Job Results:
Ligand
Structure
Job ID
5c75d0793a5c7fa3ef7fd4eba45db829
Job name
NA
Time
2026-02-27 11:58:42
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 1 | Protein cereblon (CRBN) | 5FQD | 5.24 | |
Target general information Gen name CRBN Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Protein cereblon Protein family CRBN family Biochemical class NA Function Substrate recognition component of a DCX (DDB1-CUL4-X-box) E3 protein ligase complex that mediates the ubiquitination and subsequent proteasomal degradation of target proteins, such as MEIS2. Normal degradation of key regulatory proteins is required for normal limb outgrowth and expression of the fibroblast growth factor FGF8. May play a role in memory and learning by regulating the assembly and neuronal surface expression of large-conductance calcium-activated potassium channels in brain regions involved in memory and learning via its interaction with KCNT1. Binding of pomalidomide and other thalidomide-related drugs changes the substrate specificity of the human protein, leading to decreased degradation of MEIS2 and other target proteins and increased degradation of MYC, IRF4, IKZF1 and IKZF3. Related diseases Intellectual developmental disorder, autosomal recessive 2 (MRT2) [MIM:607417]: A disorder characterized by significantly below average general intellectual functioning associated with impairments in adaptive behavior and manifested during the developmental period. MRT2 patients display mild intellectual disability with a standard IQ ranged from 50 to 70. IQ scores are lower in males than females. Developmental milestones are mildly delayed. There are no dysmorphic or autistic features. {ECO:0000269|PubMed:15557513, ECO:0000269|PubMed:28143899}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00480; DB08910; DB01041 Interacts with Q96A83-2; P48729; Q16531; O14901; Q8IVT2; Q9P286; A0A6Q8PF08; Q93062; Q16531; Q13422-7 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; Disease variant; Intellectual disability; Membrane; Metal-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Ubl conjugation; Ubl conjugation pathway; Zinc Protein physicochemical properties Chain ID B,E Molecular weight (Da) 38245.7 Length 337 Aromaticity 0.08 Instability index 40.62 Isoelectric point 5.7 Charge (pH=7) -6.53 2D Binding mode Binding energy (Kcal/mol) -7.14 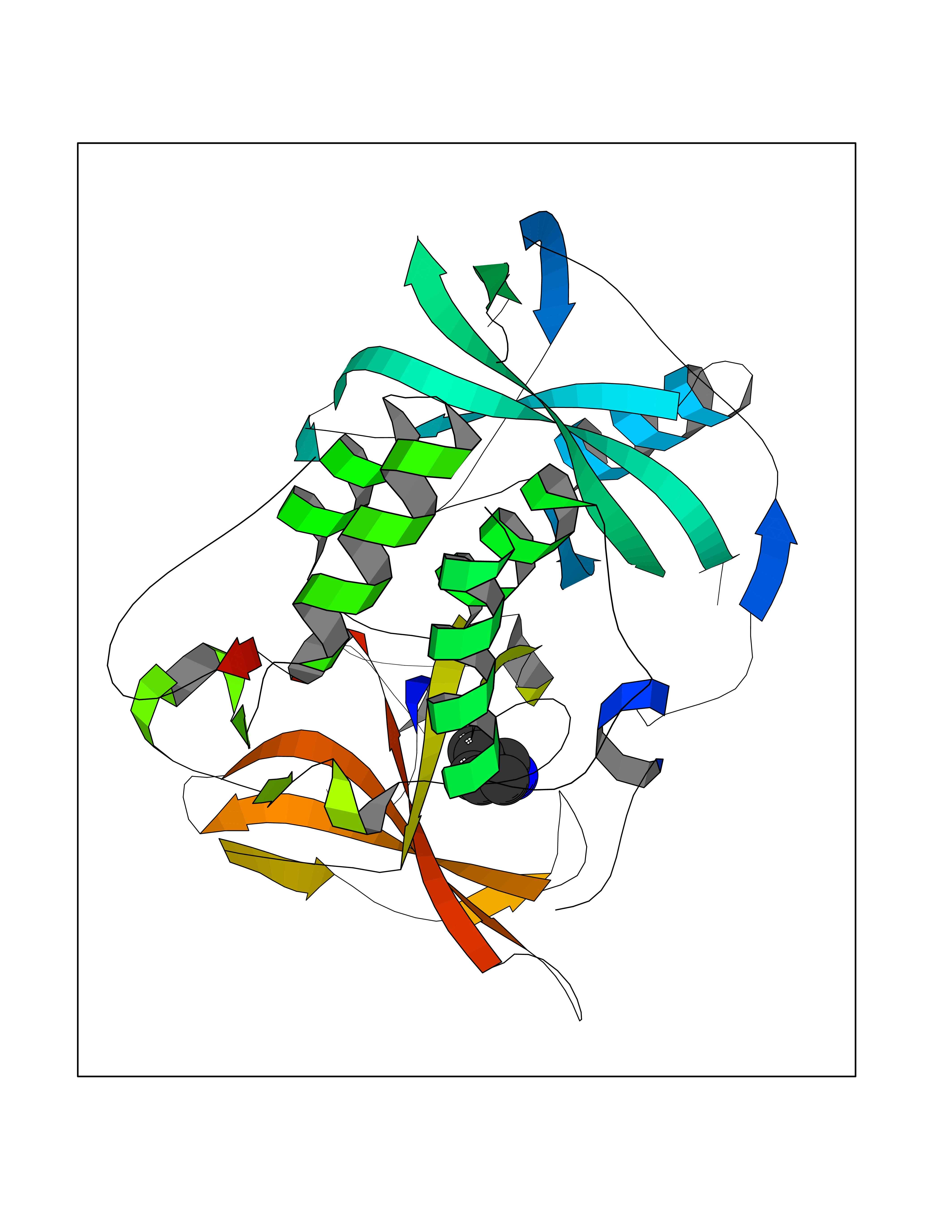 Molscript Map 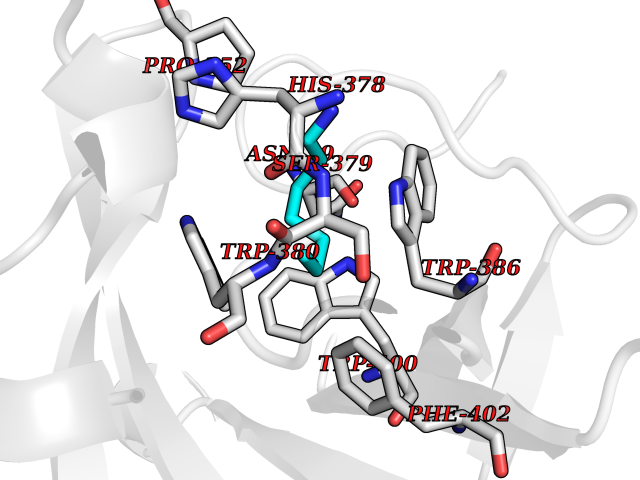 Pymol Map  Ligplot Map 3D Binding mode Sequence EFIVGGKYKLNITNGEEVAVINFDTSLPTSHTYLGADMEEFHGRTLHDDDSCQVIPVLPQVMMILIPGQTLPLQLFHPQEVSMVRNLIQKDRTFAVLAYSNVQEREAQFGTTAEIYAYREEIVKVKAIGRQRFKVLEQQAKVQILPECVLAETLMDRIKKQLREWDENLKDDSLPSNPIDFSYRVAACLPIDDVLRIQLLKIGSAIQRLRCELDIMNKCTSLCCKQCQETEITTKNEIFSLSLCGPMAAYVNPHGYVHETLTVYKACNLNLIGRPSTEHSWFPGYAWTVAQCKICASHIGWKFTATKKDMSPQKFWGLTRSALLPTIPDTEDEISPD Hydrogen bonds contact Hydrophobic contact | ||||
| 2 | Zinc finger protein Helios (IKZF2) | 7LPS | 5.21 | |
Target general information Gen name IKZF2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Ikaros family zinc finger protein 2 Protein family Ikaros C2H2-type zinc-finger protein family Biochemical class NA Function Associates with Ikaros at centromeric heterochromatin. Related diseases Developmental and epileptic encephalopathy 25, with amelogenesis imperfecta (DEE25) [MIM:615905]: An autosomal recessive disease characterized by subclinical seizures appearing in the first days of life, evolving to severe epileptic disease. Affected individuals have profound or severe delayed development with lack of speech, and most patients do not acquire the ability to sit. Additional variable features include axial hypotonia, peripheral hypertonia, and abnormal involuntary movements such as dystonia and choreoathetosis. Dental abnormalities, including delayed eruption, hypodontia, tooth hypoplasia, yellow discoloration, thin enamel, and enamel chipping are observed in most patients. {ECO:0000269|PubMed:24995870, ECO:0000269|PubMed:26384929, ECO:0000269|PubMed:30054523}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P29972; P56545; P56545-3; Q17RB8; P09022; Q8N8B7-2 EC number NA Uniprot keywords 3D-structure; Acetylation; Activator; Alternative splicing; DNA-binding; Isopeptide bond; Metal-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Transcription; Transcription regulation; Ubl conjugation; Zinc; Zinc-finger Protein physicochemical properties Chain ID B,C Molecular weight (Da) 47006.6 Length 410 Aromaticity 0.09 Instability index 44.28 Isoelectric point 7.23 Charge (pH=7) 0.69 2D Binding mode Binding energy (Kcal/mol) -7.1 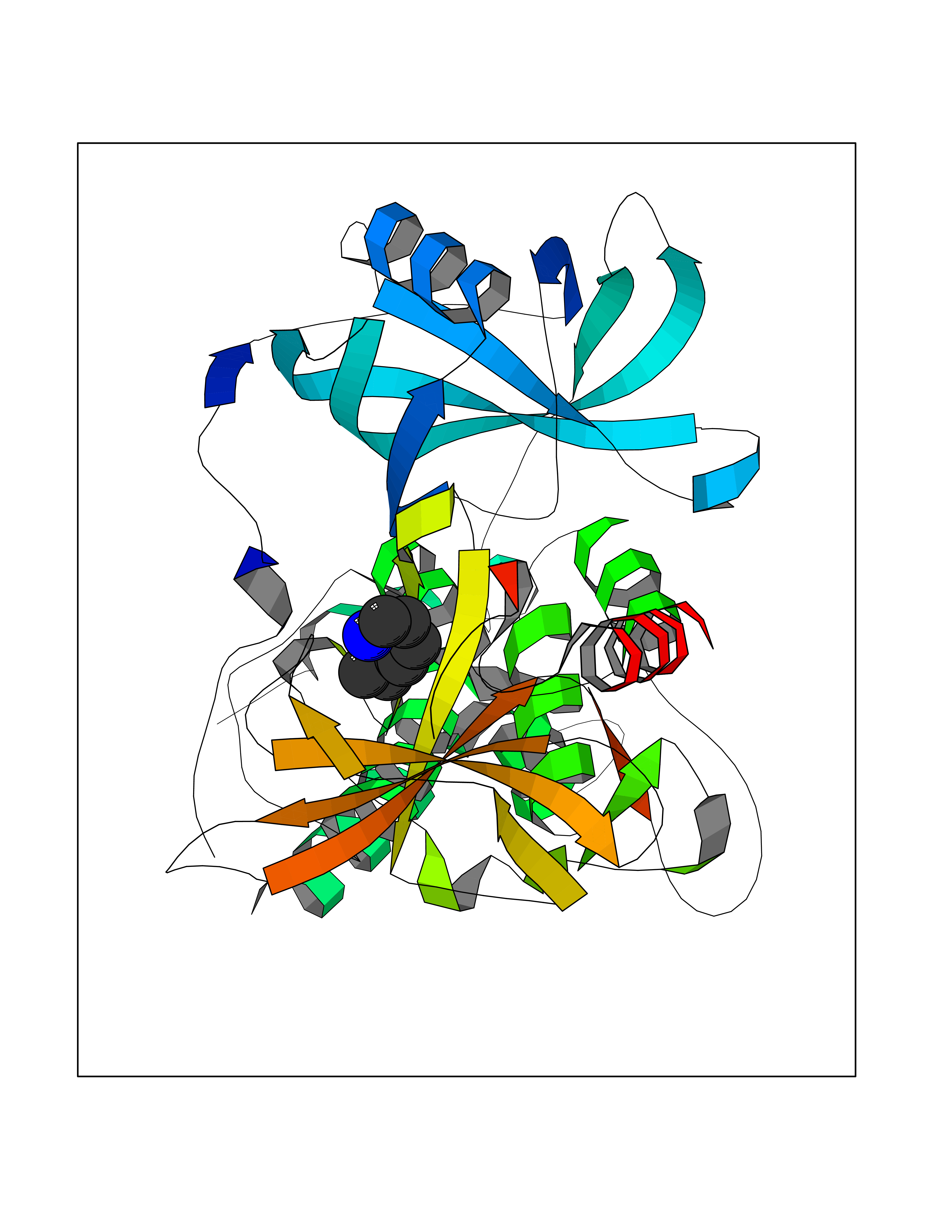 Molscript Map 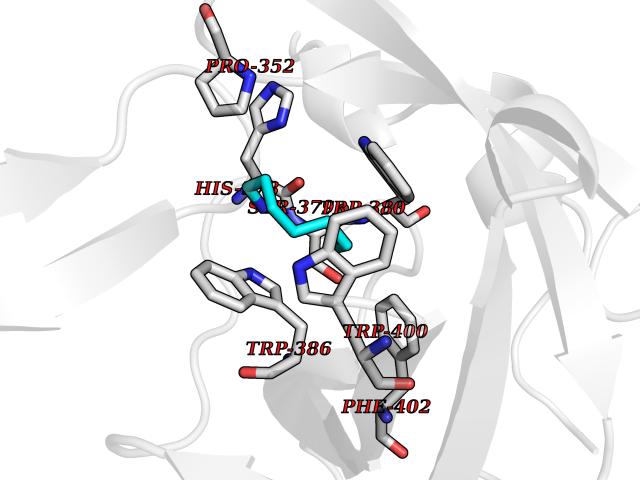 Pymol Map 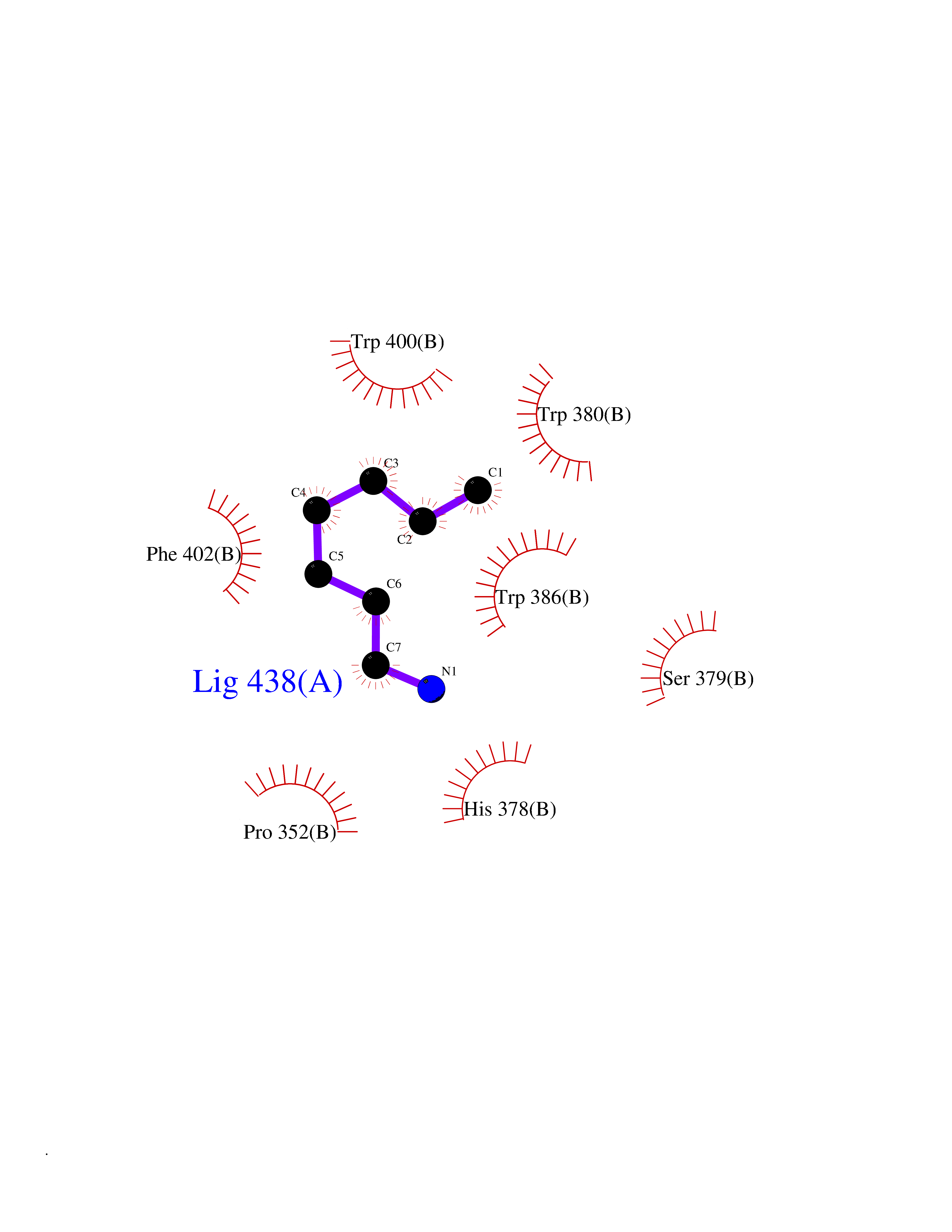 Ligplot Map 3D Binding mode Sequence INFDTSLPTSHTYLGADMEEFHGRTLHDDDSCQVIPVLPQVMMILIPGQTLPLQLFHPQEVSMVRNLIQKDRTFAVLAYSNVQEREAQFGTTAEIYAYREEQDFGIEIVKVKAIGRQRFKVLELRTQSDGIQQAKVQILPECVLPSTMSAVQLESLNKCQIFPCSYKWWQKYQKRKFHCANLTSWPRWLYSLYDAETLMDRIKKQLREWDENLKDDSLPSNPIDFSYRVAACLPIDDVLRIQLLKIGSAIQRLRCELDIMNKCTSLCCKQCQETEITTKNEIFSLSLCGPMAAYVNPHGYVHETLTVYKACNLNLIGRPSTEHSWFPGYAWTVAQCKICASHIGWKFTATKKDMSPQKFWGLTRSALLPTIPDTEDEISPDGERPFHCNQCGASFTQKGNLLRHIKLHSG Hydrogen bonds contact Hydrophobic contact | ||||
| 3 | SET domain containing 8 (KMT5A) | 5TEG | 5.20 | |
Target general information Gen name KMT5A Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms SETD8; SET8; SET07; SET domain-containing protein 8; PRSET7; PR/SET07; PR/SET domain-containing protein 07; PR-Set7; N-lysine methyltransferase KMT5A; Lysine-specific methylase 5A; Lysine N-methyltran Protein family Class V-like SAM-binding methyltransferase superfamily, Histone-lysine methyltransferase family, PR/SET subfamily Biochemical class Methyltransferase Function Specifically monomethylates 'Lys-20' of histone H4 (H4K20me1). H4K20me1 is enriched during mitosis and represents a specific tag for epigenetic transcriptional repression. Mainly functions in euchromatin regions, thereby playing a central role in the silencing of euchromatic genes. Required for cell proliferation, probably by contributing to the maintenance of proper higher-order structure of DNA during mitosis. Involved in chromosome condensation and proper cytokinesis. Nucleosomes are preferred as substrate compared to free histones. Mediates monomethylation of p53/TP53 at 'Lys-382', leading to repress p53/TP53-target genes. Plays a negative role in TGF-beta response regulation and a positive role in cell migration. Protein-lysine N-methyltransferase that monomethylates both histones and non-histone proteins. Related diseases Sick sinus syndrome 2 (SSS2) [MIM:163800]: The term 'sick sinus syndrome' encompasses a variety of conditions caused by sinus node dysfunction. The most common clinical manifestations are syncope, presyncope, dizziness, and fatigue. Electrocardiogram typically shows sinus bradycardia, sinus arrest, and/or sinoatrial block. Episodes of atrial tachycardias coexisting with sinus bradycardia ('tachycardia-bradycardia syndrome') are also common in this disorder. SSS occurs most often in the elderly associated with underlying heart disease or previous cardiac surgery, but can also occur in the fetus, infant, or child without heart disease or other contributing factors. SSS2 onset is in utero or at