Ligand
Structure
Job ID
7083c90105499e655fafcdb3ccf0ad5e
Job name
Wu_Test37
Time
2024-10-01 14:35:27
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 1 | Purine nucleoside phosphorylase (PNP) | 4EAR | 7.43 | |
Target general information Gen name PNP Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms PNP; Inosine phosphorylase Protein family PNP/MTAP phosphorylase family Biochemical class Pentosyltransferase Function The purine nucleoside phosphorylases catalyze the phosphorolytic breakdown of the N-glycosidic bond in the beta- (deoxy)ribonucleoside molecules, with the formation of the corresponding free purine bases and pentose-1-phosphate. Related diseases Purine nucleoside phosphorylase deficiency (PNPD) [MIM:613179]: A disorder that interrupts both the catabolism of inosine into hypoxanthine and guanosine into guanine, and leads to the accumulation of guanosine, inosine, and their deoxified by-products. The main clinical presentation is recurrent infections due to severe T-cell immunodeficiency. Some patients also have neurologic impairment. {ECO:0000269|PubMed:1384322, ECO:0000269|PubMed:3029074, ECO:0000269|PubMed:8931706}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03881; DB03551; DB02222; DB02391; DB03609; DB01667; DB04260; DB02796; DB04753; DB00640; DB00242; DB00900; DB06185; DB02377; DB02857; DB04754; DB04757; DB04076; DB02230; DB04335; DB02568; DB03101 Interacts with P05067; Q9UQM7; O14576-2; P06241; P14136; Q92993-2; Q9BXM7; P00491; P17612; P63000; Q92673; Q15583 EC number EC 2.4.2.1 Uniprot keywords 3D-structure; Acetylation; Cytoplasm; Direct protein sequencing; Disease variant; Glycosyltransferase; Phosphoprotein; Proteomics identification; Purine salvage; Reference proteome; Transferase Protein physicochemical properties Chain ID A,B,C Molecular weight (Da) 31849.2 Length 288 Aromaticity 0.1 Instability index 34.77 Isoelectric point 6.42 Charge (pH=7) -1.63 2D Binding mode Binding energy (Kcal/mol) -8.18  Molscript Map 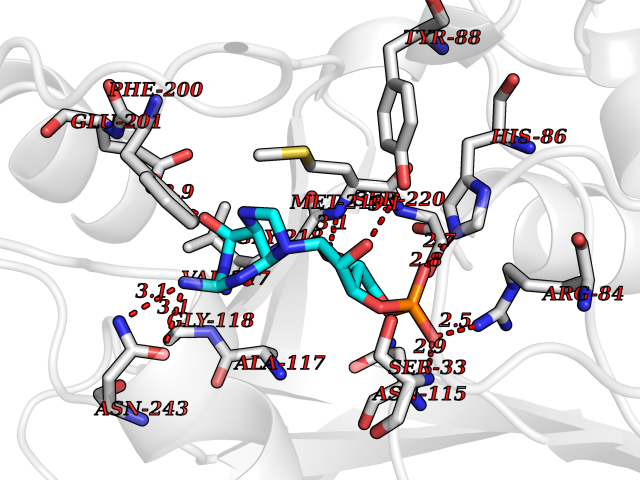 Pymol Map 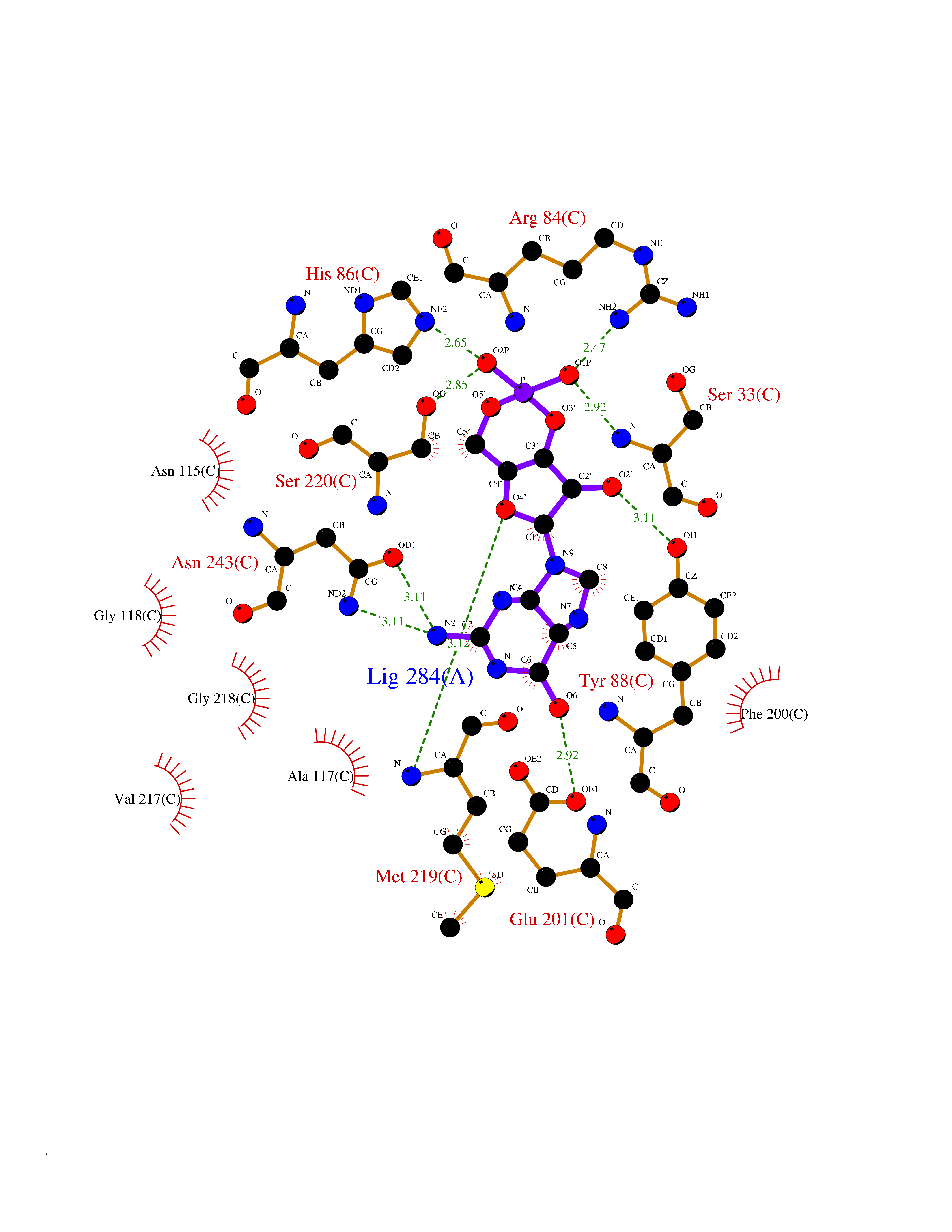 Ligplot Map 3D Binding mode Sequence GYTYEDYKNTAEYLLSHTKHRPQVAIICGSGLGGLTDKLTQAQIFDYSEIPNFPRSTVPGHAGRLVFGFLNGRACVMMQGRFHMYEGYPLYKVTFPVRVFHLLGVDTLVVTNAAGGLNPKFEVGDIMLIRDHINLPGFSGQNPLRGPNDERFGDRFPAMSDAYDRTMRQRALSTYKQMGEQRELQEGTYVMVAGPSFETVAECRVLQKLGADAVGMSTVPEVIVARHCGLRVFGFSLITNKVIMDYESLEKANXEEVLAAGKQAAQKLEQFVSILMASIDRFPAMSDA Hydrogen bonds contact Hydrophobic contact | ||||
| 2 | Phosphodiesterase 4A (PDE4A) | 2QYK | 7.30 | |
Target general information Gen name PDE4A Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms cAMP-specific 3',5'-cyclic phosphodiesterase 4A; Type 4A cAMP phosphodiesterase; PDE46; DPDE2 Protein family Cyclic nucleotide phosphodiesterase family, PDE4 subfamily Biochemical class Phosphoric diester hydrolase Function Hydrolyzes the second messenger cAMP, which is a key regulator of many important physiological processes. Related diseases Developmental and epileptic encephalopathy 50 (DEE50) [MIM:616457]: A form of epileptic encephalopathy, a heterogeneous group of severe early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE50 is an autosomal recessive, progressive disease with onset in infancy and favorable response to treatment with oral uridine. {ECO:0000269|PubMed:25678555, ECO:0000269|PubMed:28087732}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB06842; DB08299; DB01427; DB00201; DB05219; DB00975; DB06751; DB00651; DB00824; DB05266; DB01088; DB01303; DB01791; DB06479; DB01656; DB01954; DB00277; DB08811; DB09283 Interacts with P55212; O14569; P13473-2; Q9UJX0; P16118; O75400-2; Q9Y371 EC number EC 3.1.4.53 Uniprot keywords 3D-structure; Alternative splicing; cAMP; Cell membrane; Cell projection; Cytoplasm; Hydrolase; Isopeptide bond; Manganese; Membrane; Metal-binding; Phosphoprotein; Proteomics identification; Reference proteome; Ubl conjugation; Zinc Protein physicochemical properties Chain ID A,B Molecular weight (Da) 38579.6 Length 335 Aromaticity 0.08 Instability index 37.43 Isoelectric point 5.01 Charge (pH=7) -19.92 2D Binding mode Binding energy (Kcal/mol) -7.94  Molscript Map 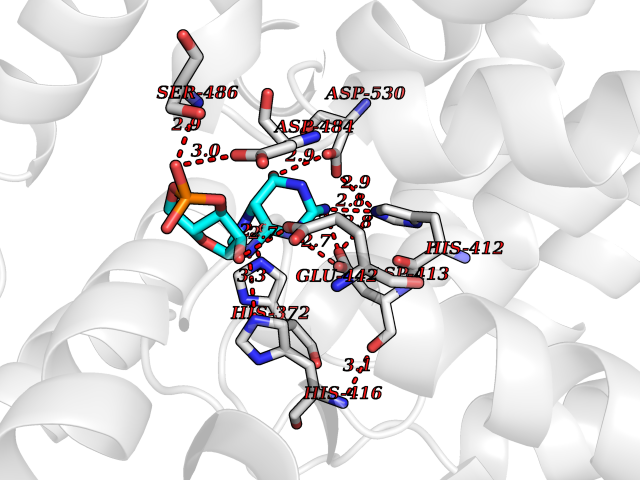 Pymol Map 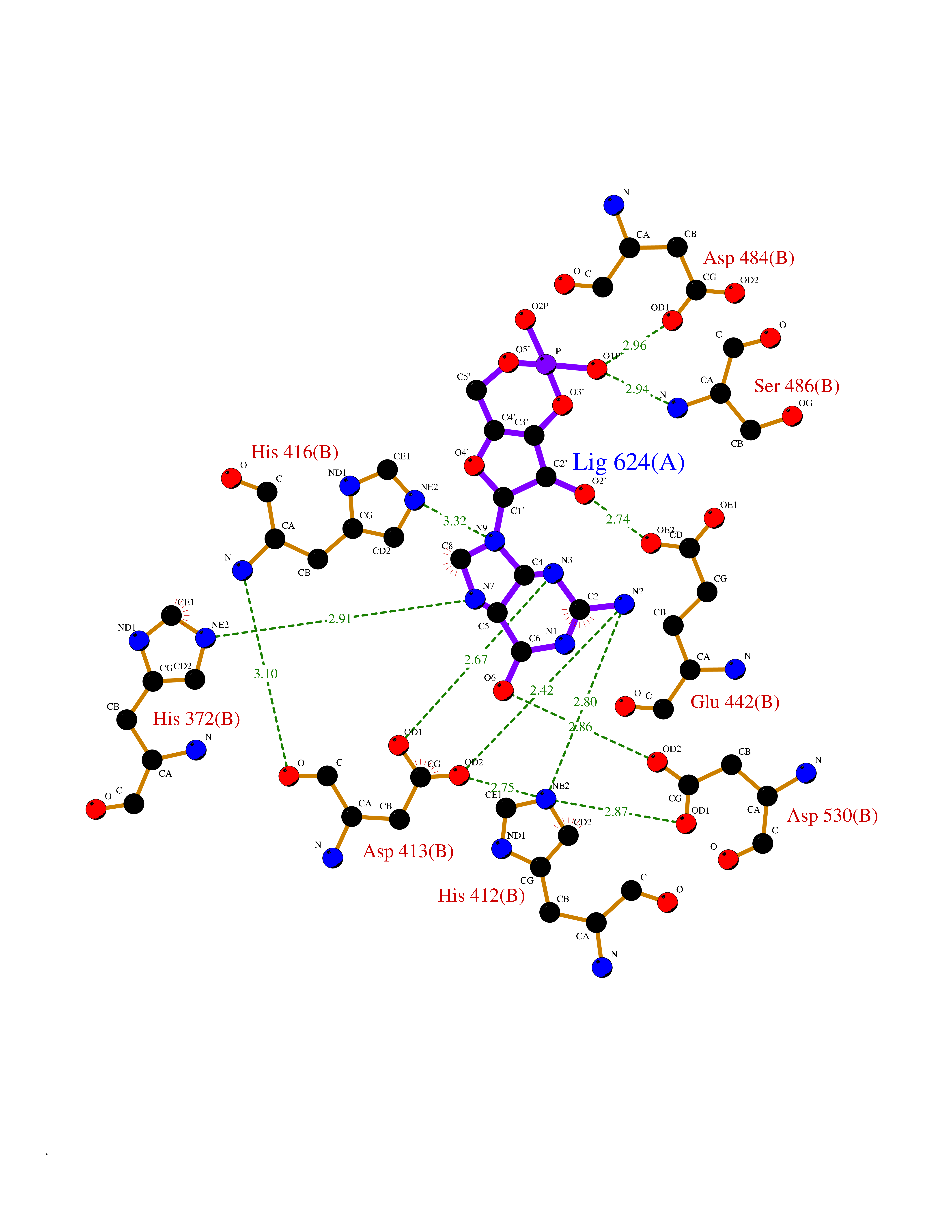 Ligplot Map 3D Binding mode Sequence HMNIPRFGVKTDQEELLAQELENLNKWGLNIFCVSDYAGGRSLTCIMYMIFQERDLLKKFRIPVDTMVTYMLTLEDHYHADVAYHNSLHAADVLQSTHVLLATPALDAVFTDLEILAALFAAAIHDVDHPGVSNQFLINTNSELALMYNDESVLENHHLAVGFKLLQEDNCDIFQNLSKRQRQSLRKMVIDMVLATDMSKHMTLLADLKTMVETKKVTSSGVLLLDNYSDRIQVLRNMVHCADLSNPTKPLELYRQWTDRIMAEFFQQGDRERERGMEISPMCDKHTASVEKSQVGFIDYIVHPLWETWADLVHPDAQEILDTLEDNRDWYYSAI Hydrogen bonds contact Hydrophobic contact | ||||
| 3 | Solute carrier family 19 member 1 (SLC19A1) | 8GOF | 7.25 | |
Target general information Gen name SLC19A1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Reduced folate carrier protein; RFC1; RFC; Placental folate transporter; Intestinal folate carrier 1; IFC-1; Folate transporter 1; FOLT; FLOT1 Protein family Reduced folate carrier (RFC) transporter (TC 2.A.48) family Biochemical class NA Function Transporter for the intake of folate. Uptake of folate in human placental choriocarcinoma cells occurs by a novel mechanism called potocytosis which functionally couples three components, namely the folate receptor, the folate transporter, and a V-type H(+)-pump. Related diseases Megaloblastic anemia, folate-responsive (MEGAF) [MIM:601775]: An autosomal recessive metabolic disorder characterized by megaloblastic anemia resulting from decreased folate transport into erythrocytes. Disease manifestations include hemolytic anemia, hyperhomocysteinemia, and low vitamin B12. Serum folate levels are normal, but erythrocyte folate levels are decreased. Treatment with oral folate corrects the anemia and normalizes homocysteine. {ECO:0000269|PubMed:32276275}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Immunodeficiency 114, folate-responsive (IMD114) [MIM:620603]: An autosomal recessive immunologic disorder manifesting in early infancy and characterized by recurrent skin and respiratory infections, mucosal bleeding, oral ulcers, chronic diarrhea, and poor overall growth. Affected individuals have lymphopenia, low serum immunoglobulins, and impaired T cell proliferation. Some patients have global developmental delay. {ECO:0000269|PubMed:36517554, ECO:0000269|PubMed:36745868}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB11256; DB00563; DB00642; DB06813; DB01157 Interacts with Q7Z3Y9 EC number NA Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Antiport; Cell membrane; Disease variant; Folate-binding; Glycoprotein; Hereditary hemolytic anemia; Membrane; Phosphoprotein; Proteomics identification; Reference proteome; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A Molecular weight (Da) 46087.7 Length 407 Aromaticity 0.15 Instability index 34.62 Isoelectric point 9.82 Charge (pH=7) 17.33 2D Binding mode Binding energy (Kcal/mol) -7.81  Molscript Map 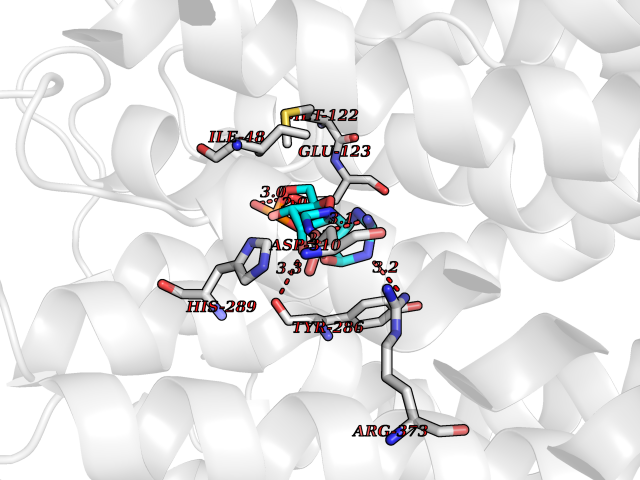 Pymol Map 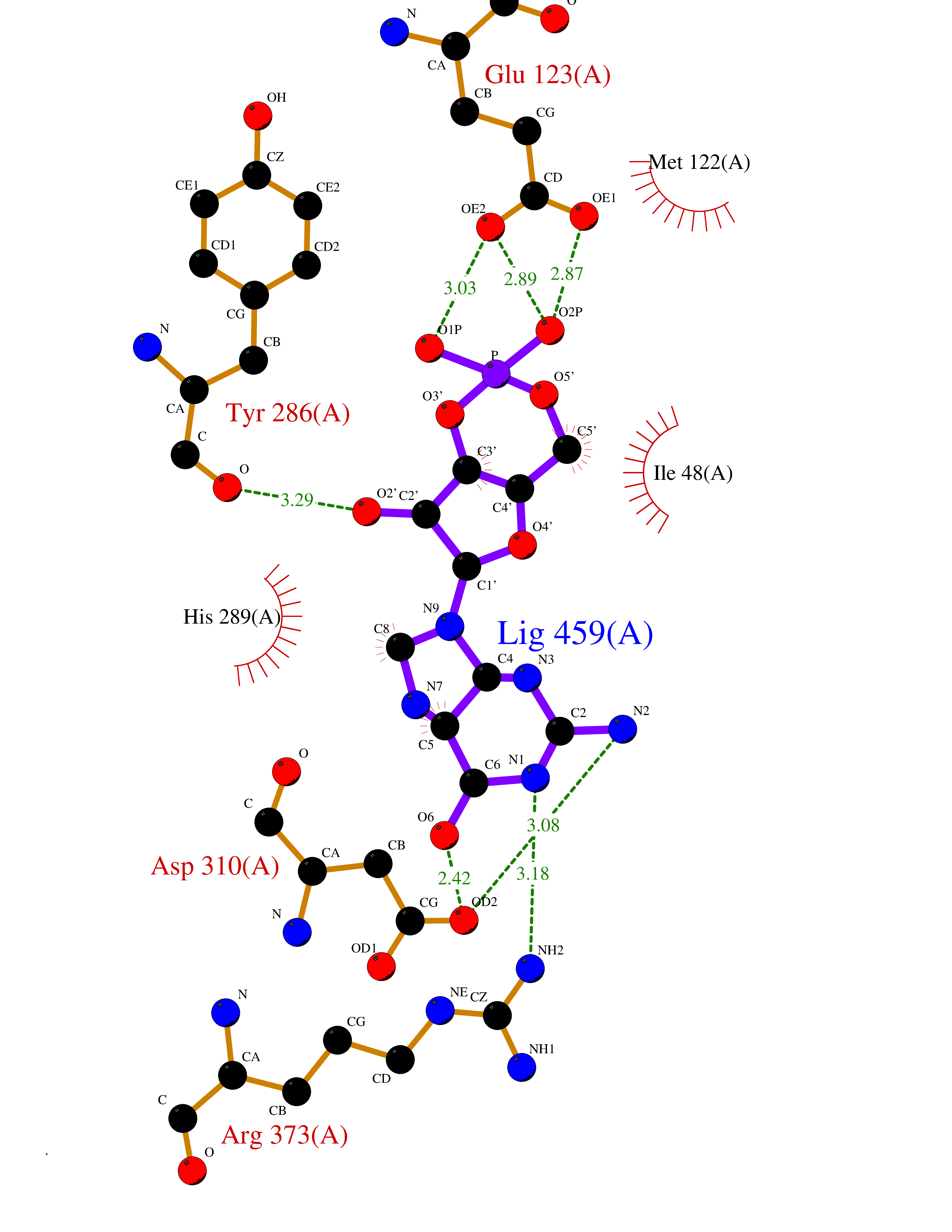 Ligplot Map 3D Binding mode Sequence DPELRSWRHLVCYLCFYGFMAQIRPGESFITPYLLGPDKNFTREQVTNEITPVLSYSYLAVLVPVFLLTDYLRYTPVLLLQGLSFVSVWLLLLLGHSVAHMQLMELFYSVTMAARIAYSSYIFSLVRPARYQRVAGYSRAAVLLGVFTSSVLGQLLVTVGRVSFSTLNYISLAFLTFSVVLALFLKRPKRSLFFNRDDSVLARMLRELGDSLRRPQLRLWSLWWVFNSAGYYLVVYYVHILWNEVDPTTNSARVYNGAADAASTLLGAITSFAAGFVKIRWARWSKLLIAGVTATQAGLVFLLAHTRHPSSIWLCYAAFVLFRGSYQFLVPIATFQIASSLSKELCALVFGVNTFFATIVKTIITFIVSDVRGLGLPVRKQFQLYSVYFLILSIIYFLGAMLDGLRH Hydrogen bonds contact Hydrophobic contact | ||||
