Job Results:
Ligand
Structure
Job ID
d1681d1a563fba4481a5af742fb3bb0c
Job name
NA
Time
2026-02-27 11:58:22
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 1 | Penicillin-binding protein 1A | 2ZC6 | 4.74 | |
Target general information Gen name ponA Organism Streptococcus pneumoniae serotype 4 (strain ATCC BAA-334 / TIGR4) Uniprot ID TTD ID NA Synonyms SP_0369 Protein family Glycosyltransferase 51 family; Transpeptidase family Biochemical class Biosynthetic protein Function Penicillin binding.Peptidoglycan glycosyltransferase activity.Serine-type D-Ala-D-Ala carboxypeptidase activity. Related diseases Aminoacylase-1 deficiency (ACY1D) [MIM:609924]: An enzymatic deficiency resulting in encephalopathy, unspecific psychomotor delay, psychomotor delay with atrophy of the vermis and syringomyelia, marked muscular hypotonia or normal clinical features. Epileptic seizures are a frequent feature. All affected individuals exhibit markedly increased urinary excretion of several N-acetylated amino acids. {ECO:0000269|PubMed:16274666, ECO:0000269|PubMed:16465618, ECO:0000269|PubMed:17562838, ECO:0000269|PubMed:21414403}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01150; DB05659 Interacts with NA EC number 2.4.99.28; 3.4.16.4 Uniprot keywords 3D-structure; Antibiotic resistance; Carboxypeptidase; Cell shape; Cell wall biogenesis/degradation; Glycosyltransferase; Hydrolase; Multifunctional enzyme; Peptidoglycan synthesis; Protease; Reference proteome; Secreted; Transferase Protein physicochemical properties Chain ID A,C Molecular weight (Da) 44805.1 Length 400 Aromaticity 0.12 Instability index 31.82 Isoelectric point 4.88 Charge (pH=7) -14.68 2D Binding mode Binding energy (Kcal/mol) -6.46 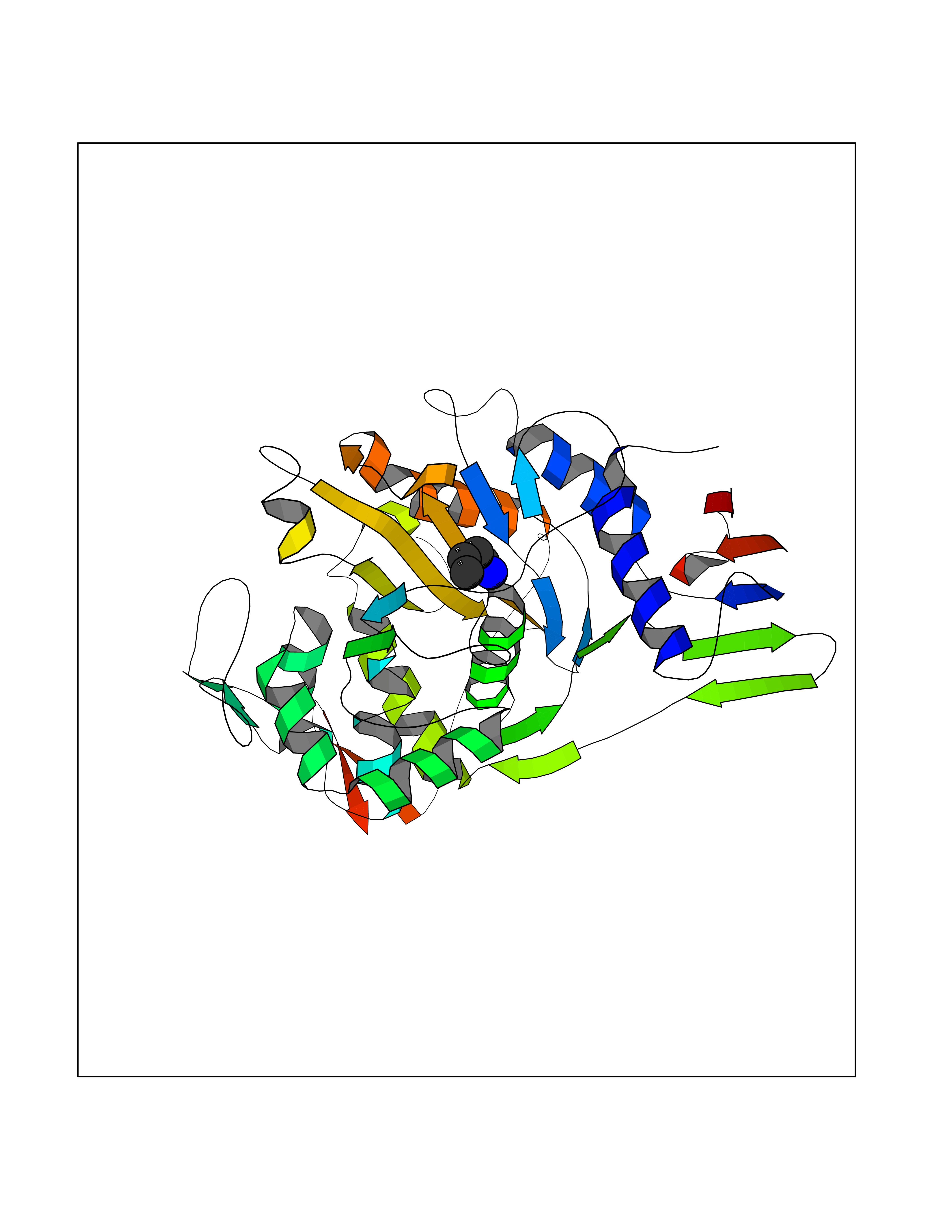 Molscript Map 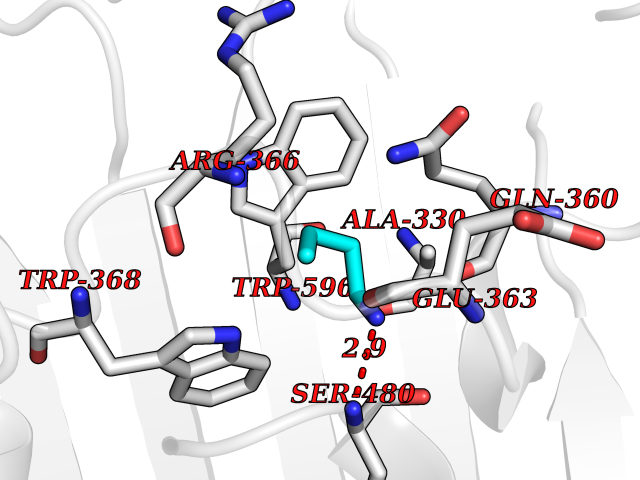 Pymol Map 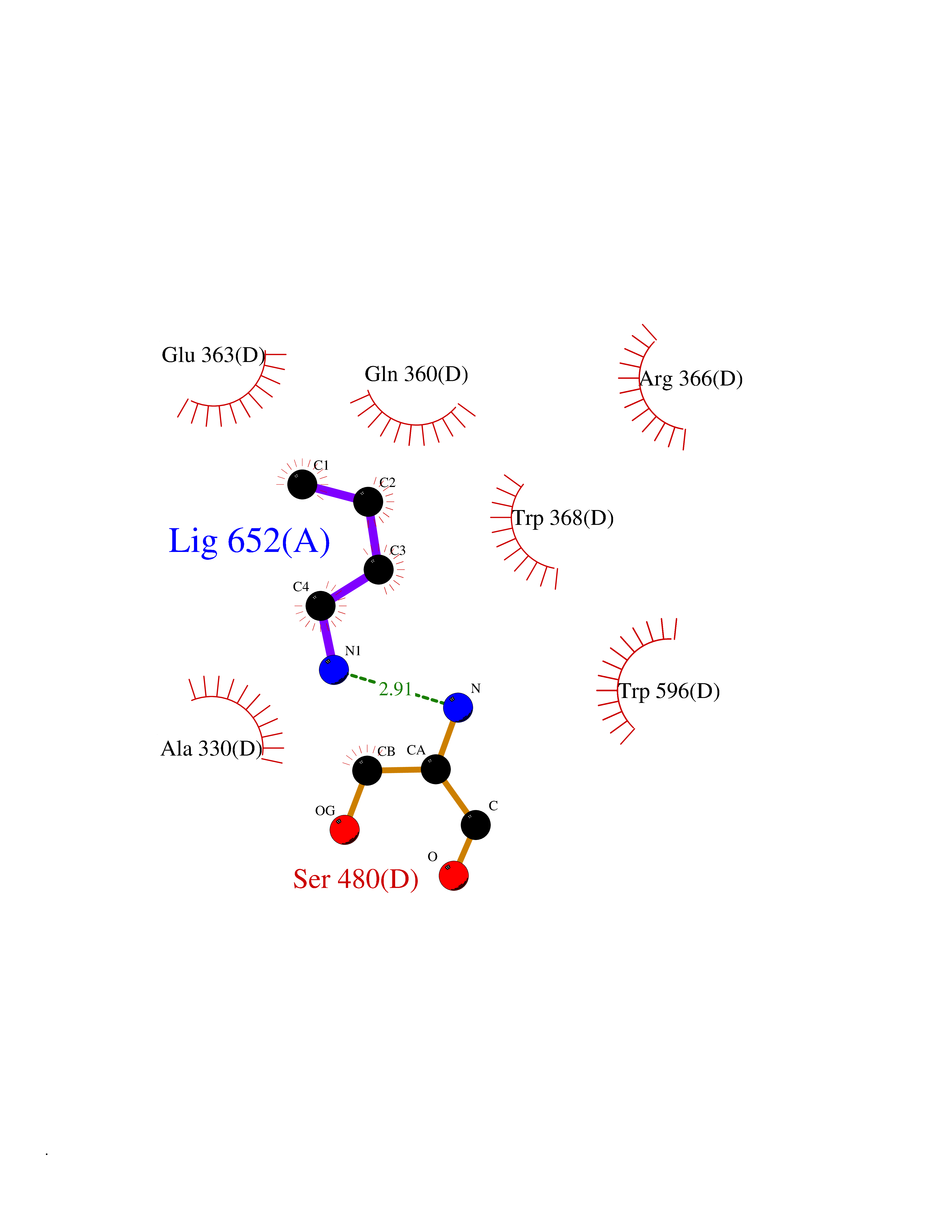 Ligplot Map 3D Binding mode Sequence NYPAYMDNYLKEVINQVEEETGYNLLTTGMDVYTNVDQEAQKHLWDIYNTDEYVAYPDDELQVASTIVDVSNGKVIAQLGARHQSSNVSFGINQAVETNRDWGSTMKPITDYAPALEYGVYDSTATIVHDEPYNYPGTNTPVYNWDRGYFGNITLQYALQQSRNVPAVETLNKVGLNRAKTFLNGLGIDYPSIHYSNAISSNTTESDKKYGASSEKMAAAYAAFANGGTYYKPMYIHKVVFSDGSEKEFSNVGTRAMKETTAYMMTDMMKTVLSYGTGQNAYLAWLPQAGKTGTSNYTDEEIENHIKTSQFVAPDELFAGYTRKYSMAVWTGYSNRLTPLVGNGLTVAAKVYRSMMTYLSEGSNPEDWNIPEGLYRNGEFVFKNTSSKIYDNKNQLIADL Hydrogen bonds contact Hydrophobic contact | ||||
| 2 | Lysine-specific demethylase 4C (KDM4C) | 4XDO | 4.74 | |
Target general information Gen name KDM4C Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms KIAA0780; Jumonji domain-containing protein 2C; JmjC domain-containing histone demethylation protein 3C; JMJD2C; JHDM3C; Gene amplified in squamous cell carcinoma 1 protein; GASC1; GASC-1 protein Protein family JHDM3 histone demethylase family Biochemical class Paired donor oxygen oxidoreductase Function Does not demethylate histone H3 'Lys-4', H3 'Lys-27' nor H4 'Lys-20'. Demethylates trimethylated H3 'Lys-9' and H3 'Lys-36' residue, while it has no activity on mono- and dimethylated residues. Demethylation of Lys residue generates formaldehyde and succinate. Histone demethylase that specifically demethylates 'Lys-9' and 'Lys-36' residues of histone H3, thereby playing a central role in histone code. Related diseases Renal cell carcinoma (RCC) [MIM:144700]: Renal cell carcinoma is a heterogeneous group of sporadic or hereditary carcinoma derived from cells of the proximal renal tubular epithelium. It is subclassified into clear cell renal carcinoma (non-papillary carcinoma), papillary renal cell carcinoma, chromophobe renal cell carcinoma, collecting duct carcinoma with medullary carcinoma of the kidney, and unclassified renal cell carcinoma. Clear cell renal cell carcinoma is the most common subtype. {ECO:0000269|PubMed:20054297, ECO:0000269|PubMed:23622243, ECO:0000269|PubMed:23792563, ECO:0000269|PubMed:25728682}. The disease may be caused by variants affecting the gene represented in this entry. Defects of SETD2 are associated with loss of DNA methylation at non-promoter regions (PubMed:23792563). SETD2 defects lead to aberrant and reduced nucleosome compaction and chromatin association of key replication proteins, such as MCM7 and DNA polymerase delta, leading to hinder replication fork progression and prevent loading of RAD51 homologous recombination repair factor at DNA breaks (PubMed:25728682). {ECO:0000269|PubMed:23792563, ECO:0000269|PubMed:25728682}.; DISEASE: Luscan-Lumish syndrome (LLS) [MIM:616831]: An autosomal dominant syndrome with a variable phenotype. Clinical features include macrocephaly, distinctive facial appearance, postnatal overgrowth, various degrees of learning difficulties, autism spectrum disorder, and intellectual disability. {ECO:0000269|PubMed:23160955, ECO:0000269|PubMed:24852293, ECO:0000269|PubMed:26084711, ECO:0000269|PubMed:27317772}. The disease may be caused by variants affecting the gene represented in this entry.; DISEASE: Leukemia, acute lymphoblastic (ALL) [MIM:613065]: A subtype of acute leukemia, a cancer of the white blood cells. ALL is a malignant disease of bone marrow and the most common malignancy diagnosed in children. The malignant cells are lymphoid precursor cells (lymphoblasts) that are arrested in an early stage of development. The lymphoblasts replace the normal marrow elements, resulting in a marked decrease in the production of normal blood cells. Consequently, anemia, thrombocytopenia, and neutropenia occur to varying degrees. The lymphoblasts also proliferate in organs other than the marrow, particularly the liver, spleen, and lymphnodes. {ECO:0000269|PubMed:24509477, ECO:0000269|PubMed:24662245}. The disease may be caused by variants affecting distinct genetic loci, including the gene represented in this entry.; DISEASE: Leukemia, acute myelogenous (AML) [MIM:601626]: A subtype of acute leukemia, a cancer of the white blood cells. AML is a malignant disease of bone marrow characterized by maturational arrest of hematopoietic precursors at an early stage of development. Clonal expansion of myeloid blasts occurs in bone marrow, blood, and other tissue. Myelogenous leukemias develop from changes in cells that normally produce neutrophils, basophils, eosinophils and monocytes. {ECO:0000269|PubMed:16314571, ECO:0000269|PubMed:24509477}. The disease may be caused by variants affecting distinct genetic loci, including the gene represented in this entry.; DISEASE: Intellectual developmental disorder, autosomal dominant 70 (MRD70) [MIM:620157]: An autosomal dominant disorder characterized by mild global developmental delay, moderately impaired intellectual disability with speech difficulties, and behavioral abnormalities. {ECO:0000269|PubMed:32710489}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Rabin-Pappas syndrome (RAPAS) [MIM:620155]: An autosomal dominant neurodevelopmental disorder characterized by severely impaired global development, intellectual disability, microcephaly, facial dysmorphism, and variable congenital anomalies affecting the skeletal, genitourinary, cardiac, and other organ systems. {ECO:0000269|PubMed:32710489}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number EC 1.14.11.- Uniprot keywords 3D-structure; Alternative splicing; Chromatin regulator; Dioxygenase; Iron; Metal-binding; Nucleus; Oxidoreductase; Proteomics identification; Reference proteome; Repeat; Transcription; Transcription regulation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A Molecular weight (Da) 39355.6 Length 338 Aromaticity 0.14 Instability index 38.34 Isoelectric point 8.04 Charge (pH=7) 2.41 2D Binding mode Binding energy (Kcal/mol) -6.46  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence LNPSCKIMTFRPSMEEFREFNKYLAYMESKGAHRAGLAKVIPPKEWKPRQCYDDIDNLLIPAPIQQMVTGQSGLFTQYNIQKKAMTVKEFRQLANSGKYCTPRYLDYEDLERKYWKNLTFVAPIYGADINGSIYDEGVDEWNIARLNTVLDVVEEECGISIEGVNTPYLYFGMWKTTFAWHTEDMDLYSINYLHFGEPKSWYAIPPEHGKRLERLAQGFFPSSSQGCDAFLRHKMTLISPSVLKKYGIPFDKITQEAGEFMITFPYGYHAGFNHGFNCAESTNFATVRWIDYGKVAKLCTCRKDMVKISMDIFVRKFQPDRYQLWKQGKDIYTIDHTK Hydrogen bonds contact Hydrophobic contact | ||||
