Job Results:
Ligand
Structure
Job ID
6035497cf04470e01d5d7dbbed13b512
Job name
NA
Time
2025-10-13 17:35:32
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 1 | Gamma-aminobutyric acid type B receptor subunit 1 | 4MS4 | 6.01 | |
Target general information Gen name GABBR1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms GPRC3B;GPR51 Protein family G-protein coupled receptor 3 family, GABA-B receptor subfamily Biochemical class Signaling protein / antagonist Function G-protein coupled GABA receptor activity. Related diseases Neurodevelopmental disorder with poor language and loss of hand skills (NDPLHS) [MIM:617903]: An autosomal dominant disorder characterized by psychomotor developmental stagnation or regression. NDPLHS manifest in the first years of life as loss of purposeful hand movements, loss of language, and intellectual disability. {ECO:0000269|PubMed:26740508, ECO:0000269|PubMed:28856709, ECO:0000269|PubMed:29369404}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Developmental and epileptic encephalopathy 59 (DEE59) [MIM:617904]: A form of epileptic encephalopathy, a heterogeneous group of severe early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE59 is an autosomal dominant condition characterized by onset of refractory seizures in early infancy. {ECO:0000269|PubMed:28856709, ECO:0000269|PubMed:29100083, ECO:0000269|PubMed:29369404}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08891; DB08892; DB00181; DB00363; DB02530; DB05010; DB09072 Interacts with Q9UBS5; Q9UBS5-2; P46459; Q86UR5 EC number NA Uniprot keywords 3D-structure; Cell membrane; Coiled coil; Direct protein sequencing; Disease variant; Disulfide bond; Epilepsy; G-protein coupled receptor; Glycoprotein; Intellectual disability; Membrane; Phosphoprotein; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Signal; Synapse; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 46502.1 Length 408 Aromaticity 0.12 Instability index 50.05 Isoelectric point 5.78 Charge (pH=7) -5.62 2D Binding mode Binding energy (Kcal/mol) -8.19 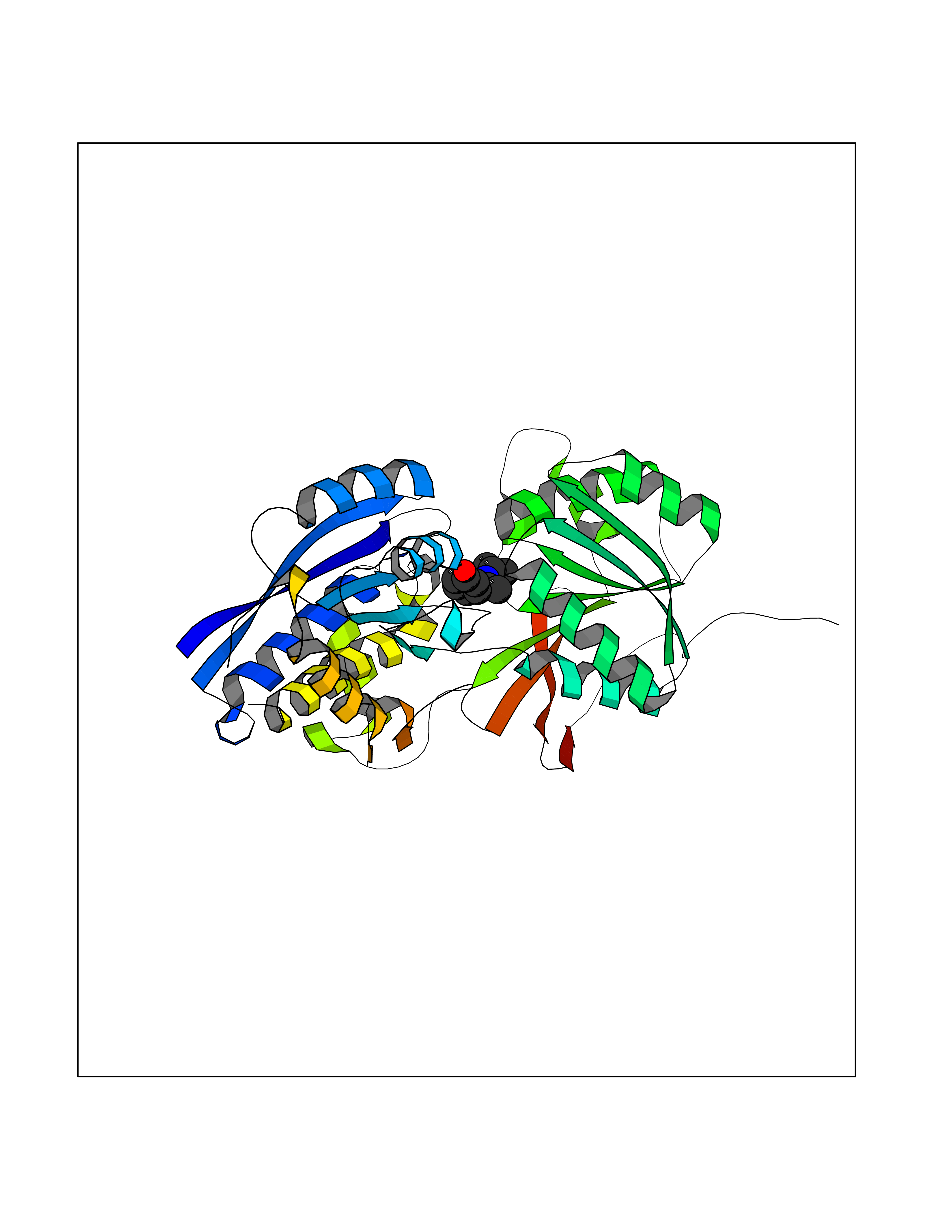 Molscript Map 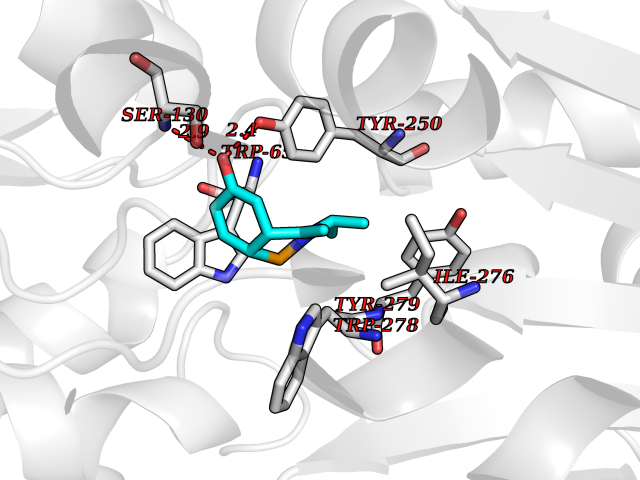 Pymol Map 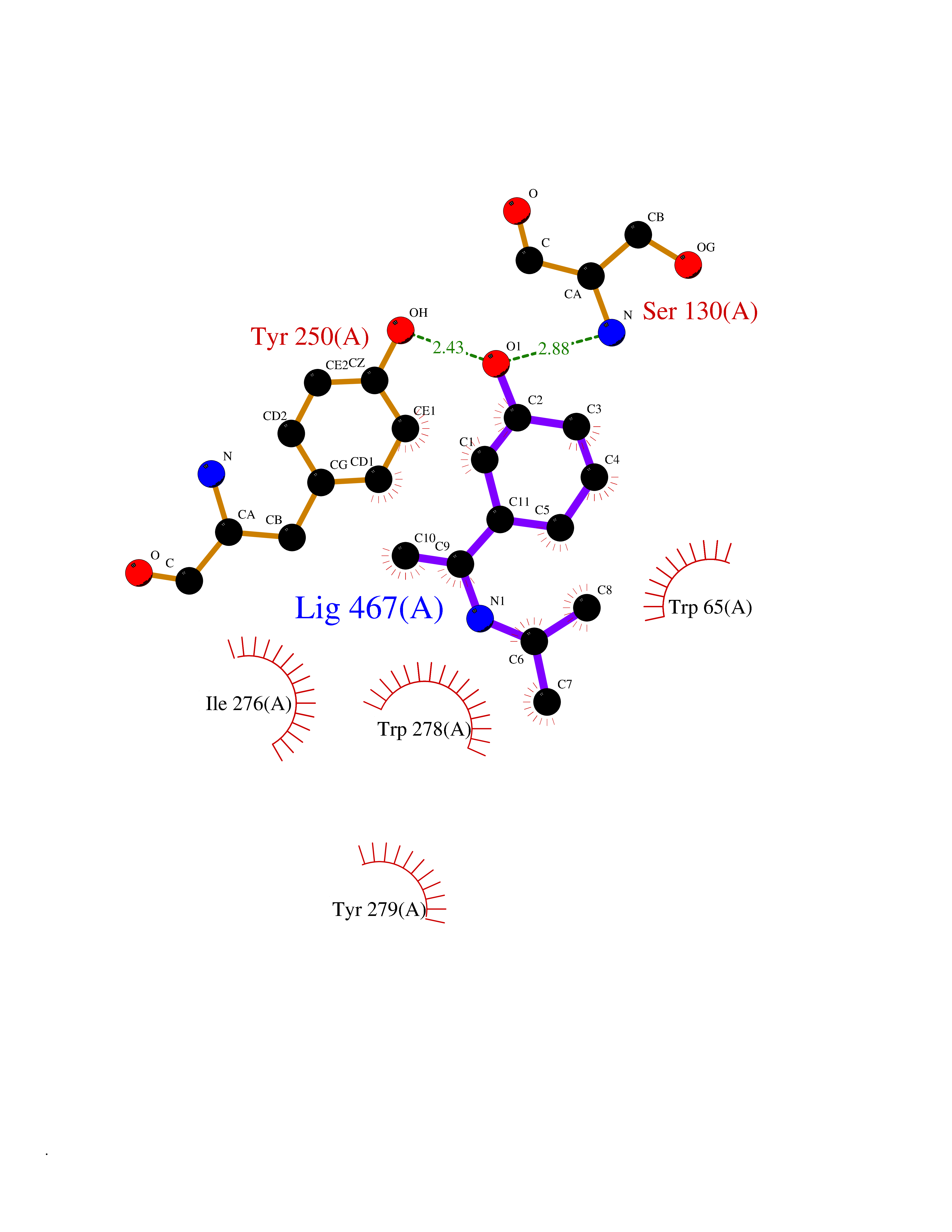 Ligplot Map 3D Binding mode Sequence RRAVYIGALFPMSGGWPGGQACQPAVEMALEDVNSRRDILPDYELKLIHHDSKCDPGQATKYLYELLYNDPIKIILMPGCSSVSTLVAEAARMWNLIVLSYGSSSPALSNRQRFPTFFRTHPSATLHNPTRVKLFEKWGWKKIATIQQTTEVFTSTLDDLEERVKEAGIEITFRQSFFSDPAVPVKNLKRQDARIIVGLFYETEARKVFCEVYKERLFGKKYVWFLIGWYADNWFKIYDPSINCTVDEMTEAVEGHITTEIVMLNPANTRSISNMTSQEFVEKLTKRLKRHPEETGGFQEAPLAYDAIWALALALNKTSRLEDFNYNNQTITDQIYRAMNSSSFEGVSGHVVFDASGSRMAWTLIEQLQGGSYKKIGYYDSTKDDLSWSKTDKWIGGSPPADDYKDDD Hydrogen bonds contact Hydrophobic contact | ||||
| 2 | Tankyrase-2 (TNKS-2) | 3U9H | 5.99 | |
Target general information Gen name TNKS2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Tankyrase-related protein; Tankyrase-like protein; Tankyrase II; TRF1-interacting ankyrin-related ADP-ribose polymerase 2; TNKL; TANK2; Protein poly-ADP-ribosyltransferase tankyrase-2; Poly [ADP-ribos Protein family ARTD/PARP family Biochemical class Glycosyltransferases Function Acts as an activator of the Wnt signaling pathway by mediating poly-ADP-ribosylation of AXIN1 and AXIN2, 2 key components of the beta-catenin destruction complex: poly-ADP-ribosylated target proteins are recognized by RNF146, which mediates their ubiquitination and subsequent degradation. Also mediates poly-ADP-ribosylation of BLZF1 and CASC3, followed by recruitment of RNF146 and subsequent ubiquitination. Mediates poly-ADP-ribosylation of TERF1, thereby contributing to the regulation of telomere length. Stimulates 26S proteasome activity. Poly-ADP-ribosyltransferase involved in various processes such as Wnt signaling pathway, telomere length and vesicle trafficking. Related diseases Intellectual developmental disorder with macrocephaly, seizures, and speech delay (IDDMSSD) [MIM:618158]: An autosomal dominant neurodevelopmental disorder characterized by impaired intellectual development, poor speech, postnatal macrocephaly, and seizures. {ECO:0000269|PubMed:30290153}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with O15084; Q7Z6K5-1; O15169; Q9NWV8; P11274; Q13698; Q9NRI5; Q6V0I7; Q9NWT6; P14652; Q9UIQ6; Q14980; Q9BZL4; Q92698; P78314; O43815; P54274; Q9C0C2; Q9UHP3; Q06649 EC number EC 2.4.2.30 Uniprot keywords 3D-structure; ADP-ribosylation; ANK repeat; Chromosome; Cytoplasm; Glycosyltransferase; Golgi apparatus; Hydroxylation; Membrane; Metal-binding; NAD; Nucleotidyltransferase; Nucleus; Proteomics identification; Reference proteome; Repeat; Telomere; Transferase; Ubl conjugation; Wnt signaling pathway; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 23695.5 Length 208 Aromaticity 0.11 Instability index 47.61 Isoelectric point 8.28 Charge (pH=7) 2.88 2D Binding mode Binding energy (Kcal/mol) -8.17 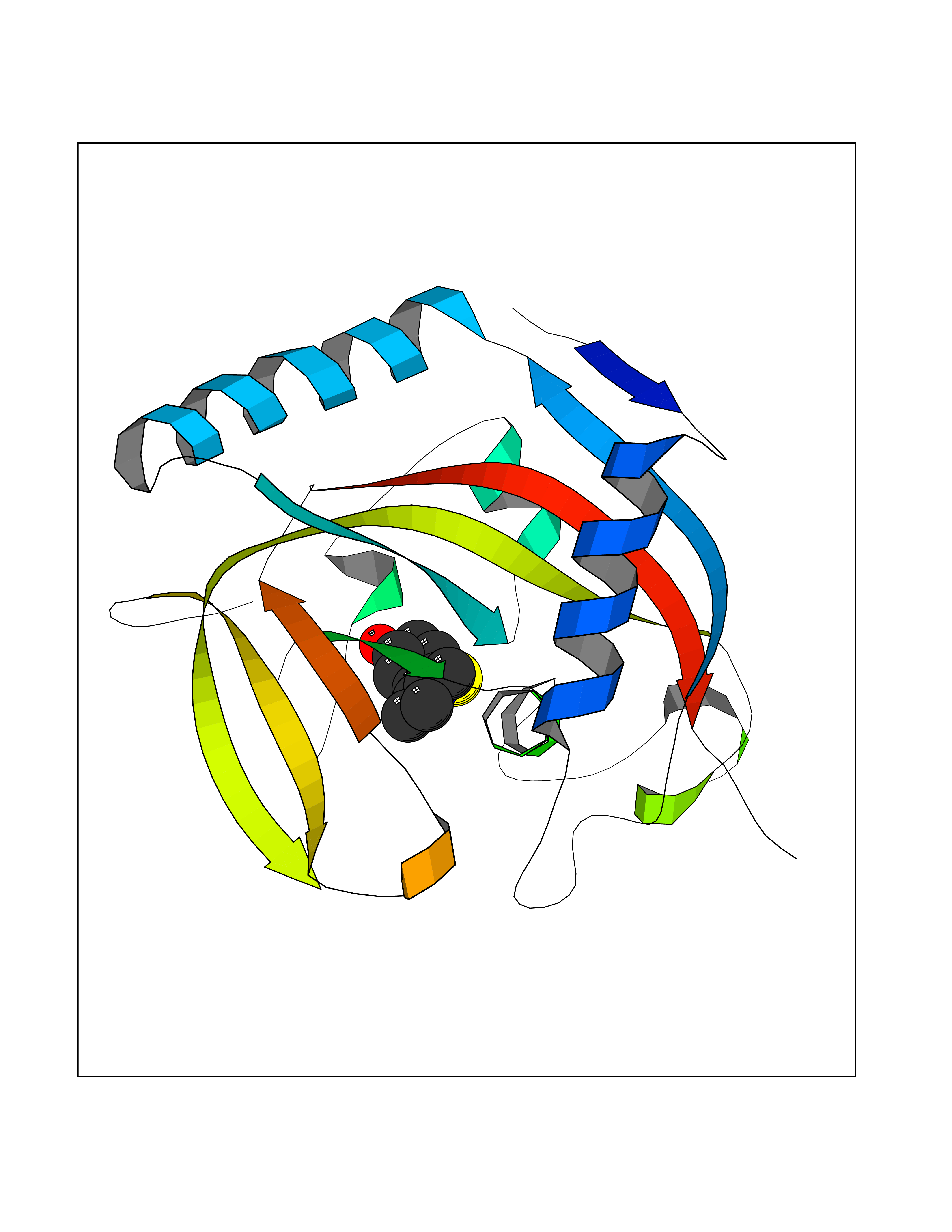 Molscript Map 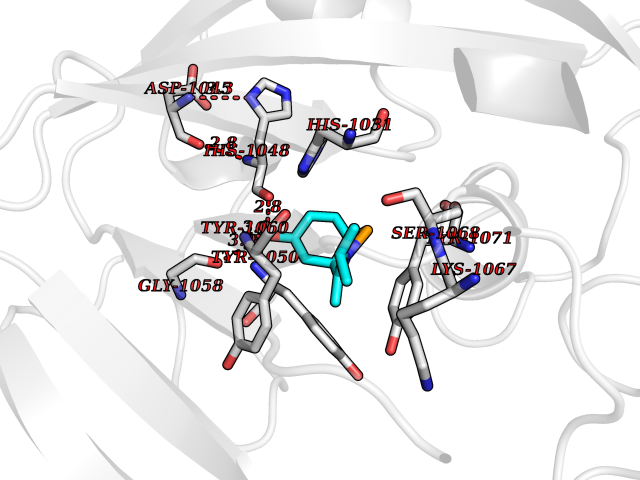 Pymol Map 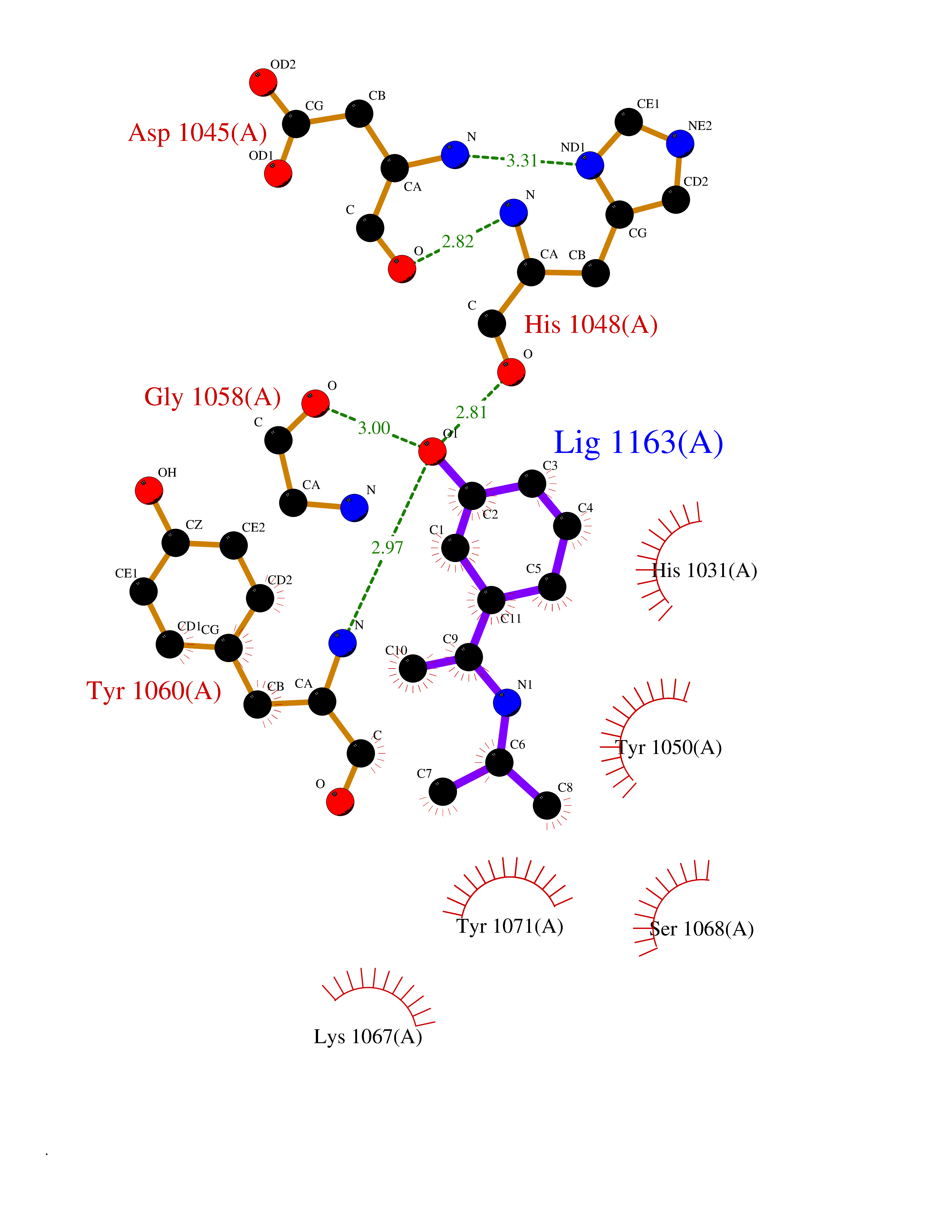 Ligplot Map 3D Binding mode Sequence GTILIDLSPDDKEFQSVEEEMQSTVREHRDGGHAGGIFNRYNILKIQKVCNKKLWERYTHRRKEVSEENHNHANERMLFHGSPFVNAIIHKGFDERHAYIGGMFGAGIYFAENSSKSNQYVYGIGGGTGCPVHKDRSCYICHRQLLFCRVTLGKSFLQFSAMAHSPPGHHSVTGRPSVNGLALAEYVIYRGEQAYPEYLITYQIMRPE Hydrogen bonds contact Hydrophobic contact | ||||
| 3 | 5'-methylthioadenosine/S-adenosylhomocysteine nucleosidase | 4WKC | 5.94 | |
Target general information Gen name mtnN Organism Escherichia coli (strain K12) Uniprot ID TTD ID NA Synonyms pfs;b0159;yadA;JW0155;mtn Protein family PNP/UDP phosphorylase family, MtnN subfamily Biochemical class hydrolase / hydrolase inhibitor Function Adenosylhomocysteine nucleosidase activity.Identical protein binding.Methylthioadenosine nucleosidase activity. Related diseases Pigmentary disorder, reticulate, with systemic manifestations, X-linked (PDR) [MIM:301220]: An X-linked recessive disorder characterized by recurrent infections and sterile inflammation in various organs. Diffuse skin hyperpigmentation with a distinctive reticulate pattern is universally evident by early childhood. This is later followed in many patients by hypohidrosis, corneal inflammation and scarring, enterocolitis that resembles inflammatory bowel disease, and recurrent urethral strictures. Melanin and amyloid deposition is present in the dermis. Affected males also have a characteristic facies with frontally upswept hair and flared eyebrows. Female