Job Results:
Ligand
Structure
Job ID
1ef4a574a09d4116e223f8984a7fcf68
Job name
yw123
Time
2025-03-21 15:49:07
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 1 | Macrophage migration inhibitory factor (MIF) | 3IJJ | 6.44 | |
Target general information Gen name MIF Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Phenylpyruvate tautomerase; MMIF; L-dopachrome tautomerase; L-dopachrome isomerase; Glycosylation-inhibiting factor; GLIF; GIF Protein family MIF family Biochemical class Intramolecular oxidoreductase Function Involved in the innate immune response to bacterial pathogens. The expression of MIF at sites of inflammation suggests a role as mediator in regulating the function of macrophages in host defense. Counteracts the anti-inflammatory activity of glucocorticoids. Has phenylpyruvate tautomerase and dopachrome tautomerase activity (in vitro), but the physiological substrate is not known. It is not clear whether the tautomerase activity has any physiological relevance, and whether it is important for cytokine activity. Pro-inflammatory cytokine. Related diseases Rheumatoid arthritis systemic juvenile (RASJ) [MIM:604302]: An inflammatory articular disorder with systemic onset beginning before the age of 16. It represents a subgroup of juvenile arthritis associated with severe extraarticular features and occasionally fatal complications. During active phases of the disorder, patients display a typical daily spiking fever, an evanescent macular rash, lymphadenopathy, hepatosplenomegaly, serositis, myalgia and arthritis. {ECO:0000269|PubMed:11508429}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01880; DB07888; DB08334; DB08335; DB08333; DB07718; DB08765; DB02728 Interacts with O43521-2; P00533; Q92743; P14174; Q96HA8 EC number EC 5.3.2.1 Uniprot keywords 3D-structure; Acetylation; Cytokine; Cytoplasm; Direct protein sequencing; Immunity; Inflammatory response; Innate immunity; Isomerase; Proteomics identification; Reference proteome; Secreted Protein physicochemical properties Chain ID A,C Molecular weight (Da) 24671.9 Length 228 Aromaticity 0.09 Instability index 31.45 Isoelectric point 8.37 Charge (pH=7) 2.26 2D Binding mode Binding energy (Kcal/mol) -8.78 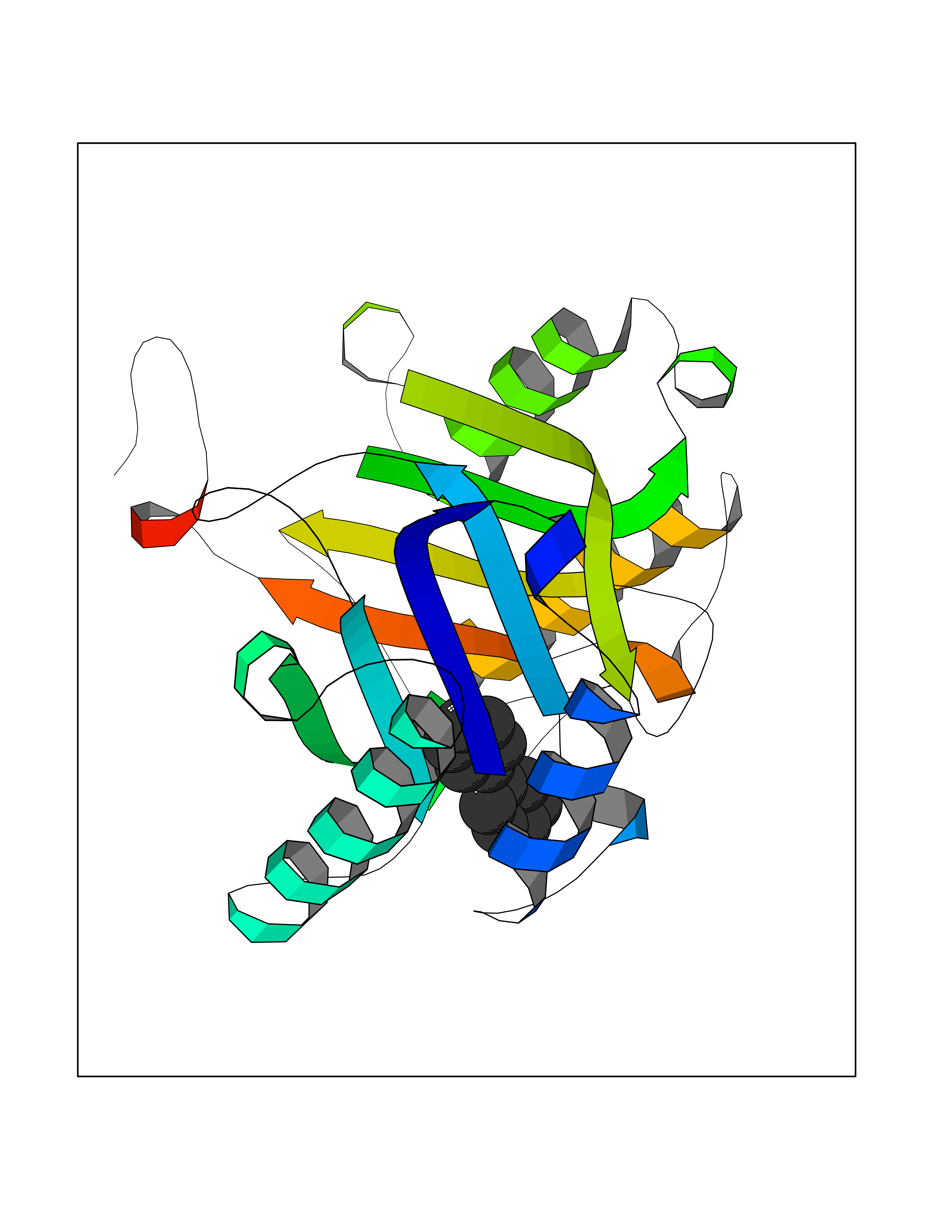 Molscript Map 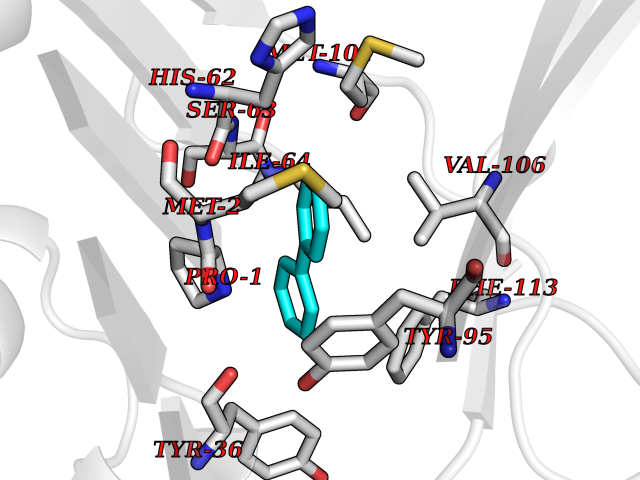 Pymol Map 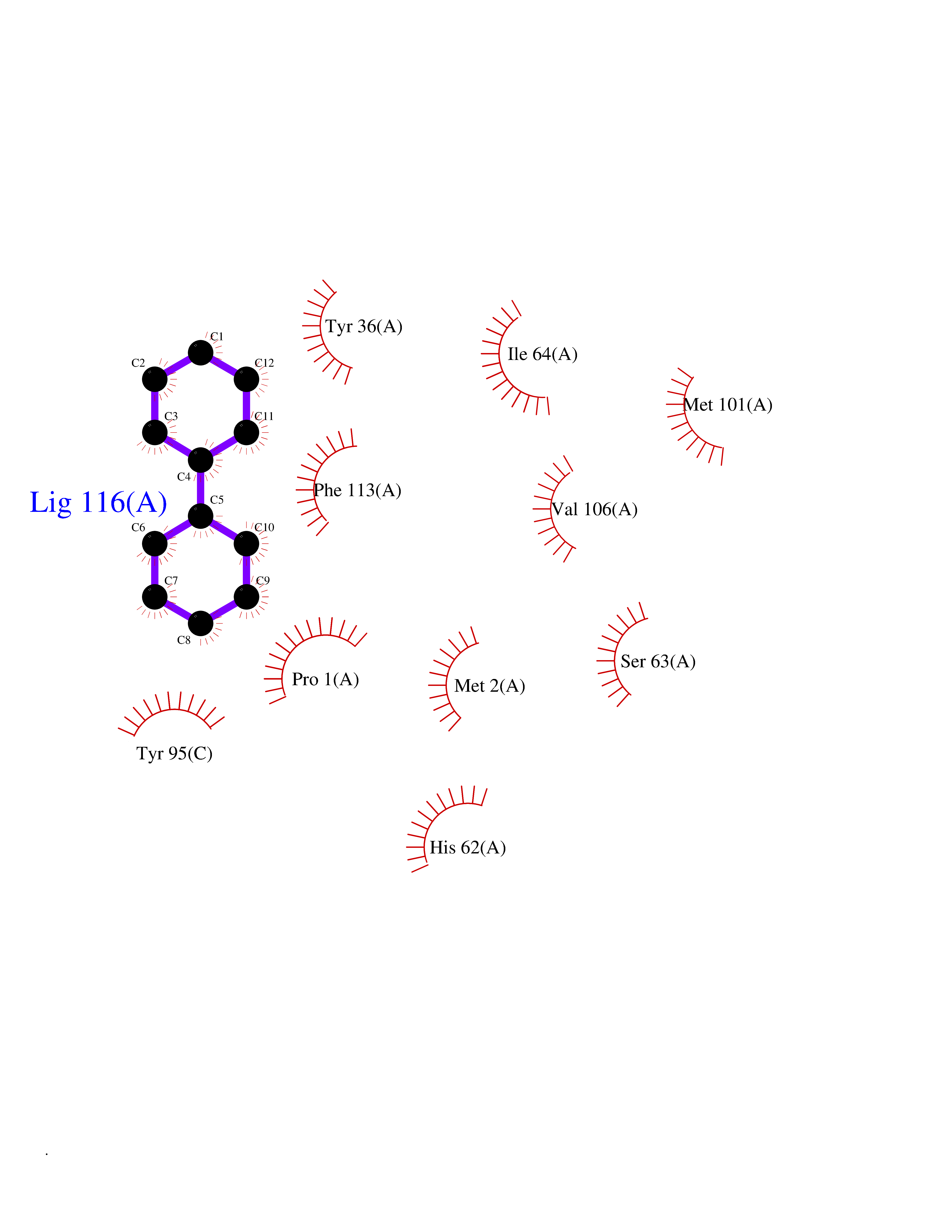 Ligplot Map 3D Binding mode Sequence PMFIVNTNVPRASVPDGFLSELTQQLAQATGKPPQYIAVHVVPDQLMAFGGSSEPCALCSLHSIGKIGGAQNRSYSKLLCGLLAERLRISPDRVYINYYDMNAANVGWNNSTFAPMFIVNTNVPRASVPDGFLSELTQQLAQATGKPPQYIAVHVVPDQLMAFGGSSEPCALCSLHSIGKIGGAQNRSYSKLLCGLLAERLRISPDRVYINYYDMNAANVGWNNSTFA Hydrogen bonds contact Hydrophobic contact | ||||
| 2 | Plasmepsin-2 | 2BJU | 6.43 | |
Target general information Gen name N/A Organism Plasmodium falciparum (isolate HB3) Uniprot ID TTD ID NA Synonyms NA Protein family Peptidase A1 family Biochemical class Hydrolase Function Aspartic-type endopeptidase activity. Related diseases Short/branched-chain acyl-CoA dehydrogenase deficiency (SBCADD) [MIM:610006]: Autosomal recessive disorder and consists of a defect in catabolism of L-isoleucine which is characterized by an increase of 2-methylbutyrylglycine and 2-methylbutyrylcarnitine in blood and urine. Affected individuals have seizures and psychomotor delay as the main clinical features. {ECO:0000269|PubMed:10832746, ECO:0000269|PubMed:11013134, ECO:0000269|PubMed:16317551}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB04378; DB04373; DB11638; DB01218; DB02505; DB03063 Interacts with NA EC number 3.4.23.39 Uniprot keywords 3D-structure; Aspartyl protease; Direct protein sequencing; Disulfide bond; Hydrolase; Membrane; Protease; Reference proteome; Signal-anchor; Transmembrane; Transmembrane helix; Vacuole; Zymogen Protein physicochemical properties Chain ID A Molecular weight (Da) 36923.5 Length 329 Aromaticity 0.13 Instability index 44.31 Isoelectric point 4.67 Charge (pH=7) -17.94 2D Binding mode Binding energy (Kcal/mol) -8.77 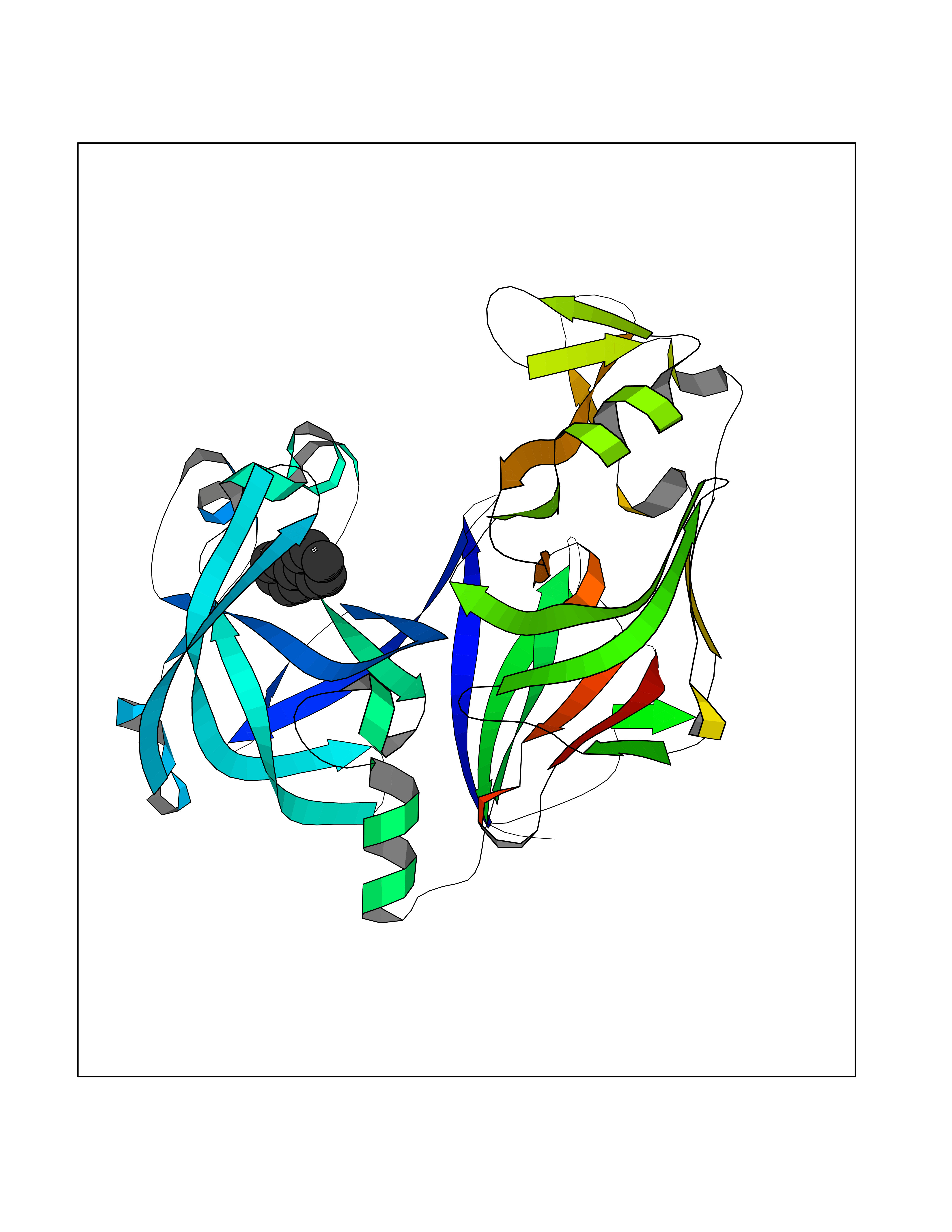 Molscript Map 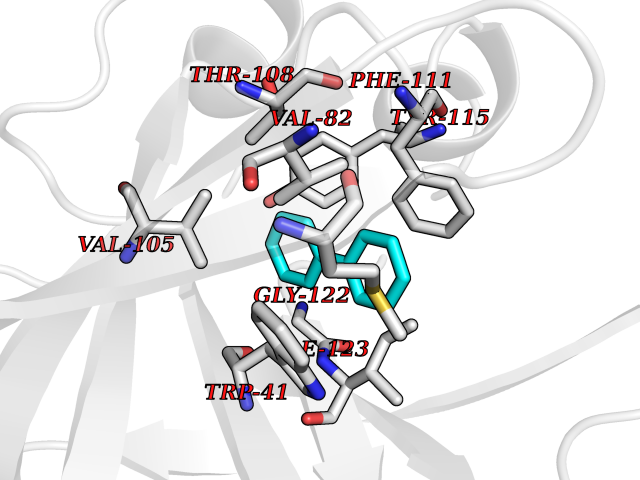 Pymol Map 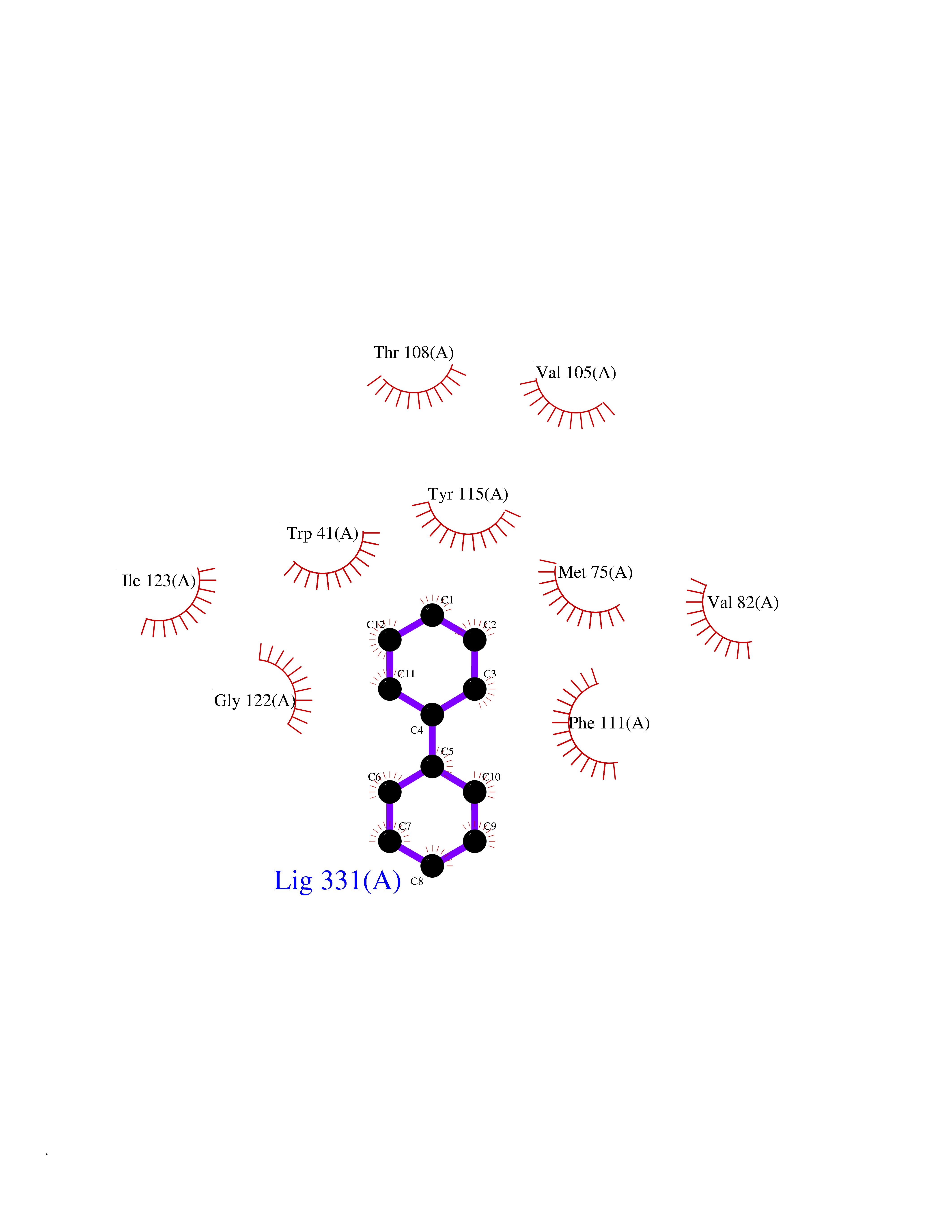 Ligplot Map 3D Binding mode Sequence SSNDNIELVDFQNIMFYGDAEVGDNQQPFTFILDTGSANLWVPSVKCTTAGCLTKHLYDSSKSRTYEKDGTKVEMNYVSGTVSGFFSKDLVTVGNLSLPYKFIEVIDTNGFEPTYTASTFDGILGLGWKDLSIGSVDPIVVELKNQNKIENALFTFYLPVHDKHTGFLTIGGIEERFYEGPLTYEKLNHDLYWQITLDAHVGNIMLEKANCIVDSGTSAITVPTDFLNKMLQNLDVIKVPFLPFYVTLCNNSKLPTFEFTSENGKYTLEPEYYLQHIEDVGPGLCMLNIIGLDFPVPTFILGDPFMRKYFTVFDYDNHSVGIALAKKNL Hydrogen bonds contact Hydrophobic contact | ||||
| 3 | Protein cereblon (CRBN) | 5FQD | 6.43 | |
Target general information Gen name CRBN Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Protein cereblon Protein family CRBN family Biochemical class NA Function Substrate recognition component of a DCX (DDB1-CUL4-X-box) E3 protein ligase complex that mediates the ubiquitination and subsequent proteasomal degradation of target proteins, such as MEIS2. Normal degradation of key regulatory proteins is required for normal limb outgrowth and expression of the fibroblast growth factor FGF8. May play a role in memory and learning by regulating the assembly and neuronal surface expression of large-conductance calcium-activated potassium channels in brain regions involved in memory and learning via its interaction with KCNT1. Binding of pomalidomide and other thalidomide-related drugs changes the substrate specificity of the human protein, leading to decreased degradation of MEIS2 and other target proteins and increased degradation of MYC, IRF4, IKZF1 and IKZF3. Related diseases Intellectual developmental disorder, autosomal recessive 2 (MRT2) [MIM:607417]: A disorder characterized by significantly below average general intellectual functioning associated with impairments in adaptive behavior and manifested during the developmental period. MRT2 patients display mild intellectual disability with a standard IQ ranged from 50 to 70. IQ scores are lower in males than females. Developmental milestones are mildly delayed. There are no dysmorphic or autistic features. {ECO:0000269|PubMed:15557513, ECO:0000269|PubMed:28143899}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00480; DB08910; DB01041 Interacts with Q96A83-2; P48729; Q16531; O14901; Q8IVT2; Q9P286; A0A6Q8PF08; Q93062; Q16531; Q13422-7 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; Disease variant; Intellectual disability; Membrane; Metal-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Ubl conjugation; Ubl conjugation pathway; Zinc Protein physicochemical properties Chain ID B,E Molecular weight (Da) 38245.7 Length 337 Aromaticity 0.08 Instability index 40.62 Isoelectric point 5.7 Charge (pH=7) -6.53 2D Binding mode Binding energy (Kcal/mol) -8.77 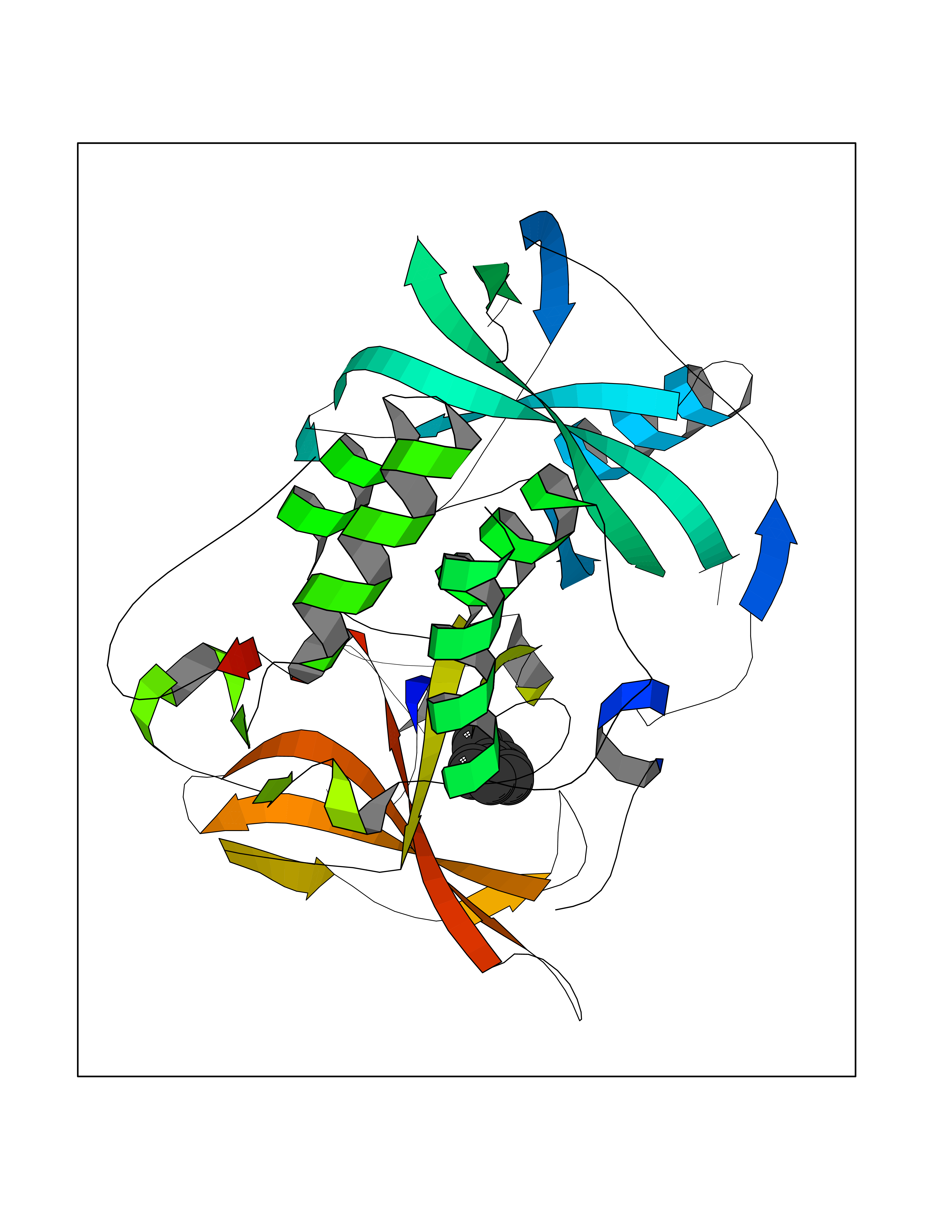 Molscript Map 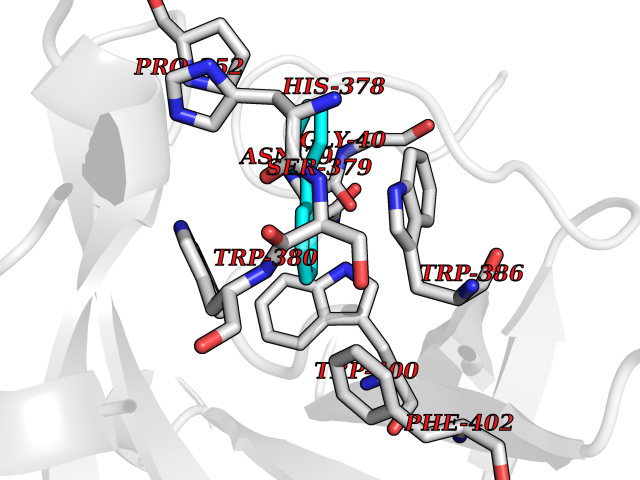 Pymol Map 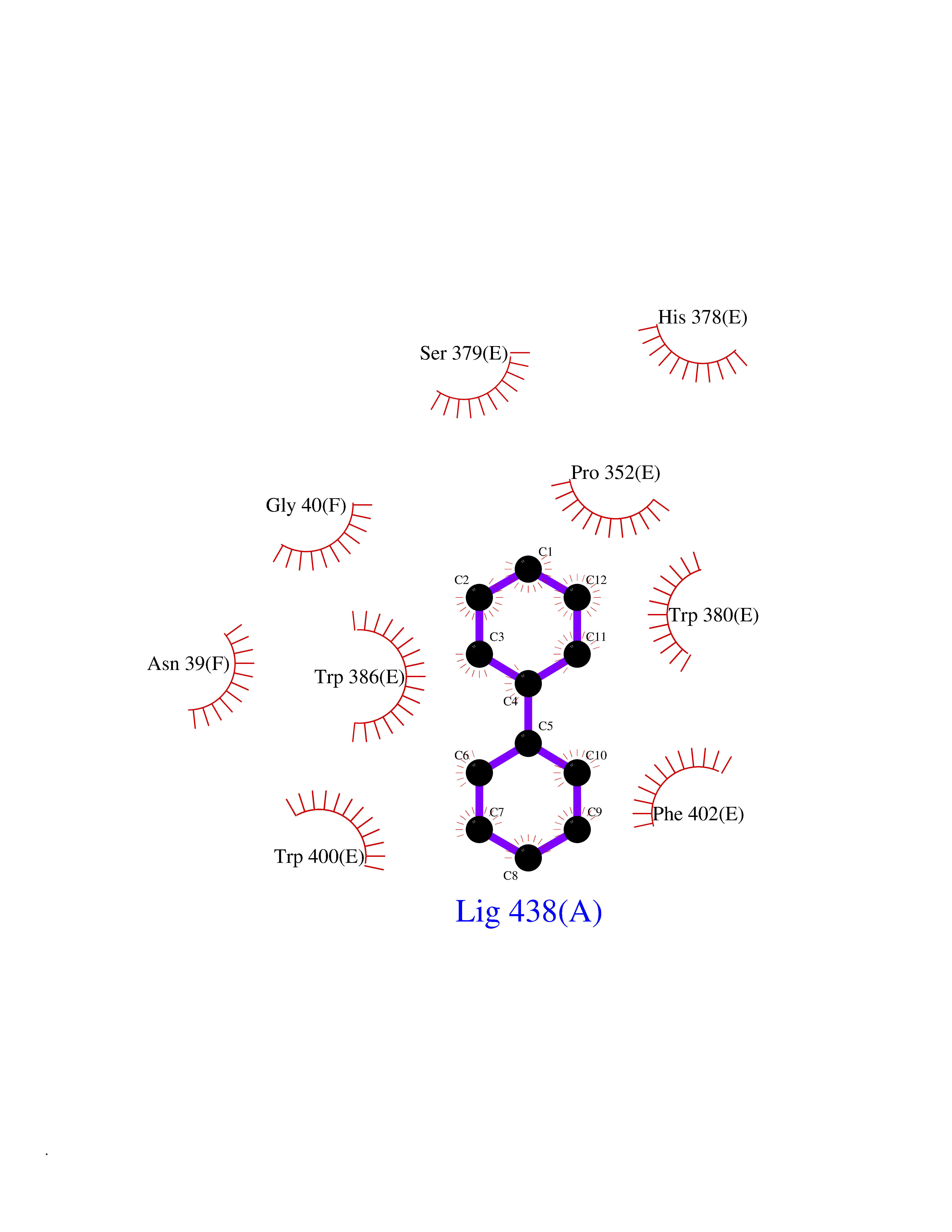 Ligplot Map 3D Binding mode Sequence EFIVGGKYKLNITNGEEVAVINFDTSLPTSHTYLGADMEEFHGRTLHDDDSCQVIPVLPQVMMILIPGQTLPLQLFHPQEVSMVRNLIQKDRTFAVLAYSNVQEREAQFGTTAEIYAYREEIVKVKAIGRQRFKVLEQQAKVQILPECVLAETLMDRIKKQLREWDENLKDDSLPSNPIDFSYRVAACLPIDDVLRIQLLKIGSAIQRLRCELDIMNKCTSLCCKQCQETEITTKNEIFSLSLCGPMAAYVNPHGYVHETLTVYKACNLNLIGRPSTEHSWFPGYAWTVAQCKICASHIGWKFTATKKDMSPQKFWGLTRSALLPTIPDTEDEISPD Hydrogen bonds contact Hydrophobic contact | ||||
| 4 | 4-hydroxyphenylpyruvate dioxygenase | 3ISQ | 6.42 | |
