Ligand
Structure
Job ID
8f839b99a76fe7cc6d814ee31801df3b
Job name
Miller_challenge18
Time
2024-11-16 15:48:56
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 1 | Endothelin-converting enzyme 1 (ECE1) | 3DWB | 8.75 | |
Target general information Gen name ECE1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms ECE-1 Protein family Peptidase M13 family Biochemical class Peptidase Function Converts big endothelin-1 to endothelin-1. Related diseases Hirschsprung disease, cardiac defects, and autonomic dysfunction (HCAD) [MIM:613870]: A disorder characterized by skip-lesions Hirschsprung disease, craniofacial abnormalities and other dysmorphic features, cardiac defects including ductus arteriosus, small subaortic ventricular septal defect, small atrial septal defect, and autonomic dysfunction. {ECO:0000269|PubMed:9915973}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07171 Interacts with P49760; A8MQ03; Q8IUG1; P60370; P60410 EC number EC 3.4.24.71 Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disease variant; Disulfide bond; Glycoprotein; Hirschsprung disease; Hydrolase; Membrane; Metal-binding; Metalloprotease; Phosphoprotein; Protease; Proteomics identification; Reference proteome; Signal-anchor; Transmembrane; Transmembrane helix; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 75247.9 Length 660 Aromaticity 0.12 Instability index 46.29 Isoelectric point 5.33 Charge (pH=7) -18.3 2D Binding mode Binding energy (Kcal/mol) -8.32 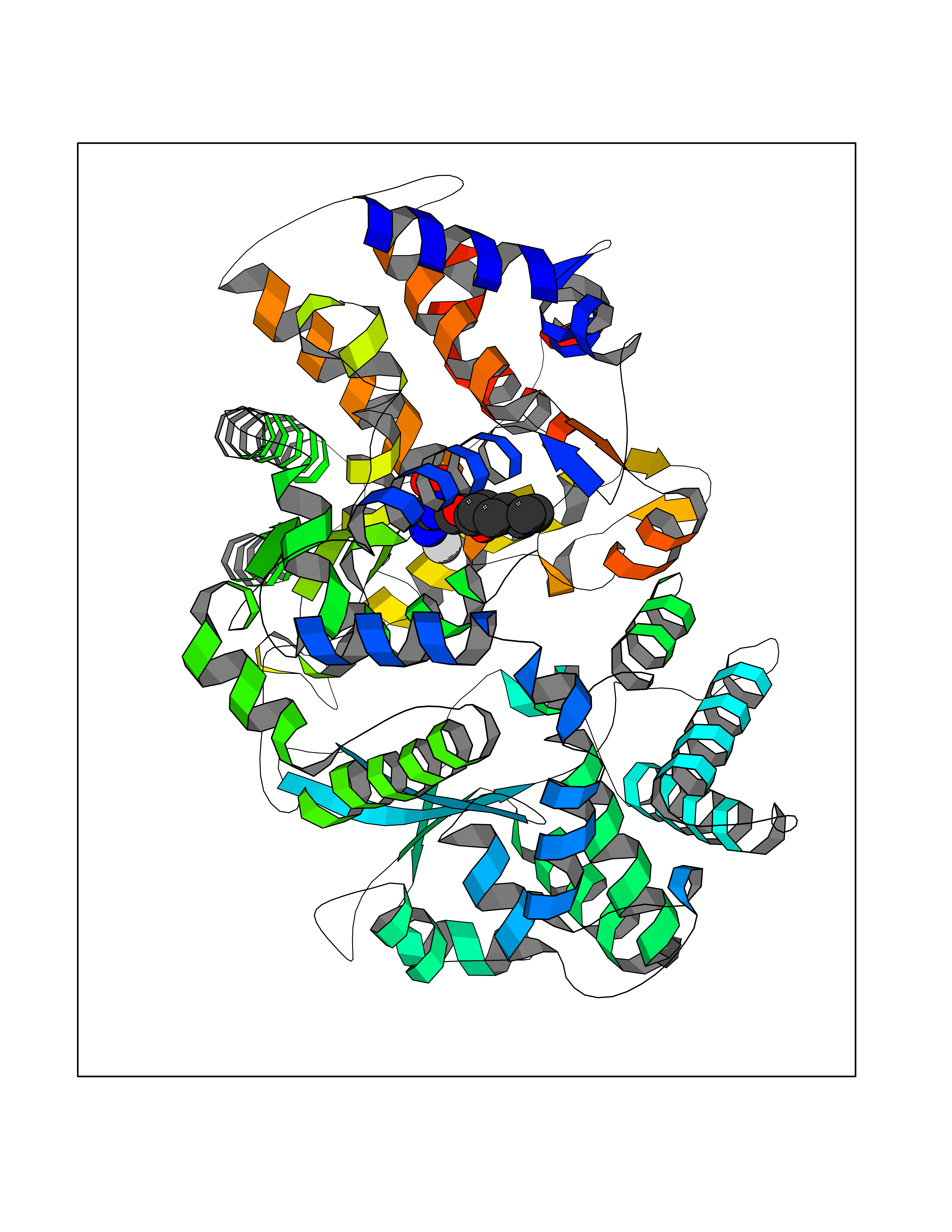 Molscript Map 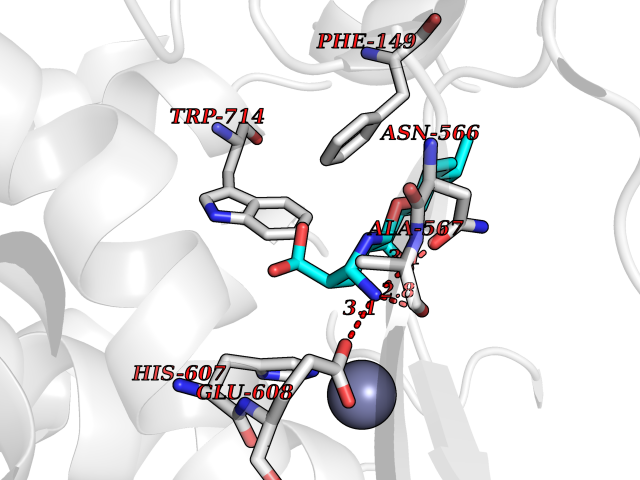 Pymol Map 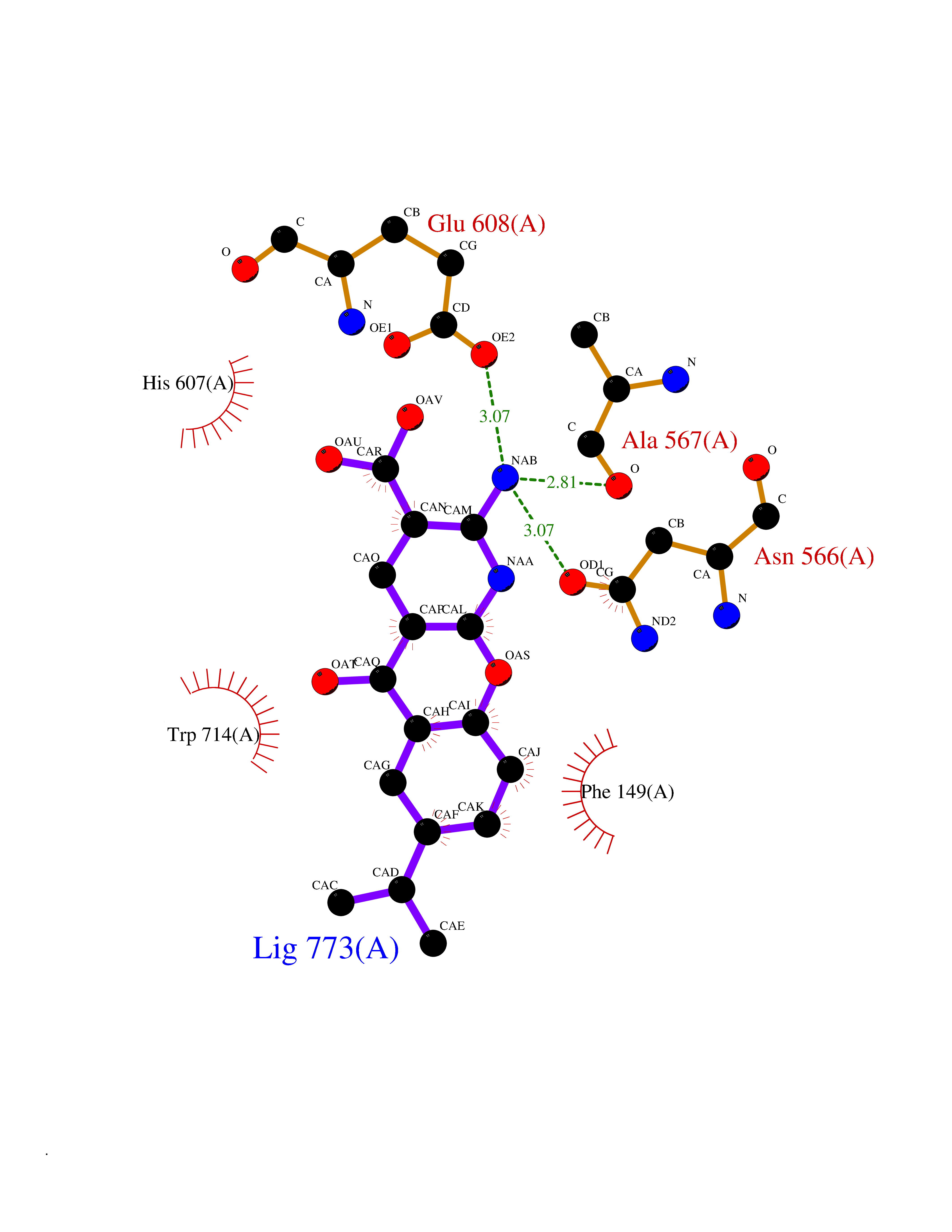 Ligplot Map 3D Binding mode Sequence SEACVSVTSSILSSMDPTVDPCHDFFSYACGGWIKANPVPDGHSRWGTFSNLWEHNQAIIKHLLENSTASVSEAERKAQVYYRACMNETRIEELRAKPLMELIERLGGWNITGPWAKDNFQDTLQVVTAHYRTSPFFSVYVSADSKNSNSNVIQVDQSGLGLPSRDYYLNKTENEKVLTGYLNYMVQLGKLLGGGDEEAIRPQMQQILDFETALANITIPQEKRRDEELIYHKVTAAELQTLAPAINWLPFLNTIFYPVEINESEPIVVYDKEYLEQISTLINTTDRCLLNNYMIWNLVRKTSSFLDQRFQDADEKFMEVMWKFCVSDTENNLGFALGPMFVKATFAEDSKSIATEIILEIKKAFEESLSTLKWMDEETRKSAKEKADAIYNMIGYPNFIMDPKELDKVFNDYTAVPDLYFENAMRFFNFSWRVTADQLRKAPNRDQWSMTPPMVNAYYSPTKNEIVFPAGILQAPFYTRSSPKALNFGGIGVVVGHELTHAFDDQGREYDKDGNLRPWWKNSSVEAFKRQTECMVEQYSNYSVNGEPVNGRHTLGENIADNGGLKAAYRAYQNWVKKNGAEHSLPTLGLTNNQLFFLGFAQVWCSVRTPESSHEGLITDPHSPSRFRVIGSLSNSKEFSEHFRCPPGSPMNPPHKCEVW Hydrogen bonds contact Hydrophobic contact | ||||
| 2 | Organic cation transporter 3 (OCT3) | 7ZH6 | 8.55 | |
Target general information Gen name SLC22A3 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Solute carrier family 22 member 3; Extraneuronal monoamine transporter; EMTH Protein family Major facilitator (TC 2.A.1) superfamily, Organic cation transporter (TC 2.A.1.19) family Biochemical class NA Function Mediates potential-dependent transport of a variety of organic cations. May play a significant role in the disposition of cationic neurotoxins and neurotransmitters in the brain. Related diseases Deafness, autosomal dominant, 2A (DFNA2A) [MIM:600101]: A form of non-syndromic sensorineural hearing loss. Sensorineural deafness results from damage to the neural receptors of the inner ear, the nerve pathways to the brain, or the area of the brain that receives sound information. {ECO:0000269|PubMed:10025409, ECO:0000269|PubMed:10369879, ECO:0000269|PubMed:10571947, ECO:0000269|PubMed:10925378, ECO:0000269|PubMed:21242547}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00718; DB08838; DB00182; DB00122; DB14006; DB00501; DB00575; DB00363; DB01151; DB00988; DB00783; DB13952; DB13953; DB13954; DB13955; DB13956; DB00983; DB00536; DB05381; DB00458; DB00762; DB00709; DB00448; DB08882; DB01042; DB01577; DB00331; DB08893; DB00184; DB00368; DB00526; DB00925; DB00413; DB00457; DB01035; DB00396; DB00938; DB00391; DB13943; DB13944; DB08837; DB08841; DB00541 Interacts with P00519 EC number NA Uniprot keywords 3D-structure; Cell membrane; Glycoprotein; Ion transport; Membrane; Mitochondrion; Nucleus; Proteomics identification; Reference proteome; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A Molecular weight (Da) 53067.4 Length 478 Aromaticity 0.13 Instability index 38.82 Isoelectric point 9.07 Charge (pH=7) 10.54 2D Binding mode Binding energy (Kcal/mol) -8.87 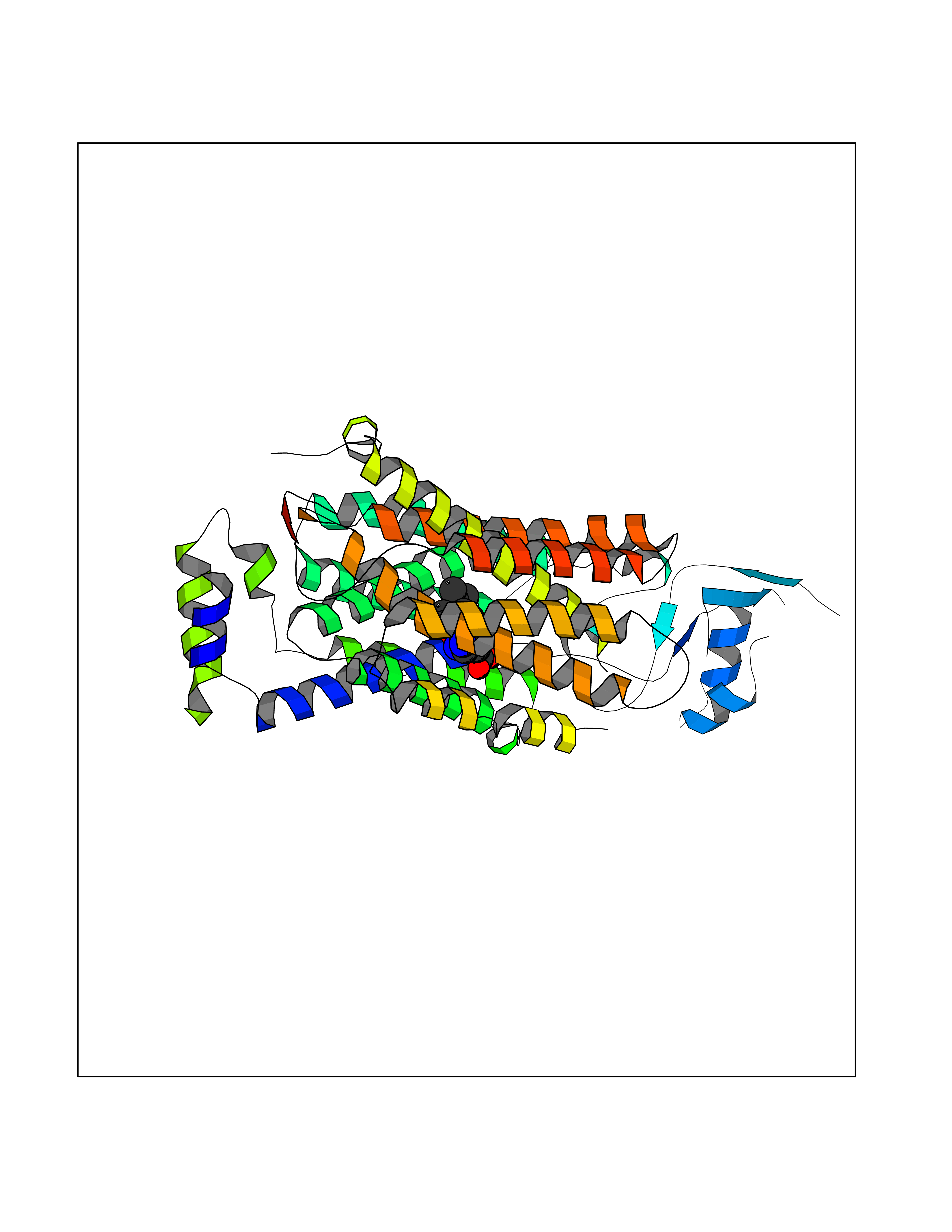 Molscript Map 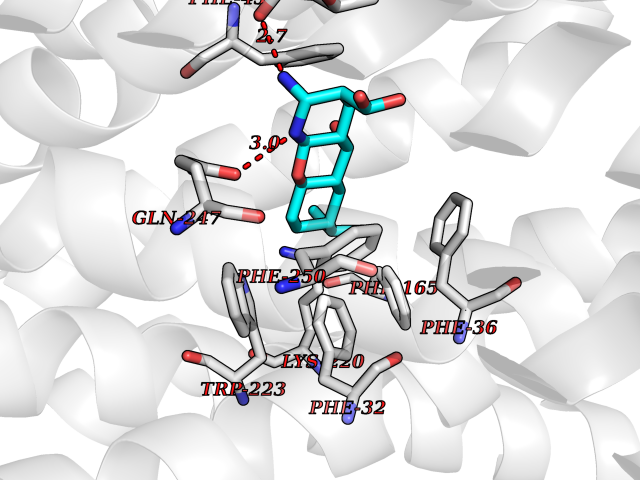 Pymol Map 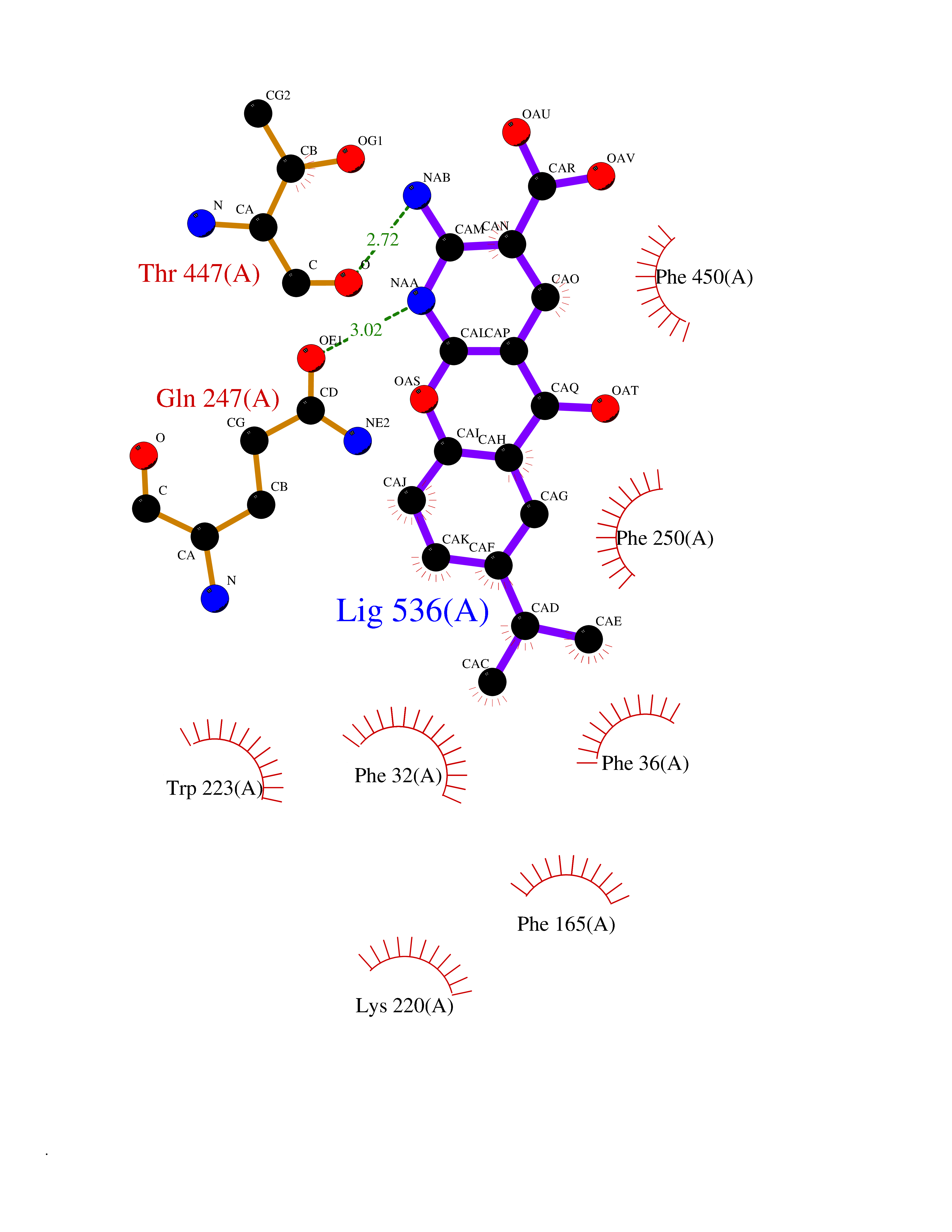 Ligplot Map 3D Binding mode Sequence SFDEALQRVGEFGRFQRRVFLLLCLTGVTFAFLFVGVVFLGTQPDHYWCRGPSAAALAERCGWSPEEEWNRTAPASRGRCQRYLLSAPLVPCRGGWRYAQAHSTIVSEFDLVCVNAWMLDLTQAILNLGFLTGAFTLGYAADRYGRIVIYLLSCLGVGVTGVVVAFAPNFPVFVIFRFLQGVFGKGTWMTCYVIVTEIVGSKQRRIVGIVIQMFFTLGIIILPGIAYFIPNWQGIQLAITLPSFLFLLYYWVVPESPRWLITRKKGDKALQILRRIAKCNVSNPSFLDLVRTPQMRKCTLILMFAWFTSAVVYQGLVMRLGNLYIDFFISGVVELPGALLILLTIERLGRRLPFAASNIVAGVACLVTAFLPEGIAWLRTTVATLGRLGITMAFEIVYLVNSELYPTTLRNFGVSLCSGLCDFGGIIAPFLLFRLAAVWLELPLIIFGILASICGGLVMLLPETKGIALPETVDDVEK Hydrogen bonds contact Hydrophobic contact | ||||
| 3 | FkbI | 1R2J | 8.47 | |
Target general information Gen name fkbI Organism Streptomyces hygroscopicus subsp. ascomyceticus Uniprot ID TTD ID NA Synonyms NA Protein family Acyl-CoA dehydrogenase family Biochemical class Oxidoreductase Function Flavin adenine dinucleotide binding.Oxidoreductase activity, acting on the CH-CH group of donors. Related diseases Thiamine metabolism dysfunction syndrome 5, episodic encephalopathy type (THMD5) [MIM:614458]: An autosomal recessive metabolic disorder due to an inborn error of thiamine metabolism. The phenotype is highly variable, but in general, affected individuals have onset in early childhood of acute encephalopathic episodes associated with increased serum and CSF lactate. These episodes result in progressive neurologic dysfunction manifest as gait disturbances, ataxia, dystonia, and spasticity, which in some cases may result in loss of ability to walk. Cognitive function is usually preserved, although mildly delayed development has been reported. These episodes are usually associated with infection and metabolic decompensation. Some patients may have recovery of some neurologic deficits. {ECO:0000269|PubMed:22152682}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03147 Interacts with NA EC number NA Uniprot keywords 3D-structure; FAD; Flavoprotein; Nucleotide-binding; Oxidoreductase Protein physicochemical properties Chain ID A Molecular weight (Da) 36670.3 Length 353 Aromaticity 0.04 Instability index 22.05 Isoelectric point 6.12 Charge (pH=7) -5.04 2D Binding mode Binding energy (Kcal/mol) -8.48 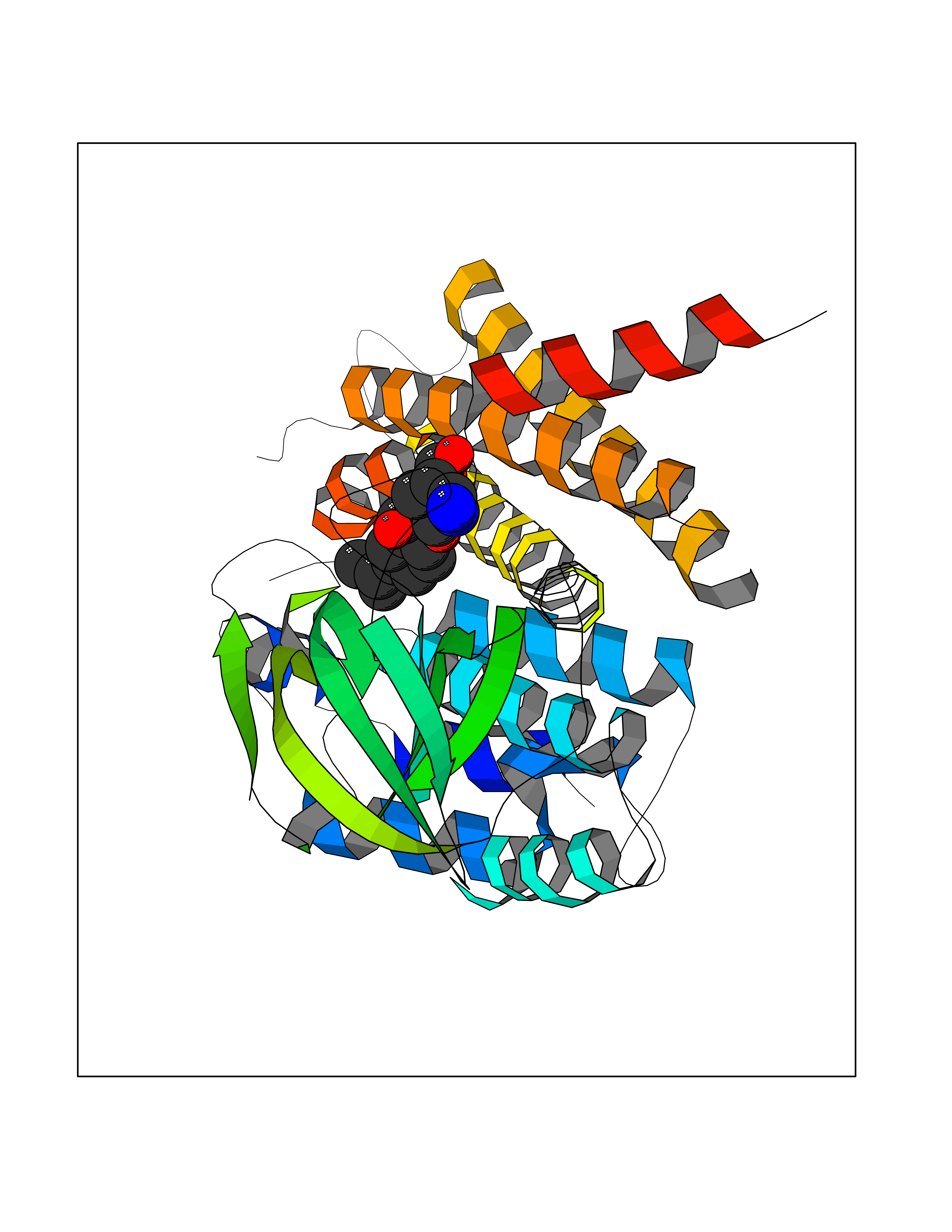 Molscript Map 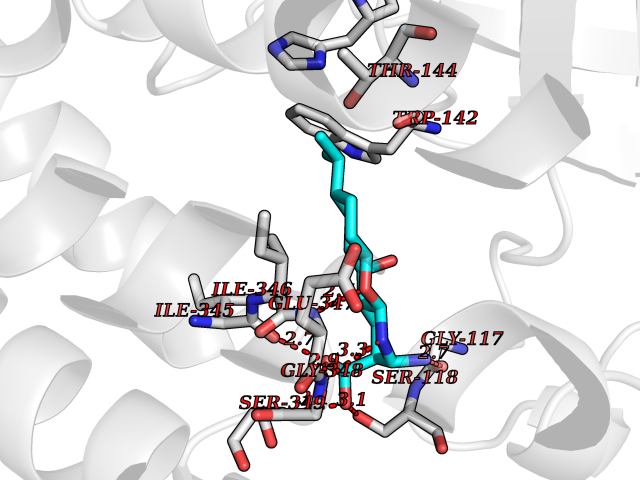 Pymol Map 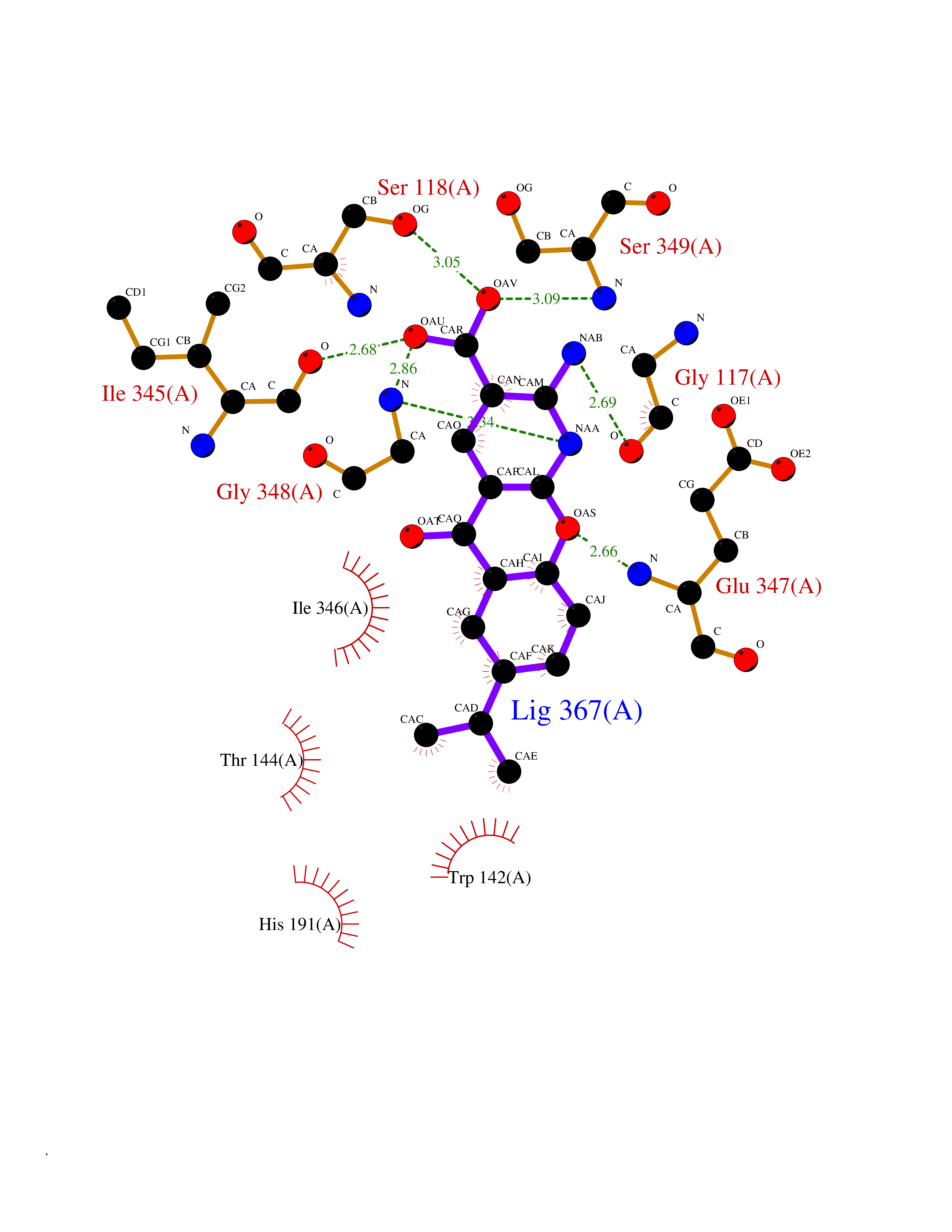 Ligplot Map 3D Binding mode Sequence ERDALLTDLVGDRAAEWDTSGELPRDLLVRLGADGLLCAEVAAEHGGLGLGSRENGEFTAHVGSLCSSLRSVMTSQGMAAWTVQRLGDAGQRATFLKELTSGLAAVGFSERQAGSDLSAMRTRVRLDGDTAVVDGHKVWTTAAAYADHLVVFGLQEDGSGAVVVVPADTPGVRVERVPKPSGCRAAGHADLHLDQVRVPAGAVLAGSGASLPMLVAASLAYGRKSVAWGCVGILRACRTAAVAHARTREQFGRPLGDHQLVAGHIADLWTAEQIAARVCEYASDHMVPATILAKHVAAERAAAGAATAAQVLASAGAGHVVERAYRDAKLMEIIEGSSEMCRVMLAQHALALP Hydrogen bonds contact Hydrophobic contact | ||||
| 4 | Vitamin D3 receptor (VDR) | 3B0T | 8.42 | |