| 3 | Medium-chain specific acyl-CoA dehydrogenase, mitochondrial | 4P13 | 4.70 | |
Target general information Gen name ACADM Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Acyl-CoA dehydrogenase family Biochemical class Oxidoreductase Function Acyl-CoA dehydrogenase activity.Flavin adenine dinucleotide binding.Identical protein binding.Medium-chain-acyl-CoA dehydrogenase activity. Related diseases Acyl-CoA dehydrogenase medium-chain deficiency (ACADMD) [MIM:201450]: An inborn error of mitochondrial fatty acid beta-oxidation which causes fasting hypoglycemia, hepatic dysfunction and encephalopathy, often resulting in death in infancy. {ECO:0000269|PubMed:10767181, ECO:0000269|PubMed:11349232, ECO:0000269|PubMed:11409868, ECO:0000269|PubMed:11486912, ECO:0000269|PubMed:1363805, ECO:0000269|PubMed:1671131, ECO:0000269|PubMed:1684086, ECO:0000269|PubMed:1902818, ECO:0000269|PubMed:2251268, ECO:0000269|PubMed:2393404, ECO:0000269|PubMed:2394825, ECO:0000269|PubMed:7603790, ECO:0000269|PubMed:7929823, ECO:0000269|PubMed:8198141, ECO:0000269|PubMed:9158144, ECO:0000269|PubMed:9882619}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03415; DB03147; DB02910 Interacts with PRO_0000000502 [P11310] EC number 1.3.8.7 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Direct protein sequencing; Disease variant; FAD; Fatty acid metabolism; Flavoprotein; Lipid metabolism; Mitochondrion; Oxidoreductase; Phosphoprotein; Proteomics identification; Reference proteome; Transit peptide Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 85080.3 Length 773 Aromaticity 0.09 Instability index 30.55 Isoelectric point 5.71 Charge (pH=7) -7.7 2D Binding mode Binding energy (Kcal/mol) -6.41  Molscript Map 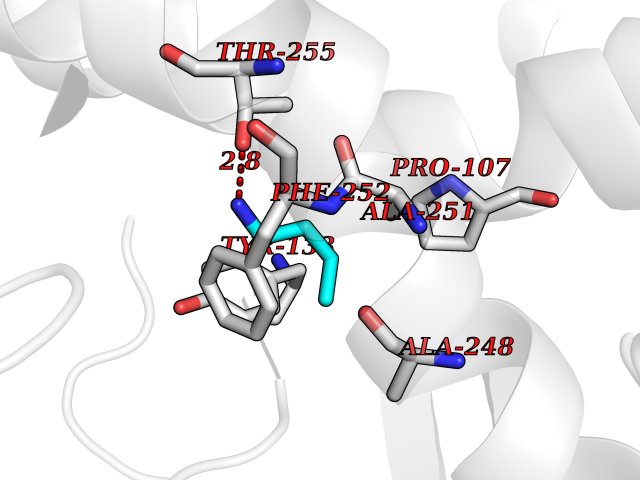 Pymol Map 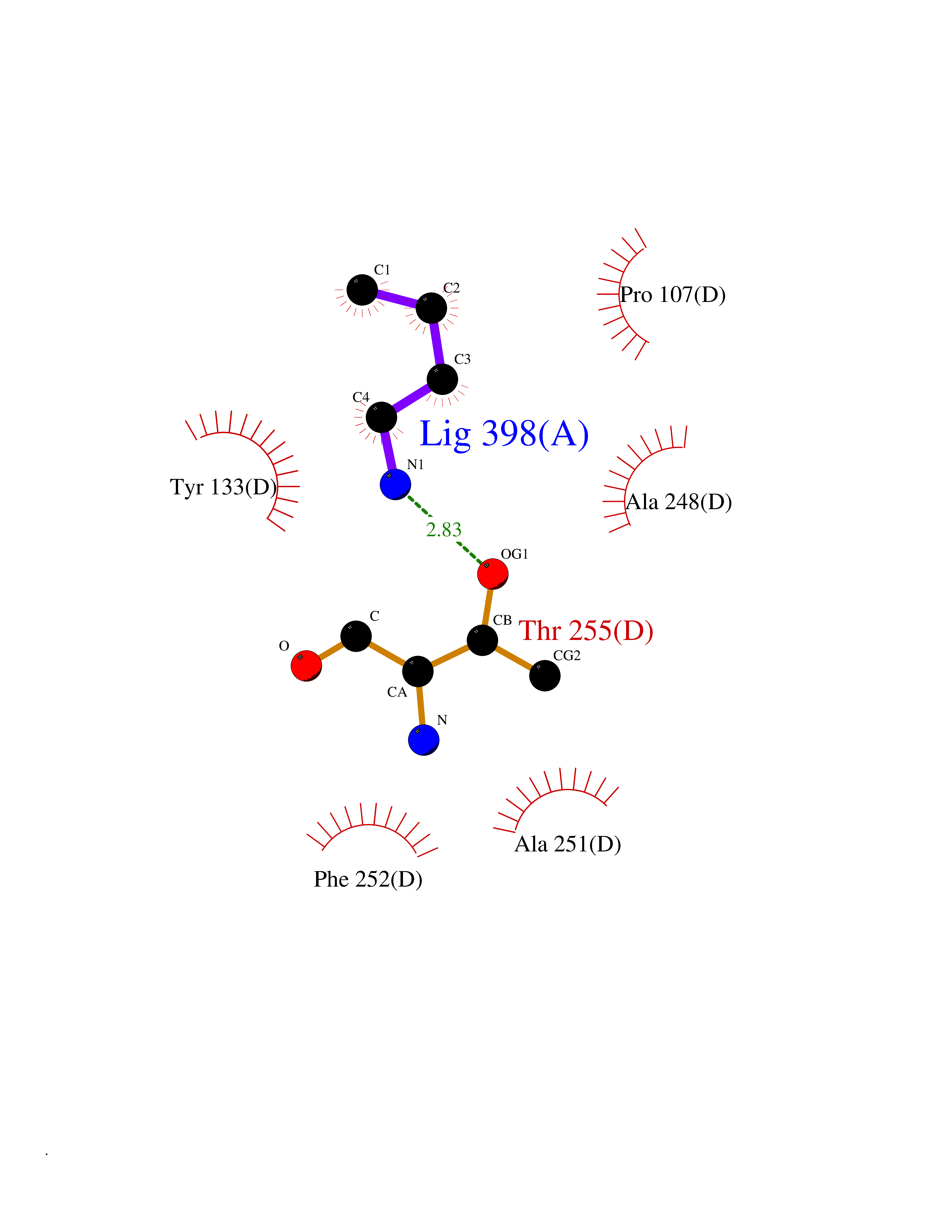 Ligplot Map 3D Binding mode Sequence LGFSFEFTEQQKEFQATARKFAREEIIPVAAEYDKTGEYPVPLIRRAWELGLMNTHIPENCGGLGLGTFDACLISEELAYGCTGVQTAIEGNSLGQMPIIIAGNDQQKKKYLGRMTEEPLMCAYCVTEPGAGSDVAGIKTKAEKKGDEYIINGQKMWITNGGKANWYFLLARSDPDPKAPANKAFTGFIVEADTPGIQIGRKELNMGQRCSDTRGIVFEDVKVPKENVLIGDGAGFKVAMGAFDKTRPVVAAGAVGLAQRALDEATKYALERKTFGKLLVEHQAISFMLAEMAMEVELARMSYQRAAWEVDSGRRNTYYASIAKAFAGDIANQLATDAVQILGGNGFNTEYPVEKLMRDAKIYQIYEGTSQIQRLIVAREHIDKYKLGFSFEFTEQQKEFQATARKFAREEIIPVAAEYDKTGEYPVPLIRRAWELGLMNTHIPENCGGLGLGTFDACLISEELAYGCTGVQTAIEGNSLGQMPIIIAGNDQQKKKYLGRMTEEPLMCAYCVTEPGAGSDVAGIKTKAEKKGDEYIINGQKMWITNGGKANWYFLLARSDPDPKAPANKAFTGFIVEADTPGIQIGRKELNMGQRCSDTRGIVFEDVKVPKENVLIGDGAGFKVAMGAFDKTRPVVAAGAVGLAQRALDEATKYALERKTFGKLLVEHQAISFMLAEMAMEVELARMSYQRAAWEVDSGRRNTYYASIAKAFAGDIANQLATDAVQILGGNGFNTEYPVEKLMRDAKIYQIYEGTSQIQRLIVAREHIDKYKN Hydrogen bonds contact Hydrophobic contact | ||||
| 4 | Ubiquitin thioesterase L3 (UCHL3) | 1XD3 | 4.69 | |
Target general information Gen name UCHL3 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Ubiquitin carboxyl-terminal hydrolase isozyme L3; UCH-L3 Protein family Peptidase C12 family Biochemical class Peptidase Function Thiol protease that recognizes and hydrolyzes a peptide bond at the C-terminal glycine of either ubiquitin or NEDD8. Has a 10-fold preference for Arg and Lys at position P3", and exhibits a preference towards 'Lys-48'-linked ubiquitin chains. Deubiquitinates ENAC in apical compartments, thereby regulating apical membrane recycling. Indirectly increases the phosphorylation of IGFIR, AKT and FOXO1 and promotes insulin-signaling and insulin-induced adipogenesis. Required for stress-response retinal, skeletal muscle and germ cell maintenance. May be involved in working memory. Can hydrolyze UBB(+1), a mutated form of ubiquitin which is not effectively degraded by the proteasome and is associated with neurogenerative disorders. Deubiquitinating enzyme (DUB) that controls levels of cellular ubiquitin through processing of ubiquitin precursors and ubiquitinated proteins. Related diseases Epilepsy, familial focal, with variable foci 4 (FFEVF4) [MIM:617935]: An autosomal dominant form of epilepsy characterized by focal seizures arising from different cortical regions, including the temporal, frontal, parietal, and occipital lobes. Seizure types commonly include temporal lobe epilepsy, frontal lobe epilepsy, and nocturnal frontal lobe epilepsy. Some patients may have intellectual disability or autism spectrum disorders. Seizure onset usually occurs in the first or second decades, although later onset has been reported, and there is phenotypic variability within families. A subset of patients have structural brain abnormalities. Penetrance of the disorder is incomplete. FFEVF4 is characterized by onset of focal seizures in the first years of life. {ECO:0000269|PubMed:24157691, ECO:0000269|PubMed:28235671}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Developmental and epileptic encephalopathy 62 (DEE62) [MIM:617938]: A form of epileptic encephalopathy, a heterogeneous group of severe early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE62 is characterized by onset of seizures in the first year of life. {ECO:0000269|PubMed:29466837}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q9H078; G5E9A7; Q15797; Q7Z699 EC number EC 3.4.19.12 Uniprot keywords 3D-structure; Cytoplasm; Hydrolase; Phosphoprotein; Protease; Proteomics identification; Reference proteome; Thiol protease; Ubl conjugation pathway Protein physicochemical properties Chain ID A,B Molecular weight (Da) 34540.8 Length 304 Aromaticity 0.07 Instability index 39.91 Isoelectric point 5.01 Charge (pH=7) -17.24 2D Binding mode Binding energy (Kcal/mol) -6.39  Molscript Map 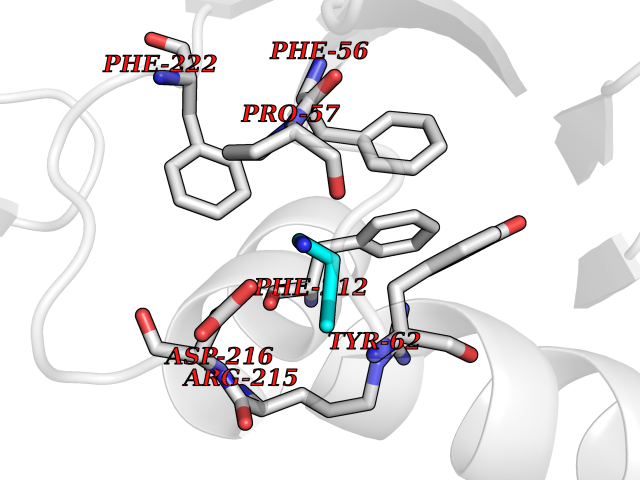 Pymol Map 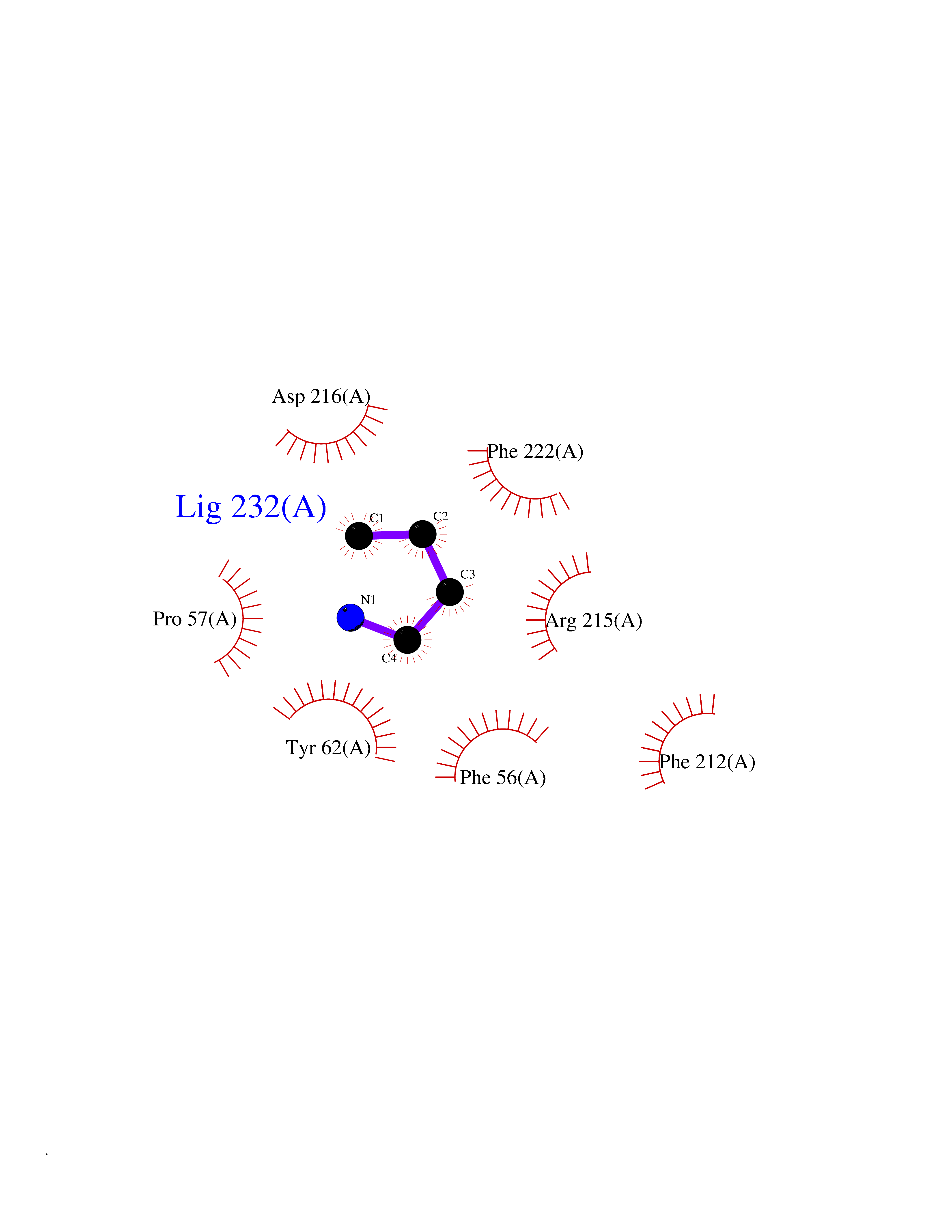 Ligplot Map 3D Binding mode Sequence EGQRWLPLEANPEVTNQFLKQLGLHPNWQFVDVYGMDPELLSMVPRPVCAVLLLFPITEKYEVFRTEEEEKIKSQGQDVTSSVYFMKQTISNACGTIGLIHAIANNKDKMHFESGSTLKKFLEESVSMSPEERARYLENYDAIRVTHETSAHEGQTEAPSIDEKVDLHFIALVHVDGHLYELDGRKPFPINHGETSDETLLEDAIEVCKKFMERDPDELRFNAIALSAAMQIFVKTLTGKTITLEVEPSDTIENVKAKIQDKEGIPPDQQRLIFAGKQLEDGRTLSDYNIQKESTLHLVLRLRG Hydrogen bonds contact Hydrophobic contact | ||||
| 5 | Ribonucleoside-diphosphate reductase subunit M2 | 3OLJ | 4.68 | |
Target general information Gen name RRM2 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms RR2 Protein family Ribonucleoside diphosphate reductase small chain family Biochemical class Oxidoreductase Function Metal ion binding.Ribonucleoside-diphosphate reductase activity, thioredoxin disulfide as acceptor. Related diseases Pyruvate kinase hyperactivity (PKHYP) [MIM:102900]: Autosomal dominant phenotype characterized by increase of red blood cell ATP. {ECO:0000269|PubMed:9090535}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Pyruvate kinase deficiency of red cells (PKRD) [MIM:266200]: A frequent cause of hereditary non-spherocytic hemolytic anemia. Clinically, pyruvate kinase-deficient patients suffer from a highly variable degree of chronic hemolysis, ranging from severe neonatal jaundice and fatal anemia at birth, severe transfusion-dependent chronic hemolysis, moderate hemolysis with exacerbation during infection, to a fully compensated hemolysis without apparent anemia. {ECO:0000269|PubMed:10087985, ECO:0000269|PubMed:10772876, ECO:0000269|PubMed:11328279, ECO:0000269|PubMed:11960989, ECO:0000269|PubMed:1536957, ECO:0000269|PubMed:1896471, ECO:0000269|PubMed:19085939, ECO:0000269|PubMed:2018831, ECO:0000269|PubMed:21794208, ECO:0000269|PubMed:7706479, ECO:0000269|PubMed:8161798, ECO:0000269|PubMed:8180378, ECO:0000269|PubMed:8476433, ECO:0000269|PubMed:8481523, ECO:0000269|PubMed:8483951, ECO:0000269|PubMed:8664896, ECO:0000269|PubMed:8807089, ECO:0000269|PubMed:9075576, ECO:0000269|PubMed:9482576, ECO:0000269|PubMed:9827908, ECO:0000269|PubMed:9886305, ECO:0000269|Ref.24}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00242; DB05260; DB05801; DB05003; DB05428 