Target general information Gen name HPD Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms PPD Protein family 4HPPD family Biochemical class Oxidoreductase Function 4-hydroxyphenylpyruvate dioxygenase activity.Metal ion binding. Related diseases Tyrosinemia 3 (TYRSN3) [MIM:276710]: An inborn error of metabolism characterized by elevations of tyrosine in the blood and urine, seizures and mild intellectual disability. {ECO:0000269|PubMed:10942115, ECO:0000269|PubMed:11073718}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Hawkinsinuria (HWKS) [MIM:140350]: An inborn error of tyrosine metabolism characterized by failure to thrive, persistent metabolic acidosis, fine and sparse hair, and excretion of the unusual cyclic amino acid metabolite, hawkinsin, in the urine. {ECO:0000269|PubMed:11073718}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02850; DB00348 Interacts with NA EC number 1.13.11.27 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Cytoplasm; Dioxygenase; Disease variant; Endoplasmic reticulum; Golgi apparatus; Intellectual disability; Iron; Membrane; Metal-binding; Oxidoreductase; Phenylalanine catabolism; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Tyrosine catabolism Protein physicochemical properties Chain ID A Molecular weight (Da) 43164.8 Length 376 Aromaticity 0.11 Instability index 32.38 Isoelectric point 6.73 Charge (pH=7) -1.04 2D Binding mode Binding energy (Kcal/mol) -8.76 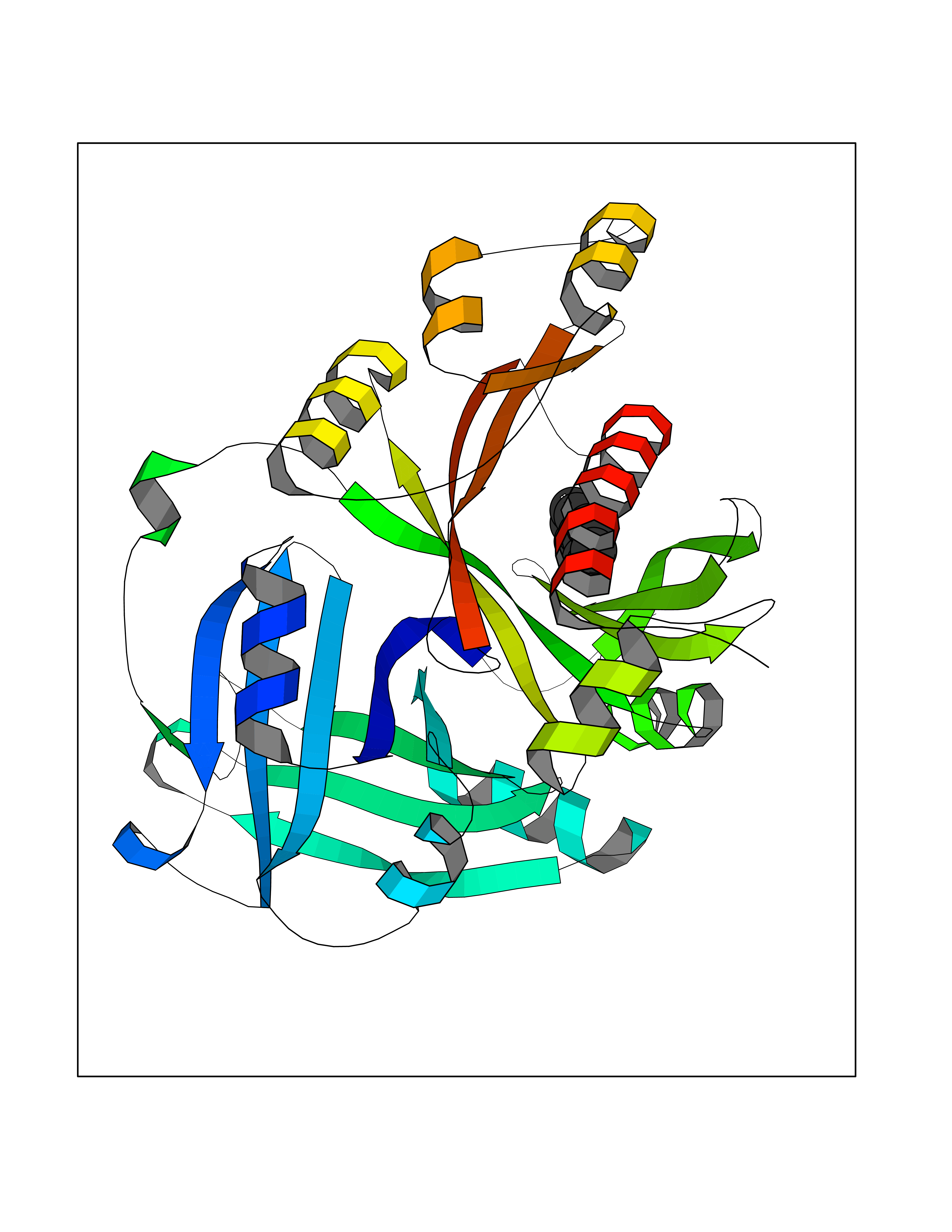 Molscript Map 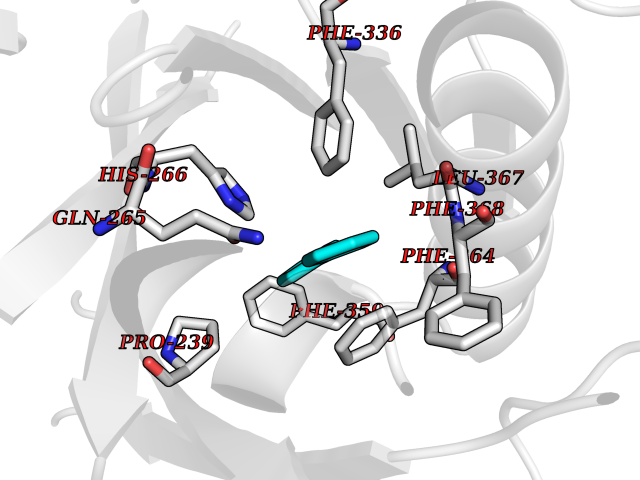 Pymol Map 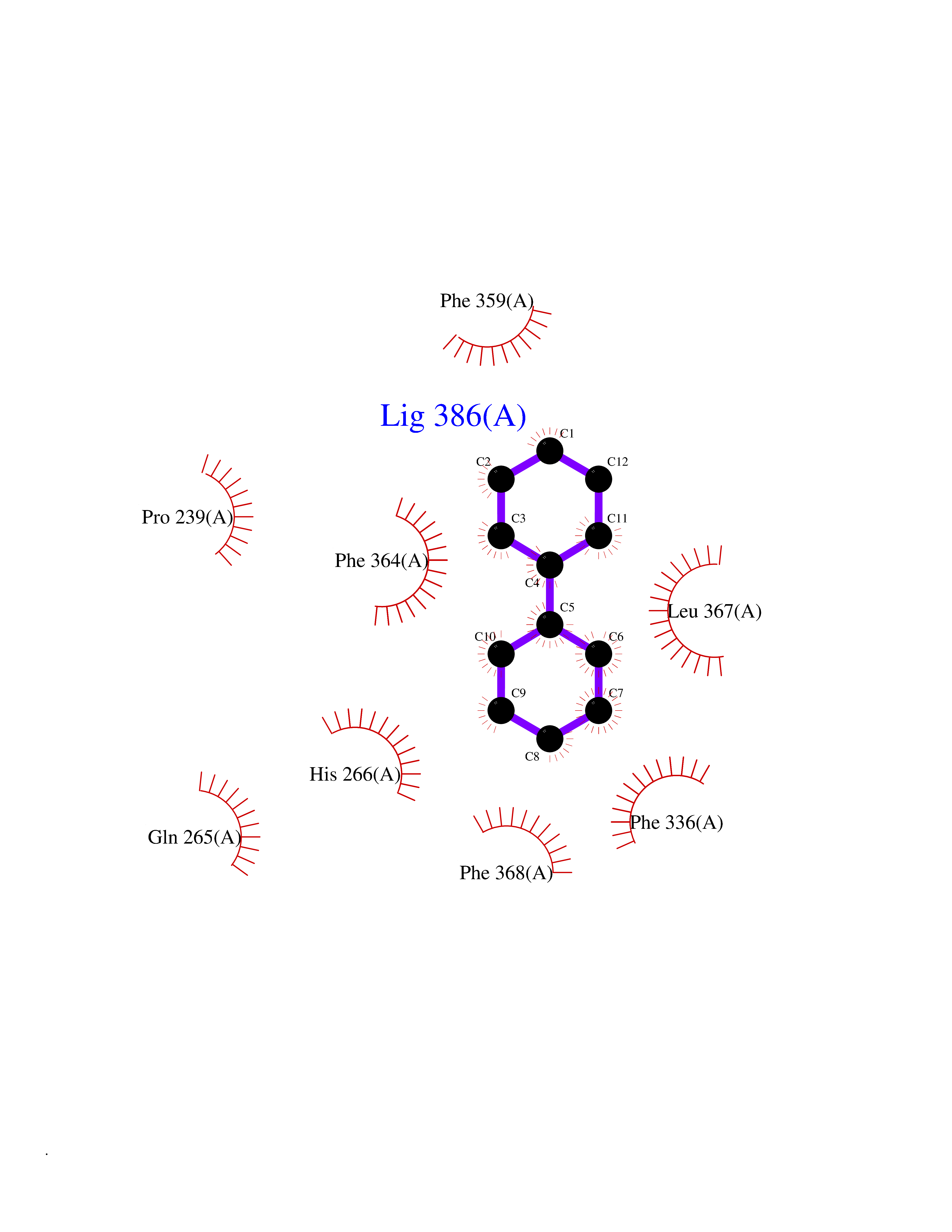 Ligplot Map 3D Binding mode Sequence AKPERGRFLHFHSVTFWVGNAKQAASFYCSKMGFEPLAYRGLETGSREVVSHVIKQGKIVFVLSSALNPWNKEMGDHLVKHGDGVKDIAFEVEDCDYIVQKARERGAKIMREPWVEQDKFGKVKFAVLQTYGDTTHTLVEKMNYIGQFLPGYEAPAFMDPLLPKLPKCSLEMIDHIVGNQPDQEMVSASEWYLKNLQFHRFWSVDDTQVHTEYSSLRSIVVANYEESIKMPINEPAPGKKKSQIQEYVDYNGGAGVQHIALKTEDIITAIRHLRERGLEFLSVPSTYYKQLREKLKTAKIKVKENIDALEELKILVDYDEKGYLLQIFTKPVQDRPTLFLEVIQRHNHQGFGAGNFNSLFKAFEEEQNLRGNLTNM Hydrogen bonds contact Hydrophobic contact | ||||
| 5 | Cocaine esterase | 3I2K | 6.42 | |
Target general information Gen name cocE Organism Rhodococcus sp. (strain MB1 Bresler) Uniprot ID TTD ID NA Synonyms NA Protein family CocE/NonD hydrolase family Biochemical class Hydrolase Function Carboxylic ester hydrolase activity.Dipeptidyl-peptidase activity. Related diseases Thiamine metabolism dysfunction syndrome 5, episodic encephalopathy type (THMD5) [MIM:614458]: An autosomal recessive metabolic disorder due to an inborn error of thiamine metabolism. The phenotype is highly variable, but in general, affected individuals have onset in early childhood of acute encephalopathic episodes associated with increased serum and CSF lactate. These episodes result in progressive neurologic dysfunction manifest as gait disturbances, ataxia, dystonia, and spasticity, which in some cases may result in loss of ability to walk. Cognitive function is usually preserved, although mildly delayed development has been reported. These episodes are usually associated with infection and metabolic decompensation. Some patients may have recovery of some neurologic deficits. {ECO:0000269|PubMed:22152682}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03793; DB01795 Interacts with NA EC number 3.1.1.84 Uniprot keywords 3D-structure; Cytoplasm; Direct protein sequencing; Hydrolase; Serine esterase Protein physicochemical properties Chain ID A Molecular weight (Da) 62127.9 Length 574 Aromaticity 0.09 Instability index 26.62 Isoelectric point 4.56 Charge (pH=7) -33.24 2D Binding mode Binding energy (Kcal/mol) -8.76 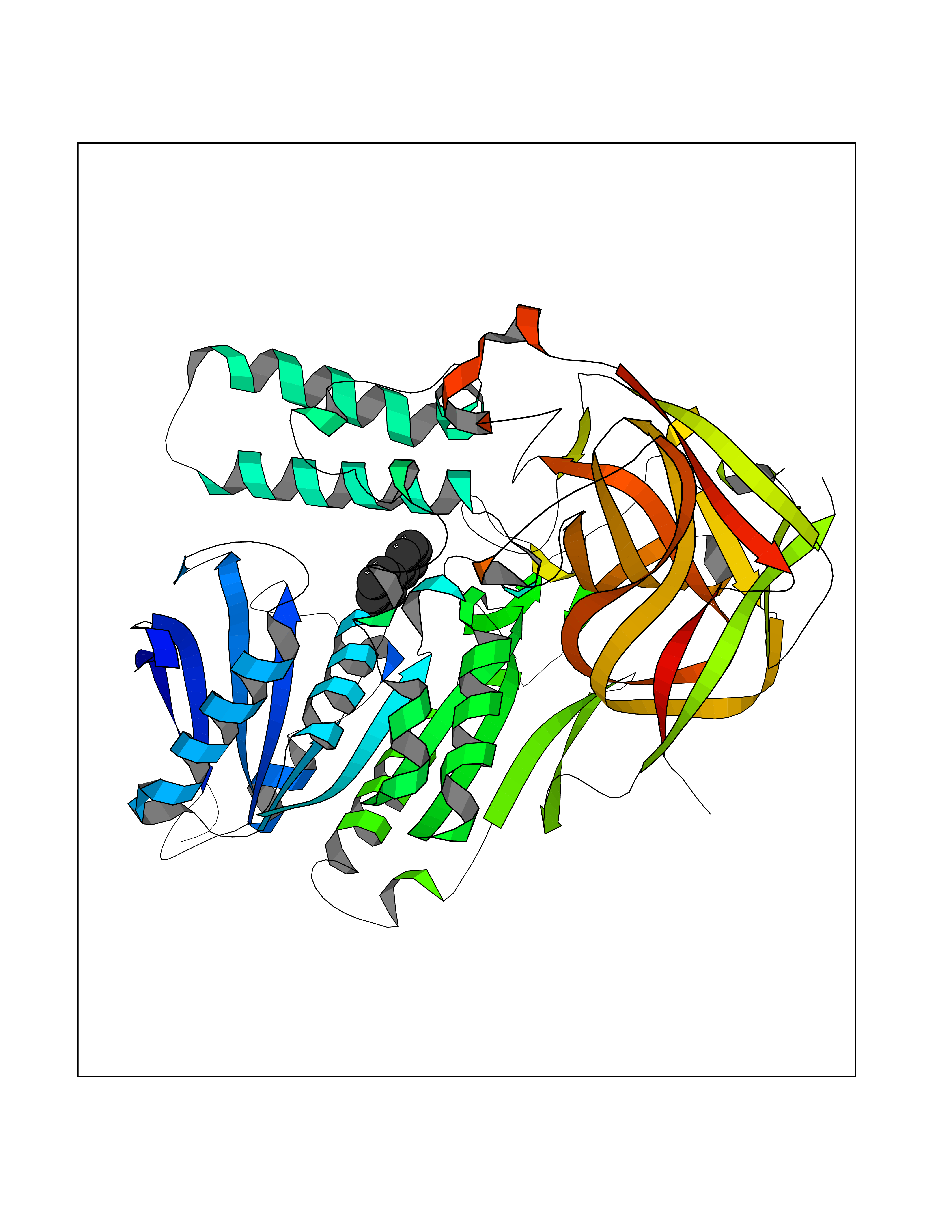 Molscript Map 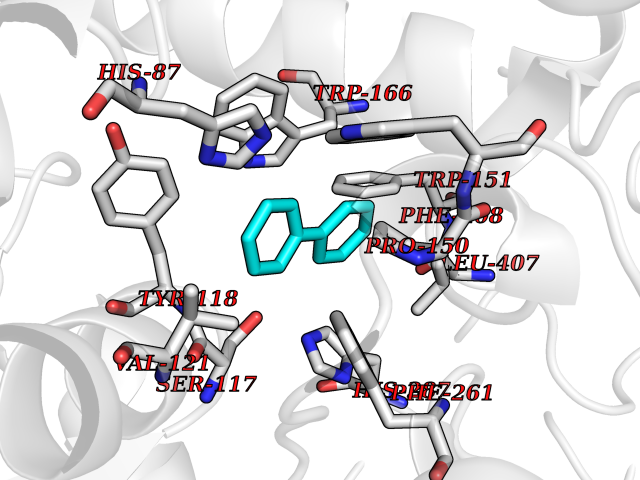 Pymol Map 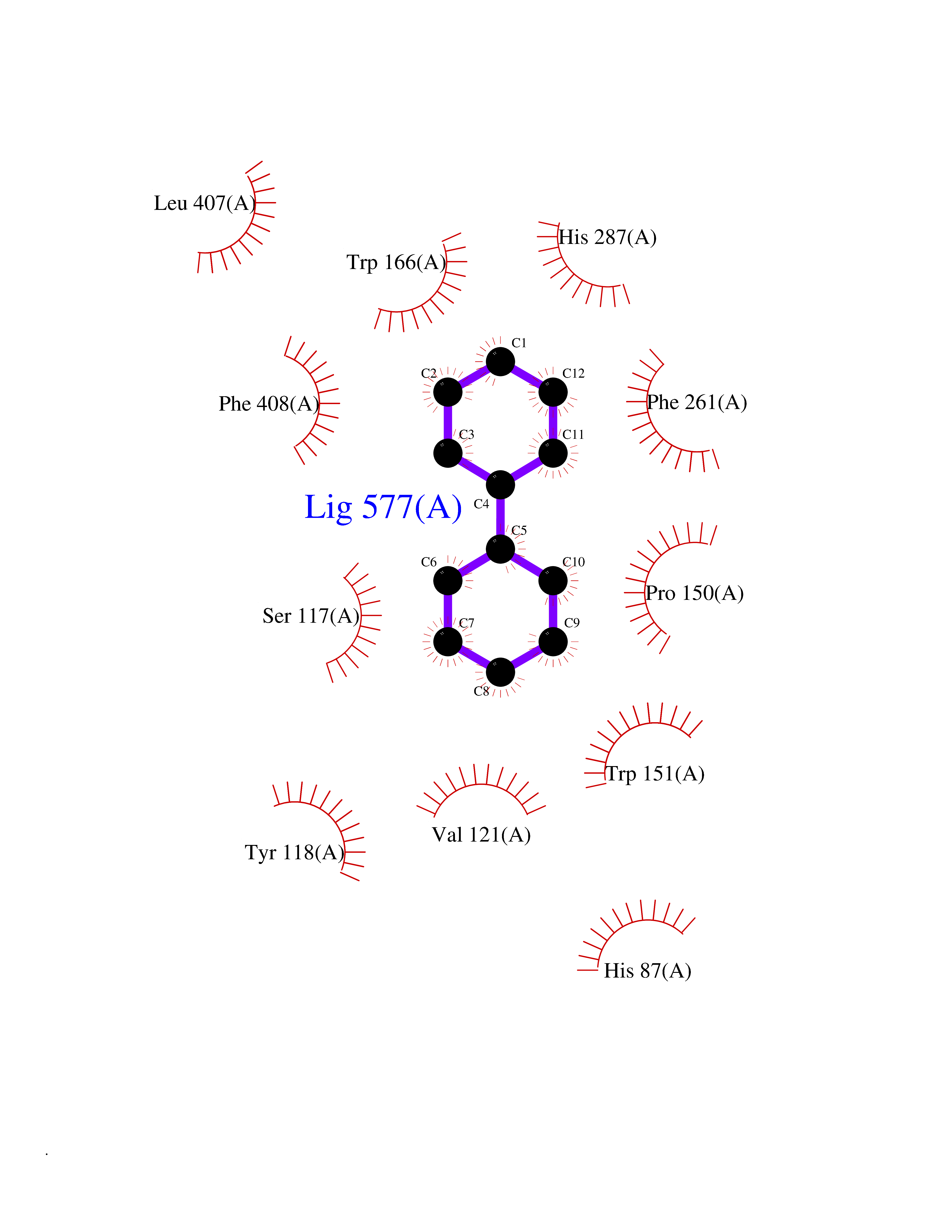 Ligplot Map 3D Binding mode Sequence VDGNYSVASNVMVPMRDGVRLAVDLYRPDADGPVPVLLVRNPYDKFDVFAWSTQSTNWLEFVRDGYAVVIQDTRGLFASEGEFVPHVDDEADAEDTLSWILEQAWCDGNVGMFGVSYLGVTQWQAAVSGVGGLKAIAPSMASADLYRAPWYGPGGALSVEALLGWSALIGTGLITSRSDARPEDAADFVQLAAILNDVAGAASVTPLAEQPLLGRLIPWVIDQVVDHPDNDESWQSISLFERLGGLATPALITAGWYDGFVGESLRTFVAVKDNADARLVVGPWSHSNLTGRNADRKFGIAATYPIQEATTMHKAFFDRHLRGETDALAGVPKVRLFVMGIDEWRDETDWPLPDTAYTPFYLGGSGAANTSTGGGTLSTSISGTESADTYLYDPADPVPSLGGTLLFHNGDNGPADQRPIHDRDDVLCYSTEVLTDPVEVTGTVSARLFVSSSAVDTDFTAKLVDVFPDGRAIALCDGIVRMRYRETLVNPTLIEAGEIYEVAIDMLATSNVFLPGHRIMVQVSSSNFPKYDRNSNTGGVIAREQLEEMCTAVNRIHRGPEHPSHIVLPIIKRK Hydrogen bonds contact Hydrophobic contact | ||||
| 6 | Zinc finger protein Helios (IKZF2) | 7LPS | 6.42 | |
Target general information Gen name IKZF2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Ikaros family zinc finger protein 2 Protein family Ikaros C2H2-type zinc-finger protein family Biochemical class NA Function Associates with Ikaros at centromeric heterochromatin. Related diseases Developmental and epileptic encephalopathy 25, with amelogenesis imperfecta (DEE25) [MIM:615905]: An autosomal recessive disease characterized by subclinical seizures appearing in the first days of life, evolving to severe epileptic disease. Affected individuals have profound or severe delayed development with lack of speech, and most patients do not acquire the ability to sit. Additional variable features include axial hypotonia, peripheral hypertonia, and abnormal involuntary movements such as dystonia and choreoathetosis. Dental abnormalities, including delayed eruption, hypodontia, tooth hypoplasia, yellow discoloration, thin enamel, and enamel chipping are observed in most patients. {ECO:0000269|PubMed:24995870, ECO:0000269|PubMed:26384929, ECO:0000269|PubMed:30054523}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P29972; P56545; P56545-3; Q17RB8; P09022; Q8N8B7-2 EC number NA Uniprot keywords 3D-structure; Acetylation; Activator; Alternative splicing; DNA-binding; Isopeptide bond; Metal-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Transcription; Transcription regulation; Ubl conjugation; Zinc; Zinc-finger Protein physicochemical properties Chain ID B,C Molecular weight (Da) 47006.6 Length 410 Aromaticity 0.09 Instability index 44.28 Isoelectric point 7.23 Charge (pH=7) 0.69 2D Binding mode Binding energy (Kcal/mol) -8.76 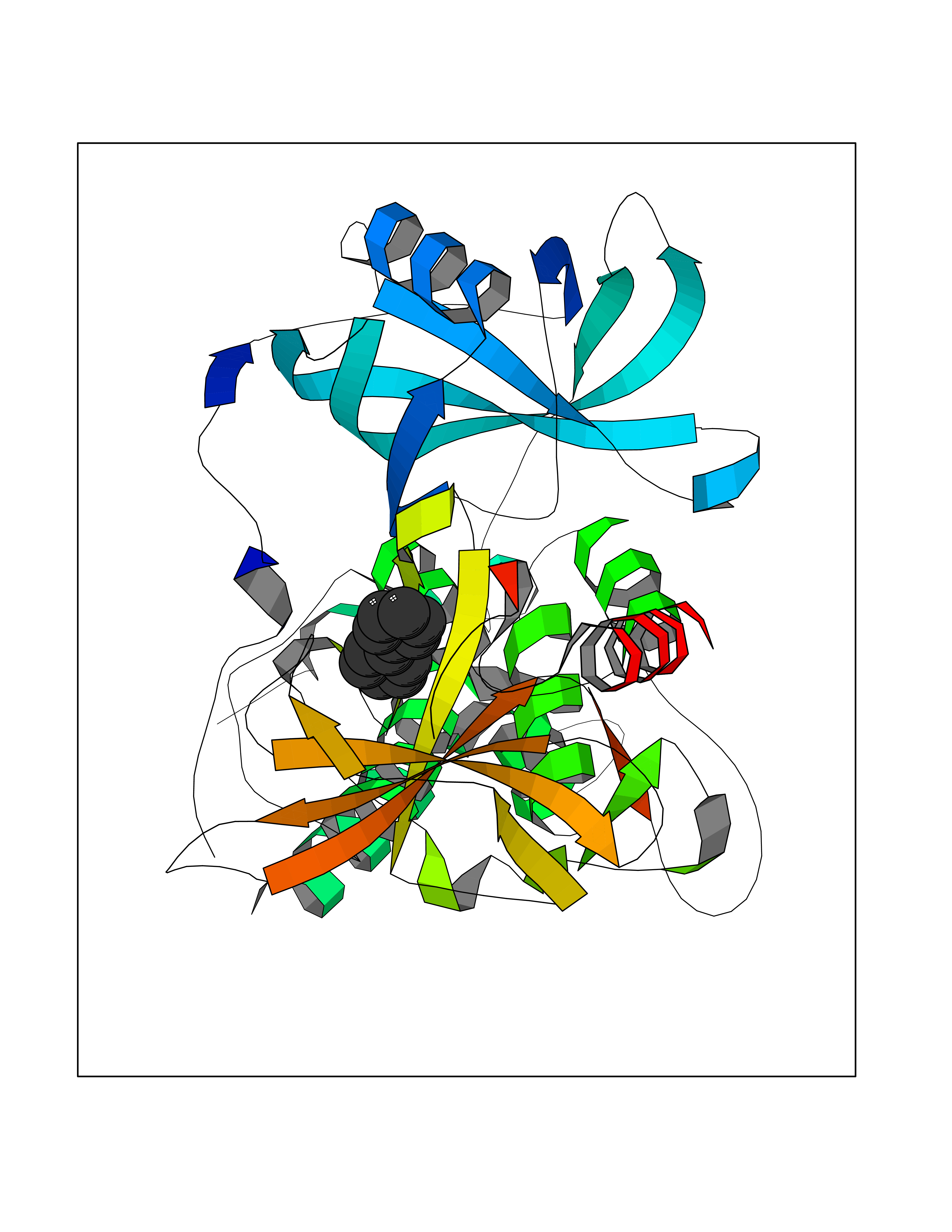 Molscript Map 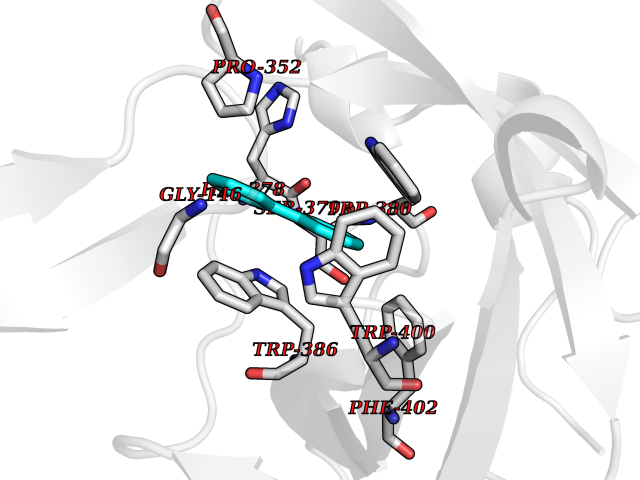 Pymol Map 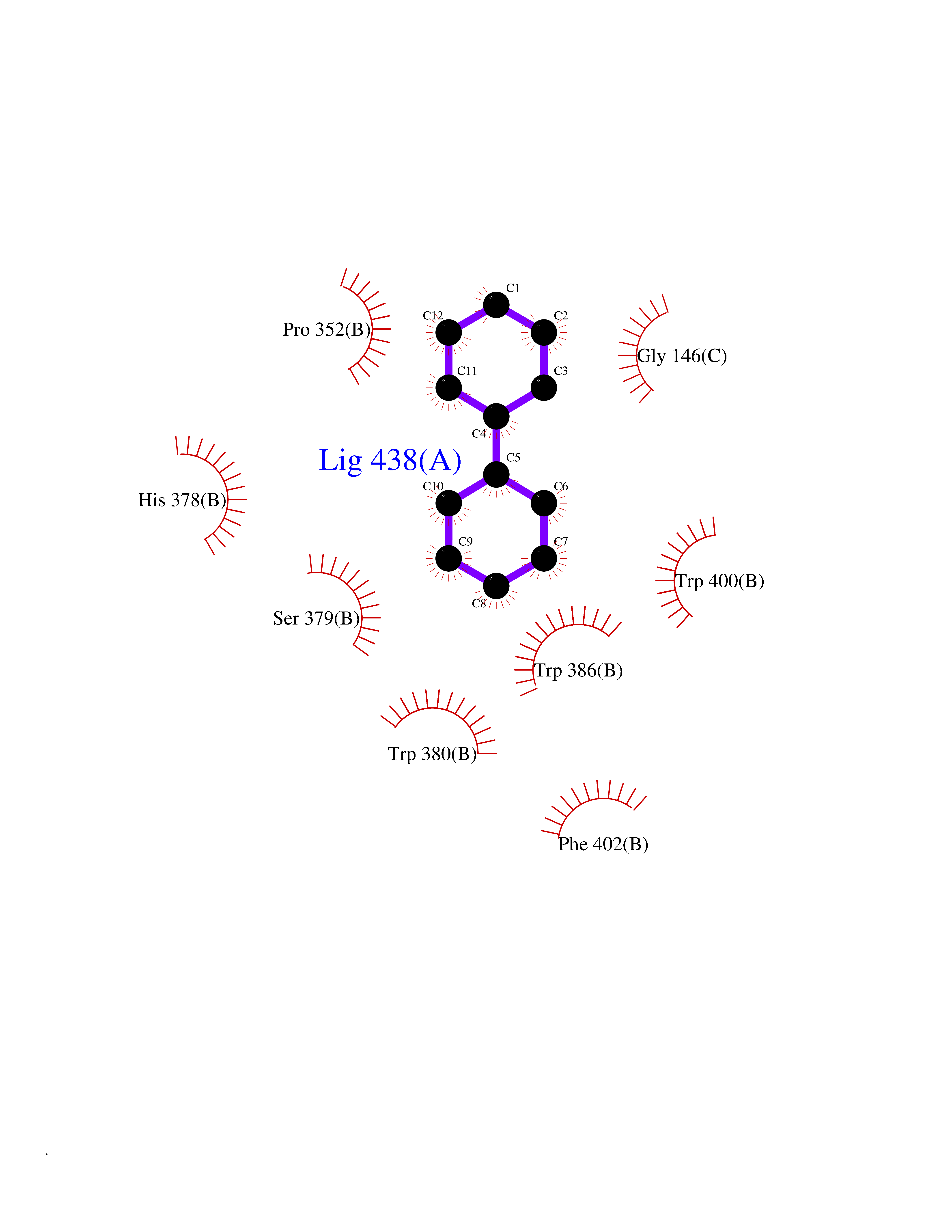 Ligplot Map 3D Binding mode Sequence INFDTSLPTSHTYLGADMEEFHGRTLHDDDSCQVIPVLPQVMMILIPGQTLPLQLFHPQEVSMVRNLIQKDRTFAVLAYSNVQEREAQFGTTAEIYAYREEQDFGIEIVKVKAIGRQRFKVLELRTQSDGIQQAKVQILPECVLPSTMSAVQLESLNKCQIFPCSYKWWQKYQKRKFHCANLTSWPRWLYSLYDAETLMDRIKKQLREWDENLKDDSLPSNPIDFSYRVAACLPIDDVLRIQLLKIGSAIQRLRCELDIMNKCTSLCCKQCQETEITTKNEIFSLSLCGPMAAYVNPHGYVHETLTVYKACNLNLIGRPSTEHSWFPGYAWTVAQCKICASHIGWKFTATKKDMSPQKFWGLTRSALLPTIPDTEDEISPDGERPFHCNQCGASFTQKGNLLRHIKLHSG Hydrogen bonds contact Hydrophobic contact | ||||
| 7 | Acetylcholine receptor subunit alpha | 4ZJS | 6.40 | |
