Job Results:
Ligand
Structure
Job ID
f5a9e34d4d6e0fad4e04e6dd0d3ae3aa
Job name
NA
Time
2026-01-10 22:50:50
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 1 | Glucose-dependent insulinotropic receptor (GPR119) | 7XZ6 | 7.32 | |
Target general information Gen name GPR119 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms GPR119; G-protein coupled receptor 119 Protein family G-protein coupled receptor 1 family Biochemical class GPCR rhodopsin Function Receptor for the endogenous fatty-acid ethanolamide oleoylethanolamide (OEA) and lysophosphatidylcholine (LPC). Functions as a glucose-dependent insulinotropic receptor. The activity of this receptor is mediated by G proteins which activate adenylate cyclase. Seems to act through a G(s) mediated pathway. Related diseases Developmental and epileptic encephalopathy 24 (DEE24) [MIM:615871]: A disease characterized by early-onset seizures, intellectual disability of varying degrees, and behavioral disturbances or autistic features in most individuals. {ECO:0000269|PubMed:24747641, ECO:0000269|PubMed:27864847, ECO:0000269|PubMed:30351409}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Generalized epilepsy with febrile seizures plus 10 (GEFSP10) [MIM:618482]: An autosomal dominant neurologic disorder with incomplete penetrance, characterized by variable types of seizures including absence, tonic-clonic, febrile, focal, and eyelid myoclonia. Some patients have normal neurologic development. Others have mild-to-moderate intellectual disability or autism spectrum disorder. {ECO:0000269|PubMed:29936235, ECO:0000269|PubMed:30351409}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB05166 Interacts with Q12797-6 EC number NA Uniprot keywords 3D-structure; Cell membrane; G-protein coupled receptor; Lipid-binding; Membrane; Receptor; Reference proteome; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID R Molecular weight (Da) 32134.1 Length 292 Aromaticity 0.12 Instability index 34.96 Isoelectric point 9.12 Charge (pH=7) 8.03 2D Binding mode Binding energy (Kcal/mol) -9.99  Molscript Map 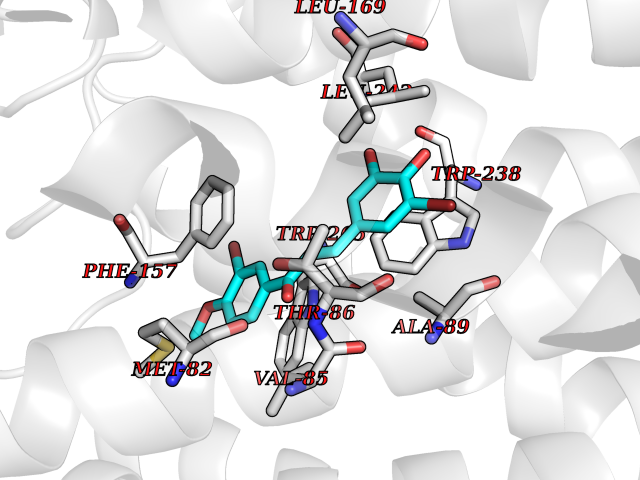 Pymol Map 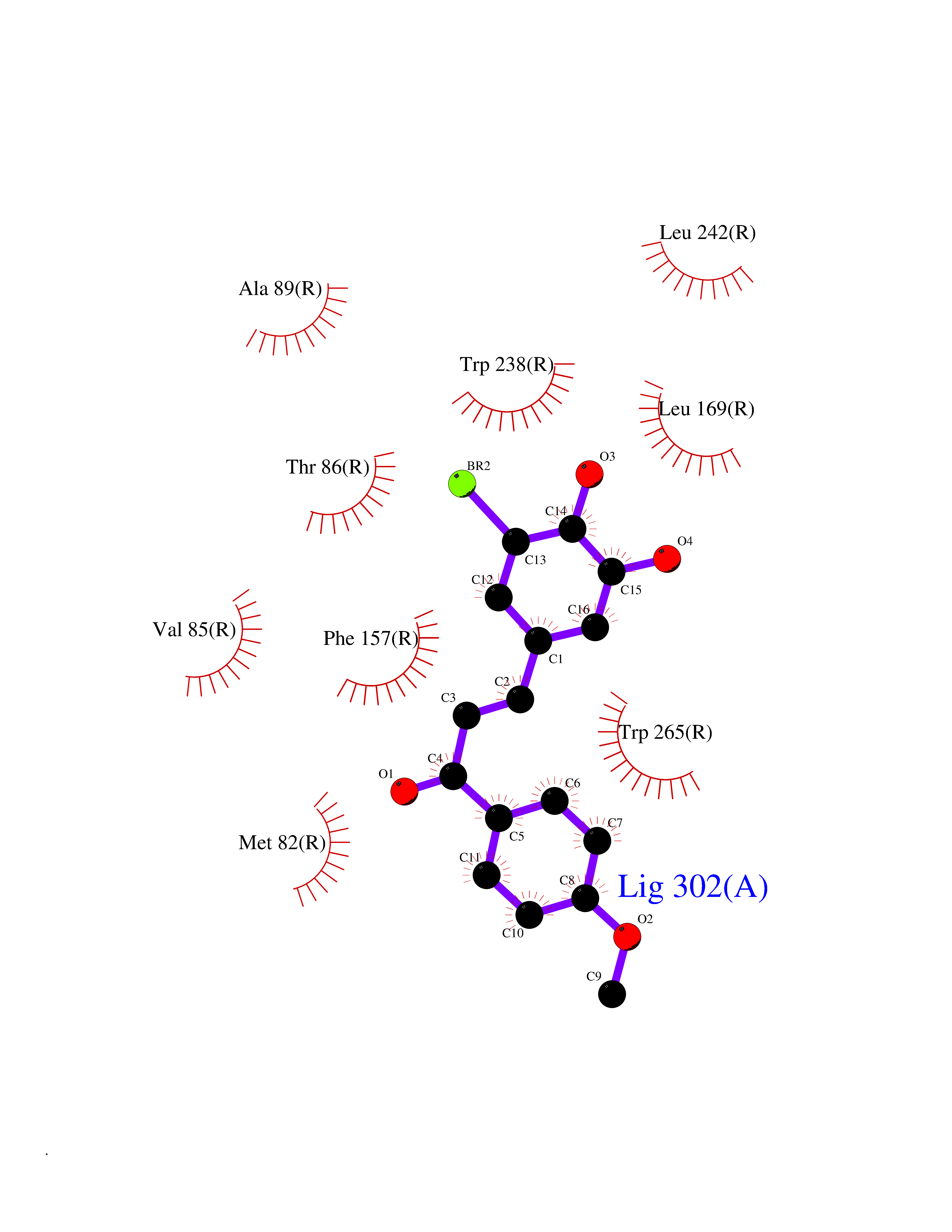 Ligplot Map 3D Binding mode Sequence MESSFSFGVILAVLASLIIATNTLVAVAVLLLIHKNDGVSLCFTLNLAVADTLIGVAISGLLTDQLSSPSRPTQKTLCSLRMAFVTSSAAASVLTVMLITFDRYLAIKQPFRYLKIMSGFVAGACIAGLWLVSYLIGFLPLGIPMFQQTAYKGQCSFFAVFHPHFVLTLSCVGFFPAMLLFVFFYCDMLKIASMHSQQIRKMEHAGAMAGSDFKALRTVSVLIGSFALSWTPFLITGIVQVACQECHLYLVLERYLWLLGVGNSLLNPLIYAYWQKEVRLQLYHMALGVKKV Hydrogen bonds contact Hydrophobic contact | ||||
| 2 | Histamine H3 receptor (H3R) | 7F61 | 7.29 | |
Target general information Gen name HRH3 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Histamine receptor 3; HH3R; GPCR97; G-protein coupled receptor 97; G protein-coupled receptor 97 Protein family G-protein coupled receptor 1 family Biochemical class GPCR rhodopsin Function Signals through the inhibition of adenylate cyclase and displays high constitutive activity (spontaneous activity in the absence of agonist). Agonist stimulation of isoform 3 neither modified adenylate cyclase activity nor induced intracellular calcium mobilization. The H3 subclass of histamine receptors could mediate the histamine signals in CNS and peripheral nervous system. Related diseases Immunodeficiency 48 (IMD48) [MIM:269840]: A form of severe immunodeficiency characterized by a selective absence of CD8+ T-cells. {ECO:0000269|PubMed:11123350, ECO:0000269|PubMed:11412303, ECO:0000269|PubMed:18509675, ECO:0000269|PubMed:8124727, ECO:0000269|PubMed:8202713}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Autoimmune disease, multisystem, infantile-onset, 2 (ADMIO2) [MIM:617006]: An autosomal recessive, autoimmune disorder characterized by systemic manifestations including blistering skin disease, uncontrollable bullous pemphigoid, inflammatory colitis, autoimmune hypothyroidism, proteinuria and nephrotic syndrome. {ECO:0000269|PubMed:26783323}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01238; DB06698; DB05381; DB17087; DB05080; DB00768; DB11642 Interacts with NA EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disulfide bond; G-protein coupled receptor; Glycoprotein; Membrane; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 34321.1 Length 301 Aromaticity 0.16 Instability index 32.22 Isoelectric point 9.63 Charge (pH=7) 15.11 2D Binding mode Binding energy (Kcal/mol) -9.94 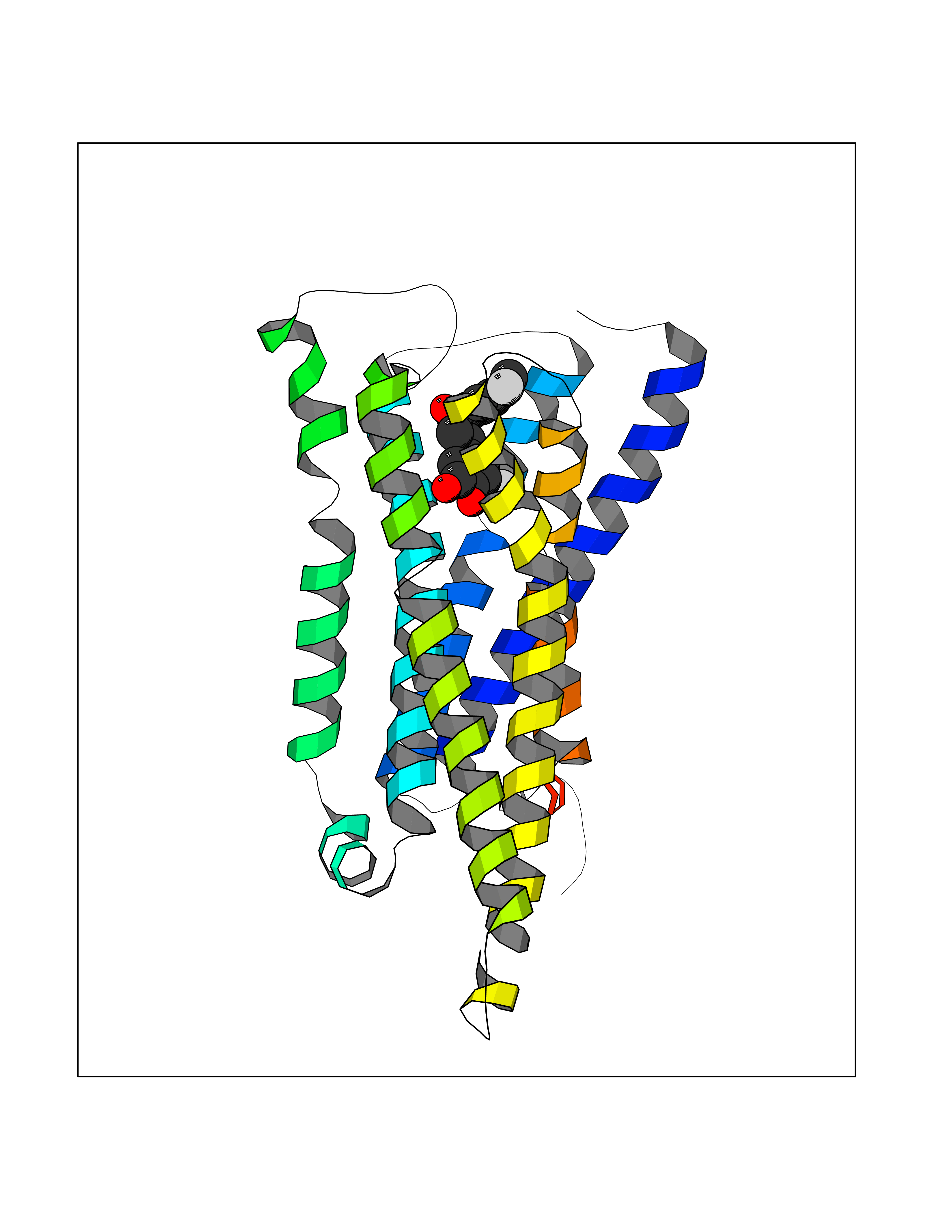 Molscript Map 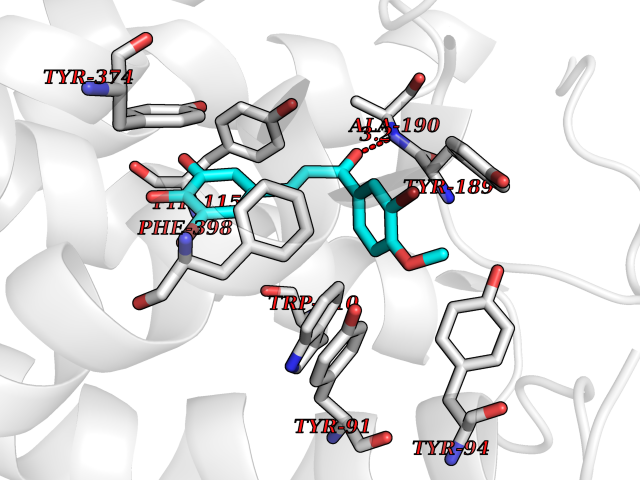 Pymol Map 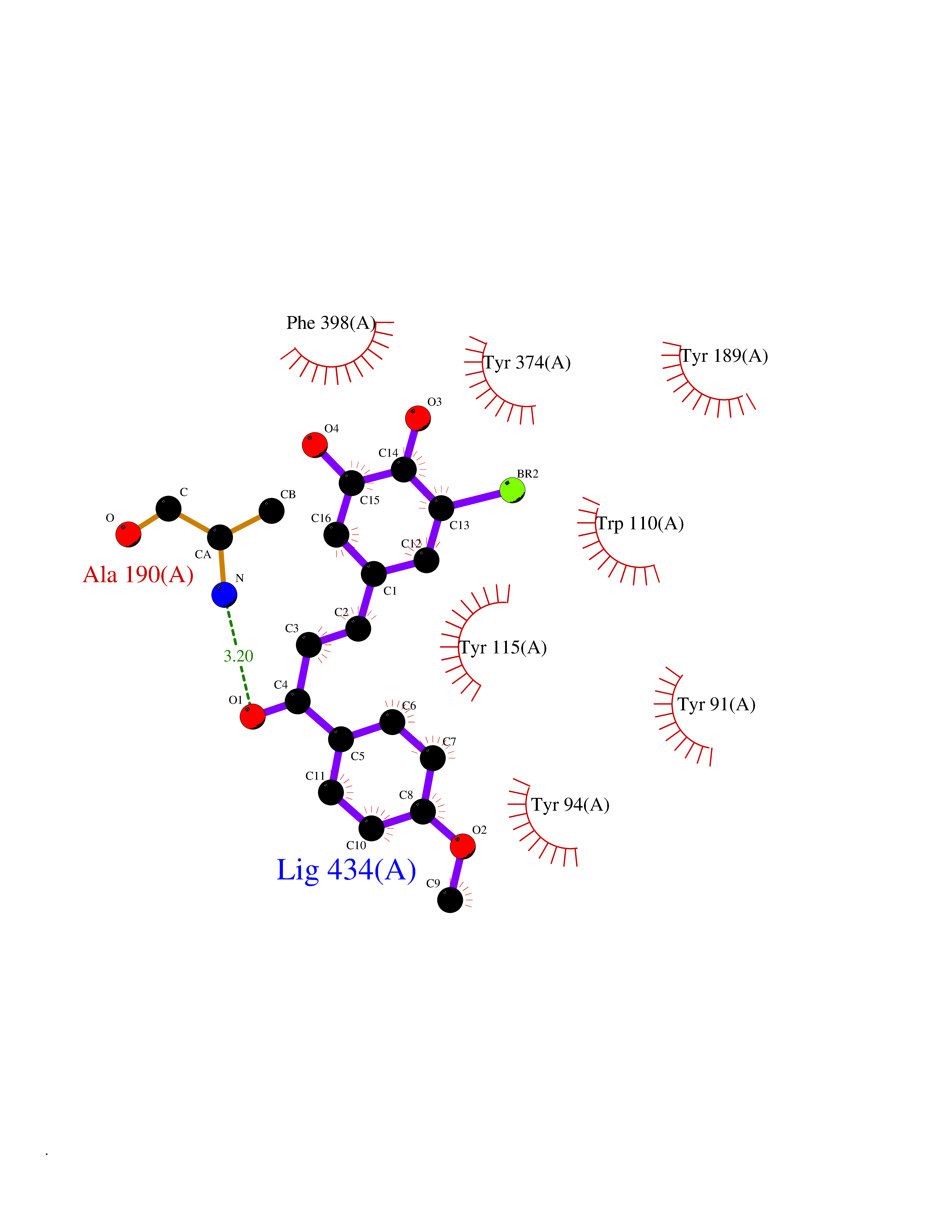 Ligplot Map 3D Binding mode Sequence RGFSAAWTAVLAALMALLIVATVLGNALVMLAFVADSSLRTQNNFFLLNLAISDFLVGAFCIPLYVPYVLTGRWTFGRGLCKLWLVVDYLLCTSKAFNIVLISYDRFLSVTRAVSYRAQQGDTRRAVRKMLLVWVLAFLLYGPAILSWEYLSGGSSIPEGHCYAEFFYNWYFLITASTLEFFTPFLSVTFFNLSIYLNIQRRTRLRLDGAREAAGRFRLSRDRKVAKSLAVIVSIFGLCWAPYTLLMIIRAACHGHCVPDYWYETSFWLLWANSAVNPVLYPLCHHSFRRAFTKLLCPQKL Hydrogen bonds contact Hydrophobic contact | ||||
| 3 | Thiopurine S-methyltransferase | 2BZG | 7.28 | |
Target general information Gen name TPMT Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Class I-like SAM-binding methyltransferase superfamily, TPMT family Biochemical class Transferase Function Thiopurine S-methyltransferase activity. Related diseases Cerebral creatine deficiency syndrome 3 (CCDS3) [MIM:612718]: An autosomal recessive disorder characterized by developmental delay/regression, intellectual disability, severe disturbance of expressive and cognitive speech, and severe depletion of creatine/phosphocreatine in the brain. Most patients develop a myopathy characterized by muscle weakness and atrophy later in life. {ECO:0000269|PubMed:11555793, ECO:0000269|PubMed:20682460, ECO:0000269|PubMed:22386973, ECO:0000269|PubMed:23660394, ECO:0000269|PubMed:23770102, ECO:0000269|PubMed:26490222, ECO:0000269|PubMed:27233232}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Fanconi renotubular syndrome 1 (FRTS1) [MIM:134600]: A form of Fanconi renotubular syndrome, a disease due to a generalized dysfunction of the proximal kidney tubule resulting in decreased solute and water reabsorption. Patients have polydipsia and polyuria with phosphaturia, glycosuria and aminoaciduria. They may develop hypophosphatemic rickets or osteomalacia, acidosis and a tendency toward dehydration. Some eventually develop renal insufficiency. FRTS1 inheritance is autosomal dominant. {ECO:0000269|PubMed:29654216}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00993; DB00436; DB01327; DB01033; DB01250; DB01021 Interacts with Q8TAP4-4; Q15047-2; P61981 EC number 2.1.1.67 Uniprot keywords 3D-structure; Acetylation; Cytoplasm; Direct protein sequencing; Methyltransferase; Phosphoprotein; Proteomics identification; Reference proteome; S-adenosyl-L-methionine; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 25971.5 Length 229 Aromaticity 0.12 Instability index 32.58 Isoelectric point 6.74 Charge (pH=7) -0.6 2D Binding mode Binding energy (Kcal/mol) -9.94 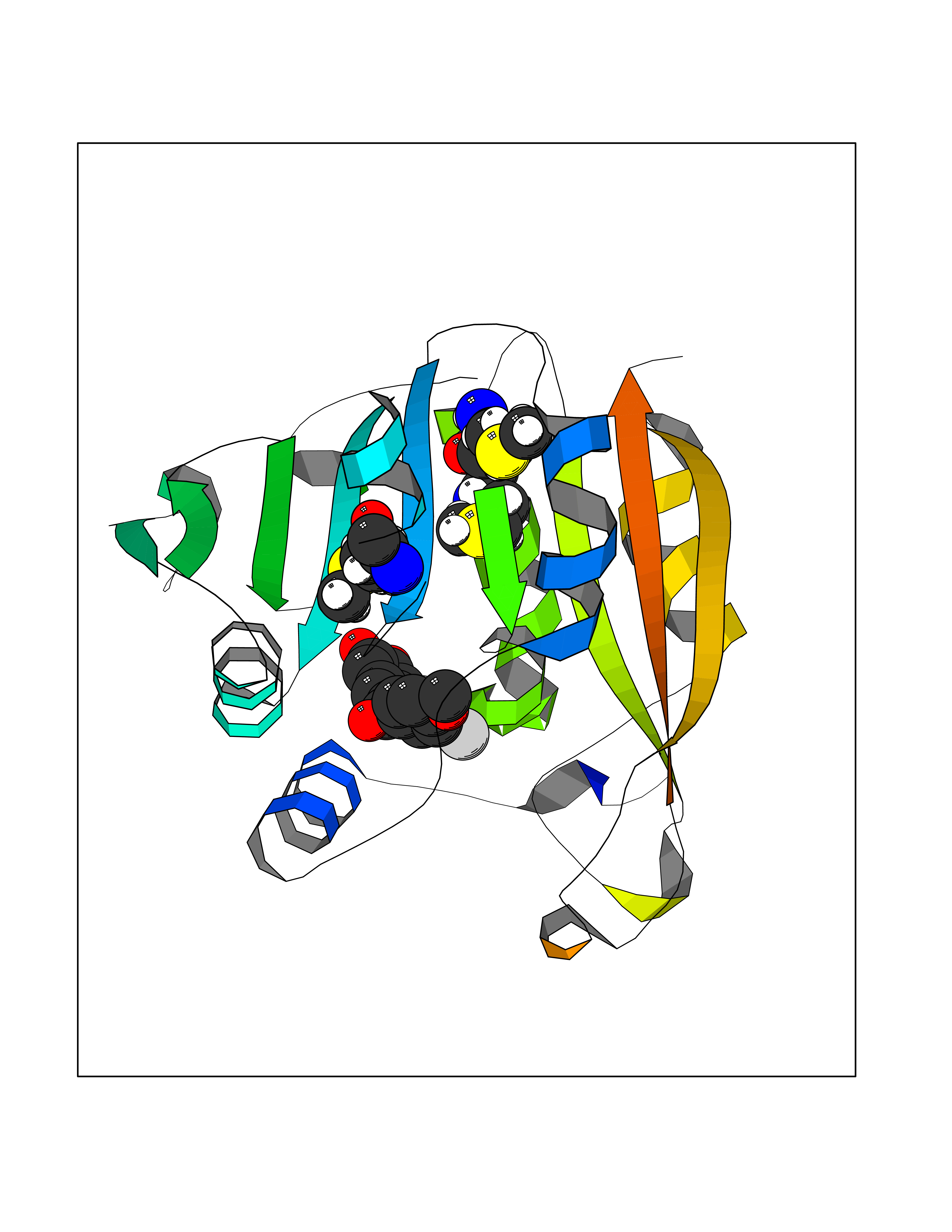 Molscript Map 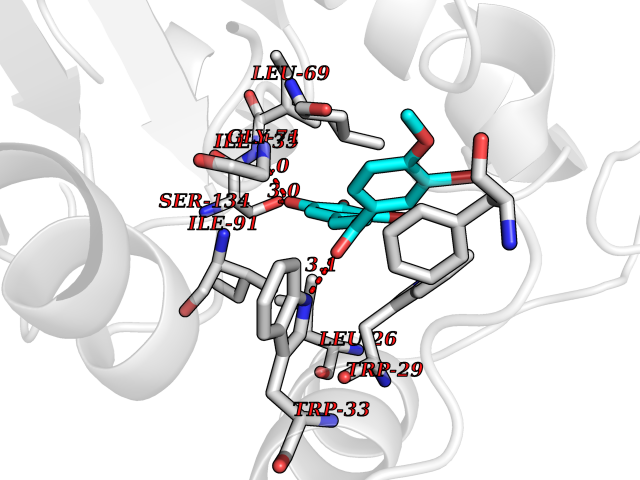 Pymol Map 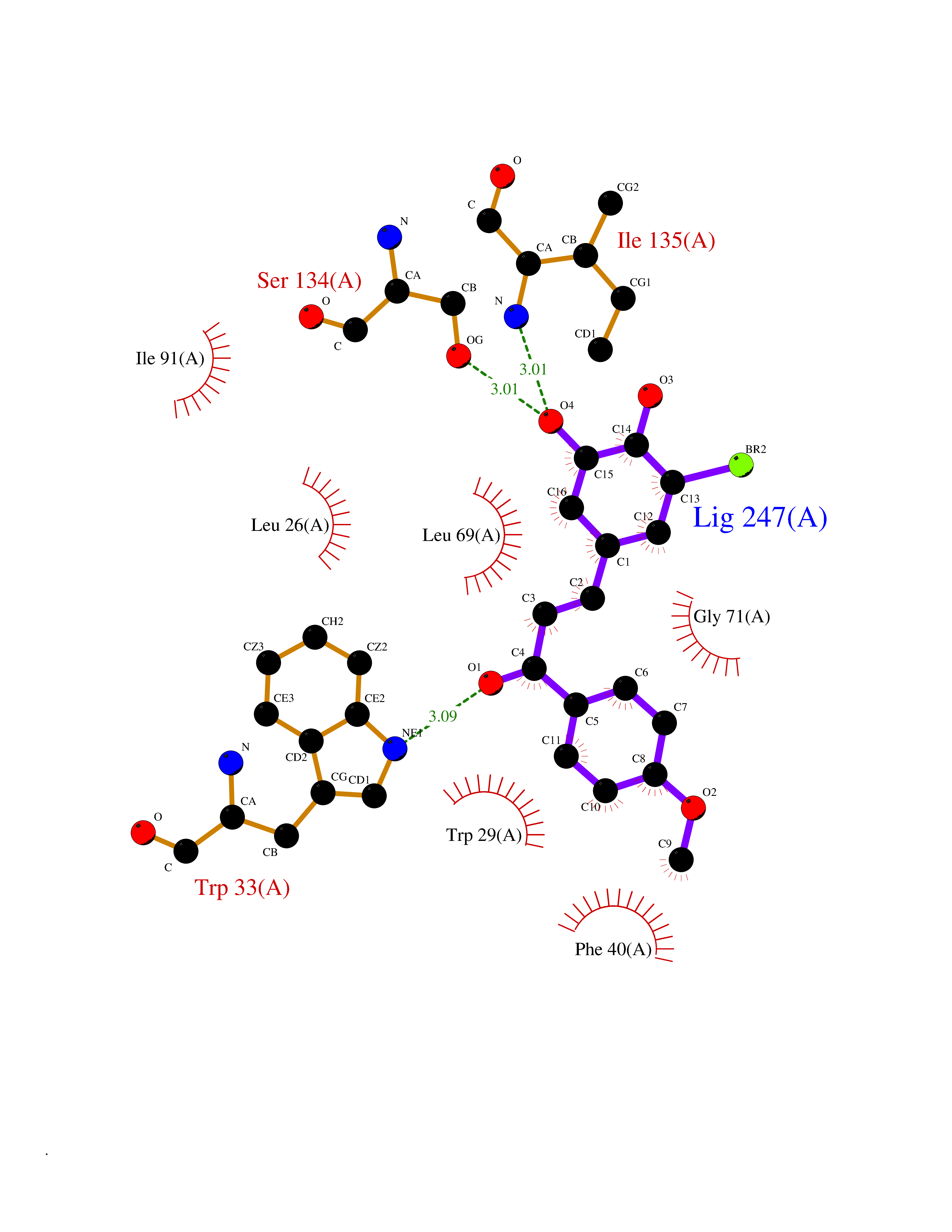 Ligplot Map 3D Binding mode Sequence EVQKNQVLTLEEWQDKWVNGKTAFHQEQGHQLLKKHLDTFLKGKSGLRVFFPLCGKAVEXKWFADRGHSVVGVEISELGIQEFFTEQNLSYSEEPITEIPGTKVFKSSSGNISLYCCSIFDLPRTNIGKFDXIWDRGALVAINPGDRKCYADTXFSLLGKKFQYLLCVLSYDPTKHPGPPFYVPHAEIERLFGKICNIRCLEKVDAFEERHKSWGIDCLFEKLYLLTEK Hydrogen bonds contact Hydrophobic contact | ||||
