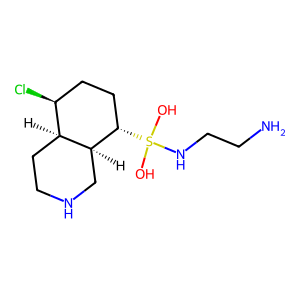
Ligand
Structure
Job ID
2a1862048f8f7aa127a9ace2cac14948
Job name
Martin_task1
Time
2024-08-25 23:04:05
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 1 | Endothelin-converting enzyme 1 (ECE1) | 3DWB | 8.52 | |
Target general information Gen name ECE1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms ECE-1 Protein family Peptidase M13 family Biochemical class Peptidase Function Converts big endothelin-1 to endothelin-1. Related diseases Hirschsprung disease, cardiac defects, and autonomic dysfunction (HCAD) [MIM:613870]: A disorder characterized by skip-lesions Hirschsprung disease, craniofacial abnormalities and other dysmorphic features, cardiac defects including ductus arteriosus, small subaortic ventricular septal defect, small atrial septal defect, and autonomic dysfunction. {ECO:0000269|PubMed:9915973}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07171 Interacts with P49760; A8MQ03; Q8IUG1; P60370; P60410 EC number EC 3.4.24.71 Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disease variant; Disulfide bond; Glycoprotein; Hirschsprung disease; Hydrolase; Membrane; Metal-binding; Metalloprotease; Phosphoprotein; Protease; Proteomics identification; Reference proteome; Signal-anchor; Transmembrane; Transmembrane helix; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 75247.9 Length 660 Aromaticity 0.12 Instability index 46.29 Isoelectric point 5.33 Charge (pH=7) -18.3 2D Binding mode Binding energy (Kcal/mol) -7.43  Molscript Map 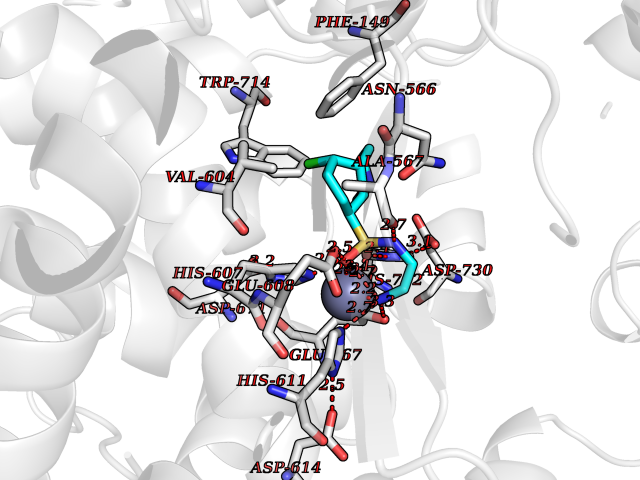 Pymol Map 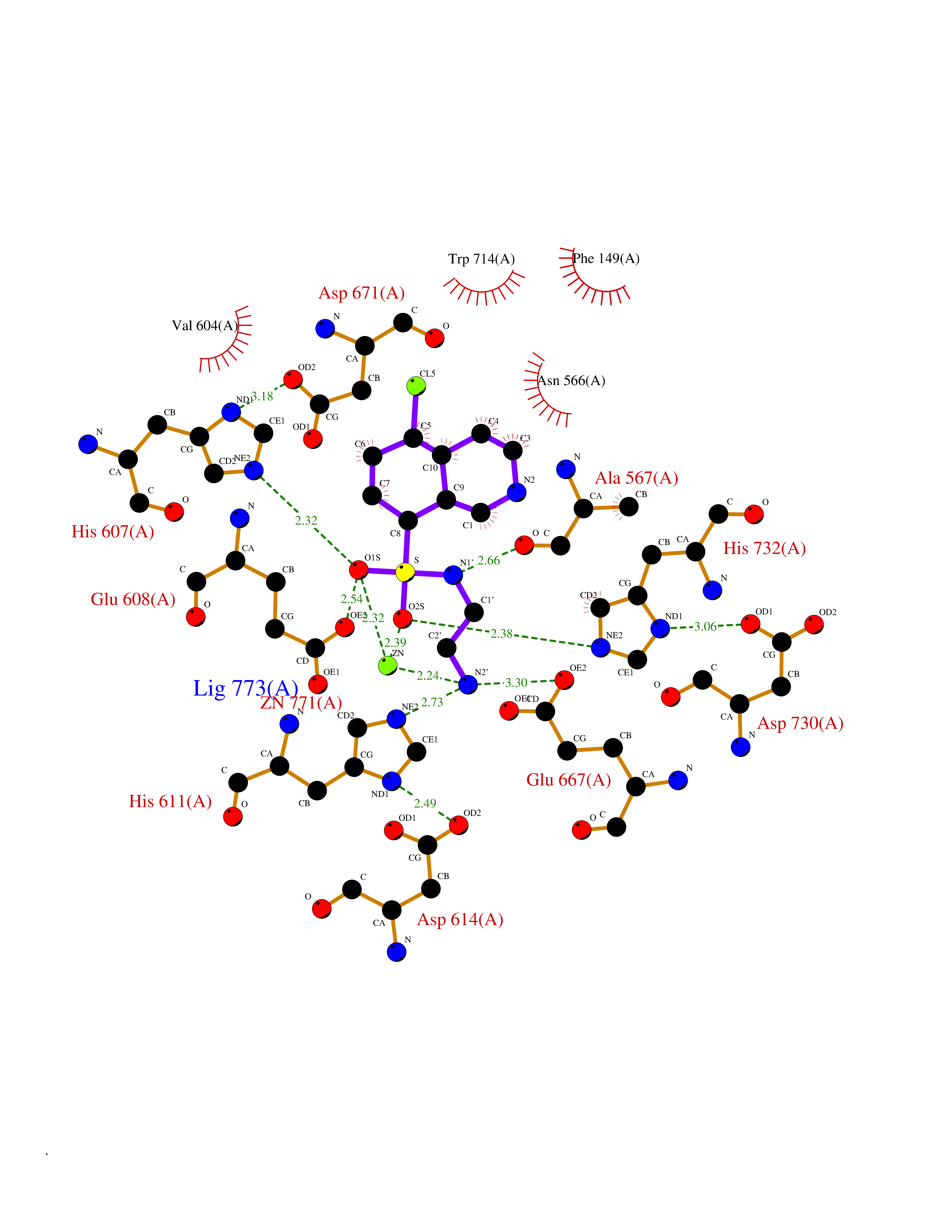 Ligplot Map 3D Binding mode Sequence SEACVSVTSSILSSMDPTVDPCHDFFSYACGGWIKANPVPDGHSRWGTFSNLWEHNQAIIKHLLENSTASVSEAERKAQVYYRACMNETRIEELRAKPLMELIERLGGWNITGPWAKDNFQDTLQVVTAHYRTSPFFSVYVSADSKNSNSNVIQVDQSGLGLPSRDYYLNKTENEKVLTGYLNYMVQLGKLLGGGDEEAIRPQMQQILDFETALANITIPQEKRRDEELIYHKVTAAELQTLAPAINWLPFLNTIFYPVEINESEPIVVYDKEYLEQISTLINTTDRCLLNNYMIWNLVRKTSSFLDQRFQDADEKFMEVMWKFCVSDTENNLGFALGPMFVKATFAEDSKSIATEIILEIKKAFEESLSTLKWMDEETRKSAKEKADAIYNMIGYPNFIMDPKELDKVFNDYTAVPDLYFENAMRFFNFSWRVTADQLRKAPNRDQWSMTPPMVNAYYSPTKNEIVFPAGILQAPFYTRSSPKALNFGGIGVVVGHELTHAFDDQGREYDKDGNLRPWWKNSSVEAFKRQTECMVEQYSNYSVNGEPVNGRHTLGENIADNGGLKAAYRAYQNWVKKNGAEHSLPTLGLTNNQLFFLGFAQVWCSVRTPESSHEGLITDPHSPSRFRVIGSLSNSKEFSEHFRCPPGSPMNPPHKCEVW Hydrogen bonds contact Hydrophobic contact | ||||
| 2 | Clostridium histolyticum Collagenase (CH colG) | 7Z5U | 8.43 | |
Target general information Gen name CH colG Organism Hathewaya histolytica (Clostridium histolyticum) Uniprot ID TTD ID Synonyms Microbial collagenase; Gelatinase ColG; Collagenase ColG; Class I collagenase Protein family Peptidase M9B family, Collagenase subfamily Biochemical class NA Function Clostridial collagenases are among the most efficient degraders of eukaryotic collagen known; saprophytes use collagen as a carbon source while pathogens additionally digest collagen to aid in host colonization. Has both tripeptidylcarboxypeptidase on Gly-X-Y and endopeptidase activities; the endopeptidase cuts within the triple helix region of collagen while tripeptidylcarboxypeptidase successively digests the exposed ends, thus clostridial collagenases can digest large sections of collagen. Active on soluble type I collagen, insoluble collagen, azocoll, soluble PZ-peptide (all collagenase substrates) and gelatin. The full-length protein has collagenase activity, while the in vivo derived C-terminally truncated shorter versions only act on gelatin. In vitro digestion of soluble calf skin collagen fibrils requires both ColG and ColH; ColG forms missing the second collagen-binding domain are also synergistic with ColH, although their overall efficiency is decreased. The activator domain (residues 119-388) and catalytic subdomain (389-670) open and close around substrate using a Gly-rich hinge (387-397), allowing digestion when the protein is closed. Binding of collagen requires Ca(2+) and is inhibited by EGTA; the collagen-binding domain (CBD, S3a plus S3b) specifically recognizes the triple-helical conformation made by 3 collagen protein chains in the triple-helical region. Isolated CBD (S3a plus S3b) binds collagen fibrils and sheets of many tissues. Related diseases Intellectual developmental disorder, X-linked, syndromic, Claes-Jensen type (MRXSCJ) [MIM:300534]: A disorder characterized by significantly below average general intellectual functioning associated with impairments in adaptive behavior and manifested during the developmental period. MRXSCJ patients manifest intellectual disability associated with variable features such as slowly progressive spastic paraplegia, seizures, facial dysmorphism. {ECO:0000269|PubMed:15586325, ECO:0000269|PubMed:16538222, ECO:0000269|PubMed:16541399, ECO:0000269|PubMed:17320160, ECO:0000269|PubMed:17468742, ECO:0000269|PubMed:23356856, ECO:0000269|PubMed:25666439}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number EC 3.4.24.3 Uniprot keywords 3D-structure; Calcium; Direct protein sequencing; Hydrolase; Metal-binding; Metalloprotease; Pharmaceutical; Protease; Repeat; Secreted; Signal; Virulence; Zinc; Zymogen Protein physicochemical properties Chain ID A Molecular weight (Da) 44751.2 Length 386 Aromaticity 0.15 Instability index 27.33 Isoelectric point 5.77 Charge (pH=7) -7.33 2D Binding mode Binding energy (Kcal/mol) -7.49 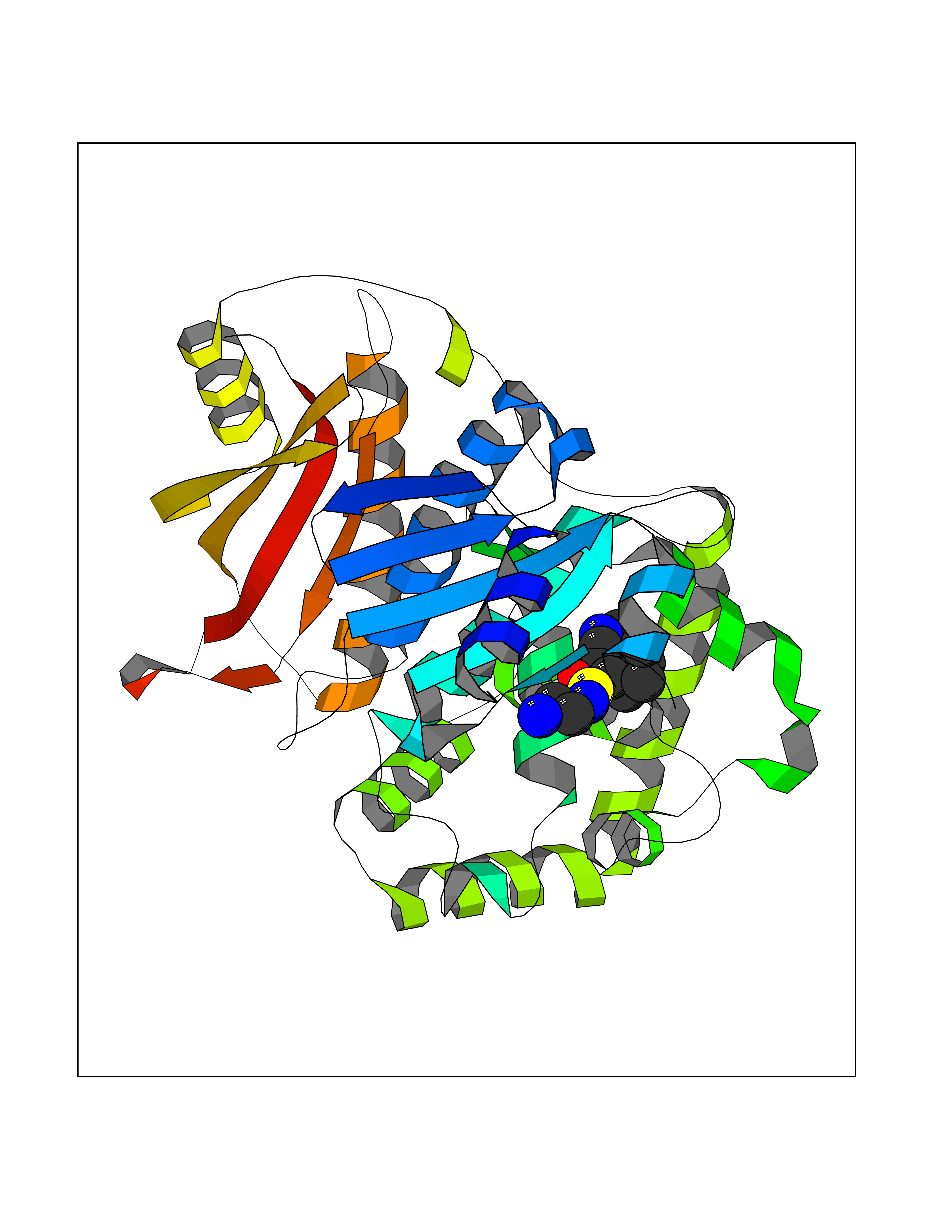 Molscript Map 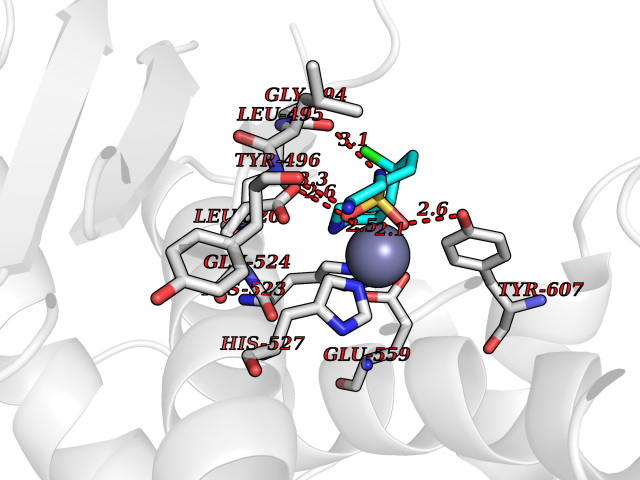 Pymol Map 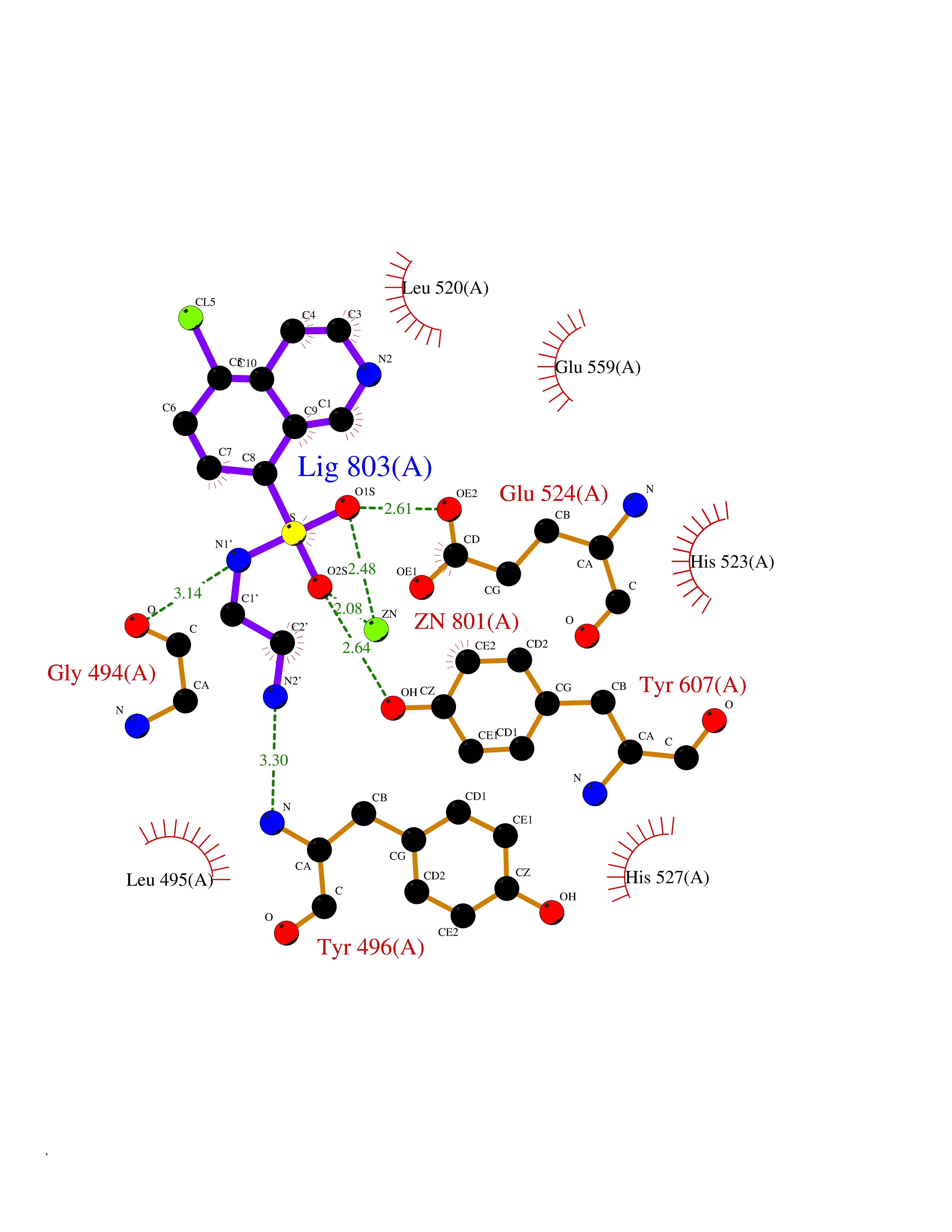 Ligplot Map 3D Binding mode Sequence DHDKFLDDAEKHYLPKTYTFDNGTFIIRAGDKVSEEKIKRLYWASREVKSQFHRVVGNDKALEVGNADDVLTMKIFNSPEEYKFNTTDNGGLYIEPRGTFYTYERTPQQSIFSLEELFRHEYTHYLQARYLVDGLWGQGPFYEKNRLTWFDEGTAEFFAGSTRTSGVLPRKLILGYLAKDKVDHRYSLKKTLNSGYDDSDWMFYNYGFAVAHYLYEKDMPTFIKMNKAILNTDVKSYDEIIKKLSDDANKNTEYQNHIQELVDKYQGAGIPLVSDDYLKDHGYKKASEVYSEISKAASLTNTSVTAEKSQYFNTFTLRGTYTGETSKGEFKDWDEMSKKLDGTLESLAKNSWSGYKTLTAYFTNYRVTSDNKVQYDVVFHGVLTDN Hydrogen bonds contact Hydrophobic contact | ||||
| 3 | Corynebacterium Pup-protein ligase (Cory pafA) | 4BJR | 8.40 | |
Target general information Gen name Cory pafA Organism Corynebacterium glutamicum (strain ATCC 13032 / DSM 20300 / JCM 1318 / BCRC 11384 / CCUG 27702 / LMG 3730 / NBRC 12168 / NCIMB 10025 / NRRL B-2784 / 534) Uniprot ID TTD ID Synonyms Pup-conjugating enzyme; Pup--protein ligase; Proteasome accessory factor A Protein family Pup ligase/Pup deamidase family, Pup-conjugating enzyme subfamily Biochemical class NA Function Catalyzes the covalent attachment of the prokaryotic ubiquitin-like protein modifier Pup to the proteasomal substrate proteins, thereby targeting them for proteasomal degradation. This tagging system is termed pupylation. The ligation reaction involves the side-chain carboxylate of the C-terminal glutamate of Pup and the side-chain amino group of a substrate lysine. Related diseases Anemia, non-spherocytic hemolytic, due to G6PD deficiency (NSHA) [MIM:300908]: A disease characterized by G6PD deficiency, acute hemolytic anemia, fatigue, back pain, and jaundice. In most patients, the disease is triggered by an exogenous agent, such as some drugs, food, or infection. Increased unconjugated bilirubin, lactate dehydrogenase, and reticulocytosis are markers of the disorder. Although G6PD deficiency can be life-threatening, most patients are asymptomatic throughout their life. {ECO:0000269|PubMed:12524354, ECO:0000269|PubMed:1303180, ECO:0000269|PubMed:1303182, ECO:0000269|PubMed:1536798, ECO:0000269|PubMed:1611091, ECO:0000269|PubMed:1889820, ECO:0000269|PubMed:1945893, ECO:0000269|PubMed:20007901, ECO:0000269|PubMed:26479991, ECO:0000269|PubMed:2836867, ECO:0000269|PubMed:2912069, ECO:0000269|PubMed:30988594, ECO:0000269|PubMed:38066190, ECO:0000269|PubMed:7858267, ECO:0000269|PubMed:7959695, ECO:0000269|PubMed:8193373, ECO:0000269|PubMed:8490627, ECO:0000269|PubMed:8533762, ECO:0000269|PubMed:8733135, ECO:0000269|PubMed:9452072}. The disease is caused by variants affecting the gene represented in this entry. Deficiency of G6PD is associated with hemolytic anemia in two different situations. First, in areas in which malaria has been endemic, G6PD-deficiency alleles have reached high frequencies (1% to 50%) and deficient individuals, though essentially asymptomatic in the steady state, have a high risk of acute hemolytic attacks. Secondly, sporadic cases of G6PD deficiency occur at a very low frequencies, and they usually present a more severe phenotype. Several types of NSHA are recognized. Class-I variants are associated with severe NSHA; class-II have an activity <10% of normal; class-III have an activity of 10% to 60% of normal; class-IV have near normal activity. Drugs (DrugBank