Job Results:
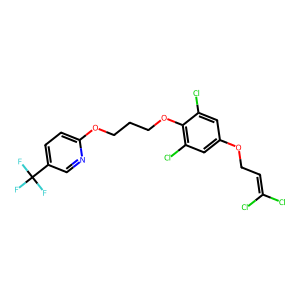
Ligand
Structure
Job ID
215921f481371e928c7811c49ba9b3ae
Job name
NA
Time
2025-01-23 16:39:47
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 1 | Histamine H3 receptor (H3R) | 7F61 | 7.40 | |
Target general information Gen name HRH3 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Histamine receptor 3; HH3R; GPCR97; G-protein coupled receptor 97; G protein-coupled receptor 97 Protein family G-protein coupled receptor 1 family Biochemical class GPCR rhodopsin Function Signals through the inhibition of adenylate cyclase and displays high constitutive activity (spontaneous activity in the absence of agonist). Agonist stimulation of isoform 3 neither modified adenylate cyclase activity nor induced intracellular calcium mobilization. The H3 subclass of histamine receptors could mediate the histamine signals in CNS and peripheral nervous system. Related diseases Immunodeficiency 48 (IMD48) [MIM:269840]: A form of severe immunodeficiency characterized by a selective absence of CD8+ T-cells. {ECO:0000269|PubMed:11123350, ECO:0000269|PubMed:11412303, ECO:0000269|PubMed:18509675, ECO:0000269|PubMed:8124727, ECO:0000269|PubMed:8202713}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Autoimmune disease, multisystem, infantile-onset, 2 (ADMIO2) [MIM:617006]: An autosomal recessive, autoimmune disorder characterized by systemic manifestations including blistering skin disease, uncontrollable bullous pemphigoid, inflammatory colitis, autoimmune hypothyroidism, proteinuria and nephrotic syndrome. {ECO:0000269|PubMed:26783323}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01238; DB06698; DB05381; DB17087; DB05080; DB00768; DB11642 Interacts with NA EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disulfide bond; G-protein coupled receptor; Glycoprotein; Membrane; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 34321.1 Length 301 Aromaticity 0.16 Instability index 32.22 Isoelectric point 9.63 Charge (pH=7) 15.11 2D Binding mode Binding energy (Kcal/mol) -10.09 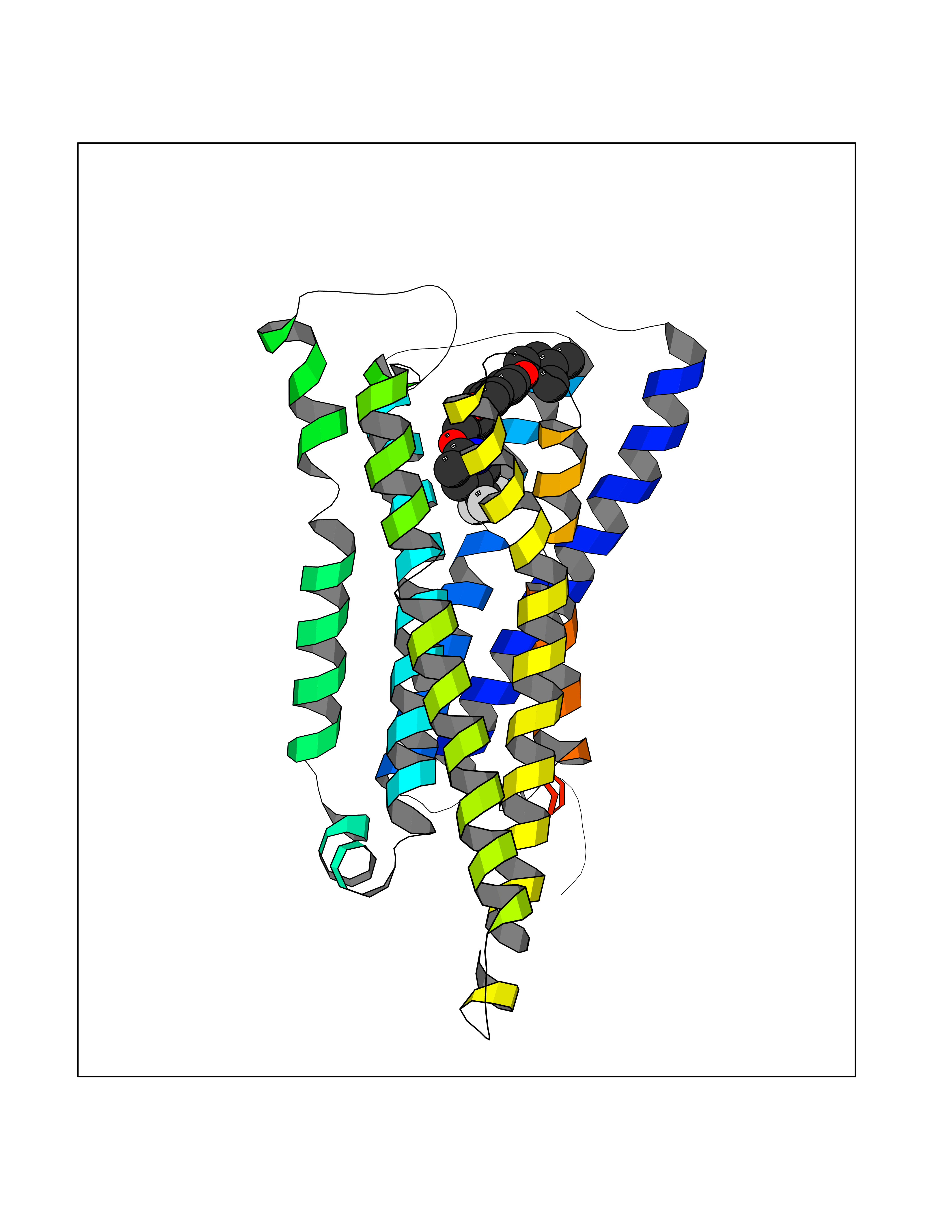 Molscript Map 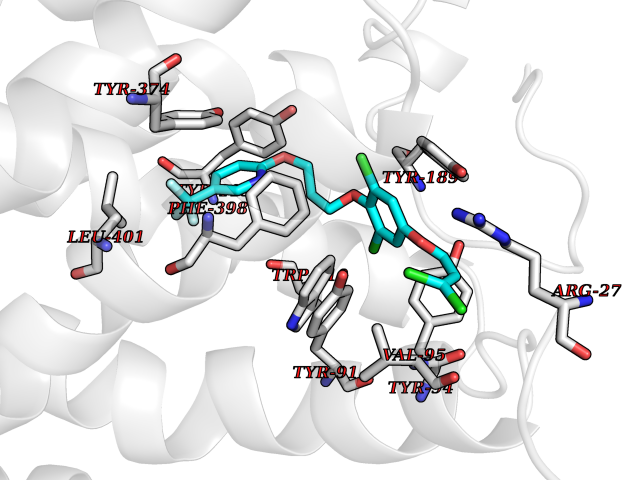 Pymol Map 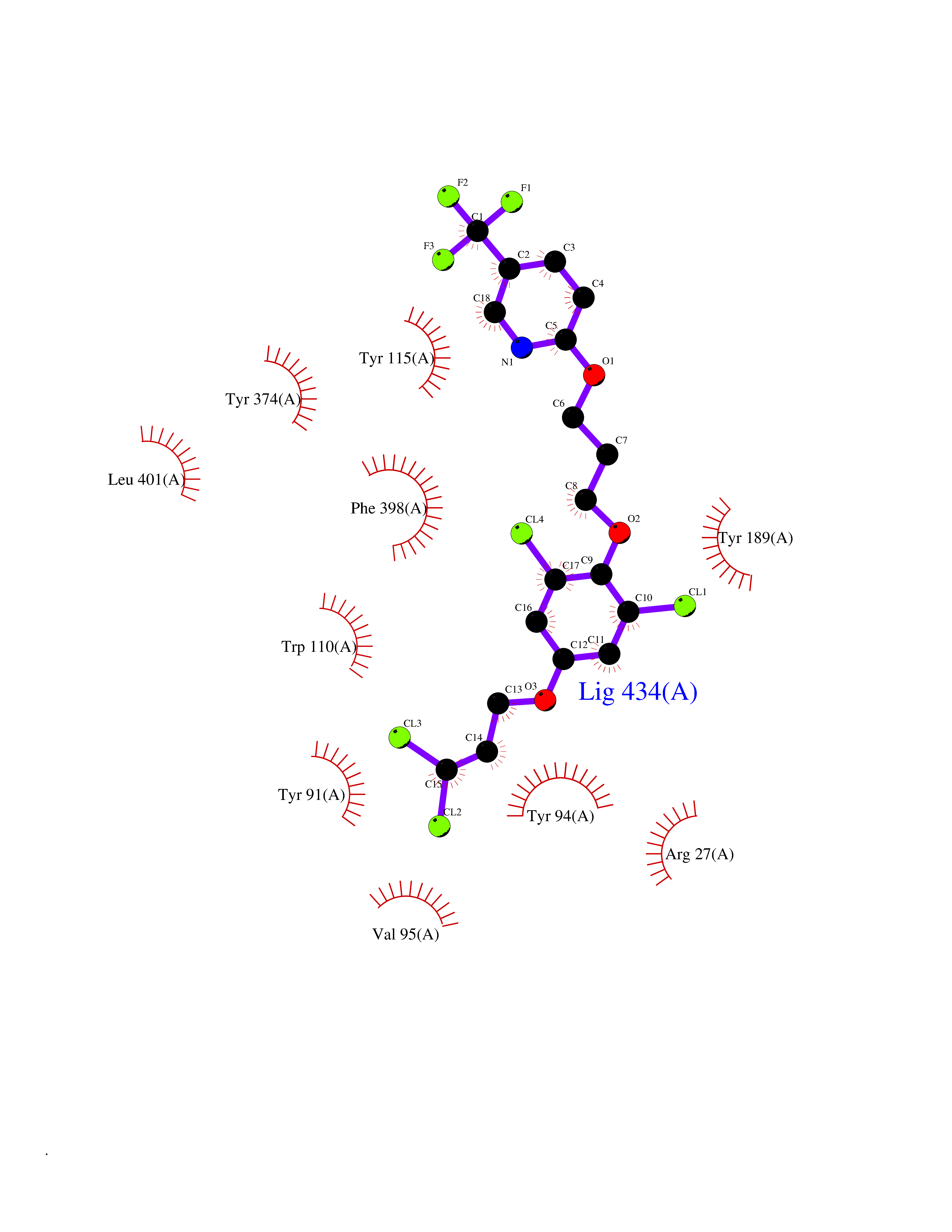 Ligplot Map 3D Binding mode Sequence RGFSAAWTAVLAALMALLIVATVLGNALVMLAFVADSSLRTQNNFFLLNLAISDFLVGAFCIPLYVPYVLTGRWTFGRGLCKLWLVVDYLLCTSKAFNIVLISYDRFLSVTRAVSYRAQQGDTRRAVRKMLLVWVLAFLLYGPAILSWEYLSGGSSIPEGHCYAEFFYNWYFLITASTLEFFTPFLSVTFFNLSIYLNIQRRTRLRLDGAREAAGRFRLSRDRKVAKSLAVIVSIFGLCWAPYTLLMIIRAACHGHCVPDYWYETSFWLLWANSAVNPVLYPLCHHSFRRAFTKLLCPQKL Hydrogen bonds contact Hydrophobic contact | ||||
| 2 | Glucose-dependent insulinotropic receptor (GPR119) | 7XZ6 | 7.38 | |
Target general information Gen name GPR119 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms GPR119; G-protein coupled receptor 119 Protein family G-protein coupled receptor 1 family Biochemical class GPCR rhodopsin Function Receptor for the endogenous fatty-acid ethanolamide oleoylethanolamide (OEA) and lysophosphatidylcholine (LPC). Functions as a glucose-dependent insulinotropic receptor. The activity of this receptor is mediated by G proteins which activate adenylate cyclase. Seems to act through a G(s) mediated pathway. Related diseases Developmental and epileptic encephalopathy 24 (DEE24) [MIM:615871]: A disease characterized by early-onset seizures, intellectual disability of varying degrees, and behavioral disturbances or autistic features in most individuals. {ECO:0000269|PubMed:24747641, ECO:0000269|PubMed:27864847, ECO:0000269|PubMed:30351409}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Generalized epilepsy with febrile seizures plus 10 (GEFSP10) [MIM:618482]: An autosomal dominant neurologic disorder with incomplete penetrance, characterized by variable types of seizures including absence, tonic-clonic, febrile, focal, and eyelid myoclonia. Some patients have normal neurologic development. Others have mild-to-moderate intellectual disability or autism spectrum disorder. {ECO:0000269|PubMed:29936235, ECO:0000269|PubMed:30351409}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB05166 Interacts with Q12797-6 EC number NA Uniprot keywords 3D-structure; Cell membrane; G-protein coupled receptor; Lipid-binding; Membrane; Receptor; Reference proteome; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID R Molecular weight (Da) 32134.1 Length 292 Aromaticity 0.12 Instability index 34.96 Isoelectric point 9.12 Charge (pH=7) 8.03 2D Binding mode Binding energy (Kcal/mol) -10.06 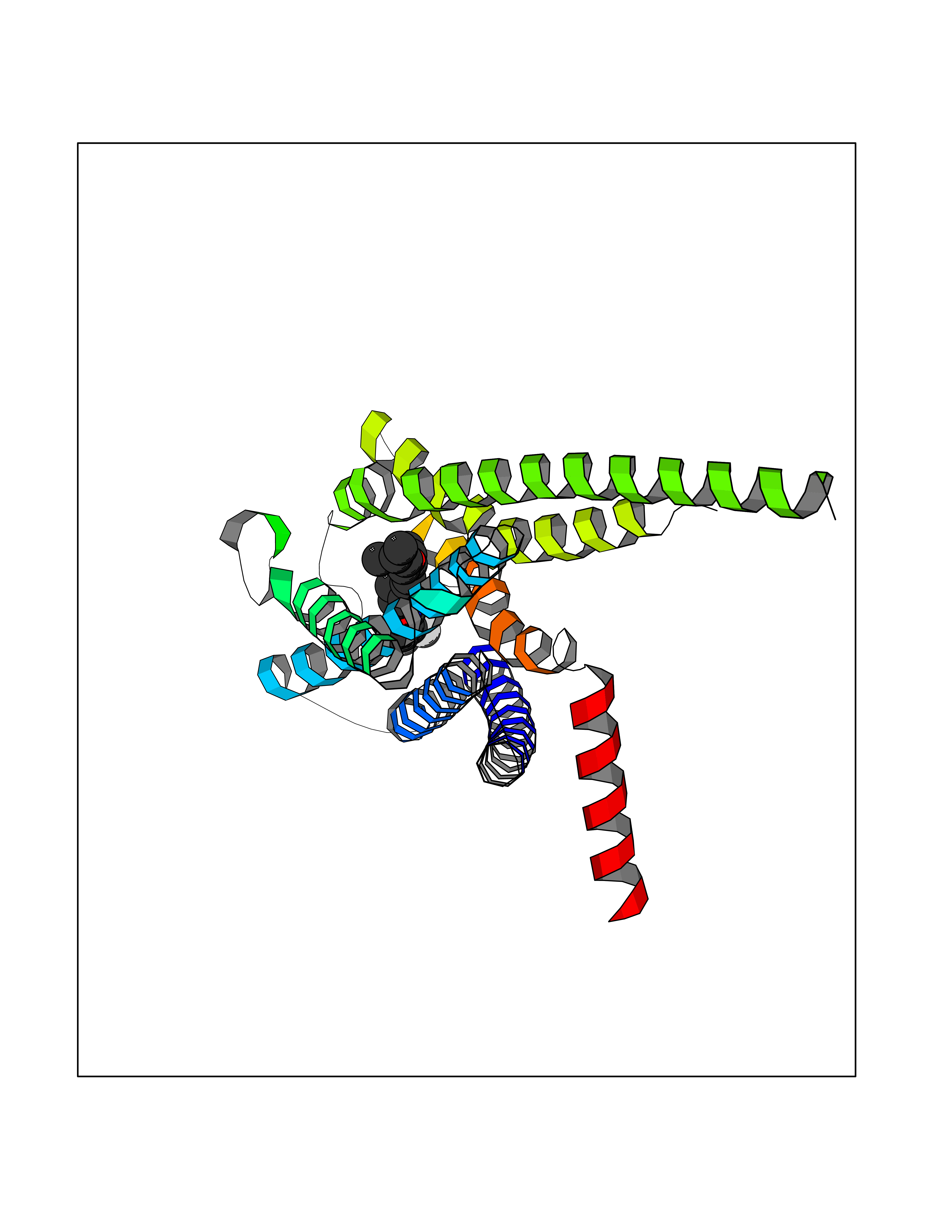 Molscript Map 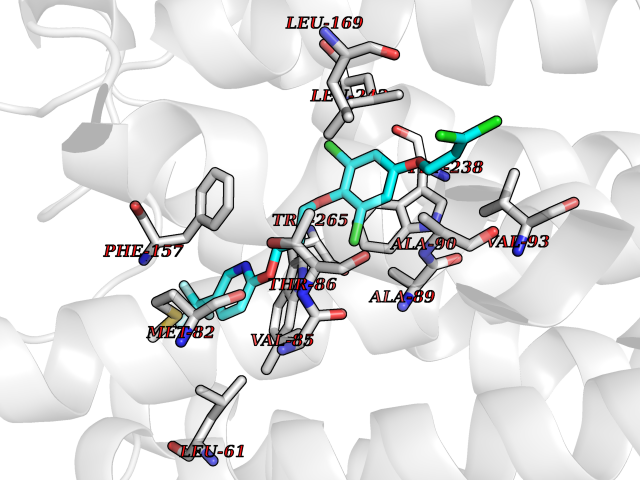 Pymol Map 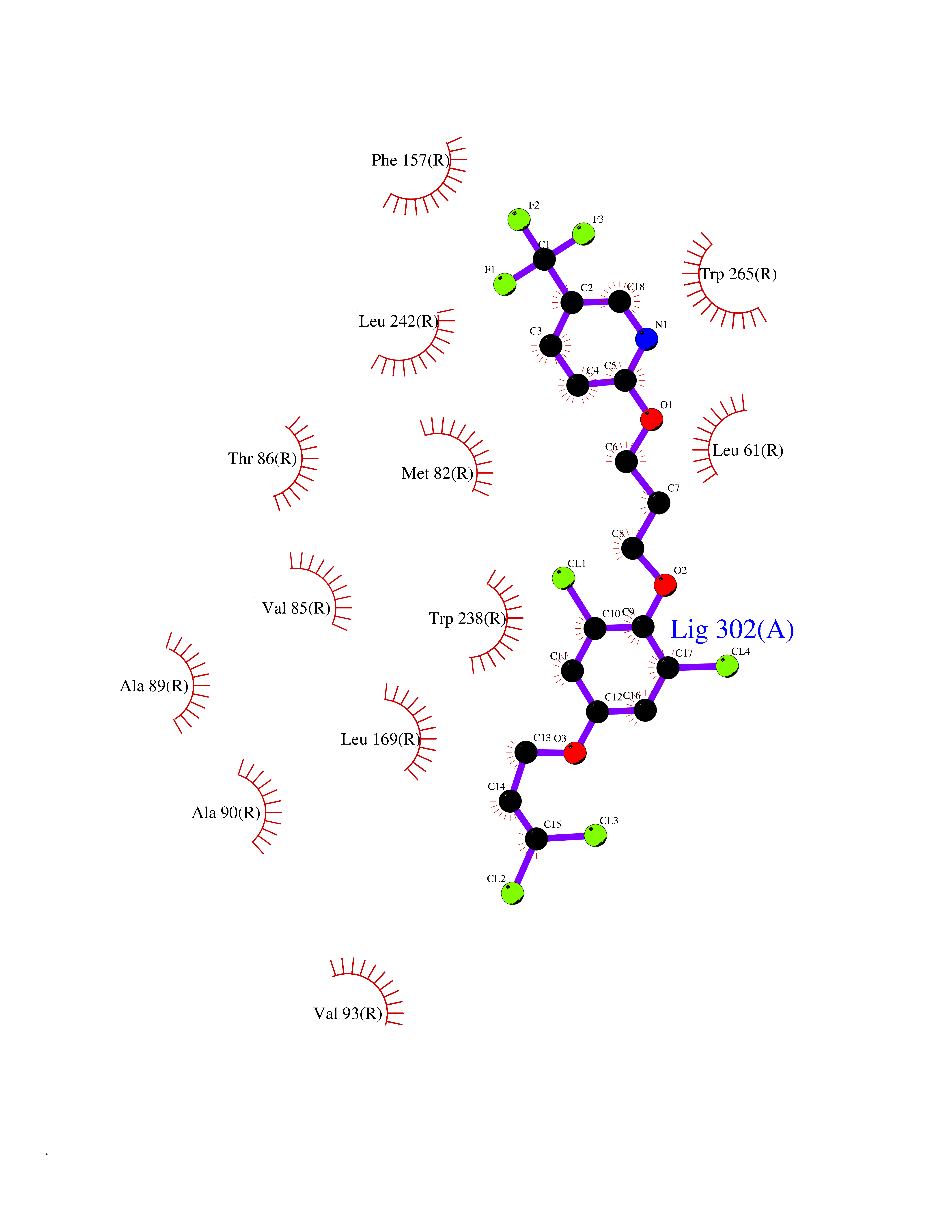 Ligplot Map 3D Binding mode Sequence MESSFSFGVILAVLASLIIATNTLVAVAVLLLIHKNDGVSLCFTLNLAVADTLIGVAISGLLTDQLSSPSRPTQKTLCSLRMAFVTSSAAASVLTVMLITFDRYLAIKQPFRYLKIMSGFVAGACIAGLWLVSYLIGFLPLGIPMFQQTAYKGQCSFFAVFHPHFVLTLSCVGFFPAMLLFVFFYCDMLKIASMHSQQIRKMEHAGAMAGSDFKALRTVSVLIGSFALSWTPFLITGIVQVACQECHLYLVLERYLWLLGVGNSLLNPLIYAYWQKEVRLQLYHMALGVKKV Hydrogen bonds contact Hydrophobic contact | ||||
| 3 | Cytoplasmic aspartate aminotransferase (GOT1) | 3II0 | 7.37 | |
Target general information Gen name GOT1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Glutamate oxaloacetate transaminase-1; GOT1 Protein family Class-I pyridoxal-phosphate-dependent aminotransferase family Biochemical class Transaminase Function Biosynthesis of L-glutamate from L-aspartate or L- cysteine. Important regulator of levels of glutamate, the major excitatory neurotransmitter of the vertebrate central nervous system. Acts as a scavenger of glutamate in brain neuroprotection. The aspartate aminotransferase activity is involved in hepatic glucose synthesis during development and in adipocyte glyceroneogenesis. Using L-cysteine as substrate, regulates levels of mercaptopyruvate, an important source of hydrogen sulfide. Mercaptopyruvate is converted into H(2)S via the action of 3- mercaptopyruvate sulfurtransferase (3MST). Hydrogen sulfide is an important synaptic modulator and neuroprotectant in the brain. Related diseases Multiple fibroadenomas of the breast (MFAB) [MIM:615554]: A benign breast disease marked by lobuloalveolar growth with abnormally high proliferation of the epithelium, and characterized by the presence of more than 3 fibroadenomas in one breast. Fibroadenomas are adenomas containing fibrous tissue. {ECO:0000269|PubMed:18779591}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Hyperprolactinemia (HPRL) [MIM:615555]: A disorder characterized by increased levels of prolactin in the blood not associated with gestation or the puerperium. HPRL may result in infertility, hypogonadism, and galactorrhea. {ECO:0000269|PubMed:24195502}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00210; DB00128; DB09130; DB00151; DB00142; DB04299; DB00114 Interacts with NA EC number EC 2.6.1.1 Uniprot keywords 3D-structure; Alternative splicing; Amino-acid biosynthesis; Aminotransferase; Cytoplasm; Direct protein sequencing; Phosphoprotein; Proteomics identification; Pyridoxal phosphate; Reference proteome; Transferase Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 51732.3 Length 462 Aromaticity 0.1 Instability index 31.83 Isoelectric point 7.48 Charge (pH=7) 0.86 2D Binding mode Binding energy (Kcal/mol) -10.05 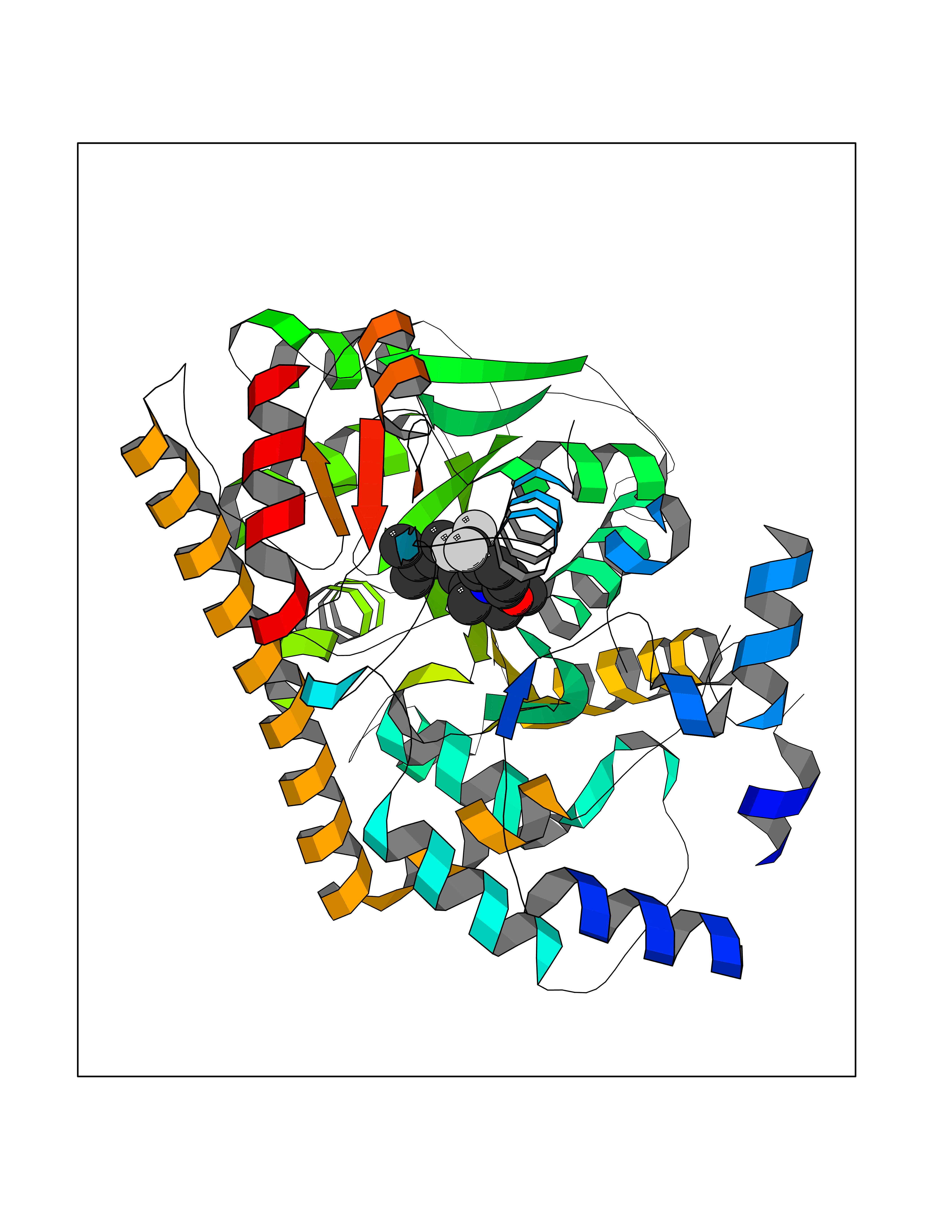 Molscript Map 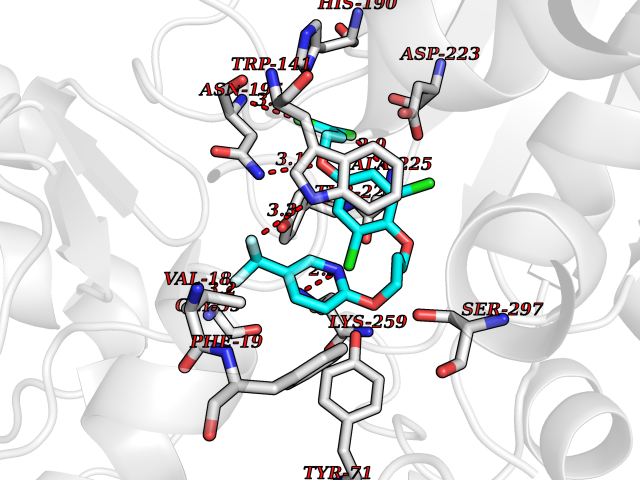 Pymol Map 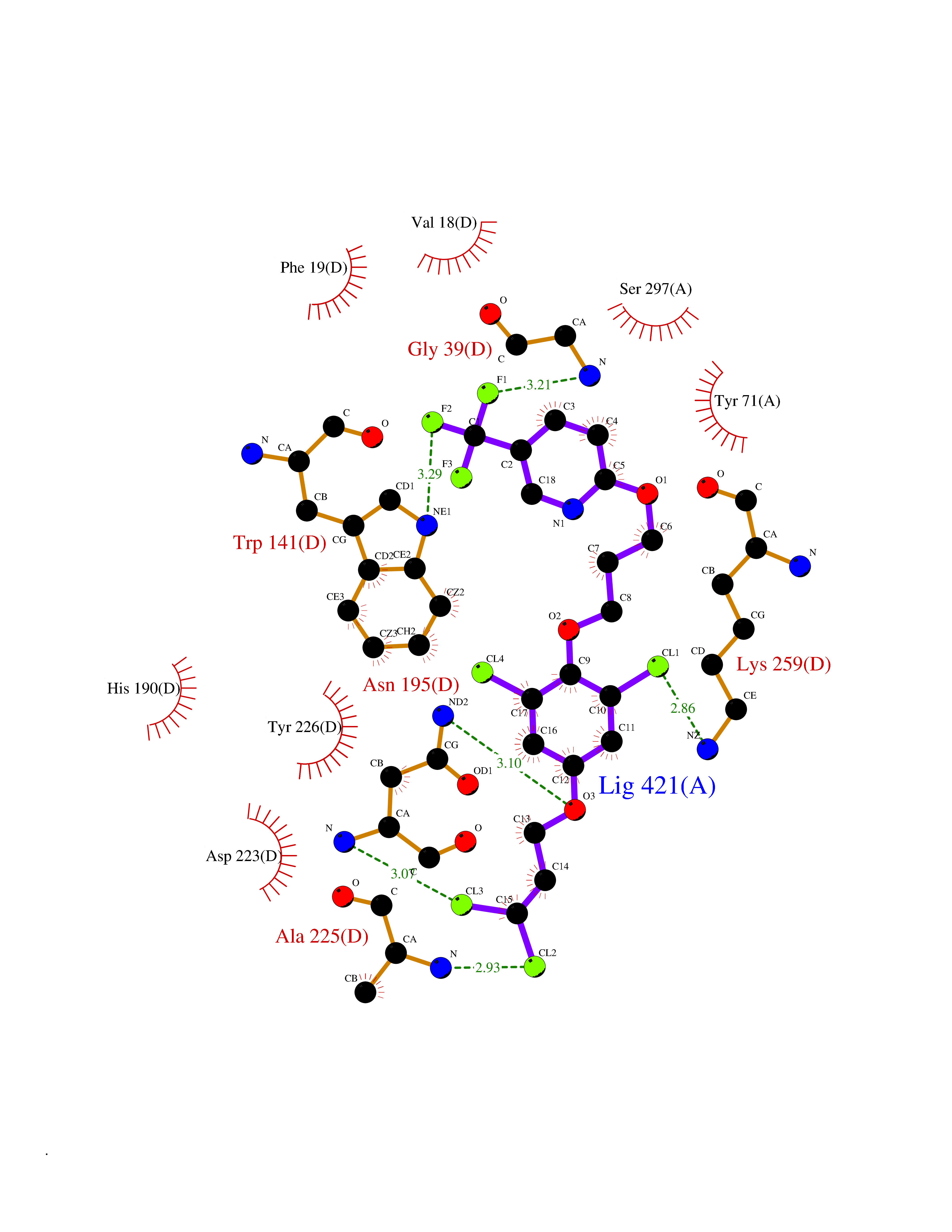 Ligplot Map 3D Binding mode Sequence PVLVFKLTVLPVVKKVEQKIANDNSLNHEYLPILGLAEFRVQSLGGTGALRIGADEKIVRITWSNPMQPVLVFKLTADFREDPDPRKVNLGVGAYRTDDCHPWVLPVVKKVEQKIANDNSLNHEYLPILGLAEFRSCASRLALGDDSPALKEKRVGGVQSLGGTGALRIGADFLARWYNGTNNKNTPVYVSSPTWENHNAVFSAAGFKDIRSYRYWDAEKRGLDLQGFLNDLENAPEFSIVVLHACAHNPTGIDPTPEQWKQIASVMKHRFLFPFFDSAYQGFASGNLERDAWAIRYFVSEGFEFFCAQSFSKNFGLYNERVGNLTVVGKEPESILQVLSQMEKIVRITWSNPPAQGARIVASTLSNPELFEEWTGNVKTMADRILTMRSELRARLEALKTPGTWNHITDQIGMFSFTGLNPKQVEYLVNEKHIYLLPSGRINVSGLTTKNLDYVATSIHEA Hydrogen bonds contact Hydrophobic contact | ||||
| 4 | Nicotinamide N-methyltransferase | 2IIP | 7.36 | |