| 4 | Pseudomonas Transcriptional activator protein LasR (Pseudo LasR) | 3IX3 | 7.27 | |
Target general information Gen name Pseudo LasR Organism Pseudomonas aeruginosa (strain ATCC 15692 / DSM 22644 / CIP 104116 / JCM 14847 / LMG 12228 / 1C / PRS 101 / PAO1) Uniprot ID TTD ID Synonyms NA Protein family Autoinducer-regulated transcriptional regulatory protein family Biochemical class NA Function Transcriptional activator of elastase structural gene (LasB). Binds to the PAI autoinducer. Related diseases Growth hormone deficiency, isolated, 1A (IGHD1A) [MIM:262400]: An autosomal recessive, severe deficiency of growth hormone leading to dwarfism. Patients often develop antibodies to administered growth hormone. {ECO:0000269|PubMed:8364549}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Growth hormone deficiency, isolated, 1B (IGHD1B) [MIM:612781]: An autosomal recessive deficiency of growth hormone leading to short stature. Patients have low but detectable levels of growth hormone, significantly retarded bone age, and a positive response and immunologic tolerance to growth hormone therapy. {ECO:0000269|PubMed:12655557}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Kowarski syndrome (KWKS) [MIM:262650]: A syndrome clinically characterized by short stature associated with bioinactive growth hormone, normal or slightly increased growth hormone secretion, pathologically low insulin-like growth factor 1 levels, and normal catch-up growth on growth hormone replacement therapy. {ECO:0000269|PubMed:17519310, ECO:0000269|PubMed:8552145, ECO:0000269|PubMed:9276733}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Growth hormone deficiency, isolated, 2 (IGHD2) [MIM:173100]: An autosomal dominant deficiency of growth hormone leading to short stature. Clinical severity is variable. Patients have a positive response and immunologic tolerance to growth hormone therapy. {ECO:0000269|PubMed:11502836, ECO:0000269|PubMed:9152628}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08324 Interacts with NA EC number NA Uniprot keywords 3D-structure; Activator; DNA-binding; Quorum sensing; Reference proteome; Transcription; Transcription regulation Protein physicochemical properties Chain ID A Molecular weight (Da) 18305.5 Length 163 Aromaticity 0.12 Instability index 46.52 Isoelectric point 5.19 Charge (pH=7) -6.78 2D Binding mode Binding energy (Kcal/mol) -9.92  Molscript Map 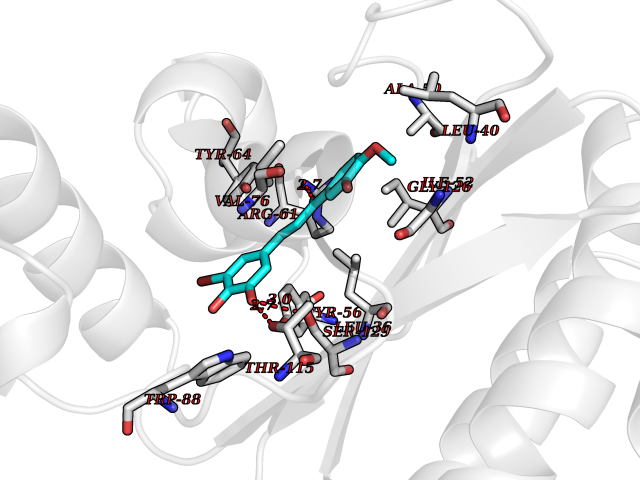 Pymol Map 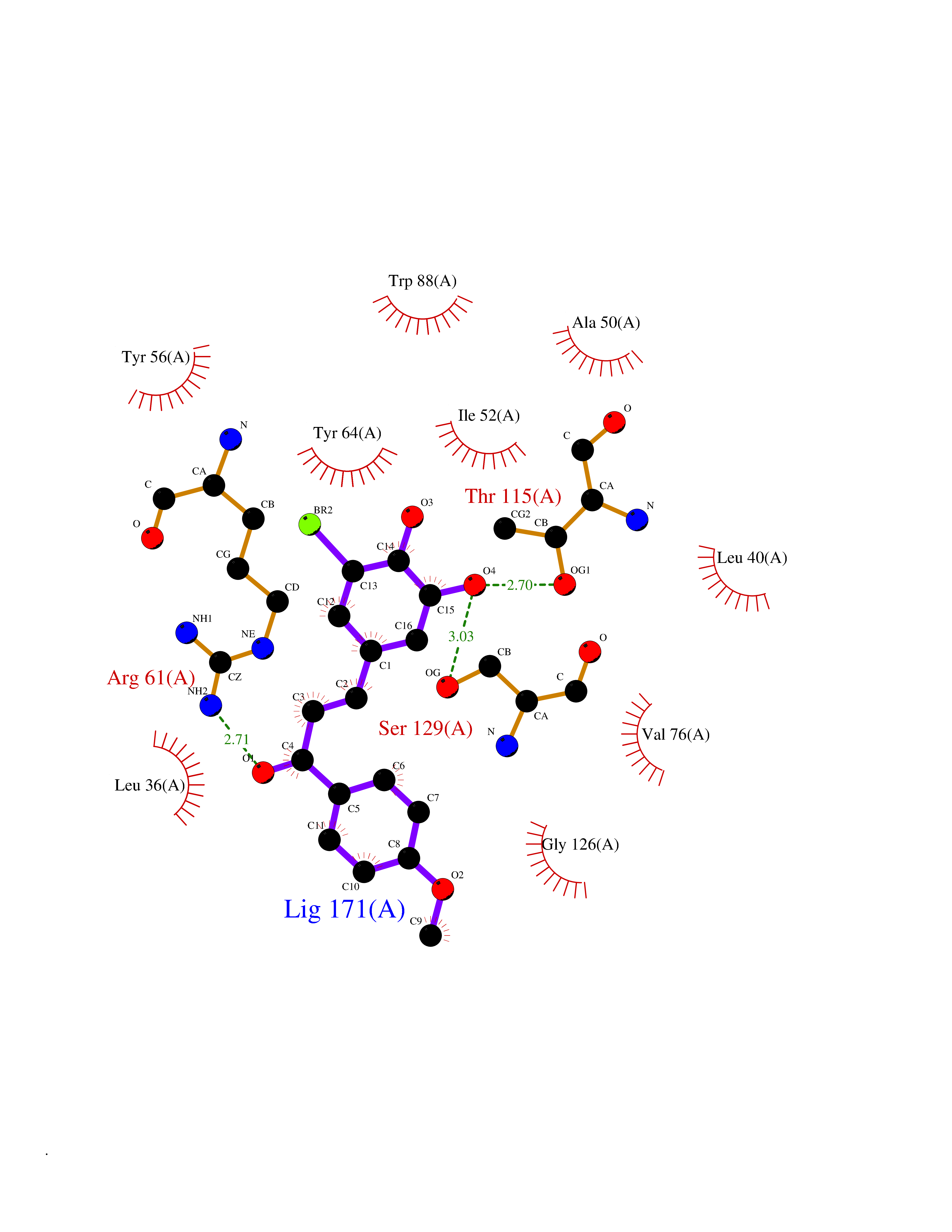 Ligplot Map 3D Binding mode Sequence FLELERSSGKLEWSAILQKMASDLGFSKILFGLLPKDSQDYENAFIVGNYPAAWREHYDRAGYARVDPTVSHCTQSVLPIFWEPSIYQTRKQHEFFEEASAAGLVYGLTMPLHGARGELGALSLSVEAENRAEANRFMESVLPTLWMLKDYALQSGAGLAFEH Hydrogen bonds contact Hydrophobic contact | ||||
| 5 | Pyruvate synthase | 2C42 | 7.26 | |
Target general information Gen name por Organism Desulfocurvibacter africanus (Desulfovibrio africanus) Uniprot ID TTD ID NA Synonyms NA Protein family Pyruvate:ferredoxin/flavodoxin oxidoreductase family Biochemical class Oxidoreductase Function 4 iron, 4 sulfur cluster binding.Iron ion binding.Pyruvate synthase activity.Thiamine pyrophosphate binding. Related diseases Intellectual developmental disorder, autosomal dominant 62 (MRD62) [MIM:618793]: An autosomal dominant form of intellectual disability, a disorder characterized by significantly below average general intellectual functioning associated with impairments in adaptive behavior and manifested during the developmental period. MRD62 is characterized by mild to moderately impaired intellectual development. {ECO:0000269|PubMed:27479843, ECO:0000269|PubMed:29460436}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02410; DB01987; DB00507 Interacts with NA EC number 1.2.7.1 Uniprot keywords 3D-structure; 4Fe-4S; Calcium; Cytoplasm; Direct protein sequencing; Disulfide bond; Electron transport; Iron; Iron-sulfur; Magnesium; Metal-binding; Oxidoreductase; Pyruvate; Thiamine pyrophosphate; Transport Protein physicochemical properties Chain ID A,B Molecular weight (Da) 115569 Length 1065 Aromaticity 0.09 Instability index 31.51 Isoelectric point 6.32 Charge (pH=7) -5.62 2D Binding mode Binding energy (Kcal/mol) -9.9  Molscript Map  Pymol Map 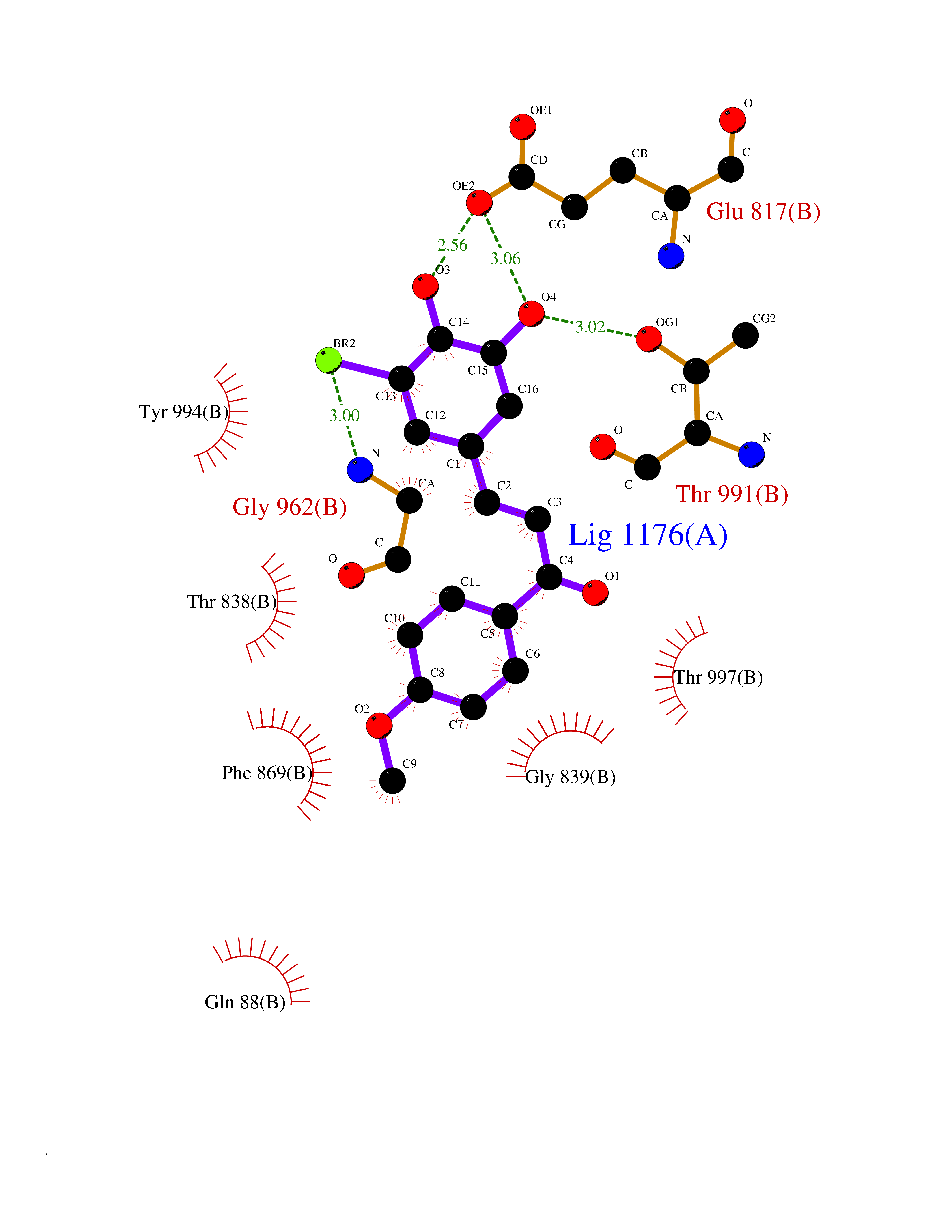 Ligplot Map 3D Binding mode Sequence GKKMMTTDGNTATAHVAYAMSEVAAIYPITPSSTMGEEADDWAAQGRKNIFGQTLTIREMQSEAGAAGAVHGALAAGALTTTFTASQGLLLMIPNMYKISGELLPGVFHVTARAIAAHALSIFGDHQDIYAARQTGFAMLASSSVQEAHDMALVAHLAAIESNVPFMHFFDGFRTSHEIQKIEVLDYADMASLVNQKALAEFRAKSPGIVAEYMQKVASLTGRSYKLFDYVGAPDAERVIVSMGSSCETIEEVINHLAAKGEKIGLIKVRLYRPFVSEAFFAALPASAKVITVLDRTKEPGAPGDPLYLDVCSAFVERGEAMPKILAGRYGLGSKEFSPAMVKSVYDNMSGAKKNHFTVGIEDDVTGTSLPVDNAFADTTPKGTIQCQFWGLGADGTVGANKQAIKIIGDNTDLFAQGYFSYDSKKSGGITISHLRFGEKPIQSTYLVNRADYVACHNPAYVGIYDILEGIKDGGTFVLNSPWSSLEDMDKHLPSGIKRTIANKKLKFYNIDAVKIATDVGLGGRINMIMQTAFFKLAGVLPFEKAVDLLKKSIHKAYGKKGEKIVKMNTDAVDQAVTSLQEFKYPDSWKDAPAETKAEPMTNEFFKNVVKPILTQQGDKLPVSAFEADGRFPLGTSQFEKRGVAINVPQWVPENCIQCNQCAFVCPHSAILPVLAKEEELVGAPANFTALEAKGKELKGYKFRIQINTLDCMGCGNCADICPPKEKALVMQPLDTQRDAQVPNLEYAARIPVKSEVLPRDSLKGSQFQEPLMEFSGACSGCGETPYVRVITQLFGERMFIANATGCSSIWGASAPSMPYKTNRLGQGPAWGNSLFEDAAEYGFGMSVWIFGGDGWAYDIGYGGLDHVLASGEDVNVFVMDTEVYSNTGGQSSKATPTGAVAKFAAAGKRTGKKDLARMVMTYGYVYVATVSMGYSKQQFLKVLKEAESFPGPSLVIAYATCINQGLRKGMGKSQDVMNTAVKSGYWPLFRYDPRLAAQGKNPFQLDSKAPDGSVEEFLMAQNRFAVLDRSFPEDAKRLRAQVAHELDVRFKELEHMAATNIFES Hydrogen bonds contact Hydrophobic contact | ||||
| 6 | Retinoic acid receptor alpha (RARA) | 3KMR | 7.25 | |
Target general information Gen name RARA Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms RAR-alpha; RAR alpha; Nuclear receptor subfamily 1 group B member 1; NR1B1 Protein family Nuclear hormone receptor family, NR1 subfamily Biochemical class Nuclear hormone receptor Function Retinoic acid receptors bind as heterodimers to their target response elements in response to their ligands, all-trans or 9-cis retinoic acid, and regulate gene expression in various biological processes. The RXR/RAR heterodimers bind to the retinoic acid response elements (RARE) composed of tandem 5'-AGGTCA-3' sites known as DR1-DR5. In the absence of ligand, the RXR-RAR heterodimers associate with a multiprotein complex containing transcription corepressors that induce histone acetylation, chromatin condensation and transcriptional suppression. On ligand binding, the corepressors dissociate from the receptors and associate with the coactivators leading to transcriptional activation. RARA plays an essential role in the regulation of retinoic acid-induced germ cell development during spermatogenesis. Has a role in the survival of early spermatocytes at the beginning prophase of meiosis. In Sertoli cells, may promote the survival and development of early meiotic prophase spermatocytes. In concert with RARG, required for skeletal growth, matrix homeostasis and growth plate function. Receptor for retinoic acid. Related