carriers have only restricted pigmentary changes along Blaschko's lines. {ECO:0000269|PubMed:27019227}. The disease is caused by variants affecting the gene represented in this entry. XLPDR is caused by a recurrent intronic mutation that results in missplicing and reduced POLA1 expression. This leads to a decrease in cytosolic RNA:DNA hybrids and constitutive activation of type I interferon responses, but has no effect on cell replication. {ECO:0000269|PubMed:27019227}.; DISEASE: Van Esch-O'Driscoll syndrome (VEODS) [MIM:301030]: An X-linked recessive syndrome characterized by different degrees of intellectual disability, moderate to severe short stature, microcephaly, hypogonadism, and variable congenital malformations. {ECO:0000269|PubMed:31006512}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02158; DB08606; DB02933; DB00173; DB02281 Interacts with P0AF12 EC number 3.2.2.9 Uniprot keywords 3D-structure; Amino-acid biosynthesis; Hydrolase; Methionine biosynthesis; Reference proteome Protein physicochemical properties Chain ID A Molecular weight (Da) 24353.7 Length 232 Aromaticity 0.05 Instability index 22.1 Isoelectric point 5.09 Charge (pH=7) -9.9 2D Binding mode Binding energy (Kcal/mol) -8.1 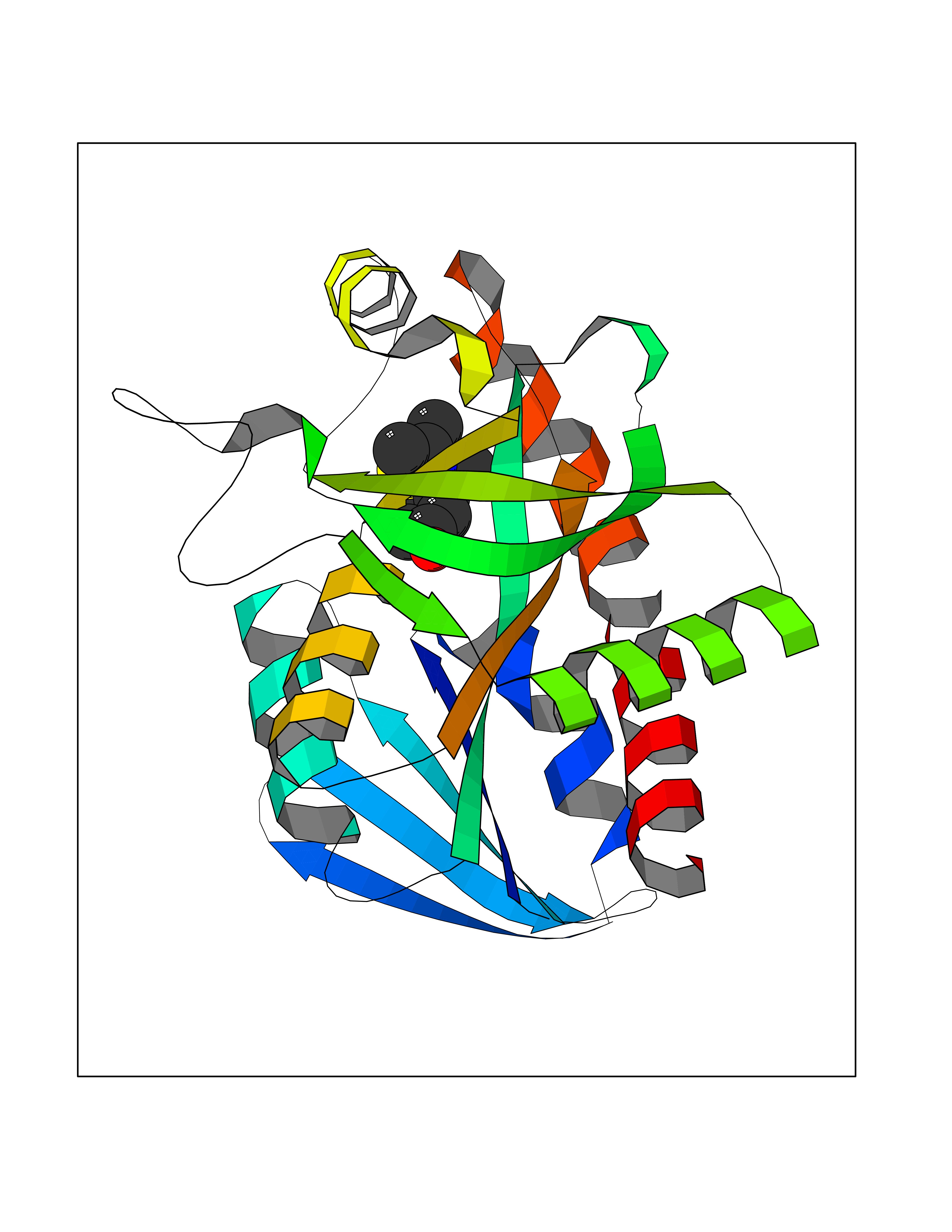 Molscript Map 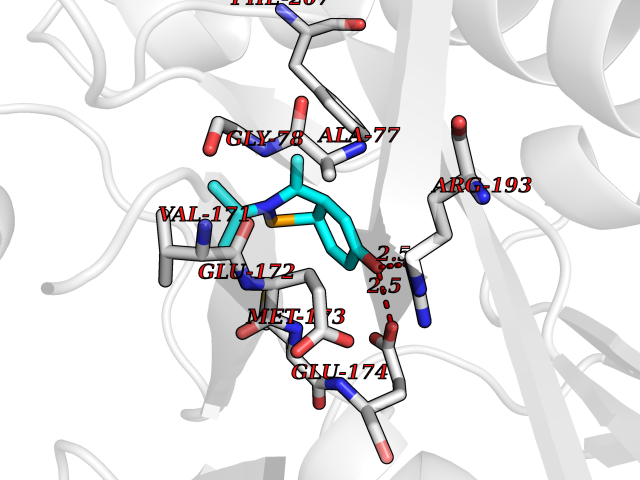 Pymol Map 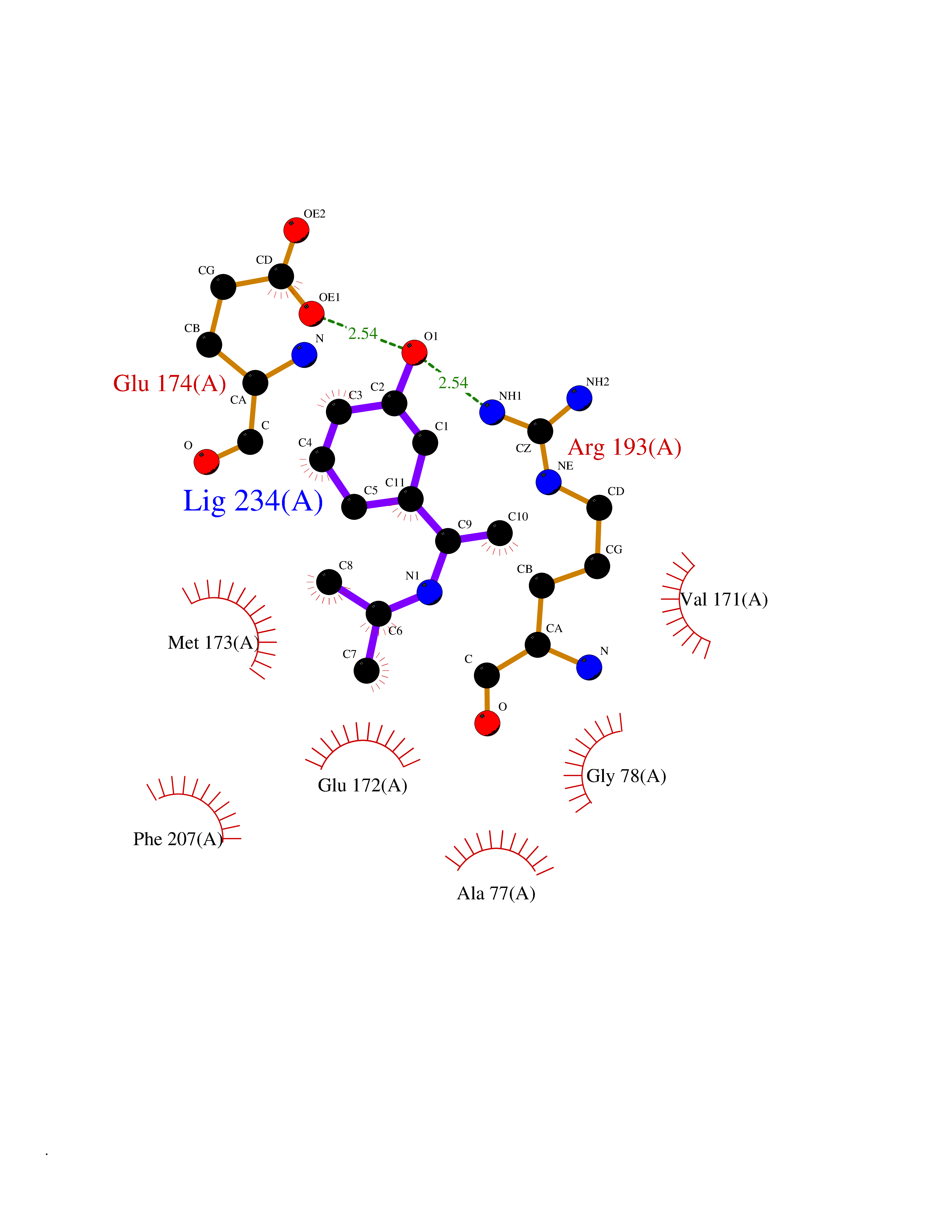 Ligplot Map 3D Binding mode Sequence MKIGIIGAMEEEVTLLRDKIENRQTISLGGCEIYTGQLNGTEVALLKSGIGKVAAALGATLLLEHCKPDVIINTGSAGGLAPTLKVGDIVVSDEARYHDADVTAFGYEYGQLPGCPAGFKADDKLIAAAEACIAELNLNAVRGLIVSGDAFINGSVGLAKIRHNFPQAIAVEMEATAIAHVCHNFNVPFVVVRAISDVADQQSHLSFDEFLAVAAKQSSLMVESLVQKLAHG Hydrogen bonds contact Hydrophobic contact | ||||
| 4 | 4-hydroxyphenylpyruvate dioxygenase | 3ISQ | 5.94 | |
Target general information Gen name HPD Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms PPD Protein family 4HPPD family Biochemical class Oxidoreductase Function 4-hydroxyphenylpyruvate dioxygenase activity.Metal ion binding. Related diseases Tyrosinemia 3 (TYRSN3) [MIM:276710]: An inborn error of metabolism characterized by elevations of tyrosine in the blood and urine, seizures and mild intellectual disability. {ECO:0000269|PubMed:10942115, ECO:0000269|PubMed:11073718}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Hawkinsinuria (HWKS) [MIM:140350]: An inborn error of tyrosine metabolism characterized by failure to thrive, persistent metabolic acidosis, fine and sparse hair, and excretion of the unusual cyclic amino acid metabolite, hawkinsin, in the urine. {ECO:0000269|PubMed:11073718}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02850; DB00348 Interacts with NA EC number 1.13.11.27 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Cytoplasm; Dioxygenase; Disease variant; Endoplasmic reticulum; Golgi apparatus; Intellectual disability; Iron; Membrane; Metal-binding; Oxidoreductase; Phenylalanine catabolism; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Tyrosine catabolism Protein physicochemical properties Chain ID A Molecular weight (Da) 43164.8 Length 376 Aromaticity 0.11 Instability index 32.38 Isoelectric point 6.73 Charge (pH=7) -1.04 2D Binding mode Binding energy (Kcal/mol) -8.1  Molscript Map 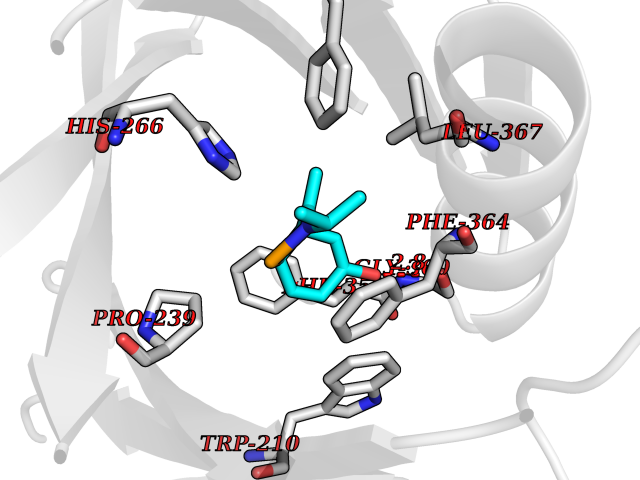 Pymol Map 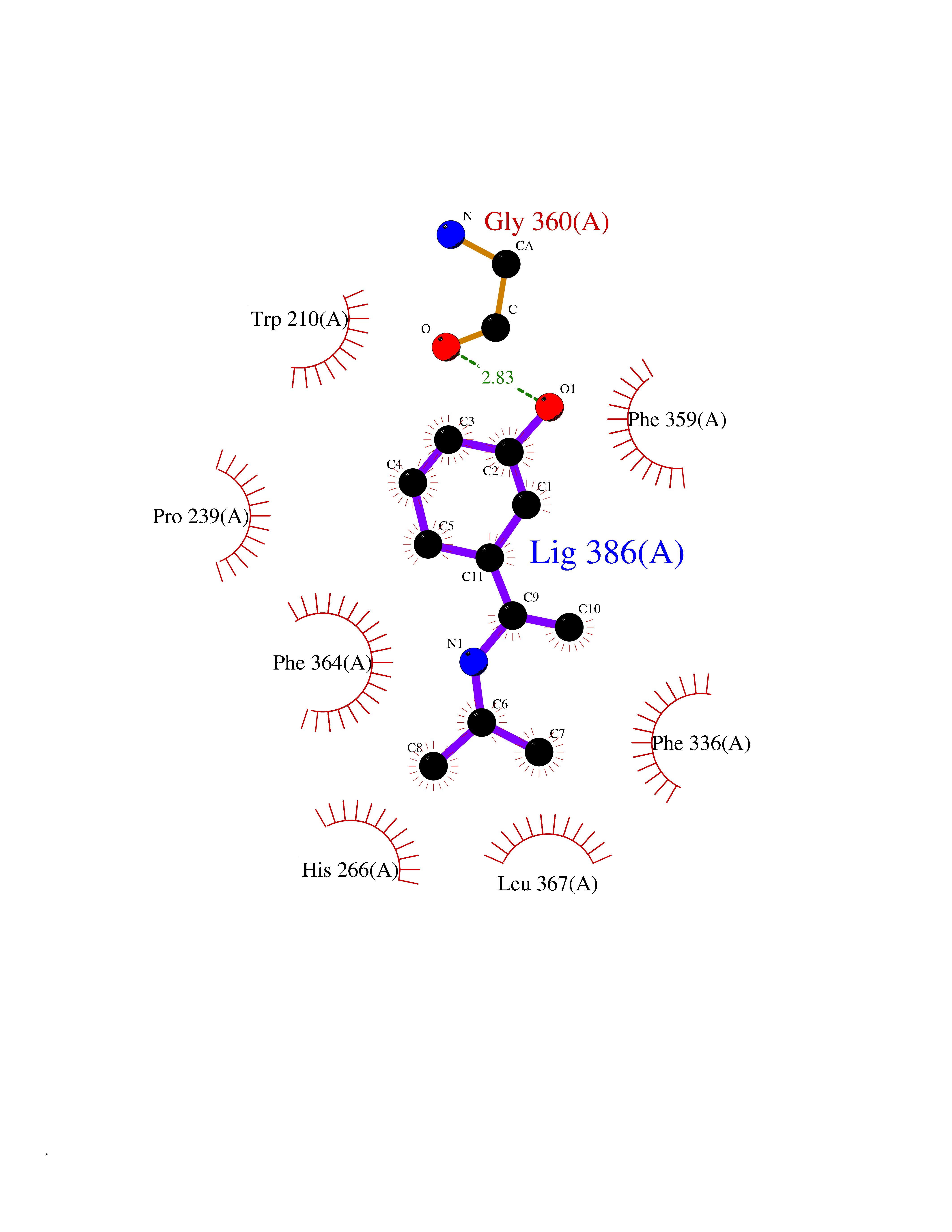 Ligplot Map 3D Binding mode Sequence AKPERGRFLHFHSVTFWVGNAKQAASFYCSKMGFEPLAYRGLETGSREVVSHVIKQGKIVFVLSSALNPWNKEMGDHLVKHGDGVKDIAFEVEDCDYIVQKARERGAKIMREPWVEQDKFGKVKFAVLQTYGDTTHTLVEKMNYIGQFLPGYEAPAFMDPLLPKLPKCSLEMIDHIVGNQPDQEMVSASEWYLKNLQFHRFWSVDDTQVHTEYSSLRSIVVANYEESIKMPINEPAPGKKKSQIQEYVDYNGGAGVQHIALKTEDIITAIRHLRERGLEFLSVPSTYYKQLREKLKTAKIKVKENIDALEELKILVDYDEKGYLLQIFTKPVQDRPTLFLEVIQRHNHQGFGAGNFNSLFKAFEEEQNLRGNLTNM Hydrogen bonds contact Hydrophobic contact | ||||
| 5 | Neuronal acetylcholine receptor alpha-2 (CHRNA2) | 5FJV | 5.92 | |
Target general information Gen name CHRNA2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms CHRNA2 Protein family Ligand-gated ion channel (TC 1.A.9) family, Acetylcholine receptor (TC 1.A.9.1) subfamily, Alpha-2/CHRNA2 sub-subfamily Biochemical class Neurotransmitter receptor Function After binding acetylcholine, the AChR responds by an extensive change in conformation that affects all subunits and leads to opening of an ion-conducting channel across the plasma membrane. Related diseases Epilepsy, nocturnal frontal lobe, 4 (ENFL4) [MIM:610353]: An autosomal dominant focal epilepsy characterized by nocturnal seizures associated with fear sensation, tongue movements, and nocturnal wandering, closely resembling nightmares and sleep walking. {ECO:0000269|PubMed:16826524}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Seizures, benign familial infantile, 6 (BFIS6) [MIM:610353]: A form of benign familial infantile epilepsy, a neurologic disorder characterized by afebrile seizures occurring in clusters during the first year of life, without neurologic sequelae. BFIS6 inheritance is autosomal dominant. {ECO:0000269|PubMed:25847220}. The disease may be caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00732; DB00237; DB00411; DB00565; DB01245; DB00514; DB01135; DB07720; DB00898; DB00472; DB00483; DB08960; DB00657; DB01336; DB00416; DB01226; DB00184; DB01337; DB01338; DB00721; DB00728; DB05740; DB00202; DB01199; DB01339 Interacts with NA EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disease variant; Disulfide bond; Epilepsy; Glycoprotein; Ion channel; Ion transport; Ligand-gated ion channel; Membrane; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Signal; Synapse; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A,B Molecular weight (Da) 48323.4 Length 413 Aromaticity 0.15 Instability index 32 Isoelectric point 5.69 Charge (pH=7) -6.58 2D Binding mode Binding energy (Kcal/mol) -8.08 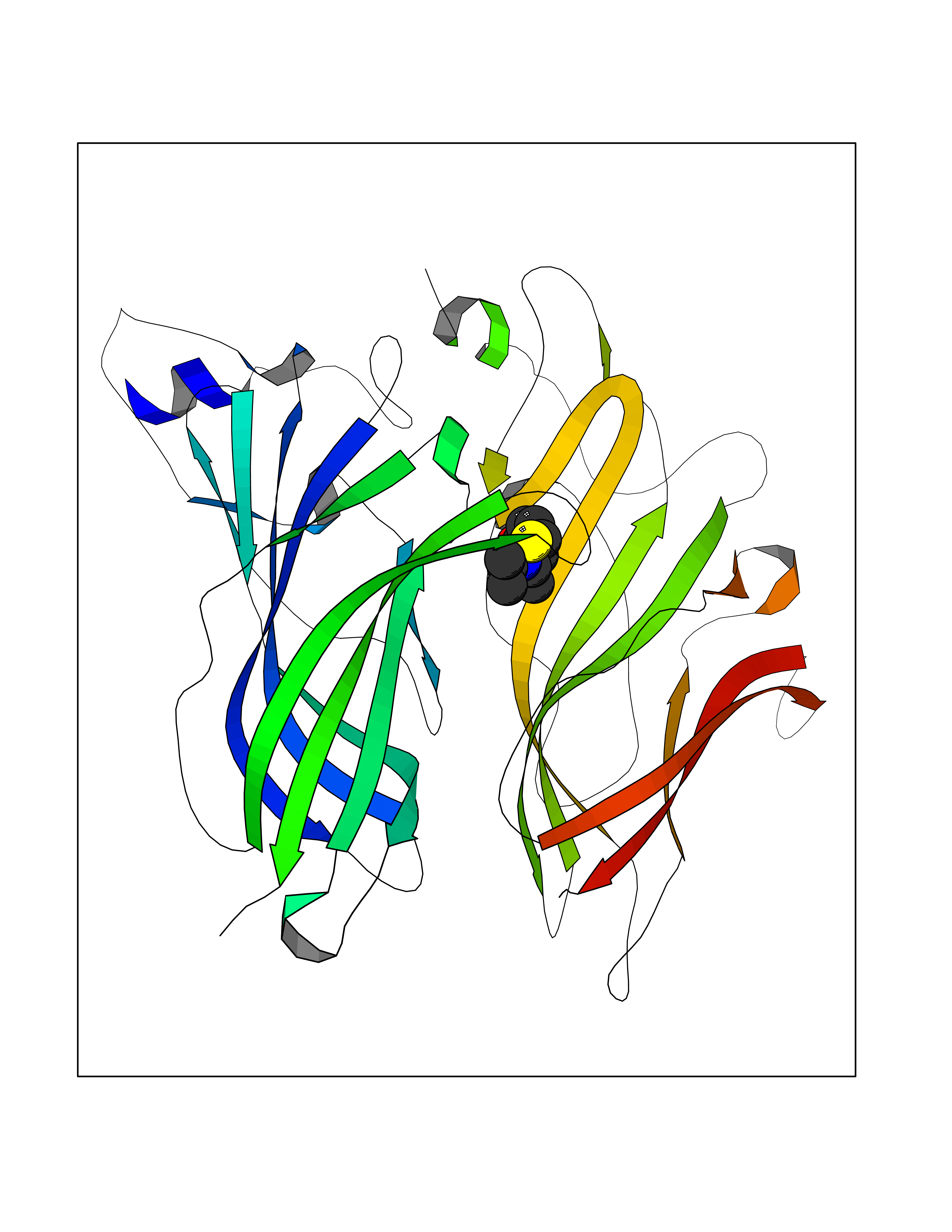 Molscript Map 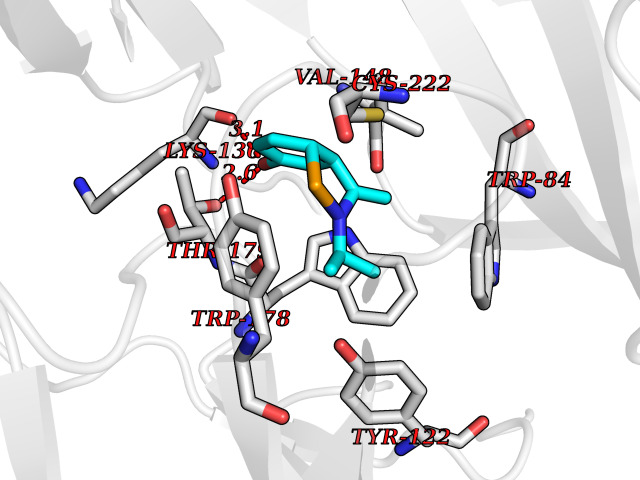 Pymol Map 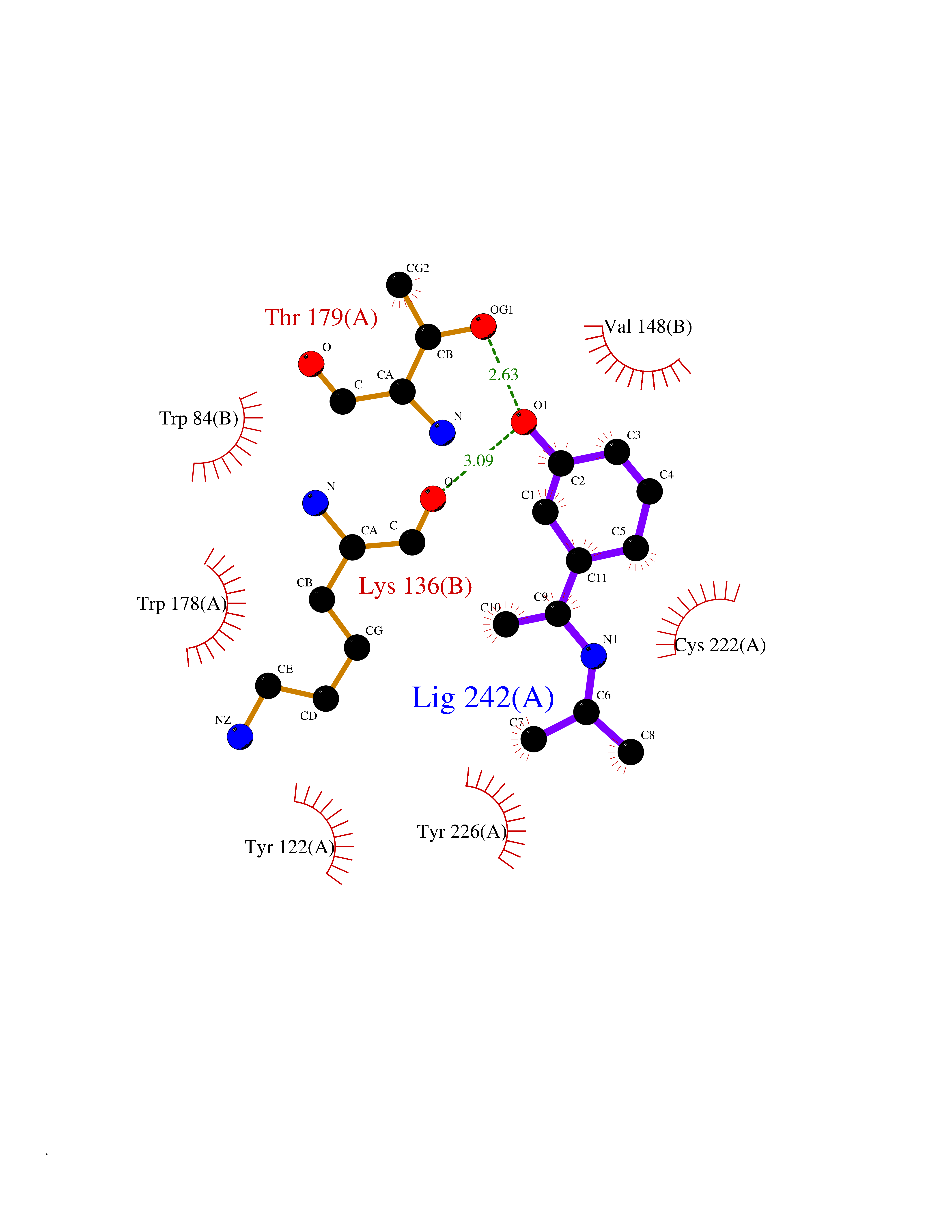 Ligplot Map 3D Binding mode Sequence DRLFKHLFRGYNRWARPVPNTSDVVIVRFGLSIAQLIDVDEKNQMMTTNVWLKQEWSDYKLRWNPTDFGNITSLRVPSEMIWIPDIVLYNNADGEFAVTHMTKAHLFSTGTVHWVPPAIYKSSCSIDVTFFPFDQQNCKMKFGSWTYDKAKIDLEQMEQTVDLKDYWESGEWAIVNATGTYNSKKYDCCAEIYPDVTYAFVIRRLPEDRLFKHLFRGYNRWARPVPNTSDVVIVRFGLSIAQLIDVDEKNQMMTTNVWLKQEWSDYKLRWNPTDFGNITSLRVPSEMIWIPDIVLYNNADGEFAVTHMTKAHLFSTGTVHWVPPAIYKSSCSIDVTFFPFDQQNCKMKFGSWTYDKAKIDLEQMEQTVDLKDYWESGEWAIVNATGTYNSKKYDCCAEIYPDVTYAFVIRRLP Hydrogen bonds contact Hydrophobic contact | ||||