Target general information Gen name CHRNA1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms ACHRA;CHNRA Protein family Ligand-gated ion channel (TC 1.A.9) family, Acetylcholine receptor (TC 1.A.9.1) subfamily, Alpha-1/CHRNA1 sub-subfamily Biochemical class Immune system Function Acetylcholine binding.Acetylcholine-gated cation-selective channel activity.Acetylcholine receptor activity.Ion channel activity.Ligand-gated ion channel activity. Related diseases Multiple pterygium syndrome, lethal type (LMPS) [MIM:253290]: Multiple pterygia are found infrequently in children with arthrogryposis and in fetuses with fetal akinesia syndrome. In lethal multiple pterygium syndrome there is intrauterine growth retardation, multiple pterygia, and flexion contractures causing severe arthrogryposis and fetal akinesia. Subcutaneous edema can be severe, causing fetal hydrops with cystic hygroma and lung hypoplasia. Oligohydramnios and facial anomalies are frequent. {ECO:0000269|PubMed:18252226}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: The alpha subunit is the main focus for antibody binding in myasthenia gravis. Myasthenia gravis is characterized by sporadic muscular fatigability and weakness, occurring chiefly in muscles innervated by cranial nerves, and characteristically improved by cholinesterase-inhibiting drugs.; DISEASE: Myasthenic syndrome, congenital, 1A, slow-channel (CMS1A) [MIM:601462]: A common congenital myasthenic syndrome. Congenital myasthenic syndromes are characterized by muscle weakness affecting the axial and limb muscles (with hypotonia in early-onset forms), the ocular muscles (leading to ptosis and ophthalmoplegia), and the facial and bulbar musculature (affecting sucking and swallowing, and leading to dysphonia). The symptoms fluctuate and worsen with physical effort. CMS1A is a slow-channel myasthenic syndrome. It is caused by kinetic abnormalities of the AChR, resulting in prolonged AChR channel opening episodes, prolonged endplate currents, and depolarization block. This is associated with calcium overload, which may contribute to subsequent degeneration of the endplate and postsynaptic membrane. {ECO:0000269|PubMed:16685696, ECO:0000269|PubMed:7619526, ECO:0000269|PubMed:8872460, ECO:0000269|PubMed:9158151, ECO:0000269|PubMed:9221765}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Myasthenic syndrome, congenital, 1B, fast-channel (CMS1B) [MIM:608930]: A form of congenital myasthenic syndrome, a group of disorders characterized by failure of neuromuscular transmission, including pre-synaptic, synaptic, and post-synaptic disorders that are not of autoimmune origin. Clinical features are easy fatigability and muscle weakness affecting the axial and limb muscles (with hypotonia in early-onset forms), the ocular muscles (leading to ptosis and ophthalmoplegia), and the facial and bulbar musculature (affecting sucking and swallowing, and leading to dysphonia). The symptoms fluctuate and worsen with physical effort. CMS1B is a fast-channel myasthenic syndrome. It is caused by kinetic abnormalities of the AChR, resulting in brief opening and activity of the channel, with a rapid decay in endplate current, failure to achieve threshold depolarization of the endplate and consequent failure to fire an action potential. {ECO:0000269|PubMed:10195214, ECO:0000269|PubMed:12588888, ECO:0000269|PubMed:15079006}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08838; DB00565; DB00555 Interacts with NA EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Congenital myasthenic syndrome; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; Ion channel; Ion transport; Ligand-gated ion channel; Membrane; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Signal; Synapse; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A,B,C,D,E Molecular weight (Da) 46717.8 Length 411 Aromaticity 0.11 Instability index 38.02 Isoelectric point 4.77 Charge (pH=7) -22.31 2D Binding mode Binding energy (Kcal/mol) -8.73 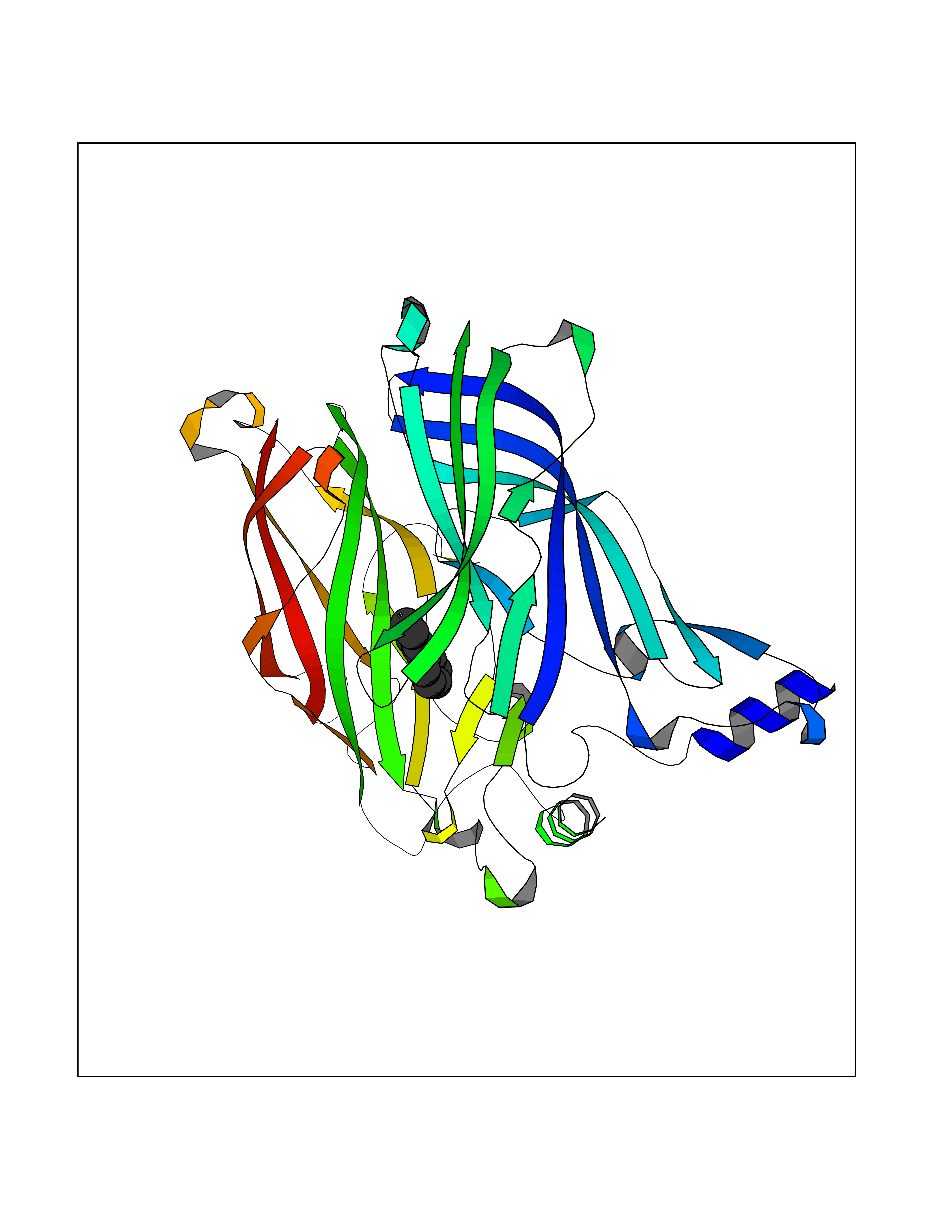 Molscript Map 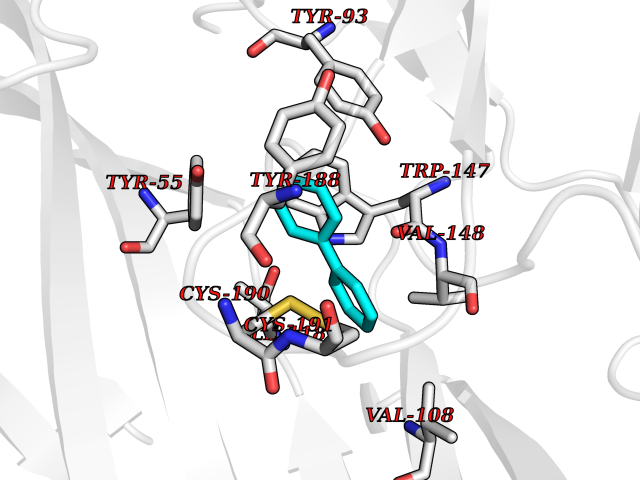 Pymol Map 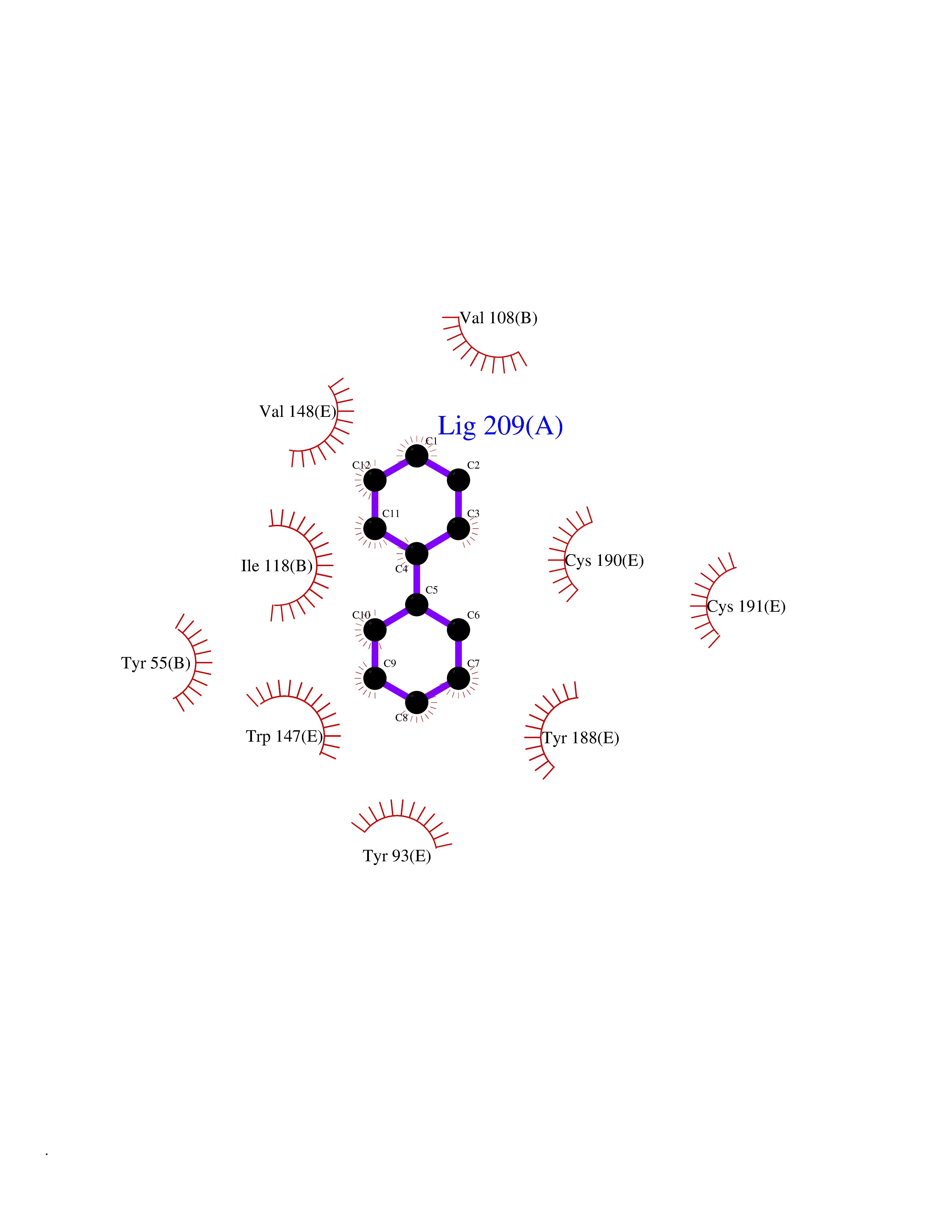 Ligplot Map 3D Binding mode Sequence EHETRLVAKLFKDYSSVVRPVEDHRQVVEVTLGFTLQDIVKADSSTNEVDLVYYEQQRWVDYNLKWNPDDYGGVKKIHIPAADIWTPDITAYSSTRPVQVLSPQIAVVTHDGSVMFIPAQRLSFMCDPTGVDSEEGATCAVKFGSWVYSGFEIDLKTDTDQVDLSSYYASSKYEILSATQTRQVQHYSCCPEPYIDVNLVVKFREEHETRLVAKLFKDYSSVVRPVEDHRQVVEVTLGFTLQDIVKADSSTNEVDLVYYEQQRWVDYNLKWNPDDYGGVKKIHIPAADIWTPDITAYSSTRPVQVLSPQIAVVTHDGSVMFIPAQRLSFMCDPTGVDSEEGATCAVKFGSWVYSGFEIDLKTDTDQVDLSSYYASSKYEILSATQTRQVQHYSCCPEPYIDVNLVVKFRER Hydrogen bonds contact Hydrophobic contact | ||||
| 8 | Helicobacter pylori Methylthioadenosine nucleosidase (HELPY mtnN) | 4BMZ | 6.40 | |
Target general information Gen name HELPY mtnN Organism Helicobacter pylori (strain ATCC 700392 / 26695) (Campylobacter pylori) Uniprot ID TTD ID Synonyms MTAN; MTA/SAH nucleosidase; Aminofutalosine nucleosidase; Aminodeoxyfutalosine nucleosidase; AFL nucleosidase; 6-amino-6-deoxyfutalosine N-ribosylhydrolase; 5'-methylthioadenosine/S-adenosylhomocystei Protein family PNP/UDP phosphorylase family Biochemical class NA Function Catalyzes the direct conversion of aminodeoxyfutalosine (AFL) into dehypoxanthine futalosine (DHFL) and adenine via the hydrolysis of the N-glycosidic bond; this reaction seems to represent an essential step in the menaquinone biosynthesis pathway in Helicobacter species. Can also probably catalyzes the hydrolysis of 5'-methylthioadenosine (MTA) and S-adenosylhomocysteine (SAH) to adenine and the corresponding thioribose, 5'-methylthioribose and S-ribosylhomocysteine, respectively. These other activities highlight the tremendous versatility of the enzyme, which also plays key roles in S-adenosylmethionine recycling and in the biosynthesis of the quorum-sensing molecule autoinducer-2. Does not act on futalosine (FL) as substrate. Related diseases Progressive familial heart block 1B (PFHB1B) [MIM:604559]: A cardiac bundle branch disorder characterized by progressive alteration of cardiac conduction through the His-Purkinje system, with a pattern of a right bundle-branch block and/or left anterior hemiblock occurring individually or together. It leads to complete atrio-ventricular block causing syncope and sudden death. {ECO:0000269|PubMed:19726882, ECO:0000269|PubMed:20562447, ECO:0000269|PubMed:21887725}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Erythrokeratodermia variabilis et progressiva 6 (EKVP6) [MIM:618531]: A form of erythrokeratodermia variabilis et progressiva, a genodermatosis characterized by the coexistence of two independent skin lesions: transient erythema and hyperkeratosis that is usually localized but occasionally occurs in its generalized form. Clinical presentation varies significantly within a family and from one family to another. Palmoplantar keratoderma is present in around 50% of cases. EKVP6 inheritance is autosomal dominant. {ECO:0000269|PubMed:30528822}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number NA Uniprot keywords 3D-structure; Amino-acid biosynthesis; Hydrolase; Menaquinone biosynthesis; Methionine biosynthesis; Reference proteome Protein physicochemical properties Chain ID A,B Molecular weight (Da) 50547.6 Length 464 Aromaticity 0.08 Instability index 26.92 Isoelectric point 5.13 Charge (pH=7) -20.92 2D Binding mode Binding energy (Kcal/mol) -8.73 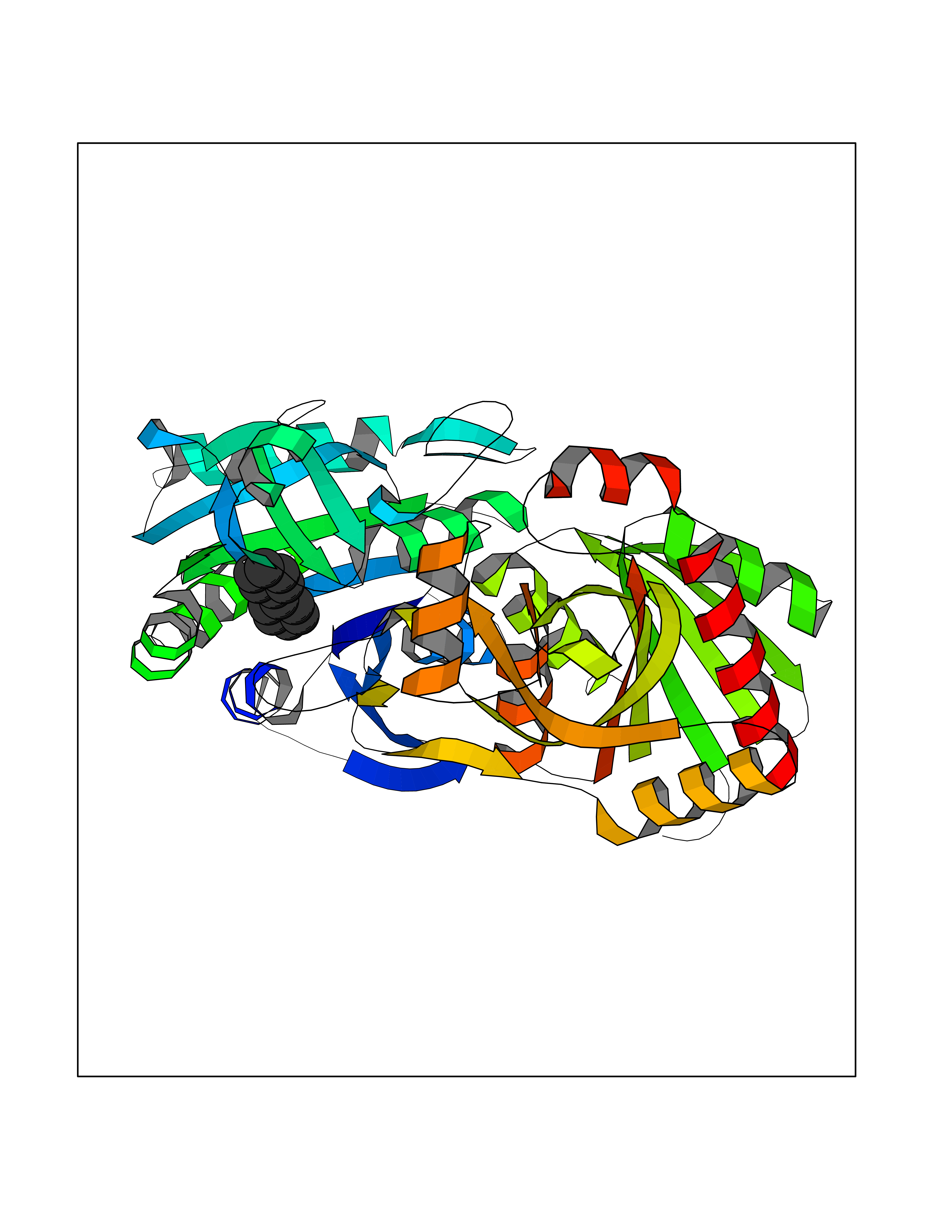 Molscript Map 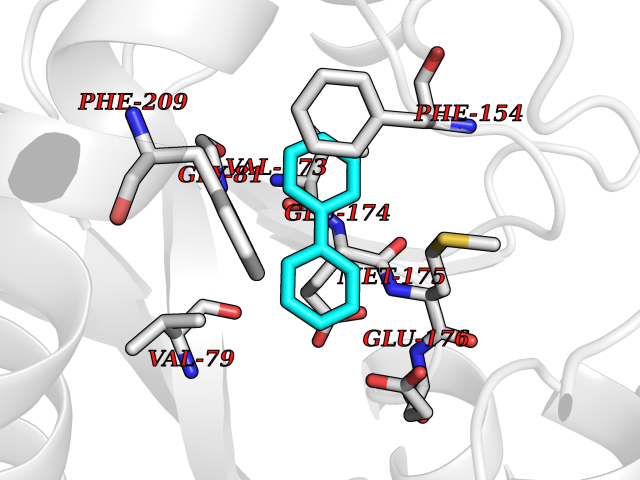 Pymol Map 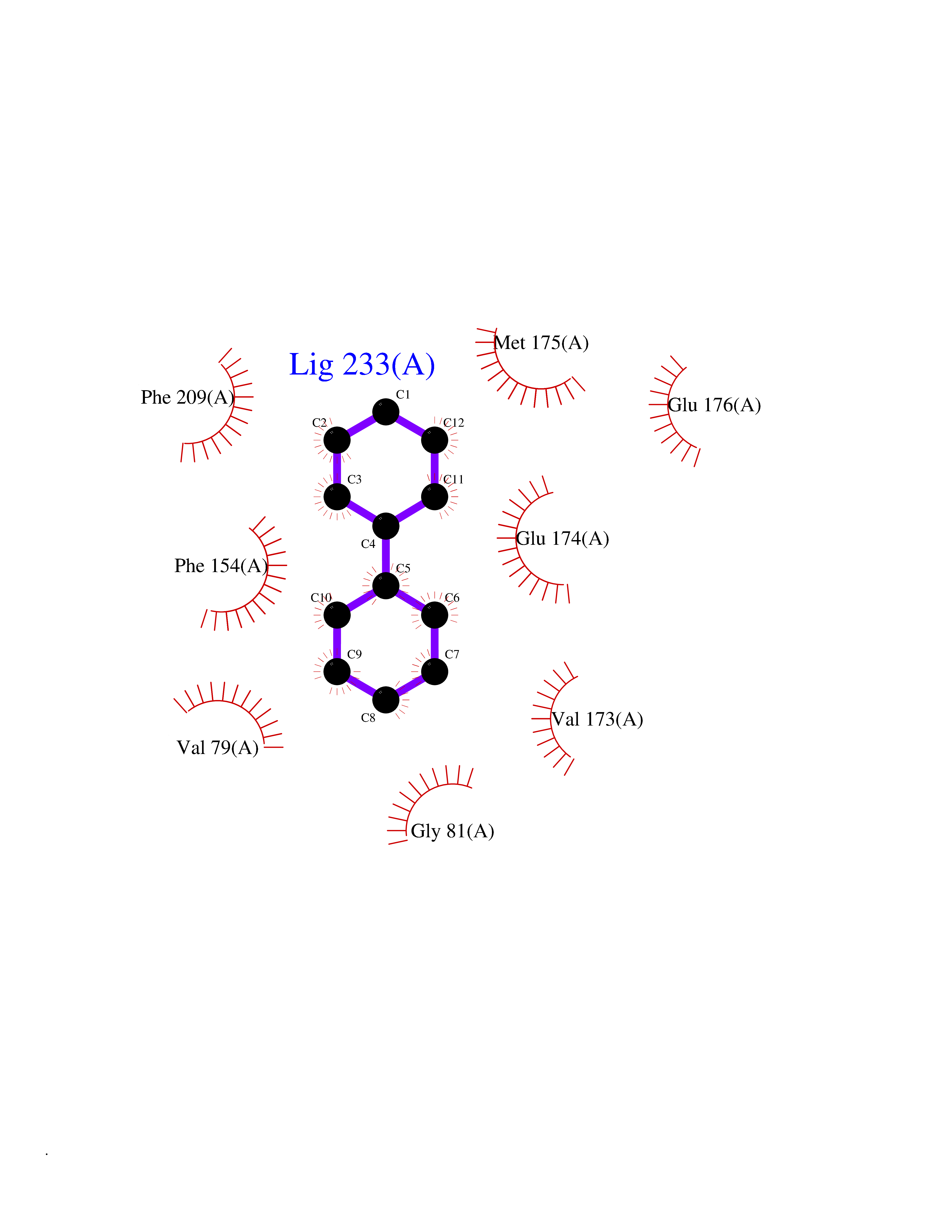 Ligplot Map 3D Binding mode Sequence VQKIGILGAMREEITPILELFGVDFEEIPLGGNVFHKGVYHNKEIIVAYSKIGKVHSTLTTTSMILAFGVQKVLFSGVAGSLVKDLKINDLLVAIQLVQHDVDLSAFDHPLGFIPESAIFIETSESLNALAKEVANEQHIVLKEGVIASGDQFVHSKERKEFLVSEFKASAVEMEGASVAFVCQKFGVPCCVLRSISNNADEEANMSFDAFLEKSAQTSAKFLKSMVDELGSHMVQKIGILGAMREEITPILELFGVDFEEIPLGGNVFHKGVYHNKEIIVAYSKIGKVHSTLTTTSMILAFGVQKVLFSGVAGSLVKDLKINDLLVAIQLVQHDVDLSAFDHPLGFIPESAIFIETSESLNALAKEVANEQHIVLKEGVIASGDQFVHSKERKEFLVSEFKASAVEMEGASVAFVCQKFGVPCCVLRSISNNADEEANMSFDAFLEKSAQTSAKFLKSMVDEL Hydrogen bonds contact Hydrophobic contact | ||||
