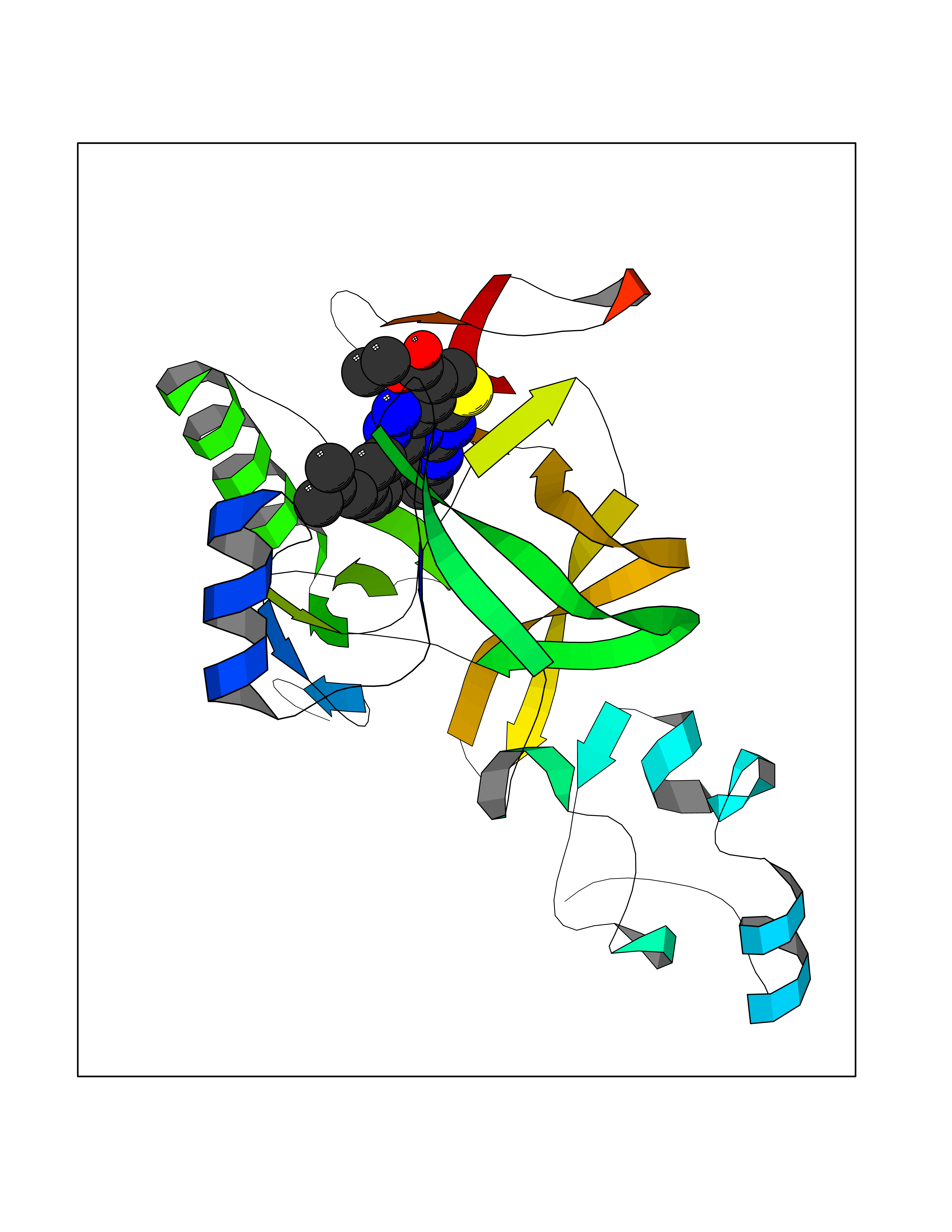 Molscript Map 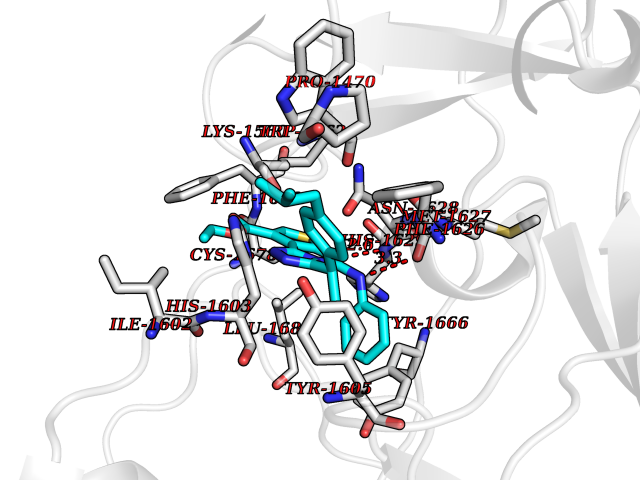 Pymol Map 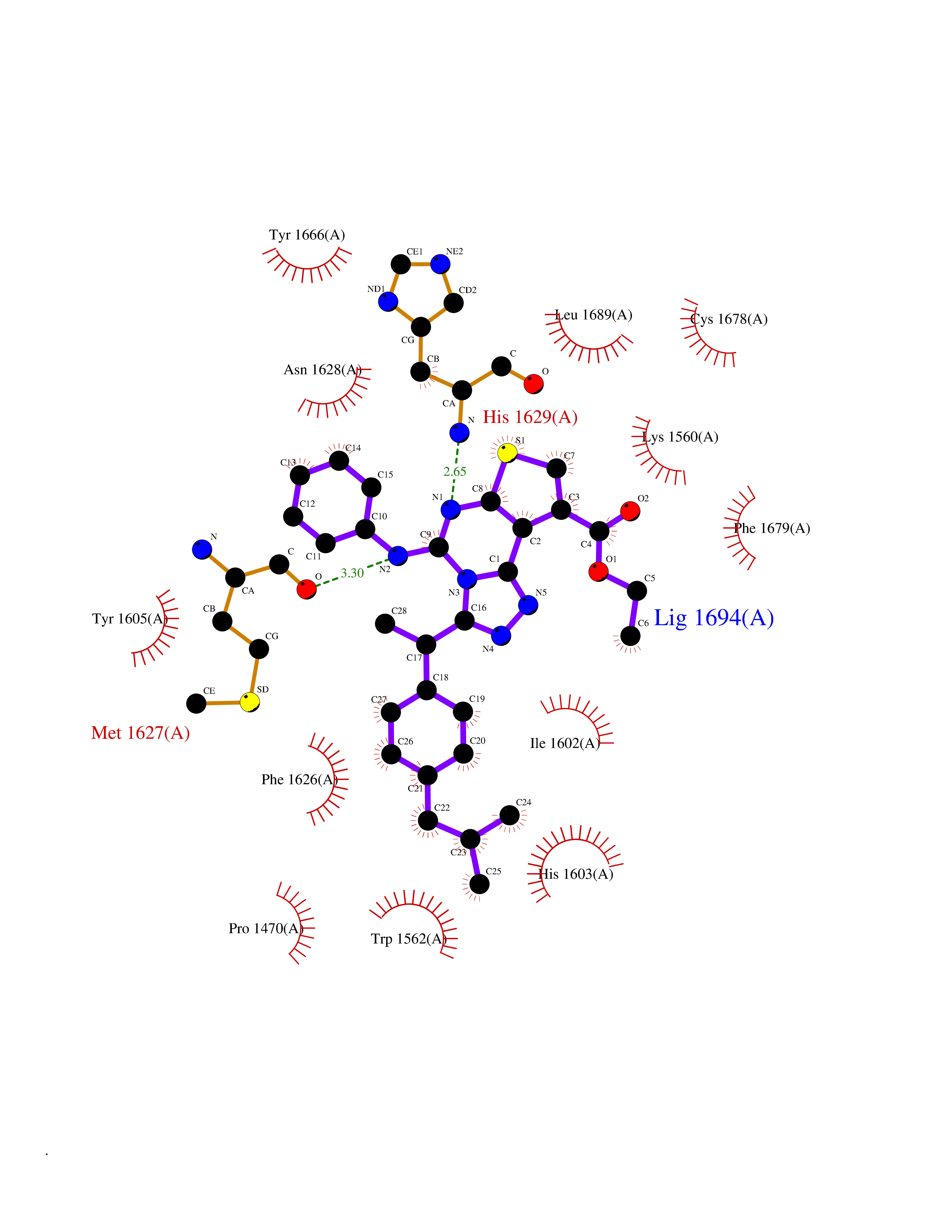 Ligplot Map 3D Binding mode Sequence GPSCVMDDFRDPQRWKECAKQGKMPCYFDLIEENVYLTERRMQCECTPLSKDERAQGEIACGEDCLNRLLMIECSSRCPNGDYCSNRRFQRKQHADVEVILTEKKGWGLRAAKDLPSNTFVLEYCGEVLDHKEFKARVKEYARNKNIHYYFMALKNDEIIDATQKGNCSRFMNHSCEPNCETQKWTVNGQLRVGFFTTKLVPSGSELTFDYQFQRYGKEAQKCFCGSANCRGYLGGE Hydrogen bonds contact Hydrophobic contact | ||||
| 13 | Aldehyde oxidoreductase | 4USA | 8.57 | |
Target general information Gen name mop Organism Megalodesulfovibrio gigas (Desulfovibrio gigas) Uniprot ID TTD ID NA Synonyms NA Protein family Xanthine dehydrogenase family Biochemical class Oxidoreductase Function 2 iron, 2 sulfur cluster binding.Aldehyde dehydrogenase (FAD-independent) activity.Electron carrier activity.Metal ion binding. Related diseases LTC4 synthase deficiency is associated with a neurometabolic developmental disorder characterized by muscular hypotonia, psychomotor retardation, failure to thrive, and microcephaly. {ECO:0000269|PubMed:10896305, ECO:0000269|PubMed:9820300}. Drugs (DrugBank ID) DB02137 Interacts with NA EC number 1.2.99.7 Uniprot keywords 2Fe-2S; 3D-structure; FAD; Flavoprotein; Iron; Iron-sulfur; Metal-binding; Molybdenum; NAD; Oxidoreductase Protein physicochemical properties Chain ID A Molecular weight (Da) 96930.4 Length 907 Aromaticity 0.07 Instability index 29.17 Isoelectric point 5.69 Charge (pH=7) -17.56 2D Binding mode Binding energy (Kcal/mol) -11.69 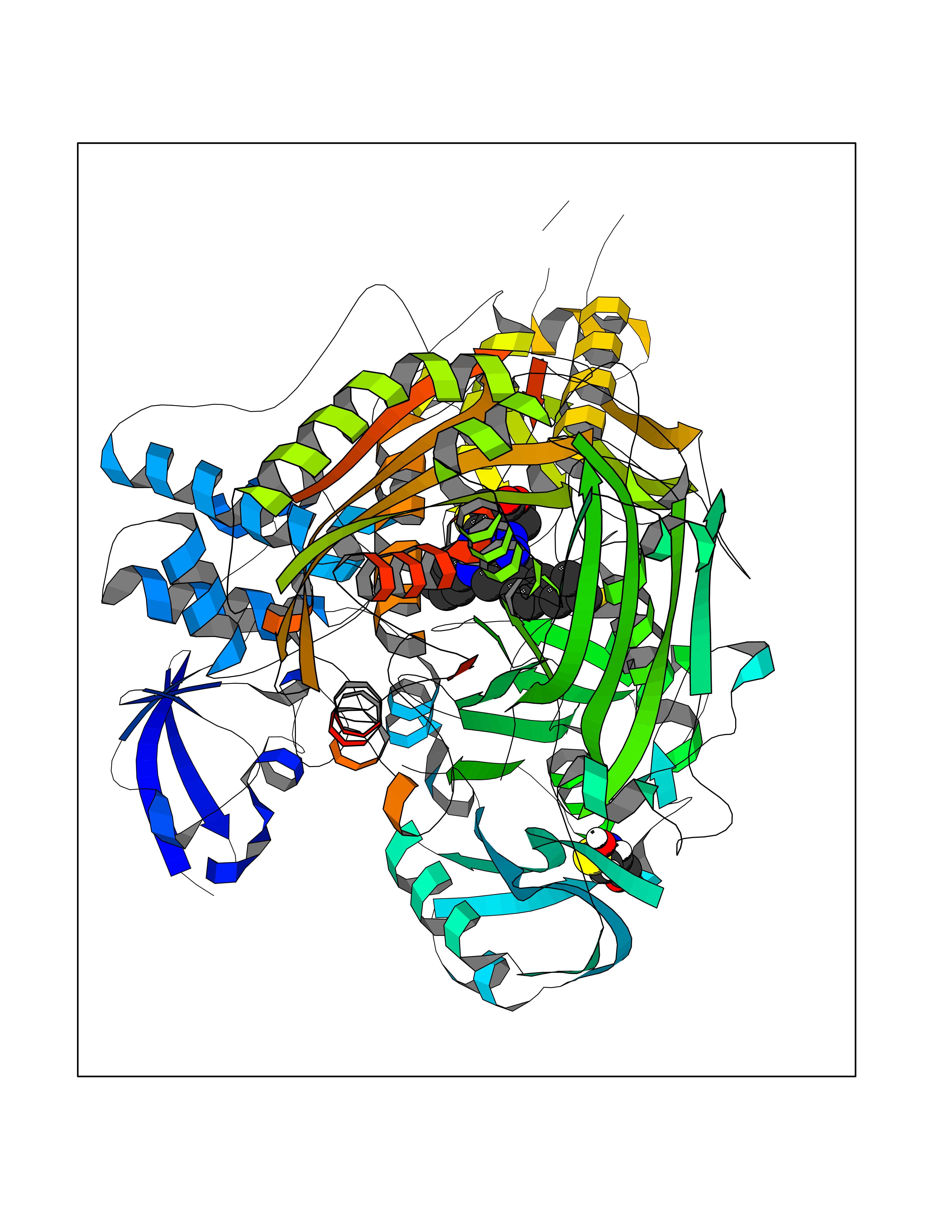 Molscript Map 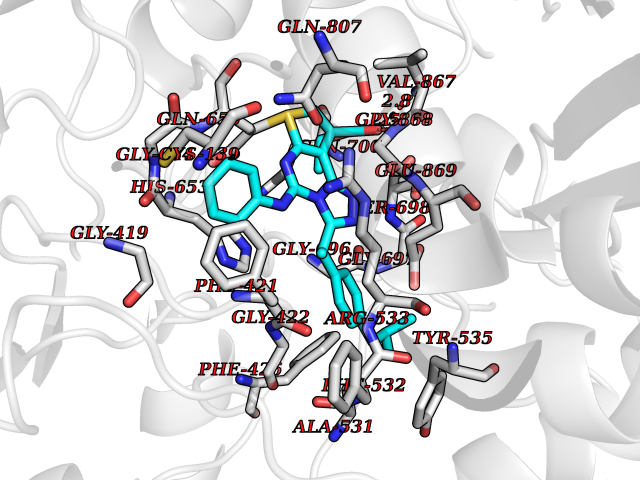 Pymol Map 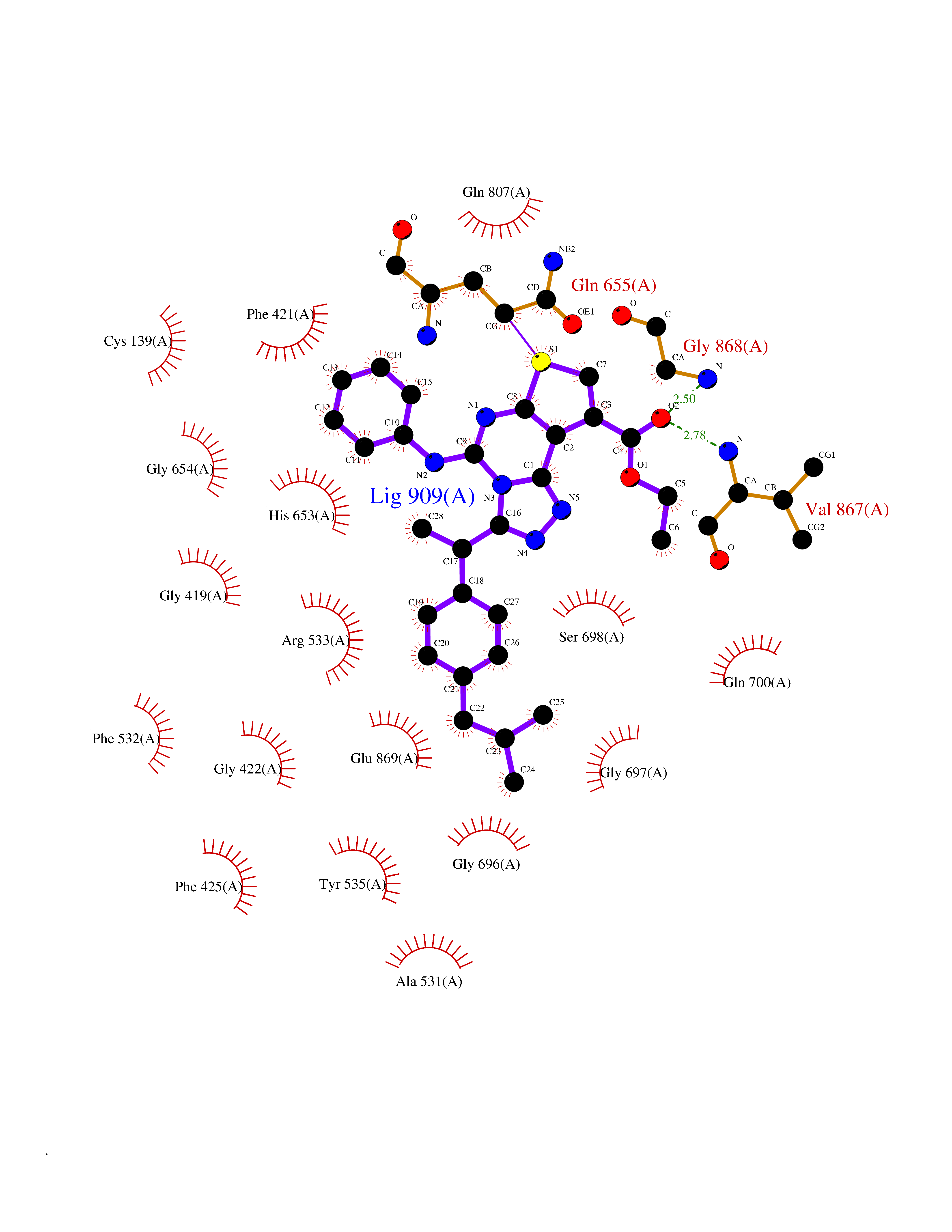 Ligplot Map 3D Binding mode Sequence MIQKVITVNGIEQNLFVDAEALLSDVLRQQLGLTGVKVGCEQGQCGACSVILDGKVVRACVTKMKRVADGAQITTIEGVGQPENLHPLQKAWVLHGGAQCGFCSPGFIVSAKGLLDTNADPSREDVRDWFQKHRNACRCTGYKPLVDAVMDAAAVINGKKPETDLEFKMPADGRIWGSKYPRPTAVAKVTGTLDYGADLGLKMPAGTLHLAMVQAKVSHANIKGIDTSEALTMPGVHSVITHKDVKGKNRITGLITFPTNKGDGWDRPILXDEKVFQYGDCIALVCADSEANARAAAEKVKVDLEELPAYMSGPAAAAEDAIEIHPGTPNVYFEQPIVKGEDTGPIFASADVTVEGDFYVGRQPHMPIEPDVAFAYMGDDGKCYIHSKSIGVHLHLYMIAPGVGLEPDQLVLVANPMGGTFGYKFSPTSEALVAVAAMATGRPVHLRYNYQQQQQYTGKRSPWEMNVKFAAKKDGTLLAMESDWLVDHGPYSEFGDLLTLRGAQFIGAGYNIPNIRGLGRTVATNHVWGSAFRGYGAPQSMFASECLMDMLAEKLGMDPLELRYKNAYRPGDTNPTGQEPEVFSLPDMIDQLRPKYQAALEKAQKESTATHKKGVGISIGVYGSGLDGPDASEAWAELNADGTITVHTAWEDHGQGADIGCVGTAHEALRPMGVAPEKIKFTWPNTATTPNSGPSGGSRQQVMTGNAIRVACENLLKACEKPGGGYYTYDELKAADKPTKITGNWTASGATHCDAVTGLGKPFVVYMYGVFMAEVTVDVATGQTTVDGMTLMADLGSLCNQLATDGQIYGGLAQGIGLALSEDFEDIKKHATLVGAGFPFIKQIPDKLDIVYVNHPRPDGPFGASGVGELPLTSPHAAIINAIKSATGVRIYRLPAYPEKVLEALKA Hydrogen bonds contact Hydrophobic contact | ||||