Interacts with P41002; Q9UM11; P23921; O00560 EC number 1.17.4.1 Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; Deoxyribonucleotide synthesis; Iron; Metal-binding; Nucleus; Oxidoreductase; Phosphoprotein; Proteomics identification; Reference proteome; Ubl conjugation Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 33579.4 Length 286 Aromaticity 0.14 Instability index 43.7 Isoelectric point 5.12 Charge (pH=7) -12.86 2D Binding mode Binding energy (Kcal/mol) -6.39 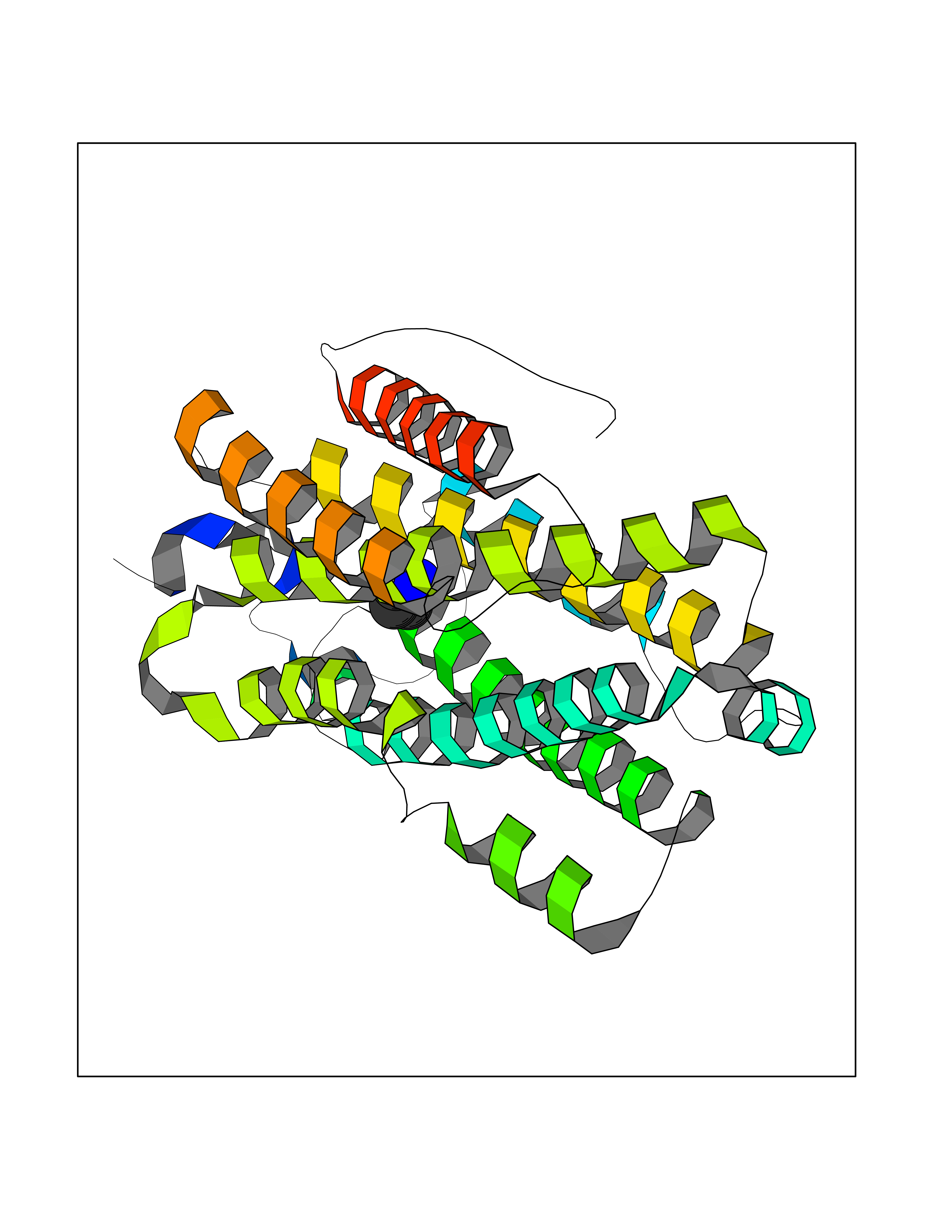 Molscript Map 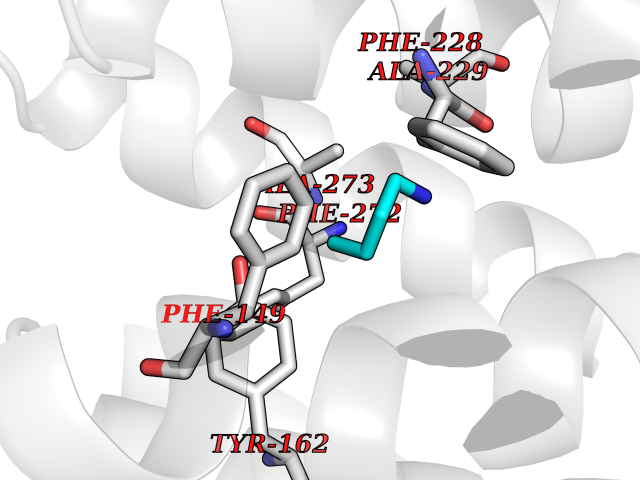 Pymol Map 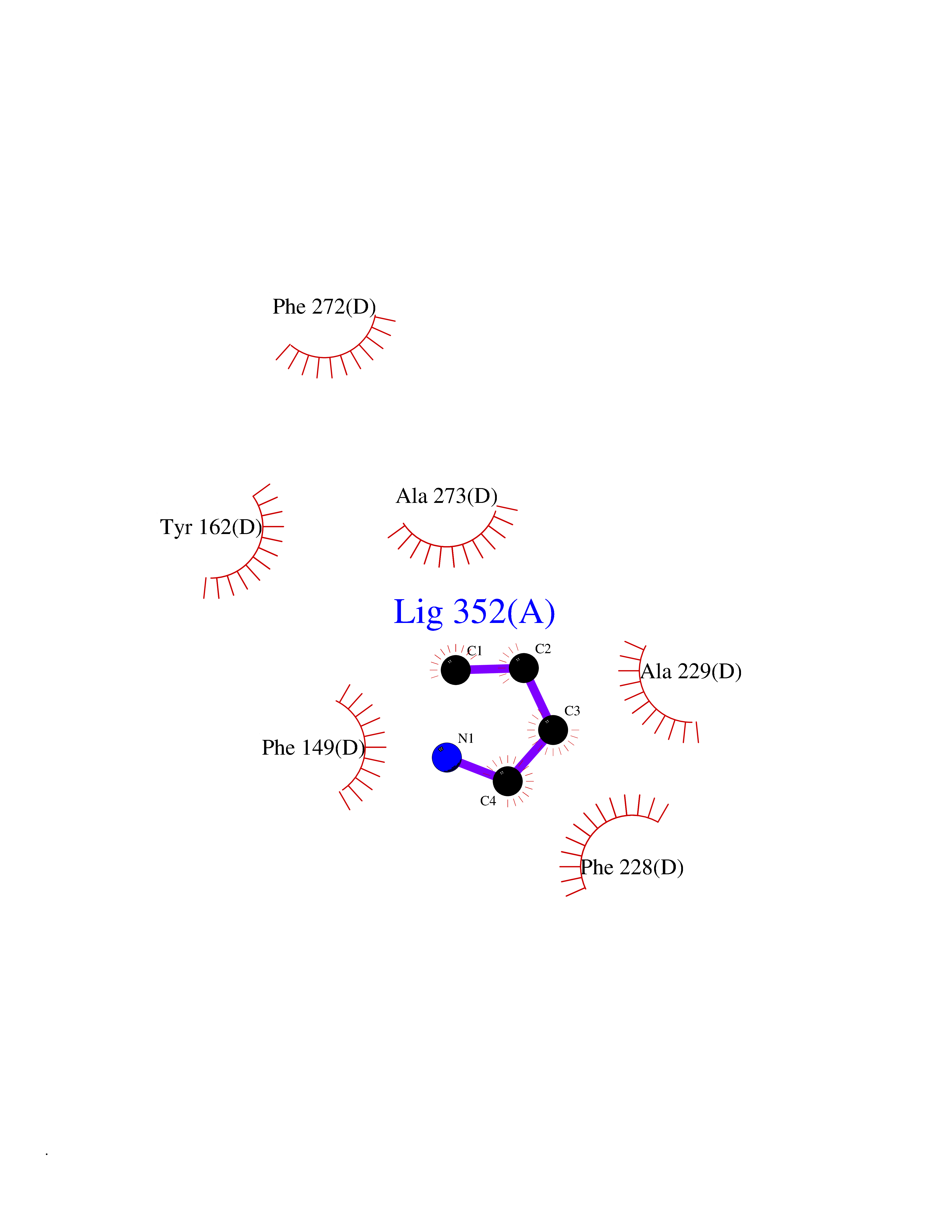 Ligplot Map 3D Binding mode Sequence MGVEDEPLLRENPRRFVIFPIEYHDIWQMYKKAEASFWTAEEVDLSKDIQHWESLKPEERYFISHVLAFFAASDGIVNENLVERFSQEVQITEARCFYGFQIAMENIHSEMYSLLIDTYIKDPKEREFLFNAIETMPCVKKKADWALRWIGDKEATYGERVVAFAAVEGIFFSGSFASIFWLKKRGLMPGLTFSNELISRDEGLHCDFACLMFKHLVHKPSEERVREIIINAVRIEQEFLTEALPVKLIGMNCTLMKQYIEFVADRLMLELGFSKVFRVENPFDFM Hydrogen bonds contact Hydrophobic contact | ||||
| 6 | Neuronal acetylcholine receptor subunit alpha-2 | 5FJV | 4.68 | |
Target general information Gen name CHRNA2 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Ligand-gated ion channel (TC 1.A.9) family, Acetylcholine receptor (TC 1.A.9.1) subfamily, Alpha-2/CHRNA2 sub-subfamily Biochemical class NA Function After binding acetylcholine, the AChR responds by an extensive change in conformation that affects all subunits and leads to opening of an ion-conducting channel across the plasma membrane." Related diseases Epilepsy, nocturnal frontal lobe, 4 (ENFL4) [MIM:610353]: An autosomal dominant focal epilepsy characterized by nocturnal seizures associated with fear sensation, tongue movements, and nocturnal wandering, closely resembling nightmares and sleep walking. {ECO:0000269|PubMed:16826524}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Seizures, benign familial infantile, 6 (BFIS6) [MIM:610353]: A form of benign familial infantile epilepsy, a neurologic disorder characterized by afebrile seizures occurring in clusters during the first year of life, without neurologic sequelae. BFIS6 inheritance is autosomal dominant. {ECO:0000269|PubMed:25847220}. The disease may be caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00732; DB00237; DB00411; DB00565; DB01245; DB00514; DB01135; DB07720; DB00898; DB00472; DB00483; DB08960; DB00657; DB01336; DB00416; DB01226; DB00184; DB01337; DB01338; DB00721; DB00728; DB05740; DB00202; DB01199; DB01339 Interacts with NA EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disease variant; Disulfide bond; Epilepsy; Glycoprotein; Ion channel; Ion transport; Ligand-gated ion channel; Membrane; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Signal; Synapse; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A,B,C,D,E Molecular weight (Da) 120584 Length 1031 Aromaticity 0.14 Instability index 32.21 Isoelectric point 5.62 Charge (pH=7) -17.58 2D Binding mode Binding energy (Kcal/mol) -6.39 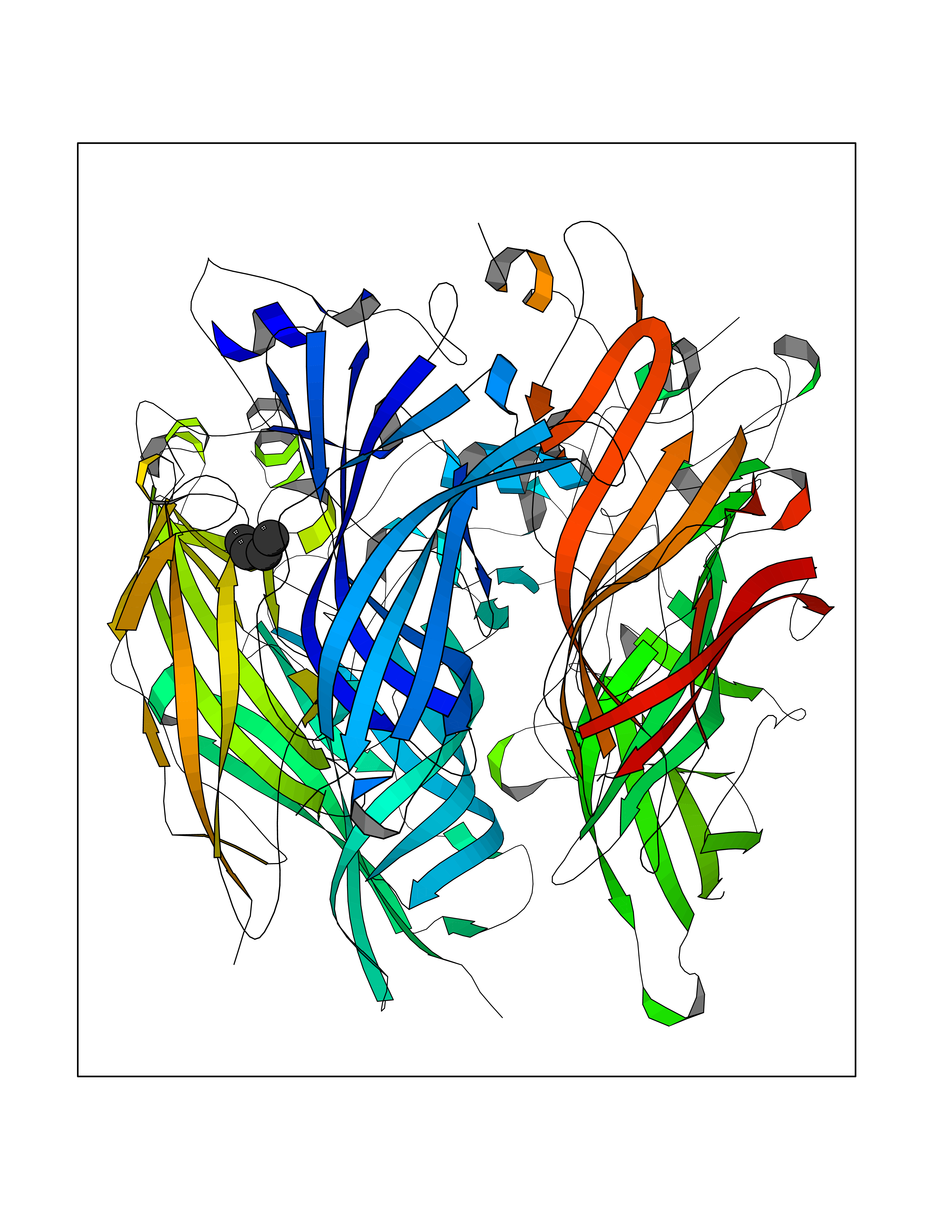 Molscript Map 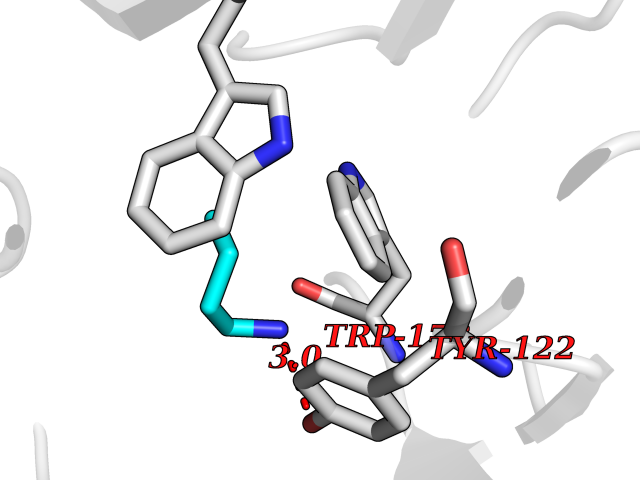 Pymol Map 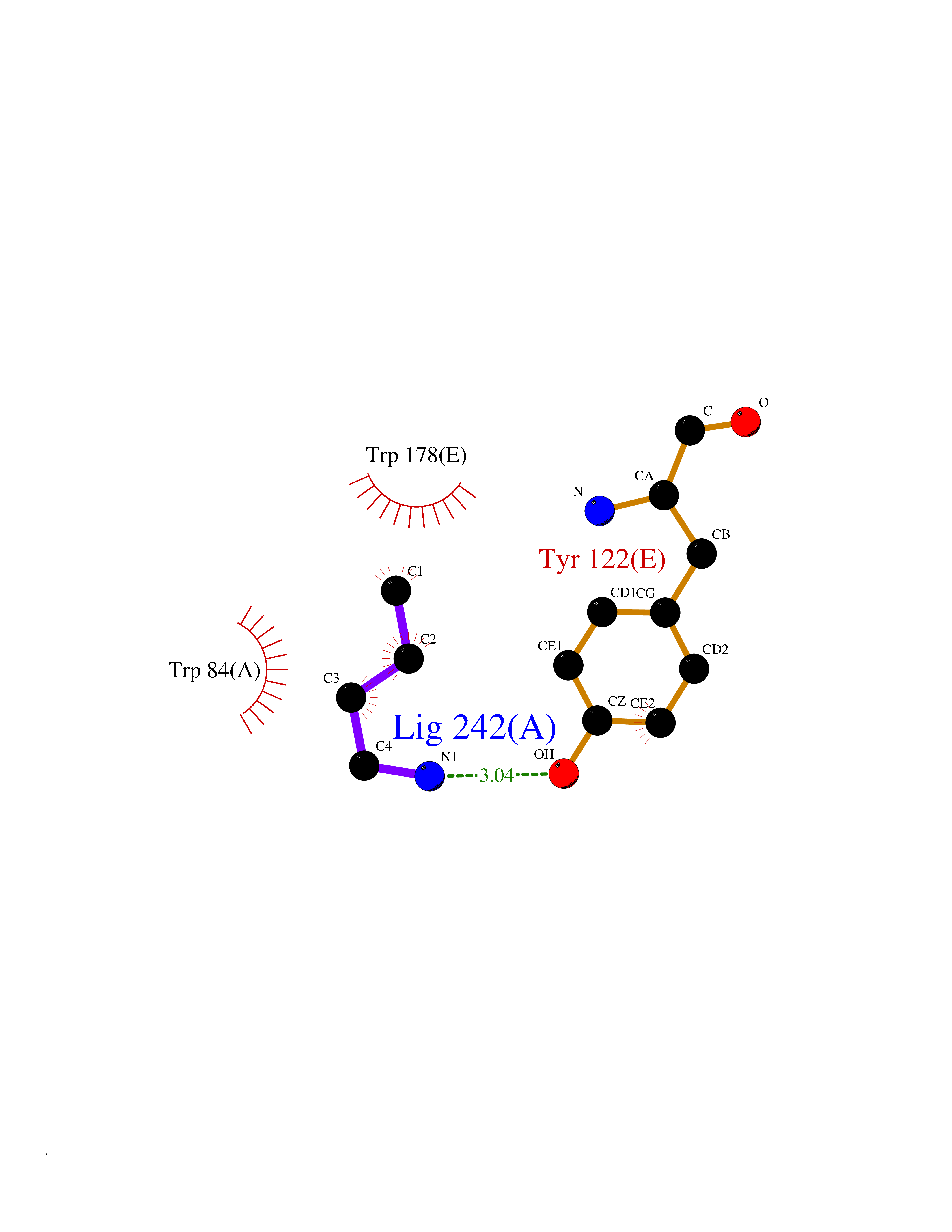 Ligplot Map 3D Binding mode Sequence DRLFKHLFRGYNRWARPVPNTSDVVIVRFGLSIAQLIDVDEKNQMMTTNVWLKQEWSDYKLRWNPTDFGNITSLRVPSEMIWIPDIVLYNNADGEFAVTHMTKAHLFSTGTVHWVPPAIYKSSCSIDVTFFPFDQQNCKMKFGSWTYDKAKIDLEQMEQTVDLKDYWESGEWAIVNATGTYNSKKYDCCAEIYPDVTYAFVIRRLPEDRLFKHLFRGYNRWARPVPNTSDVVIVRFGLSIAQLIDVDEKNQMMTTNVWLKQEWSDYKLRWNPTDFGNITSLRVPSEMIWIPDIVLYNNADGEFAVTHMTKAHLFSTGTVHWVPPAIYKSSCSIDVTFDQQNCKMKFGSWTYDKAKIDLEQMEQTVDLKDYWESGEWAIVNATGTYNSKKYDCCAEIYPDVTYAFVIRRLPEDRLFKHLFRGYNRWARPVPNTSDVVIVRFGLSIAQLIDVDEKNQMMTTNVWLKQEWSDYKLRWNPTDFGNITSLRVPSEMIWIPDIVLYNNADGEFAVTHMTKAHLFSTGTVHWVPPAIYKSSCSIDVTFFPFDQQNCKMKFGSWTYDKAKIDLEQMEQTVDLKDYWESGEWAIVNATGTYNSKKYDCCAEIYPDVTYAFVIRRLPEDRLFKHLFRGYNRWARPVPNTSDVVIVRFGLSIAQLIDVDEKNQMMTTNVWLKQEWSDYKLRWNPTDFGNITSLRVPSEMIWIPDIVLYNNADGEFAVTHMTKAHLFSTGTVHWVPPAIYKSSCSIDVTFFPFDQQNCKMKFGSWTYDKAKIDLEQMEQTVDLKDYWESGEWAIVNATGTYNSKKYDCCAEIYPDVTYAFVIRRLPEDRLFKHLFRGYNRWARPVPNTSDVVIVRFGLSIAQLIDVDEKNQMMTTNVWLKQEWSDYKLRWNPTDFGNITSLRVPSEMIWIPDIVLYNNADGEFAVTHMTKAHLFSTGTVHWVPPAIYKSSCSIDVTFFPFDQQNCKMKFGSWTYDKAKIDLEQMEQTVDLKDYWESGEWAIVNATGTYNSKKYDCCAEIYPDVTYAFVIRRLP Hydrogen bonds contact Hydrophobic contact | ||||
| 7 | Natriuretic peptides B | 1YK1 | 4.67 | |
Target general information Gen name NPPB Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Natriuretic peptide family Biochemical class Hormone / growth factor receptor Function Diuretic hormone activity.Hormone activity.Peptide hormone receptor binding.Receptor binding. Related diseases Multiple fibroadenomas of the breast (MFAB) [MIM:615554]: A benign breast disease marked by lobuloalveolar growth with abnormally high proliferation of the epithelium, and characterized by the presence of more than 3 fibroadenomas in one breast. Fibroadenomas are adenomas containing fibrous tissue. {ECO:0000269|PubMed:18779591}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Hyperprolactinemia (HPRL) [MIM:615555]: A disorder characterized by increased levels of prolactin in the blood not associated with gestation or the puerperium. HPRL may result in infertility, hypogonadism, and galactorrhea. {ECO:0000269|PubMed:24195502}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01136; DB06412 Interacts with A8MQ03; P57678; Q6A162; P60411; Q7Z3S9; P25788; Q9UJW9 EC number NA Uniprot keywords 3D-structure; Direct protein sequencing; Disulfide bond; Glycoprotein; Hormone; Pharmaceutical; Proteoglycan; Proteomics identification; Reference proteome; Secreted; Signal; Vasoactive; Vasodilator Protein physicochemical properties Chain ID E Molecular weight (Da) 46353.1 Length 415 Aromaticity 0.1 Instability index 37.91 Isoelectric point 5.51 Charge (pH=7) -12.09 2D Binding mode Binding energy (Kcal/mol) -6.37 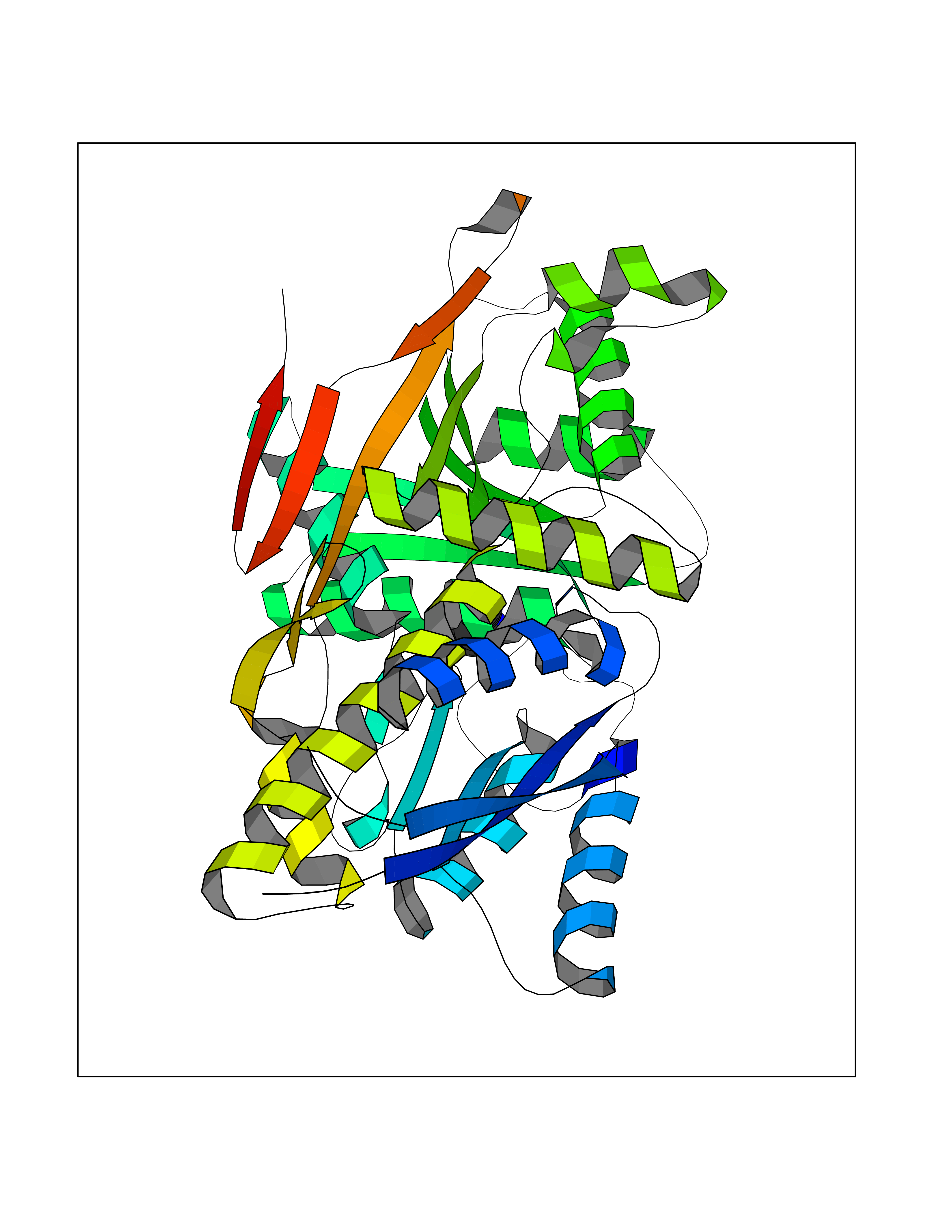 Molscript Map 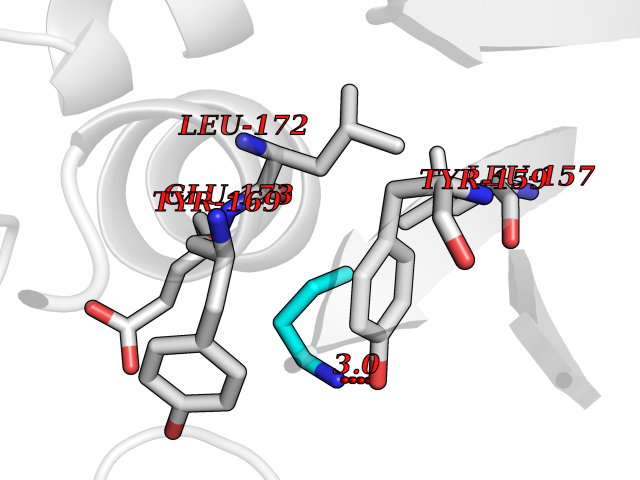 Pymol Map 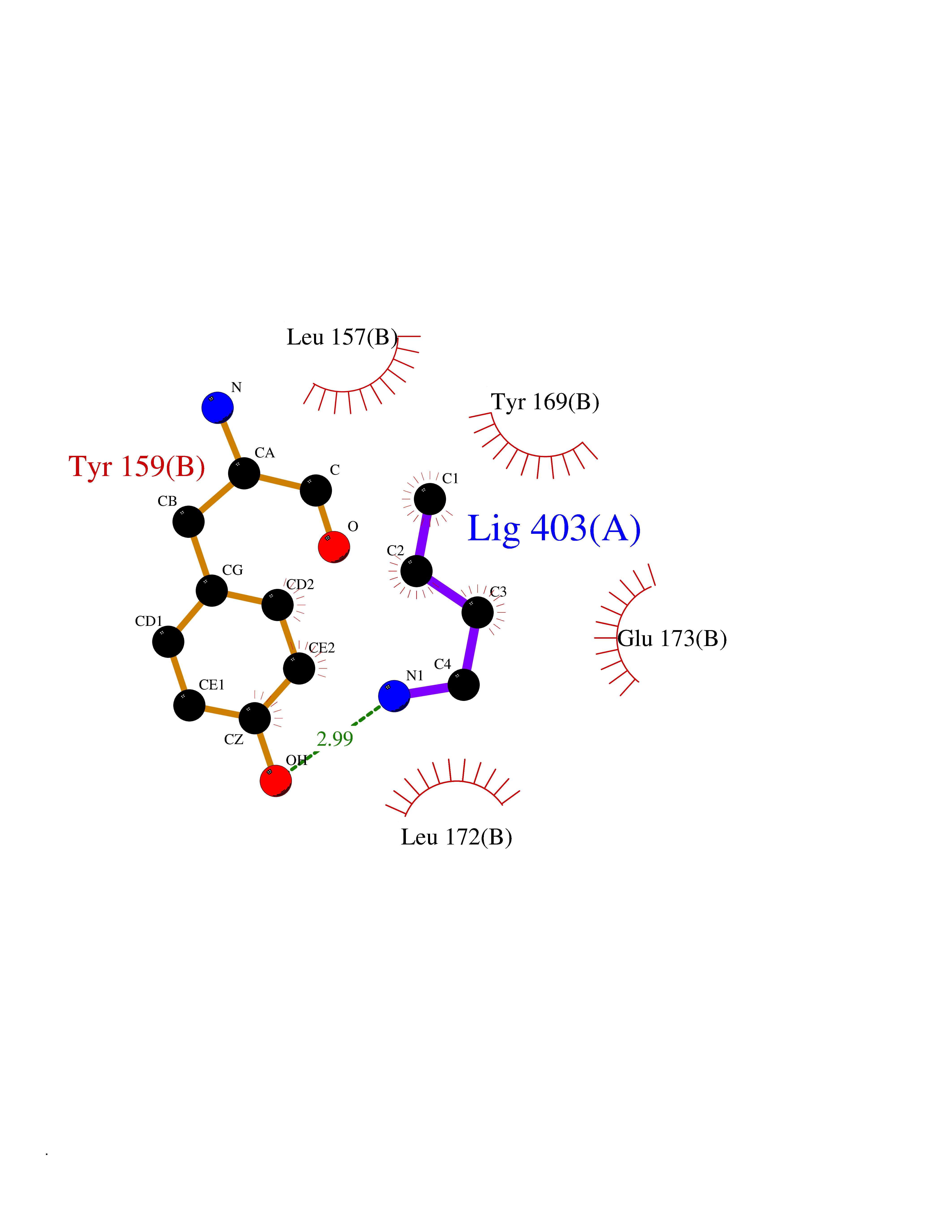 Ligplot Map 3D Binding mode Sequence GCFGRKMDRISSSSGLGCKVLALPPQKIEVLVLLPQDDSYLFSLTRVRPAIEYALRSVEGLLPPGTRFQVAYEDSDCGNRALFSLVDRVAAARGAKPDLILGPVCEYAAAPVARLASHWDLPMLSAGALAAGFQHKDSEYSHLTRVAPAYAKMGEMMLALFRHHHWSRAALVYSDDKLERNCYFTLEGVHEVFQEEGLHTSIYSFDETKDLDLEDIVRNIQASERVVIMCASSDTIRSIMLVAHRHGMTSGDYAFFNIELFNSSSYGDGSWKRGDKHDFEAKQAYSSLQTVTLLRTVKPEFEKFSMEVKSSVEKQGLNMEDYVNMFVEGFHDAILLYVLALHEVLRAGYSKKDGGKIIQQTWNRTFEGIAGQVSIDANGDRYGDFSVIAMTDVEAGTQEVIGDYFGKEGRFEMRP Hydrogen bonds contact Hydrophobic contact | ||||
| 8 | Nitric-oxide synthase endothelial (NOS3) | 4D1P | 4.67 | |
