Job Results:
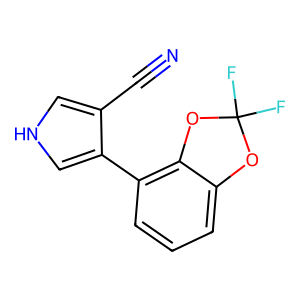
Ligand
Structure
Job ID
9af75475a67248e060ab578f15db3d0b
Job name
NA
Time
2025-04-03 17:54:57
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 1 | Gamma-aminobutyric acid type B receptor subunit 1 | 4MS4 | 6.42 | |
Target general information Gen name GABBR1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms GPRC3B;GPR51 Protein family G-protein coupled receptor 3 family, GABA-B receptor subfamily Biochemical class Signaling protein / antagonist Function G-protein coupled GABA receptor activity. Related diseases Neurodevelopmental disorder with poor language and loss of hand skills (NDPLHS) [MIM:617903]: An autosomal dominant disorder characterized by psychomotor developmental stagnation or regression. NDPLHS manifest in the first years of life as loss of purposeful hand movements, loss of language, and intellectual disability. {ECO:0000269|PubMed:26740508, ECO:0000269|PubMed:28856709, ECO:0000269|PubMed:29369404}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Developmental and epileptic encephalopathy 59 (DEE59) [MIM:617904]: A form of epileptic encephalopathy, a heterogeneous group of severe early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE59 is an autosomal dominant condition characterized by onset of refractory seizures in early infancy. {ECO:0000269|PubMed:28856709, ECO:0000269|PubMed:29100083, ECO:0000269|PubMed:29369404}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08891; DB08892; DB00181; DB00363; DB02530; DB05010; DB09072 Interacts with Q9UBS5; Q9UBS5-2; P46459; Q86UR5 EC number NA Uniprot keywords 3D-structure; Cell membrane; Coiled coil; Direct protein sequencing; Disease variant; Disulfide bond; Epilepsy; G-protein coupled receptor; Glycoprotein; Intellectual disability; Membrane; Phosphoprotein; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Signal; Synapse; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 46502.1 Length 408 Aromaticity 0.12 Instability index 50.05 Isoelectric point 5.78 Charge (pH=7) -5.62 2D Binding mode Binding energy (Kcal/mol) -8.76  Molscript Map 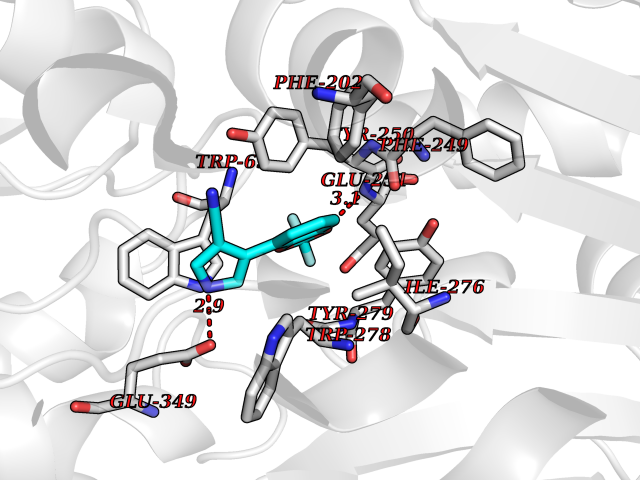 Pymol Map 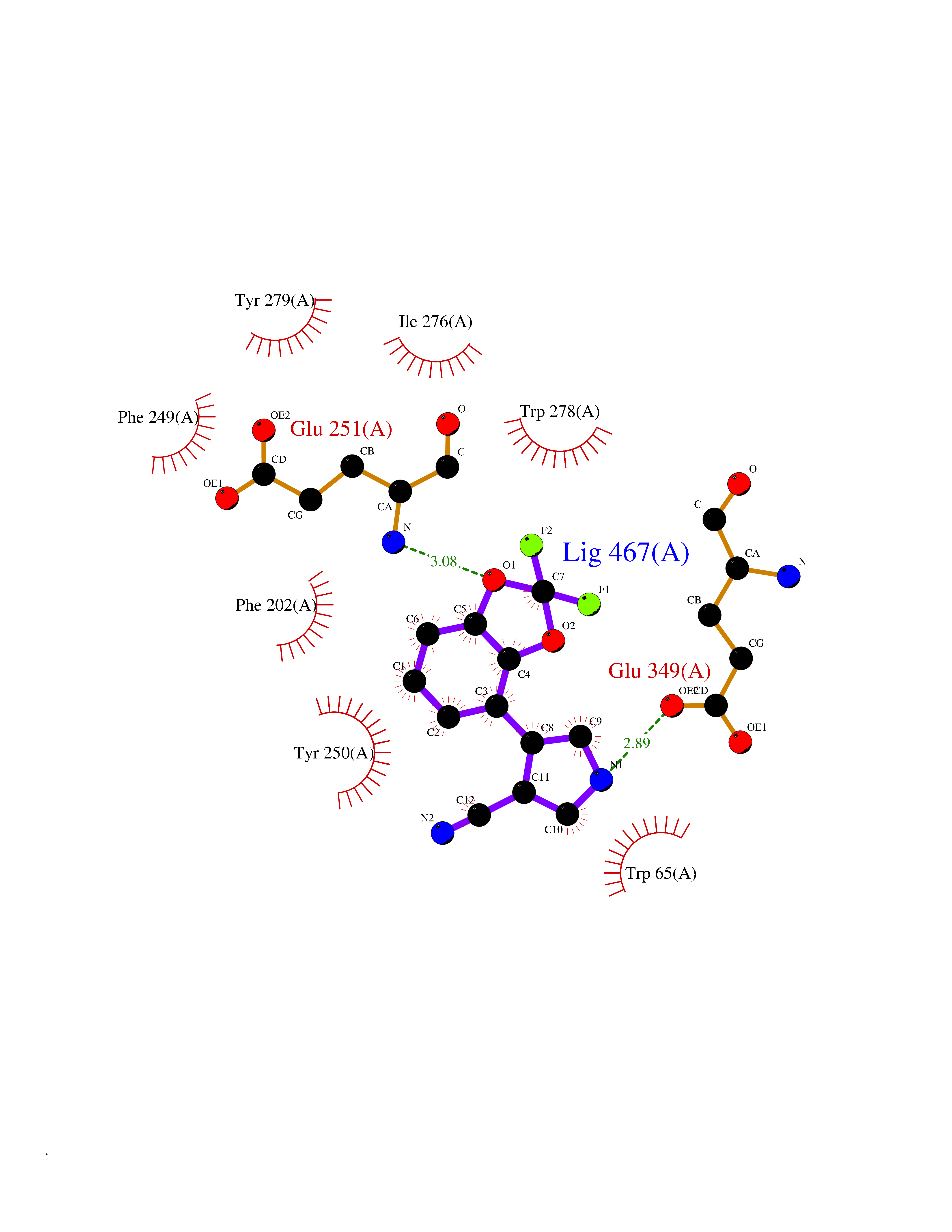 Ligplot Map 3D Binding mode Sequence RRAVYIGALFPMSGGWPGGQACQPAVEMALEDVNSRRDILPDYELKLIHHDSKCDPGQATKYLYELLYNDPIKIILMPGCSSVSTLVAEAARMWNLIVLSYGSSSPALSNRQRFPTFFRTHPSATLHNPTRVKLFEKWGWKKIATIQQTTEVFTSTLDDLEERVKEAGIEITFRQSFFSDPAVPVKNLKRQDARIIVGLFYETEARKVFCEVYKERLFGKKYVWFLIGWYADNWFKIYDPSINCTVDEMTEAVEGHITTEIVMLNPANTRSISNMTSQEFVEKLTKRLKRHPEETGGFQEAPLAYDAIWALALALNKTSRLEDFNYNNQTITDQIYRAMNSSSFEGVSGHVVFDASGSRMAWTLIEQLQGGSYKKIGYYDSTKDDLSWSKTDKWIGGSPPADDYKDDD Hydrogen bonds contact Hydrophobic contact | ||||
| 2 | Wnt-7a protein (WNT7A) | 4UZQ | 6.24 | |
Target general information Gen name WNT7A Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Protein Wnt-7a Protein family Wnt family Biochemical class NA Function Plays an important role in embryonic development, including dorsal versus ventral patterning during limb development, skeleton development and urogenital tract development. Required for central nervous system (CNS) angiogenesis and blood-brain barrier regulation. Required for normal, sexually dimorphic development of the Mullerian ducts, and for normal fertility in both sexes. Required for normal neural stem cell proliferation in the hippocampus dentate gyrus. Required for normal progress through the cell cycle in neural progenitor cells, for self-renewal of neural stem cells, and for normal neuronal differentiation and maturation. Promotes formation of synapses via its interaction with FZD5. Ligand for members of the frizzled family of seven transmembrane receptors that functions in the canonical Wnt/beta-catenin signaling pathway. Related diseases Limb pelvis hypoplasia aplasia syndrome (LPHAS) [MIM:276820]: A syndrome of severe deficiency of the extremities due to hypo- or aplasia of one or more long bones of one or more limbs. Pelvic manifestations include hip dislocation, hypoplastic iliac bone and aplastic pubic bones. Thoracic deformity, unusual facies and genitourinary anomalies can be present. {ECO:0000269|PubMed:16826533, ECO:0000269|PubMed:17431918, ECO:0000269|PubMed:20949531, ECO:0000269|PubMed:21271649, ECO:0000269|PubMed:27638328}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Fuhrmann syndrome (FUHRS) [MIM:228930]: Distinct limb-malformation disorder characterized also by various degrees of limb aplasia/hypoplasia and joint dysplasia. {ECO:0000269|PubMed:16826533}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P55212; P22607; P06396; P13473-2; Q9UMX0; Q9Y5W5; Q5T9L3; Q9Z0J1 EC number NA Uniprot keywords 3D-structure; Developmental protein; Disease variant; Disulfide bond; Extracellular matrix; Glycoprotein; Lipoprotein; Proteomics identification; Reference proteome; Secreted; Signal; Wnt signaling pathway Protein physicochemical properties Chain ID A,B Molecular weight (Da) 40475.5 Length 356 Aromaticity 0.1 Instability index 50.49 Isoelectric point 7.67 Charge (pH=7) 1.62 2D Binding mode Binding energy (Kcal/mol) -8.52  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence EDLRLHLLLNTSVTCNDGSPAGYYLKESRGSRRWLLFLEGGWYCFNRENCDSRYDTMRRLMSSRDWPRTRTGTGILSSQPEENPYWWNANMVFIPYCSSDVWSGASSKSEKNEYAFMGALIIQEVVRELLGRGLSGAKVLLLAGSAAGGTGVLLNVDRVAEQLEKLGYPAIQVRGLADSGWFLDNKQYRHTDCVDTITCAPTEAIRRGIRYWNGVVPERCRRQFQEGEEWNCFFGYKVYPTLRSPVFVVQWLFDEAQLTVDNVHLTGQPVQEGLRLYIQNLGRELRHTLKDVPASFAPACLSHEIIIRSHWTDVQVKGTSLPRALHCWDRSLHKGCPVHLVDSCPWPHCNPSCPTS Hydrogen bonds contact Hydrophobic contact | ||||
| 3 | Neuronal acetylcholine receptor subunit alpha-2 | 5FJV | 6.21 | |
Target general information Gen name CHRNA2 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Ligand-gated ion channel (TC 1.A.9) family, Acetylcholine receptor (TC 1.A.9.1) subfamily, Alpha-2/CHRNA2 sub-subfamily Biochemical class NA Function After binding acetylcholine, the AChR responds by an extensive change in conformation that affects all subunits and leads to opening of an ion-conducting channel across the plasma membrane." Related diseases Epilepsy, nocturnal frontal lobe, 4 (ENFL4) [MIM:610353]: An autosomal dominant focal epilepsy characterized by nocturnal seizures associated with fear sensation, tongue movements, and nocturnal wandering, closely resembling nightmares and sleep walking. {ECO:0000269|PubMed:16826524}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Seizures, benign familial infantile, 6 (BFIS6) [MIM:610353]: A form of benign familial infantile epilepsy, a neurologic disorder characterized by afebrile seizures occurring in clusters during the first year of life, without neurologic sequelae. BFIS6 inheritance is autosomal dominant. {ECO:0000269|PubMed:25847220}. The disease may be caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00732; DB00237; DB00411; DB00565; DB01245; DB00514; DB01135; DB07720; DB00898; DB00472; DB00483; DB08960; DB00657; DB01336; DB00416; DB01226; DB00184; DB01337; DB01338; DB00721; DB00728; DB05740; DB00202; DB01199; DB01339 Interacts with NA EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disease variant; Disulfide bond; Epilepsy; Glycoprotein; Ion channel; Ion transport; Ligand-gated ion channel; Membrane; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Signal; Synapse; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A,B,C,D,E Molecular weight (Da) 120584 Length 1031 Aromaticity 0.14 Instability index 32.21 Isoelectric point 5.62 Charge (pH=7) -17.58 2D Binding mode Binding energy (Kcal/mol) -8.47  Molscript Map 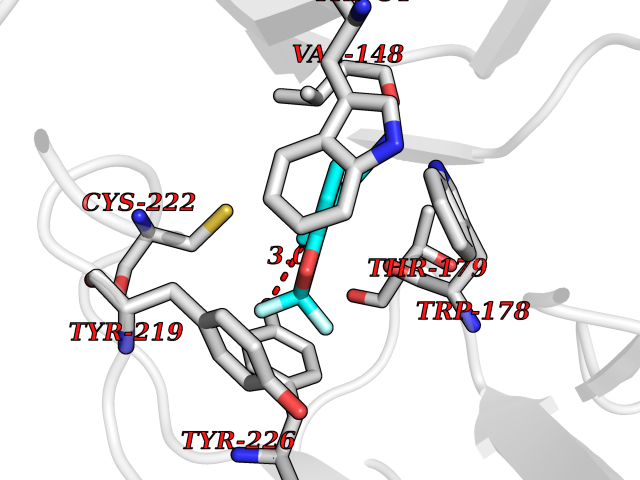 Pymol Map 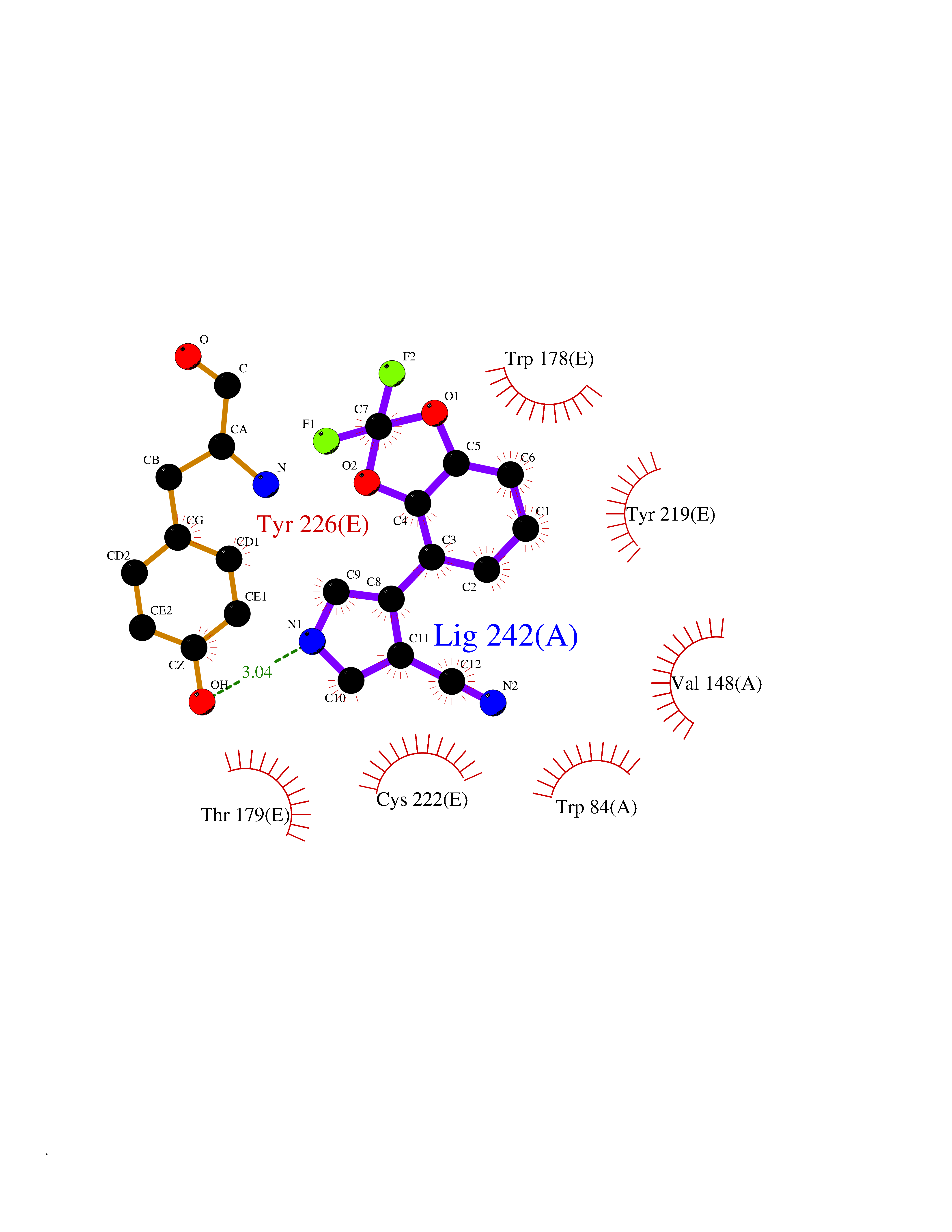 Ligplot Map 3D Binding mode Sequence DRLFKHLFRGYNRWARPVPNTSDVVIVRFGLSIAQLIDVDEKNQMMTTNVWLKQEWSDYKLRWNPTDFGNITSLRVPSEMIWIPDIVLYNNADGEFAVTHMTKAHLFSTGTVHWVPPAIYKSSCSIDVTFFPFDQQNCKMKFGSWTYDKAKIDLEQMEQTVDLKDYWESGEWAIVNATGTYNSKKYDCCAEIYPDVTYAFVIRRLPEDRLFKHLFRGYNRWARPVPNTSDVVIVRFGLSIAQLIDVDEKNQMMTTNVWLKQEWSDYKLRWNPTDFGNITSLRVPSEMIWIPDIVLYNNADGEFAVTHMTKAHLFSTGTVHWVPPAIYKSSCSIDVTFDQQNCKMKFGSWTYDKAKIDLEQMEQTVDLKDYWESGEWAIVNATGTYNSKKYDCCAEIYPDVTYAFVIRRLPEDRLFKHLFRGYNRWARPVPNTSDVVIVRFGLSIAQLIDVDEKNQMMTTNVWLKQEWSDYKLRWNPTDFGNITSLRVPSEMIWIPDIVLYNNADGEFAVTHMTKAHLFSTGTVHWVPPAIYKSSCSIDVTFFPFDQQNCKMKFGSWTYDKAKIDLEQMEQTVDLKDYWESGEWAIVNATGTYNSKKYDCCAEIYPDVTYAFVIRRLPEDRLFKHLFRGYNRWARPVPNTSDVVIVRFGLSIAQLIDVDEKNQMMTTNVWLKQEWSDYKLRWNPTDFGNITSLRVPSEMIWIPDIVLYNNADGEFAVTHMTKAHLFSTGTVHWVPPAIYKSSCSIDVTFFPFDQQNCKMKFGSWTYDKAKIDLEQMEQTVDLKDYWESGEWAIVNATGTYNSKKYDCCAEIYPDVTYAFVIRRLPEDRLFKHLFRGYNRWARPVPNTSDVVIVRFGLSIAQLIDVDEKNQMMTTNVWLKQEWSDYKLRWNPTDFGNITSLRVPSEMIWIPDIVLYNNADGEFAVTHMTKAHLFSTGTVHWVPPAIYKSSCSIDVTFFPFDQQNCKMKFGSWTYDKAKIDLEQMEQTVDLKDYWESGEWAIVNATGTYNSKKYDCCAEIYPDVTYAFVIRRLP Hydrogen bonds contact Hydrophobic contact | ||||