diseases Chromosomal aberrations involving RARA are commonly found in acute promyelocytic leukemia. Translocation t(11;17)(q32;q21) with ZBTB16/PLZF; translocation t(15;17)(q21;q21) with PML; translocation t(5;17)(q32;q11) with NPM. The PML-RARA oncoprotein requires both the PML ring structure and coiled-coil domain for both interaction with UBE2I, nuclear microspeckle location and sumoylation. In addition, the coiled-coil domain functions in blocking RA-mediated transactivation and cell differentiation. {ECO:0000269|PubMed:12691149, ECO:0000269|PubMed:8302850, ECO:0000269|PubMed:8562957}. Drugs (DrugBank ID) DB00459; DB00210; DB00523; DB00926; DB00982; DB05785; DB04942; DB00799; DB00755; DB12808 Interacts with O43707-1; O15296; Q15699; Q96RK4; O95273; P51946; Q15910; P50148; Q9UKP3; Q96EZ8; Q15648; Q71SY5; Q15788; Q9Y6Q9; O75376; Q9Y618; Q16236; P13056-2; P48552; Q9UPP1-2; Q9H8W4; P37231; P78527; P19793; P28702; P28702-3; P48443; Q96EB6; P63165; Q8WW24; Q2M1K9; Q91XC0; P59598; Q14457; P48552; Q96CV9; P28702; P48443; Q8WW24 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Chromosomal rearrangement; Cytoplasm; DNA-binding; Isopeptide bond; Metal-binding; Nucleus; Phosphoprotein; Proteomics identification; Proto-oncogene; Receptor; Reference proteome; Transcription; Transcription regulation; Ubl conjugation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A Molecular weight (Da) 27724.1 Length 244 Aromaticity 0.05 Instability index 50.8 Isoelectric point 5.82 Charge (pH=7) -3.61 2D Binding mode Binding energy (Kcal/mol) -9.89  Molscript Map 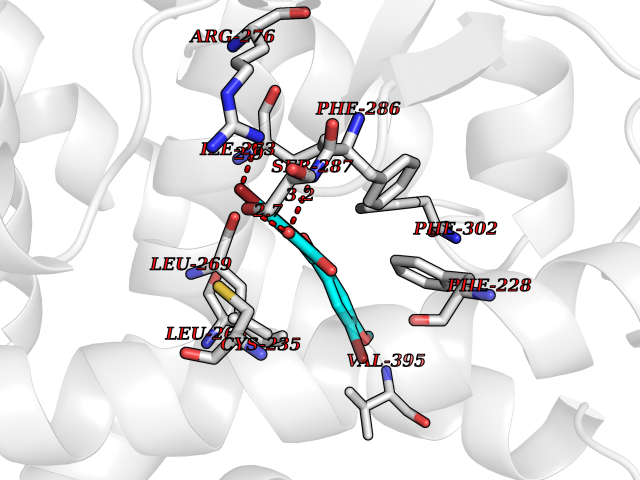 Pymol Map 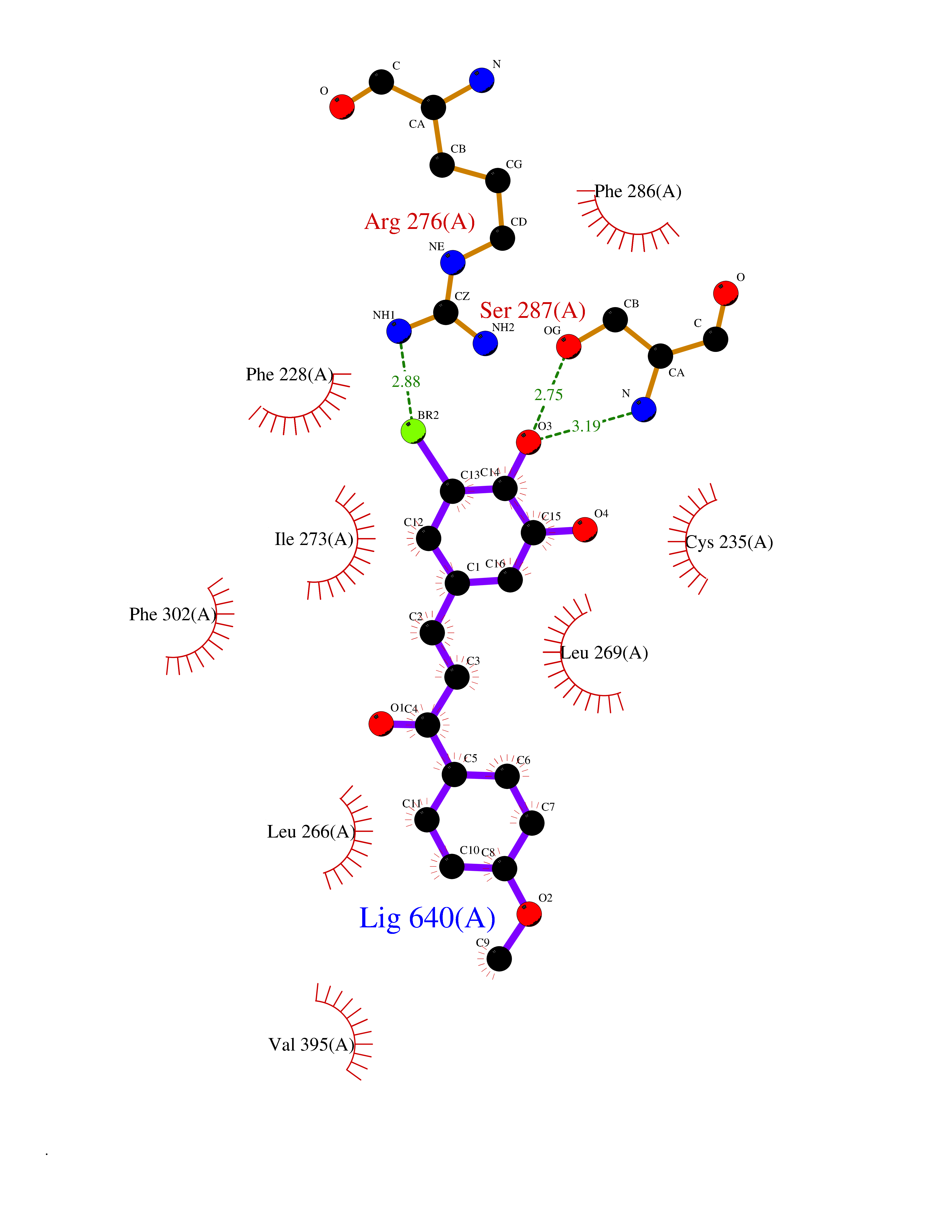 Ligplot Map 3D Binding mode Sequence PEVGELIEKVRKAHQETFPALCQLGKYTTNNSSEQRVSLDIDLWDKFSELSTKCIIKTVEFAKQLPGFTTLTIADQITLLKAACLDILILRICTRYTPEQDTMTFSDGLTLNRTQMHNAGFGPLTDLVFAFANQLLPLEMDDAETGLLSAICLICGDRQDLEQPDRVDMLQEPLLEALKVYVRKRRPSRPHMFPKMLMKITDLRSISAKGAERVITLKMEIPGSMPPLIQEMLEHKILHRLLQE Hydrogen bonds contact Hydrophobic contact | ||||
| 7 | Catechol-O-methyl-transferase (COMT) | 3BWY | 7.24 | |
Target general information Gen name COMT Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms S-COMT; MB-COMT; Catechol-O-methyltransferase; COMT Protein family Class I-like SAM-binding methyltransferase superfamily, Cation-dependent O-methyltransferase family Biochemical class Methyltransferase Function Catalyzes the O-methylation, and thereby the inactivation, of catecholamine neurotransmitters and catechol hormones. Also shortens the biological half-lives of certain neuroactive drugs, like L-DOPA, alpha-methyl DOPA and isoproterenol. Related diseases Schizophrenia (SCZD) [MIM:181500]: A complex, multifactorial psychotic disorder or group of disorders characterized by disturbances in the form and content of thought (e.g. delusions, hallucinations), in mood (e.g. inappropriate affect), in sense of self and relationship to the external world (e.g. loss of ego boundaries, withdrawal), and in behavior (e.g bizarre or apparently purposeless behavior). Although it affects emotions, it is distinguished from mood disorders in which such disturbances are primary. Similarly, there may be mild impairment of cognitive function, and it is distinguished from the dementias in which disturbed cognitive function is considered primary. Some patients manifest schizophrenic as well as bipolar disorder symptoms and are often given the diagnosis of schizoaffective disorder. {ECO:0000269|PubMed:15645182}. Disease susceptibility may be associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07462; DB02342; DB02105; DB08049; DB00118; DB00714; DB03336; DB00286; DB00255; DB00841; DB00988; DB15488; DB00494; DB00668; DB00783; DB00977; DB01064; DB00968; DB01141; DB03907; DB04820; DB06152; DB11632; DB00252; DB01420; DB00323 Interacts with Q6P5T0; P30518; Q8NFU1; Q8NHW4; P34972; Q96BA8; P50402; Q5JX71; O14843; O00258; P08034; O75712; Q9NTQ9; O95377; Q8TDT2; Q8N6U8; O15529; P31937; Q9H2F3; O95279; Q5SR56; A6NDP7; Q0D2K0; Q7RTS5; Q9UHJ9-5; Q8IY26; Q9H6H4; Q6NTF9-3; O75783; Q99500; Q9Y6D0; Q3KNW5; O60669; P22732; Q96G79; Q5T1Q4; Q9NY26; Q9NP94; Q6P1K1; P30825; Q9UHI5; B2RUZ4; Q9UPZ6; Q96MV1; Q9NV29; A0PK00; Q9NUH8; Q9P0S9; Q14656; Q6UW68; Q9H0R3; O95807; P34981; Q15645; Q15836; O95183; O76024; P30260; Q9H816; Q92997; P29323-3; P22607; P06396; Q15323; Q6A162; P26371; O15116; P20645; O14744; Q5T160; Q9UJD0; Q2MKA7; Q8N488; O75880; Q14141; Q9UNE7; Q15645; Q9NYH9; Q8NA23-2 EC number EC 2.1.1.6 Uniprot keywords 3D-structure; Alternative initiation; Catecholamine metabolism; Cell membrane; Cytoplasm; Direct protein sequencing; Lipid metabolism; Magnesium; Membrane; Metal-binding; Methyltransferase; Neurotransmitter degradation; Phosphoprotein; Proteomics identification; Reference proteome; S-adenosyl-L-methionine; Schizophrenia; Signal-anchor; Transferase; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 23851.2 Length 214 Aromaticity 0.07 Instability index 25.99 Isoelectric point 5.25 Charge (pH=7) -7.75 2D Binding mode Binding energy (Kcal/mol) -9.87  Molscript Map 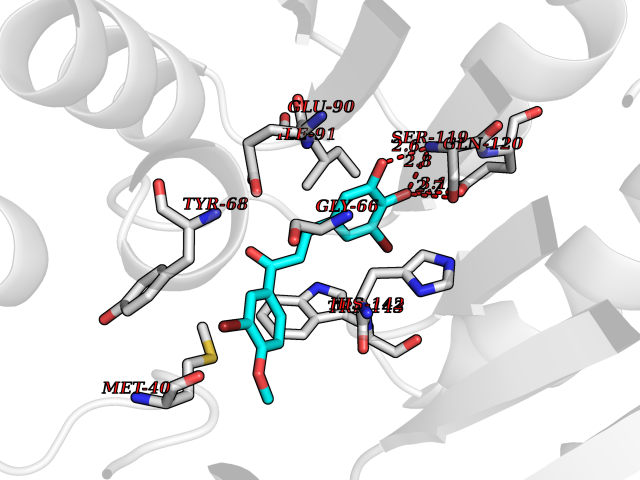 Pymol Map 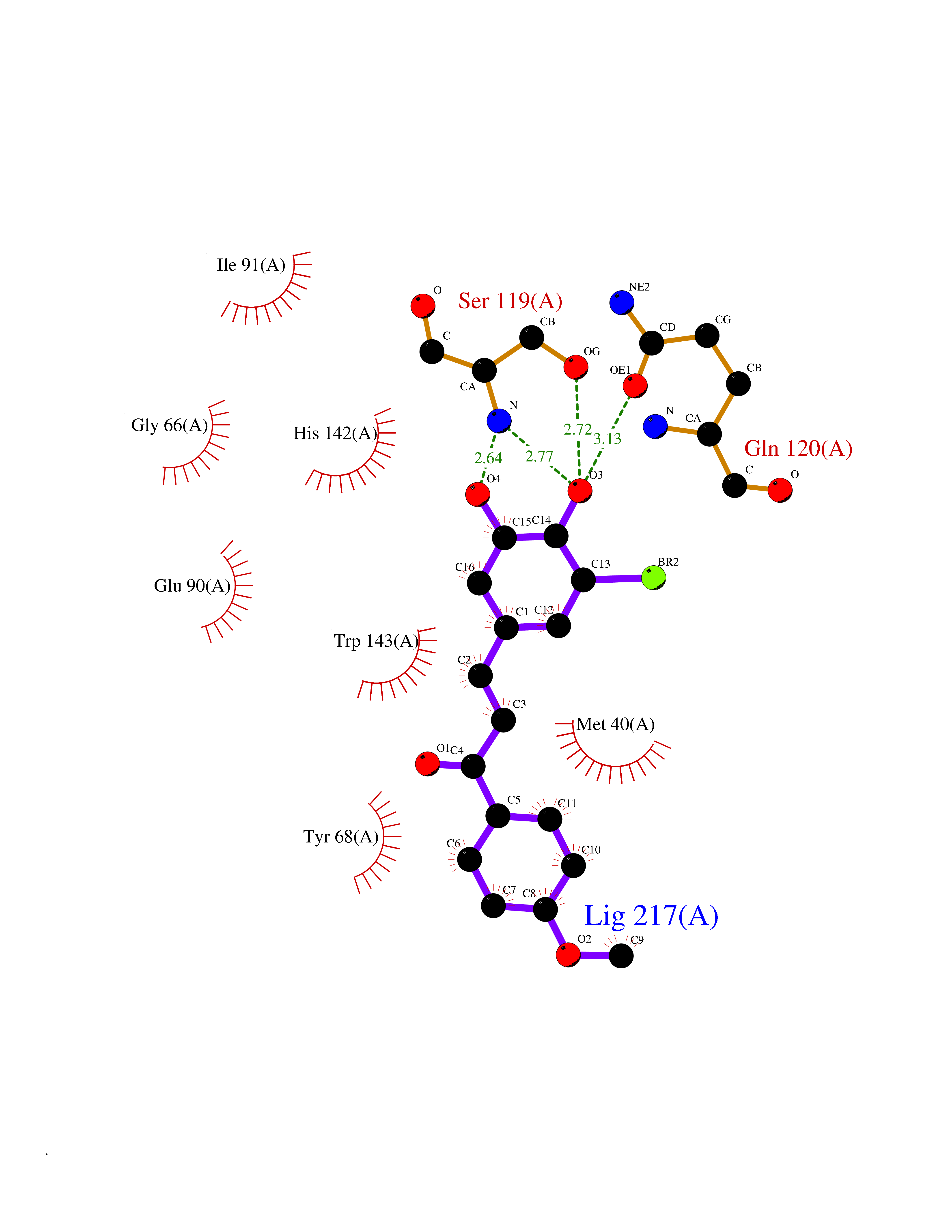 Ligplot Map 3D Binding mode Sequence GDTKEQRILNHVLQHAEPGNAQSVLEAIDTYCEQKEWAMNVGDKKGKIVDAVIQEHQPSVLLELGAYCGYSAVRMARLLSPGARLITIEINPDCAAITQRMVDFAGMKDKVTLVVGASQDIIPQLKKKYDVDTLDMVFLDHWKDRYLPDTLLLEECGLLRKGTVLLADNVICPGAPDFLAHVRGSSCFECTHYQSFLEYREVVDGLEKAIYKGP Hydrogen bonds contact Hydrophobic contact | ||||
| 8 | Metabotropic glutamate receptor 5 (mGluR5) | 4OO9 | 7.24 | |
Target general information Gen name GRM5 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms MGLUR5; GPRC1E Protein family G-protein coupled receptor 3 family Biochemical class GPCR glutamate Function G-protein coupled receptor for glutamate. Ligand binding causes a conformation change that triggers signaling via guanine nucleotide-binding proteins (G proteins) and modulates the activity of down-stream effectors. Signaling activates a phosphatidylinositol-calcium second messenger system and generates a calcium-activated chloride current. Plays an important role in the regulation of synaptic plasticity and the modulation of the neural network activity. Related diseases Charcot-Marie-Tooth disease, axonal, 2D (CMT2D) [MIM:601472]: A dominant axonal form of Charcot-Marie-Tooth disease, a disorder of the peripheral nervous system, characterized by progressive weakness and atrophy, initially of the peroneal muscles and later of the distal muscles of the arms. Charcot-Marie-Tooth disease is classified in two main groups on the basis of electrophysiologic properties and histopathology: primary peripheral demyelinating neuropathies (designated CMT1 when they are dominantly inherited) and primary peripheral axonal neuropathies (CMT2). Neuropathies of the CMT2 group are characterized by signs of axonal degeneration in the absence of obvious myelin alterations, normal or slightly reduced nerve conduction velocities, and progressive distal muscle weakness and