Ligand
Structure
Job ID
ee09f99f02be9e21dc8ef9d41d6f4c66
Job name
Lee_analyze21
Time
2024-12-01 14:54:25
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 1 | Gamma-aminobutyric acid type B receptor subunit 1 | 4MS4 | 7.28 | |
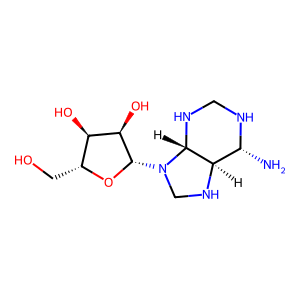
Target general information Gen name GABBR1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms GPRC3B;GPR51 Protein family G-protein coupled receptor 3 family, GABA-B receptor subfamily Biochemical class Signaling protein / antagonist Function G-protein coupled GABA receptor activity. Related diseases Neurodevelopmental disorder with poor language and loss of hand skills (NDPLHS) [MIM:617903]: An autosomal dominant disorder characterized by psychomotor developmental stagnation or regression. NDPLHS manifest in the first years of life as loss of purposeful hand movements, loss of language, and intellectual disability. {ECO:0000269|PubMed:26740508, ECO:0000269|PubMed:28856709, ECO:0000269|PubMed:29369404}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Developmental and epileptic encephalopathy 59 (DEE59) [MIM:617904]: A form of epileptic encephalopathy, a heterogeneous group of severe early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE59 is an autosomal dominant condition characterized by onset of refractory seizures in early infancy. {ECO:0000269|PubMed:28856709, ECO:0000269|PubMed:29100083, ECO:0000269|PubMed:29369404}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08891; DB08892; DB00181; DB00363; DB02530; DB05010; DB09072 Interacts with Q9UBS5; Q9UBS5-2; P46459; Q86UR5 EC number NA Uniprot keywords 3D-structure; Cell membrane; Coiled coil; Direct protein sequencing; Disease variant; Disulfide bond; Epilepsy; G-protein coupled receptor; Glycoprotein; Intellectual disability; Membrane; Phosphoprotein; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Signal; Synapse; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 46502.1 Length 408 Aromaticity 0.12 Instability index 50.05 Isoelectric point 5.78 Charge (pH=7) -5.62 2D Binding mode Binding energy (Kcal/mol) -7.79 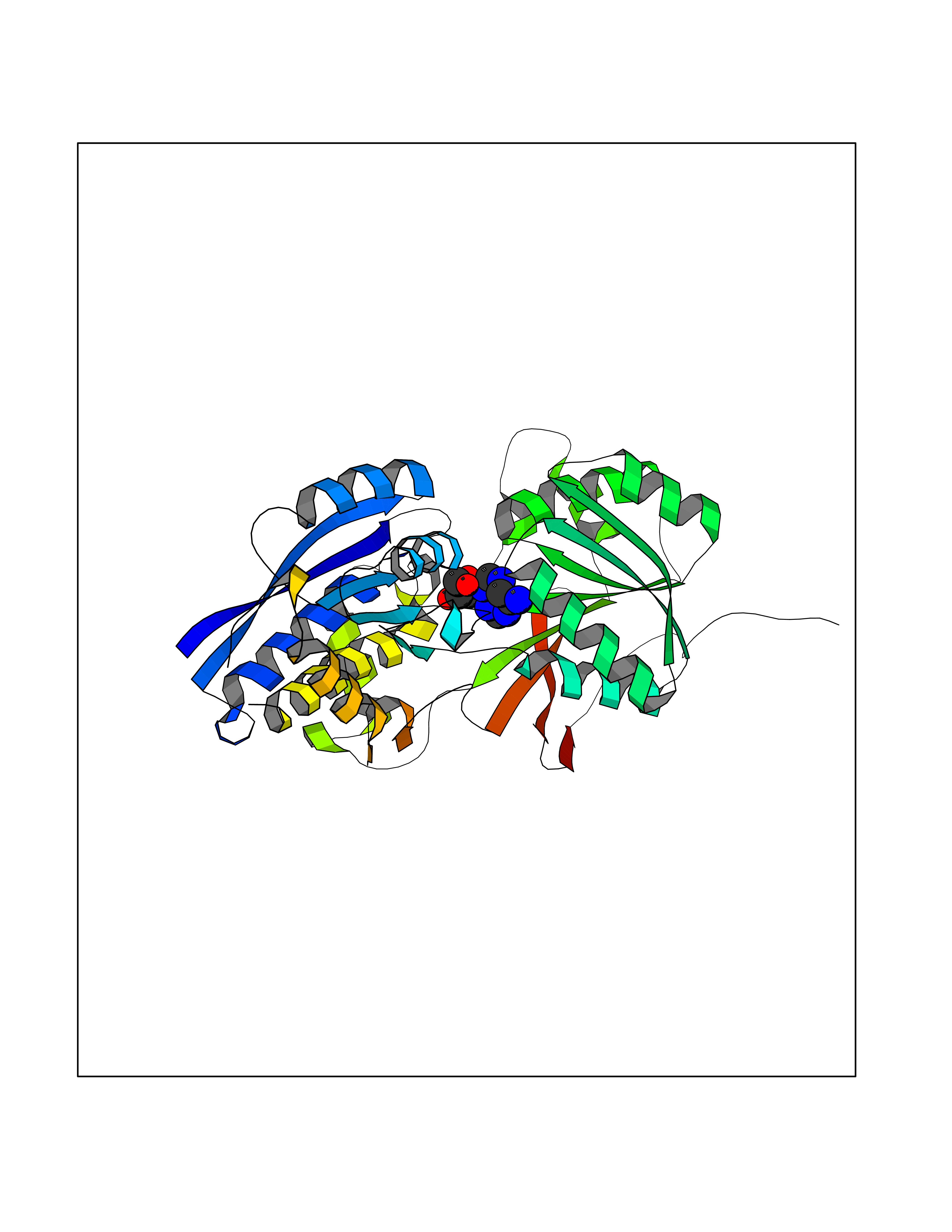 Molscript Map 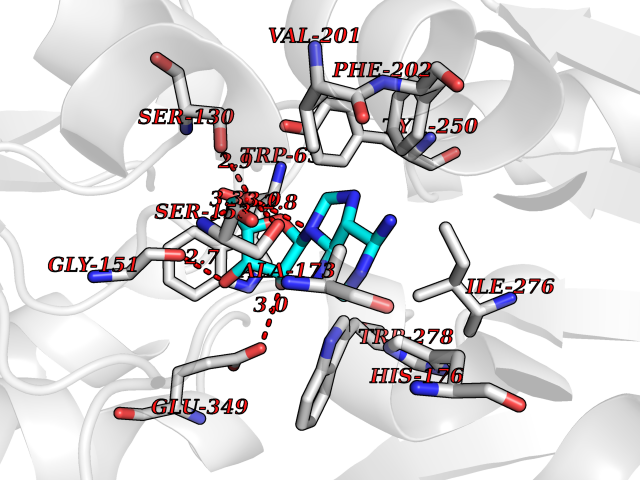 Pymol Map 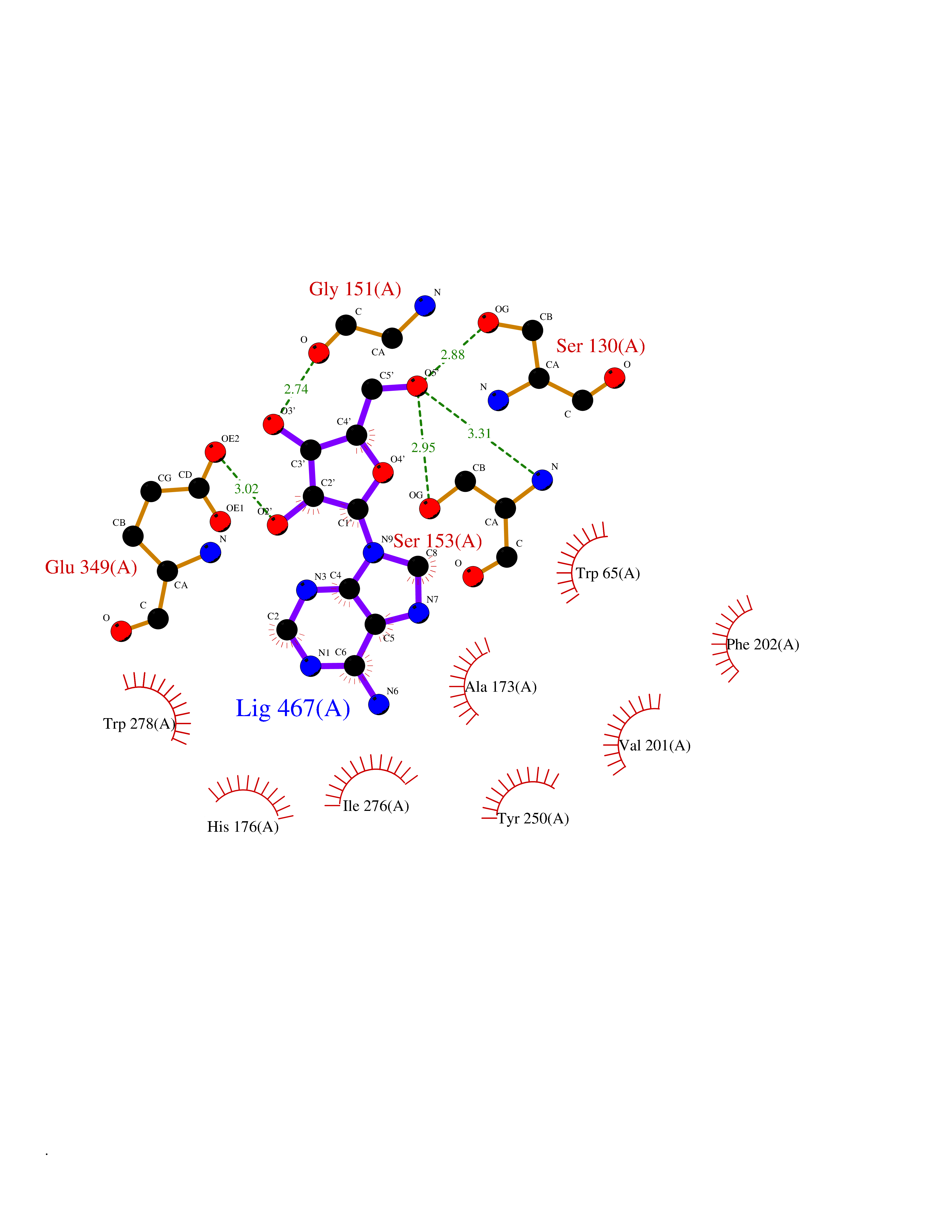 Ligplot Map 3D Binding mode Sequence RRAVYIGALFPMSGGWPGGQACQPAVEMALEDVNSRRDILPDYELKLIHHDSKCDPGQATKYLYELLYNDPIKIILMPGCSSVSTLVAEAARMWNLIVLSYGSSSPALSNRQRFPTFFRTHPSATLHNPTRVKLFEKWGWKKIATIQQTTEVFTSTLDDLEERVKEAGIEITFRQSFFSDPAVPVKNLKRQDARIIVGLFYETEARKVFCEVYKERLFGKKYVWFLIGWYADNWFKIYDPSINCTVDEMTEAVEGHITTEIVMLNPANTRSISNMTSQEFVEKLTKRLKRHPEETGGFQEAPLAYDAIWALALALNKTSRLEDFNYNNQTITDQIYRAMNSSSFEGVSGHVVFDASGSRMAWTLIEQLQGGSYKKIGYYDSTKDDLSWSKTDKWIGGSPPADDYKDDD Hydrogen bonds contact Hydrophobic contact | ||||
| 2 | Dopamine beta-hydroxylase | 4ZEL | 7.27 | |
Target general information Gen name DBH Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Copper type II ascorbate-dependent monooxygenase family Biochemical class Oxidoreductase Function Catalytic activity.Copper ion binding.Dopamine beta-monooxygenase activity.L-ascorbic acid binding. Related diseases Orthostatic hypotension 1 (ORTHYP1) [MIM:223360]: A form of orthostatic hypotension due to congenital dopamine beta-hydroxylase deficiency. Orthostatic hypotension, also known as postural hypotension, is a finding defined as a 20-mm Hg decrease in systolic pressure or a 10-mm Hg decrease in diastolic pressure occurring 3 minutes after a person has risen from supine to standing. Symptoms include dizziness, blurred vision, and sometimes syncope. ORTHYP1 is an autosomal recessive condition apparent from infancy or early childhood and characterized by low plasma and urinary levels of norepinephrine and epinephrine, and episodic hypoglycemia. {ECO:0000269|PubMed:11857564}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00126; DB06774; DB09130; DB05394; DB00822; DB00988; DB00968; DB00550 Interacts with P00352; P63010-2; Q04656; Q8WUW1; Q9UNS2; Q71DI3; P61978; Q9Y2M5; Q92876; P08727; Q14693; P0DPK4; Q6GQQ9-2; P27986-2; Q9ULX5; Q96D59; Q8N6K7-2; Q9GZS3; Q8IUW3; Q86WT6-2 EC number 1.14.17.1 Uniprot keywords 3D-structure; Catecholamine biosynthesis; Copper; Cytoplasmic vesicle; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; Membrane; Metal-binding; Monooxygenase; Oxidoreductase; Proteomics identification; Reference proteome; Secreted; Signal-anchor; Transmembrane; Transmembrane helix; Vitamin C Protein physicochemical properties Chain ID A,B Molecular weight (Da) 123694 Length 1094 Aromaticity 0.1 Instability index 51.85 Isoelectric point 5.84 Charge (pH=7) -24.5 2D Binding mode Binding energy (Kcal/mol) -7.66 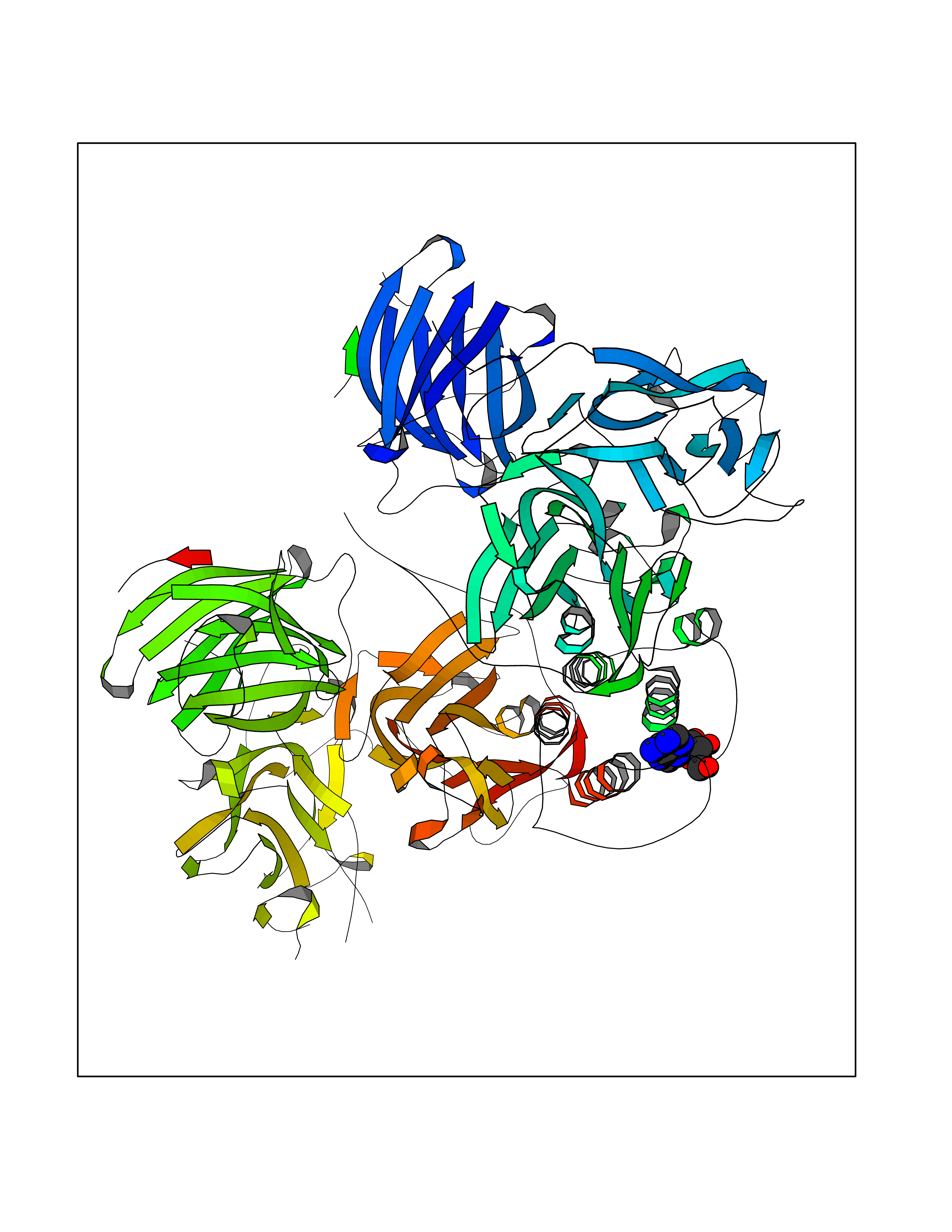 Molscript Map 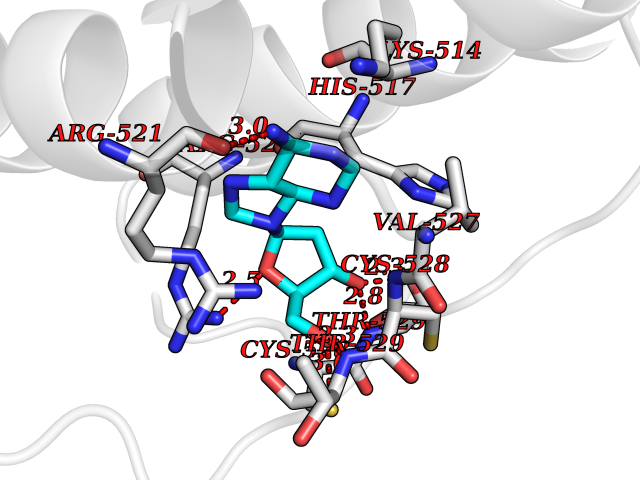 Pymol Map 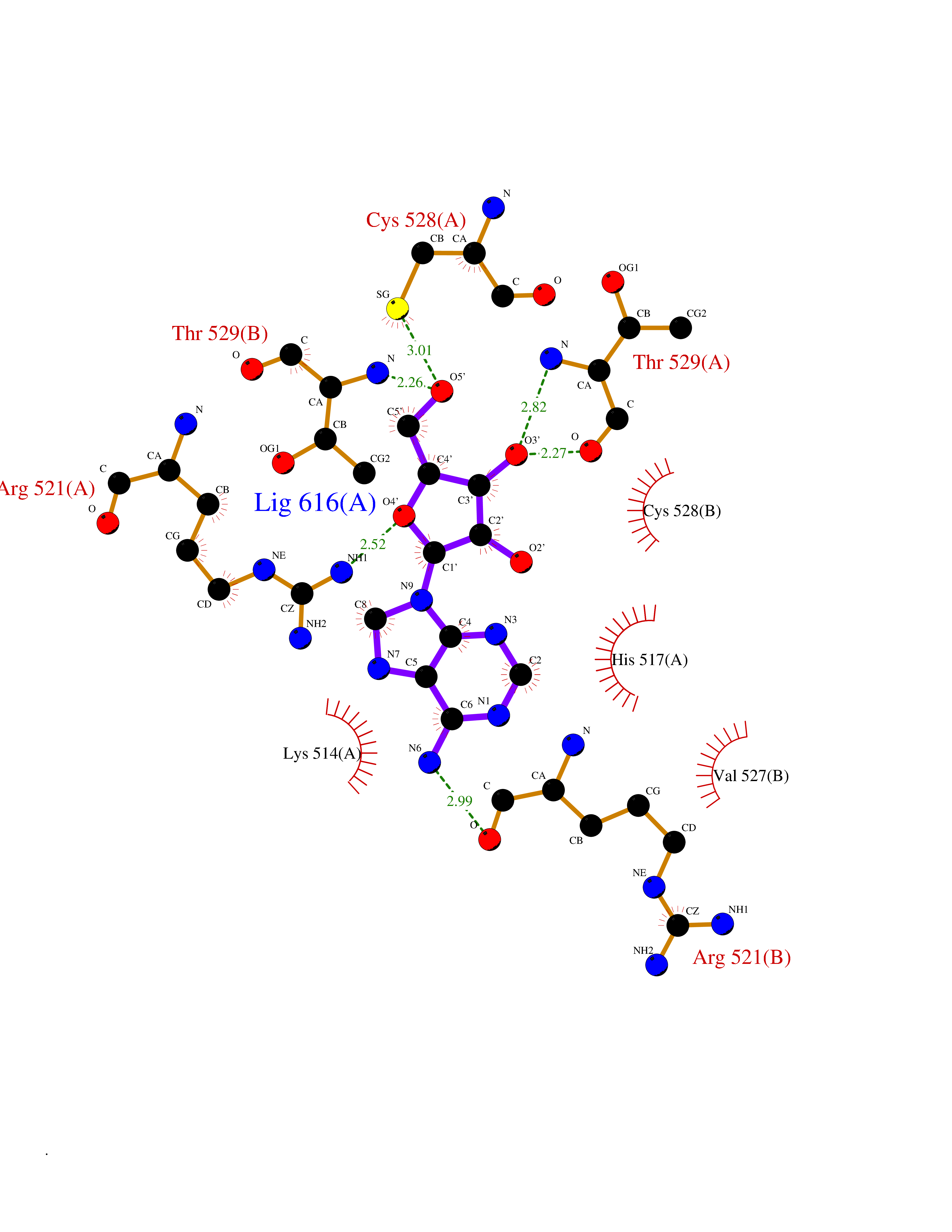 Ligplot Map 3D Binding mode Sequence PLPYHIPLDPEGSLELSWNVSYTQEAIHFQLLVRRLKAGVLFGMSDRGELENADLVVLWTDGDAYFADAWSDQKGQIHLDPQQDYQLLQVQRTPEGLTLLFKRPFGTCDPKDYLIEDGTVHLVYGILEEPFRSLEAINGSGLQMGLQRVQLLKPNIPEPELPSDACTMEVQAPNIQIPSQETTYWCYIKELPKGFSRHHIIKYEPIVTKGNEALVHHMEVFQCAPEMDSVPHFSGPCDSKMKPDRLNYCRHVLAAWALGAKAFYYPEEAGLAFGGPGSSRYLRLEVHYHNPLVIEGRNDSSGIRLYYTAKLRRFNAGIMELGLVYTPVMAIPPRETAFILTGYCTDKCTQLALPPSGIHIFASQLHTHLTGRKVVTVLVRDGREWEIVNQDNHYSPHFQEIRMLKKVVSVHPGDVLITSCTYNTEDRELATVGGFGILEEMCVNYVHYYPQTQLELCKSAVDAGFLQKYFHLINRFNNEDVCTCPQASVSQQFTSVPWNSFNRDVLKALYSFAPISMHCNKSSAVRFQGEWNLQPLPKVISTLEEPTVVSPLPYHIPLDPEGSLELSWNVSYTQEAIHFQLLVRRLKAGVLFGMSDRGELENADLVVLAYFADAWSDQKGQIHLDPQQDYQLLQVQRTPEGLTLLFKRPFGTCDPKDYLIEDGTVHLVYGILEEPFRSLEAINGSGLQMGLQRVQLLKPNIPEPELPSDACTMEVQAPNIQIPSQETTYWCYIKELPKGFSRHHIIKYEPIVTKGNEALVHHMEVFQCAPEVPHFSGPCDSKMLNYCRHVLAAWALGAKAFYYPEEAGLAFGGPGSSRYLRLEVHYHNPLVIEGRNDSSGIRLYYTAKLRRFNAGIMELGLVYTPVMAIPPRETAFILTGYCTDKCTQLALPPSGIHIFASQLHTHLTGRKVVTVLVRDGREWEIVNQDNHYSPHFQEIRMLKKVVSVHPGDVLITSCTYNTEDRELATVGGFGILEEMCVNYVHYYPQTQLELCKSAVDAGFLQKYFHLINRFNNEDVCTCPQASVSQQFTSVPWNSFNRDVLKALYSFAPISMHCNKSSAVRFQGEWNLQPLPKVISTLEEPTPQCVVSIGG Hydrogen bonds contact Hydrophobic contact | ||||
| 3 | Cytochrome P450 1A2 | 2HI4 | 7.21 | |
Target general information Gen name CYP1A2 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Cytochrome P450 family Biochemical class Oxidoreductase Function Aromatase activity.Caffeine oxidase activity.Demethylase activity.Electron carrier activity.Enzyme binding.Heme binding.Iron ion binding.Monooxygenase activity.Oxidoreductase activity.Oxidoreductase activity, acting on paired donors, with incorporation or reduction of molecular oxygen, reduced flavin or flavoprotein as one donor, and incorporation of one atom of oxygen.Oxygen binding. Related diseases Myeloperoxidase deficiency (MPOD) [MIM:254600]: A disorder characterized by decreased myeloperoxidase activity in neutrophils and monocytes that results in disseminated candidiasis. {ECO:0000269|PubMed:37198333, ECO:0000269|PubMed:7904599, ECO:0000269|PubMed:8142659, ECO:0000269|PubMed:8621627, ECO:0000269|PubMed:9354683, ECO:0000269|PubMed:9637725}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08496; DB01667; DB14132; DB04356; DB02489; DB11932; DB12001; DB05812; DB13573; DB01418; DB00316; DB15568; DB06594; DB00518; DB05396; DB00969; DB07453; DB01424; DB01223; DB01118; DB00321; DB00261; DB01217; DB01435; DB06605; DB05676; DB06413; DB06216; DB01072; DB15011; DB06442; DB06626; DB00993; DB00972; DB13203; DB05015; DB16703; DB06769; DB01086; DB06770; DB06771; DB06732; DB00195; DB04889; DB11967; DB13975; DB00188; DB12151; DB01558; DB14018; DB13812; DB00201; DB09061; DB14737; DB11791; DB06774; DB00564; DB06016; DB01136; DB12814; DB00477; DB00356; DB01166; DB00501; DB01012; DB00568; DB00827; DB00537; DB00215; DB12499; DB14025; DB00349; DB01242; DB00575; DB00758; DB00363; DB00286; DB11672; DB14635; DB00924; DB08912; DB00851; DB06292; DB01254; DB01609; DB01151; DB16650; DB12161; DB01191; DB00633; DB11994; DB00586; DB11511; DB12945; DB00280; DB01184; DB09167; DB05928; DB01142; DB09273; DB00470; DB00476; DB00625; DB15444; DB06210; DB13874; DB11718; DB00467; DB11404; DB00530; DB00783; DB13952; DB13953; DB13954; DB13955; DB13956; DB00655; DB04574; DB13592; DB00330; DB00898; DB00977; DB00773; DB01628; DB00927; DB04854