Job Results:
Ligand
Structure
Job ID
8da1e79bf3c5af0a1c99e2e2a8101bb5
Job name
NA
Time
2025-02-13 15:08:06
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 1 | Ecto-5'-nucleotidase (CD73) | 4H2G | 4.07 | |
Target general information Gen name NT5E Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms NT5; CD73 antigen; 5'-nucleotidase; 5'-NT Protein family 5'-nucleotidase family Biochemical class Phosphoric monoester hydrolase Function Exhibits AMP-, NAD-, and NMN-nucleosidase activities. Hydrolyzes extracellular nucleotides into membrane permeable nucleosides. Related diseases Calcification of joints and arteries (CALJA) [MIM:211800]: A condition characterized by adult-onset calcification of the lower extremity arteries, including the iliac, femoral and tibial arteries, and hand and foot capsule joints. Age of onset has been reported as early as the second decade of life, usually involving intense joint pain or calcification in the hands. {ECO:0000269|PubMed:21288095, ECO:0000269|PubMed:24887587}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00987; DB00806 Interacts with Q9Y225-2; Q8WWF5 EC number EC 3.1.3.5 Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; GPI-anchor; Hydrolase; Lipoprotein; Membrane; Metal-binding; Nucleotide-binding; Proteomics identification; Reference proteome; Signal; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 24417.6 Length 219 Aromaticity 0.09 Instability index 40.43 Isoelectric point 5.49 Charge (pH=7) -5.75 2D Binding mode Binding energy (Kcal/mol) -5.55  Molscript Map 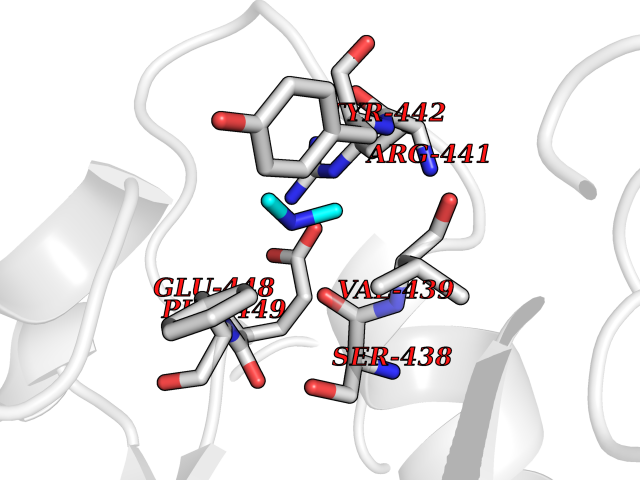 Pymol Map 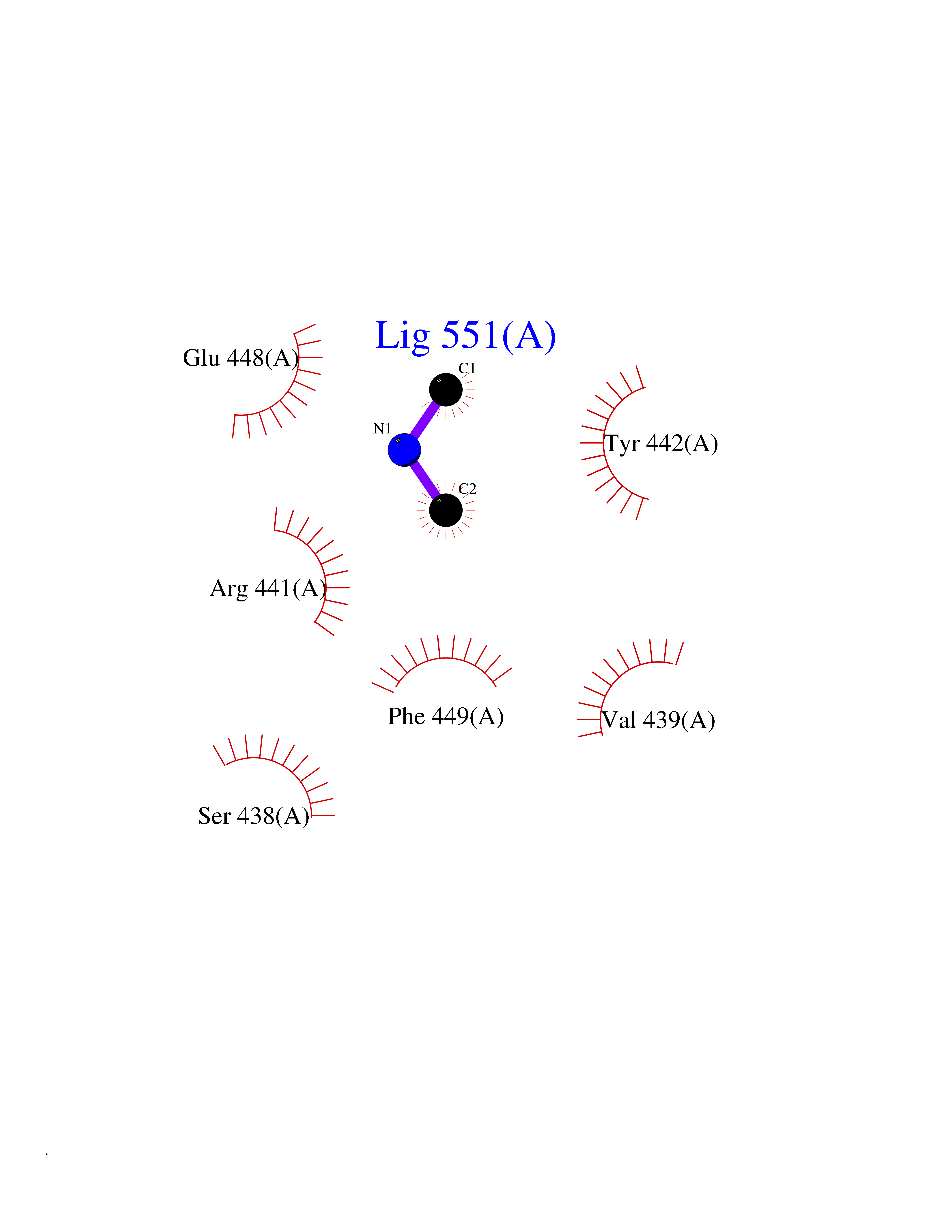 Ligplot Map 3D Binding mode Sequence LDDYSTQELGKTIVYLDGSSQSCRFRECNMGNLICDAMINNNLRHADEMFWNHVSMCILNGGGIRSPIDERNDGTITWENLAAVLPFGGTFDLVQLKGSTLKKAFEHSVHRYGQSTGEFLQVGGIHVVYDLSRKPGDRVVKLDVLCTACAVPSYDPLKMDEVYKVILPNFLANGGDGFQMIKDELLRHDSGDQDINVVSTYISKMKVIYPAVEGRIKFS Hydrogen bonds contact Hydrophobic contact | ||||
| 2 | Lysine-specific demethylase 2A (KDM2A) | 2YU1 | 4.07 | |
Target general information Gen name KDM2A Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms FBXL11; FBL7; F-box/LRR-repeat protein 11; F-box protein Lilina; F-box protein FBL7; F-box and leucine-rich repeat protein 11; CXXC8; CXXC-type zinc finger protein 8; [Histone-H3]-lysine-36 demethylas Protein family JHDM1 histone demethylase family Biochemical class NA Function Histone demethylase that specifically demethylates 'Lys-36' of histone H3, thereby playing a central role in histone code. Preferentially demethylates dimethylated H3 'Lys-36' residue while it has weak or no activity for mono- and tri-methylated H3 'Lys-36'. May also recognize and bind to some phosphorylated proteins and promote their ubiquitination and degradation. Required to maintain the heterochromatic state. Associates with centromeres and represses transcription of small non-coding RNAs that are encoded by the clusters of satellite repeats at the centromere. Required to sustain centromeric integrity and genomic stability, particularly during mitosis. Regulates circadian gene expression by repressing the transcriptional activator activity of CLOCK-ARNTL/BMAL1 heterodimer and RORA in a catalytically-independent manner. Related diseases Pseudovaginal perineoscrotal hypospadias (PPSH) [MIM:264600]: A form of male pseudohermaphroditism in which 46,XY males show ambiguous genitalia at birth, including perineal hypospadias and a blind perineal pouch, and develop masculinization at puberty. The name of the disorder stems from the finding of a blind-ending perineal opening resembling a vagina and a severely hypospadiac penis with the urethra opening onto the perineum. {ECO:0000269|PubMed:10718838, ECO:0000269|PubMed:10898110, ECO:0000269|PubMed:10999800, ECO:0000269|PubMed:12843198, ECO:0000269|PubMed:15064320, ECO:0000269|PubMed:1522235, ECO:0000269|PubMed:15528927, ECO:0000269|PubMed:15770495, ECO:0000269|PubMed:16098368, ECO:0000269|PubMed:16181229, ECO:0000269|PubMed:7554313, ECO:0000269|PubMed:8626825, ECO:0000269|PubMed:8768837, ECO:0000269|PubMed:9208814, ECO:0000269|PubMed:9745434, ECO:0000269|PubMed:9843052}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q04206; P63208 EC number EC 1.14.11.27 Uniprot keywords 3D-structure; ADP-ribosylation; Alternative splicing; Biological rhythms; Chromatin regulator; Chromosome; Dioxygenase; DNA-binding; Iron; Isopeptide bond; Leucine-rich repeat; Metal-binding; Nucleus; Oxidoreductase; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Repressor; Transcription; Transcription regulation; Ubl conjugation; Ubl conjugation pathway; Zinc; Zinc-finger Protein physicochemical properties Chain ID A Molecular weight (Da) 45337.4 Length 386 Aromaticity 0.12 Instability index 31.19 Isoelectric point 5.5 Charge (pH=7) -12.12 2D Binding mode Binding energy (Kcal/mol) -5.55 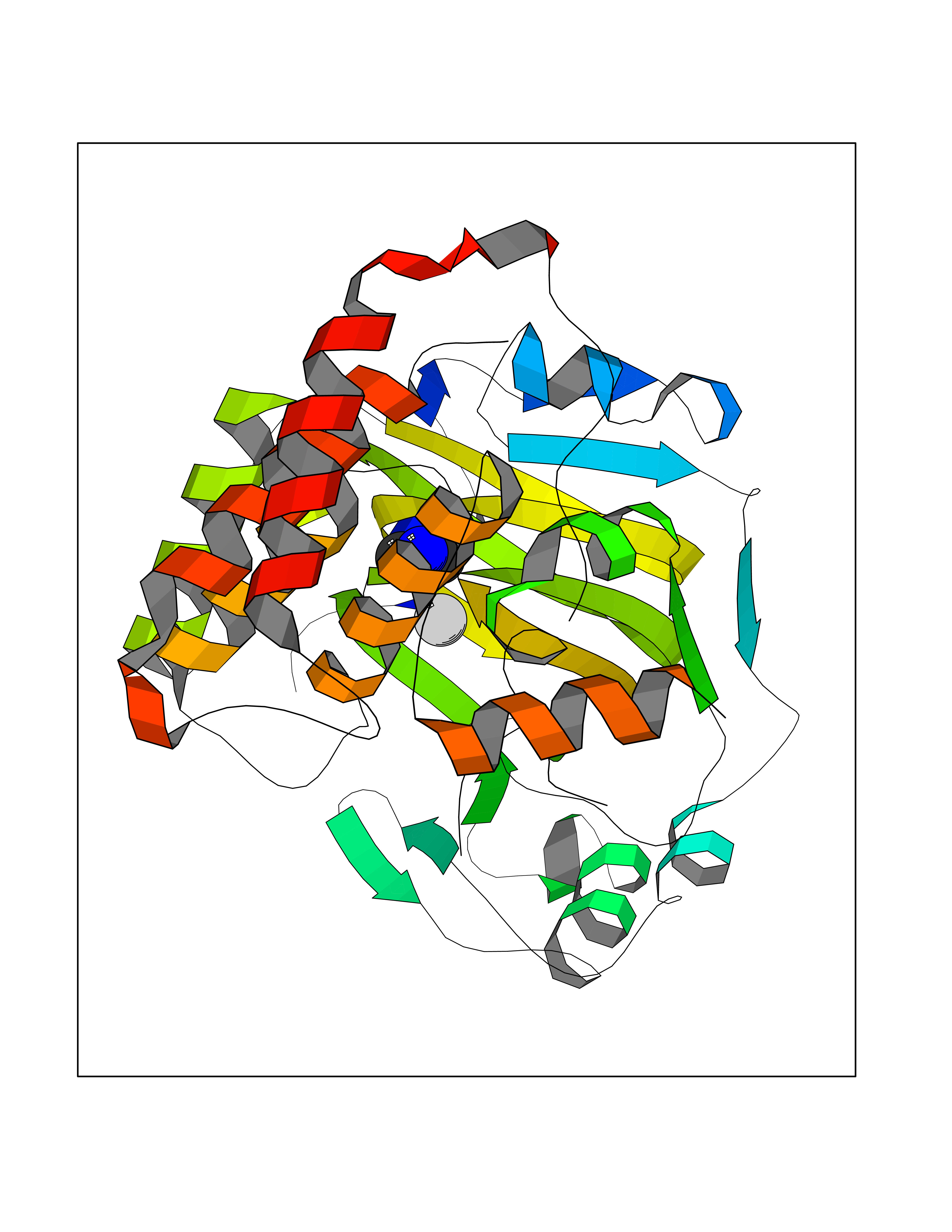 Molscript Map 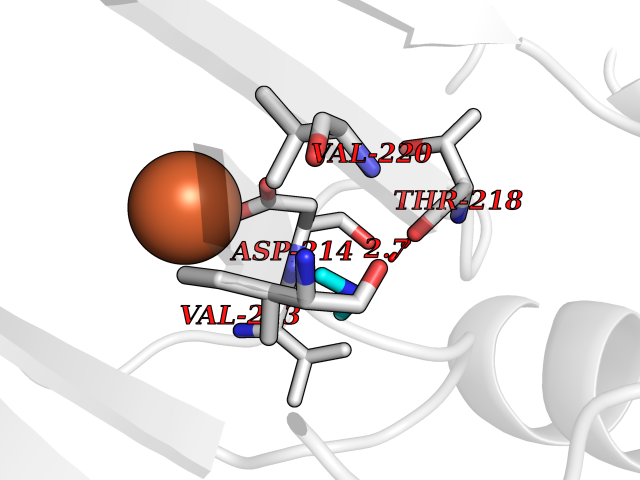 Pymol Map 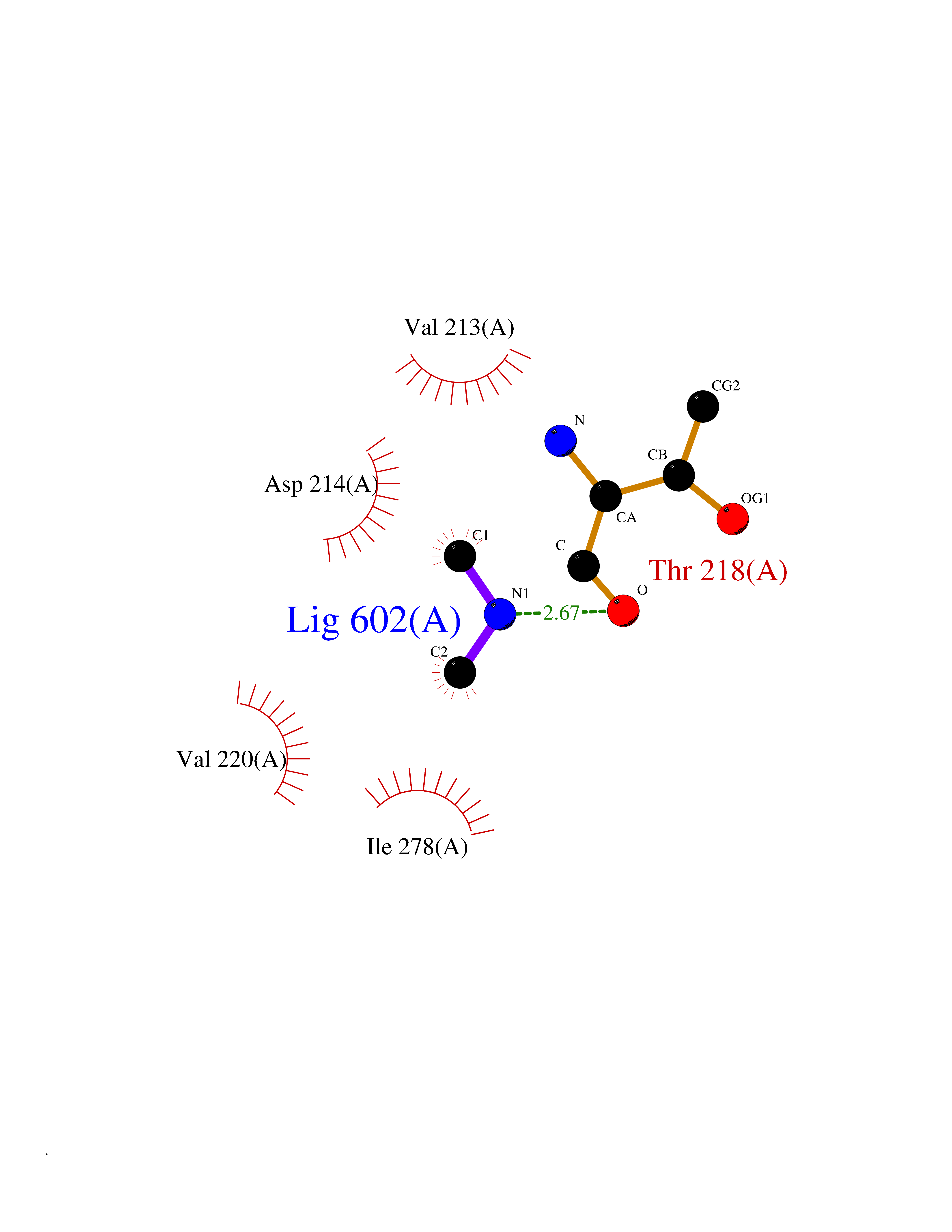 Ligplot Map 3D Binding mode Sequence RTFDLEEKLHTNKYNANFVTFMEGKDFNVEYIQRGGLRDPLIFKNSDGLGIKMPDPDFTVNDVKMCVGSRRMVDVMDVNTQKGIEMTMAQWTRYYETPEEEREKLYNVISLEFSHTRLENMVQRPSTVDFIDWVDNMWPRHLKEMQYPKVQKYCLMSVRGCYTDFHVDFGGTSVWYHIHQGGKVFWLIPPTAHNLELYENWLLSGSQGDIFLGDRVSDCQRIELKQGYTFVIPSGWIHAVYTPTDTLVFGGNFLHSFNIPMQLKIYNIEDRTRVPNKFRYPFYYEMCWYVLERYVYCITNRSHLTKEFQKESLSMDLEQVHLTHFELEGLRCLVDKLESLPLHKKCVPTGIEDEDALIADVKILLEELANSDPKLALTGVPIVQWP Hydrogen bonds contact Hydrophobic contact | ||||
| 3 | Ubiquitin thioesterase L3 (UCHL3) | 1XD3 | 4.07 | |
Target general information Gen name UCHL3 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Ubiquitin carboxyl-terminal hydrolase isozyme L3; UCH-L3 Protein family Peptidase C12 family Biochemical class Peptidase Function Thiol protease that recognizes and hydrolyzes a peptide bond at the C-terminal glycine of either ubiquitin or NEDD8. Has a 10-fold preference for Arg and Lys at position P3", and exhibits a preference towards 'Lys-48'-linked ubiquitin chains. Deubiquitinates ENAC in apical compartments, thereby regulating apical membrane recycling. Indirectly increases the phosphorylation of IGFIR, AKT and FOXO1 and promotes insulin-signaling and insulin-induced adipogenesis. Required for stress-response retinal, skeletal muscle and germ cell maintenance. May be involved in working memory. Can hydrolyze UBB(+1), a mutated form of ubiquitin which is not effectively degraded by the proteasome and is associated with neurogenerative disorders. Deubiquitinating enzyme (DUB) that controls levels of cellular ubiquitin through processing of ubiquitin precursors and ubiquitinated proteins. Related diseases Epilepsy, familial focal, with variable foci 4 (FFEVF4) [MIM:617935]: An autosomal dominant form of epilepsy characterized by focal seizures arising from different cortical regions, including the temporal, frontal, parietal, and occipital lobes. Seizure types commonly include temporal lobe epilepsy, frontal lobe epilepsy, and nocturnal frontal lobe epilepsy. Some patients may have intellectual disability or autism spectrum disorders. Seizure onset usually occurs in the first or second decades, although later onset has been reported, and there is phenotypic variability within families. A subset of patients have structural brain abnormalities. Penetrance of the disorder is incomplete. FFEVF4 is characterized by onset of focal seizures in the first years of life. {ECO:0000269|PubMed:24157691, ECO:0000269|PubMed:28235671}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Developmental and epileptic encephalopathy 62 (DEE62) [MIM:617938]: A form of epileptic encephalopathy, a heterogeneous group of severe early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE62 is characterized by onset of seizures in the first year of life. {ECO:0000269|PubMed:29466837}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q9H078; G5E9A7; Q15797; Q7Z699 EC number EC 3.4.19.12 Uniprot keywords 3D-structure; Cytoplasm; Hydrolase; Phosphoprotein; Protease; Proteomics identification; Reference proteome; Thiol protease; Ubl conjugation pathway Protein physicochemical properties Chain ID A,B Molecular weight (Da) 34540.8 Length 304 Aromaticity 0.07 Instability index 39.91 Isoelectric point 5.01 Charge (pH=7) -17.24 2D Binding mode Binding energy (Kcal/mol) -5.55 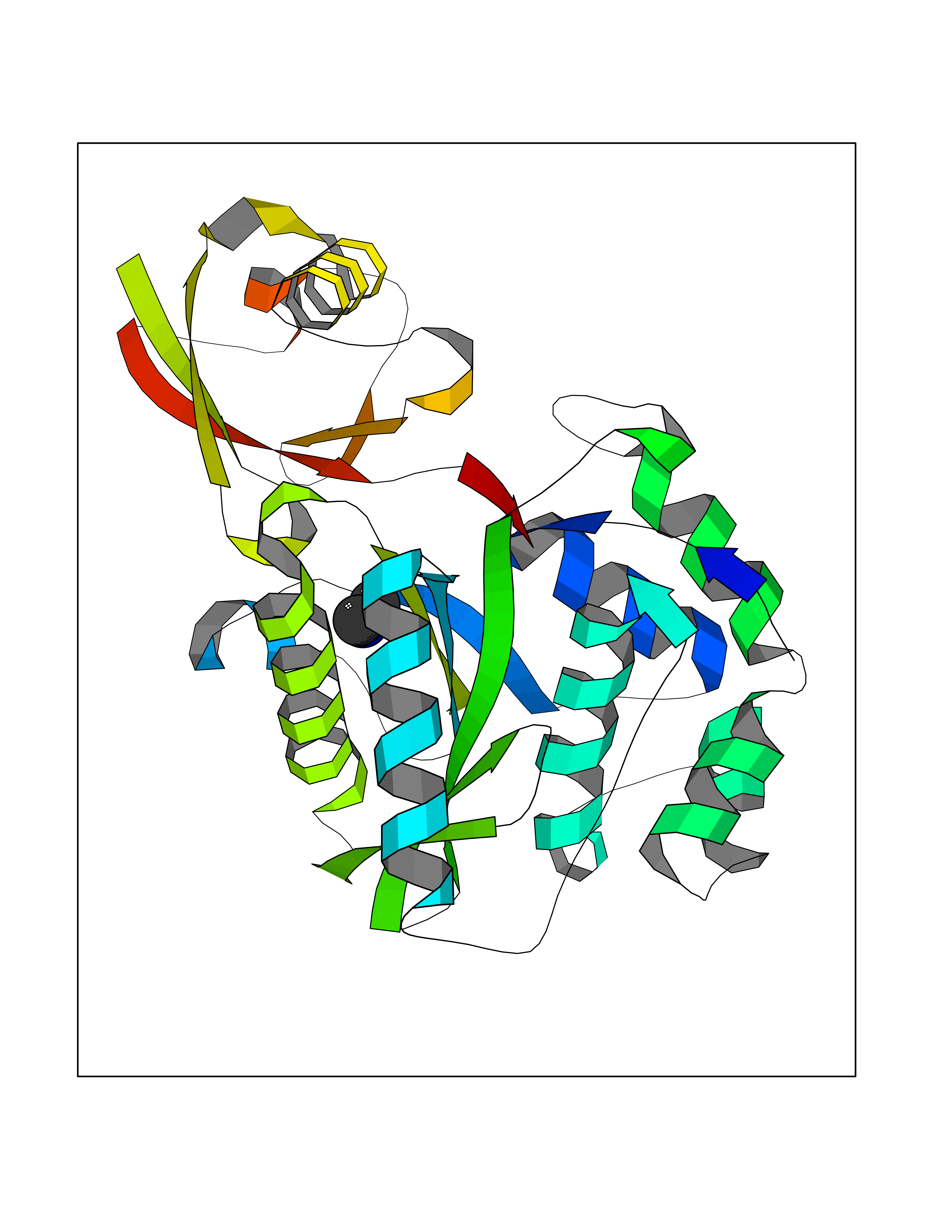 Molscript Map 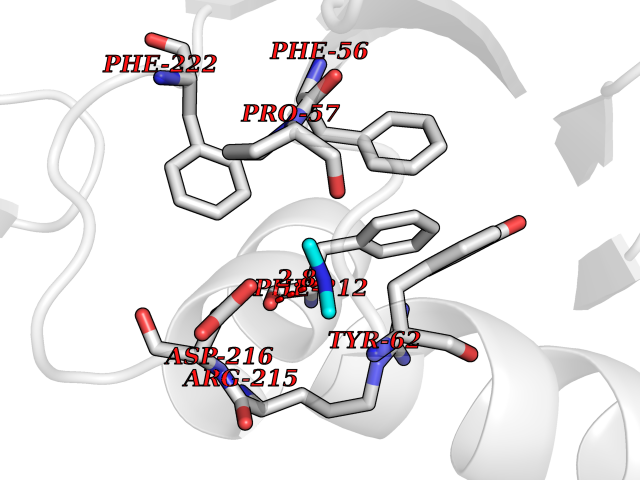 Pymol Map 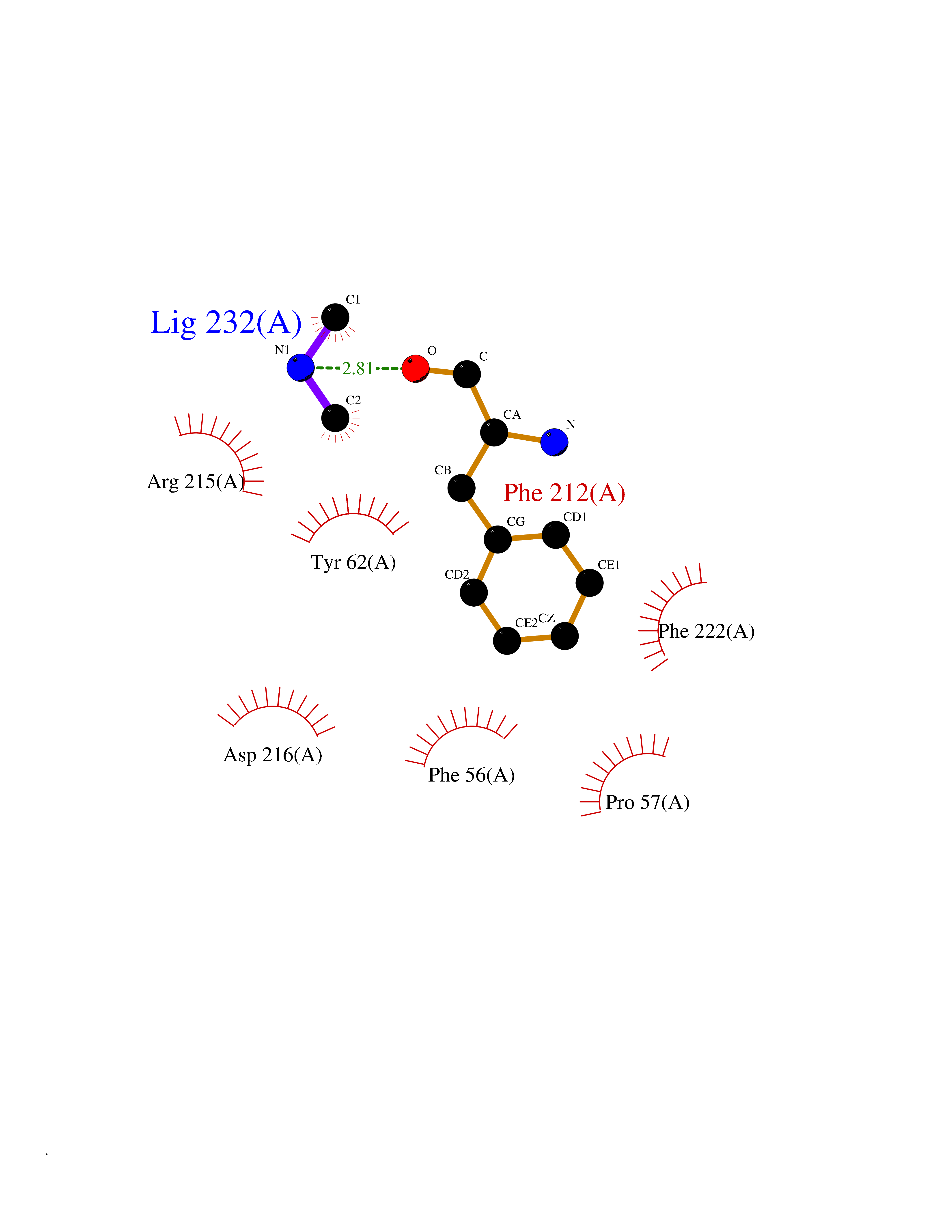 Ligplot Map 3D Binding mode Sequence EGQRWLPLEANPEVTNQFLKQLGLHPNWQFVDVYGMDPELLSMVPRPVCAVLLLFPITEKYEVFRTEEEEKIKSQGQDVTSSVYFMKQTISNACGTIGLIHAIANNKDKMHFESGSTLKKFLEESVSMSPEERARYLENYDAIRVTHETSAHEGQTEAPSIDEKVDLHFIALVHVDGHLYELDGRKPFPINHGETSDETLLEDAIEVCKKFMERDPDELRFNAIALSAAMQIFVKTLTGKTITLEVEPSDTIENVKAKIQDKEGIPPDQQRLIFAGKQLEDGRTLSDYNIQKESTLHLVLRLRG Hydrogen bonds contact Hydrophobic contact | ||||
