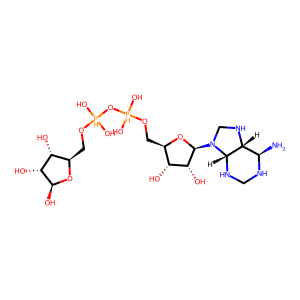
Ligand
Structure
Job ID
0675052c5ef38fae220c9665a1631b66
Job name
Wu_assess10
Time
2024-10-25 06:02:41
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 1 | Suppressor of tumorigenicity 14 protein (ST14) | 3P8G | 6.87 | |
Target general information Gen name ST14 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Tumor-associated differentially-expressed gene 15 protein; Tumor associated differentially-expressed gene-15 protein; TADG15; Serine protease TADG-15; Serine protease 14; SNC19; Prostamin; PRSS14; Mem Protein family Peptidase S1 family Biochemical class Peptidase Function Proposed to play a role in breast cancer invasion and metastasis. Exhibits trypsin-like activity as defined by cleavage of synthetic substrates with Arg or Lys as the P1 site. Involved in the terminal differentiation of keratinocytes through prostasin (PRSS8) activation and filaggrin (FLG) processing. Degrades extracellular matrix. Related diseases Ichthyosis, congenital, autosomal recessive 11 (ARCI11) [MIM:602400]: A form of autosomal recessive congenital ichthyosis, a disorder of keratinization with abnormal differentiation and desquamation of the epidermis, resulting in abnormal skin scaling over the whole body. The main skin phenotypes are lamellar ichthyosis (LI) and non-bullous congenital ichthyosiform erythroderma (NCIE), although phenotypic overlap within the same patient or among patients from the same family can occur. Lamellar ichthyosis is a condition often associated with an embedment in a collodion-like membrane at birth; skin scales later develop, covering the entire body surface. Non-bullous congenital ichthyosiform erythroderma characterized by fine whitish scaling on an erythrodermal background; larger brownish scales are present on the buttocks, neck and legs. {ECO:0000269|PubMed:17273967, ECO:0000269|PubMed:18843291}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03127; DB13729; DB00013 Interacts with NA EC number EC 3.4.21.109 Uniprot keywords 3D-structure; Disease variant; Disulfide bond; Glycoprotein; Hydrolase; Hypotrichosis; Ichthyosis; Membrane; Protease; Proteomics identification; Reference proteome; Repeat; Serine protease; Signal-anchor; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 26451.5 Length 241 Aromaticity 0.1 Instability index 30.45 Isoelectric point 5.6 Charge (pH=7) -5.69 2D Binding mode Binding energy (Kcal/mol) -7.06 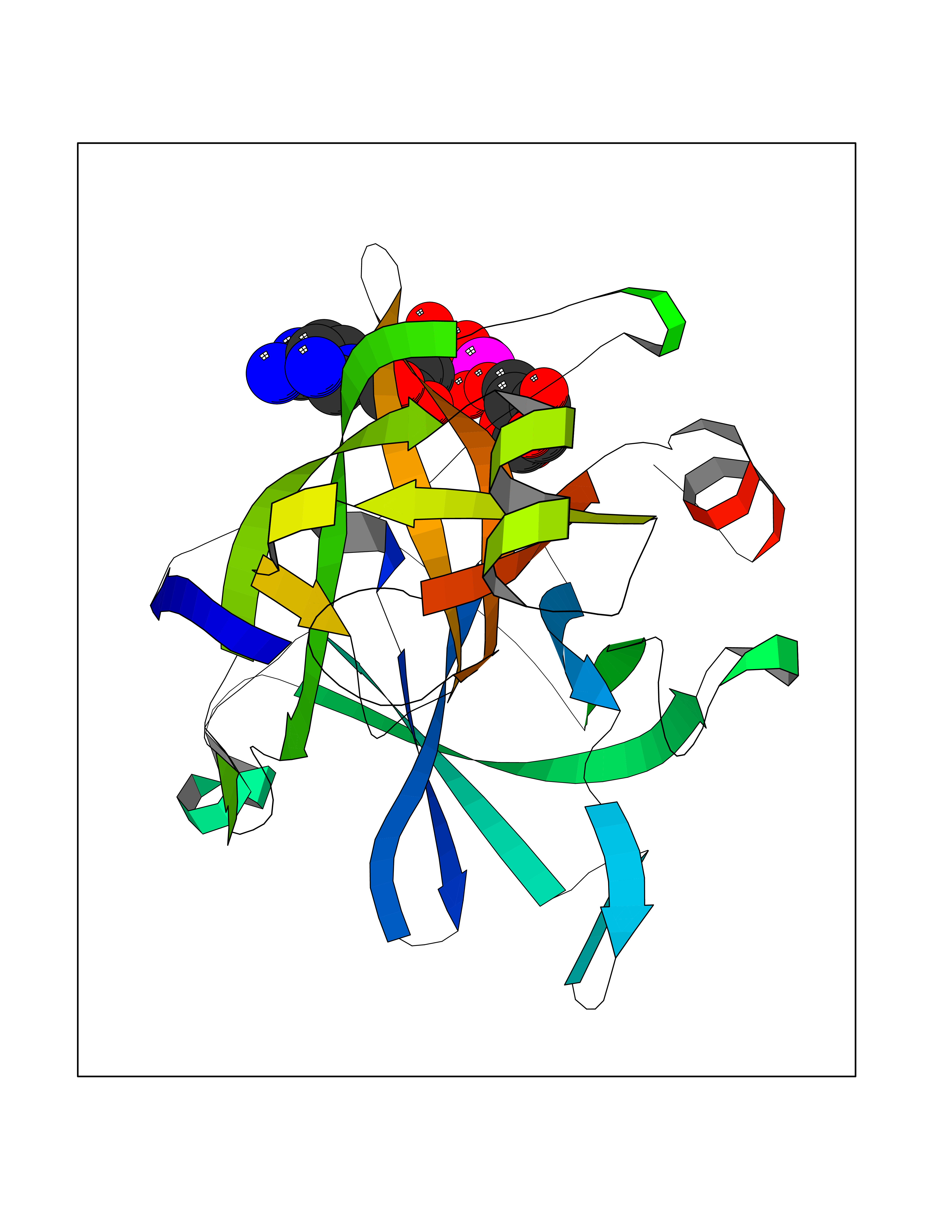 Molscript Map 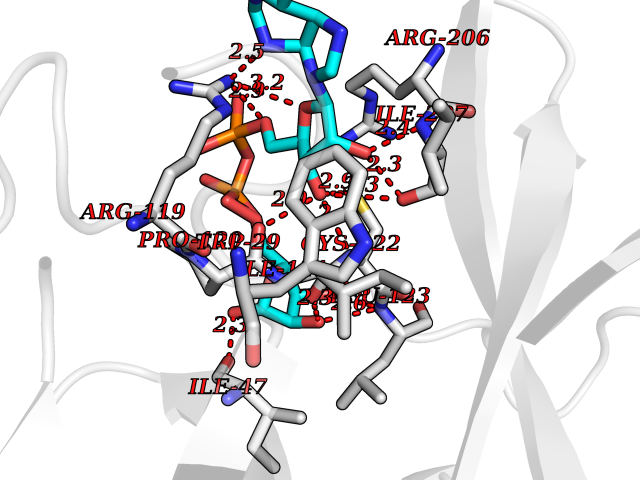 Pymol Map 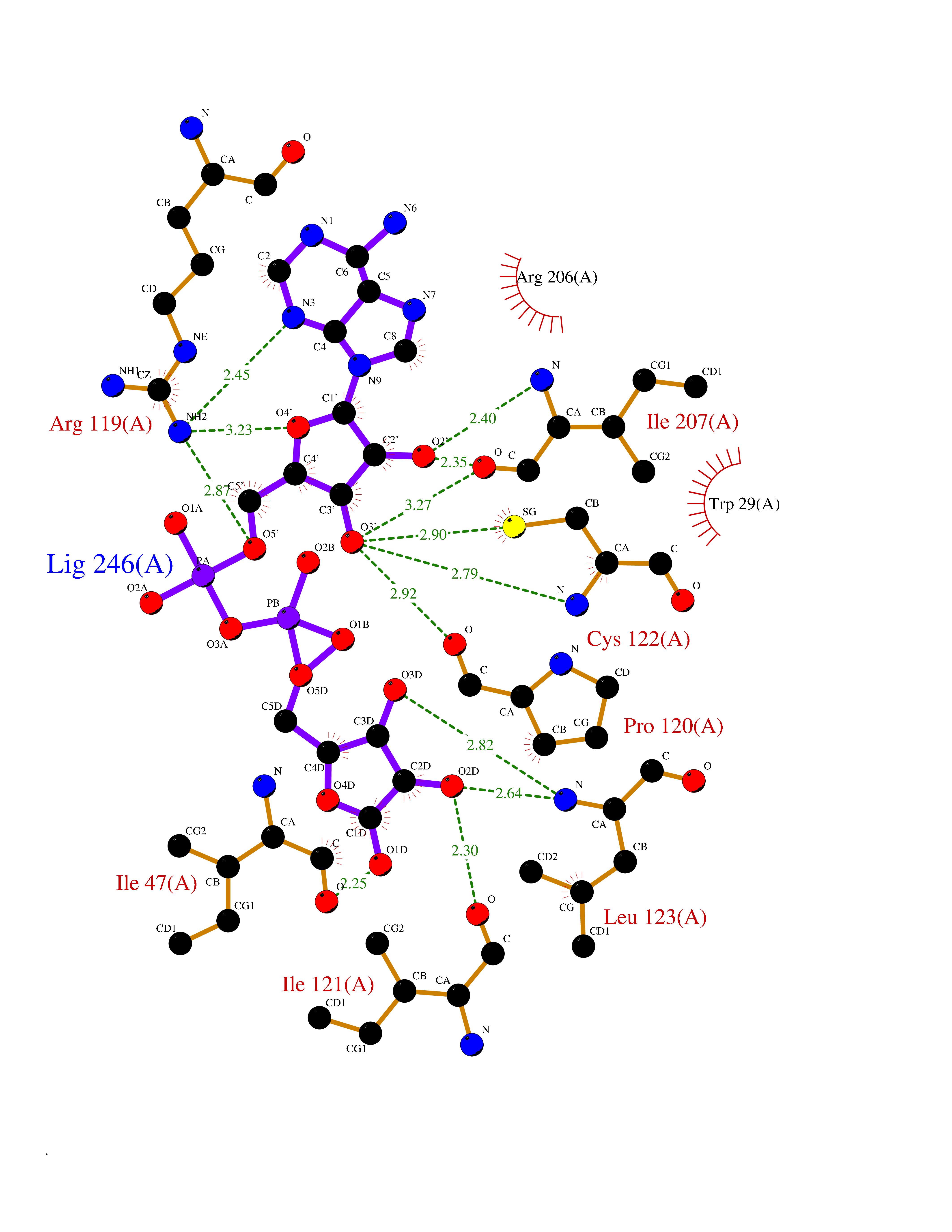 Ligplot Map 3D Binding mode Sequence VVGGTDADEGEWPWQVSLHALGQGHICGASLISPNWLVSAAHCYIDDRGFRYSDPTQWTAFLGLHDQSQRSAPGVQERRLKRIISHPFFNDFTFDYDIALLELEKPAEYSSMVRPICLPDASHVFPAGKAIWVTGWGHTQYGGTGALILQKGEIRVIQQTTCENLLPQQITPRMMCVGFLSGGVDSCQGDSGGPLSSVEADGRIFQAGVVSWGDGCAQRNKPGVYTRLPLFRDWIKENTGV Hydrogen bonds contact Hydrophobic contact | ||||
| 2 | Fibrinogen beta chain | 1FZB | 6.69 | |
Target general information Gen name FGB Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family NA Biochemical class Blood coagulation Function Chaperone binding.Protein binding, bridging.Receptor binding.Structural molecule activity. Related diseases Congenital afibrinogenemia (CAFBN) [MIM:202400]: Rare autosomal recessive disorder is characterized by bleeding that varies from mild to severe and by complete absence or extremely low levels of plasma and platelet fibrinogen. {ECO:0000269|PubMed:10666208, ECO:0000269|PubMed:11468164, ECO:0000269|PubMed:15070683, ECO:0000269|PubMed:25427968}. The disease is caused by variants affecting the gene represented in this entry. Patients with congenital fibrinogen abnormalities can manifest different clinical pictures. Some cases are clinically silent, some show a tendency toward bleeding and some show a predisposition for thrombosis with or without bleeding.; DISEASE: Dysfibrinogenemia, congenital (DYSFIBRIN) [MIM:616004]: A disorder characterized by qualitative abnormalities (dysfibrinogenemia) of the circulating fibrinogen. Affected individuals are frequently asymptomatic, but some patients have bleeding diathesis, thromboembolic complications, or both. In some cases, dysfibrinogenemia is associated with low circulating fibrinogen levels (hypodysfibrinogenemia). {ECO:0000269|PubMed:1634610}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB04919; DB13151; DB11571; DB11311; DB00364; DB11300; DB11572 Interacts with PRO_0000037566 [P27958] EC number NA Uniprot keywords 3D-structure; Adaptive immunity; Blood coagulation; Coiled coil; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; Hemostasis; Immunity; Innate immunity; Proteomics identification; Pyrrolidone carboxylic acid; Reference proteome; Secreted; Signal Protein physicochemical properties Chain ID B,E Molecular weight (Da) 29738.9 Length 260 Aromaticity 0.13 Instability index 32.58 Isoelectric point 6.87 Charge (pH=7) -0.23 2D Binding mode Binding energy (Kcal/mol) -6.38 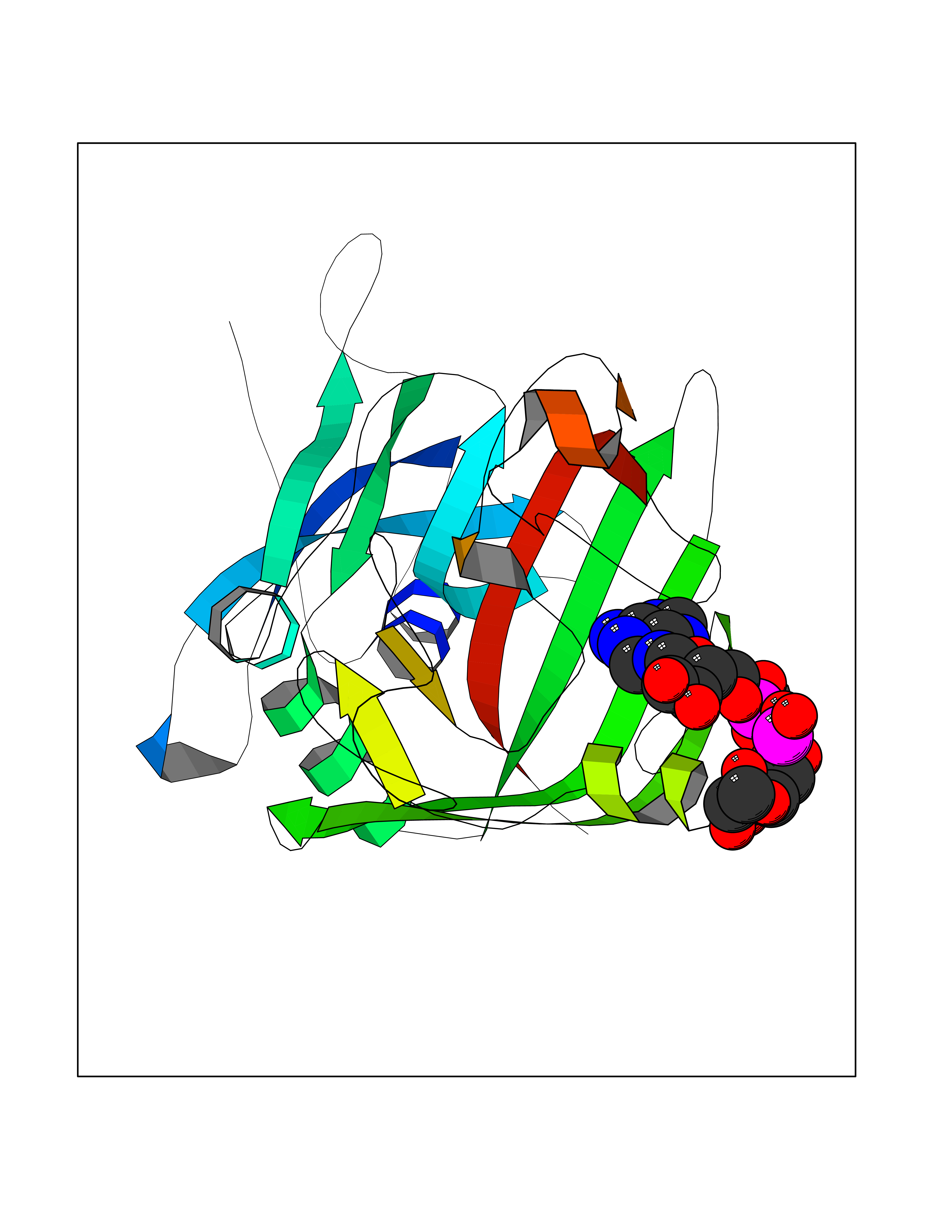 Molscript Map 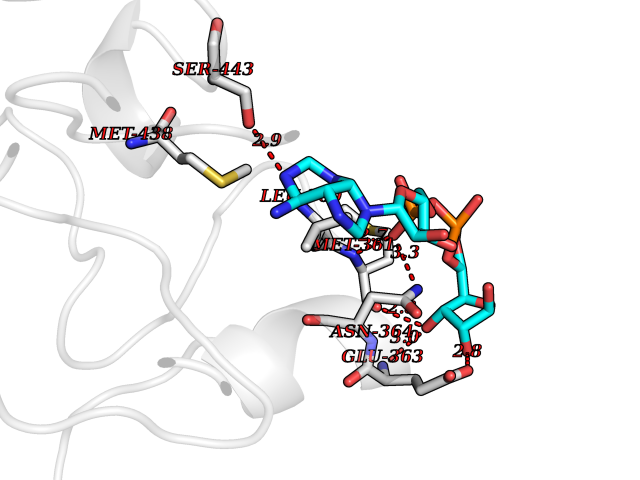 Pymol Map 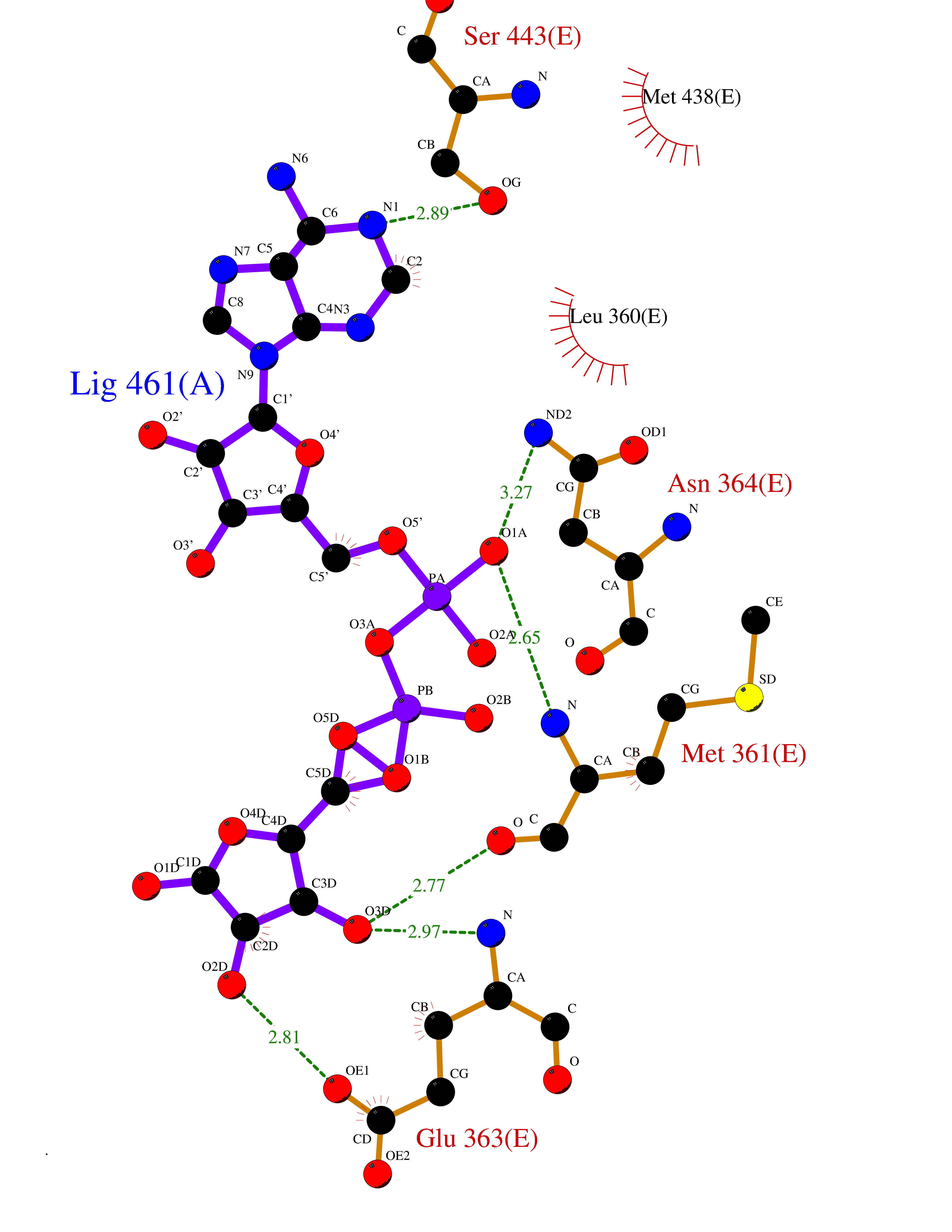 Ligplot Map 3D Binding mode Sequence SCNIPVVSGKECEEIIRKGGETSEMYLIQPDSSVKPYRVYCDMNTENGGWTVIQNRQDGSVDFGRKWDPYKQGFGNVATNTDGKNYCGLPGEYWLGNDKISQLTRMGPTELLIEMEDWKGDKVKAHYGGFTVQNEANKYQISVNKYRGTAGNALMDGASQLMGENRTMTIHNGMFFSTYDRDNDGWLTSDPRKQCSKEDGGGWWYNRCHAANPNGRYYWGGQYTWDMAKHGTDDGVVWMNWKGSWYSMRKMSMKIRPFFP Hydrogen bonds contact Hydrophobic contact | ||||
| 3 | Probable glutathione peroxidase 8 | 3KIJ | 6.67 | |
Target general information Gen name GPX8 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms UNQ847/PRO1785 Protein family Glutathione peroxidase family Biochemical class Oxidoreductase Function Glutathione peroxidase activity.Peroxidase activity. Related diseases Neurodevelopmental disorder with spastic paraplegia and microcephaly (NEDSPM) [MIM:616281]: An autosomal recessive syndrome characterized by severe psychomotor developmental delay, dysarthria, walking difficulties, moderately to severely impaired intellectual development, poor or absent speech, and progressive microcephaly. {ECO:0000269|PubMed:25758935}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB09096; DB00143; DB03310 Interacts with Q6RW13-2; Q9NVV5-2; Q9BVK2; P02656; P05090; P29972; P41181; Q92482; P07306; Q12797-6; Q92843; O15155; Q13323; O95393; Q12982; Q8WVV5; Q06432; Q8IX05; P19397; P60033; O14735; O95674; Q9BXR6; O43916; Q8N6F1-2; Q8NC01; Q6UVW9; Q96DZ9-2; O95406; Q8TBE1; P29400-2; Q4VAQ0; Q9Y5Q5; P49447; Q8NBI2; O14569; Q96LL9; Q9UPQ8; P56851; Q9UKR5; Q7L5A8; Q92520; Q96IV6; O14556; O43681; O14653; Q8TDV0; O60883; Q7Z429; P02724; Q9HCP6; P30519; P24593; Q9Y5U4; P43628; Q96E93; Q86VI4; O95214; Q8TAF8; Q9UIQ6-2; Q9UBY5; Q9Y2E5; Q9P0N8; Q9NX47; Q6N075; Q6ZSS7; Q99735; O14880; P30301; Q15546; A6NDP7; Q99519; Q92982; Q6P499; Q16617; Q8N912; Q8IXM6; Q16625; P09466; Q9NXK6; Q6TCH4; Q9UHJ9-5; Q9Y342; P26678; Q04941; Q5VZY2; Q8IY26; P54315; A5D903; Q8N0V3; Q92730; Q5QGT7; Q14108; Q14162; O00767; O75396; Q9Y6D0; Q9NRX5; Q9Y6X1; A2A2V5; P11686; Q8NHU3; Q8WWT9; Q99726; Q8N130; P78382; Q969S0; Q96JW4; Q0VAQ4; Q6UX34; Q96JF0-2; Q13277; Q9UNK0; Q9BQS2-2; P02787; P07204; Q9NPL8; P48230; P55061; Q6UX40; Q9BVC6; A0PK00; Q5BJH2-2; Q9NUH8; Q96HH6; A2RU14; Q8NBD8; Q9BU79; Q8TBM7; Q69YG0; P56557; Q9H2L4; Q8N661; Q5BJF2; Q9NRS4; Q71RG4; Q8N609; Q86UF1; Q53HI1; O75841; Q15836; O75379; O95183; Q8N0U8; Q6UX27-3; Q9UEU0; O95070; Q96EC8; Q8N966 EC number 1.11.1.9 Uniprot keywords 3D-structure; Acetylation; Membrane; Oxidoreductase; Peroxidase; Proteomics identification; Reference proteome; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A,B,C Molecular weight (Da) 18580.2 Length 161 Aromaticity 0.13 Instability index 38.6 Isoelectric point 9.39 Charge (pH=7) 6.73 2D Binding mode Binding energy (Kcal/mol) -6.67 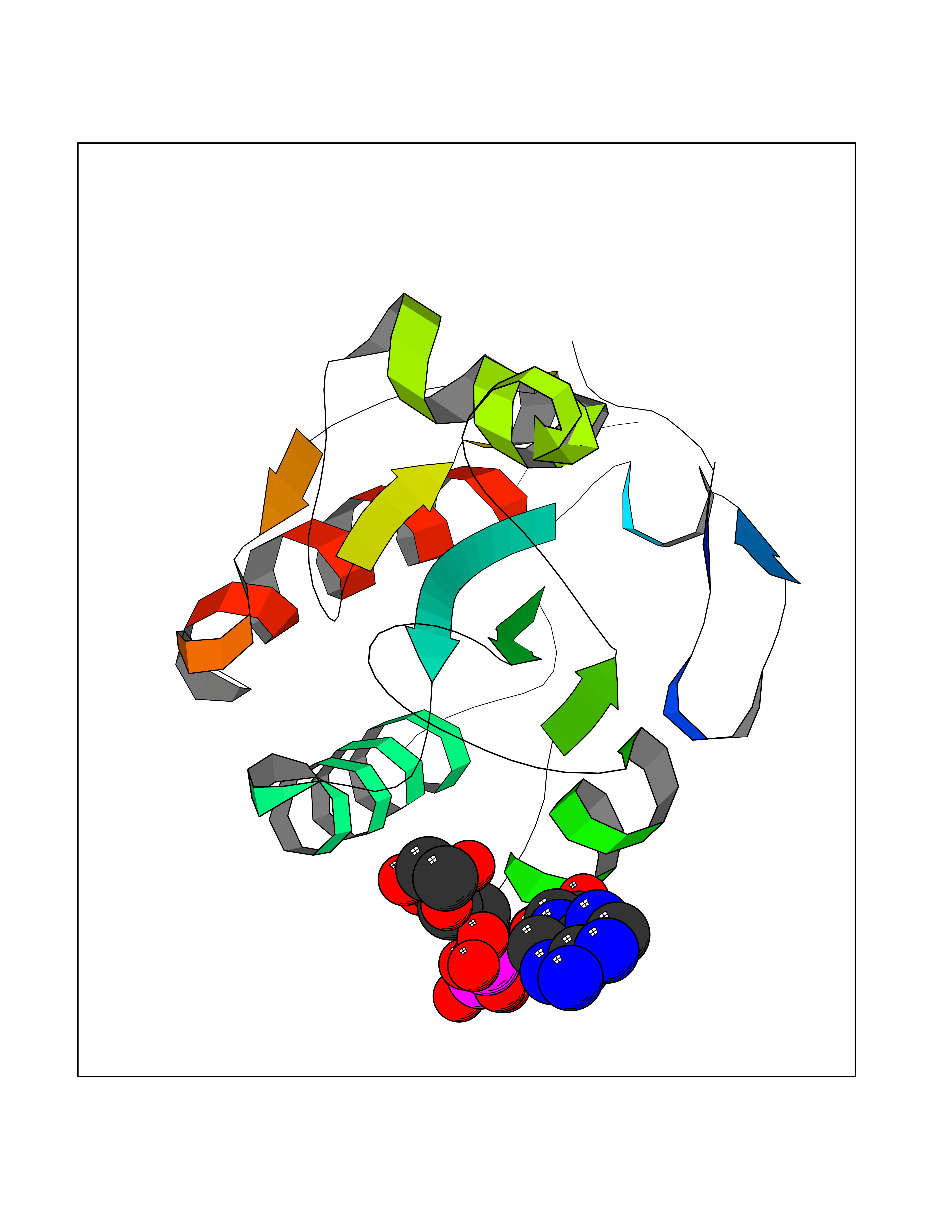 Molscript Map 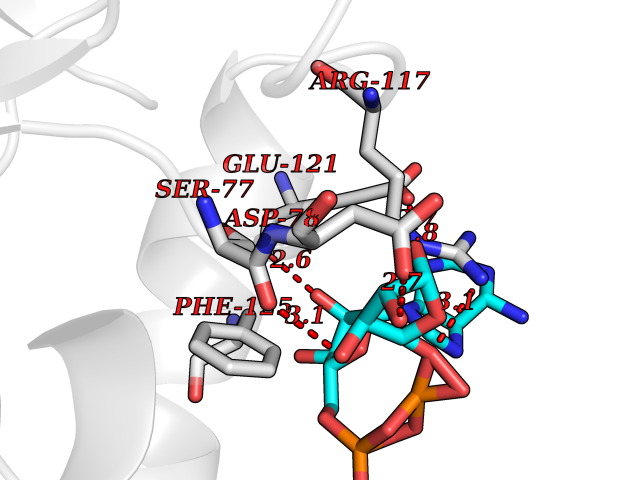 Pymol Map 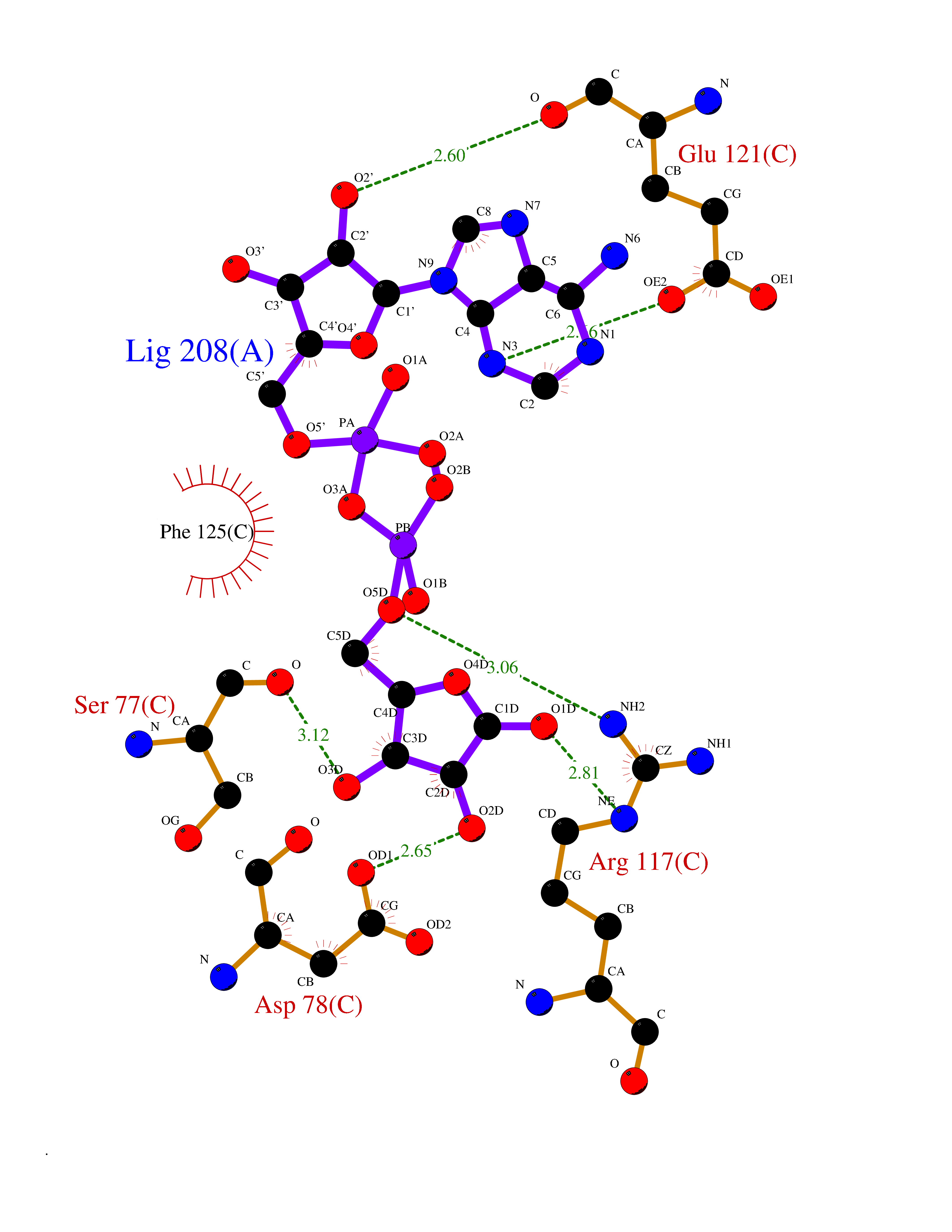 Ligplot Map 3D Binding mode Sequence SFYAFEVKDAKGRTVSLEKYKGKVSLVVNVASDCQLTDRNYLGLKELHKEFGPSHFSVLAFPCNQFGESEPRPSKEVESFARKNYGVTFPIFHKIKILGSEGEPAFRFLVDSSKKEPRWNFWKYLVNPEGQVVKFWRPEEPIEVIRPDIAALVRQVIIKKK Hydrogen bonds contact Hydrophobic contact | ||||
| 4 | Suppressor of tumorigenicity 14 protein (ST14) | 3P8G | 6.66 | |