atrophy. {ECO:0000269|PubMed:12690580, ECO:0000269|PubMed:17035524, ECO:0000269|PubMed:17101916, ECO:0000269|PubMed:17663003, ECO:0000269|PubMed:20169446, ECO:0000269|PubMed:24604904, ECO:0000269|PubMed:25168514, ECO:0000269|PubMed:26244500, ECO:0000269|PubMed:26503042, ECO:0000269|PubMed:31173493}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Neuronopathy, distal hereditary motor, autosomal dominant 5 (HMND5) [MIM:600794]: A form of distal hereditary motor neuronopathy, a heterogeneous group of neuromuscular disorders caused by selective degeneration of motor neurons in the anterior horn of the spinal cord, without sensory deficit in the posterior horn. The overall clinical picture consists of a classical distal muscular atrophy syndrome in the legs without clinical sensory loss. The disease starts with weakness and wasting of distal muscles of the anterior tibial and peroneal compartments of the legs. Later on, weakness and atrophy may expand to the proximal muscles of the lower limbs and/or to the distal upper limbs. {ECO:0000269|PubMed:12690580, ECO:0000269|PubMed:17035524, ECO:0000269|PubMed:23279345, ECO:0000269|PubMed:24627108, ECO:0000269|PubMed:26503042}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Spinal muscular atrophy, infantile, James type (SMAJI) [MIM:619042]: An autosomal dominant form of spinal muscular atrophy, a group of neuromuscular disorders characterized by degeneration of the anterior horn cells of the spinal cord, leading to symmetrical muscle weakness and atrophy. SMAJI is a severe disease characterized by hypotonia manifesting in the first weeks or months of life, delayed motor development, motor regression, and muscle weakness and atrophy primarily affecting distal muscles. Additional variable features include feeding difficulties, poor overall growth, foot deformities, kyphosis, hyperlordosis, scoliosis, vocal cord dysfunction, and respiratory insufficiency. {ECO:0000269|PubMed:32181591}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00659; DB05070; DB12733; DB06201 Interacts with P41594; Q7Z6G3 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disulfide bond; G-protein coupled receptor; Glycoprotein; Membrane; Methylation; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Signal; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 27065.4 Length 247 Aromaticity 0.13 Instability index 42.92 Isoelectric point 9.24 Charge (pH=7) 11.34 2D Binding mode Binding energy (Kcal/mol) -9.88 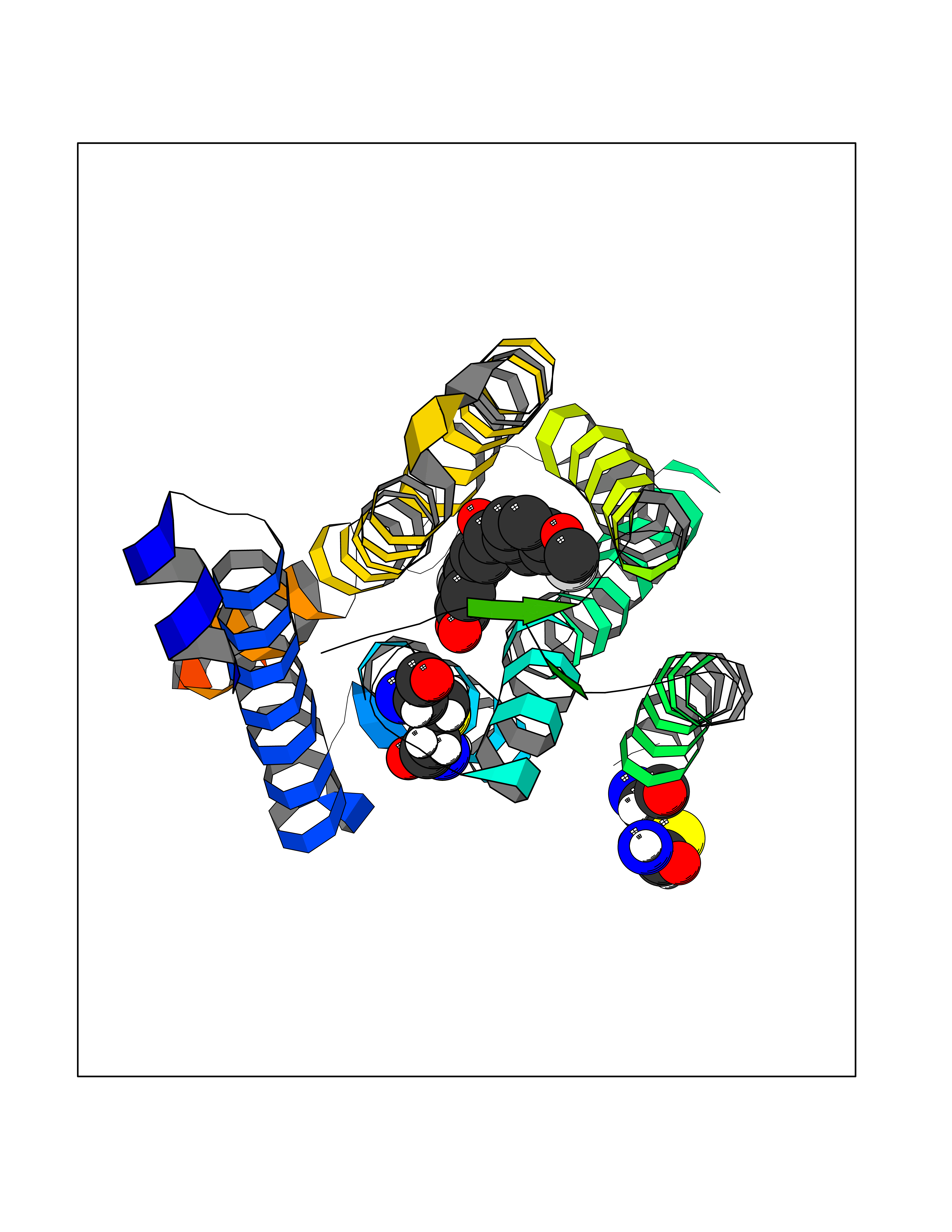 Molscript Map 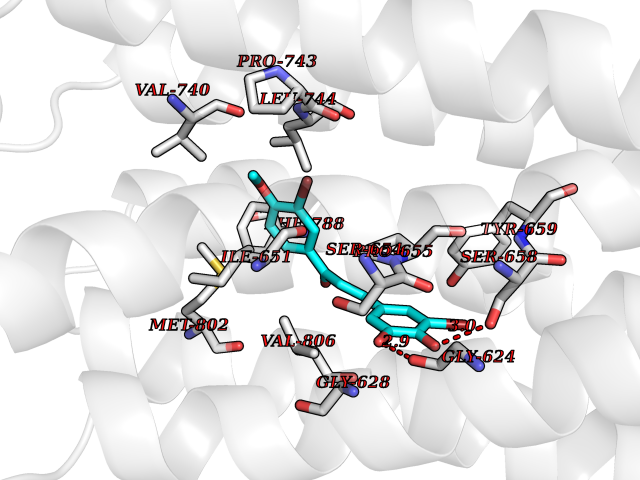 Pymol Map 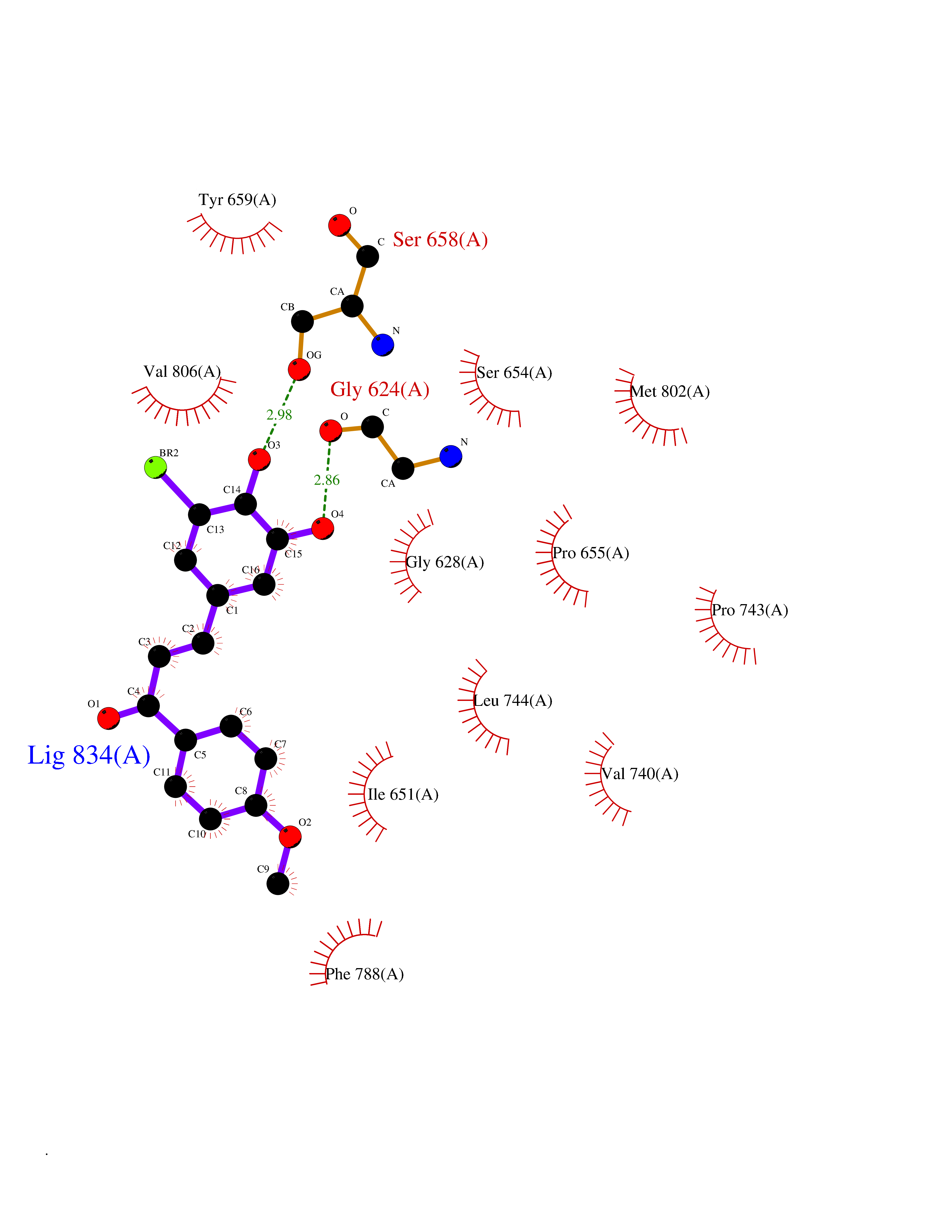 Ligplot Map 3D Binding mode Sequence SPVQYLRWGDPAPIAAVVFACLGLLATLFVTVVFIIYRDTPVVKSSSRELCYIILAGICLGYLCTFXLIAKPKQIYCYLQRIGIGLSPAMSYSALVTKTYRAARILAMSKKSAXAQLVIAFILICIQLGIIVALFIMEPPDIMVYLICNTTNLGVVAPLGYNGLLILACTFYAFKTRNVPANFNEAKYIAFTMYTTCIIWLAFVPIYFGSNYKIITMCFSVSLSATVALGCMFVPKVYIILAKPERN Hydrogen bonds contact Hydrophobic contact | ||||
| 9 | Spermine synthase | 3C6K | 7.23 | |
Target general information Gen name SMS Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Spermidine/spermine synthase family Biochemical class Transferase Function Spermine synthase activity. Related diseases Intellectual developmental disorder, X-linked, syndromic, Snyder-Robinson type (MRXSSR) [MIM:309583]: An X-linked intellectual disability syndrome characterized by a collection of clinical features including facial asymmetry, marfanoid habitus, hypertonia, osteoporosis and unsteady gait. {ECO:0000269|PubMed:14508504, ECO:0000269|PubMed:18550699, ECO:0000269|PubMed:19206178, ECO:0000269|PubMed:22612257, ECO:0000269|PubMed:23696453, ECO:0000269|PubMed:23897707}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00127 Interacts with NA EC number 2.5.1.22 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Direct protein sequencing; Intellectual disability; Phosphoprotein; Polyamine biosynthesis; Proteomics identification; Reference proteome; Transferase Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 27177.1 Length 238 Aromaticity 0.11 Instability index 41.31 Isoelectric point 4.82 Charge (pH=7) -9.19 2D Binding mode Binding energy (Kcal/mol) -9.87  Molscript Map 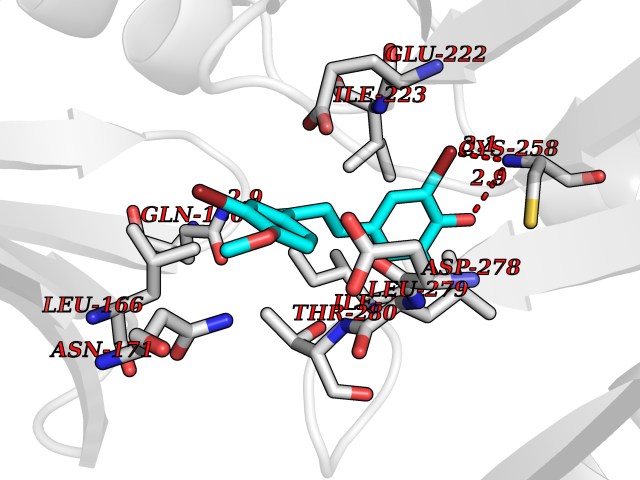 Pymol Map 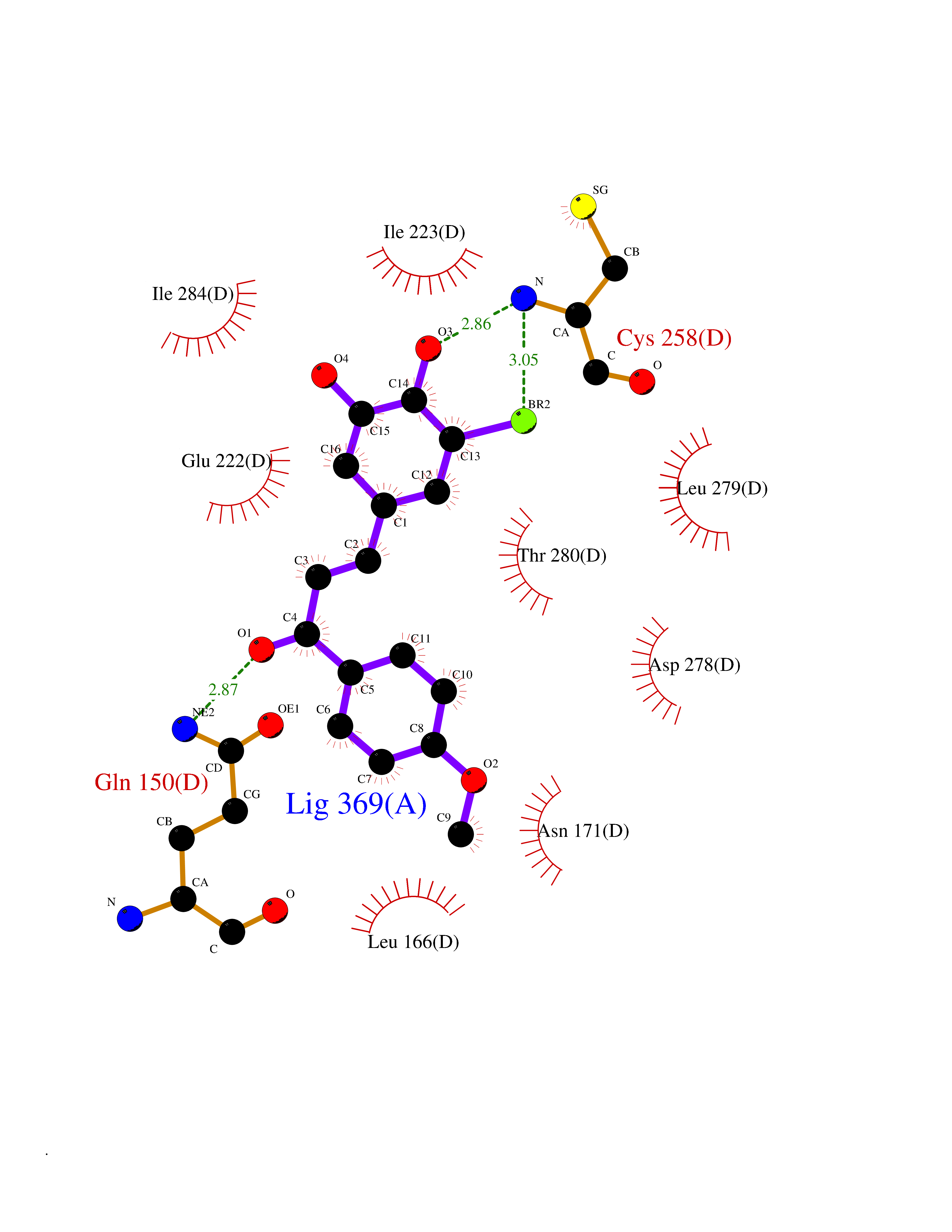 Ligplot Map 3D Binding mode Sequence RYWPTADGRLVEYDIDEVVYDEDSPYQNIKILHSKQFGNILILSGDVNLAESDLAYTRAIMGSGKEDYTGKDVLILGGGDGGILCEIVKLKPKMVTMVEIDQMVIDGCKKYMRKDVLDNLKGDCYQVLIEDCIPVLKRYAKEGREFDYVINDLTAVPISTSPSTWEFLRLILDLSMKVLKQDGKYFTQGNCVNLTEALSLYEEQLGRLYCPVEFSKEIVCVPSYLELWVFYTVWKKAK Hydrogen bonds contact Hydrophobic contact | ||||
| 10 | Phenylethanolamine N-methyltransferase (PNMT) | 2G72 | 7.22 | |
Target general information Gen name PNMT Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms PNMTase; PENT; Noradrenaline N-methyltransferase Protein family Class I-like SAM-binding methyltransferase superfamily, NNMT/PNMT/TEMT family Biochemical class NA Function Converts noradrenaline to adrenaline. Related diseases A chromosomal aberration involving TRIM24/TIF1 is found in papillary thyroid carcinomas (PTCs). Translocation t(7;10)(q32;q11) with RET. The translocation generates the TRIM24/RET (PTC6) oncogene. {ECO:0000269|PubMed:10439047}. Drugs (DrugBank ID) DB08129; DB08128; DB07739; DB07798; DB07747; DB03468; DB08550; DB03824; DB04273; DB07906; DB07597; DB09571; DB00968; DB08631; DB01752; DB08654 Interacts with Q9P2G9-2; Q8TBB1 EC number EC 2.1.1.28 Uniprot keywords 3D-structure; Catecholamine biosynthesis; Direct protein sequencing; Methyltransferase; Phosphoprotein; Proteomics identification; Reference proteome; S-adenosyl-L-methionine; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 29198.9 Length 264 Aromaticity 0.09 Instability index 54.33 Isoelectric point 5.91 Charge (pH=7) -3.69 2D Binding mode Binding energy (Kcal/mol) -9.85 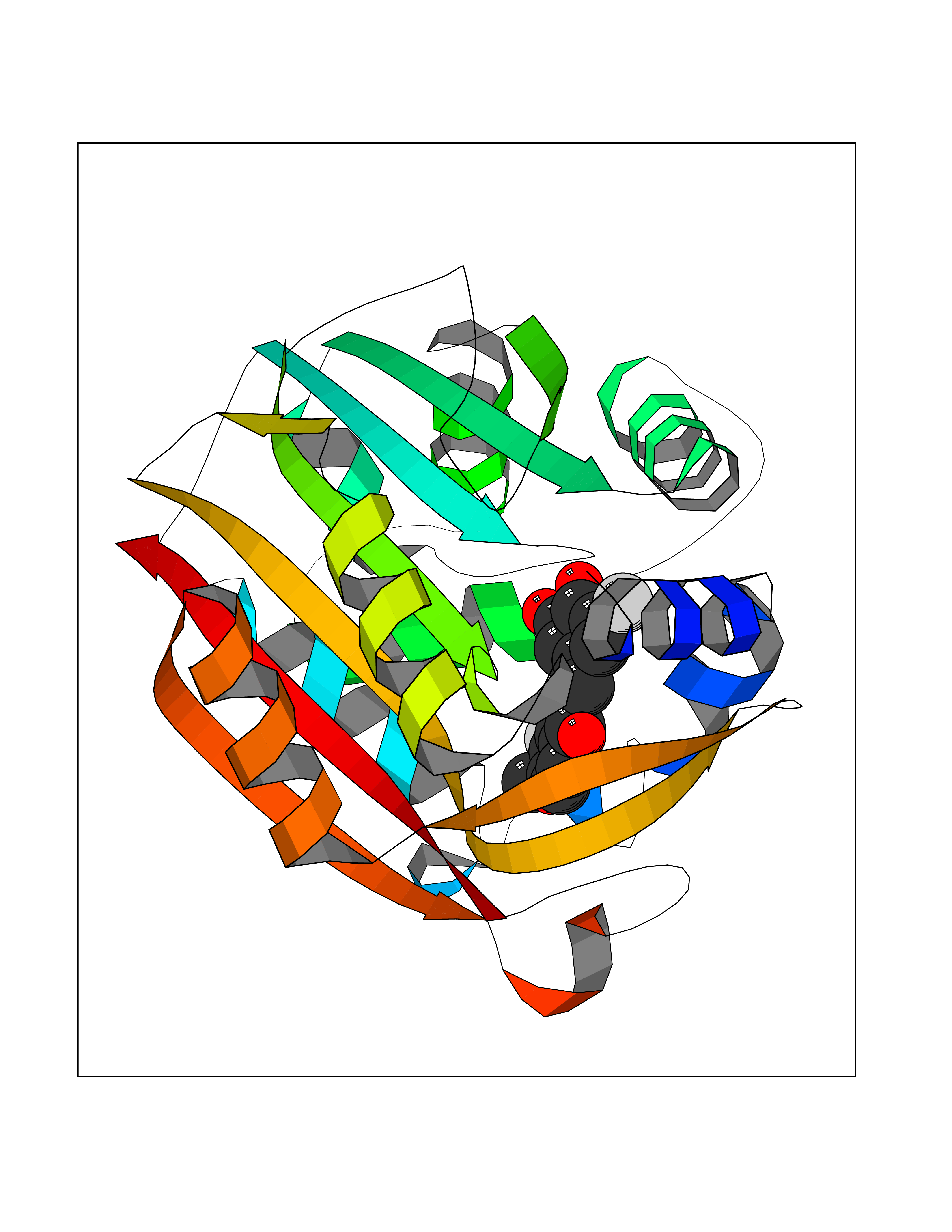 Molscript Map 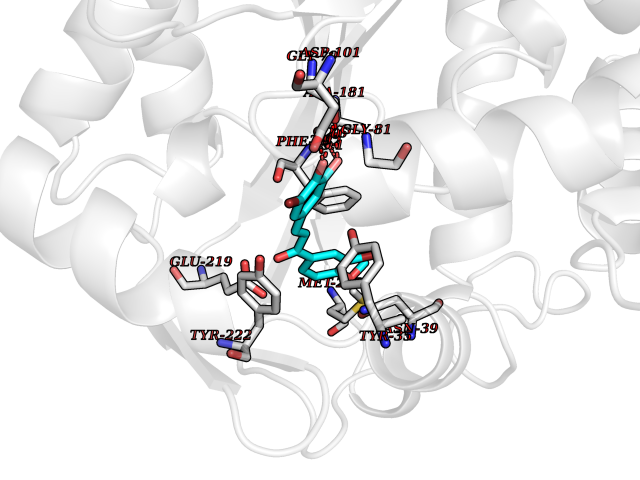 Pymol Map 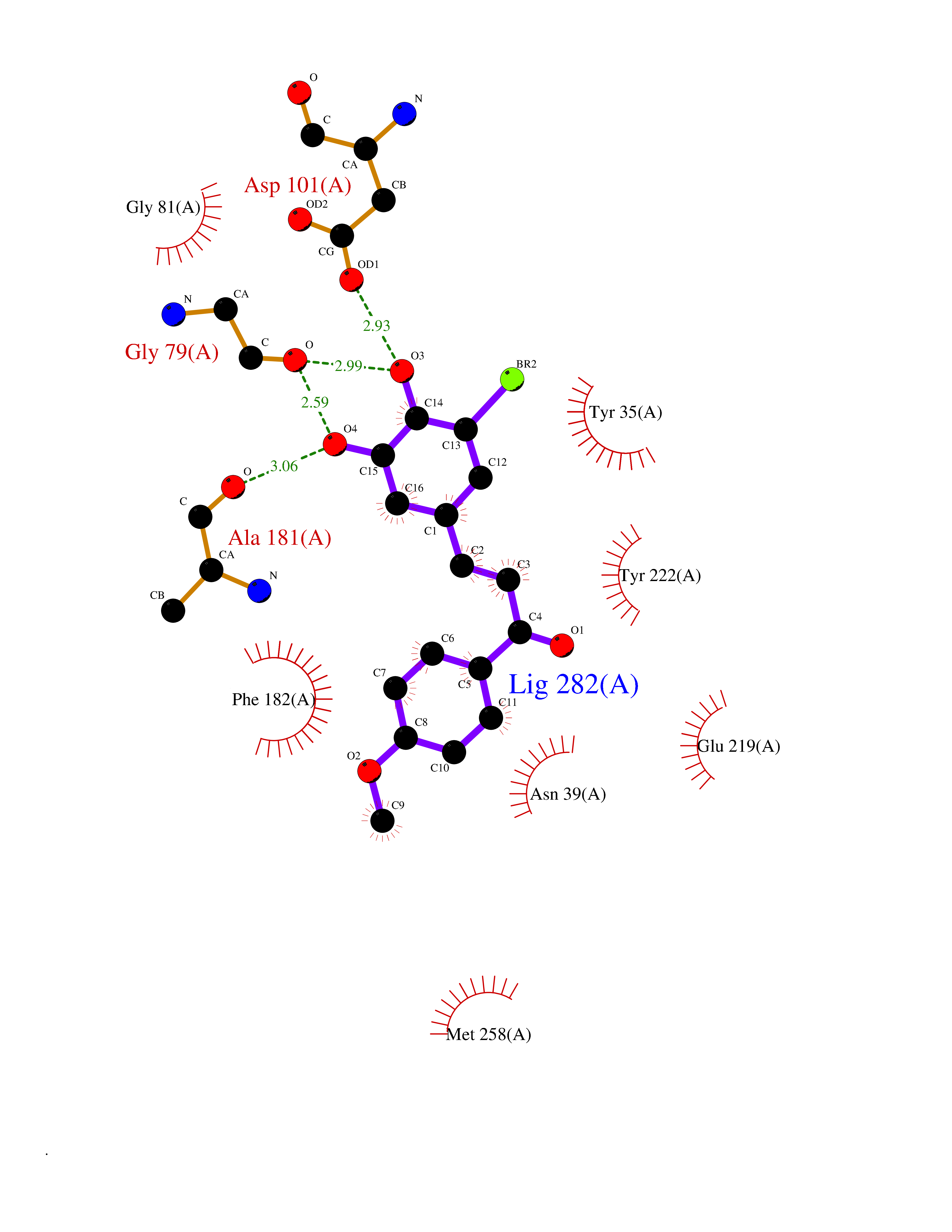 Ligplot Map 3D Binding mode Sequence APGQAAVASAYQRFEPRAYLRNNYAPPRGDLCNPNGVGPWKLRCLAQTFATGEVSGRTLIDIGSGPTVYQLLSACSHFEDITMTDFLEVNRQELGRWLQEEPGAFNWSMYSQHACLIEGKGECWQDKERQLRARVKRVLPIDVHQPQPLGAGSPAPLPADALVSAFCLEAVSPDLASFQRALDHITTLLRPGGHLLLIGALEESWYLAGEARLTVVPVSEEEVREALVRSGYKVRDLRTYIMPAHLQTGVDDVKGVFFAWAQKV Hydrogen bonds contact Hydrophobic contact | ||||
| 11 | Cyclopropane mycolic acid synthase MmaA2 | 1TPY | 7.21 | |
Target general information Gen name mmaA2 Organism Mycobacterium tuberculosis (strain ATCC 25618 / H37Rv) Uniprot ID TTD ID NA Synonyms Rv0644c;mma2 Protein family CFA/CMAS family Biochemical class Transferase Function Cyclopropane-fatty-acyl-phospholipid synthase activity.Methyltransferase activity. Related diseases Oocyte/zygote/embryo maturation arrest 16 (OZEMA16) [MIM:617234]: A rare cause of female primary infertility. In affected women, ovulation and fertilization proceed normally but embryos are arrested at early stages of development. Inheritance is autosomal recessive. {ECO:0000269|PubMed:27545678}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01718; DB01752 Interacts with NA EC number 2.1.1.79 Uniprot keywords 3D-structure; Acetylation; Lipid biosynthesis; Lipid metabolism; Methyltransferase; Reference proteome; S-adenosyl-L-methionine; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 32493.6 Length 285 Aromaticity 0.1 Instability index 43.61 Isoelectric point 5.53 Charge (pH=7) -10.17 2D Binding mode Binding energy (Kcal/mol) -9.84 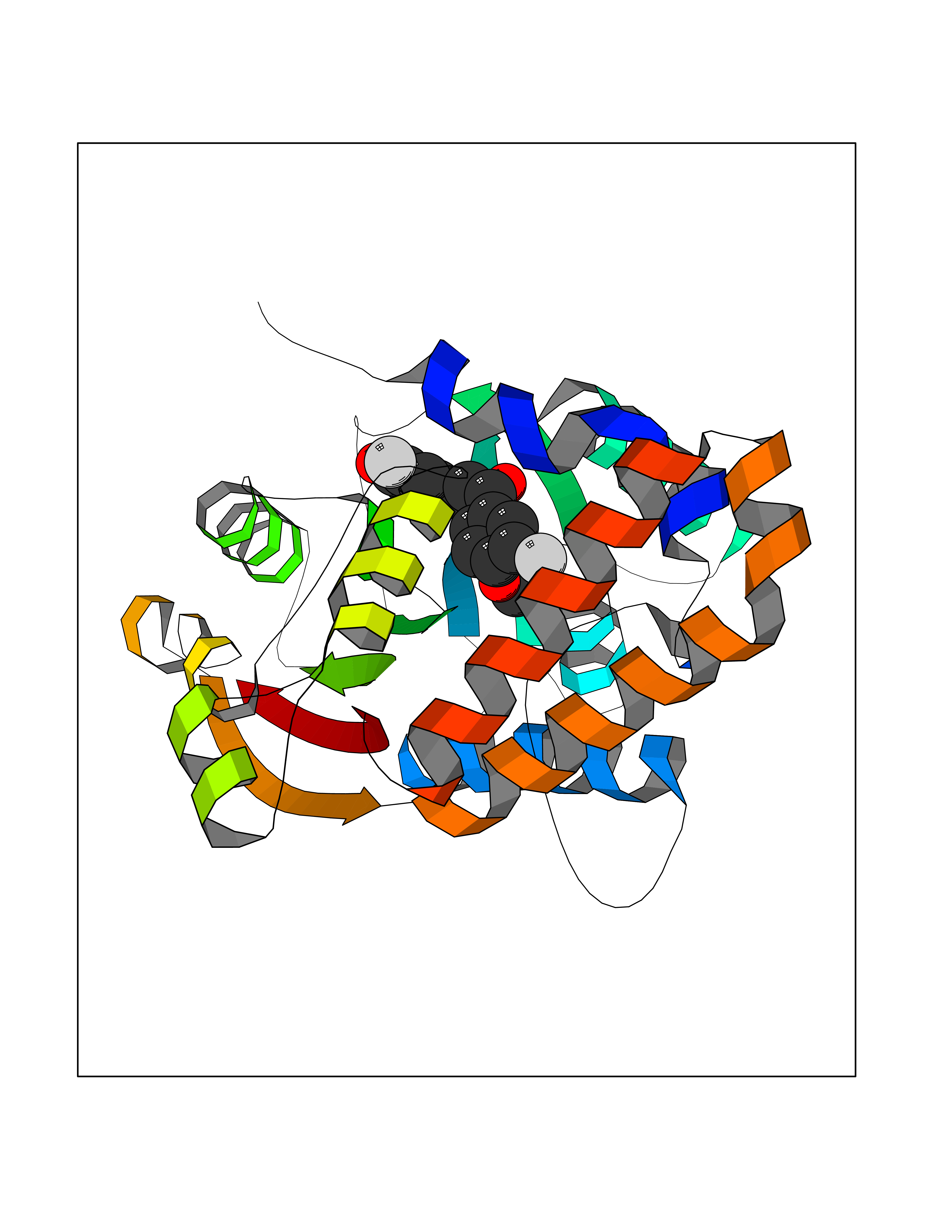 Molscript Map 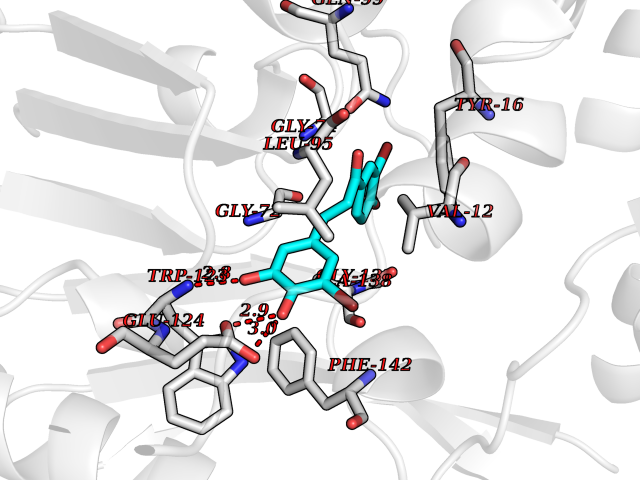 Pymol Map 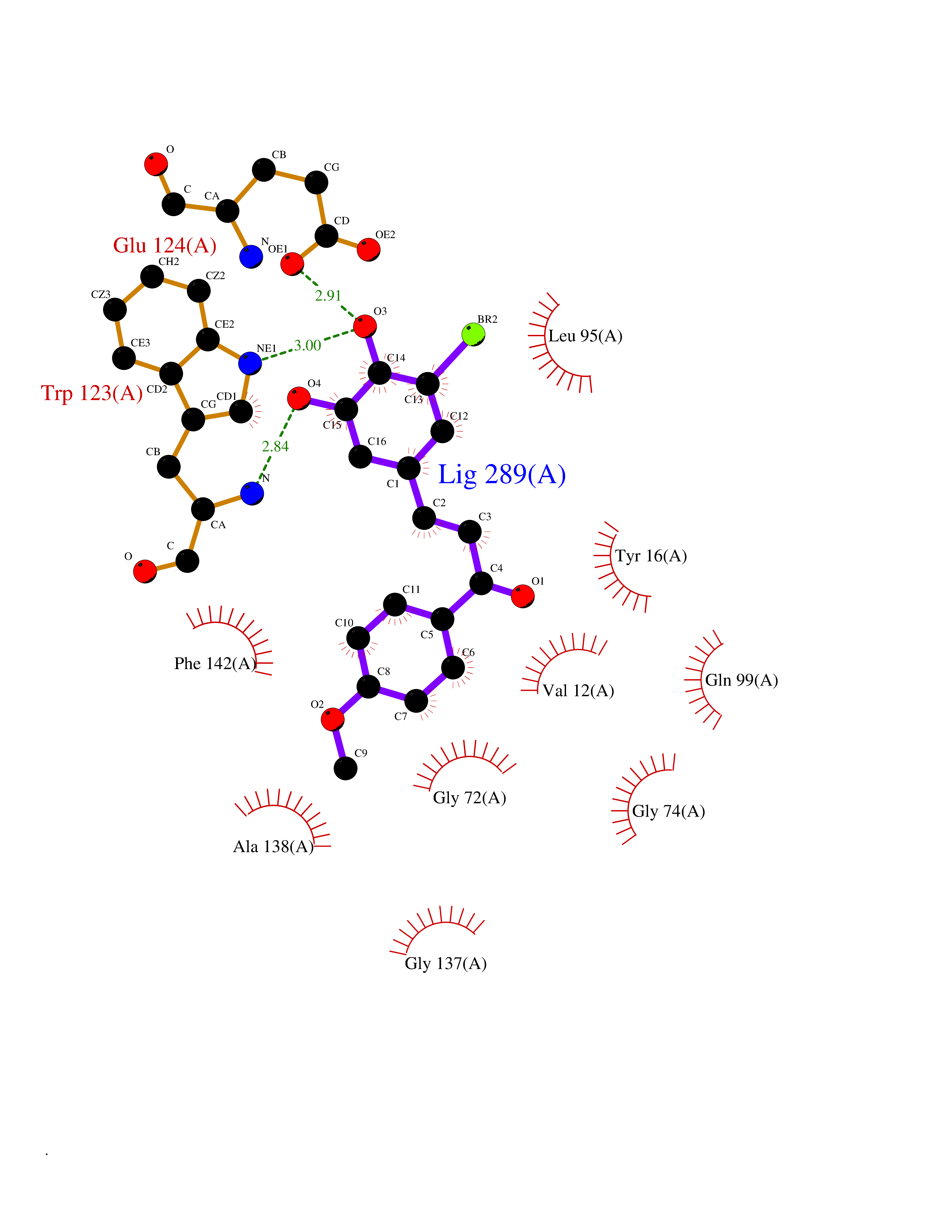 Ligplot Map 3D Binding mode Sequence NDLTPHFEDVQAHYDLSDDFFRLFLDPTQTYSCAHFEREDMTLEEAQIAKIDLALGKLGLQPGMTLLDIGCGWGATMRRAIAQYDVNVVGLTLSKNQAAHVQKSFDEMDTPRDRRVLLAGWEQFNEPVDRIVSIGAFEHFGHDRHADFFARAHKILPPDGVLLLHTITGLTRQQMVDHGLPLTLWLARFLKFIATEIFPGGQPPTIEMVEEQSAKTGFTLTRRQSLQPHYARTLDLWAEALQEHKSEAIAIQSEEVYERYMKYLTGCAKLFRVGYIDVNQFTLAK Hydrogen bonds contact Hydrophobic contact | ||||