birth. {ECO:0000269|PubMed:15123648, ECO:0000269|PubMed:16407510, ECO:0000269|PubMed:20662977, ECO:0000269|PubMed:23103389}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Brugada syndrome 8 (BRGDA8) [MIM:613123]: A tachyarrhythmia characterized by right bundle branch block and ST segment elevation on an electrocardiogram (ECG). It can cause the ventricles to beat so fast that the blood is prevented from circulating efficiently in the body. When this situation occurs, the individual will faint and may die in a few minutes if the heart is not reset. {ECO:0000269|PubMed:19165230}. The gene represented in this entry may be involved in disease pathogenesis.; DISEASE: Epilepsy, idiopathic generalized 18 (EIG18) [MIM:619521]: An autosomal dominant form of idiopathic generalized epilepsy, a disorder characterized by recurring generalized seizures in the absence of detectable brain lesions and/or metabolic abnormalities. Generalized seizures arise diffusely and simultaneously from both hemispheres of the brain. Seizure types include juvenile myoclonic seizures, absence seizures, and generalized tonic-clonic seizures. EIG18 is characterized by onset of myoclonic seizures in infancy. Although the seizures remit, some patients may have later speech or cognitive impairment. {ECO:0000269|PubMed:30127718}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P62805; P07910; Q15672 EC number EC 2.1.1.- Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Cell cycle; Cell division; Chromatin regulator; Chromosome; Coiled coil; Direct protein sequencing; Methyltransferase; Mitosis; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Repressor; S-adenosyl-L-methionine; Transcription; Transcription regulation; Transferase; Ubl conjugation Protein physicochemical properties Chain ID A,D Molecular weight (Da) 19129.4 Length 167 Aromaticity 0.08 Instability index 49.18 Isoelectric point 7.88 Charge (pH=7) 1.37 2D Binding mode Binding energy (Kcal/mol) -7.09  Molscript Map 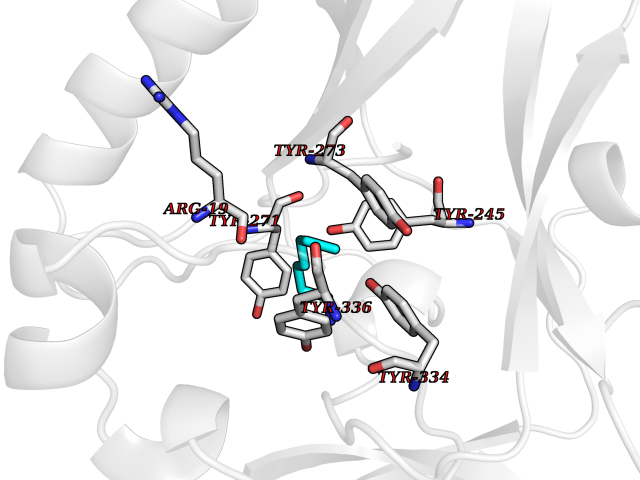 Pymol Map 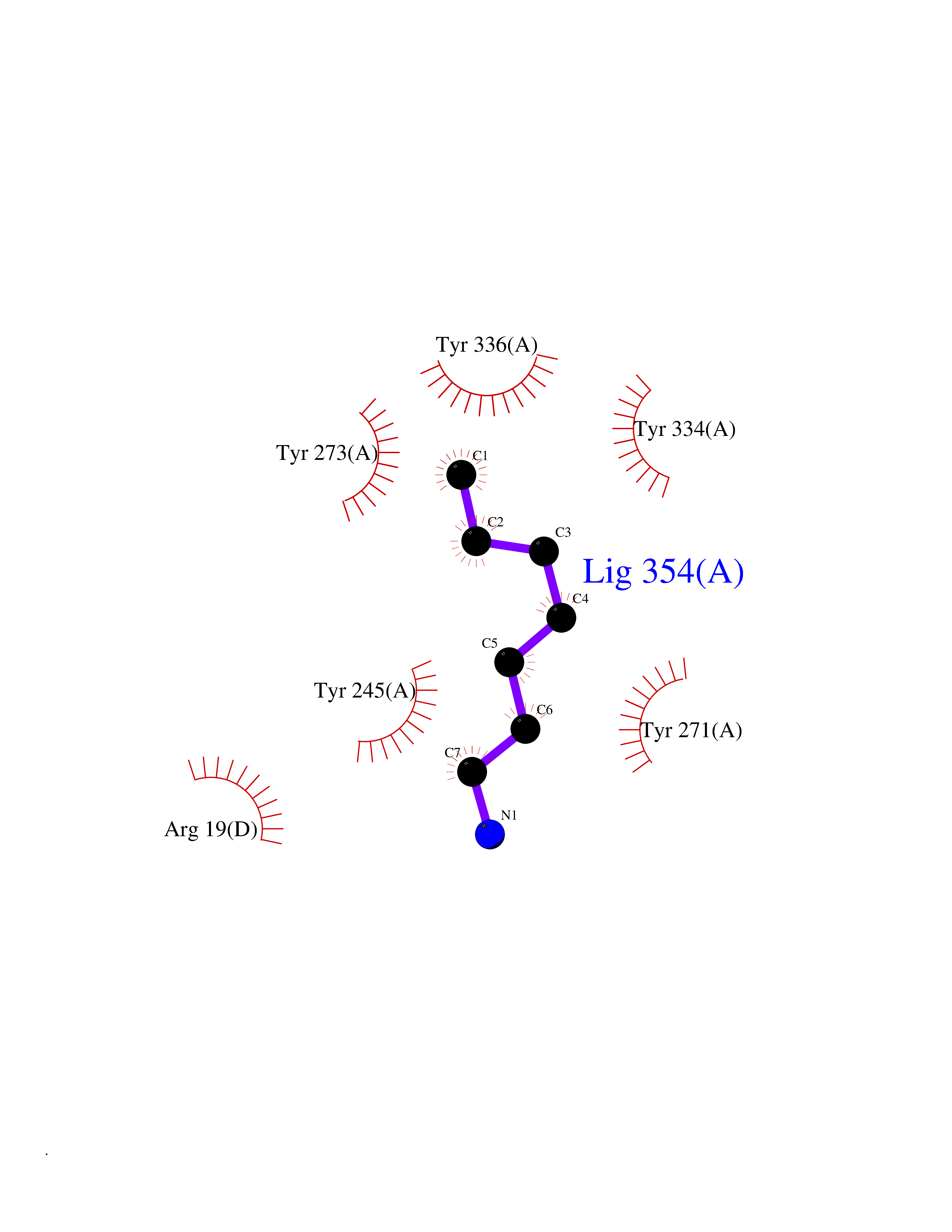 Ligplot Map 3D Binding mode Sequence KSKAELQSEERKRIDELIESGKEEGMKIDLIDGKGRGVIATKQFSRGDFVVEYHGDLIEITDAKKREALYAQDPSTGCYMYYFQYLSKTYCVDATRETNRLGRLINHSKSGNCQTKLHDIDGVPHLILIASRDIAAGEELLYDYGDRSKASIEAHPWLKHKRHRVLR Hydrogen bonds contact Hydrophobic contact | ||||
| 4 | Cyclin-dependent kinase 4 | 2W96 | 5.18 | |
Target general information Gen name CDK4 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Protein kinase superfamily, CMGC Ser/Thr protein kinase family, CDC2/CDKX subfamily Biochemical class Cell cycle Function ATP binding.Cyclin binding.Cyclin-dependent protein serine/threonine kinase activity.Cyclin-dependent protein serine/threonine kinase regulator activity.Protein complex binding. Related diseases Melanoma, cutaneous malignant 3 (CMM3) [MIM:609048]: A malignant neoplasm of melanocytes, arising de novo or from a pre-existing benign nevus, which occurs most often in the skin but may also involve other sites. {ECO:0000269|PubMed:7652577, ECO:0000269|PubMed:8528263, ECO:0000269|PubMed:9311594, ECO:0000269|PubMed:9425228}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB12001; DB03496; DB12010; DB09073; DB02733; DB11730; DB15442 Interacts with Q9UH17; P24385; P30279; P30281; Q16543; P50613; P38936; P46527; P49918; P42771; P42772; P42773; P55273; Q9UJC3; P08238; Q9UKT9; Q0VD86; P01106; Q9ULD0; P28749; Q08999; P09936; Q8N720 EC number 2.7.11.22 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; ATP-binding; Cell cycle; Cell division; Cytoplasm; Disease variant; Kinase; Membrane; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Serine/threonine-protein kinase; Transferase Protein physicochemical properties Chain ID B Molecular weight (Da) 30138.4 Length 267 Aromaticity 0.09 Instability index 36.2 Isoelectric point 5.78 Charge (pH=7) -5.83 2D Binding mode Binding energy (Kcal/mol) -7.06 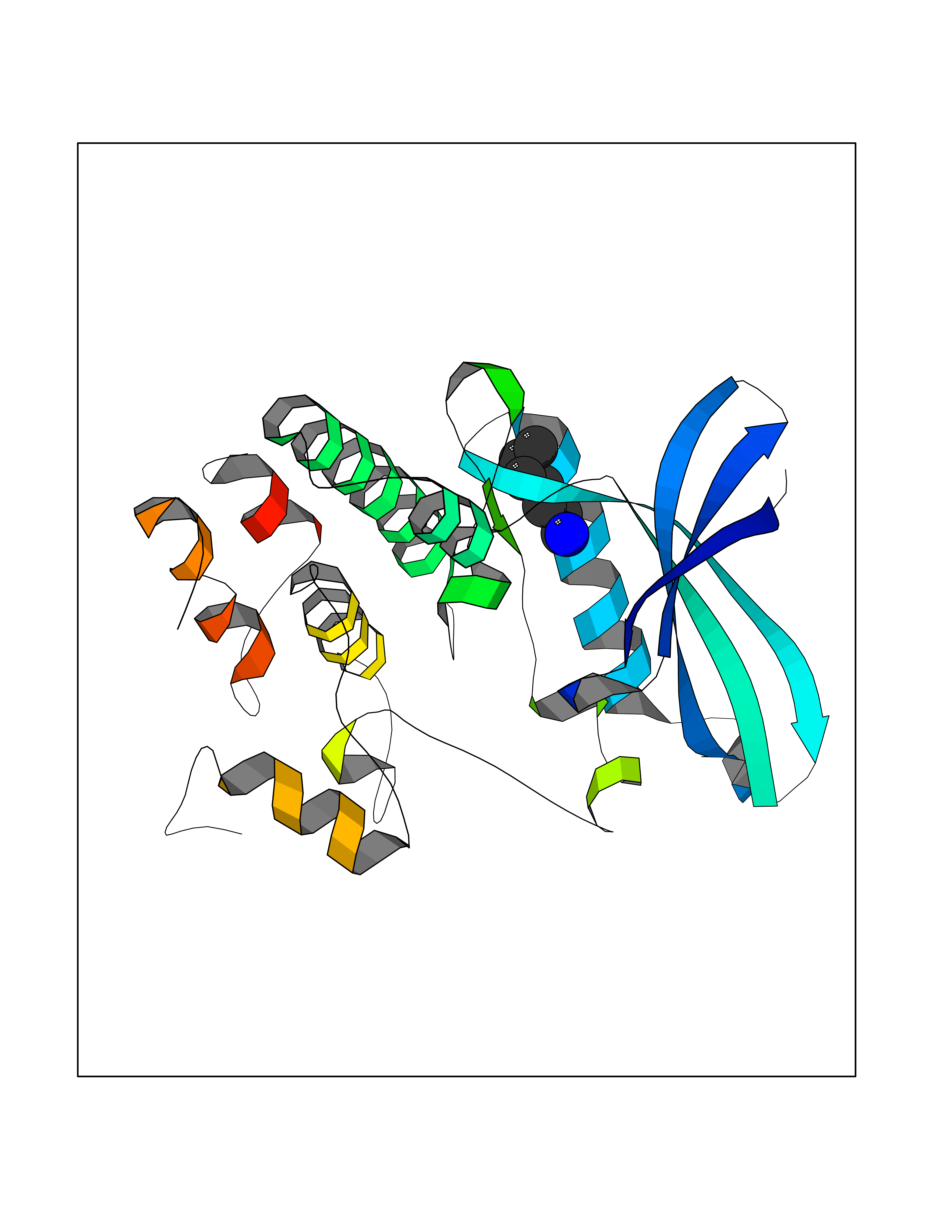 Molscript Map 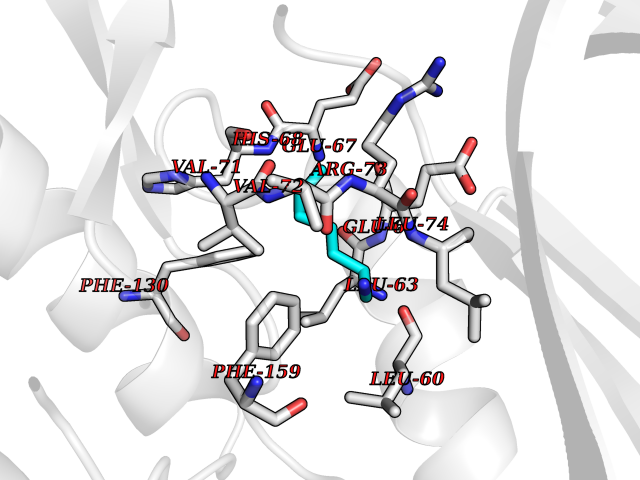 Pymol Map 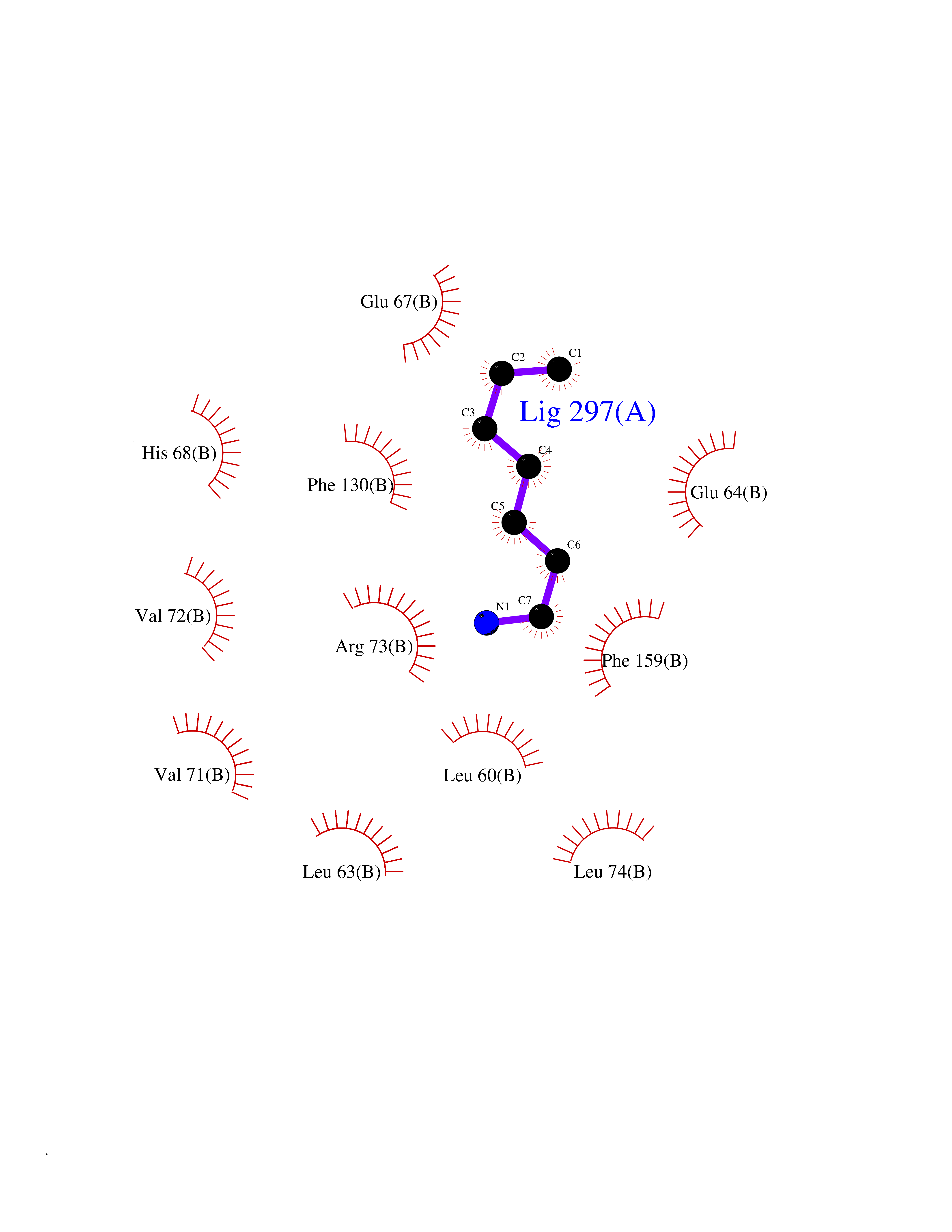 Ligplot Map 3D Binding mode Sequence SRYEPVAEIGVGAYGTVYKARDPHSGHFVALKSVRVPNGEEGLPISTVREVALLRRLEAFEHPNVVRLMDVCATSRTDREIKVTLVFEHVDQDLRTYLDKAPPPGLPAETIKDLMRQFLRGLDFLHANCIVHRDLKPENILVTSGGTVKLADFGLARIYSYQMALDPVVVTLWYRAPEVLLQSTYATPVDMWSVGCIFAEMFRRKPLFCGNSEADQLGKIFDLIGLPPEDDWVPEMEESGAQLLLEMLTFNPHKRISAFRALQHSYL Hydrogen bonds contact Hydrophobic contact | ||||
| 5 | Fms-like tyrosine kinase 3 (FLT-3) | 1RJB | 5.18 | |
Target general information Gen name FLT3 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Stem cell tyrosine kinase 1; STK1; STK-1; Receptor-type tyrosine-protein kinase FLT3; Fetal liver kinase-2; FLT-3; FLK2; FLK-2; FL cytokine receptor; CD135 Protein family Protein kinase superfamily, Tyr protein kinase family, CSF-1/PDGF receptor subfamily Biochemical class Kinase Function Tyrosine-protein kinase that acts as cell-surface receptor for the cytokine FLT3LG and regulates differentiation, proliferation and survival of hematopoietic progenitor cells and of dendritic cells. Promotes phosphorylation of SHC1 and AKT1, and activation of the downstream effector MTOR. Promotes activation of RAS signaling and phosphorylation of downstream kinases, including MAPK1/ERK2 and/or MAPK3/ERK1. Promotes phosphorylation of FES, FER, PTPN6/SHP, PTPN11/SHP-2, PLCG1, and STAT5A and/or STAT5B. Activation of wild-type FLT3 causes only marginal activation of STAT5A or STAT5B. Mutations that cause constitutive kinase activity promote cell proliferation and resistance to apoptosis via the activation of multiple signaling pathways. Related diseases Leukemia, acute myelogenous (AML) [MIM:601626]: A subtype of acute leukemia, a cancer of the white blood cells. AML is a malignant disease of bone marrow characterized by maturational arrest of hematopoietic precursors at an early stage of development. Clonal expansion of myeloid blasts occurs in bone marrow, blood, and other tissue. Myelogenous leukemias develop from changes in cells that normally produce neutrophils, basophils, eosinophils and monocytes. {ECO:0000269|PubMed:11090077, ECO:0000269|PubMed:11290608, ECO:0000269|PubMed:11442493, ECO:0000269|PubMed:14504097, ECO:0000269|PubMed:16266983, ECO:0000269|PubMed:18305215, ECO:0000269|PubMed:8946930, ECO:0000269|PubMed:9737679}. The gene represented in this entry may be involved in disease pathogenesis. Somatic mutations that lead to constitutive activation of FLT3 are frequent in AML patients. These mutations fall into two classes, the most common being in-frame internal tandem duplications of variable length in the juxtamembrane region that disrupt the normal regulation of the kinase activity. Likewise, point mutations in the activation loop of the kinase domain can result in a constitutively activated kinase. Drugs (DrugBank ID) DB12742; DB12267; DB12500; DB12010; DB12141; DB06469; DB06080; DB06595; DB11763; DB09079; DB11697; DB12978; DB08901; DB15822; DB12874; DB00398; DB01268; DB05465; DB11800; DB05014 Interacts with P00519; P42684; P46108; P46109; P06241; Q13322; Q9Y6K9; P06239; P27986; P20936; P43405; Q8R4L0 EC number EC 2.7.10.1 Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Disease variant; Disulfide bond; Endoplasmic reticulum; Glycoprotein; Host-virus interaction; Immunoglobulin domain; Kinase; Membrane; Nucleotide-binding; Phosphoprotein; Proteomics identification; Proto-oncogene; Receptor; Reference proteome; Signal; Transferase; Transmembrane; Transmembrane helix; Tyrosine-protein kinase; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 34209.2 Length 298 Aromaticity 0.14 Instability index 39.68 Isoelectric point 5.57 Charge (pH=7) -4.02 2D Binding mode Binding energy (Kcal/mol) -7.06  Molscript Map 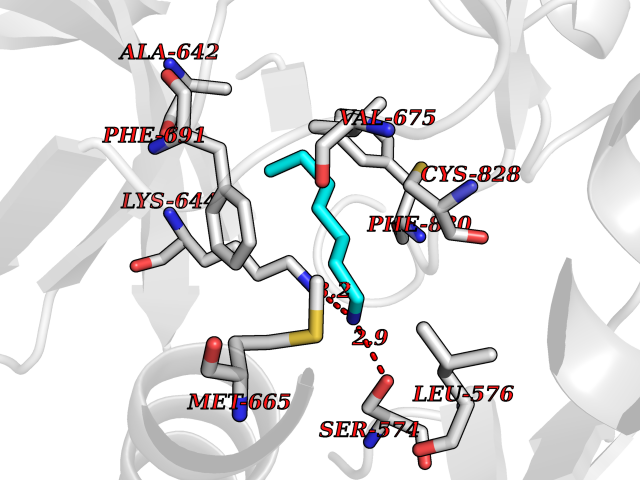 Pymol Map 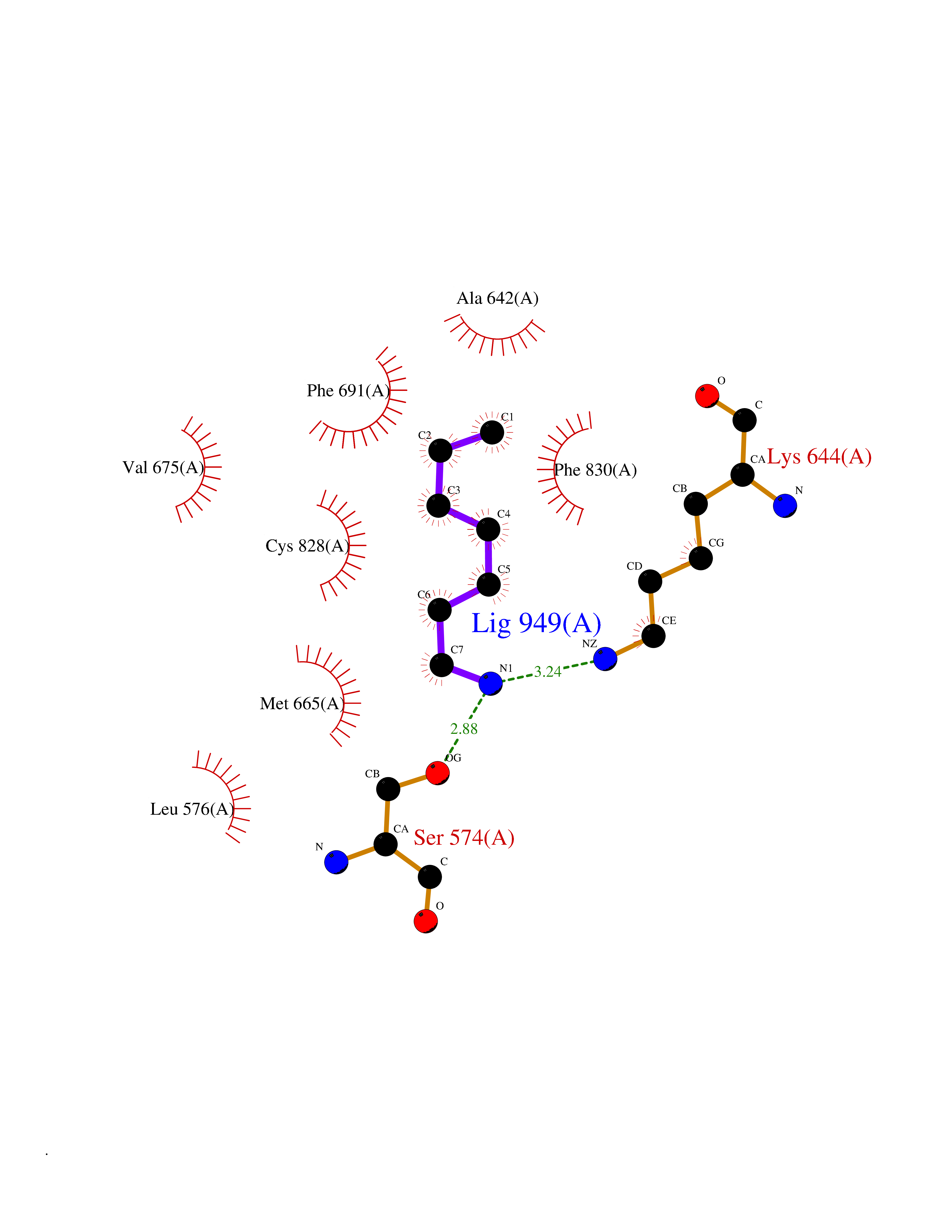 Ligplot Map 3D Binding mode Sequence YESQLQMVQVTGSSDNEYFYVDFREYEYDLKWEFPRENLEFGKVLGSGAFGKVMNATAYGISKTGVSIQVAVKMLKEREALMSELKMMTQLGSHENIVNLLGACTLSGPIYLIFEYCCYGDLLNYLRSKREKFLTFEDLLCFAYQVAKGMEFLEFKSCVHRDLAARNVLVTHGKVVKICDFGLARDIMSDSNYVVRGNARLPVKWMAPESLFEGIYTIKSDVWSYGILLWEIFSLGVNPYPGIPVDANFYKLIQNGFKMDQPFYATEEIYIIMQSCWAFDSRKRPSFPNLTSFLGCQL Hydrogen bonds contact Hydrophobic contact | ||||