Target general information Gen name VDR Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Vitamin D(3) receptor; Nuclear vitamin D receptor; Nuclear receptor subfamily 1 group I member 1; NR1I1; 1,25-dihydroxyvitamin D3 receptor Protein family Nuclear hormone receptor family, NR1 subfamily Biochemical class Nuclear hormone receptor Function Enters the nucleus upon vitamin D3 binding where it forms heterodimers with the retinoid X receptor/RXR. The VDR-RXR heterodimers bind to specific response elements on DNA and activate the transcription of vitamin D3-responsive target genes. Plays a central role in calcium homeostasis. Nuclear receptor for calcitriol, the active form of vitamin D3 which mediates the action of this vitamin on cells. Related diseases Rickets vitamin D-dependent 2A (VDDR2A) [MIM:277440]: A disorder of vitamin D metabolism resulting in severe rickets, hypocalcemia and secondary hyperparathyroidism. Most patients have total alopecia in addition to rickets. {ECO:0000269|PubMed:1652893, ECO:0000269|PubMed:17970811, ECO:0000269|PubMed:2177843, ECO:0000269|PubMed:2849209, ECO:0000269|PubMed:28698609, ECO:0000269|PubMed:7828346, ECO:0000269|PubMed:8106618, ECO:0000269|PubMed:8381803, ECO:0000269|PubMed:8392085, ECO:0000269|PubMed:8675579, ECO:0000269|PubMed:8961271, ECO:0000269|PubMed:9005998}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07530; DB08742; DB01436; DB04891; DB00146; DB02300; DB00136; DB00169; DB04540; DB05024; DB11672; DB14635; DB01070; DB06410; DB05295; DB06194; DB00153; DB04796; DB03451; DB00910; DB04258; DB11094 Interacts with P35222; Q09472; Q15648; P50222; Q15788; P26045; P19793; Q13573; Q13501; P04637; Q15645; Q9JLI4; P28700; X5D778; Q96HA8; Q01804; Q96S38; P48443 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; Disease variant; DNA-binding; Metal-binding; Nucleus; Proteomics identification; Receptor; Reference proteome; Transcription; Transcription regulation; Ubl conjugation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A Molecular weight (Da) 28781 Length 254 Aromaticity 0.07 Instability index 47.69 Isoelectric point 6.15 Charge (pH=7) -3.44 2D Binding mode Binding energy (Kcal/mol) -9.29 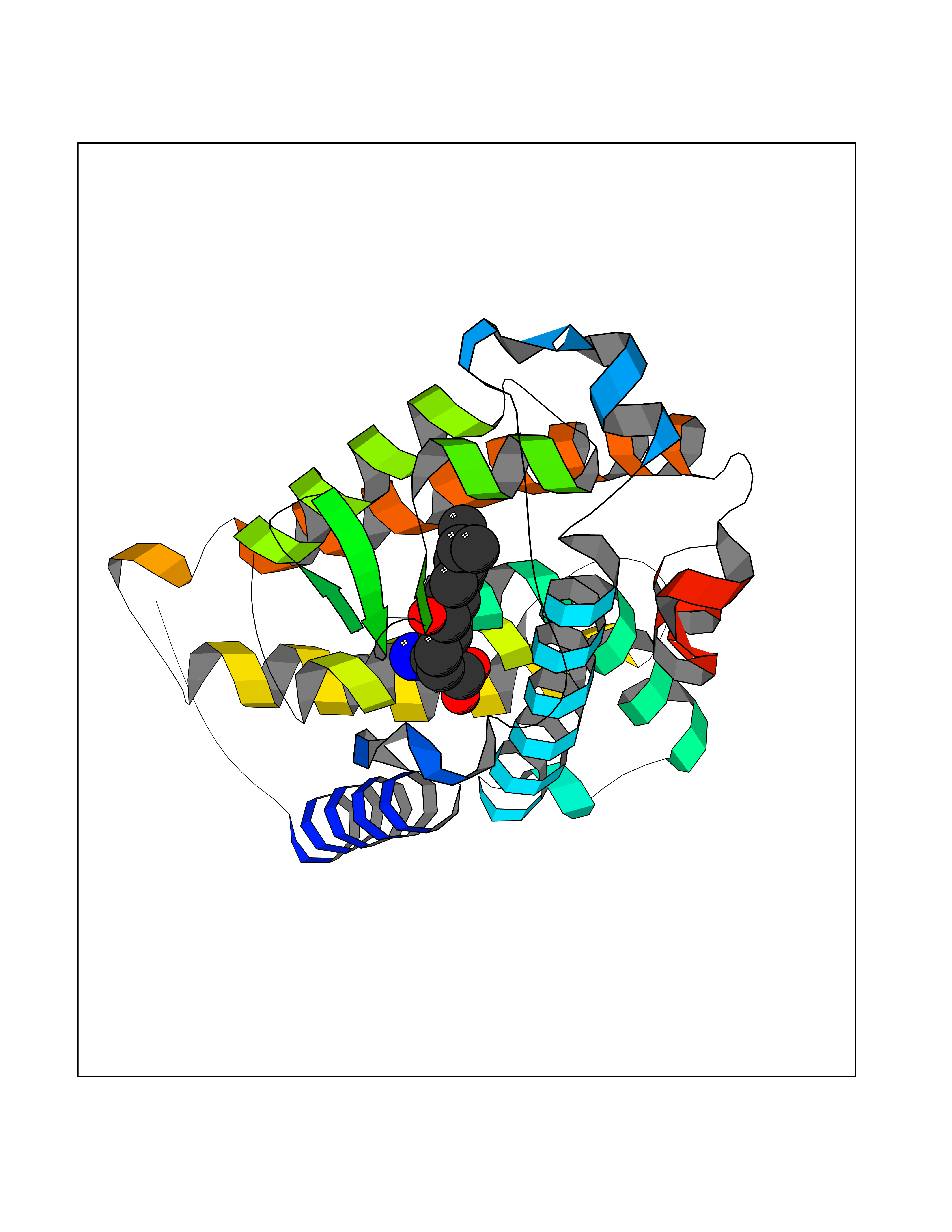 Molscript Map 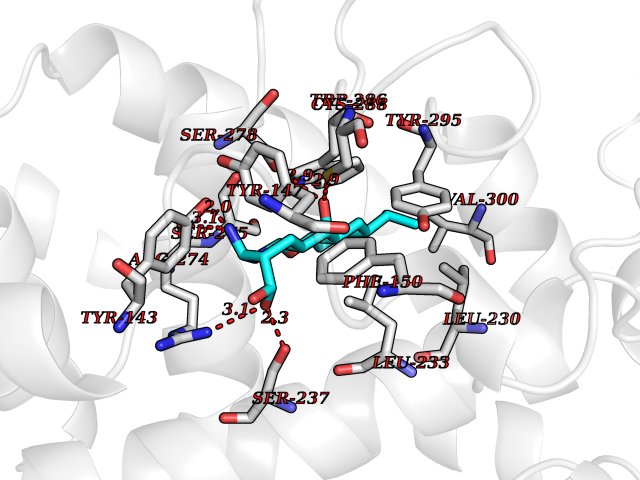 Pymol Map 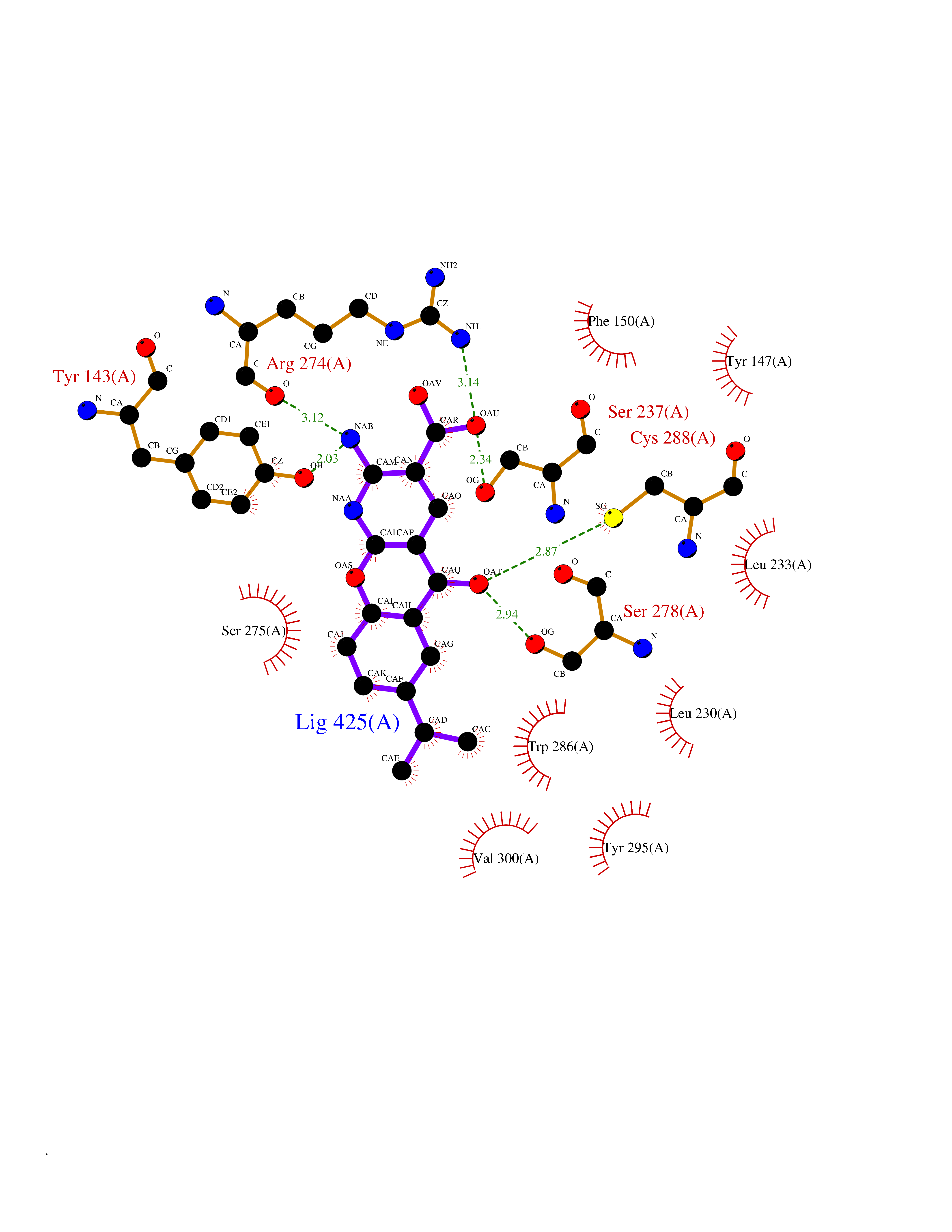 Ligplot Map 3D Binding mode Sequence ALRPKLSEEQQRIIAILLDAHHKTYDPTYSDFCQFRPPVRVNDGGGSVTLELSQLSMLPHLADLVSYSIQKVIGFAKMIPGFRDLTSEDQIVLLKSSAIEVIMLRSNESFTMDDMSWTCGNQDYKYRVSDVTKAGHSLELIEPLIKFQVGLKKLNLHEEEHVLLMAICIVSPDRPGVQDAALIEAIQDRLSNTLQTYIRCRHPPPGSHLLYAKMIQKLADLRSLNEEHSKQYRCLSFQPECSMKLTPLVLEVFG Hydrogen bonds contact Hydrophobic contact | ||||
| 5 | Retinoic acid receptor RXR-beta (RXRB) | 5HJP | 8.40 | |
Target general information Gen name RXRB Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Retinoid X receptor beta; Nuclear receptor subfamily 2 group B member 2; NR2B2 Protein family Nuclear hormone receptor family, NR2 subfamily Biochemical class Nuclear hormone receptor Function Retinoic acid receptors bind as heterodimers to their target response elements in response to their ligands, all-trans or 9-cis retinoic acid, and regulate gene expression in various biological processes. The RAR/RXR heterodimers bind to the retinoic acid response elements (RARE). Receptor for retinoic acid. Related diseases Noonan syndrome 13 (NS13) [MIM:619087]: A form of Noonan syndrome, a disease characterized by short stature, facial dysmorphic features such as hypertelorism, a downward eyeslant and low-set posteriorly rotated ears, and a high incidence of congenital heart defects and hypertrophic cardiomyopathy. Other features can include a short neck with webbing or redundancy of skin, deafness, motor delay, variable intellectual deficits, multiple skeletal defects, cryptorchidism, and bleeding diathesis. Individuals with Noonan syndrome are at risk of juvenile myelomonocytic leukemia, a myeloproliferative disorder characterized by excessive production of myelomonocytic cells. NS13 inheritance is autosomal dominant. There is considerable variability in severity. {ECO:0000269|PubMed:32721402}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08175; DB00459; DB00210; DB00523; DB00307; DB01393; DB03756; DB00926; DB01941; DB07929; DB02746; DB00412; DB00799; DB07080; DB00755 Interacts with Q00975; Q9HB07; F1D8P7; Q13133; Q13133-3; Q96RI1-1; P04150; Q9NRD5; P37231; P10276; P10276-2; P10826-2; P13631; Q6IQ16; Q13137; Q96B26; Q08379; Q6A162; Q9UJV3-2; Q13133-3; Q96RI1-1; O43586; P10276; P10826-2; Q8IUQ4-2; O75528; Q12800; Q9UBB9; Q05BL1; P14373; O94972; Q96S82 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; DNA-binding; Metal-binding; Methylation; Nucleus; Proteomics identification; Receptor; Reference proteome; Transcription; Transcription regulation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A,C Molecular weight (Da) 28845.8 Length 251 Aromaticity 0.08 Instability index 54.86 Isoelectric point 6.74 Charge (pH=7) -0.6 2D Binding mode Binding energy (Kcal/mol) -9.21  Molscript Map 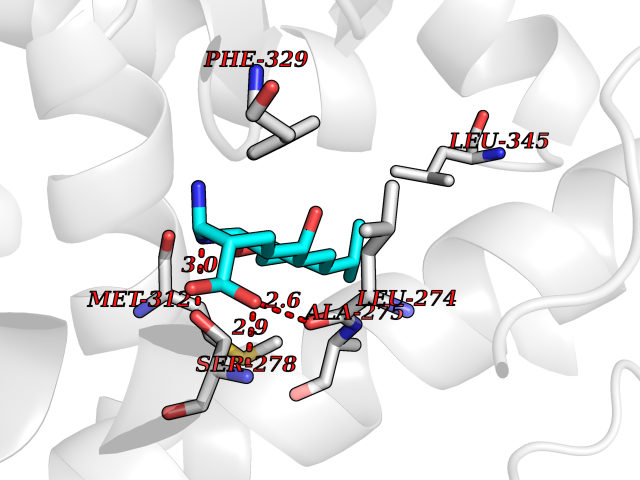 Pymol Map  Ligplot Map 3D Binding mode Sequence QLTAAQELMIQQLVAAQLQCNKRSFSDQPKVTPWPSASQQRFAHFTELAIISVQEIVDFAKQVPGFLQLGREDQIALLKASTIEIMLLETARRYNHETECITFLKDFTYSKDDFHRAGLQVEFINPIFEFSRAMRRLGLDDAEYALLIAINIFSADRPNVQEPGRVEALQQPYVEALLSYTRIKRPQDQLRFPRMLMKLVSLRTLSSVHSEQVFALRLQDKKLPPLLSEIWDVHEGSGSGSHKILHRLLQD Hydrogen bonds contact Hydrophobic contact | ||||
| 6 | Purine nucleoside phosphorylase (PNP) | 4EAR | 8.39 | |
Target general information Gen name PNP Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms PNP; Inosine phosphorylase Protein family PNP/MTAP phosphorylase family Biochemical class Pentosyltransferase Function The purine nucleoside phosphorylases catalyze the phosphorolytic breakdown of the N-glycosidic bond in the beta- (deoxy)ribonucleoside molecules, with the formation of the corresponding free purine bases and pentose-1-phosphate. Related diseases Purine nucleoside phosphorylase deficiency (PNPD) [MIM:613179]: A disorder that interrupts both the catabolism of inosine into hypoxanthine and guanosine into guanine, and leads to the accumulation of guanosine, inosine, and their deoxified by-products. The main clinical presentation is recurrent infections due to severe T-cell immunodeficiency. Some patients also have neurologic impairment. {ECO:0000269|PubMed:1384322, ECO:0000269|PubMed:3029074, ECO:0000269|PubMed:8931706}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03881; DB03551; DB02222; DB02391; DB03609; DB01667; DB04260; DB02796; DB04753; DB00640; DB00242; DB00900; DB06185; DB02377; DB02857; DB04754; DB04757; DB04076; DB02230; DB04335; DB02568; DB03101 Interacts with P05067; Q9UQM7; O14576-2; P06241; P14136; Q92993-2; Q9BXM7; P00491; P17612; P63000; Q92673; Q15583 EC number EC 2.4.2.1 Uniprot keywords 3D-structure; Acetylation; Cytoplasm; Direct protein sequencing; Disease variant; Glycosyltransferase; Phosphoprotein; Proteomics identification; Purine salvage; Reference proteome; Transferase Protein physicochemical properties Chain ID A,B,C Molecular weight (Da) 31849.2 Length 288 Aromaticity 0.1 Instability index 34.77 Isoelectric point 6.42 Charge (pH=7) -1.63 2D Binding mode Binding energy (Kcal/mol) -8.8  Molscript Map 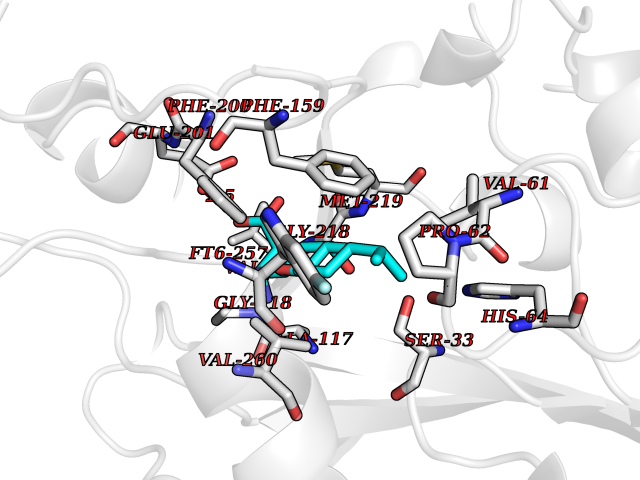 Pymol Map 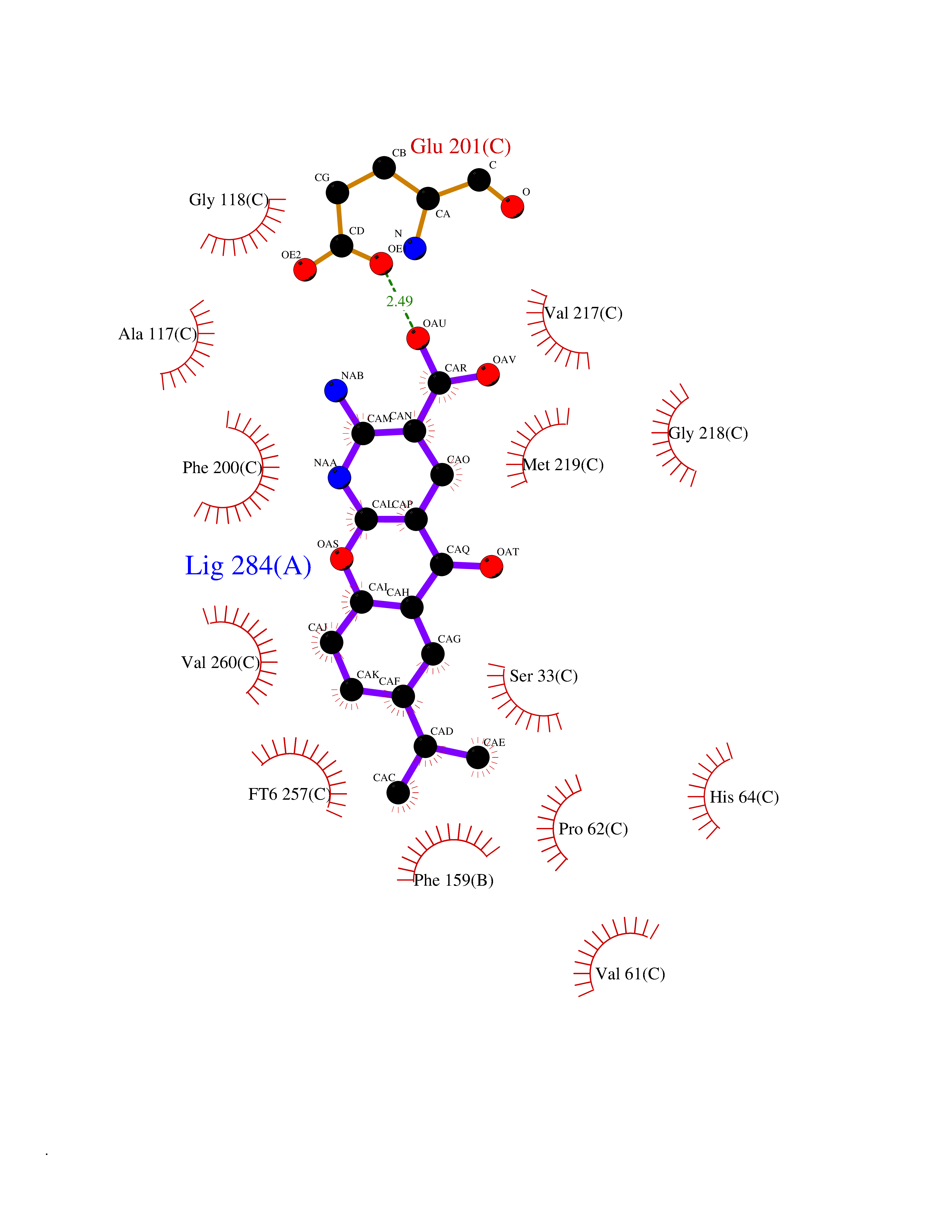 Ligplot Map 3D Binding mode Sequence GYTYEDYKNTAEYLLSHTKHRPQVAIICGSGLGGLTDKLTQAQIFDYSEIPNFPRSTVPGHAGRLVFGFLNGRACVMMQGRFHMYEGYPLYKVTFPVRVFHLLGVDTLVVTNAAGGLNPKFEVGDIMLIRDHINLPGFSGQNPLRGPNDERFGDRFPAMSDAYDRTMRQRALSTYKQMGEQRELQEGTYVMVAGPSFETVAECRVLQKLGADAVGMSTVPEVIVARHCGLRVFGFSLITNKVIMDYESLEKANXEEVLAAGKQAAQKLEQFVSILMASIDRFPAMSDA Hydrogen bonds contact Hydrophobic contact | ||||
