Ligand
Structure
Job ID
3cc3f250ba10c44a59af4a44b579343d
Job name
Johnson_project43
Time
2024-10-14 12:06:25
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 1 | Mucin-1 (MUC1) | 6KX1 | 6.81 | |
Target general information Gen name MUC1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Tumour-associated antigen mucin 1; Tumor-associated mucin; Tumor-associated epithelial membraneantigen; Tumor-associated epithelial membrane antigen; Polymorphic epithelial mucin; Peanut-reactive urin Protein family NA Biochemical class NA Function Can act both as an adhesion and an anti-adhesion protein. May provide a protective layer on epithelial cells against bacterial and enzyme attack. The alpha subunit has cell adhesive properties. Related diseases MUC1/CA 15-3 is used as a serological clinical marker of breast cancer to monitor response to breast cancer treatment and disease recurrence (PubMed:20816948). Decreased levels over time may be indicative of a positive response to treatment. Conversely, increased levels may indicate disease progression. At an early stage disease, only 21% of patients exhibit high MUC1/CA 15-3 levels, that is why CA 15-3 is not a useful screening test. Most antibodies target the highly immunodominant core peptide domain of 20 amino acid (APDTRPAPGSTAPPAHGVTS) tandem repeats. Some antibodies recognize glycosylated epitopes. {ECO:0000269|PubMed:20816948}.; DISEASE: Tubulointerstitial kidney disease, autosomal dominant, 2 (ADTKD2) [MIM:174000]: A form of autosomal dominant tubulointerstitial kidney disease, a genetically heterogeneous disorder characterized by slowly progressive loss of kidney function, bland urinary sediment, hyperuricemia, absent or mildly increased albuminuria, lack of severe hypertension during the early stages, and normal or small kidneys on ultrasound. Renal histology shows variable abnormalities including interstitial fibrosis with tubular atrophy, microcystic dilatation of the tubules, thickening of tubular basement membranes, medullary cysts, and secondary glomerulosclerotic or glomerulocystic changes with abnormal glomerular tufting. There is significant variability, as well as