| 6 | Wnt-7a protein (WNT7A) | 4UZQ | 5.91 | |
Target general information Gen name WNT7A Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Protein Wnt-7a Protein family Wnt family Biochemical class NA Function Plays an important role in embryonic development, including dorsal versus ventral patterning during limb development, skeleton development and urogenital tract development. Required for central nervous system (CNS) angiogenesis and blood-brain barrier regulation. Required for normal, sexually dimorphic development of the Mullerian ducts, and for normal fertility in both sexes. Required for normal neural stem cell proliferation in the hippocampus dentate gyrus. Required for normal progress through the cell cycle in neural progenitor cells, for self-renewal of neural stem cells, and for normal neuronal differentiation and maturation. Promotes formation of synapses via its interaction with FZD5. Ligand for members of the frizzled family of seven transmembrane receptors that functions in the canonical Wnt/beta-catenin signaling pathway. Related diseases Limb pelvis hypoplasia aplasia syndrome (LPHAS) [MIM:276820]: A syndrome of severe deficiency of the extremities due to hypo- or aplasia of one or more long bones of one or more limbs. Pelvic manifestations include hip dislocation, hypoplastic iliac bone and aplastic pubic bones. Thoracic deformity, unusual facies and genitourinary anomalies can be present. {ECO:0000269|PubMed:16826533, ECO:0000269|PubMed:17431918, ECO:0000269|PubMed:20949531, ECO:0000269|PubMed:21271649, ECO:0000269|PubMed:27638328}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Fuhrmann syndrome (FUHRS) [MIM:228930]: Distinct limb-malformation disorder characterized also by various degrees of limb aplasia/hypoplasia and joint dysplasia. {ECO:0000269|PubMed:16826533}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P55212; P22607; P06396; P13473-2; Q9UMX0; Q9Y5W5; Q5T9L3; Q9Z0J1 EC number NA Uniprot keywords 3D-structure; Developmental protein; Disease variant; Disulfide bond; Extracellular matrix; Glycoprotein; Lipoprotein; Proteomics identification; Reference proteome; Secreted; Signal; Wnt signaling pathway Protein physicochemical properties Chain ID A,B Molecular weight (Da) 40475.5 Length 356 Aromaticity 0.1 Instability index 50.49 Isoelectric point 7.67 Charge (pH=7) 1.62 2D Binding mode Binding energy (Kcal/mol) -8.06 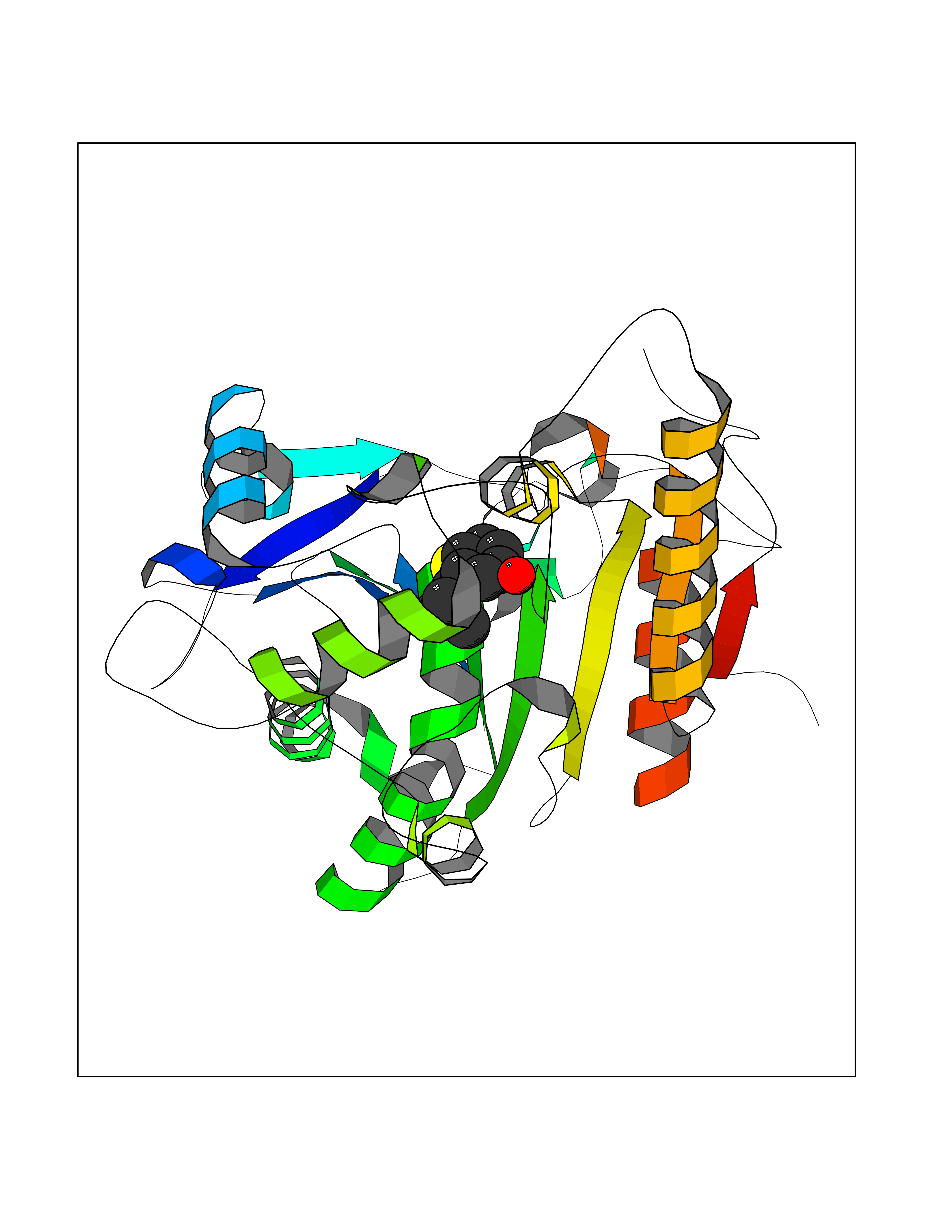 Molscript Map 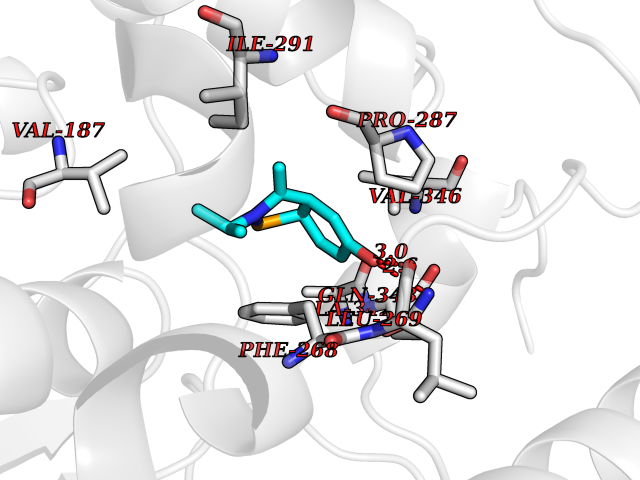 Pymol Map 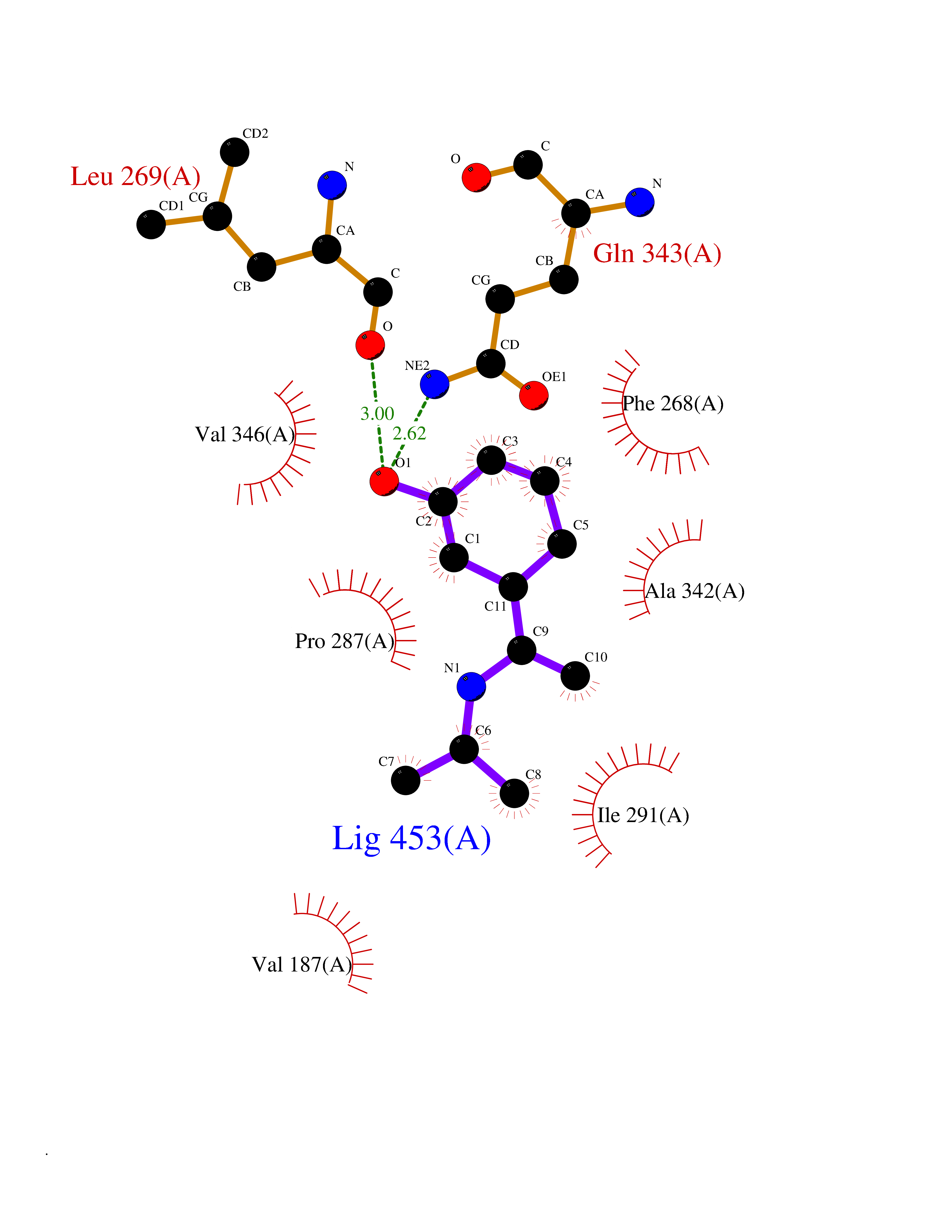 Ligplot Map 3D Binding mode Sequence EDLRLHLLLNTSVTCNDGSPAGYYLKESRGSRRWLLFLEGGWYCFNRENCDSRYDTMRRLMSSRDWPRTRTGTGILSSQPEENPYWWNANMVFIPYCSSDVWSGASSKSEKNEYAFMGALIIQEVVRELLGRGLSGAKVLLLAGSAAGGTGVLLNVDRVAEQLEKLGYPAIQVRGLADSGWFLDNKQYRHTDCVDTITCAPTEAIRRGIRYWNGVVPERCRRQFQEGEEWNCFFGYKVYPTLRSPVFVVQWLFDEAQLTVDNVHLTGQPVQEGLRLYIQNLGRELRHTLKDVPASFAPACLSHEIIIRSHWTDVQVKGTSLPRALHCWDRSLHKGCPVHLVDSCPWPHCNPSCPTS Hydrogen bonds contact Hydrophobic contact | ||||
| 7 | Free fatty acid receptor 1 | 4PHU | 5.90 | |
Target general information Gen name FFAR1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms GPR40 Protein family G-protein coupled receptor 1 family Biochemical class Fatty acid binding protein / hydrolase Function Bioactive lipid receptor activity.G-protein coupled receptor activity.Guanyl-nucleotide exchange factor activity.Lipid binding. Related diseases Refsum disease (RD) [MIM:266500]: A rare autosomal recessive peroxisomal disorder characterized by the accumulation of the branched-chain fatty acid, phytanic acid, in blood and tissues. Cardinal clinical features are retinitis pigmentosa, peripheral neuropathy, cerebellar ataxia, and elevated protein levels in the cerebrospinal fluid (CSF). Half of all patients exhibit generalized, mild to moderate ichthyosis resembling ichthyosis vulgaris. Less constant features are nerve deafness, anosmia, skeletal abnormalities, cataracts and cardiac impairment. {ECO:0000269|PubMed:10709665, ECO:0000269|PubMed:10767344, ECO:0000269|PubMed:14974078, ECO:0000269|PubMed:9326939, ECO:0000269|PubMed:9326940}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00159 Interacts with NA EC number NA Uniprot keywords 3D-structure; Cell membrane; Disulfide bond; G-protein coupled receptor; Glycoprotein; Lipid-binding; Membrane; Receptor; Reference proteome; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 28319.1 Length 272 Aromaticity 0.11 Instability index 27.3 Isoelectric point 9.07 Charge (pH=7) 6.85 2D Binding mode Binding energy (Kcal/mol) -8.05 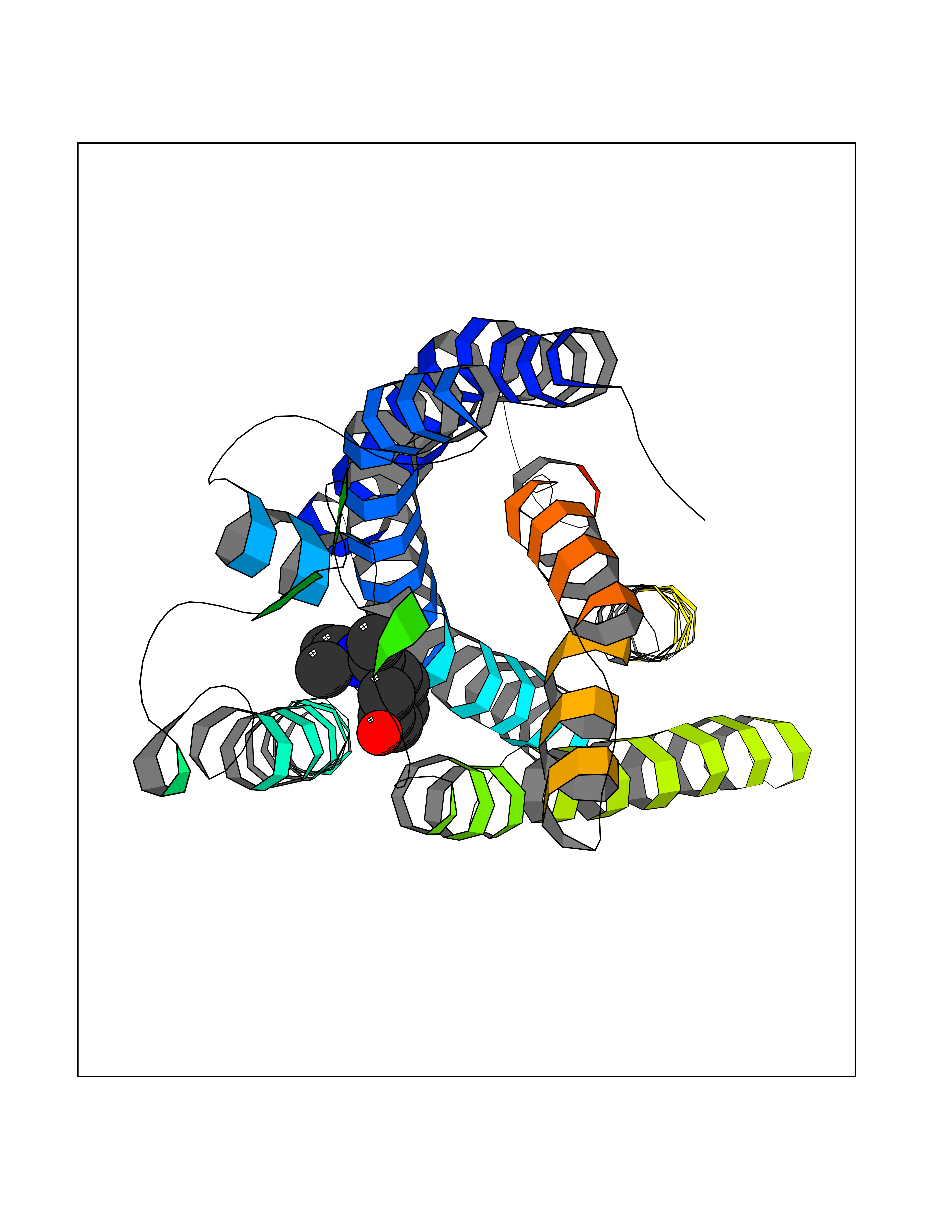 Molscript Map 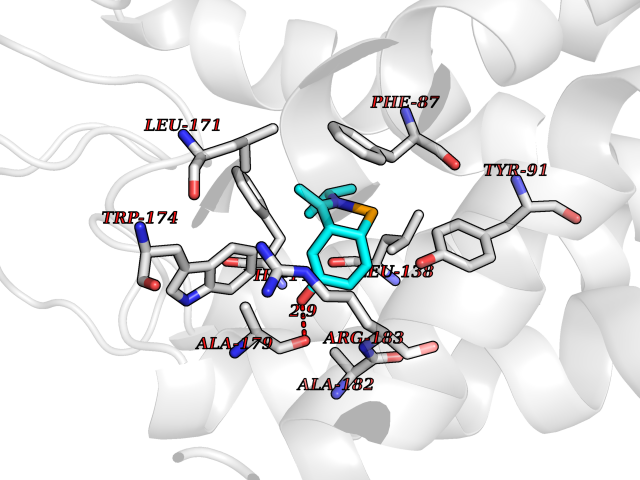 Pymol Map 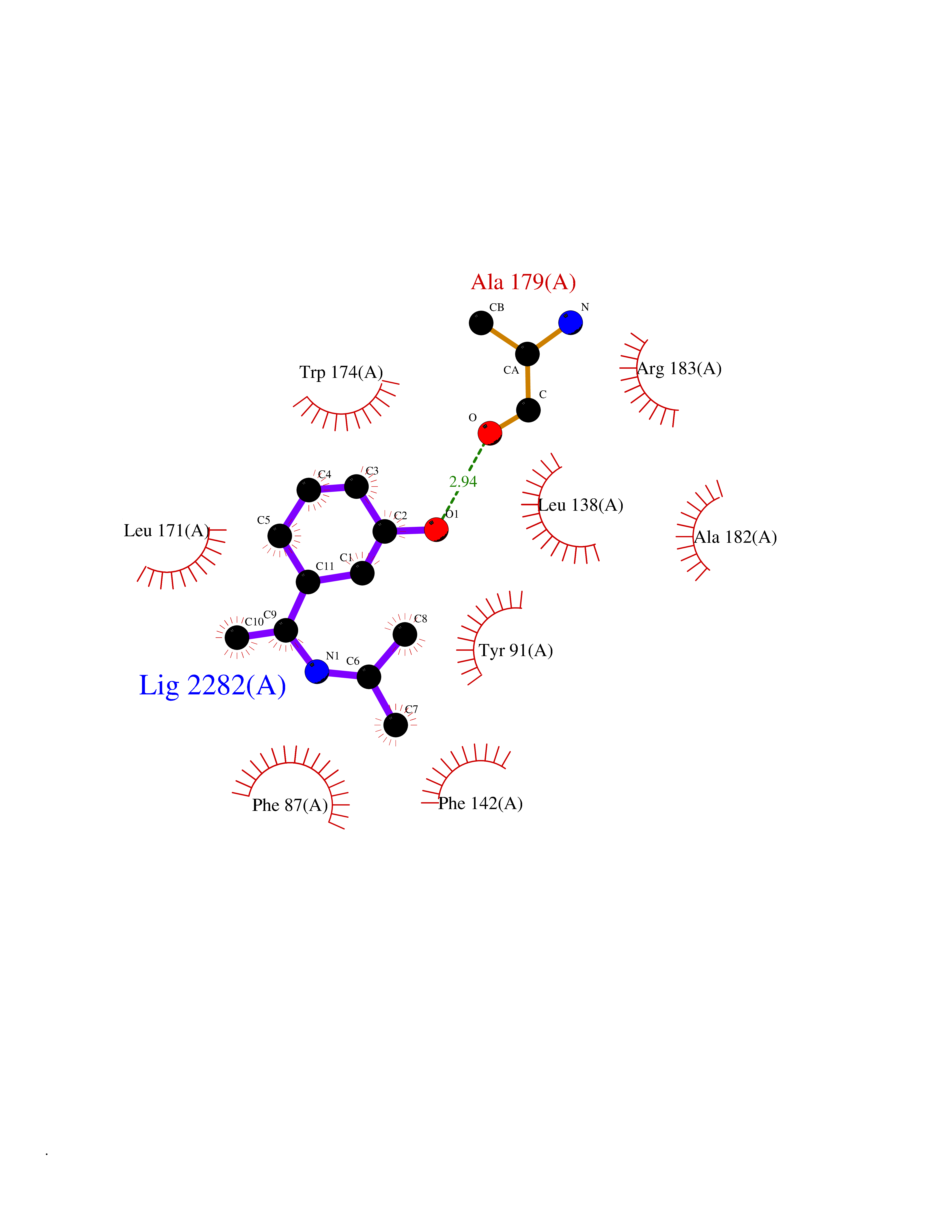 Ligplot Map 3D Binding mode Sequence MDLPPQLSFGLYVAAFALGFPLNVLAIRGATAHARLRLTPSAVYALNLGCSDLLLTVSLPLKAVEALASGAWPLPASLCPVFAVAHFAPLYAGGGFLAALSAARYLGAAFPPCYSWGVCAAIWALVLCHLGLVFGLEAPGGWLDHSNTSLGINTPVNGSPVCLEAWDPASAGPARFSLSLLLFFLPLAITAFCFVGCLRALARGSLTHRRKLRAAWVAGGALLTLLLCVGPYNASNVASFLYPNLGGSWRKLGLITGAWSVVLNPLVTGYLG Hydrogen bonds contact Hydrophobic contact | ||||
| 8 | Leukotriene A-4 hydrolase (LTA4H) | 3U9W | 5.90 | |
Target general information Gen name LTA4H Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Leukotriene A4 hydrolase; Leukotriene A(4)Leukotriene A-4 hydrolase hydrolase; Leukotriene A(4) hydrolase; LTA4; LTA-H; LTA-4hydrolase; LTA-4 hydrolase Protein family Peptidase M1 family Biochemical class Ether bond hydrolase Function Has also aminopeptidase activity. Epoxide hydrolase that catalyzes the final step in the biosynthesis of the proinflammatory mediator leukotriene B4. Related diseases Pigmentary disorder, reticulate, with systemic manifestations, X-linked (PDR) [MIM:301220]: An X-linked recessive disorder characterized by recurrent infections and sterile inflammation in various organs. Diffuse skin hyperpigmentation with a distinctive reticulate pattern is universally evident by early childhood. This is later followed in many patients by hypohidrosis, corneal inflammation and scarring, enterocolitis that resembles inflammatory bowel disease, and recurrent urethral strictures. Melanin and amyloid deposition is present in the dermis. Affected males also have a characteristic facies with frontally upswept hair and flared eyebrows. Female carriers have only restricted pigmentary changes along Blaschko's lines. {ECO:0000269|PubMed:27019227}. The disease is caused by variants affecting the