| 7 | Platelet glycoprotein VI (GP6) | 5OU7 | 8.37 | |
Target general information Gen name GP6 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Glycoprotein 6; GPVI Protein family NA Biochemical class NA Function Collagen receptor involved in collagen-induced platelet adhesion and activation. Plays a key role in platelet procoagulant activity and subsequent thrombin and fibrin formation. This procoagulant function may contribute to arterial and venous thrombus formation. The signaling pathway involves the FcR gamma-chain, the Src kinases (likely FYN or LYN) and SYK, the adapter protein LAT and leads to the activation of PLCG2. Related diseases Bleeding disorder, platelet-type, 11 (BDPLT11) [MIM:614201]: A mild to moderate bleeding disorder caused by defective platelet activation and aggregation in response to collagen. {ECO:0000269|PubMed:19549989, ECO:0000269|PubMed:19552682}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P06241; P07948; P06241; P07948 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Blood coagulation; Cell membrane; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; Hemostasis; Immunoglobulin domain; Membrane; Proteomics identification; Receptor; Reference proteome; Repeat; Signal; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 19027.4 Length 173 Aromaticity 0.11 Instability index 37.14 Isoelectric point 8.68 Charge (pH=7) 2.52 2D Binding mode Binding energy (Kcal/mol) -7.82 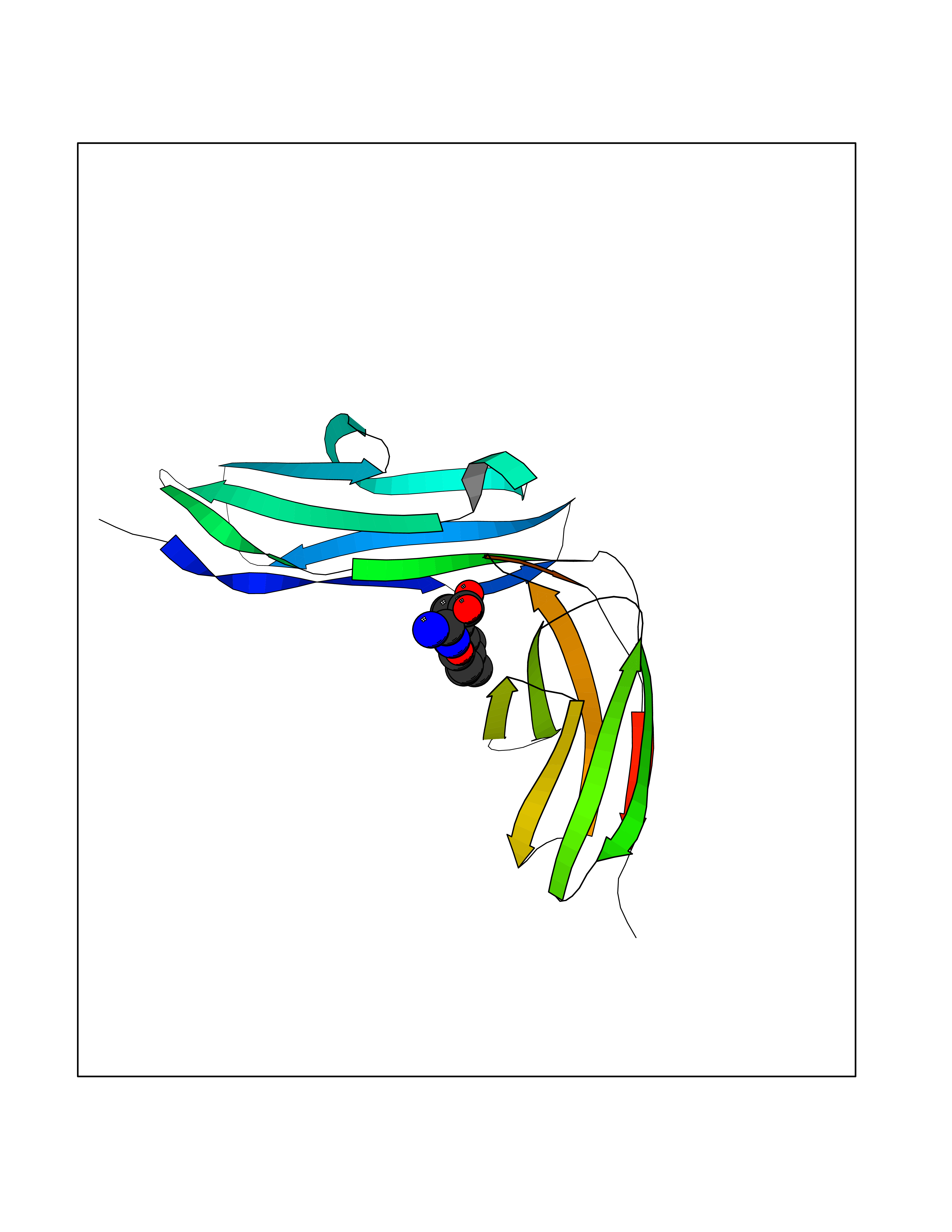 Molscript Map 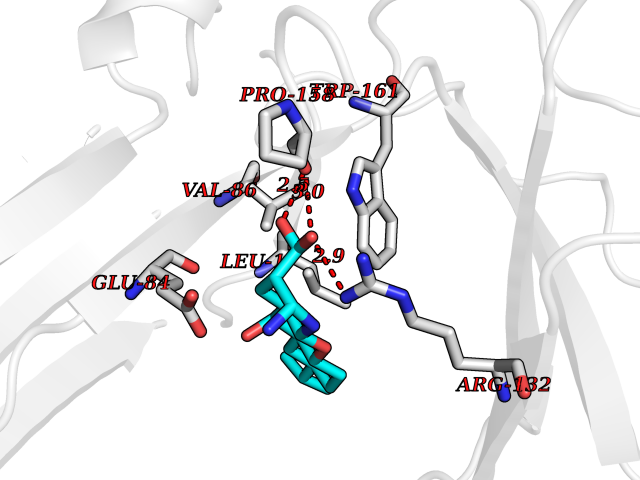 Pymol Map 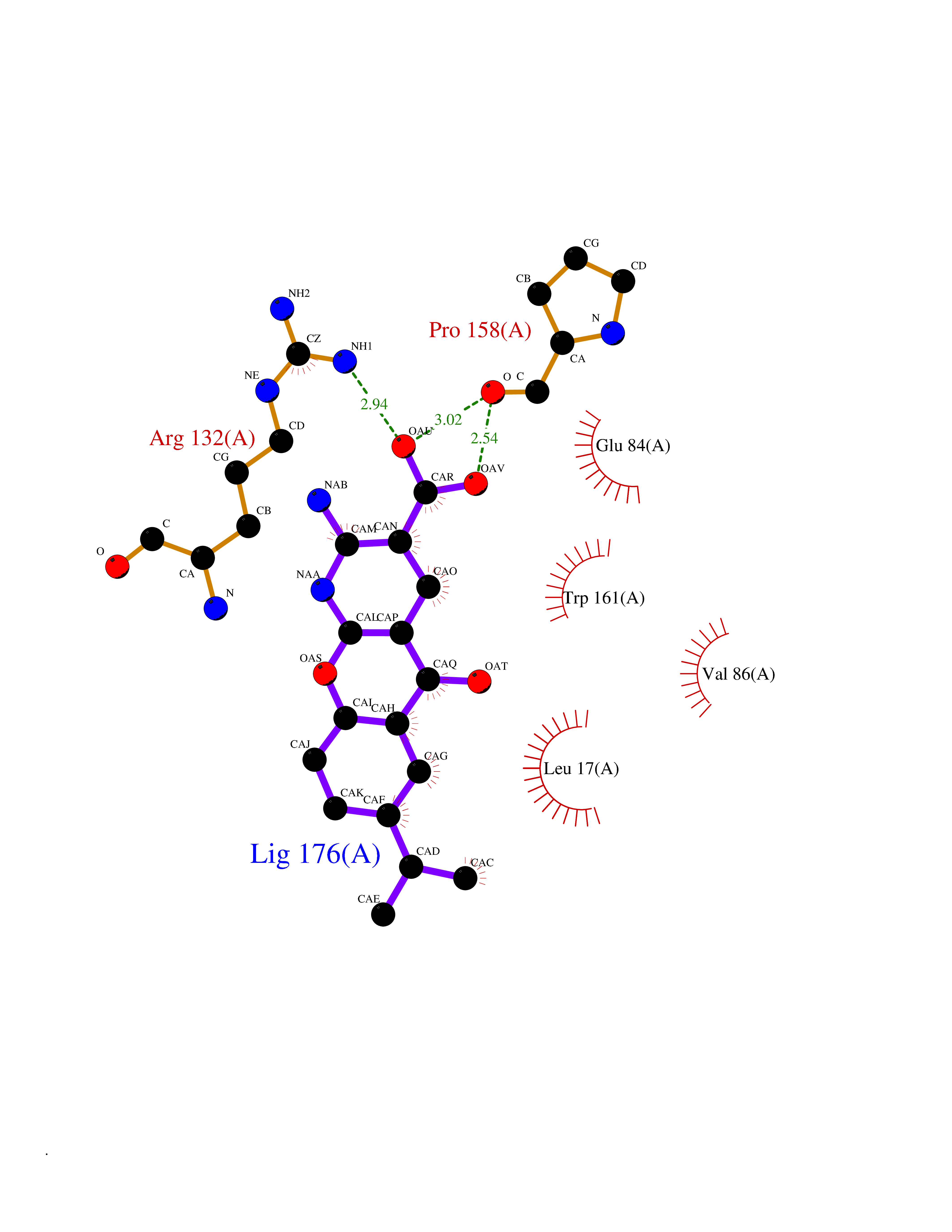 Ligplot Map 3D Binding mode Sequence SGPLPKPSLQALPSSLVPLEKPVTLRCQGPPGVDLYRLEKLSSSRYQDQAVLFIPAMKRSLAGRYRCSYQNGSLWSLPSDQLELVATGVFAKPSLSAQPGSGGDVTLQCQTRYGFDQFALYKEGDPERWYRASFPIITVTAAHSGTYRCYSFSSRDPYLWSAPSDPLELVVTG Hydrogen bonds contact Hydrophobic contact | ||||
| 8 | Fatty acid synthase (FASN) | 3TJM | 8.32 | |
Target general information Gen name FASN Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Yeast fatty acid synthase; Fatty-acyl-CoA synthase; Fatty acyl-CoA synthetase enzyme; FAS Protein family NA Biochemical class Acyltransferase Function Fatty acid synthetase catalyzes the formation of long-chain fatty acids from acetyl-CoA, malonyl-CoA and NADPH. This multifunctional protein has 7 catalytic activities as an acyl carrier protein. Related diseases Glycine encephalopathy 2 (GCE2) [MIM:620398]: A form of glycine encephalopathy, a metabolic disorder characterized by a high concentration of glycine in the body fluids. Affected individuals typically have severe neurological symptoms, including seizure, lethargy, and muscular hypotonia soon after birth. Most of them die within the neonatal period. Atypical cases have later disease onset and less severely affected psychomotor development. {ECO:0000269|PubMed:10873393, ECO:0000269|PubMed:11286506, ECO:0000269|PubMed:16051266, ECO:0000269|PubMed:26371980, ECO:0000269|PubMed:28244183, ECO:0000269|PubMed:8005589, ECO:0000269|PubMed:9600239, ECO:0000269|PubMed:9621520}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01034; DB01083 Interacts with Q15848; Q16665; P42858; Q8IV20; Q8TBB1; PRO_0000045603 [Q99IB8] EC number EC 2.3.1.85 Uniprot keywords 3D-structure; Acetylation; Cytoplasm; Direct protein sequencing; Fatty acid biosynthesis; Fatty acid metabolism; Hydrolase; Isopeptide bond; Lipid biosynthesis; Lipid metabolism; Lyase; Multifunctional enzyme; NAD; NADP; Oxidoreductase; Phosphopantetheine; Phosphoprotein; Proteomics identification; Pyridoxal phosphate; Reference proteome; S-nitrosylation; Transferase; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 30174.9 Length 275 Aromaticity 0.09 Instability index 43.28 Isoelectric point 5.92 Charge (pH=7) -5.4 2D Binding mode Binding energy (Kcal/mol) -8.88 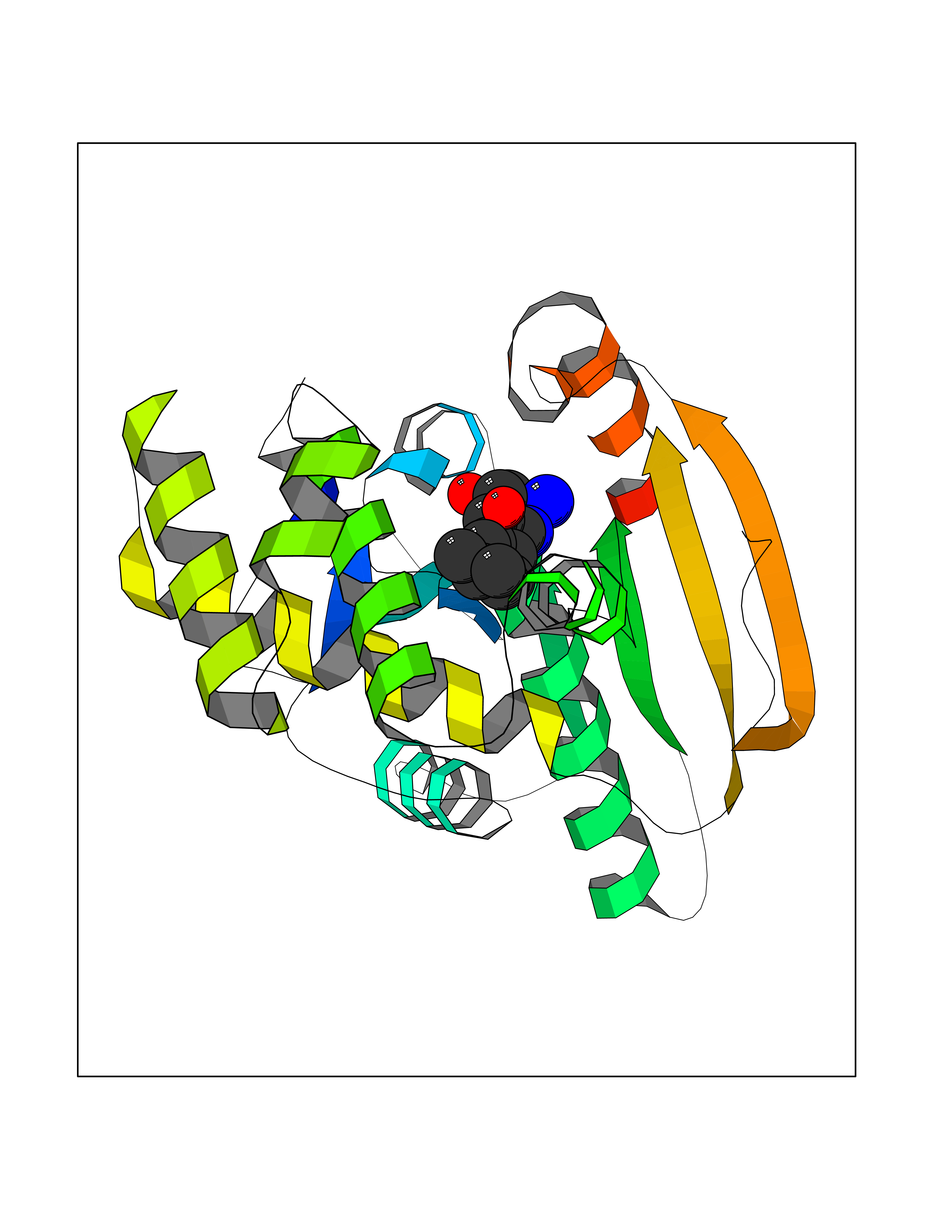 Molscript Map 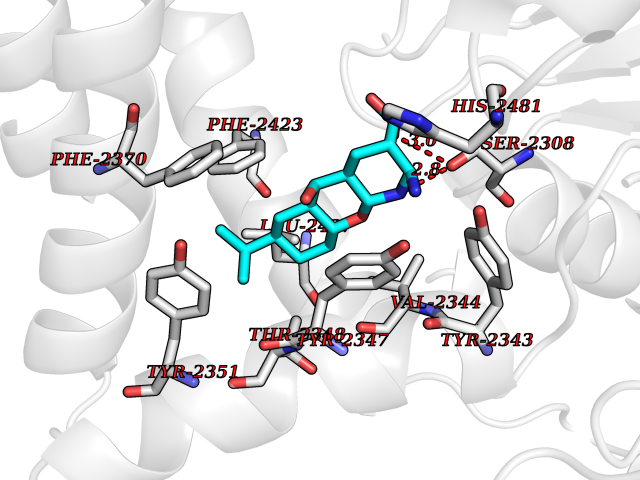 Pymol Map 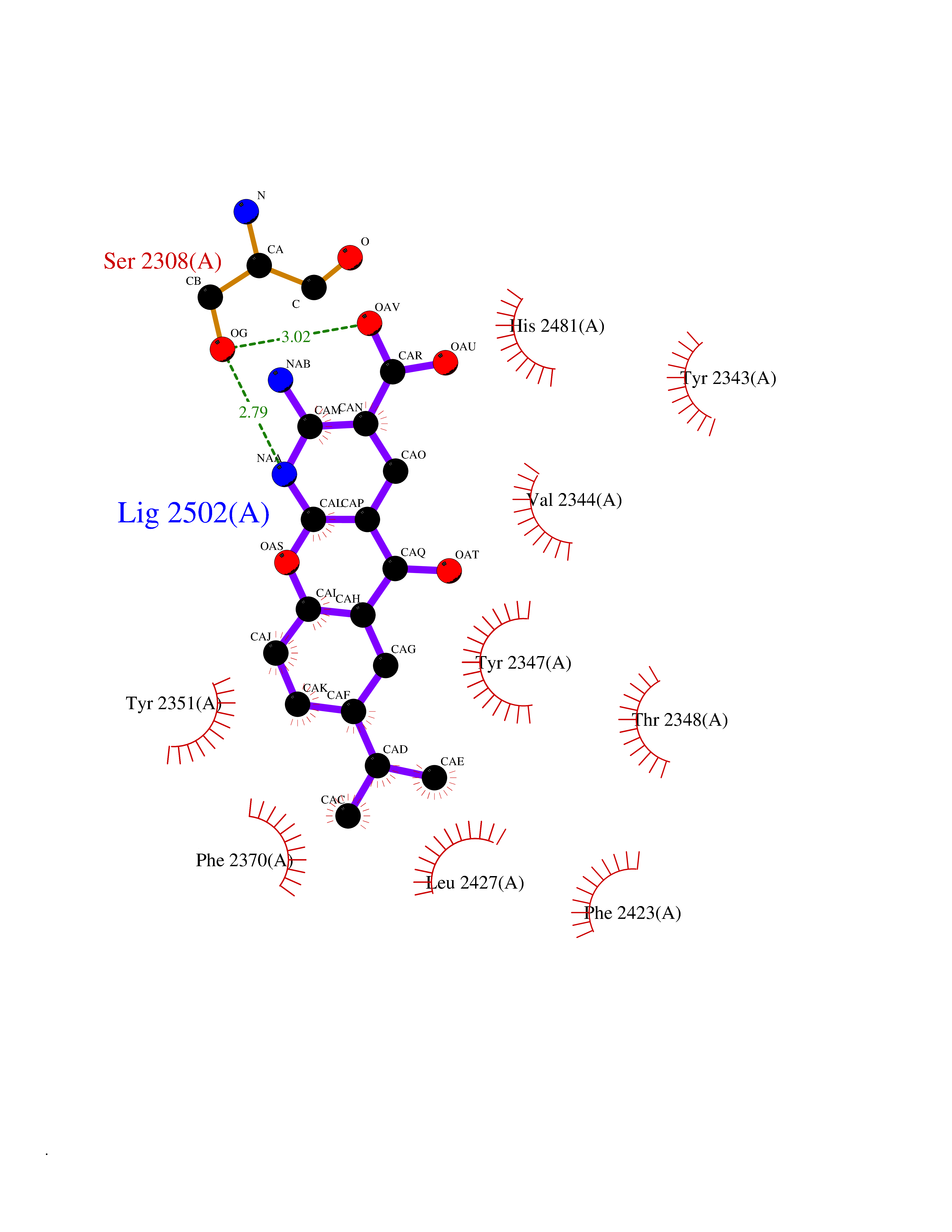 Ligplot Map 3D Binding mode Sequence NLRSLLVNPEGPTLMRLNSVQSSERPLFLVHPIEGSTTVFHSLASRLSIPTYGLQCTRAAPLDSIHSLAAYYIDCIRQVQPEGPYRVAGYSYGACVAFEMCSQLQAQQSPAPTHNSLFLFDGSPTYVLAYTGSYRAKLTPGCEAEAETEAICFFVQQFTDMEHNRVLEALLPLKGLEERVAAAVDLIIKSHQGLDRQELSFAARSFYYKLRAAEQYTPKAKYHGNVMLLRAAAGADYNLSQVCDGKVSVHVIEGDHATLLEGSGLESIISIIHSS Hydrogen bonds contact Hydrophobic contact | ||||
| 9 | Clostridium histolyticum Collagenase (CH colG) | 7Z5U | 8.28 | |
Target general information Gen name CH colG Organism Hathewaya histolytica (Clostridium histolyticum) Uniprot ID TTD ID Synonyms Microbial collagenase; Gelatinase ColG; Collagenase ColG; Class I collagenase Protein family Peptidase M9B family, Collagenase subfamily Biochemical class NA Function Clostridial collagenases are among the most efficient degraders of eukaryotic collagen known; saprophytes use collagen as a carbon source while pathogens additionally digest collagen to aid in host colonization. Has both tripeptidylcarboxypeptidase on Gly-X-Y and endopeptidase activities; the endopeptidase cuts within the triple helix region of collagen while tripeptidylcarboxypeptidase successively digests the exposed ends, thus clostridial collagenases can digest large sections of collagen. Active on soluble type I collagen, insoluble collagen, azocoll, soluble PZ-peptide (all collagenase substrates) and gelatin. The full-length protein has collagenase activity, while the in vivo derived C-terminally truncated shorter versions only act on gelatin. In vitro digestion of soluble calf skin collagen fibrils requires both ColG and ColH; ColG forms missing the second collagen-binding domain are also synergistic with ColH, although their overall efficiency is decreased. The activator domain (residues 119-388) and catalytic subdomain (389-670) open and close around substrate using a Gly-rich hinge (387-397), allowing digestion when the protein is closed. Binding of collagen requires Ca(2+) and is inhibited by EGTA; the collagen-binding domain (CBD, S3a plus S3b) specifically recognizes the triple-helical conformation made by 3 collagen protein chains in the triple-helical region. Isolated CBD (S3a plus S3b) binds collagen fibrils and sheets of many tissues. Related diseases Intellectual developmental disorder, X-linked, syndromic, Claes-Jensen type (MRXSCJ) [MIM:300534]: A disorder characterized by significantly below average general intellectual functioning associated with impairments in adaptive behavior and manifested during the developmental period. MRXSCJ patients manifest intellectual disability associated with variable features such as slowly progressive spastic paraplegia, seizures, facial dysmorphism. {ECO:0000269|PubMed:15586325, ECO:0000269|PubMed:16538222, ECO:0000269|PubMed:16541399, ECO:0000269|PubMed:17320160, ECO:0000269|PubMed:17468742, ECO:0000269|PubMed:23356856, ECO:0000269|PubMed:25666439}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number EC 3.4.24.3 Uniprot keywords 3D-structure; Calcium; Direct protein sequencing; Hydrolase; Metal-binding; Metalloprotease; Pharmaceutical; Protease; Repeat; Secreted; Signal; Virulence; Zinc; Zymogen Protein physicochemical properties Chain ID A Molecular weight (Da) 44751.2 Length 386 Aromaticity 0.15 Instability index 27.33 Isoelectric point 5.77 Charge (pH=7) -7.33 2D Binding mode Binding energy (Kcal/mol) -8.89 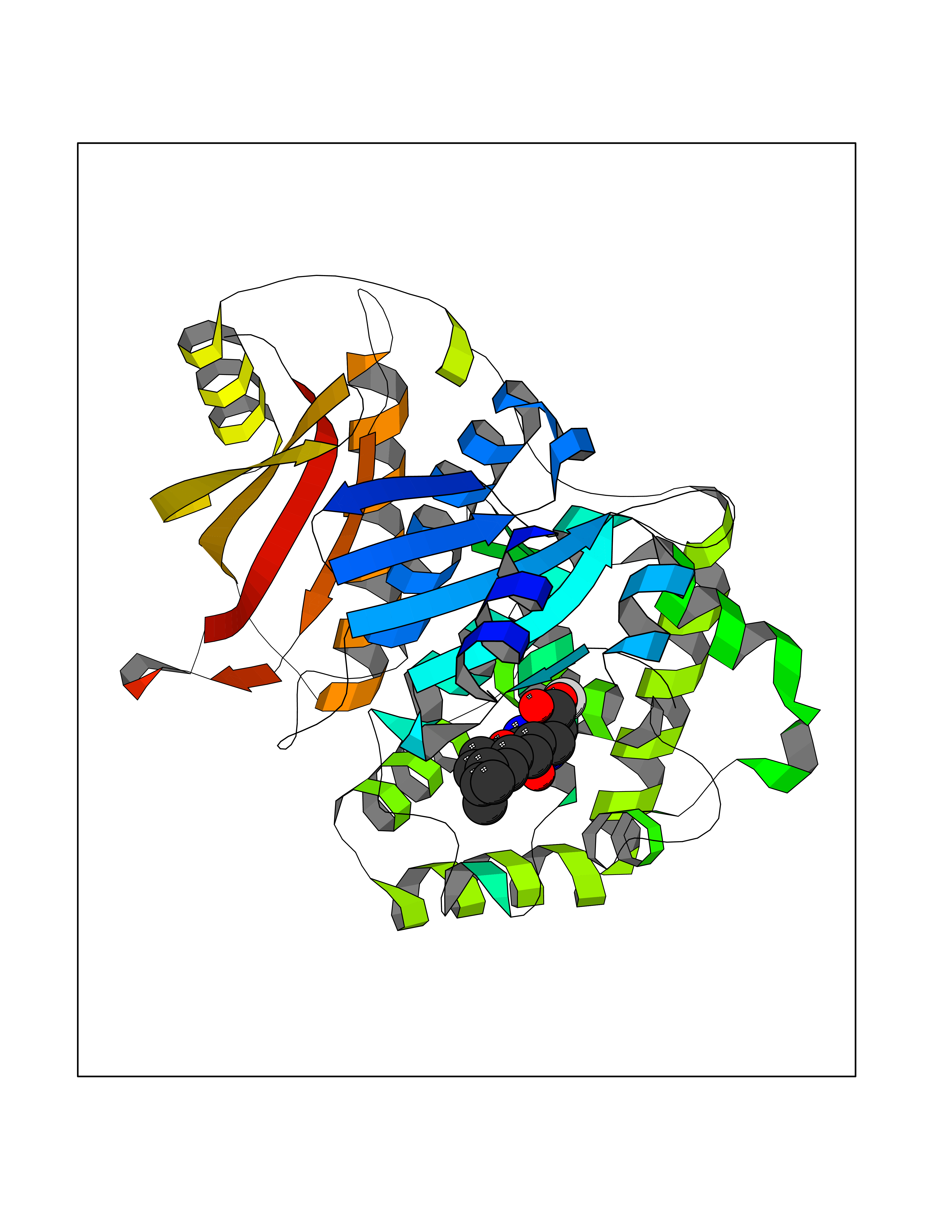 Molscript Map 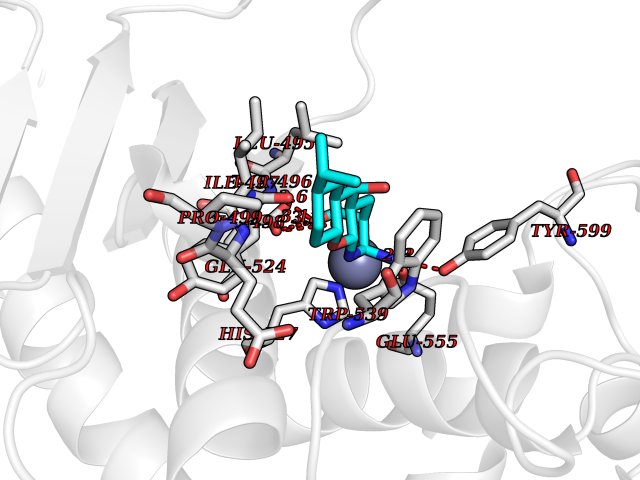 Pymol Map 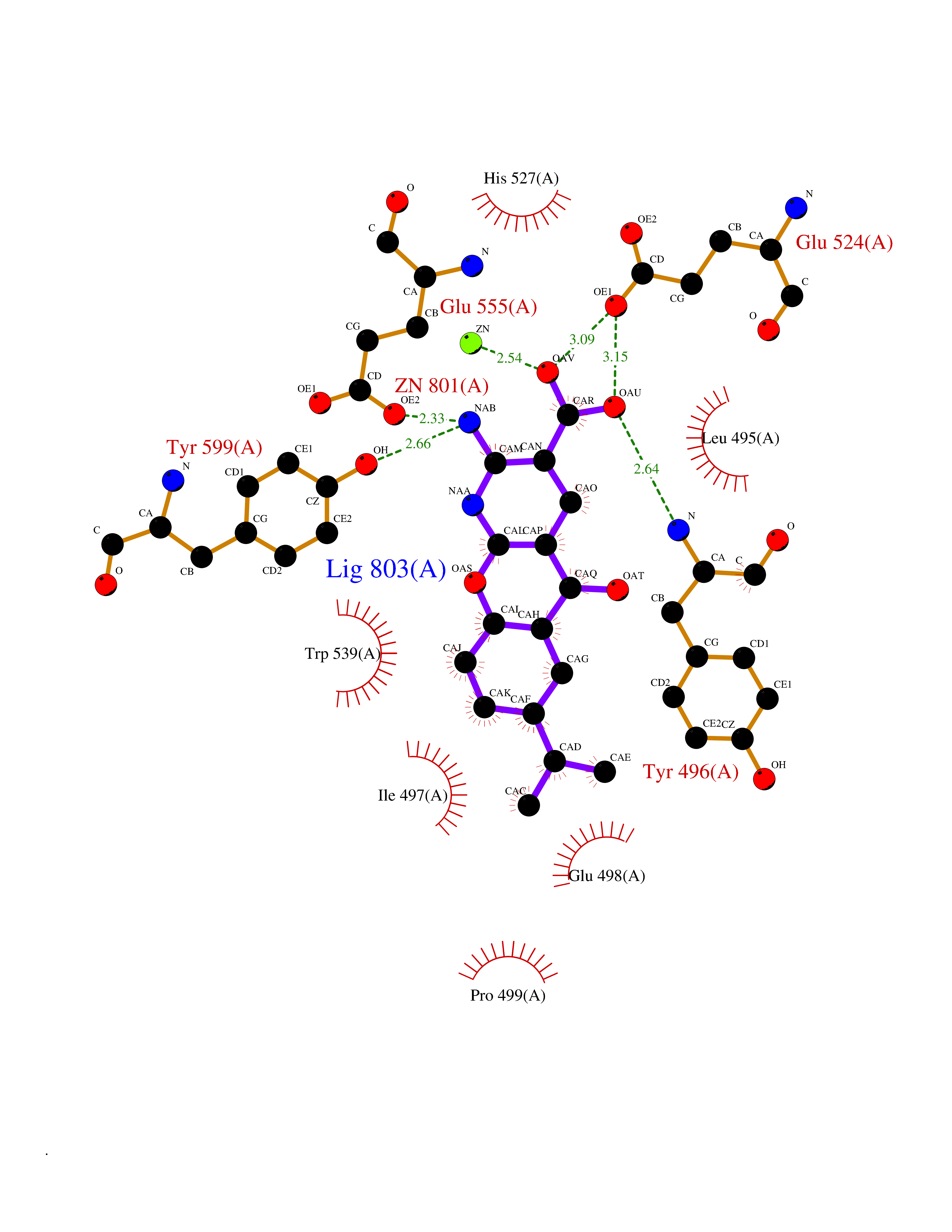 Ligplot Map 3D Binding mode Sequence DHDKFLDDAEKHYLPKTYTFDNGTFIIRAGDKVSEEKIKRLYWASREVKSQFHRVVGNDKALEVGNADDVLTMKIFNSPEEYKFNTTDNGGLYIEPRGTFYTYERTPQQSIFSLEELFRHEYTHYLQARYLVDGLWGQGPFYEKNRLTWFDEGTAEFFAGSTRTSGVLPRKLILGYLAKDKVDHRYSLKKTLNSGYDDSDWMFYNYGFAVAHYLYEKDMPTFIKMNKAILNTDVKSYDEIIKKLSDDANKNTEYQNHIQELVDKYQGAGIPLVSDDYLKDHGYKKASEVYSEISKAASLTNTSVTAEKSQYFNTFTLRGTYTGETSKGEFKDWDEMSKKLDGTLESLAKNSWSGYKTLTAYFTNYRVTSDNKVQYDVVFHGVLTDN Hydrogen bonds contact Hydrophobic contact | ||||
