Job Results:
Ligand
Structure
Job ID
7eae9f0de32fdedcd28c1404448b5ea8
Job name
NA
Time
2025-04-07 15:33:07
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 1 | Endolysin | 1AM7 | 4.34 | |
Target general information Gen name R Organism Escherichia phage lambda (Bacteriophage lambda) Uniprot ID TTD ID NA Synonyms NA Protein family Glycosyl hydrolase 24 family Biochemical class Glycosidase Function Lyase activity.Lysozyme activity.Lytic transglycosylase activity. Related diseases Estrogen resistance (ESTRR) [MIM:615363]: A disorder characterized by partial or complete resistance to estrogens, in the presence of elevated estrogen serum levels. Clinical features include absence of the pubertal growth spurt, delayed bone maturation, unfused epiphyses, reduced bone mineral density, osteoporosis, continued growth into adulthood and very tall adult stature. Glucose intolerance, hyperinsulinemia and lipid abnormalities may also be present. {ECO:0000269|PubMed:23841731, ECO:0000269|PubMed:27754803}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB04206 Interacts with NA EC number 4.2.2.n2 Uniprot keywords 3D-structure; Antimicrobial; Bacteriolytic enzyme; Cytolysis; Direct protein sequencing; Host cell lysis by virus; Host cytoplasm; Lyase; Reference proteome; Viral release from host cell Protein physicochemical properties Chain ID A,B,C Molecular weight (Da) 49834.9 Length 462 Aromaticity 0.07 Instability index 18.78 Isoelectric point 9.6 Charge (pH=7) 18.29 2D Binding mode Binding energy (Kcal/mol) -5.92  Molscript Map 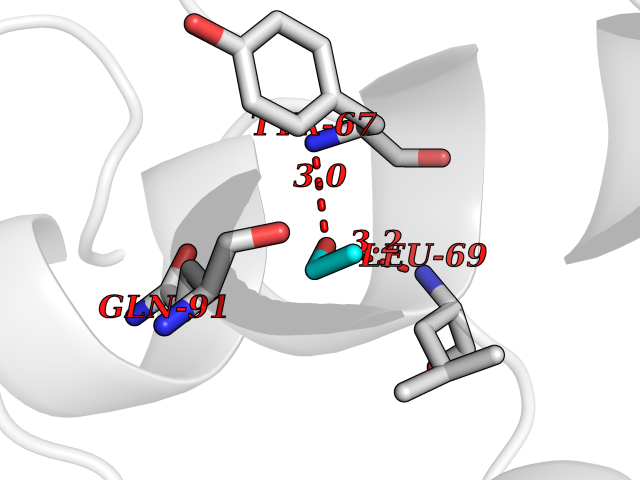 Pymol Map 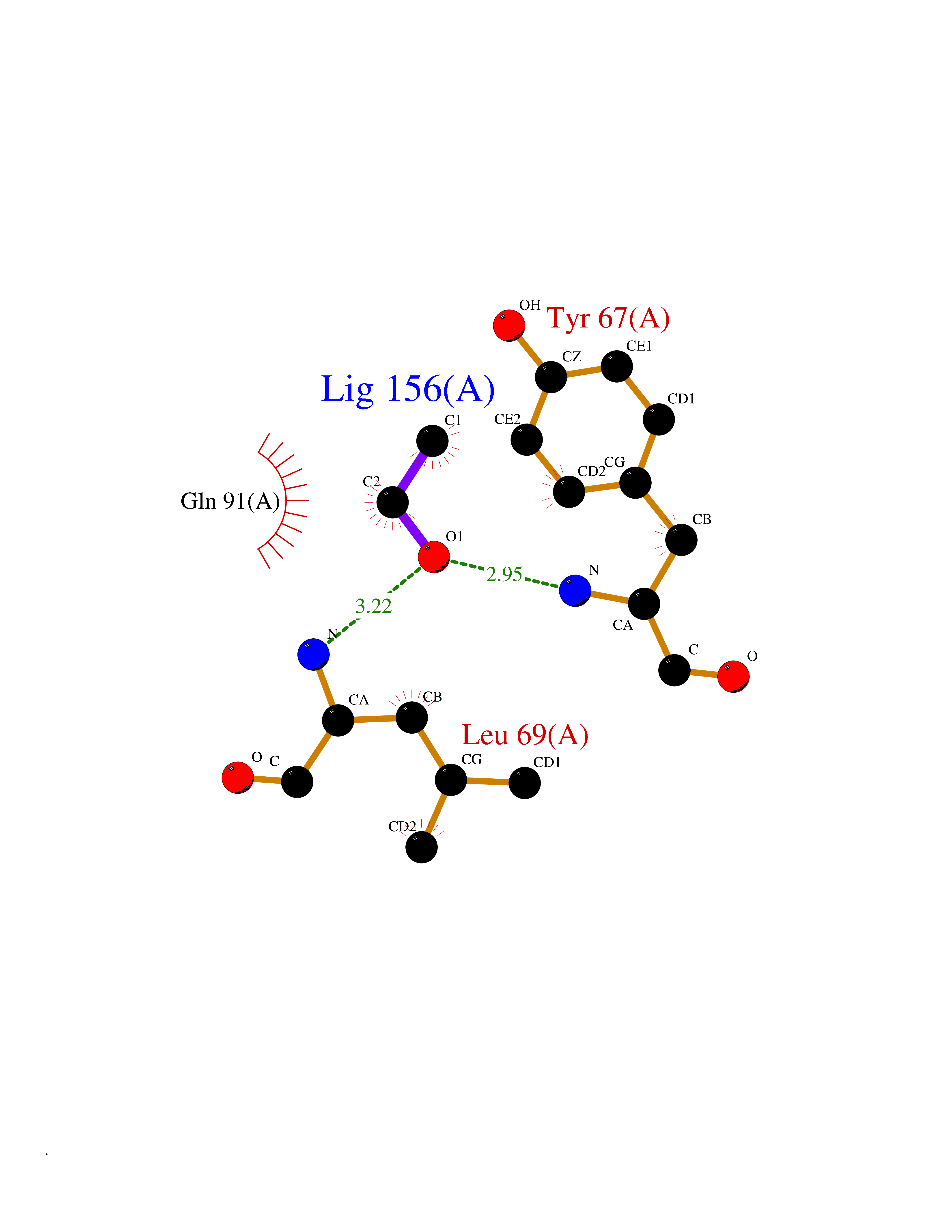 Ligplot Map 3D Binding mode Sequence MVEINNQRKAFLDMLAXSEGTDNGRQKTRNHGYDVIVGGELFTDYSDHPRKLVTLNPKLKSTGAGRYQLLSRXXDAYRKQLGLKDFSPKSQDAVALQQIKERGALPMIDRGDIRQAIDRCSNIXASLPGAGYGQFEHKADSLIAKFKEAGGTVRMVEINNQRKAFLDMLAXSEGTDNGRQKTRNHGYDVIVGGELFTDYSDHPRKLVTLNPKLKSTGAGRYQLLSRXXDAYRKQLGLKDFSPKSQDAVALQQIKERGALPMIDRGDIRQAIDRCSNIXASLPGAGYGQFEHKADSLIAKFKEAGGTVRMVEINNQRKAFLDMLAXSEGTDNGRQKTRNHGYDVIVGGELFTDYSDHPRKLVTLNPKLKSTGAGRYQLLSRXXDAYRKQLGLKDFSPKSQDAVALQQIKERGALPMIDRGDIRQAIDRCSNIXASLPGAGYGQFEHKADSLIAKFKEAGGTVR Hydrogen bonds contact Hydrophobic contact | ||||
| 2 | Penicillin-binding protein 1A | 2ZC6 | 4.34 | |
Target general information Gen name ponA Organism Streptococcus pneumoniae serotype 4 (strain ATCC BAA-334 / TIGR4) Uniprot ID TTD ID NA Synonyms SP_0369 Protein family Glycosyltransferase 51 family; Transpeptidase family Biochemical class Biosynthetic protein Function Penicillin binding.Peptidoglycan glycosyltransferase activity.Serine-type D-Ala-D-Ala carboxypeptidase activity. Related diseases Aminoacylase-1 deficiency (ACY1D) [MIM:609924]: An enzymatic deficiency resulting in encephalopathy, unspecific psychomotor delay, psychomotor delay with atrophy of the vermis and syringomyelia, marked muscular hypotonia or normal clinical features. Epileptic seizures are a frequent feature. All affected individuals exhibit markedly increased urinary excretion of several N-acetylated amino acids. {ECO:0000269|PubMed:16274666, ECO:0000269|PubMed:16465618, ECO:0000269|PubMed:17562838, ECO:0000269|PubMed:21414403}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01150; DB05659 Interacts with NA EC number 2.4.99.28; 3.4.16.4 Uniprot keywords 3D-structure; Antibiotic resistance; Carboxypeptidase; Cell shape; Cell wall biogenesis/degradation; Glycosyltransferase; Hydrolase; Multifunctional enzyme; Peptidoglycan synthesis; Protease; Reference proteome; Secreted; Transferase Protein physicochemical properties Chain ID A,C Molecular weight (Da) 44805.1 Length 400 Aromaticity 0.12 Instability index 31.82 Isoelectric point 4.88 Charge (pH=7) -14.68 2D Binding mode Binding energy (Kcal/mol) -5.92 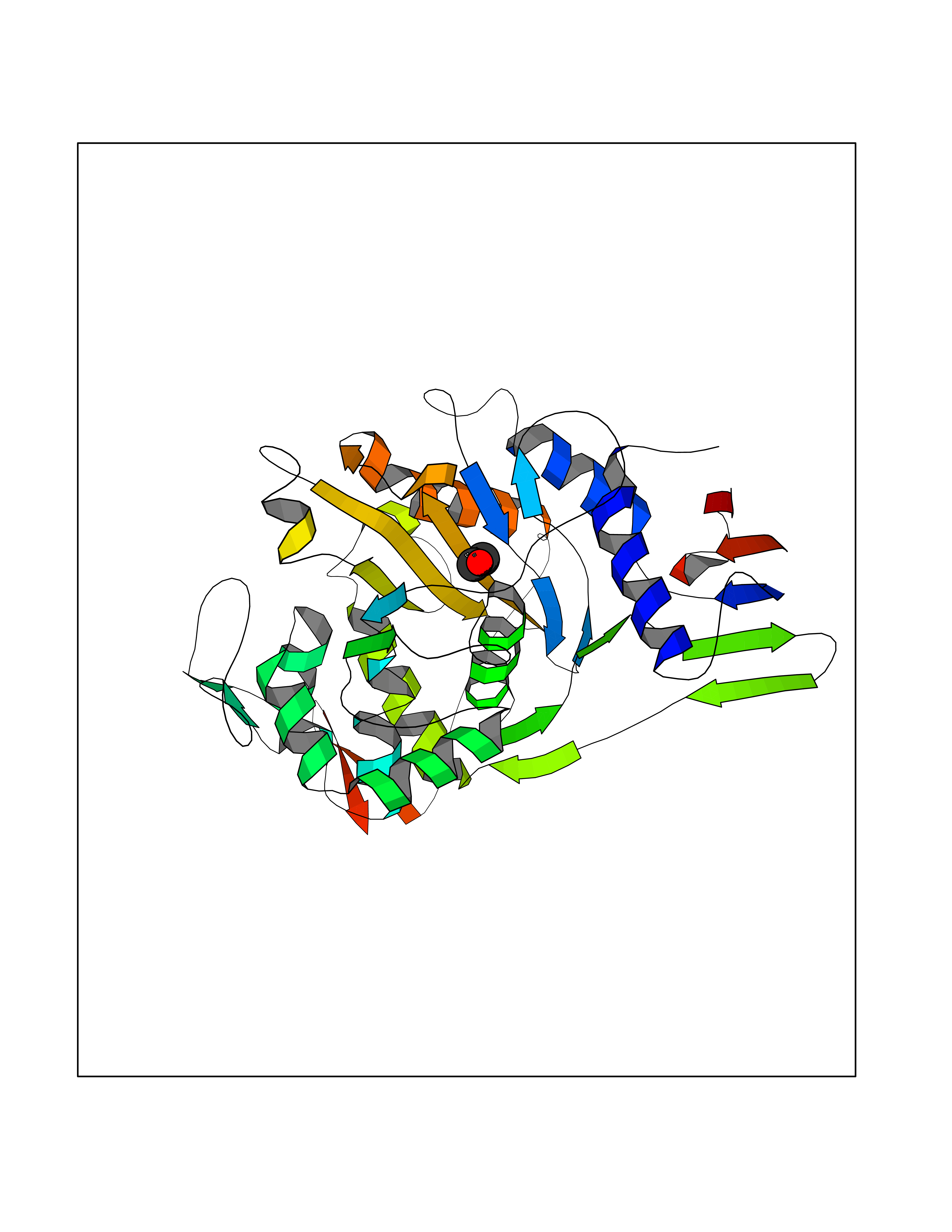 Molscript Map 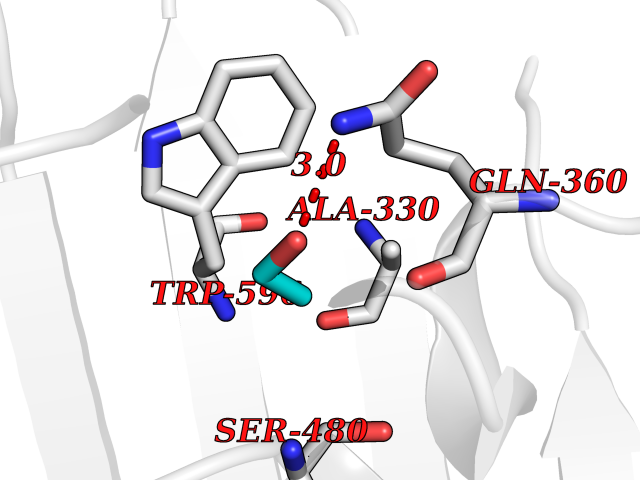 Pymol Map 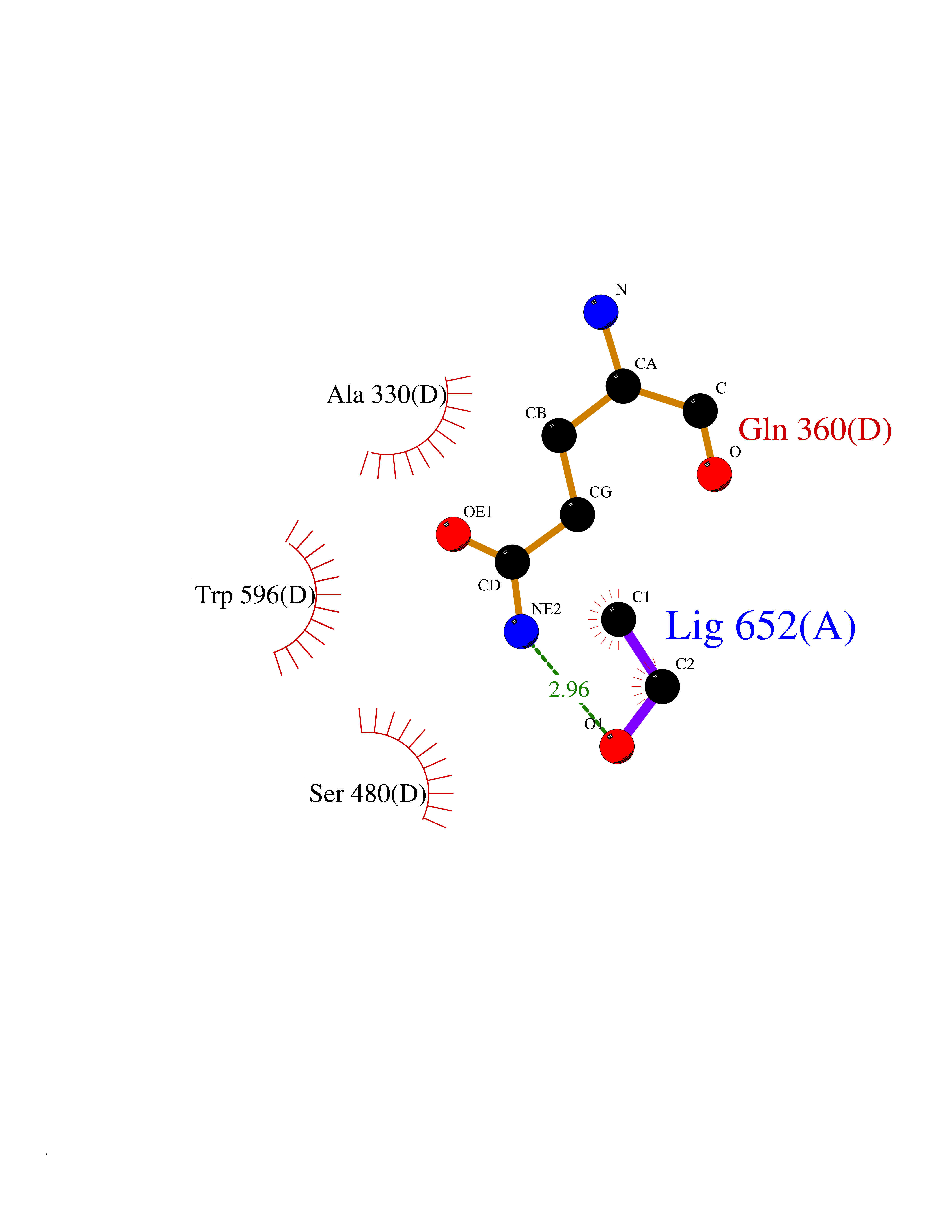 Ligplot Map 3D Binding mode Sequence NYPAYMDNYLKEVINQVEEETGYNLLTTGMDVYTNVDQEAQKHLWDIYNTDEYVAYPDDELQVASTIVDVSNGKVIAQLGARHQSSNVSFGINQAVETNRDWGSTMKPITDYAPALEYGVYDSTATIVHDEPYNYPGTNTPVYNWDRGYFGNITLQYALQQSRNVPAVETLNKVGLNRAKTFLNGLGIDYPSIHYSNAISSNTTESDKKYGASSEKMAAAYAAFANGGTYYKPMYIHKVVFSDGSEKEFSNVGTRAMKETTAYMMTDMMKTVLSYGTGQNAYLAWLPQAGKTGTSNYTDEEIENHIKTSQFVAPDELFAGYTRKYSMAVWTGYSNRLTPLVGNGLTVAAKVYRSMMTYLSEGSNPEDWNIPEGLYRNGEFVFKNTSSKIYDNKNQLIADL Hydrogen bonds contact Hydrophobic contact | ||||
| 3 | Monoglyceride lipase (MAGL) | 3PE6 | 4.34 | |
Target general information Gen name MGLL Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Monoacylglycerol lipase; MGL; Lysophospholipaselike; Lysophospholipase-like; Lysophospholipase homolog; HUK5; HU-K5 Protein family AB hydrolase superfamily, Monoacylglycerol lipase family Biochemical class Carboxylic ester hydrolase Function Hydrolyzes the endocannabinoid 2-arachidonoylglycerol, and thereby contributes to the regulation of endocannabinoid signaling, nociperception and perception of pain. Regulates the levels of fatty acids that serve as signaling molecules and promote cancer cell migration, invasion and tumor growth. Converts monoacylglycerides to free fatty acids and glycerol. Related diseases Systemic lupus erythematosus 9 (SLEB9) [MIM:610927]: A chronic, relapsing, inflammatory, and often febrile multisystemic disorder of connective tissue, characterized principally by involvement of the skin, joints, kidneys and serosal membranes. It is of unknown etiology, but is thought to represent a failure of the regulatory mechanisms of the autoimmune system. The disease is marked by a wide range of system dysfunctions, an elevated erythrocyte sedimentation rate, and the formation of LE cells in the blood or bone marrow. {ECO:0000269|PubMed:17360460}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Immunodeficiency, common variable, 7 (CVID7) [MIM:614699]: A primary immunodeficiency characterized by antibody deficiency, hypogammaglobulinemia, recurrent bacterial infections and an inability to mount an antibody response to antigen. The defect results from a failure of B-cell differentiation and impaired secretion of immunoglobulins; the numbers of circulating B-cells is usually in the normal range, but can be low. {ECO:0000269|PubMed:22035880}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P07550; P37235 EC number EC 3.1.1.23 Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; Fatty acid biosynthesis; Fatty acid metabolism; Hydrolase; Lipid biosynthesis; Lipid degradation; Lipid metabolism; Membrane; Nitration; Phosphoprotein; Proteomics identification; Reference proteome; Serine esterase Protein physicochemical properties Chain ID A Molecular weight (Da) 31808.4 Length 289 Aromaticity 0.08 Instability index 29.7 Isoelectric point 6.73 Charge (pH=7) -0.91 2D Binding mode Binding energy (Kcal/mol) -5.92 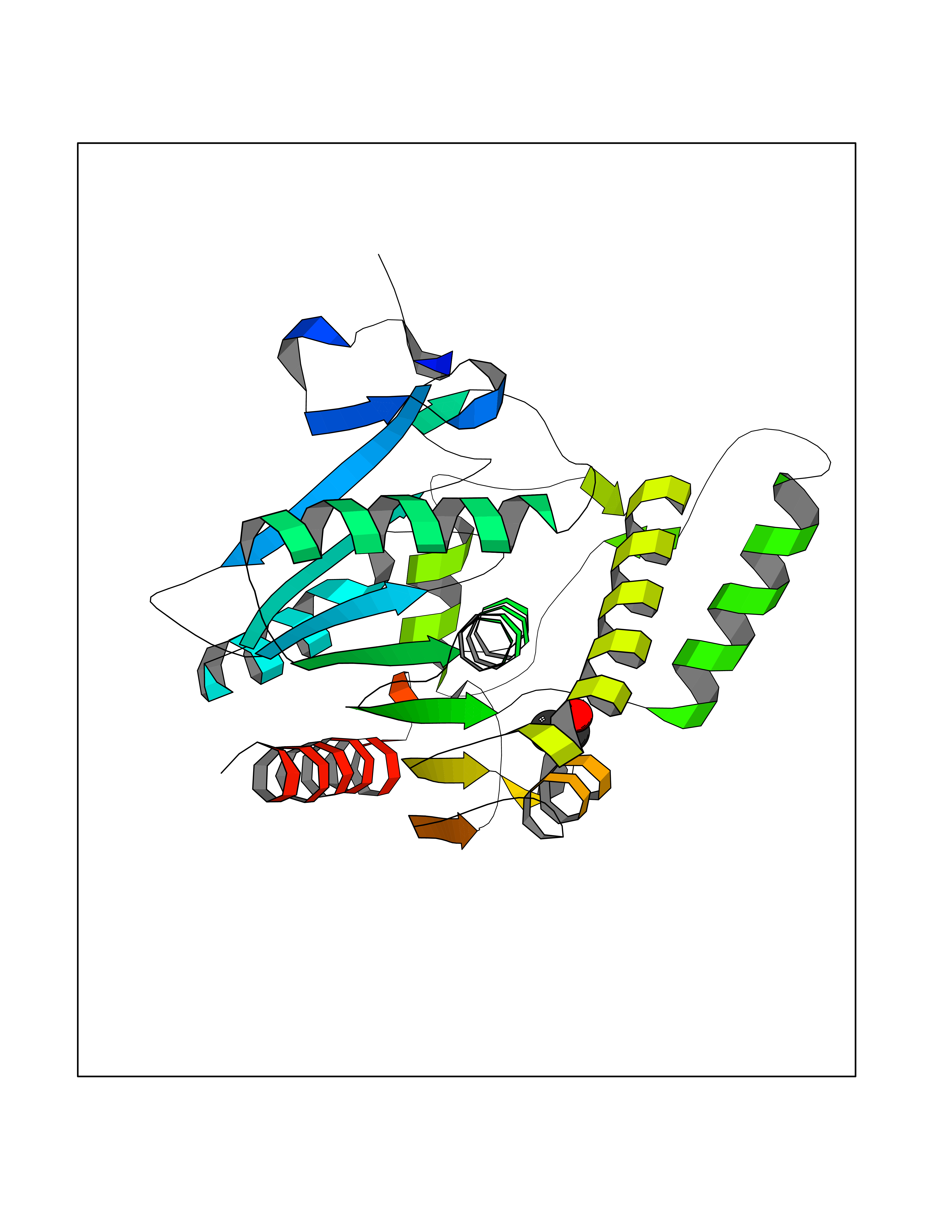 Molscript Map 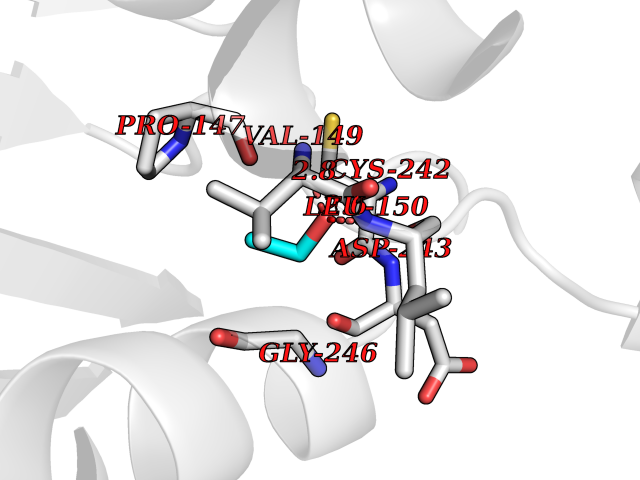 Pymol Map 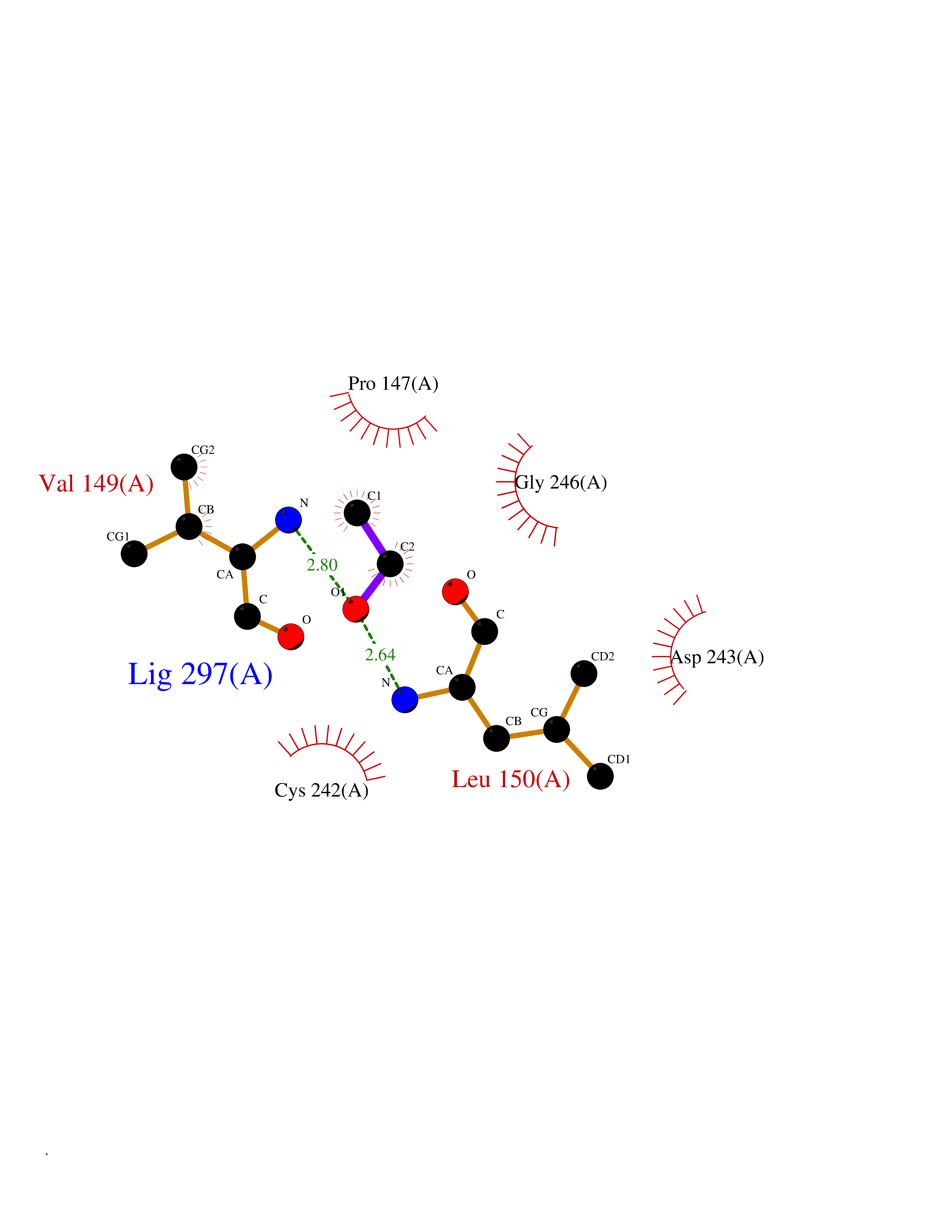 Ligplot Map 3D Binding mode Sequence PRRTPQSIPYQDLPHLVNADGQYLFCRYWAPTGTPKALIFVSHGAGEHSGRYEELARMLMGLDLLVFAHDHVGHGQSEGERMVVSDFHVFVRDVLQHVDSMQKDYPGLPVFLLGHSMGGAIAILTAAERPGHFAGMVLISPLVLANPESATTFKVLAAKVLNSVLPNLSSGPIDSSVLSRNKTEVDIYNSDPLICRAGLKVCFGIQLLNAVSRVERALPKLTVPFLLLQGSADRLCDSKGAYLLMELAKSQDKTLKIYEGAYHVLHKELPEVTNSVFHEINMWVSQRTA Hydrogen bonds contact Hydrophobic contact | ||||
