Job Results:
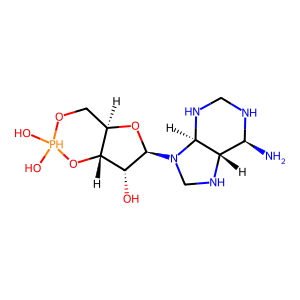
Ligand
Structure
Job ID
e848ab91d8ae28235d6e66c6c8c71011
Job name
NA
Time
2024-09-30 12:19:50
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 1 | Dopamine beta-hydroxylase | 4ZEL | 7.89 | |
Target general information Gen name DBH Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Copper type II ascorbate-dependent monooxygenase family Biochemical class Oxidoreductase Function Catalytic activity.Copper ion binding.Dopamine beta-monooxygenase activity.L-ascorbic acid binding. Related diseases Orthostatic hypotension 1 (ORTHYP1) [MIM:223360]: A form of orthostatic hypotension due to congenital dopamine beta-hydroxylase deficiency. Orthostatic hypotension, also known as postural hypotension, is a finding defined as a 20-mm Hg decrease in systolic pressure or a 10-mm Hg decrease in diastolic pressure occurring 3 minutes after a person has risen from supine to standing. Symptoms include dizziness, blurred vision, and sometimes syncope. ORTHYP1 is an autosomal recessive condition apparent from infancy or early childhood and characterized by low plasma and urinary levels of norepinephrine and epinephrine, and episodic hypoglycemia. {ECO:0000269|PubMed:11857564}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00126; DB06774; DB09130; DB05394; DB00822; DB00988; DB00968; DB00550 Interacts with P00352; P63010-2; Q04656; Q8WUW1; Q9UNS2; Q71DI3; P61978; Q9Y2M5; Q92876; P08727; Q14693; P0DPK4; Q6GQQ9-2; P27986-2; Q9ULX5; Q96D59; Q8N6K7-2; Q9GZS3; Q8IUW3; Q86WT6-2 EC number 1.14.17.1 Uniprot keywords 3D-structure; Catecholamine biosynthesis; Copper; Cytoplasmic vesicle; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; Membrane; Metal-binding; Monooxygenase; Oxidoreductase; Proteomics identification; Reference proteome; Secreted; Signal-anchor; Transmembrane; Transmembrane helix; Vitamin C Protein physicochemical properties Chain ID A,B Molecular weight (Da) 123694 Length 1094 Aromaticity 0.1 Instability index 51.85 Isoelectric point 5.84 Charge (pH=7) -24.5 2D Binding mode Binding energy (Kcal/mol) -8.29  Molscript Map 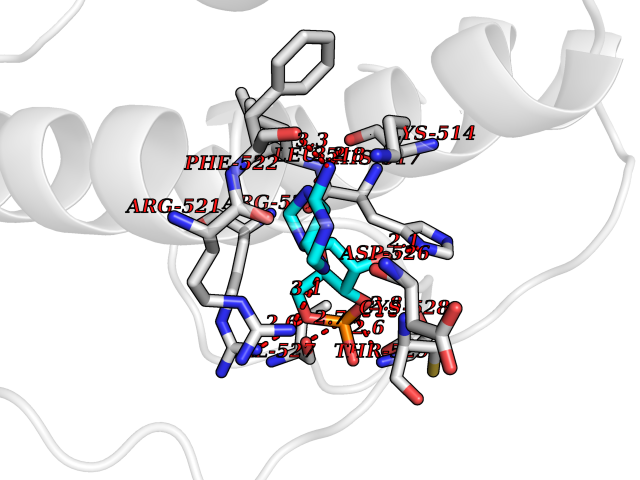 Pymol Map 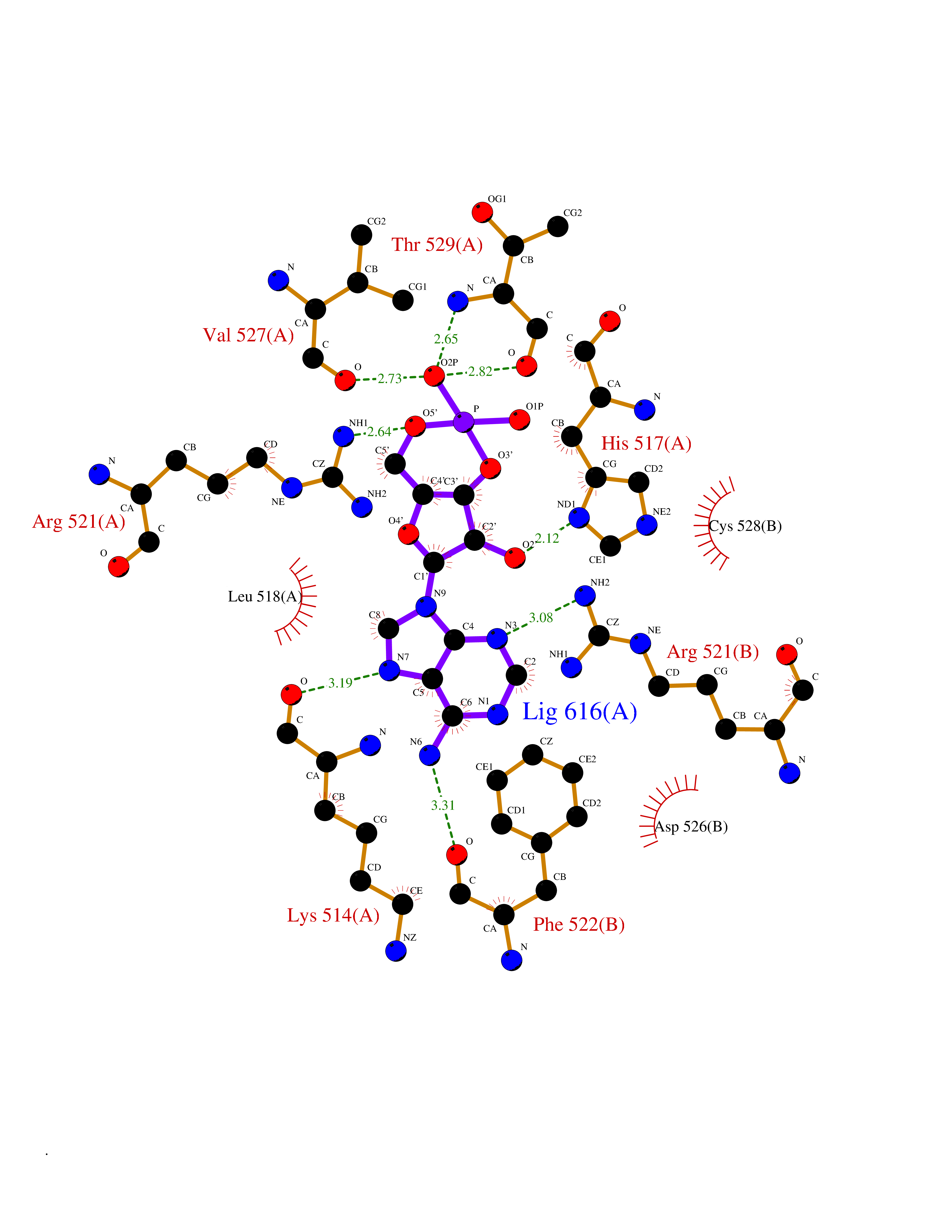 Ligplot Map 3D Binding mode Sequence PLPYHIPLDPEGSLELSWNVSYTQEAIHFQLLVRRLKAGVLFGMSDRGELENADLVVLWTDGDAYFADAWSDQKGQIHLDPQQDYQLLQVQRTPEGLTLLFKRPFGTCDPKDYLIEDGTVHLVYGILEEPFRSLEAINGSGLQMGLQRVQLLKPNIPEPELPSDACTMEVQAPNIQIPSQETTYWCYIKELPKGFSRHHIIKYEPIVTKGNEALVHHMEVFQCAPEMDSVPHFSGPCDSKMKPDRLNYCRHVLAAWALGAKAFYYPEEAGLAFGGPGSSRYLRLEVHYHNPLVIEGRNDSSGIRLYYTAKLRRFNAGIMELGLVYTPVMAIPPRETAFILTGYCTDKCTQLALPPSGIHIFASQLHTHLTGRKVVTVLVRDGREWEIVNQDNHYSPHFQEIRMLKKVVSVHPGDVLITSCTYNTEDRELATVGGFGILEEMCVNYVHYYPQTQLELCKSAVDAGFLQKYFHLINRFNNEDVCTCPQASVSQQFTSVPWNSFNRDVLKALYSFAPISMHCNKSSAVRFQGEWNLQPLPKVISTLEEPTVVSPLPYHIPLDPEGSLELSWNVSYTQEAIHFQLLVRRLKAGVLFGMSDRGELENADLVVLAYFADAWSDQKGQIHLDPQQDYQLLQVQRTPEGLTLLFKRPFGTCDPKDYLIEDGTVHLVYGILEEPFRSLEAINGSGLQMGLQRVQLLKPNIPEPELPSDACTMEVQAPNIQIPSQETTYWCYIKELPKGFSRHHIIKYEPIVTKGNEALVHHMEVFQCAPEVPHFSGPCDSKMLNYCRHVLAAWALGAKAFYYPEEAGLAFGGPGSSRYLRLEVHYHNPLVIEGRNDSSGIRLYYTAKLRRFNAGIMELGLVYTPVMAIPPRETAFILTGYCTDKCTQLALPPSGIHIFASQLHTHLTGRKVVTVLVRDGREWEIVNQDNHYSPHFQEIRMLKKVVSVHPGDVLITSCTYNTEDRELATVGGFGILEEMCVNYVHYYPQTQLELCKSAVDAGFLQKYFHLINRFNNEDVCTCPQASVSQQFTSVPWNSFNRDVLKALYSFAPISMHCNKSSAVRFQGEWNLQPLPKVISTLEEPTPQCVVSIGG Hydrogen bonds contact Hydrophobic contact | ||||
| 2 | Endothelin-converting enzyme 1 (ECE1) | 3DWB | 7.35 | |
Target general information Gen name ECE1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms ECE-1 Protein family Peptidase M13 family Biochemical class Peptidase Function Converts big endothelin-1 to endothelin-1. Related diseases Hirschsprung disease, cardiac defects, and autonomic dysfunction (HCAD) [MIM:613870]: A disorder characterized by skip-lesions Hirschsprung disease, craniofacial abnormalities and other dysmorphic features, cardiac defects including ductus arteriosus, small subaortic ventricular septal defect, small atrial septal defect, and autonomic dysfunction. {ECO:0000269|PubMed:9915973}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07171 Interacts with P49760; A8MQ03; Q8IUG1; P60370; P60410 EC number EC 3.4.24.71 Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disease variant; Disulfide bond; Glycoprotein; Hirschsprung disease; Hydrolase; Membrane; Metal-binding; Metalloprotease; Phosphoprotein; Protease; Proteomics identification; Reference proteome; Signal-anchor; Transmembrane; Transmembrane helix; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 75247.9 Length 660 Aromaticity 0.12 Instability index 46.29 Isoelectric point 5.33 Charge (pH=7) -18.3 2D Binding mode Binding energy (Kcal/mol) -7.28 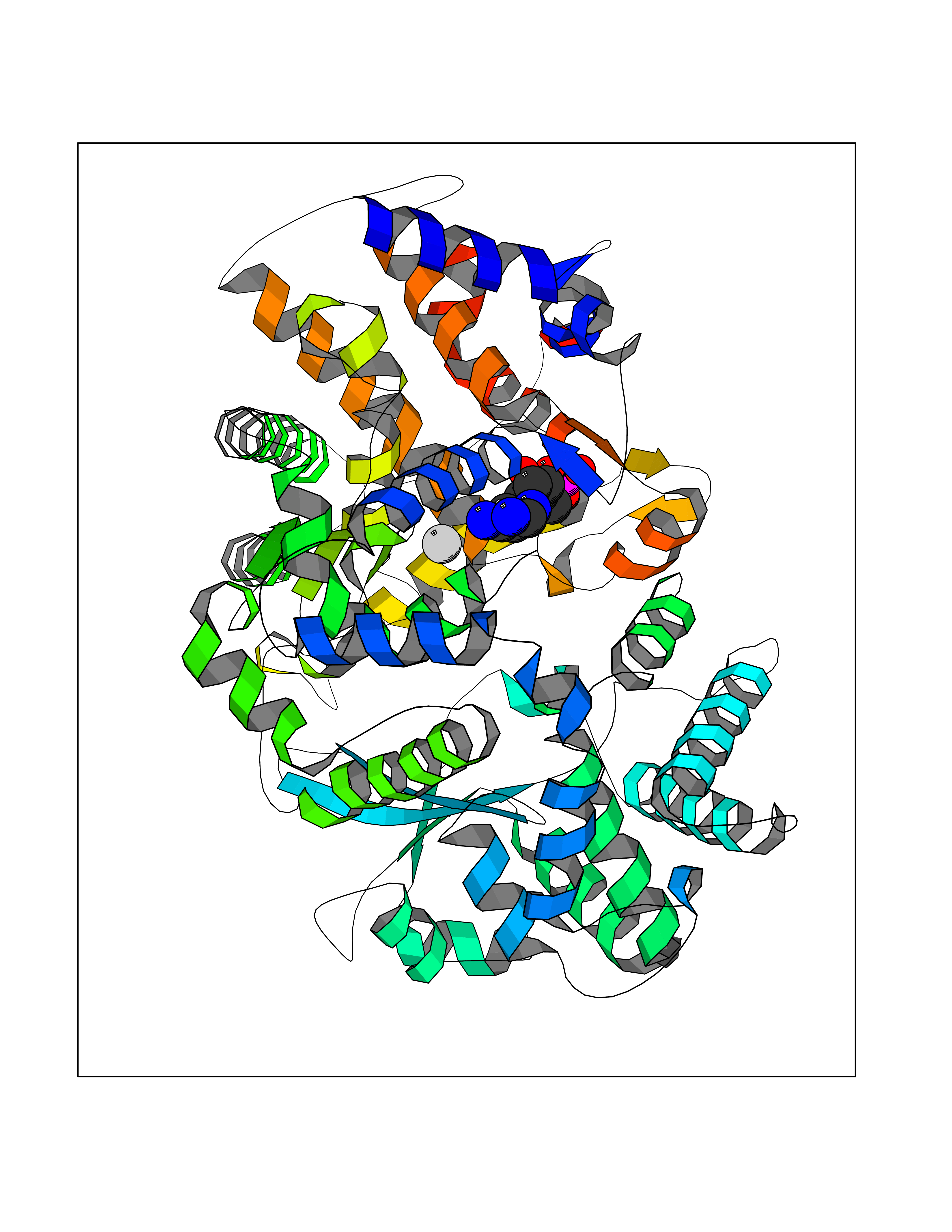 Molscript Map 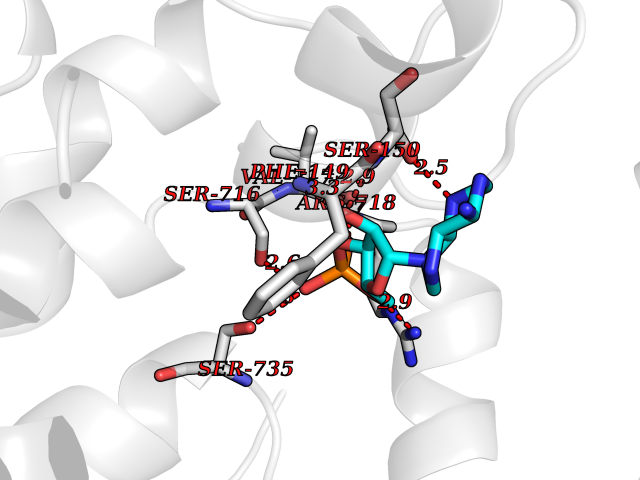 Pymol Map 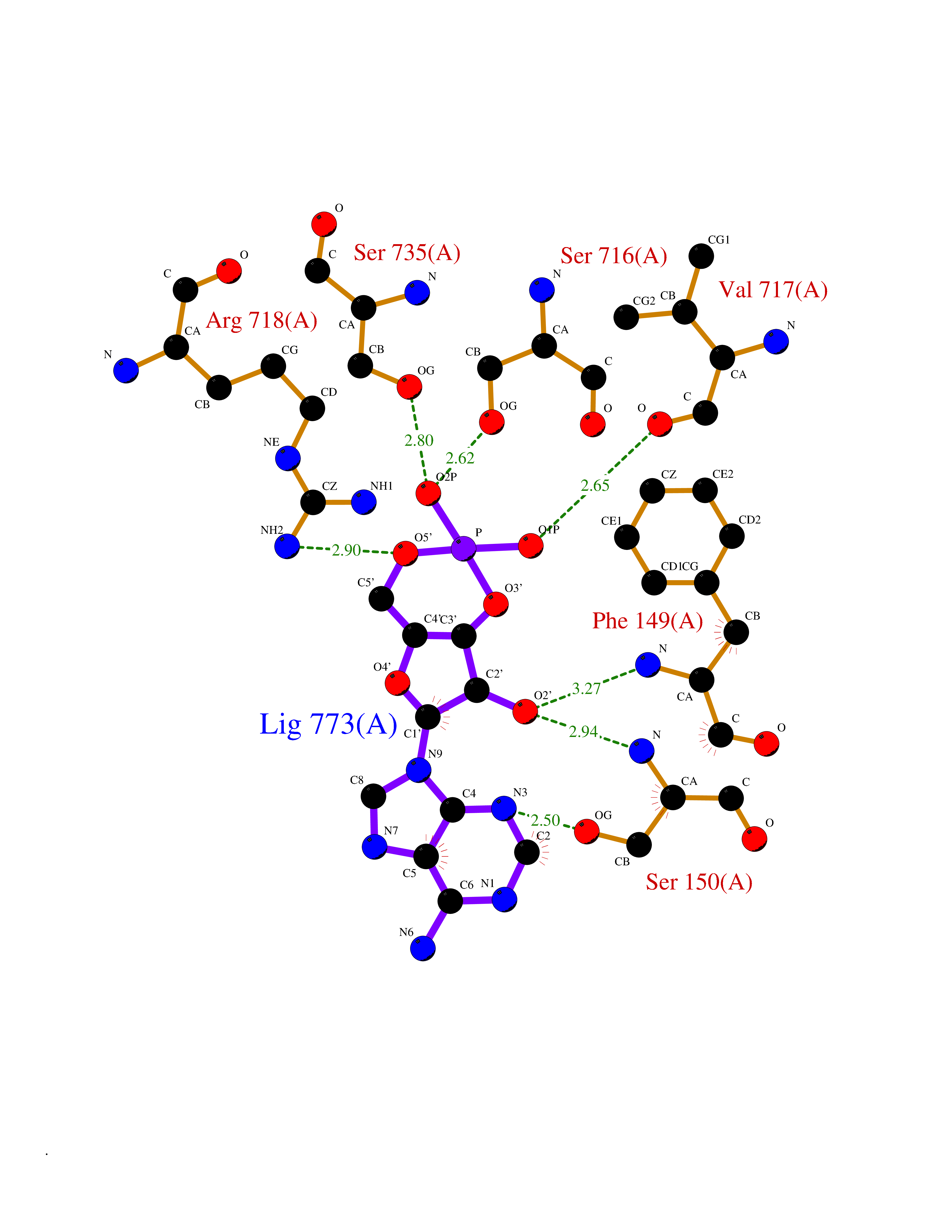 Ligplot Map 3D Binding mode Sequence SEACVSVTSSILSSMDPTVDPCHDFFSYACGGWIKANPVPDGHSRWGTFSNLWEHNQAIIKHLLENSTASVSEAERKAQVYYRACMNETRIEELRAKPLMELIERLGGWNITGPWAKDNFQDTLQVVTAHYRTSPFFSVYVSADSKNSNSNVIQVDQSGLGLPSRDYYLNKTENEKVLTGYLNYMVQLGKLLGGGDEEAIRPQMQQILDFETALANITIPQEKRRDEELIYHKVTAAELQTLAPAINWLPFLNTIFYPVEINESEPIVVYDKEYLEQISTLINTTDRCLLNNYMIWNLVRKTSSFLDQRFQDADEKFMEVMWKFCVSDTENNLGFALGPMFVKATFAEDSKSIATEIILEIKKAFEESLSTLKWMDEETRKSAKEKADAIYNMIGYPNFIMDPKELDKVFNDYTAVPDLYFENAMRFFNFSWRVTADQLRKAPNRDQWSMTPPMVNAYYSPTKNEIVFPAGILQAPFYTRSSPKALNFGGIGVVVGHELTHAFDDQGREYDKDGNLRPWWKNSSVEAFKRQTECMVEQYSNYSVNGEPVNGRHTLGENIADNGGLKAAYRAYQNWVKKNGAEHSLPTLGLTNNQLFFLGFAQVWCSVRTPESSHEGLITDPHSPSRFRVIGSLSNSKEFSEHFRCPPGSPMNPPHKCEVW Hydrogen bonds contact Hydrophobic contact | ||||
| 3 | Phosphodiesterase 4D (PDE4D) | 1Y2K | 7.26 | |
Target general information Gen name PDE4D Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms cAMP-specific 3',5'-cyclic phosphodiesterase 4D; PDE43; DPDE3 Protein family Cyclic nucleotide phosphodiesterase family, PDE4 subfamily Biochemical class Phosphoric diester hydrolase Function Hydrolyzes the second messenger cAMP, which is a key regulator of many important physiological processes. Related diseases Genetic variations in PDE4D might be associated with susceptibility to stroke. PubMed:17006457 states that association with stroke has to be considered with caution. {ECO:0000269|PubMed:17006457}.; DISEASE: Acrodysostosis 2, with or without hormone resistance (ACRDYS2) [MIM:614613]: A pleiotropic disorder characterized by skeletal, endocrine, and neurological abnormalities. Skeletal features include brachycephaly, midface hypoplasia with a small upturned nose, brachydactyly, and lumbar spinal stenosis. Endocrine abnormalities include hypothyroidism and hypogonadism in males and irregular menses in females. Developmental disability is a common finding but is variable in severity and can be associated with significant behavioral problems. {ECO:0000269|PubMed:22464250, ECO:0000269|PubMed:22464252, ECO:0000269|PubMed:23033274, ECO:0000269|PubMed:23043190}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB06842; DB04149; DB03606; DB03183; DB04469; DB02676; DB01959; DB07051; DB04271; DB07954; DB08299; DB00131; DB01427; DB00201; DB03849; DB05219; DB00651; DB06246; DB05266; DB01088; DB01113; DB01791; DB01656; DB01954; DB05298; DB09283; DB02918 Interacts with P32121; P38432; Q0D2H9; Q08AF8; P43360; Q07343; Q13077; P32121; P26769; P38432; Q96CV9; Q8IUH5 EC number EC 3.1.4.53 Uniprot keywords 3D-structure; Alternative splicing; cAMP; Cell membrane; Cytoplasm; Cytoskeleton; Disease variant; Hydrolase; Isopeptide bond; Manganese; Membrane; Metal-binding; Phosphoprotein; Proteomics identification; Reference proteome; Ubl conjugation; Zinc Protein physicochemical properties Chain ID A,B Molecular weight (Da) 37201.9 Length 322 Aromaticity 0.07 Instability index 35.83 Isoelectric point 5.02 Charge (pH=7) -21.16 2D Binding mode Binding energy (Kcal/mol) -7.85 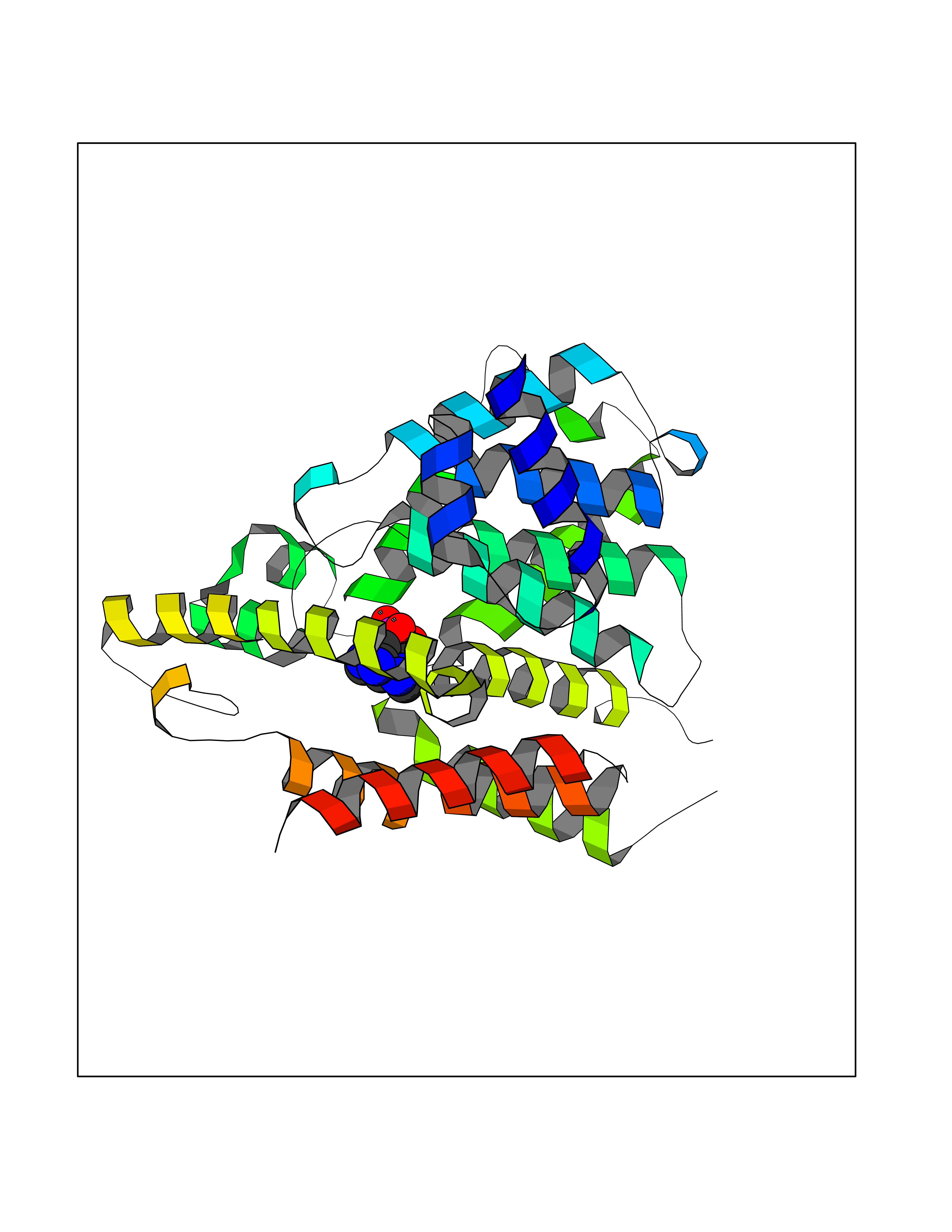 Molscript Map 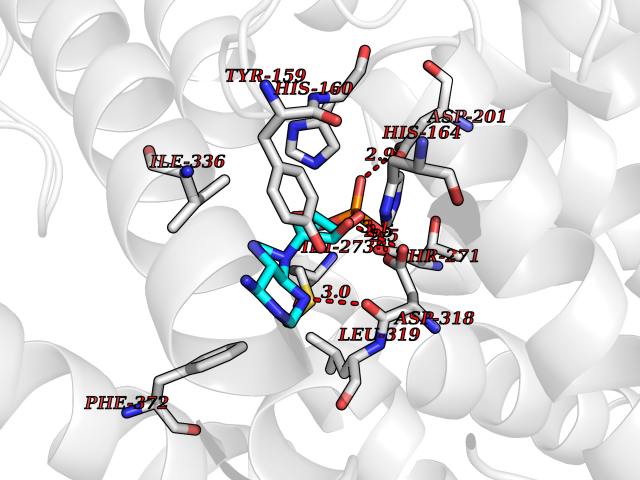 Pymol Map 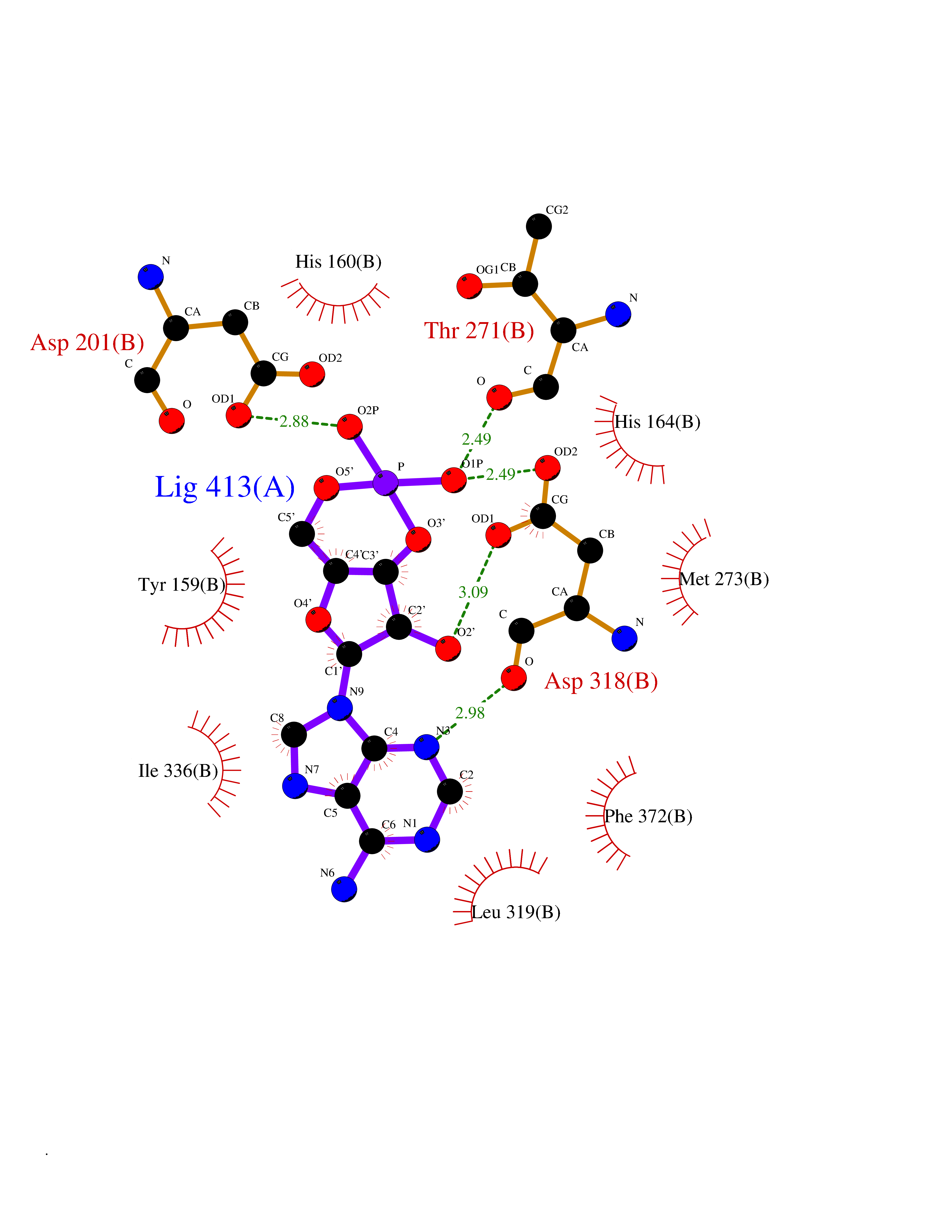 Ligplot Map 3D Binding mode Sequence TEQEDVLAKELEDVNKWGLHVFRIAELSGNRPLTVIMHTIFQERDLLKTFKIPVDTLITYLMTLEDHYHADVAYHNNIHAADVVQSTHVLLSTPALEAVFTDLEILAAIFASAIHDVDHPGVSNQFLINTNSELALMYNDSSVLENHHLAVGFKLLQEENCDIFQNLTKKQRQSLRKMVIDIVLATDMSKHMNLLADLKTMVETKKVVLLLDNYSDRIQVLQNMVHCADLSNPTKPLQLYRQWTDRIMEEFFRQGDRERERGMEISPMCDKHNASVEKSQVGFIDYIVHPLWETWADLVHPDAQDILDTLEDNREWYQSTIP Hydrogen bonds contact Hydrophobic contact | ||||
| 4 | Adrenergic receptor beta-2 (ADRB2) | 2RH1 | 7.25 | |