| 6 | S-adenosylmethionine decarboxylase proenzyme (AMD1) | 1JL0 | 5.16 | |
Target general information Gen name AMD1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms SamDC; S-adenosylmethioninedecarboxylase; AdoMetDC; AMD Protein family Eukaryotic AdoMetDC family Biochemical class Carbon-carbon lyase Function Promotes maintenance and self-renewal of embryonic stem cells, by maintaining spermine levels. Essential for biosynthesis of the polyamines spermidine and spermine. Related diseases Niemann-Pick disease A (NPDA) [MIM:257200]: An early-onset lysosomal storage disorder caused by failure to hydrolyze sphingomyelin to ceramide. It results in the accumulation of sphingomyelin and other metabolically related lipids in reticuloendothelial and other cell types throughout the body, leading to cell death. Niemann-Pick disease type A is a primarily neurodegenerative disorder characterized by onset within the first year of life, intellectual disability, digestive disorders, failure to thrive, major hepatosplenomegaly, and severe neurologic symptoms. The severe neurological disorders and pulmonary infections lead to an early death, often around the age of four. Clinical features are variable. A phenotypic continuum exists between type A (basic neurovisceral) and type B (purely visceral) forms of Niemann-Pick disease, and the intermediate types encompass a cluster of variants combining clinical features of both types A and B. {ECO:0000269|PubMed:12556236, ECO:0000269|PubMed:1391960, ECO:0000269|PubMed:15221801, ECO:0000269|PubMed:15877209, ECO:0000269|PubMed:1618760, ECO:0000269|PubMed:1718266, ECO:0000269|PubMed:18815062, ECO:0000269|PubMed:19405096, ECO:0000269|PubMed:2023926, ECO:0000269|PubMed:20386867, ECO:0000269|PubMed:22818240, ECO:0000269|PubMed:23252888, ECO:0000269|PubMed:23430884, ECO:0000269|PubMed:26499107, ECO:0000269|PubMed:27338287, ECO:0000269|PubMed:8680412, ECO:0000269|PubMed:8693491, ECO:0000269|PubMed:9266408, ECO:0000269|PubMed:9660788}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Niemann-Pick disease B (NPDB) [MIM:607616]: A late-onset lysosomal storage disorder caused by failure to hydrolyze sphingomyelin to ceramide. It results in the accumulation of sphingomyelin and other metabolically related lipids in reticuloendothelial and other cell types throughout the body, leading to cell death. Clinical signs involve only visceral organs. The most constant sign is hepatosplenomegaly which can be associated with pulmonary symptoms. Patients remain free of neurologic manifestations. However, a phenotypic continuum exists between type A (basic neurovisceral) and type B (purely visceral) forms of Niemann-Pick disease, and the intermediate types encompass a cluster of variants combining clinical features of both types A and B. In Niemann-Pick disease type B, onset of the first symptoms occurs in early childhood and patients can survive into adulthood. {ECO:0000269|PubMed:12369017, ECO:0000269|PubMed:12556236, ECO:0000269|PubMed:1301192, ECO:0000269|PubMed:15241805, ECO:0000269|PubMed:16010684, ECO:0000269|PubMed:1618760, ECO:0000269|PubMed:16472269, ECO:0000269|PubMed:18815062, ECO:0000269|PubMed:1885770, ECO:0000269|PubMed:19050888, ECO:0000269|PubMed:19405096, ECO:0000269|PubMed:20386867, ECO:0000269|PubMed:21098024, ECO:0000269|PubMed:21621718, ECO:0000269|PubMed:22613662, ECO:0000269|PubMed:22818240, ECO:0000269|PubMed:23252888, ECO:0000269|PubMed:23430512, ECO:0000269|PubMed:25920558, ECO:0000269|PubMed:26084044, ECO:0000269|PubMed:26499107, ECO:0000269|PubMed:27338287, ECO:0000269|PubMed:27659707, ECO:0000269|PubMed:8051942, ECO:0000269|PubMed:8664904}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08163; DB00118; DB01917 Interacts with P17707; Q96A98; Q8WY91 EC number EC 4.1.1.50 Uniprot keywords 3D-structure; Alternative splicing; Autocatalytic cleavage; Decarboxylase; Direct protein sequencing; Lyase; Phosphoprotein; Polyamine biosynthesis; Proteomics identification; Pyruvate; Reference proteome; S-adenosyl-L-methionine; Schiff base; Spermidine biosynthesis; Zymogen Protein physicochemical properties Chain ID A,B Molecular weight (Da) 35790.5 Length 311 Aromaticity 0.14 Instability index 39.47 Isoelectric point 6.03 Charge (pH=7) -2.01 2D Binding mode Binding energy (Kcal/mol) -7.04  Molscript Map 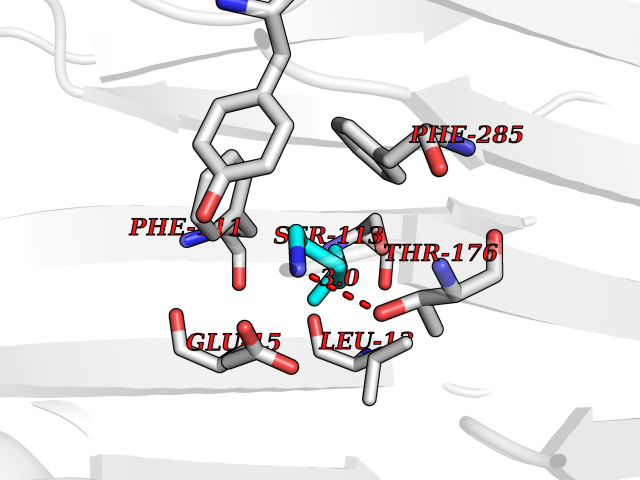 Pymol Map 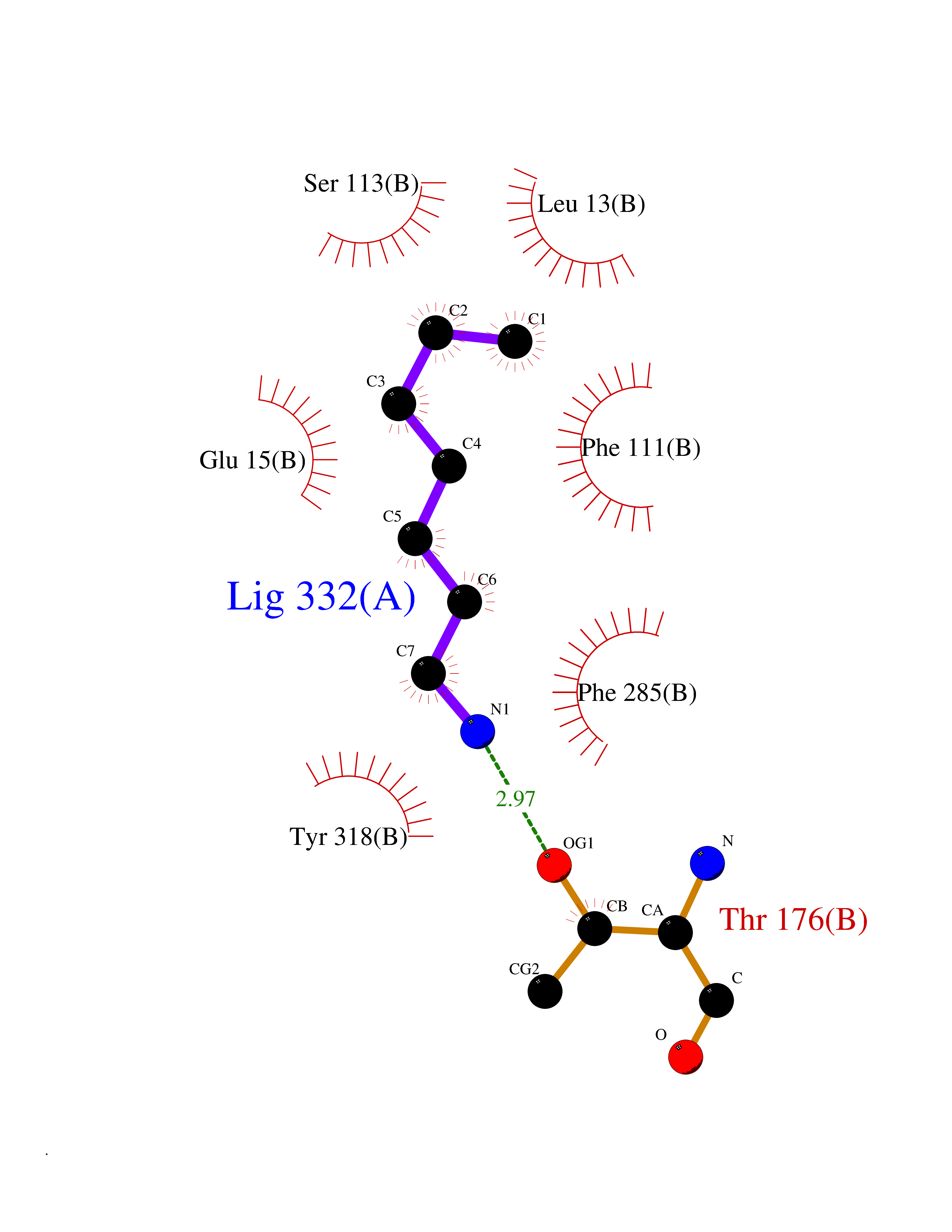 Ligplot Map 3D Binding mode Sequence HFFEGTEKLLEVWFSRQGSGDLRTIPRSEWDILLKDVQCSIISVTKTDKQEAYVLSESSMFVSKRRFILKTCGTTLLLKALVPLLKLARDYSGFDSIQSFFYSRKNFMKPSHQGYPHRNFQEEIEFLNAIFPNGAGYCMGRMNSDCWYLYTLDFRVISQPDQTLEILMSELDPAVMDQFYMKDGVTAKDVTRESGIRDLIPGSVIDATMFNPCGYSMNGMKSDGTYWTIAITPEPEFSYVSFETNLSQTSYDDLIRKVVEVFKPGKFVTTLFVNQSSKCPQKIEGFKRLDCQSAMFNDYNFVFTSFAKKQQ Hydrogen bonds contact Hydrophobic contact | ||||
| 7 | Helicobacter pylori Methylthioadenosine nucleosidase (HELPY mtnN) | 4BMZ | 5.15 | |
Target general information Gen name HELPY mtnN Organism Helicobacter pylori (strain ATCC 700392 / 26695) (Campylobacter pylori) Uniprot ID TTD ID Synonyms MTAN; MTA/SAH nucleosidase; Aminofutalosine nucleosidase; Aminodeoxyfutalosine nucleosidase; AFL nucleosidase; 6-amino-6-deoxyfutalosine N-ribosylhydrolase; 5'-methylthioadenosine/S-adenosylhomocystei Protein family PNP/UDP phosphorylase family Biochemical class NA Function Catalyzes the direct conversion of aminodeoxyfutalosine (AFL) into dehypoxanthine futalosine (DHFL) and adenine via the hydrolysis of the N-glycosidic bond; this reaction seems to represent an essential step in the menaquinone biosynthesis pathway in Helicobacter species. Can also probably catalyzes the hydrolysis of 5'-methylthioadenosine (MTA) and S-adenosylhomocysteine (SAH) to adenine and the corresponding thioribose, 5'-methylthioribose and S-ribosylhomocysteine, respectively. These other activities highlight the tremendous versatility of the enzyme, which also plays key roles in S-adenosylmethionine recycling and in the biosynthesis of the quorum-sensing molecule autoinducer-2. Does not act on futalosine (FL) as substrate. Related diseases Progressive familial heart block 1B (PFHB1B) [MIM:604559]: A cardiac bundle branch disorder characterized by progressive alteration of cardiac conduction through the His-Purkinje system, with a pattern of a right bundle-branch block and/or left anterior hemiblock occurring individually or together. It leads to complete atrio-ventricular block causing syncope and sudden death. {ECO:0000269|PubMed:19726882, ECO:0000269|PubMed:20562447, ECO:0000269|PubMed:21887725}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Erythrokeratodermia variabilis et progressiva 6 (EKVP6) [MIM:618531]: A form of