; DB01482; DB00574; DB12265; DB15669; DB01195; DB08972; DB04841; DB00544; DB00472; DB00499; DB00176; DB01320; DB00998; DB14029; DB06160; DB01044; DB01241; DB01155; DB01645; DB01381; DB00986; DB00365; DB00400; DB05708; DB00629; DB00502; DB01094; DB14999; DB04076; DB11737; DB00619; DB00458; DB11564; DB01306; DB09456; DB09564; DB01307; DB00047; DB01309; DB00030; DB00046; DB11567; DB00071; DB11568; DB05258; DB00034; DB00105; DB15131; DB00011; DB00018; DB00069; DB00060; DB00068; DB00033; DB00951; DB11757; DB09570; DB01026; DB01097; DB16217; DB09078; DB01002; DB05667; DB00281; DB12406; DB09198; DB04948; DB00978; DB06448; DB16220; DB01601; DB00455; DB04871; DB06077; DB01283; DB00772; DB00934; DB06234; DB14009; DB00784; DB01065; DB00170; DB00454; DB00532; DB00333; DB00763; DB00553; DB01028; DB09241; DB01233; DB00379; DB06148; DB01388; DB06595; DB00370; DB16236; DB00745; DB11763; DB00218; DB06510; DB14011; DB00461; DB00607; DB00779; DB00788; DB06600; DB00238; DB06803; DB00184; DB01115; DB11793; DB00435; DB05115; DB00717; DB01059; DB00540; DB05990; DB01165; DB00334; DB16267; DB00338; DB00904; DB11632; DB11443; DB01173; DB11837; DB09330; DB01303; DB11697; DB00377; DB00715; DB06589; DB11774; DB00487; DB00008; DB00022; DB09122; DB13634; DB00806; DB11198; DB08883; DB00850; DB03783; DB01174; DB00388; DB00252; DB11450; DB01100; DB13823; DB04951; DB17472; DB11642; DB08910; DB15822; DB01058; DB01087; DB00794; DB00420; DB09288; DB01182; DB06479; DB00818; DB00571; DB13449; DB11892; DB04216; DB00908; DB00468; DB01129; DB00980; DB09290; DB00863; DB01367; DB00409; DB02709; DB13174; DB01045; DB11753; DB00740; DB14924; DB00503; DB00533; DB01656; DB15119; DB00268; DB00296; DB00412; DB00817; DB12332; DB13772; DB06654; DB11491; DB00418; DB01037; DB11689; DB06290; DB13261; DB15093; DB00052; DB00398; DB01208; DB09118; DB00428; DB06820; DB00382; DB00675; DB06083; DB09071; DB05488; DB09256; DB01079; DB01405; DB00857; DB08880; DB11712; DB01412; DB00277; DB00730; DB01623; DB00208; DB06137; DB00697; DB01056; DB06264; DB00752; DB00384; DB12245; DB00831; DB15442; DB00440; DB00685; DB08867; DB14989; DB13609; DB06235; DB00313; DB08881; DB00661; DB09185; DB12026; DB00682; DB02134; DB00549; DB00744; DB00315; DB00425; DB09225; DB09120 Interacts with O95870 EC number 1.14.14.1; 4.2.1.152 Uniprot keywords 3D-structure; Direct protein sequencing; Endoplasmic reticulum; Fatty acid metabolism; Glycoprotein; Heme; Iron; Lipid metabolism; Lyase; Membrane; Metal-binding; Microsome; Monooxygenase; Oxidoreductase; Proteomics identification; Reference proteome; Steroid metabolism; Sterol metabolism Protein physicochemical properties Chain ID A Molecular weight (Da) 54475 Length 480 Aromaticity 0.1 Instability index 40.43 Isoelectric point 9.16 Charge (pH=7) 9.89 2D Binding mode Binding energy (Kcal/mol) -7.61 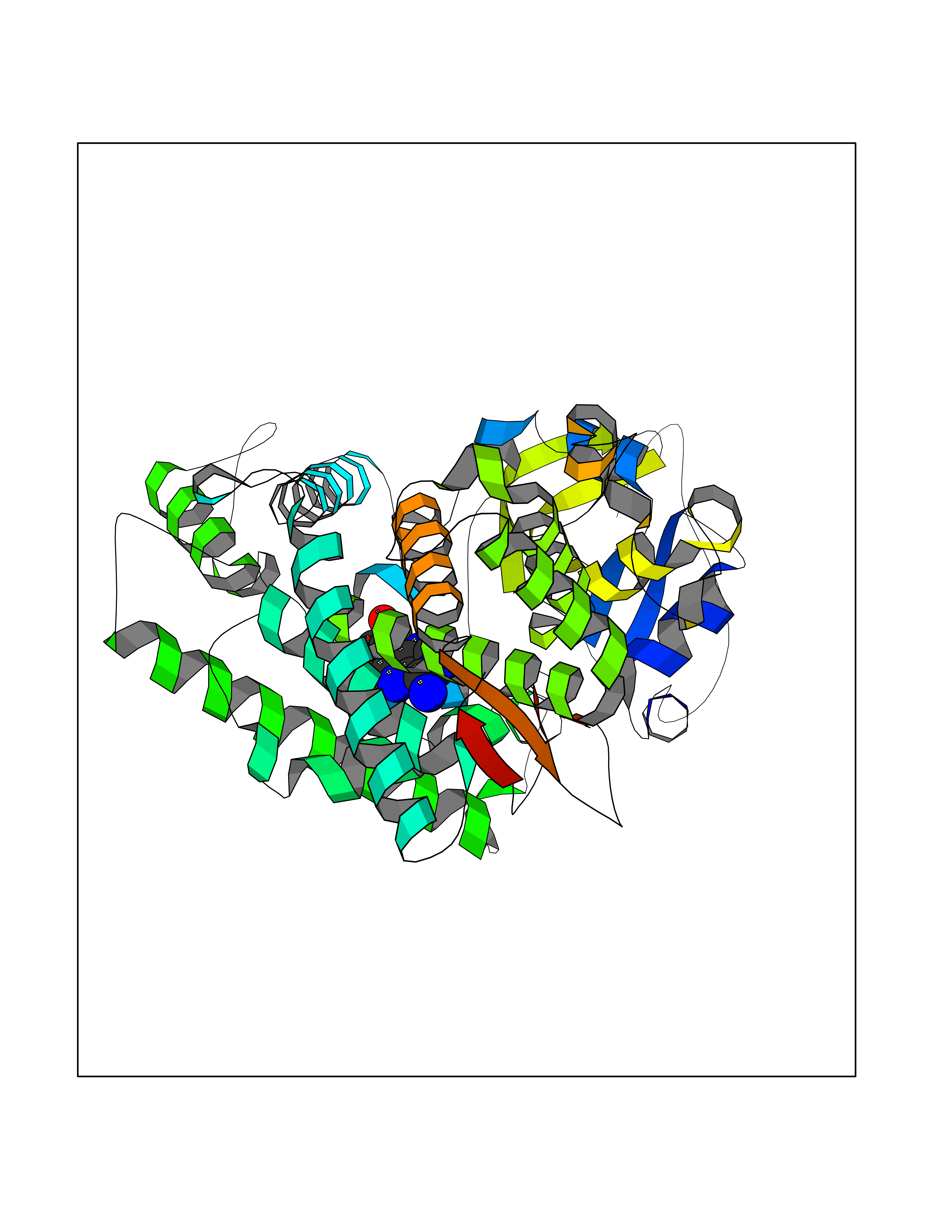 Molscript Map 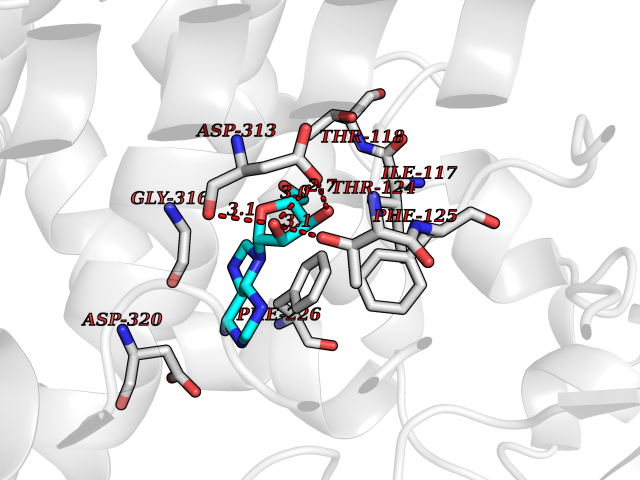 Pymol Map 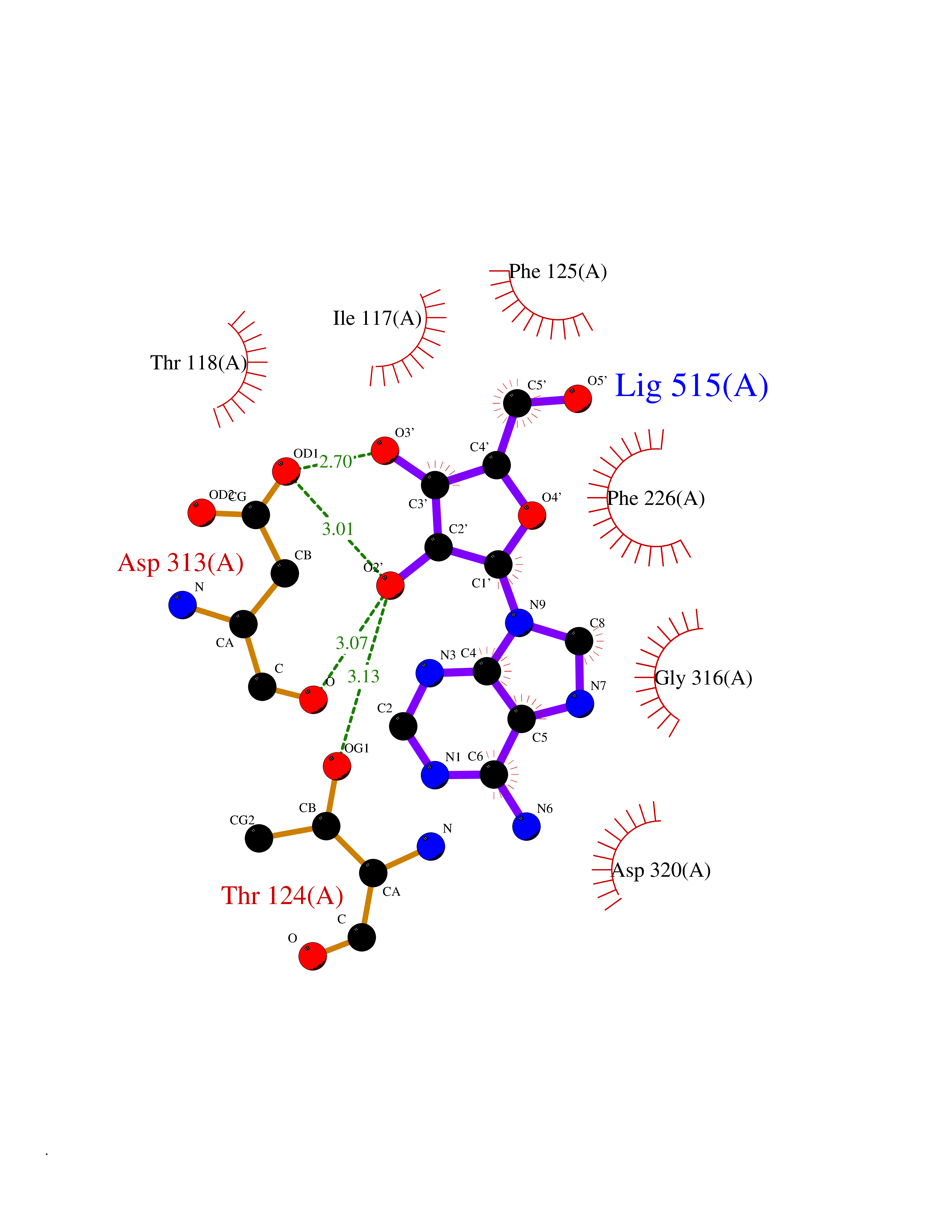 Ligplot Map 3D Binding mode Sequence RVPKGLKSPPEPWGWPLLGHVLTLGKNPHLALSRMSQRYGDVLQIRIGSTPVLVLSRLDTIRQALVRQGDDFKGRPDLYTSTLITDGQSLTFSTDSGPVWAARRRLAQNALNTFSIASDPASSSSCYLEEHVSKEAKALISRLQELMAGPGHFDPYNQVVVSVANVIGAMCFGQHFPESSDEMLSLVKNTHEFVETASSGNPLDFFPILRYLPNPALQRFKAFNQRFLWFLQKTVQEHYQDFDKNSVRDITGALFKHSKKGPRASGNLIPQEKIVNLVNDIFGAGFDTVTTAISWSLMYLVTKPEIQRKIQKELDTVIGRERRPRLSDRPQLPYLEAFILETFRHSSFLPFTIPHSTTRDTTLNGFYIPKKCCVFVNQWQVNHDPELWEDPSEFRPERFLTADGTAINKPLSEKMMLFGMGKRRCIGEVLAKWEIFLFLAILLQQLEFSVPPGVKVDLTPIYGLTMKHARCEHVQARRFS Hydrogen bonds contact Hydrophobic contact | ||||
| 4 | Pseudomonas Phosphomannomutase/phosphoglucomutase (Pseudo algC) | 1P5D | 7.19 | |
Target general information Gen name Pseudo algC Organism Pseudomonas aeruginosa (strain ATCC 15692 / DSM 22644 / CIP 104116 / JCM 14847 / LMG 12228 / 1C / PRS 101 / PAO1) Uniprot ID TTD ID Synonyms algC; PMM/PGM; PMM / PGM Protein family Phosphohexose mutase family Biochemical class Intramolecular transferases Function Highly reversible phosphoryltransferase. The phosphomannomutase activity produces a precursor for alginate polymerization, the alginate layer causes a mucoid phenotype and provides a protective barrier against host immune defenses and antibiotics. Also involved in core lipopolysaccaride (LPS) biosynthesis due to its phosphoglucomutase activity. Essential for rhamnolipid production, an exoproduct correlated with pathogenicity (PubMed:10481091). Required for biofilm production. The reaction proceeds via 2 processive phosphoryl transferase reactions; first from enzyme-phospho-Ser-108 to the substrate (generatinga bisphosphorylated substrate intermediate and a dephosphorylated enzyme), a 180 degree rotation of the intermediate (probably aided by movement of domain 4), and subsequent transfer of phosphate back to the enzyme (PubMed:11716469, PubMed:16880541, PubMed:16595672, PubMed:22242625). Related diseases Intellectual developmental disorder, X-linked, syndromic, Claes-Jensen type (MRXSCJ) [MIM:300534]: A disorder characterized by significantly below average general intellectual functioning associated with impairments in adaptive behavior and manifested during the developmental period. MRXSCJ patients manifest intellectual disability associated with variable features such as slowly progressive spastic paraplegia, seizures, facial dysmorphism. {ECO:0000269|PubMed:15586325, ECO:0000269|PubMed:16538222, ECO:0000269|PubMed:16541399, ECO:0000269|PubMed:17320160, ECO:0000269|PubMed:17468742, ECO:0000269|PubMed:23356856, ECO:0000269|PubMed:25666439}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02007; DB02843; DB02900; DB02867; DB04522 Interacts with NA EC number NA Uniprot keywords 3D-structure; Alginate biosynthesis; Direct protein sequencing; Isomerase; Lipopolysaccharide biosynthesis; Magnesium; Metal-binding; Multifunctional enzyme; Phosphoprotein; Reference proteome; Virulence Protein physicochemical properties Chain ID X Molecular weight (Da) 49419.9 Length 455 Aromaticity 0.07 Instability index 28.92 Isoelectric point 5.11 Charge (pH=7) -12.7 2D Binding mode Binding energy (Kcal/mol) -7.42 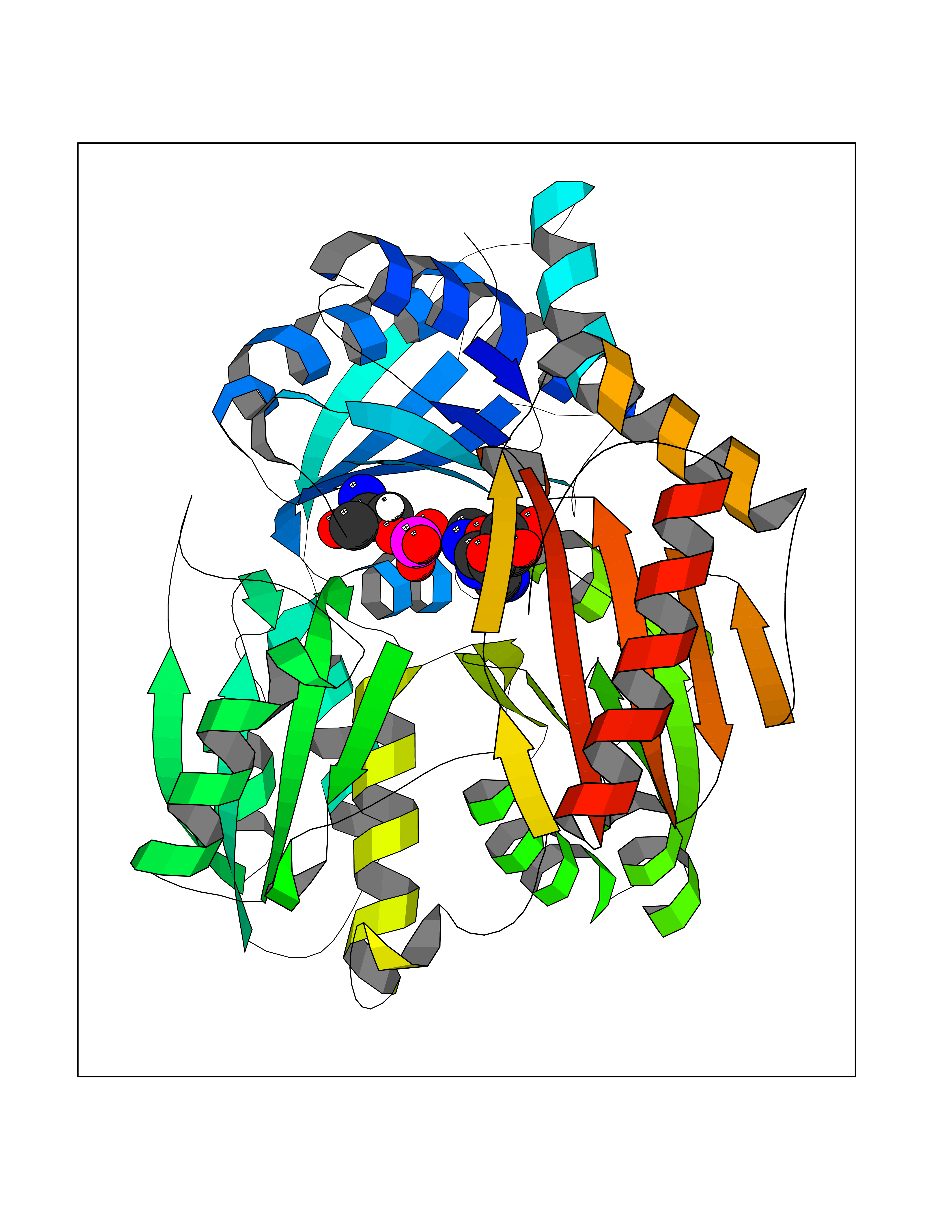 Molscript Map  Pymol Map 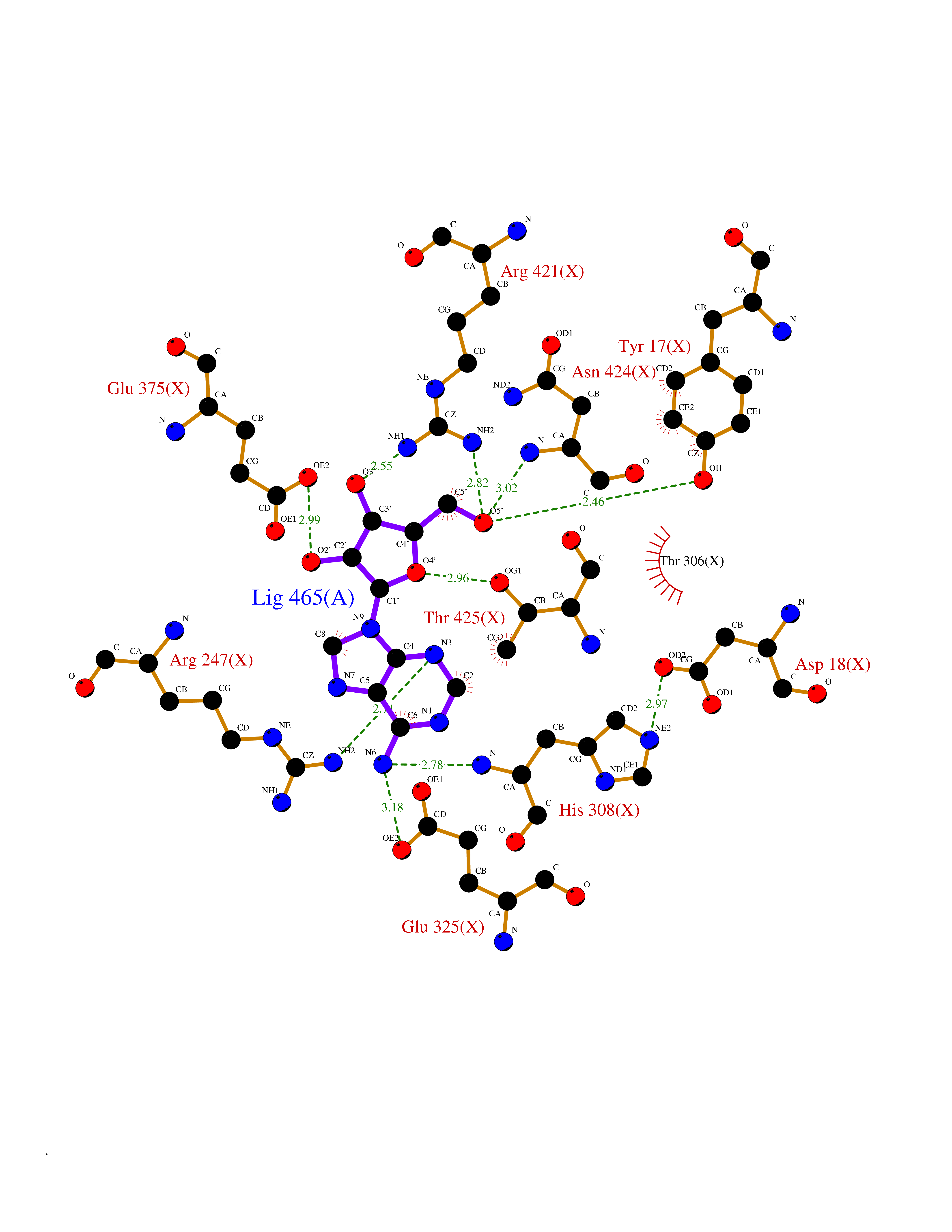 Ligplot Map 3D Binding mode Sequence LPASIFRAYDIRGVVGDTLTAETAYWIGRAIGSESLARGEPCVAVGRDGRLSGPELVKQLIQGLVDCGCQVSDVGMVPTPVLYYAANVLEGKSGVMLTGXHNPPDYNGFKIVVAGETLANEQIQALRERIEKNDLASGVGSVEQVDILPRYFKQIRDDIAMAKPMKVVVDCGNGVAGVIAPQLIEALGCSVIPLYCEVDGNFPNHHPDPGKPENLKDLIAKVKAENADLGLAFDGDGDRVGVVTNTGTIIYPDRLLMLFAKDVVSRNPGADIIFDVKCTRRLIALISGYGGRPVMWKTGHSLIKKKMKETGALLAGEMSGHVFFKERWFGFDDGIYSAARLLEILSQDQRDSEHVFSAFPSDISTPEINITVTEDSKFAIIEALQRDAQWGEGNITTLDGVRVDYPKGWGLVRASNTTPVLVLRFEADTEEELERIKTVFRNQLKAVDSSLPVPF Hydrogen bonds contact Hydrophobic contact | ||||
| 5 | Adrenergic receptor beta-2 (ADRB2) | 2RH1 | 7.14 | |
Target general information Gen name ADRB2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Beta-2 adrenoreceptor; Beta-2 adrenoceptor; Beta-2 adrenergic receptor; B2AR; ADRB2R Protein family G-protein coupled receptor 1 family, Adrenergic receptor subfamily, ADRB2 sub-subfamily Biochemical class GPCR rhodopsin Function The beta-2-adrenergic receptor binds epinephrine with an approximately 30-fold greater affinity than it does norepinephrine. Beta-adrenergic receptors mediate the catecholamine-induced activation of adenylate cyclase through the action of G proteins. Related diseases Cortical dysplasia, complex, with other brain malformations 6 (CDCBM6) [MIM:615771]: A disorder of aberrant neuronal migration and disturbed axonal guidance. Affected individuals have microcephaly, ataxia, and severe delayed psychomotor development. Brain imaging shows variable malformations of cortical development, including white matter streaks, dysmorphic basal ganglia, corpus callosum abnormalities, brainstem and cerebellar hypoplasia, cortical dysplasia, polymicrogyria. {ECO:0000269|PubMed:23246003}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Skin creases, congenital symmetric circumferential, 1 (CSCSC1) [MIM:156610]: An autosomal dominant disease characterized by multiple, symmetric, circumferential rings of folded