Target general information Gen name ADRB2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Beta-2 adrenoreceptor; Beta-2 adrenoceptor; Beta-2 adrenergic receptor; B2AR; ADRB2R Protein family G-protein coupled receptor 1 family, Adrenergic receptor subfamily, ADRB2 sub-subfamily Biochemical class GPCR rhodopsin Function The beta-2-adrenergic receptor binds epinephrine with an approximately 30-fold greater affinity than it does norepinephrine. Beta-adrenergic receptors mediate the catecholamine-induced activation of adenylate cyclase through the action of G proteins. Related diseases Cortical dysplasia, complex, with other brain malformations 6 (CDCBM6) [MIM:615771]: A disorder of aberrant neuronal migration and disturbed axonal guidance. Affected individuals have microcephaly, ataxia, and severe delayed psychomotor development. Brain imaging shows variable malformations of cortical development, including white matter streaks, dysmorphic basal ganglia, corpus callosum abnormalities, brainstem and cerebellar hypoplasia, cortical dysplasia, polymicrogyria. {ECO:0000269|PubMed:23246003}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Skin creases, congenital symmetric circumferential, 1 (CSCSC1) [MIM:156610]: An autosomal dominant disease characterized by multiple, symmetric, circumferential rings of folded skin, affecting primarily the limbs. Affected individuals also exhibit intellectual disability, cleft palate, and dysmorphic features. {ECO:0000269|PubMed:26637975}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07543; DB01193; DB00866; DB01118; DB00182; DB01102; DB01274; DB01238; DB09204; DB06216; DB00335; DB01408; DB05590; DB09013; DB00195; DB00217; DB01295; DB00612; DB00901; DB08807; DB06726; DB08808; DB00248; DB00521; DB01136; DB04846; DB01407; DB00785; DB01151; DB11273; DB13345; DB00449; DB11278; DB00841; DB09273; DB06262; DB01363; DB01364; DB00668; DB01049; DB11587; DB01288; DB00983; DB05039; DB00221; DB01064; DB00598; DB01210; DB13139; DB01365; DB13624; DB01214; DB00264; DB01203; DB05849; DB04861; DB00368; DB00540; DB00334; DB09080; DB00816; DB01580; DB00715; DB01359; DB00925; DB00397; DB00960; DB01291; DB01366; DB01182; DB00571; DB06814; DB00852; DB01917; DB11124; DB00867; DB01001; DB00938; DB00489; DB03566; DB00127; DB00871; DB00373; DB00726; DB12248; DB09082; DB09185 Interacts with P30542; P07550; P32121; Q96B67; Q9UII2; Q9ULD4-2; Q9NSI6-4; Q5M9N0-2; A0AVK6; Q658K8; O00472; Q15910-2; Q15486; P61978; Q5TCQ9; Q99685; O14745; Q9NR21-5; Q8WVD3; Q9H0X6; Q13573; P12931; Q5T0J7-2; Q8N0U2 EC number NA Uniprot keywords 3D-structure; Cell membrane; Disulfide bond; Endosome; G-protein coupled receptor; Glycoprotein; Golgi apparatus; Hydroxylation; Lipoprotein; Membrane; Palmitate; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Transducer; Transmembrane; Transmembrane helix; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 32266.1 Length 282 Aromaticity 0.15 Instability index 36.1 Isoelectric point 8.02 Charge (pH=7) 2.1 2D Binding mode Binding energy (Kcal/mol) -7.71 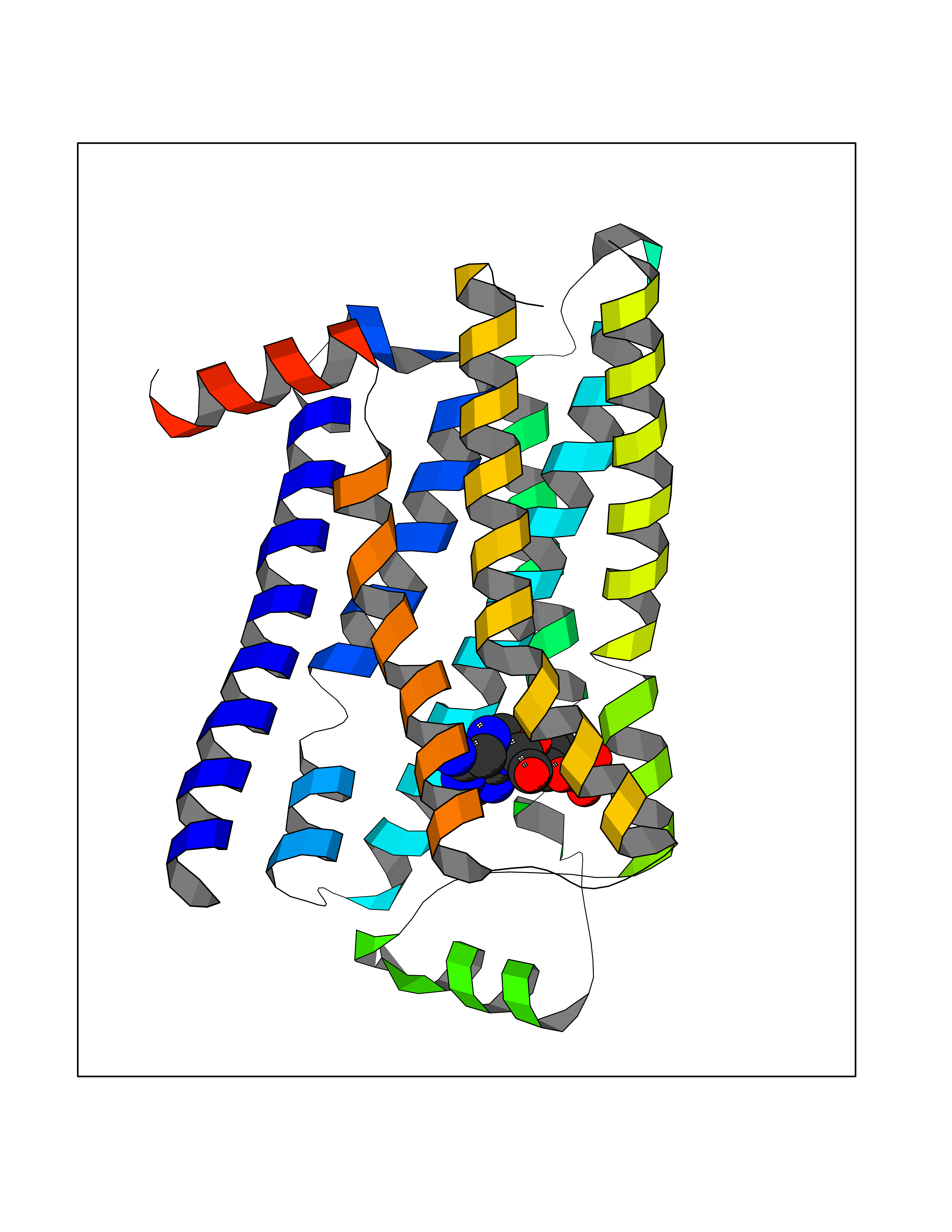 Molscript Map 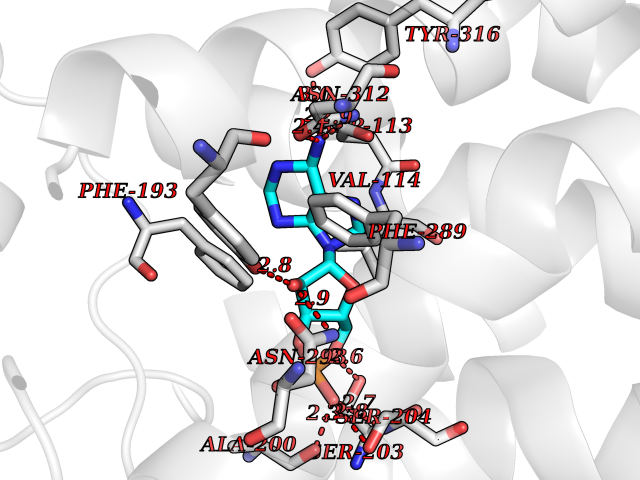 Pymol Map 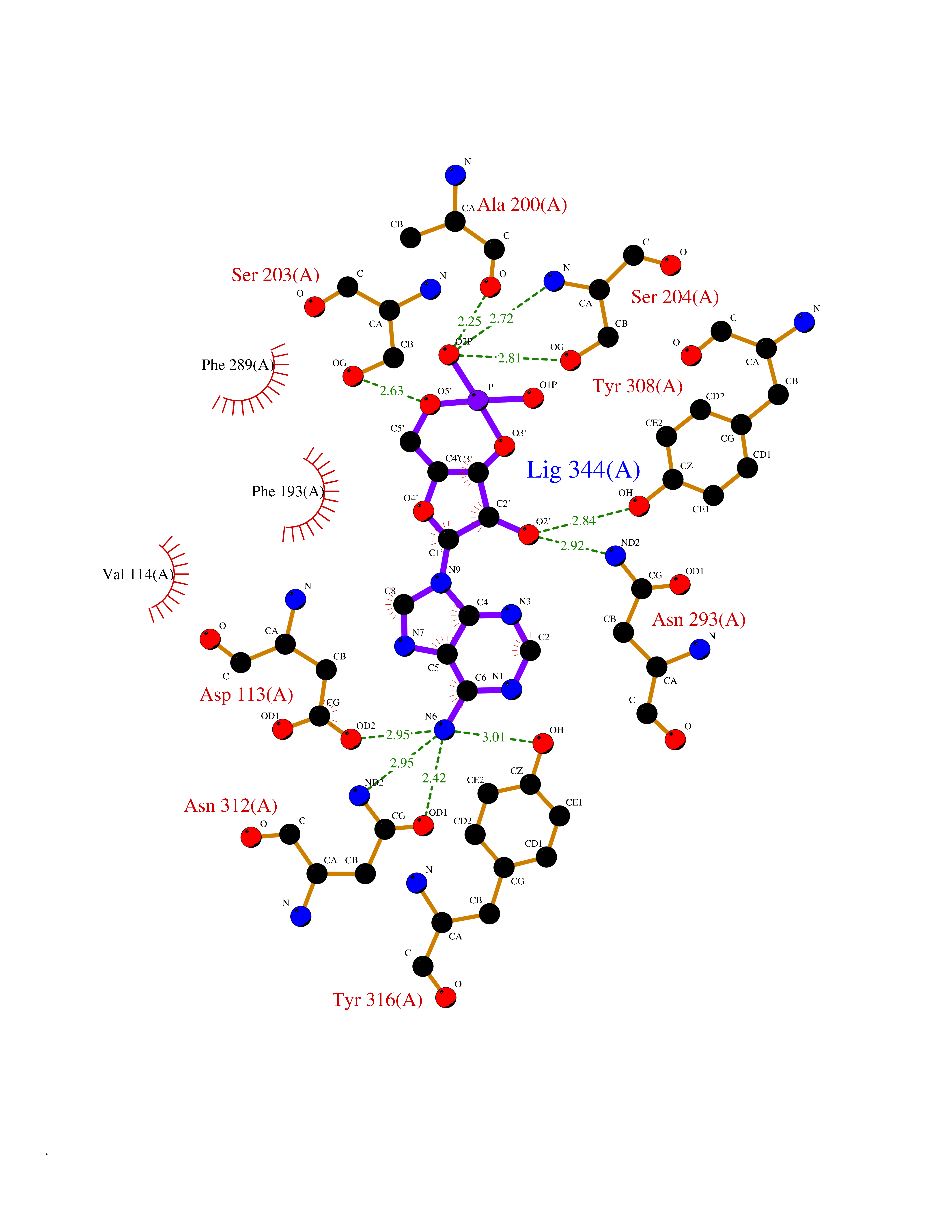 Ligplot Map 3D Binding mode Sequence DEVWVVGMGIVMSLIVLAIVFGNVLVITAIAKFERLQTVTNYFITSLACADLVMGLAVVPFGAAHILMKMWTFGNFWCEFWTSIDVLCVTASIETLCVIAVDRYFAITSPFKYQSLLTKNKARVIILMVWIVSGLTSFLPIQMHWYRATHQEAINCYAEETCCDFFTNQAYAIASSIVSFYVPLVIMVFVYSRVFQEAKRQLKFCLKEHKALKTLGIIMGTFTLCWLPFFIVNIVHVIQDNLIRKEVYILLNWIGYVNSGFNPLIYCRSPDFRIAFQELLCL Hydrogen bonds contact Hydrophobic contact | ||||
| 5 | Human immunodeficiency virus Protease (HIV PR) | 3TL9 | 7.17 | |
Target general information Gen name HIV PR Organism Human immunodeficiency virus type 1 group M subtype B (isolate BH10) (HIV-1) Uniprot ID TTD ID Synonyms HIV Retropepsin; HIV PR Protein family NA Biochemical class Peptidase Function Gag-Pol polyprotein: Mediates, with Gag polyprotein, the essential events in virion assembly, including binding the plasma membrane, making the protein-protein interactions necessary to create spherical particles, recruiting the viral Env proteins, and packaging the genomic RNA via direct interactions with the RNA packaging sequence (Psi). Gag-Pol polyprotein may regulate its own translation, by the binding genomic RNA in the 5'-UTR. At low concentration, the polyprotein would promote translation, whereas at high concentration, the polyprotein would encapsidate genomic RNA and then shut off translation. Related diseases Sitosterolemia 2 (STSL2) [MIM:618666]: A form of sitosterolemia, an autosomal recessive metabolic disorder characterized by unregulated intestinal absorption of cholesterol, phytosterols and shellfish sterols, and decreased biliary excretion of dietary sterols into bile. Patients have hypercholesterolemia, very high levels of plant sterols in the plasma, and frequently develop tendon and tuberous xanthomas, accelerated atherosclerosis and premature coronary artery disease. {ECO:0000269|PubMed:11138003, ECO:0000269|PubMed:11452359, ECO:0000269|PubMed:11668628, ECO:0000269|PubMed:15054092, ECO:0000269|PubMed:35557526}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07035; DB02704; DB07806; DB02785; DB01824; DB01732; DB06874; DB08034; DB07961; DB07451; DB08212; DB08372; DB02972; DB04190; DB04042; DB08428; DB03076; DB03141; DB08457; DB07343; DB07337; DB07018; DB07332; DB05398; DB07578; DB08639; DB06414; DB04255; DB04547; DB02683; DB02009; DB03908; DB02629; DB01887; DB03803; DB02033; DB08281; DB08282; DB08284; DB08414; DB08598; DB07327; DB07885; DB02768; DB08600; DB01891; DB05871 Interacts with NA EC number EC 3.4.23.16 Uniprot keywords 3D-structure; Activation of host caspases by virus; AIDS; Aspartyl protease; Capsid protein; Direct protein sequencing; DNA integration; DNA recombination; DNA-binding; DNA-directed DNA polymerase; Endonuclease; Eukaryotic host gene expression shutoff by virus; Eukaryotic host translation shutoff by virus; Host cell membrane; Host cytoplasm; Host endosome; Host gene expression shutoff by virus; Host membrane; Host nucleus; Host-virus interaction; Hydrolase; Lipid-binding; Lipoprotein; Magnesium; Membrane; Metal-binding; Methylation; Modulation of host cell apoptosis by virus; Multifunctional enzyme; Myristate; Nuclease; Nucleotidyltransferase; Phosphoprotein; Protease; Repeat; Ribosomal frameshifting; RNA-binding; RNA-directed DNA polymerase; Transferase; Viral genome integration; Viral nucleoprotein; Viral penetration into host nucleus; Viral release from host cell; Virion; Virion maturation; Virus entry into host cell; Zinc; Zinc-finger Protein physicochemical properties Chain ID A,B Molecular weight (Da) 21934.7 Length 202 Aromaticity 0.06 Instability index 48.65 Isoelectric point 9.66 Charge (pH=7) 6.15 2D Binding mode Binding energy (Kcal/mol) -6.95 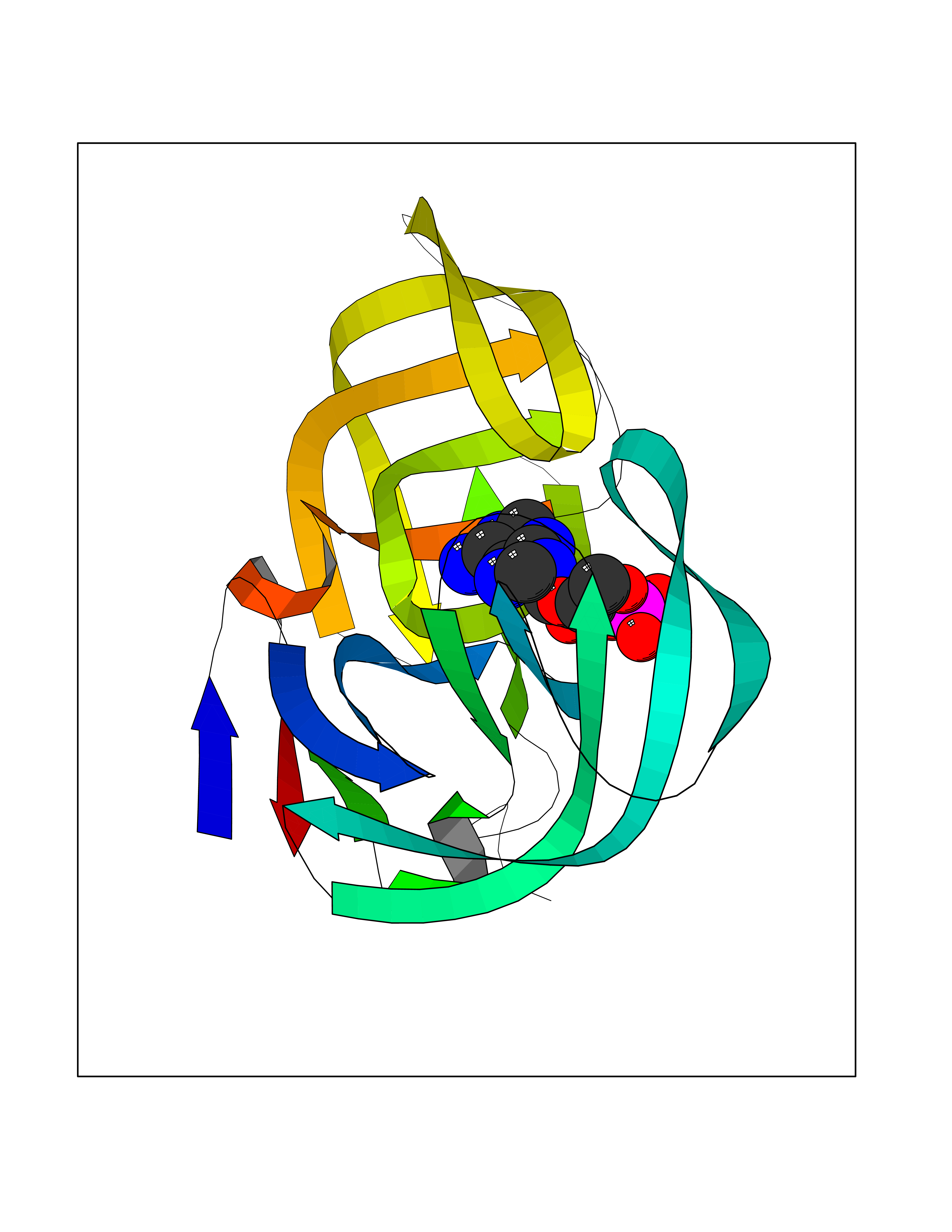 Molscript Map 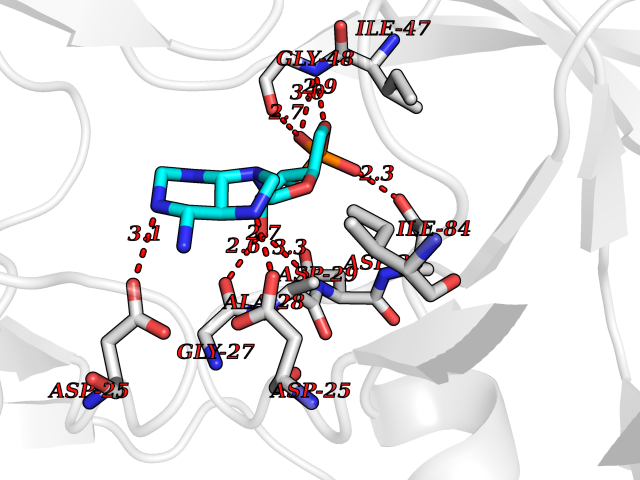 Pymol Map 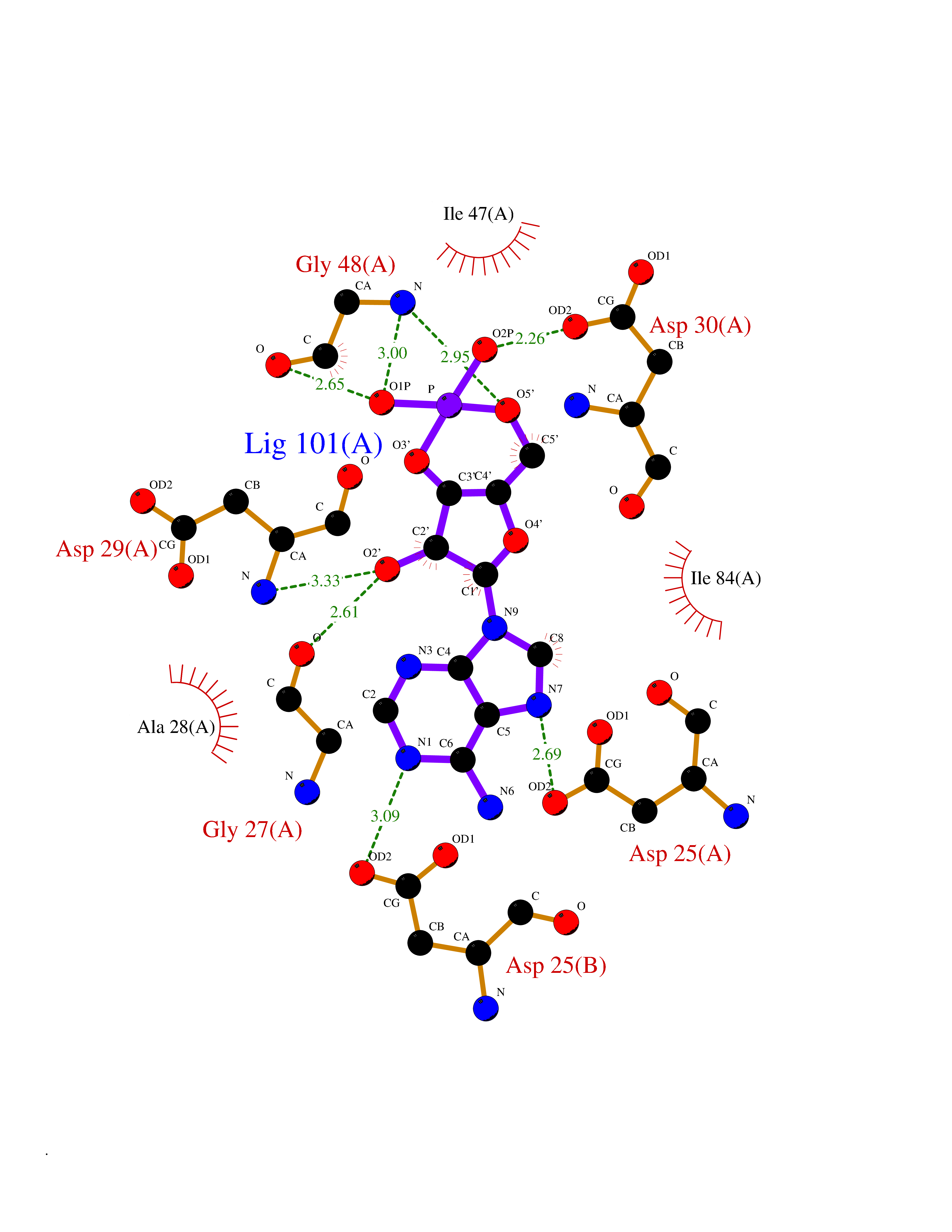 Ligplot Map 3D Binding mode Sequence PQITLWKRPLVTIKIGGQLKEALLDTGADDTVIEEMSLPGRWKPKMIGGIGGFIKVRQYDQIIIEIAGHKAIGTVLVGPTPVNIIGRNLLTQIGATLNFSFNFPQITLWKRPLVTIKIGGQLKEALLDTGADDTVIEEMSLPGRWKPKMIGGIGGFIKVRQYDQIIIEIAGHKAIGTVLVGPTPVNIIGRNLLTQIGATLNF Hydrogen bonds contact Hydrophobic contact | ||||
| 6 | Purine nucleoside phosphorylase (PNP) | 4EAR | 7.12 | |
Target general information Gen name PNP Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms PNP; Inosine phosphorylase Protein family PNP/MTAP phosphorylase family Biochemical class Pentosyltransferase Function The purine nucleoside phosphorylases catalyze the phosphorolytic breakdown of the N-glycosidic bond in the beta- (deoxy)ribonucleoside molecules, with the formation of the corresponding free purine bases and pentose-1-phosphate. Related diseases Purine nucleoside phosphorylase deficiency (PNPD) [MIM:613179]: A disorder that interrupts both the catabolism of inosine into hypoxanthine and guanosine into guanine, and leads to the accumulation of guanosine, inosine, and their deoxified by-products. The main clinical presentation is recurrent infections due to severe T-cell immunodeficiency. Some patients also have neurologic impairment. {ECO:0000269|PubMed:1384322, ECO:0000269|PubMed:3029074, ECO:0000269|PubMed:8931706}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03881; DB03551; DB02222; DB02391; DB03609; DB01667; DB04260; DB02796; DB04753; DB00640; DB00242; DB00900; DB06185; DB02377; DB02857; DB04754; DB04757; DB04076; DB02230; DB04335; DB02568; DB03101 Interacts with P05067; Q9UQM7; O14576-2; P06241; P14136; Q92993-2; Q9BXM7; P00491; P17612; P63000; Q92673; Q15583 EC number EC 2.4.2.1 Uniprot keywords 3D-structure; Acetylation; Cytoplasm; Direct protein sequencing; Disease variant; Glycosyltransferase; Phosphoprotein; Proteomics identification; Purine salvage; Reference proteome; Transferase Protein physicochemical properties Chain ID A,B,C Molecular weight (Da) 31849.2 Length 288 Aromaticity 0.1 Instability index 34.77 Isoelectric point 6.42 Charge (pH=7) -1.63 2D Binding mode Binding energy (Kcal/mol) -8.18 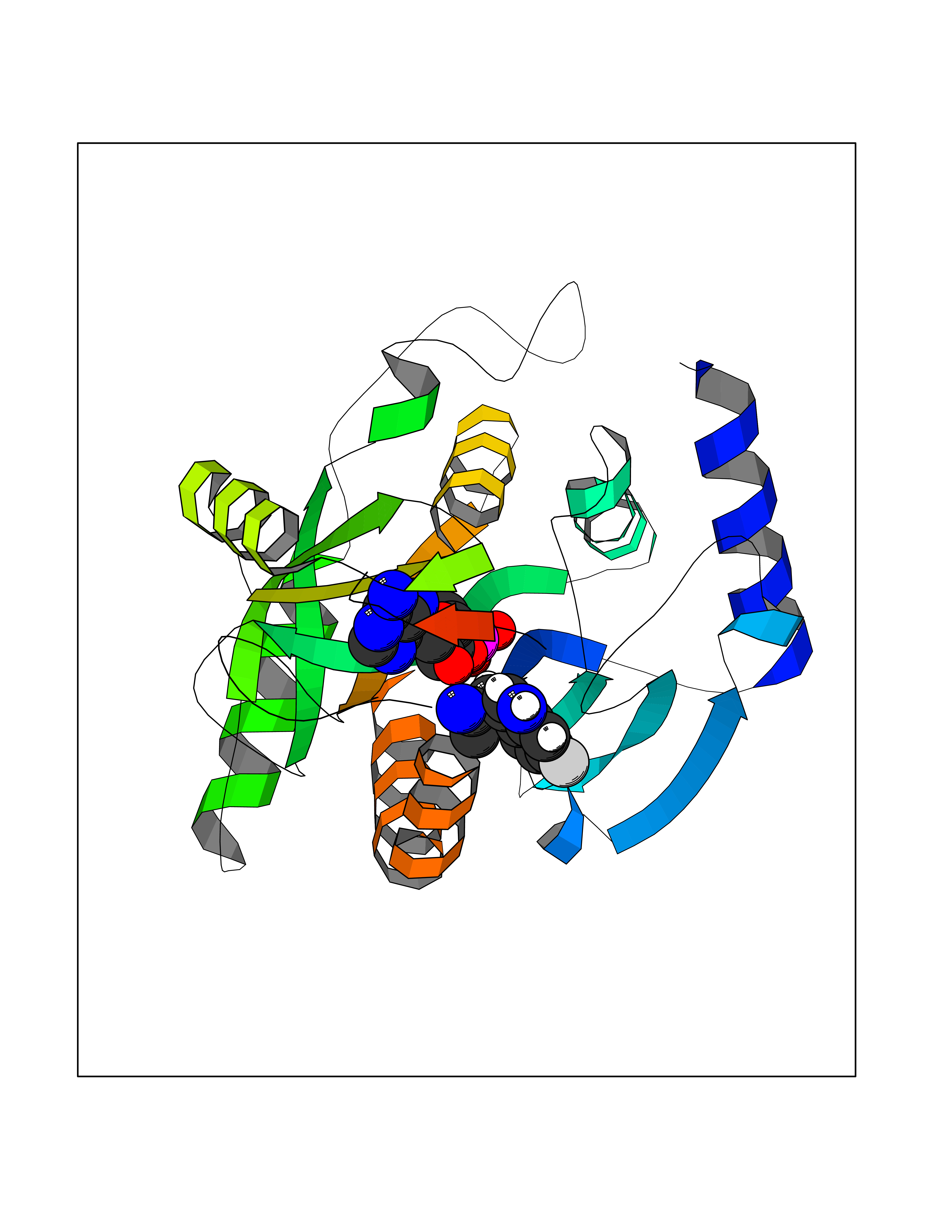 Molscript Map 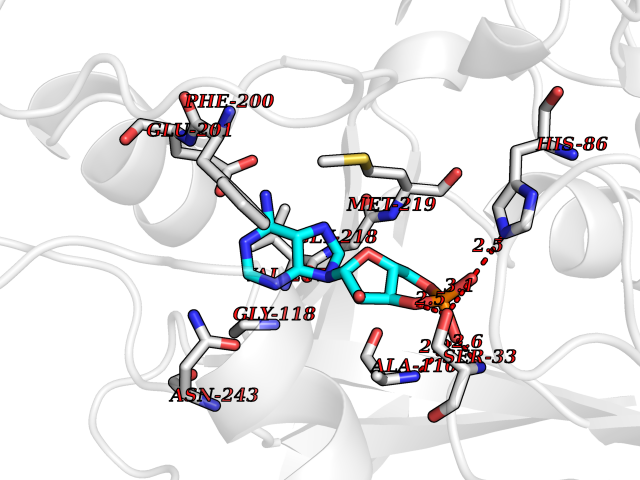 Pymol Map 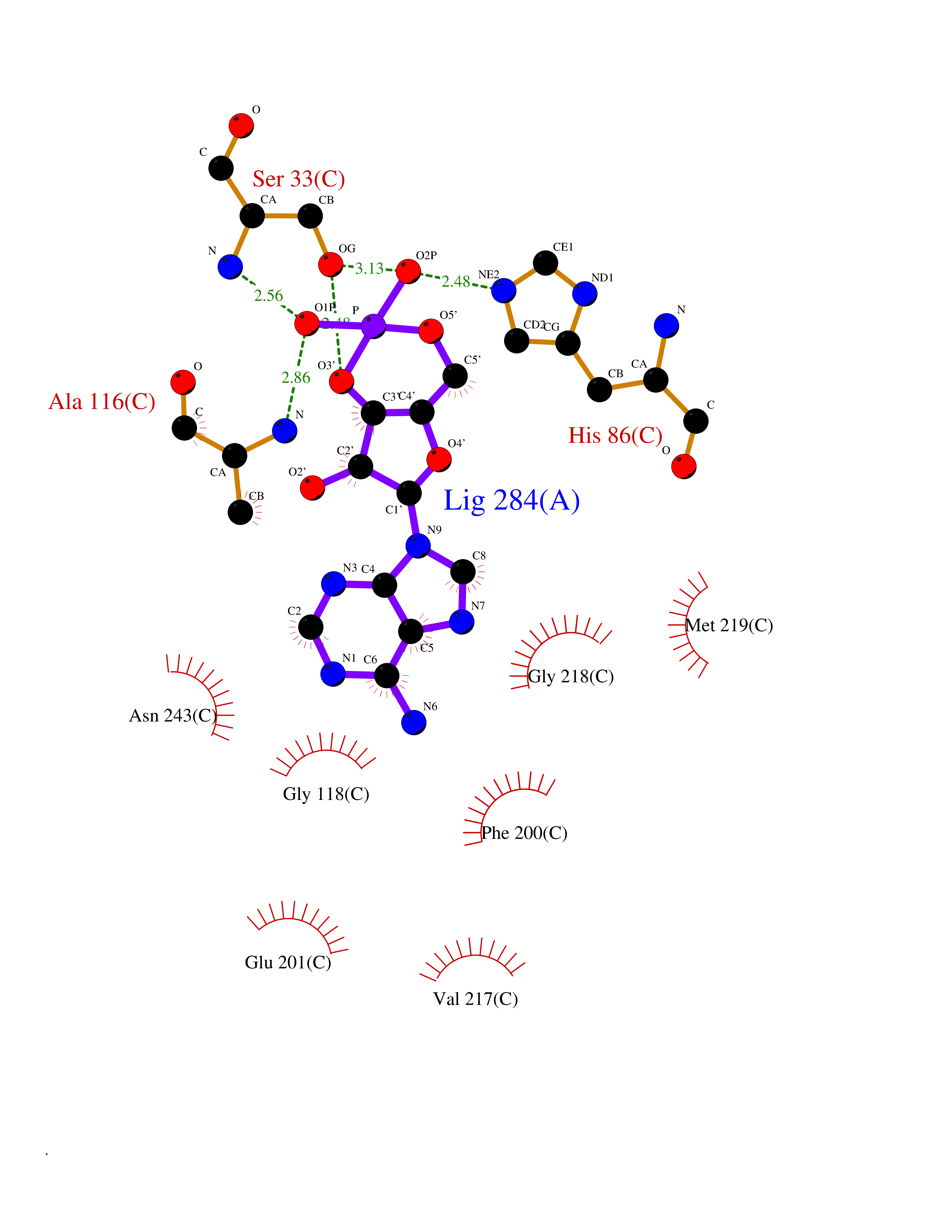 Ligplot Map 3D Binding mode Sequence GYTYEDYKNTAEYLLSHTKHRPQVAIICGSGLGGLTDKLTQAQIFDYSEIPNFPRSTVPGHAGRLVFGFLNGRACVMMQGRFHMYEGYPLYKVTFPVRVFHLLGVDTLVVTNAAGGLNPKFEVGDIMLIRDHINLPGFSGQNPLRGPNDERFGDRFPAMSDAYDRTMRQRALSTYKQMGEQRELQEGTYVMVAGPSFETVAECRVLQKLGADAVGMSTVPEVIVARHCGLRVFGFSLITNKVIMDYESLEKANXEEVLAAGKQAAQKLEQFVSILMASIDRFPAMSDA Hydrogen bonds contact Hydrophobic contact | ||||