| 12 | Thyroid hormone receptor alpha (THRA) | 3ILZ | 7.20 | |
Target general information Gen name THRA Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms V-erbA-related protein 7; THRA2; THRA1; Nuclear receptor subfamily 1 group A member 1; NR1A1; ERBA1; EAR7; EAR-7; C-erbA-alpha; C-erbA-1 Protein family Nuclear hormone receptor family, NR1 subfamily Biochemical class Nuclear hormone receptor Function High affinity receptor for thyroid hormones, including triiodothyronine and thyroxine. Isoform Alpha-1: Nuclear hormone receptor that can act as a repressor or activator of transcription. Related diseases Hypothyroidism, congenital, non-goitrous, 6 (CHNG6) [MIM:614450]: A disease characterized by growth retardation, developmental retardation, skeletal dysplasia, borderline low thyroxine levels and high triiodothyronine levels. There is differential sensitivity to thyroid hormone action, with retention of hormone responsiveness in the hypothalamic pituitary axis and liver but skeletal, gastrointestinal, and myocardial resistance. {ECO:0000269|PubMed:22168587, ECO:0000269|PubMed:24969835, ECO:0000269|PubMed:25670821, ECO:0000269|PubMed:26037512}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01118; DB00509; DB04855; DB05035; DB03176; DB00451; DB00279; DB01583; DB05235; DB09100 Interacts with Q9Y2J4; Q9Y2J4-4; O95971; Q8TAP6; Q96JM7; Q15648; Q6FHY5; P31321; Q96A49; O75410-7; Q9JLI4 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Congenital hypothyroidism; Cytoplasm; Disease variant; DNA-binding; Metal-binding; Nucleus; Proteomics identification; Receptor; Reference proteome; Transcription; Transcription regulation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A Molecular weight (Da) 29910.1 Length 267 Aromaticity 0.07 Instability index 52.75 Isoelectric point 5.31 Charge (pH=7) -11.32 2D Binding mode Binding energy (Kcal/mol) -9.83 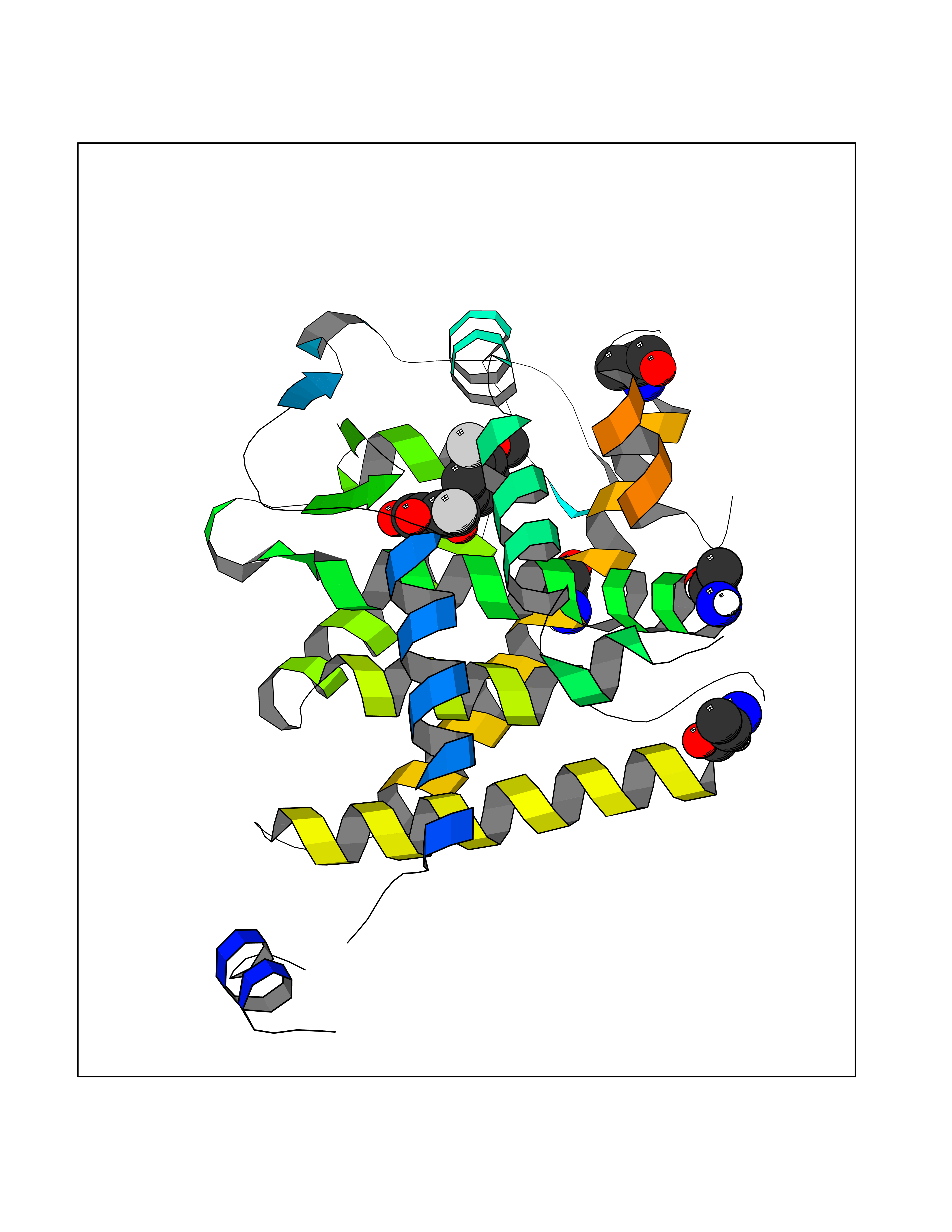 Molscript Map 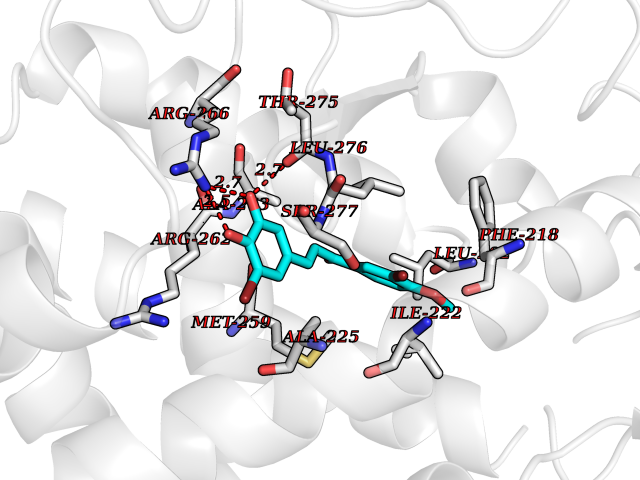 Pymol Map 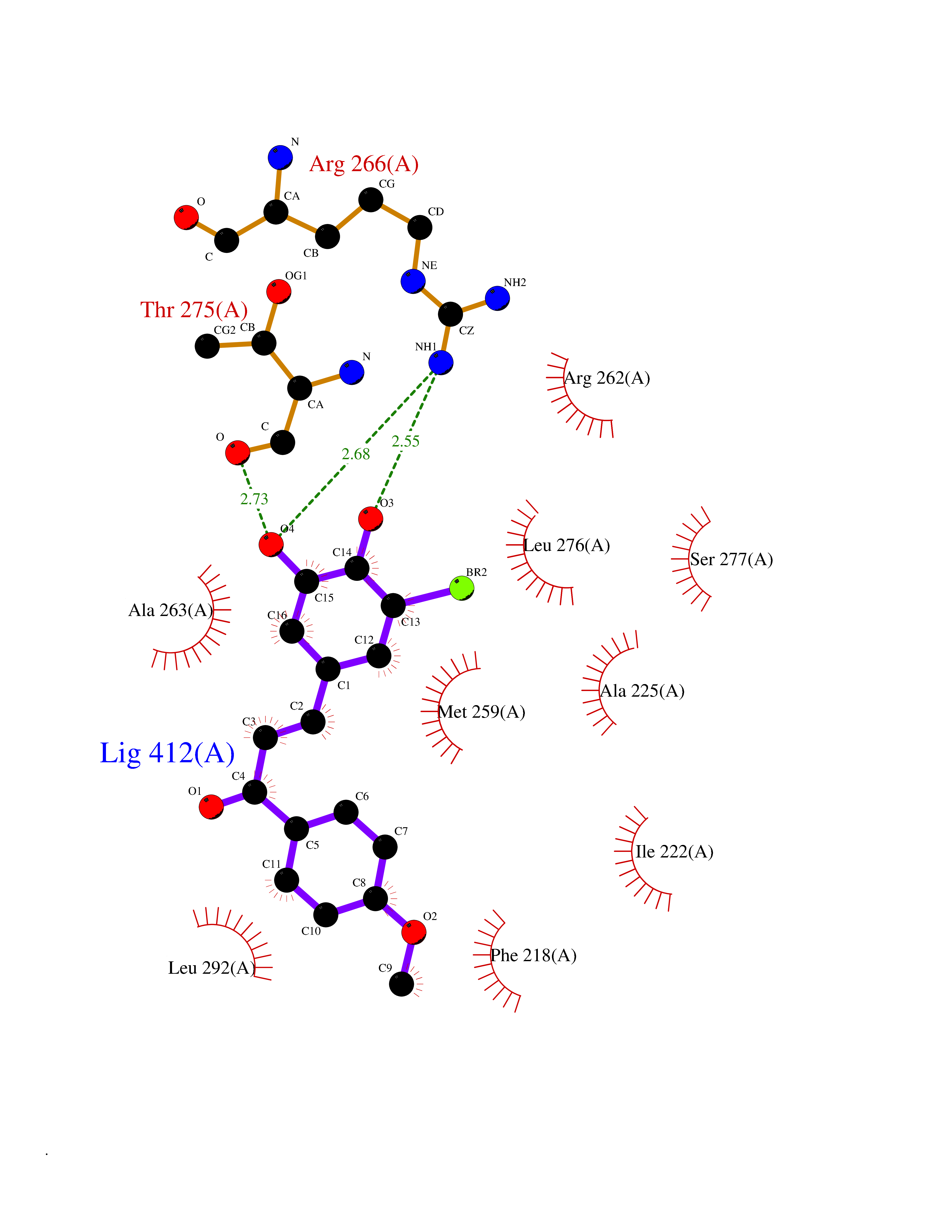 Ligplot Map 3D Binding mode Sequence GSHMEEMIRSLQQRPEPTPEEWDLIHIATEAHRSTNAQGSHWKQRRKFLPDDIGQSPIVSMPDGDKVDLEAFSEFTKIITPAITRVVDFAKKLPMFSELPXEDQIILLKGCCMEIMSLRAAVRYDPESDTLTLSGEMAVKREQLKNGGLGVVSDAIFELGKSLSAFNLDDTEVALLQAVLLMSTDRSGLLXVDKIEKSQEAYLLAFEHYVNHRKHNIPHFWPKLLMKVTDLRMIGAXHASRFLHMKVEXPTELFPPLFLEVFEDQEV Hydrogen bonds contact Hydrophobic contact | ||||
| 13 | Riboflavin kinase | 1NB0 | 7.20 | |
Target general information Gen name RFK Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family NA Biochemical class Transferase Function ATP binding.Metal ion binding.Riboflavin kinase activity. Related diseases Glutaric aciduria 1 (GA1) [MIM:231670]: An autosomal recessive metabolic disorder characterized by progressive dystonia and athetosis due to gliosis and neuronal loss in the basal ganglia. {ECO:0000269|PubMed:14707522, ECO:0000269|PubMed:18775954, ECO:0000269|PubMed:24973495, ECO:0000269|PubMed:8541831, ECO:0000269|PubMed:8900227, ECO:0000269|PubMed:8900228, ECO:0000269|PubMed:9600243, ECO:0000269|PubMed:9711871}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03247; DB00140 Interacts with Q9NXG0-2; P19438; P19438-1 EC number 2.7.1.26 Uniprot keywords 3D-structure; ATP-binding; Cytoplasm; Flavoprotein; FMN; Kinase; Magnesium; Metal-binding; Nucleotide-binding; Proteomics identification; Reference proteome; Transferase; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 16749.9 Length 147 Aromaticity 0.12 Instability index 41.55 Isoelectric point 7.09 Charge (pH=7) 0.12 2D Binding mode Binding energy (Kcal/mol) -9.82 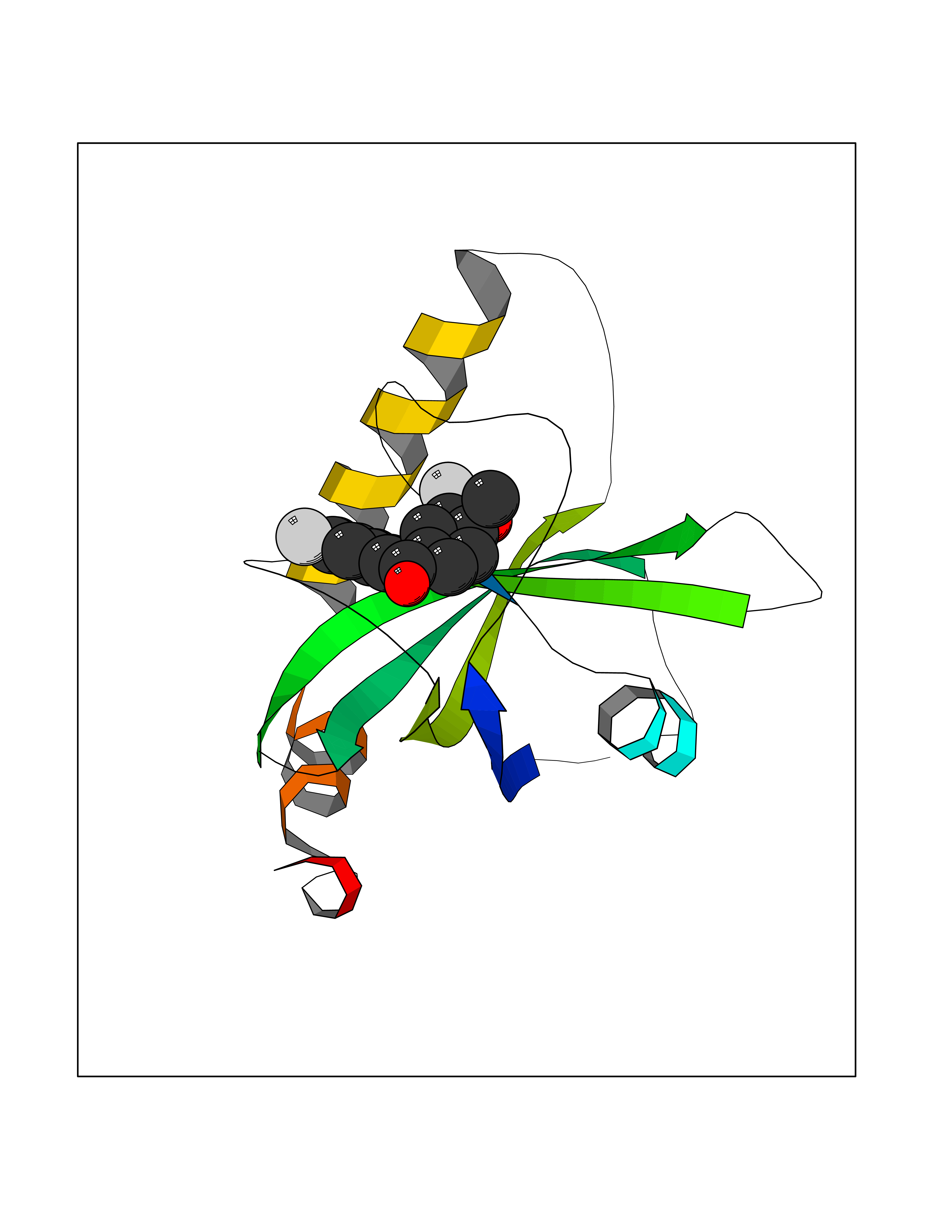 Molscript Map 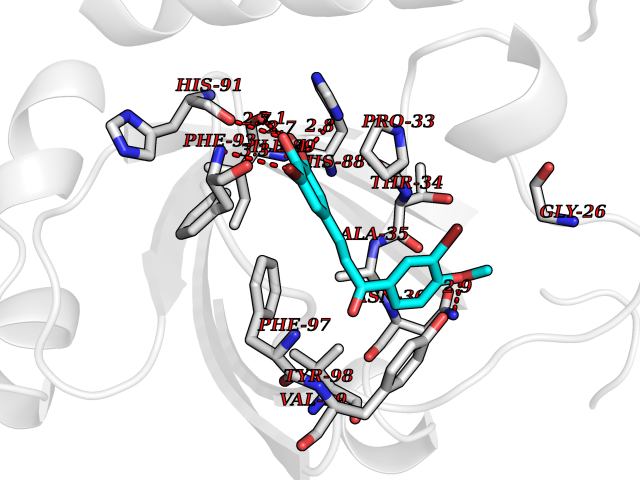 Pymol Map 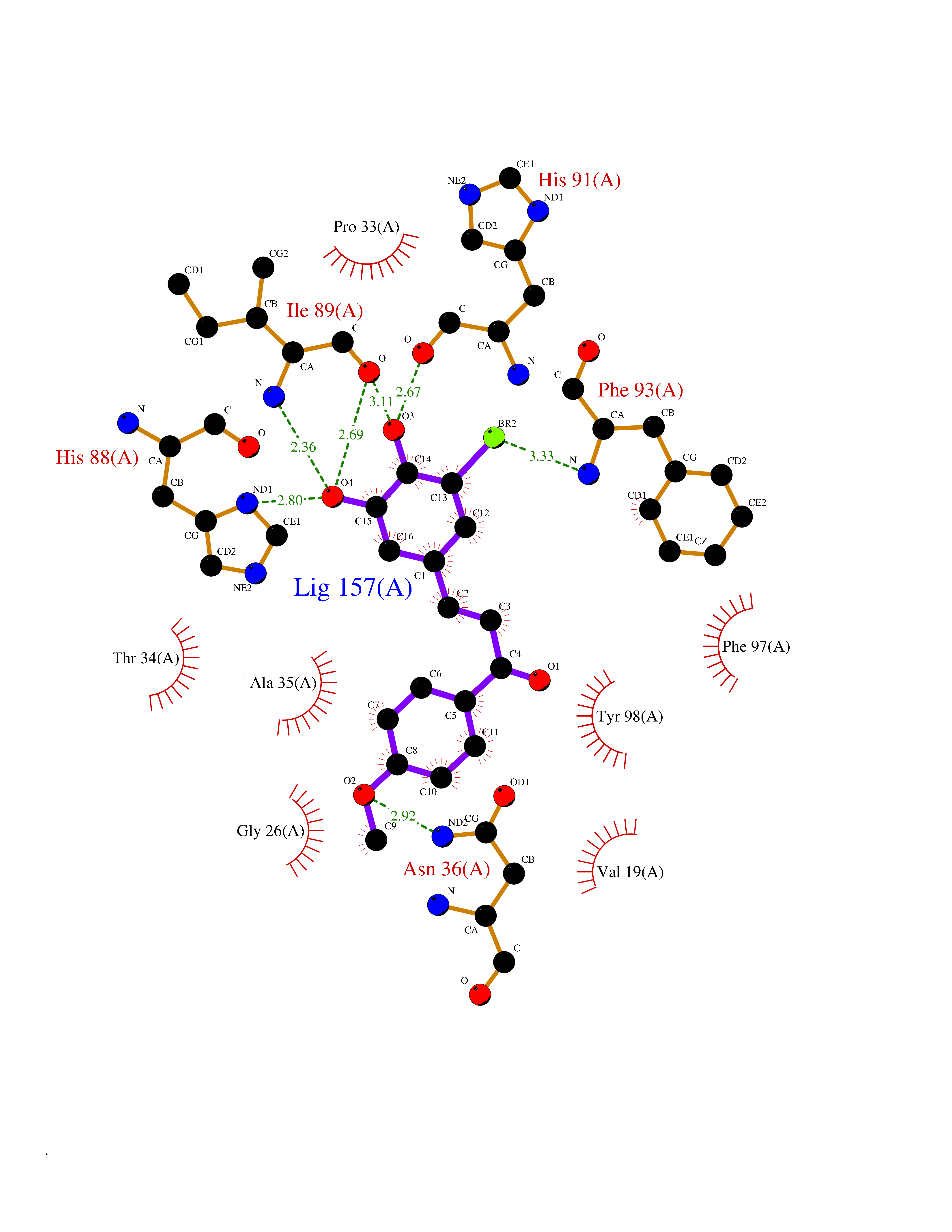 Ligplot Map 3D Binding mode Sequence RHLPYFCRGQVVRGFGRGSKQLGIPTANFPEQVVDNLPADISTGIYYGWASVGSGDVHKMVVSIGWNPYYKNTKKSMETHIMHTFKEDFYGEILNVAIVGYLRPEKNFDSLESLISAIQGDIEEAKKRLELPEYLKIKEDNFFQVSK Hydrogen bonds contact Hydrophobic contact | ||||
| 14 | Cholesterol 24-hydroxylase (CYP46A1) | 3MDR | 7.19 | |
Target general information Gen name CYP46A1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Cytochrome P450 46A1; CYP46; Cholesterol 24S-hydroxylase; Cholesterol 24-monooxygenase; CH24H Protein family Cytochrome P450 family Biochemical class Paired donor oxygen oxidoreductase Function Primarily catalyzes the hydroxylation (with S stereochemistry) at C-24 of cholesterol side chain, triggering cholesterol diffusion out of neurons and its further degradation. By promoting constant cholesterol elimination in neurons, may activate the mevalonate pathway and coordinate the synthesis of new cholesterol and nonsterol isoprenoids involved in synaptic activity and learning. Further hydroxylates cholesterol derivatives and hormone steroids on both the ring and side chain of these molecules, converting them into active oxysterols involved in lipid signaling and biosynthesis. Acts as an epoxidase converting cholesta-5,24-dien-3beta-ol/desmosterol into (24S),25-epoxycholesterol, an abundant lipid ligand of nuclear NR1H2 and NR1H3 receptors shown to promote neurogenesis in developing brain. May also catalyze the oxidative metabolism of xenobiotics, such as clotrimazole. P450 monooxygenase that plays a major role in cholesterol homeostasis in the brain. Related diseases Spinocerebellar ataxia, autosomal recessive, with axonal neuropathy 1 (SCAN1) [MIM:607250]: A form of spinocerebellar ataxia, a clinically and genetically heterogeneous group of cerebellar disorders. Patients show progressive incoordination of gait and often poor coordination of hands, speech and eye movements, due to degeneration of the cerebellum with variable involvement of the brainstem and spinal cord. SCAN1 is an autosomal recessive cerebellar ataxia (ARCA) associated with peripheral axonal motor and sensory neuropathy, distal muscular atrophy, pes cavus and steppage gait as seen in Charcot-Marie-Tooth neuropathy. All affected individuals have normal intelligence. {ECO:0000269|PubMed:12244316, ECO:0000269|PubMed:15647511, ECO:0000269|PubMed:15920477, ECO:0000269|PubMed:16141202, ECO:0000269|PubMed:17948061}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number EC 1.14.14.25 Uniprot keywords 3D-structure; Alternative splicing; Cell projection; Cholesterol metabolism; Endoplasmic reticulum; Heme; Iron; Lipid metabolism; Membrane; Metal-binding; Microsome; Monooxygenase; Oxidoreductase; Proteomics identification; Reference proteome; Steroid metabolism; Sterol metabolism; Synapse; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 48977 Length 427 Aromaticity 0.1 Instability index 49.59 Isoelectric point 9.04 Charge (pH=7) 6.25 2D Binding mode Binding energy (Kcal/mol) -9.81 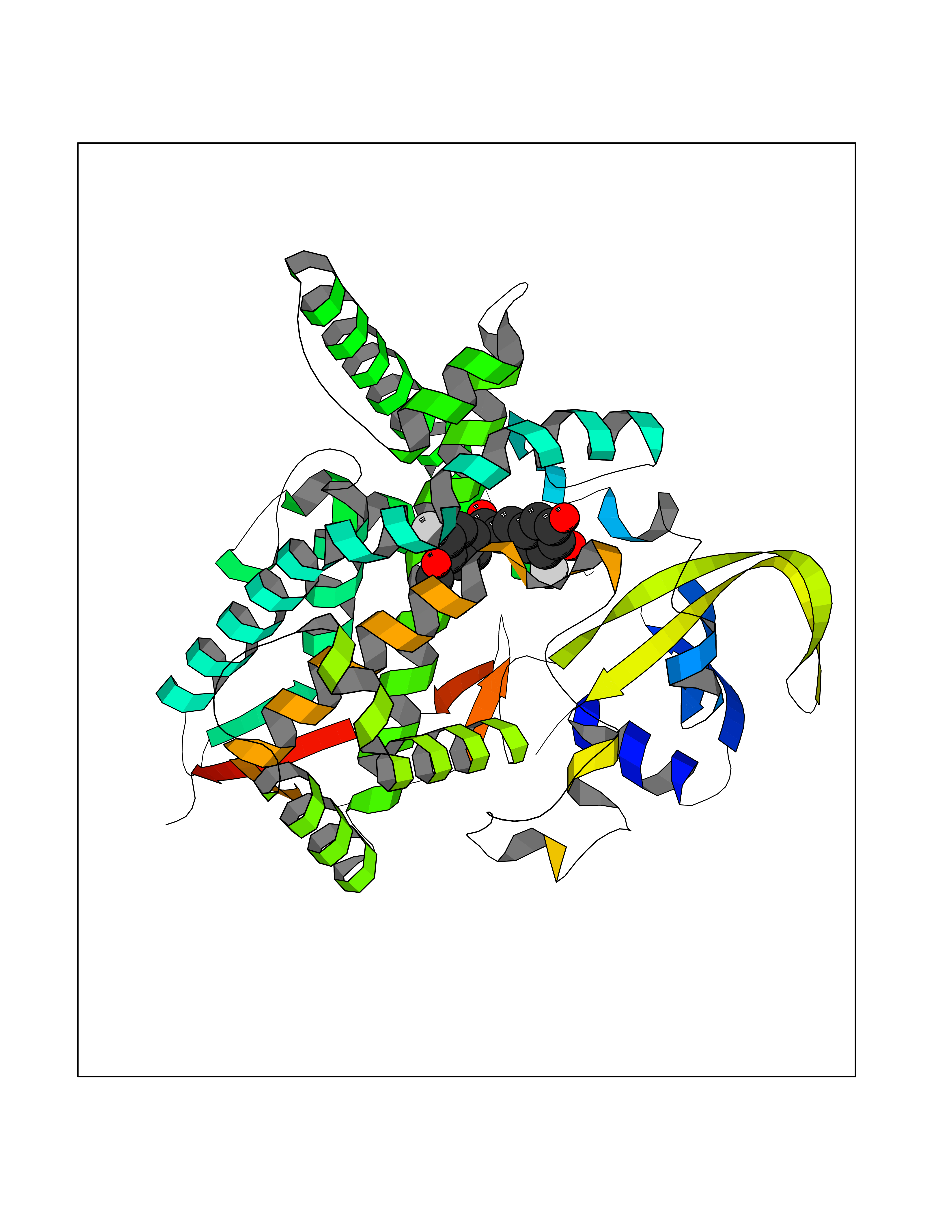 Molscript Map 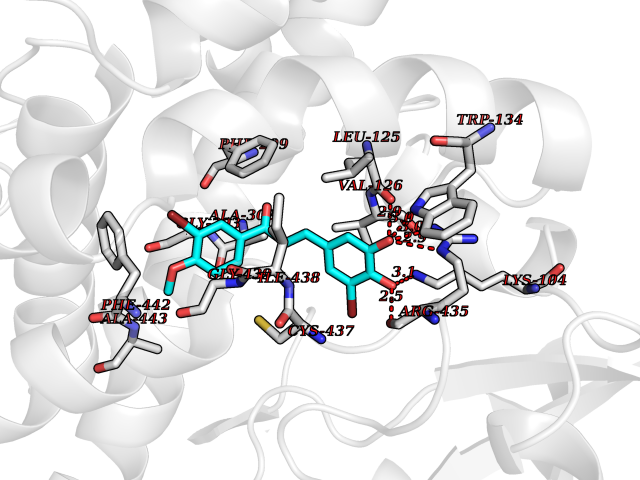 Pymol Map 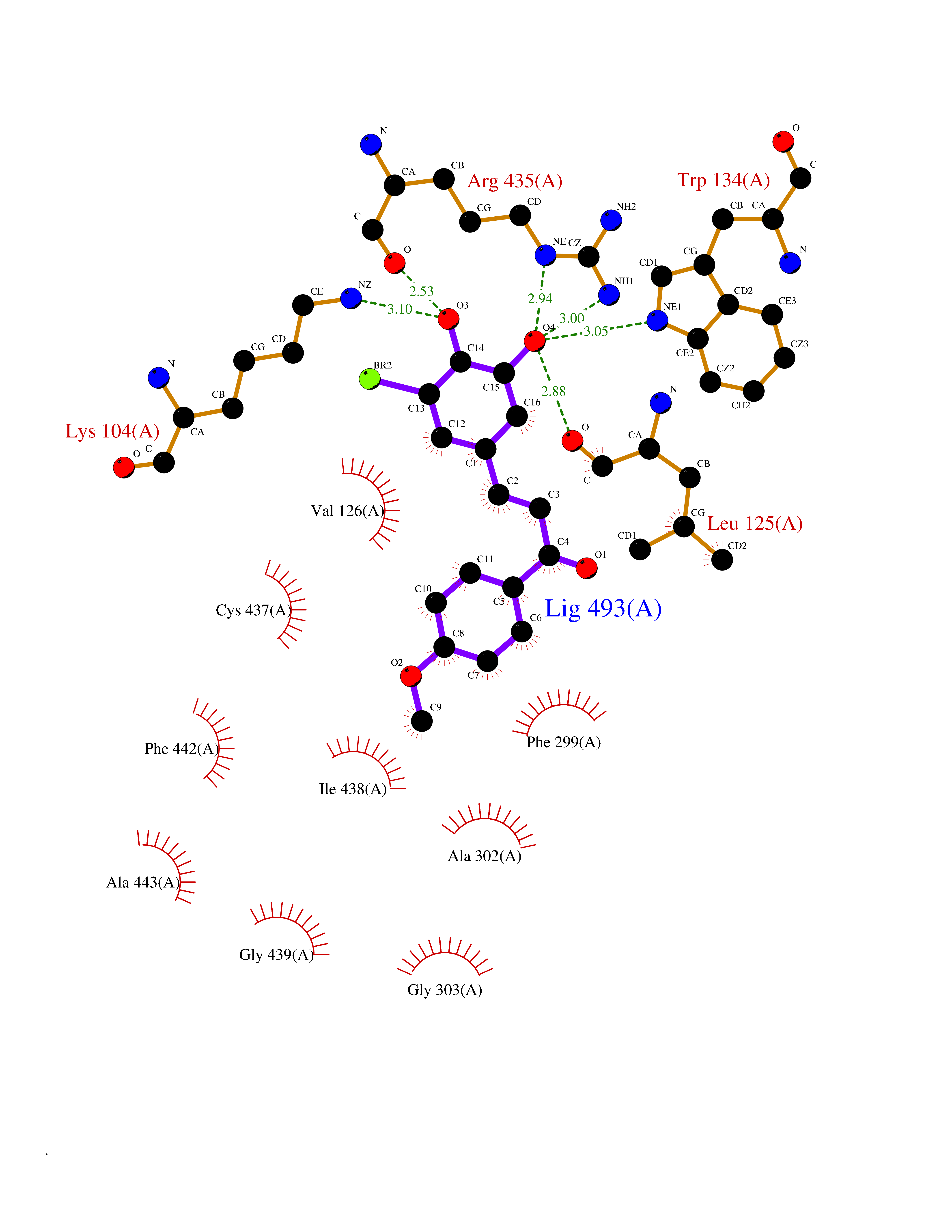 Ligplot Map 3D Binding mode Sequence RVLQDVFLDWAKKYGPVVRVNVFHKTSVIVTSPESVKKFLMSTKYNKDSKMYRALQTVFGERLFGQGLVSECNYERWHKQRRVIDLAFSRSSLVSLMETFNEKAEQLVEILEAKADGQTPVSMQDMLTYTAMDILAKAAFGMETSMLLGAQKPLSQAVKLMLEGITASRNTKRKQLREVRESIRFLRQVGRDWVQRRREALKRGEEVPADILTQILKAEEGAQDDEGLLDNFVTFFIAGHETSANHLAFTVMELSRQPEIVARLQAEVDEVIGSKRYLDFEDLGRLQYLSQVLKESLRLYPPAWGTFRLLEEETLIDGVRVPGNTPLLFSTYVMGRMDTYFEDPLTFNPDRFGPGAPKPRFTYFPFSLGHRSCIGQQFAQMEVKVVMAKLLQRLEFRLVPGQRFGLQEQATLKPLDPVLCTLRPRGW Hydrogen bonds contact Hydrophobic contact | ||||