erythrokeratodermia variabilis et progressiva, a genodermatosis characterized by the coexistence of two independent skin lesions: transient erythema and hyperkeratosis that is usually localized but occasionally occurs in its generalized form. Clinical presentation varies significantly within a family and from one family to another. Palmoplantar keratoderma is present in around 50% of cases. EKVP6 inheritance is autosomal dominant. {ECO:0000269|PubMed:30528822}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number NA Uniprot keywords 3D-structure; Amino-acid biosynthesis; Hydrolase; Menaquinone biosynthesis; Methionine biosynthesis; Reference proteome Protein physicochemical properties Chain ID A,B Molecular weight (Da) 50547.6 Length 464 Aromaticity 0.08 Instability index 26.92 Isoelectric point 5.13 Charge (pH=7) -20.92 2D Binding mode Binding energy (Kcal/mol) -7.03  Molscript Map 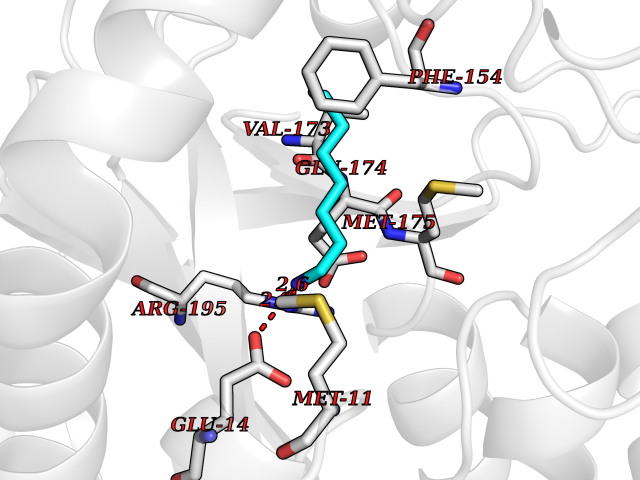 Pymol Map 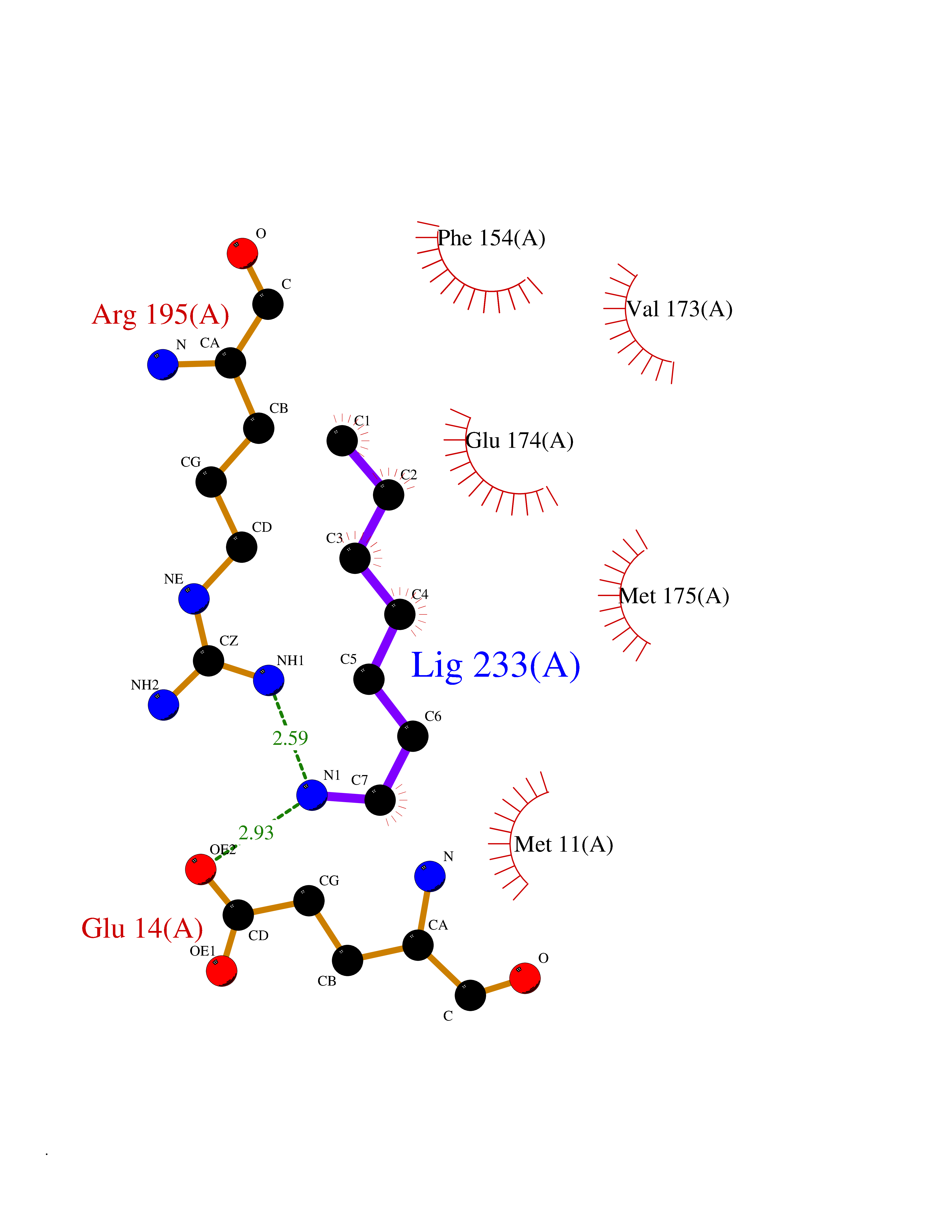 Ligplot Map 3D Binding mode Sequence VQKIGILGAMREEITPILELFGVDFEEIPLGGNVFHKGVYHNKEIIVAYSKIGKVHSTLTTTSMILAFGVQKVLFSGVAGSLVKDLKINDLLVAIQLVQHDVDLSAFDHPLGFIPESAIFIETSESLNALAKEVANEQHIVLKEGVIASGDQFVHSKERKEFLVSEFKASAVEMEGASVAFVCQKFGVPCCVLRSISNNADEEANMSFDAFLEKSAQTSAKFLKSMVDELGSHMVQKIGILGAMREEITPILELFGVDFEEIPLGGNVFHKGVYHNKEIIVAYSKIGKVHSTLTTTSMILAFGVQKVLFSGVAGSLVKDLKINDLLVAIQLVQHDVDLSAFDHPLGFIPESAIFIETSESLNALAKEVANEQHIVLKEGVIASGDQFVHSKERKEFLVSEFKASAVEMEGASVAFVCQKFGVPCCVLRSISNNADEEANMSFDAFLEKSAQTSAKFLKSMVDEL Hydrogen bonds contact Hydrophobic contact | ||||
| 8 | Thymidine kinase 1 (TK1) | 1W4R | 5.14 | |
Target general information Gen name TK1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Thymidine kinase, cytosolic Protein family Thymidine kinase family Biochemical class Kinase Function cytosol, identical protein binding, thymidine kinase activity, zinc ion binding, DNA metabolic process, nucleobase-containing compound metabolic process, protein homotetramerization, pyrimidine nucleoside salvage, thymidine metabolic process Related diseases Seizures, benign familial infantile, 3 (BFIS3) [MIM:607745]: A form of benign familial infantile epilepsy, a neurologic disorder characterized by afebrile seizures occurring in clusters during the first year of life, without neurologic sequelae. BFIS3 inheritance is autosomal dominant. {ECO:0000269|PubMed:11371648, ECO:0000269|PubMed:12243921, ECO:0000269|PubMed:15048894, ECO:0000269|PubMed:16417554, ECO:0000269|PubMed:17021166, ECO:0000269|PubMed:17386050, ECO:0000269|PubMed:18479388, ECO:0000269|PubMed:20371507, ECO:0000269|PubMed:22612257, ECO:0000269|PubMed:23360469, ECO:0000269|PubMed:23758435, ECO:0000269|PubMed:25982755, ECO:0000269|PubMed:26291284, ECO:0000269|PubMed:29844171, ECO:0000269|PubMed:30144217}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Developmental and epileptic encephalopathy 11 (DEE11) [MIM:613721]: An autosomal dominant seizure disorder characterized by neonatal or infantile onset of refractory seizures with resultant delayed neurologic development and persistent neurologic abnormalities. Patients may progress to West syndrome, which is characterized by tonic spasms with clustering, arrest of psychomotor development, and hypsarrhythmia on EEG. {ECO:0000269|PubMed:19783390, ECO:0000269|PubMed:19786696, ECO:0000269|PubMed:20956790, ECO:0000269|PubMed:22677033, ECO:0000269|PubMed:23033978, ECO:0000269|PubMed:23195492, ECO:0000269|PubMed:23550958, ECO:0000269|PubMed:23662938, ECO:0000269|PubMed:23708187, ECO:0000269|PubMed:23935176, ECO:0000269|PubMed:23988467, ECO:0000269|PubMed:24463883, ECO:0000269|PubMed:24579881, ECO:0000269|PubMed:24659627, ECO:0000269|PubMed:24710820, ECO:0000269|PubMed:25457084, ECO:0000269|PubMed:25459969, ECO:0000269|PubMed:25772804, ECO:0000269|PubMed:25818041, ECO:0000269|PubMed:26138355, ECO:0000269|PubMed:26291284, ECO:0000269|PubMed:26993267, ECO:0000269|PubMed:27864847, ECO:0000269|PubMed:29625812, ECO:0000269|PubMed:29844171, ECO:0000269|PubMed:30144217, ECO:0000269|PubMed:30415926}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Defects in SCN2A are associated with genetic epilepsy with febrile seizures plus (GEFS+), a familial autosomal dominant epilepsy syndrome, a clinical subset of febrile seizures, characterized by frequent episodes after 6 years of age and various types of subsequent epilepsy. {ECO:0000269|PubMed:29635106}.; DISEASE: Defects in SCN2A are associated with autism spectrum disorders (ASD). It seems that mutations resulting in sodium channel gain of function and increased neuron excitability lead to infantile seizures, whereas variants resulting in sodium channel loss of function and decrease neuron excitability are associated with ASD. {ECO:0000269|PubMed:28256214}.; DISEASE: Episodic ataxia 9 (EA9) [MIM:618924]: An autosomal dominant neurologic disorder characterized by episodic ataxia manifesting in the first years of life, early-onset seizures, difficulty walking, dizziness, slurred speech, headache, vomiting, and pain. The duration of ataxic episodes is heterogeneous. Most patients show episodes lasting minutes to maximum several hours, but periods lasting days up to weeks have been reported. Some patients have mildly delayed development with speech delay and/or autistic features or mildly impaired intellectual development. {ECO:0000269|PubMed:26645390, ECO:0000269|PubMed:27159988, ECO:0000269|PubMed:27328862, ECO:0000269|PubMed:28065826}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01692; DB04485; DB02452; DB00432; DB00495 Interacts with P05067; A0A087WZT3; Q92993; Q1RN33; P04183 EC number EC 2.7.1.21 Uniprot keywords 3D-structure; Acetylation; ATP-binding; Cytoplasm; DNA synthesis; Kinase; Metal-binding; Nucleotide-binding; Phosphoprotein; Proteomics identification; Reference proteome; Transferase; Ubl conjugation; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 19373.5 Length 174 Aromaticity 0.09 Instability index 36.21 Isoelectric point 8.63 Charge (pH=7) 3.88 2D Binding mode Binding energy (Kcal/mol) -7.01 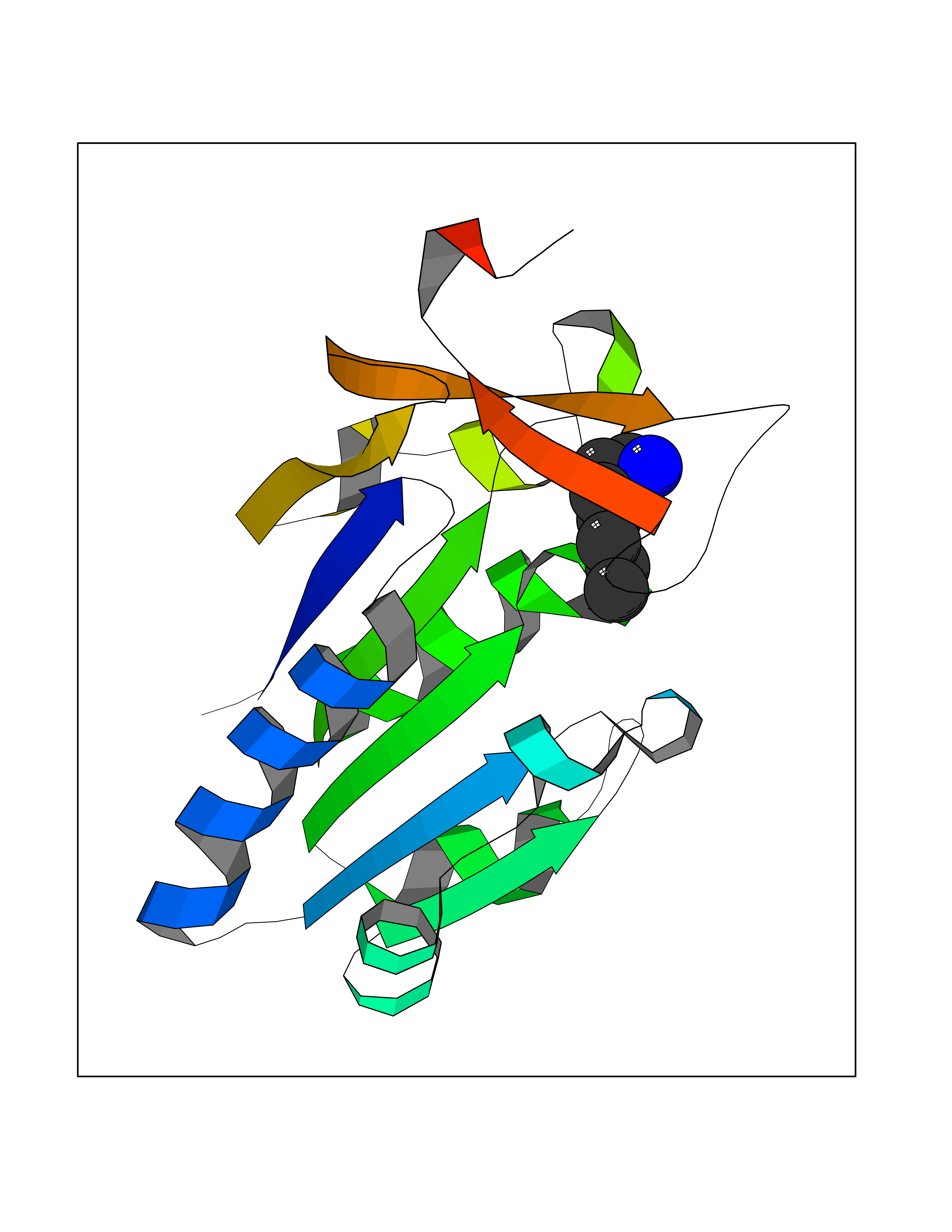 Molscript Map 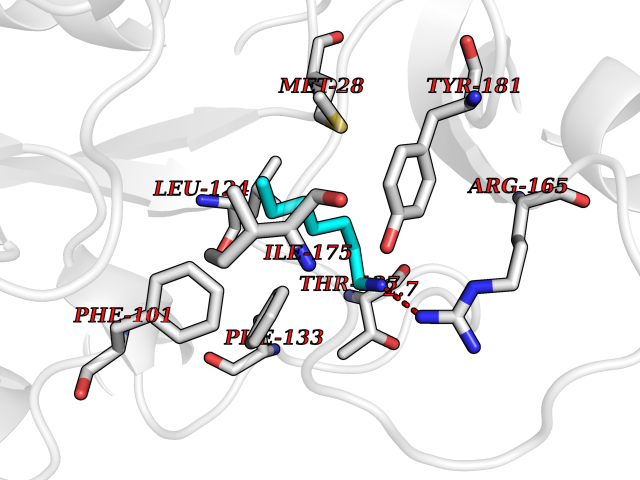 Pymol Map 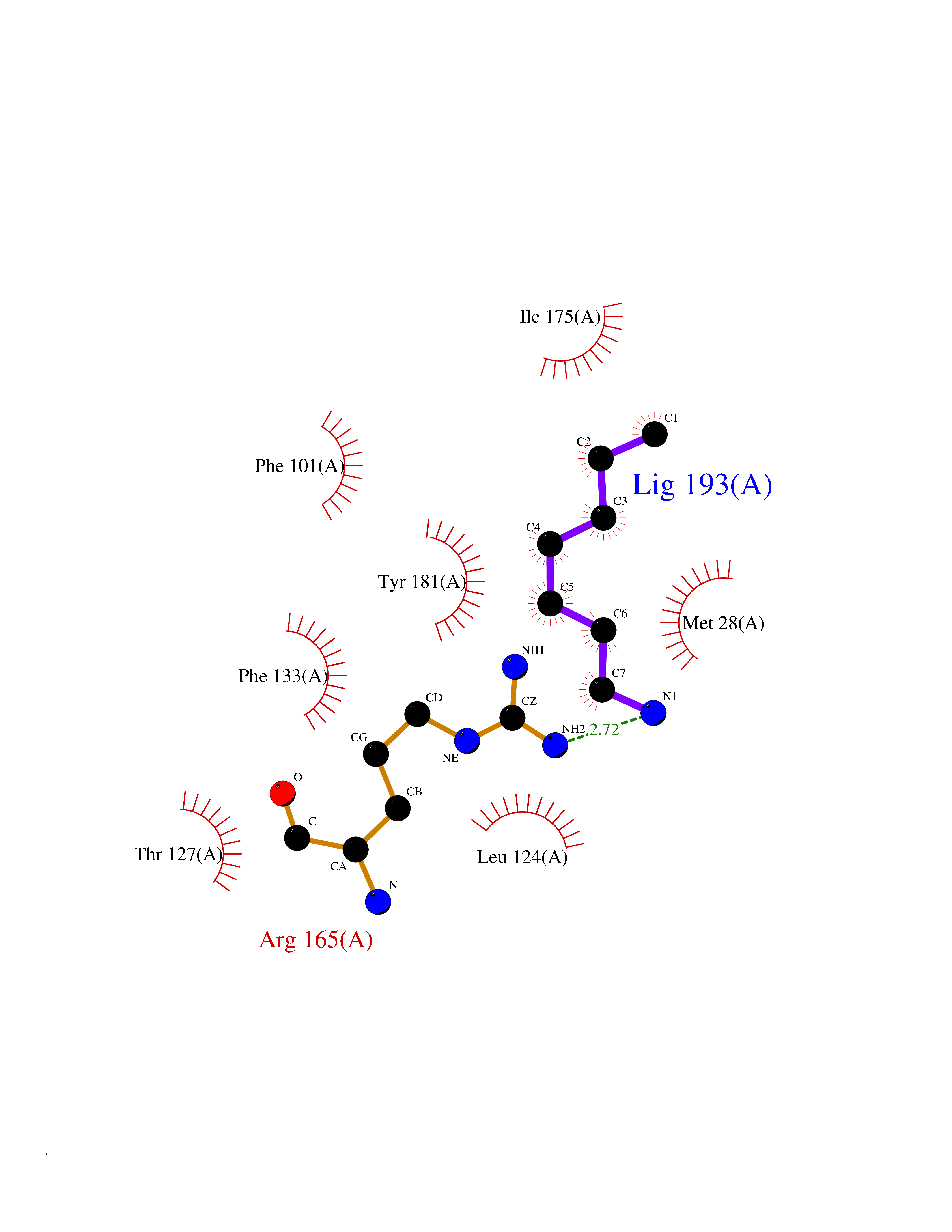 Ligplot Map 3D Binding mode Sequence RGQIQVILGPMFSGKSTELMRRVRRFQIAQYKCLVIKYAKDTRYSSSFCTHDRNTMEALPACLLRDVAQEALGVAVIGIDEGQFFPDIVEFCEAMANAGKTVIVAALDGTFQRKPFGAILNLVPLAESVVKLTAVCMECFREAAYTKRLGTEKEVEVIGGADKYHSVCRLCYFK Hydrogen bonds contact Hydrophobic contact | ||||
| 9 | Neuronal acetylcholine receptor subunit alpha-3 | 4ZK4 | 5.12 | |