| 7 | FkbI | 1R2J | 7.09 | |
Target general information Gen name fkbI Organism Streptomyces hygroscopicus subsp. ascomyceticus Uniprot ID TTD ID NA Synonyms NA Protein family Acyl-CoA dehydrogenase family Biochemical class Oxidoreductase Function Flavin adenine dinucleotide binding.Oxidoreductase activity, acting on the CH-CH group of donors. Related diseases Thiamine metabolism dysfunction syndrome 5, episodic encephalopathy type (THMD5) [MIM:614458]: An autosomal recessive metabolic disorder due to an inborn error of thiamine metabolism. The phenotype is highly variable, but in general, affected individuals have onset in early childhood of acute encephalopathic episodes associated with increased serum and CSF lactate. These episodes result in progressive neurologic dysfunction manifest as gait disturbances, ataxia, dystonia, and spasticity, which in some cases may result in loss of ability to walk. Cognitive function is usually preserved, although mildly delayed development has been reported. These episodes are usually associated with infection and metabolic decompensation. Some patients may have recovery of some neurologic deficits. {ECO:0000269|PubMed:22152682}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03147 Interacts with NA EC number NA Uniprot keywords 3D-structure; FAD; Flavoprotein; Nucleotide-binding; Oxidoreductase Protein physicochemical properties Chain ID A Molecular weight (Da) 36670.3 Length 353 Aromaticity 0.04 Instability index 22.05 Isoelectric point 6.12 Charge (pH=7) -5.04 2D Binding mode Binding energy (Kcal/mol) -7.41 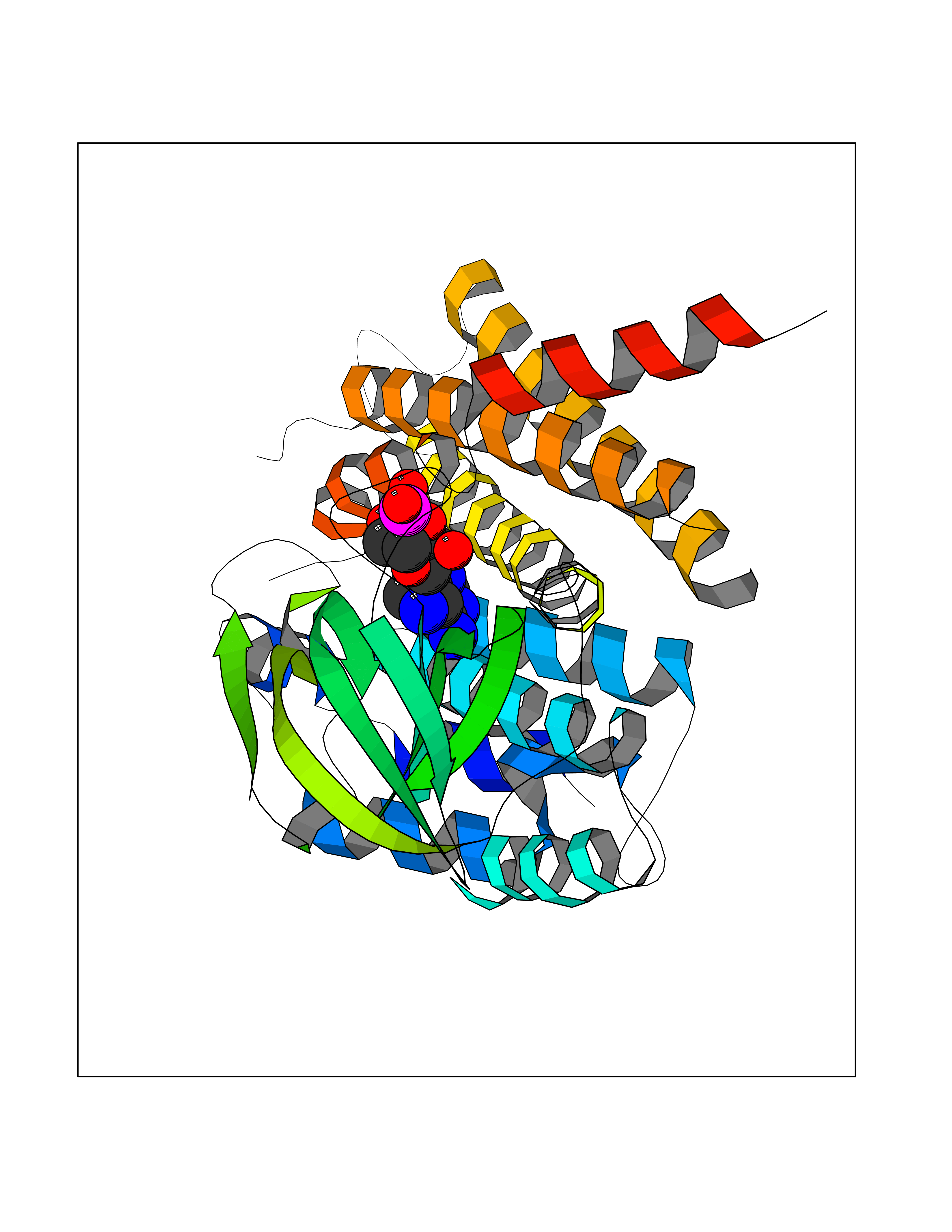 Molscript Map 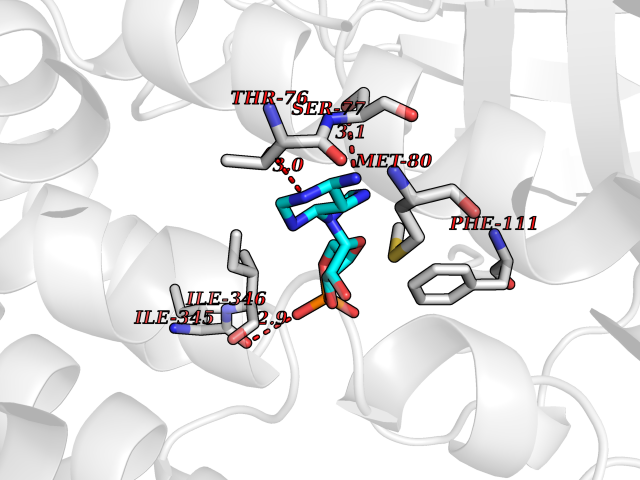 Pymol Map 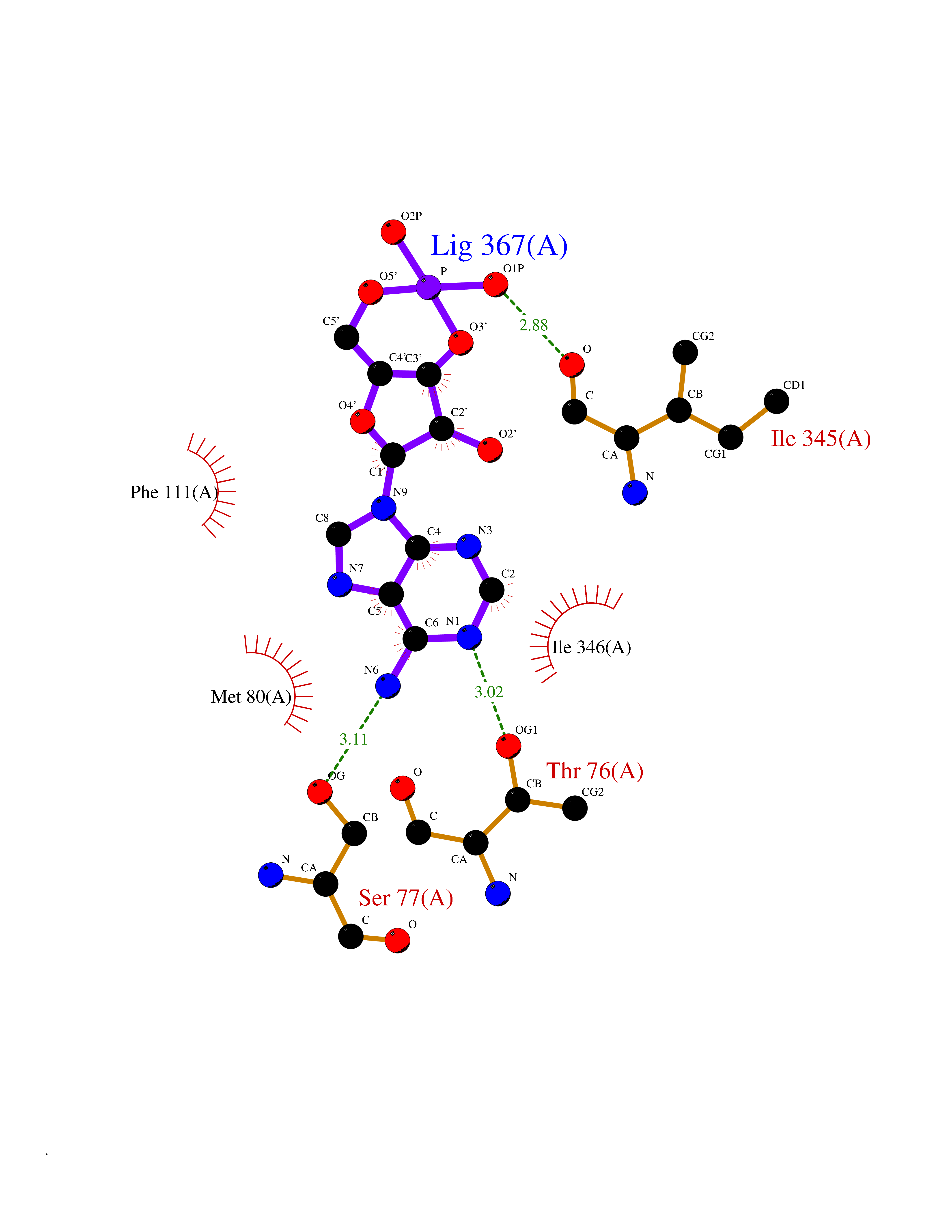 Ligplot Map 3D Binding mode Sequence ERDALLTDLVGDRAAEWDTSGELPRDLLVRLGADGLLCAEVAAEHGGLGLGSRENGEFTAHVGSLCSSLRSVMTSQGMAAWTVQRLGDAGQRATFLKELTSGLAAVGFSERQAGSDLSAMRTRVRLDGDTAVVDGHKVWTTAAAYADHLVVFGLQEDGSGAVVVVPADTPGVRVERVPKPSGCRAAGHADLHLDQVRVPAGAVLAGSGASLPMLVAASLAYGRKSVAWGCVGILRACRTAAVAHARTREQFGRPLGDHQLVAGHIADLWTAEQIAARVCEYASDHMVPATILAKHVAAERAAAGAATAAQVLASAGAGHVVERAYRDAKLMEIIEGSSEMCRVMLAQHALALP Hydrogen bonds contact Hydrophobic contact | ||||
| 8 | Enteropeptidase (TMPRSS15) | 6ZOV | 7.04 | |
Target general information Gen name TMPRSS15 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Transmembrane protease serine 15; TMPRSS15; Serine protease 7; Enterokinase Protein family Peptidase S1 family Biochemical class Peptidase Function Responsible for initiating activation of pancreatic proteolytic proenzymes (trypsin, chymotrypsin and carboxypeptidase A). It catalyzes the conversion of trypsinogen to trypsin which in turn activates other proenzymes including chymotrypsinogen, procarboxypeptidases, and proelastases. Related diseases Enterokinase deficiency (ENTKD) [MIM:226200]: Life-threatening intestinal malabsorption disorder characterized by diarrhea and failure to thrive. {ECO:0000269|PubMed:11719902}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number EC 3.4.21.9 Uniprot keywords 3D-structure; Disulfide bond; Glycoprotein; Hydrolase; Lipoprotein; Membrane; Myristate; Protease; Proteomics identification; Reference proteome; Repeat; Serine protease; Signal-anchor; Transmembrane; Transmembrane helix; Zymogen Protein physicochemical properties Chain ID A Molecular weight (Da) 26220.3 Length 234 Aromaticity 0.1 Instability index 50.13 Isoelectric point 4.82 Charge (pH=7) -9.93 2D Binding mode Binding energy (Kcal/mol) -7.56 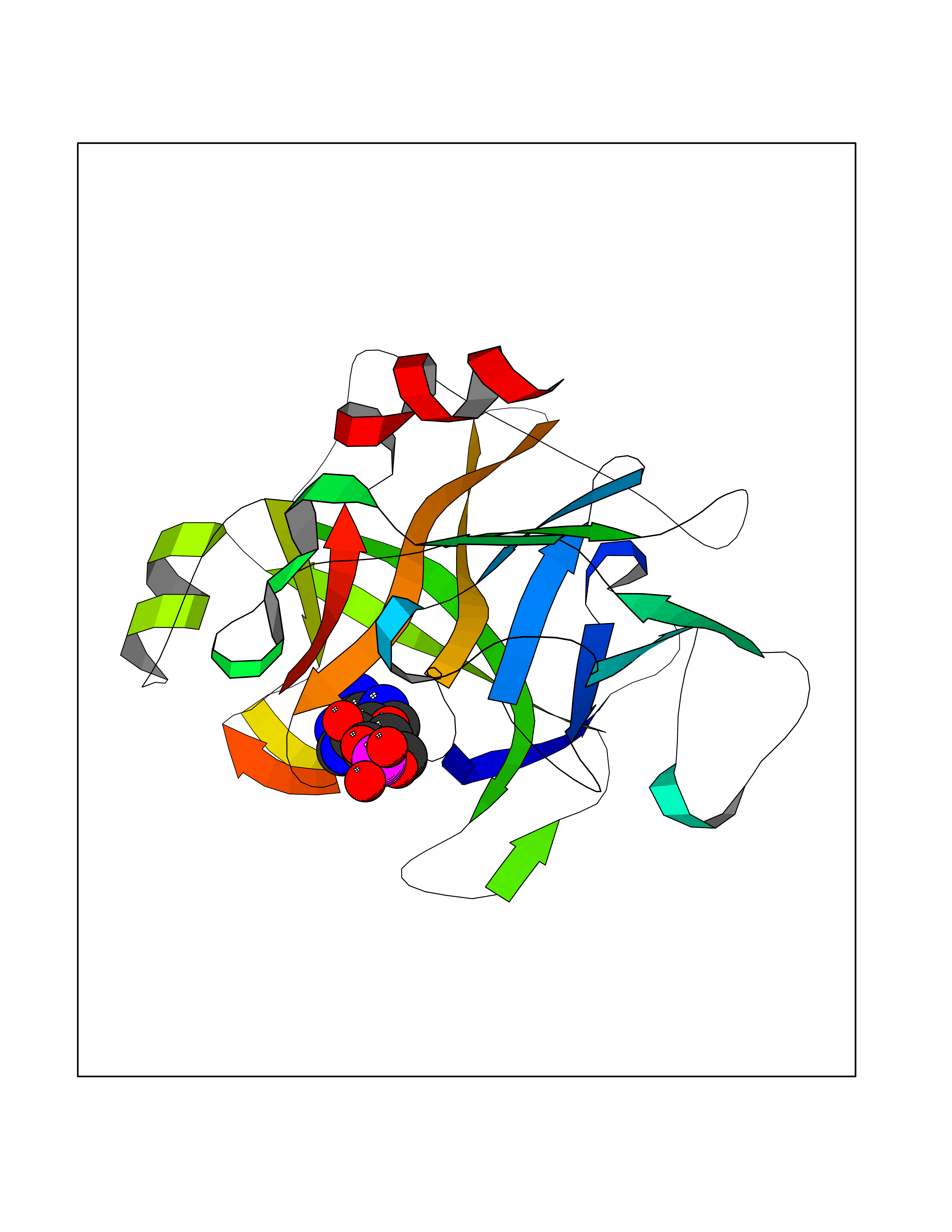 Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence IVGGSDAKEGAWPWVVGLYYDDRLLCGASLVSSDWLVSAAHCVYGRNLEPSKWTAILGLHMKSNLTSPQTVPRLIDEIVINPHYNRRRKDNDIAMMHLEFKVNYTDYIQPISLPEENQVFPPGRNCSIAGWGTVVYQGTTADILQEADVPLLSNERCQQQMPEYNITENMICAGYEEGGIDSCQGDSGGPLMCQENNRWFLAGVTSFGYECALPNRPGVYARVSRFTEWIQSFL Hydrogen bonds contact Hydrophobic contact | ||||
| 9 | Pol polyprotein | 5KAO | 7.02 | |
Target general information Gen name pol Organism Human immunodeficiency virus type 1 (HIV-1) Uniprot ID TTD ID NA Synonyms NA Protein family NA Biochemical class Hydrolase / hydrolase inhibitor Function Aspartic-type endopeptidase activity. Related diseases Oocyte/zygote/embryo maturation arrest 16 (OZEMA16) [MIM:617234]: A rare cause of female primary infertility. In affected women, ovulation and fertilization proceed normally but embryos are arrested at early stages of development. Inheritance is autosomal recessive. {ECO:0000269|PubMed:27545678}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00701; DB01072; DB04887; DB01264; DB01319; DB00224; DB01601; DB00503; DB01232; DB00932 Interacts with NA EC number NA Uniprot keywords 3D-structure; Aspartyl protease; Hydrolase; Protease Protein physicochemical properties Chain ID A,B Molecular weight (Da) 21411 Length 198 Aromaticity 0.05 Instability index 42.78 Isoelectric point 9.45 Charge (pH=7) 4.15 2D Binding mode Binding energy (Kcal/mol) -7.31  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence PQVTLWQRPLVTIKIGGQLKEALLDTGADDTVLEEMSLPGRWKPKMIGGIGGFIKVRQYDQILIEIAGHKAIGTVLVGPTPVNIIGRNLLTQIGATLNFPQVTLWQRPLVTIKIGGQLKEALLDTGADDTVLEEMSLPGRWKPKMIGGIGGFIKVRQYDQILIEIAGHKAIGTVLVGPTPVNIIGRNLLTQIGATLNF Hydrogen bonds contact Hydrophobic contact | ||||
| 10 | Vitamin D3 receptor (VDR) | 3B0T | 7.01 | |
Target general information Gen name VDR Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Vitamin D(3) receptor; Nuclear vitamin D receptor; Nuclear receptor subfamily 1 group I member 1; NR1I1; 1,25-dihydroxyvitamin D3 receptor Protein family Nuclear hormone receptor family, NR1 subfamily Biochemical class Nuclear hormone receptor Function Enters the nucleus upon vitamin D3 binding where it forms heterodimers with the retinoid X receptor/RXR. The VDR-RXR heterodimers bind to specific response elements on DNA and activate the transcription of vitamin D3-responsive target genes. Plays a central role in calcium homeostasis. Nuclear receptor for calcitriol, the active form of vitamin D3 which mediates the action of this vitamin on cells. Related diseases Rickets vitamin D-dependent 2A (VDDR2A) [MIM:277440]: A disorder of vitamin D metabolism resulting in severe rickets, hypocalcemia and secondary hyperparathyroidism. Most patients have total alopecia in addition to rickets. {ECO:0000269|PubMed:1652893, ECO:0000269|PubMed:17970811, ECO:0000269|PubMed:2177843, ECO:0000269|PubMed:2849209, ECO:0000269|PubMed:28698609, ECO:0000269|PubMed:7828346, ECO:0000269|PubMed:8106618, ECO:0000269|PubMed:8381803, ECO:0000269|PubMed:8392085, ECO:0000269|PubMed:8675579, ECO:0000269|PubMed:8961271, ECO:0000269|PubMed:9005998}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07530; DB08742; DB01436; DB04891; DB00146; DB02300; DB00136; DB00169; DB04540; DB05024; DB11672; DB14635; DB01070; DB06410; DB05295; DB06194; DB00153; DB04796; DB03451; DB00910; DB04258; DB11094 Interacts with P35222; Q09472; Q15648; P50222; Q15788; P26045; P19793; Q13573; Q13501; P04637; Q15645; Q9JLI4; P28700; X5D778; Q96HA8; Q01804; Q96S38; P48443 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; Disease variant; DNA-binding; Metal-binding; Nucleus; Proteomics identification; Receptor; Reference proteome; Transcription; Transcription regulation; Ubl conjugation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A Molecular weight (Da) 28781 Length 254 Aromaticity 0.07 Instability index 47.69 Isoelectric point 6.15 Charge (pH=7) -3.44 2D Binding mode Binding energy (Kcal/mol) -7.5 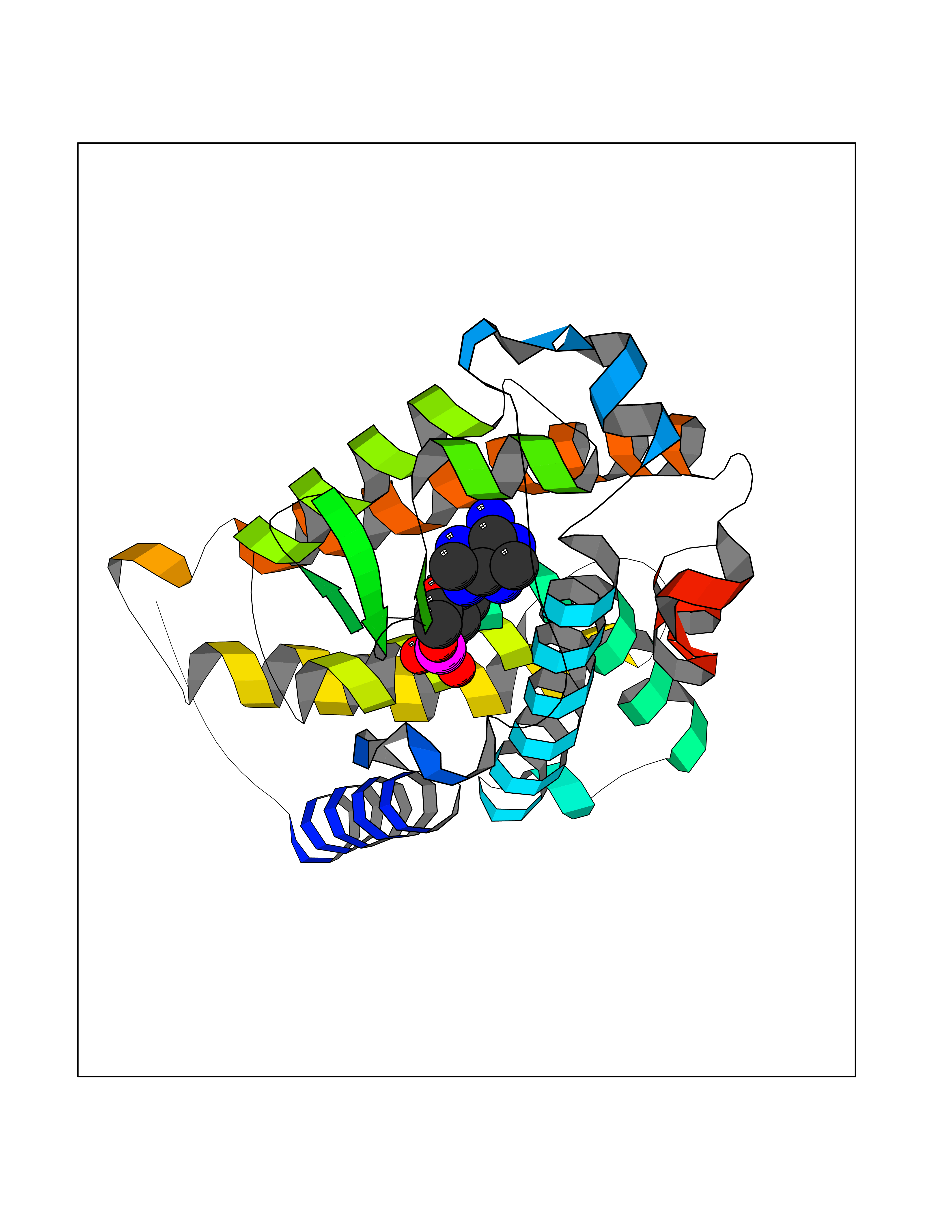 Molscript Map 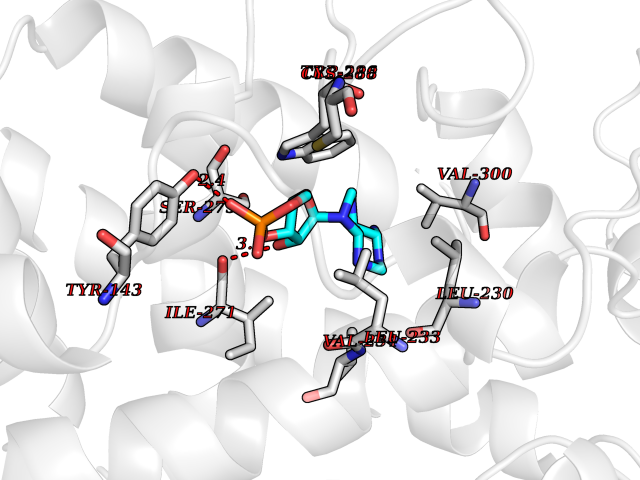 Pymol Map 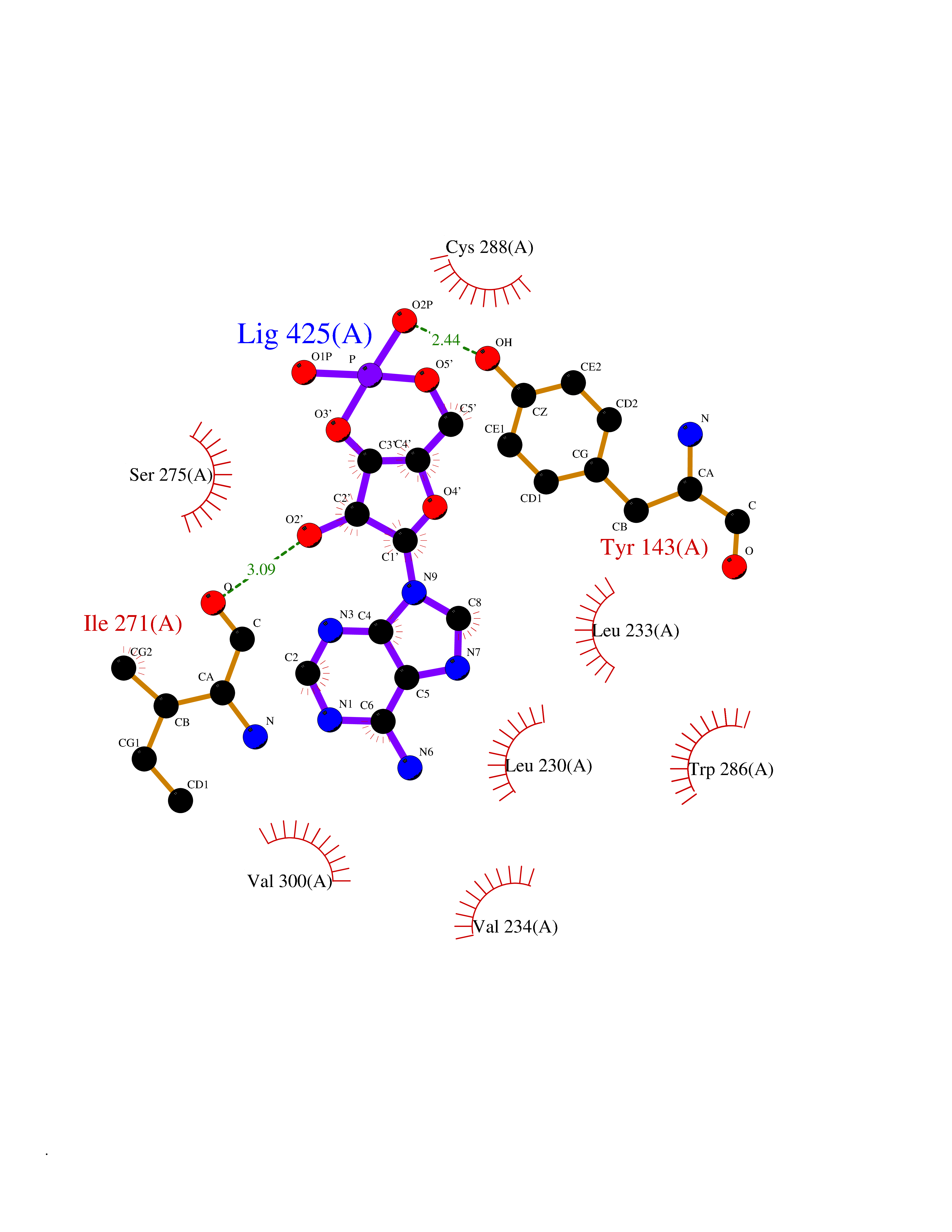 Ligplot Map 3D Binding mode Sequence ALRPKLSEEQQRIIAILLDAHHKTYDPTYSDFCQFRPPVRVNDGGGSVTLELSQLSMLPHLADLVSYSIQKVIGFAKMIPGFRDLTSEDQIVLLKSSAIEVIMLRSNESFTMDDMSWTCGNQDYKYRVSDVTKAGHSLELIEPLIKFQVGLKKLNLHEEEHVLLMAICIVSPDRPGVQDAALIEAIQDRLSNTLQTYIRCRHPPPGSHLLYAKMIQKLADLRSLNEEHSKQYRCLSFQPECSMKLTPLVLEVFG Hydrogen bonds contact Hydrophobic contact | ||||