Target general information Gen name NNMT Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Class I-like SAM-binding methyltransferase superfamily, NNMT/PNMT/TEMT family Biochemical class Transferase Function Nicotinamide N-methyltransferase activity.Pyridine N-methyltransferase activity. Related diseases Defects in PPARG can lead to type 2 insulin-resistant diabetes and hyptertension. PPARG mutations may be associated with colon cancer. {ECO:0000269|PubMed:10394368}.; DISEASE: Obesity (OBESITY) [MIM:601665]: A condition characterized by an increase of body weight beyond the limitation of skeletal and physical requirements, as the result of excessive accumulation of body fat. {ECO:0000269|PubMed:9753710}. Disease susceptibility may be associated with variants affecting the gene represented in this entry.; DISEASE: Lipodystrophy, familial partial, 3 (FPLD3) [MIM:604367]: A form of lipodystrophy characterized by marked loss of subcutaneous fat from the extremities. Facial adipose tissue may be increased, decreased or normal. Affected individuals show an increased preponderance of insulin resistance, diabetes mellitus and dyslipidemia. {ECO:0000269|PubMed:11788685, ECO:0000269|PubMed:12453919}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Glioma 1 (GLM1) [MIM:137800]: Gliomas are benign or malignant central nervous system neoplasms derived from glial cells. They comprise astrocytomas and glioblastoma multiforme that are derived from astrocytes, oligodendrogliomas derived from oligodendrocytes and ependymomas derived from ependymocytes. {ECO:0000269|PubMed:10851250}. Disease susceptibility may be associated with variants affecting the gene represented in this entry. Polymorphic PPARG alleles have been found to be significantly over-represented among a cohort of American patients with sporadic glioblastoma multiforme suggesting a possible contribution to disease susceptibility. Drugs (DrugBank ID) DB00627 Interacts with NA EC number 2.1.1.1 Uniprot keywords 3D-structure; Acetylation; Citrullination; Cytoplasm; Direct protein sequencing; Methyltransferase; Proteomics identification; Reference proteome; S-adenosyl-L-methionine; Transferase Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 27886.8 Length 251 Aromaticity 0.1 Instability index 40.66 Isoelectric point 5.23 Charge (pH=7) -5.11 2D Binding mode Binding energy (Kcal/mol) -10.04  Molscript Map 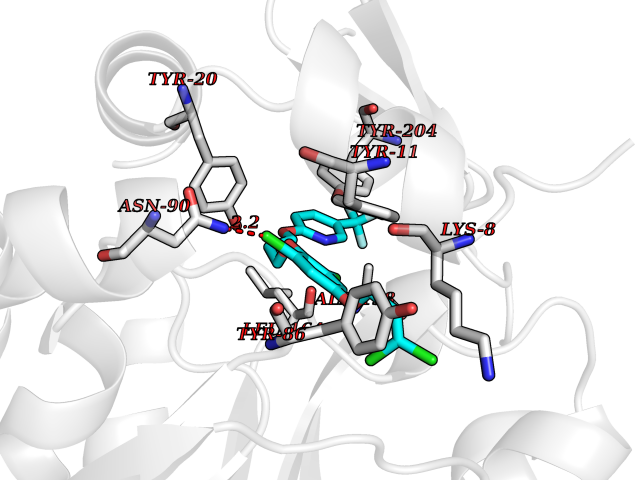 Pymol Map 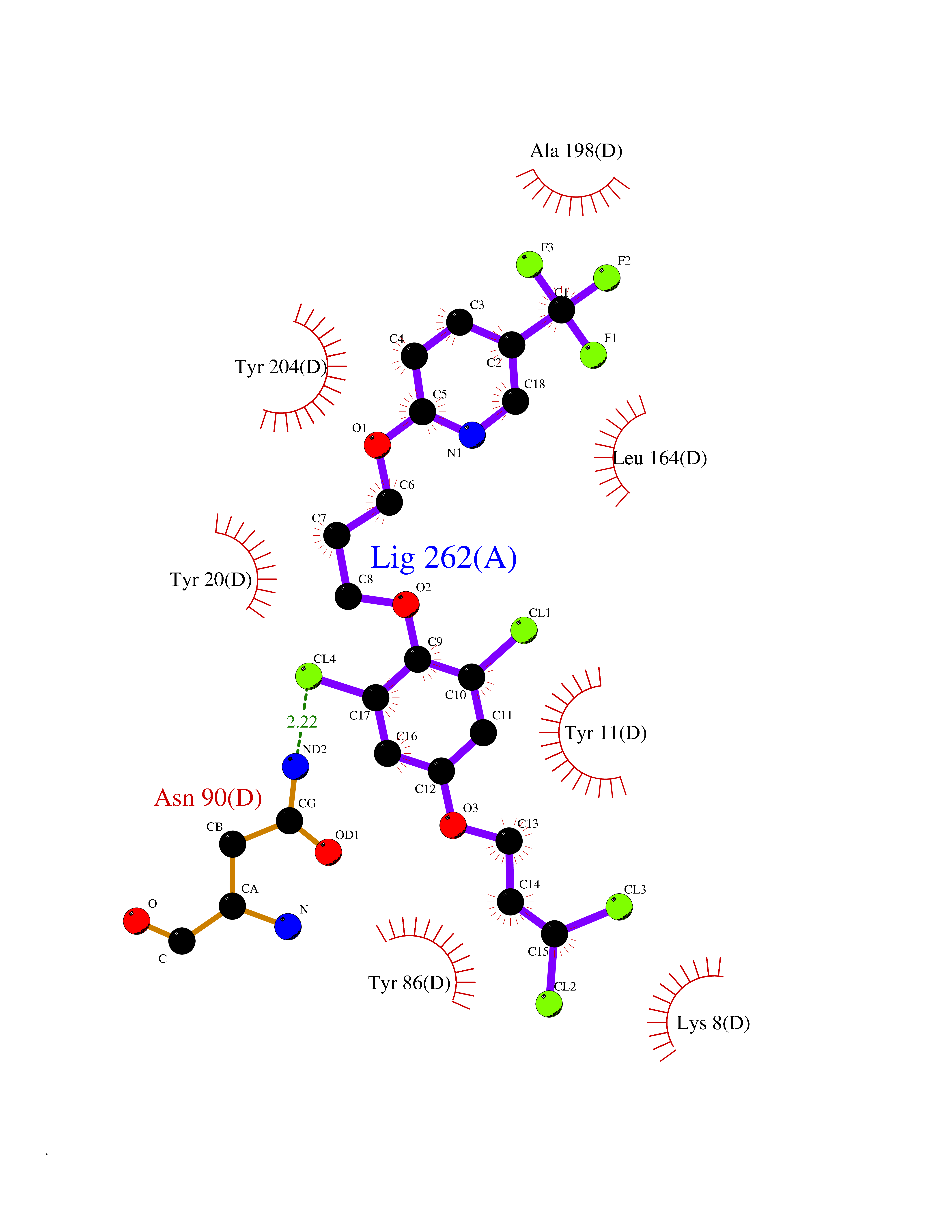 Ligplot Map 3D Binding mode Sequence GFTSKDTYLSHFNPRDYLEKYYSAESQILKHLLKNLFKIFCLDGVKGDLLIDIGSGPTIYQLLSACESFKEIVVTDYSDQNLQELEKWLKAAPAAFDWSPVVTYVCDLEGNRVKGPEKEEKLRQAVKQVLKCDVTQSQPLGAVPLPPADCVLSTLCLDAACPDLPTYCRALRNLGSLLKPGGFLVIMDALKSSYYMIGEQKFSSLPLGREAVEAAVKEAGYTIEWFEVISQSYSSTMANNEGLFSLVARKL Hydrogen bonds contact Hydrophobic contact | ||||
| 5 | Pseudomonas Transcriptional activator protein LasR (Pseudo LasR) | 3IX3 | 7.35 | |
Target general information Gen name Pseudo LasR Organism Pseudomonas aeruginosa (strain ATCC 15692 / DSM 22644 / CIP 104116 / JCM 14847 / LMG 12228 / 1C / PRS 101 / PAO1) Uniprot ID TTD ID Synonyms NA Protein family Autoinducer-regulated transcriptional regulatory protein family Biochemical class NA Function Transcriptional activator of elastase structural gene (LasB). Binds to the PAI autoinducer. Related diseases Growth hormone deficiency, isolated, 1A (IGHD1A) [MIM:262400]: An autosomal recessive, severe deficiency of growth hormone leading to dwarfism. Patients often develop antibodies to administered growth hormone. {ECO:0000269|PubMed:8364549}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Growth hormone deficiency, isolated, 1B (IGHD1B) [MIM:612781]: An autosomal recessive deficiency of growth hormone leading to short stature. Patients have low but detectable levels of growth hormone, significantly retarded bone age, and a positive response and immunologic tolerance to growth hormone therapy. {ECO:0000269|PubMed:12655557}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Kowarski syndrome (KWKS) [MIM:262650]: A syndrome clinically characterized by short stature associated with bioinactive growth hormone, normal or slightly increased growth hormone secretion, pathologically low insulin-like growth factor 1 levels, and normal catch-up growth on growth hormone replacement therapy. {ECO:0000269|PubMed:17519310, ECO:0000269|PubMed:8552145, ECO:0000269|PubMed:9276733}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Growth hormone deficiency, isolated, 2 (IGHD2) [MIM:173100]: An autosomal dominant deficiency of growth hormone leading to short stature. Clinical severity is variable. Patients have a positive response and immunologic tolerance to growth hormone therapy. {ECO:0000269|PubMed:11502836, ECO:0000269|PubMed:9152628}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08324 Interacts with NA EC number NA Uniprot keywords 3D-structure; Activator; DNA-binding; Quorum sensing; Reference proteome; Transcription; Transcription regulation Protein physicochemical properties Chain ID A Molecular weight (Da) 18305.5 Length 163 Aromaticity 0.12 Instability index 46.52 Isoelectric point 5.19 Charge (pH=7) -6.78 2D Binding mode Binding energy (Kcal/mol) -10.02  Molscript Map 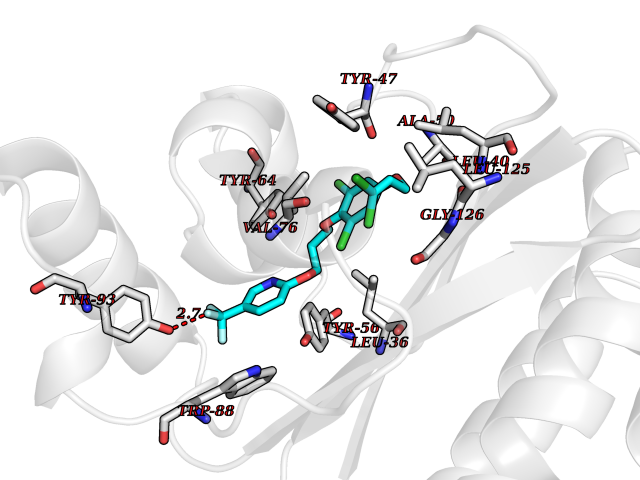 Pymol Map 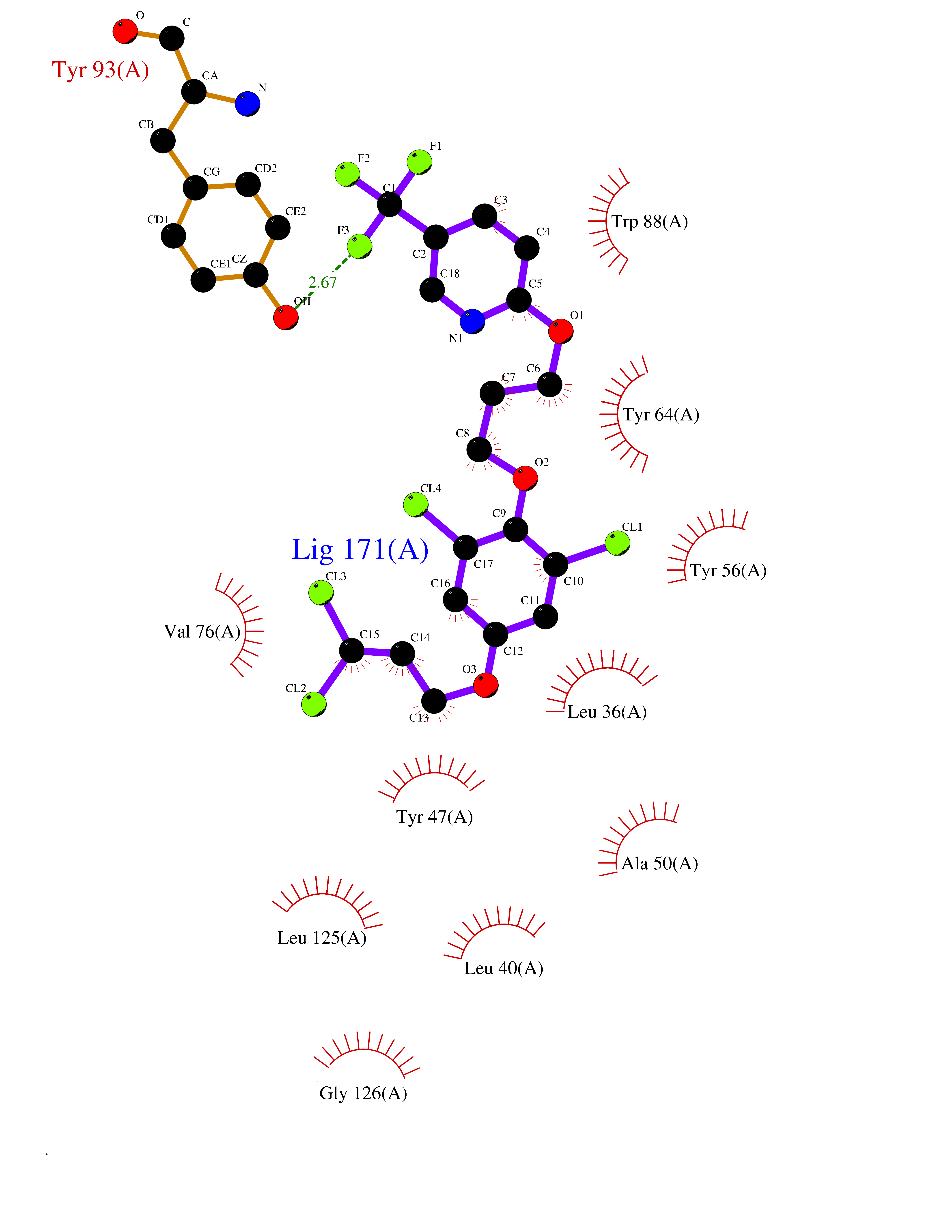 Ligplot Map 3D Binding mode Sequence FLELERSSGKLEWSAILQKMASDLGFSKILFGLLPKDSQDYENAFIVGNYPAAWREHYDRAGYARVDPTVSHCTQSVLPIFWEPSIYQTRKQHEFFEEASAAGLVYGLTMPLHGARGELGALSLSVEAENRAEANRFMESVLPTLWMLKDYALQSGAGLAFEH Hydrogen bonds contact Hydrophobic contact | ||||
| 6 | Folate receptor beta (FOLR2) | 4KN0 | 7.34 | |
Target general information Gen name FOLR2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Placental folate-binding protein; Folate receptor, fetal/placental; Folate receptor type-beta; Folate receptor 2; FR-beta; FOLR2 Protein family Folate receptor family Biochemical class Folate receptor Function Binds to folate and reduced folic acid derivatives and mediates delivery of 5-methyltetrahydrofolate and folate analogs into the interior of cells. Has high affinity for folate and folic acid analogs at neutral pH. Exposure to slightly acidic pH after receptor endocytosis triggers a conformation change that strongly reduces its affinity for folates and mediates their release. Related diseases Acute hepatic porphyria (AHEPP) [MIM:612740]: A form of porphyria. Porphyrias are inherited defects in the biosynthesis of heme, resulting in the accumulation and increased excretion of porphyrins or porphyrin precursors. They are classified as erythropoietic or hepatic, depending on whether the enzyme deficiency occurs in red blood cells or in the liver. AHP is characterized by attacks of gastrointestinal disturbances, abdominal colic, paralyses and peripheral neuropathy. Most attacks are precipitated by drugs, alcohol, caloric deprivation, infections, or endocrine factors. {ECO:0000269|PubMed:10706561, ECO:0000269|PubMed:1309003, ECO:0000269|PubMed:1569184, ECO:0000269|PubMed:17236137, ECO:0000269|PubMed:2063868}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00158; DB00563; DB05168 Interacts with NA EC number NA Uniprot keywords 3D-structure; Cell membrane; Direct protein sequencing; Disulfide bond; Folate-binding; Glycoprotein; GPI-anchor; Lipoprotein; Membrane; Proteomics identification; Receptor; Reference proteome; Secreted; Signal; Transport Protein physicochemical properties Chain ID A Molecular weight (Da) 23841.6 Length 205 Aromaticity 0.12 Instability index 56.78 Isoelectric point 7.92 Charge (pH=7) 2.58 2D Binding mode Binding energy (Kcal/mol) -10.01  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence RTDLLNVCMDAKHHKTKPGPEDKLHDQCSPWKKNACCTASTSQELHKDTSRLYNFNWDHCGKMEPACKRHFIQDTCLYECSPNLGPWIQQVNQSWRKERFLDVPLCKEDCQRWWEDCHTSHTCKSNWHRGWDWTSGVNKCPAGALCRTFESYFPTPAALCEGLWSHSYKVSNYSRGSGRCIQMWFDSAQGNPNEEVARFYAAAMH Hydrogen bonds contact Hydrophobic contact | ||||
| 7 | Lysine-specific histone demethylase 1B (KDM1B) | 4HSU | 7.34 | |