Target general information Gen name CHRNA3 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NACHRA3 Protein family Ligand-gated ion channel (TC 1.A.9) family, Acetylcholine receptor (TC 1.A.9.1) subfamily, Alpha-3/CHRNA3 sub-subfamily Biochemical class Acetylcholine binding protein Function Acetylcholine binding.Acetylcholine-gated cation-selective channel activity.Acetylcholine receptor activity.Ligand-gated ion channel activity.Serotonin-gated cation-selective channel activity. Related diseases Bladder dysfunction, autonomic, with impaired pupillary reflex and secondary CAKUT (BAIPRCK) [MIM:191800]: An autosomal recessive disease characterized by impaired innervation and autonomic dysfunction of the urinary bladder, hydronephrosis, vesicoureteral reflux, small kidneys, recurrent urinary tract infections, and progressive renal insufficiency. Additional autonomic features are impaired pupillary reflex and orthostatic hypotension. The disease manifests in utero or early childhood. {ECO:0000269|PubMed:31708116}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00915; DB01156; DB00237; DB00565; DB09028; DB00514; DB07720; DB00898; DB00472; DB05710; DB01227; DB00848; DB00333; DB00184; DB01090; DB00202; DB01273 Interacts with NA EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disease variant; Disulfide bond; Endoplasmic reticulum; Glycoprotein; Golgi apparatus; Ion channel; Ion transport; Ligand-gated ion channel; Membrane; Phosphoprotein; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Signal; Synapse; Transmembrane; Transmembrane helix; Transport; Ubl conjugation Protein physicochemical properties Chain ID A,B,C,D,E Molecular weight (Da) 46391.5 Length 408 Aromaticity 0.12 Instability index 30.23 Isoelectric point 4.6 Charge (pH=7) -22.73 2D Binding mode Binding energy (Kcal/mol) -6.99 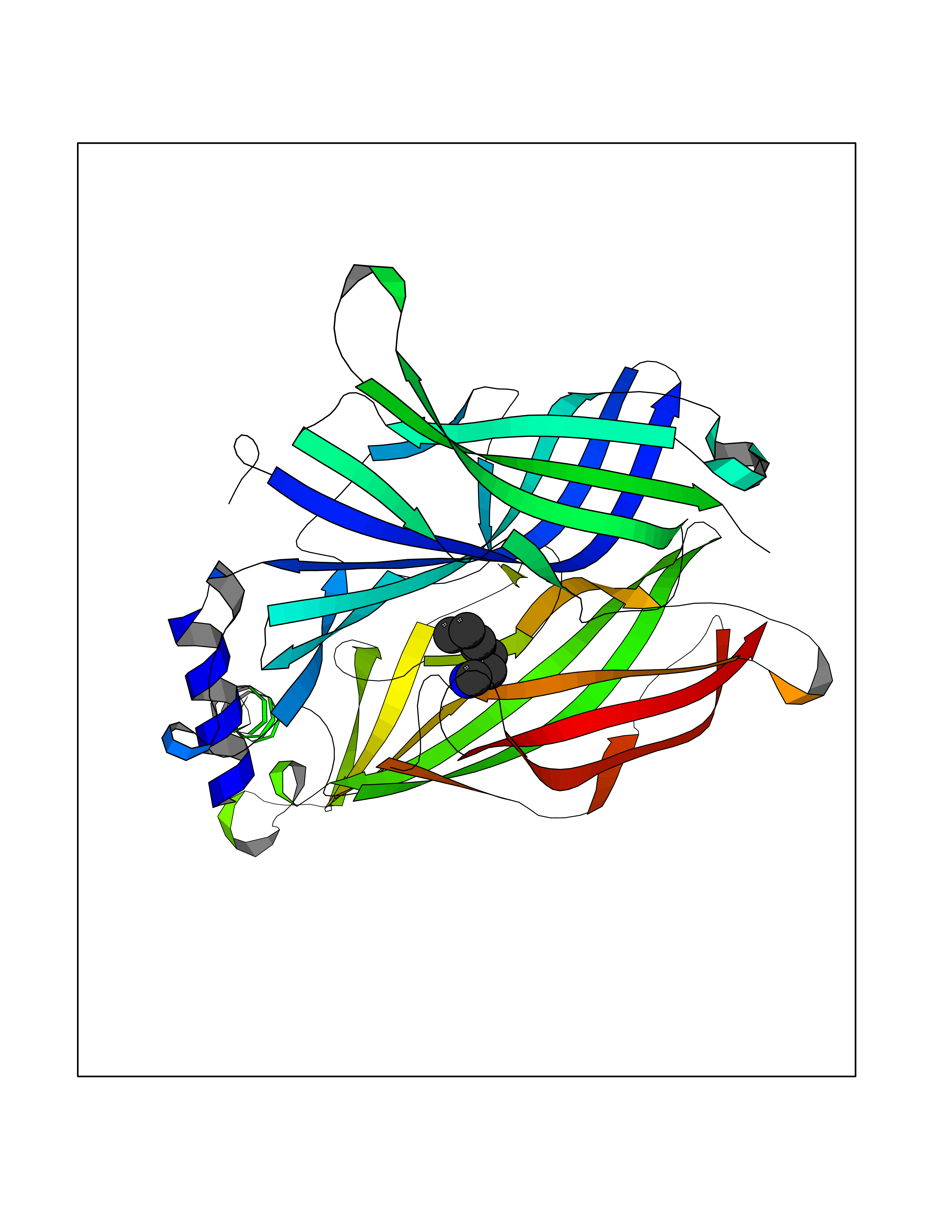 Molscript Map 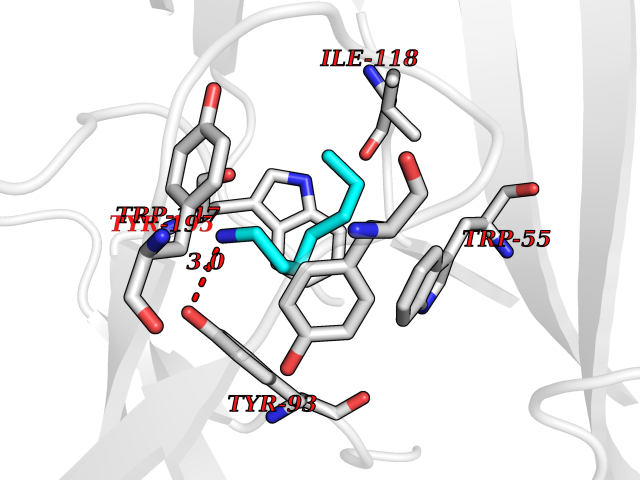 Pymol Map 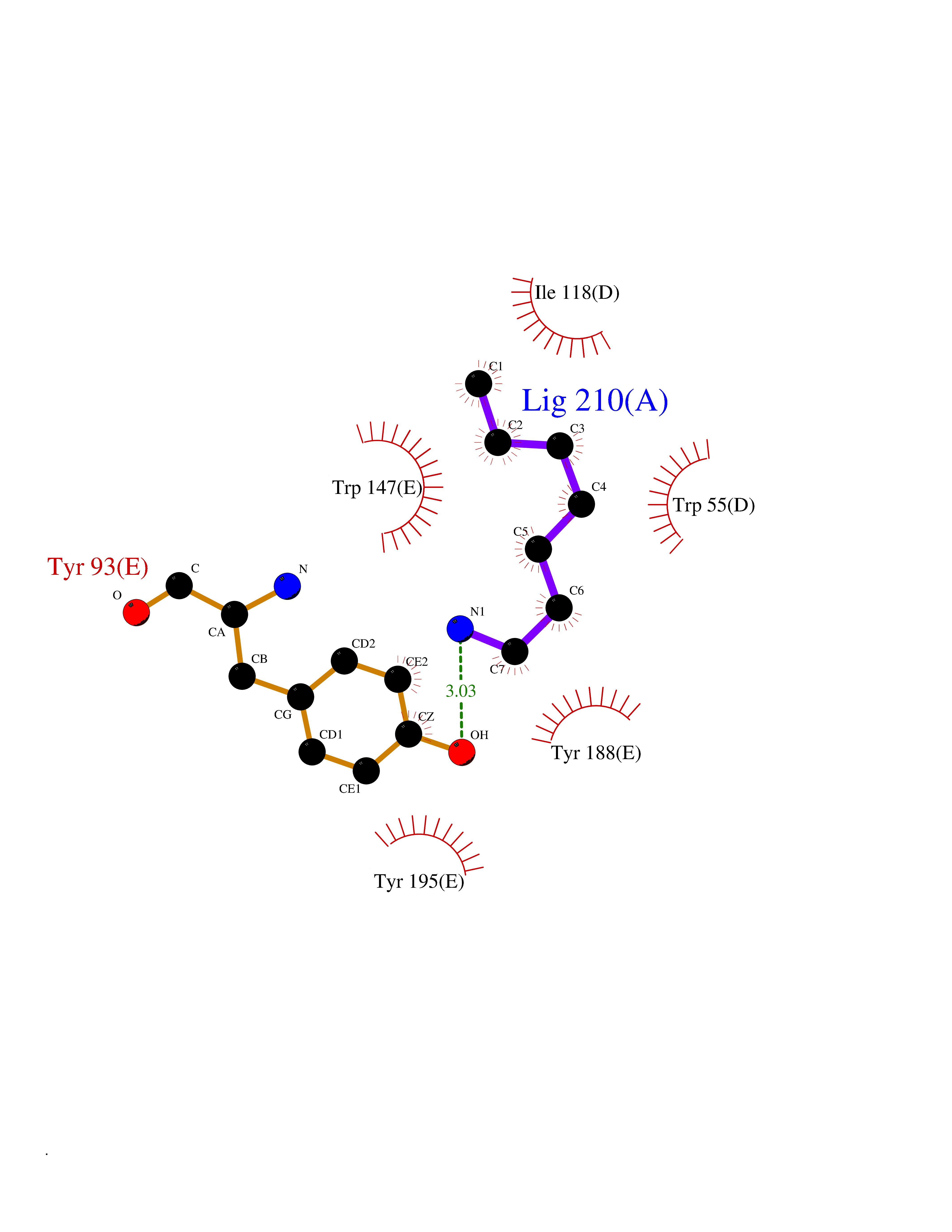 Ligplot Map 3D Binding mode Sequence LHSQANLMRLKSDLFYPGPTKDDPLTVTLGFTLQDIVKADSSTNEVDLVYWEQQRWKLNSLMWDPNEYGNITDFRTSAADIWTPDITAYSSTRPVQVLSPQIAVVTHDGSVMFIPAQRLSFMCDPTGVDSEEGATCAVKFGSWVYSGFEIDLKTDTDQVDLSSYYASSKYEILSATQYKHDIKYNCCEEIYPDVVLVVKFRERRLHSQANLMRLKSDLFNRYPGPTKDDPLTVTLGFTLQDIVKADSSTNEVDLVYWEQQRWKLNSLMWDPNEYGNITDFRTSAADIWTPDITAYSSTRPVQVLSPQIAVVTHDGSVMFIPAQRLSFMCDPTGVDSEEGATCAVKFGSWVYSGFEIDLKTDTDQVDLSSYYASSKYEILSATQYKHDIKYNCCEEIYPDVVLVVKFRE Hydrogen bonds contact Hydrophobic contact | ||||
| 10 | 4-hydroxyphenylpyruvate dioxygenase | 3ISQ | 5.11 | |
Target general information Gen name HPD Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms PPD Protein family 4HPPD family Biochemical class Oxidoreductase Function 4-hydroxyphenylpyruvate dioxygenase activity.Metal ion binding. Related diseases Tyrosinemia 3 (TYRSN3) [MIM:276710]: An inborn error of metabolism characterized by elevations of tyrosine in the blood and urine, seizures and mild intellectual disability. {ECO:0000269|PubMed:10942115, ECO:0000269|PubMed:11073718}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Hawkinsinuria (HWKS) [MIM:140350]: An inborn error of tyrosine metabolism characterized by failure to thrive, persistent metabolic acidosis, fine and sparse hair, and excretion of the unusual cyclic amino acid metabolite, hawkinsin, in the urine. {ECO:0000269|PubMed:11073718}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02850; DB00348 Interacts with NA EC number 1.13.11.27 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Cytoplasm; Dioxygenase; Disease variant; Endoplasmic reticulum; Golgi apparatus; Intellectual disability; Iron; Membrane; Metal-binding; Oxidoreductase; Phenylalanine catabolism; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Tyrosine catabolism Protein physicochemical properties Chain ID A Molecular weight (Da) 43164.8 Length 376 Aromaticity 0.11 Instability index 32.38 Isoelectric point 6.73 Charge (pH=7) -1.04 2D Binding mode Binding energy (Kcal/mol) -6.96 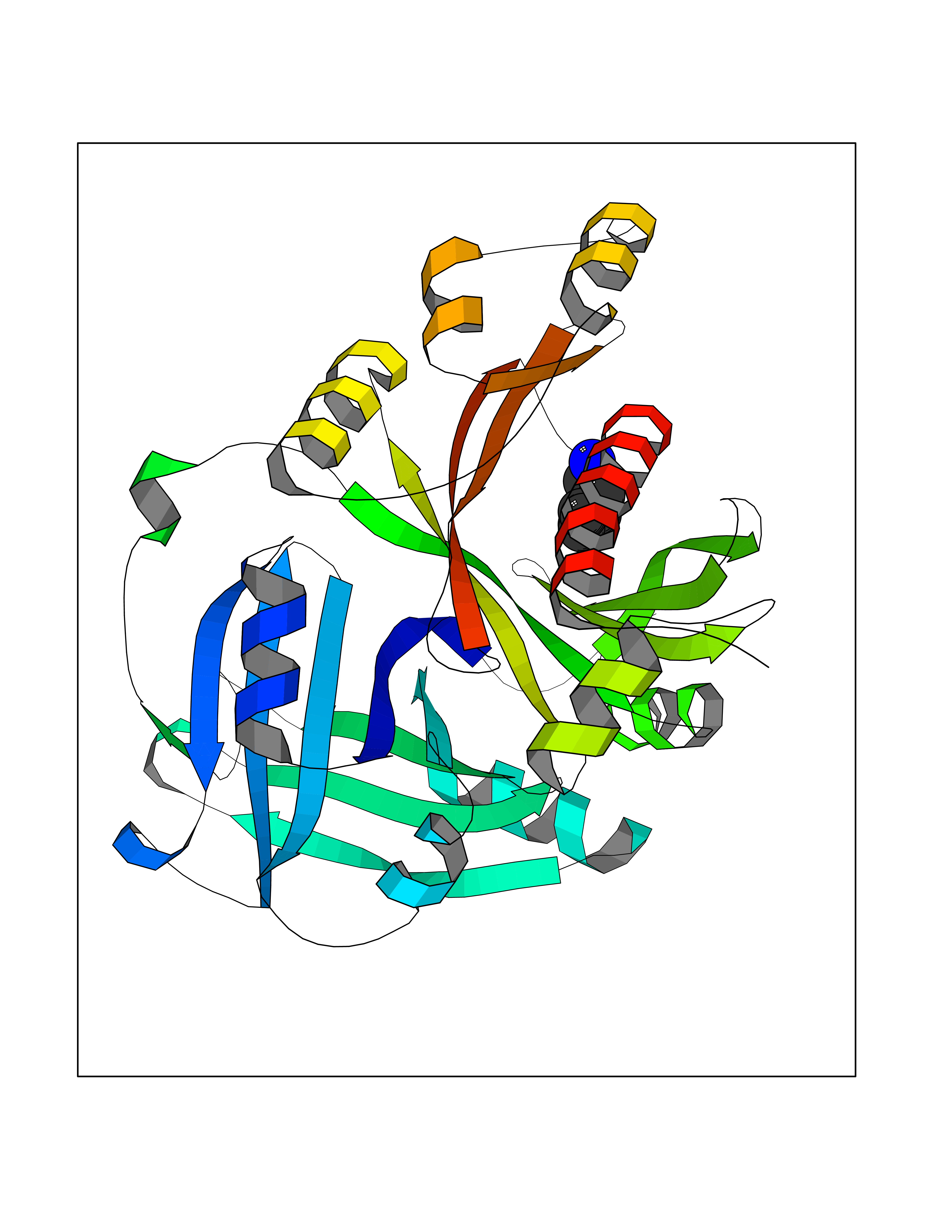 Molscript Map 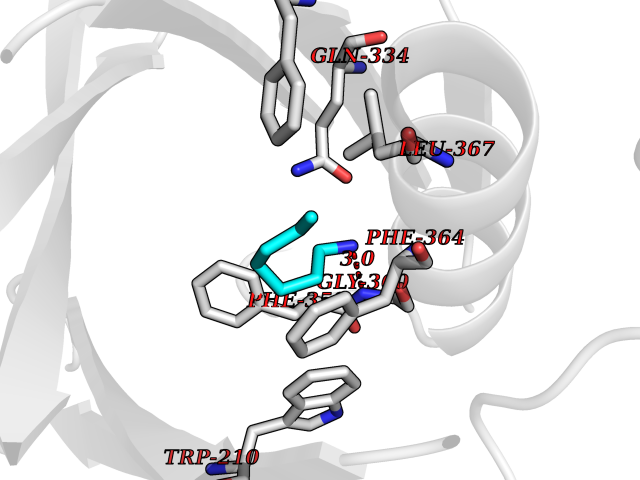 Pymol Map 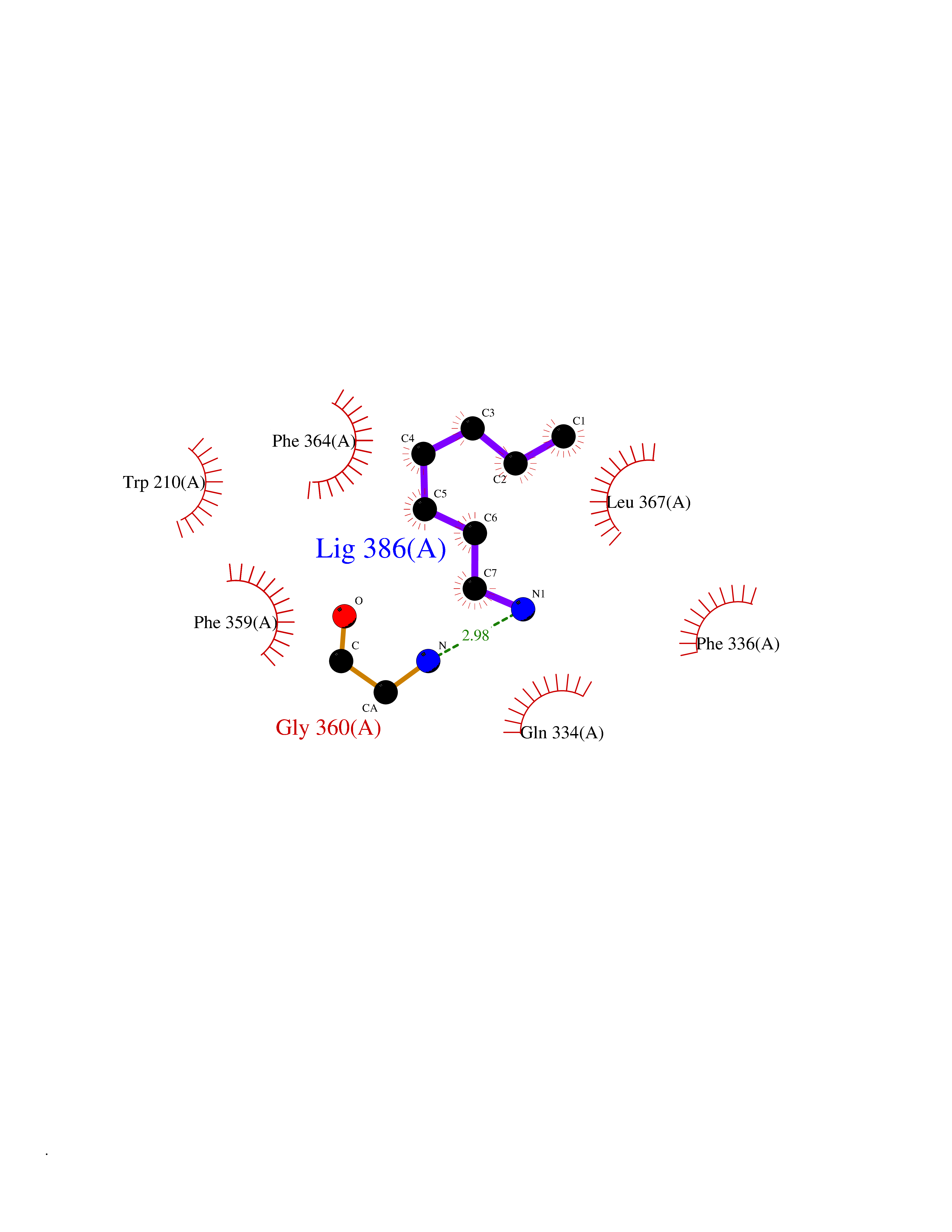 Ligplot Map 3D Binding mode Sequence AKPERGRFLHFHSVTFWVGNAKQAASFYCSKMGFEPLAYRGLETGSREVVSHVIKQGKIVFVLSSALNPWNKEMGDHLVKHGDGVKDIAFEVEDCDYIVQKARERGAKIMREPWVEQDKFGKVKFAVLQTYGDTTHTLVEKMNYIGQFLPGYEAPAFMDPLLPKLPKCSLEMIDHIVGNQPDQEMVSASEWYLKNLQFHRFWSVDDTQVHTEYSSLRSIVVANYEESIKMPINEPAPGKKKSQIQEYVDYNGGAGVQHIALKTEDIITAIRHLRERGLEFLSVPSTYYKQLREKLKTAKIKVKENIDALEELKILVDYDEKGYLLQIFTKPVQDRPTLFLEVIQRHNHQGFGAGNFNSLFKAFEEEQNLRGNLTNM Hydrogen bonds contact Hydrophobic contact | ||||
| 11 | Neuronal acetylcholine receptor subunit alpha-2 | 5FJV | 5.11 | |
Target general information Gen name CHRNA2 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Ligand-gated ion channel (TC 1.A.9) family, Acetylcholine receptor (TC 1.A.9.1) subfamily, Alpha-2/CHRNA2 sub-subfamily Biochemical class NA Function After binding acetylcholine, the AChR responds by an extensive change in conformation that affects all subunits and leads to opening of an ion-conducting channel across the plasma membrane." Related diseases Epilepsy, nocturnal frontal lobe, 4 (ENFL4) [MIM:610353]: An autosomal dominant focal epilepsy characterized by nocturnal seizures associated with fear sensation, tongue movements, and nocturnal wandering, closely resembling nightmares and sleep walking. {ECO:0000269|PubMed:16826524}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Seizures, benign familial infantile, 6 (BFIS6) [MIM:610353]: A form of benign familial infantile epilepsy, a neurologic disorder characterized by afebrile seizures occurring in clusters during the first year of life, without neurologic sequelae. BFIS6 inheritance is autosomal dominant. {ECO:0000269|PubMed:25847220}. The disease may be caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00732; DB00237; DB00411; DB00565; DB01245; DB00514; DB01135; DB07720; DB00898; DB00472; DB00483; DB08960; DB00657; DB01336; DB00416; DB01226; DB00184; DB01337; DB01338; DB00721; DB00728; DB05740; DB00202; DB01199; DB01339 Interacts with NA EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disease variant; Disulfide bond; Epilepsy; Glycoprotein; Ion channel; Ion transport; Ligand-gated ion channel; Membrane; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Signal; Synapse; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A,B,C,D,E Molecular weight (Da) 120584 Length 1031 Aromaticity 0.14 Instability index 32.21 Isoelectric point 5.62 Charge (pH=7) -17.58 2D Binding mode Binding energy (Kcal/mol) -6.97 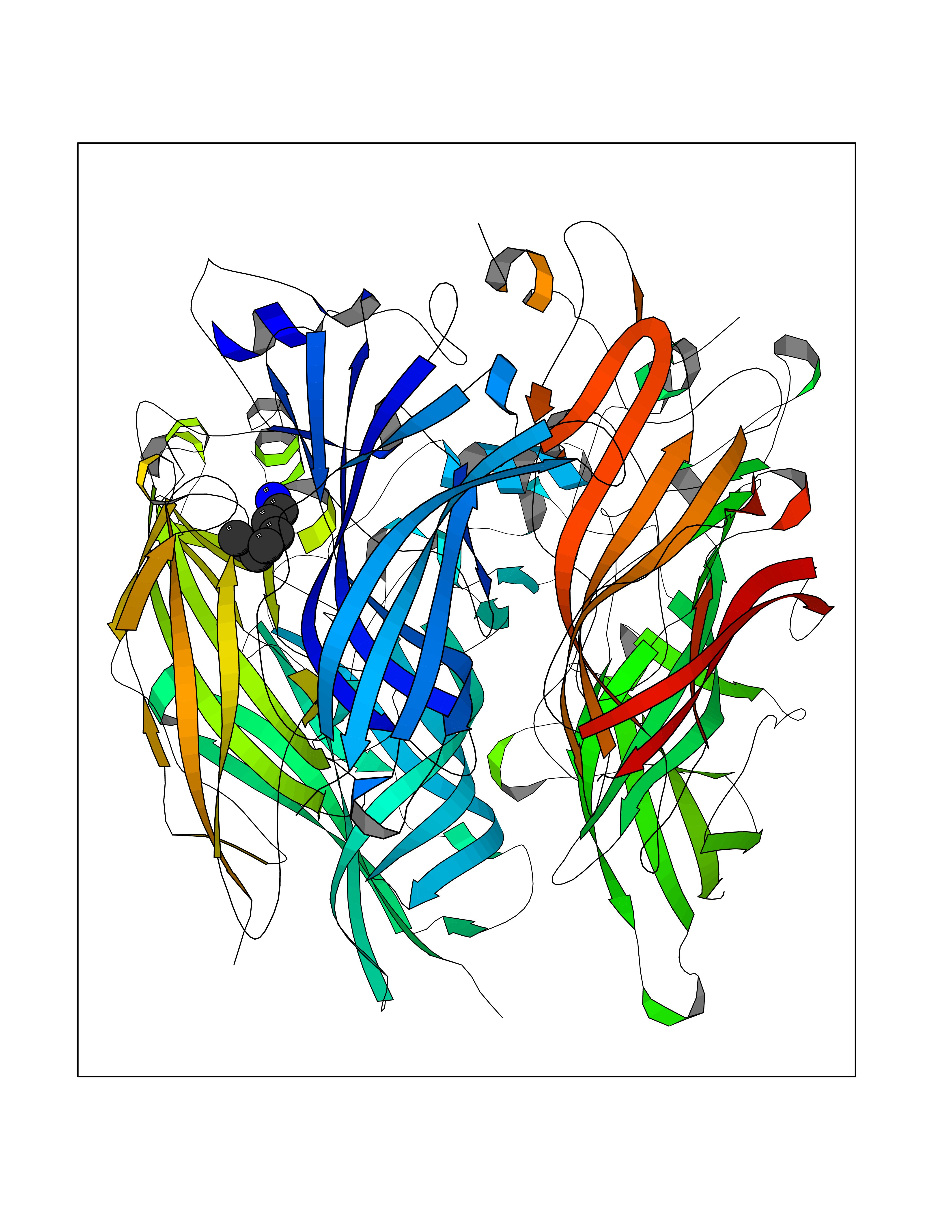 Molscript Map 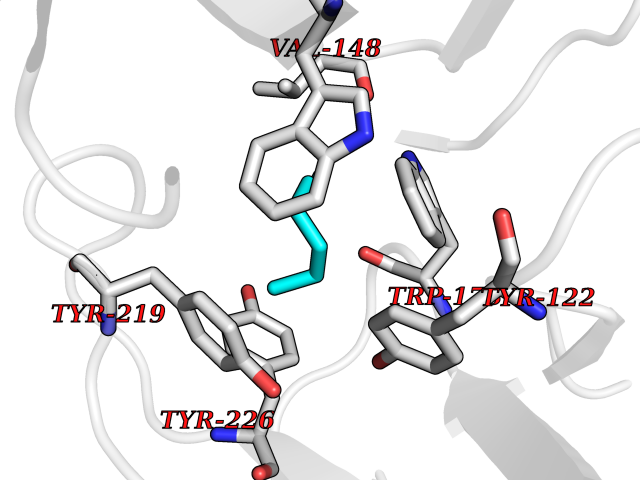 Pymol Map 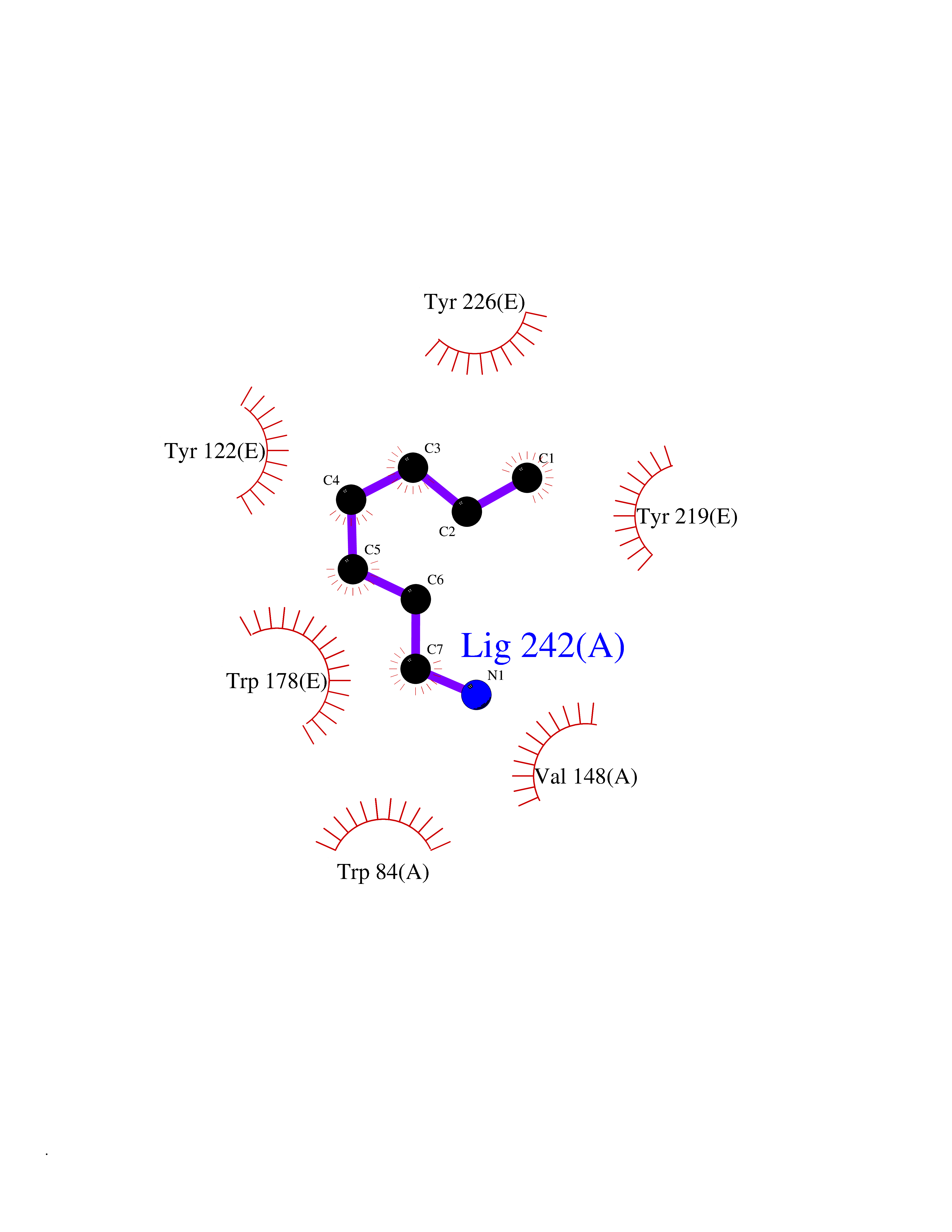 Ligplot Map 3D Binding mode Sequence DRLFKHLFRGYNRWARPVPNTSDVVIVRFGLSIAQLIDVDEKNQMMTTNVWLKQEWSDYKLRWNPTDFGNITSLRVPSEMIWIPDIVLYNNADGEFAVTHMTKAHLFSTGTVHWVPPAIYKSSCSIDVTFFPFDQQNCKMKFGSWTYDKAKIDLEQMEQTVDLKDYWESGEWAIVNATGTYNSKKYDCCAEIYPDVTYAFVIRRLPEDRLFKHLFRGYNRWARPVPNTSDVVIVRFGLSIAQLIDVDEKNQMMTTNVWLKQEWSDYKLRWNPTDFGNITSLRVPSEMIWIPDIVLYNNADGEFAVTHMTKAHLFSTGTVHWVPPAIYKSSCSIDVTFDQQNCKMKFGSWTYDKAKIDLEQMEQTVDLKDYWESGEWAIVNATGTYNSKKYDCCAEIYPDVTYAFVIRRLPEDRLFKHLFRGYNRWARPVPNTSDVVIVRFGLSIAQLIDVDEKNQMMTTNVWLKQEWSDYKLRWNPTDFGNITSLRVPSEMIWIPDIVLYNNADGEFAVTHMTKAHLFSTGTVHWVPPAIYKSSCSIDVTFFPFDQQNCKMKFGSWTYDKAKIDLEQMEQTVDLKDYWESGEWAIVNATGTYNSKKYDCCAEIYPDVTYAFVIRRLPEDRLFKHLFRGYNRWARPVPNTSDVVIVRFGLSIAQLIDVDEKNQMMTTNVWLKQEWSDYKLRWNPTDFGNITSLRVPSEMIWIPDIVLYNNADGEFAVTHMTKAHLFSTGTVHWVPPAIYKSSCSIDVTFFPFDQQNCKMKFGSWTYDKAKIDLEQMEQTVDLKDYWESGEWAIVNATGTYNSKKYDCCAEIYPDVTYAFVIRRLPEDRLFKHLFRGYNRWARPVPNTSDVVIVRFGLSIAQLIDVDEKNQMMTTNVWLKQEWSDYKLRWNPTDFGNITSLRVPSEMIWIPDIVLYNNADGEFAVTHMTKAHLFSTGTVHWVPPAIYKSSCSIDVTFFPFDQQNCKMKFGSWTYDKAKIDLEQMEQTVDLKDYWESGEWAIVNATGTYNSKKYDCCAEIYPDVTYAFVIRRLP Hydrogen bonds contact Hydrophobic contact | ||||
| 12 | Cyclopropane mycolic acid synthase MmaA2 | 1TPY | 5.11 | |