| 14 | Sphingosine-1-phosphate receptor 2 (S1PR2) | 7T6B | 8.57 | |
Target general information Gen name S1PR2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Sphingosine 1-phosphate receptor Edg-5; S1PR2; S1P2; S1P receptor Edg-5; S1P receptor 2; Endothelial differentiation G-protein coupled receptor 5 Protein family G-protein coupled receptor 1 family Biochemical class GPCR rhodopsin Function Receptor for the lysosphingolipid sphingosine 1- phosphate (S1P). S1P is a bioactive lysophospholipid that elicits diverse physiological effect on most types of cells and tissues. When expressed in rat HTC4 hepatoma cells, is capable of mediating S1P-induced cell proliferation and suppression of apoptosis. Related diseases Deafness, autosomal recessive, 68 (DFNB68) [MIM:610419]: A form of non-syndromic sensorineural hearing loss. Sensorineural deafness results from damage to the neural receptors of the inner ear, the nerve pathways to the brain, or the area of the brain that receives sound information. {ECO:0000269|PubMed:26805784}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P16144; Q9JK11-1 EC number NA Uniprot keywords 3D-structure; Cell membrane; Deafness; Disease variant; G-protein coupled receptor; Glycoprotein; Lipoprotein; Membrane; Non-syndromic deafness; Palmitate; Proteomics identification; Receptor; Reference proteome; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID R Molecular weight (Da) 28917.3 Length 264 Aromaticity 0.11 Instability index 38.95 Isoelectric point 9.11 Charge (pH=7) 9.27 2D Binding mode Binding energy (Kcal/mol) -11.69 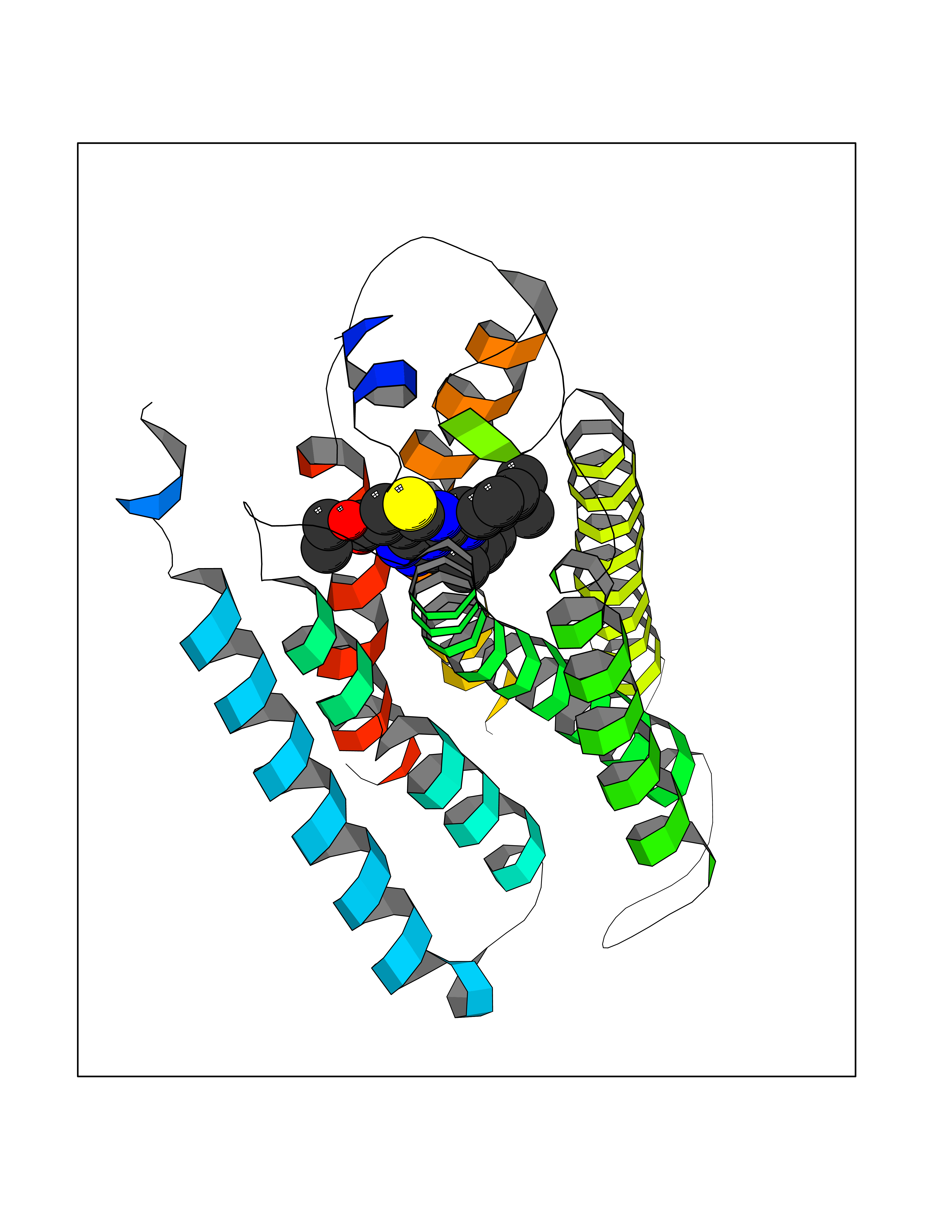 Molscript Map 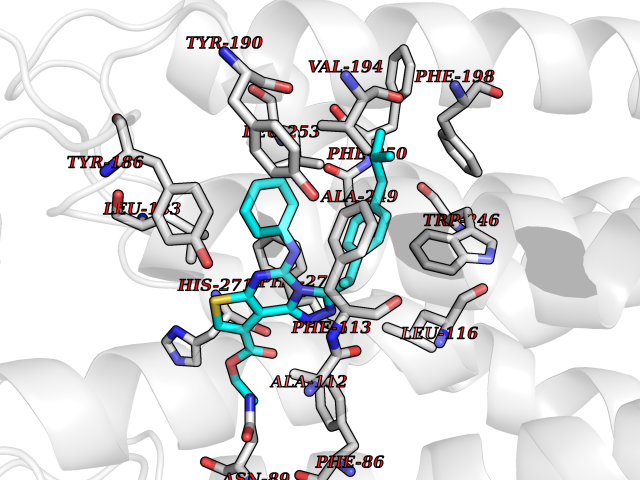 Pymol Map 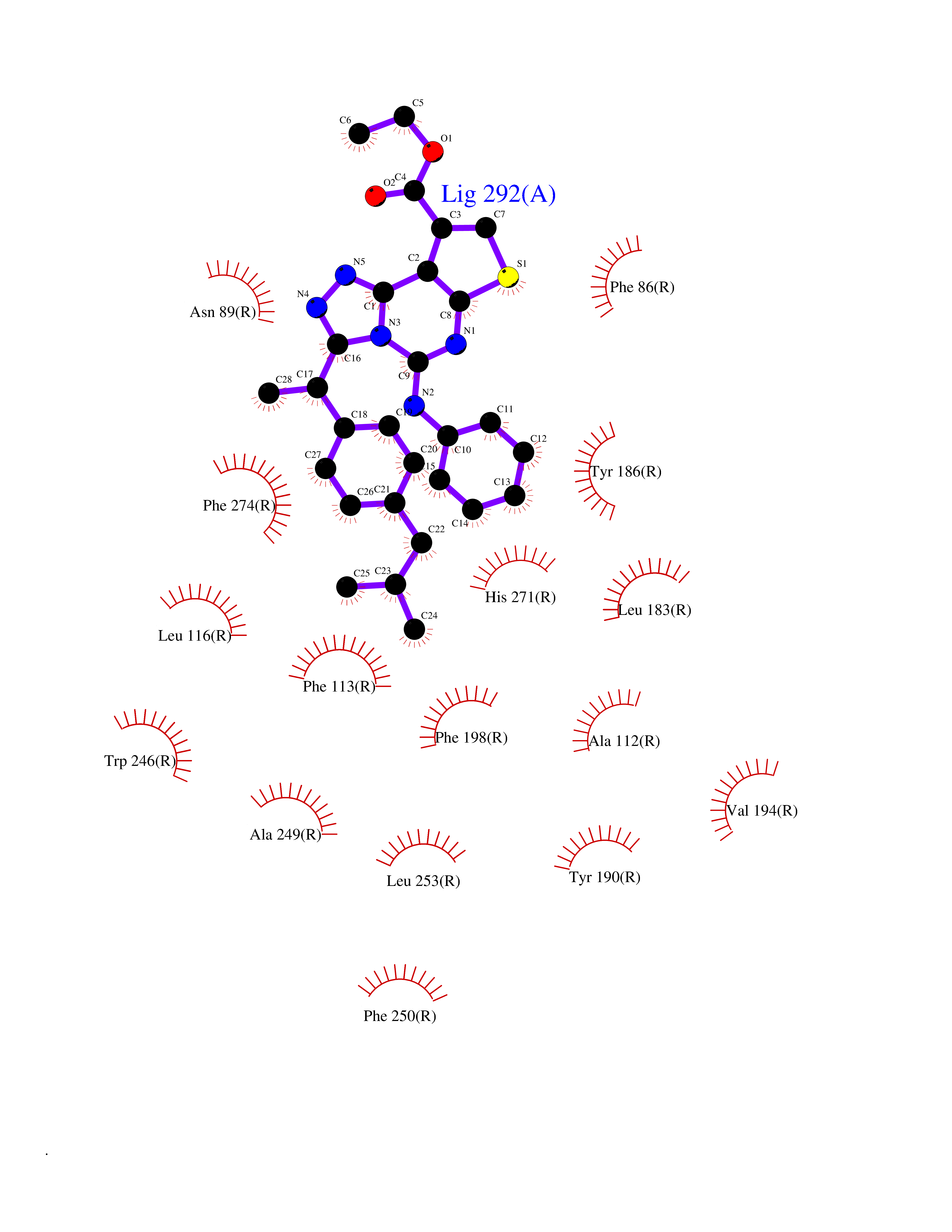 Ligplot Map 3D Binding mode Sequence NKVQEHYNYTKTSRQVASAFIVILCCAIVVENLLVLIAVARNSKFHSAMYLFLGNLAASDLLAGVAFVANTLLSGSVTLRLTPVQWFAREGSAFITLSASVFSLLAIAIERHVAIAKVKLYGSDKSCRMLLLIGASWLISLVLGGLPILGWNCLGHLEACSTVLPLYAKHYVLCVVTIFSIILLAIVALYVRIYCVVRSSQTLALLKTVTIVLGVFIVCWLPAFSILLLDYACPVHSCPILYKAHYFFAVSTLNSLLNPVIYTW Hydrogen bonds contact Hydrophobic contact | ||||
| 15 | Melatonin receptor type 1B (MTNR1B) | 6ME9 | 8.57 | |
Target general information Gen name MTNR1B Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Mel1b receptor; Mel1b melatonin receptor; Mel-1B-R Protein family G-protein coupled receptor 1 family Biochemical class GPCR rhodopsin Function Likely to mediate the reproductive and circadian actions of melatonin. The activity of this receptor is mediated by pertussis toxin sensitive G proteins that inhibit adenylate cyclase activity. High affinity receptor for melatonin. Related diseases Insulin-like growth