Target general information Gen name KDM1B Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Lysine-specific histone demethylase 2; LSD2; Flavin-containing amine oxidase domain-containing protein 1; C6orf193; AOF1 Protein family Flavin monoamine oxidase family Biochemical class CH-NH(2) donor oxidoreductase Function Required for de novo DNA methylation of a subset of imprinted genes during oogenesis. Acts by oxidizing the substrate by FAD to generate the corresponding imine that is subsequently hydrolyzed. Demethylates both mono- and di-methylated 'Lys-4' of histone H3. Has no effect on tri-methylated 'Lys-4', mono-, di- or tri-methylated 'Lys-9', mono-, di- or tri-methylated 'Lys-27', mono-, di- or tri-methylated 'Lys-36' of histone H3, or on mono-, di- or tri-methylated 'Lys-20' of histone H4. Histone demethylase that demethylates 'Lys-4' of histone H3, a specific tag for epigenetic transcriptional activation, thereby acting as a corepressor. Related diseases Angioedema, hereditary, 1 (HAE1) [MIM:106100]: An autosomal dominant disorder characterized by episodic local swelling involving subcutaneous or submucous tissue of the upper respiratory and gastrointestinal tracts, face, extremities, and genitalia. Hereditary angioedema due to C1 esterase inhibitor deficiency is comprised of two clinically indistinguishable forms. In hereditary angioedema type 1, serum levels of C1 esterase inhibitor are decreased, while in type 2, the levels are normal or elevated, but the protein is non-functional. {ECO:0000269|PubMed:12773530, ECO:0000269|PubMed:1363816, ECO:0000269|PubMed:1451784, ECO:0000269|PubMed:14635117, ECO:0000269|PubMed:16409206, ECO:0000269|PubMed:2118657, ECO:0000269|PubMed:2296585, ECO:0000269|PubMed:22994404, ECO:0000269|PubMed:2365061, ECO:0000269|PubMed:24456027, ECO:0000269|PubMed:3178731, ECO:0000269|PubMed:7814636, ECO:0000269|PubMed:7883978, ECO:0000269|PubMed:8172583, ECO:0000269|PubMed:8529136, ECO:0000269|PubMed:8755917, ECO:0000269|Ref.41}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q96L03 EC number EC 1.-.-.- Uniprot keywords 3D-structure; Alternative splicing; Chromatin regulator; Chromosome; Developmental protein; FAD; Flavoprotein; Metal-binding; Nucleus; Oxidoreductase; Phosphoprotein; Proteomics identification; Reference proteome; Repressor; Transcription; Transcription regulation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A,C Molecular weight (Da) 85795.5 Length 763 Aromaticity 0.1 Instability index 37.87 Isoelectric point 8.41 Charge (pH=7) 9.16 2D Binding mode Binding energy (Kcal/mol) -10.01  Molscript Map 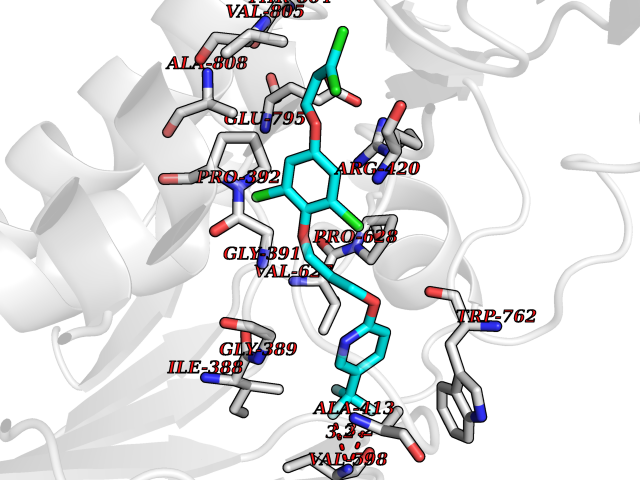 Pymol Map 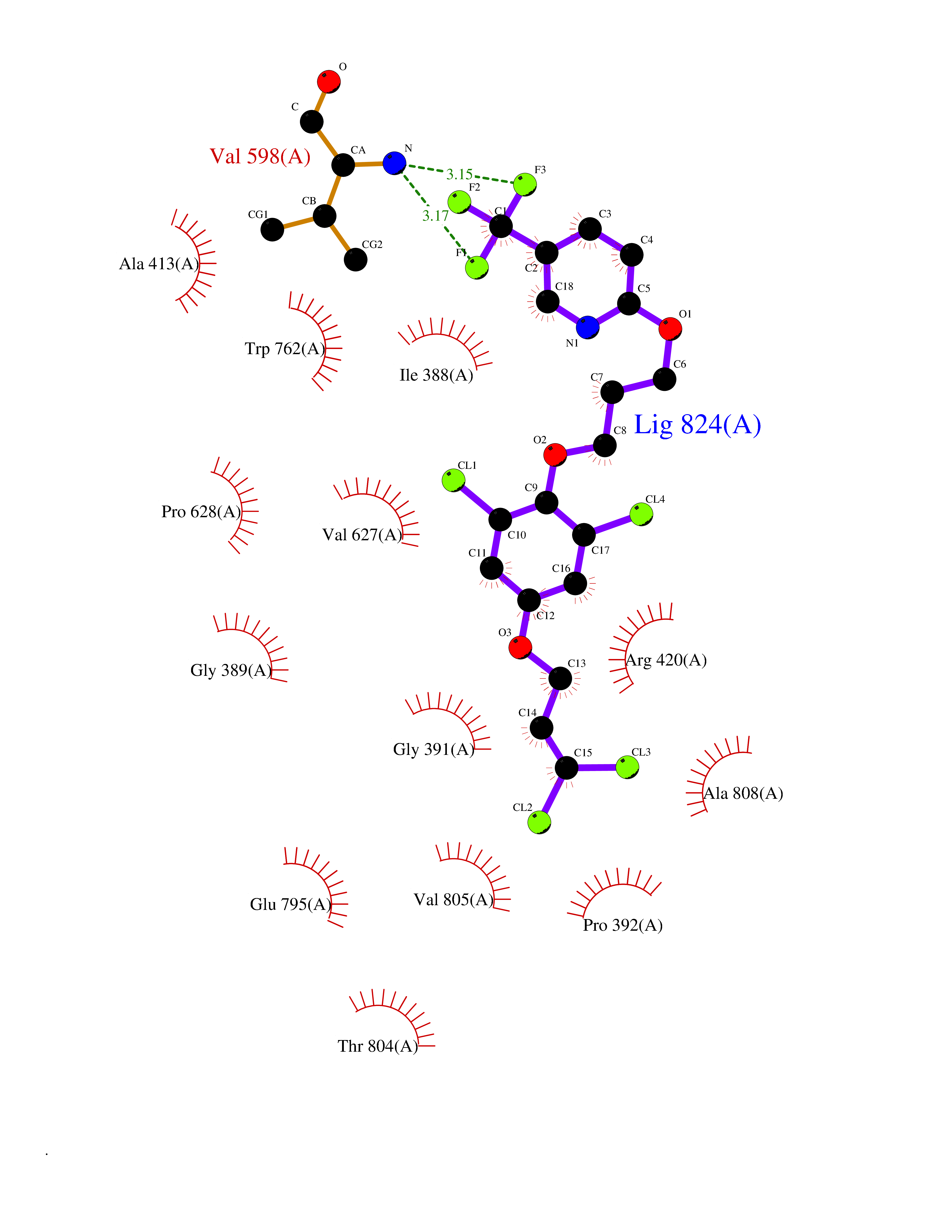 Ligplot Map 3D Binding mode Sequence GSRKCEKAGCTATCPVCFASASERCAKNGYTSRWYHLSCGEHFCNECFDHYYRSHKDGYDKYTTWKKIWTSNGKTEPSPKAFMADQQLPYWVQCTKPECRKWRQLTKEIQLTPQIAKTYRCGMKSDHCSLPEDLRVLEVSNHWWYSMLILPPLLKDSVAAPLLSAYYPDCVGMSPSCTGMNRYFQPFYQPNECGKALCVRPDVMELDELYEFPEYSRDPTMYLALRNLILALWYTNCKEALTPQKCIPHIIVRGLVRIRCVQEVERILYFMTRKGLINTGVLSVGADQYLLPKDYHNKSVIIIGAGPAGLAAARQLHNFGIKVTVLEAKDRIGGRVWDDKSFKGVTVGRGAQIVNGCINNPVALMCEQLGISMHKFGERCDLIQEGGRITDPTIDKRMDFHFNALLDVVSEWRKDKTQLQDVPLGEKIEEIYKAFIKESGIQFSELEGQVLQFHLSNLEYACGSNLHQVSARSWDHNEFFAQFAGDHTLLTPGYSVIIEKLAEGLDIQLKSPVQCIDYSGDEVQVTTTDGTGYSAQKVLVTVPLALLQKGAIQFNPPLSEKKMKAINSLGAGIIEKIALQFPYRFWDSKVQGADFFGHVPPSASKRGLFAVFYDMDPQKKHSVLMSVIAGEAVASVRTLDDKQVLQQCMATLRELFKEQEVPDPTKYFVTRWSTDPWIQMAYSFVKTGGSGEAYDIIAEDIQGTVFFAGEATNRHFPQTVTGAYLSGVREASKIAAFARTMQTARKSTGGKAPRKQLATKAAR Hydrogen bonds contact Hydrophobic contact | ||||
| 8 | Metabotropic glutamate receptor 3 (mGluR3) | 4XAR | 7.32 | |
Target general information Gen name GRM3 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms mGLUR3; Group III metabotropic glutamate receptor; GPRC1C Protein family G-protein coupled receptor 3 family Biochemical class GPCR glutamate Function Ligand binding causes a conformation change that triggers signaling via guanine nucleotide-binding proteins (G proteins) and modulates the activity of down-stream effectors. Signaling inhibits adenylate cyclase activity. G-protein coupled receptor for glutamate. Related diseases Paramyotonia congenita (PMC) [MIM:168300]: An autosomal dominant channelopathy characterized by myotonia, increased by exposure to cold, intermittent flaccid paresis, not necessarily dependent on cold or myotonia, lability of serum potassium, non-progressive nature and lack of atrophy or hypertrophy of muscles. In some patients, myotonia is not increased by cold exposure (paramyotonia without cold paralysis). Patients may have a combination phenotype of PMC and HYPP. {ECO:0000269|PubMed:10369308, ECO:0000269|PubMed:10727489, ECO:0000269|PubMed:1310898, ECO:0000269|PubMed:1316765, ECO:0000269|PubMed:1338909, ECO:0000269|PubMed:15318338, ECO:0000269|PubMed:15790667, ECO:0000269|PubMed:16786525, ECO:0000269|PubMed:18166706, ECO:0000269|PubMed:18690054, ECO:0000269|PubMed:19077043, ECO:0000269|PubMed:20076800, ECO:0000269|PubMed:8242056, ECO:0000269|PubMed:8308722, ECO:0000269|PubMed:8388676, ECO:0000269|PubMed:8580427}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Periodic paralysis hypokalemic 2 (HOKPP2) [MIM:613345]: An autosomal dominant disorder manifested by episodic flaccid generalized muscle weakness associated with falls of serum potassium levels. {ECO:0000269|PubMed:10599760, ECO:0000269|PubMed:10851391, ECO:0000269|PubMed:10944223, ECO:0000269|PubMed:11558801, ECO:0000269|PubMed:11591859, ECO:0000269|PubMed:16890191, ECO:0000269|PubMed:17898326, ECO:0000269|PubMed:18162704, ECO:0000269|PubMed:19118277, ECO:0000269|PubMed:20522878, ECO:0000269|PubMed:21043388, ECO:0000269|PubMed:24549961}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Periodic paralysis hyperkalemic (HYPP) [MIM:170500]: An autosomal dominant channelopathy characterized by episodic flaccid generalized muscle weakness associated with high levels of serum potassium. Concurrence of myotonia is found in HYPP patients. {ECO:0000269|PubMed:1659668, ECO:0000269|PubMed:1659948, ECO:0000269|PubMed:20076800}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Periodic paralysis normokalemic (NKPP) [MIM:170500]: A disorder closely related to hyperkalemic periodic paralysis, but marked by a lack of alterations in potassium levels during attacks of muscle weakness. {ECO:0000269|PubMed:15596759, ECO:0000269|PubMed:18046642, ECO:0000269|PubMed:20522878}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Myotonia SCN4A-related (MYOSCN4A) [MIM:608390]: A phenotypically highly variable myotonia aggravated by potassium loading, and sometimes by cold. Myotonia is characterized by sustained muscle tensing that prevents muscles from relaxing normally. It causes muscle stiffness that can interfere with movement. In some people the stiffness is very mild, while in other cases it may be severe enough to interfere with walking, running, and other activities of daily life. Myotonia SCN4A-related includes myotonia permanens and myotonia fluctuans. In myotonia permanens, the myotonia is generalized and there is a hypertrophy of the muscle, particularly in the neck and the shoulder. Attacks of severe muscle stiffness of the thoracic muscles may be life threatening due to impaired ventilation. In myotonia fluctuans, the muscle stiffness may fluctuate from day to day, provoked by exercise. {ECO:0000269|PubMed:10218481, ECO:0000269|PubMed:16786525, ECO:0000269|PubMed:16832098, ECO:0000269|PubMed:17212350, ECO:0000269|PubMed:17998485, ECO:0000269|PubMed:18203179, ECO:0000269|PubMed:18337100, ECO:0000269|PubMed:19015483, ECO:0000269|PubMed:19347921, ECO:0000269|PubMed:20076800, ECO:0000269|PubMed:27653901, ECO:0000269|PubMed:8058156, ECO:0000269|PubMed:9392583}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Myasthenic syndrome, congenital, 16 (CMS16) [MIM:614198]: A form of congenital myasthenic syndrome, a group of disorders characterized by failure of neuromuscular transmission, including pre-synaptic, synaptic, and post-synaptic disorders that are not of autoimmune origin. Clinical features are easy fatigability and muscle weakness. CMS16 is characterized by fatigable generalized weakness and recurrent attacks of respiratory and bulbar paralysis since birth. The fatigable weakness involves lid-elevator, external ocular, facial, limb and truncal muscles and an decremental response of the compound muscle action potential on repetitive stimulation. {ECO:0000269|PubMed:12766226, ECO:0000269|PubMed:25707578, ECO:0000269|PubMed:26659129}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Congenital myopathy 22A, classic (CMYO22A) [MIM:620351]: A form of congenital myopathy, a clinically and genetically heterogeneous group of muscle disorders characterized by hypotonia and muscle weakness apparent at birth, and specific pathological features on muscle biopsy. CMYO22A is an autosomal recessive form characterized by fetal