| 10 | Oxygen-insensitive NADPH nitroreductase | 3QDL | 8.27 | |
Target general information Gen name rdxA Organism Helicobacter pylori (strain ATCC 700392 / 26695) (Campylobacter pylori) Uniprot ID TTD ID NA Synonyms HP_0954 Protein family Nitroreductase family Biochemical class Oxidoreductase Function Oxidoreductase activity. Related diseases Hypervalinemia and hyperleucine-isoleucinemia (HVLI) [MIM:618850]: An autosomal recessive metabolic disorder characterized by highly elevated plasma concentrations of valine and leucine/isoleucine. Affected individuals suffer from headache and mild memory impairment. {ECO:0000269|PubMed:25653144}. The disease is caused by variants affecting the gene represented in this entry. A patient with hypervalinemia and hyperleucine-isoleucinemia was identified as compound heterozygote for Gln-170 (inherited from his father) and Lys-264 (inherited from his mother), both variants reduced the catalytic activity of the enzyme. After treatment with vitamin B6, a precursor of pyridoxal 5'-phosphate, a BCAT2 cofactor, the blood levels of branched chain amino acids, especially valine, were decreased and brain lesions were improved. {ECO:0000269|PubMed:25653144}. Drugs (DrugBank ID) DB00916 Interacts with NA EC number 1.-.-.- Uniprot keywords 3D-structure; Antibiotic resistance; NADP; Oxidoreductase; Reference proteome Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 40094.3 Length 352 Aromaticity 0.08 Instability index 55.15 Isoelectric point 6.72 Charge (pH=7) -0.86 2D Binding mode Binding energy (Kcal/mol) -8.51 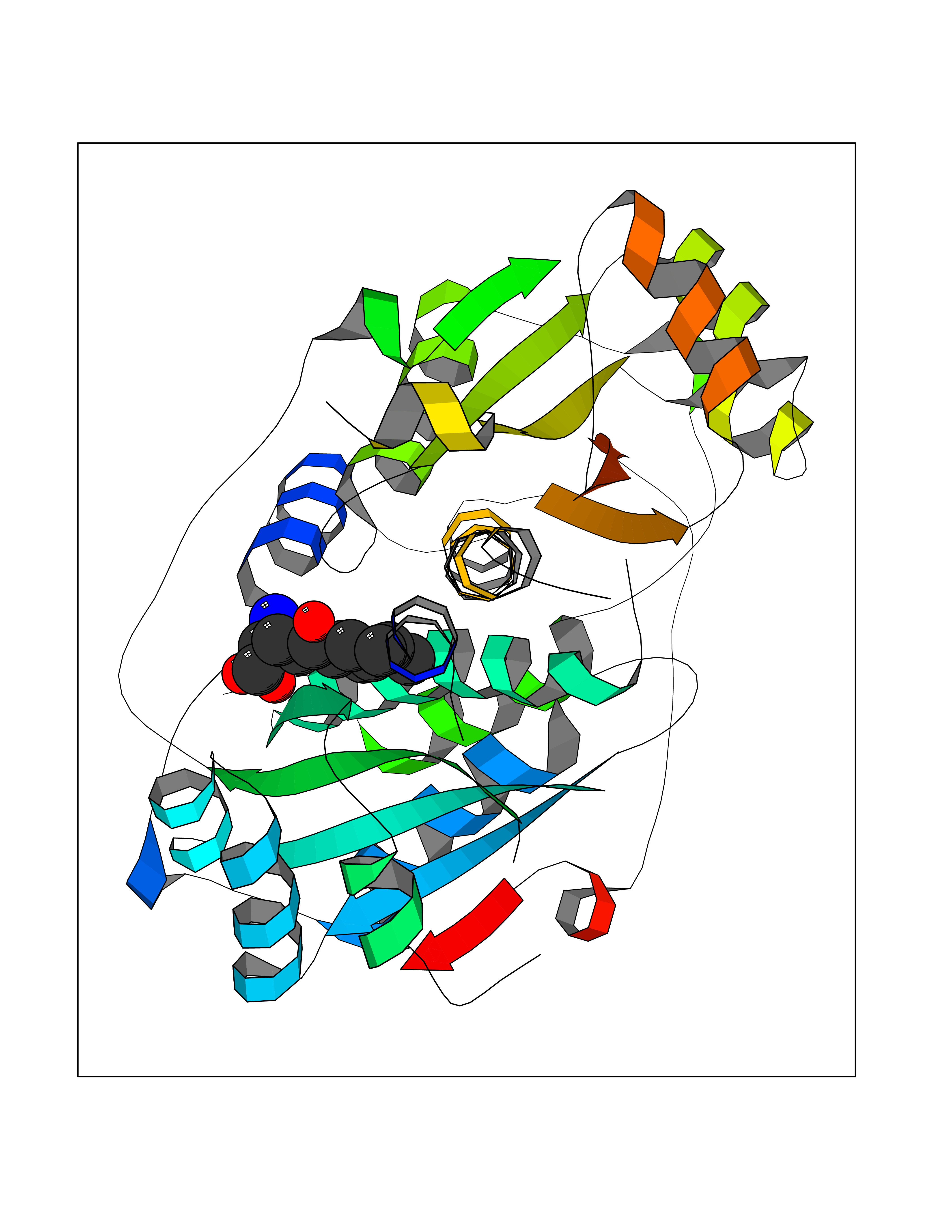 Molscript Map 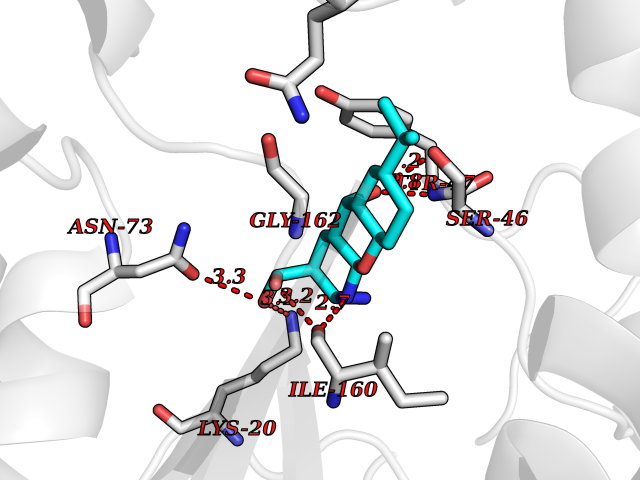 Pymol Map 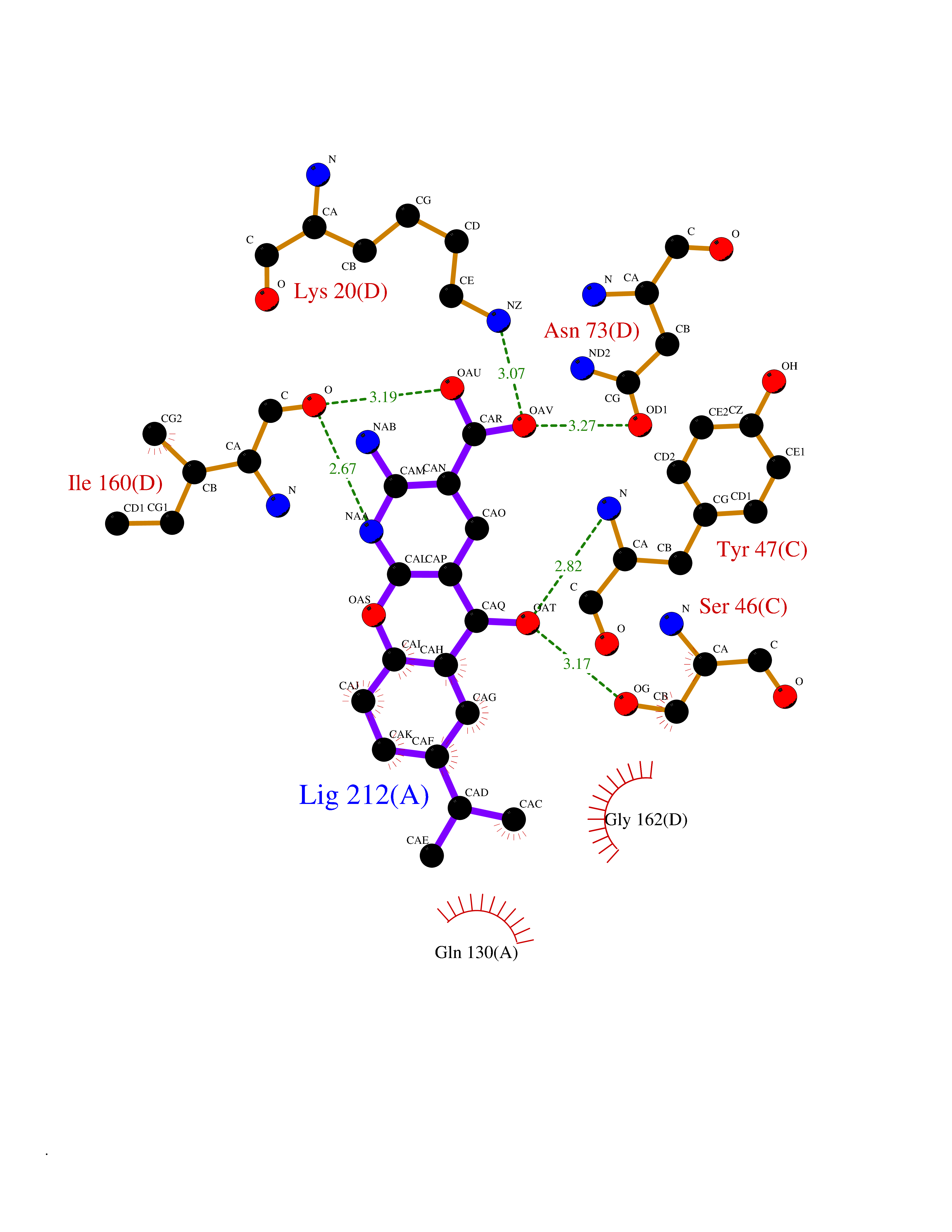 Ligplot Map 3D Binding mode Sequence MQRLESYILMKFLDQEKRRQLLNERHSCKMFDSHYEFSSTELEEIAEIARLSPSSYNTQPWHFVMVTDKDLKKQIAAHSYFNEEMIKSASALMVVCSLSYILEQCYIAVGQICMGVSLMGLDSCIIGGFDPLKVGEVLEERINPKIACLIALGKRVAEASQKSRKSKVDAITWLMKFLDQEKRRQLLNERHSCKMFDSHYEFSSTELEEIAEIARLSPSSYNTQPWHFVMVTDKDLKKQIAAHSYFNEEMIKSASALMVVCSLRPSELLPMQRLESYILEQCYIAVGQICMGVSLMGLDSCIIGGFDPLKVGEVLEERINKPKIACLIALGKRVAEASQKSRKSKVDAITWL Hydrogen bonds contact Hydrophobic contact | ||||
| 11 | Retinal rod rhodopsin-sensitive cGMP 3',5'-cyclic phosphodiesterase subunit gamma | 3JWR | 8.27 | |
Target general information Gen name PDE6G Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms PDEG Protein family Rod/cone cGMP-PDE gamma subunit family Biochemical class Hydrolase Function 3',5'-cyclic-GMP phosphodiesterase activity.CGMP binding.Enzyme inhibitor activity.Spectrin binding. Related diseases Retinitis pigmentosa 57 (RP57) [MIM:613582]: A retinal dystrophy belonging to the group of pigmentary retinopathies. Retinitis pigmentosa is characterized by retinal pigment deposits visible on fundus examination and primary loss of rod photoreceptor cells followed by secondary loss of cone photoreceptors. Patients typically have night vision blindness and loss of midperipheral visual field. As their condition progresses, they lose their far peripheral visual field and eventually central vision as well. {ECO:0000269|PubMed:20655036}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07954; DB00203; DB00820; DB00862 Interacts with O14503; Q96JM7; A0A6Q8PF08; O43741; Q8R511; P62994; Q9QY17; Q63787 EC number 3.1.4.35 Uniprot keywords 3D-structure; Acetylation; cGMP; Hydrolase; Reference proteome; Retinitis pigmentosa; Sensory transduction; Vision Protein physicochemical properties Chain ID C,D Molecular weight (Da) 40027.7 Length 345 Aromaticity 0.09 Instability index 37.44 Isoelectric point 6.02 Charge (pH=7) -6.88 2D Binding mode Binding energy (Kcal/mol) -8.49  Molscript Map 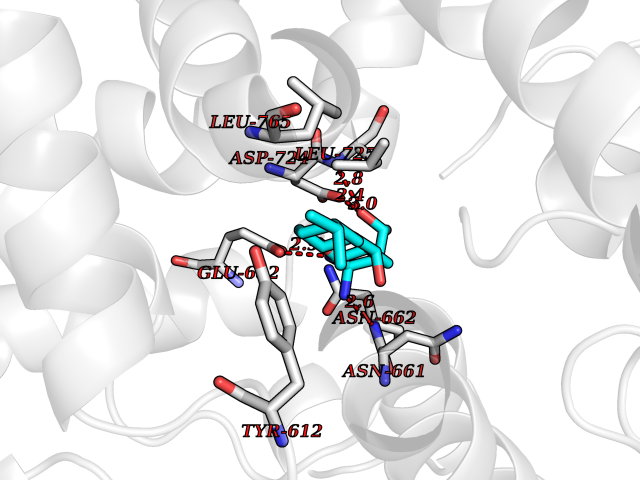 Pymol Map 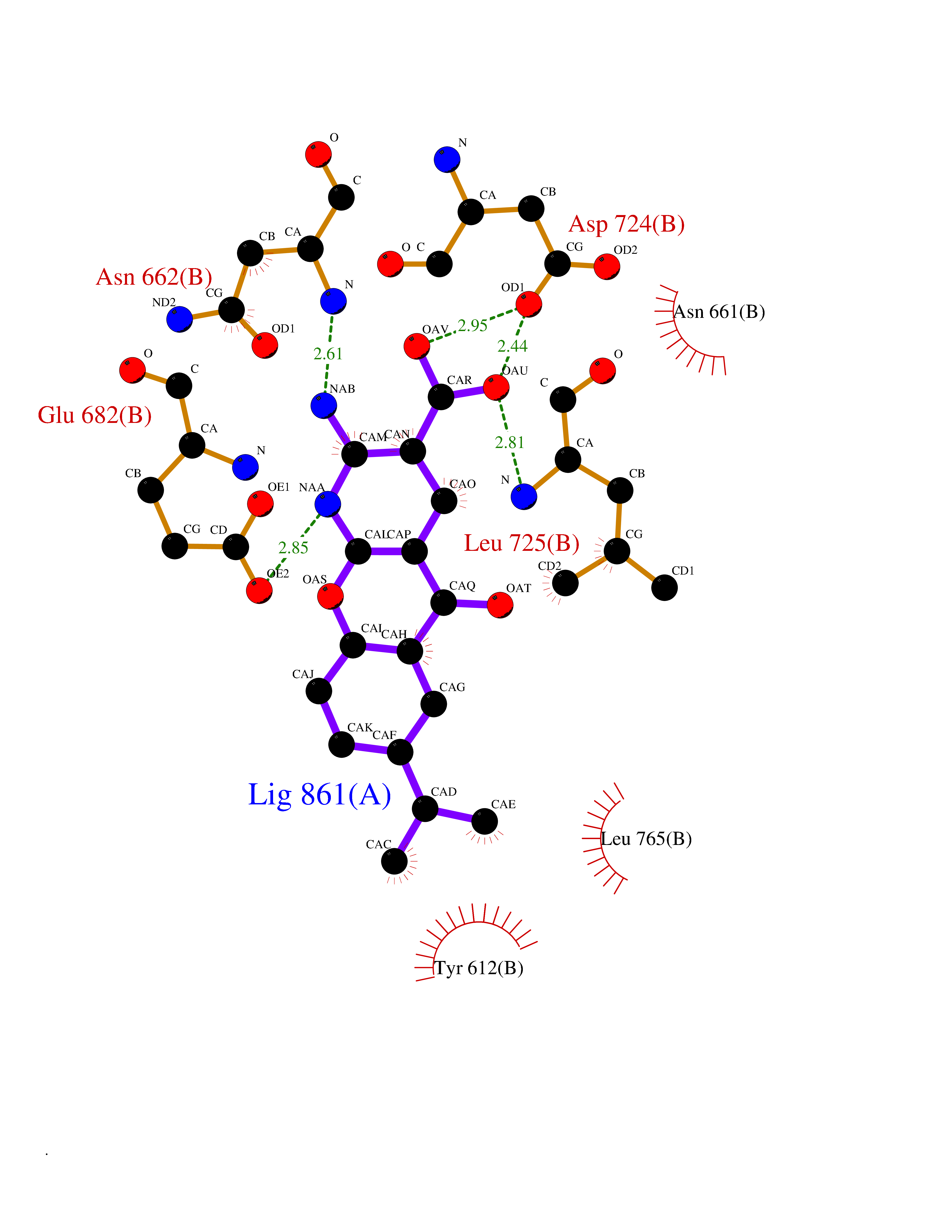 Ligplot Map 3D Binding mode Sequence EAFNHLELHELAQYGIISHMEETRELQSLAAAVVPSAQTLKITDFSFSDFELSDLETALCTIRMFTDLNLVQNFQMKHEVLCRWILSVKKNYRKNVAYHNWRHAFNTAQCMFAALKAGKIQNKLTDLEILALLIAALSHDLDHRGVNNSYIQRSEHPLAQLYCHSIMEHHHFDQCLMILNSPGNQILSGLSIEEYKTTLKIIKQAILATDLALYIKRRGEFFELIRKNQFNLEDPHQKELFLAMLMTACDLSAITKPWPIQQRIAELVATEFWEQGDLERTVLQQQPIPMMDRNKRDELPKLQVGFIDFVCTQLYEALTHVSEDCFPLLDGCRKNRQKWQALAEQ Hydrogen bonds contact Hydrophobic contact | ||||
| 12 | Oxalosuccinate decarboxylase (IDH1) | 6ADG | 8.24 | |
Target general information Gen name IDH1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms PICD; NADP(+)-specific ICDH; Isocitrate dehydrogenase [NADP] cytoplasmic; IDP; IDH; Cytosolic NADP-isocitrate dehydrogenase Protein family Isocitrate and isopropylmalate dehydrogenases family Biochemical class Short-chain dehydrogenases reductase Function Catalyses the NADPH-dependent reduction of alpha-ketoglutarate to R(-)-2-hydroxyglutarate (2HG). Related diseases Glioma (GLM) [MIM:137800]: Gliomas are benign or malignant central nervous system neoplasms derived from glial cells. They comprise astrocytomas and glioblastoma multiforme that are derived from astrocytes, oligodendrogliomas derived from oligodendrocytes and ependymomas derived from ependymocytes. {ECO:0000269|PubMed:19117336, ECO:0000269|PubMed:19935646}. The gene represented in this entry is involved in disease pathogenesis. Mutations affecting Arg-132 are tissue-specific, and suggest that this residue plays a unique role in the development of high-grade gliomas. Mutations of Arg-132 to Cys, His, Leu or Ser abolish magnesium binding and abolish the conversion of isocitrate to alpha-ketoglutarate. Instead, alpha-ketoglutarate is converted to R(-)-2-hydroxyglutarate. Elevated levels of R(-)-2-hydroxyglutarate are correlated with an elevated risk of malignant brain tumors. {ECO:0000269|PubMed:19935646}.; DISEASE: Genetic variations are associated with cartilaginous tumors such as enchondroma or chondrosarcoma. Mutations of Arg-132 to Cys, Gly or His abolish the conversion of isocitrate to alpha-ketoglutarate. Instead, alpha-ketoglutarate is converted to R(-)-2-hydroxyglutarate. {ECO:0000269|PubMed:26161668}. Drugs (DrugBank ID) DB09374; DB01727; DB14568; DB03461; DB16267 Interacts with P0DP23; P27797; P36957; O75874; Q8TDX7; P16284; P17612; P50454; P37173; Q05086-3 EC number EC 1.1.1.42 Uniprot keywords 3D-structure; Acetylation; Cytoplasm; Direct protein sequencing; Glyoxylate bypass; Magnesium; Manganese; Metal-binding; NADP; Oxidoreductase; Peroxisome; Phosphoprotein; Proteomics identification; Reference proteome; Tricarboxylic acid cycle Protein physicochemical properties Chain ID A,B Molecular weight (Da) 92711.7 Length 823 Aromaticity 0.1 Instability index 26.74 Isoelectric point 6.42 Charge (pH=7) -4.48 2D Binding mode Binding energy (Kcal/mol) -9.22 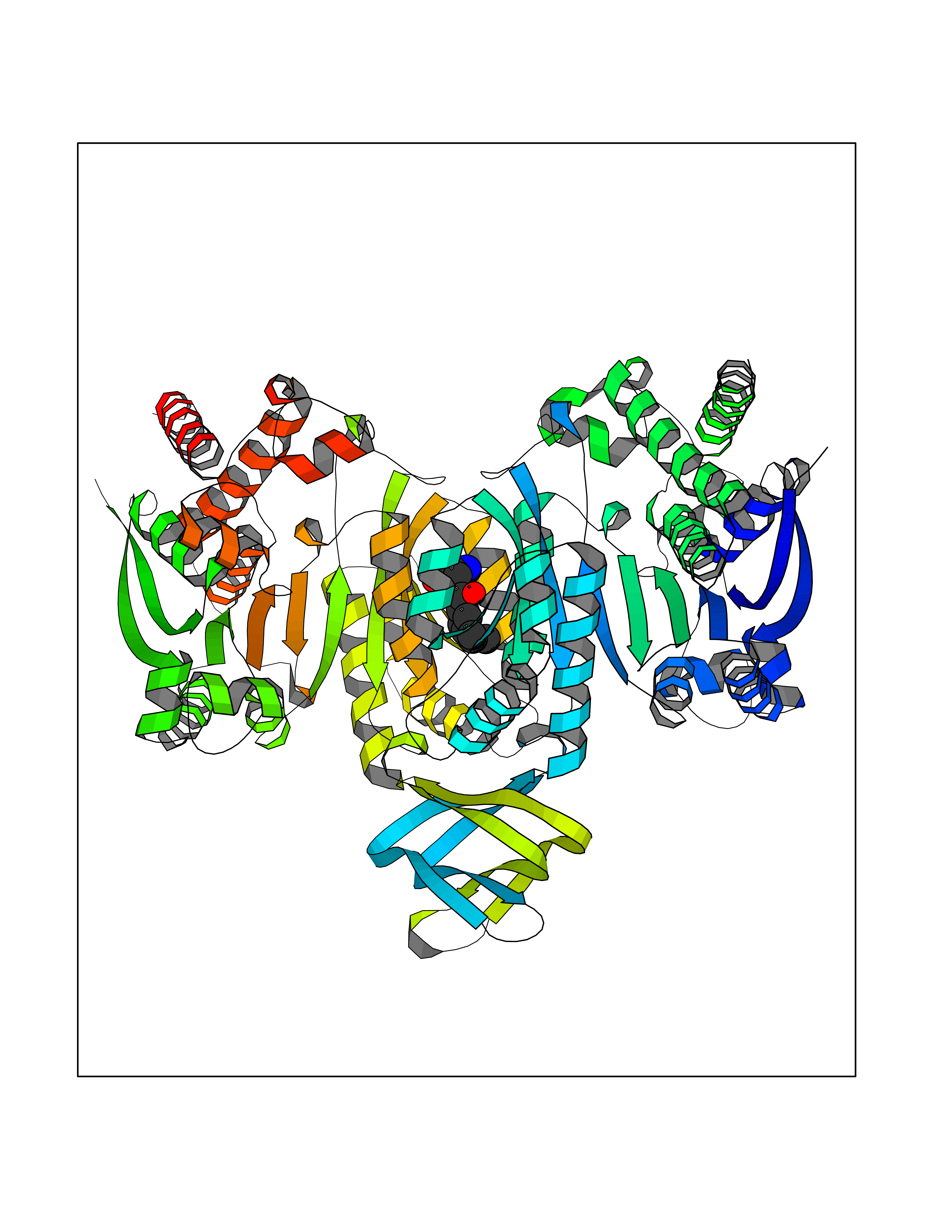 Molscript Map 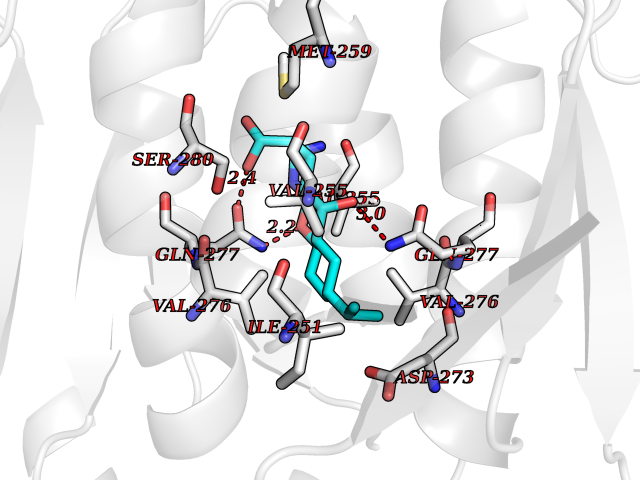 Pymol Map 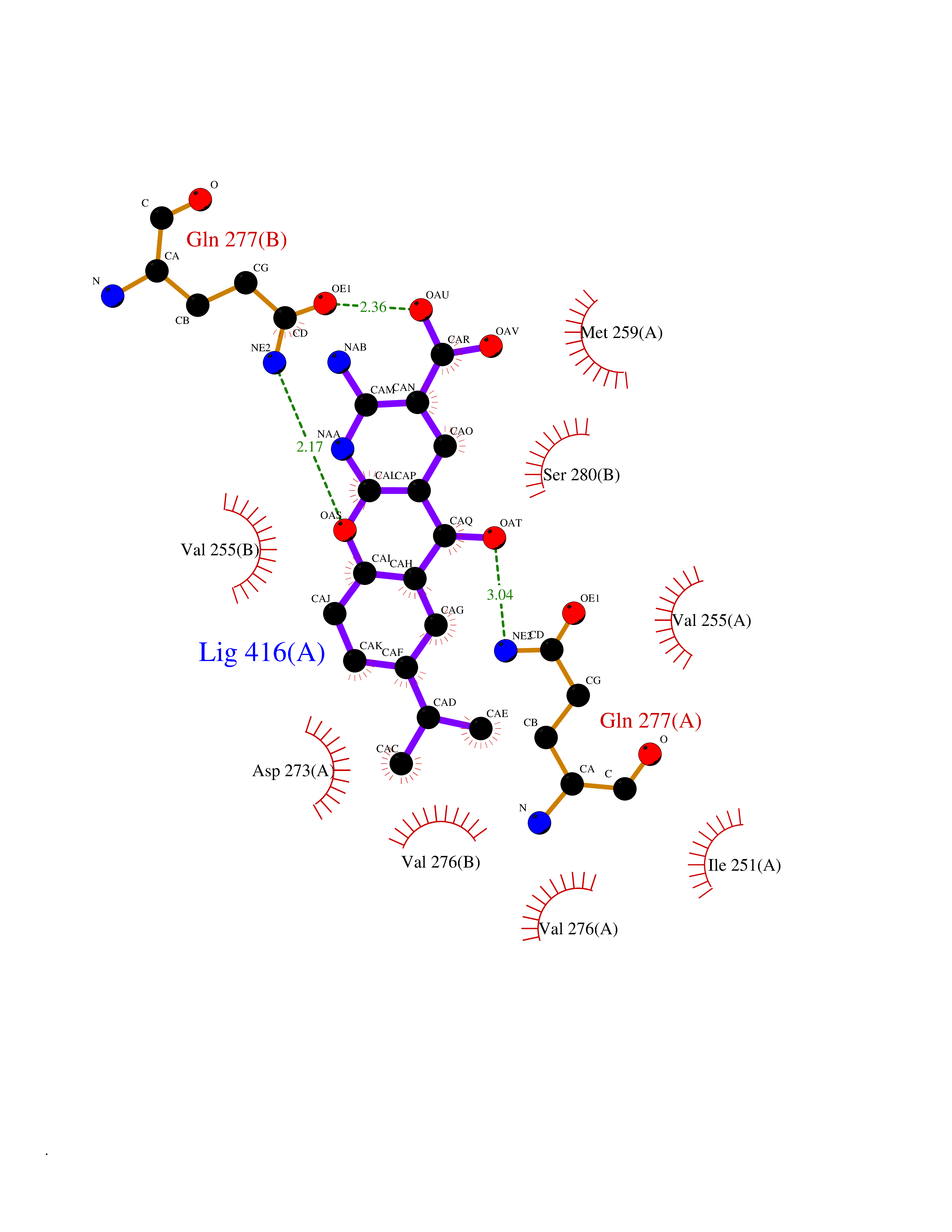 Ligplot Map 3D Binding mode Sequence KKISGGSVVEMQGDEMTRIIWELIKEKLIFPYVELDLHSYDLGIENRDATNDQVTKDAAEAIKKHNVGVKCATITPDEKRVEEFKLKQMWKSPNGTIRNILGGTVFREAIICKNIPRLVSGWVKPIIIGHHAYGDQYRATDFVVPGPGKVEITYTPSDGTQKVTYLVHNFEEGGGVAMGMYNQDKSIEDFAHSSFQMALSKGWPLYLSTKNTILKKYDGRFKDIFQEIYDKQYKSQFEAQKIWYEHRLIDDMVAQAMKSEGGFIWACKNYDGDVQSDSVAQGYGSLGMMTSVLVCPDGKTVEAEAAHGTVTRHYRMYQKGQETSTNPIASIFAWTRGLAHRAKLDNNKELAFFANALEEVSIETIEAGFMTKDLAACIKGLPNVQRSDYLNTFEFMDKLGENLKIKLAQAKLKKISGGSVVEMQGDEMTRIIWELIKEKLIFPYVELDLHSYDLGIENRDATNDQVTKDAAEAIKKHNVGVKCATITPDEKRVEEFKLKQMWKSPNGTIRNILGGTVFREAIICKNIPRLVSGWVKPIIIGHHAYGDQYRATDFVVPGPGKVEITYTPSDGTQKVTYLVHNFEEGGGVAMGMYNQDKSIEDFAHSSFQMALSKGWPLYLSTKNTILKKYDGRFKDIFQEIYDKQYKSQFEAQKIWYEHRLIDDMVAQAMKSEGGFIWACKNYDGDVQSDSVAQGYGSLGMMTSVLVCPDGKTVEAEAAHGTVTRHYRMYQKGQETSTNPIASIFAWTRGLAHRAKLDNNKELAFFANALEEVSIETIEAGFMTKDLAACIKGLPNVQRSDYLNTFEFMDKLGENLKIKLAQAK Hydrogen bonds contact Hydrophobic contact | ||||