Target general information Gen name ST14 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Tumor-associated differentially-expressed gene 15 protein; Tumor associated differentially-expressed gene-15 protein; TADG15; Serine protease TADG-15; Serine protease 14; SNC19; Prostamin; PRSS14; Mem Protein family Peptidase S1 family Biochemical class Peptidase Function Proposed to play a role in breast cancer invasion and metastasis. Exhibits trypsin-like activity as defined by cleavage of synthetic substrates with Arg or Lys as the P1 site. Involved in the terminal differentiation of keratinocytes through prostasin (PRSS8) activation and filaggrin (FLG) processing. Degrades extracellular matrix. Related diseases Ichthyosis, congenital, autosomal recessive 11 (ARCI11) [MIM:602400]: A form of autosomal recessive congenital ichthyosis, a disorder of keratinization with abnormal differentiation and desquamation of the epidermis, resulting in abnormal skin scaling over the whole body. The main skin phenotypes are lamellar ichthyosis (LI) and non-bullous congenital ichthyosiform erythroderma (NCIE), although phenotypic overlap within the same patient or among patients from the same family can occur. Lamellar ichthyosis is a condition often associated with an embedment in a collodion-like membrane at birth; skin scales later develop, covering the entire body surface. Non-bullous congenital ichthyosiform erythroderma characterized by fine whitish scaling on an erythrodermal background; larger brownish scales are present on the buttocks, neck and legs. {ECO:0000269|PubMed:17273967, ECO:0000269|PubMed:18843291}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03127; DB13729; DB00013 Interacts with NA EC number EC 3.4.21.109 Uniprot keywords 3D-structure; Disease variant; Disulfide bond; Glycoprotein; Hydrolase; Hypotrichosis; Ichthyosis; Membrane; Protease; Proteomics identification; Reference proteome; Repeat; Serine protease; Signal-anchor; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 26451.5 Length 241 Aromaticity 0.1 Instability index 30.45 Isoelectric point 5.6 Charge (pH=7) -5.69 2D Binding mode Binding energy (Kcal/mol) -7.85 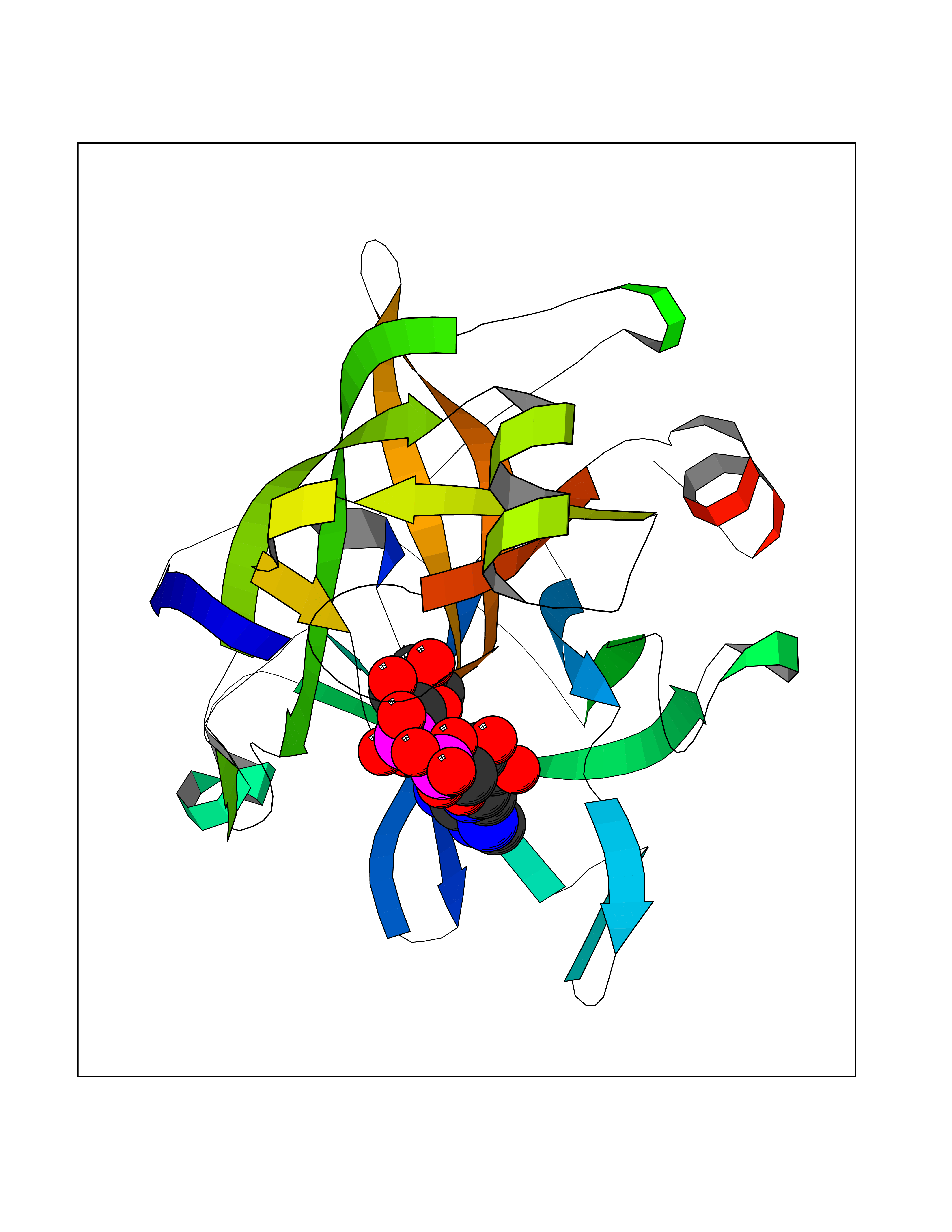 Molscript Map 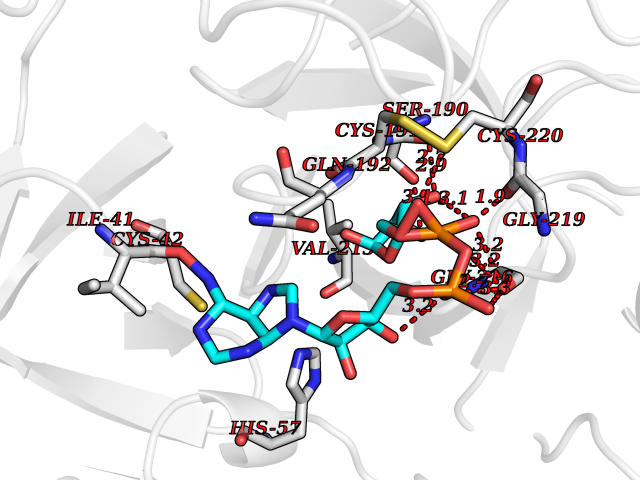 Pymol Map 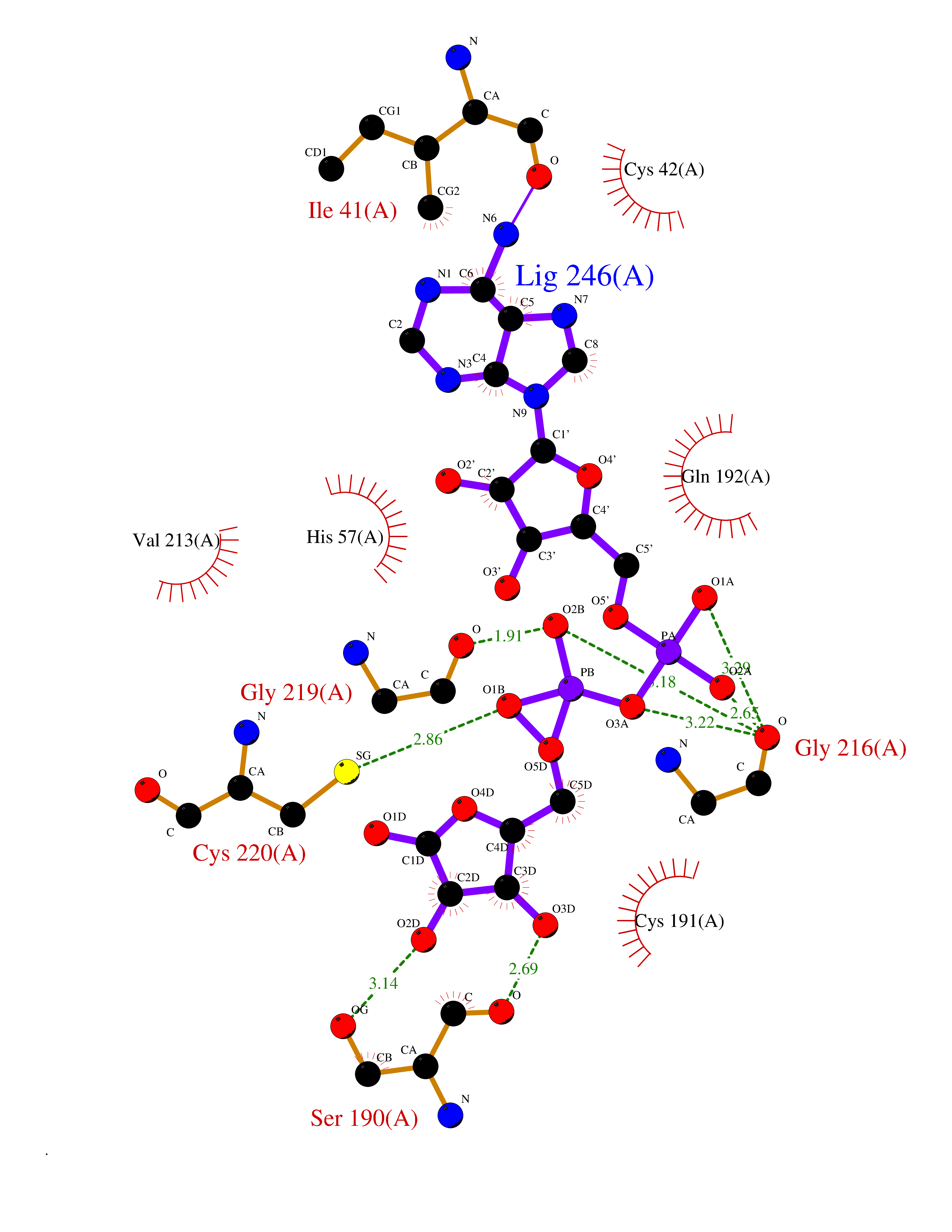 Ligplot Map 3D Binding mode Sequence VVGGTDADEGEWPWQVSLHALGQGHICGASLISPNWLVSAAHCYIDDRGFRYSDPTQWTAFLGLHDQSQRSAPGVQERRLKRIISHPFFNDFTFDYDIALLELEKPAEYSSMVRPICLPDASHVFPAGKAIWVTGWGHTQYGGTGALILQKGEIRVIQQTTCENLLPQQITPRMMCVGFLSGGVDSCQGDSGGPLSSVEADGRIFQAGVVSWGDGCAQRNKPGVYTRLPLFRDWIKENTGV Hydrogen bonds contact Hydrophobic contact | ||||
| 5 | Prothrombin | 4UD9 | 6.66 | |
Target general information Gen name F2 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Peptidase S1 family Biochemical class Hydrolase Function Calcium ion binding.Growth factor activity.Heparin binding.Lipopolysaccharide binding.Receptor binding.Serine-type endopeptidase activity.Thrombospondin receptor activity. Related diseases Factor II deficiency (FA2D) [MIM:613679]: A very rare blood coagulation disorder characterized by mucocutaneous bleeding symptoms. The severity of the bleeding manifestations correlates with blood factor II levels. {ECO:0000269|PubMed:1349838, ECO:0000269|PubMed:1354985, ECO:0000269|PubMed:1421398, ECO:0000269|PubMed:14962227, ECO:0000269|PubMed:2719946, ECO:0000269|PubMed:3242619, ECO:0000269|PubMed:3567158, ECO:0000269|PubMed:3771562, ECO:0000269|PubMed:3801671, ECO:0000269|PubMed:6405779, ECO:0000269|PubMed:7792730, ECO:0000269|PubMed:7865694}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Ischemic stroke (ISCHSTR) [MIM:601367]: A stroke is an acute neurologic event leading to death of neural tissue of the brain and resulting in loss of motor, sensory and/or cognitive function. Ischemic strokes, resulting from vascular occlusion, is considered to be a highly complex disease consisting of a group of heterogeneous disorders with multiple genetic and environmental risk factors. {ECO:0000269|PubMed:15534175}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Thrombophilia due to thrombin defect (THPH1) [MIM:188050]: A multifactorial disorder of hemostasis characterized by abnormal platelet aggregation in response to various agents and recurrent thrombi formation. {ECO:0000269|PubMed:2825773}. The disease is caused by variants affecting the gene represented in this entry. A common genetic variation in the 3-prime untranslated region of the prothrombin gene is associated with elevated plasma prothrombin levels and an increased risk of venous thrombosis.; DISEASE: Pregnancy loss, recurrent, 2 (RPRGL2) [MIM:614390]: A common complication of pregnancy, resulting in spontaneous abortion before the fetus has reached viability. The term includes all miscarriages from the time of conception until 24 weeks of gestation. Recurrent pregnancy loss is defined as 3 or more consecutive spontaneous abortions. {ECO:0000269|PubMed:11506076}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07211; DB07796; DB07016; DB07521; DB06850; DB07091; DB06845; DB07088; DB07131; DB07095; DB07515; DB07897; DB06878; DB06947; DB08624; DB06869; DB06929; DB07400; DB04771; DB04772; DB02287; DB07277; DB07550; DB07549; DB07548; DB07105; DB04722; DB07366; DB08254; DB01725; DB08062; DB07639; DB07461; DB07120; DB07190; DB07741; DB07353; DB07508; DB07809; DB08546; DB08061; DB07718; DB03136; DB02723; DB07440; DB07376; DB06861; DB06866; DB06865; DB03865; DB06841; DB07934; DB08422; DB07659; DB07660; DB07658; DB13151; DB00025; DB11166; DB00278; DB01766; DB07083; DB00006; DB00100; DB13152; DB09228; DB09130; DB03159; DB06911; DB06996; DB06919; DB07027; DB07133; DB07143; DB07005; DB06695; DB00055; DB01225; DB05714; DB12831; DB03847; DB07278; DB01767; DB06404; DB09332; DB00001; DB13998; DB04136; DB00170; DB06838; DB13999; DB06868; DB06942; DB06936; DB07165; DB07527; DB07522; DB07665; DB07946; DB06859; DB06853; DB06858; DB07279; DB08187; DB04591; DB07944; DB07128; DB12598; DB01123; DB04786; DB05777; DB04697; DB09109; DB14738; DB04898; DB01593; DB14487; DB08152 Interacts with P05067; P07204; Q846V4; PRO_0000032489 [P01008] EC number 3.4.21.5 Uniprot keywords 3D-structure; Acute phase; Blood coagulation; Calcium; Cleavage on pair of basic residues; Direct protein sequencing; Disease variant; Disulfide bond; Gamma-carboxyglutamic acid; Glycoprotein; Hemostasis; Hydrolase; Kringle; Pharmaceutical; Protease; Proteomics identification; Reference proteome; Repeat; Secreted; Serine protease; Signal; Thrombophilia; Zymogen Protein physicochemical properties Chain ID H Molecular weight (Da) 29321.6 Length 254 Aromaticity 0.1 Instability index 39.57 Isoelectric point 8.56 Charge (pH=7) 4.16 2D Binding mode Binding energy (Kcal/mol) -8  Molscript Map 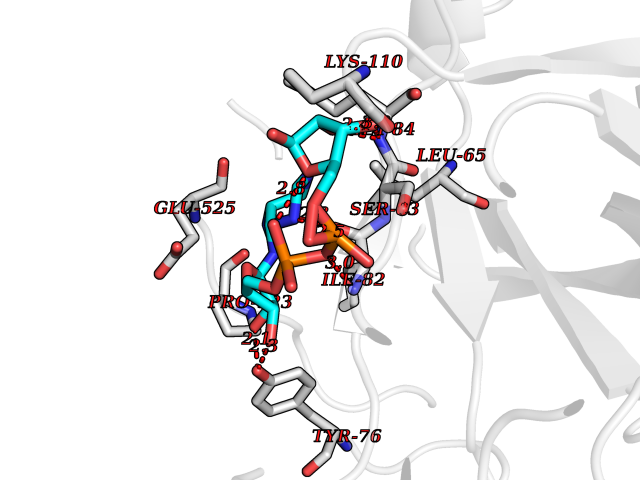 Pymol Map 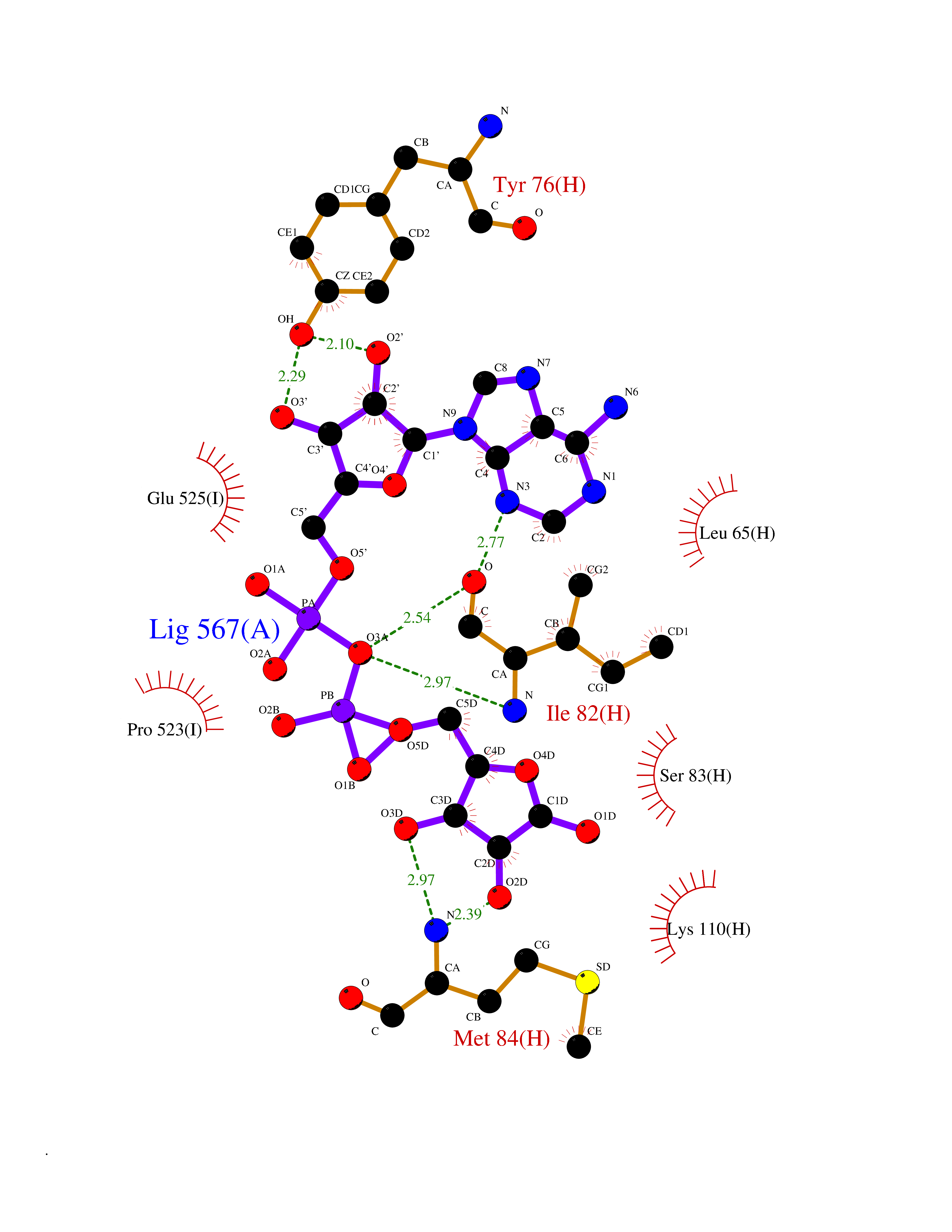 Ligplot Map 3D Binding mode Sequence IVEGSDAEIGMSPWQVMLFRSPQELLCGASLISDRWVLTAAHCLLTENDLLVRIGKHSRTRYRNIEKISMLEKIYIHPRYNWENLDRDIALMKLKKPVAFSDYIHPVCLPDRETLLQAGYKGRVTGWGNLKETWGQPSVLQVVNLPIVERPVCKDSTRIRITDNMFCAYKKRGDACEGDSGGPFVMKSNNRWYQMGIVSWGEGCRDGKYGFYTHVFRLKKWIQKVIDQFGGDFEEIPEELQCGLRPLFEKKSLE Hydrogen bonds contact Hydrophobic contact | ||||
| 6 | Enteropeptidase (TMPRSS15) | 6ZOV | 6.60 | |
Target general information Gen name TMPRSS15 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Transmembrane protease serine 15; TMPRSS15; Serine protease 7; Enterokinase Protein family Peptidase S1 family Biochemical class Peptidase Function Responsible for initiating activation of pancreatic proteolytic proenzymes (trypsin, chymotrypsin and carboxypeptidase A). It catalyzes the conversion of trypsinogen to trypsin which in turn activates other proenzymes including chymotrypsinogen, procarboxypeptidases, and proelastases. Related diseases Enterokinase deficiency (ENTKD) [MIM:226200]: Life-threatening intestinal malabsorption disorder characterized by diarrhea and failure to thrive. {ECO:0000269|PubMed:11719902}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number EC 3.4.21.9 Uniprot keywords 3D-structure; Disulfide bond; Glycoprotein; Hydrolase; Lipoprotein; Membrane; Myristate; Protease; Proteomics identification; Reference proteome; Repeat; Serine protease; Signal-anchor; Transmembrane; Transmembrane helix; Zymogen Protein physicochemical properties Chain ID A Molecular weight (Da) 26220.3 Length 234 Aromaticity 0.1 Instability index 50.13 Isoelectric point 4.82 Charge (pH=7) -9.93 2D Binding mode Binding energy (Kcal/mol) -7.85 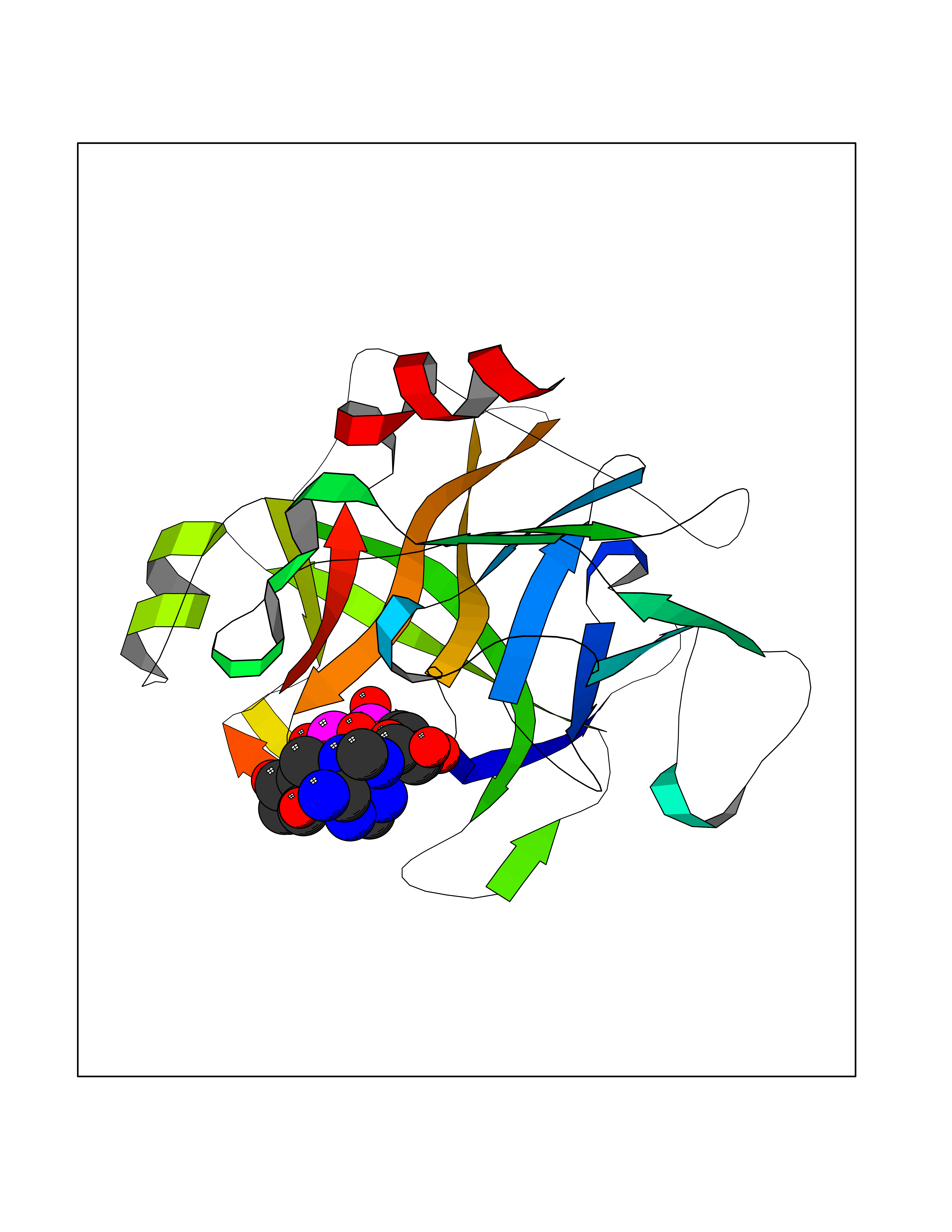 Molscript Map 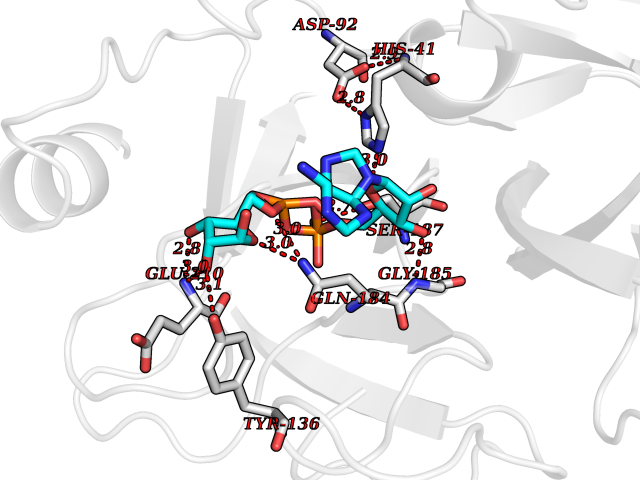 Pymol Map 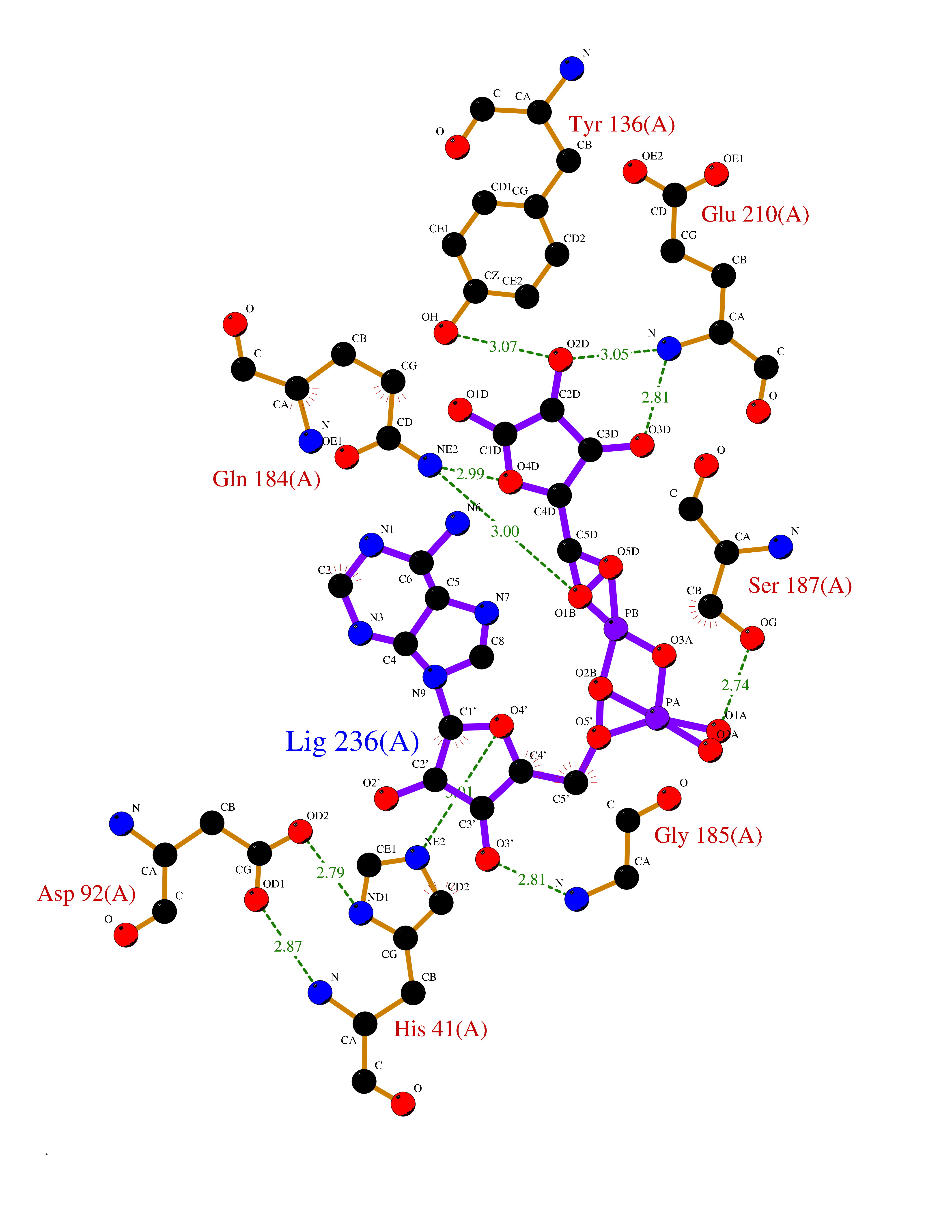 Ligplot Map 3D Binding mode Sequence IVGGSDAKEGAWPWVVGLYYDDRLLCGASLVSSDWLVSAAHCVYGRNLEPSKWTAILGLHMKSNLTSPQTVPRLIDEIVINPHYNRRRKDNDIAMMHLEFKVNYTDYIQPISLPEENQVFPPGRNCSIAGWGTVVYQGTTADILQEADVPLLSNERCQQQMPEYNITENMICAGYEEGGIDSCQGDSGGPLMCQENNRWFLAGVTSFGYECALPNRPGVYARVSRFTEWIQSFL Hydrogen bonds contact Hydrophobic contact | ||||