Target general information Gen name NOS3 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Nitric oxide synthase, endothelial; NOSIII; NOS,type III; NOS type III; Endothelial nitric oxide synthase; Endothelial NOS; ENOS; EC-NOS; Constitutive NOS; CNOS Protein family NOS family Biochemical class Paired donor oxygen oxidoreductase Function NO mediates vascular endothelial growth factor (VEGF)-induced angiogenesis in coronary vessels and promotes blood clotting through the activation of platelets. Produces nitric oxide (NO) which is implicated in vascular smooth muscle relaxation through a cGMP-mediated signal transduction pathway. Related diseases Variation Asp-298 in NOS3 may be associated with susceptibility to coronary spasm. {ECO:0000269|PubMed:11740345, ECO:0000269|PubMed:9737779}. Drugs (DrugBank ID) DB07001; DB02048; DB02911; DB02335; DB01997; DB03332; DB04534; DB07244; DB03100; DB03918; DB02207; DB03065; DB00125; DB02994; DB01833; DB00155; DB00997; DB07388; DB03974; DB02077; DB01821; DB09237; DB01110; DB03144; DB03305; DB01686; DB04559; DB02044; DB08019; DB08018; DB02027; DB02979; DB00435; DB04223; DB06154; DB03910; DB02141; DB03963; DB03707; DB02234; DB04018; DB00360; DB02589 Interacts with P60709; P63010-2; Q8N6T3-3; Q9Y575-3; Q96FT7-4; Q5SZD1; Q16543; Q9UNS2; Q8IUI8; P35222; Q05193; O15287; Q08379; Q71DI3; P69905; P61978; Q12891; Q9UKT9; Q9Y2M5; Q14525; Q6DKI2; P43364-2; Q8N6F8; O94851; A4FUJ8; Q8N594; Q8IVI9; Q6X4W1-6; O15381-5; Q9NV79; Q16549; Q5T2W1; O75925; Q96I34; Q6ZMI0-5; P57052; Q9GZR2; Q96D59; Q8N6K7-2; Q9GZS3; Q8IUW3; Q7Z699; Q7Z698; P50502; Q9BR01-2; Q9NVV9; Q86WT6-2; Q9H347; P58304; Q9NZC7-5; Q9UNY5; P14079 EC number EC 1.14.13.39 Uniprot keywords 3D-structure; Alternative splicing; Calcium; Calmodulin-binding; Cell membrane; Cytoplasm; Cytoskeleton; Direct protein sequencing; FAD; Flavoprotein; FMN; Golgi apparatus; Heme; Iron; Lipoprotein; Membrane; Metal-binding; Myristate; NADP; Oxidoreductase; Palmitate; Phosphoprotein; Proteomics identification; Reference proteome; Zinc Protein physicochemical properties Chain ID A,B Molecular weight (Da) 90790.1 Length 803 Aromaticity 0.11 Instability index 50.67 Isoelectric point 6.03 Charge (pH=7) -9.56 2D Binding mode Binding energy (Kcal/mol) -6.37  Molscript Map 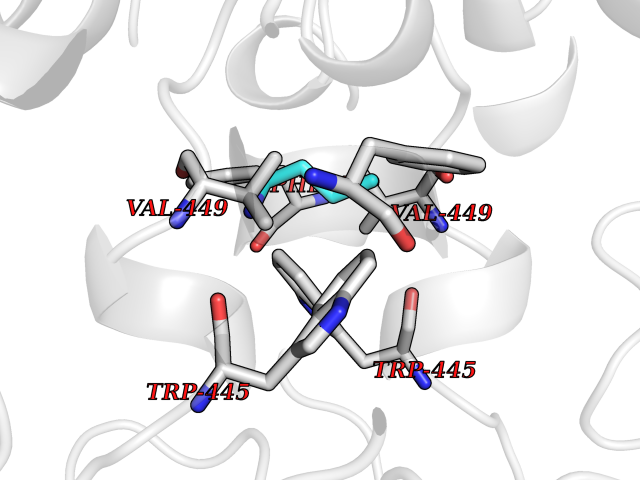 Pymol Map  Ligplot Map 3D Binding mode Sequence FPRVKNWEVGSITYDTLSAQAQQDGPCTPRRCLGSLVFPAPEQLLSQARDFINQYYSSIKRSGSQAHEQRLQEVEAEVAATGTYQLRESELVFGAKQAWRNAPRCVGRIQWGKLQVFDARDCRSAQEMFTYICNHIKYATNRGNLRSAITVFPQRCPGRGDFRIWNSQLVRYAGYRQQDGSVRGDPANVEITELCIQHGWTPGNGRFDVLPLLLQAPDEPPELFLLPPELVLEVPLEHPTLEWFAALGLRWYALPAVSNMLLEIGGLEFPAAPFSGWYMSTEIGTRNLCDPHRYNILEDVAVCMDLDTRTTSSLWKDKAAVEINVAVLHSYQLAKVTIVDHHAATASFMKHLENEQKARGGCPADWAWIVPPISGSLTPVFHQEMVNYFLSPAFRYQPDPWKFPRVKNWEVGSITYDTLSAQAQQDGPCTPRRCLGSLVFPAPEQLLSQARDFINQYYSSIKRSGSQAHEQRLQEVEAEVAATGTYQLRESELVFGAKQAWRNAPRCVGRIQWGKLQVFDARDCRSAQEMFTYICNHIKYATNRGNLRSAITVFPQRCPGRGDFRIWNSQLVRYAGYRQQDGSVRGDPANVEITELCIQHGWTPGNGRFDVLPLLLQAPDEPPELFLLPPELVLEVPLEHPTLEWFAALGLRWYALPAVSNMLLEIGGLEFPAAPFSGWYMSTEIGTRNLCDPHRYNILEDVAVCMDLDTRTTSSLWKDKAAVEINVAVLHSYQLAKVTIVDHHAATASFMKHLENEQKARGGCPADWAWIVPPISGSLTPVFHQEMVNYFLSPAFRYQPDPW Hydrogen bonds contact Hydrophobic contact | ||||
| 9 | Protein cereblon (CRBN) | 5FQD | 4.67 | |
Target general information Gen name CRBN Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Protein cereblon Protein family CRBN family Biochemical class NA Function Substrate recognition component of a DCX (DDB1-CUL4-X-box) E3 protein ligase complex that mediates the ubiquitination and subsequent proteasomal degradation of target proteins, such as MEIS2. Normal degradation of key regulatory proteins is required for normal limb outgrowth and expression of the fibroblast growth factor FGF8. May play a role in memory and learning by regulating the assembly and neuronal surface expression of large-conductance calcium-activated potassium channels in brain regions involved in memory and learning via its interaction with KCNT1. Binding of pomalidomide and other thalidomide-related drugs changes the substrate specificity of the human protein, leading to decreased degradation of MEIS2 and other target proteins and increased degradation of MYC, IRF4, IKZF1 and IKZF3. Related diseases Intellectual developmental disorder, autosomal recessive 2 (MRT2) [MIM:607417]: A disorder characterized by significantly below average general intellectual functioning associated with impairments in adaptive behavior and manifested during the developmental period. MRT2 patients display mild intellectual disability with a standard IQ ranged from 50 to 70. IQ scores are lower in males than females. Developmental milestones are mildly delayed. There are no dysmorphic or autistic features. {ECO:0000269|PubMed:15557513, ECO:0000269|PubMed:28143899}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00480; DB08910; DB01041 Interacts with Q96A83-2; P48729; Q16531; O14901; Q8IVT2; Q9P286; A0A6Q8PF08; Q93062; Q16531; Q13422-7 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; Disease variant; Intellectual disability; Membrane; Metal-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Ubl conjugation; Ubl conjugation pathway; Zinc Protein physicochemical properties Chain ID B,E Molecular weight (Da) 38245.7 Length 337 Aromaticity 0.08 Instability index 40.62 Isoelectric point 5.7 Charge (pH=7) -6.53 2D Binding mode Binding energy (Kcal/mol) -6.37  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence EFIVGGKYKLNITNGEEVAVINFDTSLPTSHTYLGADMEEFHGRTLHDDDSCQVIPVLPQVMMILIPGQTLPLQLFHPQEVSMVRNLIQKDRTFAVLAYSNVQEREAQFGTTAEIYAYREEIVKVKAIGRQRFKVLEQQAKVQILPECVLAETLMDRIKKQLREWDENLKDDSLPSNPIDFSYRVAACLPIDDVLRIQLLKIGSAIQRLRCELDIMNKCTSLCCKQCQETEITTKNEIFSLSLCGPMAAYVNPHGYVHETLTVYKACNLNLIGRPSTEHSWFPGYAWTVAQCKICASHIGWKFTATKKDMSPQKFWGLTRSALLPTIPDTEDEISPD Hydrogen bonds contact Hydrophobic contact | ||||
| 10 | Zinc finger protein Helios (IKZF2) | 7LPS | 4.66 | |
Target general information Gen name IKZF2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Ikaros family zinc finger protein 2 Protein family Ikaros C2H2-type zinc-finger protein family Biochemical class NA Function Associates with Ikaros at centromeric heterochromatin. Related diseases Developmental and epileptic encephalopathy 25, with amelogenesis imperfecta (DEE25) [MIM:615905]: An autosomal recessive disease characterized by subclinical seizures appearing in the first days of life, evolving to severe epileptic disease. Affected individuals have profound or severe delayed development with lack of speech, and most patients do not acquire the ability to sit. Additional variable features include axial hypotonia, peripheral hypertonia, and abnormal involuntary movements such as dystonia and choreoathetosis. Dental abnormalities, including delayed eruption, hypodontia, tooth hypoplasia, yellow discoloration, thin enamel, and enamel chipping are observed in most patients. {ECO:0000269|PubMed:24995870, ECO:0000269|PubMed:26384929, ECO:0000269|PubMed:30054523}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P29972; P56545; P56545-3; Q17RB8; P09022; Q8N8B7-2 EC number NA Uniprot keywords 3D-structure; Acetylation; Activator; Alternative splicing; DNA-binding; Isopeptide bond; Metal-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Transcription; Transcription regulation; Ubl conjugation; Zinc; Zinc-finger Protein physicochemical properties Chain ID B,C Molecular weight (Da) 47006.6 Length 410 Aromaticity 0.09 Instability index 44.28 Isoelectric point 7.23 Charge (pH=7) 0.69 2D Binding mode Binding energy (Kcal/mol) -6.36 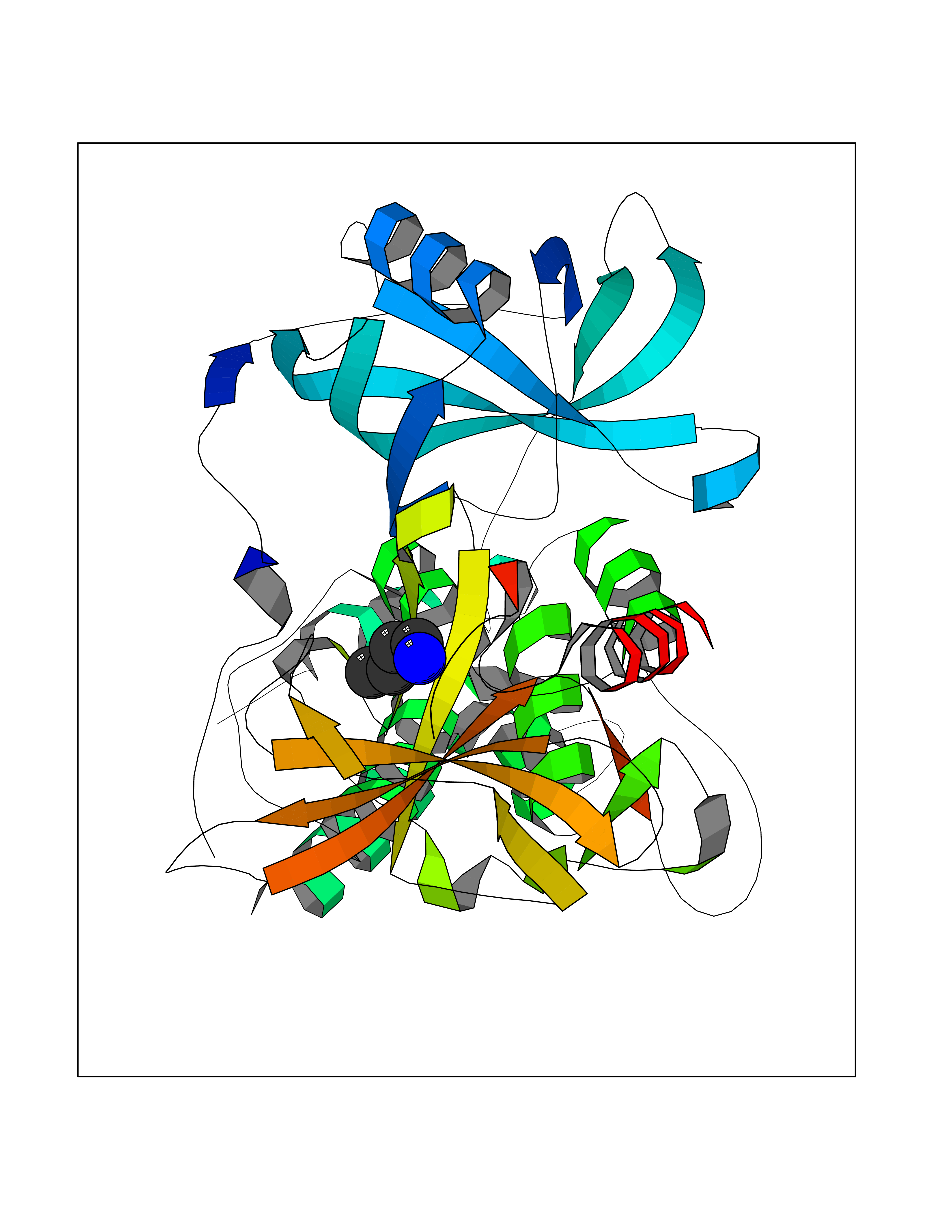 Molscript Map 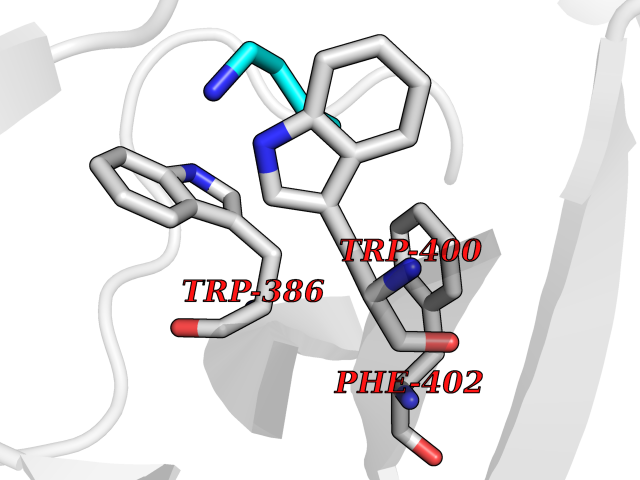 Pymol Map 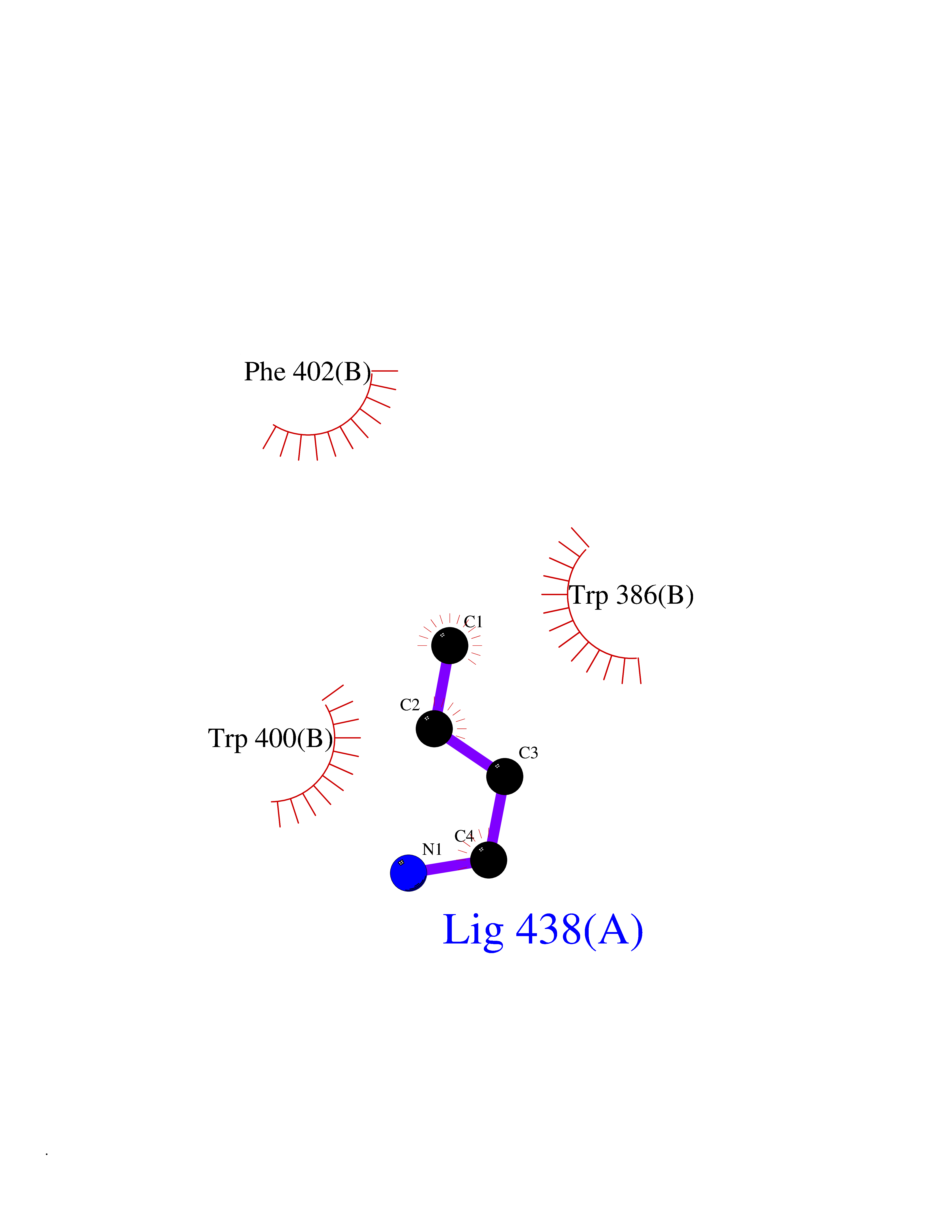 Ligplot Map 3D Binding mode Sequence INFDTSLPTSHTYLGADMEEFHGRTLHDDDSCQVIPVLPQVMMILIPGQTLPLQLFHPQEVSMVRNLIQKDRTFAVLAYSNVQEREAQFGTTAEIYAYREEQDFGIEIVKVKAIGRQRFKVLELRTQSDGIQQAKVQILPECVLPSTMSAVQLESLNKCQIFPCSYKWWQKYQKRKFHCANLTSWPRWLYSLYDAETLMDRIKKQLREWDENLKDDSLPSNPIDFSYRVAACLPIDDVLRIQLLKIGSAIQRLRCELDIMNKCTSLCCKQCQETEITTKNEIFSLSLCGPMAAYVNPHGYVHETLTVYKACNLNLIGRPSTEHSWFPGYAWTVAQCKICASHIGWKFTATKKDMSPQKFWGLTRSALLPTIPDTEDEISPDGERPFHCNQCGASFTQKGNLLRHIKLHSG Hydrogen bonds contact Hydrophobic contact | ||||
| 11 | Ecto-5'-nucleotidase (CD73) | 4H2G | 4.65 | |