ID) NA Interacts with NA EC number NA Uniprot keywords 3D-structure; ATP-binding; Ligase; Magnesium; Metal-binding; Nucleotide-binding; Reference proteome; Ubl conjugation pathway Protein physicochemical properties Chain ID A,B Molecular weight (Da) 110572 Length 994 Aromaticity 0.07 Instability index 42.33 Isoelectric point 5.26 Charge (pH=7) -34.19 2D Binding mode Binding energy (Kcal/mol) -7.83 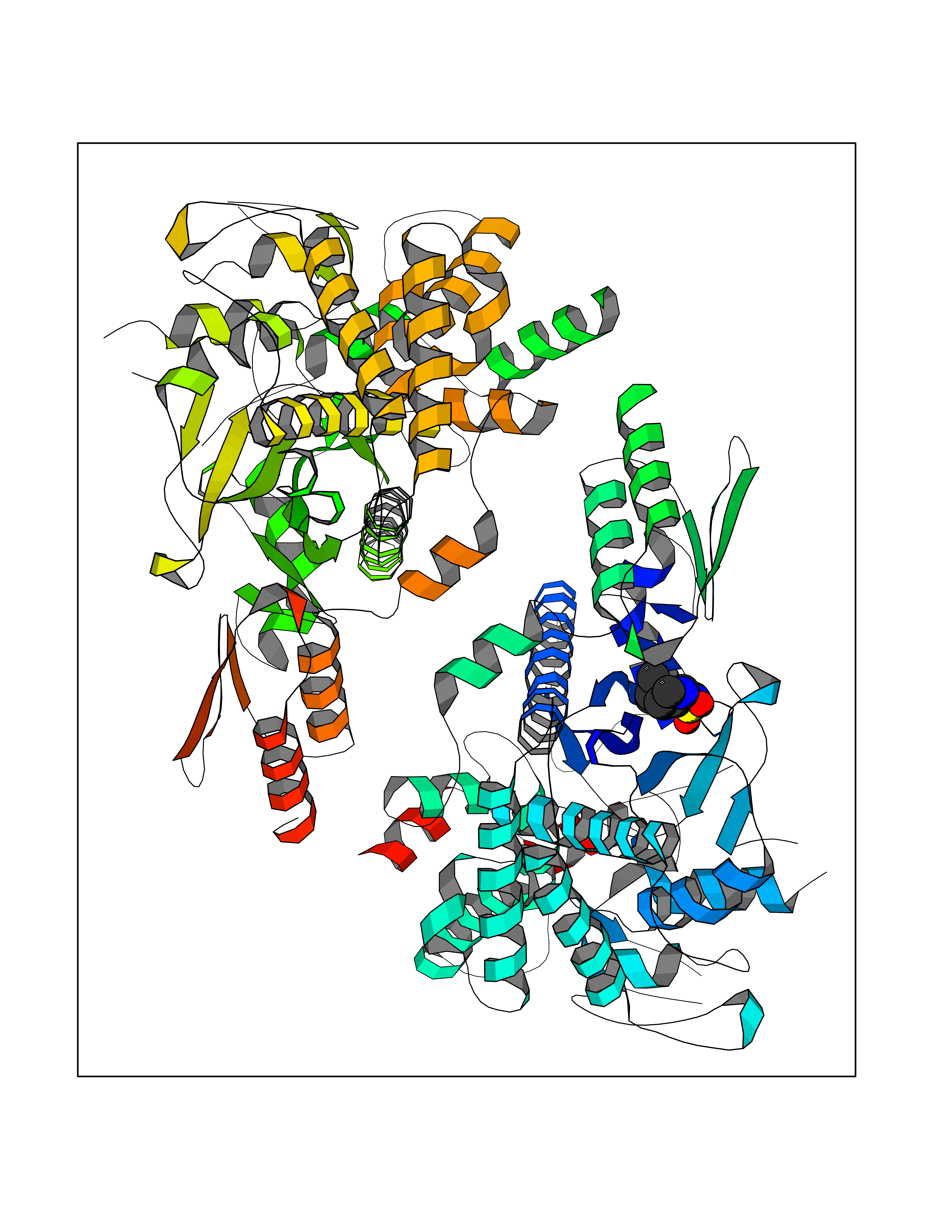 Molscript Map 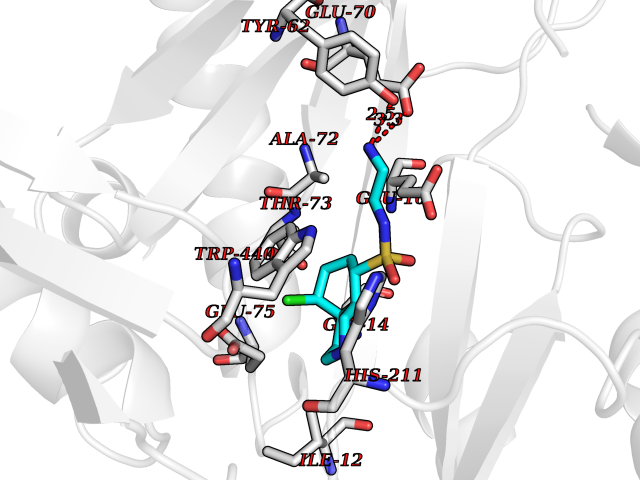 Pymol Map 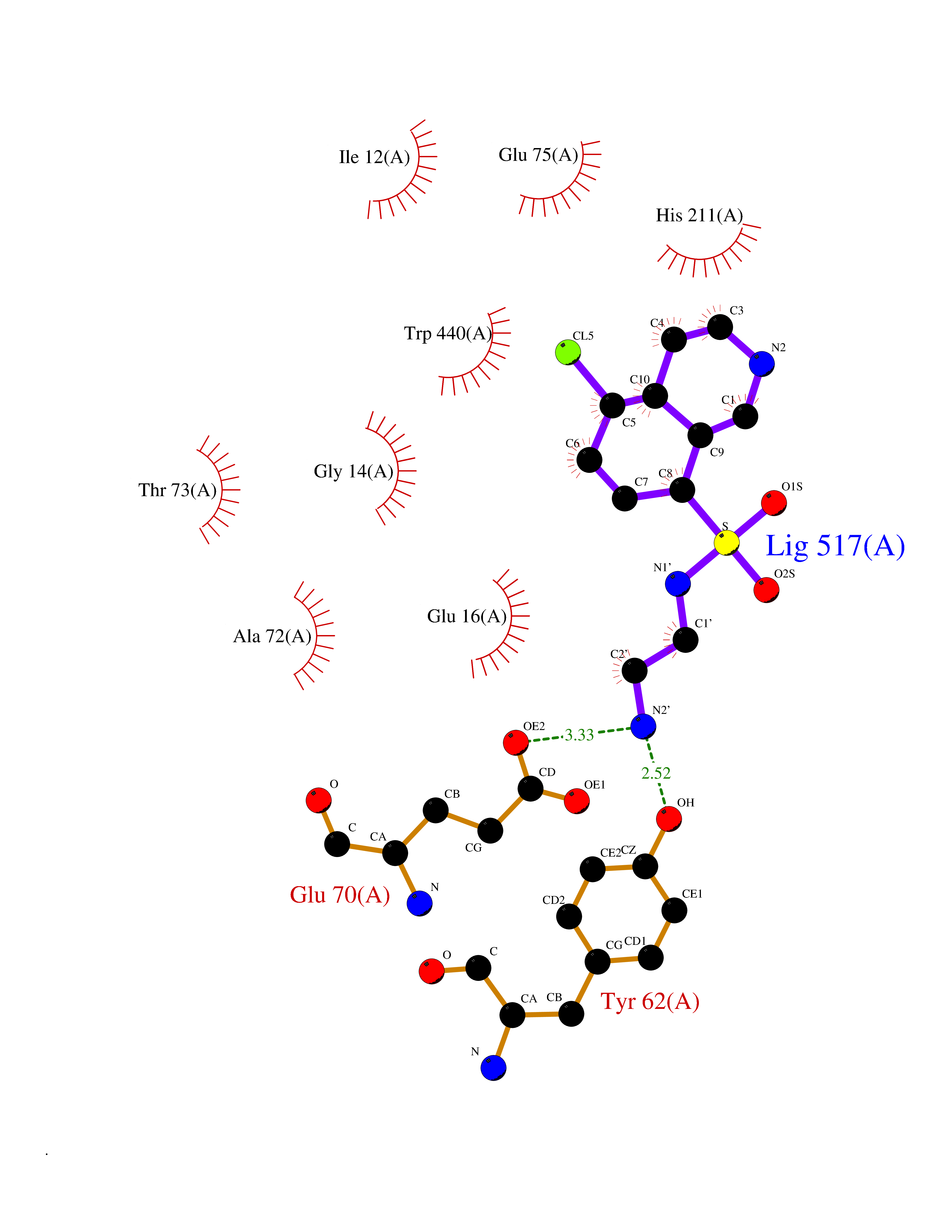 Ligplot Map 3D Binding mode Sequence TVESALTRRIMGIETEYGLTFVDGDSKKLRPDEIARRMFRPIVEKYSSSNIFIPNGSRLYLNVGSHPEYATAECDNLTQLINFEKAGDVIADRMAVDAEESLAKEDIAGQVYLFKNNVDSVGNSYGCHENYLVGRSMPLKALGKRLMPFLITRQLICGAGRIHHPNPSFPLGYCISQRSDHVWEGVSSASRPIINTRDEPHADSHSYRRLHVIVGDANMAEPSIALKVGSTLLVLEMIEADFGLPSLELANDIASIREISRDATGSTLLSLKDGTTMTALQIQQVVFEHASKWLEQRPEPEFSGTSNTEMARVLDLWGRMLKAIESGDFSEVDTEIDWVIKKKLIDRFIQRGNLGLDDPKLAQVDLTYHDIRPGRGLFSVLQSRGMIKRWTTDEAILAAVDTAPDTTRAHLRGRILKAADTLGVPVTVDWMRHKVNRPEPQSVELGDPFSAVNSEVDQLIEYMTVHAGSASGTSLLDEIDGLLENNAEEFVRSYVQKGGETVESALTRRIMGIETEYGLTFVDGDSKKLRPDEIARRMFRPIVEKYSSSNIFIPNGSRLYLNVGSHPEYATAECDNLTQLINFEKAGDVIADRMAVDAEESLAKEDIAGQVYLFKNNVDSVGNSYGCHENYLVGRSMPLKALGKRLMPFLITRQLICGAGRIHHPNPSFPLGYCISQRSDHVWEGVSSASRPIINTRDEPHADSHSYRRLHVIVGDANMAEPSIALKVGSTLLVLEMIEADFGLPSLELANDIASIREISRDATGSTLLSLKDGTTMTALQIQQVVFEHASKWLEQRPEPEFSGTSNTEMARVLDLWGRMLKAIESGDFSEVDTEIDWVIKKKLIDRFIQRGNLGLDDPKLAQVDLTYHDIRPGRGLFSVLQSRGMIKRWTTDEAILAAVDTAPDTTRAHLRGRILKAADTLGVPVTVDWMRHKVNRPEPQSVELGDPFSAVNSEVDQLIEYMTVHASLLDEIDGLLENNAEEFVRSYVQKGGE Hydrogen bonds contact Hydrophobic contact | ||||
| 4 | Metabotropic glutamate receptor 3 (mGluR3) | 4XAR | 8.30 | |
Target general information Gen name GRM3 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms mGLUR3; Group III metabotropic glutamate receptor; GPRC1C Protein family G-protein coupled receptor 3 family Biochemical class GPCR glutamate Function Ligand binding causes a conformation change that triggers signaling via guanine nucleotide-binding proteins (G proteins) and modulates the activity of down-stream effectors. Signaling inhibits adenylate cyclase activity. G-protein coupled receptor for glutamate. Related diseases Paramyotonia congenita (PMC) [MIM:168300]: An autosomal dominant channelopathy characterized by myotonia, increased by exposure to cold, intermittent flaccid paresis, not necessarily dependent on cold or myotonia, lability of serum potassium, non-progressive nature and lack of atrophy or hypertrophy of muscles. In some patients, myotonia is not increased by cold exposure (paramyotonia without cold paralysis). Patients may have a combination phenotype of PMC and HYPP. {ECO:0000269|PubMed:10369308, ECO:0000269|PubMed:10727489, ECO:0000269|PubMed:1310898, ECO:0000269|PubMed:1316765, ECO:0000269|PubMed:1338909, ECO:0000269|PubMed:15318338, ECO:0000269|PubMed:15790667, ECO:0000269|PubMed:16786525, ECO:0000269|PubMed:18166706, ECO:0000269|PubMed:18690054, ECO:0000269|PubMed:19077043, ECO:0000269|PubMed:20076800, ECO:0000269|PubMed:8242056, ECO:0000269|PubMed:8308722, ECO:0000269|PubMed:8388676, ECO:0000269|PubMed:8580427}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Periodic paralysis hypokalemic 2 (HOKPP2) [MIM:613345]: An autosomal dominant disorder manifested by episodic flaccid generalized muscle weakness associated with falls of serum potassium levels. {ECO:0000269|PubMed:10599760, ECO:0000269|PubMed:10851391, ECO:0000269|PubMed:10944223, ECO:0000269|PubMed:11558801, ECO:0000269|PubMed:11591859, ECO:0000269|PubMed:16890191, ECO:0000269|PubMed:17898326, ECO:0000269|PubMed:18162704, ECO:0000269|PubMed:19118277, ECO:0000269|PubMed:20522878, ECO:0000269|PubMed:21043388, ECO:0000269|PubMed:24549961}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Periodic paralysis hyperkalemic (HYPP) [MIM:170500]: An autosomal dominant channelopathy characterized by episodic flaccid generalized muscle weakness associated with high levels of serum potassium. Concurrence of myotonia is found in HYPP patients. {ECO:0000269|PubMed:1659668, ECO:0000269|PubMed:1659948, ECO:0000269|PubMed:20076800}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Periodic paralysis normokalemic (NKPP) [MIM:170500]: A disorder closely related to hyperkalemic periodic paralysis, but marked by a lack of alterations in potassium levels during attacks of muscle weakness. {ECO:0000269|PubMed:15596759, ECO:0000269|PubMed:18046642, ECO:0000269|PubMed:20522878}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Myotonia SCN4A-related (MYOSCN4A) [MIM:608390]: A phenotypically highly variable myotonia aggravated by potassium loading, and sometimes by cold. Myotonia is characterized by sustained muscle tensing that prevents muscles from relaxing normally. It causes muscle stiffness that can interfere with movement. In some people the stiffness is very mild, while in other cases it may be severe enough to interfere with walking, running, and other activities of daily life. Myotonia SCN4A-related includes myotonia permanens and myotonia fluctuans. In myotonia permanens, the myotonia is generalized and there is a hypertrophy of the muscle, particularly in the neck and the shoulder. Attacks of severe muscle stiffness of the thoracic muscles may be life threatening due to impaired ventilation. In myotonia fluctuans, the muscle stiffness may fluctuate from day to day, provoked by exercise. {ECO:0000269|PubMed:10218481, ECO:0000269|PubMed:16786525, ECO:0000269|PubMed:16832098, ECO:0000269|PubMed:17212350, ECO:0000269|PubMed:17998485, ECO:0000269|PubMed:18203179, ECO:0000269|PubMed:18337100, ECO:0000269|PubMed:19015483, ECO:0000269|PubMed:19347921, ECO:0000269|PubMed:20076800, ECO:0000269|PubMed:27653901, ECO:0000269|PubMed:8058156, ECO:0000269|PubMed:9392583}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Myasthenic syndrome, congenital, 16 (CMS16) [MIM:614198]: A form of congenital myasthenic syndrome, a group of disorders characterized by failure of neuromuscular transmission, including pre-synaptic, synaptic, and post-synaptic disorders that are not of autoimmune origin. Clinical features are easy fatigability and muscle weakness. CMS16 is characterized by fatigable generalized weakness and recurrent attacks of respiratory and bulbar paralysis since birth. The fatigable weakness involves lid-elevator, external ocular, facial, limb and truncal muscles and an decremental response of the compound muscle action potential on repetitive stimulation. {ECO:0000269|PubMed:12766226, ECO:0000269|PubMed:25707578, ECO:0000269|PubMed:26659129}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Congenital myopathy 22A, classic (CMYO22A) [MIM:620351]: A form of congenital myopathy, a clinically and genetically heterogeneous group of muscle disorders characterized by hypotonia and muscle weakness apparent at birth, and specific pathological features on muscle biopsy. CMYO22A is an autosomal recessive form characterized by fetal hypokinesia, polyhydramnios, and severe neonatal hypotonia associated with respiratory insufficiency. Affected individuals who survive the neonatal period have delayed motor development, difficulty walking, proximal muscle weakness of the upper and lower limbs, facial and neck muscle weakness, easy fatigability, and mild limb contractures or foot deformities. {ECO:0000269|PubMed:26700687, ECO:0000269|PubMed:28262468, ECO:0000269|PubMed:36090556}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Congenital myopathy 22B, severe fetal (CMYO22B) [MIM:620369]: A severe congenital myopathy, a clinically and genetically heterogeneous group of muscle disorders characterized by hypotonia and muscle weakness apparent at birth, and specific pathological features on muscle biopsy. CMYO22B is an autosomal recessive form characterized by onset in utero. Affected individuals show fetal akinesia, and develop fetal hydrops with pulmonary hypoplasia, severe joint contractures, and generalized muscle hypoplasia. Death occurs in utero or soon after birth. {ECO:0000269|PubMed:26700687}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB05096 Interacts with NA EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disulfide bond; G-protein coupled receptor; Glycoprotein; Membrane; Proteomics identification; Receptor; Reference proteome; Signal; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 50355.5 Length 445 Aromaticity 0.11 Instability index 38.26 Isoelectric point 6.52 Charge (pH=7) -1.53 2D Binding mode Binding energy (Kcal/mol) -7.75 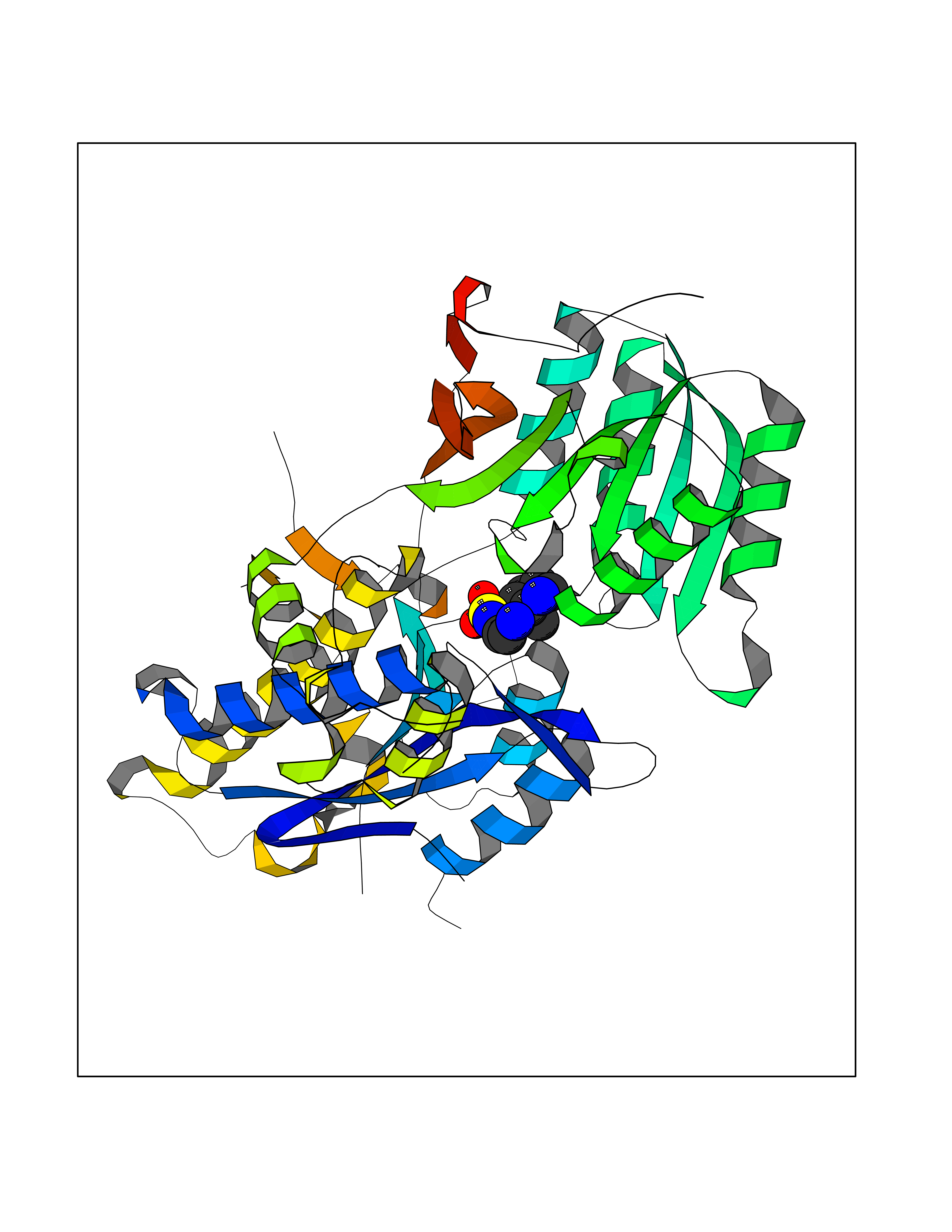 Molscript Map 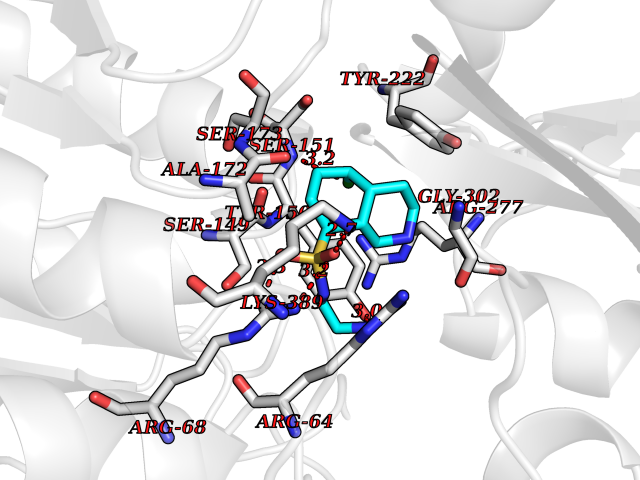 Pymol Map 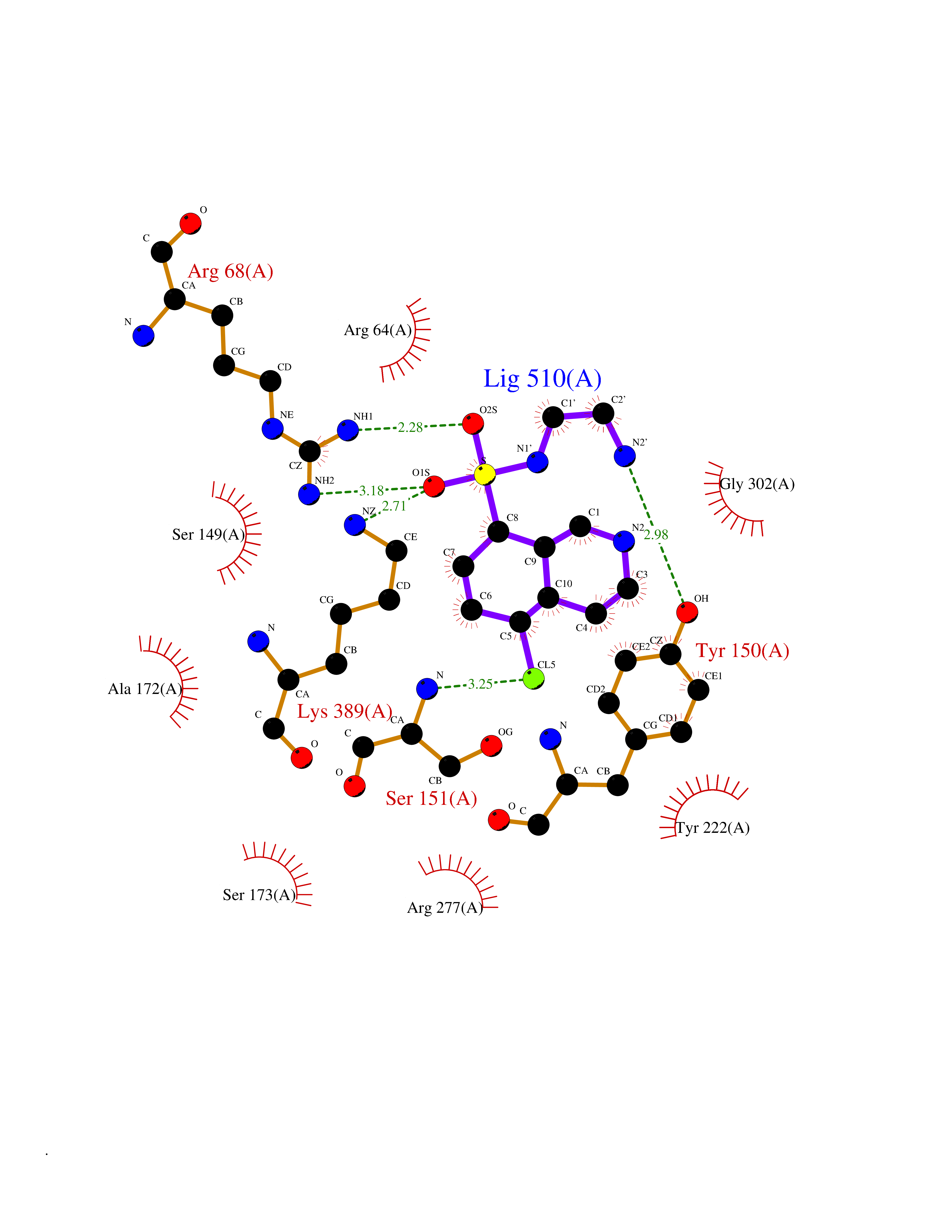 Ligplot Map 3D Binding mode Sequence RREIKIEGDLVLGGLFPINEKGTGTEECGRINEDRGIQRLEAMLFAIDEINKDDYLLPGVKLGVHILDTCSRDTYALEQSLEFVRASLLLIAGVIGGSYSSVSIQVANLLRLFQIPQISYASTSAKLSDKSRYDYFARTVPPDFYQAKAMAEILRFFNWTYVSTVASEGDYGETGIEAFEQEARLRNISIATAEKVGRSNIRKSYDSVIRELLQKPNARVVVLFMRSDDSRELIAAASRANASFTWVASDGWGAQESIIKGSEHVAYGAITLELASQPVRQFDRYFQSLNPYNNHRNPWFRDFWEQKFQCSLRVCDKHLAIDSSNYEQESKIMFVVNAVYAMAHALHKMQRTLCPNTTKLCDAMKILDGKKLYKDYLLKINFTAPDADSIVKFDTFGDGMGRYNVFNFQNVGGKYSYLKVGHWAETLSLDVNSIHWSRNSVPTSE Hydrogen bonds contact Hydrophobic contact | ||||