skin, affecting primarily the limbs. Affected individuals also exhibit intellectual disability, cleft palate, and dysmorphic features. {ECO:0000269|PubMed:26637975}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07543; DB01193; DB00866; DB01118; DB00182; DB01102; DB01274; DB01238; DB09204; DB06216; DB00335; DB01408; DB05590; DB09013; DB00195; DB00217; DB01295; DB00612; DB00901; DB08807; DB06726; DB08808; DB00248; DB00521; DB01136; DB04846; DB01407; DB00785; DB01151; DB11273; DB13345; DB00449; DB11278; DB00841; DB09273; DB06262; DB01363; DB01364; DB00668; DB01049; DB11587; DB01288; DB00983; DB05039; DB00221; DB01064; DB00598; DB01210; DB13139; DB01365; DB13624; DB01214; DB00264; DB01203; DB05849; DB04861; DB00368; DB00540; DB00334; DB09080; DB00816; DB01580; DB00715; DB01359; DB00925; DB00397; DB00960; DB01291; DB01366; DB01182; DB00571; DB06814; DB00852; DB01917; DB11124; DB00867; DB01001; DB00938; DB00489; DB03566; DB00127; DB00871; DB00373; DB00726; DB12248; DB09082; DB09185 Interacts with P30542; P07550; P32121; Q96B67; Q9UII2; Q9ULD4-2; Q9NSI6-4; Q5M9N0-2; A0AVK6; Q658K8; O00472; Q15910-2; Q15486; P61978; Q5TCQ9; Q99685; O14745; Q9NR21-5; Q8WVD3; Q9H0X6; Q13573; P12931; Q5T0J7-2; Q8N0U2 EC number NA Uniprot keywords 3D-structure; Cell membrane; Disulfide bond; Endosome; G-protein coupled receptor; Glycoprotein; Golgi apparatus; Hydroxylation; Lipoprotein; Membrane; Palmitate; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Transducer; Transmembrane; Transmembrane helix; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 32266.1 Length 282 Aromaticity 0.15 Instability index 36.1 Isoelectric point 8.02 Charge (pH=7) 2.1 2D Binding mode Binding energy (Kcal/mol) -7.16 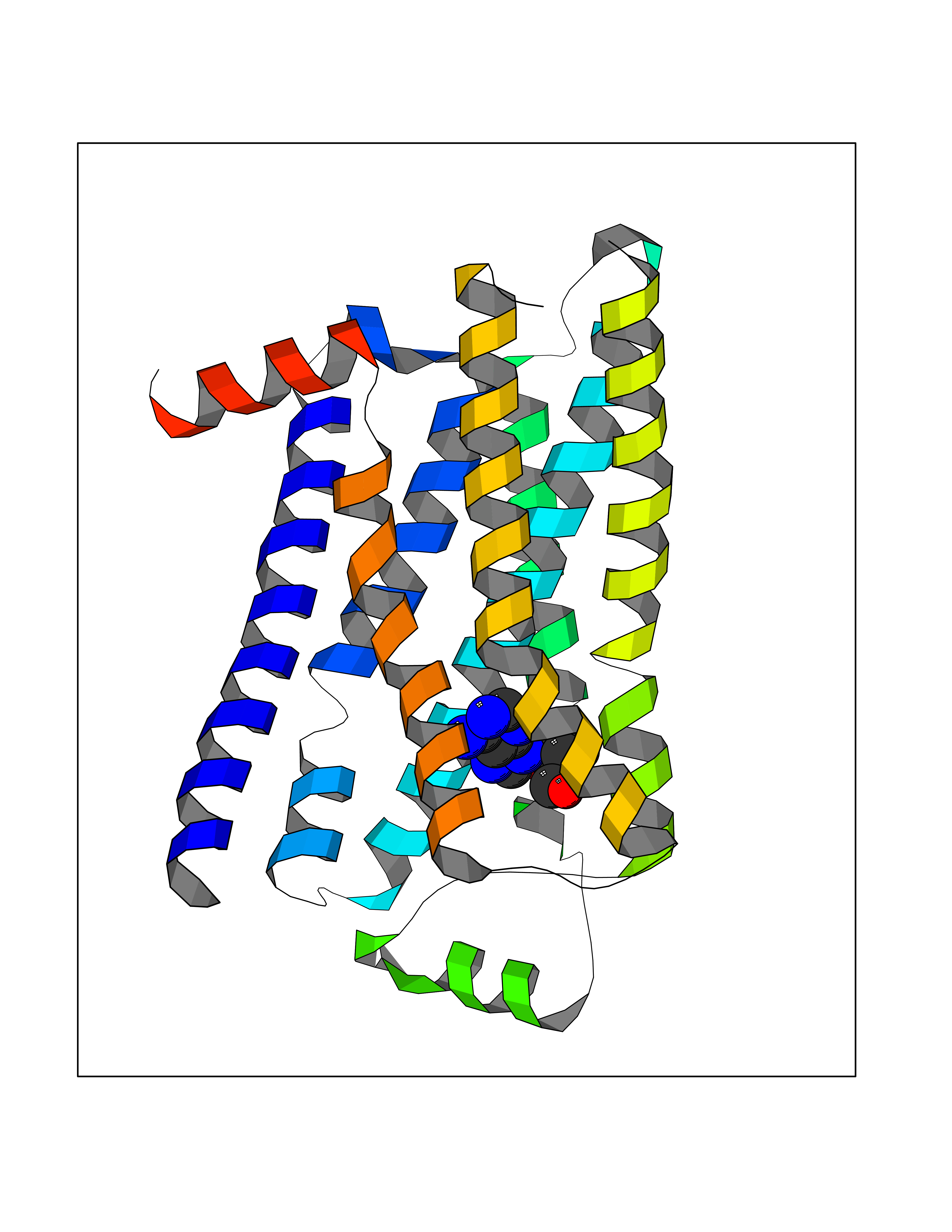 Molscript Map 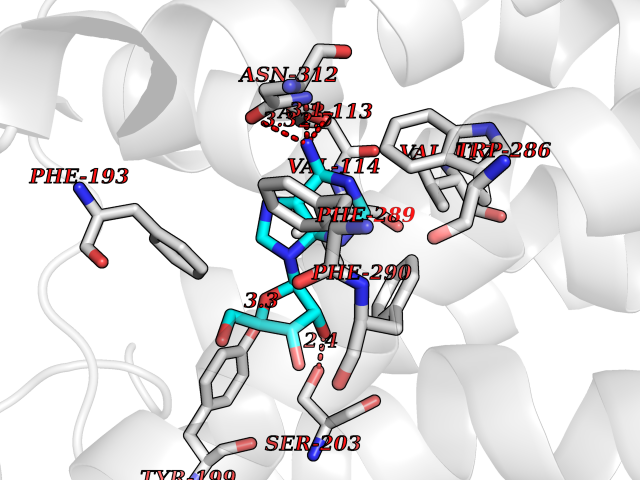 Pymol Map 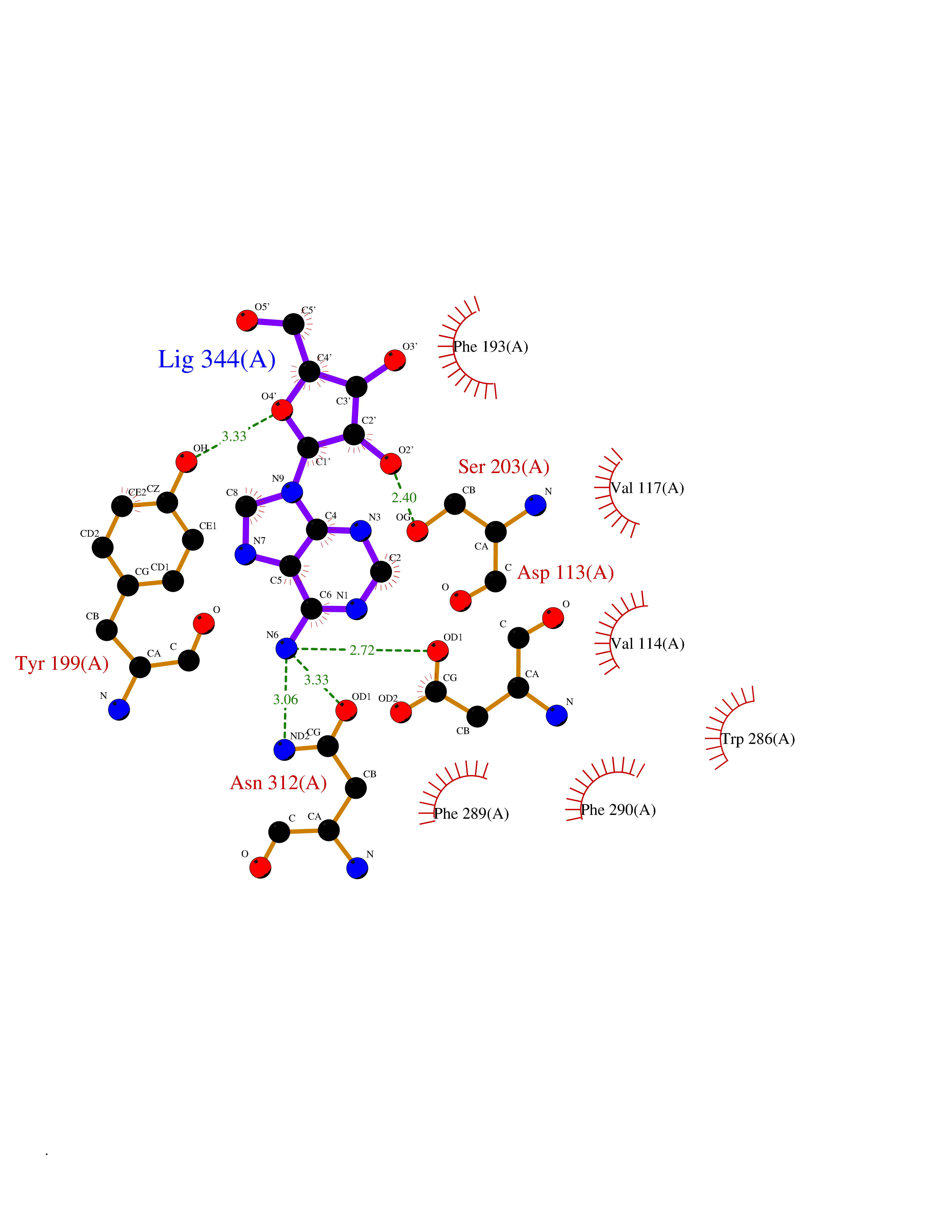 Ligplot Map 3D Binding mode Sequence DEVWVVGMGIVMSLIVLAIVFGNVLVITAIAKFERLQTVTNYFITSLACADLVMGLAVVPFGAAHILMKMWTFGNFWCEFWTSIDVLCVTASIETLCVIAVDRYFAITSPFKYQSLLTKNKARVIILMVWIVSGLTSFLPIQMHWYRATHQEAINCYAEETCCDFFTNQAYAIASSIVSFYVPLVIMVFVYSRVFQEAKRQLKFCLKEHKALKTLGIIMGTFTLCWLPFFIVNIVHVIQDNLIRKEVYILLNWIGYVNSGFNPLIYCRSPDFRIAFQELLCL Hydrogen bonds contact Hydrophobic contact | ||||
| 6 | Dopamine beta-hydroxylase | 4ZEL | 7.12 | |
Target general information Gen name DBH Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Copper type II ascorbate-dependent monooxygenase family Biochemical class Oxidoreductase Function Catalytic activity.Copper ion binding.Dopamine beta-monooxygenase activity.L-ascorbic acid binding. Related diseases Orthostatic hypotension 1 (ORTHYP1) [MIM:223360]: A form of orthostatic hypotension due to congenital dopamine beta-hydroxylase deficiency. Orthostatic hypotension, also known as postural hypotension, is a finding defined as a 20-mm Hg decrease in systolic pressure or a 10-mm Hg decrease in diastolic pressure occurring 3 minutes after a person has risen from supine to standing. Symptoms include dizziness, blurred vision, and sometimes syncope. ORTHYP1 is an autosomal recessive condition apparent from infancy or early childhood and characterized by low plasma and urinary levels of norepinephrine and epinephrine, and episodic hypoglycemia. {ECO:0000269|PubMed:11857564}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00126; DB06774; DB09130; DB05394; DB00822; DB00988; DB00968; DB00550 Interacts with P00352; P63010-2; Q04656; Q8WUW1; Q9UNS2; Q71DI3; P61978; Q9Y2M5; Q92876; P08727; Q14693; P0DPK4; Q6GQQ9-2; P27986-2; Q9ULX5; Q96D59; Q8N6K7-2; Q9GZS3; Q8IUW3; Q86WT6-2 EC number 1.14.17.1 Uniprot keywords 3D-structure; Catecholamine biosynthesis; Copper; Cytoplasmic vesicle; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; Membrane; Metal-binding; Monooxygenase; Oxidoreductase; Proteomics identification; Reference proteome; Secreted; Signal-anchor; Transmembrane; Transmembrane helix; Vitamin C Protein physicochemical properties Chain ID A,B Molecular weight (Da) 123694 Length 1094 Aromaticity 0.1 Instability index 51.85 Isoelectric point 5.84 Charge (pH=7) -24.5 2D Binding mode Binding energy (Kcal/mol) -6.96 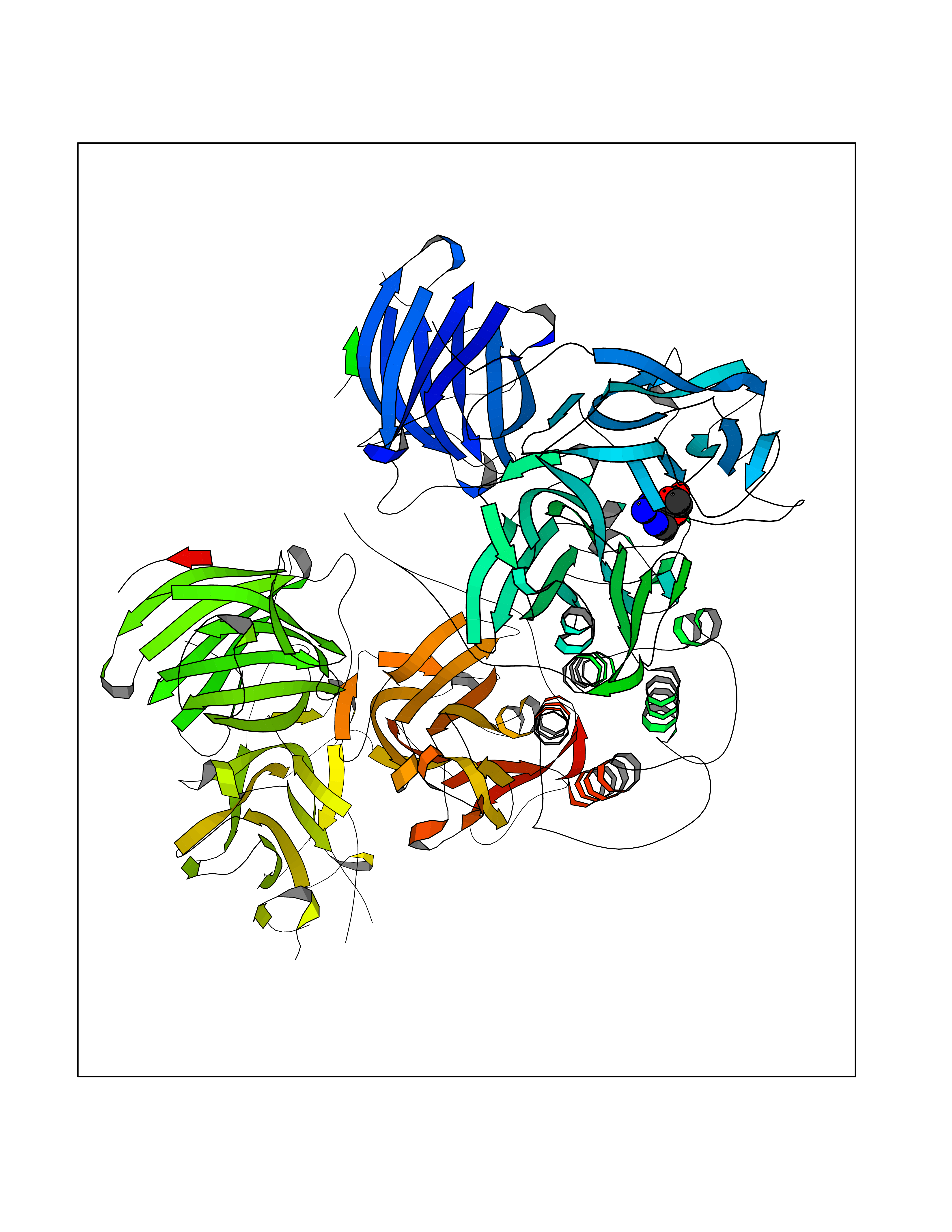 Molscript Map 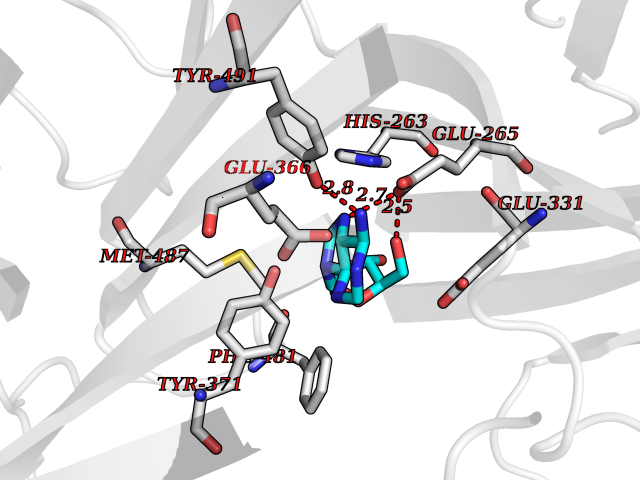 Pymol Map 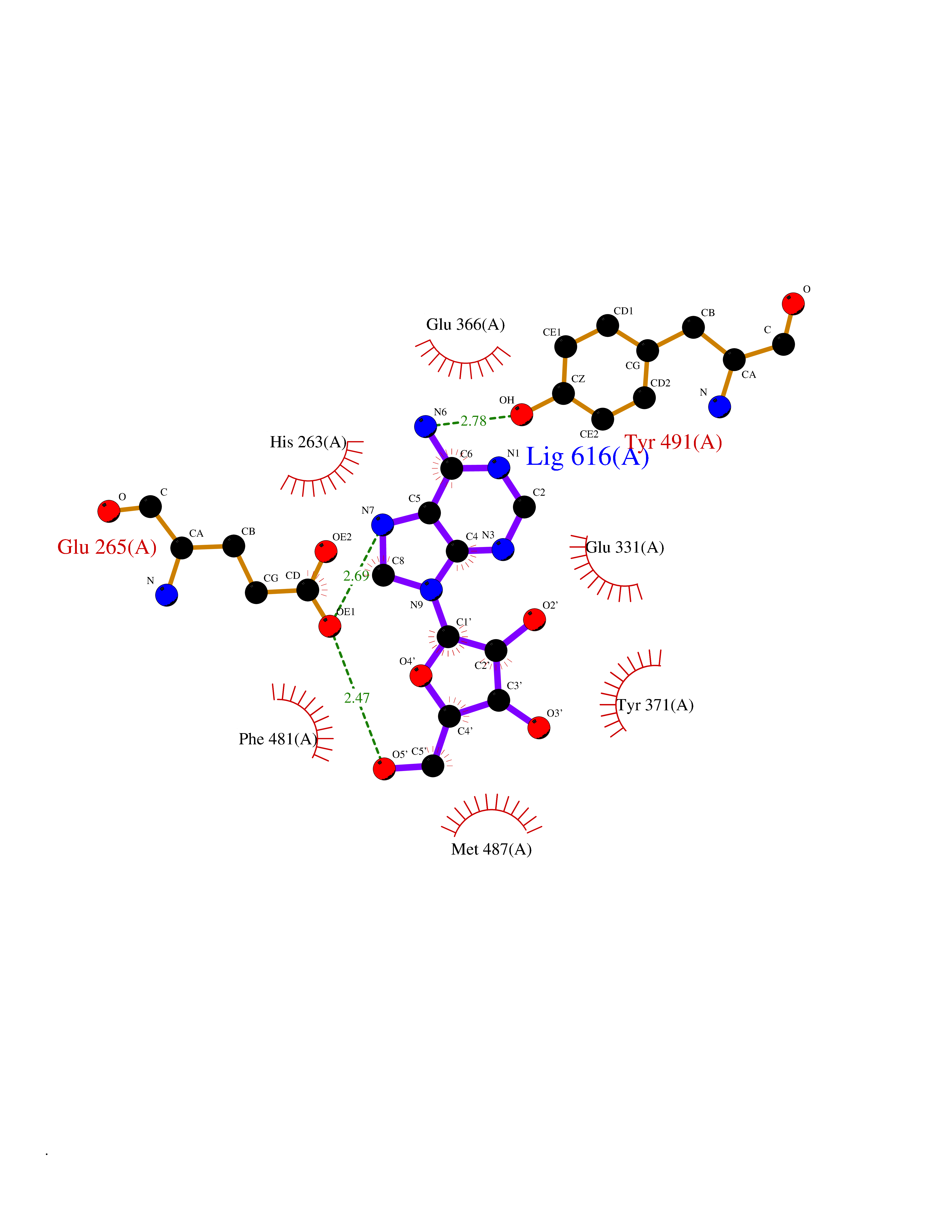 Ligplot Map 3D Binding mode Sequence PLPYHIPLDPEGSLELSWNVSYTQEAIHFQLLVRRLKAGVLFGMSDRGELENADLVVLWTDGDAYFADAWSDQKGQIHLDPQQDYQLLQVQRTPEGLTLLFKRPFGTCDPKDYLIEDGTVHLVYGILEEPFRSLEAINGSGLQMGLQRVQLLKPNIPEPELPSDACTMEVQAPNIQIPSQETTYWCYIKELPKGFSRHHIIKYEPIVTKGNEALVHHMEVFQCAPEMDSVPHFSGPCDSKMKPDRLNYCRHVLAAWALGAKAFYYPEEAGLAFGGPGSSRYLRLEVHYHNPLVIEGRNDSSGIRLYYTAKLRRFNAGIMELGLVYTPVMAIPPRETAFILTGYCTDKCTQLALPPSGIHIFASQLHTHLTGRKVVTVLVRDGREWEIVNQDNHYSPHFQEIRMLKKVVSVHPGDVLITSCTYNTEDRELATVGGFGILEEMCVNYVHYYPQTQLELCKSAVDAGFLQKYFHLINRFNNEDVCTCPQASVSQQFTSVPWNSFNRDVLKALYSFAPISMHCNKSSAVRFQGEWNLQPLPKVISTLEEPTVVSPLPYHIPLDPEGSLELSWNVSYTQEAIHFQLLVRRLKAGVLFGMSDRGELENADLVVLAYFADAWSDQKGQIHLDPQQDYQLLQVQRTPEGLTLLFKRPFGTCDPKDYLIEDGTVHLVYGILEEPFRSLEAINGSGLQMGLQRVQLLKPNIPEPELPSDACTMEVQAPNIQIPSQETTYWCYIKELPKGFSRHHIIKYEPIVTKGNEALVHHMEVFQCAPEVPHFSGPCDSKMLNYCRHVLAAWALGAKAFYYPEEAGLAFGGPGSSRYLRLEVHYHNPLVIEGRNDSSGIRLYYTAKLRRFNAGIMELGLVYTPVMAIPPRETAFILTGYCTDKCTQLALPPSGIHIFASQLHTHLTGRKVVTVLVRDGREWEIVNQDNHYSPHFQEIRMLKKVVSVHPGDVLITSCTYNTEDRELATVGGFGILEEMCVNYVHYYPQTQLELCKSAVDAGFLQKYFHLINRFNNEDVCTCPQASVSQQFTSVPWNSFNRDVLKALYSFAPISMHCNKSSAVRFQGEWNLQPLPKVISTLEEPTPQCVVSIGG Hydrogen bonds contact Hydrophobic contact | ||||
| 7 | Endothelin-converting enzyme 1 (ECE1) | 3DWB | 7.10 | |
Target general information Gen name ECE1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms ECE-1 Protein family Peptidase M13 family Biochemical class Peptidase Function Converts big endothelin-1 to endothelin-1. Related diseases Hirschsprung disease, cardiac defects, and autonomic dysfunction (HCAD) [MIM:613870]: A disorder characterized by skip-lesions Hirschsprung disease, craniofacial abnormalities and other dysmorphic features, cardiac defects including ductus arteriosus, small subaortic ventricular septal defect, small atrial septal defect, and autonomic dysfunction. {ECO:0000269|PubMed:9915973}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07171 Interacts with P49760; A8MQ03; Q8IUG1; P60370; P60410 EC number EC 3.4.24.71 Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disease variant; Disulfide bond; Glycoprotein; Hirschsprung disease; Hydrolase; Membrane; Metal-binding; Metalloprotease; Phosphoprotein; Protease; Proteomics identification; Reference proteome; Signal-anchor; Transmembrane; Transmembrane helix; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 75247.9 Length 660 Aromaticity 0.12 Instability index 46.29 Isoelectric point 5.33 Charge (pH=7) -18.3 2D Binding mode Binding energy (Kcal/mol) -7.09 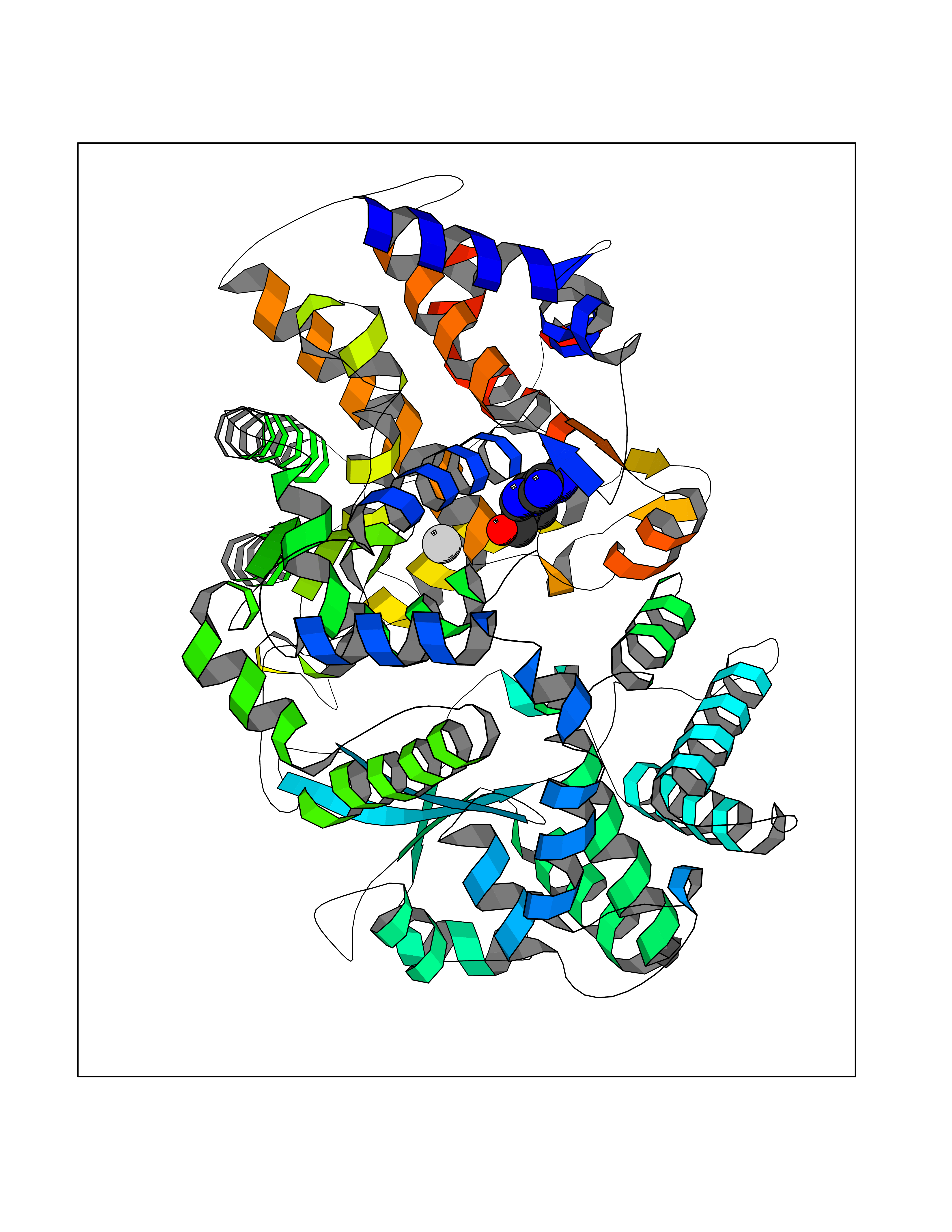 Molscript Map 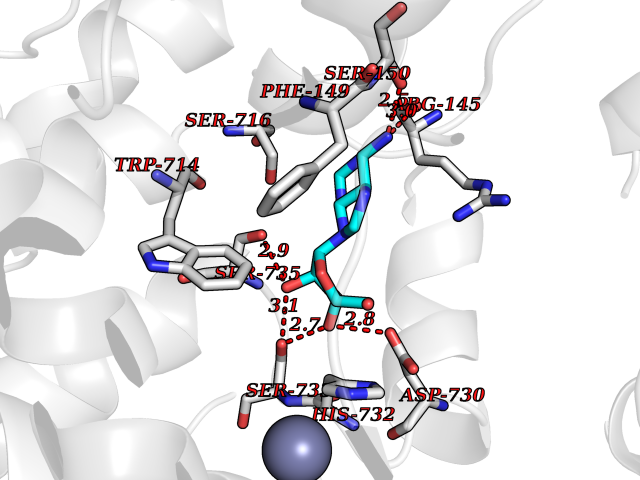 Pymol Map 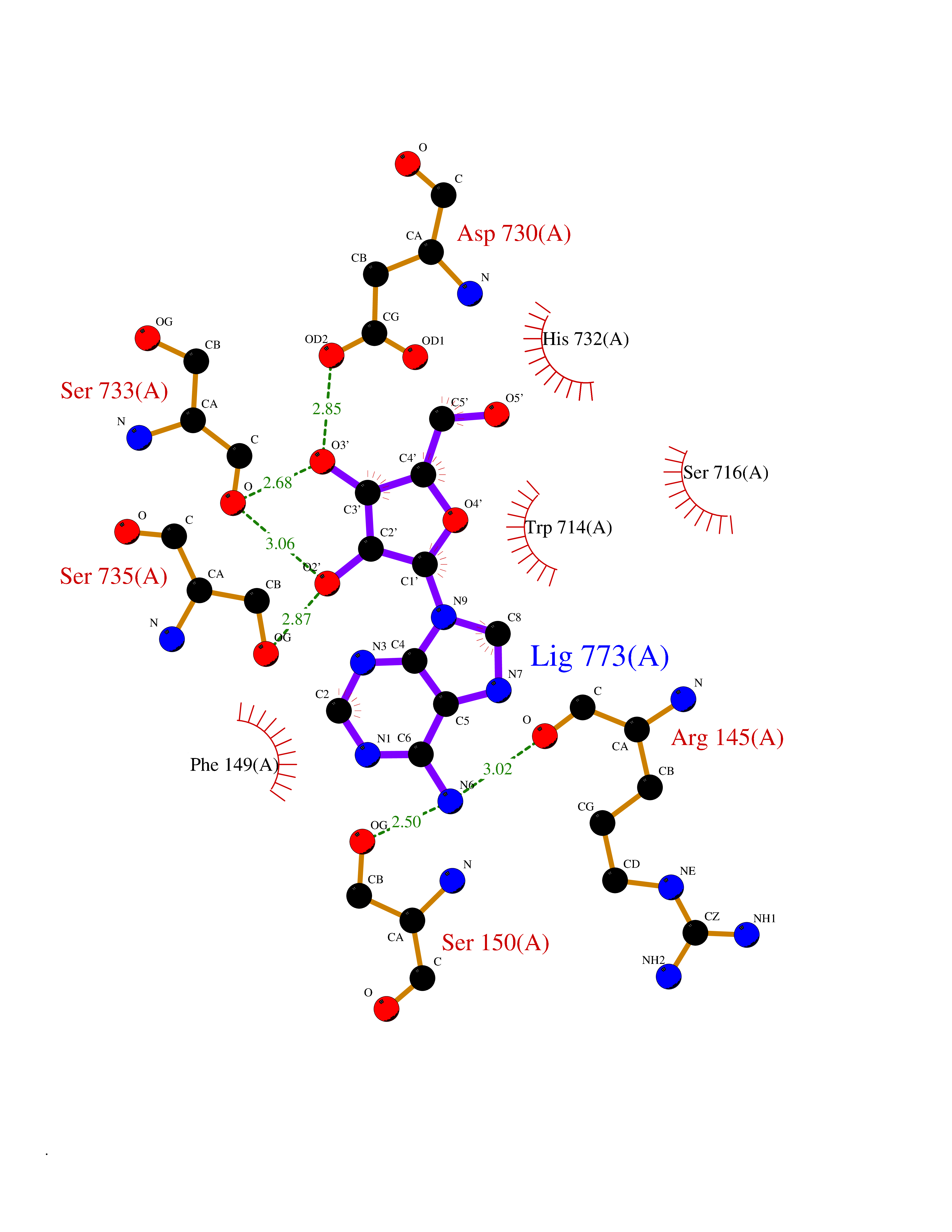 Ligplot Map 3D Binding mode Sequence SEACVSVTSSILSSMDPTVDPCHDFFSYACGGWIKANPVPDGHSRWGTFSNLWEHNQAIIKHLLENSTASVSEAERKAQVYYRACMNETRIEELRAKPLMELIERLGGWNITGPWAKDNFQDTLQVVTAHYRTSPFFSVYVSADSKNSNSNVIQVDQSGLGLPSRDYYLNKTENEKVLTGYLNYMVQLGKLLGGGDEEAIRPQMQQILDFETALANITIPQEKRRDEELIYHKVTAAELQTLAPAINWLPFLNTIFYPVEINESEPIVVYDKEYLEQISTLINTTDRCLLNNYMIWNLVRKTSSFLDQRFQDADEKFMEVMWKFCVSDTENNLGFALGPMFVKATFAEDSKSIATEIILEIKKAFEESLSTLKWMDEETRKSAKEKADAIYNMIGYPNFIMDPKELDKVFNDYTAVPDLYFENAMRFFNFSWRVTADQLRKAPNRDQWSMTPPMVNAYYSPTKNEIVFPAGILQAPFYTRSSPKALNFGGIGVVVGHELTHAFDDQGREYDKDGNLRPWWKNSSVEAFKRQTECMVEQYSNYSVNGEPVNGRHTLGENIADNGGLKAAYRAYQNWVKKNGAEHSLPTLGLTNNQLFFLGFAQVWCSVRTPESSHEGLITDPHSPSRFRVIGSLSNSKEFSEHFRCPPGSPMNPPHKCEVW Hydrogen bonds contact Hydrophobic contact | ||||
| 8 | Adenosine deaminase (ADA) | 3IAR | 7.10 | |
