Ligand
Structure
Job ID
b13b9e525e5aff18220368891dd7239c
Job name
Taylor_quest26
Time
2024-11-21 01:26:53
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 1 | Opioid receptor delta (OPRD1) | 4N6H | 6.81 | |
Target general information Gen name OPRD1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms OPRD; Delta-type opioid receptor; Delta opioid receptor; DOR-1; D-OR-1 Protein family G-protein coupled receptor 1 family Biochemical class GPCR rhodopsin Function Ligand binding causes a conformation change that triggers signaling via guanine nucleotide-binding proteins (G proteins) and modulates the activity of down-stream effectors, such as adenylate cyclase. Signaling leads to the inhibition of adenylate cyclase activity. Inhibits neurotransmitter release by reducing calcium ion currents and increasing potassium ion conductance. Plays a role in the perception of pain and in opiate-mediated analgesia. Plays a role in developing analgesic tolerance to morphine. G-protein coupled receptor that functions as receptor for endogenous enkephalins and for a subset of other opioids. Related diseases Defects in PPARG can lead to type 2 insulin-resistant diabetes and hyptertension. PPARG mutations may be associated with colon cancer. {ECO:0000269|PubMed:10394368}.; DISEASE: Obesity (OBESITY) [MIM:601665]: A condition characterized by an increase of body weight beyond the limitation of skeletal and physical requirements, as the result of excessive accumulation of body fat. {ECO:0000269|PubMed:9753710}. Disease susceptibility may be associated with variants affecting the gene represented in this entry.; DISEASE: Lipodystrophy, familial partial, 3 (FPLD3) [MIM:604367]: A form of lipodystrophy characterized by marked loss of subcutaneous fat from the extremities. Facial adipose tissue may be increased, decreased or normal. Affected individuals show an increased preponderance of insulin resistance, diabetes mellitus and dyslipidemia. {ECO:0000269|PubMed:11788685, ECO:0000269|PubMed:12453919}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Glioma 1 (GLM1) [MIM:137800]: Gliomas are benign or malignant central nervous system neoplasms derived from glial cells. They comprise astrocytomas and glioblastoma multiforme that are derived from astrocytes, oligodendrogliomas derived from oligodendrocytes and ependymomas derived from ependymocytes. {ECO:0000269|PubMed:10851250}. Disease susceptibility may be associated with variants affecting the gene represented in this entry. Polymorphic PPARG alleles have been found to be significantly over-represented among a cohort of American patients with sporadic glioblastoma multiforme suggesting a possible contribution to disease susceptibility. Drugs (DrugBank ID) DB01571; DB01439; DB05050; DB06274; DB06288; DB00321; DB01238; DB00921; DB00611; DB09173; DB09061; DB01535; DB00318; DB00514; DB00647; DB01452; DB01565; DB01444; DB01081; DB01548; DB09272; DB01497; DB00813; DB00956; DB00327; DB01221; DB06738; DB00854; DB00836; DB14146; DB14009; DB12668; DB00333; DB00295; DB06409; DB14011; DB00844; DB11691; DB06230; DB01183; DB00704; DB11130; DB00497; DB01192; DB09209; DB00899; DB12543; DB00708; DB06204; DB00193 Interacts with P16615; P27824; Q4LDR2; Q5JY77; Q9NS64; Q9Y666-2; Q9UKG4; Q0VAQ4; Q96Q45-2; P11607 EC number NA Uniprot keywords 3D-structure; Cell membrane; Disulfide bond; G-protein coupled receptor; Glycoprotein; Lipoprotein; Membrane; Palmitate; Proteomics identification; Receptor; Reference proteome; Transducer; Transmembrane; Transmembrane helix; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 32859.3 Length 294 Aromaticity 0.11 Instability index 33.86 Isoelectric point 9.38 Charge (pH=7) 13.6 2D Binding mode Binding energy (Kcal/mol) -5.65  Molscript Map 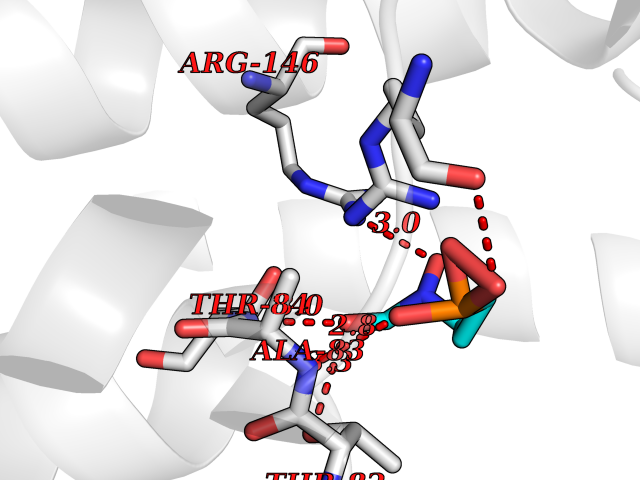 Pymol Map 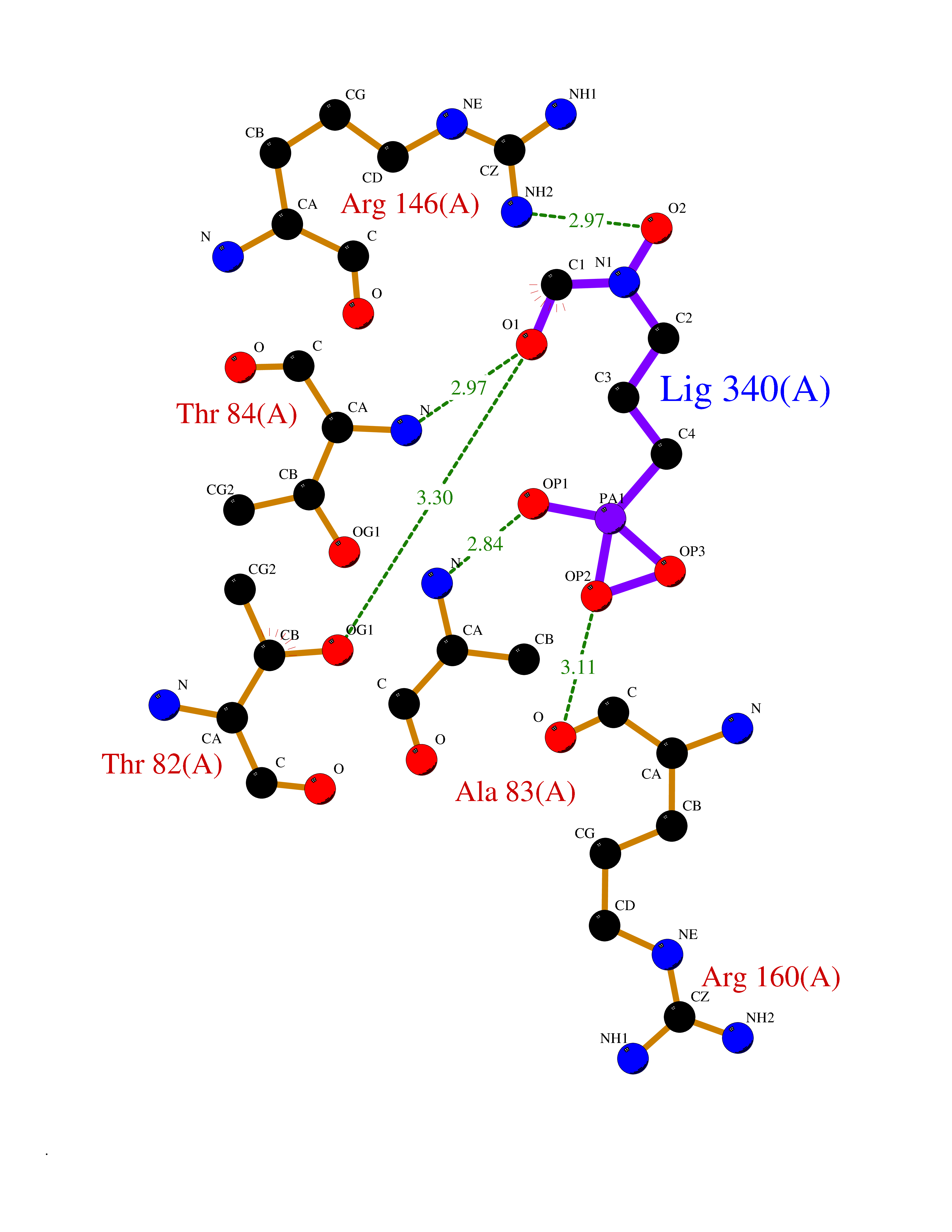 Ligplot Map 3D Binding mode Sequence SLALAIAITALYSAVCAVGLLGNVLVMFGIVRYTKMKTATNIYIFNLALADALATSTLPFQSAKYLMETWPFGELLCKAVLSIDYYNMFTSIFTLTMMSVDRYIAVCHPVKALDFRTPAKAKLINICIWVLASGVGVPIMVMAVTRPRDGAVVCMLQFPSPSWYWDTVTKICVFLFAFVVPILIITVCYGLMLLRLRSVRLLSGSKEKDRSLRRITRMVLVVVGAFVVCWAPIHIFVIVWTLVDIDRRDPLVVAALHLCIALGYANSSLNPVLYAFLDENFKRCFRQLCRKPCG Hydrogen bonds contact Hydrophobic contact | ||||
| 2 | Pyruvate kinase PKLR | 4IP7 | 6.74 | |
Target general information Gen name PKLR Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms PKL;PK1 Protein family Pyruvate kinase family Biochemical class Transferase Function ATP binding.Kinase activity.Magnesium ion binding.Potassium ion binding.Pyruvate kinase activity. Related diseases Pyruvate kinase hyperactivity (PKHYP) [MIM:102900]: Autosomal dominant phenotype characterized by increase of red blood cell ATP. {ECO:0000269|PubMed:9090535}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Pyruvate kinase deficiency of red cells (PKRD) [MIM:266200]: A frequent cause of hereditary non-spherocytic hemolytic anemia. Clinically, pyruvate kinase-deficient patients suffer from a highly variable degree of chronic hemolysis, ranging from severe neonatal jaundice and fatal anemia at birth, severe transfusion-dependent chronic hemolysis, moderate hemolysis with exacerbation during infection, to a fully compensated hemolysis without apparent anemia. {ECO:0000269|PubMed:10087985, ECO:0000269|PubMed:10772876, ECO:0000269|PubMed:11328279, ECO:0000269|PubMed:11960989, ECO:0000269|PubMed:1536957, ECO:0000269|PubMed:1896471, ECO:0000269|PubMed:19085939, ECO:0000269|PubMed:2018831, ECO:0000269|PubMed:21794208, ECO:0000269|PubMed:7706479, ECO:0000269|PubMed:8161798, ECO:0000269|PubMed:8180378, ECO:0000269|PubMed:8476433, ECO:0000269|PubMed:8481523, ECO:0000269|PubMed:8483951, ECO:0000269|PubMed:8664896, ECO:0000269|PubMed:8807089, ECO:0000269|PubMed:9075576, ECO:0000269|PubMed:9482576, ECO:0000269|PubMed:9827908, ECO:0000269|PubMed:9886305, ECO:0000269|Ref.24}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02726; DB00787; DB04551; DB16236; DB00119 Interacts with Q9UBL6-2 EC number 2.7.1.40 Uniprot keywords 3D-structure; Allosteric enzyme; Alternative splicing; ATP-binding; Disease variant; Glycolysis; Hereditary hemolytic anemia; Kinase; Magnesium; Manganese; Metal-binding; Nucleotide-binding; Phosphoprotein; Potassium; Proteomics identification; Pyruvate; Reference proteome; Transferase Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 45695.1 Length 421 Aromaticity 0.06 Instability index 34.44 Isoelectric point 6.88 Charge (pH=7) -0.35 2D Binding mode Binding energy (Kcal/mol) -6.73 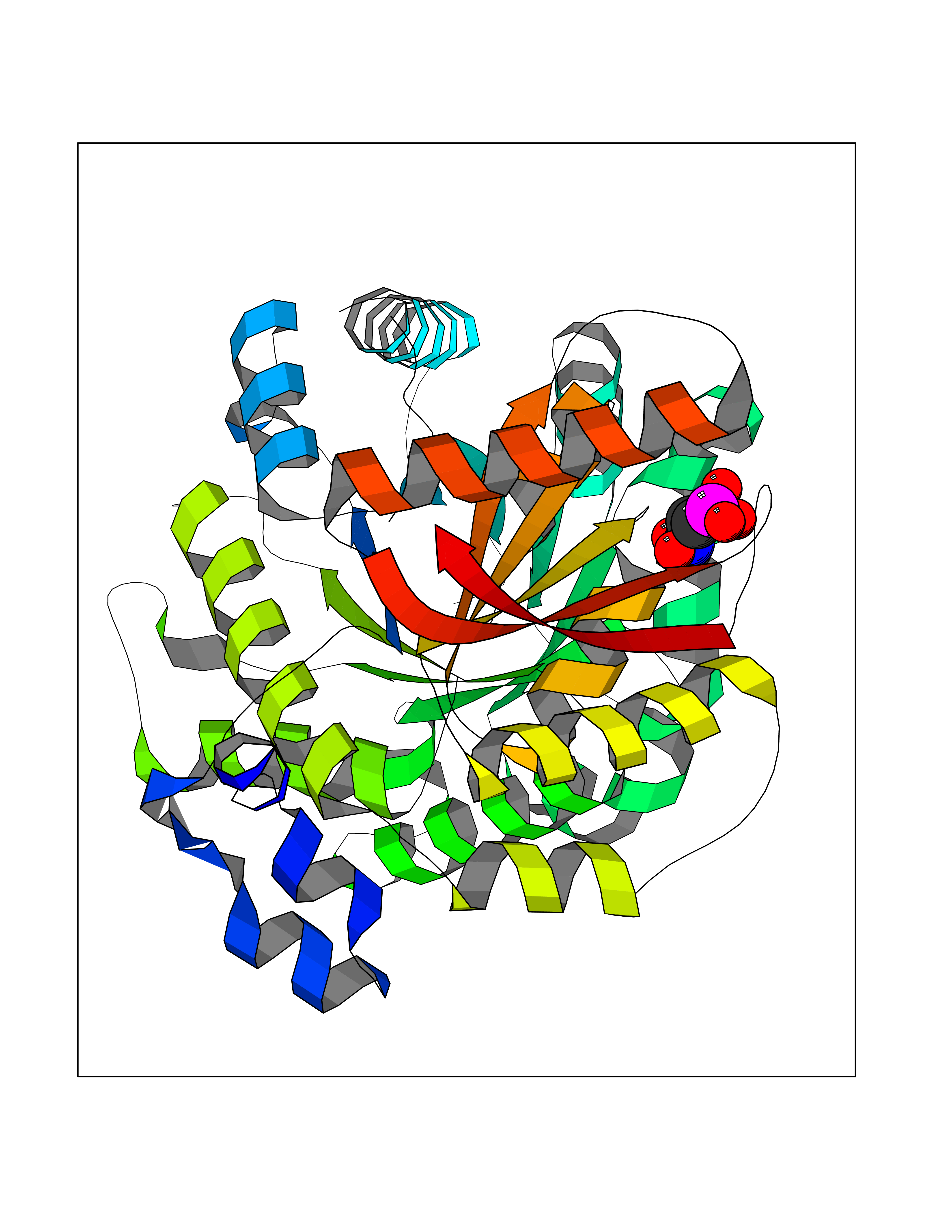 Molscript Map 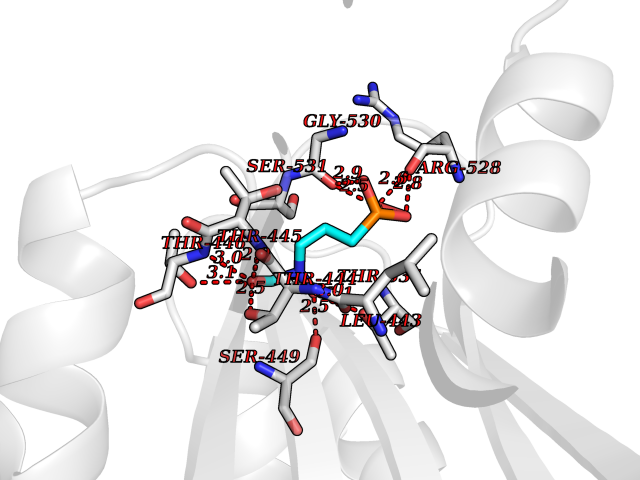 Pymol Map 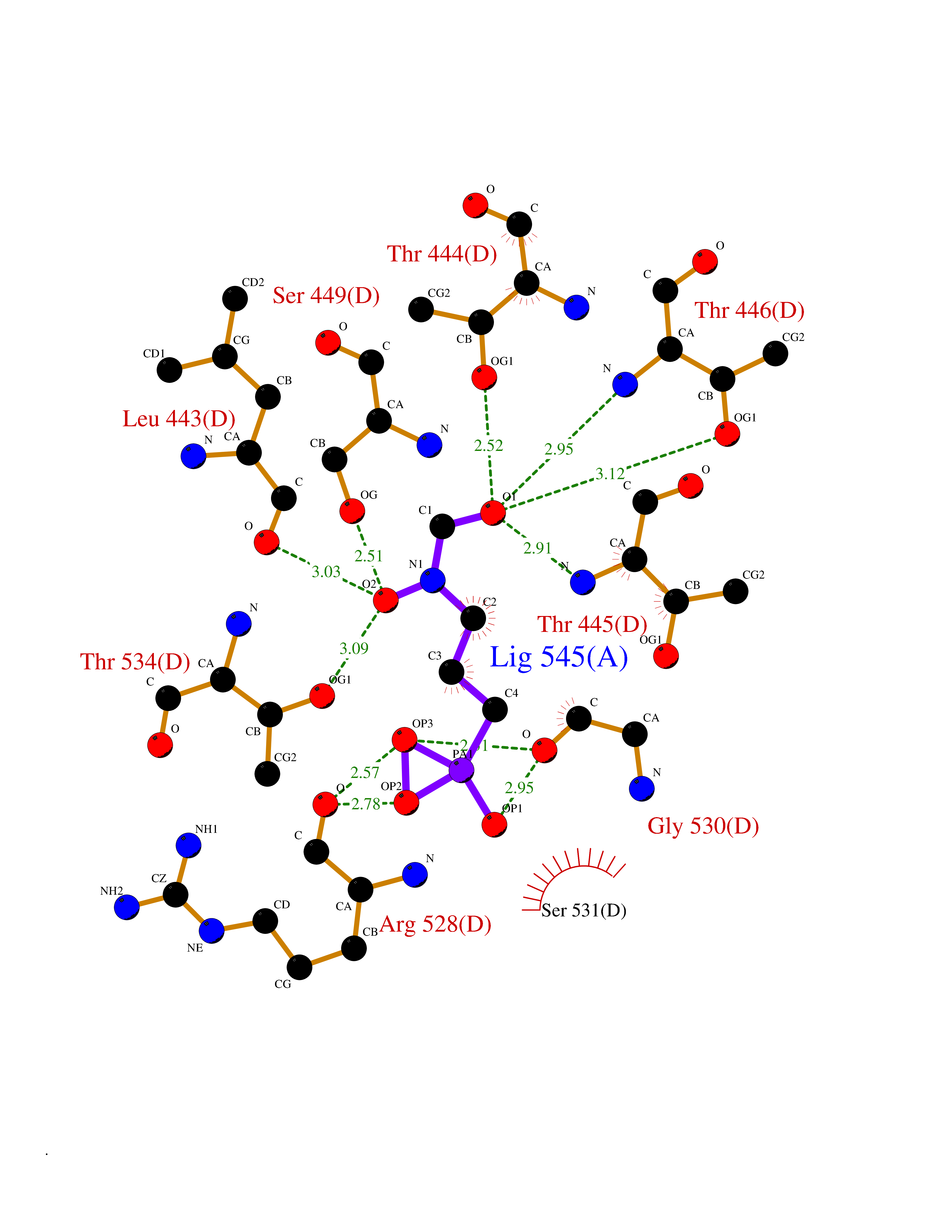 Ligplot Map 3D Binding mode Sequence GTAFFQQQQLPAAMADTFLEHLCLLDIDSEPVAARSTSIIATIGPASRSVERLKEMIKAGMNIARLNFSHGSHEYHAESIANVREAVESFSPLSYRPVAIALDTKGPEIGLSEQDVRDLRFGVEHGVDIVFASFVRKASDVAAVRAALGPEGHGIKIISKIENHEGVKRFDEILEVSDGIMVARGDLGIEIPAEKVFLAQKMMIGRCNLAGKPVVCATQMLESMITKPRPTRAETSDVANAVLDGADCIMLSGETAKGNFPVEAVKMQHAIAREAEAAVYHRQLFEELRRAAPLSRDPTEVTAIGAVEAAFKCCAAAIIVLTTTGRSAQLLSRYRPRAAVIAVTRSAQAARQVHLCRGVFPLLYREPPEAIWADDVDRRVQFGIESGKLRGFLRVGDLVIVVTGWRPGSGYTNIMRVLSIS Hydrogen bonds contact Hydrophobic contact | ||||
| 3 | Cathepsin K (CTSK) | 6QM0 | 6.73 | |