| 5 | Solute carrier family 19 member 1 (SLC19A1) | 8GOF | 8.26 | |
Target general information Gen name SLC19A1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Reduced folate carrier protein; RFC1; RFC; Placental folate transporter; Intestinal folate carrier 1; IFC-1; Folate transporter 1; FOLT; FLOT1 Protein family Reduced folate carrier (RFC) transporter (TC 2.A.48) family Biochemical class NA Function Transporter for the intake of folate. Uptake of folate in human placental choriocarcinoma cells occurs by a novel mechanism called potocytosis which functionally couples three components, namely the folate receptor, the folate transporter, and a V-type H(+)-pump. Related diseases Megaloblastic anemia, folate-responsive (MEGAF) [MIM:601775]: An autosomal recessive metabolic disorder characterized by megaloblastic anemia resulting from decreased folate transport into erythrocytes. Disease manifestations include hemolytic anemia, hyperhomocysteinemia, and low vitamin B12. Serum folate levels are normal, but erythrocyte folate levels are decreased. Treatment with oral folate corrects the anemia and normalizes homocysteine. {ECO:0000269|PubMed:32276275}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Immunodeficiency 114, folate-responsive (IMD114) [MIM:620603]: An autosomal recessive immunologic disorder manifesting in early infancy and characterized by recurrent skin and respiratory infections, mucosal bleeding, oral ulcers, chronic diarrhea, and poor overall growth. Affected individuals have lymphopenia, low serum immunoglobulins, and impaired T cell proliferation. Some patients have global developmental delay. {ECO:0000269|PubMed:36517554, ECO:0000269|PubMed:36745868}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB11256; DB00563; DB00642; DB06813; DB01157 Interacts with Q7Z3Y9 EC number NA Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Antiport; Cell membrane; Disease variant; Folate-binding; Glycoprotein; Hereditary hemolytic anemia; Membrane; Phosphoprotein; Proteomics identification; Reference proteome; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A Molecular weight (Da) 46087.7 Length 407 Aromaticity 0.15 Instability index 34.62 Isoelectric point 9.82 Charge (pH=7) 17.33 2D Binding mode Binding energy (Kcal/mol) -7.72  Molscript Map 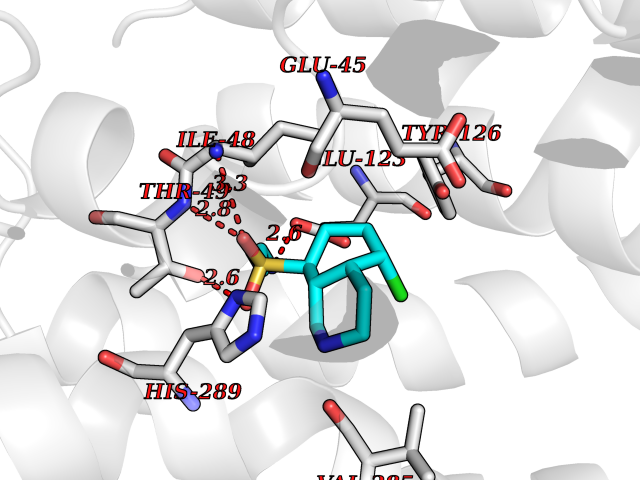 Pymol Map 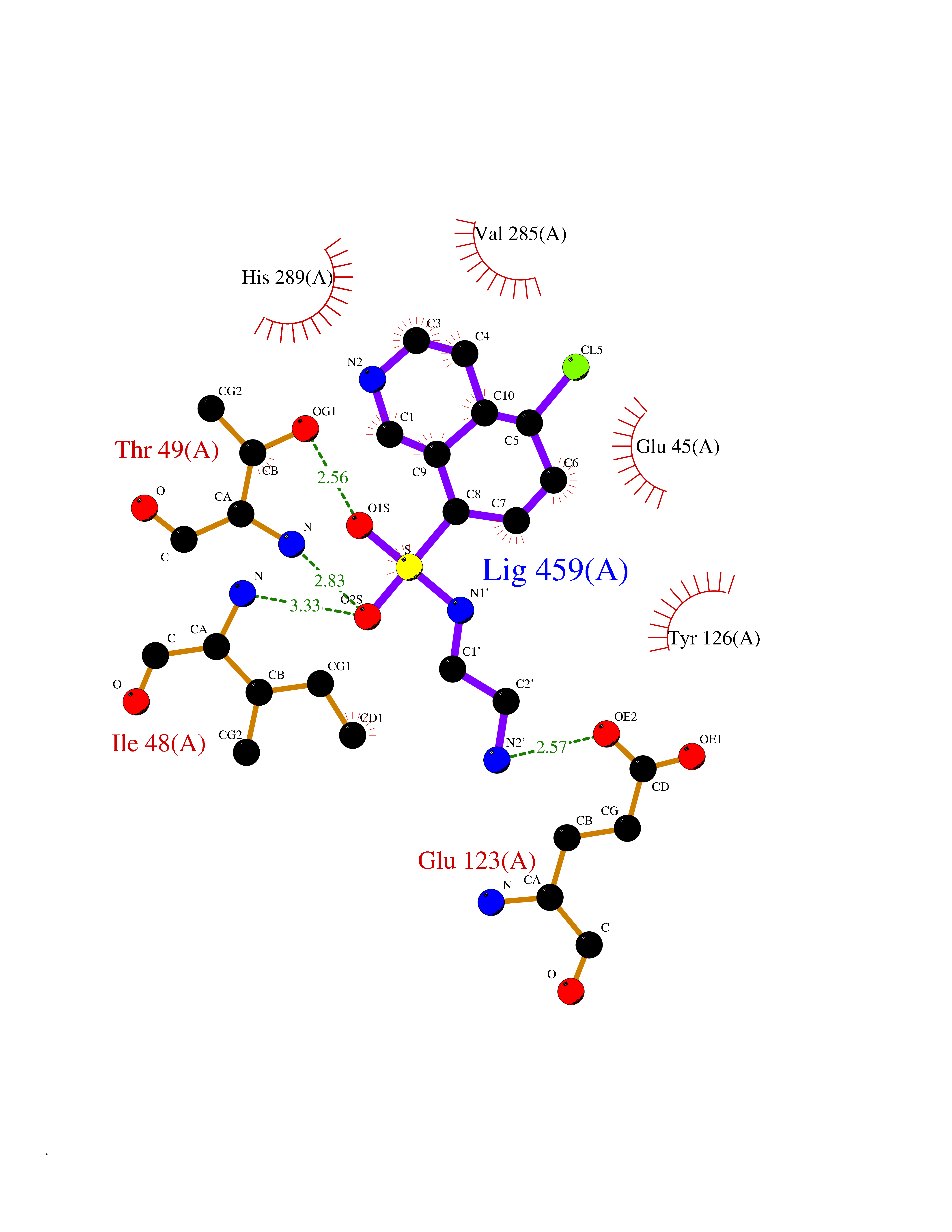 Ligplot Map 3D Binding mode Sequence DPELRSWRHLVCYLCFYGFMAQIRPGESFITPYLLGPDKNFTREQVTNEITPVLSYSYLAVLVPVFLLTDYLRYTPVLLLQGLSFVSVWLLLLLGHSVAHMQLMELFYSVTMAARIAYSSYIFSLVRPARYQRVAGYSRAAVLLGVFTSSVLGQLLVTVGRVSFSTLNYISLAFLTFSVVLALFLKRPKRSLFFNRDDSVLARMLRELGDSLRRPQLRLWSLWWVFNSAGYYLVVYYVHILWNEVDPTTNSARVYNGAADAASTLLGAITSFAAGFVKIRWARWSKLLIAGVTATQAGLVFLLAHTRHPSSIWLCYAAFVLFRGSYQFLVPIATFQIASSLSKELCALVFGVNTFFATIVKTIITFIVSDVRGLGLPVRKQFQLYSVYFLILSIIYFLGAMLDGLRH Hydrogen bonds contact Hydrophobic contact | ||||
| 6 | Cytochrome P450 1A2 | 2HI4 | 8.21 | |
Target general information Gen name CYP1A2 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Cytochrome P450 family Biochemical class Oxidoreductase Function Aromatase activity.Caffeine oxidase activity.Demethylase activity.Electron carrier activity.Enzyme binding.Heme binding.Iron ion binding.Monooxygenase activity.Oxidoreductase activity.Oxidoreductase activity, acting on paired donors, with incorporation or reduction of molecular oxygen, reduced flavin or flavoprotein as one donor, and incorporation of one atom of oxygen.Oxygen binding. Related diseases Myeloperoxidase deficiency (MPOD) [MIM:254600]: A disorder characterized by decreased myeloperoxidase activity in neutrophils and monocytes that results in disseminated candidiasis. {ECO:0000269|PubMed:37198333, ECO:0000269|PubMed:7904599, ECO:0000269|PubMed:8142659, ECO:0000269|PubMed:8621627, ECO:0000269|PubMed:9354683, ECO:0000269|PubMed:9637725}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08496; DB01667; DB14132; DB04356; DB02489; DB11932; DB12001; DB05812; DB13573; DB01418; DB00316; DB15568; DB06594; DB00518; DB05396; DB00969; DB07453; DB01424; DB01223; DB01118; DB00321; DB00261; DB01217; DB01435; DB06605; DB05676; DB06413; DB06216; DB01072; DB15011; DB06442; DB06626; DB00993; DB00972; DB13203; DB05015; DB16703; DB06769; DB01086; DB06770; DB06771; DB06732; DB00195; DB04889; DB11967; DB13975; DB00188; DB12151; DB01558; DB14018; DB13812; DB00201; DB09061; DB14737; DB11791; DB06774; DB00564; DB06016; DB01136; DB12814; DB00477; DB00356; DB01166; DB00501; DB01012; DB00568; DB00827; DB00537; DB00215; DB12499; DB14025; DB00349; DB01242; DB00575; DB00758; DB00363; DB00286; DB11672; DB14635; DB00924; DB08912; DB00851; DB06292; DB01254; DB01609; DB01151; DB16650; DB12161; DB01191; DB00633; DB11994; DB00586; DB11511; DB12945; DB00280; DB01184; DB09167; DB05928; DB01142; DB09273; DB00470; DB00476; DB00625; DB15444; DB06210; DB13874; DB11718; DB00467; DB11404; DB00530; DB00783; DB13952; DB13953; DB13954; DB13955; DB13956; DB00655; DB04574; DB13592; DB00330; DB00898; DB00977; DB00773; DB01628; DB00927; DB04854; DB01482; DB00574; DB12265; DB15669; DB01195; DB08972; DB04841; DB00544; DB00472; DB00499; DB00176; DB01320; DB00998; DB14029; DB06160; DB01044; DB01241; DB01155; DB01645; DB01381; DB00986; DB00365; DB00400; DB05708; DB00629; DB00502; DB01094; DB14999; DB04076; DB11737; DB00619; DB00458; DB11564; DB01306; DB09456; DB09564; DB01307; DB00047; DB01309; DB00030; DB00046; DB11567; DB00071; DB11568; DB05258; DB00034; DB00105; DB15131; DB00011; DB00018; DB00069; DB00060; DB00068; DB00033; DB00951; DB11757; DB09570; DB01026; DB01097; DB16217; DB09078; DB01002; DB05667; DB00281; DB12406; DB09198; DB04948; DB00978; DB06448; DB16220; DB01601; DB00455; DB04871; DB06077; DB01283; DB00772; DB00934; DB06234; DB14009; DB00784; DB01065; DB00170; DB00454; DB00532; DB00333; DB00763; DB00553; DB01028; DB09241; DB01233; DB00379; DB06148; DB01388; DB06595; DB00370; DB16236; DB00745; DB11763; DB00218; DB06510; DB14011; DB00461; DB00607; DB00779; DB00788; DB06600; DB00238; DB06803; DB00184; DB01115; DB11793; DB00435; DB05115; DB00717; DB01059; DB00540; DB05990; DB01165; DB00334; DB16267; DB00338; DB00904; DB11632; DB11443; DB01173; DB11837; DB09330; DB01303; DB11697; DB00377; DB00715; DB06589; DB11774; DB00487; DB00008; DB00022; DB09122; DB13634; DB00806; DB11198; DB08883; DB00850; DB03783; DB01174; DB00388; DB00252; DB11450; DB01100; DB13823; DB04951; DB17472; DB11642; DB08910; DB15822; DB01058; DB01087; DB00794; DB00420; DB09288; DB01182; DB06479; DB00818; DB00571; DB13449; DB11892; DB04216; DB00908; DB00468; DB01129; DB00980; DB09290; DB00863; DB01367; DB00409; DB02709; DB13174; DB01045; DB11753; DB00740; DB14924; DB00503; DB00533; DB01656; DB15119; DB00268; DB00296; DB00412; DB00817; DB12332; DB13772; DB06654; DB11491; DB00418; DB01037; DB11689; DB06290; DB13261; DB15093; DB00052; DB00398; DB01208; DB09118; DB00428; DB06820; DB00382; DB00675; DB06083; DB09071; DB05488; DB09256; DB01079; DB01405; DB00857; DB08880; DB11712; DB01412; DB00277; DB00730; DB01623; DB00208; DB06137; DB00697; DB01056; DB06264; DB00752; DB00384; DB12245; DB00831; DB15442; DB00440; DB00685; DB08867; DB14989; DB13609; DB06235; DB00313; DB08881; DB00661; DB09185; DB12026; DB00682; DB02134; DB00549; DB00744; DB00315; DB00425; DB09225; DB09120 Interacts with O95870 EC number 1.14.14.1; 4.2.1.152 Uniprot keywords 3D-structure; Direct protein sequencing; Endoplasmic reticulum; Fatty acid metabolism; Glycoprotein; Heme; Iron; Lipid metabolism; Lyase; Membrane; Metal-binding; Microsome; Monooxygenase; Oxidoreductase; Proteomics identification; Reference proteome; Steroid metabolism; Sterol metabolism Protein physicochemical properties Chain ID A Molecular weight (Da) 54475 Length 480 Aromaticity 0.1 Instability index 40.43 Isoelectric point 9.16 Charge (pH=7) 9.89 2D Binding mode Binding energy (Kcal/mol) -8.08  Molscript Map 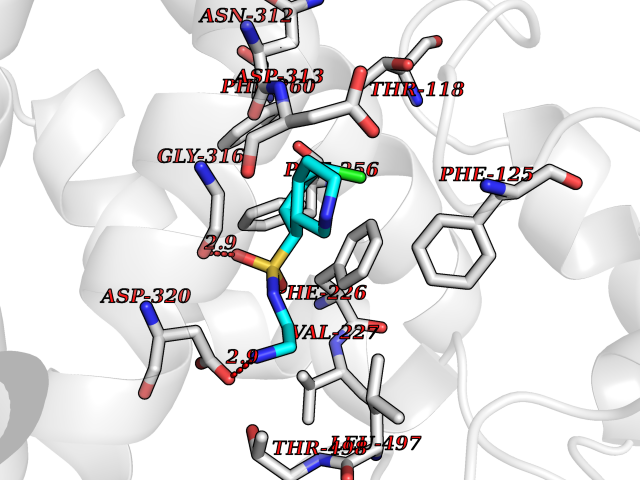 Pymol Map 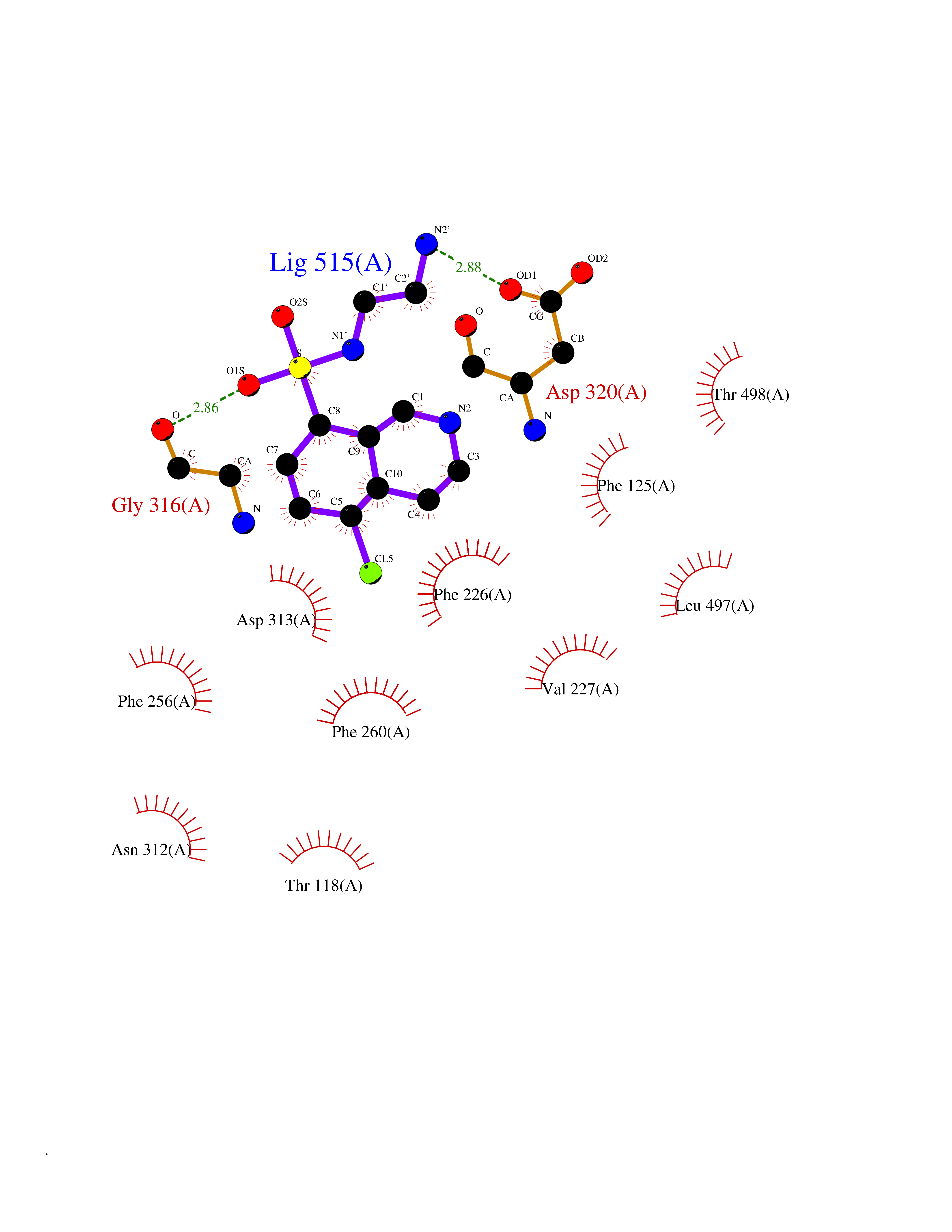 Ligplot Map 3D Binding mode Sequence RVPKGLKSPPEPWGWPLLGHVLTLGKNPHLALSRMSQRYGDVLQIRIGSTPVLVLSRLDTIRQALVRQGDDFKGRPDLYTSTLITDGQSLTFSTDSGPVWAARRRLAQNALNTFSIASDPASSSSCYLEEHVSKEAKALISRLQELMAGPGHFDPYNQVVVSVANVIGAMCFGQHFPESSDEMLSLVKNTHEFVETASSGNPLDFFPILRYLPNPALQRFKAFNQRFLWFLQKTVQEHYQDFDKNSVRDITGALFKHSKKGPRASGNLIPQEKIVNLVNDIFGAGFDTVTTAISWSLMYLVTKPEIQRKIQKELDTVIGRERRPRLSDRPQLPYLEAFILETFRHSSFLPFTIPHSTTRDTTLNGFYIPKKCCVFVNQWQVNHDPELWEDPSEFRPERFLTADGTAINKPLSEKMMLFGMGKRRCIGEVLAKWEIFLFLAILLQQLEFSVPPGVKVDLTPIYGLTMKHARCEHVQARRFS Hydrogen bonds contact Hydrophobic contact | ||||