| 13 | Phenylethanolamine N-methyltransferase (PNMT) | 2G72 | 8.21 | |
Target general information Gen name PNMT Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms PNMTase; PENT; Noradrenaline N-methyltransferase Protein family Class I-like SAM-binding methyltransferase superfamily, NNMT/PNMT/TEMT family Biochemical class NA Function Converts noradrenaline to adrenaline. Related diseases A chromosomal aberration involving TRIM24/TIF1 is found in papillary thyroid carcinomas (PTCs). Translocation t(7;10)(q32;q11) with RET. The translocation generates the TRIM24/RET (PTC6) oncogene. {ECO:0000269|PubMed:10439047}. Drugs (DrugBank ID) DB08129; DB08128; DB07739; DB07798; DB07747; DB03468; DB08550; DB03824; DB04273; DB07906; DB07597; DB09571; DB00968; DB08631; DB01752; DB08654 Interacts with Q9P2G9-2; Q8TBB1 EC number EC 2.1.1.28 Uniprot keywords 3D-structure; Catecholamine biosynthesis; Direct protein sequencing; Methyltransferase; Phosphoprotein; Proteomics identification; Reference proteome; S-adenosyl-L-methionine; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 29198.9 Length 264 Aromaticity 0.09 Instability index 54.33 Isoelectric point 5.91 Charge (pH=7) -3.69 2D Binding mode Binding energy (Kcal/mol) -9.6  Molscript Map 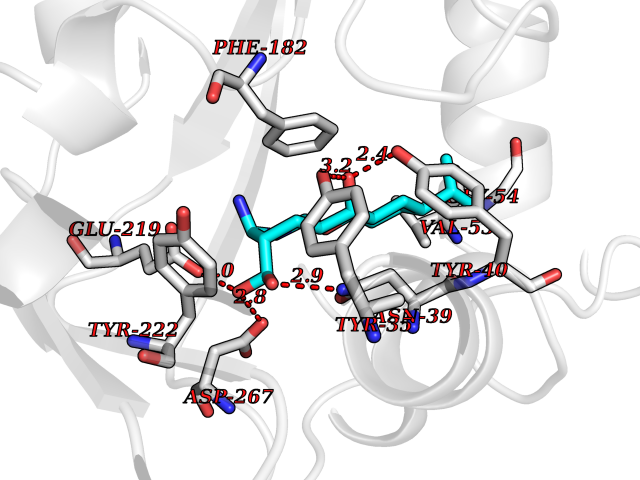 Pymol Map 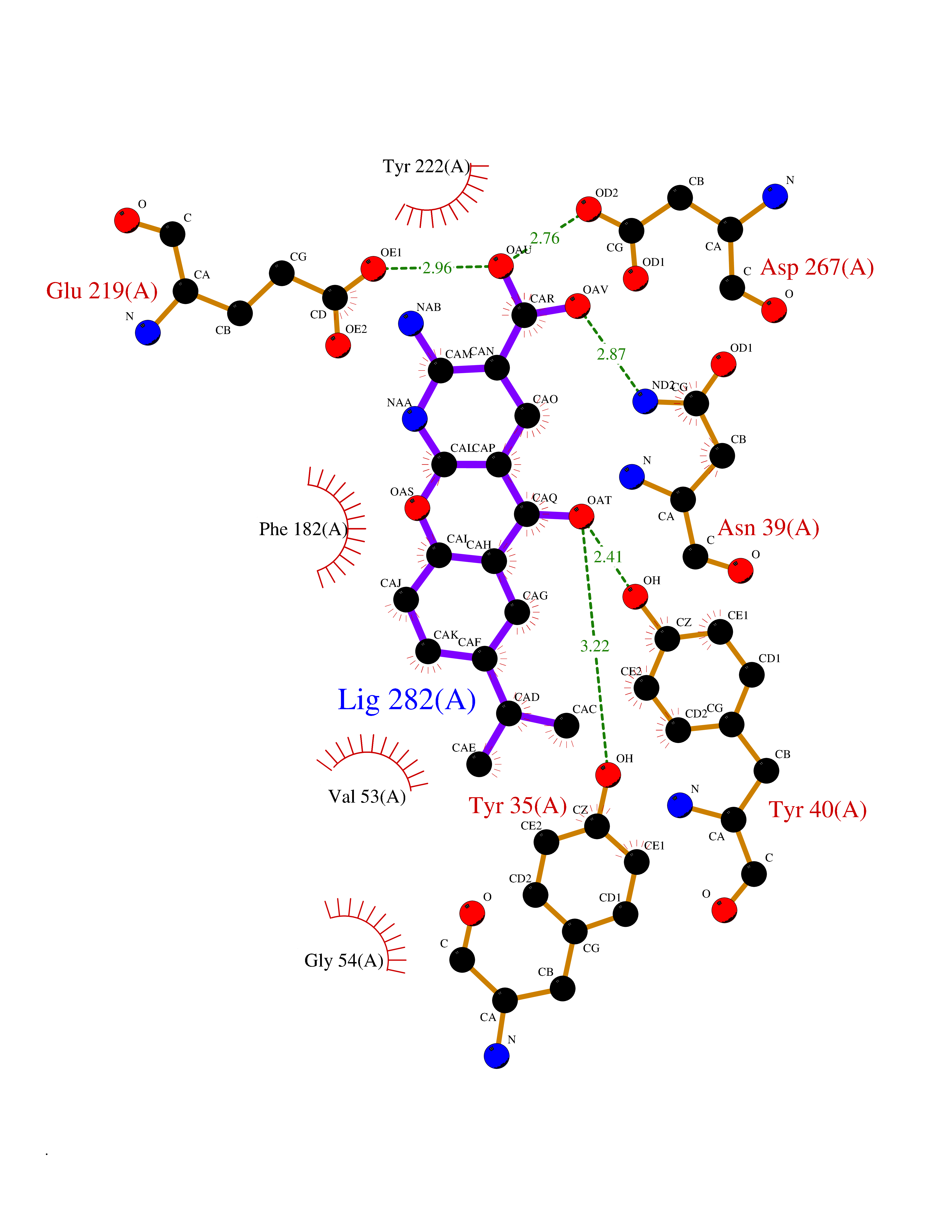 Ligplot Map 3D Binding mode Sequence APGQAAVASAYQRFEPRAYLRNNYAPPRGDLCNPNGVGPWKLRCLAQTFATGEVSGRTLIDIGSGPTVYQLLSACSHFEDITMTDFLEVNRQELGRWLQEEPGAFNWSMYSQHACLIEGKGECWQDKERQLRARVKRVLPIDVHQPQPLGAGSPAPLPADALVSAFCLEAVSPDLASFQRALDHITTLLRPGGHLLLIGALEESWYLAGEARLTVVPVSEEEVREALVRSGYKVRDLRTYIMPAHLQTGVDDVKGVFFAWAQKV Hydrogen bonds contact Hydrophobic contact | ||||
| 14 | Protein cereblon (CRBN) | 5FQD | 8.21 | |
Target general information Gen name CRBN Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Protein cereblon Protein family CRBN family Biochemical class NA Function Substrate recognition component of a DCX (DDB1-CUL4-X-box) E3 protein ligase complex that mediates the ubiquitination and subsequent proteasomal degradation of target proteins, such as MEIS2. Normal degradation of key regulatory proteins is required for normal limb outgrowth and expression of the fibroblast growth factor FGF8. May play a role in memory and learning by regulating the assembly and neuronal surface expression of large-conductance calcium-activated potassium channels in brain regions involved in memory and learning via its interaction with KCNT1. Binding of pomalidomide and other thalidomide-related drugs changes the substrate specificity of the human protein, leading to decreased degradation of MEIS2 and other target proteins and increased degradation of MYC, IRF4, IKZF1 and IKZF3. Related diseases Intellectual developmental disorder, autosomal recessive 2 (MRT2) [MIM:607417]: A disorder characterized by significantly below average general intellectual functioning associated with impairments in adaptive behavior and manifested during the developmental period. MRT2 patients display mild intellectual disability with a standard IQ ranged from 50 to 70. IQ scores are lower in males than females. Developmental milestones are mildly delayed. There are no dysmorphic or autistic features. {ECO:0000269|PubMed:15557513, ECO:0000269|PubMed:28143899}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00480; DB08910; DB01041 Interacts with Q96A83-2; P48729; Q16531; O14901; Q8IVT2; Q9P286; A0A6Q8PF08; Q93062; Q16531; Q13422-7 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; Disease variant; Intellectual disability; Membrane; Metal-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Ubl conjugation; Ubl conjugation pathway; Zinc Protein physicochemical properties Chain ID B,E Molecular weight (Da) 38245.7 Length 337 Aromaticity 0.08 Instability index 40.62 Isoelectric point 5.7 Charge (pH=7) -6.53 2D Binding mode Binding energy (Kcal/mol) -9.65 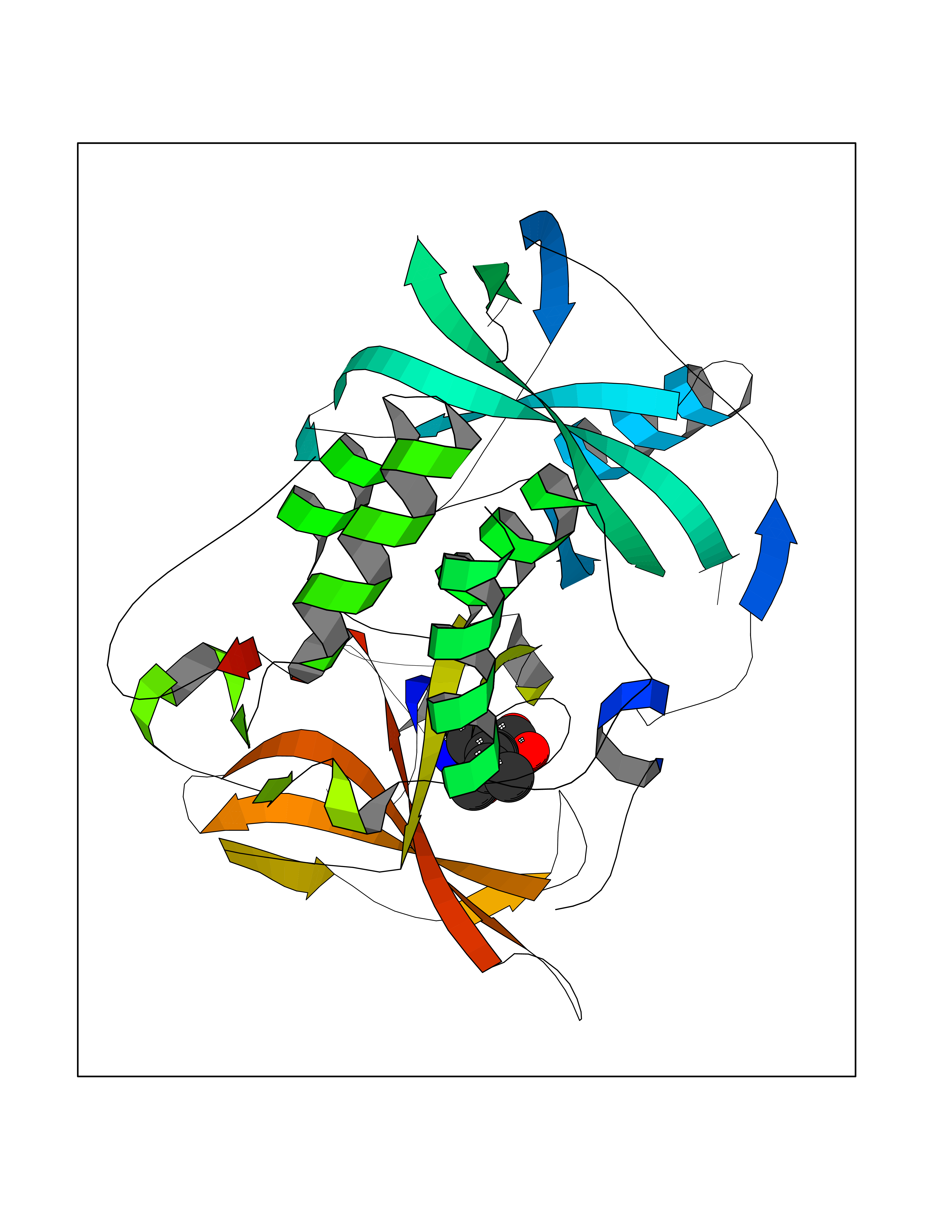 Molscript Map 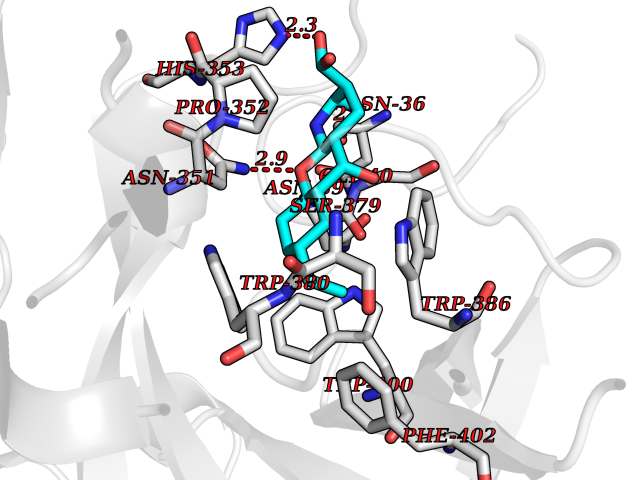 Pymol Map 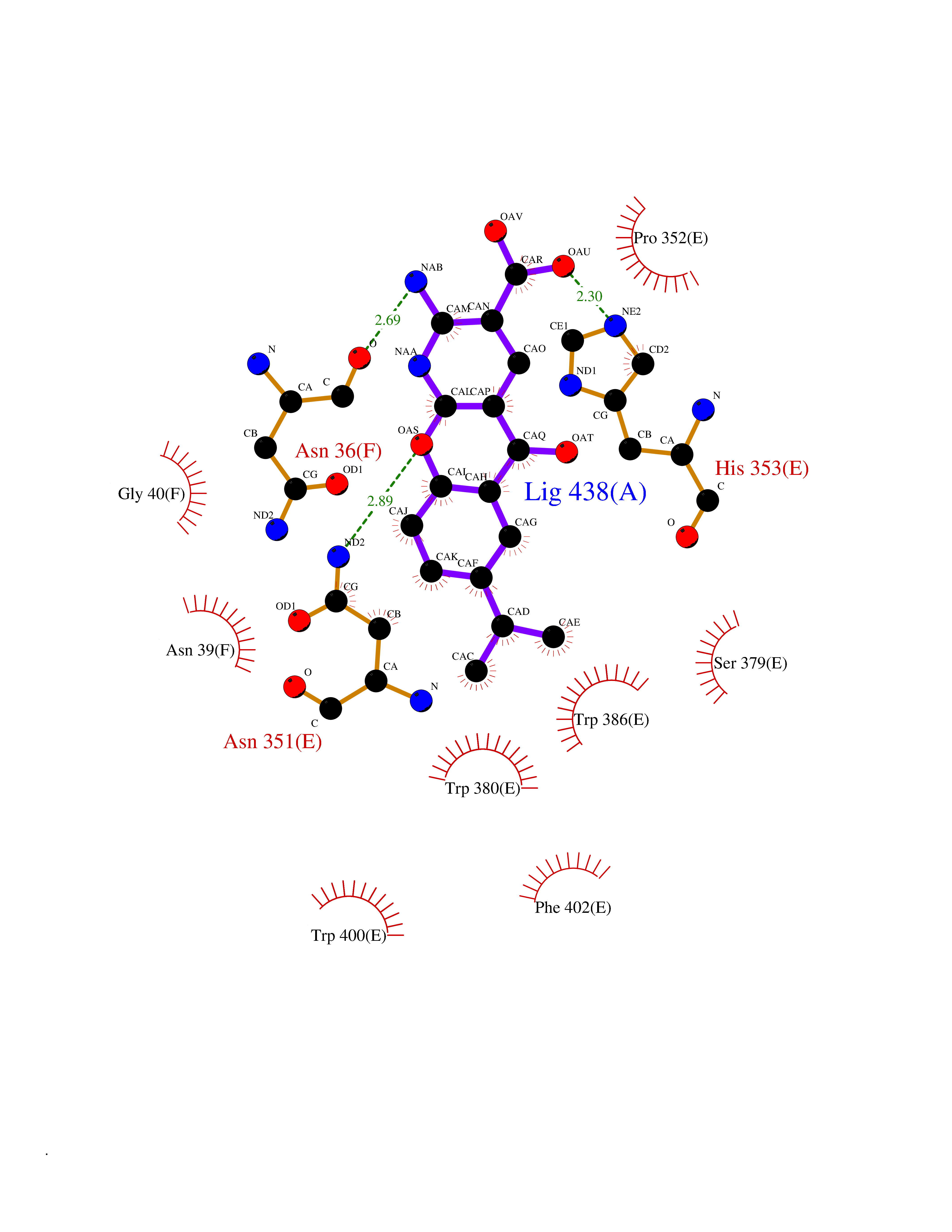 Ligplot Map 3D Binding mode Sequence EFIVGGKYKLNITNGEEVAVINFDTSLPTSHTYLGADMEEFHGRTLHDDDSCQVIPVLPQVMMILIPGQTLPLQLFHPQEVSMVRNLIQKDRTFAVLAYSNVQEREAQFGTTAEIYAYREEIVKVKAIGRQRFKVLEQQAKVQILPECVLAETLMDRIKKQLREWDENLKDDSLPSNPIDFSYRVAACLPIDDVLRIQLLKIGSAIQRLRCELDIMNKCTSLCCKQCQETEITTKNEIFSLSLCGPMAAYVNPHGYVHETLTVYKACNLNLIGRPSTEHSWFPGYAWTVAQCKICASHIGWKFTATKKDMSPQKFWGLTRSALLPTIPDTEDEISPD Hydrogen bonds contact Hydrophobic contact | ||||
| 15 | Adrenergic receptor beta-2 (ADRB2) | 2RH1 | 8.20 | |
Target general information Gen name ADRB2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Beta-2 adrenoreceptor; Beta-2 adrenoceptor; Beta-2 adrenergic receptor; B2AR; ADRB2R Protein family G-protein coupled receptor 1 family, Adrenergic receptor subfamily, ADRB2 sub-subfamily Biochemical class GPCR rhodopsin Function The beta-2-adrenergic receptor binds epinephrine with an approximately 30-fold greater affinity than it does norepinephrine. Beta-adrenergic receptors mediate the catecholamine-induced activation of adenylate cyclase through the action of G proteins. Related diseases Cortical dysplasia, complex, with other brain malformations 6 (CDCBM6) [MIM:615771]: A disorder of aberrant neuronal migration and disturbed axonal guidance. Affected individuals have microcephaly, ataxia, and severe delayed psychomotor development. Brain imaging shows variable malformations of cortical development, including white matter streaks, dysmorphic basal ganglia, corpus callosum abnormalities, brainstem and cerebellar hypoplasia, cortical dysplasia, polymicrogyria. {ECO:0000269|PubMed:23246003}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Skin creases, congenital symmetric circumferential, 1 (CSCSC1) [MIM:156610]: An autosomal dominant disease characterized by multiple, symmetric, circumferential rings of folded skin, affecting primarily the limbs. Affected individuals also exhibit intellectual disability, cleft palate, and dysmorphic