| 4 | Plasmodium Dihydroorotate dehydrogenase (Malaria DHOdehase) | 1TV5 | 6.20 | |
Target general information Gen name Malaria DHOdehase Organism Plasmodium falciparum (isolate 3D7) Uniprot ID TTD ID Synonyms PFF0160c; Mitochondrially bound dihydroorotate-ubiqui oxidoreductase; Dihydroorotate oxidase of Plasmodium falciparum; Dihydroorotate dehydrogenase of Plasmodium falciparum; DHOdehase of Plasmodium fa Protein family Dihydroorotate dehydrogenase family, Type 2 subfamily Biochemical class CH-CH donor oxidoreductase Function Catalyzes the conversion of dihydroorotate to orotate with quinone as electron acceptor. Related diseases Combined oxidative phosphorylation deficiency 33 (COXPD33) [MIM:617713]: An autosomal recessive disorder caused by multiple mitochondrial respiratory chain defects and impaired mitochondrial energy metabolism. Clinical manifestations are highly variable. Affected infants present with cardiomyopathy accompanied by multisystemic features involving liver, kidney, and brain. Death in infancy is observed in some patients. Children and adults present with myopathy and progressive external ophthalmoplegia. {ECO:0000269|PubMed:28942965}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01117 Interacts with NA EC number EC 1.3.5.2 Uniprot keywords 3D-structure; Flavoprotein; FMN; Membrane; Mitochondrion; Mitochondrion inner membrane; Oxidoreductase; Pyrimidine biosynthesis; Reference proteome; Transit peptide; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 41846.8 Length 371 Aromaticity 0.1 Instability index 37.25 Isoelectric point 8.21 Charge (pH=7) 3.13 2D Binding mode Binding energy (Kcal/mol) -8.45 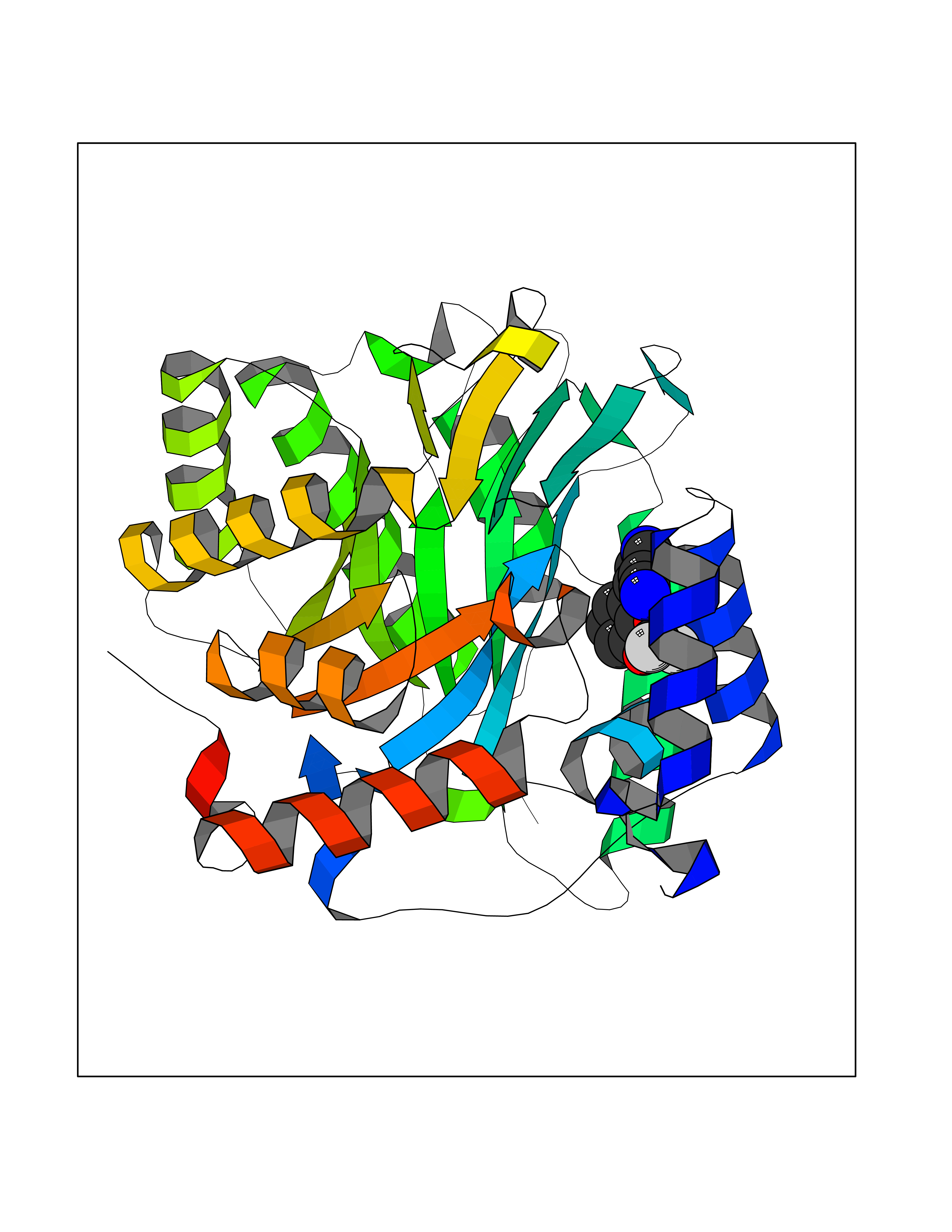 Molscript Map 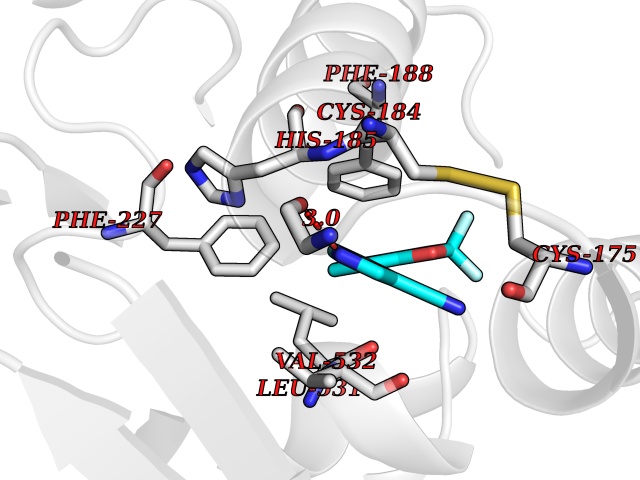 Pymol Map 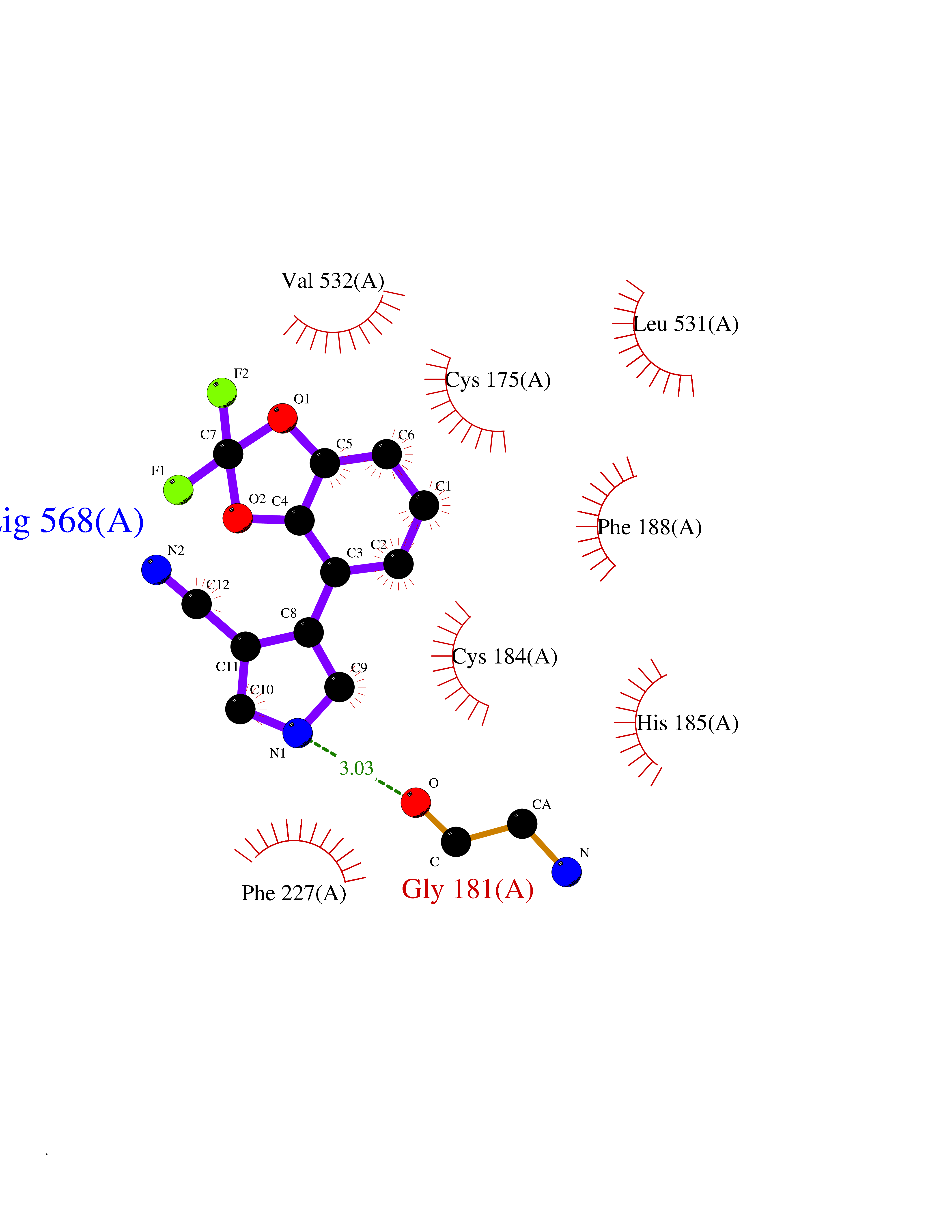 Ligplot Map 3D Binding mode Sequence FESYNPEFFLYDIFLKFCLKYIDGEICHDLFLLLGKYNILPYDTSNDSIYACTNIKHLDFINPFGVAAGFDKNGVCIDSILKLGFSFIEIGTITPRGQTGNAKPRIFRDVESRSIINSCGFNNMGCDKVTENLILFRKRQEEDKLLSKHIVGVSIGKNKDTVNIVDDLKYCINKIGRYADYIAINVSSPNTPGLRDNQEAGKLKNIILSVKEEIDNLEFLWFNTTKKKPLVFVKLAPDLNQEQKKEIADVLLETNIDGMIISNTTTQINDIKSFENKKGGVSGAKLKDISTKFICEMYNYTNKQIPIIASGGIFSGLDALEKIEAGASVCQLYSCLVFNGMKSAVQIKRELNHLLYQRGYYNLKEAIGRKH Hydrogen bonds contact Hydrophobic contact | ||||
| 5 | Tankyrase-2 (TNKS-2) | 3U9H | 6.19 | |
Target general information Gen name TNKS2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Tankyrase-related protein; Tankyrase-like protein; Tankyrase II; TRF1-interacting ankyrin-related ADP-ribose polymerase 2; TNKL; TANK2; Protein poly-ADP-ribosyltransferase tankyrase-2; Poly [ADP-ribos Protein family ARTD/PARP family Biochemical class Glycosyltransferases Function Acts as an activator of the Wnt signaling pathway by mediating poly-ADP-ribosylation of AXIN1 and AXIN2, 2 key components of the beta-catenin destruction complex: poly-ADP-ribosylated target proteins are recognized by RNF146, which mediates their ubiquitination and subsequent degradation. Also mediates poly-ADP-ribosylation of BLZF1 and CASC3, followed by recruitment of RNF146 and subsequent ubiquitination. Mediates poly-ADP-ribosylation of TERF1, thereby contributing to the regulation of telomere length. Stimulates 26S proteasome activity. Poly-ADP-ribosyltransferase involved in various processes such as Wnt signaling pathway, telomere length and vesicle trafficking. Related diseases Intellectual developmental disorder with macrocephaly, seizures, and speech delay (IDDMSSD) [MIM:618158]: An autosomal dominant neurodevelopmental disorder characterized by impaired intellectual development, poor speech, postnatal macrocephaly, and seizures. {ECO:0000269|PubMed:30290153}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with O15084; Q7Z6K5-1; O15169; Q9NWV8; P11274; Q13698; Q9NRI5; Q6V0I7; Q9NWT6; P14652; Q9UIQ6; Q14980; Q9BZL4; Q92698; P78314; O43815; P54274; Q9C0C2; Q9UHP3; Q06649 EC number EC 2.4.2.30 Uniprot keywords 3D-structure; ADP-ribosylation; ANK repeat; Chromosome; Cytoplasm; Glycosyltransferase; Golgi apparatus; Hydroxylation; Membrane; Metal-binding; NAD; Nucleotidyltransferase; Nucleus; Proteomics identification; Reference proteome; Repeat; Telomere; Transferase; Ubl conjugation; Wnt signaling pathway; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 23695.5 Length 208 Aromaticity 0.11 Instability index 47.61 Isoelectric point 8.28 Charge (pH=7) 2.88 2D Binding mode Binding energy (Kcal/mol) -8.45  Molscript Map  Pymol Map 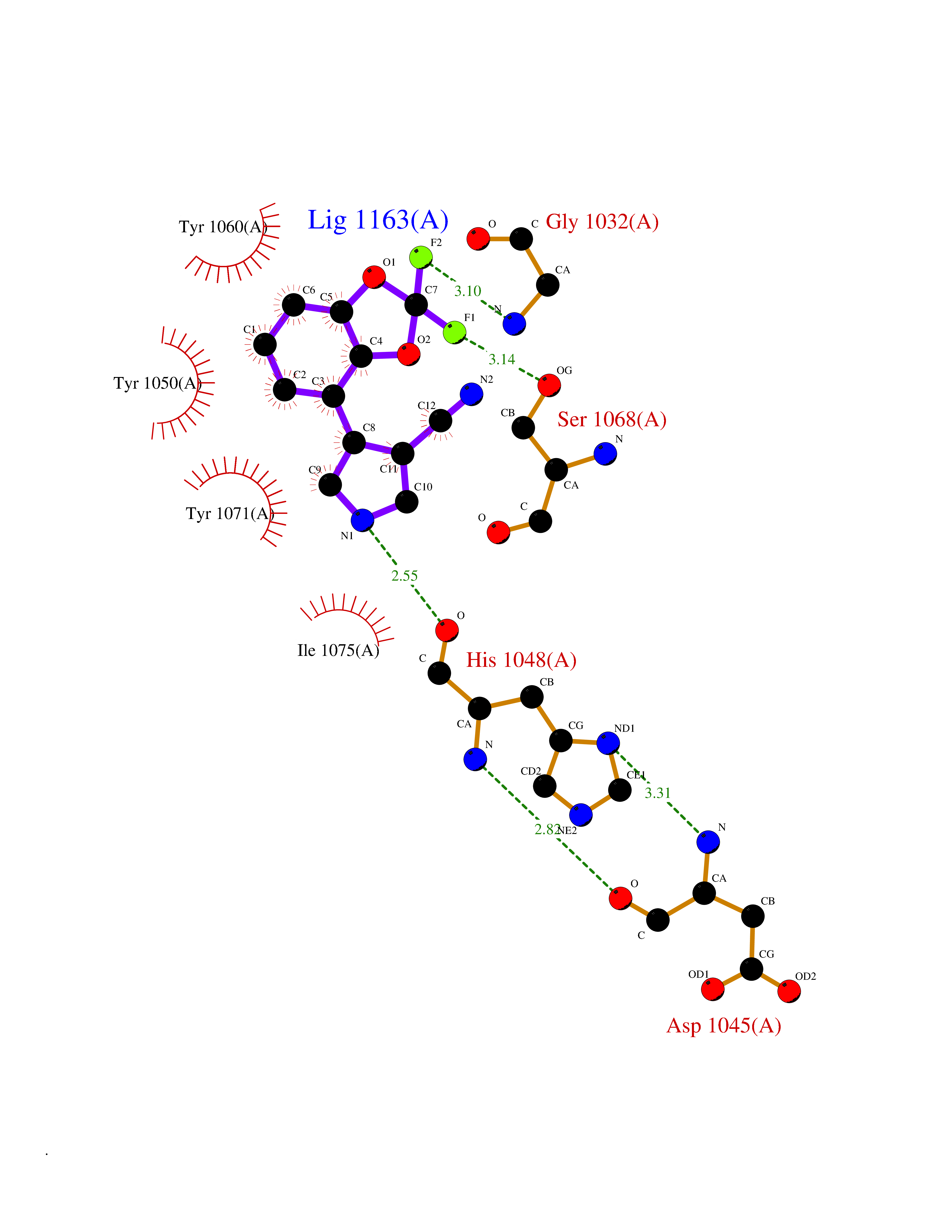 Ligplot Map 3D Binding mode Sequence GTILIDLSPDDKEFQSVEEEMQSTVREHRDGGHAGGIFNRYNILKIQKVCNKKLWERYTHRRKEVSEENHNHANERMLFHGSPFVNAIIHKGFDERHAYIGGMFGAGIYFAENSSKSNQYVYGIGGGTGCPVHKDRSCYICHRQLLFCRVTLGKSFLQFSAMAHSPPGHHSVTGRPSVNGLALAEYVIYRGEQAYPEYLITYQIMRPE Hydrogen bonds contact Hydrophobic contact | ||||
| 6 | Free fatty acid receptor 1 | 4PHU | 6.18 | |
Target general information Gen name FFAR1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms GPR40 Protein family G-protein coupled receptor 1 family Biochemical class Fatty acid binding protein / hydrolase Function Bioactive lipid receptor activity.G-protein coupled receptor activity.Guanyl-nucleotide exchange factor activity.Lipid binding. Related diseases Refsum disease (RD) [MIM:266500]: A rare autosomal recessive peroxisomal disorder characterized by the accumulation of the branched-chain fatty acid, phytanic acid, in blood and tissues. Cardinal clinical features are retinitis pigmentosa, peripheral neuropathy, cerebellar ataxia, and elevated protein levels in the cerebrospinal fluid (CSF). Half of all patients exhibit generalized, mild to moderate ichthyosis resembling ichthyosis vulgaris. Less constant features are nerve deafness, anosmia, skeletal abnormalities, cataracts and cardiac impairment. {ECO:0000269|PubMed:10709665, ECO:0000269|PubMed:10767344, ECO:0000269|PubMed:14974078, ECO:0000269|PubMed:9326939, ECO:0000269|PubMed:9326940}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00159 Interacts with NA EC number NA Uniprot keywords 3D-structure; Cell membrane; Disulfide bond; G-protein coupled receptor; Glycoprotein; Lipid-binding; Membrane; Receptor; Reference proteome; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 28319.1 Length 272 Aromaticity 0.11 Instability index 27.3 Isoelectric point 9.07 Charge (pH=7) 6.85 2D Binding mode Binding energy (Kcal/mol) -8.42 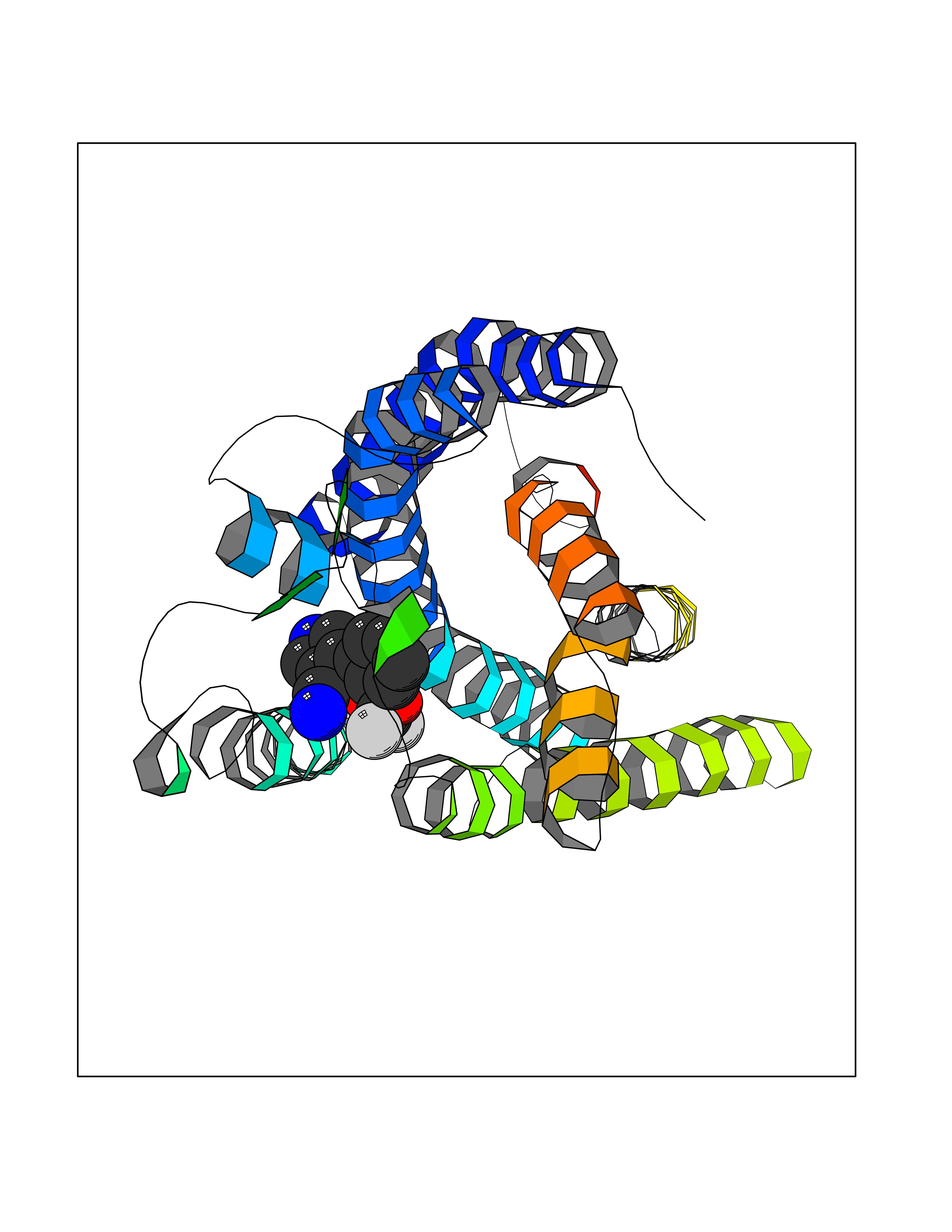 Molscript Map 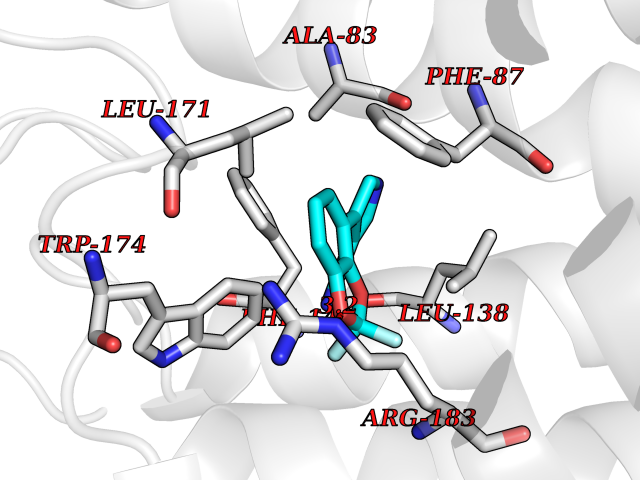 Pymol Map 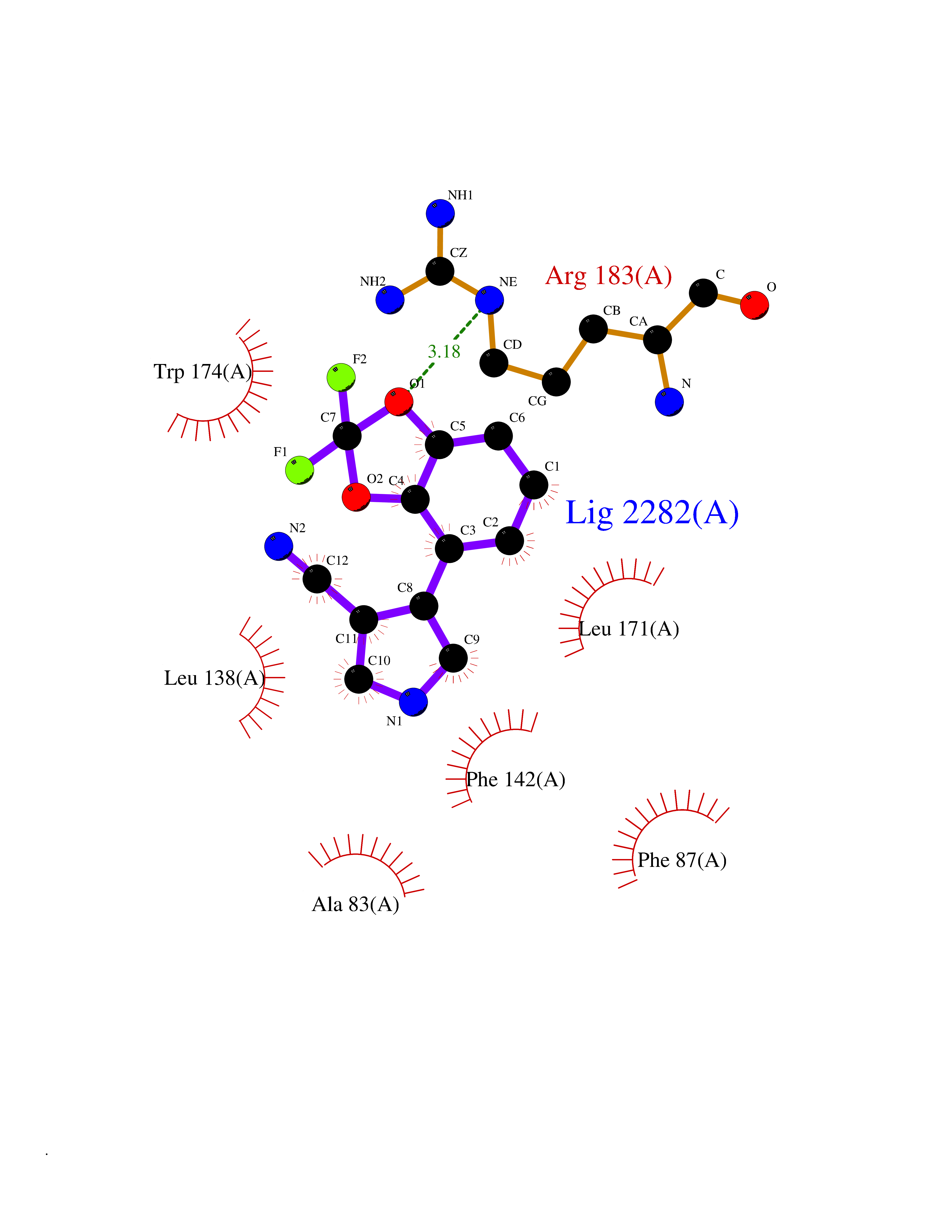 Ligplot Map 3D Binding mode Sequence MDLPPQLSFGLYVAAFALGFPLNVLAIRGATAHARLRLTPSAVYALNLGCSDLLLTVSLPLKAVEALASGAWPLPASLCPVFAVAHFAPLYAGGGFLAALSAARYLGAAFPPCYSWGVCAAIWALVLCHLGLVFGLEAPGGWLDHSNTSLGINTPVNGSPVCLEAWDPASAGPARFSLSLLLFFLPLAITAFCFVGCLRALARGSLTHRRKLRAAWVAGGALLTLLLCVGPYNASNVASFLYPNLGGSWRKLGLITGAWSVVLNPLVTGYLG Hydrogen bonds contact Hydrophobic contact | ||||
| 7 | Neprilysin | 1R1H | 6.18 | |