Target general information Gen name ADA Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Adenosine aminohydrolase; ADA1 Protein family Metallo-dependent hydrolases superfamily, Adenosine and AMP deaminases family Biochemical class Carbon-nitrogen hydrolase Function Plays an important role in purine metabolism and in adenosine homeostasis. Modulates signaling by extracellular adenosine, and so contributes indirectly to cellular signaling events. Acts as a positive regulator of T-cell coactivation, by binding DPP4. Its interaction with DPP4 regulates lymphocyte-epithelial cell adhesion. Enhances dendritic cell immunogenicity by affecting dendritic cell costimulatory molecule expression and cytokines and chemokines secretion. Enhances CD4+ T-cell differentiation and proliferation. Acts as a positive modulator of adenosine receptors ADORA1 and ADORA2A, by enhancing their ligand affinity via conformational change. Stimulates plasminogen activation. Plays a role in male fertility. Plays a protective role in early postimplantation embryonic development. Catalyzes the hydrolytic deamination of adenosine and 2-deoxyadenosine. Related diseases Severe combined immunodeficiency autosomal recessive T-cell-negative/B-cell-negative/NK-cell-negative due to adenosine deaminase deficiency (ADASCID) [MIM:102700]: An autosomal recessive disorder accounting for about 50% of non-X-linked SCIDs. SCID refers to a genetically and clinically heterogeneous group of rare congenital disorders characterized by impairment of both humoral and cell-mediated immunity, leukopenia, and low or absent antibody levels. Patients with SCID present in infancy with recurrent, persistent infections by opportunistic organisms. The common characteristic of all types of SCID is absence of T-cell-mediated cellular immunity due to a defect in T-cell development. ADA deficiency has been diagnosed in chronically ill teenagers and adults (late or adult onset). Population and newborn screening programs have also identified several healthy individuals with normal immunity who have partial ADA deficiency. {ECO:0000269|PubMed:10200056, ECO:0000269|PubMed:1284479, ECO:0000269|PubMed:2166947, ECO:0000269|PubMed:2783588, ECO:0000269|PubMed:3182793, ECO:0000269|PubMed:3839802, ECO:0000269|PubMed:6208479, ECO:0000269|PubMed:7599635, ECO:0000269|PubMed:8227344, ECO:0000269|PubMed:8299233, ECO:0000269|PubMed:9361033}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07711; DB07783; DB07786; DB04218; DB07785; DB03015; DB02472; DB00640; DB00975; DB14598; DB14600; DB00974; DB05057; DB03220; DB02616; DB02096; DB03572; DB02830; DB03370; DB04440; DB01280; DB00552; DB03000; DB00277; DB00194 Interacts with A5A3E0 EC number EC 3.5.4.4 Uniprot keywords 3D-structure; Acetylation; Cell adhesion; Cell junction; Cell membrane; Cytoplasm; Cytoplasmic vesicle; Direct protein sequencing; Disease variant; Hereditary hemolytic anemia; Hydrolase; Lysosome; Membrane; Metal-binding; Nucleotide metabolism; Proteomics identification; Reference proteome; SCID; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 40403.5 Length 360 Aromaticity 0.08 Instability index 38.7 Isoelectric point 5.63 Charge (pH=7) -9.06 2D Binding mode Binding energy (Kcal/mol) -7.35  Molscript Map 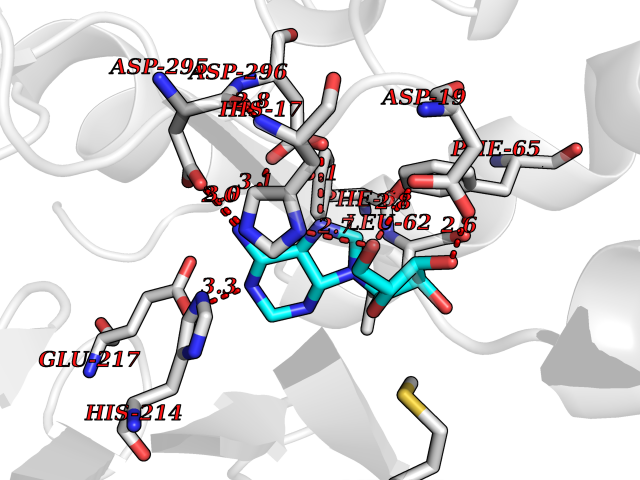 Pymol Map 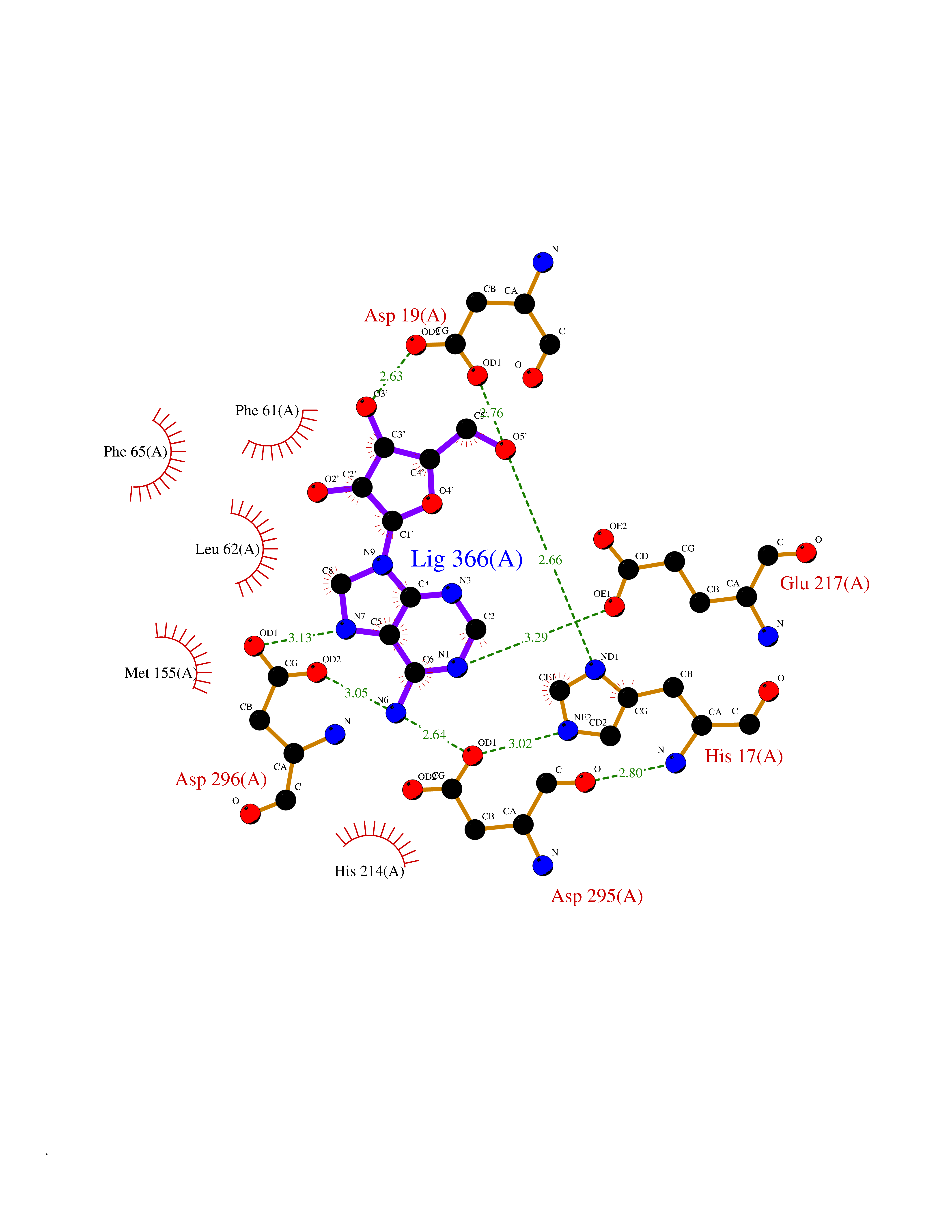 Ligplot Map 3D Binding mode Sequence PAFDKPKVELHVHLDGSIKPETILYYGRRRGIALPANTAEGLLNVIGMDKPLTLPDFLAKFDYYMPAIAGCREAIKRIAYEFVEMKAKEGVVYVEVRYSPHLLANSKVEPIPWNQAEGDLTPDEVVALVGQGLQEGERDFGVKARSILCCMRHQPNWSPKVVELCKKYQQQTVVAIDLAGDETIPGSSLLPGHVQAYQEAVKSGIHRTVHAGEVGSAEVVKEAVDILKTERLGHGYHTLEDQALYNRLRQENMHFEICPWSSYLTGAWKPDTEHAVIRLKNDQANYSLNTDDPLIFKSTLDTDYQMTKRDMGFTEEEFKRLNINAAKSSFLPEDEKRELLDLLYKAYGMPPSASAGQNLA Hydrogen bonds contact Hydrophobic contact | ||||
| 9 | SET domain containing 8 (KMT5A) | 5TEG | 7.08 | |
Target general information Gen name KMT5A Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms SETD8; SET8; SET07; SET domain-containing protein 8; PRSET7; PR/SET07; PR/SET domain-containing protein 07; PR-Set7; N-lysine methyltransferase KMT5A; Lysine-specific methylase 5A; Lysine N-methyltran Protein family Class V-like SAM-binding methyltransferase superfamily, Histone-lysine methyltransferase family, PR/SET subfamily Biochemical class Methyltransferase Function Specifically monomethylates 'Lys-20' of histone H4 (H4K20me1). H4K20me1 is enriched during mitosis and represents a specific tag for epigenetic transcriptional repression. Mainly functions in euchromatin regions, thereby playing a central role in the silencing of euchromatic genes. Required for cell proliferation, probably by contributing to the maintenance of proper higher-order structure of DNA during mitosis. Involved in chromosome condensation and proper cytokinesis. Nucleosomes are preferred as substrate compared to free histones. Mediates monomethylation of p53/TP53 at 'Lys-382', leading to repress p53/TP53-target genes. Plays a negative role in TGF-beta response regulation and a positive role in cell migration. Protein-lysine N-methyltransferase that monomethylates both histones and non-histone proteins. Related diseases Sick sinus syndrome 2 (SSS2) [MIM:163800]: The term 'sick sinus syndrome' encompasses a variety of conditions caused by sinus node dysfunction. The most common clinical manifestations are syncope, presyncope, dizziness, and fatigue. Electrocardiogram typically shows sinus bradycardia, sinus arrest, and/or sinoatrial block. Episodes of atrial tachycardias coexisting with sinus bradycardia ('tachycardia-bradycardia syndrome') are also common in this disorder. SSS occurs most often in the elderly associated with underlying heart disease or previous cardiac surgery, but can also occur in the fetus, infant, or child without heart disease or other contributing factors. SSS2 onset is in utero or at birth. {ECO:0000269|PubMed:15123648, ECO:0000269|PubMed:16407510, ECO:0000269|PubMed:20662977, ECO:0000269|PubMed:23103389}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Brugada syndrome 8 (BRGDA8) [MIM:613123]: A tachyarrhythmia characterized by right bundle branch block and ST segment elevation on an electrocardiogram (ECG). It can cause the ventricles to beat so fast that the blood is prevented from circulating efficiently in the body. When this situation occurs, the individual will faint and may die in a few minutes if the heart is not reset. {ECO:0000269|PubMed:19165230}. The gene represented in this entry may be involved in disease pathogenesis.; DISEASE: Epilepsy, idiopathic generalized 18 (EIG18) [MIM:619521]: An autosomal dominant form of idiopathic generalized epilepsy, a disorder characterized by recurring generalized seizures in the absence of detectable brain lesions and/or metabolic abnormalities. Generalized seizures arise diffusely and simultaneously from both hemispheres of the brain. Seizure types include juvenile myoclonic seizures, absence seizures, and generalized tonic-clonic seizures. EIG18 is characterized by onset of myoclonic seizures in infancy. Although the seizures remit, some patients may have later speech or cognitive impairment. {ECO:0000269|PubMed:30127718}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P62805; P07910; Q15672 EC number EC 2.1.1.- Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Cell cycle; Cell division; Chromatin regulator; Chromosome; Coiled coil; Direct protein sequencing; Methyltransferase; Mitosis; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Repressor; S-adenosyl-L-methionine; Transcription; Transcription regulation; Transferase; Ubl conjugation Protein physicochemical properties Chain ID A,D Molecular weight (Da) 19129.4 Length 167 Aromaticity 0.08 Instability index 49.18 Isoelectric point 7.88 Charge (pH=7) 1.37 2D Binding mode Binding energy (Kcal/mol) -7.98 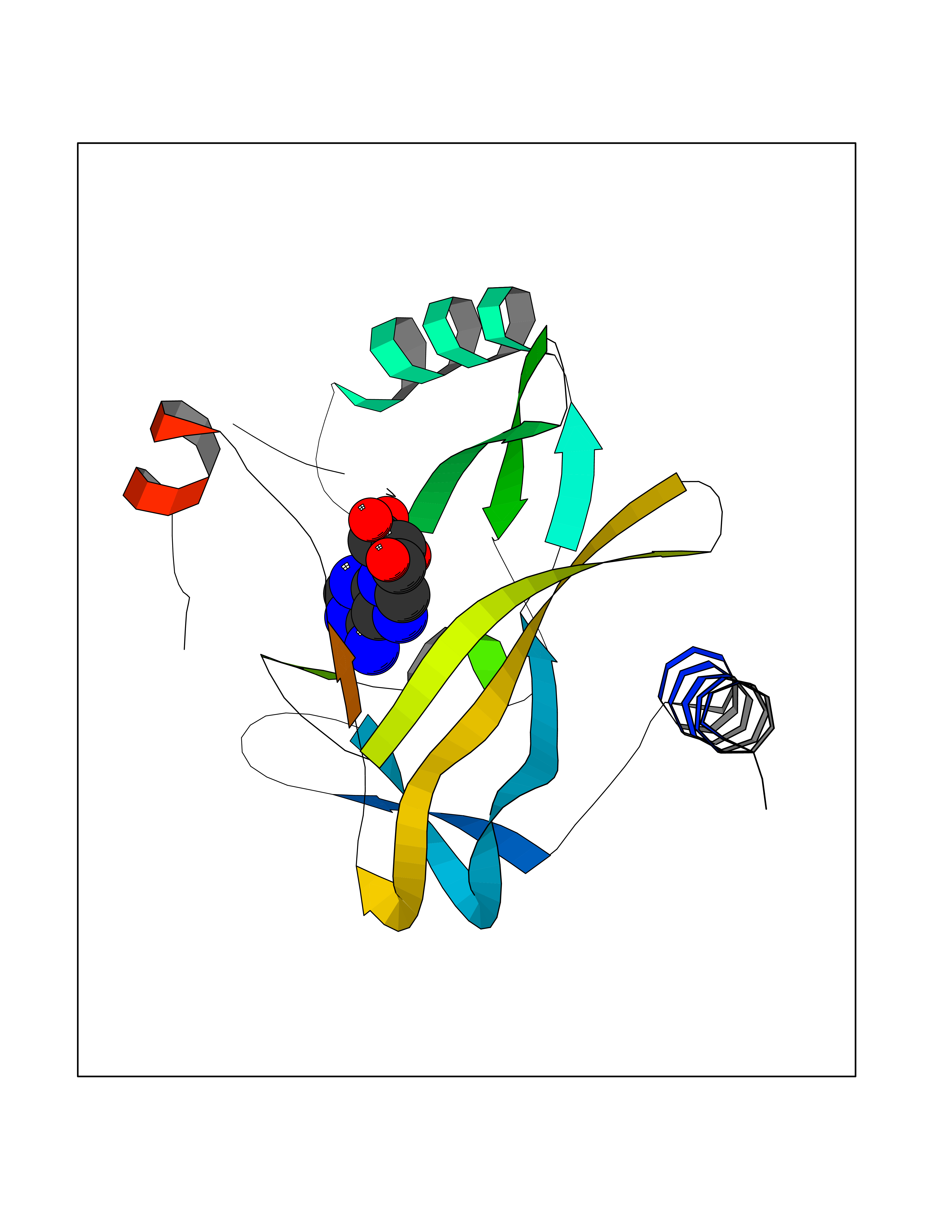 Molscript Map 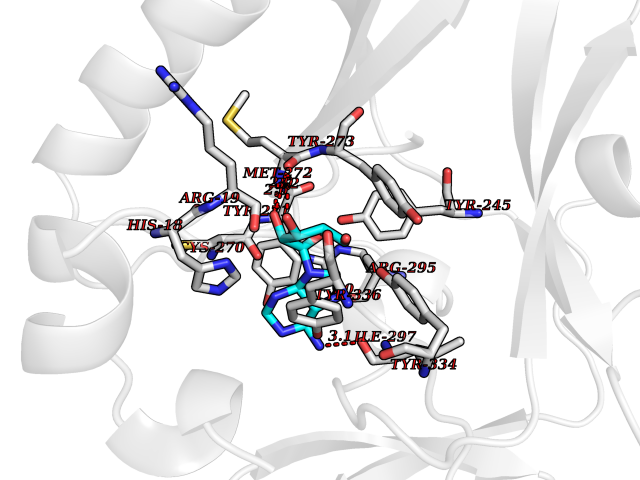 Pymol Map 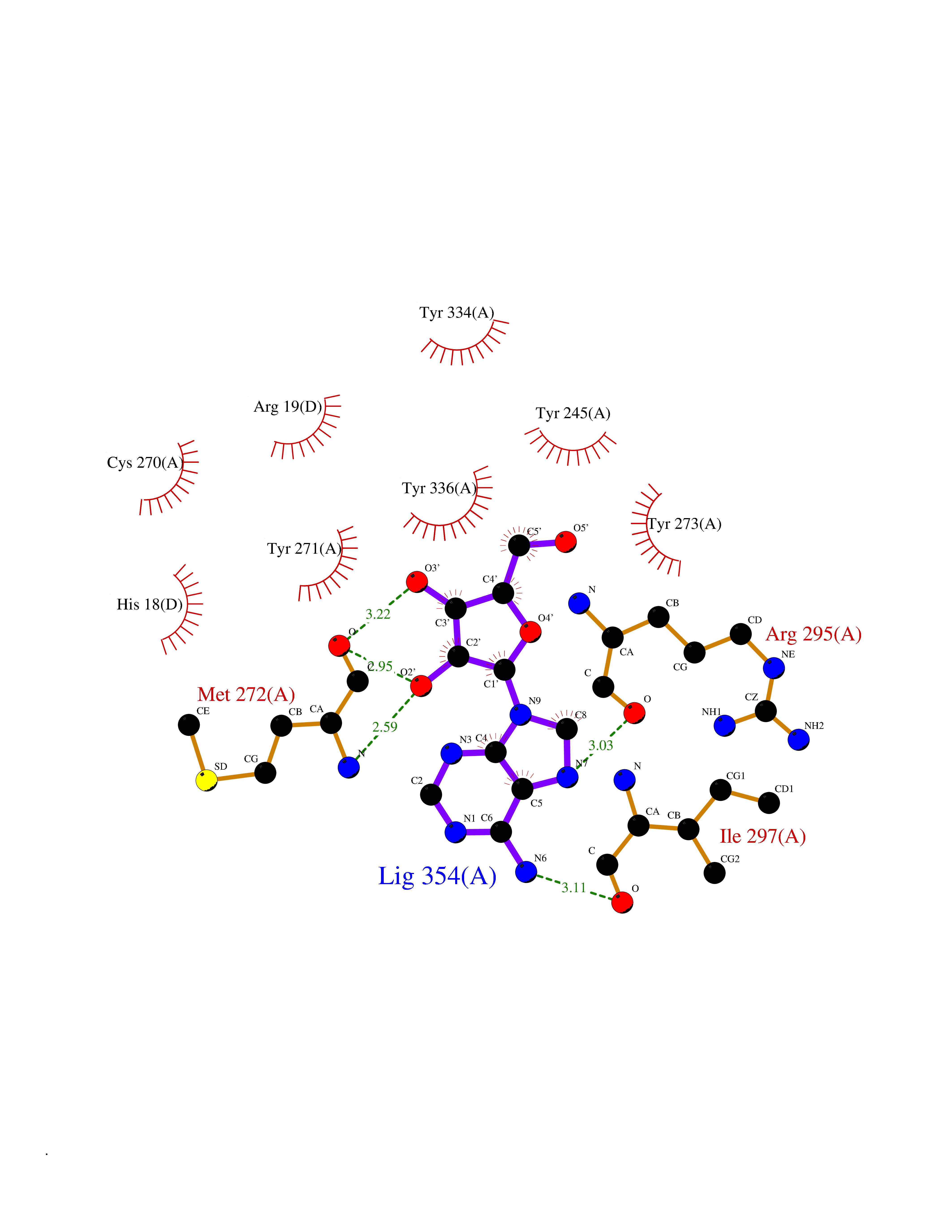 Ligplot Map 3D Binding mode Sequence KSKAELQSEERKRIDELIESGKEEGMKIDLIDGKGRGVIATKQFSRGDFVVEYHGDLIEITDAKKREALYAQDPSTGCYMYYFQYLSKTYCVDATRETNRLGRLINHSKSGNCQTKLHDIDGVPHLILIASRDIAAGEELLYDYGDRSKASIEAHPWLKHKRHRVLR Hydrogen bonds contact Hydrophobic contact | ||||
| 10 | Alpha-N-acetylglucosaminidase (NAGLU) | 4XWH | 7.07 | |