Target general information Gen name mmaA2 Organism Mycobacterium tuberculosis (strain ATCC 25618 / H37Rv) Uniprot ID TTD ID NA Synonyms Rv0644c;mma2 Protein family CFA/CMAS family Biochemical class Transferase Function Cyclopropane-fatty-acyl-phospholipid synthase activity.Methyltransferase activity. Related diseases Oocyte/zygote/embryo maturation arrest 16 (OZEMA16) [MIM:617234]: A rare cause of female primary infertility. In affected women, ovulation and fertilization proceed normally but embryos are arrested at early stages of development. Inheritance is autosomal recessive. {ECO:0000269|PubMed:27545678}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01718; DB01752 Interacts with NA EC number 2.1.1.79 Uniprot keywords 3D-structure; Acetylation; Lipid biosynthesis; Lipid metabolism; Methyltransferase; Reference proteome; S-adenosyl-L-methionine; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 32493.6 Length 285 Aromaticity 0.1 Instability index 43.61 Isoelectric point 5.53 Charge (pH=7) -10.17 2D Binding mode Binding energy (Kcal/mol) -6.97 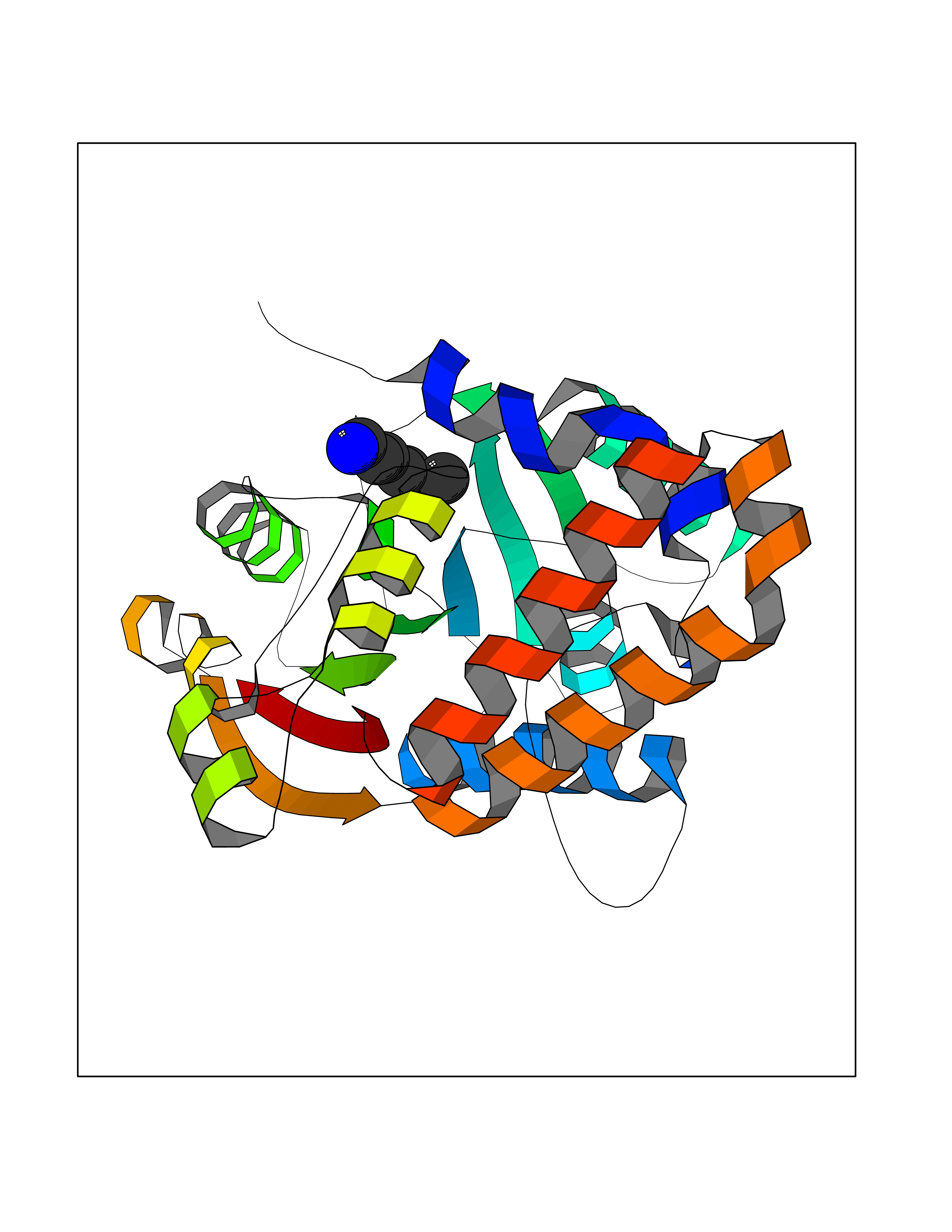 Molscript Map 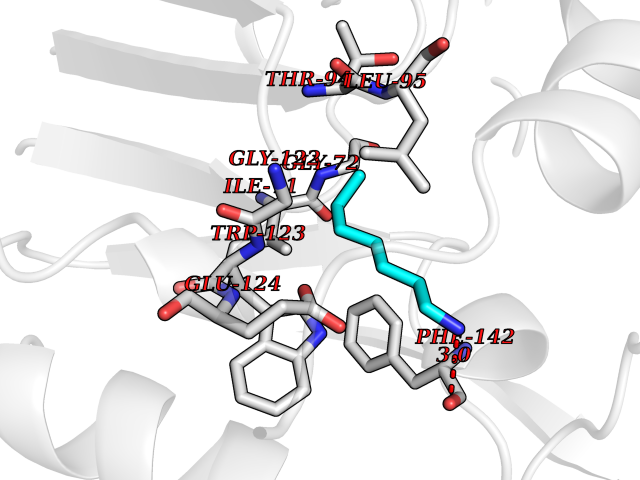 Pymol Map 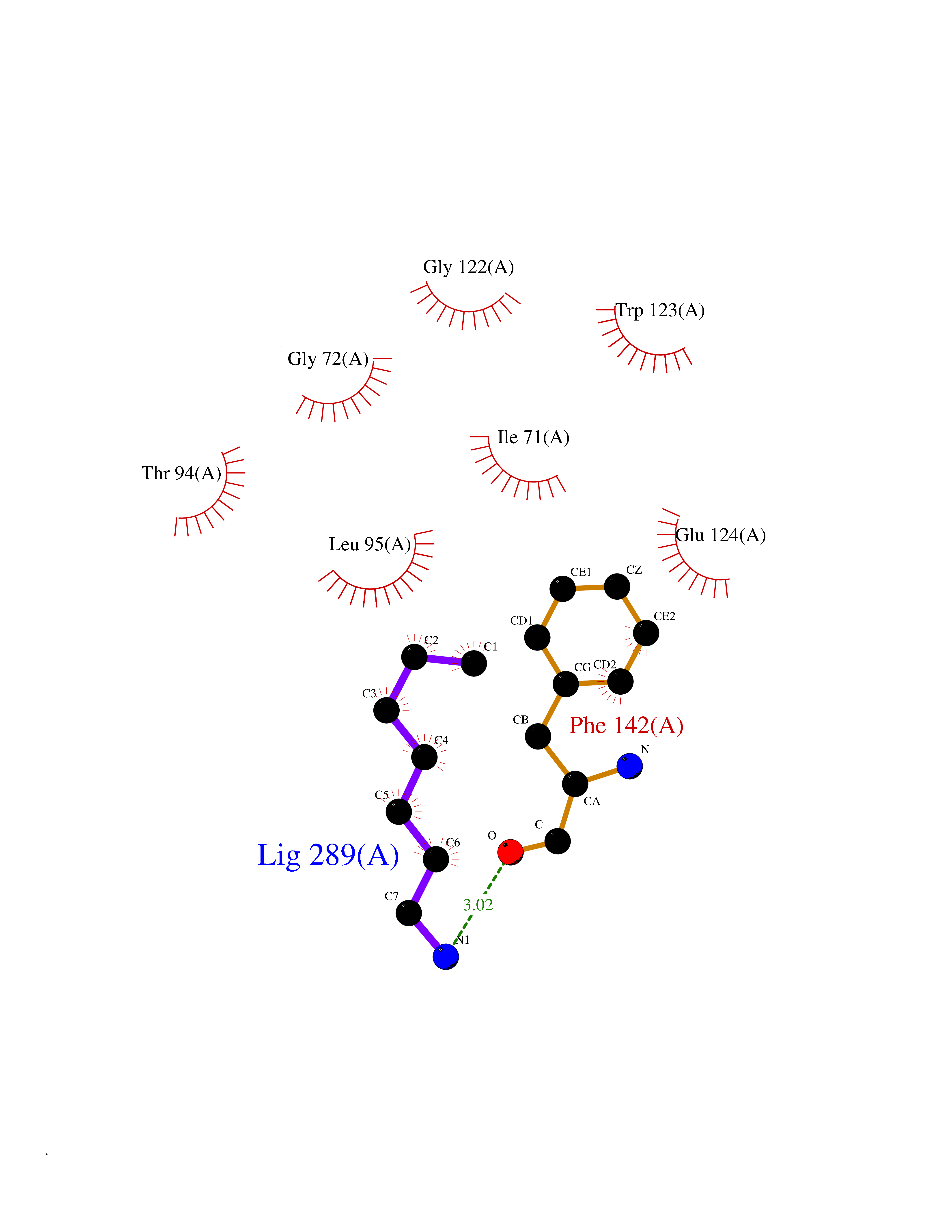 Ligplot Map 3D Binding mode Sequence NDLTPHFEDVQAHYDLSDDFFRLFLDPTQTYSCAHFEREDMTLEEAQIAKIDLALGKLGLQPGMTLLDIGCGWGATMRRAIAQYDVNVVGLTLSKNQAAHVQKSFDEMDTPRDRRVLLAGWEQFNEPVDRIVSIGAFEHFGHDRHADFFARAHKILPPDGVLLLHTITGLTRQQMVDHGLPLTLWLARFLKFIATEIFPGGQPPTIEMVEEQSAKTGFTLTRRQSLQPHYARTLDLWAEALQEHKSEAIAIQSEEVYERYMKYLTGCAKLFRVGYIDVNQFTLAK Hydrogen bonds contact Hydrophobic contact | ||||
| 13 | Cocaine esterase | 3I2K | 5.10 | |
Target general information Gen name cocE Organism Rhodococcus sp. (strain MB1 Bresler) Uniprot ID TTD ID NA Synonyms NA Protein family CocE/NonD hydrolase family Biochemical class Hydrolase Function Carboxylic ester hydrolase activity.Dipeptidyl-peptidase activity. Related diseases Thiamine metabolism dysfunction syndrome 5, episodic encephalopathy type (THMD5) [MIM:614458]: An autosomal recessive metabolic disorder due to an inborn error of thiamine metabolism. The phenotype is highly variable, but in general, affected individuals have onset in early childhood of acute encephalopathic episodes associated with increased serum and CSF lactate. These episodes result in progressive neurologic dysfunction manifest as gait disturbances, ataxia, dystonia, and spasticity, which in some cases may result in loss of ability to walk. Cognitive function is usually preserved, although mildly delayed development has been reported. These episodes are usually associated with infection and metabolic decompensation. Some patients may have recovery of some neurologic deficits. {ECO:0000269|PubMed:22152682}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03793; DB01795 Interacts with NA EC number 3.1.1.84 Uniprot keywords 3D-structure; Cytoplasm; Direct protein sequencing; Hydrolase; Serine esterase Protein physicochemical properties Chain ID A Molecular weight (Da) 62127.9 Length 574 Aromaticity 0.09 Instability index 26.62 Isoelectric point 4.56 Charge (pH=7) -33.24 2D Binding mode Binding energy (Kcal/mol) -6.96 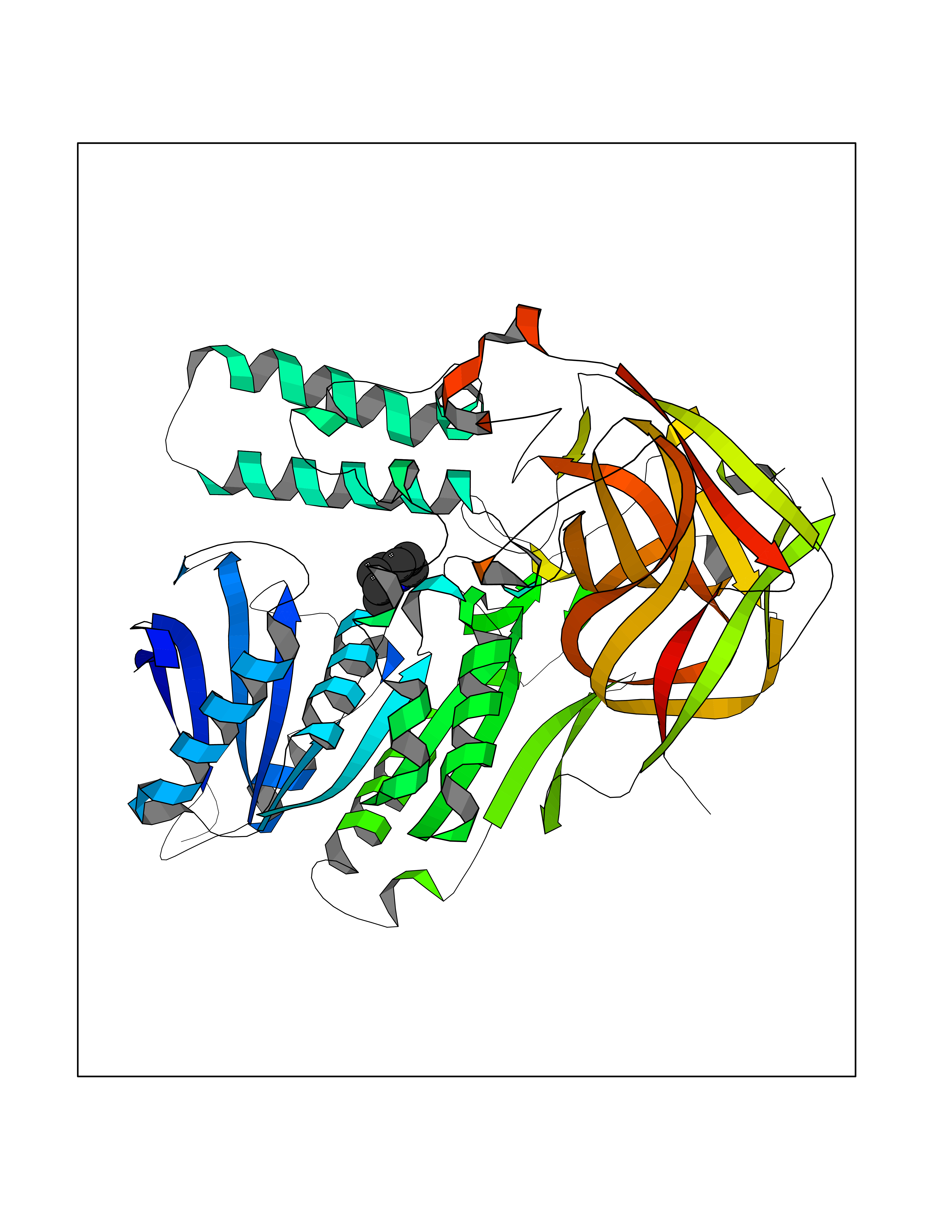 Molscript Map 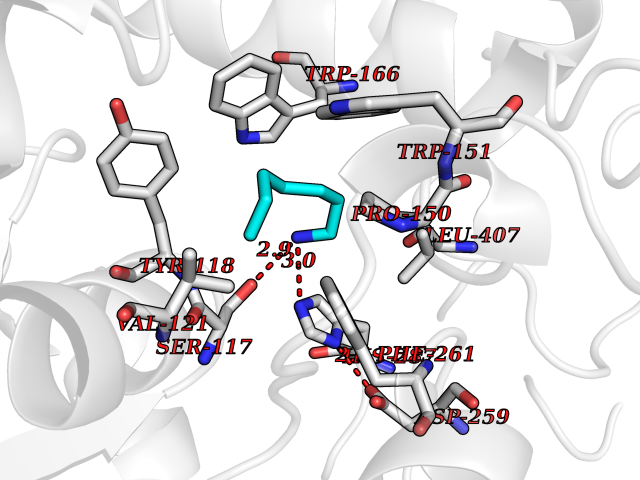 Pymol Map 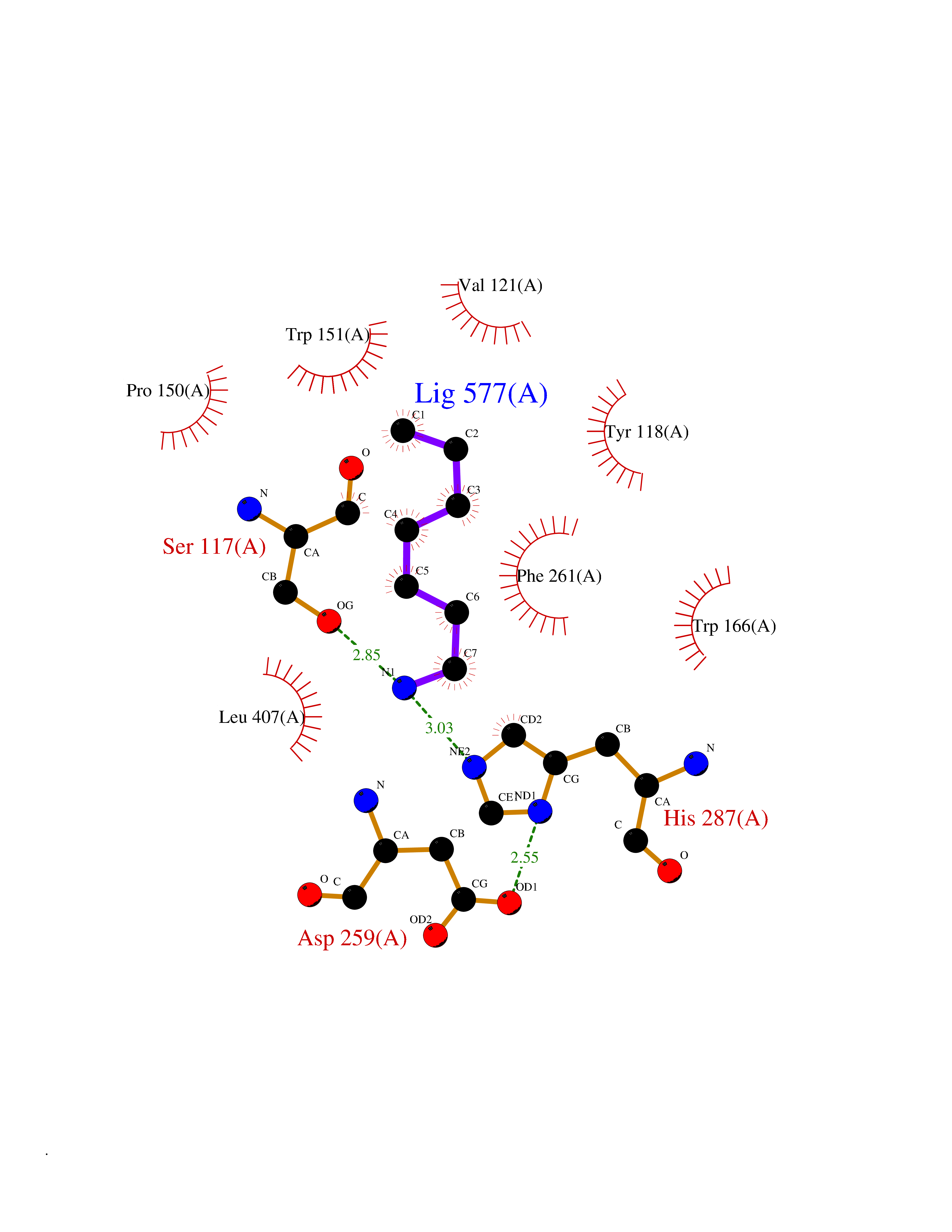 Ligplot Map 3D Binding mode Sequence VDGNYSVASNVMVPMRDGVRLAVDLYRPDADGPVPVLLVRNPYDKFDVFAWSTQSTNWLEFVRDGYAVVIQDTRGLFASEGEFVPHVDDEADAEDTLSWILEQAWCDGNVGMFGVSYLGVTQWQAAVSGVGGLKAIAPSMASADLYRAPWYGPGGALSVEALLGWSALIGTGLITSRSDARPEDAADFVQLAAILNDVAGAASVTPLAEQPLLGRLIPWVIDQVVDHPDNDESWQSISLFERLGGLATPALITAGWYDGFVGESLRTFVAVKDNADARLVVGPWSHSNLTGRNADRKFGIAATYPIQEATTMHKAFFDRHLRGETDALAGVPKVRLFVMGIDEWRDETDWPLPDTAYTPFYLGGSGAANTSTGGGTLSTSISGTESADTYLYDPADPVPSLGGTLLFHNGDNGPADQRPIHDRDDVLCYSTEVLTDPVEVTGTVSARLFVSSSAVDTDFTAKLVDVFPDGRAIALCDGIVRMRYRETLVNPTLIEAGEIYEVAIDMLATSNVFLPGHRIMVQVSSSNFPKYDRNSNTGGVIAREQLEEMCTAVNRIHRGPEHPSHIVLPIIKRK Hydrogen bonds contact Hydrophobic contact | ||||
| 14 | Ghrelin (GHRL) | 7F9Y | 5.10 | |
Target general information Gen name GHRL Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms UNQ524/PRO1066; Motilin-related peptide; M46 protein; Growth hormone secretagogue; Growth hormone releasing peptide; Gastric peptide ghrelin; GHRL Protein family Motilin family Biochemical class NA Function Specific ligand for the growth hormone secretagogue receptor type 1 (ghsr) inducing the release of growth hormone from the pituitary. Has an appetite-stimulating effect, induces adiposity and stimulates gastric acid secretion.Involved in growth regulation. Related diseases Neurodevelopmental disorder with structural brain anomalies and dysmorphic facies (NEDBAF) [MIM:618577]: An autosomal dominant neurodevelopmental disorder characterized by global developmental delay, severe intellectual disability, poor language, seizures, dysmorphic features, and thin corpus callosum. {ECO:0000269|PubMed:29276006, ECO:0000269|PubMed:30293988}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q9UMX0; Q9UMX0-2; Q9UHD9 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Amidation; Direct protein sequencing; Hormone; Lipoprotein; Proteomics identification; Reference proteome; Secreted; Signal Protein physicochemical properties Chain ID C,R Molecular weight (Da) 34053.1 Length 300 Aromaticity 0.14 Instability index 38.34 Isoelectric point 9.3 Charge (pH=7) 11.02 2D Binding mode Binding energy (Kcal/mol) -6.95 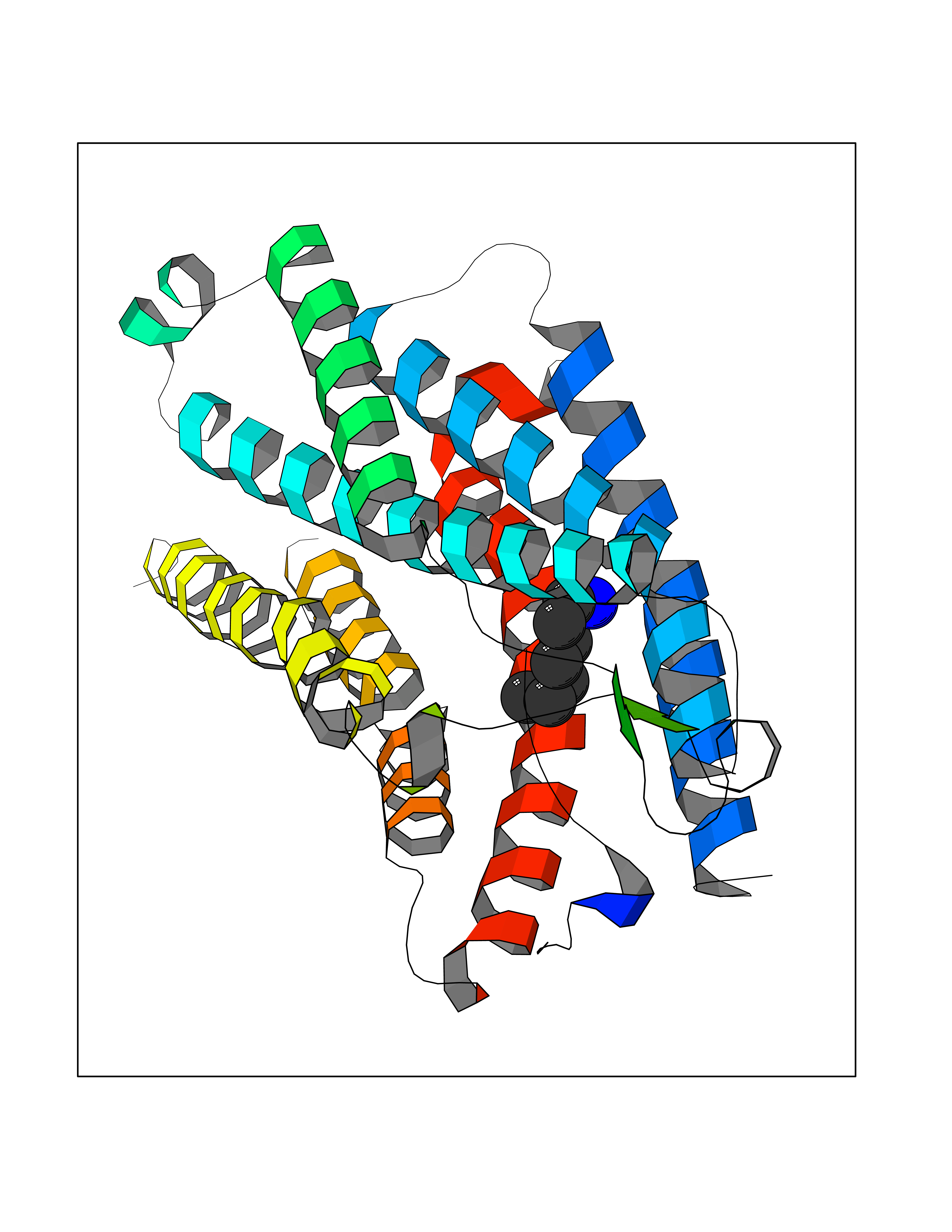 Molscript Map 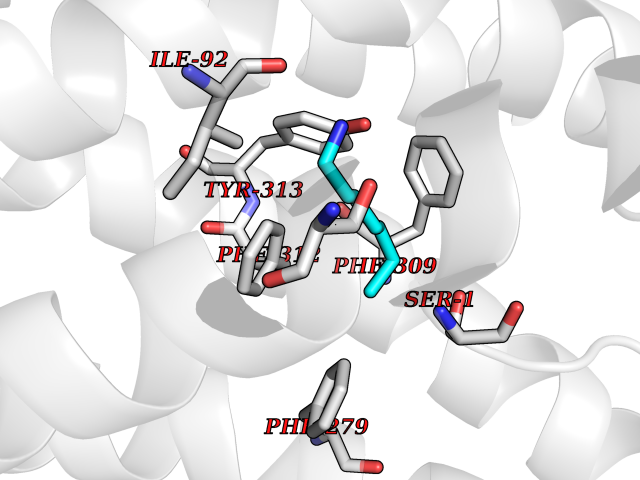 Pymol Map 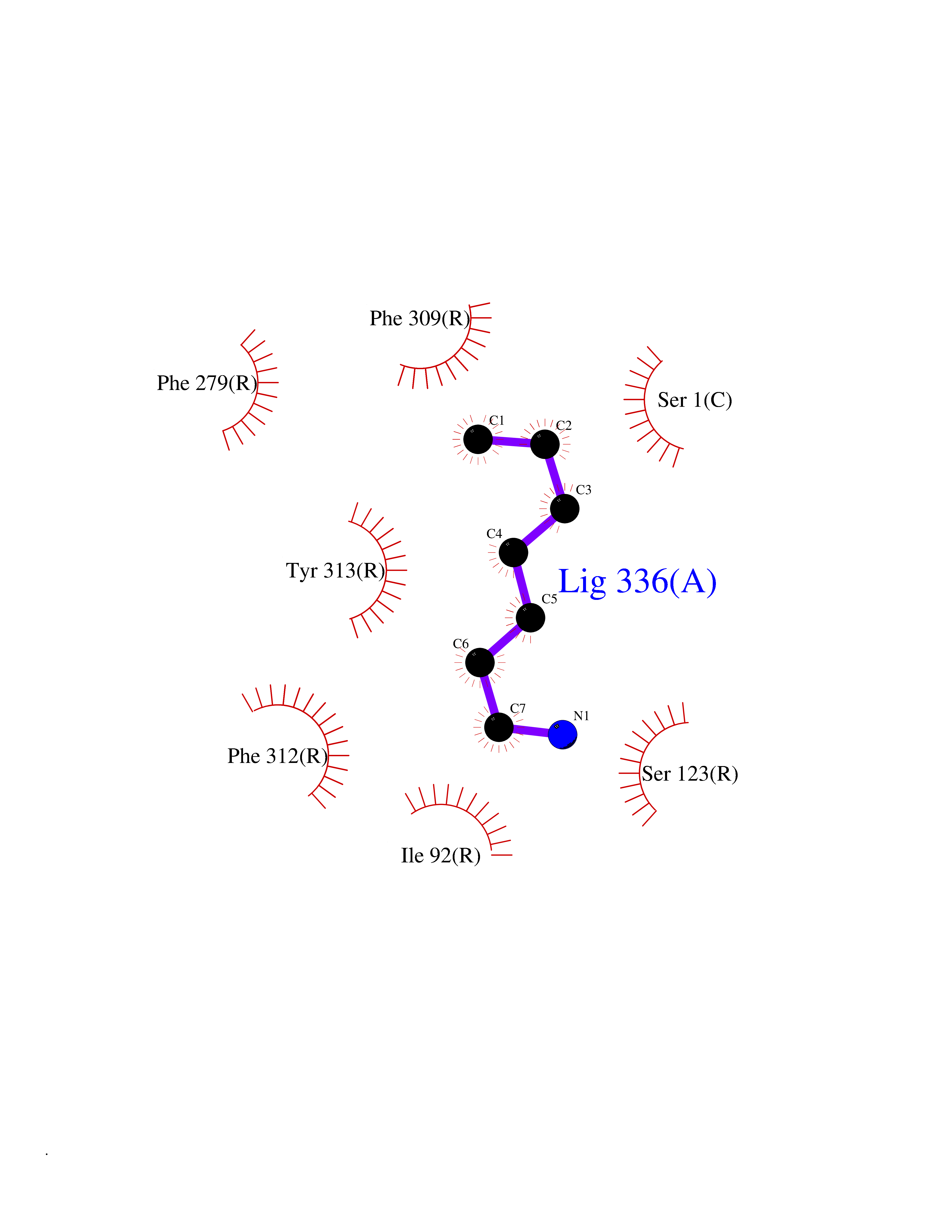 Ligplot Map 3D Binding mode Sequence GSSFLSPEHQRVQQRLQLFPAPLLAGVTATCVALFVVGIAGNLLTMLVVSRFRELRTTTNLYLSSMAFSDLLIFLCMPLDLVRLWQYRPWNFGDLLCKLFQFVSESCTYATVLTITALSVERYFAICFPLRAKVVVTKGRVKLVIFVIWAVAFCSAGPIFVLVGVEHENGTDPWDTNECRPTEFAVRSGLLTVMVWVSSIFFFLPVFCLTVLYSLIGRKLWRNHKQTVKMLAVVVFAFILCWLPFHVGRYLFSKSFEPGSLEIAQISQYCNLVSFVLFYLSAAINPILYNIMSKKYRVAV Hydrogen bonds contact Hydrophobic contact | ||||
| 15 | Haemophilus influenzae NadR protein (Hae-influ nadR) | 1LW7 | 5.10 | |
Target general information Gen name Hae-influ nadR Organism Haemophilus influenzae (strain ATCC 51907 / DSM 11121 / KW20 / Rd) Uniprot ID TTD ID Synonyms nadR; Transcriptional regulator nadR Protein family Bacterial NMN adenylyltransferase family; Bacterial RNK family Biochemical class Nicotinamide ribonucleoside uptake permease Function This enzyme has twoactivities: nicotinamide mononucleotide (NMN) adenylyltransferase and ribosylnicotinamide (RN) kinase. The RN kinase activity catalyzes the phosphorylation of RN to form nicotinamide ribonucleotide. The NMN adenylyltransferase activity catalyzes the transfer of the AMP moiety of ATP to nicotinamide ribonucleotide to form NAD(+). Related diseases Involved in the epigenetic regulation of ESR1 expression in breast cancer in a TFAP2C, IFI16 and HDAC4/5/6-dependent manner. {ECO:0000269|PubMed:24413532}. Drugs (DrugBank ID) NA Interacts with NA EC number NA Uniprot keywords 3D-structure; Alternative initiation; ATP-binding; Cell membrane; Cytoplasm; Kinase; Membrane; Multifunctional enzyme; NAD; Nucleotide-binding; Pyridine nucleotide biosynthesis; Reference proteome; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 39581.5 Length 344 Aromaticity 0.14 Instability index 41.39 Isoelectric point 6.94 Charge (pH=7) -0.18 2D Binding mode Binding energy (Kcal/mol) -6.95  Molscript Map 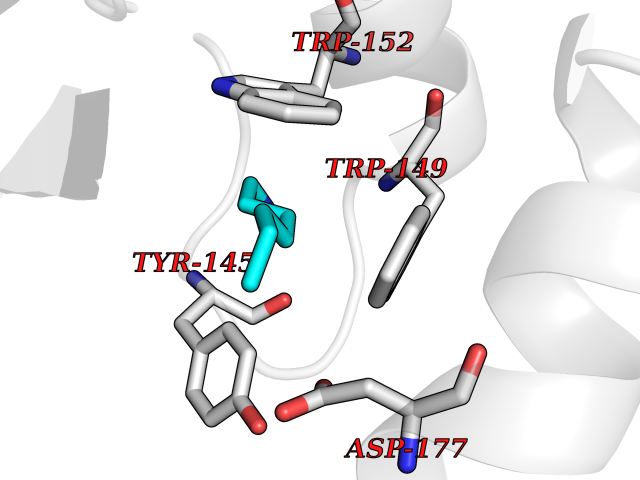 Pymol Map 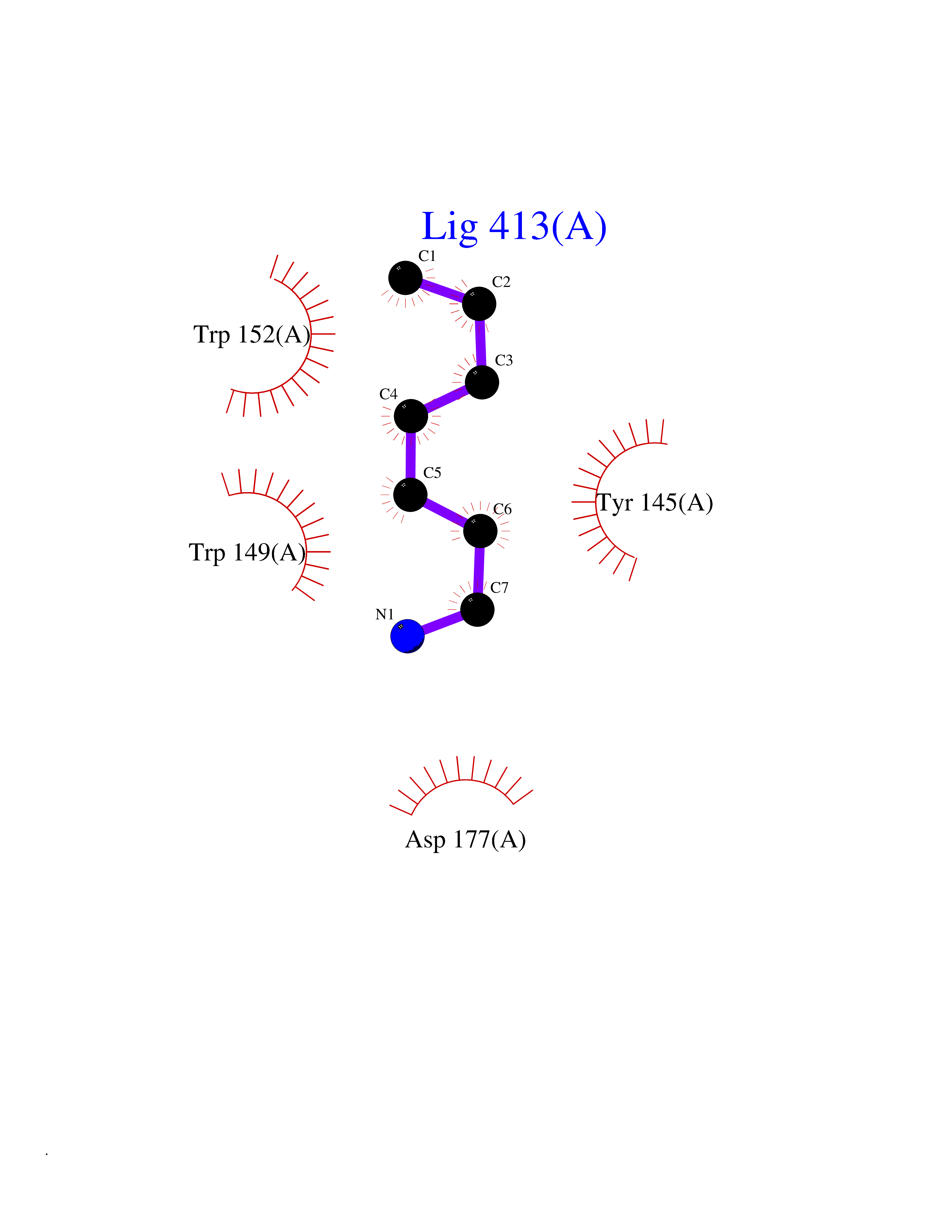 Ligplot Map 3D Binding mode Sequence EKKVGVIFGKFYPVHTGHINXIYEAFSKVDELHVIVCSDTVRDLKLFYDSKXKRXPTVQDRLRWXQQIFKYQKNQIFIHHLVEDGIPSYPNGWQSWSEAVKTLFHEKHFEPSIVFSSEPQDKAPYEKYLGLEVSLVDPDRTFFNVSATKIRTTPFQYWKFIPKEARPFFAKTVAILGGESSGKSVLVNKLAAVFNTTSAWEYGREFVFEKLGGDEQAMQYSDYPQXALGHQRYIDYAVRHSHKIAFIDTDFITTQAFCIQYEGKAHPFLDSXIKEYPFDVTILLKNNTEQKQRQQFQQLLKKLLDKYKVPYIEIESPSYLDRYNQVKAVIEKVLNEEEISELQN Hydrogen bonds contact Hydrophobic contact | ||||