incomplete penetrance. {ECO:0000269|PubMed:23396133}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB11090; DB06584 Interacts with P00519; P00533; P08581; P15941-7; Q08AM2; O60242; Q15848; Q86W74-2; P02652; P05067-2; P29972; P41181; Q92482; Q9H2C2; Q92843; Q6PL45-2; Q8WVV5; P06681; O14523; Q06432; Q9P0B6; Q08722-3; P19397; P34810; Q8N6F1-2; P56747; Q8NHS1; Q96FZ5; Q4VAQ0; Q8N6G5; Q07325; O43169; P78329; P56851; Q9BV81; P54852; O75355-2; Q9UKR5; P01350; P39905-3; Q9Y3E0; Q9NPR9; Q9HCP6; O60725; Q9Y5U4; P11215; Q969L2; Q13021; Q9P0N8; Q6N075; P30301; Q96S97; O95167; Q99519; Q92982; Q9NZG7; Q16617; Q8N912; Q8NH19; Q6TCH4; P26678; P60201-2; Q8IY26; P54315; Q59EV6; P30405; Q96AA3; Q02161-2; Q8TAC9; Q9Y6D0; Q8N6R1; P11686; Q8IWU4; Q969S0; Q6ICL7; Q9NVC3; Q9NRQ5; B2RUZ4; Q9NZ01; P07204; Q9BZW4; P17152; A0PK00; Q9BTD3; Q5BJH2-2; Q9BVK8; Q9Y6G1; Q9P0S9; Q14656; Q8NBD8; Q9BU79; Q8N2M4; Q8N661; Q5BJF2; Q9Y2Y6; O14763; Q8N609; Q5BVD1; Q53HI1; O95183; Q9BQB6; Q8IVQ6; P00519; P17676 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Autocatalytic cleavage; Cell