Ligand
Structure
Job ID
3e9a1e6c4d6c315fe174d22ce9f4a70c
Job name
Rodriguez_task5
Time
2024-06-13 17:24:44
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 1 | Endothelin-converting enzyme 1 (ECE1) | 3DWB | 8.95 | |
Target general information Gen name ECE1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms ECE-1 Protein family Peptidase M13 family Biochemical class Peptidase Function Converts big endothelin-1 to endothelin-1. Related diseases Hirschsprung disease, cardiac defects, and autonomic dysfunction (HCAD) [MIM:613870]: A disorder characterized by skip-lesions Hirschsprung disease, craniofacial abnormalities and other dysmorphic features, cardiac defects including ductus arteriosus, small subaortic ventricular septal defect, small atrial septal defect, and autonomic dysfunction. {ECO:0000269|PubMed:9915973}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07171 Interacts with P49760; A8MQ03; Q8IUG1; P60370; P60410 EC number EC 3.4.24.71 Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disease variant; Disulfide bond; Glycoprotein; Hirschsprung disease; Hydrolase; Membrane; Metal-binding; Metalloprotease; Phosphoprotein; Protease; Proteomics identification; Reference proteome; Signal-anchor; Transmembrane; Transmembrane helix; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 75247.9 Length 660 Aromaticity 0.12 Instability index 46.29 Isoelectric point 5.33 Charge (pH=7) -18.3 2D Binding mode Binding energy (Kcal/mol) -8.67 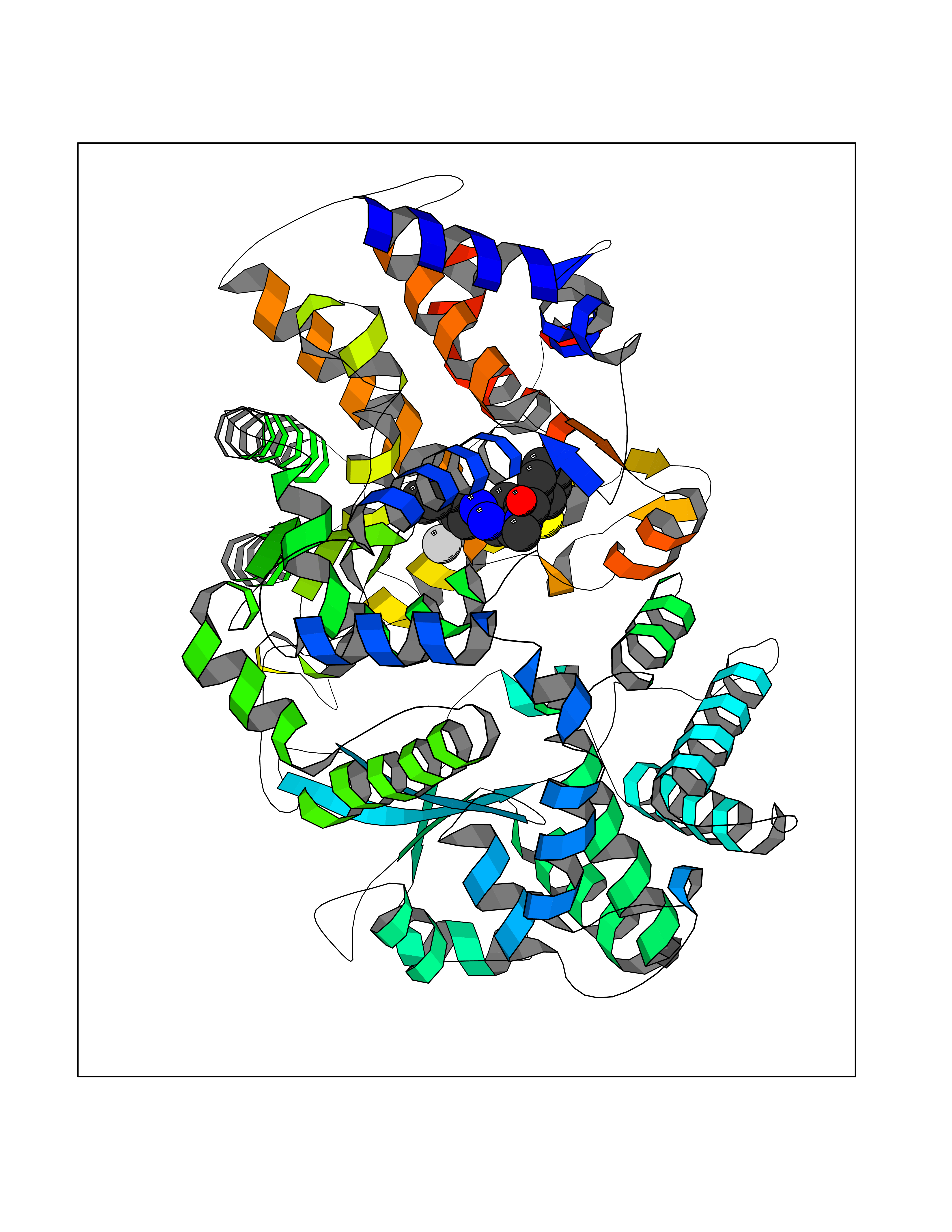 Molscript Map 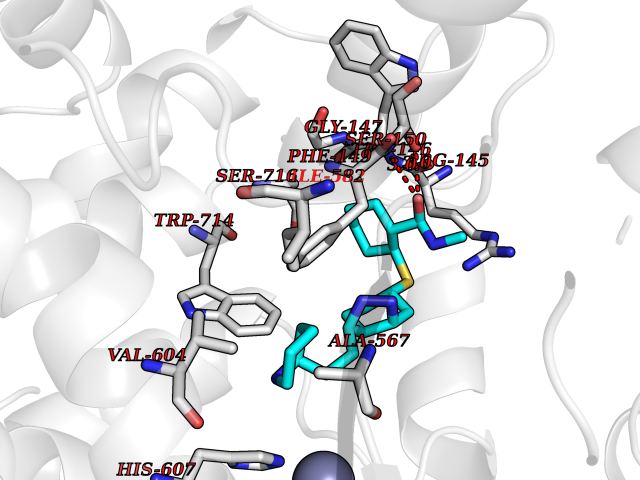 Pymol Map 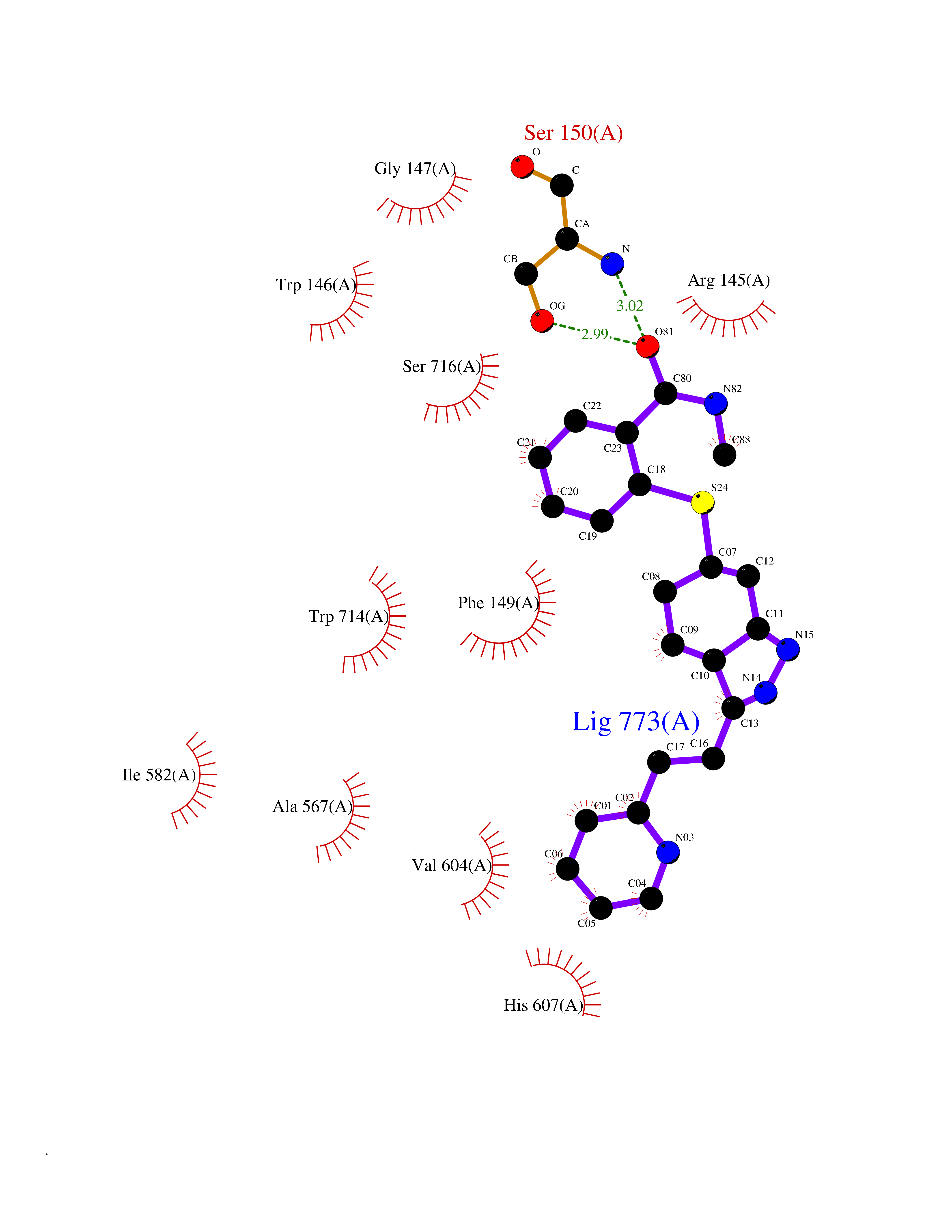 Ligplot Map 3D Binding mode Sequence SEACVSVTSSILSSMDPTVDPCHDFFSYACGGWIKANPVPDGHSRWGTFSNLWEHNQAIIKHLLENSTASVSEAERKAQVYYRACMNETRIEELRAKPLMELIERLGGWNITGPWAKDNFQDTLQVVTAHYRTSPFFSVYVSADSKNSNSNVIQVDQSGLGLPSRDYYLNKTENEKVLTGYLNYMVQLGKLLGGGDEEAIRPQMQQILDFETALANITIPQEKRRDEELIYHKVTAAELQTLAPAINWLPFLNTIFYPVEINESEPIVVYDKEYLEQISTLINTTDRCLLNNYMIWNLVRKTSSFLDQRFQDADEKFMEVMWKFCVSDTENNLGFALGPMFVKATFAEDSKSIATEIILEIKKAFEESLSTLKWMDEETRKSAKEKADAIYNMIGYPNFIMDPKELDKVFNDYTAVPDLYFENAMRFFNFSWRVTADQLRKAPNRDQWSMTPPMVNAYYSPTKNEIVFPAGILQAPFYTRSSPKALNFGGIGVVVGHELTHAFDDQGREYDKDGNLRPWWKNSSVEAFKRQTECMVEQYSNYSVNGEPVNGRHTLGENIADNGGLKAAYRAYQNWVKKNGAEHSLPTLGLTNNQLFFLGFAQVWCSVRTPESSHEGLITDPHSPSRFRVIGSLSNSKEFSEHFRCPPGSPMNPPHKCEVW Hydrogen bonds contact Hydrophobic contact | ||||
| 2 | Purine nucleoside phosphorylase (PNP) | 4EAR | 8.84 | |
Target general information Gen name PNP Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms PNP; Inosine phosphorylase Protein family PNP/MTAP phosphorylase family Biochemical class Pentosyltransferase Function The purine nucleoside phosphorylases catalyze the phosphorolytic breakdown of the N-glycosidic bond in the beta- (deoxy)ribonucleoside molecules, with the formation of the corresponding free purine bases and pentose-1-phosphate. Related diseases Purine nucleoside phosphorylase deficiency (PNPD) [MIM:613179]: A disorder that interrupts both the catabolism of inosine into hypoxanthine and guanosine into guanine, and leads to the accumulation of guanosine, inosine, and their deoxified by-products. The main clinical presentation is recurrent infections due to severe T-cell immunodeficiency. Some patients also have neurologic impairment. {ECO:0000269|PubMed:1384322, ECO:0000269|PubMed:3029074, ECO:0000269|PubMed:8931706}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03881; DB03551; DB02222; DB02391; DB03609; DB01667; DB04260; DB02796; DB04753; DB00640; DB00242; DB00900; DB06185; DB02377; DB02857; DB04754; DB04757; DB04076; DB02230; DB04335; DB02568; DB03101 Interacts with P05067; Q9UQM7; O14576-2; P06241; P14136; Q92993-2; Q9BXM7; P00491; P17612; P63000; Q92673; Q15583 EC number EC 2.4.2.1 Uniprot keywords 3D-structure; Acetylation; Cytoplasm; Direct protein sequencing; Disease variant; Glycosyltransferase; Phosphoprotein; Proteomics identification; Purine salvage; Reference proteome; Transferase Protein physicochemical properties Chain ID A,B,C Molecular weight (Da) 31849.2 Length 288 Aromaticity 0.1 Instability index 34.77 Isoelectric point 6.42 Charge (pH=7) -1.63 2D Binding mode Binding energy (Kcal/mol) -10.36  Molscript Map 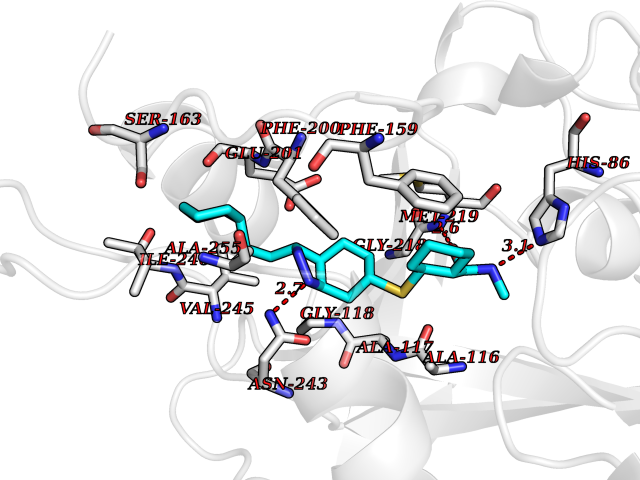 Pymol Map 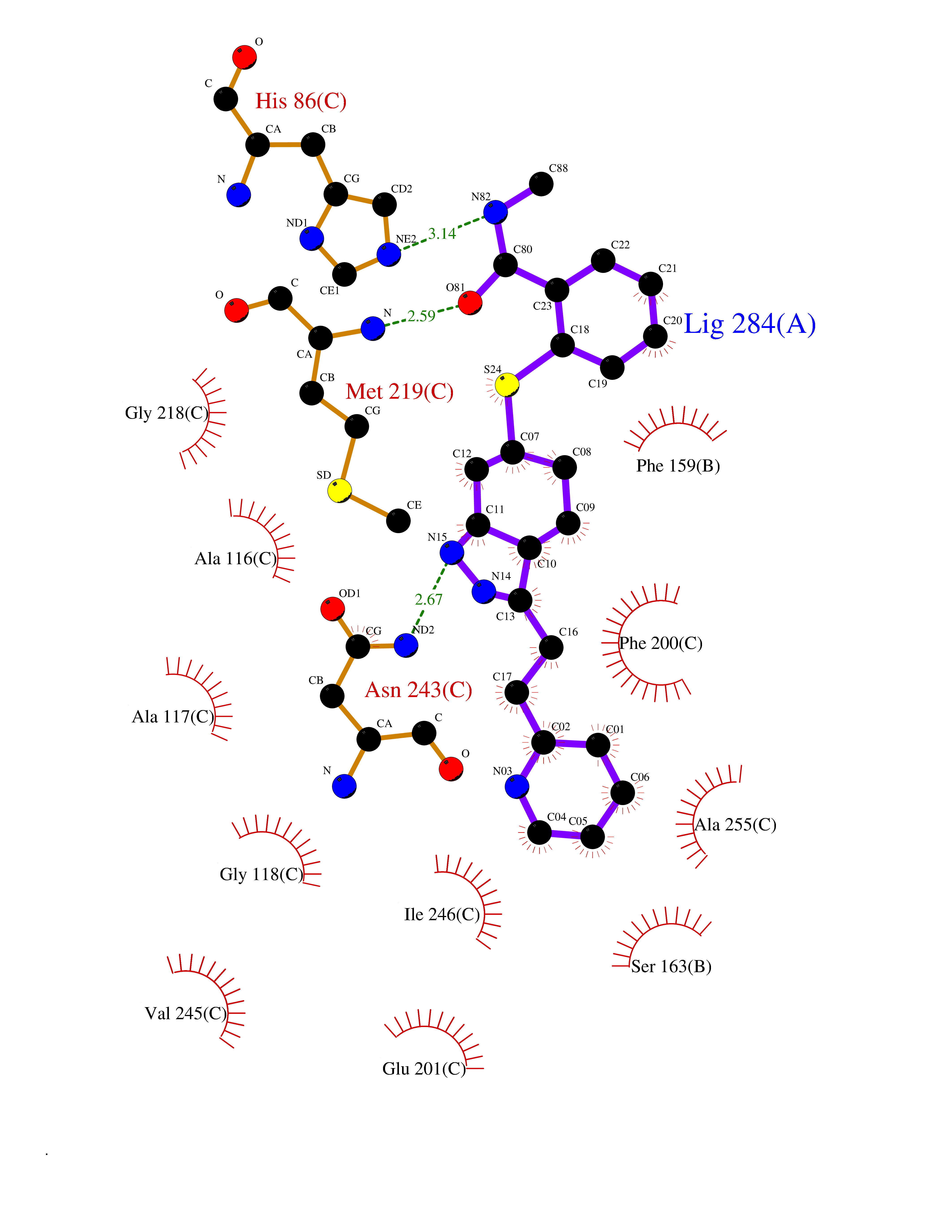 Ligplot Map 3D Binding mode Sequence GYTYEDYKNTAEYLLSHTKHRPQVAIICGSGLGGLTDKLTQAQIFDYSEIPNFPRSTVPGHAGRLVFGFLNGRACVMMQGRFHMYEGYPLYKVTFPVRVFHLLGVDTLVVTNAAGGLNPKFEVGDIMLIRDHINLPGFSGQNPLRGPNDERFGDRFPAMSDAYDRTMRQRALSTYKQMGEQRELQEGTYVMVAGPSFETVAECRVLQKLGADAVGMSTVPEVIVARHCGLRVFGFSLITNKVIMDYESLEKANXEEVLAAGKQAAQKLEQFVSILMASIDRFPAMSDA Hydrogen bonds contact Hydrophobic contact | ||||
| 3 | Enteropeptidase (TMPRSS15) | 6ZOV | 8.82 | |
Target general information Gen name TMPRSS15 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Transmembrane protease serine 15; TMPRSS15; Serine protease 7; Enterokinase Protein family Peptidase S1 family Biochemical class Peptidase Function Responsible for initiating activation of pancreatic proteolytic proenzymes (trypsin, chymotrypsin and carboxypeptidase A). It catalyzes the conversion of trypsinogen to trypsin which in turn activates other proenzymes including chymotrypsinogen, procarboxypeptidases, and proelastases. Related diseases Enterokinase deficiency (ENTKD) [MIM:226200]: Life-threatening intestinal malabsorption disorder characterized by diarrhea and failure to thrive. {ECO:0000269|PubMed:11719902}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number EC 3.4.21.9 Uniprot keywords 3D-structure; Disulfide bond; Glycoprotein; Hydrolase; Lipoprotein; Membrane; Myristate; Protease; Proteomics identification; Reference proteome; Repeat; Serine protease; Signal-anchor; Transmembrane; Transmembrane helix; Zymogen Protein physicochemical properties Chain ID A Molecular weight (Da) 26220.3 Length 234 Aromaticity 0.1 Instability index 50.13 Isoelectric point 4.82 Charge (pH=7) -9.93 2D Binding mode Binding energy (Kcal/mol) -8.68  Molscript Map 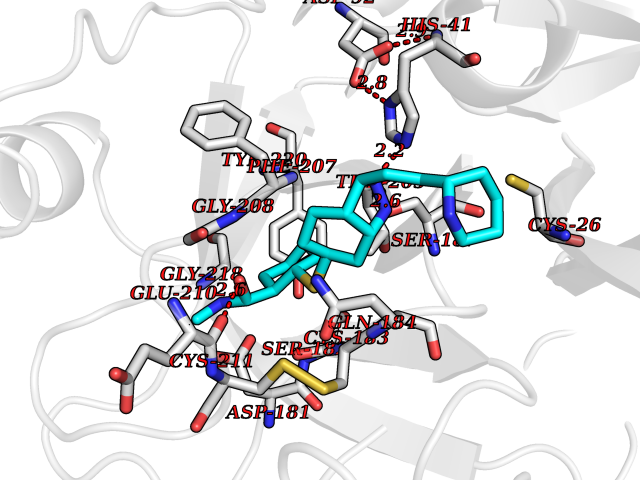 Pymol Map 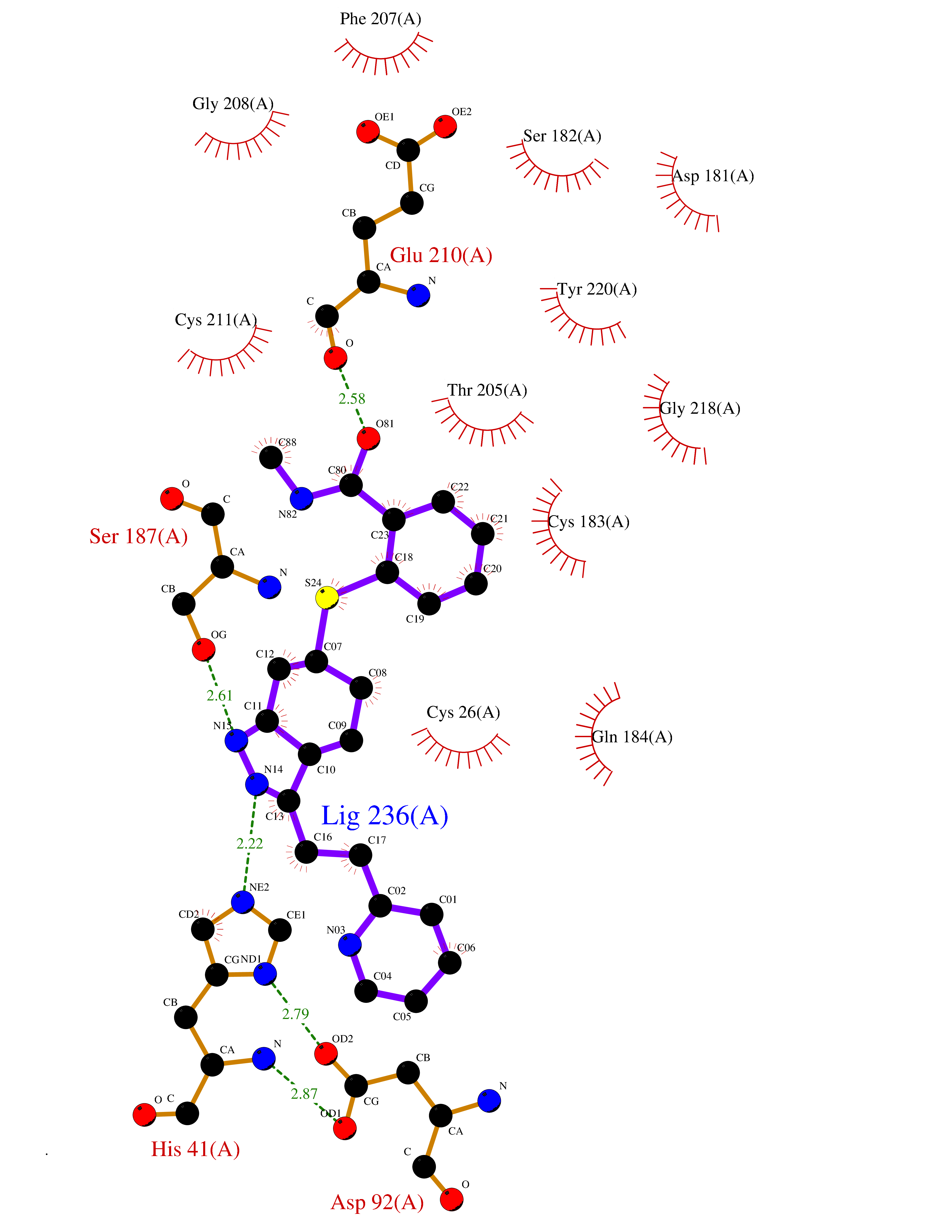 Ligplot Map 3D Binding mode Sequence IVGGSDAKEGAWPWVVGLYYDDRLLCGASLVSSDWLVSAAHCVYGRNLEPSKWTAILGLHMKSNLTSPQTVPRLIDEIVINPHYNRRRKDNDIAMMHLEFKVNYTDYIQPISLPEENQVFPPGRNCSIAGWGTVVYQGTTADILQEADVPLLSNERCQQQMPEYNITENMICAGYEEGGIDSCQGDSGGPLMCQENNRWFLAGVTSFGYECALPNRPGVYARVSRFTEWIQSFL Hydrogen bonds contact Hydrophobic contact | ||||
| 4 | Cytochrome P450 1A2 | 2HI4 | 8.71 | |
Target general information Gen name CYP1A2 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Cytochrome P450 family Biochemical class Oxidoreductase Function Aromatase activity.Caffeine oxidase activity.Demethylase activity.Electron carrier activity.Enzyme binding.Heme binding.Iron ion binding.Monooxygenase activity.Oxidoreductase activity.Oxidoreductase activity, acting on paired donors, with incorporation or reduction of molecular oxygen, reduced flavin or flavoprotein as one donor, and incorporation of one atom of oxygen.Oxygen binding. Related diseases Myeloperoxidase deficiency (MPOD) [MIM:254600]: A disorder characterized by decreased myeloperoxidase activity in neutrophils and monocytes that results in disseminated candidiasis. {ECO:0000269|PubMed:37198333, ECO:0000269|PubMed:7904599, ECO:0000269|PubMed:8142659, ECO:0000269|PubMed:8621627, ECO:0000269|PubMed:9354683, ECO:0000269|PubMed:9637725}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08496; DB01667; DB14132; DB04356; DB02489; DB11932; DB12001; DB05812; DB13573; DB01418; DB00316; DB15568; DB06594; DB00518; DB05396; DB00969; DB07453; DB01424; DB01223; DB01118; DB00321; DB00261; DB01217; DB01435; DB06605; DB05676; DB06413; DB06216; DB01072; DB15011; DB06442; DB06626; DB00993; DB00972; DB13203; DB05015; DB16703; DB06769; DB01086; DB06770; DB06771; DB06732; DB00195; DB04889; DB11967; DB13975; DB00188; DB12151; DB01558; DB14018; DB13812; DB00201; DB09061; DB14737; DB11791; DB06774; DB00564; DB06016; DB01136; DB12814; DB00477; DB00356; DB01166; DB00501; DB01012; DB00568; DB00827; DB00537; DB00215; DB12499; DB14025; DB00349; DB01242; DB00575; DB00758; DB00363; DB00286; DB11672; DB14635; DB00924; DB08912; DB00851; DB06292; DB01254; DB01609; DB01151; DB16650; DB12161; DB01191; DB00633; DB11994; DB00586; DB11511; DB12945; DB00280; DB01184; DB09167; DB05928; DB01142; DB09273; DB00470; DB00476; DB00625; DB15444; DB06210; DB13874; DB11718; DB00467; DB11404; DB00530; DB00783; DB13952; DB13953; DB13954; DB13955; DB13956