Target general information Gen name CTSK Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Cathepsin X; Cathepsin O2; Cathepsin O; CTSO2; CTSO Protein family Peptidase C1 family Biochemical class Peptidase Function Displays potent endoprotease activity against fibrinogen at acid pH. May play an important role in extracellular matrix degradation. Closely involved in osteoclastic bone resorption and may participate partially in the disorder of bone remodeling. Related diseases Pycnodysostosis (PKND) [MIM:265800]: A rare autosomal recessive bone disorder characterized by deformity of the skull, maxilla and phalanges, osteosclerosis, and fragility of bone. {ECO:0000269|PubMed:10491211, ECO:0000269|PubMed:10878663, ECO:0000269|PubMed:22822386, ECO:0000269|PubMed:25731711, ECO:0000269|PubMed:8703060, ECO:0000269|PubMed:9529353}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08287; DB04244; DB07592; DB07593; DB07563; DB02869; DB07965; DB07967; DB01858; DB12239; DB02679; DB03891; DB05736; DB15599; DB08270; DB03642; DB04234; DB03405; DB03456; DB06670; DB06367; DB08594; DB04523 Interacts with NA EC number EC 3.4.22.38 Uniprot keywords 3D-structure; Cell membrane; Disease variant; Disulfide bond; Glycoprotein; Hydrolase; Lysosome; Membrane; Protease; Proteomics identification; Reference proteome; Secreted; Signal; Thiol protease; Zymogen Protein physicochemical properties Chain ID A Molecular weight (Da) 23651.4 Length 216 Aromaticity 0.1 Instability index 31.38 Isoelectric point 9.01 Charge (pH=7) 7.86 2D Binding mode Binding energy (Kcal/mol) -5.39 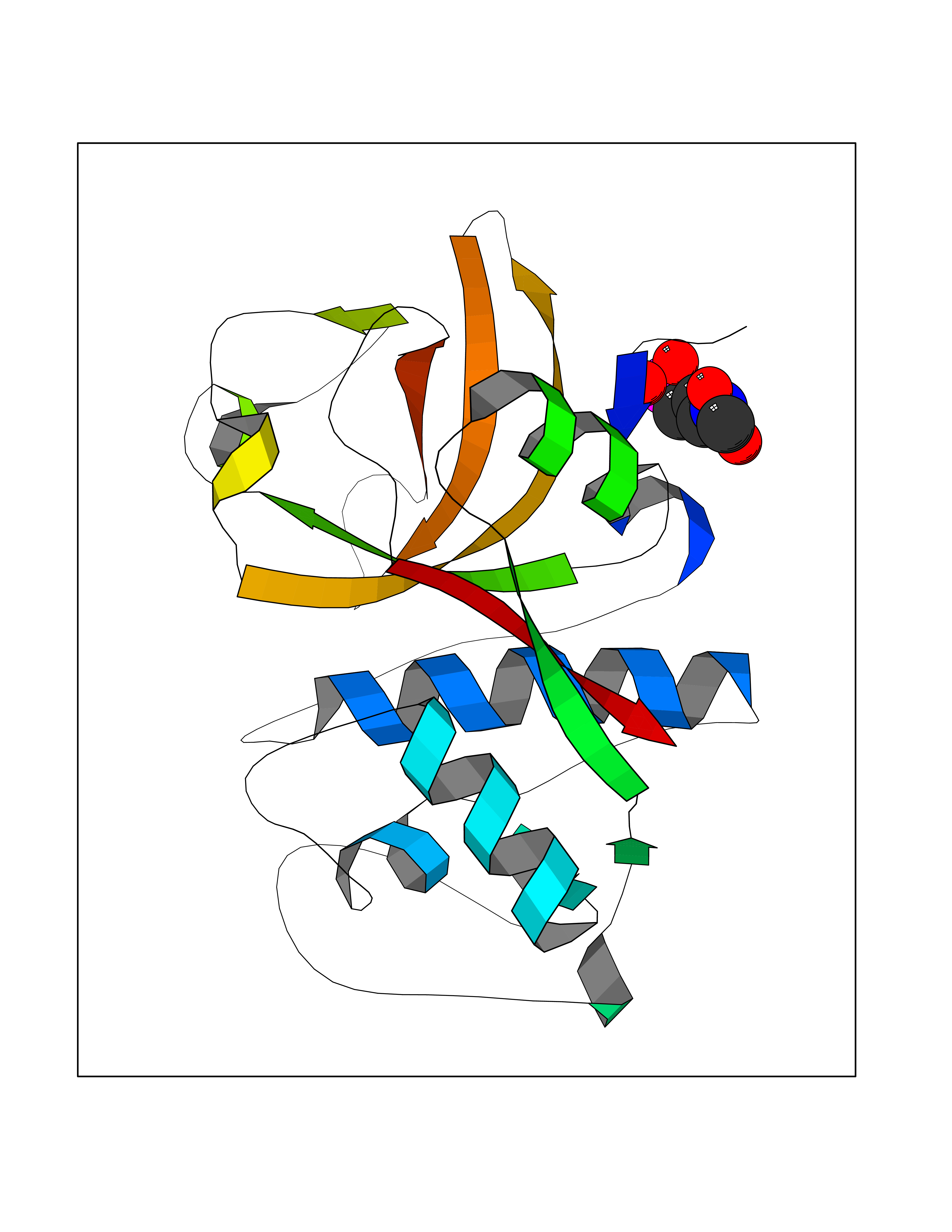 Molscript Map 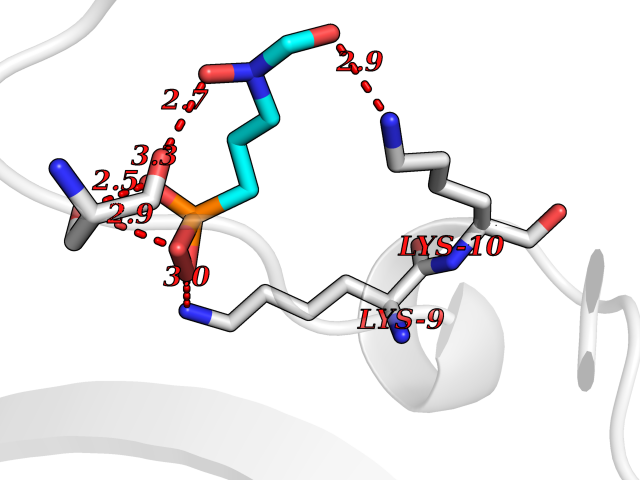 Pymol Map  Ligplot Map 3D Binding mode Sequence RAPDSVDYRKKGYVTPVKNQGQCGSCWAFSSVGALEGQLKKKTGKLLNLSPQNLVDCVSENDGCGGGYMTNAFQYVQKNRGIDSEDAYPYVGQEESCMYNPTGKAAKCRGYREIPEGNEKALKRAVARVGPVSVAIDASLTSFQFYSKGVYYDESCNSDNLNHAVLAVGYGIQKGNKHWIIKNSWGENWGNKGYILMARNKNNACGIANLASFPKM Hydrogen bonds contact Hydrophobic contact | ||||
| 4 | Endonuclease 8-like 1 | 5ITQ | 6.64 | |
Target general information Gen name NEIL1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family FPG family Biochemical class Dna binding protein / dna Function Class I DNA-(apurinic or apyrimidinic site) lyase activity.Class III/IV DNA-(apurinic or apyrimidinic site) lyase activity.Damaged DNA binding.DNA-(apurinic or apyrimidinic site) lyase activity.DNA N-glycosylase activity.Hydrolase activity, acting on glycosyl bonds.Protein C-terminus binding.Zinc ion binding. Related diseases Glutaric aciduria 1 (GA1) [MIM:231670]: An autosomal recessive metabolic disorder characterized by progressive dystonia and athetosis due to gliosis and neuronal loss in the basal ganglia. {ECO:0000269|PubMed:14707522, ECO:0000269|PubMed:18775954, ECO:0000269|PubMed:24973495, ECO:0000269|PubMed:8541831, ECO:0000269|PubMed:8900227, ECO:0000269|PubMed:8900228, ECO:0000269|PubMed:9600243, ECO:0000269|PubMed:9711871}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB09130; DB14490; DB14491; DB14488; DB14501; DB14489; DB01592 Interacts with NA EC number 3.2.2.-; 4.2.99.18 Uniprot keywords 3D-structure; Chromosome; Cytoplasm; Cytoskeleton; DNA damage; DNA repair; DNA-binding; Glycosidase; Hydrolase; Lyase; Multifunctional enzyme; Nucleus; Proteomics identification; Reference proteome; RNA editing Protein physicochemical properties Chain ID A Molecular weight (Da) 32177.5 Length 286 Aromaticity 0.1 Instability index 57.82 Isoelectric point 8.89 Charge (pH=7) 5.38 2D Binding mode Binding energy (Kcal/mol) -5.43 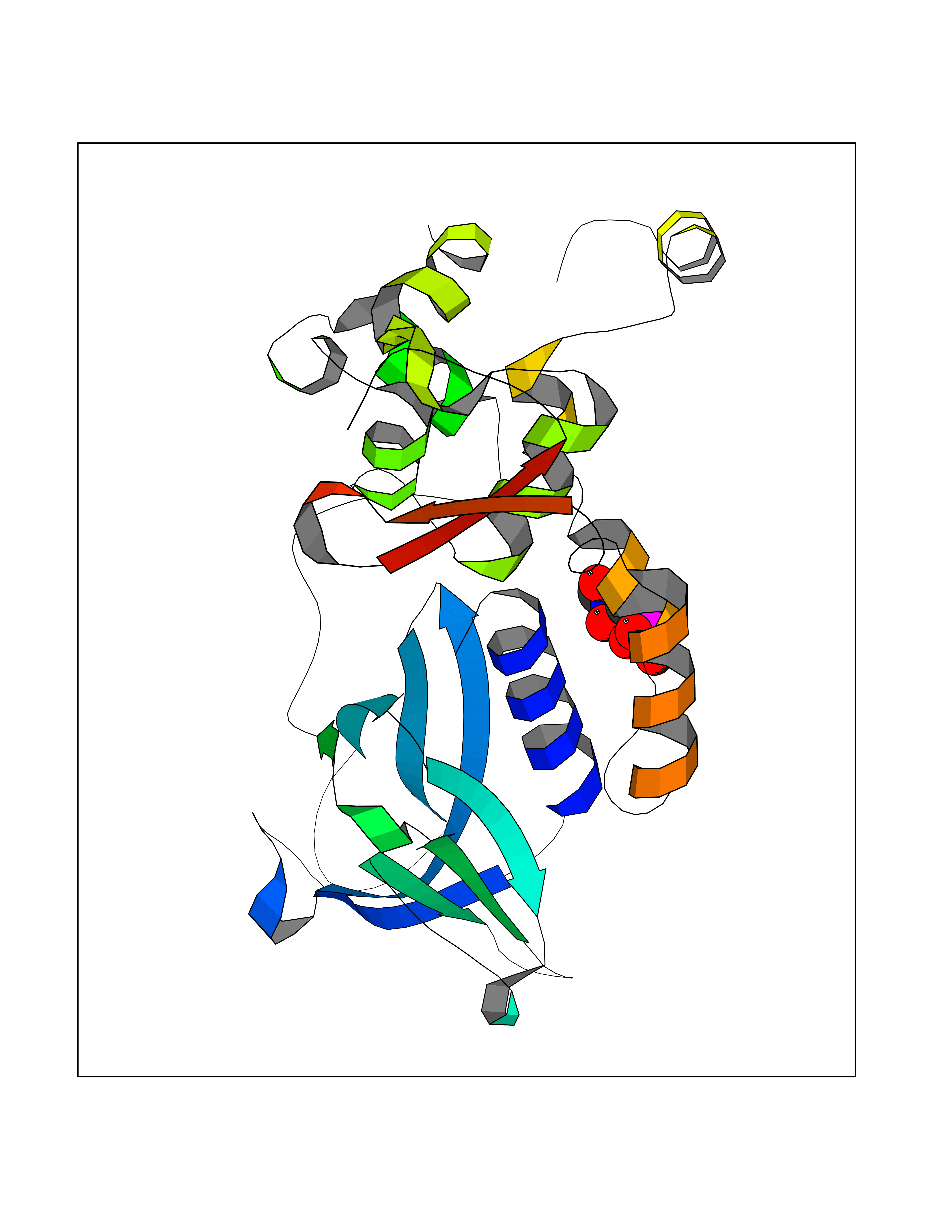 Molscript Map 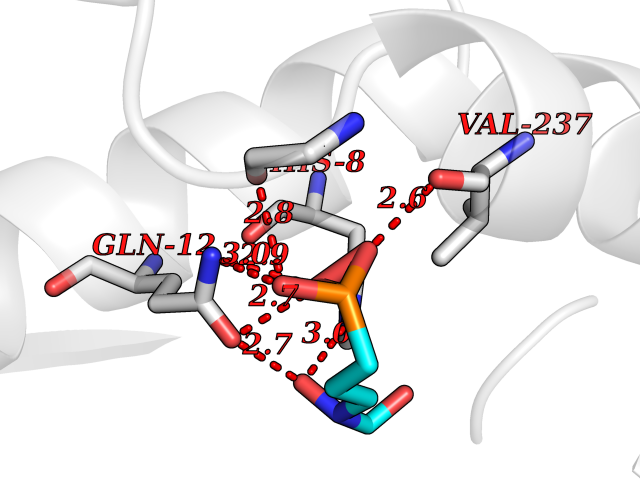 Pymol Map 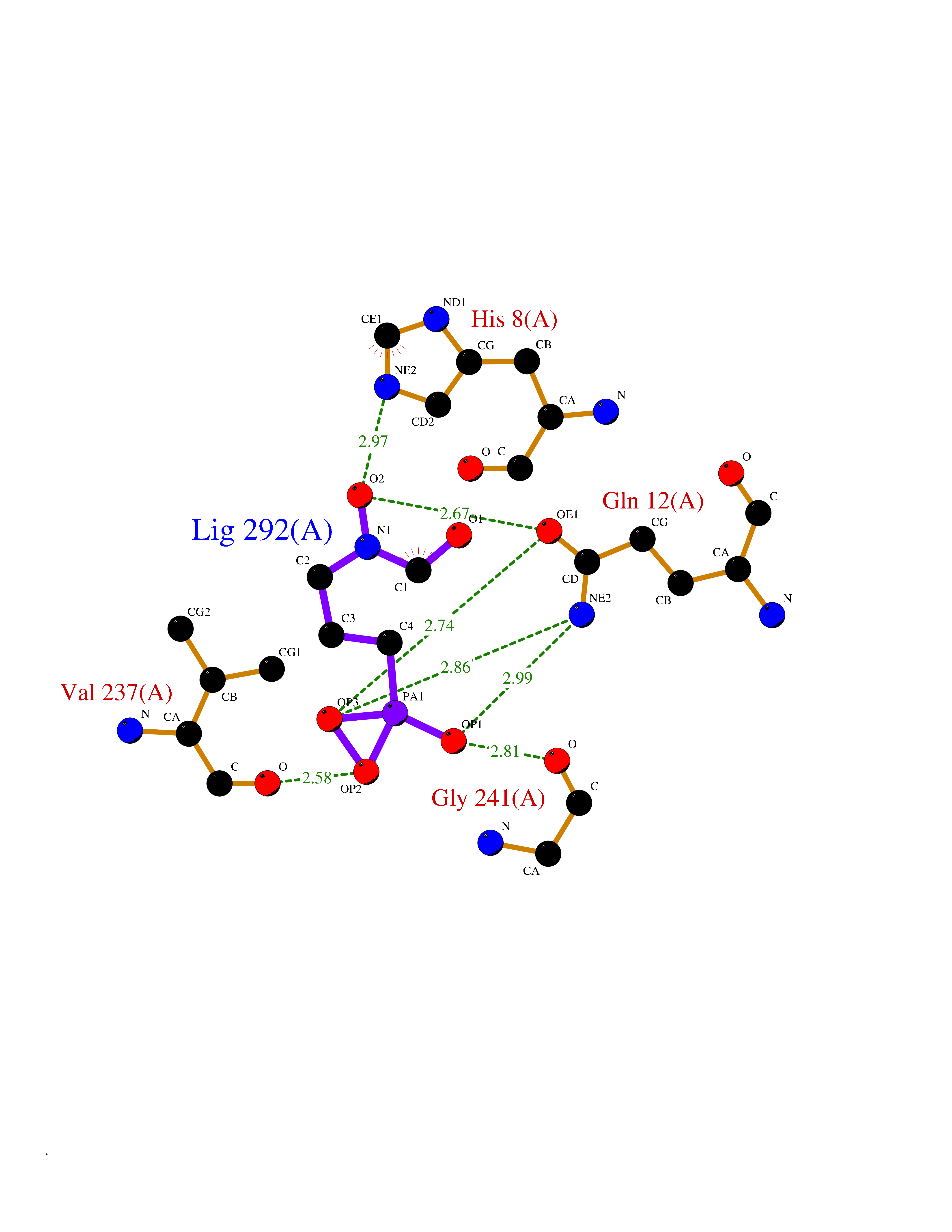 Ligplot Map 3D Binding mode Sequence PEGPELHLASQFVNEACRALVFGGCVEKSSVSRNPEVPFESSAYRISASARGKELRLILSPLPGAQPQQEPLALVFRFGMSGSFQLVPREELPRHAHLRFYTAPPGPRLALCFVDIRRFGRWDLGGKWQPGRGPCVLQEYQQFRESVLRNLADKAFDRPICEALLDQRFFNGIGNYLRAEILYRLKIPPFEKARSVLEALQQSPELTLSQKIRTKLQNPDLLELCHSVPKEVVQLGGRGYGSESGEEDFAAFRAWLRCYGMPGMSSLQDRHGRTIWFQGDPGPLAP Hydrogen bonds contact Hydrophobic contact | ||||
| 5 | Platelet glycoprotein VI (GP6) | 5OU7 | 6.63 | |
Target general information Gen name GP6 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Glycoprotein 6; GPVI Protein family NA Biochemical class NA Function Collagen receptor involved in collagen-induced platelet adhesion and activation. Plays a key role in platelet procoagulant activity and subsequent thrombin and fibrin formation. This procoagulant function may contribute to arterial and venous thrombus formation. The signaling pathway involves the FcR gamma-chain, the Src kinases (likely FYN or LYN) and SYK, the adapter protein LAT and leads to the activation of PLCG2. Related diseases Bleeding disorder, platelet-type, 11 (BDPLT11) [MIM:614201]: A mild to moderate bleeding disorder caused by defective platelet activation and aggregation in response to collagen. {ECO:0000269|PubMed:19549989, ECO:0000269|PubMed:19552682}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P06241; P07948; P06241; P07948 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Blood coagulation; Cell membrane; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; Hemostasis; Immunoglobulin domain; Membrane; Proteomics identification; Receptor; Reference proteome; Repeat; Signal; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 19027.4 Length 173 Aromaticity 0.11 Instability index 37.14 Isoelectric point 8.68 Charge (pH=7) 2.52 2D Binding mode Binding energy (Kcal/mol) -5.89  Molscript Map 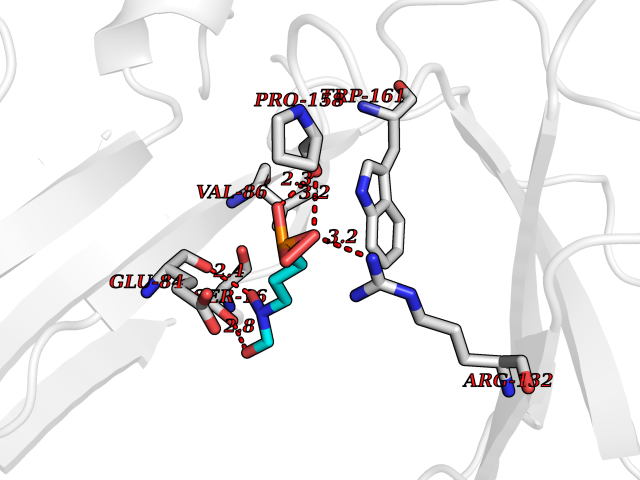 Pymol Map 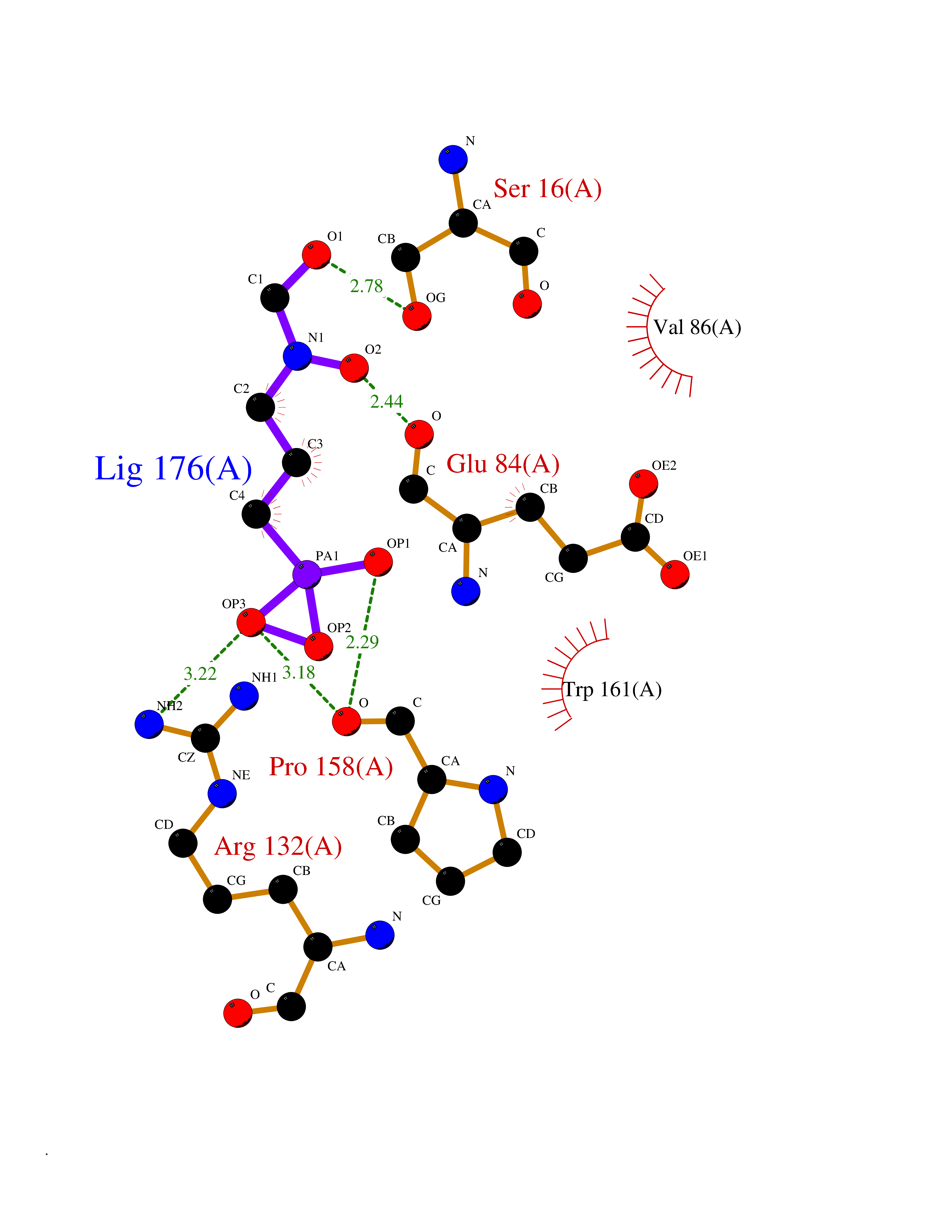 Ligplot Map 3D Binding mode Sequence SGPLPKPSLQALPSSLVPLEKPVTLRCQGPPGVDLYRLEKLSSSRYQDQAVLFIPAMKRSLAGRYRCSYQNGSLWSLPSDQLELVATGVFAKPSLSAQPGSGGDVTLQCQTRYGFDQFALYKEGDPERWYRASFPIITVTAAHSGTYRCYSFSSRDPYLWSAPSDPLELVVTG Hydrogen bonds contact Hydrophobic contact | ||||
| 6 | DNA topoisomerase 4 subunit A | 1ZVT | 6.62 | |
Target general information Gen name parC Organism Escherichia coli (strain K12) Uniprot ID TTD ID NA Synonyms b3019;JW2987 Protein family Type II topoisomerase GyrA/ParC subunit family, ParC type 1 subfamily Biochemical class Isomerase Function ATP binding.DNA binding.DNA topoisomerase type II (ATP-hydrolyzing) activity. Related diseases Pigmentary disorder, reticulate, with systemic manifestations, X-linked (PDR) [MIM:301220]: An X-linked recessive disorder characterized by recurrent infections and sterile inflammation in various organs. Diffuse skin hyperpigmentation with a distinctive reticulate pattern is universally evident by early childhood. This is later followed in many patients by hypohidrosis, corneal inflammation and scarring, enterocolitis that resembles inflammatory bowel disease, and recurrent urethral strictures. Melanin and amyloid deposition is present in the dermis. Affected males also have a characteristic facies with frontally upswept hair and flared eyebrows. Female carriers have only restricted pigmentary changes along Blaschko's lines. {ECO:0000269|PubMed:27019227}. The disease is caused by variants affecting the gene represented in this entry. XLPDR is caused by a recurrent intronic mutation that results in missplicing and reduced POLA1 expression. This leads to a decrease in cytosolic RNA:DNA hybrids and constitutive activation of type I interferon responses, but has