| 7 | Coagulation factor XII (F12) | 4XDE | 6.58 | |
Target general information Gen name F12 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Hageman factor; HAF Protein family Peptidase S1 family Biochemical class Peptidase Function NA Related diseases Factor XII deficiency (FA12D) [MIM:234000]: An asymptomatic anomaly of in vitro blood coagulation. Its diagnosis is based on finding a low plasma activity of the factor in coagulating assays. It is usually only accidentally discovered through pre-operative blood tests. Factor XII deficiency is divided into two categories, a cross-reacting material (CRM)-negative group (negative F12 antigen detection) and a CRM-positive group (positive F12 antigen detection). {ECO:0000269|PubMed:10361128, ECO:0000269|PubMed:11776307, ECO:0000269|PubMed:15205584, ECO:0000269|PubMed:15617741, ECO:0000269|PubMed:2510163, ECO:0000269|PubMed:2882793, ECO:0000269|PubMed:8049433, ECO:0000269|PubMed:8528215, ECO:0000269|PubMed:9354665}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Angioedema, hereditary, 3 (HAE3) [MIM:610618]: A hereditary angioedema occurring only in women. Hereditary angioedema is an autosomal dominant disorder characterized by episodic local swelling involving subcutaneous or submucous tissue of the upper respiratory and gastrointestinal tracts, face, extremities, and genitalia. Hereditary angioedema type 3 differs from types 1 and 2 in that both concentration and function of C1 esterase inhibitor are normal. Hereditary angioedema type 3 is precipitated or worsened by high estrogen levels (e.g., during pregnancy or treatment with oral contraceptives). {ECO:0000269|PubMed:16638441, ECO:0000269|PubMed:17186468}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB09228; DB06689; DB06404; DB12598; DB01593; DB14487 Interacts with P05067; Q07021; P13473-2 EC number EC 3.4.21.38 Uniprot keywords 3D-structure; Blood coagulation; Direct protein sequencing; Disease variant; Disulfide bond; EGF-like domain; Fibrinolysis; Glycoprotein; Hemostasis; Hydrolase; Kringle; Protease; Proteomics identification; Reference proteome; Repeat; Secreted; Serine protease; Signal; Zymogen Protein physicochemical properties Chain ID A Molecular weight (Da) 26038 Length 241 Aromaticity 0.08 Instability index 50.34 Isoelectric point 5.25 Charge (pH=7) -11.38 2D Binding mode Binding energy (Kcal/mol) -6.48 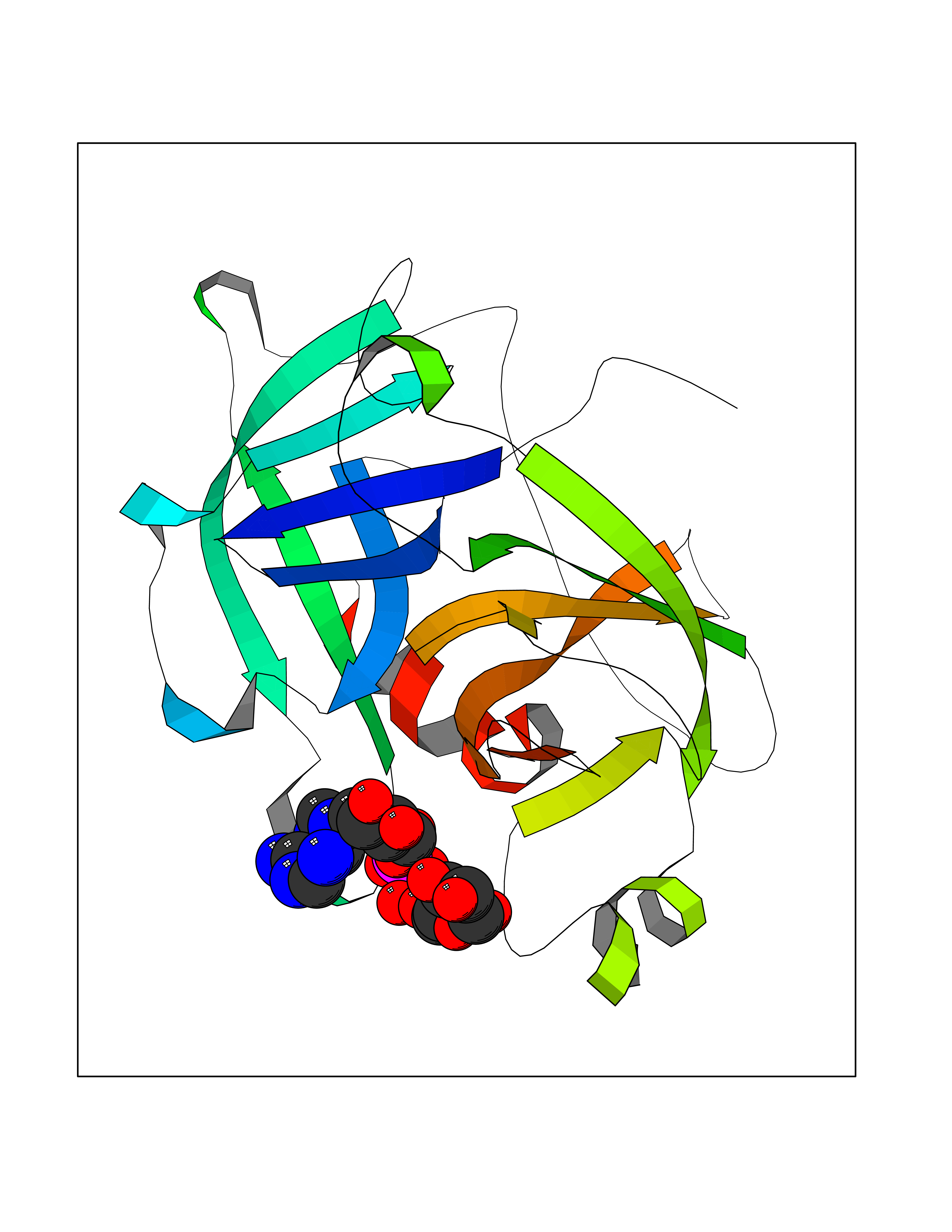 Molscript Map 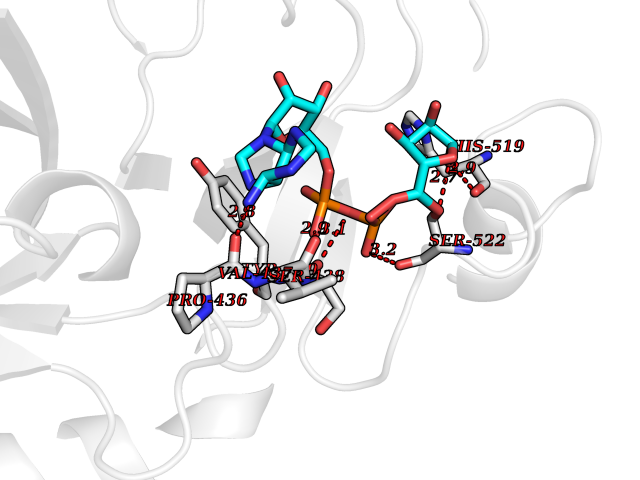 Pymol Map 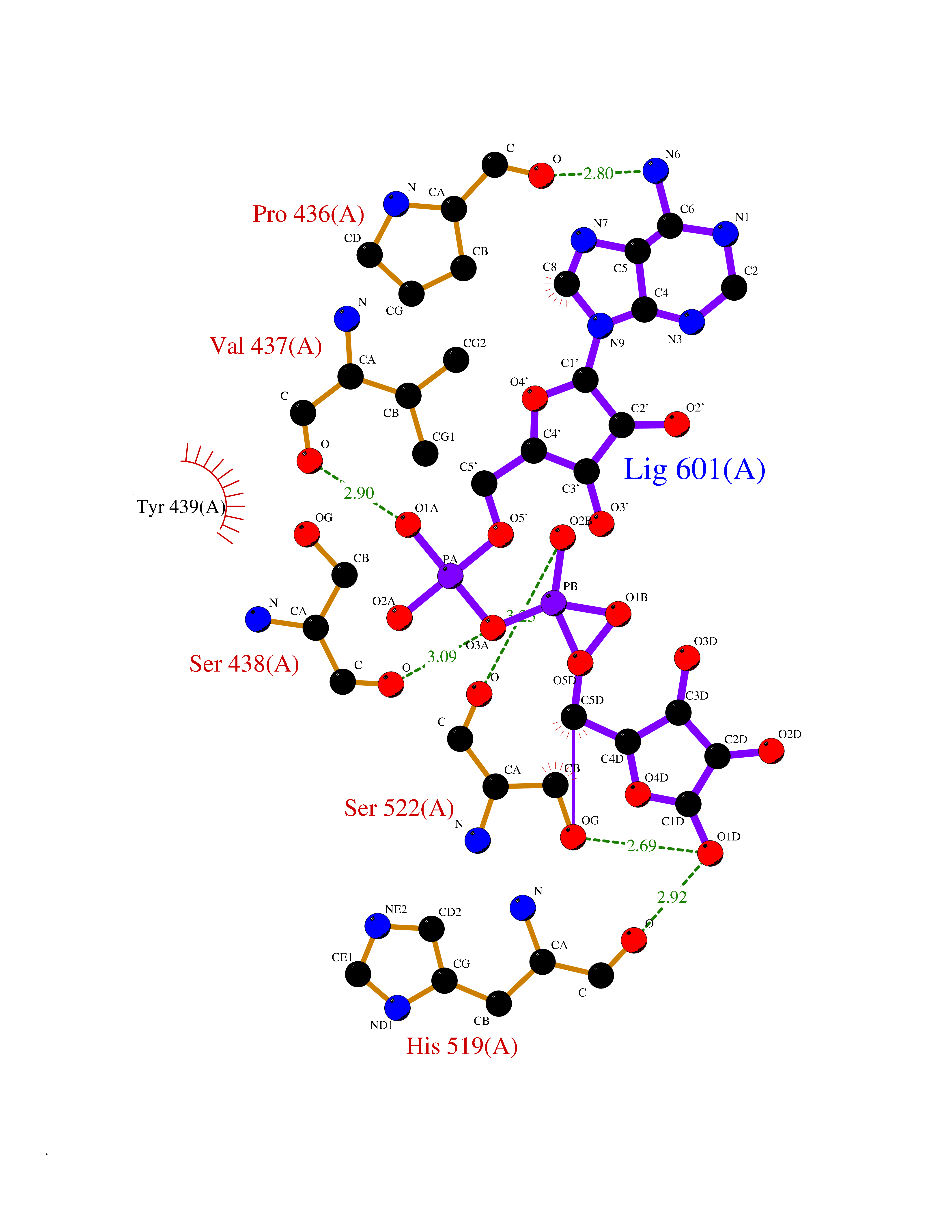 Ligplot Map 3D Binding mode Sequence VALRGAHPYIAALYWGHSFCAGSLIAPCWVLTAAHCLQDRPAPEDLTVVLGQERRNHSCEPCQTLAVRSYRLHEAFSPVSYQHDLALLRLQEDADGSCALLSPYVQPVSLPSGAARPSETTLCQVAGWGHQFEGAEEYASFLQEAQVPFLSLERCSAPDVHGSSILPGMLCAGFLEGGTDACQGDSGGPLVCEDQAAERRLTLQGIISWGSGCGDRNKPGVYTDVAYYLAWIREHTVSHHT Hydrogen bonds contact Hydrophobic contact | ||||
| 8 | Dopamine beta-hydroxylase | 4ZEL | 6.56 | |
Target general information Gen name DBH Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Copper type II ascorbate-dependent monooxygenase family Biochemical class Oxidoreductase Function Catalytic activity.Copper ion binding.Dopamine beta-monooxygenase activity.L-ascorbic acid binding. Related diseases Orthostatic hypotension 1 (ORTHYP1) [MIM:223360]: A form of orthostatic hypotension due to congenital dopamine beta-hydroxylase deficiency. Orthostatic hypotension, also known as postural hypotension, is a finding defined as a 20-mm Hg decrease in systolic pressure or a 10-mm Hg decrease in diastolic pressure occurring 3 minutes after a person has risen from supine to standing. Symptoms include dizziness, blurred vision, and sometimes syncope. ORTHYP1 is an autosomal recessive condition apparent from infancy or early childhood and characterized by low plasma and urinary levels of norepinephrine and epinephrine, and episodic hypoglycemia. {ECO:0000269|PubMed:11857564}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00126; DB06774; DB09130; DB05394; DB00822; DB00988; DB00968; DB00550 Interacts with P00352; P63010-2; Q04656; Q8WUW1; Q9UNS2; Q71DI3; P61978; Q9Y2M5; Q92876; P08727; Q14693; P0DPK4; Q6GQQ9-2; P27986-2; Q9ULX5; Q96D59; Q8N6K7-2; Q9GZS3; Q8IUW3; Q86WT6-2 EC number 1.14.17.1 Uniprot keywords 3D-structure; Catecholamine biosynthesis; Copper; Cytoplasmic vesicle; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; Membrane; Metal-binding; Monooxygenase; Oxidoreductase; Proteomics identification; Reference proteome; Secreted; Signal-anchor; Transmembrane; Transmembrane helix; Vitamin C Protein physicochemical properties Chain ID A,B Molecular weight (Da) 123694 Length 1094 Aromaticity 0.1 Instability index 51.85 Isoelectric point 5.84 Charge (pH=7) -24.5 2D Binding mode Binding energy (Kcal/mol) -7.7 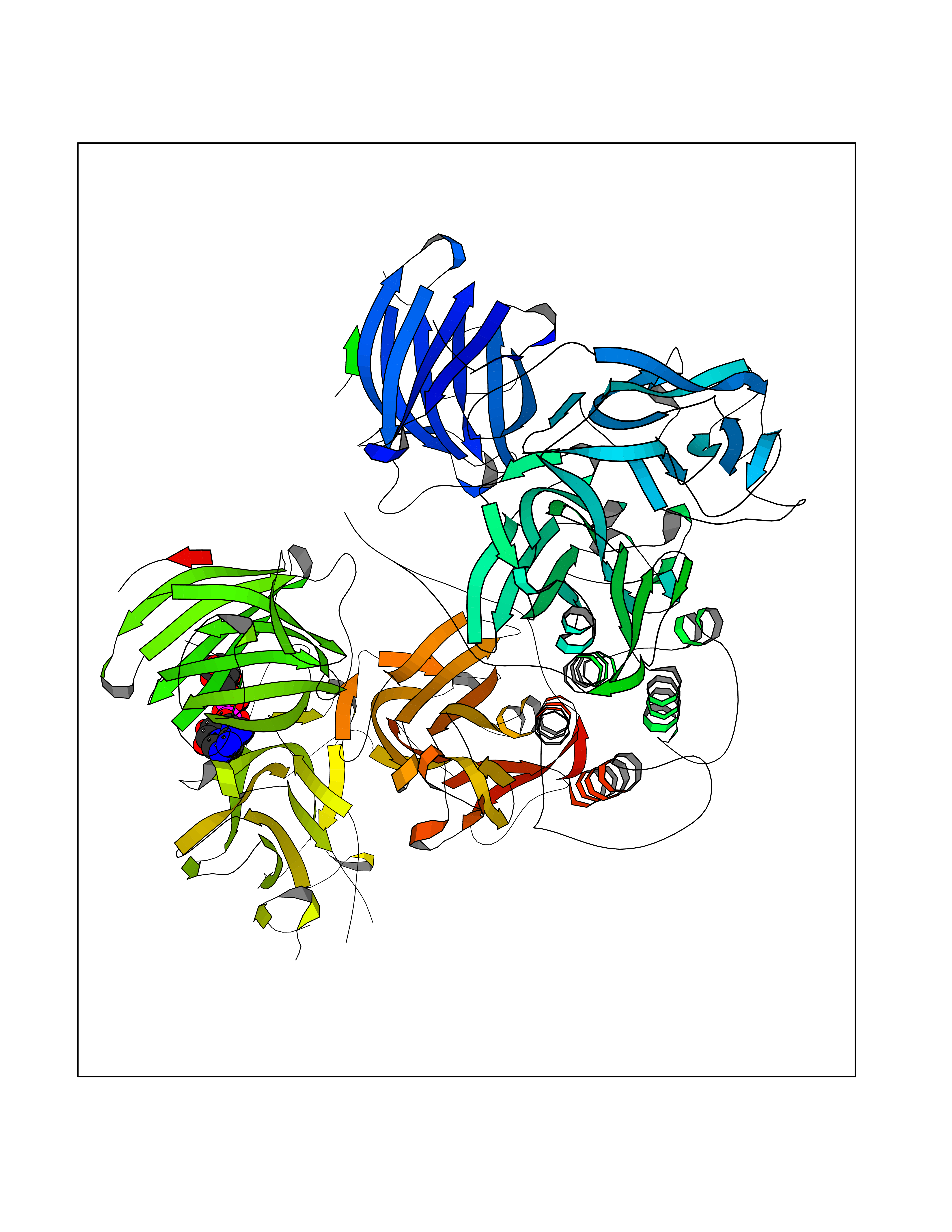 Molscript Map 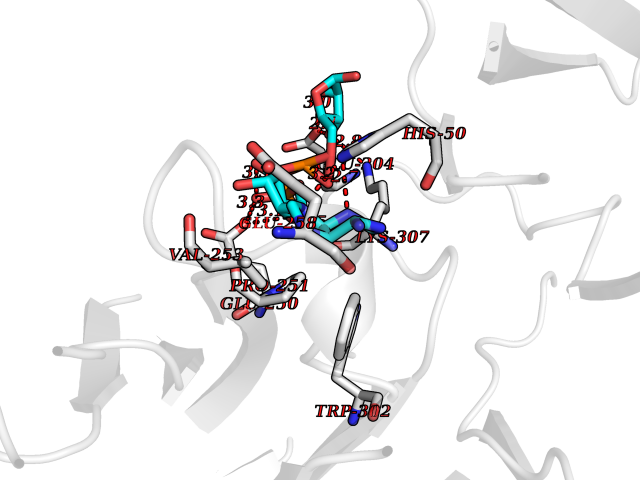 Pymol Map 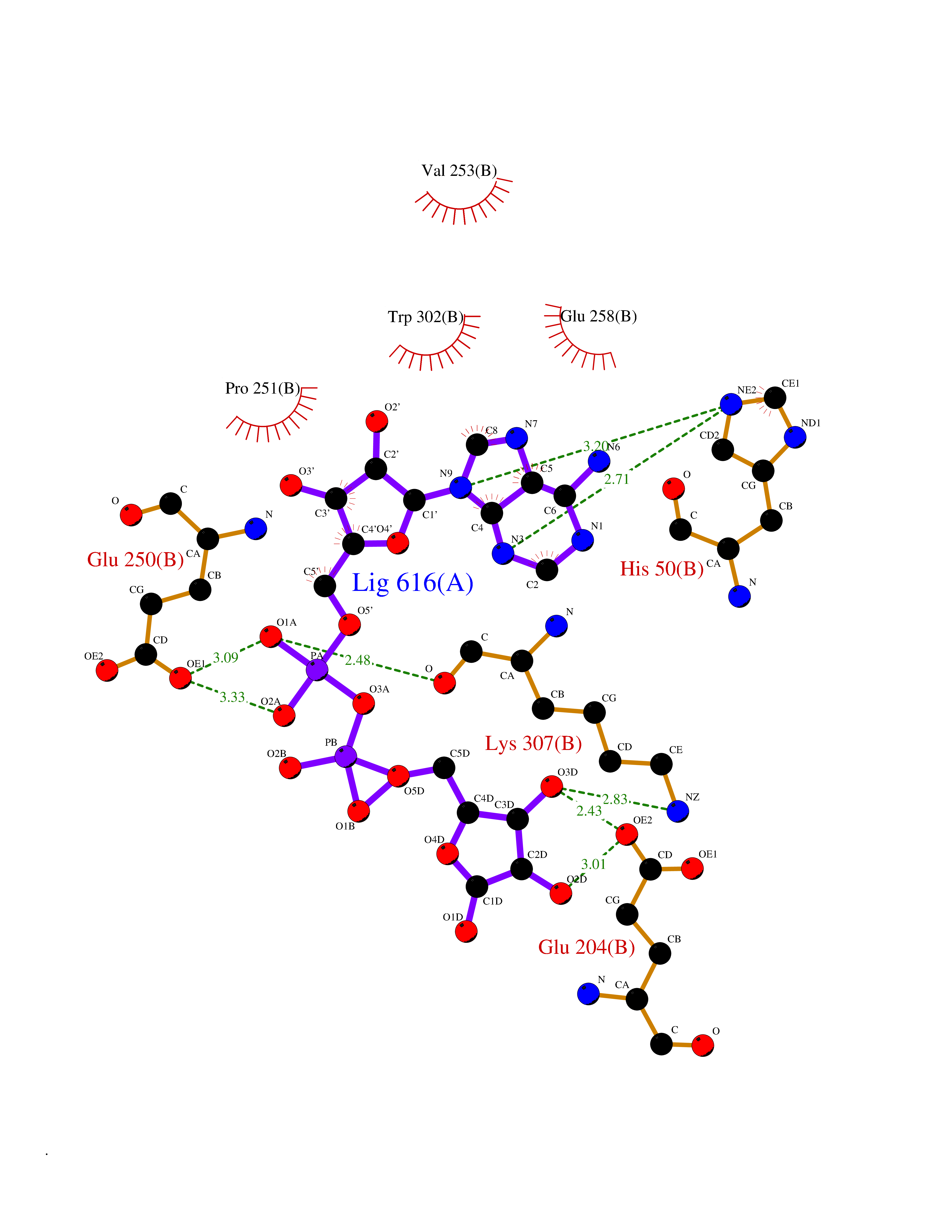 Ligplot Map 3D Binding mode Sequence PLPYHIPLDPEGSLELSWNVSYTQEAIHFQLLVRRLKAGVLFGMSDRGELENADLVVLWTDGDAYFADAWSDQKGQIHLDPQQDYQLLQVQRTPEGLTLLFKRPFGTCDPKDYLIEDGTVHLVYGILEEPFRSLEAINGSGLQMGLQRVQLLKPNIPEPELPSDACTMEVQAPNIQIPSQETTYWCYIKELPKGFSRHHIIKYEPIVTKGNEALVHHMEVFQCAPEMDSVPHFSGPCDSKMKPDRLNYCRHVLAAWALGAKAFYYPEEAGLAFGGPGSSRYLRLEVHYHNPLVIEGRNDSSGIRLYYTAKLRRFNAGIMELGLVYTPVMAIPPRETAFILTGYCTDKCTQLALPPSGIHIFASQLHTHLTGRKVVTVLVRDGREWEIVNQDNHYSPHFQEIRMLKKVVSVHPGDVLITSCTYNTEDRELATVGGFGILEEMCVNYVHYYPQTQLELCKSAVDAGFLQKYFHLINRFNNEDVCTCPQASVSQQFTSVPWNSFNRDVLKALYSFAPISMHCNKSSAVRFQGEWNLQPLPKVISTLEEPTVVSPLPYHIPLDPEGSLELSWNVSYTQEAIHFQLLVRRLKAGVLFGMSDRGELENADLVVLAYFADAWSDQKGQIHLDPQQDYQLLQVQRTPEGLTLLFKRPFGTCDPKDYLIEDGTVHLVYGILEEPFRSLEAINGSGLQMGLQRVQLLKPNIPEPELPSDACTMEVQAPNIQIPSQETTYWCYIKELPKGFSRHHIIKYEPIVTKGNEALVHHMEVFQCAPEVPHFSGPCDSKMLNYCRHVLAAWALGAKAFYYPEEAGLAFGGPGSSRYLRLEVHYHNPLVIEGRNDSSGIRLYYTAKLRRFNAGIMELGLVYTPVMAIPPRETAFILTGYCTDKCTQLALPPSGIHIFASQLHTHLTGRKVVTVLVRDGREWEIVNQDNHYSPHFQEIRMLKKVVSVHPGDVLITSCTYNTEDRELATVGGFGILEEMCVNYVHYYPQTQLELCKSAVDAGFLQKYFHLINRFNNEDVCTCPQASVSQQFTSVPWNSFNRDVLKALYSFAPISMHCNKSSAVRFQGEWNLQPLPKVISTLEEPTPQCVVSIGG Hydrogen bonds contact Hydrophobic contact | ||||
| 9 | Prothrombin | 4UD9 | 6.52 | |
Target general information Gen name F2 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Peptidase S1 family Biochemical class Hydrolase Function Calcium ion binding.Growth factor activity.Heparin binding.Lipopolysaccharide binding.Receptor binding.Serine-type endopeptidase activity.Thrombospondin receptor activity. Related diseases Factor II deficiency (FA2D) [MIM:613679]: A very rare blood coagulation disorder characterized by mucocutaneous bleeding symptoms. The severity of the bleeding manifestations correlates with blood factor II levels. {ECO:0000269|PubMed:1349838, ECO:0000269|PubMed:1354985, ECO:0000269|PubMed:1421398, ECO:0000269|PubMed:14962227, ECO:0000269|PubMed:2719946, ECO:0000269|PubMed:3242619, ECO:0000269|PubMed:3567158, ECO:0000269|PubMed:3771562, ECO:0000269|PubMed:3801671, ECO:0000269|PubMed:6405779, ECO:0000269|PubMed:7792730, ECO:0000269|PubMed:7865694}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Ischemic stroke (ISCHSTR) [MIM:601367]: A stroke is an acute neurologic event leading to death of neural tissue of the brain and resulting in loss of motor, sensory and/or cognitive function. Ischemic strokes, resulting from vascular occlusion, is considered to be a highly complex disease consisting of a group of heterogeneous disorders with multiple genetic and environmental risk factors. {ECO:0000269|PubMed:15534175}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Thrombophilia due to thrombin defect (THPH1) [MIM:188050]: A