| 4 | Endothelin-converting enzyme 1 (ECE1) | 3DWB | 7.24 | |
Target general information Gen name ECE1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms ECE-1 Protein family Peptidase M13 family Biochemical class Peptidase Function Converts big endothelin-1 to endothelin-1. Related diseases Hirschsprung disease, cardiac defects, and autonomic dysfunction (HCAD) [MIM:613870]: A disorder characterized by skip-lesions Hirschsprung disease, craniofacial abnormalities and other dysmorphic features, cardiac defects including ductus arteriosus, small subaortic ventricular septal defect, small atrial septal defect, and autonomic dysfunction. {ECO:0000269|PubMed:9915973}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07171 Interacts with P49760; A8MQ03; Q8IUG1; P60370; P60410 EC number EC 3.4.24.71 Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disease variant; Disulfide bond; Glycoprotein; Hirschsprung disease; Hydrolase; Membrane; Metal-binding; Metalloprotease; Phosphoprotein; Protease; Proteomics identification; Reference proteome; Signal-anchor; Transmembrane; Transmembrane helix; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 75247.9 Length 660 Aromaticity 0.12 Instability index 46.29 Isoelectric point 5.33 Charge (pH=7) -18.3 2D Binding mode Binding energy (Kcal/mol) -7.49 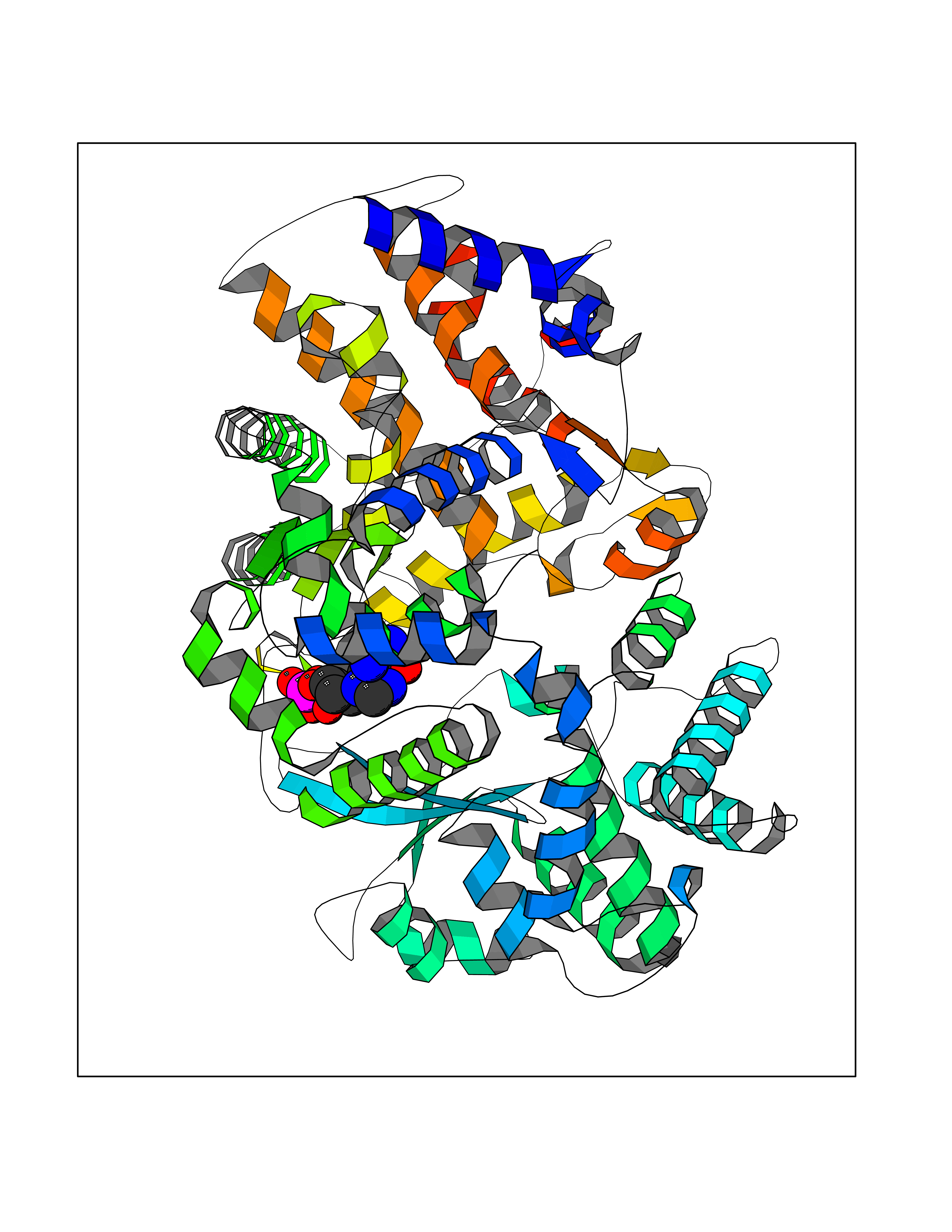 Molscript Map 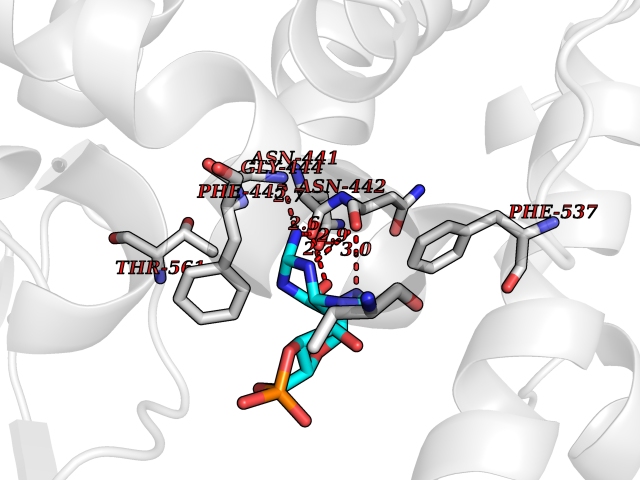 Pymol Map 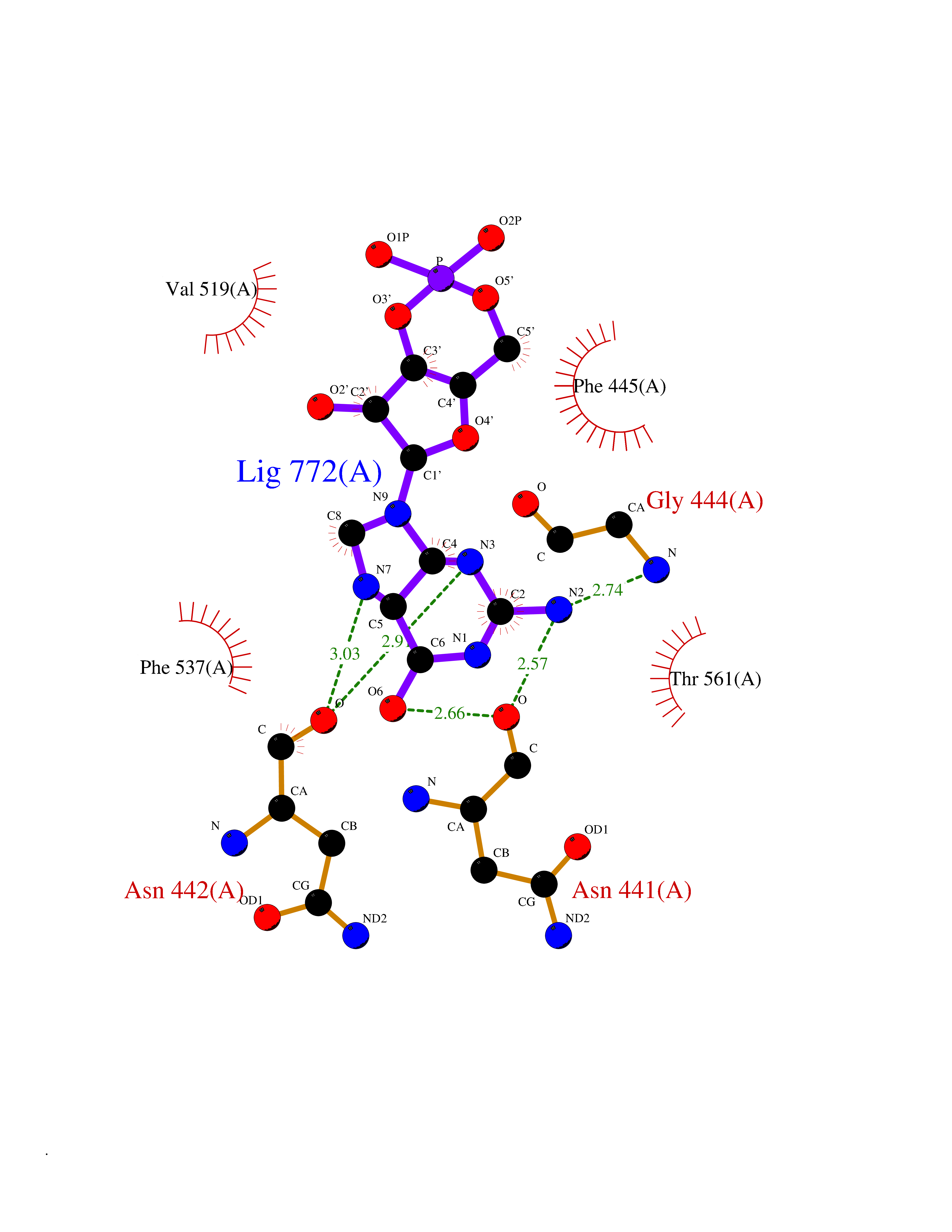 Ligplot Map 3D Binding mode Sequence SEACVSVTSSILSSMDPTVDPCHDFFSYACGGWIKANPVPDGHSRWGTFSNLWEHNQAIIKHLLENSTASVSEAERKAQVYYRACMNETRIEELRAKPLMELIERLGGWNITGPWAKDNFQDTLQVVTAHYRTSPFFSVYVSADSKNSNSNVIQVDQSGLGLPSRDYYLNKTENEKVLTGYLNYMVQLGKLLGGGDEEAIRPQMQQILDFETALANITIPQEKRRDEELIYHKVTAAELQTLAPAINWLPFLNTIFYPVEINESEPIVVYDKEYLEQISTLINTTDRCLLNNYMIWNLVRKTSSFLDQRFQDADEKFMEVMWKFCVSDTENNLGFALGPMFVKATFAEDSKSIATEIILEIKKAFEESLSTLKWMDEETRKSAKEKADAIYNMIGYPNFIMDPKELDKVFNDYTAVPDLYFENAMRFFNFSWRVTADQLRKAPNRDQWSMTPPMVNAYYSPTKNEIVFPAGILQAPFYTRSSPKALNFGGIGVVVGHELTHAFDDQGREYDKDGNLRPWWKNSSVEAFKRQTECMVEQYSNYSVNGEPVNGRHTLGENIADNGGLKAAYRAYQNWVKKNGAEHSLPTLGLTNNQLFFLGFAQVWCSVRTPESSHEGLITDPHSPSRFRVIGSLSNSKEFSEHFRCPPGSPMNPPHKCEVW Hydrogen bonds contact Hydrophobic contact | ||||
| 5 | Protein cereblon (CRBN) | 5FQD | 7.15 | |
Target general information Gen name CRBN Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Protein cereblon Protein family CRBN family Biochemical class NA Function Substrate recognition component of a DCX (DDB1-CUL4-X-box) E3 protein ligase complex that mediates the ubiquitination and subsequent proteasomal degradation of target proteins, such as MEIS2. Normal degradation of key regulatory proteins is required for normal limb outgrowth and expression of the fibroblast growth factor FGF8. May play a role in memory and learning by regulating the assembly and neuronal surface expression of large-conductance calcium-activated potassium channels in brain regions involved in memory and learning via its interaction with KCNT1. Binding of pomalidomide and other thalidomide-related drugs changes the substrate specificity of the human protein, leading to decreased degradation of MEIS2 and other target proteins and increased degradation of MYC, IRF4, IKZF1 and IKZF3. Related diseases Intellectual developmental disorder, autosomal recessive 2 (MRT2) [MIM:607417]: A disorder characterized by significantly below average general intellectual functioning associated with impairments in adaptive behavior and manifested during the developmental period. MRT2 patients display mild intellectual disability with a standard IQ ranged from 50 to 70. IQ scores are lower in males than females. Developmental milestones are mildly delayed. There are no dysmorphic or autistic features. {ECO:0000269|PubMed:15557513, ECO:0000269|PubMed:28143899}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00480; DB08910; DB01041 Interacts with Q96A83-2; P48729; Q16531; O14901; Q8IVT2; Q9P286; A0A6Q8PF08; Q93062; Q16531; Q13422-7 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; Disease variant; Intellectual disability; Membrane; Metal-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Ubl conjugation; Ubl conjugation pathway; Zinc Protein physicochemical properties Chain ID B,E Molecular weight (Da) 38245.7 Length 337 Aromaticity 0.08 Instability index 40.62 Isoelectric point 5.7 Charge (pH=7) -6.53 2D Binding mode Binding energy (Kcal/mol) -8.18  Molscript Map 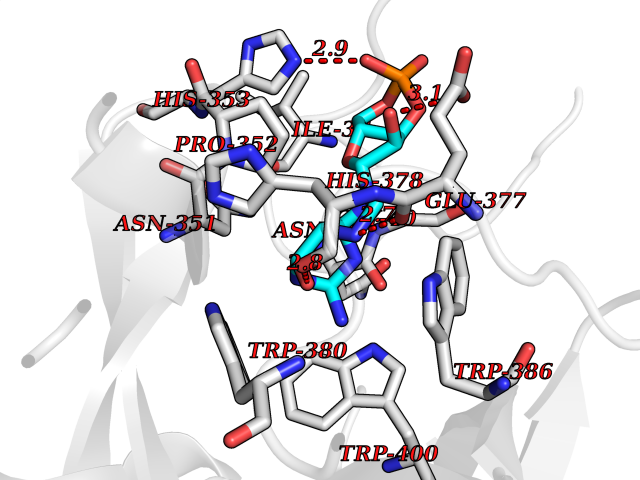 Pymol Map 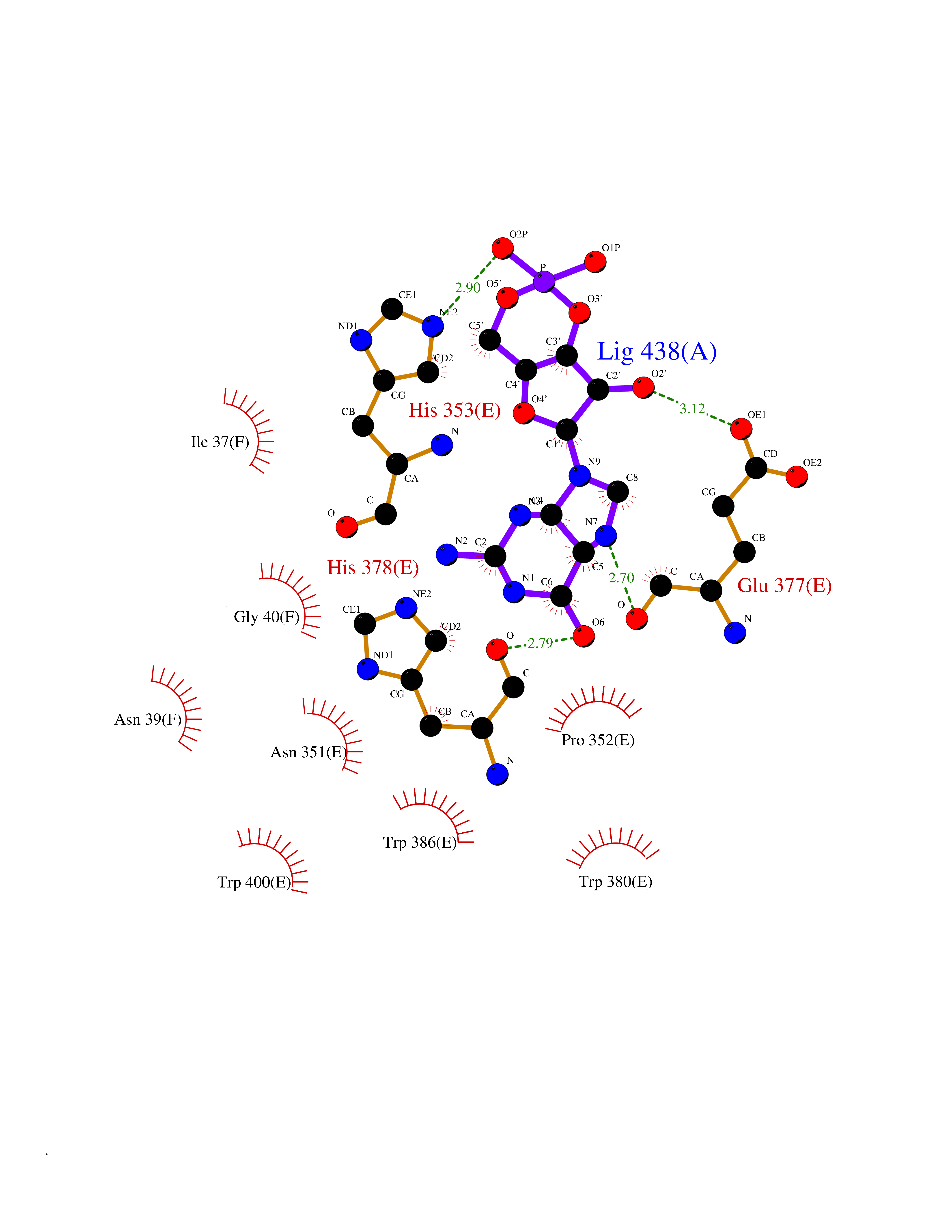 Ligplot Map 3D Binding mode Sequence EFIVGGKYKLNITNGEEVAVINFDTSLPTSHTYLGADMEEFHGRTLHDDDSCQVIPVLPQVMMILIPGQTLPLQLFHPQEVSMVRNLIQKDRTFAVLAYSNVQEREAQFGTTAEIYAYREEIVKVKAIGRQRFKVLEQQAKVQILPECVLAETLMDRIKKQLREWDENLKDDSLPSNPIDFSYRVAACLPIDDVLRIQLLKIGSAIQRLRCELDIMNKCTSLCCKQCQETEITTKNEIFSLSLCGPMAAYVNPHGYVHETLTVYKACNLNLIGRPSTEHSWFPGYAWTVAQCKICASHIGWKFTATKKDMSPQKFWGLTRSALLPTIPDTEDEISPD Hydrogen bonds contact Hydrophobic contact | ||||
| 6 | Dopamine beta-hydroxylase | 4ZEL | 7.13 | |
Target general information Gen name DBH Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Copper type II ascorbate-dependent monooxygenase family Biochemical class Oxidoreductase Function Catalytic activity.Copper ion binding.Dopamine beta-monooxygenase activity.L-ascorbic acid binding. Related diseases Orthostatic hypotension 1 (ORTHYP1) [MIM:223360]: A form of orthostatic hypotension due to congenital dopamine beta-hydroxylase deficiency. Orthostatic hypotension, also known as postural hypotension, is a finding defined as a 20-mm Hg decrease in systolic pressure or a 10-mm Hg decrease in diastolic pressure occurring 3 minutes after a person has risen from supine to standing. Symptoms include dizziness, blurred vision, and sometimes syncope. ORTHYP1 is an autosomal recessive condition apparent from infancy or early childhood and characterized by low plasma and urinary levels of norepinephrine and epinephrine, and episodic hypoglycemia. {ECO:0000269|PubMed:11857564}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00126; DB06774; DB09130; DB05394; DB00822; DB00988; DB00968; DB00550 Interacts with P00352; P63010-2; Q04656; Q8WUW1; Q9UNS2; Q71DI3; P61978; Q9Y2M5; Q92876; P08727; Q14693; P0DPK4; Q6GQQ9-2; P27986-2; Q9ULX5; Q96D59; Q8N6K7-2; Q9GZS3; Q8IUW3; Q86WT6-2 EC number 1.14.17.1 Uniprot keywords 3D-structure; Catecholamine biosynthesis; Copper; Cytoplasmic vesicle; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; Membrane; Metal-binding; Monooxygenase; Oxidoreductase; Proteomics identification; Reference proteome; Secreted; Signal-anchor; Transmembrane; Transmembrane helix; Vitamin C Protein physicochemical properties Chain ID A,B Molecular weight (Da) 123694 Length 1094 Aromaticity 0.1 Instability index 51.85 Isoelectric point 5.84 Charge (pH=7) -24.5 2D Binding mode Binding energy (Kcal/mol) -7.89  Molscript Map 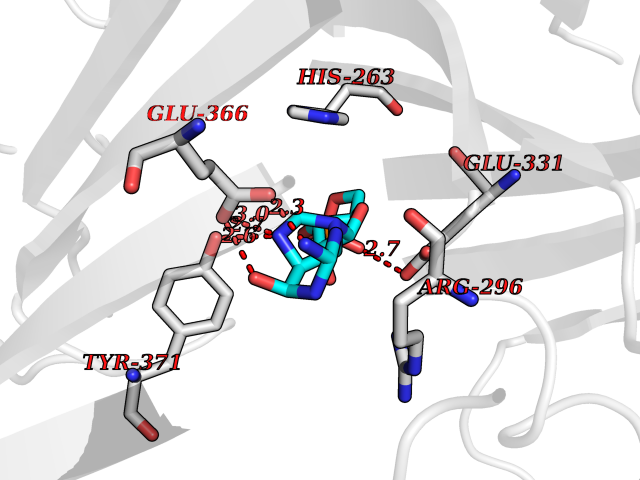 Pymol Map 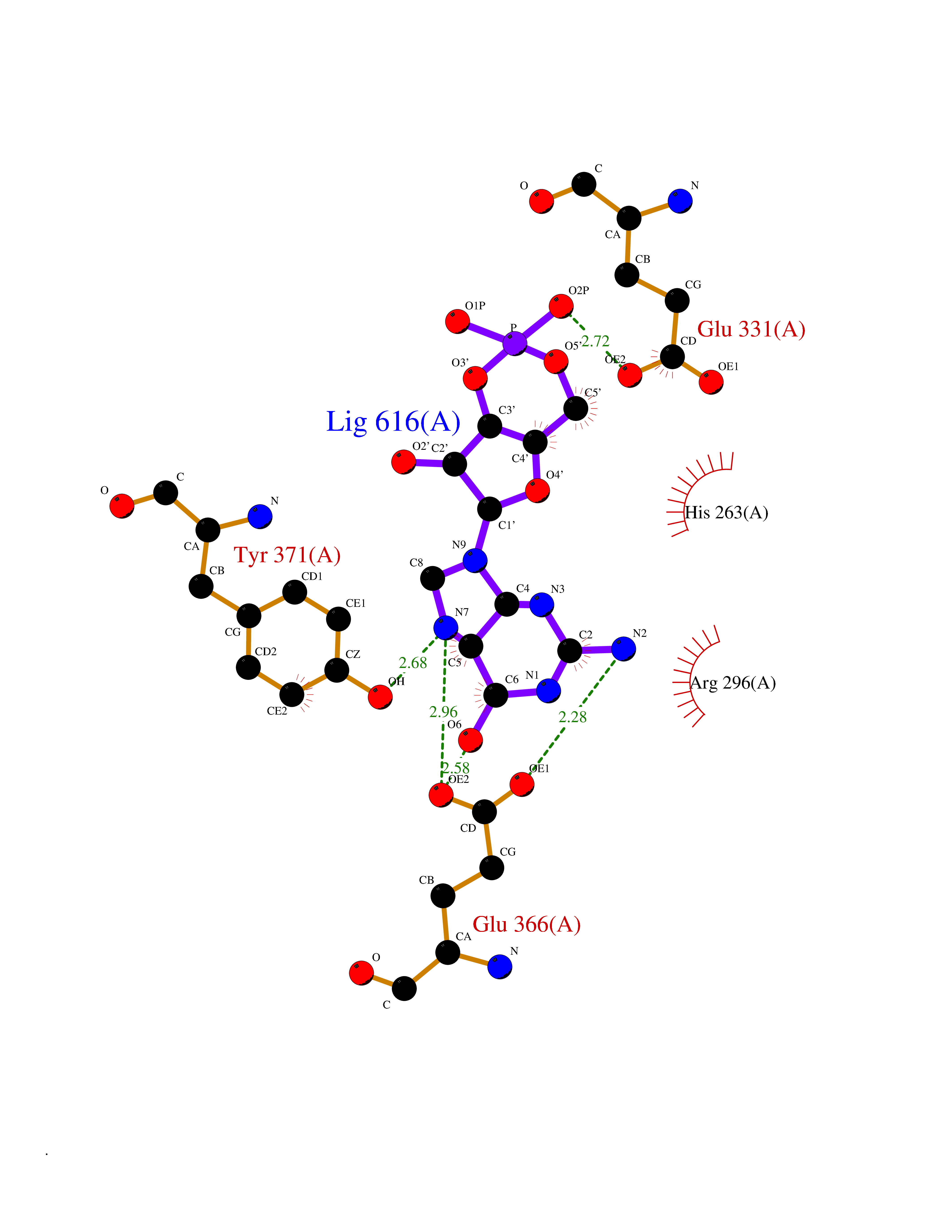 Ligplot Map 3D Binding mode Sequence PLPYHIPLDPEGSLELSWNVSYTQEAIHFQLLVRRLKAGVLFGMSDRGELENADLVVLWTDGDAYFADAWSDQKGQIHLDPQQDYQLLQVQRTPEGLTLLFKRPFGTCDPKDYLIEDGTVHLVYGILEEPFRSLEAINGSGLQMGLQRVQLLKPNIPEPELPSDACTMEVQAPNIQIPSQETTYWCYIKELPKGFSRHHIIKYEPIVTKGNEALVHHMEVFQCAPEMDSVPHFSGPCDSKMKPDRLNYCRHVLAAWALGAKAFYYPEEAGLAFGGPGSSRYLRLEVHYHNPLVIEGRNDSSGIRLYYTAKLRRFNAGIMELGLVYTPVMAIPPRETAFILTGYCTDKCTQLALPPSGIHIFASQLHTHLTGRKVVTVLVRDGREWEIVNQDNHYSPHFQEIRMLKKVVSVHPGDVLITSCTYNTEDRELATVGGFGILEEMCVNYVHYYPQTQLELCKSAVDAGFLQKYFHLINRFNNEDVCTCPQASVSQQFTSVPWNSFNRDVLKALYSFAPISMHCNKSSAVRFQGEWNLQPLPKVISTLEEPTVVSPLPYHIPLDPEGSLELSWNVSYTQEAIHFQLLVRRLKAGVLFGMSDRGELENADLVVLAYFADAWSDQKGQIHLDPQQDYQLLQVQRTPEGLTLLFKRPFGTCDPKDYLIEDGTVHLVYGILEEPFRSLEAINGSGLQMGLQRVQLLKPNIPEPELPSDACTMEVQAPNIQIPSQETTYWCYIKELPKGFSRHHIIKYEPIVTKGNEALVHHMEVFQCAPEVPHFSGPCDSKMLNYCRHVLAAWALGAKAFYYPEEAGLAFGGPGSSRYLRLEVHYHNPLVIEGRNDSSGIRLYYTAKLRRFNAGIMELGLVYTPVMAIPPRETAFILTGYCTDKCTQLALPPSGIHIFASQLHTHLTGRKVVTVLVRDGREWEIVNQDNHYSPHFQEIRMLKKVVSVHPGDVLITSCTYNTEDRELATVGGFGILEEMCVNYVHYYPQTQLELCKSAVDAGFLQKYFHLINRFNNEDVCTCPQASVSQQFTSVPWNSFNRDVLKALYSFAPISMHCNKSSAVRFQGEWNLQPLPKVISTLEEPTPQCVVSIGG Hydrogen bonds contact Hydrophobic contact | ||||