hypokinesia, polyhydramnios, and severe neonatal hypotonia associated with respiratory insufficiency. Affected individuals who survive the neonatal period have delayed motor development, difficulty walking, proximal muscle weakness of the upper and lower limbs, facial and neck muscle weakness, easy fatigability, and mild limb contractures or foot deformities. {ECO:0000269|PubMed:26700687, ECO:0000269|PubMed:28262468, ECO:0000269|PubMed:36090556}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Congenital myopathy 22B, severe fetal (CMYO22B) [MIM:620369]: A severe congenital myopathy, a clinically and genetically heterogeneous group of muscle disorders characterized by hypotonia and muscle weakness apparent at birth, and specific pathological features on muscle biopsy. CMYO22B is an autosomal recessive form characterized by onset in utero. Affected individuals show fetal akinesia, and develop fetal hydrops with pulmonary hypoplasia, severe joint contractures, and generalized muscle hypoplasia. Death occurs in utero or soon after birth. {ECO:0000269|PubMed:26700687}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB05096 Interacts with NA EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disulfide bond; G-protein coupled receptor; Glycoprotein; Membrane; Proteomics identification; Receptor; Reference proteome; Signal; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 50355.5 Length 445 Aromaticity 0.11 Instability index 38.26 Isoelectric point 6.52 Charge (pH=7) -1.53 2D Binding mode Binding energy (Kcal/mol) -9.98 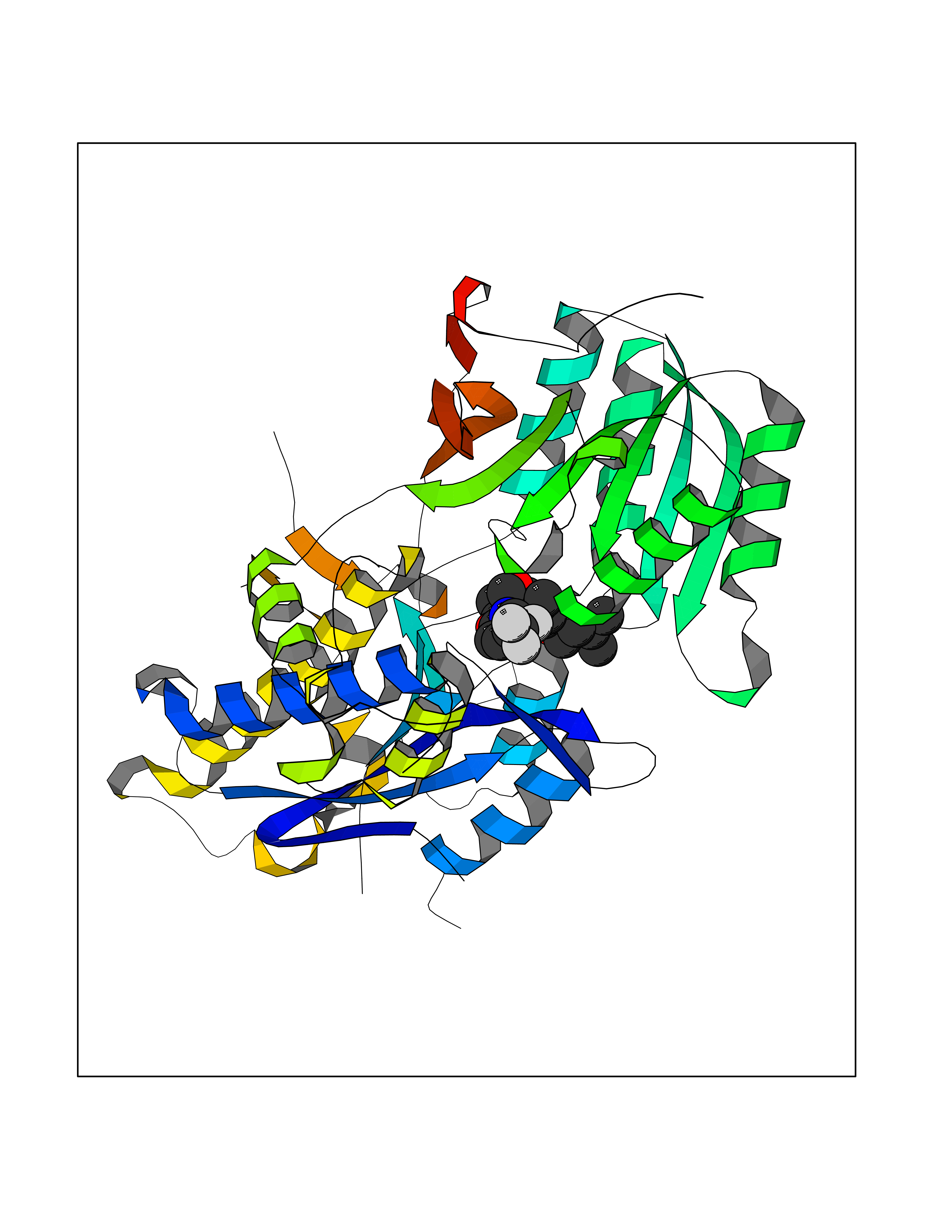 Molscript Map 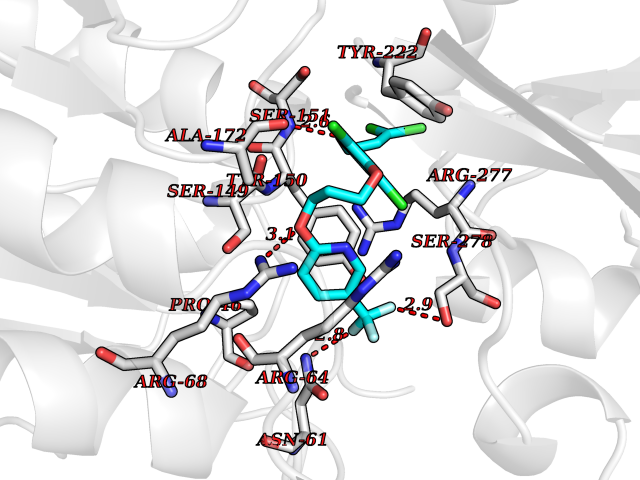 Pymol Map 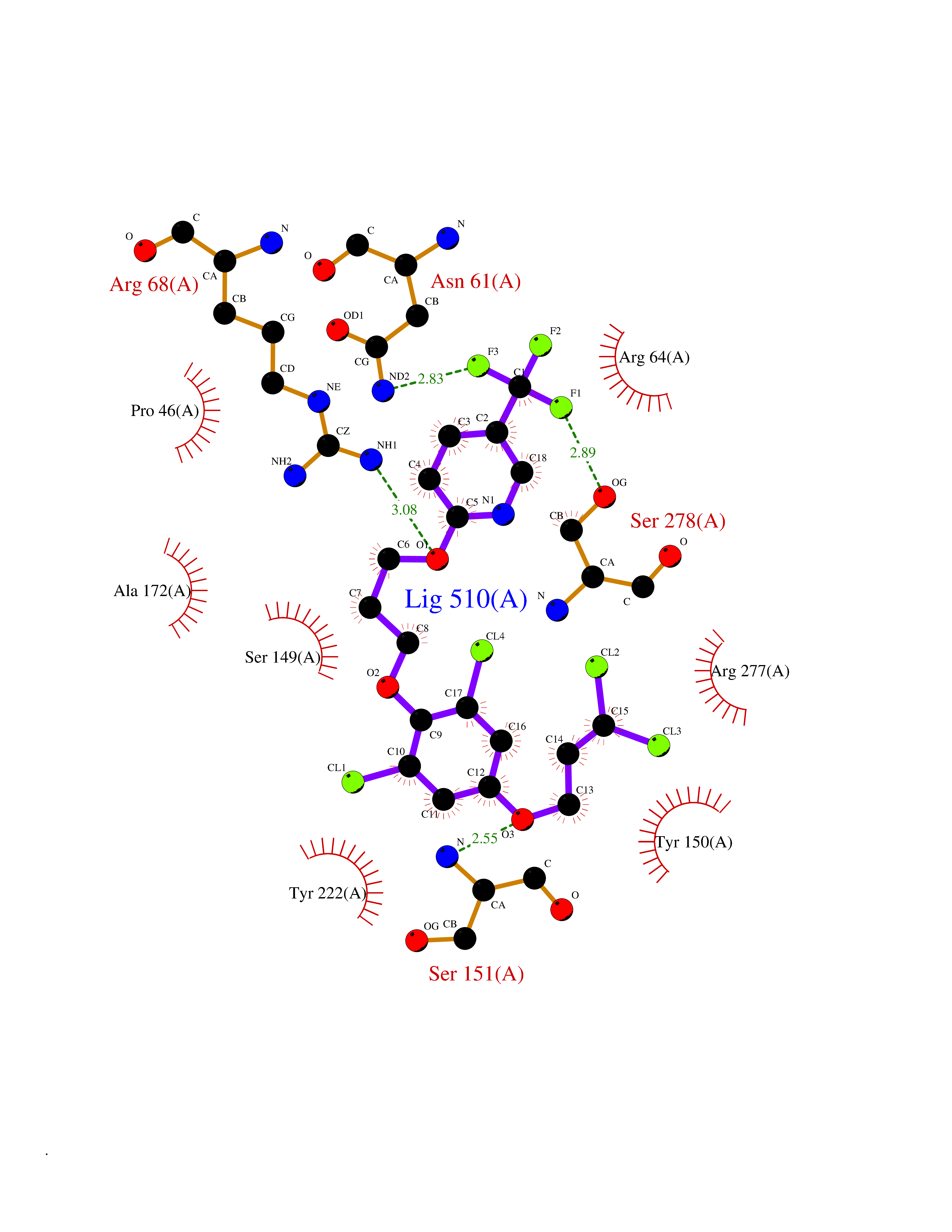 Ligplot Map 3D Binding mode Sequence RREIKIEGDLVLGGLFPINEKGTGTEECGRINEDRGIQRLEAMLFAIDEINKDDYLLPGVKLGVHILDTCSRDTYALEQSLEFVRASLLLIAGVIGGSYSSVSIQVANLLRLFQIPQISYASTSAKLSDKSRYDYFARTVPPDFYQAKAMAEILRFFNWTYVSTVASEGDYGETGIEAFEQEARLRNISIATAEKVGRSNIRKSYDSVIRELLQKPNARVVVLFMRSDDSRELIAAASRANASFTWVASDGWGAQESIIKGSEHVAYGAITLELASQPVRQFDRYFQSLNPYNNHRNPWFRDFWEQKFQCSLRVCDKHLAIDSSNYEQESKIMFVVNAVYAMAHALHKMQRTLCPNTTKLCDAMKILDGKKLYKDYLLKINFTAPDADSIVKFDTFGDGMGRYNVFNFQNVGGKYSYLKVGHWAETLSLDVNSIHWSRNSVPTSE Hydrogen bonds contact Hydrophobic contact | ||||
| 9 | Phenylethanolamine N-methyltransferase (PNMT) | 2G72 | 7.31 | |
Target general information Gen name PNMT Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms PNMTase; PENT; Noradrenaline N-methyltransferase Protein family Class I-like SAM-binding methyltransferase superfamily, NNMT/PNMT/TEMT family Biochemical class NA Function Converts noradrenaline to adrenaline. Related diseases A chromosomal aberration involving TRIM24/TIF1 is found in papillary thyroid carcinomas (PTCs). Translocation t(7;10)(q32;q11) with RET. The translocation generates the TRIM24/RET (PTC6) oncogene. {ECO:0000269|PubMed:10439047}. Drugs (DrugBank ID) DB08129; DB08128; DB07739; DB07798; DB07747; DB03468; DB08550; DB03824; DB04273; DB07906; DB07597; DB09571; DB00968; DB08631; DB01752; DB08654 Interacts with Q9P2G9-2; Q8TBB1 EC number EC 2.1.1.28 Uniprot keywords 3D-structure; Catecholamine biosynthesis; Direct protein sequencing; Methyltransferase; Phosphoprotein; Proteomics identification; Reference proteome; S-adenosyl-L-methionine; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 29198.9 Length 264 Aromaticity 0.09 Instability index 54.33 Isoelectric point 5.91 Charge (pH=7) -3.69 2D Binding mode Binding energy (Kcal/mol) -9.97  Molscript Map 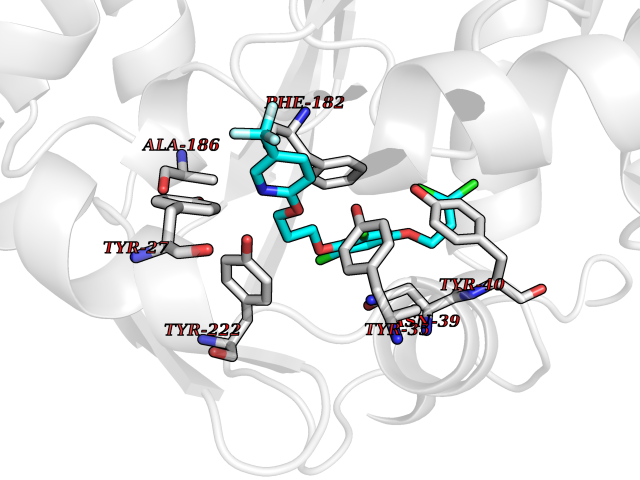 Pymol Map 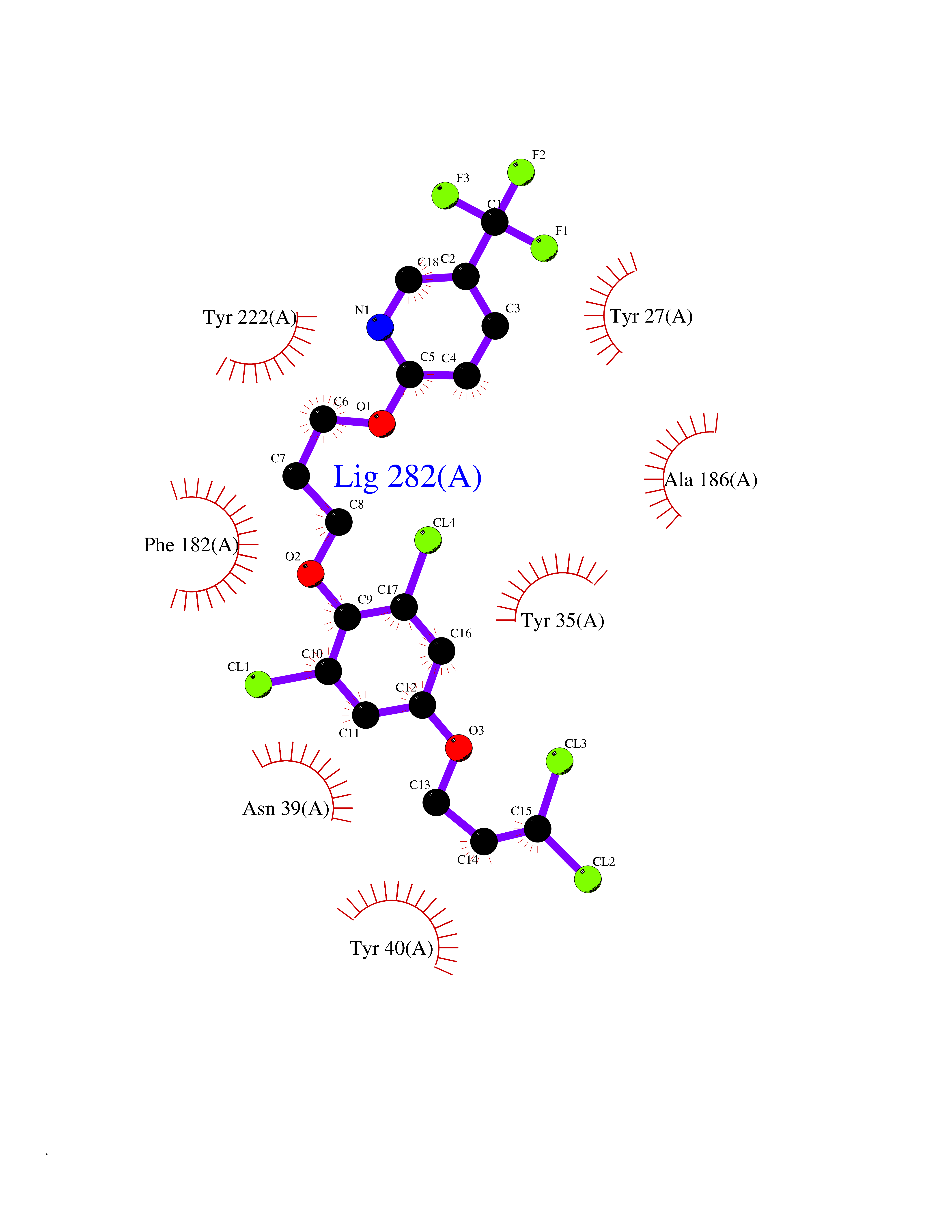 Ligplot Map 3D Binding mode Sequence APGQAAVASAYQRFEPRAYLRNNYAPPRGDLCNPNGVGPWKLRCLAQTFATGEVSGRTLIDIGSGPTVYQLLSACSHFEDITMTDFLEVNRQELGRWLQEEPGAFNWSMYSQHACLIEGKGECWQDKERQLRARVKRVLPIDVHQPQPLGAGSPAPLPADALVSAFCLEAVSPDLASFQRALDHITTLLRPGGHLLLIGALEESWYLAGEARLTVVPVSEEEVREALVRSGYKVRDLRTYIMPAHLQTGVDDVKGVFFAWAQKV Hydrogen bonds contact Hydrophobic contact | ||||
| 10 | Aldose reductase (AKR1B1) | 1US0 | 7.30 | |
Target general information Gen name AKR1B1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Aldehyde reductase; AKR1B1 Protein family Aldo/keto reductase family Biochemical class Short-chain dehydrogenases reductase Function Catalyzes the NADPH-dependent reduction of a wide variety of carbonyl-containing compounds to their corresponding alcohols with a broad range of catalytic efficiencies. Related diseases Glutamine deficiency, congenital (GLND) [MIM:610015]: An autosomal recessive disorder characterized by variable brain malformations, encephalopathy, severe developmental delay, seizures, and decreased glutamine levels in bodily fluids. Death in early infancy may occur. {ECO:0000269|PubMed:16267323, ECO:0000269|PubMed:26711351, ECO:0000269|PubMed:38579670}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Developmental and epileptic encephalopathy 116 (DEE116) [MIM:620806]: A form of epileptic encephalopathy, a heterogeneous group of early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE116 is autosomal dominant form characterized by severe developmental delay, seizures, and white matter abnormalities. {ECO:0000269|PubMed:38579670}. The disease is caused by variants affecting the gene represented in this entry. DEE116 is caused by variants that disrupt the canonical translation start codon in GLUL resulting in initiation of translation at Met-18 (PubMed:38579670). The resulting protein is enzymatically competent but insensitive to negative feedback regulation via glutamine-induced degradation. {ECO:0000269|PubMed:38579670}. Drugs (DrugBank ID) DB07028; DB07030; DB07450; DB02101; DB08449; DB08000; DB07139; DB07498; DB02007; DB02020; DB11859; DB02994; DB04272; DB07187; DB00694; DB00997; DB06246; DB01039; DB02021; DB16707; DB00143; DB02834; DB08084; DB01689; DB07063; DB06077; DB02518; DB00157; DB03461; DB05383; DB05533; DB05327; DB02712; DB00605; DB02383; DB02132; DB08772; DB07093; DB07999; DB08098 Interacts with Q9BUY7 EC number EC 1.1.1.300 Uniprot keywords 3D-structure; Acetylation; Cytoplasm; Direct protein sequencing; Lipid metabolism; NADP; Oxidoreductase; Phosphoprotein; Proteomics identification; Reference proteome Protein physicochemical properties Chain ID A Molecular weight (Da) 35447.6 Length 313 Aromaticity 0.09 Instability index 36.41 Isoelectric point 7.1 Charge (pH=7) 0.26 2D Binding mode Binding energy (Kcal/mol) -9.96 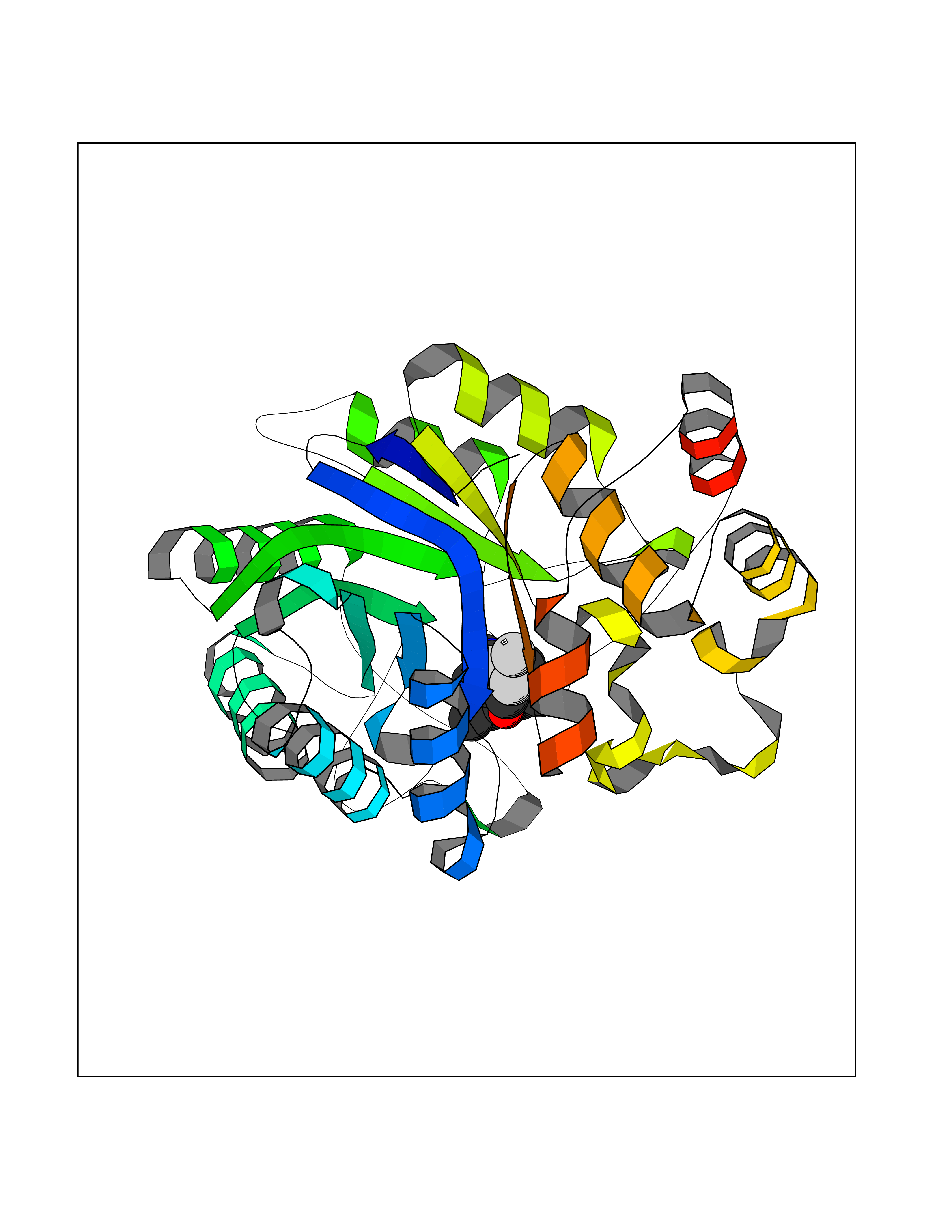 Molscript Map 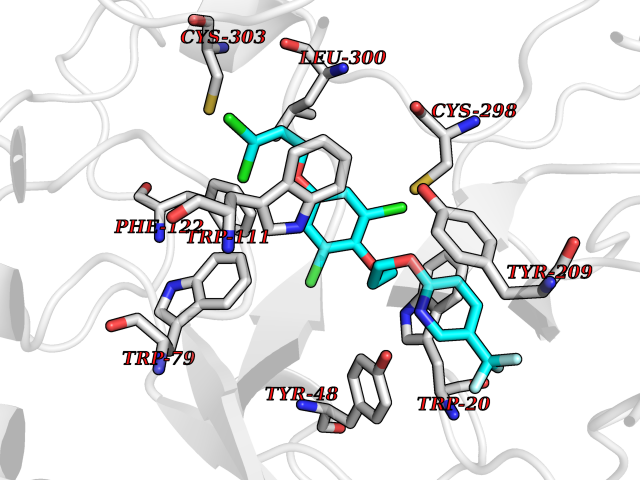 Pymol Map 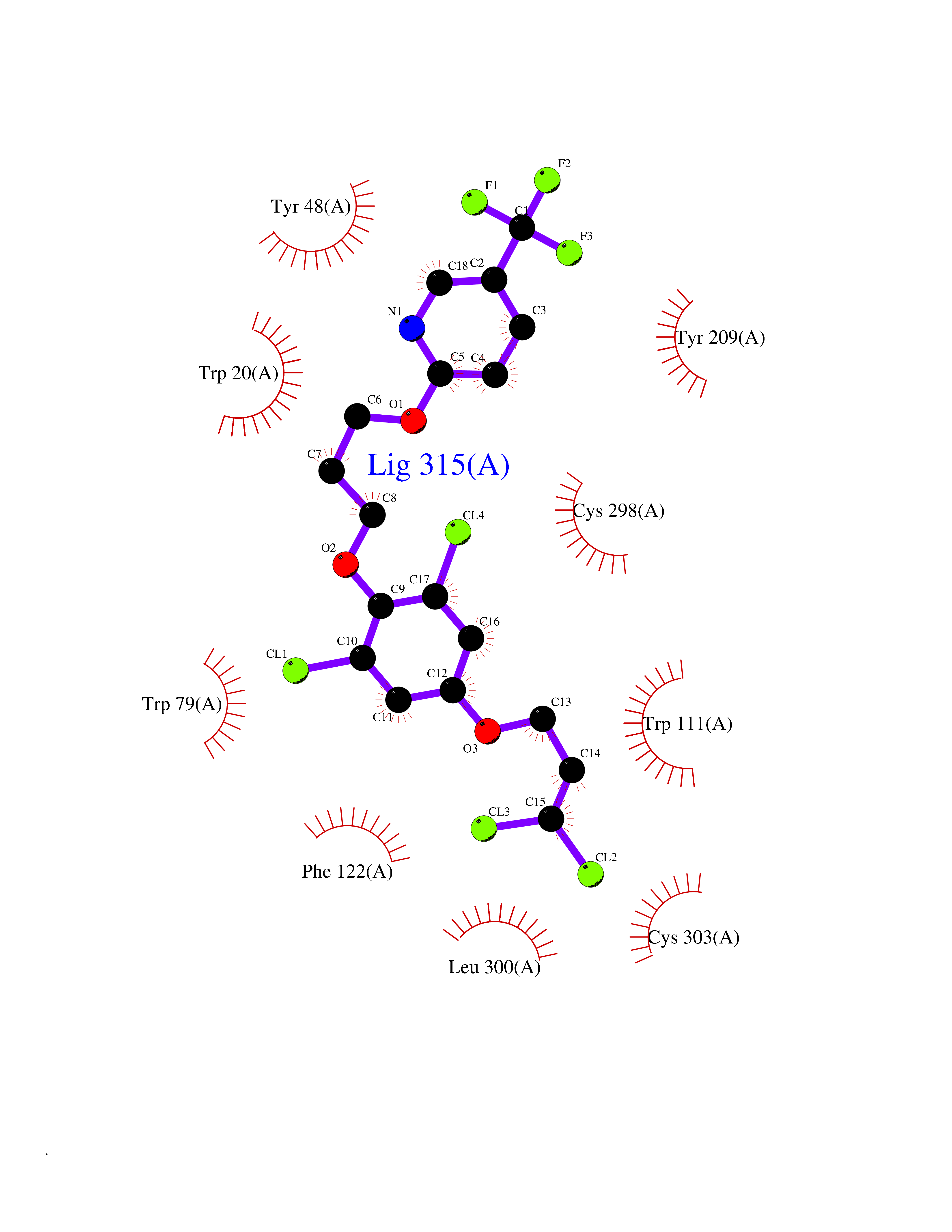 Ligplot Map 3D Binding mode Sequence MASRILLNNGAKMPILGLGTWKSPPGQVTEAVKVAIDVGYRHIDCAHVYQNENEVGVAIQEKLREQVVKREELFIVSKLWCTYHEKGLVKGACQKTLSDLKLDYLDLYLIHWPTGFKPGKEFFPLDESGNVVPSDTNILDTWAAMEELVDEGLVKAIGISNFNHLQVEMILNKPGLKYKPAVNQIECHPYLTQEKLIQYCQSKGIVVTAYSPLGSPDRPWAKPEDPSLLEDPRIKAIAAKHNKTTAQVLIRFPMQRNLVVIPKSVTPERIAENFKVFDFELSSQDMTTLLSYNRNWRVCALLSCTSHKDYPFH Hydrogen bonds contact Hydrophobic contact | ||||
| 11 | Metabotropic glutamate receptor 5 (mGluR5) | 4OO9 | 7.30 | |
Target general information Gen name GRM5 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms MGLUR5; GPRC1E Protein family G-protein coupled receptor 3 family Biochemical class GPCR glutamate Function G-protein coupled receptor for glutamate. Ligand binding causes a conformation change that triggers signaling via guanine nucleotide-binding proteins (G proteins) and modulates the activity of down-stream effectors. Signaling activates a phosphatidylinositol-calcium second messenger system and generates a calcium-activated chloride current. Plays an important role in the regulation of synaptic plasticity and the modulation of the neural network activity. Related diseases Charcot-Marie-Tooth disease, axonal, 2D (CMT2D) [MIM:601472]: A dominant axonal form of Charcot-Marie-Tooth disease, a disorder of the peripheral nervous system, characterized by progressive weakness and atrophy, initially of the peroneal muscles and later of the distal muscles of the arms. Charcot-Marie-Tooth disease is classified in two main groups on the basis of electrophysiologic properties and histopathology: primary peripheral demyelinating neuropathies (designated CMT1 when they are dominantly inherited) and primary peripheral axonal neuropathies (CMT2). Neuropathies of the CMT2 group are characterized by signs of axonal degeneration in the