factor 1 resistance (IGF1RES) [MIM:270450]: A disorder characterized by intrauterine growth retardation, poor postnatal growth and increased plasma IGF1 levels. {ECO:0000269|PubMed:14657428, ECO:0000269|PubMed:15928254, ECO:0000269|PubMed:25040157}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB06594; DB01065; DB00980; DB02709; DB09071; DB15133 Interacts with P28335; P48039; O76081; Q14669 EC number NA Uniprot keywords 3D-structure; Cell membrane; Disulfide bond; G-protein coupled receptor; Glycoprotein; Membrane; Receptor; Reference proteome; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 50184.9 Length 448 Aromaticity 0.11 Instability index 37.2 Isoelectric point 5.72 Charge (pH=7) -5.68 2D Binding mode Binding energy (Kcal/mol) -11.69 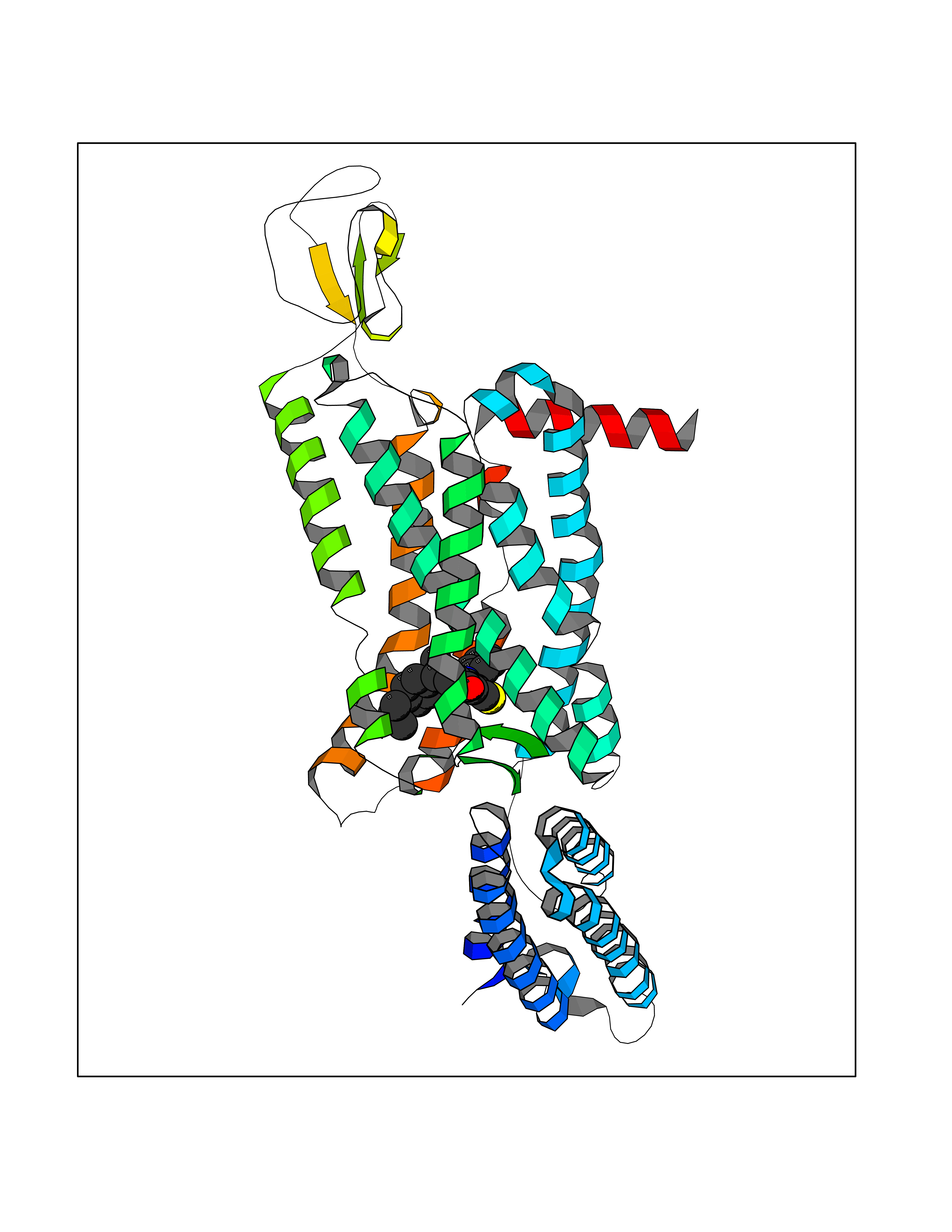 Molscript Map 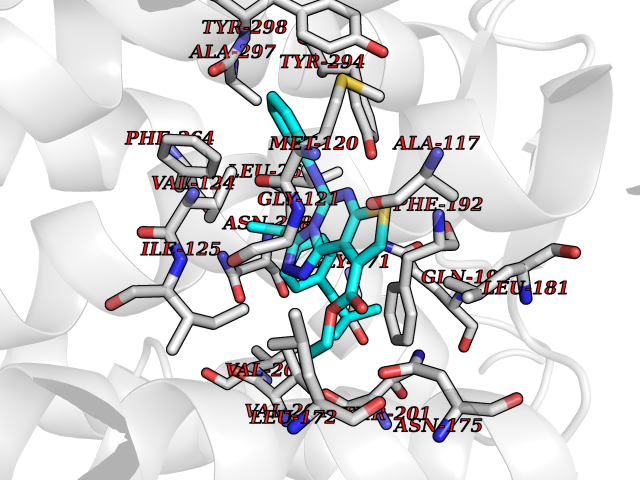 Pymol Map 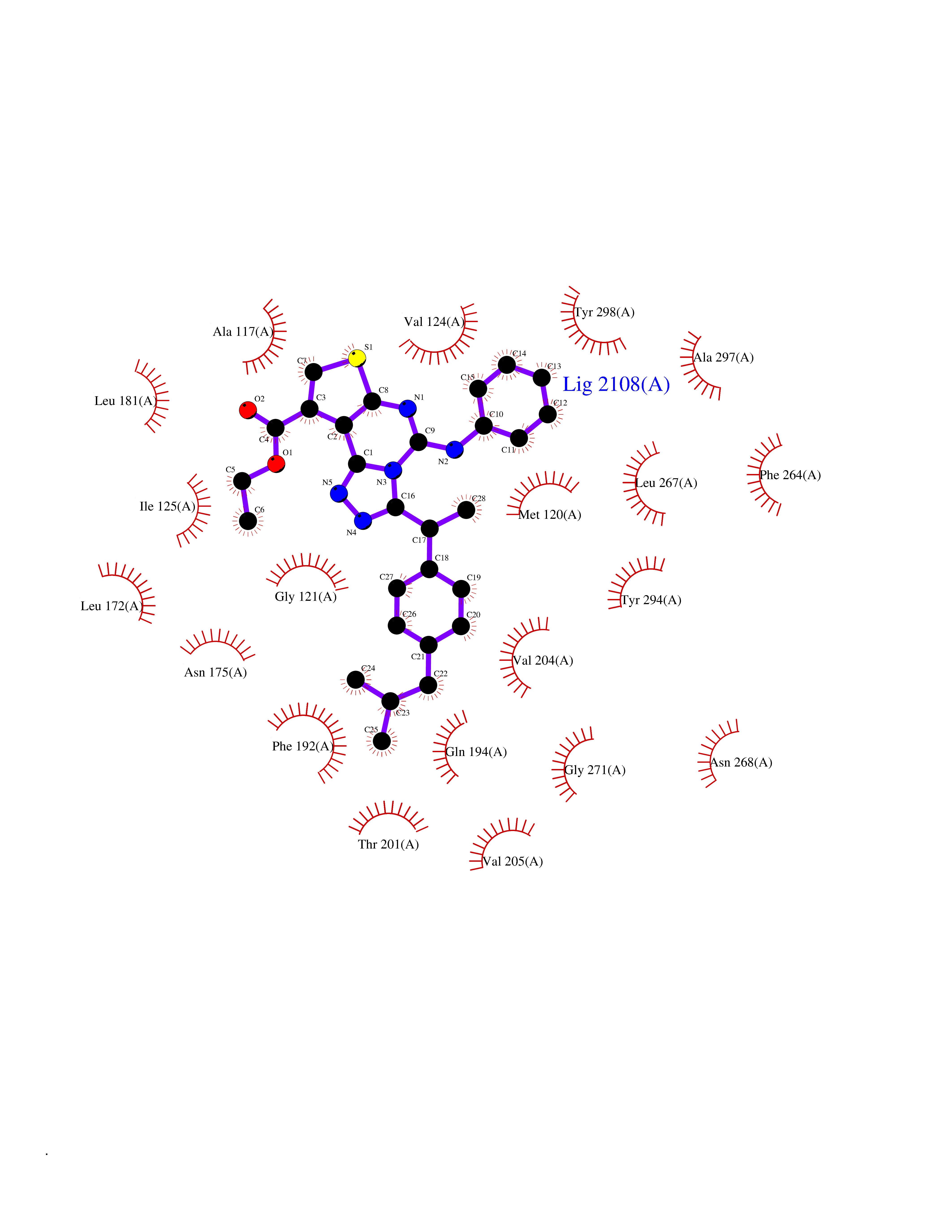 Ligplot Map 3D Binding mode Sequence ADLEDNWETLNDNLKVIEKADNAAQVKDALTKMRAAALDAQKATPPKLEDKSPDSPEMKDFRHGFDILVGQIDDALKLANEGKVKEAQAAAEQLKTTRNAYIQKYLGDGARPSWVAPALSAVLIVTTAVDVVGNLLVILSVLRNRKLRNAGNLFLVSLALANLVVAFYPYPLILVAIFYDGWAFGEEHCKASAFVMGLSVIGSVWNITAIAIDRYLYICHSMAYHRIYRRWHTPLHICLIWLLTVVALLPNFFVGSLEYDPRIYSCTFIQTASTQYTAAVVVIHFLLPIAVVSFCYLRIWVLVLQARMKKYTCTVCGYIYNPEDGDPDNGVNPGTDFKDIPDDWVCPLCGVGKDQFEEVECLKPSDLRSFLTMFVVFVIFAICFAPLNCIGLAVAINPQEMAPQIPEGLFVTSYLLAYFNSCLNPIVYGLLDQNFRREYKRILLALWN Hydrogen bonds contact Hydrophobic contact | ||||
| 16 | Histamine N-methyltransferase (HNMT) | 2AOT | 8.56 | |
Target general information Gen name HNMT Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Histamine-N-methyltransferase; HNMT; HMT Protein family Class I-like SAM-binding methyltransferase superfamily, HNMT family Biochemical class Methyltransferase Function Inactivates histamine by N-methylation. Plays an important role in degrading histamine and in regulating the airway response to histamine. Related diseases Intellectual developmental disorder, autosomal recessive 51 (MRT51) [MIM:616739]: A disorder characterized by significantly below average general intellectual functioning associated with impairments in adaptive behavior and manifested during the developmental period. {ECO:0000269|PubMed:26206890}. The disease is caused by variants affecting distinct genetic loci, including the gene represented in this entry. Drugs (DrugBank ID) DB00613; DB13875; DB05381; DB04655; DB01103; DB01752; DB07106 Interacts with NA EC number EC 2.1.1.8 Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; Disease variant; Intellectual disability; Methyltransferase; Proteomics identification; Reference proteome; S-adenosyl-L-methionine; Transferase Protein