| 4 | Prolyl endopeptidase (PREP) | 3DDU | 4.34 | |
Target general information Gen name PREP Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Post-proline cleaving enzyme; PREP; PE Protein family Peptidase S9A family Biochemical class Peptidase Function Cleaves peptide bonds on the C-terminal side of prolyl residues within peptides that are up to approximately 30 amino acids long. Related diseases Intellectual developmental disorder, X-linked, syndromic, Claes-Jensen type (MRXSCJ) [MIM:300534]: A disorder characterized by significantly below average general intellectual functioning associated with impairments in adaptive behavior and manifested during the developmental period. MRXSCJ patients manifest intellectual disability associated with variable features such as slowly progressive spastic paraplegia, seizures, facial dysmorphism. {ECO:0000269|PubMed:15586325, ECO:0000269|PubMed:16538222, ECO:0000269|PubMed:16541399, ECO:0000269|PubMed:17320160, ECO:0000269|PubMed:17468742, ECO:0000269|PubMed:23356856, ECO:0000269|PubMed:25666439}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07148; DB01684; DB08738; DB08739; DB03864; DB00107; DB03382; DB03535 Interacts with P04406 EC number EC 3.4.21.26 Uniprot keywords 3D-structure; Acetylation; Cytoplasm; Direct protein sequencing; Hydrolase; Protease; Proteomics identification; Reference proteome; Serine protease Protein physicochemical properties Chain ID A Molecular weight (Da) 80424.2 Length 707 Aromaticity 0.13 Instability index 35.02 Isoelectric point 5.52 Charge (pH=7) -20.31 2D Binding mode Binding energy (Kcal/mol) -5.92  Molscript Map 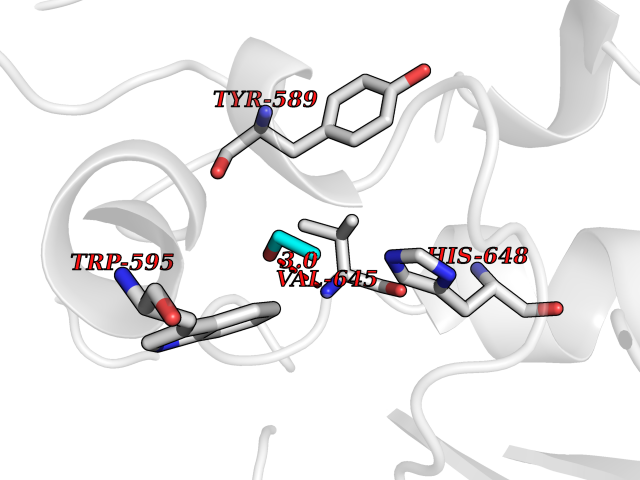 Pymol Map 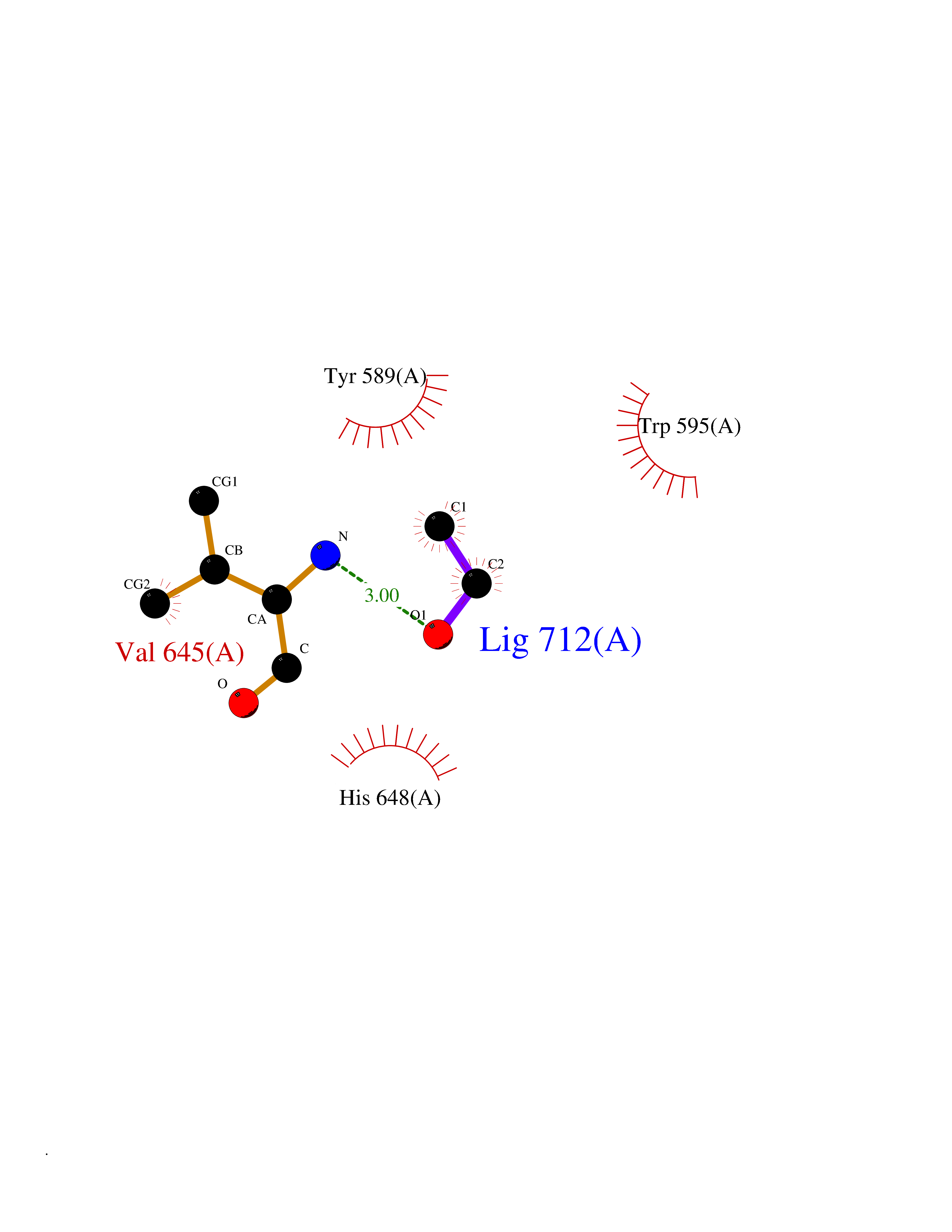 Ligplot Map 3D Binding mode Sequence LSFQYPDVYRDETAVQDYHGHKICDPYAWLEDPDSEQTKAFVEAQNKITVPFLEQCPIRGLYKERMTELYDYPKYSCHFKKGKRYFYFYNTGLQNQRVLYVQDSLEGEARVFLDPNILSDDGTVALRGYAFSEDGEYFAYGLSASGSDWVTIKFMKVDGAKELPDVLERVKFSCMAWTHDGKGMFYNSYPQQDGKSDGTETSTNLHQKLYYHVLGTDQSEDILCAEFPDEPKWMGGAELSDDGRYVLLSIREGCDPVNRLWYCDLQQESSGIAGILKWVKLIDNFEGEYDYVTNEGTVFTFKTNRHSPNYRVINIDFTDPEESKWKVLVPEHEKDVLEWIACVRSNFLVLCYLHDVKNTLQLHDLTTGALLKIFPLDVGSIVGYSGQKKDTEIFYQFTSFLSPGIIYHCDLTKEELEPRVFREVVKIDASDYQTVQIFYPSKDGTKIPMFIVHKKGIKLDGSHPAFLYGYGGFNISITPNYSVSRLIFVRHMGGILAVANIRGGGEYGETWHKGGILANKQNCFDDFQCAAEYLIKEGYTSPKRLTINGGSNGGLLVAACANQRPDLFGCVIAQVGVMDMLKFHKYTIGHAWTTDYGCSDSKQHFEWLVKYSPLHNVKLPEADDIQYPSMLLLTADHDDRVVPLHSLKFIATLQYIVGRSRKQNNPLLIHVDTKAGHGAGKPTAKVIEEVSDMFAFIARCLNVDWIP Hydrogen bonds contact Hydrophobic contact | ||||
| 5 | Aldehyde oxidoreductase | 4USA | 4.33 | |
Target general information Gen name mop Organism Megalodesulfovibrio gigas (Desulfovibrio gigas) Uniprot ID TTD ID NA Synonyms NA Protein family Xanthine dehydrogenase family Biochemical class Oxidoreductase Function 2 iron, 2 sulfur cluster binding.Aldehyde dehydrogenase (FAD-independent) activity.Electron carrier activity.Metal ion binding. Related diseases LTC4 synthase deficiency is associated with a neurometabolic developmental disorder characterized by muscular hypotonia, psychomotor retardation, failure to thrive, and microcephaly. {ECO:0000269|PubMed:10896305, ECO:0000269|PubMed:9820300}. Drugs (DrugBank ID) DB02137 Interacts with NA EC number 1.2.99.7 Uniprot keywords 2Fe-2S; 3D-structure; FAD; Flavoprotein; Iron; Iron-sulfur; Metal-binding; Molybdenum; NAD; Oxidoreductase Protein physicochemical properties Chain ID A Molecular weight (Da) 96930.4 Length 907 Aromaticity 0.07 Instability index 29.17 Isoelectric point 5.69 Charge (pH=7) -17.56 2D Binding mode Binding energy (Kcal/mol) -5.9 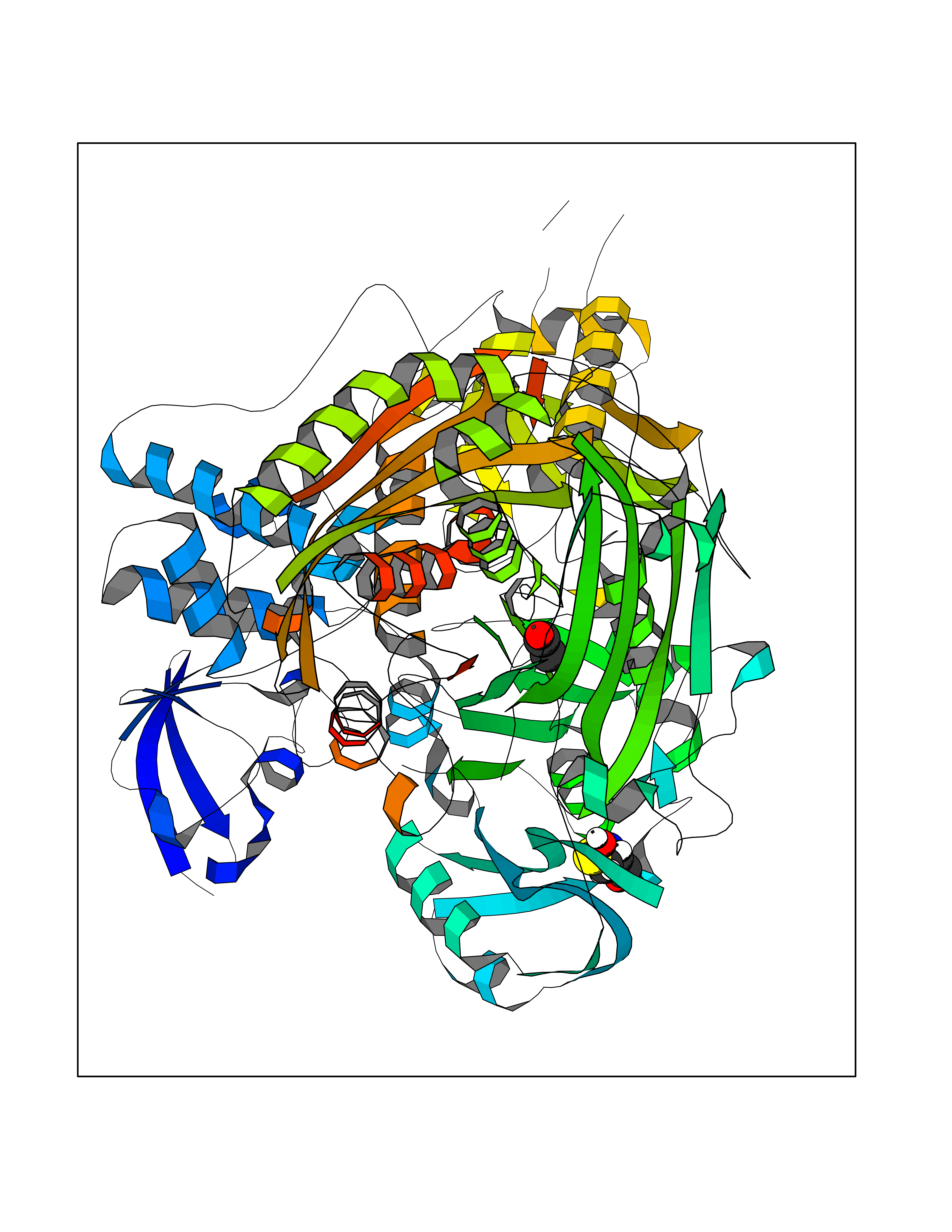 Molscript Map  Pymol Map 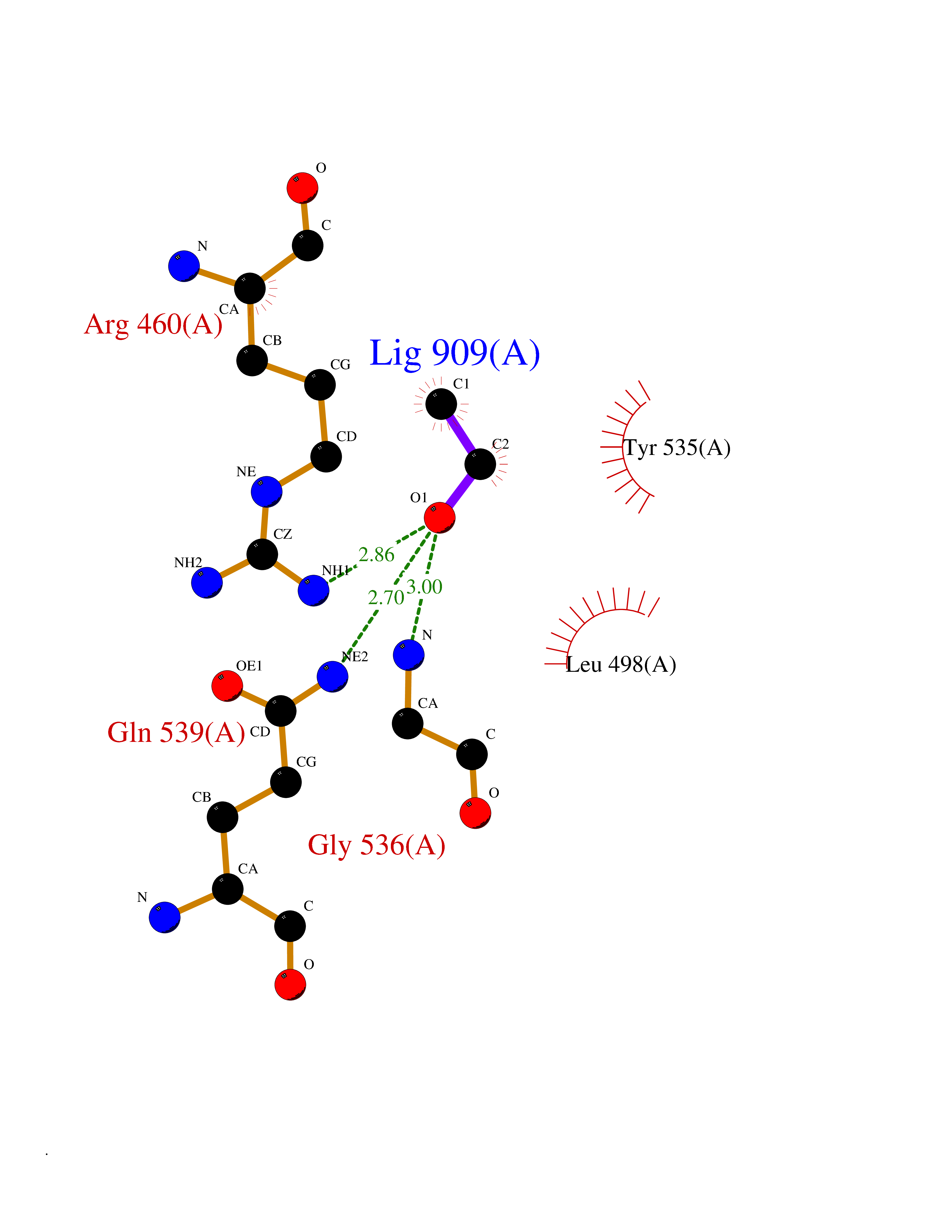 Ligplot Map 3D Binding mode Sequence MIQKVITVNGIEQNLFVDAEALLSDVLRQQLGLTGVKVGCEQGQCGACSVILDGKVVRACVTKMKRVADGAQITTIEGVGQPENLHPLQKAWVLHGGAQCGFCSPGFIVSAKGLLDTNADPSREDVRDWFQKHRNACRCTGYKPLVDAVMDAAAVINGKKPETDLEFKMPADGRIWGSKYPRPTAVAKVTGTLDYGADLGLKMPAGTLHLAMVQAKVSHANIKGIDTSEALTMPGVHSVITHKDVKGKNRITGLITFPTNKGDGWDRPILXDEKVFQYGDCIALVCADSEANARAAAEKVKVDLEELPAYMSGPAAAAEDAIEIHPGTPNVYFEQPIVKGEDTGPIFASADVTVEGDFYVGRQPHMPIEPDVAFAYMGDDGKCYIHSKSIGVHLHLYMIAPGVGLEPDQLVLVANPMGGTFGYKFSPTSEALVAVAAMATGRPVHLRYNYQQQQQYTGKRSPWEMNVKFAAKKDGTLLAMESDWLVDHGPYSEFGDLLTLRGAQFIGAGYNIPNIRGLGRTVATNHVWGSAFRGYGAPQSMFASECLMDMLAEKLGMDPLELRYKNAYRPGDTNPTGQEPEVFSLPDMIDQLRPKYQAALEKAQKESTATHKKGVGISIGVYGSGLDGPDASEAWAELNADGTITVHTAWEDHGQGADIGCVGTAHEALRPMGVAPEKIKFTWPNTATTPNSGPSGGSRQQVMTGNAIRVACENLLKACEKPGGGYYTYDELKAADKPTKITGNWTASGATHCDAVTGLGKPFVVYMYGVFMAEVTVDVATGQTTVDGMTLMADLGSLCNQLATDGQIYGGLAQGIGLALSEDFEDIKKHATLVGAGFPFIKQIPDKLDIVYVNHPRPDGPFGASGVGELPLTSPHAAIINAIKSATGVRIYRLPAYPEKVLEALKA Hydrogen bonds contact Hydrophobic contact | ||||
| 6 | Cyclin-dependent kinase 4 | 2W96 | 4.32 | |
Target general information Gen name CDK4 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Protein kinase superfamily, CMGC Ser/Thr protein kinase family, CDC2/CDKX subfamily Biochemical class Cell cycle Function ATP binding.Cyclin binding.Cyclin-dependent protein serine/threonine kinase activity.Cyclin-dependent protein serine/threonine kinase regulator activity.Protein complex binding. Related diseases Melanoma, cutaneous malignant 3 (CMM3) [MIM:609048]: A malignant neoplasm of melanocytes, arising de novo or from a pre-existing benign nevus, which occurs most often in the skin but may also involve other sites. {ECO:0000269|PubMed:7652577, ECO:0000269|PubMed:8528263, ECO:0000269|PubMed:9311594, ECO:0000269|PubMed:9425228}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB12001; DB03496; DB12010; DB09073; DB02733; DB11730; DB15442 Interacts with Q9UH17; P24385; P30279; P30281; Q16543; P50613; P38936; P46527; P49918; P42771; P42772; P42773; P55273; Q9UJC3; P08238; Q9UKT9; Q0VD86; P01106; Q9ULD0; P28749; Q08999; P09936; Q8N720 EC number 2.7.11.22 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; ATP-binding; Cell cycle; Cell division; Cytoplasm; Disease variant; Kinase; Membrane; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Serine/threonine-protein kinase; Transferase Protein physicochemical properties Chain ID B Molecular weight (Da) 30138.4 Length 267 Aromaticity 0.09 Instability index 36.2 Isoelectric point 5.78 Charge (pH=7) -5.83 2D Binding mode Binding energy (Kcal/mol) -5.89  Molscript Map 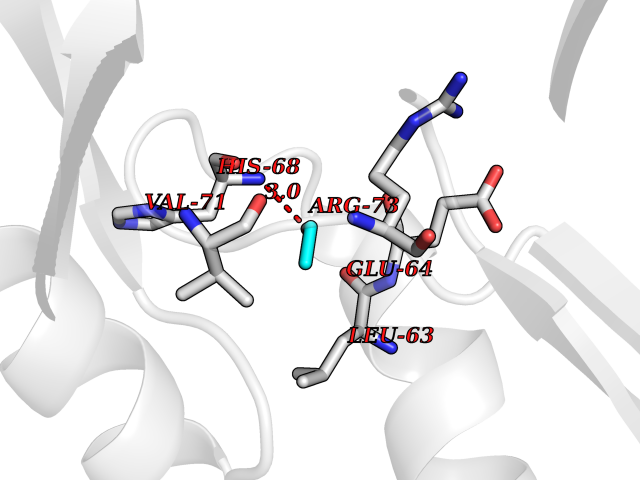 Pymol Map 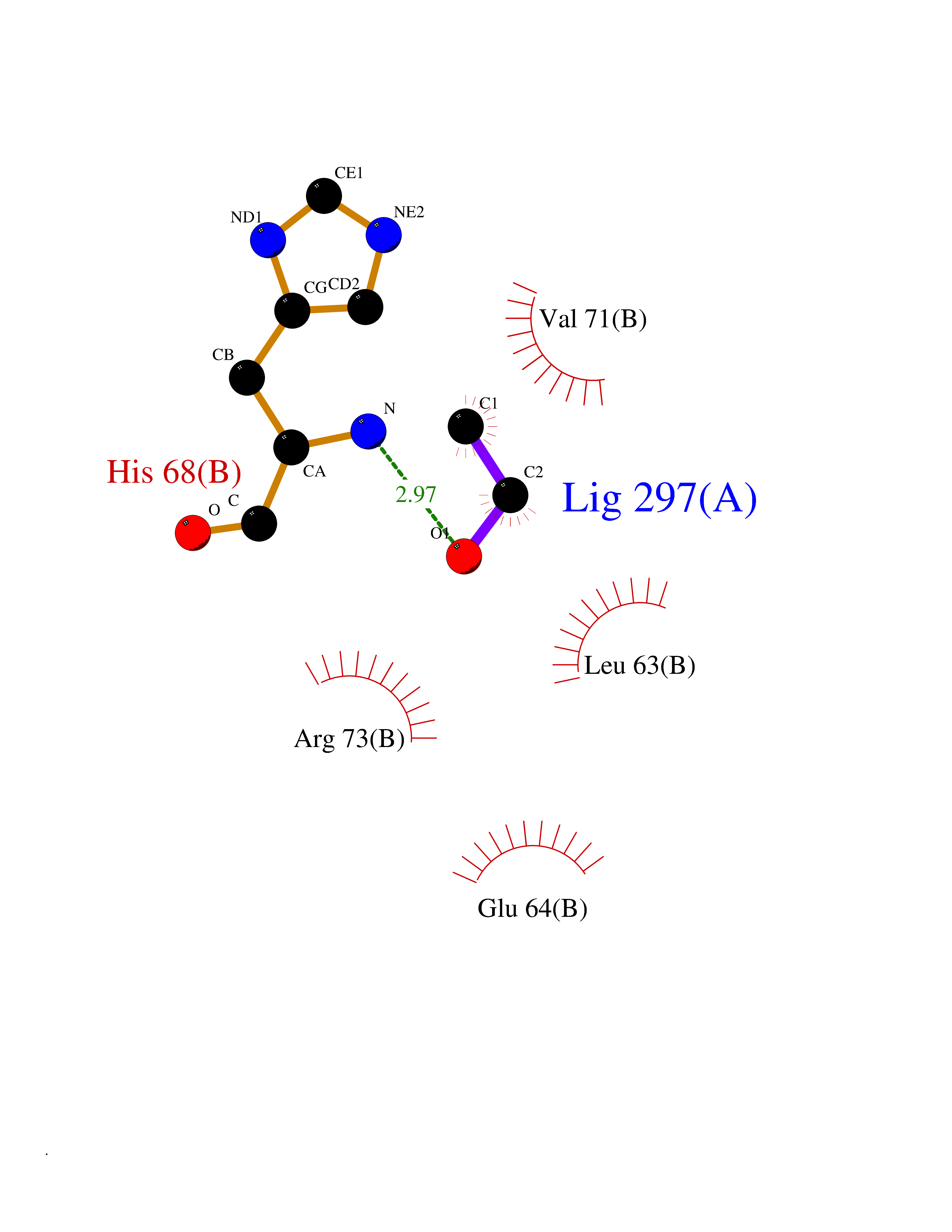 Ligplot Map 3D Binding mode Sequence SRYEPVAEIGVGAYGTVYKARDPHSGHFVALKSVRVPNGEEGLPISTVREVALLRRLEAFEHPNVVRLMDVCATSRTDREIKVTLVFEHVDQDLRTYLDKAPPPGLPAETIKDLMRQFLRGLDFLHANCIVHRDLKPENILVTSGGTVKLADFGLARIYSYQMALDPVVVTLWYRAPEVLLQSTYATPVDMWSVGCIFAEMFRRKPLFCGNSEADQLGKIFDLIGLPPEDDWVPEMEESGAQLLLEMLTFNPHKRISAFRALQHSYL Hydrogen bonds contact Hydrophobic contact | ||||