multifactorial disorder of hemostasis characterized by abnormal platelet aggregation in response to various agents and recurrent thrombi formation. {ECO:0000269|PubMed:2825773}. The disease is caused by variants affecting the gene represented in this entry. A common genetic variation in the 3-prime untranslated region of the prothrombin gene is associated with elevated plasma prothrombin levels and an increased risk of venous thrombosis.; DISEASE: Pregnancy loss, recurrent, 2 (RPRGL2) [MIM:614390]: A common complication of pregnancy, resulting in spontaneous abortion before the fetus has reached viability. The term includes all miscarriages from the time of conception until 24 weeks of gestation. Recurrent pregnancy loss is defined as 3 or more consecutive spontaneous abortions. {ECO:0000269|PubMed:11506076}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07211; DB07796; DB07016; DB07521; DB06850; DB07091; DB06845; DB07088; DB07131; DB07095; DB07515; DB07897; DB06878; DB06947; DB08624; DB06869; DB06929; DB07400; DB04771; DB04772; DB02287; DB07277; DB07550; DB07549; DB07548; DB07105; DB04722; DB07366; DB08254; DB01725; DB08062; DB07639; DB07461; DB07120; DB07190; DB07741; DB07353; DB07508; DB07809; DB08546; DB08061; DB07718; DB03136; DB02723; DB07440; DB07376; DB06861; DB06866; DB06865; DB03865; DB06841; DB07934; DB08422; DB07659; DB07660; DB07658; DB13151; DB00025; DB11166; DB00278; DB01766; DB07083; DB00006; DB00100; DB13152; DB09228; DB09130; DB03159; DB06911; DB06996; DB06919; DB07027; DB07133; DB07143; DB07005; DB06695; DB00055; DB01225; DB05714; DB12831; DB03847; DB07278; DB01767; DB06404; DB09332; DB00001; DB13998; DB04136; DB00170; DB06838; DB13999; DB06868; DB06942; DB06936; DB07165; DB07527; DB07522; DB07665; DB07946; DB06859; DB06853; DB06858; DB07279; DB08187; DB04591; DB07944; DB07128; DB12598; DB01123; DB04786; DB05777; DB04697; DB09109; DB14738; DB04898; DB01593; DB14487; DB08152 Interacts with P05067; P07204; Q846V4; PRO_0000032489 [P01008] EC number 3.4.21.5 Uniprot keywords 3D-structure; Acute phase; Blood coagulation; Calcium; Cleavage on pair of basic residues; Direct protein sequencing; Disease variant; Disulfide bond; Gamma-carboxyglutamic acid; Glycoprotein; Hemostasis; Hydrolase; Kringle; Pharmaceutical; Protease; Proteomics identification; Reference proteome; Repeat; Secreted; Serine protease; Signal; Thrombophilia; Zymogen Protein physicochemical properties Chain ID H Molecular weight (Da) 29321.6 Length 254 Aromaticity 0.1 Instability index 39.57 Isoelectric point 8.56 Charge (pH=7) 4.16 2D Binding mode Binding energy (Kcal/mol) -8.18  Molscript Map 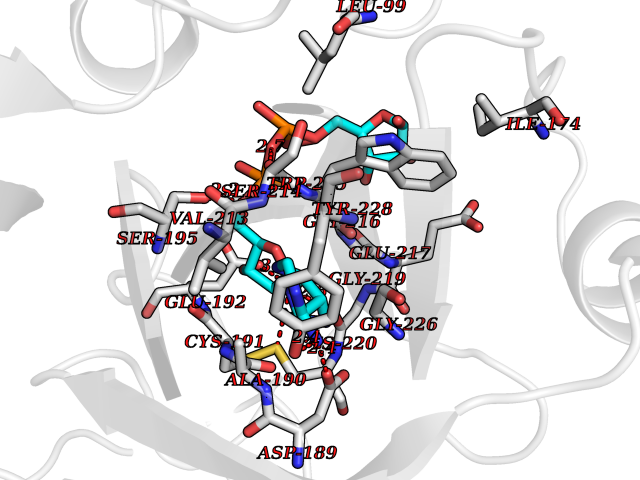 Pymol Map 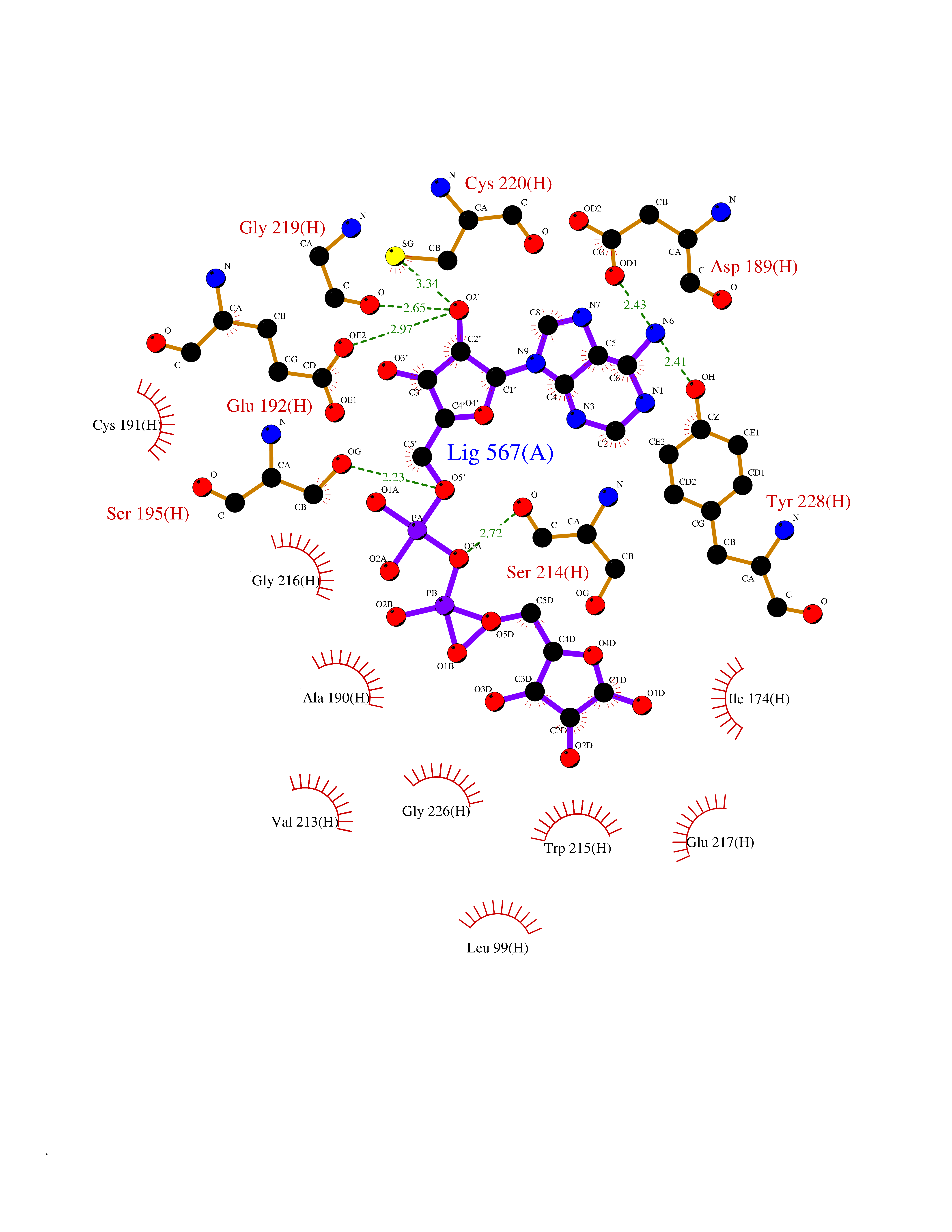 Ligplot Map 3D Binding mode Sequence IVEGSDAEIGMSPWQVMLFRSPQELLCGASLISDRWVLTAAHCLLTENDLLVRIGKHSRTRYRNIEKISMLEKIYIHPRYNWENLDRDIALMKLKKPVAFSDYIHPVCLPDRETLLQAGYKGRVTGWGNLKETWGQPSVLQVVNLPIVERPVCKDSTRIRITDNMFCAYKKRGDACEGDSGGPFVMKSNNRWYQMGIVSWGEGCRDGKYGFYTHVFRLKKWIQKVIDQFGGDFEEIPEELQCGLRPLFEKKSLE Hydrogen bonds contact Hydrophobic contact | ||||
| 10 | Opioid receptor delta (OPRD1) | 4N6H | 6.51 | |
Target general information Gen name OPRD1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms OPRD; Delta-type opioid receptor; Delta opioid receptor; DOR-1; D-OR-1 Protein family G-protein coupled receptor 1 family Biochemical class GPCR rhodopsin Function Ligand binding causes a conformation change that triggers signaling via guanine nucleotide-binding proteins (G proteins) and modulates the activity of down-stream effectors, such as adenylate cyclase. Signaling leads to the inhibition of adenylate cyclase activity. Inhibits neurotransmitter release by reducing calcium ion currents and increasing potassium ion conductance. Plays a role in the perception of pain and in opiate-mediated analgesia. Plays a role in developing analgesic tolerance to morphine. G-protein coupled receptor that functions as receptor for endogenous enkephalins and for a subset of other opioids. Related diseases Defects in PPARG can lead to type 2 insulin-resistant diabetes and hyptertension. PPARG mutations may be associated with colon cancer. {ECO:0000269|PubMed:10394368}.; DISEASE: Obesity (OBESITY) [MIM:601665]: A condition characterized by an increase of body weight beyond the limitation of skeletal and physical requirements, as the result of excessive accumulation of body fat. {ECO:0000269|PubMed:9753710}. Disease susceptibility may be associated with variants affecting the gene represented in this entry.; DISEASE: Lipodystrophy, familial partial, 3 (FPLD3) [MIM:604367]: A form of lipodystrophy characterized by marked loss of subcutaneous fat from the extremities. Facial adipose tissue may be increased, decreased or normal. Affected individuals show an increased preponderance of insulin resistance, diabetes mellitus and dyslipidemia. {ECO:0000269|PubMed:11788685, ECO:0000269|PubMed:12453919}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Glioma 1 (GLM1) [MIM:137800]: Gliomas are benign or malignant central nervous system neoplasms derived from glial cells. They comprise astrocytomas and glioblastoma multiforme that are derived from astrocytes, oligodendrogliomas derived from oligodendrocytes and ependymomas derived from ependymocytes. {ECO:0000269|PubMed:10851250}. Disease susceptibility may be associated with variants affecting the gene represented in this entry. Polymorphic PPARG alleles have been found to be significantly over-represented among a cohort of American patients with sporadic glioblastoma multiforme suggesting a possible contribution to disease susceptibility. Drugs (DrugBank ID) DB01571; DB01439; DB05050; DB06274; DB06288; DB00321; DB01238; DB00921; DB00611; DB09173; DB09061; DB01535; DB00318; DB00514; DB00647; DB01452; DB01565; DB01444; DB01081; DB01548; DB09272; DB01497; DB00813; DB00956; DB00327; DB01221; DB06738; DB00854; DB00836; DB14146; DB14009; DB12668; DB00333; DB00295; DB06409; DB14011; DB00844; DB11691; DB06230; DB01183; DB00704; DB11130; DB00497; DB01192; DB09209; DB00899; DB12543; DB00708; DB06204; DB00193 Interacts with P16615; P27824; Q4LDR2; Q5JY77; Q9NS64; Q9Y666-2; Q9UKG4; Q0VAQ4; Q96Q45-2; P11607 EC number NA Uniprot keywords 3D-structure; Cell membrane; Disulfide bond; G-protein coupled receptor; Glycoprotein; Lipoprotein; Membrane; Palmitate; Proteomics identification; Receptor; Reference proteome; Transducer; Transmembrane; Transmembrane helix; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 32859.3 Length 294 Aromaticity 0.11 Instability index 33.86 Isoelectric point 9.38 Charge (pH=7) 13.6 2D Binding mode Binding energy (Kcal/mol) -6.85 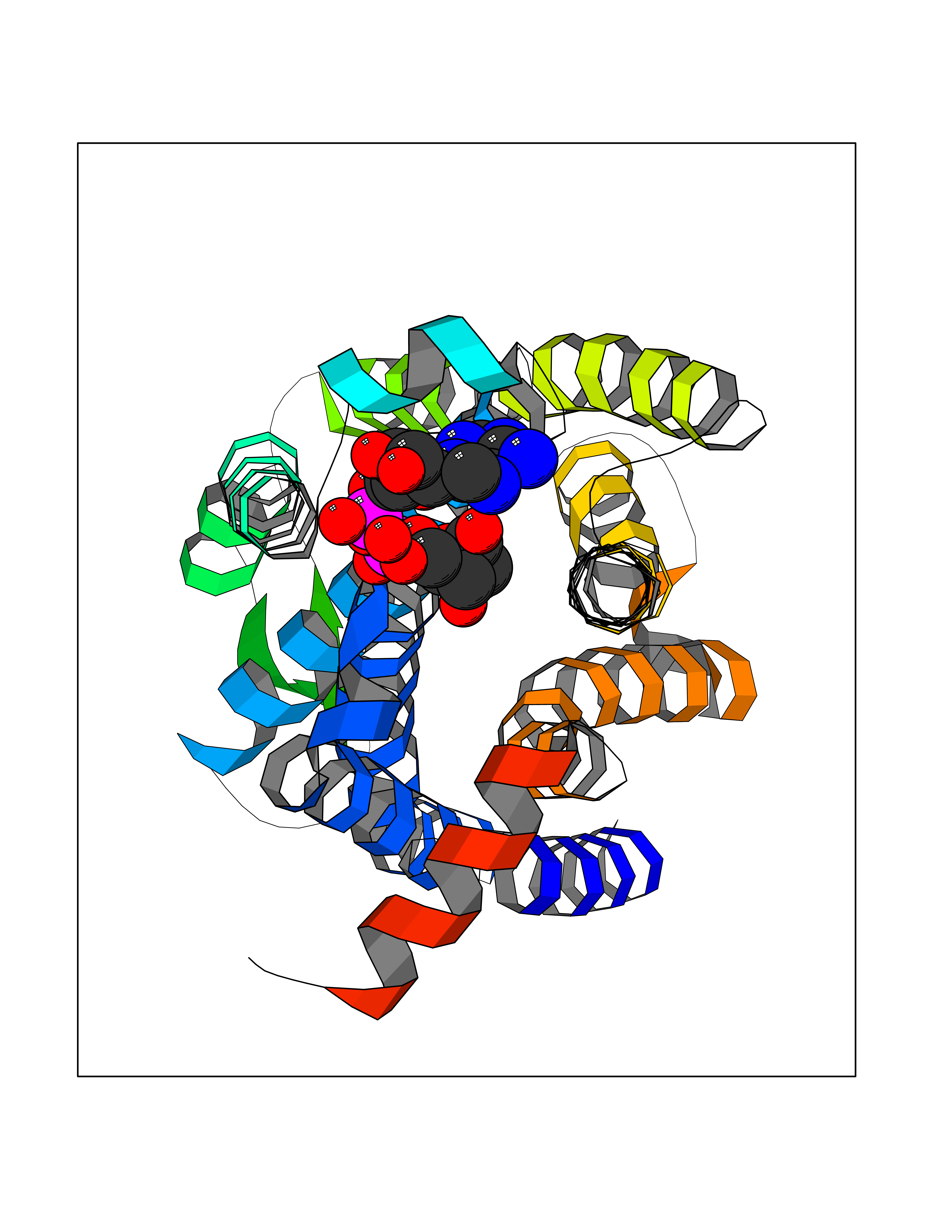 Molscript Map 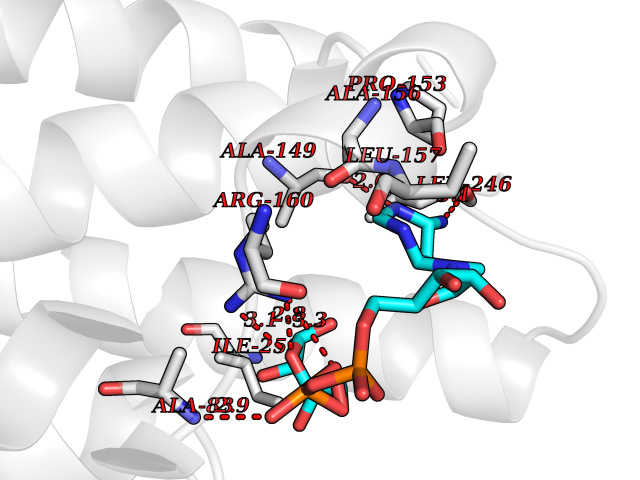 Pymol Map 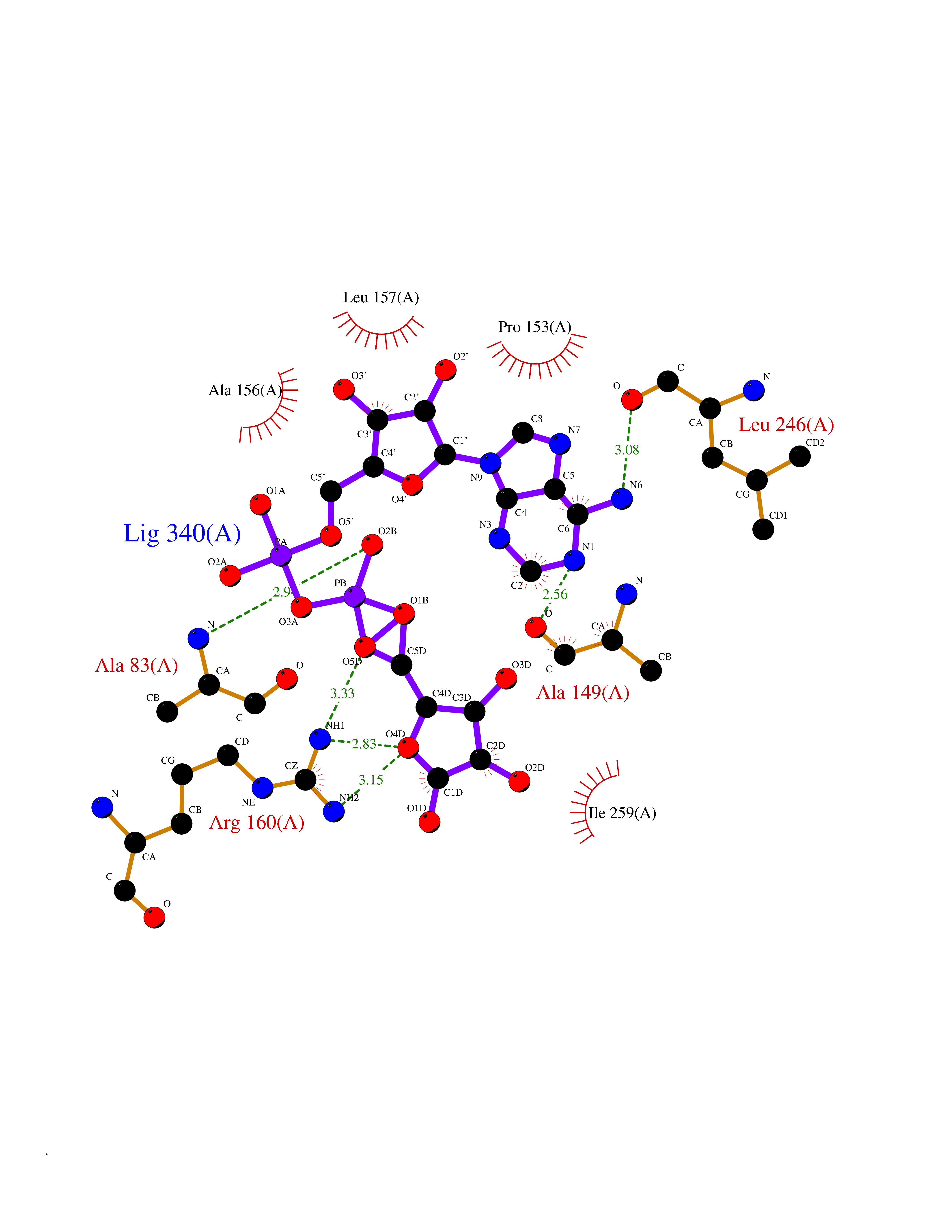 Ligplot Map 3D Binding mode Sequence SLALAIAITALYSAVCAVGLLGNVLVMFGIVRYTKMKTATNIYIFNLALADALATSTLPFQSAKYLMETWPFGELLCKAVLSIDYYNMFTSIFTLTMMSVDRYIAVCHPVKALDFRTPAKAKLINICIWVLASGVGVPIMVMAVTRPRDGAVVCMLQFPSPSWYWDTVTKICVFLFAFVVPILIITVCYGLMLLRLRSVRLLSGSKEKDRSLRRITRMVLVVVGAFVVCWAPIHIFVIVWTLVDIDRRDPLVVAALHLCIALGYANSSLNPVLYAFLDENFKRCFRQLCRKPCG Hydrogen bonds contact Hydrophobic contact | ||||
| 11 | T-cell surface glycoprotein CD1a (CD1A) | 6NUX | 6.51 | |