Target general information Gen name MME Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms EPN Protein family Peptidase M13 family Biochemical class Hydrolase Function Endopeptidase activity.Exopeptidase activity.Metalloendopeptidase activity.Metallopeptidase activity.Peptide binding.Zinc ion binding. Related diseases Charcot-Marie-Tooth disease, axonal, 2T (CMT2T) [MIM:617017]: An axonal form of Charcot-Marie-Tooth disease, a disorder of the peripheral nervous system, characterized by progressive weakness and atrophy, initially of the peroneal muscles and later of the distal muscles of the arms. Charcot-Marie-Tooth disease is classified in two main groups on the basis of electrophysiologic properties and histopathology: primary peripheral demyelinating neuropathies (designated CMT1 when they are dominantly inherited) and primary peripheral axonal neuropathies (CMT2). Neuropathies of the CMT2 group are characterized by signs of axonal degeneration in the absence of obvious myelin alterations, normal or slightly reduced nerve conduction velocities, and progressive distal muscle weakness and atrophy. {ECO:0000269|PubMed:26991897, ECO:0000269|PubMed:27588448}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Spinocerebellar ataxia 43 (SCA43) [MIM:617018]: A form of spinocerebellar ataxia, a clinically and genetically heterogeneous group of cerebellar disorders. Patients show progressive incoordination of gait and often poor coordination of hands, speech and eye movements, due to degeneration of the cerebellum with variable involvement of the brainstem and spinal cord. SCA43 is a slowly progressive, autosomal dominant form. {ECO:0000269|PubMed:27583304}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08575; DB02597; DB00616; DB11623; DB05796; DB06655; DB02558; DB02062; DB00886; DB02557; DB09292; DB13928; DB08626 Interacts with P05067; P21926; Q06787-7; P08107; P04792 EC number 3.4.24.11 Uniprot keywords 3D-structure; Cell membrane; Charcot-Marie-Tooth disease; Disease variant; Disulfide bond; Glycoprotein; Hydrolase; Lipoprotein; Membrane; Metal-binding; Metalloprotease; Myristate; Neurodegeneration; Neuropathy; Phosphoprotein; Protease; Proteomics identification; Reference proteome; Signal-anchor; Spinocerebellar ataxia; Transmembrane; Transmembrane helix; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 79435.8 Length 696 Aromaticity 0.11 Instability index 37.5 Isoelectric point 5.53 Charge (pH=7) -11.46 2D Binding mode Binding energy (Kcal/mol) -8.42  Molscript Map 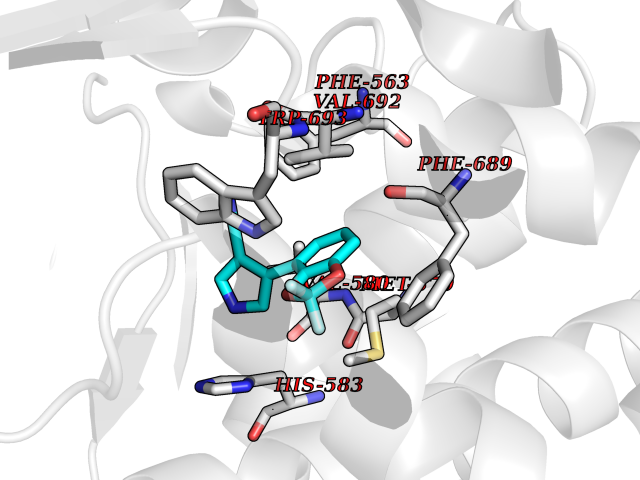 Pymol Map 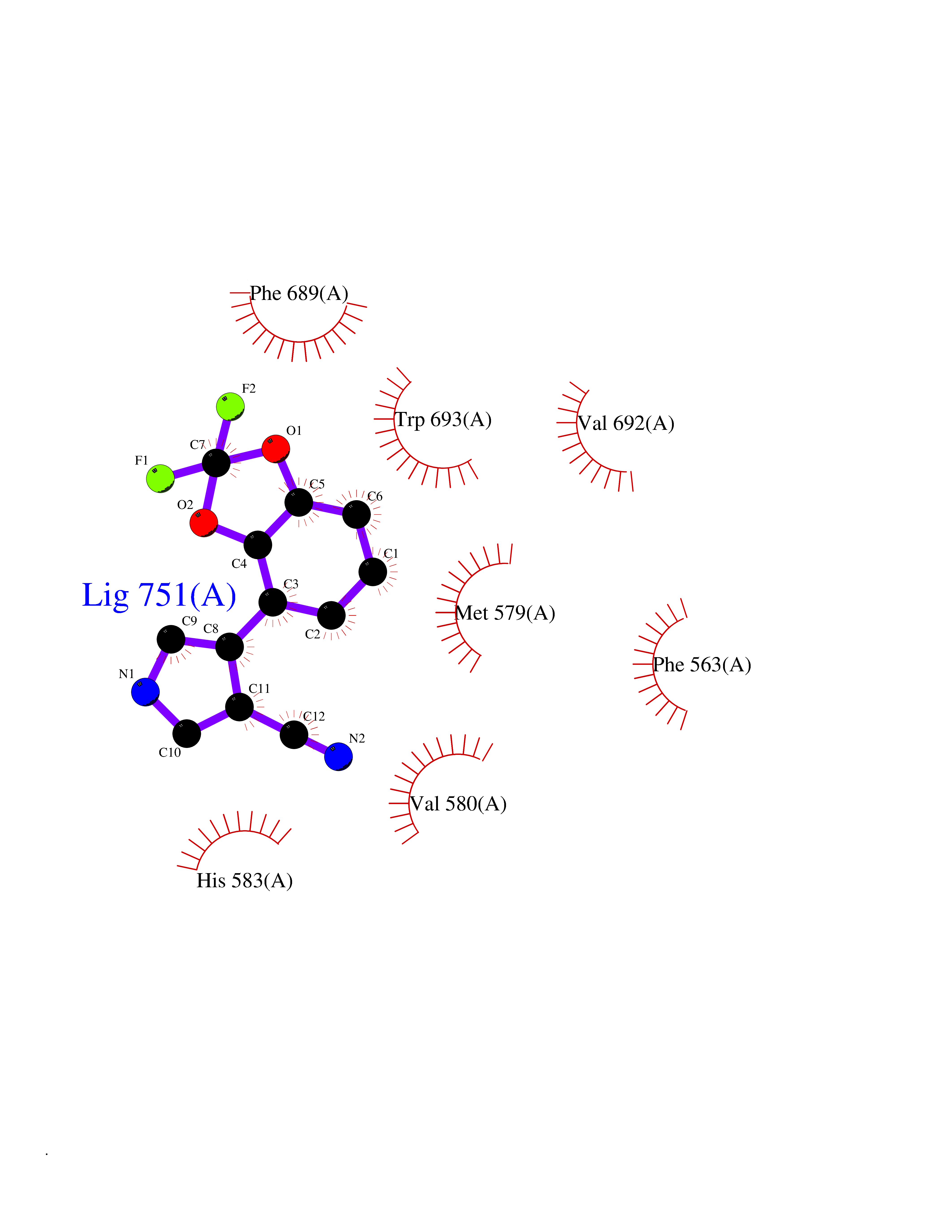 Ligplot Map 3D Binding mode Sequence GICKSSDCIKSAARLIQNMDATTEPCTDFFKYACGGWLKRNVIPETSSRYGNFDILRDELEVVLKDVLQEPKTEDIVAVQKAKALYRSCINESAIDSRGGEPLLKLLPDIYGWPVATENWEQKYGASWTAEKAIAQLNSKYGKKVLINLFVGTDDKNSVNHVIHIDQPRLGLPSRDYYECTGIYKEACTAYVDFMISVARLIRQEERLPIDENQLALEMNKVMELEKEIANATAKPEDRNDPMLLYNKMTLAQIQNNFSLEINGKPFSWLNFTNEIMSTVNISITNEEDVVVYAPEYLTKLKPILTKYSARDLQNLMSWRFIMDLVSSLSRTYKESRNAFRKALYGTTSETATWRRCANYVNGNMENAVGRLYVEAAFAGESKHVVEDLIAQIREVFIQTLDDLTWMDAETKKRAEEKALAIKERIGYPDDIVSNDNKLNNEYLELNYKEDEYFENIIQNLKFSQSKQLKKLREKVDKDEWISGAAVVNAFYSSGRNQIVFPAGILQPPFFSAQQSNSLNYGGIGMVIGHEITHGFDDNGRNFNKDGDLVDWWTQQSASNFKEQSQCMVYQYGNFSWDLAGGQHLNGINTLGENIADNGGLGQAYRAYQNYIKKNGEEKLLPGLDLNHKQLFFLNFAQVWCGTYRPEYAVNSIKTDVHSPGNFRIIGTLQNSAEFSEAFHCRKNSYMNPEKKCRVW Hydrogen bonds contact Hydrophobic contact | ||||
| 8 | Thiopurine S-methyltransferase | 2BZG | 6.16 | |
Target general information Gen name TPMT Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Class I-like SAM-binding methyltransferase superfamily, TPMT family Biochemical class Transferase Function Thiopurine S-methyltransferase activity. Related diseases Cerebral creatine deficiency syndrome 3 (CCDS3) [MIM:612718]: An autosomal recessive disorder characterized by developmental delay/regression, intellectual disability, severe disturbance of expressive and cognitive speech, and severe depletion of creatine/phosphocreatine in the brain. Most patients develop a myopathy characterized by muscle weakness and atrophy later in life. {ECO:0000269|PubMed:11555793, ECO:0000269|PubMed:20682460, ECO:0000269|PubMed:22386973, ECO:0000269|PubMed:23660394, ECO:0000269|PubMed:23770102, ECO:0000269|PubMed:26490222, ECO:0000269|PubMed:27233232}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Fanconi renotubular syndrome 1 (FRTS1) [MIM:134600]: A form of Fanconi renotubular syndrome, a disease due to a generalized dysfunction of the proximal kidney tubule resulting in decreased solute and water reabsorption. Patients have polydipsia and polyuria with phosphaturia, glycosuria and aminoaciduria. They may develop hypophosphatemic rickets or osteomalacia, acidosis and a tendency toward dehydration. Some eventually develop renal insufficiency. FRTS1 inheritance is autosomal dominant. {ECO:0000269|PubMed:29654216}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00993; DB00436; DB01327; DB01033; DB01250; DB01021 Interacts with Q8TAP4-4; Q15047-2; P61981 EC number 2.1.1.67 Uniprot keywords 3D-structure; Acetylation; Cytoplasm; Direct protein sequencing; Methyltransferase; Phosphoprotein; Proteomics identification; Reference proteome; S-adenosyl-L-methionine; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 25971.5 Length 229 Aromaticity 0.12 Instability index 32.58 Isoelectric point 6.74 Charge (pH=7) -0.6 2D Binding mode Binding energy (Kcal/mol) -8.4 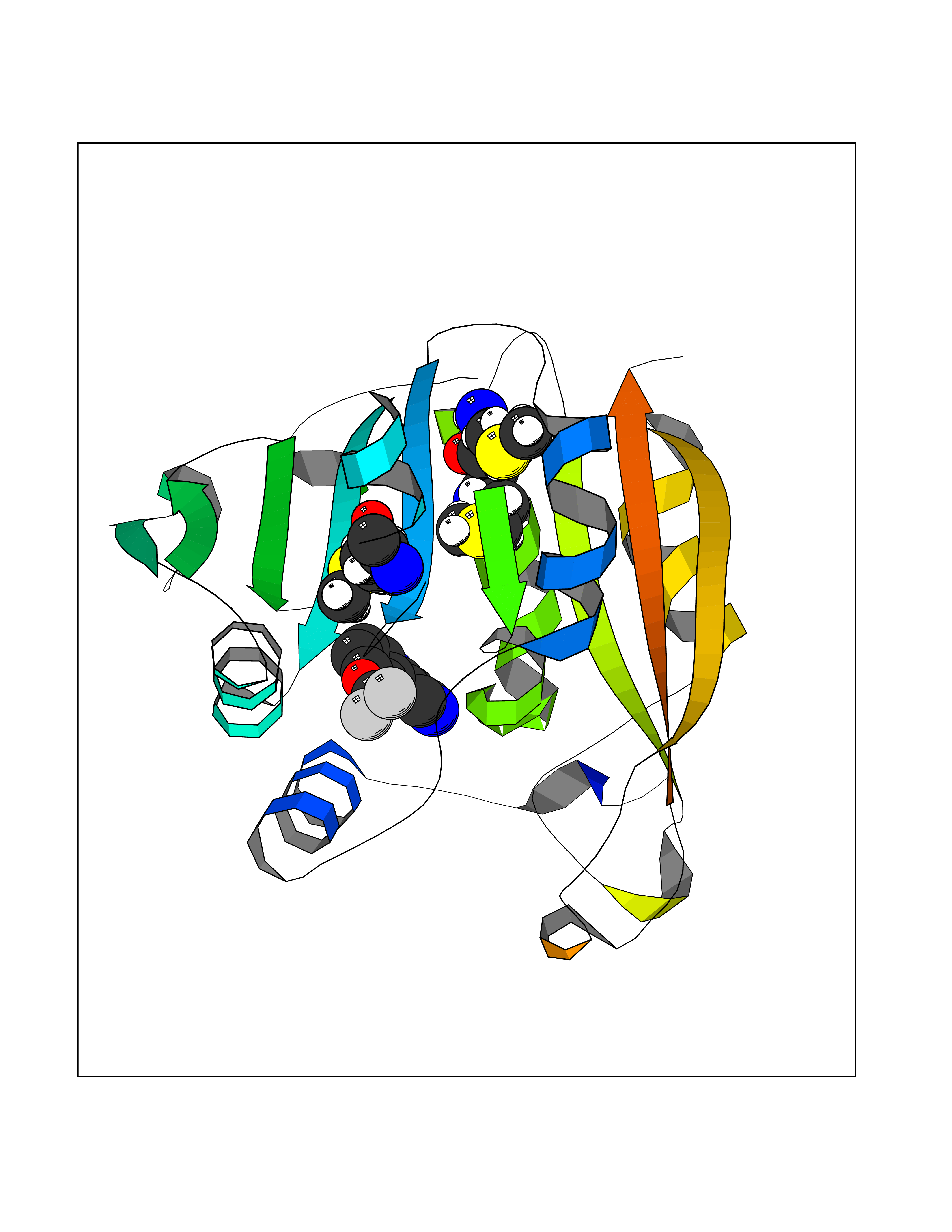 Molscript Map 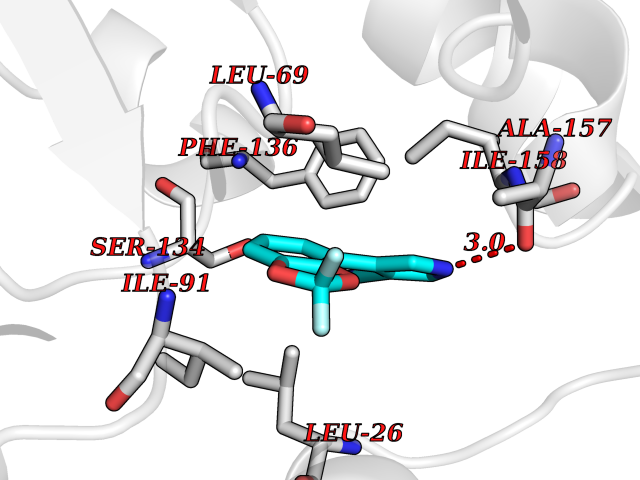 Pymol Map 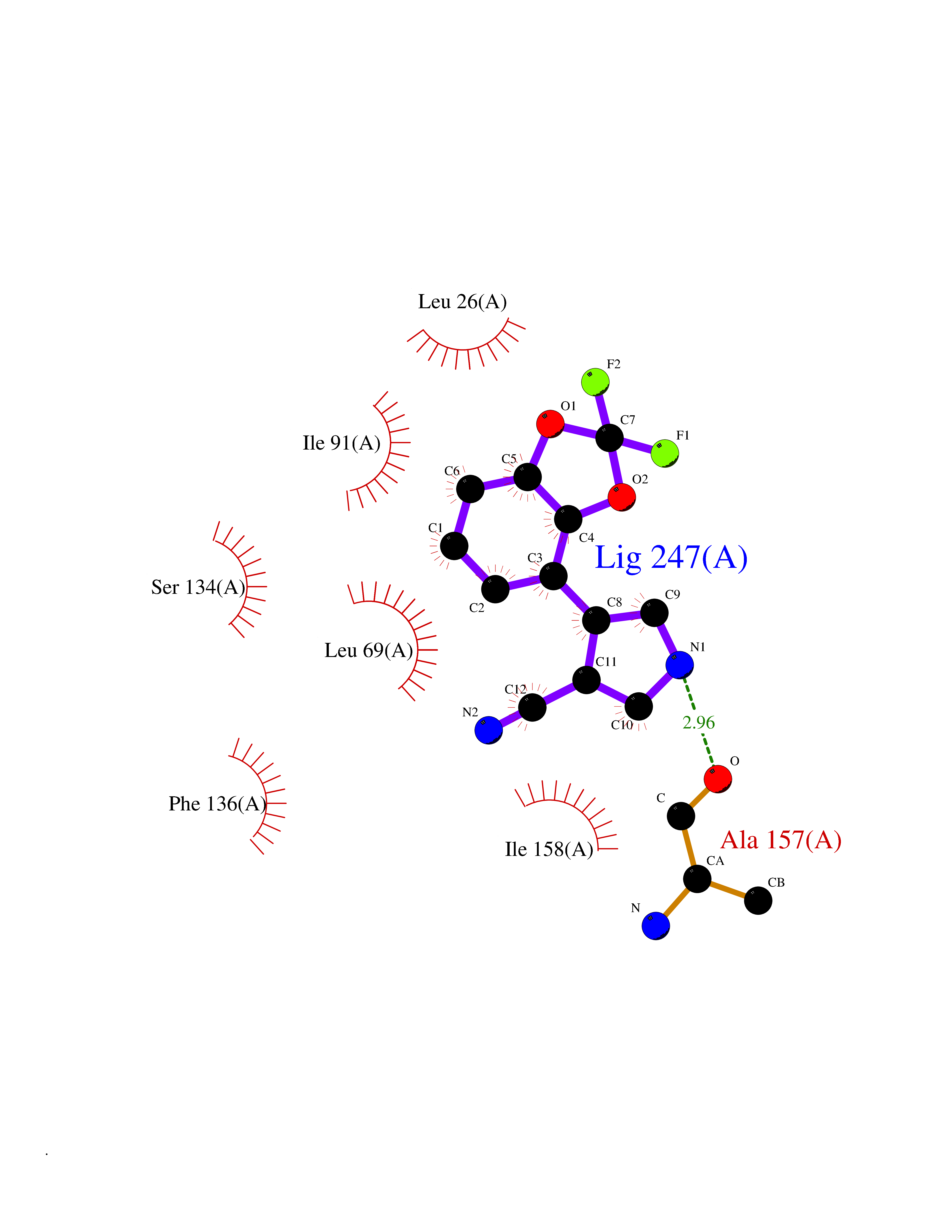 Ligplot Map 3D Binding mode Sequence EVQKNQVLTLEEWQDKWVNGKTAFHQEQGHQLLKKHLDTFLKGKSGLRVFFPLCGKAVEXKWFADRGHSVVGVEISELGIQEFFTEQNLSYSEEPITEIPGTKVFKSSSGNISLYCCSIFDLPRTNIGKFDXIWDRGALVAINPGDRKCYADTXFSLLGKKFQYLLCVLSYDPTKHPGPPFYVPHAEIERLFGKICNIRCLEKVDAFEERHKSWGIDCLFEKLYLLTEK Hydrogen bonds contact Hydrophobic contact | ||||
| 9 | Scavenger decapping enzyme DcpS (DCPS) | 1ST4 | 6.16 | |
Target general information Gen name DCPS Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Scavenger mRNA-decapping enzyme DcpS; Histidine triad protein member5; Hint-related 7meGMP-directed hydrolase; HINT-5; DCS-1; DCPS Protein family HIT family Biochemical class Acid anhydrides hydrolase Function Decapping scavenger enzyme that catalyzes the cleavage of a residual cap structure following the degradation of mRNAs by the 3'->5' exosome-mediated mRNA decay pathway. Hydrolyzes cap analog structures like 7-methylguanosine nucleoside triphosphate (m7GpppG) with up to 10 nucleotide substrates (small capped oligoribonucleotides) and specifically releases 5'-phosphorylated RNA fragments and 7-methylguanosine monophosphate (m7GMP). Cleaves cap analog structures like tri-methyl guanosine nucleoside triphosphate (m3(2,2,7)GpppG) with very poor efficiency. Does not hydrolyze unmethylated cap analog (GpppG) and shows no decapping activity on intact m7GpppG-capped mRNA molecules longer than 25 nucleotides. Does not hydrolyze 7-methylguanosine diphosphate (m7GDP) to m7GMP (PubMed:22985415). May also play a role in the 5'->3 mRNA decay pathway; m7GDP, the downstream product released by the 5'->3' mRNA mediated decapping activity, may be also converted by DCPS to m7GMP (PubMed:14523240). Binds to m7GpppG and strongly to m7GDP. Plays a role in first intron splicing of pre- mRNAs. Inhibits activation-induced cell death. Related diseases Al-Raqad syndrome (ARS) [MIM:616459]: A syndrome characterized by delayed psychomotor development, moderate to severe intellectual disability, poor or absent speech, microcephaly, congenital hypotonia, and severe growth delay. {ECO:0000269|PubMed:25701870, ECO:0000269|PubMed:25712129}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07644; DB07643; DB07642; DB03593; DB01960; DB01649; DB03958 Interacts with Q96C86; P52292; O15131; O60684 EC number EC 3.6.1.59 Uniprot keywords 3D-structure; Acetylation; Cytoplasm; Direct