gene represented in this entry. XLPDR is caused by a recurrent intronic mutation that results in missplicing and reduced POLA1 expression. This leads to a decrease in cytosolic RNA:DNA hybrids and constitutive activation of type I interferon responses, but has no effect on cell replication. {ECO:0000269|PubMed:27019227}.; DISEASE: Van Esch-O'Driscoll syndrome (VEODS) [MIM:301030]: An X-linked recessive syndrome characterized by different degrees of intellectual disability, moderate to severe short stature, microcephaly, hypogonadism, and variable congenital malformations. {ECO:0000269|PubMed:31006512}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07102; DB06917; DB07258; DB07094; DB07259; DB02352; DB07292; DB07104; DB06828; DB08466; DB01197; DB05177; DB03366; DB08040; DB06851; DB02062; DB07099; DB07260; DB07196; DB11781; DB03424; DB07237 Interacts with Q9BSI4 EC number EC 3.3.2.6 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Cytoplasm; Direct protein sequencing; Hydrolase; Leukotriene biosynthesis; Lipid metabolism; Metal-binding; Metalloprotease; Phosphoprotein; Protease; Proteomics identification; Reference proteome; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 68927 Length 608 Aromaticity 0.1 Instability index 38.84 Isoelectric point 5.87 Charge (pH=7) -9.86 2D Binding mode Binding energy (Kcal/mol) -8.05  Molscript Map 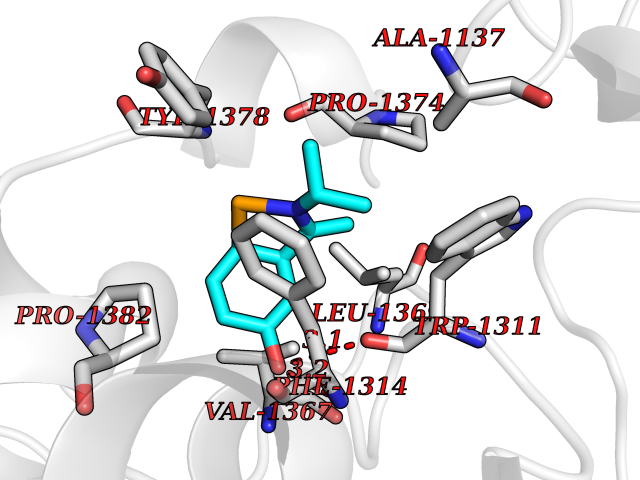 Pymol Map 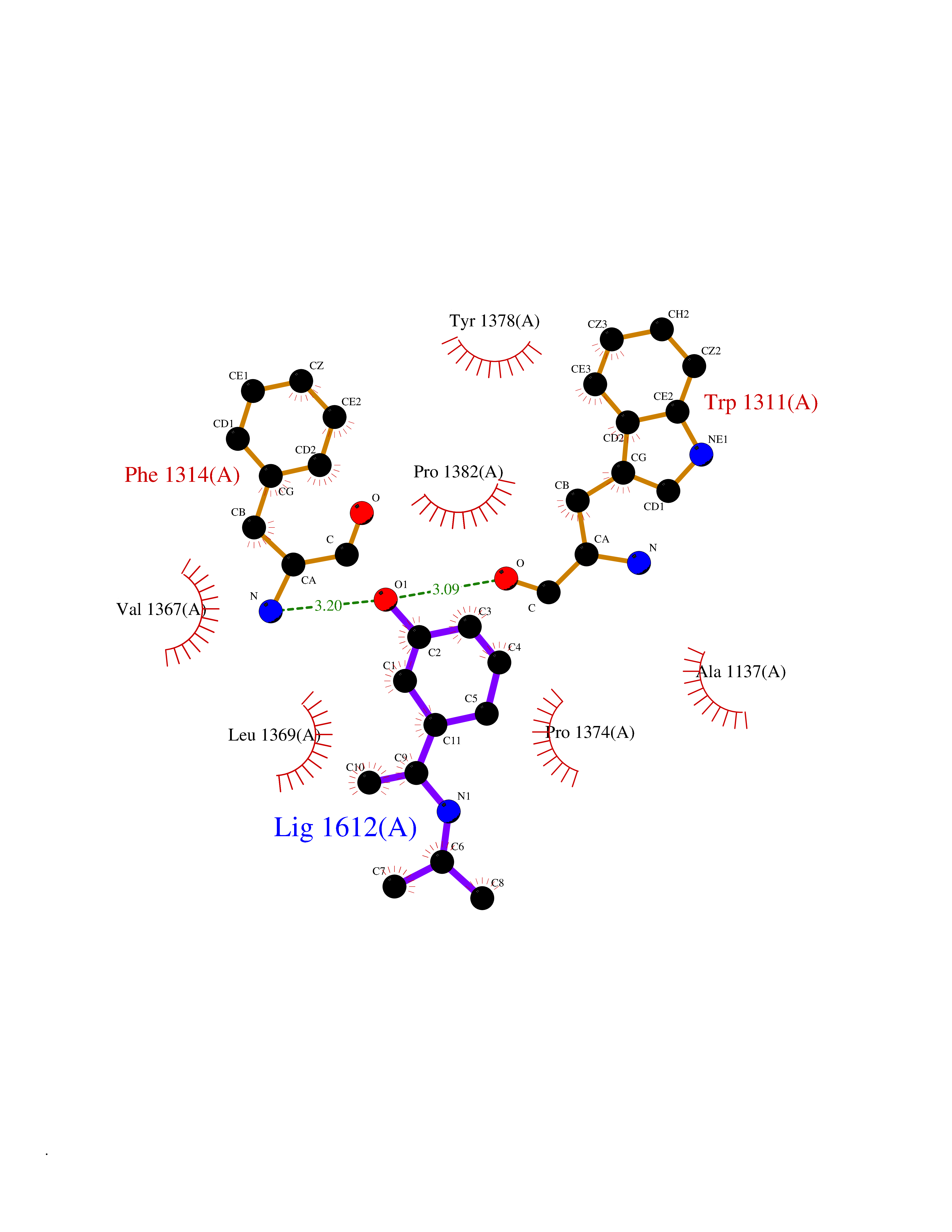 Ligplot Map 3D Binding mode Sequence IVDTCSLASPASVCRTKHLHLRCSVDFTRRTLTGTAALTVQSQEDNLRSLVLDTKDLTIEKVVINGQEVKYALGERQSYKGSPMEISLPIALSKNQEIVIEISFETSPKSSALQWLTPEQTSGKEHPYLFSQCQAIHCRAILPCQDTPSVKLTYTAEVSVPKELVALMSAIRDGETPDPEDPSRKIYKFIQKVPIPCYLIALVVGALESRQIGPRTLVWSEKEQVEKSAYEFSETESMLKIAEDLGGPYVWGQYDLLVLPPSFPYGGMENPCLTFVTPTLLAGDKSLSNVIAHEISHSWTGNLVTNKTWDHFWLNEGHTVYLERHICGRLFGEKFRHFNALGGWGELQNSVKTFGETHPFTKLVVDLTDIDPDVAYSSVPYEKGFALLFYLEQLLGGPEIFLGFLKAYVEKFSYKSITTDDWKDFLYSYFKDKVDVLNQVDWNAWLYSPGLPPIKPNYDMTLTNACIALSQRWITAKEDDLNSFNATDLKDLSSHQLNEFLAQTLQRAPLPLGHIKRMQEVYNFNAINNSEIRFRWLRLCIQSKWEDAIPLALKMATEQGRMKFTRPLFKDLAAFDKSHDQAVRTYQEHKASMHPVTAMLVGKDLKVD Hydrogen bonds contact Hydrophobic contact | ||||
| 9 | TRANSPORT INHIBITOR RESPONSE 1 protein | 2P1Q | 5.89 | |
Target general information Gen name IAA7 Organism Arabidopsis thaliana (Mouse-ear cress) Uniprot ID TTD ID NA Synonyms AXR2;At3g23050;MXC7.8 Protein family Aux/IAA family Biochemical class Signaling protein Function DNA binding transcription factor activity. Related diseases LTC4 synthase deficiency is associated with a neurometabolic developmental disorder characterized by muscular hypotonia, psychomotor retardation, failure to thrive, and microcephaly. {ECO:0000269|PubMed:10896305, ECO:0000269|PubMed:9820300}. Drugs (DrugBank ID) NA Interacts with Q9LW29; Q9C5W9; Q8RYC8; Q94AH6; Q9ZR12; P49677; Q38828; Q38829; Q38830; Q38831; O24407; O24408; O24409; P49678; O24410; Q8LAL2; Q9XFM0; Q38822; Q9M1R4; Q9C5X0; Q9C8Y3; Q39255; Q570C0 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Auxin signaling pathway; Nucleus; Reference proteome; Repressor; Transcription; Transcription regulation Protein physicochemical properties Chain ID B,C Molecular weight (Da) 65385.2 Length 581 Aromaticity 0.09 Instability index 47.83 Isoelectric point 7.46 Charge (pH=7) 1.23 2D Binding mode Binding energy (Kcal/mol) -8.03  Molscript Map  Pymol Map 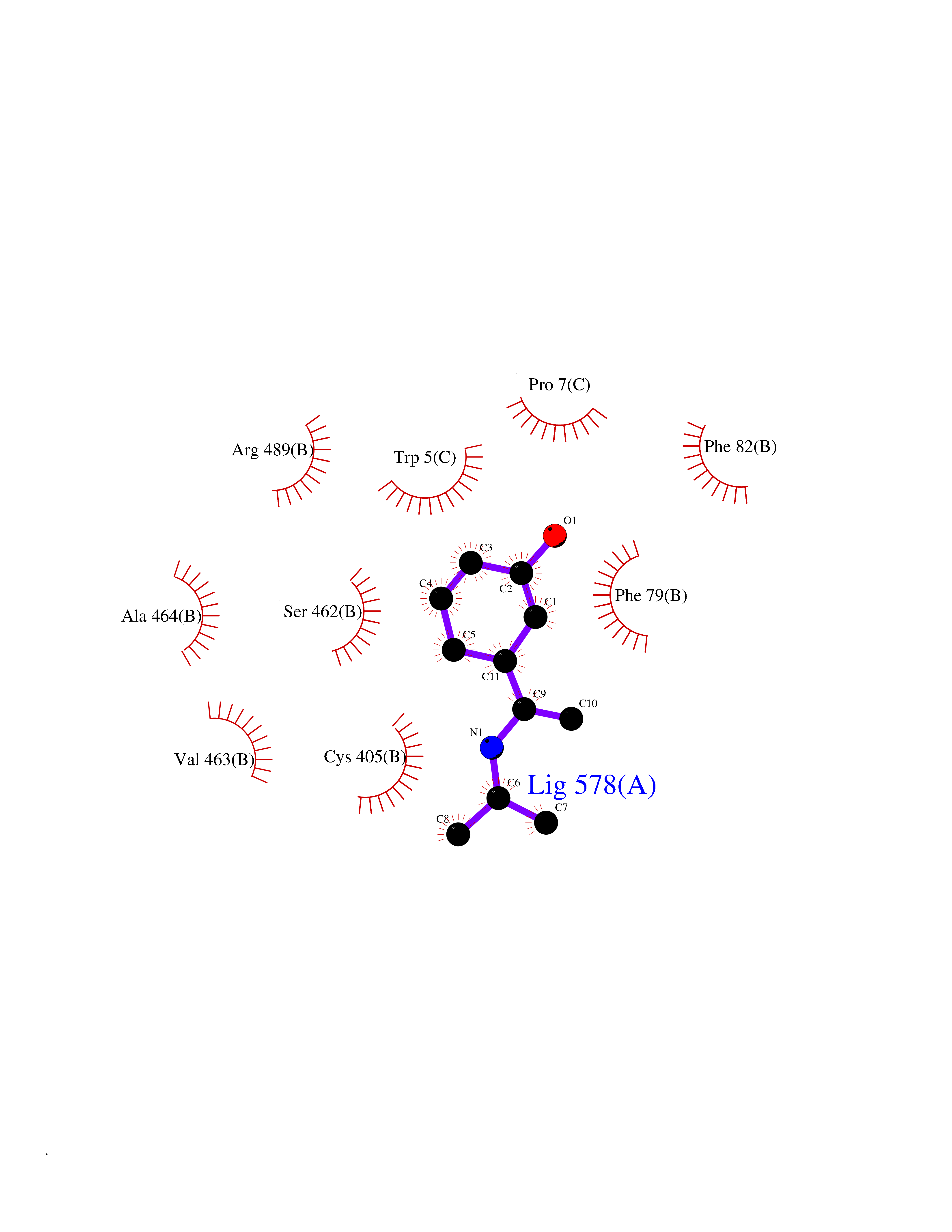 Ligplot Map 3D Binding mode Sequence QVVGWPPVRNYRKFPEEVLEHVFSFIQLDKDRNSVSLVCKSWYEIERWCRRKVFIGNCYAVSPATVIRRFPKVRSVELKGKPHFADFNLVPDGWGGYVYPWIEAMSSSYTWLEEIRLKRMVVTDDCLELIAKSFKNFKVLVLSSCEGFSTDGLAAIAATCRNLKELDLRESDVDDVSGHWLSHFPDTYTSLVSLNISCLASEVSFSALERLVTRCPNLKSLKLNRAVPLEKLATLLQRAPQLEELGTGGYTAEVRPDVYSGLSVALSGCKELRCLSGFWDAVPAYLPAVYSVCSRLTTLNLSYATVQSYDLVKLLCQCPKLQRLWVLDYIEDAGLEVLASTCKDLRELRVFPSEPFVMEPNVALTEQGLVSVSMGCPKLESVLYFCRQMTNAALITIARNRPNMTRFRLCIIEPKAPDYLTLEPLDIGFGAIVEHCKDLRRLSLSGLLTDKVFEYIGTYAKKMEMLSVAFAGDSDLGMHHVLSGCDSLRKLEIRDCPFGDKALLANASKLETMRSLWMSSCSVSFGACKLLGQKMPKLNVEVIDERGAPDSRPESCPVERVFIYRTVAGPRFDMPGFVWNM Hydrogen bonds contact Hydrophobic contact | ||||
| 10 | Plasmodium Dihydroorotate dehydrogenase (Malaria DHOdehase) | 1TV5 | 5.89 | |
Target general information Gen name Malaria DHOdehase Organism Plasmodium falciparum (isolate 3D7) Uniprot ID TTD ID Synonyms PFF0160c; Mitochondrially bound dihydroorotate-ubiqui oxidoreductase; Dihydroorotate oxidase of Plasmodium falciparum; Dihydroorotate dehydrogenase of Plasmodium falciparum; DHOdehase of Plasmodium fa Protein family Dihydroorotate dehydrogenase family, Type 2 subfamily Biochemical class CH-CH donor oxidoreductase Function Catalyzes the conversion of dihydroorotate to orotate with quinone as electron acceptor. Related diseases Combined oxidative phosphorylation deficiency 33 (COXPD33) [MIM:617713]: An autosomal recessive disorder caused by multiple mitochondrial respiratory chain defects and impaired mitochondrial energy metabolism. Clinical manifestations are highly variable. Affected infants present with cardiomyopathy accompanied by multisystemic features involving liver, kidney, and brain. Death in infancy is observed in some patients. Children and adults present with myopathy and progressive external ophthalmoplegia. {ECO:0000269|PubMed:28942965}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01117 Interacts with NA EC number EC 1.3.5.2 Uniprot keywords 3D-structure; Flavoprotein; FMN; Membrane; Mitochondrion; Mitochondrion inner membrane; Oxidoreductase; Pyrimidine biosynthesis; Reference proteome; Transit peptide; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 41846.8 Length 371 Aromaticity 0.1 Instability index 37.25 Isoelectric point 8.21 Charge (pH=7) 3.13 2D Binding mode Binding energy (Kcal/mol) -8.03 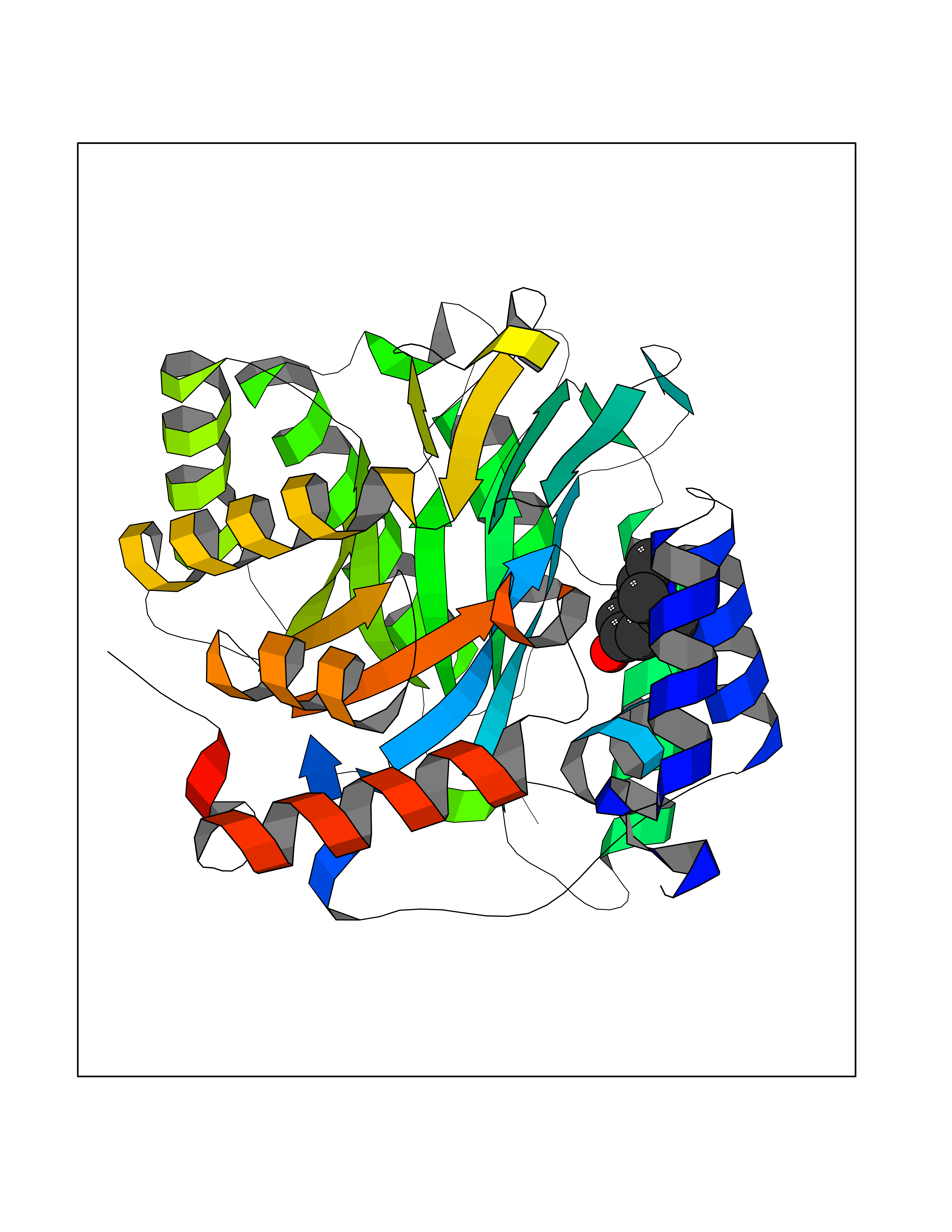 Molscript Map 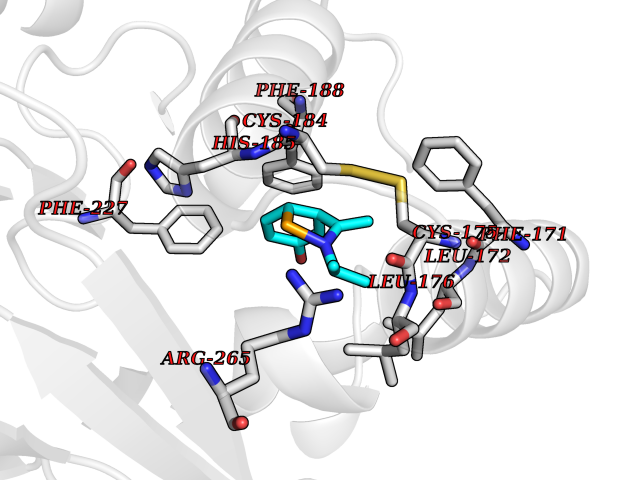 Pymol Map 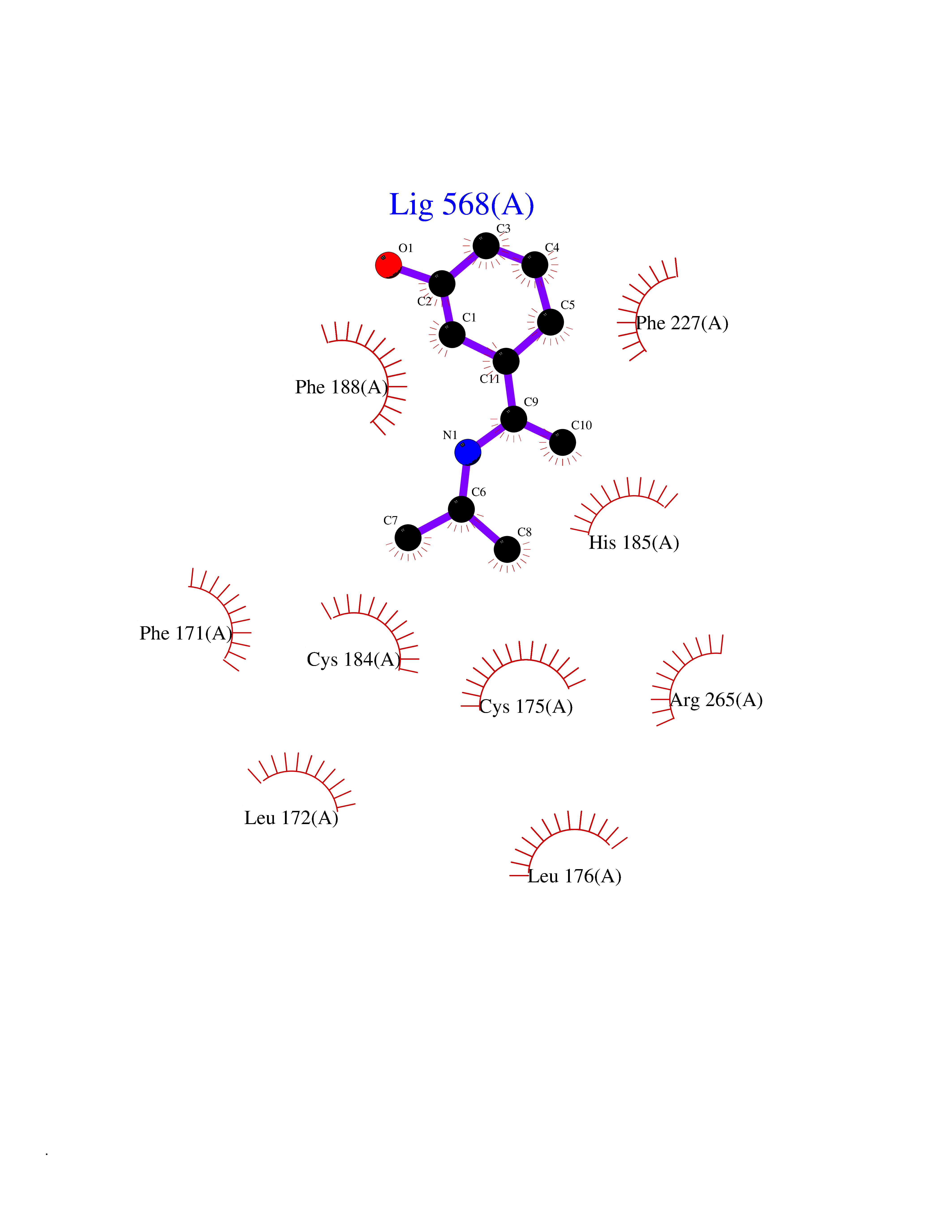 Ligplot Map 3D Binding mode Sequence FESYNPEFFLYDIFLKFCLKYIDGEICHDLFLLLGKYNILPYDTSNDSIYACTNIKHLDFINPFGVAAGFDKNGVCIDSILKLGFSFIEIGTITPRGQTGNAKPRIFRDVESRSIINSCGFNNMGCDKVTENLILFRKRQEEDKLLSKHIVGVSIGKNKDTVNIVDDLKYCINKIGRYADYIAINVSSPNTPGLRDNQEAGKLKNIILSVKEEIDNLEFLWFNTTKKKPLVFVKLAPDLNQEQKKEIADVLLETNIDGMIISNTTTQINDIKSFENKKGGVSGAKLKDISTKFICEMYNYTNKQIPIIASGGIFSGLDALEKIEAGASVCQLYSCLVFNGMKSAVQIKRELNHLLYQRGYYNLKEAIGRKH Hydrogen bonds contact Hydrophobic contact | ||||
| 11 | Neuronal acetylcholine receptor alpha-4 (CHRNA4) | 6CNJ | 5.87 | |