| 7 | Metabotropic glutamate receptor 2 (mGluR2) | 7E9G | 7.13 | |
Target general information Gen name GRM2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms mGLUR2; Group II metabotropic glutamate receptor; Glutamate receptor mGLU2; GPRC1B Protein family G-protein coupled receptor 3 family Biochemical class GPCR glutamate Function Ligand binding causes a conformation change that triggers signaling via guanine nucleotide-binding proteins (G proteins) and modulates the activity of down-stream effectors, such as adenylate cyclase. Signaling inhibits adenylate cyclase activity. May mediate suppression of neurotransmission or may be involved in synaptogenesis or synaptic stabilization. G-protein coupled receptor for glutamate. Related diseases Oocyte/zygote/embryo maturation arrest 21 (OZEMA21) [MIM:620610]: An autosomal dominant, female infertility disorder characterized by zygote development arrest due to failure of pronuclei fusion. {ECO:0000269|PubMed:33948904, ECO:0000269|PubMed:33953335}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB05096 Interacts with Q5T8D3-2; Q9BYF1; Q13520; Q13323; Q8WV48; P57739; O95484; Q7Z7G2; P00387; P27487; P28223-1; Q5SR56; O14880; Q8N4V1; Q58DX5; Q13113; Q9NR31; Q8IWU4; Q9H2H9; P27105; Q8N3G9; Q96Q45-2; Q9NWD8; Q8WUV1; Q9UMX0-2; P0DTC2 EC number NA Uniprot keywords 3D-structure; Cell membrane; Cell projection; Disulfide bond; G-protein coupled receptor; Glycoprotein; Membrane; Proteomics identification; Receptor; Reference proteome; Signal; Synapse; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID R Molecular weight (Da) 85146.2 Length 769 Aromaticity 0.11 Instability index 36.84 Isoelectric point 8.48 Charge (pH=7) 9.7 2D Binding mode Binding energy (Kcal/mol) -8.05  Molscript Map 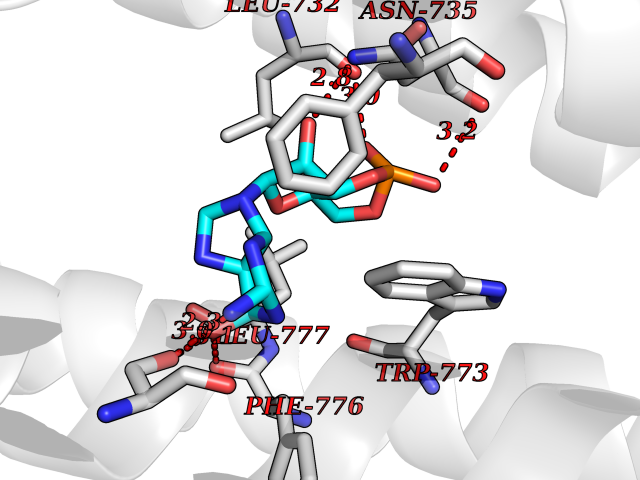 Pymol Map 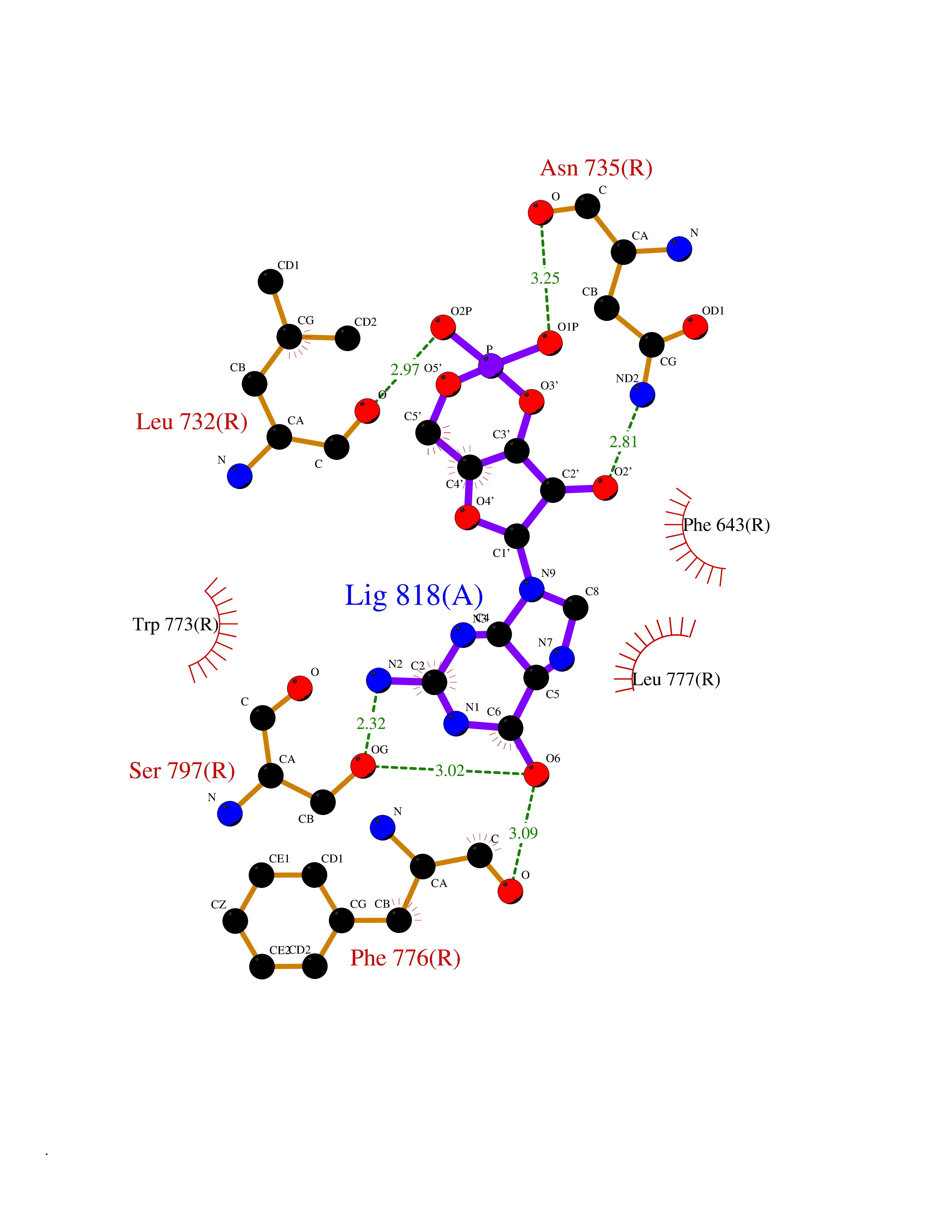 Ligplot Map 3D Binding mode Sequence KKVLTLEGDLVLGGLFPVHQKGGPAEDCGPVNEHRGIQRLEAMLFALDRINRDPHLLPGVRLGAHILDSCSKDTHALEQALDFVRASLITGVIGGSYSDVSIQVANLLRLFQIPQISYASTSAKLSDKSRYDYFARTVPPDFFQAKAMAEILRFFNWTYVSTVASEGDYGETGIEAFELEARARNICVATSEKVGRAMSRAAFEGVVRALLQKPSARVAVLFTRSEDARELLAASQRLNASFTWVASDGWGALESVVAGSEGAAEGAITIELASYPISDFASYFQSLDPWNNSRNPWFREFWEQRFRCSFRQRDCAAHSLRAVPFEQESKIMFVVNAVYAMAHALHNMHRALCPNTTRLCDAMRPVNGRRLYKDFVLNVKFDAPFRPADTHNEVRFDRFGDGIGRYNIFTYLRAGSGRYRYQKVGYWAEGLTLDTSLIPWASPSAGPLPASRCSEPCLQNEVKSVQPGEVCCWLCIPCQPYEYRLDEFTCADCGLGYWPNASLTGCFELPQEYIRWGDAWAVGPVTIACLGALATLFVLGVFVRHNATPVVKAAGRELCYILLGGVFLCYCMTFIFIAKPSTAVCTLRRLGLGTAFSVCYSALLTKTNRIARIFGGAREGAQRPRFISPASQVAICLALISGQLLIVVAWLVVEAPGTGKETAPERREVVTLRCNHRDASMLGSLAYNVLLIALCTLYAFKTRKCPENFNEAKFIGFTMYTTCIIWLAFLPIFYVTSSDYRVQTTTMCVSVSLSGSVVLGCLFAPKLHI Hydrogen bonds contact Hydrophobic contact | ||||
| 8 | Glutamate carboxypeptidase III (NAALAD2) | 3FED | 7.11 | |
Target general information Gen name NAALAD2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms NAALADase II; NAALAD2 Protein family Peptidase M28 family, M28B subfamily Biochemical class Peptidase Function Has N-acetylated-alpha-linked-acidic dipeptidase (NAALADase) activity. Also exhibits a dipeptidyl-peptidase IV type activity. Inactivate the peptide neurotransmitter N- acetylaspartylglutamate. Related diseases Dystonia 1, torsion, autosomal dominant (DYT1) [MIM:128100]: A primary torsion dystonia, and the most common and severe form. Dystonia is defined by the presence of sustained involuntary muscle contractions, often leading to abnormal postures. Dystonia type 1 is characterized by involuntary, repetitive, sustained muscle contractions or postures involving one or more sites of the body, in the absence of other neurological symptoms. Typically, symptoms develop first in an arm or leg in middle to late childhood and progress in approximately 30% of patients to other body regions (generalized dystonia) within about five years. 'Torsion' refers to the twisting nature of body movements observed in DYT1, often affecting the trunk. Distribution and severity of symptoms vary widely between affected individuals, ranging from mild focal dystonia to severe generalized dystonia, even within families. {ECO:0000269|PubMed:14970196, ECO:0000269|PubMed:15505207, ECO:0000269|PubMed:16361107, ECO:0000269|PubMed:17428918, ECO:0000269|PubMed:18167355, ECO:0000269|PubMed:18477710, ECO:0000269|PubMed:18827015, ECO:0000269|PubMed:19955557, ECO:0000269|PubMed:20169475, ECO:0000269|PubMed:21102408, ECO:0000269|PubMed:24930953, ECO:0000269|PubMed:27490483, ECO:0000269|PubMed:9288096}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Arthrogryposis multiplex congenita 5 (AMC5) [MIM:618947]: A form of arthrogryposis multiplex congenita, a developmental condition characterized by multiple joint contractures resulting from reduced or absent fetal movements. AMC5 is an autosomal recessive form characterized by severe congenital contractures, developmental delay, strabismus and tremor. {ECO:0000269|PubMed:28516161, ECO:0000269|PubMed:29053766, ECO:0000269|PubMed:30244176}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q9UHD4; Q6NTF9-3; B2RUZ4; O76024 EC number EC 3.4.17.21 Uniprot keywords 3D-structure; Alternative splicing; Calcium; Carboxypeptidase; Cell membrane; Dipeptidase; Glycoprotein; Hydrolase; Membrane; Metal-binding; Metalloprotease; Multifunctional enzyme; Protease; Proteomics identification; Reference proteome; Signal-anchor; Transmembrane; Transmembrane helix; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 77761.6 Length 690 Aromaticity 0.12 Instability index 39.42 Isoelectric point 8.48 Charge (pH=7) 4.65 2D Binding mode Binding energy (Kcal/mol) -7.25  Molscript Map 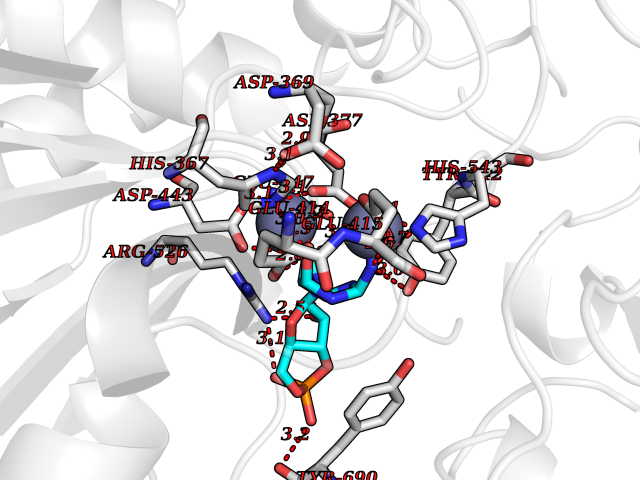 Pymol Map 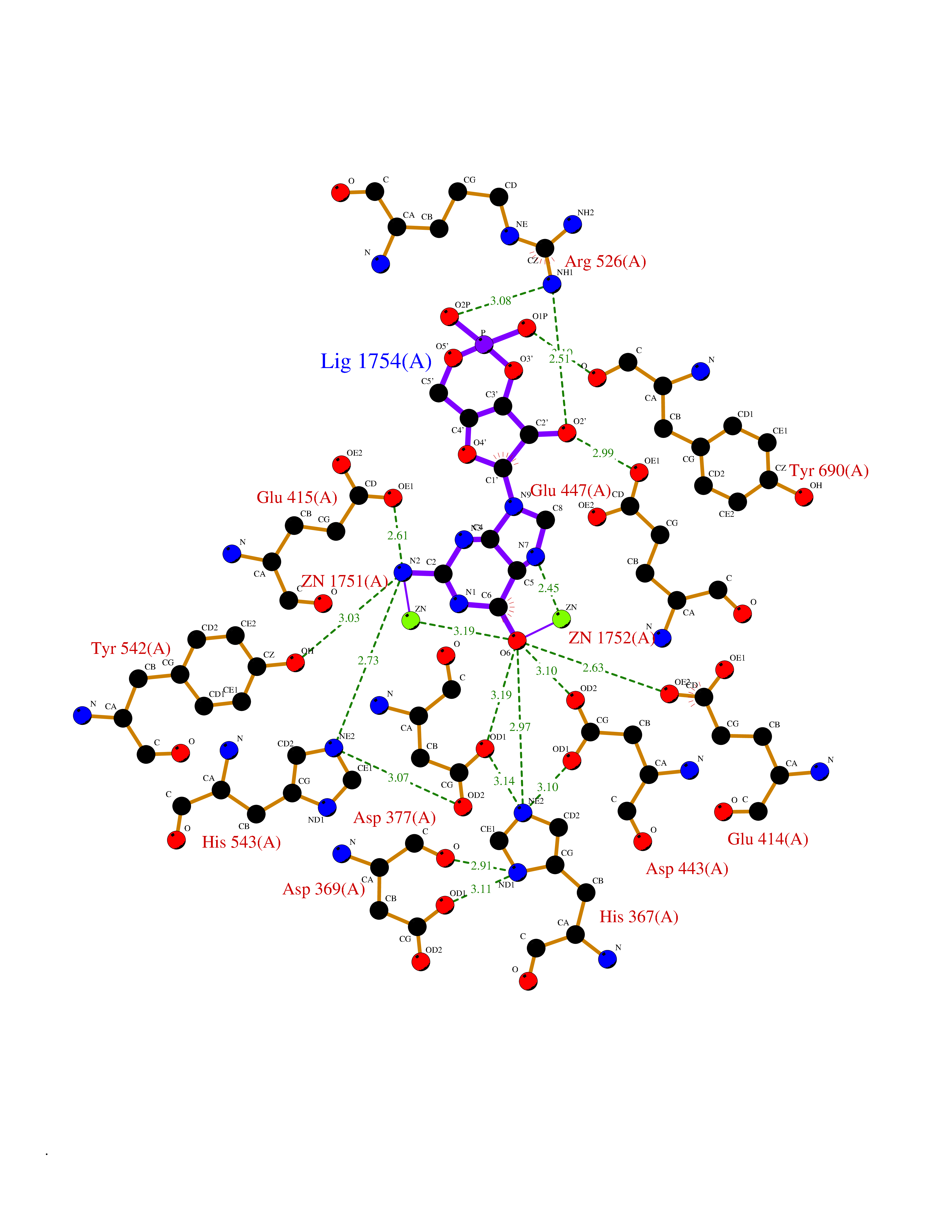 Ligplot Map 3D Binding mode Sequence SIRWKLVSEMKAENIKSFLRSFTKLPHLAGTEQNFLLAKKIQTQWKKFGLDSAKLVHYDVLLSYPNETNANYISIVDEHETEIFKTSPPPDGYENVTNIVPPYNAFSAQGMPEGDLVYVNYARTEDFFKLEREMGINCTGKIVIARYGKIFRGNKVKNAMLAGAIGIILYSDPADYFAPEVQPYPKGWNLPGTAAQRGNVLNLNGAGDPLTPGYPAKEYTFRLDVEEGVGIPRIPVHPIGYNDAEILLRYLGGIAPPDKSWKGALNVSYSIGPGFTGSSFRKVRMHVYNINKITRIYNVVGTIRGSVEPDRYVILGGHRDSWVFGAIDPTSGVAVLQEIARSFGKLMSKGWRPRRTIIFASWDAEEFGLLGSTEWAEENVKILQERSIAYINSDSSIEGNYTLRVDCTPLLYQLVYKLTKEIPSPDDGFESKSLYESWLEKDPSPENKNLPRINKLGSGSDFEAYFQRLGIASGRARYTKNKKTDKYSSYPVYHTIYETFELVEKFYDPTFKKQLSVAQLRGALVYELVDSKIIPFNIQDYAEALKNYAASIYNLSKKHDQQLTDHGVSFDSLFSAVKNFSEAASDFHKRLIQVDLNNPIAVRMMNDQLMLLERAFIDPLGLPGKLFYRHIIFAPSSHNKYAGESFPGIYDAIFDIENKANSRLAWKEVKKHISIAAFTIQAAAGTLKEV Hydrogen bonds contact Hydrophobic contact | ||||
| 9 | Human immunodeficiency virus Protease (HIV PR) | 3TL9 | 7.07 | |