absence of obvious myelin alterations, normal or slightly reduced nerve conduction velocities, and progressive distal muscle weakness and atrophy. {ECO:0000269|PubMed:12690580, ECO:0000269|PubMed:17035524, ECO:0000269|PubMed:17101916, ECO:0000269|PubMed:17663003, ECO:0000269|PubMed:20169446, ECO:0000269|PubMed:24604904, ECO:0000269|PubMed:25168514, ECO:0000269|PubMed:26244500, ECO:0000269|PubMed:26503042, ECO:0000269|PubMed:31173493}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Neuronopathy, distal hereditary motor, autosomal dominant 5 (HMND5) [MIM:600794]: A form of distal hereditary motor neuronopathy, a heterogeneous group of neuromuscular disorders caused by selective degeneration of motor neurons in the anterior horn of the spinal cord, without sensory deficit in the posterior horn. The overall clinical picture consists of a classical distal muscular atrophy syndrome in the legs without clinical sensory loss. The disease starts with weakness and wasting of distal muscles of the anterior tibial and peroneal compartments of the legs. Later on, weakness and atrophy may expand to the proximal muscles of the lower limbs and/or to the distal upper limbs. {ECO:0000269|PubMed:12690580, ECO:0000269|PubMed:17035524, ECO:0000269|PubMed:23279345, ECO:0000269|PubMed:24627108, ECO:0000269|PubMed:26503042}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Spinal muscular atrophy, infantile, James type (SMAJI) [MIM:619042]: An autosomal dominant form of spinal muscular atrophy, a group of neuromuscular disorders characterized by degeneration of the anterior horn cells of the spinal cord, leading to symmetrical muscle weakness and atrophy. SMAJI is a severe disease characterized by hypotonia manifesting in the first weeks or months of life, delayed motor development, motor regression, and muscle weakness and atrophy primarily affecting distal muscles. Additional variable features include feeding difficulties, poor overall growth, foot deformities, kyphosis, hyperlordosis, scoliosis, vocal cord dysfunction, and respiratory insufficiency. {ECO:0000269|PubMed:32181591}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00659; DB05070; DB12733; DB06201 Interacts with P41594; Q7Z6G3 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disulfide bond; G-protein coupled receptor; Glycoprotein; Membrane; Methylation; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Signal; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 27065.4 Length 247 Aromaticity 0.13 Instability index 42.92 Isoelectric point 9.24 Charge (pH=7) 11.34 2D Binding mode Binding energy (Kcal/mol) -9.96 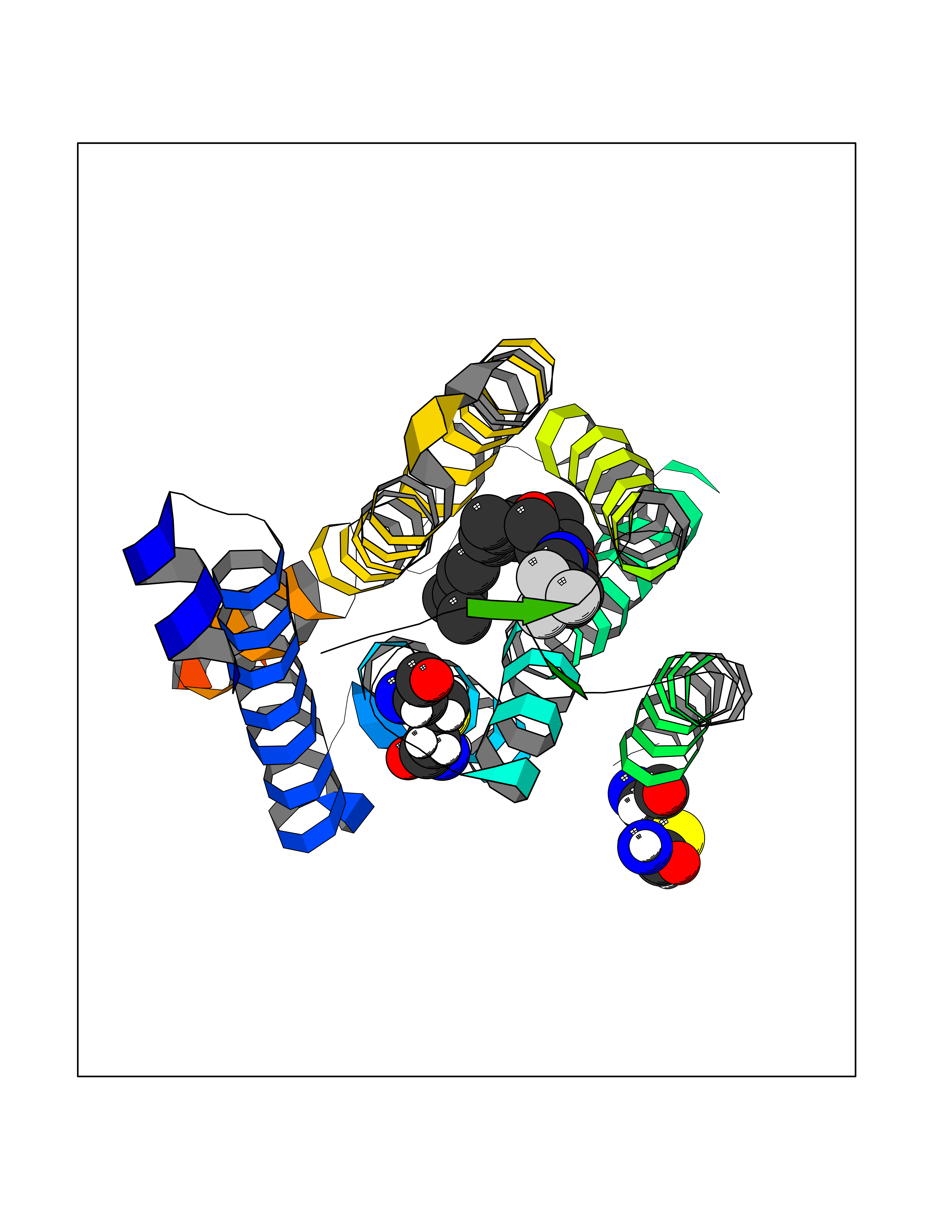 Molscript Map 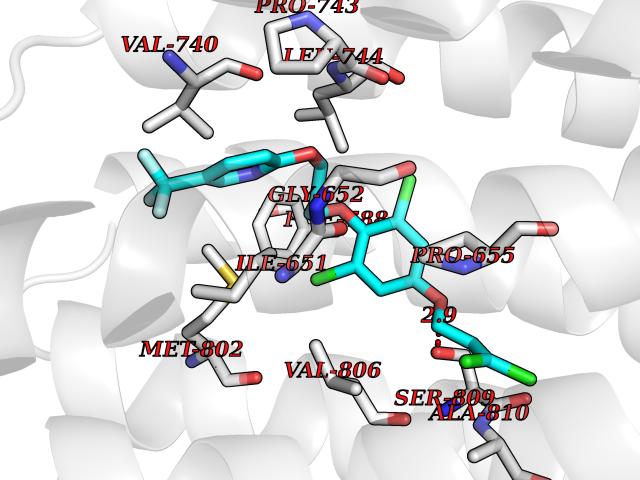 Pymol Map 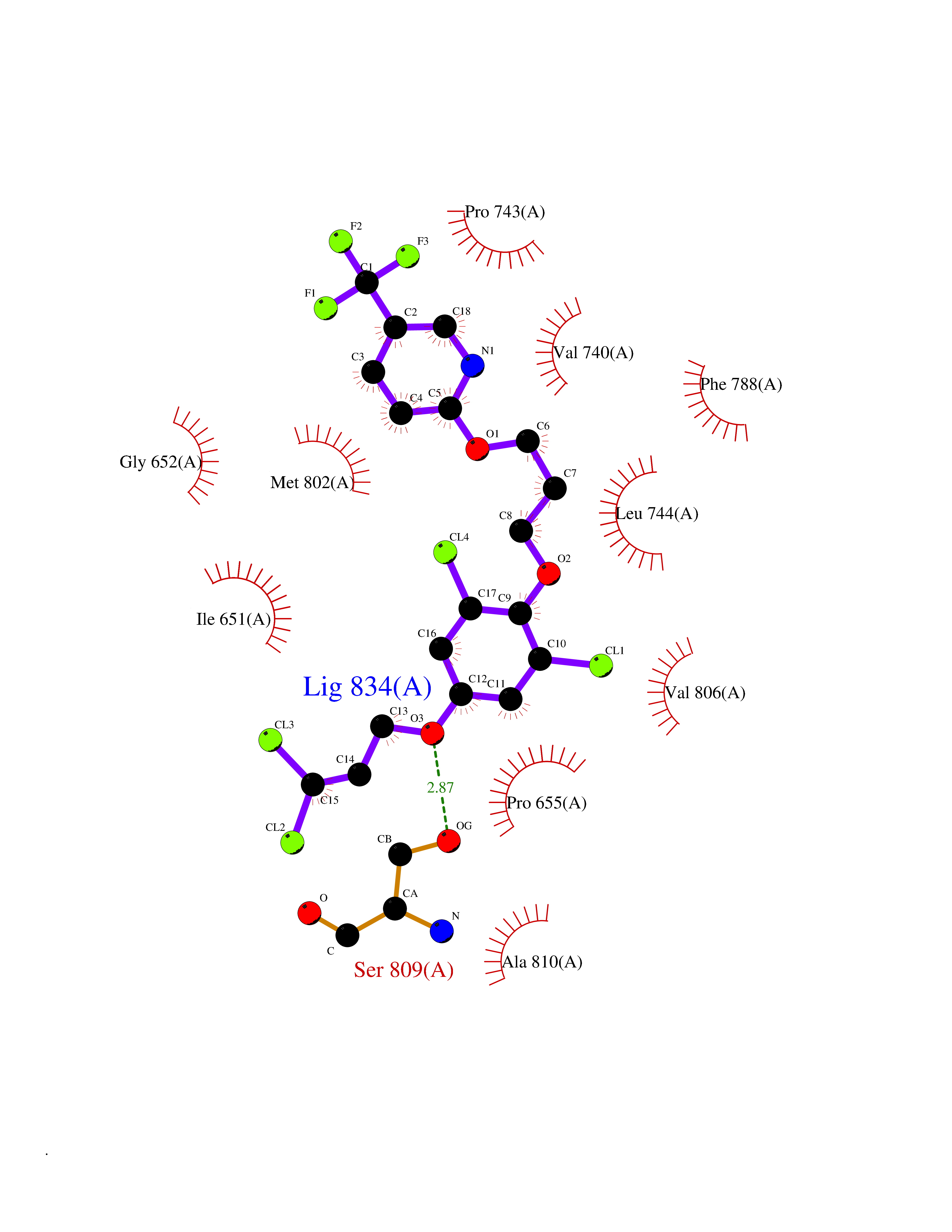 Ligplot Map 3D Binding mode Sequence SPVQYLRWGDPAPIAAVVFACLGLLATLFVTVVFIIYRDTPVVKSSSRELCYIILAGICLGYLCTFXLIAKPKQIYCYLQRIGIGLSPAMSYSALVTKTYRAARILAMSKKSAXAQLVIAFILICIQLGIIVALFIMEPPDIMVYLICNTTNLGVVAPLGYNGLLILACTFYAFKTRNVPANFNEAKYIAFTMYTTCIIWLAFVPIYFGSNYKIITMCFSVSLSATVALGCMFVPKVYIILAKPERN Hydrogen bonds contact Hydrophobic contact | ||||
| 12 | Glutamate receptor ionotropic NMDA 2A (NMDAR2A) | 5KCJ | 7.30 | |
Target general information Gen name GRIN2A Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms NR2A; NMDA receptor NR2A; N-methyl D-aspartate receptor subtype 2A; HNR2A; Glutamate receptor ionotropic, NMDA 2A; Glutamate [NMDA] receptor subunit epsilon-1; GluN2A Protein family Glutamate-gated ion channel (TC 1.A.10.1) family, NR1/GRIN1 subfamily Biochemical class Glutamate-gated ion channel Function Channel activation requires binding of the neurotransmitter glutamate to the epsilon subunit, glycine binding to the zeta subunit, plus membrane depolarization to eliminate channel inhibition by Mg(2+). Sensitivity to glutamate and channel kinetics depend on the subunit composition; channels containing GRIN1 and GRIN2A have higher sensitivity to glutamate and faster kinetics than channels formed by GRIN1 and GRIN2B. Contributes to the slow phase of excitatory postsynaptic current, long-term synaptic potentiation, and learning. Component of NMDA receptor complexes that function as heterotetrameric, ligand-gated ion channels with high calcium permeability and voltage-dependent sensitivity to magnesium. Related diseases Neurodevelopmental disorder with or without hyperkinetic movements and seizures, autosomal dominant (NDHMSD) [MIM:614254]: An autosomal dominant neurodevelopmental disorder characterized by severe intellectual disability and developmental delay, absent speech, muscular hypotonia, dyskinesia, and hyperkinetic movements. Cortical blindness, cerebral atrophy, and seizures are present in some patients. {ECO:0000269|PubMed:21376300, ECO:0000269|PubMed:25167861, ECO:0000269|PubMed:25864721, ECO:0000269|PubMed:27164704, ECO:0000269|PubMed:28095420, ECO:0000269|PubMed:28228639, ECO:0000269|PubMed:28389307, ECO:0000269|PubMed:38538865}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Neurodevelopmental disorder with or without hyperkinetic movements and seizures, autosomal recessive (NDHMSR) [MIM:617820]: An autosomal recessive neurodevelopmental disorder characterized by severe intellectual disability and psychomotor developmental delay, involuntary and stereotypic movements, spasticity, and inability to walk without support. Intractable seizures manifest in some patients. {ECO:0000269|PubMed:27164704, ECO:0000269|PubMed:28051072}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Developmental and epileptic encephalopathy 101 (DEE101) [MIM:619814]: A form of epileptic encephalopathy, a heterogeneous group of early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE101 is an autosomal recessive, severe form characterized by onset of seizures in early infancy. Death in infancy may occur. {ECO:0000269|PubMed:27164704, ECO:0000269|PubMed:34611970}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01931; DB00659; DB06151; DB08838; DB01238; DB00289; DB05824; DB04620; DB03929; DB00647; DB00843; DB00228; DB11823; DB13146; DB06741; DB00142; DB00874; DB08954; DB06738; DB09409; DB09481; DB01043; DB00454; DB00333; DB04896; DB01173; DB00312; DB01174; DB01708; DB00418; DB00193 Interacts with P05067; P35637; Q12879-1; Q13224; Q62936 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Calcium; Cell membrane; Disease variant; Disulfide bond; Epilepsy; Glycoprotein; Intellectual disability; Ion channel; Ion transport; Ligand-gated ion channel; Magnesium; Membrane; Metal-binding; Phosphoprotein; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Signal; Synapse; Transmembrane; Transmembrane helix; Transport; Zinc Protein physicochemical properties Chain ID B Molecular weight (Da) 53395.6 Length 469 Aromaticity 0.11 Instability index 29.84 Isoelectric point 8.72 Charge (pH=7) 5.65 2D Binding mode Binding energy (Kcal/mol) -9.96  Molscript Map 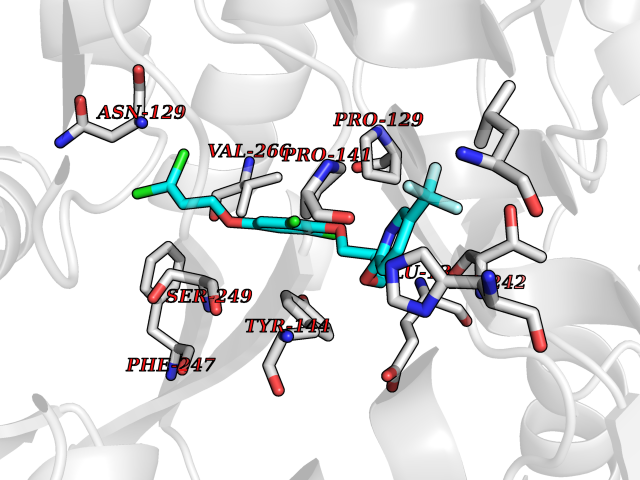 Pymol Map 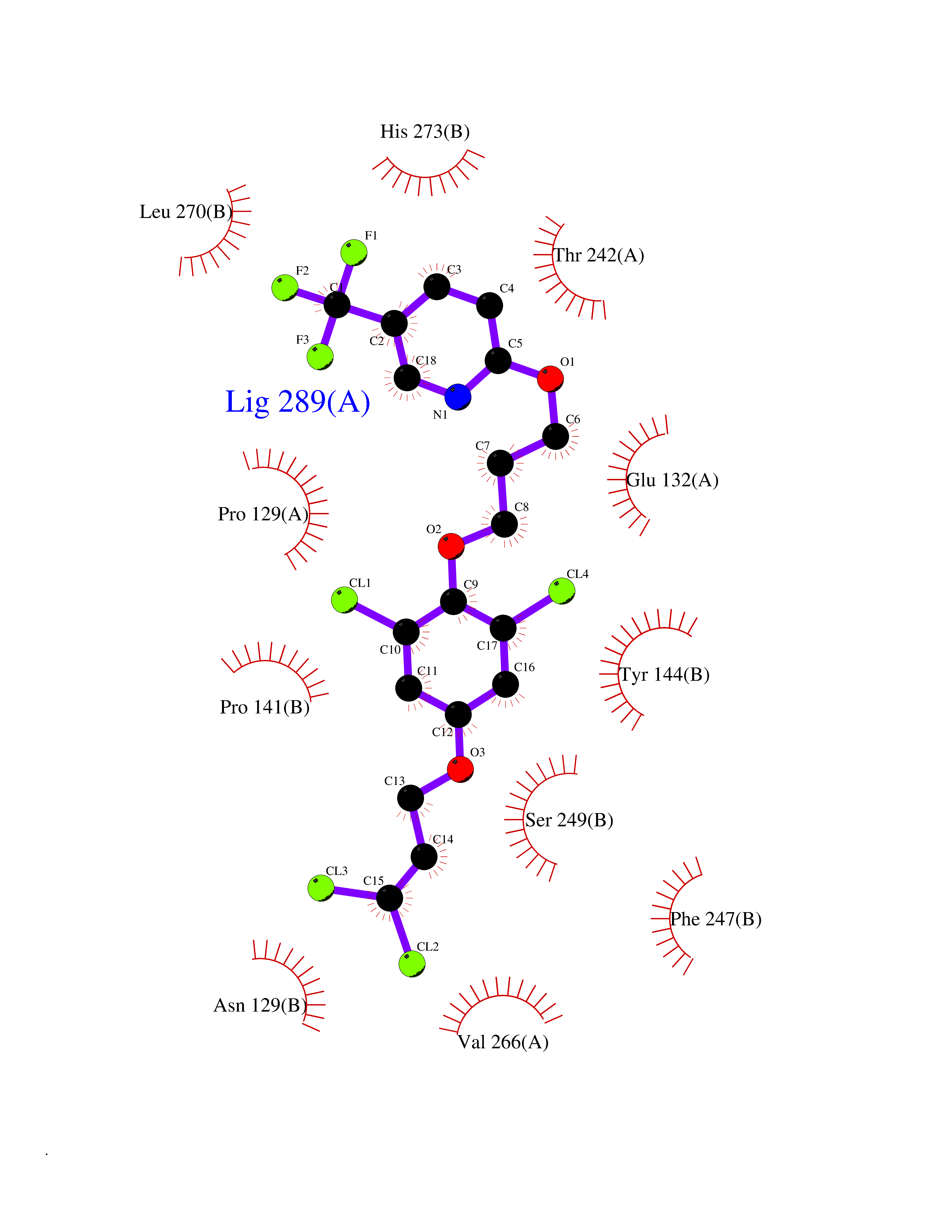 Ligplot Map 3D Binding mode Sequence DNHLSIVTLEEAPFVILKKLSRTVKFTYDLYLVTNGKHGKKVNNVWNGMIGEVVYQRAVMAVGSLTINEERSEVVDFSVPFVETGISVMVSRGTQVTGLSDKKFQRPHDYSPPFRFGTVPNGSTERNIRNNYPYMHQYMTKFNQKGVEDALVSLKTGKLDAFIYDAAVLNYKAGRDEGCKLVTIGSGYIFATTGYGIALQKGSPWKRQIDLALLQFVGDGEMEELETLWLTGICTRLKIVTIHQEPFVYYGFCIDLLIKLARTMNFTYEVHLVADGKFGTQERVNKKEWNGMMGELLSGQADMIVAPLTINNERAQYIEFSKPFKYQGLTILVKKGTRITGINDPRLRNPSDKFIYATVKQSSVDIYFRRQVELSTMYRHMEKHNYESAAEAIQAVRDNKLHAFIWDSAVLEFEASQKCDLVTTGELFFRSGFGIGMRKDSPWKQNVSLSILKSHENGFMEDLDKTWVR Hydrogen bonds contact Hydrophobic contact | ||||
| 13 | Retinoic acid receptor alpha (RARA) | 3KMR | 7.28 | |