| 16 | Acetylcholine receptor subunit alpha | 4ZJS | 5.09 | |
Target general information Gen name CHRNA1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms ACHRA;CHNRA Protein family Ligand-gated ion channel (TC 1.A.9) family, Acetylcholine receptor (TC 1.A.9.1) subfamily, Alpha-1/CHRNA1 sub-subfamily Biochemical class Immune system Function Acetylcholine binding.Acetylcholine-gated cation-selective channel activity.Acetylcholine receptor activity.Ion channel activity.Ligand-gated ion channel activity. Related diseases Multiple pterygium syndrome, lethal type (LMPS) [MIM:253290]: Multiple pterygia are found infrequently in children with arthrogryposis and in fetuses with fetal akinesia syndrome. In lethal multiple pterygium syndrome there is intrauterine growth retardation, multiple pterygia, and flexion contractures causing severe arthrogryposis and fetal akinesia. Subcutaneous edema can be severe, causing fetal hydrops with cystic hygroma and lung hypoplasia. Oligohydramnios and facial anomalies are frequent. {ECO:0000269|PubMed:18252226}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: The alpha subunit is the main focus for antibody binding in myasthenia gravis. Myasthenia gravis is characterized by sporadic muscular fatigability and weakness, occurring chiefly in muscles innervated by cranial nerves, and characteristically improved by cholinesterase-inhibiting drugs.; DISEASE: Myasthenic syndrome, congenital, 1A, slow-channel (CMS1A) [MIM:601462]: A common congenital myasthenic syndrome. Congenital myasthenic syndromes are characterized by muscle weakness affecting the axial and limb muscles (with hypotonia in early-onset forms), the ocular muscles (leading to ptosis and ophthalmoplegia), and the facial and bulbar musculature (affecting sucking and swallowing, and leading to dysphonia). The symptoms fluctuate and worsen with physical effort. CMS1A is a slow-channel myasthenic syndrome. It is caused by kinetic abnormalities of the AChR, resulting in prolonged AChR channel opening episodes, prolonged endplate currents, and depolarization block. This is associated with calcium overload, which may contribute to subsequent degeneration of the endplate and postsynaptic membrane. {ECO:0000269|PubMed:16685696, ECO:0000269|PubMed:7619526, ECO:0000269|PubMed:8872460, ECO:0000269|PubMed:9158151, ECO:0000269|PubMed:9221765}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Myasthenic syndrome, congenital, 1B, fast-channel (CMS1B) [MIM:608930]: A form of congenital myasthenic syndrome, a group of disorders characterized by failure of neuromuscular transmission, including pre-synaptic, synaptic, and post-synaptic disorders that are not of autoimmune origin. Clinical features are easy fatigability and muscle weakness affecting the axial and limb muscles (with hypotonia in early-onset forms), the ocular muscles (leading to ptosis and ophthalmoplegia), and the facial and bulbar musculature (affecting sucking and swallowing, and leading to dysphonia). The symptoms fluctuate and worsen with physical effort. CMS1B is a fast-channel myasthenic syndrome. It is caused by kinetic abnormalities of the AChR, resulting in brief opening and activity of the channel, with a rapid decay in endplate current, failure to achieve threshold depolarization of the endplate and consequent failure to fire an action potential. {ECO:0000269|PubMed:10195214, ECO:0000269|PubMed:12588888, ECO:0000269|PubMed:15079006}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08838; DB00565; DB00555 Interacts with NA EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Congenital myasthenic syndrome; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; Ion channel; Ion transport; Ligand-gated ion channel; Membrane; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Signal; Synapse; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A,B,C,D,E Molecular weight (Da) 46717.8 Length 411 Aromaticity 0.11 Instability index 38.02 Isoelectric point 4.77 Charge (pH=7) -22.31 2D Binding mode Binding energy (Kcal/mol) -6.95 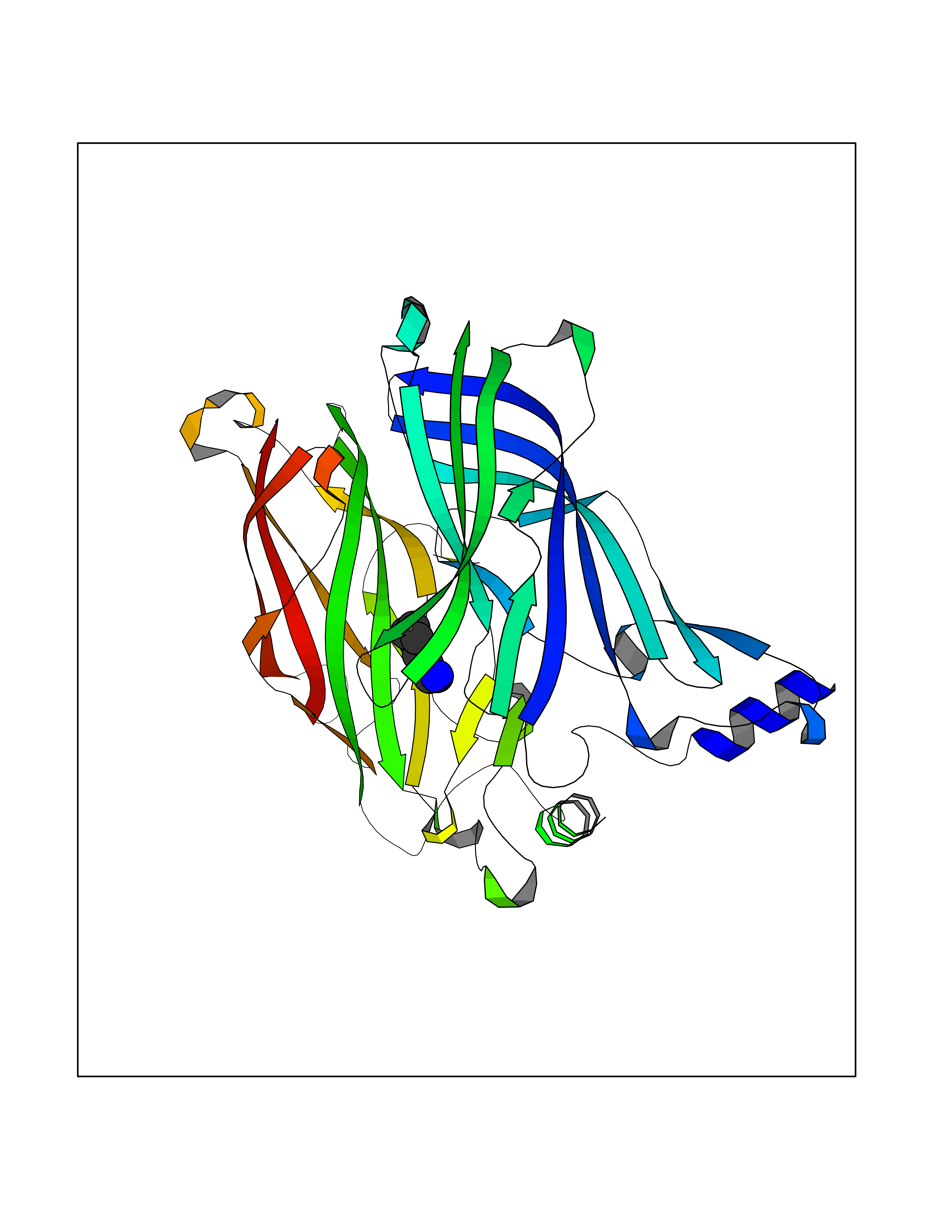 Molscript Map 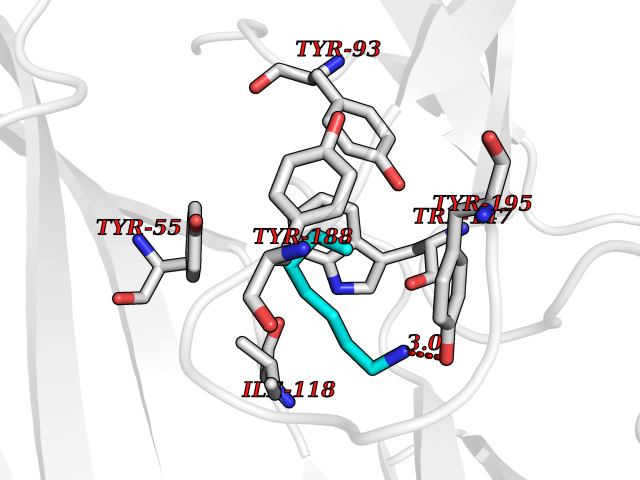 Pymol Map 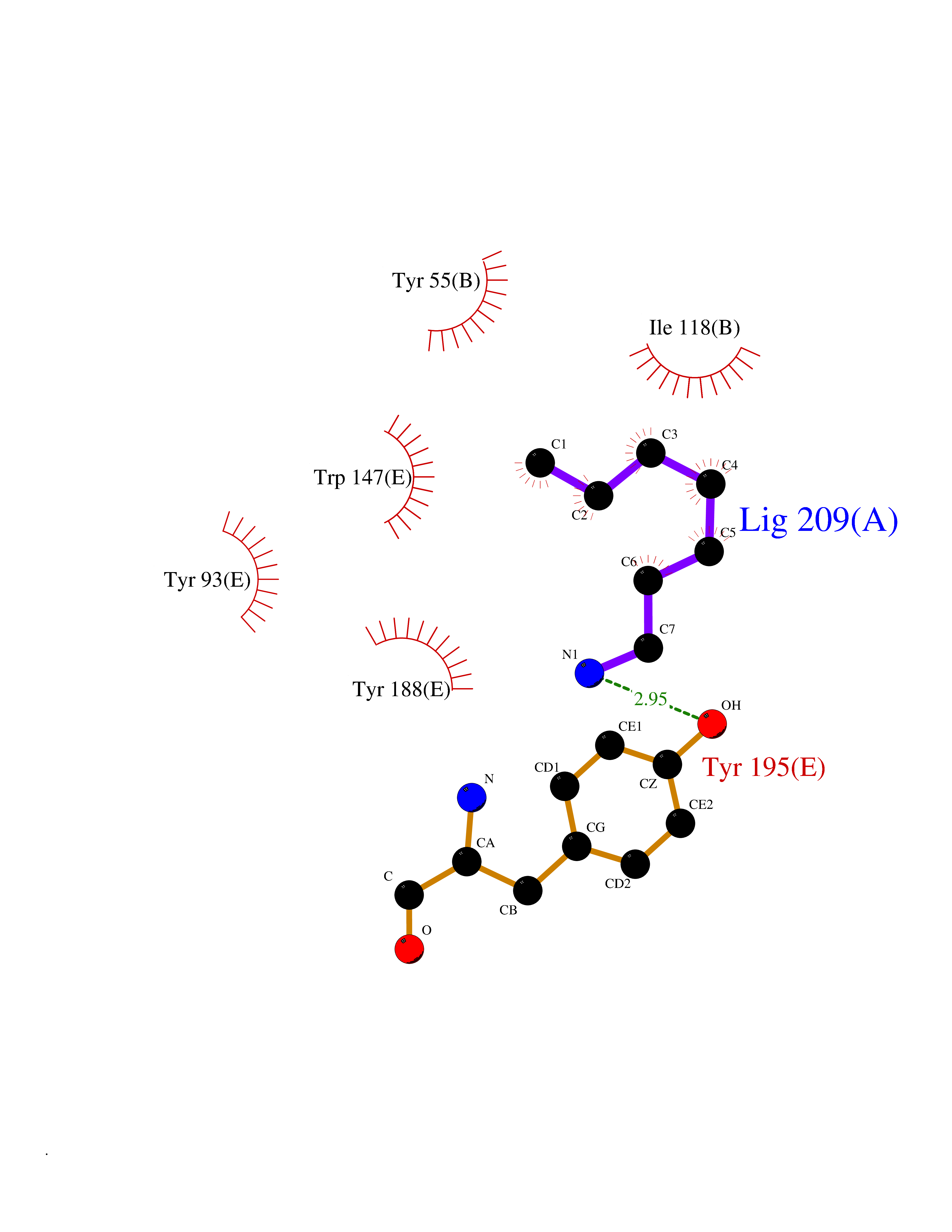 Ligplot Map 3D Binding mode Sequence EHETRLVAKLFKDYSSVVRPVEDHRQVVEVTLGFTLQDIVKADSSTNEVDLVYYEQQRWVDYNLKWNPDDYGGVKKIHIPAADIWTPDITAYSSTRPVQVLSPQIAVVTHDGSVMFIPAQRLSFMCDPTGVDSEEGATCAVKFGSWVYSGFEIDLKTDTDQVDLSSYYASSKYEILSATQTRQVQHYSCCPEPYIDVNLVVKFREEHETRLVAKLFKDYSSVVRPVEDHRQVVEVTLGFTLQDIVKADSSTNEVDLVYYEQQRWVDYNLKWNPDDYGGVKKIHIPAADIWTPDITAYSSTRPVQVLSPQIAVVTHDGSVMFIPAQRLSFMCDPTGVDSEEGATCAVKFGSWVYSGFEIDLKTDTDQVDLSSYYASSKYEILSATQTRQVQHYSCCPEPYIDVNLVVKFRER Hydrogen bonds contact Hydrophobic contact | ||||
| 17 | Vitamin D3 receptor (VDR) | 3B0T | 5.09 | |
Target general information Gen name VDR Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Vitamin D(3) receptor; Nuclear vitamin D receptor; Nuclear receptor subfamily 1 group I member 1; NR1I1; 1,25-dihydroxyvitamin D3 receptor Protein family Nuclear hormone receptor family, NR1 subfamily Biochemical class Nuclear hormone receptor Function Enters the nucleus upon vitamin D3 binding where it forms heterodimers with the retinoid X receptor/RXR. The VDR-RXR heterodimers bind to specific response elements on DNA and activate the transcription of vitamin D3-responsive target genes. Plays a central role in calcium homeostasis. Nuclear receptor for calcitriol, the active form of vitamin D3 which mediates the action of this vitamin on cells. Related diseases Rickets vitamin D-dependent 2A (VDDR2A) [MIM:277440]: A disorder of vitamin D metabolism resulting in severe rickets, hypocalcemia and secondary hyperparathyroidism. Most patients have total alopecia in addition to rickets. {ECO:0000269|PubMed:1652893, ECO:0000269|PubMed:17970811, ECO:0000269|PubMed:2177843, ECO:0000269|PubMed:2849209, ECO:0000269|PubMed:28698609, ECO:0000269|PubMed:7828346, ECO:0000269|PubMed:8106618, ECO:0000269|PubMed:8381803, ECO:0000269|PubMed:8392085, ECO:0000269|PubMed:8675579, ECO:0000269|PubMed:8961271, ECO:0000269|PubMed:9005998}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07530; DB08742; DB01436; DB04891; DB00146; DB02300; DB00136; DB00169; DB04540; DB05024; DB11672; DB14635; DB01070; DB06410; DB05295; DB06194; DB00153; DB04796; DB03451; DB00910; DB04258; DB11094 Interacts with P35222; Q09472; Q15648; P50222; Q15788; P26045; P19793; Q13573; Q13501; P04637; Q15645; Q9JLI4; P28700; X5D778; Q96HA8; Q01804; Q96S38; P48443 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; Disease variant; DNA-binding; Metal-binding; Nucleus; Proteomics identification; Receptor; Reference proteome; Transcription; Transcription regulation; Ubl conjugation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A Molecular weight (Da) 28781 Length 254 Aromaticity 0.07 Instability index 47.69 Isoelectric point 6.15 Charge (pH=7) -3.44 2D Binding mode Binding energy (Kcal/mol) -6.94 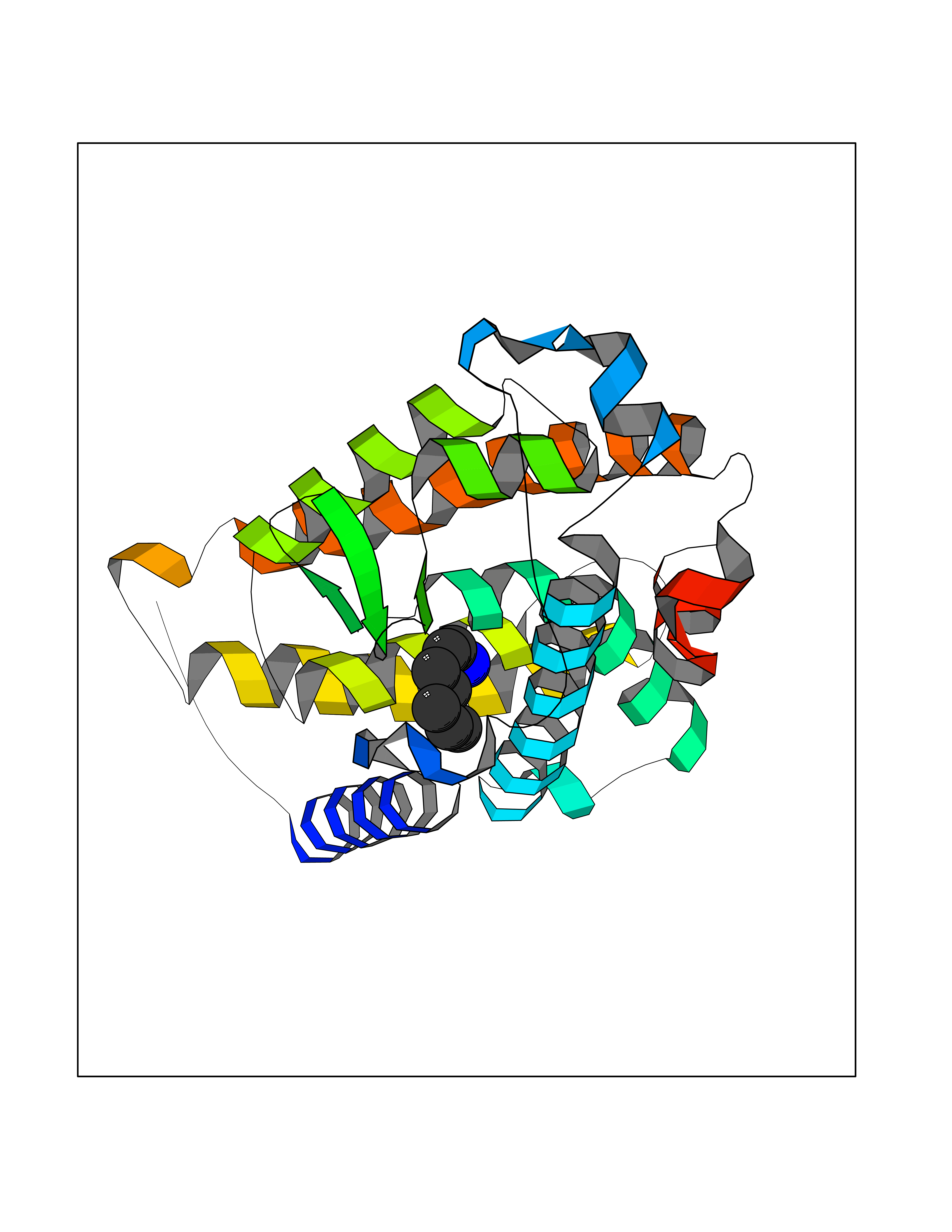 Molscript Map 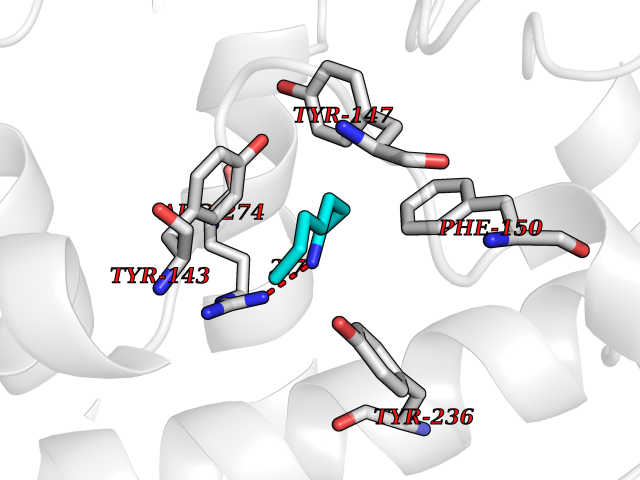 Pymol Map 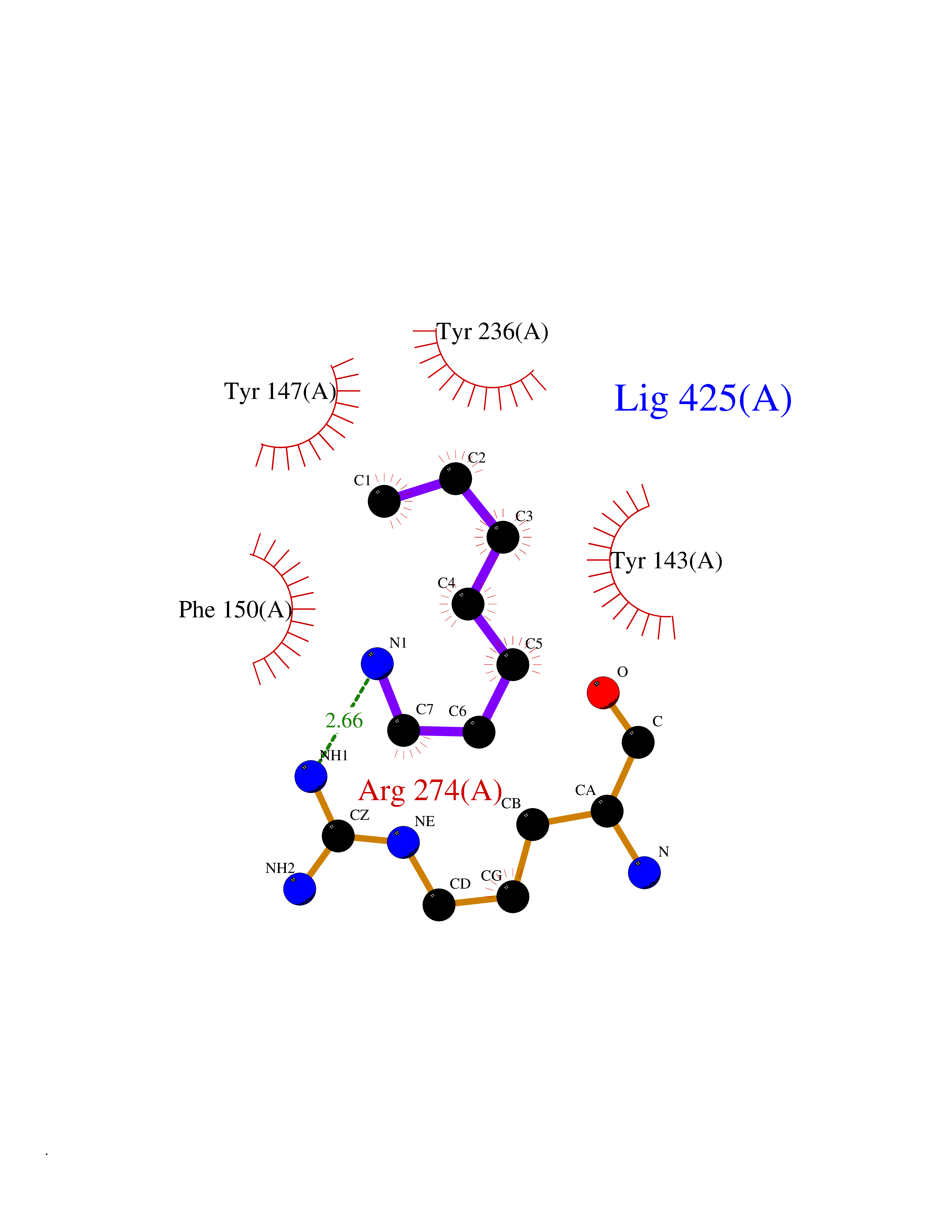 Ligplot Map 3D Binding mode Sequence ALRPKLSEEQQRIIAILLDAHHKTYDPTYSDFCQFRPPVRVNDGGGSVTLELSQLSMLPHLADLVSYSIQKVIGFAKMIPGFRDLTSEDQIVLLKSSAIEVIMLRSNESFTMDDMSWTCGNQDYKYRVSDVTKAGHSLELIEPLIKFQVGLKKLNLHEEEHVLLMAICIVSPDRPGVQDAALIEAIQDRLSNTLQTYIRCRHPPPGSHLLYAKMIQKLADLRSLNEEHSKQYRCLSFQPECSMKLTPLVLEVFG Hydrogen bonds contact Hydrophobic contact | ||||