| 15 | Receptor-interacting protein 1 (RIPK1) | 5TX5 | 7.19 | |
Target general information Gen name RIPK1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Receptor-interacting serine/threonine-protein kinase 1; RIP1; RIP-1; RIP; Cell death protein RIP Protein family Protein kinase superfamily, TKL Ser/Thr protein kinase family Biochemical class Kinase Function Upon activation of TNFR1 by the TNF-alpha family cytokines, TRADD and TRAF2 are recruited to the receptor. Phosphorylates DAB2IP at 'Ser-728' in a TNF-alpha-dependent manner, and thereby activates the MAP3K5-JNK apoptotic cascade. Ubiquitination by TRAF2 via 'Lys-63'-link chains acts as a critical enhancer of communication with downstream signal transducers in the mitogen-activated protein kinase pathway and the NF-kappa-B pathway, which in turn mediate downstream events including the activation of genes encoding inflammatory molecules. Polyubiquitinated protein binds to IKBKG/NEMO, the regulatory subunit of the IKK complex, a critical event for NF-kappa-B activation. Interaction with other cellular RHIM-containing adapters initiates gene activation and cell death. RIPK1 and RIPK3 association, in particular, forms a necrosis-inducing complex. Serine-threonine kinase which transduces inflammatory and cell-death signals (programmed necrosis) following death receptors ligation, activation of pathogen recognition receptors (PRRs), and DNA damage. Related diseases Immunodeficiency 57 with autoinflammation (IMD57) [MIM:618108]: An autosomal recessive primary immunodeficiency characterized by lymphopenia and recurrent viral, bacterial, and fungal infections. Patients exhibit early-onset inflammatory bowel disease involving the upper and lower gastrointestinal tract, and develop progressive polyarthritis. {ECO:0000269|PubMed:30026316}. The disease is caused by variants affecting the gene represented in this entry. RIPK1-deficient immune cells from IMD57 patients have impaired proinflammatory signaling leading to dysregulated cytokine secretion and are prone to necroptosis. {ECO:0000269|PubMed:30026316}.; DISEASE: Autoinflammation with episodic fever and lymphadenopathy (AIEFL) [MIM:618852]: An autosomal dominant immunologic disorder characterized by early onset of recurrent episodes of unexplained fever, lymphadenopathy, hepatosplenomegaly, and increased levels of inflammatory cytokines and chemokines in patient serum. {ECO:0000269|PubMed:31827280, ECO:0000269|PubMed:31827281}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB12010 Interacts with P04083; Q13490; Q13489; Q92851; Q14790; Q8IVM0; P48729; Q13158; Q9Y6K9; Q96AB6; Q9ULZ3; Q13546; Q9Y572; P19438; Q13077; Q12933; Q13114; Q13107; B7UI21; PRO_0000449629 [P0DTD1]; U5TQE9 EC number EC 2.7.11.1 Uniprot keywords 3D-structure; Alternative splicing; Apoptosis; ATP-binding; Cell membrane; Cytoplasm; Disease variant; Glycoprotein; Host-virus interaction; Inflammatory response; Isopeptide bond; Kinase; Membrane; Necrosis; Nucleotide-binding; Phosphoprotein; Proteomics identification; Reference proteome; Serine/threonine-protein kinase; Transferase; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 29554.2 Length 259 Aromaticity 0.08 Instability index 48.26 Isoelectric point 6.29 Charge (pH=7) -2.52 2D Binding mode Binding energy (Kcal/mol) -9.81 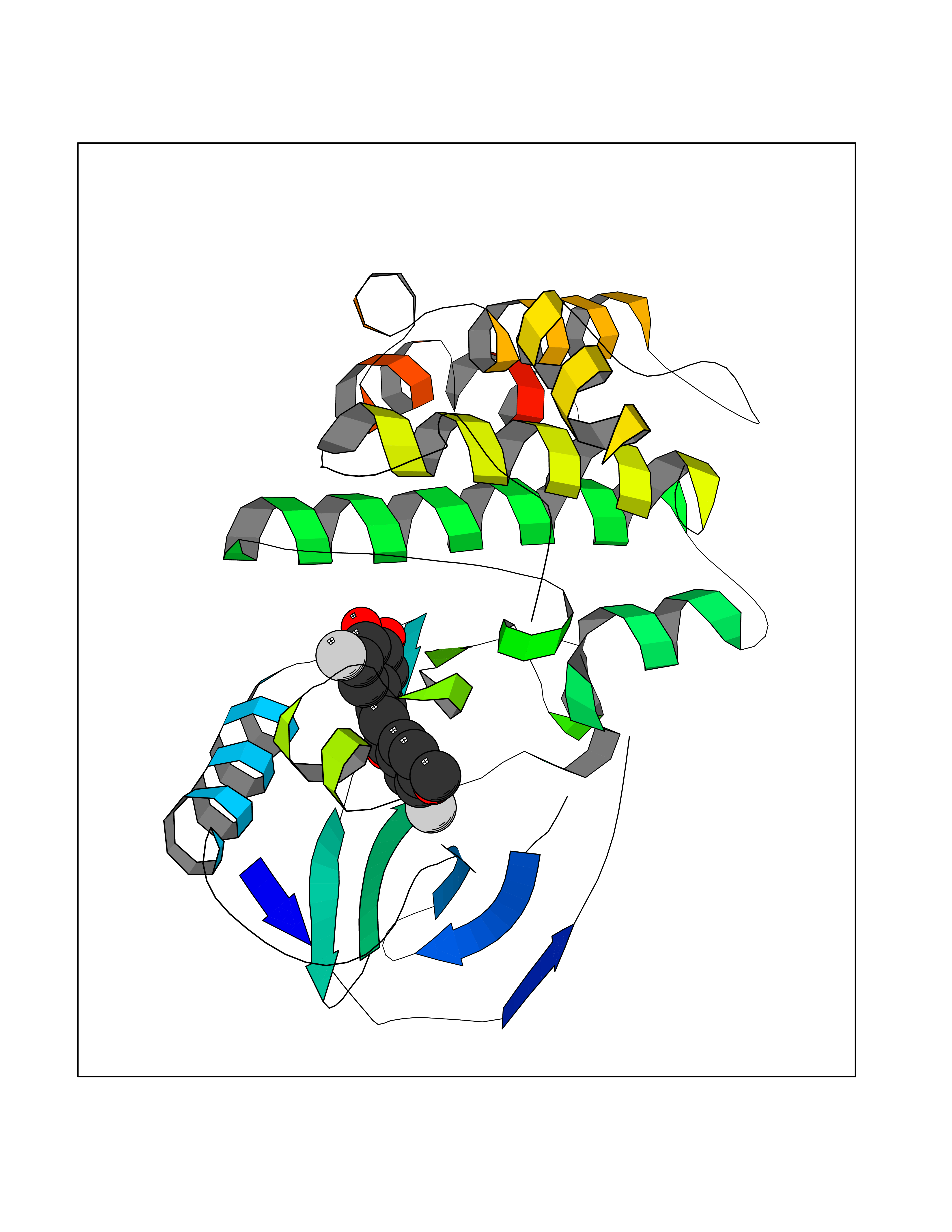 Molscript Map 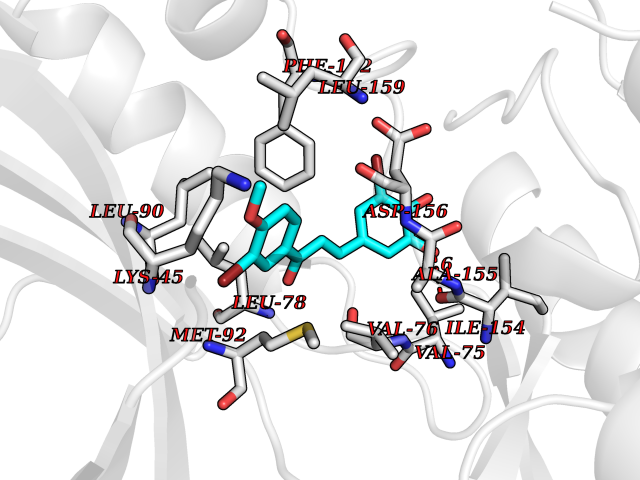 Pymol Map 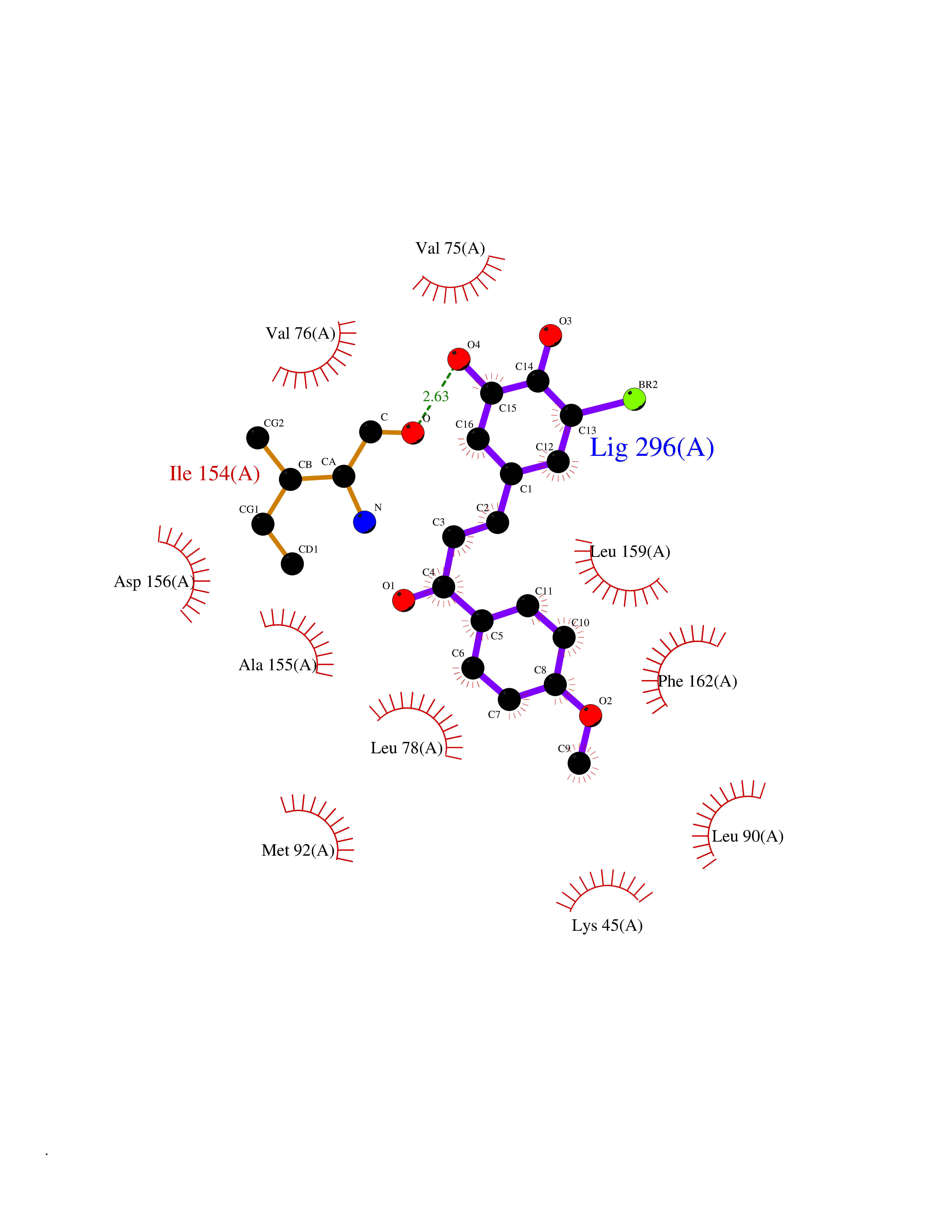 Ligplot Map 3D Binding mode Sequence IKMKSSDFLESAELDSGGKVSLAFHRTQGLMIMKTVYKGPNCIEHNEALLEEAKMMNRLRHSRVVKLLGVIIEEGKYSLVMEYMEKGNLMHVLKAEMSTPLSVKGRIILEIIEGMAYLHGKGVIHKDLKPENILVDNDFHIKIADLGLASFKMWSKLNGTLYYMAPEHLNDVNAKPTEKSDVYSFAVVLWAIFANKEPYQQLIMAIKSGNRPDVDDITEYCPREIISLMKLCWEANPEARPTFPGIEEKFRPFYLSQLE Hydrogen bonds contact Hydrophobic contact | ||||
| 16 | "Periplasmic trehalase (EC 3.2.1.28) (Alpha,alpha-trehalase) (Alpha,alpha-trehalose glucohydrolase) (Tre37A)" | 2JG0 | 7.17 | |
Target general information Gen name treA Organism Escherichia coli (strain K12) Uniprot ID TTD ID NA Synonyms JW1186;osmA;b1197 Protein family Glycosyl hydrolase 37 family Biochemical class NA Function Provides the cells with the ability to utilize trehalose at high osmolarity by splitting it into glucose molecules that can subsequently be taken up by the phosphotransferase-mediated uptake system. Related diseases SRC kinase activity has been shown to be increased in several tumor tissues and tumor cell lines such as colon carcinoma cells. {ECO:0000269|PubMed:2498394, ECO:0000269|PubMed:3093483}.; DISEASE: Thrombocytopenia 6 (THC6) [MIM:616937]: A form of thrombocytopenia, a hematologic disorder defined by a decrease in the number of platelets in circulating blood, resulting in the potential for increased bleeding and decreased ability for clotting. THC6 is an autosomal dominant form. Affected individuals may also have bone abnormalities and an increased risk for myelofibrosis. {ECO:0000269|PubMed:26936507}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number 3.2.1.28 Uniprot keywords 3D-structure; Direct protein sequencing; Glycosidase; Hydrolase; Periplasm; Reference proteome; Signal Protein physicochemical properties Chain ID A Molecular weight (Da) 57508.9 Length 507 Aromaticity 0.11 Instability index 48.32 Isoelectric point 5.48 Charge (pH=7) -10.13 2D Binding mode Binding energy (Kcal/mol) -9.78 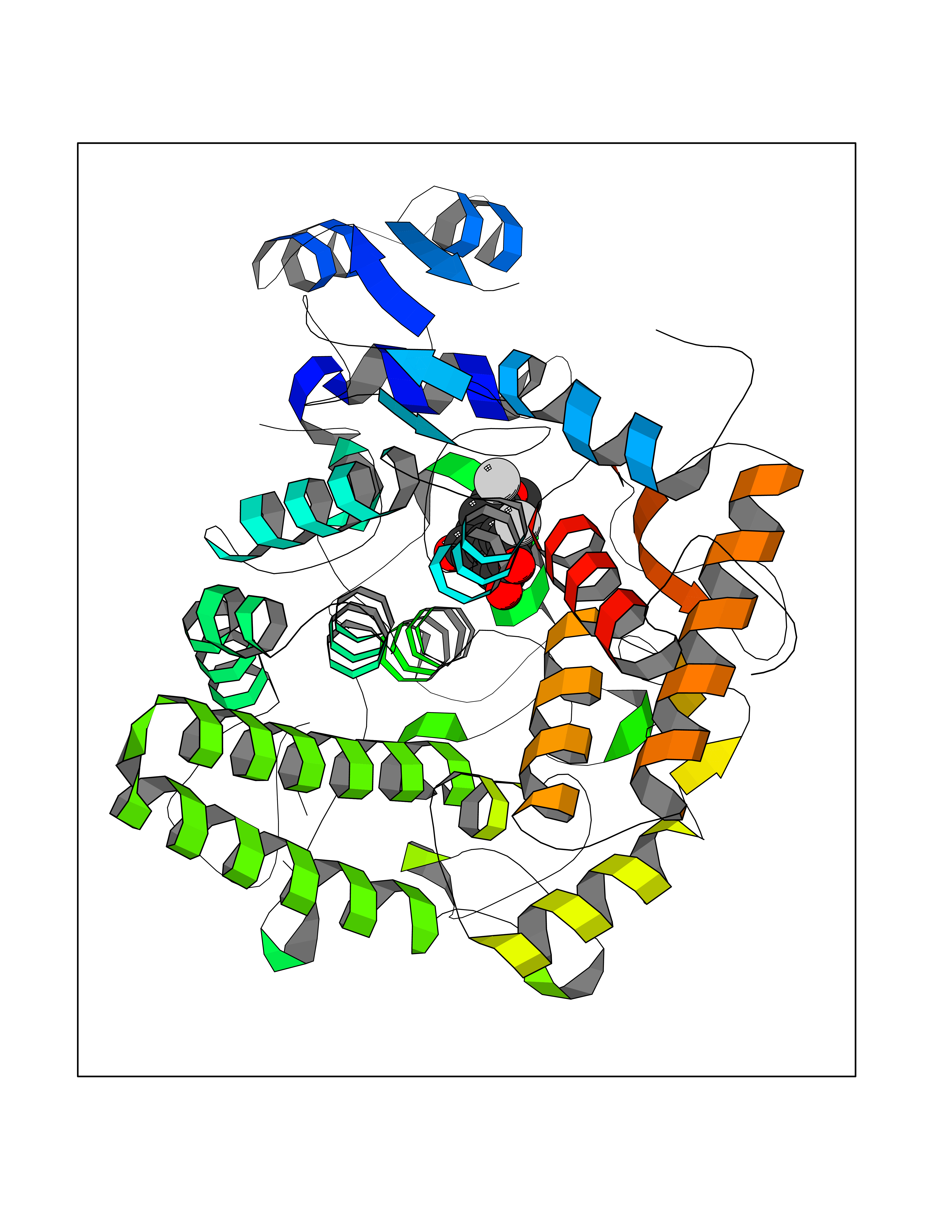 Molscript Map 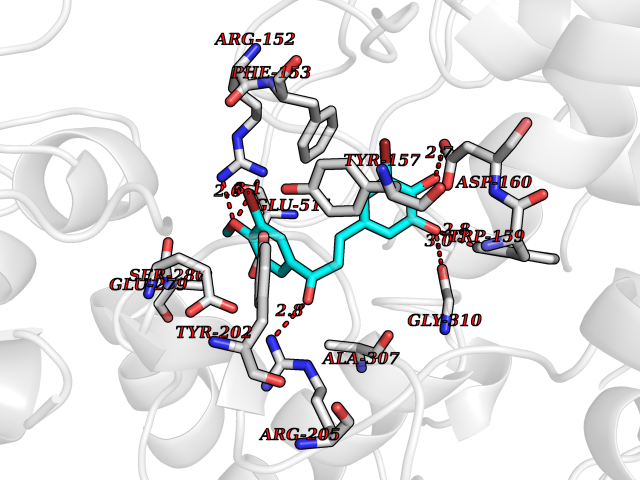 Pymol Map 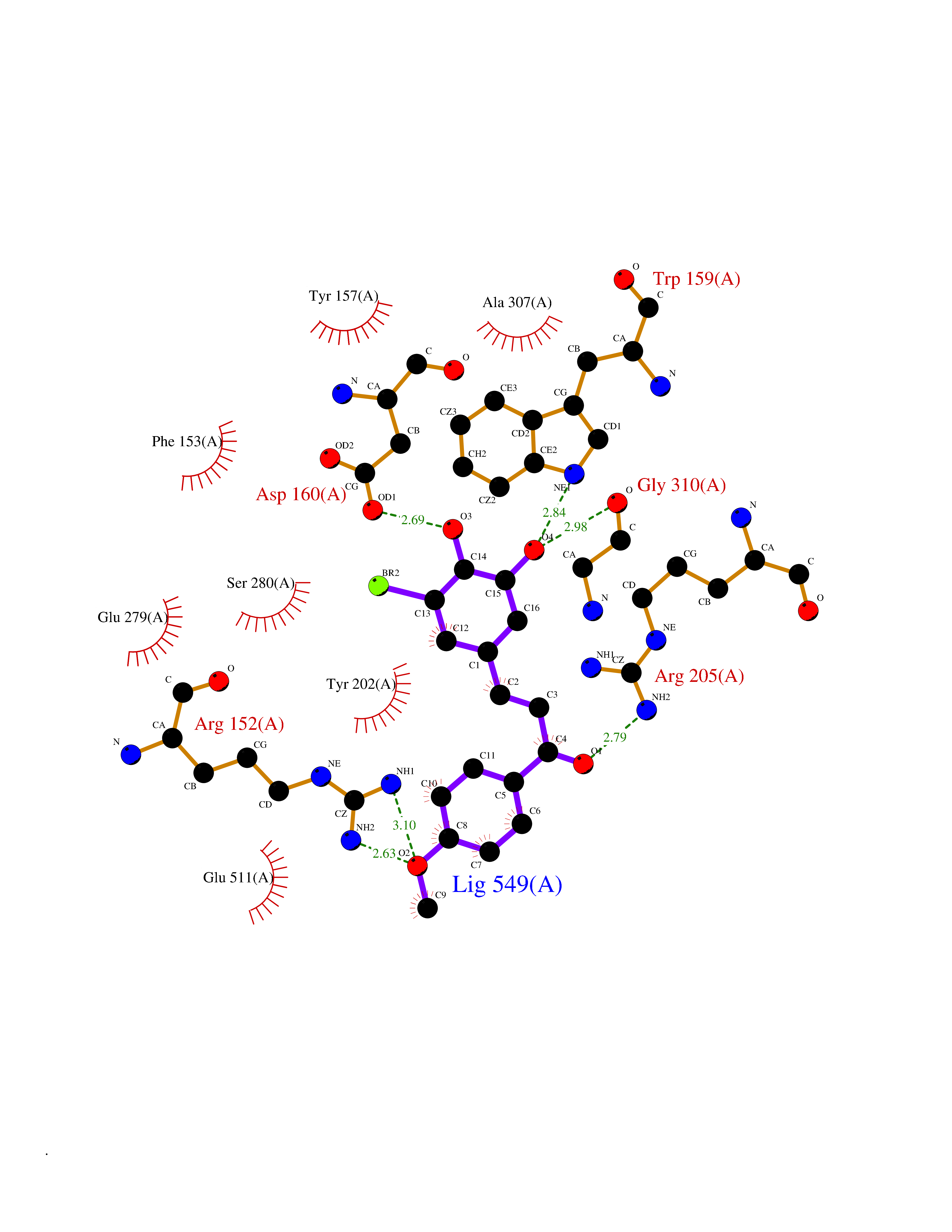 Ligplot Map 3D Binding mode Sequence PQPPDILLGPLFNDVQNAKLFPDQKTFADAVPNSDPLMILADYRMQQNQSGFDLRHFVNVNFTLPKYVPPEGQSLREHIDGLWPVLTRSTENTEKWDSLLPLPEPYVVPGGRFREVYYWDSYFTMLGLAESGHWDKVADMVANFAHEIDTYGHIPNGNRSYYLSRSQPPFFALMVELLAQHEGDAALKQYLPQMQKEYAYWMDGVENLQAGQQEKRVVKLQDGTLLNRYWDDRDTPRPESWVEDIATAKSNPNRPATEIYRDLRSAAASGWDFSSRWMDNPQQLNTLRTTSIVPVDLNSLMFKMEKILARASKAAGDNAMANQYETLANARQKGIEKYLWNDQQGWYADYDLKSHKVRNQLTAAALFPLYVNAAAKDRANKMATATKTHLLQPGGLNTTSVKSGQQWDAPNGWAPLQWVATEGLQNYGQKEVAMDISWHFLTNVQHTYDREKKLVEKYDVSTTGTGGGGGEYPLQDGFGWTNGVTLKMLDLICPKEQPCDNVPATRP Hydrogen bonds contact Hydrophobic contact | ||||
| 17 | SEC14-like protein 2 | 4OMJ | 7.17 | |