| 4 | Serum paraoxonase/arylesterase 1 | 1V04 | 4.06 | |
Target general information Gen name PON1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms PON Protein family Paraoxonase family Biochemical class Hydrolase Function Acyl-L-homoserine-lactone lactonohydrolase activity.Aryldialkylphosphatase activity.Arylesterase activity.Calcium ion binding.Phospholipid binding.Protein homodimerization activity. Related diseases Microvascular complications of diabetes 5 (MVCD5) [MIM:612633]: Pathological conditions that develop in numerous tissues and organs as a consequence of diabetes mellitus. They include diabetic retinopathy, diabetic nephropathy leading to end-stage renal disease, and diabetic neuropathy. Diabetic retinopathy remains the major cause of new-onset blindness among diabetic adults. It is characterized by vascular permeability and increased tissue ischemia and angiogenesis. Disease susceptibility is associated with variants affecting the gene represented in this entry. Homozygosity for the Leu-55 allele is strongly associated with the development of retinal disease in diabetic patients. Drugs (DrugBank ID) DB01327; DB09130; DB01395; DB14598; DB14600; DB14596; DB00218; DB01085; DB01593; DB14487; DB14533; DB14548 Interacts with NA EC number 3.1.1.2; 3.1.1.81; 3.1.8.1 Uniprot keywords 3D-structure; Calcium; Direct protein sequencing; Disulfide bond; Glycoprotein; HDL; Hydrolase; Metal-binding; Proteomics identification; Reference proteome; Secreted; Signal Protein physicochemical properties Chain ID A Molecular weight (Da) 37232.8 Length 332 Aromaticity 0.11 Instability index 35.09 Isoelectric point 5.06 Charge (pH=7) -17.08 2D Binding mode Binding energy (Kcal/mol) -5.53  Molscript Map 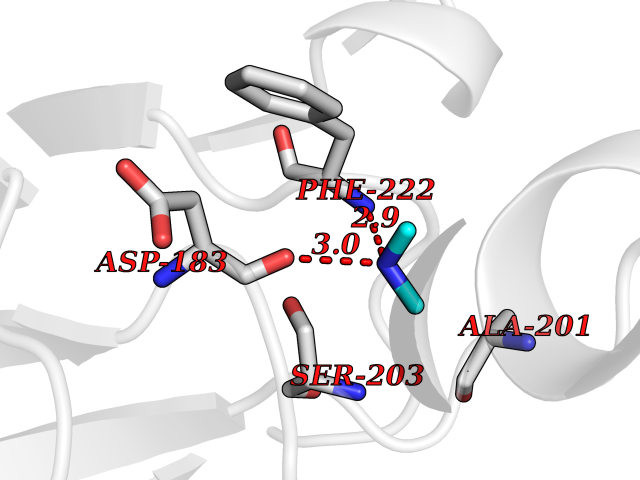 Pymol Map 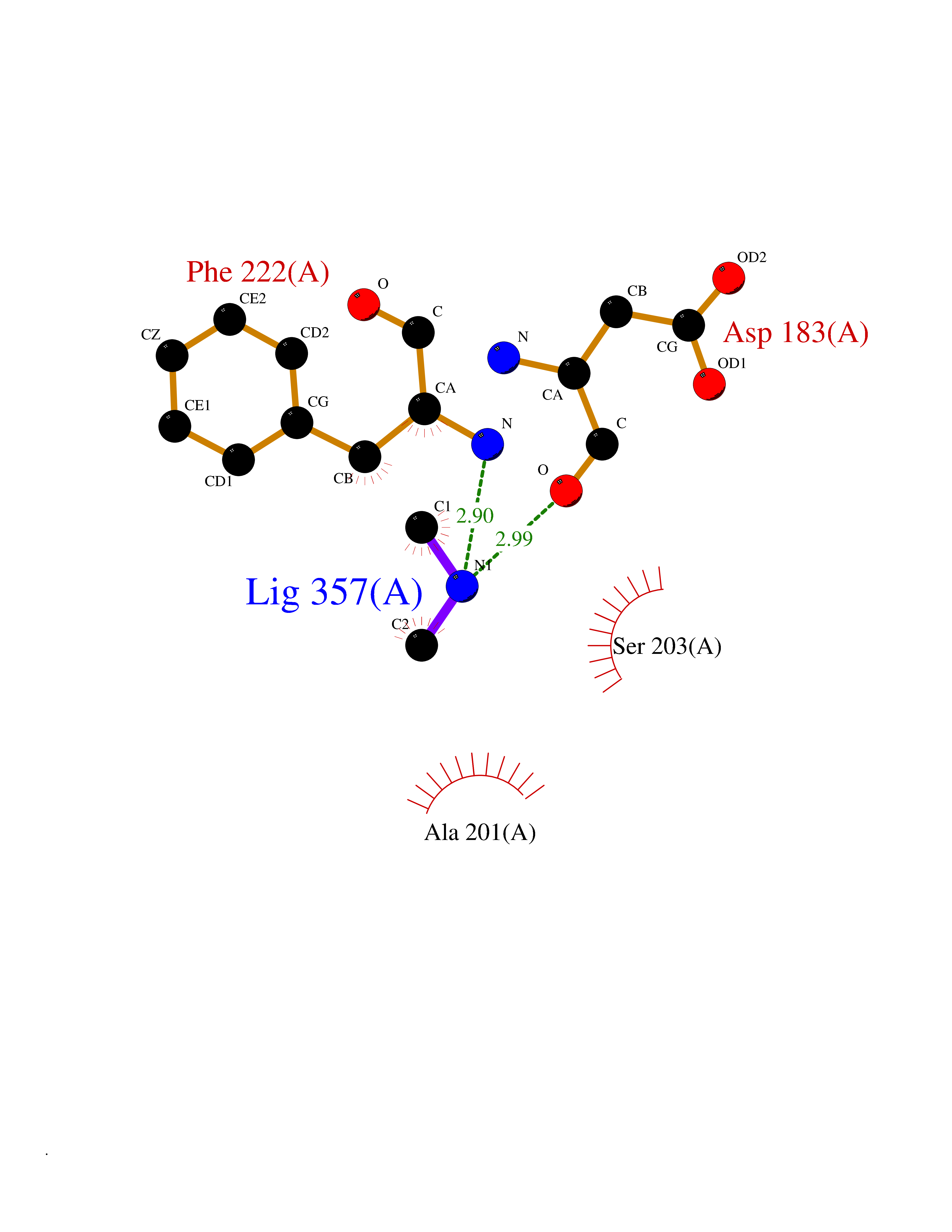 Ligplot Map 3D Binding mode Sequence LFDRQKSSFQTRFNVHREVTPVELPNCNLVKGIDNGSEDLEILPNGLAFISSGLKYDKSGKILLMDLNEKEPAVSELEIIGNTLDISSFNPHGISTFIDDDNTVYLLVVNHPGSSSTVEVFKFQEEEKSLLHLKTIRHKLLPSVNDIVAVGPEHFYATNDHYFIDPYLKSWEMHLGLAWSFVTYYSPNDVRVVAEGFDFANGINISPDGKYVYIAELLAHKIHVYEKHANWTLTPLRVLSFDTLVDNISVDPVTGDLWVGCHPNGMRIFFYDAENPPGSEVLRIQDILSEEPKVTVVYAENGTVLQGSTVAAVYKGKLLIGTVFHKALYCDL Hydrogen bonds contact Hydrophobic contact | ||||
| 5 | Penicillin-binding protein 1A | 2ZC6 | 4.06 | |
Target general information Gen name ponA Organism Streptococcus pneumoniae serotype 4 (strain ATCC BAA-334 / TIGR4) Uniprot ID TTD ID NA Synonyms SP_0369 Protein family Glycosyltransferase 51 family; Transpeptidase family Biochemical class Biosynthetic protein Function Penicillin binding.Peptidoglycan glycosyltransferase activity.Serine-type D-Ala-D-Ala carboxypeptidase activity. Related diseases Aminoacylase-1 deficiency (ACY1D) [MIM:609924]: An enzymatic deficiency resulting in encephalopathy, unspecific psychomotor delay, psychomotor delay with atrophy of the vermis and syringomyelia, marked muscular hypotonia or normal clinical features. Epileptic seizures are a frequent feature. All affected individuals exhibit markedly increased urinary excretion of several N-acetylated amino acids. {ECO:0000269|PubMed:16274666, ECO:0000269|PubMed:16465618, ECO:0000269|PubMed:17562838, ECO:0000269|PubMed:21414403}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01150; DB05659 Interacts with NA EC number 2.4.99.28; 3.4.16.4 Uniprot keywords 3D-structure; Antibiotic resistance; Carboxypeptidase; Cell shape; Cell wall biogenesis/degradation; Glycosyltransferase; Hydrolase; Multifunctional enzyme; Peptidoglycan synthesis; Protease; Reference proteome; Secreted; Transferase Protein physicochemical properties Chain ID A,C Molecular weight (Da) 44805.1 Length 400 Aromaticity 0.12 Instability index 31.82 Isoelectric point 4.88 Charge (pH=7) -14.68 2D Binding mode Binding energy (Kcal/mol) -5.54 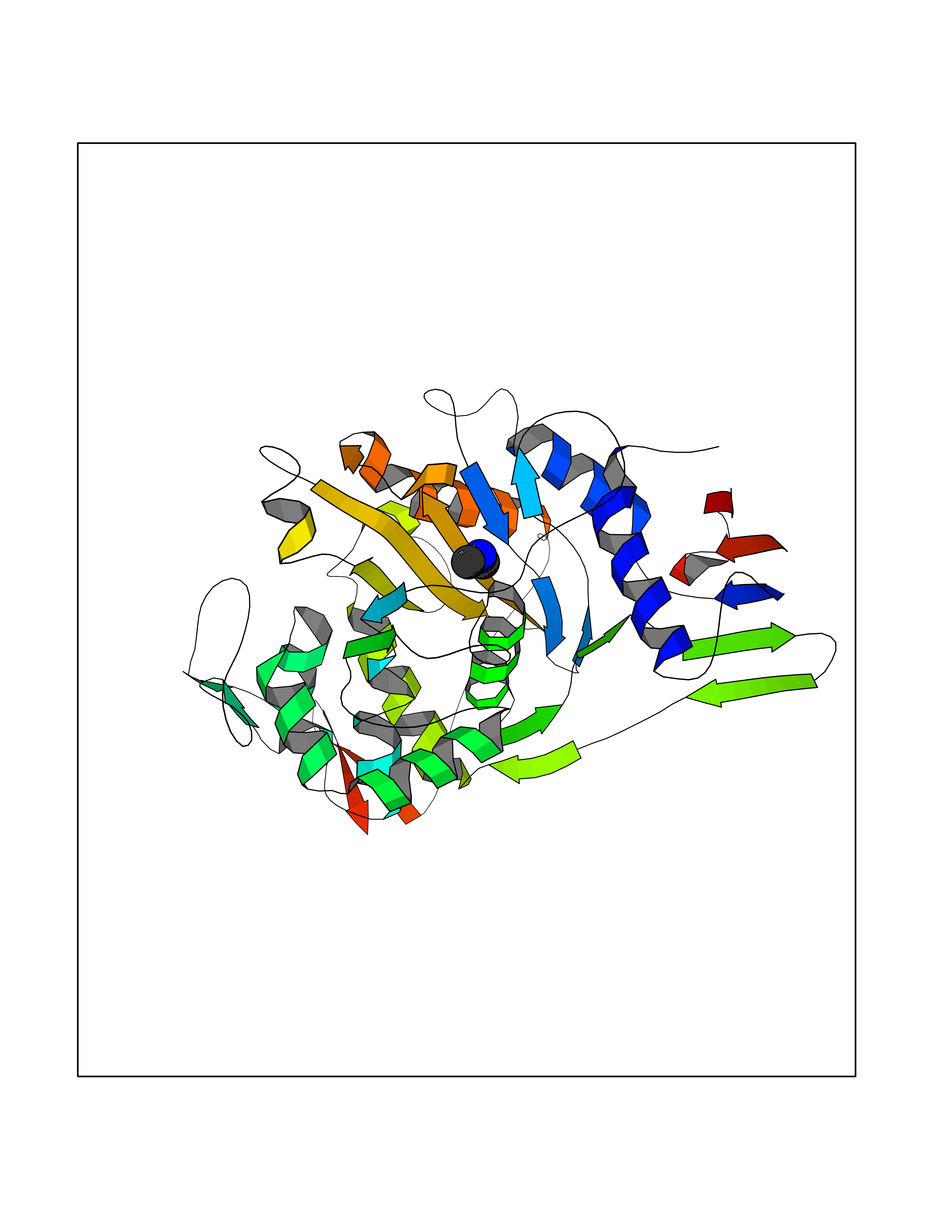 Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence NYPAYMDNYLKEVINQVEEETGYNLLTTGMDVYTNVDQEAQKHLWDIYNTDEYVAYPDDELQVASTIVDVSNGKVIAQLGARHQSSNVSFGINQAVETNRDWGSTMKPITDYAPALEYGVYDSTATIVHDEPYNYPGTNTPVYNWDRGYFGNITLQYALQQSRNVPAVETLNKVGLNRAKTFLNGLGIDYPSIHYSNAISSNTTESDKKYGASSEKMAAAYAAFANGGTYYKPMYIHKVVFSDGSEKEFSNVGTRAMKETTAYMMTDMMKTVLSYGTGQNAYLAWLPQAGKTGTSNYTDEEIENHIKTSQFVAPDELFAGYTRKYSMAVWTGYSNRLTPLVGNGLTVAAKVYRSMMTYLSEGSNPEDWNIPEGLYRNGEFVFKNTSSKIYDNKNQLIADL Hydrogen bonds contact Hydrophobic contact | ||||
| 6 | Pectate lyase | 1R76 | 4.06 | |
Target general information Gen name pelA Organism Niveispirillum irakense (Azospirillum irakense) Uniprot ID TTD ID NA Synonyms NA Protein family NA Biochemical class Lyase Function Lyase activity. Related diseases A chromosomal aberration involving ALK is found in a form of non-Hodgkin lymphoma. Translocation t(2;5)(p23;q35) with NPM1. The resulting chimeric NPM1-ALK protein homodimerize and the kinase becomes constitutively activated. The constitutively active fusion proteins are responsible for 5-10% of non-Hodgkin lymphomas. {ECO:0000269|PubMed:15938644}.; DISEASE: A chromosomal aberration involving ALK is associated with inflammatory myofibroblastic tumors (IMTs). Translocation t(2;11)(p23;p15) with CARS; translocation t(2;4)(p23;q21) with SEC31A. {ECO:0000269|PubMed:12112524, ECO:0000269|PubMed:16161041}.; DISEASE: A chromosomal aberration involving ALK is associated with anaplastic large-cell lymphoma (ALCL). Translocation t(2;17)(p23;q25) with ALO17. {ECO:0000269|PubMed:12112524}.; DISEASE: Neuroblastoma 3 (NBLST3) [MIM:613014]: A common neoplasm of early childhood arising from embryonic cells that form the primitive neural crest and give rise to the adrenal medulla and the sympathetic nervous system. {ECO:0000269|PubMed:18724359, ECO:0000269|PubMed:18923523, ECO:0000269|PubMed:18923525, ECO:0000269|PubMed:21242967, ECO:0000269|PubMed:22932897}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: The ALK signaling pathway plays an important role in glioblastoma, the most common malignant brain tumor of adults and one of the most lethal cancers. It regulates both glioblastoma migration and growth. {ECO:0000269|PubMed:15908427}.; DISEASE: A chromosomal aberration involving ALK is found in one subject with colorectal cancer. Translocation t(2;2)(p23.1;p23.3). A 5 million base pair tandem duplication generates an in-frame WDCP-ALK gene fusion. {ECO:0000269|PubMed:22327622}.; DISEASE: A chromosomal aberration involving ALK has been identified in a subset of patients with non-small-cell lung carcinoma. This aberration leads to the production of a fusion protein between the N-terminus of EML4 et the C-terminus of ALK. It is unclear whether the fusion protein