Target general information Gen name NT5E Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms NT5; CD73 antigen; 5'-nucleotidase; 5'-NT Protein family 5'-nucleotidase family Biochemical class Phosphoric monoester hydrolase Function Exhibits AMP-, NAD-, and NMN-nucleosidase activities. Hydrolyzes extracellular nucleotides into membrane permeable nucleosides. Related diseases Calcification of joints and arteries (CALJA) [MIM:211800]: A condition characterized by adult-onset calcification of the lower extremity arteries, including the iliac, femoral and tibial arteries, and hand and foot capsule joints. Age of onset has been reported as early as the second decade of life, usually involving intense joint pain or calcification in the hands. {ECO:0000269|PubMed:21288095, ECO:0000269|PubMed:24887587}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00987; DB00806 Interacts with Q9Y225-2; Q8WWF5 EC number EC 3.1.3.5 Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; GPI-anchor; Hydrolase; Lipoprotein; Membrane; Metal-binding; Nucleotide-binding; Proteomics identification; Reference proteome; Signal; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 24417.6 Length 219 Aromaticity 0.09 Instability index 40.43 Isoelectric point 5.49 Charge (pH=7) -5.75 2D Binding mode Binding energy (Kcal/mol) -6.34 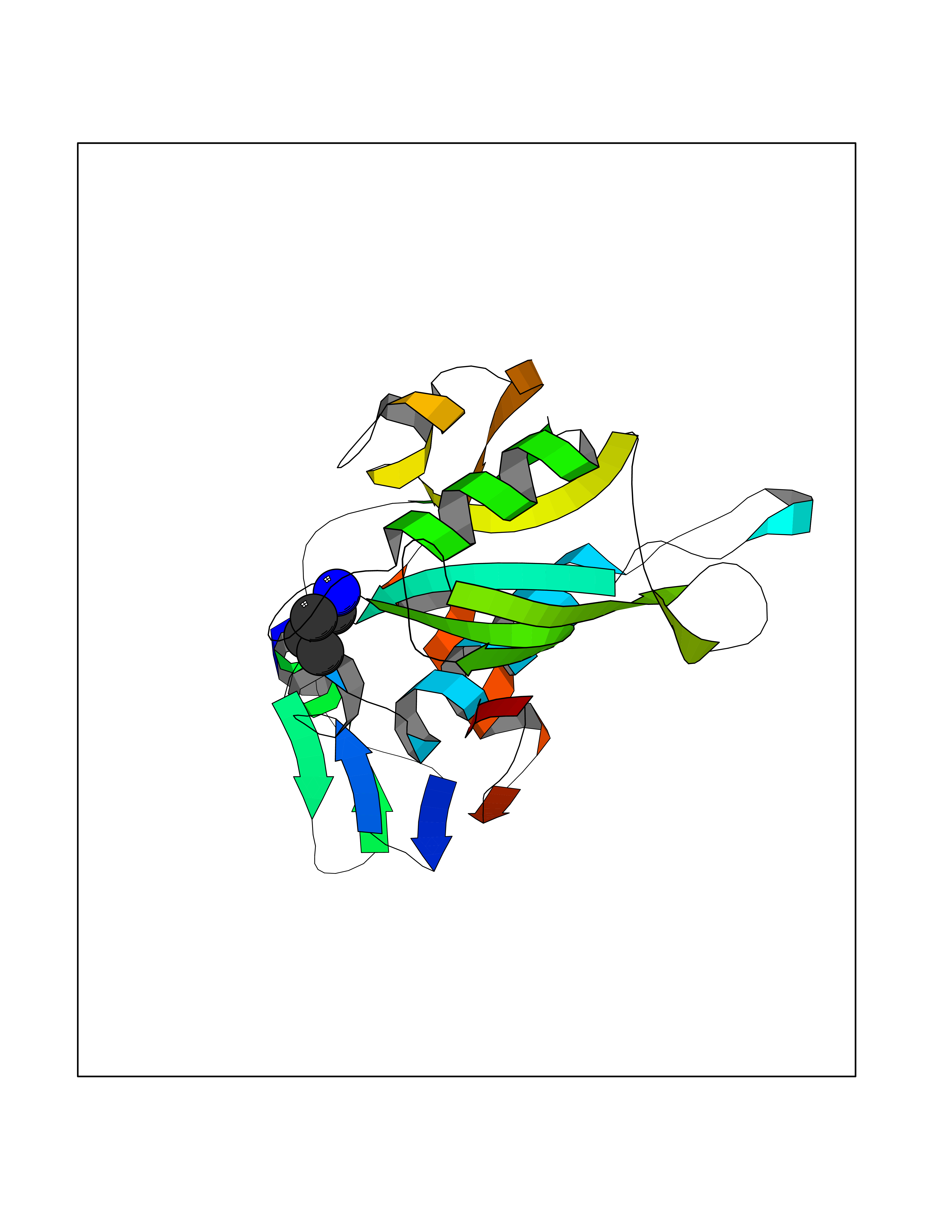 Molscript Map 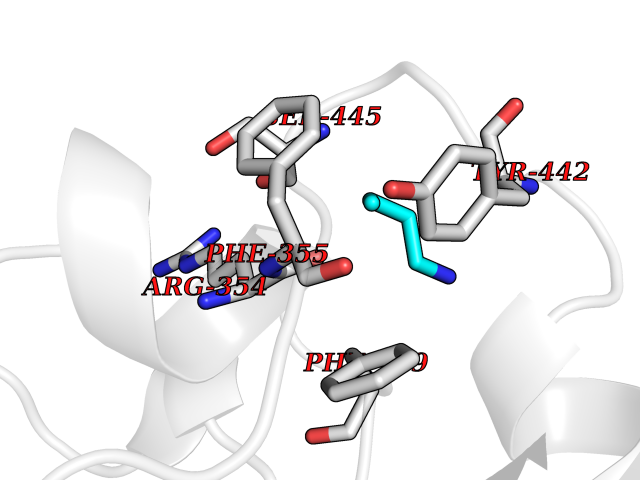 Pymol Map 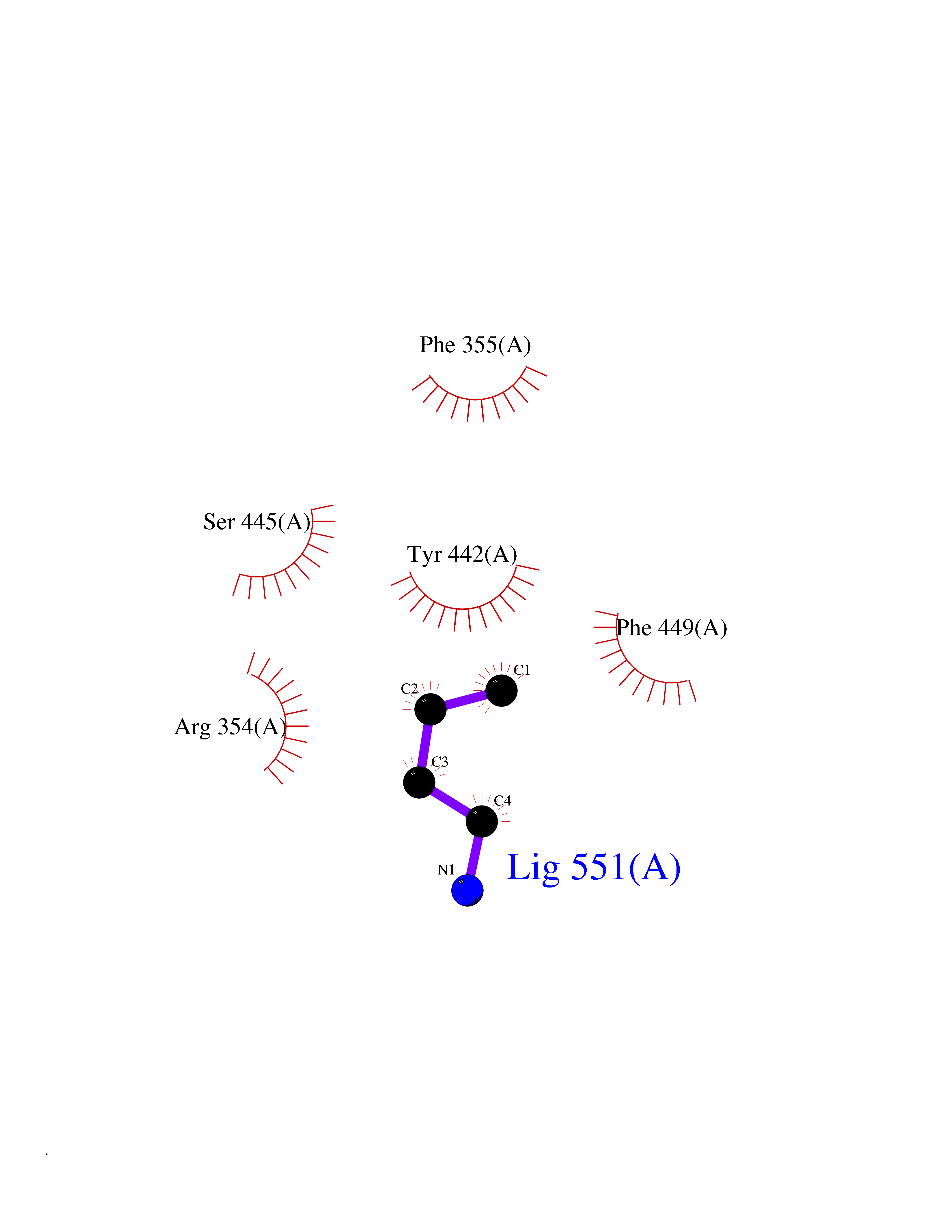 Ligplot Map 3D Binding mode Sequence LDDYSTQELGKTIVYLDGSSQSCRFRECNMGNLICDAMINNNLRHADEMFWNHVSMCILNGGGIRSPIDERNDGTITWENLAAVLPFGGTFDLVQLKGSTLKKAFEHSVHRYGQSTGEFLQVGGIHVVYDLSRKPGDRVVKLDVLCTACAVPSYDPLKMDEVYKVILPNFLANGGDGFQMIKDELLRHDSGDQDINVVSTYISKMKVIYPAVEGRIKFS Hydrogen bonds contact Hydrophobic contact | ||||
| 12 | Cerebron E3 ubiquitin ligase complex (CRL4-CRBN E3 ubiquitin ligase) | 4CI1 | 4.65 | |
Target general information Gen name CUL4A/CUL4B-DDB1-CRBN Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms NA Protein family Cullin family Biochemical class NA Function NA Related diseases Orotic aciduria 1 (ORAC1) [MIM:258900]: A disorder of pyrimidine metabolism resulting in megaloblastic anemia and orotic acid crystalluria that is frequently associated with some degree of physical and intellectual disability. A minority of cases have additional features, particularly congenital malformations and immune deficiencies. {ECO:0000269|PubMed:9042911}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P54253; Q86VP6; Q16531; Q92466; P08238; O94888; P55072 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Biological rhythms; DNA damage; DNA repair; Host-virus interaction; Isopeptide bond; Phosphoprotein; Proteomics identification; Reference proteome; Ubl conjugation; Ubl conjugation pathway Protein physicochemical properties Chain ID B Molecular weight (Da) 42669.7 Length 368 Aromaticity 0.1 Instability index 44.94 Isoelectric point 8.72 Charge (pH=7) 6.58 2D Binding mode Binding energy (Kcal/mol) -6.34  Molscript Map 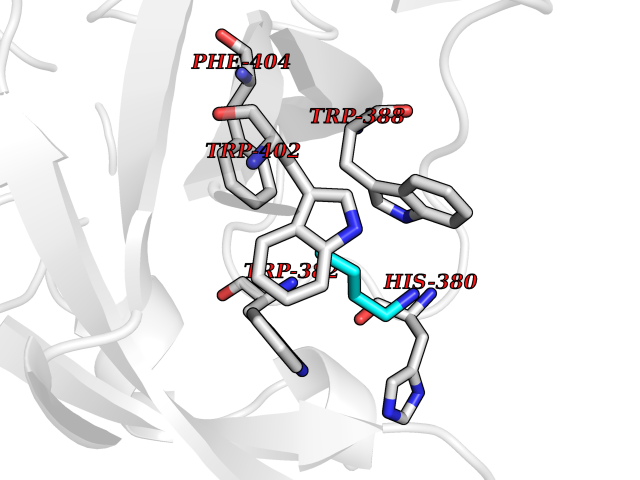 Pymol Map 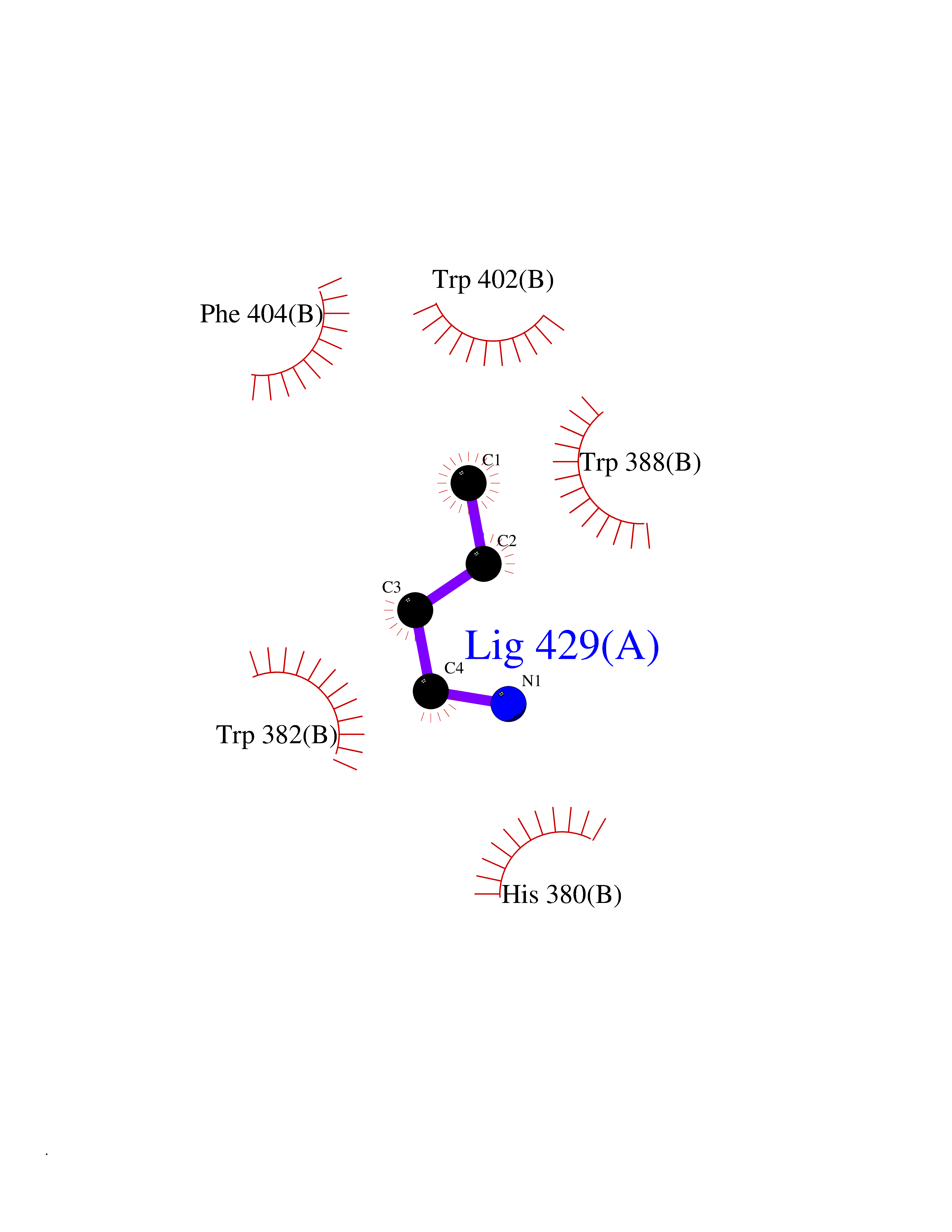 Ligplot Map 3D Binding mode Sequence MINFDTSLPTSHMYLGSDMEEFHGRTLHDDDSCQVIPVLPHVMVMLIPGQTLPLQLFHPQEVSMVRNLIQKDRTFAVLAYSNVREREAHFGTTAEIYAYREEQEYGIETVKVKAIGRQRFKVLEIRTQSDGIQQAKVQILPERVLPSTMSAVQLQSLSRRHIRAFRQWWQKYQKRKFHCASLTSWPPWLYSLYDAETLMERVKRQLHEWDENLKDESLPTNPIDFSYRVAACLPIDDALRIQLLKIGSAIQRLRELDIMNKTSLCCKQCQDTEITTKNEIFSLSLCGPMAAYVNPHGYIHETLTVYKACNLNLSGRPSTEHSWFPGYAWTIAQCRICGNHMGWKFTATKKDMSPQKFWGLTRSALLPR Hydrogen bonds contact Hydrophobic contact | ||||
| 13 | Matrix metalloproteinase-13 (MMP-13) | 2OW9 | 4.65 | |
Target general information Gen name MMP13 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Matrix metalloproteinase 13; Collagenase-3; Collagenase 3 Protein family Peptidase M10A family Biochemical class Peptidase Function Cleaves triple helical collagens, including type I, type II and type III collagen, but has the highest activity with soluble type II collagen. Can also degrade collagen type IV, type XIV and type X. May also function by activating or degrading key regulatory proteins, such as TGFB1 and CTGF. Plays a role in wound healing, tissue remodeling, cartilage degradation, bone development, bone mineralization and ossification. Required for normal embryonic bone development and ossification. Plays a role in the healing of bone fractures via endochondral ossification. Plays a role in wound healing, probably by a mechanism that involves proteolytic activation of TGFB1 and degradation of CTGF. Plays a role in keratinocyte migration during wound healing. May play a role in cell migration and in tumor cell invasion. Plays a role in the degradation of extracellular matrix proteins including fibrillar collagen, fibronectin, TNC and ACAN. Related diseases Spondyloepimetaphyseal dysplasia, Missouri type (SEMDM) [MIM:602111]: A bone disease characterized by moderate to severe metaphyseal changes, mild epiphyseal involvement, rhizomelic shortening of the lower limbs with bowing of the femora and/or tibiae, coxa vara, genu varum and pear-shaped vertebrae in childhood. Epimetaphyseal changes improve with age. {ECO:0000269|PubMed:16167086}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Metaphyseal anadysplasia 1 (MANDP1) [MIM:602111]: A bone development disorder characterized by skeletal anomalies that resolve spontaneously with age. Clinical characteristics are evident from the first months of life and include slight shortness of stature and a mild varus deformity of the legs. Patients attain a normal stature in adolescence and show improvement or complete resolution of varus deformity of the legs and rhizomelic micromelia. {ECO:0000269|PubMed:19615667}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Metaphyseal dysplasia, Spahr type (MDST) [MIM:250400]: An autosomal recessive, rare disease characterized by moderate short stature, mild genua vara, and radiographic signs of metaphyseal dysplasia, but no biochemical signs of rickets. {ECO:0000269|PubMed:24648384, ECO:0000269|PubMed:24781753}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02049; DB01996; DB03033; DB07827; DB08388; DB08561; DB08490; DB06423; DB02697; DB00786; DB04759; DB04760; DB04761; DB07013; DB02071 Interacts with NA EC number EC 3.4.24.- Uniprot keywords 3D-structure; Calcium; Collagen degradation; Direct protein sequencing; Disease variant; Disulfide bond; Dwarfism; Extracellular matrix; Glycoprotein; Hydrolase; Metal-binding; Metalloprotease; Phosphoprotein; Protease; Proteomics identification; Reference proteome; Repeat; Secreted; Signal; Zinc; Zymogen Protein physicochemical properties Chain ID A Molecular weight (Da) 18757.8 Length 167 Aromaticity 0.15 Instability index 9.97 Isoelectric point 5.47 Charge (pH=7) -7.44 2D Binding mode Binding energy (Kcal/mol) -6.34  Molscript Map 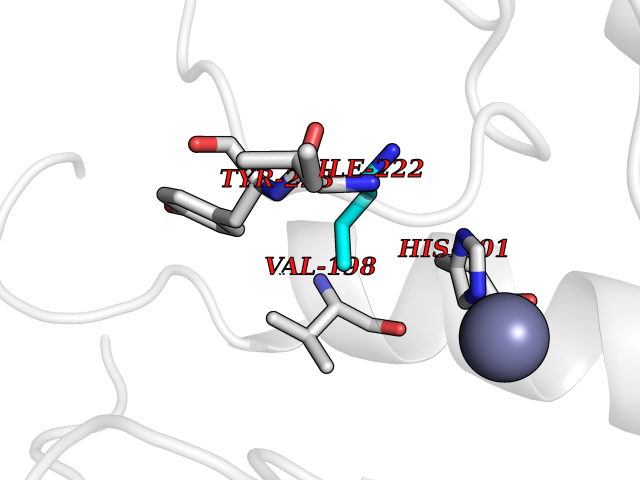 Pymol Map 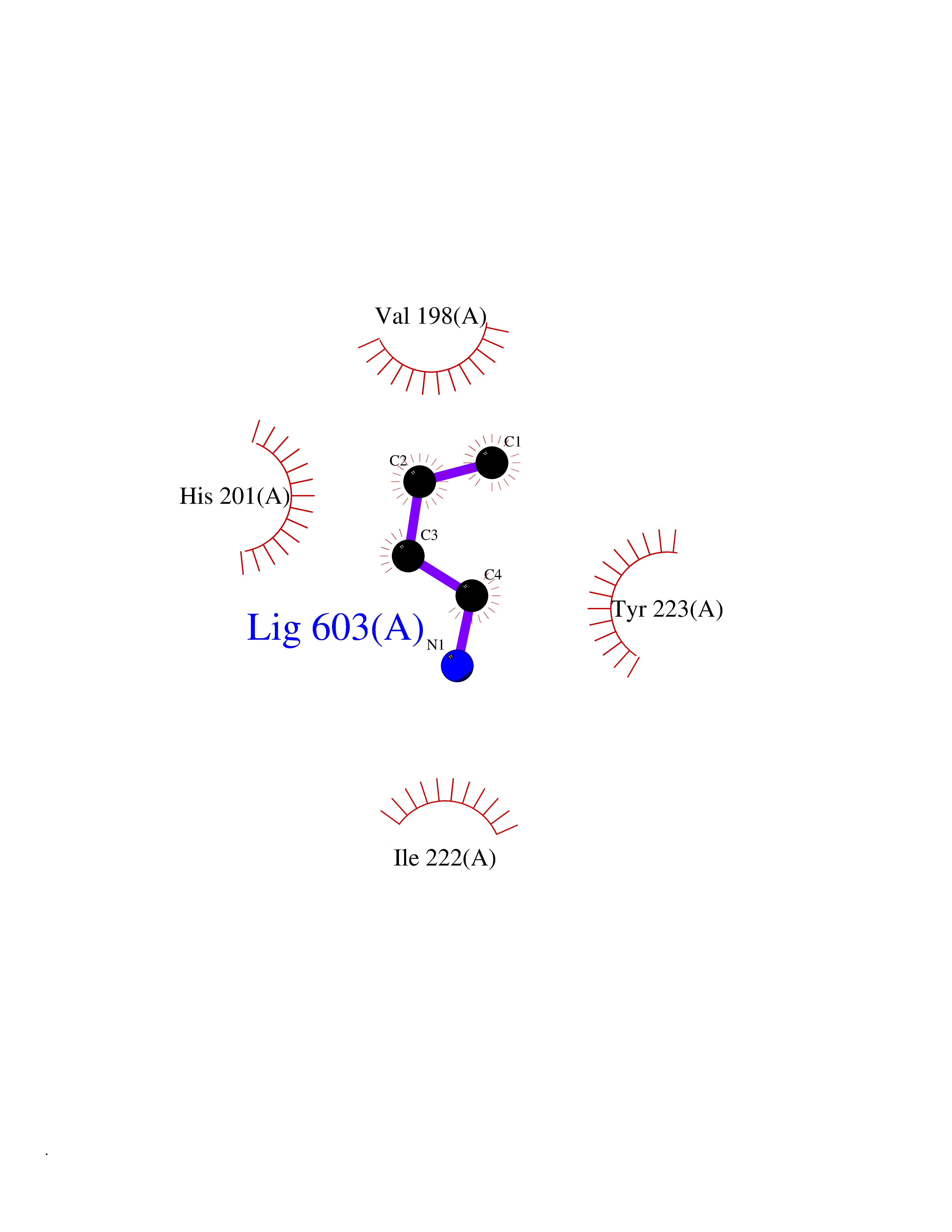 Ligplot Map 3D Binding mode Sequence YNVFPRTLKWSKMNLTYRIVNYTPDMTHSEVEKAFKKAFKVWSDVTPLNFTRLHDGIADIMISFGIKEHGDFYPFDGPSGLLAHAFPPGPNYGGDAHFDDDETWTSSSKGYNLFLVAAHEFGHSLGLDHSKDPGALMFPIYTYTGKSHFMLPDDDVQGIQSLYGPGD Hydrogen bonds contact Hydrophobic contact | ||||
| 14 | DNA-dependent protein kinase catalytic (PRKDC) | 7SGL | 4.65 | |