| 11 | Endothelin-converting enzyme 1 (ECE1) | 3DWB | 6.95 | |
Target general information Gen name ECE1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms ECE-1 Protein family Peptidase M13 family Biochemical class Peptidase Function Converts big endothelin-1 to endothelin-1. Related diseases Hirschsprung disease, cardiac defects, and autonomic dysfunction (HCAD) [MIM:613870]: A disorder characterized by skip-lesions Hirschsprung disease, craniofacial abnormalities and other dysmorphic features, cardiac defects including ductus arteriosus, small subaortic ventricular septal defect, small atrial septal defect, and autonomic dysfunction. {ECO:0000269|PubMed:9915973}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07171 Interacts with P49760; A8MQ03; Q8IUG1; P60370; P60410 EC number EC 3.4.24.71 Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disease variant; Disulfide bond; Glycoprotein; Hirschsprung disease; Hydrolase; Membrane; Metal-binding; Metalloprotease; Phosphoprotein; Protease; Proteomics identification; Reference proteome; Signal-anchor; Transmembrane; Transmembrane helix; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 75247.9 Length 660 Aromaticity 0.12 Instability index 46.29 Isoelectric point 5.33 Charge (pH=7) -18.3 2D Binding mode Binding energy (Kcal/mol) -7 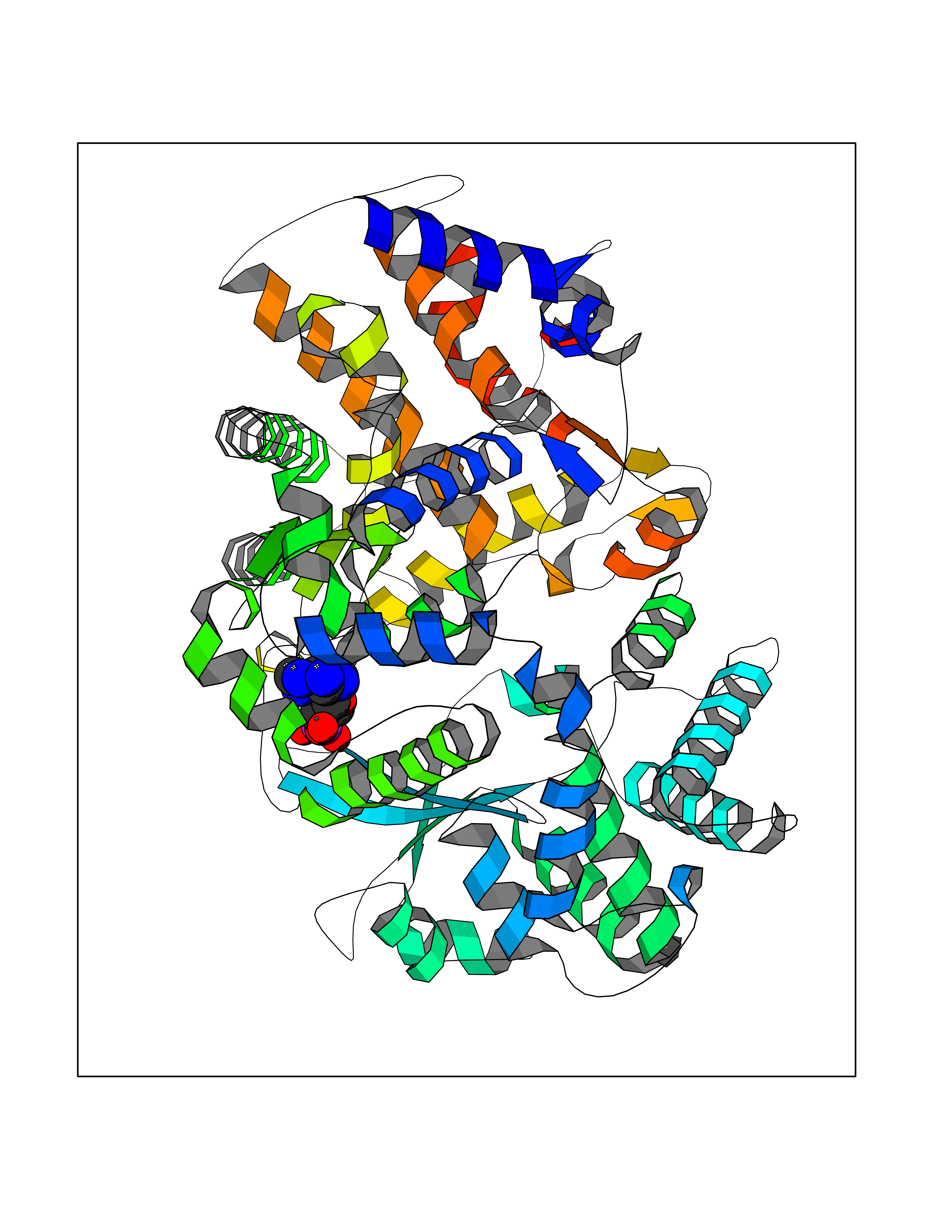 Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence SEACVSVTSSILSSMDPTVDPCHDFFSYACGGWIKANPVPDGHSRWGTFSNLWEHNQAIIKHLLENSTASVSEAERKAQVYYRACMNETRIEELRAKPLMELIERLGGWNITGPWAKDNFQDTLQVVTAHYRTSPFFSVYVSADSKNSNSNVIQVDQSGLGLPSRDYYLNKTENEKVLTGYLNYMVQLGKLLGGGDEEAIRPQMQQILDFETALANITIPQEKRRDEELIYHKVTAAELQTLAPAINWLPFLNTIFYPVEINESEPIVVYDKEYLEQISTLINTTDRCLLNNYMIWNLVRKTSSFLDQRFQDADEKFMEVMWKFCVSDTENNLGFALGPMFVKATFAEDSKSIATEIILEIKKAFEESLSTLKWMDEETRKSAKEKADAIYNMIGYPNFIMDPKELDKVFNDYTAVPDLYFENAMRFFNFSWRVTADQLRKAPNRDQWSMTPPMVNAYYSPTKNEIVFPAGILQAPFYTRSSPKALNFGGIGVVVGHELTHAFDDQGREYDKDGNLRPWWKNSSVEAFKRQTECMVEQYSNYSVNGEPVNGRHTLGENIADNGGLKAAYRAYQNWVKKNGAEHSLPTLGLTNNQLFFLGFAQVWCSVRTPESSHEGLITDPHSPSRFRVIGSLSNSKEFSEHFRCPPGSPMNPPHKCEVW Hydrogen bonds contact Hydrophobic contact | ||||
| 12 | Platelet-derived growth factor receptor alpha (PDGFRA) | 5K5X | 6.94 | |
Target general information Gen name PDGFRA Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms RHEPDGFRA; Platelet-derived growth factor receptor 2; Platelet-derived growth factor alpha receptor; PDGFR2; PDGFR-alpha; PDGFR-2; PDGF-R-alpha; CD140a antigen; CD140a; CD140 antigen-like family membe Protein family Protein kinase superfamily, Tyr protein kinase family, CSF-1/PDGF receptor subfamily Biochemical class Kinase Function Depending on the context, promotes or inhibits cell proliferation and cell migration. Plays an important role in the differentiation of bone marrow-derived mesenchymal stem cells. Required for normal skeleton development and cephalic closure during embryonic development. Required for normal development of the mucosa lining the gastrointestinal tract, and for recruitment of mesenchymal cells and normal development of intestinal villi. Plays a role in cell migration and chemotaxis in wound healing. Plays a role in platelet activation, secretion of agonists from platelet granules, and in thrombin-induced platelet aggregation. Binding of its cognate ligands - homodimeric PDGFA, homodimeric PDGFB, heterodimers formed by PDGFA and PDGFB or homodimeric PDGFC -leads to the activation of several signaling cascades; the response depends on the nature of the bound ligand and is modulated by the formation of heterodimers between PDGFRA and PDGFRB. Phosphorylates PIK3R1, PLCG1, and PTPN11. Activation of PLCG1 leads to the production of the cellular signaling molecules diacylglycerol and inositol 1,4,5-trisphosphate, mobilization of cytosolic Ca(2+) and the activation of protein kinase C. Phosphorylates PIK3R1, the regulatory subunit of phosphatidylinositol 3-kinase, and thereby mediates activation of the AKT1 signaling pathway. Mediates activation of HRAS and of the MAP kinases MAPK1/ERK2 and/or MAPK3/ERK1. Promotes activation of STAT family members STAT1, STAT3 and STAT5A and/or STAT5B. Receptor signaling is down-regulated by protein phosphatases that dephosphorylate the receptor and its down-stream effectors, and by rapid internalization of the activated receptor. Tyrosine-protein kinase that acts as a cell-surface receptor for PDGFA, PDGFB and PDGFC and plays an essential role in the regulation of embryonic development, cell proliferation, survival and chemotaxis. Related diseases A chromosomal aberration involving PDGFRA is found in some cases of hypereosinophilic syndrome. Interstitial chromosomal deletion del(4)(q12q12) causes the fusion of FIP1L1 and PDGFRA (FIP1L1-PDGFRA). Mutations that cause overexpression and/or constitutive activation of PDGFRA may be a cause of hypereosinophilic syndrome. {ECO:0000269|PubMed:12808148}.; DISEASE: Gastrointestinal stromal tumor (GIST) [MIM:606764]: Common mesenchymal neoplasms arising in the gastrointestinal tract, most often in the stomach. They are histologically, immunohistochemically, and genetically different from typical leiomyomas, leiomyosarcomas, and schwannomas. Most GISTs are composed of a fairly uniform population of spindle-shaped cells. Some tumors are dominated by epithelioid cells or contain a mixture of spindle and epithelioid morphologies. Primary GISTs in the gastrointestinal tract commonly metastasize in the omentum and mesenteries, often as multiple nodules. However, primary tumors may also occur outside of the gastrointestinal tract, in other intra-abdominal locations, especially in the omentum and mesentery. {ECO:0000269|PubMed:12522257, ECO:0000269|PubMed:15928335}. The gene represented in this entry may be involved in disease pathogenesis. Mutations causing PDGFRA constitutive activation have been found in gastrointestinal stromal tumors lacking KIT mutations (PubMed:12522257). {ECO:0000269|PubMed:12522257}.; DISEASE: GIST-plus syndrome (GISTPS) [MIM:175510]: A disorder characterized by multiple mesenchymal tumors of the gastrointestinal tract, including gastrointestinal stromal tumor, inflammatory fibroid polyps, and fibroid tumors. Additional features are coarse facies and skin, broad hands and feet, and premature tooth loss. GISTPS is an autosomal dominant disease with incomplete penetrance. Gastrointestinal stromal tumor and inflammatory fibroid polyps may also occur in isolation. {ECO:0000269|PubMed:14699510, ECO:0000269|PubMed:17087943, ECO:0000269|PubMed:25975287}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB12742; DB00102; DB12147; DB10772; DB12010; DB00619; DB09078; DB06595; DB09079; DB06043; DB06589; DB08901; DB08896; DB14840; DB01268; DB11800; DB05146 Interacts with P46108; P46109; P00533; Q8N6L0; P04085; P01127; Q9NRA1; P31947; P62258; Q9NRA1-1; A8T7D5; P05067; Q8IY26 EC number EC 2.7.10.1 Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Cell membrane; Cell projection; Chemotaxis; Developmental protein; Disease variant; Disulfide bond; Glycoprotein; Golgi apparatus; Host-virus interaction; Immunoglobulin domain; Kinase; Membrane; Nucleotide-binding; Phosphoprotein; Proteomics identification; Proto-oncogene; Receptor; Reference proteome; Repeat; Signal; Transferase; Transmembrane; Transmembrane helix; Tyrosine-protein kinase; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 39294.8 Length 345 Aromaticity 0.11 Instability index 46.74 Isoelectric point 6.6 Charge (pH=7) -1.38 2D Binding mode Binding energy (Kcal/mol) -6.84  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence RYEIRWRVIESISPDGHEYIYVDPMQLPYDSRWEFPRDGLVLGRVLGSGAFGKVVEGTAYGLSRSQPVMKVAVKMLKPTARSSEKQALMSELKIMTHLGPHLNIVNLLGACTKSGPIYIITEYCFYGDLVNYLHKNRDSFLSHSMLDSEVKNLLSDDNSEGLTLLDLLSFTYQVARGMEFLASKNCVHRDLAARNVLLAQGKIVKICDFGLARDIMHDSNYVSKGSTFLPVKWMAPESIFDNLYTTLSDVWSYGILLWEIFSLGGTPYPGMMVDSTFYNKIKSGYRMAKPDHATSEVYEIMVKCWNSEPEKRPSFYHLSEIVENLLPGQYKKSYEKIHLDFLKSD Hydrogen bonds contact Hydrophobic contact | ||||