is caused by a simple inversion within 2p (inv(2)(p21p23)) or whether the chromosome translocation involving 2p is more complex. When tested in a heterologous system, the fusion protein EML4-ALK possesses transforming activity that is dependent on ALK catalytic activity, possibly due to spontaneous dimerization mediated by the EML4 moiety, leading to ALK kinase activation. {ECO:0000269|PubMed:17625570}. Drugs (DrugBank ID) NA Interacts with NA EC number NA Uniprot keywords 3D-structure; Lyase; Signal Protein physicochemical properties Chain ID A Molecular weight (Da) 41907.5 Length 384 Aromaticity 0.08 Instability index 43.72 Isoelectric point 6.11 Charge (pH=7) -3.46 2D Binding mode Binding energy (Kcal/mol) -5.54  Molscript Map 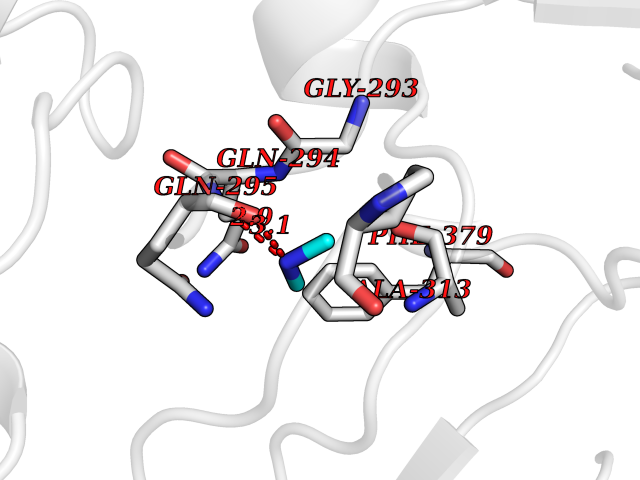 Pymol Map 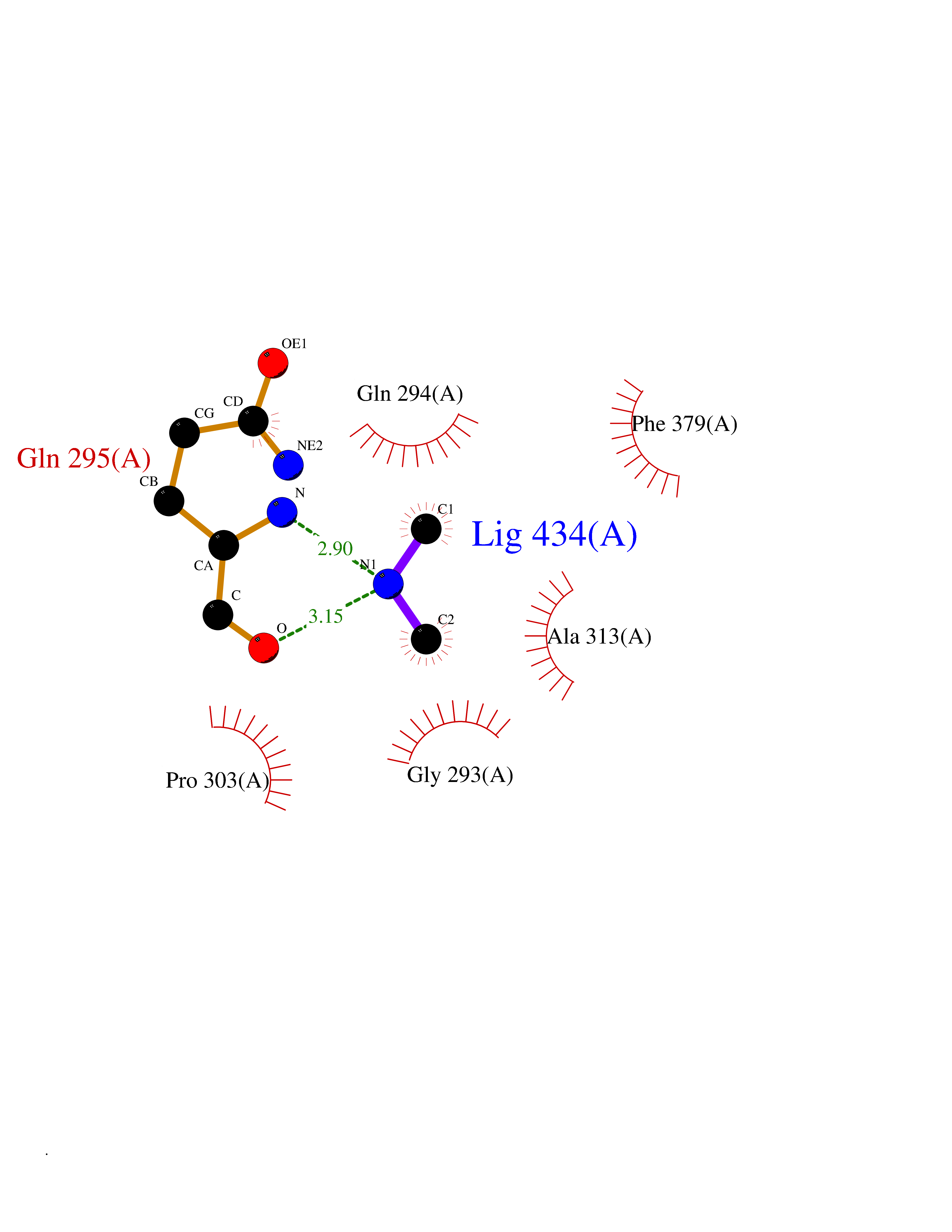 Ligplot Map 3D Binding mode Sequence AVIGMNEAASALTPSRVSSLPDTQRAAWQEYLARSEAQLSRDKASLAAELAPGQPLPPPPAEGKGADTMPLDKPAAWYTSKAARHVADVIVSFQTPAGGWGKNQPRDGALRLPGQHYTGENVAKVKRDRDWHYVGTIDNDATVTEIRFLAQVVSQLAPEEAAPYRDAALKGIEYLLASQFPNGGWPQVWPLEGGYHDAITYNDDALVHVAELLSDIAAGRDGFGFVPPAIRTRALEATNAAIHCIVETQVVQDGKRLGWGQQHDALTLRPTSARNFEPAALSSTESARILLFLMEIEAPSDAVKQAIRGGVAWLNTSVIRDQGAKPLWSRFYSLDGNKPVFGDRDKTIHDDVMGISQERRTGYAWYTTSPQKALSAFTKWEKRS Hydrogen bonds contact Hydrophobic contact | ||||
| 7 | Histone deacetylase 8 (HDAC8) | 5BWZ | 4.06 | |
Target general information Gen name HDAC8 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Histone deacetylase-8; HDACL1; HD8; CDA07 Protein family Histone deacetylase family, HD type 1 subfamily Biochemical class Carbon-nitrogen hydrolase Function Gives a tag for epigenetic repression and plays an important role in transcriptional regulation, cell cycle progression and developmental events. Histone deacetylases act via the formation of large multiprotein complexes. Also involved in the deacetylation of cohesin complex protein SMC3 regulating release of cohesin complexes from chromatin. May play a role in smooth muscle cell contractility. Responsible for the deacetylation of lysine residues on the N-terminal part of the core histones (H2A, H2B, H3 and H4). Related diseases Cornelia de Lange syndrome 5 (CDLS5) [MIM:300882]: A form of Cornelia de Lange syndrome, a clinically heterogeneous developmental disorder associated with malformations affecting multiple systems. It is characterized by facial dysmorphisms, abnormal hands and feet, growth delay, cognitive retardation, hirsutism, gastroesophageal dysfunction and cardiac, ophthalmologic and genitourinary anomalies. {ECO:0000269|PubMed:22885700, ECO:0000269|PubMed:22889856}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07350; DB02565; DB07586; DB12565; DB05015; DB08168; DB01262; DB11841; DB14490; DB14491; DB14488; DB14501; DB14489; DB12645; DB01592; DB02917; DB06603; DB06819; DB03766; DB12847; DB06176; DB04297; DB00313; DB02546; DB01593; DB14487; DB14533; DB14548 Interacts with NA EC number EC 3.5.1.98 Uniprot keywords 3D-structure; Alternative splicing; Chromatin regulator; Chromosome; Cytoplasm; Disease variant; Hydrolase; Intellectual disability; Metal-binding; Nucleus; Obesity; Phosphoprotein; Proteomics identification; Reference proteome; Repressor; Transcription; Transcription regulation Protein physicochemical properties Chain ID A,B Molecular weight (Da) 39018.4 Length 351 Aromaticity 0.11 Instability index 38.57 Isoelectric point 6.06 Charge (pH=7) -5.26 2D Binding mode Binding energy (Kcal/mol) -5.53  Molscript Map 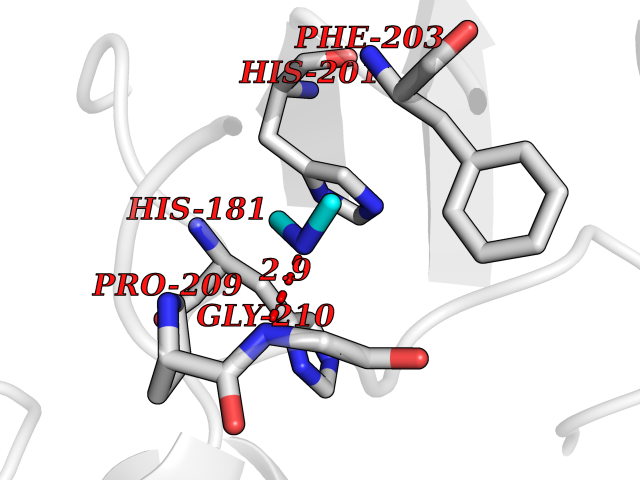 Pymol Map 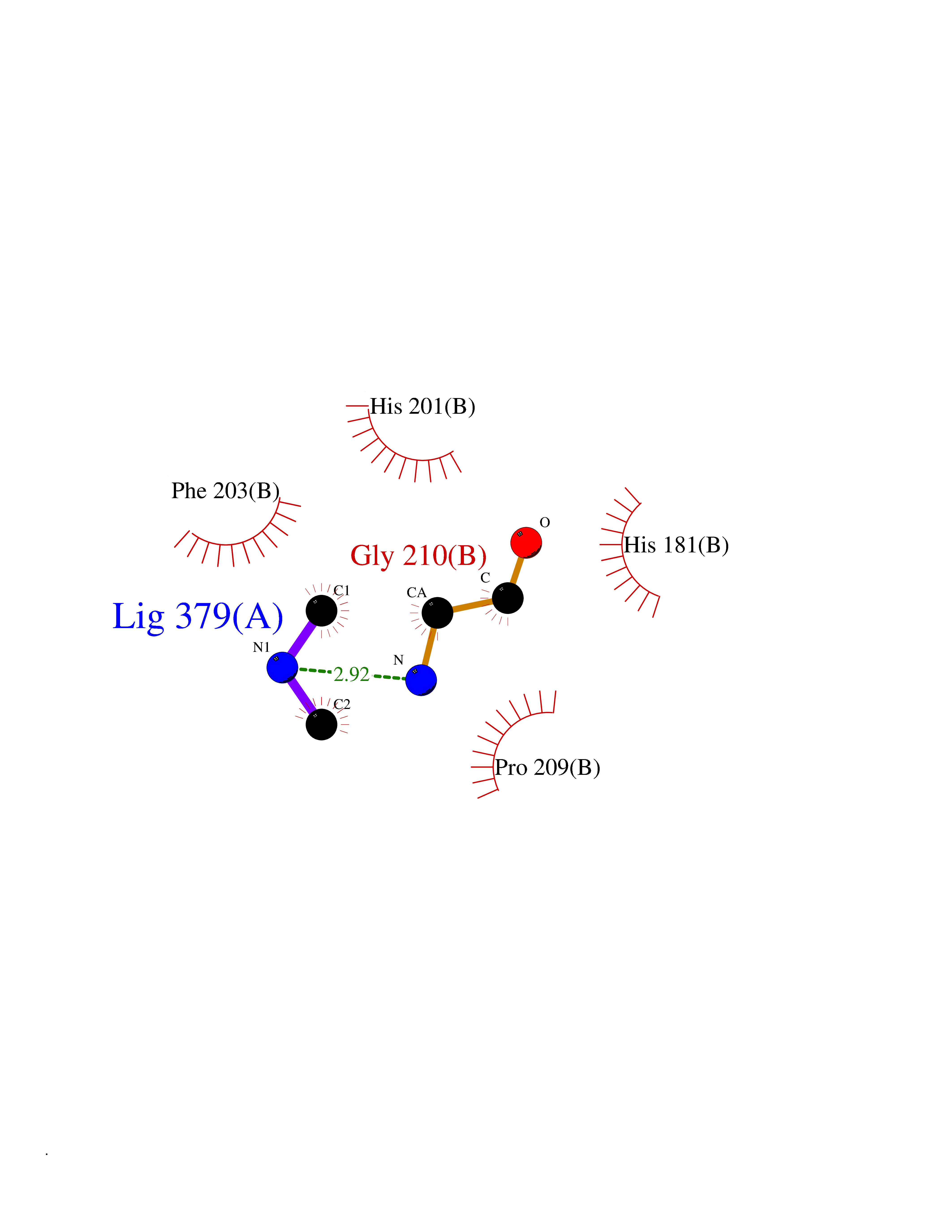 Ligplot Map 3D Binding mode Sequence LVPVYIYSPEYVSMCDSLPKRAEMVHSLIEAYALHKQMRIVKPKVASMEEMATFHTDAYLQHLQKVSQEYGLGYDCPATEGIFDYAAAIGGATITAAQCLIDGMCKVAINWSGGWHHAKKDEASGFCYLNDAVLGILRLRRKFERILYVDLDLHHGDGVEDAFSFTSKVMTVSLHKFSPGFFPGTGDVSDVGLGKGRYYSVNVPIQDGIQDEKYYQICESVLKEVYQAFNPKAVVLQLGADTIAGDPMCSFNMTPVGIGKCLKYILQWQLATLILGGGGYNLANTARCWTYLTGVILGKTLSSEIPDHEFFTAYGPDYVLEITPSCRPDRNEPHRIQQILNYIKGNLKHVV Hydrogen bonds contact Hydrophobic contact | ||||
| 8 | Aldehyde dehydrogenase family 1 member A3 | 5FHZ | 4.05 | |
Target general information Gen name ALDH1A3 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms ALDH6 Protein family Aldehyde dehydrogenase family Biochemical class Oxidoreductase Function Aldehyde dehydrogenase (NAD) activity.Aldehyde dehydrogenase [NAD(P)+] activity.NAD+ binding.Protein homodimerization activity.Retinal dehydrogenase activity.Thyroid hormone binding. Related diseases Microphthalmia, isolated, 8 (MCOP8) [MIM:615113]: A disorder of eye formation, ranging from small size of a single eye to complete bilateral absence of ocular tissues. Ocular abnormalities like opacities of the cornea and lens, scaring of the retina and choroid, and other abnormalities may also be present. {ECO:0000269|PubMed:23312594, ECO:0000269|PubMed:23591992, ECO:0000269|PubMed:23646827, ECO:0000269|PubMed:23881059, ECO:0000269|PubMed:24024553, ECO:0000269|PubMed:24568872, ECO:0000269|PubMed:24777706}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00157; DB00162 Interacts with NA EC number 1.2.1.36 Uniprot keywords 3D-structure; Acetylation; Cytoplasm; Direct protein sequencing; Disease variant; Lipid metabolism; Microphthalmia; NAD; Oxidoreductase; Proteomics identification; Reference proteome Protein physicochemical properties Chain ID A,B,C,D,E,F,G,H Molecular weight (Da) 50635.7 Length 461 Aromaticity 0.09 Instability index 33.75 Isoelectric point 7.07 Charge (pH=7) 0.15 2D Binding mode Binding energy (Kcal/mol) -5.52  Molscript Map 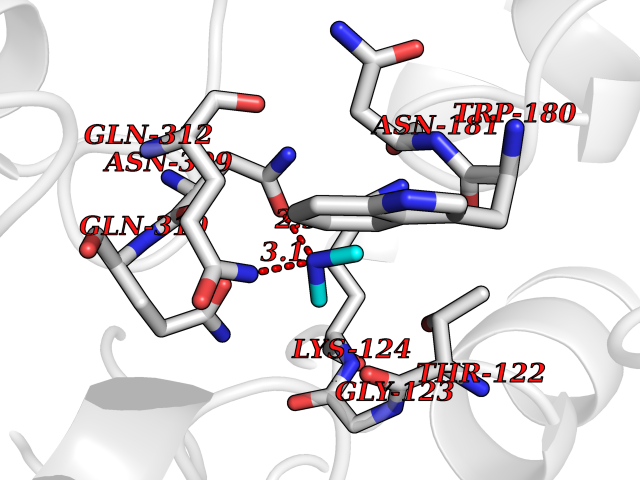 Pymol Map 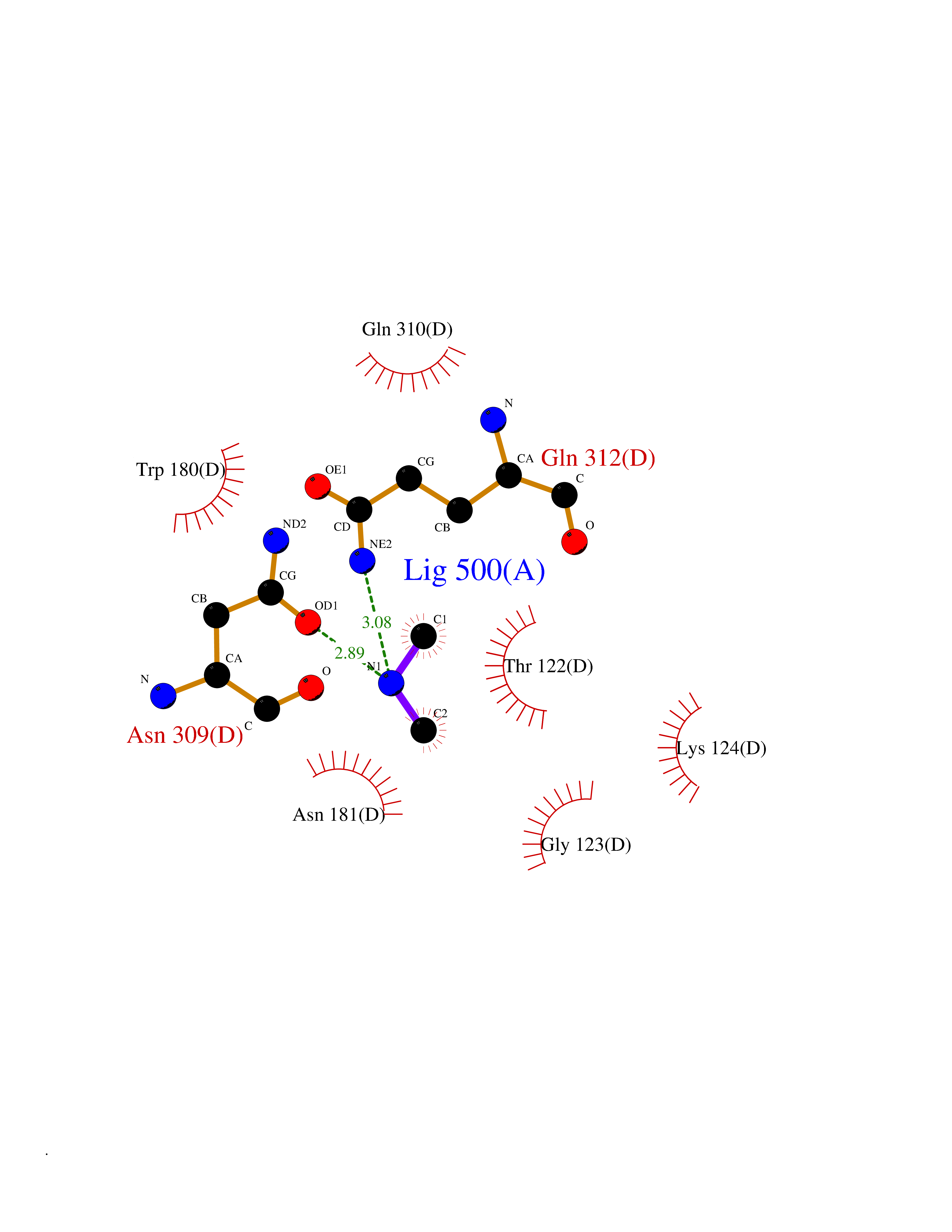 Ligplot Map 3D Binding mode Sequence LPRPIRNLEVKFTKIFINNEWHESKSGKKFATCNPSTREQICEVEEGDKPDVDKAVEAAQVAFQRGSPWRRLDALSRGRLLHQLADLVERDRATLAALETMDTGKPFLHAFFIDLEGCIRTLRYFAGWADKIPIGVCGAITPWNFPLLMLVWKLAPALCCGNTMVLKPAEQTPLTALYLGSLIKEAGFPPGVVNIVPGFGPTVGAAISSHPQINKIAFTGSTEVGKLVKEAASRSNLKRVTLELGGKNPCIVCADADLDLAVECAHQGVFFNQGQCCTAASRVFVEEQVYSEFVRRSVEYAKKRPVGDPFDVKTEQGPQIDQKQFDKILELIESGKKEGAKLECGGSAMEDKGLFIKPTVFSEVTDNMRIAKEEIFGPVQPILKFKSIEEVIKRANSTDYGLTAAVFTKNLDKALKLASALESGTVWINCYNALYAQAPFGGFKMSGNGRELGEYALAEYT Hydrogen bonds contact Hydrophobic contact | ||||
| 9 | Histone lysine demethylase PHF8 (PHF8) | 3KV4 | 4.05 | |
Target general information Gen name PHF8 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms ZNF422; PHD finger protein 8; KIAA1111 Protein family JHDM1 histone demethylase family, JHDM1D subfamily Biochemical class Paired donor oxygen oxidoreductase Function Demethylates mono- and dimethylated histone H3 'Lys-9' residue (H3K9Me1 and H3K9Me2), dimethylated H3 'Lys-27' (H3K27Me2) and monomethylated histone H4 'Lys-20' residue (H4K20Me1). Acts as a transcription activator as H3K9Me1, H3K9Me2, H3K27Me2 and H4K20Me1 are epigenetic repressive marks. Involved in cell cycle progression by being required to control G1-S transition. Acts as a coactivator of rDNA transcription, by activating polymerase I (pol I) mediated transcription of rRNA genes. Required for brain development, probably by regulating expression of neuron-specific genes. Only has activity toward H4K20Me1 when nucleosome is used as a substrate and when not histone octamer is used as substrate. May also have weak activity toward dimethylated H3 'Lys-36' (H3K36Me2), however, the relevance of this result remains unsure in vivo. Specifically binds trimethylated 'Lys-4' of histone H3 (H3K4me3), affecting histone demethylase specificity: has weak activity toward H3K9Me2 in absence of H3K4me3, while it has high activity toward H3K9me2 when binding H3K4me3. Histone lysine demethylase with selectivity for the di- and monomethyl states that plays a key role cell cycle progression, rDNA transcription and