| 7 | Tartrate-resistant acid phosphatase type 5 | 2BQ8 | 4.32 | |
Target general information Gen name ACP5 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Metallophosphoesterase superfamily, Purple acid phosphatase family Biochemical class Hydrolase Function Acid phosphatase activity.Ferric iron binding.Ferrous iron binding. Related diseases Spondyloenchondrodysplasia with immune dysregulation (SPENCDI) [MIM:607944]: A disease characterized by vertebral and metaphyseal dysplasia, spasticity with cerebral calcifications, and strong predisposition to autoimmune diseases. The skeletal dysplasia is characterized by radiolucent and irregular spondylar and metaphyseal lesions that represent islands of chondroid tissue within bone. {ECO:0000269|PubMed:21217752, ECO:0000269|PubMed:21217755}. The disease is caused by variants affecting the gene represented in this entry. ACP5 inactivating mutations result in a functional excess of phosphorylated osteopontin causing deregulation of osteopontin signaling and consequential autoimmune disease. Drugs (DrugBank ID) NA Interacts with NA EC number 3.1.3.2 Uniprot keywords 3D-structure; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; Hydrolase; Iron; Lysosome; Metal-binding; Proteomics identification; Reference proteome; Signal Protein physicochemical properties Chain ID X Molecular weight (Da) 34330.6 Length 304 Aromaticity 0.12 Instability index 42.3 Isoelectric point 9.11 Charge (pH=7) 6.75 2D Binding mode Binding energy (Kcal/mol) -5.89  Molscript Map 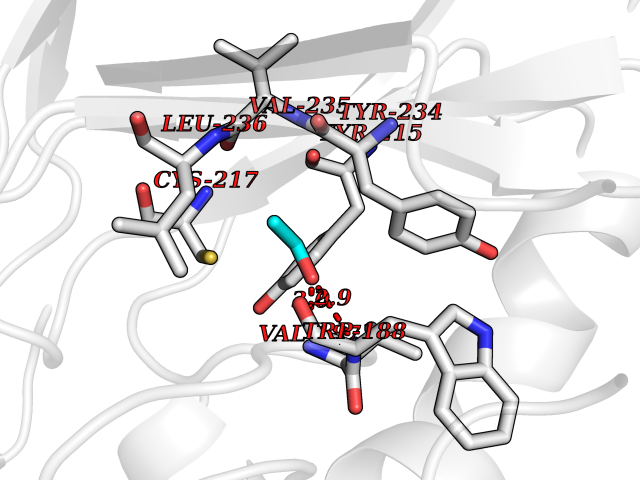 Pymol Map 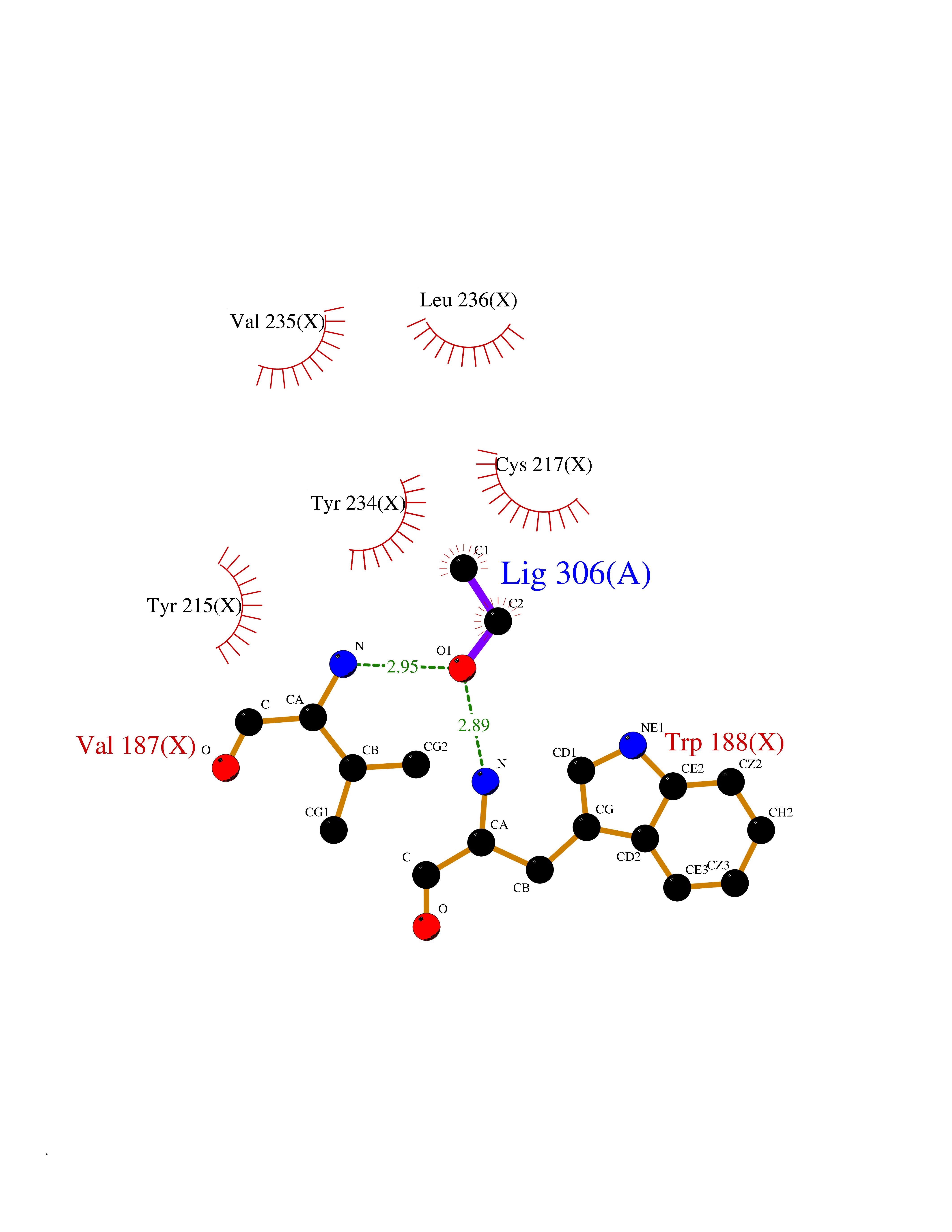 Ligplot Map 3D Binding mode Sequence ATPALRFVAVGDWGGVPNAPFHTAREMANAKEIARTVQILGADFILSLGDNFYFTGVQDINDKRFQETFEDVFSDRSLRKVPWYVLAGNHDHLGNVSAQIAYSKISKRWNFPSPFYRLHFKIPQTNVSVAIFMLDTVTLCGNSDDFLSQQPERPRDVKLARTQLSWLKKQLAAAREDYVLVAGHYPVWSIAEHGPTHCLVKQLRPLLATYGVTAYLCGHDHNLQYLQDENGVGYVLSGAGNFMDPSKRHQRKVPNGYLRFHYGTEDSLGGFAYVEISSKEMTVTYIEASGKSLFKTRLPRRARP Hydrogen bonds contact Hydrophobic contact | ||||
| 8 | Ornithine delta-aminotransferase (OAT) | 2OAT | 4.32 | |
Target general information Gen name OAT Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Ornithine--oxo-acid aminotransferase; Ornithine aminotransferase, mitochondrial Protein family Class-III pyridoxal-phosphate-dependent aminotransferase family Biochemical class Transaminase Function Catalyzes the transfer of the delta-amino group from L-ornithine. Related diseases Hyperornithinemia with gyrate atrophy of choroid and retina (HOGA) [MIM:258870]: A disorder clinically characterized by a triad of progressive chorioretinal degeneration, early cataract formation, and type II muscle fiber atrophy. Characteristic chorioretinal atrophy with progressive constriction of the visual fields leads to blindness at the latest during the sixth decade of life. Patients generally have normal intelligence. {ECO:0000269|PubMed:1612597, ECO:0000269|PubMed:1737786, ECO:0000269|PubMed:23076989, ECO:0000269|PubMed:2793865, ECO:0000269|PubMed:3375240, ECO:0000269|PubMed:7668253, ECO:0000269|PubMed:7887415}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02821; DB02054; DB00129; DB00114 Interacts with P05067 EC number EC 2.6.1.13 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Aminotransferase; Direct protein sequencing; Disease variant; Mitochondrion; Proteomics identification; Pyridoxal phosphate; Reference proteome; Transferase; Transit peptide Protein physicochemical properties Chain ID A,B,C Molecular weight (Da) 44807.9 Length 404 Aromaticity 0.09 Instability index 26.67 Isoelectric point 5.72 Charge (pH=7) -6.54 2D Binding mode Binding energy (Kcal/mol) -5.89  Molscript Map 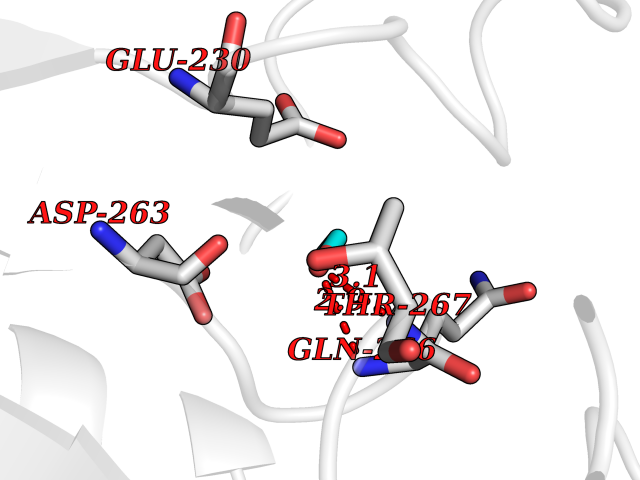 Pymol Map 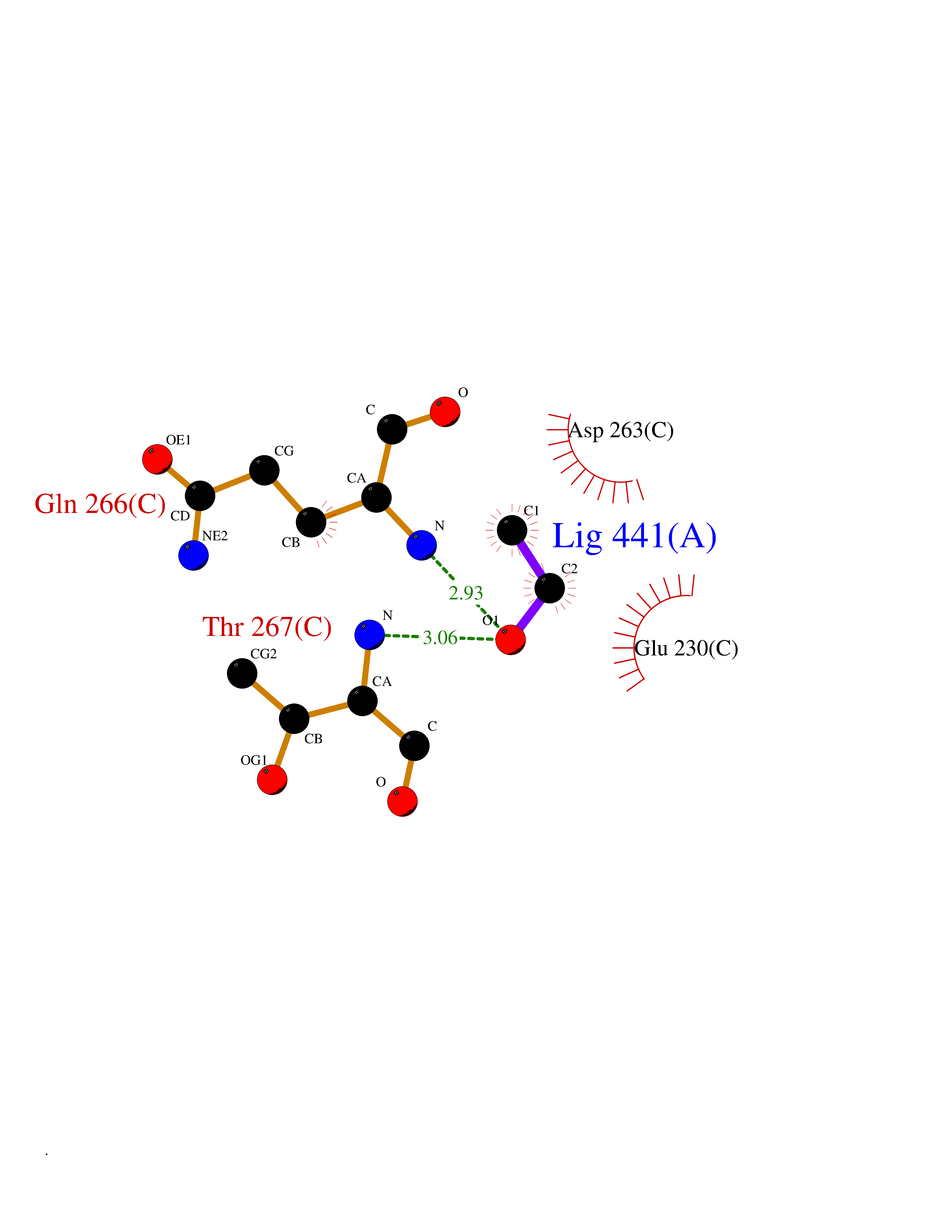 Ligplot Map 3D Binding mode Sequence GPPTSDDIFEREYKYGAHNYHPLPVALERGKGIYLWDVEGRKYFDFLSSYSAVNQGHCHPKIVNALKSQVDKLTLTSRAFYNNVLGEYEEYITKLFNYHKVLPMNTGVEAGETACKLARKWGYTVKGIQKYKAKIVFAAGNFWGRTLSAISSSTDPTSYDGFGPFMPGFDIIPYNDLPALERALQDPNVAAFMVEPIQGEAGVVVPDPGYLMGVRELCTRHQVLFIADEIQTGLARTGRWLAVDYENVRPDIVLLGKALSGGLYPVSAVLCDDDIMLTIKPGEHGSTYGGNPLGCRVAIAALEVLEEENLAENADKLGIILRNELMKLPSDVVTAVRGKGLLNAIVIKETKDWDAWKVCLRLRDNGLLAKPTHGDIIRFAPPLVIKEDELRESIEIINKTILSF Hydrogen bonds contact Hydrophobic contact | ||||
| 9 | Pyruvate oxidase | 2EZ9 | 4.32 | |
Target general information Gen name pox5 Organism Lactiplantibacillus plantarum (strain ATCC BAA-793 / NCIMB 8826 / WCFS1) (Lactobacillus plantarum) Uniprot ID TTD ID NA Synonyms lp_3589 Protein family TPP enzyme family Biochemical class Oxidoreductase Function Magnesium ion binding.Pyruvate oxidase activity.Thiamine pyrophosphate binding. Related diseases Telangiectasia, hereditary hemorrhagic, 2 (HHT2) [MIM:600376]: A multisystemic vascular dysplasia leading to dilation of permanent blood vessels and arteriovenous malformations of skin, mucosa, and viscera. The disease is characterized by recurrent epistaxis and gastro-intestinal hemorrhage. Visceral involvement includes arteriovenous malformations of the lung, liver, and brain. {ECO:0000269|PubMed:10694922, ECO:0000269|PubMed:10767348, ECO:0000269|PubMed:11170071, ECO:0000269|PubMed:11484689, ECO:0000269|PubMed:14684682, ECO:0000269|PubMed:15024723, ECO:0000269|PubMed:15712270, ECO:0000269|PubMed:16525724, ECO:0000269|PubMed:16752392, ECO:0000269|PubMed:20414677, ECO:0000269|PubMed:26176610, ECO:0000269|PubMed:8640225, ECO:0000269|PubMed:9245985}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01987; DB03147 Interacts with NA EC number 1.2.3.3 Uniprot keywords 3D-structure; FAD; Flavoprotein; Magnesium; Metal-binding; Oxidoreductase; Reference proteome; Thiamine pyrophosphate Protein physicochemical