Target general information Gen name RARA Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms RAR-alpha; RAR alpha; Nuclear receptor subfamily 1 group B member 1; NR1B1 Protein family Nuclear hormone receptor family, NR1 subfamily Biochemical class Nuclear hormone receptor Function Retinoic acid receptors bind as heterodimers to their target response elements in response to their ligands, all-trans or 9-cis retinoic acid, and regulate gene expression in various biological processes. The RXR/RAR heterodimers bind to the retinoic acid response elements (RARE) composed of tandem 5'-AGGTCA-3' sites known as DR1-DR5. In the absence of ligand, the RXR-RAR heterodimers associate with a multiprotein complex containing transcription corepressors that induce histone acetylation, chromatin condensation and transcriptional suppression. On ligand binding, the corepressors dissociate from the receptors and associate with the coactivators leading to transcriptional activation. RARA plays an essential role in the regulation of retinoic acid-induced germ cell development during spermatogenesis. Has a role in the survival of early spermatocytes at the beginning prophase of meiosis. In Sertoli cells, may promote the survival and development of early meiotic prophase spermatocytes. In concert with RARG, required for skeletal growth, matrix homeostasis and growth plate function. Receptor for retinoic acid. Related diseases Chromosomal aberrations involving RARA are commonly found in acute promyelocytic leukemia. Translocation t(11;17)(q32;q21) with ZBTB16/PLZF; translocation t(15;17)(q21;q21) with PML; translocation t(5;17)(q32;q11) with NPM. The PML-RARA oncoprotein requires both the PML ring structure and coiled-coil domain for both interaction with UBE2I, nuclear microspeckle location and sumoylation. In addition, the coiled-coil domain functions in blocking RA-mediated transactivation and cell differentiation. {ECO:0000269|PubMed:12691149, ECO:0000269|PubMed:8302850, ECO:0000269|PubMed:8562957}. Drugs (DrugBank ID) DB00459; DB00210; DB00523; DB00926; DB00982; DB05785; DB04942; DB00799; DB00755; DB12808 Interacts with O43707-1; O15296; Q15699; Q96RK4; O95273; P51946; Q15910; P50148; Q9UKP3; Q96EZ8; Q15648; Q71SY5; Q15788; Q9Y6Q9; O75376; Q9Y618; Q16236; P13056-2; P48552; Q9UPP1-2; Q9H8W4; P37231; P78527; P19793; P28702; P28702-3; P48443; Q96EB6; P63165; Q8WW24; Q2M1K9; Q91XC0; P59598; Q14457; P48552; Q96CV9; P28702; P48443; Q8WW24 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Chromosomal rearrangement; Cytoplasm; DNA-binding; Isopeptide bond; Metal-binding; Nucleus; Phosphoprotein; Proteomics identification; Proto-oncogene; Receptor; Reference proteome; Transcription; Transcription regulation; Ubl conjugation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A Molecular weight (Da) 27724.1 Length 244 Aromaticity 0.05 Instability index 50.8 Isoelectric point 5.82 Charge (pH=7) -3.61 2D Binding mode Binding energy (Kcal/mol) -9.93  Molscript Map 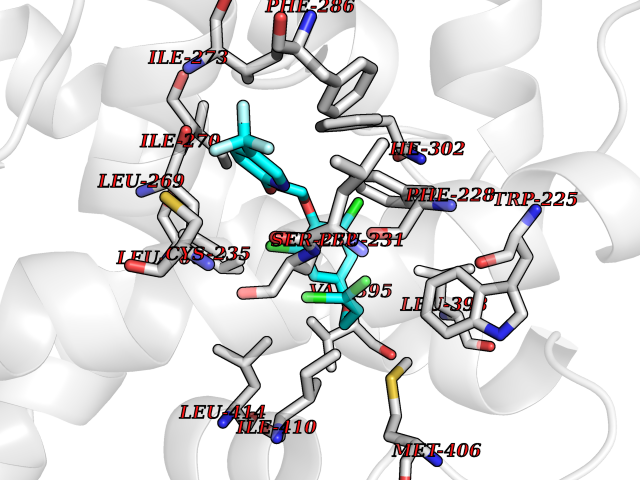 Pymol Map 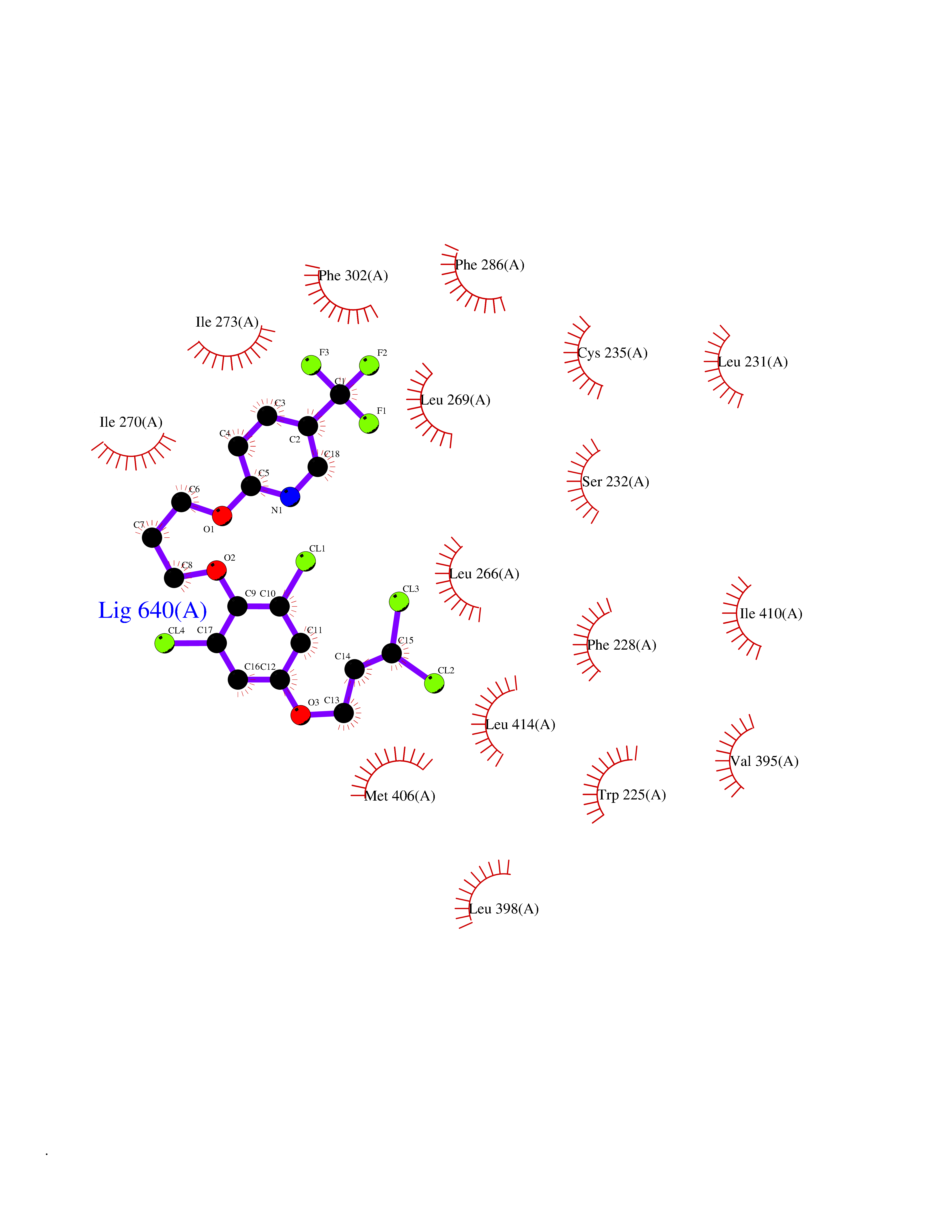 Ligplot Map 3D Binding mode Sequence PEVGELIEKVRKAHQETFPALCQLGKYTTNNSSEQRVSLDIDLWDKFSELSTKCIIKTVEFAKQLPGFTTLTIADQITLLKAACLDILILRICTRYTPEQDTMTFSDGLTLNRTQMHNAGFGPLTDLVFAFANQLLPLEMDDAETGLLSAICLICGDRQDLEQPDRVDMLQEPLLEALKVYVRKRRPSRPHMFPKMLMKITDLRSISAKGAERVITLKMEIPGSMPPLIQEMLEHKILHRLLQE Hydrogen bonds contact Hydrophobic contact | ||||
| 14 | Opioid receptor sigma 1 (OPRS1) | 6DJZ | 7.28 | |
Target general information Gen name SIGMAR1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms hSigmaR1; Sigma1R; Sigma1-receptor; Sigma non-opioid intracellular receptor 1; Sigma 1-type opioid receptor; SRBP; SR31747-binding protein; SR31747 binding protein 1; SR-BP; SIG-1R; Opioid receptor, s Protein family ERG2 family Biochemical class GPCR rhodopsin Function Involved in the regulation of different receptors it plays a role in BDNF signaling and EGF signaling. Also regulates ion channels like the potassium channel and could modulate neurotransmitter release. Plays a role in calcium signaling through modulation together with ANK2 of the ITP3R-dependent calcium efflux at the endoplasmic reticulum. Plays a role in several other cell functions including proliferation, survival and death. Originally identified for its ability to bind various psychoactive drugs it is involved in learning processes, memory and mood alteration. Necessary for proper mitochondrial axonal transport in motor neurons, in particular the retrograde movement of mitochondria. Plays a role in protecting cells against oxidative stress-induced cell death via its interaction with RNF112. Functions in lipid transport from the endoplasmic reticulum and is involved in a wide array of cellular functions probably through regulation of the biogenesis of lipid microdomains at the plasma membrane. Related diseases Amyotrophic lateral sclerosis 16, juvenile (ALS16) [MIM:614373]: A neurodegenerative disorder affecting upper motor neurons in the brain and lower motor neurons in the brain stem and spinal cord, resulting in fatal paralysis. Sensory abnormalities are absent. The pathologic hallmarks of the disease include pallor of the corticospinal tract due to loss of motor neurons, presence of ubiquitin-positive inclusions within surviving motor neurons, and deposition of pathologic aggregates. The etiology of amyotrophic lateral sclerosis is likely to be multifactorial, involving both genetic and environmental factors. The disease is inherited in 5-10% of the cases. {ECO:0000269|PubMed:21842496}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Neuronopathy, distal hereditary motor, autosomal recessive 2 (HMNR2) [MIM:605726]: A form of distal hereditary motor neuronopathy, a heterogeneous group of neuromuscular disorders caused by selective degeneration of motor neurons in the anterior horn of the spinal cord, without sensory deficit in the posterior horn. The overall clinical picture consists of a classical distal muscular atrophy syndrome in the legs without clinical sensory loss. The disease starts with weakness and wasting of distal muscles of the anterior tibial and peroneal compartments of the legs. Later on, weakness and atrophy may expand to the proximal muscles of the lower limbs and/or to the distal upper limbs. HMNR2 is characterized by onset of distal muscle weakness and wasting affecting the lower and upper limbs in the first decade. {ECO:0000269|PubMed:26078401, ECO:0000269|PubMed:27629094}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00321; DB09014; DB00907; DB00514; DB01488; DB00574; DB00502; DB00956; DB00704; DB00540; DB06174; DB00652; DB11186; DB03575; DB05316; DB01708; DB00409; DB01104 Interacts with Q92847-1; Q99720-1; O00213-2; P17612; P50454; P37173 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Amyotrophic lateral sclerosis; Cell junction; Cell membrane; Cell projection; Cytoplasmic vesicle; Direct protein sequencing; Disease variant; Endoplasmic reticulum; Lipid droplet; Lipid transport; Membrane; Neurodegeneration; Neuropathy; Nucleus; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Synapse; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A Molecular weight (Da) 23901 Length 212 Aromaticity 0.14 Instability index 33.12 Isoelectric point 5.61 Charge (pH=7) -5.6 2D Binding mode Binding energy (Kcal/mol) -9.93 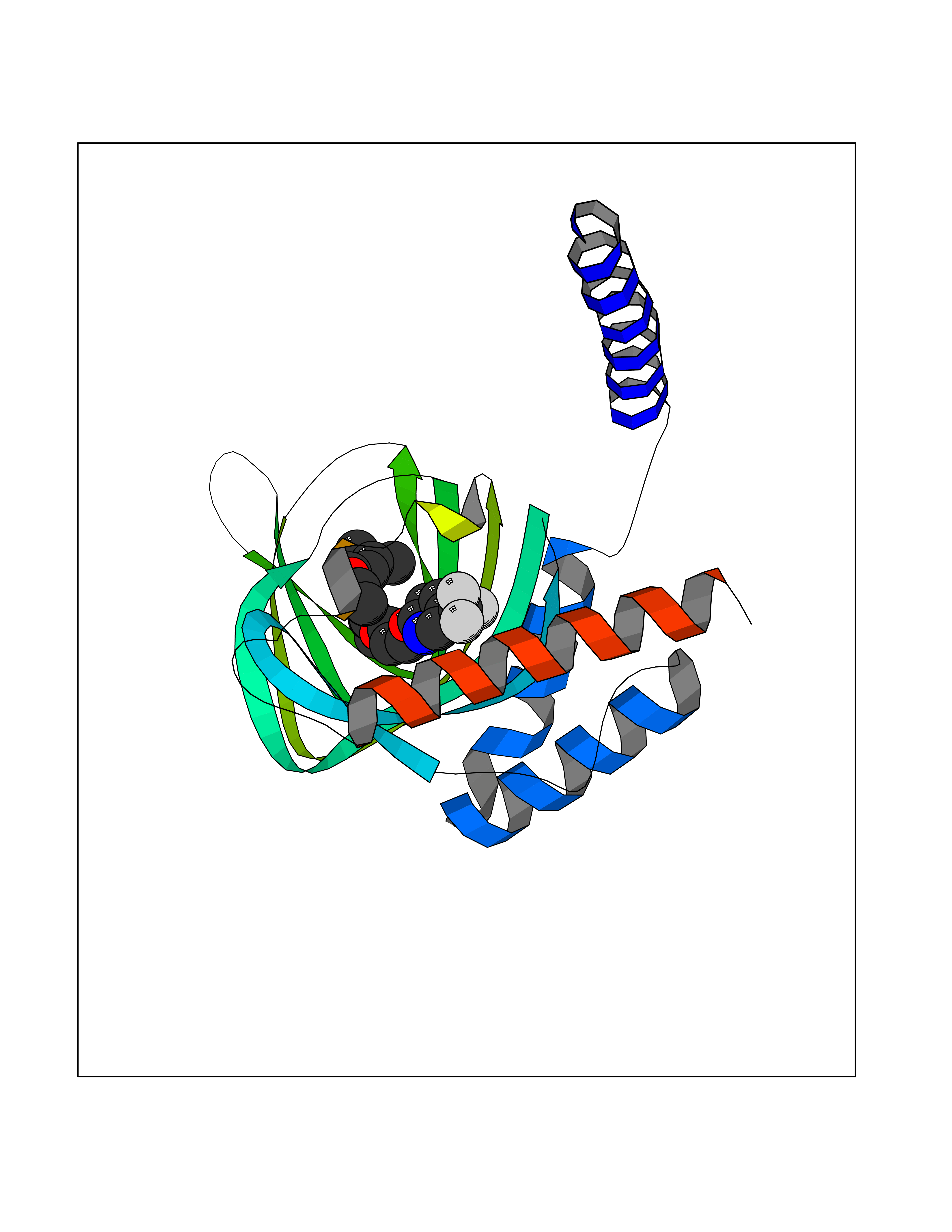 Molscript Map 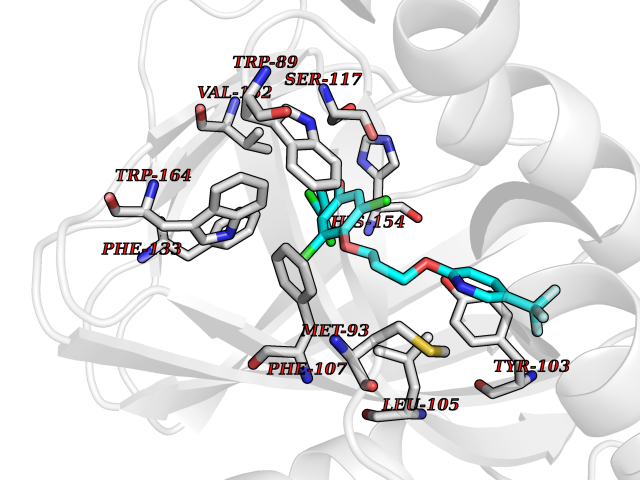 Pymol Map 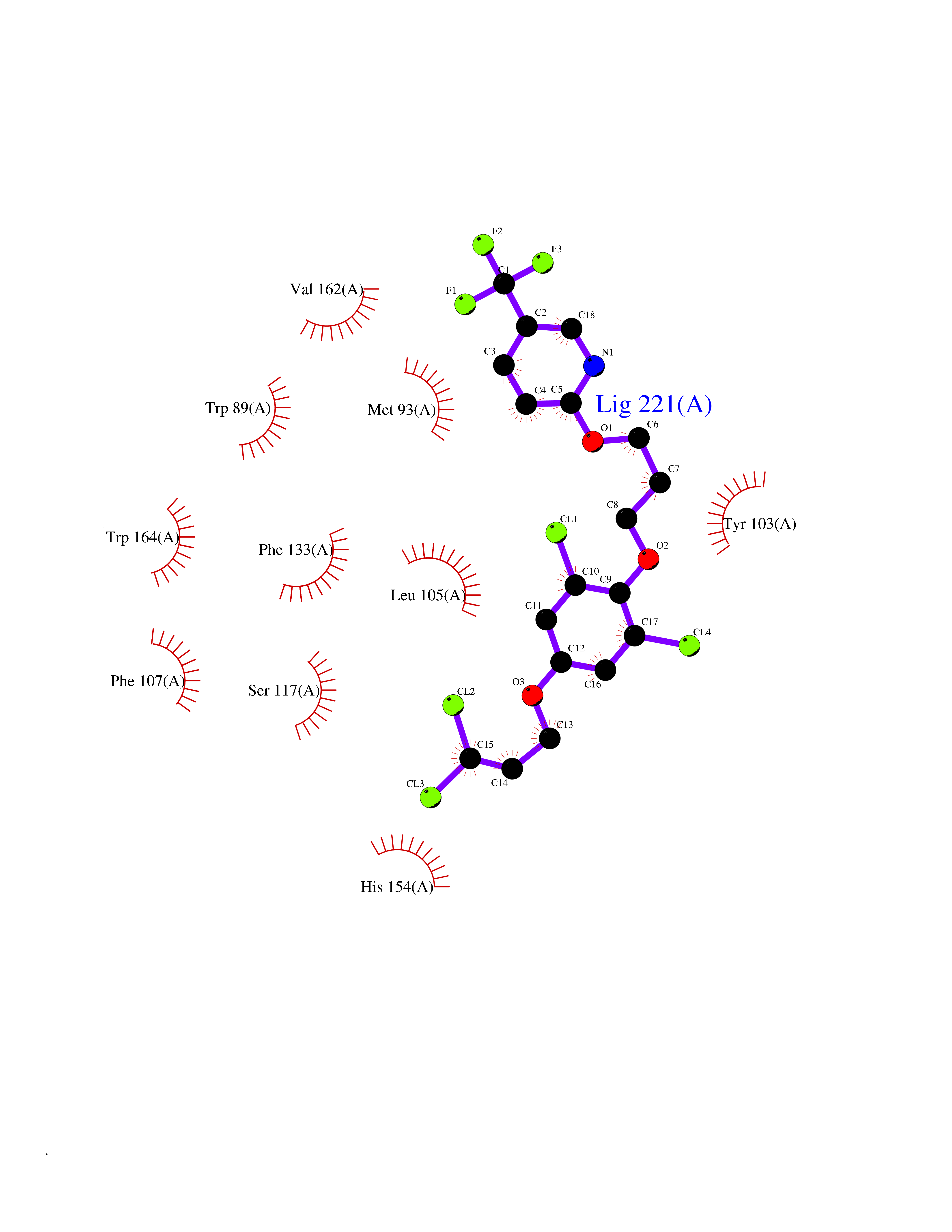 Ligplot Map 3D Binding mode Sequence RWAWAALLLAVAAVLTQVVWLWLGTQSFVFQREEIAQLARQYAGLDHELAFSRLIVELRRLHPGHVLPDEELQWVFVNAGGWMGAMCLLHASLSEYVLLFGTALGSRGHSGRYWAEISDTIISGTFHQWREGTTKSEVFYPGETVVHGPGEATAVEWGPNTWMVEYGRGVIPSTLAFALADTVFSTQDFLTLFYTLRSYARGLRLELTTYLF Hydrogen bonds contact Hydrophobic contact | ||||
| 15 | Monoamine oxidase type B (MAO-B) | 2V5Z | 7.27 | |