membrane; Cytoplasm; Direct protein sequencing; Disulfide bond; Glycoprotein; Lipoprotein; Membrane; Nucleus; Palmitate; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Secreted; Signal; Transmembrane; Transmembrane helix; Tumor suppressor Protein physicochemical properties Chain ID B,C Molecular weight (Da) 25132.6 Length 230 Aromaticity 0.09 Instability index 44.8 Isoelectric point 7.12 Charge (pH=7) 0.18 2D Binding mode Binding energy (Kcal/mol) -6.51 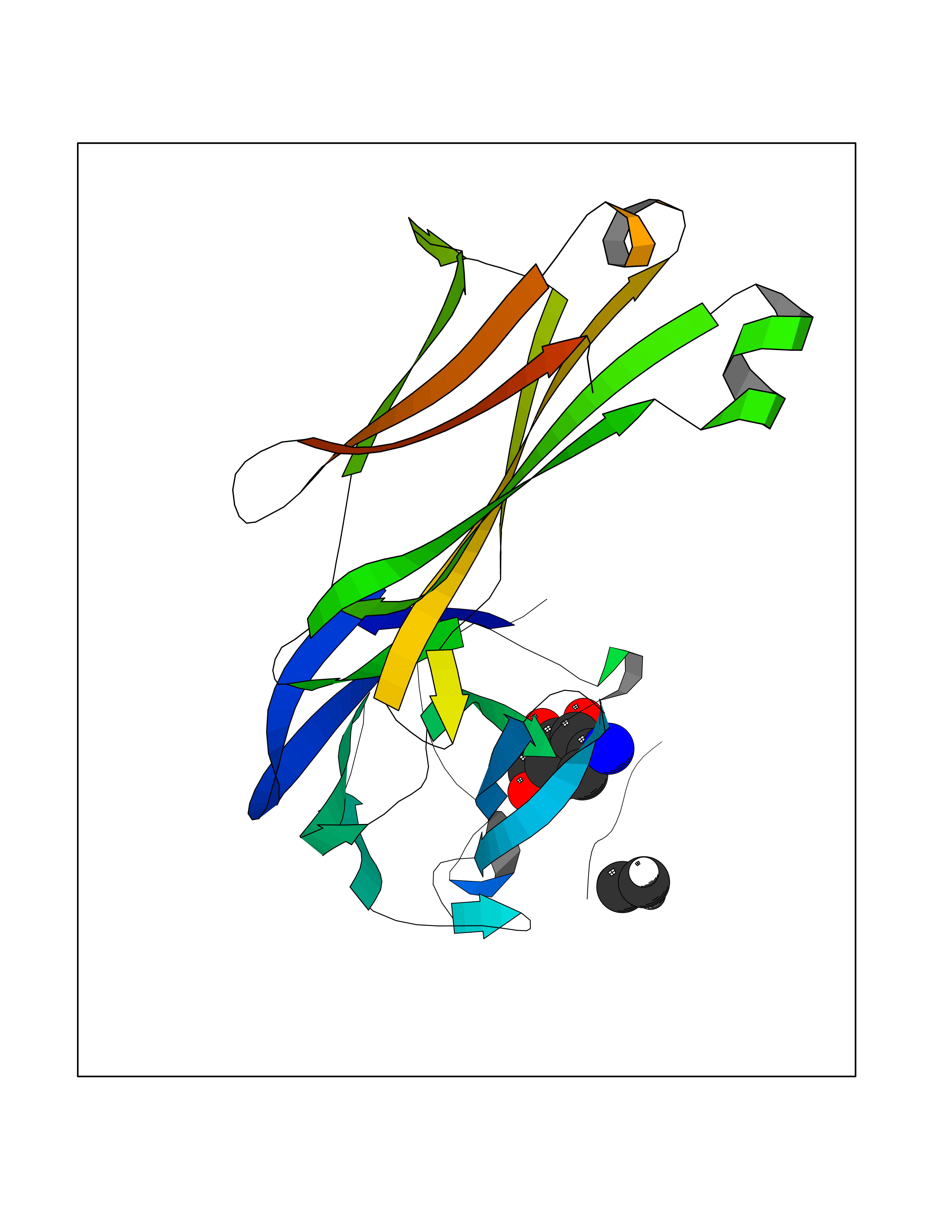 Molscript Map 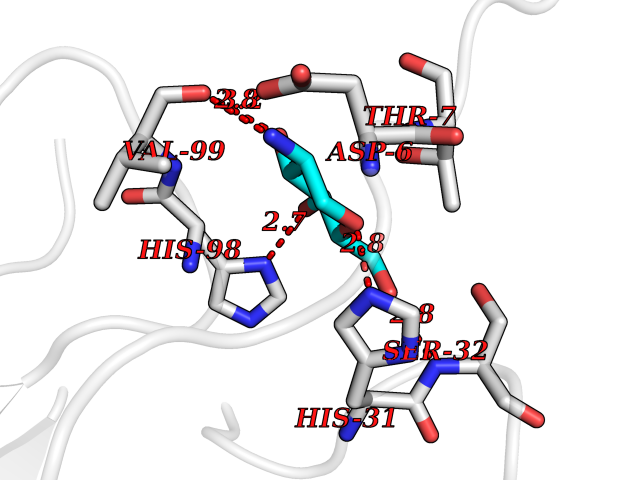 Pymol Map 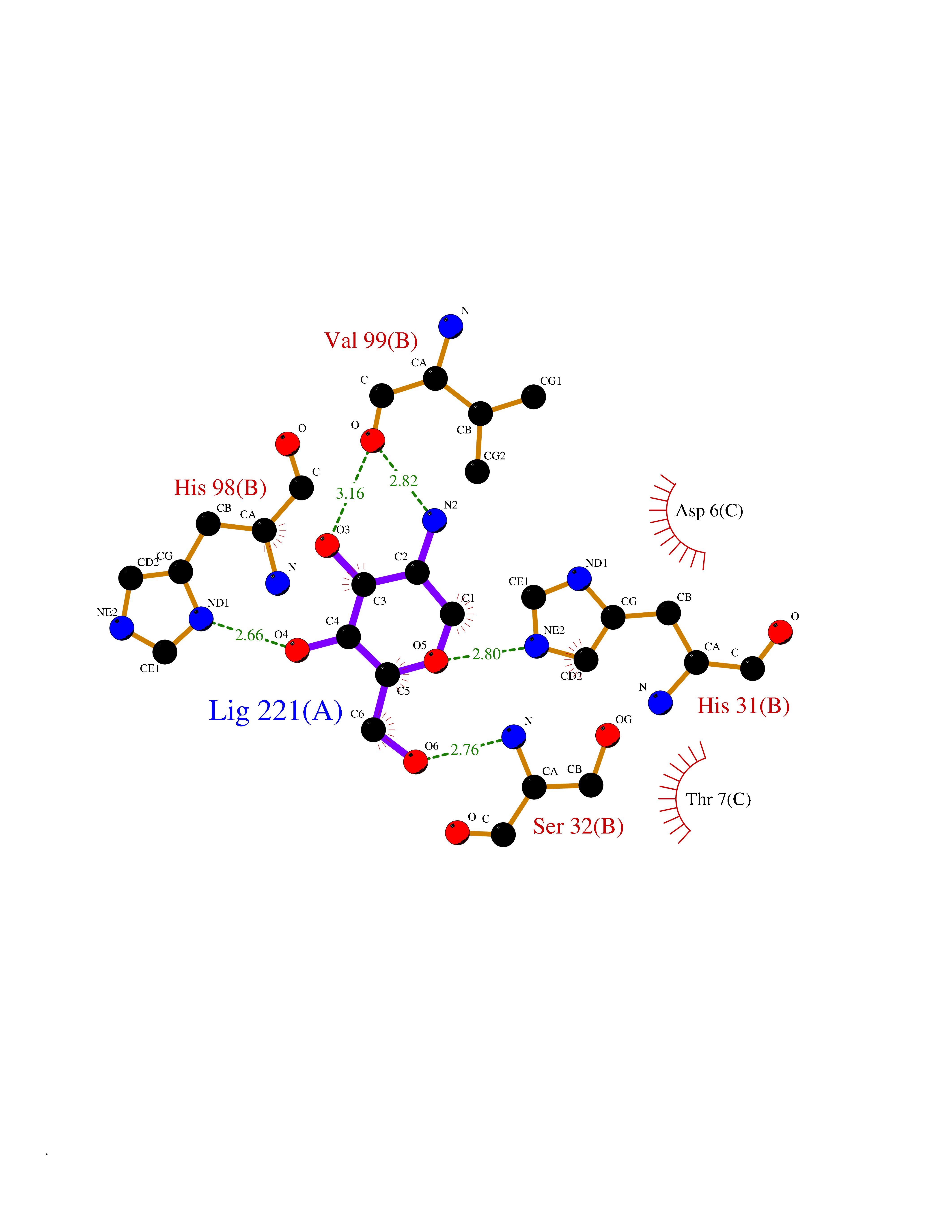 Ligplot Map 3D Binding mode Sequence DVVMTQTPLSLPVSLGDQASISCRSSQSLVHSNGNTYLHWYLQKPGQSPKLLIYKVSNRFSGVPDRFSGSGSGTDFTLKISRVEAEDLGVYFCSQSTHVPPWTFGGGTKLEIKRADAAPTVSIFPPSSEQLTSGGASVVCFLNNFYPKDINVKWKIDGSERQNGVLNSWTDQDSKDSTYSMSSTLTLTKDEYERHNSYTCEATHKTSTSPIVKSFNRNEXVTSAPDTRPA Hydrogen bonds contact Hydrophobic contact | ||||
| 2 | Interleukin 21 receptor (IL21R) | 6PLH | 6.77 | |