properties Chain ID A,B Molecular weight (Da) 64137.9 Length 585 Aromaticity 0.08 Instability index 28.31 Isoelectric point 5.17 Charge (pH=7) -18.41 2D Binding mode Binding energy (Kcal/mol) -5.89 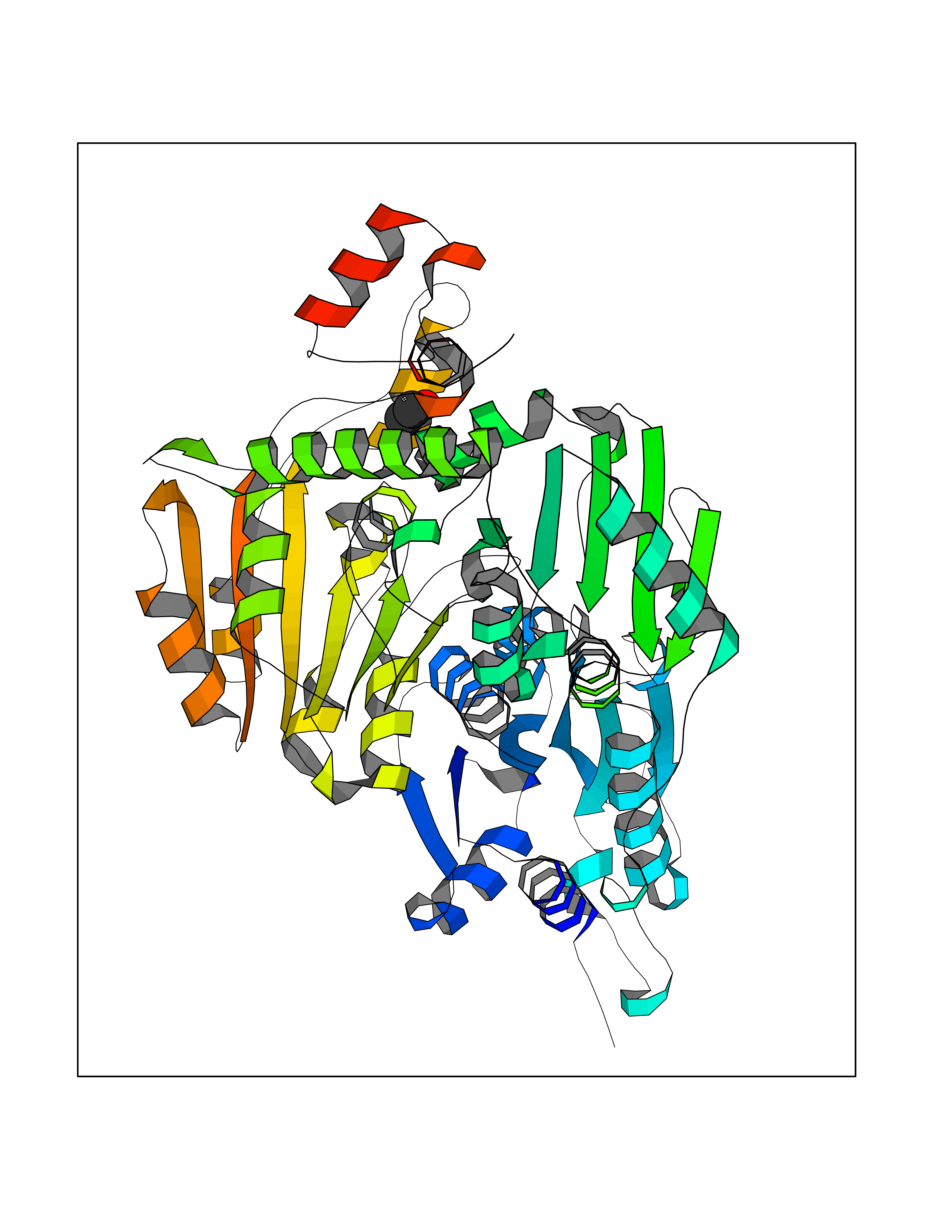 Molscript Map 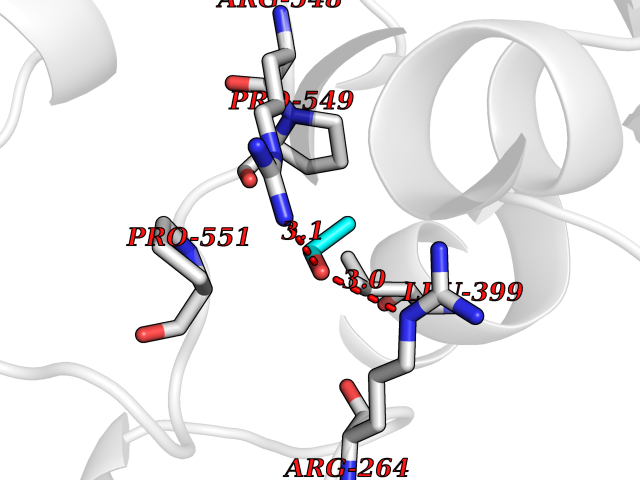 Pymol Map 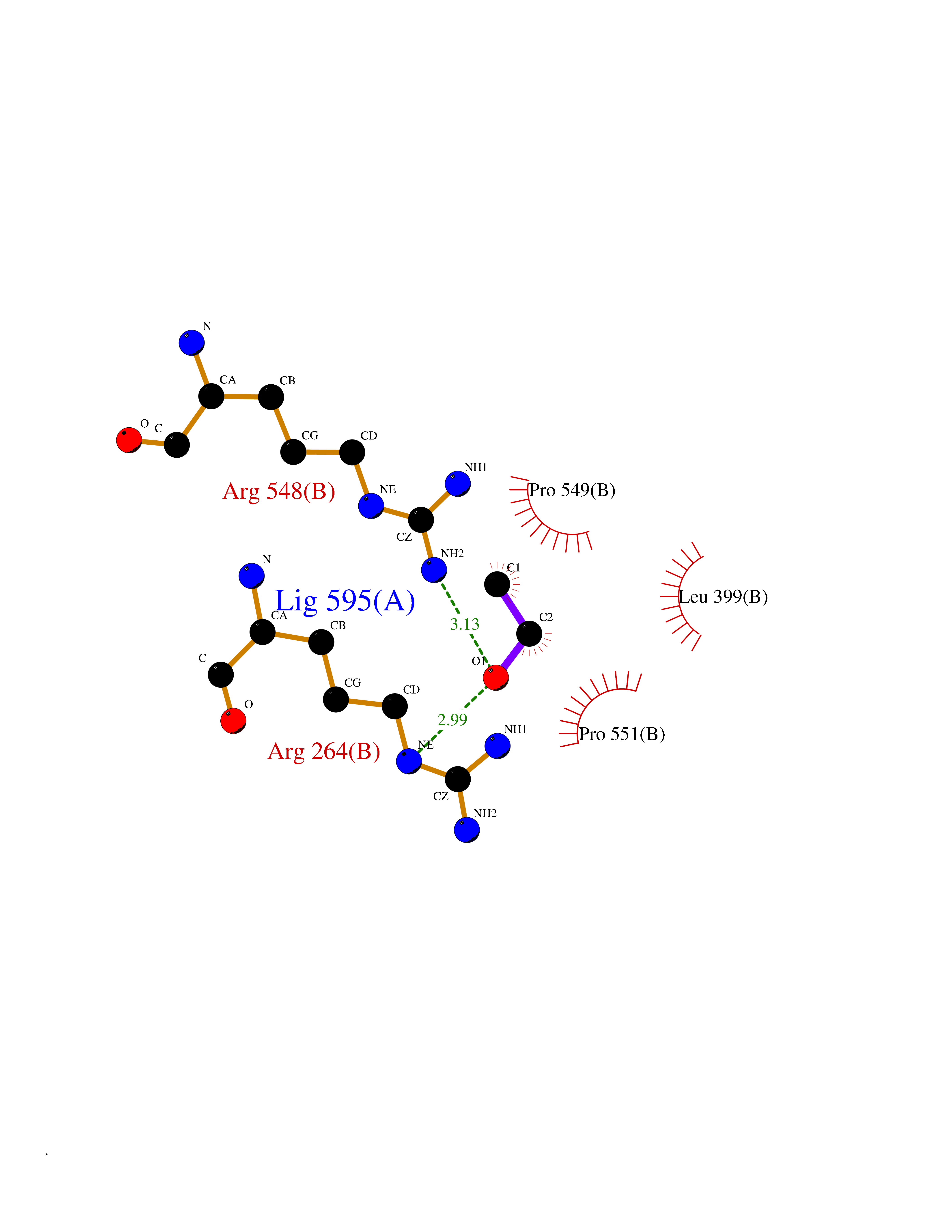 Ligplot Map 3D Binding mode Sequence TNILAGAAVIKVLEAWGVDHLYGIPGGSINSIMDALSAERDRIHYIQVRHEEVGAMAAAADAKLTGKIGVCFGSAGPGGTHLMNGLYDAREDHVPVLALIGQFGTTGMNMDTFQEMNENPIYADVADYNVTAVNAATLPHVIDEAIRRAYAHQGVAVVQIPVDLPWQQIPAEDWYASANSYQTPLLPEPDVQAVTRLTQTLLAAERPLIYYGIGARKAGKELEQLSKTLKIPLMSTYPAKGIVADRYPAYLGSANRVAQKPANEALAQADVVLFVGNNYPFAEVSKAFKNTRYFLQIDIDPAKLGKRHKTDIAVLADAQKTLAAILAQVSERESTPWWQANLANVKNWRAYLASLEDKQEGPLQAYQVLRAVNKIAEPDAIYSIDVGDINLNANRHLKLTPSNRHITSNLFATMGVGIPGAIAAKLNYPERQVFNLAGDGGASMTMQDLATQVQYHLPVINVVFTNCQYGWIKDEQEDTNQNDFIGVEFNDIDFSKIADGVHMQAFRVNKIEQLPDVFEQAKAIAQHEPVLIDAVITGDRPLPAEKLRLDSAMSSAADIEAFKQRYEAQDLQPLSTYLKQFGLDD Hydrogen bonds contact Hydrophobic contact | ||||
| 10 | Histone deacetylase 8 (HDAC8) | 5BWZ | 4.32 | |
Target general information Gen name HDAC8 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Histone deacetylase-8; HDACL1; HD8; CDA07 Protein family Histone deacetylase family, HD type 1 subfamily Biochemical class Carbon-nitrogen hydrolase Function Gives a tag for epigenetic repression and plays an important role in transcriptional regulation, cell cycle progression and developmental events. Histone deacetylases act via the formation of large multiprotein complexes. Also involved in the deacetylation of cohesin complex protein SMC3 regulating release of cohesin complexes from chromatin. May play a role in smooth muscle cell contractility. Responsible for the deacetylation of lysine residues on the N-terminal part of the core histones (H2A, H2B, H3 and H4). Related diseases Cornelia de Lange syndrome 5 (CDLS5) [MIM:300882]: A form of Cornelia de Lange syndrome, a clinically heterogeneous developmental disorder associated with malformations affecting multiple systems. It is characterized by facial dysmorphisms, abnormal hands and feet, growth delay, cognitive retardation, hirsutism, gastroesophageal dysfunction and cardiac, ophthalmologic and genitourinary anomalies. {ECO:0000269|PubMed:22885700, ECO:0000269|PubMed:22889856}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07350; DB02565; DB07586; DB12565; DB05015; DB08168; DB01262; DB11841; DB14490; DB14491; DB14488; DB14501; DB14489; DB12645; DB01592; DB02917; DB06603; DB06819; DB03766; DB12847; DB06176; DB04297; DB00313; DB02546; DB01593; DB14487; DB14533; DB14548 Interacts with NA EC number EC 3.5.1.98 Uniprot keywords 3D-structure; Alternative splicing; Chromatin regulator; Chromosome; Cytoplasm; Disease variant; Hydrolase; Intellectual disability; Metal-binding; Nucleus; Obesity; Phosphoprotein; Proteomics identification; Reference proteome; Repressor; Transcription; Transcription regulation Protein physicochemical properties Chain ID A,B Molecular weight (Da) 39018.4 Length 351 Aromaticity 0.11 Instability index 38.57 Isoelectric point 6.06 Charge (pH=7) -5.26 2D Binding mode Binding energy (Kcal/mol) -5.89 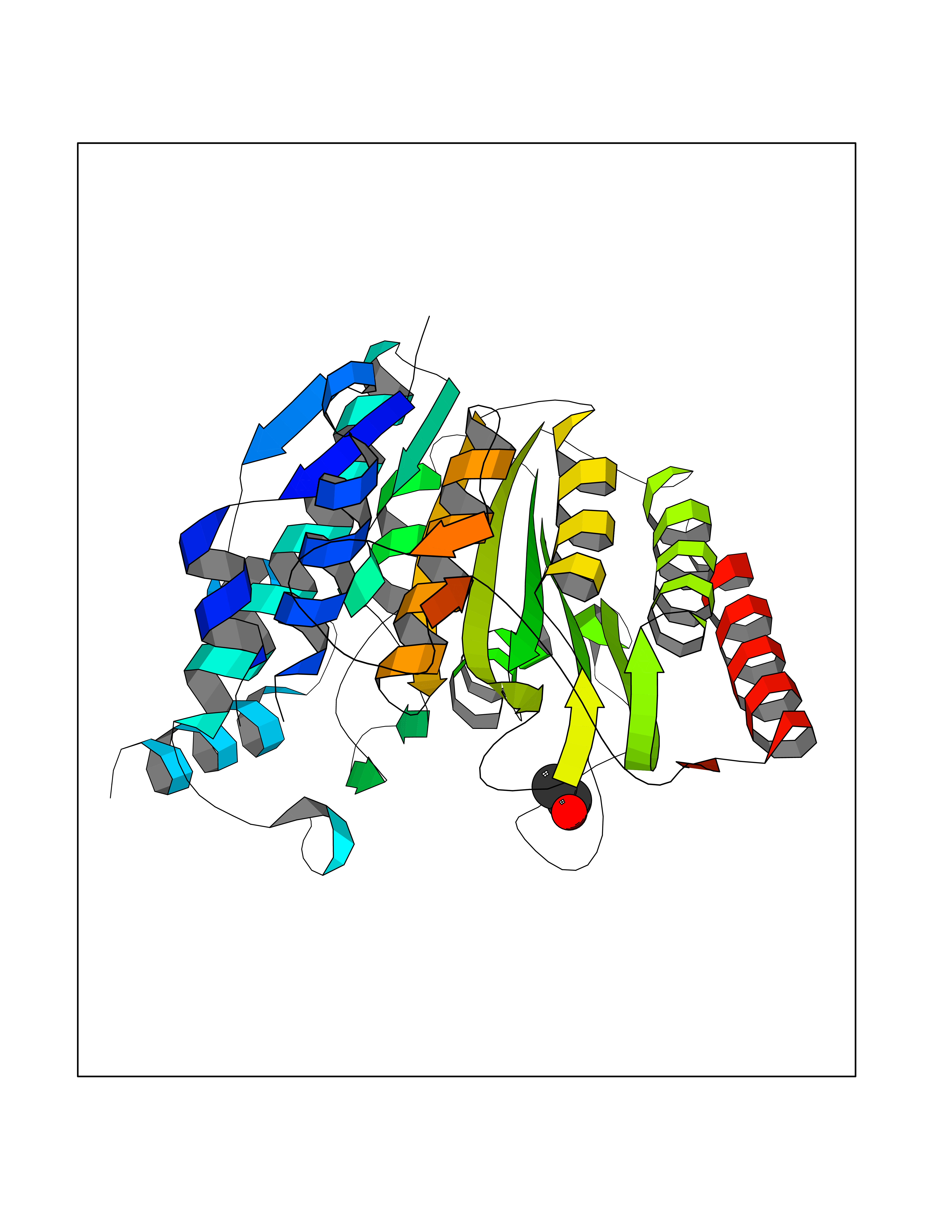 Molscript Map 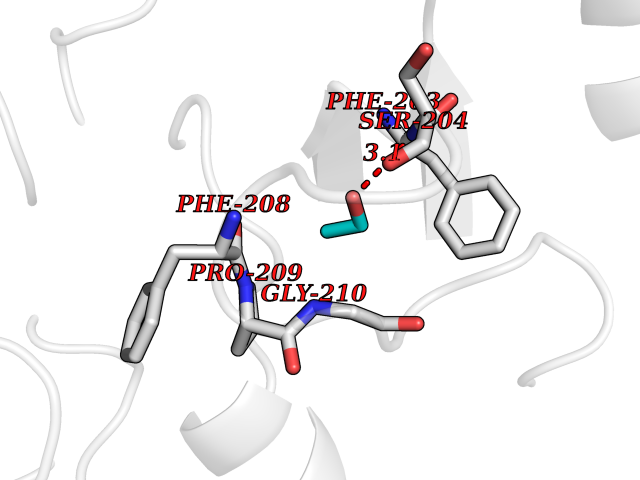 Pymol Map 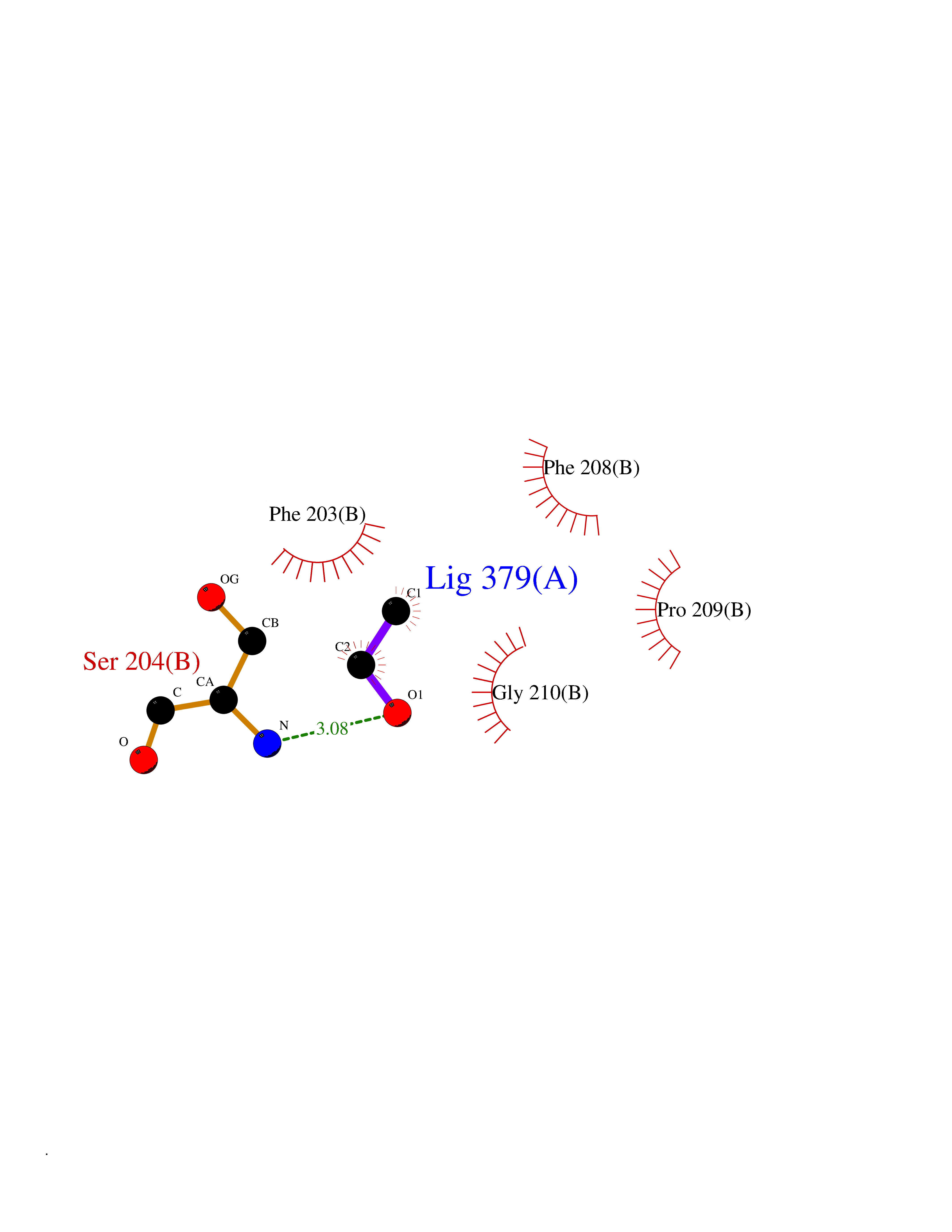 Ligplot Map 3D Binding mode Sequence LVPVYIYSPEYVSMCDSLPKRAEMVHSLIEAYALHKQMRIVKPKVASMEEMATFHTDAYLQHLQKVSQEYGLGYDCPATEGIFDYAAAIGGATITAAQCLIDGMCKVAINWSGGWHHAKKDEASGFCYLNDAVLGILRLRRKFERILYVDLDLHHGDGVEDAFSFTSKVMTVSLHKFSPGFFPGTGDVSDVGLGKGRYYSVNVPIQDGIQDEKYYQICESVLKEVYQAFNPKAVVLQLGADTIAGDPMCSFNMTPVGIGKCLKYILQWQLATLILGGGGYNLANTARCWTYLTGVILGKTLSSEIPDHEFFTAYGPDYVLEITPSCRPDRNEPHRIQQILNYIKGNLKHVV Hydrogen bonds contact Hydrophobic contact | ||||
| 11 | Glutamine amidotransferase (GMPS) | 2VXO | 4.32 | |
Target general information Gen name GMPS Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms GMP synthetase; GMP synthase [glutamine-hydrolyzing] Protein family NA Biochemical class Carbon-nitrogen ligase Function Involved in the de novo synthesis of guanine nucleotides which are not only essential for DNA and RNA synthesis, but also provide GTP, which is involved in a number of cellular processes important for cell division. Related diseases A chromosomal aberration involving GMPS is found in acute myeloid leukemias. Translocation t(3,11)(q25,q23) with KMT2A/MLL1. Drugs (DrugBank ID) DB00993; DB00142; DB00130 Interacts with NA EC number EC 6.3.5.2 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; ATP-binding; Chromosomal rearrangement; Cytoplasm; Direct protein sequencing; Glutamine amidotransferase; GMP biosynthesis; Ligase; Nucleotide-binding; Phosphoprotein; Proteomics identification; Proto-oncogene; Purine biosynthesis; Reference proteome Protein physicochemical properties Chain ID A,B Molecular weight (Da) 144051 Length 1301 Aromaticity 0.07 Instability index 41.55 Isoelectric point 6.8 Charge (pH=7) -1.64 2D Binding mode Binding energy (Kcal/mol) -5.89 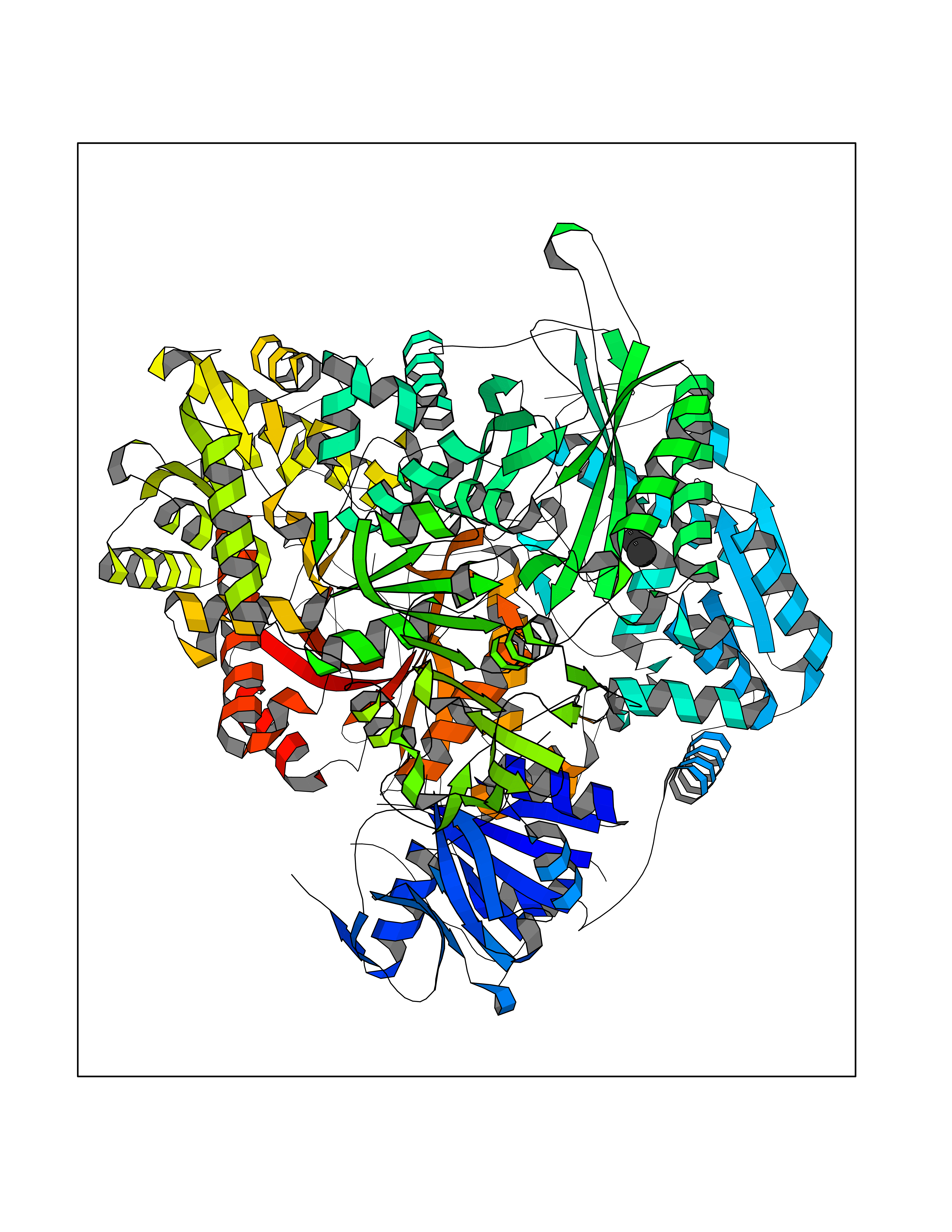 Molscript Map 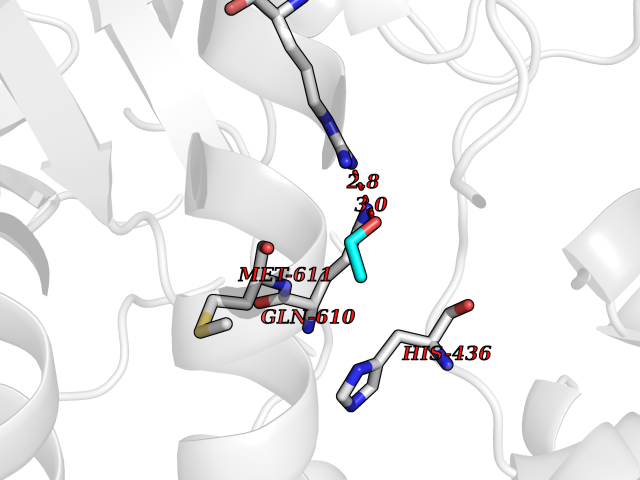 Pymol Map 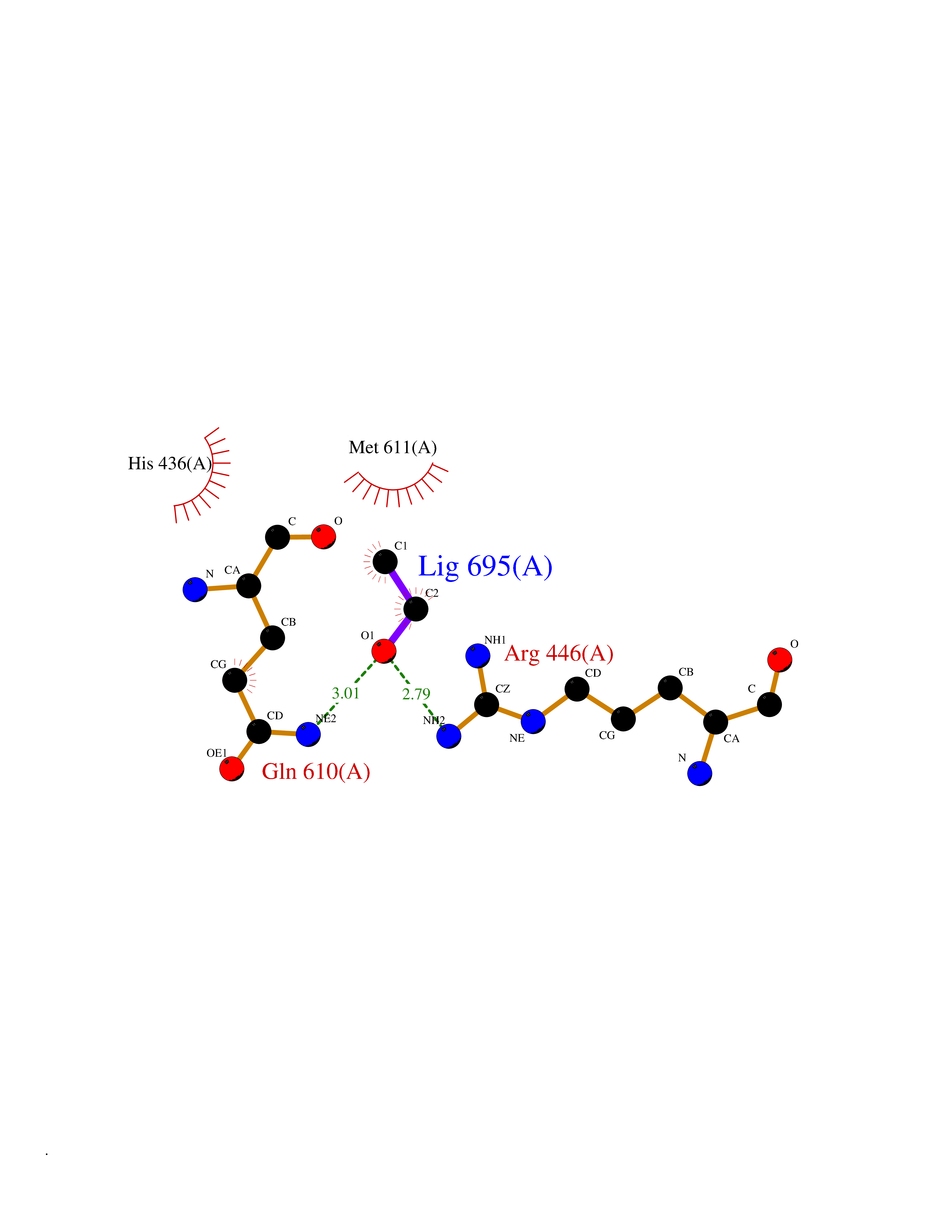 Ligplot Map 3D Binding mode Sequence EGAVVILDAGYGKVIDRRVRELFVQSEIFPLETPAFAIKEQGFRAIIISGGPAPWFDPAIFTIGKPVLGICYGMQMMNKVFGGTVHKVFNISVDNTCSLFRGLQKEEVVLLTHGDSVDKVADGFKVVARSGNIVAGIANESKKLYGAQFHPEVGLTENGKVILKNFLYDIAGCSGTFTVQNRELECIREIKERVGTSKVLVLLSGGVDSTVCTALLNRALNQEQVIAVHIDNGFMRKRESQSVEEALKKLGIQVKVINAAHSFYNGTTTLPRISKTLNMTTSPEEKRKIIGDTFVKIANEVIGEMNLKPEEVFLAQGTLRPDLIESASLVASGKAELIKTHHNDTELIRKLREEGKVIEPLKDFHKDEVRILGRELGLPEELVSRHPFPGPGLAIRVICAEEPYICKDFPETNNILKIVADFSASVKKPHTLLQRVKACTTEEDQEKLMQITSLHSLNAFLLPIKTVGVQGDCRSYSYVCGISSKDEPDWESLIFLARLIPRMCHNVNRVVYIFGPPVKEPPTDVTPTFLTTGVLSTLRQADFEAHNILRESGYAGKISQMPVILTPLHFDRDPLQKQPSCQRSVVIRTFITSDFMTGIPATPGNEIPVEVVLKMVTEIKKIPGISRIMYDLTSKPPGTTEWEHYEGAVVILDAGAQYGKVIDRRVRELFVQSEIFPLETPAFAIKEQGFRAIIISGGPNSVYAEDAPWFDPAIFTIGKPVLGICYGMQMMNKVFGGTVHKKSDGVFNISVDNTCSLFRGLQKEEVVLLTHGDSVDKVADGFKVVARSGNIVAGIANESKKLYGAQFHPEVGLTENGKVILKNFLYDIAGCSGTFTVQNRELECIREIKERVGTSKVLVLLSGGVDSTVCTALLNRALNQEQVIAVHIDNGFMRKRESQSVEEALKKLGIQVKVINAAHSFYNGTTTLPRISKTLNMTTSPEEKRKIIGDTFVKIANEVIGEMNLKPEEVFLAQGTLRPDLIESASLVASGKAELIKTHHNDTELIRKLREEGKVIEPLKDFHKDEVRILGRELGLPEELVSRHPFPGPGLAIRVICAEEPYICKDFPETNNILKIVADFSASVKKPHTLLQRVKACTTEEDQEKLMQITSLHSLNAFLLPIKTVGVQGDCRSYSYVCGISSKDEPDWESLIFLARLIPRMCHNVNRVVYIFGPPVKEPPTDVTPTFLTTGVLSTLRQADFEAHNILRESGYAGKISQMPVILTPLHFDRDPLQKQPSCQRSVVIRTFITSDFMTGIPATPGNEIPVEVVLKMVTEIKKIPGISRIMYDLTSKPPGTTEWE Hydrogen bonds contact Hydrophobic contact | ||||