Target general information Gen name PRKDC Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms p460; HYRC1; HYRC; DNPK1; DNA-dependent protein kinase catalytic subunit; DNA-PKcs; DNA-PK catalytic subunit Protein family PI3/PI4-kinase family Biochemical class Kinase Function Serine/threonine-protein kinase that acts as a molecular sensor for DNA damage. Involved in DNA non-homologous end joining (NHEJ) required for double-strand break (DSB) repair and V(D)J recombination. Must be bound to DNA to express its catalytic properties. Promotes processing of hairpin DNA structures in V(D)J recombination by activation of the hairpin endonuclease artemis (DCLRE1C). The assembly of the DNA-PK complex at DNA ends is also required for the NHEJ ligation step. Required to protect and align broken ends of DNA. May also act as a scaffold protein to aid the localization of DNA repair proteins to the site of damage. Found at the ends of chromosomes, suggesting a further role in the maintenance of telomeric stability and the prevention of chromosomal end fusion. Also involved in modulation of transcription. Recognizes the substrate consensus sequence [ST]-Q. Phosphorylates 'Ser-139' of histone variant H2AX/H2AFX, thereby regulating DNA damage response mechanism. Phosphorylates DCLRE1C, c-Abl/ABL1, histone H1, HSPCA, c-jun/JUN, p53/TP53, PARP1, POU2F1, DHX9, FH, SRF, XRCC1, XRCC1, XRCC4, XRCC5, XRCC6, WRN, MYC and RFA2. Can phosphorylate C1D not only in the presence of linear DNA but also in the presence of supercoiled DNA. Ability to phosphorylate p53/TP53 in the presence of supercoiled DNA is dependent on C1D. Contributes to the determination of the circadian period length by antagonizing phosphorylation of CRY1 'Ser-588' and increasing CRY1 protein stability, most likely through an indirect mechanism (By similarity). Plays a role in the regulation of DNA virus-mediated innate immune response by assembling into the HDP-RNP complex, a complex that serves as a platform for IRF3 phosphorylation and subsequent innate immune response activation through the cGAS-STING pathway. Related diseases Immunodeficiency 26 with or without neurologic abnormalities (IMD26) [MIM:615966]: A form of severe combined immunodeficiency characterized by reduced or absent T and B cells, recurrent candidiasis, and lower respiratory tract infections. Some patients show dysmorphic features, severe growth failure, microcephaly, seizures, and impaired neurological functions. {ECO:0000269|PubMed:19075392, ECO:0000269|PubMed:23722905}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00201; DB05210 Interacts with O43918; P10275; Q96SD1; P14921; P50549; P09629; Q9BPZ7; Q96RI1-2; P10276; P17947; P13010; P12956; P25490 EC number EC 2.7.11.1 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; ATP-binding; Biological rhythms; Disease variant; DNA damage; DNA recombination; DNA repair; DNA-binding; Immunity; Innate immunity; Kinase; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Ribosome biogenesis; SCID; Serine/threonine-protein kinase; TPR repeat; Transferase; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 437836 Length 3852 Aromaticity 0.09 Instability index 47.88 Isoelectric point 6.63 Charge (pH=7) -10.35 2D Binding mode Binding energy (Kcal/mol) -6.34 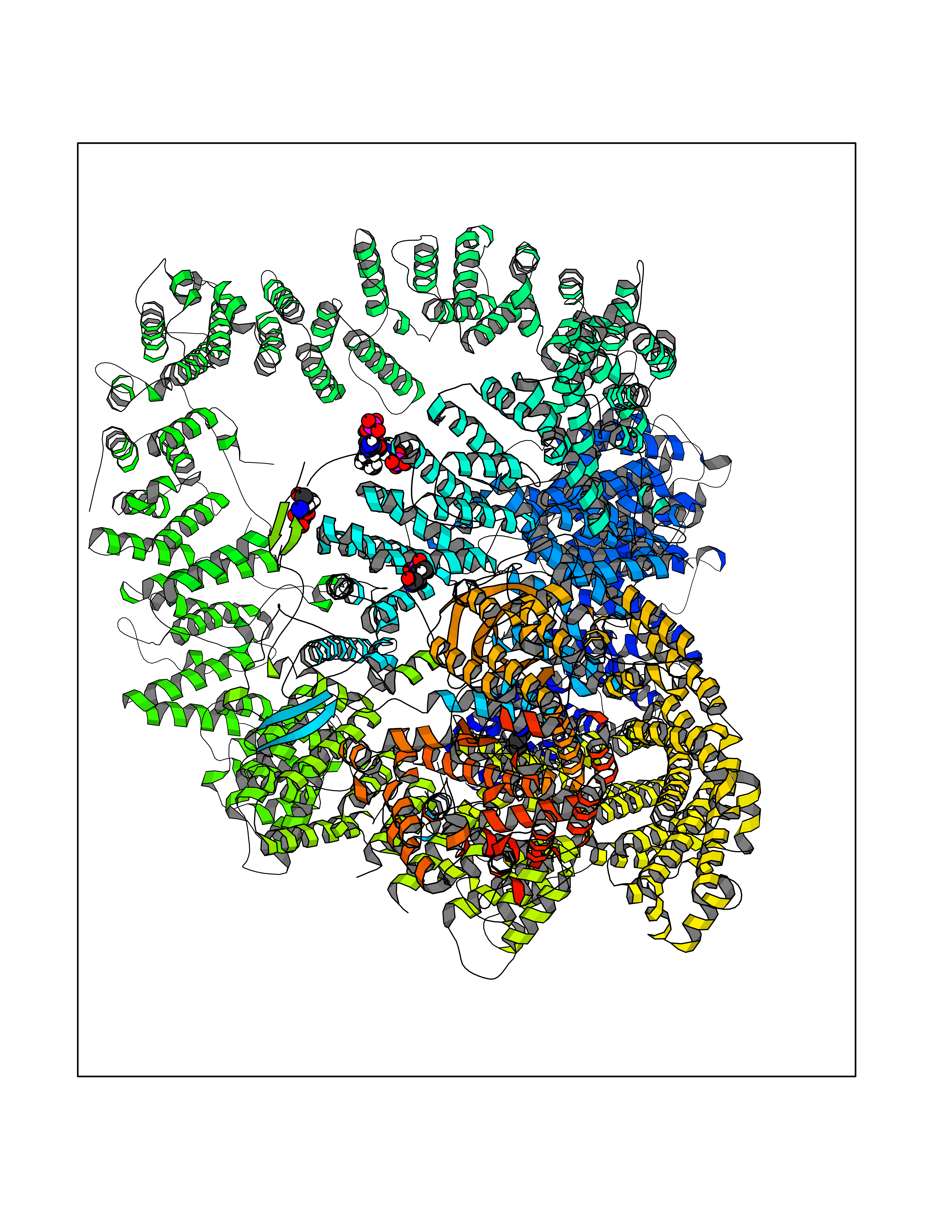 Molscript Map 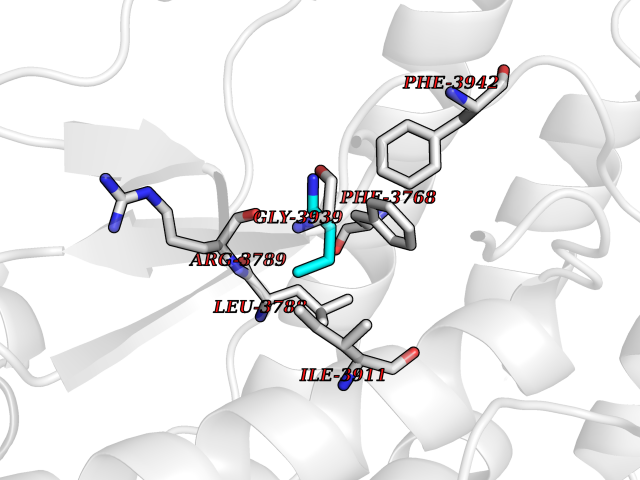 Pymol Map 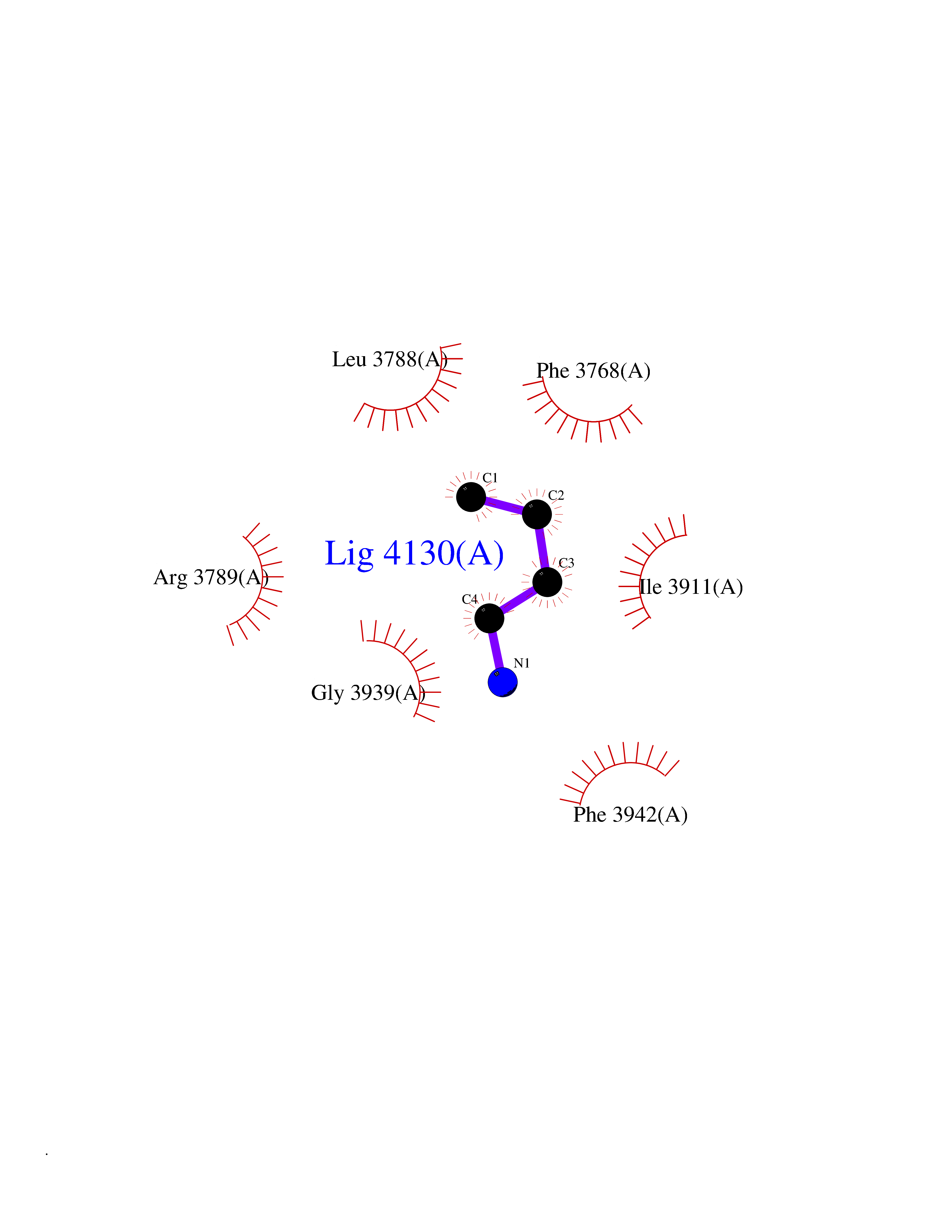 Ligplot Map 3D Binding mode Sequence MAGSGAGVRCSLLRLQETLSAADRCGAALAGHQLIRGLGQECVLSSSPAVLALQTSLVFSRDFGLLVFVRKSLNSIEFRECREEILKFLCIFLEKMGQKIAPYSVEIKNTCTSVYTKDRAAKCKIPALDLLIKLLQTFRSSRLMDEFKIGELFSKFYGELALKKKIPDTVLEKVYELLGLLGEVHPSEMINNAENLFRAFLGELKTQMTSAVREPKLPVLAGCLKGLSSLLCNFTKSMEEDPQTSREIFNFVLKAIRPQIDLKRYAVPSAGLRLFALHASQFSTCLLDNYVSLFEVLLKWCAHTNVELKKAALSALESFLKQVSNMVAKNAEMHKNKLQYFMEQFYGIIRNVDSNNKELSIAIRGYGLFAGPCKVINAKDVDFMYVELIQRCKQMFLTQTDTGDDRVYQMPSFLQSVASVLLYLDTVPEVYTPVLEHLVVMQIDSFPQYSPKMQLVCCRAIVKVFLALAAKGPVLRNCISTVVHQGLIRICSKPVVLPKGPESESEDHRASGEVRTGKWKVPTYKDYVDLFRHLLSSDQMMDSILADEAFFSVNSSSESLNHLLYDEFVKSVLKIVEKLDLTLEIDPAANLHPAKPKDFSAFINLVEFCREILPEKQAEFFEPWVYSFSYELILQSTRLPLISGFYKLLSITVRNAKKIKYFEGVSDPEKYSCFALFVKFGKEVAVKMKQYKDELLASCLTFLLSLPHNIIELDVRAYVPALQMAFKLGLSYTPLAEVGLNALEEWSIYIDRHVMQPYYKDILPCLDGYLKTNWEVSALSRAAAISLEEIRIRVVQMLGSLGGQINKNLLTVTSSDEMMKSYVAWDREKRLSFAVPFREMKPVIFLDVFLPRVTELALTASDRQTKVAACELLHSMVMFMLGKATQMPEGGQGAPPMYQLYKRTFPVLLRLACDVDQVTRQLYEPLVMQLIHWFTNNKKFESQDTVALLEAILDGIVDPVDSTLRDFCGRCIREFLKWSIKQITPQQQEKSPVNTKSLFKRLYSLALHPNAFKRLGASLAFNNIYREFREEESLVEQFVFEALVIYMESLALAHADEKSLGTIQQCCDAIDHLCRIIEKKHVSLNKAKKRRLPRGFPPSASLCLLDLVKWLLAHCGRPQTECRHKSIELFYKFVPLLPGNRSPNLWLKDVLKEEGVSFLINTFEGGGCGQPSGILAQPTLLYLRGPFSLQATLCWLDLLLAALECYNTFIGERTVGALQVLGTEAQSSLLKAVAFFLESIAMHDIIAAEKCFGTGAAGNRTSPQEGERYNYSKCTVVVRIMEFTTTLLNTSPEGWKLLKKDLCNTHLMRVLVQTLCEPASIGFNIGDVQVMAHLPDVCVNLMKALKMSPYKDILETHLREKITAQSIEELCAVNLYGPDAQVDRSRLAAVVSACKQLHRAGLLHNILPSQSTDLHHSVGTELLSLVYKGIAPGDERQCLPSLDLSCKQLASGLLELAFAFGGLCERLVSLLLNPAVLSTSVIHFSHGEYFYSLFSETINTELLKNLDLAVLELMQSSVDNTKMVSAVLNGMLDQSFRERANQKHQGLKLATTILQHWKKCDSWWAKDSPLETKMAVLALLAKILQIDSSVSFNTSHGSFPEVFTTYISLLADTKLDLHLKGQAVTLLPFFTSLTGGSLEELRRVLEQLIVAHFPMQSREFPPGTPRFNNYVDCMKKFLDALELSQSPMLLELMTEVLCREQQHVMEELFQSSFRRIARRGSCVTQVGLLESVYEMFRKDDPRLSFTRQSFVDRSLLTLLWHCSLDALREFFSTIVVDAIDVLKSRFTKLNESTFDTQITKKMGYYKILDVMYSRLPKDDVHAKESKINQVFHGSCITEGNELTKTLIKLCYDAFTENMAGENQLLERRRLYHCAAYNCAISVICCVFNELKFYQGFLFSEKPEKNLLIFENLIDLKRRYNFPVEVEVPMERKKKYIEIRKEAREAANGDSDGPSYMSSLSYLADSTLSEEMSQFDFSTGVQSYSYSSQLEMDELNRHECMAPLTALVKHMHRSPRDLPSWMKFLHGKLGNPIVPLNIRLFLAKLVINTEEVFRPYAKHWLSPLLQLAASENNGGEGIHYMVVEIVATILSWTGLATPTGVPKDEVLANRLLNFLMKHVFHPKRAVFRHNLEIIKTLVECWKDCLSIPYRLIFEKFSGKDPNSKDNSVGIQLLGIVMANDLPPYDPQCGIQSSEYFQALVNNMSFVRYKEVYAAAAEVLGLILRYVMERKNILEESLCELVAKQLKQHQNTMEDKFIVCLNKVTKSFPPLADRFMNAVFFLLPKFHGVLKTLCLEVVLCRVEGMTELYFQLKSKDFVQVMRHRDDERQKVCLDIIYKMMPKLKPVELRELLNPVVEFVSHPSTTCREQMYNILMWIHDNYRDPESETDNDSQEIFKLAKDVLIQGLIDENPGLQLIIRNFWSHETRLPSNTLDRLLALNSLYSPKIEVHFLSLATNFLLEMTSMSPDYPNPMFEHPLSECEFQEYTIDSDWRFRSTVLTPMFVEXQASQGPVAGQIRAXQQQHDFXLXQTQVVLYRSYRHGDLPDIQIKHSSLITPLQAVAQRDPIIAKQLFSSLFSGILKEMDKFKTLSEKNNITQKLLQDFNRFLNTTFSFFPPFVSCIQDISCQHAALLSLDPAAVSAGCLASLQQPVGIRLLEEALLRLLPALPPDVLRWVELAKLYRSIGEYDVLRGIFTSEIGTKQITQSALLAEARSDYSEAAKQYDEALNKQDWVDGEPTEAEKDFWELASLDCYNHLAEWKSLEYCSTASIDSENPPDLNKIWSEPFYQETYLPYMIRSKLKLLLQGEADQSLLTFIDKAMHGELQKAILELHYSQELSLLYLLQDDVDRAKYYIQNGIQSFMQNYSSIDVLLHQSRLTKLQSVQALTEIQEFISFISKQGNLSSQVPLKRLLNTWTNRYPDAKMDPMNIWDDIITNRCFFLSKIEEKLTEDISSLIRSCKFSMKMKMIDSARKQNNFSLAMKLLKELHKESKTRDDWLVSWVQSYCRLSHCRSRSQGCSEQVLTVLKTVSLLDENNVSSYLSKNILAFRDQNILLGTTYRIIANALSSEPACLAEIEEDKARRILELSGSSSEDSEKVIAGLYQRAFQHLSEAVQAAEEEAQPPSWSCGPAAGVIDAYMTLADFCDQQLRKEEENASVIDSAELQAYPALVVEKMLKALKLNSNEARLKFPRLLQIIERYPEETLSLMTKEISSVPCWQFISWISHMVALLDKDQAVAVQHSVEEITDNYPQAIVYPFIISSESYSFKDTSTGHKNKEFVARIKSKLDQGGVIQDFINALDQLSNPELLFKDWSNDVRAELAKTPVNKKNIEKMYERMYAALGDPKAPGLGAFRRKFIQTFGKEFDKHFGKGGSKLLRMKLSDFNDITNMLLLKMNKDSKPPGNLKECSPWMSDFKVEFLRNELEIPGQYDGRGKPLPEYHVRIAGFDERVTVMASLRRPKRIIIRGHDEREHPFLVKGGEDLRQDQRVEQLFQVMNGILAQDSACSQRALQLRTYSVVPMTSRLGLIEWLENTVTLKDLLLNTMSQEEKAAYLSDPRAPPCEYKDWLTKMSGKHDVGAYMLMYKGANRTETVTSFRKRESKVPADLLKRAFVRMSTSPEAFLALRSHFASSHALICISHWILGIGDRHLNNFMVAMETGGVIGIDFGHAFGSATQFLPVPELMPFRLTRQFINLMLPMKETGLMYSIMVHALRAFRSDPGLLTNTMDVFVKEPSFDWKNFEQKMLKKGGSWIQEINVAEKNWYPRQKICYAKRKLAGANPAVITCDELLLGHEKAPAFRDYVAVARGSKDHNIRAQEPESGLSEETQVKCLMDQATDPNILGRTWEGWEPWM Hydrogen bonds contact Hydrophobic contact | ||||
| 15 | Hydroxymethylglutaryl-CoA synthase 2 (HMGCS2) | 2WYA | 4.65 | |
Target general information Gen name HMGCS2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms HMGCS2; HMG-CoAsynthase; 3-hydroxy-3-methylglutaryl coenzyme A synthase 2 Protein family Thiolase-like superfamily, HMG-CoA synthase family Biochemical class Acyltransferase Function This enzyme condenses acetyl-CoA with acetoacetyl-CoA to form HMG-CoA, which is the substrate for HMG-CoA reductase. Related diseases 3-hydroxy-3-methylglutaryl-CoA synthase-2 deficiency (HMGCS2D) [MIM:605911]: A metabolic disorder characterized by severe hypoketotic hypoglycemia, encephalopathy, and hepatomegaly. {ECO:0000269|PubMed:11228257, ECO:0000269|PubMed:11479731, ECO:0000269|PubMed:12647205, ECO:0000269|PubMed:16601895, ECO:0000269|PubMed:23751782, ECO:0000269|PubMed:25511235, ECO:0000269|PubMed:29597274}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number EC 2.3.3.10 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Cholesterol biosynthesis; Cholesterol metabolism; Disease variant; Lipid biosynthesis; Lipid metabolism; Mitochondrion; Phosphoprotein; Proteomics identification; Reference proteome; Steroid biosynthesis; Steroid metabolism; Sterol biosynthesis; Sterol