; DB00655; DB04574; DB13592; DB00330; DB00898; DB00977; DB00773; DB01628; DB00927; DB04854; DB01482; DB00574; DB12265; DB15669; DB01195; DB08972; DB04841; DB00544; DB00472; DB00499; DB00176; DB01320; DB00998; DB14029; DB06160; DB01044; DB01241; DB01155; DB01645; DB01381; DB00986; DB00365; DB00400; DB05708; DB00629; DB00502; DB01094; DB14999; DB04076; DB11737; DB00619; DB00458; DB11564; DB01306; DB09456; DB09564; DB01307; DB00047; DB01309; DB00030; DB00046; DB11567; DB00071; DB11568; DB05258; DB00034; DB00105; DB15131; DB00011; DB00018; DB00069; DB00060; DB00068; DB00033; DB00951; DB11757; DB09570; DB01026; DB01097; DB16217; DB09078; DB01002; DB05667; DB00281; DB12406; DB09198; DB04948; DB00978; DB06448; DB16220; DB01601; DB00455; DB04871; DB06077; DB01283; DB00772; DB00934; DB06234; DB14009; DB00784; DB01065; DB00170; DB00454; DB00532; DB00333; DB00763; DB00553; DB01028; DB09241; DB01233; DB00379; DB06148; DB01388; DB06595; DB00370; DB16236; DB00745; DB11763; DB00218; DB06510; DB14011; DB00461; DB00607; DB00779; DB00788; DB06600; DB00238; DB06803; DB00184; DB01115; DB11793; DB00435; DB05115; DB00717; DB01059; DB00540; DB05990; DB01165; DB00334; DB16267; DB00338; DB00904; DB11632; DB11443; DB01173; DB11837; DB09330; DB01303; DB11697; DB00377; DB00715; DB06589; DB11774; DB00487; DB00008; DB00022; DB09122; DB13634; DB00806; DB11198; DB08883; DB00850; DB03783; DB01174; DB00388; DB00252; DB11450; DB01100; DB13823; DB04951; DB17472; DB11642; DB08910; DB15822; DB01058; DB01087; DB00794; DB00420; DB09288; DB01182; DB06479; DB00818; DB00571; DB13449; DB11892; DB04216; DB00908; DB00468; DB01129; DB00980; DB09290; DB00863; DB01367; DB00409; DB02709; DB13174; DB01045; DB11753; DB00740; DB14924; DB00503; DB00533; DB01656; DB15119; DB00268; DB00296; DB00412; DB00817; DB12332; DB13772; DB06654; DB11491; DB00418; DB01037; DB11689; DB06290; DB13261; DB15093; DB00052; DB00398; DB01208; DB09118; DB00428; DB06820; DB00382; DB00675; DB06083; DB09071; DB05488; DB09256; DB01079; DB01405; DB00857; DB08880; DB11712; DB01412; DB00277; DB00730; DB01623; DB00208; DB06137; DB00697; DB01056; DB06264; DB00752; DB00384; DB12245; DB00831; DB15442; DB00440; DB00685; DB08867; DB14989; DB13609; DB06235; DB00313; DB08881; DB00661; DB09185; DB12026; DB00682; DB02134; DB00549; DB00744; DB00315; DB00425; DB09225; DB09120 Interacts with O95870 EC number 1.14.14.1; 4.2.1.152 Uniprot keywords 3D-structure; Direct protein sequencing; Endoplasmic reticulum; Fatty acid metabolism; Glycoprotein; Heme; Iron; Lipid metabolism; Lyase; Membrane; Metal-binding; Microsome; Monooxygenase; Oxidoreductase; Proteomics identification; Reference proteome; Steroid metabolism; Sterol metabolism Protein physicochemical properties Chain ID A Molecular weight (Da) 54475 Length 480 Aromaticity 0.1 Instability index 40.43 Isoelectric point 9.16 Charge (pH=7) 9.89 2D Binding mode Binding energy (Kcal/mol) -9.56 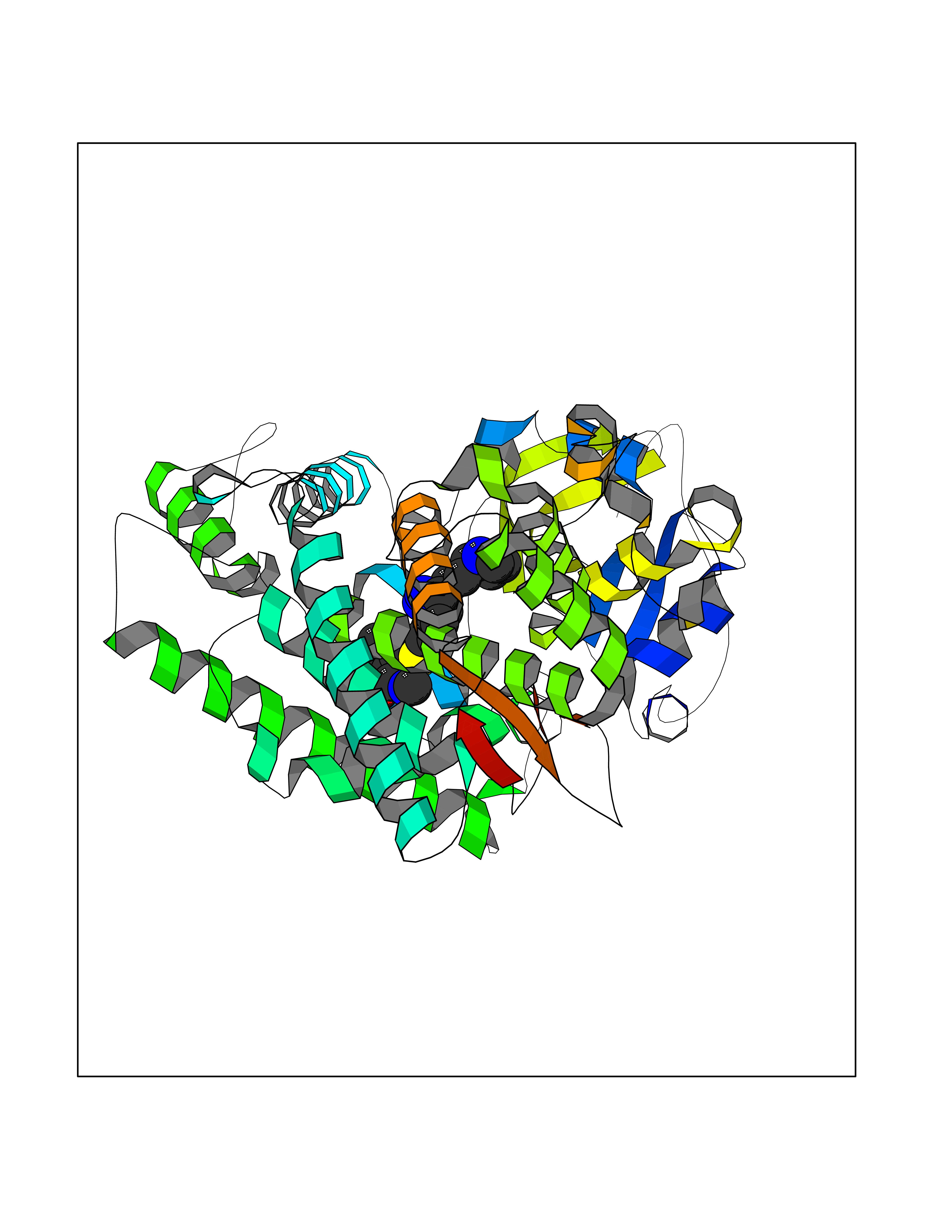 Molscript Map 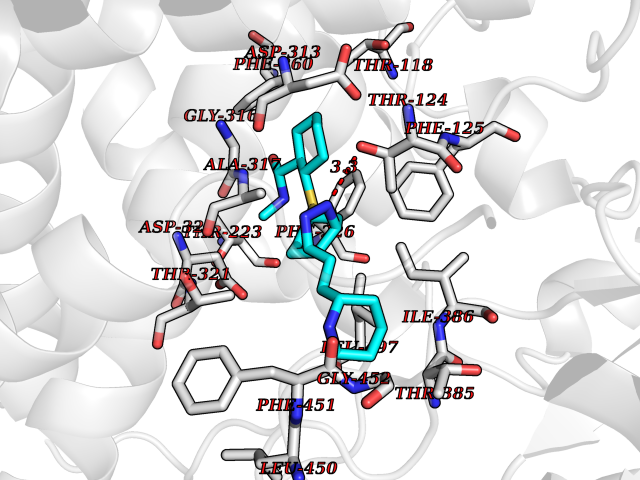 Pymol Map 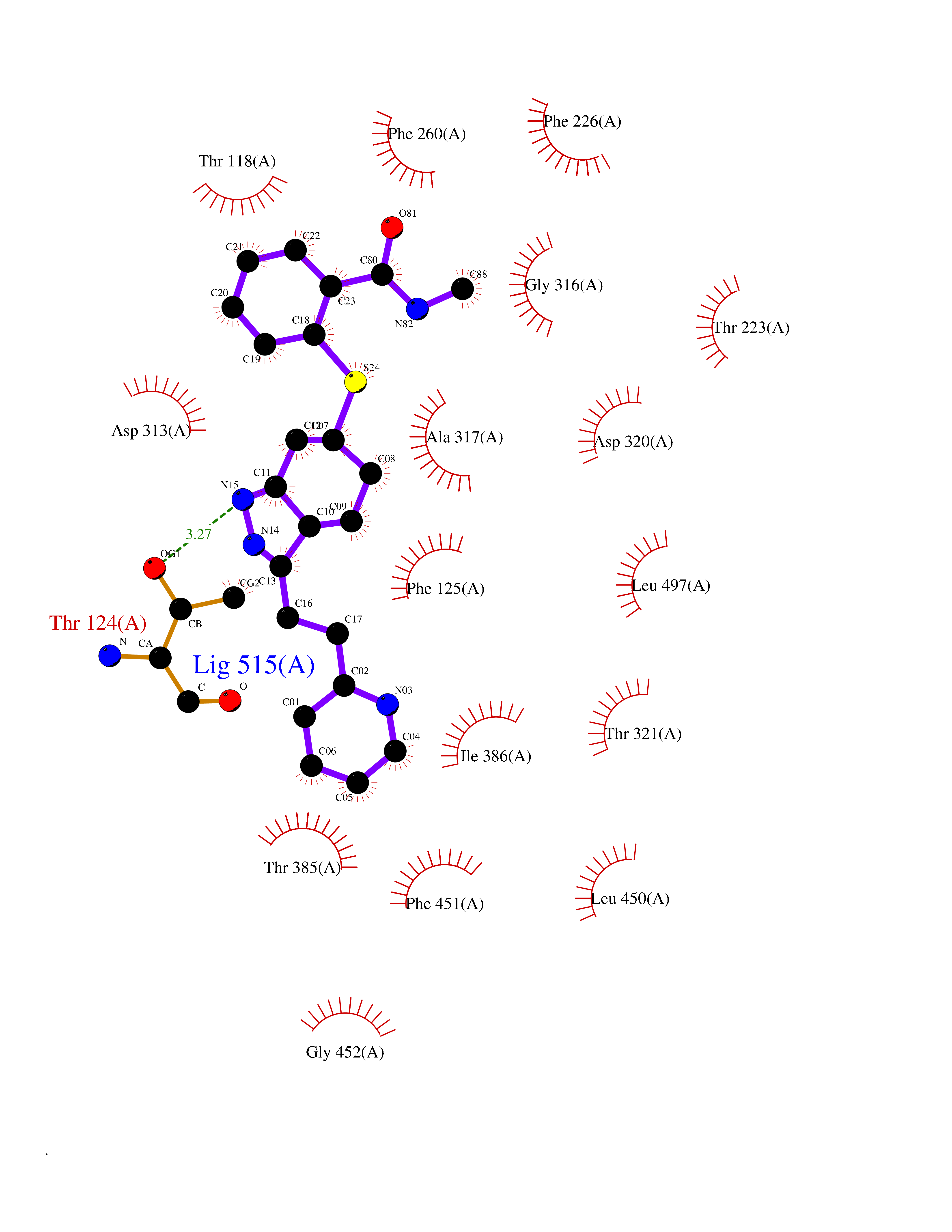 Ligplot Map 3D Binding mode Sequence RVPKGLKSPPEPWGWPLLGHVLTLGKNPHLALSRMSQRYGDVLQIRIGSTPVLVLSRLDTIRQALVRQGDDFKGRPDLYTSTLITDGQSLTFSTDSGPVWAARRRLAQNALNTFSIASDPASSSSCYLEEHVSKEAKALISRLQELMAGPGHFDPYNQVVVSVANVIGAMCFGQHFPESSDEMLSLVKNTHEFVETASSGNPLDFFPILRYLPNPALQRFKAFNQRFLWFLQKTVQEHYQDFDKNSVRDITGALFKHSKKGPRASGNLIPQEKIVNLVNDIFGAGFDTVTTAISWSLMYLVTKPEIQRKIQKELDTVIGRERRPRLSDRPQLPYLEAFILETFRHSSFLPFTIPHSTTRDTTLNGFYIPKKCCVFVNQWQVNHDPELWEDPSEFRPERFLTADGTAINKPLSEKMMLFGMGKRRCIGEVLAKWEIFLFLAILLQQLEFSVPPGVKVDLTPIYGLTMKHARCEHVQARRFS Hydrogen bonds contact Hydrophobic contact | ||||
| 5 | Fatty acid synthase (FASN) | 3TJM | 8.71 | |
Target general information Gen name FASN Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Yeast fatty acid synthase; Fatty-acyl-CoA synthase; Fatty acyl-CoA synthetase enzyme; FAS Protein family NA Biochemical class Acyltransferase Function Fatty acid synthetase catalyzes the formation of long-chain fatty acids from acetyl-CoA, malonyl-CoA and NADPH. This multifunctional protein has 7 catalytic activities as an acyl carrier protein. Related diseases Glycine encephalopathy 2 (GCE2) [MIM:620398]: A form of glycine encephalopathy, a metabolic disorder characterized by a high concentration of glycine in the body fluids. Affected individuals typically have severe neurological symptoms, including seizure, lethargy, and muscular hypotonia soon after birth. Most of them die within the neonatal period. Atypical cases have later disease onset and less severely affected psychomotor development. {ECO:0000269|PubMed:10873393, ECO:0000269|PubMed:11286506, ECO:0000269|PubMed:16051266, ECO:0000269|PubMed:26371980, ECO:0000269|PubMed:28244183, ECO:0000269|PubMed:8005589, ECO:0000269|PubMed:9600239, ECO:0000269|PubMed:9621520}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01034; DB01083 Interacts with Q15848; Q16665; P42858; Q8IV20; Q8TBB1; PRO_0000045603 [Q99IB8] EC number EC 2.3.1.85 Uniprot keywords 3D-structure; Acetylation; Cytoplasm; Direct protein sequencing; Fatty acid biosynthesis; Fatty acid metabolism; Hydrolase; Isopeptide bond; Lipid biosynthesis; Lipid metabolism; Lyase; Multifunctional enzyme; NAD; NADP; Oxidoreductase; Phosphopantetheine; Phosphoprotein; Proteomics identification; Pyridoxal phosphate; Reference proteome; S-nitrosylation; Transferase; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 30174.9 Length 275 Aromaticity 0.09 Instability index 43.28 Isoelectric point 5.92 Charge (pH=7) -5.4 2D Binding mode Binding energy (Kcal/mol) -10.02 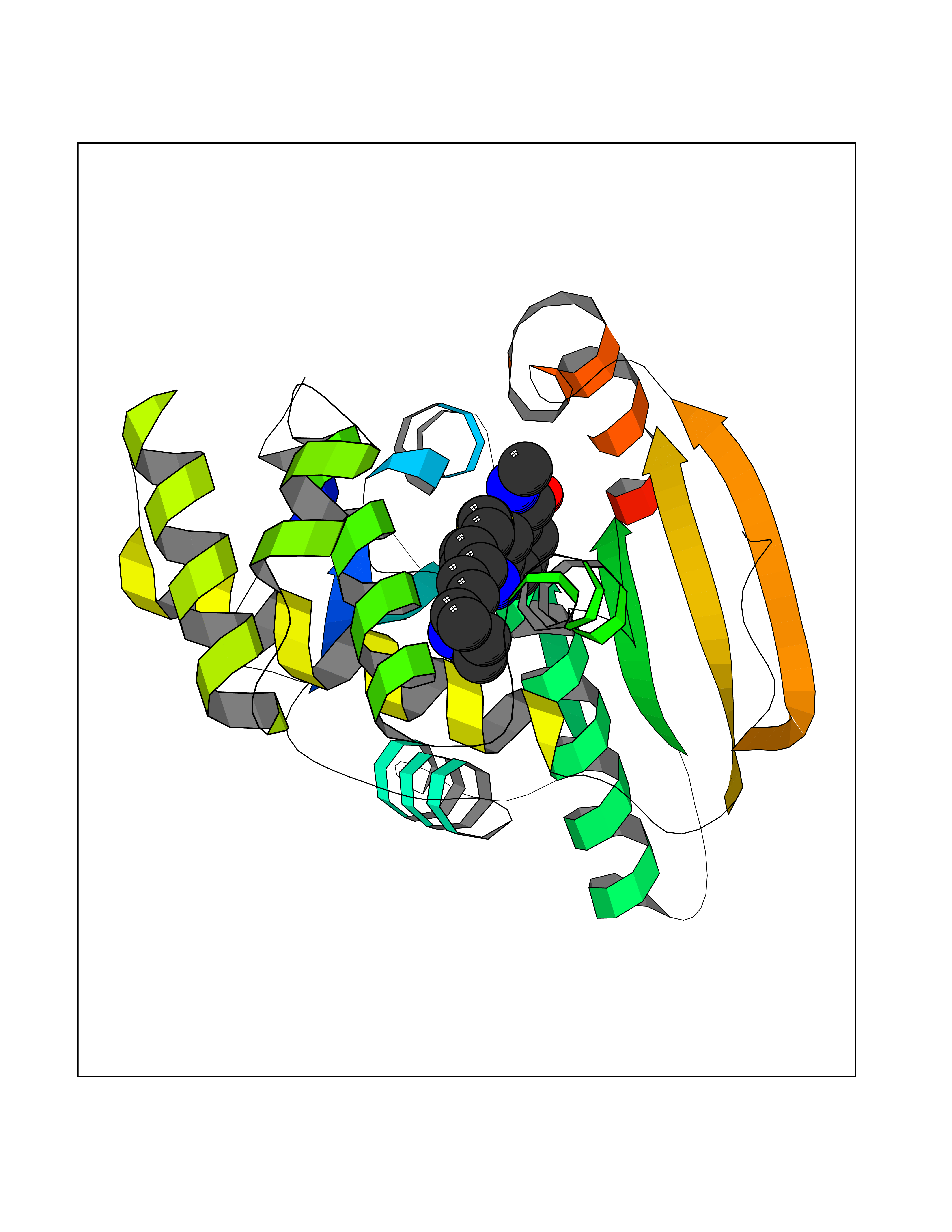 Molscript Map 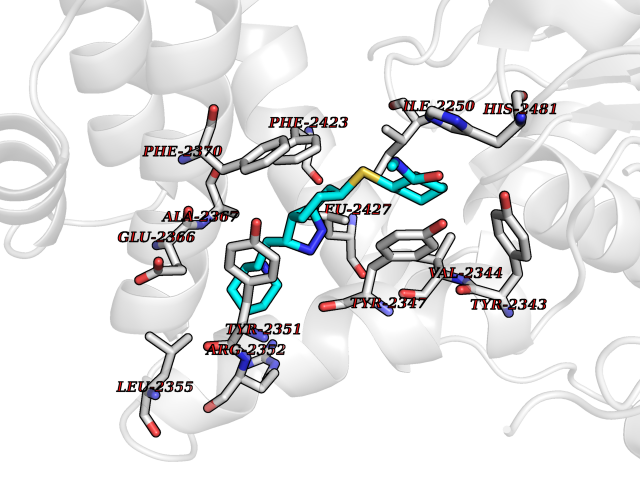 Pymol Map 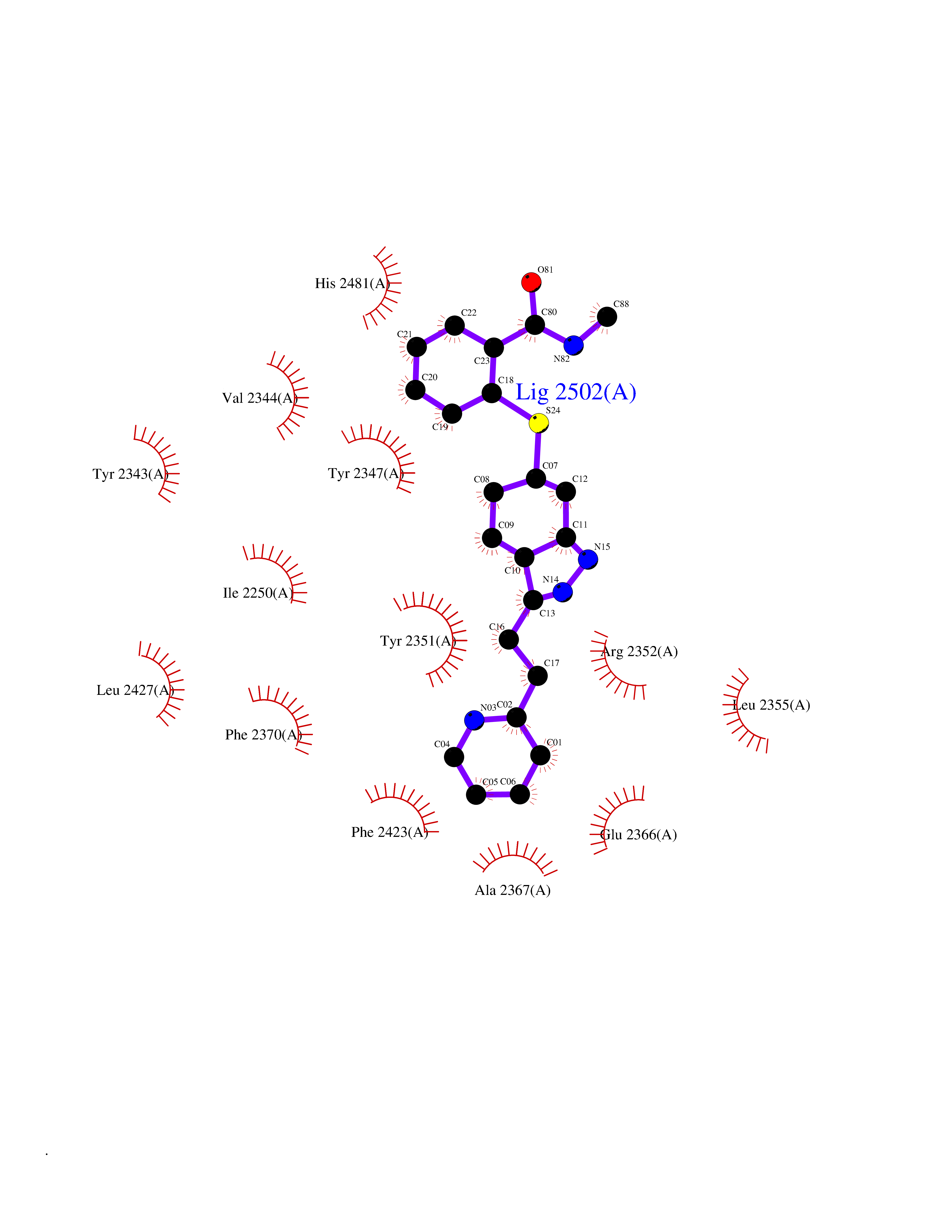 Ligplot Map 3D Binding mode Sequence NLRSLLVNPEGPTLMRLNSVQSSERPLFLVHPIEGSTTVFHSLASRLSIPTYGLQCTRAAPLDSIHSLAAYYIDCIRQVQPEGPYRVAGYSYGACVAFEMCSQLQAQQSPAPTHNSLFLFDGSPTYVLAYTGSYRAKLTPGCEAEAETEAICFFVQQFTDMEHNRVLEALLPLKGLEERVAAAVDLIIKSHQGLDRQELSFAARSFYYKLRAAEQYTPKAKYHGNVMLLRAAAGADYNLSQVCDGKVSVHVIEGDHATLLEGSGLESIISIIHSS Hydrogen bonds contact Hydrophobic contact | ||||
| 6 | Organic cation transporter 3 (OCT3) | 7ZH6 | 8.67 | |
Target general information Gen name SLC22A3 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Solute carrier family 22 member 3; Extraneuronal monoamine transporter; EMTH Protein family Major facilitator (TC 2.A.1) superfamily, Organic cation transporter (TC 2.A.1.19) family Biochemical class NA Function Mediates potential-dependent transport of a variety of organic cations. May play a significant role in the disposition of cationic neurotoxins and neurotransmitters in the brain. Related diseases Deafness, autosomal dominant, 2A (DFNA2A) [MIM:600101]: A form of non-syndromic sensorineural hearing loss. Sensorineural deafness results from damage to the neural receptors of the inner ear, the nerve pathways to the brain, or the area of the brain that receives sound information. {ECO:0000269|PubMed:10025409, ECO:0000269|PubMed:10369879, ECO:0000269|PubMed:10571947, ECO:0000269|PubMed:10925378, ECO:0000269|PubMed:21242547}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00718; DB08838; DB00182; DB00122; DB14006; DB00501; DB00575; DB00363; DB01151; DB00988; DB00783; DB13952; DB13953; DB13954; DB13955; DB13956; DB00983; DB00536; DB05381; DB00458; DB00762; DB00709; DB00448; DB08882; DB01042; DB01577; DB00331; DB08893; DB00184; DB00368; DB00526; DB00925; DB00413; DB00457; DB01035; DB00396; DB00938; DB00391; DB13943; DB13944; DB08837; DB08841; DB00541 Interacts with P00519 EC number NA Uniprot keywords 3D-structure; Cell membrane; Glycoprotein; Ion transport; Membrane; Mitochondrion; Nucleus; Proteomics identification; Reference proteome; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A Molecular weight (Da) 53067.4 Length 478 Aromaticity 0.13 Instability index 38.82 Isoelectric point 9.07 Charge (pH=7) 10.54 2D Binding mode Binding energy (Kcal/mol) -9.49 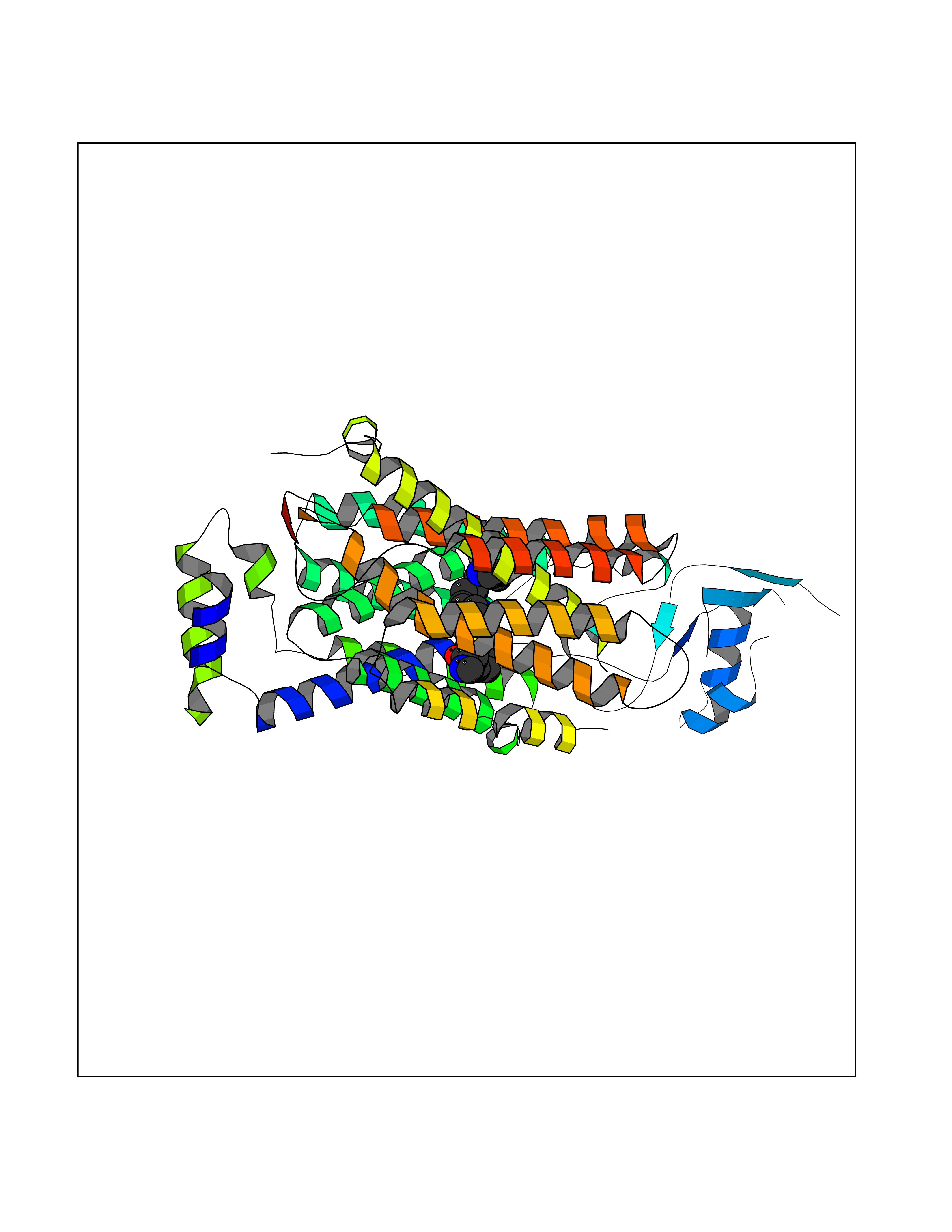 Molscript Map 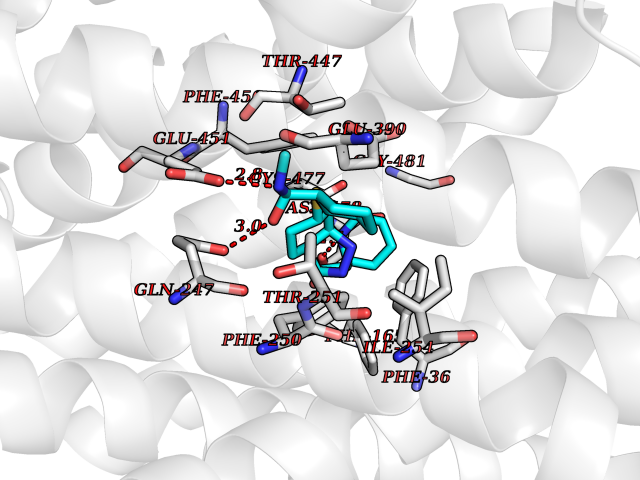 Pymol Map 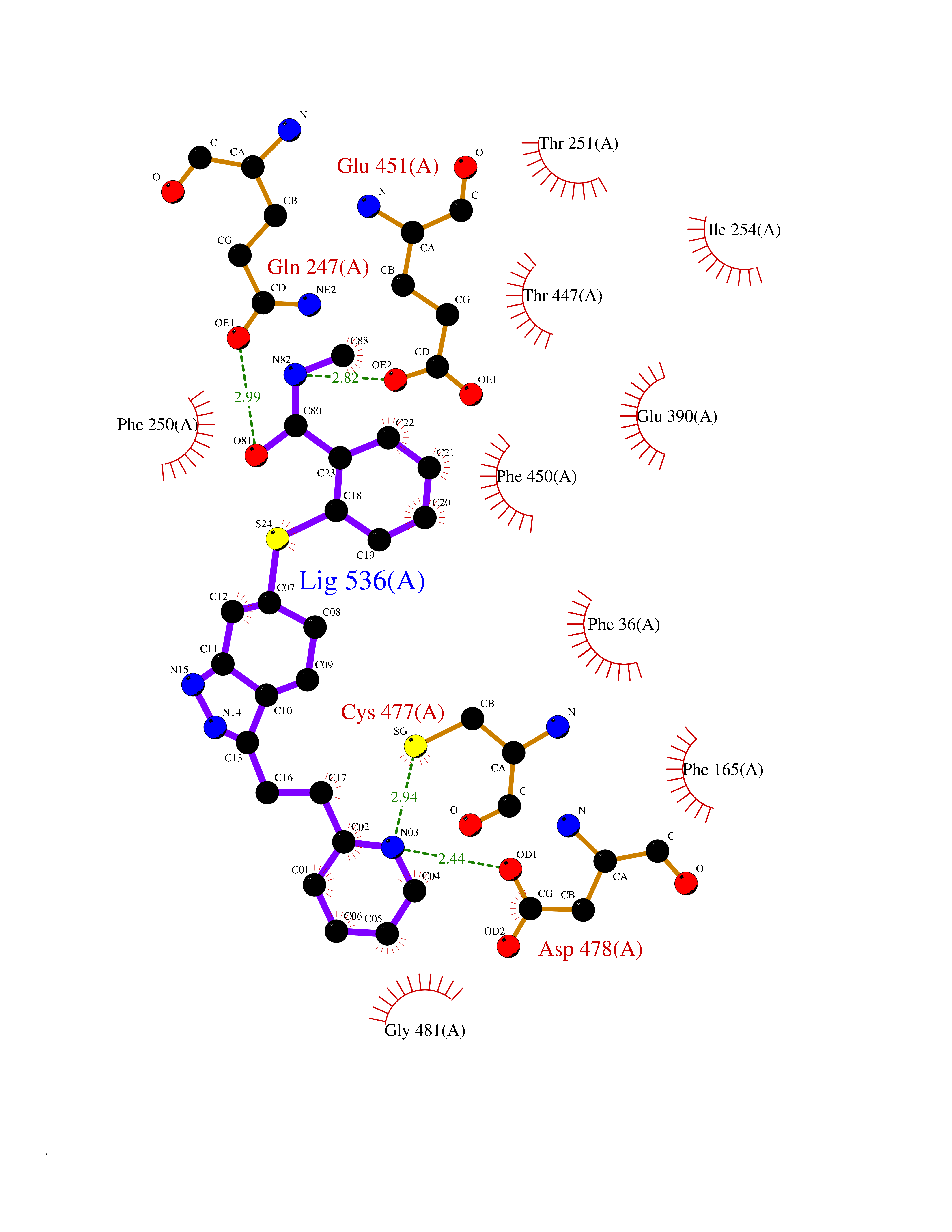 Ligplot Map 3D Binding mode Sequence SFDEALQRVGEFGRFQRRVFLLLCLTGVTFAFLFVGVVFLGTQPDHYWCRGPSAAALAERCGWSPEEEWNRTAPASRGRCQRYLLSAPLVPCRGGWRYAQAHSTIVSEFDLVCVNAWMLDLTQAILNLGFLTGAFTLGYAADRYGRIVIYLLSCLGVGVTGVVVAFAPNFPVFVIFRFLQGVFGKGTWMTCYVIVTEIVGSKQRRIVGIVIQMFFTLGIIILPGIAYFIPNWQGIQLAITLPSFLFLLYYWVVPESPRWLITRKKGDKALQILRRIAKCNVSNPSFLDLVRTPQMRKCTLILMFAWFTSAVVYQGLVMRLGNLYIDFFISGVVELPGALLILLTIERLGRRLPFAASNIVAGVACLVTAFLPEGIAWLRTTVATLGRLGITMAFEIVYLVNSELYPTTLRNFGVSLCSGLCDFGGIIAPFLLFRLAAVWLELPLIIFGILASICGGLVMLLPETKGIALPETVDDVEK Hydrogen bonds contact Hydrophobic contact | ||||