protein sequencing; Disease variant; Hydrolase; Intellectual disability; mRNA processing; mRNA splicing; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome Protein physicochemical properties Chain ID B,A Molecular weight (Da) 69192.9 Length 597 Aromaticity 0.09 Instability index 54.62 Isoelectric point 6.12 Charge (pH=7) -9.94 2D Binding mode Binding energy (Kcal/mol) -8.4  Molscript Map 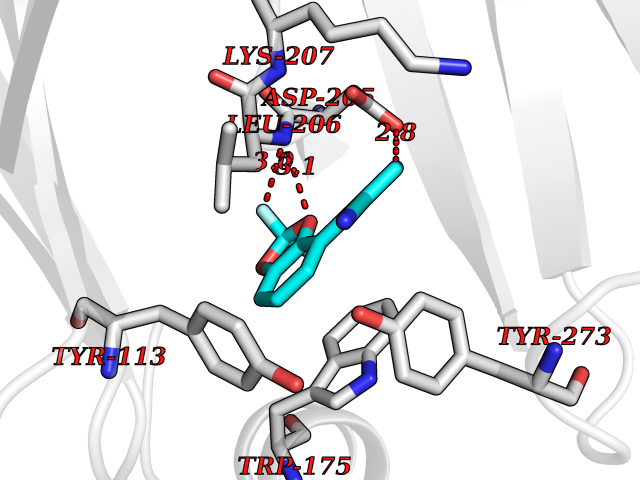 Pymol Map 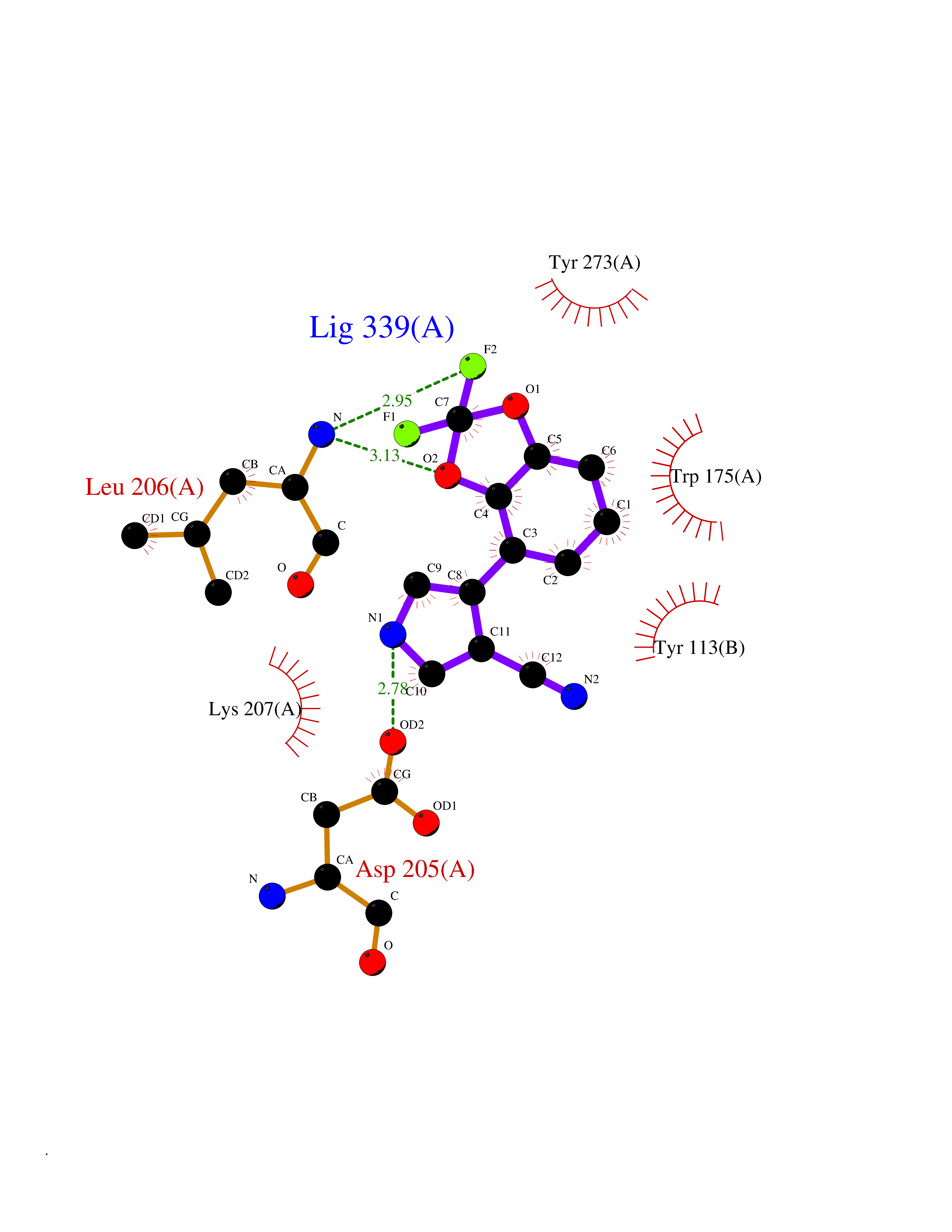 Ligplot Map 3D Binding mode Sequence VRLPFSGFRLQKVLRESARDKIIFLHGKVNEASGDGDGEDAVVILEKTPFQVEQVAQLLTGSPELQLQFSNDIYSTYHLFPPRQLNDVKTTVVYPATEKHLQKYLRQDLRLIRETGDDYRNITLPHLESQSLSIQWVYNILDKKAEADRIVFENPDPSDGFVLIPDLKWNQQQLDDLYLIAICHRRGIRSLRDLTPEHLPLLRNILHQGQEAILQRYRMKGDHLRVYLHYLPSYYHLNVHFTALGFEAPGSGVERAHLLAEVIENLECDPRHYQQRTLTFALRADDPLLKLLQEAQQAPVRLPFSGFRLQKVLRESARDKIIFLHGKVNEASGDGDGEDAVVILEKTPFQVEQVAQLLTGSPELQLQFSNDIYSTYHLFPPRQLNDVKTTVVYPATEKHLQKYLRQDLRLIRETGDDYRNITLPHLESQSLSIQWVYNILDKKAEADRIVFENPDPSDGFVLIPDLKWNQQQLDDLYLIAICHRRGIRSLRDLTPEHLPLLRNILHQGQEAILQRYRMKGDHLRVYLHYLPSYYHLNVHFTALGFEAPGSGVERAHLLAEVIENLECDPRHYQQRTLTFALRADDPLLKLLQEAQQS Hydrogen bonds contact Hydrophobic contact | ||||
| 10 | Hypoxia-inducible factor 1 alpha (HIF-1A) | 5L9B | 6.15 | |
Target general information Gen name HIF1A Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms bHLHe78; Transcription factor HIF-1; PASD8; PAS domain-containing protein 8; Member of PAS protein 1; MOP1; Hypoxia-inducible transcription factor (HIF)-1; Hypoxia-inducible factor 1-alpha; Hypoxia-in Protein family NA Biochemical class NA Function Under hypoxic conditions, activates the transcription of over 40 genes, including erythropoietin, glucose transporters, glycolytic enzymes, vascular endothelial growth factor, HILPDA, and other genes whose protein products increase oxygen delivery or facilitate metabolic adaptation to hypoxia. Plays an essential role in embryonic vascularization, tumor angiogenesis and pathophysiology of ischemic disease. Heterodimerizes with ARNT; heterodimer binds to core DNA sequence 5'-TACGTG-3' within the hypoxia response element (HRE) of target gene promoters. Activation requires recruitment of transcriptional coactivators such as CREBBP and EP300. Activity is enhanced by interaction with both, NCOA1 or NCOA2. Interaction with redox regulatory protein APEX seems to activate CTAD and potentiates activation by NCOA1 and CREBBP. Involved in the axonal distribution and transport of mitochondria in neurons during hypoxia. Functions as a master transcriptional regulator of the adaptive response to hypoxia. Related diseases Essential hypertension (EHT) [MIM:145500]: A condition in which blood pressure is consistently higher than normal with no identifiable cause. {ECO:0000269|PubMed:12372404}. The disease may be caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02342; DB01136; DB05959; DB08687; DB01275; DB06082; DB12255 Interacts with P10275; P27540; P49407; Q9C0J9; O00257; Q92793; P35222; Q96CJ1; Q9GZT9; Q96KS0; Q9H6Z9; Q09472; P11474; P49327; P09467; Q8N461; P23771; Q9NWT6; Q9Y2N7-4; Q92831; Q13330; P46531; Q13438; Q8N2W9; P14618-1; P25789; Q9UHD8-1; P84022; P08047; O60224; P63165; O94888; P40818; P40337; P17861; Q99PV5; Q5RIX9; Q6S7F2; Q61539; Q8BIF2; P51450-2 EC number NA Uniprot keywords 3D-structure; Acetylation; Activator; Alternative splicing; Cytoplasm; Direct protein sequencing; DNA-binding; Glycoprotein; Host-virus interaction; Hydroxylation; Isopeptide bond; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; S-nitrosylation; Transcription; Transcription regulation; Ubl conjugation Protein physicochemical properties Chain ID C,D Molecular weight (Da) 26523.9 Length 236 Aromaticity 0.1 Instability index 24.97 Isoelectric point 5.58 Charge (pH=7) -4.83 2D Binding mode Binding energy (Kcal/mol) -8.38  Molscript Map 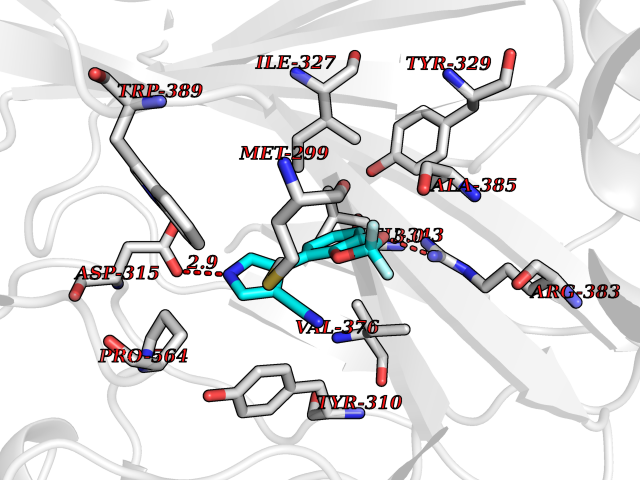 Pymol Map 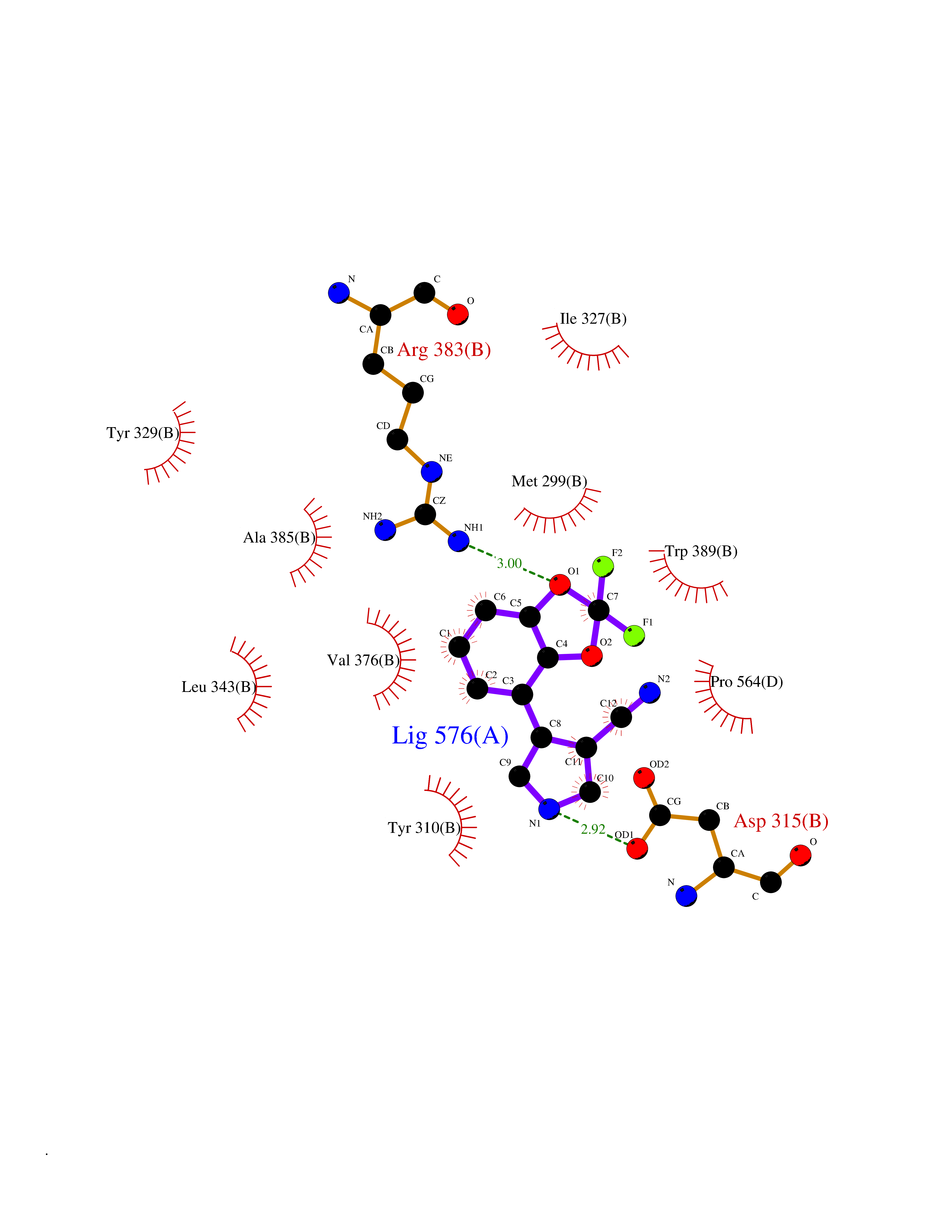 Ligplot Map 3D Binding mode Sequence LDLEMLAPYIPMDDDFQLLPALKLALEYIVPAMNKHGICVVDDFLGKETGQQIGDEVRALHDTGKFTDGQLVSQKSDSSKDIRGDKITWIEGKEPGCETIGLLMSSMDDLIRHCNGKLGSYKINGRTKAMVACYPGNGTGYVRHVDNPNGDGRCVTCIYYLNKDWDAKVSGGILRIFPEGKAQFADIEPKFDRLLFFWSDRRNPHEVQPAYATRYAITVWYFDADERAAAKVKYLT Hydrogen bonds contact Hydrophobic contact | ||||
| 11 | Histone acetyltransferase p300 (EP300) | 5LKX | 6.15 | |
Target general information Gen name EP300 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms p300 HAT; Protein propionyltransferase p300; P300; Histone crotonyltransferase p300; Histone butyryltransferase p300; E1Aassociated protein p300; E1A-associated protein p300 Protein family NA Biochemical class Acyltransferase Function Acetylates all four core histones in nucleosomes. Histone acetylation gives an epigenetic tag for transcriptional activation. Mediates cAMP-gene regulation by binding specifically to phosphorylated CREB protein. Mediates acetylation of histone H3 at 'Lys-122' (H3K122ac), a modification that localizes at the surface of the histone octamer and stimulates transcription, possibly by promoting nucleosome instability. Mediates acetylation of histone H3 at 'Lys-27' (H3K27ac). Also functions as acetyltransferase for non-histone targets, such as ALX1, HDAC1, PRMT1 or SIRT2. Acetylates 'Lys-131' of ALX1 and acts as its coactivator. Acetylates SIRT2 and is proposed to indirectly increase the transcriptional activity of TP53 through acetylation and subsequent attenuation of SIRT2 deacetylase function. Acetylates HDAC1 leading to its inactivation and modulation of transcription. Acts as a TFAP2A-mediated transcriptional coactivator in presence of CITED2. Plays a role as a coactivator of NEUROD1-dependent transcription of the secretin and p21 genes and controls terminal differentiation of cells in the intestinal epithelium. Promotes cardiac myocyte enlargement. Can also mediate transcriptional repression. Acetylates FOXO1 and enhances its transcriptional activity. Acetylates BCL6 wich disrupts its ability to recruit histone deacetylases and hinders its transcriptional repressor activity. Participates in CLOCK or NPAS2-regulated rhythmic gene transcription; exhibits a circadian association with CLOCK or NPAS2, correlating with increase in PER1/2 mRNA and histone H3 acetylation on the PER1/2 promoter. Acetylates MTA1 at 'Lys-626' which is essential for its transcriptional coactivator activity. Acetylates XBP1 isoform 2; acetylation increases protein stability of XBP1 isoform 2 and enhances its transcriptional activity. Acetylates PCNA; acetylation promotes removal of chromatin-bound PCNA and its degradation during nucleotide excision repair (NER). Acetylates MEF2D. Acetylates and stabilizes ZBTB7B protein by antagonizing ubiquitin conjugation and degragation, this mechanism may be involved in CD4/CD8 lineage differentiation. In addition to protein acetyltransferase, can use different acyl-CoA substrates, such as (2E)-butenoyl-CoA (crotonyl-CoA), butanoyl-CoA (butyryl-CoA) or propanoyl-CoA (propionyl-CoA), and is able to mediate protein crotonylation, butyrylation or propionylation, respectively. Acts as a histone crotonyltransferase; crotonylation marks active promoters and enhancers and confers resistance to transcriptional repressors. Histone crotonyltransferase activity is dependent on the concentration of (2E)-butenoyl-CoA (crotonyl-CoA) substrate and such activity is weak when (E)-but-2-enoyl-CoA (crotonyl-CoA) concentration is low. Also acts as a histone butyryltransferase; butyrylation marks active promoters. Functions as a transcriptional coactivator for SMAD4 in the TGF-beta signaling pathway. Acetylates PCK1 and promotes PCK1 anaplerotic activity. Functions as histone acetyltransferase and regulates transcription via chromatin remodeling. Related diseases Defects in EP300 may play a role in epithelial cancer.; DISEASE: Chromosomal