Target general information Gen name SEC14L2 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms KIAA1658;C22orf6;KIAA1186 Protein family NA Biochemical class Transport protein Function Phospholipid binding.Transporter activity.Vitamin E binding. Related diseases Neurodevelopmental disorder with poor language and loss of hand skills (NDPLHS) [MIM:617903]: An autosomal dominant disorder characterized by psychomotor developmental stagnation or regression. NDPLHS manifest in the first years of life as loss of purposeful hand movements, loss of language, and intellectual disability. {ECO:0000269|PubMed:26740508, ECO:0000269|PubMed:28856709, ECO:0000269|PubMed:29369404}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Developmental and epileptic encephalopathy 59 (DEE59) [MIM:617904]: A form of epileptic encephalopathy, a heterogeneous group of severe early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE59 is an autosomal dominant condition characterized by onset of refractory seizures in early infancy. {ECO:0000269|PubMed:28856709, ECO:0000269|PubMed:29100083, ECO:0000269|PubMed:29369404}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB14003; DB14001; DB14002; DB11635; DB11251; DB00163 Interacts with NA EC number NA Uniprot keywords 3D-structure; Activator; Alternative splicing; Cytoplasm; Lipid-binding; Nucleus; Proteomics identification; Reference proteome; Transcription; Transcription regulation; Transport Protein physicochemical properties Chain ID A,B Molecular weight (Da) 31533.3 Length 274 Aromaticity 0.1 Instability index 49.26 Isoelectric point 8.34 Charge (pH=7) 2.81 2D Binding mode Binding energy (Kcal/mol) -9.78 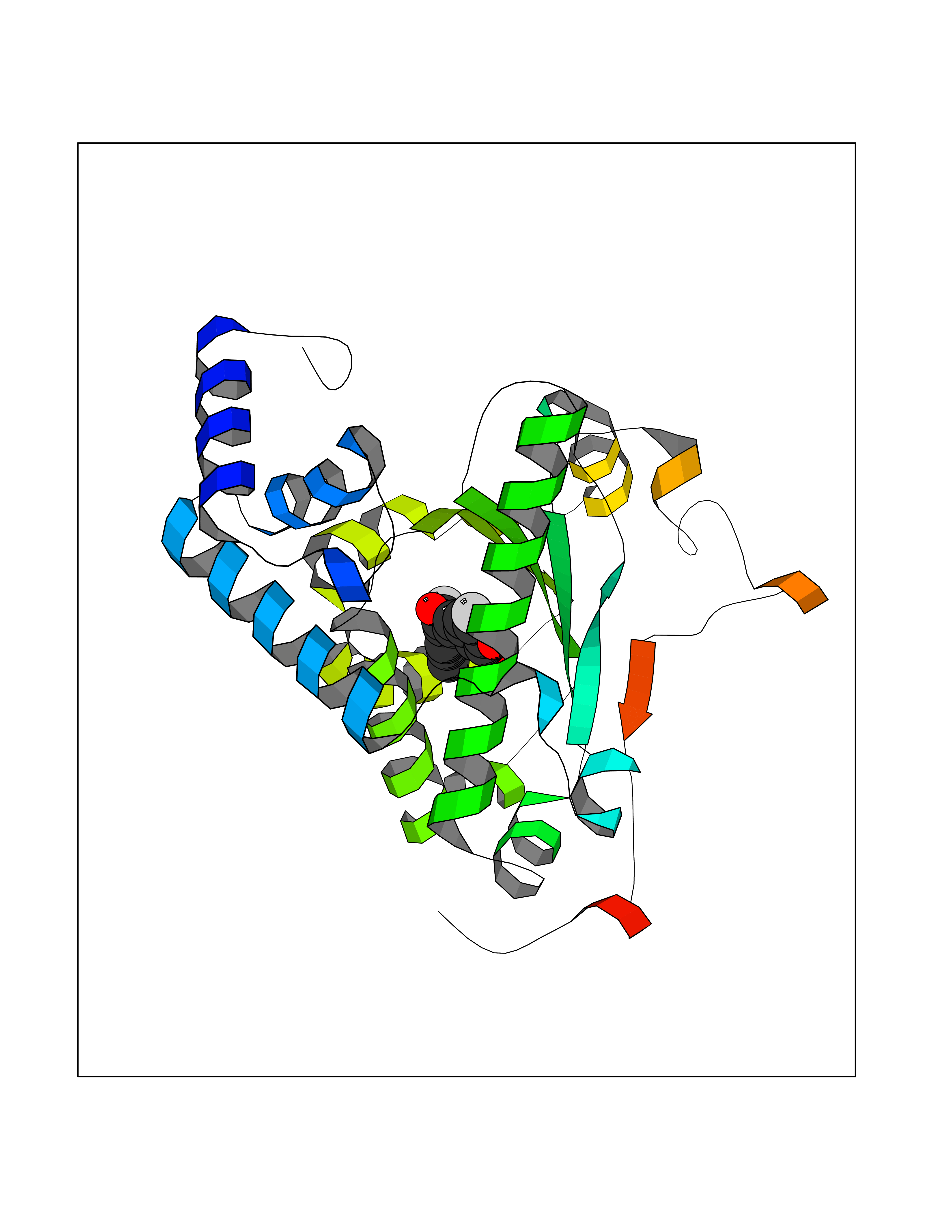 Molscript Map 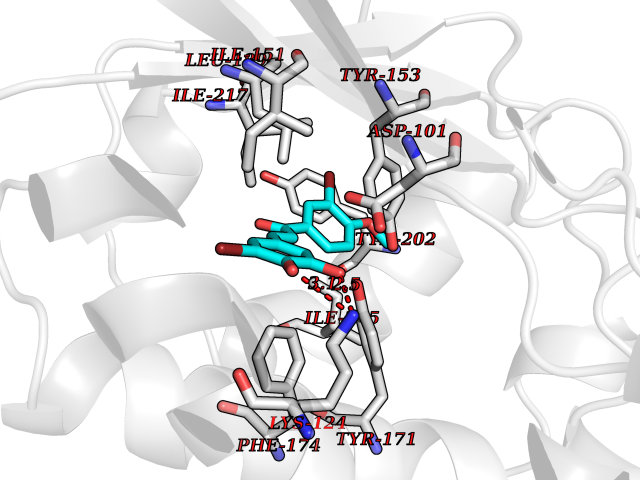 Pymol Map 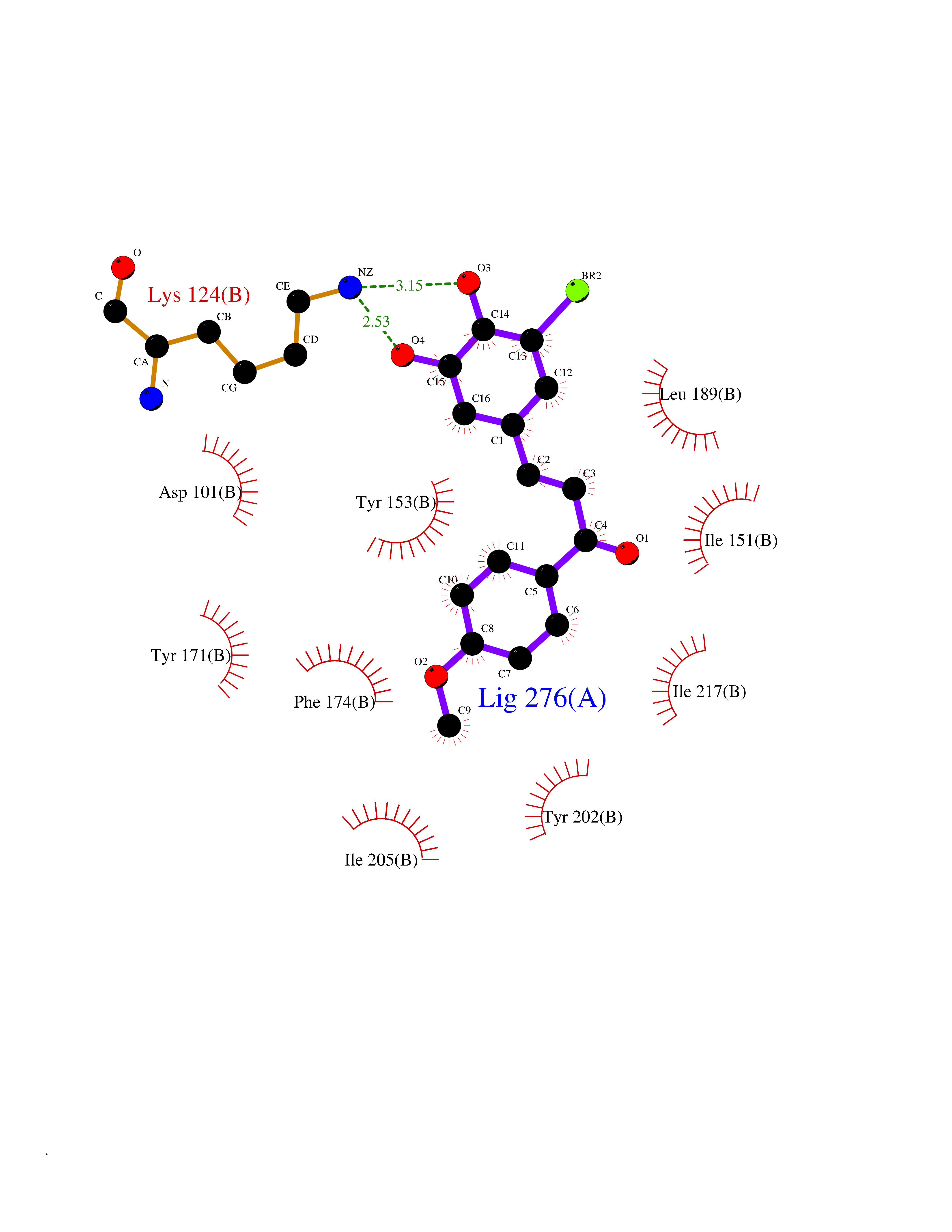 Ligplot Map 3D Binding mode Sequence MSGRVGDLSPRQKEALAKFRENVQDVLPALPNPDDYFLLRWLRARSFDLQKSEAMLRKHVEFRKQKDIDNIISWQPPEVIQQYLSGGMCGYDLDGCPVWYDIIGPLDAKGLLFSASKQDLLRTKMRECELLLQECAHQTTKLGRKVETITIIYDCEGLGLKHLWKPAVEAYGEFLCMFEENYPETLKRLFVVKAPKLFPVAYNLIKPFLSEDTRKKIMVLGANWKEVLLKHISPDQVPVEYGGTMTDPDGNPKCKSKINYGGDIPRKYYVRDQV Hydrogen bonds contact Hydrophobic contact | ||||
| 18 | Glutamate receptor ionotropic NMDA 2A (NMDAR2A) | 5KCJ | 7.17 | |
Target general information Gen name GRIN2A Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms NR2A; NMDA receptor NR2A; N-methyl D-aspartate receptor subtype 2A; HNR2A; Glutamate receptor ionotropic, NMDA 2A; Glutamate [NMDA] receptor subunit epsilon-1; GluN2A Protein family Glutamate-gated ion channel (TC 1.A.10.1) family, NR1/GRIN1 subfamily Biochemical class Glutamate-gated ion channel Function Channel activation requires binding of the neurotransmitter glutamate to the epsilon subunit, glycine binding to the zeta subunit, plus membrane depolarization to eliminate channel inhibition by Mg(2+). Sensitivity to glutamate and channel kinetics depend on the subunit composition; channels containing GRIN1 and GRIN2A have higher sensitivity to glutamate and faster kinetics than channels formed by GRIN1 and GRIN2B. Contributes to the slow phase of excitatory postsynaptic current, long-term synaptic potentiation, and learning. Component of NMDA receptor complexes that function as heterotetrameric, ligand-gated ion channels with high calcium permeability and voltage-dependent sensitivity to magnesium. Related diseases Neurodevelopmental disorder with or without hyperkinetic movements and seizures, autosomal dominant (NDHMSD) [MIM:614254]: An autosomal dominant neurodevelopmental disorder characterized by severe intellectual disability and developmental delay, absent speech, muscular hypotonia, dyskinesia, and hyperkinetic movements. Cortical blindness, cerebral atrophy, and seizures are present in some patients. {ECO:0000269|PubMed:21376300, ECO:0000269|PubMed:25167861, ECO:0000269|PubMed:25864721, ECO:0000269|PubMed:27164704, ECO:0000269|PubMed:28095420, ECO:0000269|PubMed:28228639, ECO:0000269|PubMed:28389307, ECO:0000269|PubMed:38538865}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Neurodevelopmental disorder with or without hyperkinetic movements and seizures, autosomal recessive (NDHMSR) [MIM:617820]: An autosomal recessive neurodevelopmental disorder characterized by severe intellectual disability and psychomotor developmental delay, involuntary and stereotypic movements, spasticity, and inability to walk without support. Intractable seizures manifest in some patients. {ECO:0000269|PubMed:27164704, ECO:0000269|PubMed:28051072}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Developmental and epileptic encephalopathy 101 (DEE101) [MIM:619814]: A form of epileptic encephalopathy, a heterogeneous group of early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE101 is an autosomal recessive, severe form characterized by onset of seizures in early infancy. Death in infancy may occur. {ECO:0000269|PubMed:27164704, ECO:0000269|PubMed:34611970}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01931; DB00659; DB06151; DB08838; DB01238; DB00289; DB05824; DB04620; DB03929; DB00647; DB00843; DB00228; DB11823; DB13146; DB06741; DB00142; DB00874; DB08954; DB06738; DB09409; DB09481; DB01043; DB00454; DB00333; DB04896; DB01173; DB00312; DB01174; DB01708; DB00418; DB00193 Interacts with P05067; P35637; Q12879-1; Q13224; Q62936 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Calcium; Cell membrane; Disease variant; Disulfide bond; Epilepsy; Glycoprotein; Intellectual disability; Ion channel; Ion transport; Ligand-gated ion channel; Magnesium; Membrane; Metal-binding; Phosphoprotein; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Signal; Synapse; Transmembrane; Transmembrane helix; Transport; Zinc Protein physicochemical properties Chain ID B Molecular weight (Da) 53395.6 Length 469 Aromaticity 0.11 Instability index 29.84 Isoelectric point 8.72 Charge (pH=7) 5.65 2D Binding mode Binding energy (Kcal/mol) -9.79  Molscript Map 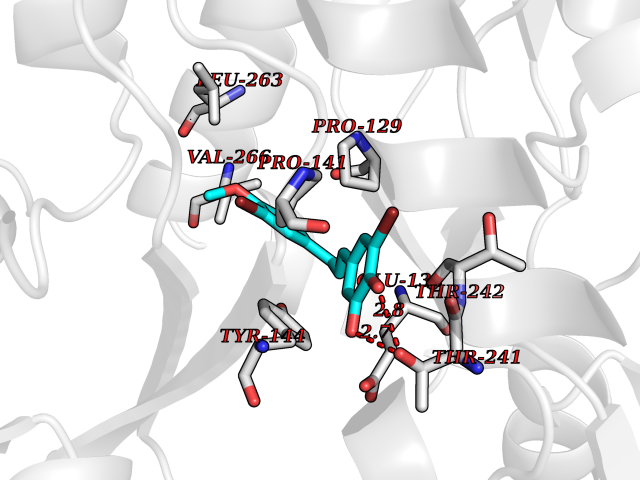 Pymol Map 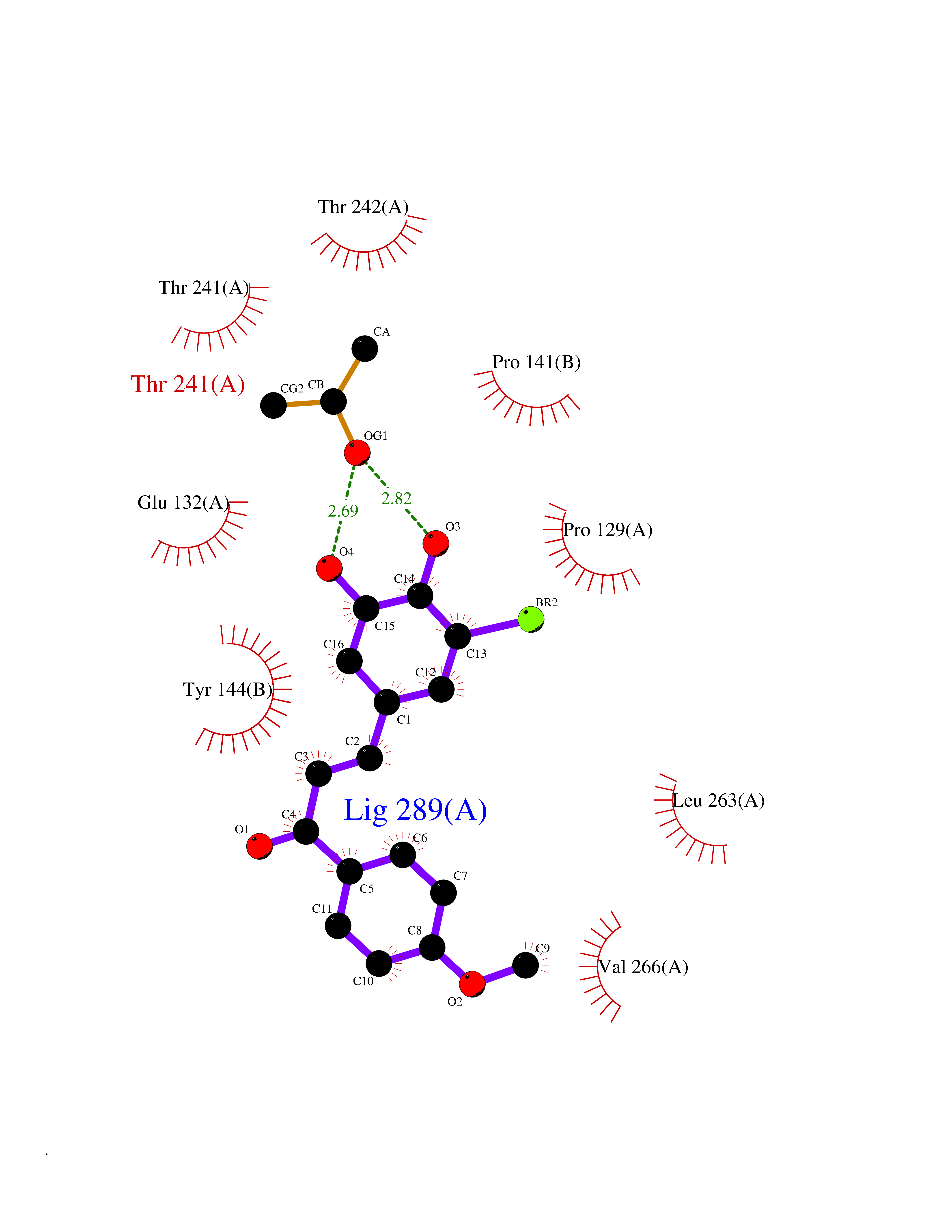 Ligplot Map 3D Binding mode Sequence DNHLSIVTLEEAPFVILKKLSRTVKFTYDLYLVTNGKHGKKVNNVWNGMIGEVVYQRAVMAVGSLTINEERSEVVDFSVPFVETGISVMVSRGTQVTGLSDKKFQRPHDYSPPFRFGTVPNGSTERNIRNNYPYMHQYMTKFNQKGVEDALVSLKTGKLDAFIYDAAVLNYKAGRDEGCKLVTIGSGYIFATTGYGIALQKGSPWKRQIDLALLQFVGDGEMEELETLWLTGICTRLKIVTIHQEPFVYYGFCIDLLIKLARTMNFTYEVHLVADGKFGTQERVNKKEWNGMMGELLSGQADMIVAPLTINNERAQYIEFSKPFKYQGLTILVKKGTRITGINDPRLRNPSDKFIYATVKQSSVDIYFRRQVELSTMYRHMEKHNYESAAEAIQAVRDNKLHAFIWDSAVLEFEASQKCDLVTTGELFFRSGFGIGMRKDSPWKQNVSLSILKSHENGFMEDLDKTWVR Hydrogen bonds contact Hydrophobic contact | ||||
| 19 | Plasmodium Dihydroorotate dehydrogenase (Malaria DHOdehase) | 1TV5 | 7.17 | |