metabolism; Transferase; Transit peptide Protein physicochemical properties Chain ID A,D Molecular weight (Da) 102499 Length 920 Aromaticity 0.1 Instability index 33.48 Isoelectric point 6.72 Charge (pH=7) -1.41 2D Binding mode Binding energy (Kcal/mol) -6.34 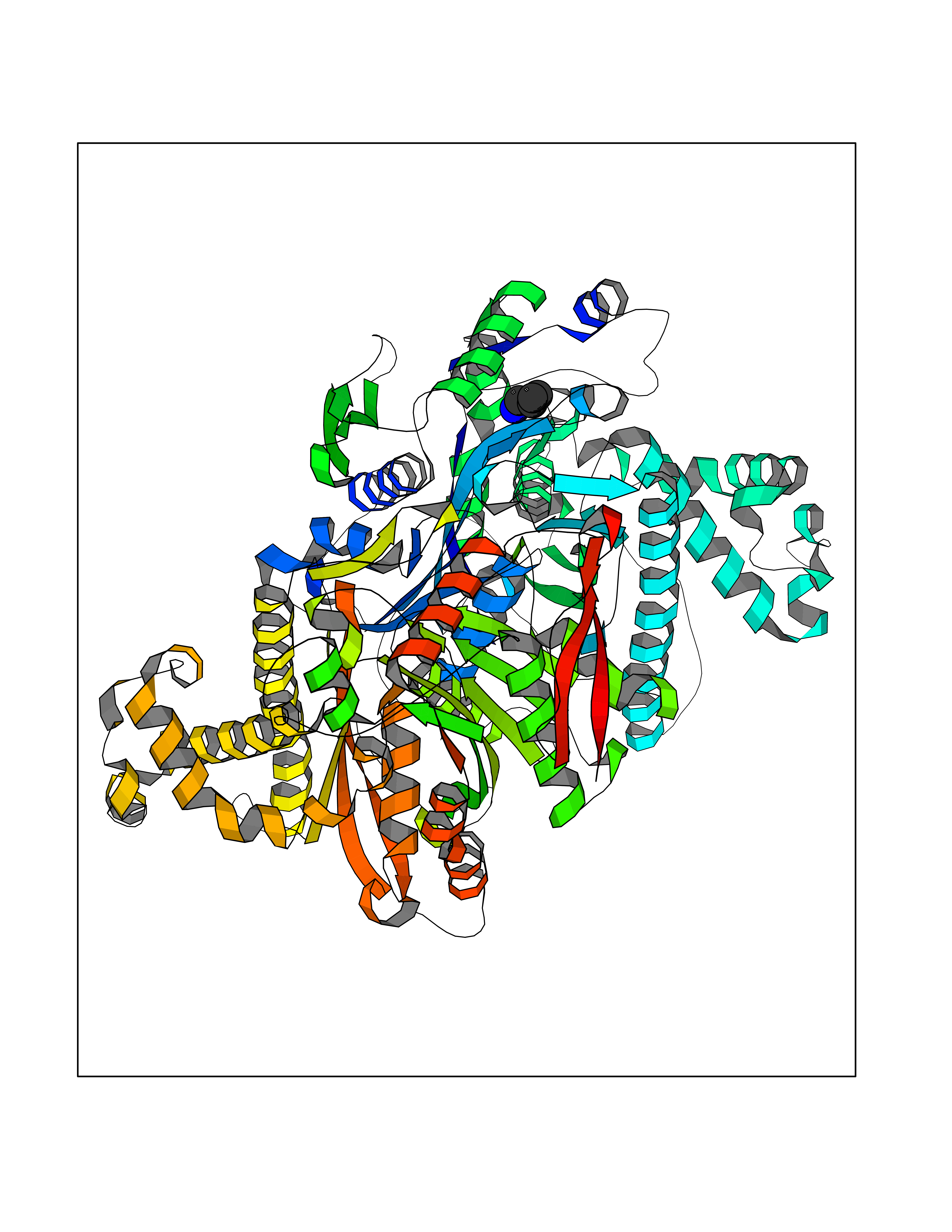 Molscript Map 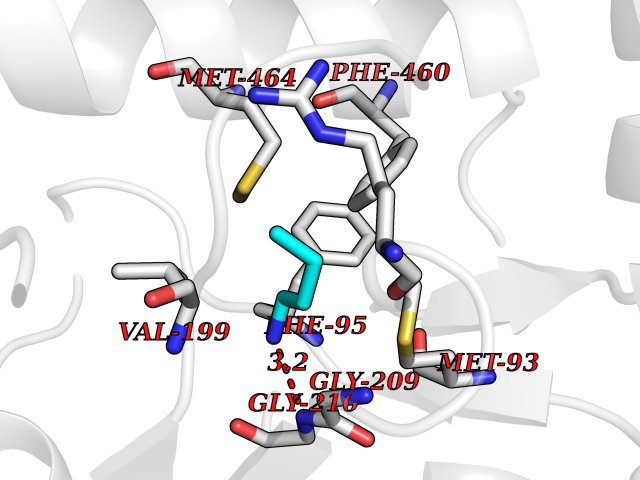 Pymol Map 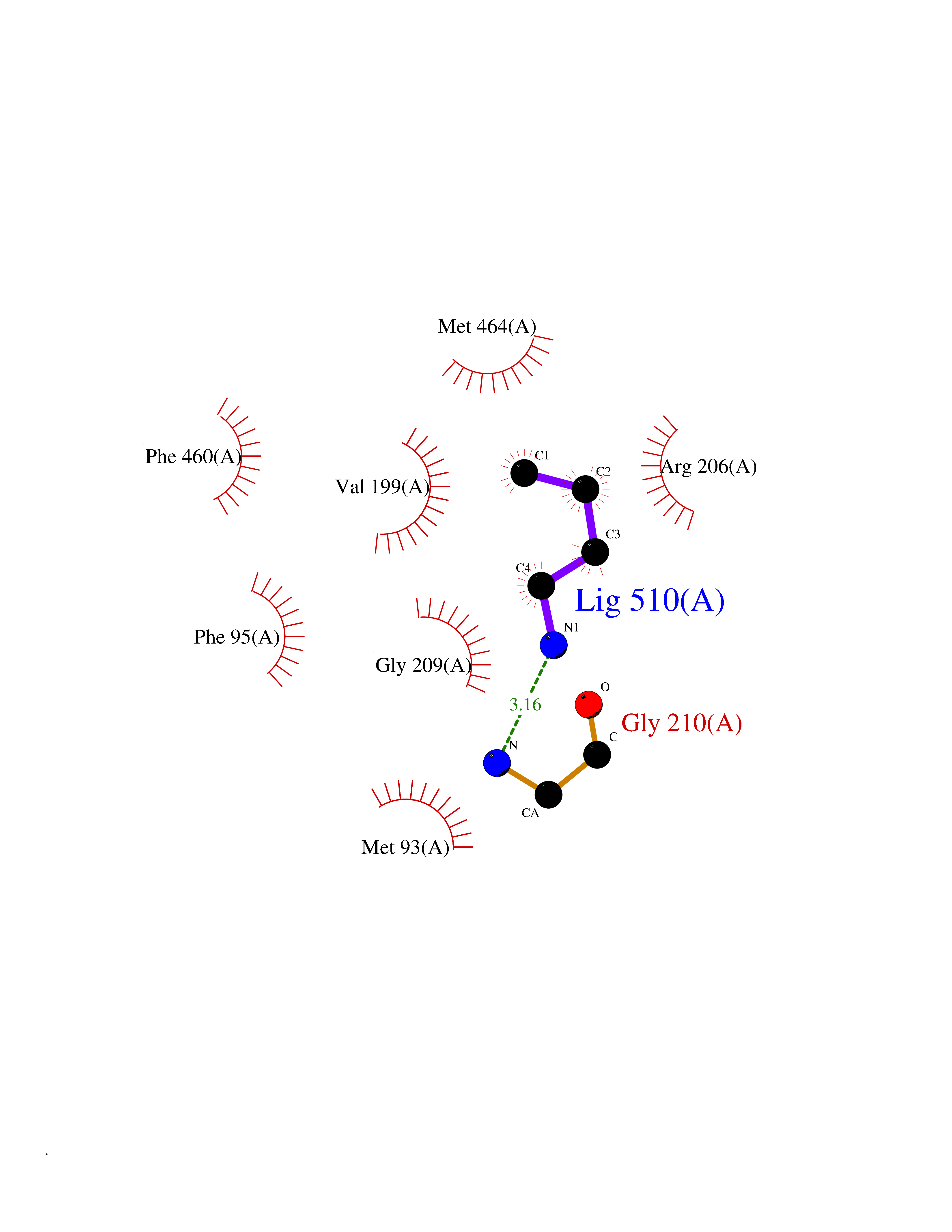 Ligplot Map 3D Binding mode Sequence SMPKDVGILALEVYFPAQYVDQTDLEKYNNVEAGKYTVGLGQTRMGFCSVQEDINSLCLTVVQRLMERIQLPWDSVGRLEVGTETIIDKSKAVKTVLMELFQDSGNTDIEGIDTTNACYGGTASLFNAANWMESSSWDGRYAMVVCGDIAVYPSGNARPTGGAGAVAMLIGPKAPLALERGLRGTHMENVYDFYKPNLASEYPIVDGKLSIQCYLRALDRCYTSYRKKIQNQWKQAGSDRPFTLDDLQYMIFHTPFCKMVQKSLARLMFNDFLSASSDTQTSLYKGLEAFGGLKLEDTYTNKDLDKALLKASQDMFDKKTKASLYLSTHNGNMYTSSLYGCLASLLSHHSAQELAGSRIGAFSYGSGLAASFFSFRVSQDAAPGSPLDKLVSSTSDLPKRLASRKCVSPEEFTEIMNQREQFYHKVNFSPPGDTNSLFPGTWYLERVDEQHRRKYARRPVSMPKDVGILALEVYFPAQYVDQTDLEKYNNVEAGKYTVGLGQTRMGFCSVQEDINSLCLTVVQRLMERIQLPWDSVGRLEVGTETIIDKSKAVKTVLMELFQDSGNTDIEGIDTTNACYGGTASLFNAANWMESSSWDGRYAMVVCGDIAVYPSGNARPTGGAGAVAMLIGPKAPLALERGLRGTHMENVYDFYKPNLASEYPIVDGKLSIQCYLRALDRCYTSYRKKIQNQWKQAGSDRPFTLDDLQYMIFHTPFCKMVQKSLARLMFNDFLSASSDTQTSLYKGLEAFGGLKLEDTYTNKDLDKALLKASQDMFDKKTKASLYLSTHNGNMYTSSLYGCLASLLSHHSAQELAGSRIGAFSYGSGLAASFFSFRVSQDAAPGSPLDKLVSSTSDLPKRLASRKCVSPEEFTEIMNQREQFYHKVNFSPPGDTNSLFPGTWYLERVDEQHRRKYARRPV Hydrogen bonds contact Hydrophobic contact | ||||
| 16 | Folate receptor beta (FOLR2) | 4KN0 | 4.64 | |
Target general information Gen name FOLR2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Placental folate-binding protein; Folate receptor, fetal/placental; Folate receptor type-beta; Folate receptor 2; FR-beta; FOLR2 Protein family Folate receptor family Biochemical class Folate receptor Function Binds to folate and reduced folic acid derivatives and mediates delivery of 5-methyltetrahydrofolate and folate analogs into the interior of cells. Has high affinity for folate and folic acid analogs at neutral pH. Exposure to slightly acidic pH after receptor endocytosis triggers a conformation change that strongly reduces its affinity for folates and mediates their release. Related diseases Acute hepatic porphyria (AHEPP) [MIM:612740]: A form of porphyria. Porphyrias are inherited defects in the biosynthesis of heme, resulting in the accumulation and increased excretion of porphyrins or porphyrin precursors. They are classified as erythropoietic or hepatic, depending on whether the enzyme deficiency occurs in red blood cells or in the liver. AHP is characterized by attacks of gastrointestinal disturbances, abdominal colic, paralyses and peripheral neuropathy. Most attacks are precipitated by drugs, alcohol, caloric deprivation, infections, or endocrine factors. {ECO:0000269|PubMed:10706561, ECO:0000269|PubMed:1309003, ECO:0000269|PubMed:1569184, ECO:0000269|PubMed:17236137, ECO:0000269|PubMed:2063868}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00158; DB00563; DB05168 Interacts with NA EC number NA Uniprot keywords 3D-structure; Cell membrane; Direct protein sequencing; Disulfide bond; Folate-binding; Glycoprotein; GPI-anchor; Lipoprotein; Membrane; Proteomics identification; Receptor; Reference proteome; Secreted; Signal; Transport Protein physicochemical properties Chain ID A Molecular weight (Da) 23841.6 Length 205 Aromaticity 0.12 Instability index 56.78 Isoelectric point 7.92 Charge (pH=7) 2.58 2D Binding mode Binding energy (Kcal/mol) -6.33  Molscript Map 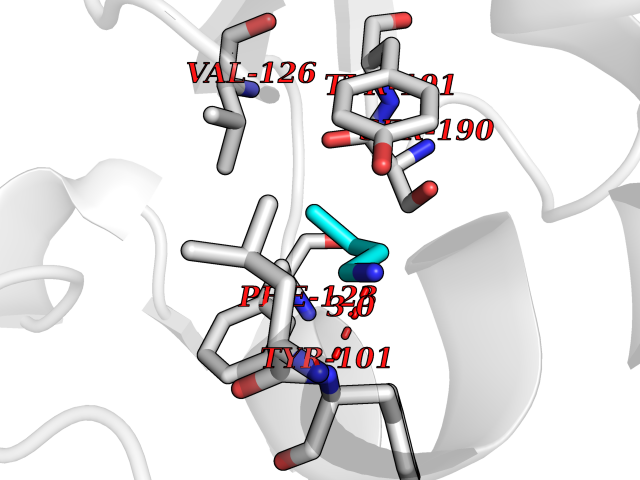 Pymol Map 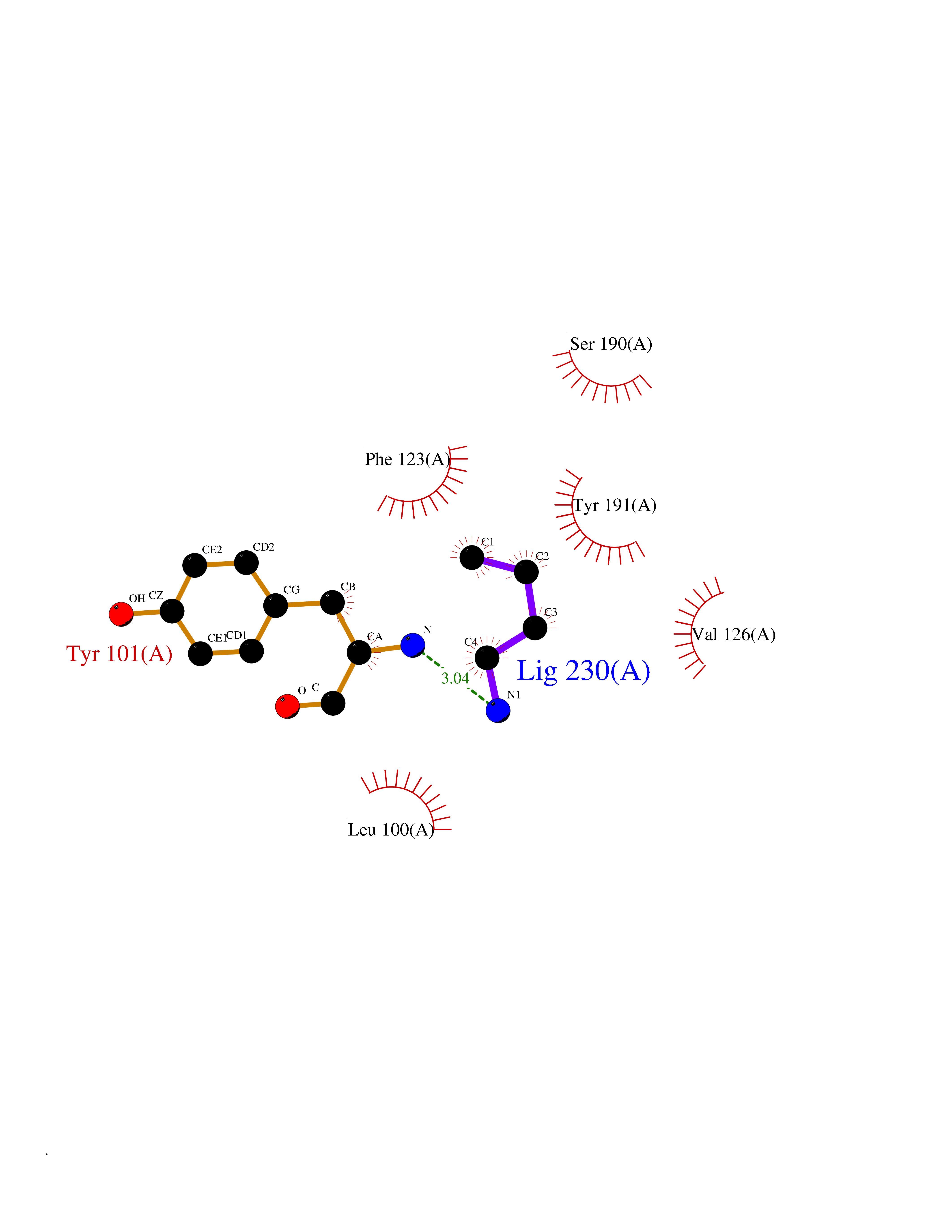 Ligplot Map 3D Binding mode Sequence RTDLLNVCMDAKHHKTKPGPEDKLHDQCSPWKKNACCTASTSQELHKDTSRLYNFNWDHCGKMEPACKRHFIQDTCLYECSPNLGPWIQQVNQSWRKERFLDVPLCKEDCQRWWEDCHTSHTCKSNWHRGWDWTSGVNKCPAGALCRTFESYFPTPAALCEGLWSHSYKVSNYSRGSGRCIQMWFDSAQGNPNEEVARFYAAAMH Hydrogen bonds contact Hydrophobic contact | ||||
| 17 | Non-heme chloroperoxidase | 1A8U | 4.64 | |
Target general information Gen name cpo Organism Kitasatospora aureofaciens (Streptomyces aureofaciens) Uniprot ID TTD ID NA Synonyms cpoT Protein family AB hydrolase superfamily, Bacterial non-heme haloperoxidase / perhydrolase family Biochemical class Haloperoxidase Function Chloride peroxidase activity. Related diseases Hypervalinemia and hyperleucine-isoleucinemia (HVLI) [MIM:618850]: An autosomal recessive metabolic disorder characterized by highly elevated plasma concentrations of valine and leucine/isoleucine. Affected individuals suffer from headache and mild memory impairment. {ECO:0000269|PubMed:25653144}. The disease is caused by variants affecting the gene represented in this entry. A patient with hypervalinemia and hyperleucine-isoleucinemia was identified as compound heterozygote for Gln-170 (inherited from his father) and Lys-264 (inherited from his mother), both variants reduced the catalytic activity of the enzyme. After treatment with vitamin B6, a precursor of pyridoxal 5'-phosphate, a BCAT2 cofactor, the blood levels of branched chain amino acids, especially valine, were decreased and brain lesions were improved. {ECO:0000269|PubMed:25653144}. Drugs (DrugBank ID) DB03793 Interacts with NA EC number 1.11.1.- Uniprot keywords 3D-structure; Chloride; Oxidoreductase; Peroxidase Protein physicochemical properties Chain ID A,B Molecular weight (Da) 60428.4 Length 554 Aromaticity 0.13 Instability index 31.26 Isoelectric point 4.65 Charge (pH=7) -32.72 2D Binding mode Binding energy (Kcal/mol) -6.33 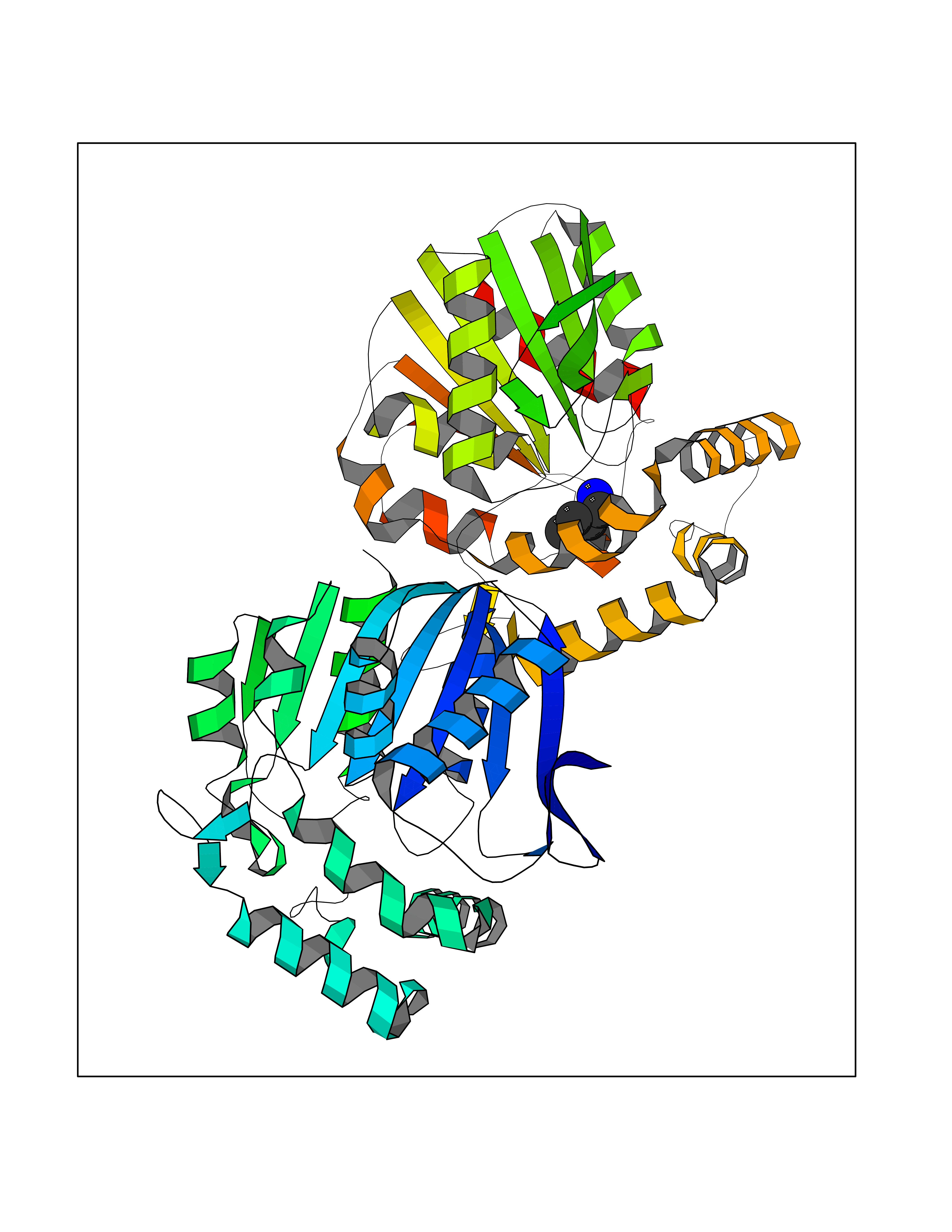 Molscript Map 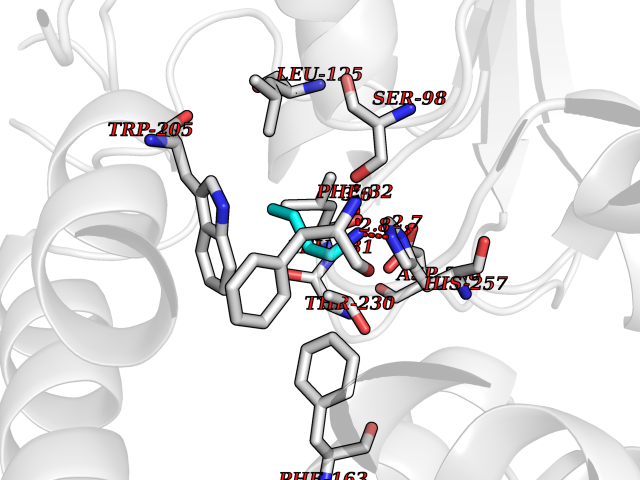 Pymol Map 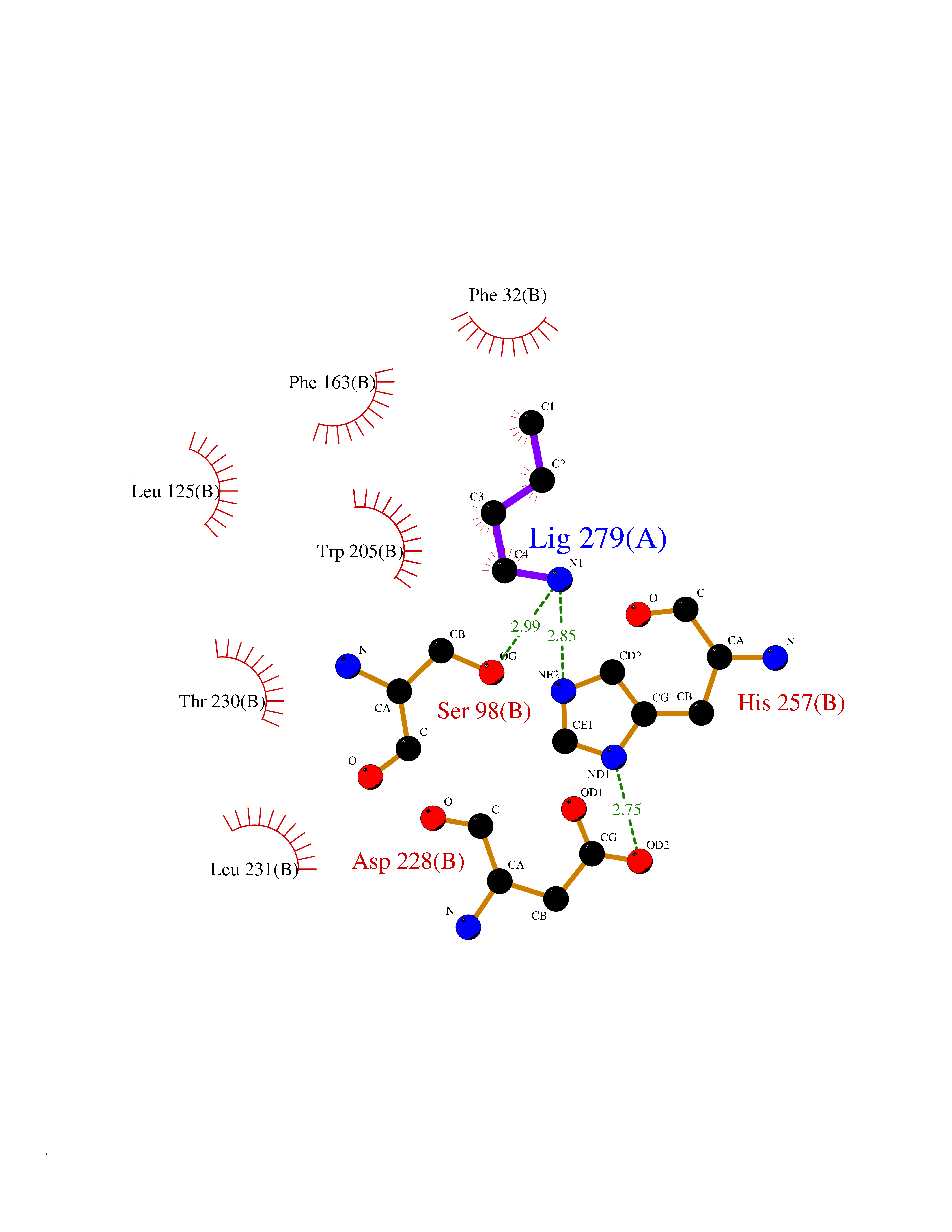 Ligplot Map 3D Binding mode Sequence PFITVGQENSTSIDLYYEDHGAGQPVVLIHGFPLSGHSWERQSAALLDAGYRVITYDRRGFGQSSQPTTGYDYDTFAADLNTVLETLDLQDAVLVGFSMGTGEVARYVSSYGTARIAKVAFLASLEPFLLKTDDNPDGAAPKEFFDGIVAAVKADRYAFYTGFFNDFYNLDENLGTRISEEAVRNSWNTAASGGFFAAAAAPTTWYTDFRADIPRIDVPALILHGTGDRTLPIENTARVFHKALPSAEYVEVEGAPHGLLWTHAEEVNTALLAFLAKPFITVGQENSTSIDLYYEDHGAGQPVVLIHGFPLSGHSWERQSAALLDAGYRVITYDRRGFGQSSQPTTGYDYDTFAADLNTVLETLDLQDAVLVGFSMGTGEVARYVSSYGTARIAKVAFLASLEPFLLKTDDNPDGAAPKEFFDGIVAAVKADRYAFYTGFFNDFYNLDENLGTRISEEAVRNSWNTAASGGFFAAAAAPTTWYTDFRADIPRIDVPALILHGTGDRTLPIENTARVFHKALPSAEYVEVEGAPHGLLWTHAEEVNTALLAFLAK Hydrogen bonds contact Hydrophobic contact | ||||
| 18 | Dopamine D2 receptor (D2R) | 5AER | 4.64 | |
Target general information Gen name DRD2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Dopamine receptor 2; D(2) dopamine receptor Protein family G-protein coupled receptor 1 family Biochemical class GPCR rhodopsin Function Dopamine receptor whose activity is mediated by G proteins which inhibit adenylyl cyclase. Related diseases Congenital sucrase-isomaltase deficiency (CSID) [MIM:222900]: Autosomal recessive intestinal disorder that is clinically characterized by fermentative diarrhea, abdominal pain, and cramps upon ingestion of sugar. The symptoms are the consequence of absent or drastically reduced enzymatic activities of sucrase and isomaltase. The prevalence of CSID is 0.02 % in individuals of European descent and appears to be much higher in Greenland, Alaskan, and Canadian native people. CSID arises due to post-translational perturbations in the intracellular transport, polarized sorting, aberrant processing, and defective function of