no effect on cell replication. {ECO:0000269|PubMed:27019227}.; DISEASE: Van Esch-O'Driscoll syndrome (VEODS) [MIM:301030]: An X-linked recessive syndrome characterized by different degrees of intellectual disability, moderate to severe short stature, microcephaly, hypogonadism, and variable congenital malformations. {ECO:0000269|PubMed:31006512}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB11943; DB12924; DB00817 Interacts with P22523; P0A7K2 EC number 5.6.2.2 Uniprot keywords 3D-structure; Cell membrane; Direct protein sequencing; DNA-binding; Isomerase; Membrane; Reference proteome; Topoisomerase Protein physicochemical properties Chain ID A,B Molecular weight (Da) 26490.3 Length 246 Aromaticity 0.04 Instability index 46.03 Isoelectric point 8.94 Charge (pH=7) 2.83 2D Binding mode Binding energy (Kcal/mol) -6.18  Molscript Map 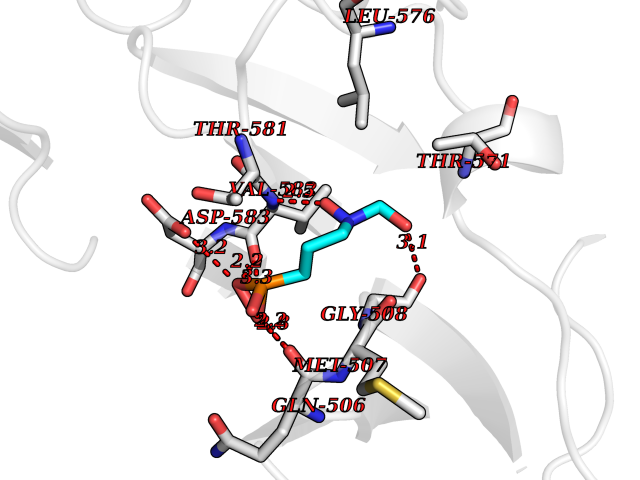 Pymol Map 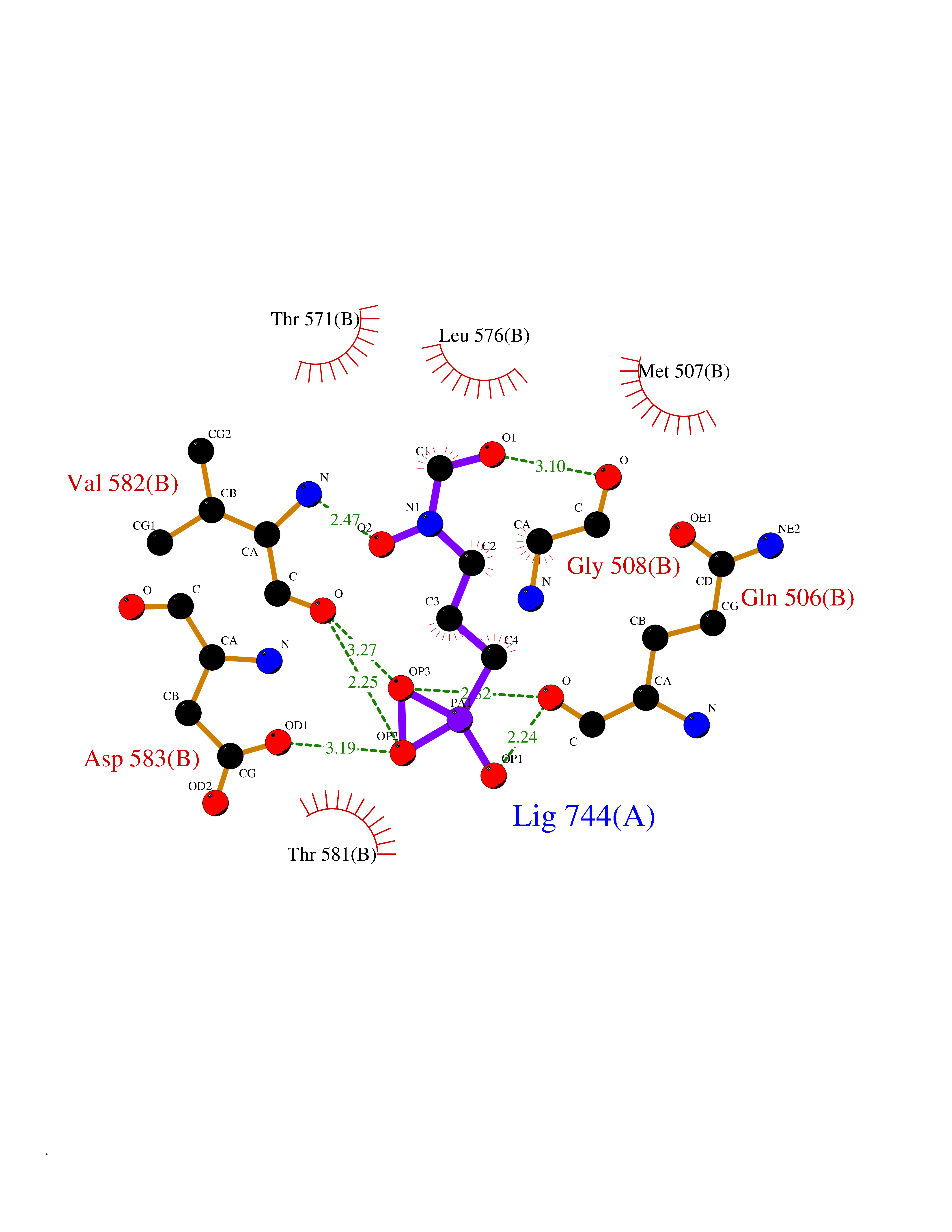 Ligplot Map 3D Binding mode Sequence SEPVTIVLSQMGWVRSAKGHDIDAPGLNYKAGDSFKAAVKGKSNQPVVFVDSTGRSYAIDPITLPSARGQGEPLTGKLTLPPGATVDHMLMESDDQKLLMASDAGYGFVCTFNDLVARNRAGKALITLPENAHVMPPVVIEDASDMLLAITQAGRMLMFPVSDLPQLSKGKGNKIINIPSAEAARGEDGLAQLYVLPPQSTLTIHVGKRKIKLRPEELQKVTGERGRRGTLMRGLQRIDRVEIDSP Hydrogen bonds contact Hydrophobic contact | ||||
| 7 | Endothelin-converting enzyme 1 (ECE1) | 3DWB | 6.60 | |
Target general information Gen name ECE1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms ECE-1 Protein family Peptidase M13 family Biochemical class Peptidase Function Converts big endothelin-1 to endothelin-1. Related diseases Hirschsprung disease, cardiac defects, and autonomic dysfunction (HCAD) [MIM:613870]: A disorder characterized by skip-lesions Hirschsprung disease, craniofacial abnormalities and other dysmorphic features, cardiac defects including ductus arteriosus, small subaortic ventricular septal defect, small atrial septal defect, and autonomic dysfunction. {ECO:0000269|PubMed:9915973}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07171 Interacts with P49760; A8MQ03; Q8IUG1; P60370; P60410 EC number EC 3.4.24.71 Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disease variant; Disulfide bond; Glycoprotein; Hirschsprung disease; Hydrolase; Membrane; Metal-binding; Metalloprotease; Phosphoprotein; Protease; Proteomics identification; Reference proteome; Signal-anchor; Transmembrane; Transmembrane helix; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 75247.9 Length 660 Aromaticity 0.12 Instability index 46.29 Isoelectric point 5.33 Charge (pH=7) -18.3 2D Binding mode Binding energy (Kcal/mol) -6.03 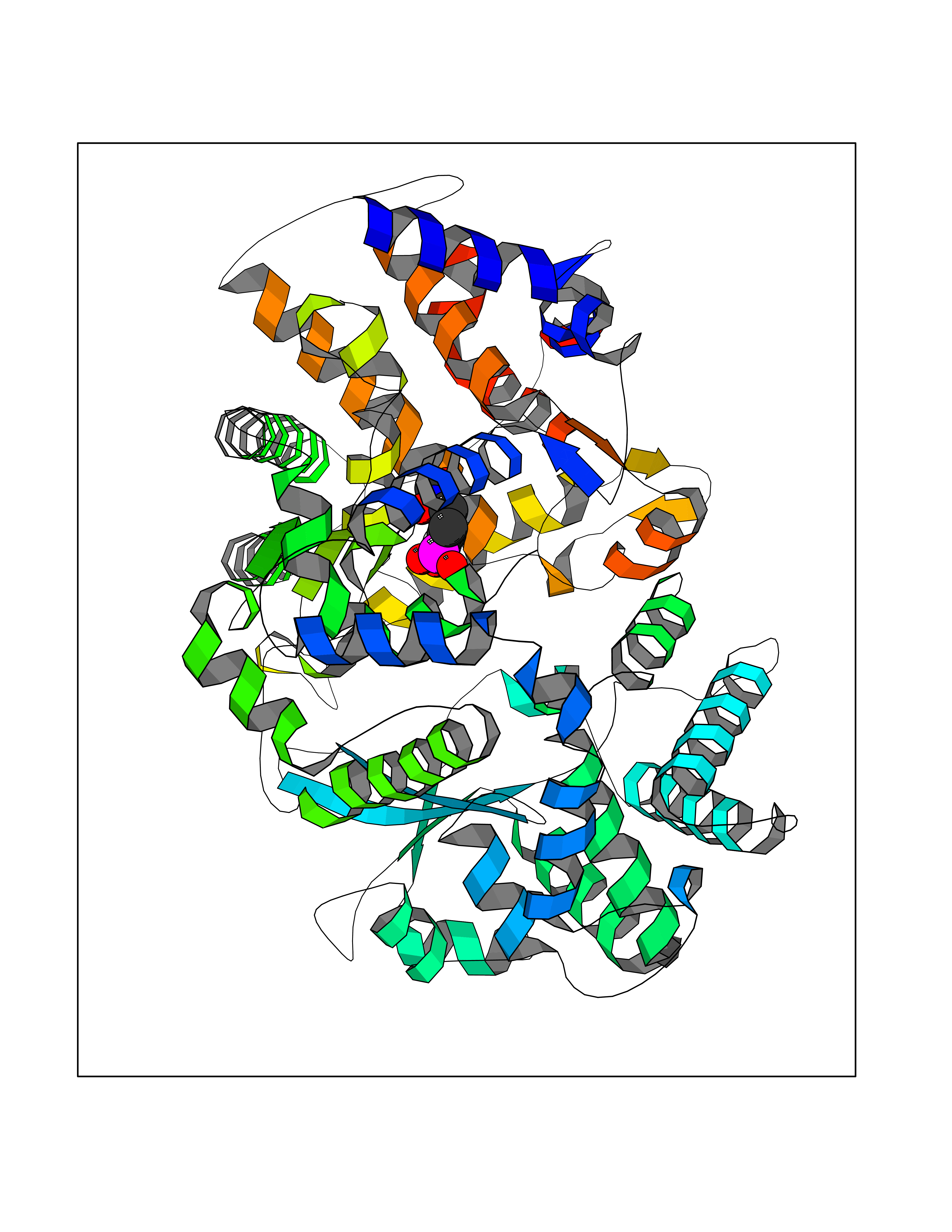 Molscript Map 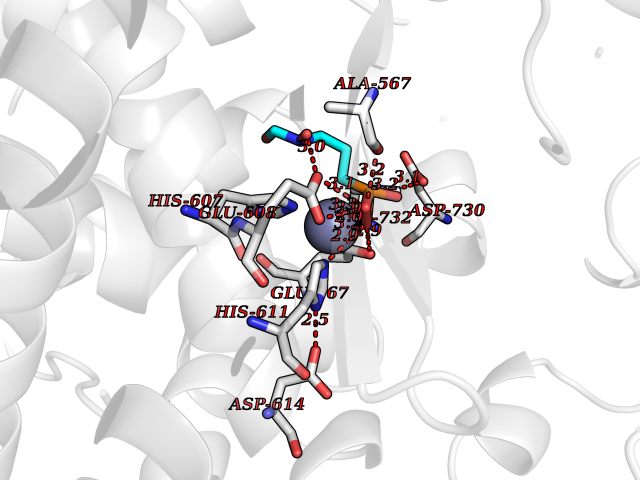 Pymol Map  Ligplot Map 3D Binding mode Sequence SEACVSVTSSILSSMDPTVDPCHDFFSYACGGWIKANPVPDGHSRWGTFSNLWEHNQAIIKHLLENSTASVSEAERKAQVYYRACMNETRIEELRAKPLMELIERLGGWNITGPWAKDNFQDTLQVVTAHYRTSPFFSVYVSADSKNSNSNVIQVDQSGLGLPSRDYYLNKTENEKVLTGYLNYMVQLGKLLGGGDEEAIRPQMQQILDFETALANITIPQEKRRDEELIYHKVTAAELQTLAPAINWLPFLNTIFYPVEINESEPIVVYDKEYLEQISTLINTTDRCLLNNYMIWNLVRKTSSFLDQRFQDADEKFMEVMWKFCVSDTENNLGFALGPMFVKATFAEDSKSIATEIILEIKKAFEESLSTLKWMDEETRKSAKEKADAIYNMIGYPNFIMDPKELDKVFNDYTAVPDLYFENAMRFFNFSWRVTADQLRKAPNRDQWSMTPPMVNAYYSPTKNEIVFPAGILQAPFYTRSSPKALNFGGIGVVVGHELTHAFDDQGREYDKDGNLRPWWKNSSVEAFKRQTECMVEQYSNYSVNGEPVNGRHTLGENIADNGGLKAAYRAYQNWVKKNGAEHSLPTLGLTNNQLFFLGFAQVWCSVRTPESSHEGLITDPHSPSRFRVIGSLSNSKEFSEHFRCPPGSPMNPPHKCEVW Hydrogen bonds contact Hydrophobic contact | ||||
| 8 | Oxygen-insensitive NADPH nitroreductase | 3QDL | 6.60 | |
Target general information Gen name rdxA Organism Helicobacter pylori (strain ATCC 700392 / 26695) (Campylobacter pylori) Uniprot ID TTD ID NA Synonyms HP_0954 Protein family Nitroreductase family Biochemical class Oxidoreductase Function Oxidoreductase activity. Related diseases Hypervalinemia and hyperleucine-isoleucinemia (HVLI) [MIM:618850]: An autosomal recessive metabolic disorder characterized by highly elevated plasma concentrations of valine and leucine/isoleucine. Affected individuals suffer from headache and mild memory impairment. {ECO:0000269|PubMed:25653144}. The disease is caused by variants affecting the gene represented in this entry. A patient with hypervalinemia and hyperleucine-isoleucinemia was identified as compound heterozygote for Gln-170 (inherited from his father) and Lys-264 (inherited from his mother), both variants reduced the catalytic activity of the enzyme. After treatment with vitamin B6, a precursor of pyridoxal 5'-phosphate, a BCAT2 cofactor, the blood levels of branched chain amino acids, especially valine, were decreased and brain lesions were improved. {ECO:0000269|PubMed:25653144}. Drugs (DrugBank ID) DB00916 Interacts with NA EC number 1.-.-.- Uniprot keywords 3D-structure; Antibiotic resistance; NADP; Oxidoreductase; Reference proteome Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 40094.3 Length 352 Aromaticity 0.08 Instability index 55.15 Isoelectric point 6.72 Charge (pH=7) -0.86 2D Binding mode Binding energy (Kcal/mol) -6.06  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence MQRLESYILMKFLDQEKRRQLLNERHSCKMFDSHYEFSSTELEEIAEIARLSPSSYNTQPWHFVMVTDKDLKKQIAAHSYFNEEMIKSASALMVVCSLSYILEQCYIAVGQICMGVSLMGLDSCIIGGFDPLKVGEVLEERINPKIACLIALGKRVAEASQKSRKSKVDAITWLMKFLDQEKRRQLLNERHSCKMFDSHYEFSSTELEEIAEIARLSPSSYNTQPWHFVMVTDKDLKKQIAAHSYFNEEMIKSASALMVVCSLRPSELLPMQRLESYILEQCYIAVGQICMGVSLMGLDSCIIGGFDPLKVGEVLEERINKPKIACLIALGKRVAEASQKSRKSKVDAITWL Hydrogen bonds contact Hydrophobic contact | ||||
| 9 | DNA topoisomerase 4 subunit A | 1ZVT | 6.57 | |
Target general information Gen name parC Organism Escherichia coli (strain K12) Uniprot ID TTD ID NA Synonyms b3019;JW2987 Protein family Type II topoisomerase GyrA/ParC subunit family, ParC type 1 subfamily Biochemical class Isomerase Function ATP binding.DNA binding.DNA topoisomerase type II (ATP-hydrolyzing) activity. Related diseases Pigmentary disorder, reticulate, with systemic manifestations, X-linked (PDR) [MIM:301220]: An X-linked recessive disorder characterized by recurrent infections and sterile inflammation in various organs. Diffuse skin hyperpigmentation with a distinctive reticulate pattern is universally evident by early childhood. This is later followed in many patients by hypohidrosis, corneal inflammation and scarring, enterocolitis that resembles inflammatory bowel disease, and recurrent urethral strictures. Melanin and amyloid deposition is present in the dermis. Affected males also have a characteristic facies with frontally upswept hair and flared eyebrows. Female carriers have only restricted pigmentary changes along Blaschko's lines. {ECO:0000269|PubMed:27019227}. The disease is caused by variants affecting the gene represented in this entry. XLPDR is caused by a recurrent intronic mutation that results in missplicing and reduced POLA1 expression. This leads to a decrease in cytosolic RNA:DNA hybrids and constitutive activation of type I interferon responses, but has no effect on cell replication. {ECO:0000269|PubMed:27019227}.; DISEASE: Van Esch-O'Driscoll syndrome (VEODS) [MIM:301030]: An X-linked recessive syndrome characterized by different degrees of intellectual disability, moderate to severe short stature, microcephaly, hypogonadism, and variable congenital malformations. {ECO:0000269|PubMed:31006512}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB11943; DB12924; DB00817 Interacts with P22523; P0A7K2 EC number 5.6.2.2 Uniprot keywords 3D-structure; Cell membrane; Direct protein sequencing; DNA-binding; Isomerase; Membrane; Reference proteome; Topoisomerase Protein physicochemical properties Chain ID A,B Molecular weight (Da) 26490.3 Length 246 Aromaticity 0.04 Instability index 46.03 Isoelectric point 8.94 Charge (pH=7) 2.83 2D Binding mode Binding energy (Kcal/mol) -6.01 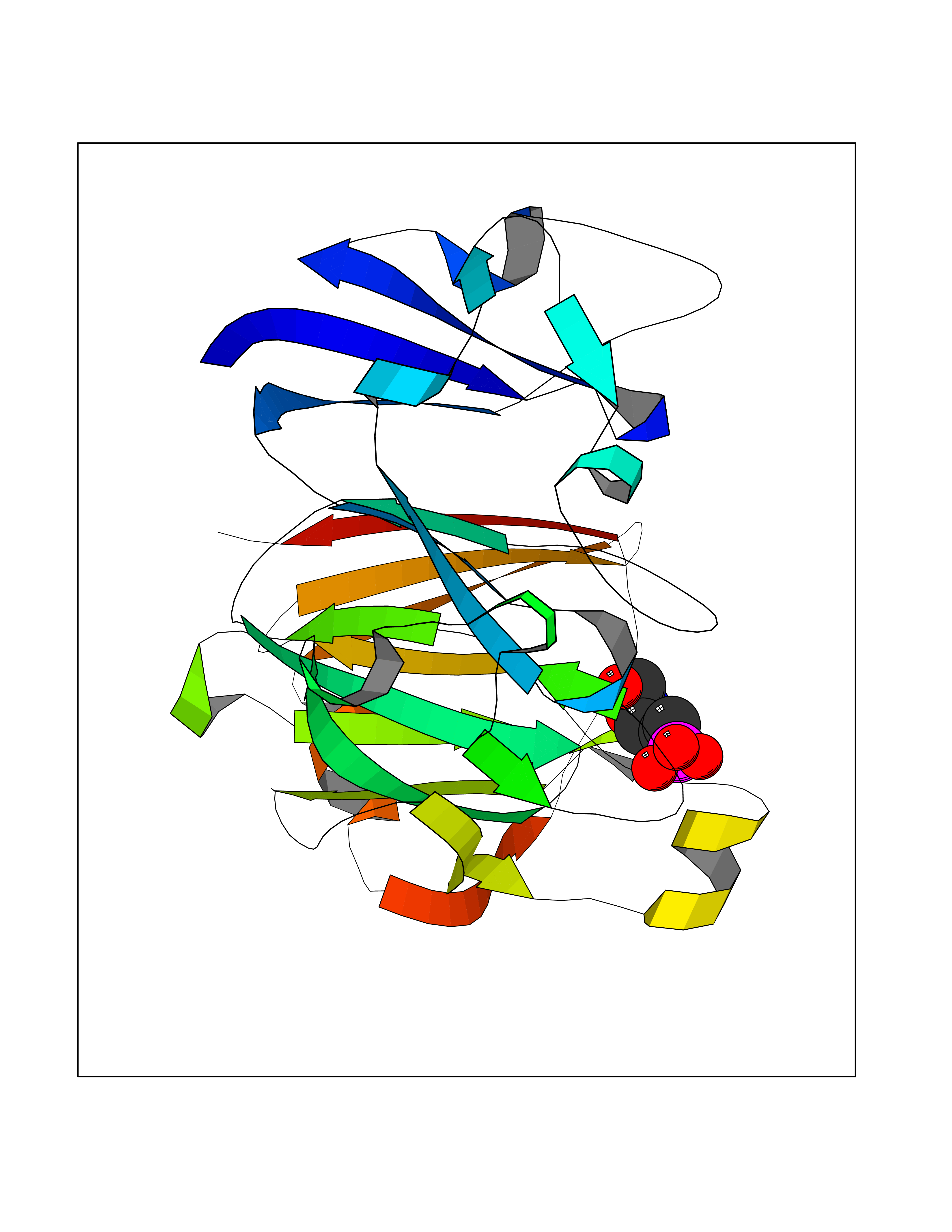 Molscript Map 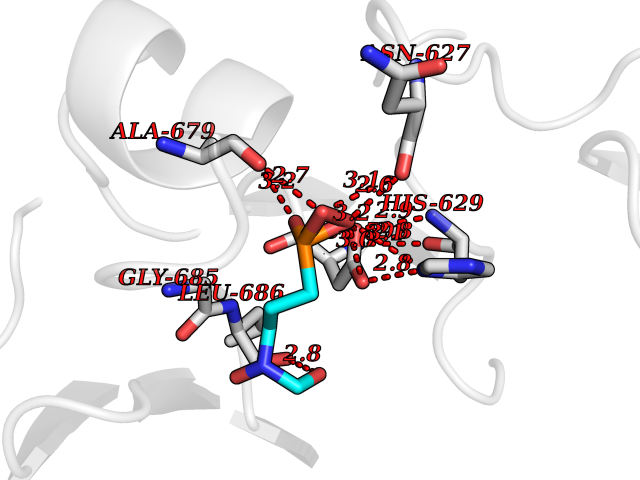 Pymol Map 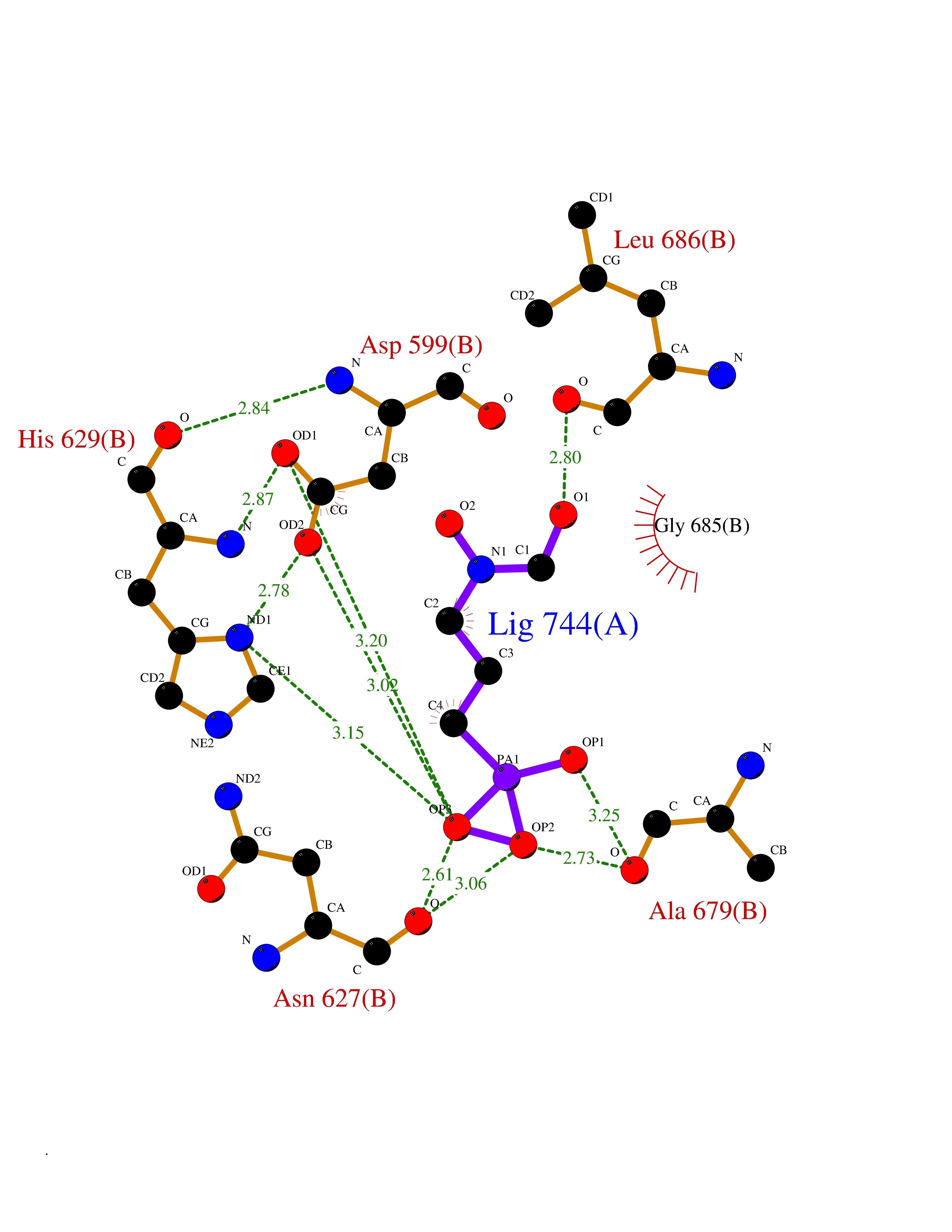 Ligplot Map 3D Binding mode Sequence SEPVTIVLSQMGWVRSAKGHDIDAPGLNYKAGDSFKAAVKGKSNQPVVFVDSTGRSYAIDPITLPSARGQGEPLTGKLTLPPGATVDHMLMESDDQKLLMASDAGYGFVCTFNDLVARNRAGKALITLPENAHVMPPVVIEDASDMLLAITQAGRMLMFPVSDLPQLSKGKGNKIINIPSAEAARGEDGLAQLYVLPPQSTLTIHVGKRKIKLRPEELQKVTGERGRRGTLMRGLQRIDRVEIDSP Hydrogen bonds contact Hydrophobic contact | ||||