| 7 | Choline O-acetyltransferase | 2FY3 | 8.20 | |
Target general information Gen name CHAT Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Carnitine/choline acetyltransferase family Biochemical class Transferase Function Choline O-acetyltransferase activity. Related diseases Myasthenic syndrome, congenital, 6, presynaptic (CMS6) [MIM:254210]: A form of congenital myasthenic syndrome, a group of disorders characterized by failure of neuromuscular transmission, including pre-synaptic, synaptic, and post-synaptic disorders that are not of autoimmune origin. Clinical features are easy fatigability and muscle weakness affecting the axial and limb muscles (with hypotonia in early-onset forms), the ocular muscles (leading to ptosis and ophthalmoplegia), and the facial and bulbar musculature (affecting sucking and swallowing, and leading to dysphonia). The symptoms fluctuate and worsen with physical effort. CMS6 affected individuals have myasthenic symptoms since birth or early infancy, negative tests for anti-AChR antibodies, and abrupt episodic crises with increased weakness, bulbar paralysis, and apnea precipitated by undue exertion, fever, or excitement. CMS6 inheritance is autosomal recessive. {ECO:0000269|PubMed:11172068, ECO:0000269|PubMed:12756141}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00122; DB14006; DB00184 Interacts with Q6H8Q1-8; Q8N302-2; Q9NXL2-1; Q6XD76; Q9UII2; Q8TBE0; Q9UQB8-6; Q9ULD4-2; Q9NSI6-4; Q6P5X5; Q96LL4; P20807-4; O00257-3; Q6ZP82-1; O95674; Q9H3R5; Q8WUX9; Q9H2A9; Q3SX64; Q92782-2; Q14117; O14641; Q658K8; Q6UXG2-3; O00472; Q6NXG1; Q15910-2; Q8IZU1; P15407; P55318; Q06547-3; P23769-2; P23771; Q15486; Q8IV36; Q4VB01; Q53GQ0; P10809; P41134; Q9NZH6; Q8NA54; Q86U28; P17275; Q8N5Z5; Q6P597; P08727; Q14525; Q8IUC2; Q6IAA8; Q14847-2; P27338; Q9GZQ8; Q53S70; Q5JXC2; A0A0A0MR05; Q8NEH6; Q8TCY5; Q6IN84-2; Q96H12; P01106; P41271-2; P14598; Q9GZM8; Q5BJF6-2; Q9H8K7; Q9NR21-5; Q5VU43-8; Q13956; Q5SXH7-1; Q96T60; Q96I34; Q86UA1; Q15311; Q8TBY0; Q04206; P47804-3; Q9H0X6; P62899; Q66K80; Q9BY12-3; Q86SQ7-2; Q7Z6I5; Q496A3; Q7Z698; Q9C004; Q92783-2; Q8N4C7; O75528; Q15814; O15273; Q96A09; Q8WTV1; Q53NU3; Q71RG4-4; Q86WT6-2; Q9Y3Q8; Q99598; P49459; P11441; Q9H270; P19544-6; Q53FD0-2; Q3KNS6-3 EC number 2.3.1.6 Uniprot keywords 3D-structure; Acyltransferase; Alternative splicing; Congenital myasthenic syndrome; Direct protein sequencing; Disease variant; Neurotransmitter biosynthesis; Phosphoprotein; Proteomics identification; Reference proteome; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 66365.9 Length 595 Aromaticity 0.08 Instability index 53.36 Isoelectric point 8.16 Charge (pH=7) 4.64 2D Binding mode Binding energy (Kcal/mol) -7.85 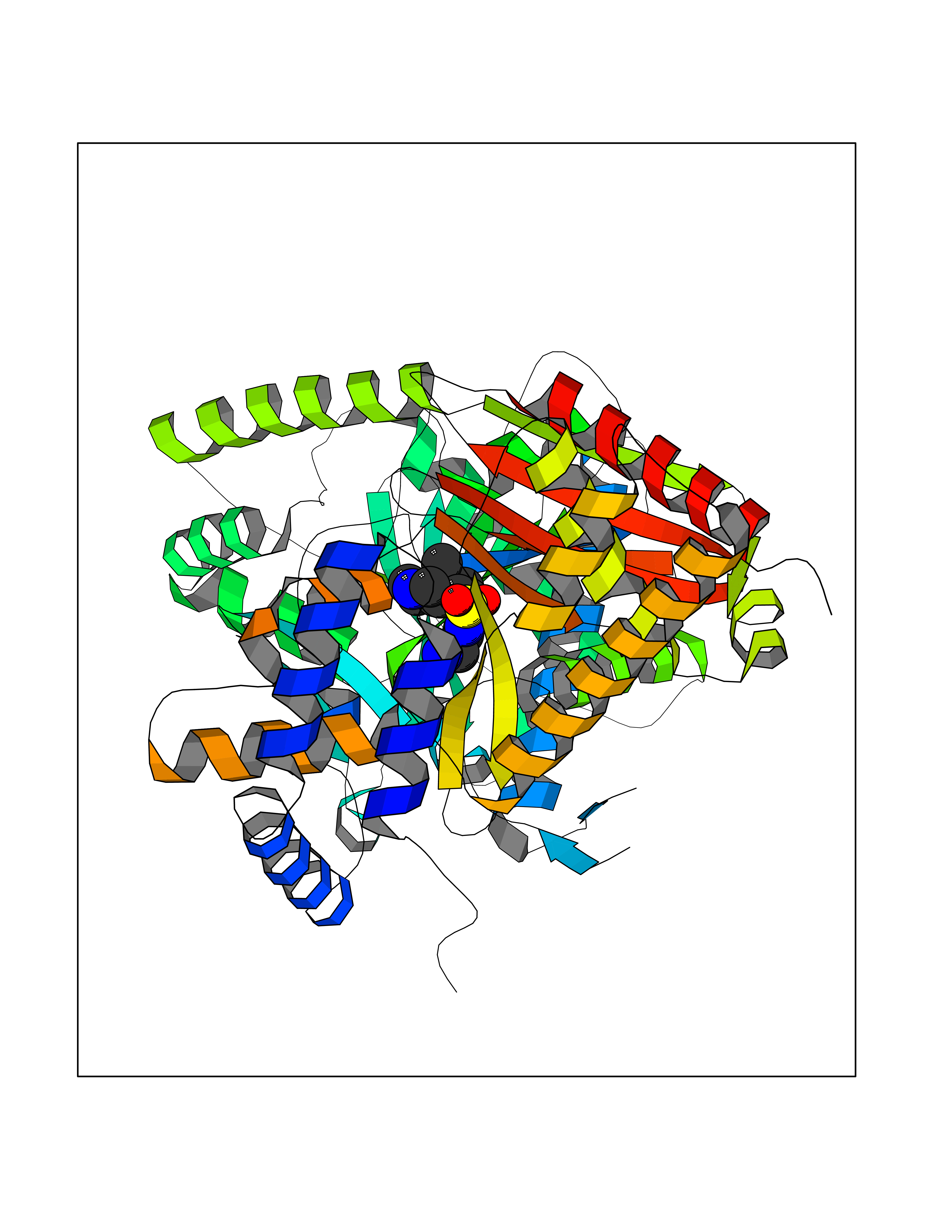 Molscript Map 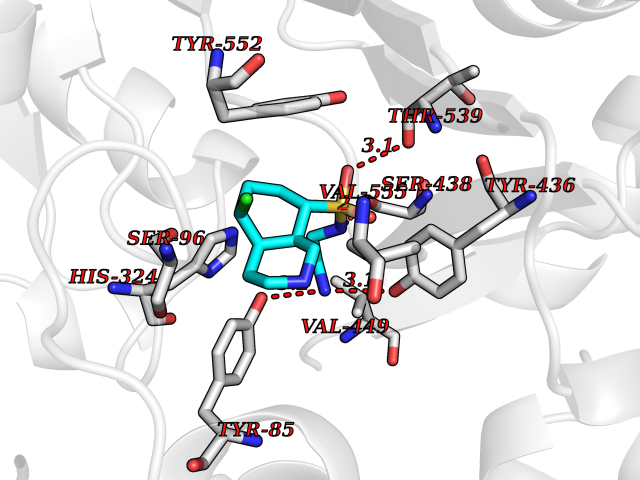 Pymol Map 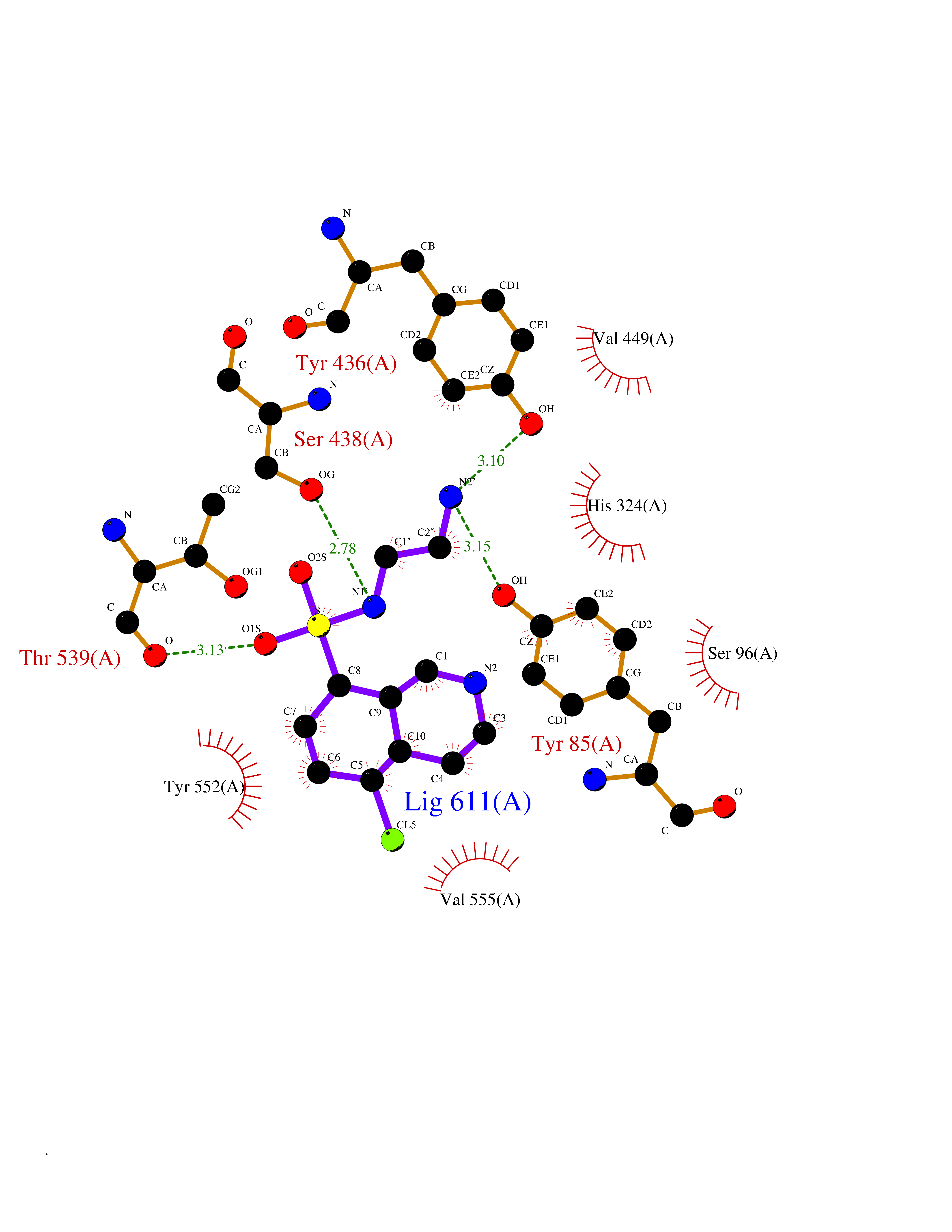 Ligplot Map 3D Binding mode Sequence SEESGLPKLPVPPLQQTLATYLQCMRHLVSEEQFRKSQAIVQQFGAPGGLGETLQQKLLERQEKTANWVSEYWLNDMYLNNRLALPVNSSPAVIFARQHFPGTDDQLRFAASLISGVLSYKALLDSHSIPTDCAKGQPLCMKQYYGLFSSYRLPGHTQDTLVAQNSSIMPEPEHVIVACCNQFFVLDVVINFRRLSEGDLFTQLRKIVKMASNAAARLPPIGLLTSDGRSEWAEARTVLVKDSTNRDSLDMIERCICLVCLDAPGGVELSDTHRALQLLHGGGYSKNGANRWYDKSLQFVVGRDGTCGVVCEHSPFDGIVLVQCTEHLLKHMTQPELVRSPMVPLPAPRRLRWKCSPEIQGHLASSAEKLQRIVKNLDFIVYKFDNYGKTFIKKQKCSPDAFIQVALQLAFYRLHRRLVPTYESASIRRFQEGRVDNIRSATPEALAFVRAVTDHKAAVPASEKLLLLKDAIRAQTAYTVMAITGMAIDNHLLALRELARAMCAALPEMFMDETYLMSNRFVLSTSQVPTTTEMFCCYGPVVPNGYGACYNPQPETILFCISSFHSCAATSSSKFAKAVEESLIDMRDLCSLLPP Hydrogen bonds contact Hydrophobic contact | ||||
| 8 | Lysine-specific demethylase 4E (KDM4E) | 2W2I | 8.12 | |
Target general information Gen name KDM4E Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Lysine-specific demethylase 4D-like; KDM4DL; KDM4D-like protein Protein family JHDM3 histone demethylase family Biochemical class Paired donor oxygen oxidoreductase Function Histone demethylase that specifically demethylates 'Lys-9' of histone H3, thereby playing a central role in histone code. Related diseases Defects in KAT2B has been found in a patient with isolated coloboma, a defect of the eye characterized by the absence of ocular structures due to abnormal morphogenesis of the optic cup and stalk, and the fusion of the fetal fissure (optic fissure). Isolated colobomas may be associated with an abnormally small eye (microphthalmia) or small cornea. {ECO:0000269|PubMed:28493397}. Drugs (DrugBank ID) NA Interacts with NA EC number EC 1.14.11.- Uniprot keywords 3D-structure; Chromatin regulator; Dioxygenase; Iron; Metal-binding; Nucleus; Oxidoreductase; Reference proteome; Transcription; Transcription regulation; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 35131.5 Length 305 Aromaticity 0.14 Instability index 39.34 Isoelectric point 6 Charge (pH=7) -6.1 2D Binding mode Binding energy (Kcal/mol) -7.87 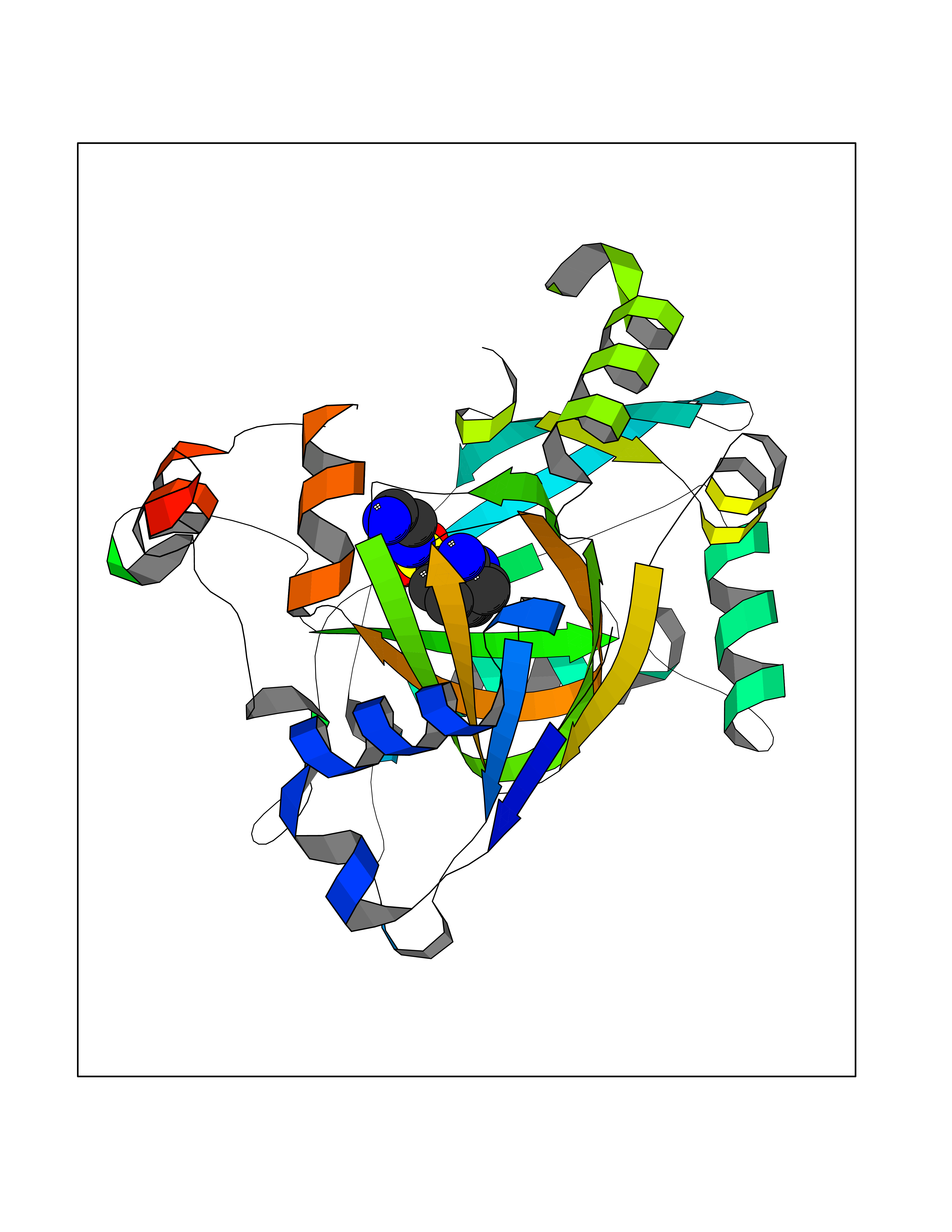 Molscript Map 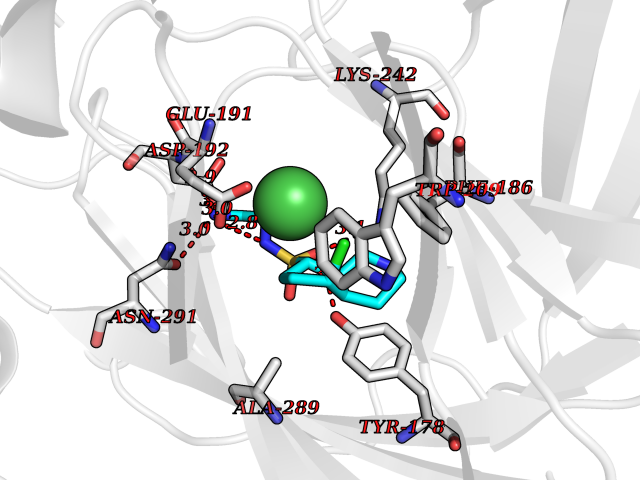 Pymol Map 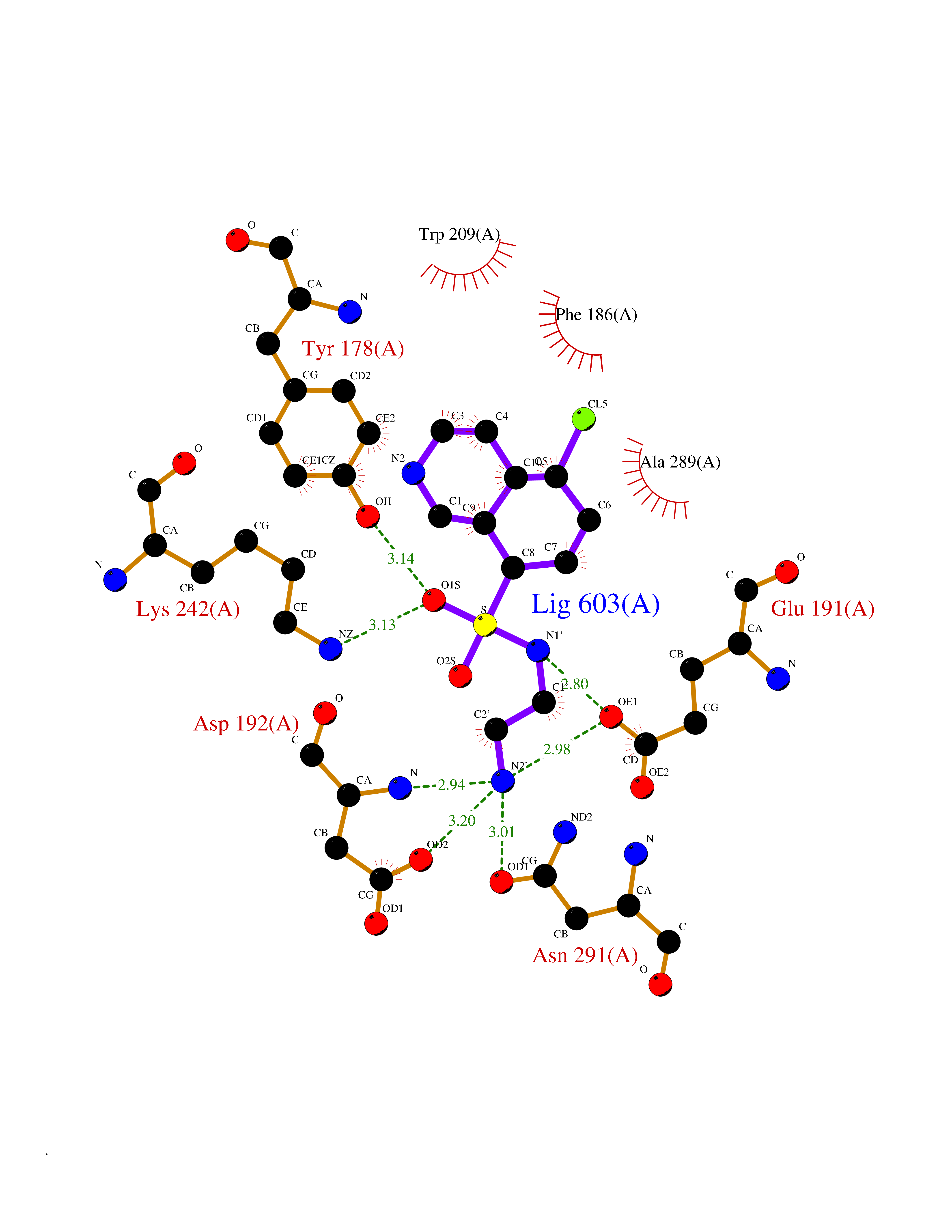 Ligplot Map 3D Binding mode Sequence HTIMTFYPTMEEFADFNTYVAYMESQGAHQAGLAKVIPPKEWKARQMYDDIEDILIATPLQQVTSGQGGVFTQYHKKKKAMRVGQYRRLANSKKYQTPPHQNFADLEQRYWKSHPGNPPIYGADISGSLFEESTKQWNLGHLGTILDLLEQECGVVIEGVNTPYLYFGMWKTTFAWHTEDMDLYSINYLHFGEPKTWYVVPPEHGQHLERLARELFPDISAFLRHKVALISPTVLKENGIPFNCMTQEAGEFMVTFPYGYHAGFNHGFNCAEAINFATPRWIDYGKMAVTFSMDPFVRIVQPESY Hydrogen bonds contact Hydrophobic contact | ||||
| 9 | Mycobacterium Isocitrate lyase (MycB icl) | 1F8M | 8.07 | |
Target general information Gen name MycB icl Organism Mycobacterium tuberculosis (strain ATCC 25618 / H37Rv) Uniprot ID TTD ID Synonyms Isocitratase; Isocitrase; ICL Protein family Isocitrate lyase/PEP mutase superfamily, Isocitrate lyase family Biochemical class Carbon-carbon lyase Function Catalyzes the formation of succinate and glyoxylate from isocitrate, a key step of the glyoxylate cycle. May be involved in the assimilation of one-carbon compounds via the isocitrate lyase- positive serine pathway. Related diseases Intellectual developmental disorder, autosomal recessive 80, with variant lissencephaly (MRT80) [MIM:620653]: An autosomal recessive disorder characterized by global developmental delay, mildly to moderately impaired intellectual development, attention deficit-hyperactivity disorder, hypotonia, seizure, poor social skills, and autistic traits. Brain imaging shows fronto-temporal lissencephaly and pachygyria. {ECO:0000269|PubMed:37880421}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB04343 Interacts with NA EC number EC 4.1.3.1 Uniprot keywords 3D-structure; Glyoxylate bypass; Isopeptide bond; Lyase; Magnesium; Manganese; Metal-binding; Reference proteome; Tricarboxylic acid cycle; Ubl conjugation Protein physicochemical properties Chain ID A,B Molecular weight (Da) 93759.5 Length 854 Aromaticity 0.09 Instability index 28.02 Isoelectric point 4.98 Charge (pH=7) -34.53 2D Binding mode Binding energy (Kcal/mol) -7.17 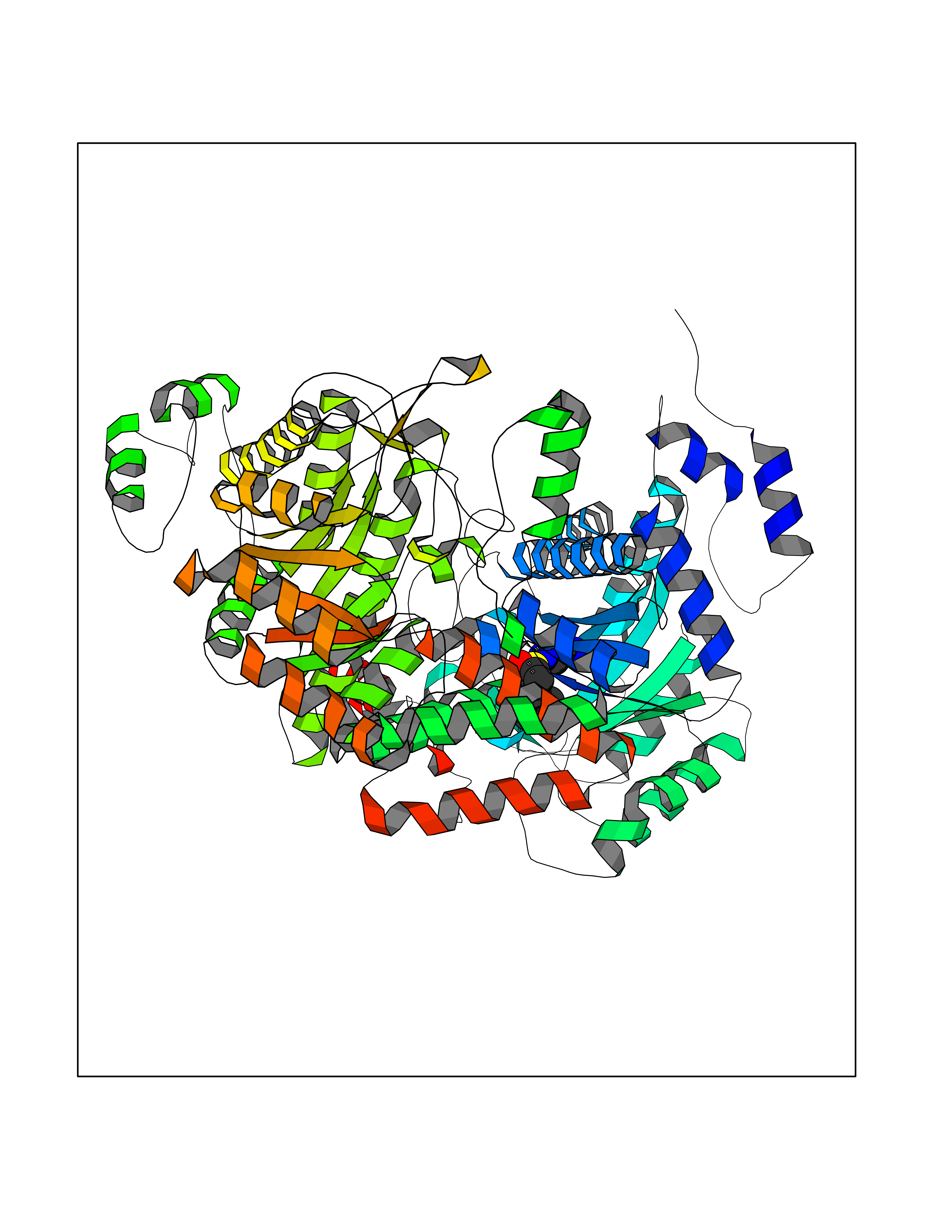 Molscript Map 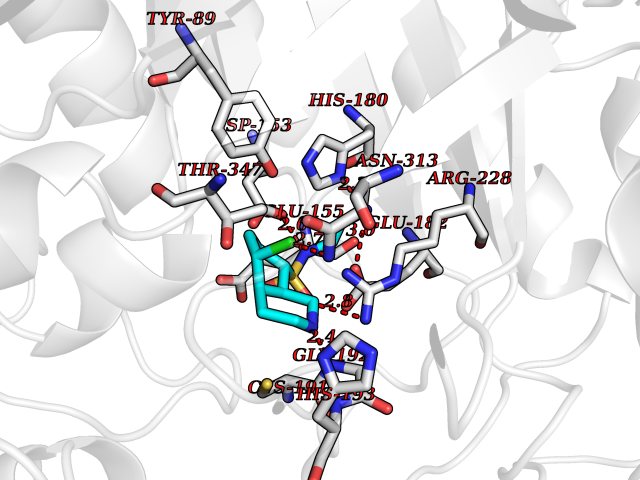 Pymol Map 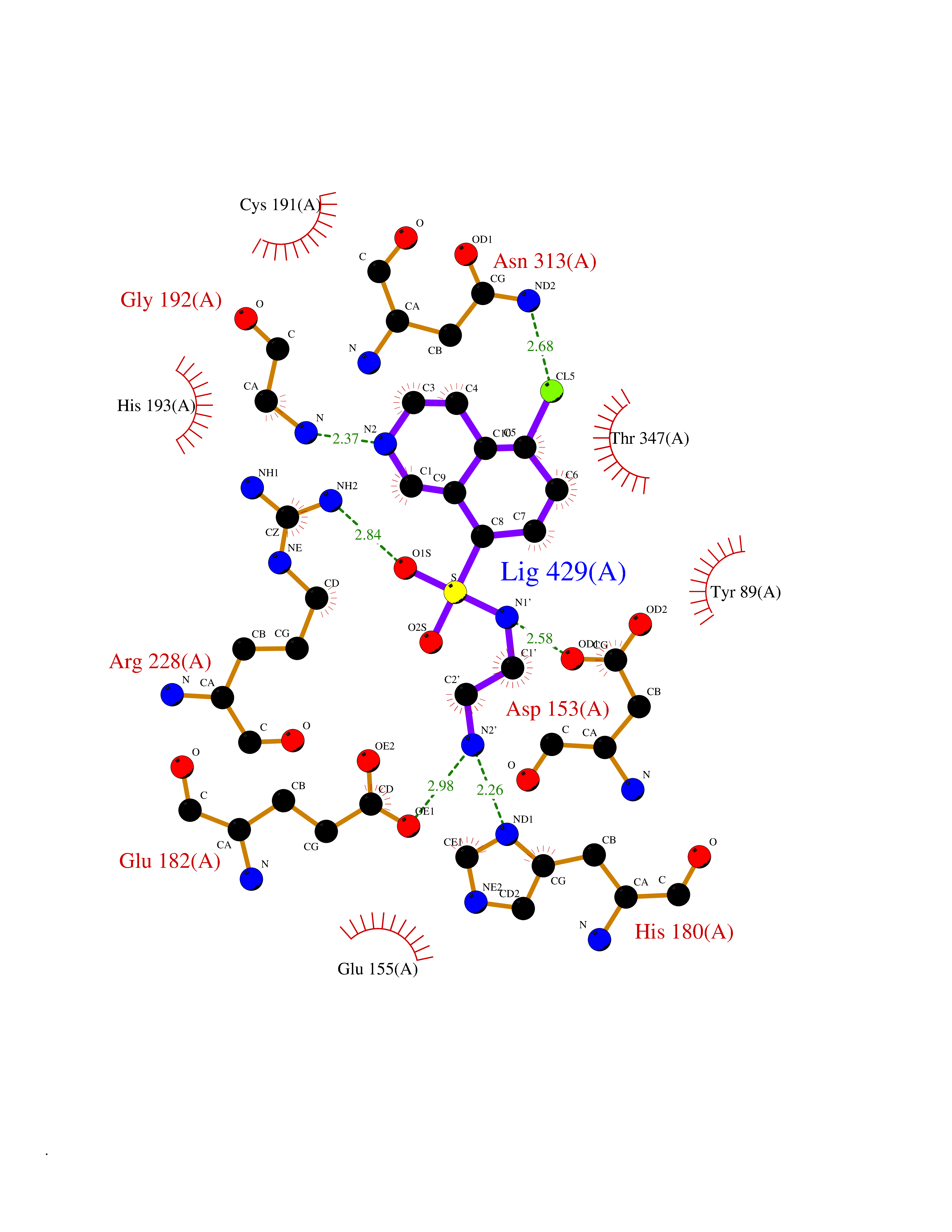 Ligplot Map 3D Binding mode Sequence ASVVGTPKSAEQIQQEWDTNPRWKDVTRTYSAEDVVALQGSVVEEHTLARRGAEVLWEQLHDLEWVNALGALTGNMAVQQVRAGLKAIYLSGWQVAGDANLSGHTYPDQSLYPANSVPQVVRRINNALQRADQIAKIEGDTSVENWLAPIVADGEAGFGGALNVYELQKALIAAGVAGSHWEDQLASEKKCGHLGGKVLIPTQQHIRTLTSARLAADVADVPTVVIARTDAEAATLITSDVDERDQPFITGERTREGFYRTKNGIEPCIARAKAYAPFADLIWMETGTPDLEAARQFSEAVKAEYPDQMLAYNCSPSFNWKKHLDDATIAKFQKELAAMGFKFQFITLAGFHALNYSMFDLAYGYAQNQMSAYVELQEREFAAEERGYTATKHQREVGAGYFDRIATTVDPNSSTTALTGSTEEGQFASVVGTPKSAEQIQQEWDTNPRWKDVTRTYSAEDVVALQGSVVEEHTLARRGAEVLWEQLHDLEWVNALGALTGNMAVQQVRAGLKAIYLSGWQVAGDANLSGHTYPDQSLYPANSVPQVVRRINNALQRADQIAKIEGDTSVENWLAPIVADGEAGFGGALNVYELQKALIAAGVAGSHWEDQLASEKKCGHLGGKVLIPTQQHIRTLTSARLAADVADVPTVVIARTDAEAATLITSDVDERDQPFITGERTREGFYRTKNGIEPCIARAKAYAPFADLIWMETGTPDLEAARQFSEAVKAEYPDQMLAYNCSPSFNWKKHLDDATIAKFQKELAAMGFKFQFITLAGFHALNYSMFDLAYGYAQNQMSAYVELQEREFAAEERGYTATKHQREVGAGYFDRIATTVDPNSSTTALTGSTEEGQF Hydrogen bonds contact Hydrophobic contact | ||||
| 10 | "Periplasmic trehalase (EC 3.2.1.28) (Alpha,alpha-trehalase) (Alpha,alpha-trehalose glucohydrolase) (Tre37A)" | 2JG0 | 8.05 | |