| 7 | SET domain containing 8 (KMT5A) | 5TEG | 8.67 | |
Target general information Gen name KMT5A Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms SETD8; SET8; SET07; SET domain-containing protein 8; PRSET7; PR/SET07; PR/SET domain-containing protein 07; PR-Set7; N-lysine methyltransferase KMT5A; Lysine-specific methylase 5A; Lysine N-methyltran Protein family Class V-like SAM-binding methyltransferase superfamily, Histone-lysine methyltransferase family, PR/SET subfamily Biochemical class Methyltransferase Function Specifically monomethylates 'Lys-20' of histone H4 (H4K20me1). H4K20me1 is enriched during mitosis and represents a specific tag for epigenetic transcriptional repression. Mainly functions in euchromatin regions, thereby playing a central role in the silencing of euchromatic genes. Required for cell proliferation, probably by contributing to the maintenance of proper higher-order structure of DNA during mitosis. Involved in chromosome condensation and proper cytokinesis. Nucleosomes are preferred as substrate compared to free histones. Mediates monomethylation of p53/TP53 at 'Lys-382', leading to repress p53/TP53-target genes. Plays a negative role in TGF-beta response regulation and a positive role in cell migration. Protein-lysine N-methyltransferase that monomethylates both histones and non-histone proteins. Related diseases Sick sinus syndrome 2 (SSS2) [MIM:163800]: The term 'sick sinus syndrome' encompasses a variety of conditions caused by sinus node dysfunction. The most common clinical manifestations are syncope, presyncope, dizziness, and fatigue. Electrocardiogram typically shows sinus bradycardia, sinus arrest, and/or sinoatrial block. Episodes of atrial tachycardias coexisting with sinus bradycardia ('tachycardia-bradycardia syndrome') are also common in this disorder. SSS occurs most often in the elderly associated with underlying heart disease or previous cardiac surgery, but can also occur in the fetus, infant, or child without heart disease or other contributing factors. SSS2 onset is in utero or at birth. {ECO:0000269|PubMed:15123648, ECO:0000269|PubMed:16407510, ECO:0000269|PubMed:20662977, ECO:0000269|PubMed:23103389}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Brugada syndrome 8 (BRGDA8) [MIM:613123]: A tachyarrhythmia characterized by right bundle branch block and ST segment elevation on an electrocardiogram (ECG). It can cause the ventricles to beat so fast that the blood is prevented from circulating efficiently in the body. When this situation occurs, the individual will faint and may die in a few minutes if the heart is not reset. {ECO:0000269|PubMed:19165230}. The gene represented in this entry may be involved in disease pathogenesis.; DISEASE: Epilepsy, idiopathic generalized 18 (EIG18) [MIM:619521]: An autosomal dominant form of idiopathic generalized epilepsy, a disorder characterized by recurring generalized seizures in the absence of detectable brain lesions and/or metabolic abnormalities. Generalized seizures arise diffusely and simultaneously from both hemispheres of the brain. Seizure types include juvenile myoclonic seizures, absence seizures, and generalized tonic-clonic seizures. EIG18 is characterized by onset of myoclonic seizures in infancy. Although the seizures remit, some patients may have later speech or cognitive impairment. {ECO:0000269|PubMed:30127718}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P62805; P07910; Q15672 EC number EC 2.1.1.- Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Cell cycle; Cell division; Chromatin regulator; Chromosome; Coiled coil; Direct protein sequencing; Methyltransferase; Mitosis; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Repressor; S-adenosyl-L-methionine; Transcription; Transcription regulation; Transferase; Ubl conjugation Protein physicochemical properties Chain ID A,D Molecular weight (Da) 19129.4 Length 167 Aromaticity 0.08 Instability index 49.18 Isoelectric point 7.88 Charge (pH=7) 1.37 2D Binding mode Binding energy (Kcal/mol) -9.23  Molscript Map 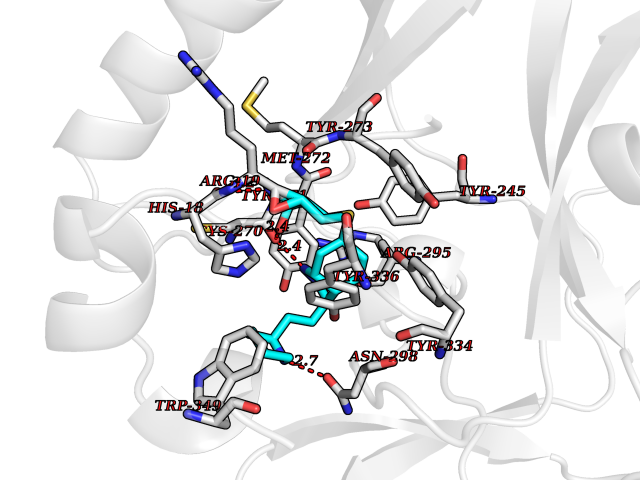 Pymol Map 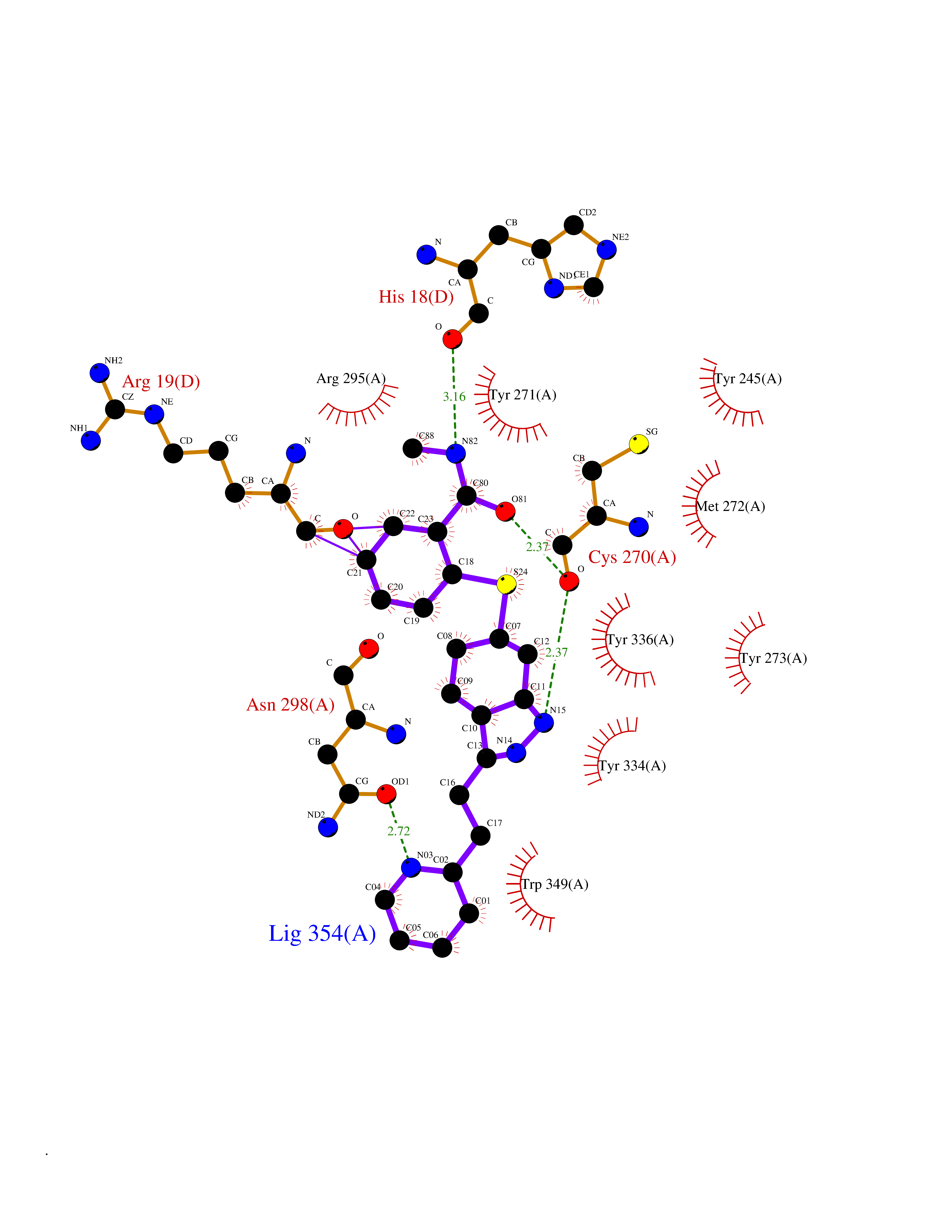 Ligplot Map 3D Binding mode Sequence KSKAELQSEERKRIDELIESGKEEGMKIDLIDGKGRGVIATKQFSRGDFVVEYHGDLIEITDAKKREALYAQDPSTGCYMYYFQYLSKTYCVDATRETNRLGRLINHSKSGNCQTKLHDIDGVPHLILIASRDIAAGEELLYDYGDRSKASIEAHPWLKHKRHRVLR Hydrogen bonds contact Hydrophobic contact | ||||
| 8 | Extracellular calcium-sensing receptor (CASR) | 5FBK | 8.67 | |
Target general information Gen name CASR Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms hCasR; Parathyroid cell calciumreceptor; Parathyroid cell calcium-sensing receptor 1; Parathyroid calcium receptor; Parathyroid Cell calcium-sensing receptor; PCaR1; GPRC2A; CaSR Protein family G-protein coupled receptor 3 family Biochemical class GPCR glutamate Function Senses fluctuations in the circulating calcium concentration and modulates the production of parathyroid hormone (PTH) in parathyroid glands. The activity of this receptor is mediated by a G-protein that activates a phosphatidylinositol-calcium second messenger system. The G-protein-coupled receptor activity is activated by a co-agonist mechanism: aromatic amino acids, such as Trp or Phe, act concertedly with divalent cations, such as calcium or magnesium, to achieve full receptor activation. G-protein-coupled receptor that senses changes in the extracellular concentration of calcium ions and plays a key role in maintaining calcium homeostasis. Related diseases Hypocalciuric hypercalcemia, familial 1 (HHC1) [MIM:145980]: A form of hypocalciuric hypercalcemia, a disorder of mineral homeostasis that is transmitted as an autosomal dominant trait with a high degree of penetrance. It is characterized biochemically by lifelong elevation of serum calcium concentrations and is associated with inappropriately low urinary calcium excretion and a normal or mildly elevated circulating parathyroid hormone level. Hypermagnesemia is typically present. Affected individuals are usually asymptomatic and the disorder is considered benign. However, chondrocalcinosis and pancreatitis occur in some adults. {ECO:0000269|PubMed:11762699, ECO:0000269|PubMed:15572418, ECO:0000269|PubMed:15579740, ECO:0000269|PubMed:15879434, ECO:0000269|PubMed:16598859, ECO:0000269|PubMed:16740594, ECO:0000269|PubMed:17473068, ECO:0000269|PubMed:17698911, ECO:0000269|PubMed:19179454, ECO:0000269|PubMed:19789209, ECO:0000269|PubMed:21566075, ECO:0000269|PubMed:21643651, ECO:0000269|PubMed:22114145, ECO:0000269|PubMed:23169696, ECO:0000269|PubMed:23966241, ECO:0000269|PubMed:25104082, ECO:0000269|PubMed:25292184, ECO:0000269|PubMed:26386835, ECO:0000269|PubMed:27434672, ECO:0000269|PubMed:7673400, ECO:0000269|PubMed:7726161, ECO:0000269|PubMed:7916660, ECO:0000269|PubMed:8636323, ECO:0000269|PubMed:8702647, ECO:0000269|PubMed:8878438, ECO:0000269|PubMed:9298824}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Hyperparathyroidism, neonatal severe (NSHPT) [MIM:239200]: A disorder characterized by severe hypercalcemia, bone demineralization, and failure to thrive usually manifesting in the first 6 months of life. If untreated, NSHPT can be a devastating neurodevelopmental disorder, which in some cases is lethal without parathyroidectomy. {ECO:0000269|PubMed:14985373, ECO:0000269|PubMed:15572418, ECO:0000269|PubMed:17555508, ECO:0000269|PubMed:27434672, ECO:0000269|PubMed:8675635, ECO:0000269|PubMed:8878438, ECO:0000269|PubMed:9253359}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Hypocalcemia, autosomal dominant 1 (HYPOC1) [MIM:601198]: A disorder of mineral homeostasis characterized by blood calcium levels below normal, and low or normal serum parathyroid hormone concentrations. Disease manifestations include mild or asymptomatic hypocalcemia, paresthesias, carpopedal spasm, seizures, hypercalciuria with nephrocalcinosis or kidney stones, and ectopic and basal ganglia calcifications. Few patients manifest hypocalcemia and features of Bartter syndrome, including hypomagnesemia, hypokalemia, metabolic alkalosis, hyperreninemia, and