| 10 | Retinal rod rhodopsin-sensitive cGMP 3',5'-cyclic phosphodiesterase subunit gamma | 3JWR | 6.57 | |
Target general information Gen name PDE6G Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms PDEG Protein family Rod/cone cGMP-PDE gamma subunit family Biochemical class Hydrolase Function 3',5'-cyclic-GMP phosphodiesterase activity.CGMP binding.Enzyme inhibitor activity.Spectrin binding. Related diseases Retinitis pigmentosa 57 (RP57) [MIM:613582]: A retinal dystrophy belonging to the group of pigmentary retinopathies. Retinitis pigmentosa is characterized by retinal pigment deposits visible on fundus examination and primary loss of rod photoreceptor cells followed by secondary loss of cone photoreceptors. Patients typically have night vision blindness and loss of midperipheral visual field. As their condition progresses, they lose their far peripheral visual field and eventually central vision as well. {ECO:0000269|PubMed:20655036}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07954; DB00203; DB00820; DB00862 Interacts with O14503; Q96JM7; A0A6Q8PF08; O43741; Q8R511; P62994; Q9QY17; Q63787 EC number 3.1.4.35 Uniprot keywords 3D-structure; Acetylation; cGMP; Hydrolase; Reference proteome; Retinitis pigmentosa; Sensory transduction; Vision Protein physicochemical properties Chain ID C,D Molecular weight (Da) 40027.7 Length 345 Aromaticity 0.09 Instability index 37.44 Isoelectric point 6.02 Charge (pH=7) -6.88 2D Binding mode Binding energy (Kcal/mol) -6.38  Molscript Map 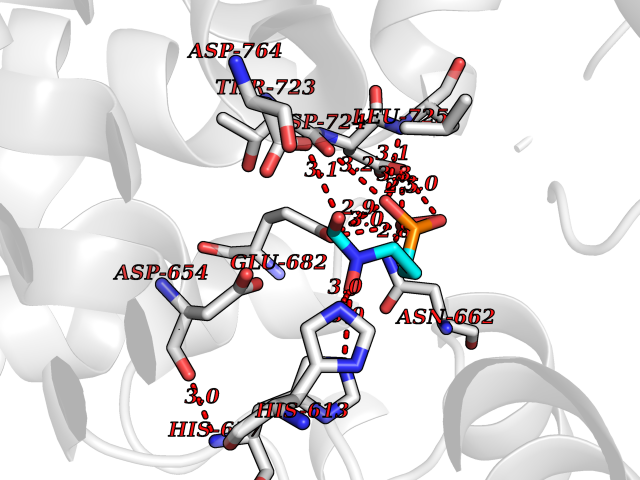 Pymol Map 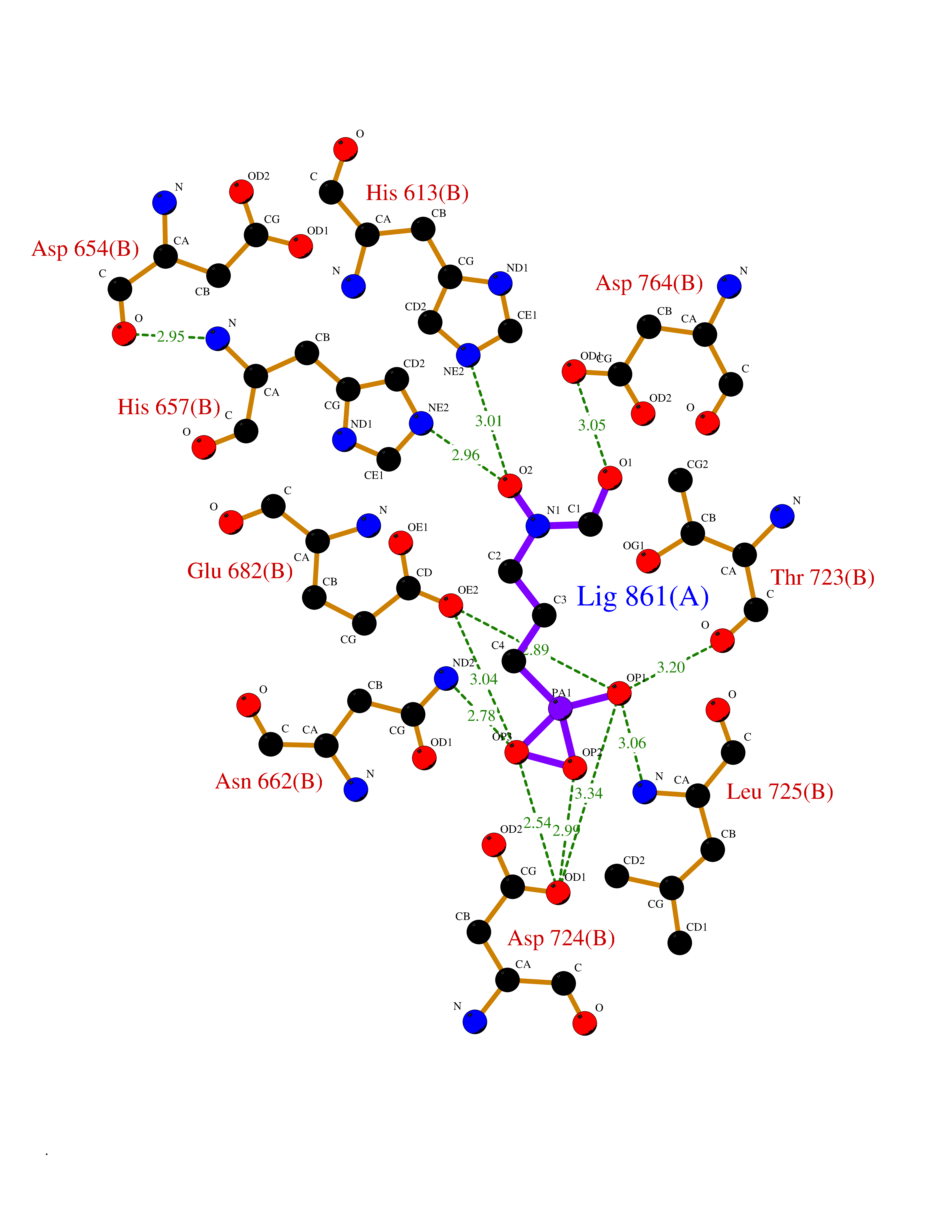 Ligplot Map 3D Binding mode Sequence EAFNHLELHELAQYGIISHMEETRELQSLAAAVVPSAQTLKITDFSFSDFELSDLETALCTIRMFTDLNLVQNFQMKHEVLCRWILSVKKNYRKNVAYHNWRHAFNTAQCMFAALKAGKIQNKLTDLEILALLIAALSHDLDHRGVNNSYIQRSEHPLAQLYCHSIMEHHHFDQCLMILNSPGNQILSGLSIEEYKTTLKIIKQAILATDLALYIKRRGEFFELIRKNQFNLEDPHQKELFLAMLMTACDLSAITKPWPIQQRIAELVATEFWEQGDLERTVLQQQPIPMMDRNKRDELPKLQVGFIDFVCTQLYEALTHVSEDCFPLLDGCRKNRQKWQALAEQ Hydrogen bonds contact Hydrophobic contact | ||||
| 11 | Carbonic anhydrase II (CA-II) | 3K34 | 6.56 | |
Target general information Gen name CA2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Carbonic anhydrase C; Carbonic anhydrase 2; Carbonate dehydratase II; CAC Protein family Alpha-carbonic anhydrase family Biochemical class Alpha-carbonic anhydrase Function Reversible hydration of carbon dioxide. Can hydrate cyanamide to urea. Involved in the regulation of fluid secretion into the anterior chamber of the eye. Contributes to intracellular pH regulation in the duodenal upper villous epithelium during proton-coupled peptide absorption. Stimulates the chloride-bicarbonate exchange activity of SLC26A6. Essential for bone resorption and osteoclast differentiation. Related diseases Osteopetrosis, autosomal recessive 3 (OPTB3) [MIM:259730]: A rare genetic disease characterized by abnormally dense bone, due to defective resorption of immature bone. Osteopetrosis occurs in two forms: a severe autosomal recessive form occurring in utero, infancy, or childhood, and a benign autosomal dominant form occurring in adolescence or adulthood. Recessive osteopetrosis commonly manifests in early infancy with macrocephaly, feeding difficulties, evolving blindness and deafness, bone marrow failure, severe anemia, and hepatosplenomegaly. Deafness and blindness are generally thought to represent effects of pressure on nerves. OPTB3 is associated with renal tubular acidosis, cerebral calcification (marble brain disease) and in some cases with intellectual disability. {ECO:0000269|PubMed:15300855, ECO:0000269|PubMed:1542674, ECO:0000269|PubMed:1928091, ECO:0000269|PubMed:8834238, ECO:0000269|PubMed:9143915}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07596; DB03333; DB08418; DB04081; DB08416; DB02479; DB07467; DB03950; DB03594; DB03294; DB04763; DB03270; DB08083; DB06954; DB08659; DB08046; DB02087; DB08156; DB04203; DB04394; DB08782; DB04549; DB02221; DB04180; DB03039; DB02861; DB02429; DB08202; DB04600; DB04601; DB01784; DB03385; DB03697; DB04002; DB07632; DB07050; DB06891; DB08645; DB08765; DB00819; DB03877; DB03262; DB03598; DB04089; DB01964; DB03526; DB04371; DB02220; DB03221; DB02602; DB02535; DB00436; DB00562; DB01194; DB00482; DB00880; DB02679; DB00606; DB02866; DB01144; DB00869; DB08846; DB01031; DB00311; DB08157; DB01942; DB00695; DB00774; DB08165; DB02292; DB03975; DB00703; DB00232; DB02610; DB07742; DB03844; DB02069; DB02986; DB08301; DB07048; DB03596; DB07476; DB01748; DB08155; DB01671; DB07710; DB01325; DB09460; DB09472; DB02894; DB00391; DB08329; DB07363; DB00273; DB01021; DB03904; DB00580; DB14533; DB14548; DB00909 Interacts with NA EC number EC 4.2.1.1 Uniprot keywords 3D-structure; Acetylation; Cell membrane; Cytoplasm; Direct protein sequencing; Disease variant; Lyase; Membrane; Metal-binding; Osteopetrosis; Phosphoprotein; Proteomics identification; Reference proteome; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 29027.4 Length 258 Aromaticity 0.1 Instability index 20.07 Isoelectric point 6.94 Charge (pH=7) -0.18 2D Binding mode Binding energy (Kcal/mol) -6.53 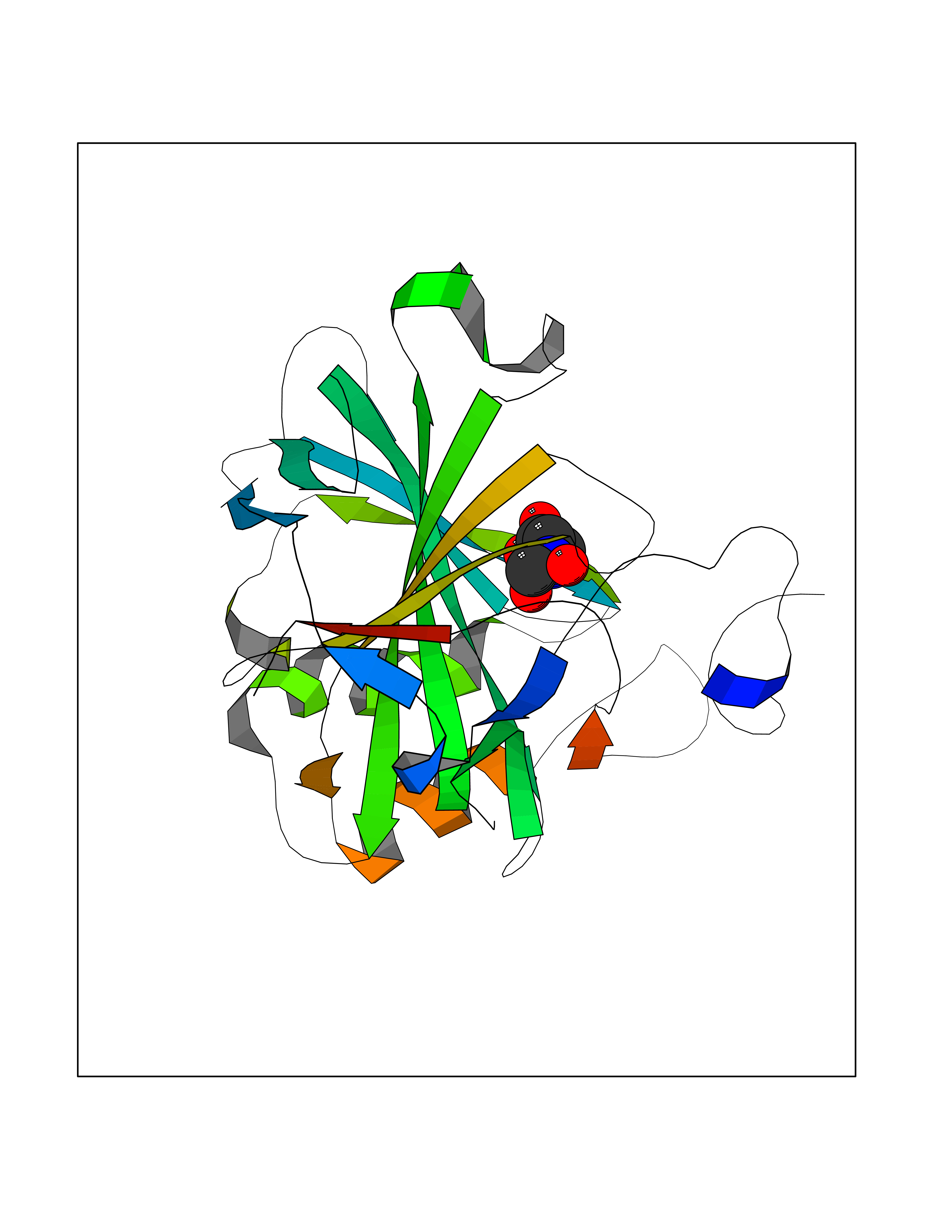 Molscript Map 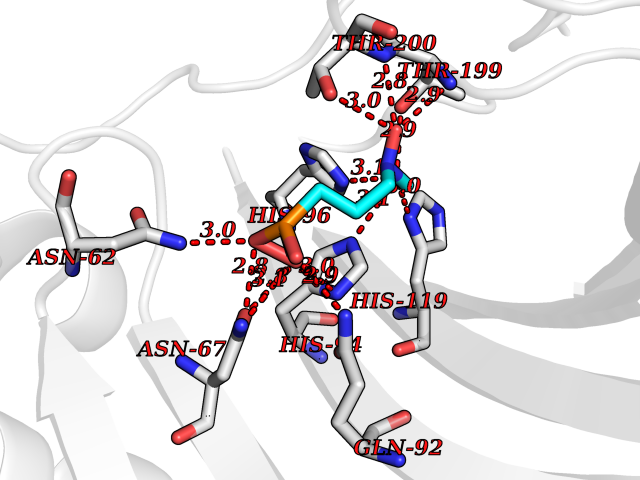 Pymol Map 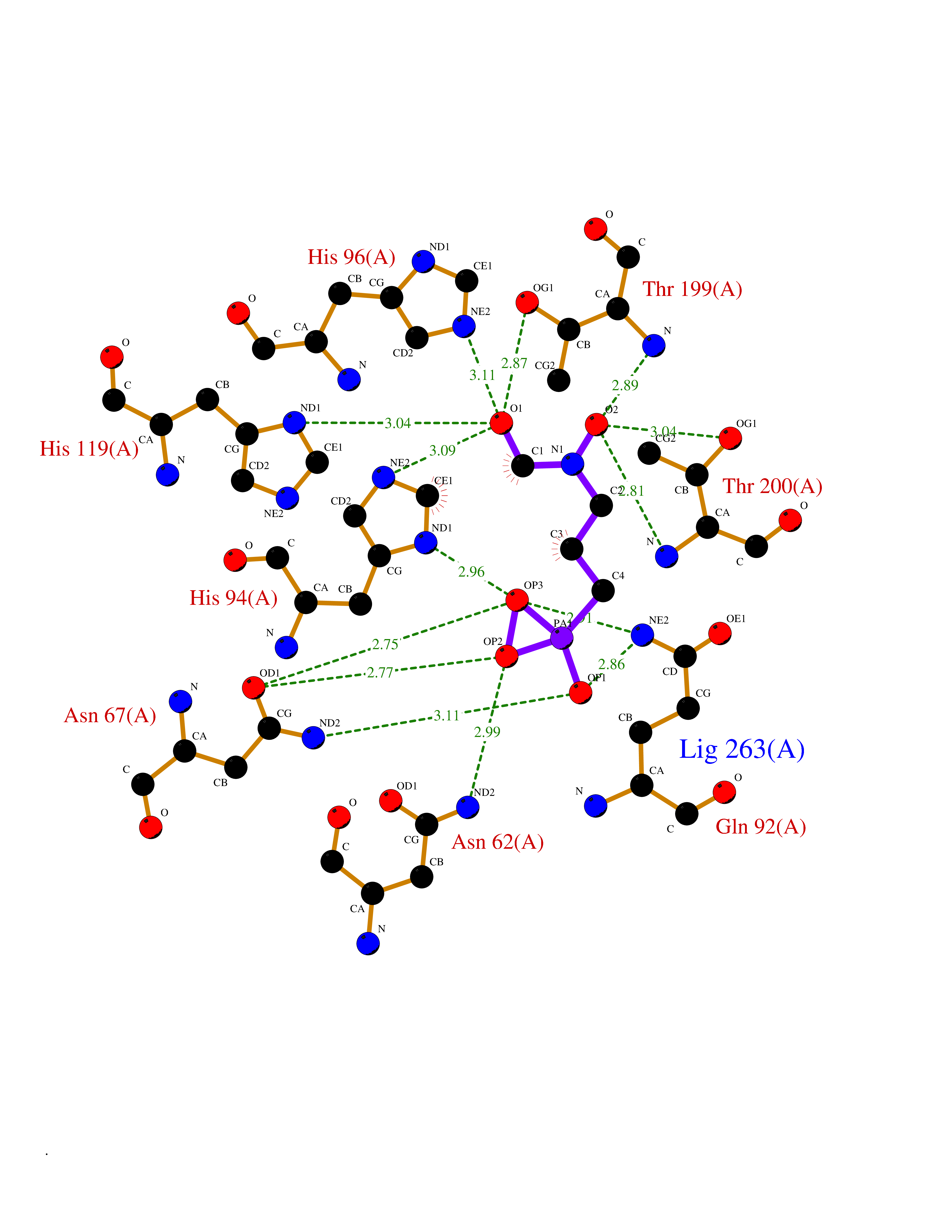 Ligplot Map 3D Binding mode Sequence HHWGYGKHNGPEHWHKDFPIAKGERQSPVDIDTHTAKYDPSLKPLSVSYDQATSLRILNNGHAFNVEFDDSQDKAVLKGGPLDGTYRLIQFHFHWGSLDGQGSEHTVDKKKYAAELHLVHWNTKYGDFGKAVQQPDGLAVLGIFLKVGSAKPGLQKVVDVLDSIKTKGKSADFTNFDPRGLLPESLDYWTYPGSLTTPPLLECVTWIVLKEPISVSSEQVLKFRKLNFNGEGEPEELMVDNWRPAQPLKNRQIKASFK Hydrogen bonds contact Hydrophobic contact | ||||
| 12 | Amylin receptor (IAPPR) | 6ZIS | 6.55 | |