aberrations involving EP300 may be a cause of acute myeloid leukemias. Translocation t(8;22)(p11;q13) with KAT6A.; DISEASE: Rubinstein-Taybi syndrome 2 (RSTS2) [MIM:613684]: A disorder characterized by craniofacial abnormalities, postnatal growth deficiency, broad thumbs, broad big toes, intellectual disability and a propensity for development of malignancies. Some individuals with RSTS2 have less severe mental impairment, more severe microcephaly, and a greater degree of changes in facial bone structure than RSTS1 patients. {ECO:0000269|PubMed:15706485}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Menke-Hennekam syndrome 2 (MKHK2) [MIM:618333]: A form of Menke-Hennekam syndrome, a congenital autosomal dominant disease characterized by developmental delay, growth retardation, and craniofacial dysmorphism. Patients have intellectual disability of variable severity, speech delay, autistic behavior, short stature and microcephaly. Main facial characteristics include short palpebral fissures, telecanthi, depressed nasal ridge, short nose, anteverted nares, short columella and long philtrum. {ECO:0000269|PubMed:29460469}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q9NXW9; P27695; Q9UBL3; Q8WXX7; Q9NPI1; P24941; Q99967; P61201; P16220-1; P17844; Q01844; P35637; Q00403; Q16665; Q9H2X6; Q92831; P55209; O60934; P20265; Q96KQ4; Q8WUF5; Q13761; Q96EB6; Q13309; O95863; P42226; Q9UL17; P56279; P05549; P04637; Q13625; O15350; P11473; P67809; K4P3M7; P03122; P06422; P06790; Q61221; Q9QXM1; P04608; P03070; P03255; P03255-2; P03259 EC number EC 2.3.1.48 Uniprot keywords 3D-structure; Acetylation; Acyltransferase; Biological rhythms; Bromodomain; Cell cycle; Chromosomal rearrangement; Chromosome; Citrullination; Cytoplasm; Direct protein sequencing; Disease variant; Host-virus interaction; Intellectual disability; Isopeptide bond; Metal-binding; Methylation; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Transcription; Transcription regulation; Transferase; Ubl conjugation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A Molecular weight (Da) 64477.2 Length 554 Aromaticity 0.12 Instability index 45.78 Isoelectric point 7.01 Charge (pH=7) 0.05 2D Binding mode Binding energy (Kcal/mol) -8.39  Molscript Map 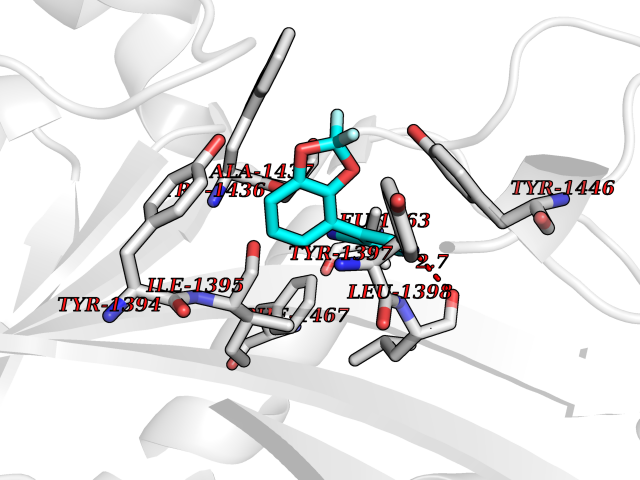 Pymol Map 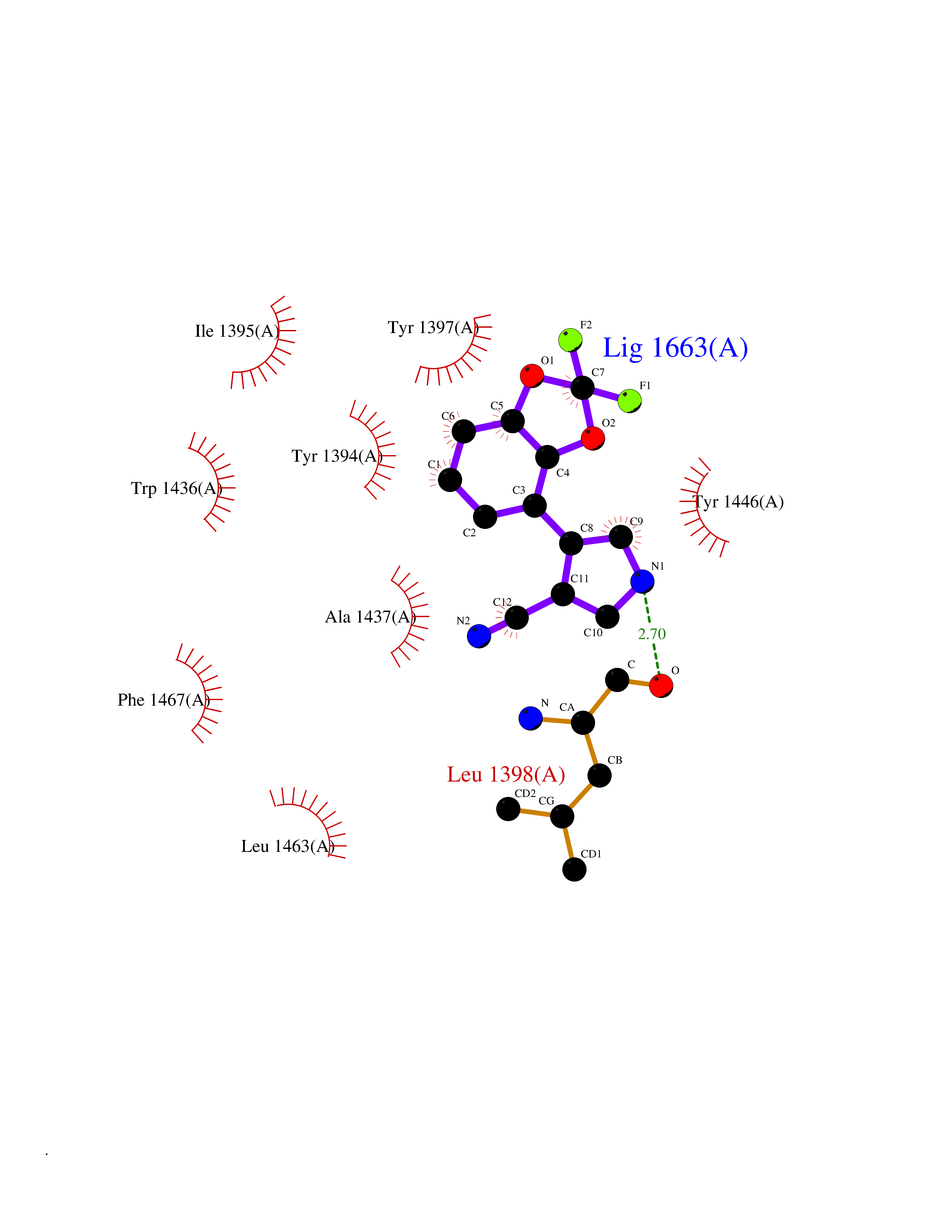 Ligplot Map 3D Binding mode Sequence KKIFKPEELRQALMPTLEALYRQDPESLPFRQPVDPQLLGIPDYFDIVKSPMDLSTIKRKLDTGQYQEPWQYVDDIWLMFNNAWLYNRKTSRVYKYCSKLSEVFEQEIDPVMQSLGYCCGRKLEFSPQTLCCYGKQLCTIPRDATYYSYQNRYHFCEKCFNEIQGESVSLGQTTINKEQFSKRKNDTLDPELFVECTECGRKMHQICVLHHEIIWPAGFVCDGCLKKSARTRKENKFSAKRLPSTRLGTFLENRVNDFLRRQNHPESGEVTVRVVHASDKTVEVKPGMKARFVDSGEMAESFPYRTKALFAFEEIDGVDLCFFGMHVQEYGSDCPPPNQRRVYISYLDSVHFFRPKCLRTAVYHEILIGYLEYVKKLGYTTGHIWACPPSEGDDYIFHCHPPDQKIPKPKRLQEWFKKMLDKAVSERIVHDYKDIFKQATEDRLTSAKELPYFEGDFWPNVLEESIKESGGSGSQKLYATMEKHKEVFFVIRLIAGPAANSLPPIVDPDPLIPCDLMDGRDAFLTLARDKHLEFSSLRRAQWSTMCMLVELHTQ Hydrogen bonds contact Hydrophobic contact | ||||
| 12 | Folate receptor alpha (FOLR1) | 4LRH | 6.15 | |
Target general information Gen name FOLR1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Ovarian tumorassociated antigen MOv18; KB cells FBP; Folate receptor, adult; Folate receptor 1; FRalpha; FOLR1; Adult folatebinding protein Protein family Folate receptor family Biochemical class Folate receptor Function Binds to folate and reduced folic acid derivatives and mediates delivery of 5-methyltetrahydrofolate and folate analogs into the interior of cells. Has high affinity for folate and folic acid analogs at neutral pH. Exposure to slightly acidic pHafter receptor endocytosis triggers a conformation change that strongly reduces its affinity for folates and mediates their release. Required for normal embryonic development and normal cell proliferation. Related diseases Neurodegeneration due to cerebral folate transport deficiency (NCFTD) [MIM:613068]: An autosomal recessive neurodegenerative disorder resulting from brain-specific folate deficiency early in life. Onset is apparent in late infancy with severe developmental regression, movement disturbances, epilepsy and leukodystrophy. {ECO:0000269|PubMed:19732866}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB05595; DB00158; DB00563; DB12489; DB15413; DB05168 Interacts with Q8N357 EC number NA Uniprot keywords 3D-structure; Cell membrane; Cytoplasmic vesicle; Direct protein sequencing; Disulfide bond; Endosome; Folate-binding; Glycoprotein; GPI-anchor; Lipoprotein; Membrane; Neurodegeneration; Proteomics identification; Receptor; Reference proteome; Secreted; Signal; Transport Protein physicochemical properties Chain ID A Molecular weight (Da) 24216 Length 207 Aromaticity 0.13 Instability index 49.36 Isoelectric point 8.14 Charge (pH=7) 3.41 2D Binding mode Binding energy (Kcal/mol) -8.39  Molscript Map 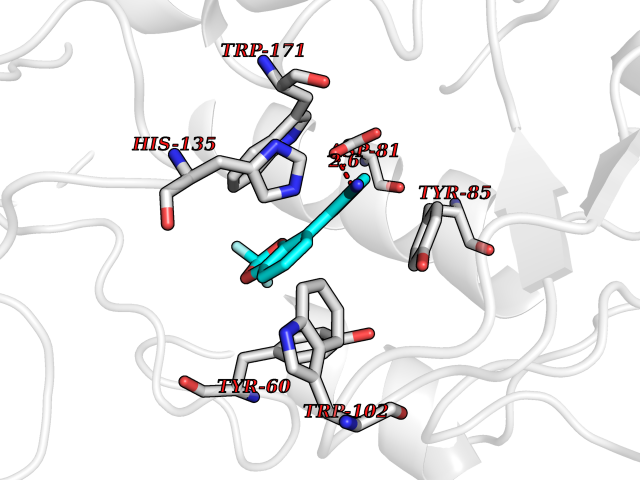 Pymol Map 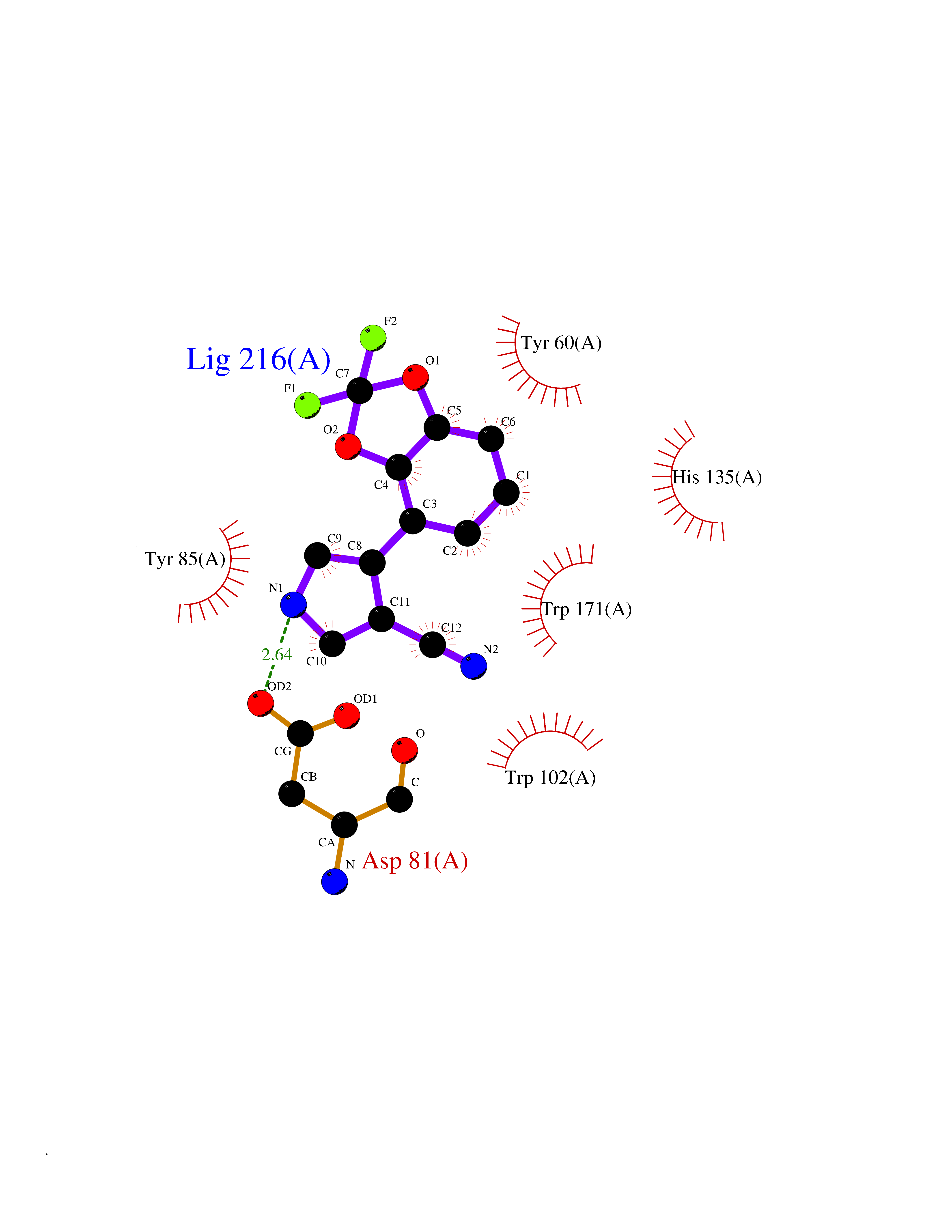 Ligplot Map 3D Binding mode Sequence RTELLNVCMNAKHHKEKPGPEDKLHEQCRPWRKNACCSTNTSQEAHKDVSYLYRFNWNHCGEMAPACKRHFIQDTCLYECSPNLGPWIQQVDQSWRKERVLNVPLCKEDCEQWWEDCRTSYTCKSNWHKGWNWTSGFNKCAVGAACQPFHFYFPTPTVLCNEIWTHSYKVSNYSRGSGRCIQMWFDPAQGNPNEEVARFYAAAMSGT Hydrogen bonds contact Hydrophobic contact | ||||
| 13 | Dihydroorotate dehydrogenase (quinone), mitochondrial | 4CQ8 | 6.13 | |
Target general information Gen name PFF0160c Organism Plasmodium falciparum (isolate 3D7) Uniprot ID TTD ID NA Synonyms NA Protein family Dihydroorotate dehydrogenase family, Type 2 subfamily Biochemical class Oxidoreductase Function Dihydroorotate dehydrogenase activity. Related diseases Combined oxidative phosphorylation deficiency 33 (COXPD33) [MIM:617713]: An autosomal recessive disorder caused by multiple mitochondrial respiratory chain defects and impaired mitochondrial energy metabolism. Clinical manifestations are highly variable. Affected infants present with cardiomyopathy accompanied by multisystemic features involving liver, kidney, and brain. Death in infancy is observed in some patients. Children and adults present with myopathy and progressive external ophthalmoplegia. {ECO:0000269|PubMed:28942965}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01117 Interacts with NA EC number 1.3.5.2 Uniprot keywords 3D-structure; Flavoprotein; FMN; Membrane; Mitochondrion; Mitochondrion inner membrane; Oxidoreductase; Pyrimidine biosynthesis; Reference proteome; Transit peptide; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A,B Molecular weight (Da) 42573.5 Length 378 Aromaticity 0.1 Instability index 36.63 Isoelectric point 8.21 Charge (pH=7) 3.17 2D Binding mode Binding energy (Kcal/mol) -8.36 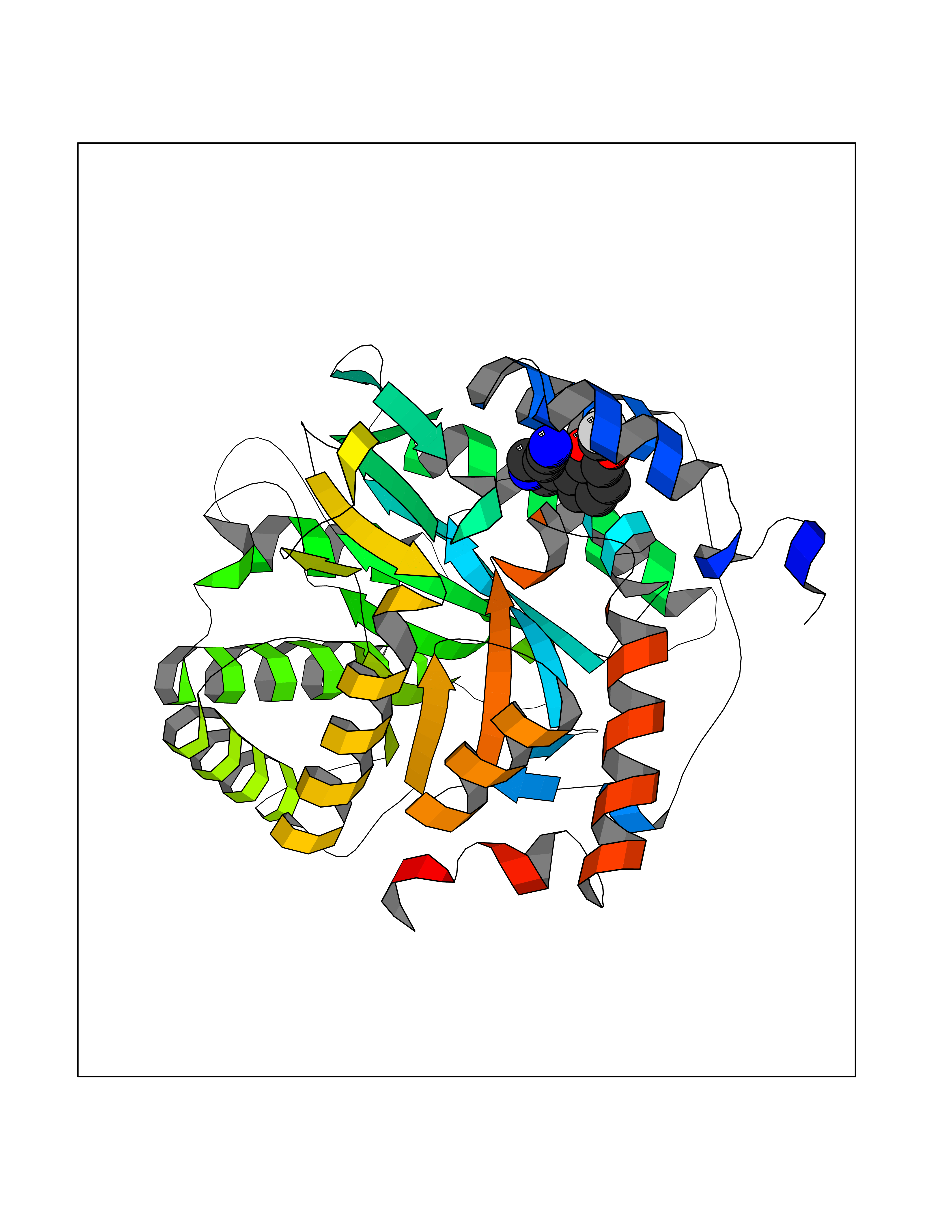 Molscript Map 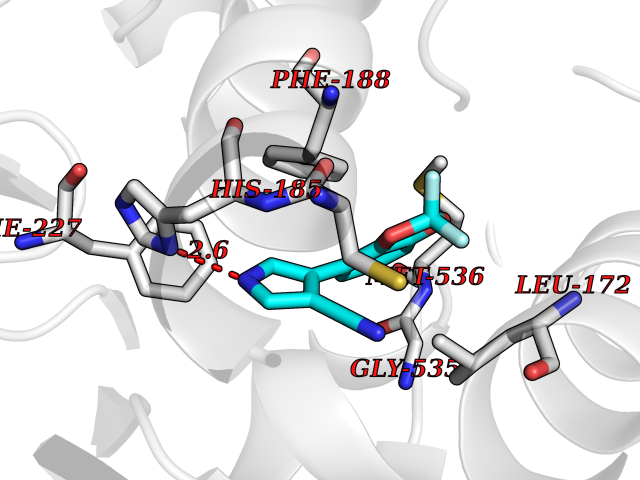 Pymol Map 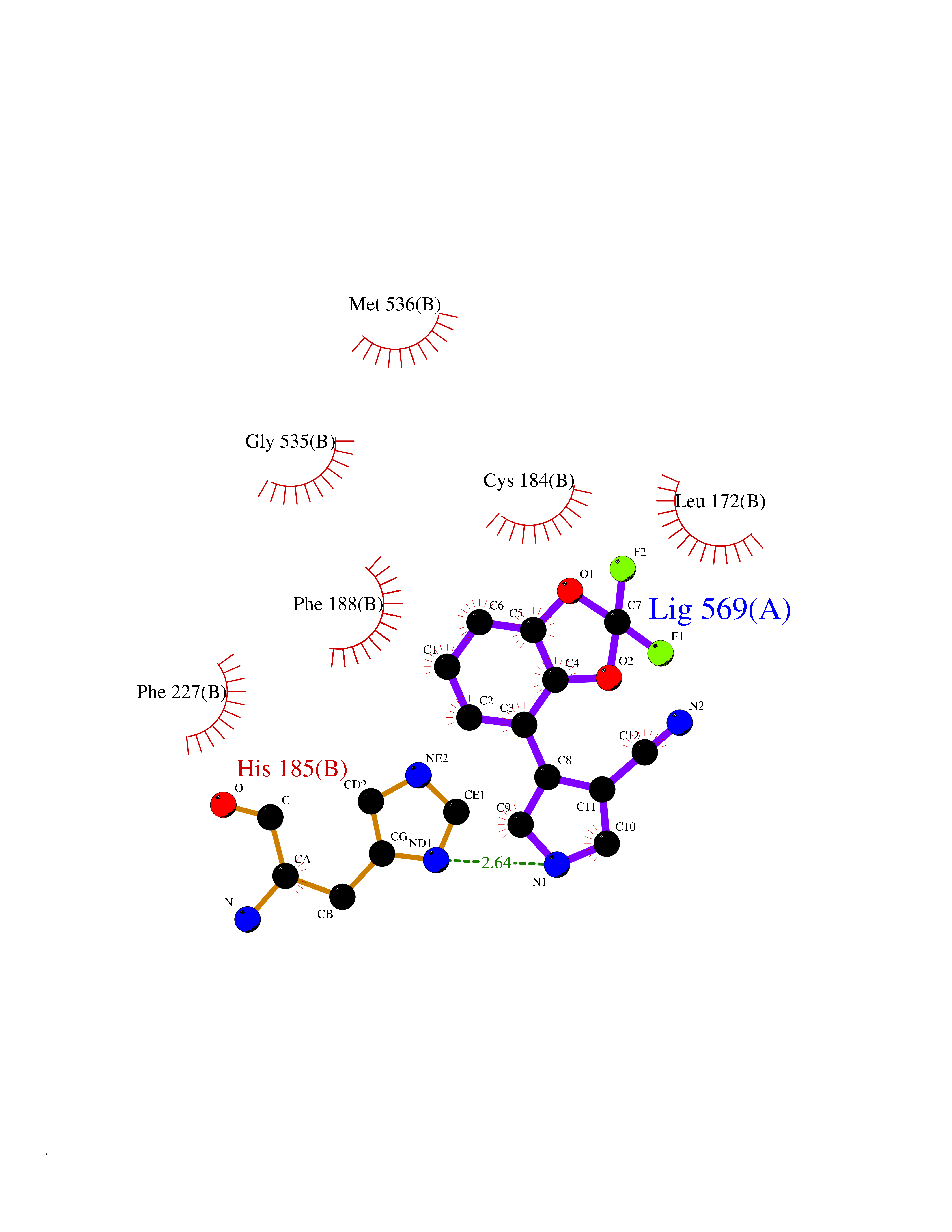 Ligplot Map 3D Binding mode Sequence ADPFESYNPEFFLYDIFLKFCLKYIDGEICHDLFLLLGKYNILPYDTSNDSIYACTNIKHLDFINPFGVAAGFDKNGVCIDSILKLGFSFIEIGTITPRGQTGNAKPRIFRDVESRSIINSCGFNNMGCDKVTENLILFRKRQEEDKLLSKHIVGVSIGKNKDTVNIVDDLKYCINKIGRYADYIAINVSSPNTPGLRDNQEAGKLKNIILSVKEEIDNLEKNNFLWFNTTKKKPLVFVKLAPDLNQEQKKEIADVLLETNIDGMIISNTTTQINDIKSFENKKGGVSGAKLKDISTKFICEMYNYTNKQIPIIASGGIFSGLDALEKIEAGASVCQLYSCLVFNGMKSAVQIKRELNHLLYQRGYYNLKEAIGRKHS Hydrogen bonds contact Hydrophobic contact | ||||
| 14 | Acetylcholine receptor subunit alpha | 4ZJS | 6.12 | |