brain development. Related diseases Intellectual developmental disorder, X-linked, syndromic, Siderius type (MRXSSD) [MIM:300263]: A syndrome characterized by mild to borderline intellectual disability with or without cleft lip/cleft palate. {ECO:0000269|PubMed:16199551, ECO:0000269|PubMed:17661819, ECO:0000269|PubMed:20101266, ECO:0000269|PubMed:20208542, ECO:0000269|PubMed:20346720, ECO:0000269|PubMed:20421419, ECO:0000269|PubMed:20548336, ECO:0000269|PubMed:20622853, ECO:0000269|PubMed:20622854, ECO:0000269|PubMed:31691806}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q96QS3; Q06330; Q9Y462; P51610-1; Q15156; P10276 EC number EC 1.14.11.27 Uniprot keywords 3D-structure; Activator; Alternative splicing; Cell cycle; Chromatin regulator; Dioxygenase; Disease variant; Intellectual disability; Iron; Metal-binding; Nucleus; Oxidoreductase; Phosphoprotein; Proteomics identification; Reference proteome; Transcription; Transcription regulation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A,B Molecular weight (Da) 51141.1 Length 446 Aromaticity 0.11 Instability index 44.47 Isoelectric point 6.21 Charge (pH=7) -4.56 2D Binding mode Binding energy (Kcal/mol) -5.52  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence SVPVYCLCRLPYDVTRFMIECDMCQDWFHGSCVGVEEEKAADIDLYHCPNCEVLHGPSIMKKKPVKTGSPTFVRELRSRTFDSSDEVILKPTGNQLTVEFLEENSFSVPILVLKKDGLGMTLPSPSFTVRDVEHYVGSDKEIDVIDVTRQADCKMKLGDFVKYYYSGKREKVLNVISLEFSDTRLSNLVETPKIVRKLSWVENLWPEECVFERPNVQKYCLMSVRDSYTDFHIDFGGTSVWYHVLKGEKIFYLIRPTNANLTLFECWSSSSNQNEMFFGDQVDKCYKCSVKQGQTLFIPTGWIHAVLTPVDCLAFGGNFLHSLNIEMQLKAYEIEKRLSTADLFRFPNFETICWYVGKHILDIFRGLRENRRHPASYLVHGGKALNLAFRAWTRKEALPDHEDEIPETVRTVQLIKDLAREIRLVEDIFQQNARTXQTARXSTGGK Hydrogen bonds contact Hydrophobic contact | ||||
| 10 | Ubiquitin thioesterase L1 (UCHL1) | 3IFW | 4.05 | |
Target general information Gen name UCHL1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Ubiquitin thiolesterase L1; Ubiquitin carboxyl-terminal hydrolase isozyme L1; Ubiquitin carboxy-terminal hydrolase L1; UCH-L1; PGP9.5; PGP 9.5; Neuron cytoplasmic protein 9.5 Protein family Peptidase C12 family Biochemical class Peptidase Function Ubiquitin-protein hydrolase involved both in the processing of ubiquitin precursors and of ubiquitinated proteins. This enzyme is a thiol protease that recognizes and hydrolyzes a peptide bond at the C-terminal glycine of ubiquitin. Also binds to free monoubiquitin and may prevent its degradation in lysosomes. The homodimer may have ATP-independent ubiquitin ligase activity. Related diseases Parkinson disease 5 (PARK5) [MIM:613643]: A complex neurodegenerative disorder with manifestations ranging from typical Parkinson disease to dementia with Lewy bodies. Clinical features include parkinsonian symptoms (resting tremor, rigidity, postural instability and bradykinesia), dementia, diffuse Lewy body pathology, autonomic dysfunction, hallucinations and paranoia. {ECO:0000269|PubMed:12408865, ECO:0000269|PubMed:12705903, ECO:0000269|PubMed:9774100}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Spastic paraplegia 79A, autosomal dominant, with ataxia (SPG79A) [MIM:620221]: A form of spastic paraplegia, a neurodegenerative disorder characterized by a slow, gradual, progressive weakness and spasticity of the lower limbs. Rate of progression and the severity of symptoms are quite variable. Initial symptoms may include difficulty with balance, weakness and stiffness in the legs, muscle spasms, and dragging the toes when walking. In some forms of the disorder, bladder symptoms (such as incontinence) may appear, or the weakness and stiffness may spread to other parts of the body. SPG79A is a slowly progressive form characterized by late-onset spastic ataxia, neuropathy, and often optic atrophy. {ECO:0000269|PubMed:35986737}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Spastic paraplegia 79B, autosomal recessive (SPG79B) [MIM:615491]: A form of spastic paraplegia, a neurodegenerative disorder characterized by a slow, gradual, progressive weakness and spasticity of the lower limbs. Rate of progression and the severity of symptoms are quite variable. Initial symptoms may include difficulty with balance, weakness and stiffness in the legs, muscle spasms, and dragging the toes when walking. In some forms of the disorder, bladder symptoms (such as incontinence) may appear, or the weakness and stiffness may spread to other parts of the body. SPG79B is characterized by childhood onset blindness, cerebellar ataxia, nystagmus, dorsal column dysfunction, and spasticity with upper motor neuron dysfunction. {ECO:0000269|PubMed:23359680, ECO:0000269|PubMed:28007905}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB12695 Interacts with P63010-2; P05067; P05067-2; Q8N6T3-3; P18847; Q9H1Y0; O15392; Q8WUW1; P83916; P11802; Q00535; Q9UNS2; Q92905; P00533; O60739; Q8TC29; Q9UI08-2; Q8WVV9-3; Q14164; Q6DN90-2; Q96JM7-2; P13473-2; Q9BYZ2; O95777; A4FUJ8; Q15843; O15381-5; Q9BR81; Q13113; P62826; Q8TAI7; Q9ULX5; Q15554-4; Q9NYB0; P04637; Q9Y4K3; P19474; Q9BSL1; Q7KZS0; P61086; Q9UK80; Q86WB0-2 EC number EC 3.4.19.12 Uniprot keywords 3D-structure; Cytoplasm; Direct protein sequencing; Disease variant; Endoplasmic reticulum; Glycoprotein; Hereditary spastic paraplegia; Hydrolase; Lipoprotein; Membrane; Neurodegeneration; Oxidation; Parkinson disease; Parkinsonism; Phosphoprotein; Prenylation; Protease; Proteomics identification; Reference proteome; Thiol protease; Ubl conjugation pathway Protein physicochemical properties Chain ID A,B Molecular weight (Da) 33389.8 Length 298 Aromaticity 0.07 Instability index 38.69 Isoelectric point 5.51 Charge (pH=7) -7.88 2D Binding mode Binding energy (Kcal/mol) -5.52 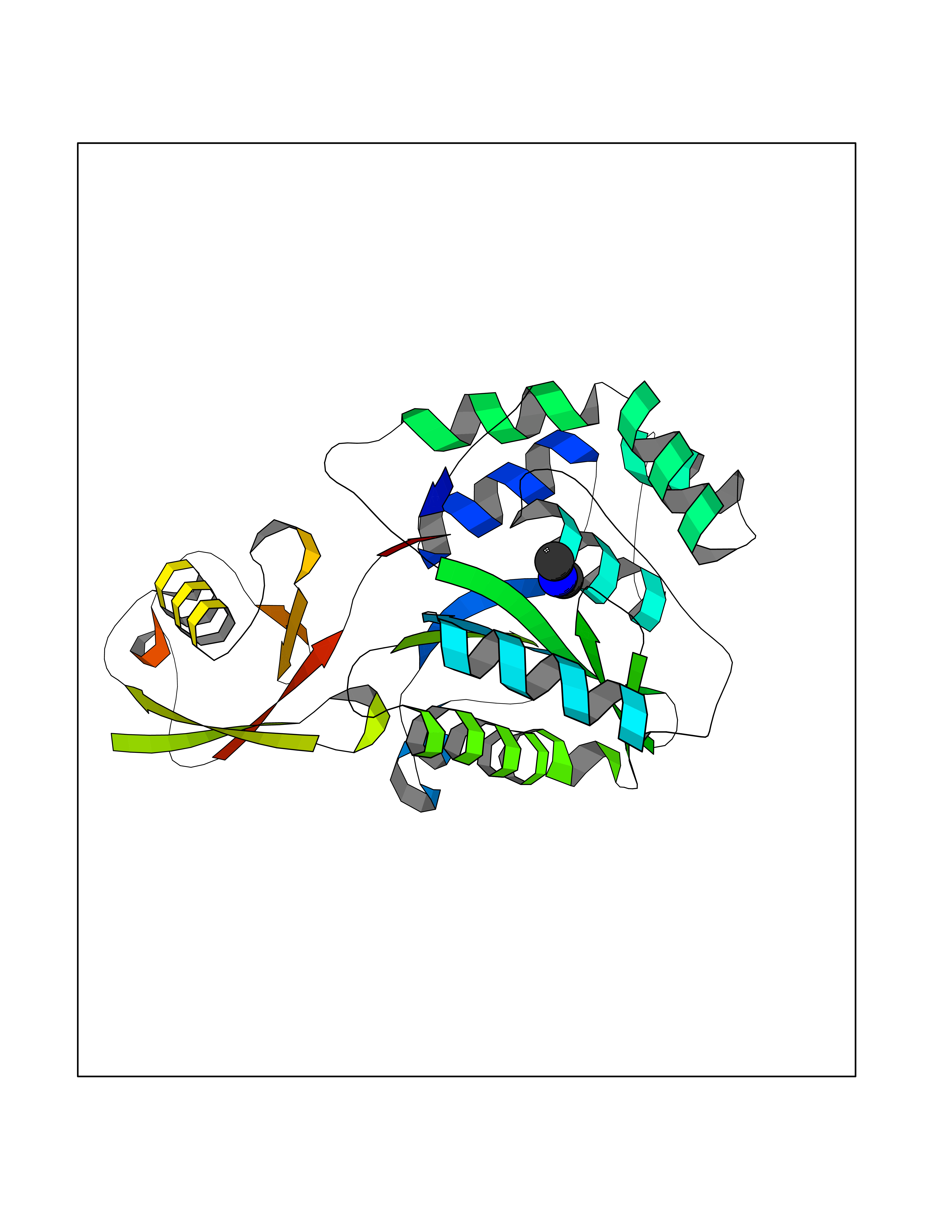 Molscript Map 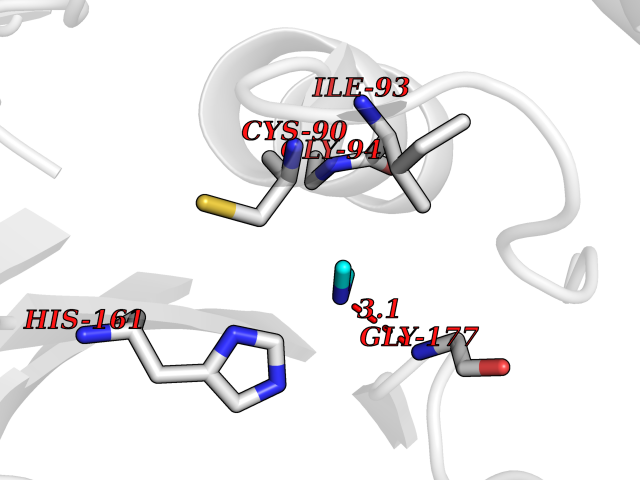 Pymol Map 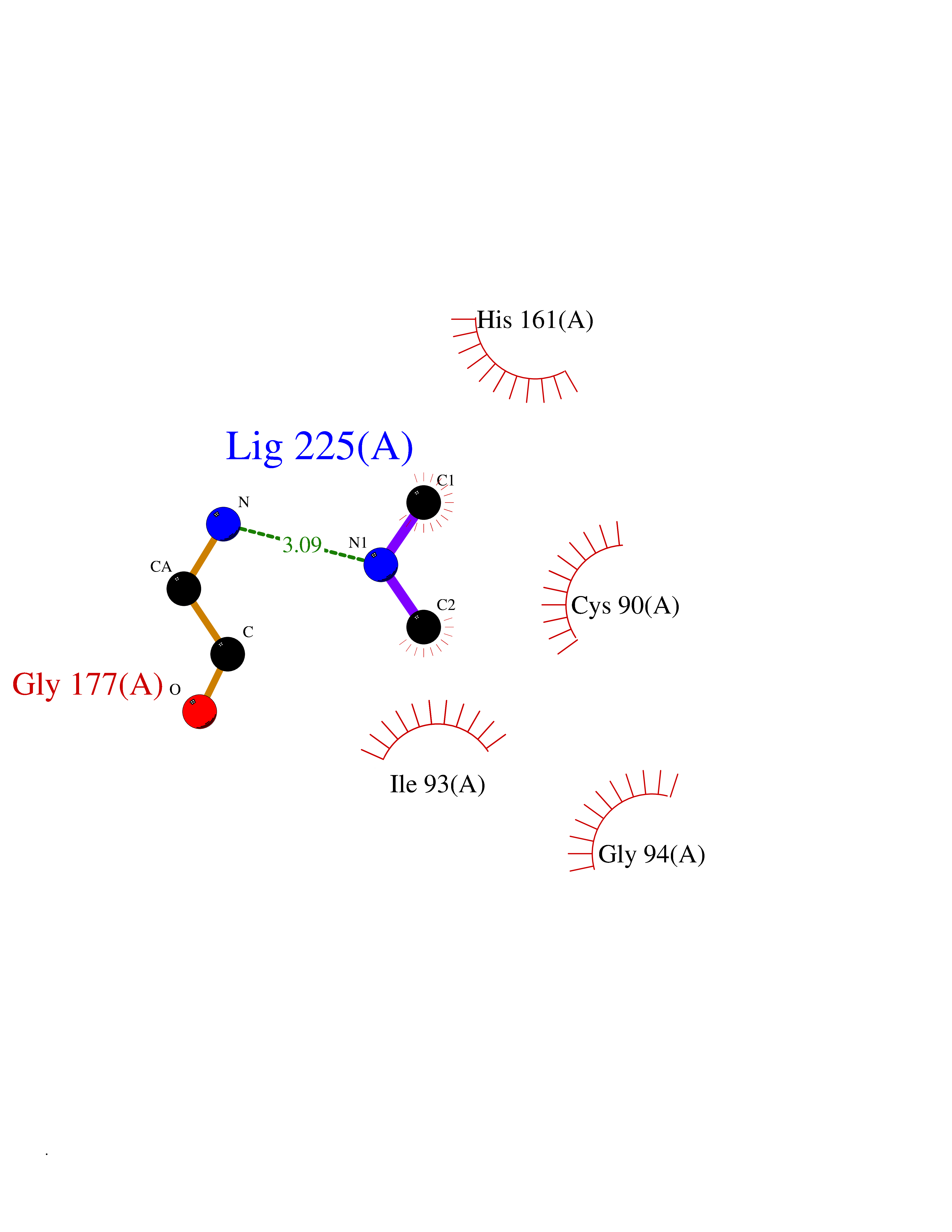 Ligplot Map 3D Binding mode Sequence MQLKPMEINPEMLNKVLYRLGVAGQWRFVDVLGLEEESLGSVPAPACALLLLFPLTAQHENFRKKQIEELKGQEVSPKVYFMKQTIGNSCGTIGLIHAVANNQDKLGFEDGSVLKQFLSETEKMSPEDRAKCFEKNEAIQAAHDAVAQEGQCRVDDKVNFHFILFNNVDGHLYELDGRMPFPVNHGASSEDTLLKDAAKVCREFTEREQGEVRFSAVALCKAAMQIFVKTLTGKTITLEVEPSDTIENVKAKIQDKEGIPPDQQRLIFAGKQLEDGRTLSDYNIQKESTLHLVLRLRG Hydrogen bonds contact Hydrophobic contact | ||||
| 11 | Histone deacetylase 7 (HDAC7) | 3C0Z | 4.05 | |
Target general information Gen name HDAC7 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Histone deacetylase 7A; HDAC7A; HD7a; HD7 Protein family Histone deacetylase family, HD type 2 subfamily Biochemical class Carbon-nitrogen hydrolase Function Gives a tag for epigenetic repression and plays an important role in transcriptional regulation, cell cycle progression and developmental events. Histone deacetylases act via the formation of large multiprotein complexes. Involved in muscle maturation by repressing transcription of myocyte enhancer factors such as MEF2A, MEF2B and MEF2C. During muscle differentiation, it shuttles into the cytoplasm, allowing the expression of myocyte enhancer factors. May be involved in Epstein-Barr virus (EBV) latency, possibly by repressing the viral BZLF1 gene. Positively regulates the transcriptional repressor activity of FOXP3. Responsible for the deacetylation of lysine residues on the N-terminal part of the core histones (H2A, H2B, H3 and H4). Related diseases Mucopolysaccharidosis 1H (MPS1H) [MIM:607014]: A severe form of mucopolysaccharidosis type 1, a rare lysosomal storage disease characterized by progressive physical deterioration with urinary excretion of dermatan sulfate and heparan sulfate. Patients with MPS1H usually present, within the first year of life, a combination of hepatosplenomegaly, skeletal deformities, corneal clouding and severe intellectual