Target general information Gen name CALCR-RAMP1/RAMP2/RAMP3 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Complex of Calcitonin receptor and Receptor activity-modifying protein Protein family RAMP family Biochemical class NA Function Transports the calcitonin gene-related peptide type 1 receptor (CALCRL) to the plasma membrane. Acts as a receptor for calcitonin-gene-related peptide (CGRP) together with CALCRL. Related diseases Immunodeficiency 9 (IMD9) [MIM:612782]: An immune disorder characterized by recurrent infections, impaired activation and proliferative response of T-cells, decreased T-cell production of cytokines, and normal lymphocytes counts and serum immunoglobulin levels. In surviving patients ectodermal dysplasia with anhidrosis and non-progressive myopathy may be observed. {ECO:0000269|PubMed:16147976, ECO:0000269|PubMed:16582901}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Myopathy, tubular aggregate, 2 (TAM2) [MIM:615883]: A rare congenital myopathy characterized by regular arrays of membrane tubules on muscle biopsies without additional histopathological hallmarks. Tubular aggregates in muscle are structures of variable appearance consisting of an outer tubule containing either one or more microtubule-like structures or amorphous material. TAM2 patients have myopathy and pupillary abnormalities. {ECO:0000269|PubMed:24591628, ECO:0000269|PubMed:28058752}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01278 Interacts with Q16602; P21145; Q5J8X5; Q16617 EC number NA Uniprot keywords 3D-structure; Cell membrane; Disulfide bond; Membrane; Proteomics identification; Receptor; Reference proteome; Signal; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A Molecular weight (Da) 63832.6 Length 569 Aromaticity 0.12 Instability index 22.42 Isoelectric point 5.07 Charge (pH=7) -16.96 2D Binding mode Binding energy (Kcal/mol) -6.04 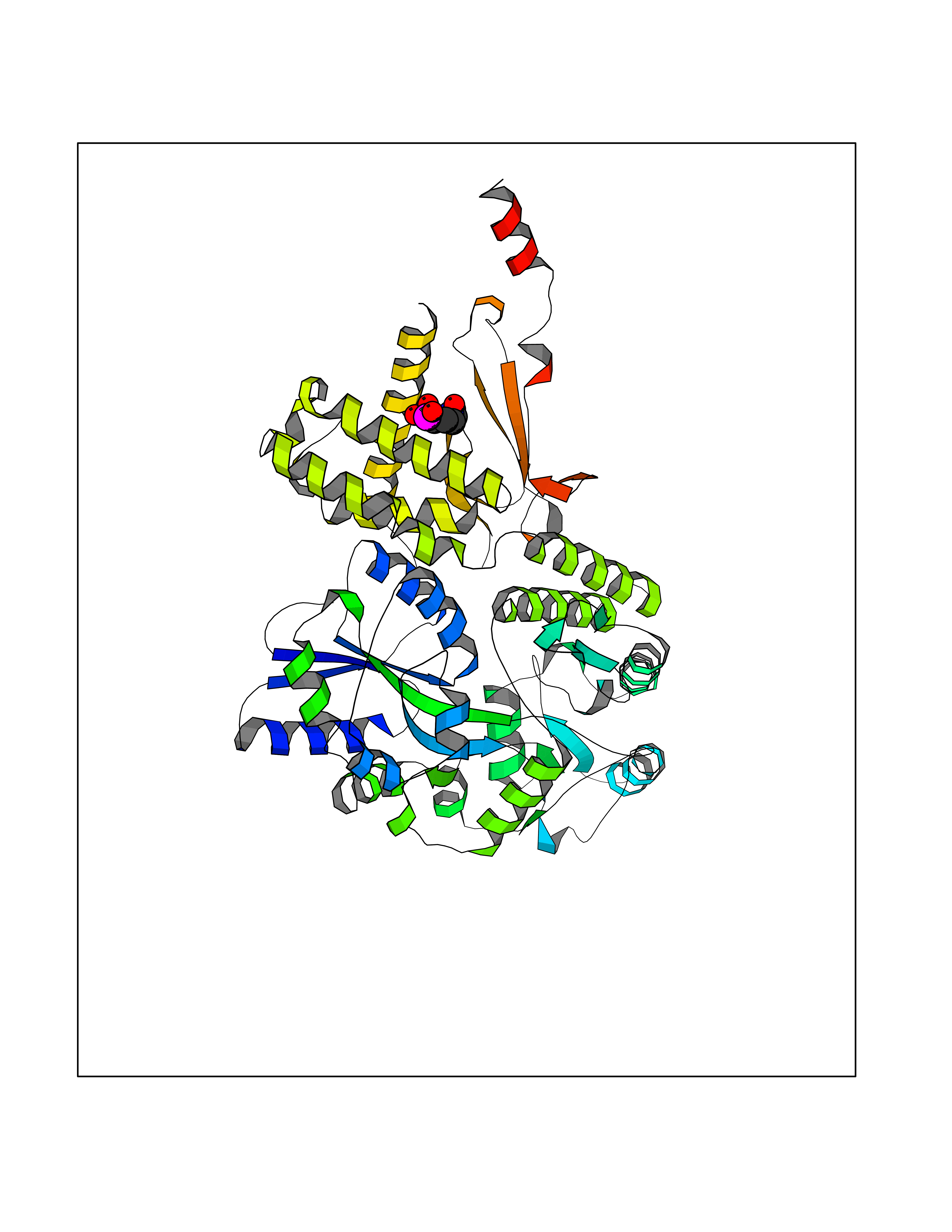 Molscript Map 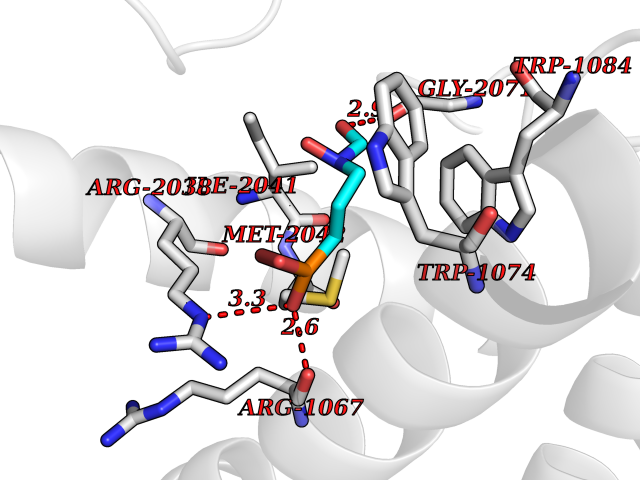 Pymol Map 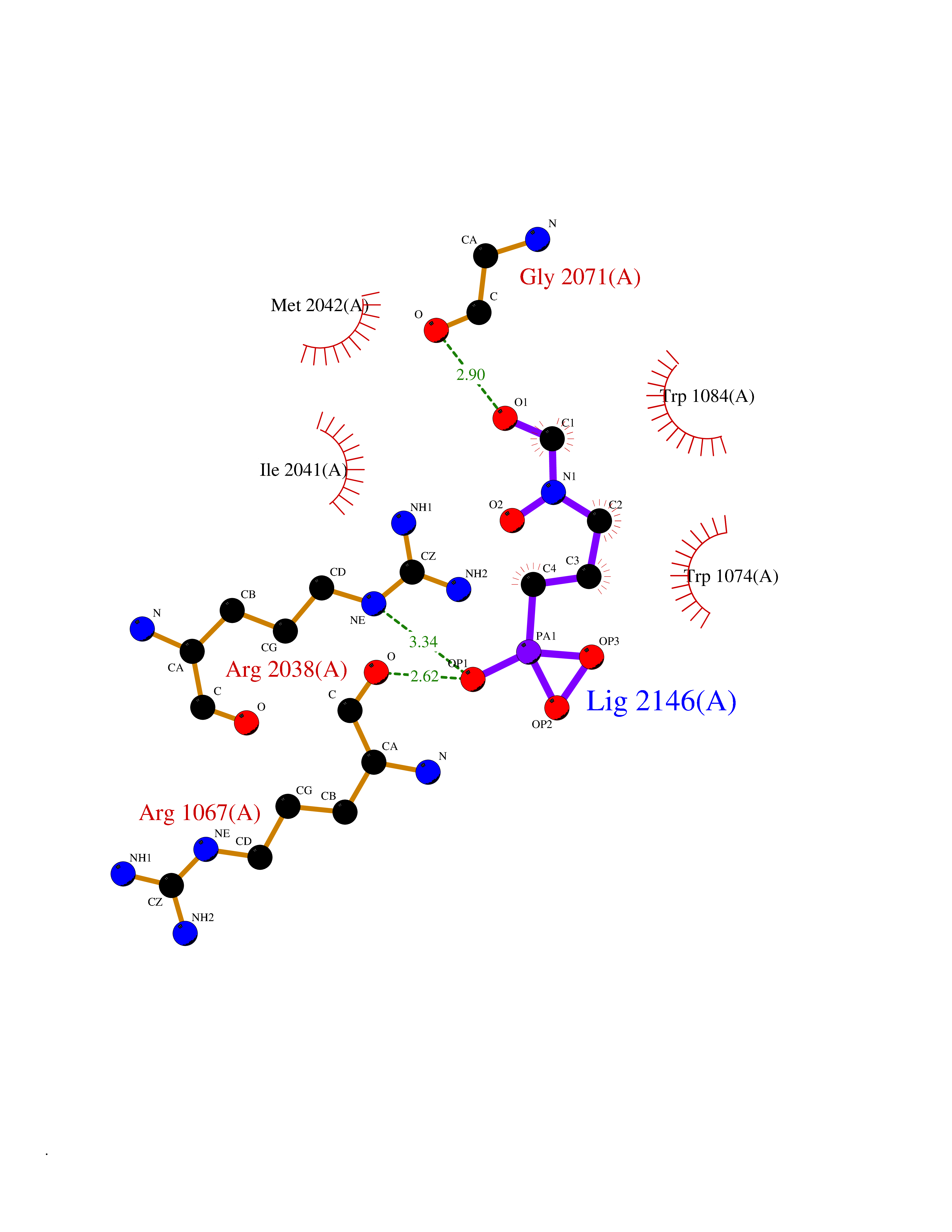 Ligplot Map 3D Binding mode Sequence SAKIEEGKLVIWINGDKGYNGLAEVGKKFEKDTGIKVTVEHPDKLEEKFPQVAATGDGPDIIFWAHDRFGGYAQSGLLAEITPDKAFQDKLYPFTWDAVRYNGKLIAYPIAVEALSLIYNKDLLPNPPKTWEEIPALDKELKAKGKSALMFNLQEPYFTWPLIAADGGYAFKYENGKYDIKDVGVDNAGAKAGLTFLVDLIKNKHMNADTDYSIAEAAFNKGETAMTINGPWAWSNIDTSKVNYGVTVLPTFKGQPSKPFVGVLSAGINAASPNKELAKEFLENYLLTDEGLEAVNKDKPLGAVALKSYEEELAKDPRIAATMENAQKGEIMPNIPQMSAFWYAVRTAVINAASGRQTVDEALKDAQTNAAAEFTTACQEANYGALLRELCLTQFQVDMEAVGETLWCDWGRTIRSYRELADCTWHMAEKLGCFWPNAEVDRFFLAVHGRYFRSCPISIQLGVTRNKIMTAQYECYQKIMQDPIQQGVYCQRTWDGWLCWNDVAAGTESMQLCPDYFQDFDPSEKVTKICDQDGNWFRHPASQRTWTDYTQCNVNTHEKVKTALNLFYL Hydrogen bonds contact Hydrophobic contact | ||||
| 13 | Alanine--tRNA ligase, cytoplasmic | 4XEM | 6.53 | |
Target general information Gen name AARS Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms AARS Protein family Class-II aminoacyl-tRNA synthetase family Biochemical class Ligase Function Alanine-tRNA ligase activity.Amino acid binding.Aminoacyl-tRNA editing activity.ATP binding.Metal ion binding.TRNA binding. Related diseases Charcot-Marie-Tooth disease, axonal, 2N (CMT2N) [MIM:613287]: An axonal form of Charcot-Marie-Tooth disease, a disorder of the peripheral nervous system, characterized by progressive weakness and atrophy, initially of the peroneal muscles and later of the distal muscles of the arms. Charcot-Marie-Tooth disease is classified in two main groups on the basis of electrophysiologic properties and histopathology: primary peripheral demyelinating neuropathies (designated CMT1 when they are dominantly inherited) and primary peripheral axonal neuropathies (CMT2). Neuropathies of the CMT2 group are characterized by signs of axonal degeneration in the absence of obvious myelin alterations, normal or slightly reduced nerve conduction velocities, and progressive distal muscle weakness and atrophy. {ECO:0000269|PubMed:20045102, ECO:0000269|PubMed:22009580, ECO:0000269|PubMed:22206013, ECO:0000269|PubMed:35911843, ECO:0000269|PubMed:35971119}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Developmental and epileptic encephalopathy 29 (DEE29) [MIM:616339]: A form of epileptic encephalopathy, a heterogeneous group of severe early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE29 patients manifest severe infantile epileptic encephalopathy, clubfoot, absent deep tendon reflexes, extrapyramidal symptoms, and persistently deficient myelination. {ECO:0000269|PubMed:25817015, ECO:0000269|PubMed:28493438}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Leukoencephalopathy, hereditary diffuse, with spheroids 2 (HDLS2) [MIM:619661]: An autosomal dominant neurodegenerative disorder characterized by progressive cognitive and executive dysfunction, psychiatric disturbances, and neurologic symptoms, such as gait abnormalities, paresis, seizures, and rigidity. Symptom onset is usually in adulthood, although earlier onset has been reported. Some patients have an acute encephalopathic course with severe neurologic decline resulting in early death, whereas other patients have a more protracted and chronic disease course. Neuropathologic examination shows a leukoencephalopathy with axonal spheroids and myelination defects. {ECO:0000269|PubMed:31775912, ECO:0000269|PubMed:37106376}. The disease may be caused by variants affecting the gene represented in this entry.; DISEASE: Trichothiodystrophy 8, non-photosensitive (TTD8) [MIM:619691]: A form of trichothiodystrophy, a disease characterized by sulfur-deficient brittle hair and multisystem variable abnormalities. The spectrum of clinical features varies from mild disease with only hair involvement to severe disease with cutaneous, neurologic and profound developmental defects. Ichthyosis, intellectual and developmental disabilities, decreased fertility, abnormal characteristics at birth, ocular abnormalities, short stature, and infections are common manifestations. There are both photosensitive and non-photosensitive forms of the disorder. TTD8 is an autosomal recessive, non-photosensitive form characterized by brittle hair and nails, scaly skin, accompanied by failure to thrive, microcephaly, and neuromotor developmental delay. {ECO:0000269|PubMed:33909043}. The disease may be caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00160 Interacts with NA EC number 6.-.-.-; 6.1.1.7 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Aminoacyl-tRNA synthetase; ATP-binding; Charcot-Marie-Tooth disease; Cytoplasm; Direct protein sequencing; Disease variant; Epilepsy; Ligase; Metal-binding; Methylation; Neurodegeneration; Neuropathy; Nucleotide-binding; Nucleus; Phosphoprotein; Protein biosynthesis; Proteomics identification; Reference proteome; RNA-binding; tRNA-binding; Ubl conjugation; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 42385.6 Length 377 Aromaticity 0.1 Instability index 27.33 Isoelectric point 5.15 Charge (pH=7) -13.8 2D Binding mode Binding energy (Kcal/mol) -6.04  Molscript Map 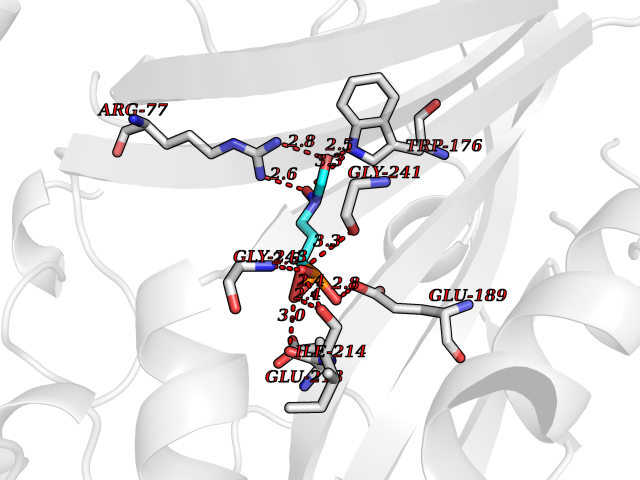 Pymol Map 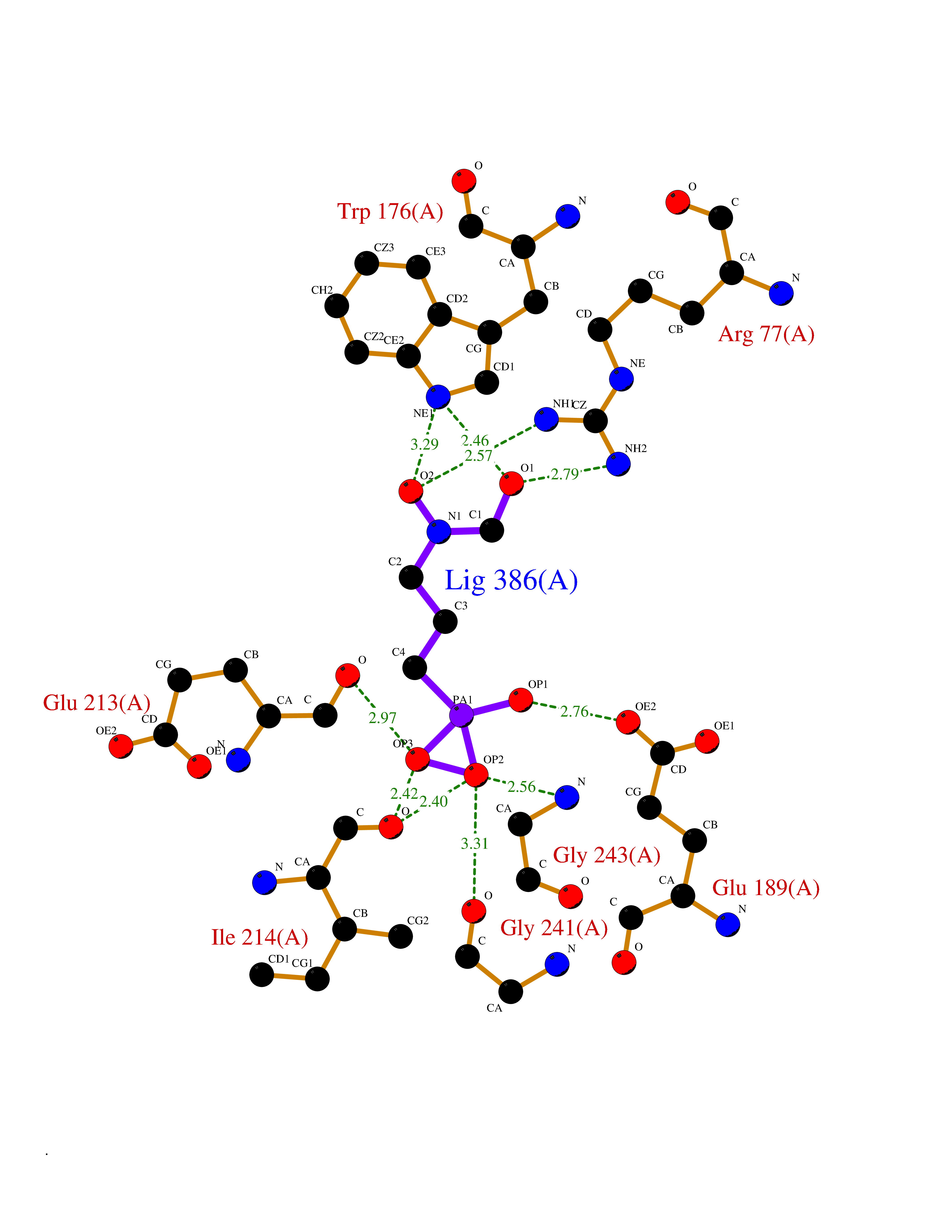 Ligplot Map 3D Binding mode Sequence TLTASEIRQRFIDFFKRNEHTYVHSSATIPLDDPTLLFANAGMNQFKPIFLNTIDPSHPMAKLSRAANTQKCIRAGDLDDVGKDVYHHTFFEMLGSWSFGDYFKELACKMALELLTQEFGIPIERLYVTYFGGDEAAGLEADLECKQIWQNLGLDDTKILPGNMKDNFWEMGDTGPCGPCSEIHYDRIGGRDAAHLVNQDDPNVLEIWNLVFIQYNREADGILKPLPKKSIDTGMGLERLVSVLQNKMSNYDTDLFVPYFEAIQKGTGARPYTGKVGAEDADGIDMAYRVLADHARTITVALADGGRPDNTGRGYVLRRILRRAVRYAHEKLNASRGFFATLVDVVVQSLGDAFPELKKDPDMVKDIINEEEVQFLK Hydrogen bonds contact Hydrophobic contact | ||||
| 14 | Vasopressin V1a receptor | 1YTV | 6.51 | |