| 12 | Bacterial Cystathionine beta-lyase (Bact metC) | 4ITX | 4.31 | |
Target general information Gen name Bact metC Organism Escherichia coli (strain K12) Uniprot ID TTD ID Synonyms Cysteine-S-conjugate beta-lyase MetC; Cysteine lyase MetC; Cysteine desulfhydrase MetC; Cystathionine beta-lyase MetC; CL; CBL; Beta-cystathionase MetC; Bacterial CD Protein family Trans-sulfuration enzymes family Biochemical class Carbon-sulfur lyases Function Primarily catalyzes the cleavage of cystathionine to homocysteine, pyruvate and ammonia during methionine biosynthesis. Also exhibits cysteine desulfhydrase activity, producing sulfide from cysteine. In addition, under certain growth conditions, exhibits significant alanine racemase coactivity. Related diseases Coronary artery disease, autosomal dominant, 2 (ADCAD2) [MIM:610947]: A common heart disease characterized by reduced or absent blood flow in one or more of the arteries that encircle and supply the heart. Its most important complication is acute myocardial infarction. {ECO:0000269|PubMed:17332414, ECO:0000269|PubMed:23703864}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Tooth agenesis, selective, 7 (STHAG7) [MIM:616724]: An autosomal dominant form of selective tooth agenesis, a common anomaly characterized by the congenital absence of one or more teeth. Selective tooth agenesis without associated systemic disorders has sometimes been divided into 2 types: oligodontia, defined as agenesis of 6 or more permanent teeth, and hypodontia, defined as agenesis of less than 6 teeth. The number in both cases does not include absence of third molars (wisdom teeth). {ECO:0000269|PubMed:26387593}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number EC 4.4.1.13 Uniprot keywords 3D-structure; Amino-acid biosynthesis; Cytoplasm; Direct protein sequencing; Lyase; Methionine biosynthesis; Pyridoxal phosphate; Reference proteome Protein physicochemical properties Chain ID A Molecular weight (Da) 42756.3 Length 391 Aromaticity 0.08 Instability index 27.11 Isoelectric point 6.01 Charge (pH=7) -6.11 2D Binding mode Binding energy (Kcal/mol) -5.88  Molscript Map 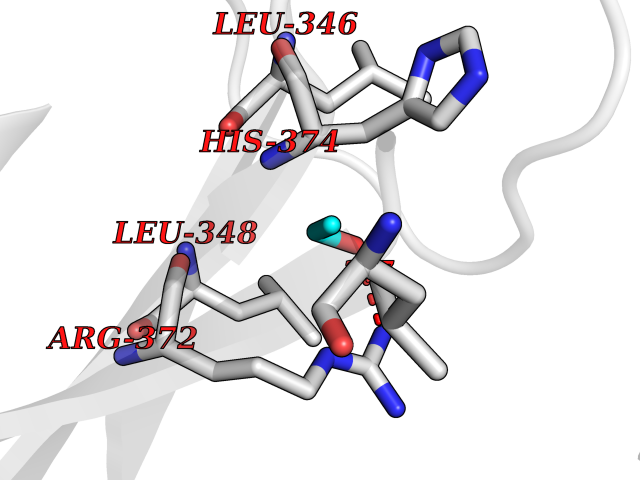 Pymol Map 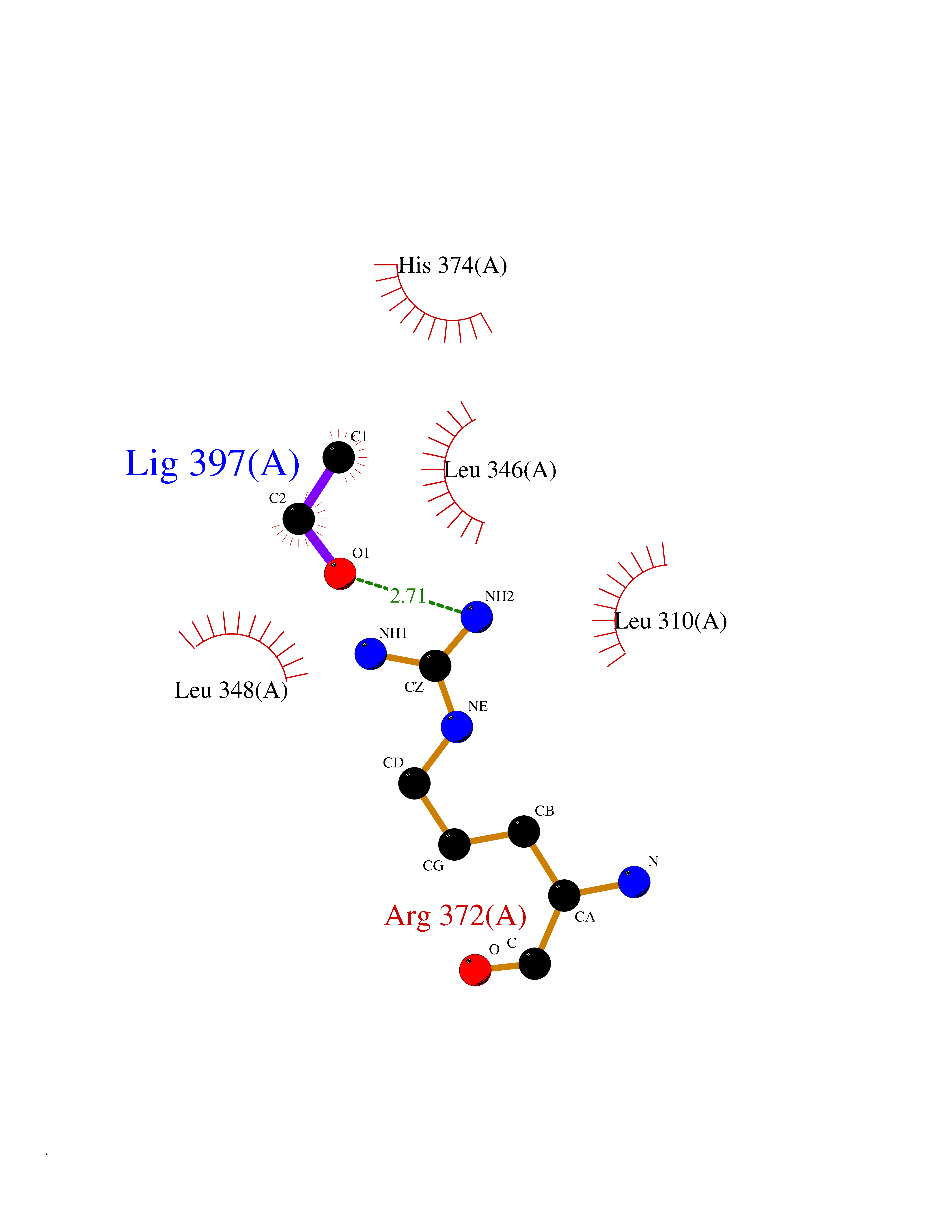 Ligplot Map 3D Binding mode Sequence KLDTQLVNAGRSKKYTLGAVNSVIQRASSLVFDSVEAKKHATRNRANGELFYGRRGTLTHFSLQQAMCELEGGAGCVLFPCGAAAVANSILAFIEQGDHVLMTNTAYESSQDFCSKILSKLGVTTSWFDPLIGADIVKHLQPNTKIVFLESPGSITMEVHDVPAIVAAVRSVVPDAIIMIDNTWAAGVLFKALDFGIDVSIQAATKYLVGHSDAMIGTAVCNARCWEQLRENAYLMGQMVDADTAYITSRGLRTLGVRLRQHHESSLKVAEWLAEHPQVARVNHPALPGSKGHEFWKRDFTGSSGLFSFVLKKKLNNEELANYLDNFSLFSMAYSWGGYESLILANQPEHIAAIRPQGEIDFSGTLIRLHIGLEDVDDLIADLDAGFARIV Hydrogen bonds contact Hydrophobic contact | ||||
| 13 | ADAM metallopeptidase with thrombospondin 1 (ADAMTS1) | 2JIH | 4.31 | |
Target general information Gen name ADAMTS1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms METH1; METH-1; KIAA1346; ADAMTS-1; ADAM-TS1; ADAM-TS 1 Protein family NA Biochemical class Peptidase Function Has angiogenic inhibitor activity. Active metalloprotease, which may be associated with various inflammatory processes as well as development of cancer cachexia. May play a critical role in follicular rupture. Cleaves aggrecan, a cartilage proteoglycan, at the '1938-Glu-|-Leu-1939' site (within the chondroitin sulfate attachment domain), and may be involved in its turnover. Related diseases Spermatogenic failure 21 (SPGF21) [MIM:617644]: An infertility disorder caused by spermatogenesis defects and characterized by acephalic spermatozoa in the semen of affected individuals. SPGF21 inheritance is autosomal recessive. {ECO:0000269|PubMed:28199965}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q6A162; P60410; O00233; P0DTC8 EC number EC 3.4.24.- Uniprot keywords 3D-structure; Calcium; Cleavage on pair of basic residues; Disulfide bond; Extracellular matrix; Glycoprotein; Heparin-binding; Hydrolase; Metal-binding; Metalloprotease; Protease; Proteomics identification; Reference proteome; Repeat; Secreted; Signal; Zinc; Zymogen Protein physicochemical properties Chain ID A Molecular weight (Da) 31341 Length 284 Aromaticity 0.07 Instability index 42.81 Isoelectric point 5.68 Charge (pH=7) -10.36 2D Binding mode Binding energy (Kcal/mol) -5.88 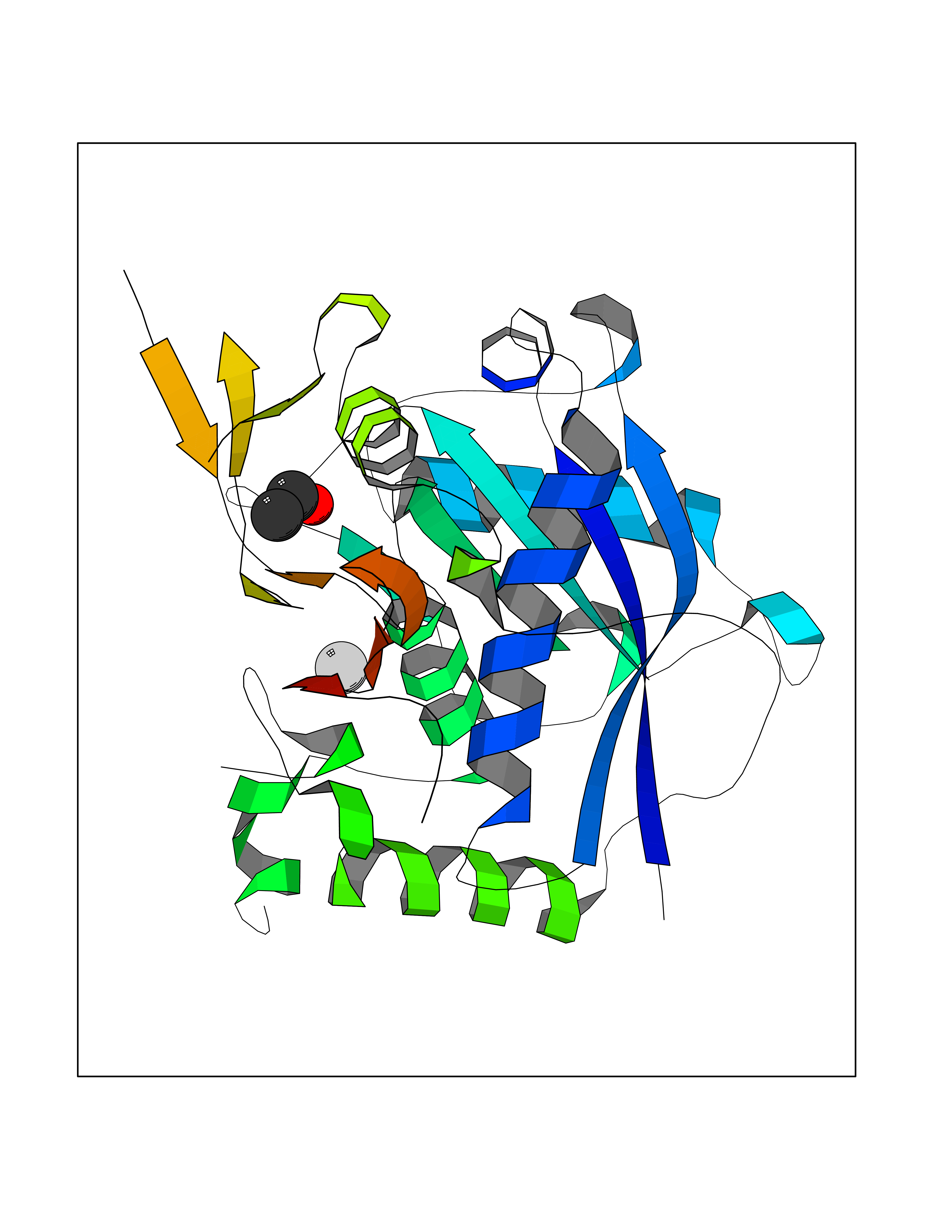 Molscript Map 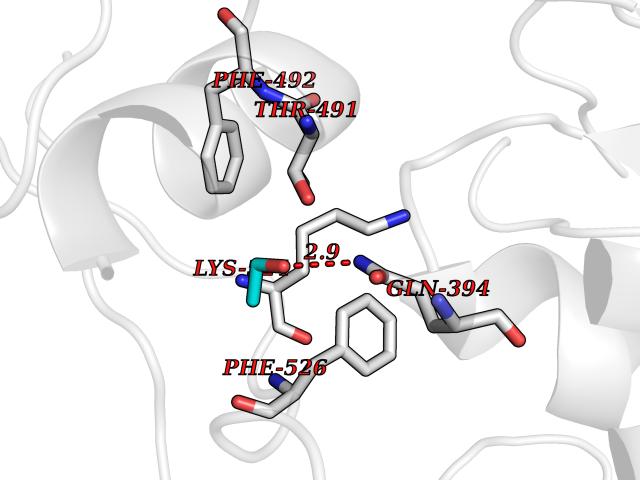 Pymol Map 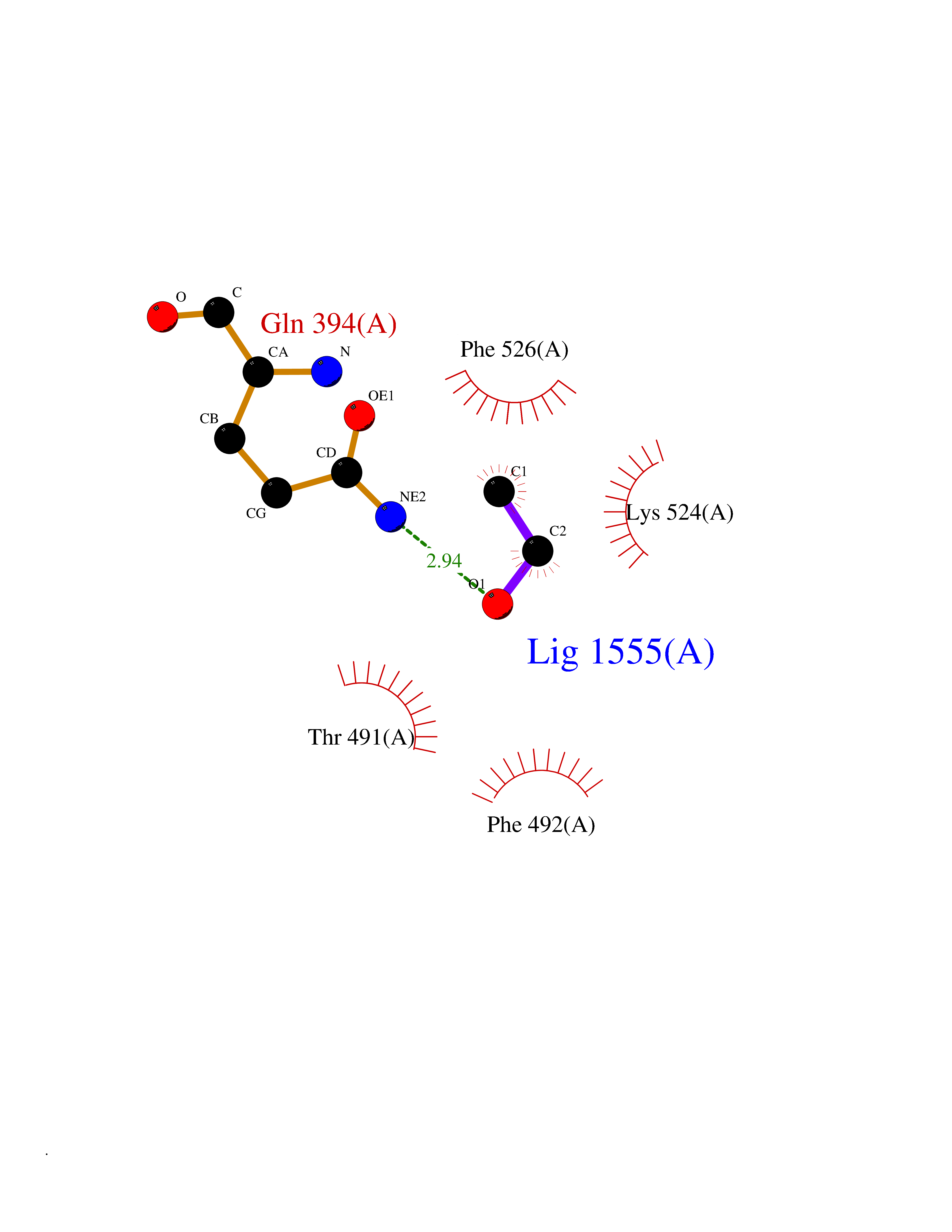 Ligplot Map 3D Binding mode Sequence SHRYVETMLVADQSMAEFHGSGLKHYLLTLFSVAARLYKHPSIRNSVSLVVVKILVIHDEQKGPEVTSNAALTLRNFCNWQKQHNPPSDRDAEHYDTAILFTRQDLCGSQTCDTLGMADVGTVCDPSRSCSVIEDDGLQAAFTTAHELGHVFNMPHDDAKQCASLNDSHMMASMLSNLDHSQPWSPCSAYMITSFLDNGHGECLMDKPQNPIQLPGDLPGTSYDANRQCQFTFGEDSKHCPTCSTLWCTGVLVCQTKHFPWADGTSCGEGKWCINGKCVNKLVP Hydrogen bonds contact Hydrophobic contact | ||||
| 14 | Coagulation factor Xa (F10) | 2JKH | 4.30 | |
Target general information Gen name F10 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Fxa; Factor Xa; F10; Activated coagulation factor X Protein family Peptidase S1 family Biochemical class Peptidase Function Factor Xa is avitamin K-dependent glycoprotein that converts prothrombin to thrombin in the presence of factor Va, calcium and phospholipid during blood clotting. Related diseases Factor X deficiency (FA10D) [MIM:227600]: A hemorrhagic disease with variable presentation. Affected individuals can manifest prolonged nasal and mucosal hemorrhage, menorrhagia, hematuria, and occasionally hemarthrosis. Some patients do not have clinical bleeding diathesis. {ECO:0000269|PubMed:10468877, ECO:0000269|PubMed:10739379, ECO:0000269|PubMed:10746568, ECO:0000269|PubMed:11248282, ECO:0000269|PubMed:11728527, ECO:0000269|PubMed:12574802, ECO:0000269|PubMed:12945883, ECO:0000269|PubMed:15075089, ECO:0000269|PubMed:15650540, ECO:0000269|PubMed:17393015, ECO:0000269|PubMed:19135706, ECO:0000269|PubMed:1973167, ECO:0000269|PubMed:1985698, ECO:0000269|PubMed:25313940, ECO:0000269|PubMed:26222694, ECO:0000269|PubMed:2790181, ECO:0000269|PubMed:7669671, ECO:0000269|PubMed:7860069, ECO:0000269|PubMed:8529633, ECO:0000269|PubMed:8845463, ECO:0000269|PubMed:8910490}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07211; DB08746; DB07974; DB07277; DB07605; DB08487; DB08495; DB04673; DB08745; DB08488; DB07804; DB08174; DB08173; DB07872; DB07843; DB07848; DB07875; DB08143; DB07847; DB07844; DB13884; DB06552; DB13151; DB13192; DB00025; DB11166; DB06605; DB09258; DB12364; DB00100; DB13152; DB13150; DB00036; DB09075; DB16662; DB13923; DB01225; DB06920; DB00569; DB03847; DB07278; DB01109; DB06406; DB09332; DB06245; DB13998; DB05713; DB13999; DB07630; DB07629; DB07973; DB07800; DB12598; DB13933; DB06635; DB09141; DB13149; DB11311; DB06228; DB05362; DB07261; DB08426; DB09109; DB14738 