| 9 | Cytochrome b (Complex III subunit 3) (Complex III subunit III) (Cytochrome b-c1 complex subunit 3) (Ubiquinol-cytochrome-c reductase complex cytochrome b subunit) | 1SQB | 6.37 | |
Target general information Gen name MT-CYB Organism Bos taurus (Bovine) Uniprot ID TTD ID NA Synonyms CYTB;MTCYB;COB Protein family Cytochrome b family Biochemical class NA Function Component of the ubiquinol-cytochrome c reductase complex (complex III or cytochrome b-c1 complex) that is part of the mitochondrial respiratory chain. The b-c1 complex mediates electron transfer from ubiquinol to cytochrome c. Contributes to the generation of a proton gradient across the mitochondrial membrane that is then used for ATP synthesis. {ECO:0000269|PubMed:1327781, ECO:0000269|PubMed:20025846, ECO:0000269|PubMed:9485330, ECO:0000305|PubMed:189810}." Related diseases Combined oxidative phosphorylation deficiency 6 (COXPD6) [MIM:300816]: A mitochondrial disease resulting in a neurodegenerative disorder characterized by psychomotor delay, hypotonia, areflexia, muscle weakness and wasting. Some patients manifest prenatal ventriculomegaly and severe postnatal encephalomyopathy. {ECO:0000269|PubMed:20362274, ECO:0000269|PubMed:22019070, ECO:0000269|PubMed:25583628, ECO:0000269|PubMed:26004228, ECO:0000269|PubMed:26173962, ECO:0000269|PubMed:27178839}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Charcot-Marie-Tooth disease, X-linked recessive, 4, with or without cerebellar ataxia (CMTX4) [MIM:310490]: A neuromuscular disorder characterized by progressive sensorimotor axonal neuropathy, distal sensory impairment, difficulty walking due to peripheral neuropathy and/or cerebellar ataxia, and deafness due to auditory neuropathy. Additional features include cognitive impairment, cerebellar atrophy, dysarthria, abnormal extraocular movements, tremor, dysmetria and spasticity. The age at onset ranges from infancy to young adulthood. {ECO:0000269|PubMed:23217327, ECO:0000269|PubMed:26004228}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Deafness, X-linked, 5, with peripheral neuropathy (DFNX5) [MIM:300614]: A form of hearing loss characterized by absent or severely abnormal auditory brainstem response, abnormal middle ear reflexes, abnormal speech discrimination, loss of outer hair cell function, and cochlear nerve hypoplasia. DFNX5 patients manifest auditory neuropathy with childhood onset, associated with distal sensory impairment affecting the peripheral nervous system. {ECO:0000269|PubMed:25986071}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Spondyloepimetaphyseal dysplasia, X-linked, with hypomyelinating leukodystrophy (SEMDHL) [MIM:300232]: An X-linked recessive developmental disorder characterized by slowly progressive skeletal and neurologic abnormalities, including short stature, large and deformed joints, significant motor impairment, visual defects, and sometimes cognitive deficits. Affected individuals typically have normal early development in the first year or so of life, followed by development regression and the development of symptoms. Brain imaging shows white matter abnormalities consistent with hypomyelinating leukodystrophy. {ECO:0000269|PubMed:28842795}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number NA Uniprot keywords 3D-structure; Electron transport; Heme; Iron; Membrane; Metal-binding; Mitochondrion; Mitochondrion inner membrane; Reference proteome; Respiratory chain; Transmembrane; Transmembrane helix; Transport; Ubiquinone Protein physicochemical properties Chain ID C,D,F,G,H Molecular weight (Da) 99281.2 Length 866 Aromaticity 0.12 Instability index 43.81 Isoelectric point 8.32 Charge (pH=7) 7.12 2D Binding mode Binding energy (Kcal/mol) -8.69  Molscript Map 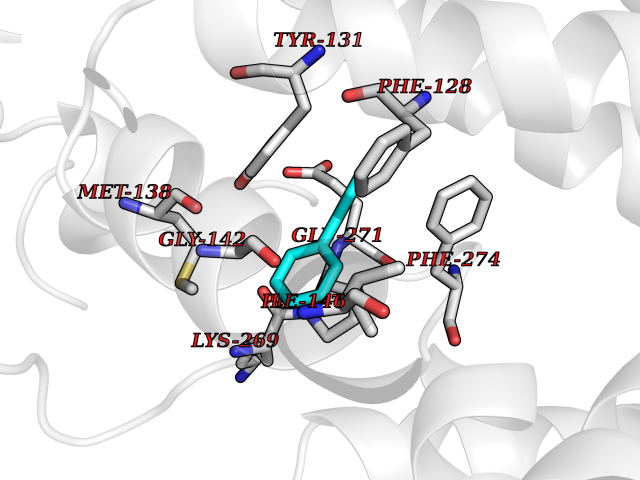 Pymol Map 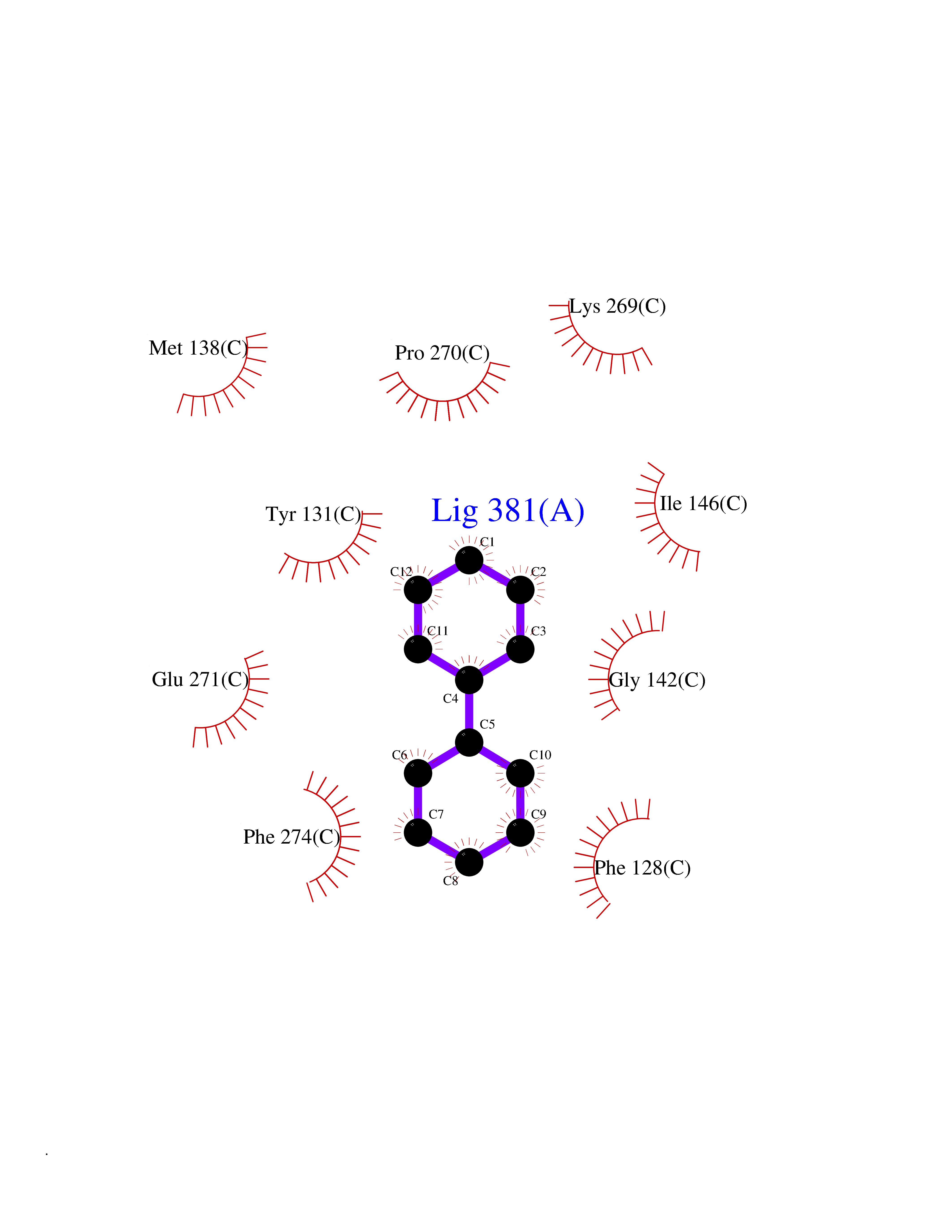 Ligplot Map 3D Binding mode Sequence VSASSRWLEGIRKWYYNAAGFNKLGLMRDDTIHENDDVKEAIRRLPENLYDDRVFRIKRALDLSMRQQILPKEQWTKYEEDKSYLEPYLKEVIRERKEREEWAKKELVDPLTTVREQCEQLEKCVKARERLELCDERVSSRSQTEEDCTEELLDFLHARDHCVAHKLFNSLKTNIRKSHPLMKIVNNAFIDLPAPSNISSWWNFGSLLGICLILQILTGLFLAMHYTSDTTTAFSSVTHICRDVNYGWIIRYMHANGASMFFICLYMHVGRGLYYGSYTFLETWNIGVILLLTVMATAFMGYVLPWGQMSFWGATVITNLLSAIPYIGTNLVEWIWGGFSVDKATLTRFFAFHFILPFIIMAIAMVHLLFLHETGSNNPTGISSDVDKIPFHPYYTIKDILGALLLILALMLLVLFAPDLLGDPDNYTPANPLNTPPHIKPEWYFLFAYAILRSIPNKLGGVLALAFSILILALIPLLHTSKQRSMMFRPLSQCLFWALVADLLTLTWIGGQPVEHPYITIGQLASVLYFLLILVLMPTAGTIENKLLKWSDLELHPPSYPWSHRGLLSSLDHTSIRRGFQVYKQVCSSCHSMDYVAYRHLVGVCYTEDEAKALAEEVEVQDGPNEDGEMFMRPGKLSDYFPKPYPNPEAARAANNGALPPDLSYIVRARHGGEDYVFSLLTGYCEPPTGVSLREGLYFNPYFPGQAIGMAPPIYNEVLEFDDGTPATMSQVAKDVCTFLRWAAEPEHDHRKRMGLKMLLMMGLLLPLVYAMKRHKWSVLKSRKLAYRPPKGRQFGHLTRVRHVITYSLSPFEQRAFPHYFSKGIPNVLRRTRACILRVAPPFVAFYLVYTWGTQEFEKSKRKNPA Hydrogen bonds contact Hydrophobic contact | ||||
| 10 | Leukotriene A-4 hydrolase (LTA4H) | 3U9W | 6.36 | |
Target general information Gen name LTA4H Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Leukotriene A4 hydrolase; Leukotriene A(4)Leukotriene A-4 hydrolase hydrolase; Leukotriene A(4) hydrolase; LTA4; LTA-H; LTA-4hydrolase; LTA-4 hydrolase Protein family Peptidase M1 family Biochemical class Ether bond hydrolase Function Has also aminopeptidase activity. Epoxide hydrolase that catalyzes the final step in the biosynthesis of the proinflammatory mediator leukotriene B4. Related diseases Pigmentary disorder, reticulate, with systemic manifestations, X-linked (PDR) [MIM:301220]: An X-linked recessive disorder characterized by recurrent infections and sterile inflammation in various organs. Diffuse skin hyperpigmentation with a distinctive reticulate pattern is universally evident by early childhood. This is later followed in many patients by hypohidrosis, corneal inflammation and scarring, enterocolitis that resembles inflammatory bowel disease, and recurrent urethral strictures. Melanin and amyloid deposition is present in the dermis. Affected males also have a characteristic facies with frontally upswept hair and flared eyebrows. Female carriers have only restricted pigmentary changes along Blaschko's lines. {ECO:0000269|PubMed:27019227}. The disease is caused by variants affecting the gene represented in this entry. XLPDR is caused by a recurrent intronic mutation that results in missplicing and reduced POLA1 expression. This leads to a decrease in cytosolic RNA:DNA hybrids and constitutive activation of type I interferon responses, but has no effect on cell replication. {ECO:0000269|PubMed:27019227}.; DISEASE: Van Esch-O'Driscoll syndrome (VEODS) [MIM:301030]: An X-linked recessive syndrome characterized by different degrees of intellectual disability, moderate to severe short stature, microcephaly, hypogonadism, and variable congenital malformations. {ECO:0000269|PubMed:31006512}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07102; DB06917; DB07258; DB07094; DB07259; DB02352; DB07292; DB07104; DB06828; DB08466; DB01197; DB05177; DB03366; DB08040; DB06851; DB02062; DB07099; DB07260; DB07196; DB11781; DB03424; DB07237 Interacts with Q9BSI4 EC number EC 3.3.2.6 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Cytoplasm; Direct protein sequencing; Hydrolase; Leukotriene biosynthesis; Lipid metabolism; Metal-binding; Metalloprotease; Phosphoprotein; Protease; Proteomics identification; Reference proteome; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 68927 Length 608 Aromaticity 0.1 Instability index 38.84 Isoelectric point 5.87 Charge (pH=7) -9.86 2D Binding mode Binding energy (Kcal/mol) -8.67 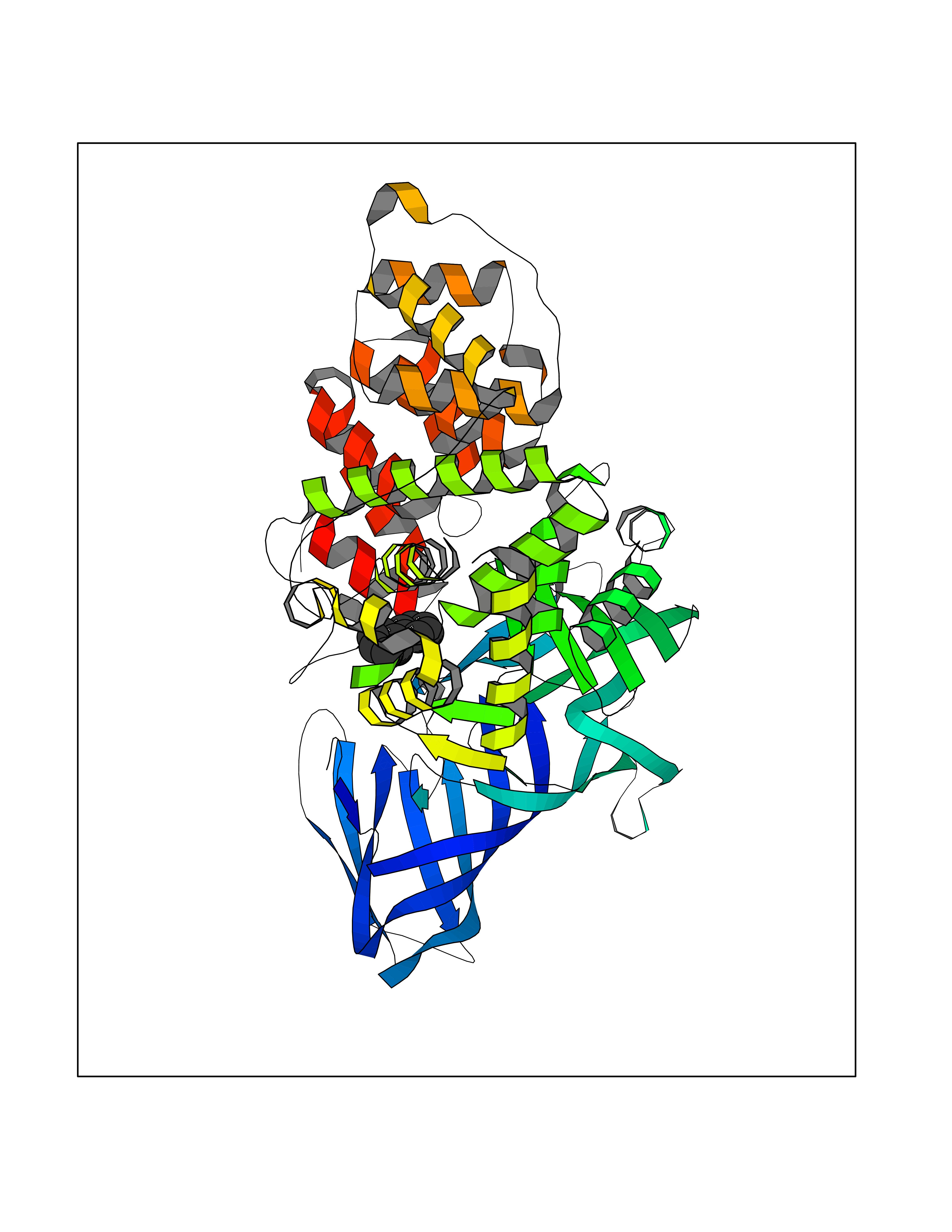 Molscript Map 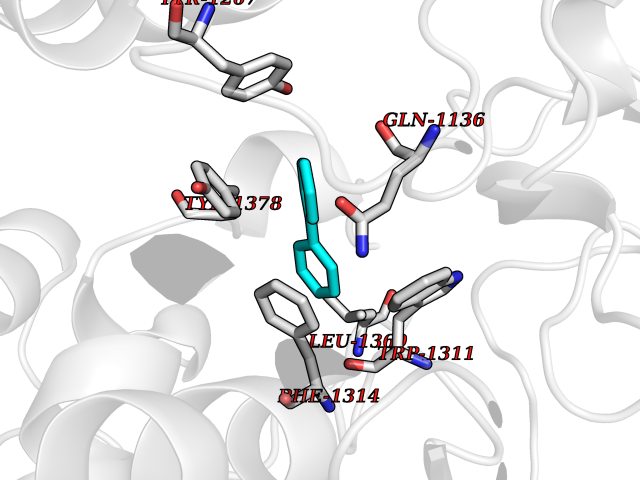 Pymol Map 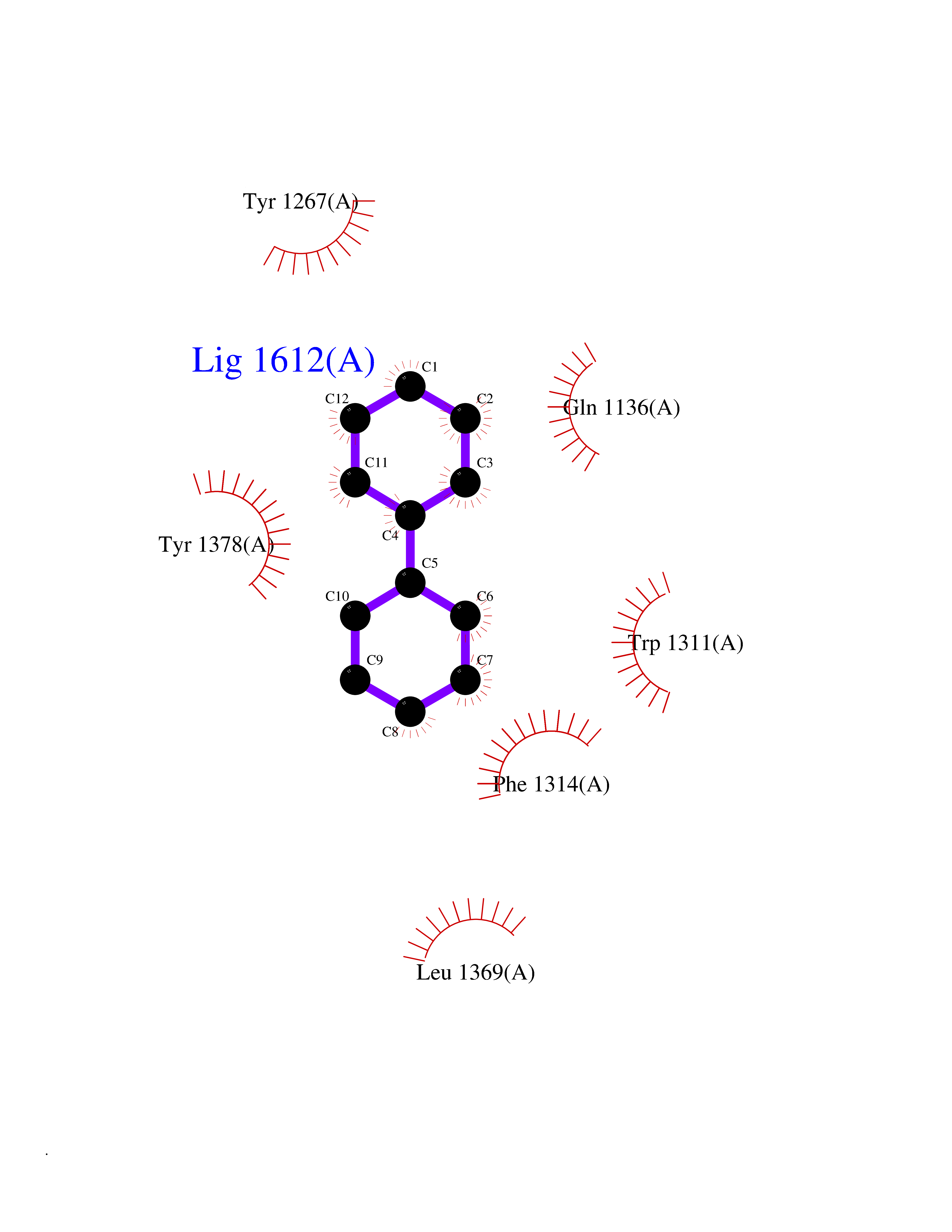 Ligplot Map 3D Binding mode Sequence IVDTCSLASPASVCRTKHLHLRCSVDFTRRTLTGTAALTVQSQEDNLRSLVLDTKDLTIEKVVINGQEVKYALGERQSYKGSPMEISLPIALSKNQEIVIEISFETSPKSSALQWLTPEQTSGKEHPYLFSQCQAIHCRAILPCQDTPSVKLTYTAEVSVPKELVALMSAIRDGETPDPEDPSRKIYKFIQKVPIPCYLIALVVGALESRQIGPRTLVWSEKEQVEKSAYEFSETESMLKIAEDLGGPYVWGQYDLLVLPPSFPYGGMENPCLTFVTPTLLAGDKSLSNVIAHEISHSWTGNLVTNKTWDHFWLNEGHTVYLERHICGRLFGEKFRHFNALGGWGELQNSVKTFGETHPFTKLVVDLTDIDPDVAYSSVPYEKGFALLFYLEQLLGGPEIFLGFLKAYVEKFSYKSITTDDWKDFLYSYFKDKVDVLNQVDWNAWLYSPGLPPIKPNYDMTLTNACIALSQRWITAKEDDLNSFNATDLKDLSSHQLNEFLAQTLQRAPLPLGHIKRMQEVYNFNAINNSEIRFRWLRLCIQSKWEDAIPLALKMATEQGRMKFTRPLFKDLAAFDKSHDQAVRTYQEHKASMHPVTAMLVGKDLKVD Hydrogen bonds contact Hydrophobic contact | ||||