Target general information Gen name CHRNA4 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Nicotinic acetylcholine receptor alpha4; CHRNA4; Alpha-4 nAChR Protein family Ligand-gated ion channel (TC 1.A.9) family, Acetylcholine receptor (TC 1.A.9.1) subfamily, Alpha-4/CHRNA4 sub-subfamily Biochemical class Neurotransmitter receptor Function After binding acetylcholine, the AChR responds by an extensive change in conformation that affects all subunits and leads to opening of an ion-conducting channel across the plasmamembrane permeable to sodium ions. Related diseases Epilepsy, nocturnal frontal lobe, 1 (ENFL1) [MIM:600513]: An autosomal dominant focal epilepsy characterized by nocturnal seizures with hyperkinetic automatisms and poorly organized stereotyped movements. {ECO:0000269|PubMed:10563623, ECO:0000269|PubMed:14623738, ECO:0000269|PubMed:7550350}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00915; DB01351; DB01352; DB00572; DB01483; DB00237; DB00241; DB01353; DB00564; DB00565; DB09028; DB01245; DB00514; DB01496; DB07720; DB00783; DB13952; DB13953; DB13954; DB13955; DB13956; DB00898; DB01354; DB01355; DB00753; DB00657; DB00333; DB00463; DB00849; DB00184; DB00312; DB01174; DB00981; DB05458; DB00794; DB05740; DB00747; DB00418; DB00202; DB00306; DB00599; DB01273 Interacts with Q6UY14-3; P05067; P83916; Q6UXH1-1; Q6UXH1-3; P20042; Q9NZR2; Q92673; P17787 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disease variant; Disulfide bond; Epilepsy; Glycoprotein; Ion channel; Ion transport; Ligand-gated ion channel; Lipoprotein; Membrane; Palmitate; Phosphoprotein; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Signal; Synapse; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A,B Molecular weight (Da) 84601.2 Length 728 Aromaticity 0.13 Instability index 39.72 Isoelectric point 5.86 Charge (pH=7) -9.84 2D Binding mode Binding energy (Kcal/mol) -8 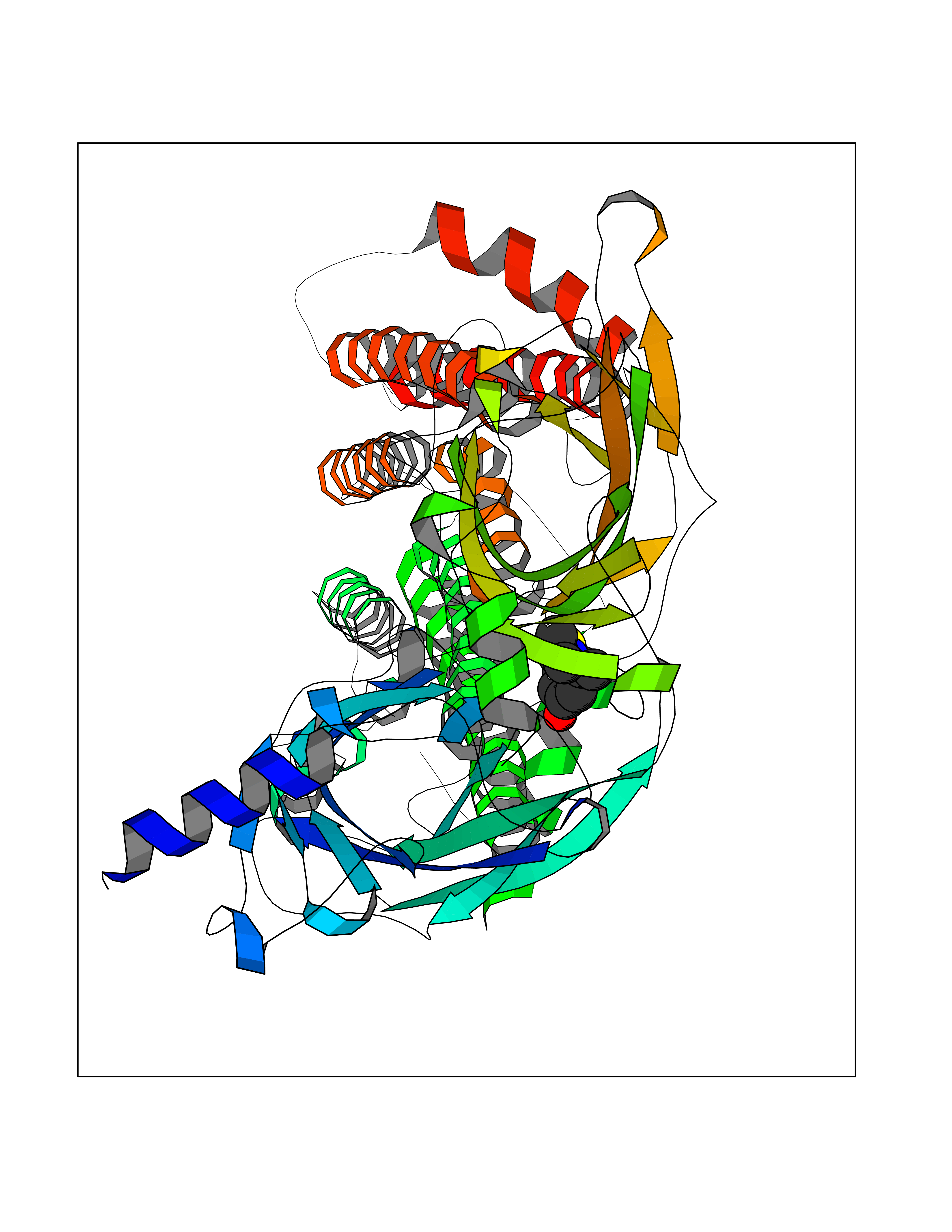 Molscript Map 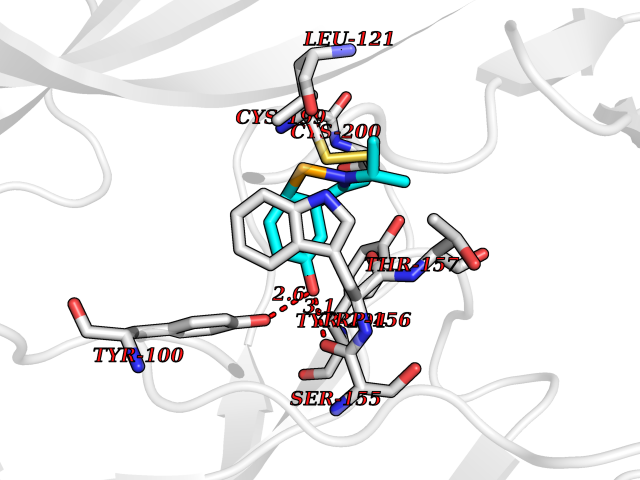 Pymol Map 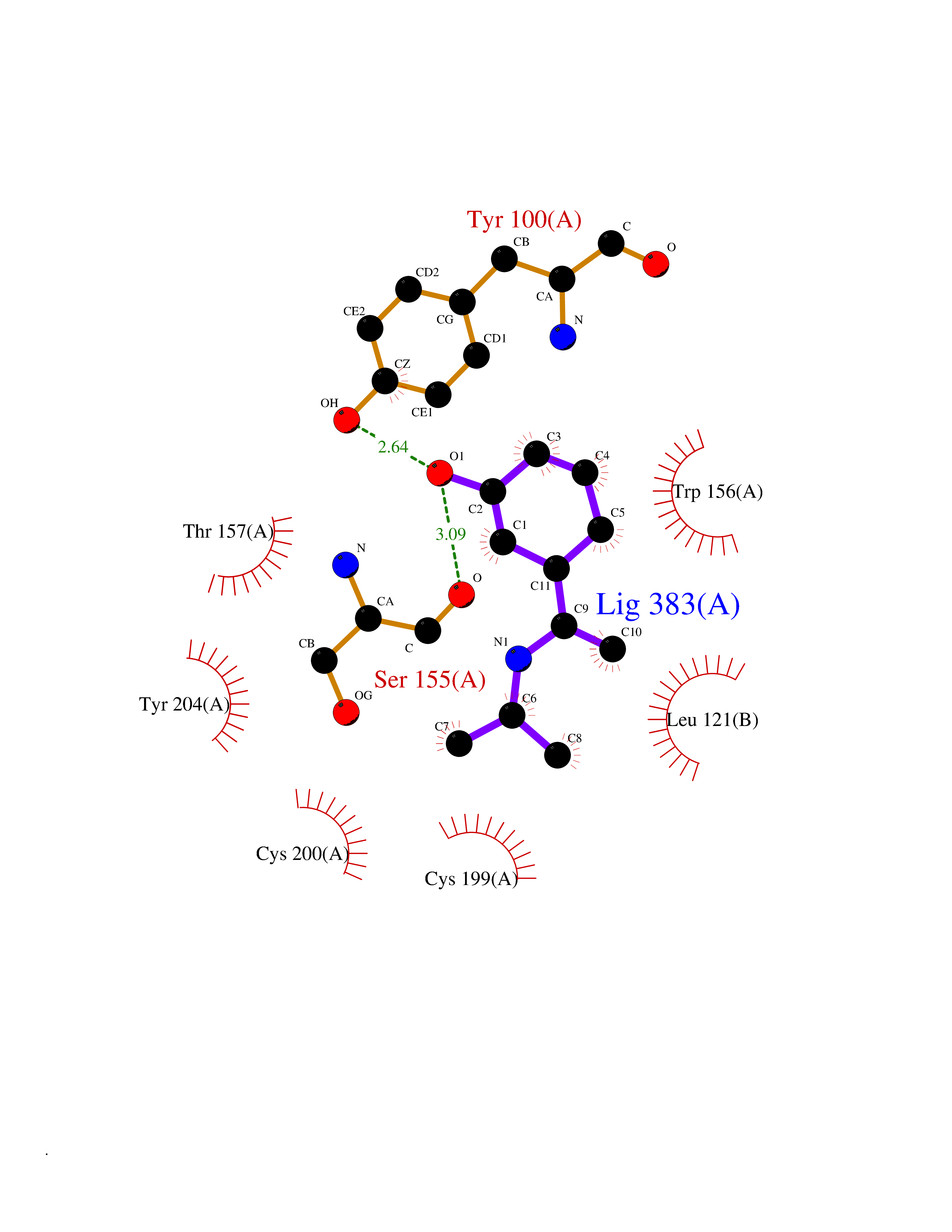 Ligplot Map 3D Binding mode Sequence ETRAHAEERLLKKLFSGYNKWSRPVANISDVVLVRFGLSIAQLIDVDEKNQMMTTNVWVKQEWHDYKLRWDPADYENVTSIRIPSELIWRPDIVLYNNADGDFAVTHLTKAHLFHDGRVQWTPPAIYKSSCSIDVTFFPFDQQNCTMKFGSWTYDKAKIDLVNMHSRVDQLDFWESGEWVIVDAVGTYNTRKYECCAEIYPDITYAFVIRRLPLFYTINLIIPCLLISCLTVLVFYLPSECGEKITLCISVLLSLTVFLLLITEIIPSTSLVIPLIGEYLLFTMIFVTLSIVITVFVLNVHHRSPRTHTMPTWVRRVFLDIVPRLLLMKRFERSVKEDWKYVAMVIDRIFLWMFIIVCLLGTVGLFLPPWDTEERLVEHLLDPSRYNKLIRPATNGSELVTVQLMVSLAQLISVHEREQIMTTNVWLTQEWEDYRLTWKPEEFDNMKKVRLPSKHIWLPDVVLYNNADGMYEVSFYSNAVVSYDGSIFWLPPAIYKSACKIEVKHFPFDQQNCTMKFRSWTYDRTEIDLVLKSEVASLDDFTPSGEWDIVALPGRRNENPDDSTYVDITYDFIIRRKPLFYTINLIIPCVLITSLAILVFYLPSDCGEKMTLCISVLLALTVFLLLISKIVPPTSLDVPLVGKYLMFTMVLVTFSIVTSVCVLNVHHRSPTTHTMAPWVKVVFLEKLPALLFMQQSVSEDWKYVAMVIDRLFLWIFVFVCVFGTIGMF Hydrogen bonds contact Hydrophobic contact | ||||
| 12 | "Periplasmic trehalase (EC 3.2.1.28) (Alpha,alpha-trehalase) (Alpha,alpha-trehalose glucohydrolase) (Tre37A)" | 2JG0 | 5.85 | |
Target general information Gen name treA Organism Escherichia coli (strain K12) Uniprot ID TTD ID NA Synonyms JW1186;osmA;b1197 Protein family Glycosyl hydrolase 37 family Biochemical class NA Function Provides the cells with the ability to utilize trehalose at high osmolarity by splitting it into glucose molecules that can subsequently be taken up by the phosphotransferase-mediated uptake system. Related diseases SRC kinase activity has been shown to be increased in several tumor tissues and tumor cell lines such as colon carcinoma cells. {ECO:0000269|PubMed:2498394, ECO:0000269|PubMed:3093483}.; DISEASE: Thrombocytopenia 6 (THC6) [MIM:616937]: A form of thrombocytopenia, a hematologic disorder defined by a decrease in the number of platelets in circulating blood, resulting in the potential for increased bleeding and decreased ability for clotting. THC6 is an autosomal dominant form. Affected individuals may also have bone abnormalities and an increased risk for myelofibrosis. {ECO:0000269|PubMed:26936507}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number 3.2.1.28 Uniprot keywords 3D-structure; Direct protein sequencing; Glycosidase; Hydrolase; Periplasm; Reference proteome; Signal Protein physicochemical properties Chain ID A Molecular weight (Da) 57508.9 Length 507 Aromaticity 0.11 Instability index 48.32 Isoelectric point 5.48 Charge (pH=7) -10.13 2D Binding mode Binding energy (Kcal/mol) -7.98 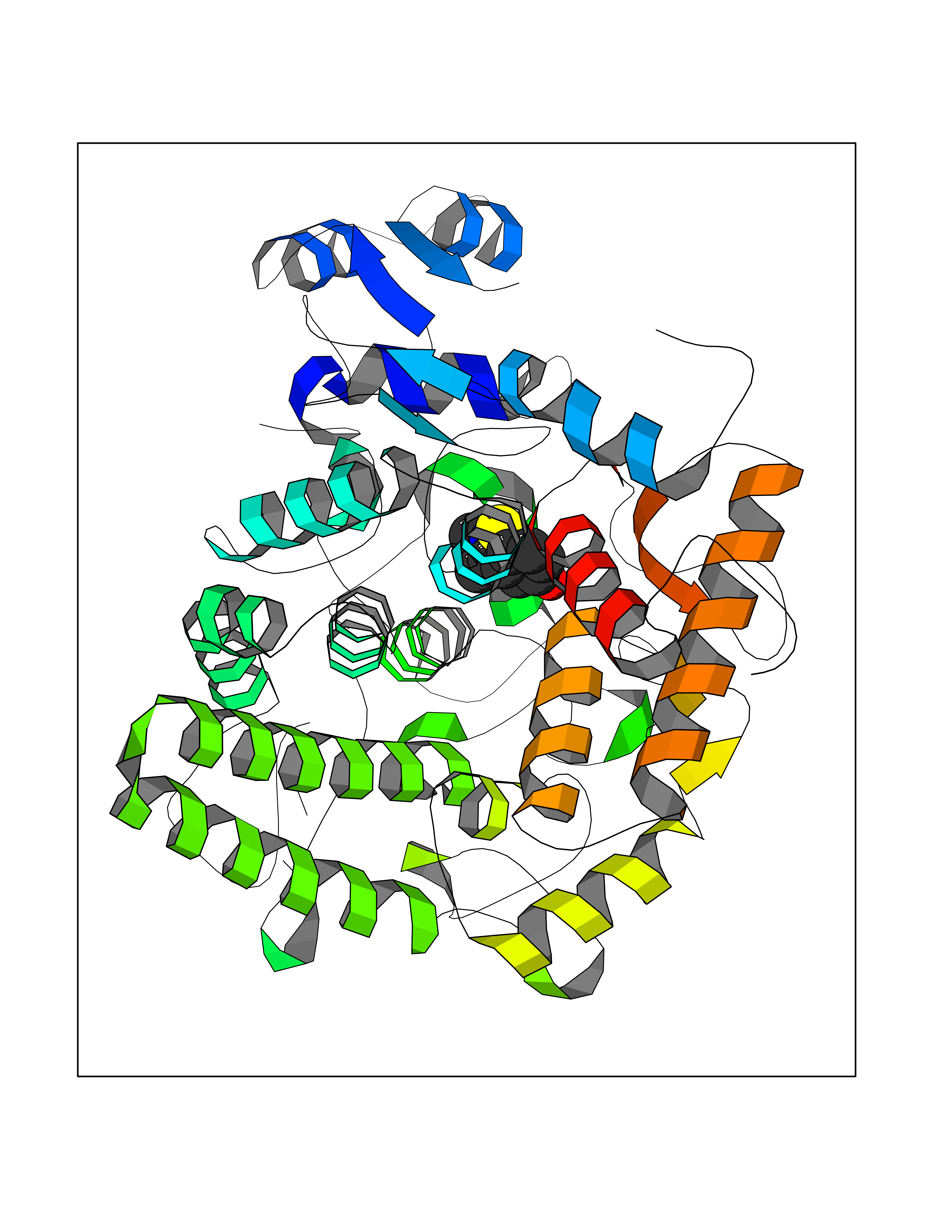 Molscript Map 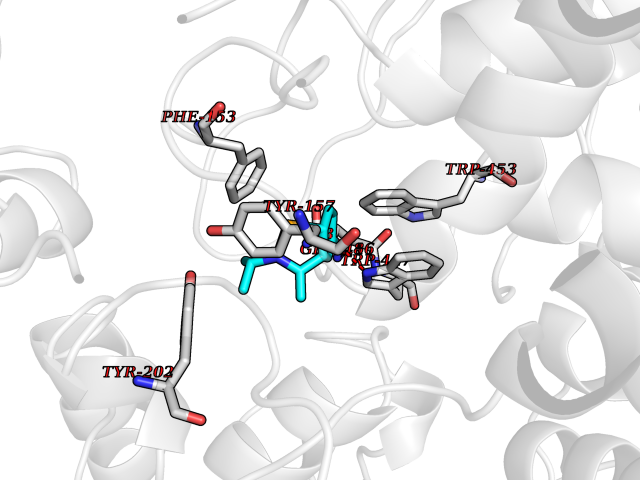 Pymol Map 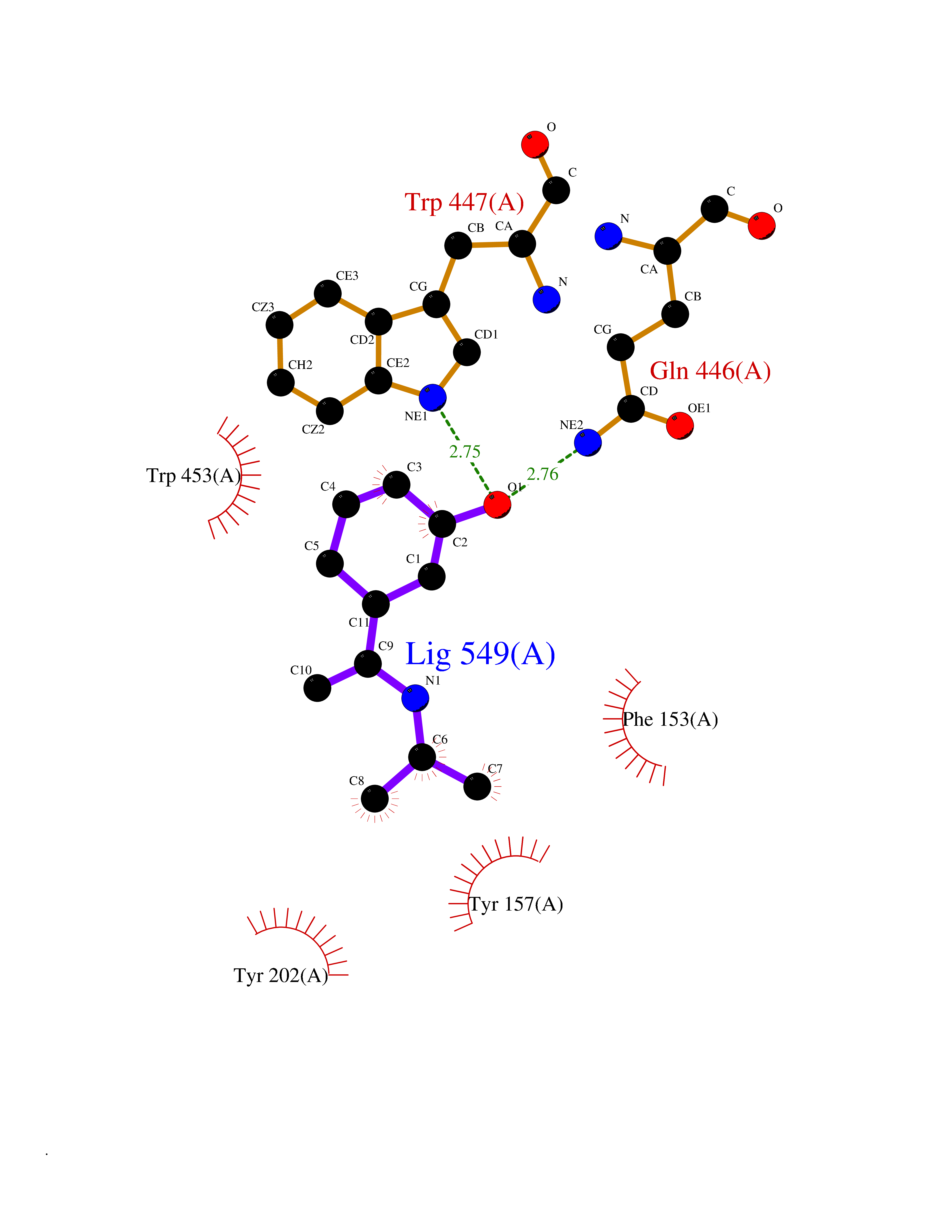 Ligplot Map 3D Binding mode Sequence PQPPDILLGPLFNDVQNAKLFPDQKTFADAVPNSDPLMILADYRMQQNQSGFDLRHFVNVNFTLPKYVPPEGQSLREHIDGLWPVLTRSTENTEKWDSLLPLPEPYVVPGGRFREVYYWDSYFTMLGLAESGHWDKVADMVANFAHEIDTYGHIPNGNRSYYLSRSQPPFFALMVELLAQHEGDAALKQYLPQMQKEYAYWMDGVENLQAGQQEKRVVKLQDGTLLNRYWDDRDTPRPESWVEDIATAKSNPNRPATEIYRDLRSAAASGWDFSSRWMDNPQQLNTLRTTSIVPVDLNSLMFKMEKILARASKAAGDNAMANQYETLANARQKGIEKYLWNDQQGWYADYDLKSHKVRNQLTAAALFPLYVNAAAKDRANKMATATKTHLLQPGGLNTTSVKSGQQWDAPNGWAPLQWVATEGLQNYGQKEVAMDISWHFLTNVQHTYDREKKLVEKYDVSTTGTGGGGGEYPLQDGFGWTNGVTLKMLDLICPKEQPCDNVPATRP Hydrogen bonds contact Hydrophobic contact | ||||
| 13 | Helicobacter pylori Methylthioadenosine nucleosidase (HELPY mtnN) | 4BMZ | 5.85 | |
Target general information Gen name HELPY mtnN Organism Helicobacter pylori (strain ATCC 700392 / 26695) (Campylobacter pylori) Uniprot ID TTD ID Synonyms MTAN; MTA/SAH nucleosidase; Aminofutalosine nucleosidase; Aminodeoxyfutalosine nucleosidase; AFL nucleosidase; 6-amino-6-deoxyfutalosine N-ribosylhydrolase; 5'-methylthioadenosine/S-adenosylhomocystei Protein family PNP/UDP phosphorylase family Biochemical class NA Function Catalyzes the direct conversion of aminodeoxyfutalosine (AFL) into dehypoxanthine futalosine (DHFL) and adenine via the hydrolysis of the N-glycosidic bond; this reaction seems to represent an essential step in the menaquinone biosynthesis pathway in Helicobacter species. Can also probably catalyzes the hydrolysis of 5'-methylthioadenosine (MTA) and S-adenosylhomocysteine (SAH) to adenine and the corresponding thioribose, 5'-methylthioribose and S-ribosylhomocysteine, respectively. These other activities highlight the tremendous versatility of the enzyme, which also plays key roles in S-adenosylmethionine recycling and in the biosynthesis of the quorum-sensing molecule autoinducer-2. Does not act on futalosine (FL) as substrate. Related diseases