hyperaldosteronemia. {ECO:0000269|PubMed:10487661, ECO:0000269|PubMed:12050233, ECO:0000269|PubMed:12107202, ECO:0000269|PubMed:12241879, ECO:0000269|PubMed:12574188, ECO:0000269|PubMed:12915654, ECO:0000269|PubMed:15551332, ECO:0000269|PubMed:16608894, ECO:0000269|PubMed:19179454, ECO:0000269|PubMed:22789683, ECO:0000269|PubMed:23169696, ECO:0000269|PubMed:23966241, ECO:0000269|PubMed:25766501, ECO:0000269|PubMed:7874174, ECO:0000269|PubMed:8702647, ECO:0000269|PubMed:8733126, ECO:0000269|PubMed:8813042, ECO:0000269|PubMed:8878438, ECO:0000269|PubMed:9253358, ECO:0000269|PubMed:9661634, ECO:0000269|PubMed:9920108}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Epilepsy, idiopathic generalized 8 (EIG8) [MIM:612899]: A disorder characterized by recurring generalized seizures in the absence of detectable brain lesions and/or metabolic abnormalities. Seizure types are variable, but include myoclonic seizures, absence seizures, febrile seizures, complex partial seizures, and generalized tonic-clonic seizures. {ECO:0000269|PubMed:18756473}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB11093; DB11348; DB14481; DB01012; DB12865; DB00994; DB05695; DB05255; DB00127 Interacts with Q15363; P41180-1 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Calcium; Cell membrane; Disease variant; Disulfide bond; Epilepsy; G-protein coupled receptor; Glycoprotein; Membrane; Metal-binding; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Signal; Transducer; Transmembrane; Transmembrane helix; Ubl conjugation Protein physicochemical properties Chain ID A,B Molecular weight (Da) 52438.9 Length 467 Aromaticity 0.12 Instability index 39.62 Isoelectric point 5.63 Charge (pH=7) -10.18 2D Binding mode Binding energy (Kcal/mol) -9.08 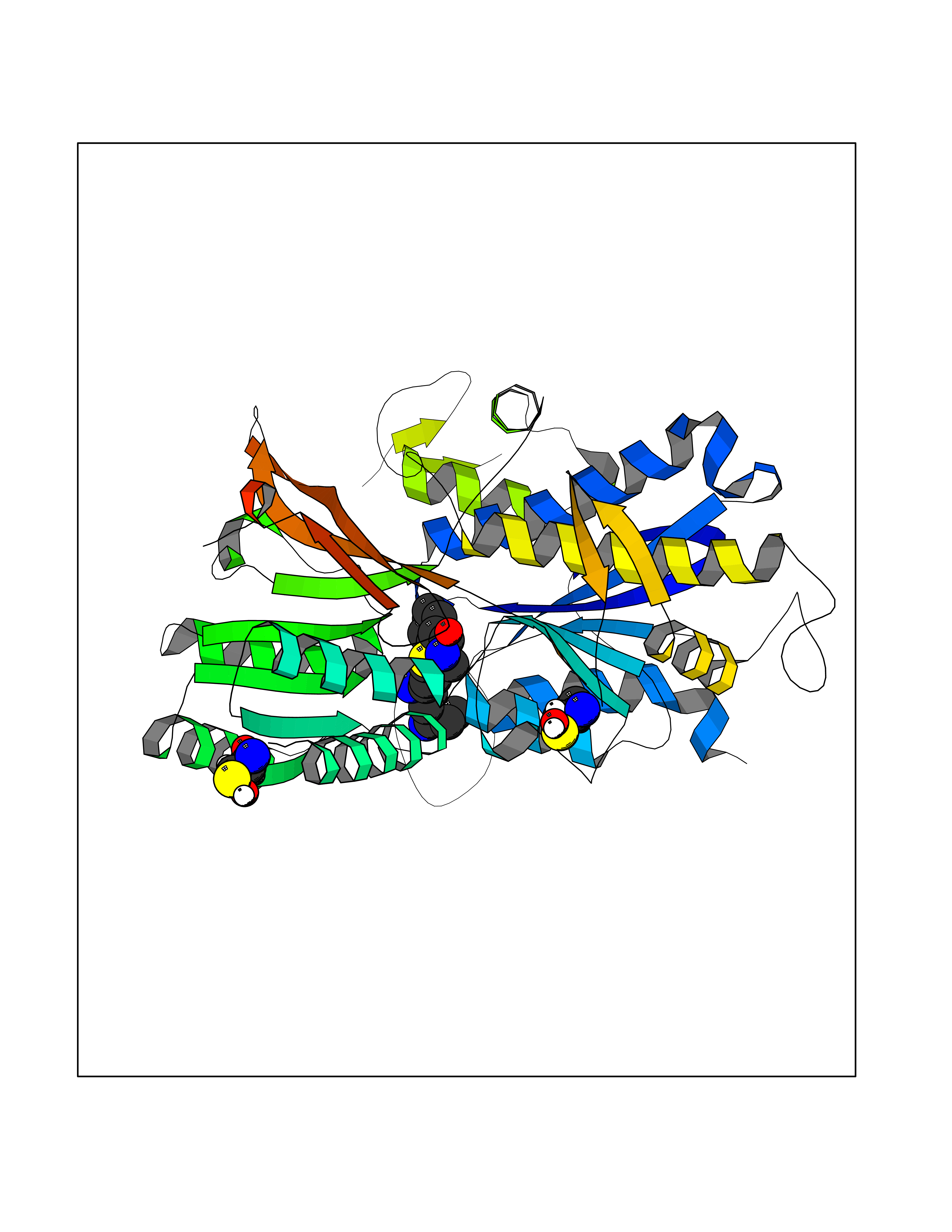 Molscript Map 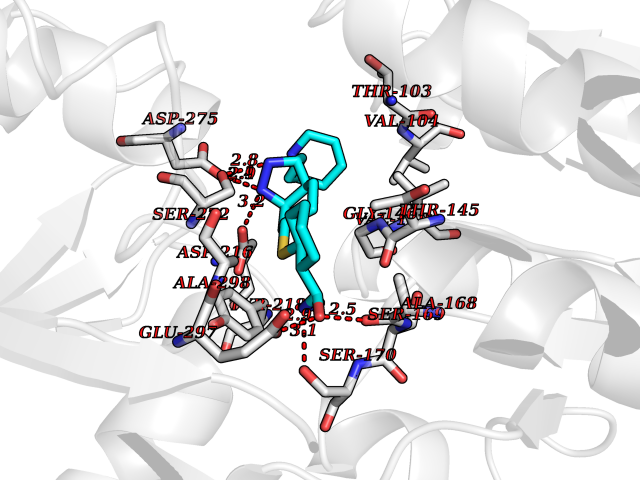 Pymol Map 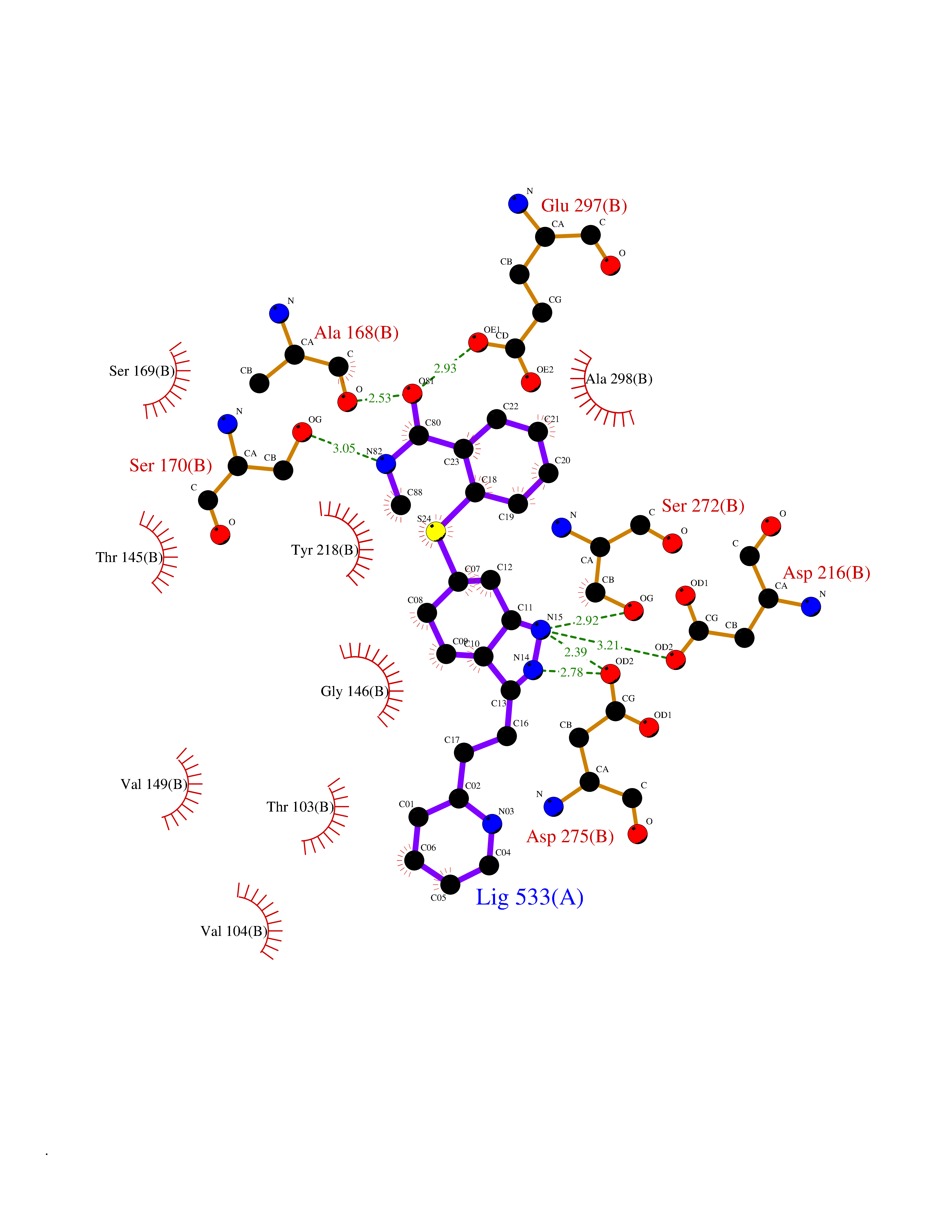 Ligplot Map 3D Binding mode Sequence GPDQRAQKKGDIILGGLFPIHFGVAAKDQDLKSRPESVECIRYNFRGFRWLQAMIFAIEEINSSPALLPNLTLGYRIFDTCNTVSKALEATLSFVAQNKIDSTIAVVGATGSGVSTAVANLLGLFYIPQVSYASSSRLLSNKNQFKSFLRTIPNDEHQATAMADIIEYFRWNWVGTIAADDDYGRPGIEKFREEAEERDIXIDFSELISQYSDEEEIQHVVEVIQNSTAKVIVVFSSGPDLEPLIKEIVRRNITGKIWLASEAWASSSLIAMPQYFHVVGGTIGFALKAGQIPGFREFLKKVHPRKSVHNGFAKEFWEETFNCHLQFRPLCTGDENISSVETPYIDYTHLRISYNVYLAVYSIAHALQDIYTCLPGRGLFTNGSCADIKKVEAWQVLKHLRHLNFTNNMGEQVTFDEXGDLVGNYSIINWHLSPEDGSIVFKEVGYYNVYAKKGERLFINEEKILWS Hydrogen bonds contact Hydrophobic contact | ||||
| 9 | Human immunodeficiency virus Protease (HIV PR) | 3TL9 | 8.60 | |
Target general information Gen name HIV PR Organism Human immunodeficiency virus type 1 group M subtype B (isolate BH10) (HIV-1) Uniprot ID TTD ID Synonyms HIV Retropepsin; HIV PR Protein family NA Biochemical class Peptidase Function Gag-Pol polyprotein: Mediates, with Gag polyprotein, the essential events in virion assembly, including binding the plasma membrane, making the protein-protein interactions necessary to create spherical particles, recruiting the viral Env proteins, and packaging the genomic RNA via direct interactions with the RNA packaging sequence (Psi). Gag-Pol polyprotein may regulate its own translation, by the binding genomic RNA in the 5'-UTR. At low concentration, the polyprotein would promote translation, whereas at high concentration, the polyprotein would encapsidate genomic RNA and then shut off translation. Related diseases Sitosterolemia 2 (STSL2) [MIM:618666]: A form of sitosterolemia, an autosomal recessive metabolic disorder characterized by unregulated intestinal absorption of cholesterol, phytosterols and shellfish sterols, and decreased biliary excretion of dietary sterols into bile. Patients have hypercholesterolemia, very high levels of plant sterols in the plasma, and frequently develop tendon and tuberous xanthomas, accelerated atherosclerosis and premature coronary artery disease. {ECO:0000269|PubMed:11138003, ECO:0000269|PubMed:11452359, ECO:0000269|PubMed:11668628, ECO:0000269|PubMed:15054092, ECO:0000269|PubMed:35557526}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07035; DB02704; DB07806; DB02785; DB01824; DB01732; DB06874; DB08034; DB07961; DB07451; DB08212; DB08372; DB02972; DB04190; DB04042; DB08428; DB03076; DB03141; DB08457; DB07343; DB07337; DB07018; DB07332; DB05398; DB07578; DB08639; DB06414; DB04255; DB04547; DB02683; DB02009; DB03908; DB02629; DB01887; DB03803; DB02033; DB08281; DB08282; DB08284; DB08414; DB08598; DB07327; DB07885; DB02768; DB08600; DB01891; DB05871 Interacts with NA EC number EC 3.4.23.16 Uniprot keywords 3D-structure; Activation of host caspases by virus; AIDS; Aspartyl protease; Capsid protein; Direct protein sequencing; DNA integration; DNA recombination; DNA-binding; DNA-directed DNA polymerase; Endonuclease; Eukaryotic host gene expression shutoff by virus; Eukaryotic host translation shutoff by virus; Host cell membrane; Host cytoplasm; Host endosome; Host gene expression shutoff by virus; Host membrane; Host nucleus; Host-virus interaction; Hydrolase; Lipid-binding; Lipoprotein; Magnesium; Membrane; Metal-binding; Methylation; Modulation of host cell apoptosis by virus; Multifunctional enzyme; Myristate; Nuclease; Nucleotidyltransferase; Phosphoprotein; Protease; Repeat; Ribosomal frameshifting; RNA-binding; RNA-directed DNA polymerase; Transferase; Viral genome integration; Viral nucleoprotein; Viral penetration into host nucleus; Viral release from host cell; Virion; Virion maturation; Virus entry into host cell; Zinc; Zinc-finger Protein physicochemical properties Chain ID A,B Molecular weight (Da) 21934.7 Length 202 Aromaticity 0.06 Instability index 48.65 Isoelectric point 9.66 Charge (pH=7) 6.15 2D Binding mode Binding energy (Kcal/mol) -8.55 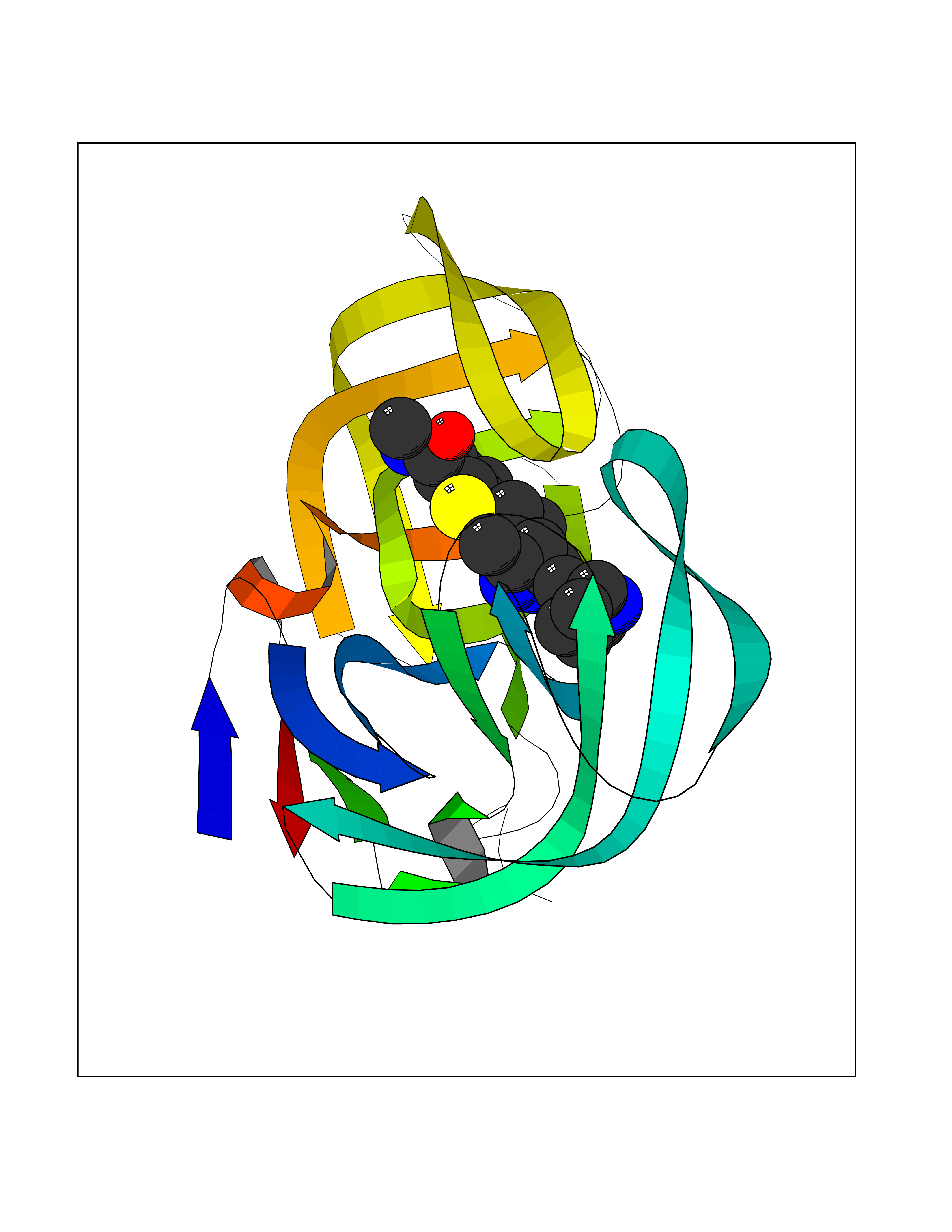 Molscript Map 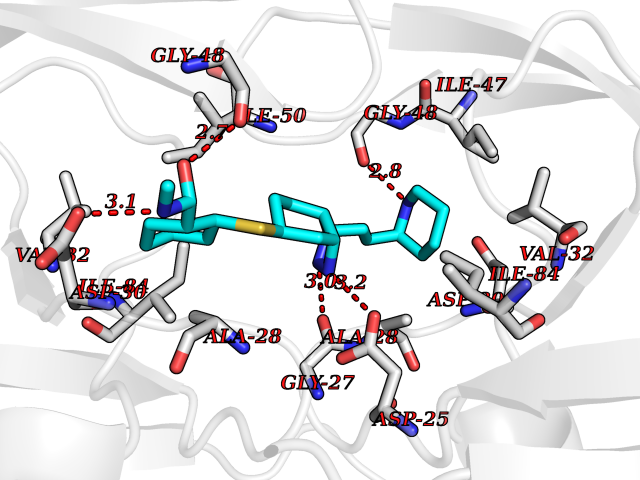 Pymol Map 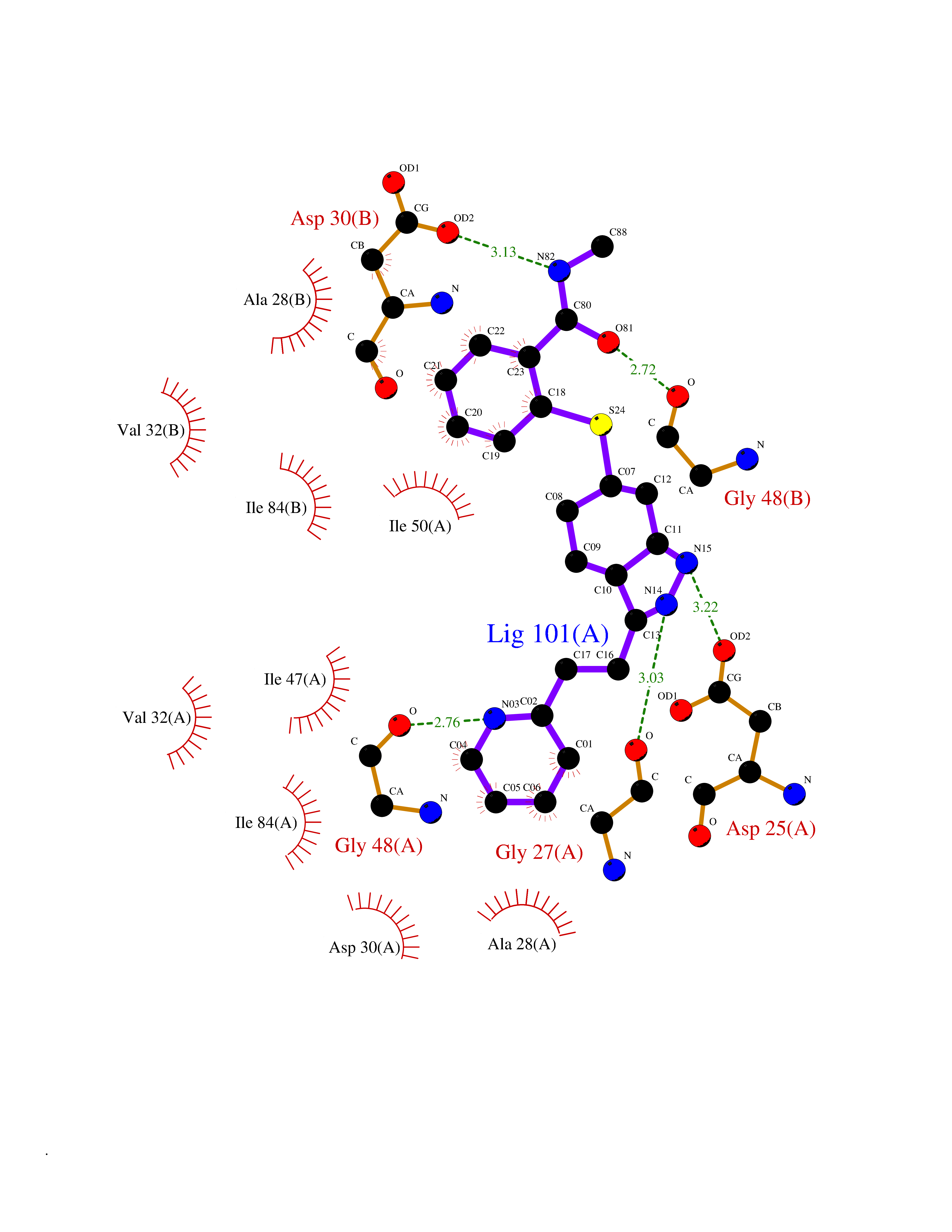 Ligplot Map 3D Binding mode Sequence PQITLWKRPLVTIKIGGQLKEALLDTGADDTVIEEMSLPGRWKPKMIGGIGGFIKVRQYDQIIIEIAGHKAIGTVLVGPTPVNIIGRNLLTQIGATLNFSFNFPQITLWKRPLVTIKIGGQLKEALLDTGADDTVIEEMSLPGRWKPKMIGGIGGFIKVRQYDQIIIEIAGHKAIGTVLVGPTPVNIIGRNLLTQIGATLNF Hydrogen bonds contact Hydrophobic contact | ||||
| 10 | Lanosterol 14-alpha demethylase (CYP51A1) | 4UHI | 8.56 | |
Target general information Gen name CYP51A1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Cytochrome P450LI; Cytochrome P45014DM; Cytochrome P450-14DM; Cytochrome P450 51A1 Protein family Cytochrome P450 family Biochemical class Cytochrome P450 family Function Catalyzes C14-demethylation of lanosterol; it transforms lanosterol into 4,4'-dimethyl cholesta-8,14,24-triene-3-beta-ol. Related diseases Spondyloepimetaphyseal dysplasia, short limb-hand type (SEMD-SL) [MIM:271665]: A bone disease characterized by short-limbed dwarfism, a narrow chest with pectus excavatum, brachydactyly in the hands and feet, a characteristic craniofacial appearance and premature calcifications. The radiological findings are distinctive and comprise short long bones throughout the skeleton with striking epiphyses that are stippled, flattened and fragmented and flared, irregular metaphyses. Platyspondyly in the spine with wide intervertebral spaces is observed and some vertebral bodies are pear-shaped with central humps, anterior protrusions and posterior scalloping. {ECO:0000269|PubMed:19110212, ECO:0000269|PubMed:20223752, ECO:0000269|PubMed:26463668}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Warburg-Cinotti syndrome (WRCN) [MIM:618175]: An autosomal dominant disease characterized by progressive corneal neovascularization, keloid formation, chronic skin ulcers, wasting of subcutaneous tissue, flexion contractures of the fingers, and acro-osteolysis. {ECO:0000269|PubMed:30449416}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07705; DB05667; DB01110; DB01007 Interacts with NA EC number EC 1.14.14.154 Uniprot keywords 3D-structure; Alternative splicing; Cholesterol biosynthesis; Cholesterol metabolism; Endoplasmic reticulum; Heme; Iron; Lipid biosynthesis; Lipid metabolism; Membrane; Metal-binding; Microsome; Monooxygenase; Oxidoreductase; Proteomics identification; Reference proteome; Steroid biosynthesis; Steroid metabolism; Sterol biosynthesis; Sterol metabolism; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 53013.3 Length 462 Aromaticity 0.11 Instability index 47.66 Isoelectric point 8.8 Charge (pH=7) 7 2D Binding mode Binding energy (Kcal/mol) -8.86  Molscript Map 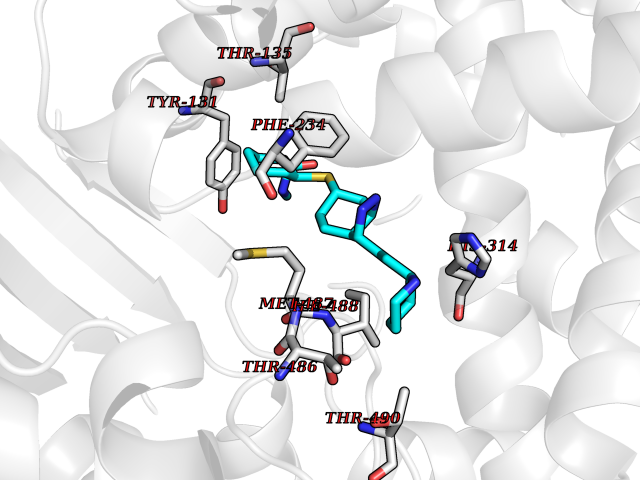 Pymol Map 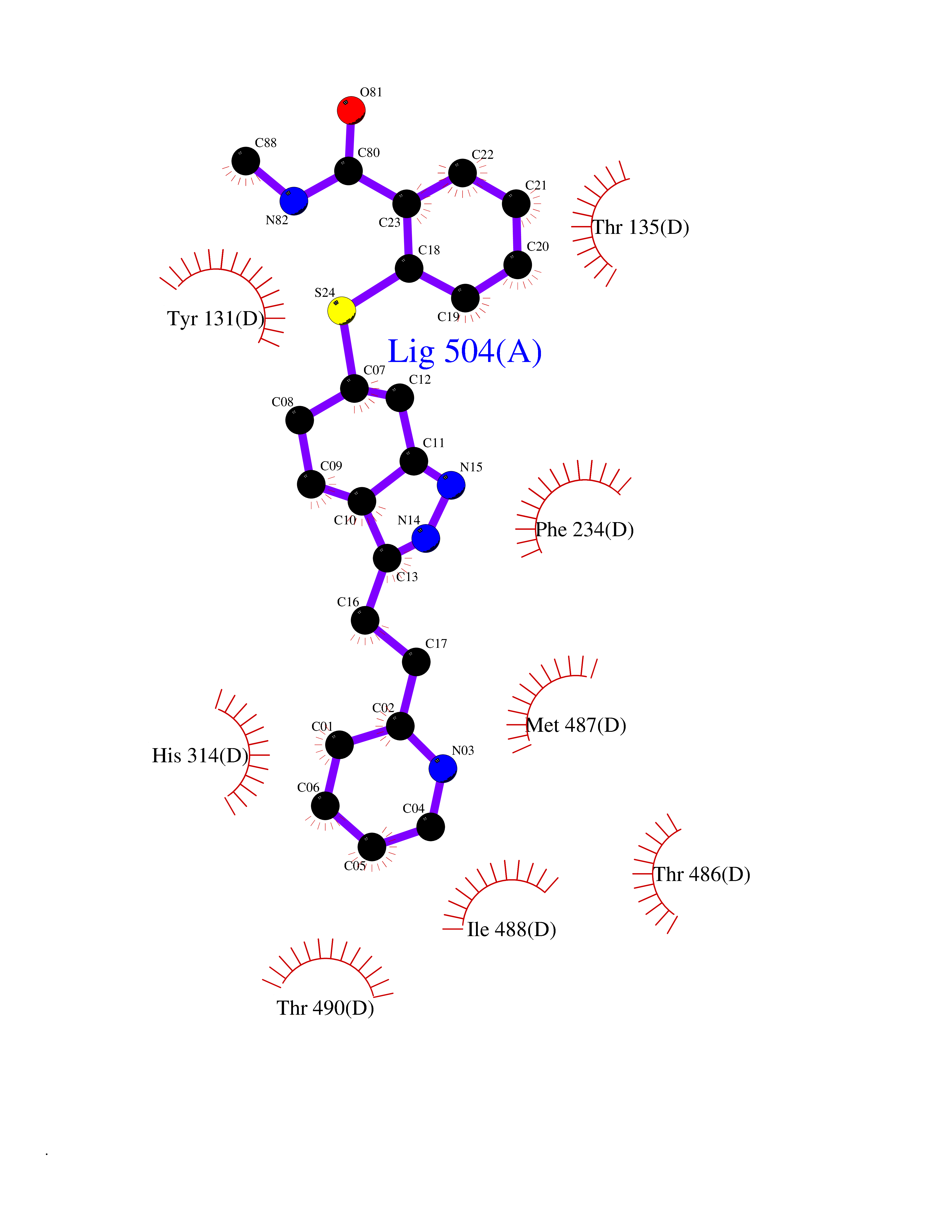 Ligplot Map 3D Binding mode Sequence PPYIFSPIPFLGHAIAFGKSPIEFLENAYEKYGPVFSFTMVGKTFTYLLGSDAAALLFNSKNEDLNAEDVYSRLTTPVFGKGVAYDVPNPVFLEQKKMLKSGLNIAHFKQHVSIIEKETKEYFESWGESGEKNVFEALSELIILTASHCLHGKEIRSQLNEKVAQLYADLDGGFSHAAWLLPGWLPLPSFRRRDRAHREIKDIFYKAIQKRRQSQEKIDDILQTLLDATYKDGRPLTDDEVAGMLIGLLLAGQHTSSTTSAWMGFFLARDKTLQKKCYLEQKTVCGENLPPLTYDQLKDLNLLDRCIKETLRLRPPIMIMMRMARTPQTVAGYTIPPGHQVCVSPTVNQRLKDSWVERLDFNPDRYLQDNPASGEKFAYVPFGAGRHRCIGENFAYVQIKTIWSTMLRLYEFDLIDGYFPTVNYTTMIHTPENPVIRYKRRSLPGWLPLPSFRRRDRAHREI Hydrogen bonds contact Hydrophobic contact | ||||