Target general information Gen name IL21R Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms UNQ3121/PRO10273; Novel interleukin receptor; NILR; Interleukin-21 receptor; IL21 receptor; IL-21R; IL-21 receptor; CD360 Protein family Type I cytokine receptor family, Type 4 subfamily Biochemical class Cytokine receptor Function This is a receptor for interleukin-21. Related diseases Immunodeficiency 56 (IMD56) [MIM:615207]: An autosomal recessive primary immunodeficiency characterized by B- and T-cell defects and variable dysfunction of NK cells. Patients tend to have normal numbers of lymphocytes, but show defective class-switched B-cells, low IgG, defective antibody response, and defective T-cell responses to certain antigens. {ECO:0000269|PubMed:23440042}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Chromosomal aberrations involving IL21R is a cause of B-cell non-Hodgkin lymphomas (B-cell NHL). Translocation t(3;16)(q27;p11), with BCL6. Drugs (DrugBank ID) NA Interacts with P29972 EC number NA Uniprot keywords 3D-structure; Chromosomal rearrangement; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; Membrane; Proteomics identification; Receptor; Reference proteome; Repeat; Signal; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A,C,B Molecular weight (Da) 48376.5 Length 446 Aromaticity 0.1 Instability index 43.94 Isoelectric point 8.24 Charge (pH=7) 3.56 2D Binding mode Binding energy (Kcal/mol) -6.82 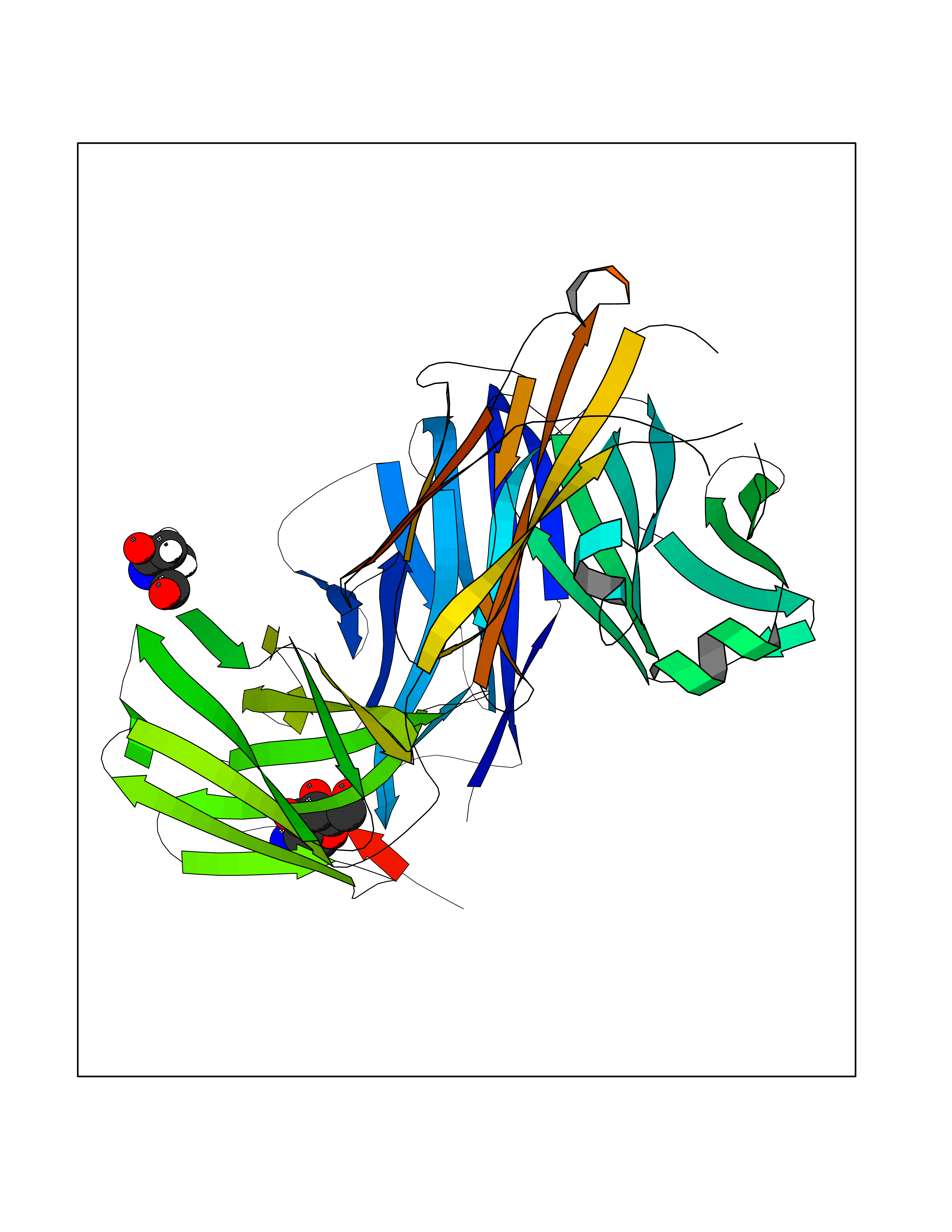 Molscript Map 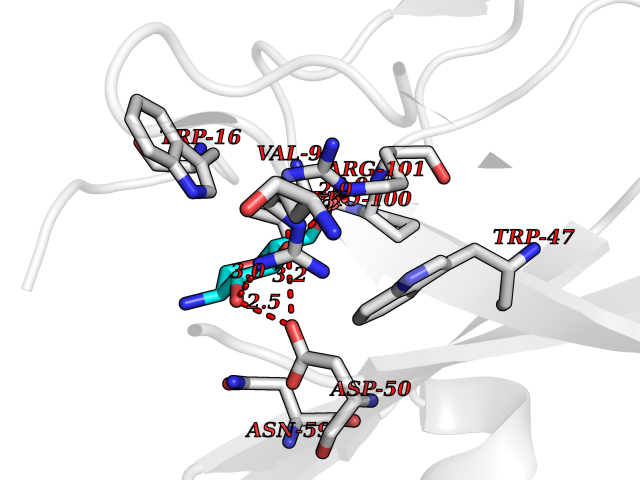 Pymol Map 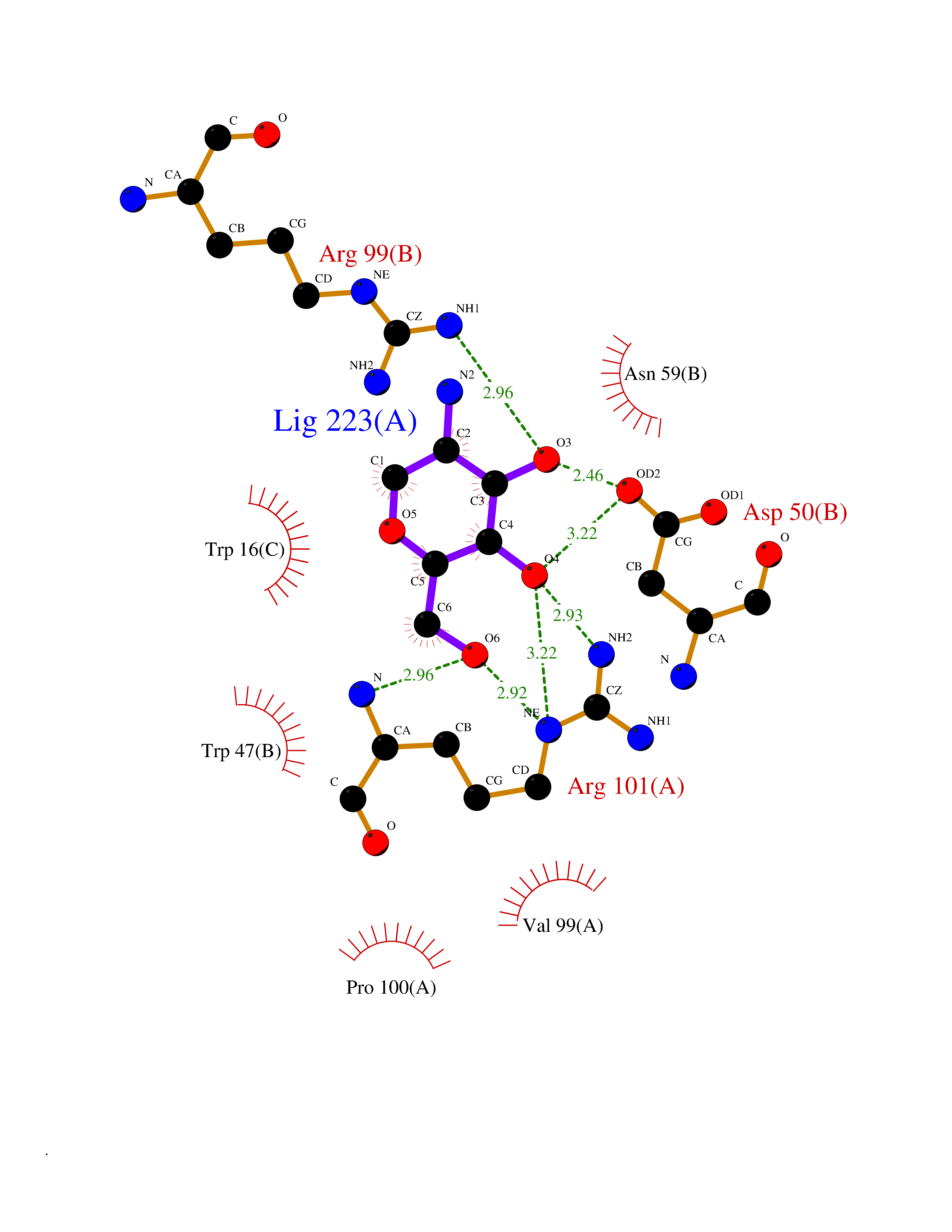 Ligplot Map 3D Binding mode Sequence DVVMTHTPLSLPVSLGDQASISCRSSQSLVHSNGNTYLHWYLQKPGQSPKLLIYKVSNRFSGVPDRFSGSGSGADFTLKISRVEAEDLGVYFCSQSTHVPRTFGGGTKLEIKRADAAPTVSIFPPSSEQLTSGGASVVCFLNNFYPKDINVKWKIDGSERQNGVLNSWTDQDSKDSTYSMSSTLTLTKDEYERHNSYTCEATHKTSTSPIVKSFNRNECXVHLQQPGADLVKPGASVKMSCKASGYTFTSYWITWVKLRPGQGLEWIGDIYPGSGSTNFIEKFKSKATLTVDTSSSTAYMQLRSLTSEDSAVYYCARRGHGNYEDYWGQGTTLIVSSAKTTAPSVYPLAPVCGTGSSVTLGCLVKGYFPEPVTLTWNSGSLSSGVHTFPAVLQSDLYTLSSSVTVTSSTWPSQSITCNVAHPASSTKVDKKIEPRGPTTWSEWSDP Hydrogen bonds contact Hydrophobic contact | ||||
| 3 | Myeloperoxidase (MPO) | 4DL1 | 6.74 | |