SI. {ECO:0000269|PubMed:10903344, ECO:0000269|PubMed:11340066, ECO:0000269|PubMed:14724820, ECO:0000269|PubMed:16329100, ECO:0000269|PubMed:8609217}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01614; DB01063; DB01425; DB00915; DB06288; DB05964; DB00543; DB00182; DB04599; DB00714; DB01238; DB14185; DB09207; DB06216; DB04889; DB04888; DB05687; DB09223; DB04857; DB09128; DB01200; DB09018; DB00490; DB00248; DB06016; DB01038; DB00477; DB01239; DB00568; DB00363; DB01151; DB11274; DB13345; DB00320; DB01184; DB00988; DB00450; DB11275; DB01049; DB00696; DB01175; DB09194; DB00875; DB00623; DB04842; DB00502; DB04946; DB00458; DB04924; DB12579; DB01221; DB00555; DB01235; DB00589; DB00408; DB06077; DB08815; DB00934; DB09224; DB01043; DB00933; DB01403; DB01233; DB06148; DB00805; DB01618; DB08804; DB05766; DB00540; DB06229; DB00334; DB01267; DB12061; DB00715; DB01186; DB08922; DB00850; DB01100; DB09286; DB01621; DB12478; DB00413; DB00433; DB00420; DB01069; DB00777; DB01224; DB09097; DB12518; DB00409; DB00734; DB01549; DB00268; DB05271; DB06454; DB06144; DB00391; DB06477; DB04844; DB12093; DB00372; DB01622; DB00679; DB01623; DB13025; DB00831; DB00508; DB00726; DB06109; DB01392; DB00246; DB09225; DB01624 Interacts with Q9NRI5; P14416; Q01959 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disulfide bond; G-protein coupled receptor; Glycoprotein; Golgi apparatus; Lipoprotein; Membrane; Palmitate; Receptor; Reference proteome; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID B,C Molecular weight (Da) 24300.3 Length 209 Aromaticity 0.13 Instability index 40.14 Isoelectric point 4.97 Charge (pH=7) -7.83 2D Binding mode Binding energy (Kcal/mol) -6.33  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence PEVVEELTRKTYFTEKEVQQWYKGFIKDCPSGQLDAAGFQKIYKQFFPFGDPTKFATFVFNVFDENKDGRIEFSEFIQALSVTSRGTLDEKLRWAFKLYDLDNDGYITRNEMLDIVDAIYQMVGNTVELPEEENTPEKRVDRIFAMMDKNADGKLTLQEFQEGSKADPSIVQALSLYDGLVNIEFRKAFLKILHSNIEFRKAFLKILHS Hydrogen bonds contact Hydrophobic contact | ||||
| 19 | S-adenosylmethionine decarboxylase proenzyme (AMD1) | 1JL0 | 4.64 | |
Target general information Gen name AMD1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms SamDC; S-adenosylmethioninedecarboxylase; AdoMetDC; AMD Protein family Eukaryotic AdoMetDC family Biochemical class Carbon-carbon lyase Function Promotes maintenance and self-renewal of embryonic stem cells, by maintaining spermine levels. Essential for biosynthesis of the polyamines spermidine and spermine. Related diseases Niemann-Pick disease A (NPDA) [MIM:257200]: An early-onset lysosomal storage disorder caused by failure to hydrolyze sphingomyelin to ceramide. It results in the accumulation of sphingomyelin and other metabolically related lipids in reticuloendothelial and other cell types throughout the body, leading to cell death. Niemann-Pick disease type A is a primarily neurodegenerative disorder characterized by onset within the first year of life, intellectual disability, digestive disorders, failure to thrive, major hepatosplenomegaly, and severe neurologic symptoms. The severe neurological disorders and pulmonary infections lead to an early death, often around the age of four. Clinical features are variable. A phenotypic continuum exists between type A (basic neurovisceral) and type B (purely visceral) forms of Niemann-Pick disease, and the intermediate types encompass a cluster of variants combining clinical features of both types A and B. {ECO:0000269|PubMed:12556236, ECO:0000269|PubMed:1391960, ECO:0000269|PubMed:15221801, ECO:0000269|PubMed:15877209, ECO:0000269|PubMed:1618760, ECO:0000269|PubMed:1718266, ECO:0000269|PubMed:18815062, ECO:0000269|PubMed:19405096, ECO:0000269|PubMed:2023926, ECO:0000269|PubMed:20386867, ECO:0000269|PubMed:22818240, ECO:0000269|PubMed:23252888, ECO:0000269|PubMed:23430884, ECO:0000269|PubMed:26499107, ECO:0000269|PubMed:27338287, ECO:0000269|PubMed:8680412, ECO:0000269|PubMed:8693491, ECO:0000269|PubMed:9266408, ECO:0000269|PubMed:9660788}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Niemann-Pick disease B (NPDB) [MIM:607616]: A late-onset lysosomal storage disorder caused by failure to hydrolyze sphingomyelin to ceramide. It results in the accumulation of sphingomyelin and other metabolically related lipids in reticuloendothelial and other cell types throughout the body, leading to cell death. Clinical signs involve only visceral organs. The most constant sign is hepatosplenomegaly which can be associated with pulmonary symptoms. Patients remain free of neurologic manifestations. However, a phenotypic continuum exists between type A (basic neurovisceral) and type B (purely visceral) forms of Niemann-Pick disease, and the intermediate types encompass a cluster of variants combining clinical features of both types A and B. In Niemann-Pick disease type B, onset of the first symptoms occurs in early childhood and patients can survive into adulthood. {ECO:0000269|PubMed:12369017, ECO:0000269|PubMed:12556236, ECO:0000269|PubMed:1301192, ECO:0000269|PubMed:15241805, ECO:0000269|PubMed:16010684, ECO:0000269|PubMed:1618760, ECO:0000269|PubMed:16472269, ECO:0000269|PubMed:18815062, ECO:0000269|PubMed:1885770, ECO:0000269|PubMed:19050888, ECO:0000269|PubMed:19405096, ECO:0000269|PubMed:20386867, ECO:0000269|PubMed:21098024, ECO:0000269|PubMed:21621718, ECO:0000269|PubMed:22613662, ECO:0000269|PubMed:22818240, ECO:0000269|PubMed:23252888, ECO:0000269|PubMed:23430512, ECO:0000269|PubMed:25920558, ECO:0000269|PubMed:26084044, ECO:0000269|PubMed:26499107, ECO:0000269|PubMed:27338287, ECO:0000269|PubMed:27659707, ECO:0000269|PubMed:8051942, ECO:0000269|PubMed:8664904}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08163; DB00118; DB01917 Interacts with P17707; Q96A98; Q8WY91 EC number EC 4.1.1.50 Uniprot keywords 3D-structure; Alternative splicing; Autocatalytic cleavage; Decarboxylase; Direct protein sequencing; Lyase; Phosphoprotein; Polyamine biosynthesis; Proteomics identification; Pyruvate; Reference proteome; S-adenosyl-L-methionine; Schiff base; Spermidine biosynthesis; Zymogen Protein physicochemical properties Chain ID A,B Molecular weight (Da) 35790.5 Length 311 Aromaticity 0.14 Instability index 39.47 Isoelectric point 6.03 Charge (pH=7) -2.01 2D Binding mode Binding energy (Kcal/mol) -6.32  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence HFFEGTEKLLEVWFSRQGSGDLRTIPRSEWDILLKDVQCSIISVTKTDKQEAYVLSESSMFVSKRRFILKTCGTTLLLKALVPLLKLARDYSGFDSIQSFFYSRKNFMKPSHQGYPHRNFQEEIEFLNAIFPNGAGYCMGRMNSDCWYLYTLDFRVISQPDQTLEILMSELDPAVMDQFYMKDGVTAKDVTRESGIRDLIPGSVIDATMFNPCGYSMNGMKSDGTYWTIAITPEPEFSYVSFETNLSQTSYDDLIRKVVEVFKPGKFVTTLFVNQSSKCPQKIEGFKRLDCQSAMFNDYNFVFTSFAKKQQ Hydrogen bonds contact Hydrophobic contact | ||||
| 20 | 2-hydroxy-6-oxo-7-methylocta-2,4-dienoate hydrolase | 1UK8 | 4.64 | |
Target general information Gen name cumD Organism Pseudomonas fluorescens Uniprot ID TTD ID NA Synonyms NA Protein family NA Biochemical class Hydrolase Function Hydrolase activity. Related diseases Intellectual developmental disorder, autosomal dominant 62 (MRD62) [MIM:618793]: An autosomal dominant form of intellectual disability, a disorder characterized by significantly below average general intellectual functioning associated with impairments in adaptive behavior and manifested during the developmental period. MRD62 is characterized by mild to moderately impaired intellectual development. {ECO:0000269|PubMed:27479843, ECO:0000269|PubMed:29460436}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03741; DB03793; DB03568; DB02531; DB03750; DB02406; DB03766 Interacts with NA EC number NA Uniprot keywords 3D-structure; Hydrolase Protein physicochemical properties Chain ID A Molecular weight (Da) 30307.9 Length 271 Aromaticity 0.1 Instability index 37.49 Isoelectric point 5.02 Charge (pH=7) -11.58 2D Binding mode Binding energy (Kcal/mol) -6.32  Molscript Map  Pymol Map 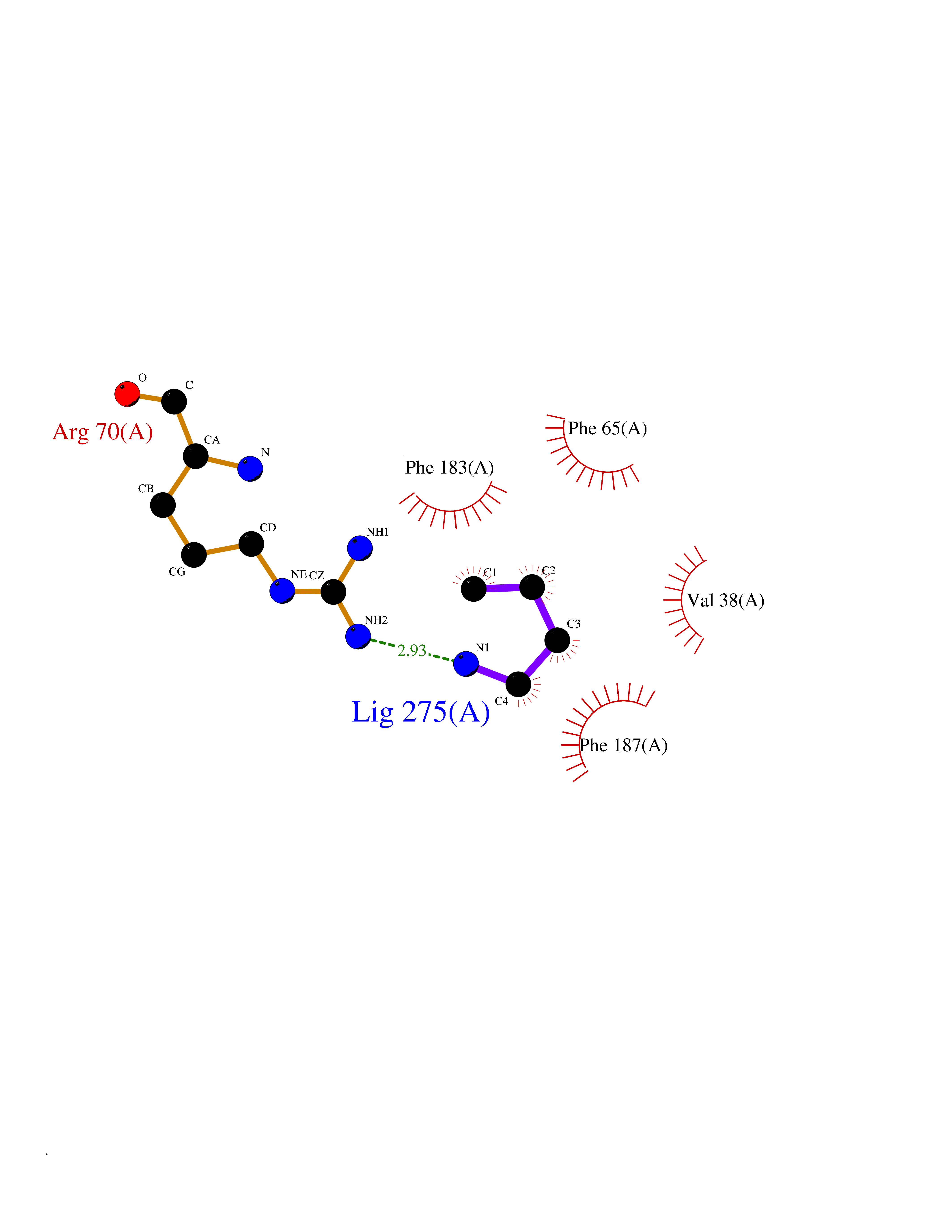 Ligplot Map 3D Binding mode Sequence NLEIGKSILAAGVLTNYHDVGEGQPVILIHGSGPGVSAYANWRLTIPALSKFYRVIAPDMVGFGFTDRPENYNYSKDSWVDHIIGIMDALEIEKAHIVGNAFGGGLAIATALRYSERVDRMVLMGAAGTRFDVTEGLNAVWGYTPSIENMRNLLDIFAYDRSLVTDELARLRYEASIQPGFQESFSSMFPEPRQRWIDALASSDEDIKTLPNETLIIHGREDQVVPLSSSLRLGELIDRAQLHVFGRCGHWTQIEQTDRFNRLVVEFFNEA Hydrogen bonds contact Hydrophobic contact | ||||