Progressive familial heart block 1B (PFHB1B) [MIM:604559]: A cardiac bundle branch disorder characterized by progressive alteration of cardiac conduction through the His-Purkinje system, with a pattern of a right bundle-branch block and/or left anterior hemiblock occurring individually or together. It leads to complete atrio-ventricular block causing syncope and sudden death. {ECO:0000269|PubMed:19726882, ECO:0000269|PubMed:20562447, ECO:0000269|PubMed:21887725}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Erythrokeratodermia variabilis et progressiva 6 (EKVP6) [MIM:618531]: A form of erythrokeratodermia variabilis et progressiva, a genodermatosis characterized by the coexistence of two independent skin lesions: transient erythema and hyperkeratosis that is usually localized but occasionally occurs in its generalized form. Clinical presentation varies significantly within a family and from one family to another. Palmoplantar keratoderma is present in around 50% of cases. EKVP6 inheritance is autosomal dominant. {ECO:0000269|PubMed:30528822}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number NA Uniprot keywords 3D-structure; Amino-acid biosynthesis; Hydrolase; Menaquinone biosynthesis; Methionine biosynthesis; Reference proteome Protein physicochemical properties Chain ID A,B Molecular weight (Da) 50547.6 Length 464 Aromaticity 0.08 Instability index 26.92 Isoelectric point 5.13 Charge (pH=7) -20.92 2D Binding mode Binding energy (Kcal/mol) -7.98  Molscript Map 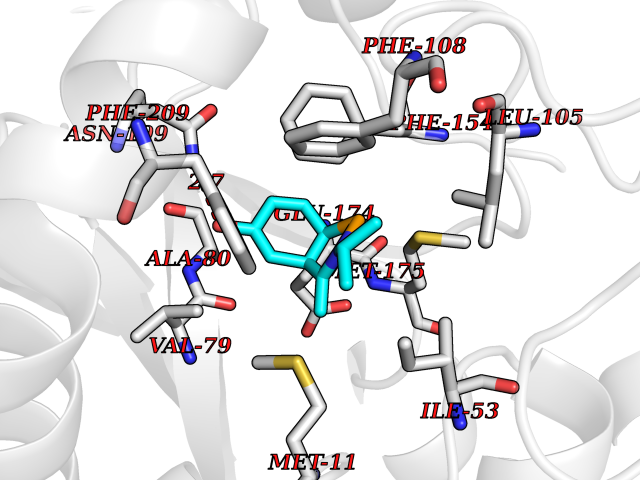 Pymol Map 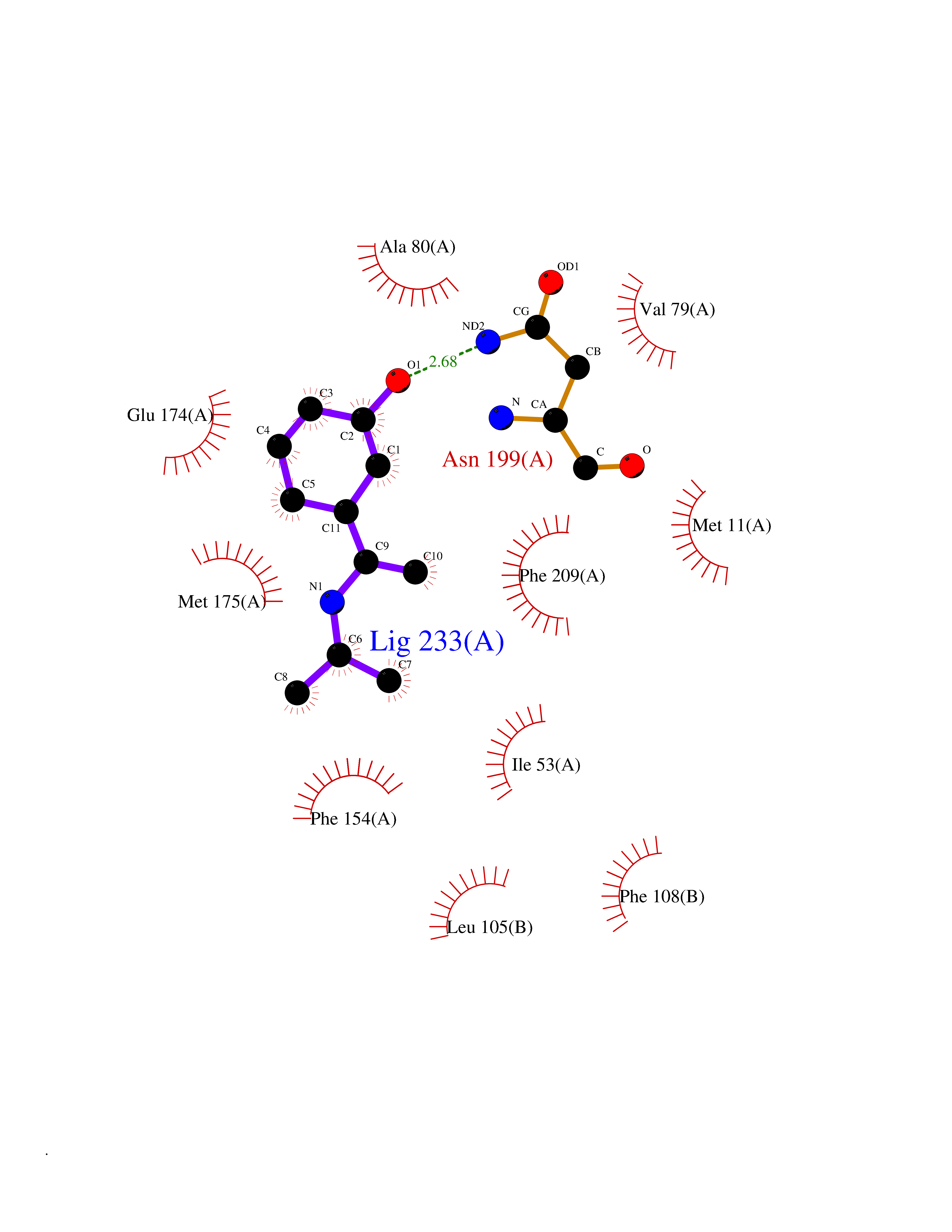 Ligplot Map 3D Binding mode Sequence VQKIGILGAMREEITPILELFGVDFEEIPLGGNVFHKGVYHNKEIIVAYSKIGKVHSTLTTTSMILAFGVQKVLFSGVAGSLVKDLKINDLLVAIQLVQHDVDLSAFDHPLGFIPESAIFIETSESLNALAKEVANEQHIVLKEGVIASGDQFVHSKERKEFLVSEFKASAVEMEGASVAFVCQKFGVPCCVLRSISNNADEEANMSFDAFLEKSAQTSAKFLKSMVDELGSHMVQKIGILGAMREEITPILELFGVDFEEIPLGGNVFHKGVYHNKEIIVAYSKIGKVHSTLTTTSMILAFGVQKVLFSGVAGSLVKDLKINDLLVAIQLVQHDVDLSAFDHPLGFIPESAIFIETSESLNALAKEVANEQHIVLKEGVIASGDQFVHSKERKEFLVSEFKASAVEMEGASVAFVCQKFGVPCCVLRSISNNADEEANMSFDAFLEKSAQTSAKFLKSMVDEL Hydrogen bonds contact Hydrophobic contact | ||||
| 14 | Gamma-butyrobetaine dioxygenase | 4C5W | 5.84 | |
Target general information Gen name BBOX1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms BBH;BBOX Protein family Gamma-BBH/TMLD family Biochemical class Oxidoreductase Function Gamma-butyrobetaine dioxygenase activity.Identical protein binding.Iron ion binding.Zinc ion binding. Related diseases Neurodevelopmental disorder with poor language and loss of hand skills (NDPLHS) [MIM:617903]: An autosomal dominant disorder characterized by psychomotor developmental stagnation or regression. NDPLHS manifest in the first years of life as loss of purposeful hand movements, loss of language, and intellectual disability. {ECO:0000269|PubMed:26740508, ECO:0000269|PubMed:28856709, ECO:0000269|PubMed:29369404}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Developmental and epileptic encephalopathy 59 (DEE59) [MIM:617904]: A form of epileptic encephalopathy, a heterogeneous group of severe early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE59 is an autosomal dominant condition characterized by onset of refractory seizures in early infancy. {ECO:0000269|PubMed:28856709, ECO:0000269|PubMed:29100083, ECO:0000269|PubMed:29369404}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00126 Interacts with O75936; A0MZ66-7 EC number 1.14.11.1 Uniprot keywords 3D-structure; Carnitine biosynthesis; Cytoplasm; Dioxygenase; Iron; Metal-binding; Oxidoreductase; Phosphoprotein; Proteomics identification; Reference proteome; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 31642.5 Length 275 Aromaticity 0.12 Instability index 35.68 Isoelectric point 6.33 Charge (pH=7) -2.46 2D Binding mode Binding energy (Kcal/mol) -7.96 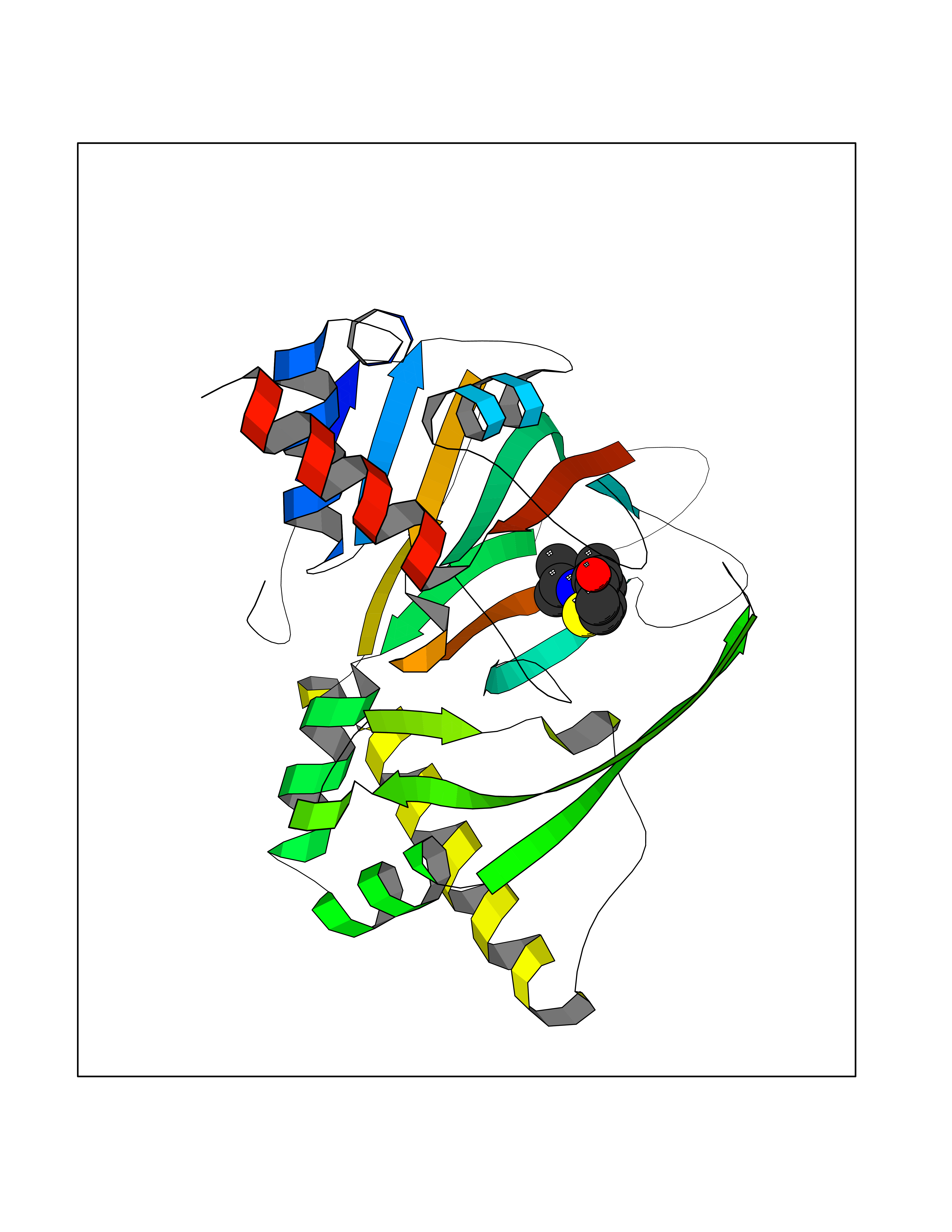 Molscript Map 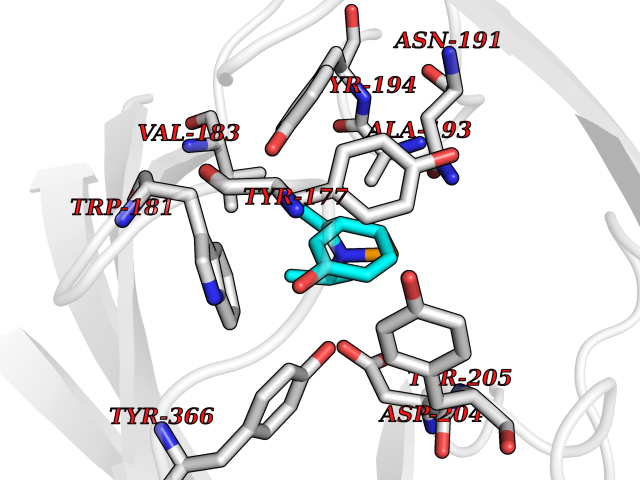 Pymol Map 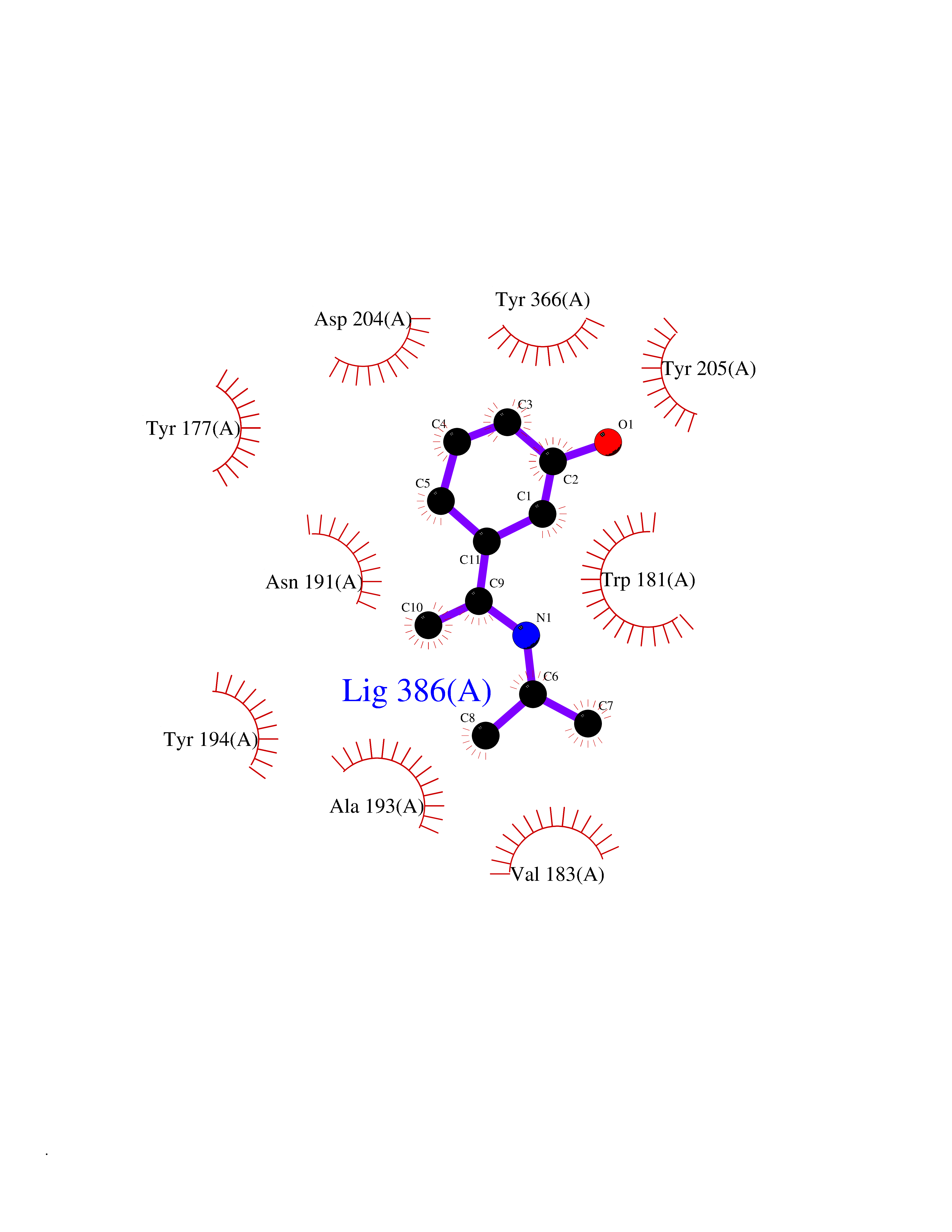 Ligplot Map 3D Binding mode Sequence FPECQYWGSELQLPTLDFEDVLRYDEHAYKWLSTLKKVGIVRLTGASDKPGEVSKLGKRMGFLYLTFYGHTWQVQDKIDANNVAYTTGKLSFHTDYPALHHPPGVQLLHCIKQTVTGGDSEIVDGFNVCQKLKKNNPQAFQILSSTFVDFTDIGVDYCDFSVQSKHKIIELDDKGQVVRINFNNATRDTIFDVPVERVQPFYAALKEFVDLMNSKESKFTFKMNPGDVITFDNWRLLHGRRSYEAGTEISRHLEGAYADWDVVMSRLRILRQRVE Hydrogen bonds contact Hydrophobic contact | ||||
| 15 | Plasmepsin-2 | 2BJU | 5.84 | |
Target general information Gen name N/A Organism Plasmodium falciparum (isolate HB3) Uniprot ID TTD ID NA Synonyms NA Protein family Peptidase A1 family Biochemical class Hydrolase Function Aspartic-type endopeptidase activity. Related diseases Short/branched-chain acyl-CoA dehydrogenase deficiency (SBCADD) [MIM:610006]: Autosomal recessive disorder and consists of a defect in catabolism of L-isoleucine which is characterized by an increase of 2-methylbutyrylglycine and 2-methylbutyrylcarnitine in blood and urine. Affected individuals have seizures and psychomotor delay as the main clinical features. {ECO:0000269|PubMed:10832746, ECO:0000269|PubMed:11013134, ECO:0000269|PubMed:16317551}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB04378; DB04373; DB11638; DB01218; DB02505; DB03063 Interacts with NA EC number 3.4.23.39 Uniprot keywords 3D-structure; Aspartyl protease; Direct protein sequencing; Disulfide bond; Hydrolase; Membrane; Protease; Reference proteome; Signal-anchor; Transmembrane; Transmembrane helix; Vacuole; Zymogen Protein physicochemical properties Chain ID A Molecular weight (Da) 36923.5 Length 329 Aromaticity 0.13 Instability index 44.31 Isoelectric point 4.67 Charge (pH=7) -17.94 2D Binding mode Binding energy (Kcal/mol) -7.96 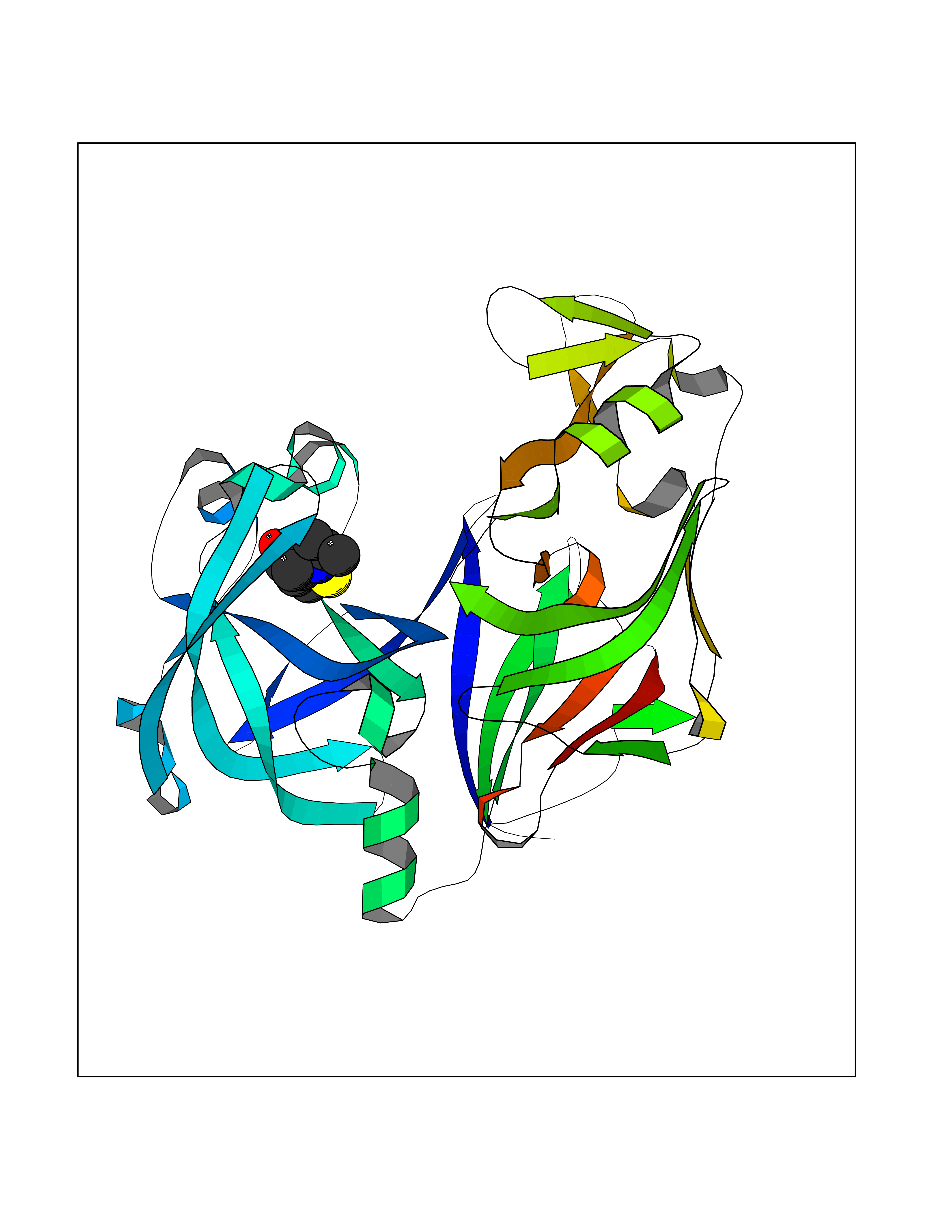 Molscript Map 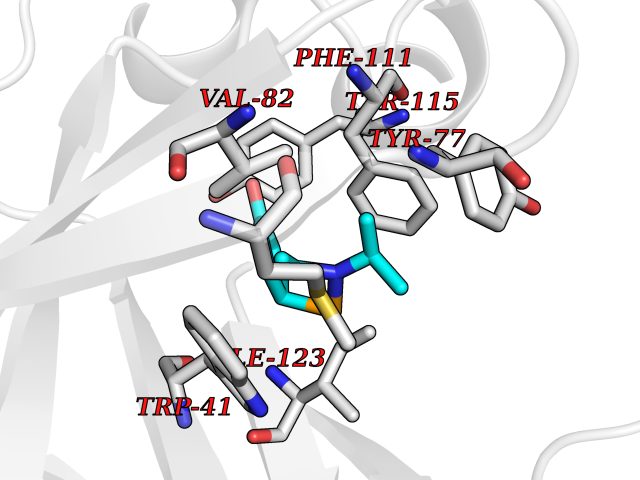 Pymol Map 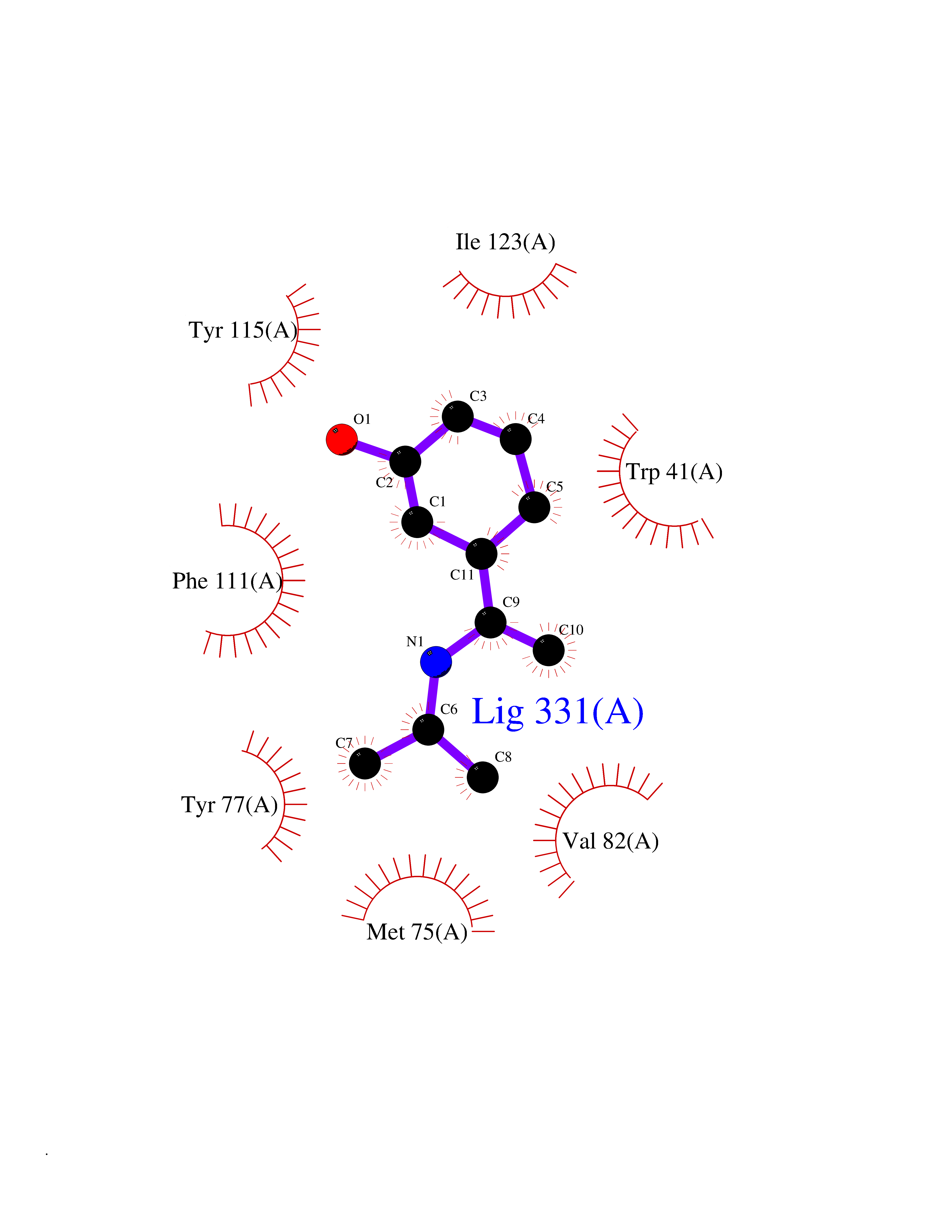 Ligplot Map 3D Binding mode Sequence SSNDNIELVDFQNIMFYGDAEVGDNQQPFTFILDTGSANLWVPSVKCTTAGCLTKHLYDSSKSRTYEKDGTKVEMNYVSGTVSGFFSKDLVTVGNLSLPYKFIEVIDTNGFEPTYTASTFDGILGLGWKDLSIGSVDPIVVELKNQNKIENALFTFYLPVHDKHTGFLTIGGIEERFYEGPLTYEKLNHDLYWQITLDAHVGNIMLEKANCIVDSGTSAITVPTDFLNKMLQNLDVIKVPFLPFYVTLCNNSKLPTFEFTSENGKYTLEPEYYLQHIEDVGPGLCMLNIIGLDFPVPTFILGDPFMRKYFTVFDYDNHSVGIALAKKNL Hydrogen bonds contact Hydrophobic contact | ||||
| 16 | Gamma-aminobutyric acid receptor subunit beta-3 | 4COF | 5.83 | |