Target general information Gen name HIV PR Organism Human immunodeficiency virus type 1 group M subtype B (isolate BH10) (HIV-1) Uniprot ID TTD ID Synonyms HIV Retropepsin; HIV PR Protein family NA Biochemical class Peptidase Function Gag-Pol polyprotein: Mediates, with Gag polyprotein, the essential events in virion assembly, including binding the plasma membrane, making the protein-protein interactions necessary to create spherical particles, recruiting the viral Env proteins, and packaging the genomic RNA via direct interactions with the RNA packaging sequence (Psi). Gag-Pol polyprotein may regulate its own translation, by the binding genomic RNA in the 5'-UTR. At low concentration, the polyprotein would promote translation, whereas at high concentration, the polyprotein would encapsidate genomic RNA and then shut off translation. Related diseases Sitosterolemia 2 (STSL2) [MIM:618666]: A form of sitosterolemia, an autosomal recessive metabolic disorder characterized by unregulated intestinal absorption of cholesterol, phytosterols and shellfish sterols, and decreased biliary excretion of dietary sterols into bile. Patients have hypercholesterolemia, very high levels of plant sterols in the plasma, and frequently develop tendon and tuberous xanthomas, accelerated atherosclerosis and premature coronary artery disease. {ECO:0000269|PubMed:11138003, ECO:0000269|PubMed:11452359, ECO:0000269|PubMed:11668628, ECO:0000269|PubMed:15054092, ECO:0000269|PubMed:35557526}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07035; DB02704; DB07806; DB02785; DB01824; DB01732; DB06874; DB08034; DB07961; DB07451; DB08212; DB08372; DB02972; DB04190; DB04042; DB08428; DB03076; DB03141; DB08457; DB07343; DB07337; DB07018; DB07332; DB05398; DB07578; DB08639; DB06414; DB04255; DB04547; DB02683; DB02009; DB03908; DB02629; DB01887; DB03803; DB02033; DB08281; DB08282; DB08284; DB08414; DB08598; DB07327; DB07885; DB02768; DB08600; DB01891; DB05871 Interacts with NA EC number EC 3.4.23.16 Uniprot keywords 3D-structure; Activation of host caspases by virus; AIDS; Aspartyl protease; Capsid protein; Direct protein sequencing; DNA integration; DNA recombination; DNA-binding; DNA-directed DNA polymerase; Endonuclease; Eukaryotic host gene expression shutoff by virus; Eukaryotic host translation shutoff by virus; Host cell membrane; Host cytoplasm; Host endosome; Host gene expression shutoff by virus; Host membrane; Host nucleus; Host-virus interaction; Hydrolase; Lipid-binding; Lipoprotein; Magnesium; Membrane; Metal-binding; Methylation; Modulation of host cell apoptosis by virus; Multifunctional enzyme; Myristate; Nuclease; Nucleotidyltransferase; Phosphoprotein; Protease; Repeat; Ribosomal frameshifting; RNA-binding; RNA-directed DNA polymerase; Transferase; Viral genome integration; Viral nucleoprotein; Viral penetration into host nucleus; Viral release from host cell; Virion; Virion maturation; Virus entry into host cell; Zinc; Zinc-finger Protein physicochemical properties Chain ID A,B Molecular weight (Da) 21934.7 Length 202 Aromaticity 0.06 Instability index 48.65 Isoelectric point 9.66 Charge (pH=7) 6.15 2D Binding mode Binding energy (Kcal/mol) -7.39  Molscript Map 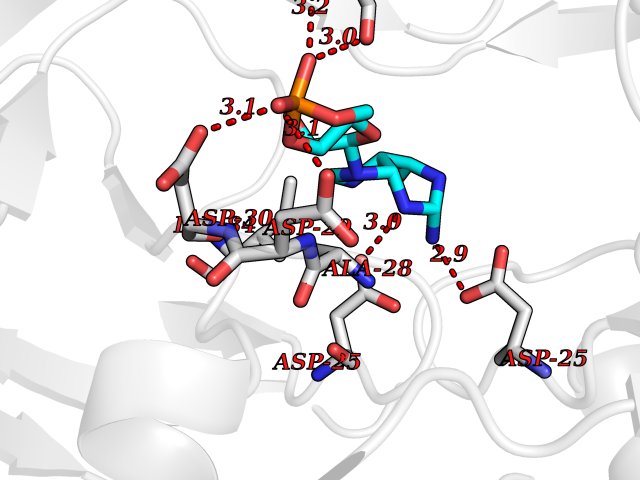 Pymol Map 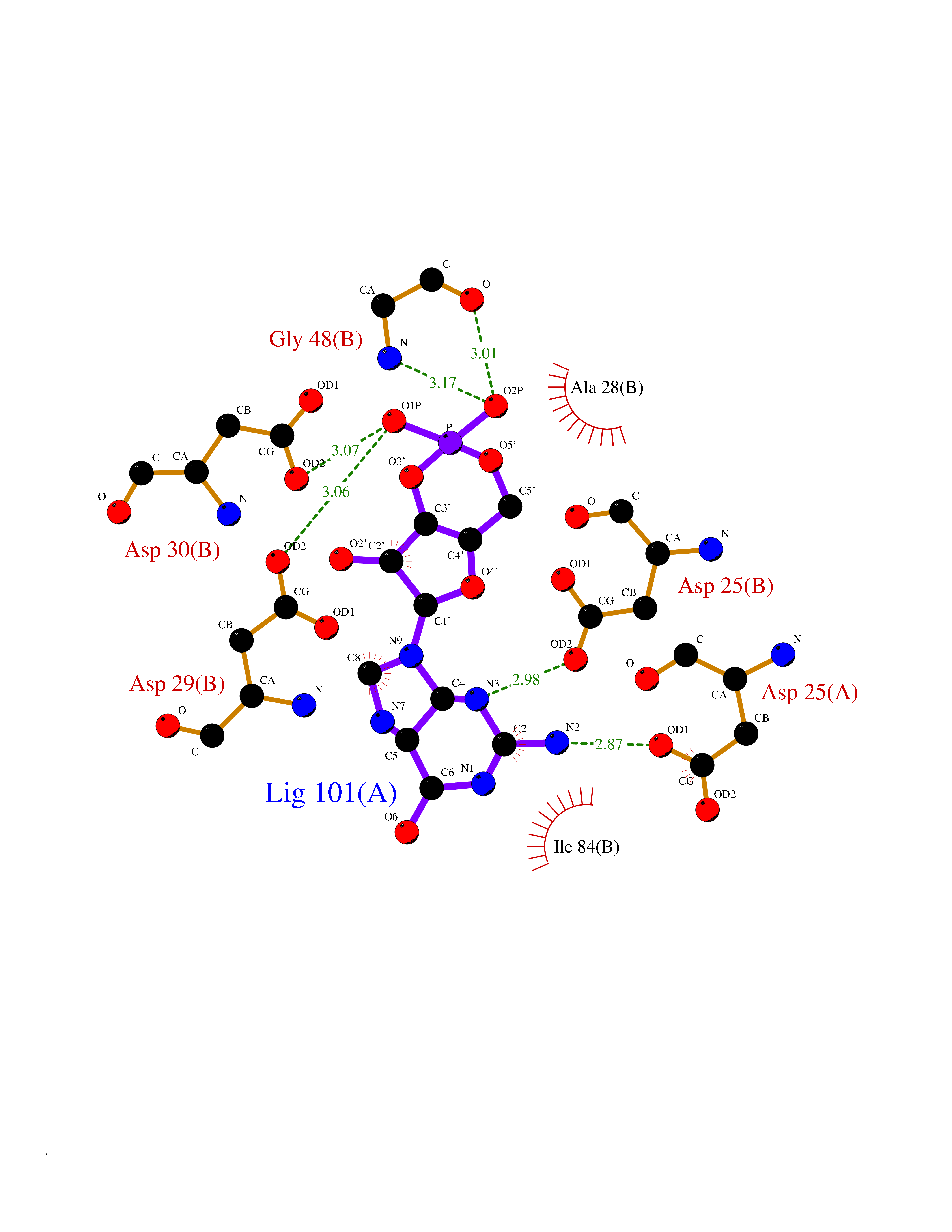 Ligplot Map 3D Binding mode Sequence PQITLWKRPLVTIKIGGQLKEALLDTGADDTVIEEMSLPGRWKPKMIGGIGGFIKVRQYDQIIIEIAGHKAIGTVLVGPTPVNIIGRNLLTQIGATLNFSFNFPQITLWKRPLVTIKIGGQLKEALLDTGADDTVIEEMSLPGRWKPKMIGGIGGFIKVRQYDQIIIEIAGHKAIGTVLVGPTPVNIIGRNLLTQIGATLNF Hydrogen bonds contact Hydrophobic contact | ||||
| 10 | Serine/threonine PP1-alpha (PPP1CA) | 3E7B | 7.06 | |
Target general information Gen name PPP1CA Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Serine/threonine-protein phosphatase PP1-alpha catalytic subunit; Protein phosphatase 1alpha; PPP1A; PP-1A Protein family PPP phosphatase family, PP-1 subfamily Biochemical class Phosphoric monoester hydrolase Function Protein phosphatase 1 (PP1) is essential for cell division, and participates in the regulation of glycogen metabolism, muscle contractility and protein synthesis. Involved in regulation of ionic conductances and long-term synaptic plasticity. May play an important role in dephosphorylating substrates such as the postsynaptic density-associated Ca(2+)/calmodulin dependent protein kinase II. Component of the PTW/PP1 phosphatase complex, which plays a role in the control of chromatin structure and cell cycle progression during the transition from mitosis into interphase. Regulates NEK2 function in terms of kinase activity and centrosome number and splitting, both in the presence and absence of radiation-induced DNA damage. Regulator of neural tube and optic fissure closure, and enteric neural crest cell (ENCCs) migration during development. In balance with CSNK1D and CSNK1E, determines the circadian period length, through the regulation of the speed and rhythmicity of PER1 and PER2 phosphorylation. May dephosphorylate CSNK1D and CSNK1E. Dephosphorylates the 'Ser-418' residue of FOXP3 in regulatory T-cells (Treg) from patients with rheumatoid arthritis, thereby inactivating FOXP3 and rendering Treg cells functionally defective. Dephosphorylates CENPA. Dephosphorylates the 'Ser-139' residue of ATG16L1 causing dissociation of ATG12-ATG5-ATG16L1 complex, thereby inhibiting autophagy. Protein phosphatase that associates with over 200 regulatory proteins to form highly specific holoenzymes which dephosphorylate hundreds of biological targets. Related diseases A chromosomal aberration involving FHIT has been found in a lymphoblastoid cell line established from a family with renal cell carcinoma and thyroid carcinoma. Translocation t(3;8)(p14.2;q24.1) with RNF139. Although the 3p14.2 breakpoint has been shown to interrupt FHIT in its 5-prime non-coding region, it is unlikely that FHIT is causally related to renal or other malignancies. {ECO:0000269|PubMed:15007172}.; DISEASE: Associated with digestive tract cancers. Numerous tumor types are found to have aberrant forms of FHIT protein due to deletions in a coding region of chromosome 3p14.2 including the fragile site locus FRA3B. {ECO:0000269|PubMed:15007172}. Drugs (DrugBank ID) DB02506 Interacts with Q6ZMQ8; P31749; O14727; P05067; O15169; P38398; O95400; Q99459; P12830; Q8TEP8; Q9NX63; Q6PJW8; Q96S65; Q9H175; Q92796; P05198; P55199; Q9BZS1; P42858; Q8NI77; Q8NG31; Q5S007; O00566; Q9UPR0; Q96QC0; Q96KQ4; Q8WUF5; O75807; Q5SWA1; Q96T49; Q6NYC8; P41236; Q5T8A7; Q86WC6; Q6NXS1; O14990; O75864; Q86XI6; Q9UQK1; O95685; Q15435; Q12972; Q12972-1; Q12972-2; Q96SB3; P60484; P06400; Q5UIP0; Q14684; P04271; Q7Z5V6; A8K8P3; Q562F6; Q9H788; Q8TEC5; P63208; Q7Z699; P43405-2; Q9HCH5; Q14C87; Q5JTV8; Q05BL1; Q13625; Q4KMQ1; Q4KMQ1-2; Q8TEL6; P49815; P55072; Q9Y2W2; Q9H4A3; P16989; P49750; Q9HBF4; Q7Z3T8; O95405; O08785; P36313; K9N4V7; Q76TK5; O35867; O35274 EC number EC 3.1.3.16 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Carbohydrate metabolism; Cell cycle; Cell division; Cytoplasm; Direct protein sequencing; Glycogen metabolism; Host-virus interaction; Hydrolase; Manganese; Metal-binding; Nucleus; Phosphoprotein; Protein phosphatase; Proteomics identification; Reference proteome Protein physicochemical properties Chain ID A Molecular weight (Da) 33626.3 Length 294 Aromaticity 0.11 Instability index 44.81 Isoelectric point 5.16 Charge (pH=7) -10.09 2D Binding mode Binding energy (Kcal/mol) -7.22  Molscript Map 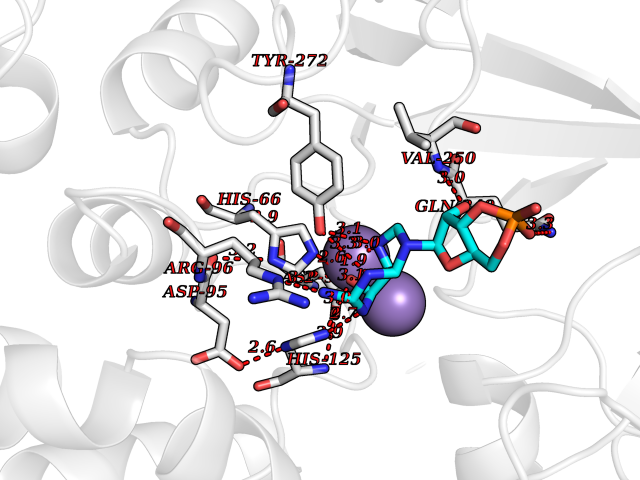 Pymol Map 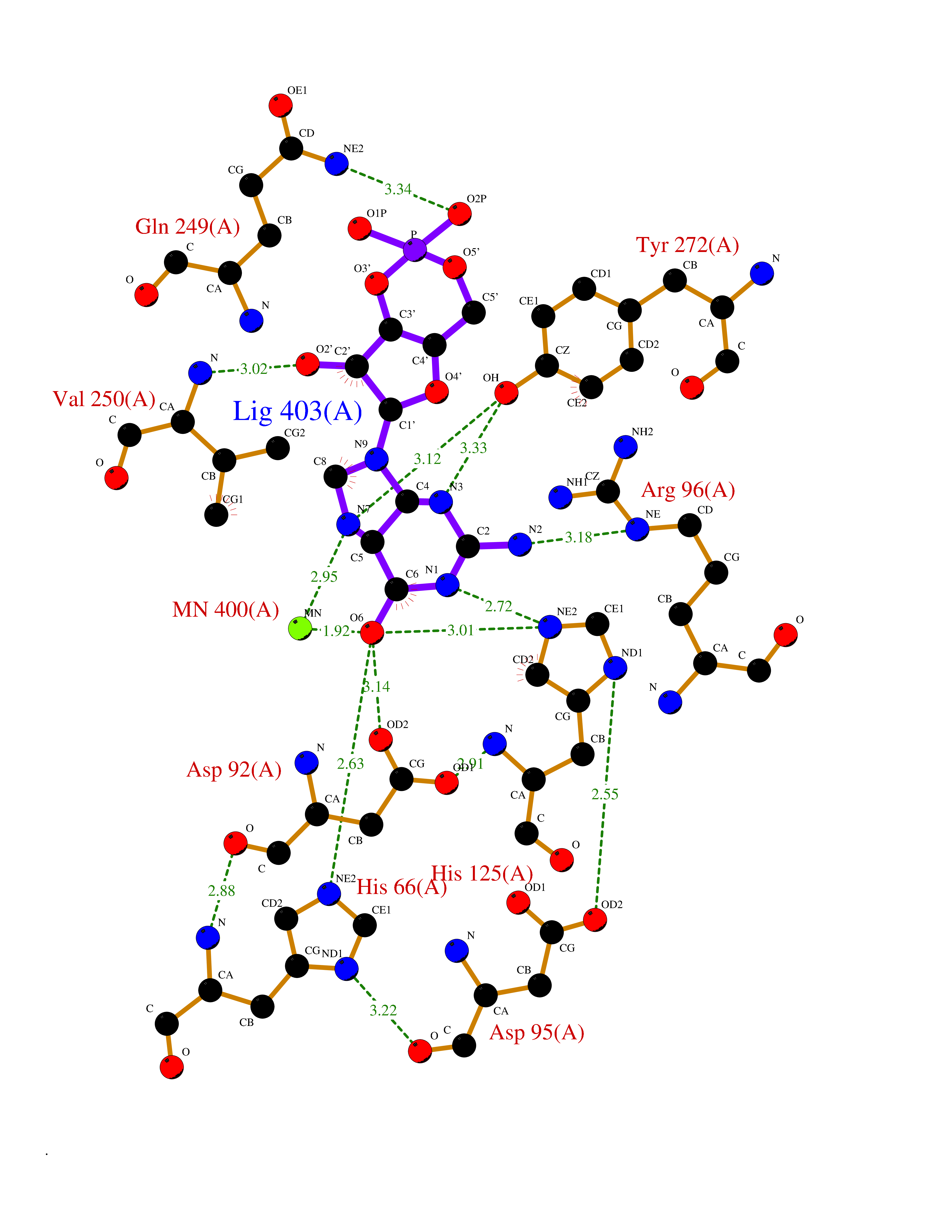 Ligplot Map 3D Binding mode Sequence SLNLDSIIGRLLEVQGSRPGKNVQLTENEIRGLCLKSREIFLSQPILLELEAPLKICGDIHGQYYDLLRLFEYGGFPPESNYLFLGDYVDRGKQSLETICLLLAYKIKYPENFFLLRGNHECASINRIYGFYDECKRRYNIKLWKTFTDCFNCLPIAAIVDEKIFCCHGGLSPDLQSMEQIRRIMRPTDVPDQGLLCDLLWSDPDKDVQGWGENDRGVSFTFGAEVVAKFLHKHDLDLICRAHQVVEDGYEFFAKRQLVTLFSAPNYCGEFDNAGAMMSVDETLMCSFQILKPA Hydrogen bonds contact Hydrophobic contact | ||||