Target general information Gen name AVPR1A Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms AVPR1 Protein family G-protein coupled receptor 1 family, Vasopressin/oxytocin receptor subfamily Biochemical class Sugar binding protein Function Peptide binding.Peptide hormone binding.Protein kinase C binding.V1A vasopressin receptor binding.Vasopressin receptor activity. Related diseases Defects in PPARG can lead to type 2 insulin-resistant diabetes and hyptertension. PPARG mutations may be associated with colon cancer. {ECO:0000269|PubMed:10394368}.; DISEASE: Obesity (OBESITY) [MIM:601665]: A condition characterized by an increase of body weight beyond the limitation of skeletal and physical requirements, as the result of excessive accumulation of body fat. {ECO:0000269|PubMed:9753710}. Disease susceptibility may be associated with variants affecting the gene represented in this entry.; DISEASE: Lipodystrophy, familial partial, 3 (FPLD3) [MIM:604367]: A form of lipodystrophy characterized by marked loss of subcutaneous fat from the extremities. Facial adipose tissue may be increased, decreased or normal. Affected individuals show an increased preponderance of insulin resistance, diabetes mellitus and dyslipidemia. {ECO:0000269|PubMed:11788685, ECO:0000269|PubMed:12453919}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Glioma 1 (GLM1) [MIM:137800]: Gliomas are benign or malignant central nervous system neoplasms derived from glial cells. They comprise astrocytomas and glioblastoma multiforme that are derived from astrocytes, oligodendrogliomas derived from oligodendrocytes and ependymomas derived from ependymocytes. {ECO:0000269|PubMed:10851250}. Disease susceptibility may be associated with variants affecting the gene represented in this entry. Polymorphic PPARG alleles have been found to be significantly over-represented among a cohort of American patients with sporadic glioblastoma multiforme suggesting a possible contribution to disease susceptibility. Drugs (DrugBank ID) DB09059; DB00872; DB00035; DB00093; DB14642; DB16279; DB05452; DB13929; DB02638; DB06212; DB00067 Interacts with P25106 EC number NA Uniprot keywords 3D-structure; Cell membrane; Disulfide bond; G-protein coupled receptor; Glycoprotein; Lipoprotein; Membrane; Palmitate; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID M,N Molecular weight (Da) 40697.6 Length 371 Aromaticity 0.1 Instability index 20.94 Isoelectric point 5.07 Charge (pH=7) -9.93 2D Binding mode Binding energy (Kcal/mol) -5.57  Molscript Map 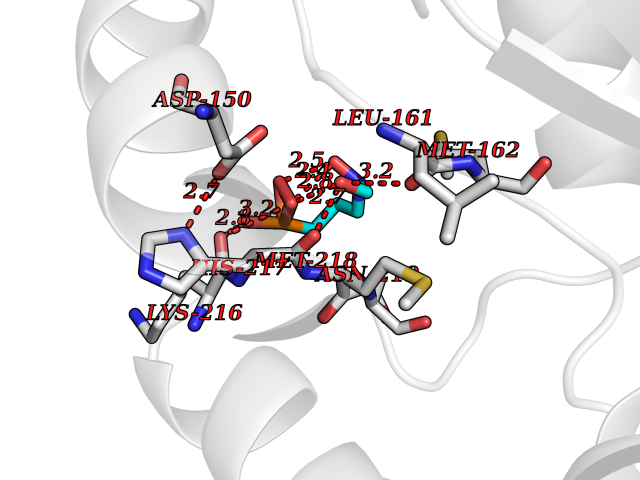 Pymol Map  Ligplot Map 3D Binding mode Sequence NSSSNKIEEGKLVIWINGDKGYNGLAEVGKKFEKDTGIKVTVEHPDKLEEKFPQVAATGDGPDIIFWAHDRFGGYAQSGLLAEITPDKAFQDKLYPFTWDAVRYNGKLIAYPIAVEALSLIYNKDLLPNPPKTWEEIPALDKELKAKGKSALMFNLQEPYFTWPLIAADGGYAFKYENGKYDIKDVGVDNAGAKAGLTFLVDLIKNKHMNADTDYSIAEAAFNKGETAMTINGPWAWSNIDTSKVNYGVTVLPTFKGQPSKPFVGVLSAGINAASPNKELAKEFLENYLLTDEGLEAVNKDKPLGAVALKSYEEELAKDPRIAATMENAQKGEIMPNIPQMSAFWYAVRTAVINAASGRQTVDEALKDAQT Hydrogen bonds contact Hydrophobic contact | ||||
| 15 | Endonuclease 8-like 1 | 5ITQ | 6.50 | |
Target general information Gen name NEIL1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family FPG family Biochemical class Dna binding protein / dna Function Class I DNA-(apurinic or apyrimidinic site) lyase activity.Class III/IV DNA-(apurinic or apyrimidinic site) lyase activity.Damaged DNA binding.DNA-(apurinic or apyrimidinic site) lyase activity.DNA N-glycosylase activity.Hydrolase activity, acting on glycosyl bonds.Protein C-terminus binding.Zinc ion binding. Related diseases Glutaric aciduria 1 (GA1) [MIM:231670]: An autosomal recessive metabolic disorder characterized by progressive dystonia and athetosis due to gliosis and neuronal loss in the basal ganglia. {ECO:0000269|PubMed:14707522, ECO:0000269|PubMed:18775954, ECO:0000269|PubMed:24973495, ECO:0000269|PubMed:8541831, ECO:0000269|PubMed:8900227, ECO:0000269|PubMed:8900228, ECO:0000269|PubMed:9600243, ECO:0000269|PubMed:9711871}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB09130; DB14490; DB14491; DB14488; DB14501; DB14489; DB01592 Interacts with NA EC number 3.2.2.-; 4.2.99.18 Uniprot keywords 3D-structure; Chromosome; Cytoplasm; Cytoskeleton; DNA damage; DNA repair; DNA-binding; Glycosidase; Hydrolase; Lyase; Multifunctional enzyme; Nucleus; Proteomics identification; Reference proteome; RNA editing Protein physicochemical properties Chain ID A Molecular weight (Da) 32177.5 Length 286 Aromaticity 0.1 Instability index 57.82 Isoelectric point 8.89 Charge (pH=7) 5.38 2D Binding mode Binding energy (Kcal/mol) -5.34  Molscript Map 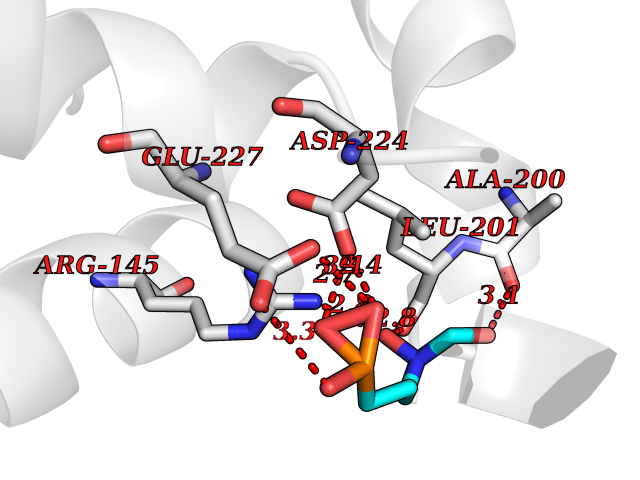 Pymol Map 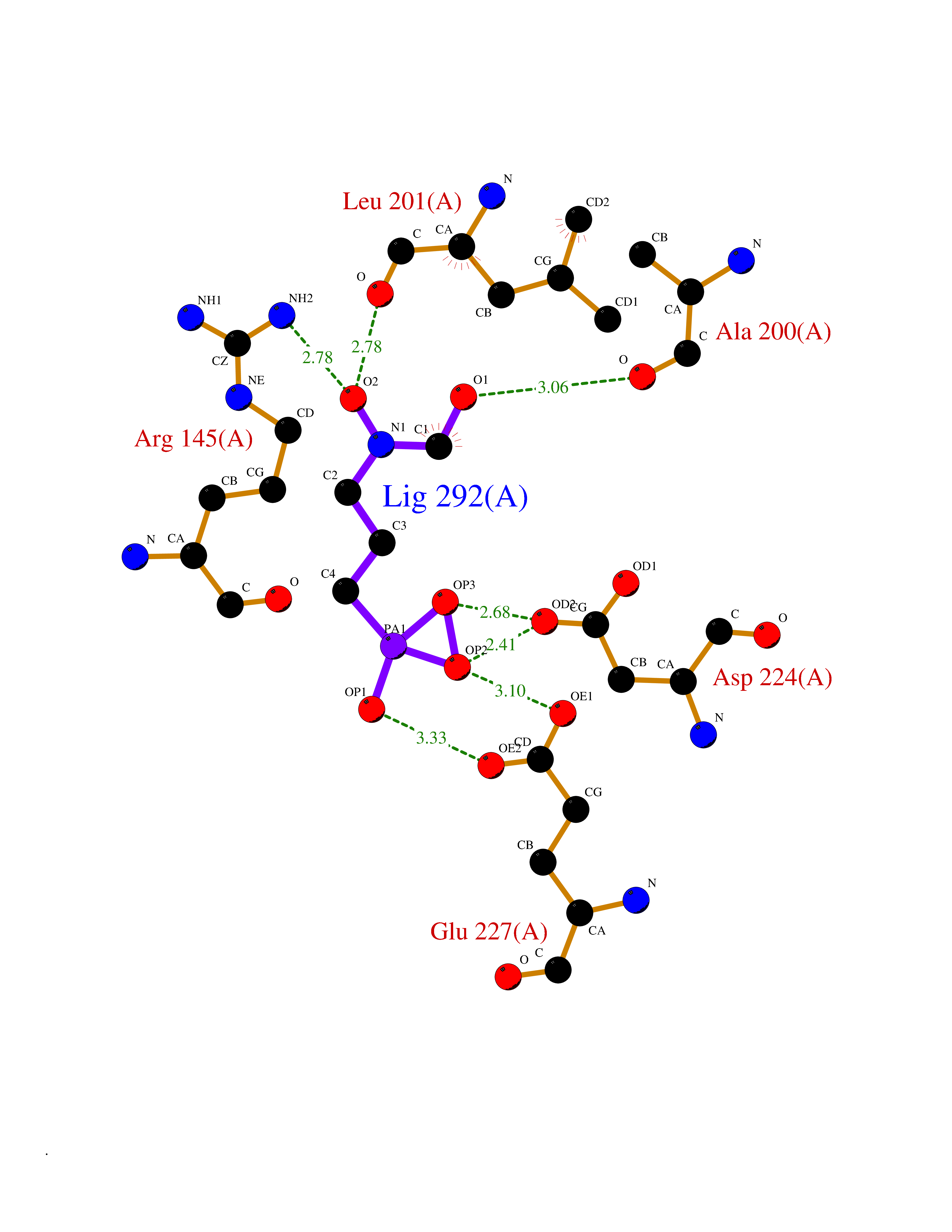 Ligplot Map 3D Binding mode Sequence PEGPELHLASQFVNEACRALVFGGCVEKSSVSRNPEVPFESSAYRISASARGKELRLILSPLPGAQPQQEPLALVFRFGMSGSFQLVPREELPRHAHLRFYTAPPGPRLALCFVDIRRFGRWDLGGKWQPGRGPCVLQEYQQFRESVLRNLADKAFDRPICEALLDQRFFNGIGNYLRAEILYRLKIPPFEKARSVLEALQQSPELTLSQKIRTKLQNPDLLELCHSVPKEVVQLGGRGYGSESGEEDFAAFRAWLRCYGMPGMSSLQDRHGRTIWFQGDPGPLAP Hydrogen bonds contact Hydrophobic contact | ||||
| 16 | Carbonic anhydrase IX (CA-IX) | 5FL4 | 6.48 | |
Target general information Gen name CA9 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Renal cell carcinoma-associated antigen G250; RCC-associated antigen G250; PMW1; P54/58N; Membrane antigen MN; MN; G250 antigen (MN/CA IX/G250); G250; Carbonic anhydrase 9; Carbonate dehydratase IX; C Protein family Alpha-carbonic anhydrase family Biochemical class Alpha-carbonic anhydrase Function Participates in pH regulation. May be involved in the control of cell proliferation and transformation. Appears to be a novel specific biomarker for a cervical neoplasia. Reversible hydration of carbon dioxide. Related diseases Hydroxykynureninuria (HYXKY) [MIM:236800]: An inborn error of amino acid metabolism characterized by massive urinary excretion of large amounts of kynurenine, 3-hydroxykynurenine and xanthurenic acid. Affected individuals manifest renal tubular dysfunction, metabolic acidosis, psychomotor retardation, non-progressive encephalopathy, and muscular hypertonia. {ECO:0000269|PubMed:17334708, ECO:0000269|PubMed:28792876}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Vertebral, cardiac, renal, and limb defects syndrome 2 (VCRL2) [MIM:617661]: An autosomal recessive congenital malformation syndrome characterized by vertebral segmentation abnormalities, congenital cardiac defects, renal defects, and distal mild limb defects. {ECO:0000269|PubMed:28792876}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00562; DB00606; DB12741; DB08846; DB05304; DB00774; DB09460; DB00909 Interacts with P21291; O76003 EC number EC 4.2.1.1 Uniprot keywords 3D-structure; Cell membrane; Cell projection; Direct protein sequencing; Disulfide bond; Glycoprotein; Lyase; Membrane; Metal-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Signal; Transmembrane; Transmembrane helix; Zinc Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 27522.8 Length 251 Aromaticity 0.08 Instability index 48.97 Isoelectric point 5.48 Charge (pH=7) -7.5 2D Binding mode Binding energy (Kcal/mol) -6.15 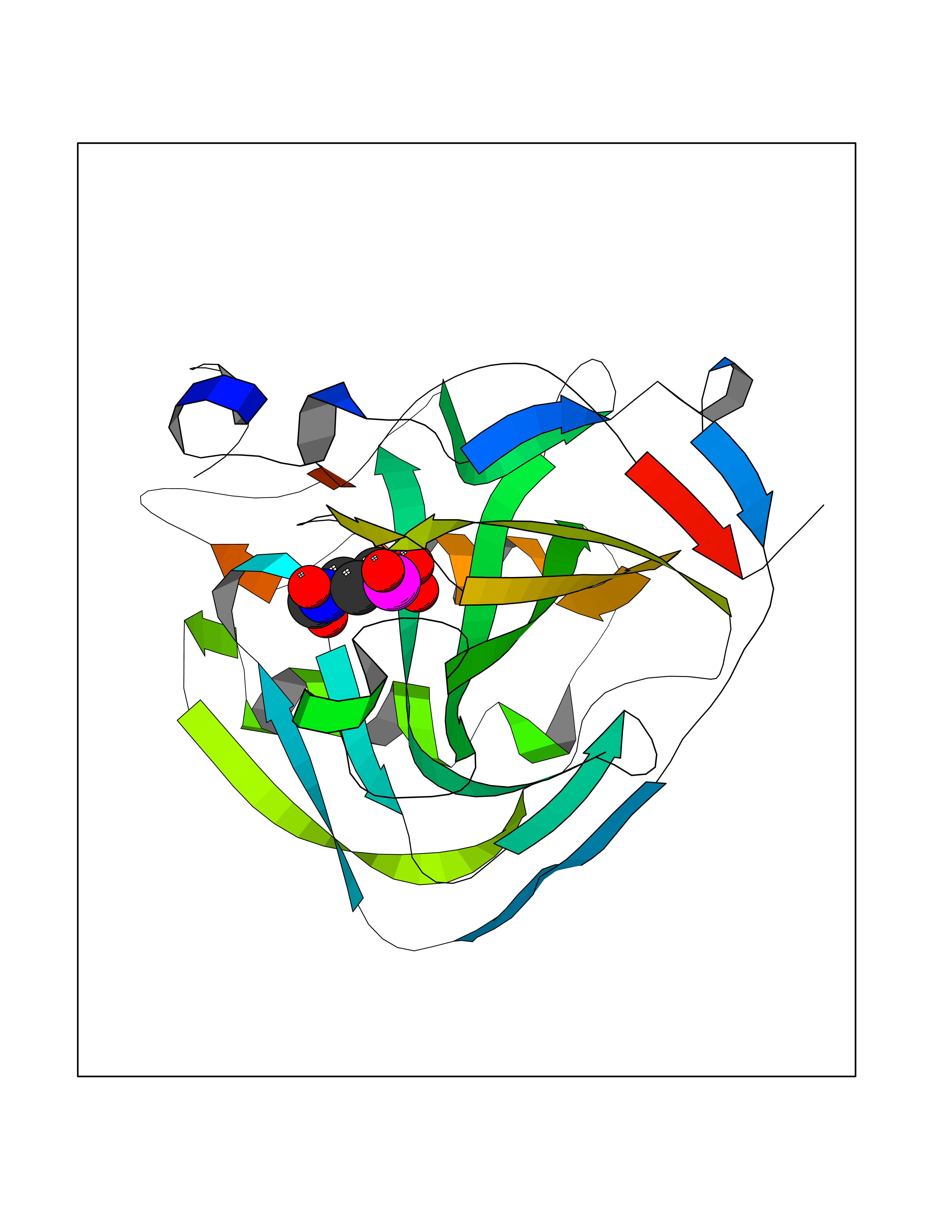 Molscript Map 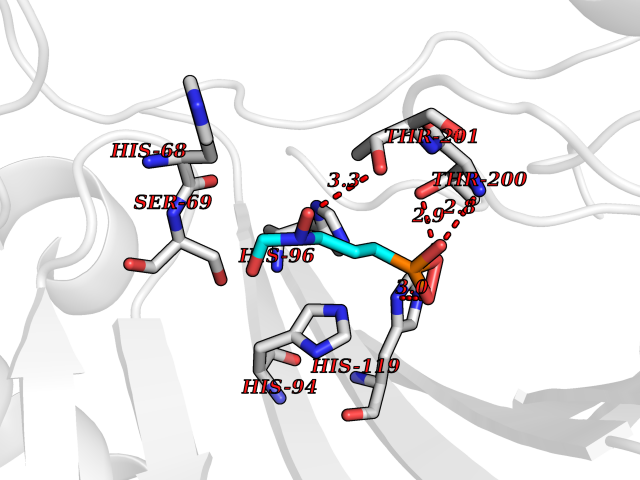 Pymol Map 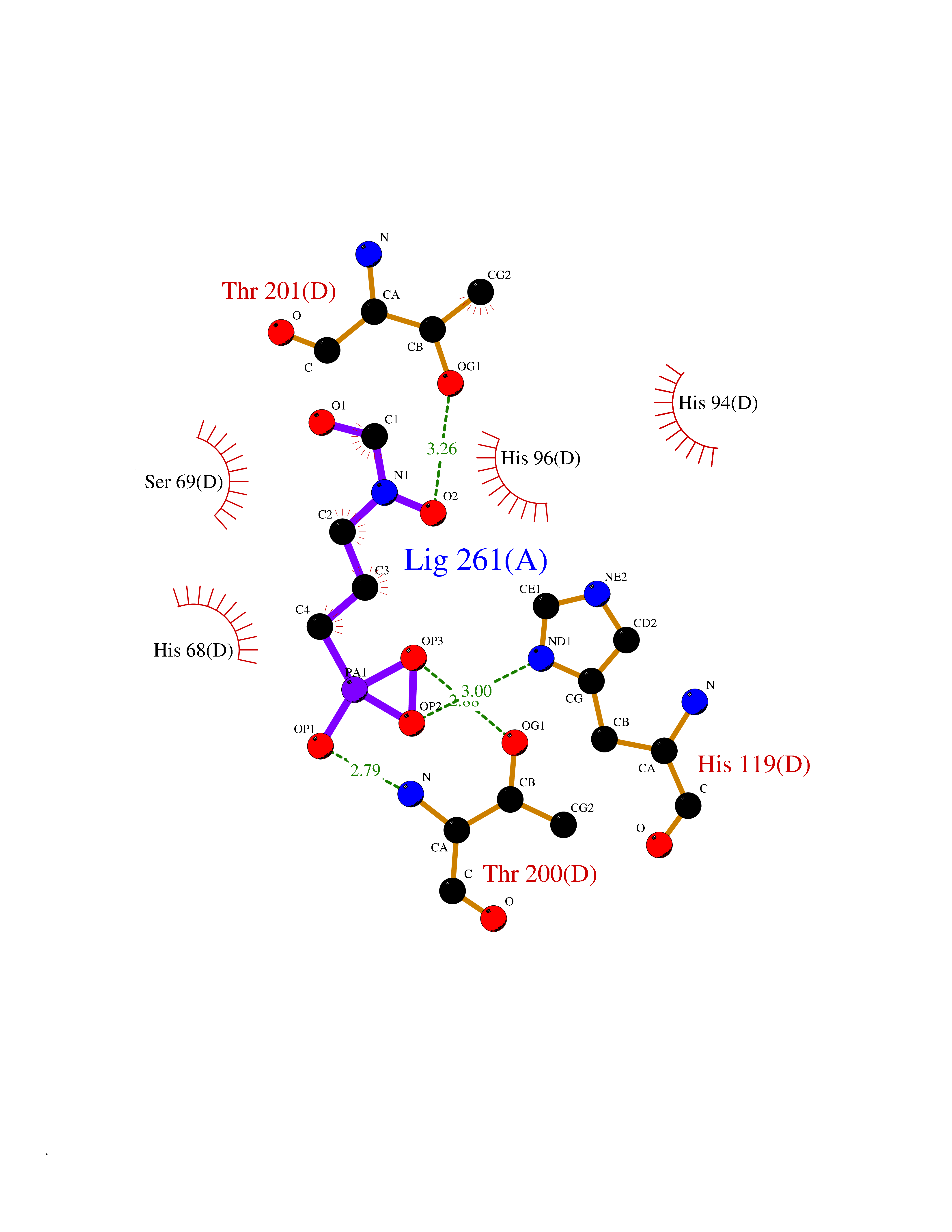 Ligplot Map 3D Binding mode Sequence WRYGGDPPWPRVSPACAGRFQSPVDIRPQLAAFSPALRPLELLGFQLPPLPELRLRNNGHSVQLTLPPGLEMALGPGREYRALQLHLHWGAAGRPGSEHTVEGHRFPAEIHVVHLSTAFARVDEALGRPGGLAVLAAFLEEGPEENSAYEQLLSRLEEIAEEGSETQVPGLDISALLPSDFSRYFQYEGSLTTPPCAQGVIWTVFNQTVMLSAKQLHTLSDTLWGPGDSRLQLNFRATQPLNGRVIEASFP Hydrogen bonds contact Hydrophobic contact | ||||