physicochemical properties Chain ID A,B Molecular weight (Da) 32712 Length 288 Aromaticity 0.1 Instability index 36.38 Isoelectric point 5.18 Charge (pH=7) -9.97 2D Binding mode Binding energy (Kcal/mol) -11.68 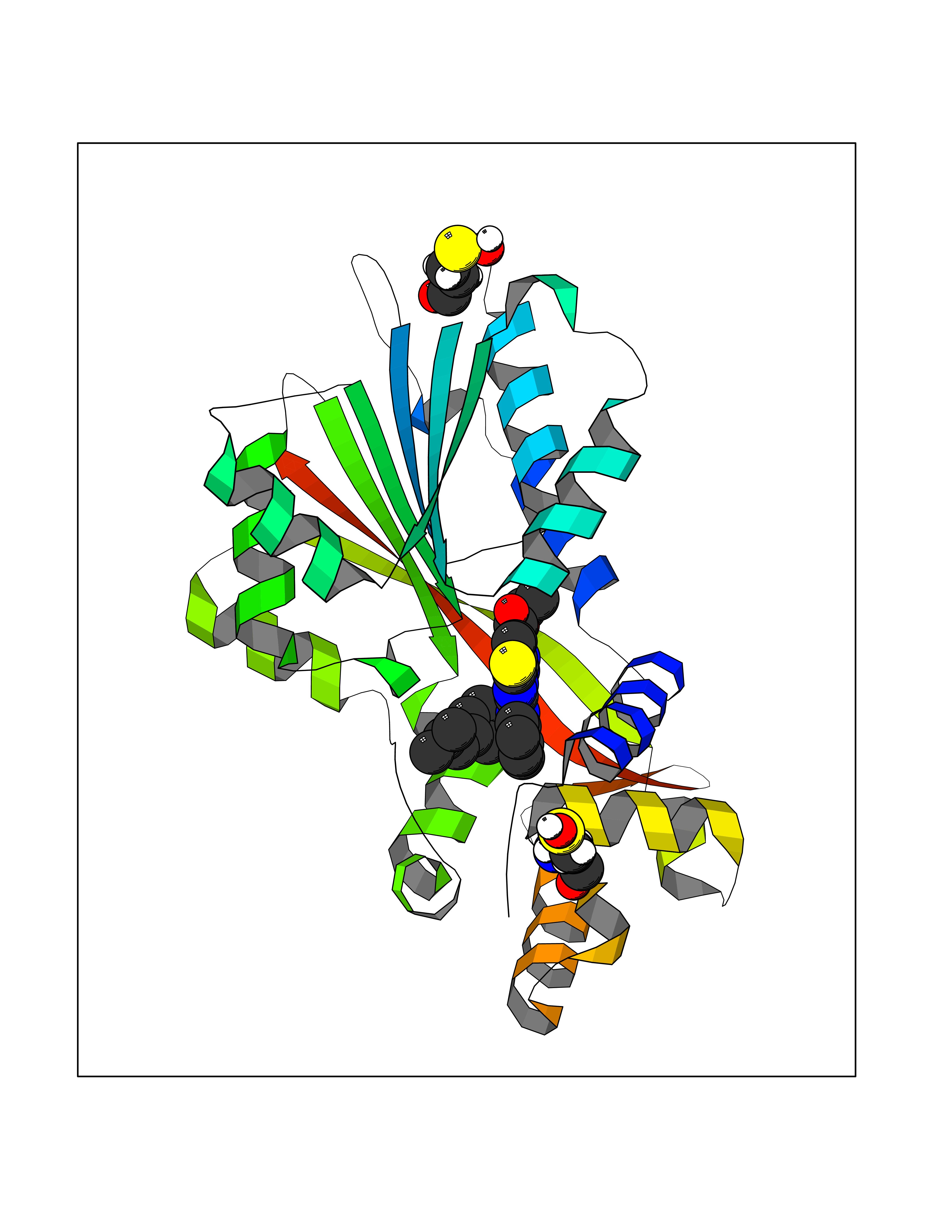 Molscript Map 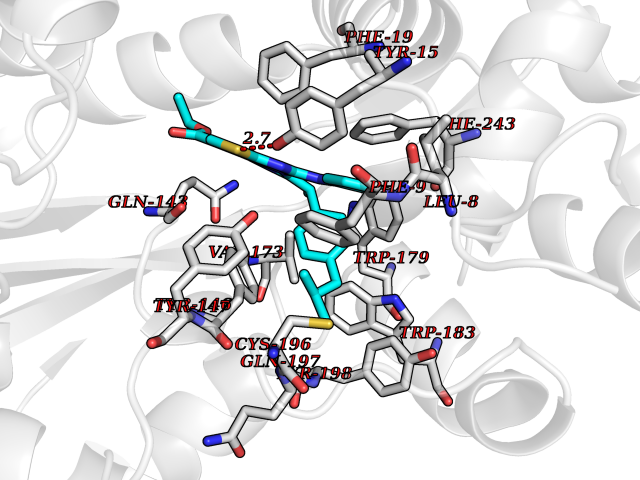 Pymol Map 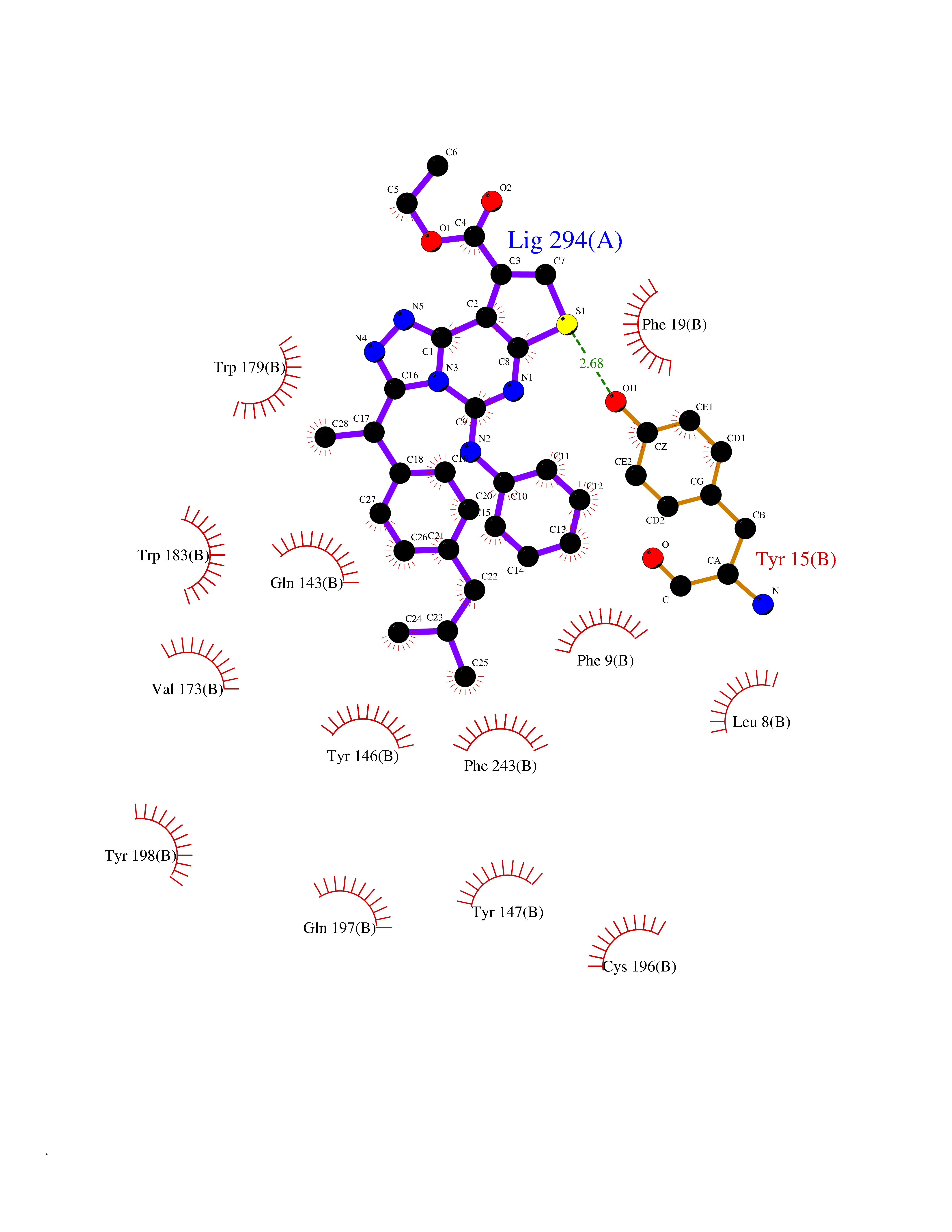 Ligplot Map 3D Binding mode Sequence MRSLFSDHGKYVESFRRFLNHSTEHQCMQEFMDKKLPGIIGRIGDTKSEIKILSIGGGAGEIDLQILSKVQAQYPGVXINNEVVEPSAEQIAKYKELVAKTSNLENVKFAWHKETSSEYQSRMLEKKELQKWDFIHMIQMLYYVKDIPATLKFFHSLLGTNAKMLIIVVSGSSGWDKLWKKYGSRFPQDDLCQYITSDDLTQMLDNLGLKYECYDLLSTMDISDCFIDGNENGDLLWDFLTETXNFNATAPPDLRAELGKDLQEPEFSAKKEGKVLFNNTLSFIVIEA Hydrogen bonds contact Hydrophobic contact | ||||
| 17 | Lysine-specific histone demethylase 1B (KDM1B) | 4HSU | 8.56 | |
Target general information Gen name KDM1B Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Lysine-specific histone demethylase 2; LSD2; Flavin-containing amine oxidase domain-containing protein 1; C6orf193; AOF1 Protein family Flavin monoamine oxidase family Biochemical class CH-NH(2) donor oxidoreductase Function Required for de novo DNA methylation of a subset of imprinted genes during oogenesis. Acts by oxidizing the substrate by FAD to generate the corresponding imine that is subsequently hydrolyzed. Demethylates both mono- and di-methylated 'Lys-4' of histone H3. Has no effect on tri-methylated 'Lys-4', mono-, di- or tri-methylated 'Lys-9', mono-, di- or tri-methylated 'Lys-27', mono-, di- or tri-methylated 'Lys-36' of histone H3, or on mono-, di- or tri-methylated 'Lys-20' of histone H4. Histone demethylase that demethylates 'Lys-4' of histone H3, a specific tag for epigenetic transcriptional activation, thereby acting as a corepressor. Related diseases Angioedema, hereditary, 1 (HAE1) [MIM:106100]: An autosomal dominant disorder characterized by episodic local swelling involving subcutaneous or submucous tissue of the upper respiratory and gastrointestinal tracts, face, extremities, and genitalia. Hereditary angioedema due to C1 esterase inhibitor deficiency is comprised of two clinically indistinguishable forms. In hereditary angioedema type 1, serum levels of C1 esterase inhibitor are decreased, while in type 2, the levels are normal or elevated, but the protein is non-functional. {ECO:0000269|PubMed:12773530, ECO:0000269|PubMed:1363816, ECO:0000269|PubMed:1451784, ECO:0000269|PubMed:14635117, ECO:0000269|PubMed:16409206, ECO:0000269|PubMed:2118657, ECO:0000269|PubMed:2296585, ECO:0000269|PubMed:22994404, ECO:0000269|PubMed:2365061, ECO:0000269|PubMed:24456027, ECO:0000269|PubMed:3178731, ECO:0000269|PubMed:7814636, ECO:0000269|PubMed:7883978, ECO:0000269|PubMed:8172583, ECO:0000269|PubMed:8529136, ECO:0000269|PubMed:8755917, ECO:0000269|Ref.41}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q96L03 EC number EC 1.