Target general information Gen name MAOB Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms MAO-B; Amine oxidase [flavin-containing] B Protein family Flavin monoamine oxidase family Biochemical class CH-NH(2) donor oxidoreductase Function Catalyzes the oxidative deamination of biogenic and xenobiotic amines and has important functions in the metabolism of neuroactive and vasoactive amines in the central nervous system and peripheral tissues. MAOB preferentially degrades benzylamine and phenylethylamine. Related diseases Microvascular complications of diabetes 5 (MVCD5) [MIM:612633]: Pathological conditions that develop in numerous tissues and organs as a consequence of diabetes mellitus. They include diabetic retinopathy, diabetic nephropathy leading to end-stage renal disease, and diabetic neuropathy. Diabetic retinopathy remains the major cause of new-onset blindness among diabetic adults. It is characterized by vascular permeability and increased tissue ischemia and angiogenesis. Disease susceptibility is associated with variants affecting the gene represented in this entry. Homozygosity for the Leu-55 allele is strongly associated with the development of retinal disease in diabetic patients. Drugs (DrugBank ID) DB08176; DB02211; DB08516; DB08480; DB01472; DB04307; DB07512; DB07513; DB00915; DB00182; DB06698; DB04889; DB00215; DB09130; DB04147; DB00988; DB01363; DB00668; DB01175; DB02509; DB03147; DB14914; DB00614; DB04818; DB02095; DB01247; DB00601; DB01577; DB01442; DB01171; DB08082; DB02643; DB04677; DB03894; DB08804; DB04820; DB00184; DB04821; DB12612; DB01626; DB00780; DB00191; DB00388; DB01132; DB00721; DB01168; DB01367; DB09363; DB06654; DB01037; DB01104; DB14569; DB09042; DB00752; DB16446; DB09185; DB04832; DB00909 Interacts with P55212; P28329-3; Q8NI60; Q5RI15; Q92915-2; P22607; Q53GS7; P06396; P01112; O14901; P13473-2; P21397; Q9BVL2; O75400-2; P62826; Q6NTF9-3; Q9Y371; Q7Z699; Q9UMX0; Q9Y649 EC number EC 1.4.3.4 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Direct protein sequencing; FAD; Flavoprotein; Membrane; Mitochondrion; Mitochondrion outer membrane; Oxidoreductase; Proteomics identification; Reference proteome; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A,B Molecular weight (Da) 56019.9 Length 494 Aromaticity 0.09 Instability index 34.81 Isoelectric point 6.51 Charge (pH=7) -2.2 2D Binding mode Binding energy (Kcal/mol) -9.92 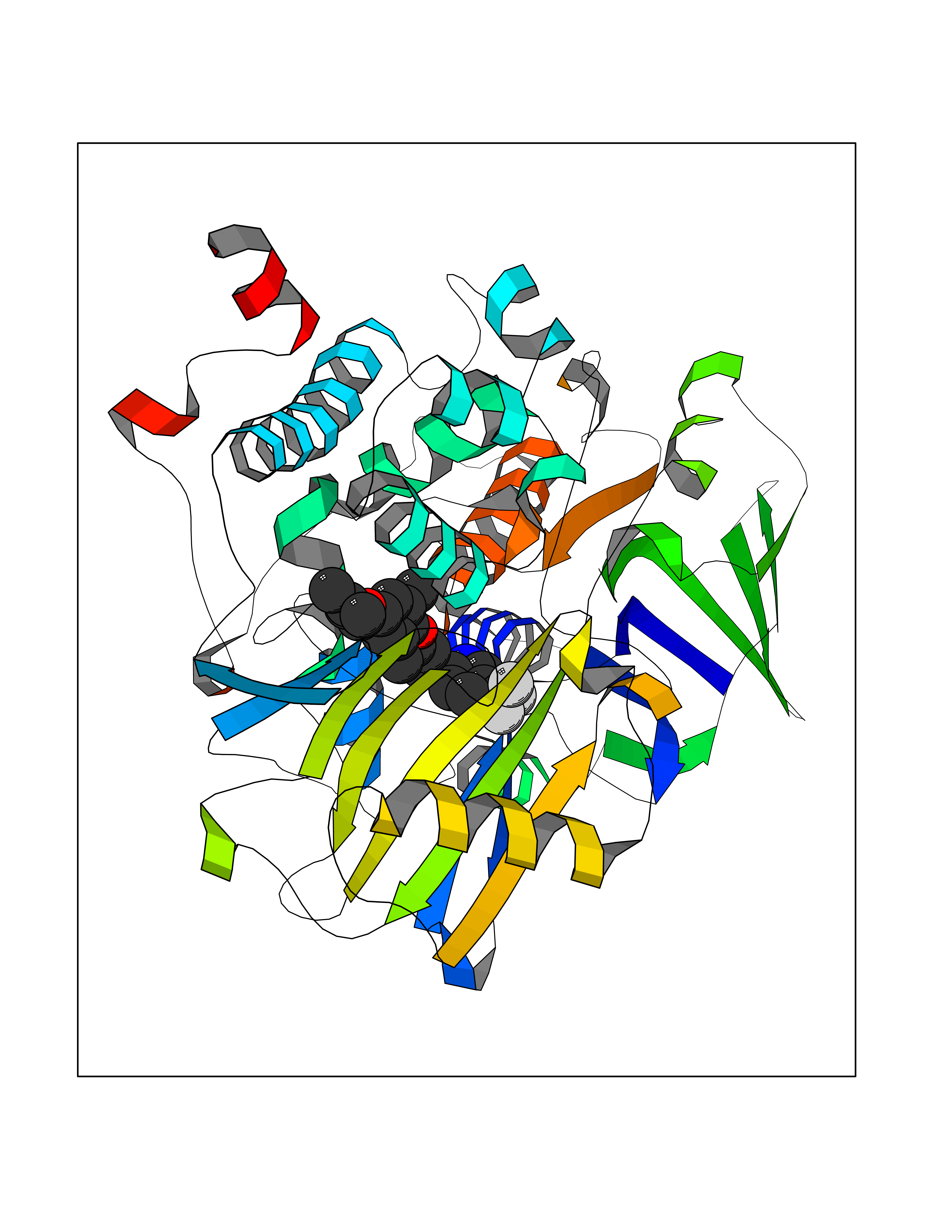 Molscript Map 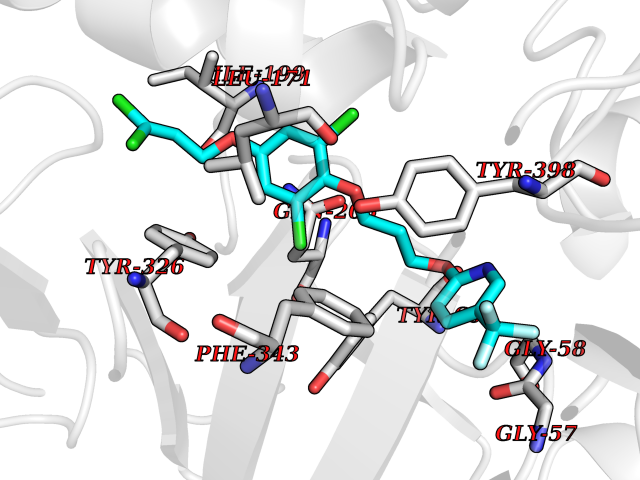 Pymol Map 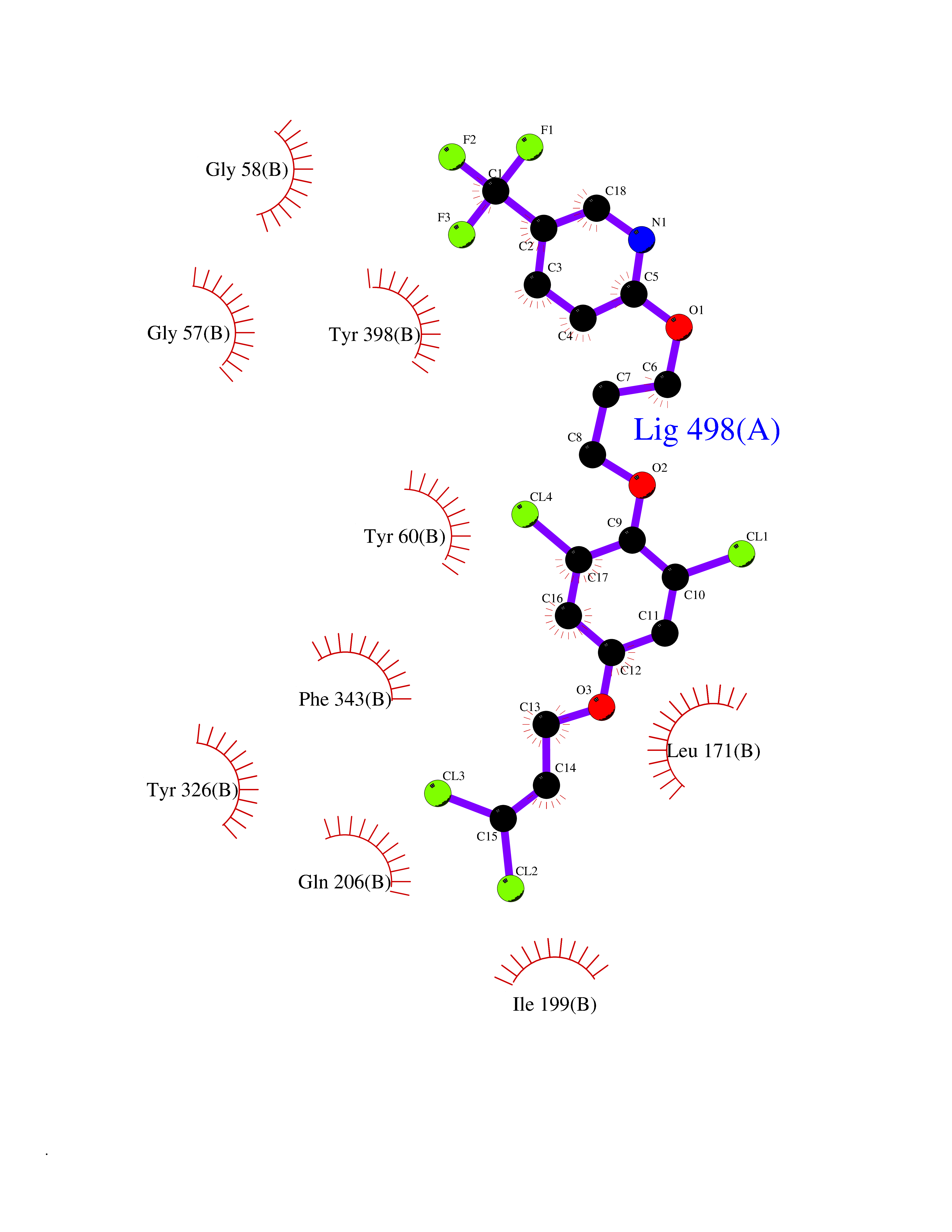 Ligplot Map 3D Binding mode Sequence NKCDVVVVGGGISGMAAAKLLHDSGLNVVVLEARDRVGGRTYTLRNQKVKYVDLGGSYVGPTQNRILRLAKELGLETYKVNEVERLIHHVKGKSYPFRGPFPPVWNPITYLDHNNFWRTMDDMGREIPSDAPWKAPLAEEWDNMTMKELLDKLCWTESAKQLATLFVNLCVTAETHEVSALWFLWYVKQCGGTTRIISTTNGGQERKFVGGSGQVSERIMDLLGDRVKLERPVIYIDQTRENVLVETLNHEMYEAKYVISAIPPTLGMKIHFNPPLPMMRNQMITRVPLGSVIKCIVYYKEPFWRKKDYCGTMIIDGEEAPVAYTLDDTKPEGNYAAIMGFILAHKARKLARLTKEERLKKLCELYAKVLGSLEALEPVHYEEKNWCEEQYSGGCYTTYFPPGILTQYGRVLRQPVDRIYFAGTETATHWSGYMEGAVEAGERAAREILHAMGKIPEDEIWQSEPESVDVPAQPITTTFLERHLPSVPGLLRLI Hydrogen bonds contact Hydrophobic contact | ||||
| 16 | SEC14-like protein 4 | 4TLG | 7.24 | |
Target general information Gen name SEC14L4 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms TAP3 Protein family NA Biochemical class Transport protein Function Lipid binding.Transporter activity. Related diseases Chondrodysplasia with platyspondyly, distinctive brachydactyly, hydrocephaly, and microphthalmia (CDP-PBHM) [MIM:300863]: A disease characterized by chondrodysplasia, severe platyspondyly, hydrocephaly, and facial features with microphthalmia. Bone abnormalities include a distinctive metaphyseal cupping of the metacarpals, metatarsals, and phalanges. Affected females show a milder phenotype with small stature, sometimes associated with body asymmetry and mild intellectual disability. {ECO:0000269|PubMed:20181727}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB14003; DB11635; DB11251; DB00163 Interacts with Q96LC9; O43186; P78358; Q9NYQ3; Q0VD86; Q15323; O76011; P50221; Q6FHY5; Q02548; P26367; Q9H8W4; Q04864; Q04864-2; Q9UHV2; P15884; P15884-3; Q96N21; Q9BYV2; Q8N6Y0; Q9H0C1 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Lipid-binding; Proteomics identification; Reference proteome; Transport Protein physicochemical properties Chain ID A,B Molecular weight (Da) 23947.6 Length 210 Aromaticity 0.1 Instability index 50.84 Isoelectric point 5.55 Charge (pH=7) -3.11 2D Binding mode Binding energy (Kcal/mol) -9.88 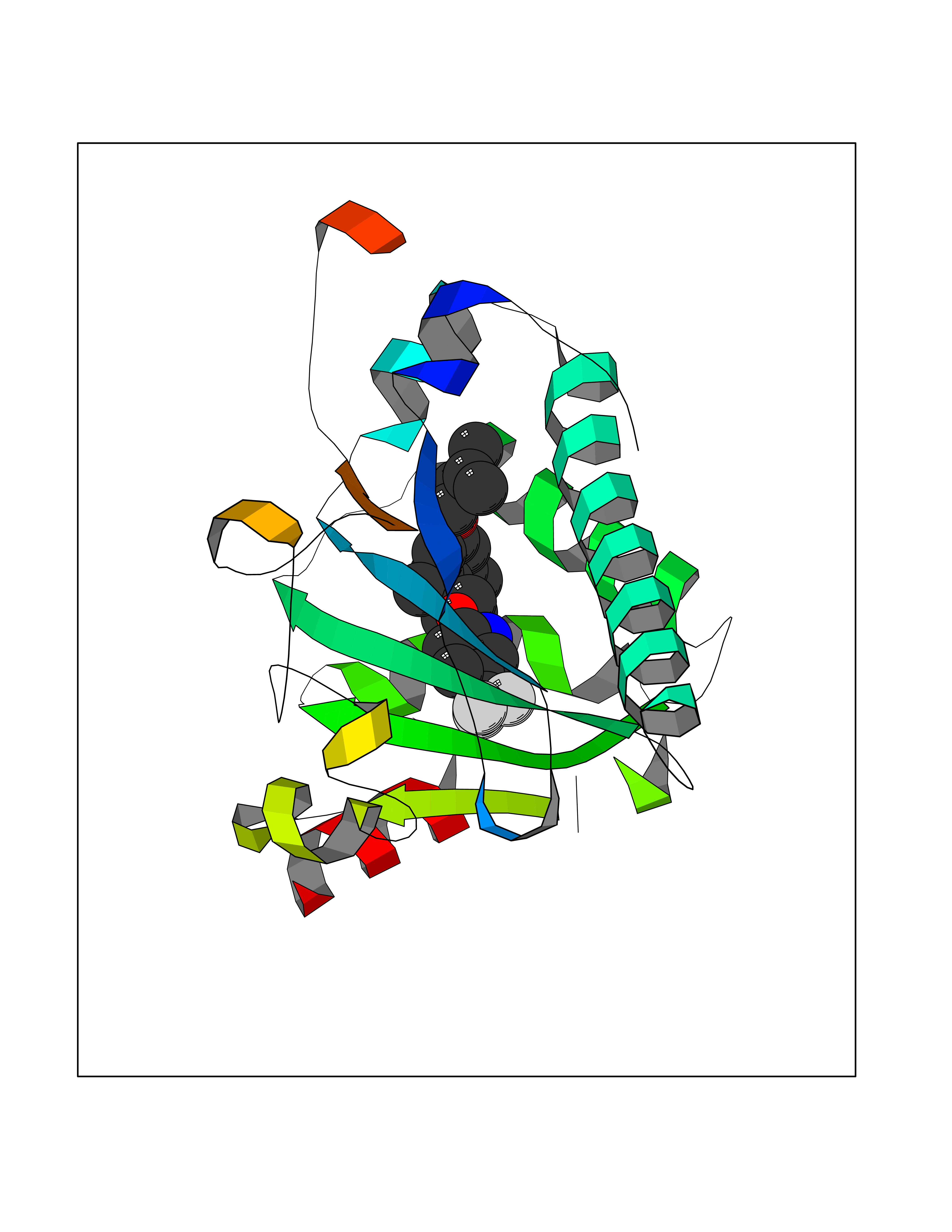 Molscript Map 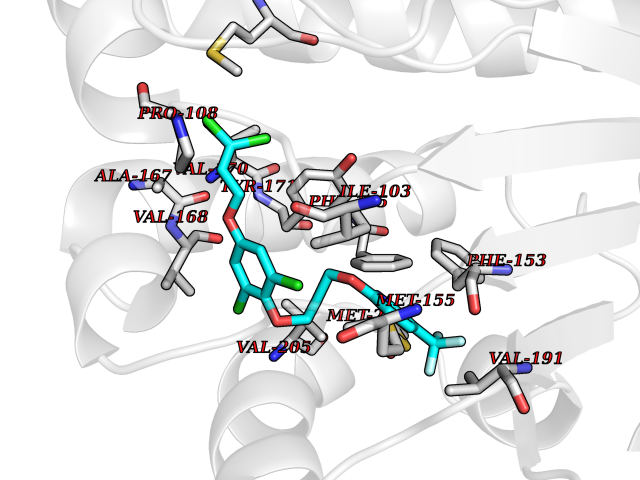 Pymol Map 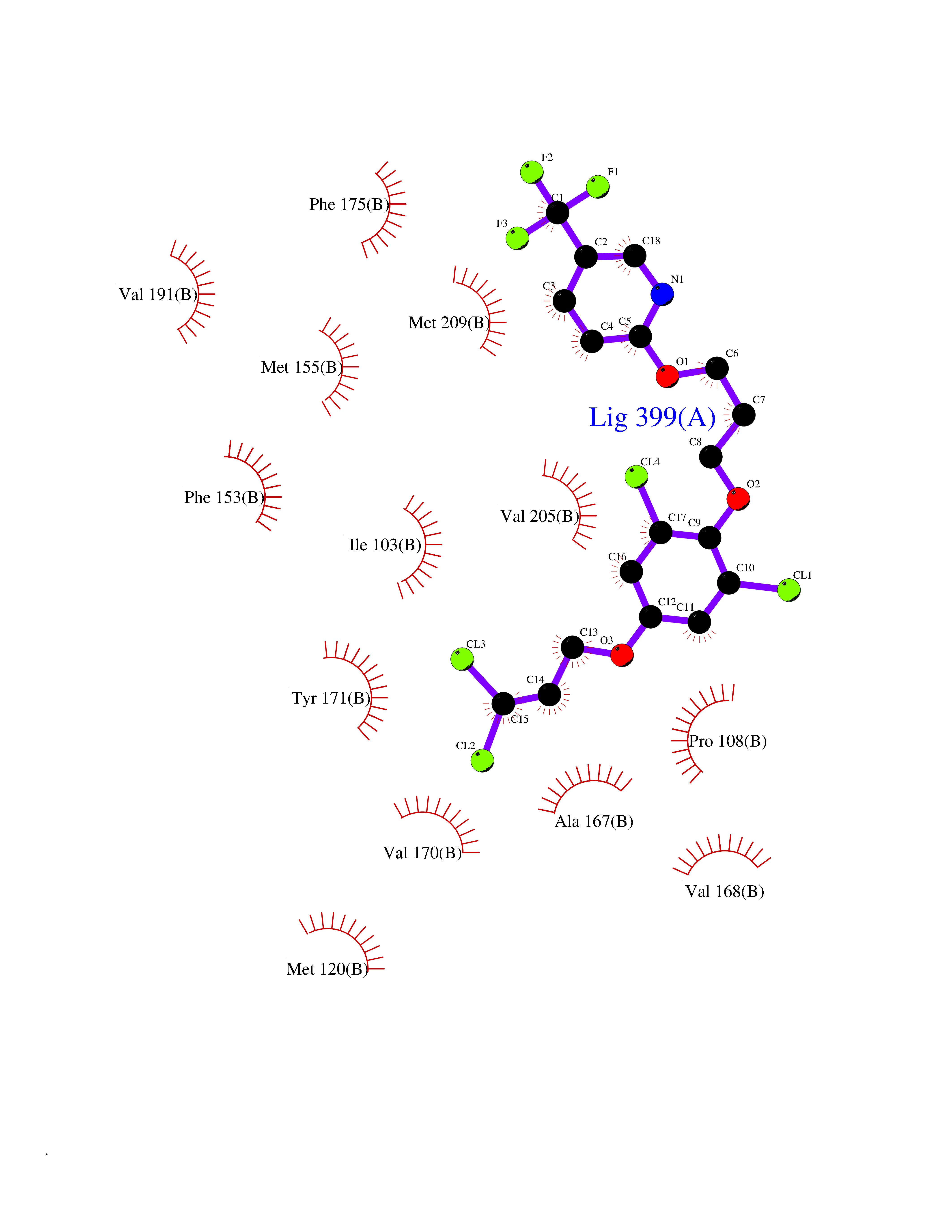 Ligplot Map 3D Binding mode Sequence VTWQPPEVIQLYDSGGLCGYDYEGCPVYFNIIGSLDPKGLLLSASKQDMIRKRIKVCELLLHECELQTQKLGRKIEMALMVFDMEGLSLKHLWKPAVEVYQQFFSILEANYPETLKNLIVIRAPKLFPVAFNLVKSFMSEETRRKIVILGDNWKQELTKFISPDQLPVEFGGTMTDPDGNPKCLTKINYGGEVPKSYYPDKASEETLQSM Hydrogen bonds contact Hydrophobic contact | ||||
| 17 | UDP-N-acetylglucosamine 1-carboxyvinyltransferase | 1C3R | 7.23 | |
Target general information Gen name murA Organism Aquifex aeolicus (strain VF5) Uniprot ID TTD ID NA Synonyms aq_1023 Protein family Histone deacetylase family Biochemical class Lyase Function UDP-N-acetylglucosamine 1-carboxyvinyltransferase activity. Related diseases Atrial fibrillation, familial, 14 (ATFB14) [MIM:615378]: A familial form of atrial fibrillation, a common sustained cardiac rhythm disturbance. Atrial fibrillation is characterized by disorganized atrial electrical activity and ineffective atrial contraction promoting blood stasis in the atria and reduces ventricular filling. It can result in palpitations, syncope, thromboembolic stroke, and congestive heart failure. {ECO:0000269|PubMed:19808477}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Genetic variations in SCN2B may be involved in Brugada syndrome (PubMed:23559163). This tachyarrhythmia is characterized by right bundle branch block and ST segment elevation on an electrocardiogram (ECG). It can cause the ventricles to beat so fast that the blood is prevented from circulating efficiently in the body. When this situation occurs, the individual will faint and may die in a few minutes if the heart is not reset. {ECO:0000269|PubMed:23559163}. Drugs (DrugBank ID) DB04297; DB02546 Interacts with NA EC number NA Uniprot keywords 3D-structure; Acetoin catabolism; Metal-binding; Reference proteome; Zinc Protein physicochemical properties Chain ID A,B Molecular weight (Da) 42338.9 Length 372 Aromaticity 0.12 Instability index 35.74 Isoelectric point 5.77 Charge (pH=7) -6.5 2D Binding mode Binding energy (Kcal/mol) -9.86 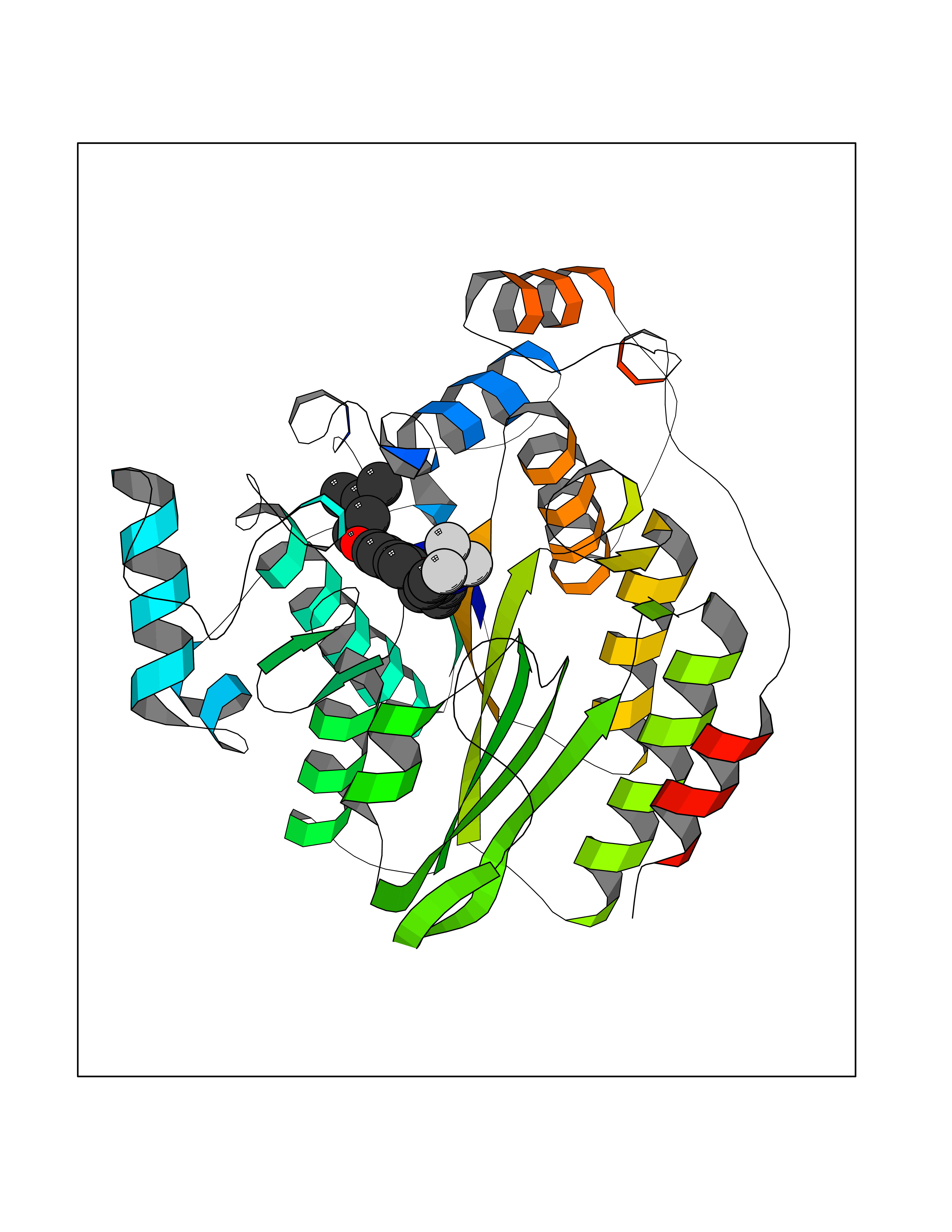 Molscript Map 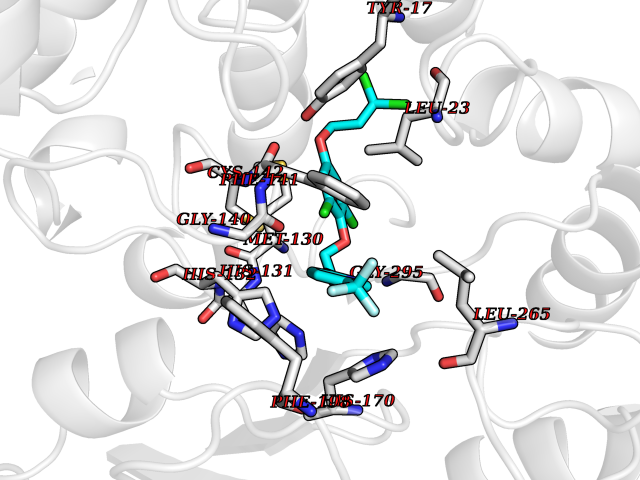 Pymol Map 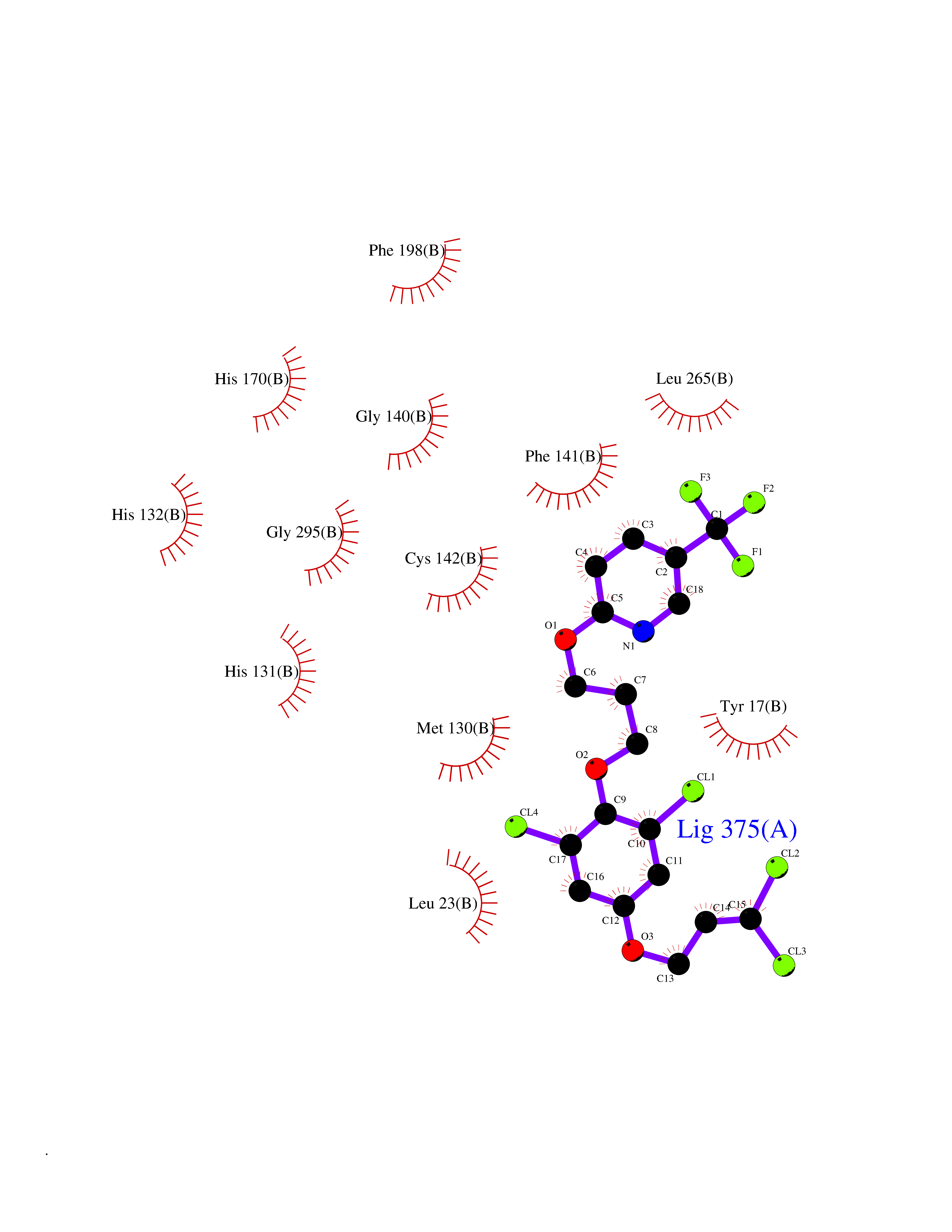 Ligplot Map 3D Binding mode Sequence KKVKLIGTLDYGKYRYPKNHPLKIPRVSLLLRFKDAMNLIDEKELIKSRPATKEELLLFHTEDYINTLMEAERSQSVPKGAREKYNIGGYENPVSYAMFTGSSLATGSTVQAIEEFLKGNVAFNPAGGMHHAFKSRANGFCYINNPAVGIEYLRKKGFKRILYIDLDAHHCDGVQEAFYDTDQVFVLSLHQSPEYAFPFEKGFLEEIGEGKGKGYNLNIPLPKGLNDNEFLFALEKSLEIVKEVFEPEVYLLQLGTDPLLEDYLSKFNLSNVAFLKAFNIVREVFGEGVYLGGGGYHPYALARAWTLIWCELSGREVPEKLNNKAKELLKSIDFEEFDDEVDRSYMLETLKDPWRGGEVRKEVKDTLEKAKA Hydrogen bonds contact Hydrophobic contact | ||||
| 18 | Retinoic acid receptor beta (RARB) | 4DM6 | 7.22 | |
Target general information Gen name RARB Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms RAR-epsilon; RAR-beta; Nuclear receptor subfamily 1 group B member 2; NR1B2; HBV-activated protein; HAP Protein family Nuclear hormone receptor family, NR1 subfamily Biochemical class Nuclear hormone receptor Function Retinoic acid receptors bind as heterodimers to their target response elements in response to their ligands, all-trans or 9-cis retinoic acid, and regulate gene expression in various biological processes. The RXR/RAR heterodimers bind to the retinoic acid response elements (RARE) composed of tandem 5'-AGGTCA-3' sites known as DR1-DR5. In the absence or presence of hormone ligand, acts mainly as an activator of gene expression due to weak binding to corepressors. In concert with RARG, required for skeletal growth, matrix homeostasis and growth plate function. Receptor for retinoic acid. Related diseases Microphthalmia, syndromic, 12 (MCOPS12) [MIM:615524]: A form of microphthalmia, a disorder of eye formation, ranging from small size of a single eye to complete bilateral absence of ocular tissues (anophthalmia). In many cases, microphthalmia/anophthalmia occurs in association with syndromes that include non-ocular abnormalities. MCOPS12 patients manifest variable features, including diaphragmatic hernia, pulmonary hypoplasia, and cardiac abnormalities. {ECO:0000269|PubMed:24075189, ECO:0000269|PubMed:27120018}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00459; DB00210; DB00523; DB02877; DB00926; DB05785; DB04942; DB00799; DB00755; DB12808 Interacts with O95273; P50222; Q9UBK2; P62195; P28702; P28702-3; P48443; P03255 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; Disease variant; DNA-binding; Metal-binding; Microphthalmia; Nucleus; Phosphoprotein; Proto-oncogene; Receptor; Reference proteome; Transcription; Transcription regulation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A,B Molecular weight (Da) 25904.1 Length 229 Aromaticity 0.06 Instability index 44.34 Isoelectric point 7.55 Charge (pH=7) 0.73 2D Binding mode Binding energy (Kcal/mol) -9.85  Molscript Map 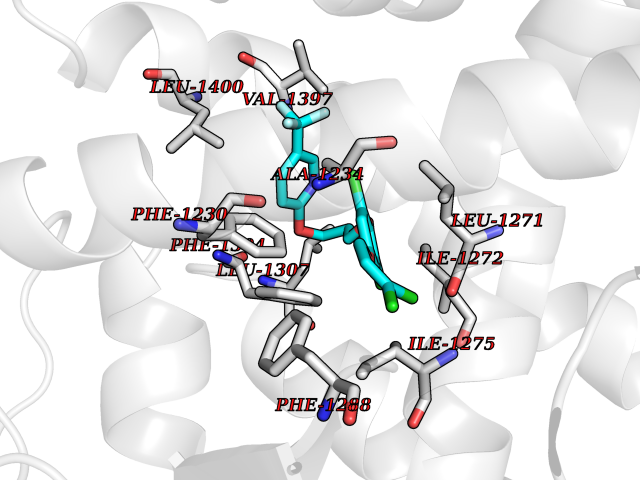 Pymol Map 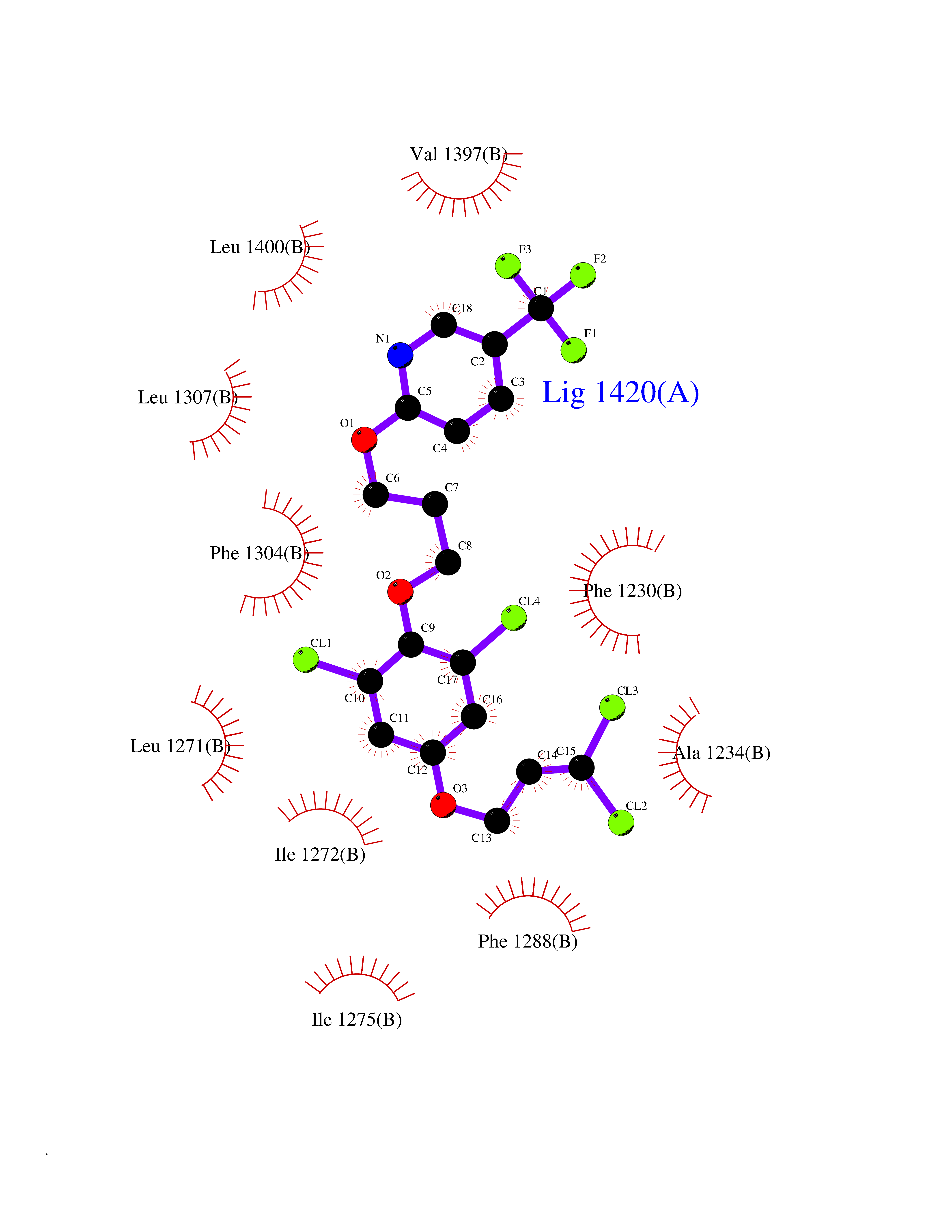 Ligplot Map 3D Binding mode Sequence TEKIRKAHQETFPSLCQLGKYTTNSSADHRVRLDLGLWDKFSELATKCIIKIVEFAKRLPGFTGLTIADQITLLKAACLDILILRICTRYTPEQDTMTFSDGLTLNRTQMHNAGFGPLTDLVFTFANQLLPLEMDDTETGLLSAICLICGDRQDLEEPTKVDKLQEPLLEALKIYIRKRRPSKPHMFPKILMKITDLRSISAKGAERVITLKMEIPGSMPPLIQEMLEN Hydrogen bonds contact Hydrophobic contact | ||||