Target general information Gen name CHRNA1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms ACHRA;CHNRA Protein family Ligand-gated ion channel (TC 1.A.9) family, Acetylcholine receptor (TC 1.A.9.1) subfamily, Alpha-1/CHRNA1 sub-subfamily Biochemical class Immune system Function Acetylcholine binding.Acetylcholine-gated cation-selective channel activity.Acetylcholine receptor activity.Ion channel activity.Ligand-gated ion channel activity. Related diseases Multiple pterygium syndrome, lethal type (LMPS) [MIM:253290]: Multiple pterygia are found infrequently in children with arthrogryposis and in fetuses with fetal akinesia syndrome. In lethal multiple pterygium syndrome there is intrauterine growth retardation, multiple pterygia, and flexion contractures causing severe arthrogryposis and fetal akinesia. Subcutaneous edema can be severe, causing fetal hydrops with cystic hygroma and lung hypoplasia. Oligohydramnios and facial anomalies are frequent. {ECO:0000269|PubMed:18252226}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: The alpha subunit is the main focus for antibody binding in myasthenia gravis. Myasthenia gravis is characterized by sporadic muscular fatigability and weakness, occurring chiefly in muscles innervated by cranial nerves, and characteristically improved by cholinesterase-inhibiting drugs.; DISEASE: Myasthenic syndrome, congenital, 1A, slow-channel (CMS1A) [MIM:601462]: A common congenital myasthenic syndrome. Congenital myasthenic syndromes are characterized by muscle weakness affecting the axial and limb muscles (with hypotonia in early-onset forms), the ocular muscles (leading to ptosis and ophthalmoplegia), and the facial and bulbar musculature (affecting sucking and swallowing, and leading to dysphonia). The symptoms fluctuate and worsen with physical effort. CMS1A is a slow-channel myasthenic syndrome. It is caused by kinetic abnormalities of the AChR, resulting in prolonged AChR channel opening episodes, prolonged endplate currents, and depolarization block. This is associated with calcium overload, which may contribute to subsequent degeneration of the endplate and postsynaptic membrane. {ECO:0000269|PubMed:16685696, ECO:0000269|PubMed:7619526, ECO:0000269|PubMed:8872460, ECO:0000269|PubMed:9158151, ECO:0000269|PubMed:9221765}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Myasthenic syndrome, congenital, 1B, fast-channel (CMS1B) [MIM:608930]: A form of congenital myasthenic syndrome, a group of disorders characterized by failure of neuromuscular transmission, including pre-synaptic, synaptic, and post-synaptic disorders that are not of autoimmune origin. Clinical features are easy fatigability and muscle weakness affecting the axial and limb muscles (with hypotonia in early-onset forms), the ocular muscles (leading to ptosis and ophthalmoplegia), and the facial and bulbar musculature (affecting sucking and swallowing, and leading to dysphonia). The symptoms fluctuate and worsen with physical effort. CMS1B is a fast-channel myasthenic syndrome. It is caused by kinetic abnormalities of the AChR, resulting in brief opening and activity of the channel, with a rapid decay in endplate current, failure to achieve threshold depolarization of the endplate and consequent failure to fire an action potential. {ECO:0000269|PubMed:10195214, ECO:0000269|PubMed:12588888, ECO:0000269|PubMed:15079006}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08838; DB00565; DB00555 Interacts with NA EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Congenital myasthenic syndrome; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; Ion channel; Ion transport; Ligand-gated ion channel; Membrane; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Signal; Synapse; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A,B,C,D,E Molecular weight (Da) 46717.8 Length 411 Aromaticity 0.11 Instability index 38.02 Isoelectric point 4.77 Charge (pH=7) -22.31 2D Binding mode Binding energy (Kcal/mol) -8.35 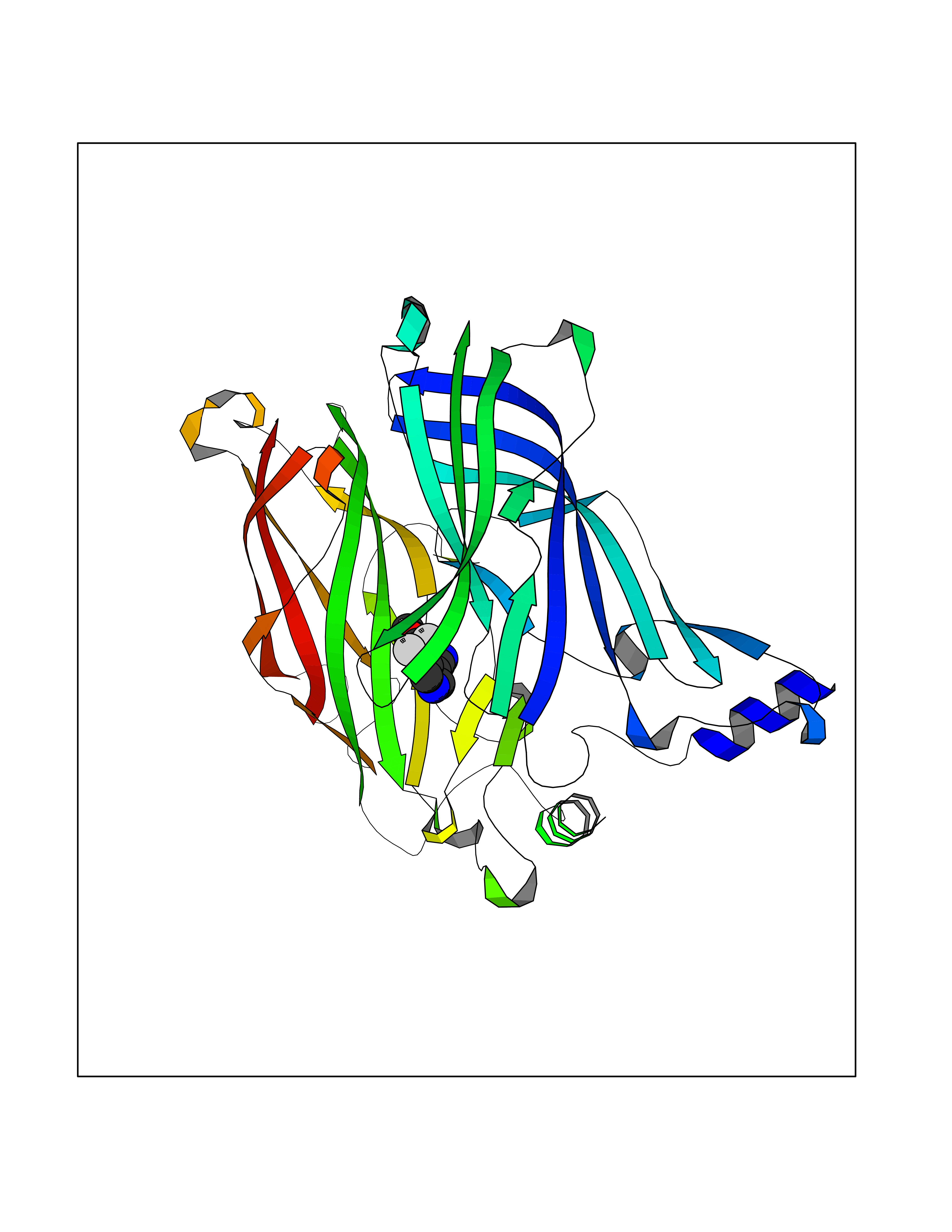 Molscript Map 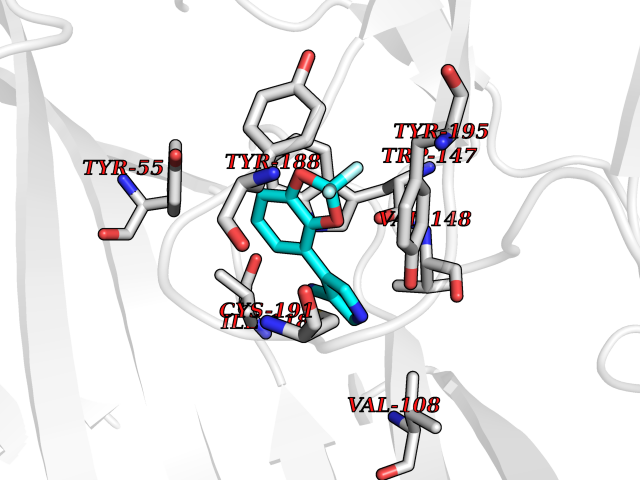 Pymol Map 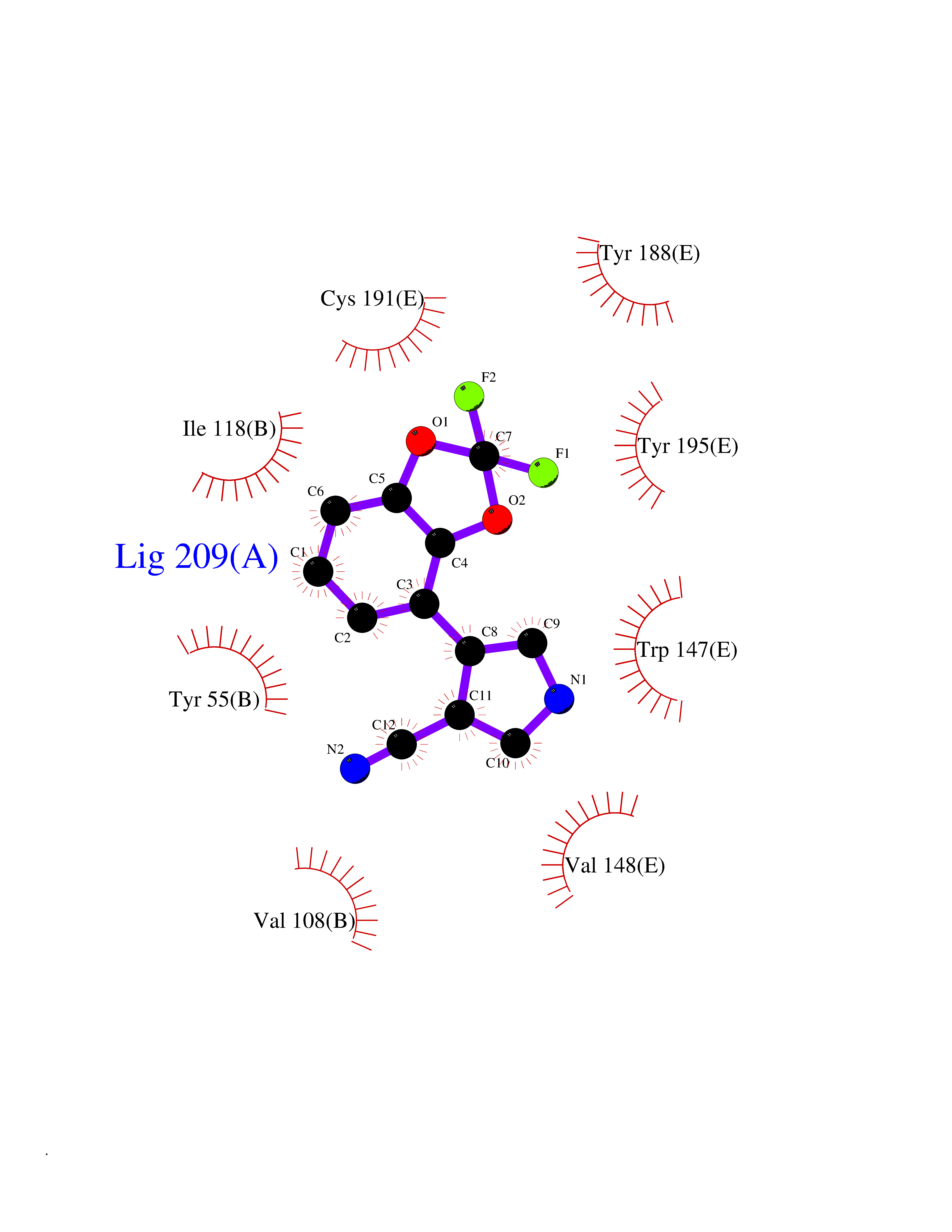 Ligplot Map 3D Binding mode Sequence EHETRLVAKLFKDYSSVVRPVEDHRQVVEVTLGFTLQDIVKADSSTNEVDLVYYEQQRWVDYNLKWNPDDYGGVKKIHIPAADIWTPDITAYSSTRPVQVLSPQIAVVTHDGSVMFIPAQRLSFMCDPTGVDSEEGATCAVKFGSWVYSGFEIDLKTDTDQVDLSSYYASSKYEILSATQTRQVQHYSCCPEPYIDVNLVVKFREEHETRLVAKLFKDYSSVVRPVEDHRQVVEVTLGFTLQDIVKADSSTNEVDLVYYEQQRWVDYNLKWNPDDYGGVKKIHIPAADIWTPDITAYSSTRPVQVLSPQIAVVTHDGSVMFIPAQRLSFMCDPTGVDSEEGATCAVKFGSWVYSGFEIDLKTDTDQVDLSSYYASSKYEILSATQTRQVQHYSCCPEPYIDVNLVVKFRER Hydrogen bonds contact Hydrophobic contact | ||||
| 15 | Helicobacter pylori Methylthioadenosine nucleosidase (HELPY mtnN) | 4BMZ | 6.11 | |
Target general information Gen name HELPY mtnN Organism Helicobacter pylori (strain ATCC 700392 / 26695) (Campylobacter pylori) Uniprot ID TTD ID Synonyms MTAN; MTA/SAH nucleosidase; Aminofutalosine nucleosidase; Aminodeoxyfutalosine nucleosidase; AFL nucleosidase; 6-amino-6-deoxyfutalosine N-ribosylhydrolase; 5'-methylthioadenosine/S-adenosylhomocystei Protein family PNP/UDP phosphorylase family Biochemical class NA Function Catalyzes the direct conversion of aminodeoxyfutalosine (AFL) into dehypoxanthine futalosine (DHFL) and adenine via the hydrolysis of the N-glycosidic bond; this reaction seems to represent an essential step in the menaquinone biosynthesis pathway in Helicobacter species. Can also probably catalyzes the hydrolysis of 5'-methylthioadenosine (MTA) and S-adenosylhomocysteine (SAH) to adenine and the corresponding thioribose, 5'-methylthioribose and S-ribosylhomocysteine, respectively. These other activities highlight the tremendous versatility of the enzyme, which also plays key roles in S-adenosylmethionine recycling and in the biosynthesis of the quorum-sensing molecule autoinducer-2. Does not act on futalosine (FL) as substrate. Related diseases Progressive familial heart block 1B (PFHB1B) [MIM:604559]: A cardiac bundle branch disorder characterized by progressive alteration of cardiac conduction through the His-Purkinje system, with a pattern of a right bundle-branch block and/or left anterior hemiblock occurring individually or together. It leads to complete atrio-ventricular block causing syncope and sudden death. {ECO:0000269|PubMed:19726882, ECO:0000269|PubMed:20562447, ECO:0000269|PubMed:21887725}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Erythrokeratodermia variabilis et progressiva 6 (EKVP6) [MIM:618531]: A form of erythrokeratodermia variabilis et progressiva, a genodermatosis characterized by the coexistence of two independent skin lesions: transient erythema and hyperkeratosis that is usually localized but occasionally occurs in its generalized form. Clinical presentation varies significantly within a family and from one family to another. Palmoplantar keratoderma is present in around 50% of cases. EKVP6 inheritance is autosomal dominant. {ECO:0000269|PubMed:30528822}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number NA Uniprot keywords 3D-structure; Amino-acid biosynthesis; Hydrolase; Menaquinone biosynthesis; Methionine biosynthesis; Reference proteome Protein physicochemical properties Chain ID A,B Molecular weight (Da) 50547.6 Length 464 Aromaticity 0.08 Instability index 26.92 Isoelectric point 5.13 Charge (pH=7) -20.92 2D Binding mode Binding energy (Kcal/mol) -8.33  Molscript Map 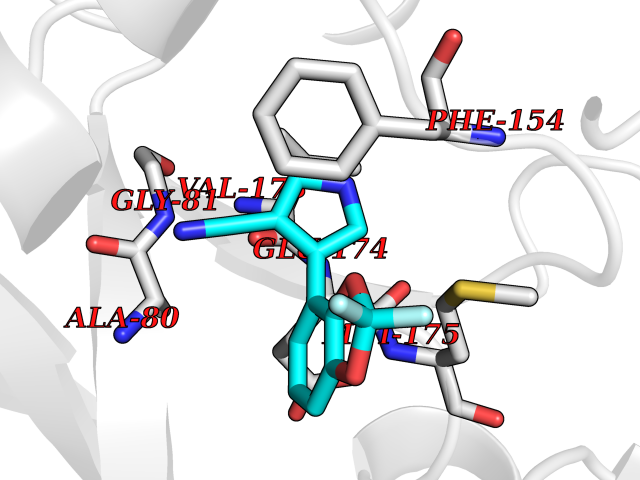 Pymol Map 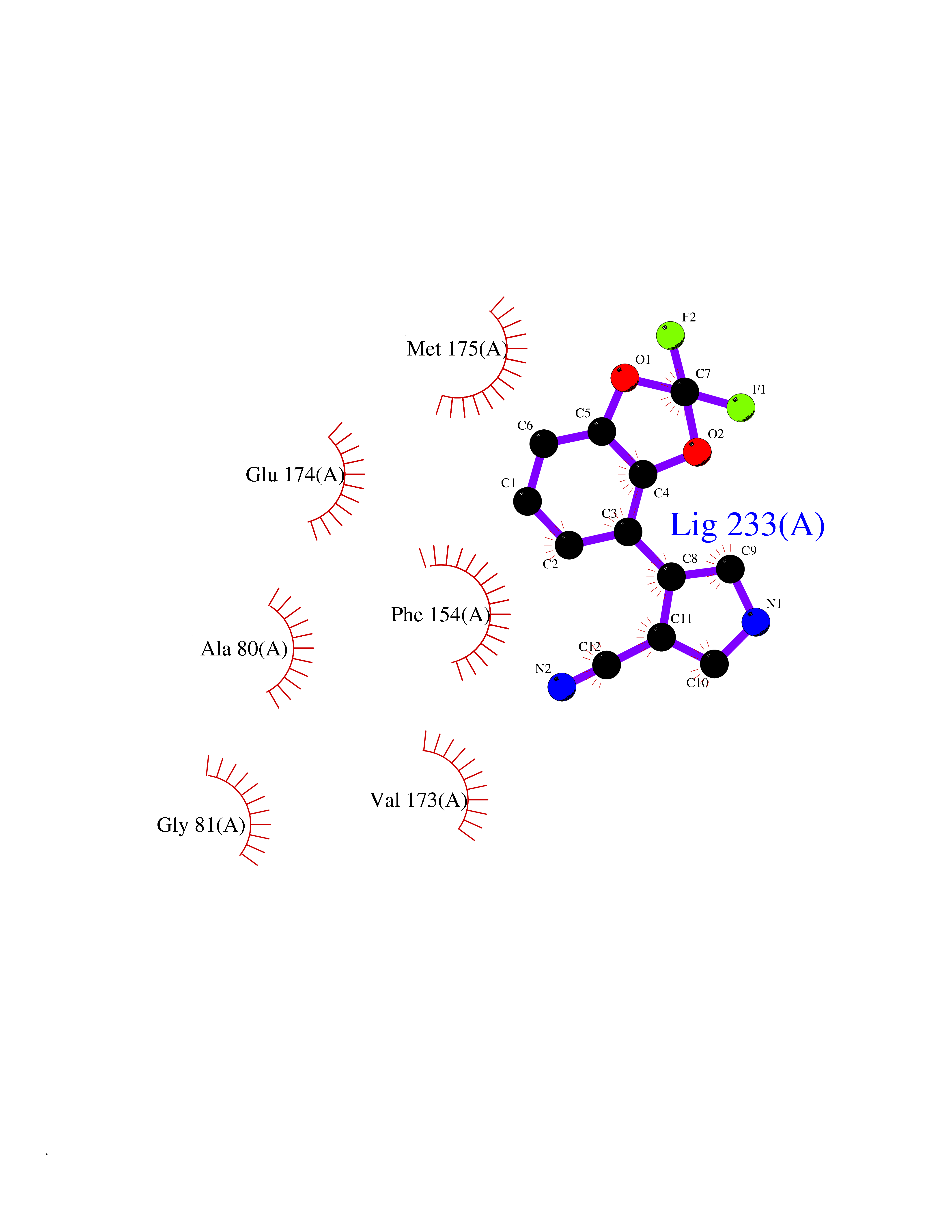 Ligplot Map 3D Binding mode Sequence VQKIGILGAMREEITPILELFGVDFEEIPLGGNVFHKGVYHNKEIIVAYSKIGKVHSTLTTTSMILAFGVQKVLFSGVAGSLVKDLKINDLLVAIQLVQHDVDLSAFDHPLGFIPESAIFIETSESLNALAKEVANEQHIVLKEGVIASGDQFVHSKERKEFLVSEFKASAVEMEGASVAFVCQKFGVPCCVLRSISNNADEEANMSFDAFLEKSAQTSAKFLKSMVDELGSHMVQKIGILGAMREEITPILELFGVDFEEIPLGGNVFHKGVYHNKEIIVAYSKIGKVHSTLTTTSMILAFGVQKVLFSGVAGSLVKDLKINDLLVAIQLVQHDVDLSAFDHPLGFIPESAIFIETSESLNALAKEVANEQHIVLKEGVIASGDQFVHSKERKEFLVSEFKASAVEMEGASVAFVCQKFGVPCCVLRSISNNADEEANMSFDAFLEKSAQTSAKFLKSMVDEL Hydrogen bonds contact Hydrophobic contact | ||||
| 16 | Neuronal acetylcholine receptor subunit alpha-3 | 4ZK4 | 6.10 | |