Target general information Gen name NAGLU Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms UFHSD1; NAGLU; NAG; N-acetyl-alpha-glucosaminidase; Alpha-N-acetylglucosaminidase82 kDa form; Alpha-N-acetylglucosaminidase77 kDa form Protein family Glycosyl hydrolase 89 family Biochemical class Glycosylase Function Involved in the degradation of heparan sulfate. Related diseases Mucopolysaccharidosis 3B (MPS3B) [MIM:252920]: A form of mucopolysaccharidosis type 3, an autosomal recessive lysosomal storage disease due to impaired degradation of heparan sulfate. MPS3 is characterized by severe central nervous system degeneration, but only mild somatic disease. Onset of clinical features usually occurs between 2 and 6 years; severe neurologic degeneration occurs in most patients between 6 and 10 years of age, and death occurs typically during the second or third decade of life. {ECO:0000269|PubMed:10094189, ECO:0000269|PubMed:11068184, ECO:0000269|PubMed:11153910, ECO:0000269|PubMed:11286389, ECO:0000269|PubMed:11793481, ECO:0000269|PubMed:11836372, ECO:0000269|PubMed:12202988, ECO:0000269|PubMed:14984474, ECO:0000269|PubMed:15933803, ECO:0000269|PubMed:16151907, ECO:0000269|PubMed:28101780, ECO:0000269|PubMed:9443875, ECO:0000269|PubMed:9443878, ECO:0000269|PubMed:9832037, ECO:0000269|PubMed:9950362}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Charcot-Marie-Tooth disease, axonal, 2V (CMT2V) [MIM:616491]: An axonal form of Charcot-Marie-Tooth disease, a disorder of the peripheral nervous system, characterized by progressive weakness and atrophy, initially of the peroneal muscles and later of the distal muscles of the arms. Charcot-Marie-Tooth disease is classified in two main groups on the basis of electrophysiologic properties and histopathology: primary peripheral demyelinating neuropathies (designated CMT1 when they are dominantly inherited) and primary peripheral axonal neuropathies (CMT2). Neuropathies of the CMT2 group are characterized by signs of axonal degeneration in the absence of obvious myelin alterations, normal or slightly reduced nerve conduction velocities, and progressive distal muscle weakness and atrophy. CMT2V is an autosomal dominant sensory neuropathy with late onset. The main clinical feature is recurrent leg pain that progresses to constant painful paraesthesias in the feet and later the hands. As it evolves, some patients develop a mild sensory ataxia. {ECO:0000269|PubMed:25818867}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB06773; DB00141 Interacts with NA EC number EC 3.2.1.50 Uniprot keywords 3D-structure; Charcot-Marie-Tooth disease; Direct protein sequencing; Disease variant; Glycoprotein; Glycosidase; Hydrolase; Lysosome; Mucopolysaccharidosis; Neurodegeneration; Neuropathy; Proteomics identification; Reference proteome; Signal Protein physicochemical properties Chain ID A Molecular weight (Da) 80206.3 Length 720 Aromaticity 0.13 Instability index 41.59 Isoelectric point 6.28 Charge (pH=7) -4.09 2D Binding mode Binding energy (Kcal/mol) -7.17 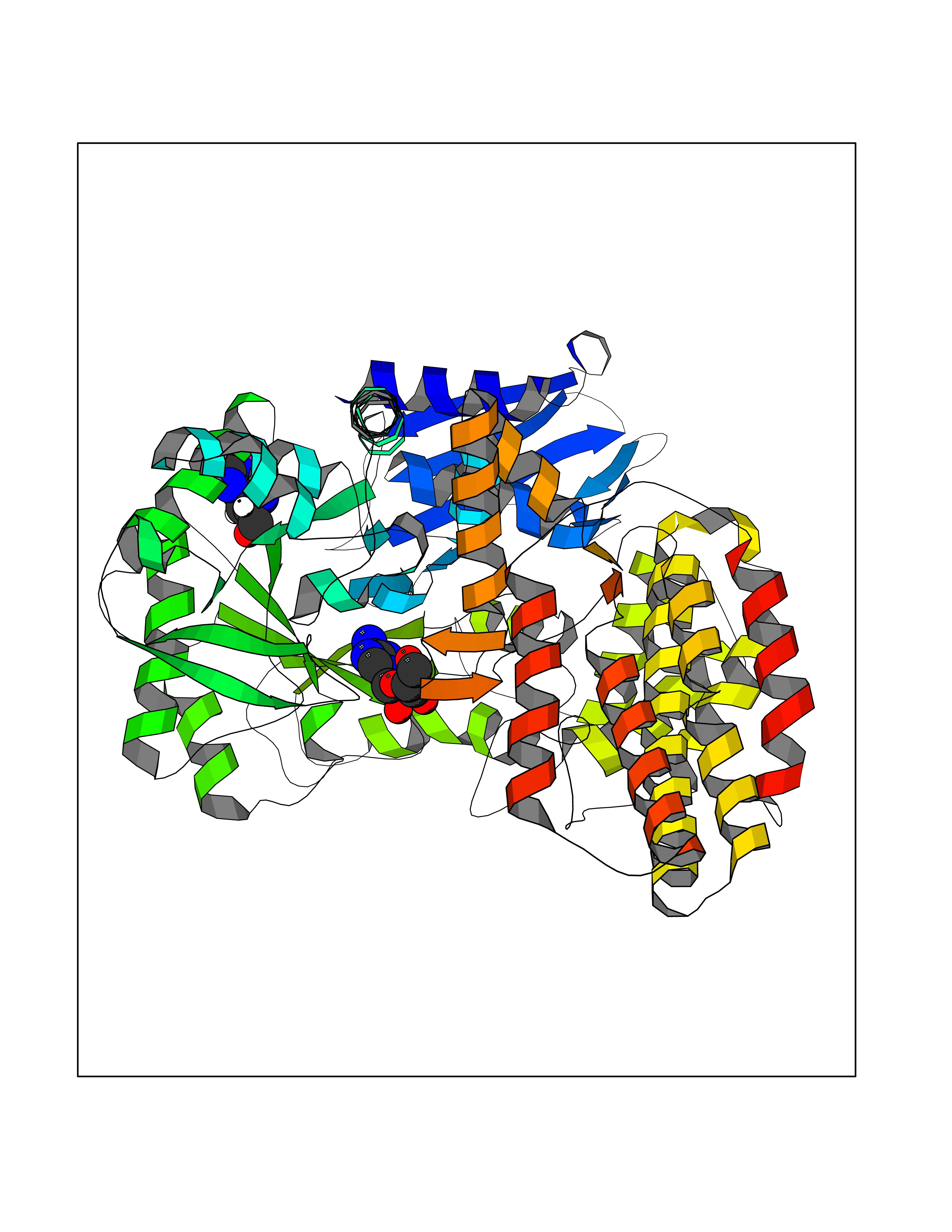 Molscript Map 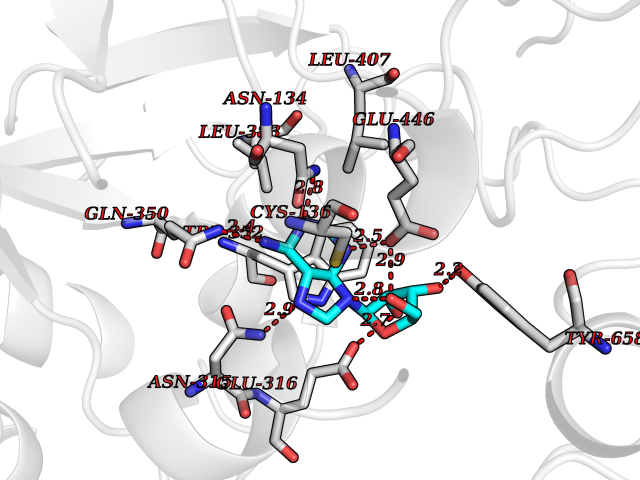 Pymol Map 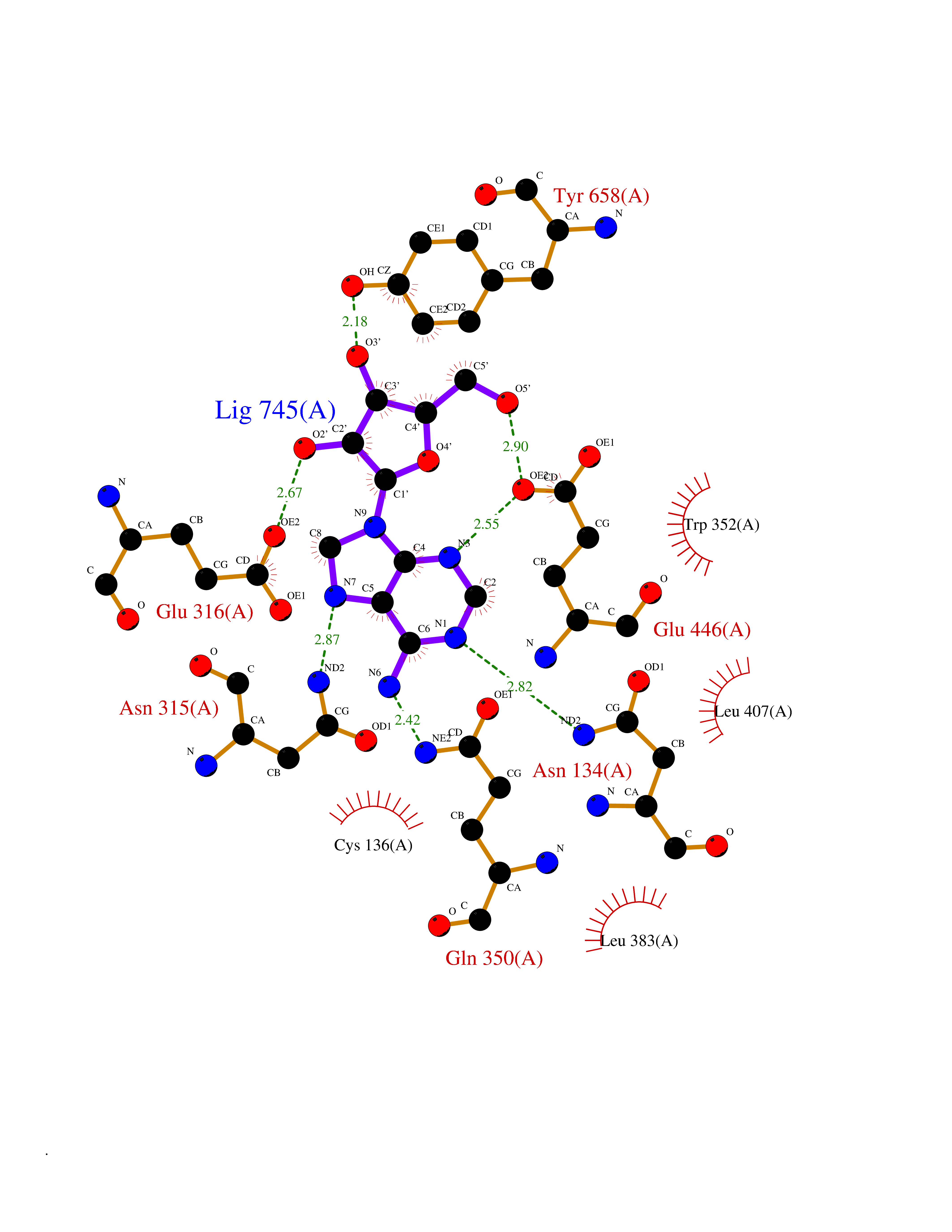 Ligplot Map 3D Binding mode Sequence DEAREAAAVRALVARLLGPGPAADFSVSVERALAAKPGLDTYSLGGGGAARVRVRGSTGVAAAAGLHRYLRDFCGCHVAWSGSQLRLPRPLPAVPGELTEATPNRYRYYQNVCTQSYSFVWWDWARWEREIDWMALNGINLALAWSGQEAIWQRVYLALGLTQAEINEFFTGPAFLAWGRMGNLHTWDGPLPPSWHIKQLYLQHRVLDQMRSFGMTPVLPAFAGHVPEAVTRVFPQVNVTKMGSWGHFNCSYSCSFLLAPEDPIFPIIGSLFLRELIKEFGTDXIYGADTFNEMQPPSSEPSYLAAATTAVYEAMTAVDTEAVWLLQGWLFQHQPQFWGPAQIRAVLGAVPRGRLLVLDLFAESQPVYTRTASFQGQPFIWCMLHNFGGNHGLFGALEAVNGGPEAARLFPNSTMVGTGMAPEGISQNEVVYSLMAELGWRKDPVPDLAAWVTSFAARRYGVSHPDAGAAWRLLLRSVYNCSGEACRGHNRSPLVRRPSLQMNTSIWYNRSDVFEAWRLLLTSAPSLATSPAFRYDLLDLTRQAVQELVSLYYEEARSAYLSKELASLLRAGGVLAYELLPALDEVLASDSRFLLGSWLEQARAAAVSEAEADFYEQNSRYQLTLWGPEGNILDYANKQLAGLVANYYTPRWRLFLEALVDSVAQGIPFQQHQFDKNVFQLEQAFVLSKQRYPSQPRGDTVDLAKKIFLKYYPRWVAGSW Hydrogen bonds contact Hydrophobic contact | ||||
| 11 | Purine nucleoside phosphorylase (PNP) | 4EAR | 7.06 | |
Target general information Gen name PNP Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms PNP; Inosine phosphorylase Protein family PNP/MTAP phosphorylase family Biochemical class Pentosyltransferase Function The purine nucleoside phosphorylases catalyze the phosphorolytic breakdown of the N-glycosidic bond in the beta- (deoxy)ribonucleoside molecules, with the formation of the corresponding free purine bases and pentose-1-phosphate. Related diseases Purine nucleoside phosphorylase deficiency (PNPD) [MIM:613179]: A disorder that interrupts both the catabolism of inosine into hypoxanthine and guanosine into guanine, and leads to the accumulation of guanosine, inosine, and their deoxified by-products. The main clinical presentation is recurrent infections due to severe T-cell immunodeficiency. Some patients also have neurologic impairment. {ECO:0000269|PubMed:1384322, ECO:0000269|PubMed:3029074, ECO:0000269|PubMed:8931706}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03881; DB03551; DB02222; DB02391; DB03609; DB01667; DB04260; DB02796; DB04753; DB00640; DB00242; DB00900; DB06185; DB02377; DB02857; DB04754; DB04757; DB04076; DB02230; DB04335; DB02568; DB03101 Interacts with P05067; Q9UQM7; O14576-2; P06241; P14136; Q92993-2; Q9BXM7; P00491; P17612; P63000; Q92673; Q15583 EC number EC 2.4.2.1 Uniprot keywords 3D-structure; Acetylation; Cytoplasm; Direct protein sequencing; Disease variant; Glycosyltransferase; Phosphoprotein; Proteomics identification; Purine salvage; Reference proteome; Transferase Protein physicochemical properties Chain ID A,B,C Molecular weight (Da) 31849.2 Length 288 Aromaticity 0.1 Instability index 34.77 Isoelectric point 6.42 Charge (pH=7) -1.63 2D Binding mode Binding energy (Kcal/mol) -7.53 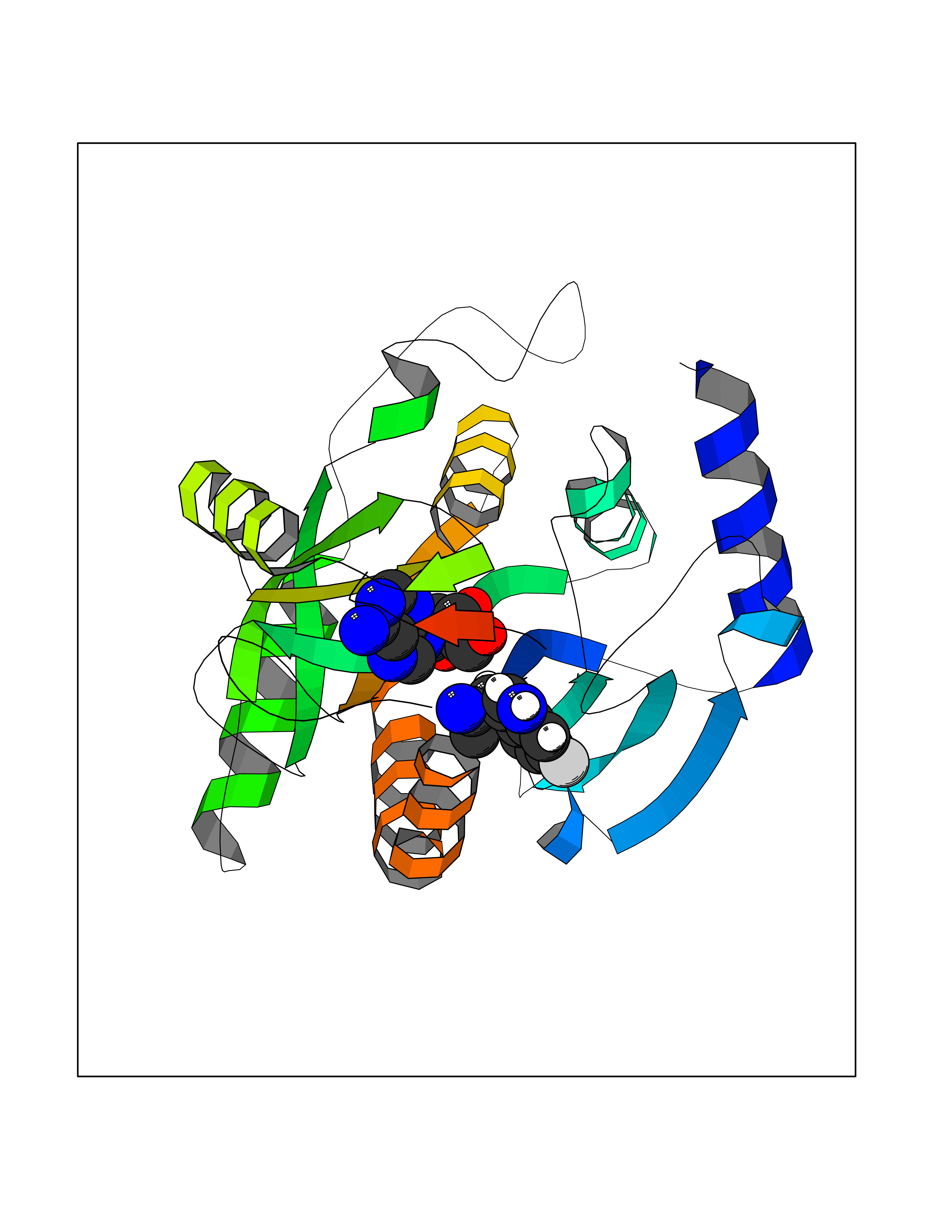 Molscript Map 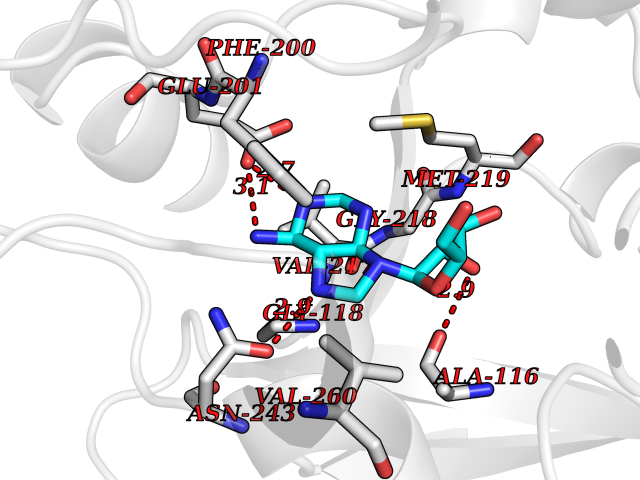 Pymol Map 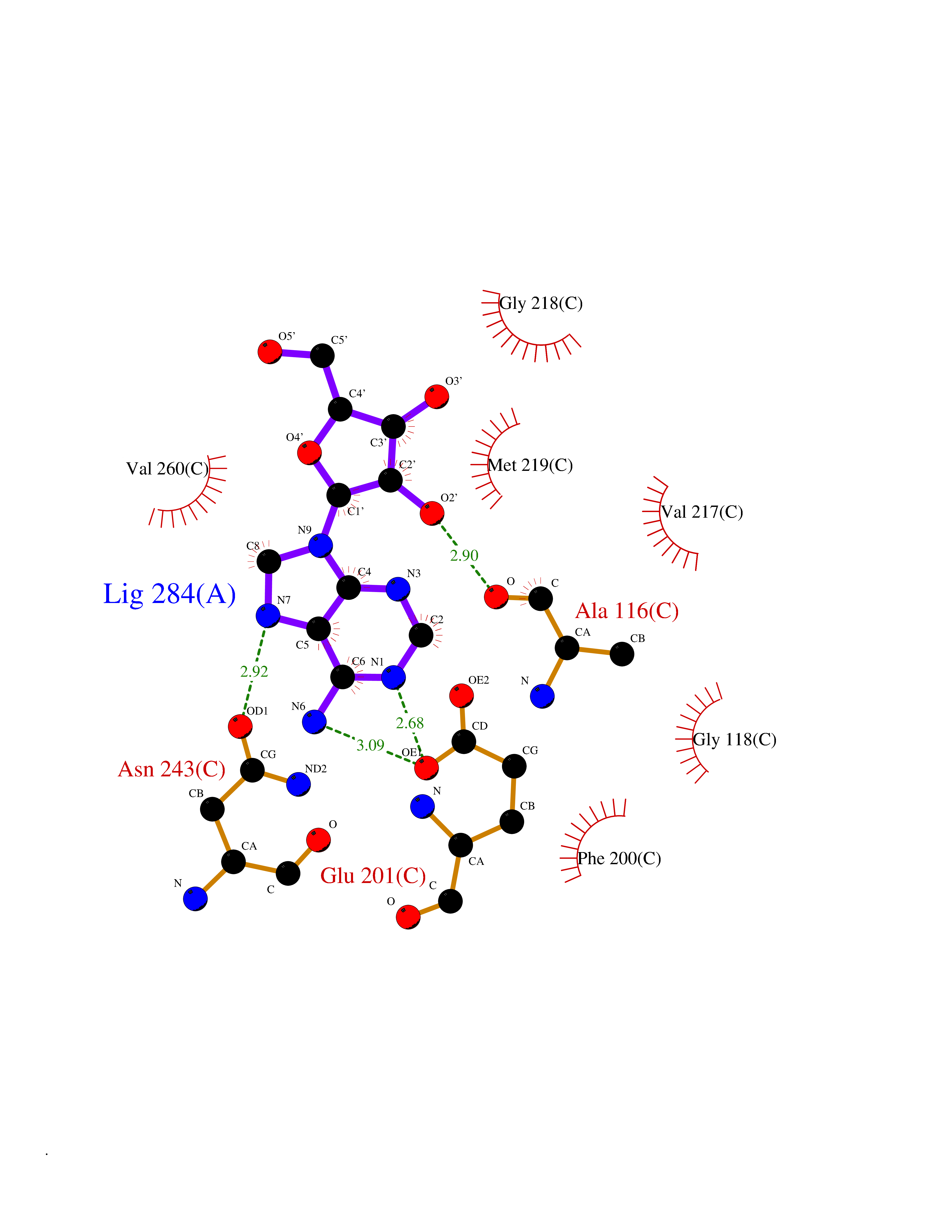 Ligplot Map 3D Binding mode Sequence GYTYEDYKNTAEYLLSHTKHRPQVAIICGSGLGGLTDKLTQAQIFDYSEIPNFPRSTVPGHAGRLVFGFLNGRACVMMQGRFHMYEGYPLYKVTFPVRVFHLLGVDTLVVTNAAGGLNPKFEVGDIMLIRDHINLPGFSGQNPLRGPNDERFGDRFPAMSDAYDRTMRQRALSTYKQMGEQRELQEGTYVMVAGPSFETVAECRVLQKLGADAVGMSTVPEVIVARHCGLRVFGFSLITNKVIMDYESLEKANXEEVLAAGKQAAQKLEQFVSILMASIDRFPAMSDA Hydrogen bonds contact Hydrophobic contact | ||||
| 12 | Carbonic anhydrase IV (CA-IV) | 3FW3 | 7.04 | |
Target general information Gen name CA4 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Carbonic anhydrase 4; Carbonate dehydratase IV; CAIV Protein family Alpha-carbonic anhydrase family Biochemical class Alpha-carbonic anhydrase Function May stimulate the sodium/bicarbonate transporter activity of SLC4A4 that acts in pH homeostasis. It is essential for acid overload removal from the retina and retina epithelium, and acid release in the choriocapillaris in the choroid. Reversible hydration of carbon dioxide. Related diseases Retinitis pigmentosa 17 (RP17) [MIM:600852]: A retinal dystrophy belonging to the group of pigmentary retinopathies. Retinitis pigmentosa is characterized by retinal pigment deposits visible on fundus examination and primary loss of rod photoreceptor cells followed by secondary loss of cone photoreceptors. Patients typically have night vision blindness and loss of midperipheral visual field. As their condition progresses, they lose their far peripheral visual field and eventually central vision as well. {ECO:0000269|PubMed:15563508, ECO:0000269|PubMed:17652713, ECO:0000269|PubMed:20450258}. The disease is caused by variants affecting the gene represented in this entry. Defective acid overload removal from retina and retinal epithelium, due to mutant CA4, is responsible for photoreceptor degeneration, indicating that impaired pH homeostasis is the most likely cause underlying the RP17 phenotype. Drugs (DrugBank ID) DB00819; DB00436; DB00562; DB01194; DB00606; DB01144; DB00869; DB08846; DB00311; DB00774; DB00703; DB00232; DB09460; DB00273; DB01021; DB00909 Interacts with NA EC number EC 4.2.1.1 Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; GPI-anchor; Lipoprotein; Lyase; Membrane; Metal-binding; Proteomics identification; Reference proteome; Retinitis pigmentosa; Signal; Zinc Protein physicochemical properties Chain ID A,B Molecular weight (Da) 27055.7 Length 235 Aromaticity 0.09 Instability index 44.3 Isoelectric point 6.87 Charge (pH=7) -0.36 2D Binding mode Binding energy (Kcal/mol) -6.97 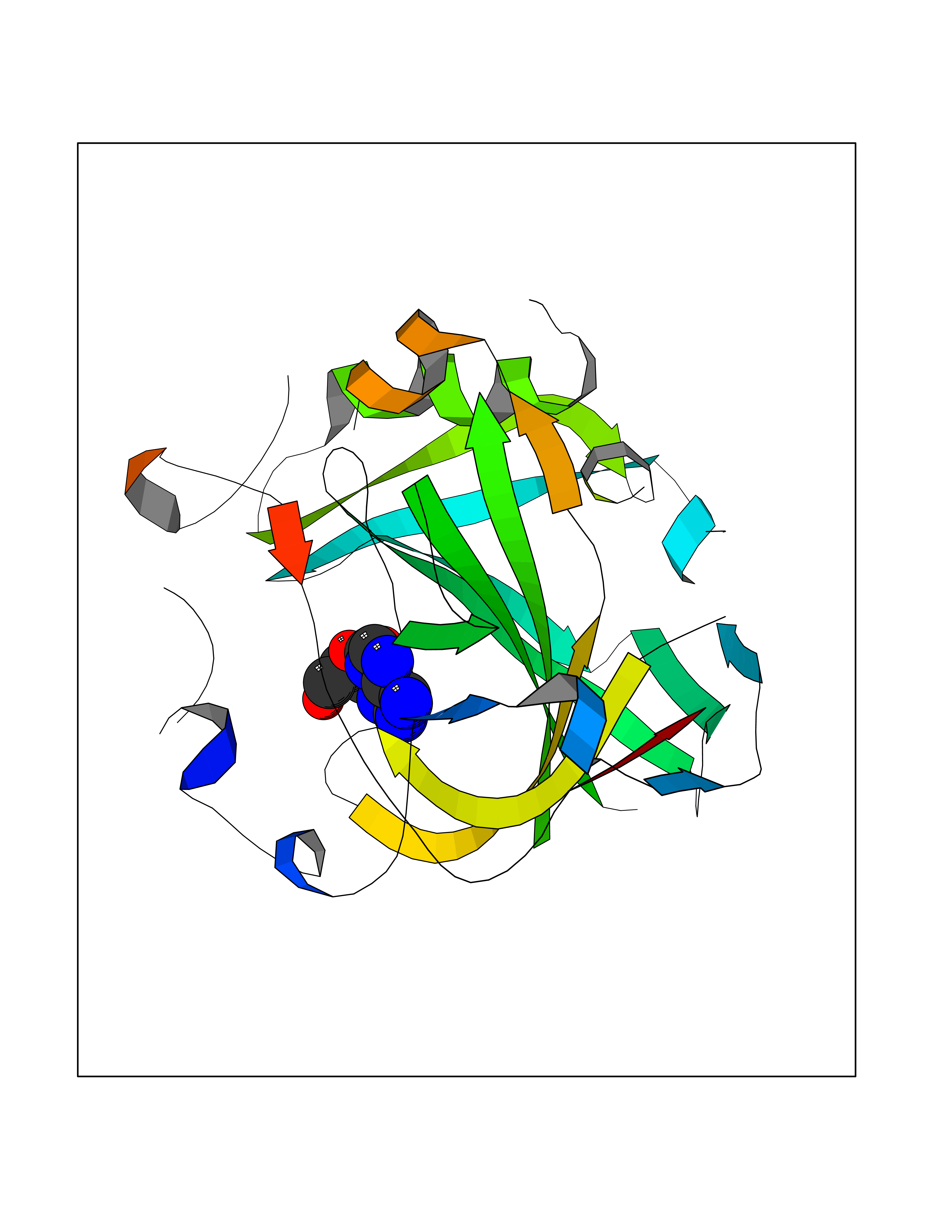 Molscript Map 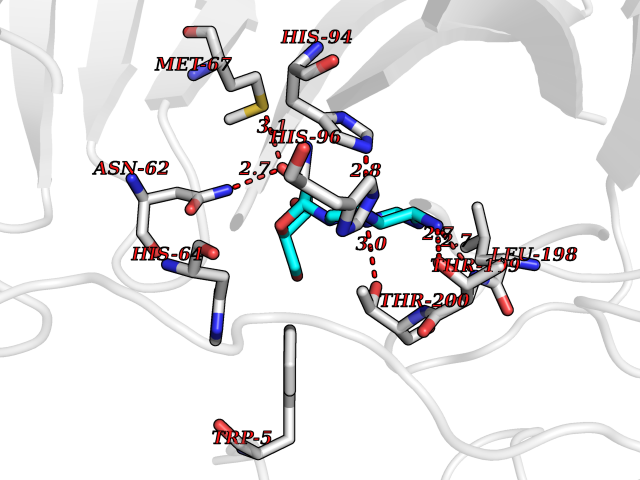 Pymol Map 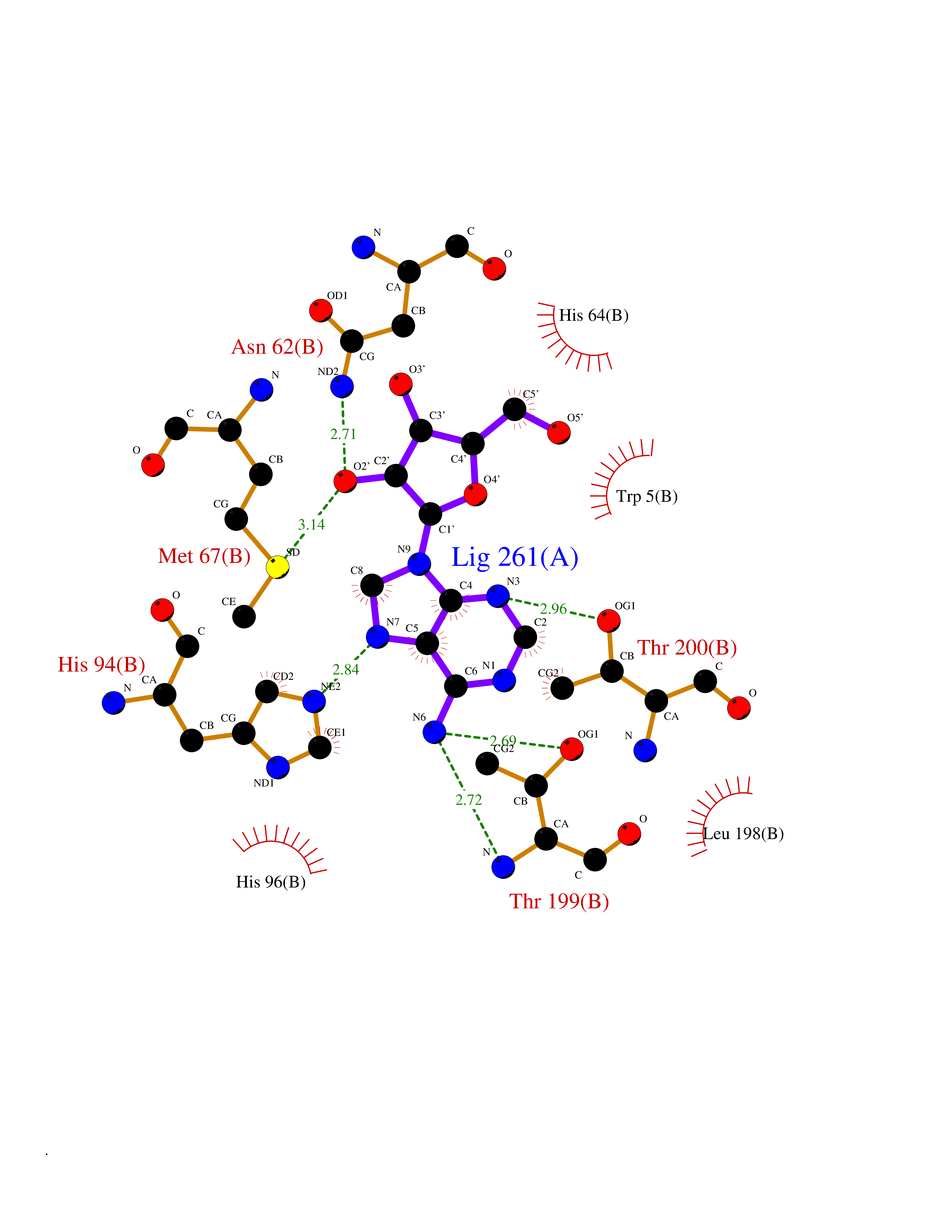 Ligplot Map 3D Binding mode Sequence HWCYEVQLVPVKWGGNCQKDRQSPINIVTTKAKVDKKLGRFFFGYDKKQTWTVQNNGHSVMMLLENKASISGGGLPAPYQAKQLHLHWSDLPYKGSEHSLDGEHFAMEMHIVHEKEEIAVLAFLVEATQVNEGFQPLVEALSNIPKPEMSTTMAESSLLDLLPEEKRHYFRYLGSLTTPTCDEKVVWTVFREPIQLHREQILAFQKLYYDKEQTVSMKDNVRPLQQLGQRTVIKS Hydrogen bonds contact Hydrophobic contact | ||||