| 11 | Glycolipid transfer protein | 3RZN | 6.36 | |
Target general information Gen name GLTP Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family GLTP family Biochemical class Lipid transport Function Glycolipid binding.Glycolipid transporter activity.Identical protein binding.Intermembrane lipid transfer activity.Lipid binding. Related diseases Brugada syndrome 7 (BRGDA7) [MIM:613120]: A tachyarrhythmia characterized by right bundle branch block and ST segment elevation on an electrocardiogram (ECG). It can cause the ventricles to beat so fast that the blood is prevented from circulating efficiently in the body. When this situation occurs, the individual will faint and may die in a few minutes if the heart is not reset. {ECO:0000269|PubMed:20031595}. The gene represented in this entry may be involved in disease pathogenesis.; DISEASE: Atrial fibrillation, familial, 16 (ATFB16) [MIM:613120]: A familial form of atrial fibrillation, a common sustained cardiac rhythm disturbance. Atrial fibrillation is characterized by disorganized atrial electrical activity and ineffective atrial contraction promoting blood stasis in the atria and reduces ventricular filling. It can result in palpitations, syncope, thromboembolic stroke, and congestive heart failure. {ECO:0000269|PubMed:20558140, ECO:0000269|PubMed:21051419}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03600; DB04465; DB03017; DB03203 Interacts with Q96DZ9; Q9NZD2 EC number NA Uniprot keywords 3D-structure; Acetylation; Cytoplasm; Lipid transport; Proteomics identification; Reference proteome; Repeat; Transport Protein physicochemical properties Chain ID A Molecular weight (Da) 23534.1 Length 206 Aromaticity 0.11 Instability index 36.45 Isoelectric point 7.08 Charge (pH=7) 0.1 2D Binding mode Binding energy (Kcal/mol) -8.67 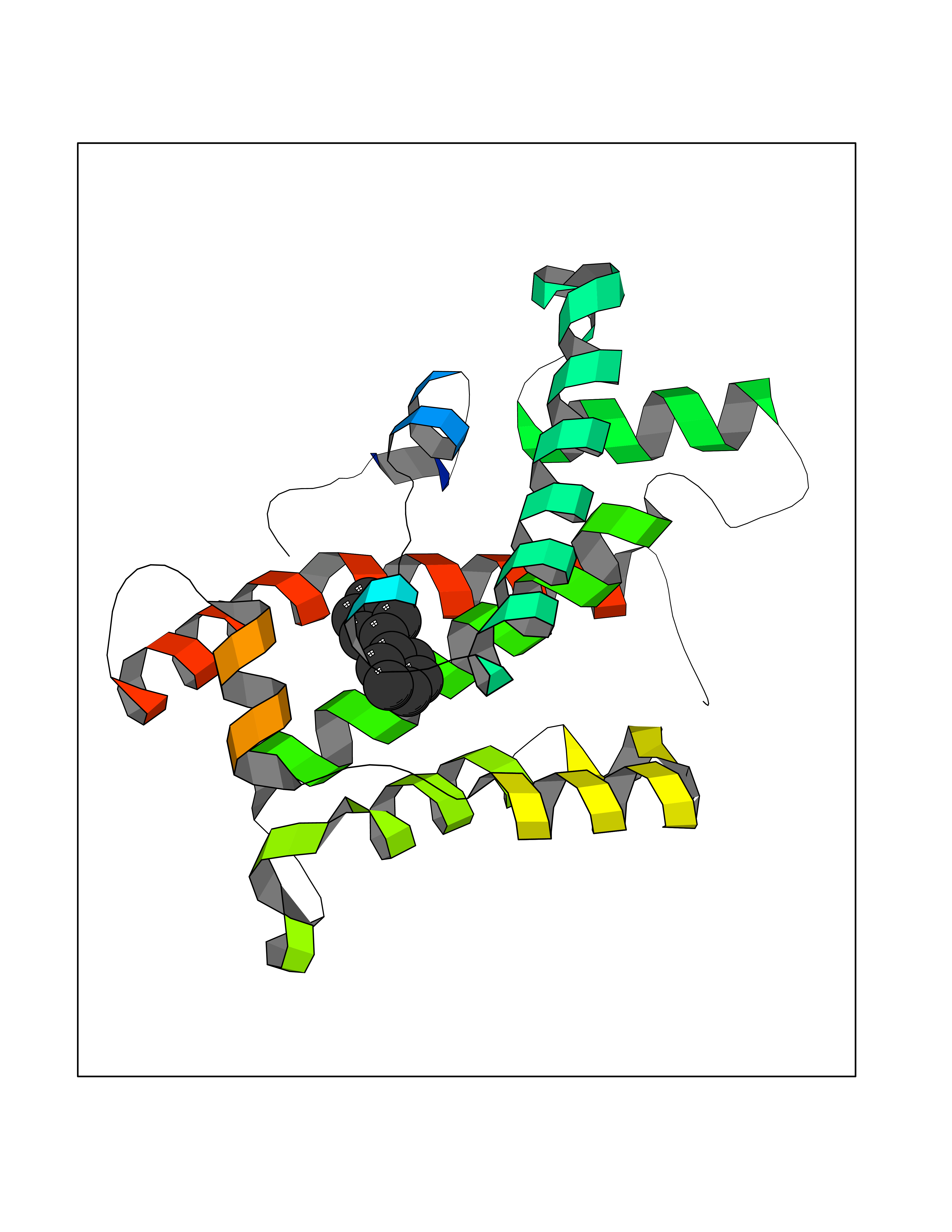 Molscript Map 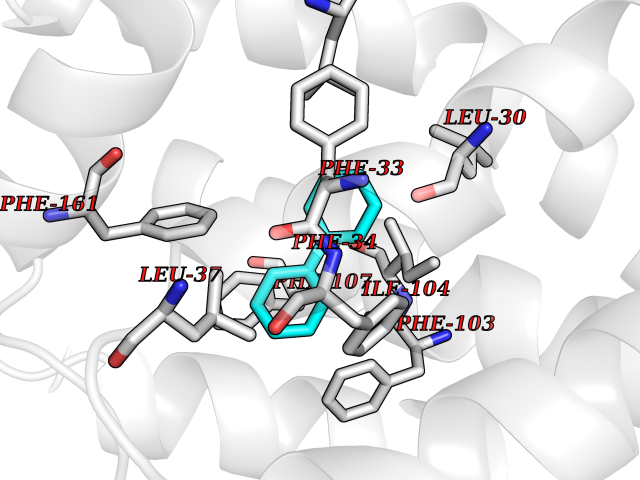 Pymol Map 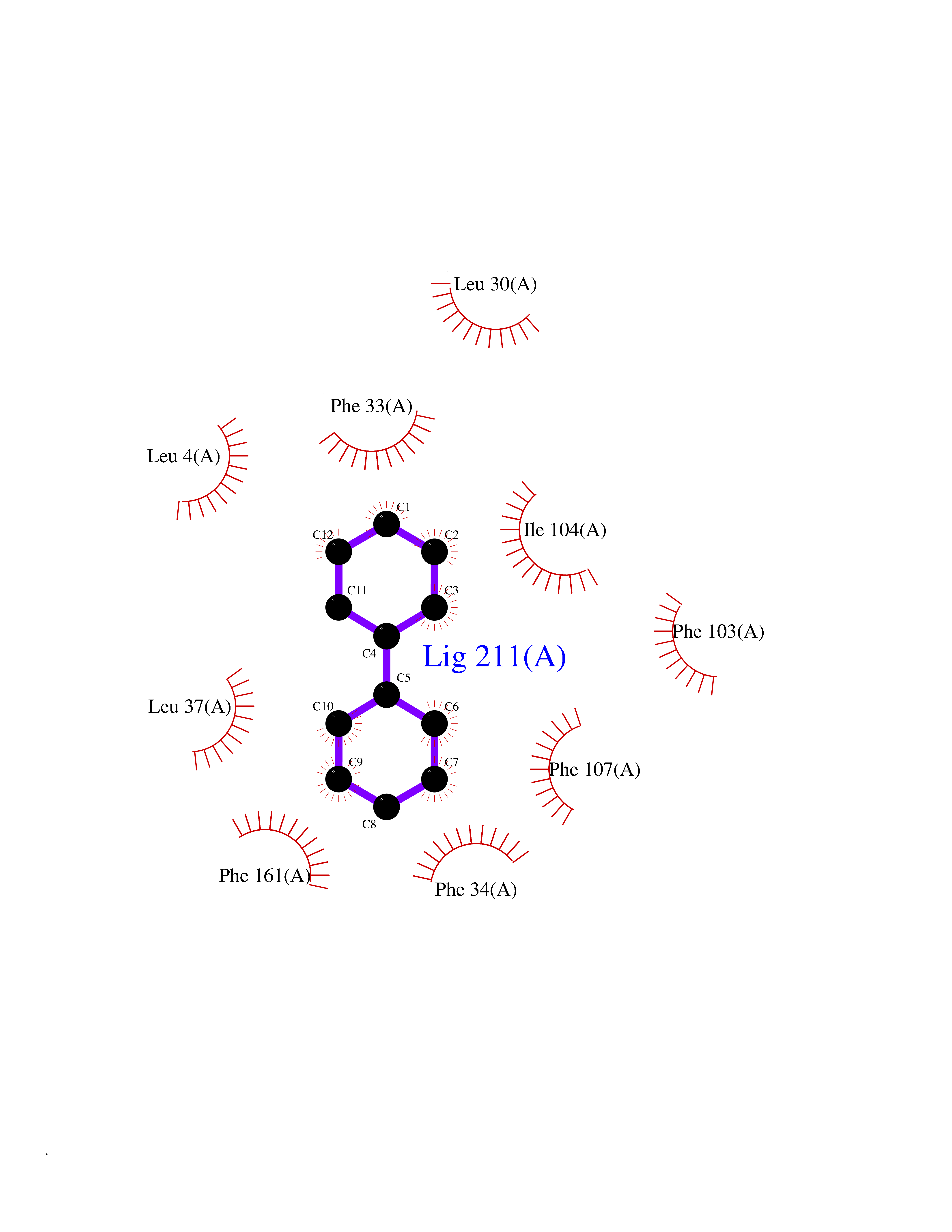 Ligplot Map 3D Binding mode Sequence LAEHLLKPLPADKQIETGPFLEAVSHLPPFFDCLGSPVFTPIKADISGNITKIKAVYDTNPAKFRTLQNILEVEKEMYGAEWPKVGATLALMWLKRGLRFIQVFLQSICDGERDENHPNLIRVNATKAYEMALKKYHGWIVQKIFQAALYAAPYKSDFLKALSKGQNVTEEECLEKIRLFLVNYTATIDVIYEMYTQMNAELNYKV Hydrogen bonds contact Hydrophobic contact | ||||
| 12 | MAPK signal-integrating kinase 1 (MKNK1) | 5WVD | 6.36 | |
Target general information Gen name MKNK1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Mnk1; MAP kinase signal-integrating kinase 1 Protein family Protein kinase superfamily, CAMK Ser/Thr protein kinase family Biochemical class Protein kinase superfamily. CAMK Ser/Thr protein kinase family Function May play a role in the response to environmental stress and cytokines. Appears to regulate translation by phosphorylating EIF4E, thus increasing the affinity of this protein for the 7-methylguanosine-containing mRNA cap. Related diseases Defects in MELK are associated with some cancers, such as brain or breast cancers. Expression is dramatically increased in aggressive undifferentiated tumors, correlating with poor patient outcome in breast and brain cancers, suggesting a role in tumor-initiating cells and proliferation via its function in cell proliferation regulation. Drugs (DrugBank ID) DB12010 Interacts with P54253; Q03060-25; P42858; P28482; Q16539; Q96CV9 EC number EC 2.7.11.1 Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Cytoplasm; Kinase; Magnesium; Metal-binding; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Serine/threonine-protein kinase; Transferase; Translation regulation Protein physicochemical properties Chain ID A Molecular weight (Da) 27536.2 Length 241 Aromaticity 0.11 Instability index 50.42 Isoelectric point 6.02 Charge (pH=7) -3.43 2D Binding mode Binding energy (Kcal/mol) -8.67  Molscript Map 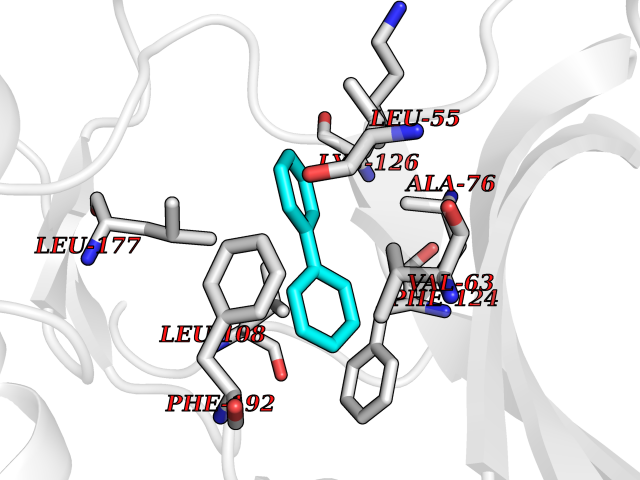 Pymol Map 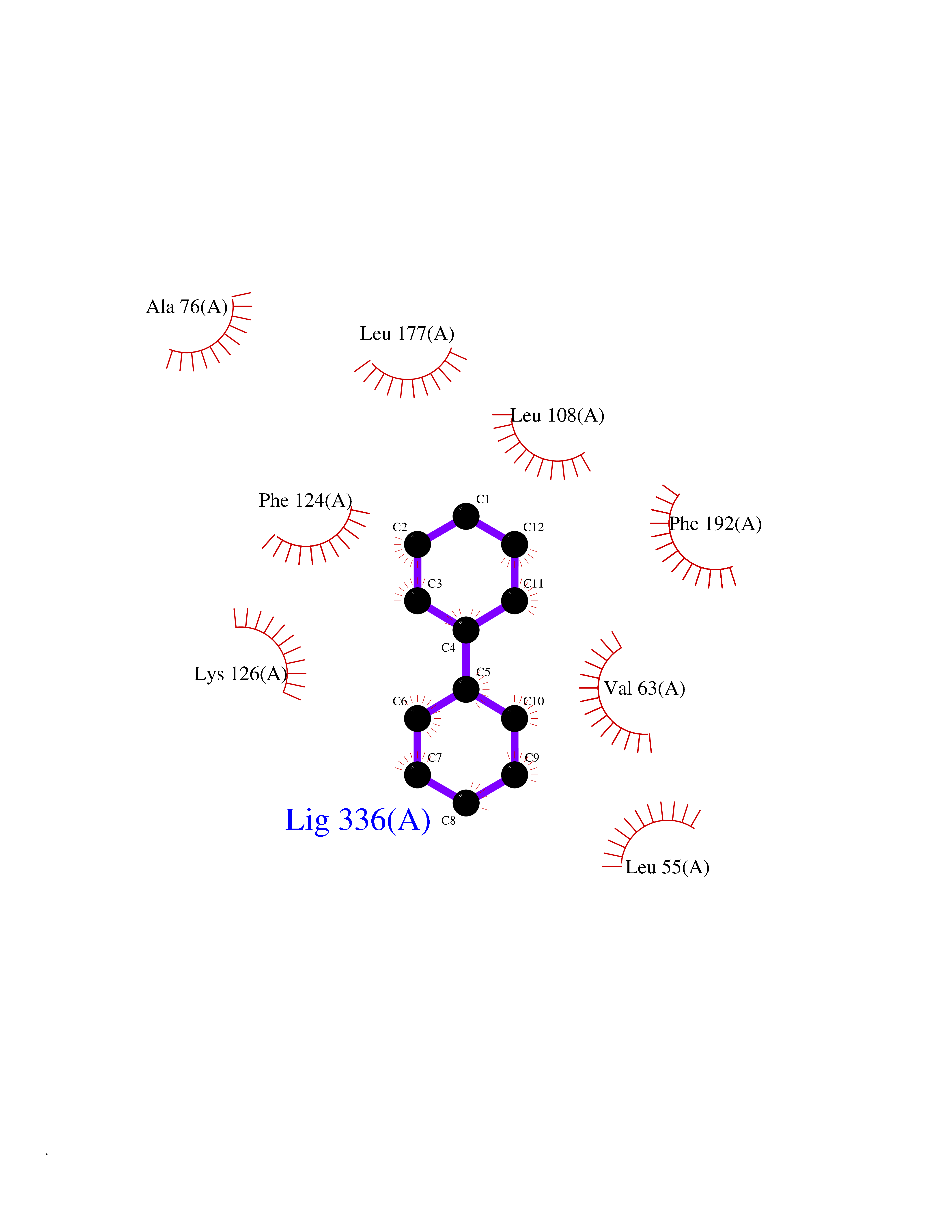 Ligplot Map 3D Binding mode Sequence PGKFEDMYKLTSELLGEGAYAKVQGAVSLQNGKEYAVKIIEKQAGHSRSRVFREVETLYQCQGNKNILELIEFFEDDTRFYLVFEKLQGGSILAHIQKQKHFNEREASRVVRDVAAALDFLHTKGIAHRDLKPENILCESPEKVSPVKICDFDLGSGYMAPEVVEVFTDQATFYDKRCDLWSLGVVLYIMLSGYPPFKYEFPDKDWAHISSEAKDLISKLLVRDAKQRLSAAQVLQHPWVQ Hydrogen bonds contact Hydrophobic contact | ||||
| 13 | Programmed cell death 1 ligand 1 (PD-L1) | 5J89 | 6.36 | |
Target general information Gen name CD274 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms hPD-L1; Programmed death ligand 1; PDL1; PDCD1LG1; PDCD1L1; PDCD1 ligand 1; B7H1; B7-H1; B7 homolog 1 Protein family Immunoglobulin superfamily, BTN/MOG family Biochemical class Immunoglobulin Function As a ligand for the inhibitory receptor PDCD1/PD-1, modulates the activation threshold of T-cells and limits T-cell effector response. Through a yet unknown activating receptor, may costimulate T-cell subsets that predominantly produce interleukin-10 (IL10). Plays a critical role in induction and maintenance of immune tolerance to self. Related diseases Truncation of the 3'-untranslated (3'-UTR) region of CD274 transcripts leads to elevated expression of CD274 in multiple cancers including T-cell leukemia, diffuse large B-cell lymphoma and stomach adenocarcinoma (PubMed:27281199). Disruption of 3'-UTR region is caused by structural variants that stabilize CD274 transcripts, leading to overexpression (PubMed:27281199). Increased expression in tumors promotes immune evasion and tumor cell growth by allowing malignant cells to escape destruction by the immune system (PubMed:27281199). {ECO:0000269|PubMed:27281199}. Drugs (DrugBank ID) DB15773; DB11595; DB15771; DB11945; DB15772; DB14776; DB15770; DB11714; DB15769; DB09035; DB09037; DB00203; DB00313 Interacts with P33681; Q8IZR5; Q9NX76; Q15116; Q15116 EC number NA Uniprot keywords 3D-structure; Adaptive immunity; Alternative splicing; Cell membrane; Disulfide bond; Endosome; Glycoprotein; Immunity; Immunoglobulin domain; Membrane; Nucleus; Proteomics identification; Receptor; Reference proteome; Repeat; Secreted; Signal; Transmembrane; Transmembrane helix; Ubl conjugation Protein physicochemical properties Chain ID C,D Molecular weight (Da) 28335.2 Length 249 Aromaticity 0.1 Instability index 35.39 Isoelectric point 6.15 Charge (pH=7) -3.43 2D Binding mode Binding energy (Kcal/mol) -8.68 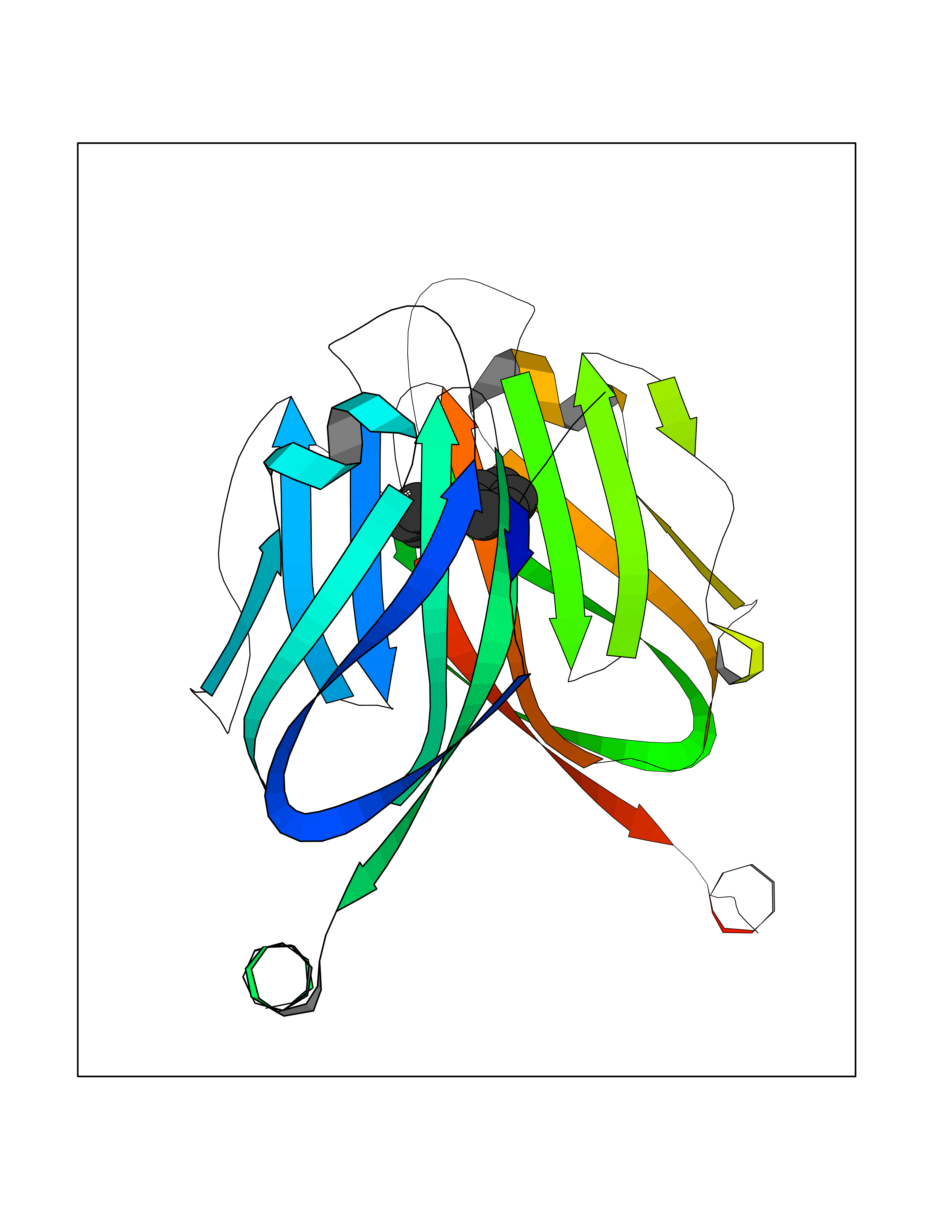 Molscript Map 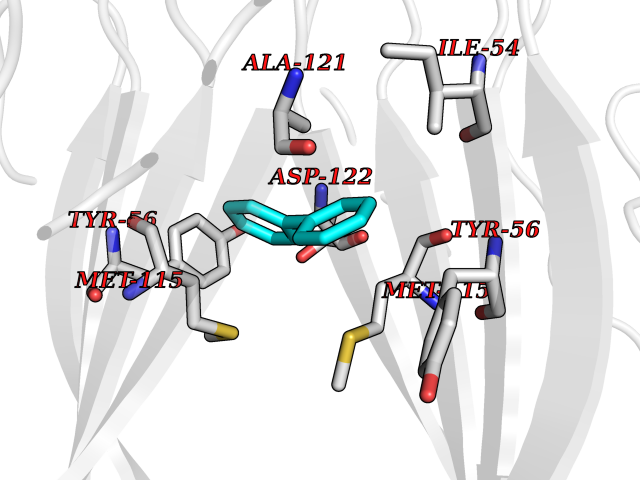 Pymol Map 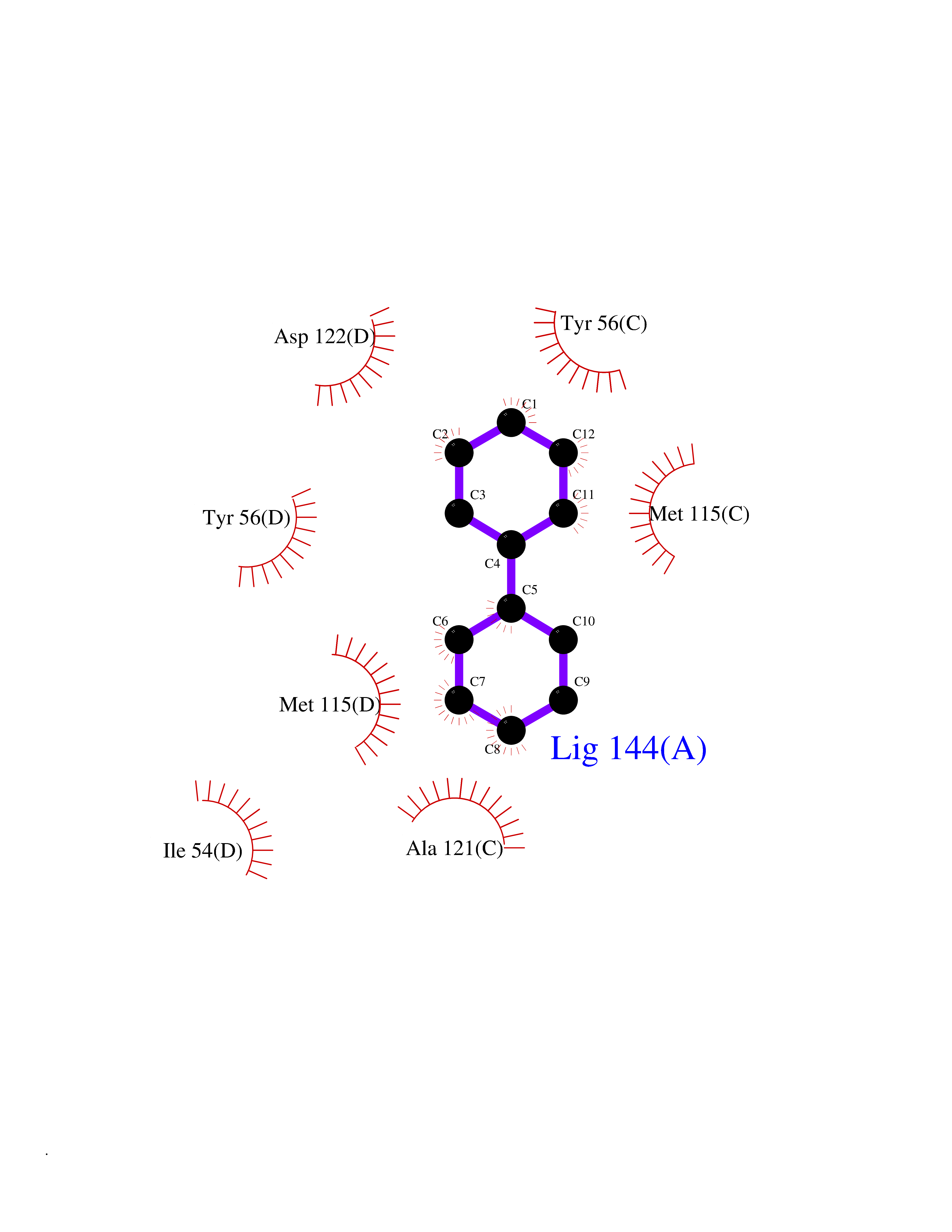 Ligplot Map 3D Binding mode Sequence AFTVTVPKDLYVVEYGSNMTIECKFPVEKQLDLAALIVYWEMEDKNIIQFVHGEEDLKVQHSSYRQRARLLKDQLSLGNAALQITDVKLQDAGVYRCMISYGGADYKRITVKVNAPYAAALEHHHAFTVTVPKDLYVVEYGSNMTIECKFPVEKQLDLAALIVYWEMEDKNIIQFVHGEEDLKVQHSSYRQRARLLKDQLSLGNAALQITDVKLQDAGVYRCMISYGGADYKRITVKVNAPYAAALEHH Hydrogen bonds contact Hydrophobic contact | ||||