disability. Obstructive airways disease, respiratory infection and cardiac complications usually result in death before 10 years of age. {ECO:0000269|PubMed:10466419, ECO:0000269|PubMed:10735634, ECO:0000269|PubMed:12559846, ECO:0000269|PubMed:1301941, ECO:0000269|PubMed:15300847, ECO:0000269|PubMed:19396826, ECO:0000269|PubMed:21394825, ECO:0000269|PubMed:24036510, ECO:0000269|PubMed:31194252, ECO:0000269|PubMed:7550232, ECO:0000269|PubMed:7550242, ECO:0000269|PubMed:7951228, ECO:0000269|PubMed:8019563, ECO:0000269|PubMed:8328452, ECO:0000269|PubMed:8401515, ECO:0000269|Ref.20}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Mucopolysaccharidosis 1H/S (MPS1H/S) [MIM:607015]: A form of mucopolysaccharidosis type 1, a rare lysosomal storage disease characterized by progressive physical deterioration with urinary excretion of dermatan sulfate and heparan sulfate. MPS1H/S represents an intermediate phenotype of the MPS1 clinical spectrum. It is characterized by relatively little neurological involvement, but most of the somatic symptoms described for severe MPS1 develop in the early to mid-teens, causing considerable loss of mobility. {ECO:0000269|PubMed:10466419, ECO:0000269|PubMed:10735634, ECO:0000269|PubMed:12559846, ECO:0000269|PubMed:15300847, ECO:0000269|PubMed:21394825, ECO:0000269|PubMed:7550232, ECO:0000269|PubMed:7550242, ECO:0000269|PubMed:8401515}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Mucopolysaccharidosis 1S (MPS1S) [MIM:607016]: A mild form of mucopolysaccharidosis type 1, a rare lysosomal storage disease characterized by progressive physical deterioration with urinary excretion of dermatan sulfate and heparan sulfate. Patients with MPS1S may have little or no neurological involvement, normal stature and life span, but present development of joints stiffness, mild hepatosplenomegaly, aortic valve disease and corneal clouding. {ECO:0000269|PubMed:12559846, ECO:0000269|PubMed:15300847, ECO:0000269|PubMed:19396826, ECO:0000269|PubMed:21394825, ECO:0000269|PubMed:25256405, ECO:0000269|PubMed:7550232, ECO:0000269|PubMed:7550242, ECO:0000269|PubMed:8213840}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB12565; DB05015; DB01262; DB11841; DB12645; DB06603; DB06819; DB03766; DB12847; DB06176; DB04297; DB00313; DB02546 Interacts with P00533; Q9BZS1-1; Q9BZS1-2; Q9BZL6; P31947; P63104; P08393; Q8CFN5; Q13137; Q04864; Q0D2K3; Q8WXI4-2; Q9BQD7; Q03989; Q9NSI6-4; Q3SXR2; Q13137; P60953; Q7L2Z9; Q96D03; Q9NQ30; Q9UBI6; A6NEM1; A5PKX9; Q9BXK1; Q6ZNG9; O43679; Q6FHY5; Q9BRT3; O94964-4; O95411; O00746; Q9BQI9; B7ZLY0; Q96I34; P63000; P15153; P60763; Q04864-2; Q0D2K3; P62070; O15427; O95164 EC number EC 3.5.1.98 Uniprot keywords 3D-structure; Alternative splicing; Chromatin regulator; Cytoplasm; Hydrolase; Metal-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Repressor; Transcription; Transcription regulation; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 41454.5 Length 383 Aromaticity 0.08 Instability index 38.49 Isoelectric point 6.26 Charge (pH=7) -5.18 2D Binding mode Binding energy (Kcal/mol) -5.52  Molscript Map 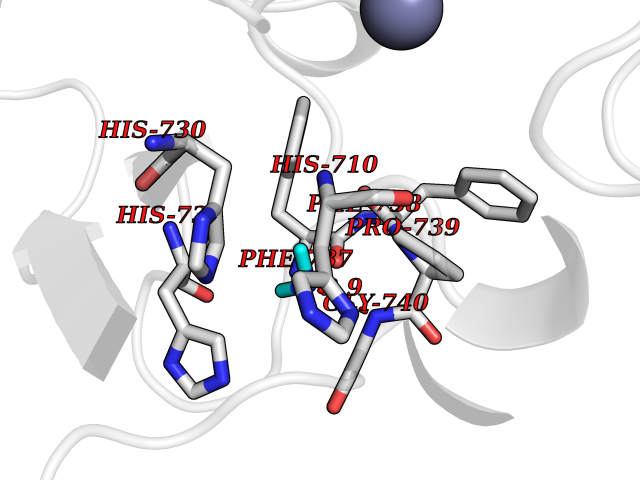 Pymol Map 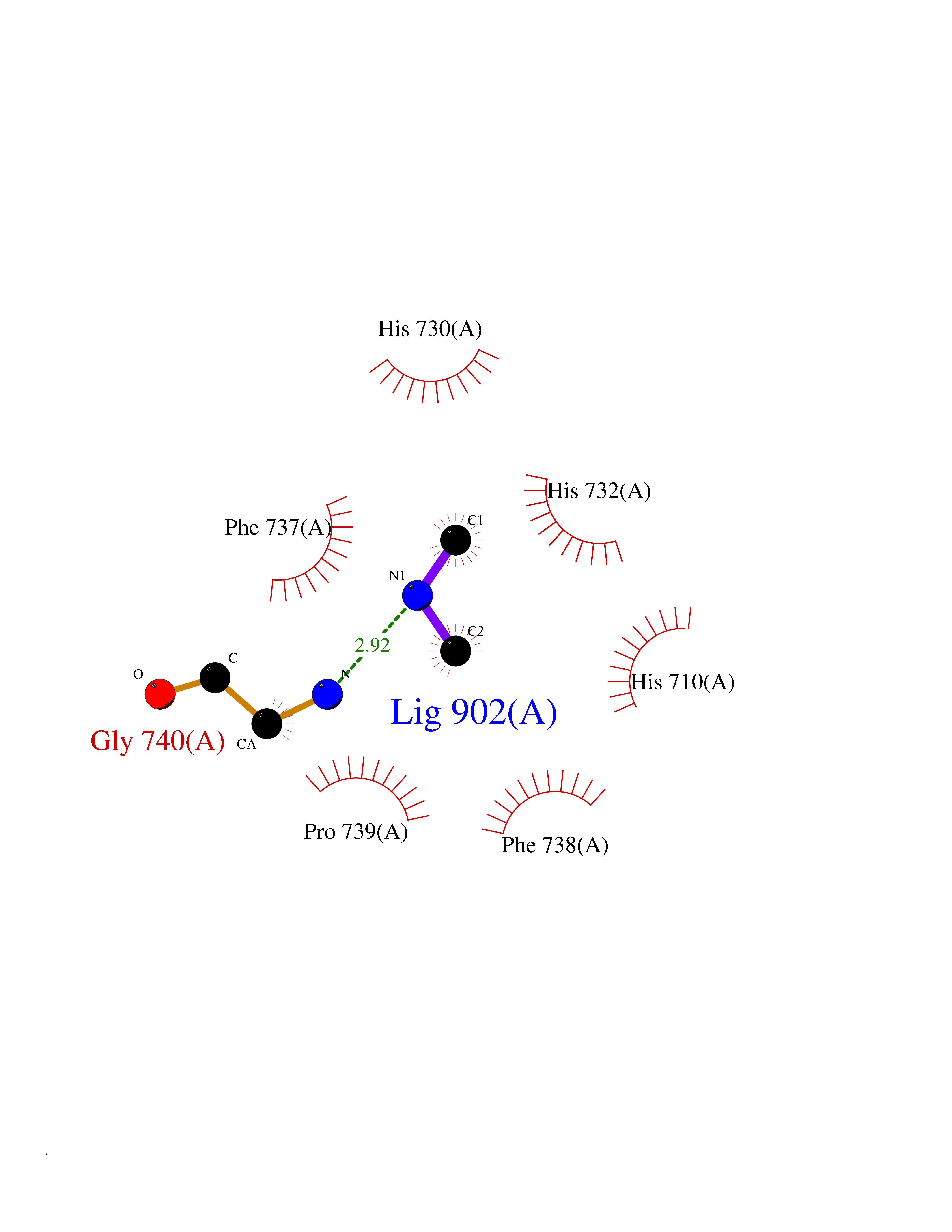 Ligplot Map 3D Binding mode Sequence TLPFTTGLIYDSVMLKHQCSCGDNSRHPEHAGRIQSIWSRLQERGLRSQCECLRGRKASLEELQSVHSERHVLLYGTNPLSRLKLDNGKLAGLLAQVMLPCGGVGVDTDTIWNELHSSNAARWAAGSVTDLAFKVASRELKNGFAVVRPPGHHADHSTAMGFCFFNSVAIACRQLQQQSKASKILIVDWDVHHGNGTQQTFYQDPSVLYISLHRHDDGNFFPGSGAVDEVGAGSGEGFNVNVAWAGGLDPPMGDPEYLAAFRIVVMPIAREFSPDLVLVSAGFDAAEGHPAPLGGYHVSAKCFGYMTQQLMNLAGGAVVLALEGGHDLTAICDASEACVAALLGNRVDPLSEEGWKQKPNLNAIRSLEAVIRVHSKYWGCMQR Hydrogen bonds contact Hydrophobic contact | ||||
| 12 | Cocaine esterase | 3I2K | 4.04 | |
Target general information Gen name cocE Organism Rhodococcus sp. (strain MB1 Bresler) Uniprot ID TTD ID NA Synonyms NA Protein family CocE/NonD hydrolase family Biochemical class Hydrolase Function Carboxylic ester hydrolase activity.Dipeptidyl-peptidase activity. Related diseases Thiamine metabolism dysfunction syndrome 5, episodic encephalopathy type (THMD5) [MIM:614458]: An autosomal recessive metabolic disorder due to an inborn error of thiamine metabolism. The phenotype is highly variable, but in general, affected individuals have onset in early childhood of acute encephalopathic episodes associated with increased serum and CSF lactate. These episodes result in progressive neurologic dysfunction manifest as gait disturbances, ataxia, dystonia, and spasticity, which in some cases may result in loss of ability to walk. Cognitive function is usually preserved, although mildly delayed development has been reported. These episodes are usually associated with infection and metabolic decompensation. Some patients may have recovery of some neurologic deficits. {ECO:0000269|PubMed:22152682}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03793; DB01795 Interacts with NA EC number 3.1.1.84 Uniprot keywords 3D-structure; Cytoplasm; Direct protein sequencing; Hydrolase; Serine esterase Protein physicochemical properties Chain ID A Molecular weight (Da) 62127.9 Length 574 Aromaticity 0.09 Instability index 26.62 Isoelectric point 4.56 Charge (pH=7) -33.24 2D Binding mode Binding energy (Kcal/mol) -5.51  Molscript Map 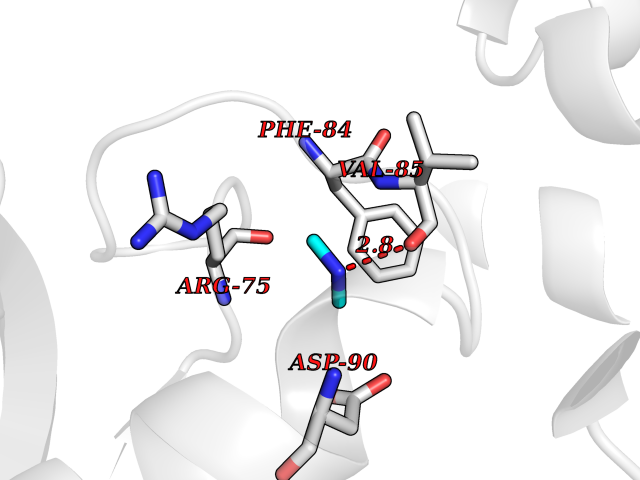 Pymol Map 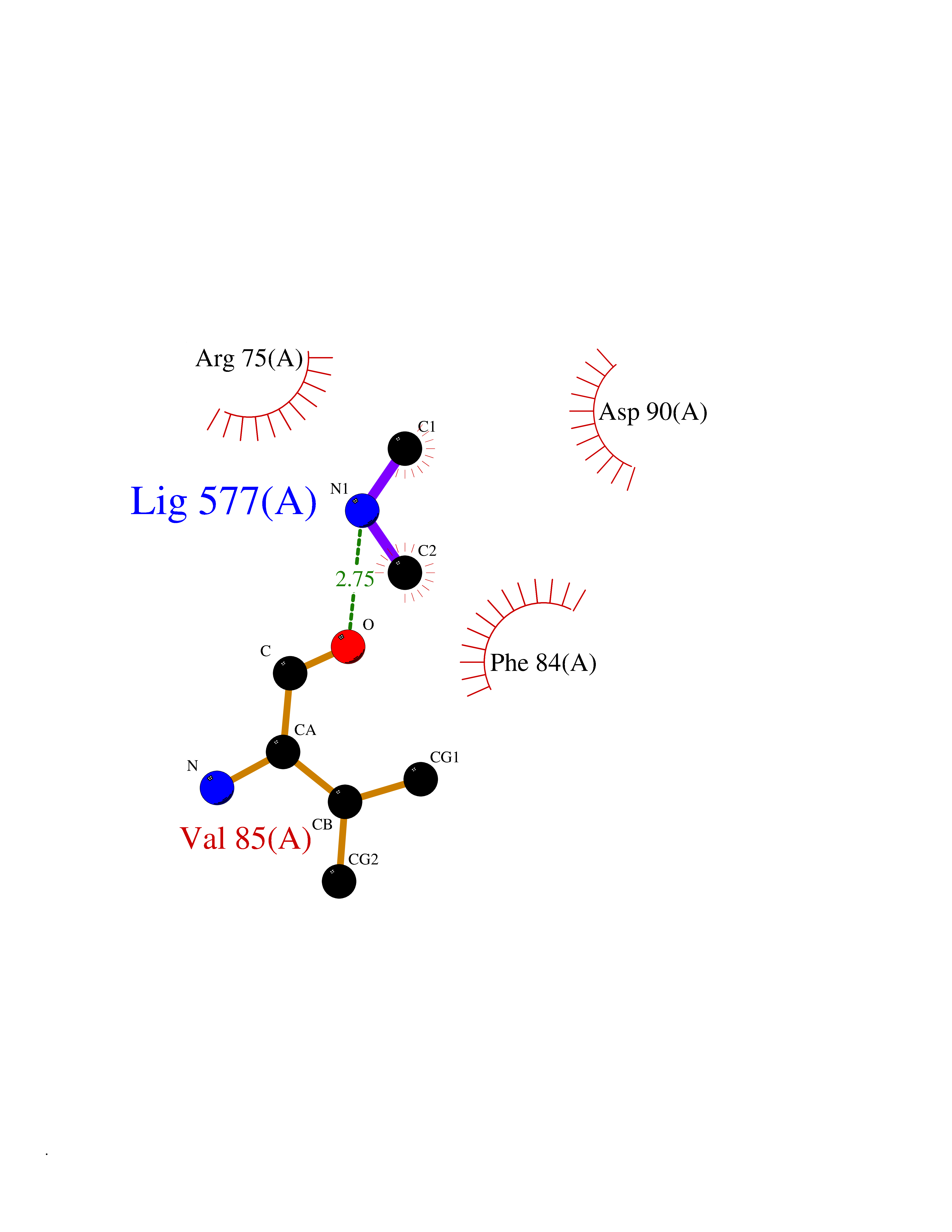 Ligplot Map 3D Binding mode Sequence VDGNYSVASNVMVPMRDGVRLAVDLYRPDADGPVPVLLVRNPYDKFDVFAWSTQSTNWLEFVRDGYAVVIQDTRGLFASEGEFVPHVDDEADAEDTLSWILEQAWCDGNVGMFGVSYLGVTQWQAAVSGVGGLKAIAPSMASADLYRAPWYGPGGALSVEALLGWSALIGTGLITSRSDARPEDAADFVQLAAILNDVAGAASVTPLAEQPLLGRLIPWVIDQVVDHPDNDESWQSISLFERLGGLATPALITAGWYDGFVGESLRTFVAVKDNADARLVVGPWSHSNLTGRNADRKFGIAATYPIQEATTMHKAFFDRHLRGETDALAGVPKVRLFVMGIDEWRDETDWPLPDTAYTPFYLGGSGAANTSTGGGTLSTSISGTESADTYLYDPADPVPSLGGTLLFHNGDNGPADQRPIHDRDDVLCYSTEVLTDPVEVTGTVSARLFVSSSAVDTDFTAKLVDVFPDGRAIALCDGIVRMRYRETLVNPTLIEAGEIYEVAIDMLATSNVFLPGHRIMVQVSSSNFPKYDRNSNTGGVIAREQLEEMCTAVNRIHRGPEHPSHIVLPIIKRK Hydrogen bonds contact Hydrophobic contact | ||||
| 13 | S-adenosylmethionine synthase type-2 (MAT2A) | 5A1I | 4.03 | |
Target general information Gen name MAT2A Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Methionine adenosyltransferase II; Methionine adenosyltransferase 2; MAT-II; MAT 2; AdoMet synthase 2 Protein family AdoMet synthase family Biochemical class AdoMet synthase family Function Catalyzes the formation of S-adenosylmethionine from methionine and ATP. The reaction comprises two steps that are both catalyzed by the same enzyme: formation of S-adenosylmethionine (AdoMet) and triphosphate, and subsequent hydrolysis of the triphosphate. Related diseases Pyruvate kinase hyperactivity (PKHYP) [MIM:102900]: Autosomal dominant phenotype characterized by increase of red blood cell ATP. {ECO:0000269|PubMed:9090535}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Pyruvate kinase deficiency of red cells (PKRD) [MIM:266200]: A frequent cause of hereditary non-spherocytic hemolytic anemia. Clinically, pyruvate kinase-deficient patients suffer from a highly variable degree of chronic hemolysis, ranging from severe neonatal jaundice and fatal anemia at birth, severe transfusion-dependent chronic hemolysis, moderate hemolysis with exacerbation during infection, to a fully compensated hemolysis without apparent anemia. {ECO:0000269|PubMed:10087985, ECO:0000269|PubMed:10772876, ECO:0000269|PubMed:11328279, ECO:0000269|PubMed:11960989, ECO:0000269|PubMed:1536957, ECO:0000269|PubMed:1896471, ECO:0000269|PubMed:19085939, ECO:0000269|PubMed:2018831, ECO:0000269|PubMed:21794208, ECO:0000269|PubMed:7706479, ECO:0000269|PubMed:8161798, ECO:0000269|PubMed:8180378, ECO:0000269|PubMed:8476433, ECO:0000269|PubMed:8481523, ECO:0000269|PubMed:8483951, ECO:0000269|PubMed:8664896, ECO:0000269|PubMed:8807089, ECO:0000269|PubMed:9075576, ECO:0000269|PubMed:9482576, ECO:0000269|PubMed:9827908, ECO:0000269|PubMed:9886305, ECO:0000269|Ref.24}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00118; DB00134 