Interacts with P0DPK4; Q9UK55; Q9UHD9 EC number EC 3.4.21.6 Uniprot keywords 3D-structure; Blood coagulation; Calcium; Cleavage on pair of basic residues; Direct protein sequencing; Disease variant; Disulfide bond; EGF-like domain; Gamma-carboxyglutamic acid; Glycoprotein; Hemostasis; Hydrolase; Hydroxylation; Protease; Proteomics identification; Reference proteome; Repeat; Secreted; Serine protease; Signal; Zymogen Protein physicochemical properties Chain ID A Molecular weight (Da) 31315.2 Length 280 Aromaticity 0.09 Instability index 38.33 Isoelectric point 6.36 Charge (pH=7) -1.82 2D Binding mode Binding energy (Kcal/mol) -5.86 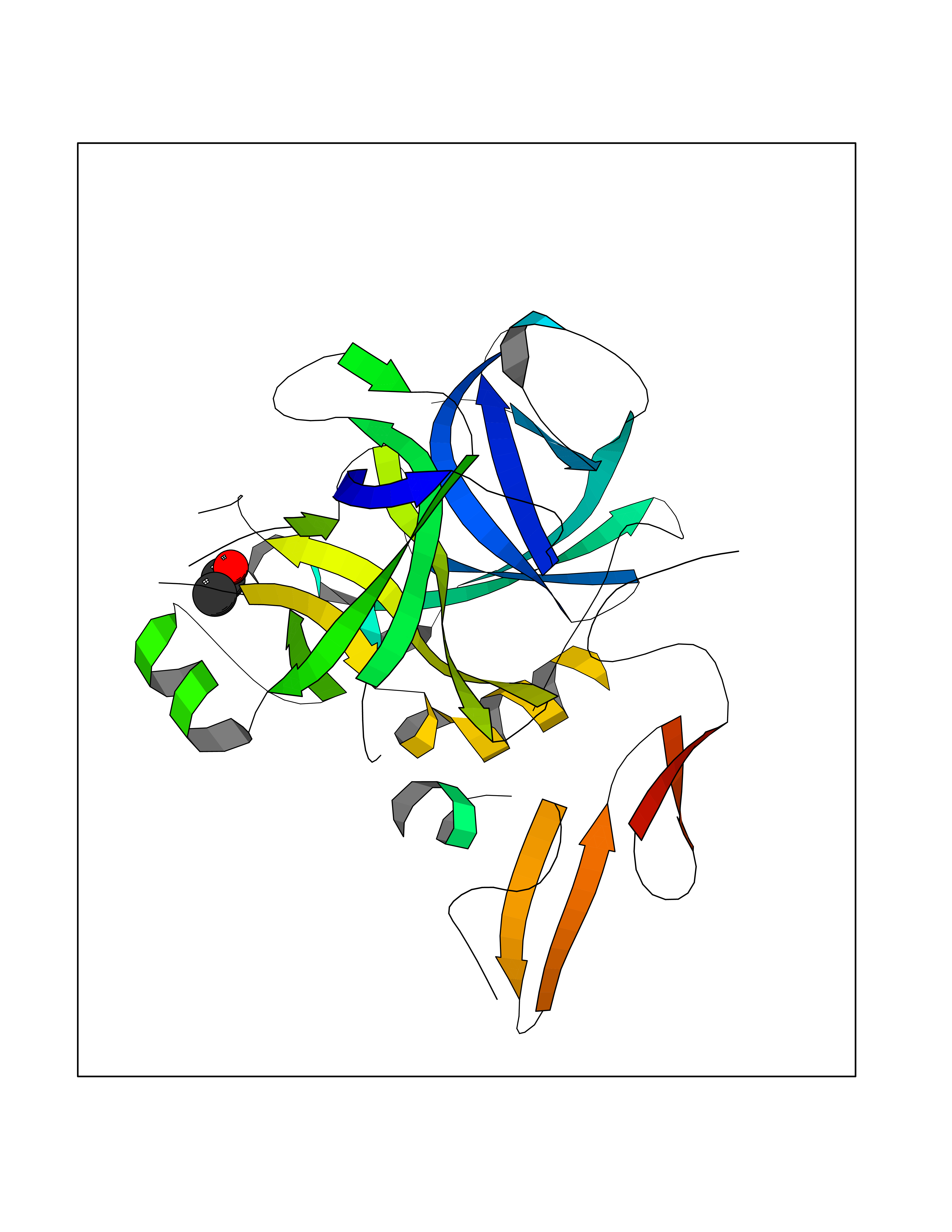 Molscript Map 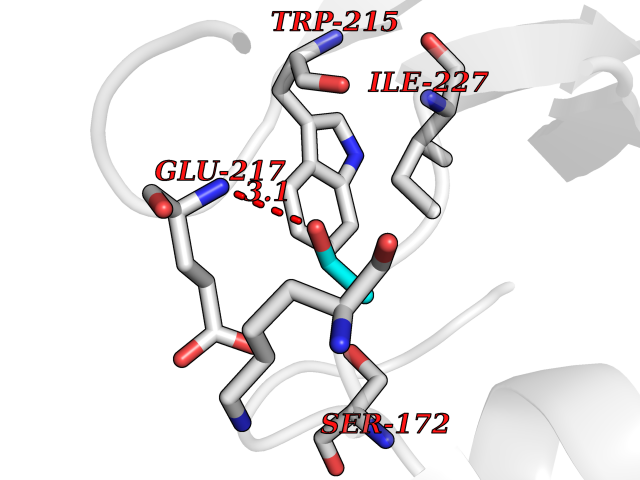 Pymol Map 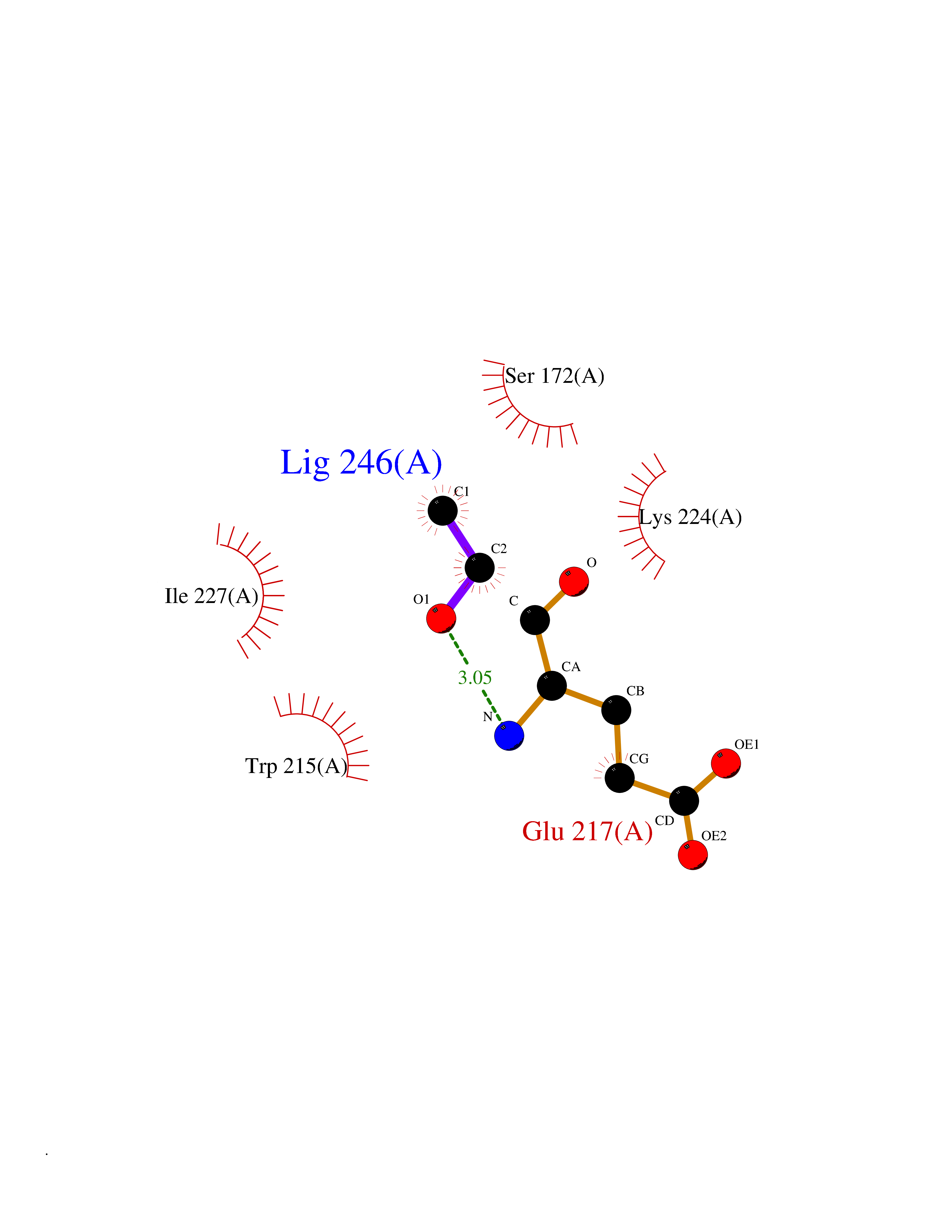 Ligplot Map 3D Binding mode Sequence IVGGQECKDGECPWQALLINEENEGFCGGTILSEFYILTAAHCLYAKRFKVRVGDRNTEQEEGGEAVHEVEVVIKHNRFTKETYDFDIAVLRLKTPITFRMNVAPACLERDWAESMTQKTGIVSGFGRTHEKGEQSTRLKMLEVPYVDRNSCKLSSSFIITQNMFCAGTKQEDACQGDSGGPHVTRFKDTYFVTGIVSWGEGCARGKYGIYTKVTAFLKWIDRSMKKLCSLDNGDCDQFCHEEQNSVVCSCARGYTLADNGKACIPTGPYPCGKQTLERR Hydrogen bonds contact Hydrophobic contact | ||||
| 15 | Fms-like tyrosine kinase 3 (FLT-3) | 1RJB | 4.30 | |
Target general information Gen name FLT3 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Stem cell tyrosine kinase 1; STK1; STK-1; Receptor-type tyrosine-protein kinase FLT3; Fetal liver kinase-2; FLT-3; FLK2; FLK-2; FL cytokine receptor; CD135 Protein family Protein kinase superfamily, Tyr protein kinase family, CSF-1/PDGF receptor subfamily Biochemical class Kinase Function Tyrosine-protein kinase that acts as cell-surface receptor for the cytokine FLT3LG and regulates differentiation, proliferation and survival of hematopoietic progenitor cells and of dendritic cells. Promotes phosphorylation of SHC1 and AKT1, and activation of the downstream effector MTOR. Promotes activation of RAS signaling and phosphorylation of downstream kinases, including MAPK1/ERK2 and/or MAPK3/ERK1. Promotes phosphorylation of FES, FER, PTPN6/SHP, PTPN11/SHP-2, PLCG1, and STAT5A and/or STAT5B. Activation of wild-type FLT3 causes only marginal activation of STAT5A or STAT5B. Mutations that cause constitutive kinase activity promote cell proliferation and resistance to apoptosis via the activation of multiple signaling pathways. Related diseases Leukemia, acute myelogenous (AML) [MIM:601626]: A subtype of acute leukemia, a cancer of the white blood cells. AML is a malignant disease of bone marrow characterized by maturational arrest of hematopoietic precursors at an early stage of development. Clonal expansion of myeloid blasts occurs in bone marrow, blood, and other tissue. Myelogenous leukemias develop from changes in cells that normally produce neutrophils, basophils, eosinophils and monocytes. {ECO:0000269|PubMed:11090077, ECO:0000269|PubMed:11290608, ECO:0000269|PubMed:11442493, ECO:0000269|PubMed:14504097, ECO:0000269|PubMed:16266983, ECO:0000269|PubMed:18305215, ECO:0000269|PubMed:8946930, ECO:0000269|PubMed:9737679}. The gene represented in this entry may be involved in disease pathogenesis. Somatic mutations that lead to constitutive activation of FLT3 are frequent in AML patients. These mutations fall into two classes, the most common being in-frame internal tandem duplications of variable length in the juxtamembrane region that disrupt the normal regulation of the kinase activity. Likewise, point mutations in the activation loop of the kinase domain can result in a constitutively activated kinase. Drugs (DrugBank ID) DB12742; DB12267; DB12500; DB12010; DB12141; DB06469; DB06080; DB06595; DB11763; DB09079; DB11697; DB12978; DB08901; DB15822; DB12874; DB00398; DB01268; DB05465; DB11800; DB05014 Interacts with P00519; P42684; P46108; P46109; P06241; Q13322; Q9Y6K9; P06239; P27986; P20936; P43405; Q8R4L0 EC number EC 2.7.10.1 Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Disease variant; Disulfide bond; Endoplasmic reticulum; Glycoprotein; Host-virus interaction; Immunoglobulin domain; Kinase; Membrane; Nucleotide-binding; Phosphoprotein; Proteomics identification; Proto-oncogene; Receptor; Reference proteome; Signal; Transferase; Transmembrane; Transmembrane helix; Tyrosine-protein kinase; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 34209.2 Length 298 Aromaticity 0.14 Instability index 39.68 Isoelectric point 5.57 Charge (pH=7) -4.02 2D Binding mode Binding energy (Kcal/mol) -5.87 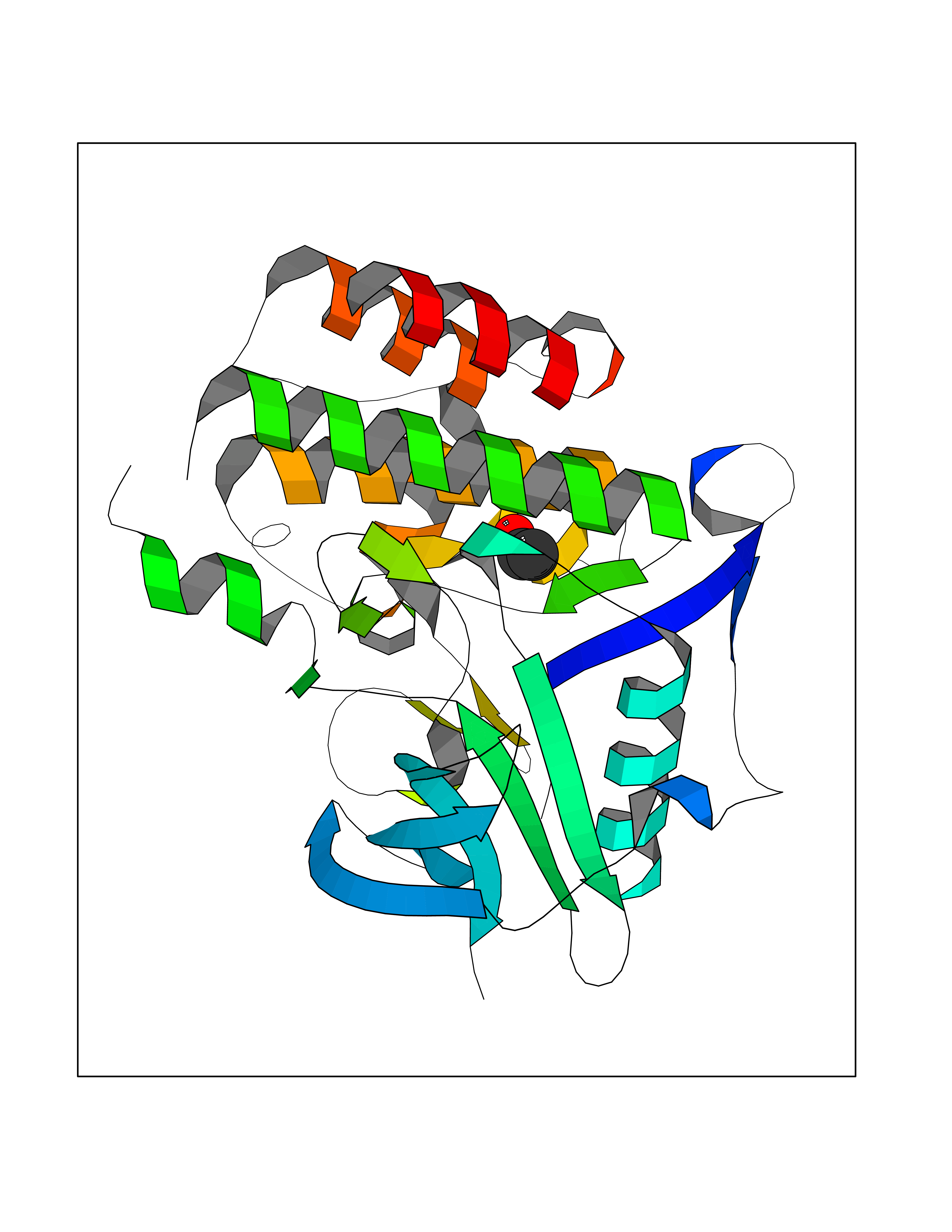 Molscript Map 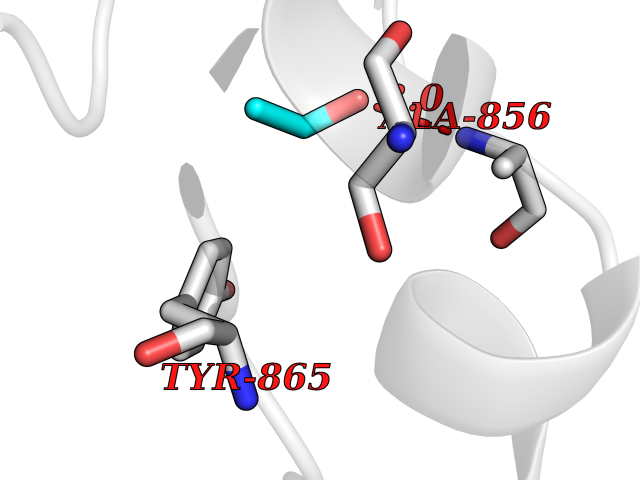 Pymol Map 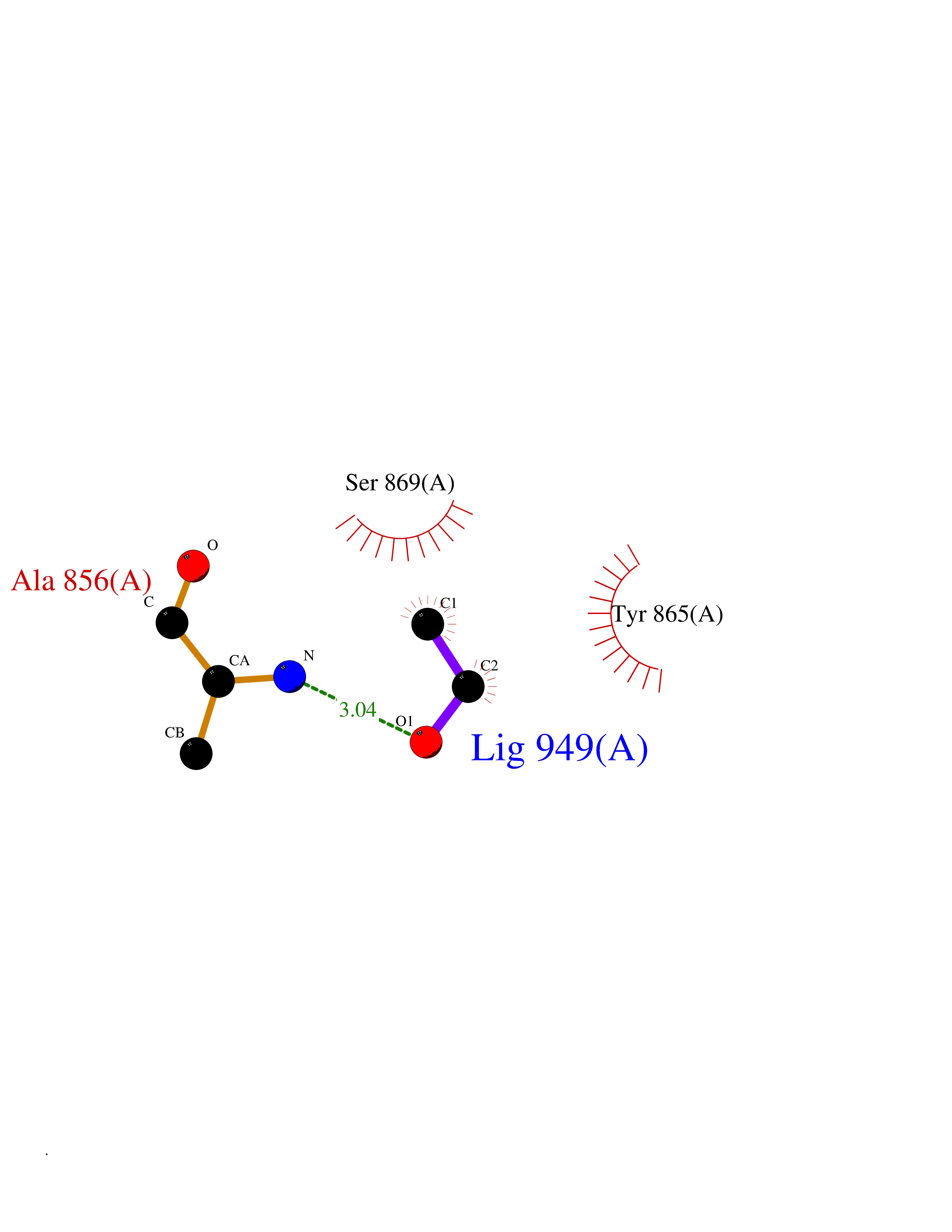 Ligplot Map 3D Binding mode Sequence YESQLQMVQVTGSSDNEYFYVDFREYEYDLKWEFPRENLEFGKVLGSGAFGKVMNATAYGISKTGVSIQVAVKMLKEREALMSELKMMTQLGSHENIVNLLGACTLSGPIYLIFEYCCYGDLLNYLRSKREKFLTFEDLLCFAYQVAKGMEFLEFKSCVHRDLAARNVLVTHGKVVKICDFGLARDIMSDSNYVVRGNARLPVKWMAPESLFEGIYTIKSDVWSYGILLWEIFSLGVNPYPGIPVDANFYKLIQNGFKMDQPFYATEEIYIIMQSCWAFDSRKRPSFPNLTSFLGCQL Hydrogen bonds contact Hydrophobic contact | ||||
| 16 | Dihydroorotate dehydrogenase (quinone), mitochondrial | 4CQ8 | 4.30 | |
Target general information Gen name PFF0160c Organism Plasmodium falciparum (isolate 3D7) Uniprot ID TTD ID NA Synonyms NA Protein family Dihydroorotate dehydrogenase family, Type 2 subfamily Biochemical class Oxidoreductase Function Dihydroorotate dehydrogenase activity. Related diseases Combined oxidative phosphorylation deficiency 33 (COXPD33) [MIM:617713]: An autosomal recessive disorder caused by multiple mitochondrial respiratory chain defects and impaired mitochondrial energy metabolism. Clinical manifestations are highly variable. Affected infants present with cardiomyopathy accompanied by multisystemic features involving liver, kidney, and brain. Death in infancy is observed in some patients. Children and adults present with myopathy and progressive external ophthalmoplegia. {ECO:0000269|PubMed:28942965}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01117 Interacts with NA EC number 1.3.5.2 Uniprot keywords 3D-structure; Flavoprotein; FMN; Membrane; Mitochondrion; Mitochondrion inner membrane; Oxidoreductase; Pyrimidine biosynthesis; Reference proteome; Transit peptide; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A,B Molecular weight (Da) 42573.5 Length 378 Aromaticity 0.1 Instability index 36.63 Isoelectric point 8.21 Charge (pH=7) 3.17 2D Binding mode Binding energy (Kcal/mol) -5.86 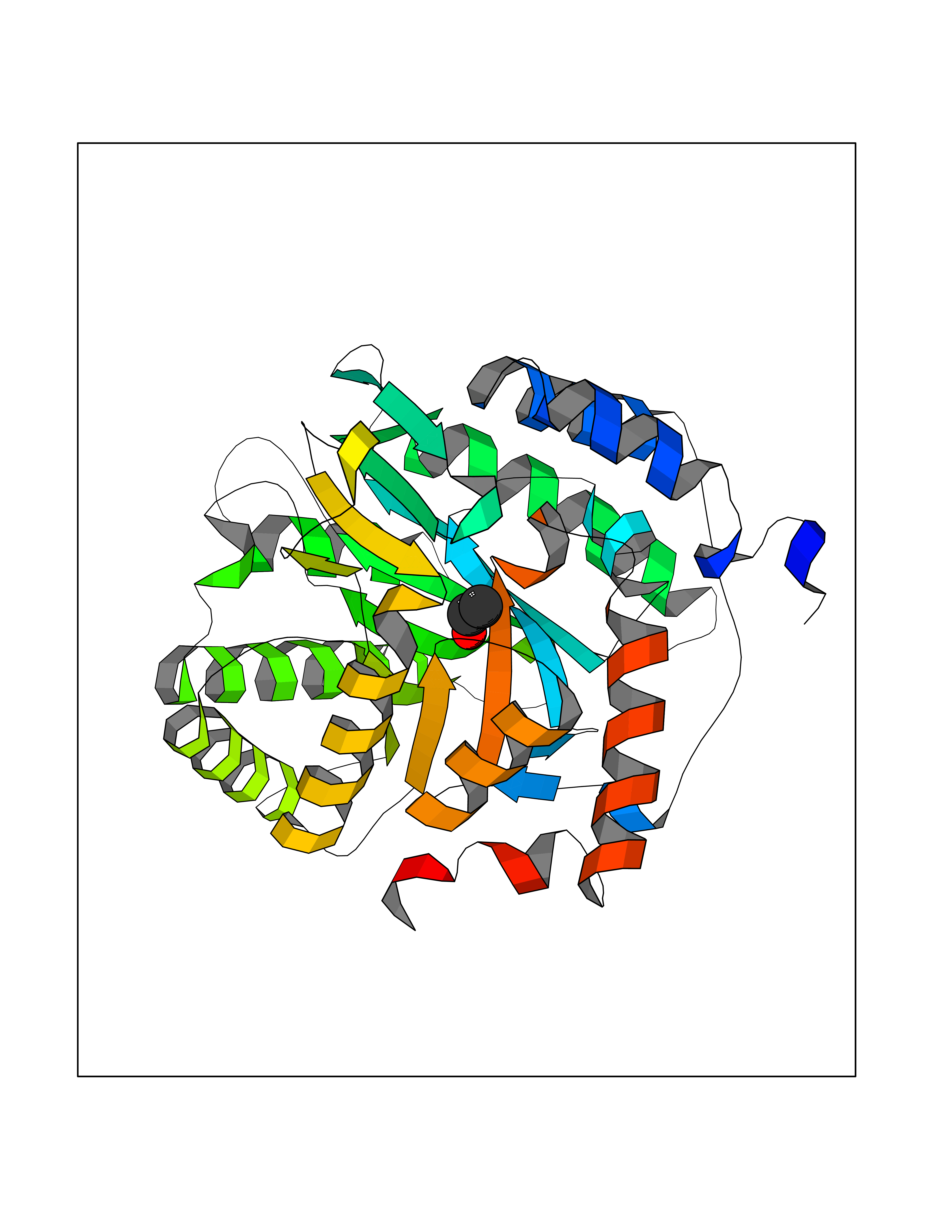 Molscript Map 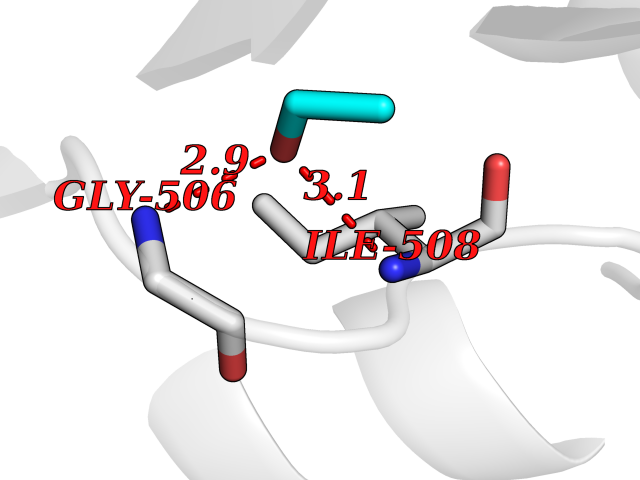 Pymol Map 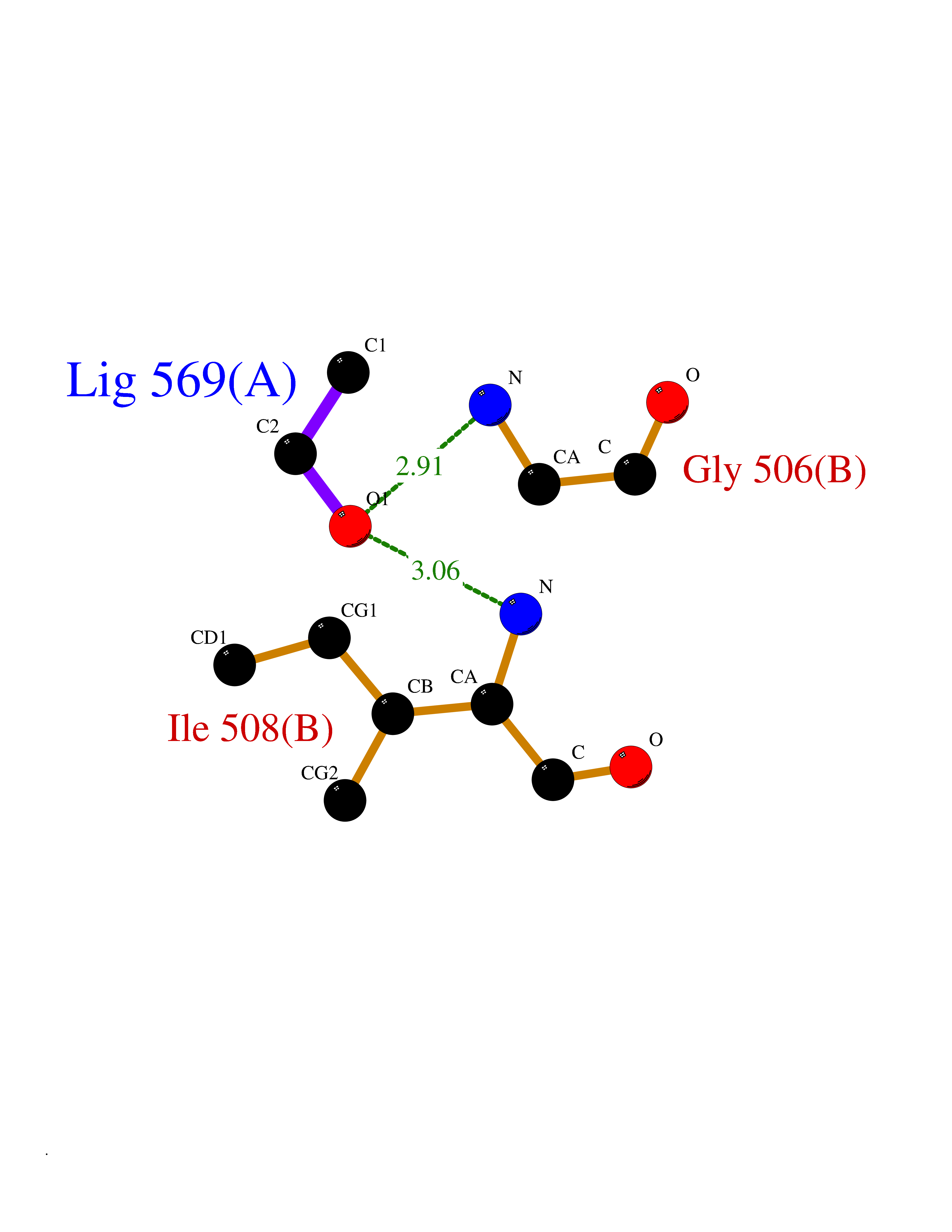 Ligplot Map 3D Binding mode Sequence ADPFESYNPEFFLYDIFLKFCLKYIDGEICHDLFLLLGKYNILPYDTSNDSIYACTNIKHLDFINPFGVAAGFDKNGVCIDSILKLGFSFIEIGTITPRGQTGNAKPRIFRDVESRSIINSCGFNNMGCDKVTENLILFRKRQEEDKLLSKHIVGVSIGKNKDTVNIVDDLKYCINKIGRYADYIAINVSSPNTPGLRDNQEAGKLKNIILSVKEEIDNLEKNNFLWFNTTKKKPLVFVKLAPDLNQEQKKEIADVLLETNIDGMIISNTTTQINDIKSFENKKGGVSGAKLKDISTKFICEMYNYTNKQIPIIASGGIFSGLDALEKIEAGASVCQLYSCLVFNGMKSAVQIKRELNHLLYQRGYYNLKEAIGRKHS Hydrogen bonds contact Hydrophobic contact | ||||