| 11 | Endothelin-converting enzyme 1 (ECE1) | 3DWB | 8.54 | |
Target general information Gen name ECE1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms ECE-1 Protein family Peptidase M13 family Biochemical class Peptidase Function Converts big endothelin-1 to endothelin-1. Related diseases Hirschsprung disease, cardiac defects, and autonomic dysfunction (HCAD) [MIM:613870]: A disorder characterized by skip-lesions Hirschsprung disease, craniofacial abnormalities and other dysmorphic features, cardiac defects including ductus arteriosus, small subaortic ventricular septal defect, small atrial septal defect, and autonomic dysfunction. {ECO:0000269|PubMed:9915973}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07171 Interacts with P49760; A8MQ03; Q8IUG1; P60370; P60410 EC number EC 3.4.24.71 Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disease variant; Disulfide bond; Glycoprotein; Hirschsprung disease; Hydrolase; Membrane; Metal-binding; Metalloprotease; Phosphoprotein; Protease; Proteomics identification; Reference proteome; Signal-anchor; Transmembrane; Transmembrane helix; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 75247.9 Length 660 Aromaticity 0.12 Instability index 46.29 Isoelectric point 5.33 Charge (pH=7) -18.3 2D Binding mode Binding energy (Kcal/mol) -8.81 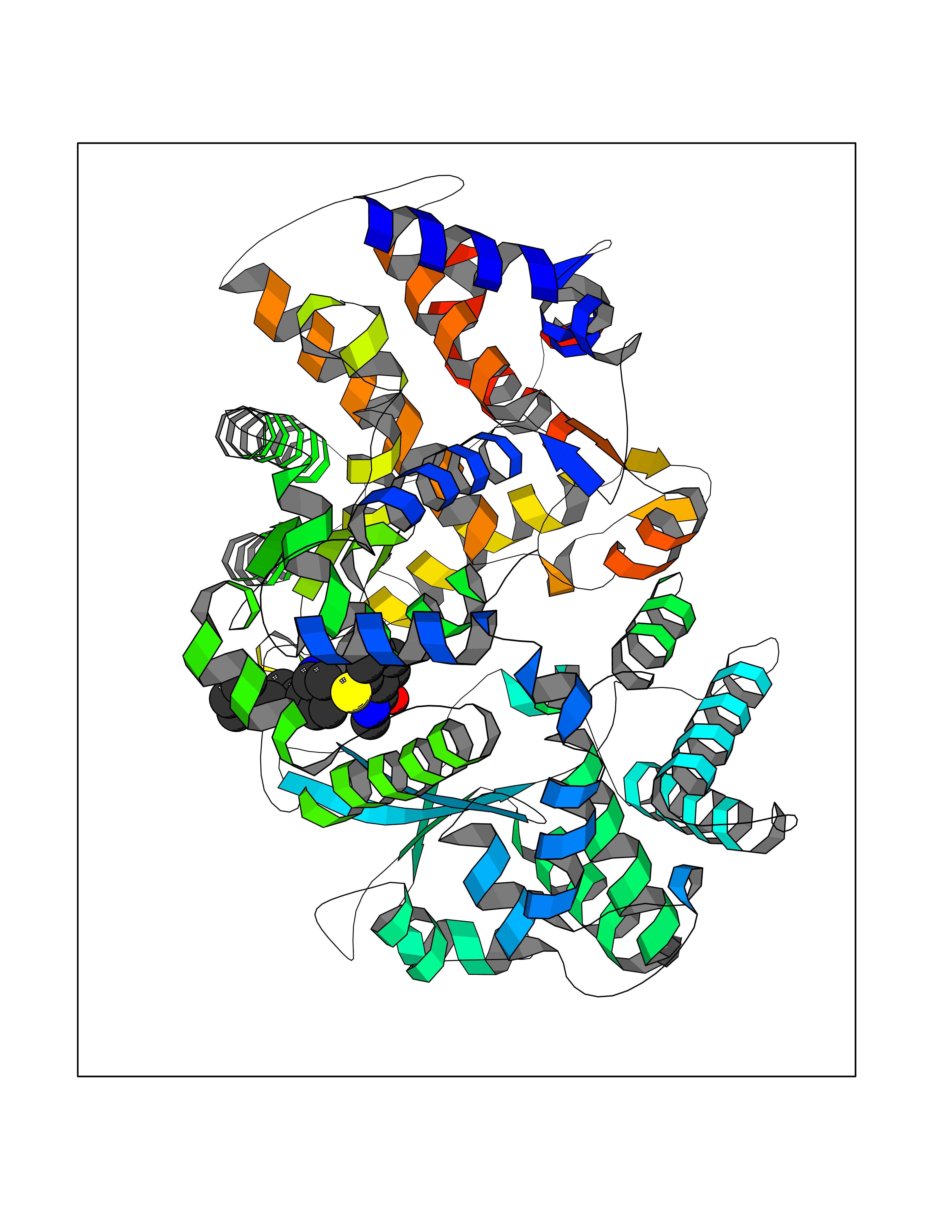 Molscript Map 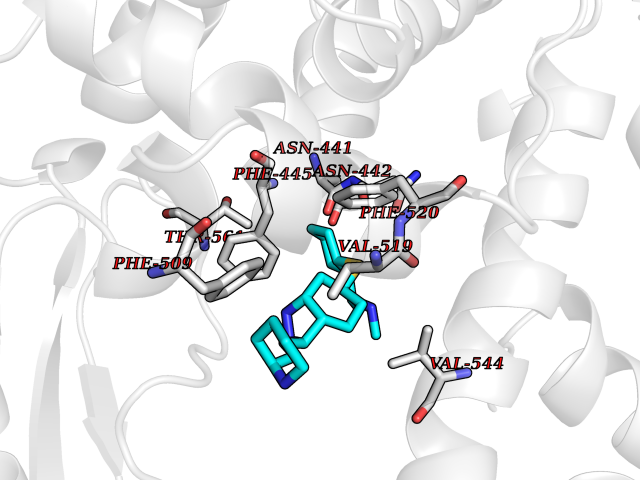 Pymol Map 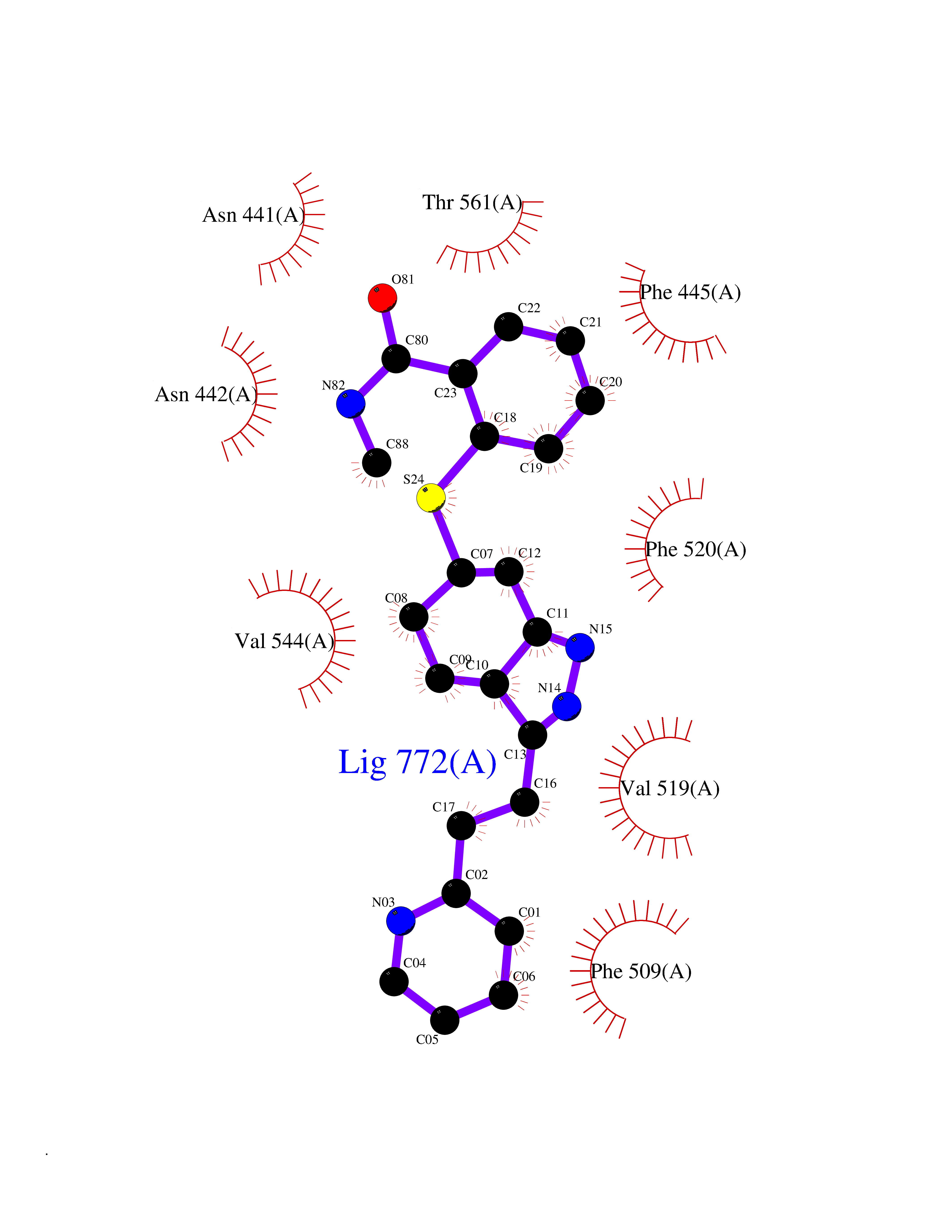 Ligplot Map 3D Binding mode Sequence SEACVSVTSSILSSMDPTVDPCHDFFSYACGGWIKANPVPDGHSRWGTFSNLWEHNQAIIKHLLENSTASVSEAERKAQVYYRACMNETRIEELRAKPLMELIERLGGWNITGPWAKDNFQDTLQVVTAHYRTSPFFSVYVSADSKNSNSNVIQVDQSGLGLPSRDYYLNKTENEKVLTGYLNYMVQLGKLLGGGDEEAIRPQMQQILDFETALANITIPQEKRRDEELIYHKVTAAELQTLAPAINWLPFLNTIFYPVEINESEPIVVYDKEYLEQISTLINTTDRCLLNNYMIWNLVRKTSSFLDQRFQDADEKFMEVMWKFCVSDTENNLGFALGPMFVKATFAEDSKSIATEIILEIKKAFEESLSTLKWMDEETRKSAKEKADAIYNMIGYPNFIMDPKELDKVFNDYTAVPDLYFENAMRFFNFSWRVTADQLRKAPNRDQWSMTPPMVNAYYSPTKNEIVFPAGILQAPFYTRSSPKALNFGGIGVVVGHELTHAFDDQGREYDKDGNLRPWWKNSSVEAFKRQTECMVEQYSNYSVNGEPVNGRHTLGENIADNGGLKAAYRAYQNWVKKNGAEHSLPTLGLTNNQLFFLGFAQVWCSVRTPESSHEGLITDPHSPSRFRVIGSLSNSKEFSEHFRCPPGSPMNPPHKCEVW Hydrogen bonds contact Hydrophobic contact | ||||
| 12 | Retinal rod rhodopsin-sensitive cGMP 3',5'-cyclic phosphodiesterase subunit gamma | 3JWR | 8.51 | |
Target general information Gen name PDE6G Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms PDEG Protein family Rod/cone cGMP-PDE gamma subunit family Biochemical class Hydrolase Function 3',5'-cyclic-GMP phosphodiesterase activity.CGMP binding.Enzyme inhibitor activity.Spectrin binding. Related diseases Retinitis pigmentosa 57 (RP57) [MIM:613582]: A retinal dystrophy belonging to the group of pigmentary retinopathies. Retinitis pigmentosa is characterized by retinal pigment deposits visible on fundus examination and primary loss of rod photoreceptor cells followed by secondary loss of cone photoreceptors. Patients typically have night vision blindness and loss of midperipheral visual field. As their condition progresses, they lose their far peripheral visual field and eventually central vision as well. {ECO:0000269|PubMed:20655036}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07954; DB00203; DB00820; DB00862 Interacts with O14503; Q96JM7; A0A6Q8PF08; O43741; Q8R511; P62994; Q9QY17; Q63787 EC number 3.1.4.35 Uniprot keywords 3D-structure; Acetylation; cGMP; Hydrolase; Reference proteome; Retinitis pigmentosa; Sensory transduction; Vision Protein physicochemical properties Chain ID C,D Molecular weight (Da) 40027.7 Length 345 Aromaticity 0.09 Instability index 37.44 Isoelectric point 6.02 Charge (pH=7) -6.88 2D Binding mode Binding energy (Kcal/mol) -9.03  Molscript Map 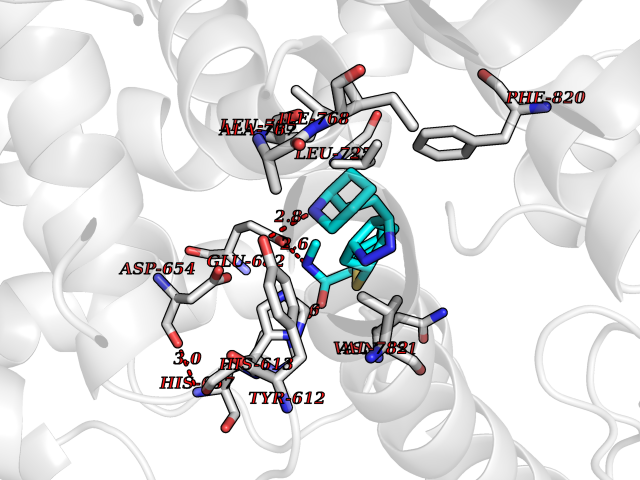 Pymol Map 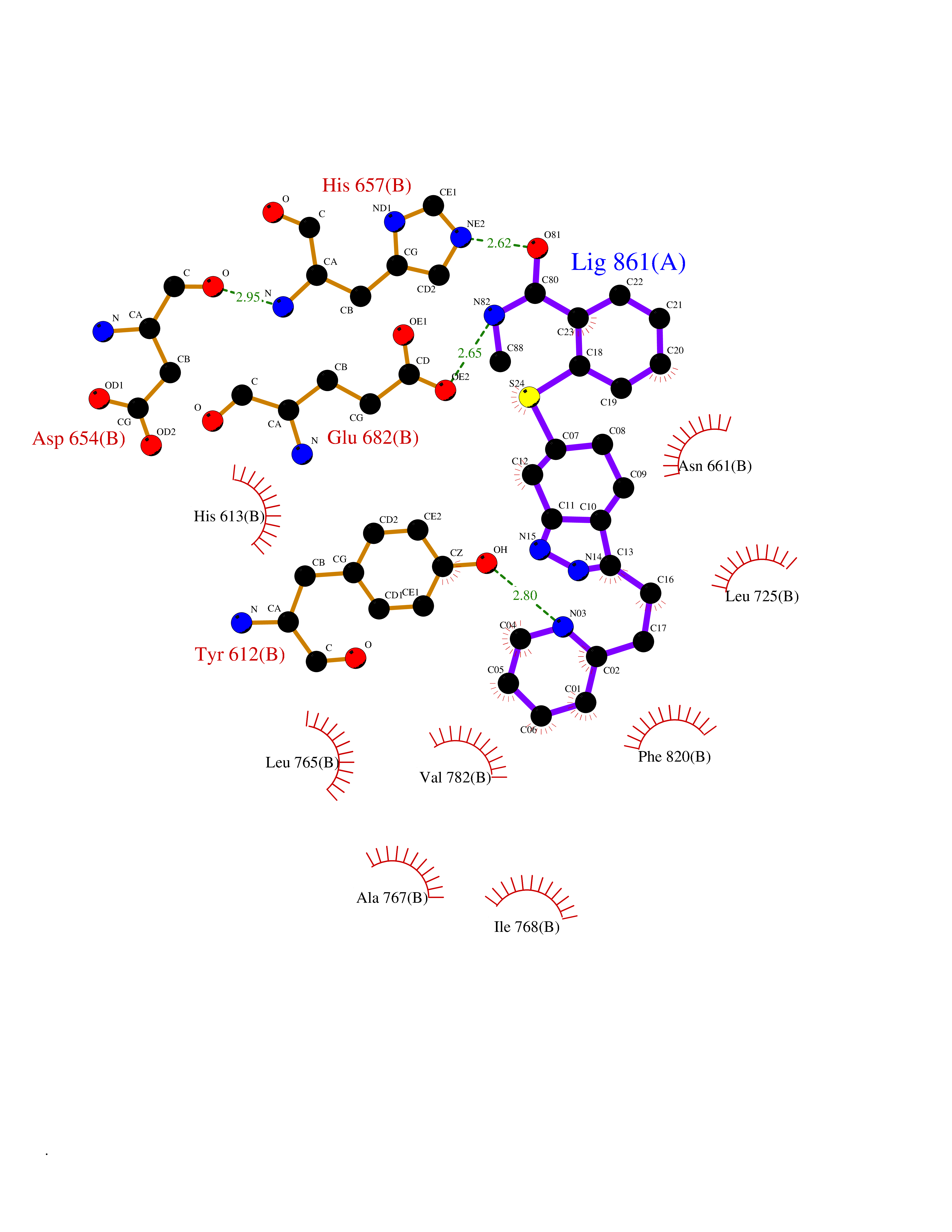 Ligplot Map 3D Binding mode Sequence EAFNHLELHELAQYGIISHMEETRELQSLAAAVVPSAQTLKITDFSFSDFELSDLETALCTIRMFTDLNLVQNFQMKHEVLCRWILSVKKNYRKNVAYHNWRHAFNTAQCMFAALKAGKIQNKLTDLEILALLIAALSHDLDHRGVNNSYIQRSEHPLAQLYCHSIMEHHHFDQCLMILNSPGNQILSGLSIEEYKTTLKIIKQAILATDLALYIKRRGEFFELIRKNQFNLEDPHQKELFLAMLMTACDLSAITKPWPIQQRIAELVATEFWEQGDLERTVLQQQPIPMMDRNKRDELPKLQVGFIDFVCTQLYEALTHVSEDCFPLLDGCRKNRQKWQALAEQ Hydrogen bonds contact Hydrophobic contact | ||||
| 13 | FkbI | 1R2J | 8.50 | |
Target general information Gen name fkbI Organism Streptomyces hygroscopicus subsp. ascomyceticus Uniprot ID TTD ID NA Synonyms NA Protein family Acyl-CoA dehydrogenase family Biochemical class Oxidoreductase Function Flavin adenine dinucleotide binding.Oxidoreductase activity, acting on the CH-CH group of donors. Related diseases Thiamine metabolism dysfunction syndrome 5, episodic encephalopathy type (THMD5) [MIM:614458]: An autosomal recessive metabolic disorder due to an inborn error of thiamine metabolism. The phenotype is highly variable, but in general, affected individuals have onset in early childhood of acute encephalopathic episodes associated with increased serum and CSF lactate. These episodes result in progressive neurologic dysfunction manifest as gait disturbances, ataxia, dystonia, and spasticity, which in some cases may result in loss of ability to walk. Cognitive function is usually preserved, although mildly delayed development has been reported. These episodes are usually associated with infection and metabolic decompensation. Some patients may have recovery of some neurologic deficits. {ECO:0000269|PubMed:22152682}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03147 Interacts with NA EC number NA Uniprot keywords 3D-structure; FAD; Flavoprotein; Nucleotide-binding; Oxidoreductase Protein physicochemical properties Chain ID A Molecular weight (Da) 36670.3 Length 353 Aromaticity 0.04 Instability index 22.05 Isoelectric point 6.12 Charge (pH=7) -5.04 2D Binding mode Binding energy (Kcal/mol) -8.29 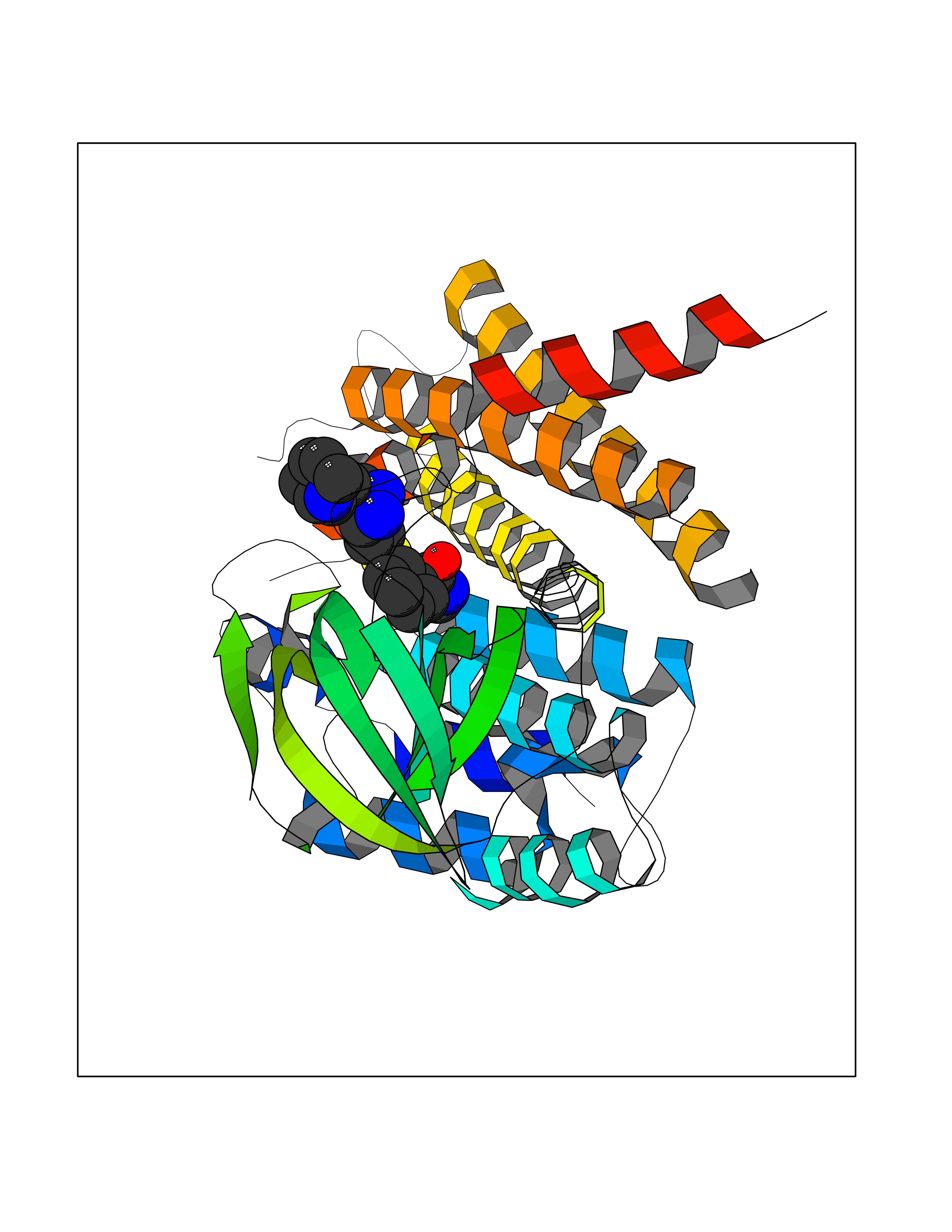 Molscript Map 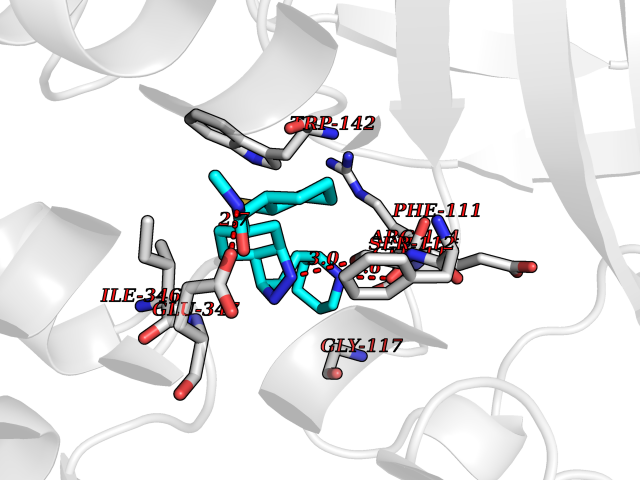 Pymol Map  Ligplot Map 3D Binding mode Sequence ERDALLTDLVGDRAAEWDTSGELPRDLLVRLGADGLLCAEVAAEHGGLGLGSRENGEFTAHVGSLCSSLRSVMTSQGMAAWTVQRLGDAGQRATFLKELTSGLAAVGFSERQAGSDLSAMRTRVRLDGDTAVVDGHKVWTTAAAYADHLVVFGLQEDGSGAVVVVPADTPGVRVERVPKPSGCRAAGHADLHLDQVRVPAGAVLAGSGASLPMLVAASLAYGRKSVAWGCVGILRACRTAAVAHARTREQFGRPLGDHQLVAGHIADLWTAEQIAARVCEYASDHMVPATILAKHVAAERAAAGAATAAQVLASAGAGHVVERAYRDAKLMEIIEGSSEMCRVMLAQHALALP Hydrogen bonds contact Hydrophobic contact | ||||
| 14 | Vitamin D3 receptor (VDR) | 3B0T | 8.48 | |
Target general information Gen name VDR Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Vitamin D(3) receptor; Nuclear vitamin D receptor; Nuclear receptor subfamily 1 group I member 1; NR1I1; 1,25-dihydroxyvitamin D3 receptor Protein family Nuclear hormone receptor family, NR1 subfamily Biochemical class Nuclear hormone receptor Function Enters the nucleus upon vitamin D3 binding where it forms heterodimers with the retinoid X receptor/RXR. The VDR-RXR heterodimers bind to specific response elements on DNA and activate the transcription of vitamin D3-responsive target genes. Plays a central role in calcium homeostasis. Nuclear receptor for calcitriol, the active form of vitamin D3 which mediates the action of this vitamin on cells. Related diseases Rickets vitamin D-dependent 2A (VDDR2A) [MIM:277440]: A disorder of vitamin D metabolism resulting in severe rickets, hypocalcemia and secondary hyperparathyroidism. Most patients have total alopecia in addition to rickets. {ECO:0000269|PubMed:1652893, ECO:0000269|PubMed:17970811, ECO:0000269|PubMed:2177843, ECO:0000269|PubMed:2849209, ECO:0000269|PubMed:28698609, ECO:0000269|PubMed:7828346, ECO:0000269|PubMed:8106618, ECO:0000269|PubMed:8381803, ECO:0000269|PubMed:8392085, ECO:0000269|PubMed:8675579, ECO:0000269|PubMed:8961271, ECO:0000269|PubMed:9005998}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07530; DB08742; DB01436; DB04891; DB00146; DB02300; DB00136; DB00169; DB04540; DB05024; DB11672; DB14635; DB01070; DB06410; DB05295; DB06194; DB00153; DB04796; DB03451; DB00910; DB04258; DB11094 Interacts with P35222; Q09472; Q15648; P50222; Q15788; P26045; P19793; Q13573; Q13501; P04637; Q15645; Q9JLI4; P28700; X5D778; Q96HA8; Q01804; Q96S38; P48443 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; Disease variant; DNA-binding; Metal-binding; Nucleus; Proteomics identification; Receptor; Reference proteome; Transcription; Transcription regulation; Ubl conjugation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A Molecular weight (Da) 28781 Length 254 Aromaticity 0.07 Instability index 47.69 Isoelectric point 6.15 Charge (pH=7) -3.44 2D Binding mode Binding energy (Kcal/mol) -10.11 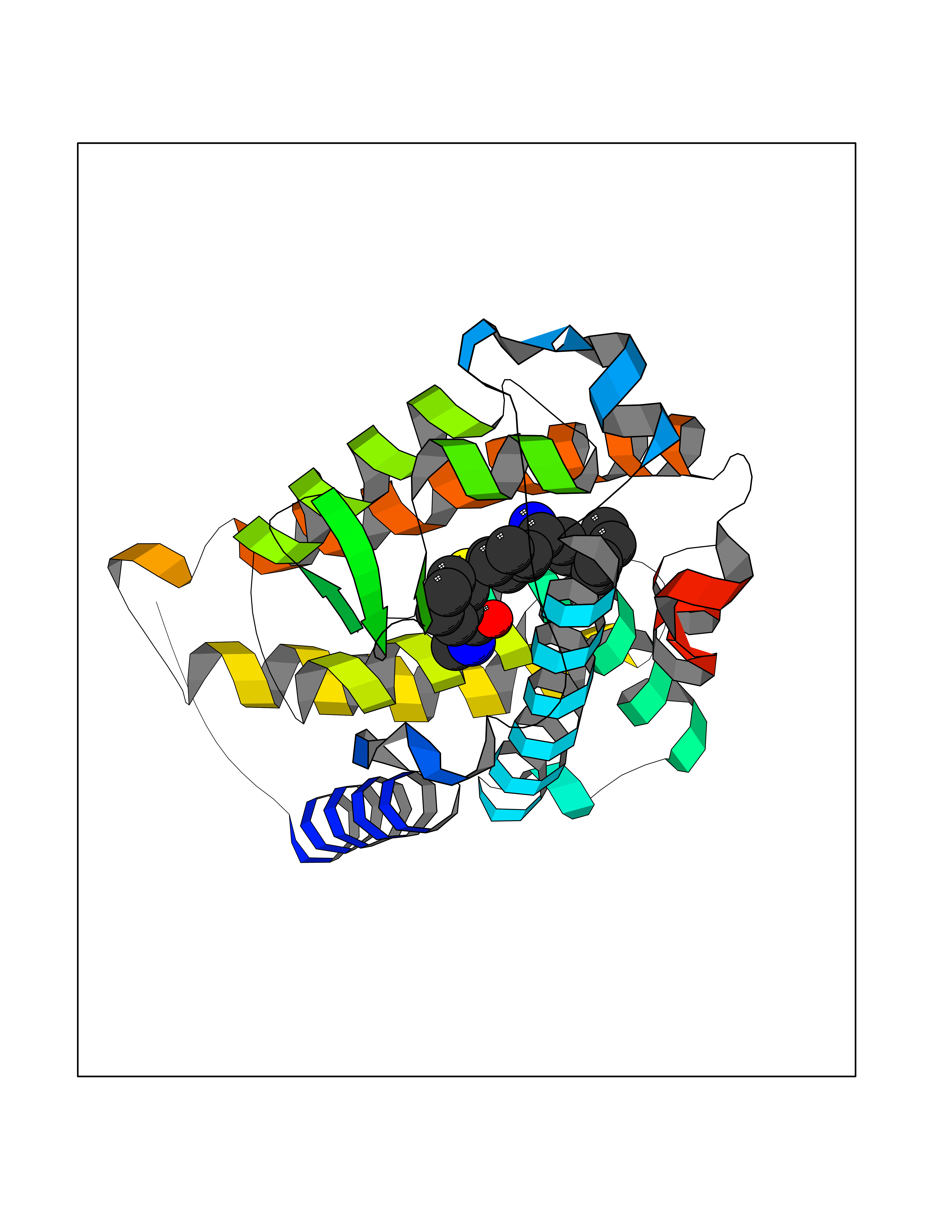 Molscript Map 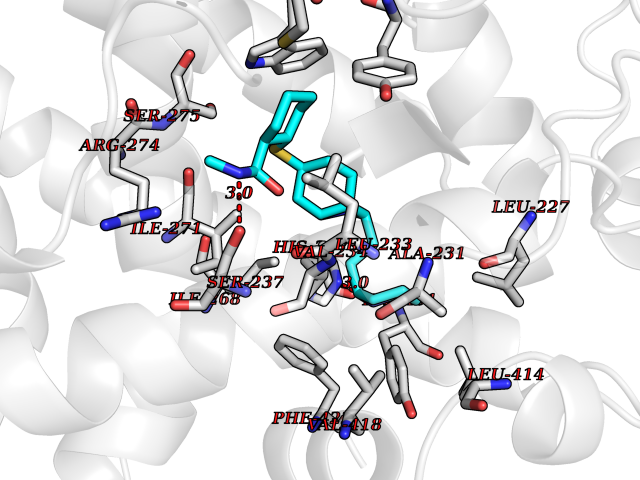 Pymol Map 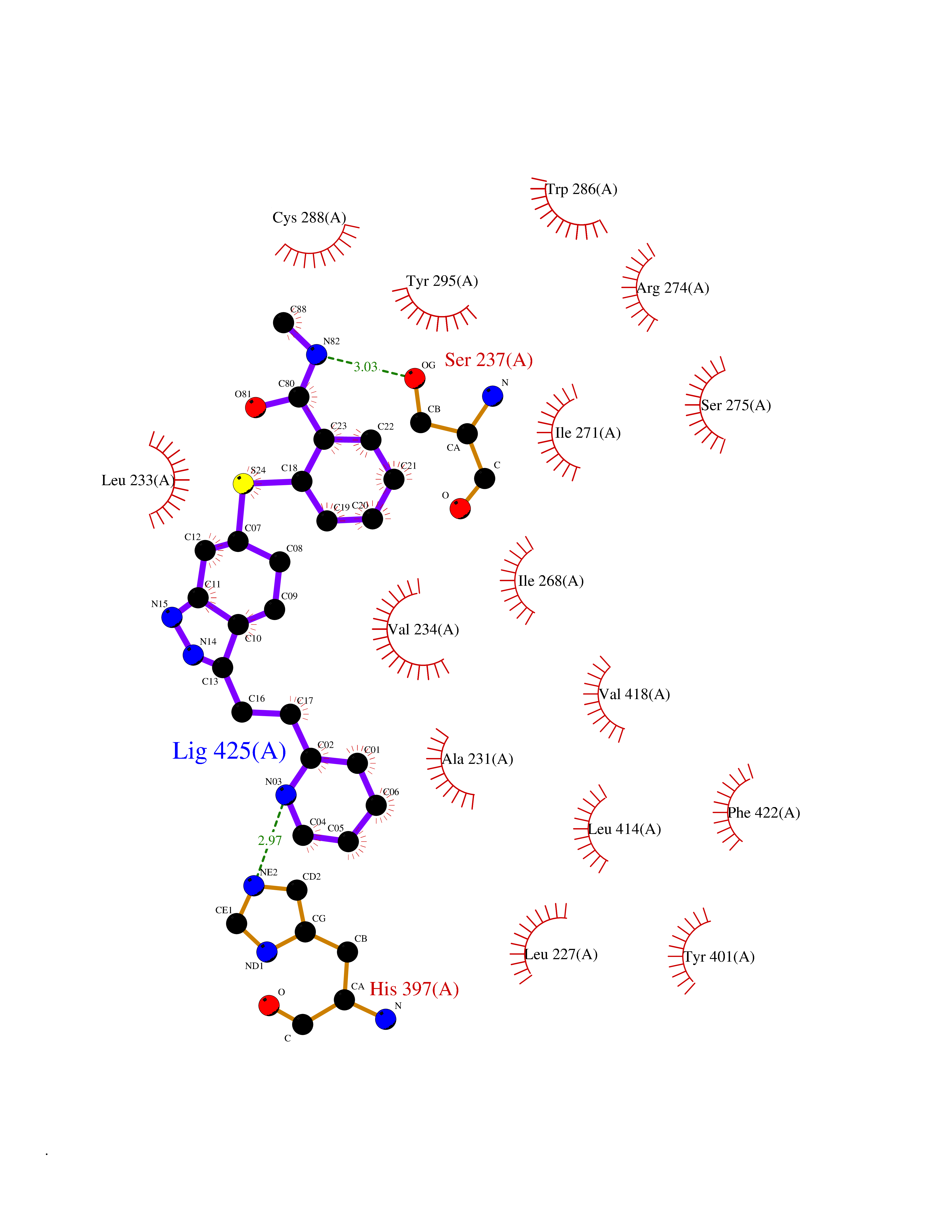 Ligplot Map 3D Binding mode Sequence ALRPKLSEEQQRIIAILLDAHHKTYDPTYSDFCQFRPPVRVNDGGGSVTLELSQLSMLPHLADLVSYSIQKVIGFAKMIPGFRDLTSEDQIVLLKSSAIEVIMLRSNESFTMDDMSWTCGNQDYKYRVSDVTKAGHSLELIEPLIKFQVGLKKLNLHEEEHVLLMAICIVSPDRPGVQDAALIEAIQDRLSNTLQTYIRCRHPPPGSHLLYAKMIQKLADLRSLNEEHSKQYRCLSFQPECSMKLTPLVLEVFG Hydrogen bonds contact Hydrophobic contact | ||||