-.-.- Uniprot keywords 3D-structure; Alternative splicing; Chromatin regulator; Chromosome; Developmental protein; FAD; Flavoprotein; Metal-binding; Nucleus; Oxidoreductase; Phosphoprotein; Proteomics identification; Reference proteome; Repressor; Transcription; Transcription regulation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A,C Molecular weight (Da) 85795.5 Length 763 Aromaticity 0.1 Instability index 37.87 Isoelectric point 8.41 Charge (pH=7) 9.16 2D Binding mode Binding energy (Kcal/mol) -11.67 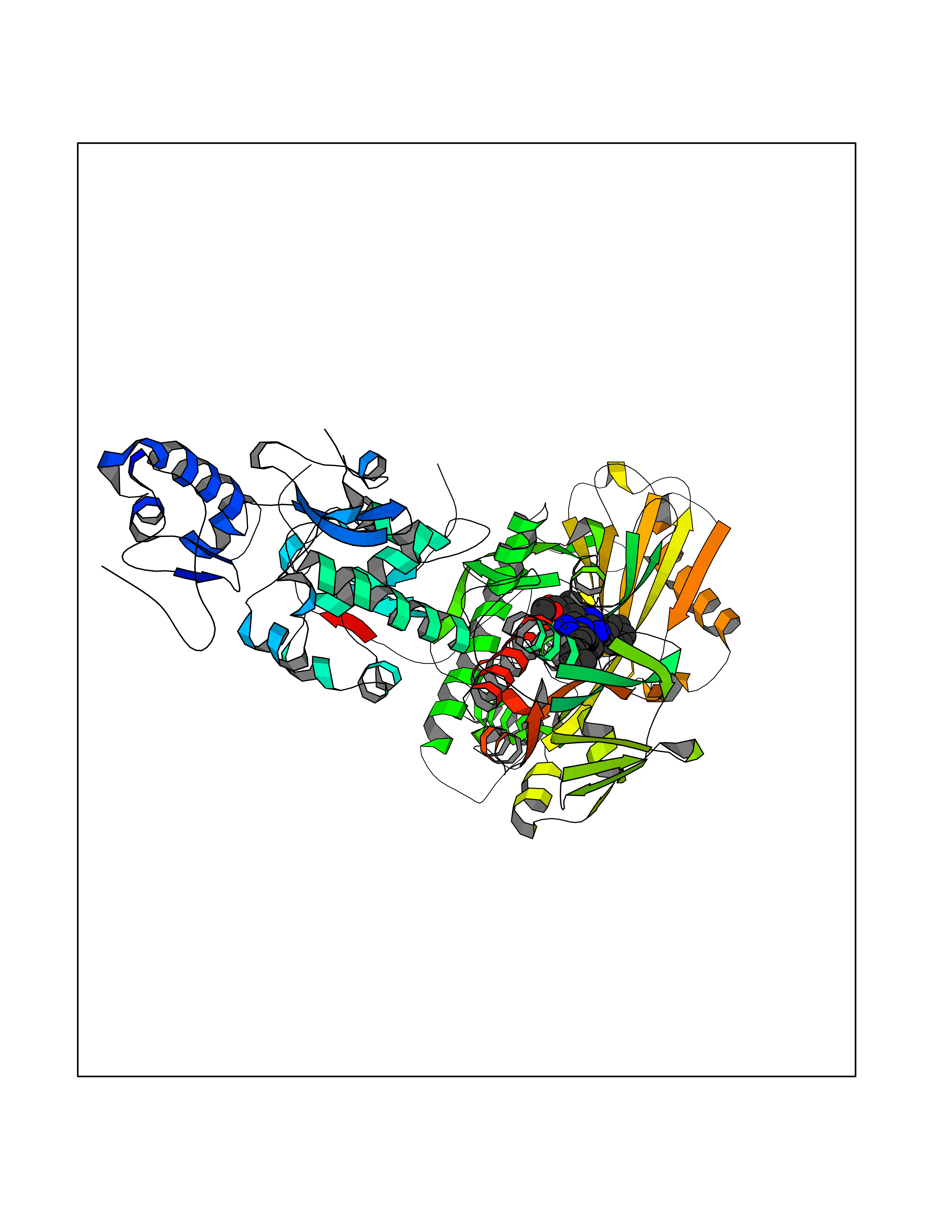 Molscript Map 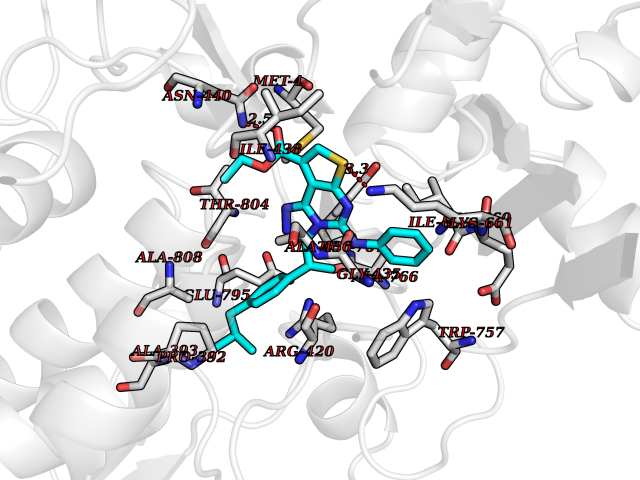 Pymol Map 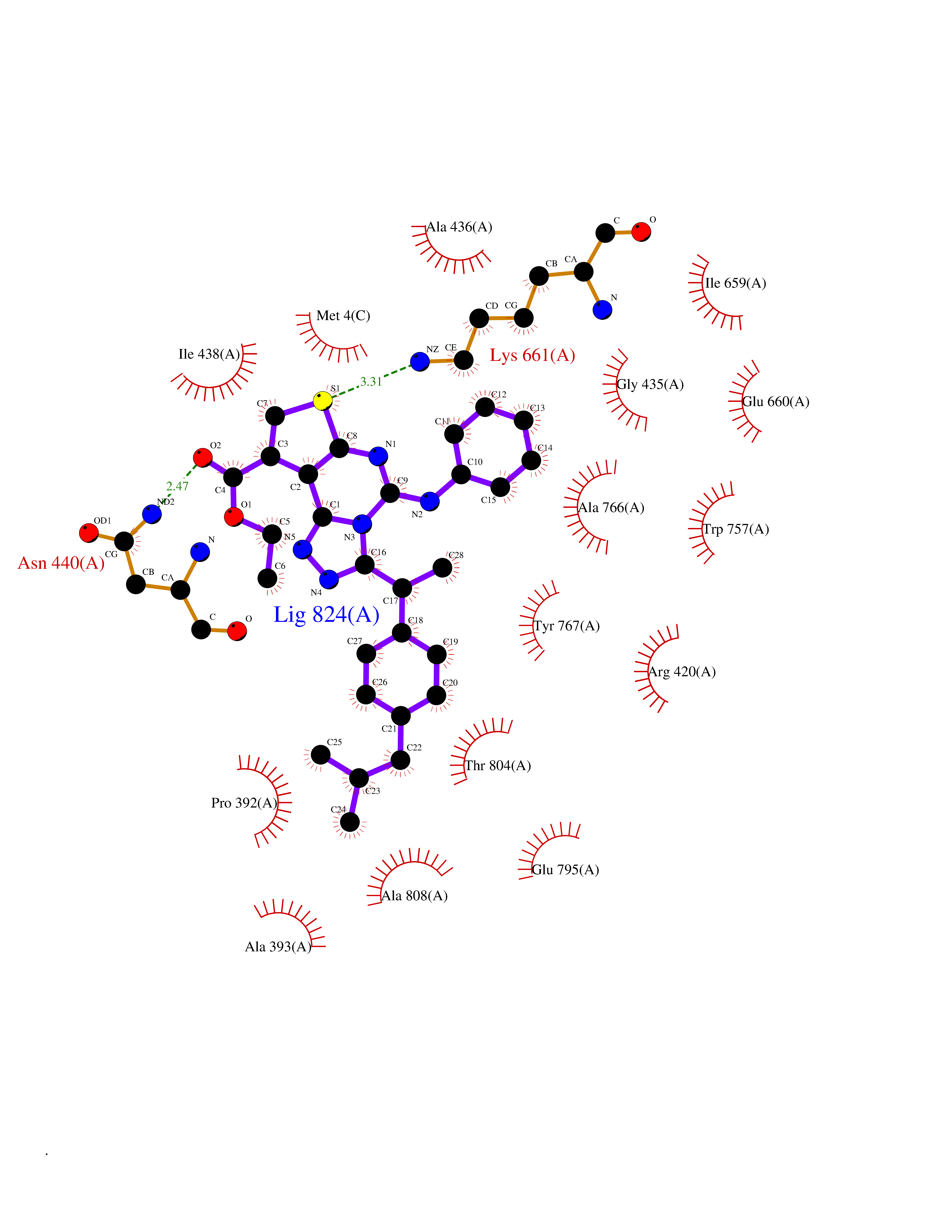 Ligplot Map 3D Binding mode Sequence GSRKCEKAGCTATCPVCFASASERCAKNGYTSRWYHLSCGEHFCNECFDHYYRSHKDGYDKYTTWKKIWTSNGKTEPSPKAFMADQQLPYWVQCTKPECRKWRQLTKEIQLTPQIAKTYRCGMKSDHCSLPEDLRVLEVSNHWWYSMLILPPLLKDSVAAPLLSAYYPDCVGMSPSCTGMNRYFQPFYQPNECGKALCVRPDVMELDELYEFPEYSRDPTMYLALRNLILALWYTNCKEALTPQKCIPHIIVRGLVRIRCVQEVERILYFMTRKGLINTGVLSVGADQYLLPKDYHNKSVIIIGAGPAGLAAARQLHNFGIKVTVLEAKDRIGGRVWDDKSFKGVTVGRGAQIVNGCINNPVALMCEQLGISMHKFGERCDLIQEGGRITDPTIDKRMDFHFNALLDVVSEWRKDKTQLQDVPLGEKIEEIYKAFIKESGIQFSELEGQVLQFHLSNLEYACGSNLHQVSARSWDHNEFFAQFAGDHTLLTPGYSVIIEKLAEGLDIQLKSPVQCIDYSGDEVQVTTTDGTGYSAQKVLVTVPLALLQKGAIQFNPPLSEKKMKAINSLGAGIIEKIALQFPYRFWDSKVQGADFFGHVPPSASKRGLFAVFYDMDPQKKHSVLMSVIAGEAVASVRTLDDKQVLQQCMATLRELFKEQEVPDPTKYFVTRWSTDPWIQMAYSFVKTGGSGEAYDIIAEDIQGTVFFAGEATNRHFPQTVTGAYLSGVREASKIAAFARTMQTARKSTGGKAPRKQLATKAAR Hydrogen bonds contact Hydrophobic contact | ||||
| 18 | Retinoic acid receptor RXR-alpha (RXRA) | 2P1T | 8.55 | |
Target general information Gen name RXRA Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Retinoid X receptor alpha; RXRalpha; Nuclear receptor subfamily 2 group B member 1; NR2B1 Protein family Nuclear hormone receptor family, NR2 subfamily Biochemical class Nuclear hormone receptor Function Retinoic acid receptors bind as heterodimers to their target response elements in response to their ligands, all-trans or 9-cis retinoic acid, and regulate gene expression in various biological processes. The RAR/RXR heterodimers bind to the retinoic acid response elements (RARE) composed of tandem 5'-AGGTCA-3' sites known as DR1-DR5. The high affinity ligand for RXRs is 9-cis retinoic acid. RXRA serves as a common heterodimeric partner for a number of nuclear receptors. In the absence of ligand, the RXR-RAR heterodimers associate with a multiprotein complex containing