| 17 | Leucine carboxyl methyltransferase 1 | 3IEI | 4.30 | |
Target general information Gen name LCMT1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms CGI-68;LCMT Protein family Methyltransferase superfamily, LCMT family Biochemical class Transferase Function Protein C-terminal carboxyl O-methyltransferase activity.Protein C-terminal leucine carboxyl O-methyltransferase activity.S-adenosylmethionine-dependent methyltransferase activity. Related diseases Neurodevelopmental disorder, mitochondrial, with abnormal movements and lactic acidosis, with or without seizures (NEMMLAS) [MIM:617710]: An autosomal recessive, mitochondrial disorder with a broad phenotypic spectrum ranging from severe neonatal lactic acidosis, encephalomyopathy and early death to an attenuated course with milder manifestations. Clinical features include delayed psychomotor development, intellectual disability, hypotonia, dystonia, ataxia, and spasticity. Severe combined respiratory chain deficiency may be found in severely affected individuals. {ECO:0000269|PubMed:28236339, ECO:0000269|PubMed:28650581, ECO:0000269|PubMed:28905505, ECO:0000269|PubMed:30920170, ECO:0000269|PubMed:35074316}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Parkinsonism-dystonia 3, childhood-onset (PKDYS3) [MIM:619738]: An autosomal recessive neurodegenerative disorder with onset in infancy or early childhood. Affected individuals present with progressive movement abnormalities, including parkinsonism with tremor, dystonia, myoclonus ataxia, and hyperkinetic movements such as ballismus. The parkinsonism features may be responsive to treatment with levodopa, although many patients develop levodopa-induced dyskinesia. Some patients may have mild cognitive impairment or psychiatric disturbances. {ECO:0000269|PubMed:29120065, ECO:0000269|PubMed:31970218, ECO:0000269|PubMed:34890876}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00149 Interacts with P51116 EC number 2.1.1.233 Uniprot keywords 3D-structure; Alternative splicing; Methyltransferase; Proteomics identification; Reference proteome; S-adenosyl-L-methionine; Transferase Protein physicochemical properties Chain ID A,B,C,D,E,F,G,H Molecular weight (Da) 35803 Length 310 Aromaticity 0.08 Instability index 42.77 Isoelectric point 6.13 Charge (pH=7) -3.58 2D Binding mode Binding energy (Kcal/mol) -5.87  Molscript Map 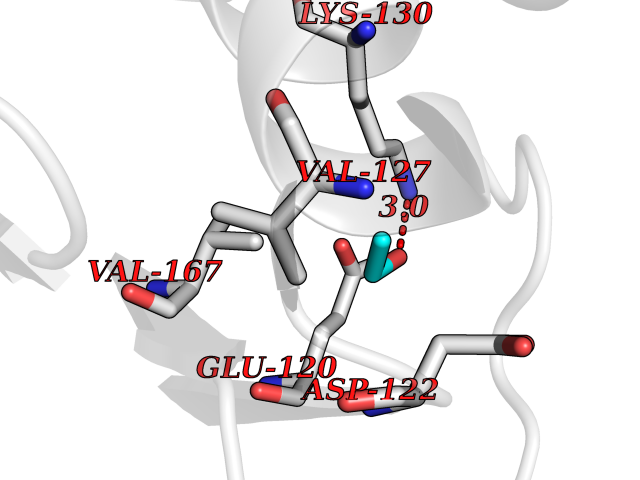 Pymol Map 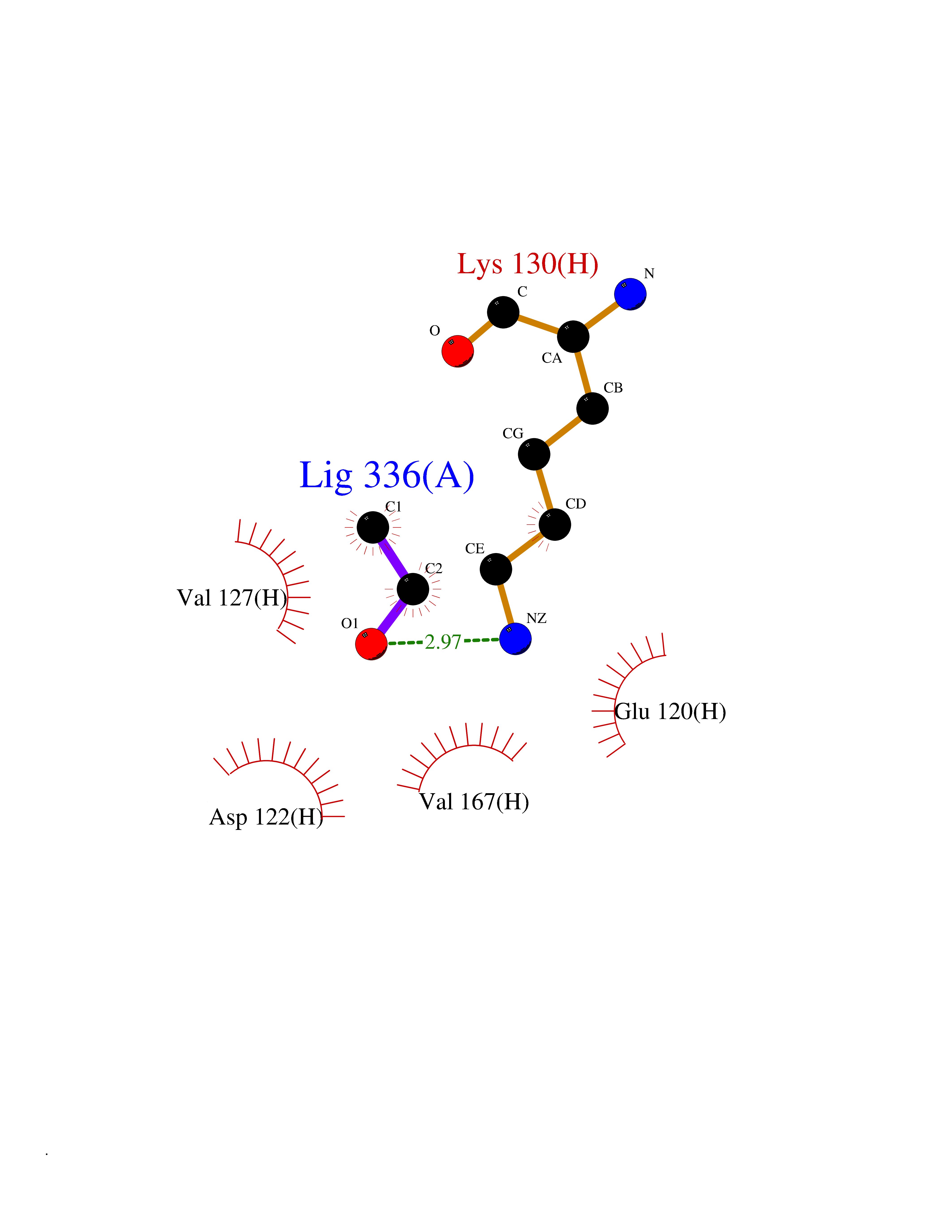 Ligplot Map 3D Binding mode Sequence GVRGTCEDASLCKRFAVSIGYWHDPYIQHFVRLSKERKAPEINRGYFARVHGVSQLIKAFLRKTECHCQIVNLGAGMDTTFWRLKDEDLLSSKYFEVDFPMIVTRKLHSIKCKPPLSSPILELHSEDTLQMDGHILDSKRYAVIGADLRDLSELEEKLKKCNMNTQLPTLLIAECVLVYMTPEQSANLLKWAANSFERAMFINYEQVNMGDRFGQIMIENLRRRQCDLAGVETCKSLESQKERLLSNGWETASAVDMMELYNRLPRAEVSRIESLEFLDEMELLEQLMRHYCLCWATKGGNELGLKEITY Hydrogen bonds contact Hydrophobic contact | ||||
| 18 | Phosphoribosylaminoimidazolecarboxamide formyltransferase (ATIC) | 1P4R | 4.30 | |
Target general information Gen name ATIC Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms PURH; OK/SW-cl.86; Bifunctional purine biosynthesis protein PURH Protein family PurH family Biochemical class Methyltransferase Function Bifunctional enzyme that catalyzes 2 steps in purine biosynthesis. Related diseases AICA-ribosuria due to ATIC deficiency (AICAR) [MIM:608688]: A neurologically devastating inborn error of purine biosynthesis. Patients excrete massive amounts of AICA-riboside in the urine and accumulate AICA-ribotide and its derivatives in erythrocytes and fibroblasts. Clinical features include profound intellectual disability, epilepsy, dysmorphic features and congenital blindness. AICAR inheritance is autosomal recessive. {ECO:0000269|PubMed:15114530}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02309; DB03442; DB01700; DB01972; DB00563; DB04057; DB00642; DB00116 Interacts with NA EC number NA Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Cytoplasm; Direct protein sequencing; Disease variant; Epilepsy; Hydrolase; Intellectual disability; Multifunctional enzyme; Proteomics identification; Purine biosynthesis; Reference proteome; Transferase Protein physicochemical properties Chain ID A,B Molecular weight (Da) 128556 Length 1177 Aromaticity 0.07 Instability index 38.21 Isoelectric point 6.28 Charge (pH=7) -7.98 2D Binding mode Binding energy (Kcal/mol) -5.87  Molscript Map 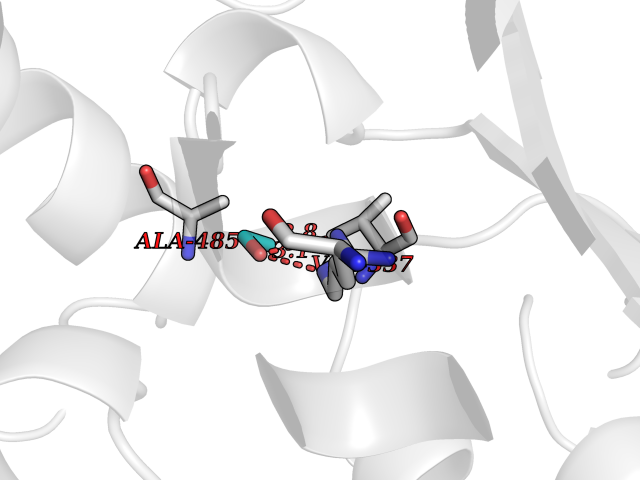 Pymol Map 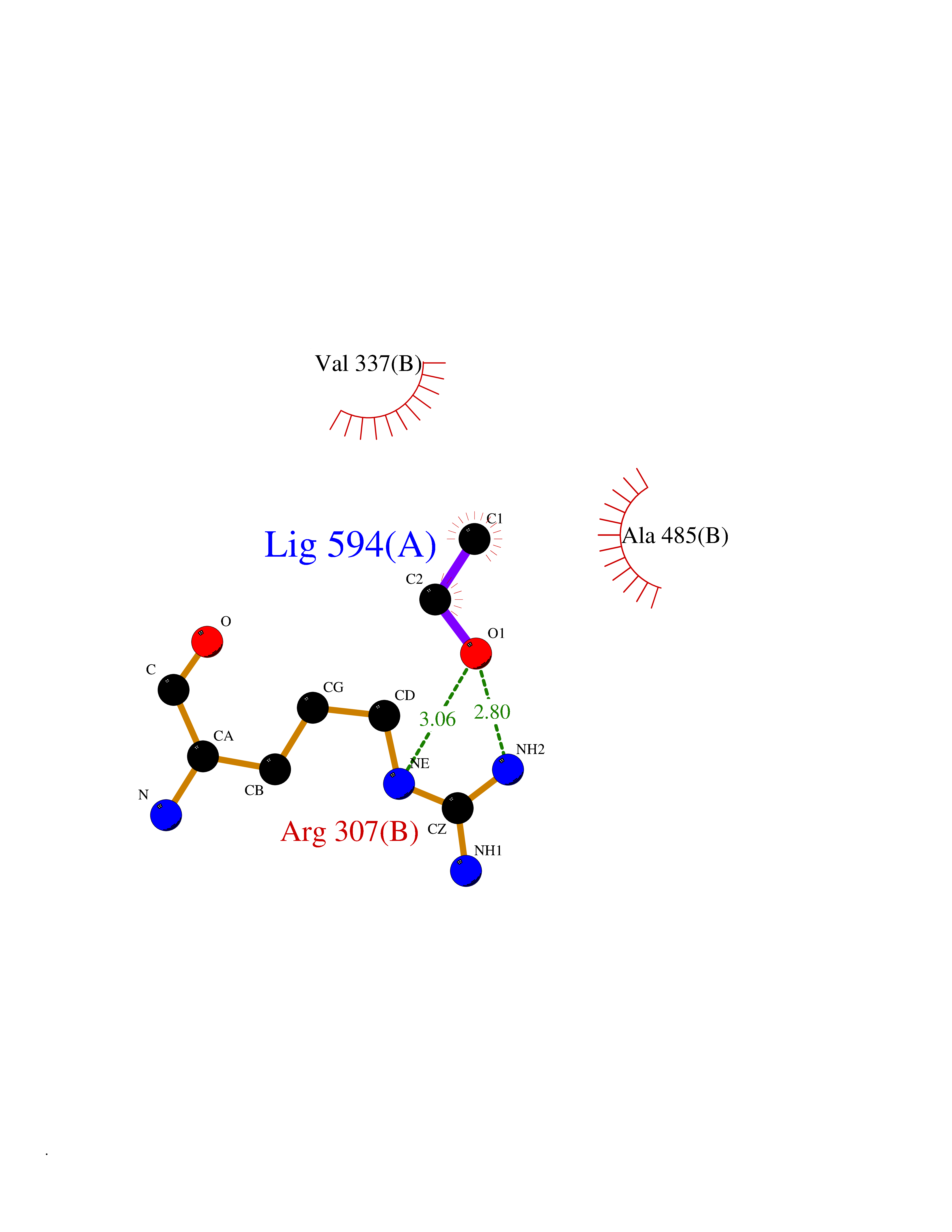 Ligplot Map 3D Binding mode Sequence GQLALFSVSDKTGLVEFARNLTALGLNLVASGGTAKALRDAGLAVRDVSELTGFPEMLGGRVKTLHPAVHAGILARNIPEDNADMARLDFNLIRVVACNLYPFVKTVASPGVTVEEAVEQIDIGGVTLLRAAAKNHARVTVVCEPEDYVVVSTEMQSSESKDTSLETRRQLALKAFTHTAQYDEAISDYFRKQYSKGVSQMPLRYGMNPHQTPAQLYTLQPKLPITVLNGAPGFINLCDALNAWQLVKELKEALGIPAAASFKHVSPAGAAVGIPLSEDEAKVCMVYDLYKTLTPISAAYARARGADRMSSFGDFVALSDVCDVPTAKIISREVSDGIIAPGYEEEALTILSKKKNGNYCVLQMDQSYKPDENEVRTLFGLHLSQKRNNGVVDKSLFSNVVTKNKDLPESALRDLIVATIAVKYTQSNSVCYAKNGQVIGIGAGQQSRIHCTRLAGDKANYWWLRHHPQVLSMKFKTGVKRAEISNAIDQYVTGTIGEDEDLIKWKALFEEVPELLTEAEKKEWVEKLTEVSISSDAFFPFRDNVDRAKRSGVAYIAAPSGSAADKVVIEACDELGIILAHTNLRLFHHQLALFSVSDKTGLVEFARNLTALGLNLVASGGTAKALRDAGLAVRDVSELTGFPEMLGGRVKTLHPAVHAGILARNIPEDNADMARLDFNLIRVVACNLYPFVKTVASPGVTVEEAVEQIDIGGVTLLRAAAKNHARVTVVCEPEDYVVVSTEMQSSESKDTSLETRRQLALKAFTHTAQYDEAISDYFRKQYSKGVSQMPLRYGMNPHQTPAQLYTLQPKLPITVLNGAPGFINLCDALNAWQLVKELKEALGIPAAASFKHVSPAGAAVGIPLSEDEAKVCMVYDLYKTLTPISAAYARARGADRMSSFGDFVALSDVCDVPTAKIISREVSDGIIAPGYEEEALTILSKKKNGNYCVLQMDQSYKPDENEVRTLFGLHLSQKRNNGVVDKSLFSNVVTKNKDLPESALRDLIVATIAVKYTQSNSVCYAKNGQVIGIGAGQQSRIHCTRLAGDKANYWWLRHHPQVLSMKFKTGVKRAEISNAIDQYVTGTIGEDEDLIKWKALFEEVPELLTEAEKKEWVEKLTEVSISSDAFFPFRDNVDRAKRSGVAYIAAPSGSAADKVVIEACDELGIILAHTNLRLFHH Hydrogen bonds contact Hydrophobic contact | ||||
| 19 | Thymidine kinase 1 (TK1) | 1W4R | 4.30 | |