| 19 | Retinoic acid receptor gamma (RARG) | 1FCY | 7.22 | |
Target general information Gen name RARG Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms RAR-gamma; Nuclear receptor subfamily 1 group B member 3; NR1B3 Protein family Nuclear hormone receptor family, NR1 subfamily Biochemical class Nuclear hormone receptor Function Receptor for retinoic acid. Retinoic acid receptors bind as heterodimers to their target response elements in response to their ligands, all-trans or 9-cis retinoic acid, and regulate gene expression in various biological processes. The RAR/RXR heterodimers bind to the retinoic acid response elements (RARE) composed of tandem 5'-AGGTCA-3' sites known as DR1-DR5. In the absence of ligand, acts mainly as an activator of gene expression due to weak binding to corepressors. Required for limb bud development. In concert with RARA or RARB, required for skeletal growth, matrix homeostasis and growth plate function (By similarity). Related diseases Cystic fibrosis (CF) [MIM:219700]: A common generalized disorder of the exocrine glands which impairs clearance of secretions in a variety of organs. It is characterized by the triad of chronic bronchopulmonary disease (with recurrent respiratory infections), pancreatic insufficiency (which leads to malabsorption and growth retardation) and elevated sweat electrolytes. It is the most common genetic disease in Caucasians, with a prevalence of about 1 in 2'000 live births. Inheritance is autosomal recessive. {ECO:0000269|PubMed:10094564, ECO:0000269|PubMed:10869121, ECO:0000269|PubMed:10923036, ECO:0000269|PubMed:11242048, ECO:0000269|PubMed:12167682, ECO:0000269|PubMed:12394343, ECO:0000269|PubMed:12529365, ECO:0000269|PubMed:1284466, ECO:0000269|PubMed:1284468, ECO:0000269|PubMed:1284529, ECO:0000269|PubMed:1284530, ECO:0000269|PubMed:1284548, ECO:0000269|PubMed:1379210, ECO:0000269|PubMed:15528182, ECO:0000269|PubMed:15716351, ECO:0000269|PubMed:16822950, ECO:0000269|PubMed:1695717, ECO:0000269|PubMed:1699669, ECO:0000269|PubMed:17098864, ECO:0000269|PubMed:1710600, ECO:0000269|PubMed:1712898, ECO:0000269|PubMed:17182731, ECO:0000269|PubMed:20008117, ECO:0000269|PubMed:20150177, ECO:0000269|PubMed:20691141, ECO:0000269|PubMed:21884936, ECO:0000269|PubMed:2236053, ECO:0000269|PubMed:23818989, ECO:0000269|PubMed:25330774, ECO:0000269|PubMed:26846474, ECO:0000269|PubMed:27241308, ECO:0000269|PubMed:28001373, ECO:0000269|PubMed:28067262, ECO:0000269|PubMed:28087700, ECO:0000269|PubMed:32026723, ECO:0000269|PubMed:33572515, ECO:0000269|PubMed:7504969, ECO:0000269|PubMed:7505694, ECO:0000269|PubMed:7505767, ECO:0000269|PubMed:7508414, ECO:0000269|PubMed:7513296, ECO:0000269|PubMed:7517264, ECO:0000269|PubMed:7520022, ECO:0000269|PubMed:7522211, ECO:0000269|PubMed:7524909, ECO:0000269|PubMed:7524913, ECO:0000269|PubMed:7525450, ECO:0000269|PubMed:7537150, ECO:0000269|PubMed:7541273, ECO:0000269|PubMed:7541510, ECO:0000269|PubMed:7543567, ECO:0000269|PubMed:7544319, ECO:0000269|PubMed:7581407, ECO:0000269|PubMed:7606851, ECO:0000269|PubMed:7680525, ECO:0000269|PubMed:7683628, ECO:0000269|PubMed:7683954, ECO:0000269|PubMed:8081395, ECO:0000269|PubMed:8406518, ECO:0000269|PubMed:8522333, ECO:0000269|PubMed:8723693, ECO:0000269|PubMed:8723695, ECO:0000269|PubMed:8800923, ECO:0000269|PubMed:8829633, ECO:0000269|PubMed:8910473, ECO:0000269|PubMed:8956039, ECO:0000269|PubMed:9101301, ECO:0000269|PubMed:9222768, ECO:0000269|PubMed:9375855, ECO:0000269|PubMed:9401006, ECO:0000269|PubMed:9443874, ECO:0000269|PubMed:9452048, ECO:0000269|PubMed:9452054, ECO:0000269|PubMed:9452073, ECO:0000269|PubMed:9482579, ECO:0000269|PubMed:9507391, ECO:0000269|PubMed:9521595, ECO:0000269|PubMed:9554753, ECO:0000269|PubMed:9736778, ECO:0000269|PubMed:9804160, ECO:0000269|PubMed:9921909}. The disease is caused by variants affecting the gene represented in this entry. There is some evidence that the functional defect caused by the most common variant Phe-508 DEL can be corrected by the binding to the snake phospholipase A2 crotoxin basic subunit CB. This toxin both disrupts the Phe-508 DEL-cytokeratin 8 complex, allowing for the escape from degradation, and increases the chloride channel current (PubMed:27241308). {ECO:0000269|PubMed:27241308}.; DISEASE: Congenital bilateral absence of the vas deferens (CBAVD) [MIM:277180]: An autosomal recessive disease characterized by vas deferens aplasia resulting in azoospermia and male infertility. CBAVD may occur in isolation or as a manifestation of cystic fibrosis. {ECO:0000269|PubMed:10066035, ECO:0000269|PubMed:10651488, ECO:0000269|PubMed:17329263, ECO:0000269|PubMed:7529962, ECO:0000269|PubMed:7539342, ECO:0000269|PubMed:9067761, ECO:0000269|PubMed:9736778, ECO:0000269|Ref.117}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07294; DB07031; DB00459; DB00210; DB00523; DB02466; DB03466; DB02741; DB03279; DB00926; DB00982; DB05785; DB05467; DB02258; DB00799; DB00755; DB12808 Interacts with Q96RK4; P13349; P31321; P28702; P48443; O60504-2 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; DNA-binding; Isopeptide bond; Metal-binding; Methylation; Nucleus; Proteomics identification; Receptor; Reference proteome; Transcription; Transcription regulation; Ubl conjugation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A Molecular weight (Da) 26574.9 Length 236 Aromaticity 0.06 Instability index 49.98 Isoelectric point 5.76 Charge (pH=7) -2.95 2D Binding mode Binding energy (Kcal/mol) -9.85  Molscript Map 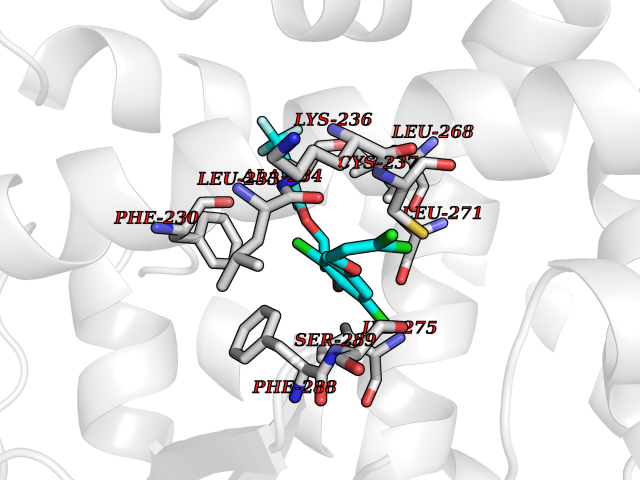 Pymol Map 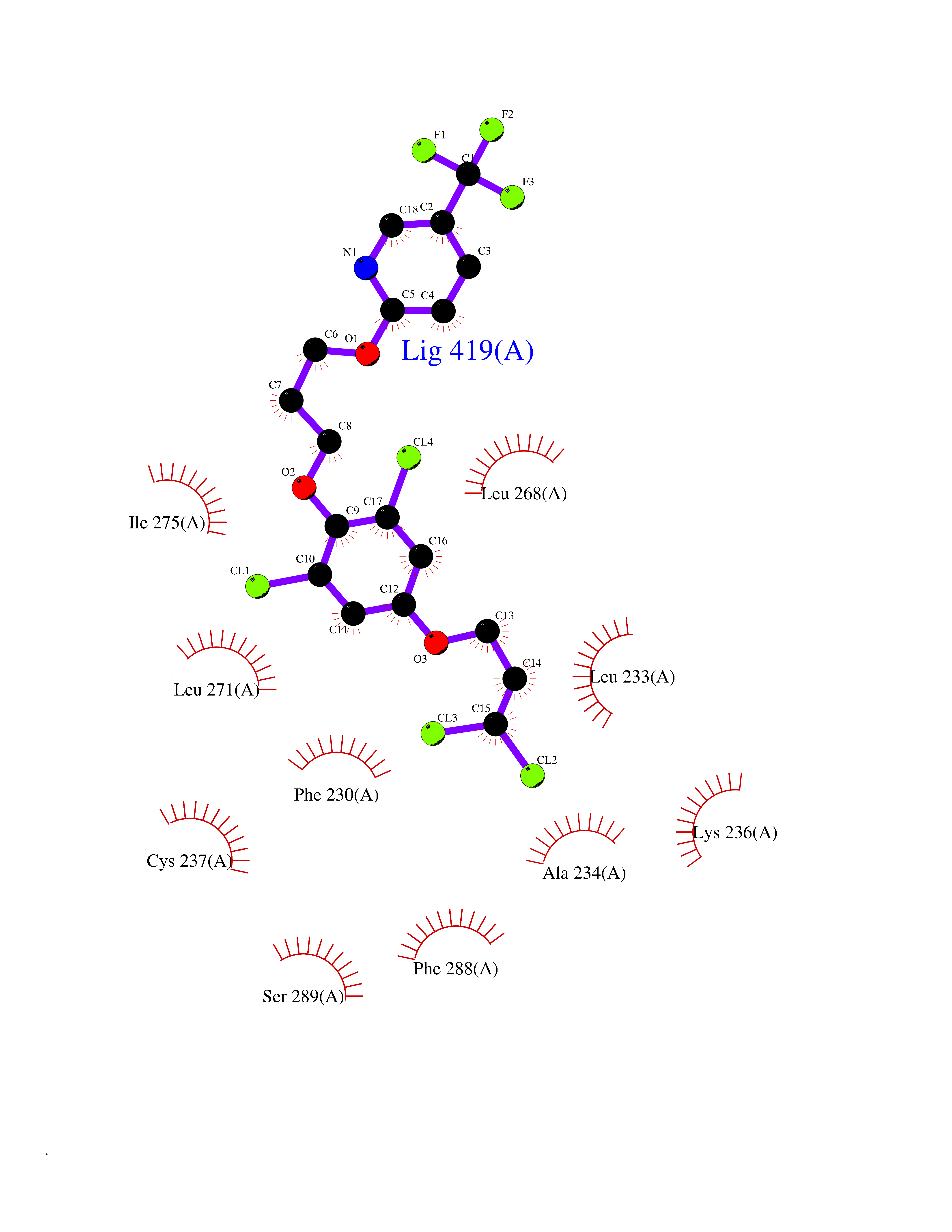 Ligplot Map 3D Binding mode Sequence ASPQLEELITKVSKAHQETFPSLCQLGKYTTNSSADHRVQLDLGLWDKFSELATKCIIKIVEFAKRLPGFTGLSIADQITLLKAACLDILMLRICTRYTPEQDTMTFSDGLTLNRTQMHNAGFGPLTDLVFAFAGQLLPLEMDDTETGLLSAICLICGDRMDLEEPEKVDKLQEPLLEALRLYARRRRPSQPYMFPRMLMKITDLRGISTKGAERAITLKMEIPGPMPPLIREMLE Hydrogen bonds contact Hydrophobic contact | ||||
| 20 | Cytochrome c oxidase subunit 2 | 3VRJ | 7.22 | |
Target general information Gen name MT-CO2 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms MTCO2;COXII;COII;COX2 Protein family Cytochrome c oxidase subunit 2 family Biochemical class Immune system Function Copper ion binding.Cytochrome-c oxidase activity. Related diseases Mitochondrial complex IV deficiency (MT-C4D) [MIM:220110]: A disorder of the mitochondrial respiratory chain with heterogeneous clinical manifestations, ranging from isolated myopathy to severe multisystem disease affecting several tissues and organs. Features include hypertrophic cardiomyopathy, hepatomegaly and liver dysfunction, hypotonia, muscle weakness, exercise intolerance, developmental delay, delayed motor development and intellectual disability. Some affected individuals manifest a fatal hypertrophic cardiomyopathy resulting in neonatal death. A subset of patients manifest Leigh syndrome. {ECO:0000269|PubMed:10486321}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02659; DB04464; DB05412 Interacts with Q9NZ94-2; P49281-3 EC number 7.1.1.9 Uniprot keywords 3D-structure; Copper; Disease variant; Electron transport; Magnesium; Membrane; Metal-binding; Mitochondrion; Mitochondrion inner membrane; Primary mitochondrial disease; Proteomics identification; Reference proteome; Respiratory chain; Translocase; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID C Molecular weight (Da) 21687.9 Length 189 Aromaticity 0.11 Instability index 38 Isoelectric point 5.68 Charge (pH=7) -3.26 2D Binding mode Binding energy (Kcal/mol) -9.85 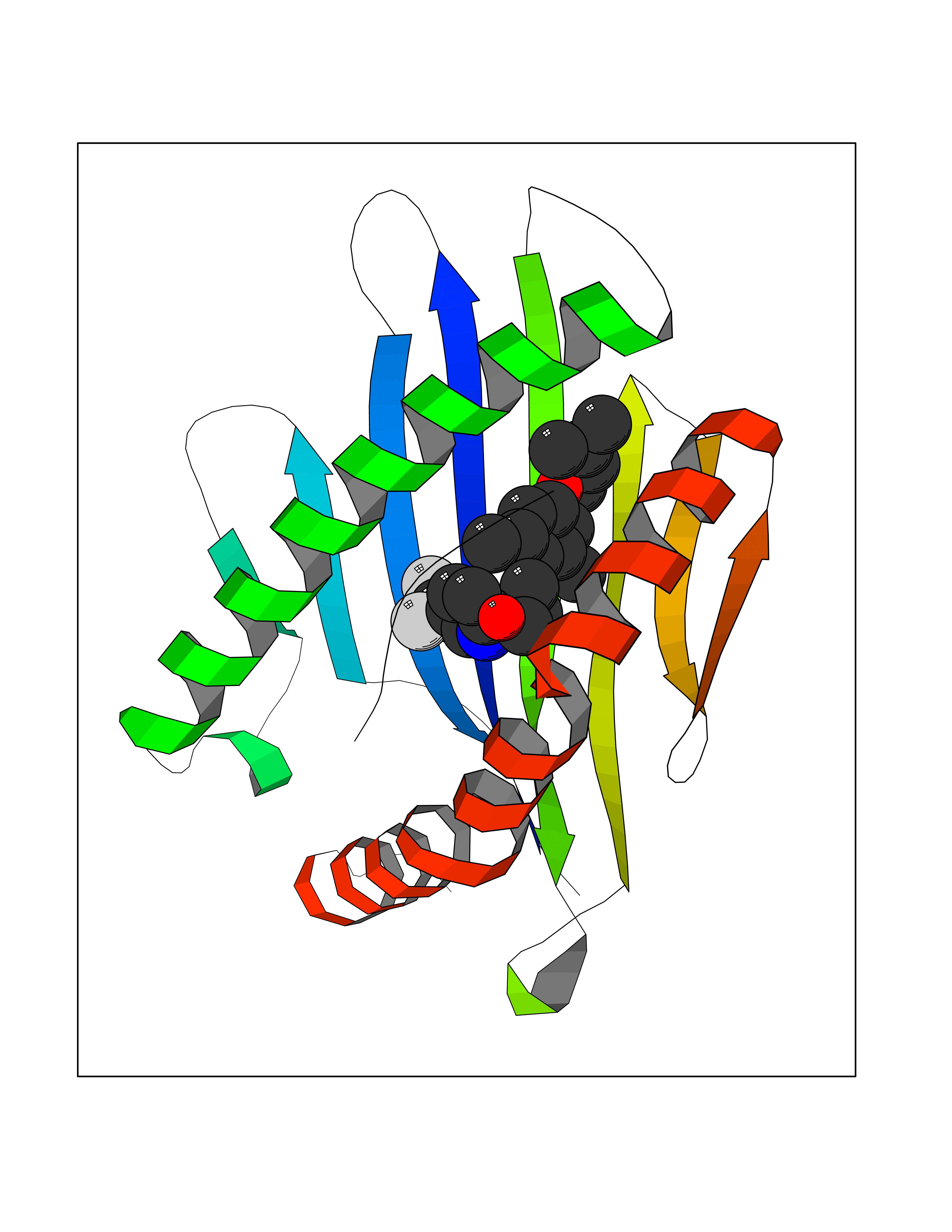 Molscript Map 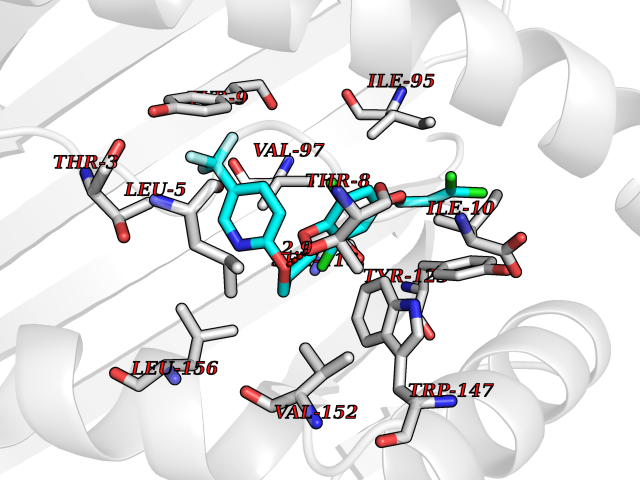 Pymol Map 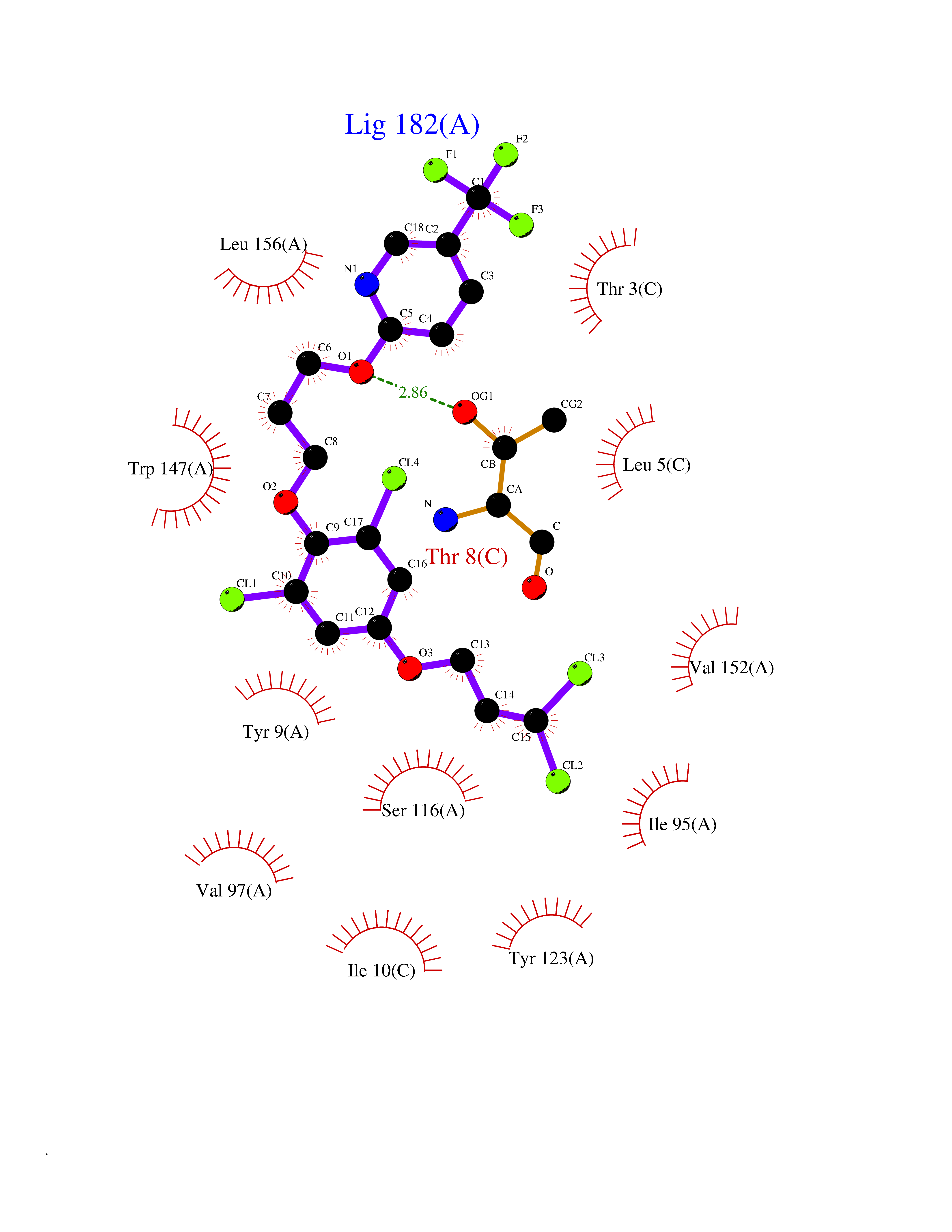 Ligplot Map 3D Binding mode Sequence SHSMRYFYTAMSRPGRGEPRFIAVGYVDDTQFVRFDSDAASPRMAPRAPWIEQEGPEYWDGETRNMKASAQTYRENLRIALRYYNQSEAGSHIIQVMYGCDVGPDGRLLRGHDQSAYDGKDYIALNEDLSSWTAADTAAQITQRKWEAARVAEQLRAYLEGLCVEWLRRYLENGKETLQLTTKLTNTNI Hydrogen bonds contact Hydrophobic contact | ||||