| 13 | Interleukin 21 receptor (IL21R) | 6PLH | 6.92 | |
Target general information Gen name IL21R Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms UNQ3121/PRO10273; Novel interleukin receptor; NILR; Interleukin-21 receptor; IL21 receptor; IL-21R; IL-21 receptor; CD360 Protein family Type I cytokine receptor family, Type 4 subfamily Biochemical class Cytokine receptor Function This is a receptor for interleukin-21. Related diseases Immunodeficiency 56 (IMD56) [MIM:615207]: An autosomal recessive primary immunodeficiency characterized by B- and T-cell defects and variable dysfunction of NK cells. Patients tend to have normal numbers of lymphocytes, but show defective class-switched B-cells, low IgG, defective antibody response, and defective T-cell responses to certain antigens. {ECO:0000269|PubMed:23440042}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Chromosomal aberrations involving IL21R is a cause of B-cell non-Hodgkin lymphomas (B-cell NHL). Translocation t(3;16)(q27;p11), with BCL6. Drugs (DrugBank ID) NA Interacts with P29972 EC number NA Uniprot keywords 3D-structure; Chromosomal rearrangement; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; Membrane; Proteomics identification; Receptor; Reference proteome; Repeat; Signal; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A,C,B Molecular weight (Da) 48376.5 Length 446 Aromaticity 0.1 Instability index 43.94 Isoelectric point 8.24 Charge (pH=7) 3.56 2D Binding mode Binding energy (Kcal/mol) -7.7  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence DVVMTHTPLSLPVSLGDQASISCRSSQSLVHSNGNTYLHWYLQKPGQSPKLLIYKVSNRFSGVPDRFSGSGSGADFTLKISRVEAEDLGVYFCSQSTHVPRTFGGGTKLEIKRADAAPTVSIFPPSSEQLTSGGASVVCFLNNFYPKDINVKWKIDGSERQNGVLNSWTDQDSKDSTYSMSSTLTLTKDEYERHNSYTCEATHKTSTSPIVKSFNRNECXVHLQQPGADLVKPGASVKMSCKASGYTFTSYWITWVKLRPGQGLEWIGDIYPGSGSTNFIEKFKSKATLTVDTSSSTAYMQLRSLTSEDSAVYYCARRGHGNYEDYWGQGTTLIVSSAKTTAPSVYPLAPVCGTGSSVTLGCLVKGYFPEPVTLTWNSGSLSSGVHTFPAVLQSDLYTLSSSVTVTSSTWPSQSITCNVAHPASSTKVDKKIEPRGPTTWSEWSDP Hydrogen bonds contact Hydrophobic contact | ||||
| 14 | Pseudomonas Phosphomannomutase/phosphoglucomutase (Pseudo algC) | 1P5D | 6.91 | |
Target general information Gen name Pseudo algC Organism Pseudomonas aeruginosa (strain ATCC 15692 / DSM 22644 / CIP 104116 / JCM 14847 / LMG 12228 / 1C / PRS 101 / PAO1) Uniprot ID TTD ID Synonyms algC; PMM/PGM; PMM / PGM Protein family Phosphohexose mutase family Biochemical class Intramolecular transferases Function Highly reversible phosphoryltransferase. The phosphomannomutase activity produces a precursor for alginate polymerization, the alginate layer causes a mucoid phenotype and provides a protective barrier against host immune defenses and antibiotics. Also involved in core lipopolysaccaride (LPS) biosynthesis due to its phosphoglucomutase activity. Essential for rhamnolipid production, an exoproduct correlated with pathogenicity (PubMed:10481091). Required for biofilm production. The reaction proceeds via 2 processive phosphoryl transferase reactions; first from enzyme-phospho-Ser-108 to the substrate (generatinga bisphosphorylated substrate intermediate and a dephosphorylated enzyme), a 180 degree rotation of the intermediate (probably aided by movement of domain 4), and subsequent transfer of phosphate back to the enzyme (PubMed:11716469, PubMed:16880541, PubMed:16595672, PubMed:22242625). Related diseases Intellectual developmental disorder, X-linked, syndromic, Claes-Jensen type (MRXSCJ) [MIM:300534]: A disorder characterized by significantly below average general intellectual functioning associated with impairments in adaptive behavior and manifested during the developmental period. MRXSCJ patients manifest intellectual disability associated with variable features such as slowly progressive spastic paraplegia, seizures, facial dysmorphism. {ECO:0000269|PubMed:15586325, ECO:0000269|PubMed:16538222, ECO:0000269|PubMed:16541399, ECO:0000269|PubMed:17320160, ECO:0000269|PubMed:17468742, ECO:0000269|PubMed:23356856, ECO:0000269|PubMed:25666439}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02007; DB02843; DB02900; DB02867; DB04522 Interacts with NA EC number NA Uniprot keywords 3D-structure; Alginate biosynthesis; Direct protein sequencing; Isomerase; Lipopolysaccharide biosynthesis; Magnesium; Metal-binding; Multifunctional enzyme; Phosphoprotein; Reference proteome; Virulence Protein physicochemical properties Chain ID X Molecular weight (Da) 49419.9 Length 455 Aromaticity 0.07 Instability index 28.92 Isoelectric point 5.11 Charge (pH=7) -12.7 2D Binding mode Binding energy (Kcal/mol) -7.02 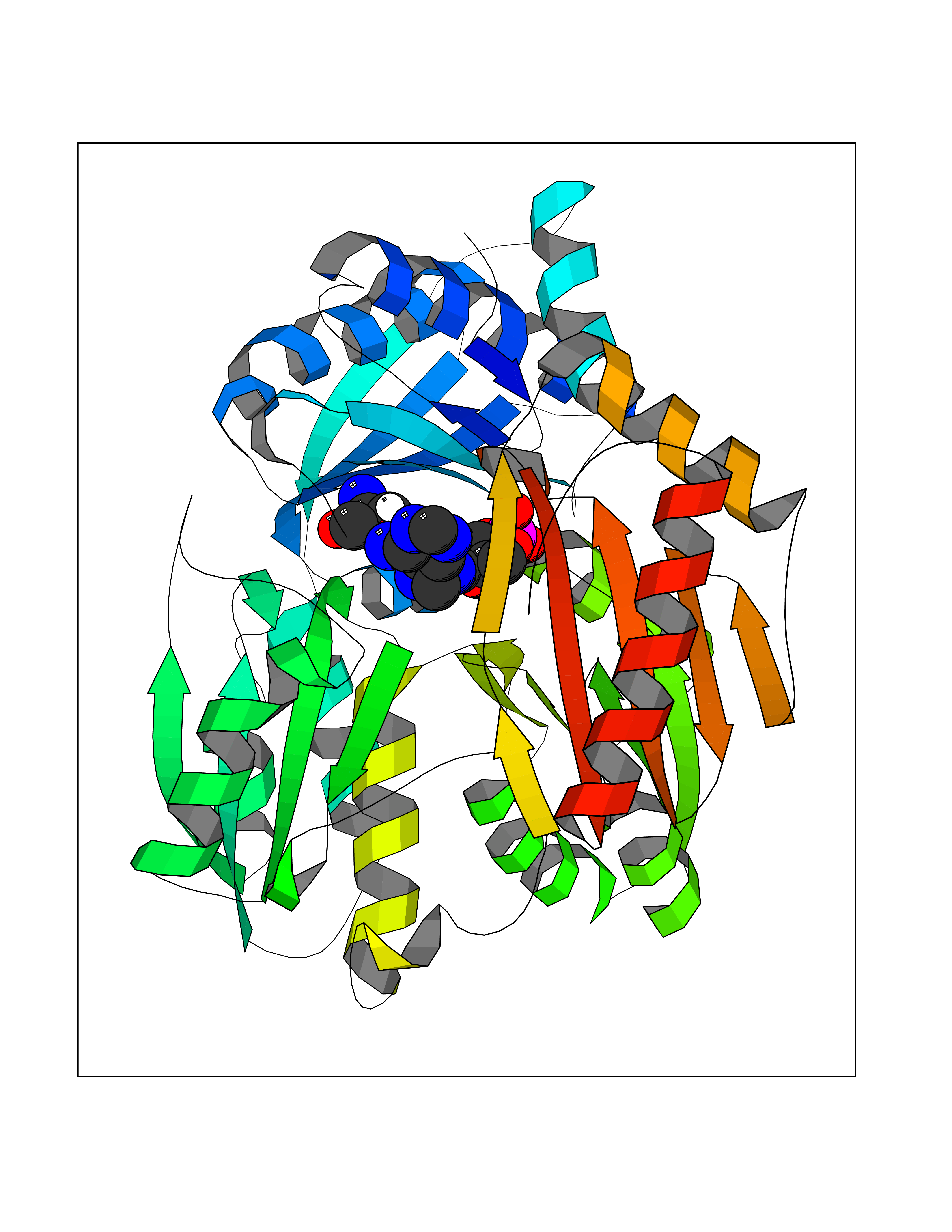 Molscript Map 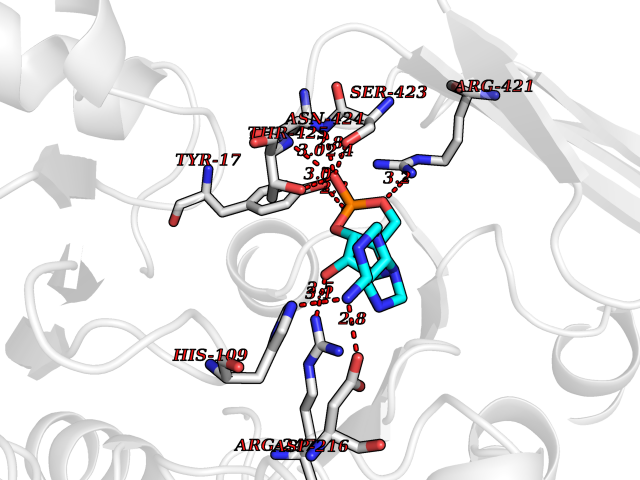 Pymol Map 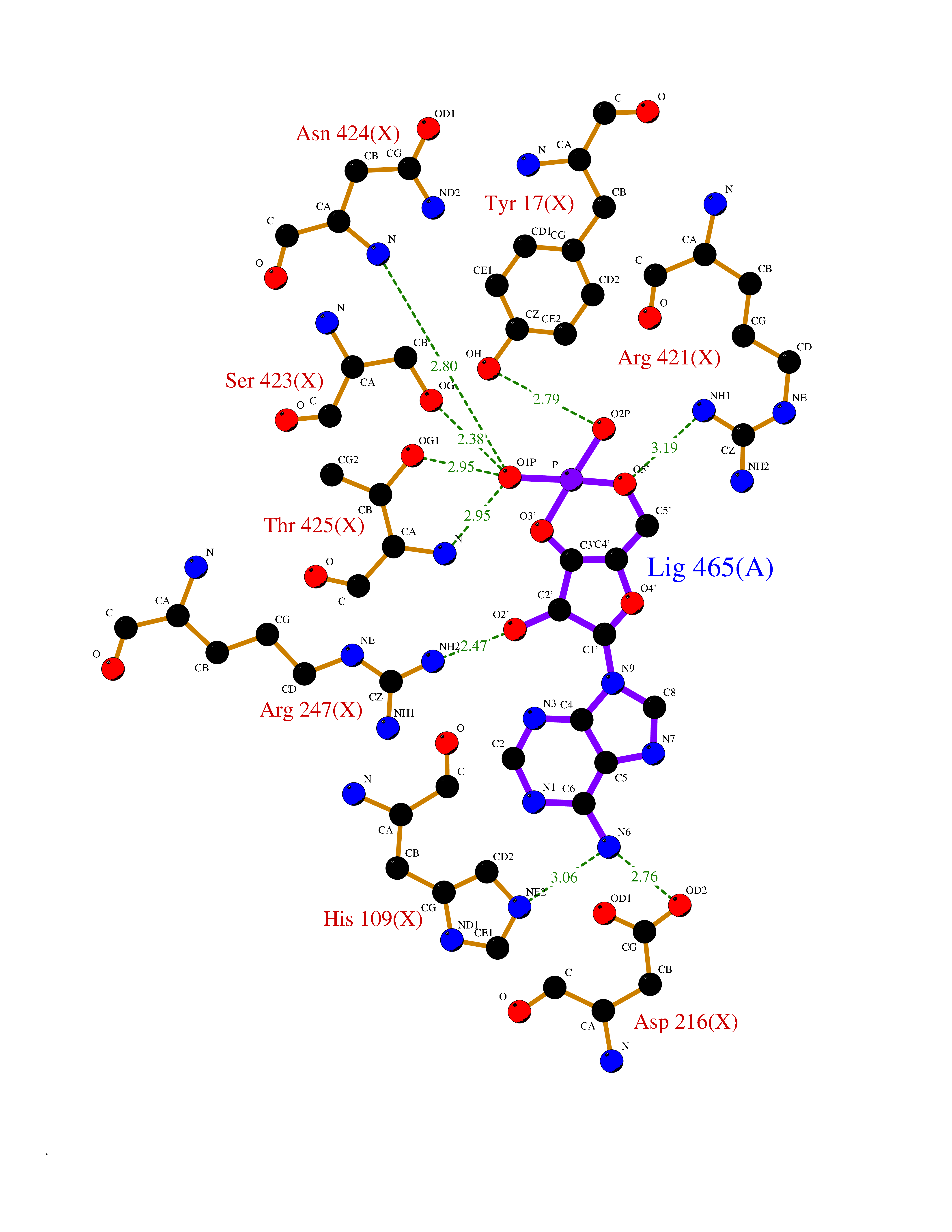 Ligplot Map 3D Binding mode Sequence LPASIFRAYDIRGVVGDTLTAETAYWIGRAIGSESLARGEPCVAVGRDGRLSGPELVKQLIQGLVDCGCQVSDVGMVPTPVLYYAANVLEGKSGVMLTGXHNPPDYNGFKIVVAGETLANEQIQALRERIEKNDLASGVGSVEQVDILPRYFKQIRDDIAMAKPMKVVVDCGNGVAGVIAPQLIEALGCSVIPLYCEVDGNFPNHHPDPGKPENLKDLIAKVKAENADLGLAFDGDGDRVGVVTNTGTIIYPDRLLMLFAKDVVSRNPGADIIFDVKCTRRLIALISGYGGRPVMWKTGHSLIKKKMKETGALLAGEMSGHVFFKERWFGFDDGIYSAARLLEILSQDQRDSEHVFSAFPSDISTPEINITVTEDSKFAIIEALQRDAQWGEGNITTLDGVRVDYPKGWGLVRASNTTPVLVLRFEADTEEELERIKTVFRNQLKAVDSSLPVPF Hydrogen bonds contact Hydrophobic contact | ||||
| 15 | Intestinal maltase-glucoamylase (MGAM) | 3L4Y | 6.90 | |
Target general information Gen name MGAM Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms MGAM Protein family Glycosyl hydrolase 31 family Biochemical class Glycosylase Function May serve as an alternate pathway for starch digestion when luminal alpha-amylase activity is reduced because of immaturity or malnutrition. May play a unique role in the digestion of malted dietary oligosaccharides used in food manufacturing. Related diseases Hypervalinemia and hyperleucine-isoleucinemia (HVLI) [MIM:618850]: An autosomal recessive metabolic disorder characterized by highly elevated plasma concentrations of valine and leucine/isoleucine. Affected individuals suffer from headache and mild memory impairment. {ECO:0000269|PubMed:25653144}. The disease is caused by variants affecting the gene represented in this entry. A patient with hypervalinemia and hyperleucine-isoleucinemia was identified as compound heterozygote for Gln-170 (inherited from his father) and Lys-264 (inherited from his mother), both variants reduced the catalytic activity of the enzyme. After treatment with vitamin B6, a precursor of pyridoxal 5'-phosphate, a BCAT2 cofactor, the blood levels of branched chain amino acids, especially valine, were decreased and brain lesions were improved. {ECO:0000269|PubMed:25653144}. Drugs (DrugBank ID) DB00284; DB00491; DB04878 Interacts with Q13520; Q7Z7G2; Q96BA8; O15529; P14410; P54219-3; Q9NUH8 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Direct protein sequencing; Disulfide bond; Glycoprotein; Glycosidase; Hydrolase; Membrane; Multifunctional enzyme; Proteomics identification; Reference proteome; Repeat; Signal-anchor; Sulfation; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 97779.4 Length 863 Aromaticity 0.12 Instability index 32.47 Isoelectric point 5.2 Charge (pH=7) -28.27 2D Binding mode Binding energy (Kcal/mol) -7.49 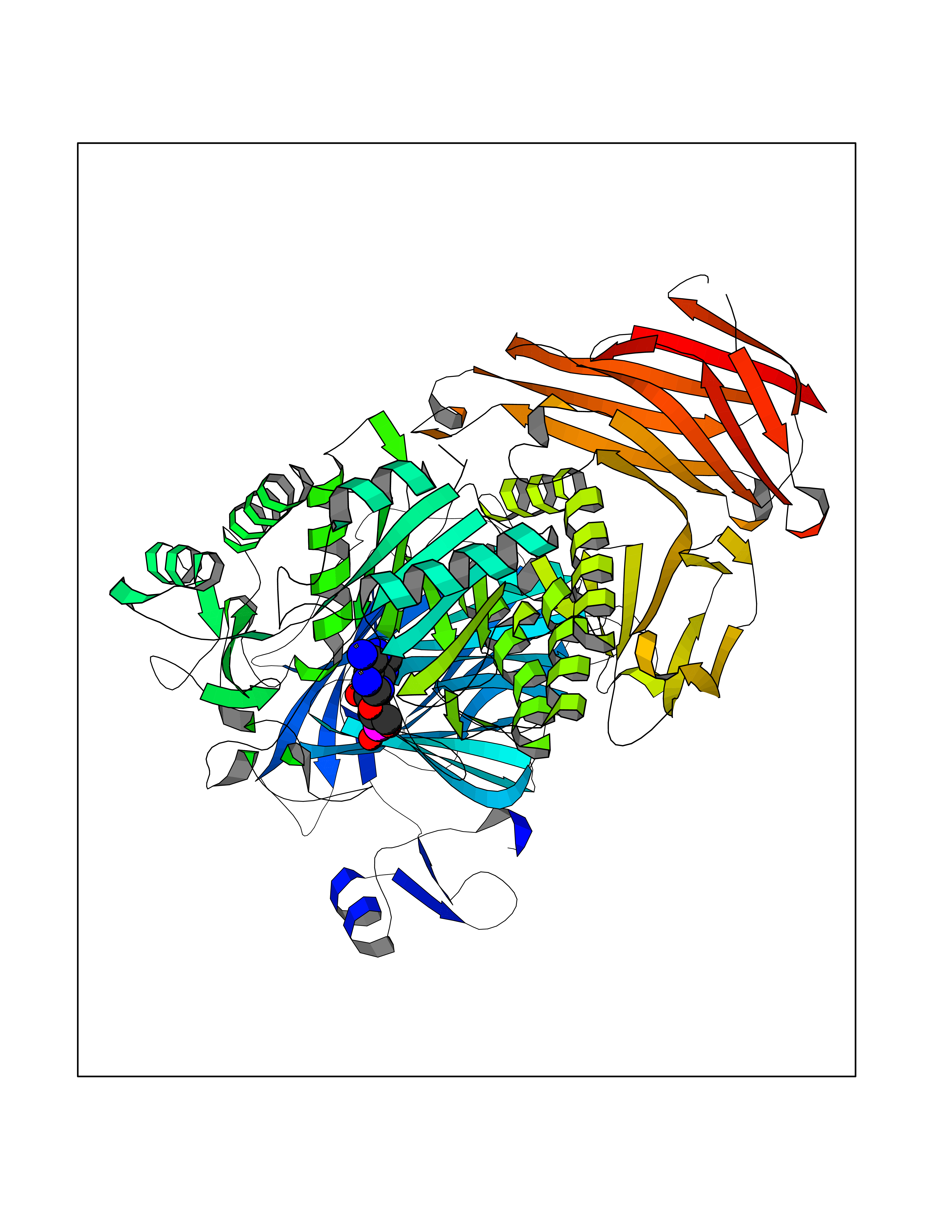 Molscript Map 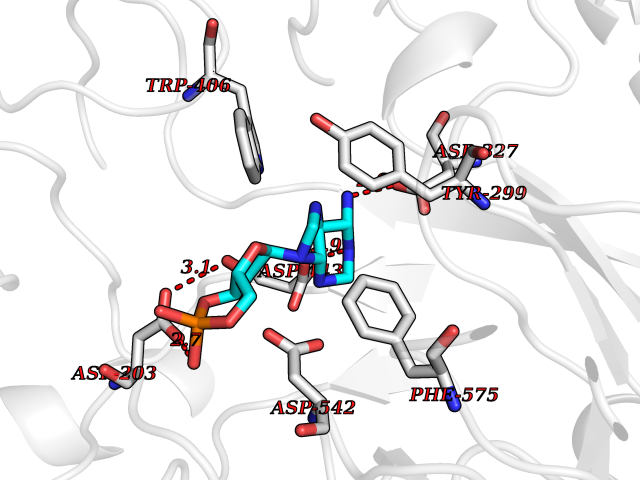 Pymol Map 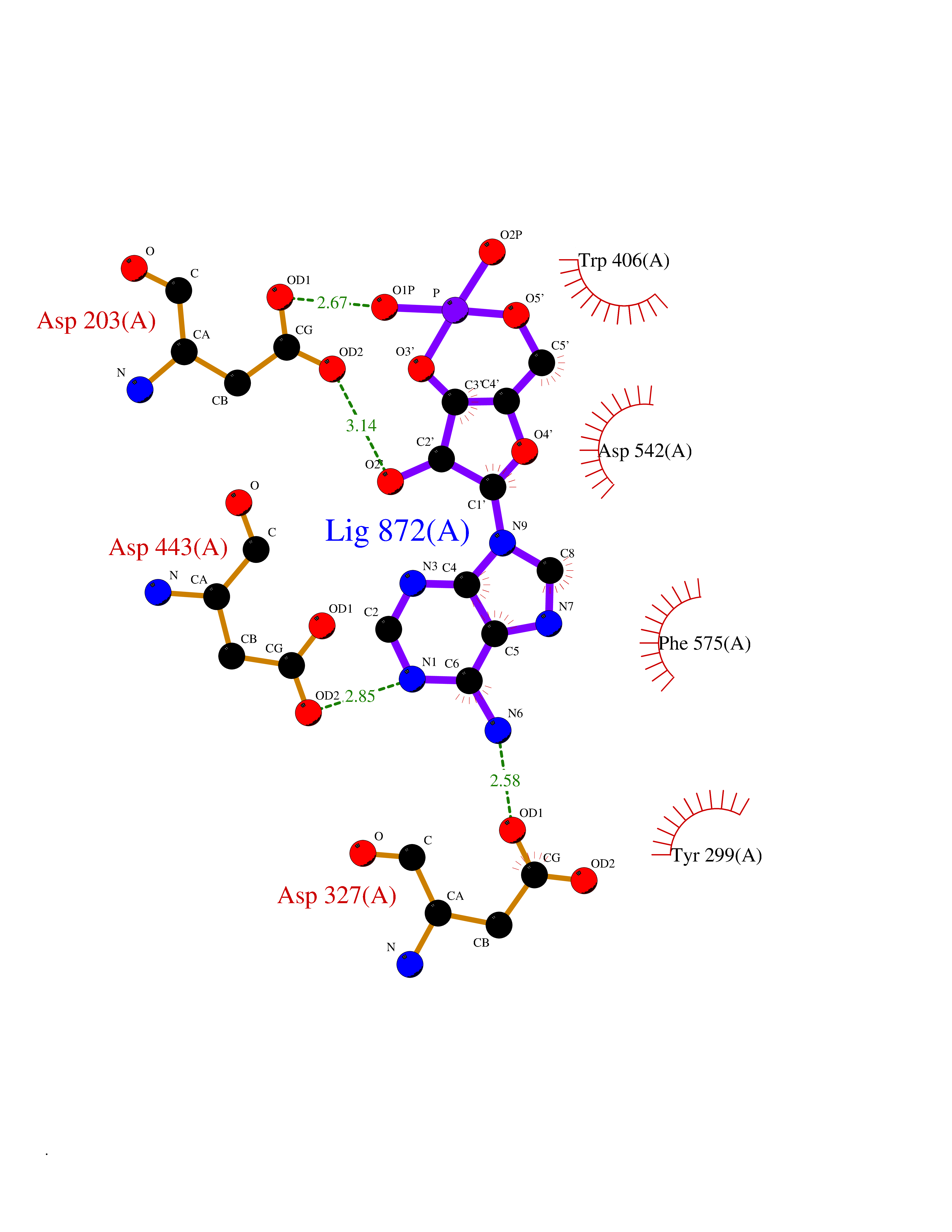 Ligplot Map 3D Binding mode Sequence VNELERINCIPDQPPTKATCDQRGCCWNPQGAVSVPWCYYSKNHSYHVEGNLVNTNAGFTARLKNLPSSPVFGSNVDNVLLTAEYQTSNRFHFKLTDQTNNRFEVPHEHVQSFSGNAAASLTYQVEISRQPFSIKVTRRSNNRVLFDSSIGPLLFADQFLQLSTRLPSTNVYGLGEHVHQQYRHDMNWKTWPIFNRDTTPNGNGTNLYGAQTFFLCLEDASGLSFGVFLMNSNAMEVVLQPAPAITYRTIGGILDFYVFLGNTPEQVVQEYLELIGRPALPSYWALGFHLSRYEYGTLDNMREVVERNRAAQLPYDVQHADIDYMDERRDFTYDSVDFKGFPEFVNELHNNGQKLVIIVDPAISNNSSSSKPYGPYDRGSDMKIWVNSSDGVTPLIGEVWPGQTVFPDYTNPNCAVWWTKEFELFHNQVEFDGIWIDMNEVSNFVDGSVSGCSTNNLNNPPFTPRILDGYLFCKTLCMDAVQHWGKQYDIHNLYGYSMAVATAEAAKTVFPNKRSFILTRSTFAGSGKFAAHWLGDNTATWDDLRWSIPGVLEFNLFGIPMVGPDICGFALDTPEELCRRWMQLGAFYPFSRNHNGQGYKDQDPASFGADSLLLNSSRHYLNIRYTLLPYLYTLFFRAHSRGDTVARPLLHEFYEDNSTWDVHQQFLWGPGLLITPVLDEGAEKVMAYVPDAVWYDYETGSQVRWRKQKVEMELPGDKIGLHLRGGYIFPTQQPNTTTLASRKNPLGLIIALDENKEAKGELFWDDGETKDTVANKVYLLCEFSVTQNRLEVNISQSTYKDPNNLAFNEIKILGTEEPSNVTVKHNGVPSTSPTVTYDSNLKVAIITDIDLLLGEAYTVEWAH Hydrogen bonds contact Hydrophobic contact | ||||
| 16 | DNA mismatch repair protein MSH2 (MSH2) | 3THX | 6.84 | |