| 13 | Myeloperoxidase (MPO) | 4DL1 | 7.03 | |
Target general information Gen name MPO Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms MPO Protein family Peroxidase family, XPO subfamily Biochemical class Peroxidases Function Part of the host defense system of polymorphonuclear leukocytes. It is responsible for microbicidal activity against a wide range of organisms. In the stimulated PMN, MPO catalyzes the production of hypohalous acids, primarily hypochlorous acidin physiologic situations, and other toxic intermediates that greatly enhance PMN microbicidal activity. Related diseases Myeloperoxidase deficiency (MPOD) [MIM:254600]: A disorder characterized by decreased myeloperoxidase activity in neutrophils and monocytes that results in disseminated candidiasis. {ECO:0000269|PubMed:37198333, ECO:0000269|PubMed:7904599, ECO:0000269|PubMed:8142659, ECO:0000269|PubMed:8621627, ECO:0000269|PubMed:9354683, ECO:0000269|PubMed:9637725}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB06111; DB00233; DB00006; DB02300; DB06774; DB00958; DB06468; DB00833; DB00535; DB00515; DB00847; DB00250; DB05161; DB01225; DB00583; DB01065; DB00461; DB04821; DB00104; DB00526; DB00550; DB00208; DB06823; DB00500; DB04827 Interacts with P27918; Q9UNE7 EC number EC 1.11.2.2 Uniprot keywords 3D-structure; Alternative splicing; Calcium; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; Heme; Hydrogen peroxide; Iron; Lysosome; Metal-binding; Oxidation; Oxidoreductase; Peroxidase; Proteomics identification; Reference proteome; Signal Protein physicochemical properties Chain ID A,B,E,F,I,J,M,N Molecular weight (Da) 53052.6 Length 466 Aromaticity 0.08 Instability index 40.64 Isoelectric point 9.48 Charge (pH=7) 15.12 2D Binding mode Binding energy (Kcal/mol) -6.52 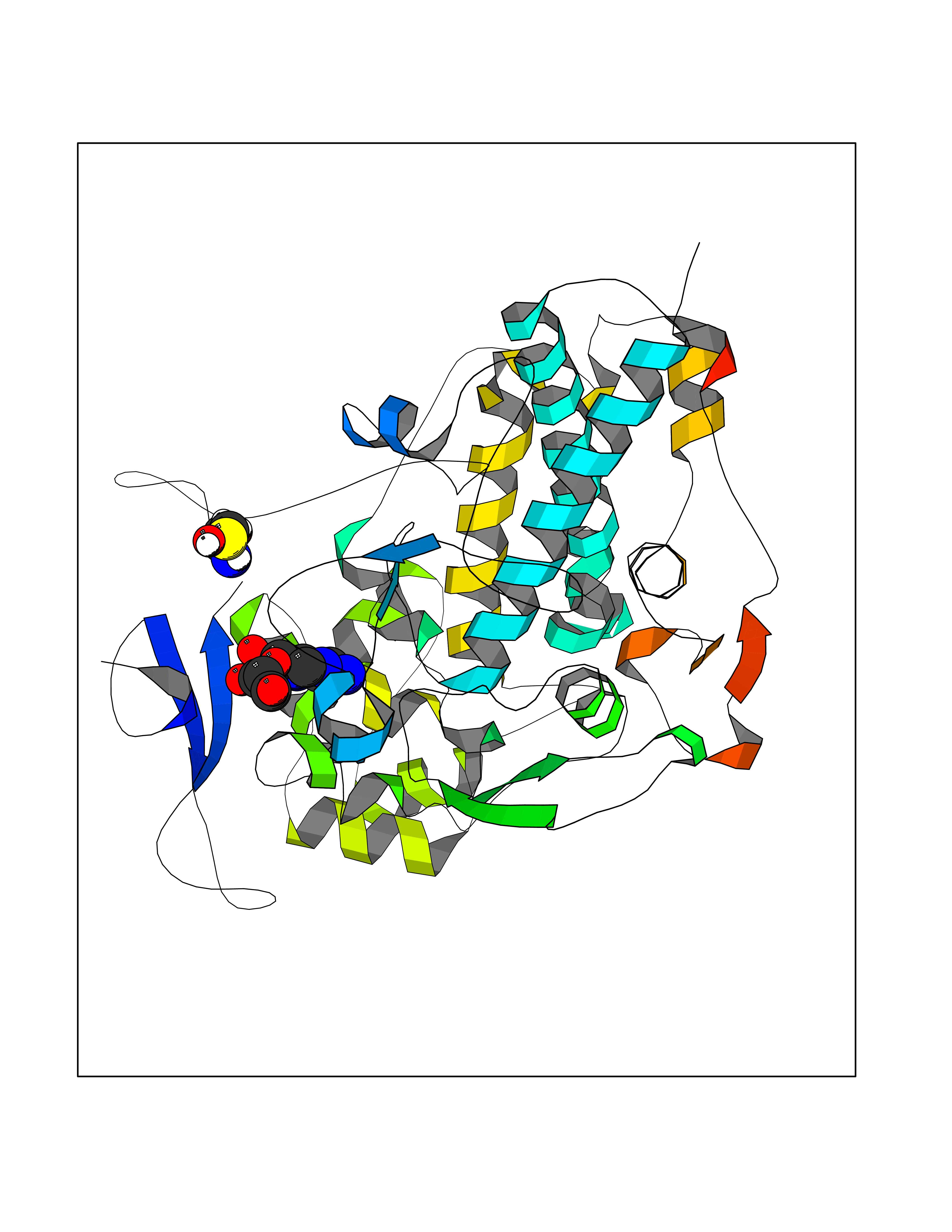 Molscript Map 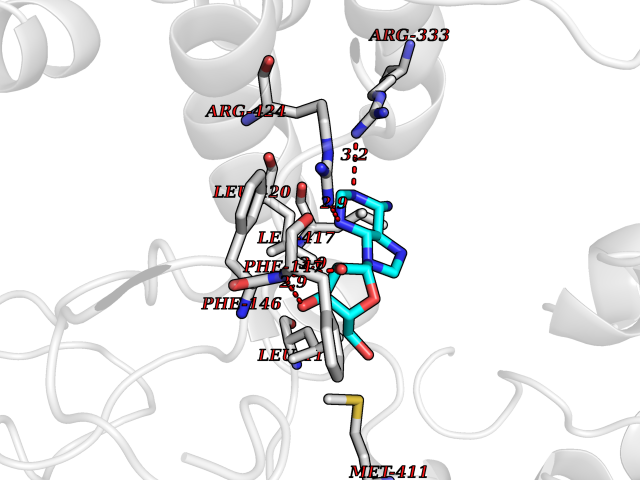 Pymol Map 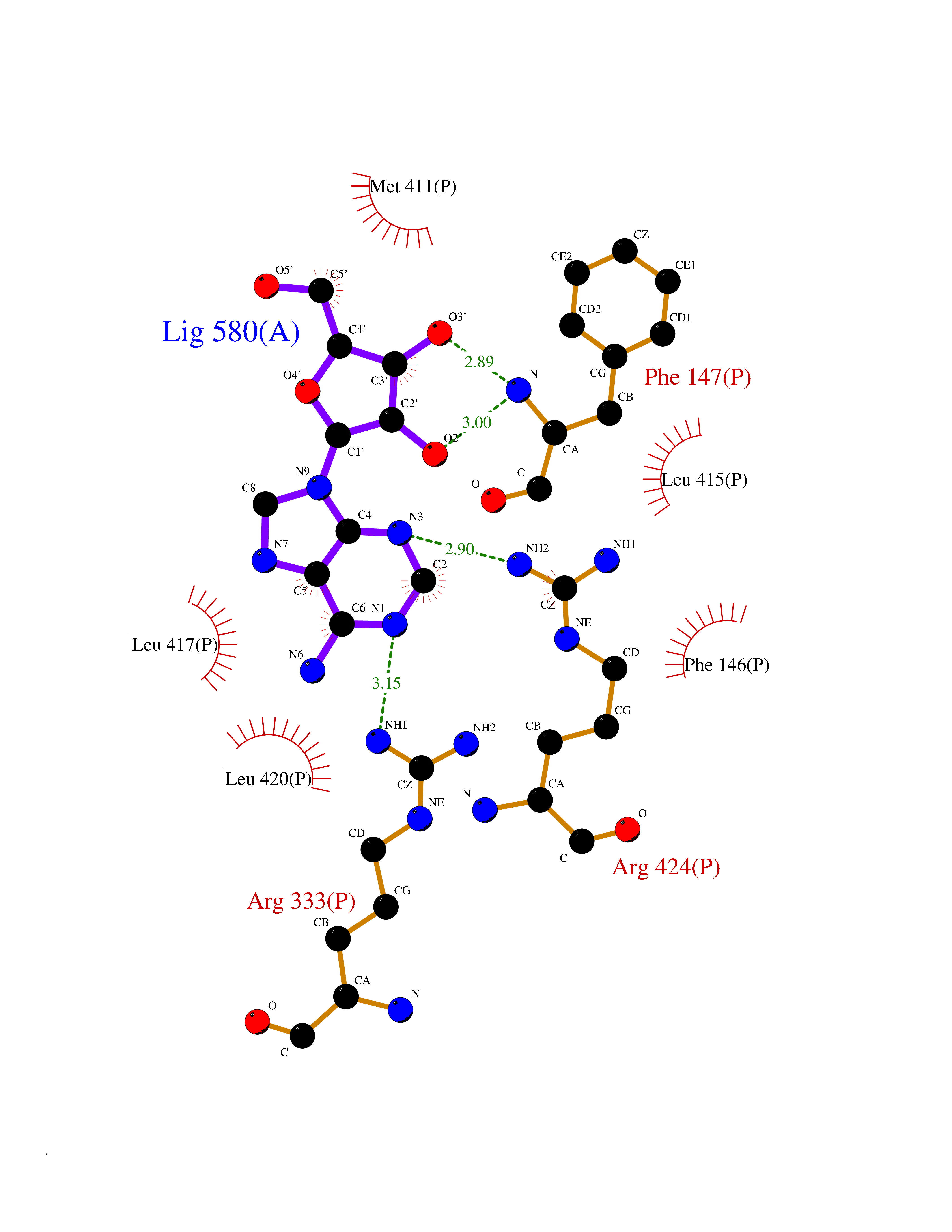 Ligplot Map 3D Binding mode Sequence VNCETSCVQQPPCFPLKIPPNDPRIKNQADCIPFFRSXPACPGSNITIRNQINALTSFVDASMVYGSEEPLARNLRNMSNQLGLLAVNQRFQDNGRALLPFDNLHDDPCLLTNRSARIPCFLAGDTRSSEMPELTSMHTLLLREHNRLATELKSLNPRWDGERLYQEARKIVGAMVQIITYRDYLPLVLGPTAMRKYLPTYRSYNDSVDPRIANVFTNAFRYGHTLIQPFMFRLDNRYQPMEPNPRVPLSRVFFASWRVVLEGGIDPILRGLMATPAKLNRQNQIAVDEIRERLFEQVMRIGLDLPALNMQRSRDHGLPGYNAWRRFCGLPQPETVGQLGTVLRNLKLARKLMEQYGTPNNIDIWMGGVSEPLKRKGRVGPLLACIIGTQFRKLRDGDRFWWENEGVFSMQQRQALAQISLPRIICDNTGITTVSKNNIFMSNSYPRDFVNCSTLPALNLASWREA Hydrogen bonds contact Hydrophobic contact | ||||
| 14 | Amylin receptor (IAPPR) | 6ZIS | 7.01 | |
Target general information Gen name CALCR-RAMP1/RAMP2/RAMP3 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Complex of Calcitonin receptor and Receptor activity-modifying protein Protein family RAMP family Biochemical class NA Function Transports the calcitonin gene-related peptide type 1 receptor (CALCRL) to the plasma membrane. Acts as a receptor for calcitonin-gene-related peptide (CGRP) together with CALCRL. Related diseases Immunodeficiency 9 (IMD9) [MIM:612782]: An immune disorder characterized by recurrent infections, impaired activation and proliferative response of T-cells, decreased T-cell production of cytokines, and normal lymphocytes counts and serum immunoglobulin levels. In surviving patients ectodermal dysplasia with anhidrosis and non-progressive myopathy may be observed. {ECO:0000269|PubMed:16147976, ECO:0000269|PubMed:16582901}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Myopathy, tubular aggregate, 2 (TAM2) [MIM:615883]: A rare congenital myopathy characterized by regular arrays of membrane tubules on muscle biopsies without additional histopathological hallmarks. Tubular aggregates in muscle are structures of variable appearance consisting of an outer tubule containing either one or more microtubule-like structures or amorphous material. TAM2 patients have myopathy and pupillary abnormalities. {ECO:0000269|PubMed:24591628, ECO:0000269|PubMed:28058752}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01278 Interacts with Q16602; P21145; Q5J8X5; Q16617 EC number NA Uniprot keywords 3D-structure; Cell membrane; Disulfide bond; Membrane; Proteomics identification; Receptor; Reference proteome; Signal; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A Molecular weight (Da) 63832.6 Length 569 Aromaticity 0.12 Instability index 22.42 Isoelectric point 5.07 Charge (pH=7) -16.96 2D Binding mode Binding energy (Kcal/mol) -7.12 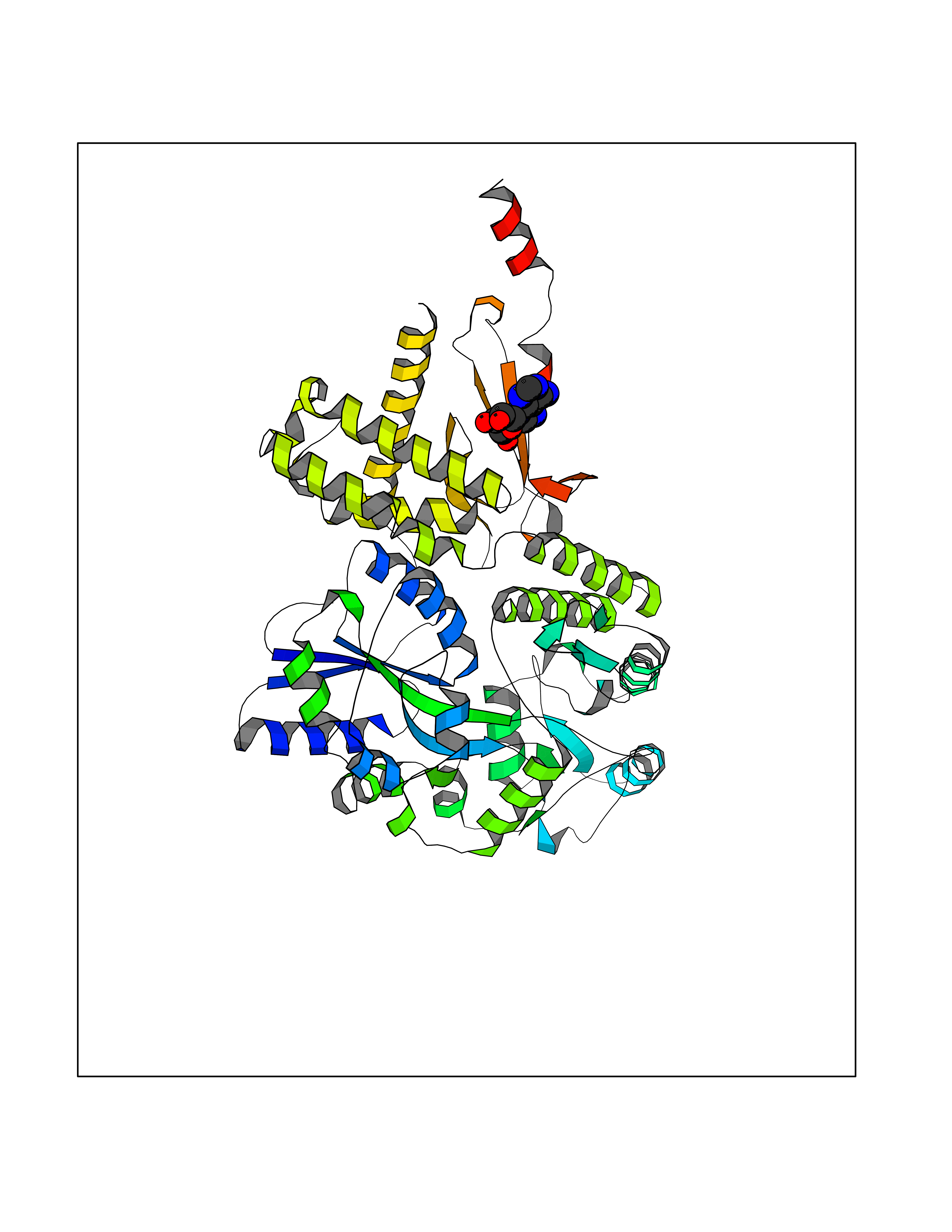 Molscript Map 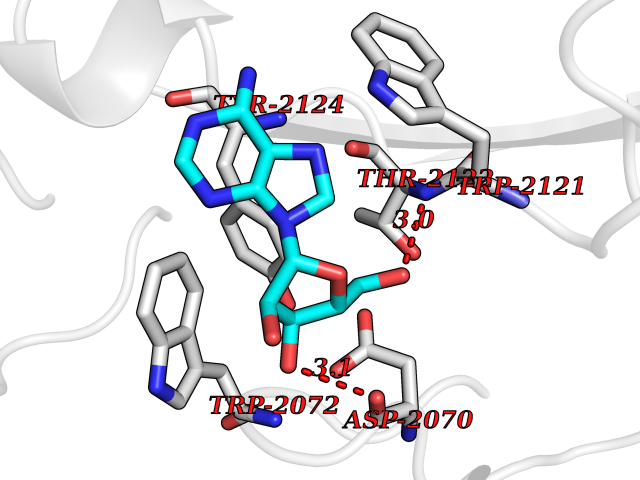 Pymol Map 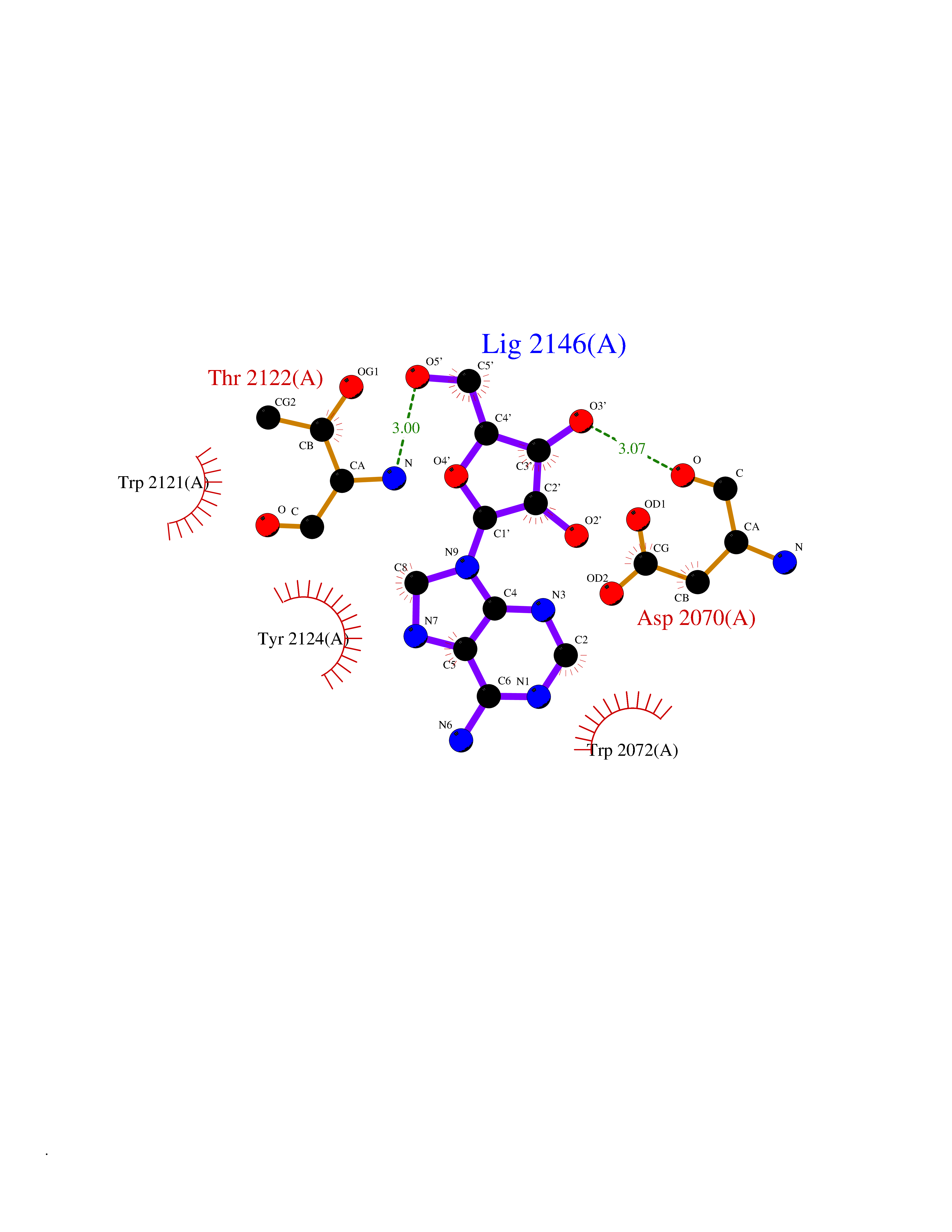 Ligplot Map 3D Binding mode Sequence SAKIEEGKLVIWINGDKGYNGLAEVGKKFEKDTGIKVTVEHPDKLEEKFPQVAATGDGPDIIFWAHDRFGGYAQSGLLAEITPDKAFQDKLYPFTWDAVRYNGKLIAYPIAVEALSLIYNKDLLPNPPKTWEEIPALDKELKAKGKSALMFNLQEPYFTWPLIAADGGYAFKYENGKYDIKDVGVDNAGAKAGLTFLVDLIKNKHMNADTDYSIAEAAFNKGETAMTINGPWAWSNIDTSKVNYGVTVLPTFKGQPSKPFVGVLSAGINAASPNKELAKEFLENYLLTDEGLEAVNKDKPLGAVALKSYEEELAKDPRIAATMENAQKGEIMPNIPQMSAFWYAVRTAVINAASGRQTVDEALKDAQTNAAAEFTTACQEANYGALLRELCLTQFQVDMEAVGETLWCDWGRTIRSYRELADCTWHMAEKLGCFWPNAEVDRFFLAVHGRYFRSCPISIQLGVTRNKIMTAQYECYQKIMQDPIQQGVYCQRTWDGWLCWNDVAAGTESMQLCPDYFQDFDPSEKVTKICDQDGNWFRHPASQRTWTDYTQCNVNTHEKVKTALNLFYL Hydrogen bonds contact Hydrophobic contact | ||||
| 15 | Clostridium histolyticum Collagenase (CH colG) | 7Z5U | 7.00 | |
Target general information Gen name CH colG Organism Hathewaya histolytica (Clostridium histolyticum) Uniprot ID TTD ID Synonyms Microbial collagenase; Gelatinase ColG; Collagenase ColG; Class I collagenase Protein family Peptidase M9B family, Collagenase subfamily Biochemical class NA Function Clostridial collagenases are among the most efficient degraders of eukaryotic collagen known; saprophytes use collagen as a carbon source while pathogens additionally digest collagen to aid in host colonization. Has both tripeptidylcarboxypeptidase on Gly-X-Y and endopeptidase activities; the endopeptidase cuts within the triple helix region of collagen while tripeptidylcarboxypeptidase successively digests the exposed ends, thus clostridial collagenases can digest large sections of collagen. Active on soluble type I collagen, insoluble collagen, azocoll, soluble PZ-peptide (all collagenase substrates) and gelatin. The full-length protein has collagenase activity, while the in vivo derived C-terminally truncated shorter versions only act on gelatin. In vitro digestion of soluble calf skin collagen fibrils requires both ColG and ColH; ColG forms missing the second collagen-binding domain are also synergistic with ColH, although their overall efficiency is decreased. The activator domain (residues 119-388) and catalytic subdomain (389-670) open and close around substrate using a Gly-rich hinge (387-397), allowing digestion when the protein is closed. Binding of collagen requires Ca(2+) and is inhibited by EGTA; the collagen-binding domain (CBD, S3a plus S3b) specifically recognizes the triple-helical conformation made by 3 collagen protein chains in the triple-helical region. Isolated CBD (S3a plus S3b) binds collagen fibrils and sheets of many tissues. Related diseases Intellectual developmental disorder, X-linked, syndromic, Claes-Jensen type (MRXSCJ) [MIM:300534]: A disorder characterized by significantly below average general intellectual functioning associated with impairments in adaptive behavior and manifested during the developmental period. MRXSCJ patients manifest intellectual disability associated with variable features such as slowly progressive spastic