| 11 | Endothelin-converting enzyme 1 (ECE1) | 3DWB | 6.98 | |
Target general information Gen name ECE1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms ECE-1 Protein family Peptidase M13 family Biochemical class Peptidase Function Converts big endothelin-1 to endothelin-1. Related diseases Hirschsprung disease, cardiac defects, and autonomic dysfunction (HCAD) [MIM:613870]: A disorder characterized by skip-lesions Hirschsprung disease, craniofacial abnormalities and other dysmorphic features, cardiac defects including ductus arteriosus, small subaortic ventricular septal defect, small atrial septal defect, and autonomic dysfunction. {ECO:0000269|PubMed:9915973}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07171 Interacts with P49760; A8MQ03; Q8IUG1; P60370; P60410 EC number EC 3.4.24.71 Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disease variant; Disulfide bond; Glycoprotein; Hirschsprung disease; Hydrolase; Membrane; Metal-binding; Metalloprotease; Phosphoprotein; Protease; Proteomics identification; Reference proteome; Signal-anchor; Transmembrane; Transmembrane helix; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 75247.9 Length 660 Aromaticity 0.12 Instability index 46.29 Isoelectric point 5.33 Charge (pH=7) -18.3 2D Binding mode Binding energy (Kcal/mol) -7.66  Molscript Map 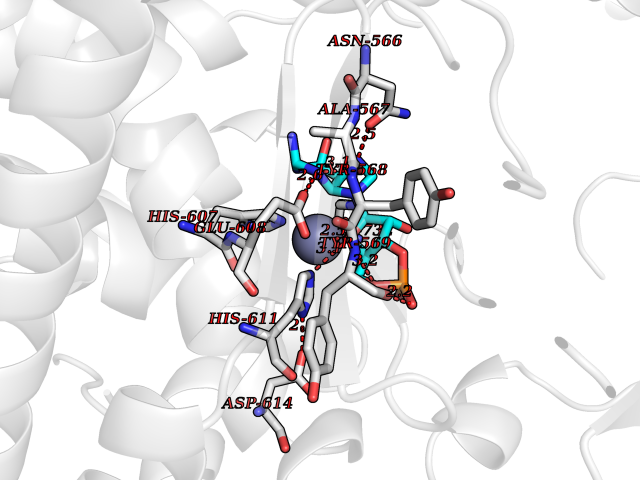 Pymol Map 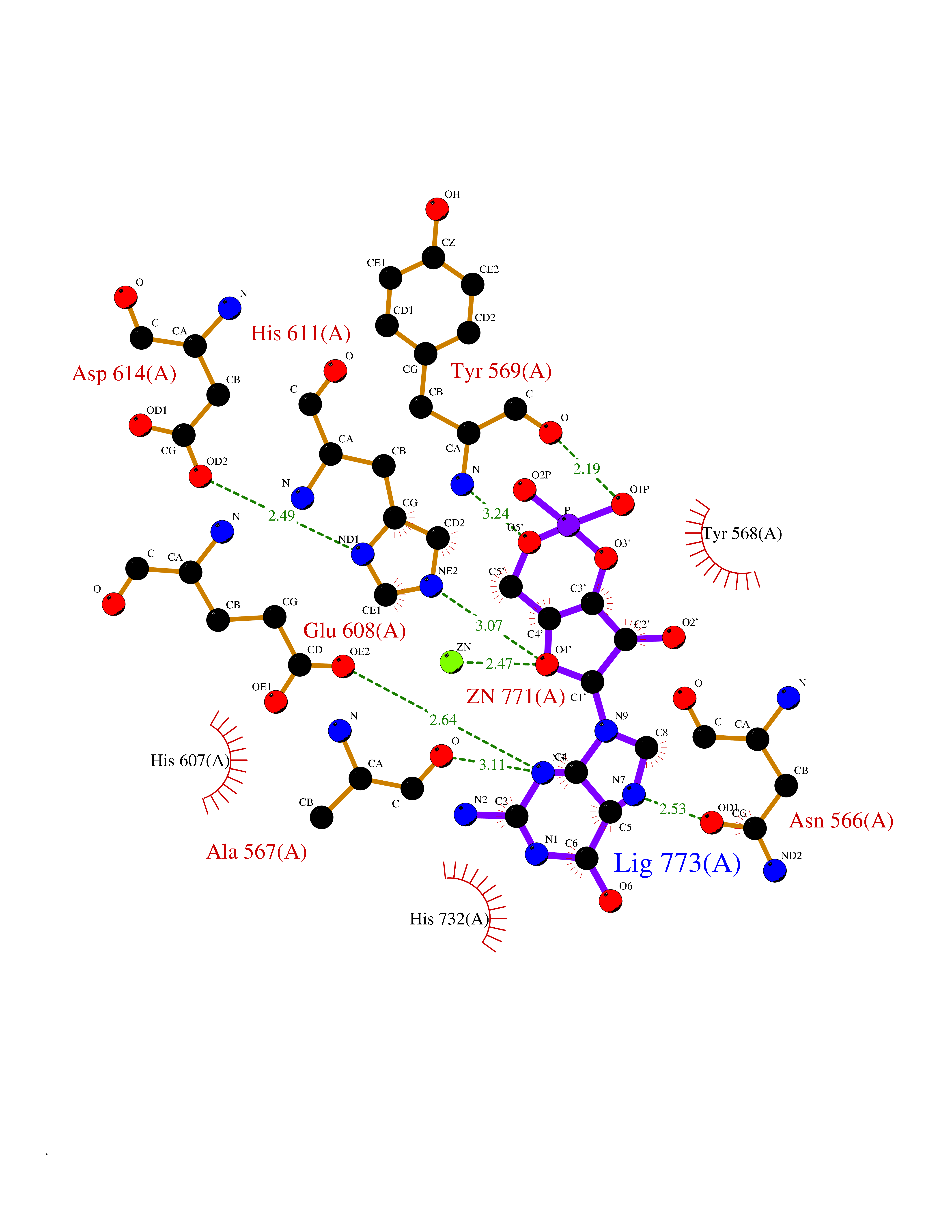 Ligplot Map 3D Binding mode Sequence SEACVSVTSSILSSMDPTVDPCHDFFSYACGGWIKANPVPDGHSRWGTFSNLWEHNQAIIKHLLENSTASVSEAERKAQVYYRACMNETRIEELRAKPLMELIERLGGWNITGPWAKDNFQDTLQVVTAHYRTSPFFSVYVSADSKNSNSNVIQVDQSGLGLPSRDYYLNKTENEKVLTGYLNYMVQLGKLLGGGDEEAIRPQMQQILDFETALANITIPQEKRRDEELIYHKVTAAELQTLAPAINWLPFLNTIFYPVEINESEPIVVYDKEYLEQISTLINTTDRCLLNNYMIWNLVRKTSSFLDQRFQDADEKFMEVMWKFCVSDTENNLGFALGPMFVKATFAEDSKSIATEIILEIKKAFEESLSTLKWMDEETRKSAKEKADAIYNMIGYPNFIMDPKELDKVFNDYTAVPDLYFENAMRFFNFSWRVTADQLRKAPNRDQWSMTPPMVNAYYSPTKNEIVFPAGILQAPFYTRSSPKALNFGGIGVVVGHELTHAFDDQGREYDKDGNLRPWWKNSSVEAFKRQTECMVEQYSNYSVNGEPVNGRHTLGENIADNGGLKAAYRAYQNWVKKNGAEHSLPTLGLTNNQLFFLGFAQVWCSVRTPESSHEGLITDPHSPSRFRVIGSLSNSKEFSEHFRCPPGSPMNPPHKCEVW Hydrogen bonds contact Hydrophobic contact | ||||
| 12 | Amylin receptor (IAPPR) | 6ZIS | 6.98 | |
Target general information Gen name CALCR-RAMP1/RAMP2/RAMP3 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Complex of Calcitonin receptor and Receptor activity-modifying protein Protein family RAMP family Biochemical class NA Function Transports the calcitonin gene-related peptide type 1 receptor (CALCRL) to the plasma membrane. Acts as a receptor for calcitonin-gene-related peptide (CGRP) together with CALCRL. Related diseases Immunodeficiency 9 (IMD9) [MIM:612782]: An immune disorder characterized by recurrent infections, impaired activation and proliferative response of T-cells, decreased T-cell production of cytokines, and normal lymphocytes counts and serum immunoglobulin levels. In surviving patients ectodermal dysplasia with anhidrosis and non-progressive myopathy may be observed. {ECO:0000269|PubMed:16147976, ECO:0000269|PubMed:16582901}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Myopathy, tubular aggregate, 2 (TAM2) [MIM:615883]: A rare congenital myopathy characterized by regular arrays of membrane tubules on muscle biopsies without additional histopathological hallmarks. Tubular aggregates in muscle are structures of variable appearance consisting of an outer tubule containing either one or more microtubule-like structures or amorphous material. TAM2 patients have myopathy and pupillary abnormalities. {ECO:0000269|PubMed:24591628, ECO:0000269|PubMed:28058752}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01278 Interacts with Q16602; P21145; Q5J8X5; Q16617 EC number NA Uniprot keywords 3D-structure; Cell membrane; Disulfide bond; Membrane; Proteomics identification; Receptor; Reference proteome; Signal; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A Molecular weight (Da) 63832.6 Length 569 Aromaticity 0.12 Instability index 22.42 Isoelectric point 5.07 Charge (pH=7) -16.96 2D Binding mode Binding energy (Kcal/mol) -7.45 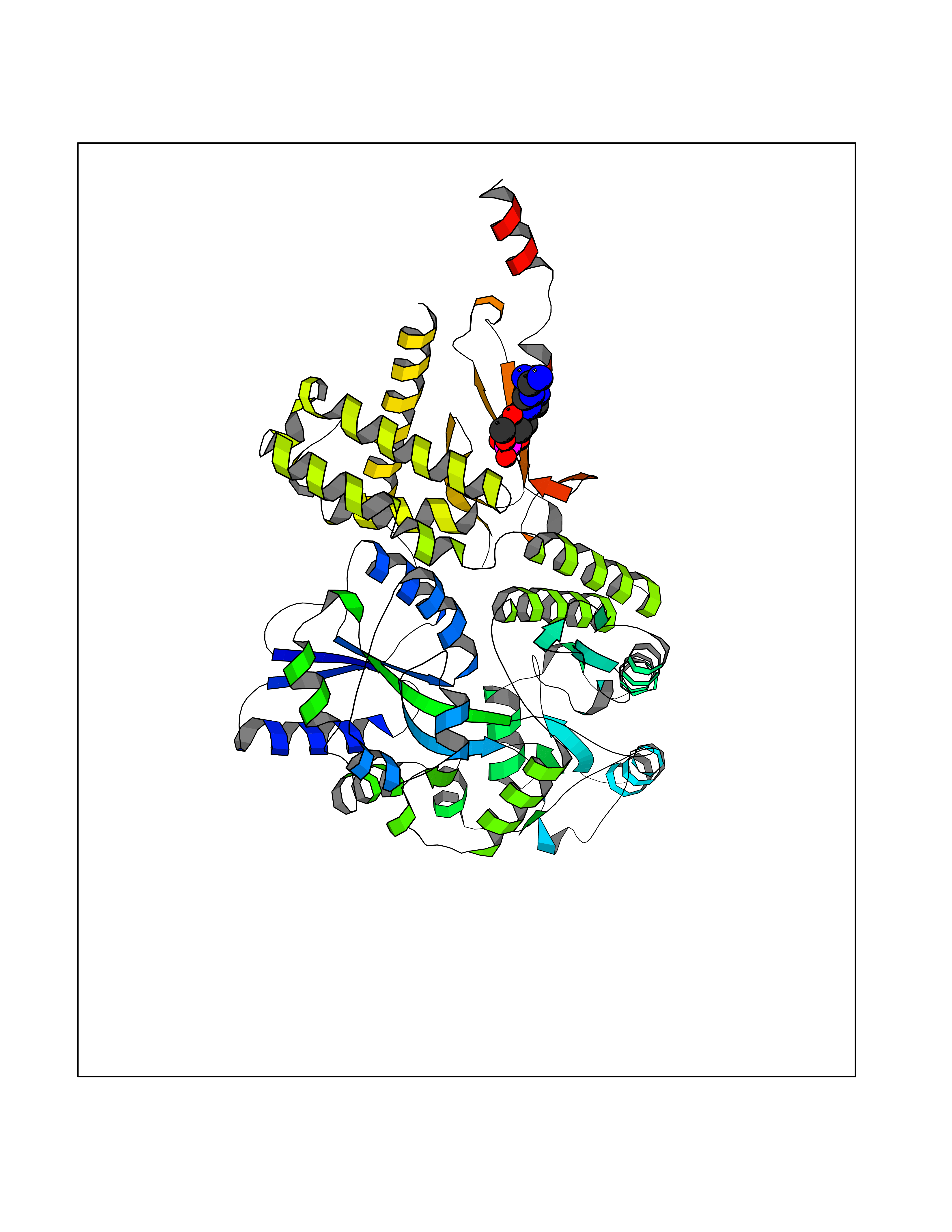 Molscript Map  Pymol Map 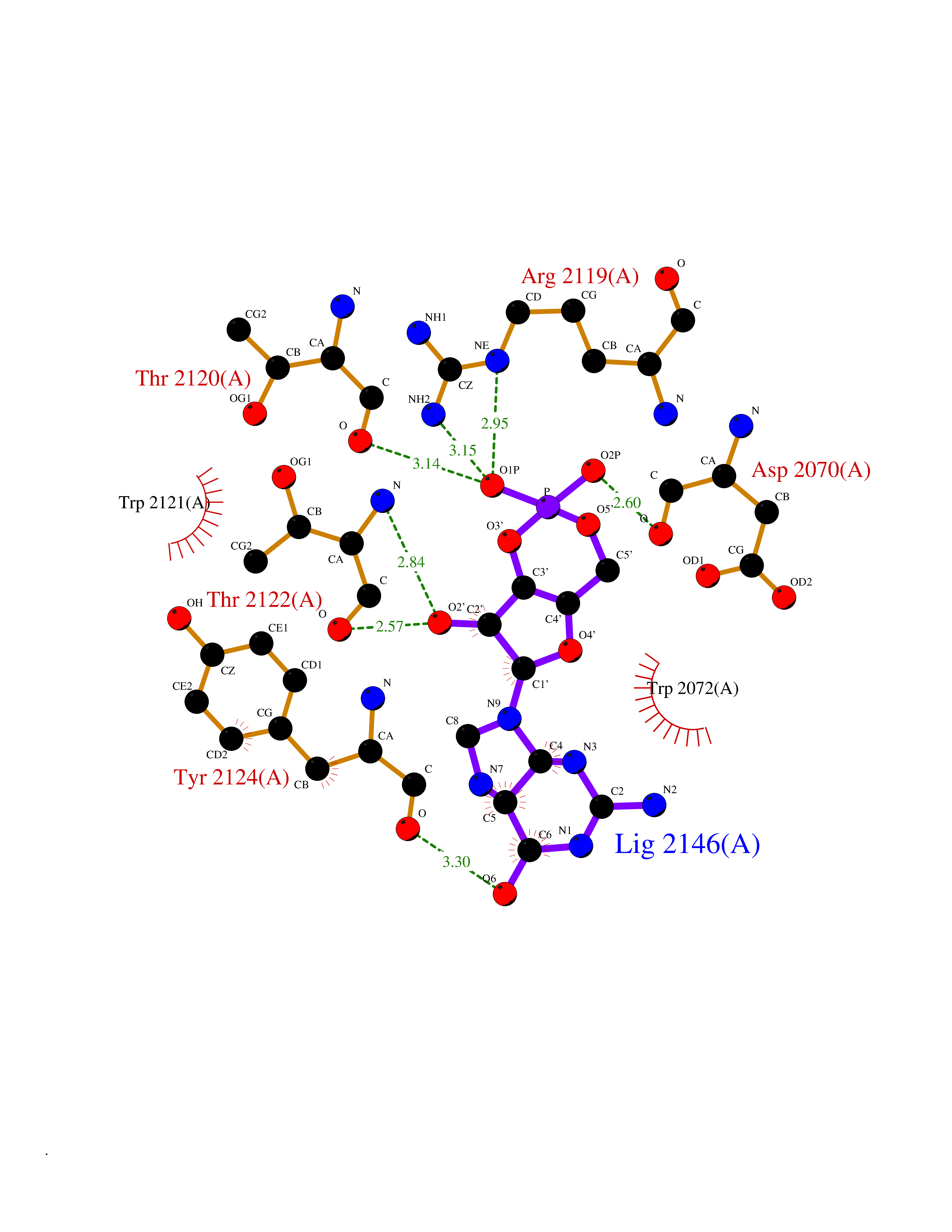 Ligplot Map 3D Binding mode Sequence SAKIEEGKLVIWINGDKGYNGLAEVGKKFEKDTGIKVTVEHPDKLEEKFPQVAATGDGPDIIFWAHDRFGGYAQSGLLAEITPDKAFQDKLYPFTWDAVRYNGKLIAYPIAVEALSLIYNKDLLPNPPKTWEEIPALDKELKAKGKSALMFNLQEPYFTWPLIAADGGYAFKYENGKYDIKDVGVDNAGAKAGLTFLVDLIKNKHMNADTDYSIAEAAFNKGETAMTINGPWAWSNIDTSKVNYGVTVLPTFKGQPSKPFVGVLSAGINAASPNKELAKEFLENYLLTDEGLEAVNKDKPLGAVALKSYEEELAKDPRIAATMENAQKGEIMPNIPQMSAFWYAVRTAVINAASGRQTVDEALKDAQTNAAAEFTTACQEANYGALLRELCLTQFQVDMEAVGETLWCDWGRTIRSYRELADCTWHMAEKLGCFWPNAEVDRFFLAVHGRYFRSCPISIQLGVTRNKIMTAQYECYQKIMQDPIQQGVYCQRTWDGWLCWNDVAAGTESMQLCPDYFQDFDPSEKVTKICDQDGNWFRHPASQRTWTDYTQCNVNTHEKVKTALNLFYL Hydrogen bonds contact Hydrophobic contact | ||||
| 13 | Short transient receptor potential channel 5 (TRPC5) | 7WDB | 6.97 | |
Target general information Gen name TRPC5 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms hTRP5; hTRP-5; TrpC5; Transient receptor protein 5; TRP-5 Protein family Transient receptor (TC 1.A.4) family, STrpC subfamily, TRPC5 sub-subfamily Biochemical class Transient receptor potential catioin channel Function Thought to form a receptor-activated non-selective calcium permeant cation channel. Probably is operated by a phosphatidylinositol second messenger system activated by receptor tyrosine kinases or G-protein coupled receptors. Has also been shown to be calcium-selective. May also be activated by intracellular calcium store depletion. Related diseases Loss-of-function variants in TRPC5 may be involved in a mental disorder characterized by maladaptive behavior, anxiety, autism, postpartum depression, extreme food-seeking and hoarding behavior, hyperphagia and obesity. {ECO:0000269|PubMed:38959890}. Drugs (DrugBank ID) NA Interacts with NA EC number NA Uniprot keywords 3D-structure; ANK repeat; Calcium; Calcium channel; Calcium transport; Cell membrane; Disease variant; Glycoprotein; Ion channel; Ion transport; Membrane; Proteomics identification; Reference proteome; Repeat; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A Molecular weight (Da) 76850.6 Length 665 Aromaticity 0.12 Instability index 40.05 Isoelectric point 6.16 Charge (pH=7) -5.94 2D Binding mode Binding energy (Kcal/mol) -7.49 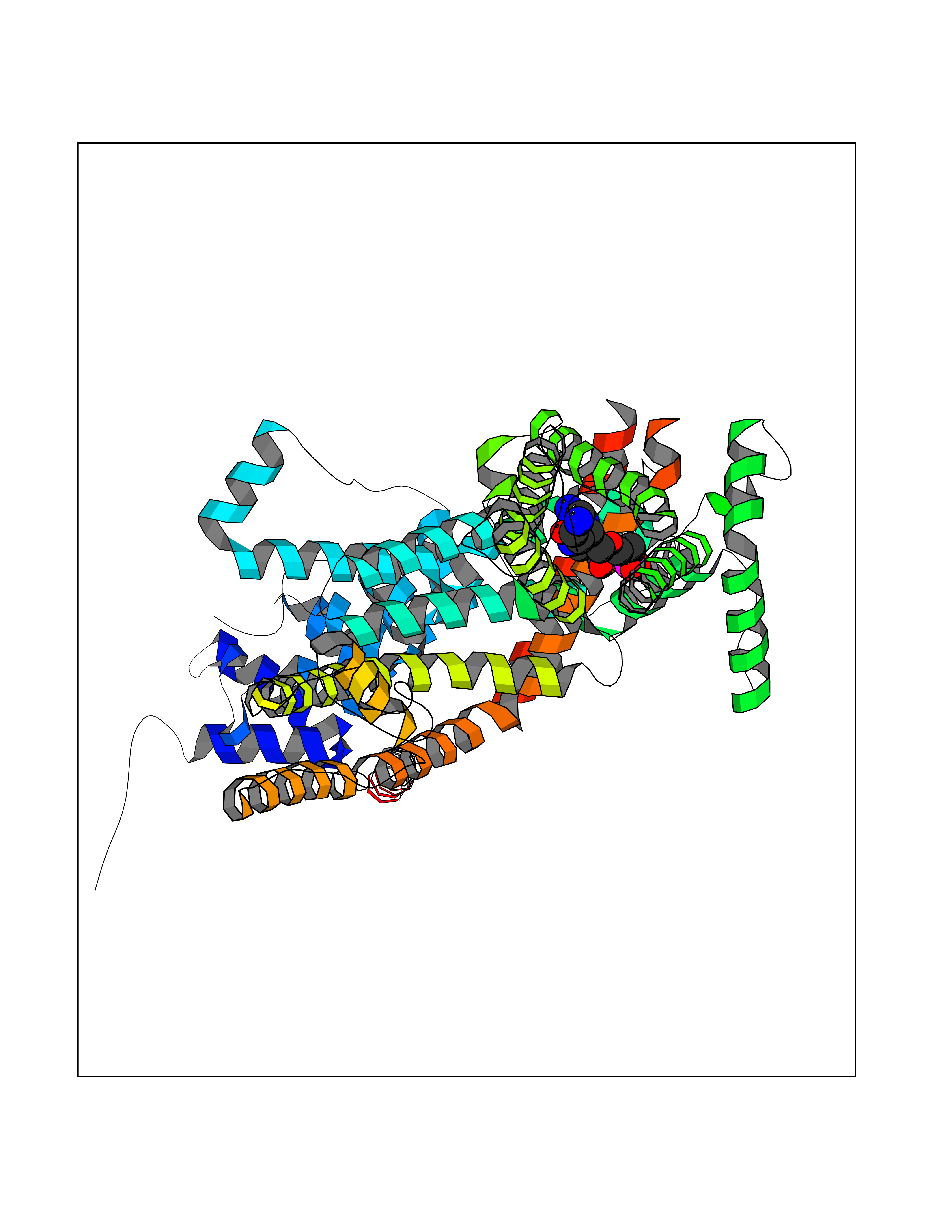 Molscript Map  Pymol Map 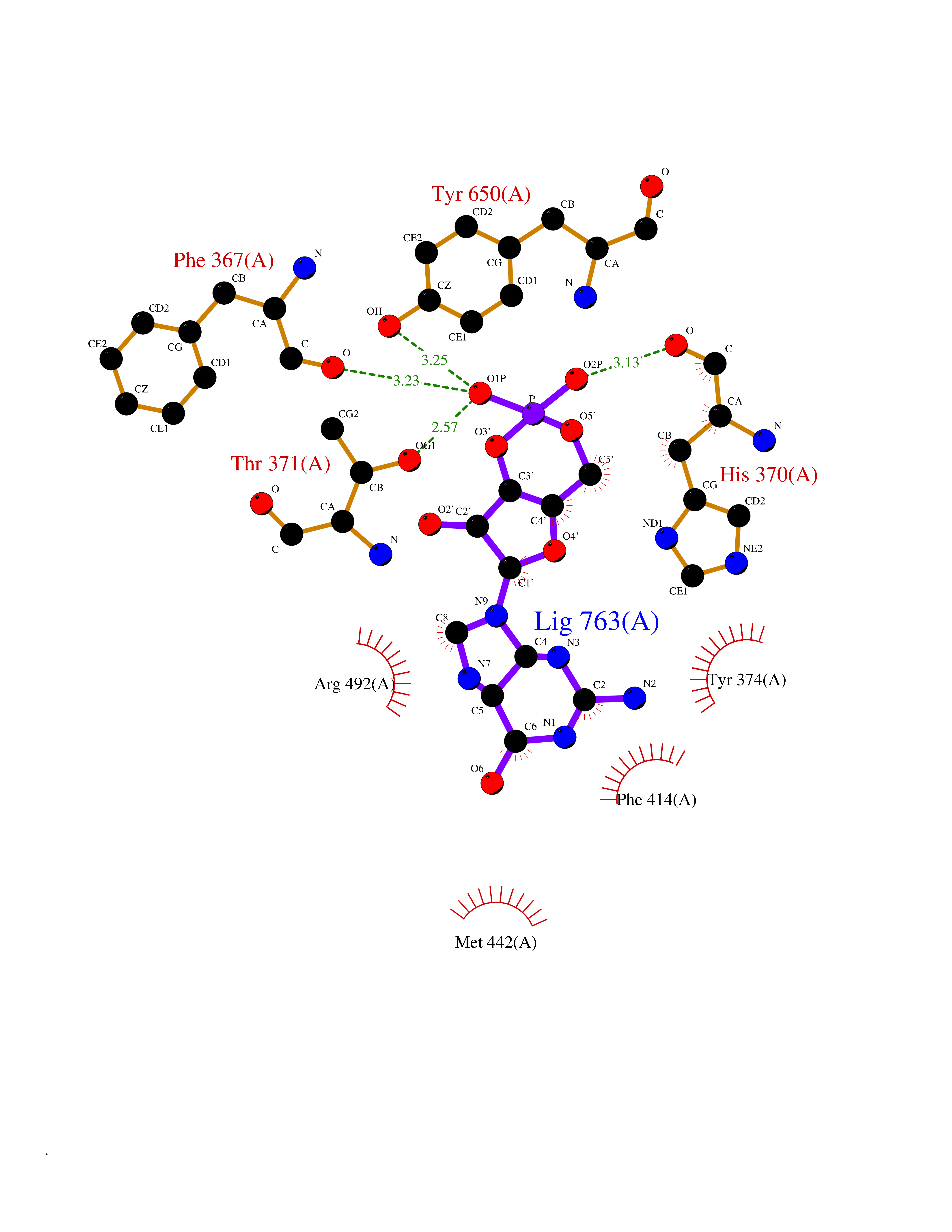 Ligplot Map 3D Binding mode Sequence RIPLQIVRAETELSAEEKAFLNAVEKGDYATVKQALQEAEIYYNVNINCMDPLGRSALLIAIENENLEIMELLLNHSVYVGDALLYAIRKEVVGAVELLLSYQFSEFTPDITPIMLAAHTNNYEIIKLLVQKRVTIPRPHQIRCNCVECVSSSEVDSLRHSRSRLNIYKALASPSLIALSSEDPILTAFRLGWELKELSKVENEFKAEYEELSQQCKLFAKDLLDQARSSRELEIILNHRDDLAKLKVAIKYHQKEFVAQPNCQQLLATLWYDGFPGWRRKHWVVKLLTCMTIGFLFPMLSIAYLISPRSNLGLFIKKPFIKFICHTASYLTFLFMLLLASQHVQGPPPTVVEWMILPWVLGFIWGEIKEMWDGGFTEYIHDWWNLMDFAMNSLYLATISLKIVAYVKYNGSRPREEWEMWHPTLIAEALFAISNILSSLRLISLFTANSHLGPLQISLGRMLLDILKFLFIYCLVLLAFANGLNQLYFYYETRAIDEPNNCKGIRCEKQNNAFSTLFETLQSLFWSVFGLLNLYVTNVKARHEFTEFVGATMFGTYNVISLVVLLNMLIAMMNNSYQLIADHADIEWKFARTKLWMSYFDEGGTLPPPFNIISLIQNQHYQEVIRNLVKRYVAAMIRNSKTTEENFKELKQDISSFRYEVLDLL Hydrogen bonds contact Hydrophobic contact | ||||