| 14 | Acetyl-CoA carboxylase (ACC) (EC 6.4.1.2) (Fatty acid synthetase 3) (mRNA transport-defective protein 7) [Includes: Biotin carboxylase (EC 6.3.4.14)] | 1UYS | 6.35 | |
Target general information Gen name ACC1 Organism Saccharomyces cerevisiae (strain ATCC 204508 / S288c) (Baker's yeast) Uniprot ID TTD ID NA Synonyms MTR7;YNR016C;N3175;ABP2;FAS3 Protein family NA Biochemical class NA Function Carries out three functions: biotin carboxyl carrier protein, biotin carboxylase and carboxyltransferase. Involved in the synthesis of very-long-chain fatty acid synthesis which is required to maintain a functional nuclear envelope. Required for acylation and vacuolar membrane association of VAC8 which is necessary to maintain a normal morphology of the vacuole. {ECO:0000269|PubMed:10757783, ECO:0000269|PubMed:12730220, ECO:0000269|PubMed:6103540, ECO:0000269|PubMed:6108218, ECO:0000269|PubMed:8943372}." Related diseases A chromosomal aberration involving NFKB2 is found in a case of B-cell non Hodgkin lymphoma (B-NHL). Translocation t(10;14)(q24;q32) with IGHA1. The resulting oncogene is also called Lyt-10C alpha variant.; DISEASE: A chromosomal aberration involving NFKB2 is found in a cutaneous T-cell leukemia (C-TCL) cell line. This rearrangement produces the p80HT gene which codes for a truncated 80 kDa protein (p80HT).; DISEASE: In B-cell leukemia (B-CLL) cell line, LB40 and EB308, can be found after heterogeneous chromosomal aberrations, such as internal deletions.; DISEASE: Immunodeficiency, common variable, 10 (CVID10) [MIM:615577]: A primary immunodeficiency characterized by childhood-onset of recurrent infections, hypogammaglobulinemia, and decreased numbers of memory and marginal zone B-cells. Some patients may develop autoimmune features and have circulating autoantibodies. An unusual feature is central adrenal insufficiency. {ECO:0000269|PubMed:24140114, ECO:0000269|PubMed:25524009}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q00955 EC number 6.3.4.14; 6.4.1.2 Uniprot keywords 3D-structure; Acetylation; ATP-binding; Biotin; Cytoplasm; Direct protein sequencing; Endoplasmic reticulum; Fatty acid biosynthesis; Fatty acid metabolism; Ligase; Lipid biosynthesis; Lipid metabolism; Manganese; Membrane; Metal-binding; Multifunctional enzyme; Nucleotide-binding; Phosphoprotein; Reference proteome Protein physicochemical properties Chain ID B,C Molecular weight (Da) 145619 Length 1328 Aromaticity 0.1 Instability index 30.31 Isoelectric point 5.32 Charge (pH=7) -26.79 2D Binding mode Binding energy (Kcal/mol) -8.67  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence WLQPKRYKAHLXGTTYVYDFPELFRQASSSQWKNFSADVKLTDDFFISNELIEDENGELTEVEREPGANAIGXVAFKITVKTPEYPRGRQFVVVANDITFKIGSFGPQEDEFFNKVTEYARKRGIPRIYLAANSGARIGXAEEIVPLFQVAWNDAANPDKGFQYLYLTSEGXETLKKFDKENSVLTERTVINGEERFVIKTIIGSEDGLGVECLRGSGLIAGATSRAYHDIFTITLVTCRSVGIGAYLVRLGQRAIQVEGQPIILTGAPAINKXLGREVYTSNLQLGGTQIXYNNGVSHLTAVDDLAGVEKIVEWXSYVPAKRNXPVPILETKDTWDRPVDFTPTNDETYDVRWXIEGRETESGFEYGLFDKGSFFETLSGWAKGVVVGRARLGGIPLGVIGVETRTVENLIPADPANPNSAETLIQEPGQVWHPNSAFKTAQAINDFNNGEQLPXXILANWRGFSGNEVLKYGSFIVDALVDYKQPIIIYIPPTGELRGGSWVVVDPTINADQXEXYADVNARAGVLEPQGXVGIKFRREKLLDTXNRLELLPIYGQISLQFADLHDRSSRXVAKGVISKELEWTEARRFFFWRLRRRLNEEYLIKRLSHQVGEASRLEKIARIRSWYPASVDHEDDRQVATWIEENYKTLDDKLKGLPIATPYPVKEWLQPKRYKAHLXGTTYVYDFPELFRQASSSQWKNFSADVKLTDDFFISNELIEDENGELTEVEREPGANAIGXVAFKITVKTPEYPRGRQFVVVANDITFKIGSFGPQEDEFFNKVTEYARKRGIPRIYLAANSGARIGXAEEIVPLFQVAWNDAANPDKGFQYLYLTSEGXETLKKFDKENSVLTERTVINGEERFVIKTIIGSEDGLGVECLRGSGLIAGATSRAYHDIFTITLVTCRSVGIGAYLVRLGQRAIQVEGQPIILTGAPAINKXLGREVYTSNLQLGGTQIXYNNGVSHLTAVDDLAGVEKIVEWXSYVPAKRNXPVPILETKDTWDRPVDFTPTNDETYDVRWXIEGRETESGFEYGLFDKGSFFETLSGWAKGVVVGRARLGGIPLGVIGVETRTVENLIPADPANPNSAETLIQEPGQVWHPNSAFKTAQAINDFNNGEQLPXXILANWRGFSGNEVLKYGSFIVDALVDYKQPIIIYIPPTGELRGGSWVVVDPTINADQXEXYADVNARAGVLEPQGXVGIKFRREKLLDTXNRLELLPIYGQISLQFADLHDRSSRXVAKGVISKELEWTEARRFFFWRLRRRLNEEYLIKRLSHQVGEASRLEKIARIRSWYPASVDHEDDRQVATWIEENYKTLDDKLKGL Hydrogen bonds contact Hydrophobic contact | ||||
| 15 | Peptostreptococcal albumin-binding protein | 2VDB | 6.35 | |
Target general information Gen name pab Organism Finegoldia magna (Peptostreptococcus magnus) Uniprot ID TTD ID NA Synonyms NA Protein family NA Biochemical class Protein binding Function Binds serum albumin. Related diseases LTC4 synthase deficiency is associated with a neurometabolic developmental disorder characterized by muscular hypotonia, psychomotor retardation, failure to thrive, and microcephaly. {ECO:0000269|PubMed:10896305, ECO:0000269|PubMed:9820300}. Drugs (DrugBank ID) DB03600; DB00788 Interacts with NA EC number NA Uniprot keywords 3D-structure; Cell wall; Peptidoglycan-anchor; Secreted; Signal Protein physicochemical properties Chain ID B Molecular weight (Da) 21751.6 Length 189 Aromaticity 0.1 Instability index 49.17 Isoelectric point 5.44 Charge (pH=7) -6.7 2D Binding mode Binding energy (Kcal/mol) -8.66  Molscript Map 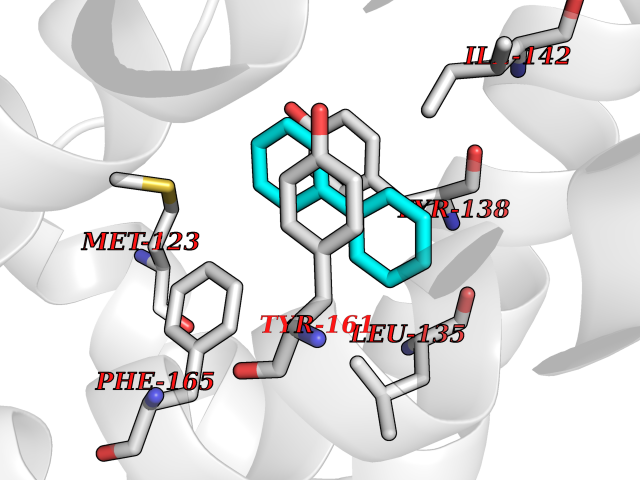 Pymol Map 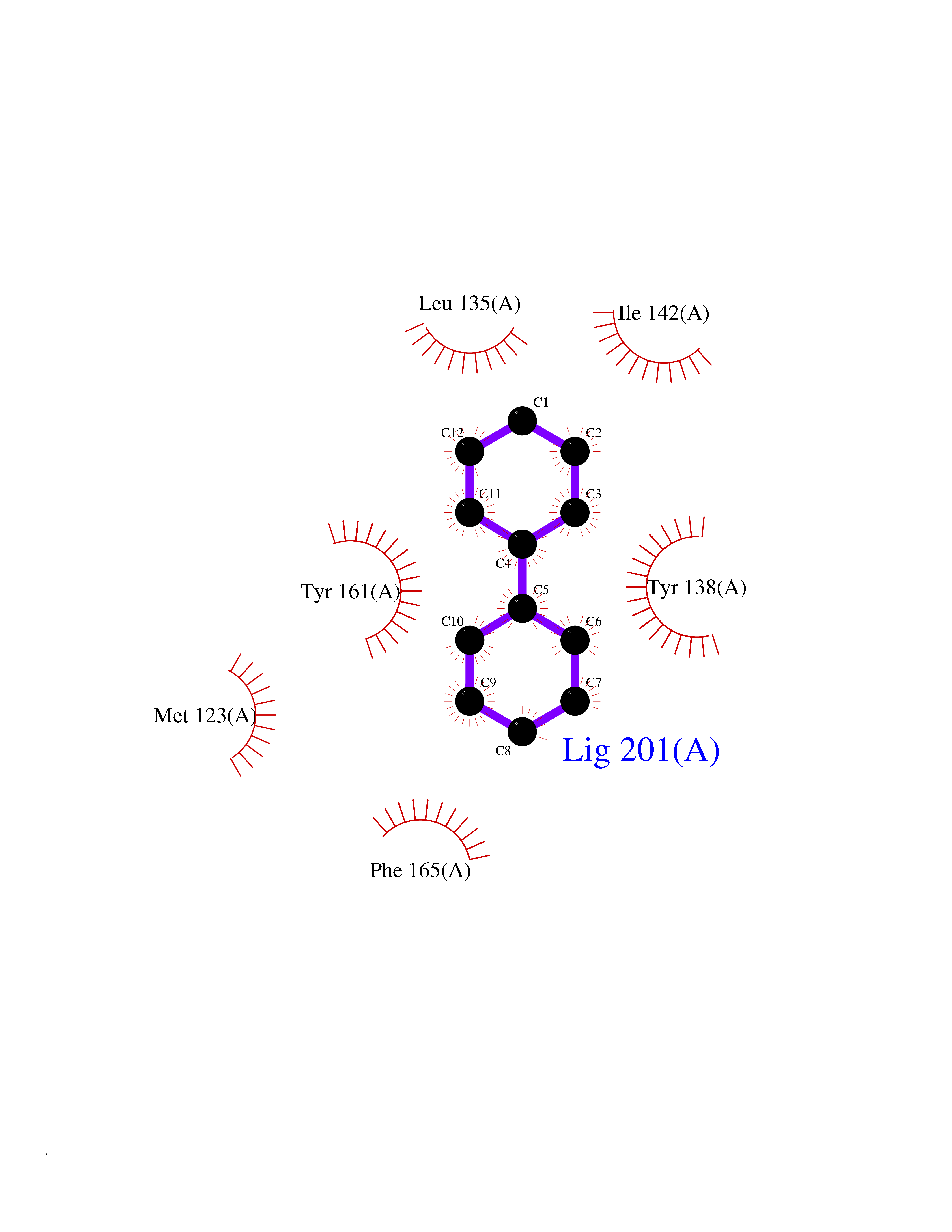 Ligplot Map 3D Binding mode Sequence EVAHRFKDLGEENFKALVLIAFAQYLQQCPFEDHVKLVNEVTEFAKTCVADESAENCDKSLHTLFGDKLCTVATLEMADCCAKQEPERNECFLQHKDDNPNLPRLVRPEVDVMCTAFHDNEETFLKKYLYEIARRHPYFYAPELLFFAKRYKAAFTECCQAADKAACLLPKLDELRDEGKASSAKQRLK Hydrogen bonds contact Hydrophobic contact | ||||
| 16 | Glucose-dependent insulinotropic receptor (GPR119) | 7XZ6 | 6.34 | |
Target general information Gen name GPR119 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms GPR119; G-protein coupled receptor 119 Protein family G-protein coupled receptor 1 family Biochemical class GPCR rhodopsin Function Receptor for the endogenous fatty-acid ethanolamide oleoylethanolamide (OEA) and lysophosphatidylcholine (LPC). Functions as a glucose-dependent insulinotropic receptor. The activity of this receptor is mediated by G proteins which activate adenylate cyclase. Seems to act through a G(s) mediated pathway. Related diseases Developmental and epileptic encephalopathy 24 (DEE24) [MIM:615871]: A disease characterized by early-onset seizures, intellectual disability of varying degrees, and behavioral disturbances or autistic features in most individuals. {ECO:0000269|PubMed:24747641, ECO:0000269|PubMed:27864847, ECO:0000269|PubMed:30351409}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Generalized epilepsy with febrile seizures plus 10 (GEFSP10) [MIM:618482]: An autosomal dominant neurologic disorder with incomplete penetrance, characterized by variable types of seizures including absence, tonic-clonic, febrile, focal, and eyelid myoclonia. Some patients have normal neurologic development. Others have mild-to-moderate intellectual disability or autism spectrum disorder. {ECO:0000269|PubMed:29936235, ECO:0000269|PubMed:30351409}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB05166 Interacts with Q12797-6 EC number NA Uniprot keywords 3D-structure; Cell membrane; G-protein coupled receptor; Lipid-binding; Membrane; Receptor; Reference proteome; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID R Molecular weight (Da) 32134.1 Length 292 Aromaticity 0.12 Instability index 34.96 Isoelectric point 9.12 Charge (pH=7) 8.03 2D Binding mode Binding energy (Kcal/mol) -8.64  Molscript Map 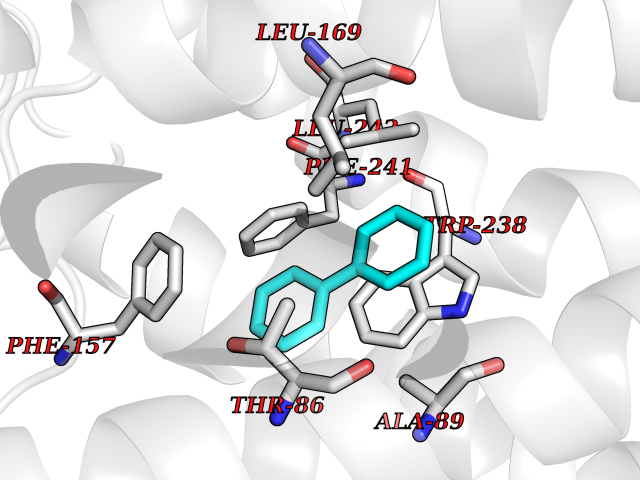 Pymol Map  Ligplot Map 3D Binding mode Sequence MESSFSFGVILAVLASLIIATNTLVAVAVLLLIHKNDGVSLCFTLNLAVADTLIGVAISGLLTDQLSSPSRPTQKTLCSLRMAFVTSSAAASVLTVMLITFDRYLAIKQPFRYLKIMSGFVAGACIAGLWLVSYLIGFLPLGIPMFQQTAYKGQCSFFAVFHPHFVLTLSCVGFFPAMLLFVFFYCDMLKIASMHSQQIRKMEHAGAMAGSDFKALRTVSVLIGSFALSWTPFLITGIVQVACQECHLYLVLERYLWLLGVGNSLLNPLIYAYWQKEVRLQLYHMALGVKKV Hydrogen bonds contact Hydrophobic contact | ||||