Target general information Gen name treA Organism Escherichia coli (strain K12) Uniprot ID TTD ID NA Synonyms JW1186;osmA;b1197 Protein family Glycosyl hydrolase 37 family Biochemical class NA Function Provides the cells with the ability to utilize trehalose at high osmolarity by splitting it into glucose molecules that can subsequently be taken up by the phosphotransferase-mediated uptake system. Related diseases SRC kinase activity has been shown to be increased in several tumor tissues and tumor cell lines such as colon carcinoma cells. {ECO:0000269|PubMed:2498394, ECO:0000269|PubMed:3093483}.; DISEASE: Thrombocytopenia 6 (THC6) [MIM:616937]: A form of thrombocytopenia, a hematologic disorder defined by a decrease in the number of platelets in circulating blood, resulting in the potential for increased bleeding and decreased ability for clotting. THC6 is an autosomal dominant form. Affected individuals may also have bone abnormalities and an increased risk for myelofibrosis. {ECO:0000269|PubMed:26936507}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number 3.2.1.28 Uniprot keywords 3D-structure; Direct protein sequencing; Glycosidase; Hydrolase; Periplasm; Reference proteome; Signal Protein physicochemical properties Chain ID A Molecular weight (Da) 57508.9 Length 507 Aromaticity 0.11 Instability index 48.32 Isoelectric point 5.48 Charge (pH=7) -10.13 2D Binding mode Binding energy (Kcal/mol) -8.43 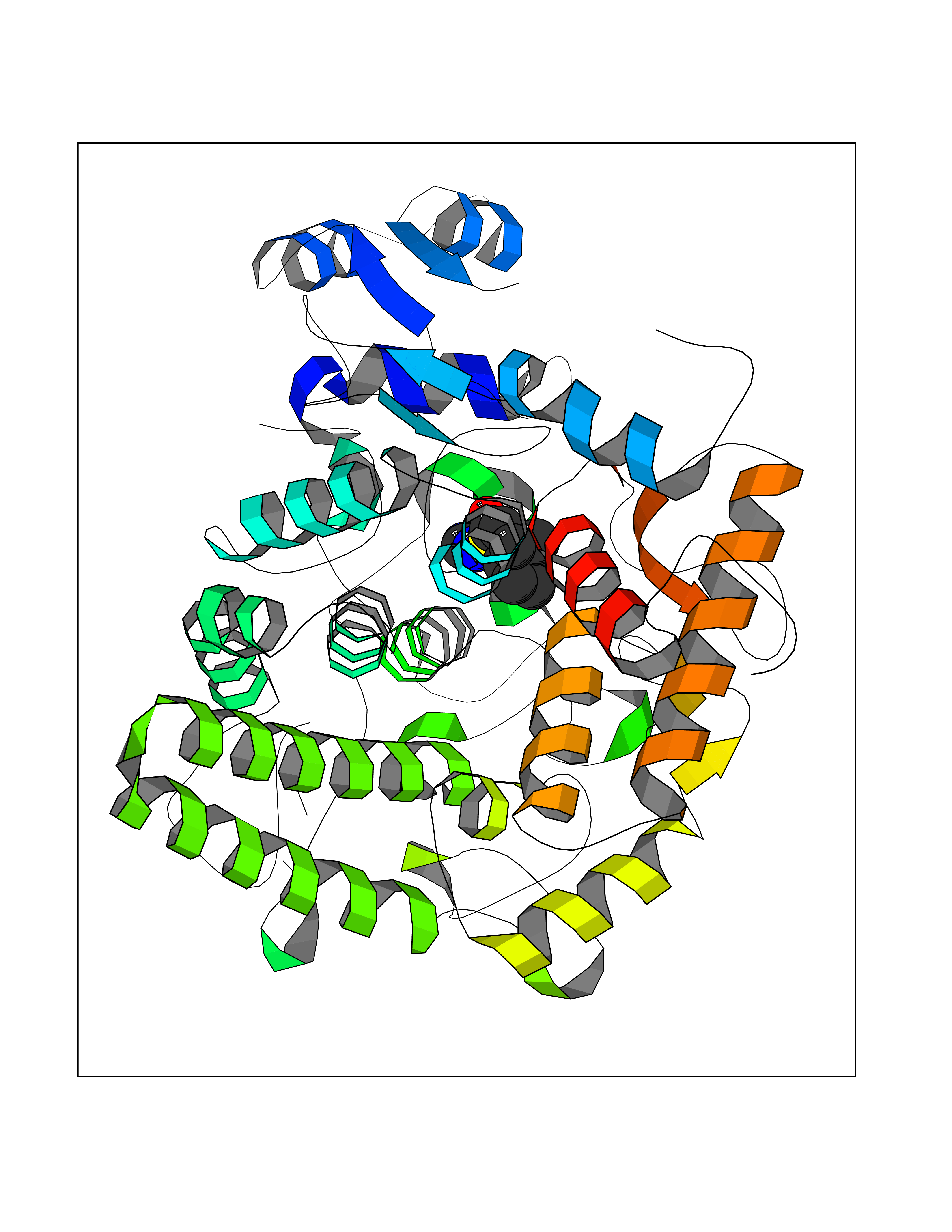 Molscript Map 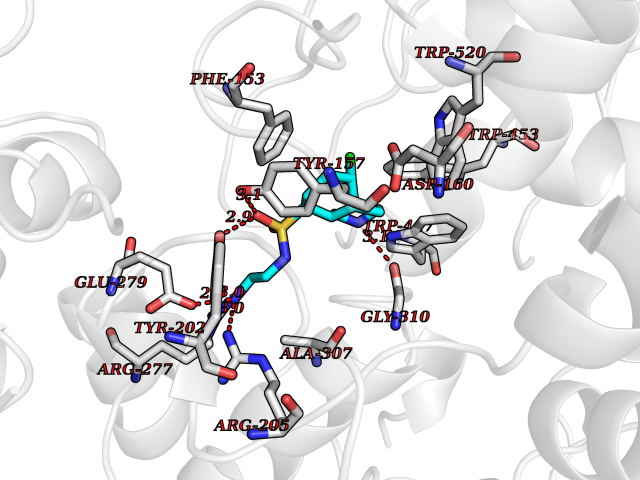 Pymol Map 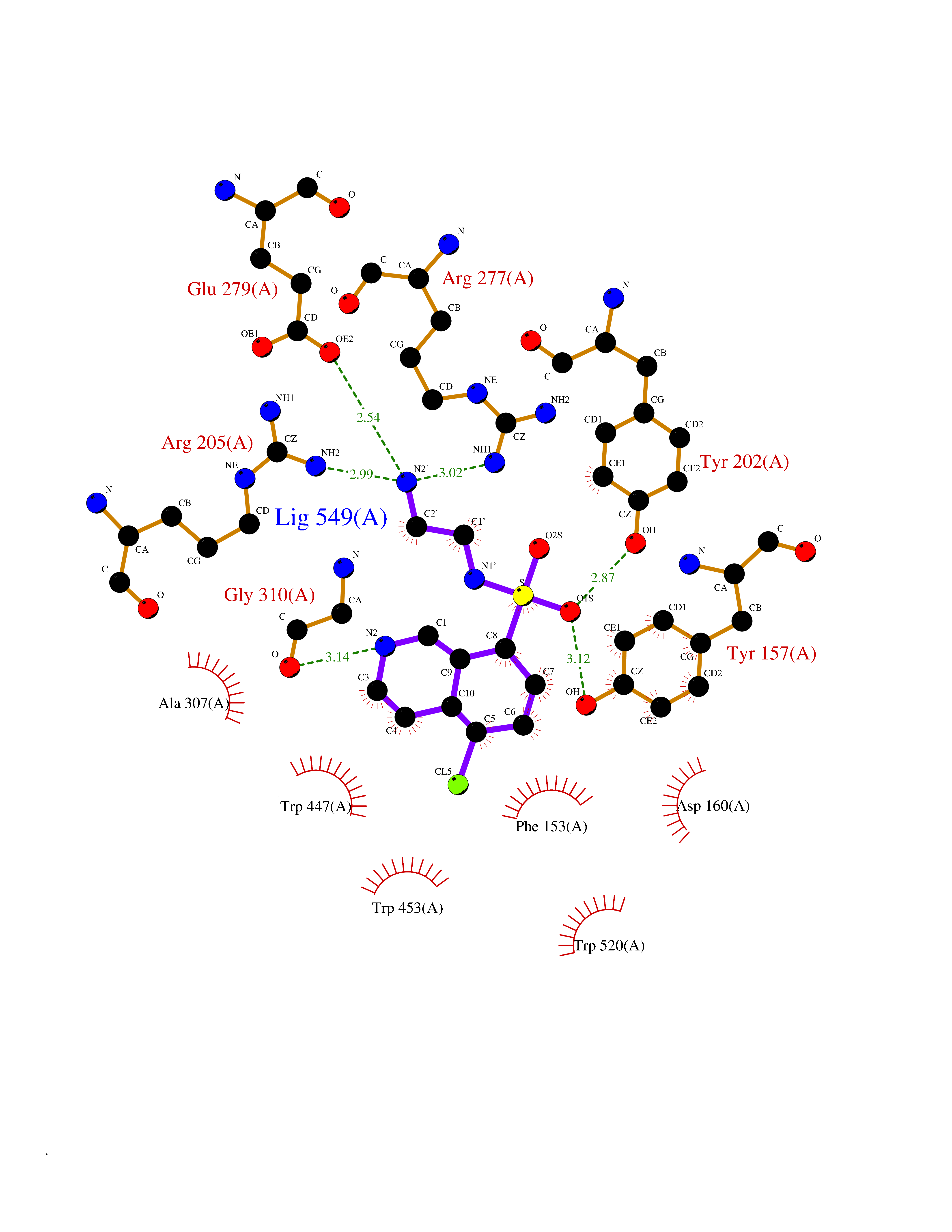 Ligplot Map 3D Binding mode Sequence PQPPDILLGPLFNDVQNAKLFPDQKTFADAVPNSDPLMILADYRMQQNQSGFDLRHFVNVNFTLPKYVPPEGQSLREHIDGLWPVLTRSTENTEKWDSLLPLPEPYVVPGGRFREVYYWDSYFTMLGLAESGHWDKVADMVANFAHEIDTYGHIPNGNRSYYLSRSQPPFFALMVELLAQHEGDAALKQYLPQMQKEYAYWMDGVENLQAGQQEKRVVKLQDGTLLNRYWDDRDTPRPESWVEDIATAKSNPNRPATEIYRDLRSAAASGWDFSSRWMDNPQQLNTLRTTSIVPVDLNSLMFKMEKILARASKAAGDNAMANQYETLANARQKGIEKYLWNDQQGWYADYDLKSHKVRNQLTAAALFPLYVNAAAKDRANKMATATKTHLLQPGGLNTTSVKSGQQWDAPNGWAPLQWVATEGLQNYGQKEVAMDISWHFLTNVQHTYDREKKLVEKYDVSTTGTGGGGGEYPLQDGFGWTNGVTLKMLDLICPKEQPCDNVPATRP Hydrogen bonds contact Hydrophobic contact | ||||
| 11 | Amylin receptor (IAPPR) | 6ZIS | 8.04 | |
Target general information Gen name CALCR-RAMP1/RAMP2/RAMP3 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Complex of Calcitonin receptor and Receptor activity-modifying protein Protein family RAMP family Biochemical class NA Function Transports the calcitonin gene-related peptide type 1 receptor (CALCRL) to the plasma membrane. Acts as a receptor for calcitonin-gene-related peptide (CGRP) together with CALCRL. Related diseases Immunodeficiency 9 (IMD9) [MIM:612782]: An immune disorder characterized by recurrent infections, impaired activation and proliferative response of T-cells, decreased T-cell production of cytokines, and normal lymphocytes counts and serum immunoglobulin levels. In surviving patients ectodermal dysplasia with anhidrosis and non-progressive myopathy may be observed. {ECO:0000269|PubMed:16147976, ECO:0000269|PubMed:16582901}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Myopathy, tubular aggregate, 2 (TAM2) [MIM:615883]: A rare congenital myopathy characterized by regular arrays of membrane tubules on muscle biopsies without additional histopathological hallmarks. Tubular aggregates in muscle are structures of variable appearance consisting of an outer tubule containing either one or more microtubule-like structures or amorphous material. TAM2 patients have myopathy and pupillary abnormalities. {ECO:0000269|PubMed:24591628, ECO:0000269|PubMed:28058752}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01278 Interacts with Q16602; P21145; Q5J8X5; Q16617 EC number NA Uniprot keywords 3D-structure; Cell membrane; Disulfide bond; Membrane; Proteomics identification; Receptor; Reference proteome; Signal; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A Molecular weight (Da) 63832.6 Length 569 Aromaticity 0.12 Instability index 22.42 Isoelectric point 5.07 Charge (pH=7) -16.96 2D Binding mode Binding energy (Kcal/mol) -7.83 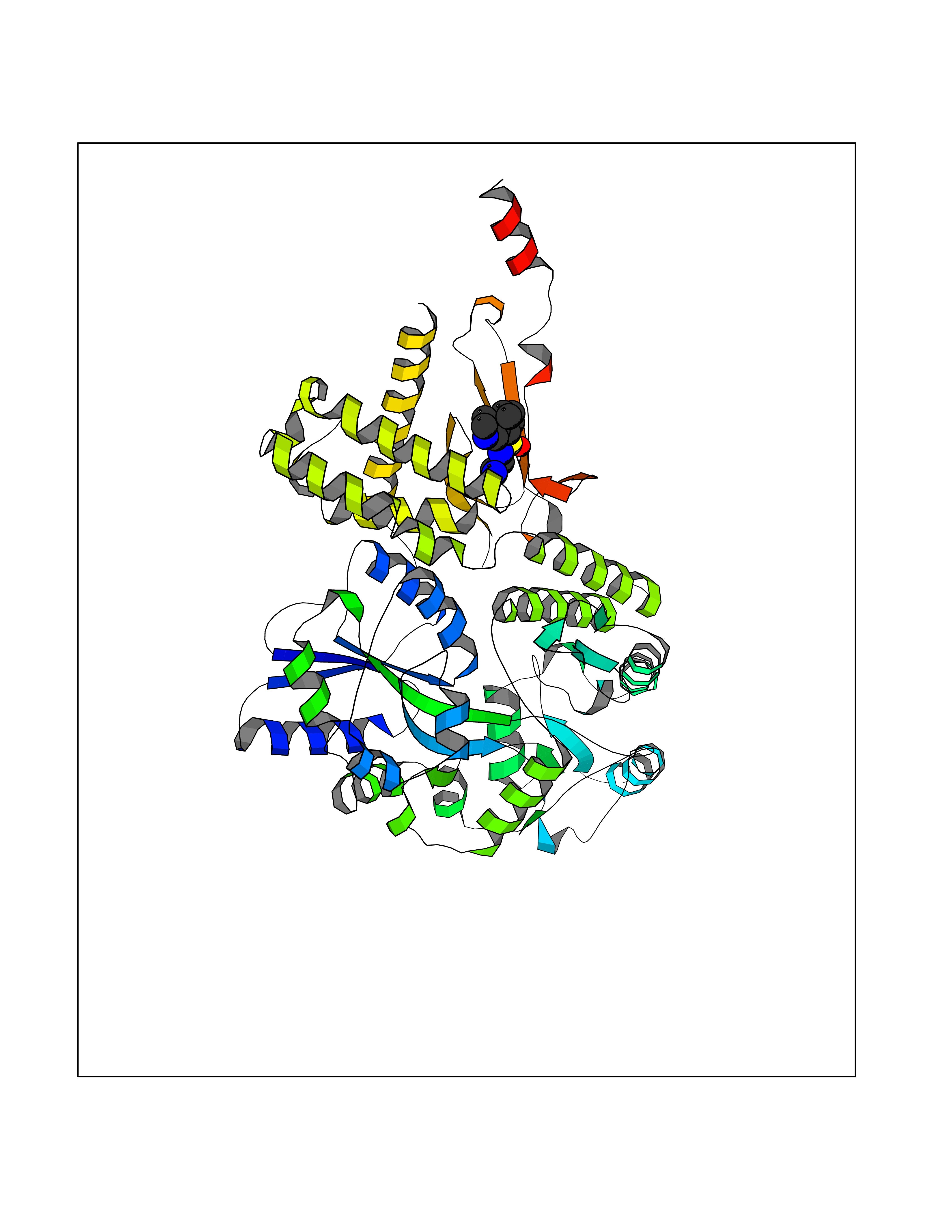 Molscript Map 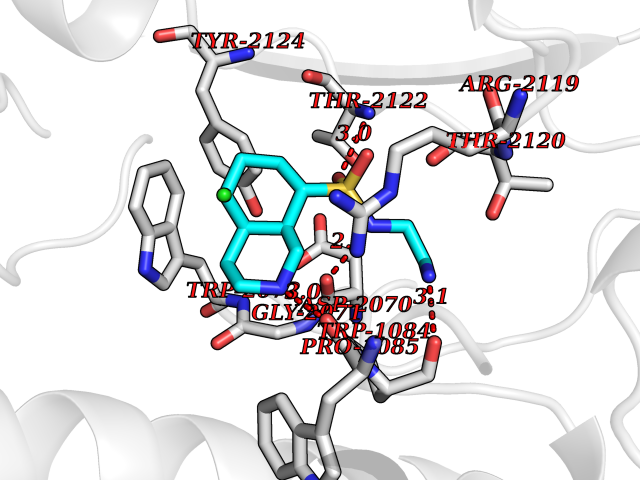 Pymol Map 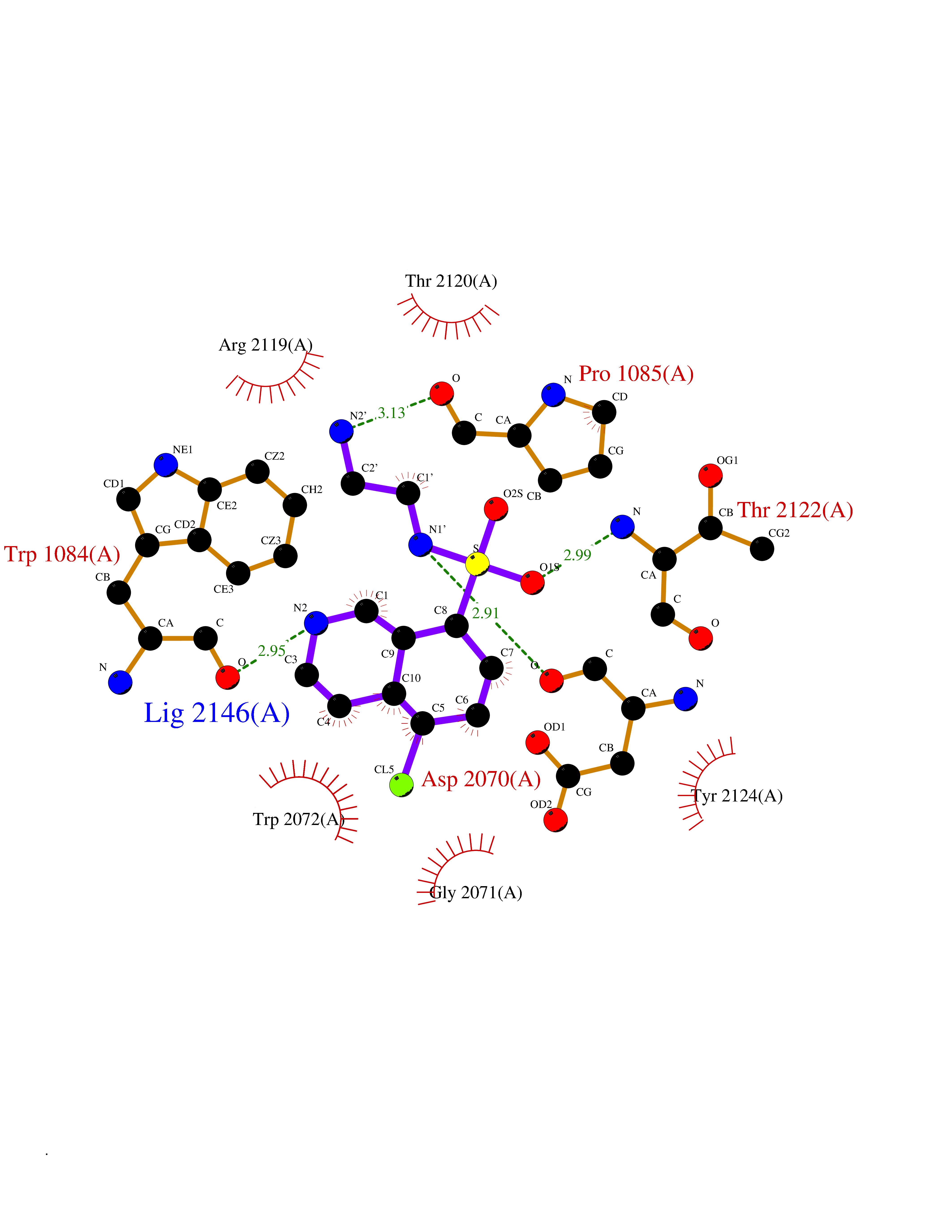 Ligplot Map 3D Binding mode Sequence SAKIEEGKLVIWINGDKGYNGLAEVGKKFEKDTGIKVTVEHPDKLEEKFPQVAATGDGPDIIFWAHDRFGGYAQSGLLAEITPDKAFQDKLYPFTWDAVRYNGKLIAYPIAVEALSLIYNKDLLPNPPKTWEEIPALDKELKAKGKSALMFNLQEPYFTWPLIAADGGYAFKYENGKYDIKDVGVDNAGAKAGLTFLVDLIKNKHMNADTDYSIAEAAFNKGETAMTINGPWAWSNIDTSKVNYGVTVLPTFKGQPSKPFVGVLSAGINAASPNKELAKEFLENYLLTDEGLEAVNKDKPLGAVALKSYEEELAKDPRIAATMENAQKGEIMPNIPQMSAFWYAVRTAVINAASGRQTVDEALKDAQTNAAAEFTTACQEANYGALLRELCLTQFQVDMEAVGETLWCDWGRTIRSYRELADCTWHMAEKLGCFWPNAEVDRFFLAVHGRYFRSCPISIQLGVTRNKIMTAQYECYQKIMQDPIQQGVYCQRTWDGWLCWNDVAAGTESMQLCPDYFQDFDPSEKVTKICDQDGNWFRHPASQRTWTDYTQCNVNTHEKVKTALNLFYL Hydrogen bonds contact Hydrophobic contact | ||||
| 12 | Adrenergic receptor beta-2 (ADRB2) | 2RH1 | 8.00 | |
Target general information Gen name ADRB2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Beta-2 adrenoreceptor; Beta-2 adrenoceptor; Beta-2 adrenergic receptor; B2AR; ADRB2R Protein family G-protein coupled receptor 1 family, Adrenergic receptor subfamily, ADRB2 sub-subfamily Biochemical class GPCR rhodopsin Function The beta-2-adrenergic receptor binds epinephrine with an approximately 30-fold greater affinity than it does norepinephrine. Beta-adrenergic receptors mediate the catecholamine-induced activation of adenylate cyclase through the action of G proteins. Related diseases Cortical dysplasia, complex, with other brain malformations 6 (CDCBM6) [MIM:615771]: A disorder of aberrant neuronal migration and disturbed axonal guidance. Affected individuals have microcephaly, ataxia, and severe delayed psychomotor development. Brain imaging shows variable malformations of cortical development, including white matter streaks, dysmorphic basal ganglia, corpus callosum abnormalities, brainstem and cerebellar hypoplasia, cortical dysplasia, polymicrogyria. {ECO:0000269|PubMed:23246003}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Skin creases, congenital symmetric circumferential, 1 (CSCSC1) [MIM:156610]: An autosomal dominant disease characterized by multiple, symmetric, circumferential rings of folded skin, affecting primarily the limbs. Affected individuals also exhibit intellectual disability, cleft palate, and dysmorphic features. {ECO:0000269|PubMed:26637975}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07543; DB01193; DB00866; DB01118; DB00182; DB01102; DB01274; DB01238; DB09204; DB06216; DB00335; DB01408; DB05590; DB09013; DB00195; DB00217; DB01295; DB00612; DB00901; DB08807; DB06726; DB08808; DB00248; DB00521; DB01136; DB04846; DB01407; DB00785; DB01151; DB11273; DB13345; DB00449; DB11278; DB00841; DB09273; DB06262; DB01363; DB01364; DB00668; DB01049; DB11587; DB01288; DB00983; DB05039; DB00221; DB01064; DB00598; DB01210; DB13139; DB01365; DB13624; DB01214; DB00264; DB01203; DB05849; DB04861; DB00368; DB00540; DB00334; DB09080; DB00816; DB01580; DB00715; DB01359; DB00925; DB00397; DB00960; DB01291; DB01366; DB01182; DB00571; DB06814; DB00852; DB01917; DB11124; DB00867; DB01001; DB00938; DB00489; DB03566; DB00127; DB00871; DB00373; DB00726; DB12248; DB09082; DB09185 Interacts with P30542; P07550; P32121; Q96B67; Q9UII2; Q9ULD4-2; Q9NSI6-4; Q5M9N0-2; A0AVK6; Q658K8; O00472; Q15910-2; Q15486; P61978; Q5TCQ9; Q99685; O14745; Q9NR21-5; Q8WVD3; Q9H0X6; Q13573; P12931; Q5T0J7-2; Q8N0U2 EC number NA Uniprot keywords 3D-structure; Cell membrane; Disulfide bond; Endosome; G-protein coupled receptor; Glycoprotein; Golgi apparatus; Hydroxylation; Lipoprotein; Membrane; Palmitate; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Transducer; Transmembrane; Transmembrane helix; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 32266.1 Length 282 Aromaticity 0.15 Instability index 36.1 Isoelectric point 8.02 Charge (pH=7) 2.1 2D Binding mode Binding energy (Kcal/mol) -7.82 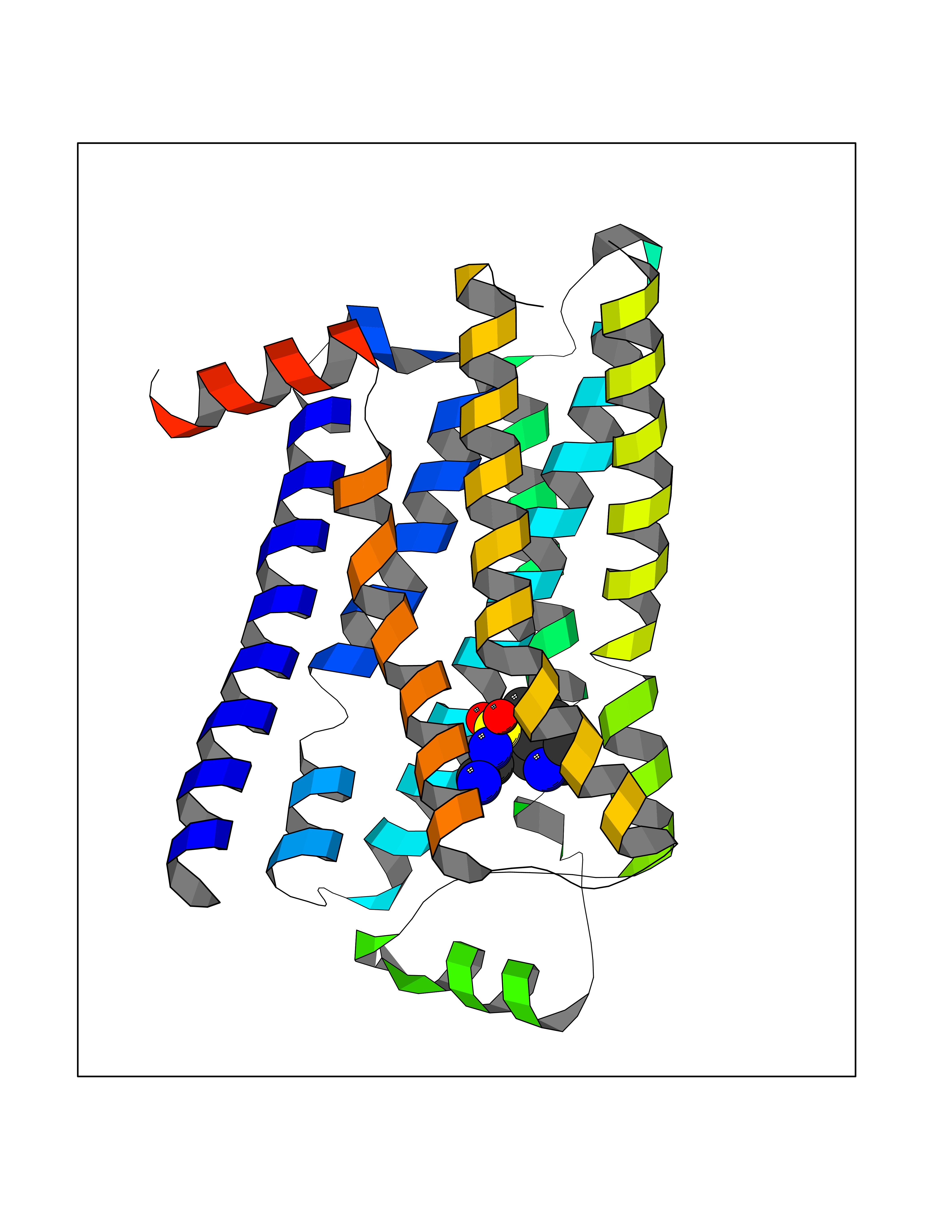 Molscript Map 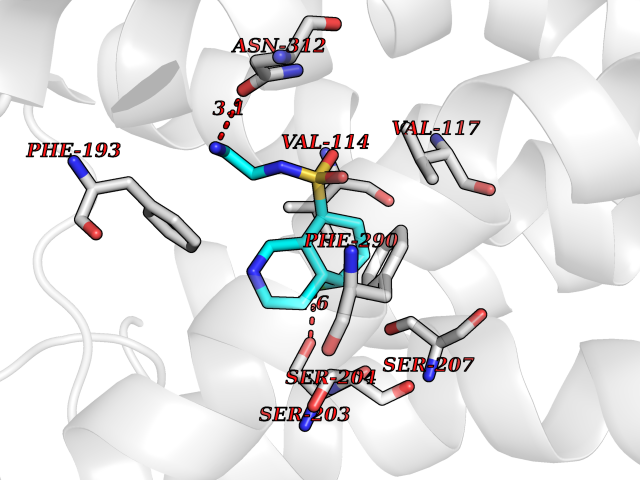 Pymol Map 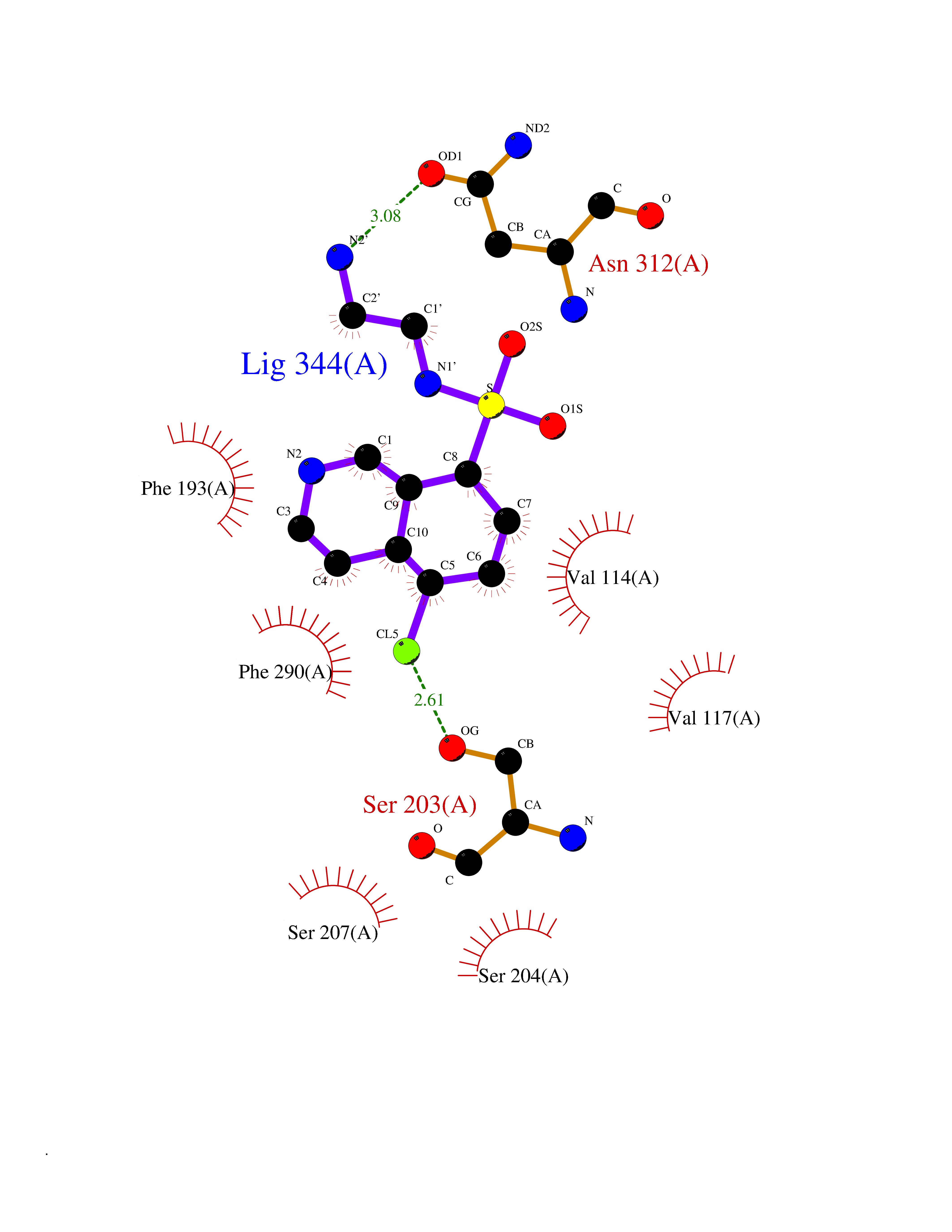 Ligplot Map 3D Binding mode Sequence DEVWVVGMGIVMSLIVLAIVFGNVLVITAIAKFERLQTVTNYFITSLACADLVMGLAVVPFGAAHILMKMWTFGNFWCEFWTSIDVLCVTASIETLCVIAVDRYFAITSPFKYQSLLTKNKARVIILMVWIVSGLTSFLPIQMHWYRATHQEAINCYAEETCCDFFTNQAYAIASSIVSFYVPLVIMVFVYSRVFQEAKRQLKFCLKEHKALKTLGIIMGTFTLCWLPFFIVNIVHVIQDNLIRKEVYILLNWIGYVNSGFNPLIYCRSPDFRIAFQELLCL Hydrogen bonds contact Hydrophobic contact | ||||
| 13 | Phenylethanolamine N-methyltransferase (PNMT) | 2G72 | 7.96 | |