Target general information Gen name CD1A Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms hTa1 thymocyteantigen; hTa1 thymocyte antigen; T-cell surfaceantigen T6/Leu-6; T-cell surface antigen T6/Leu-6; CD1a Protein family NA Biochemical class Immunoglobulin Function Antigen-presenting protein that binds self and non-self lipid and glycolipid antigens and presents them to T-cell receptors on natural killer T-cells. Related diseases Pulmonary hypertension, primary, 1 (PPH1) [MIM:178600]: A rare disorder characterized by plexiform lesions of proliferating endothelial cells in pulmonary arterioles. The lesions lead to elevated pulmonary arterial pression, right ventricular failure, and death. The disease can occur from infancy throughout life and it has a mean age at onset of 36 years. Penetrance is reduced. Although familial pulmonary hypertension is rare, cases secondary to known etiologies are more common and include those associated with the appetite-suppressant drugs. {ECO:0000269|PubMed:10903931, ECO:0000269|PubMed:10973254, ECO:0000269|PubMed:11015450, ECO:0000269|PubMed:11115378, ECO:0000269|PubMed:12045205, ECO:0000269|PubMed:12358323, ECO:0000269|PubMed:15965979, ECO:0000269|PubMed:24936649, ECO:0000269|PubMed:25187962, ECO:0000269|PubMed:28507310}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Pulmonary venoocclusive disease 1, autosomal dominant (PVOD1) [MIM:265450]: A disease characterized by widespread fibrous obstruction and intimal thickening of septal veins and preseptal venules, a low diffusing capacity for carbon monoxide, occult alveolar hemorrhage, and nodular ground-glass opacities, septal lines and lymph node enlargement showed by high-resolution computed tomography of the chest. It is frequently associated with pulmonary capillary dilatation and proliferation, and is a rare and devastating cause of pulmonary hypertension. {ECO:0000269|PubMed:12446270, ECO:0000269|PubMed:16429395}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00098 Interacts with P61769 EC number NA Uniprot keywords 3D-structure; Adaptive immunity; Cell membrane; Disulfide bond; Endosome; Glycoprotein; Immunity; Immunoglobulin domain; Membrane; Proteomics identification; Reference proteome; Signal; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 30867.3 Length 270 Aromaticity 0.13 Instability index 37.45 Isoelectric point 6.16 Charge (pH=7) -4.5 2D Binding mode Binding energy (Kcal/mol) -8.5 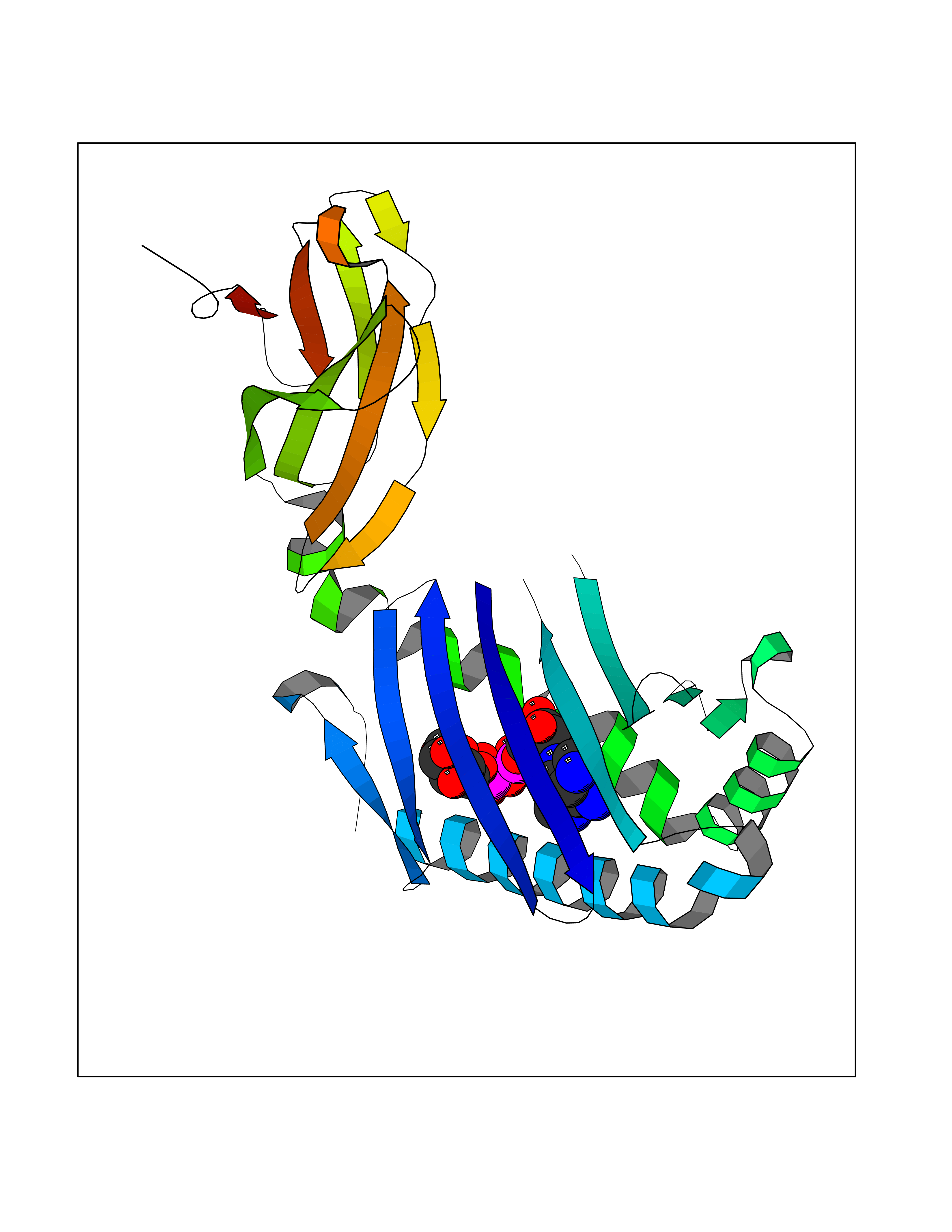 Molscript Map 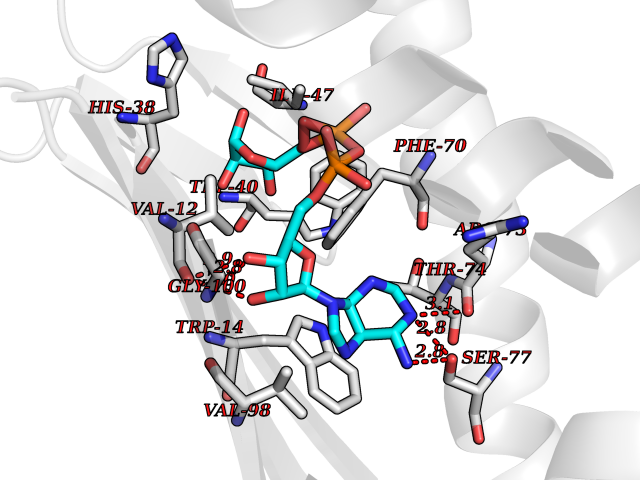 Pymol Map 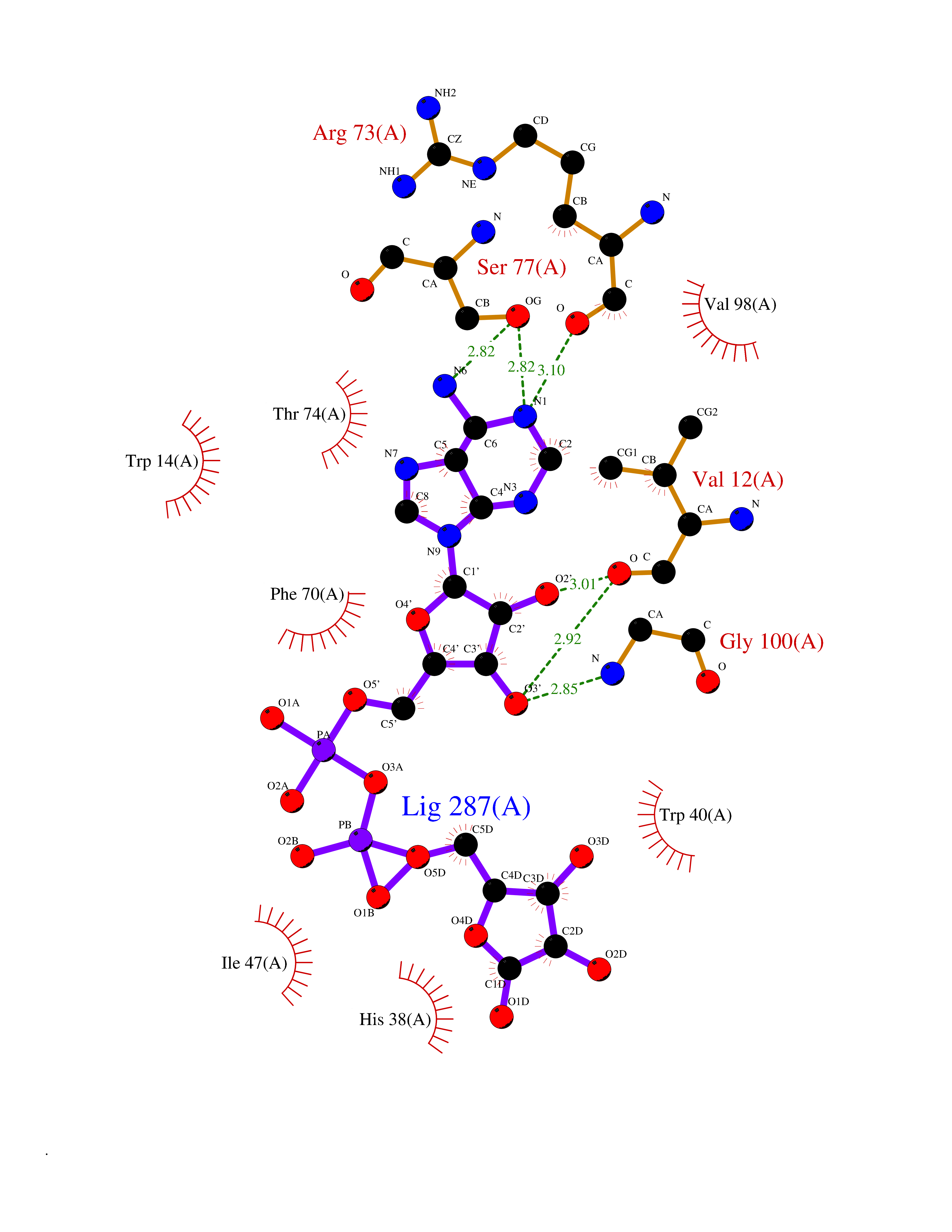 Ligplot Map 3D Binding mode Sequence SFHVIWIASFYNHSWKQNLVSGWLSDLQTHTWDSNSSTIVFLWPWSRGNFSNEWKELETLFRIRTIRSFEGIRRYAHELQFEYPFEIQVTGGCESGSFLQLAYQGSDFVSFQNNSWLPYPVAGNMAKHFCKVLNQNQHENDITHNLLSDTCPRFILGLLDAGKAHLQRQVKPEAWLSHGPSPGPGHLQLVCHVSGFYPKPVWVMWMRGEQEQQGTQRGDILPSADGTWYLRATLEVAAGEAADLSCRVKHSSLEGQDIVLYWEGSLVPRG Hydrogen bonds contact Hydrophobic contact | ||||
| 12 | Interleukin 21 receptor (IL21R) | 6PLH | 6.44 | |
Target general information Gen name IL21R Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms UNQ3121/PRO10273; Novel interleukin receptor; NILR; Interleukin-21 receptor; IL21 receptor; IL-21R; IL-21 receptor; CD360 Protein family Type I cytokine receptor family, Type 4 subfamily Biochemical class Cytokine receptor Function This is a receptor for interleukin-21. Related diseases Immunodeficiency 56 (IMD56) [MIM:615207]: An autosomal recessive primary immunodeficiency characterized by B- and T-cell defects and variable dysfunction of NK cells. Patients tend to have normal numbers of lymphocytes, but show defective class-switched B-cells, low IgG, defective antibody response, and defective T-cell responses to certain antigens. {ECO:0000269|PubMed:23440042}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Chromosomal aberrations involving IL21R is a cause of B-cell non-Hodgkin lymphomas (B-cell NHL). Translocation t(3;16)(q27;p11), with BCL6. Drugs (DrugBank ID) NA Interacts with P29972 EC number NA Uniprot keywords 3D-structure; Chromosomal rearrangement; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; Membrane; Proteomics identification; Receptor; Reference proteome; Repeat; Signal; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A,C,B Molecular weight (Da) 48376.5 Length 446 Aromaticity 0.1 Instability index 43.94 Isoelectric point 8.24 Charge (pH=7) 3.56 2D Binding mode Binding energy (Kcal/mol) -7.98 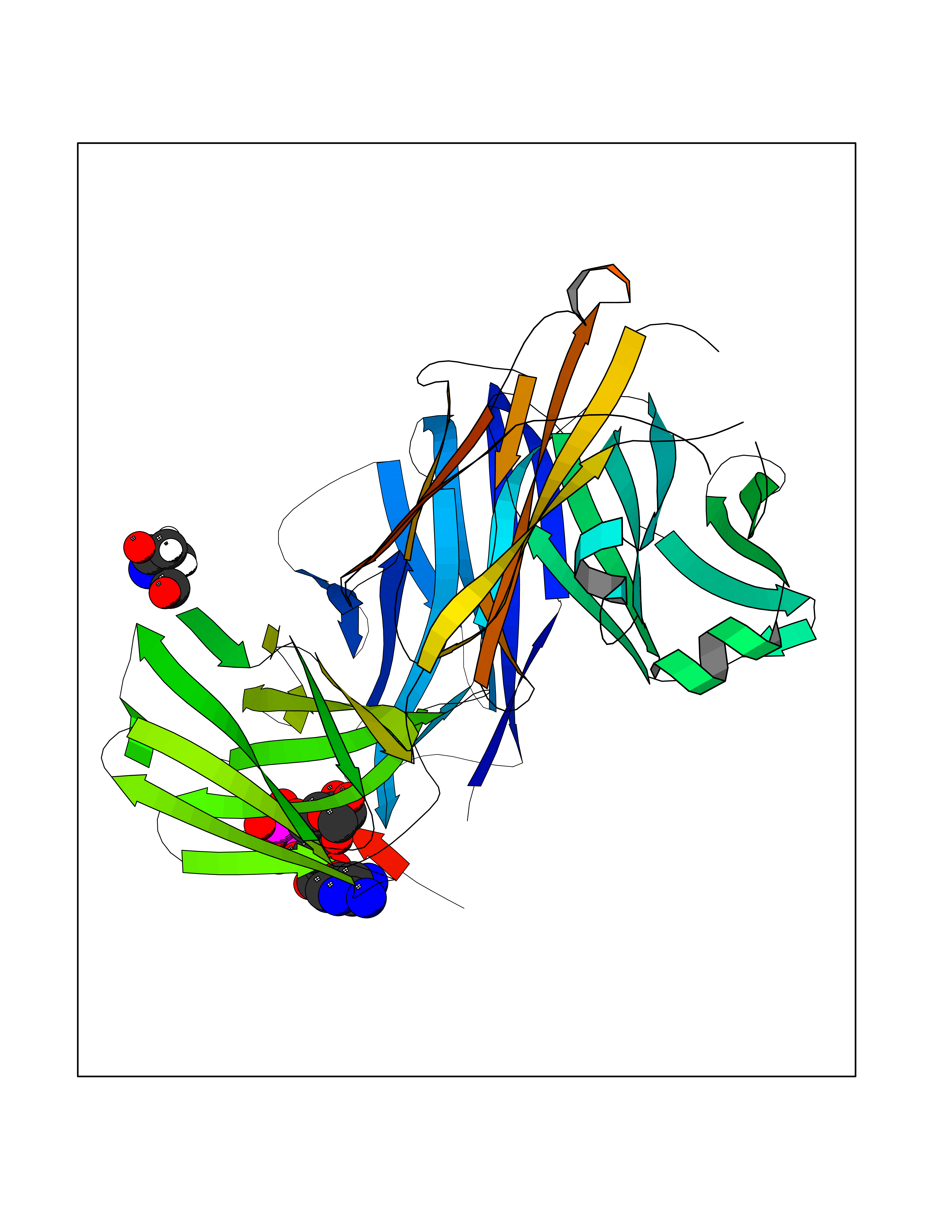 Molscript Map 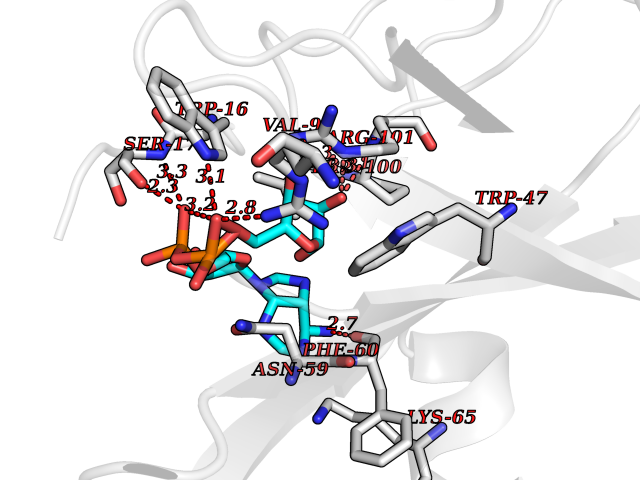 Pymol Map 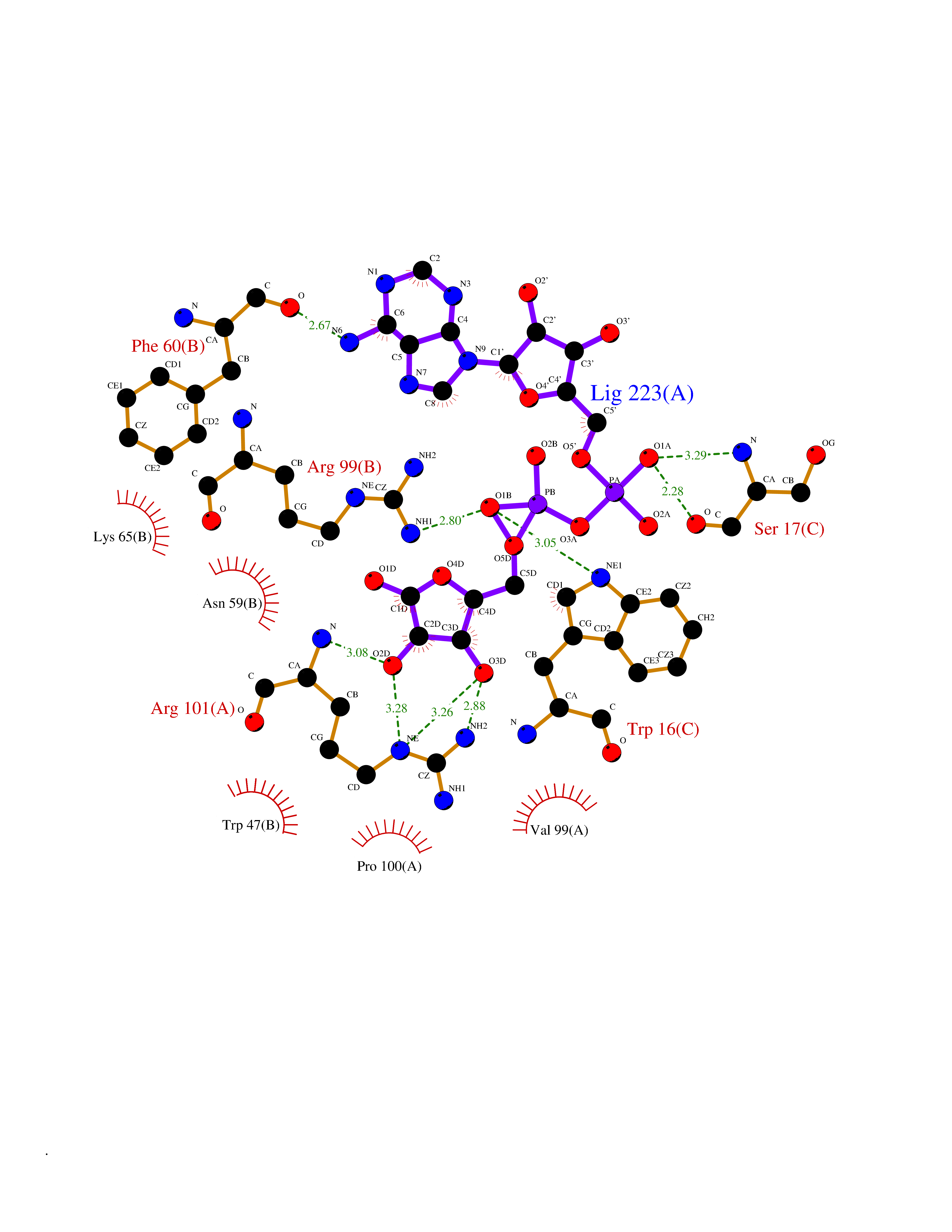 Ligplot Map 3D Binding mode Sequence DVVMTHTPLSLPVSLGDQASISCRSSQSLVHSNGNTYLHWYLQKPGQSPKLLIYKVSNRFSGVPDRFSGSGSGADFTLKISRVEAEDLGVYFCSQSTHVPRTFGGGTKLEIKRADAAPTVSIFPPSSEQLTSGGASVVCFLNNFYPKDINVKWKIDGSERQNGVLNSWTDQDSKDSTYSMSSTLTLTKDEYERHNSYTCEATHKTSTSPIVKSFNRNECXVHLQQPGADLVKPGASVKMSCKASGYTFTSYWITWVKLRPGQGLEWIGDIYPGSGSTNFIEKFKSKATLTVDTSSSTAYMQLRSLTSEDSAVYYCARRGHGNYEDYWGQGTTLIVSSAKTTAPSVYPLAPVCGTGSSVTLGCLVKGYFPEPVTLTWNSGSLSSGVHTFPAVLQSDLYTLSSSVTVTSSTWPSQSITCNVAHPASSTKVDKKIEPRGPTTWSEWSDP Hydrogen bonds contact Hydrophobic contact | ||||
| 13 | Kallikrein-5 (KLK5) | 6QFE | 6.38 | |
Target general information Gen name KLK5 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms UNQ570/PRO1132; Stratum corneum tryptic enzyme; SCTE; Kallikrein-like protein 2; KLK-L2 Protein family Peptidase S1 family, Kallikrein subfamily Biochemical class Peptidase Function May be involved in desquamation. Related diseases Lipodystrophy, familial partial, 8 (FPLD8) [MIM:620679]: An autosomal dominant form of partial lipodystrophy, a disorder characterized by abnormal subcutaneous fat distribution. FPLD8 patients show selective loss of subcutaneous adipose tissue from the limbs, beginning around 13 to 15 years of age, and abnormal accumulation of subcutaneous adipose tissue in the dorsal neck and face, as well as in the posterior thoracic and abdominal regions. The disorder is associated with metabolic abnormalities, including diabetes mellitus and hyperlipidemia. {ECO:0000269|PubMed:27376152}. The disease may be caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P20930; Q9NQG1 EC number EC 3.4.21.- Uniprot keywords 3D-structure; Disulfide bond; Glycoprotein; Hydrolase; Protease; Proteomics identification; Reference proteome; Secreted; Serine protease; Signal Protein physicochemical properties Chain ID A,B Molecular weight (Da) 50299.2 Length 454 Aromaticity 0.07 Instability index 40.74 Isoelectric point 9.25 Charge (pH=7) 23.09 2D Binding mode Binding energy (Kcal/mol) -8.75 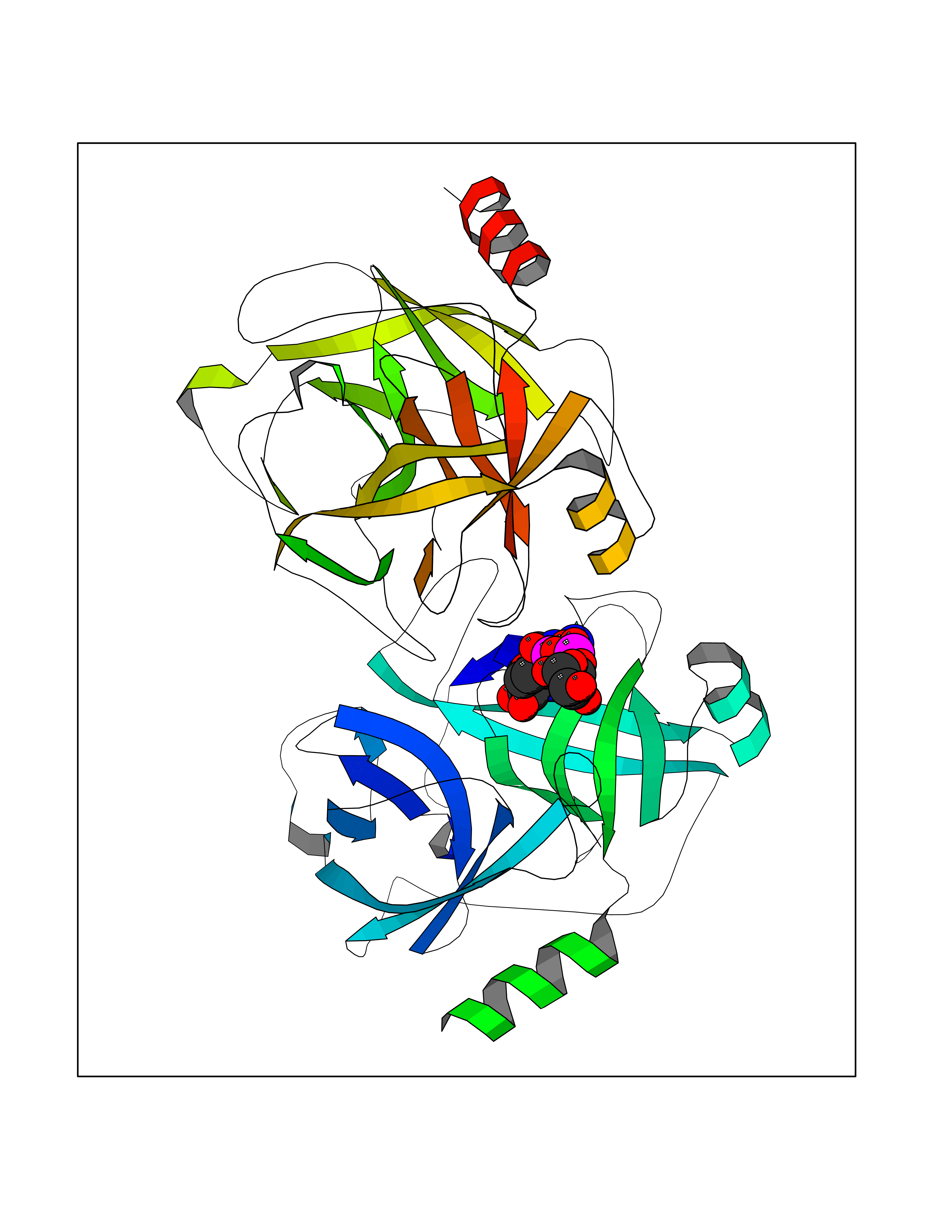 Molscript Map 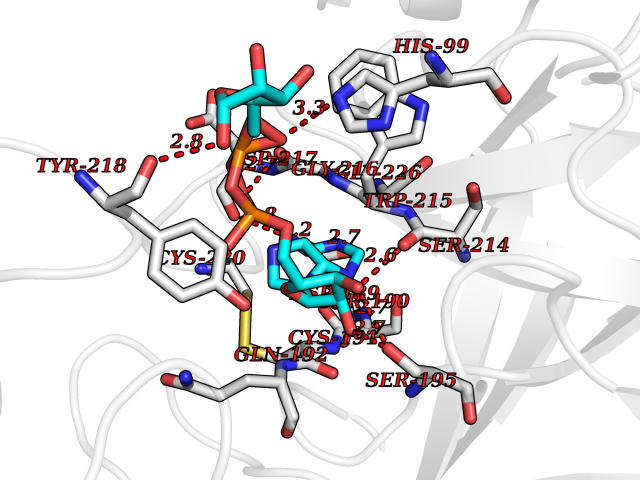 Pymol Map 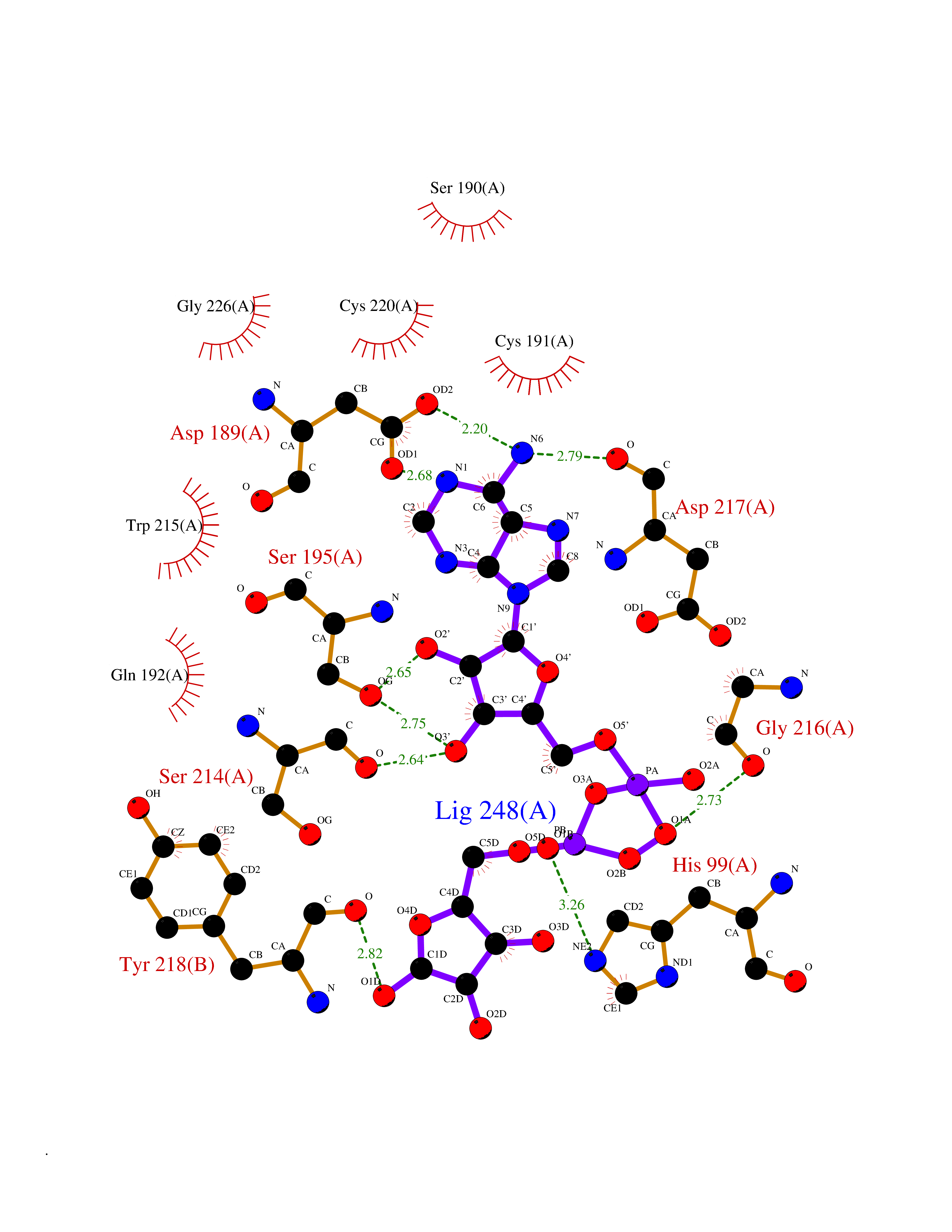 Ligplot Map 3D Binding mode Sequence IINGSDCDMHTQPWQAALLLRPNQLYCGAVLVHPQWLLTAAHCRKKVFRVRLGHYSLSPVYESGQQMFQGVKSIPHPGYSHPGHSNDLMLIKLNRRIRPTKDVRPINVSSHCPSAGTKCLVSGWGTTKSPQVHFPKVLQCLNISVLSQKRCEDAYPRQIDDTMFCAGDKAGRDSCQGDSGGPVVCNGSLQGLVSWGDYPCARPNRPGVYTNLCKFTKWIQETIQANSIINGSDCDMHTQPWQAALLLRPNQLYCGAVLVHPQWLLTAAHCRKKVFRVRLGHYSLSPVYESGQQMFQGVKSIPHPGYSHPGHSNDLMLIKLNRRIRPTKDVRPINVSSHCPSAGTKCLVSGWGTTKSPQVHFPKVLQCLNISVLSQKRCEDAYPRQIDDTMFCAGDKAGRDSCQGDSGGPVVCNGSLQGLVSWGDYPCARPNRPGVYTNLCKFTKWIQETIQANS Hydrogen bonds contact Hydrophobic contact | ||||
| 14 | Macrophage colony-stimulating factor 1 receptor (CSF1R) | 2I1M | 6.38 | |
Target general information Gen name CSF1R Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Proto-oncogene c-Fms; M-CSF-R; FMS; CSF-1R; CSF-1-R; CSF-1 receptor; CD115 Protein family Protein kinase superfamily, Tyr protein kinase family, CSF-1/PDGF receptor subfamily Biochemical class Kinase Function Promotes the release of proinflammatory chemokines in response to IL34 and CSF1, and thereby plays an important role in innate immunity and in inflammatory processes. Plays an important role in the regulation of osteoclast proliferation and differentiation, the regulation of bone resorption, and is required for normal bone and tooth development. Required for normal male and female fertility, and for normal development of milk ducts and acinar structures in the mammary gland during pregnancy. Promotes reorganization of the actin cytoskeleton, regulates formation of membrane ruffles, cell adhesion and cell migration, and promotes cancer cell invasion. Activates several signaling pathways in response to ligand binding. Phosphorylates PIK3R1, PLCG2, GRB2, SLA2 and CBL. Activation of PLCG2 leads to the production of the cellular signaling molecules diacylglycerol and inositol 1,4,5-trisphosphate, that then lead to the activation of protein kinase C family members, especially PRKCD. Phosphorylation of PIK3R1, the regulatory subunit of phosphatidylinositol 3-kinase, leads to activation of the AKT1 signaling pathway. Activated CSF1R also mediates activation of the MAP kinases MAPK1/ERK2 and/or MAPK3/ERK1, and of the SRC family kinases SRC, FYN and YES1. Activated CSF1R transmits signals both via proteins that directly interact with phosphorylated tyrosine residues in its intracellular domain, or via adapter proteins, such as GRB2. Promotes activation of STAT family members STAT3, STAT5A and/or STAT5B. Promotes tyrosine phosphorylation of SHC1 and INPP5D/SHIP-1. Receptor signaling is down-regulated by protein phosphatases, such as INPP5D/SHIP-1, that dephosphorylate the receptor