| 17 | Retinoic acid receptor gamma (RARG) | 1FCY | 6.33 | |
Target general information Gen name RARG Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms RAR-gamma; Nuclear receptor subfamily 1 group B member 3; NR1B3 Protein family Nuclear hormone receptor family, NR1 subfamily Biochemical class Nuclear hormone receptor Function Receptor for retinoic acid. Retinoic acid receptors bind as heterodimers to their target response elements in response to their ligands, all-trans or 9-cis retinoic acid, and regulate gene expression in various biological processes. The RAR/RXR heterodimers bind to the retinoic acid response elements (RARE) composed of tandem 5'-AGGTCA-3' sites known as DR1-DR5. In the absence of ligand, acts mainly as an activator of gene expression due to weak binding to corepressors. Required for limb bud development. In concert with RARA or RARB, required for skeletal growth, matrix homeostasis and growth plate function (By similarity). Related diseases Cystic fibrosis (CF) [MIM:219700]: A common generalized disorder of the exocrine glands which impairs clearance of secretions in a variety of organs. It is characterized by the triad of chronic bronchopulmonary disease (with recurrent respiratory infections), pancreatic insufficiency (which leads to malabsorption and growth retardation) and elevated sweat electrolytes. It is the most common genetic disease in Caucasians, with a prevalence of about 1 in 2'000 live births. Inheritance is autosomal recessive. {ECO:0000269|PubMed:10094564, ECO:0000269|PubMed:10869121, ECO:0000269|PubMed:10923036, ECO:0000269|PubMed:11242048, ECO:0000269|PubMed:12167682, ECO:0000269|PubMed:12394343, ECO:0000269|PubMed:12529365, ECO:0000269|PubMed:1284466, ECO:0000269|PubMed:1284468, ECO:0000269|PubMed:1284529, ECO:0000269|PubMed:1284530, ECO:0000269|PubMed:1284548, ECO:0000269|PubMed:1379210, ECO:0000269|PubMed:15528182, ECO:0000269|PubMed:15716351, ECO:0000269|PubMed:16822950, ECO:0000269|PubMed:1695717, ECO:0000269|PubMed:1699669, ECO:0000269|PubMed:17098864, ECO:0000269|PubMed:1710600, ECO:0000269|PubMed:1712898, ECO:0000269|PubMed:17182731, ECO:0000269|PubMed:20008117, ECO:0000269|PubMed:20150177, ECO:0000269|PubMed:20691141, ECO:0000269|PubMed:21884936, ECO:0000269|PubMed:2236053, ECO:0000269|PubMed:23818989, ECO:0000269|PubMed:25330774, ECO:0000269|PubMed:26846474, ECO:0000269|PubMed:27241308, ECO:0000269|PubMed:28001373, ECO:0000269|PubMed:28067262, ECO:0000269|PubMed:28087700, ECO:0000269|PubMed:32026723, ECO:0000269|PubMed:33572515, ECO:0000269|PubMed:7504969, ECO:0000269|PubMed:7505694, ECO:0000269|PubMed:7505767, ECO:0000269|PubMed:7508414, ECO:0000269|PubMed:7513296, ECO:0000269|PubMed:7517264, ECO:0000269|PubMed:7520022, ECO:0000269|PubMed:7522211, ECO:0000269|PubMed:7524909, ECO:0000269|PubMed:7524913, ECO:0000269|PubMed:7525450, ECO:0000269|PubMed:7537150, ECO:0000269|PubMed:7541273, ECO:0000269|PubMed:7541510, ECO:0000269|PubMed:7543567, ECO:0000269|PubMed:7544319, ECO:0000269|PubMed:7581407, ECO:0000269|PubMed:7606851, ECO:0000269|PubMed:7680525, ECO:0000269|PubMed:7683628, ECO:0000269|PubMed:7683954, ECO:0000269|PubMed:8081395, ECO:0000269|PubMed:8406518, ECO:0000269|PubMed:8522333, ECO:0000269|PubMed:8723693, ECO:0000269|PubMed:8723695, ECO:0000269|PubMed:8800923, ECO:0000269|PubMed:8829633, ECO:0000269|PubMed:8910473, ECO:0000269|PubMed:8956039, ECO:0000269|PubMed:9101301, ECO:0000269|PubMed:9222768, ECO:0000269|PubMed:9375855, ECO:0000269|PubMed:9401006, ECO:0000269|PubMed:9443874, ECO:0000269|PubMed:9452048, ECO:0000269|PubMed:9452054, ECO:0000269|PubMed:9452073, ECO:0000269|PubMed:9482579, ECO:0000269|PubMed:9507391, ECO:0000269|PubMed:9521595, ECO:0000269|PubMed:9554753, ECO:0000269|PubMed:9736778, ECO:0000269|PubMed:9804160, ECO:0000269|PubMed:9921909}. The disease is caused by variants affecting the gene represented in this entry. There is some evidence that the functional defect caused by the most common variant Phe-508 DEL can be corrected by the binding to the snake phospholipase A2 crotoxin basic subunit CB. This toxin both disrupts the Phe-508 DEL-cytokeratin 8 complex, allowing for the escape from degradation, and increases the chloride channel current (PubMed:27241308). {ECO:0000269|PubMed:27241308}.; DISEASE: Congenital bilateral absence of the vas deferens (CBAVD) [MIM:277180]: An autosomal recessive disease characterized by vas deferens aplasia resulting in azoospermia and male infertility. CBAVD may occur in isolation or as a manifestation of cystic fibrosis. {ECO:0000269|PubMed:10066035, ECO:0000269|PubMed:10651488, ECO:0000269|PubMed:17329263, ECO:0000269|PubMed:7529962, ECO:0000269|PubMed:7539342, ECO:0000269|PubMed:9067761, ECO:0000269|PubMed:9736778, ECO:0000269|Ref.117}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07294; DB07031; DB00459; DB00210; DB00523; DB02466; DB03466; DB02741; DB03279; DB00926; DB00982; DB05785; DB05467; DB02258; DB00799; DB00755; DB12808 Interacts with Q96RK4; P13349; P31321; P28702; P48443; O60504-2 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; DNA-binding; Isopeptide bond; Metal-binding; Methylation; Nucleus; Proteomics identification; Receptor; Reference proteome; Transcription; Transcription regulation; Ubl conjugation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A Molecular weight (Da) 26574.9 Length 236 Aromaticity 0.06 Instability index 49.98 Isoelectric point 5.76 Charge (pH=7) -2.95 2D Binding mode Binding energy (Kcal/mol) -8.64  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence ASPQLEELITKVSKAHQETFPSLCQLGKYTTNSSADHRVQLDLGLWDKFSELATKCIIKIVEFAKRLPGFTGLSIADQITLLKAACLDILMLRICTRYTPEQDTMTFSDGLTLNRTQMHNAGFGPLTDLVFAFAGQLLPLEMDDTETGLLSAICLICGDRMDLEEPEKVDKLQEPLLEALRLYARRRRPSQPYMFPRMLMKITDLRGISTKGAERAITLKMEIPGPMPPLIREMLE Hydrogen bonds contact Hydrophobic contact | ||||
| 18 | Aldose reductase (AKR1B1) | 1US0 | 6.33 | |