Target general information Gen name MSH2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms hMSH2; MutS protein homolog 2; Mismatch repair gene Msh2 Protein family DNA mismatch repair MutS family Biochemical class NA Function Forms two different heterodimers: MutS alpha (MSH2-MSH6 heterodimer) and MutS beta (MSH2-MSH3 heterodimer) which binds to DNA mismatches thereby initiating DNA repair. When bound, heterodimers bend the DNA helix and shields approximately 20 base pairs. MutS alpha recognizes single base mismatches and dinucleotide insertion-deletion loops (IDL) in the DNA. MutS beta recognizes larger insertion-deletion loops up to 13 nucleotides long. After mismatch binding, MutS alpha or beta forms a ternary complex with the MutL alpha heterodimer, which is thought to be responsible for directing the downstream MMR events, including strand discrimination, excision, and resynthesis. Recruits DNA helicase MCM9 to chromatin which unwinds the mismatch containg DNA strand. ATP binding and hydrolysis play a pivotal role in mismatch repair functions. The ATPase activity associated with MutS alpha regulates binding similar to a molecular switch: mismatched DNA provokes ADP-->ATP exchange, resulting in a discernible conformational transition that converts MutS alpha into a sliding clamp capable of hydrolysis-independent diffusion along the DNA backbone. This transition is crucial for mismatch repair. MutS alpha may also play a role in DNA homologous recombination repair. In melanocytes may modulate both UV-B-induced cell cycle regulation and apoptosis. Component of the post-replicative DNA mismatch repair system (MMR). Related diseases Lynch syndrome 1 (LYNCH1) [MIM:120435]: A form of Lynch syndrome, an autosomal dominant disease associated with marked increase in cancer susceptibility. It is characterized by a familial predisposition to early-onset colorectal carcinoma (CRC) and extra-colonic tumors of the gastrointestinal, urological and female reproductive tracts. Lynch syndrome is reported to be the most common form of inherited colorectal cancer in the Western world. Clinically, it is often divided into two subgroups. Type I is characterized by hereditary predisposition to colorectal cancer, a young age of onset, and carcinoma observed in the proximal colon. Type II is characterized by increased risk for cancers in certain tissues such as the uterus, ovary, breast, stomach, small intestine, skin, and larynx in addition to the colon. Diagnosis of classical Lynch syndrome is based on the Amsterdam criteria: 3 or more relatives affected by colorectal cancer, one a first degree relative of the other two; 2 or more generation affected; 1 or more colorectal cancers presenting before 50 years of age; exclusion of hereditary polyposis syndromes. The term 'suspected Lynch syndrome' or 'incomplete Lynch syndrome' can be used to describe families who do not or only partially fulfill the Amsterdam criteria, but in whom a genetic basis for colon cancer is strongly suspected. {ECO:0000269|PubMed:10375096, ECO:0000269|PubMed:10386556, ECO:0000269|PubMed:10528862, ECO:0000269|PubMed:10573010, ECO:0000269|PubMed:10612836, ECO:0000269|PubMed:10777691, ECO:0000269|PubMed:10829038, ECO:0000269|PubMed:11726306, ECO:0000269|PubMed:11870161, ECO:0000269|PubMed:11920458, ECO:0000269|PubMed:12112654, ECO:0000269|PubMed:12124176, ECO:0000269|PubMed:12132870, ECO:0000269|PubMed:12200596, ECO:0000269|PubMed:12362047, ECO:0000269|PubMed:12373605, ECO:0000269|PubMed:12655564, ECO:0000269|PubMed:12655568, ECO:0000269|PubMed:12658575, ECO:0000269|PubMed:14635101, ECO:0000269|PubMed:15046096, ECO:0000269|PubMed:15300854, ECO:0000269|PubMed:15342696, ECO:0000269|PubMed:15365995, ECO:0000269|PubMed:15613555, ECO:0000269|PubMed:15870828, ECO:0000269|PubMed:15896463, ECO:0000269|PubMed:15991316, ECO:0000269|PubMed:15996210, ECO:0000269|PubMed:16451135, ECO:0000269|PubMed:17101317, ECO:0000269|PubMed:17128465, ECO:0000269|PubMed:18561205, ECO:0000269|PubMed:18625694, ECO:0000269|PubMed:18781619, ECO:0000269|PubMed:18822302, ECO:0000269|PubMed:18951462, ECO:0000269|PubMed:21120944, ECO:0000269|PubMed:22102614, ECO:0000269|PubMed:22371642, ECO:0000269|PubMed:7874129, ECO:0000269|PubMed:8261515, ECO:0000269|PubMed:8700523, ECO:0000269|PubMed:8797773, ECO:0000269|PubMed:8872463, ECO:0000269|PubMed:9048925, ECO:0000269|PubMed:9240418, ECO:0000269|PubMed:9298827, ECO:0000269|PubMed:9311737, ECO:0000269|PubMed:9419403, ECO:0000269|PubMed:9559627, ECO:0000269|PubMed:9621522, ECO:0000269|PubMed:9718327, ECO:0000269|PubMed:9889267}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Muir-Torre syndrome (MRTES) [MIM:158320]: Rare autosomal dominant disorder characterized by sebaceous neoplasms and visceral malignancy. {ECO:0000269|PubMed:7713503}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Endometrial cancer (ENDMC) [MIM:608089]: A malignancy of endometrium, the mucous lining of the uterus. Most endometrial cancers are adenocarcinomas, cancers that begin in cells that make and release mucus and other fluids. {ECO:0000305|PubMed:11306449, ECO:0000305|PubMed:21642682}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Mismatch repair cancer syndrome 2 (MMRCS2) [MIM:619096]: An autosomal recessive form of mismatch repair cancer syndrome, a childhood cancer predisposition syndrome encompassing a broad tumor spectrum. This includes hematological malignancies, central nervous system tumors, Lynch syndrome-associated malignancies such as colorectal tumors as well as multiple intestinal polyps, embryonic tumors and rhabdomyosarcoma. Multiple cafe-au-lait macules, a feature reminiscent of neurofibromatosis type 1, are often found as first manifestation of the underlying cancer. {ECO:0000269|PubMed:12549480, ECO:0000269|PubMed:16372347}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Colorectal cancer (CRC) [MIM:114500]: A complex disease characterized by malignant lesions arising from the inner wall of the large intestine (the colon) and the rectum. Genetic alterations are often associated with progression from premalignant lesion (adenoma) to invasive adenocarcinoma. Risk factors for cancer of the colon and rectum include colon polyps, long-standing ulcerative colitis, and genetic family history. {ECO:0000269|PubMed:12792735, ECO:0000269|PubMed:14504054, ECO:0000269|PubMed:15996210, ECO:0000269|PubMed:9559627}. Disease susceptibility may be associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q92624; Q9UQ84-1; P09429; P20585; P52701; Q8IY92; P39875 EC number NA Uniprot keywords 3D-structure; Acetylation; Alternative splicing; ATP-binding; Chromosome; Disease variant; DNA damage; DNA repair; DNA-binding; Hereditary nonpolyposis colorectal cancer; Isopeptide bond; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Tumor suppressor; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 97883.4 Length 868 Aromaticity 0.08 Instability index 36.78 Isoelectric point 5.79 Charge (pH=7) -10.09 2D Binding mode Binding energy (Kcal/mol) -7.44  Molscript Map 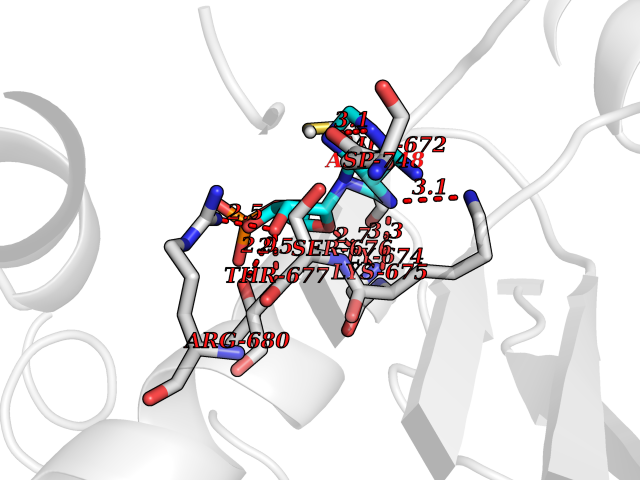 Pymol Map 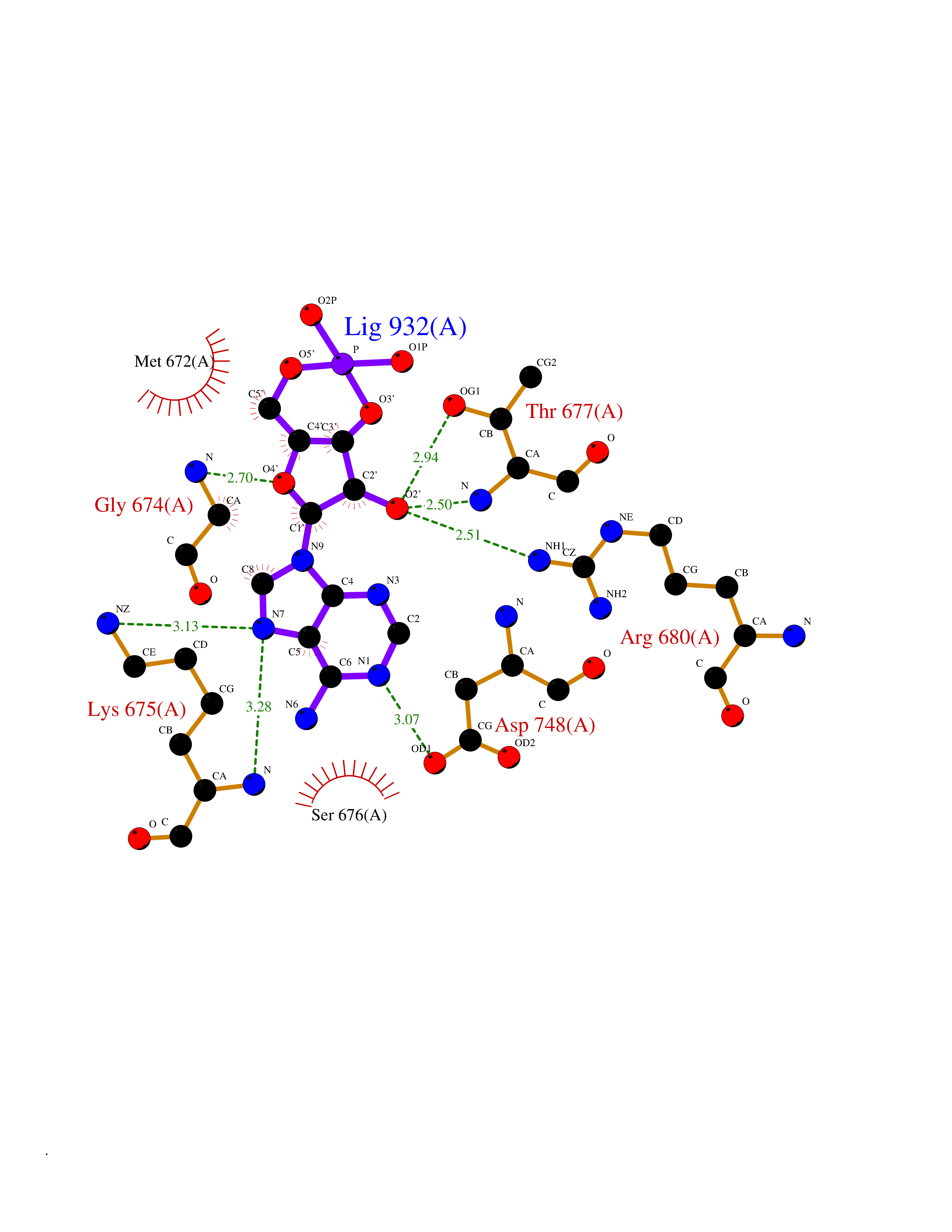 Ligplot Map 3D Binding mode Sequence ESAAEVGFVRFFQGMPEKPTTTVRLFDRGDFYTAHGEDALLAAREVFKTQGVIKYMGPAGAKNLQSVVLSKMNFESFVKDLLLVRQYRVEVYKNRASKENDWYLAYKASPGNLSQFEDILFIGVVGVKMSAVDGQRQVGVGYVDSIQRKLGLCEFPDNDQFSNLEALLIQIGPKECVLPGGETAGDMGKLRQIIQRGGILITERKKADFSTKDIYQDLNRLLKGKKGEQMNSAVLPEMENQVAVSSLSAVIKFLELLSDDSNFGQFELTTFDFSQYMKLDIAAVRALNLFQQSLAALLNKCKTPQGQRLVNQWIKQPLMDKNRIEERLNLVEAFVEDAELRQTLQEDLLRRFPDLNRLAKKFQRQAANLQDCYRLYQGINQLPNVIQALEKHEGKHQKLLLAVFVTPLTDLRSDFSKFQEMIETTLDMDQVENHEFLVKPSFDPNLSELREIMNDLEKKMQSTLISAARDLGLDPGKQIKLDSSAGYYFRVTCKEEKVLRNNKNFSTVDIQGVKFTNSKLTSLNEEYTKNKTEYEEAQDAIVKEIVNISSGYVEPMQTLNDVLAQLDAVVSFAHVSNGAPVPYVRPAILEKGQGRIILKASRHACVEVQIAFIPNDVYFEKDKQMFHIITGPNMGGKSTYIRQTGVIVLMAQIGCFVPCESAEVSIVDCILARVGSTFMAEMLETASILRSATKDSLIIIDELGRGTSTYDGFGLAWAISEYIATKIGAFCMFATHFHELTALANQIPTVNNLHVTALTTEETLTMLYQVKKGVCDQSFGIHVAELANFPKHVIECAKQKALELEEFQYKCYLEREQGEKIIQEFLSKVKQMPFTEMSEENITIKLKQLKAEVIAKNNSFVNEIISRI Hydrogen bonds contact Hydrophobic contact | ||||
| 17 | HMG-CoA reductase (HMGCR) | 2R4F | 6.83 | |