| 17 | Coagulation factor VIII (F8) | 3HNB | 6.47 | |
Target general information Gen name F8 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Procoagulant component; F8C; Antihemophilic factor; AHF Protein family Multicopper oxidase family Biochemical class NA Function Factor VIII, along with calcium and phospholipid, acts as a cofactor for F9/factor IXa when it converts F10/factor X to the activated form, factor Xa. Related diseases Hemophilia A (HEMA) [MIM:306700]: A disorder of blood coagulation characterized by a permanent tendency to hemorrhage. About 50% of patients have severe hemophilia resulting in frequent spontaneous bleeding into joints, muscles and internal organs. Less severe forms are characterized by bleeding after trauma or surgery. {ECO:0000269|PubMed:10215414, ECO:0000269|PubMed:10338101, ECO:0000269|PubMed:10404764, ECO:0000269|PubMed:10408784, ECO:0000269|PubMed:10554831, ECO:0000269|PubMed:10612839, ECO:0000269|PubMed:10691849, ECO:0000269|PubMed:10800171, ECO:0000269|PubMed:10886198, ECO:0000269|PubMed:10896236, ECO:0000269|PubMed:10910910, ECO:0000269|PubMed:10910913, ECO:0000269|PubMed:11298607, ECO:0000269|PubMed:11341489, ECO:0000269|PubMed:11410838, ECO:0000269|PubMed:11442643, ECO:0000269|PubMed:11442647, ECO:0000269|PubMed:11554935, ECO:0000269|PubMed:11748850, ECO:0000269|PubMed:11857744, ECO:0000269|PubMed:11858487, ECO:0000269|PubMed:12195713, ECO:0000269|PubMed:12199686, ECO:0000269|PubMed:12203998, ECO:0000269|PubMed:12325022, ECO:0000269|PubMed:12351418, ECO:0000269|PubMed:12406074, ECO:0000269|PubMed:12614369, ECO:0000269|PubMed:12871415, ECO:0000269|PubMed:12930394, ECO:0000269|PubMed:1301194, ECO:0000269|PubMed:1301932, ECO:0000269|PubMed:1301960, ECO:0000269|PubMed:1349567, ECO:0000269|PubMed:1356412, ECO:0000269|PubMed:15682412, ECO:0000269|PubMed:15810915, ECO:0000269|PubMed:1639429, ECO:0000269|PubMed:16805874, ECO:0000269|PubMed:18184865, ECO:0000269|PubMed:1851341, ECO:0000269|PubMed:1908096, ECO:0000269|PubMed:1908817, ECO:0000269|PubMed:1924291, ECO:0000269|PubMed:1973901, ECO:0000269|PubMed:2104766, ECO:0000269|PubMed:2105106, ECO:0000269|PubMed:2105906, ECO:0000269|PubMed:2106480, ECO:0000269|PubMed:2107542, ECO:0000269|PubMed:21371196, ECO:0000269|PubMed:2495245, ECO:0000269|PubMed:2498882, ECO:0000269|PubMed:2499363, ECO:0000269|PubMed:2506948, ECO:0000269|PubMed:2510835, ECO:0000269|PubMed:25550078, ECO:0000269|PubMed:26278069, ECO:0000269|PubMed:2833855, ECO:0000269|PubMed:2835904, ECO:0000269|PubMed:29357978, ECO:0000269|PubMed:3012775, ECO:0000269|PubMed:3122181, ECO:0000269|PubMed:7579394, ECO:0000269|PubMed:7759074, ECO:0000269|PubMed:7794769, ECO:0000269|PubMed:8322269, ECO:0000269|PubMed:8449505, ECO:0000269|PubMed:8639447, ECO:0000269|PubMed:8644728, ECO:0000269|PubMed:8759905, ECO:0000269|PubMed:9029040, ECO:0000269|PubMed:9326186, ECO:0000269|PubMed:9341862, ECO:0000269|PubMed:9450898, ECO:0000269|PubMed:9452104, ECO:0000269|PubMed:9569180, ECO:0000269|PubMed:9569189, ECO:0000269|PubMed:9603440, ECO:0000269|PubMed:9792405, ECO:0000269|PubMed:9829908, ECO:0000269|PubMed:9886318}. The disease is caused by variants affecting the gene represented in this entry. Of particular interest for the understanding of the function of F8 is the category of CRM (cross-reacting material) positive patients (approximately 5%) that have considerable amount of F8 in their plasma (at least 30% of normal), but the protein is non-functional; i.e. the F8 activity is much less than the plasma protein level. CRM-reduced is another category of patients in which the F8C antigen and activity are reduced to approximately the same level. Most mutations are CRM negative, and probably affect the folding and stability of the protein.; DISEASE: Thrombophilia 13, X-linked, due to factor VIII defect (THPH13) [MIM:301071]: An X-linked dominant, hemostatic disorder associated with markedly elevated F8 levels, and characterized by severe thrombophilia. {ECO:0000269|PubMed:33275657}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB13884; DB13151; DB00100; DB13152; DB09130; DB14700; DB00055; DB11571; DB13933; DB11312; DB16007; DB06050; DB11300; DB11572; DB13133; DB12872 Interacts with P04275; Q8N7X4; Q8N488; P04275; P00740 EC number NA Uniprot keywords 3D-structure; Acute phase; Alternative splicing; Blood coagulation; Calcium; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; Hemophilia; Hemostasis; Metal-binding; Pharmaceutical; Proteomics identification; Reference proteome; Repeat; Secreted; Signal; Sulfation; Thrombophilia Protein physicochemical properties Chain ID M Molecular weight (Da) 17668 Length 156 Aromaticity 0.1 Instability index 35.82 Isoelectric point 9.43 Charge (pH=7) 4.8 2D Binding mode Binding energy (Kcal/mol) -5.41 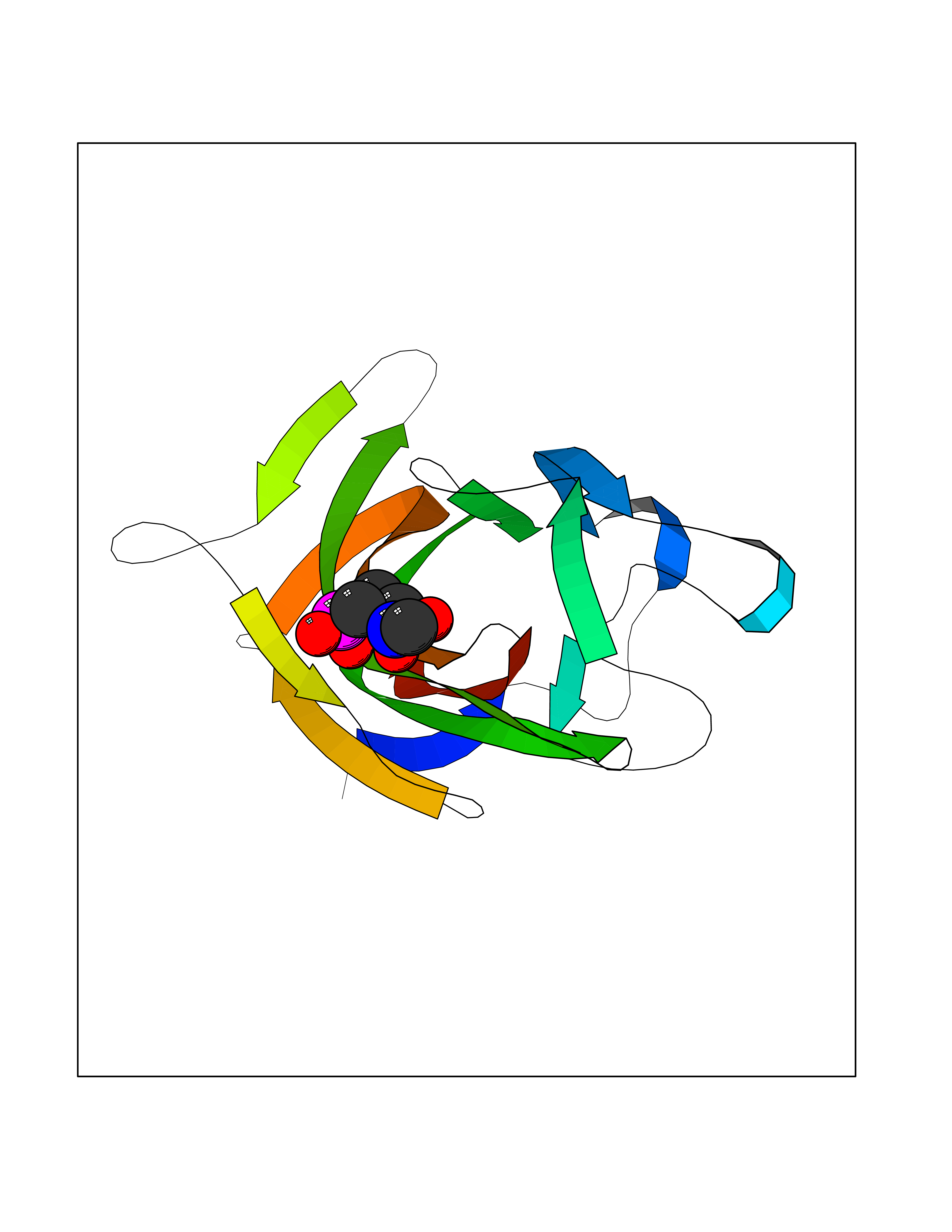 Molscript Map  Pymol Map 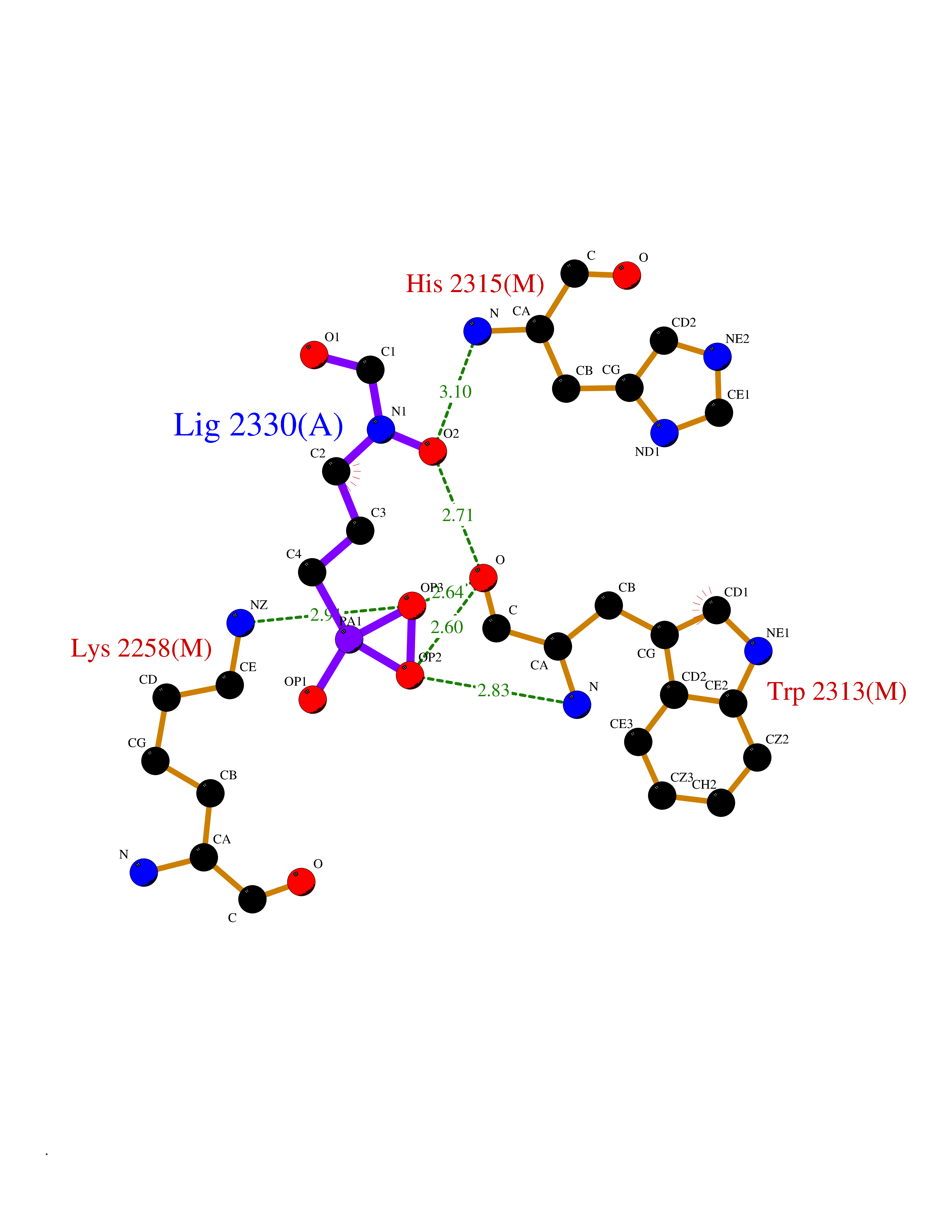 Ligplot Map 3D Binding mode Sequence SCSMPLGMESKAISDAQITASSYFTNMFATWSPSKARLHLQGRSNAWRPQVNNPKEWLQVDFQKTMKVTGVTTQGVKSLLTSMYVKEFLISSSQDGHQWTLFFQNGKVKVFQGNQDSFTPVVNSLDPPLLTRYLRIHPQSWVHQIALRMEVLGCEA Hydrogen bonds contact Hydrophobic contact | ||||
| 18 | HMG-CoA reductase (HMGCR) | 2R4F | 6.46 | |
Target general information Gen name HMGCR Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms 3-hydroxy-3-methylglutaryl-coenzyme A reductase Protein family HMG-CoA reductase family Biochemical class CH-OH donor oxidoreductase Function Transmembrane glycoprotein that is the rate-limiting enzyme in cholesterol biosynthesis as well as in the biosynthesis of nonsterol isoprenoids that are essential for normal cell function including ubiquinone and geranylgeranyl proteins. Related diseases Muscular dystrophy, limb-girdle, autosomal recessive 28 (LGMDR28) [MIM:620375]: An autosomal recessive form of limb girdle muscular dystrophy, a group of genetically heterogeneous muscular disorders that share proximal muscle weakness as the major attribute. Most limb girdle muscular dystrophies present with elevated creatinine kinase and myopathic electromyographic features. Disease is usually progressive to a variable degree, ranging from minor disability to complete inability to ambulate, and can involve the large proximal muscles, as well as axial and facial muscles. Different disease forms may exhibit skeletal muscle hypertrophy, kyphoscoliosis, and contractures or involve other muscle groups and manifest with distal weakness, cardiomyopathy, dysphagia, and respiratory difficulties. LGMDR28 is characterized by progressive muscle weakness affecting the proximal and axial muscles of the upper and lower limbs, and highly variable age at onset. Most patients have limited ambulation or become wheelchair-bound within a few decades, and respiratory insufficiency commonly occurs. {ECO:0000269|PubMed:36745799, ECO:0000269|PubMed:37167966}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03169; DB04447; DB01076; DB09061; DB00439; DB01992; DB01095; DB00227; DB14009; DB04377; DB06693; DB14011; DB00157; DB03461; DB08860; DB00175; DB01098; DB00641; DB05317; DB09270 Interacts with Q9Y5Z9; Q9Y5Z9-1 EC number EC 1.1.1.34 Uniprot keywords 3D-structure; Alternative splicing; Cholesterol biosynthesis; Cholesterol metabolism; Disease variant; Endoplasmic