| 14 | Lanosterol 14-alpha demethylase (CYP51A1) | 4UHI | 6.95 | |
Target general information Gen name CYP51A1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Cytochrome P450LI; Cytochrome P45014DM; Cytochrome P450-14DM; Cytochrome P450 51A1 Protein family Cytochrome P450 family Biochemical class Cytochrome P450 family Function Catalyzes C14-demethylation of lanosterol; it transforms lanosterol into 4,4'-dimethyl cholesta-8,14,24-triene-3-beta-ol. Related diseases Spondyloepimetaphyseal dysplasia, short limb-hand type (SEMD-SL) [MIM:271665]: A bone disease characterized by short-limbed dwarfism, a narrow chest with pectus excavatum, brachydactyly in the hands and feet, a characteristic craniofacial appearance and premature calcifications. The radiological findings are distinctive and comprise short long bones throughout the skeleton with striking epiphyses that are stippled, flattened and fragmented and flared, irregular metaphyses. Platyspondyly in the spine with wide intervertebral spaces is observed and some vertebral bodies are pear-shaped with central humps, anterior protrusions and posterior scalloping. {ECO:0000269|PubMed:19110212, ECO:0000269|PubMed:20223752, ECO:0000269|PubMed:26463668}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Warburg-Cinotti syndrome (WRCN) [MIM:618175]: An autosomal dominant disease characterized by progressive corneal neovascularization, keloid formation, chronic skin ulcers, wasting of subcutaneous tissue, flexion contractures of the fingers, and acro-osteolysis. {ECO:0000269|PubMed:30449416}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07705; DB05667; DB01110; DB01007 Interacts with NA EC number EC 1.14.14.154 Uniprot keywords 3D-structure; Alternative splicing; Cholesterol biosynthesis; Cholesterol metabolism; Endoplasmic reticulum; Heme; Iron; Lipid biosynthesis; Lipid metabolism; Membrane; Metal-binding; Microsome; Monooxygenase; Oxidoreductase; Proteomics identification; Reference proteome; Steroid biosynthesis; Steroid metabolism; Sterol biosynthesis; Sterol metabolism; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 53013.3 Length 462 Aromaticity 0.11 Instability index 47.66 Isoelectric point 8.8 Charge (pH=7) 7 2D Binding mode Binding energy (Kcal/mol) -6.82  Molscript Map 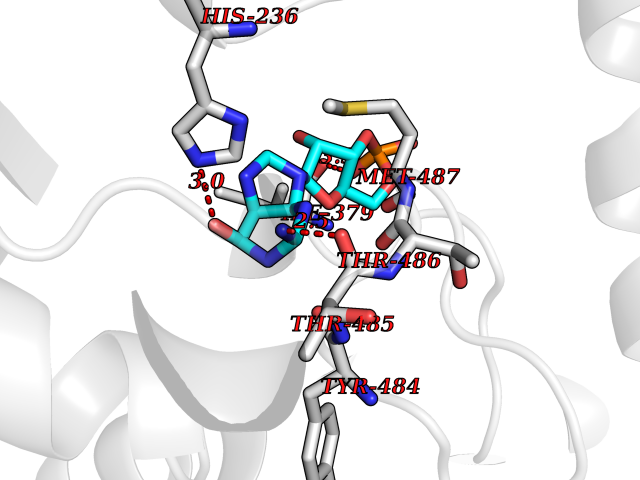 Pymol Map 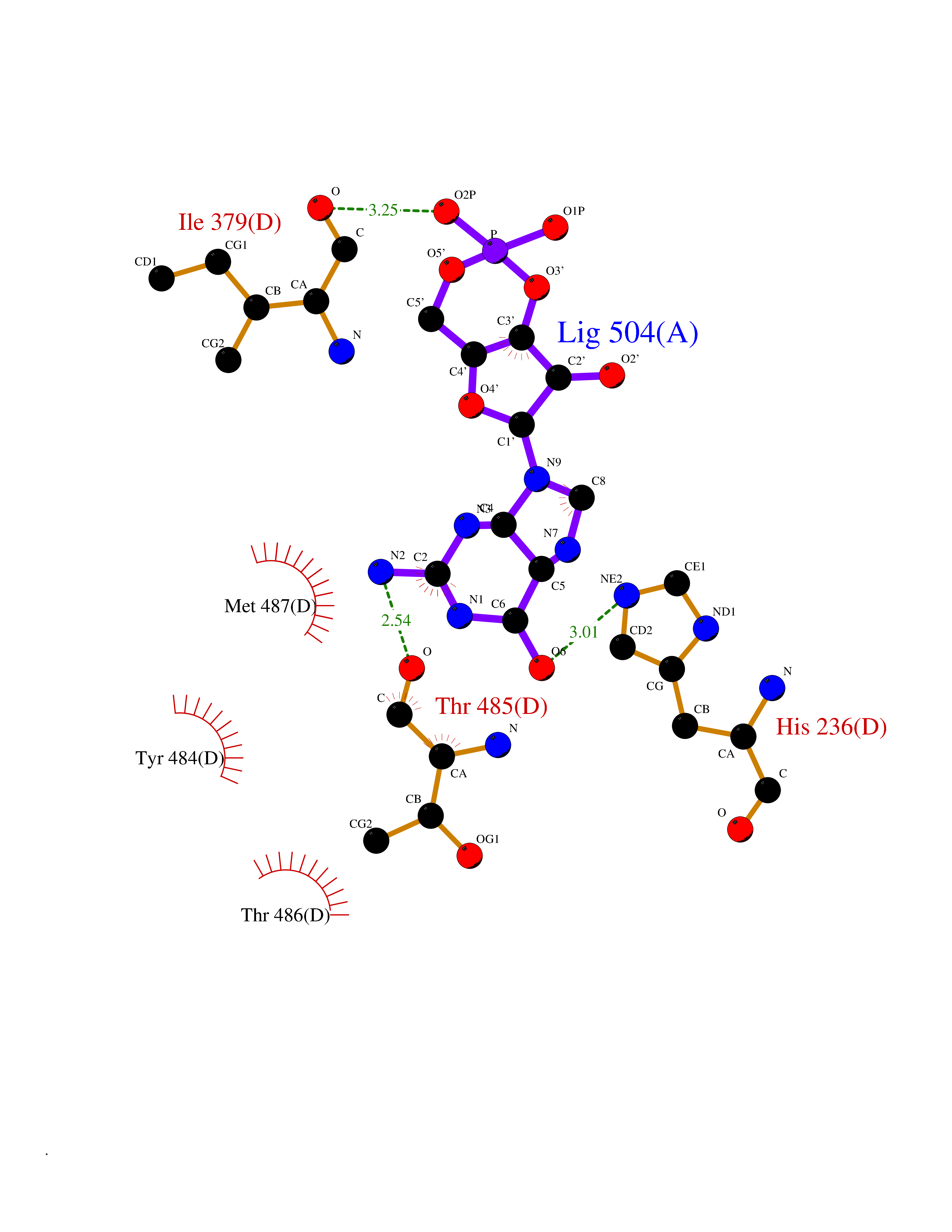 Ligplot Map 3D Binding mode Sequence PPYIFSPIPFLGHAIAFGKSPIEFLENAYEKYGPVFSFTMVGKTFTYLLGSDAAALLFNSKNEDLNAEDVYSRLTTPVFGKGVAYDVPNPVFLEQKKMLKSGLNIAHFKQHVSIIEKETKEYFESWGESGEKNVFEALSELIILTASHCLHGKEIRSQLNEKVAQLYADLDGGFSHAAWLLPGWLPLPSFRRRDRAHREIKDIFYKAIQKRRQSQEKIDDILQTLLDATYKDGRPLTDDEVAGMLIGLLLAGQHTSSTTSAWMGFFLARDKTLQKKCYLEQKTVCGENLPPLTYDQLKDLNLLDRCIKETLRLRPPIMIMMRMARTPQTVAGYTIPPGHQVCVSPTVNQRLKDSWVERLDFNPDRYLQDNPASGEKFAYVPFGAGRHRCIGENFAYVQIKTIWSTMLRLYEFDLIDGYFPTVNYTTMIHTPENPVIRYKRRSLPGWLPLPSFRRRDRAHREI Hydrogen bonds contact Hydrophobic contact | ||||
| 15 | Phosphodiesterase 4D (PDE4D) | 1Y2K | 6.95 | |
Target general information Gen name PDE4D Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms cAMP-specific 3',5'-cyclic phosphodiesterase 4D; PDE43; DPDE3 Protein family Cyclic nucleotide phosphodiesterase family, PDE4 subfamily Biochemical class Phosphoric diester hydrolase Function Hydrolyzes the second messenger cAMP, which is a key regulator of many important physiological processes. Related diseases Genetic variations in PDE4D might be associated with susceptibility to stroke. PubMed:17006457 states that association with stroke has to be considered with caution. {ECO:0000269|PubMed:17006457}.; DISEASE: Acrodysostosis 2, with or without hormone resistance (ACRDYS2) [MIM:614613]: A pleiotropic disorder characterized by skeletal, endocrine, and neurological abnormalities. Skeletal features include brachycephaly, midface hypoplasia with a small upturned nose, brachydactyly, and lumbar spinal stenosis. Endocrine abnormalities include hypothyroidism and hypogonadism in males and irregular menses in females. Developmental disability is a common finding but is variable in severity and can be associated with significant behavioral problems. {ECO:0000269|PubMed:22464250, ECO:0000269|PubMed:22464252, ECO:0000269|PubMed:23033274, ECO:0000269|PubMed:23043190}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB06842; DB04149; DB03606; DB03183; DB04469; DB02676; DB01959; DB07051; DB04271; DB07954; DB08299; DB00131; DB01427; DB00201; DB03849; DB05219; DB00651; DB06246; DB05266; DB01088; DB01113; DB01791; DB01656; DB01954; DB05298; DB09283; DB02918 Interacts with P32121; P38432; Q0D2H9; Q08AF8; P43360; Q07343; Q13077; P32121; P26769; P38432; Q96CV9; Q8IUH5 EC number EC 3.1.4.53 Uniprot keywords 3D-structure; Alternative splicing; cAMP; Cell membrane; Cytoplasm; Cytoskeleton; Disease variant; Hydrolase; Isopeptide bond; Manganese; Membrane; Metal-binding; Phosphoprotein; Proteomics identification; Reference proteome; Ubl conjugation; Zinc Protein physicochemical properties Chain ID A,B Molecular weight (Da) 37201.9 Length 322 Aromaticity 0.07 Instability index 35.83 Isoelectric point 5.02 Charge (pH=7) -21.16 2D Binding mode Binding energy (Kcal/mol) -7.85  Molscript Map 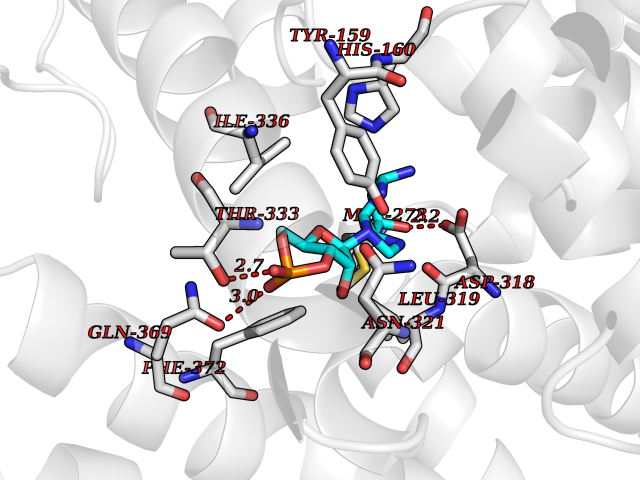 Pymol Map 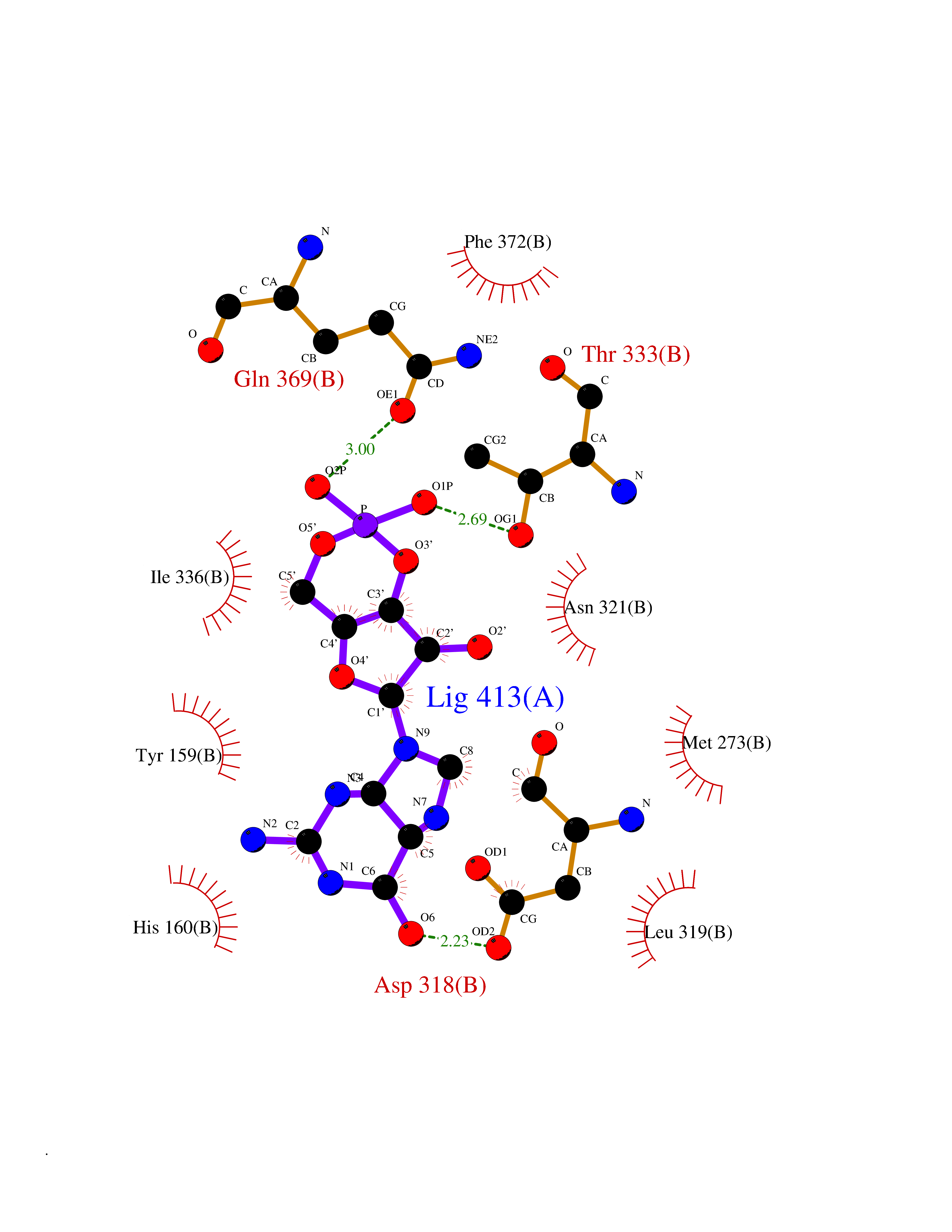 Ligplot Map 3D Binding mode Sequence TEQEDVLAKELEDVNKWGLHVFRIAELSGNRPLTVIMHTIFQERDLLKTFKIPVDTLITYLMTLEDHYHADVAYHNNIHAADVVQSTHVLLSTPALEAVFTDLEILAAIFASAIHDVDHPGVSNQFLINTNSELALMYNDSSVLENHHLAVGFKLLQEENCDIFQNLTKKQRQSLRKMVIDIVLATDMSKHMNLLADLKTMVETKKVVLLLDNYSDRIQVLQNMVHCADLSNPTKPLQLYRQWTDRIMEEFFRQGDRERERGMEISPMCDKHNASVEKSQVGFIDYIVHPLWETWADLVHPDAQDILDTLEDNREWYQSTIP Hydrogen bonds contact Hydrophobic contact | ||||
| 16 | Endothelin receptor type B | 5GLI | 6.93 | |
Target general information Gen name EDNRB Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms ETRB Protein family G-protein coupled receptor 1 family, Endothelin receptor subfamily, EDNRB sub-subfamily Biochemical class Signaling protein Function Endothelin receptor activity.Peptide hormone binding.Type 1 angiotensin receptor binding. Related diseases Waardenburg syndrome 4A (WS4A) [MIM:277580]: A disorder characterized by the association of Waardenburg features (depigmentation and deafness) with the absence of enteric ganglia in the distal part of the intestine (Hirschsprung disease). {ECO:0000269|PubMed:12189494, ECO:0000269|PubMed:8634719}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Hirschsprung disease 2 (HSCR2) [MIM:600155]: A disorder of neural crest development characterized by absence of enteric ganglia along a variable length of the intestine. It is the most common cause of congenital intestinal obstruction. Early symptoms range from complete acute neonatal obstruction, characterized by vomiting, abdominal distention and failure to pass stool, to chronic constipation in the older child. {ECO:0000269|PubMed:11471546, ECO:0000269|PubMed:28236341, ECO:0000269|PubMed:8001158, ECO:0000269|PubMed:8630503, ECO:0000269|PubMed:8852660}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: ABCD syndrome (ABCDS) [MIM:600501]: An autosomal recessive syndrome characterized by albinism, black lock at temporal occipital region, bilateral deafness, aganglionosis of the large intestine and total absence of neurocytes and nerve fibers in the small