Target general information Gen name Malaria DHOdehase Organism Plasmodium falciparum (isolate 3D7) Uniprot ID TTD ID Synonyms PFF0160c; Mitochondrially bound dihydroorotate-ubiqui oxidoreductase; Dihydroorotate oxidase of Plasmodium falciparum; Dihydroorotate dehydrogenase of Plasmodium falciparum; DHOdehase of Plasmodium fa Protein family Dihydroorotate dehydrogenase family, Type 2 subfamily Biochemical class CH-CH donor oxidoreductase Function Catalyzes the conversion of dihydroorotate to orotate with quinone as electron acceptor. Related diseases Combined oxidative phosphorylation deficiency 33 (COXPD33) [MIM:617713]: An autosomal recessive disorder caused by multiple mitochondrial respiratory chain defects and impaired mitochondrial energy metabolism. Clinical manifestations are highly variable. Affected infants present with cardiomyopathy accompanied by multisystemic features involving liver, kidney, and brain. Death in infancy is observed in some patients. Children and adults present with myopathy and progressive external ophthalmoplegia. {ECO:0000269|PubMed:28942965}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01117 Interacts with NA EC number EC 1.3.5.2 Uniprot keywords 3D-structure; Flavoprotein; FMN; Membrane; Mitochondrion; Mitochondrion inner membrane; Oxidoreductase; Pyrimidine biosynthesis; Reference proteome; Transit peptide; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 41846.8 Length 371 Aromaticity 0.1 Instability index 37.25 Isoelectric point 8.21 Charge (pH=7) 3.13 2D Binding mode Binding energy (Kcal/mol) -9.77  Molscript Map 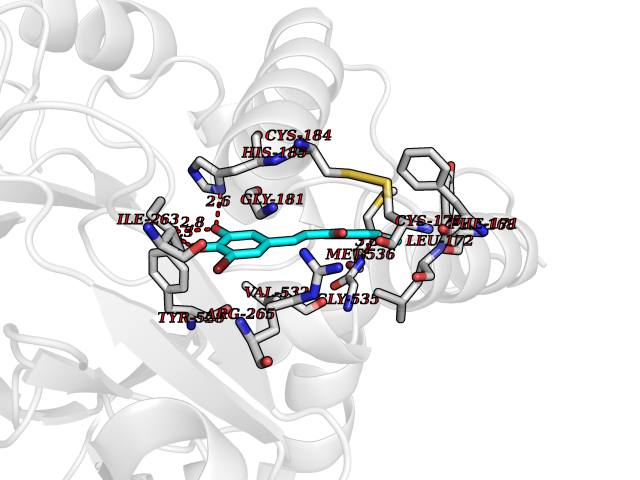 Pymol Map 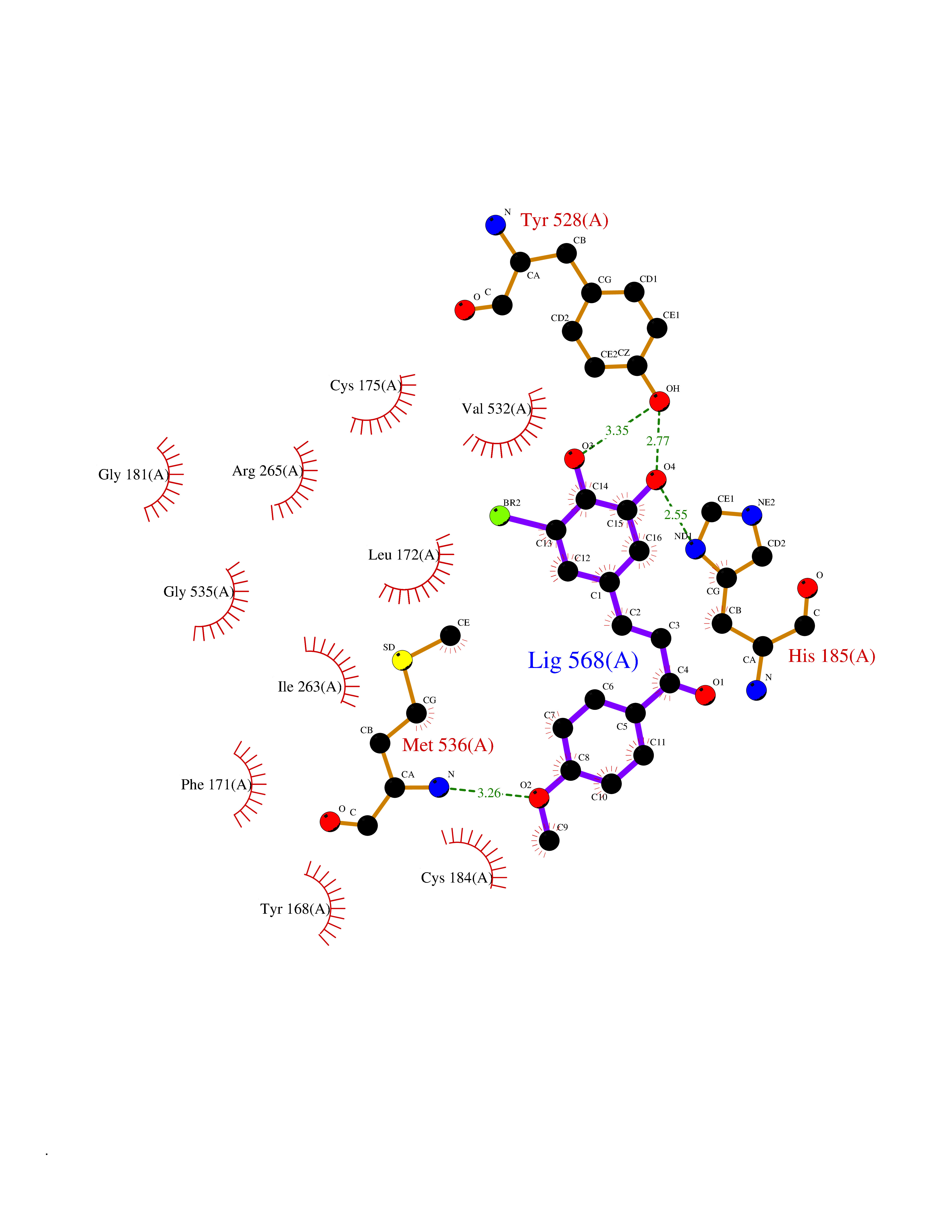 Ligplot Map 3D Binding mode Sequence FESYNPEFFLYDIFLKFCLKYIDGEICHDLFLLLGKYNILPYDTSNDSIYACTNIKHLDFINPFGVAAGFDKNGVCIDSILKLGFSFIEIGTITPRGQTGNAKPRIFRDVESRSIINSCGFNNMGCDKVTENLILFRKRQEEDKLLSKHIVGVSIGKNKDTVNIVDDLKYCINKIGRYADYIAINVSSPNTPGLRDNQEAGKLKNIILSVKEEIDNLEFLWFNTTKKKPLVFVKLAPDLNQEQKKEIADVLLETNIDGMIISNTTTQINDIKSFENKKGGVSGAKLKDISTKFICEMYNYTNKQIPIIASGGIFSGLDALEKIEAGASVCQLYSCLVFNGMKSAVQIKRELNHLLYQRGYYNLKEAIGRKH Hydrogen bonds contact Hydrophobic contact | ||||
| 20 | GTPase HRas (HRAS) | 7L0F | 7.17 | |
Target general information Gen name HRAS Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms p21ras; cHras; c-H-ras; Transforming protein p21; HaRas; Ha-Ras; H-Ras-1; GTPase HRas, Nterminally processed Protein family Small GTPase superfamily, Ras family Biochemical class Small GTPase Function Ras proteins bind GDP/GTP and possess intrinsic GTPase activity. Involved in the activation of Ras protein signal transduction. Related diseases Costello syndrome (CSTLO) [MIM:218040]: A rare condition characterized by prenatally increased growth, postnatal growth deficiency, intellectual disability, distinctive facial appearance, cardiovascular abnormalities (typically pulmonic stenosis, hypertrophic cardiomyopathy and/or atrial tachycardia), tumor predisposition, skin and musculoskeletal abnormalities. {ECO:0000269|PubMed:16170316, ECO:0000269|PubMed:16329078, ECO:0000269|PubMed:16443854, ECO:0000269|PubMed:17054105, ECO:0000269|PubMed:18039947, ECO:0000269|PubMed:18247425, ECO:0000269|PubMed:19995790}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Congenital myopathy with excess of muscle spindles (CMEMS) [MIM:218040]: Variant of Costello syndrome. {ECO:0000269|PubMed:17412879}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Thyroid cancer, non-medullary, 2 (NMTC2) [MIM:188470]: A form of non-medullary thyroid cancer (NMTC), a cancer characterized by tumors originating from the thyroid follicular cells. NMTCs represent approximately 95% of all cases of thyroid cancer and are classified into papillary, follicular, Hurthle cell, and anaplastic neoplasms. {ECO:0000269|PubMed:12727991}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Mutations which change positions 12, 13 or 61 activate the potential of HRAS to transform cultured cells and are implicated in a variety of human tumors. {ECO:0000269|PubMed:3670300}.; DISEASE: Bladder cancer (BLC) [MIM:109800]: A malignancy originating in tissues of the urinary bladder. It often presents with multiple tumors appearing at different times and at different sites in the bladder. Most bladder cancers are transitional cell carcinomas that begin in cells that normally make up the inner lining of the bladder. Other types of bladder cancer include squamous cell carcinoma (cancer that begins in thin, flat cells) and adenocarcinoma (cancer that begins in cells that make and release mucus and other fluids). Bladder cancer is a complex disorder with both genetic and environmental influences. {ECO:0000269|PubMed:6298635, ECO:0000269|PubMed:6844927}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Schimmelpenning-Feuerstein-Mims syndrome (SFM) [MIM:163200]: A disease characterized by sebaceous nevi, often on the face, associated with variable ipsilateral abnormalities of the central nervous system, ocular anomalies, and skeletal defects. Many oral manifestations have been reported, not only including hypoplastic and malformed teeth, and mucosal papillomatosis, but also ankyloglossia, hemihyperplastic tongue, intraoral nevus, giant cell granuloma, ameloblastoma, bone cysts, follicular cysts, oligodontia, and odontodysplasia. Sebaceous nevi follow the lines of Blaschko and these can continue as linear intraoral lesions, as in mucosal papillomatosis. {ECO:0000269|PubMed:22683711}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB04315; DB04137; DB02210; DB08751; DB03226; DB15588 Interacts with Q99996-3; P53677-2; P10398; Q9NXL2-1; Q9UII2; Q9H7T9; Q00994; Q9H2G9; P15056; Q7Z569; Q5PSV4; Q9ULD4-2; Q96LL4; Q96HB5; Q49A88-3; Q96GN5-2; P24941; O95674; Q9H3R5; Q9Y4F5-3; Q86XR8; Q494V2-2; Q8WUX9; Q14117; Q9Y6W6; O14641; A0AVK6; Q8NB25; Q8IZU1; O94868-3; P15407; P15408; P52655; Q96CS2; Q9BT25; Q8IV36; O43248; Q53GQ0; P10809; Q8NDH6-2; Q8IY31-2; Q8NA54; Q13352; P28290-2; Q9BVG8-5; Q2M2Z5; Q6P597; P57682; Q9UH77; P08727; Q14525; Q14847-2; Q96LR2; P27338; Q99558; Q96EZ8; Q8TAC0; Q5JXC2; Q8NEH6; Q9Y605; Q96HT8; Q9GZM8; P21359; Q8N5V2; Q6PHZ7; Q9BZ95-3; A5D8V7; O43482; Q9BR81; O15534; Q9BUL5; O00329; O00329-2; Q9UPR0; Q96I34; Q15435-3; P04049; P11233; Q15311; Q12967; Q9NS23-2; Q9NS23-4; Q8WWW0; Q8TBY0; Q9P2K3-2; Q9NZL6; O15211; Q8IXN7; Q13671; Q13671-1; Q8WVD3; Q9BY12-3; Q13435; Q12824; Q13573; Q07889; Q86W54-2; Q92783-2; O75886; Q13586; Q8N4C7; O75528; P54274-2; Q9BXU0; Q5T0J7-2; Q5T1C6; Q8IUR5-4; P36406; Q86WT6-2; Q99598; Q6PF05; Q9UGJ1-2; Q9Y5Z9; P22415; Q495M9; Q9H270; Q8NEZ2; P19544-6; O43829; Q9C0F3; Q7Z637; Q86V28; P42337; Q9Z0S9; Q9EQZ6; P27671; Q5EBH1; Q5EBH1-1; P52306-5 EC number NA Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Cell membrane; Cytoplasm; Direct protein sequencing; Disease variant; Glycoprotein; Golgi apparatus; GTP-binding; Hydrolase; Isopeptide bond; Lipoprotein; Membrane; Methylation; Nucleotide-binding; Nucleus; Palmitate; Prenylation; Proteomics identification; Proto-oncogene; Reference proteome; S-nitrosylation; Ubl conjugation Protein physicochemical properties Chain ID E,F Molecular weight (Da) 28737.2 Length 259 Aromaticity 0.1 Instability index 30.69 Isoelectric point 5.64 Charge (pH=7) -4.15 2D Binding mode Binding energy (Kcal/mol) -9.78 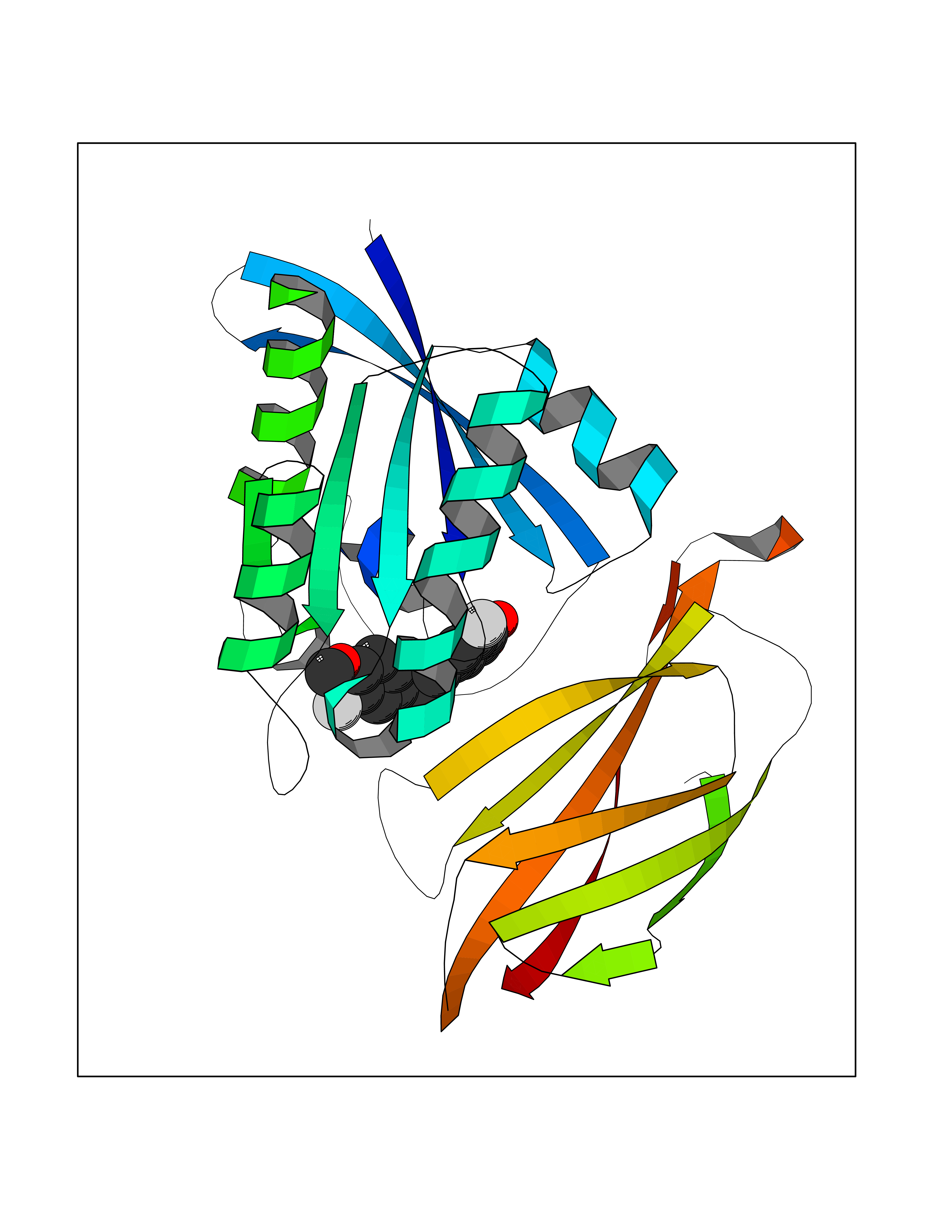 Molscript Map 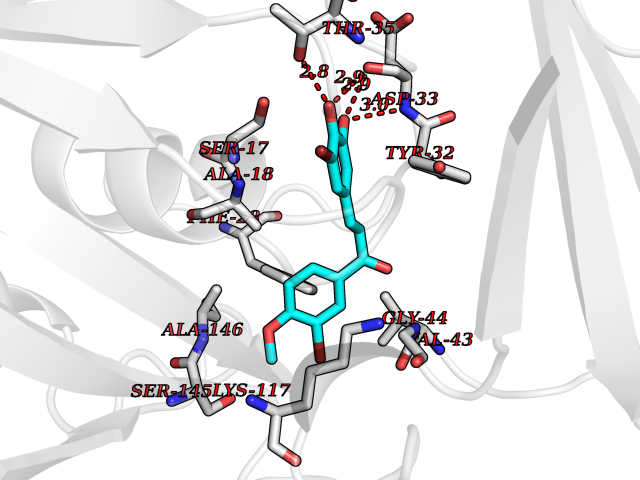 Pymol Map 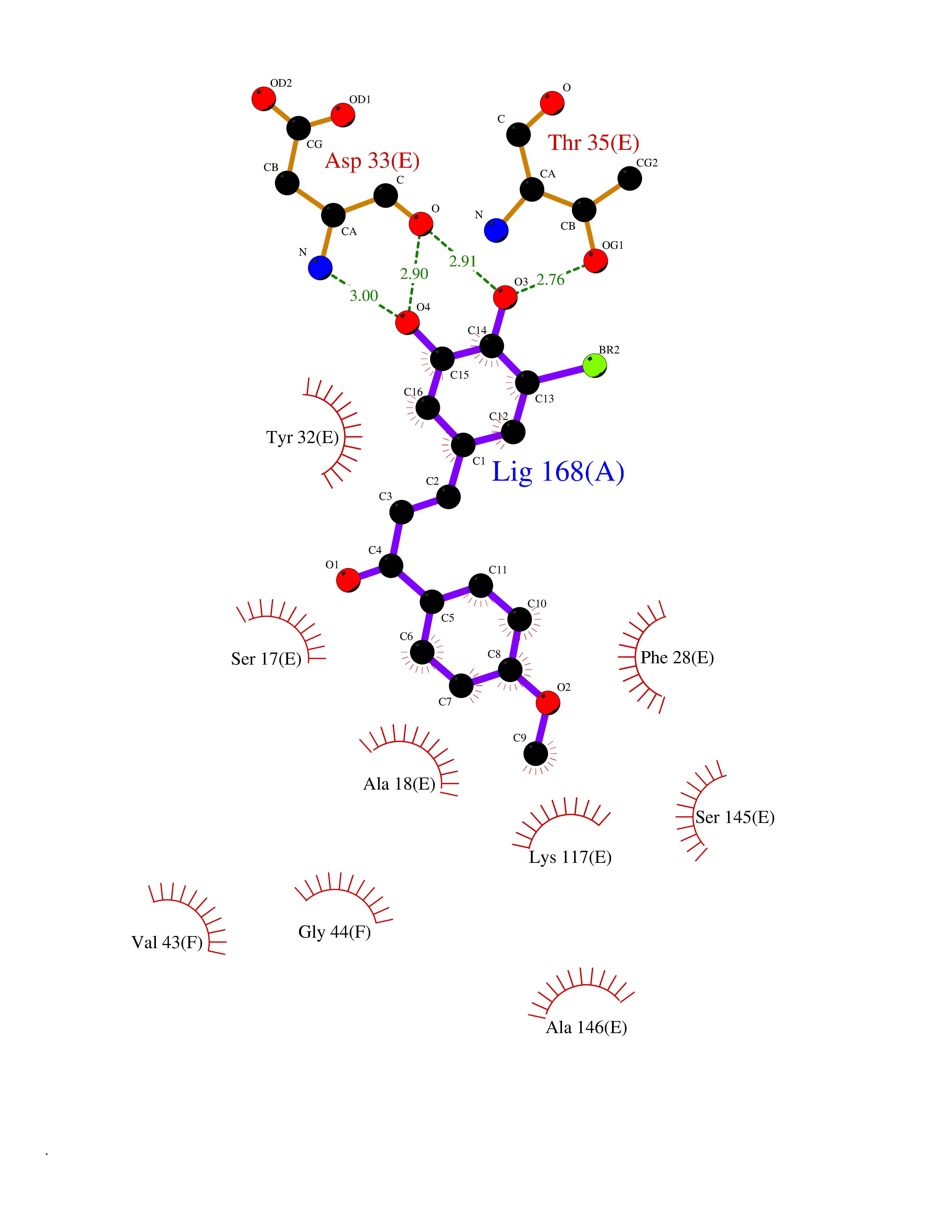 Ligplot Map 3D Binding mode Sequence MTEYKLVVVGAGGVGKSALTIQLIQNHFVDEYDPTIEDSYRKQVVIDGETCLLDILDTAGQEEYSAMRDQYMRTGEGFLCVFAINNTKSFEDIHQYREQIKRVKDSDDVPMVLVGNKCDLAARTVESRQAQDLARSYGIPYIETSAKTRQGVEDAFYTLVREIRQHSVPTKLEVVAATPTSLLISWDAPAVTVFFYIIAYGETGHGVGAFQAFRVPGSKSTATISGLKPGVDYTITVYARGYSKQGPYKPSPISINYRT Hydrogen bonds contact Hydrophobic contact | ||||