Target general information Gen name TK1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Thymidine kinase, cytosolic Protein family Thymidine kinase family Biochemical class Kinase Function cytosol, identical protein binding, thymidine kinase activity, zinc ion binding, DNA metabolic process, nucleobase-containing compound metabolic process, protein homotetramerization, pyrimidine nucleoside salvage, thymidine metabolic process Related diseases Seizures, benign familial infantile, 3 (BFIS3) [MIM:607745]: A form of benign familial infantile epilepsy, a neurologic disorder characterized by afebrile seizures occurring in clusters during the first year of life, without neurologic sequelae. BFIS3 inheritance is autosomal dominant. {ECO:0000269|PubMed:11371648, ECO:0000269|PubMed:12243921, ECO:0000269|PubMed:15048894, ECO:0000269|PubMed:16417554, ECO:0000269|PubMed:17021166, ECO:0000269|PubMed:17386050, ECO:0000269|PubMed:18479388, ECO:0000269|PubMed:20371507, ECO:0000269|PubMed:22612257, ECO:0000269|PubMed:23360469, ECO:0000269|PubMed:23758435, ECO:0000269|PubMed:25982755, ECO:0000269|PubMed:26291284, ECO:0000269|PubMed:29844171, ECO:0000269|PubMed:30144217}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Developmental and epileptic encephalopathy 11 (DEE11) [MIM:613721]: An autosomal dominant seizure disorder characterized by neonatal or infantile onset of refractory seizures with resultant delayed neurologic development and persistent neurologic abnormalities. Patients may progress to West syndrome, which is characterized by tonic spasms with clustering, arrest of psychomotor development, and hypsarrhythmia on EEG. {ECO:0000269|PubMed:19783390, ECO:0000269|PubMed:19786696, ECO:0000269|PubMed:20956790, ECO:0000269|PubMed:22677033, ECO:0000269|PubMed:23033978, ECO:0000269|PubMed:23195492, ECO:0000269|PubMed:23550958, ECO:0000269|PubMed:23662938, ECO:0000269|PubMed:23708187, ECO:0000269|PubMed:23935176, ECO:0000269|PubMed:23988467, ECO:0000269|PubMed:24463883, ECO:0000269|PubMed:24579881, ECO:0000269|PubMed:24659627, ECO:0000269|PubMed:24710820, ECO:0000269|PubMed:25457084, ECO:0000269|PubMed:25459969, ECO:0000269|PubMed:25772804, ECO:0000269|PubMed:25818041, ECO:0000269|PubMed:26138355, ECO:0000269|PubMed:26291284, ECO:0000269|PubMed:26993267, ECO:0000269|PubMed:27864847, ECO:0000269|PubMed:29625812, ECO:0000269|PubMed:29844171, ECO:0000269|PubMed:30144217, ECO:0000269|PubMed:30415926}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Defects in SCN2A are associated with genetic epilepsy with febrile seizures plus (GEFS+), a familial autosomal dominant epilepsy syndrome, a clinical subset of febrile seizures, characterized by frequent episodes after 6 years of age and various types of subsequent epilepsy. {ECO:0000269|PubMed:29635106}.; DISEASE: Defects in SCN2A are associated with autism spectrum disorders (ASD). It seems that mutations resulting in sodium channel gain of function and increased neuron excitability lead to infantile seizures, whereas variants resulting in sodium channel loss of function and decrease neuron excitability are associated with ASD. {ECO:0000269|PubMed:28256214}.; DISEASE: Episodic ataxia 9 (EA9) [MIM:618924]: An autosomal dominant neurologic disorder characterized by episodic ataxia manifesting in the first years of life, early-onset seizures, difficulty walking, dizziness, slurred speech, headache, vomiting, and pain. The duration of ataxic episodes is heterogeneous. Most patients show episodes lasting minutes to maximum several hours, but periods lasting days up to weeks have been reported. Some patients have mildly delayed development with speech delay and/or autistic features or mildly impaired intellectual development. {ECO:0000269|PubMed:26645390, ECO:0000269|PubMed:27159988, ECO:0000269|PubMed:27328862, ECO:0000269|PubMed:28065826}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01692; DB04485; DB02452; DB00432; DB00495 Interacts with P05067; A0A087WZT3; Q92993; Q1RN33; P04183 EC number EC 2.7.1.21 Uniprot keywords 3D-structure; Acetylation; ATP-binding; Cytoplasm; DNA synthesis; Kinase; Metal-binding; Nucleotide-binding; Phosphoprotein; Proteomics identification; Reference proteome; Transferase; Ubl conjugation; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 19373.5 Length 174 Aromaticity 0.09 Instability index 36.21 Isoelectric point 8.63 Charge (pH=7) 3.88 2D Binding mode Binding energy (Kcal/mol) -5.86  Molscript Map 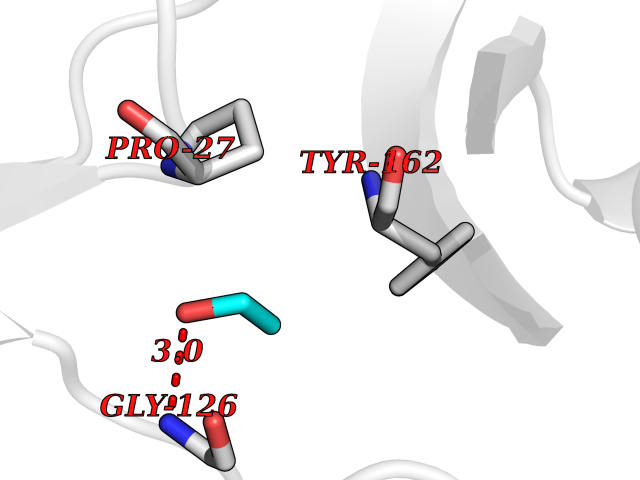 Pymol Map 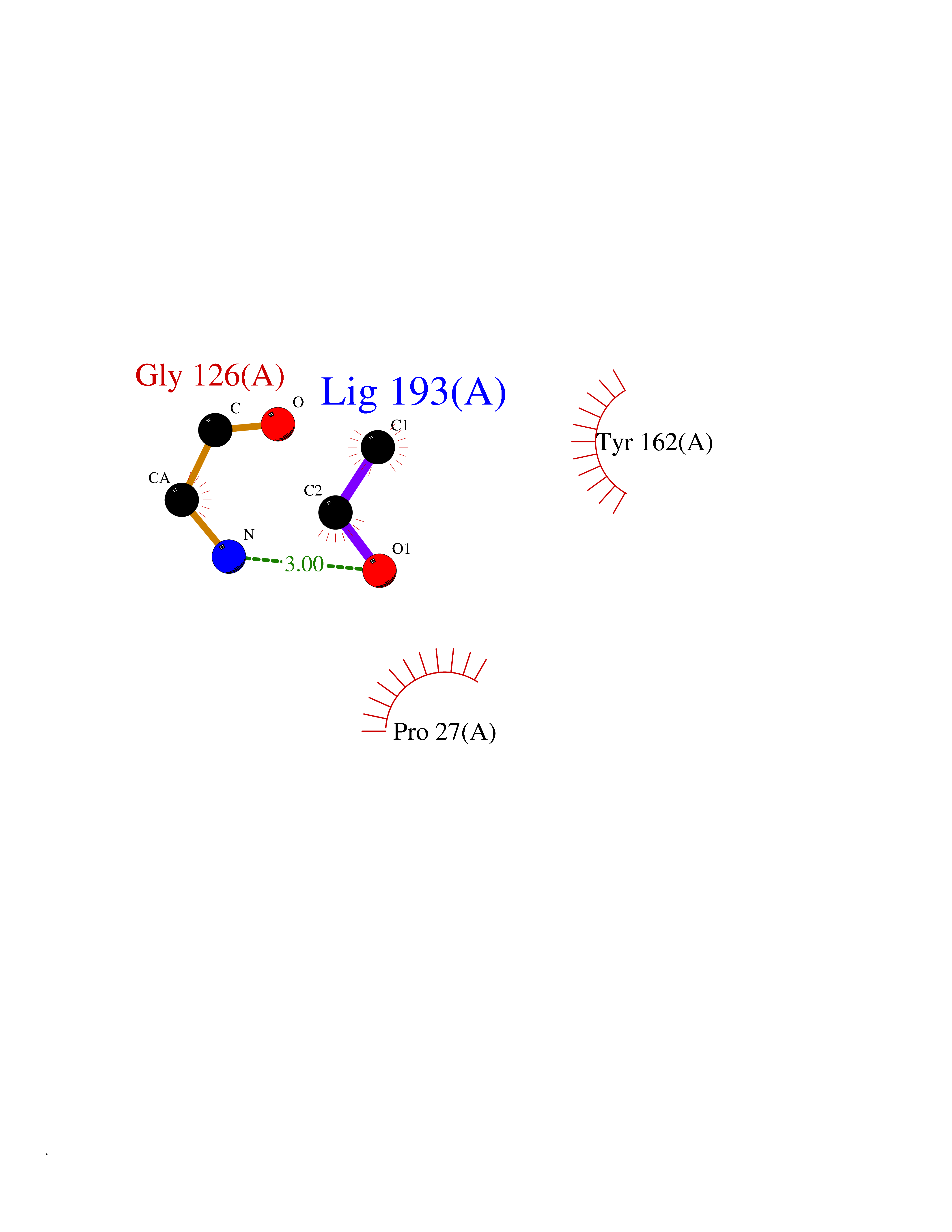 Ligplot Map 3D Binding mode Sequence RGQIQVILGPMFSGKSTELMRRVRRFQIAQYKCLVIKYAKDTRYSSSFCTHDRNTMEALPACLLRDVAQEALGVAVIGIDEGQFFPDIVEFCEAMANAGKTVIVAALDGTFQRKPFGAILNLVPLAESVVKLTAVCMECFREAAYTKRLGTEKEVEVIGGADKYHSVCRLCYFK Hydrogen bonds contact Hydrophobic contact | ||||
| 20 | Pseudomonas Omega-amino acid:pyruvate aminotransferase (Pseudo Omega-APT) | 3A8U | 4.30 | |
Target general information Gen name Pseudo Omega-APT Organism Pseudomonas putida (Arthrobacter siderocapsulatus) Uniprot ID TTD ID Synonyms Omega-APT; Beta-alanine--pyruvate aminotransferase Protein family Class-III pyridoxal-phosphate-dependent aminotransferase family Biochemical class Transaminase Function Catalyzes transamination between a variety of omega- amino acids, mono and diamines, and pyruvate. Plays a pivotal role in the metabolism of the omega amino acids. Related diseases Insulin-like growth factor 1 resistance (IGF1RES) [MIM:270450]: A disorder characterized by intrauterine growth retardation, poor postnatal growth and increased plasma IGF1 levels. {ECO:0000269|PubMed:14657428, ECO:0000269|PubMed:15928254, ECO:0000269|PubMed:25040157}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number EC 2.6.1.18 Uniprot keywords 3D-structure; Aminotransferase; Direct protein sequencing; Pyridoxal phosphate; Transferase Protein physicochemical properties Chain ID X Molecular weight (Da) 47761.1 Length 441 Aromaticity 0.09 Instability index 30.16 Isoelectric point 6.97 Charge (pH=7) -0.08 2D Binding mode Binding energy (Kcal/mol) -5.86 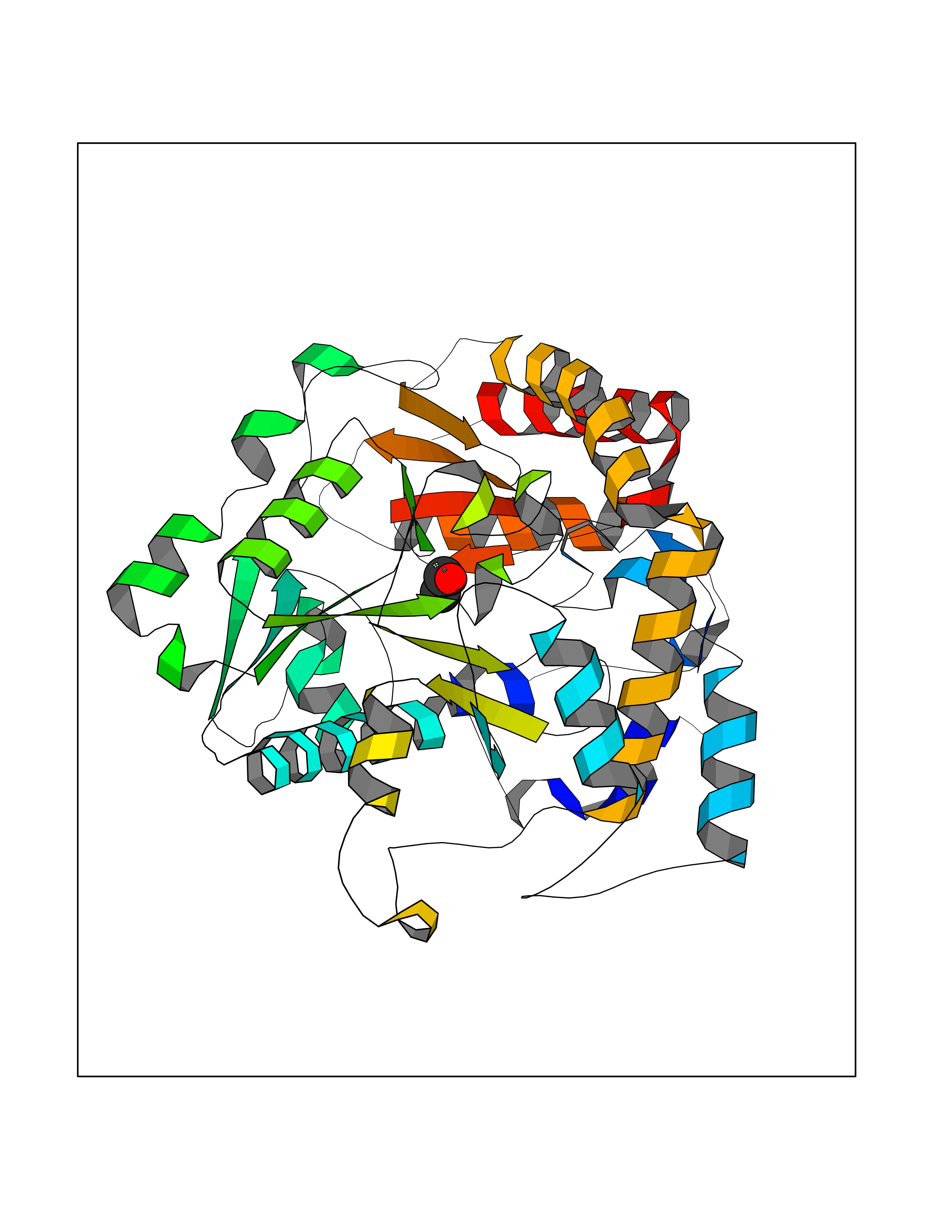 Molscript Map 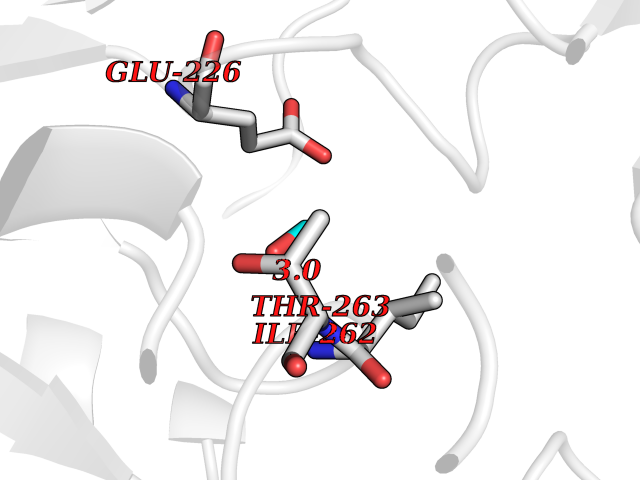 Pymol Map 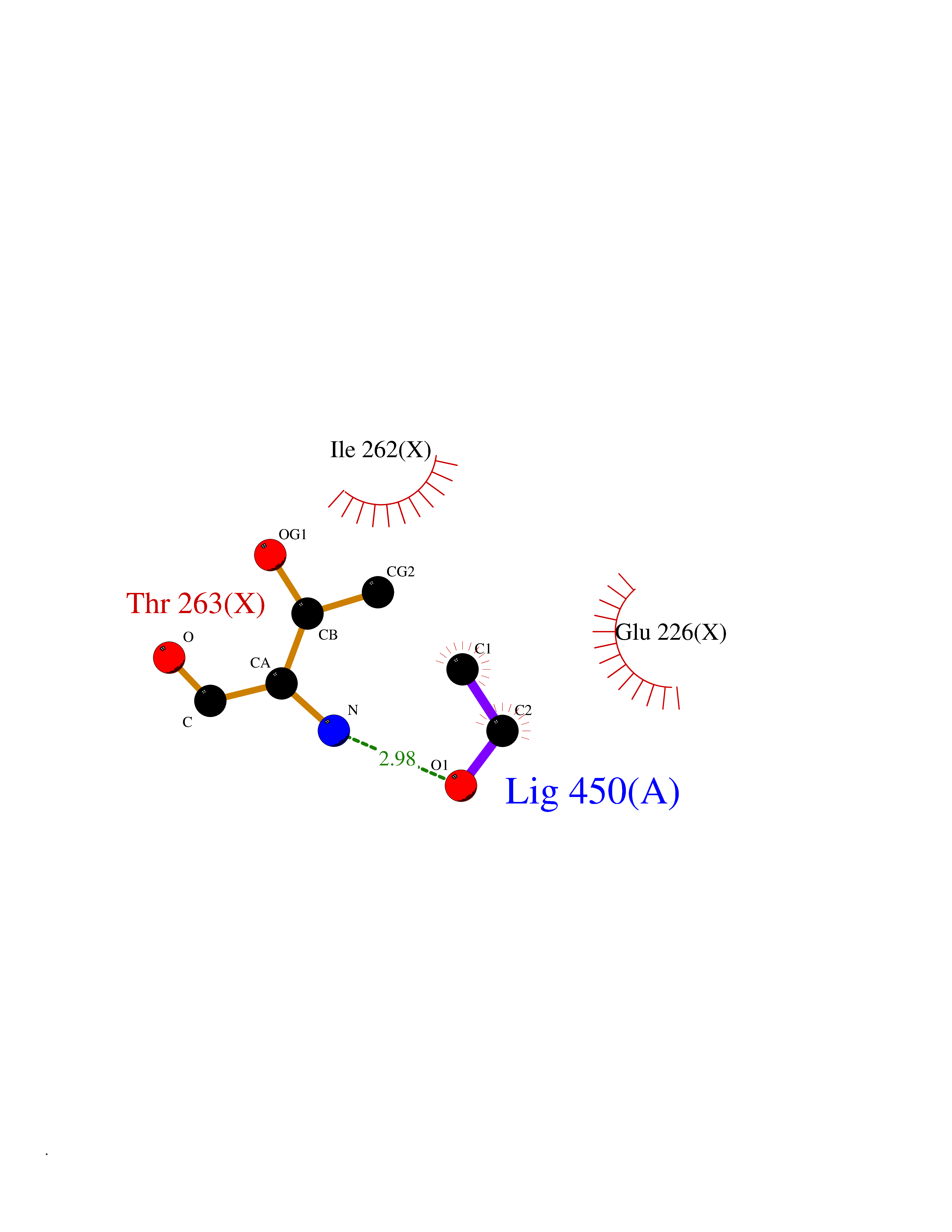 Ligplot Map 3D Binding mode Sequence ASLASQLKLDAHWMPYTANRNFLRDPRLIVAAEGSWLVDDKGRKVYDSLSGLWTCGAGHTRKEIQEAVAKQLSTLDYSPGFQYGHPLSFQLAEKITDLTPGNLNHVFFTDSGSECALTAVKMVRAYWRLKGQATKTKMIGRARGYHGVNIAGTSLGGVNGNRKLFGQPMQDVDHLPHTLLASNAYSRGMPKEGGIALADELLKLIELHDASNIAAVFVEPLAGSAGVLVPPEGYLKRNREICNQHNILLVFDEVITGFGRTGSMFGADSFGVTPDLMCIAKQVTNGAIPMGAVIASTEIYQTFMNQPTPEYAVEFPHGYTYSAHPVACAAGLAALCLLQKENLVQSVAEVAPHFEKALHGIKGAKNVIDIRNFGLAGAIQIAPRDGDAIVRPFEAGMALWKAGFYVRFGGDTLQFGPTFNSKPQDLDRLFDAVGEVLNKLL Hydrogen bonds contact Hydrophobic contact | ||||