Target general information Gen name PNMT Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms PNMTase; PENT; Noradrenaline N-methyltransferase Protein family Class I-like SAM-binding methyltransferase superfamily, NNMT/PNMT/TEMT family Biochemical class NA Function Converts noradrenaline to adrenaline. Related diseases A chromosomal aberration involving TRIM24/TIF1 is found in papillary thyroid carcinomas (PTCs). Translocation t(7;10)(q32;q11) with RET. The translocation generates the TRIM24/RET (PTC6) oncogene. {ECO:0000269|PubMed:10439047}. Drugs (DrugBank ID) DB08129; DB08128; DB07739; DB07798; DB07747; DB03468; DB08550; DB03824; DB04273; DB07906; DB07597; DB09571; DB00968; DB08631; DB01752; DB08654 Interacts with Q9P2G9-2; Q8TBB1 EC number EC 2.1.1.28 Uniprot keywords 3D-structure; Catecholamine biosynthesis; Direct protein sequencing; Methyltransferase; Phosphoprotein; Proteomics identification; Reference proteome; S-adenosyl-L-methionine; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 29198.9 Length 264 Aromaticity 0.09 Instability index 54.33 Isoelectric point 5.91 Charge (pH=7) -3.69 2D Binding mode Binding energy (Kcal/mol) -8.08  Molscript Map 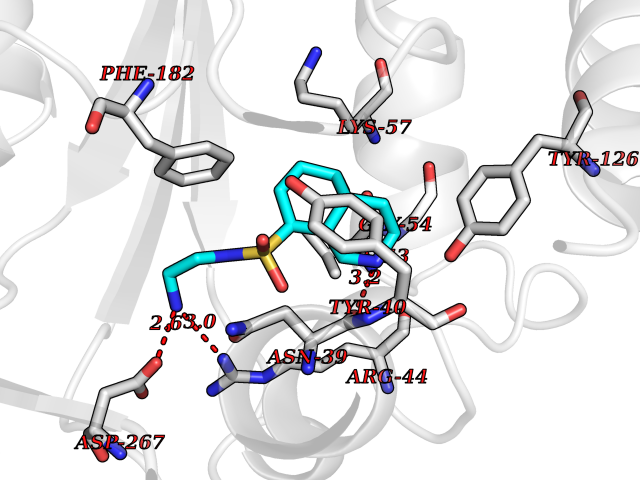 Pymol Map 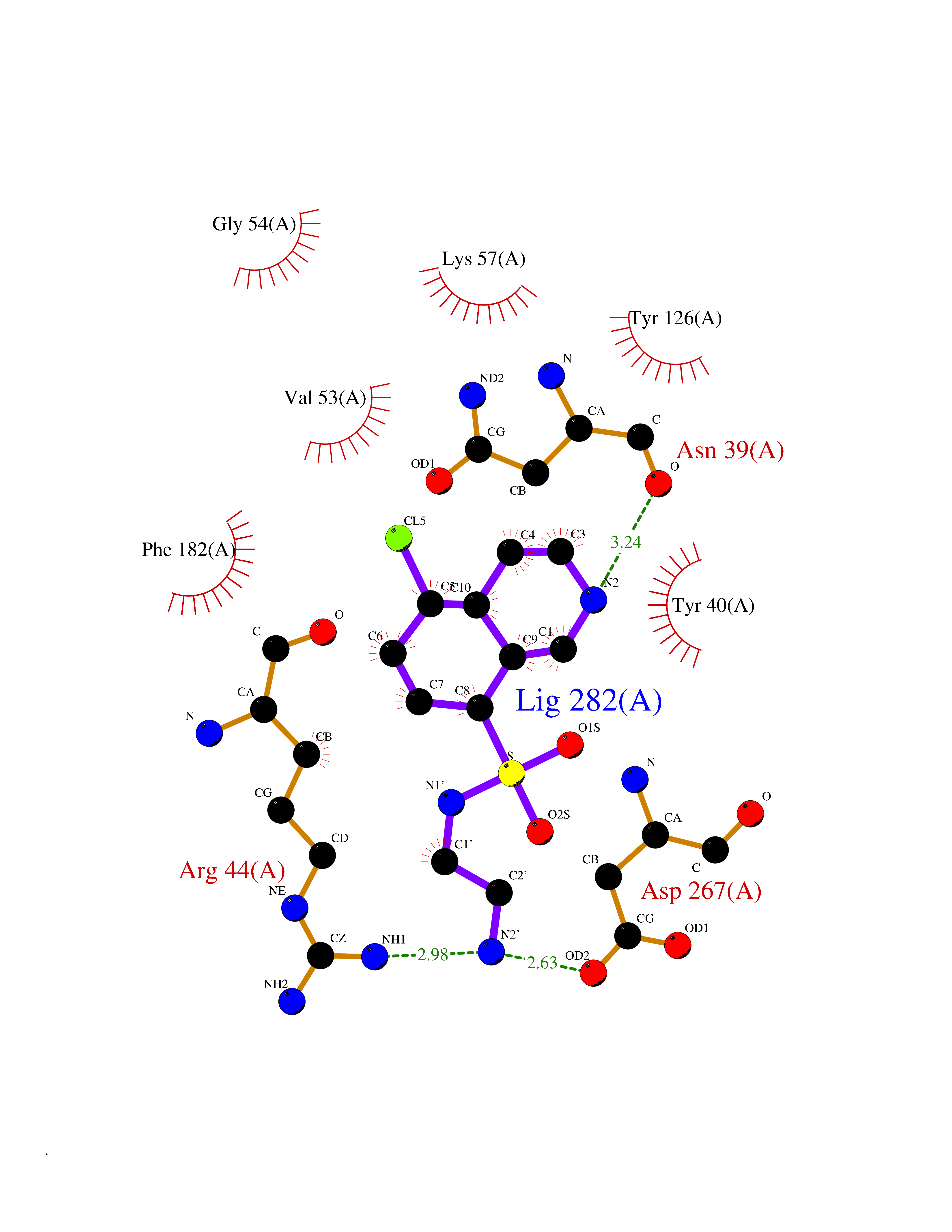 Ligplot Map 3D Binding mode Sequence APGQAAVASAYQRFEPRAYLRNNYAPPRGDLCNPNGVGPWKLRCLAQTFATGEVSGRTLIDIGSGPTVYQLLSACSHFEDITMTDFLEVNRQELGRWLQEEPGAFNWSMYSQHACLIEGKGECWQDKERQLRARVKRVLPIDVHQPQPLGAGSPAPLPADALVSAFCLEAVSPDLASFQRALDHITTLLRPGGHLLLIGALEESWYLAGEARLTVVPVSEEEVREALVRSGYKVRDLRTYIMPAHLQTGVDDVKGVFFAWAQKV Hydrogen bonds contact Hydrophobic contact | ||||
| 14 | Dopamine beta-hydroxylase | 4ZEL | 7.95 | |
Target general information Gen name DBH Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Copper type II ascorbate-dependent monooxygenase family Biochemical class Oxidoreductase Function Catalytic activity.Copper ion binding.Dopamine beta-monooxygenase activity.L-ascorbic acid binding. Related diseases Orthostatic hypotension 1 (ORTHYP1) [MIM:223360]: A form of orthostatic hypotension due to congenital dopamine beta-hydroxylase deficiency. Orthostatic hypotension, also known as postural hypotension, is a finding defined as a 20-mm Hg decrease in systolic pressure or a 10-mm Hg decrease in diastolic pressure occurring 3 minutes after a person has risen from supine to standing. Symptoms include dizziness, blurred vision, and sometimes syncope. ORTHYP1 is an autosomal recessive condition apparent from infancy or early childhood and characterized by low plasma and urinary levels of norepinephrine and epinephrine, and episodic hypoglycemia. {ECO:0000269|PubMed:11857564}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00126; DB06774; DB09130; DB05394; DB00822; DB00988; DB00968; DB00550 Interacts with P00352; P63010-2; Q04656; Q8WUW1; Q9UNS2; Q71DI3; P61978; Q9Y2M5; Q92876; P08727; Q14693; P0DPK4; Q6GQQ9-2; P27986-2; Q9ULX5; Q96D59; Q8N6K7-2; Q9GZS3; Q8IUW3; Q86WT6-2 EC number 1.14.17.1 Uniprot keywords 3D-structure; Catecholamine biosynthesis; Copper; Cytoplasmic vesicle; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; Membrane; Metal-binding; Monooxygenase; Oxidoreductase; Proteomics identification; Reference proteome; Secreted; Signal-anchor; Transmembrane; Transmembrane helix; Vitamin C Protein physicochemical properties Chain ID A,B Molecular weight (Da) 123694 Length 1094 Aromaticity 0.1 Instability index 51.85 Isoelectric point 5.84 Charge (pH=7) -24.5 2D Binding mode Binding energy (Kcal/mol) -7.72 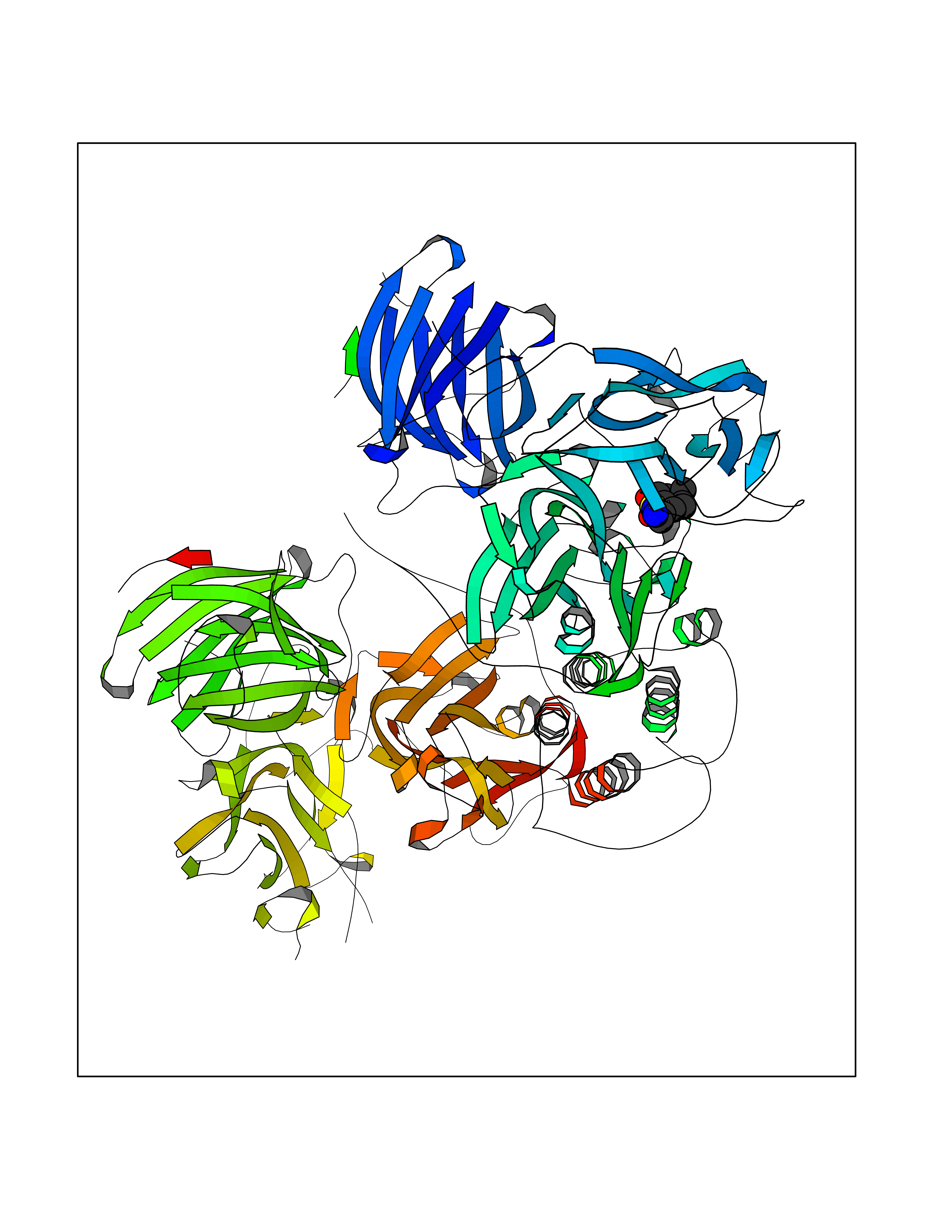 Molscript Map 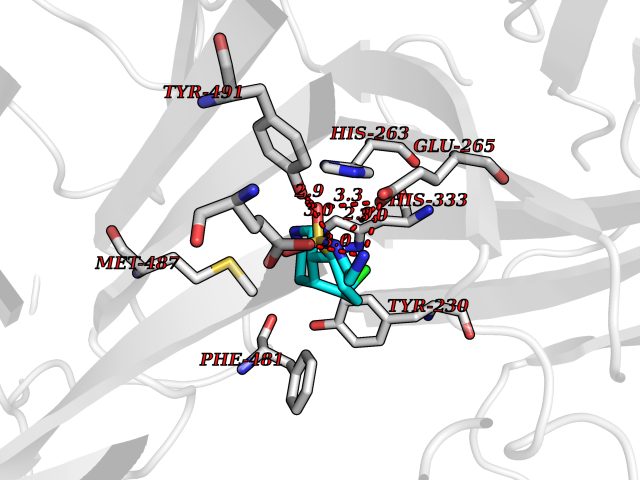 Pymol Map 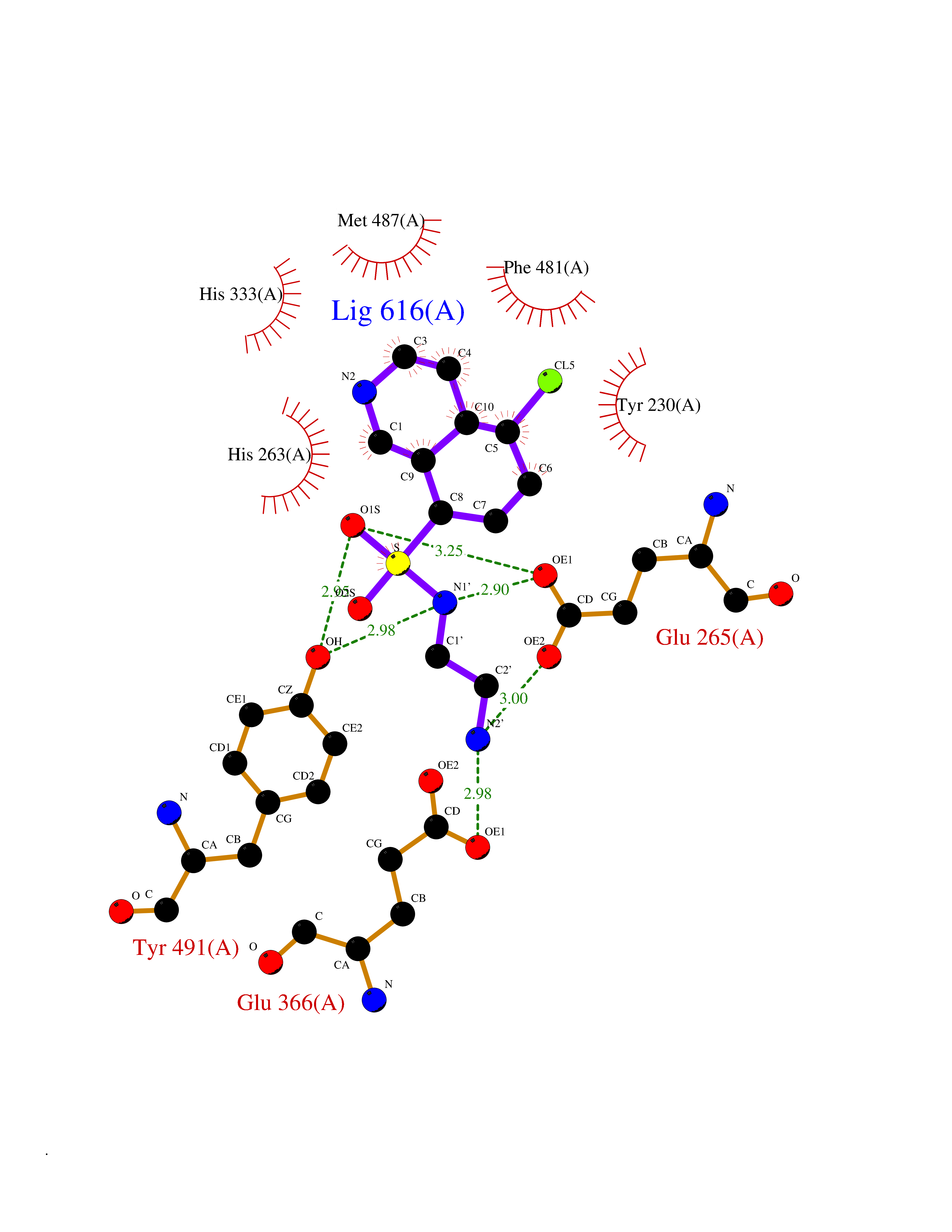 Ligplot Map 3D Binding mode Sequence PLPYHIPLDPEGSLELSWNVSYTQEAIHFQLLVRRLKAGVLFGMSDRGELENADLVVLWTDGDAYFADAWSDQKGQIHLDPQQDYQLLQVQRTPEGLTLLFKRPFGTCDPKDYLIEDGTVHLVYGILEEPFRSLEAINGSGLQMGLQRVQLLKPNIPEPELPSDACTMEVQAPNIQIPSQETTYWCYIKELPKGFSRHHIIKYEPIVTKGNEALVHHMEVFQCAPEMDSVPHFSGPCDSKMKPDRLNYCRHVLAAWALGAKAFYYPEEAGLAFGGPGSSRYLRLEVHYHNPLVIEGRNDSSGIRLYYTAKLRRFNAGIMELGLVYTPVMAIPPRETAFILTGYCTDKCTQLALPPSGIHIFASQLHTHLTGRKVVTVLVRDGREWEIVNQDNHYSPHFQEIRMLKKVVSVHPGDVLITSCTYNTEDRELATVGGFGILEEMCVNYVHYYPQTQLELCKSAVDAGFLQKYFHLINRFNNEDVCTCPQASVSQQFTSVPWNSFNRDVLKALYSFAPISMHCNKSSAVRFQGEWNLQPLPKVISTLEEPTVVSPLPYHIPLDPEGSLELSWNVSYTQEAIHFQLLVRRLKAGVLFGMSDRGELENADLVVLAYFADAWSDQKGQIHLDPQQDYQLLQVQRTPEGLTLLFKRPFGTCDPKDYLIEDGTVHLVYGILEEPFRSLEAINGSGLQMGLQRVQLLKPNIPEPELPSDACTMEVQAPNIQIPSQETTYWCYIKELPKGFSRHHIIKYEPIVTKGNEALVHHMEVFQCAPEVPHFSGPCDSKMLNYCRHVLAAWALGAKAFYYPEEAGLAFGGPGSSRYLRLEVHYHNPLVIEGRNDSSGIRLYYTAKLRRFNAGIMELGLVYTPVMAIPPRETAFILTGYCTDKCTQLALPPSGIHIFASQLHTHLTGRKVVTVLVRDGREWEIVNQDNHYSPHFQEIRMLKKVVSVHPGDVLITSCTYNTEDRELATVGGFGILEEMCVNYVHYYPQTQLELCKSAVDAGFLQKYFHLINRFNNEDVCTCPQASVSQQFTSVPWNSFNRDVLKALYSFAPISMHCNKSSAVRFQGEWNLQPLPKVISTLEEPTPQCVVSIGG Hydrogen bonds contact Hydrophobic contact | ||||
| 15 | Bifunctional aminoacyl-tRNA synthetase (EPRS) | 4HVC | 7.94 | |
Target general information Gen name EPRS Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms QPRS; QARS; ProlinetRNA ligase; PIG32; GlutamatylprolyltRNA synthetase; Glutamatyl-prolyl-tRNA synthetase; GluRS; EPRS; Cell proliferationinducing gene 32 protein; Cell proliferation-inducing gene 32 Protein family Class-I aminoacyl-tRNA synthetase family, Glutamate--tRNA ligase type 2 subfamily; Class-II aminoacyl-tRNA synthetase family Biochemical class Carbon-oxygen ligase Function The phosphorylation of EPRS, induced by interferon-gamma, dissociates the protein from the aminoacyl-tRNA synthetase multienzyme complex and recruits it to the GAIT complex that binds to stem loop-containing GAIT elements in the 3'-UTR of diverse inflammatory mRNAs (such as ceruplasmin), suppressing their translation. Interferon-gamma can therefore redirect, in specific cells, the EPRS function from protein synthesis to translation inhibition. Also functions as an effector of the mTORC1 signaling pathway by promoting, through SLC27A1, the uptake of long-chain fatty acid by adipocytes. Thereby, it also plays a role in fat metabolism and more indirectly influences lifespan. Multifunctional protein which is primarily part of the aminoacyl-tRNA synthetase multienzyme complex, also know as multisynthetase complex, that catalyzes the attachment of the cognate amino acid to the corresponding tRNA in a two-step reaction: the amino acid is first activated by ATP to form a covalent intermediate with AMP and is then transferred to the acceptor end of the cognate tRNA. Related diseases Leukodystrophy, hypomyelinating, 15 (HLD15) [MIM:617951]: An autosomal recessive disorder characterized by hypomyelinating leukodystrophy with thinning of the corpus callosum. Clinical features include motor and cognitive impairment appearing in the first or second decade of life, dystonia, ataxia, spasticity, and dysphagia. Most patients develop severe optic atrophy, and some have hearing loss. {ECO:0000269|PubMed:29576217}. The disease may be caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02684; DB02510; DB03376; DB00142; DB00172 Interacts with P07814; Q8IWL3; P41252; Q15046; P42695; P54136; O60506 EC number NA Uniprot keywords 3D-structure; Acetylation; Aminoacyl-tRNA synthetase; ATP-binding; Cytoplasm; Disease variant; Leukodystrophy; Ligase; Membrane; Metal-binding; Methylation; Multifunctional enzyme; Neurodegeneration; Nucleotide-binding; Phosphoprotein; Protein biosynthesis; Proteomics identification; Reference proteome; Repeat; RNA-binding; Translation regulation; Zinc Protein physicochemical properties Chain ID A,B Molecular weight (Da) 55039.7 Length 483 Aromaticity 0.1 Instability index 39.15 Isoelectric point 5.92 Charge (pH=7) -6.4 2D Binding mode Binding energy (Kcal/mol) -7.55 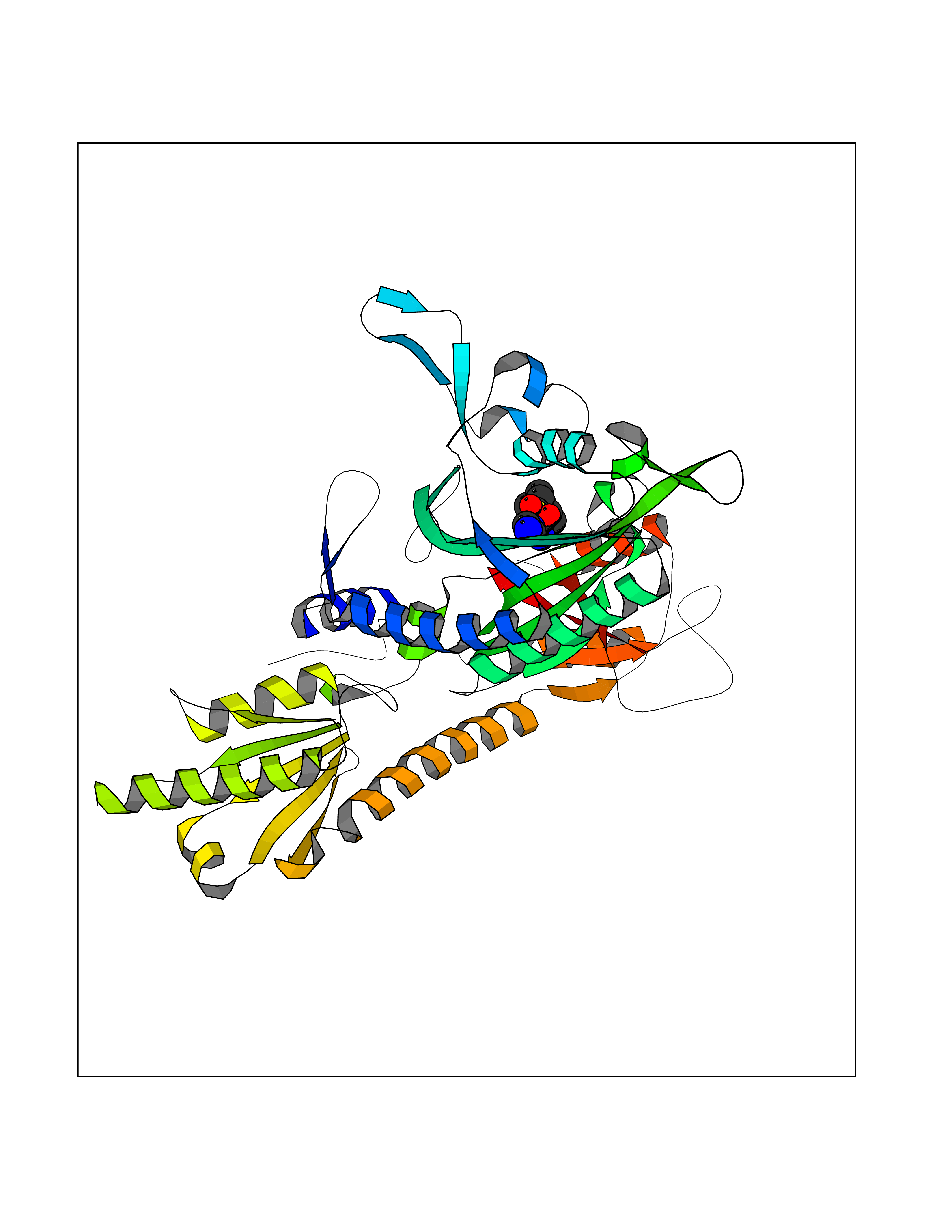 Molscript Map 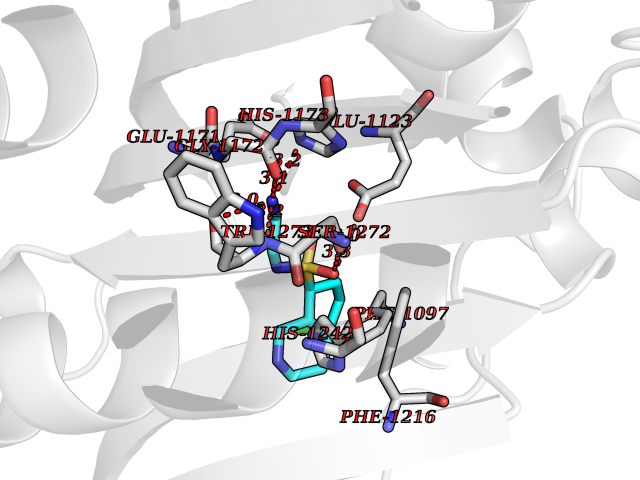 Pymol Map 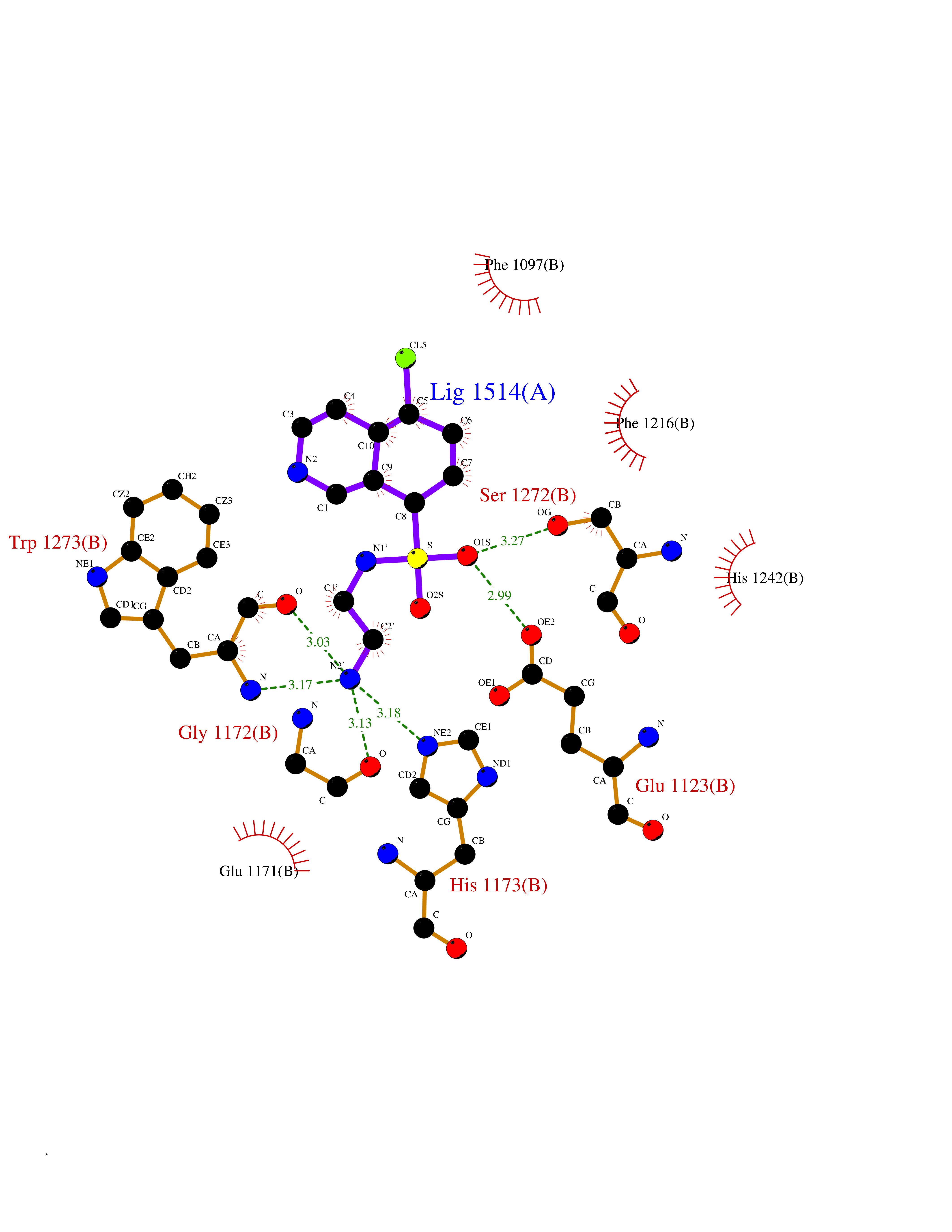 Ligplot Map 3D Binding mode Sequence GLEAKKEENLADWYSQVITKSEMIEYHDISGCYILRPWAYAIWEAIKDFFDAEIKKLGVENCYFPMFVSQSALEKEKTHVADFAPEVAWVTRSGKTELAEPIAIRPTSETVMYPAYAKWVQSHRDLPIKLNQWCNVVRWEFKHPQPFLRTREFLWQEGHSAFATMEEAAEEVLQILDLYAQVYEELLAIPVVKGRKTEKEKFAGGDYTTTIEAFISASGRAIQGGTSHHLGQNFSKMFEIVFEDPKIPGEKQFAYQNSWGLTTRTIGVMTMVHGDNMGLVLPPRVACVQVVIIPCGISEEDKEALIAKCNDYRRRLLSVNIRVRADLRDNYSPGWKFNHWELKGVPIRLEVGPRDMKSCQFVAVRRDTGEKLTVAENEAETKLQAILEDIQVTLFTRASEDLKTHMVVANTMEDFQKILDSGKIVQIPFCGEIDCEDWIKKTTAMGAKSLCIPFKPLCELQPGAKCVCGKNPAKYYTLFGRSY Hydrogen bonds contact Hydrophobic contact | ||||
| 16 | Serine/threonine PP1-alpha (PPP1CA) | 3E7B | 7.93 | |