transcription corepressors that induce histone acetylation, chromatin condensation and transcriptional suppression. On ligand binding, the corepressors dissociate from the receptors and associate with the coactivators leading to transcriptional activation. The RXRA/PPARA heterodimer is required for PPARA transcriptional activity on fatty acid oxidation genes such as ACOX1 and the P450 system genes. Receptor for retinoic acid. Related diseases Lichtenstein-Knorr syndrome (LIKNS) [MIM:616291]: An autosomal recessive neurologic disorder characterized by progressive cerebellar ataxia and severe progressive sensorineural hearing loss. {ECO:0000269|PubMed:25205112, ECO:0000269|PubMed:30237576}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08063; DB08402; DB07863; DB07557; DB00459; DB00210; DB01436; DB00523; DB00132; DB04557; DB00307; DB01393; DB03756; DB00749; DB00926; DB05956; DB04224; DB02746; DB00412; DB00755; DB08601 Interacts with O14503; P35637; Q15648; Q71SY5; Q15788; Q15596; P55055; P55055-1; Q13133; P27986; P37231; P37231-1; P10276; P42224; P11473; P97792-1; Q9JLI4; P04625; PRO_0000278730 [Q03463] EC number NA Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Cytoplasm; DNA-binding; Host-virus interaction; Isopeptide bond; Metal-binding; Mitochondrion; Nucleus; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Transcription; Transcription regulation; Ubl conjugation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A Molecular weight (Da) 24958.9 Length 221 Aromaticity 0.07 Instability index 39.47 Isoelectric point 6.16 Charge (pH=7) -3.45 2D Binding mode Binding energy (Kcal/mol) -11.66 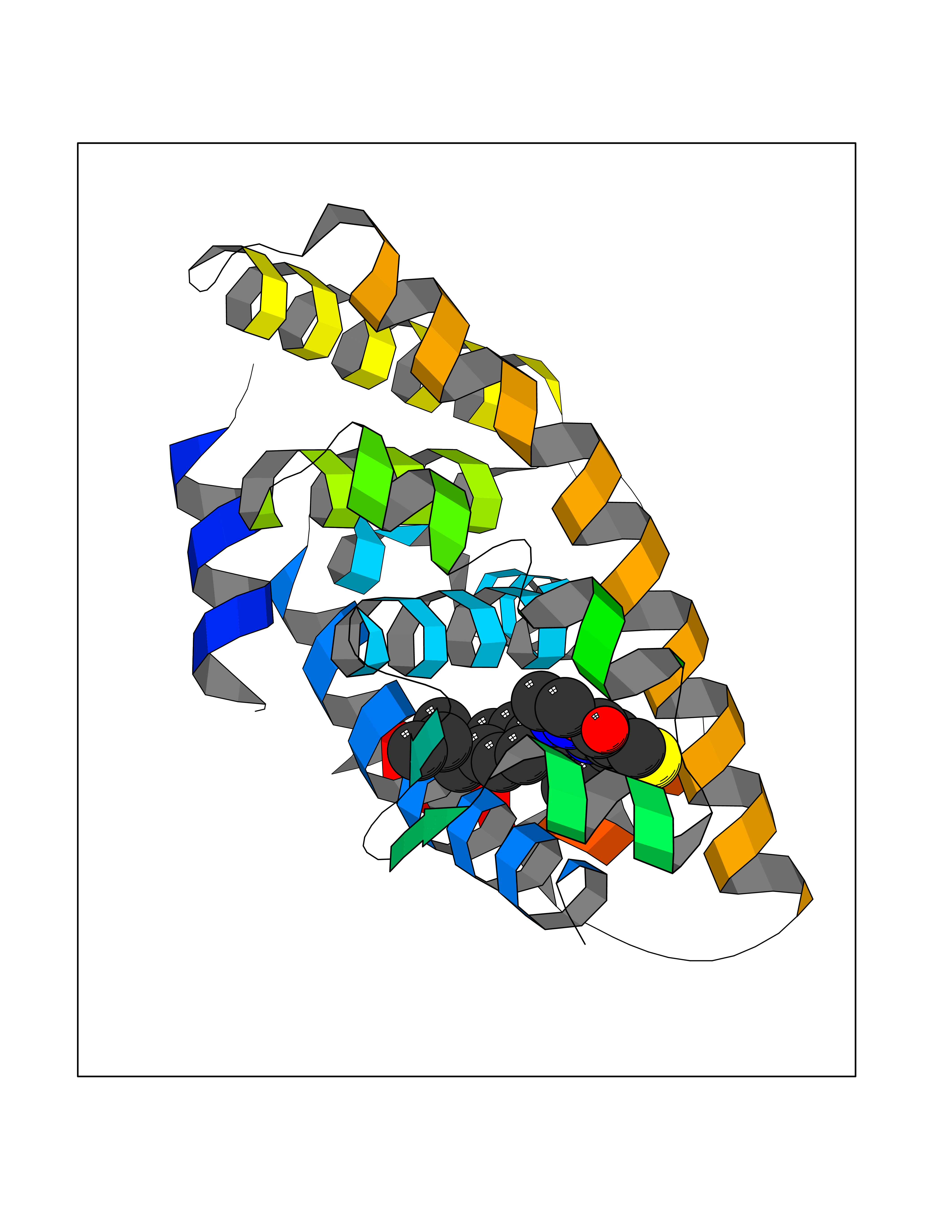 Molscript Map 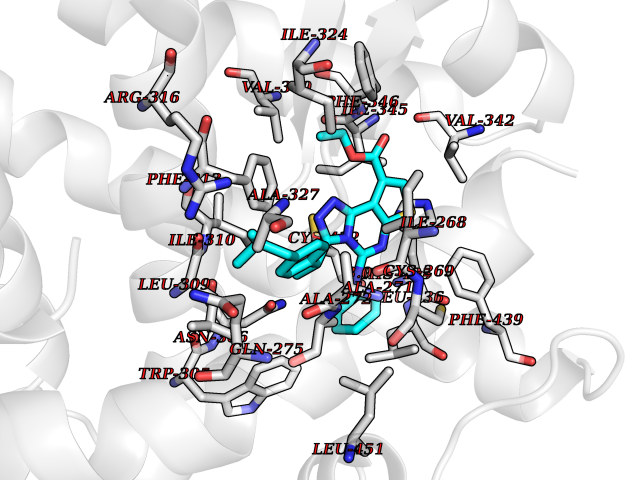 Pymol Map 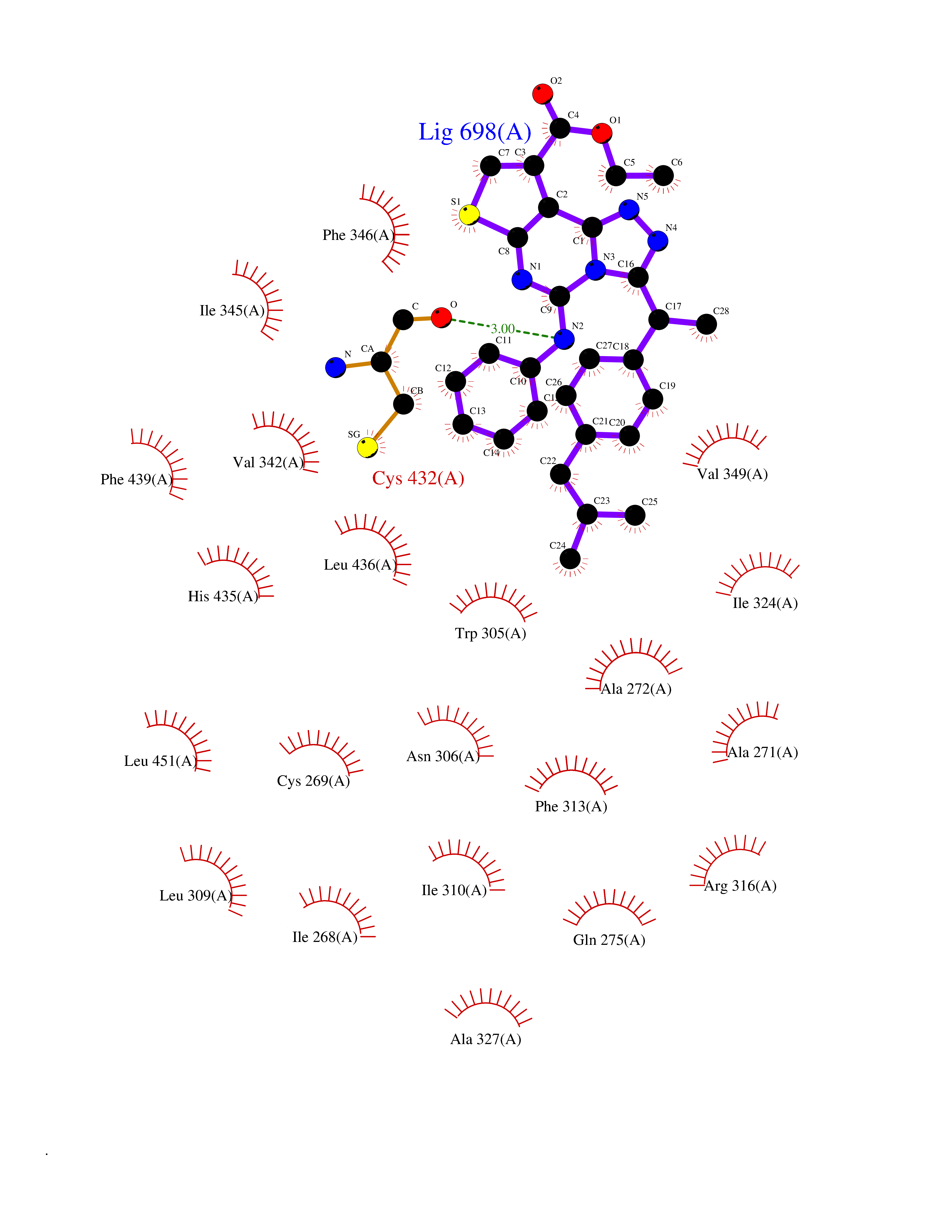 Ligplot Map 3D Binding mode Sequence DMPVERILEAELAVEDPVTNICQAADKQLFTLVEWAKRIPHFSELPLDDQVILLRAGWNELLIASFSHRSIAVKDGILLATGLHVHRNSAHSAGVGAIFDRVLTELVSKMRDMQMDKTELGCLRAIVLFNPDSKGLSNPAEVEALREKVYASLEAYCKHKYPEQPGRFAKLLLRLPALRSIGLKCLEHLFFFKLIGDTPIDTFLMEMLEAPHKILHRLLQD Hydrogen bonds contact Hydrophobic contact | ||||
| 19 | Tryptophan 2,3-dioxygenase (TDO) | 6PYZ | 8.54 | |
Target general information Gen name TDO2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Tryptophanase; Tryptophan pyrrolase; Tryptophan oxygenase; Tryptamin 2,3-dioxygenase; TRPO; TO Protein family Tryptophan 2,3-dioxygenase family Biochemical class Oxygenase Function Catalyzes the oxidative cleavage of the indole moiety. Heme-dependent dioxygenase that catalyzes the oxidative cleavage of the L-tryptophan (L-Trp) pyrrole ring and converts L-tryptophan to N-formyl-L-kynurenine. Related diseases Hypertryptophanemia (HYPTRP) [MIM:600627]: An autosomal recessive condition characterized by persistent hypertryptophanemia and hyperserotoninemia. {ECO:0000269|PubMed:28285122}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00779; DB00500; DB00150 Interacts with O43865; O95671; P27797; P12830; P36957; O60762; P06730; Q8TBB1; Q9H8S9; Q70IA8; Q8TDX7; Q9NPG2; Q9HAN9; P20393; Q9NRD5; Q8IYS1; O00560; Q9H190; P48775; Q68DK2-5 EC number EC 1.13.11.11 Uniprot keywords 3D-structure; Dioxygenase; Disease variant; Heme; Iron; Metal-binding; Oxidoreductase; Proteomics identification; Reference proteome; Tryptophan catabolism Protein physicochemical properties Chain ID A,B Molecular weight (Da) 83454.8 Length 701 Aromaticity 0.11 Instability index 43.93 Isoelectric point 6.93 Charge (pH=7) -0.48 2D Binding mode Binding energy (Kcal/mol) -11.65 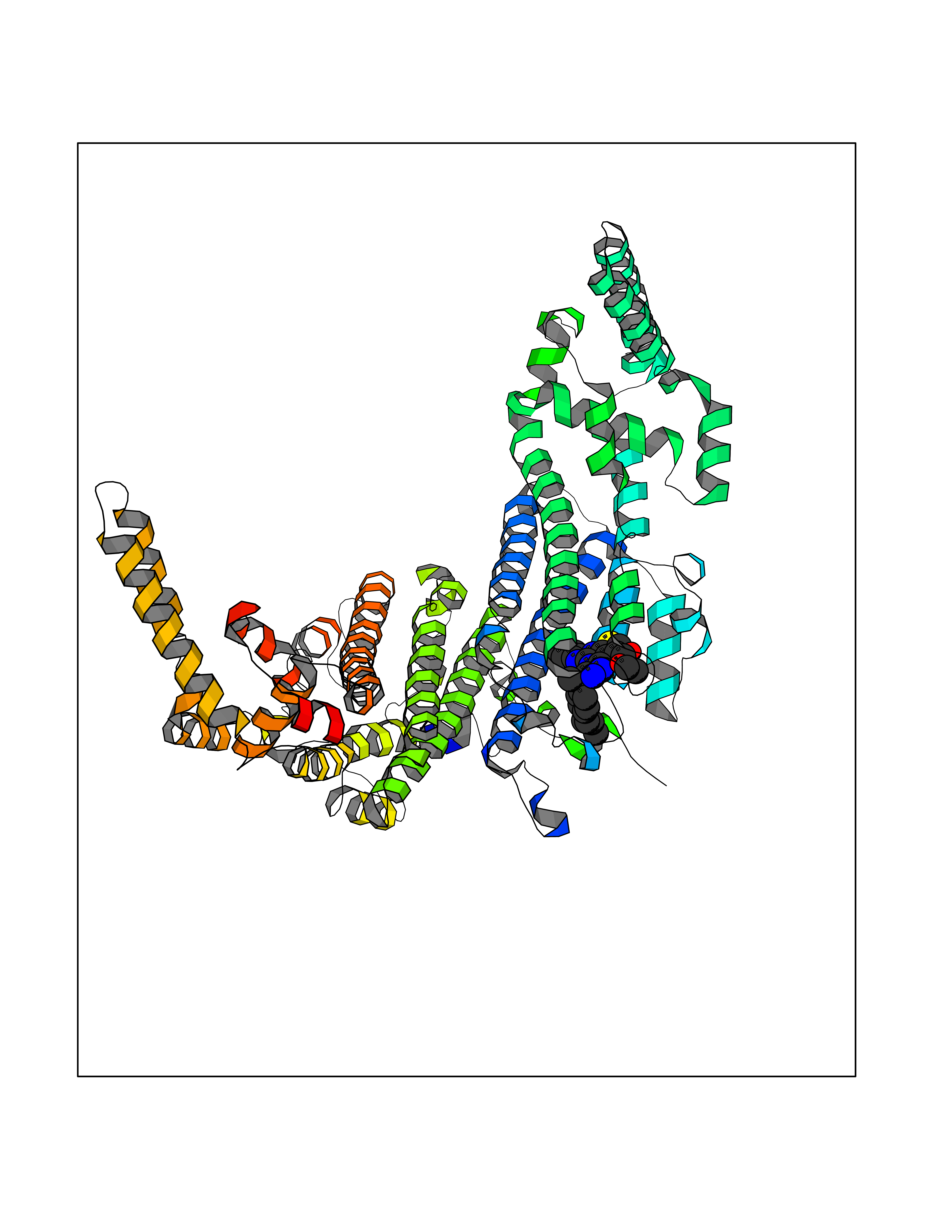 Molscript Map 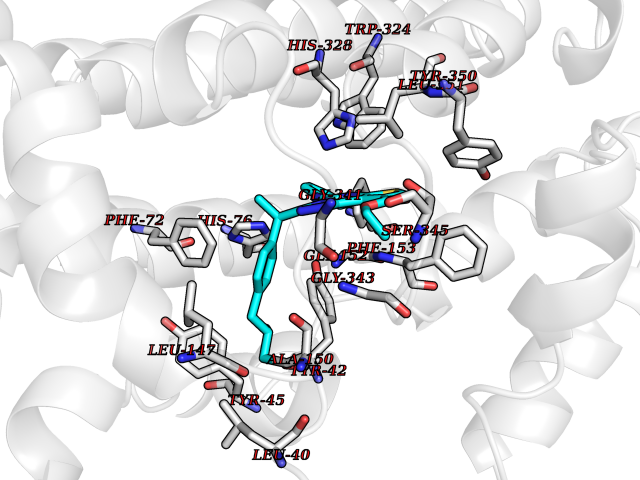 Pymol Map 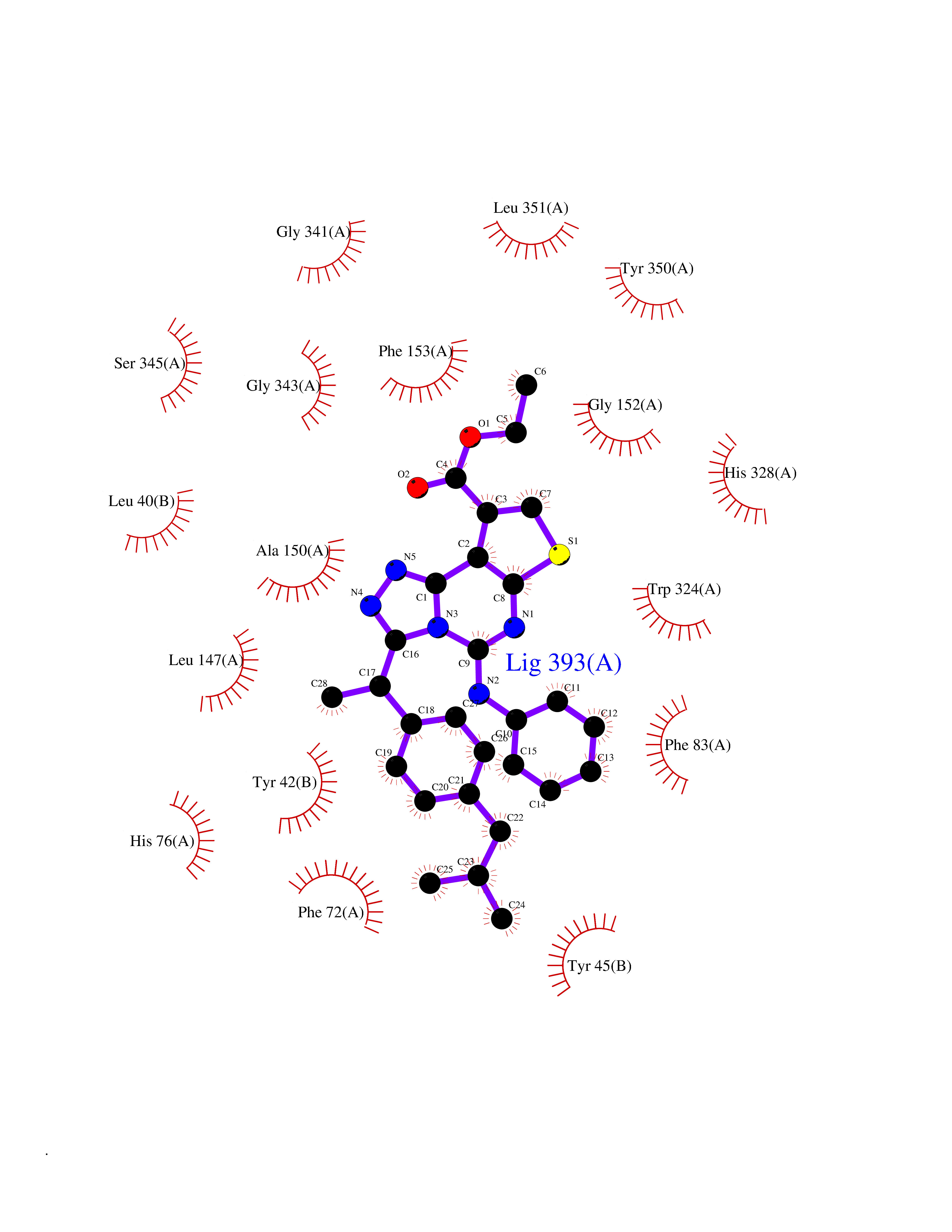 Ligplot Map 3D Binding mode Sequence GLIYGNYLHLEKVLNAQELQSETKGNKIHDEHLFIITHQAYELWFKQILWELDSVREIFQNGHVRDERNMLKVVSRMHRVSVILKLLVQQFSILETMTALDFNDFREYLSPASGFQSLQFRLLENKIGVLQNMRVPYNRRHYRDNFKGEENELLLKSEQEKTLLELVEAWLERTPGLEPHGFNFWGKLEKNITRGLEEEFIRIQAKEESEEKEEQVAEFQKQKEVLLSLFDEKRHEHLLSKGERRLSYRALQGALMIYFYREEPRFQVPFQLLTSLMDIDSLMTKWRYNHVCMVHRMLGSKAGTGGSSGYHYLRSTVSDRYKVFVDLFNLSTYLIPRHWIPKMNPTIHKFLEHGGLIYGNYLHLEKVLNAQELQSETKGNKIHDEHLFIITHQAYELWFKQILWELDSVREIFQNGHVRDERNMLKVVSRMHRVSVILKLLVQQFSILETMTALDFNDFREYLSPASGFQSLQFRLLENKIGVLQNMRVPYYRDNFKGEENELLLKSEQEKTLLELVEAWLERTPGLEPHGFNFWGKLEKNITRGLEEEFIRIQAKEESEEKEEQVAEFQKQKEVLLSLFDEKRHEHLLSKGERRLSYRALQGALMIYFYREEPRFQVPFQLLTSLMDIDSLMTKWRYNHVCMVHRMLGSKAGTGGSSGYHYLRSTVSDRYKVFVDLFNLSTYLIPRHWIPKMNPTIHKFL Hydrogen bonds contact Hydrophobic contact | ||||
| 20 | Mutated oxalosuccinate decarboxylase (mIDH1) | 6ADG | 8.54 | |
Target general information Gen name IDH1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms PICD (mutated); Oxalosuccinate decarboxylase (mutated); NADP(+)-specific ICDH (mutated); Isocitrate dehydrogenase [NADP] cytoplasmic (mutated); IDP (mutated); IDH (mutated); Cytosolic NADP-isocitrate Protein family Isocitrate and isopropylmalate dehydrogenases family Biochemical class Short-chain dehydrogenases reductase Function Catalyses the NADPH-dependent reduction of alpha-ketoglutarate to R(-)-2-hydroxyglutarate (2HG). Related diseases Glioma (GLM) [MIM:137800]: Gliomas are benign or malignant central nervous system neoplasms derived from glial cells. They comprise astrocytomas and glioblastoma multiforme that are derived from astrocytes, oligodendrogliomas derived from oligodendrocytes and ependymomas derived from