Interacts with Q96IK1-2; Q96NX5; Q6ZP82-1; Q8IUI8; Q6P1L5; P15976-2; P80217-2; Q8WZ19; Q9UIH9; Q00266; P31153; Q9NZL9; P02795; Q9BRX2; O43663; O43741; P57052; Q8N488; P08195-4; Q13573; Q86W54-2; O95789-4 EC number EC 2.5.1.6 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; ATP-binding; Isopeptide bond; Magnesium; Metal-binding; Nucleotide-binding; One-carbon metabolism; Phosphoprotein; Potassium; Proteomics identification; Reference proteome; Transferase; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 42071.4 Length 381 Aromaticity 0.08 Instability index 38.24 Isoelectric point 6.21 Charge (pH=7) -4.13 2D Binding mode Binding energy (Kcal/mol) -5.49 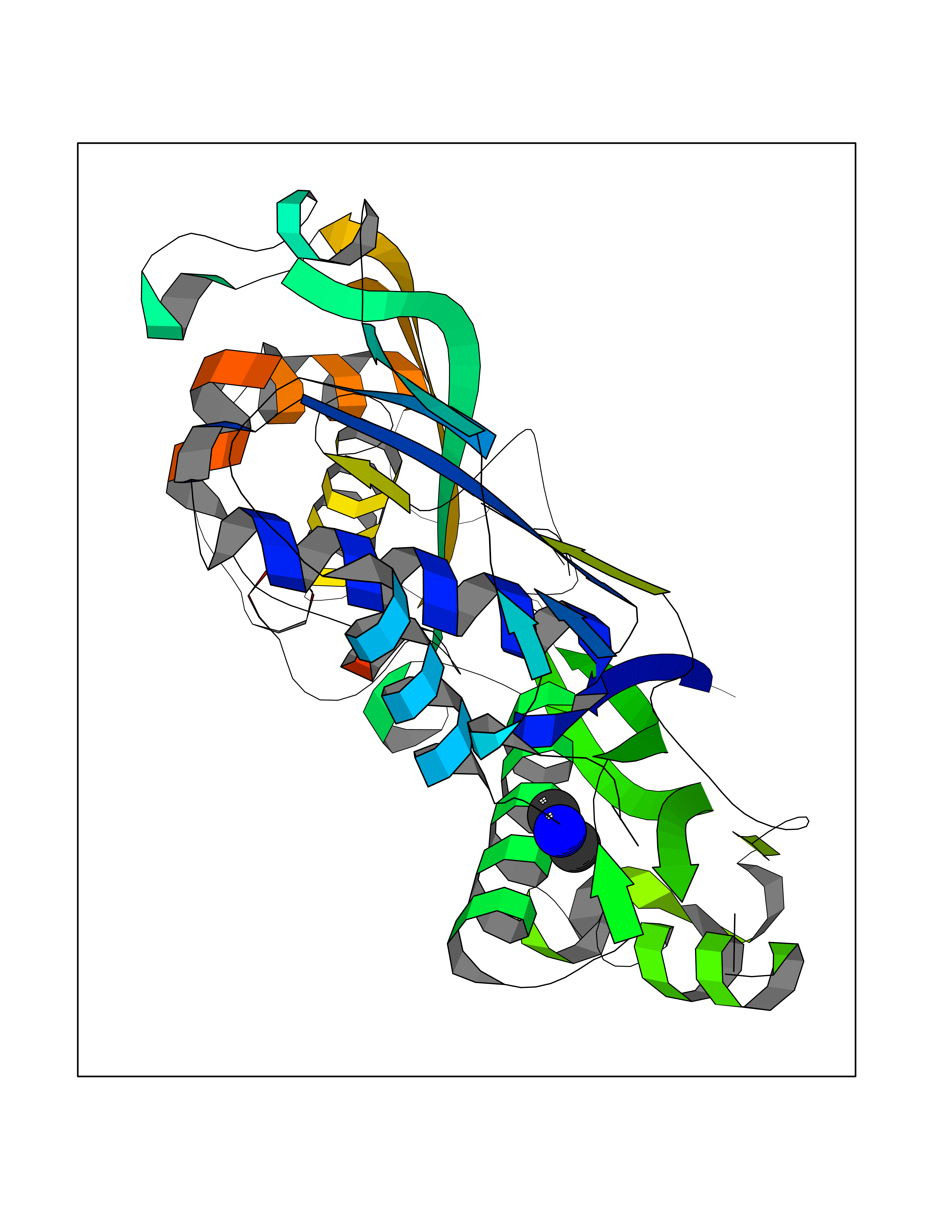 Molscript Map 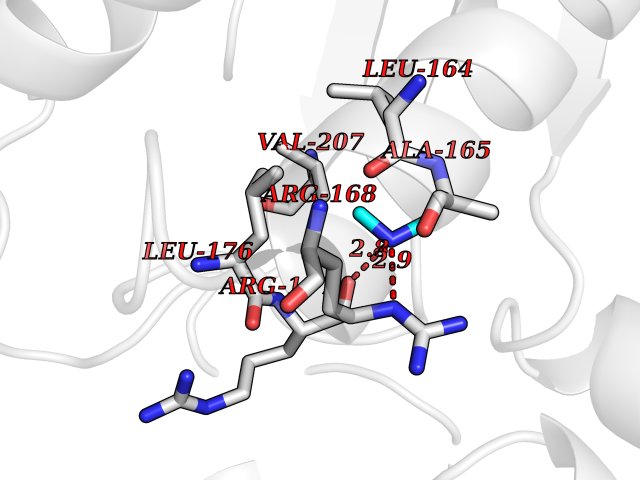 Pymol Map 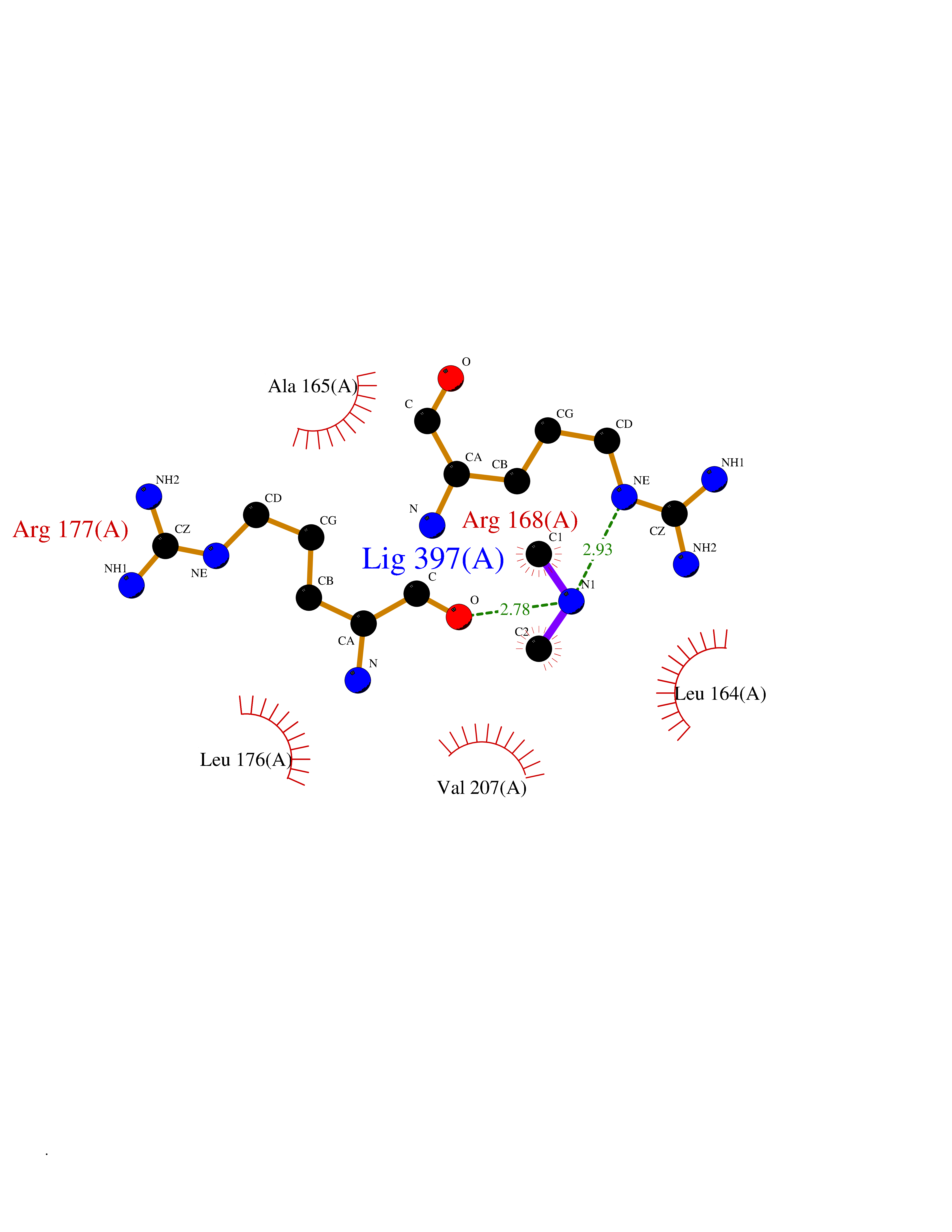 Ligplot Map 3D Binding mode Sequence EGTFLFTSESVGEGHPDKICDQISDAVLDAHLQQDPDAKVACETVAKTGMILLAGEITSRAAVDYQKVVREAVKHIGYDDSSKGFDYKTCNVLVALEQQSPDIAQGVHLDRNEEDIGAGDQGLMFGYATDETEECMPLTIVLAHKLNAKLAELRRNGTLPWLRPDSKTQVTVQYMQDRGAVLPIRVHTIVISVQHDEEVCLDEMRDALKEKVIKAVVPAKYLDEDTIYHLQPSGRFVIGGPQGDAGLTGRKIIVDTYGGWGAHGGGAFSGKDYTKVDRSAAYAARWVAKSLVKGGLCRRVLVQVSYAIGVSHPLSISIFHYGTSQKSERELLEIVKKNFDLRPGVIVRDLDLKKPIYQRTAAYGHFGRDSFPWEVPKKLKY Hydrogen bonds contact Hydrophobic contact | ||||
| 14 | Fms-like tyrosine kinase 3 (FLT-3) | 1RJB | 4.03 | |
Target general information Gen name FLT3 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Stem cell tyrosine kinase 1; STK1; STK-1; Receptor-type tyrosine-protein kinase FLT3; Fetal liver kinase-2; FLT-3; FLK2; FLK-2; FL cytokine receptor; CD135 Protein family Protein kinase superfamily, Tyr protein kinase family, CSF-1/PDGF receptor subfamily Biochemical class Kinase Function Tyrosine-protein kinase that acts as cell-surface receptor for the cytokine FLT3LG and regulates differentiation, proliferation and survival of hematopoietic progenitor cells and of dendritic cells. Promotes phosphorylation of SHC1 and AKT1, and activation of the downstream effector MTOR. Promotes activation of RAS signaling and phosphorylation of downstream kinases, including MAPK1/ERK2 and/or MAPK3/ERK1. Promotes phosphorylation of FES, FER, PTPN6/SHP, PTPN11/SHP-2, PLCG1, and STAT5A and/or STAT5B. Activation of wild-type FLT3 causes only marginal activation of STAT5A or STAT5B. Mutations that cause constitutive kinase activity promote cell proliferation and resistance to apoptosis via the activation of multiple signaling pathways. Related diseases Leukemia, acute myelogenous (AML) [MIM:601626]: A subtype of acute leukemia, a cancer of the white blood cells. AML is a malignant disease of bone marrow characterized by maturational arrest of hematopoietic precursors at an early stage of development. Clonal expansion of myeloid blasts occurs in bone marrow, blood, and other tissue. Myelogenous leukemias develop from changes in cells that normally produce neutrophils, basophils, eosinophils and monocytes. {ECO:0000269|PubMed:11090077, ECO:0000269|PubMed:11290608, ECO:0000269|PubMed:11442493, ECO:0000269|PubMed:14504097, ECO:0000269|PubMed:16266983, ECO:0000269|PubMed:18305215, ECO:0000269|PubMed:8946930, ECO:0000269|PubMed:9737679}. The gene represented in this entry may be involved in disease pathogenesis. Somatic mutations that lead to constitutive activation of FLT3 are frequent in AML patients. These mutations fall into two classes, the most common being in-frame internal tandem duplications of variable length in the juxtamembrane region that disrupt the normal regulation of the kinase activity. Likewise, point mutations in the activation loop of the kinase domain can result in a constitutively activated kinase. Drugs (DrugBank ID) DB12742; DB12267; DB12500; DB12010; DB12141; DB06469; DB06080; DB06595; DB11763; DB09079; DB11697; DB12978; DB08901; DB15822; DB12874; DB00398; DB01268; DB05465; DB11800; DB05014 Interacts with P00519; P42684; P46108; P46109; P06241; Q13322; Q9Y6K9; P06239; P27986; P20936; P43405; Q8R4L0 EC number EC 2.7.10.1 Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Disease variant; Disulfide bond; Endoplasmic reticulum; Glycoprotein; Host-virus interaction; Immunoglobulin domain; Kinase; Membrane; Nucleotide-binding; Phosphoprotein; Proteomics identification; Proto-oncogene; Receptor; Reference proteome; Signal; Transferase; Transmembrane; Transmembrane helix; Tyrosine-protein kinase; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 34209.2 Length 298 Aromaticity 0.14 Instability index 39.68 Isoelectric point 5.57 Charge (pH=7) -4.02 2D Binding mode Binding energy (Kcal/mol) -5.5 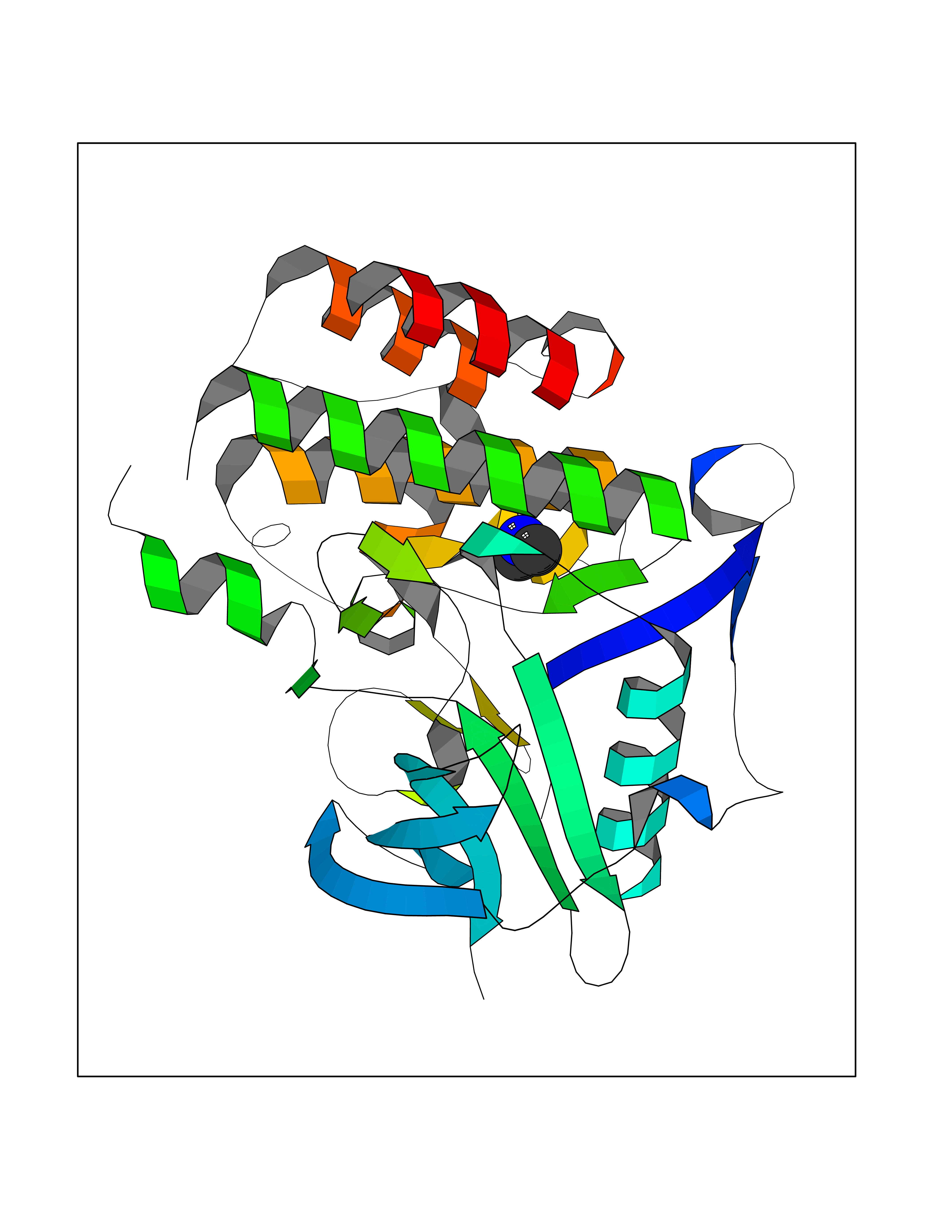 Molscript Map 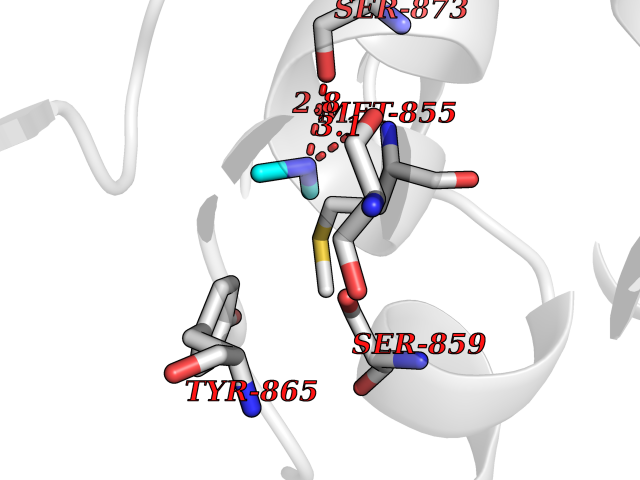 Pymol Map 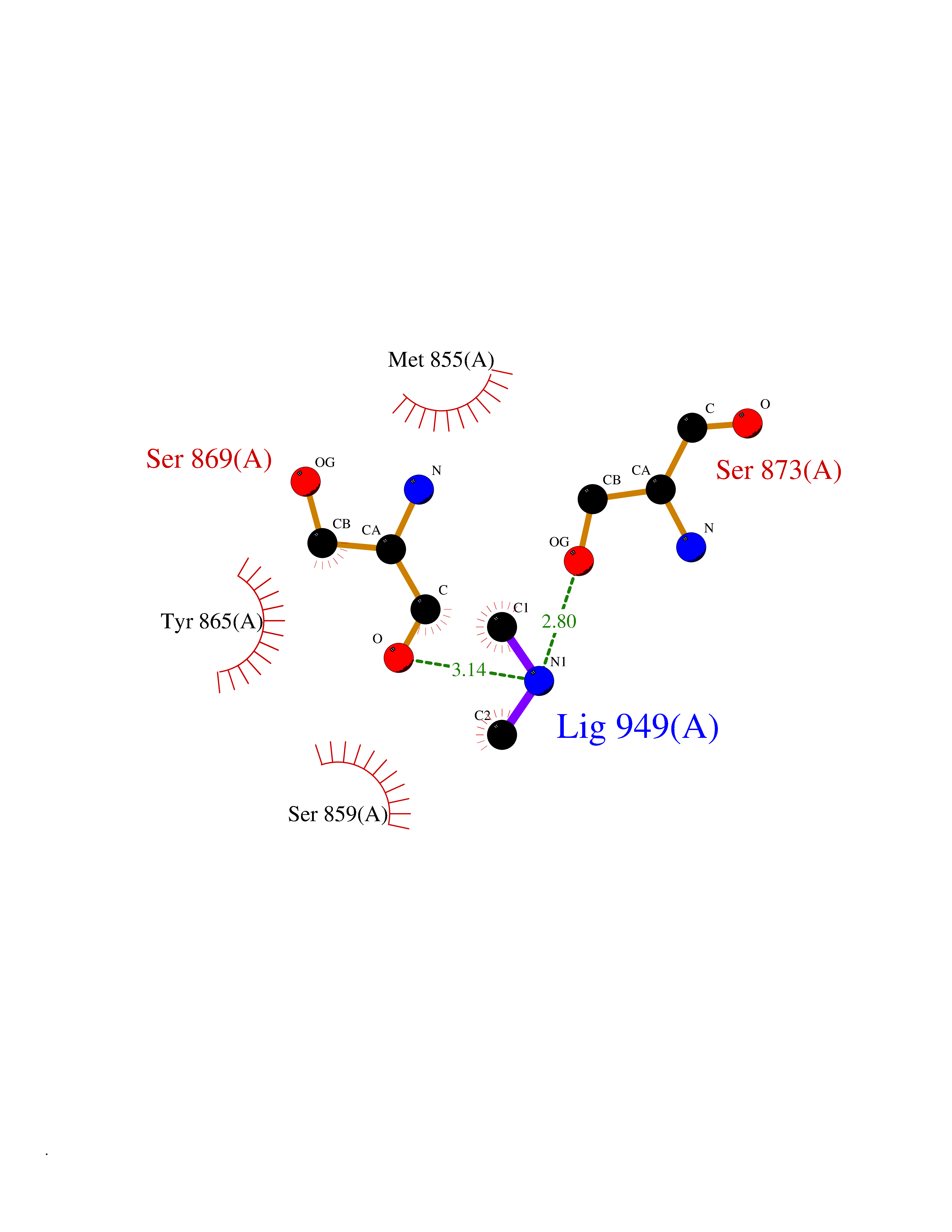 Ligplot Map 3D Binding mode Sequence YESQLQMVQVTGSSDNEYFYVDFREYEYDLKWEFPRENLEFGKVLGSGAFGKVMNATAYGISKTGVSIQVAVKMLKEREALMSELKMMTQLGSHENIVNLLGACTLSGPIYLIFEYCCYGDLLNYLRSKREKFLTFEDLLCFAYQVAKGMEFLEFKSCVHRDLAARNVLVTHGKVVKICDFGLARDIMSDSNYVVRGNARLPVKWMAPESLFEGIYTIKSDVWSYGILLWEIFSLGVNPYPGIPVDANFYKLIQNGFKMDQPFYATEEIYIIMQSCWAFDSRKRPSFPNLTSFLGCQL Hydrogen bonds contact Hydrophobic contact | ||||
| 15 | Carbapenem-hydrolyzing beta-lactamase KPC | 3RXW | 4.03 | |
Target general information Gen name bla Organism Klebsiella pneumoniae Uniprot ID TTD ID NA Synonyms kpc;kpc1 Protein family Class-A beta-lactamase family Biochemical class Hydrolase / hydrolase inhibitor Function Beta-lactamase activity. Related diseases Cornelia de Lange syndrome 5 (CDLS5) [MIM:300882]: A form of Cornelia de Lange syndrome, a clinically heterogeneous developmental disorder associated with malformations affecting multiple systems. It is characterized by facial dysmorphisms, abnormal hands and feet, growth delay, cognitive retardation, hirsutism, gastroesophageal dysfunction and cardiac, ophthalmologic and genitourinary anomalies. {ECO:0000269|PubMed:22885700, ECO:0000269|PubMed:22889856}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB09060; DB12107 Interacts with NA EC number 3.5.2.6 Uniprot keywords 3D-structure; Antibiotic resistance; Hydrolase; Plasmid; Signal Protein physicochemical properties Chain ID A Molecular weight (Da) 27965.2 Length 264 Aromaticity 0.08 Instability index 26.27 Isoelectric point 6.14 Charge (pH=7) -1.42 2D Binding mode Binding energy (Kcal/mol) -5.49  Molscript Map 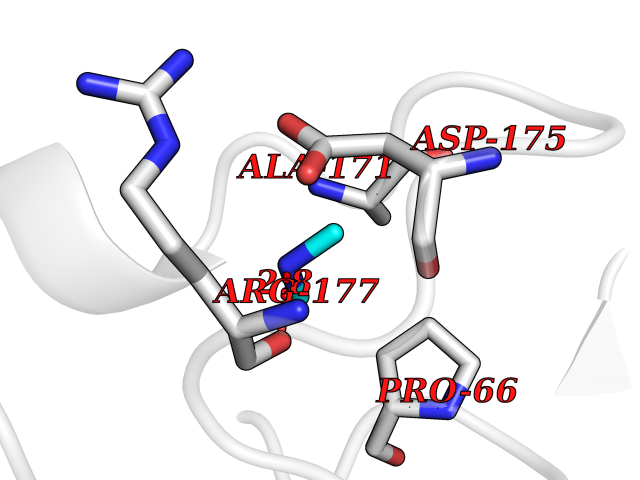 Pymol Map 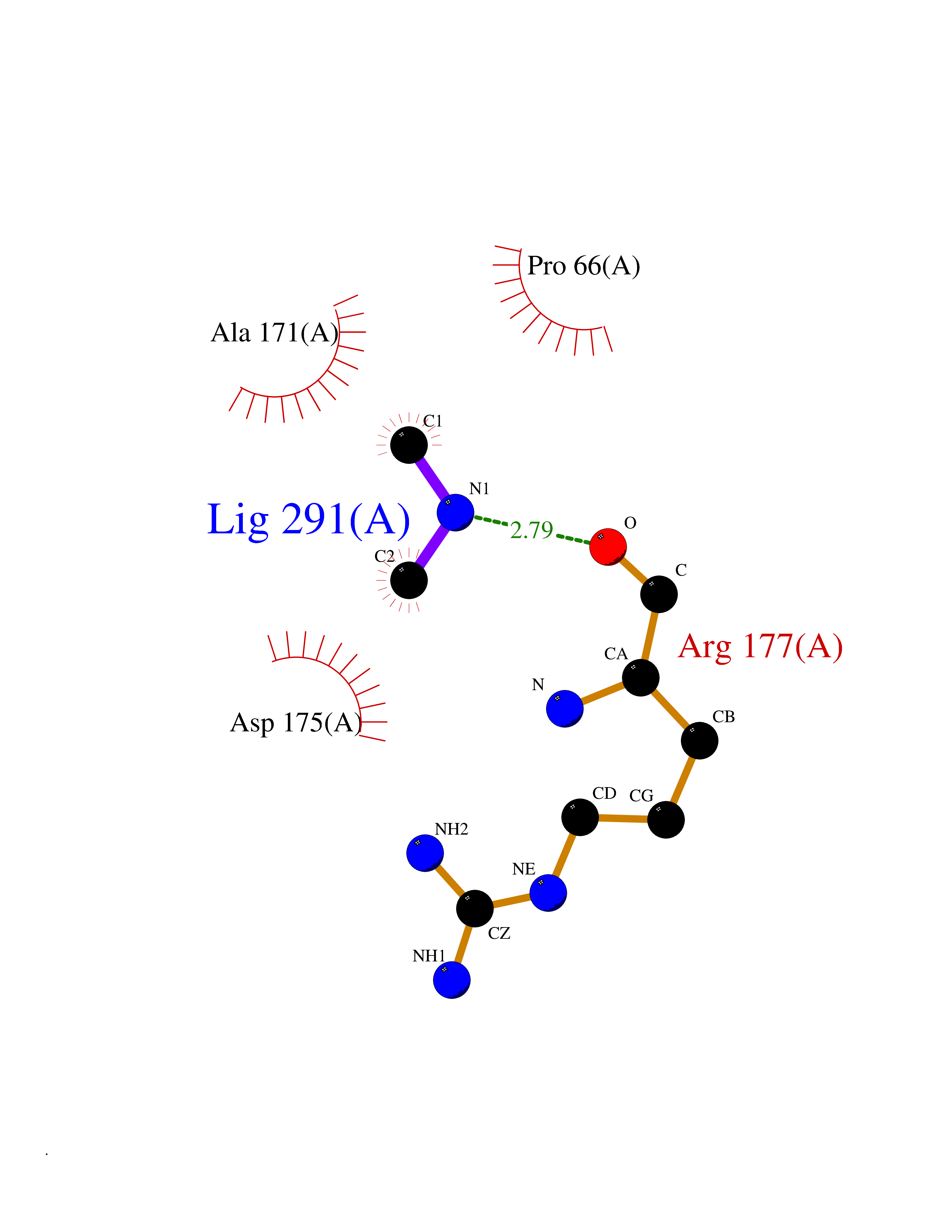 Ligplot Map 3D Binding mode Sequence TNLVAEPFAKLEQDFGGSIGVYAMDTGSGATVSYRAEERFPLCSSFKGFLAAAVLARSQQQAGLLDTPIRYGKNALVPWSPISEKYLTTGMTVAELSAAAVQYSDNAAANLLLKELGGPAGLTAFMRSIGDTTFRLDRWELELNSAIPGDARDTSSPRAVTESLQKLTLGSALAAPQRQQFVDWLKGNTTGNHRIRAAVPADWAVGDKTGTCGVYGTANDYAVVWPTGRAPIVLAVYTRAPNKDDKHSEAVIAAAARLALEGLG Hydrogen bonds contact Hydrophobic contact | ||||
| 16 | Lysine-specific demethylase 7A (KDM7A) | 3KVB | 4.03 | |