reticulum; Glycoprotein; Isopeptide bond; Limb-girdle muscular dystrophy; Lipid biosynthesis; Lipid metabolism; Membrane; NADP; Oxidoreductase; Peroxisome; Phosphoprotein; Proteomics identification; Reference proteome; Steroid biosynthesis; Steroid metabolism; Sterol biosynthesis; Sterol metabolism; Transmembrane; Transmembrane helix; Ubl conjugation Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 84796.9 Length 798 Aromaticity 0.05 Instability index 47.61 Isoelectric point 6.2 Charge (pH=7) -3.71 2D Binding mode Binding energy (Kcal/mol) -5.94 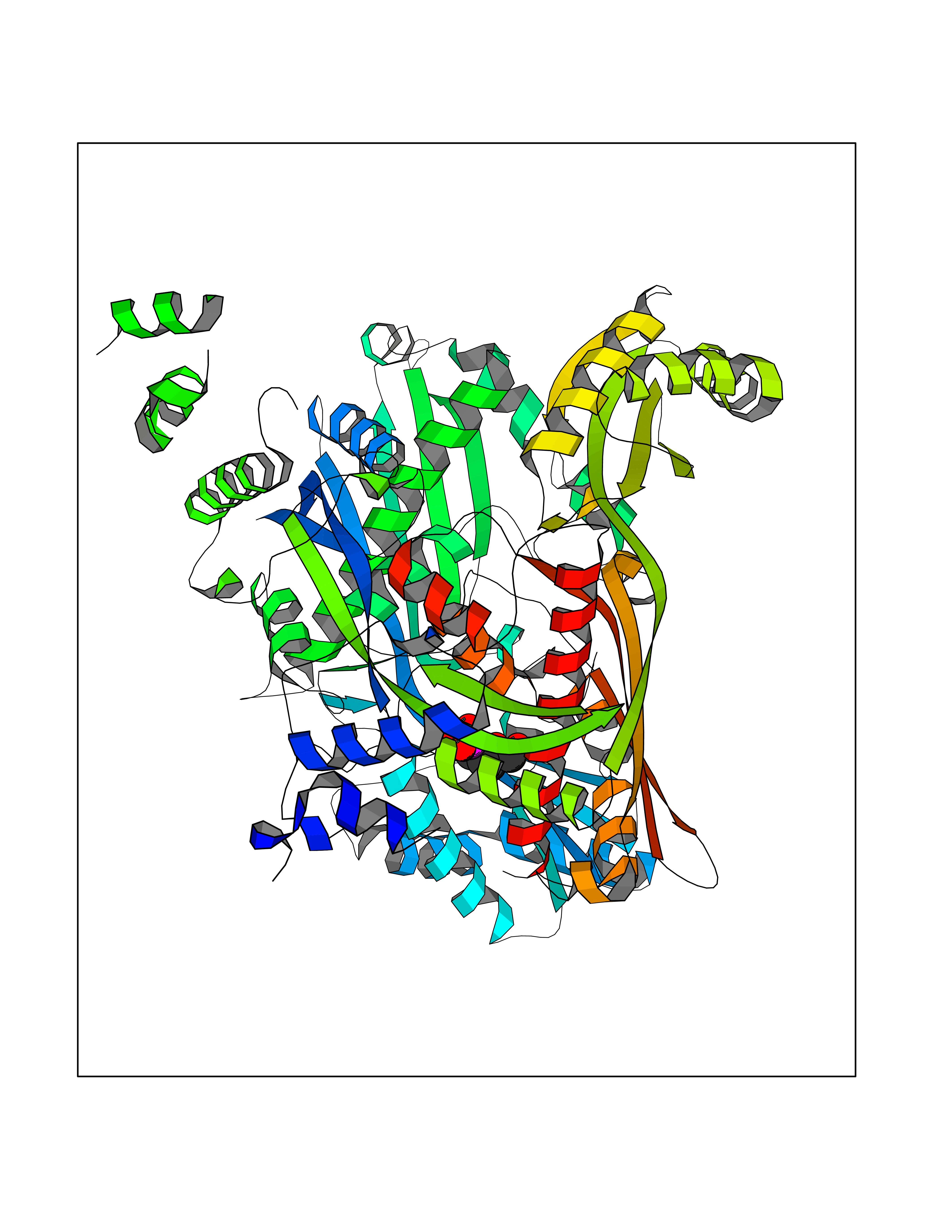 Molscript Map 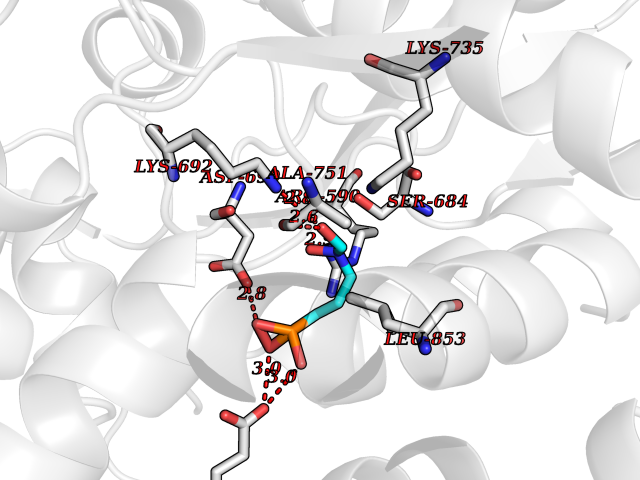 Pymol Map 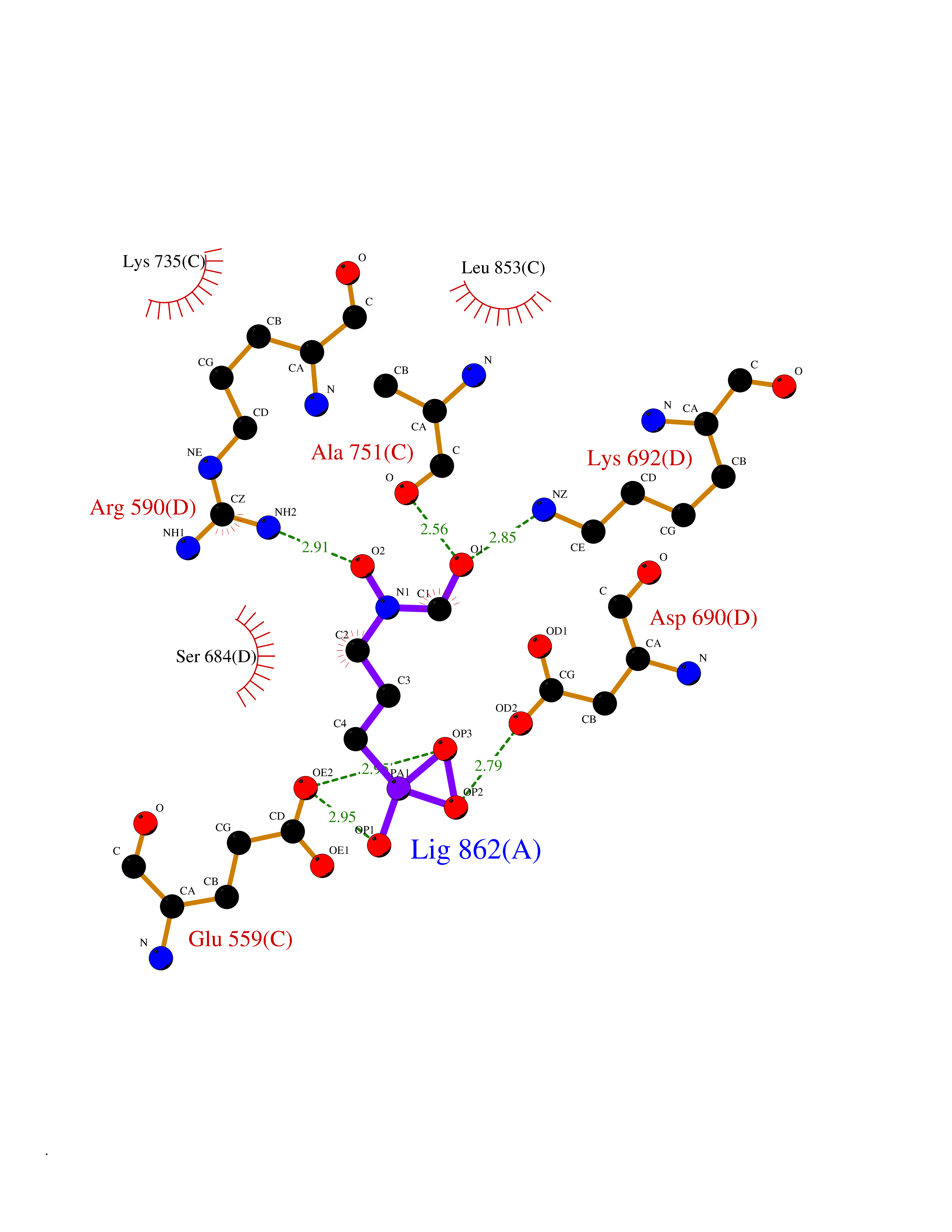 Ligplot Map 3D Binding mode Sequence GAKFLSDAEIIQLVNETLIETHERGVSIRRQLLSKKLSEPSSLQYLPYRDYNYSLVMGACCENVIGYMPIPVGVAGPLCLDEKEFQVPMATTEGCLVASTNRGCRAIGLGGGASSRVLADGMTRGPVVRLPRACDSAEVKAWLETSEGFAVIKEAFDSTSRFARLQKLHTSIAGRNLYIRFQSRSGDAMGMNMISKGTEKALSKLHEYFPEMQILAVSGNYCTDKKPAAINWIEGRGKSVVCEAVIPAKVVREVLKTTTEAMIEVNINKNLVGSAMAGSIGGYNAHAANIVTAIYIACGQDAAQNVGSSNCITLMEASGPTNEDLYISCTMPSIEIGTVGGGTNLLPQQACLQMLGVQGACKDNPGENARQLARIVCGTVMAGELSLMAALAAGPNEECLQILGNGAKFLSDAEIIQLVETLIETHERGVSIRRQLLSKKLSEPSSLQYLPYRDYNYSLVMGACCENVIGYMPIPVGVAGPLCLDEKEFQVPMATTEGCLVASTNRGCRAIGLGGGASSRVLADGMTRGPVVRLPRACDSAEVKAWLETSEGFAVIKEAFDSTSRFARLQKLHTSIAGRNLYIRFQSRSGDAMGMNMISKGTEKALSKLHEYFPEMQILAVSGNYCTDKKPAAINWIEGRGKSVVCEAVIPAKVVREVLKTTTEAMIEVNINKNLVGSAMAGSIGGYNAHAANIVTAIYIACGQDAAQNVGSSNCITLMEASGPTNEDLYISCTMPSIEIGTVGGGTNLLPQQACLQMLGVQGACKDNPGENARQLARIVCGTVMAGELSLMAALAAG Hydrogen bonds contact Hydrophobic contact | ||||
| 19 | Choline O-acetyltransferase | 2FY3 | 6.43 | |
Target general information Gen name CHAT Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Carnitine/choline acetyltransferase family Biochemical class Transferase Function Choline O-acetyltransferase activity. Related diseases Myasthenic syndrome, congenital, 6, presynaptic (CMS6) [MIM:254210]: A form of congenital myasthenic syndrome, a group of disorders characterized by failure of neuromuscular transmission, including pre-synaptic, synaptic, and post-synaptic disorders that are not of autoimmune origin. Clinical features are easy fatigability and muscle weakness affecting the axial and limb muscles (with hypotonia in early-onset forms), the ocular muscles (leading to ptosis and ophthalmoplegia), and the facial and bulbar musculature (affecting sucking and swallowing, and leading to dysphonia). The symptoms fluctuate and worsen with physical effort. CMS6 affected individuals have myasthenic symptoms since birth or early infancy, negative tests for anti-AChR antibodies, and abrupt episodic crises with increased weakness, bulbar paralysis, and apnea precipitated by undue exertion, fever, or excitement. CMS6 inheritance is autosomal recessive. {ECO:0000269|PubMed:11172068, ECO:0000269|PubMed:12756141}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00122; DB14006; DB00184 Interacts with Q6H8Q1-8; Q8N302-2; Q9NXL2-1; Q6XD76; Q9UII2; Q8TBE0; Q9UQB8-6; Q9ULD4-2; Q9NSI6-4; Q6P5X5; Q96LL4; P20807-4; O00257-3; Q6ZP82-1; O95674; Q9H3R5; Q8WUX9; Q9H2A9; Q3SX64; Q92782-2; Q14117; O14641; Q658K8; Q6UXG2-3; O00472; Q6NXG1; Q15910-2; Q8IZU1; P15407; P55318; Q06547-3; P23769-2; P23771; Q15486; Q8IV36; Q4VB01; Q53GQ0; P10809; P41134; Q9NZH6; Q8NA54; Q86U28; P17275; Q8N5Z5; Q6P597; P08727; Q14525; Q8IUC2; Q6IAA8; Q14847-2; P27338; Q9GZQ8; Q53S70; Q5JXC2; A0A0A0MR05; Q8NEH6; Q8TCY5; Q6IN84-2; Q96H12; P01106; P41271-2; P14598; Q9GZM8; Q5BJF6-2; Q9H8K7; Q9NR21-5; Q5VU43-8; Q13956; Q5SXH7-1; Q96T60; Q96I34; Q86UA1; Q15311; Q8TBY0; Q04206; P47804-3; Q9H0X6; P62899; Q66K80; Q9BY12-3; Q86SQ7-2; Q7Z6I5; Q496A3; Q7Z698; Q9C004; Q92783-2; Q8N4C7; O75528; Q15814; O15273; Q96A09; Q8WTV1; Q53NU3; Q71RG4-4; Q86WT6-2; Q9Y3Q8; Q99598; P49459; P11441; Q9H270; P19544-6; Q53FD0-2; Q3KNS6-3 EC number 2.3.1.6 Uniprot keywords 3D-structure; Acyltransferase; Alternative splicing; Congenital myasthenic syndrome; Direct protein sequencing; Disease variant; Neurotransmitter biosynthesis; Phosphoprotein; Proteomics identification; Reference proteome; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 66365.9 Length 595 Aromaticity 0.08 Instability index 53.36 Isoelectric point 8.16 Charge (pH=7) 4.64 2D Binding mode Binding energy (Kcal/mol) -6.48  Molscript Map 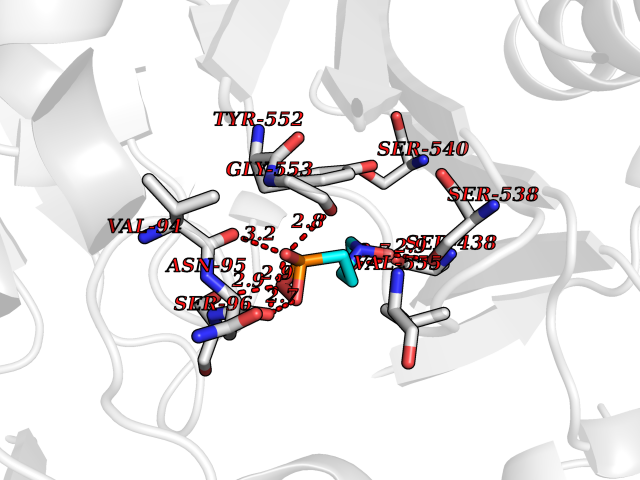 Pymol Map 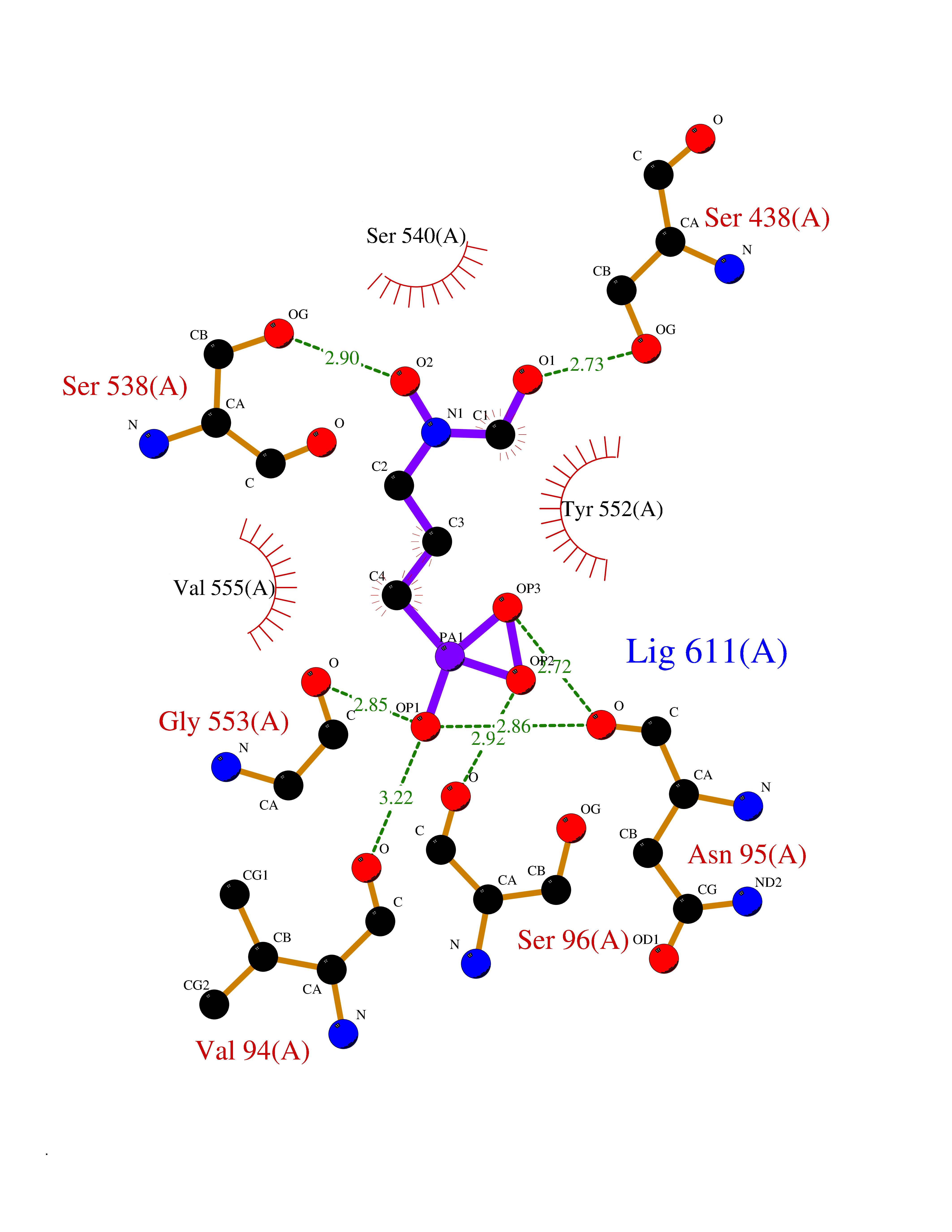 Ligplot Map 3D Binding mode Sequence SEESGLPKLPVPPLQQTLATYLQCMRHLVSEEQFRKSQAIVQQFGAPGGLGETLQQKLLERQEKTANWVSEYWLNDMYLNNRLALPVNSSPAVIFARQHFPGTDDQLRFAASLISGVLSYKALLDSHSIPTDCAKGQPLCMKQYYGLFSSYRLPGHTQDTLVAQNSSIMPEPEHVIVACCNQFFVLDVVINFRRLSEGDLFTQLRKIVKMASNAAARLPPIGLLTSDGRSEWAEARTVLVKDSTNRDSLDMIERCICLVCLDAPGGVELSDTHRALQLLHGGGYSKNGANRWYDKSLQFVVGRDGTCGVVCEHSPFDGIVLVQCTEHLLKHMTQPELVRSPMVPLPAPRRLRWKCSPEIQGHLASSAEKLQRIVKNLDFIVYKFDNYGKTFIKKQKCSPDAFIQVALQLAFYRLHRRLVPTYESASIRRFQEGRVDNIRSATPEALAFVRAVTDHKAAVPASEKLLLLKDAIRAQTAYTVMAITGMAIDNHLLALRELARAMCAALPEMFMDETYLMSNRFVLSTSQVPTTTEMFCCYGPVVPNGYGACYNPQPETILFCISSFHSCAATSSSKFAKAVEESLIDMRDLCSLLPP Hydrogen bonds contact Hydrophobic contact | ||||
| 20 | Interleukin 21 receptor (IL21R) | 6PLH | 6.43 | |
Target general information Gen name IL21R Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms UNQ3121/PRO10273; Novel interleukin receptor; NILR; Interleukin-21 receptor; IL21 receptor; IL-21R; IL-21 receptor; CD360 Protein family Type I cytokine receptor family, Type 4 subfamily Biochemical class Cytokine receptor Function This is a receptor for interleukin-21. Related diseases Immunodeficiency 56 (IMD56) [MIM:615207]: An autosomal recessive primary immunodeficiency characterized by B- and T-cell defects and variable dysfunction of NK cells. Patients tend to have normal numbers of lymphocytes, but show defective class-switched B-cells, low IgG, defective antibody response, and defective T-cell responses to certain antigens. {ECO:0000269|PubMed:23440042}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Chromosomal aberrations involving IL21R is a cause of B-cell non-Hodgkin lymphomas (B-cell NHL). Translocation t(3;16)(q27;p11), with BCL6. Drugs (DrugBank ID) NA Interacts with P29972 EC number NA Uniprot keywords 3D-structure; Chromosomal rearrangement; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; Membrane; Proteomics identification; Receptor; Reference proteome; Repeat; Signal; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A,C,B Molecular weight (Da) 48376.5 Length 446 Aromaticity 0.1 Instability index 43.94 Isoelectric point 8.24 Charge (pH=7) 3.56 2D Binding mode Binding energy (Kcal/mol) -6.56 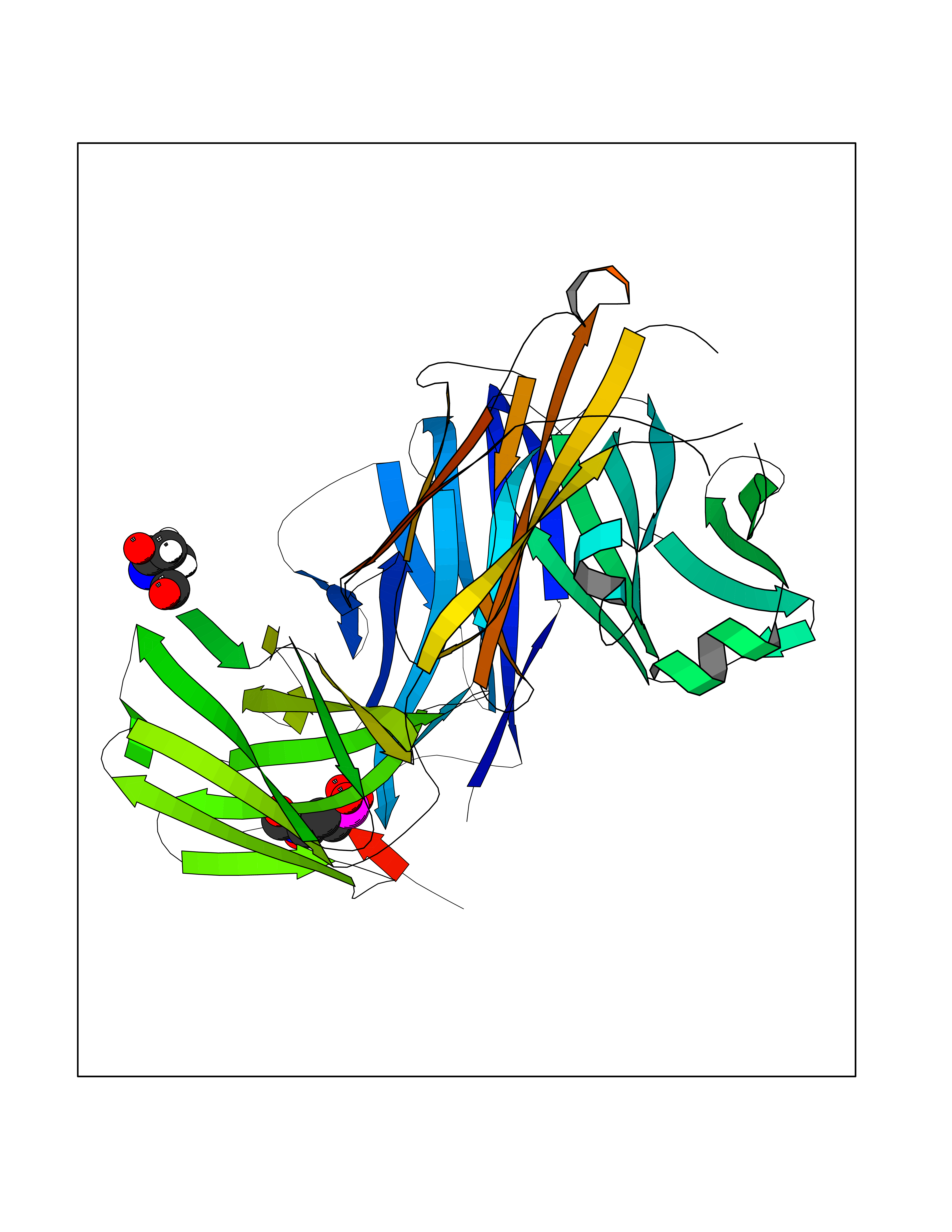 Molscript Map 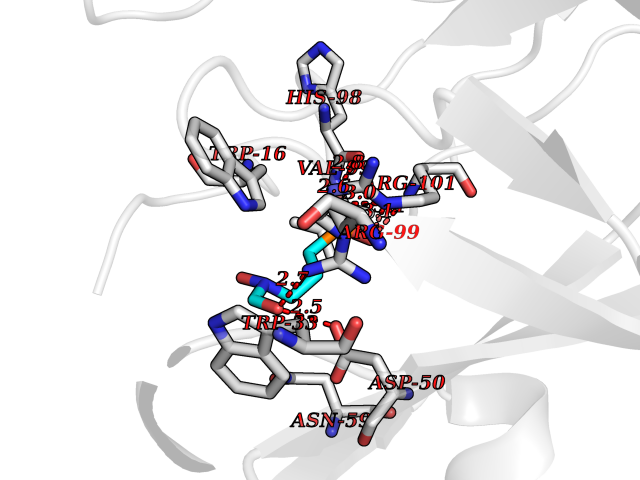 Pymol Map 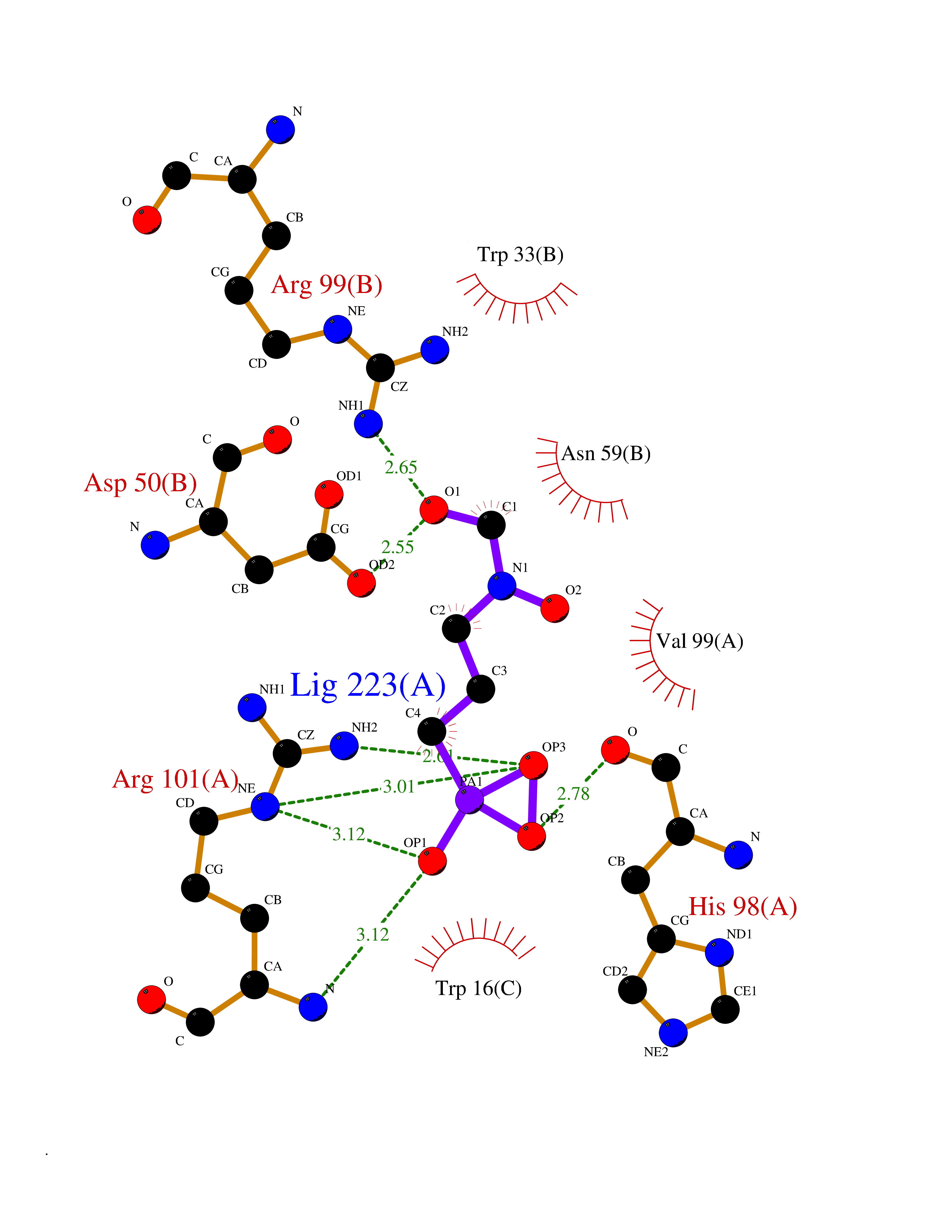 Ligplot Map 3D Binding mode Sequence DVVMTHTPLSLPVSLGDQASISCRSSQSLVHSNGNTYLHWYLQKPGQSPKLLIYKVSNRFSGVPDRFSGSGSGADFTLKISRVEAEDLGVYFCSQSTHVPRTFGGGTKLEIKRADAAPTVSIFPPSSEQLTSGGASVVCFLNNFYPKDINVKWKIDGSERQNGVLNSWTDQDSKDSTYSMSSTLTLTKDEYERHNSYTCEATHKTSTSPIVKSFNRNECXVHLQQPGADLVKPGASVKMSCKASGYTFTSYWITWVKLRPGQGLEWIGDIYPGSGSTNFIEKFKSKATLTVDTSSSTAYMQLRSLTSEDSAVYYCARRGHGNYEDYWGQGTTLIVSSAKTTAPSVYPLAPVCGTGSSVTLGCLVKGYFPEPVTLTWNSGSLSSGVHTFPAVLQSDLYTLSSSVTVTSSTWPSQSITCNVAHPASSTKVDKKIEPRGPTTWSEWSDP Hydrogen bonds contact Hydrophobic contact | ||||