Target general information Gen name AKR1B1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Aldehyde reductase; AKR1B1 Protein family Aldo/keto reductase family Biochemical class Short-chain dehydrogenases reductase Function Catalyzes the NADPH-dependent reduction of a wide variety of carbonyl-containing compounds to their corresponding alcohols with a broad range of catalytic efficiencies. Related diseases Glutamine deficiency, congenital (GLND) [MIM:610015]: An autosomal recessive disorder characterized by variable brain malformations, encephalopathy, severe developmental delay, seizures, and decreased glutamine levels in bodily fluids. Death in early infancy may occur. {ECO:0000269|PubMed:16267323, ECO:0000269|PubMed:26711351, ECO:0000269|PubMed:38579670}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Developmental and epileptic encephalopathy 116 (DEE116) [MIM:620806]: A form of epileptic encephalopathy, a heterogeneous group of early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE116 is autosomal dominant form characterized by severe developmental delay, seizures, and white matter abnormalities. {ECO:0000269|PubMed:38579670}. The disease is caused by variants affecting the gene represented in this entry. DEE116 is caused by variants that disrupt the canonical translation start codon in GLUL resulting in initiation of translation at Met-18 (PubMed:38579670). The resulting protein is enzymatically competent but insensitive to negative feedback regulation via glutamine-induced degradation. {ECO:0000269|PubMed:38579670}. Drugs (DrugBank ID) DB07028; DB07030; DB07450; DB02101; DB08449; DB08000; DB07139; DB07498; DB02007; DB02020; DB11859; DB02994; DB04272; DB07187; DB00694; DB00997; DB06246; DB01039; DB02021; DB16707; DB00143; DB02834; DB08084; DB01689; DB07063; DB06077; DB02518; DB00157; DB03461; DB05383; DB05533; DB05327; DB02712; DB00605; DB02383; DB02132; DB08772; DB07093; DB07999; DB08098 Interacts with Q9BUY7 EC number EC 1.1.1.300 Uniprot keywords 3D-structure; Acetylation; Cytoplasm; Direct protein sequencing; Lipid metabolism; NADP; Oxidoreductase; Phosphoprotein; Proteomics identification; Reference proteome Protein physicochemical properties Chain ID A Molecular weight (Da) 35447.6 Length 313 Aromaticity 0.09 Instability index 36.41 Isoelectric point 7.1 Charge (pH=7) 0.26 2D Binding mode Binding energy (Kcal/mol) -8.64 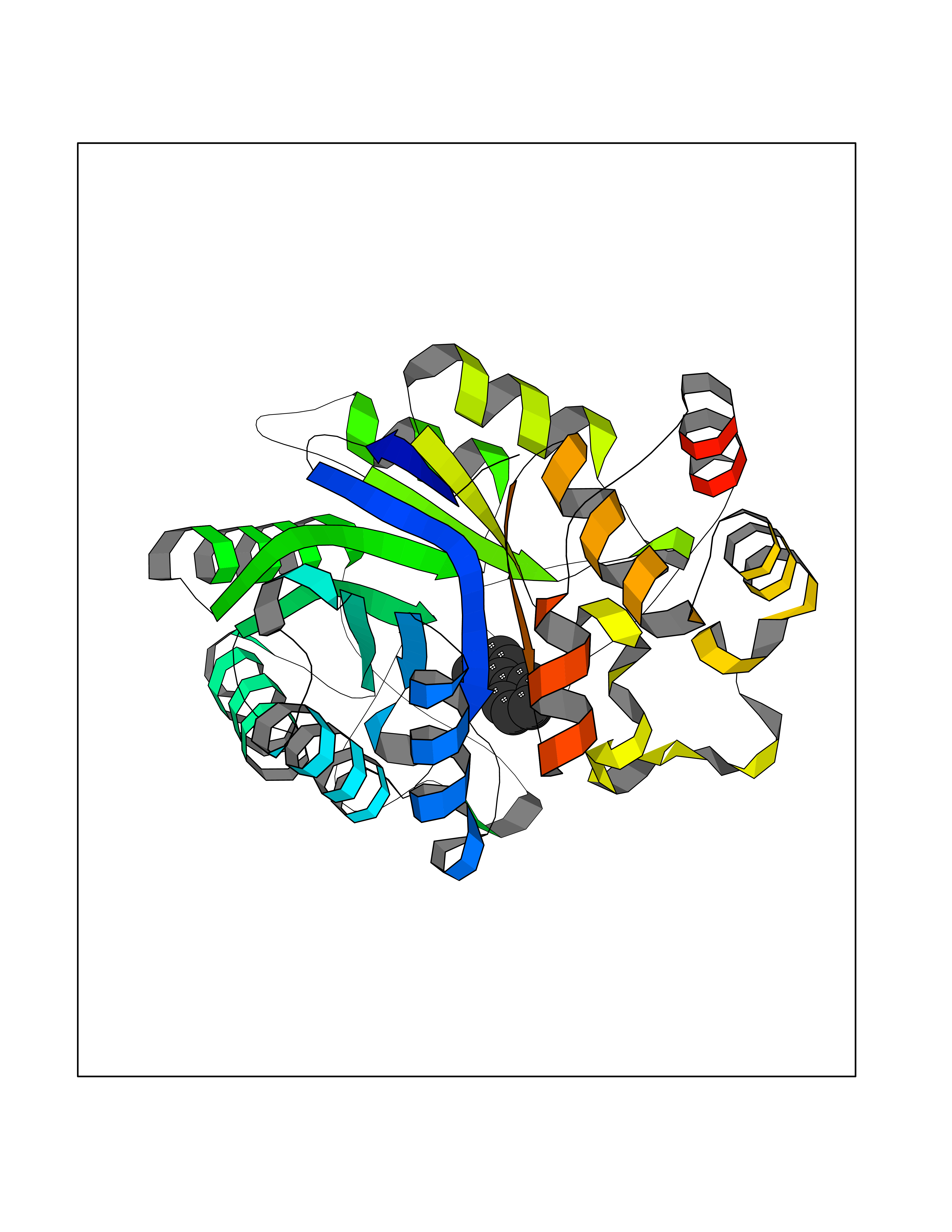 Molscript Map 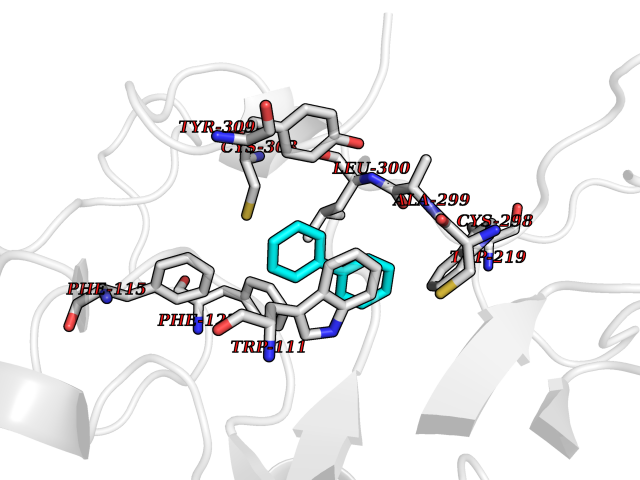 Pymol Map 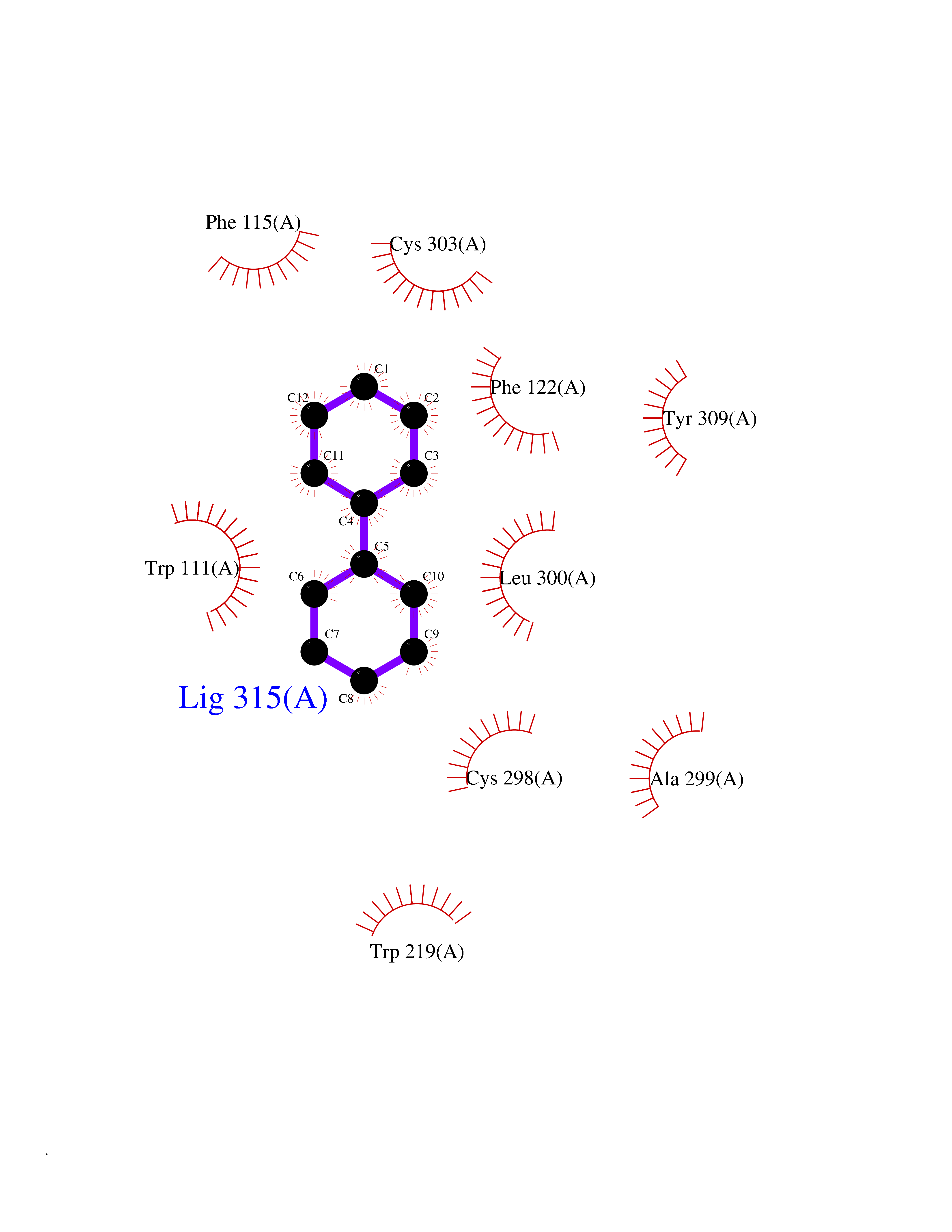 Ligplot Map 3D Binding mode Sequence MASRILLNNGAKMPILGLGTWKSPPGQVTEAVKVAIDVGYRHIDCAHVYQNENEVGVAIQEKLREQVVKREELFIVSKLWCTYHEKGLVKGACQKTLSDLKLDYLDLYLIHWPTGFKPGKEFFPLDESGNVVPSDTNILDTWAAMEELVDEGLVKAIGISNFNHLQVEMILNKPGLKYKPAVNQIECHPYLTQEKLIQYCQSKGIVVTAYSPLGSPDRPWAKPEDPSLLEDPRIKAIAAKHNKTTAQVLIRFPMQRNLVVIPKSVTPERIAENFKVFDFELSSQDMTTLLSYNRNWRVCALLSCTSHKDYPFH Hydrogen bonds contact Hydrophobic contact | ||||
| 19 | Retinoic acid receptor beta (RARB) | 4DM6 | 6.33 | |
Target general information Gen name RARB Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms RAR-epsilon; RAR-beta; Nuclear receptor subfamily 1 group B member 2; NR1B2; HBV-activated protein; HAP Protein family Nuclear hormone receptor family, NR1 subfamily Biochemical class Nuclear hormone receptor Function Retinoic acid receptors bind as heterodimers to their target response elements in response to their ligands, all-trans or 9-cis retinoic acid, and regulate gene expression in various biological processes. The RXR/RAR heterodimers bind to the retinoic acid response elements (RARE) composed of tandem 5'-AGGTCA-3' sites known as DR1-DR5. In the absence or presence of hormone ligand, acts mainly as an activator of gene expression due to weak binding to corepressors. In concert with RARG, required for skeletal growth, matrix homeostasis and growth plate function. Receptor for retinoic acid. Related diseases Microphthalmia, syndromic, 12 (MCOPS12) [MIM:615524]: A form of microphthalmia, a disorder of eye formation, ranging from small size of a single eye to complete bilateral absence of ocular tissues (anophthalmia). In many cases, microphthalmia/anophthalmia occurs in association with syndromes that include non-ocular abnormalities. MCOPS12 patients manifest variable features, including diaphragmatic hernia, pulmonary hypoplasia, and cardiac abnormalities. {ECO:0000269|PubMed:24075189, ECO:0000269|PubMed:27120018}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00459; DB00210; DB00523; DB02877; DB00926; DB05785; DB04942; DB00799; DB00755; DB12808 Interacts with O95273; P50222; Q9UBK2; P62195; P28702; P28702-3; P48443; P03255 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; Disease variant; DNA-binding; Metal-binding; Microphthalmia; Nucleus; Phosphoprotein; Proto-oncogene; Receptor; Reference proteome; Transcription; Transcription regulation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A,B Molecular weight (Da) 25904.1 Length 229 Aromaticity 0.06 Instability index 44.34 Isoelectric point 7.55 Charge (pH=7) 0.73 2D Binding mode Binding energy (Kcal/mol) -8.64 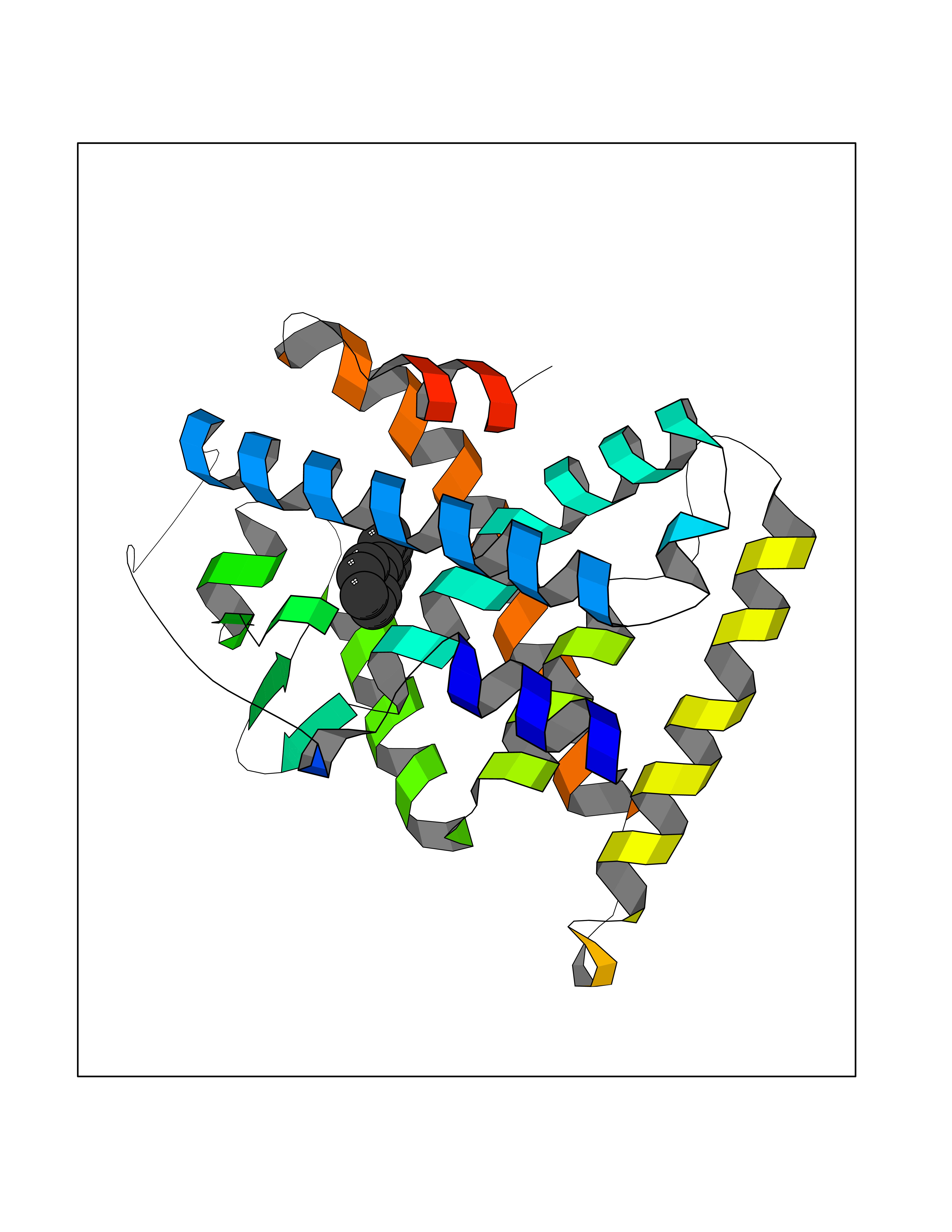 Molscript Map 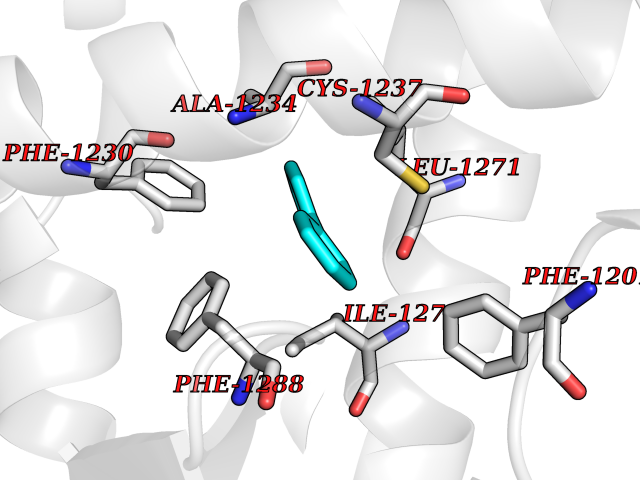 Pymol Map 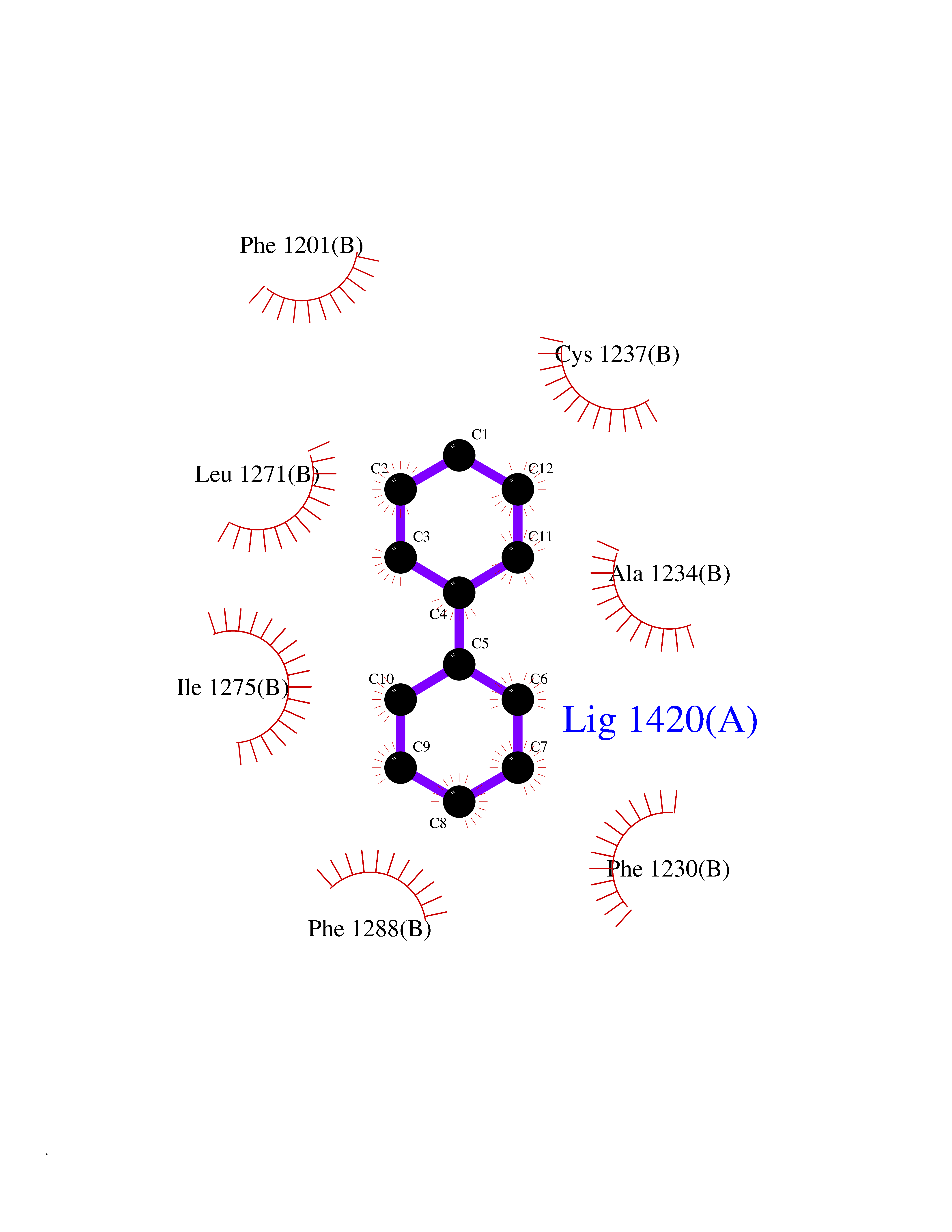 Ligplot Map 3D Binding mode Sequence TEKIRKAHQETFPSLCQLGKYTTNSSADHRVRLDLGLWDKFSELATKCIIKIVEFAKRLPGFTGLTIADQITLLKAACLDILILRICTRYTPEQDTMTFSDGLTLNRTQMHNAGFGPLTDLVFTFANQLLPLEMDDTETGLLSAICLICGDRQDLEEPTKVDKLQEPLLEALKIYIRKRRPSKPHMFPKILMKITDLRSISAKGAERVITLKMEIPGSMPPLIQEMLEN Hydrogen bonds contact Hydrophobic contact | ||||
| 20 | Wnt-7a protein (WNT7A) | 4UZQ | 6.33 | |
Target general information Gen name WNT7A Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Protein Wnt-7a Protein family Wnt family Biochemical class NA Function Plays an important role in embryonic development, including dorsal versus ventral patterning during limb development, skeleton development and urogenital tract development. Required for central nervous system (CNS) angiogenesis and blood-brain barrier regulation. Required for normal, sexually dimorphic development of the Mullerian ducts, and for normal fertility in both sexes. Required for normal neural stem cell proliferation in the hippocampus dentate gyrus. Required for normal progress through the cell cycle in neural progenitor cells, for self-renewal of neural stem cells, and for normal neuronal differentiation and maturation. Promotes formation of synapses via its interaction with FZD5. Ligand for members of the frizzled family of seven transmembrane receptors that functions in the canonical Wnt/beta-catenin signaling pathway. Related diseases Limb pelvis hypoplasia aplasia syndrome (LPHAS) [MIM:276820]: A syndrome of severe deficiency of the extremities due to hypo- or aplasia of one or more long bones of one or more limbs. Pelvic manifestations include hip dislocation, hypoplastic iliac bone and aplastic pubic bones. Thoracic deformity, unusual facies and genitourinary anomalies can be present. {ECO:0000269|PubMed:16826533, ECO:0000269|PubMed:17431918, ECO:0000269|PubMed:20949531, ECO:0000269|PubMed:21271649, ECO:0000269|PubMed:27638328}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Fuhrmann syndrome (FUHRS) [MIM:228930]: Distinct limb-malformation disorder characterized also by various degrees of limb aplasia/hypoplasia and joint dysplasia. {ECO:0000269|PubMed:16826533}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P55212; P22607; P06396; P13473-2; Q9UMX0; Q9Y5W5; Q5T9L3; Q9Z0J1 EC number NA Uniprot keywords 3D-structure; Developmental protein; Disease variant; Disulfide bond; Extracellular matrix; Glycoprotein; Lipoprotein; Proteomics identification; Reference proteome; Secreted; Signal; Wnt signaling pathway Protein physicochemical properties Chain ID A,B Molecular weight (Da) 40475.5 Length 356 Aromaticity 0.1 Instability index 50.49 Isoelectric point 7.67 Charge (pH=7) 1.62 2D Binding mode Binding energy (Kcal/mol) -8.64  Molscript Map 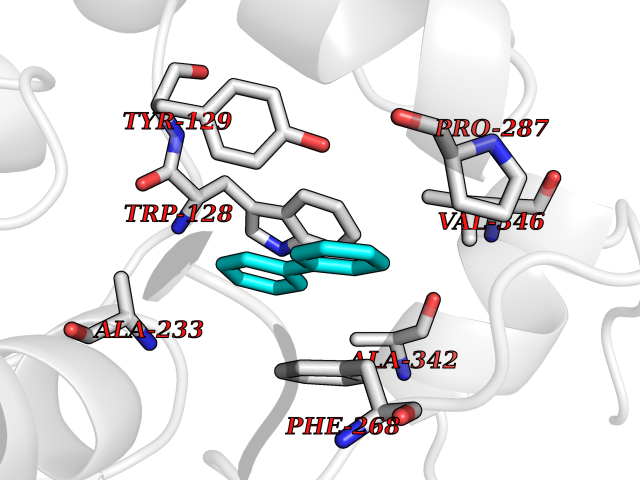 Pymol Map 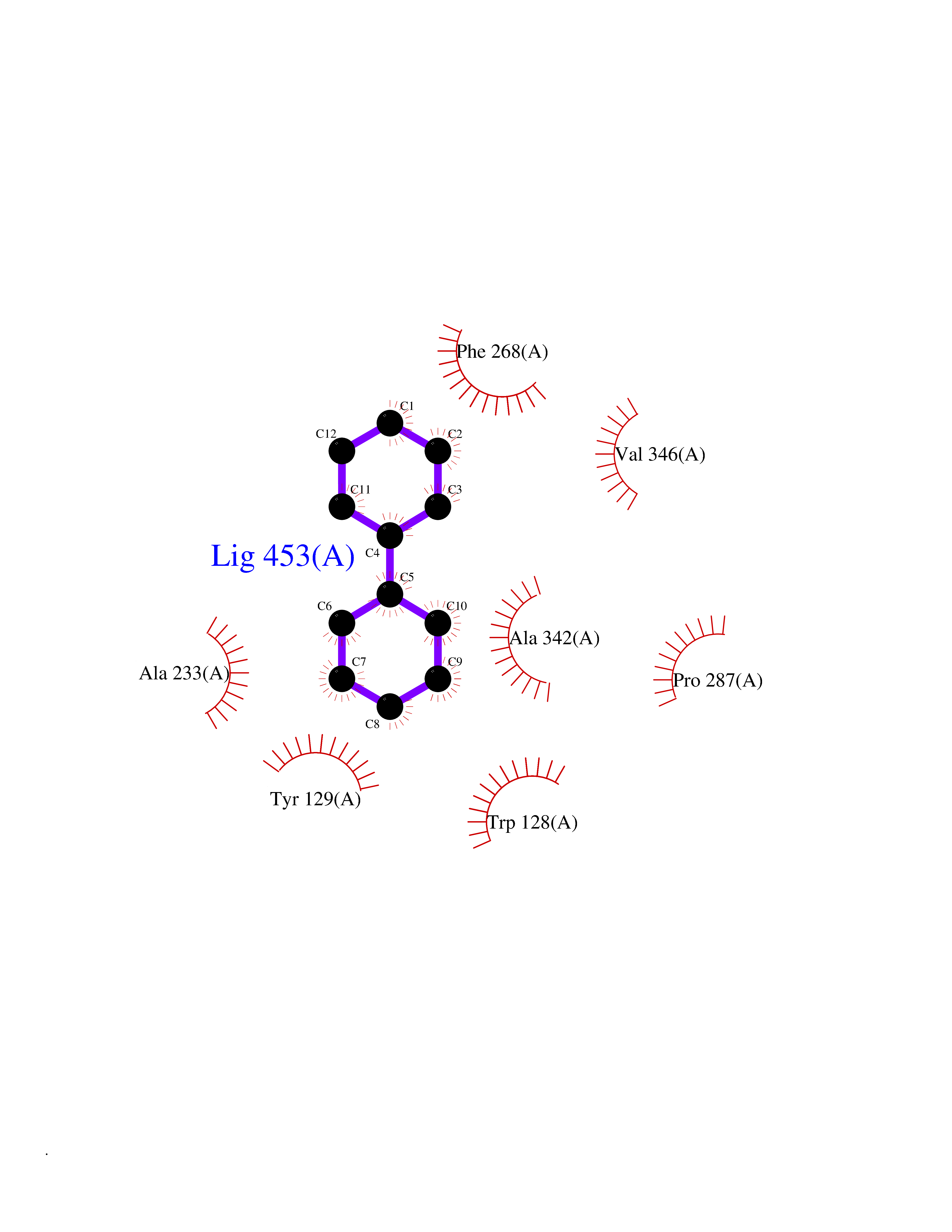 Ligplot Map 3D Binding mode Sequence EDLRLHLLLNTSVTCNDGSPAGYYLKESRGSRRWLLFLEGGWYCFNRENCDSRYDTMRRLMSSRDWPRTRTGTGILSSQPEENPYWWNANMVFIPYCSSDVWSGASSKSEKNEYAFMGALIIQEVVRELLGRGLSGAKVLLLAGSAAGGTGVLLNVDRVAEQLEKLGYPAIQVRGLADSGWFLDNKQYRHTDCVDTITCAPTEAIRRGIRYWNGVVPERCRRQFQEGEEWNCFFGYKVYPTLRSPVFVVQWLFDEAQLTVDNVHLTGQPVQEGLRLYIQNLGRELRHTLKDVPASFAPACLSHEIIIRSHWTDVQVKGTSLPRALHCWDRSLHKGCPVHLVDSCPWPHCNPSCPTS Hydrogen bonds contact Hydrophobic contact | ||||