features. {ECO:0000269|PubMed:26637975}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07543; DB01193; DB00866; DB01118; DB00182; DB01102; DB01274; DB01238; DB09204; DB06216; DB00335; DB01408; DB05590; DB09013; DB00195; DB00217; DB01295; DB00612; DB00901; DB08807; DB06726; DB08808; DB00248; DB00521; DB01136; DB04846; DB01407; DB00785; DB01151; DB11273; DB13345; DB00449; DB11278; DB00841; DB09273; DB06262; DB01363; DB01364; DB00668; DB01049; DB11587; DB01288; DB00983; DB05039; DB00221; DB01064; DB00598; DB01210; DB13139; DB01365; DB13624; DB01214; DB00264; DB01203; DB05849; DB04861; DB00368; DB00540; DB00334; DB09080; DB00816; DB01580; DB00715; DB01359; DB00925; DB00397; DB00960; DB01291; DB01366; DB01182; DB00571; DB06814; DB00852; DB01917; DB11124; DB00867; DB01001; DB00938; DB00489; DB03566; DB00127; DB00871; DB00373; DB00726; DB12248; DB09082; DB09185 Interacts with P30542; P07550; P32121; Q96B67; Q9UII2; Q9ULD4-2; Q9NSI6-4; Q5M9N0-2; A0AVK6; Q658K8; O00472; Q15910-2; Q15486; P61978; Q5TCQ9; Q99685; O14745; Q9NR21-5; Q8WVD3; Q9H0X6; Q13573; P12931; Q5T0J7-2; Q8N0U2 EC number NA Uniprot keywords 3D-structure; Cell membrane; Disulfide bond; Endosome; G-protein coupled receptor; Glycoprotein; Golgi apparatus; Hydroxylation; Lipoprotein; Membrane; Palmitate; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Transducer; Transmembrane; Transmembrane helix; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 32266.1 Length 282 Aromaticity 0.15 Instability index 36.1 Isoelectric point 8.02 Charge (pH=7) 2.1 2D Binding mode Binding energy (Kcal/mol) -8.78 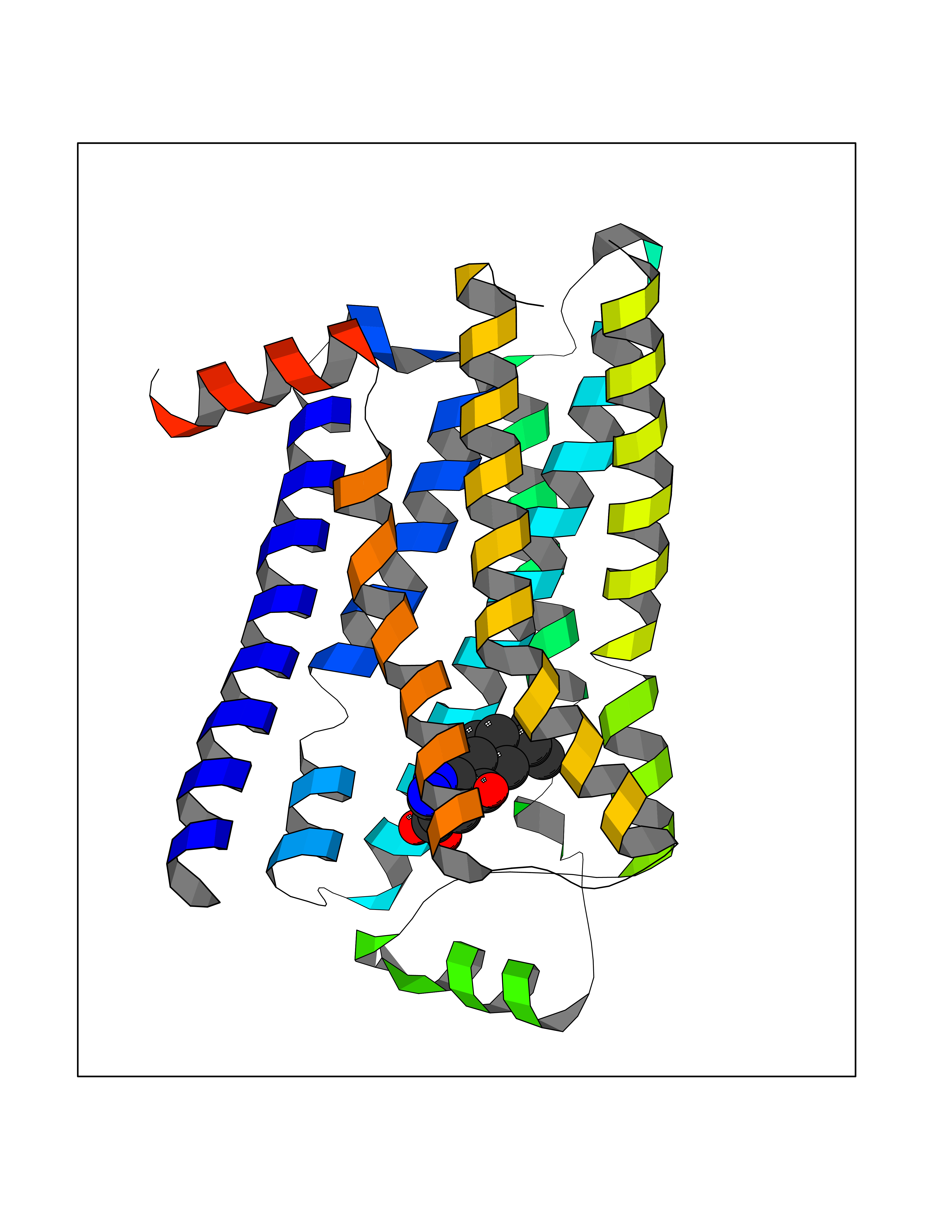 Molscript Map 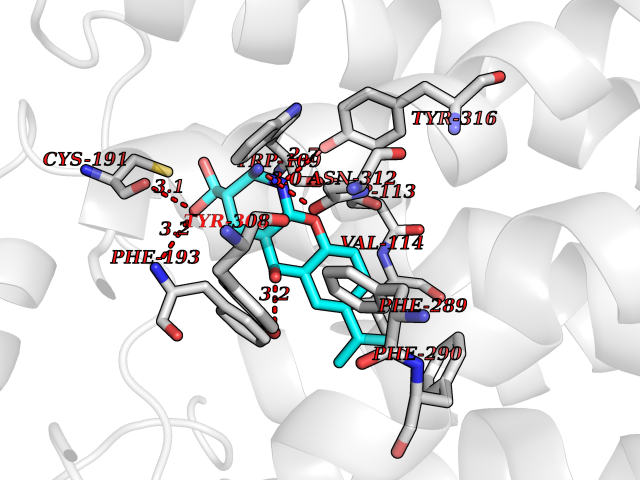 Pymol Map 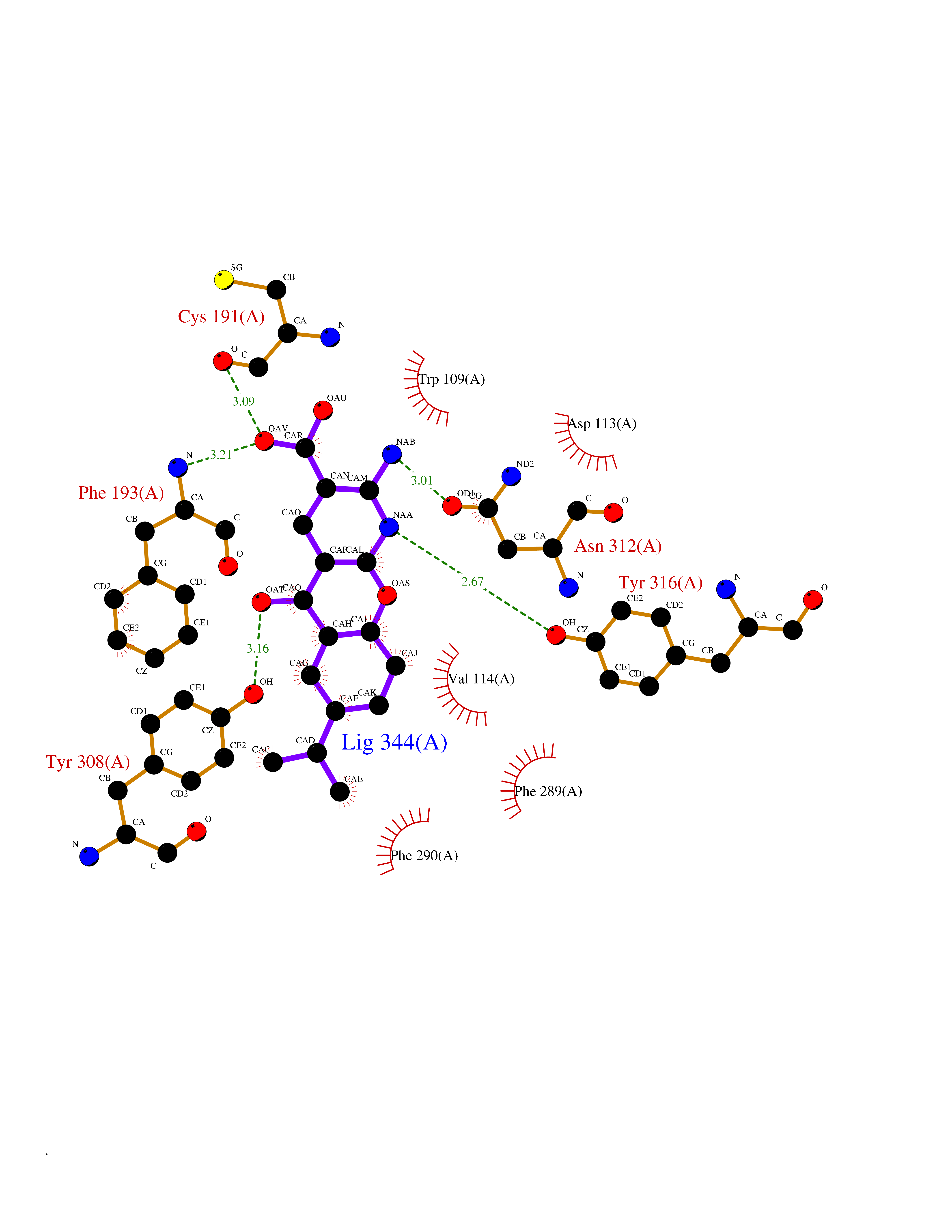 Ligplot Map 3D Binding mode Sequence DEVWVVGMGIVMSLIVLAIVFGNVLVITAIAKFERLQTVTNYFITSLACADLVMGLAVVPFGAAHILMKMWTFGNFWCEFWTSIDVLCVTASIETLCVIAVDRYFAITSPFKYQSLLTKNKARVIILMVWIVSGLTSFLPIQMHWYRATHQEAINCYAEETCCDFFTNQAYAIASSIVSFYVPLVIMVFVYSRVFQEAKRQLKFCLKEHKALKTLGIIMGTFTLCWLPFFIVNIVHVIQDNLIRKEVYILLNWIGYVNSGFNPLIYCRSPDFRIAFQELLCL Hydrogen bonds contact Hydrophobic contact | ||||
| 16 | Endothelin-converting enzyme 1 (ECE1) | 3DWB | 8.20 | |
Target general information Gen name ECE1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms ECE-1 Protein family Peptidase M13 family Biochemical class Peptidase Function Converts big endothelin-1 to endothelin-1. Related diseases Hirschsprung disease, cardiac defects, and autonomic dysfunction (HCAD) [MIM:613870]: A disorder characterized by skip-lesions Hirschsprung disease, craniofacial abnormalities and other dysmorphic features, cardiac defects including ductus arteriosus, small subaortic ventricular septal defect, small atrial septal defect, and autonomic dysfunction. {ECO:0000269|PubMed:9915973}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07171 Interacts with P49760; A8MQ03; Q8IUG1; P60370; P60410 EC number EC 3.4.24.71 Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disease variant; Disulfide bond; Glycoprotein; Hirschsprung disease; Hydrolase; Membrane; Metal-binding; Metalloprotease; Phosphoprotein; Protease; Proteomics identification; Reference proteome; Signal-anchor; Transmembrane; Transmembrane helix; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 75247.9 Length 660 Aromaticity 0.12 Instability index 46.29 Isoelectric point 5.33 Charge (pH=7) -18.3 2D Binding mode Binding energy (Kcal/mol) -8.29 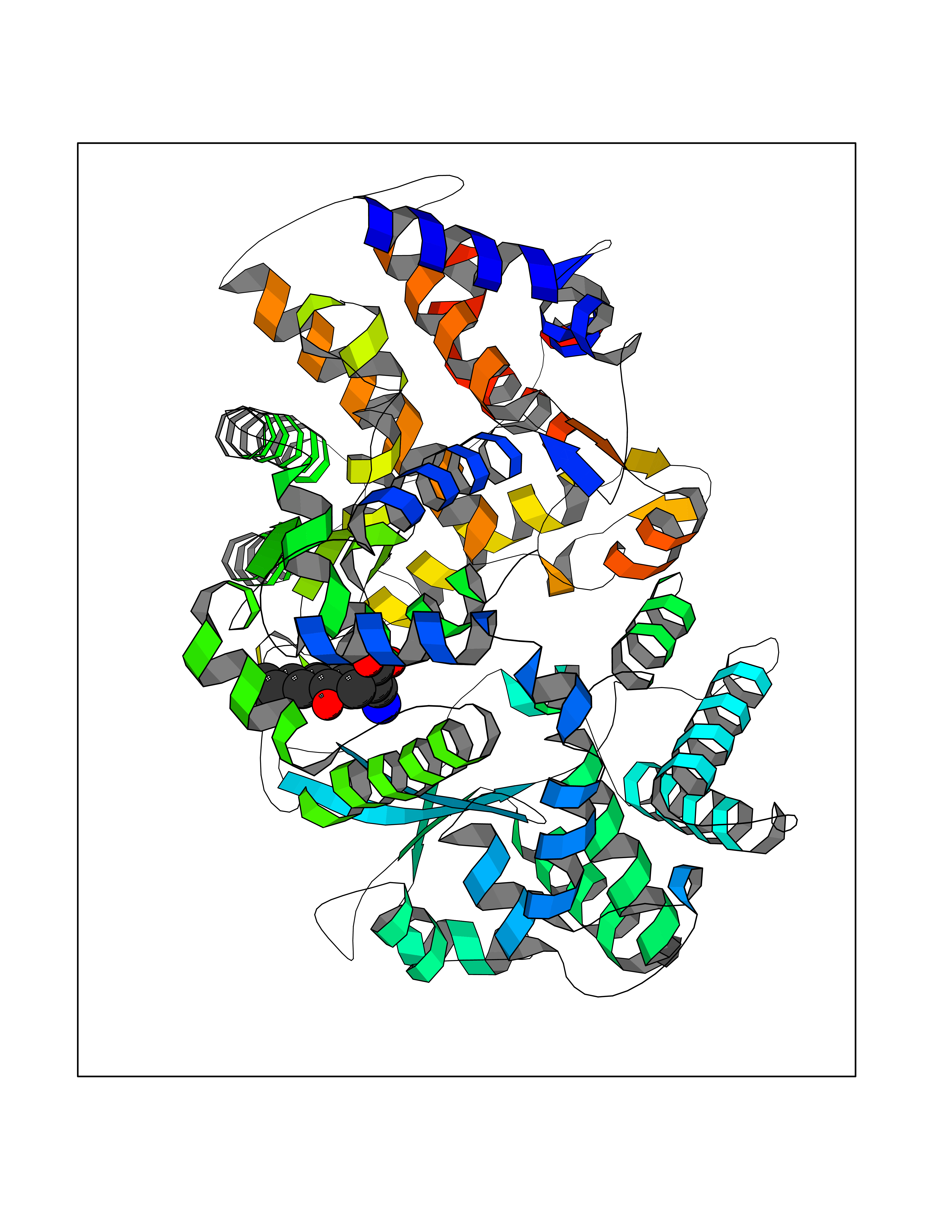 Molscript Map 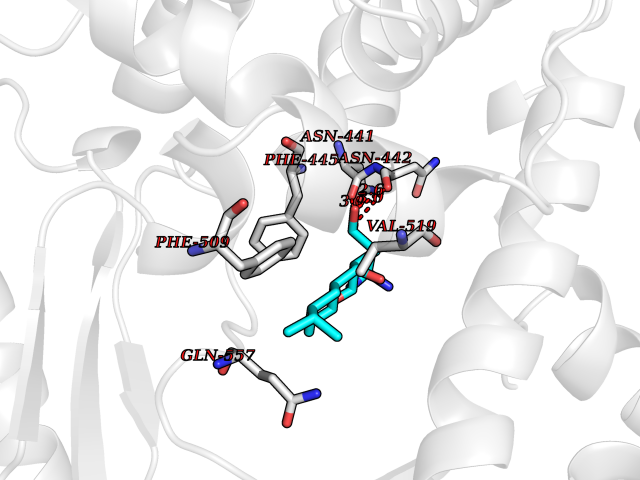 Pymol Map 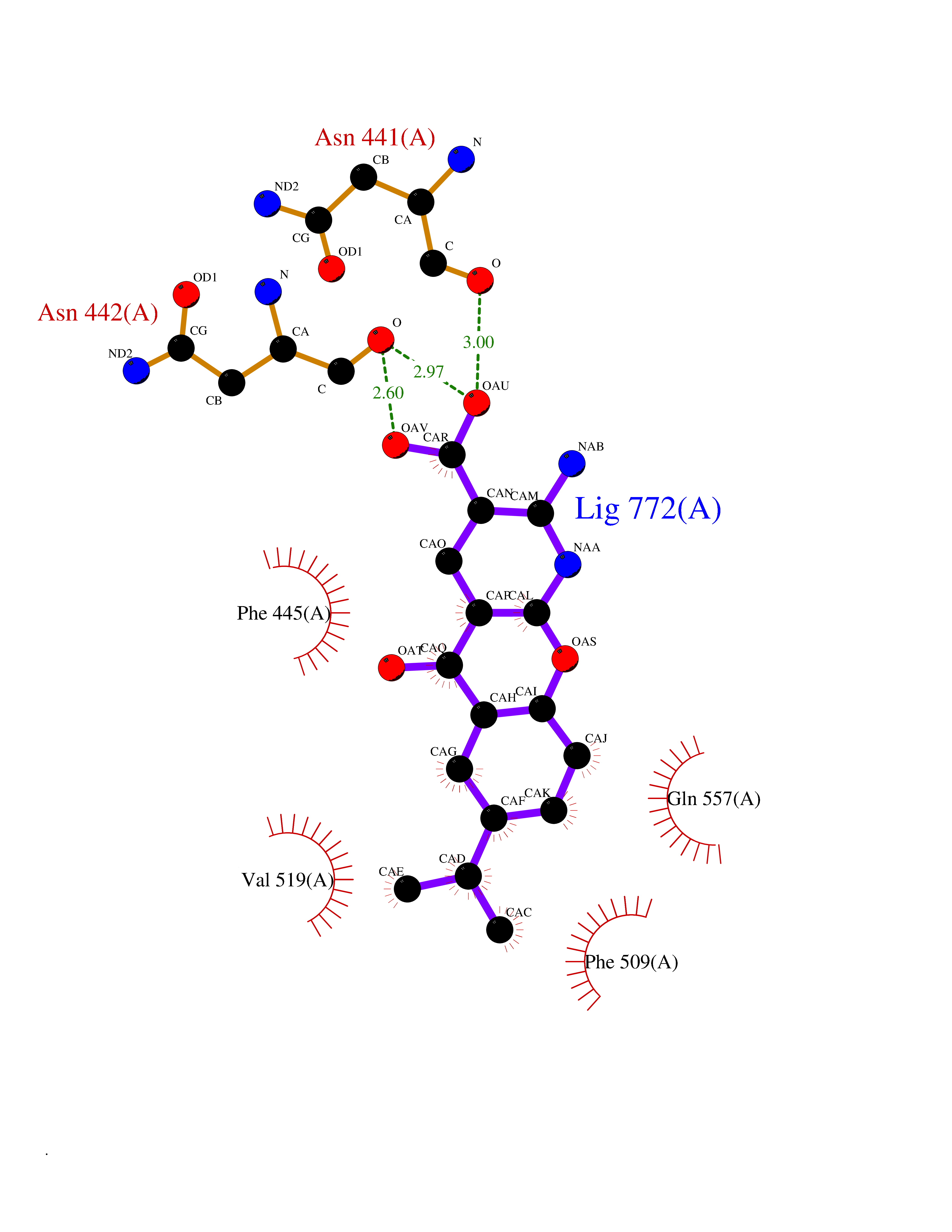 Ligplot Map 3D Binding mode Sequence SEACVSVTSSILSSMDPTVDPCHDFFSYACGGWIKANPVPDGHSRWGTFSNLWEHNQAIIKHLLENSTASVSEAERKAQVYYRACMNETRIEELRAKPLMELIERLGGWNITGPWAKDNFQDTLQVVTAHYRTSPFFSVYVSADSKNSNSNVIQVDQSGLGLPSRDYYLNKTENEKVLTGYLNYMVQLGKLLGGGDEEAIRPQMQQILDFETALANITIPQEKRRDEELIYHKVTAAELQTLAPAINWLPFLNTIFYPVEINESEPIVVYDKEYLEQISTLINTTDRCLLNNYMIWNLVRKTSSFLDQRFQDADEKFMEVMWKFCVSDTENNLGFALGPMFVKATFAEDSKSIATEIILEIKKAFEESLSTLKWMDEETRKSAKEKADAIYNMIGYPNFIMDPKELDKVFNDYTAVPDLYFENAMRFFNFSWRVTADQLRKAPNRDQWSMTPPMVNAYYSPTKNEIVFPAGILQAPFYTRSSPKALNFGGIGVVVGHELTHAFDDQGREYDKDGNLRPWWKNSSVEAFKRQTECMVEQYSNYSVNGEPVNGRHTLGENIADNGGLKAAYRAYQNWVKKNGAEHSLPTLGLTNNQLFFLGFAQVWCSVRTPESSHEGLITDPHSPSRFRVIGSLSNSKEFSEHFRCPPGSPMNPPHKCEVW Hydrogen bonds contact Hydrophobic contact | ||||
| 17 | Matrix metalloproteinase-16 (MMP-16) | 1RM8 | 8.19 | |
Target general information Gen name MMP16 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Membrane-type-3 matrix metalloproteinase; Membrane-type matrix metalloproteinase 3; MTMMP3; MT3MMP; MT3-MMP; MT-MMP 3; MMPX2; MMP-X2; C8orf57 Protein family Peptidase M10A family Biochemical class Peptidase Function Activates progelatinase A. Involved in the matrix remodeling of blood vessels. Isoform short cleaves fibronectin and also collagen type III, but at lower rate. It has no effect on type I, II, IV and V collagen. However, upon interaction with CSPG4, it may be involved in degradation and invasion of type I collagen by melanoma cells. Endopeptidase that degrades various components of the extracellular matrix, such as collagen type III and fibronectin. Related diseases Cerebral creatine deficiency syndrome 3 (CCDS3) [MIM:612718]: An autosomal recessive disorder characterized by developmental delay/regression, intellectual disability, severe disturbance of expressive and cognitive speech, and severe depletion of creatine/phosphocreatine in the brain. Most patients develop a myopathy characterized by muscle weakness and atrophy later in life. {ECO:0000269|PubMed:11555793, ECO:0000269|PubMed:20682460, ECO:0000269|PubMed:22386973, ECO:0000269|PubMed:23660394, ECO:0000269|PubMed:23770102, ECO:0000269|PubMed:26490222, ECO:0000269|PubMed:27233232}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Fanconi renotubular syndrome 1 (FRTS1) [MIM:134600]: A form of Fanconi renotubular syndrome, a disease due to a generalized dysfunction of the proximal kidney tubule resulting in decreased solute and water reabsorption. Patients have polydipsia and polyuria with phosphaturia, glycosuria and aminoaciduria. They may develop hypophosphatemic rickets or osteomalacia, acidosis and a tendency toward dehydration. Some eventually develop renal insufficiency. FRTS1 inheritance is autosomal dominant. {ECO:0000269|PubMed:29654216}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03880; DB00786 Interacts with NA EC number EC 3.4.24.- Uniprot keywords 3D-structure; Alternative splicing; Calcium; Cell membrane; Cleavage on pair of basic residues; Collagen degradation; Disulfide bond; Extracellular matrix; Glycoprotein; Hydrolase; Membrane; Metal-binding; Metalloprotease; Protease; Proteomics identification; Reference proteome; Repeat; Secreted; Signal; Transmembrane; Transmembrane helix; Zinc; Zymogen Protein physicochemical properties Chain ID A Molecular weight (Da) 18853.6 Length 169 Aromaticity 0.12 Instability index 33.65 Isoelectric point 4.88 Charge (pH=7) -12.42 2D Binding mode Binding energy (Kcal/mol) -9.42  Molscript Map  Pymol Map 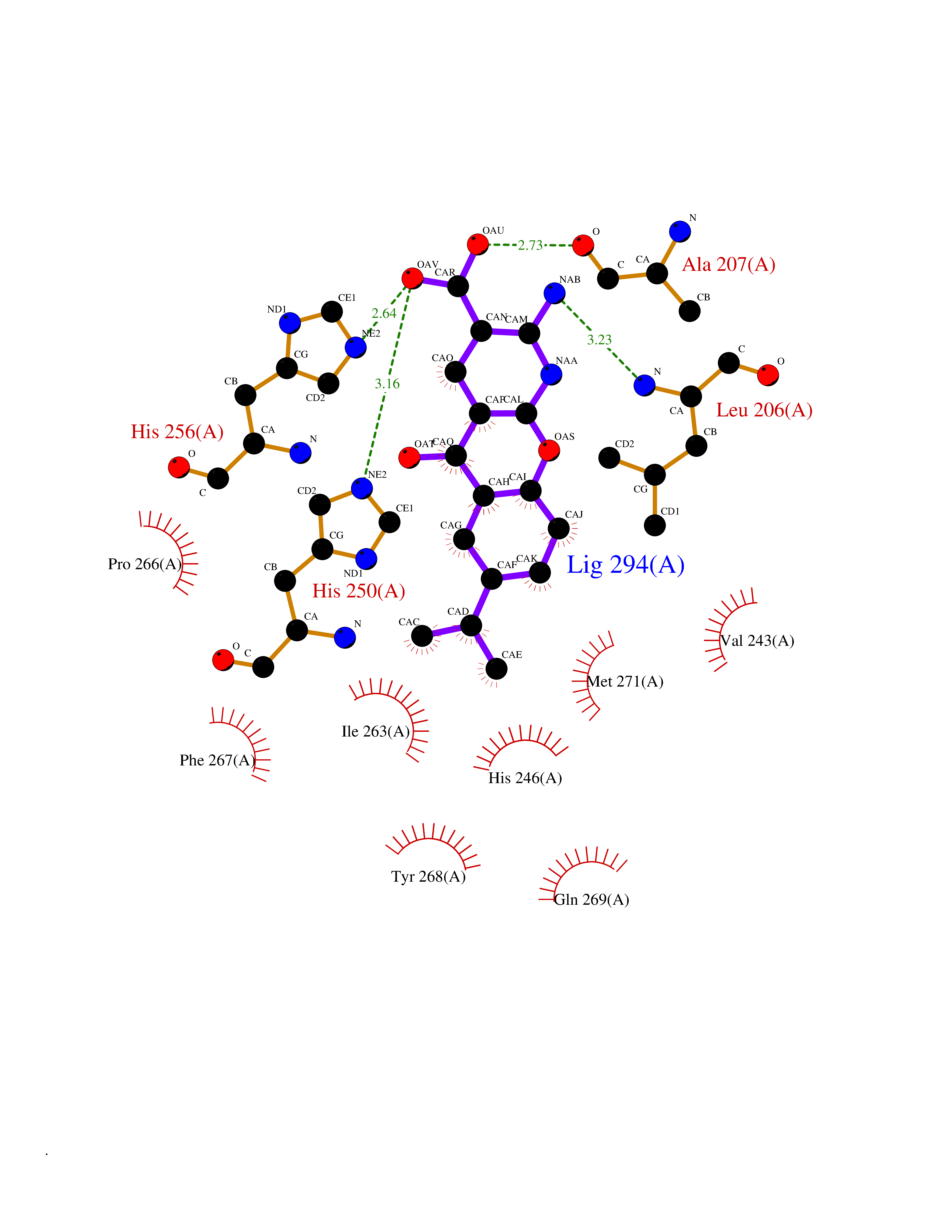 Ligplot Map 3D Binding mode Sequence GQKWQHKHITYSIKNVTPKVGDPETRKAIRRAFDVWQNVTPLTFEEVPYSELENGKRDVDITIIFASGFHGDSSPFDGEGGFLAHAYFPGPGIGGDTHFDSDEPWTLGNPNHDGNDLFLVAVHELGHALGLEHSNDPTAIMAPFYQYMETDNFKLPNDDLQGIQKIYGP Hydrogen bonds contact Hydrophobic contact | ||||
| 18 | Metabotropic glutamate receptor 3 (mGluR3) | 4XAR | 8.17 | |