and its downstream effectors, and by rapid internalization of the activated receptor. Tyrosine-protein kinase that acts as cell-surface receptor for CSF1 and IL34 and plays an essential role in the regulation of survival, proliferation and differentiation of hematopoietic precursor cells, especially mononuclear phagocytes, such as macrophages and monocytes. Related diseases Aberrant expression of CSF1 or CSF1R can promote cancer cell proliferation, invasion and formation of metastases. Overexpression of CSF1 or CSF1R is observed in a significant percentage of breast, ovarian, prostate, and endometrial cancers.; DISEASE: Aberrant expression of CSF1 or CSF1R may play a role in inflammatory diseases, such as rheumatoid arthritis, glomerulonephritis, atherosclerosis, and allograft rejection.; DISEASE: Leukoencephalopathy, hereditary diffuse, with spheroids 1 (HDLS1) [MIM:221820]: An autosomal dominant adult-onset rapidly progressive neurodegenerative disorder characterized by variable behavioral, cognitive, and motor changes. Patients often die of dementia within 6 years of onset. Brain imaging shows patchy abnormalities in the cerebral white matter, predominantly affecting the frontal and parietal lobes. {ECO:0000269|PubMed:22197934, ECO:0000269|PubMed:23408870, ECO:0000269|PubMed:24336230, ECO:0000269|PubMed:24532199}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Brain abnormalities, neurodegeneration, and dysosteosclerosis (BANDDOS) [MIM:618476]: An autosomal recessive disease with variable manifestations. Main features are brain malformations with calcifying leukoencephalopathy, progressive neurodegeneration, and bone sclerotic features. The age at onset ranges from infancy to early adulthood. Neurologic features include loss of previous motor and language skills, cognitive impairment, spasticity, and focal seizures. Brain imaging shows periventricular white matter abnormalities and calcifications, large cisterna magna or Dandy-Walker malformation, and sometimes agenesis of the corpus callosum. {ECO:0000269|PubMed:30982608, ECO:0000269|PubMed:30982609}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07167; DB07202; DB12147; DB12010; DB00619; DB06080; DB12978; DB01268 Interacts with P09603; Q15375; P29323; Q6ZMJ4-1 EC number EC 2.7.10.1 Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Cell membrane; Disease variant; Disulfide bond; Glycoprotein; Immunity; Immunoglobulin domain; Inflammatory response; Innate immunity; Kinase; Membrane; Neurodegeneration; Nucleotide-binding; Phosphoprotein; Proteomics identification; Proto-oncogene; Receptor; Reference proteome; Repeat; Signal; Transferase; Transmembrane; Transmembrane helix; Tyrosine-protein kinase; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 35082.9 Length 311 Aromaticity 0.11 Instability index 44.6 Isoelectric point 8.13 Charge (pH=7) 2.42 2D Binding mode Binding energy (Kcal/mol) -8.1 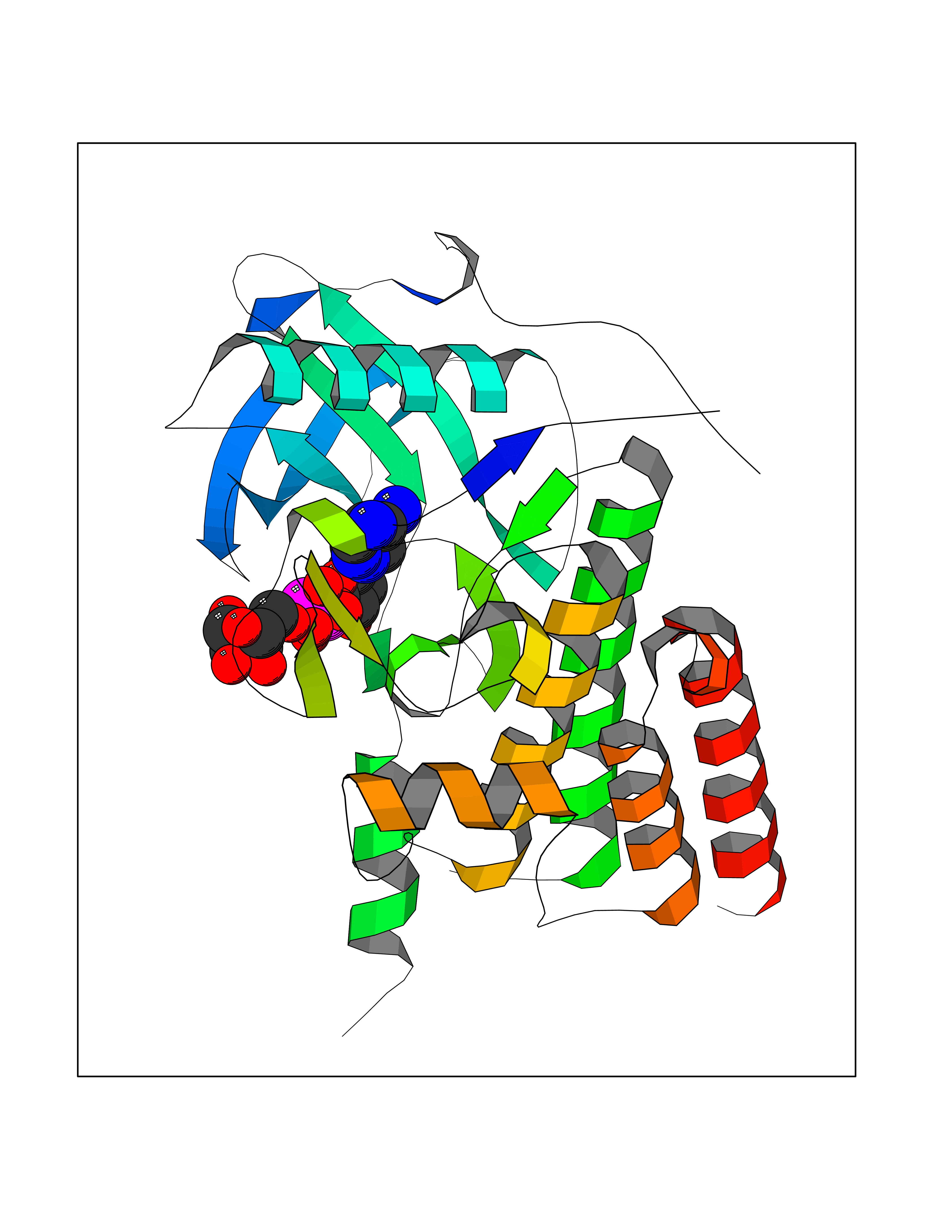 Molscript Map 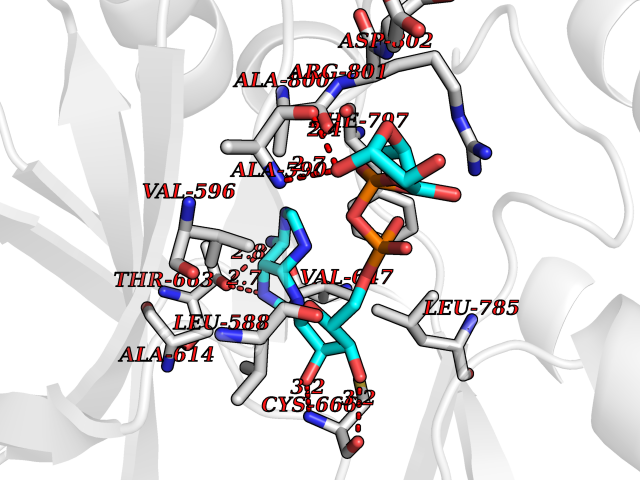 Pymol Map 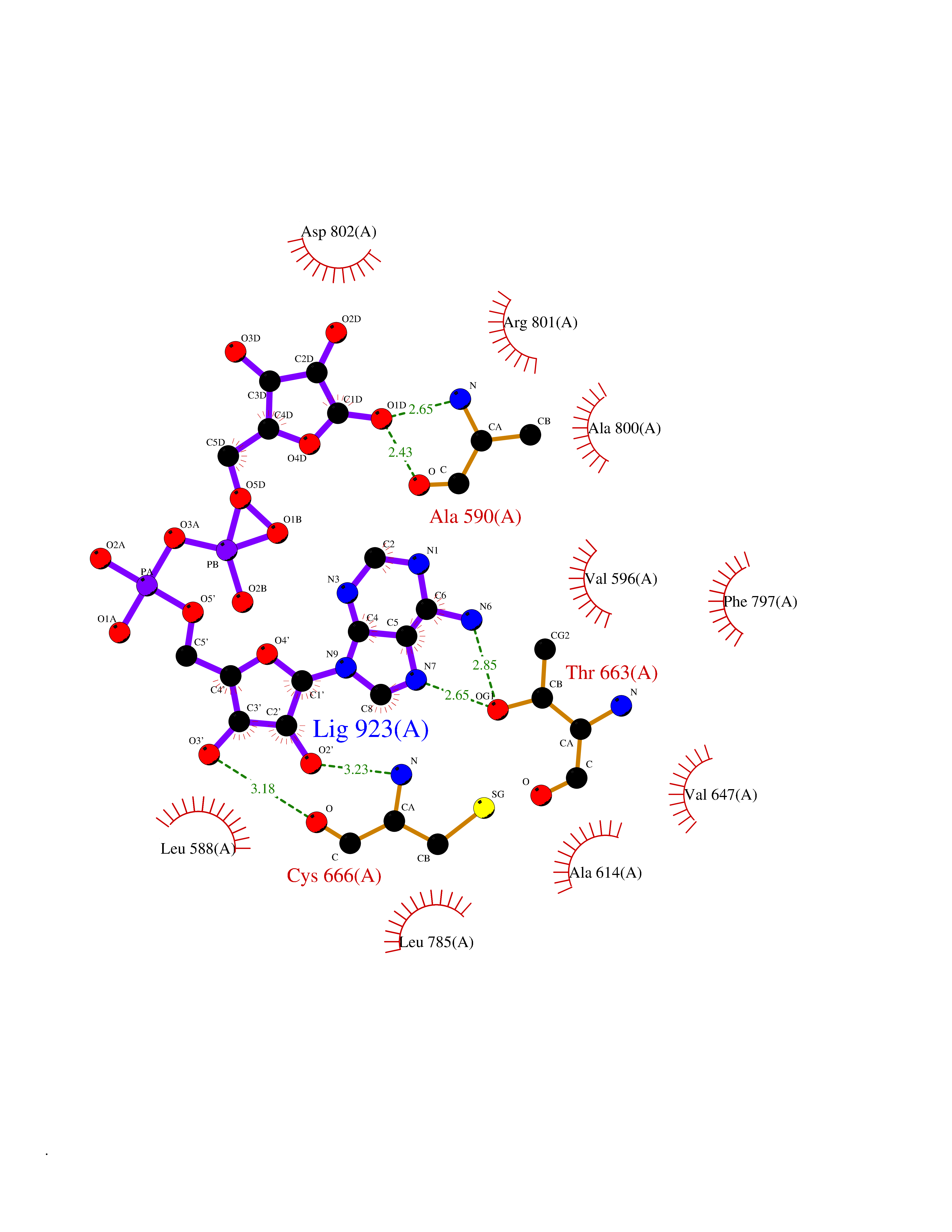 Ligplot Map 3D Binding mode Sequence QVRWKIIESYNSYTFIDPTQLPYNEKWEFPRNNLQFGKTLGAGAFGKVVEATAFGLGKEDAVLKVAVKMLKSTAHADEKEALMSELKIMSHLGQHENIVNLLGACTHGGPVLVITEYCCYGDLLNFLRRKSRVLETDSTASTRDLLHFSSQVAQGMAFLASKNCIHRDVAARNVLLTNGHVAKIGDFGLARDIMNDSNYIVKGNARLPVKWMAPESIFDCVYTVQSDVWSYGILLWEIFSLGLNPYPGILVNSKFYKLVKDGYQMAQPAFAPKNIYSIMQACWALEPTHRPTFQQICSFLQEQAQEDRRER Hydrogen bonds contact Hydrophobic contact | ||||
| 15 | Platelet glycoprotein VI (GP6) | 5OU7 | 6.37 | |
Target general information Gen name GP6 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Glycoprotein 6; GPVI Protein family NA Biochemical class NA Function Collagen receptor involved in collagen-induced platelet adhesion and activation. Plays a key role in platelet procoagulant activity and subsequent thrombin and fibrin formation. This procoagulant function may contribute to arterial and venous thrombus formation. The signaling pathway involves the FcR gamma-chain, the Src kinases (likely FYN or LYN) and SYK, the adapter protein LAT and leads to the activation of PLCG2. Related diseases Bleeding disorder, platelet-type, 11 (BDPLT11) [MIM:614201]: A mild to moderate bleeding disorder caused by defective platelet activation and aggregation in response to collagen. {ECO:0000269|PubMed:19549989, ECO:0000269|PubMed:19552682}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P06241; P07948; P06241; P07948 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Blood coagulation; Cell membrane; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; Hemostasis; Immunoglobulin domain; Membrane; Proteomics identification; Receptor; Reference proteome; Repeat; Signal; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 19027.4 Length 173 Aromaticity 0.11 Instability index 37.14 Isoelectric point 8.68 Charge (pH=7) 2.52 2D Binding mode Binding energy (Kcal/mol) -6.9 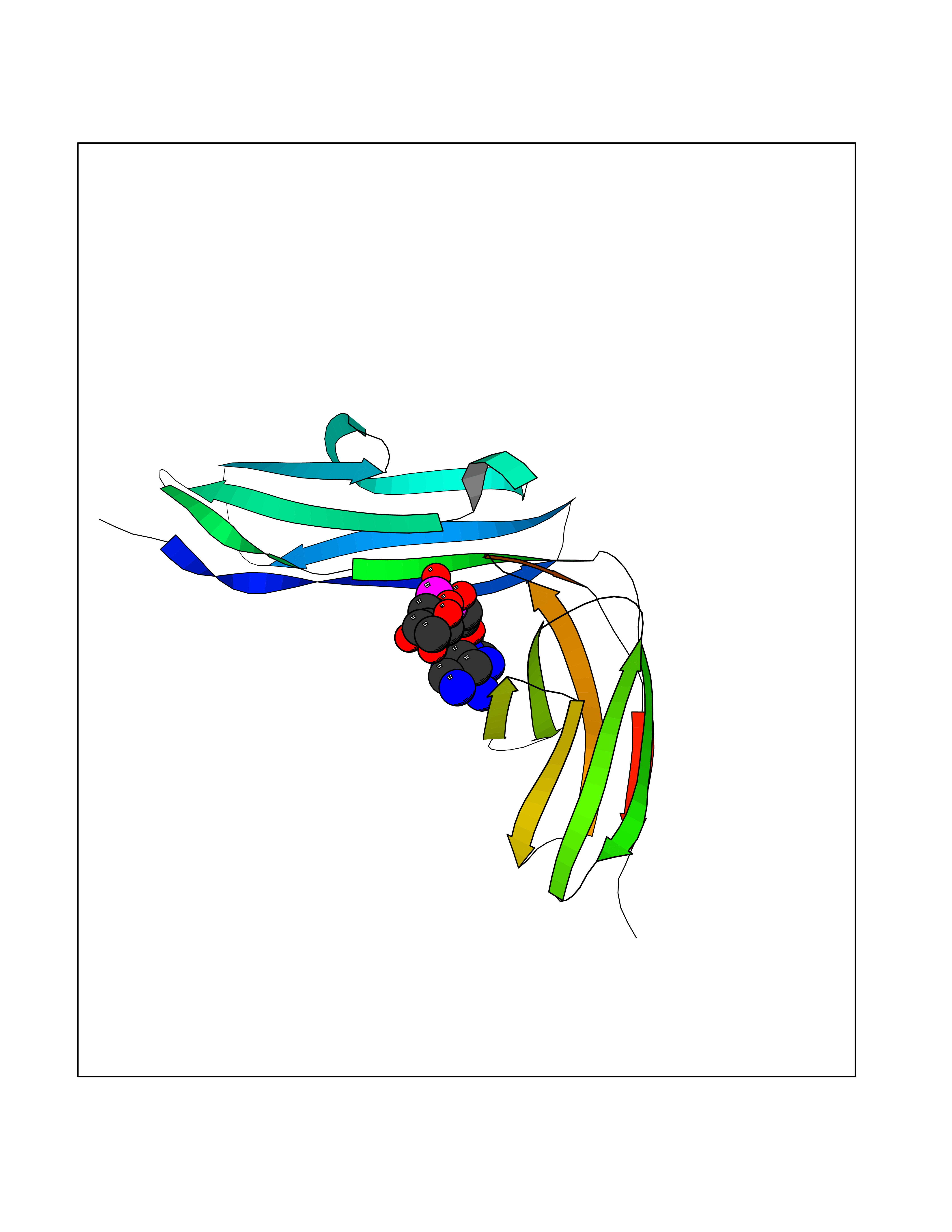 Molscript Map 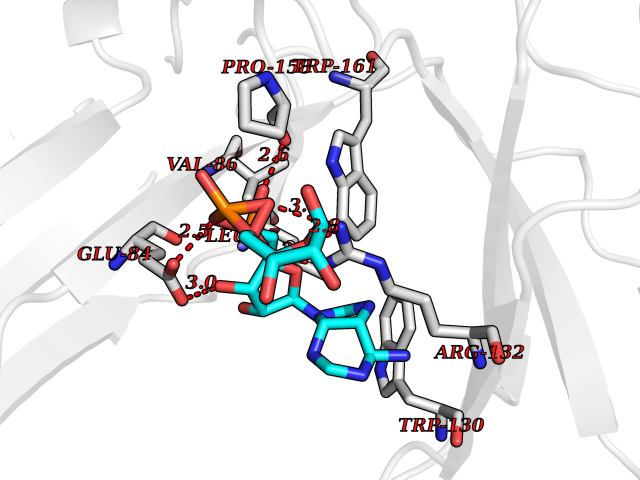 Pymol Map 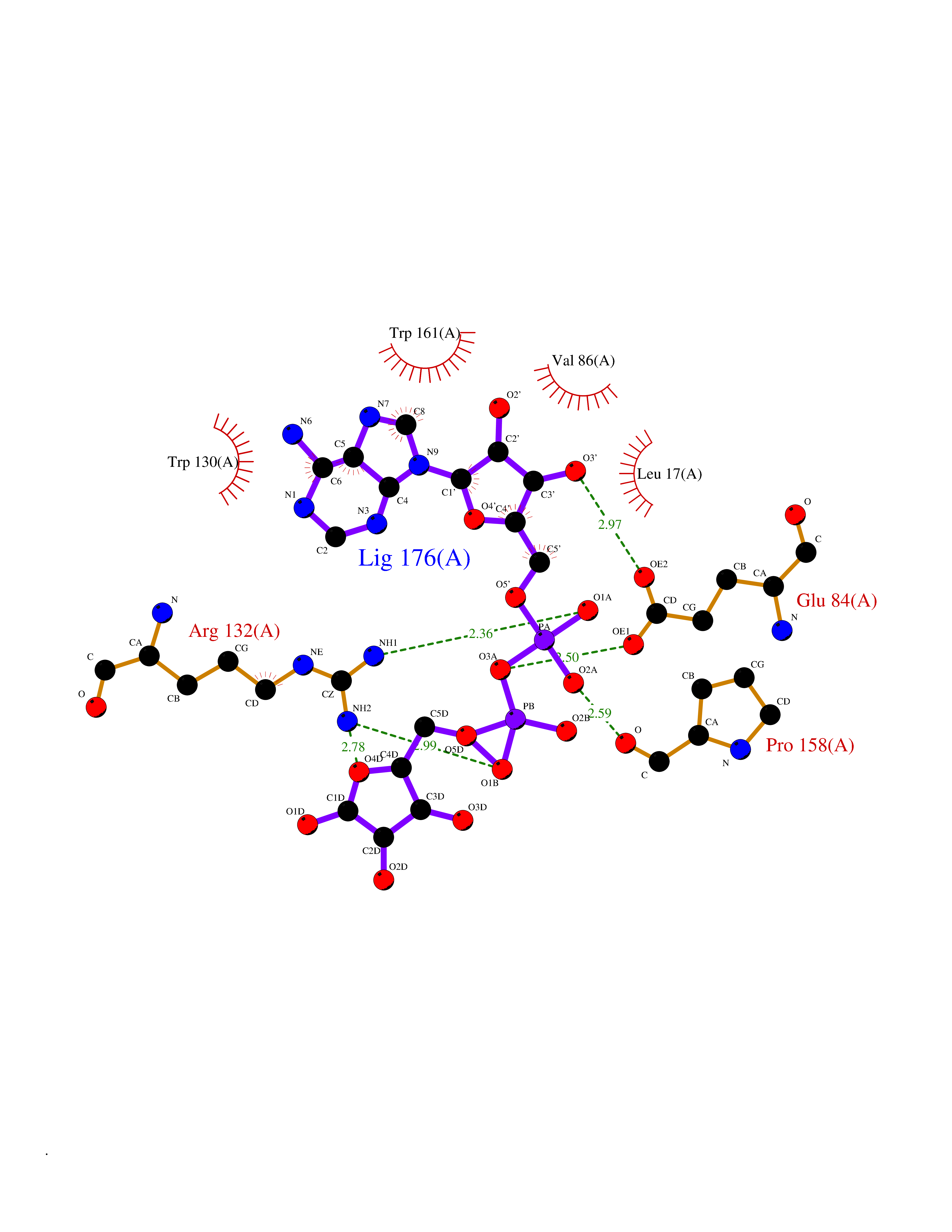 Ligplot Map 3D Binding mode Sequence SGPLPKPSLQALPSSLVPLEKPVTLRCQGPPGVDLYRLEKLSSSRYQDQAVLFIPAMKRSLAGRYRCSYQNGSLWSLPSDQLELVATGVFAKPSLSAQPGSGGDVTLQCQTRYGFDQFALYKEGDPERWYRASFPIITVTAAHSGTYRCYSFSSRDPYLWSAPSDPLELVVTG Hydrogen bonds contact Hydrophobic contact | ||||
| 16 | Carbonic anhydrase IV (CA-IV) | 3FW3 | 6.36 | |
Target general information Gen name CA4 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Carbonic anhydrase 4; Carbonate dehydratase IV; CAIV Protein family Alpha-carbonic anhydrase family Biochemical class Alpha-carbonic anhydrase Function May stimulate the sodium/bicarbonate transporter activity of SLC4A4 that acts in pH homeostasis. It is essential for acid overload removal from the retina and retina epithelium, and acid release in the choriocapillaris in the choroid. Reversible hydration of carbon dioxide. Related diseases Retinitis pigmentosa 17 (RP17) [MIM:600852]: A retinal dystrophy belonging to the group of pigmentary retinopathies. Retinitis pigmentosa is characterized by retinal pigment deposits visible on fundus examination and primary loss of rod photoreceptor cells followed by secondary loss of cone photoreceptors. Patients typically have night vision blindness and loss of midperipheral visual field. As their condition progresses, they lose their far peripheral visual field and eventually central vision as well. {ECO:0000269|PubMed:15563508, ECO:0000269|PubMed:17652713, ECO:0000269|PubMed:20450258}. The disease is caused by variants affecting the gene represented in this entry. Defective acid overload removal from retina and retinal epithelium, due to mutant CA4, is responsible for photoreceptor degeneration, indicating that impaired pH homeostasis is the most likely cause underlying the RP17 phenotype. Drugs (DrugBank ID) DB00819; DB00436; DB00562; DB01194; DB00606; DB01144; DB00869; DB08846; DB00311; DB00774; DB00703; DB00232; DB09460; DB00273; DB01021; DB00909 Interacts with NA EC number EC 4.2.1.1 Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; GPI-anchor; Lipoprotein; Lyase; Membrane; Metal-binding; Proteomics identification; Reference proteome; Retinitis pigmentosa; Signal; Zinc Protein physicochemical properties Chain ID A,B Molecular weight (Da) 27055.7 Length 235 Aromaticity 0.09 Instability index 44.3 Isoelectric point 6.87 Charge (pH=7) -0.36 2D Binding mode Binding energy (Kcal/mol) -7.31  Molscript Map 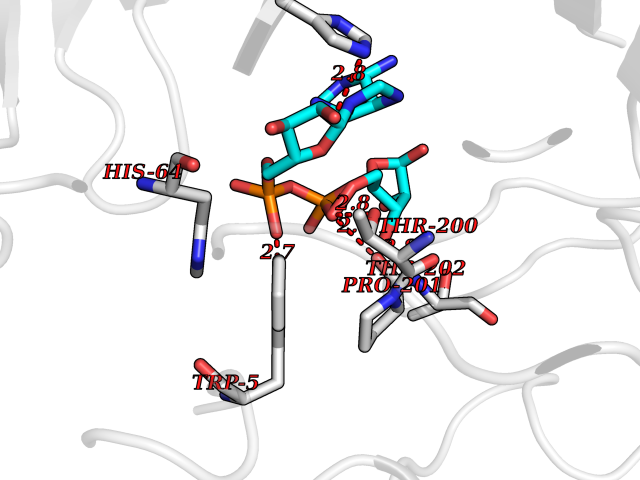 Pymol Map 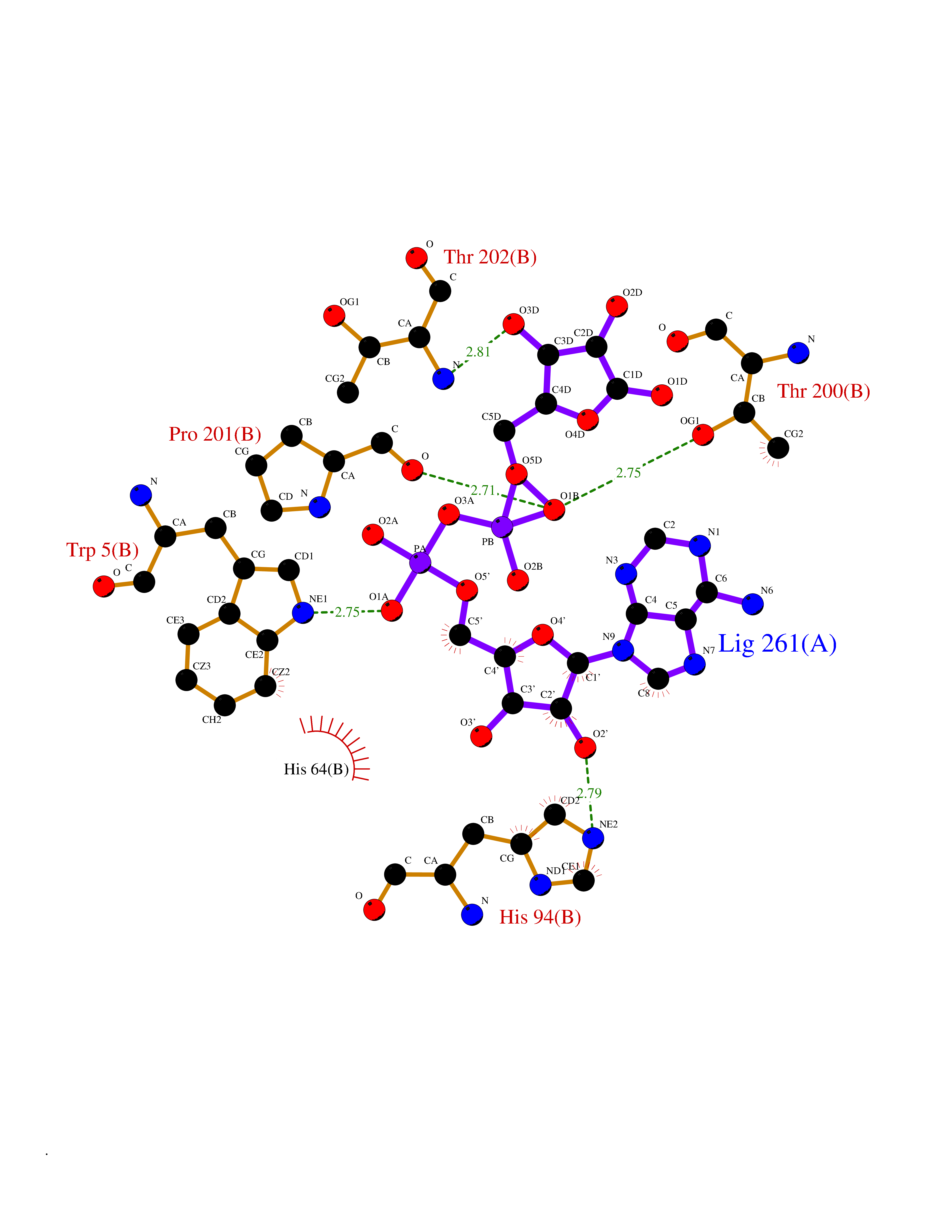 Ligplot Map 3D Binding mode Sequence HWCYEVQLVPVKWGGNCQKDRQSPINIVTTKAKVDKKLGRFFFGYDKKQTWTVQNNGHSVMMLLENKASISGGGLPAPYQAKQLHLHWSDLPYKGSEHSLDGEHFAMEMHIVHEKEEIAVLAFLVEATQVNEGFQPLVEALSNIPKPEMSTTMAESSLLDLLPEEKRHYFRYLGSLTTPTCDEKVVWTVFREPIQLHREQILAFQKLYYDKEQTVSMKDNVRPLQQLGQRTVIKS Hydrogen bonds contact Hydrophobic contact | ||||
| 17 | Platelet-derived growth factor receptor alpha (PDGFRA) | 5K5X | 6.34 | |