Target general information Gen name CHRNA3 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NACHRA3 Protein family Ligand-gated ion channel (TC 1.A.9) family, Acetylcholine receptor (TC 1.A.9.1) subfamily, Alpha-3/CHRNA3 sub-subfamily Biochemical class Acetylcholine binding protein Function Acetylcholine binding.Acetylcholine-gated cation-selective channel activity.Acetylcholine receptor activity.Ligand-gated ion channel activity.Serotonin-gated cation-selective channel activity. Related diseases Bladder dysfunction, autonomic, with impaired pupillary reflex and secondary CAKUT (BAIPRCK) [MIM:191800]: An autosomal recessive disease characterized by impaired innervation and autonomic dysfunction of the urinary bladder, hydronephrosis, vesicoureteral reflux, small kidneys, recurrent urinary tract infections, and progressive renal insufficiency. Additional autonomic features are impaired pupillary reflex and orthostatic hypotension. The disease manifests in utero or early childhood. {ECO:0000269|PubMed:31708116}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00915; DB01156; DB00237; DB00565; DB09028; DB00514; DB07720; DB00898; DB00472; DB05710; DB01227; DB00848; DB00333; DB00184; DB01090; DB00202; DB01273 Interacts with NA EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disease variant; Disulfide bond; Endoplasmic reticulum; Glycoprotein; Golgi apparatus; Ion channel; Ion transport; Ligand-gated ion channel; Membrane; Phosphoprotein; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Signal; Synapse; Transmembrane; Transmembrane helix; Transport; Ubl conjugation Protein physicochemical properties Chain ID A,B,C,D,E Molecular weight (Da) 46391.5 Length 408 Aromaticity 0.12 Instability index 30.23 Isoelectric point 4.6 Charge (pH=7) -22.73 2D Binding mode Binding energy (Kcal/mol) -8.32  Molscript Map 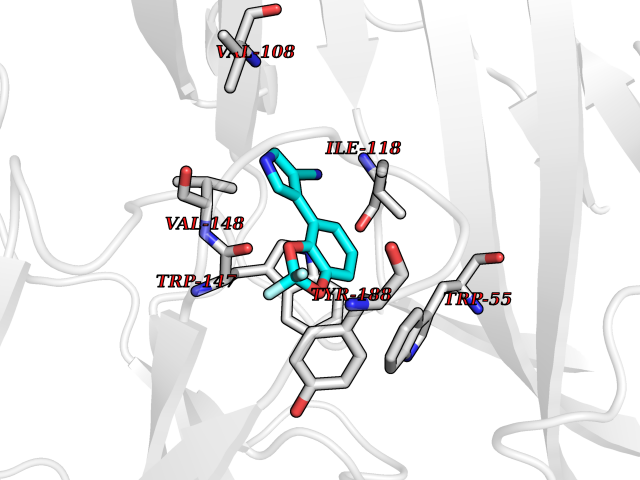 Pymol Map 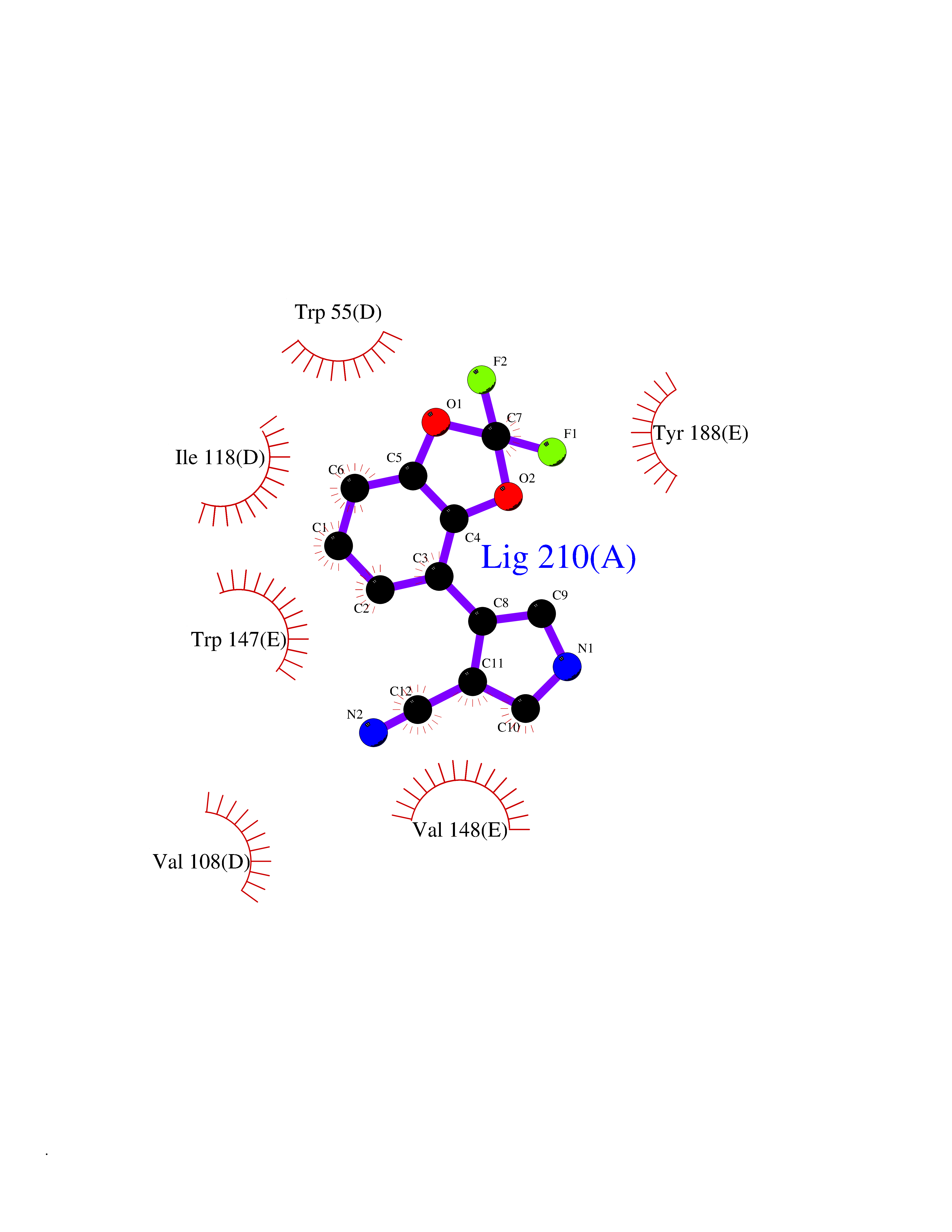 Ligplot Map 3D Binding mode Sequence LHSQANLMRLKSDLFYPGPTKDDPLTVTLGFTLQDIVKADSSTNEVDLVYWEQQRWKLNSLMWDPNEYGNITDFRTSAADIWTPDITAYSSTRPVQVLSPQIAVVTHDGSVMFIPAQRLSFMCDPTGVDSEEGATCAVKFGSWVYSGFEIDLKTDTDQVDLSSYYASSKYEILSATQYKHDIKYNCCEEIYPDVVLVVKFRERRLHSQANLMRLKSDLFNRYPGPTKDDPLTVTLGFTLQDIVKADSSTNEVDLVYWEQQRWKLNSLMWDPNEYGNITDFRTSAADIWTPDITAYSSTRPVQVLSPQIAVVTHDGSVMFIPAQRLSFMCDPTGVDSEEGATCAVKFGSWVYSGFEIDLKTDTDQVDLSSYYASSKYEILSATQYKHDIKYNCCEEIYPDVVLVVKFRE Hydrogen bonds contact Hydrophobic contact | ||||
| 17 | Neuronal acetylcholine receptor alpha-2 (CHRNA2) | 5FJV | 6.10 | |
Target general information Gen name CHRNA2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms CHRNA2 Protein family Ligand-gated ion channel (TC 1.A.9) family, Acetylcholine receptor (TC 1.A.9.1) subfamily, Alpha-2/CHRNA2 sub-subfamily Biochemical class Neurotransmitter receptor Function After binding acetylcholine, the AChR responds by an extensive change in conformation that affects all subunits and leads to opening of an ion-conducting channel across the plasma membrane. Related diseases Epilepsy, nocturnal frontal lobe, 4 (ENFL4) [MIM:610353]: An autosomal dominant focal epilepsy characterized by nocturnal seizures associated with fear sensation, tongue movements, and nocturnal wandering, closely resembling nightmares and sleep walking. {ECO:0000269|PubMed:16826524}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Seizures, benign familial infantile, 6 (BFIS6) [MIM:610353]: A form of benign familial infantile epilepsy, a neurologic disorder characterized by afebrile seizures occurring in clusters during the first year of life, without neurologic sequelae. BFIS6 inheritance is autosomal dominant. {ECO:0000269|PubMed:25847220}. The disease may be caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00732; DB00237; DB00411; DB00565; DB01245; DB00514; DB01135; DB07720; DB00898; DB00472; DB00483; DB08960; DB00657; DB01336; DB00416; DB01226; DB00184; DB01337; DB01338; DB00721; DB00728; DB05740; DB00202; DB01199; DB01339 Interacts with NA EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disease variant; Disulfide bond; Epilepsy; Glycoprotein; Ion channel; Ion transport; Ligand-gated ion channel; Membrane; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Signal; Synapse; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A,B Molecular weight (Da) 48323.4 Length 413 Aromaticity 0.15 Instability index 32 Isoelectric point 5.69 Charge (pH=7) -6.58 2D Binding mode Binding energy (Kcal/mol) -8.32 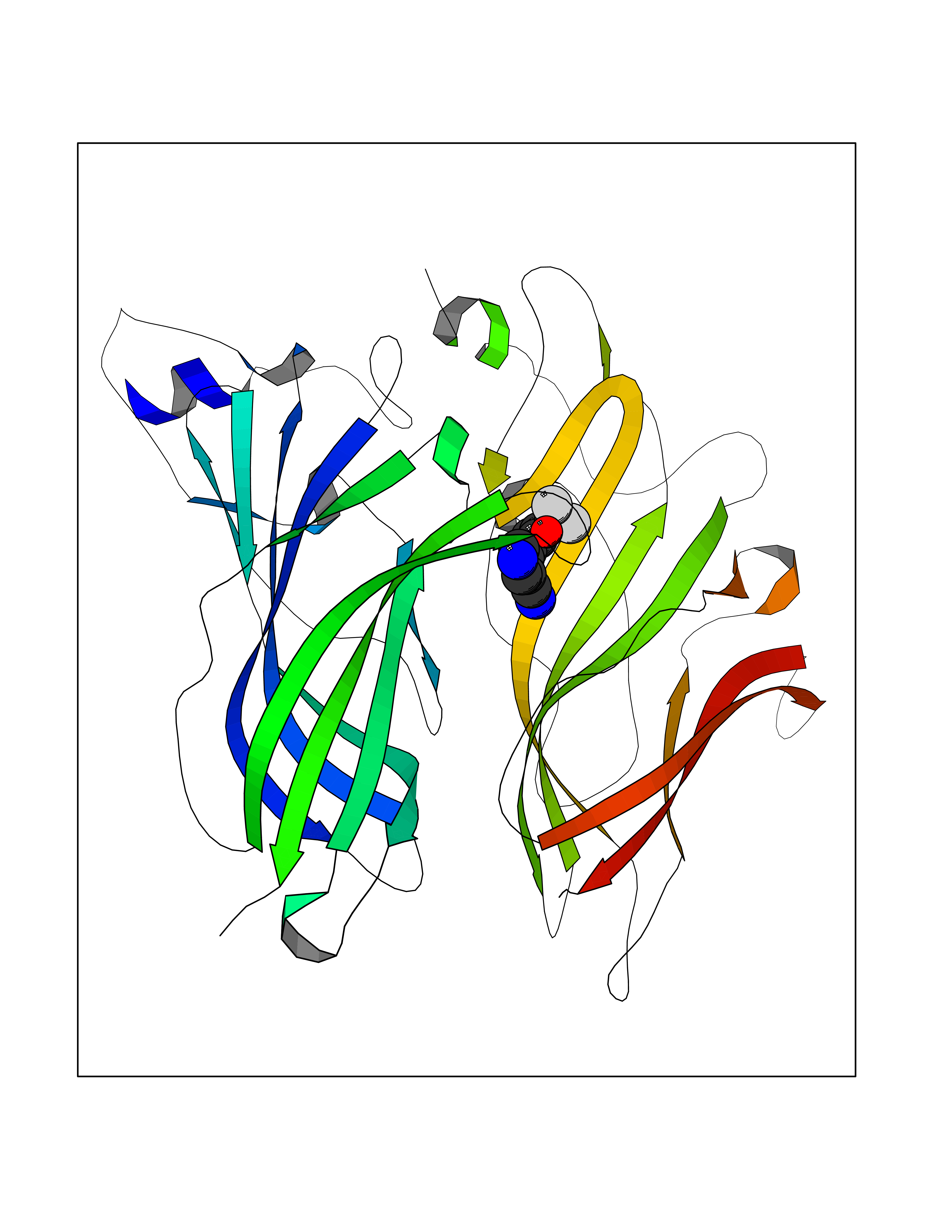 Molscript Map 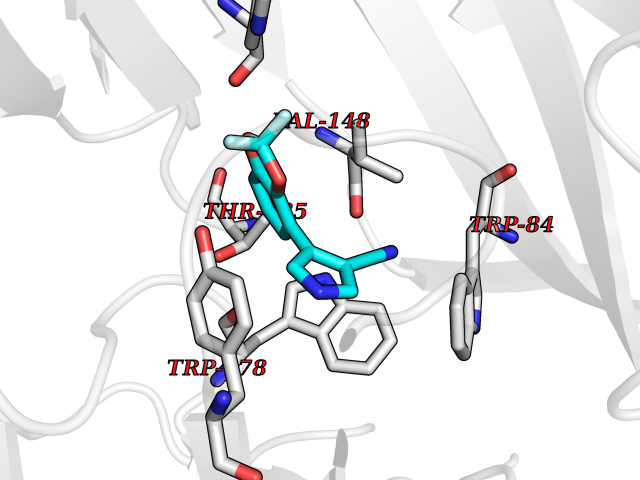 Pymol Map 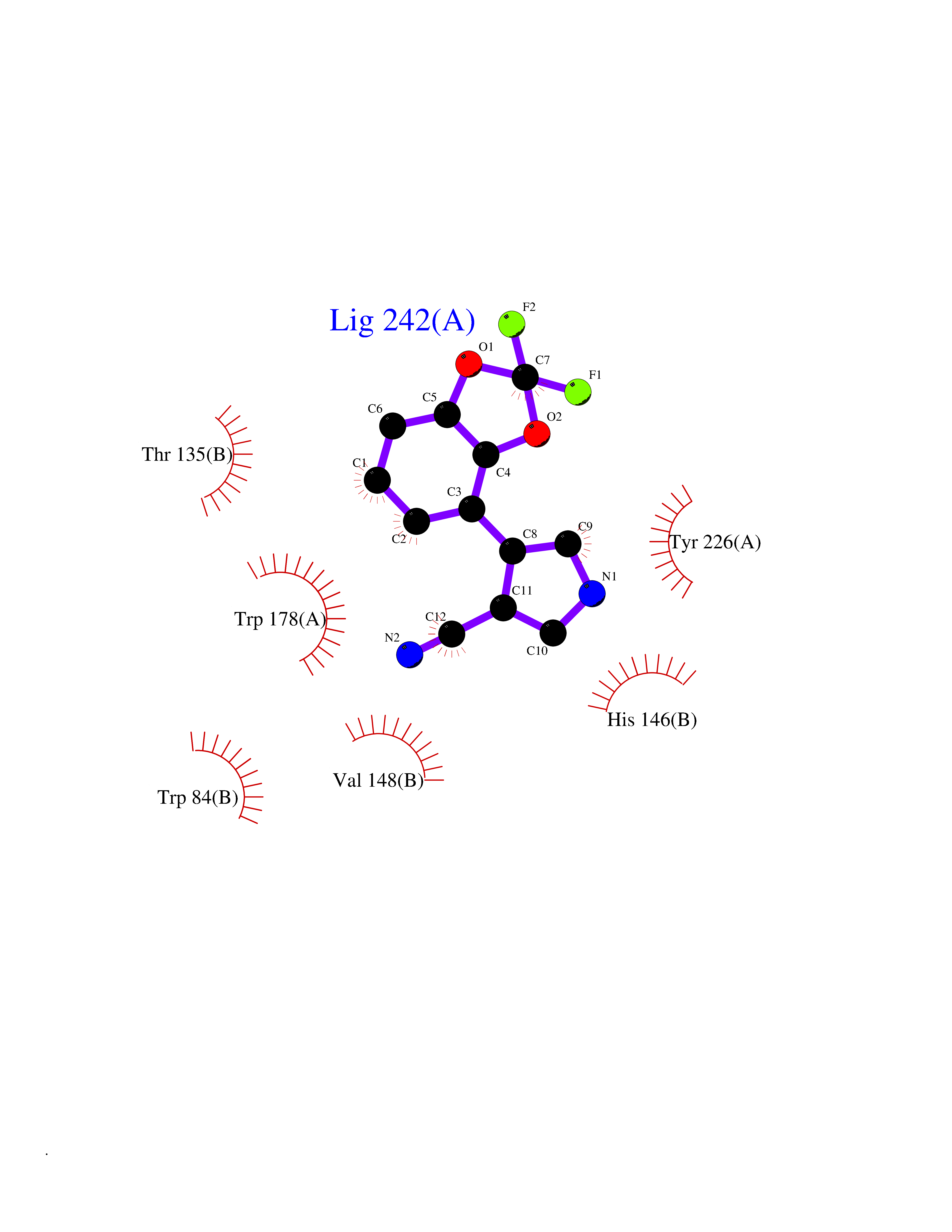 Ligplot Map 3D Binding mode Sequence DRLFKHLFRGYNRWARPVPNTSDVVIVRFGLSIAQLIDVDEKNQMMTTNVWLKQEWSDYKLRWNPTDFGNITSLRVPSEMIWIPDIVLYNNADGEFAVTHMTKAHLFSTGTVHWVPPAIYKSSCSIDVTFFPFDQQNCKMKFGSWTYDKAKIDLEQMEQTVDLKDYWESGEWAIVNATGTYNSKKYDCCAEIYPDVTYAFVIRRLPEDRLFKHLFRGYNRWARPVPNTSDVVIVRFGLSIAQLIDVDEKNQMMTTNVWLKQEWSDYKLRWNPTDFGNITSLRVPSEMIWIPDIVLYNNADGEFAVTHMTKAHLFSTGTVHWVPPAIYKSSCSIDVTFFPFDQQNCKMKFGSWTYDKAKIDLEQMEQTVDLKDYWESGEWAIVNATGTYNSKKYDCCAEIYPDVTYAFVIRRLP Hydrogen bonds contact Hydrophobic contact | ||||
| 18 | IL-1 receptor-associated kinase 1 (IRAK1) | 6BFN | 6.10 | |
Target general information Gen name IRAK1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Interleukin-1 receptor-associated kinase 1; IRAK-1; IRAK Protein family Protein kinase superfamily, TKL Ser/Thr protein kinase family, Pelle subfamily Biochemical class Kinase Function Involved in Toll-like receptor (TLR) and IL-1R signaling pathways. Is rapidly recruited by MYD88 to the receptor-signaling complex upon TLR activation. Association with MYD88 leads to IRAK1 phosphorylation by IRAK4 and subsequent autophosphorylation and kinase activation. Phosphorylates E3 ubiquitin ligases Pellino proteins (PELI1, PELI2 and PELI3) to promote pellino-mediated polyubiquitination of IRAK1. Then, the ubiquitin-binding domain of IKBKG/NEMO binds to polyubiquitinated IRAK1 bringing together the IRAK1-MAP3K7/TAK1-TRAF6 complex and the NEMO-IKKA-IKKB complex. In turn, MAP3K7/TAK1 activates IKKs (CHUK/IKKA and IKBKB/IKKB) leading to NF-kappa-B nuclear translocation and activation. Alternatively, phosphorylates TIRAP to promote its ubiquitination and subsequent degradation. Phosphorylates the interferon regulatory factor 7 (IRF7) to induce its activation and translocation to the nucleus, resulting in transcriptional activation of type I IFN genes, which drive the cell in an antiviral state. When sumoylated, translocates to the nucleus and phosphorylates STAT3. Serine/threonine-protein kinase that plays a critical role in initiating innate immune response against foreign pathogens. Related diseases Anemia, non-spherocytic hemolytic, due to G6PD deficiency (NSHA) [MIM:300908]: A disease characterized by G6PD deficiency, acute hemolytic anemia, fatigue, back pain, and jaundice. In most patients, the disease is triggered by an exogenous agent, such as some drugs, food, or infection. Increased unconjugated bilirubin, lactate dehydrogenase, and reticulocytosis are markers of the disorder. Although G6PD deficiency can be life-threatening, most patients are asymptomatic throughout their life. {ECO:0000269|PubMed:12524354, ECO:0000269|PubMed:1303180, ECO:0000269|PubMed:1303182, ECO:0000269|PubMed:1536798, ECO:0000269|PubMed:1611091, ECO:0000269|PubMed:1889820, ECO:0000269|PubMed:1945893, ECO:0000269|PubMed:20007901, ECO:0000269|PubMed:26479991, ECO:0000269|PubMed:2836867, ECO:0000269|PubMed:2912069, ECO:0000269|PubMed:30988594, ECO:0000269|PubMed:38066190, ECO:0000269|PubMed:7858267, ECO:0000269|PubMed:7959695, ECO:0000269|PubMed:8193373, ECO:0000269|PubMed:8490627, ECO:0000269|PubMed:8533762, ECO:0000269|PubMed:8733135, ECO:0000269|PubMed:9452072}. The disease is caused by variants affecting the gene represented in this entry. Deficiency of G6PD is associated with hemolytic anemia in two different situations. First, in areas in which malaria has been endemic, G6PD-deficiency alleles have reached high frequencies (1% to 50%) and deficient individuals, though essentially asymptomatic in the steady state, have a high risk of acute hemolytic