Target general information Gen name GRM3 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms mGLUR3; Group III metabotropic glutamate receptor; GPRC1C Protein family G-protein coupled receptor 3 family Biochemical class GPCR glutamate Function Ligand binding causes a conformation change that triggers signaling via guanine nucleotide-binding proteins (G proteins) and modulates the activity of down-stream effectors. Signaling inhibits adenylate cyclase activity. G-protein coupled receptor for glutamate. Related diseases Paramyotonia congenita (PMC) [MIM:168300]: An autosomal dominant channelopathy characterized by myotonia, increased by exposure to cold, intermittent flaccid paresis, not necessarily dependent on cold or myotonia, lability of serum potassium, non-progressive nature and lack of atrophy or hypertrophy of muscles. In some patients, myotonia is not increased by cold exposure (paramyotonia without cold paralysis). Patients may have a combination phenotype of PMC and HYPP. {ECO:0000269|PubMed:10369308, ECO:0000269|PubMed:10727489, ECO:0000269|PubMed:1310898, ECO:0000269|PubMed:1316765, ECO:0000269|PubMed:1338909, ECO:0000269|PubMed:15318338, ECO:0000269|PubMed:15790667, ECO:0000269|PubMed:16786525, ECO:0000269|PubMed:18166706, ECO:0000269|PubMed:18690054, ECO:0000269|PubMed:19077043, ECO:0000269|PubMed:20076800, ECO:0000269|PubMed:8242056, ECO:0000269|PubMed:8308722, ECO:0000269|PubMed:8388676, ECO:0000269|PubMed:8580427}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Periodic paralysis hypokalemic 2 (HOKPP2) [MIM:613345]: An autosomal dominant disorder manifested by episodic flaccid generalized muscle weakness associated with falls of serum potassium levels. {ECO:0000269|PubMed:10599760, ECO:0000269|PubMed:10851391, ECO:0000269|PubMed:10944223, ECO:0000269|PubMed:11558801, ECO:0000269|PubMed:11591859, ECO:0000269|PubMed:16890191, ECO:0000269|PubMed:17898326, ECO:0000269|PubMed:18162704, ECO:0000269|PubMed:19118277, ECO:0000269|PubMed:20522878, ECO:0000269|PubMed:21043388, ECO:0000269|PubMed:24549961}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Periodic paralysis hyperkalemic (HYPP) [MIM:170500]: An autosomal dominant channelopathy characterized by episodic flaccid generalized muscle weakness associated with high levels of serum potassium. Concurrence of myotonia is found in HYPP patients. {ECO:0000269|PubMed:1659668, ECO:0000269|PubMed:1659948, ECO:0000269|PubMed:20076800}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Periodic paralysis normokalemic (NKPP) [MIM:170500]: A disorder closely related to hyperkalemic periodic paralysis, but marked by a lack of alterations in potassium levels during attacks of muscle weakness. {ECO:0000269|PubMed:15596759, ECO:0000269|PubMed:18046642, ECO:0000269|PubMed:20522878}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Myotonia SCN4A-related (MYOSCN4A) [MIM:608390]: A phenotypically highly variable myotonia aggravated by potassium loading, and sometimes by cold. Myotonia is characterized by sustained muscle tensing that prevents muscles from relaxing normally. It causes muscle stiffness that can interfere with movement. In some people the stiffness is very mild, while in other cases it may be severe enough to interfere with walking, running, and other activities of daily life. Myotonia SCN4A-related includes myotonia permanens and myotonia fluctuans. In myotonia permanens, the myotonia is generalized and there is a hypertrophy of the muscle, particularly in the neck and the shoulder. Attacks of severe muscle stiffness of the thoracic muscles may be life threatening due to impaired ventilation. In myotonia fluctuans, the muscle stiffness may fluctuate from day to day, provoked by exercise. {ECO:0000269|PubMed:10218481, ECO:0000269|PubMed:16786525, ECO:0000269|PubMed:16832098, ECO:0000269|PubMed:17212350, ECO:0000269|PubMed:17998485, ECO:0000269|PubMed:18203179, ECO:0000269|PubMed:18337100, ECO:0000269|PubMed:19015483, ECO:0000269|PubMed:19347921, ECO:0000269|PubMed:20076800, ECO:0000269|PubMed:27653901, ECO:0000269|PubMed:8058156, ECO:0000269|PubMed:9392583}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Myasthenic syndrome, congenital, 16 (CMS16) [MIM:614198]: A form of congenital myasthenic syndrome, a group of disorders characterized by failure of neuromuscular transmission, including pre-synaptic, synaptic, and post-synaptic disorders that are not of autoimmune origin. Clinical features are easy fatigability and muscle weakness. CMS16 is characterized by fatigable generalized weakness and recurrent attacks of respiratory and bulbar paralysis since birth. The fatigable weakness involves lid-elevator, external ocular, facial, limb and truncal muscles and an decremental response of the compound muscle action potential on repetitive stimulation. {ECO:0000269|PubMed:12766226, ECO:0000269|PubMed:25707578, ECO:0000269|PubMed:26659129}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Congenital myopathy 22A, classic (CMYO22A) [MIM:620351]: A form of congenital myopathy, a clinically and genetically heterogeneous group of muscle disorders characterized by hypotonia and muscle weakness apparent at birth, and specific pathological features on muscle biopsy. CMYO22A is an autosomal recessive form characterized by fetal hypokinesia, polyhydramnios, and severe neonatal hypotonia associated with respiratory insufficiency. Affected individuals who survive the neonatal period have delayed motor development, difficulty walking, proximal muscle weakness of the upper and lower limbs, facial and neck muscle weakness, easy fatigability, and mild limb contractures or foot deformities. {ECO:0000269|PubMed:26700687, ECO:0000269|PubMed:28262468, ECO:0000269|PubMed:36090556}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Congenital myopathy 22B, severe fetal (CMYO22B) [MIM:620369]: A severe congenital myopathy, a clinically and genetically heterogeneous group of muscle disorders characterized by hypotonia and muscle weakness apparent at birth, and specific pathological features on muscle biopsy. CMYO22B is an autosomal recessive form characterized by onset in utero. Affected individuals show fetal akinesia, and develop fetal hydrops with pulmonary hypoplasia, severe joint contractures, and generalized muscle hypoplasia. Death occurs in utero or soon after birth. {ECO:0000269|PubMed:26700687}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB05096 Interacts with NA EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disulfide bond; G-protein coupled receptor; Glycoprotein; Membrane; Proteomics identification; Receptor; Reference proteome; Signal; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 50355.5 Length 445 Aromaticity 0.11 Instability index 38.26 Isoelectric point 6.52 Charge (pH=7) -1.53 2D Binding mode Binding energy (Kcal/mol) -8.73 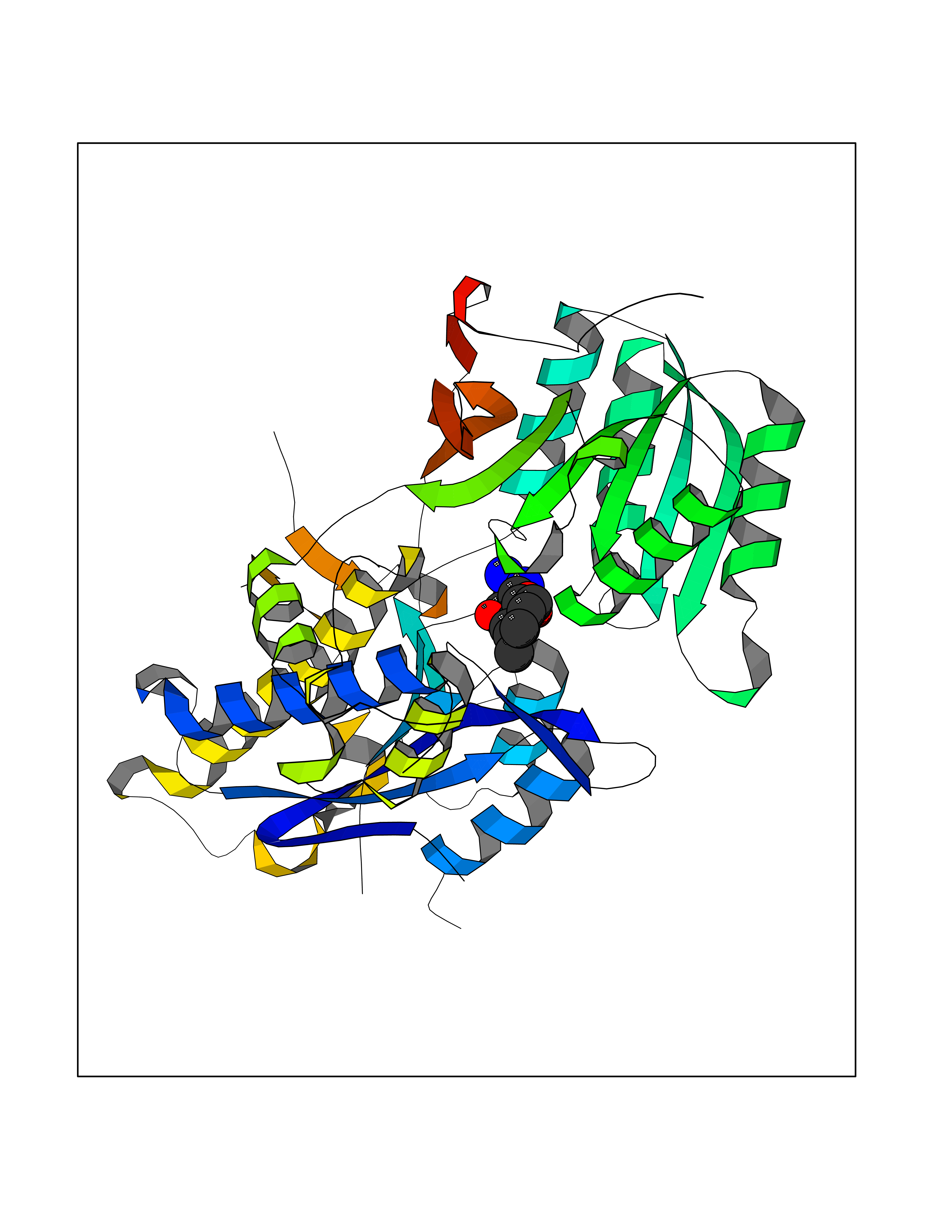 Molscript Map 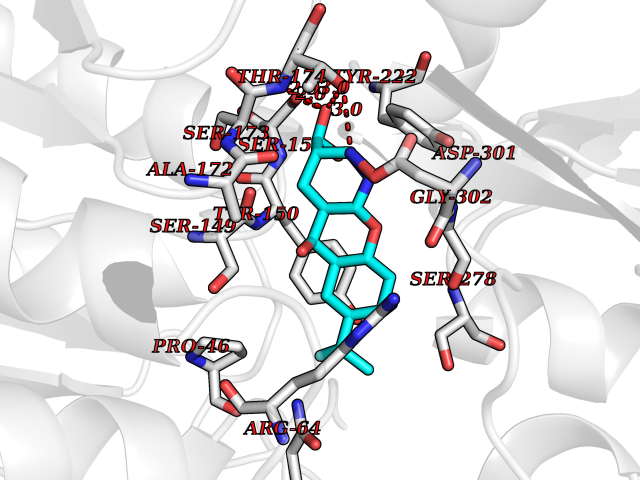 Pymol Map 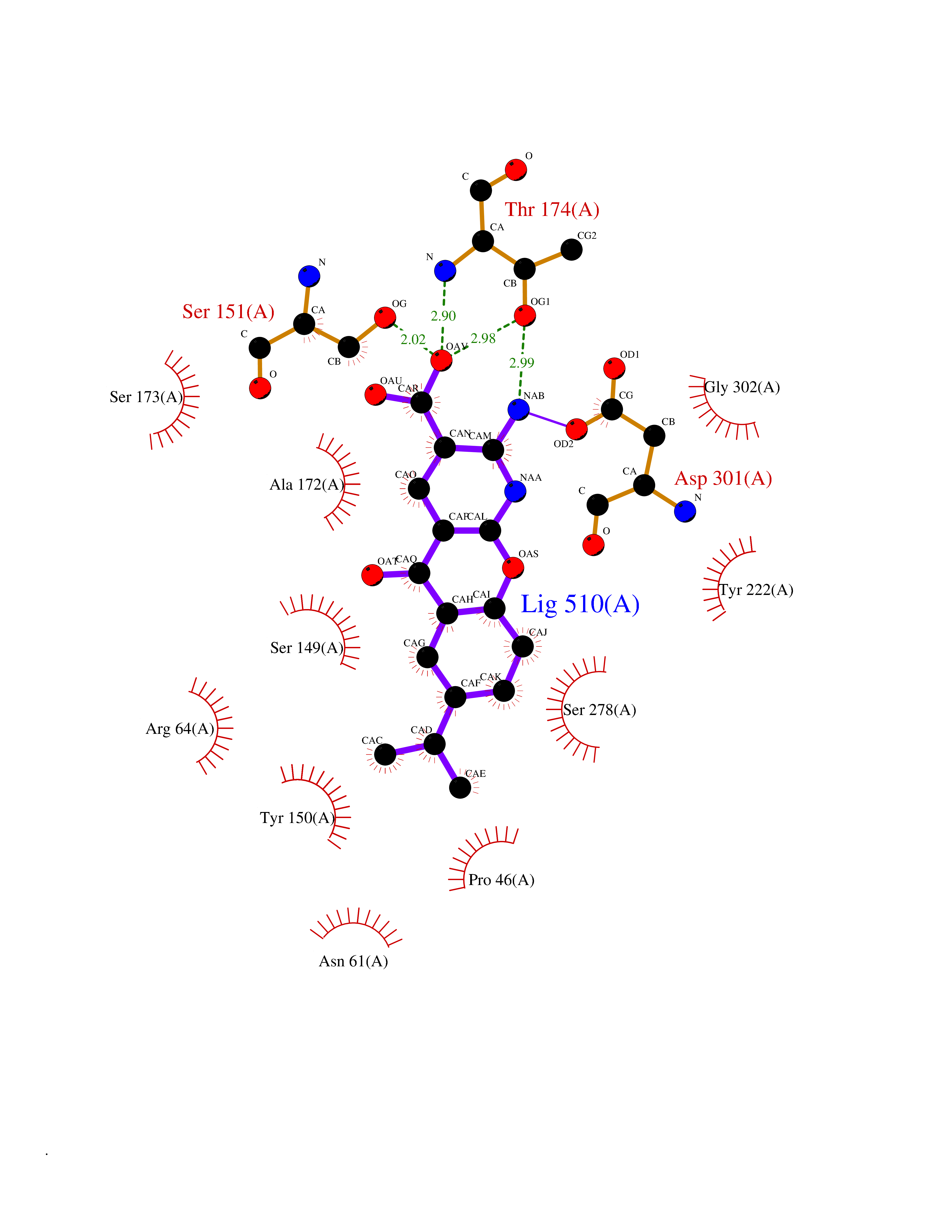 Ligplot Map 3D Binding mode Sequence RREIKIEGDLVLGGLFPINEKGTGTEECGRINEDRGIQRLEAMLFAIDEINKDDYLLPGVKLGVHILDTCSRDTYALEQSLEFVRASLLLIAGVIGGSYSSVSIQVANLLRLFQIPQISYASTSAKLSDKSRYDYFARTVPPDFYQAKAMAEILRFFNWTYVSTVASEGDYGETGIEAFEQEARLRNISIATAEKVGRSNIRKSYDSVIRELLQKPNARVVVLFMRSDDSRELIAAASRANASFTWVASDGWGAQESIIKGSEHVAYGAITLELASQPVRQFDRYFQSLNPYNNHRNPWFRDFWEQKFQCSLRVCDKHLAIDSSNYEQESKIMFVVNAVYAMAHALHKMQRTLCPNTTKLCDAMKILDGKKLYKDYLLKINFTAPDADSIVKFDTFGDGMGRYNVFNFQNVGGKYSYLKVGHWAETLSLDVNSIHWSRNSVPTSE Hydrogen bonds contact Hydrophobic contact | ||||
| 19 | SET domain containing 8 (KMT5A) | 5TEG | 8.17 | |