Target general information Gen name PDGFRA Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms RHEPDGFRA; Platelet-derived growth factor receptor 2; Platelet-derived growth factor alpha receptor; PDGFR2; PDGFR-alpha; PDGFR-2; PDGF-R-alpha; CD140a antigen; CD140a; CD140 antigen-like family membe Protein family Protein kinase superfamily, Tyr protein kinase family, CSF-1/PDGF receptor subfamily Biochemical class Kinase Function Depending on the context, promotes or inhibits cell proliferation and cell migration. Plays an important role in the differentiation of bone marrow-derived mesenchymal stem cells. Required for normal skeleton development and cephalic closure during embryonic development. Required for normal development of the mucosa lining the gastrointestinal tract, and for recruitment of mesenchymal cells and normal development of intestinal villi. Plays a role in cell migration and chemotaxis in wound healing. Plays a role in platelet activation, secretion of agonists from platelet granules, and in thrombin-induced platelet aggregation. Binding of its cognate ligands - homodimeric PDGFA, homodimeric PDGFB, heterodimers formed by PDGFA and PDGFB or homodimeric PDGFC -leads to the activation of several signaling cascades; the response depends on the nature of the bound ligand and is modulated by the formation of heterodimers between PDGFRA and PDGFRB. Phosphorylates PIK3R1, PLCG1, and PTPN11. Activation of PLCG1 leads to the production of the cellular signaling molecules diacylglycerol and inositol 1,4,5-trisphosphate, mobilization of cytosolic Ca(2+) and the activation of protein kinase C. Phosphorylates PIK3R1, the regulatory subunit of phosphatidylinositol 3-kinase, and thereby mediates activation of the AKT1 signaling pathway. Mediates activation of HRAS and of the MAP kinases MAPK1/ERK2 and/or MAPK3/ERK1. Promotes activation of STAT family members STAT1, STAT3 and STAT5A and/or STAT5B. Receptor signaling is down-regulated by protein phosphatases that dephosphorylate the receptor and its down-stream effectors, and by rapid internalization of the activated receptor. Tyrosine-protein kinase that acts as a cell-surface receptor for PDGFA, PDGFB and PDGFC and plays an essential role in the regulation of embryonic development, cell proliferation, survival and chemotaxis. Related diseases A chromosomal aberration involving PDGFRA is found in some cases of hypereosinophilic syndrome. Interstitial chromosomal deletion del(4)(q12q12) causes the fusion of FIP1L1 and PDGFRA (FIP1L1-PDGFRA). Mutations that cause overexpression and/or constitutive activation of PDGFRA may be a cause of hypereosinophilic syndrome. {ECO:0000269|PubMed:12808148}.; DISEASE: Gastrointestinal stromal tumor (GIST) [MIM:606764]: Common mesenchymal neoplasms arising in the gastrointestinal tract, most often in the stomach. They are histologically, immunohistochemically, and genetically different from typical leiomyomas, leiomyosarcomas, and schwannomas. Most GISTs are composed of a fairly uniform population of spindle-shaped cells. Some tumors are dominated by epithelioid cells or contain a mixture of spindle and epithelioid morphologies. Primary GISTs in the gastrointestinal tract commonly metastasize in the omentum and mesenteries, often as multiple nodules. However, primary tumors may also occur outside of the gastrointestinal tract, in other intra-abdominal locations, especially in the omentum and mesentery. {ECO:0000269|PubMed:12522257, ECO:0000269|PubMed:15928335}. The gene represented in this entry may be involved in disease pathogenesis. Mutations causing PDGFRA constitutive activation have been found in gastrointestinal stromal tumors lacking KIT mutations (PubMed:12522257). {ECO:0000269|PubMed:12522257}.; DISEASE: GIST-plus syndrome (GISTPS) [MIM:175510]: A disorder characterized by multiple mesenchymal tumors of the gastrointestinal tract, including gastrointestinal stromal tumor, inflammatory fibroid polyps, and fibroid tumors. Additional features are coarse facies and skin, broad hands and feet, and premature tooth loss. GISTPS is an autosomal dominant disease with incomplete penetrance. Gastrointestinal stromal tumor and inflammatory fibroid polyps may also occur in isolation. {ECO:0000269|PubMed:14699510, ECO:0000269|PubMed:17087943, ECO:0000269|PubMed:25975287}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB12742; DB00102; DB12147; DB10772; DB12010; DB00619; DB09078; DB06595; DB09079; DB06043; DB06589; DB08901; DB08896; DB14840; DB01268; DB11800; DB05146 Interacts with P46108; P46109; P00533; Q8N6L0; P04085; P01127; Q9NRA1; P31947; P62258; Q9NRA1-1; A8T7D5; P05067; Q8IY26 EC number EC 2.7.10.1 Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Cell membrane; Cell projection; Chemotaxis; Developmental protein; Disease variant; Disulfide bond; Glycoprotein; Golgi apparatus; Host-virus interaction; Immunoglobulin domain; Kinase; Membrane; Nucleotide-binding; Phosphoprotein; Proteomics identification; Proto-oncogene; Receptor; Reference proteome; Repeat; Signal; Transferase; Transmembrane; Transmembrane helix; Tyrosine-protein kinase; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 39294.8 Length 345 Aromaticity 0.11 Instability index 46.74 Isoelectric point 6.6 Charge (pH=7) -1.38 2D Binding mode Binding energy (Kcal/mol) -7.86 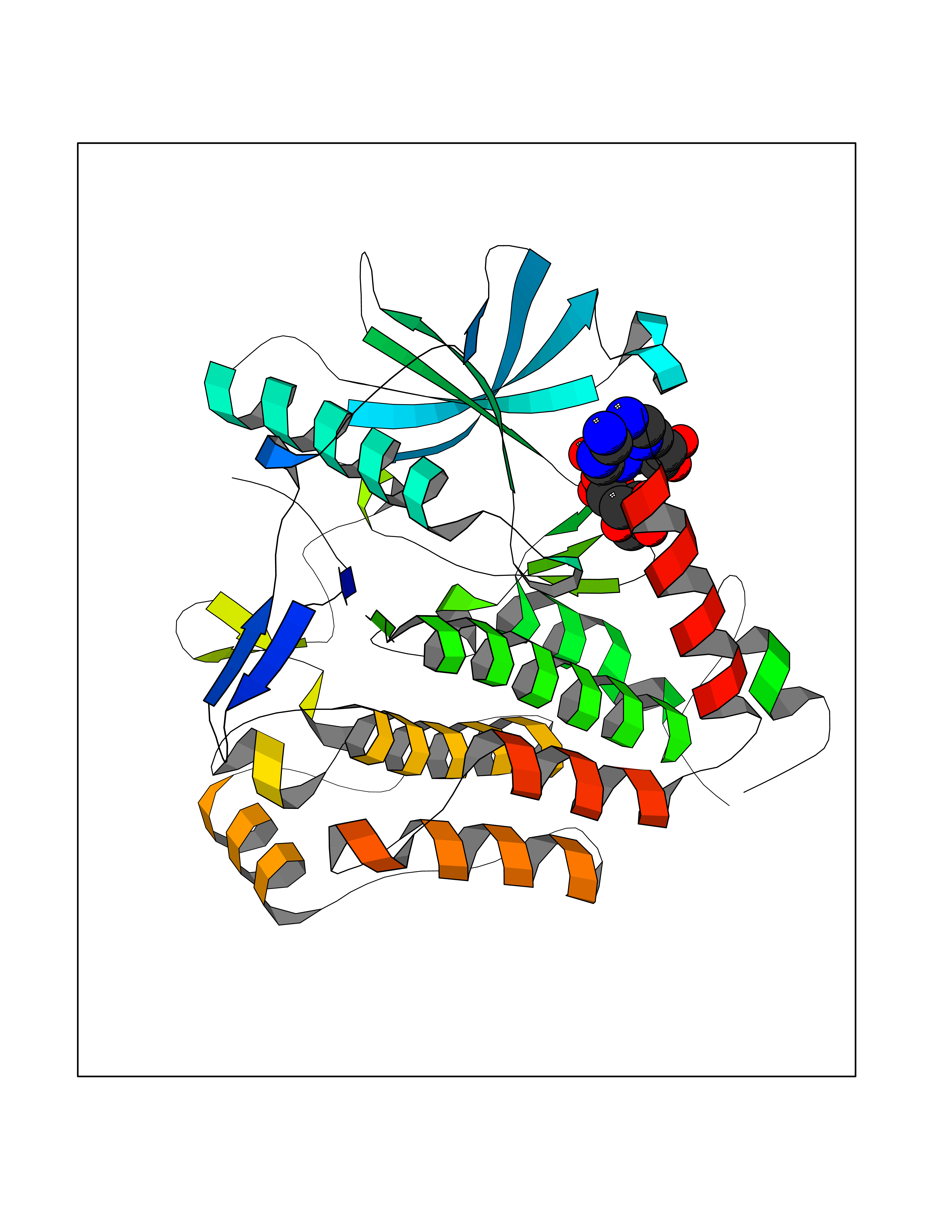 Molscript Map 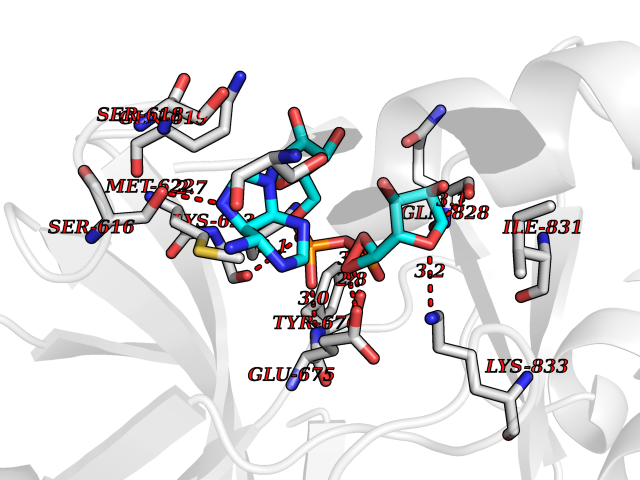 Pymol Map 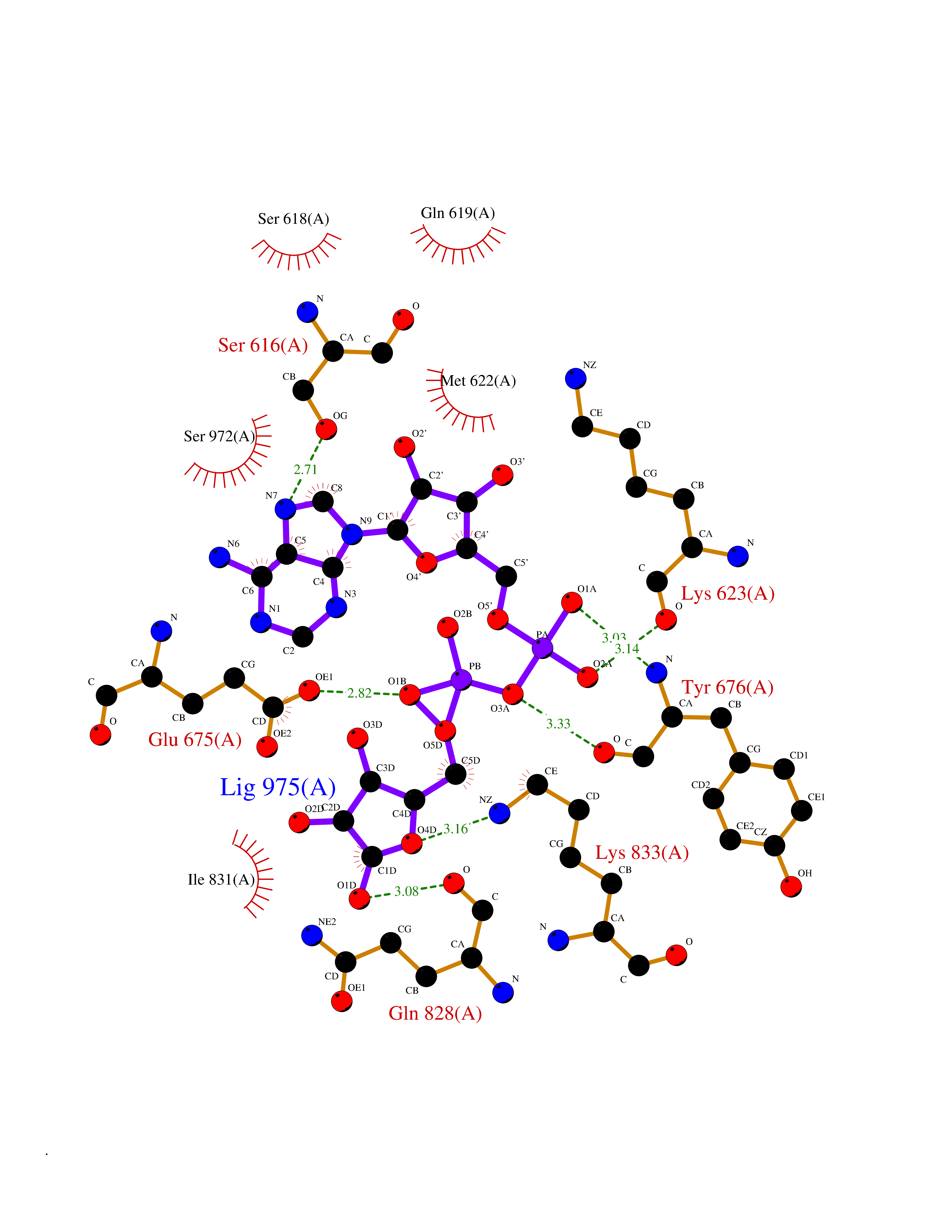 Ligplot Map 3D Binding mode Sequence RYEIRWRVIESISPDGHEYIYVDPMQLPYDSRWEFPRDGLVLGRVLGSGAFGKVVEGTAYGLSRSQPVMKVAVKMLKPTARSSEKQALMSELKIMTHLGPHLNIVNLLGACTKSGPIYIITEYCFYGDLVNYLHKNRDSFLSHSMLDSEVKNLLSDDNSEGLTLLDLLSFTYQVARGMEFLASKNCVHRDLAARNVLLAQGKIVKICDFGLARDIMHDSNYVSKGSTFLPVKWMAPESIFDNLYTTLSDVWSYGILLWEIFSLGGTPYPGMMVDSTFYNKIKSGYRMAKPDHATSEVYEIMVKCWNSEPEKRPSFYHLSEIVENLLPGQYKKSYEKIHLDFLKSD Hydrogen bonds contact Hydrophobic contact | ||||
| 18 | Mucolipin-1 (TRPML1) | 7MGL | 6.34 | |
Target general information Gen name MCOLN1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Transient receptor potential channel mucolipin 1; TRPML1; Mucolipidin; MSTP080; ML4; ML1; MG-2 Protein family Transient receptor (TC 1.A.4) family, Polycystin subfamily, MCOLN1 sub-subfamily Biochemical class NA Function Nonselective cation channel probably playing a role in the regulation of membrane trafficking events and of metal homeostasis. Proposed to play a major role in Ca(2+) release from late endosome and lysosome vesicles to the cytoplasm, which is important for many lysosome-dependent cellular events, including the fusion and trafficking of these organelles, exocytosis and autophagy. Required for efficient uptake of large particles in macrophages in which Ca(2+) release from the lysosomes triggers lysosomal exocytosis. May also play a role in phagosome-lysosome fusion (By similarity). Involved in lactosylceramide trafficking indicative for a role in the regulation of late endocytic membrane fusion/fission events. By mediating lysosomal Ca(2+) release is involved in regulation of mTORC1 signaling and in mTOR/TFEB-dependent lysosomal adaptation to environmental cues such as nutrient levels. Seems to act as lysosomal active oxygen species (ROS) sensor involved in ROS-induced TFEB activation and autophagy. Functions as a Fe(2+) permeable channel in late endosomes and lysosomes. Proposed to play a role in zinc homeostasis probably implicating its association with TMEM163 In adaptive immunity, TRPML2 and TRPML1 may play redundant roles in the function of the specialized lysosomes of B cells (By similarity). Related diseases Mucolipidosis 4 (ML4) [MIM:252650]: An autosomal recessive lysosomal storage disorder characterized by severe psychomotor retardation and ophthalmologic abnormalities, including corneal opacity, retinal degeneration and strabismus. Storage bodies of lipids and water-soluble substances are seen by electron microscopy in almost every cell type of the patients. Most patients are unable to speak or walk independently and reach a maximal developmental level of 1-2 years. All patients have constitutive achlorhydia associated with a secondary elevation of serum gastrin levels. {ECO:0000269|PubMed:10973263, ECO:0000269|PubMed:11013137, ECO:0000269|PubMed:11030752, ECO:0000269|PubMed:11317355, ECO:0000269|PubMed:12182165, ECO:0000269|PubMed:14749347, ECO:0000269|PubMed:15178326, ECO:0000269|PubMed:15523648, ECO:0000269|PubMed:16978393, ECO:0000269|PubMed:18794901, ECO:0000269|PubMed:20159435, ECO:0000269|PubMed:21256127, ECO:0000269|PubMed:28112729}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Corneal dystrophy, Lisch epithelial (LECD) [MIM:620763]: An autosomal dominant corneal dystrophy characterized by gray, band-shaped and feathery opacities in the cornea, that sometimes appear in whorled patterns. The opaque bands consist of clear, densely crowded, intra-epithelial blisters. Vision may be impaired if the bands involve the central cornea. {ECO:0000269|PubMed:37972748}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q92624; Q9GZU1; Q96IW7 EC number NA Uniprot keywords 3D-structure; Adaptive immunity; Calcium; Calcium transport; Cell membrane; Cell projection; Corneal dystrophy; Cytoplasmic vesicle; Disease variant; Disulfide bond; Endosome; Glycoprotein; Immunity; Ion channel; Ion transport; Lipid-binding; Lipoprotein; Lysosome; Membrane; Mucolipidosis; Palmitate; Phosphoprotein; Proteomics identification; Reference proteome; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A,D Molecular weight (Da) 107937 Length 944 Aromaticity 0.13 Instability index 33.13 Isoelectric point 8.76 Charge (pH=7) 17.77 2D Binding mode Binding energy (Kcal/mol) -7.73 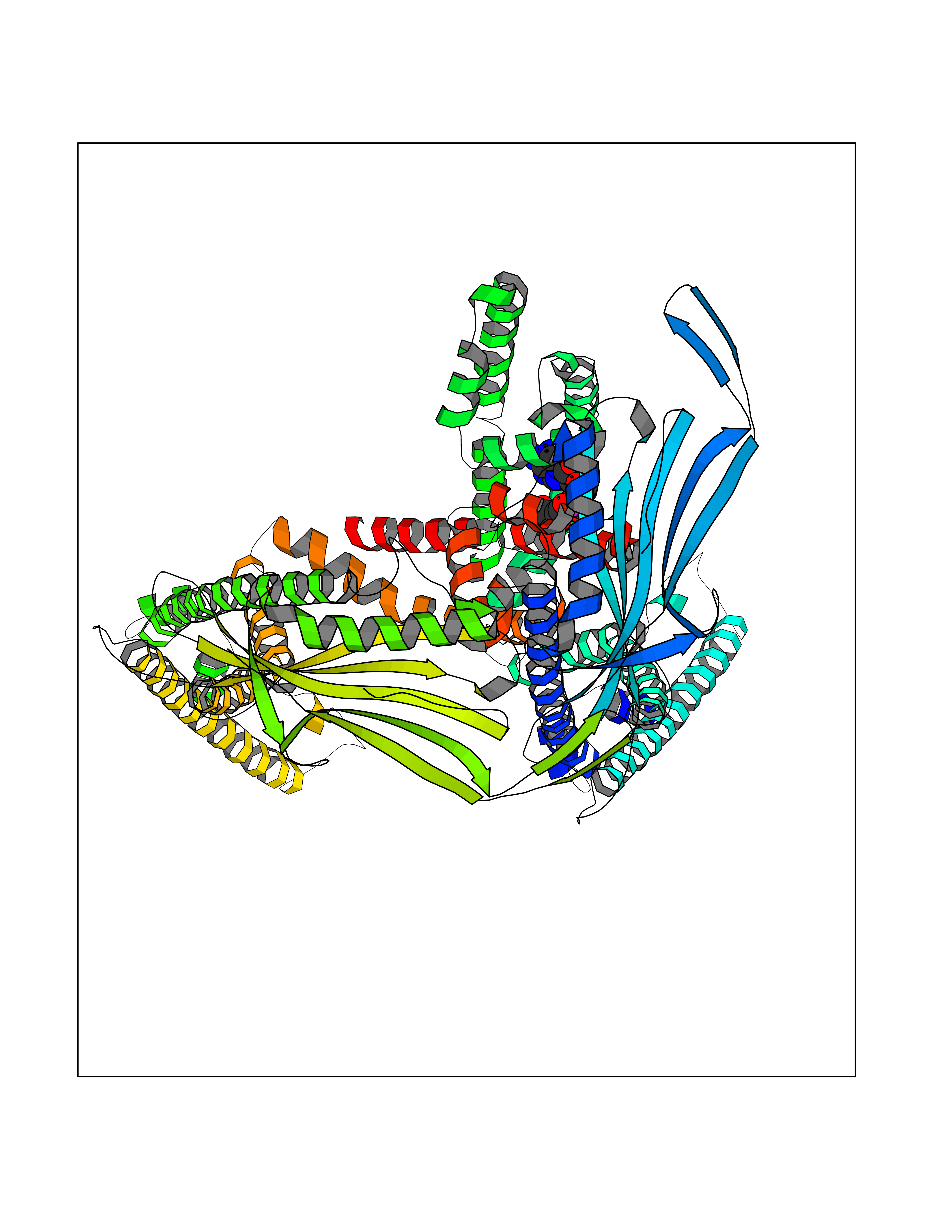 Molscript Map 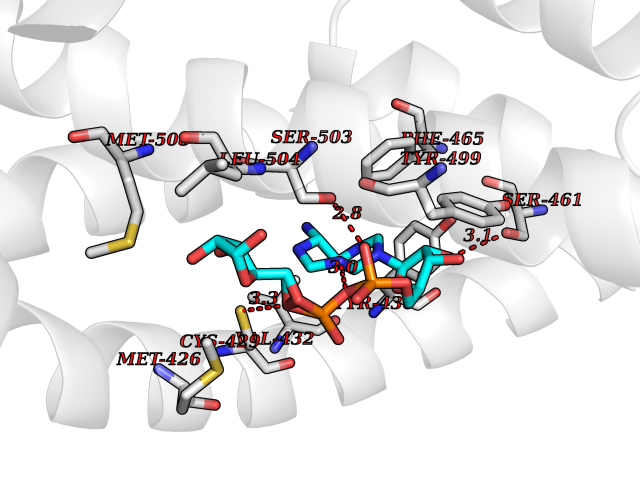 Pymol Map 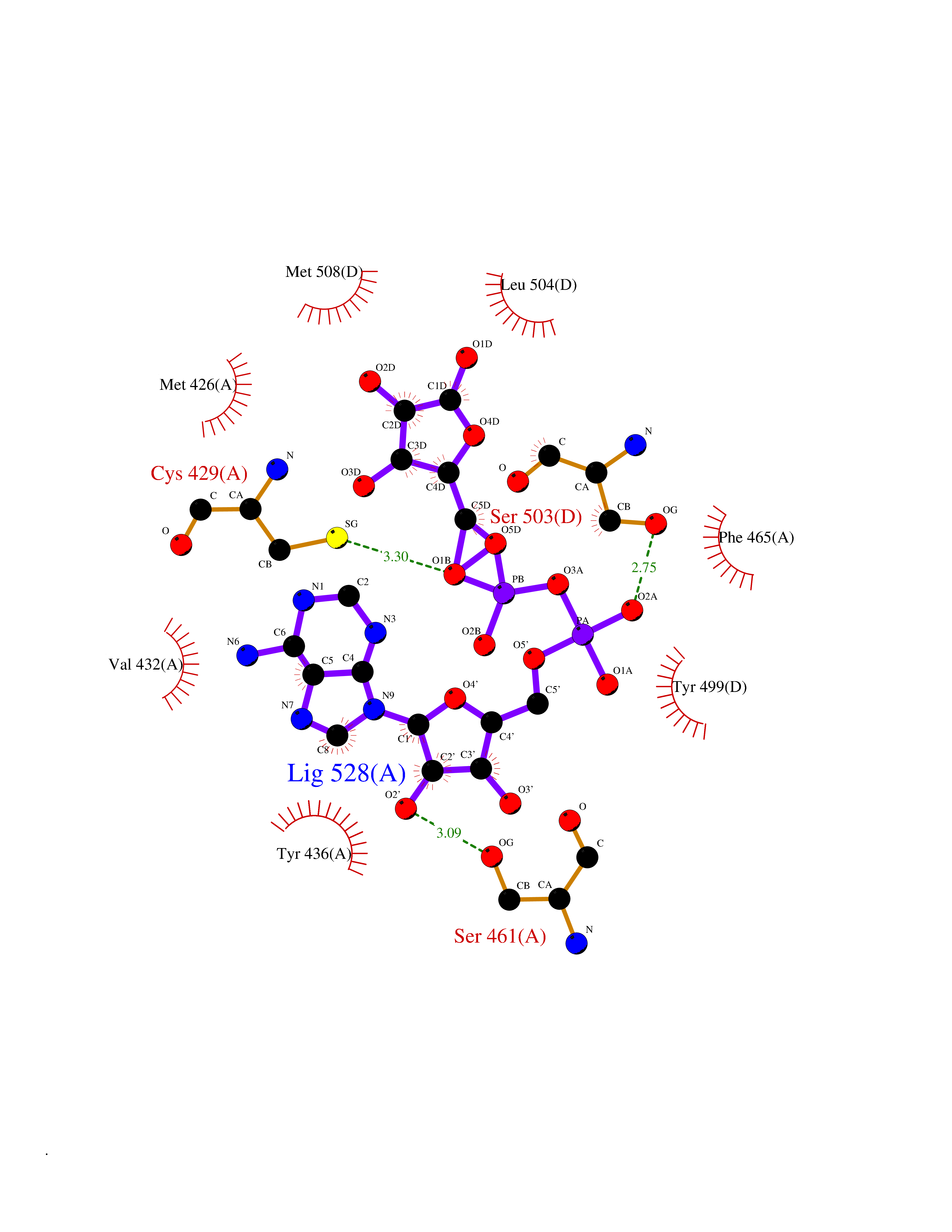 Ligplot Map 3D Binding mode Sequence DLRRRLKYFFMSPCDKFRAKGRKPCKLMLQVVKILVVTVQLILFGLSNQLAVTFREENTIAFRHLFLLGYSDGADDTFAAYTREQLYQAIFHAVDQYLALPDVSLGRYAYVRGGGDPWTNGSGLALCQRYYHRGHVDPANDTFDIDPMVVTDCIQVDPPERSSYKNLTLKFHKLVNVTIHFRLKTINLQSLINNEIPDCYTFSVLITFDNKAHSGRIPISLETQAHIQECKHPSVFQHGDNSFRLLFDVVVILTCSLSFLLCARSLLRGFLLQNEFVGFMWRQRGRVISLWERLEFVNGWYILLVTSDVLTISGTIMKIGIEAKNLASYDVCSILLGTSTLLVWVGVIRYLTFFHNYNILIATLRVALPSVMRFCCCVAVIYLGYCFCGWIVLGPYHVKFRSLSMVSECLFSLINGDDMFVTFAAMQAQQGRSSLVWLFSQLYLYSFISLFIYMVLSLFIALITGAYDTIKHDLRRRLKYFFMSPCDKFRAKGRKPCKLMLQVVKILVVTVQLILFGLSNQLAVTFREENTIAFRHLFLLGYSDGADDTFAAYTREQLYQAIFHAVDQYLALPDVSLGRYAYVRGGGDPWTNGSGLALCQRYYHRGHVDPANDTFDIDPMVVTDCIQVDPPERSSYKNLTLKFHKLVNVTIHFRLKTINLQSLINNEIPDCYTFSVLITFDNKAHSGRIPISLETQAHIQECKHPSVFQHGDNSFRLLFDVVVILTCSLSFLLCARSLLRGFLLQNEFVGFMWRQRGRVISLWERLEFVNGWYILLVTSDVLTISGTIMKIGIEAKNLASYDVCSILLGTSTLLVWVGVIRYLTFFHNYNILIATLRVALPSVMRFCCCVAVIYLGYCFCGWIVLGPYHVKFRSLSMVSECLFSLINGDDMFVTFAAMQAQQGRSSLVWLFSQLYLYSFISLFIYMVLSLFIALITGAYDTIKH Hydrogen bonds contact Hydrophobic contact | ||||
| 19 | Kinesin-like protein KIF11 (KIF11) | 6HKY | 6.34 | |
Target general information Gen name KIF11 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Thyroid receptor interacting protein 5; TRIP5; Kinesin-related motor protein Eg5; Kinesin-like spindle protein HKSP; Kinesin-like protein 1; KIF11; Eg5 Protein family TRAFAC class myosin-kinesin ATPase superfamily, Kinesin family, BimC subfamily Biochemical class Kinesin-like protein family Function Motor protein required for establishing a bipolar spindle. Blocking of KIF11 prevents centrosome migration and arrest cells in mitosis with monoastral microtubule arrays. Related diseases Microcephaly with or without chorioretinopathy, lymphedema, or impaired intellectual development (MCLMR) [MIM:152950]: An autosomal dominant disorder that involves an overlapping but variable spectrum of central nervous system and ocular developmental anomalies. Microcephaly ranges from mild to severe and is often associated with mild to moderate developmental delay and a characteristic facial phenotype with upslanting palpebral fissures, broad nose with rounded tip, long philtrum with thin upper lip, prominent chin, and prominent ears. Chorioretinopathy is the most common eye abnormality, but retinal folds, microphthalmia, and myopic and hypermetropic astigmatism have also been reported, and some individuals have no overt ocular phenotype. Congenital lymphedema, when present, is typically confined to the dorsa of the feet, and lymphoscintigraphy reveals the absence of radioactive isotope uptake from the webspaces between the toes. {ECO:0000269|PubMed:22284827}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08244; DB08246; DB08239; DB07064; DB08033; DB08250; DB03996; DB08198; DB06040; DB08037; DB04331; DB08032 Interacts with NA EC number NA Uniprot keywords 3D-structure; Acetylation; ATP-binding; Cell cycle; Cell division; Coiled coil; Cytoplasm; Cytoskeleton; Disease variant; Isopeptide bond; Microtubule; Mitosis; Motor protein; Nucleotide-binding; Phosphoprotein; Proteomics identification; Reference proteome; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 36742.4 Length 328 Aromaticity 0.06 Instability index 45.36 Isoelectric point 6.46 Charge (pH=7) -1.6 2D Binding mode Binding energy (Kcal/mol) -7.5 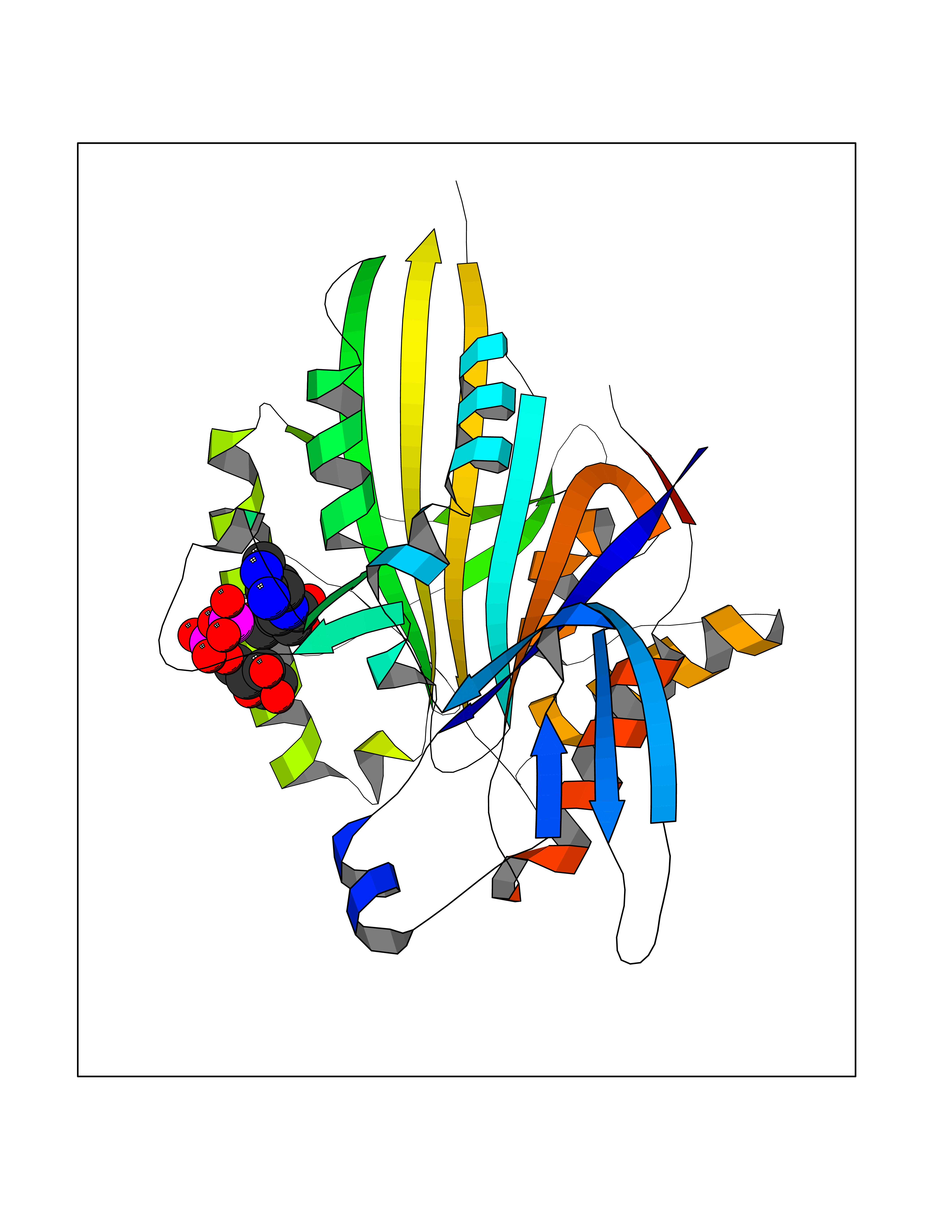 Molscript Map 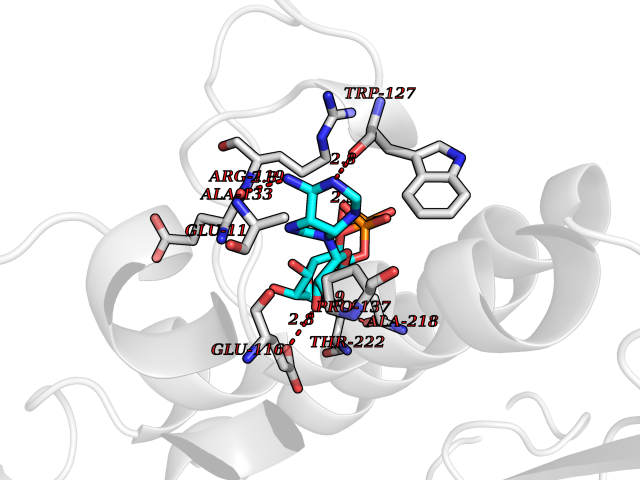 Pymol Map 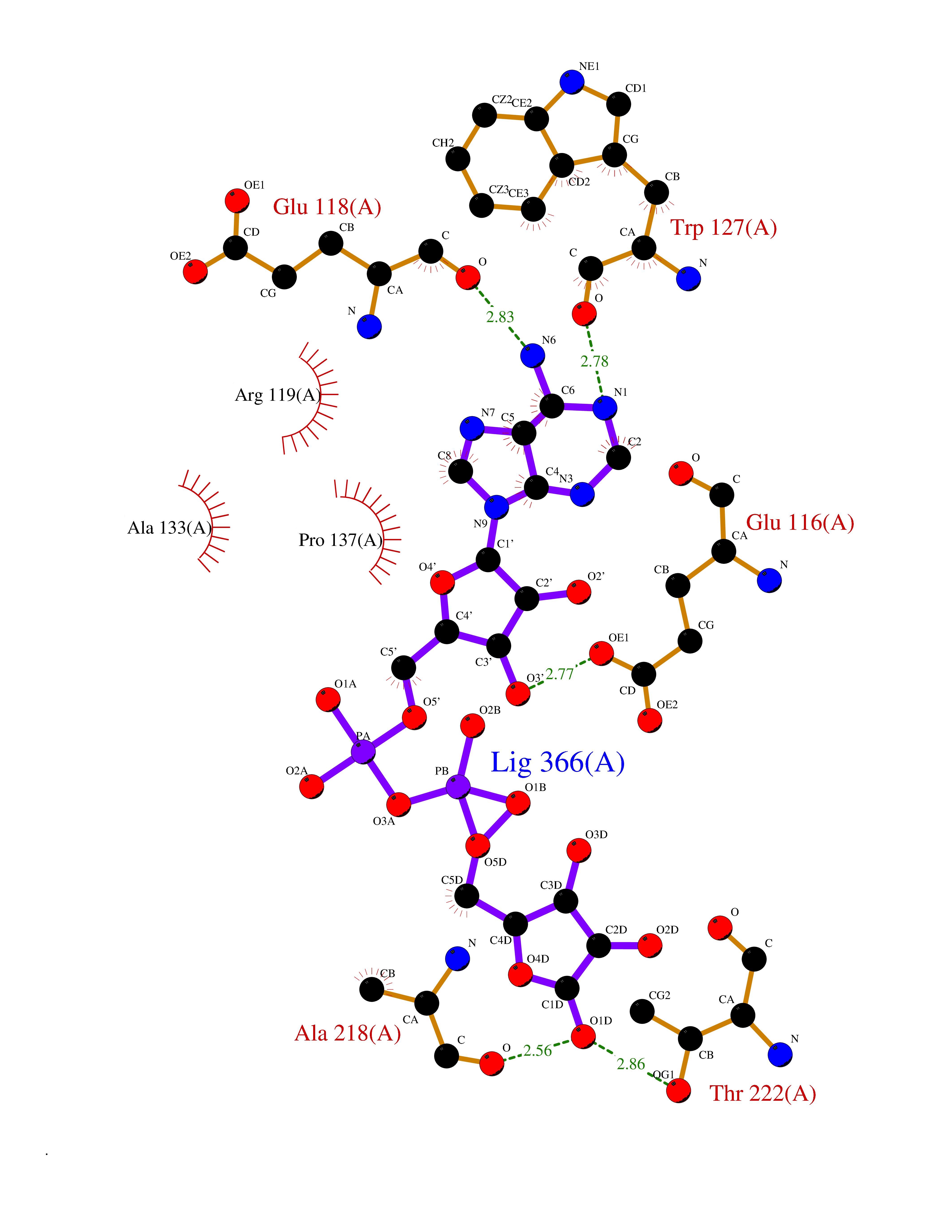 Ligplot Map 3D Binding mode Sequence GKNIQVVVRCRPFNLAERKASAHSIVECDPVRKEVSVRTGGLADKSSRKTYTFDMVFGASTKQIDVYRSVVCPILDEVIMGYNCTIFAYGQTGTGKTFTMEGERSPNEEYTWEEDPLAGIIPRTLHQIFEKLTDNGTEFSVKVSLLEIYNEELFDLLNPSSDVSERLQMFDDPRNKRGVIIKGLEEITVHNKDEVYQILEKGAAKRTTAATLMNAYSSRSHSVFSVTIHMKEELVKIGKLNLVDLAGSENNINQSLLTLGRVITALVERTPHVPYRESKLTRILQDSLGGRTRTSIIATISPASLNLEETLSTLEYAHRAKNILNKPE Hydrogen bonds contact Hydrophobic contact | ||||
| 20 | Dopamine beta-hydroxylase | 4ZEL | 6.33 | |
Target general information Gen name DBH Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Copper type II ascorbate-dependent monooxygenase family Biochemical class Oxidoreductase Function Catalytic activity.Copper ion binding.Dopamine beta-monooxygenase activity.L-ascorbic acid binding. Related diseases Orthostatic hypotension 1 (ORTHYP1) [MIM:223360]: A form of orthostatic hypotension due to congenital dopamine beta-hydroxylase deficiency. Orthostatic hypotension, also known as postural hypotension, is a finding defined as a 20-mm Hg decrease in systolic pressure or a 10-mm Hg decrease in diastolic pressure occurring 3 minutes after a person has risen from supine to standing. Symptoms include dizziness, blurred vision, and sometimes syncope. ORTHYP1 is an autosomal recessive condition apparent from infancy or early childhood and characterized by low plasma and urinary levels of norepinephrine and epinephrine, and episodic hypoglycemia. {ECO:0000269|PubMed:11857564}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00126; DB06774; DB09130; DB05394; DB00822; DB00988; DB00968; DB00550 Interacts with P00352; P63010-2; Q04656; Q8WUW1; Q9UNS2; Q71DI3; P61978; Q9Y2M5; Q92876; P08727; Q14693; P0DPK4; Q6GQQ9-2; P27986-2; Q9ULX5; Q96D59; Q8N6K7-2; Q9GZS3; Q8IUW3; Q86WT6-2 EC number 1.14.17.1 Uniprot keywords 3D-structure; Catecholamine biosynthesis; Copper; Cytoplasmic vesicle; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; Membrane; Metal-binding; Monooxygenase; Oxidoreductase; Proteomics identification; Reference proteome; Secreted; Signal-anchor; Transmembrane; Transmembrane helix; Vitamin C Protein physicochemical properties Chain ID A,B Molecular weight (Da) 123694 Length 1094 Aromaticity 0.1 Instability index 51.85 Isoelectric point 5.84 Charge (pH=7) -24.5 2D Binding mode Binding energy (Kcal/mol) -8.7 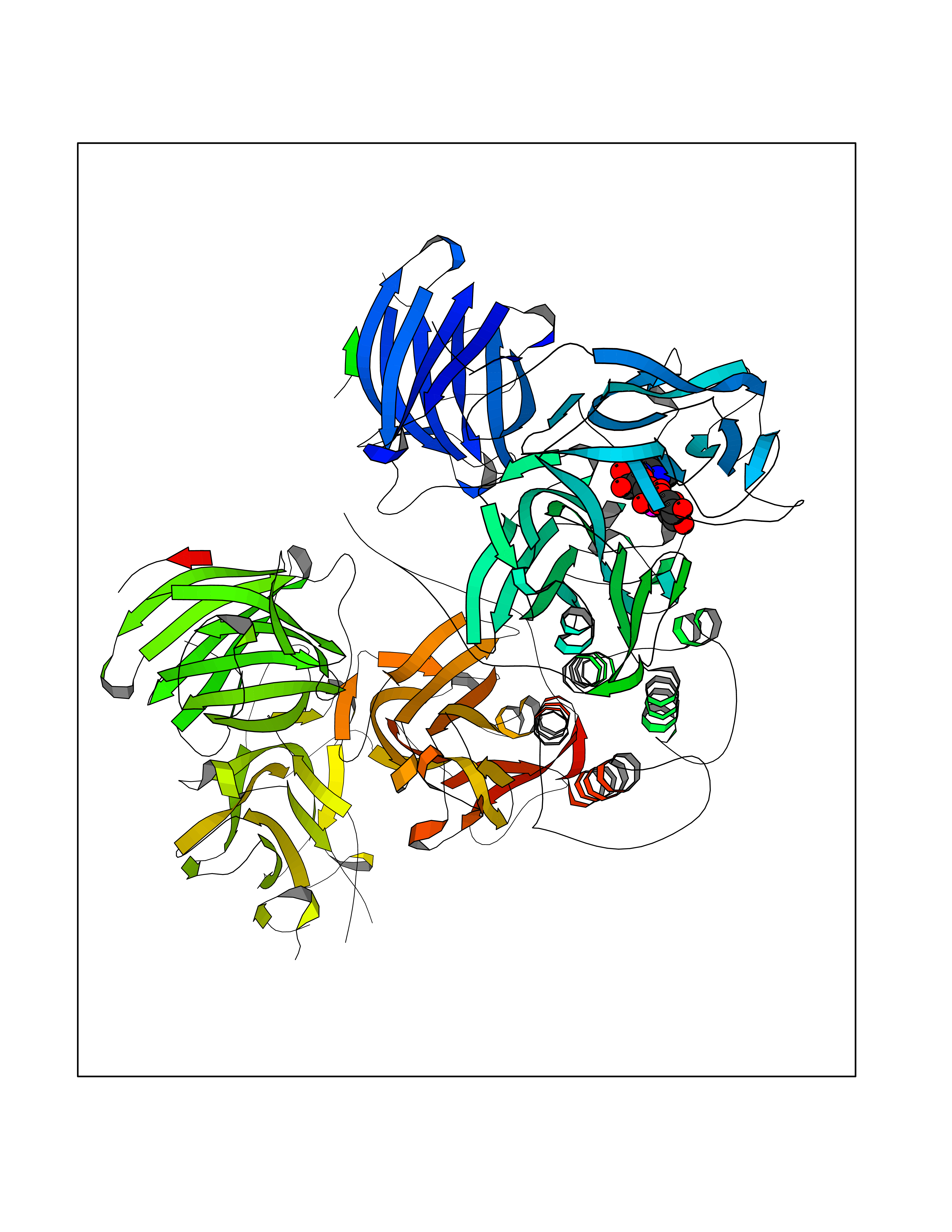 Molscript Map 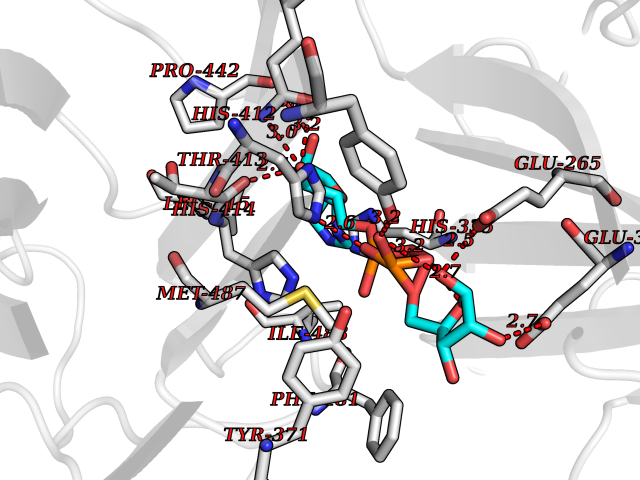 Pymol Map 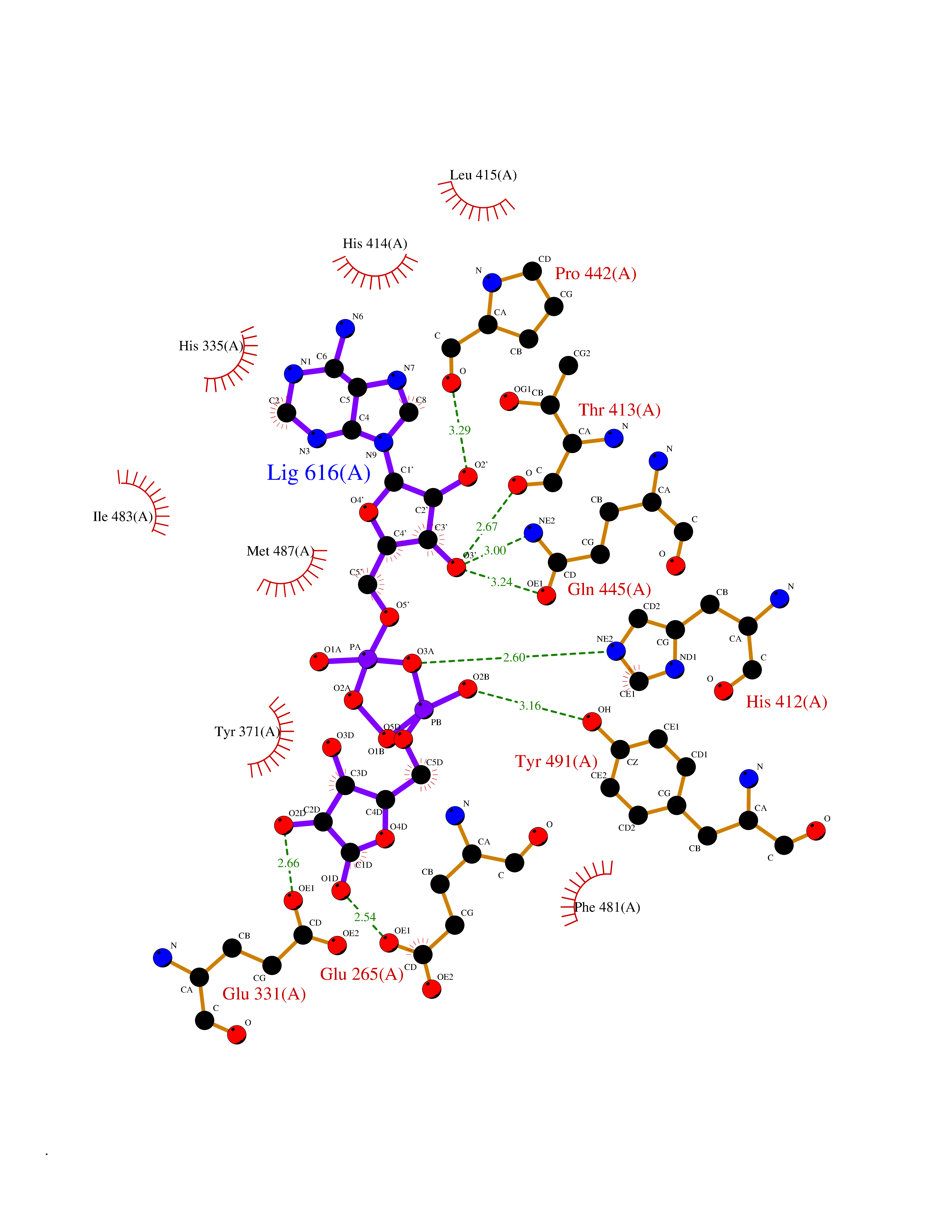 Ligplot Map 3D Binding mode Sequence PLPYHIPLDPEGSLELSWNVSYTQEAIHFQLLVRRLKAGVLFGMSDRGELENADLVVLWTDGDAYFADAWSDQKGQIHLDPQQDYQLLQVQRTPEGLTLLFKRPFGTCDPKDYLIEDGTVHLVYGILEEPFRSLEAINGSGLQMGLQRVQLLKPNIPEPELPSDACTMEVQAPNIQIPSQETTYWCYIKELPKGFSRHHIIKYEPIVTKGNEALVHHMEVFQCAPEMDSVPHFSGPCDSKMKPDRLNYCRHVLAAWALGAKAFYYPEEAGLAFGGPGSSRYLRLEVHYHNPLVIEGRNDSSGIRLYYTAKLRRFNAGIMELGLVYTPVMAIPPRETAFILTGYCTDKCTQLALPPSGIHIFASQLHTHLTGRKVVTVLVRDGREWEIVNQDNHYSPHFQEIRMLKKVVSVHPGDVLITSCTYNTEDRELATVGGFGILEEMCVNYVHYYPQTQLELCKSAVDAGFLQKYFHLINRFNNEDVCTCPQASVSQQFTSVPWNSFNRDVLKALYSFAPISMHCNKSSAVRFQGEWNLQPLPKVISTLEEPTVVSPLPYHIPLDPEGSLELSWNVSYTQEAIHFQLLVRRLKAGVLFGMSDRGELENADLVVLAYFADAWSDQKGQIHLDPQQDYQLLQVQRTPEGLTLLFKRPFGTCDPKDYLIEDGTVHLVYGILEEPFRSLEAINGSGLQMGLQRVQLLKPNIPEPELPSDACTMEVQAPNIQIPSQETTYWCYIKELPKGFSRHHIIKYEPIVTKGNEALVHHMEVFQCAPEVPHFSGPCDSKMLNYCRHVLAAWALGAKAFYYPEEAGLAFGGPGSSRYLRLEVHYHNPLVIEGRNDSSGIRLYYTAKLRRFNAGIMELGLVYTPVMAIPPRETAFILTGYCTDKCTQLALPPSGIHIFASQLHTHLTGRKVVTVLVRDGREWEIVNQDNHYSPHFQEIRMLKKVVSVHPGDVLITSCTYNTEDRELATVGGFGILEEMCVNYVHYYPQTQLELCKSAVDAGFLQKYFHLINRFNNEDVCTCPQASVSQQFTSVPWNSFNRDVLKALYSFAPISMHCNKSSAVRFQGEWNLQPLPKVISTLEEPTPQCVVSIGG Hydrogen bonds contact Hydrophobic contact | ||||