Target general information Gen name KDM7A Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Lysine-specific demethylase 7; KIAA1718; KDM7; JmjC domain-containing histone demethylation protein 1D; JHDM1D Protein family JHDM1 histone demethylase family, JHDM1D subfamily Biochemical class NA Function Histone demethylase required for brain development. Specifically demethylates dimethylated 'Lys-9' and 'Lys-27' (H3K9me2 and H3K27me2, respectively) of histone H3 and monomethylated histone H4 'Lys-20' residue (H4K20Me1), thereby playing a central role in histone code. Specifically binds trimethylated 'Lys-4' of histone H3 (H3K4me3), affecting histone demethylase specificity: in presence of H3K4me3, it has no demethylase activity toward H3K9me2, while it has high activity toward H3K27me2. Demethylates H3K9me2 in absence of H3K4me3. Has activity toward H4K20Me1 only when nucleosome is used as a substrate and when not histone octamer is used as substrate. Related diseases MUC1/CA 15-3 is used as a serological clinical marker of breast cancer to monitor response to breast cancer treatment and disease recurrence (PubMed:20816948). Decreased levels over time may be indicative of a positive response to treatment. Conversely, increased levels may indicate disease progression. At an early stage disease, only 21% of patients exhibit high MUC1/CA 15-3 levels, that is why CA 15-3 is not a useful screening test. Most antibodies target the highly immunodominant core peptide domain of 20 amino acid (APDTRPAPGSTAPPAHGVTS) tandem repeats. Some antibodies recognize glycosylated epitopes. {ECO:0000269|PubMed:20816948}.; DISEASE: Tubulointerstitial kidney disease, autosomal dominant, 2 (ADTKD2) [MIM:174000]: A form of autosomal dominant tubulointerstitial kidney disease, a genetically heterogeneous disorder characterized by slowly progressive loss of kidney function, bland urinary sediment, hyperuricemia, absent or mildly increased albuminuria, lack of severe hypertension during the early stages, and normal or small kidneys on ultrasound. Renal histology shows variable abnormalities including interstitial fibrosis with tubular atrophy, microcystic dilatation of the tubules, thickening of tubular basement membranes, medullary cysts, and secondary glomerulosclerotic or glomerulocystic changes with abnormal glomerular tufting. There is significant variability, as well as incomplete penetrance. {ECO:0000269|PubMed:23396133}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number EC 1.14.11.- Uniprot keywords 3D-structure; Alternative splicing; Chromatin regulator; Dioxygenase; Iron; Metal-binding; Neurogenesis; Nucleus; Oxidoreductase; Phosphoprotein; Proteomics identification; Reference proteome; Transcription; Transcription regulation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A Molecular weight (Da) 42365.5 Length 366 Aromaticity 0.12 Instability index 41.95 Isoelectric point 6.3 Charge (pH=7) -3.09 2D Binding mode Binding energy (Kcal/mol) -5.5  Molscript Map 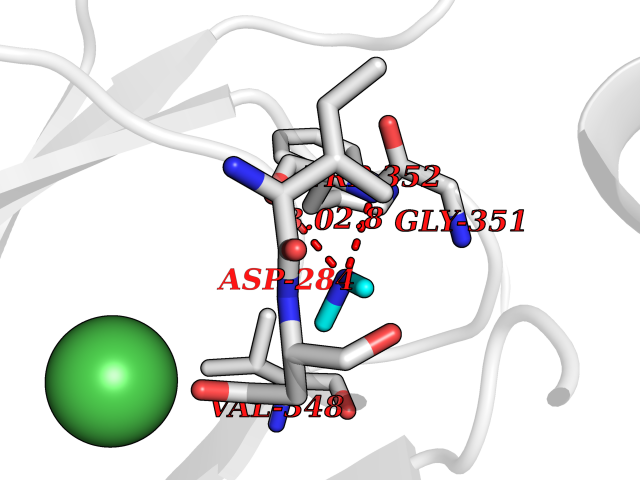 Pymol Map 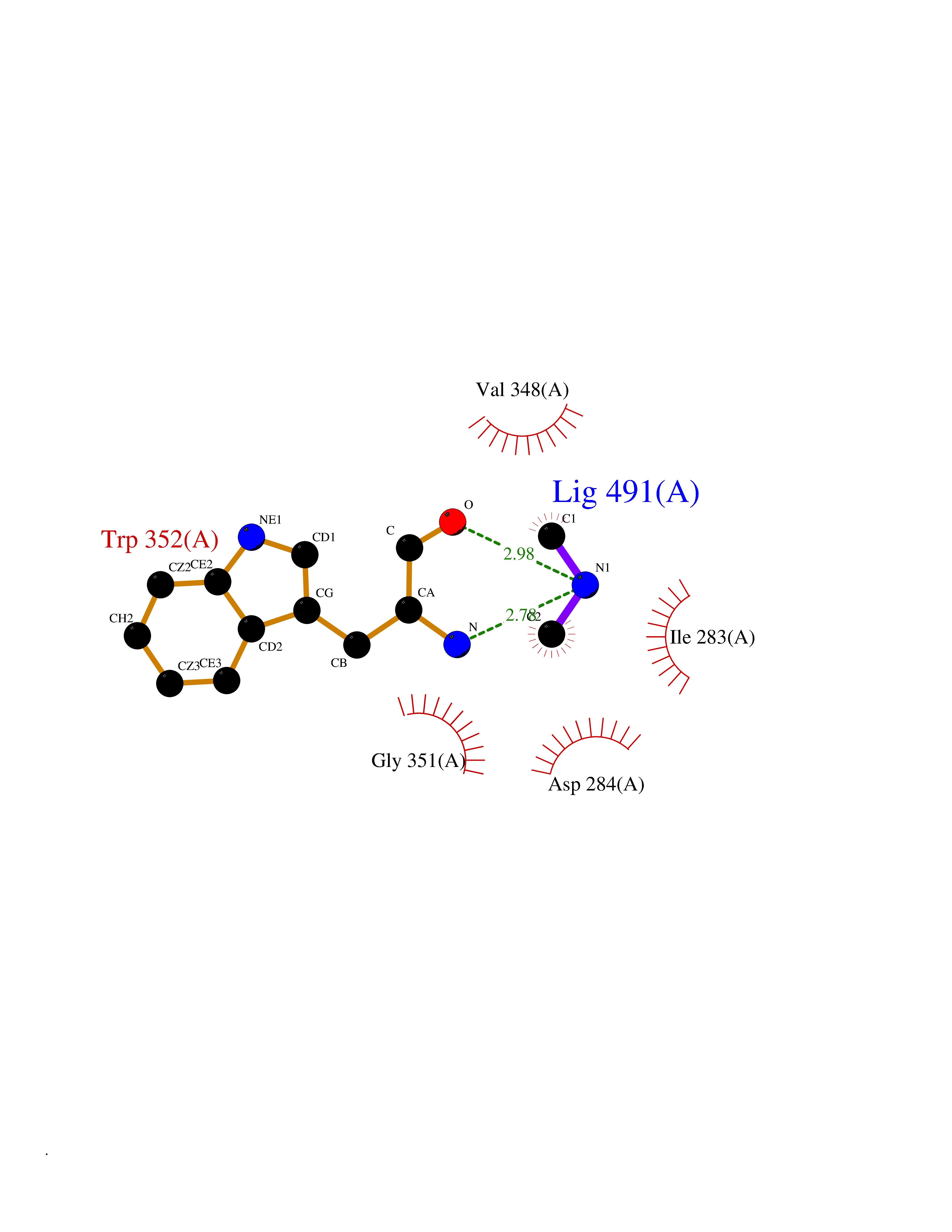 Ligplot Map 3D Binding mode Sequence PVQAGTRTFIKELRSRVFPSADEIIIKMHGSQLTQRYLEKHGFDVPIMVPKLDDLGLRLPSPTFSVMDVERYVGGDKVIDVIDVARQADSKMTLHNYVKYFMNPNRPKVLNVISLEFSDTKMSELVEVPDIAKKLSWVENYWPDDSVFPKPFVQKYCLMGVQDSYTDFHIDFGGTSVWYHVLWGEKIFYLIKPTDENLARYESWSSSVTQSEVFFGDKVDKCYKCVVKQGHTLFVPTGWIHAVLTSQDCMAFGGNFLHNLNIGMQLRCYEMEKRLKTPDLFKFPFFEAICWFVAKNLLETLKELREDGFQPQTYLVQGVKALHTALKLWMKKELVSEHAFEIPDNVRPGHLIKELSKVIRAIEEEN Hydrogen bonds contact Hydrophobic contact | ||||
| 17 | Plasmodium DOXP reductoisomerase (Malaria DXR) | 3AU9 | 4.03 | |
Target general information Gen name Malaria DXR Organism Plasmodium falciparum (isolate HB3) Uniprot ID TTD ID Synonyms IspC; DXR; DXP reductoisomerase; DOXP reductoisomerase; 2-C-Methyl-d-erythritol 4-phosphate synthase; 1-deoxyxylulose-5-phosphate reductoisomerase Protein family DXR family Biochemical class Short-chain dehydrogenases reductase Function Catalyzes the NADP-dependent rearrangement and reduction of 1-deoxy-D-xylulose-5-phosphate (DXP) to 2-C-methyl-D-erythritol 4-phosphate (MEP). Related diseases Ichthyosis, congenital, autosomal recessive 11 (ARCI11) [MIM:602400]: A form of autosomal recessive congenital ichthyosis, a disorder of keratinization with abnormal differentiation and desquamation of the epidermis, resulting in abnormal skin scaling over the whole body. The main skin phenotypes are lamellar ichthyosis (LI) and non-bullous congenital ichthyosiform erythroderma (NCIE), although phenotypic overlap within the same patient or among patients from the same family can occur. Lamellar ichthyosis is a condition often associated with an embedment in a collodion-like membrane at birth; skin scales later develop, covering the entire body surface. Non-bullous congenital ichthyosiform erythroderma characterized by fine whitish scaling on an erythrodermal background; larger brownish scales are present on the buttocks, neck and legs. {ECO:0000269|PubMed:17273967, ECO:0000269|PubMed:18843291}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number EC 1.1.1.267 Uniprot keywords 3D-structure; Apicoplast; Isoprene biosynthesis; Magnesium; Manganese; Metal-binding; NADP; Oxidoreductase; Plastid; Transit peptide Protein physicochemical properties Chain ID A Molecular weight (Da) 46644.4 Length 410 Aromaticity 0.09 Instability index 36.77 Isoelectric point 6.95 Charge (pH=7) -0.14 2D Binding mode Binding energy (Kcal/mol) -5.49 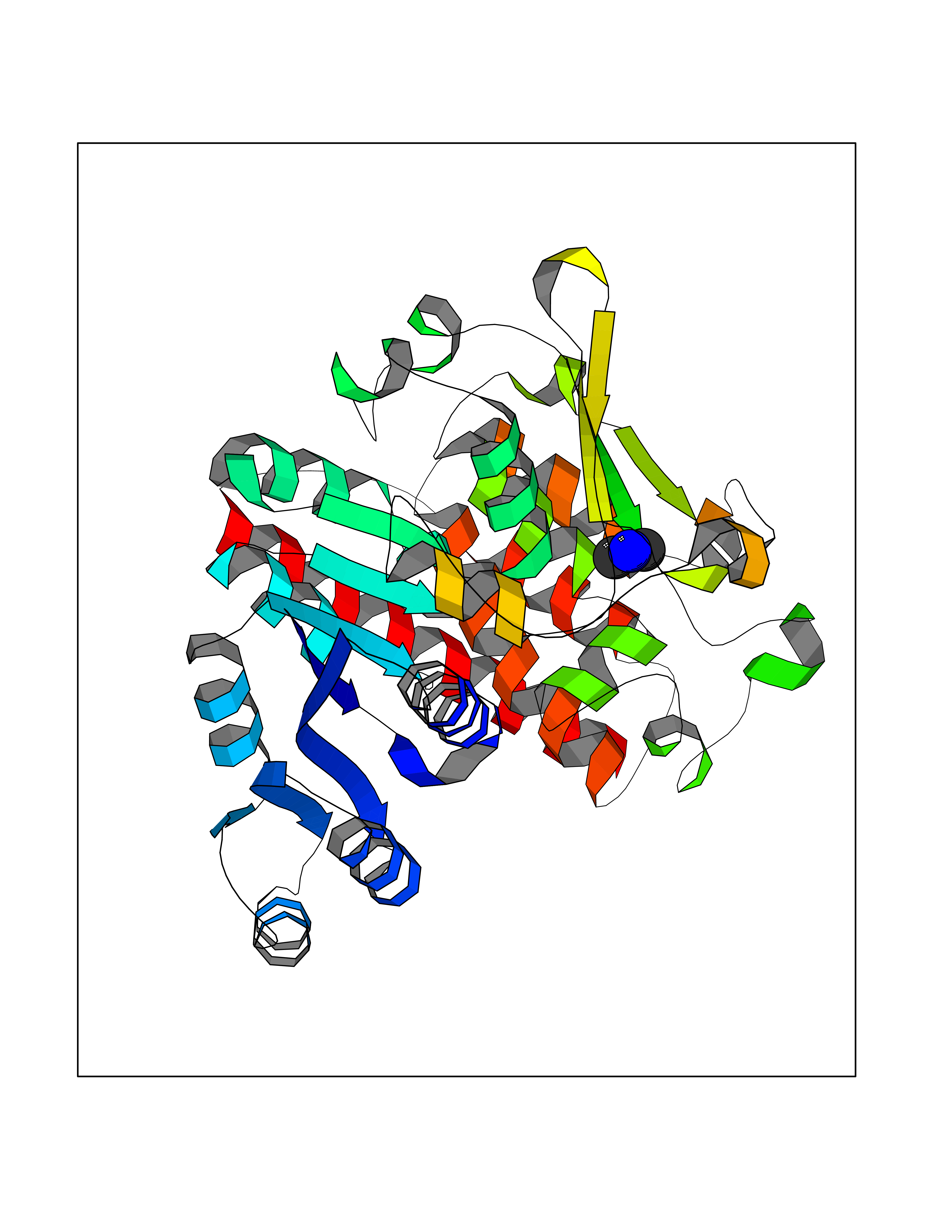 Molscript Map 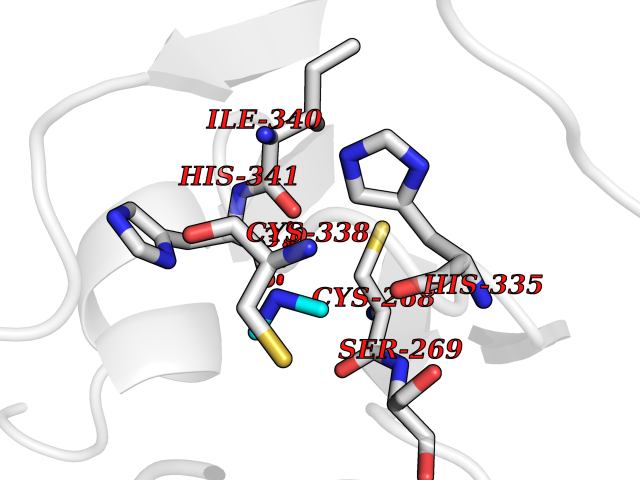 Pymol Map 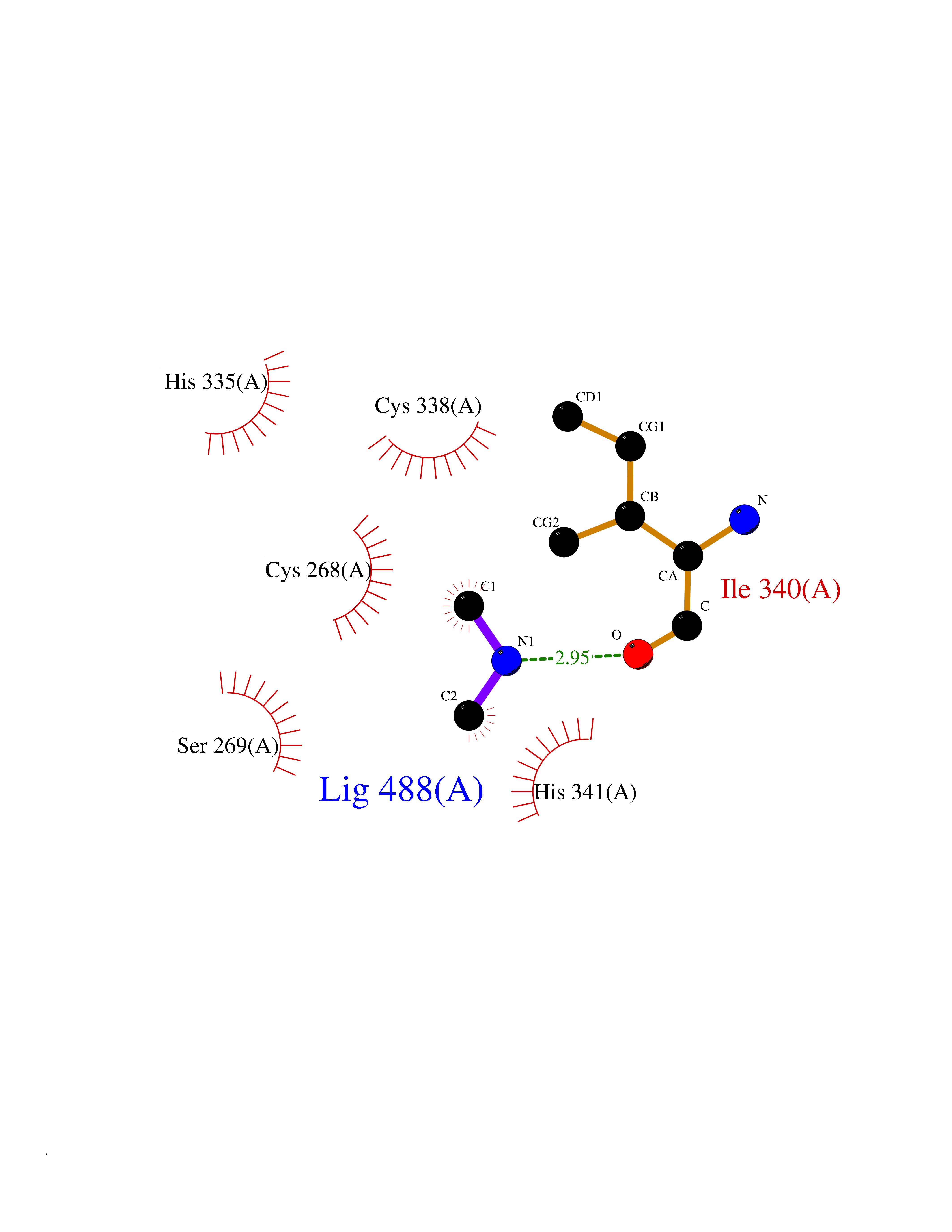 Ligplot Map 3D Binding mode Sequence PINVAIFGSTGSIGTNALNIIRECNKIENVFNVKALYVNKSVNELYEQAREFLPEYLCIHDKSVYEELKELVKNIKDYKPIILCGDEGMKEICSSNSIDKIVIGIDSFQGLYSTMYAIMNNKIVALANKESIVSAGFFLKKLLNIHKNAKIIPVDSEHSAIFQCLDNNKVLKTKCLQDNFSKINNINKIFLCSSGGPFQNLTMDELKNVTSENALKHPKWKMGKKITIDSATMMNKGLEVIETHFLFDVDYNDIEVIVHKECIIHSCVEFIDKSVISQMYYPDMQIPILYSLTWPDRIKTNLKPLDLAQVSTLTFHKPSLEHFPCIKLAYQAGIKGNFYPTVLNASNEIANNLFLNNKIKYFDISSIISQVLESFNSQKVSENSEDLMKQILQIHSWAKDKATDIYNKHN Hydrogen bonds contact Hydrophobic contact | ||||
| 18 | Protoporphyrinogen oxidase (PPOX) | 3NKS | 4.03 | |
Target general information Gen name PPOX Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms PPO Protein family Protoporphyrinogen/coproporphyrinogen oxidase family, Protoporphyrinogen oxidase subfamily Biochemical class NA Function Catalyzes the 6-electron oxidation of protoporphyrinogen-IX to form protoporphyrin-IX. Related diseases Variegate porphyria (VP) [MIM:176200]: A form of porphyria. Porphyrias are inherited defects in the biosynthesis of heme, resulting in the accumulation and increased excretion of porphyrins or porphyrin precursors. They are classified as erythropoietic or hepatic, depending on whether the enzyme deficiency occurs in red blood cells or in the liver. Variegate porphyria is an acute hepatic form characterized by partial reduction of protoporphyrinogen oxidase activity, increased photosensitivity, skin blistering and scarring of sun-exposed areas, skin hyperpigmentation, abdominal pain, and neuropsychiatric symptoms. High fecal levels of protoporphyrin and coproporphyrin, increased urine uroporphyrins and iron overload are typical markers of the disease. Inheritance is autosomal dominant with incomplete penetrance. {ECO:0000269|PubMed:10486317, ECO:0000269|PubMed:11074242, ECO:0000269|PubMed:11102990, ECO:0000269|PubMed:11348478, ECO:0000269|PubMed:11350188, ECO:0000269|PubMed:11474578, ECO:0000269|PubMed:12380696, ECO:0000269|PubMed:12655566, ECO:0000269|PubMed:12859407, ECO:0000269|PubMed:12922165, ECO:0000269|PubMed:14669009, ECO:0000269|PubMed:16433813, ECO:0000269|PubMed:16621625, ECO:0000269|PubMed:16922948, ECO:0000269|PubMed:16947091, ECO:0000269|PubMed:18350656, ECO:0000269|PubMed:18570668, ECO:0000269|PubMed:19320019, ECO:0000269|PubMed:21048046, ECO:0000269|PubMed:23430901, ECO:0000269|PubMed:23467411, ECO:0000269|PubMed:24073655, ECO:0000269|PubMed:8817334, ECO:0000269|PubMed:8852667, ECO:0000269|PubMed:9763307}. The disease is caused by variants affecting the gene represented in this entry. Mutations leading to severe PPOX deficiency cause the rare homozygous variant form of VP. Missense mutations that preserve 10%-25% of wild-type activity may not cause clinically