| 15 | Kallikrein-5 (KLK5) | 6QFE | 8.42 | |
Target general information Gen name KLK5 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms UNQ570/PRO1132; Stratum corneum tryptic enzyme; SCTE; Kallikrein-like protein 2; KLK-L2 Protein family Peptidase S1 family, Kallikrein subfamily Biochemical class Peptidase Function May be involved in desquamation. Related diseases Lipodystrophy, familial partial, 8 (FPLD8) [MIM:620679]: An autosomal dominant form of partial lipodystrophy, a disorder characterized by abnormal subcutaneous fat distribution. FPLD8 patients show selective loss of subcutaneous adipose tissue from the limbs, beginning around 13 to 15 years of age, and abnormal accumulation of subcutaneous adipose tissue in the dorsal neck and face, as well as in the posterior thoracic and abdominal regions. The disorder is associated with metabolic abnormalities, including diabetes mellitus and hyperlipidemia. {ECO:0000269|PubMed:27376152}. The disease may be caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P20930; Q9NQG1 EC number EC 3.4.21.- Uniprot keywords 3D-structure; Disulfide bond; Glycoprotein; Hydrolase; Protease; Proteomics identification; Reference proteome; Secreted; Serine protease; Signal Protein physicochemical properties Chain ID A,B Molecular weight (Da) 50299.2 Length 454 Aromaticity 0.07 Instability index 40.74 Isoelectric point 9.25 Charge (pH=7) 23.09 2D Binding mode Binding energy (Kcal/mol) -8.95 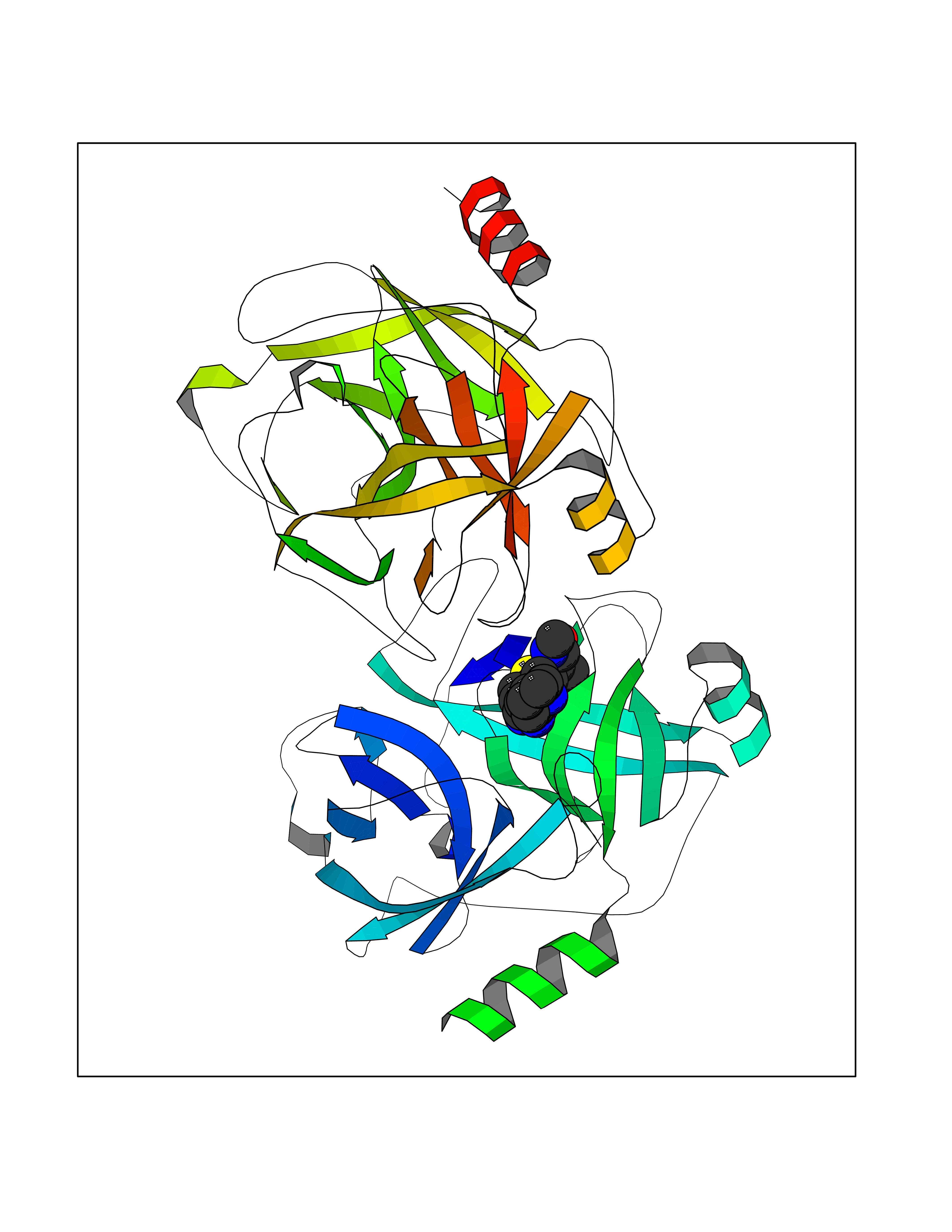 Molscript Map 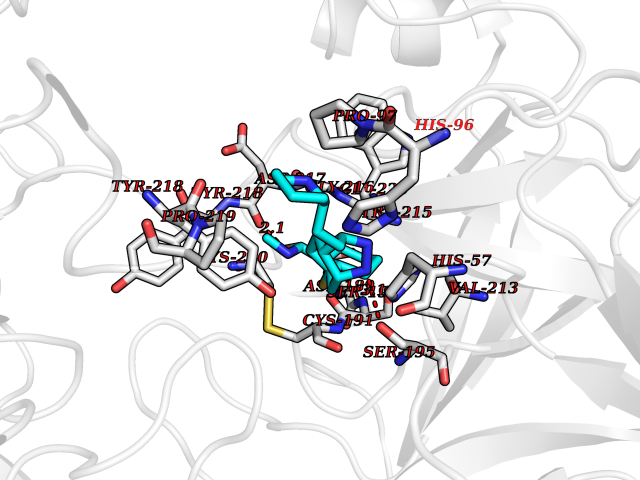 Pymol Map 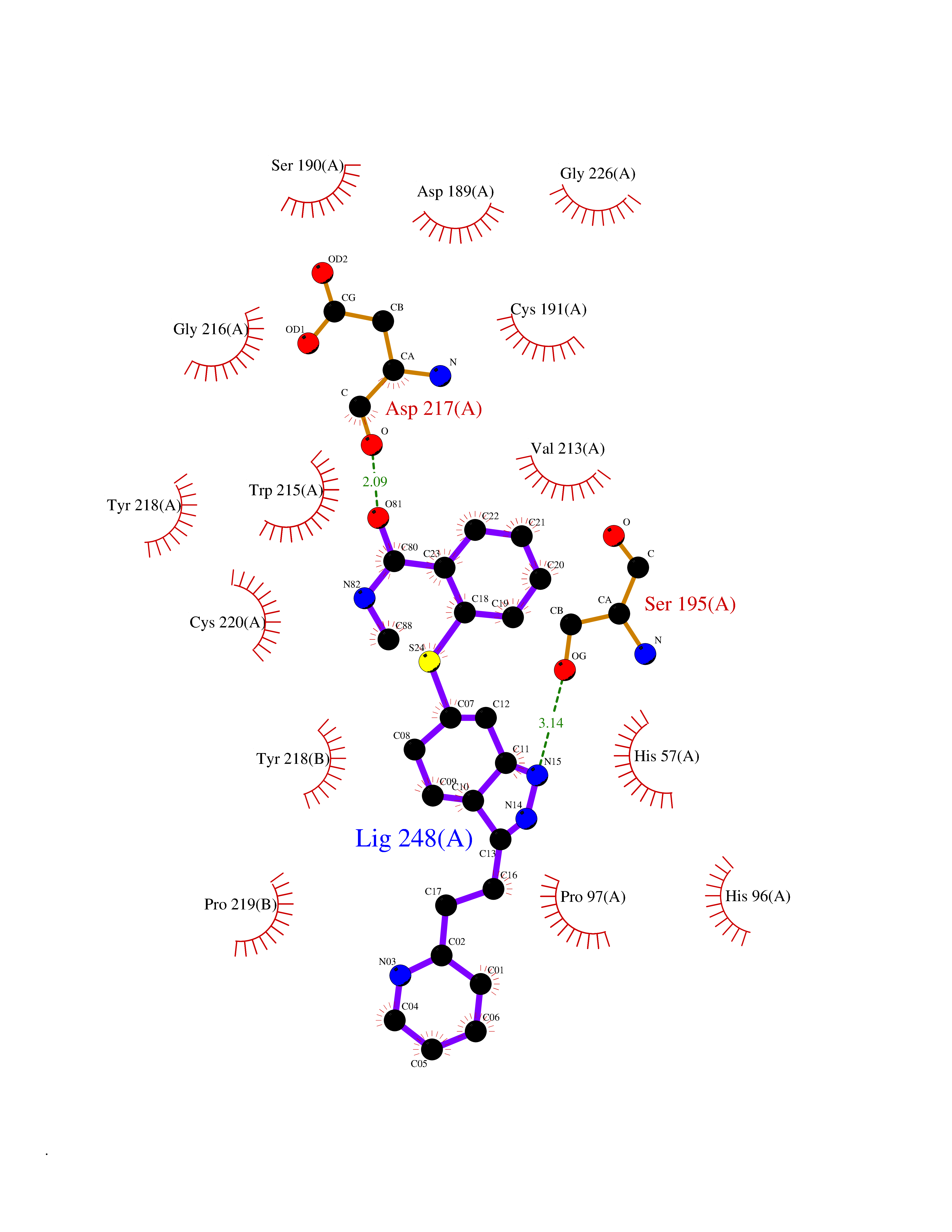 Ligplot Map 3D Binding mode Sequence IINGSDCDMHTQPWQAALLLRPNQLYCGAVLVHPQWLLTAAHCRKKVFRVRLGHYSLSPVYESGQQMFQGVKSIPHPGYSHPGHSNDLMLIKLNRRIRPTKDVRPINVSSHCPSAGTKCLVSGWGTTKSPQVHFPKVLQCLNISVLSQKRCEDAYPRQIDDTMFCAGDKAGRDSCQGDSGGPVVCNGSLQGLVSWGDYPCARPNRPGVYTNLCKFTKWIQETIQANSIINGSDCDMHTQPWQAALLLRPNQLYCGAVLVHPQWLLTAAHCRKKVFRVRLGHYSLSPVYESGQQMFQGVKSIPHPGYSHPGHSNDLMLIKLNRRIRPTKDVRPINVSSHCPSAGTKCLVSGWGTTKSPQVHFPKVLQCLNISVLSQKRCEDAYPRQIDDTMFCAGDKAGRDSCQGDSGGPVVCNGSLQGLVSWGDYPCARPNRPGVYTNLCKFTKWIQETIQANS Hydrogen bonds contact Hydrophobic contact | ||||
| 16 | Clostridium histolyticum Collagenase (CH colG) | 7Z5U | 8.41 | |
Target general information Gen name CH colG Organism Hathewaya histolytica (Clostridium histolyticum) Uniprot ID TTD ID Synonyms Microbial collagenase; Gelatinase ColG; Collagenase ColG; Class I collagenase Protein family Peptidase M9B family, Collagenase subfamily Biochemical class NA Function Clostridial collagenases are among the most efficient degraders of eukaryotic collagen known; saprophytes use collagen as a carbon source while pathogens additionally digest collagen to aid in host colonization. Has both tripeptidylcarboxypeptidase on Gly-X-Y and endopeptidase activities; the endopeptidase cuts within the triple helix region of collagen while tripeptidylcarboxypeptidase successively digests the exposed ends, thus clostridial collagenases can digest large sections of collagen. Active on soluble type I collagen, insoluble collagen, azocoll, soluble PZ-peptide (all collagenase substrates) and gelatin. The full-length protein has collagenase activity, while the in vivo derived C-terminally truncated shorter versions only act on gelatin. In vitro digestion of soluble calf skin collagen fibrils requires both ColG and ColH; ColG forms missing the second collagen-binding domain are also synergistic with ColH, although their overall efficiency is decreased. The activator domain (residues 119-388) and catalytic subdomain (389-670) open and close around substrate using a Gly-rich hinge (387-397), allowing digestion when the protein is closed. Binding of collagen requires Ca(2+) and is inhibited by EGTA; the collagen-binding domain (CBD, S3a plus S3b) specifically recognizes the triple-helical conformation made by 3 collagen protein chains in the triple-helical region. Isolated CBD (S3a plus S3b) binds collagen fibrils and sheets of many tissues. Related diseases Intellectual developmental disorder, X-linked, syndromic, Claes-Jensen type (MRXSCJ) [MIM:300534]: A disorder characterized by significantly below average general intellectual functioning associated with impairments in adaptive behavior and manifested during the developmental period. MRXSCJ patients manifest intellectual disability associated with variable features such as slowly progressive spastic paraplegia, seizures, facial dysmorphism. {ECO:0000269|PubMed:15586325, ECO:0000269|PubMed:16538222, ECO:0000269|PubMed:16541399, ECO:0000269|PubMed:17320160, ECO:0000269|PubMed:17468742, ECO:0000269|PubMed:23356856, ECO:0000269|PubMed:25666439}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number EC 3.4.24.3 Uniprot keywords 3D-structure; Calcium; Direct protein sequencing; Hydrolase; Metal-binding; Metalloprotease; Pharmaceutical; Protease; Repeat; Secreted; Signal; Virulence; Zinc; Zymogen Protein physicochemical properties Chain ID A Molecular weight (Da) 44751.2 Length 386 Aromaticity 0.15 Instability index 27.33 Isoelectric point 5.77 Charge (pH=7) -7.33 2D Binding mode Binding energy (Kcal/mol) -8.81 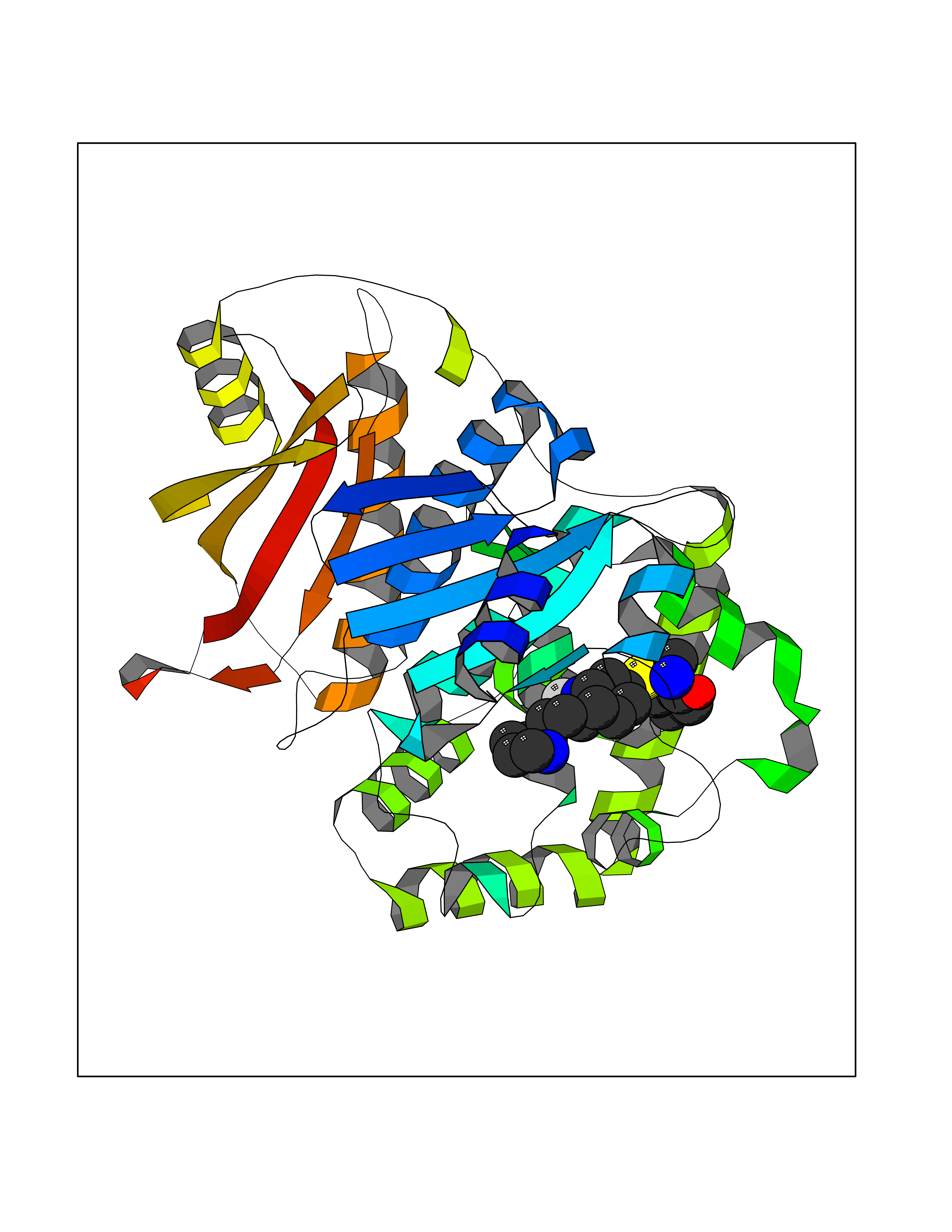 Molscript Map  Pymol Map 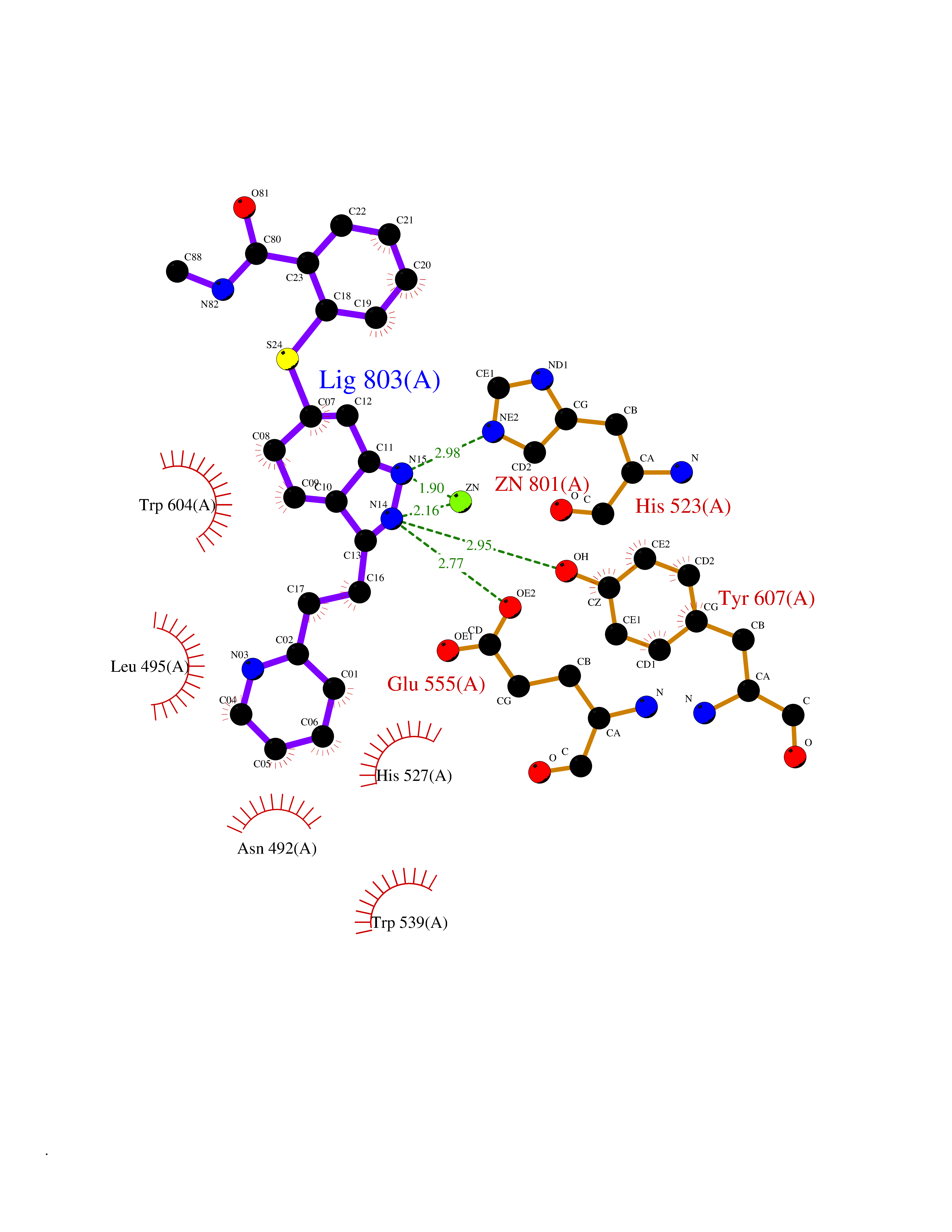 Ligplot Map 3D Binding mode Sequence DHDKFLDDAEKHYLPKTYTFDNGTFIIRAGDKVSEEKIKRLYWASREVKSQFHRVVGNDKALEVGNADDVLTMKIFNSPEEYKFNTTDNGGLYIEPRGTFYTYERTPQQSIFSLEELFRHEYTHYLQARYLVDGLWGQGPFYEKNRLTWFDEGTAEFFAGSTRTSGVLPRKLILGYLAKDKVDHRYSLKKTLNSGYDDSDWMFYNYGFAVAHYLYEKDMPTFIKMNKAILNTDVKSYDEIIKKLSDDANKNTEYQNHIQELVDKYQGAGIPLVSDDYLKDHGYKKASEVYSEISKAASLTNTSVTAEKSQYFNTFTLRGTYTGETSKGEFKDWDEMSKKLDGTLESLAKNSWSGYKTLTAYFTNYRVTSDNKVQYDVVFHGVLTDN Hydrogen bonds contact Hydrophobic contact | ||||
| 17 | Glucagon-like peptide 1 receptor (GLP1R) | 6X1A | 8.40 | |
Target general information Gen name GLP1R Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms GLP-1R; GLP-1-R; GLP-1 receptor Protein family G-protein coupled receptor 2 family Biochemical class GPCR secretin Function Ligand binding triggers activation of a signaling cascade that leads to the activation of adenylyl cyclase and increased intracellular cAMP levels. Plays a role in regulating insulin secretion in response to GLP-1. G-protein coupled receptor for glucagon-like peptide 1 (GLP-1). Related diseases Lynch syndrome 2 (LYNCH2) [MIM:609310]: A form of Lynch syndrome, an autosomal dominant disease associated with marked increase in cancer susceptibility. It is characterized by a familial predisposition to early-onset colorectal carcinoma (CRC) and extra-colonic tumors of the gastrointestinal, urological and female reproductive tracts. Lynch syndrome is reported to be the most common form of inherited colorectal cancer in the Western world. Clinically, it is often divided into two subgroups. Type I is characterized by hereditary predisposition to colorectal cancer, a young age of onset, and carcinoma observed in the proximal colon. Type II is characterized by increased risk for cancers in certain tissues such as the uterus, ovary, breast, stomach, small intestine, skin, and larynx in addition to the colon. Diagnosis of classical Lynch syndrome is based on the Amsterdam criteria: 3 or more relatives affected by colorectal cancer, one a first degree relative of the other two; 2 or more generation affected; 1 or more colorectal cancers presenting before 50 years of age; exclusion of hereditary polyposis syndromes. The term 