ependymocytes. {ECO:0000269|PubMed:19117336, ECO:0000269|PubMed:19935646}. The gene represented in this entry is involved in disease pathogenesis. Mutations affecting Arg-132 are tissue-specific, and suggest that this residue plays a unique role in the development of high-grade gliomas. Mutations of Arg-132 to Cys, His, Leu or Ser abolish magnesium binding and abolish the conversion of isocitrate to alpha-ketoglutarate. Instead, alpha-ketoglutarate is converted to R(-)-2-hydroxyglutarate. Elevated levels of R(-)-2-hydroxyglutarate are correlated with an elevated risk of malignant brain tumors. {ECO:0000269|PubMed:19935646}.; DISEASE: Genetic variations are associated with cartilaginous tumors such as enchondroma or chondrosarcoma. Mutations of Arg-132 to Cys, Gly or His abolish the conversion of isocitrate to alpha-ketoglutarate. Instead, alpha-ketoglutarate is converted to R(-)-2-hydroxyglutarate. {ECO:0000269|PubMed:26161668}. Drugs (DrugBank ID) DB09374; DB01727; DB14568; DB03461; DB16267 Interacts with P0DP23; P27797; P36957; O75874; Q8TDX7; P16284; P17612; P50454; P37173; Q05086-3 EC number EC 1.1.1.42 Uniprot keywords 3D-structure; Acetylation; Cytoplasm; Direct protein sequencing; Glyoxylate bypass; Magnesium; Manganese; Metal-binding; NADP; Oxidoreductase; Peroxisome; Phosphoprotein; Proteomics identification; Reference proteome; Tricarboxylic acid cycle Protein physicochemical properties Chain ID A,B Molecular weight (Da) 92711.7 Length 823 Aromaticity 0.1 Instability index 26.74 Isoelectric point 6.42 Charge (pH=7) -4.48 2D Binding mode Binding energy (Kcal/mol) -11.65 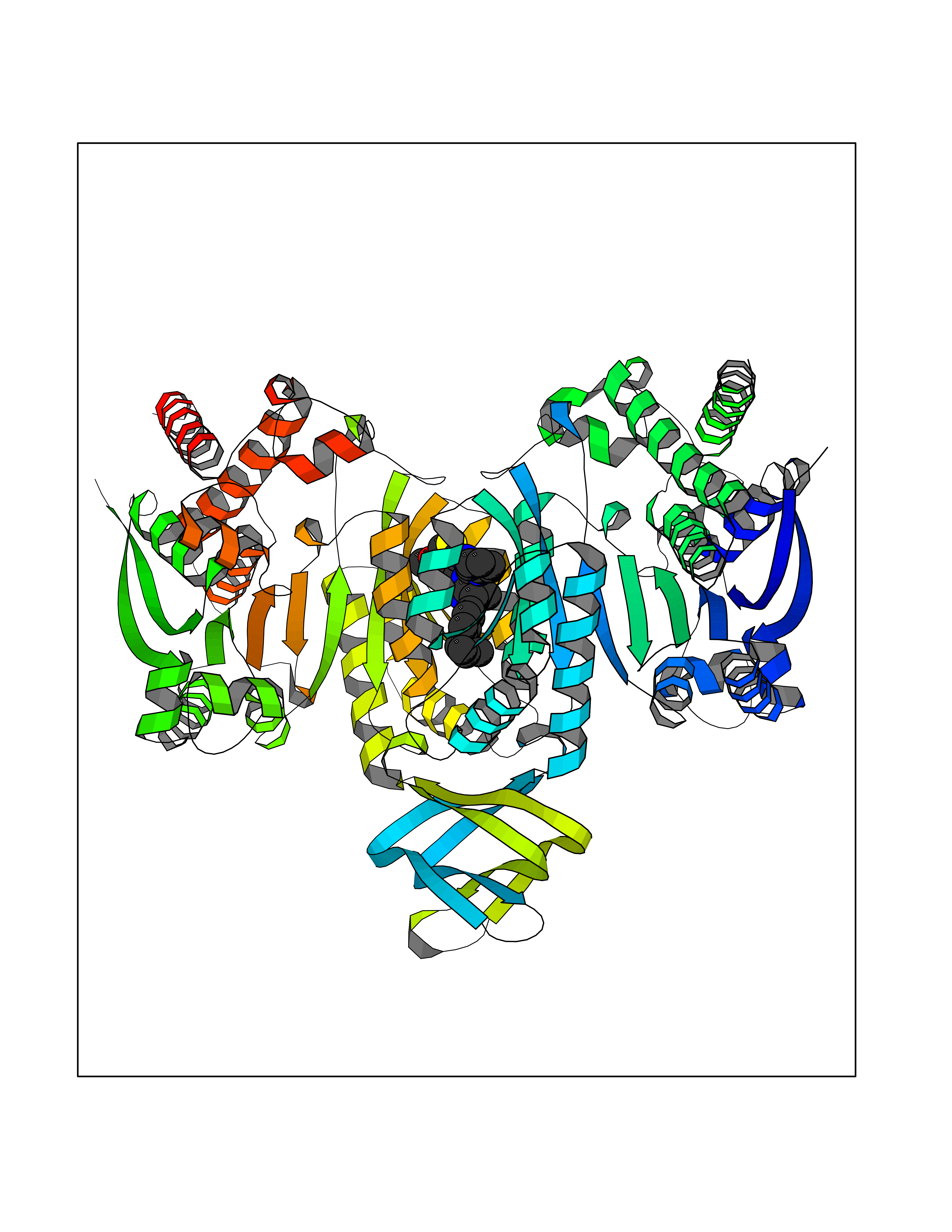 Molscript Map 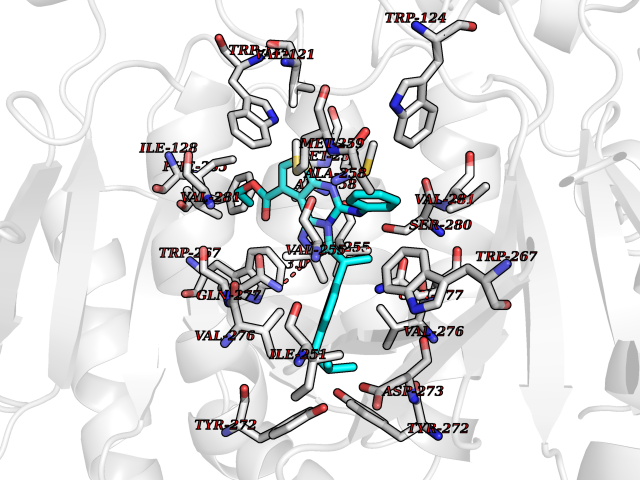 Pymol Map 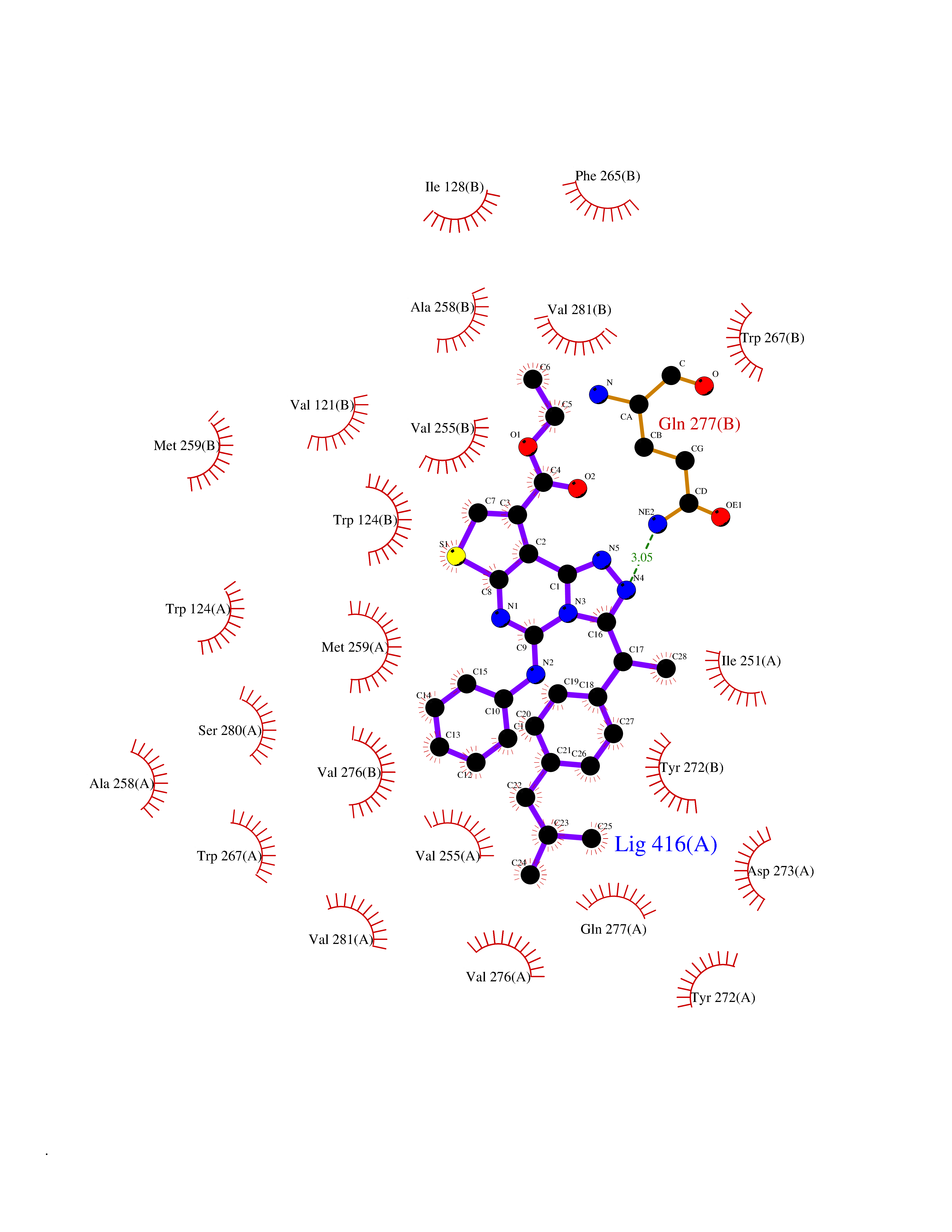 Ligplot Map 3D Binding mode Sequence KKISGGSVVEMQGDEMTRIIWELIKEKLIFPYVELDLHSYDLGIENRDATNDQVTKDAAEAIKKHNVGVKCATITPDEKRVEEFKLKQMWKSPNGTIRNILGGTVFREAIICKNIPRLVSGWVKPIIIGHHAYGDQYRATDFVVPGPGKVEITYTPSDGTQKVTYLVHNFEEGGGVAMGMYNQDKSIEDFAHSSFQMALSKGWPLYLSTKNTILKKYDGRFKDIFQEIYDKQYKSQFEAQKIWYEHRLIDDMVAQAMKSEGGFIWACKNYDGDVQSDSVAQGYGSLGMMTSVLVCPDGKTVEAEAAHGTVTRHYRMYQKGQETSTNPIASIFAWTRGLAHRAKLDNNKELAFFANALEEVSIETIEAGFMTKDLAACIKGLPNVQRSDYLNTFEFMDKLGENLKIKLAQAKLKKISGGSVVEMQGDEMTRIIWELIKEKLIFPYVELDLHSYDLGIENRDATNDQVTKDAAEAIKKHNVGVKCATITPDEKRVEEFKLKQMWKSPNGTIRNILGGTVFREAIICKNIPRLVSGWVKPIIIGHHAYGDQYRATDFVVPGPGKVEITYTPSDGTQKVTYLVHNFEEGGGVAMGMYNQDKSIEDFAHSSFQMALSKGWPLYLSTKNTILKKYDGRFKDIFQEIYDKQYKSQFEAQKIWYEHRLIDDMVAQAMKSEGGFIWACKNYDGDVQSDSVAQGYGSLGMMTSVLVCPDGKTVEAEAAHGTVTRHYRMYQKGQETSTNPIASIFAWTRGLAHRAKLDNNKELAFFANALEEVSIETIEAGFMTKDLAACIKGLPNVQRSDYLNTFEFMDKLGENLKIKLAQAK Hydrogen bonds contact Hydrophobic contact | ||||