Target general information Gen name PPP1CA Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Serine/threonine-protein phosphatase PP1-alpha catalytic subunit; Protein phosphatase 1alpha; PPP1A; PP-1A Protein family PPP phosphatase family, PP-1 subfamily Biochemical class Phosphoric monoester hydrolase Function Protein phosphatase 1 (PP1) is essential for cell division, and participates in the regulation of glycogen metabolism, muscle contractility and protein synthesis. Involved in regulation of ionic conductances and long-term synaptic plasticity. May play an important role in dephosphorylating substrates such as the postsynaptic density-associated Ca(2+)/calmodulin dependent protein kinase II. Component of the PTW/PP1 phosphatase complex, which plays a role in the control of chromatin structure and cell cycle progression during the transition from mitosis into interphase. Regulates NEK2 function in terms of kinase activity and centrosome number and splitting, both in the presence and absence of radiation-induced DNA damage. Regulator of neural tube and optic fissure closure, and enteric neural crest cell (ENCCs) migration during development. In balance with CSNK1D and CSNK1E, determines the circadian period length, through the regulation of the speed and rhythmicity of PER1 and PER2 phosphorylation. May dephosphorylate CSNK1D and CSNK1E. Dephosphorylates the 'Ser-418' residue of FOXP3 in regulatory T-cells (Treg) from patients with rheumatoid arthritis, thereby inactivating FOXP3 and rendering Treg cells functionally defective. Dephosphorylates CENPA. Dephosphorylates the 'Ser-139' residue of ATG16L1 causing dissociation of ATG12-ATG5-ATG16L1 complex, thereby inhibiting autophagy. Protein phosphatase that associates with over 200 regulatory proteins to form highly specific holoenzymes which dephosphorylate hundreds of biological targets. Related diseases A chromosomal aberration involving FHIT has been found in a lymphoblastoid cell line established from a family with renal cell carcinoma and thyroid carcinoma. Translocation t(3;8)(p14.2;q24.1) with RNF139. Although the 3p14.2 breakpoint has been shown to interrupt FHIT in its 5-prime non-coding region, it is unlikely that FHIT is causally related to renal or other malignancies. {ECO:0000269|PubMed:15007172}.; DISEASE: Associated with digestive tract cancers. Numerous tumor types are found to have aberrant forms of FHIT protein due to deletions in a coding region of chromosome 3p14.2 including the fragile site locus FRA3B. {ECO:0000269|PubMed:15007172}. Drugs (DrugBank ID) DB02506 Interacts with Q6ZMQ8; P31749; O14727; P05067; O15169; P38398; O95400; Q99459; P12830; Q8TEP8; Q9NX63; Q6PJW8; Q96S65; Q9H175; Q92796; P05198; P55199; Q9BZS1; P42858; Q8NI77; Q8NG31; Q5S007; O00566; Q9UPR0; Q96QC0; Q96KQ4; Q8WUF5; O75807; Q5SWA1; Q96T49; Q6NYC8; P41236; Q5T8A7; Q86WC6; Q6NXS1; O14990; O75864; Q86XI6; Q9UQK1; O95685; Q15435; Q12972; Q12972-1; Q12972-2; Q96SB3; P60484; P06400; Q5UIP0; Q14684; P04271; Q7Z5V6; A8K8P3; Q562F6; Q9H788; Q8TEC5; P63208; Q7Z699; P43405-2; Q9HCH5; Q14C87; Q5JTV8; Q05BL1; Q13625; Q4KMQ1; Q4KMQ1-2; Q8TEL6; P49815; P55072; Q9Y2W2; Q9H4A3; P16989; P49750; Q9HBF4; Q7Z3T8; O95405; O08785; P36313; K9N4V7; Q76TK5; O35867; O35274 EC number EC 3.1.3.16 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Carbohydrate metabolism; Cell cycle; Cell division; Cytoplasm; Direct protein sequencing; Glycogen metabolism; Host-virus interaction; Hydrolase; Manganese; Metal-binding; Nucleus; Phosphoprotein; Protein phosphatase; Proteomics identification; Reference proteome Protein physicochemical properties Chain ID A Molecular weight (Da) 33626.3 Length 294 Aromaticity 0.11 Instability index 44.81 Isoelectric point 5.16 Charge (pH=7) -10.09 2D Binding mode Binding energy (Kcal/mol) -7.27 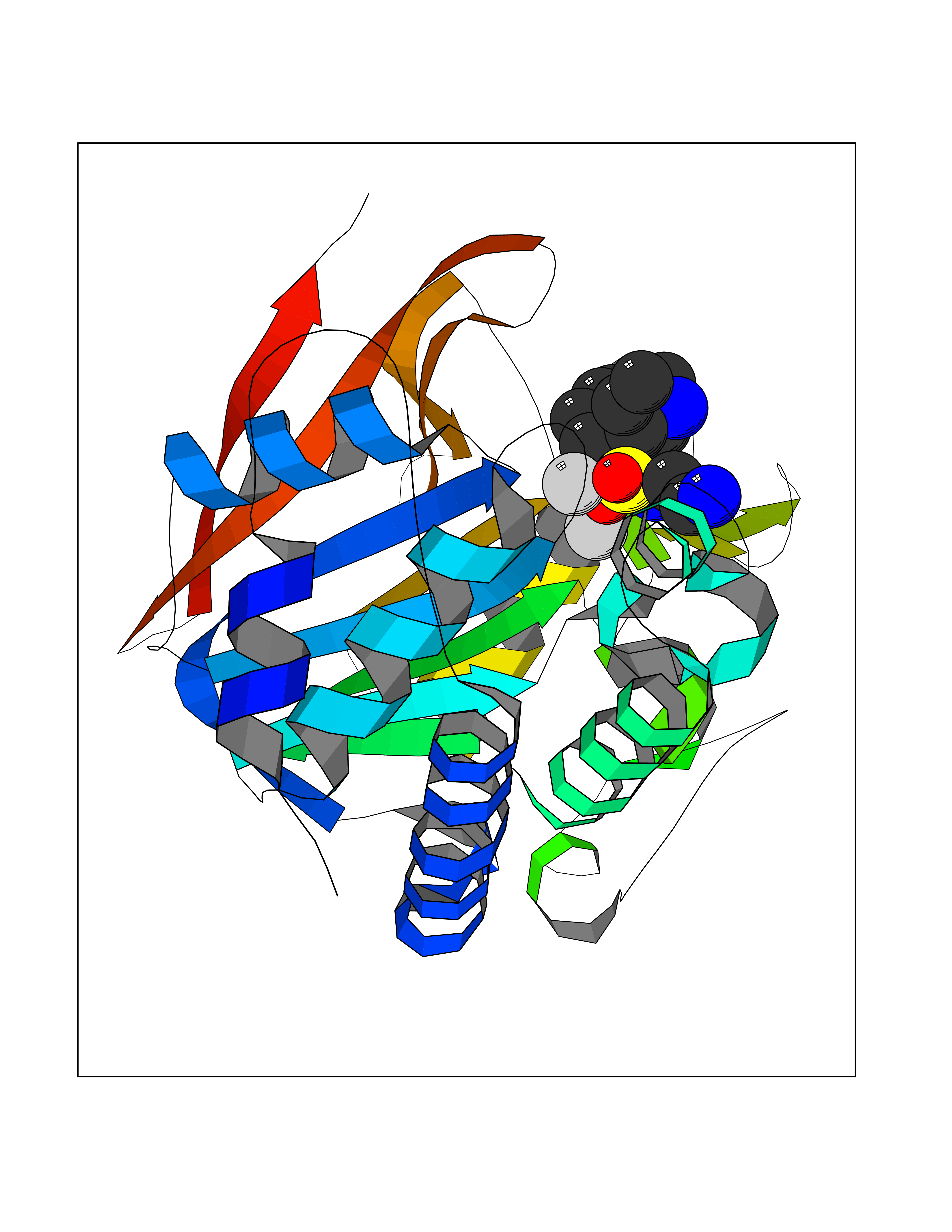 Molscript Map 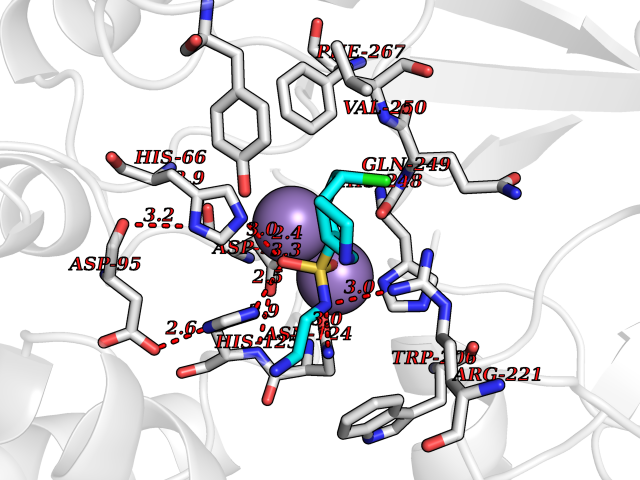 Pymol Map 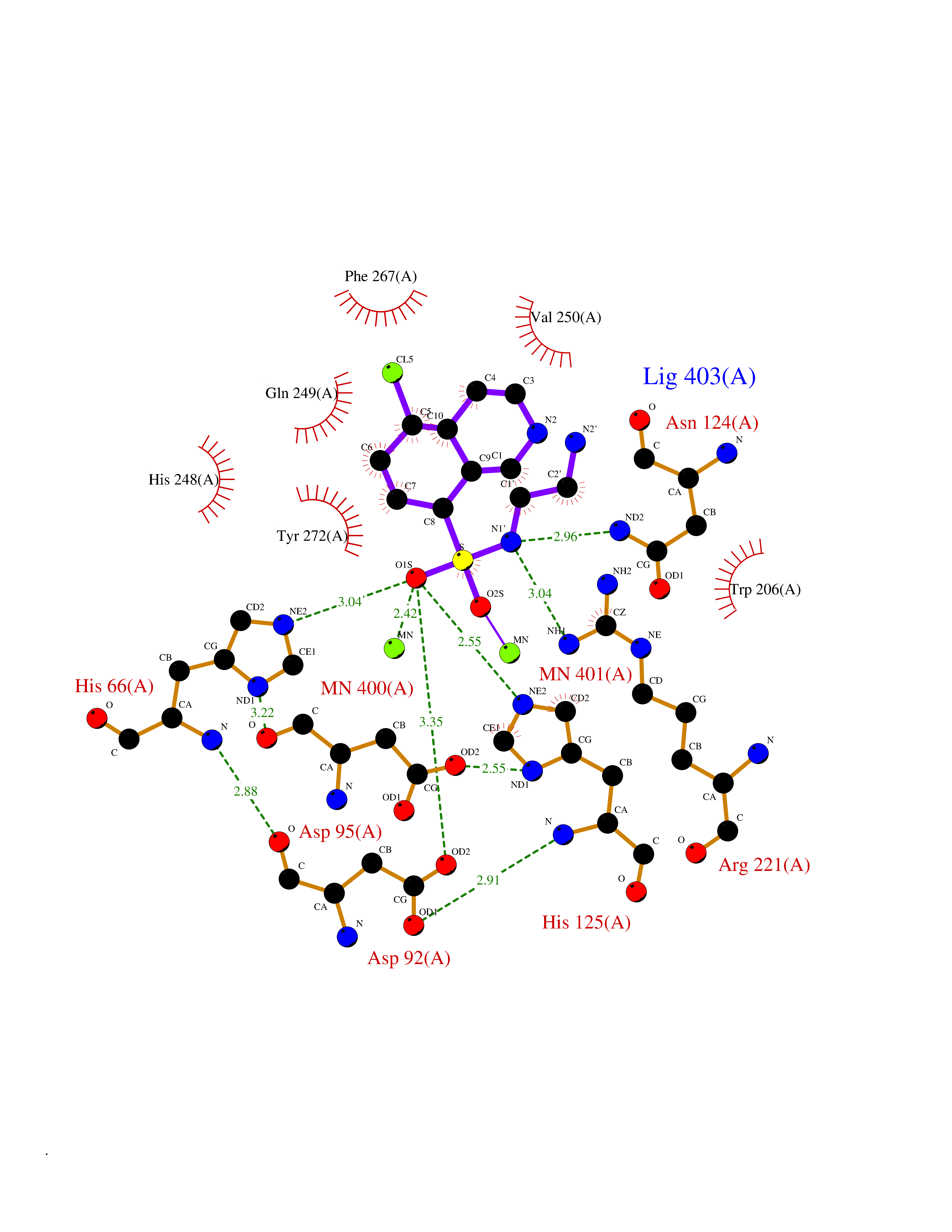 Ligplot Map 3D Binding mode Sequence SLNLDSIIGRLLEVQGSRPGKNVQLTENEIRGLCLKSREIFLSQPILLELEAPLKICGDIHGQYYDLLRLFEYGGFPPESNYLFLGDYVDRGKQSLETICLLLAYKIKYPENFFLLRGNHECASINRIYGFYDECKRRYNIKLWKTFTDCFNCLPIAAIVDEKIFCCHGGLSPDLQSMEQIRRIMRPTDVPDQGLLCDLLWSDPDKDVQGWGENDRGVSFTFGAEVVAKFLHKHDLDLICRAHQVVEDGYEFFAKRQLVTLFSAPNYCGEFDNAGAMMSVDETLMCSFQILKPA Hydrogen bonds contact Hydrophobic contact | ||||
| 17 | Aspartyl aminopeptidase (DNPEP) | 4DYO | 7.93 | |
Target general information Gen name DNPEP Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms DAP; ASPEP Protein family Peptidase M18 family Biochemical class Peptidase Function Likely to play an important role in intracellular protein and peptide metabolism. Aminopeptidase with specificity towards an acidic amino acid at the N-terminus. Related diseases Cohen-Gibson syndrome (COGIS) [MIM:617561]: An autosomal dominant overgrowth disorder characterized by accelerated osseous maturation, advanced bone age, skeletal abnormalities including flaring of the metaphyses of the long bones, large hands with long fingers and camptodactyly, scoliosis, cervical spine anomalies, dysmorphic facial features, and variable intellectual disability. {ECO:0000269|PubMed:25787343, ECO:0000269|PubMed:27193220, ECO:0000269|PubMed:27868325, ECO:0000269|PubMed:28229514, ECO:0000269|PubMed:28475857}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00142 Interacts with Q9ULA0; Q8TBB1; Q00013; Q9UPN6 EC number EC 3.4.11.21 Uniprot keywords 3D-structure; Acetylation; Alternative initiation; Aminopeptidase; Cytoplasm; Hydrolase; Metal-binding; Metalloprotease; Phosphoprotein; Protease; Proteomics identification; Reference proteome; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 50574.6 Length 457 Aromaticity 0.07 Instability index 55.73 Isoelectric point 7.73 Charge (pH=7) 2.3 2D Binding mode Binding energy (Kcal/mol) -7.35 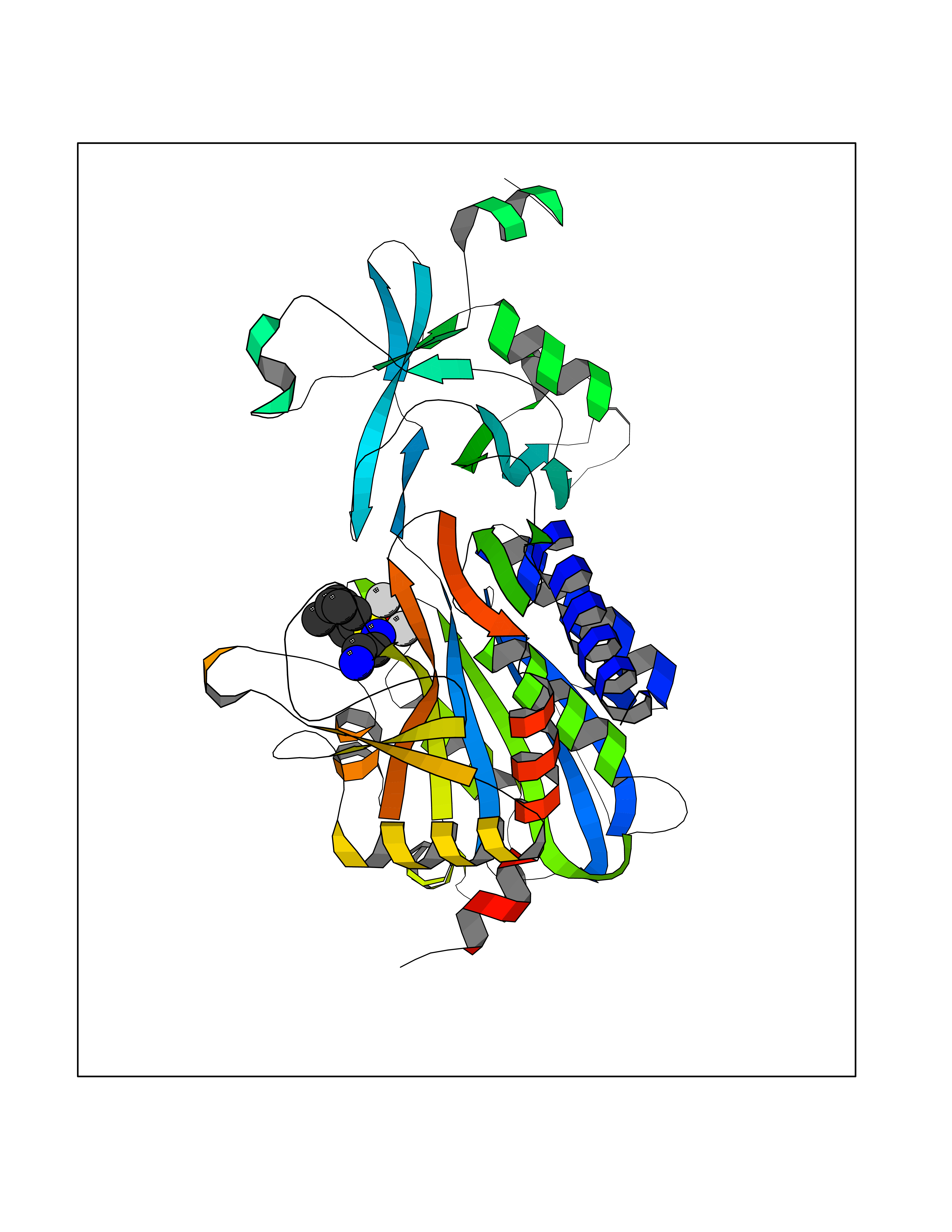 Molscript Map 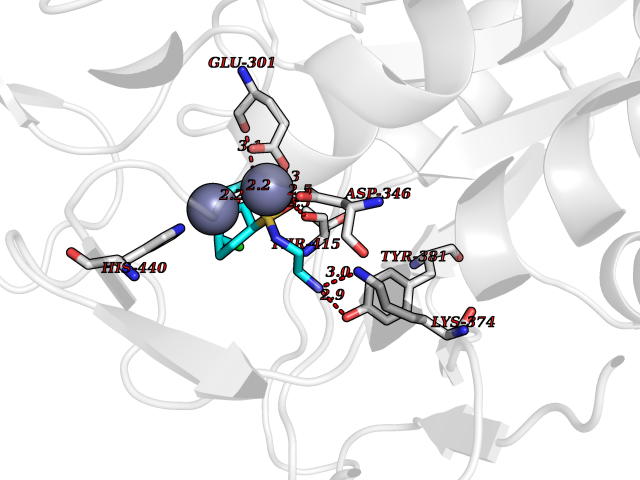 Pymol Map 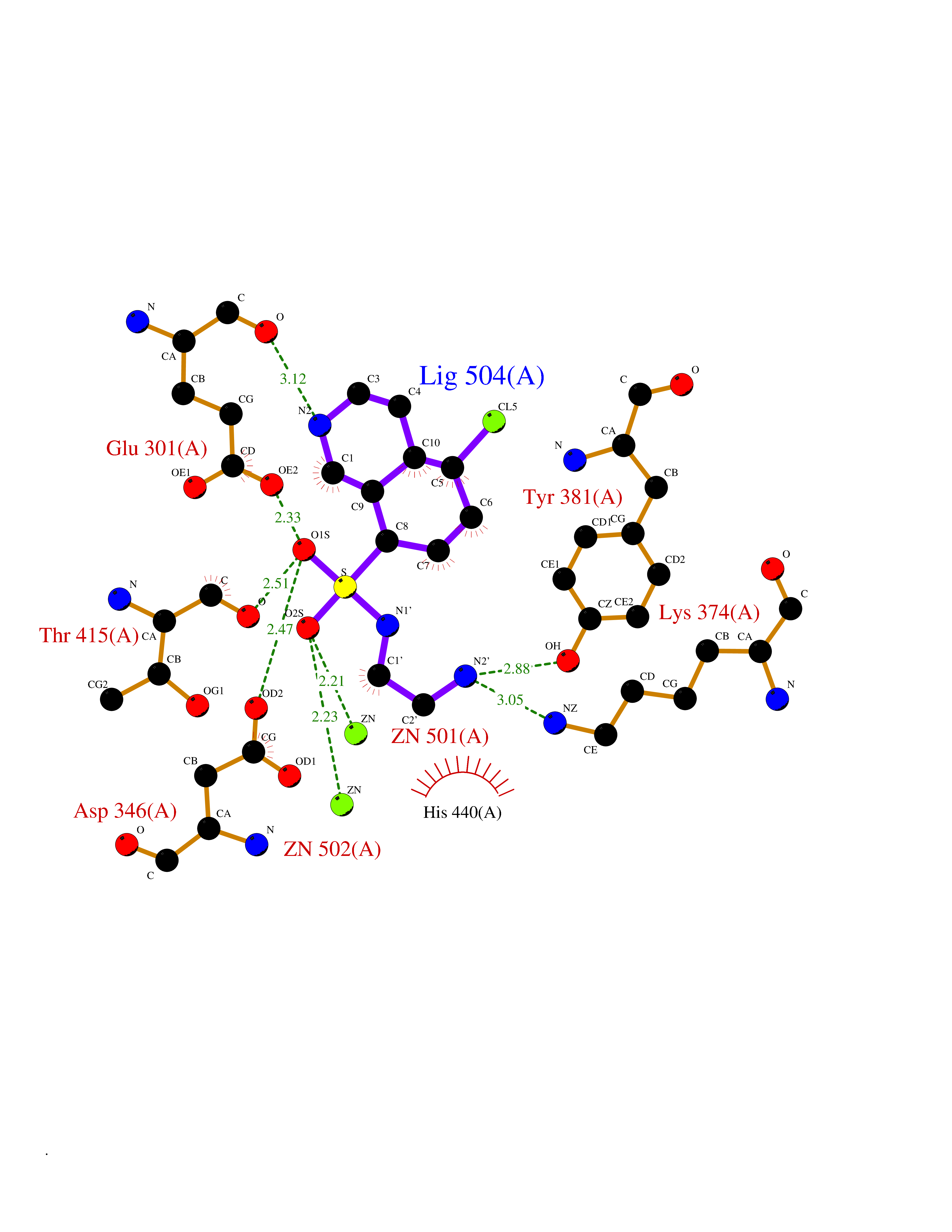 Ligplot Map 3D Binding mode Sequence GKARKEAVQTAAKELLKFVNRSPSPFHAVAECRNRLLQAGFSELKETEKWNIKPESKYFMTRNSSTIIAFAVGGQYVPGNGFSLIGAHTDSPCLRVKRRSRRSQVGFQQVGVETYGGGIWSTWFDRDLTLAGRVIVKCPTSGRLEQQLVHVERPILRIPHLAIHLQRNINENFGPNTEMHLVPILATAIQEELEKGTERHHSVLMSLLCAHLGLSPKDIVEMELCLADTQPAVLGGAYDEFIFAPRLDNLHSCFCALQALIDSCAGPGSLATEPHVRMVTLYDNEEVGSESAQGAQSLLTELVLRRISASCQHPTAFEEAIPKSFMISADMAHAVHPNYLDKHEENHRPLFHKGPVIKVNSKQRYASNAVSEALIREVANKVKVPLQDLMVRNDTPCGTTIGPILASRLGLRVLDLGSPQLAMHSIREMACTTGVLQTLTLFKGFFELFPSLAENLY Hydrogen bonds contact Hydrophobic contact | ||||
| 18 | Histone acetyltransferase p300 (EP300) | 5LKX | 7.89 | |
Target general information Gen name EP300 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms p300 HAT; Protein propionyltransferase p300; P300; Histone crotonyltransferase p300; Histone butyryltransferase p300; E1Aassociated protein p300; E1A-associated protein p300 Protein family NA Biochemical class Acyltransferase Function Acetylates all four core histones in nucleosomes. Histone acetylation gives an epigenetic tag for transcriptional activation. Mediates cAMP-gene regulation by binding specifically to phosphorylated CREB protein. Mediates acetylation of histone H3 at 'Lys-122' (H3K122ac), a modification that localizes at the surface of the histone octamer and stimulates transcription, possibly by promoting nucleosome instability. Mediates acetylation of histone H3 at 'Lys-27' (H3K27ac). Also functions as acetyltransferase for non-histone targets, such as ALX1, HDAC1, PRMT1 or SIRT2. Acetylates 'Lys-131' of ALX1 and acts as its coactivator. Acetylates SIRT2 and is proposed to indirectly increase the transcriptional activity of TP53 through acetylation and subsequent attenuation of SIRT2 deacetylase function. Acetylates HDAC1 leading to its inactivation and modulation of transcription. Acts as a TFAP2A-mediated transcriptional coactivator in presence of CITED2. Plays a role as a coactivator of NEUROD1-dependent transcription of the secretin and p21 genes and controls terminal differentiation of cells in the intestinal epithelium. Promotes cardiac myocyte enlargement. Can also mediate transcriptional repression. Acetylates FOXO1 and enhances its transcriptional activity. Acetylates BCL6 wich disrupts its ability to recruit histone deacetylases and hinders its transcriptional repressor activity. Participates in CLOCK or NPAS2-regulated rhythmic gene transcription; exhibits a circadian association with CLOCK or NPAS2, correlating with increase in PER1/2 mRNA and histone H3 acetylation on the PER1/2 promoter. Acetylates MTA1 at 'Lys-626' which is essential for its transcriptional coactivator activity. Acetylates XBP1 isoform 2; acetylation increases protein stability of XBP1 isoform 2 and enhances its transcriptional activity. Acetylates PCNA; acetylation promotes removal of chromatin-bound PCNA and its degradation during nucleotide excision repair (NER). Acetylates MEF2D. Acetylates and stabilizes ZBTB7B protein by antagonizing ubiquitin conjugation and degragation, this mechanism may be involved in CD4/CD8 lineage differentiation. In addition to protein acetyltransferase, can use different acyl-CoA substrates, such