attacks. Secondly, sporadic cases of G6PD deficiency occur at a very low frequencies, and they usually present a more severe phenotype. Several types of NSHA are recognized. Class-I variants are associated with severe NSHA; class-II have an activity <10% of normal; class-III have an activity of 10% to 60% of normal; class-IV have near normal activity. Drugs (DrugBank ID) DB12010 Interacts with Q15306; Q92985; Q99836; Q96FA3; Q9HAT8; Q8N2H9-2; Q13526; Q86WV6; P58753; Q9Y4K3; Q8VCW4; Q5D1E7 EC number EC 2.7.11.1 Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Cytoplasm; Direct protein sequencing; Host-virus interaction; Immunity; Innate immunity; Isopeptide bond; Kinase; Lipid droplet; Magnesium; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Serine/threonine-protein kinase; Transferase; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 33681.4 Length 301 Aromaticity 0.09 Instability index 39.86 Isoelectric point 8.6 Charge (pH=7) 5.09 2D Binding mode Binding energy (Kcal/mol) -8.33 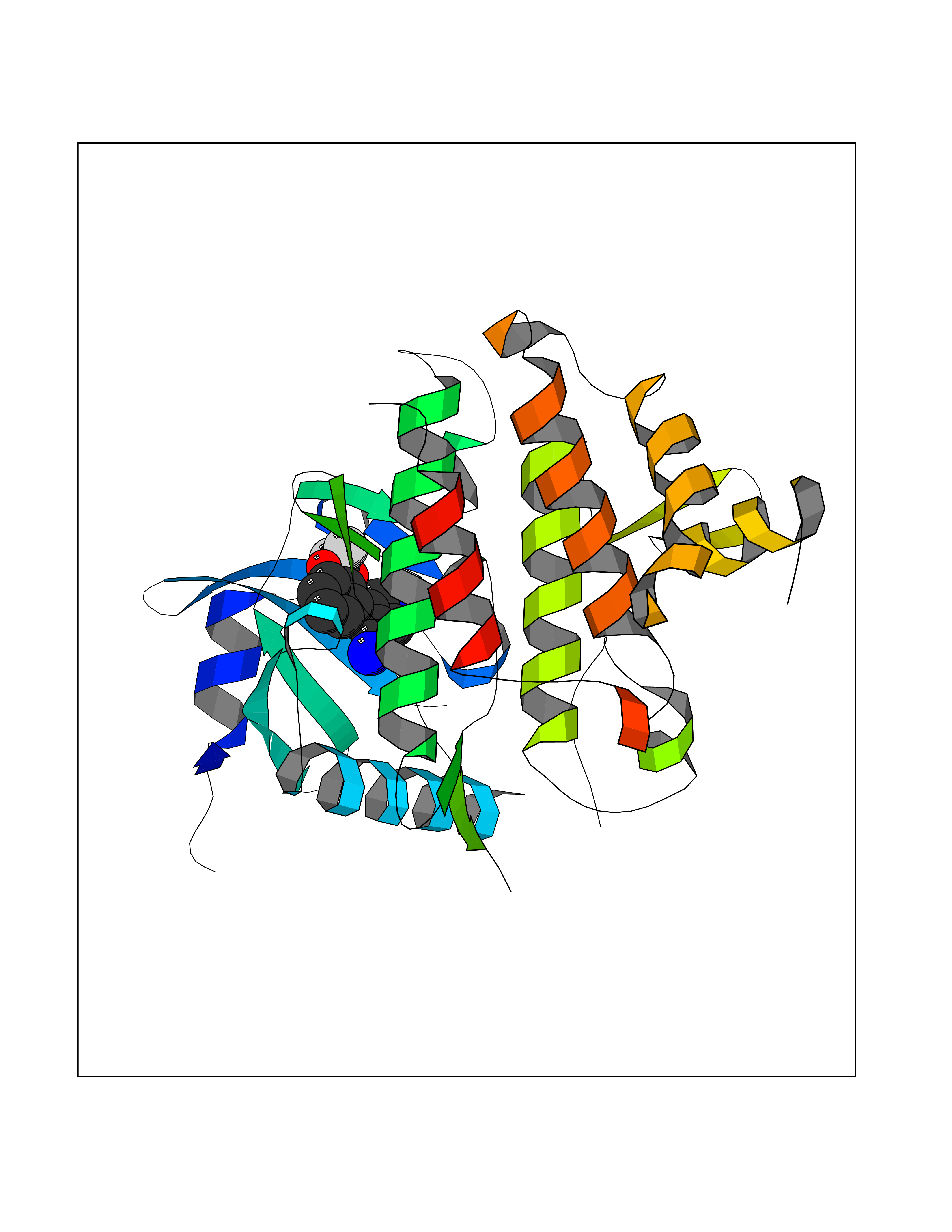 Molscript Map 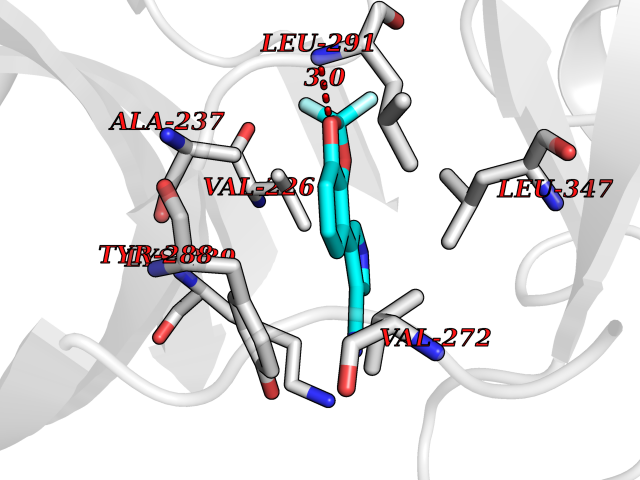 Pymol Map 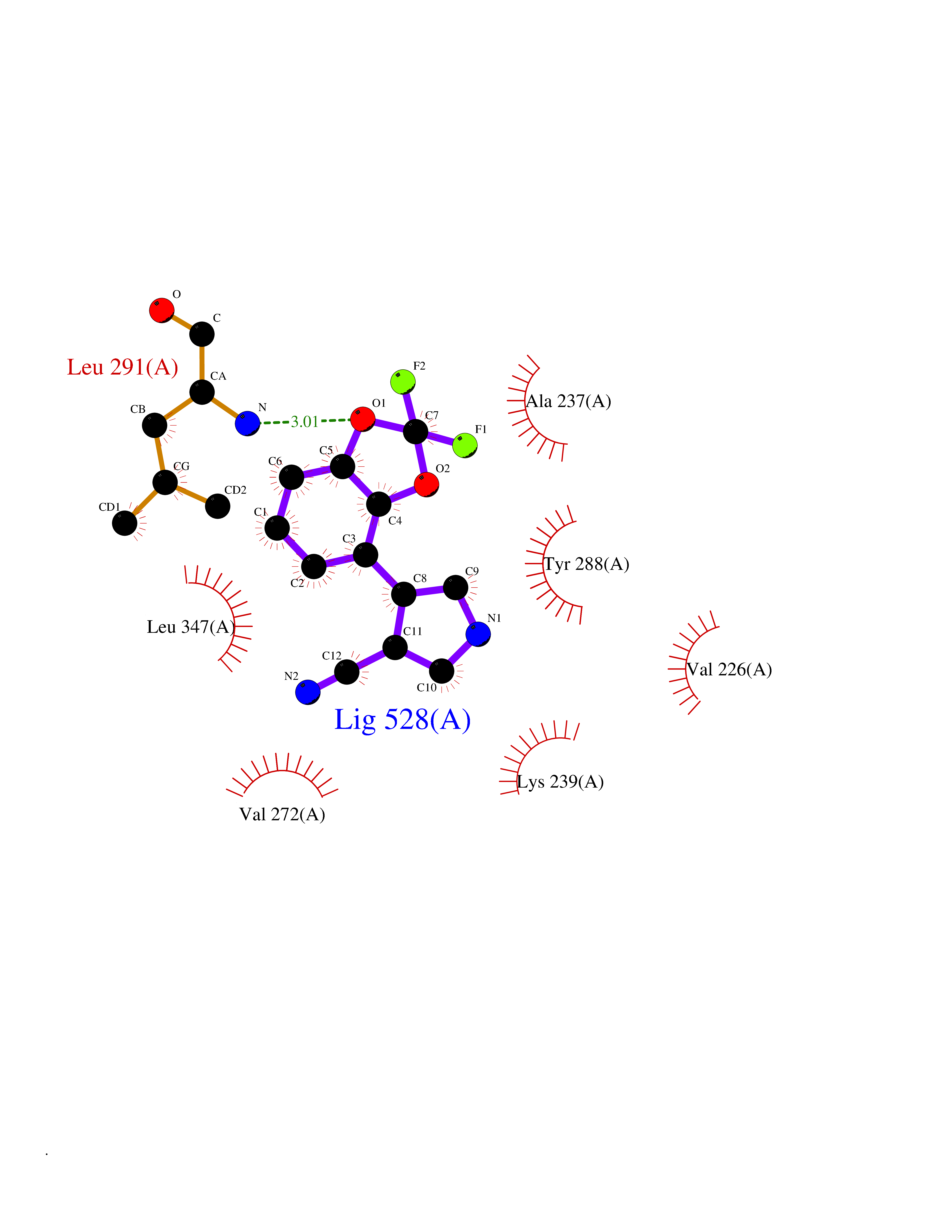 Ligplot Map 3D Binding mode Sequence SRPFPFCWPLCEISRGTHNFSEELKIGEGGFGCVYRAVMRNTVYAVKRLKEWTAVKQSFLTEVEQLSRFRHPNIVDFAGYCAQNGFYCLVYGFLPNGSLEDRLHCQTQACPPLSWPQRLDILLGTARAIQFLHQDSPSLIHGDIKSSNVLLDERLTPKLGDFGLARFSRTVRGTLAYLPEEYIKTGRLAVDTDTFSFGVVVLETLAGQRAVKTHGARTKYLKDLVEEEAEEAGVAAADAWAAPIAMQIYKKHLDPRPGPCPPELGLGLGQLACCCLHRRAKRRPPMTQVYERLEKLQAVVA Hydrogen bonds contact Hydrophobic contact | ||||
| 19 | Schistosoma Purine nucleoside phosphorylase (sch PNP) | 3F8W | 6.09 | |
Target general information Gen name sch PNP Organism Schistosoma mansoni (Blood fluke) Uniprot ID TTD ID Synonyms sch Purine nucleoside phosphorylase; sch Inosine-guanosine phosphorylase Protein family PNP/MTAP phosphorylase family Biochemical class NA Function The purine nucleoside phosphorylases catalyze the phosphorolytic breakdown of the N-glycosidic bond in the beta-(deoxy)ribonucleoside molecules, with the formation of the corresponding free purine bases and pentose-1-phosphate. Related diseases Hemolytic anemia, non-spherocytic, due to glucose phosphate isomerase deficiency (HA-GPID) [MIM:613470]: A form of anemia in which there is no abnormal hemoglobin or spherocytosis. It is caused by glucose phosphate isomerase deficiency. {ECO:0000269|PubMed:28803808, ECO:0000269|PubMed:7989588, ECO:0000269|PubMed:8499925, ECO:0000269|PubMed:8822952, ECO:0000269|PubMed:8822954, ECO:0000269|PubMed:9446754, ECO:0000269|PubMed:9856489}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number EC 2.4.2.1 Uniprot keywords 3D-structure; Glycosyltransferase; Transferase Protein physicochemical properties Chain ID A,B Molecular weight (Da) 61140.6 Length 564 Aromaticity 0.06 Instability index 29.8 Isoelectric point 7.6 Charge (pH=7) 1.82 2D Binding mode Binding energy (Kcal/mol) -8.3  Molscript Map 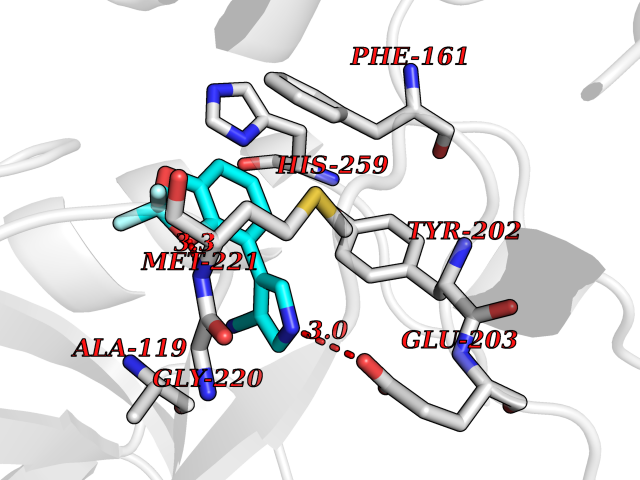 Pymol Map 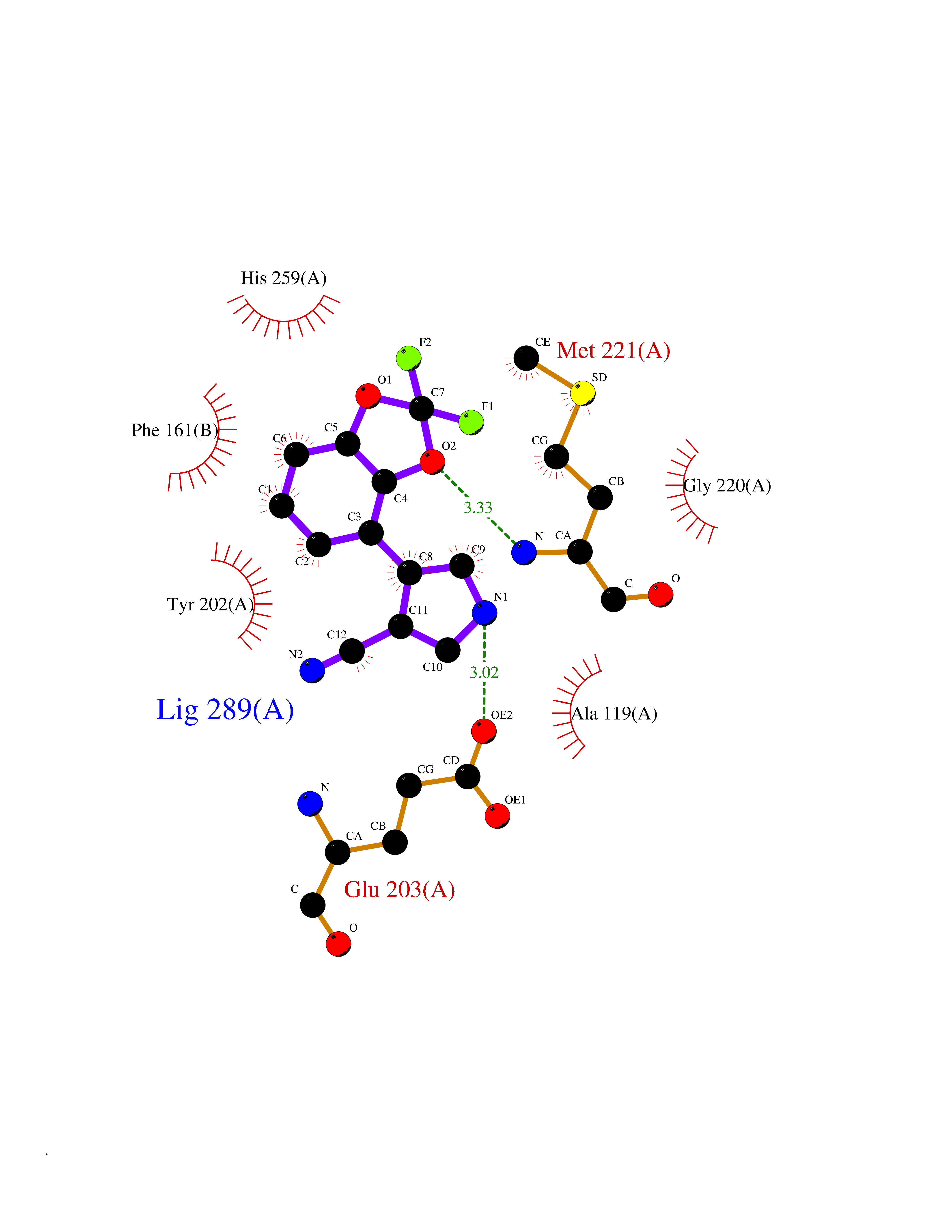 Ligplot Map 3D Binding mode Sequence SVTANIENVKKVAHHIQKLTSIVPEIGIICGSGLGKLADGVKDKITIPYTKIPNFPQTSHSGNLIFGTLSGRKVVVMQGRFHMYEGYSNDTVALPIRVMKLLGVKILMVSNAAGGLNRSLKLGDFVILKDHIYLPGLGLNNILVGPNQEAFGTRFPALSNAYDRDLRKLAVQVAEENGFGNLVHQGVYVMNGGPCYETPAECTMLLNMGCDVVGMSTIPEVVIARHCGIQVFAVSLVTNISVLDVESDLKPNHEEVLATGAQRAELMQSWFEKIIEKLPKSVTANIENVKKVAHHIQKLTSIVPEIGIICGSGLGKLADGVKDKITIPYTKIPNFPQTSVVGHSGNLIFGTLSGRKVVVMQGRFHMYEGYSNDTVALPIRVMKLLGVKILMVSNAAGGLNRSLKLGDFVILKDHIYLPGLGLNNILVGPNQEAFGTRFPALSNAYDRDLRKLAVQVAEENGFGNLVHQGVYVMNGGPCYETPAECTMLLNMGCDVVGMSTIPEVVIARHCGIQVFAVSLVTNISVLDVESDLKPNHEEVLATGAQRAELMQSWFEKIIEKLPKD Hydrogen bonds contact Hydrophobic contact | ||||
| 20 | Histone-lysine N-methyltransferase KMT5C (KMT5C) | 3RQ4 | 6.08 | |
Target general information Gen name KMT5C Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Lysine N-methyltransferase 5C; Lysine-specific methyltransferase 5C; Suppressor of variegation 4-20 homolog 2; Su(var)4-20 homolog 2; Suv4-20h2; [histone H4]-N-methyl-L-lysine20 N-methyltransferase KM Protein family Class V-like SAM-binding methyltransferase superfamily, Histone-lysine methyltransferase family, Suvar4-20 subfamily Biochemical class NA Function Histone methyltransferase that specifically methylates monomethylated 'Lys-20' (H4K20me1) and dimethylated 'Lys-20' (H4K20me2) of histone H4 to produce respectively dimethylated 'Lys-20' (H4K20me2) and trimethylated 'Lys-20' (H4K20me3) and thus regulates transcription and maintenance of genome integrity. In vitro also methylates unmodified 'Lys-20' (H4K20me0) of histone H4 and nucleosomes. H4 'Lys-20' trimethylation represents a specific tag for epigenetic transcriptional repression. Mainly functions in pericentric heterochromatin regions, thereby playing a central role in the establishment of constitutive heterochromatin in these regions. KMT5C is targeted to histone H3 via its interaction with RB1 family proteins (RB1, RBL1 and RBL2) (By similarity). Facilitates TP53BP1 foci formation upon DNA damage and proficient non-homologous end-joining (NHEJ)-directed DNA repair by catalyzing the di- and trimethylation of 'Lys-20' of histone H4. May play a role in class switch reconbination by catalyzing the di- and trimethylation of 'Lys-20' of histone H4 (By similarity). Related diseases Brachydactyly A2 (BDA2) [MIM:112600]: A form of brachydactyly. Brachydactyly defines a group of inherited malformations characterized by shortening of the digits due to abnormal development of the phalanges and/or the metacarpals. In brachydactyly type A2 shortening of the middle phalanges is confined to the index finger and the second toe, all other digits being more or less normal. Because of a rhomboid or triangular shape of the affected middle phalanx, the end of the second finger usually deviates radially. {ECO:0000269|PubMed:19327734, ECO:0000269|PubMed:21357617}. The gene represented in this entry is involved in disease pathogenesis. Duplications of a cis-regulatory element located approximately 110 kb downstream of BMP2 have been found in BDA2 families. They likely cause altered BMP2 expression with pathological consequences. {ECO:0000269|PubMed:19327734, ECO:0000269|PubMed:21357617}.; DISEASE: Short stature, facial dysmorphism, and skeletal anomalies with or without cardiac anomalies 1 (SSFSC1) [MIM:617877]: An autosomal dominant disorder characterized by short stature, facial dysmorphism, skeletal anomalies, and variable cardiac defects. Distinctive facial features include midface retrusion, short upturned nose, long philtrum, high-arched or cleft palate, and variable degrees of micrognathia and dental crowding. Skeletal anomalies include patterning defects of the axial skeleton, characterized by 11 pairs of ribs and brachydactyly of the fifth ray. Congenital heart defects are variably observed and appear to involve primarily the cardiac outflow tract. {ECO:0000269|PubMed:29198724}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q13185 EC number EC 2.1.1.361 Uniprot keywords 3D-structure; Alternative splicing; Chromatin regulator; Chromosome; Metal-binding; Methyltransferase; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Repressor; S-adenosyl-L-methionine; Transcription; Transcription regulation; Transferase; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 27285.8 Length 240 Aromaticity 0.1 Instability index 42.74 Isoelectric point 8.32 Charge (pH=7) 3.24 2D Binding mode Binding energy (Kcal/mol) -8.29  Molscript Map  Pymol Map 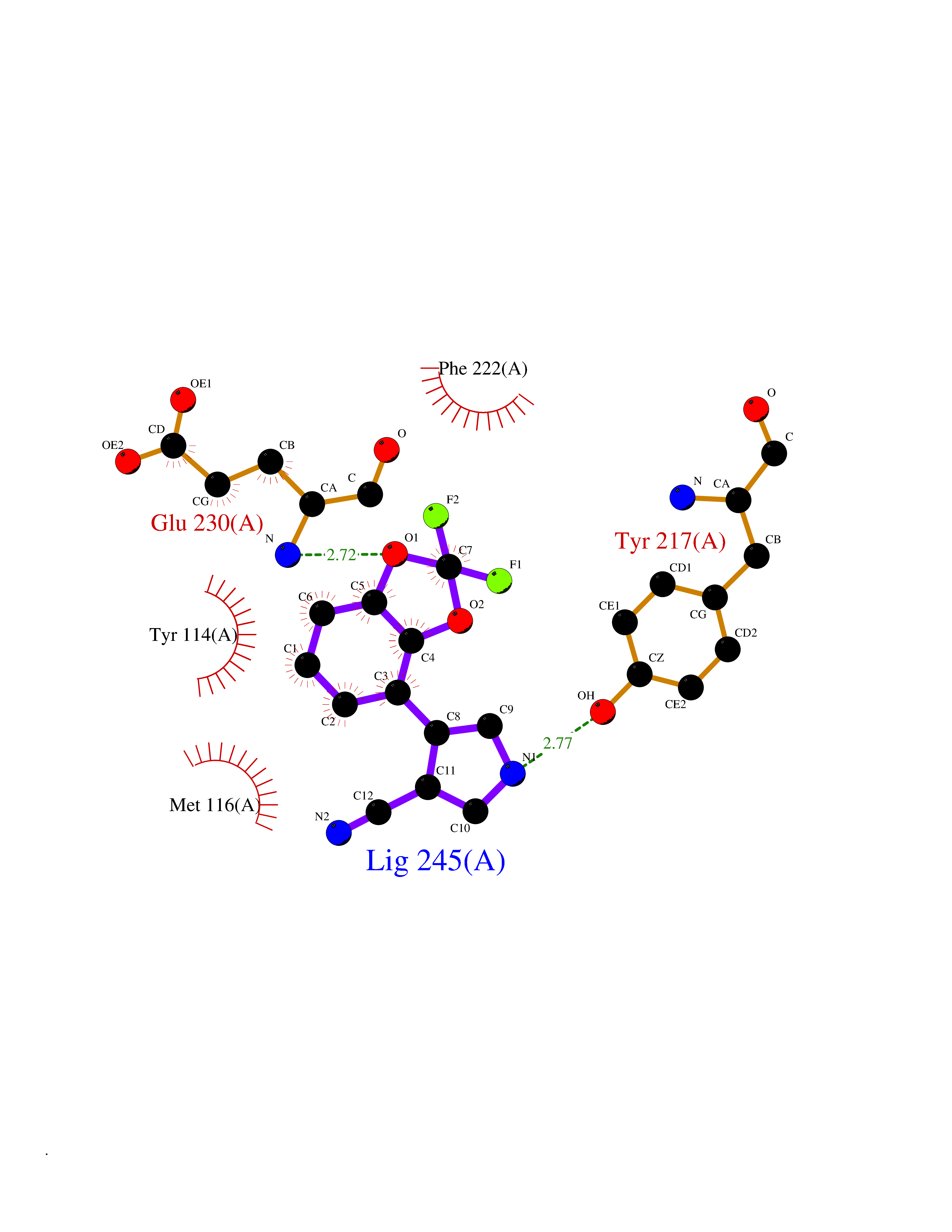 Ligplot Map 3D Binding mode Sequence DRVTARELCENDDLATSLVLDPYLGFRTHKMNVSPVPPLRRQQHLRSALETFLRQRDLEAAYRALTLGGWTARYFQSRGPRQEAALKTHVYRYLRAFLPESGFTILPCTRYSMETNGAKIVSTRAWKKNEKLELLVGCIAELREADEGLLRAGENDFSIMYSTRKRSAQLWLGPAAFINHDCKPNCKFVPADGNAACVKVLRDIEPGDEVTCFYGEGFFGEKNEHCECHTCERKGEGAFR Hydrogen bonds contact Hydrophobic contact | ||||