'suspected Lynch syndrome' or 'incomplete Lynch syndrome' can be used to describe families who do not or only partially fulfill the Amsterdam criteria, but in whom a genetic basis for colon cancer is strongly suspected. {ECO:0000269|PubMed:10323887, ECO:0000269|PubMed:10375096, ECO:0000269|PubMed:10386556, ECO:0000269|PubMed:10413423, ECO:0000269|PubMed:10480359, ECO:0000269|PubMed:10598809, ECO:0000269|PubMed:10627141, ECO:0000269|PubMed:10660333, ECO:0000269|PubMed:10671064, ECO:0000269|PubMed:10713887, ECO:0000269|PubMed:10777691, ECO:0000269|PubMed:10882759, ECO:0000269|PubMed:11139242, ECO:0000269|PubMed:11427529, ECO:0000269|PubMed:11726306, ECO:0000269|PubMed:11748856, ECO:0000269|PubMed:11754112, ECO:0000269|PubMed:11781295, ECO:0000269|PubMed:11793442, ECO:0000269|PubMed:11839723, ECO:0000269|PubMed:11870161, ECO:0000269|PubMed:12095971, ECO:0000269|PubMed:12132870, ECO:0000269|PubMed:12200596, ECO:0000269|PubMed:12362047, ECO:0000269|PubMed:12373605, ECO:0000269|PubMed:12655562, ECO:0000269|PubMed:12658575, ECO:0000269|PubMed:14635101, ECO:0000269|PubMed:14961575, ECO:0000269|PubMed:15064764, ECO:0000269|PubMed:15139004, ECO:0000269|PubMed:15365995, ECO:0000269|PubMed:15365996, ECO:0000269|PubMed:16083711, ECO:0000269|PubMed:16451135, ECO:0000269|PubMed:17301300, ECO:0000269|PubMed:17510385, ECO:0000269|PubMed:18561205, ECO:0000269|PubMed:20020535, ECO:0000269|PubMed:21120944, ECO:0000269|PubMed:22753075, ECO:0000269|PubMed:7757073, ECO:0000269|PubMed:8566964, ECO:0000269|PubMed:8571956, ECO:0000269|PubMed:8574961, ECO:0000269|PubMed:8797773, ECO:0000269|PubMed:8872463, ECO:0000269|PubMed:8993976, ECO:0000269|PubMed:9048925, ECO:0000269|PubMed:9067757, ECO:0000269|PubMed:9218993, ECO:0000269|PubMed:9272156, ECO:0000269|PubMed:9298827, ECO:0000269|PubMed:9311737, ECO:0000269|PubMed:9326924, ECO:0000269|PubMed:9399661, ECO:0000269|PubMed:9559627, ECO:0000269|PubMed:9718327, ECO:0000269|PubMed:9833759, ECO:0000269|PubMed:9927034, ECO:0000269|Ref.5}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Mismatch repair cancer syndrome 1 (MMRCS1) [MIM:276300]: An autosomal recessive form of mismatch repair cancer syndrome, a childhood cancer predisposition syndrome encompassing a broad tumor spectrum. This includes hematological malignancies, central nervous system tumors, Lynch syndrome-associated malignancies such as colorectal tumors as well as multiple intestinal polyps, embryonic tumors and rhabdomyosarcoma. Multiple cafe-au-lait macules, a feature reminiscent of neurofibromatosis type 1, are often found as first manifestation of the underlying cancer. {ECO:0000269|PubMed:11427529, ECO:0000269|PubMed:17440981, ECO:0000269|PubMed:7661930}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Muir-Torre syndrome (MRTES) [MIM:158320]: Rare autosomal dominant disorder characterized by sebaceous neoplasms and visceral malignancy. {ECO:0000269|PubMed:8751876}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Defects in MLH1 may contribute to lobular carcinoma in situ (LCIS), a non-invasive neoplastic disease of the breast.; DISEASE: Endometrial cancer (ENDMC) [MIM:608089]: A malignancy of endometrium, the mucous lining of the uterus. Most endometrial cancers are adenocarcinomas, cancers that begin in cells that make and release mucus and other fluids. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Some epigenetic changes can be transmitted unchanged through the germline (termed 'epigenetic inheritance'). Evidence that this mechanism occurs in humans is provided by the identification of individuals in whom 1 allele of the MLH1 gene is epigenetically silenced throughout the soma (implying a germline event). These individuals are affected by Lynch syndrome but does not have identifiable mutations in MLH1, even though it is silenced, which demonstrates that an epimutation can phenocopy a genetic disease.; DISEASE: Colorectal cancer (CRC) [MIM:114500]: A complex disease characterized by malignant lesions arising from the inner wall of the large intestine (the colon) and the rectum. Genetic alterations are often associated with progression from premalignant lesion (adenoma) to invasive adenocarcinoma. Risk factors for cancer of the colon and rectum include colon polyps, long-standing ulcerative colitis, and genetic family history. {ECO:0000269|PubMed:10598809, ECO:0000269|PubMed:10882759, ECO:0000269|PubMed:12132870, ECO:0000269|PubMed:12655564, ECO:0000269|PubMed:14504054, ECO:0000269|PubMed:15184898, ECO:0000269|PubMed:18033691, ECO:0000269|PubMed:8872463, ECO:0000269|PubMed:9032648, ECO:0000269|PubMed:9087566, ECO:0000269|PubMed:9611074}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB09043; DB09045; DB15650; DB01276; DB00040; DB16697; DB06655; DB09265; DB13928; DB14027; DB15171 Interacts with A8MQ03; Q07627; Q8IUG1; P60409; P60410; P60411; Q9BYP8; P26371; Q7Z3S9; P0DPK4 EC number NA Uniprot keywords 3D-structure; ADP-ribosylation; Cell membrane; Disulfide bond; G-protein coupled receptor; Glycoprotein; Membrane; Receptor; Reference proteome; Signal; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID R Molecular weight (Da) 45579.6 Length 390 Aromaticity 0.16 Instability index 39.66 Isoelectric point 6.73 Charge (pH=7) -0.68 2D Binding mode Binding energy (Kcal/mol) -9.98 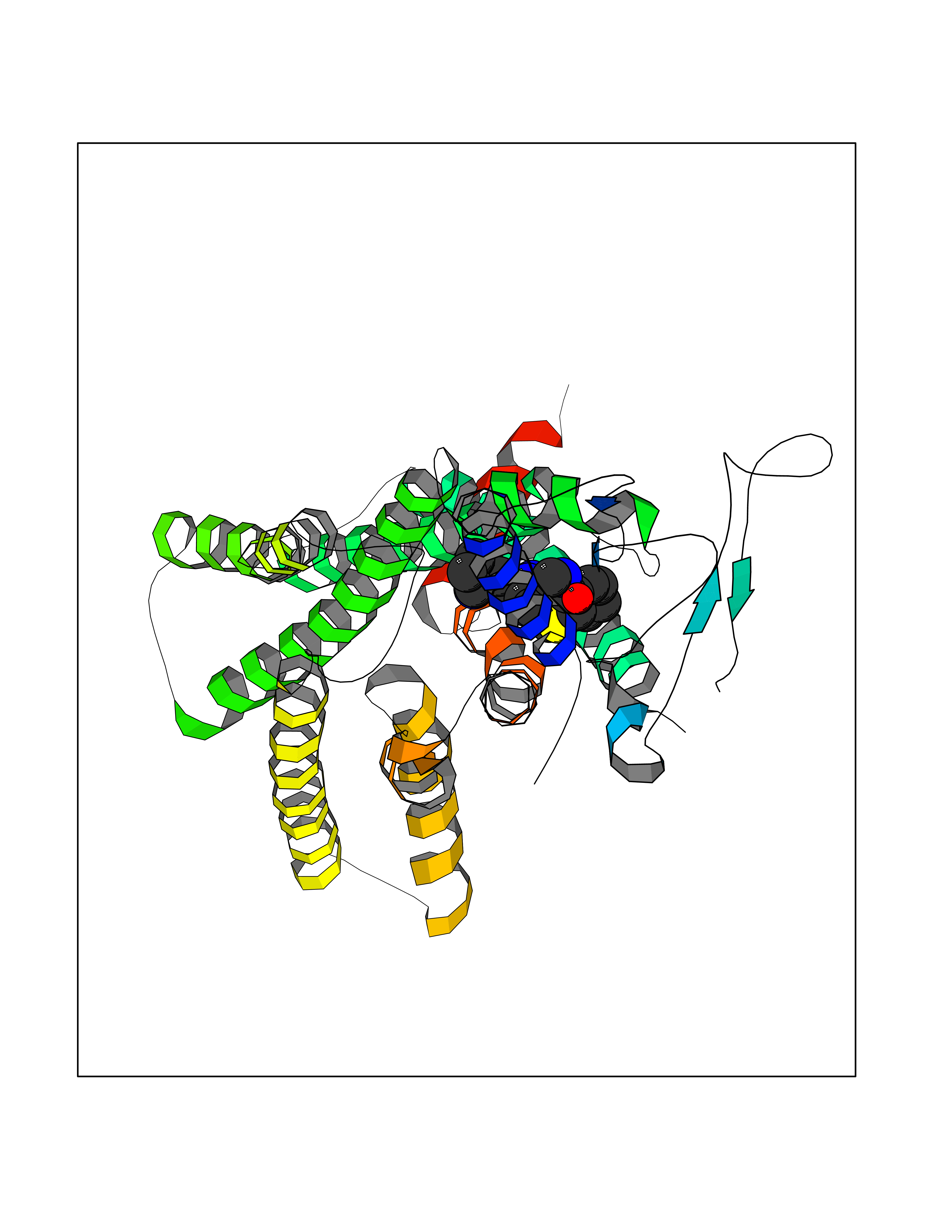 Molscript Map 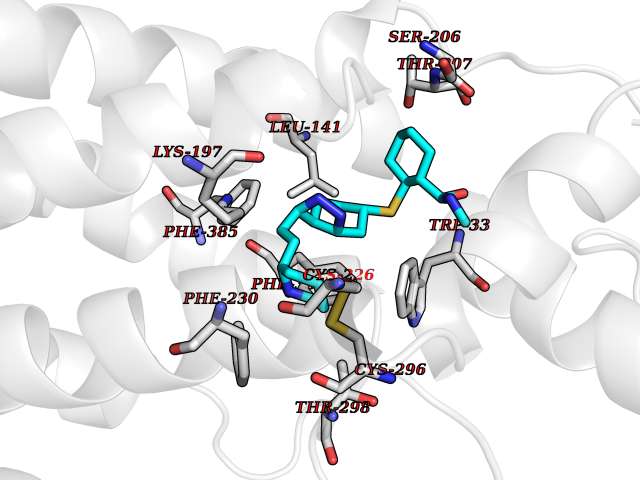 Pymol Map 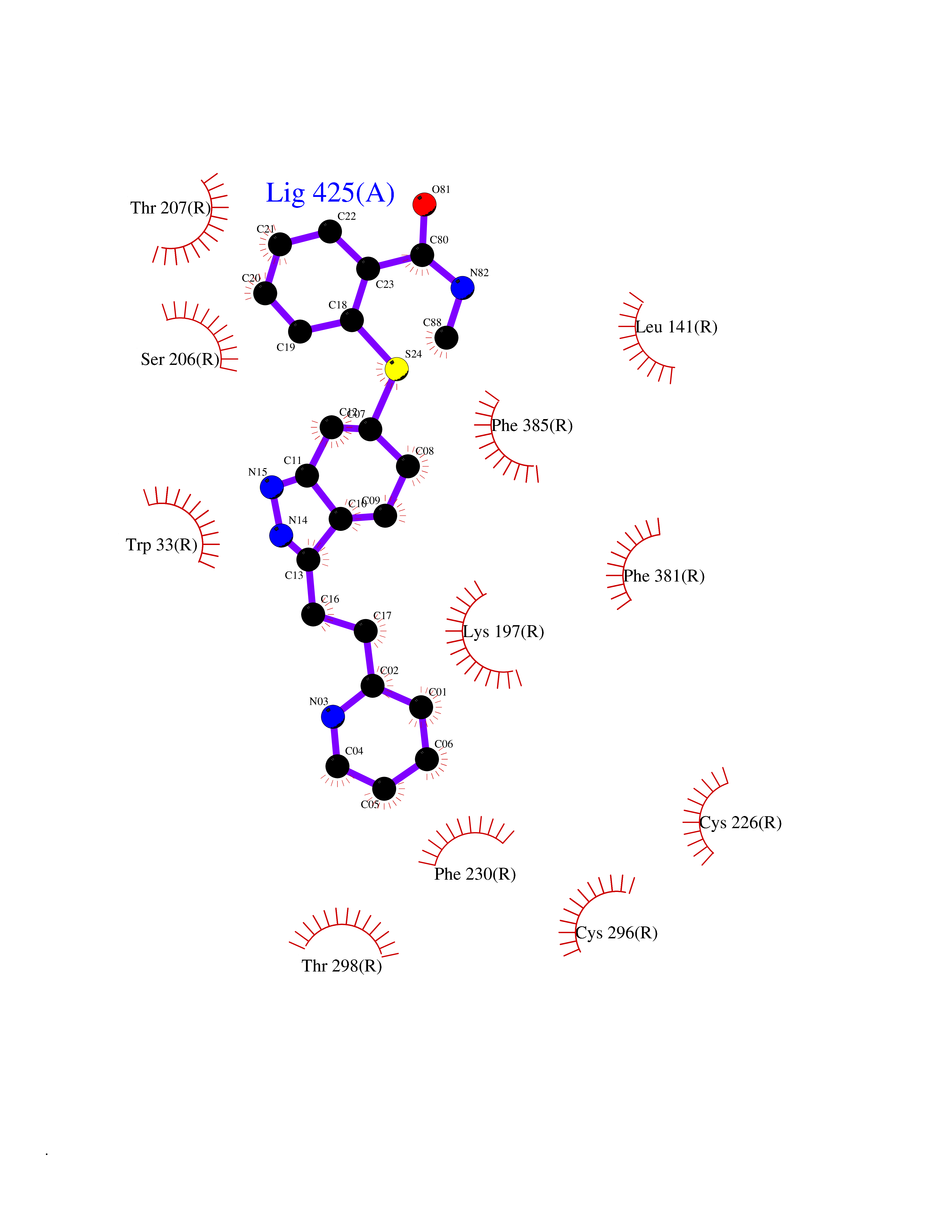 Ligplot Map 3D Binding mode Sequence ATVSLWETVQKWREYRRQCQRSLTEDPPPATDLFCNRTFDEYACWPDGEPGSFVNVSCPWYLPWASSVPQGHVYRFCTAEGLWLQKDNSSLPWRDLSECEESSPEEQLLFLYIIYTVGYALSFSALVIASAILLGFRHLHCTRNYIHLNLFASFILRALSVFIKDAALKWMYSTAAQQHQWDGLLSYQDSLSCRLVFLLMQYCVAANYYWLLVEGVYLYTLLAFSVFSEQWIFRLYVSIGWGVPLLFVVPWGIVKYLYEDEGCWTRNSNMNYWLIIRLPILFAIGVNFLIFVRVICIVVSKLKANLMCKTDIKCRLAKSTLTLIPLLGTHEVIFAFVMDEHARGTLRFIKLFTELSFTSFQGLMVAILYCFVNNEVQLEFRKSWERWRLE Hydrogen bonds contact Hydrophobic contact | ||||
| 18 | Multidrug resistance protein 3 (ABCB4) | 6S7P | 8.38 | |
Target general information Gen name ABCB4 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms PGY3; MDR3; ATP-binding cassette sub-family B member 4; ABCB4 Protein family ABC transporter superfamily, ABCB family, Multidrug resistance exporter (TC 3.A.1.201) subfamily Biochemical class Acid anhydrides hydrolase Function Mediates ATP-dependent export of organic anions and drugs from the cytoplasm. Hydrolyzes ATP with low efficiency. Not capable of conferring drug resistance. Mediates the translocation of phosphatidylcholine across the canalicular membrane of the hepatocyte. Related diseases Cholestasis, progressive familial intrahepatic, 3 (PFIC3) [MIM:602347]: A disorder characterized by early onset of cholestasis that progresses to hepatic fibrosis, cirrhosis, and end-stage liver disease before adulthood. PFIC3 inheritance is autosomal recessive. {ECO:0000269|PubMed:11313315, ECO:0000269|PubMed:12671900, ECO:0000269|PubMed:17726488, ECO:0000269|PubMed:21119540, ECO:0000269|PubMed:24045840, ECO:0000269|PubMed:24594635, ECO:0000269|PubMed:24806754, ECO:0000269|PubMed:28012258, ECO:0000269|PubMed:9419367}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Cholestasis of pregnancy, intrahepatic 3 (ICP3) [MIM:614972]: A liver disorder of pregnancy. It presents during the second or, more commonly, the third trimester of pregnancy with intense pruritus which becomes more severe with advancing gestation and cholestasis. It causes fetal distress, spontaneous premature delivery and intrauterine death. Patients have spontaneous and progressive disappearance of cholestasis after delivery. Cholestasis results from abnormal biliary transport from the liver into the small intestine. {ECO:0000269|PubMed:10767346, ECO:0000269|PubMed:12746424, ECO:0000269|PubMed:15077010}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Gallbladder disease 1 (GBD1) [MIM:600803]: One of the major digestive diseases. Gallstones composed of cholesterol (cholelithiasis) are the common manifestations in western countries. Most people with gallstones, however, remain asymptomatic through their lifetimes. {ECO:0000269|PubMed:11313316, ECO:0000269|PubMed:12891548, ECO:0000269|PubMed:22331132, ECO:0000269|PubMed:23533021, ECO:0000269|PubMed:24723470, ECO:0000269|PubMed:28012258, ECO:0000269|PubMed:28587926, ECO:0000269|Ref.2}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB06414; DB06207 Interacts with NA EC number EC 7.6.2.1 Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Cell