intestine. {ECO:0000269|PubMed:11891690}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Heterozygous mutations in EDNRB may be responsible for Waardenburg syndrome 2, an autosomal dominant disorder characterized by sensorineural deafness and pigmentary disturbances. {ECO:0000269|PubMed:28236341}. Drugs (DrugBank ID) DB06403; DB00559; DB06460; DB06138; DB08932; DB06268; DB06558 Interacts with P05305 EC number NA Uniprot keywords 3D-structure; Albinism; Alternative splicing; Cell membrane; Deafness; Disease variant; Disulfide bond; G-protein coupled receptor; Glycoprotein; Hirschsprung disease; Lipoprotein; Membrane; Palmitate; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Signal; Transducer; Transmembrane; Transmembrane helix; Waardenburg syndrome Protein physicochemical properties Chain ID A Molecular weight (Da) 35251 Length 312 Aromaticity 0.12 Instability index 32.82 Isoelectric point 9.08 Charge (pH=7) 11.97 2D Binding mode Binding energy (Kcal/mol) -7.65  Molscript Map 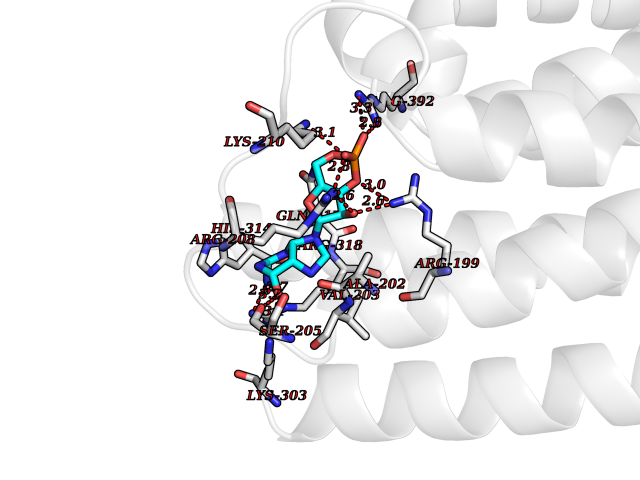 Pymol Map 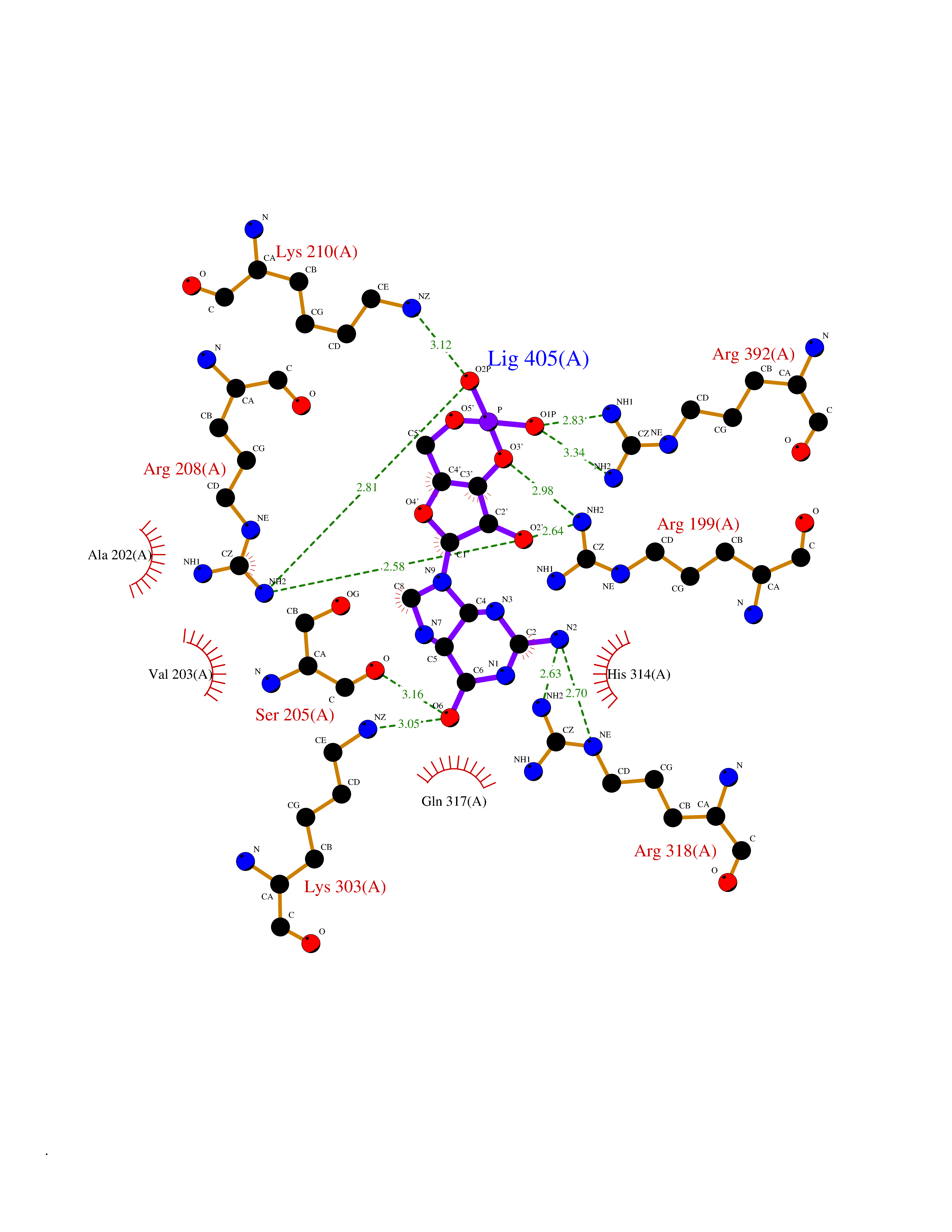 Ligplot Map 3D Binding mode Sequence ISPPPCQGPIEIKETFKYINTVVSCLVFVLGIIGNSTLLYIIYKNKCMRNGPNILIASLALGDLLHIVIAIPINVYKLLAEDWPFGAEMCKLVPFIQKASVGITVLSLCALSIDRYRAVASWSRIKGIGVPKWTAVEIVLIWVVSVVLAVPEAIGFDIITMDYKGSYLRICLLHPVQKTAFMQFYATAKDWWLFSFYFCLPLAITAFFYTLMTCEMLRKLNDHLKQRREVAKTVFCLVLVFALCWLPLHLARILKLTLYNQNDPNRCELLSFLLVLDYIGINMASLNSCANPIALYLVSKRFKNAFKSALCC Hydrogen bonds contact Hydrophobic contact | ||||
| 17 | Retinal rod rhodopsin-sensitive cGMP 3',5'-cyclic phosphodiesterase subunit gamma | 3JWR | 6.93 | |
Target general information Gen name PDE6G Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms PDEG Protein family Rod/cone cGMP-PDE gamma subunit family Biochemical class Hydrolase Function 3',5'-cyclic-GMP phosphodiesterase activity.CGMP binding.Enzyme inhibitor activity.Spectrin binding. Related diseases Retinitis pigmentosa 57 (RP57) [MIM:613582]: A retinal dystrophy belonging to the group of pigmentary retinopathies. Retinitis pigmentosa is characterized by retinal pigment deposits visible on fundus examination and primary loss of rod photoreceptor cells followed by secondary loss of cone photoreceptors. Patients typically have night vision blindness and loss of midperipheral visual field. As their condition progresses, they lose their far peripheral visual field and eventually central vision as well. {ECO:0000269|PubMed:20655036}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07954; DB00203; DB00820; DB00862 Interacts with O14503; Q96JM7; A0A6Q8PF08; O43741; Q8R511; P62994; Q9QY17; Q63787 EC number 3.1.4.35 Uniprot keywords 3D-structure; Acetylation; cGMP; Hydrolase; Reference proteome; Retinitis pigmentosa; Sensory transduction; Vision Protein physicochemical properties Chain ID C,D Molecular weight (Da) 40027.7 Length 345 Aromaticity 0.09 Instability index 37.44 Isoelectric point 6.02 Charge (pH=7) -6.88 2D Binding mode Binding energy (Kcal/mol) -7.68 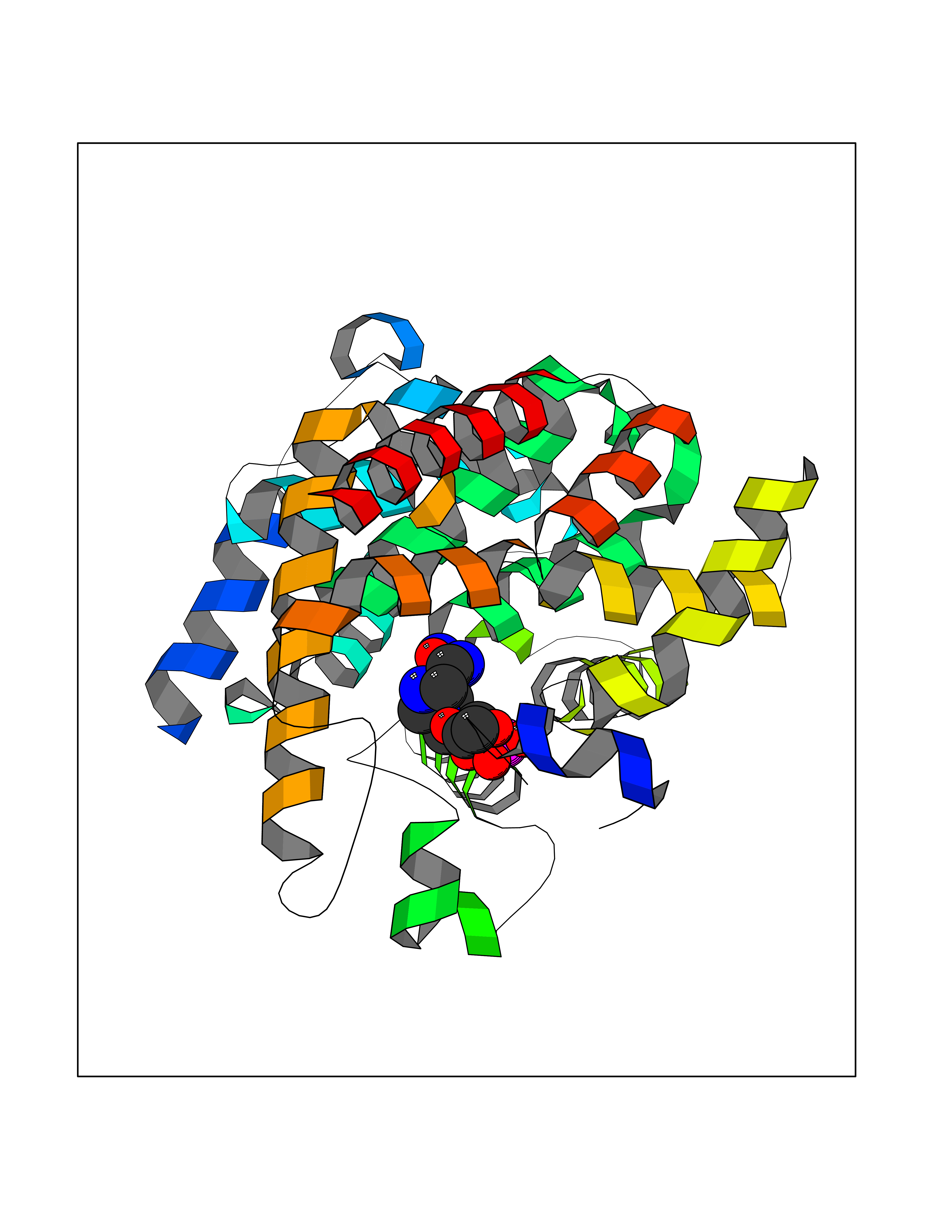 Molscript Map 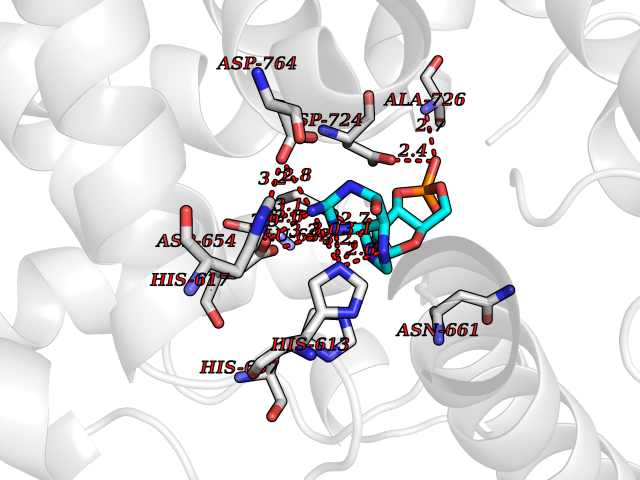 Pymol Map 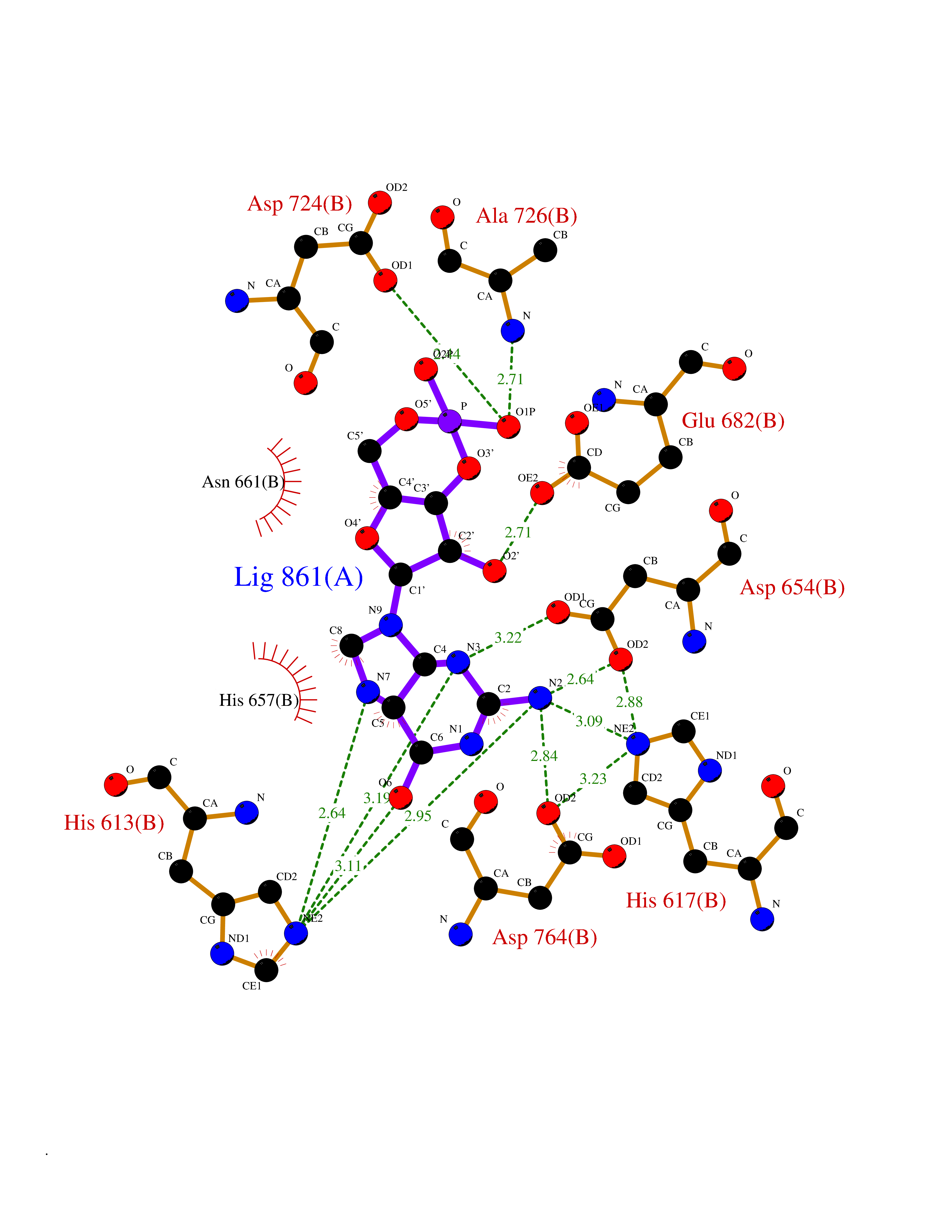 Ligplot Map 3D Binding mode Sequence EAFNHLELHELAQYGIISHMEETRELQSLAAAVVPSAQTLKITDFSFSDFELSDLETALCTIRMFTDLNLVQNFQMKHEVLCRWILSVKKNYRKNVAYHNWRHAFNTAQCMFAALKAGKIQNKLTDLEILALLIAALSHDLDHRGVNNSYIQRSEHPLAQLYCHSIMEHHHFDQCLMILNSPGNQILSGLSIEEYKTTLKIIKQAILATDLALYIKRRGEFFELIRKNQFNLEDPHQKELFLAMLMTACDLSAITKPWPIQQRIAELVATEFWEQGDLERTVLQQQPIPMMDRNKRDELPKLQVGFIDFVCTQLYEALTHVSEDCFPLLDGCRKNRQKWQALAEQ Hydrogen bonds contact Hydrophobic contact | ||||
| 18 | Intestinal maltase-glucoamylase (MGAM) | 3L4Y | 6.88 | |
Target general information Gen name MGAM Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms MGAM Protein family Glycosyl hydrolase 31 family Biochemical class Glycosylase Function May serve as an alternate pathway for starch digestion when luminal alpha-amylase activity is reduced because of immaturity or malnutrition. May play a unique role in the digestion of malted dietary oligosaccharides used in food manufacturing. Related diseases Hypervalinemia and hyperleucine-isoleucinemia (HVLI) [MIM:618850]: An autosomal recessive metabolic disorder characterized by highly elevated plasma concentrations of valine and leucine/isoleucine. Affected individuals suffer from headache and mild memory impairment. {ECO:0000269|PubMed:25653144}. The disease is caused by variants affecting the gene represented in this entry. A patient with hypervalinemia and hyperleucine-isoleucinemia was identified as compound heterozygote for Gln-170 (inherited from his father) and Lys-264 (inherited from his mother), both variants reduced the catalytic activity of the enzyme. After treatment with vitamin B6, a precursor of pyridoxal 5'-phosphate, a BCAT2 cofactor, the blood levels of branched chain amino acids, especially valine, were decreased and brain lesions were improved. {ECO:0000269|PubMed:25653144}. Drugs (DrugBank ID) DB00284; DB00491; DB04878 Interacts with Q13520; Q7Z7G2; Q96BA8; O15529; P14410; P54219-3; Q9NUH8 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Direct protein sequencing; Disulfide bond; Glycoprotein; Glycosidase; Hydrolase; Membrane; Multifunctional enzyme; Proteomics identification; Reference proteome; Repeat; Signal-anchor; Sulfation; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 97779.4 Length 863 Aromaticity 0.12 Instability index 32.47 Isoelectric point 5.2 Charge (pH=7) -28.27 2D Binding mode Binding energy (Kcal/mol) -7.31 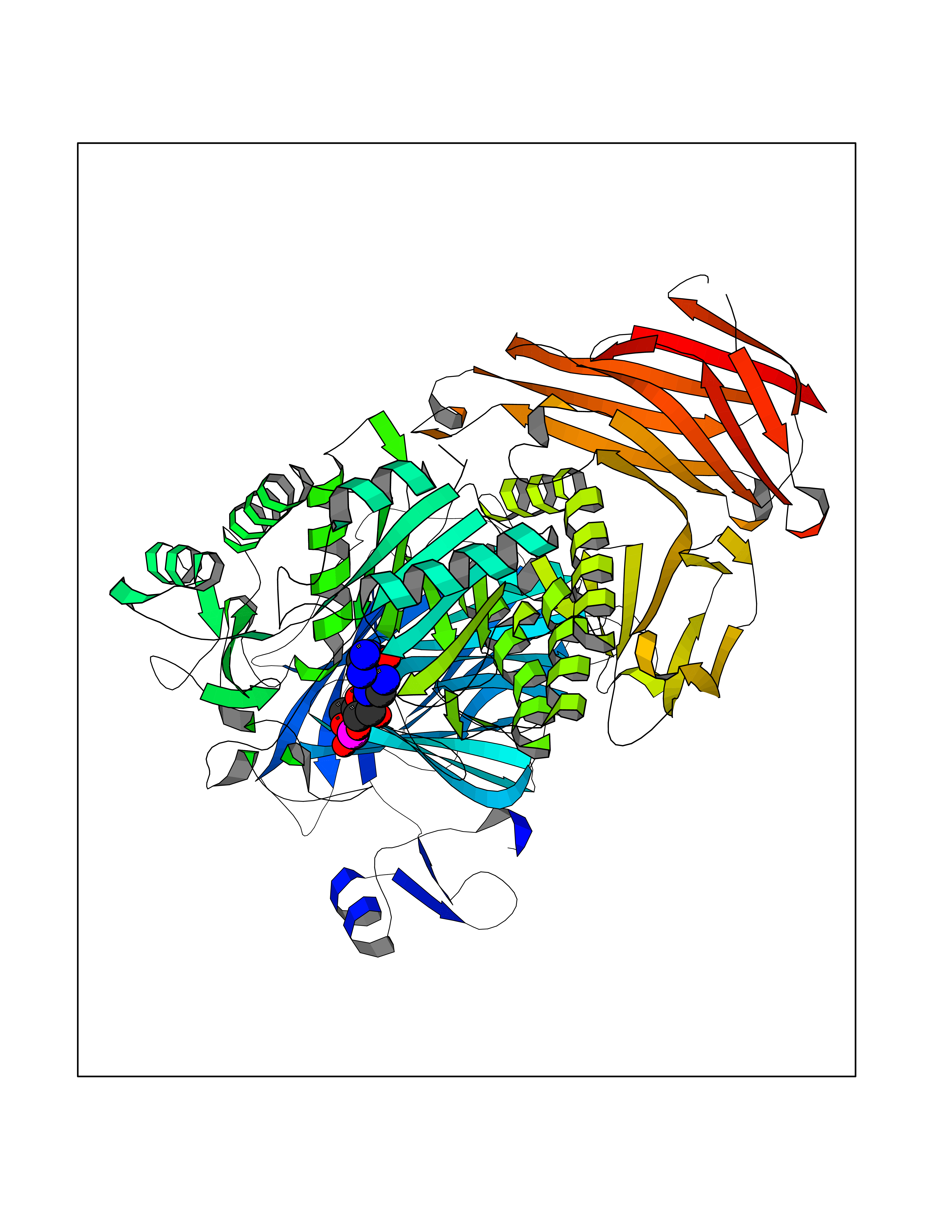 Molscript Map 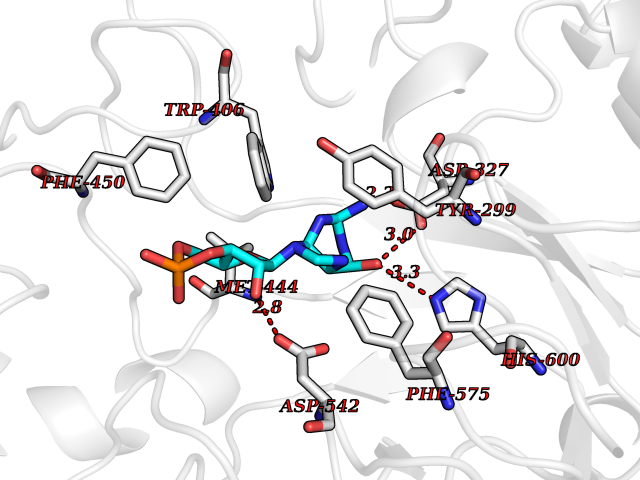 Pymol Map 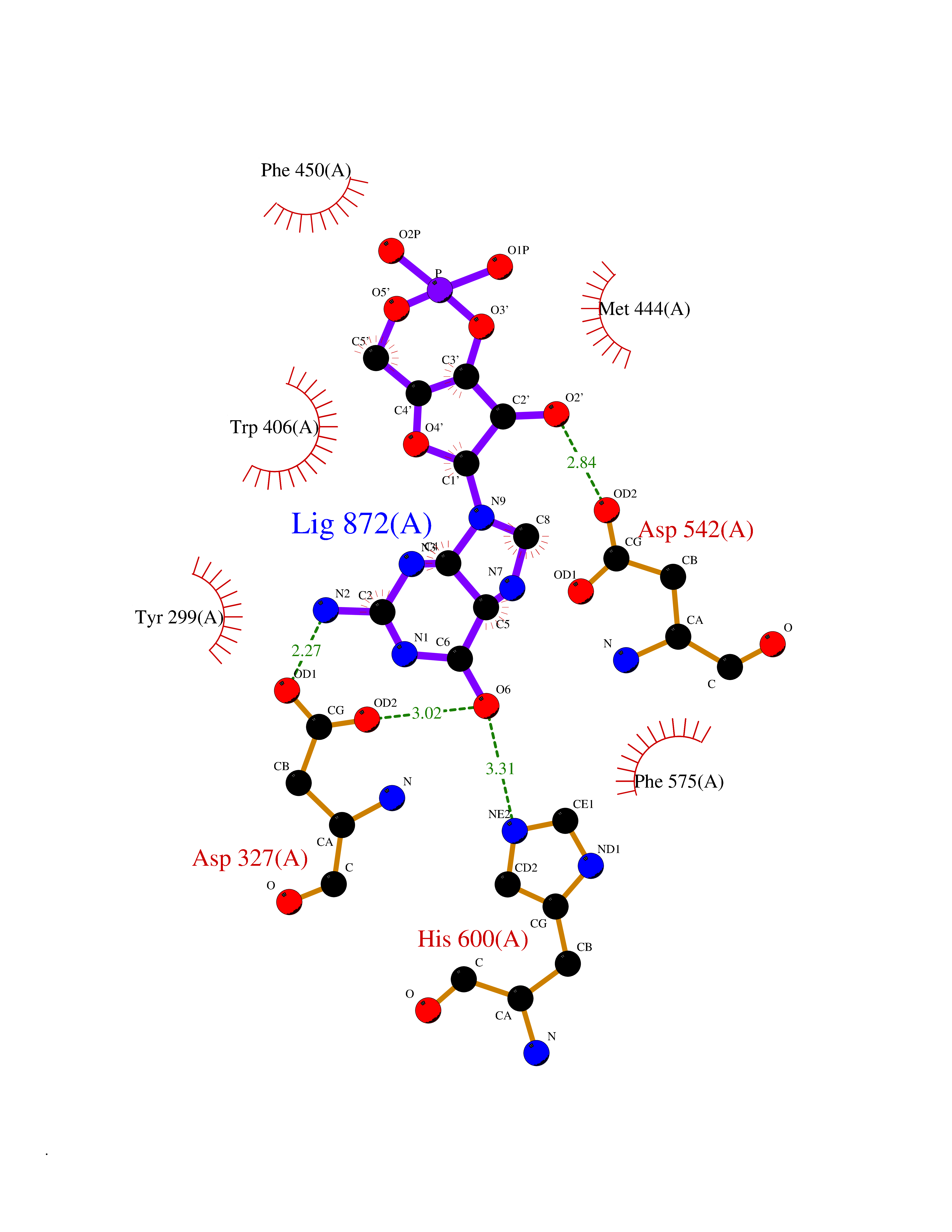 Ligplot Map 3D Binding mode Sequence VNELERINCIPDQPPTKATCDQRGCCWNPQGAVSVPWCYYSKNHSYHVEGNLVNTNAGFTARLKNLPSSPVFGSNVDNVLLTAEYQTSNRFHFKLTDQTNNRFEVPHEHVQSFSGNAAASLTYQVEISRQPFSIKVTRRSNNRVLFDSSIGPLLFADQFLQLSTRLPSTNVYGLGEHVHQQYRHDMNWKTWPIFNRDTTPNGNGTNLYGAQTFFLCLEDASGLSFGVFLMNSNAMEVVLQPAPAITYRTIGGILDFYVFLGNTPEQVVQEYLELIGRPALPSYWALGFHLSRYEYGTLDNMREVVERNRAAQLPYDVQHADIDYMDERRDFTYDSVDFKGFPEFVNELHNNGQKLVIIVDPAISNNSSSSKPYGPYDRGSDMKIWVNSSDGVTPLIGEVWPGQTVFPDYTNPNCAVWWTKEFELFHNQVEFDGIWIDMNEVSNFVDGSVSGCSTNNLNNPPFTPRILDGYLFCKTLCMDAVQHWGKQYDIHNLYGYSMAVATAEAAKTVFPNKRSFILTRSTFAGSGKFAAHWLGDNTATWDDLRWSIPGVLEFNLFGIPMVGPDICGFALDTPEELCRRWMQLGAFYPFSRNHNGQGYKDQDPASFGADSLLLNSSRHYLNIRYTLLPYLYTLFFRAHSRGDTVARPLLHEFYEDNSTWDVHQQFLWGPGLLITPVLDEGAEKVMAYVPDAVWYDYETGSQVRWRKQKVEMELPGDKIGLHLRGGYIFPTQQPNTTTLASRKNPLGLIIALDENKEAKGELFWDDGETKDTVANKVYLLCEFSVTQNRLEVNISQSTYKDPNNLAFNEIKILGTEEPSNVTVKHNGVPSTSPTVTYDSNLKVAIITDIDLLLGEAYTVEWAH Hydrogen bonds contact Hydrophobic contact | ||||
| 19 | Activin receptor type IIA (ACVR2A) | 3Q4T | 6.87 | |