| 18 | 2-hydroxy-6-oxo-7-methylocta-2,4-dienoate hydrolase | 1UK8 | 5.09 | |
Target general information Gen name cumD Organism Pseudomonas fluorescens Uniprot ID TTD ID NA Synonyms NA Protein family NA Biochemical class Hydrolase Function Hydrolase activity. Related diseases Intellectual developmental disorder, autosomal dominant 62 (MRD62) [MIM:618793]: An autosomal dominant form of intellectual disability, a disorder characterized by significantly below average general intellectual functioning associated with impairments in adaptive behavior and manifested during the developmental period. MRD62 is characterized by mild to moderately impaired intellectual development. {ECO:0000269|PubMed:27479843, ECO:0000269|PubMed:29460436}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03741; DB03793; DB03568; DB02531; DB03750; DB02406; DB03766 Interacts with NA EC number NA Uniprot keywords 3D-structure; Hydrolase Protein physicochemical properties Chain ID A Molecular weight (Da) 30307.9 Length 271 Aromaticity 0.1 Instability index 37.49 Isoelectric point 5.02 Charge (pH=7) -11.58 2D Binding mode Binding energy (Kcal/mol) -6.95 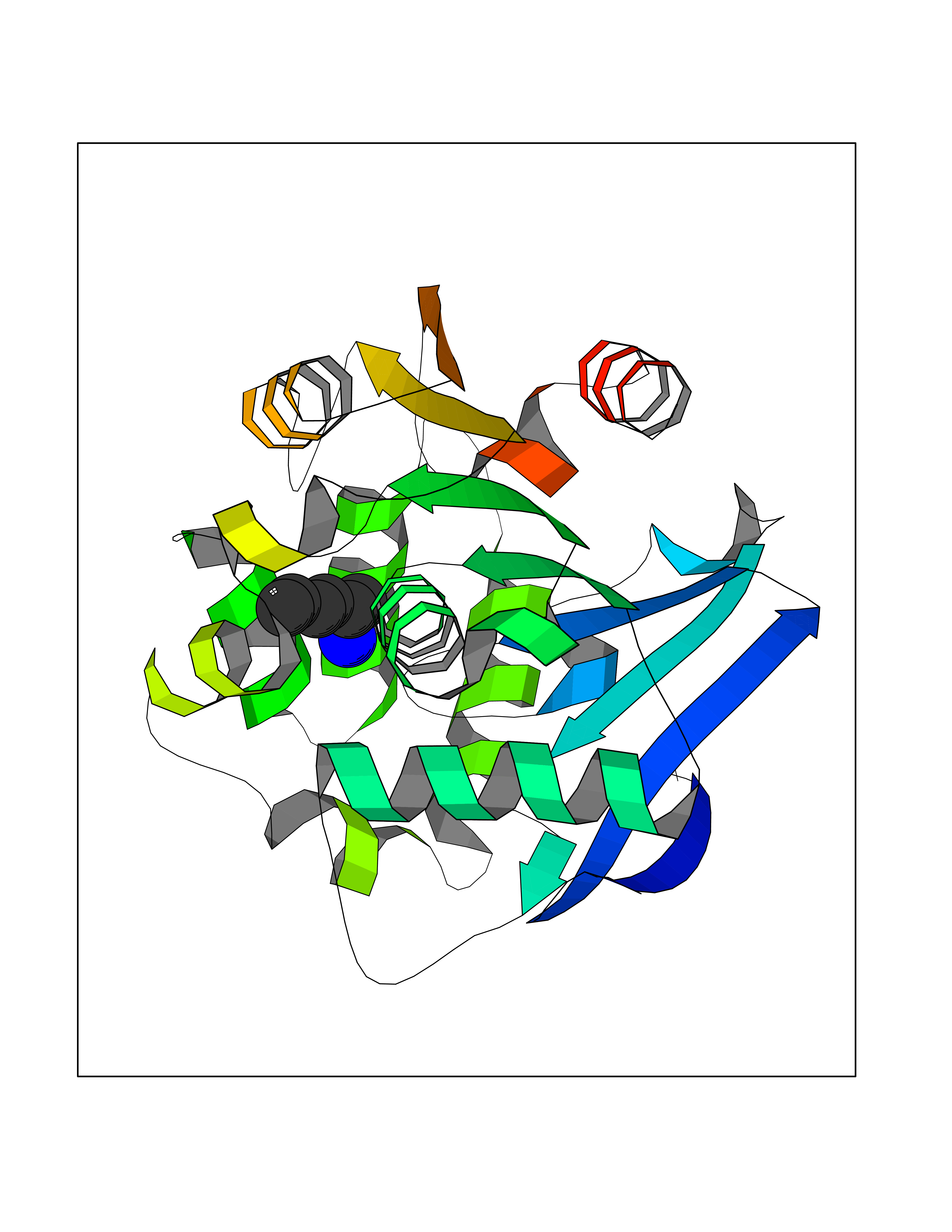 Molscript Map 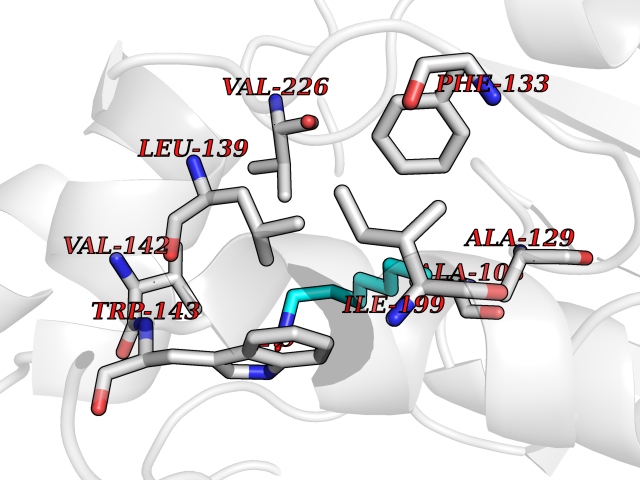 Pymol Map 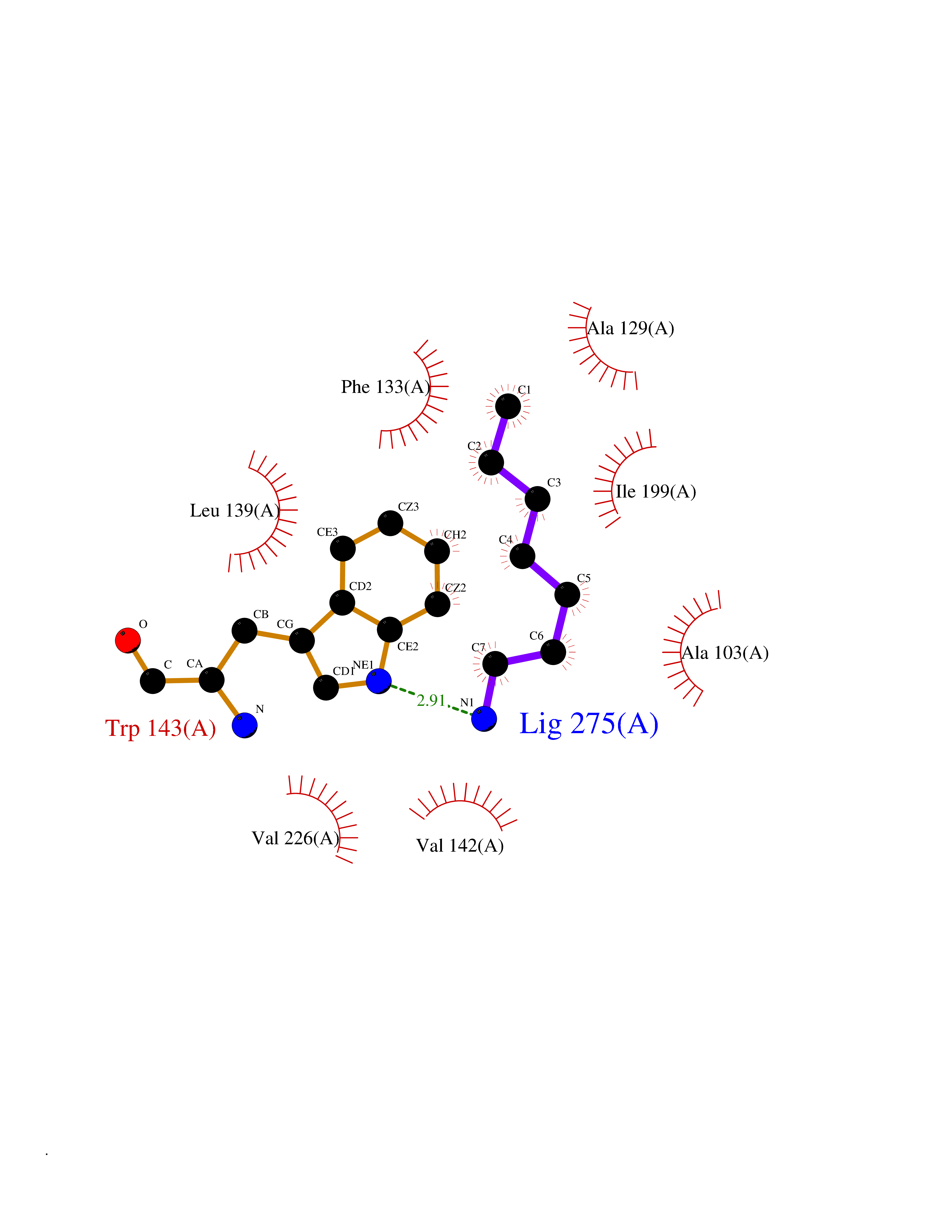 Ligplot Map 3D Binding mode Sequence NLEIGKSILAAGVLTNYHDVGEGQPVILIHGSGPGVSAYANWRLTIPALSKFYRVIAPDMVGFGFTDRPENYNYSKDSWVDHIIGIMDALEIEKAHIVGNAFGGGLAIATALRYSERVDRMVLMGAAGTRFDVTEGLNAVWGYTPSIENMRNLLDIFAYDRSLVTDELARLRYEASIQPGFQESFSSMFPEPRQRWIDALASSDEDIKTLPNETLIIHGREDQVVPLSSSLRLGELIDRAQLHVFGRCGHWTQIEQTDRFNRLVVEFFNEA Hydrogen bonds contact Hydrophobic contact | ||||
| 19 | Pseudomonas Transcriptional activator protein LasR (Pseudo LasR) | 3IX3 | 5.09 | |
Target general information Gen name Pseudo LasR Organism Pseudomonas aeruginosa (strain ATCC 15692 / DSM 22644 / CIP 104116 / JCM 14847 / LMG 12228 / 1C / PRS 101 / PAO1) Uniprot ID TTD ID Synonyms NA Protein family Autoinducer-regulated transcriptional regulatory protein family Biochemical class NA Function Transcriptional activator of elastase structural gene (LasB). Binds to the PAI autoinducer. Related diseases Growth hormone deficiency, isolated, 1A (IGHD1A) [MIM:262400]: An autosomal recessive, severe deficiency of growth hormone leading to dwarfism. Patients often develop antibodies to administered growth hormone. {ECO:0000269|PubMed:8364549}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Growth hormone deficiency, isolated, 1B (IGHD1B) [MIM:612781]: An autosomal recessive deficiency of growth hormone leading to short stature. Patients have low but detectable levels of growth hormone, significantly retarded bone age, and a positive response and immunologic tolerance to growth hormone therapy. {ECO:0000269|PubMed:12655557}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Kowarski syndrome (KWKS) [MIM:262650]: A syndrome clinically characterized by short stature associated with bioinactive growth hormone, normal or slightly increased growth hormone secretion, pathologically low insulin-like growth factor 1 levels, and normal catch-up growth on growth hormone replacement therapy. {ECO:0000269|PubMed:17519310, ECO:0000269|PubMed:8552145, ECO:0000269|PubMed:9276733}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Growth hormone deficiency, isolated, 2 (IGHD2) [MIM:173100]: An autosomal dominant deficiency of growth hormone leading to short stature. Clinical severity is variable. Patients have a positive response and immunologic tolerance to growth hormone therapy. {ECO:0000269|PubMed:11502836, ECO:0000269|PubMed:9152628}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08324 Interacts with NA EC number NA Uniprot keywords 3D-structure; Activator; DNA-binding; Quorum sensing; Reference proteome; Transcription; Transcription regulation Protein physicochemical properties Chain ID A Molecular weight (Da) 18305.5 Length 163 Aromaticity 0.12 Instability index 46.52 Isoelectric point 5.19 Charge (pH=7) -6.78 2D Binding mode Binding energy (Kcal/mol) -6.95 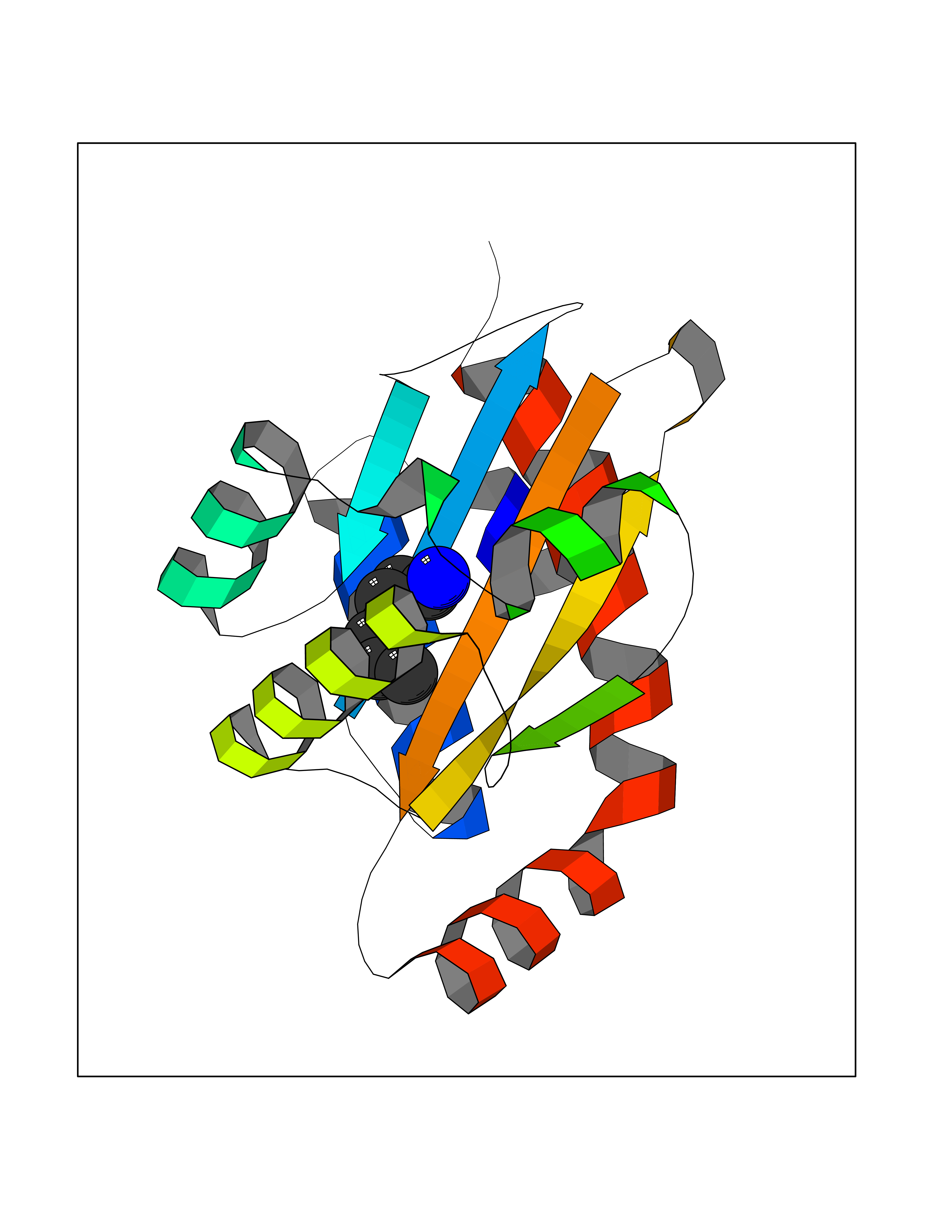 Molscript Map 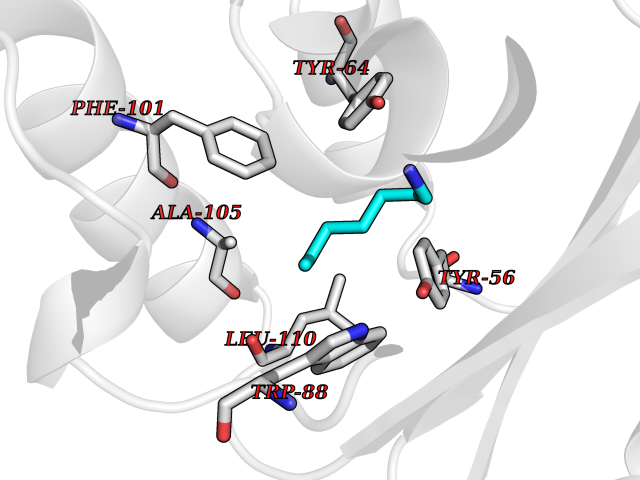 Pymol Map 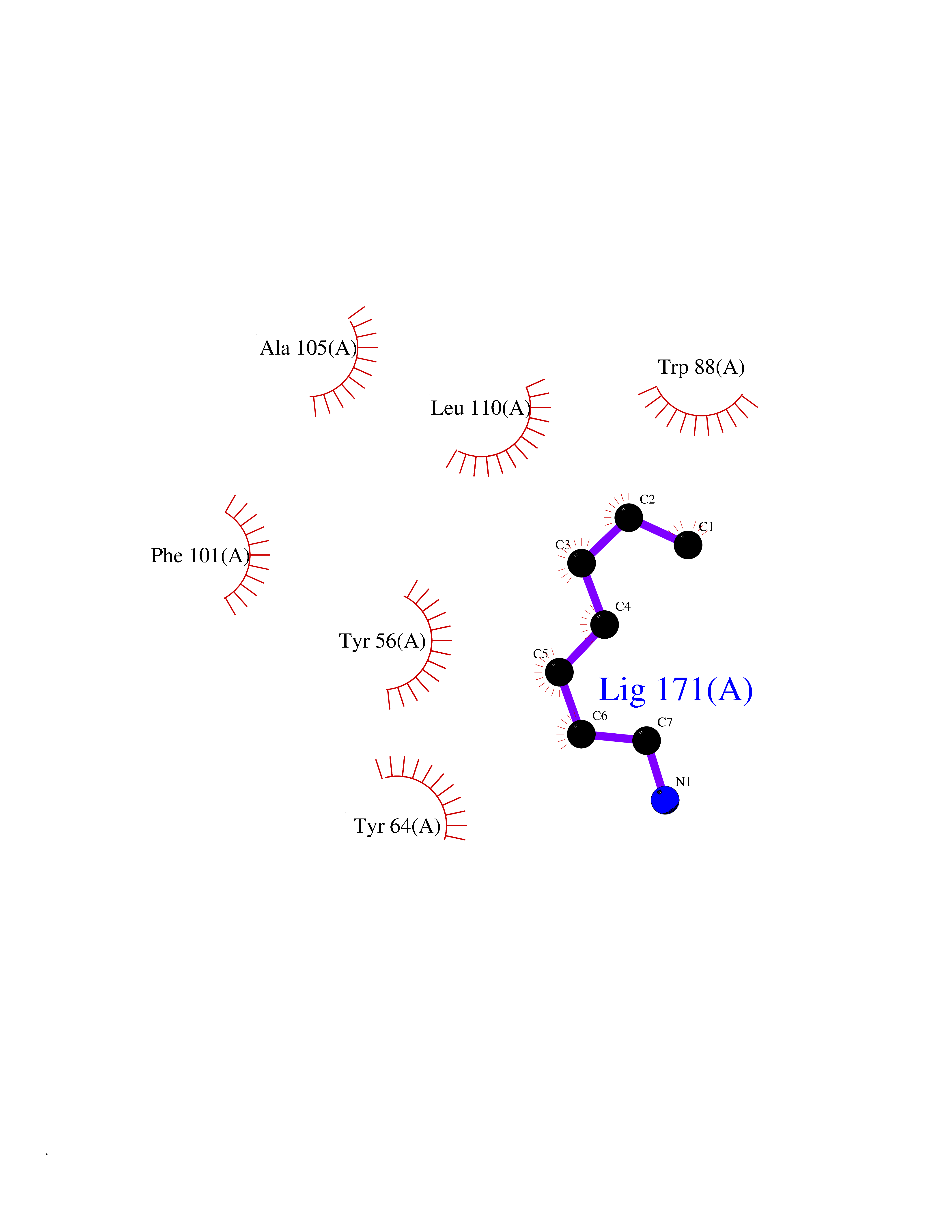 Ligplot Map 3D Binding mode Sequence FLELERSSGKLEWSAILQKMASDLGFSKILFGLLPKDSQDYENAFIVGNYPAAWREHYDRAGYARVDPTVSHCTQSVLPIFWEPSIYQTRKQHEFFEEASAAGLVYGLTMPLHGARGELGALSLSVEAENRAEANRFMESVLPTLWMLKDYALQSGAGLAFEH Hydrogen bonds contact Hydrophobic contact | ||||
| 20 | Neuronal acetylcholine receptor alpha-3/beta-4 (CHRNA3/B4) | 6PV7 | 5.09 | |
Target general information Gen name CHRNA3-CHRNB4 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Neuronal acetylcholine receptor Protein family Ligand-gated ion channel (TC 1.A.9) family, Acetylcholine receptor (TC 1.A.9.1) subfamily, Alpha-3/CHRNA3 sub-subfamily Biochemical class Neurotransmitter receptor Function A type of nicotinic acetylcholine receptor, consisting of 3 and 4 subunits. Related diseases Bladder dysfunction, autonomic, with impaired pupillary reflex and secondary CAKUT (BAIPRCK) [MIM:191800]: An autosomal recessive disease characterized by impaired innervation and autonomic dysfunction of the urinary bladder, hydronephrosis, vesicoureteral reflux, small kidneys, recurrent urinary tract infections, and progressive renal insufficiency. Additional autonomic features are impaired pupillary reflex and orthostatic hypotension. The disease manifests in utero or early childhood. {ECO:0000269|PubMed:31708116}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00915; DB01156; DB00237; DB00565; DB09028; DB00514; DB07720; DB00898; DB00472; DB05710; DB01227; DB00848; DB00333; DB00184; DB01090; DB00202; DB01273 Interacts with NA EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disease variant; Disulfide bond; Endoplasmic reticulum; Glycoprotein; Golgi apparatus; Ion channel; Ion transport; Ligand-gated ion channel; Membrane; Phosphoprotein; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Signal; Synapse; Transmembrane; Transmembrane helix; Transport; Ubl conjugation Protein physicochemical properties Chain ID A,B Molecular weight (Da) 89661.2 Length 775 Aromaticity 0.12 Instability index 35.11 Isoelectric point 6.07 Charge (pH=7) -5.67 2D Binding mode Binding energy (Kcal/mol) -6.94 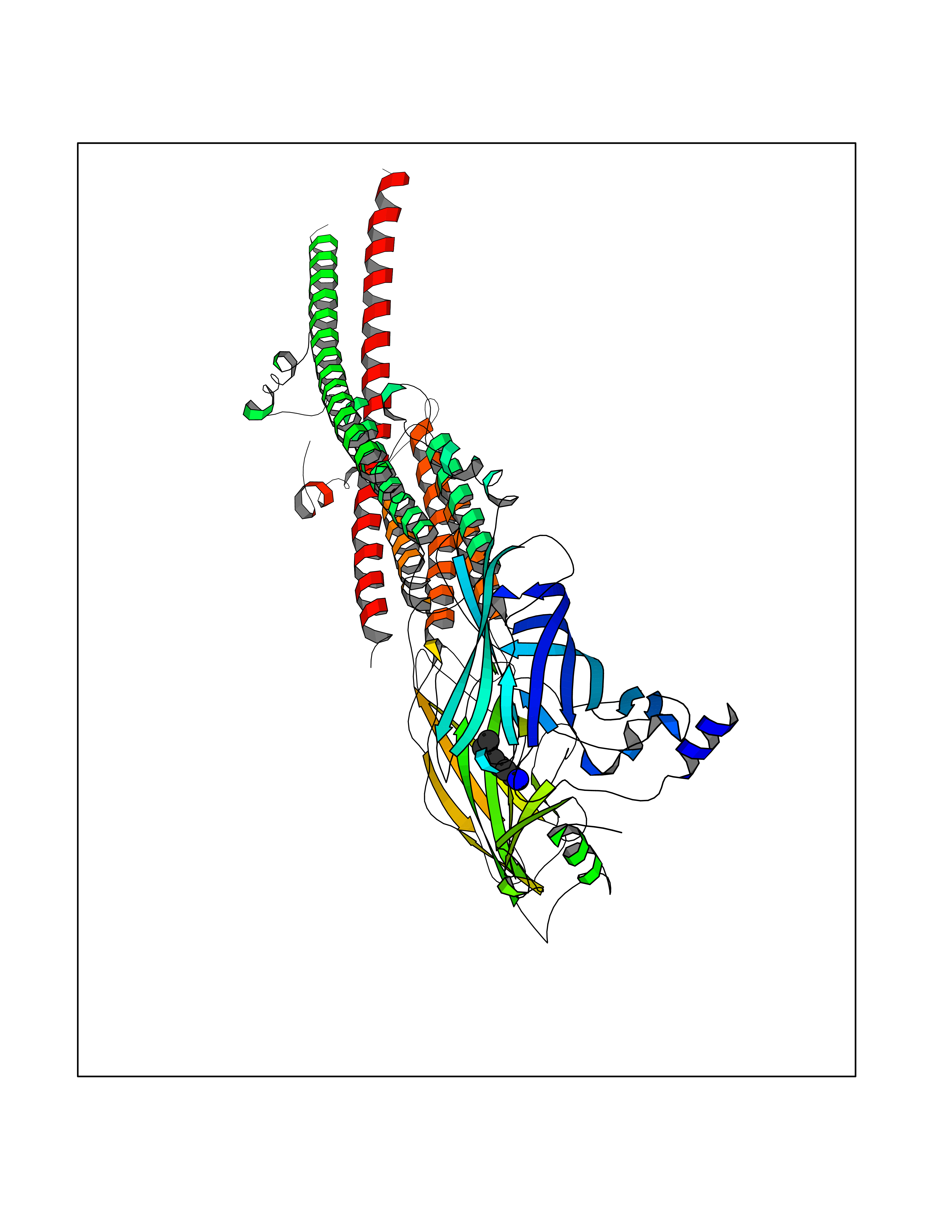 Molscript Map 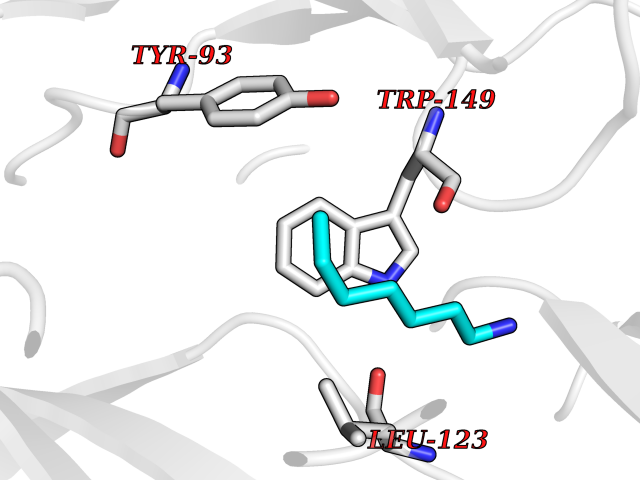 Pymol Map 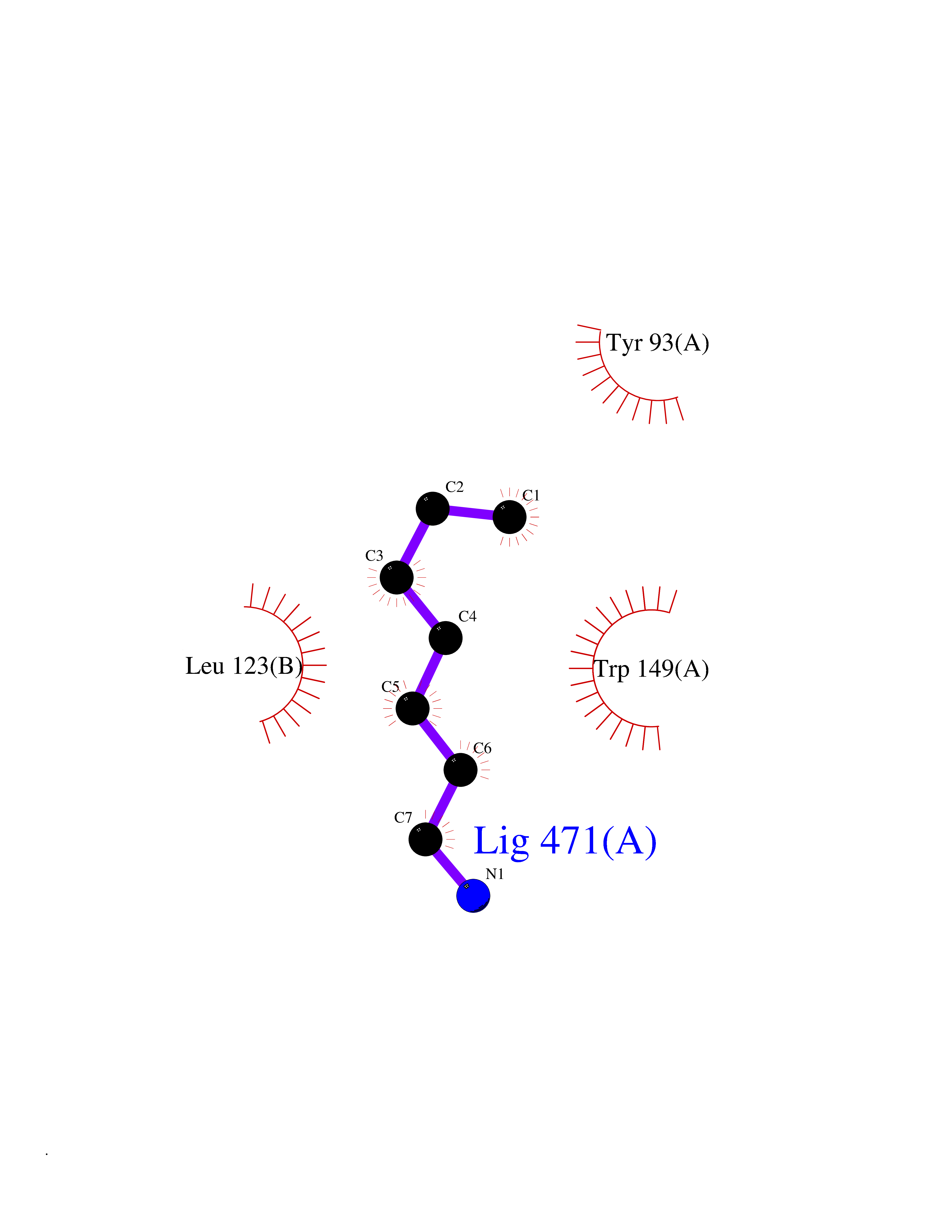 Ligplot Map 3D Binding mode Sequence SEAEHRLFERLFEDYNEIIRPVANVSDPVIIHFEVSMSQLVKVDEVNQIMETNLWLKQIWNDYKLKWNPSDYGGAEFMRVPAQKIWKPDIVLYNNAVGDFQVDDKTKALLKYTGEVTWIPPAIFKSSCKIDVTYFPFDYQNCTMKFGSWSYDKAKIDLVLIGSSMNLKDYWESGEWAIIKAPGYKHDIKYNCCEEIYPDITYSLYIRRLPLFYTINLIIPCLLISFLTVLVFYLPSDCGEKVTLCISVLLSLTVFLLVITETIPSTSLVIPLIGEYLLFTMIFVTLSIVITVFVLNVHYRTPTTHTMPSWVKTVFLNLLPRVMFMTRIKEAIQSVKYIAENMKAQNEAKEIQDDWKYVAMVIDRIFLWVFTLVCILGTAGLFLQPLMRVANAEEKLMDDLLNKTRYNNLIRPATSSSQLISIKLQLSLAQLISVNEREQIMTTNVWLKQEWTDYRLTWNSSRYEGVNILRIPAKRIWLPDIVLYNNADGTYEVSVYTNLIVRSNGSVLWLPPAIYKSACKIEVKYFPFDQQNCTLKFRSWTYDHTEIDMVLMTPTASMDDFTPSGEWDIVALPGRRTVNPQDPSYVDVTYDFIIKRKPLFYTINLIIPCVLTTLLAILVFYLPSDCGEKMTLCISVLLALTFFLLLISKIVPPTSLDVPLIGKYLMFTMVLVTFSIVTSVCVLNVHHRSPSTHTMAPWVKRCFLHKLPTFLFMKRRQDVQEALEGVSFIAQHMKNDDEDQSVVEDWKYVAMVVDRLFLWVFMFVCVLGTVGLFLP Hydrogen bonds contact Hydrophobic contact | ||||