Target general information Gen name HMGCR Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms 3-hydroxy-3-methylglutaryl-coenzyme A reductase Protein family HMG-CoA reductase family Biochemical class CH-OH donor oxidoreductase Function Transmembrane glycoprotein that is the rate-limiting enzyme in cholesterol biosynthesis as well as in the biosynthesis of nonsterol isoprenoids that are essential for normal cell function including ubiquinone and geranylgeranyl proteins. Related diseases Muscular dystrophy, limb-girdle, autosomal recessive 28 (LGMDR28) [MIM:620375]: An autosomal recessive form of limb girdle muscular dystrophy, a group of genetically heterogeneous muscular disorders that share proximal muscle weakness as the major attribute. Most limb girdle muscular dystrophies present with elevated creatinine kinase and myopathic electromyographic features. Disease is usually progressive to a variable degree, ranging from minor disability to complete inability to ambulate, and can involve the large proximal muscles, as well as axial and facial muscles. Different disease forms may exhibit skeletal muscle hypertrophy, kyphoscoliosis, and contractures or involve other muscle groups and manifest with distal weakness, cardiomyopathy, dysphagia, and respiratory difficulties. LGMDR28 is characterized by progressive muscle weakness affecting the proximal and axial muscles of the upper and lower limbs, and highly variable age at onset. Most patients have limited ambulation or become wheelchair-bound within a few decades, and respiratory insufficiency commonly occurs. {ECO:0000269|PubMed:36745799, ECO:0000269|PubMed:37167966}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03169; DB04447; DB01076; DB09061; DB00439; DB01992; DB01095; DB00227; DB14009; DB04377; DB06693; DB14011; DB00157; DB03461; DB08860; DB00175; DB01098; DB00641; DB05317; DB09270 Interacts with Q9Y5Z9; Q9Y5Z9-1 EC number EC 1.1.1.34 Uniprot keywords 3D-structure; Alternative splicing; Cholesterol biosynthesis; Cholesterol metabolism; Disease variant; Endoplasmic reticulum; Glycoprotein; Isopeptide bond; Limb-girdle muscular dystrophy; Lipid biosynthesis; Lipid metabolism; Membrane; NADP; Oxidoreductase; Peroxisome; Phosphoprotein; Proteomics identification; Reference proteome; Steroid biosynthesis; Steroid metabolism; Sterol biosynthesis; Sterol metabolism; Transmembrane; Transmembrane helix; Ubl conjugation Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 84796.9 Length 798 Aromaticity 0.05 Instability index 47.61 Isoelectric point 6.2 Charge (pH=7) -3.71 2D Binding mode Binding energy (Kcal/mol) -7.56  Molscript Map 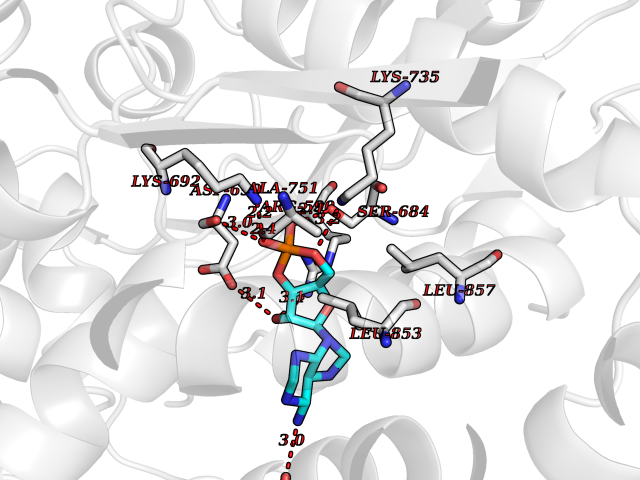 Pymol Map 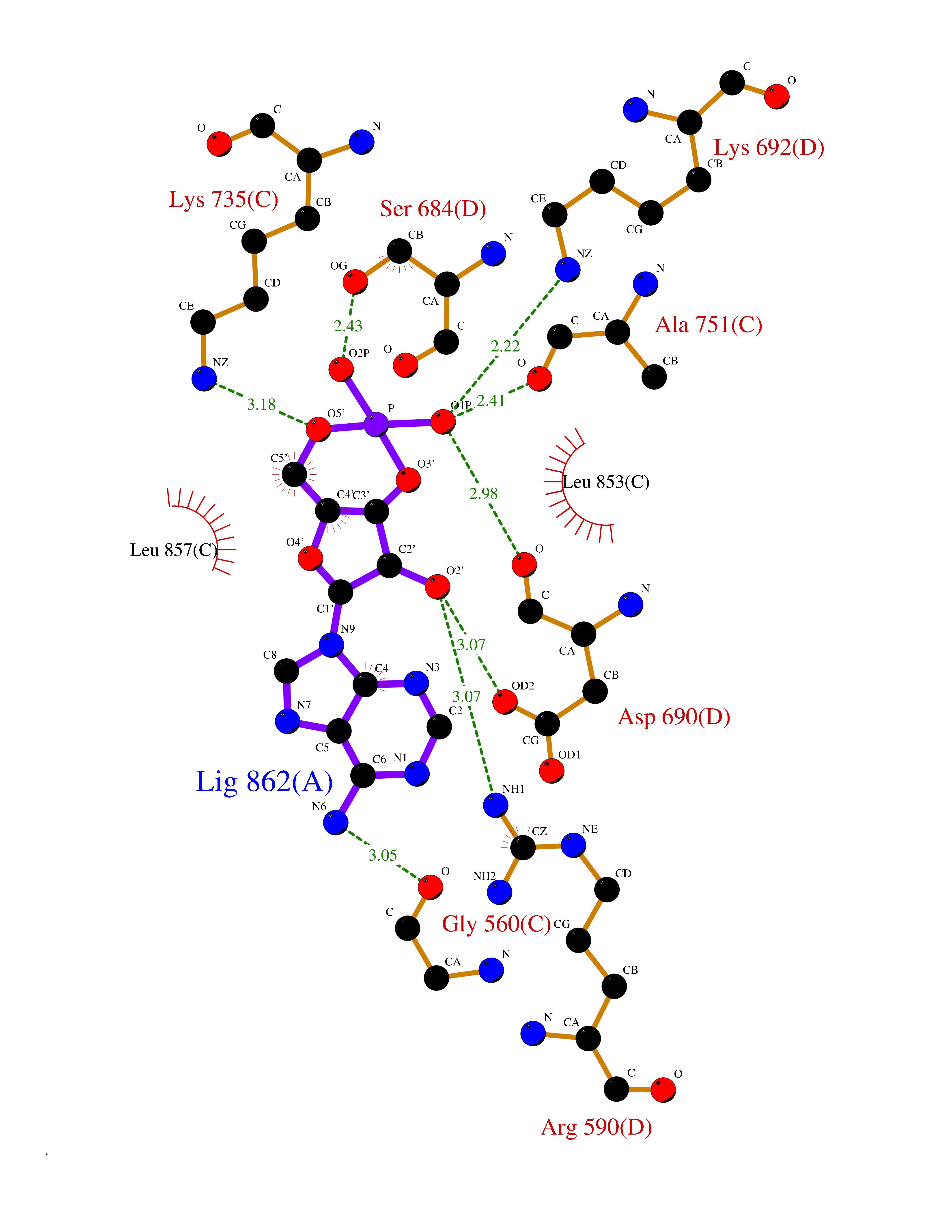 Ligplot Map 3D Binding mode Sequence GAKFLSDAEIIQLVNETLIETHERGVSIRRQLLSKKLSEPSSLQYLPYRDYNYSLVMGACCENVIGYMPIPVGVAGPLCLDEKEFQVPMATTEGCLVASTNRGCRAIGLGGGASSRVLADGMTRGPVVRLPRACDSAEVKAWLETSEGFAVIKEAFDSTSRFARLQKLHTSIAGRNLYIRFQSRSGDAMGMNMISKGTEKALSKLHEYFPEMQILAVSGNYCTDKKPAAINWIEGRGKSVVCEAVIPAKVVREVLKTTTEAMIEVNINKNLVGSAMAGSIGGYNAHAANIVTAIYIACGQDAAQNVGSSNCITLMEASGPTNEDLYISCTMPSIEIGTVGGGTNLLPQQACLQMLGVQGACKDNPGENARQLARIVCGTVMAGELSLMAALAAGPNEECLQILGNGAKFLSDAEIIQLVETLIETHERGVSIRRQLLSKKLSEPSSLQYLPYRDYNYSLVMGACCENVIGYMPIPVGVAGPLCLDEKEFQVPMATTEGCLVASTNRGCRAIGLGGGASSRVLADGMTRGPVVRLPRACDSAEVKAWLETSEGFAVIKEAFDSTSRFARLQKLHTSIAGRNLYIRFQSRSGDAMGMNMISKGTEKALSKLHEYFPEMQILAVSGNYCTDKKPAAINWIEGRGKSVVCEAVIPAKVVREVLKTTTEAMIEVNINKNLVGSAMAGSIGGYNAHAANIVTAIYIACGQDAAQNVGSSNCITLMEASGPTNEDLYISCTMPSIEIGTVGGGTNLLPQQACLQMLGVQGACKDNPGENARQLARIVCGTVMAGELSLMAALAAG Hydrogen bonds contact Hydrophobic contact | ||||
| 18 | Clostridium histolyticum Collagenase (CH colG) | 7Z5U | 6.82 | |
Target general information Gen name CH colG Organism Hathewaya histolytica (Clostridium histolyticum) Uniprot ID TTD ID Synonyms Microbial collagenase; Gelatinase ColG; Collagenase ColG; Class I collagenase Protein family Peptidase M9B family, Collagenase subfamily Biochemical class NA Function Clostridial collagenases are among the most efficient degraders of eukaryotic collagen known; saprophytes use collagen as a carbon source while pathogens additionally digest collagen to aid in host colonization. Has both tripeptidylcarboxypeptidase on Gly-X-Y and endopeptidase activities; the endopeptidase cuts within the triple helix region of collagen while tripeptidylcarboxypeptidase successively digests the exposed ends, thus clostridial collagenases can digest large sections of collagen. Active on soluble type I collagen, insoluble collagen, azocoll, soluble PZ-peptide (all collagenase substrates) and gelatin. The full-length protein has collagenase activity, while the in vivo derived C-terminally truncated shorter versions only act on gelatin. In vitro digestion of soluble calf skin collagen fibrils requires both ColG and ColH; ColG forms missing the second collagen-binding domain are also synergistic with ColH, although their overall efficiency is decreased. The activator domain (residues 119-388) and catalytic subdomain (389-670) open and close around substrate using a Gly-rich hinge (387-397), allowing digestion when the protein is closed. Binding of collagen requires Ca(2+) and is inhibited by EGTA; the collagen-binding domain (CBD, S3a plus S3b) specifically recognizes the triple-helical conformation made by 3 collagen protein chains in the triple-helical region. Isolated CBD (S3a plus S3b) binds collagen fibrils and sheets of many tissues. Related diseases Intellectual developmental disorder, X-linked, syndromic, Claes-Jensen type (MRXSCJ) [MIM:300534]: A disorder characterized by significantly below average general intellectual functioning associated with impairments in adaptive behavior and manifested during the developmental period. MRXSCJ patients manifest intellectual disability associated with variable features such as slowly progressive spastic paraplegia, seizures, facial dysmorphism. {ECO:0000269|PubMed:15586325, ECO:0000269|PubMed:16538222, ECO:0000269|PubMed:16541399, ECO:0000269|PubMed:17320160, ECO:0000269|PubMed:17468742, ECO:0000269|PubMed:23356856, ECO:0000269|PubMed:25666439}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number EC 3.4.24.3 Uniprot keywords 3D-structure; Calcium; Direct protein sequencing; Hydrolase; Metal-binding; Metalloprotease; Pharmaceutical; Protease; Repeat; Secreted; Signal; Virulence; Zinc; Zymogen Protein physicochemical properties Chain ID A Molecular weight (Da) 44751.2 Length 386 Aromaticity 0.15 Instability index 27.33 Isoelectric point 5.77 Charge (pH=7) -7.33 2D Binding mode Binding energy (Kcal/mol) -7.97 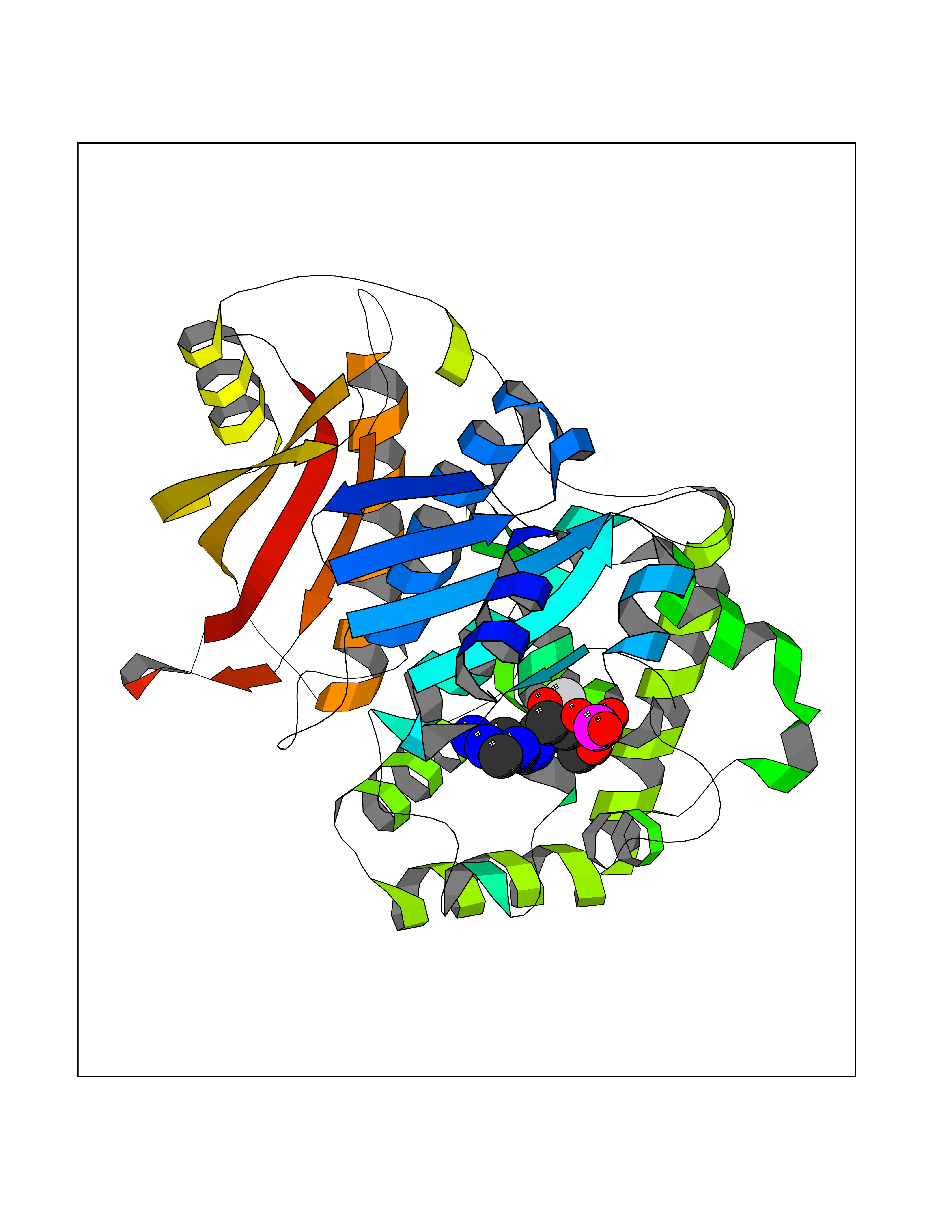 Molscript Map 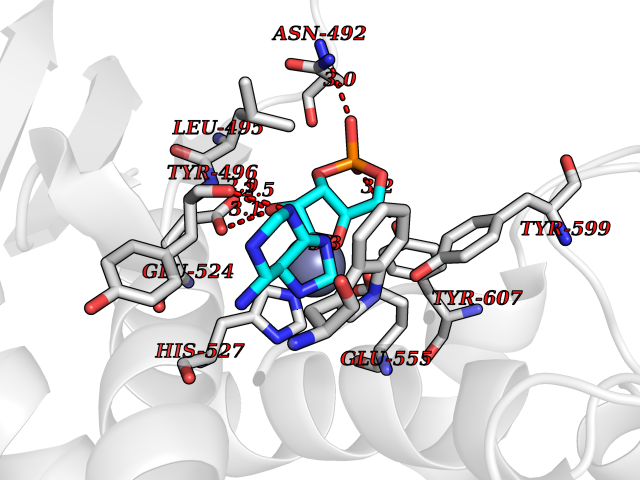 Pymol Map 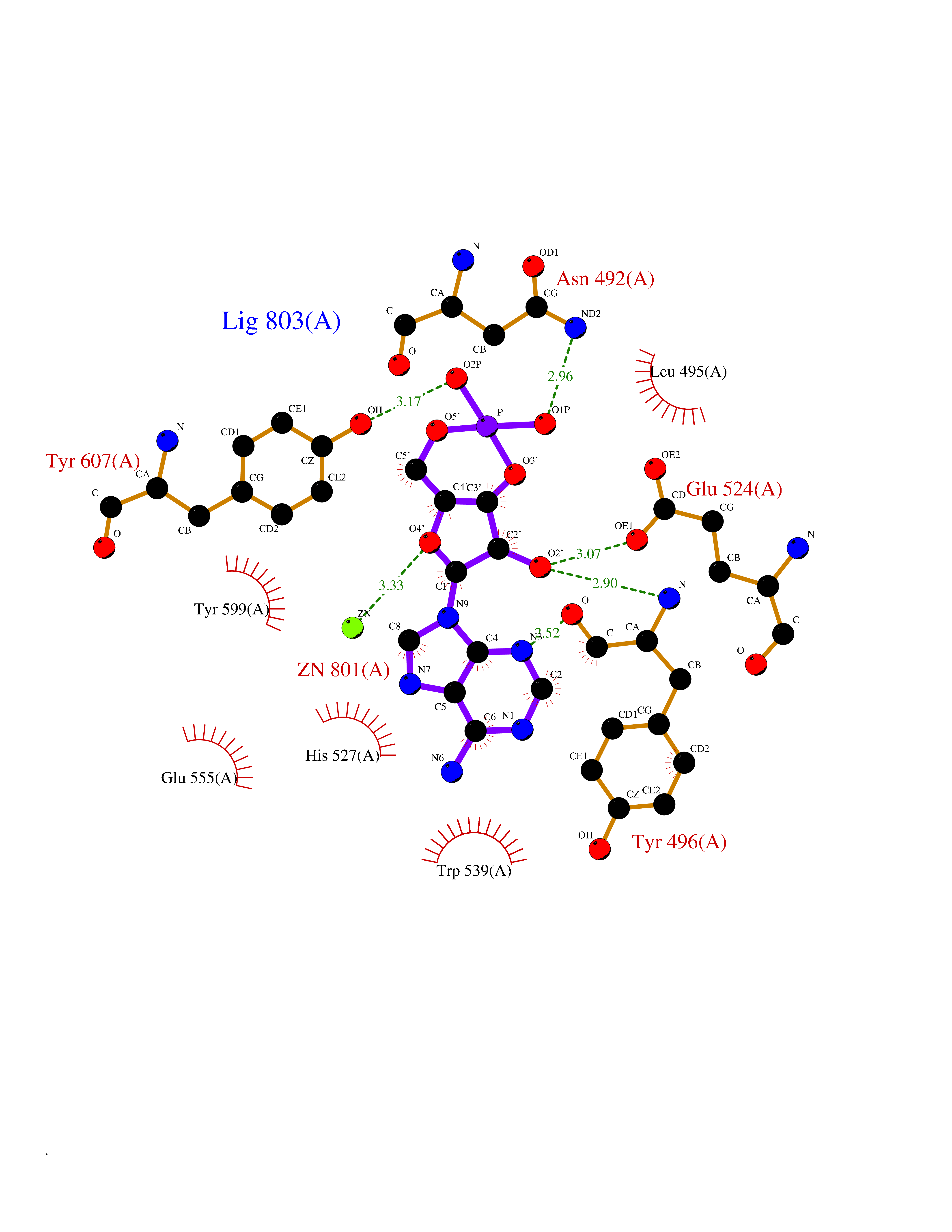 Ligplot Map 3D Binding mode Sequence DHDKFLDDAEKHYLPKTYTFDNGTFIIRAGDKVSEEKIKRLYWASREVKSQFHRVVGNDKALEVGNADDVLTMKIFNSPEEYKFNTTDNGGLYIEPRGTFYTYERTPQQSIFSLEELFRHEYTHYLQARYLVDGLWGQGPFYEKNRLTWFDEGTAEFFAGSTRTSGVLPRKLILGYLAKDKVDHRYSLKKTLNSGYDDSDWMFYNYGFAVAHYLYEKDMPTFIKMNKAILNTDVKSYDEIIKKLSDDANKNTEYQNHIQELVDKYQGAGIPLVSDDYLKDHGYKKASEVYSEISKAASLTNTSVTAEKSQYFNTFTLRGTYTGETSKGEFKDWDEMSKKLDGTLESLAKNSWSGYKTLTAYFTNYRVTSDNKVQYDVVFHGVLTDN Hydrogen bonds contact Hydrophobic contact | ||||
| 19 | Bacterial DD-carboxypeptidase (Bact vanYB) | 5ZHW | 6.80 | |
Target general information Gen name Bact vanYB Organism Enterococcus faecalis (strain ATCC 700802 / V583) Uniprot ID TTD ID Synonyms vanYB; DD-peptidase; DD-carboxypeptidase; D-alanyl-D-alanine carboxypeptidase-transpeptidase Protein family Peptidase M15B family Biochemical class Peptidase Function Vancomycin-inducible, penicillin-resistant, DD- carboxypeptidase that hydrolyzes depsipeptide- and D-alanyl-D- alanine-containing peptidoglycan precursors. Insensitive to beta- lactams. Related diseases Brachyolmia 3 (BCYM3) [MIM:113500]: A form of brachyolmia, a clinically and genetically heterogeneous skeletal dysplasia primarily affecting the spine and characterized by a short trunk, short stature, and platyspondyly. BCYM3 is an autosomal dominant form with severe scoliosis with or without kyphosis, and flattened irregular cervical vertebrae. {ECO:0000269|PubMed:18587396}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Spondylometaphyseal dysplasia Kozlowski type (SMDK) [MIM:184252]: A form of spondylometaphyseal dysplasia, a group of short stature disorders distinguished by abnormalities in the vertebrae and the metaphyses of the tubular bones. It is characterized by postnatal dwarfism, significant scoliosis and mild metaphyseal abnormalities in the pelvis. The vertebrae exhibit platyspondyly and overfaced pedicles. {ECO:0000269|PubMed:19232556, ECO:0000269|PubMed:20577006, ECO:0000269|PubMed:22702953}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Metatropic dysplasia (MTD) [MIM:156530]: A severe spondyloepimetaphyseal