paraplegia, seizures, facial dysmorphism. {ECO:0000269|PubMed:15586325, ECO:0000269|PubMed:16538222, ECO:0000269|PubMed:16541399, ECO:0000269|PubMed:17320160, ECO:0000269|PubMed:17468742, ECO:0000269|PubMed:23356856, ECO:0000269|PubMed:25666439}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number EC 3.4.24.3 Uniprot keywords 3D-structure; Calcium; Direct protein sequencing; Hydrolase; Metal-binding; Metalloprotease; Pharmaceutical; Protease; Repeat; Secreted; Signal; Virulence; Zinc; Zymogen Protein physicochemical properties Chain ID A Molecular weight (Da) 44751.2 Length 386 Aromaticity 0.15 Instability index 27.33 Isoelectric point 5.77 Charge (pH=7) -7.33 2D Binding mode Binding energy (Kcal/mol) -7.99 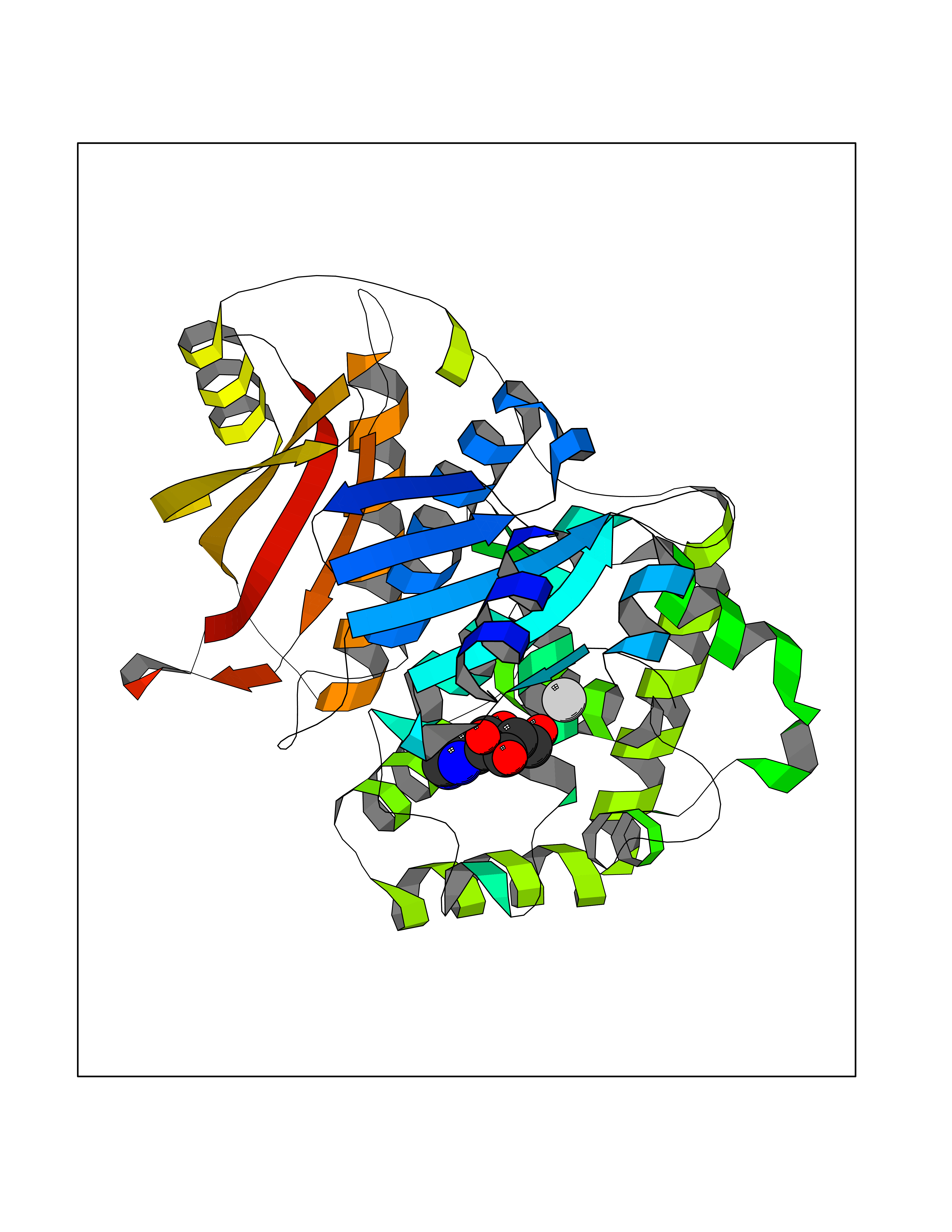 Molscript Map 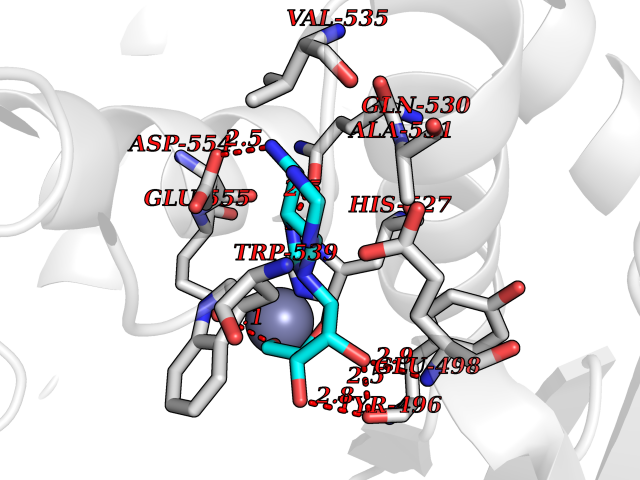 Pymol Map 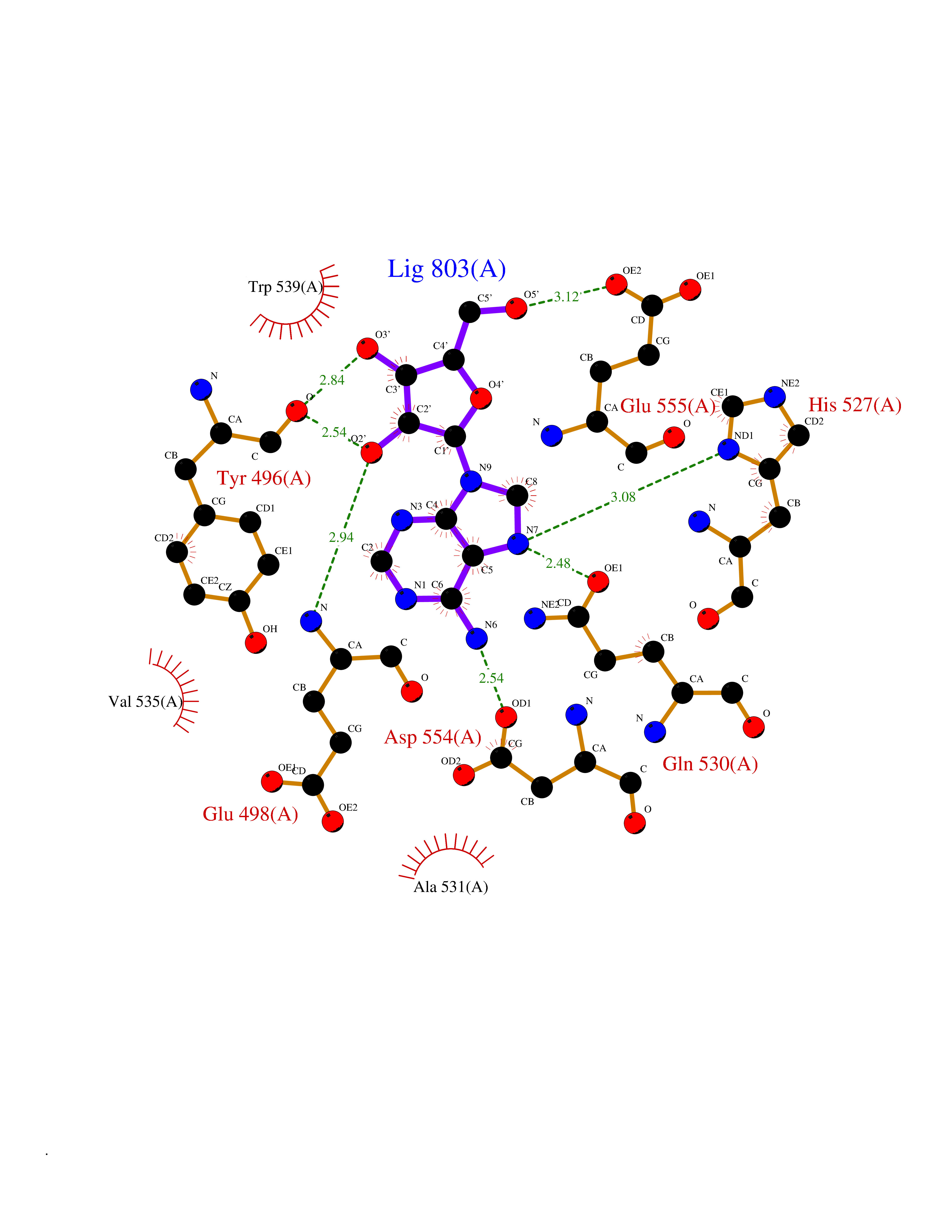 Ligplot Map 3D Binding mode Sequence DHDKFLDDAEKHYLPKTYTFDNGTFIIRAGDKVSEEKIKRLYWASREVKSQFHRVVGNDKALEVGNADDVLTMKIFNSPEEYKFNTTDNGGLYIEPRGTFYTYERTPQQSIFSLEELFRHEYTHYLQARYLVDGLWGQGPFYEKNRLTWFDEGTAEFFAGSTRTSGVLPRKLILGYLAKDKVDHRYSLKKTLNSGYDDSDWMFYNYGFAVAHYLYEKDMPTFIKMNKAILNTDVKSYDEIIKKLSDDANKNTEYQNHIQELVDKYQGAGIPLVSDDYLKDHGYKKASEVYSEISKAASLTNTSVTAEKSQYFNTFTLRGTYTGETSKGEFKDWDEMSKKLDGTLESLAKNSWSGYKTLTAYFTNYRVTSDNKVQYDVVFHGVLTDN Hydrogen bonds contact Hydrophobic contact | ||||
| 16 | Ribosomal protein S6 kinase beta-1 (S6K1) | 7N93 | 6.97 | |
Target general information Gen name RPS6KB1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms p70-S6K 1; p70 ribosomal S6 kinase alpha; p70 S6KA; p70 S6K-alpha; p70 S6 kinase alpha; Serine/threonine-protein kinase 14A; STK14A; S6K-beta-1; S6K; Ribosomal protein S6 kinase I; P70S6K1; P70-S6K; 7 Protein family Protein kinase superfamily, AGC Ser/Thr protein kinase family, S6 kinase subfamily Biochemical class Kinase Function Regulates protein synthesis through phosphorylation of EIF4B, RPS6 and EEF2K, and contributes to cell survival by repressing the pro-apoptotic function of BAD. Under conditions of nutrient depletion, the inactive form associates with the EIF3 translation initiation complex. Upon mitogenic stimulation, phosphorylation by the mammalian target of rapamycin complex 1 (mTORC1) leads to dissociation from the EIF3 complex and activation. The active form then phosphorylates and activates several substrates in the pre-initiation complex, including the EIF2B complex and the cap-binding complex component EIF4B. Also controls translation initiation by phosphorylating a negative regulator of EIF4A, PDCD4, targeting it for ubiquitination and subsequent proteolysis. Promotes initiation of the pioneer round of protein synthesis by phosphorylating POLDIP3/SKAR. In response to IGF1, activates translation elongation by phosphorylating EEF2 kinase (EEF2K), which leads to its inhibition and thus activation of EEF2. Also plays a role in feedback regulation of mTORC2 by mTORC1 by phosphorylating RICTOR, resulting in the inhibition of mTORC2 and AKT1 signaling. Mediates cell survival by phosphorylating the pro-apoptotic protein BAD and suppressing its pro-apoptotic function. Phosphorylates mitochondrial URI1 leading to dissociation of a URI1-PPP1CC complex. The free mitochondrial PPP1CC can then dephosphorylate RPS6KB1 at Thr-412, which is proposed to be a negative feedback mechanism for the RPS6KB1 anti-apoptotic function. Mediates TNF-alpha-induced insulin resistance by phosphorylating IRS1 at multiple serine residues, resulting in accelerated degradation of IRS1. In cells lacking functional TSC1-2 complex, constitutively phosphorylates and inhibits GSK3B. May be involved in cytoskeletal rearrangement through binding to neurabin. Phosphorylates and activates the pyrimidine biosynthesis enzyme CAD, downstream of MTOR. Following activation by mTORC1, phosphorylates EPRS and thereby plays a key role in fatty acid uptake by adipocytes and also most probably in interferon-gamma-induced translation inhibition. Serine/threonine-protein kinase that acts downstream of mTOR signaling in response to growth factors and nutrients to promote cell proliferation, cell growth and cell cycle progression. Related diseases Progressive familial heart block 1B (PFHB1B) [MIM:604559]: A cardiac bundle branch disorder characterized by progressive alteration of cardiac conduction through the His-Purkinje system, with a pattern of a right bundle-branch block and/or left anterior hemiblock occurring individually or together. It leads to complete atrio-ventricular block causing syncope and sudden death. {ECO:0000269|PubMed:19726882, ECO:0000269|PubMed:20562447, ECO:0000269|PubMed:21887725}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Erythrokeratodermia variabilis et progressiva 6 (EKVP6) [MIM:618531]: A form of erythrokeratodermia variabilis et progressiva, a genodermatosis characterized by the coexistence of two independent skin lesions: transient erythema and hyperkeratosis that is usually localized but occasionally occurs in its generalized form. Clinical presentation varies significantly within a family and from one family to another. Palmoplantar keratoderma is present in around 50% of cases. EKVP6 inheritance is autosomal dominant. {ECO:0000269|PubMed:30528822}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P31749; Q06481-5; P55884; P08151; Q5VY09; Q00005; P13051-2; P08151; P05067; P27986-2 EC number EC 2.7.11.1 Uniprot keywords 3D-structure; Acetylation; Alternative initiation; Alternative splicing; Apoptosis; ATP-binding; Cell cycle; Cytoplasm; Kinase; Membrane; Mitochondrion; Mitochondrion outer membrane; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Serine/threonine-protein kinase; Synapse; Synaptosome; Transferase; Translation regulation Protein physicochemical properties Chain ID A Molecular weight (Da) 35187.6 Length 312 Aromaticity 0.1 Instability index 39.58 Isoelectric point 8.8 Charge (pH=7) 5.54 2D Binding mode Binding energy (Kcal/mol) -7.07  Molscript Map 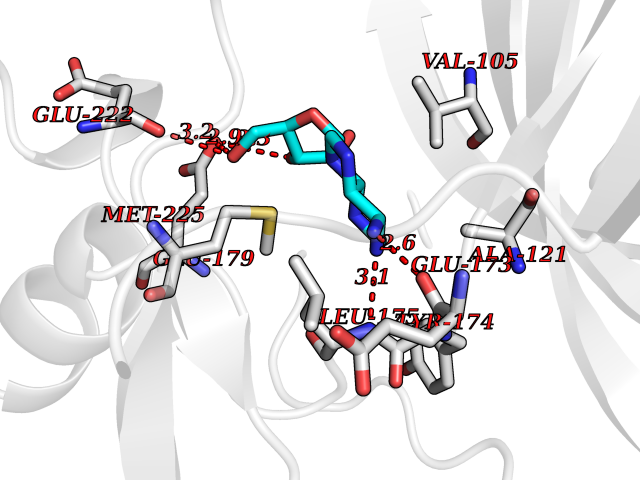 Pymol Map 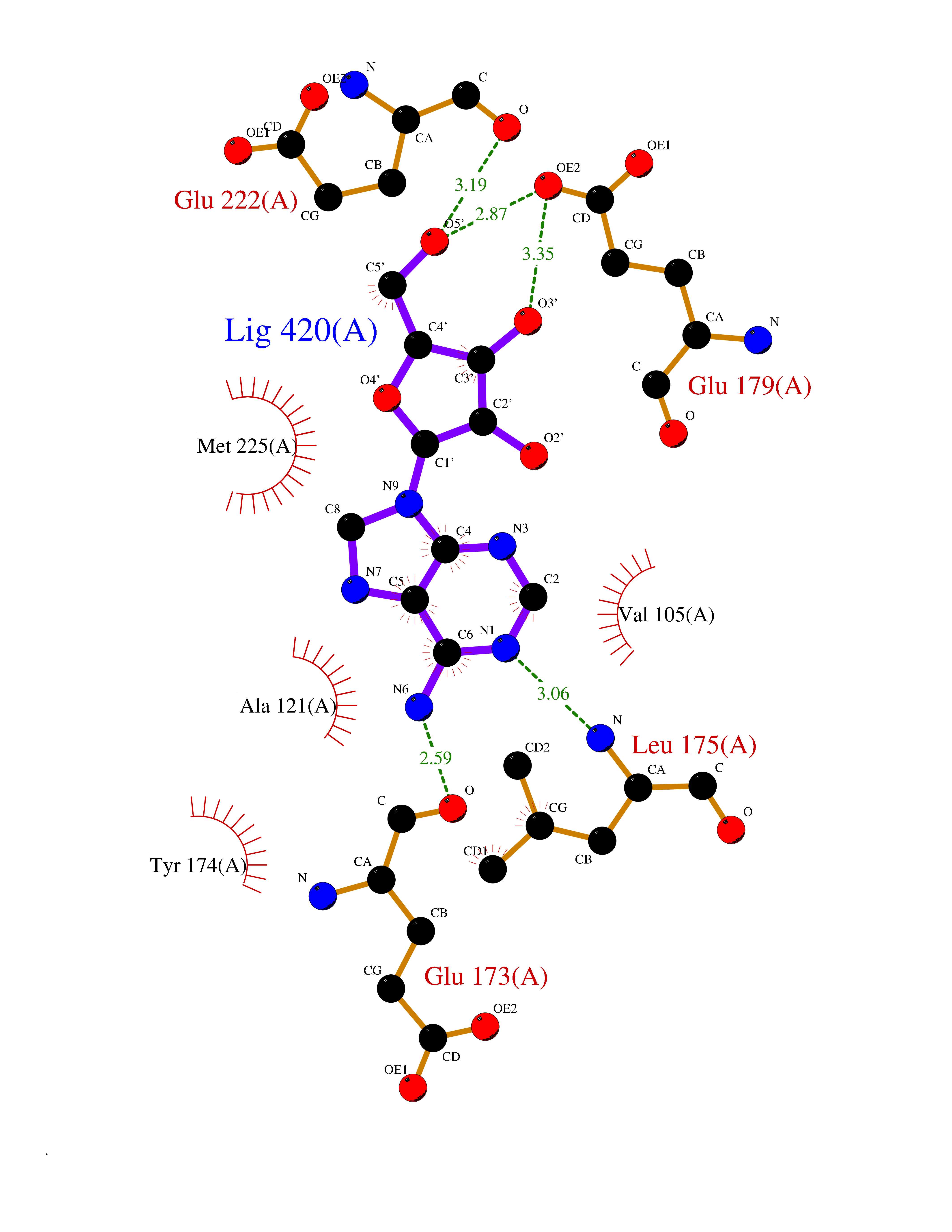 Ligplot Map 3D Binding mode Sequence RPECFELLRVLGKGGYGKVFQVRKVTGANTGKIFAMKVLKKAMIVRNAKDTAHTKAERNILEEVKHPFIVDLIYAFQTGGKLYLILEYLSGGELFMQLEREGIFMEDTACFYLAEISMALGHLHQKGIIYRDLKPENIMLNHQGHVKLTDFGLCKECGTIEYMAPEILMRSGHNRAVDWWSLGALMYDMLTGAPPFTGENRKKTIDKILKCKLNLPPYLTQEARDLLKKLLKRNAASRLGAGPGDAGEVQAHPFFRHINWEELLARKVEPPFKPLLQSEEDVSQFDSKFTRQXPVDXPNQVFLGFEYVAPSV Hydrogen bonds contact Hydrophobic contact | ||||
| 17 | Retinal rod rhodopsin-sensitive cGMP 3',5'-cyclic phosphodiesterase subunit gamma | 3JWR | 6.91 | |
Target general information Gen name PDE6G Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms PDEG Protein family Rod/cone cGMP-PDE gamma subunit family Biochemical class Hydrolase Function 3',5'-cyclic-GMP phosphodiesterase activity.CGMP binding.Enzyme inhibitor activity.Spectrin binding. Related diseases Retinitis pigmentosa 57 (RP57) [MIM:613582]: A retinal dystrophy belonging to the group of pigmentary retinopathies. Retinitis pigmentosa is characterized by retinal pigment deposits visible on fundus examination and primary loss of rod photoreceptor cells followed by secondary loss of cone photoreceptors. Patients typically have night vision blindness and loss of midperipheral visual field. As their condition progresses, they lose their far peripheral visual field and eventually central vision as well. {ECO:0000269|PubMed:20655036}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07954; DB00203; DB00820; DB00862 Interacts with O14503; Q96JM7; A0A6Q8PF08; O43741; Q8R511; P62994; Q9QY17; Q63787 EC number 3.1.4.35 Uniprot keywords 3D-structure; Acetylation; cGMP; Hydrolase; Reference proteome; Retinitis pigmentosa; Sensory transduction; Vision Protein physicochemical properties Chain ID C,D Molecular weight (Da) 40027.7 Length 345 Aromaticity 0.09 Instability index 37.44 Isoelectric point 6.02 Charge (pH=7) -6.88 2D Binding mode Binding energy (Kcal/mol) -7.45 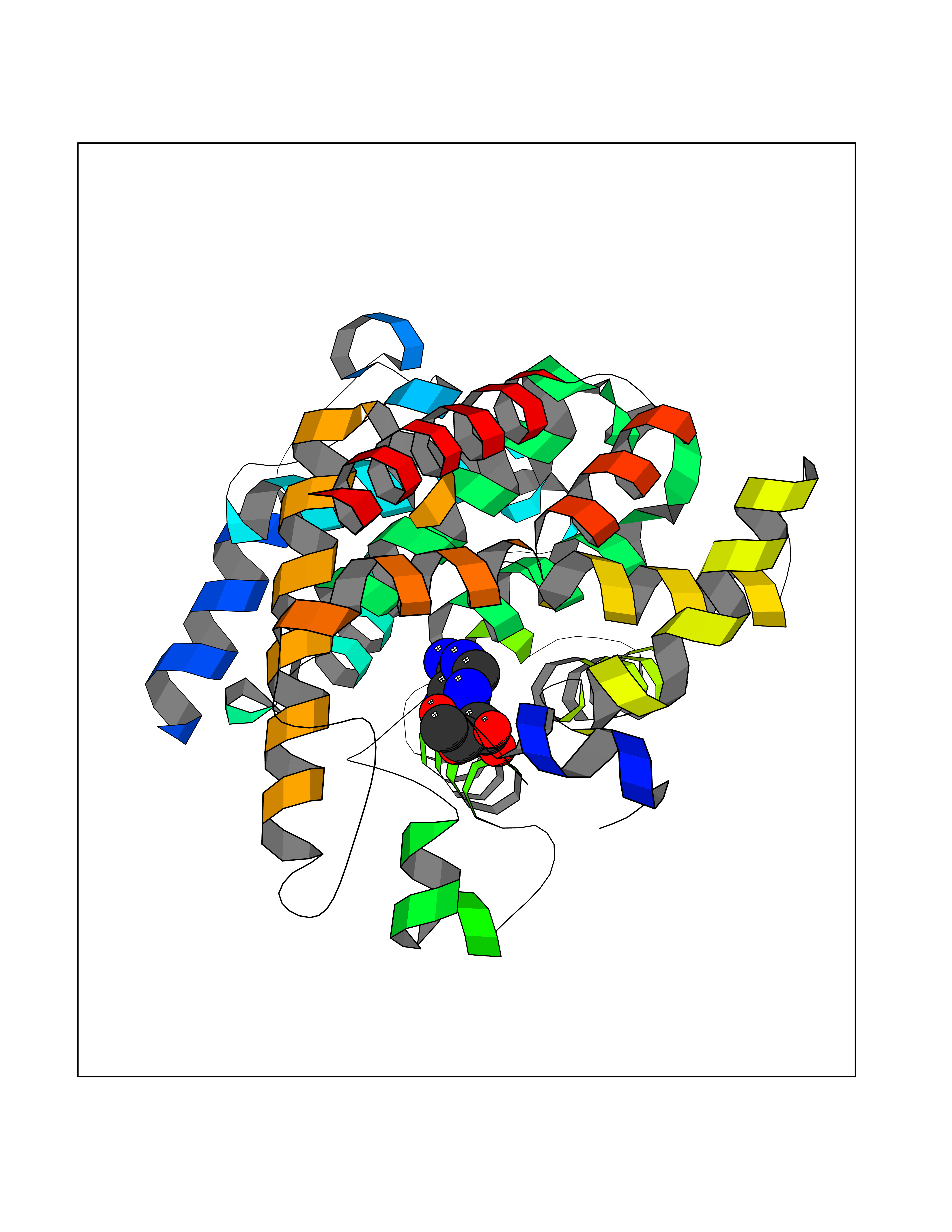 Molscript Map 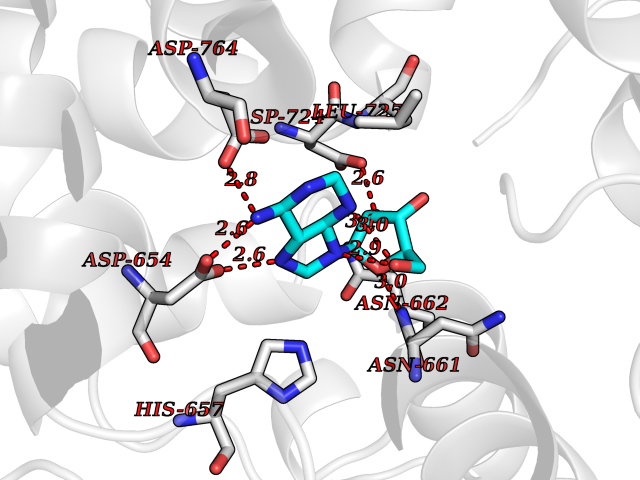 Pymol Map 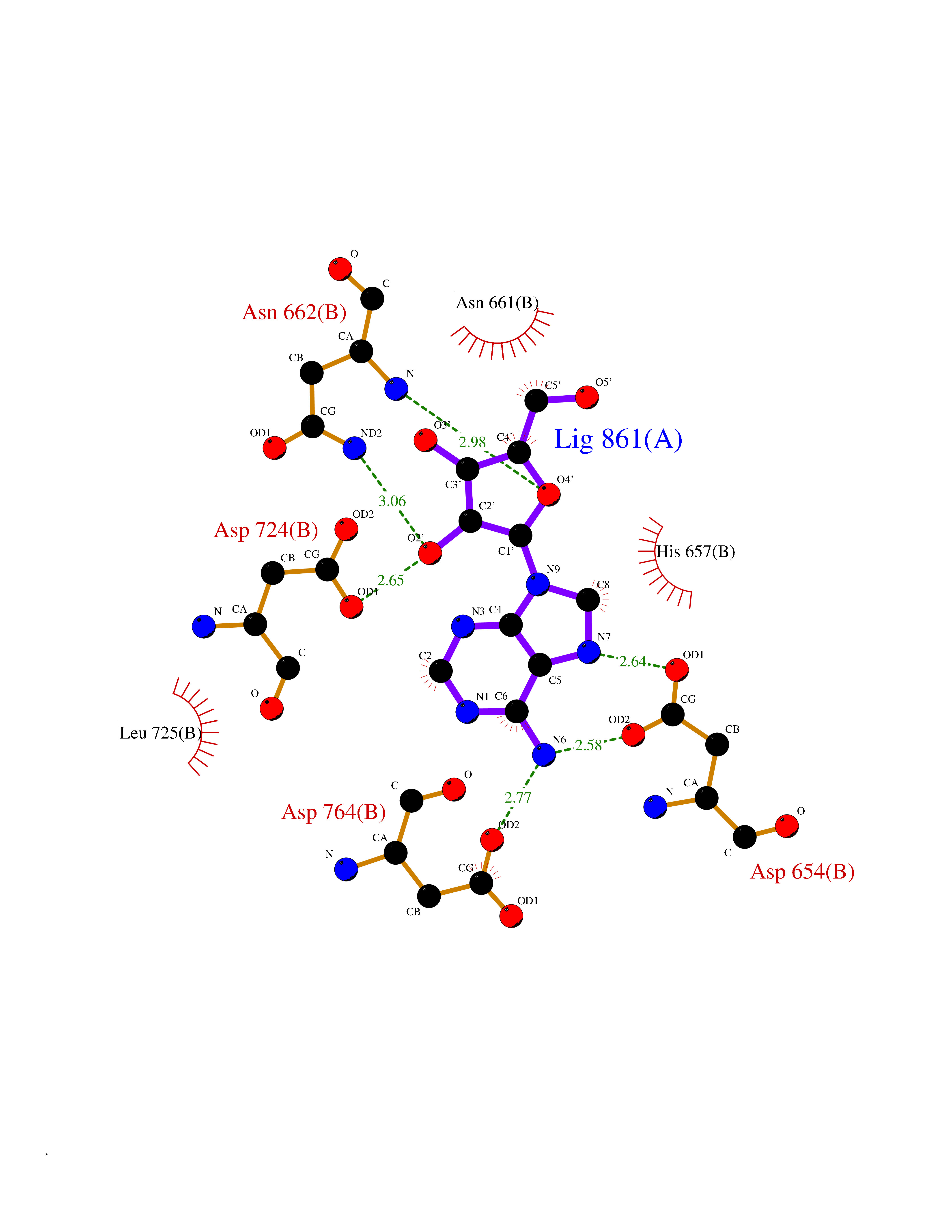 Ligplot Map 3D Binding mode Sequence EAFNHLELHELAQYGIISHMEETRELQSLAAAVVPSAQTLKITDFSFSDFELSDLETALCTIRMFTDLNLVQNFQMKHEVLCRWILSVKKNYRKNVAYHNWRHAFNTAQCMFAALKAGKIQNKLTDLEILALLIAALSHDLDHRGVNNSYIQRSEHPLAQLYCHSIMEHHHFDQCLMILNSPGNQILSGLSIEEYKTTLKIIKQAILATDLALYIKRRGEFFELIRKNQFNLEDPHQKELFLAMLMTACDLSAITKPWPIQQRIAELVATEFWEQGDLERTVLQQQPIPMMDRNKRDELPKLQVGFIDFVCTQLYEALTHVSEDCFPLLDGCRKNRQKWQALAEQ Hydrogen bonds contact Hydrophobic contact | ||||