Target general information Gen name GABRB3 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Ligand-gated ion channel (TC 1.A.9) family, Gamma-aminobutyric acid receptor (TC 1.A.9.5) subfamily, GABRB3 sub-subfamily Biochemical class Transport protein Function GABA-A receptor activity.GABA-gated chloride ion channel activity.Identical protein binding. Related diseases Epilepsy, childhood absence 5 (ECA5) [MIM:612269]: A subtype of idiopathic generalized epilepsy characterized by an onset at age 6-7 years, frequent absence seizures (several per day) and bilateral, synchronous, symmetric 3-Hz spike waves on EEG. Tonic-clonic seizures often develop in adolescence. Absence seizures may either remit or persist into adulthood. {ECO:0000269|PubMed:18514161, ECO:0000269|PubMed:22303015}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Developmental and epileptic encephalopathy 43 (DEE43) [MIM:617113]: A form of epileptic encephalopathy, a heterogeneous group of severe early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE43 inheritance is autosomal dominant. {ECO:0000269|PubMed:23934111, ECO:0000269|PubMed:25356899, ECO:0000269|PubMed:26950270, ECO:0000269|PubMed:26993267, ECO:0000269|PubMed:27476654, ECO:0000269|PubMed:27864847}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB12537; DB00546; DB00404; DB00543; DB11901; DB14719; DB11859; DB01558; DB09017; DB00237; DB00241; DB01489; DB00475; DB14715; DB01594; DB00349; DB01068; DB00628; DB01559; DB01553; DB01511; DB01189; DB00829; DB13837; DB00228; DB01215; DB00402; DB00898; DB00189; DB01545; DB09166; DB00292; DB01567; DB01205; DB01544; DB00690; DB06716; DB05087; DB01437; DB00801; DB01159; DB00753; DB01587; DB00555; DB00431; DB13643; DB00186; DB13872; DB13437; DB00603; DB01043; DB00371; DB00463; DB01028; DB01107; DB15489; DB00683; DB12458; DB01595; DB14028; DB00842; DB14672; DB00312; DB00252; DB13335; DB00592; DB01708; DB01588; DB00794; DB00818; DB01589; DB12404; DB01236; DB09118; DB00306; DB01956; DB00231; DB11582; DB00897; DB15490 Interacts with P28472 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Chloride; Chloride channel; Cytoplasmic vesicle; Direct protein sequencing; Disease variant; Disulfide bond; Epilepsy; Glycoprotein; Ion channel; Ion transport; Ligand-gated ion channel; Membrane; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Signal; Synapse; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A,B,C,D,E Molecular weight (Da) 76970.6 Length 663 Aromaticity 0.15 Instability index 39.4 Isoelectric point 8.37 Charge (pH=7) 4.12 2D Binding mode Binding energy (Kcal/mol) -7.95 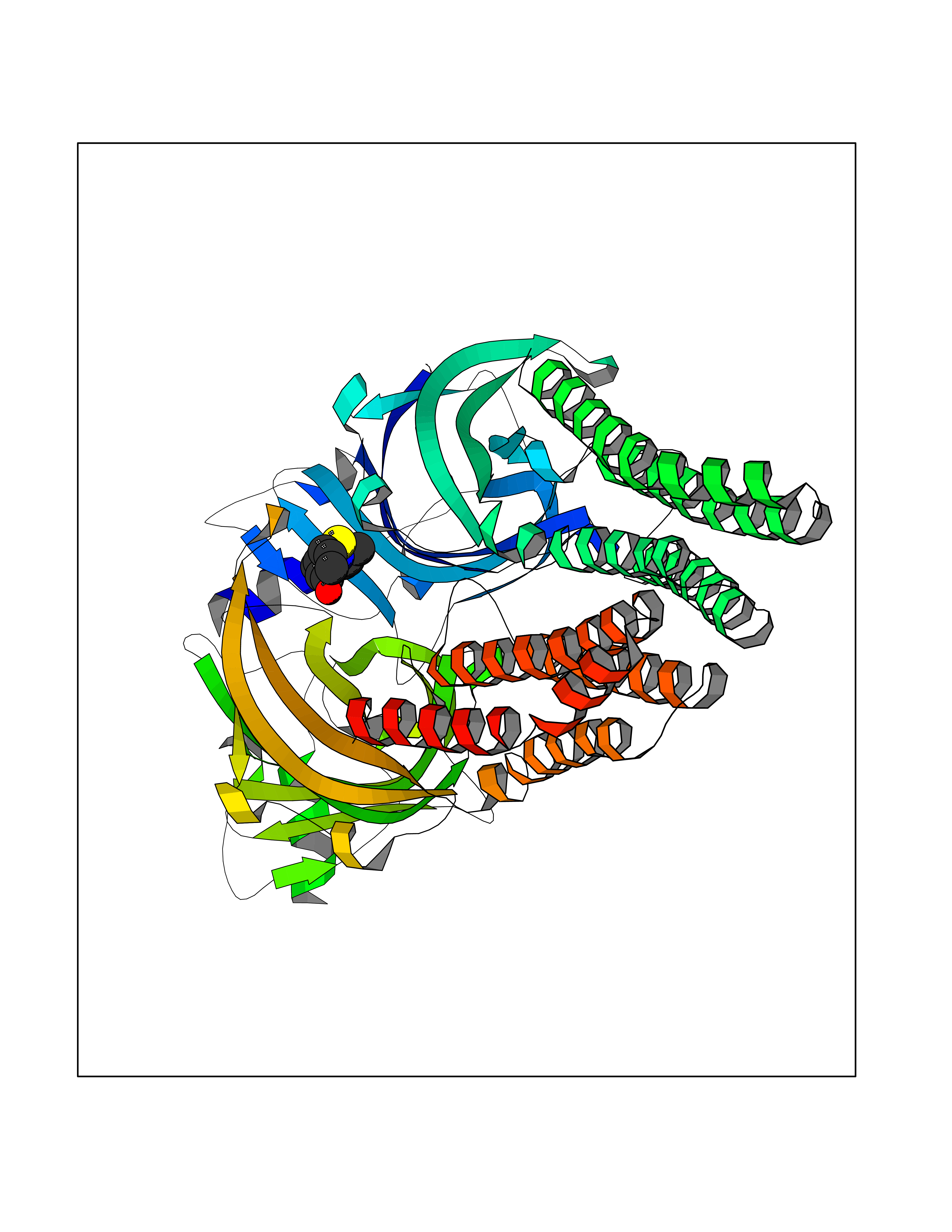 Molscript Map 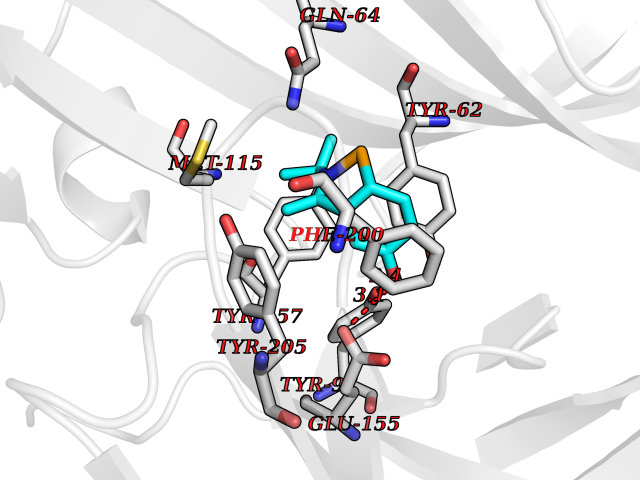 Pymol Map 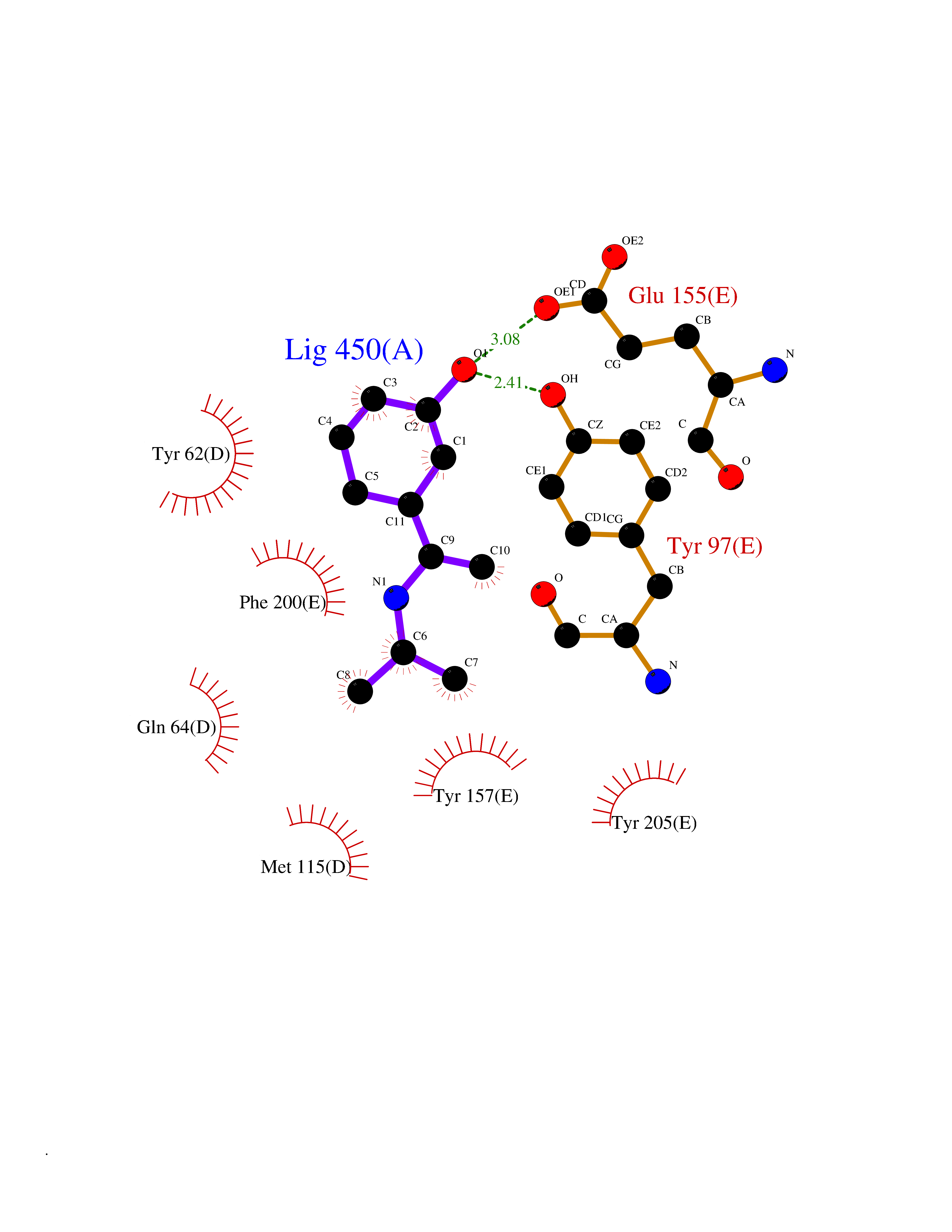 Ligplot Map 3D Binding mode Sequence SFVKETVDKLLKGYDIRLRPDFGGPPVCVGMNIDIASIDMVSEVNMDYTLTMYFQQYWRDKRLAYSGIPLNLTLDNRVADQLWVPDTYFLNDKKSFVHGVTVKNRMIRLHPDGTVLYGLRITTTAACMMDLRRYPLDEQNCTLEIESYGYTTDDIEFYWRGGDKAVTGVERIELPQFSIVEHRLVSRNVVFATGAYPRLSLSFRLKRNIGYFILQTYMPSILITILSWVSFWINYDASAARVALGITTVLTMTTINTHLRETLPKIPYVKAIDMYLMGCFVFVFLALLEYAFVNYIFFSQPARAAAIDRWSRIVFPFTFSLFNLVYWLYYVNSFVKETVDKLLKGYDIRLRPDFGGPPVCVGMNIDIASIDMVSEVNMDYTLTMYFQQYWRDKRLAYSGIPLNLTLDNRVADQLWVPDTYFLNDKKSFVHGVTVKNRMIRLHPDGTVLYGLRITTTAACMMDLRRYPLDEQNCTLEIESYGYTTDDIEFYWRGGDKAVTGVERIELPQFSIVEHRLVSRNVVFATGAYPRLSLSFRLKRNIGYFILQTYMPSILITILSWVSFWINYDASAARVALGITTVLTMTTINTHLRETLPKIPYVKAIDMYLMGCFVFVFLALLEYAFVNYIFFSQPARAAAIDRWSRIVFPFTFSLFNLVYWLYYV Hydrogen bonds contact Hydrophobic contact | ||||
| 17 | Histamine N-methyltransferase (HNMT) | 2AOT | 5.83 | |
Target general information Gen name HNMT Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Histamine-N-methyltransferase; HNMT; HMT Protein family Class I-like SAM-binding methyltransferase superfamily, HNMT family Biochemical class Methyltransferase Function Inactivates histamine by N-methylation. Plays an important role in degrading histamine and in regulating the airway response to histamine. Related diseases Intellectual developmental disorder, autosomal recessive 51 (MRT51) [MIM:616739]: A disorder characterized by significantly below average general intellectual functioning associated with impairments in adaptive behavior and manifested during the developmental period. {ECO:0000269|PubMed:26206890}. The disease is caused by variants affecting distinct genetic loci, including the gene represented in this entry. Drugs (DrugBank ID) DB00613; DB13875; DB05381; DB04655; DB01103; DB01752; DB07106 Interacts with NA EC number EC 2.1.1.8 Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; Disease variant; Intellectual disability; Methyltransferase; Proteomics identification; Reference proteome; S-adenosyl-L-methionine; Transferase Protein physicochemical properties Chain ID A,B Molecular weight (Da) 32712 Length 288 Aromaticity 0.1 Instability index 36.38 Isoelectric point 5.18 Charge (pH=7) -9.97 2D Binding mode Binding energy (Kcal/mol) -7.95  Molscript Map 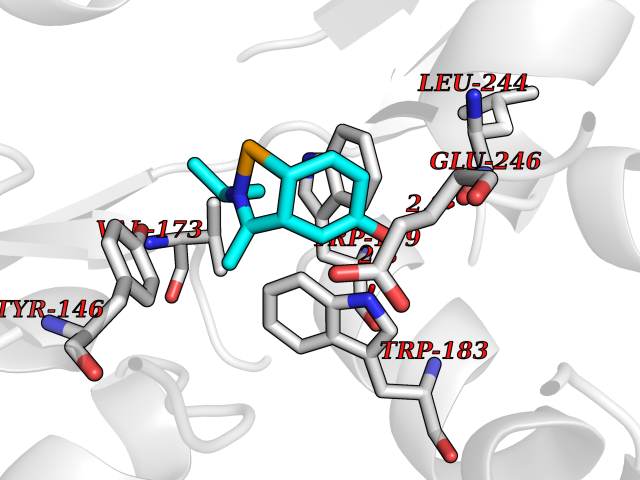 Pymol Map 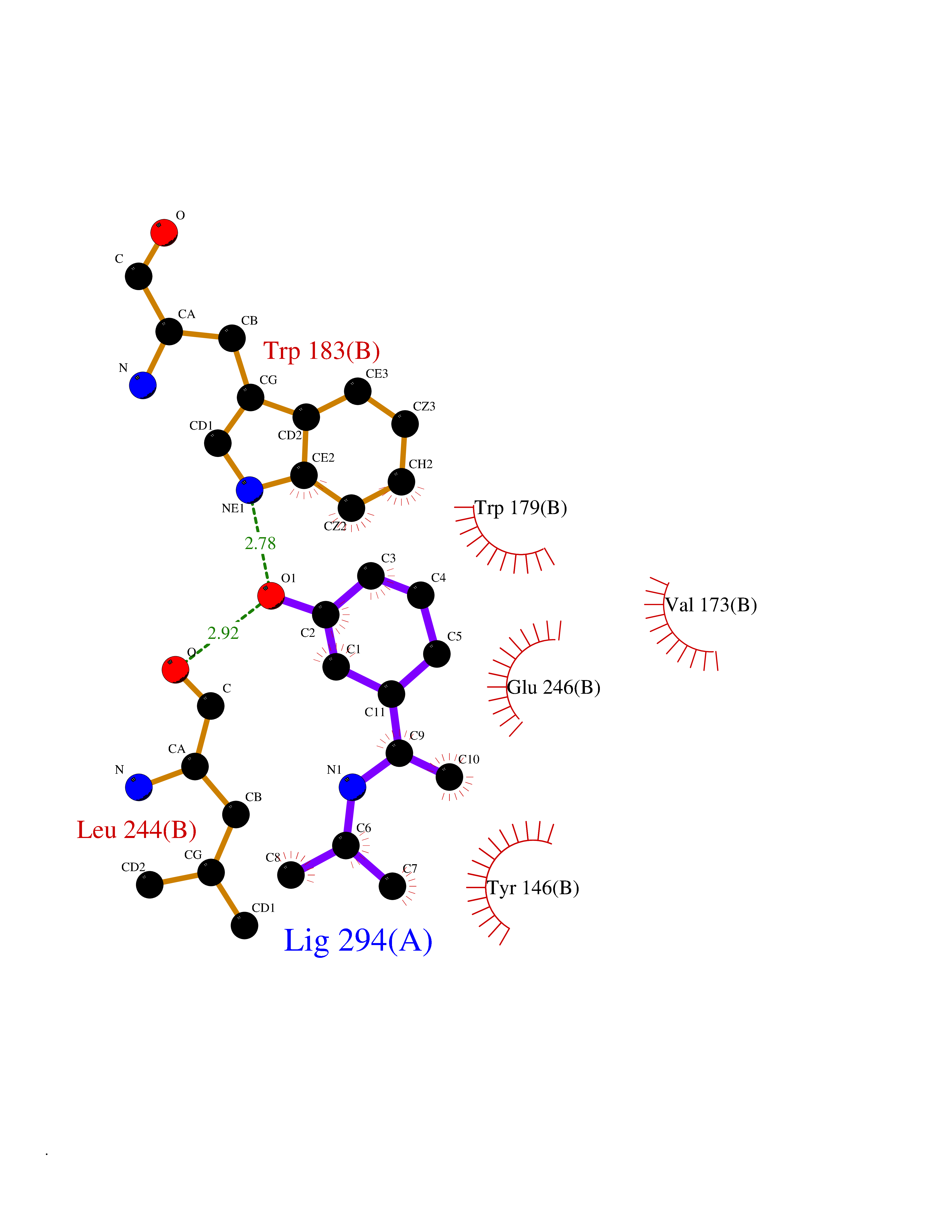 Ligplot Map 3D Binding mode Sequence MRSLFSDHGKYVESFRRFLNHSTEHQCMQEFMDKKLPGIIGRIGDTKSEIKILSIGGGAGEIDLQILSKVQAQYPGVXINNEVVEPSAEQIAKYKELVAKTSNLENVKFAWHKETSSEYQSRMLEKKELQKWDFIHMIQMLYYVKDIPATLKFFHSLLGTNAKMLIIVVSGSSGWDKLWKKYGSRFPQDDLCQYITSDDLTQMLDNLGLKYECYDLLSTMDISDCFIDGNENGDLLWDFLTETXNFNATAPPDLRAELGKDLQEPEFSAKKEGKVLFNNTLSFIVIEA Hydrogen bonds contact Hydrophobic contact | ||||
| 18 | cAMP-dependent protein kinase A type I (PRKAR1A) | 5KJZ | 5.83 | |
Target general information Gen name PRKAR1A Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms cAMP-dependent protein kinase type I-alpha regulatory subunit; Tissue-specific extinguisher 1; TSE1; PRKAR1; PKR1 Protein family CAMP-dependent kinase regulatory chain family Biochemical class Kinase Function Regulatory subunit of the cAMP-dependent protein kinases involved in cAMP signaling in cells. Related diseases Carney complex 1 (CNC1) [MIM:160980]: CNC is a multiple neoplasia syndrome characterized by spotty skin pigmentation, cardiac and other myxomas, endocrine tumors, and psammomatous melanotic schwannomas. {ECO:0000269|PubMed:15371594, ECO:0000269|PubMed:18241045, ECO:0000269|PubMed:22785148, ECO:0000269|PubMed:23323113, ECO:0000269|PubMed:26405036}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Intracardiac myxoma (INTMYX) [MIM:255960]: Inheritance is autosomal recessive. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Primary pigmented nodular adrenocortical disease 1 (PPNAD1) [MIM:610489]: A rare bilateral adrenal defect causing ACTH-independent Cushing syndrome. Macroscopic appearance of the adrenals is characteristic with small pigmented micronodules observed in the cortex. Clinical manifestations of Cushing syndrome include facial and truncal obesity, abdominal striae, muscular weakness, osteoporosis, arterial hypertension, diabetes. PPNAD1 is most often diagnosed in patients with Carney complex, a multiple neoplasia syndrome. However it can also be observed in patients without other manifestations. {ECO:0000269|PubMed:12213893}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Acrodysostosis 1, with or without hormone resistance (ACRDYS1) [MIM:101800]: A form of skeletal dysplasia characterized by short stature, severe brachydactyly, facial dysostosis, and nasal hypoplasia. Affected individuals often have advanced bone age and obesity. Laboratory studies show resistance to multiple hormones, including parathyroid, thyrotropin, calcitonin, growth hormone-releasing hormone, and gonadotropin. However, not all patients show endocrine abnormalities. {ECO:0000269|PubMed:21651393, ECO:0000269|PubMed:22464250, ECO:0000269|PubMed:22464252, ECO:0000269|PubMed:22723333, ECO:0000269|PubMed:23043190, ECO:0000269|PubMed:23425300, ECO:0000269|PubMed:26405036}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01790; DB02527; DB02315; DB05798 Interacts with Q9GZX7; P24588; O43687-2; Q9BSF0; Q9H6J7-2; Q86Y01; P0C7A2-2; Q9H0R8; Q9H8W4; P17612; P31321; P51817; P35250; Q86UC2; Q01105; Q8N0X7; O96006; P03259-2 EC number NA Uniprot keywords 3D-structure; Acetylation; Alternative splicing; cAMP; cAMP-binding; Cell membrane; Cushing syndrome; Direct protein sequencing; Disease variant; Disulfide bond; Membrane; Nucleotide-binding; Phosphoprotein; Proteomics identification; Reference proteome; Repeat Protein physicochemical properties Chain ID A Molecular weight (Da) 17007.4 Length 149 Aromaticity 0.08 Instability index 49.36 Isoelectric point 6.36 Charge (pH=7) -0.52 2D Binding mode Binding energy (Kcal/mol) -7.95 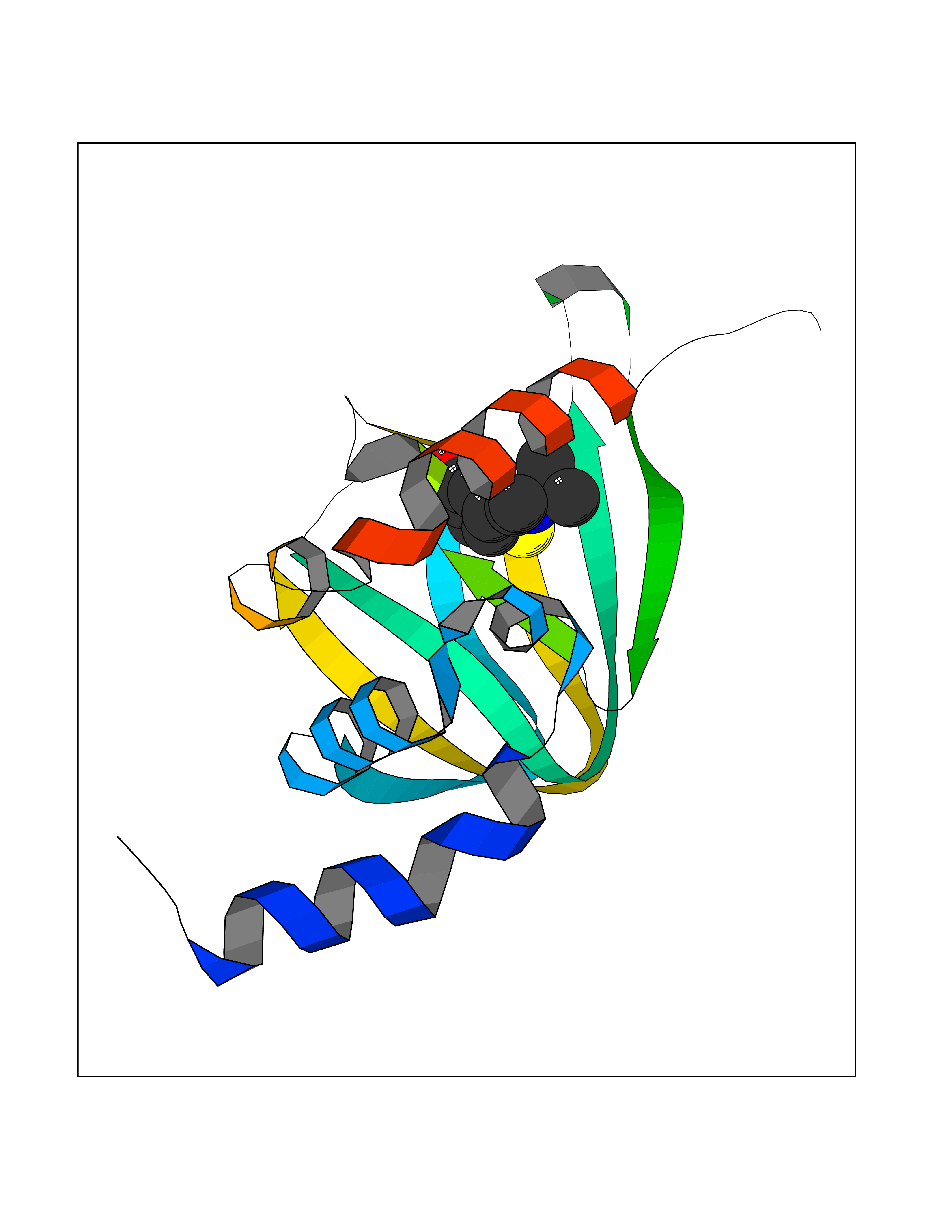 Molscript Map 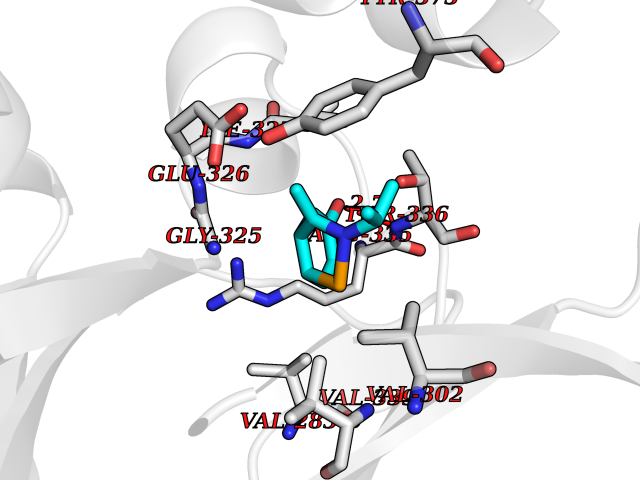 Pymol Map 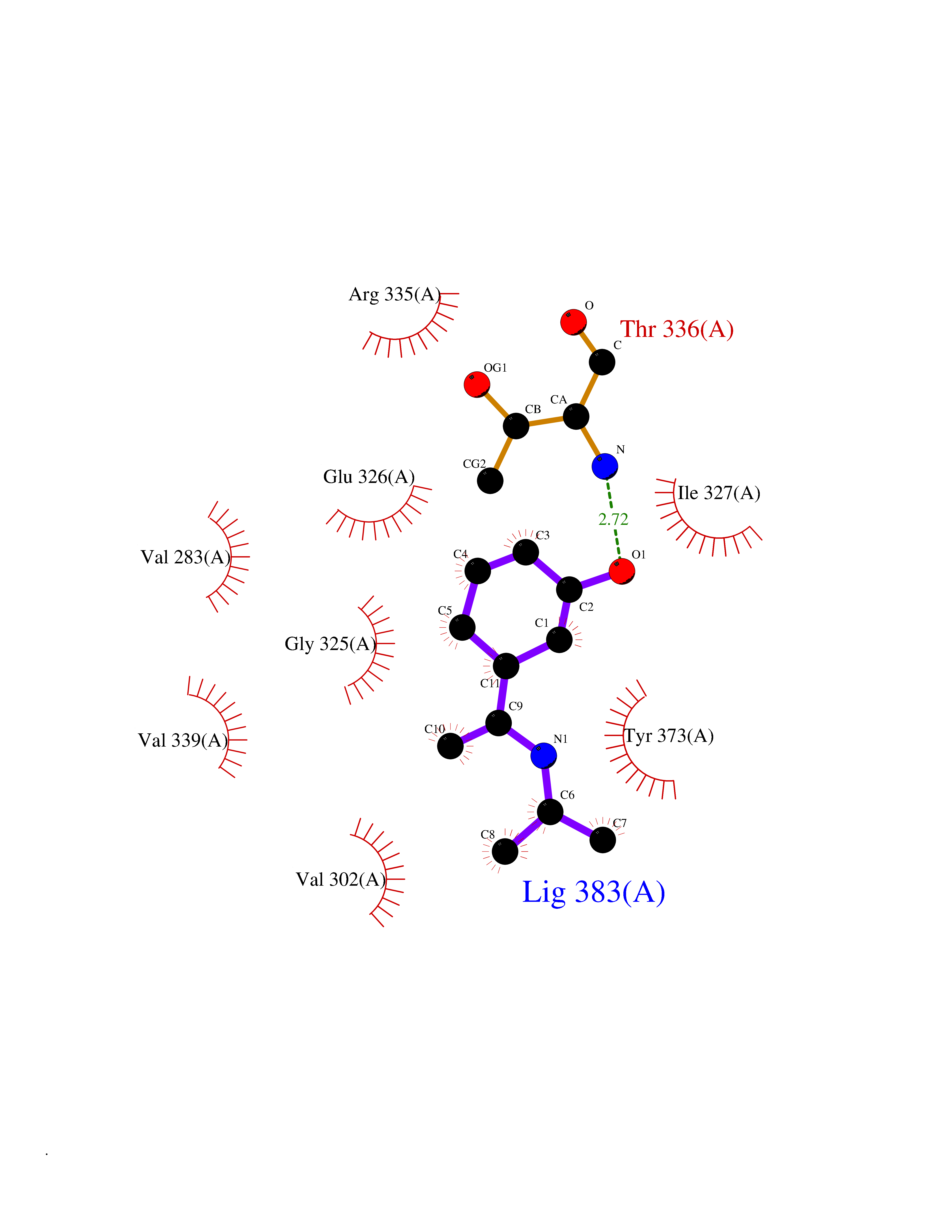 Ligplot Map 3D Binding mode Sequence SILMGSTLRKRKMYEEFLSKVSILESLDKWERLTVADALEPVQFEDGQKIVVQGEPGDEFFIILEGSAAVLQRRSENEEFVEVRRLGPSDYFGEIALLMNRPRTATVVARGPLKCVKLDRPRFERVLGPCSDILKRNIQQYNSFVSLSV Hydrogen bonds contact Hydrophobic contact | ||||
| 19 | N-acylethanolamine-hydrolyzing acidamidase (NAAA) | 6DXX | 5.83 | |
Target general information Gen name NAAA Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Nacylsphingosine amidohydrolaselike; Nacylethanolaminehydrolyzing acid amidase subunit beta; NAAA; Acid ceramidaselike protein; ASAHlike protein Protein family Acid ceramidase family Biochemical class Carbon-nitrogen hydrolase Function Degrades bioactive fatty acid amides to their corresponding acids, with the following preference: N- palmitoylethanolamine > N-myristoylethanolamine > N- lauroylethanolamine = N-stearoylethanolamine > N- arachidonoylethanolamine > N-oleoylethanolamine. Also exhibits weak hydrolytic activity against the ceramides N- lauroylsphingosine and N-palmitoylsphingosine. Related diseases Hypertriglyceridemia, transient infantile (HTGTI) [MIM:614480]: An autosomal recessive disorder characterized by onset of moderate to severe transient hypertriglyceridemia in infancy that normalizes with age. The hypertriglyceridemia is associated with hepatomegaly, moderately elevated transaminases, persistent fatty liver, and the development of hepatic fibrosis. {ECO:0000269|PubMed:22226083, ECO:0000269|PubMed:24549054}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB09061; DB14009; DB14011 Interacts with NA EC number EC 3.5.1.- Uniprot keywords 3D-structure; Alternative splicing; Autocatalytic cleavage; Direct protein sequencing; Disulfide bond; Fatty acid metabolism; Glycoprotein; Hydrolase; Lipid degradation; Lipid metabolism; Lysosome; Membrane; Proteomics identification; Reference proteome; Signal; Zymogen Protein physicochemical properties Chain ID A,B Molecular weight (Da) 36877.8 Length 328 Aromaticity 0.11 Instability index 44.37 Isoelectric point 7.72 Charge (pH=7) 1.08 2D Binding mode Binding energy (Kcal/mol) -7.95 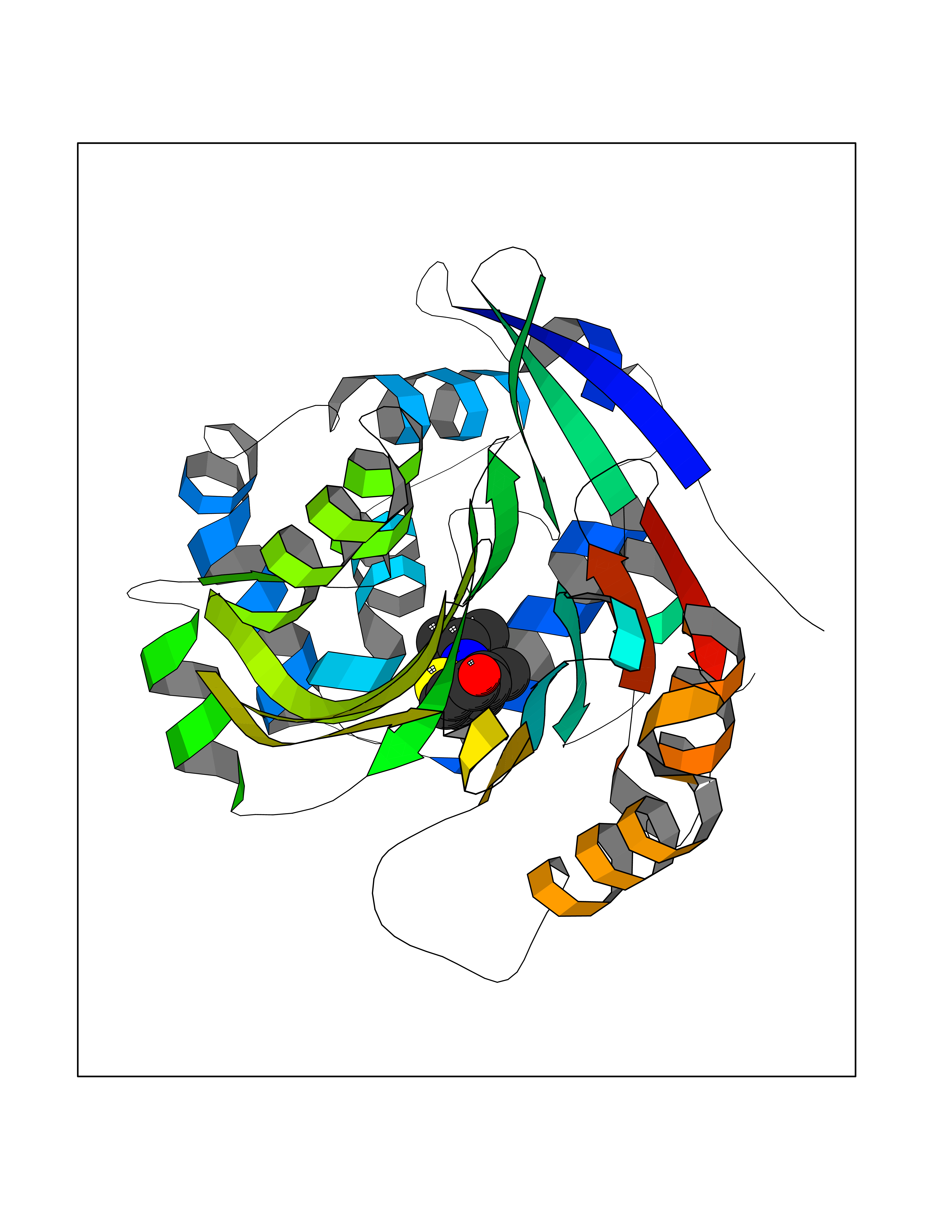 Molscript Map 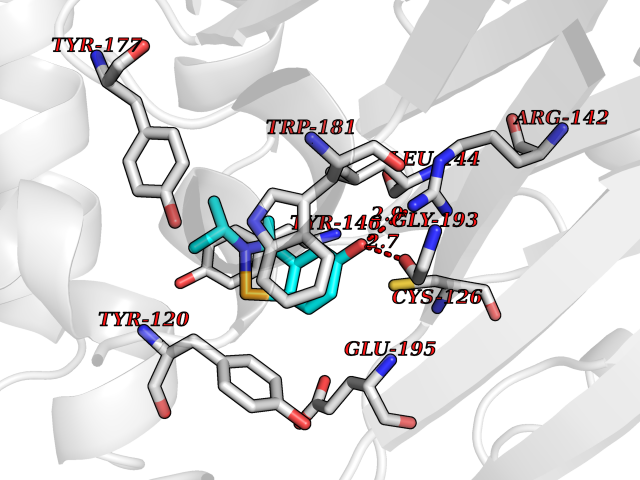 Pymol Map 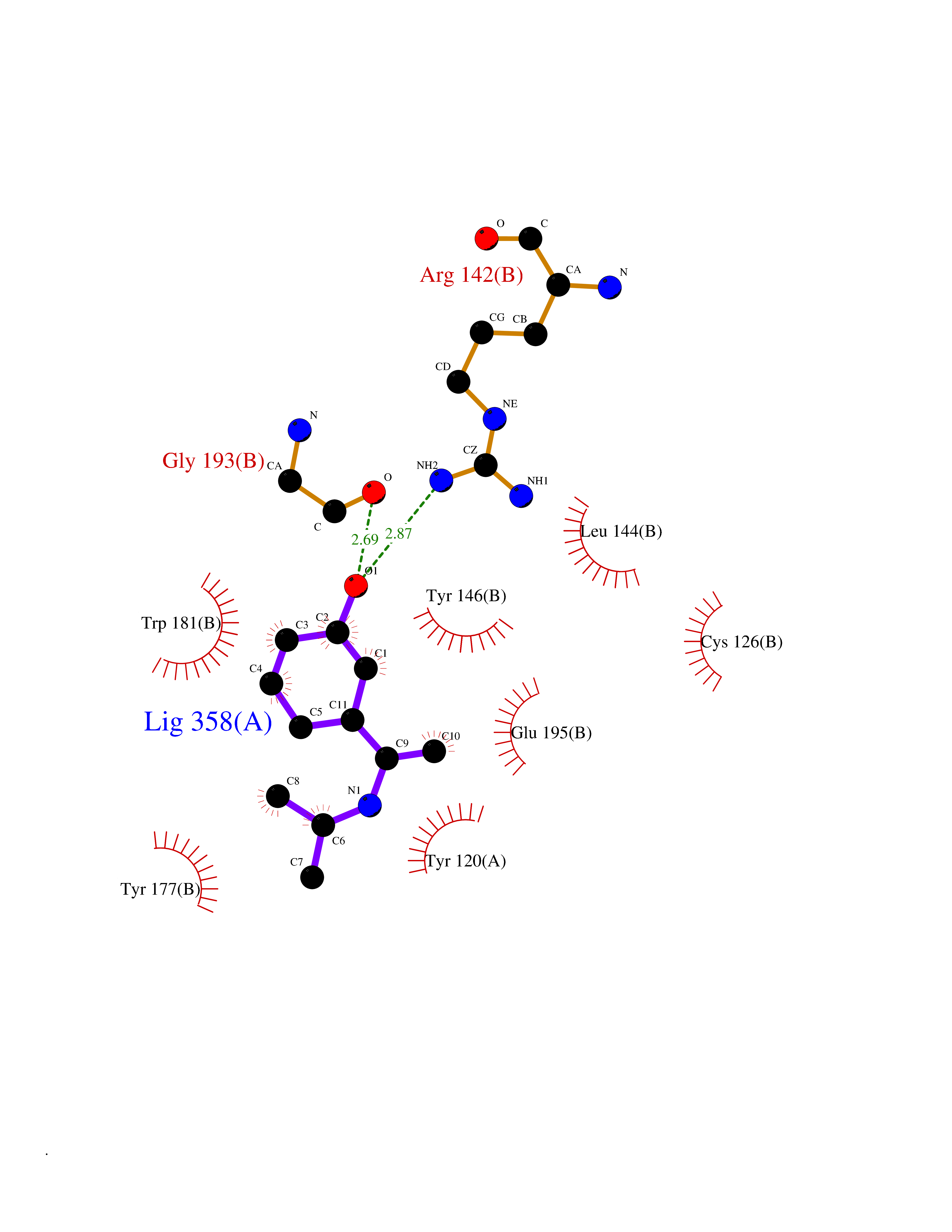 Ligplot Map 3D Binding mode Sequence SPPAAPRFNVSLDSVPELRWLPVLRHYDLDLVRAAMAQVIGDRVPKWVHVLIGKVVLELERFLPQPFTGEIRGMCDFMNLSLADCLLVNLAYESSVFCTSIVAQDSRGHIYHGRNLDYPFGNVLRKLTVDVQFLKNGQIAFTGTTFIGYVGLWTGQSPHKFTVSGDERDKGWWWENAIAALFRRHIPVSWLIRATLSESENFEAAVGKLAKTPLIADVYYIVGGTSPREGVVITRNRDGPADIWPLDPLNGAWFRVETNYDHWKPAPKEDDRRTSAIKALNATGQANLSLEALFQILSVVPVYNNFTIYTTVMSAGSPDKYMTRIRNP Hydrogen bonds contact Hydrophobic contact | ||||
| 20 | Spermine synthase | 3C6K | 5.82 | |
Target general information Gen name SMS Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Spermidine/spermine synthase family Biochemical class Transferase Function Spermine synthase activity. Related diseases Intellectual developmental disorder, X-linked, syndromic, Snyder-Robinson type (MRXSSR) [MIM:309583]: An X-linked intellectual disability syndrome characterized by a collection of clinical features including facial asymmetry, marfanoid habitus, hypertonia, osteoporosis and unsteady gait. {ECO:0000269|PubMed:14508504, ECO:0000269|PubMed:18550699, ECO:0000269|PubMed:19206178, ECO:0000269|PubMed:22612257, ECO:0000269|PubMed:23696453, ECO:0000269|PubMed:23897707}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00127 Interacts with NA EC number 2.5.1.22 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Direct protein sequencing; Intellectual disability; Phosphoprotein; Polyamine biosynthesis; Proteomics identification; Reference proteome; Transferase Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 27177.1 Length 238 Aromaticity 0.11 Instability index 41.31 Isoelectric point 4.82 Charge (pH=7) -9.19 2D Binding mode Binding energy (Kcal/mol) -7.94  Molscript Map 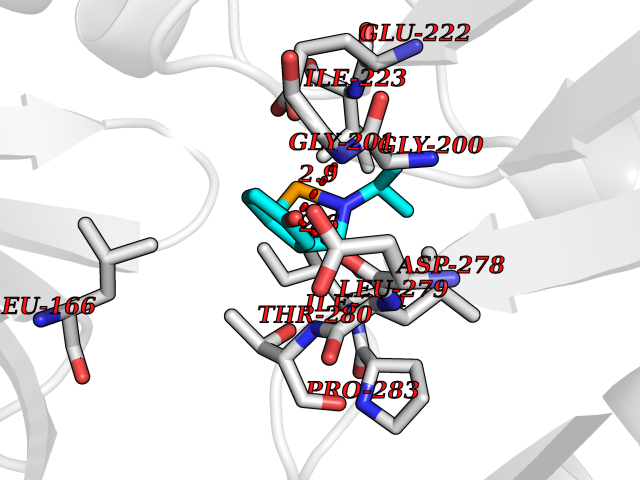 Pymol Map 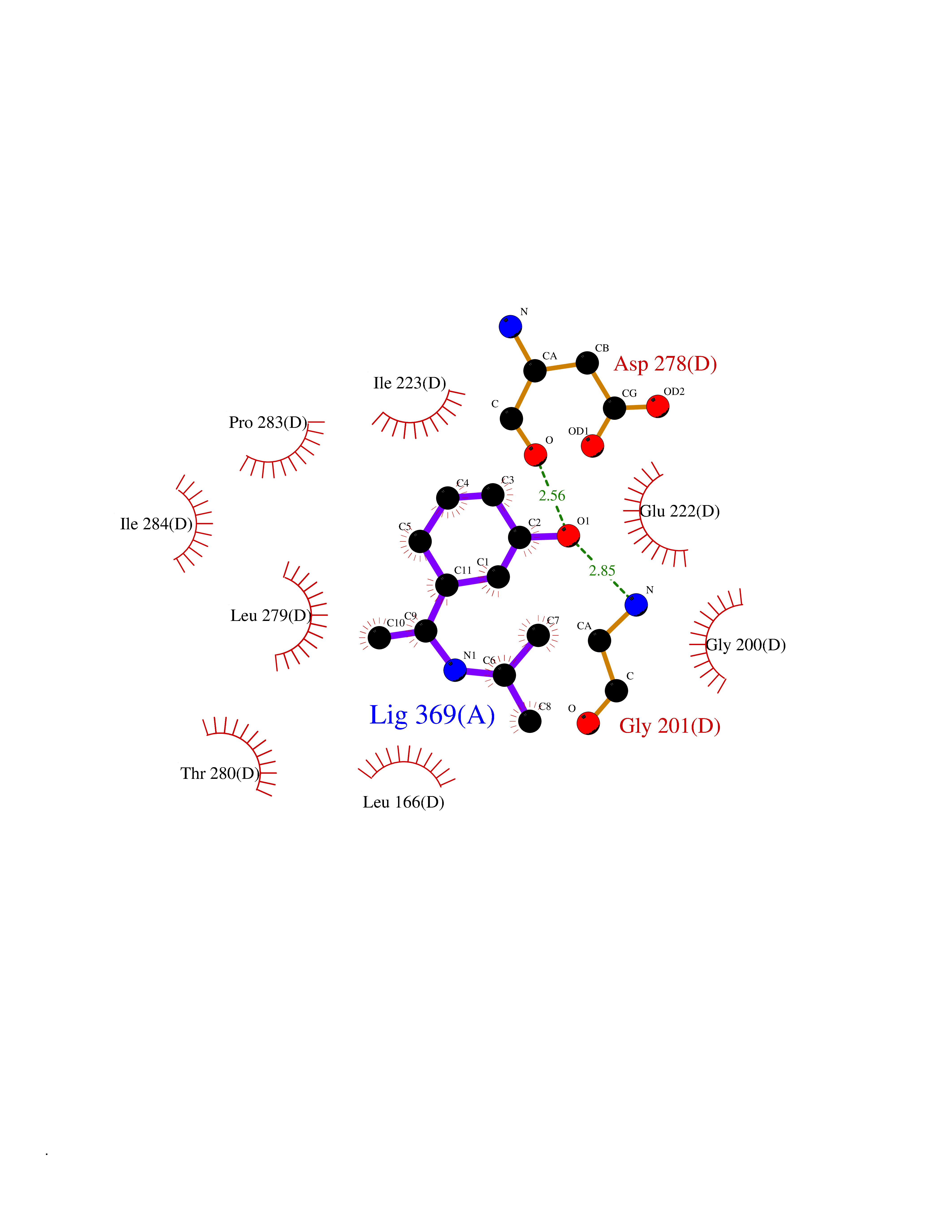 Ligplot Map 3D Binding mode Sequence RYWPTADGRLVEYDIDEVVYDEDSPYQNIKILHSKQFGNILILSGDVNLAESDLAYTRAIMGSGKEDYTGKDVLILGGGDGGILCEIVKLKPKMVTMVEIDQMVIDGCKKYMRKDVLDNLKGDCYQVLIEDCIPVLKRYAKEGREFDYVINDLTAVPISTSPSTWEFLRLILDLSMKVLKQDGKYFTQGNCVNLTEALSLYEEQLGRLYCPVEFSKEIVCVPSYLELWVFYTVWKKAK Hydrogen bonds contact Hydrophobic contact | ||||