dysplasia characterized by short limbs with limitation and enlargement of joints and usually severe kyphoscoliosis. Radiologic features include severe platyspondyly, severe metaphyseal enlargement and shortening of long bones. {ECO:0000269|PubMed:19232556, ECO:0000269|PubMed:20425821, ECO:0000269|PubMed:20577006, ECO:0000269|PubMed:22702953, ECO:0000269|PubMed:26249260, ECO:0000269|Ref.6}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Neuronopathy, distal hereditary motor, autosomal dominant 8 (HMND8) [MIM:600175]: A form of distal hereditary motor neuronopathy, a heterogeneous group of neuromuscular disorders caused by selective degeneration of motor neurons in the anterior horn of the spinal cord, without sensory deficit in the posterior horn. The overall clinical picture consists of a classical distal muscular atrophy syndrome in the legs without clinical sensory loss. The disease starts with weakness and wasting of distal muscles of the anterior tibial and peroneal compartments of the legs. Later on, weakness and atrophy may expand to the proximal muscles of the lower limbs and/or to the distal upper limbs. {ECO:0000269|PubMed:20037588, ECO:0000269|PubMed:22526352, ECO:0000269|PubMed:22702953}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Charcot-Marie-Tooth disease, axonal, 2C (CMT2C) [MIM:606071]: An axonal form of Charcot-Marie-Tooth disease, a disorder of the peripheral nervous system, characterized by progressive weakness and atrophy, initially of the peroneal muscles and later of the distal muscles of the arms. Charcot-Marie-Tooth disease is classified in two main groups on the basis of electrophysiologic properties and histopathology: primary peripheral demyelinating neuropathies (designated CMT1 when they are dominantly inherited) and primary peripheral axonal neuropathies (CMT2). Neuropathies of the CMT2 group are characterized by signs of axonal degeneration in the absence of obvious myelin alterations, normal or slightly reduced nerve conduction velocities, and progressive distal muscle weakness and atrophy. {ECO:0000269|PubMed:20037586, ECO:0000269|PubMed:20037587, ECO:0000269|PubMed:20037588, ECO:0000269|PubMed:21115951, ECO:0000269|PubMed:21288981, ECO:0000269|PubMed:22702953, ECO:0000269|PubMed:25256292}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Scapuloperoneal spinal muscular atrophy (SPSMA) [MIM:181405]: A clinically variable neuromuscular disorder characterized by neurogenic scapuloperoneal amyotrophy, laryngeal palsy, congenital absence of muscles, progressive scapuloperoneal atrophy and progressive distal weakness and amyotrophy. {ECO:0000269|PubMed:20037587, ECO:0000269|PubMed:22702953}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Spondyloepiphyseal dysplasia Maroteaux type (SEDM) [MIM:184095]: A clinically variable spondyloepiphyseal dysplasia with manifestations limited to the musculoskeletal system. Clinical features include short stature, brachydactyly, platyspondyly, short and stubby hands and feet, epiphyseal hypoplasia of the large joints, and iliac hypoplasia. Intelligence is normal. {ECO:0000269|PubMed:20503319, ECO:0000269|PubMed:22702953}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Parastremmatic dwarfism (PSTD) [MIM:168400]: A bone dysplasia characterized by severe dwarfism, kyphoscoliosis, distortion and bowing of the extremities, and contractures of the large joints. Radiographically, the disease is characterized by a combination of decreased bone density, bowing of the long bones, platyspondyly and striking irregularities of endochondral ossification with areas of calcific stippling and streaking in radiolucent epiphyses, metaphyses and apophyses. {ECO:0000269|PubMed:20503319}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Digital arthropathy-brachydactyly, familial (FDAB) [MIM:606835]: A disorder characterized by irregularities in the proximal articular surfaces of the distal interphalangeal joints of the hand. Individuals appear normal at birth, with no clinical or radiographic evidence of a developmental skeletal dysplasia. The earliest changes appear during the first decade of life. By adulthood, all interphalangeal, metacarpophalangeal, and metatarsophalangeal joints are affected by a deforming, painful osteoarthritis. The remainder of the skeleton is clinically and radiographically unaffected. {ECO:0000269|PubMed:21964574}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Avascular necrosis of the femoral head, primary 2 (ANFH2) [MIM:617383]: A disease characterized by mechanical failure of the subchondral bone, and degeneration of the hip joint. It usually leads to destruction of the hip joint in the third to fifth decade of life. The clinical manifestations, such as pain on exertion, a limping gait, and a discrepancy in leg length, cause considerable disability. {ECO:0000269|PubMed:27330106}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number EC 3.4.16.4 Uniprot keywords 3D-structure; Antibiotic resistance; Carboxypeptidase; Cell membrane; Cell shape; Cell wall biogenesis/degradation; Hydrolase; Membrane; Metal-binding; Peptidoglycan synthesis; Protease; Reference proteome; Transmembrane; Transmembrane helix; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 20547.5 Length 181 Aromaticity 0.11 Instability index 33.61 Isoelectric point 4.81 Charge (pH=7) -10.78 2D Binding mode Binding energy (Kcal/mol) -8 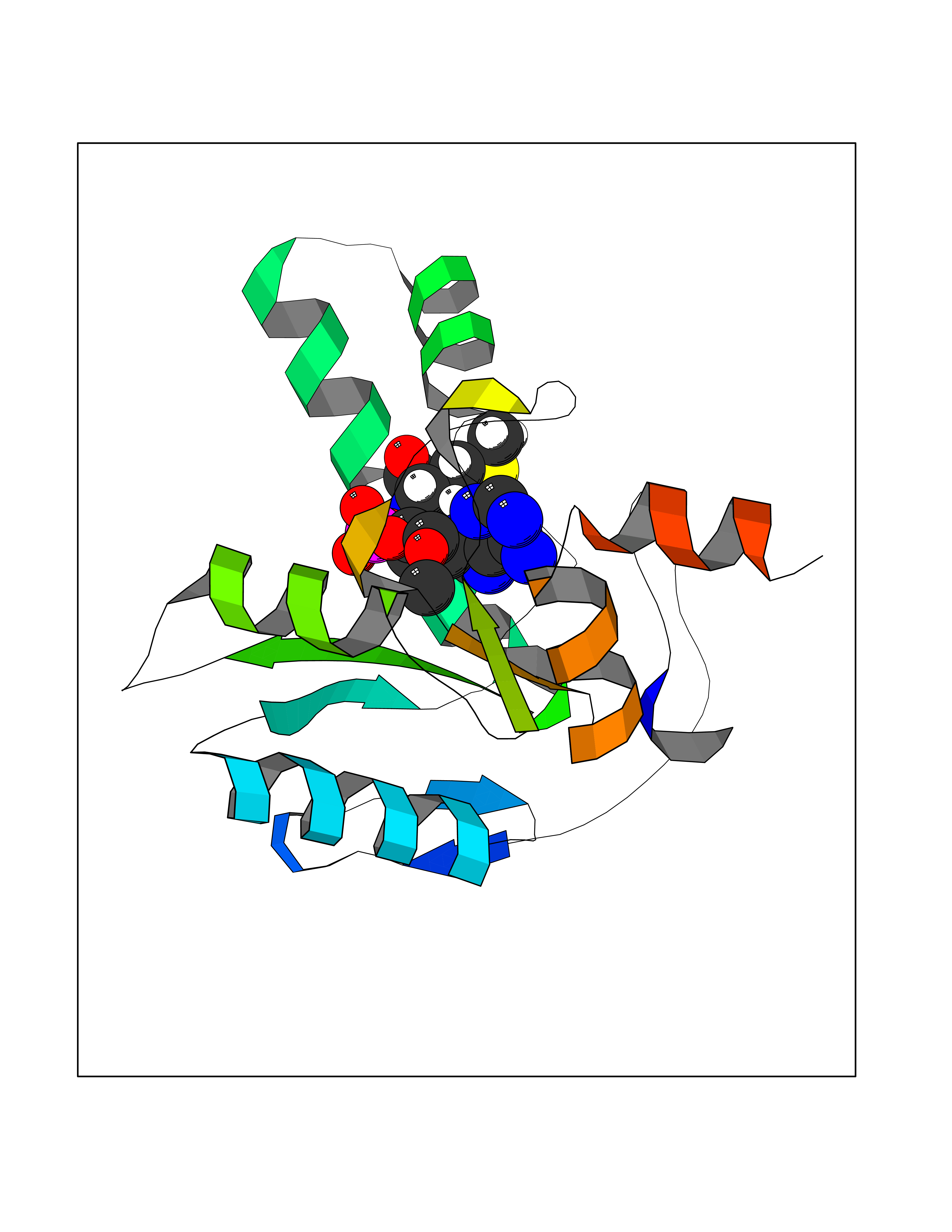 Molscript Map 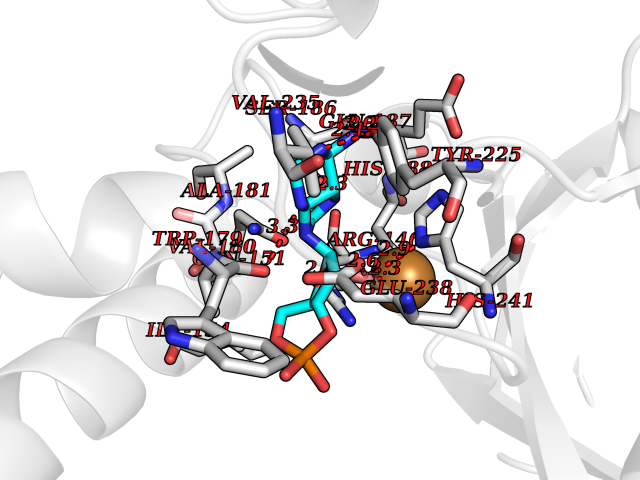 Pymol Map 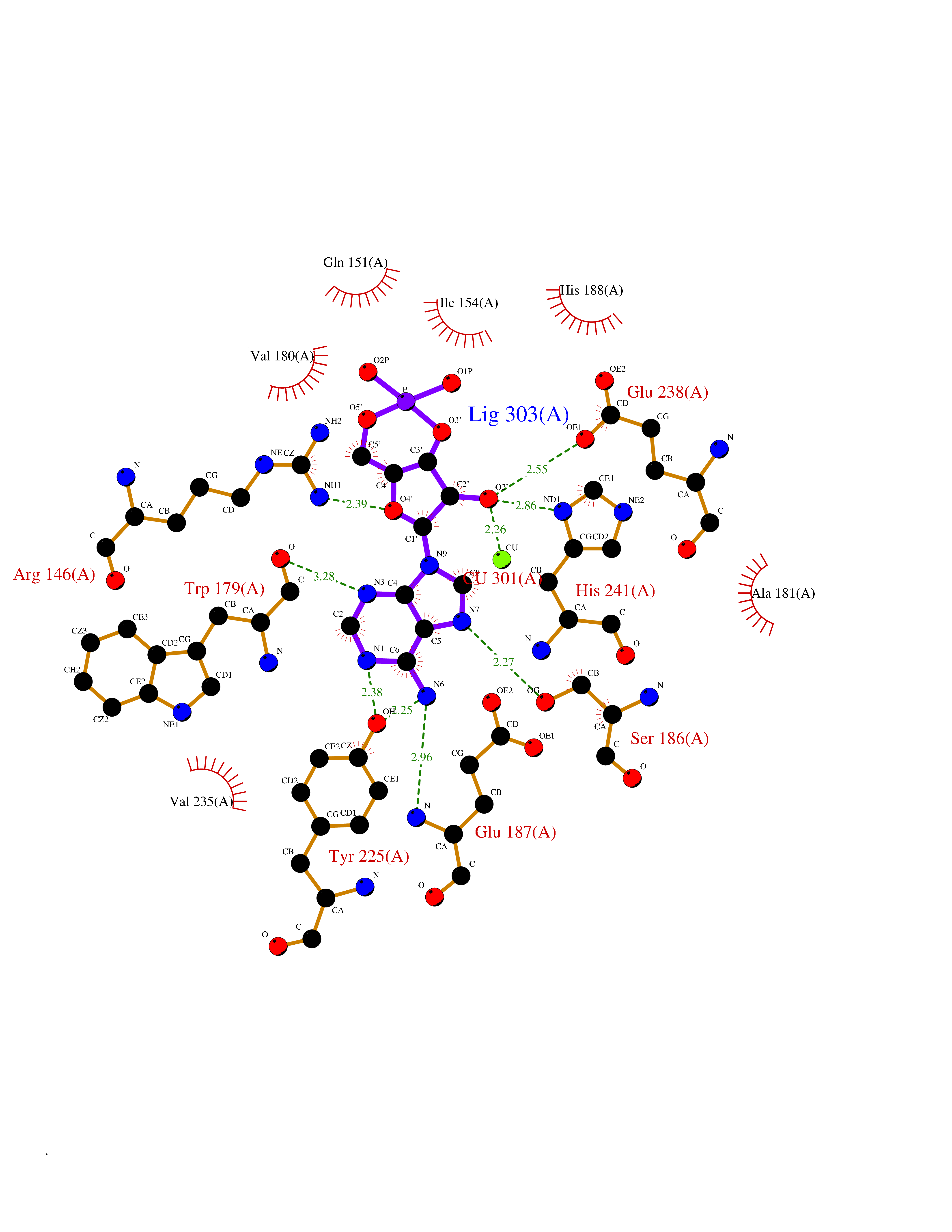 Ligplot Map 3D Binding mode Sequence EWSLILVNRQNPIPAQYDVELEQLSNGERIDIRISPYLQDLFDAARADGVYPIVASGYRTTEKQQEIXDEKVAEYKAKGYTSAQAKAEAETWVAVPGTSEHQLGLAVDINADGIHSTGNEVYRWLDENSYRFGFIRRYPPDKTEITGVSNEPWHYRYVGIEAATKIYHQGLCLEEYLNTEK Hydrogen bonds contact Hydrophobic contact | ||||
| 20 | Amylin receptor (IAPPR) | 6ZIS | 6.80 | |
Target general information Gen name CALCR-RAMP1/RAMP2/RAMP3 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Complex of Calcitonin receptor and Receptor activity-modifying protein Protein family RAMP family Biochemical class NA Function Transports the calcitonin gene-related peptide type 1 receptor (CALCRL) to the plasma membrane. Acts as a receptor for calcitonin-gene-related peptide (CGRP) together with CALCRL. Related diseases Immunodeficiency 9 (IMD9) [MIM:612782]: An immune disorder characterized by recurrent infections, impaired activation and proliferative response of T-cells, decreased T-cell production of cytokines, and normal lymphocytes counts and serum immunoglobulin levels. In surviving patients ectodermal dysplasia with anhidrosis and non-progressive myopathy may be observed. {ECO:0000269|PubMed:16147976, ECO:0000269|PubMed:16582901}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Myopathy, tubular aggregate, 2 (TAM2) [MIM:615883]: A rare congenital myopathy characterized by regular arrays of membrane tubules on muscle biopsies without additional histopathological hallmarks. Tubular aggregates in muscle are structures of variable appearance consisting of an outer tubule containing either one or more microtubule-like structures or amorphous material. TAM2 patients have myopathy and pupillary abnormalities. {ECO:0000269|PubMed:24591628, ECO:0000269|PubMed:28058752}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01278 Interacts with Q16602; P21145; Q5J8X5; Q16617 EC number NA Uniprot keywords 3D-structure; Cell membrane; Disulfide bond; Membrane; Proteomics identification; Receptor; Reference proteome; Signal; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A Molecular weight (Da) 63832.6 Length 569 Aromaticity 0.12 Instability index 22.42 Isoelectric point 5.07 Charge (pH=7) -16.96 2D Binding mode Binding energy (Kcal/mol) -7.07  Molscript Map 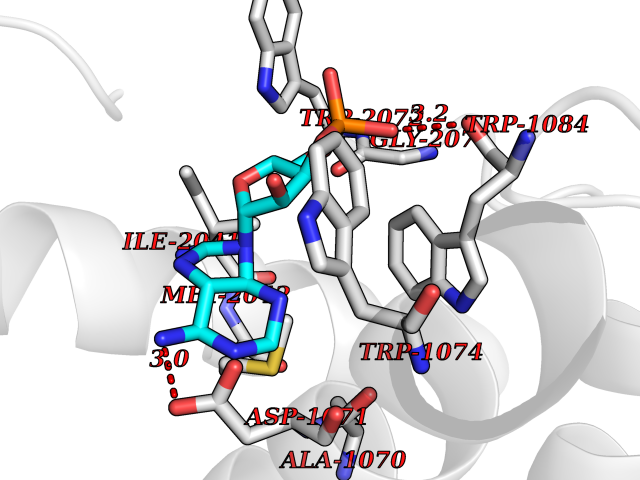 Pymol Map  Ligplot Map 3D Binding mode Sequence SAKIEEGKLVIWINGDKGYNGLAEVGKKFEKDTGIKVTVEHPDKLEEKFPQVAATGDGPDIIFWAHDRFGGYAQSGLLAEITPDKAFQDKLYPFTWDAVRYNGKLIAYPIAVEALSLIYNKDLLPNPPKTWEEIPALDKELKAKGKSALMFNLQEPYFTWPLIAADGGYAFKYENGKYDIKDVGVDNAGAKAGLTFLVDLIKNKHMNADTDYSIAEAAFNKGETAMTINGPWAWSNIDTSKVNYGVTVLPTFKGQPSKPFVGVLSAGINAASPNKELAKEFLENYLLTDEGLEAVNKDKPLGAVALKSYEEELAKDPRIAATMENAQKGEIMPNIPQMSAFWYAVRTAVINAASGRQTVDEALKDAQTNAAAEFTTACQEANYGALLRELCLTQFQVDMEAVGETLWCDWGRTIRSYRELADCTWHMAEKLGCFWPNAEVDRFFLAVHGRYFRSCPISIQLGVTRNKIMTAQYECYQKIMQDPIQQGVYCQRTWDGWLCWNDVAAGTESMQLCPDYFQDFDPSEKVTKICDQDGNWFRHPASQRTWTDYTQCNVNTHEKVKTALNLFYL Hydrogen bonds contact Hydrophobic contact | ||||