| 18 | Transferrin (TF) | 1RYO | 6.86 | |
Target general information Gen name TF Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Siderophilin; Serotransferrin; PRO1400; Beta-1 metal-binding globulin Protein family Transferrin family Biochemical class Transferrin Function It is responsible for the transport of iron from sites of absorption and heme degradation to those of storage and utilization. Serum transferrin may also have a further role in stimulating cell proliferation. Transferrins are iron binding transport proteins which can bind two Fe(3+) ions in association with the binding of an anion, usually bicarbonate. Related diseases Atransferrinemia (ATRAF) [MIM:209300]: A rare autosomal recessive disorder characterized by abnormal synthesis of transferrin leading to iron overload and microcytic hypochromic anemia. {ECO:0000269|PubMed:11110675, ECO:0000269|PubMed:15466165}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01370; DB14517; DB14518; DB01294; DB14526; DB14527; DB11136; DB14528; DB14529; DB14530; DB00515; DB09130; DB11397; DB13949; DB14490; DB14491; DB14488; DB14501; DB14489; DB13257; DB06215; DB06784; DB05260; DB01592; DB00893; DB00677; DB06757; DB11182; DB14520; DB01593; DB14487; DB14533; DB14548 Interacts with O43315; O00501; Q7Z7G2; Q9GZR5; Q9Y282; Q96KR6; P01350; P08034; Q8NBJ4; O15529; Q8TED1; Q7Z5P4; A8MZ59; O15173; Q96TC7; Q3KNW5; Q9BXS9-3; Q99523; O43278-2; Q8N9I0; P02786; Q4KMG9; Q9K0U9; Q09057; Q9K0V0; P02786 EC number NA Uniprot keywords 3D-structure; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; Ion transport; Iron; Iron transport; Metal-binding; Methylation; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Secreted; Signal; Transport Protein physicochemical properties Chain ID A Molecular weight (Da) 35854.5 Length 324 Aromaticity 0.1 Instability index 34.17 Isoelectric point 7.58 Charge (pH=7) 1.42 2D Binding mode Binding energy (Kcal/mol) -7.23  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence KTVRWCAVSEHEATKCQSFRDHMKSVIPSDGPSVACVKKASYLDCIRAIAANEADAVTLDAGLVYDAYLAPNNLKPVVAEFYGSKEDPQTFYYAVAVVKKDSGFQMNQLRGKKSCHTGLGRSAGWNIPIGLLYCDLPEPRKPLEKAVANFFSGSCAPCADGTDFPQLCQLCPGCGCSTLNQYFGYSGAFKCLKDGAGDVAFVKHSTIFENLANKADRDQYELLCLDNTRKPVDEYKDCHLAQVPSHTVVARSMGGKEDLIWELLNQAQEHFGKDKSKEFQLFSSPHGKDLLFKDSAHGFLKVPPRMDAKMYLGYEYVTAIRNLR Hydrogen bonds contact Hydrophobic contact | ||||
| 19 | Trypanosoma Dihydrolipoamide dehydrogenase (Trypano LPD) | 2QAE | 6.86 | |
Target general information Gen name Trypano LPD Organism Trypanosoma cruzi Uniprot ID TTD ID Synonyms Lipoamide dehydrogenase; LipDH; LPD; Glycine cleavage system L protein; Dihydrolipoamide dehydrogenase Protein family Class-I pyridine nucleotide-disulfide oxidoreductase family Biochemical class Sulfur donor oxidoreductase Function Lipoamide dehydrogenase is a component of the glycine cleavage system as well as of the alpha-ketoacid dehydrogenase complexes. Related diseases Focal segmental glomerulosclerosis 2 (FSGS2) [MIM:603965]: A renal pathology defined by the presence of segmental sclerosis in glomeruli and resulting in proteinuria, reduced glomerular filtration rate and progressive decline in renal function. Renal insufficiency often progresses to end-stage renal disease, a highly morbid state requiring either dialysis therapy or kidney transplantation. {ECO:0000269|PubMed:15879175, ECO:0000269|PubMed:15924139, ECO:0000269|PubMed:19458060, ECO:0000269|PubMed:19936226, ECO:0000269|PubMed:20798252, ECO:0000269|PubMed:21511817, ECO:0000269|PubMed:21734084, ECO:0000269|PubMed:22732337, ECO:0000269|PubMed:23014460, ECO:0000269|PubMed:23291369, ECO:0000269|PubMed:26892346}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number NA Uniprot keywords 3D-structure; Disulfide bond; FAD; Flavoprotein; NAD; Oxidoreductase; Redox-active center Protein physicochemical properties Chain ID A,B Molecular weight (Da) 98183.9 Length 930 Aromaticity 0.06 Instability index 29.02 Isoelectric point 6.32 Charge (pH=7) -6.37 2D Binding mode Binding energy (Kcal/mol) -7.17  Molscript Map 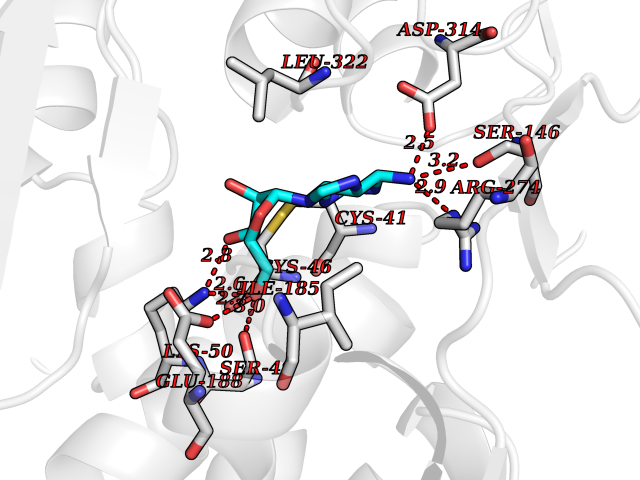 Pymol Map 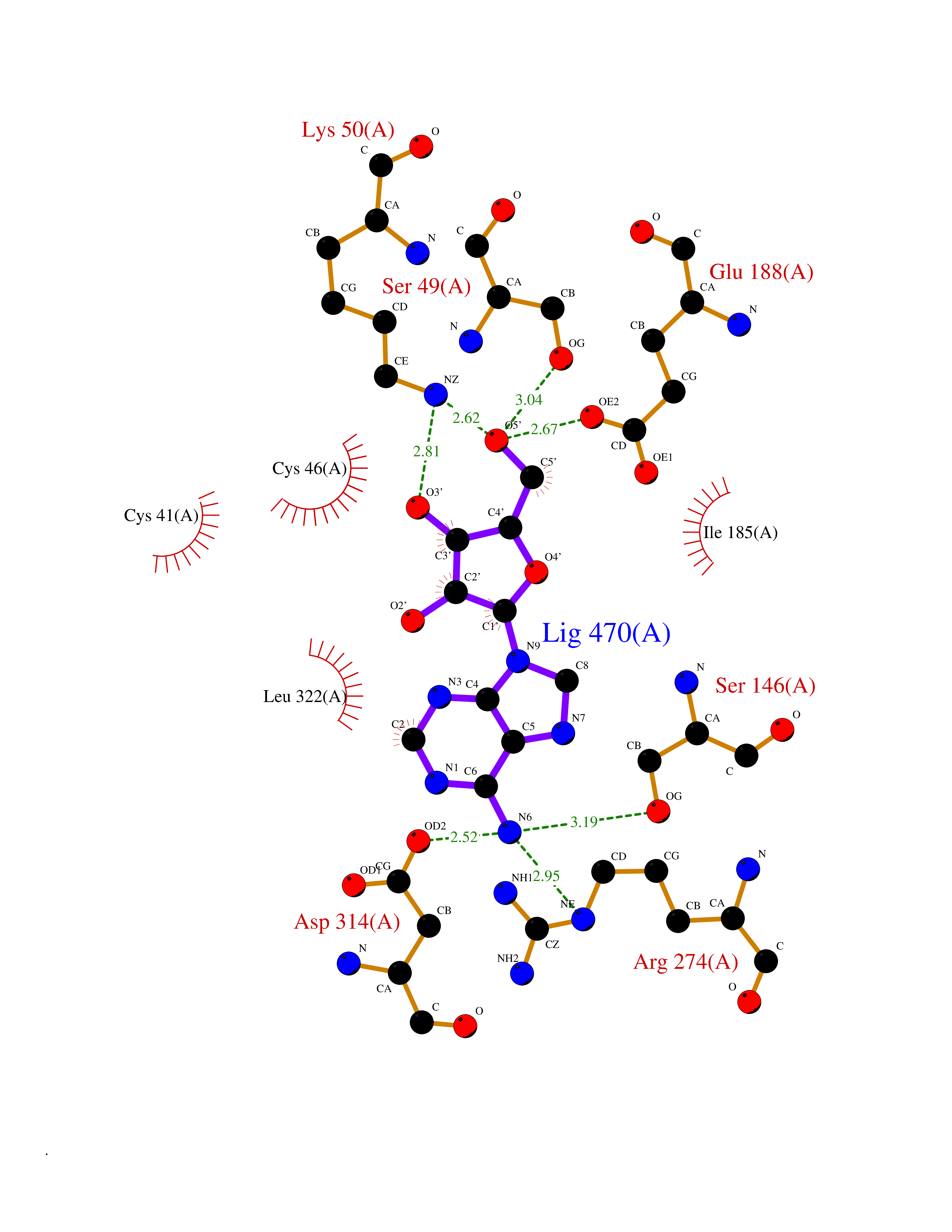 Ligplot Map 3D Binding mode Sequence NPYDVVVIGGGPGGYVASIKAAQLGMKTACVEKRGALGGTCLNVGCIPSKALLHATHLYHDAHANFARYGLMGGEGVTMDSAKMQQQKERAVKGLTGGVEYLFKKNKVTYYKGEGSFETAHSIRVNGLDGKQEMLETKKTIIATGSEPTELPFLPFDEKVVLSSTGALALPRVPKTMVVIGGGVIGLELGSVWARLGAEVTVVEFAPRCAPTLDEDVTNALVGALAKNEKMKFMTSTKVVGGTNNGDSVSLEVEGKRETVTCEALLVSVGRRPFTGGLGLDKINVAKNERGFVKIGDHFETSIPDVYAIGDVVDKGPMLAHKAEDEGVACAEILAGKPGHVNYGVIPAVIYTMPEVASVGKSEDELKKEGVAYKVGKFPFNANSRAKAVSTEDGFVKVLVDKATDRILGVHIVCTTAGELIGEACLAMEYGASSEDVGRTCHAHPTMSEALKEACMALFAKTINFNPYDVVVIGGGPGGYVASIKAAQLGMKTACVEKRGALGGTCLNVGCIPSKALLHATHLYHDAHANFARYGLMGGEGVTMDSAKMQQQKERAVKGLTGGVEYLFKKNKVTYYKGEGSFETAHSIRVNGLDGKQEMLETKKTIIATGSEPTELPFLPFDEKVVLSSTGALALPRVPKTMVVIGGGVIGLELGSVWARLGAEVTVVEFAPRCAPTLDEDVTNALVGALAKNEKMKFMTSTKVVGGTNNGDSVSLEVEGKRETVTCEALLVSVGRRPFTGGLGLDKINVAKNERGFVKIGDHFETSIPDVYAIGDVVDKGPMLAHKAEDEGVACAEILAGKPGHVNYGVIPAVIYTMPEVASVGKSEDELKKEGVAYKVGKFPFNANSRAKAVSTEDGFVKVLVDKATDRILGVHIVCTTAGELIGEACLAMEYGASSEDVGRTCHAHPTMSEALKEACMALFAKTINF Hydrogen bonds contact Hydrophobic contact | ||||
| 20 | Organic cation transporter 3 (OCT3) | 7ZH6 | 6.85 | |
Target general information Gen name SLC22A3 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Solute carrier family 22 member 3; Extraneuronal monoamine transporter; EMTH Protein family Major facilitator (TC 2.A.1) superfamily, Organic cation transporter (TC 2.A.1.19) family Biochemical class NA Function Mediates potential-dependent transport of a variety of organic cations. May play a significant role in the disposition of cationic neurotoxins and neurotransmitters in the brain. Related diseases Deafness, autosomal dominant, 2A (DFNA2A) [MIM:600101]: A form of non-syndromic sensorineural hearing loss. Sensorineural deafness results from damage to the neural receptors of the inner ear, the nerve pathways to the brain, or the area of the brain that receives sound information. {ECO:0000269|PubMed:10025409, ECO:0000269|PubMed:10369879, ECO:0000269|PubMed:10571947, ECO:0000269|PubMed:10925378, ECO:0000269|PubMed:21242547}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00718; DB08838; DB00182; DB00122; DB14006; DB00501; DB00575; DB00363; DB01151; DB00988; DB00783; DB13952; DB13953; DB13954; DB13955; DB13956; DB00983; DB00536; DB05381; DB00458; DB00762; DB00709; DB00448; DB08882; DB01042; DB01577; DB00331; DB08893; DB00184; DB00368; DB00526; DB00925; DB00413; DB00457; DB01035; DB00396; DB00938; DB00391; DB13943; DB13944; DB08837; DB08841; DB00541 Interacts with P00519 EC number NA Uniprot keywords 3D-structure; Cell membrane; Glycoprotein; Ion transport; Membrane; Mitochondrion; Nucleus; Proteomics identification; Reference proteome; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A Molecular weight (Da) 53067.4 Length 478 Aromaticity 0.13 Instability index 38.82 Isoelectric point 9.07 Charge (pH=7) 10.54 2D Binding mode Binding energy (Kcal/mol) -6.95  Molscript Map 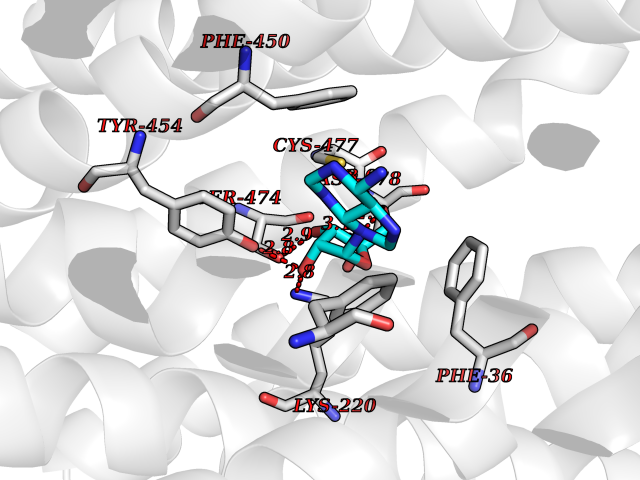 Pymol Map 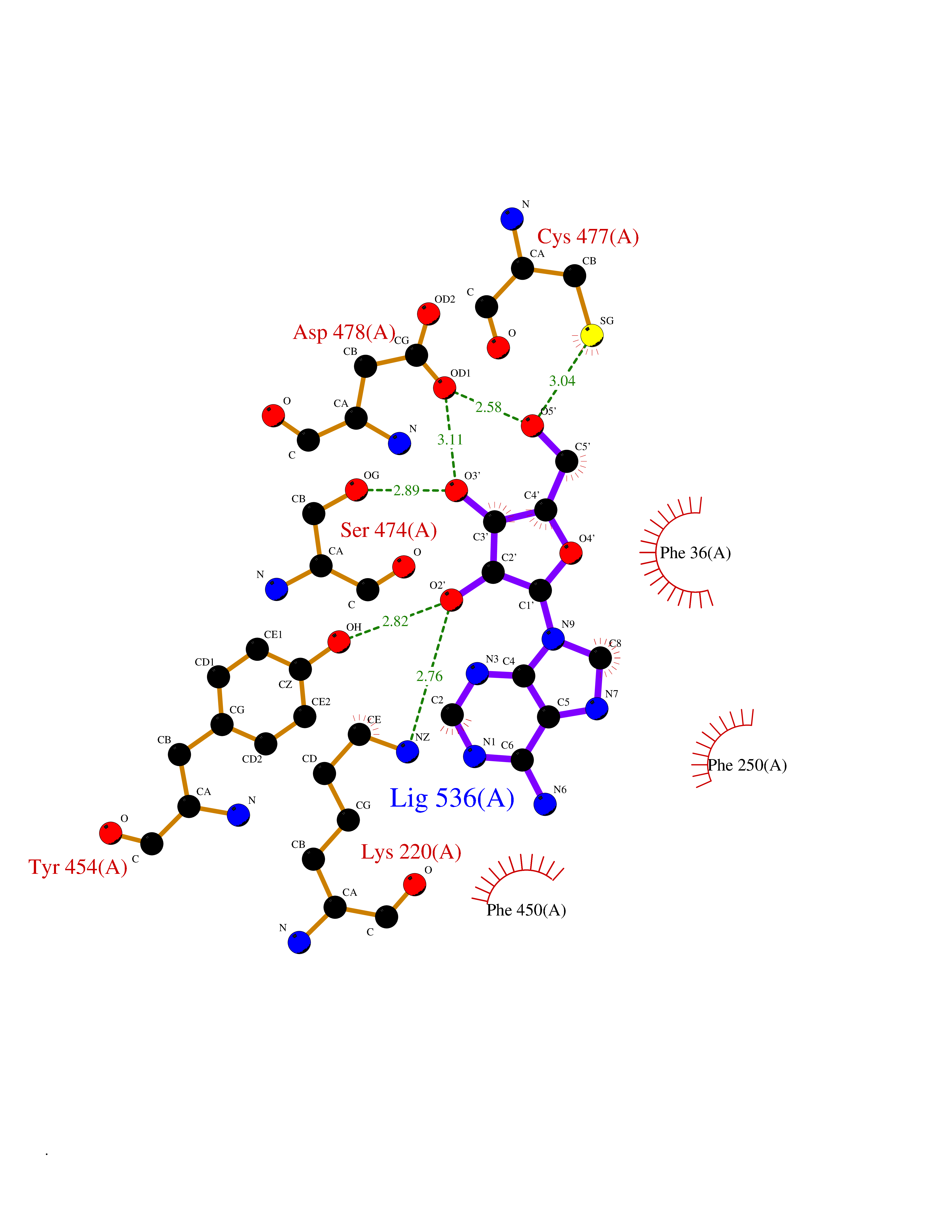 Ligplot Map 3D Binding mode Sequence SFDEALQRVGEFGRFQRRVFLLLCLTGVTFAFLFVGVVFLGTQPDHYWCRGPSAAALAERCGWSPEEEWNRTAPASRGRCQRYLLSAPLVPCRGGWRYAQAHSTIVSEFDLVCVNAWMLDLTQAILNLGFLTGAFTLGYAADRYGRIVIYLLSCLGVGVTGVVVAFAPNFPVFVIFRFLQGVFGKGTWMTCYVIVTEIVGSKQRRIVGIVIQMFFTLGIIILPGIAYFIPNWQGIQLAITLPSFLFLLYYWVVPESPRWLITRKKGDKALQILRRIAKCNVSNPSFLDLVRTPQMRKCTLILMFAWFTSAVVYQGLVMRLGNLYIDFFISGVVELPGALLILLTIERLGRRLPFAASNIVAGVACLVTAFLPEGIAWLRTTVATLGRLGITMAFEIVYLVNSELYPTTLRNFGVSLCSGLCDFGGIIAPFLLFRLAAVWLELPLIIFGILASICGGLVMLLPETKGIALPETVDDVEK Hydrogen bonds contact Hydrophobic contact | ||||