Target general information Gen name ACVR2A Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Activin receptor type2A; Activin receptor type-2A; ACVR2; ACTRIIA; ACTR-IIA Protein family Protein kinase superfamily, TKL Ser/Thr protein kinase family, TGFB receptor subfamily Biochemical class Kinase Function Type II receptors phosphorylate and activate type I receptors which autophosphorylate, then bind and activate SMAD transcriptional regulators. Receptor for activin A, activin B and inhibin A. Mediates induction of adipogenesis by GDF6. On ligand binding, forms a receptor complex consisting of two type II and two type I transmembrane serine/threonine kinases. Related diseases Periventricular nodular heterotopia 8 (PVNH8) [MIM:618185]: A form of periventricular nodular heterotopia, a disorder resulting from a defect in the pattern of neuronal migration in which ectopic collections of neurons lie along the lateral ventricles of the brain or just beneath, contiguously or in isolated patches. PVNH8 is an autosomal dominant disease characterized by developmental disabilities, speech delay, seizures and attention deficit-hyperactivity disorder. {ECO:0000269|PubMed:28868155}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB12118 Interacts with NA EC number EC 2.7.11.30 Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Cell membrane; Disulfide bond; Glycoprotein; Kinase; Magnesium; Manganese; Membrane; Metal-binding; Nucleotide-binding; Proteomics identification; Receptor; Reference proteome; Serine/threonine-protein kinase; Signal; Transferase; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 34599.3 Length 305 Aromaticity 0.09 Instability index 44.48 Isoelectric point 5.7 Charge (pH=7) -8.29 2D Binding mode Binding energy (Kcal/mol) -7.34 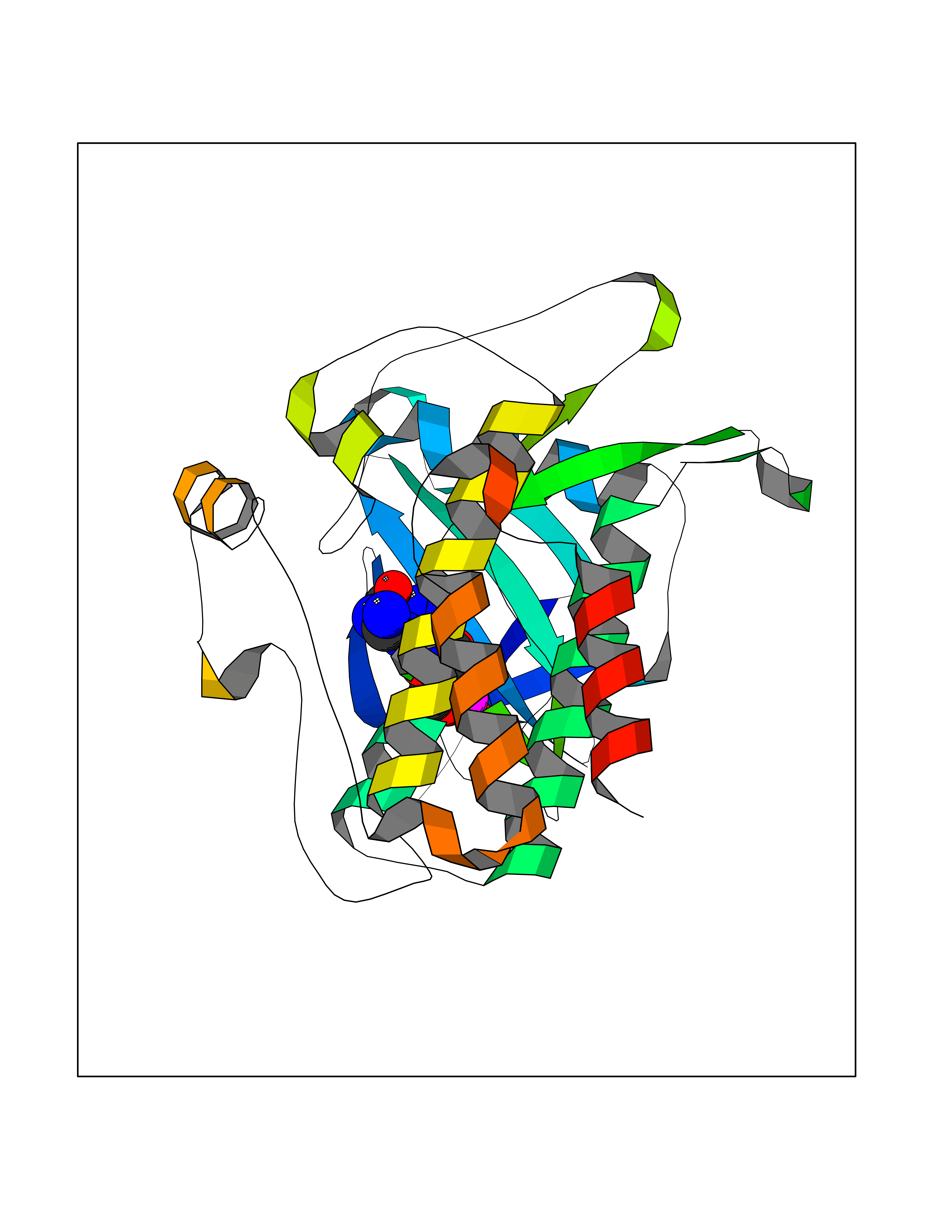 Molscript Map 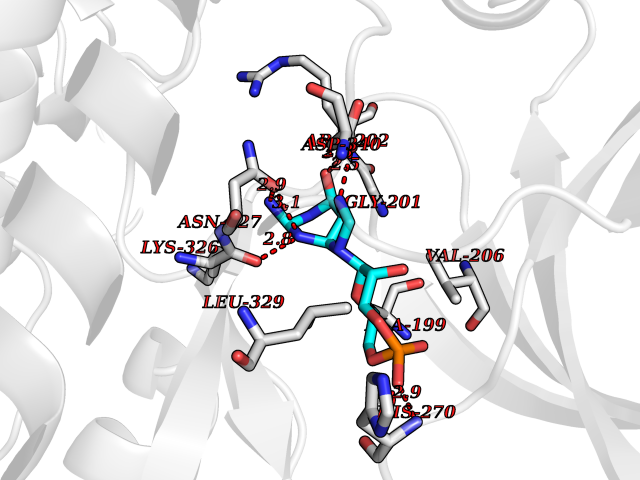 Pymol Map 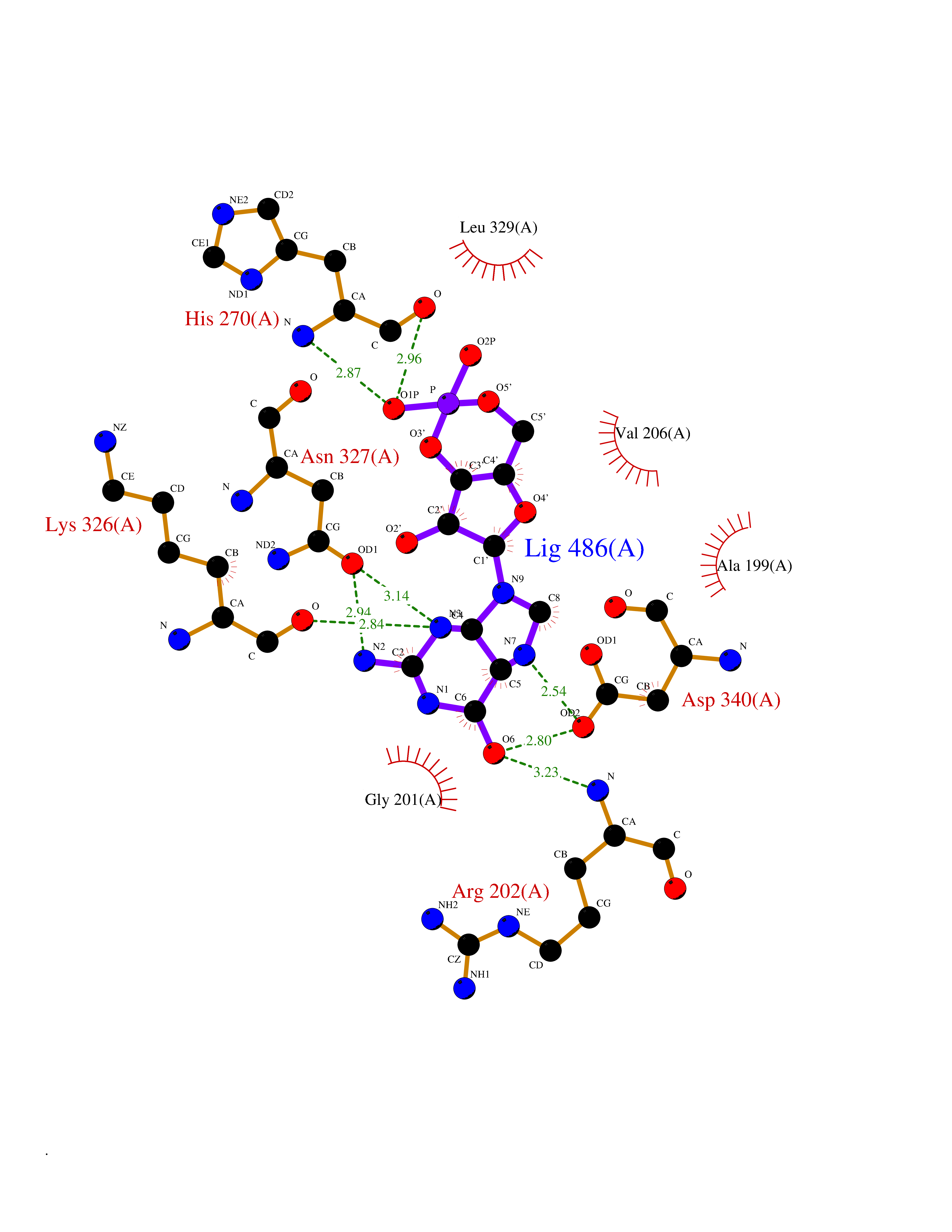 Ligplot Map 3D Binding mode Sequence LGTENLYFQSMPLQLLEVKARGRFGCVWKAQLLNEYVAVKIFPIQDKQSWQNEYEVYSLPGMKHENILQFIGAEKRGTSVDVDLWLITAFHEKGSLSDFLKANVVSWNELCHIAETMARGLAYLHEDIPGLKDGHKPAISHRDIKSKNVLLKNNLTACIADFGLALKFEAGKSAGDTHGQVGTRRYMAPEVLEGAINFQRDAFLRIDMYAMGLVLWELASRCTAADGPVDEYMLPFEEEIGQHPSLEDMQEVVVHKKKRPVLRDYWQKHAGMAMLCETIEECWDHDAEARLSAGCVGERITQMQR Hydrogen bonds contact Hydrophobic contact | ||||
| 20 | Platelet-derived growth factor receptor alpha (PDGFRA) | 5K5X | 6.86 | |
Target general information Gen name PDGFRA Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms RHEPDGFRA; Platelet-derived growth factor receptor 2; Platelet-derived growth factor alpha receptor; PDGFR2; PDGFR-alpha; PDGFR-2; PDGF-R-alpha; CD140a antigen; CD140a; CD140 antigen-like family membe Protein family Protein kinase superfamily, Tyr protein kinase family, CSF-1/PDGF receptor subfamily Biochemical class Kinase Function Depending on the context, promotes or inhibits cell proliferation and cell migration. Plays an important role in the differentiation of bone marrow-derived mesenchymal stem cells. Required for normal skeleton development and cephalic closure during embryonic development. Required for normal development of the mucosa lining the gastrointestinal tract, and for recruitment of mesenchymal cells and normal development of intestinal villi. Plays a role in cell migration and chemotaxis in wound healing. Plays a role in platelet activation, secretion of agonists from platelet granules, and in thrombin-induced platelet aggregation. Binding of its cognate ligands - homodimeric PDGFA, homodimeric PDGFB, heterodimers formed by PDGFA and PDGFB or homodimeric PDGFC -leads to the activation of several signaling cascades; the response depends on the nature of the bound ligand and is modulated by the formation of heterodimers between PDGFRA and PDGFRB. Phosphorylates PIK3R1, PLCG1, and PTPN11. Activation of PLCG1 leads to the production of the cellular signaling molecules diacylglycerol and inositol 1,4,5-trisphosphate, mobilization of cytosolic Ca(2+) and the activation of protein kinase C. Phosphorylates PIK3R1, the regulatory subunit of phosphatidylinositol 3-kinase, and thereby mediates activation of the AKT1 signaling pathway. Mediates activation of HRAS and of the MAP kinases MAPK1/ERK2 and/or MAPK3/ERK1. Promotes activation of STAT family members STAT1, STAT3 and STAT5A and/or STAT5B. Receptor signaling is down-regulated by protein phosphatases that dephosphorylate the receptor and its down-stream effectors, and by rapid internalization of the activated receptor. Tyrosine-protein kinase that acts as a cell-surface receptor for PDGFA, PDGFB and PDGFC and plays an essential role in the regulation of embryonic development, cell proliferation, survival and chemotaxis. Related diseases A chromosomal aberration involving PDGFRA is found in some cases of hypereosinophilic syndrome. Interstitial chromosomal deletion del(4)(q12q12) causes the fusion of FIP1L1 and PDGFRA (FIP1L1-PDGFRA). Mutations that cause overexpression and/or constitutive activation of PDGFRA may be a cause of hypereosinophilic syndrome. {ECO:0000269|PubMed:12808148}.; DISEASE: Gastrointestinal stromal tumor (GIST) [MIM:606764]: Common mesenchymal neoplasms arising in the gastrointestinal tract, most often in the stomach. They are histologically, immunohistochemically, and genetically different from typical leiomyomas, leiomyosarcomas, and schwannomas. Most GISTs are composed of a fairly uniform population of spindle-shaped cells. Some tumors are dominated by epithelioid cells or contain a mixture of spindle and epithelioid morphologies. Primary GISTs in the gastrointestinal tract commonly metastasize in the omentum and mesenteries, often as multiple nodules. However, primary tumors may also occur outside of the gastrointestinal tract, in other intra-abdominal locations, especially in the omentum and mesentery. {ECO:0000269|PubMed:12522257, ECO:0000269|PubMed:15928335}. The gene represented in this entry may be involved in disease pathogenesis. Mutations causing PDGFRA constitutive activation have been found in gastrointestinal stromal tumors lacking KIT mutations (PubMed:12522257). {ECO:0000269|PubMed:12522257}.; DISEASE: GIST-plus syndrome (GISTPS) [MIM:175510]: A disorder characterized by multiple mesenchymal tumors of the gastrointestinal tract, including gastrointestinal stromal tumor, inflammatory fibroid polyps, and fibroid tumors. Additional features are coarse facies and skin, broad hands and feet, and premature tooth loss. GISTPS is an autosomal dominant disease with incomplete penetrance. Gastrointestinal stromal tumor and inflammatory fibroid polyps may also occur in isolation. {ECO:0000269|PubMed:14699510, ECO:0000269|PubMed:17087943, ECO:0000269|PubMed:25975287}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB12742; DB00102; DB12147; DB10772; DB12010; DB00619; DB09078; DB06595; DB09079; DB06043; DB06589; DB08901; DB08896; DB14840; DB01268; DB11800; DB05146 Interacts with P46108; P46109; P00533; Q8N6L0; P04085; P01127; Q9NRA1; P31947; P62258; Q9NRA1-1; A8T7D5; P05067; Q8IY26 EC number EC 2.7.10.1 Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Cell membrane; Cell projection; Chemotaxis; Developmental protein; Disease variant; Disulfide bond; Glycoprotein; Golgi apparatus; Host-virus interaction; Immunoglobulin domain; Kinase; Membrane; Nucleotide-binding; Phosphoprotein; Proteomics identification; Proto-oncogene; Receptor; Reference proteome; Repeat; Signal; Transferase; Transmembrane; Transmembrane helix; Tyrosine-protein kinase; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 39294.8 Length 345 Aromaticity 0.11 Instability index 46.74 Isoelectric point 6.6 Charge (pH=7) -1.38 2D Binding mode Binding energy (Kcal/mol) -6.84 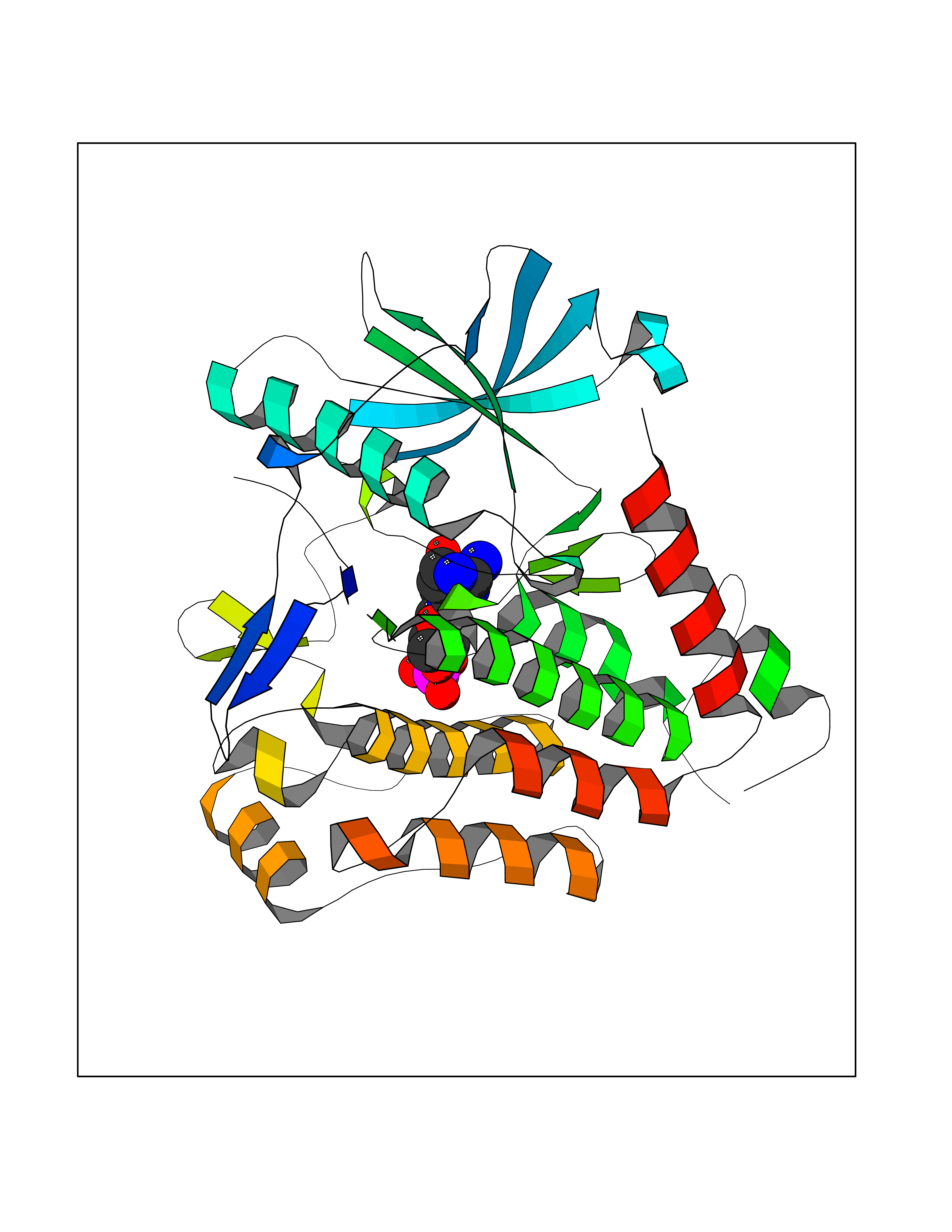 Molscript Map 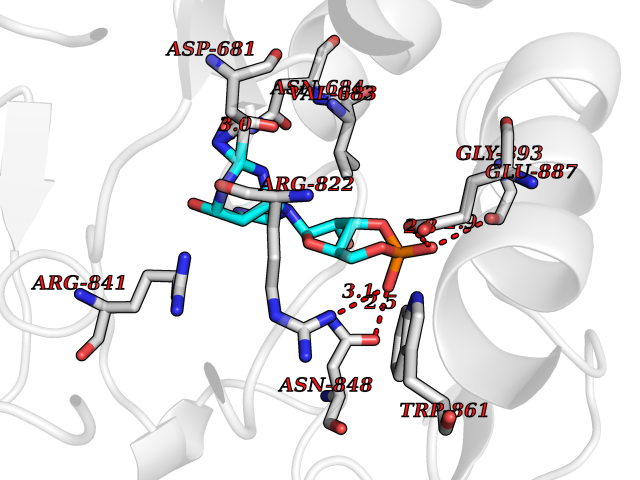 Pymol Map 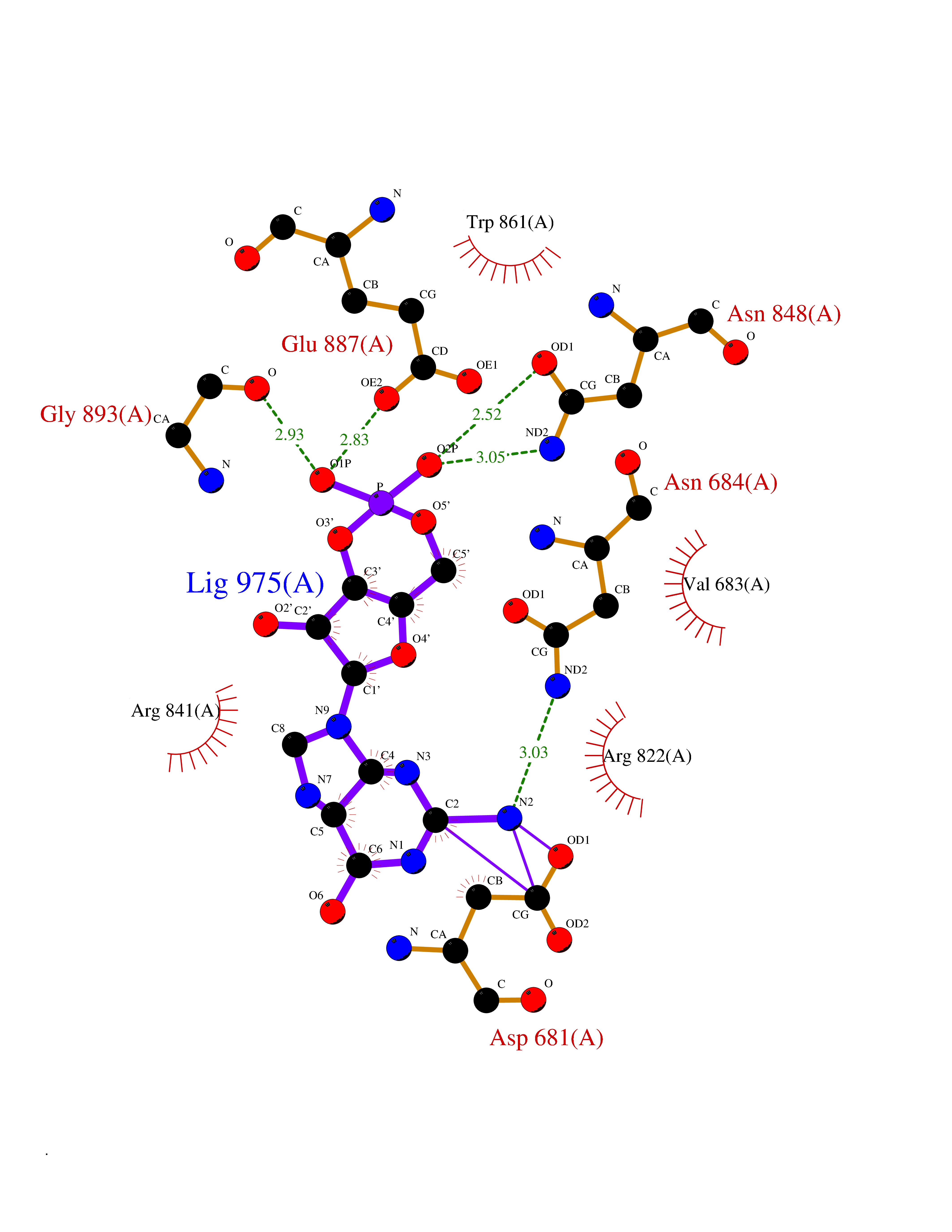 Ligplot Map 3D Binding mode Sequence RYEIRWRVIESISPDGHEYIYVDPMQLPYDSRWEFPRDGLVLGRVLGSGAFGKVVEGTAYGLSRSQPVMKVAVKMLKPTARSSEKQALMSELKIMTHLGPHLNIVNLLGACTKSGPIYIITEYCFYGDLVNYLHKNRDSFLSHSMLDSEVKNLLSDDNSEGLTLLDLLSFTYQVARGMEFLASKNCVHRDLAARNVLLAQGKIVKICDFGLARDIMHDSNYVSKGSTFLPVKWMAPESIFDNLYTTLSDVWSYGILLWEIFSLGGTPYPGMMVDSTFYNKIKSGYRMAKPDHATSEVYEIMVKCWNSEPEKRPSFYHLSEIVENLLPGQYKKSYEKIHLDFLKSD Hydrogen bonds contact Hydrophobic contact | ||||