overt VP in heterozygotes (PubMed:9811936). Mutations with intermediate effect on catalytic activity may cause VP, but with a low clinical penetrance (PubMed:10486317). {ECO:0000269|PubMed:10486317, ECO:0000269|PubMed:9811936}.; DISEASE: Variegate porphyria, childhood-onset (VPCO) [MIM:620483]: An autosomal recessive form of variegate porphyria, a disorder of heme biosynthesis that results from diminished activity of protoporphyrinogen oxidase. VPCO is characterized by severe protoporphyrinogen oxidase deficiency, onset of photosensitization by porphyrins in early childhood, skin scarring and hyperpigmentation, and skeletal abnormalities of the hand. Additional variable features are short stature, impaired intellectual development, and seizures. VPCO patients rarely experience acute neuropsychiatric or abdominal attacks. {ECO:0000269|PubMed:10870850, ECO:0000269|PubMed:11286631, ECO:0000269|PubMed:33159949, ECO:0000269|PubMed:8673113, ECO:0000269|PubMed:9541112, ECO:0000269|PubMed:9811936}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number EC 1.3.3.4 Uniprot keywords 3D-structure; Disease variant; FAD; Flavoprotein; Heme biosynthesis; Membrane; Mitochondrion; Mitochondrion inner membrane; Oxidoreductase; Porphyrin biosynthesis; Proteomics identification; Reference proteome Protein physicochemical properties Chain ID A Molecular weight (Da) 49470.2 Length 465 Aromaticity 0.05 Instability index 48.23 Isoelectric point 7.8 Charge (pH=7) 1.98 2D Binding mode Binding energy (Kcal/mol) -5.5 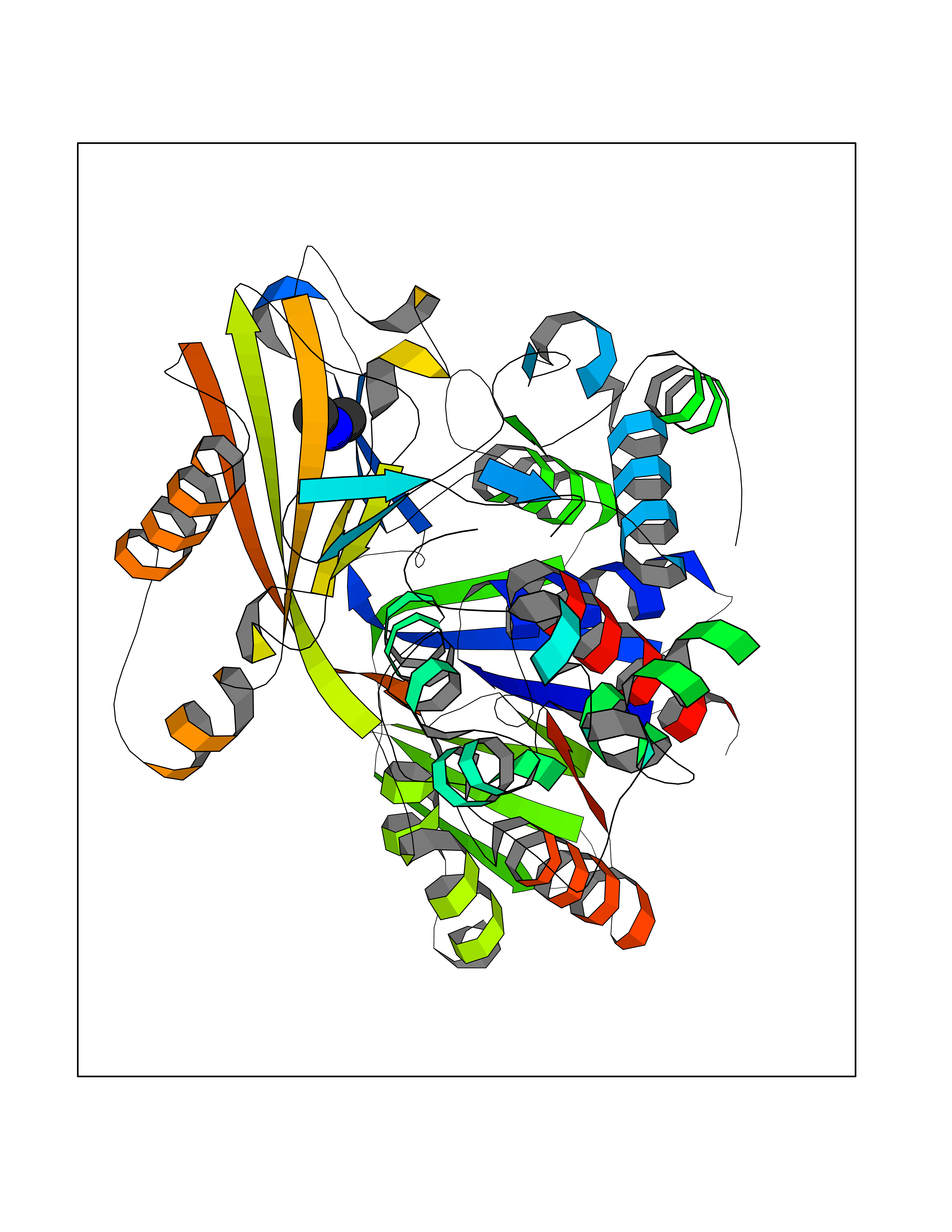 Molscript Map 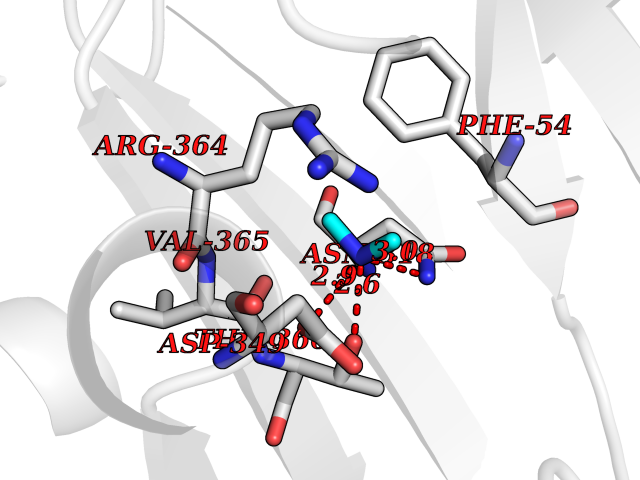 Pymol Map 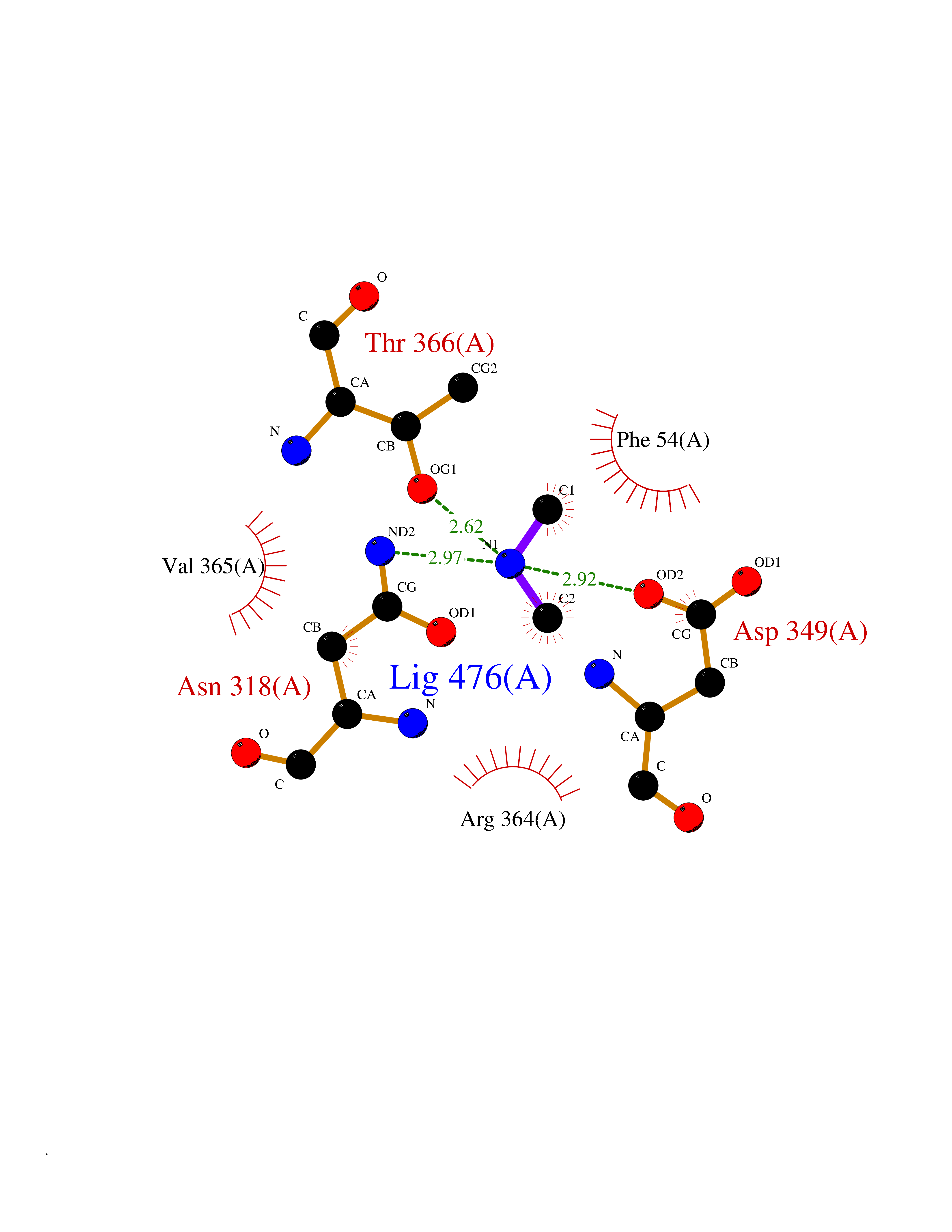 Ligplot Map 3D Binding mode Sequence GRTVVVLGGGISGLAASYHLSRAPCPPKVVLVESSERLGGWIRSVRGPNGAIFELGPRGIRPAGALGARTLLLVSELGLDSEVLPVRGDHPAAQNRFLYVGGALHALPTGLRGPSPPFSKPLFWAGLRELTKPRGKEPDETVHSFAQRRLGPEVASLAMDSLCRGVFAGNSRELSIRSCFPSLFQAEQTHRSILLGLLLGQPDSALIRQALAERWSQWSLRGGLEMLPQALETHLTSRGVSVLRGQPVCGLSLQAEGRWKVSLRDSSLEADHVISAIPASVLSELLPAEAAPLARALSAITAVSVAVVNLQYQGAHLPVQGFGHLVPSSEDPGVLGIVYDSVAFPEQDGSPPGLRVTVMLGGSWLQTLEASGCVLSQELFQQRAQEAAATQLGLKEMPSHCLVHLHKNCIPQYTLGHWQKLESARQFLTAHRLPLTLAGASYEGVAVNDCIESGRQAAVSVLGTE Hydrogen bonds contact Hydrophobic contact | ||||
| 19 | Deubiquitinating enzyme 1 (USP1) | 7ZH4 | 4.03 | |
Target general information Gen name USP1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms hUBP; Ubiquitin-specific-processing protease 1; Ubiquitin thioesterase 1; Ubiquitin carboxyl-terminal hydrolase 1 Protein family Peptidase C19 family Biochemical class Peptidase Function Involved in PCNA-mediated translesion synthesis (TLS) by deubiquitinating monoubiquitinated PCNA. Has almost no deubiquitinating activity by itself and requires the interaction with WDR48 to have a high activity. Negative regulator of DNA damage repair which specifically deubiquitinates monoubiquitinated FANCD2. Related diseases Brachydactyly A2 (BDA2) [MIM:112600]: A form of brachydactyly. Brachydactyly defines a group of inherited malformations characterized by shortening of the digits due to abnormal development of the phalanges and/or the metacarpals. In brachydactyly type A2 shortening of the middle phalanges is confined to the index finger and the second toe, all other digits being more or less normal. Because of a rhomboid or triangular shape of the affected middle phalanx, the end of the second finger usually deviates radially. {ECO:0000269|PubMed:19327734, ECO:0000269|PubMed:21357617}. The gene represented in this entry is involved in disease pathogenesis. Duplications of a cis-regulatory element located approximately 110 kb downstream of BMP2 have been found in BDA2 families. They likely cause altered BMP2 expression with pathological consequences. {ECO:0000269|PubMed:19327734, ECO:0000269|PubMed:21357617}.; DISEASE: Short stature, facial dysmorphism, and skeletal anomalies with or without cardiac anomalies 1 (SSFSC1) [MIM:617877]: An autosomal dominant disorder characterized by short stature, facial dysmorphism, skeletal anomalies, and variable cardiac defects. Distinctive facial features include midface retrusion, short upturned nose, long philtrum, high-arched or cleft palate, and variable degrees of micrognathia and dental crowding. Skeletal anomalies include patterning defects of the axial skeleton, characterized by 11 pairs of ribs and brachydactyly of the fifth ray. Congenital heart defects are variably observed and appear to involve primarily the cardiac outflow tract. {ECO:0000269|PubMed:29198724}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q8TAF3; Q8TAF3-1 EC number EC 3.4.19.12 Uniprot keywords 3D-structure; Autocatalytic cleavage; DNA damage; DNA repair; Hydrolase; Nucleus; Phosphoprotein; Protease; Proteomics identification; Reference proteome; Thiol protease; Ubl conjugation; Ubl conjugation pathway Protein physicochemical properties Chain ID D Molecular weight (Da) 32426 Length 285 Aromaticity 0.1 Instability index 50.73 Isoelectric point 5.85 Charge (pH=7) -4.67 2D Binding mode Binding energy (Kcal/mol) -5.49 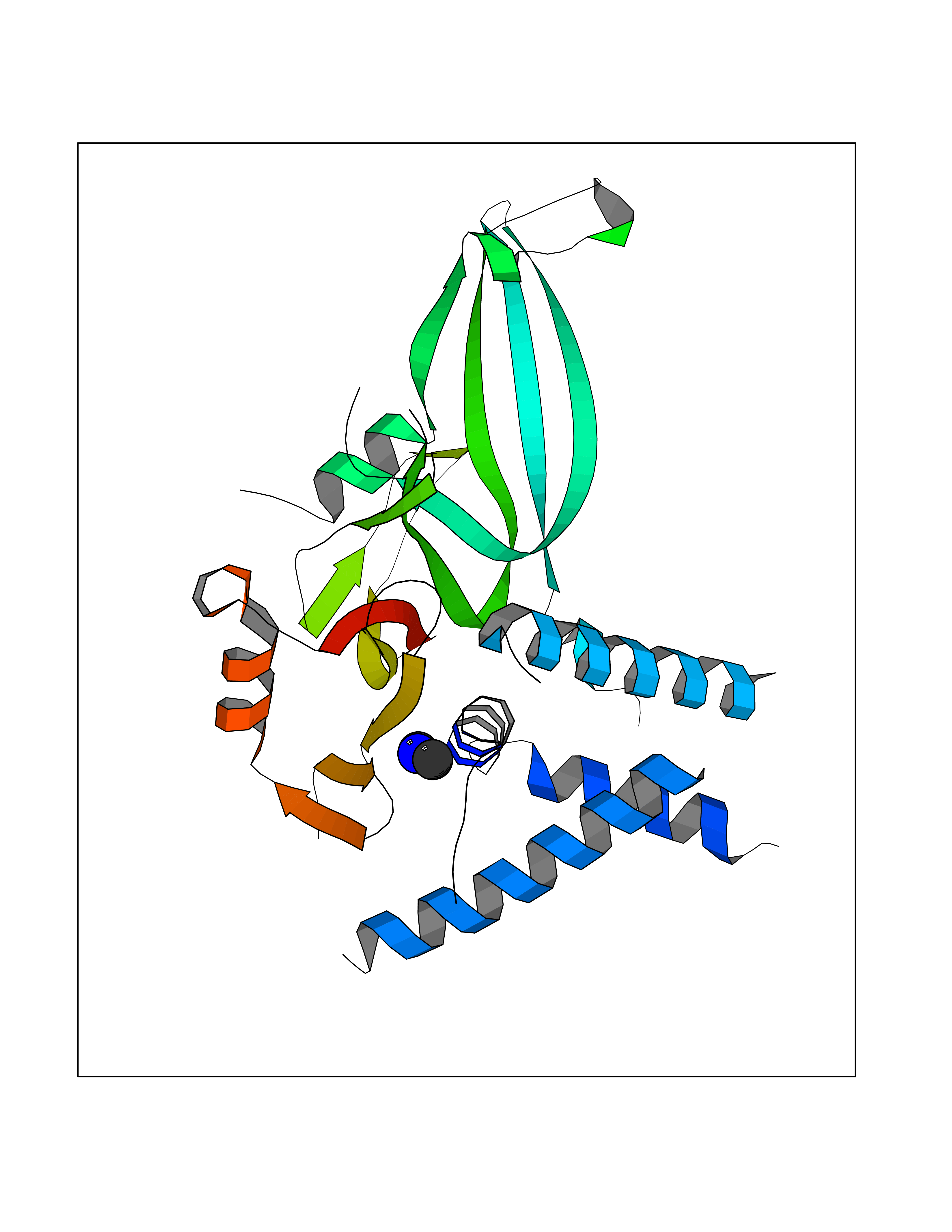 Molscript Map 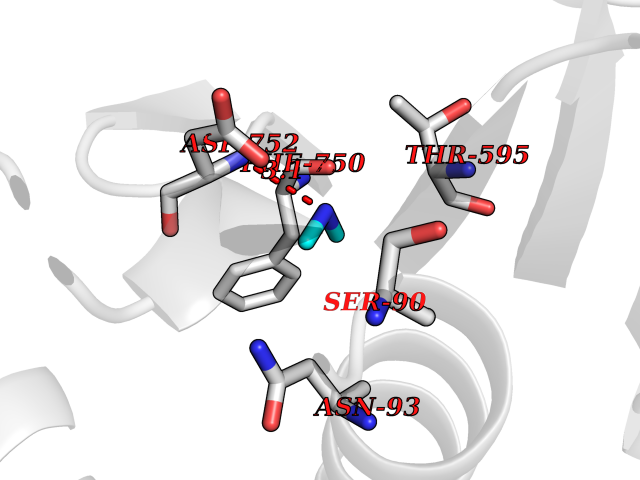 Pymol Map 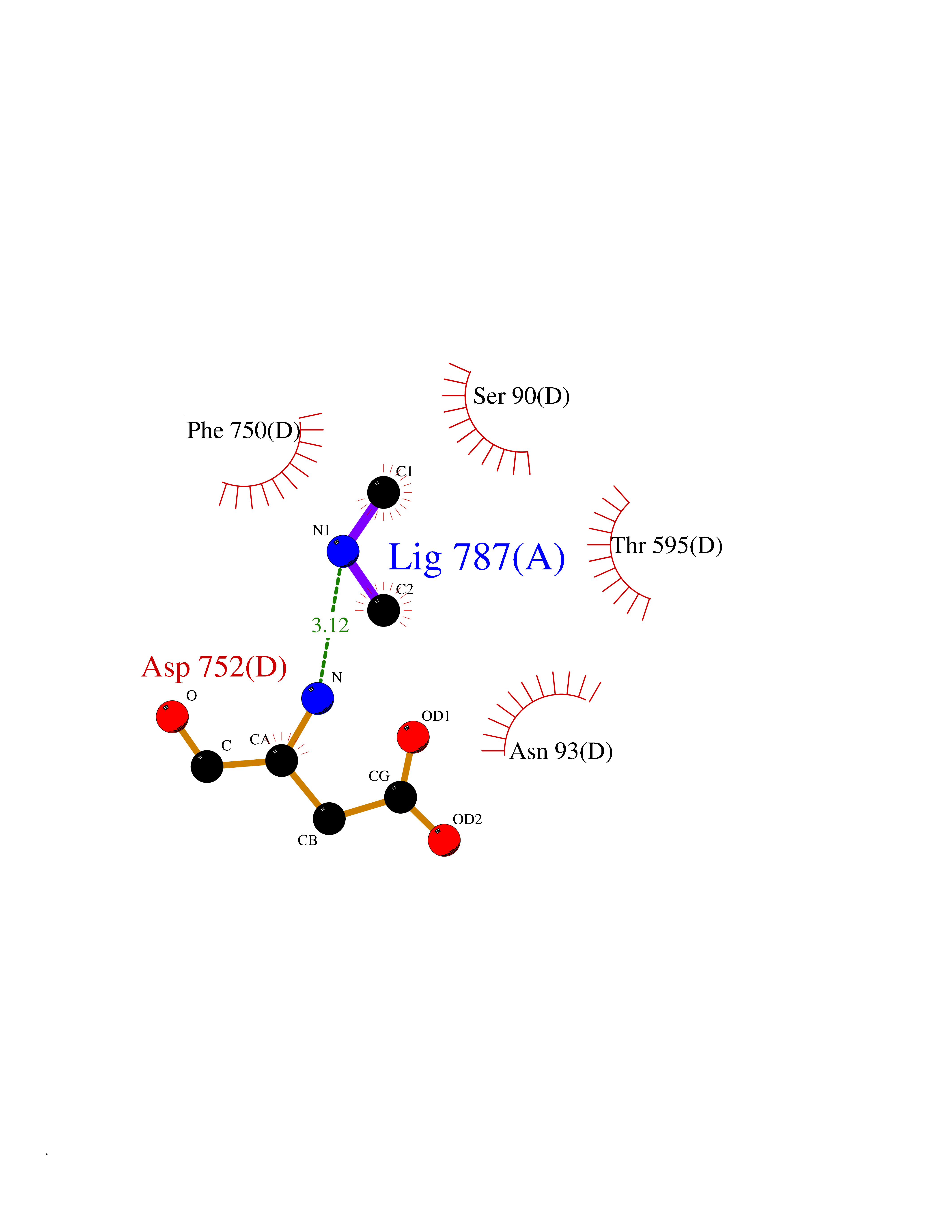 Ligplot Map 3D Binding mode Sequence GLNNLGNTSYLNSILQVLYFCPGFKSGVKHLFNIISRKKYELICSLQSLIISVEQLQASFLLNPLQHDAQEVLQCILGNIQETCQLLKKGFELVEKLFQGQLVLRTRCLECESLTERREDFQDISVPVQEDMKTLRWAISQFASVERIVGEDKYFCENCHHYTEAERSLLFDKMPEVITIHLKCFAASGLSKINTPLLTPLKLSLEEWSTKPTNDSYGLFAVVMHSGITISSGHYTASVKVTYEGKWLLFDDSEVKVTEEKDFLNSLSPSTSPTSTPYLLFYKKL Hydrogen bonds contact Hydrophobic contact | ||||
| 20 | Plasma kallikrein (KLKB1) | 6T7P | 4.03 | |
Target general information Gen name KLKB1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Plasma prekallikrein; Plasma kallikrein light chain; Plasma kallikrein heavy chain; PKK; Kininogenin; KLK3; Fletcher factor Protein family Peptidase S1 family, Plasma kallikrein subfamily Biochemical class Peptidase Function It activates, in a reciprocal reaction, factor XII after its binding to a negatively charged surface. It also releases bradykinin from HMW kininogen and may also play a role in the renin-angiotensin system by converting prorenin into renin. The enzyme cleaves Lys-Arg and Arg-Ser bonds. Related diseases Prekallikrein deficiency (PKKD) [MIM:612423]: An autosomal recessive condition characterized by a clotting defect due to prolongation of activated partial thromboplastin time. Affected individuals are clinically asymptomatic. {ECO:0000269|PubMed:14652634, ECO:0000269|PubMed:17598838, ECO:0000269|PubMed:34847617}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB15982; DB09228; DB05311; DB12831; DB06404; DB14597; DB01593; DB14487; DB14533; DB14548 Interacts with Q9UI10; O00746; C9J082; O14744; Q8TAS3; O00233; Q8IYM2; Q9UMY4; O43493-5; Q8NFB2; Q8N0U8 EC number EC 3.4.21.34 Uniprot keywords 3D-structure; Blood coagulation; Direct protein sequencing; Disease variant; Disulfide bond; Fibrinolysis; Glycoprotein; Hemostasis; Hydrolase; Inflammatory response; Protease; Proteomics identification; Reference proteome; Repeat; Secreted; Serine protease; Signal; Zymogen Protein physicochemical properties Chain ID A Molecular weight (Da) 26696.2 Length 237 Aromaticity 0.1 Instability index 34.33 Isoelectric point 8.07 Charge (pH=7) 2.21 2D Binding mode Binding energy (Kcal/mol) -5.5 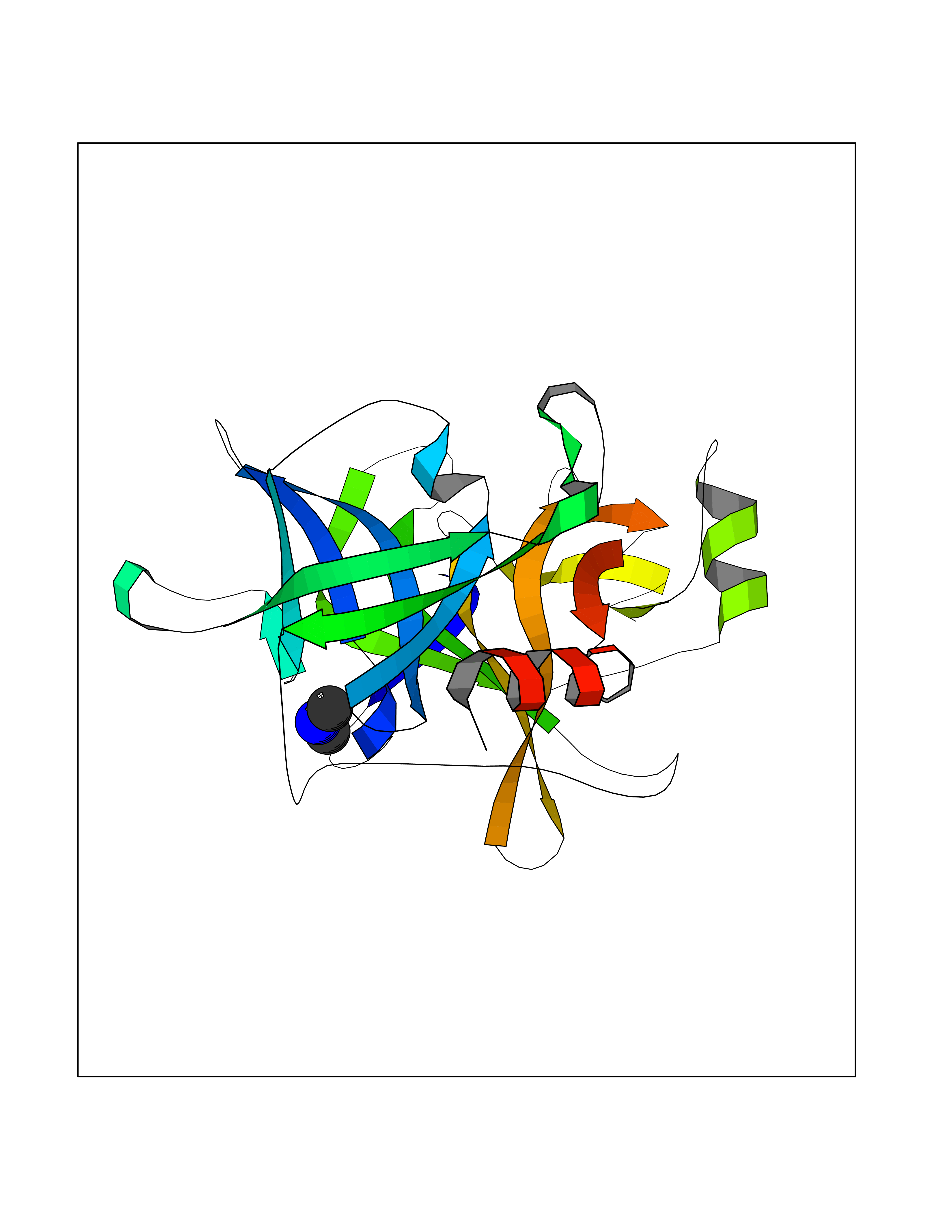 Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence IVGGTNSSWGEWPWQVSLQVKLTAQRHLCGGSLIGHQWVLTAAHCFDGLPLQDVWRIYSGILNLSDITKDTPFSQIKEIIIHQNYKVSEGNHDIALIKLQAPLNYTEFQKPICLPSKGDTSTIYTNCWVTGWGFSKEKGEIQNILQKVNIPLVTNEECQKRYQDYKITQRMVCAGYKEGGKDACKGDSGGPLVCKHNGMWRLVGITSWGEGCARREQPGVYTKVAEYMDWILEKTQS Hydrogen bonds contact Hydrophobic contact | ||||