membrane; Cytoplasm; Cytoplasmic vesicle; Disease variant; Glycoprotein; Intrahepatic cholestasis; Lipid transport; Membrane; Nucleotide-binding; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Translocase; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A Molecular weight (Da) 123919 Length 1128 Aromaticity 0.1 Instability index 29.44 Isoelectric point 8.78 Charge (pH=7) 11.08 2D Binding mode Binding energy (Kcal/mol) -8.82 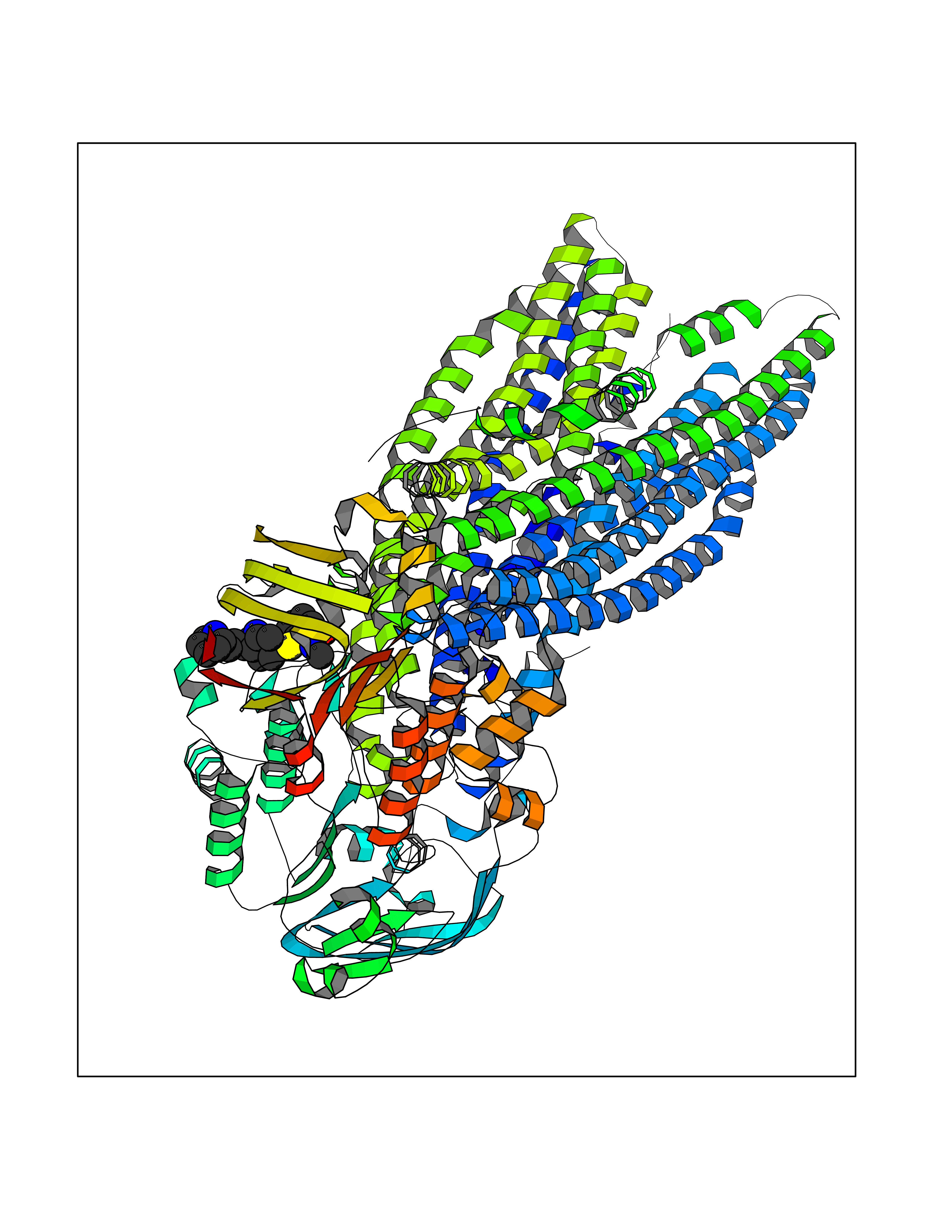 Molscript Map 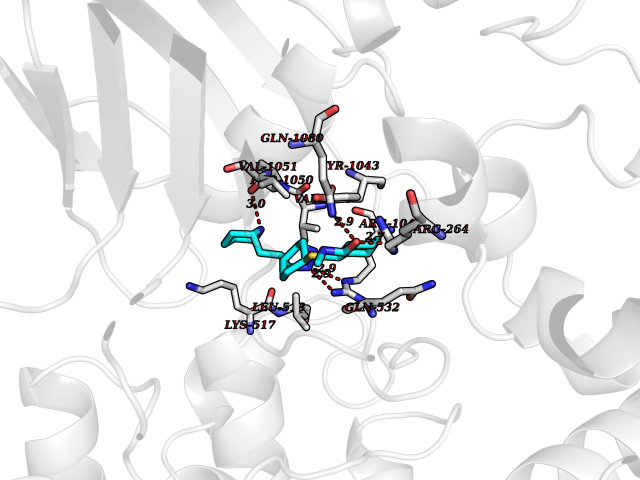 Pymol Map 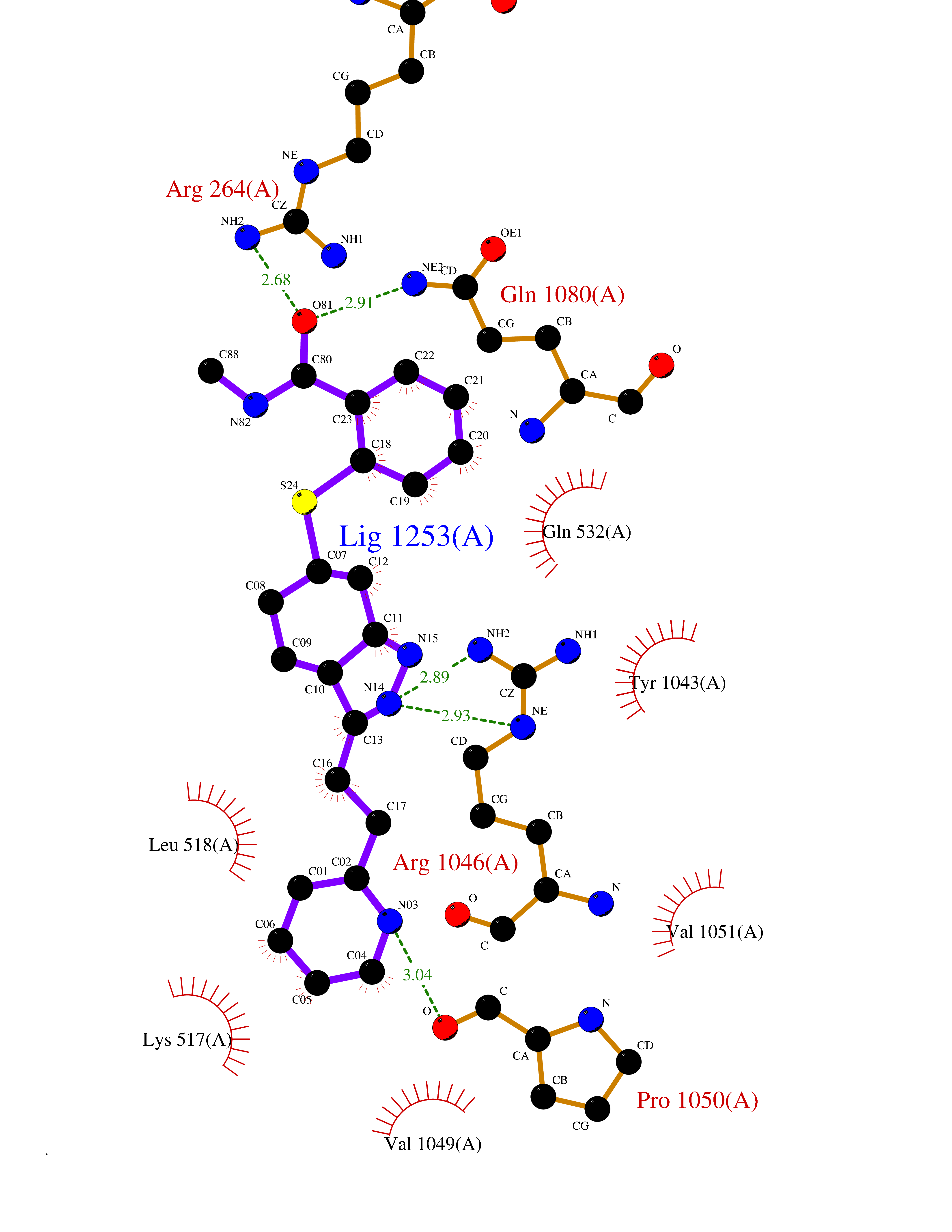 Ligplot Map 3D Binding mode Sequence VLTLFRYSDWQDKLFMSLGTIMAIAHGSGLPLMMIVFGEMTDKPGKILEEEMTRYAYYYSGLGAGVLVAAYIQVSFWTLAAGRQIRKIRQKFFHAILRQEIGWFDINDTTELNTRLTDDISKISEGIGDKVGMFFQAVATFFAGFIVGFIRGWKLTLVIMAISPILGLSAAVWAKILSAFSDKELAAYAKAGAVAEEALGAIRTVIAFGGQNKELERYQKHLENAKEIGIKKAISANISMGIAFLLIYASYALAFWYGSTLVISKEYTIGNAMTVFFSILIGAFSVGQAAPCIDAFANARGAAYVIFDIIDNNPKIDSFSERGHKPDSIKGNLEFNDVHFSYPSRANVKILKGLNLKVQSGQTVALVGSSGCGKSTTVQLIQRLYDPDEGTINIDGQDIRNFNVNYLREIIGVVSQEPVLFSTTIAENICYGRGNVTMDEIKKAVKEANAYEFIMKLPQKFDTLVGERGAQLSGGQKQRIAIARALVRNPKILLLDQATSALDTESEAEVQAALDKAREGRTTIVIAHRLSTVRNADVIAGFEDGVIVEQGSHSELMKKEGVYFKLVNVPPVSFLKVLKLNKTEWPYFVVGTVCAIANGGLQPAFSVIFSEIIAIFGPGDDAVKQQKCNIFSLIFLFLGIISFFTFFLQGFTFGKAGEILTRRLRSMAFKAMLRQDMSWFDDHKNSTGALSTRLATDAAQVQGATGTRLALIAQNIANLGTGIIISFIYGWQLTLLLLAVVPIIAVSGIVEMKLLAGNAKRDKKELEAAGKIATEAIENIRTVVSLTQERKFESMYVEKLYGPYRNSVQKAHIYGITFSISQAFMYFSYAGCFRFGAYLIVNGHMRFRDVILVFSAIVFGAVALGHASSFAPDYAKAKLSAAHLFMLFERQPLIDSYSEEGLKPDKFEGNITFNEVVFNYPTRANVPVLQGLSLEVKKGQTLALVGSSGCGKSTVVQLLERFYDPLAGTVLLDGQEAKKLNVQWLRAQLGIVSQEPILFDCSIAENIAYGDNSRVVSQDEIVSAAKAANIHPFIETLPHKYETRVGDKGTQLSGGQKQRIAIARALIRQPQILLLDQATSALDTESEKVVQEALDKAREGRTCIVIAHRLSTIQNADLIVVFQNGRVK Hydrogen bonds contact Hydrophobic contact | ||||
| 19 | Oxalosuccinate decarboxylase (IDH1) | 6ADG | 8.35 | |
Target general information Gen name IDH1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms PICD; NADP(+)-specific ICDH; Isocitrate dehydrogenase [NADP] cytoplasmic; IDP; IDH; Cytosolic NADP-isocitrate dehydrogenase Protein family Isocitrate and isopropylmalate dehydrogenases family Biochemical class Short-chain dehydrogenases reductase Function Catalyses the NADPH-dependent reduction of alpha-ketoglutarate to R(-)-2-hydroxyglutarate (2HG). Related diseases Glioma (GLM) [MIM:137800]: Gliomas are benign or malignant central nervous system neoplasms derived from glial cells. They comprise astrocytomas and glioblastoma multiforme that are derived from astrocytes, oligodendrogliomas derived from oligodendrocytes and ependymomas derived from ependymocytes. {ECO:0000269|PubMed:19117336, ECO:0000269|PubMed:19935646}. The gene represented in this entry is involved in disease pathogenesis. Mutations affecting Arg-132 are tissue-specific, and suggest that this residue plays a unique role in the development of high-grade gliomas. Mutations of Arg-132 to Cys, His, Leu or Ser abolish magnesium binding and abolish the conversion of isocitrate to alpha-ketoglutarate. Instead, alpha-ketoglutarate is converted to R(-)-2-hydroxyglutarate. Elevated levels of R(-)-2-hydroxyglutarate are correlated with an elevated risk of malignant brain tumors. {ECO:0000269|PubMed:19935646}.; DISEASE: Genetic variations are associated with cartilaginous tumors such as enchondroma or chondrosarcoma. Mutations of Arg-132 to Cys, Gly or His abolish the conversion of isocitrate to alpha-ketoglutarate. Instead, alpha-ketoglutarate is converted to R(-)-2-hydroxyglutarate. {ECO:0000269|PubMed:26161668}. Drugs (DrugBank ID) DB09374; DB01727; DB14568; DB03461; DB16267 Interacts with P0DP23; P27797; P36957; O75874; Q8TDX7; P16284; P17612; P50454; P37173; Q05086-3 EC number EC 1.1.1.42 Uniprot keywords 3D-structure; Acetylation; Cytoplasm; Direct protein sequencing; Glyoxylate bypass; Magnesium; Manganese; Metal-binding; NADP; Oxidoreductase; Peroxisome; Phosphoprotein; Proteomics identification; Reference proteome; Tricarboxylic acid cycle Protein physicochemical properties Chain ID A,B Molecular weight (Da) 92711.7 Length 823 Aromaticity 0.1 Instability index 26.74 Isoelectric point 6.42 Charge (pH=7) -4.48 2D Binding mode Binding energy (Kcal/mol) -9.5 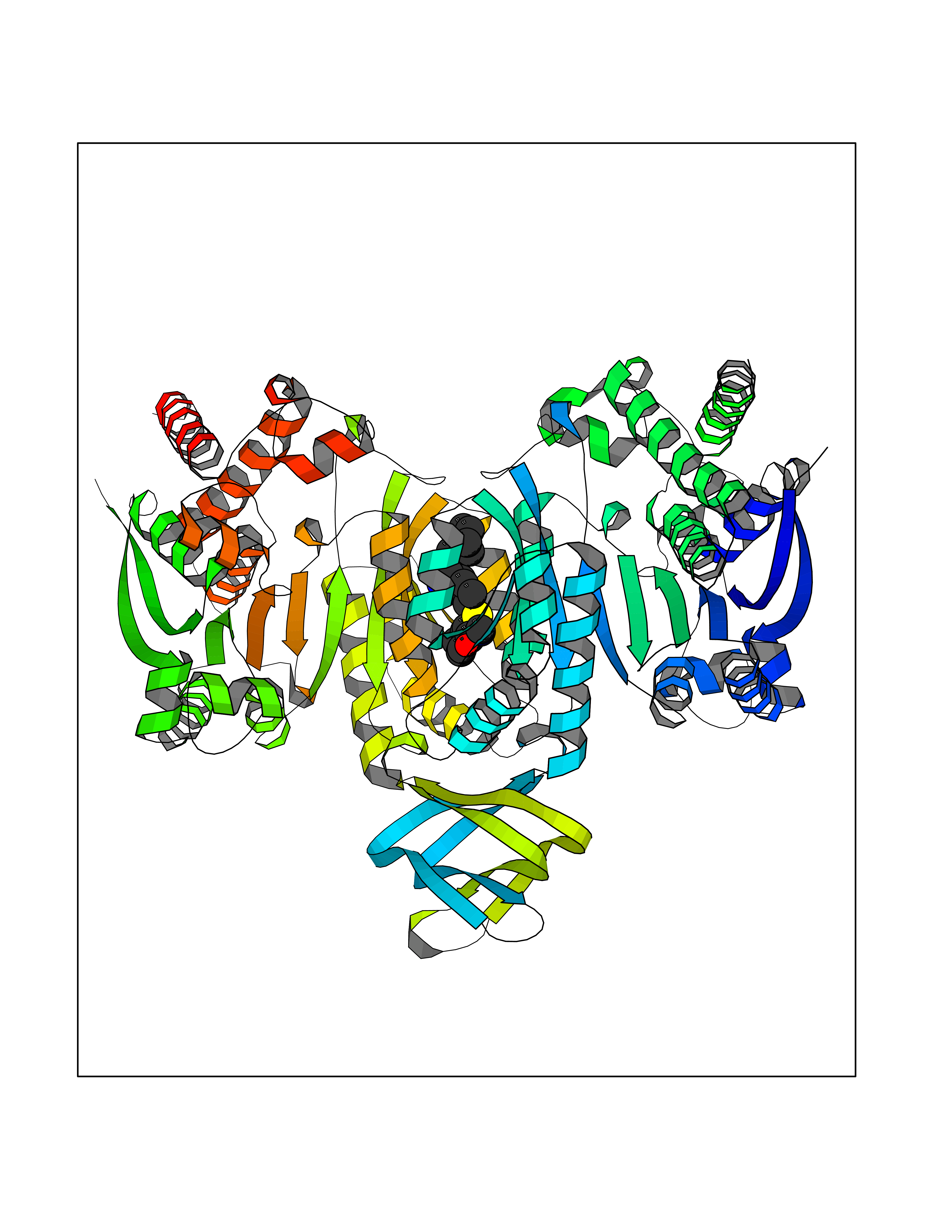 Molscript Map 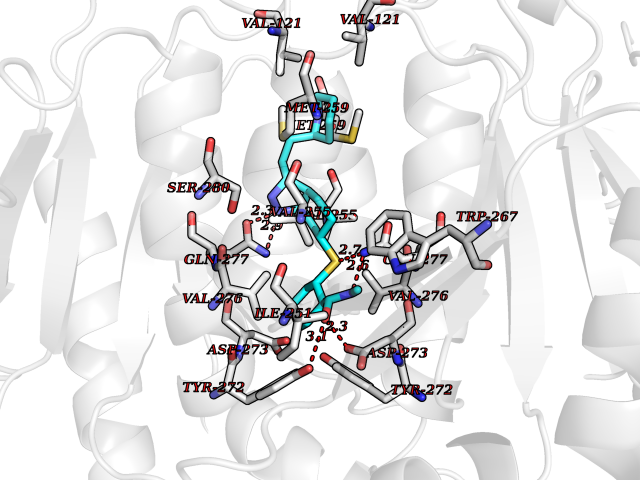 Pymol Map 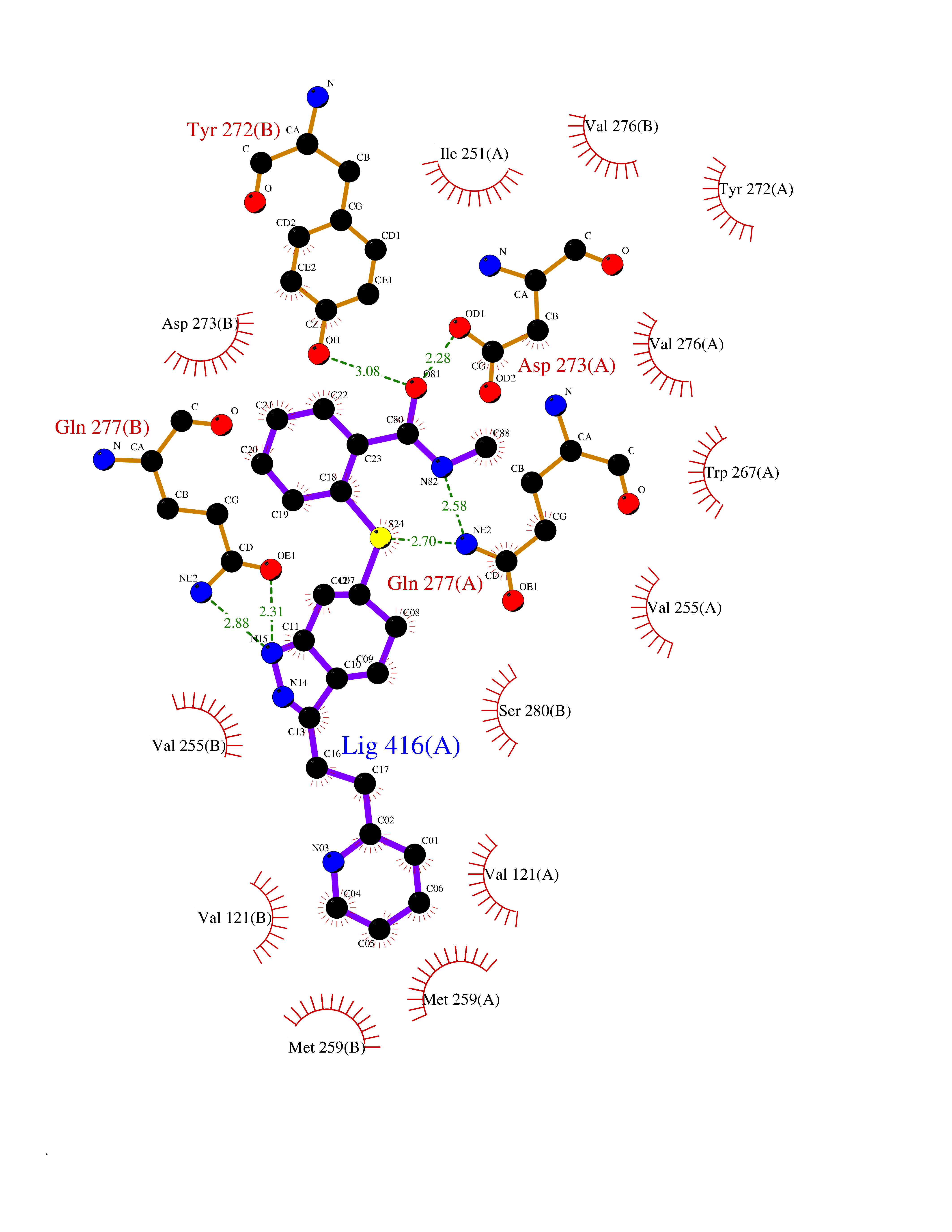 Ligplot Map 3D Binding mode Sequence KKISGGSVVEMQGDEMTRIIWELIKEKLIFPYVELDLHSYDLGIENRDATNDQVTKDAAEAIKKHNVGVKCATITPDEKRVEEFKLKQMWKSPNGTIRNILGGTVFREAIICKNIPRLVSGWVKPIIIGHHAYGDQYRATDFVVPGPGKVEITYTPSDGTQKVTYLVHNFEEGGGVAMGMYNQDKSIEDFAHSSFQMALSKGWPLYLSTKNTILKKYDGRFKDIFQEIYDKQYKSQFEAQKIWYEHRLIDDMVAQAMKSEGGFIWACKNYDGDVQSDSVAQGYGSLGMMTSVLVCPDGKTVEAEAAHGTVTRHYRMYQKGQETSTNPIASIFAWTRGLAHRAKLDNNKELAFFANALEEVSIETIEAGFMTKDLAACIKGLPNVQRSDYLNTFEFMDKLGENLKIKLAQAKLKKISGGSVVEMQGDEMTRIIWELIKEKLIFPYVELDLHSYDLGIENRDATNDQVTKDAAEAIKKHNVGVKCATITPDEKRVEEFKLKQMWKSPNGTIRNILGGTVFREAIICKNIPRLVSGWVKPIIIGHHAYGDQYRATDFVVPGPGKVEITYTPSDGTQKVTYLVHNFEEGGGVAMGMYNQDKSIEDFAHSSFQMALSKGWPLYLSTKNTILKKYDGRFKDIFQEIYDKQYKSQFEAQKIWYEHRLIDDMVAQAMKSEGGFIWACKNYDGDVQSDSVAQGYGSLGMMTSVLVCPDGKTVEAEAAHGTVTRHYRMYQKGQETSTNPIASIFAWTRGLAHRAKLDNNKELAFFANALEEVSIETIEAGFMTKDLAACIKGLPNVQRSDYLNTFEFMDKLGENLKIKLAQAK Hydrogen bonds contact Hydrophobic contact | ||||
| 20 | Dopamine beta-hydroxylase | 4ZEL | 8.33 | |
Target general information Gen name DBH Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Copper type II ascorbate-dependent monooxygenase family Biochemical class Oxidoreductase Function Catalytic activity.Copper ion binding.Dopamine beta-monooxygenase activity.L-ascorbic acid binding. Related diseases Orthostatic hypotension 1 (ORTHYP1) [MIM:223360]: A form of orthostatic hypotension due to congenital dopamine beta-hydroxylase deficiency. Orthostatic hypotension, also known as postural hypotension, is a finding defined as a 20-mm Hg decrease in systolic pressure or a 10-mm Hg decrease in diastolic pressure occurring 3 minutes after a person has risen from supine to standing. Symptoms include dizziness, blurred vision, and sometimes syncope. ORTHYP1 is an autosomal recessive condition apparent from infancy or early childhood and characterized by low plasma and urinary levels of norepinephrine and epinephrine, and episodic hypoglycemia. {ECO:0000269|PubMed:11857564}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00126; DB06774; DB09130; DB05394; DB00822; DB00988; DB00968; DB00550 Interacts with P00352; P63010-2; Q04656; Q8WUW1; Q9UNS2; Q71DI3; P61978; Q9Y2M5; Q92876; P08727; Q14693; P0DPK4; Q6GQQ9-2; P27986-2; Q9ULX5; Q96D59; Q8N6K7-2; Q9GZS3; Q8IUW3; Q86WT6-2 EC number 1.14.17.1 Uniprot keywords 3D-structure; Catecholamine biosynthesis; Copper; Cytoplasmic vesicle; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; Membrane; Metal-binding; Monooxygenase; Oxidoreductase; Proteomics identification; Reference proteome; Secreted; Signal-anchor; Transmembrane; Transmembrane helix; Vitamin C Protein physicochemical properties Chain ID A,B Molecular weight (Da) 123694 Length 1094 Aromaticity 0.1 Instability index 51.85 Isoelectric point 5.84 Charge (pH=7) -24.5 2D Binding mode Binding energy (Kcal/mol) -8.59  Molscript Map 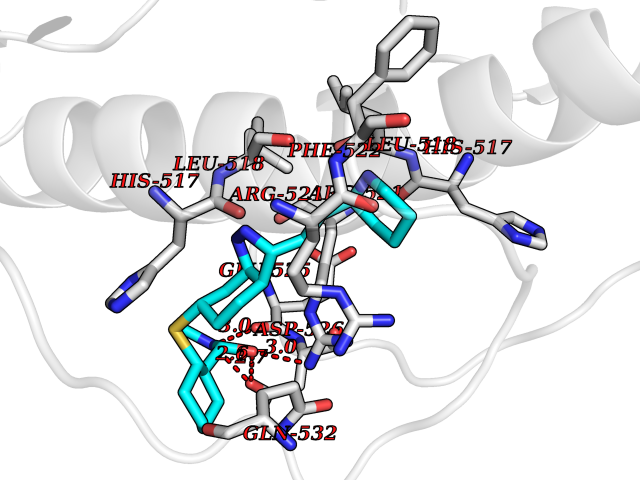 Pymol Map 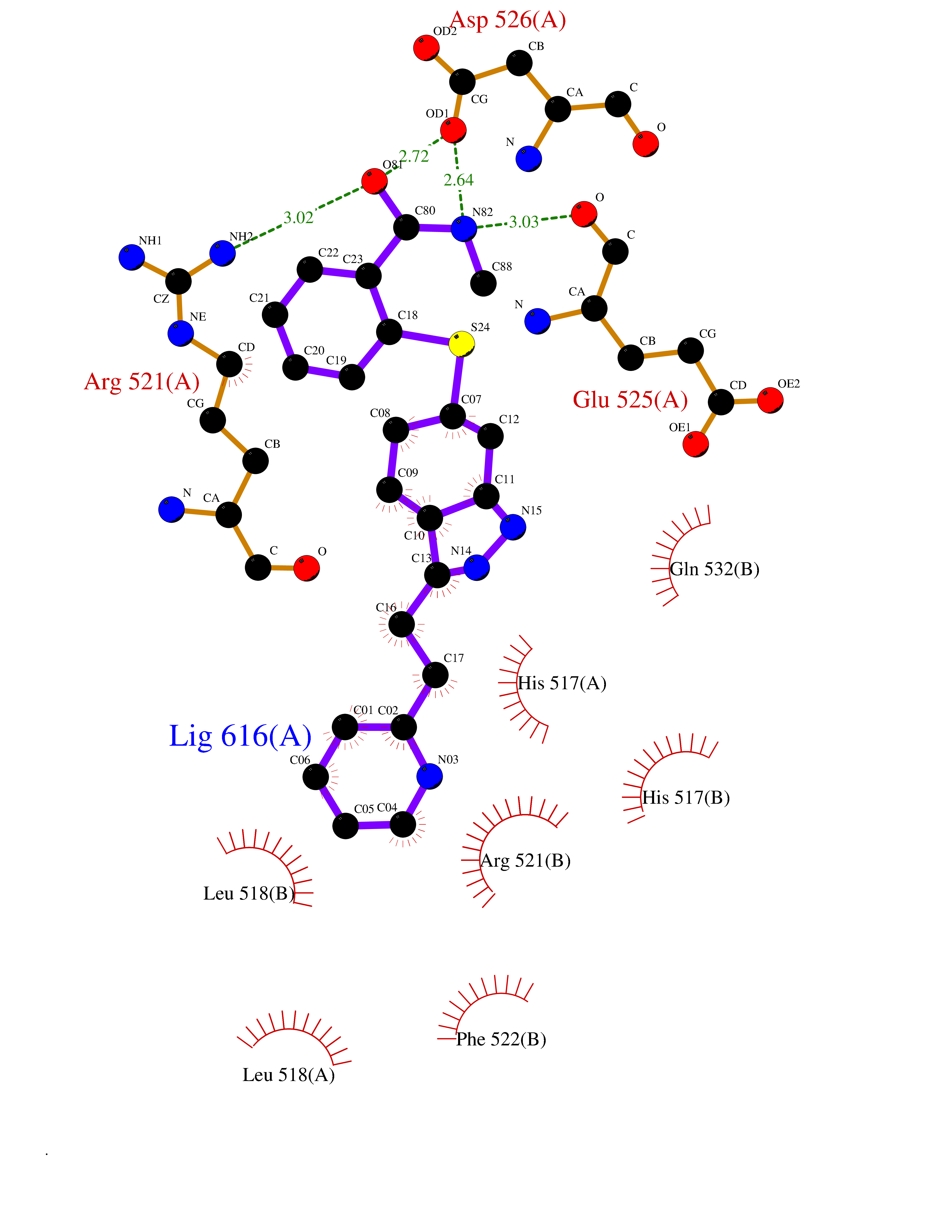 Ligplot Map 3D Binding mode Sequence PLPYHIPLDPEGSLELSWNVSYTQEAIHFQLLVRRLKAGVLFGMSDRGELENADLVVLWTDGDAYFADAWSDQKGQIHLDPQQDYQLLQVQRTPEGLTLLFKRPFGTCDPKDYLIEDGTVHLVYGILEEPFRSLEAINGSGLQMGLQRVQLLKPNIPEPELPSDACTMEVQAPNIQIPSQETTYWCYIKELPKGFSRHHIIKYEPIVTKGNEALVHHMEVFQCAPEMDSVPHFSGPCDSKMKPDRLNYCRHVLAAWALGAKAFYYPEEAGLAFGGPGSSRYLRLEVHYHNPLVIEGRNDSSGIRLYYTAKLRRFNAGIMELGLVYTPVMAIPPRETAFILTGYCTDKCTQLALPPSGIHIFASQLHTHLTGRKVVTVLVRDGREWEIVNQDNHYSPHFQEIRMLKKVVSVHPGDVLITSCTYNTEDRELATVGGFGILEEMCVNYVHYYPQTQLELCKSAVDAGFLQKYFHLINRFNNEDVCTCPQASVSQQFTSVPWNSFNRDVLKALYSFAPISMHCNKSSAVRFQGEWNLQPLPKVISTLEEPTVVSPLPYHIPLDPEGSLELSWNVSYTQEAIHFQLLVRRLKAGVLFGMSDRGELENADLVVLAYFADAWSDQKGQIHLDPQQDYQLLQVQRTPEGLTLLFKRPFGTCDPKDYLIEDGTVHLVYGILEEPFRSLEAINGSGLQMGLQRVQLLKPNIPEPELPSDACTMEVQAPNIQIPSQETTYWCYIKELPKGFSRHHIIKYEPIVTKGNEALVHHMEVFQCAPEVPHFSGPCDSKMLNYCRHVLAAWALGAKAFYYPEEAGLAFGGPGSSRYLRLEVHYHNPLVIEGRNDSSGIRLYYTAKLRRFNAGIMELGLVYTPVMAIPPRETAFILTGYCTDKCTQLALPPSGIHIFASQLHTHLTGRKVVTVLVRDGREWEIVNQDNHYSPHFQEIRMLKKVVSVHPGDVLITSCTYNTEDRELATVGGFGILEEMCVNYVHYYPQTQLELCKSAVDAGFLQKYFHLINRFNNEDVCTCPQASVSQQFTSVPWNSFNRDVLKALYSFAPISMHCNKSSAVRFQGEWNLQPLPKVISTLEEPTPQCVVSIGG Hydrogen bonds contact Hydrophobic contact | ||||