as (2E)-butenoyl-CoA (crotonyl-CoA), butanoyl-CoA (butyryl-CoA) or propanoyl-CoA (propionyl-CoA), and is able to mediate protein crotonylation, butyrylation or propionylation, respectively. Acts as a histone crotonyltransferase; crotonylation marks active promoters and enhancers and confers resistance to transcriptional repressors. Histone crotonyltransferase activity is dependent on the concentration of (2E)-butenoyl-CoA (crotonyl-CoA) substrate and such activity is weak when (E)-but-2-enoyl-CoA (crotonyl-CoA) concentration is low. Also acts as a histone butyryltransferase; butyrylation marks active promoters. Functions as a transcriptional coactivator for SMAD4 in the TGF-beta signaling pathway. Acetylates PCK1 and promotes PCK1 anaplerotic activity. Functions as histone acetyltransferase and regulates transcription via chromatin remodeling. Related diseases Defects in EP300 may play a role in epithelial cancer.; DISEASE: Chromosomal aberrations involving EP300 may be a cause of acute myeloid leukemias. Translocation t(8;22)(p11;q13) with KAT6A.; DISEASE: Rubinstein-Taybi syndrome 2 (RSTS2) [MIM:613684]: A disorder characterized by craniofacial abnormalities, postnatal growth deficiency, broad thumbs, broad big toes, intellectual disability and a propensity for development of malignancies. Some individuals with RSTS2 have less severe mental impairment, more severe microcephaly, and a greater degree of changes in facial bone structure than RSTS1 patients. {ECO:0000269|PubMed:15706485}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Menke-Hennekam syndrome 2 (MKHK2) [MIM:618333]: A form of Menke-Hennekam syndrome, a congenital autosomal dominant disease characterized by developmental delay, growth retardation, and craniofacial dysmorphism. Patients have intellectual disability of variable severity, speech delay, autistic behavior, short stature and microcephaly. Main facial characteristics include short palpebral fissures, telecanthi, depressed nasal ridge, short nose, anteverted nares, short columella and long philtrum. {ECO:0000269|PubMed:29460469}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q9NXW9; P27695; Q9UBL3; Q8WXX7; Q9NPI1; P24941; Q99967; P61201; P16220-1; P17844; Q01844; P35637; Q00403; Q16665; Q9H2X6; Q92831; P55209; O60934; P20265; Q96KQ4; Q8WUF5; Q13761; Q96EB6; Q13309; O95863; P42226; Q9UL17; P56279; P05549; P04637; Q13625; O15350; P11473; P67809; K4P3M7; P03122; P06422; P06790; Q61221; Q9QXM1; P04608; P03070; P03255; P03255-2; P03259 EC number EC 2.3.1.48 Uniprot keywords 3D-structure; Acetylation; Acyltransferase; Biological rhythms; Bromodomain; Cell cycle; Chromosomal rearrangement; Chromosome; Citrullination; Cytoplasm; Direct protein sequencing; Disease variant; Host-virus interaction; Intellectual disability; Isopeptide bond; Metal-binding; Methylation; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Transcription; Transcription regulation; Transferase; Ubl conjugation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A Molecular weight (Da) 64477.2 Length 554 Aromaticity 0.12 Instability index 45.78 Isoelectric point 7.01 Charge (pH=7) 0.05 2D Binding mode Binding energy (Kcal/mol) -7.85  Molscript Map 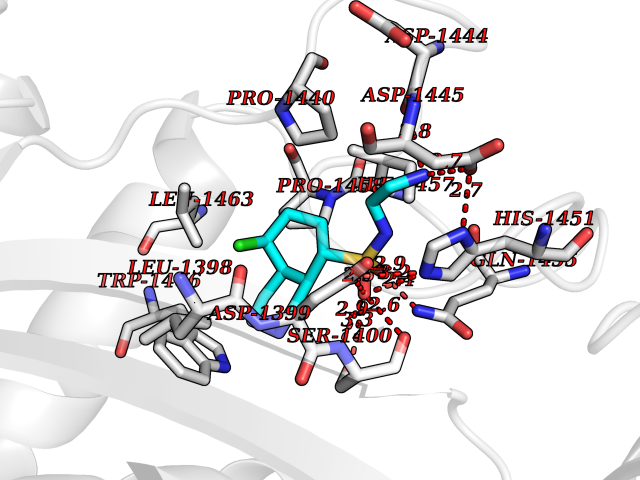 Pymol Map 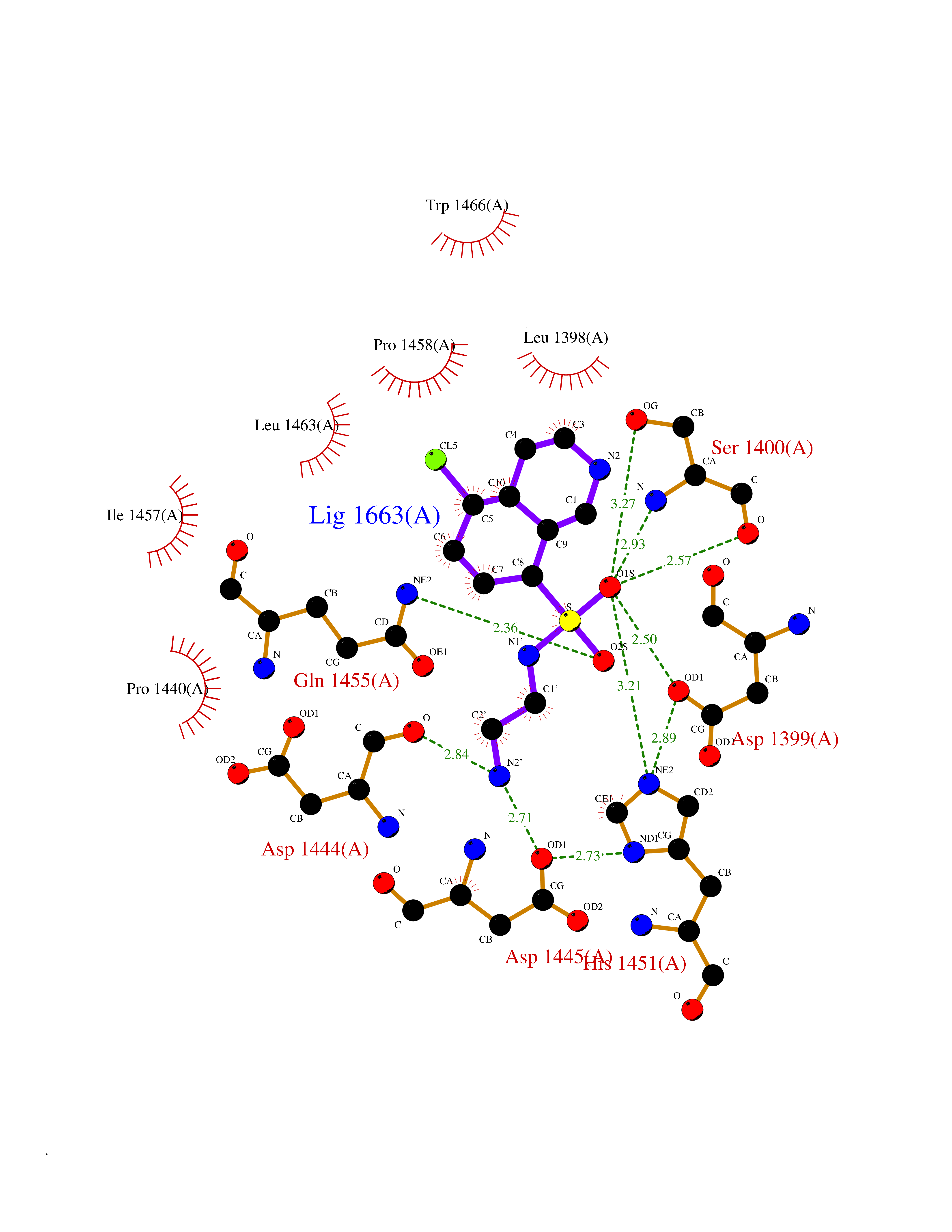 Ligplot Map 3D Binding mode Sequence KKIFKPEELRQALMPTLEALYRQDPESLPFRQPVDPQLLGIPDYFDIVKSPMDLSTIKRKLDTGQYQEPWQYVDDIWLMFNNAWLYNRKTSRVYKYCSKLSEVFEQEIDPVMQSLGYCCGRKLEFSPQTLCCYGKQLCTIPRDATYYSYQNRYHFCEKCFNEIQGESVSLGQTTINKEQFSKRKNDTLDPELFVECTECGRKMHQICVLHHEIIWPAGFVCDGCLKKSARTRKENKFSAKRLPSTRLGTFLENRVNDFLRRQNHPESGEVTVRVVHASDKTVEVKPGMKARFVDSGEMAESFPYRTKALFAFEEIDGVDLCFFGMHVQEYGSDCPPPNQRRVYISYLDSVHFFRPKCLRTAVYHEILIGYLEYVKKLGYTTGHIWACPPSEGDDYIFHCHPPDQKIPKPKRLQEWFKKMLDKAVSERIVHDYKDIFKQATEDRLTSAKELPYFEGDFWPNVLEESIKESGGSGSQKLYATMEKHKEVFFVIRLIAGPAANSLPPIVDPDPLIPCDLMDGRDAFLTLARDKHLEFSSLRRAQWSTMCMLVELHTQ Hydrogen bonds contact Hydrophobic contact | ||||
| 19 | Ferredoxin reductase | 2GQW | 7.87 | |
Target general information Gen name bphA4 Organism Pseudomonas sp. (strain KKS102) Uniprot ID TTD ID NA Synonyms NA Protein family NA Biochemical class Oxidoreductase Function Flavin adenine dinucleotide binding.Oxidoreductase activity. Related diseases LTC4 synthase deficiency is associated with a neurometabolic developmental disorder characterized by muscular hypotonia, psychomotor retardation, failure to thrive, and microcephaly. {ECO:0000269|PubMed:10896305, ECO:0000269|PubMed:9820300}. Drugs (DrugBank ID) DB03147 Interacts with NA EC number NA Uniprot keywords 3D-structure; FAD; Flavoprotein; Nucleotide-binding Protein physicochemical properties Chain ID A Molecular weight (Da) 42484 Length 401 Aromaticity 0.05 Instability index 32.45 Isoelectric point 6.13 Charge (pH=7) -2.69 2D Binding mode Binding energy (Kcal/mol) -7.58 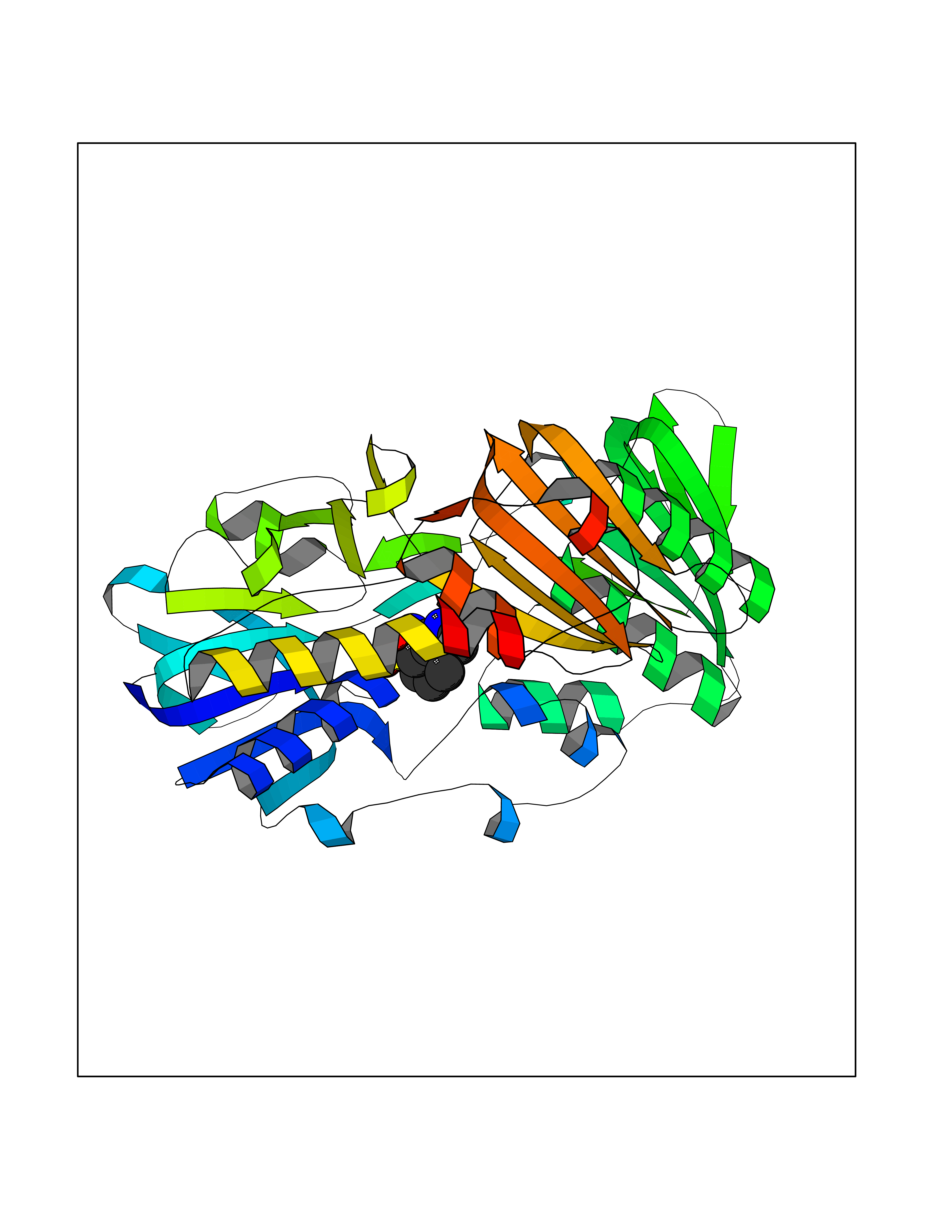 Molscript Map 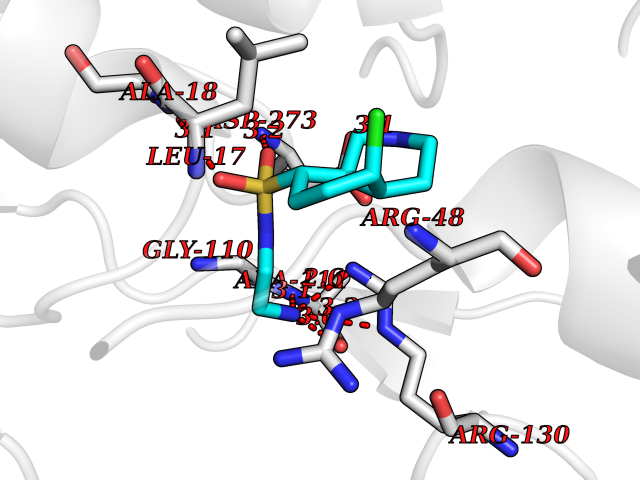 Pymol Map 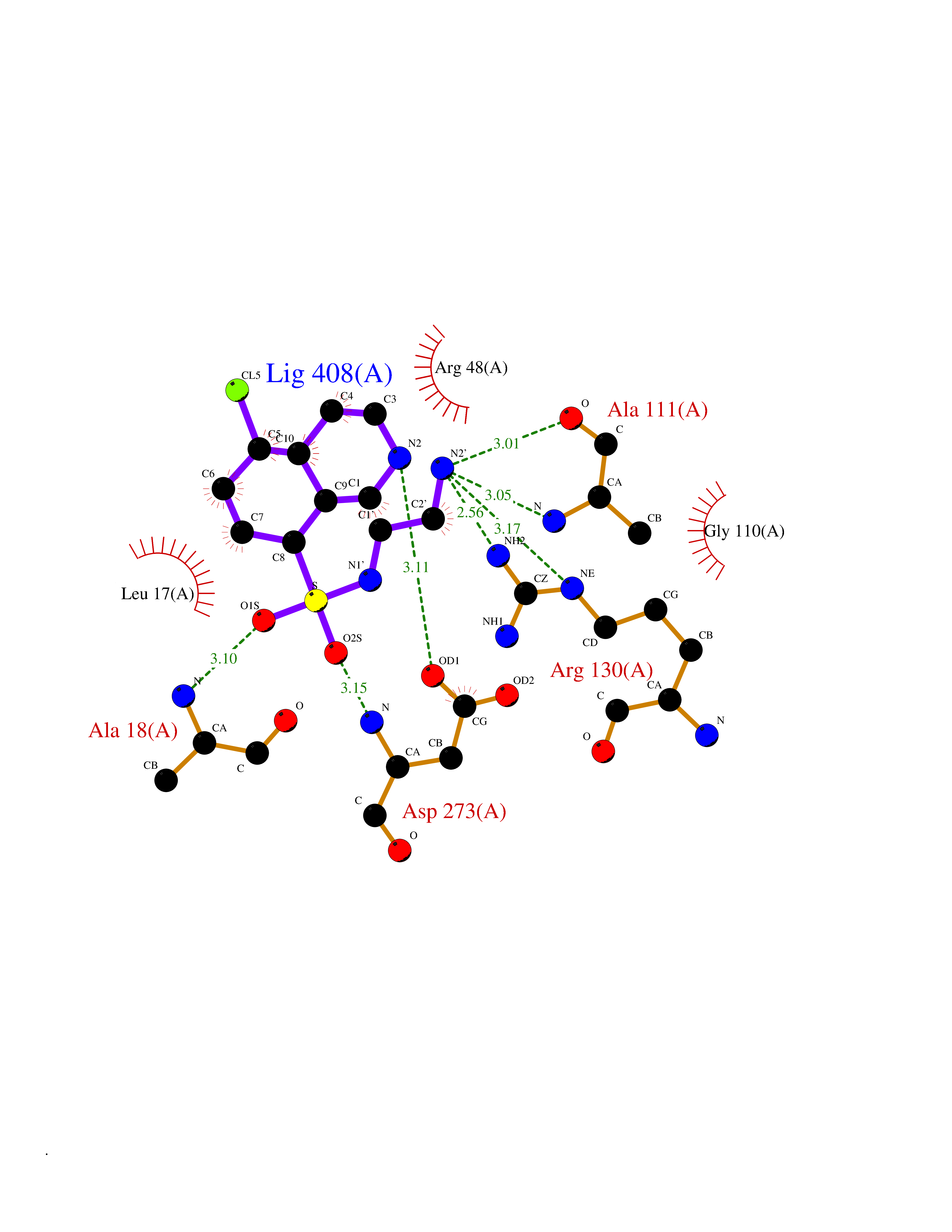 Ligplot Map 3D Binding mode Sequence LKAPVVVLGAGLASVSFVAELRQAGYQGLITVVGDEAERPYDRPPLSKDFMAHGDAEKIRLDCKRAPEVEWLLGVTAQSFDPQAHTVALSDGRTLPYGTLVLATGAAPRALPTLQGATMPVHTLRTLEDARRIQAGLRPQSRLLIVGGGVIGLELAATARTAGVHVSLVETQPRLMSRAAPATLADFVARYHAAQGVDLRFERSVTGSVDGVVLLDDGTRIAADMVVVGIGVLANDALARAAGLACDDGIFVDAYGRTTCPDVYALGDVTRQRNPLSGRFERIETWSNAQNQGIAVARHLVDPTAPGYAELPWYWSDQGALRIQVAGLASGDEEIVRGEVSLDAPKFTLIELQKGRIVGATCVNNARDFAPLRRLLAVGAKPDRAALADPATDLRKLAAAV Hydrogen bonds contact Hydrophobic contact | ||||
| 20 | Dopamine beta-hydroxylase | 4ZEL | 7.86 | |
Target general information Gen name DBH Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Copper type II ascorbate-dependent monooxygenase family Biochemical class Oxidoreductase Function Catalytic activity.Copper ion binding.Dopamine beta-monooxygenase activity.L-ascorbic acid binding. Related diseases Orthostatic hypotension 1 (ORTHYP1) [MIM:223360]: A form of orthostatic hypotension due to congenital dopamine beta-hydroxylase deficiency. Orthostatic hypotension, also known as postural hypotension, is a finding defined as a 20-mm Hg decrease in systolic pressure or a 10-mm Hg decrease in diastolic pressure occurring 3 minutes after a person has risen from supine to standing. Symptoms include dizziness, blurred vision, and sometimes syncope. ORTHYP1 is an autosomal recessive condition apparent from infancy or early childhood and characterized by low plasma and urinary levels of norepinephrine and epinephrine, and episodic hypoglycemia. {ECO:0000269|PubMed:11857564}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00126; DB06774; DB09130; DB05394; DB00822; DB00988; DB00968; DB00550 Interacts with P00352; P63010-2; Q04656; Q8WUW1; Q9UNS2; Q71DI3; P61978; Q9Y2M5; Q92876; P08727; Q14693; P0DPK4; Q6GQQ9-2; P27986-2; Q9ULX5; Q96D59; Q8N6K7-2; Q9GZS3; Q8IUW3; Q86WT6-2 EC number 1.14.17.1 Uniprot keywords 3D-structure; Catecholamine biosynthesis; Copper; Cytoplasmic vesicle; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; Membrane; Metal-binding; Monooxygenase; Oxidoreductase; Proteomics identification; Reference proteome; Secreted; Signal-anchor; Transmembrane; Transmembrane helix; Vitamin C Protein physicochemical properties Chain ID A,B Molecular weight (Da) 123694 Length 1094 Aromaticity 0.1 Instability index 51.85 Isoelectric point 5.84 Charge (pH=7) -24.5 2D Binding mode Binding energy (Kcal/mol) -7.34 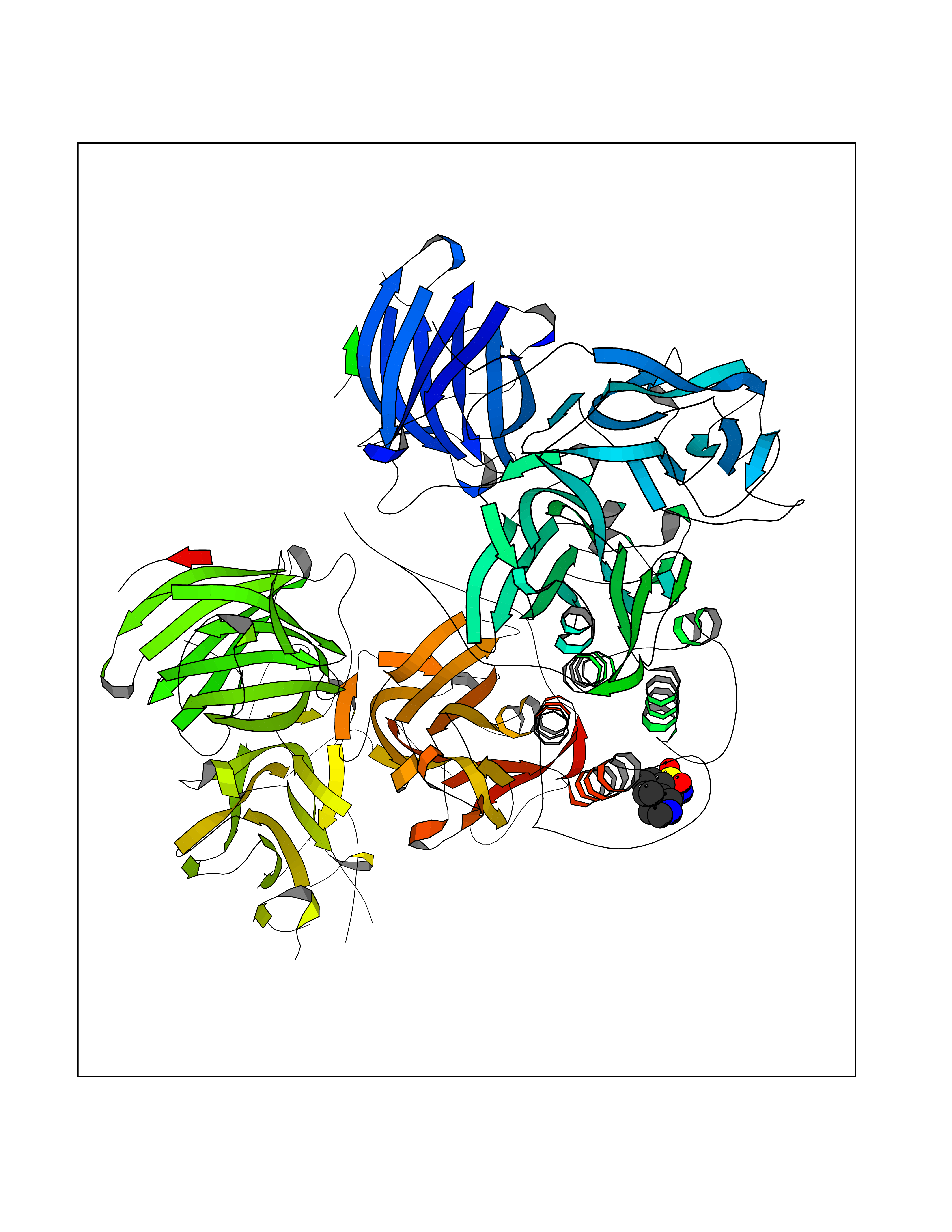 Molscript Map 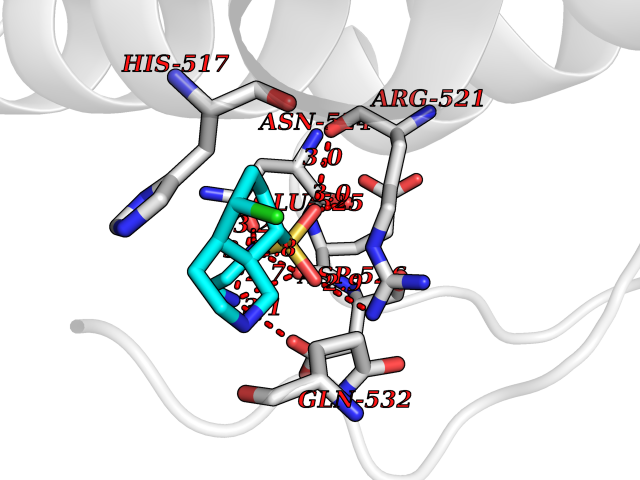 Pymol Map 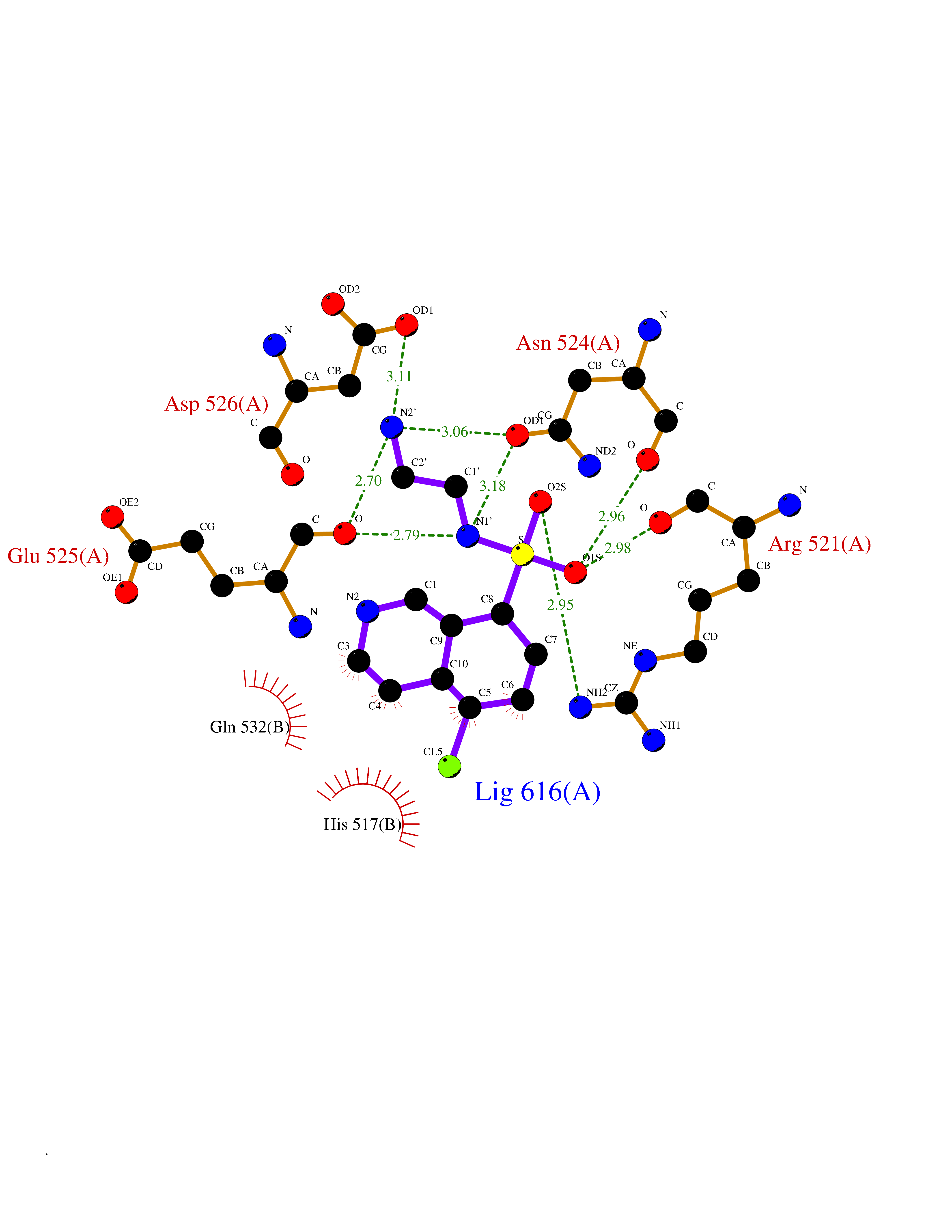 Ligplot Map 3D Binding mode Sequence PLPYHIPLDPEGSLELSWNVSYTQEAIHFQLLVRRLKAGVLFGMSDRGELENADLVVLWTDGDAYFADAWSDQKGQIHLDPQQDYQLLQVQRTPEGLTLLFKRPFGTCDPKDYLIEDGTVHLVYGILEEPFRSLEAINGSGLQMGLQRVQLLKPNIPEPELPSDACTMEVQAPNIQIPSQETTYWCYIKELPKGFSRHHIIKYEPIVTKGNEALVHHMEVFQCAPEMDSVPHFSGPCDSKMKPDRLNYCRHVLAAWALGAKAFYYPEEAGLAFGGPGSSRYLRLEVHYHNPLVIEGRNDSSGIRLYYTAKLRRFNAGIMELGLVYTPVMAIPPRETAFILTGYCTDKCTQLALPPSGIHIFASQLHTHLTGRKVVTVLVRDGREWEIVNQDNHYSPHFQEIRMLKKVVSVHPGDVLITSCTYNTEDRELATVGGFGILEEMCVNYVHYYPQTQLELCKSAVDAGFLQKYFHLINRFNNEDVCTCPQASVSQQFTSVPWNSFNRDVLKALYSFAPISMHCNKSSAVRFQGEWNLQPLPKVISTLEEPTVVSPLPYHIPLDPEGSLELSWNVSYTQEAIHFQLLVRRLKAGVLFGMSDRGELENADLVVLAYFADAWSDQKGQIHLDPQQDYQLLQVQRTPEGLTLLFKRPFGTCDPKDYLIEDGTVHLVYGILEEPFRSLEAINGSGLQMGLQRVQLLKPNIPEPELPSDACTMEVQAPNIQIPSQETTYWCYIKELPKGFSRHHIIKYEPIVTKGNEALVHHMEVFQCAPEVPHFSGPCDSKMLNYCRHVLAAWALGAKAFYYPEEAGLAFGGPGSSRYLRLEVHYHNPLVIEGRNDSSGIRLYYTAKLRRFNAGIMELGLVYTPVMAIPPRETAFILTGYCTDKCTQLALPPSGIHIFASQLHTHLTGRKVVTVLVRDGREWEIVNQDNHYSPHFQEIRMLKKVVSVHPGDVLITSCTYNTEDRELATVGGFGILEEMCVNYVHYYPQTQLELCKSAVDAGFLQKYFHLINRFNNEDVCTCPQASVSQQFTSVPWNSFNRDVLKALYSFAPISMHCNKSSAVRFQGEWNLQPLPKVISTLEEPTPQCVVSIGG Hydrogen bonds contact Hydrophobic contact | ||||