Target general information Gen name KMT5A Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms SETD8; SET8; SET07; SET domain-containing protein 8; PRSET7; PR/SET07; PR/SET domain-containing protein 07; PR-Set7; N-lysine methyltransferase KMT5A; Lysine-specific methylase 5A; Lysine N-methyltran Protein family Class V-like SAM-binding methyltransferase superfamily, Histone-lysine methyltransferase family, PR/SET subfamily Biochemical class Methyltransferase Function Specifically monomethylates 'Lys-20' of histone H4 (H4K20me1). H4K20me1 is enriched during mitosis and represents a specific tag for epigenetic transcriptional repression. Mainly functions in euchromatin regions, thereby playing a central role in the silencing of euchromatic genes. Required for cell proliferation, probably by contributing to the maintenance of proper higher-order structure of DNA during mitosis. Involved in chromosome condensation and proper cytokinesis. Nucleosomes are preferred as substrate compared to free histones. Mediates monomethylation of p53/TP53 at 'Lys-382', leading to repress p53/TP53-target genes. Plays a negative role in TGF-beta response regulation and a positive role in cell migration. Protein-lysine N-methyltransferase that monomethylates both histones and non-histone proteins. Related diseases Sick sinus syndrome 2 (SSS2) [MIM:163800]: The term 'sick sinus syndrome' encompasses a variety of conditions caused by sinus node dysfunction. The most common clinical manifestations are syncope, presyncope, dizziness, and fatigue. Electrocardiogram typically shows sinus bradycardia, sinus arrest, and/or sinoatrial block. Episodes of atrial tachycardias coexisting with sinus bradycardia ('tachycardia-bradycardia syndrome') are also common in this disorder. SSS occurs most often in the elderly associated with underlying heart disease or previous cardiac surgery, but can also occur in the fetus, infant, or child without heart disease or other contributing factors. SSS2 onset is in utero or at birth. {ECO:0000269|PubMed:15123648, ECO:0000269|PubMed:16407510, ECO:0000269|PubMed:20662977, ECO:0000269|PubMed:23103389}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Brugada syndrome 8 (BRGDA8) [MIM:613123]: A tachyarrhythmia characterized by right bundle branch block and ST segment elevation on an electrocardiogram (ECG). It can cause the ventricles to beat so fast that the blood is prevented from circulating efficiently in the body. When this situation occurs, the individual will faint and may die in a few minutes if the heart is not reset. {ECO:0000269|PubMed:19165230}. The gene represented in this entry may be involved in disease pathogenesis.; DISEASE: Epilepsy, idiopathic generalized 18 (EIG18) [MIM:619521]: An autosomal dominant form of idiopathic generalized epilepsy, a disorder characterized by recurring generalized seizures in the absence of detectable brain lesions and/or metabolic abnormalities. Generalized seizures arise diffusely and simultaneously from both hemispheres of the brain. Seizure types include juvenile myoclonic seizures, absence seizures, and generalized tonic-clonic seizures. EIG18 is characterized by onset of myoclonic seizures in infancy. Although the seizures remit, some patients may have later speech or cognitive impairment. {ECO:0000269|PubMed:30127718}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P62805; P07910; Q15672 EC number EC 2.1.1.- Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Cell cycle; Cell division; Chromatin regulator; Chromosome; Coiled coil; Direct protein sequencing; Methyltransferase; Mitosis; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Repressor; S-adenosyl-L-methionine; Transcription; Transcription regulation; Transferase; Ubl conjugation Protein physicochemical properties Chain ID A,D Molecular weight (Da) 19129.4 Length 167 Aromaticity 0.08 Instability index 49.18 Isoelectric point 7.88 Charge (pH=7) 1.37 2D Binding mode Binding energy (Kcal/mol) -9.66  Molscript Map 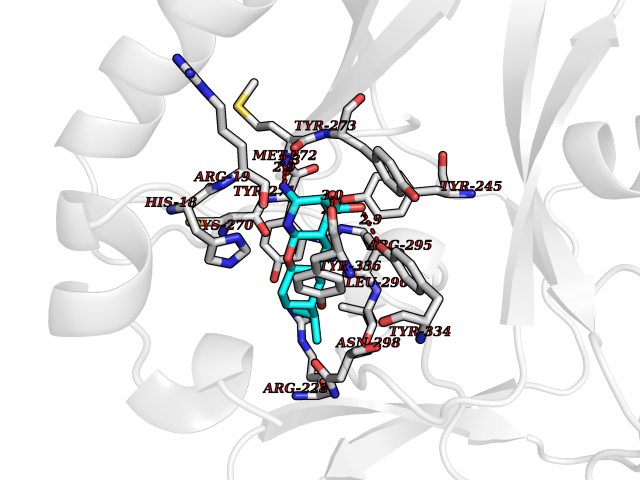 Pymol Map 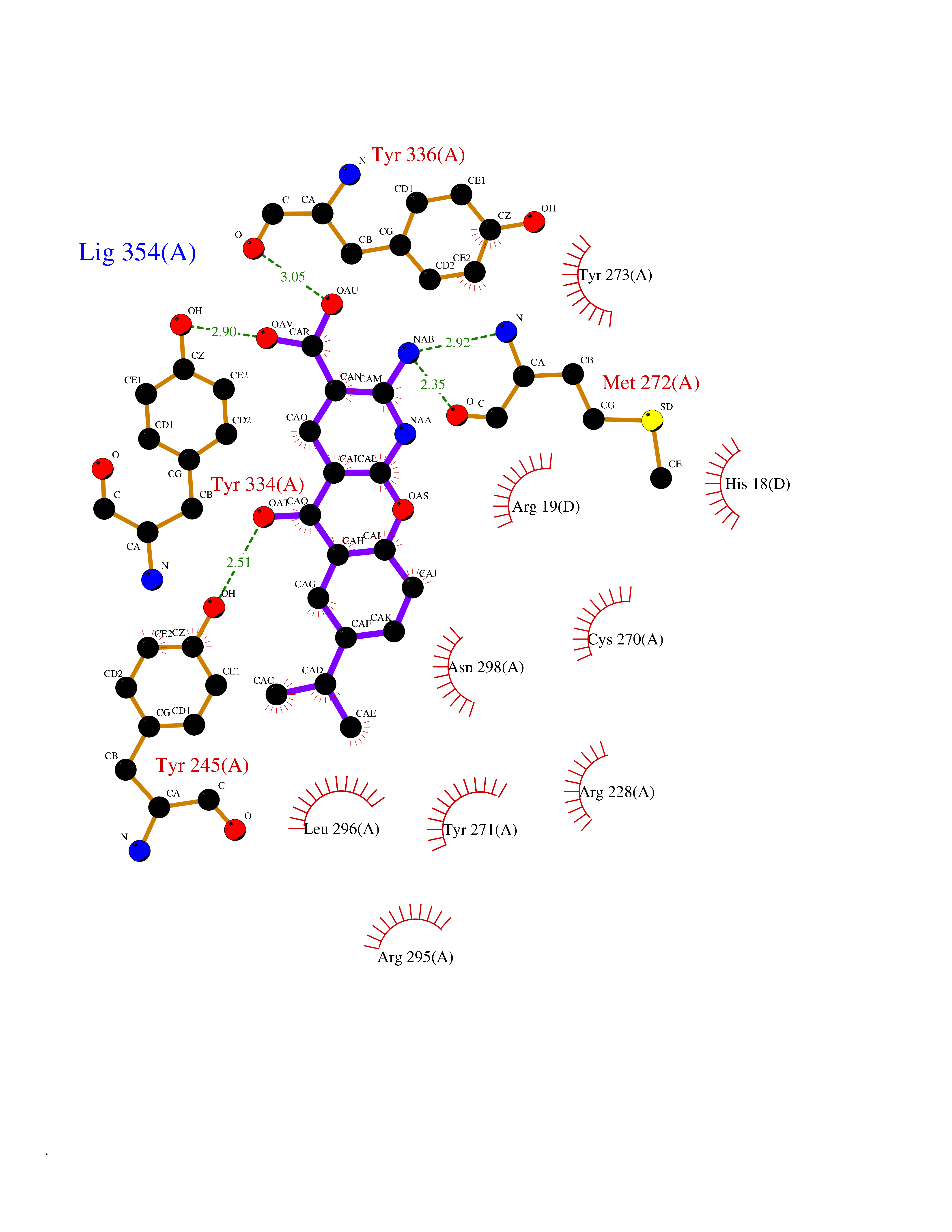 Ligplot Map 3D Binding mode Sequence KSKAELQSEERKRIDELIESGKEEGMKIDLIDGKGRGVIATKQFSRGDFVVEYHGDLIEITDAKKREALYAQDPSTGCYMYYFQYLSKTYCVDATRETNRLGRLINHSKSGNCQTKLHDIDGVPHLILIASRDIAAGEELLYDYGDRSKASIEAHPWLKHKRHRVLR Hydrogen bonds contact Hydrophobic contact | ||||
| 20 | Dopamine beta-hydroxylase | 4ZEL | 8.17 | |
Target general information Gen name DBH Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Copper type II ascorbate-dependent monooxygenase family Biochemical class Oxidoreductase Function Catalytic activity.Copper ion binding.Dopamine beta-monooxygenase activity.L-ascorbic acid binding. Related diseases Orthostatic hypotension 1 (ORTHYP1) [MIM:223360]: A form of orthostatic hypotension due to congenital dopamine beta-hydroxylase deficiency. Orthostatic hypotension, also known as postural hypotension, is a finding defined as a 20-mm Hg decrease in systolic pressure or a 10-mm Hg decrease in diastolic pressure occurring 3 minutes after a person has risen from supine to standing. Symptoms include dizziness, blurred vision, and sometimes syncope. ORTHYP1 is an autosomal recessive condition apparent from infancy or early childhood and characterized by low plasma and urinary levels of norepinephrine and epinephrine, and episodic hypoglycemia. {ECO:0000269|PubMed:11857564}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00126; DB06774; DB09130; DB05394; DB00822; DB00988; DB00968; DB00550 Interacts with P00352; P63010-2; Q04656; Q8WUW1; Q9UNS2; Q71DI3; P61978; Q9Y2M5; Q92876; P08727; Q14693; P0DPK4; Q6GQQ9-2; P27986-2; Q9ULX5; Q96D59; Q8N6K7-2; Q9GZS3; Q8IUW3; Q86WT6-2 EC number 1.14.17.1 Uniprot keywords 3D-structure; Catecholamine biosynthesis; Copper; Cytoplasmic vesicle; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; Membrane; Metal-binding; Monooxygenase; Oxidoreductase; Proteomics identification; Reference proteome; Secreted; Signal-anchor; Transmembrane; Transmembrane helix; Vitamin C Protein physicochemical properties Chain ID A,B Molecular weight (Da) 123694 Length 1094 Aromaticity 0.1 Instability index 51.85 Isoelectric point 5.84 Charge (pH=7) -24.5 2D Binding mode Binding energy (Kcal/mol) -8.83  Molscript Map 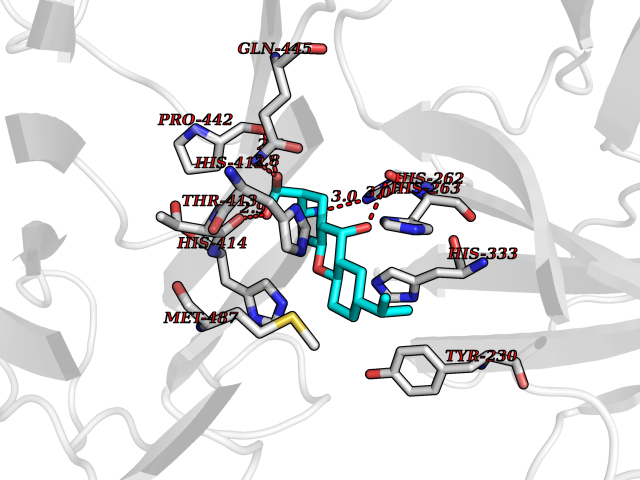 Pymol Map 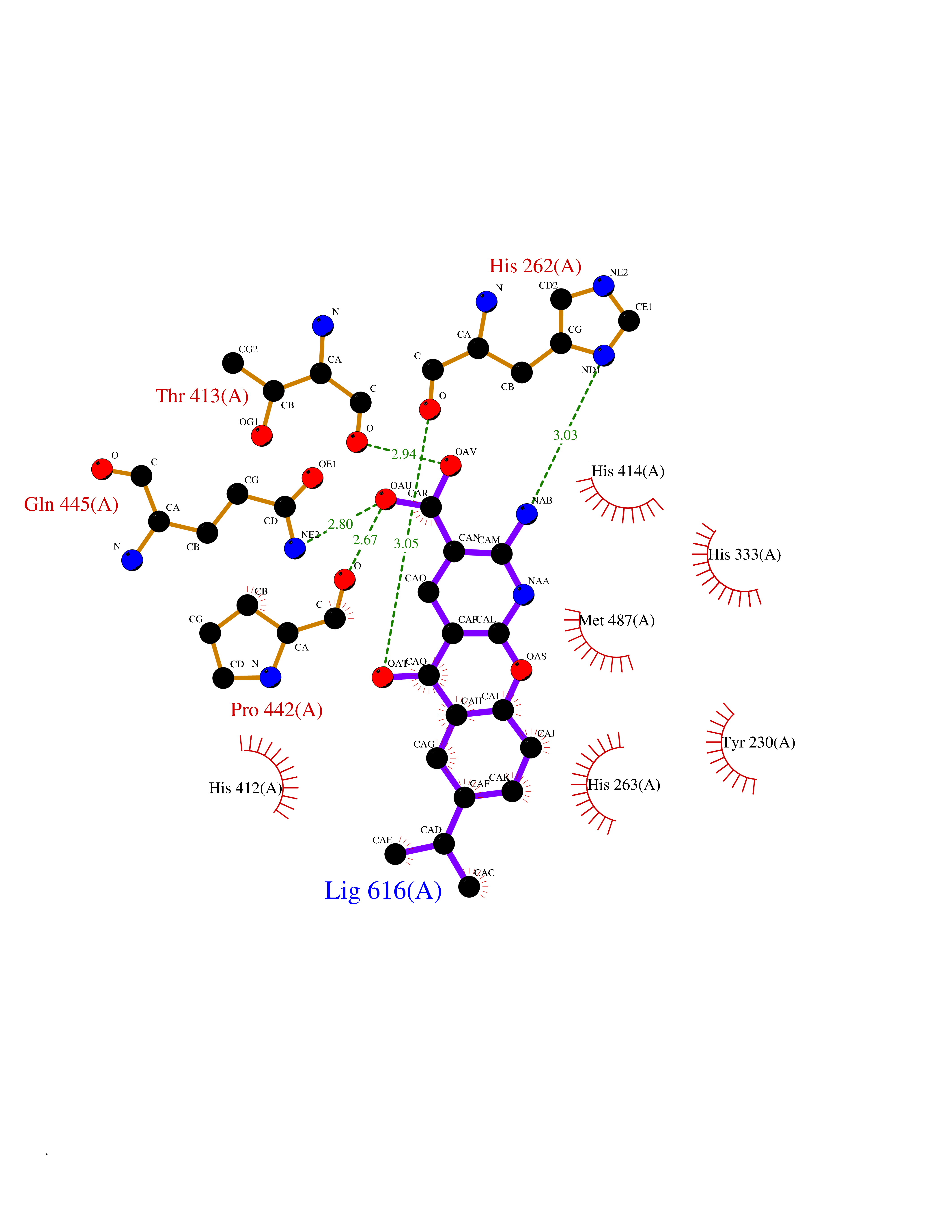 Ligplot Map 3D Binding mode Sequence PLPYHIPLDPEGSLELSWNVSYTQEAIHFQLLVRRLKAGVLFGMSDRGELENADLVVLWTDGDAYFADAWSDQKGQIHLDPQQDYQLLQVQRTPEGLTLLFKRPFGTCDPKDYLIEDGTVHLVYGILEEPFRSLEAINGSGLQMGLQRVQLLKPNIPEPELPSDACTMEVQAPNIQIPSQETTYWCYIKELPKGFSRHHIIKYEPIVTKGNEALVHHMEVFQCAPEMDSVPHFSGPCDSKMKPDRLNYCRHVLAAWALGAKAFYYPEEAGLAFGGPGSSRYLRLEVHYHNPLVIEGRNDSSGIRLYYTAKLRRFNAGIMELGLVYTPVMAIPPRETAFILTGYCTDKCTQLALPPSGIHIFASQLHTHLTGRKVVTVLVRDGREWEIVNQDNHYSPHFQEIRMLKKVVSVHPGDVLITSCTYNTEDRELATVGGFGILEEMCVNYVHYYPQTQLELCKSAVDAGFLQKYFHLINRFNNEDVCTCPQASVSQQFTSVPWNSFNRDVLKALYSFAPISMHCNKSSAVRFQGEWNLQPLPKVISTLEEPTVVSPLPYHIPLDPEGSLELSWNVSYTQEAIHFQLLVRRLKAGVLFGMSDRGELENADLVVLAYFADAWSDQKGQIHLDPQQDYQLLQVQRTPEGLTLLFKRPFGTCDPKDYLIEDGTVHLVYGILEEPFRSLEAINGSGLQMGLQRVQLLKPNIPEPELPSDACTMEVQAPNIQIPSQETTYWCYIKELPKGFSRHHIIKYEPIVTKGNEALVHHMEVFQCAPEVPHFSGPCDSKMLNYCRHVLAAWALGAKAFYYPEEAGLAFGGPGSSRYLRLEVHYHNPLVIEGRNDSSGIRLYYTAKLRRFNAGIMELGLVYTPVMAIPPRETAFILTGYCTDKCTQLALPPSGIHIFASQLHTHLTGRKVVTVLVRDGREWEIVNQDNHYSPHFQEIRMLKKVVSVHPGDVLITSCTYNTEDRELATVGGFGILEEMCVNYVHYYPQTQLELCKSAVDAGFLQKYFHLINRFNNEDVCTCPQASVSQQFTSVPWNSFNRDVLKALYSFAPISMHCNKSSAVRFQGEWNLQPLPKVISTLEEPTPQCVVSIGG Hydrogen bonds contact Hydrophobic contact | ||||