Target general information Gen name MPO Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms MPO Protein family Peroxidase family, XPO subfamily Biochemical class Peroxidases Function Part of the host defense system of polymorphonuclear leukocytes. It is responsible for microbicidal activity against a wide range of organisms. In the stimulated PMN, MPO catalyzes the production of hypohalous acids, primarily hypochlorous acidin physiologic situations, and other toxic intermediates that greatly enhance PMN microbicidal activity. Related diseases Myeloperoxidase deficiency (MPOD) [MIM:254600]: A disorder characterized by decreased myeloperoxidase activity in neutrophils and monocytes that results in disseminated candidiasis. {ECO:0000269|PubMed:37198333, ECO:0000269|PubMed:7904599, ECO:0000269|PubMed:8142659, ECO:0000269|PubMed:8621627, ECO:0000269|PubMed:9354683, ECO:0000269|PubMed:9637725}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB06111; DB00233; DB00006; DB02300; DB06774; DB00958; DB06468; DB00833; DB00535; DB00515; DB00847; DB00250; DB05161; DB01225; DB00583; DB01065; DB00461; DB04821; DB00104; DB00526; DB00550; DB00208; DB06823; DB00500; DB04827 Interacts with P27918; Q9UNE7 EC number EC 1.11.2.2 Uniprot keywords 3D-structure; Alternative splicing; Calcium; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; Heme; Hydrogen peroxide; Iron; Lysosome; Metal-binding; Oxidation; Oxidoreductase; Peroxidase; Proteomics identification; Reference proteome; Signal Protein physicochemical properties Chain ID A,B,E,F,I,J,M,N Molecular weight (Da) 53052.6 Length 466 Aromaticity 0.08 Instability index 40.64 Isoelectric point 9.48 Charge (pH=7) 15.12 2D Binding mode Binding energy (Kcal/mol) -6.03 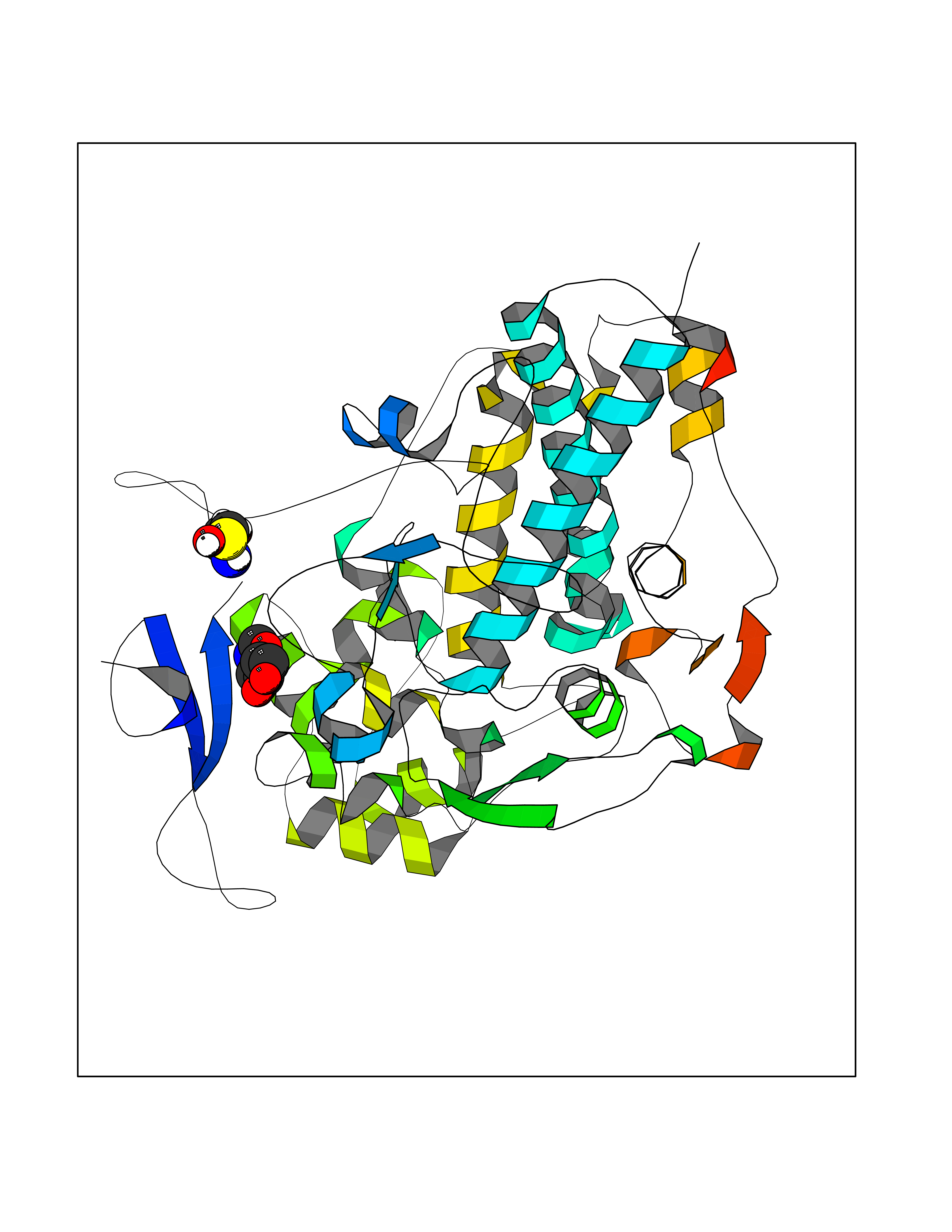 Molscript Map 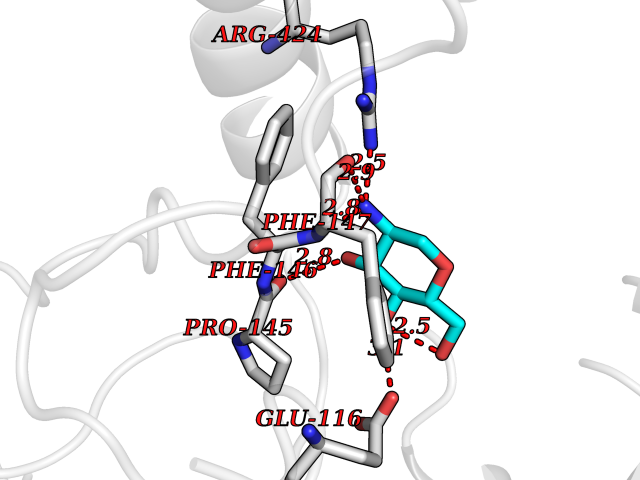 Pymol Map 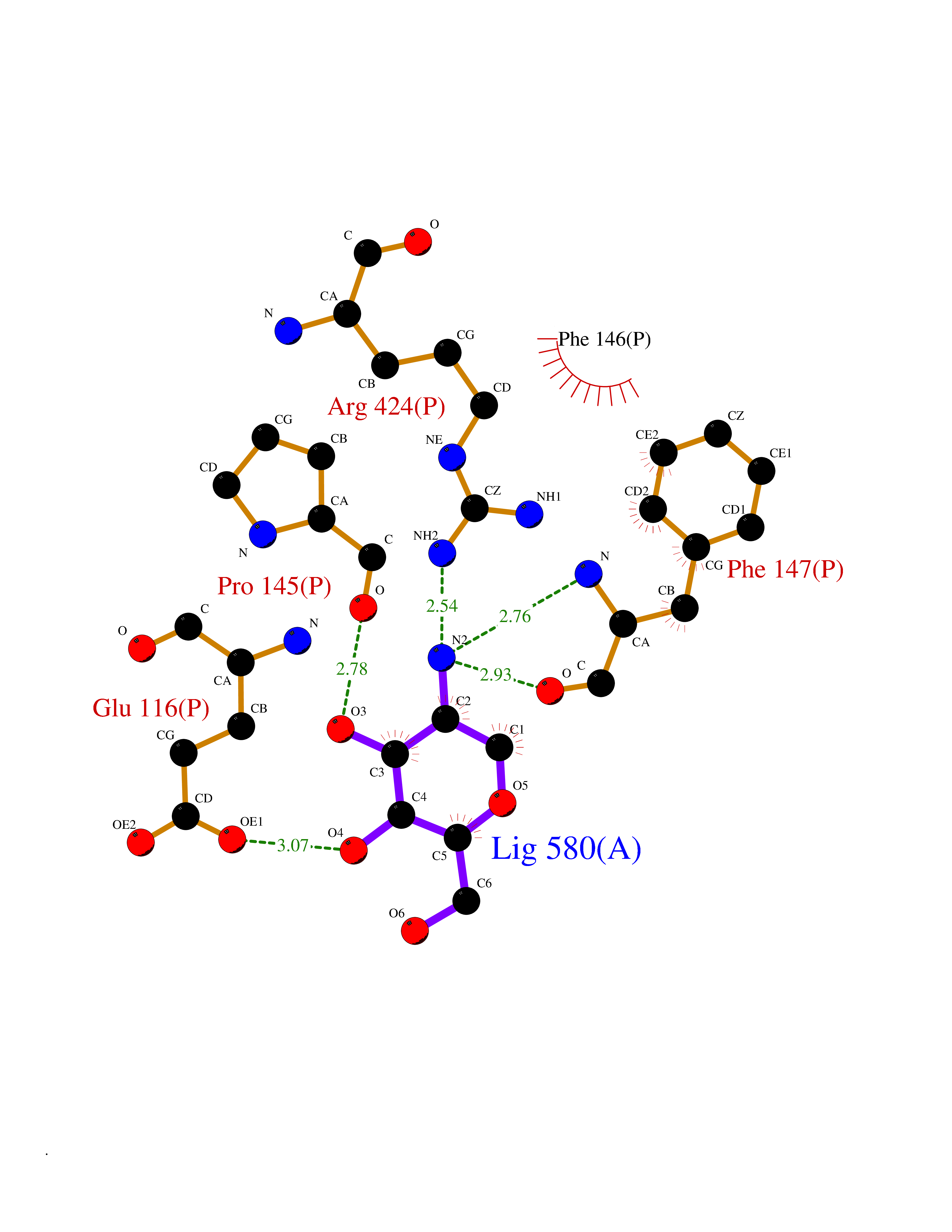 Ligplot Map 3D Binding mode Sequence VNCETSCVQQPPCFPLKIPPNDPRIKNQADCIPFFRSXPACPGSNITIRNQINALTSFVDASMVYGSEEPLARNLRNMSNQLGLLAVNQRFQDNGRALLPFDNLHDDPCLLTNRSARIPCFLAGDTRSSEMPELTSMHTLLLREHNRLATELKSLNPRWDGERLYQEARKIVGAMVQIITYRDYLPLVLGPTAMRKYLPTYRSYNDSVDPRIANVFTNAFRYGHTLIQPFMFRLDNRYQPMEPNPRVPLSRVFFASWRVVLEGGIDPILRGLMATPAKLNRQNQIAVDEIRERLFEQVMRIGLDLPALNMQRSRDHGLPGYNAWRRFCGLPQPETVGQLGTVLRNLKLARKLMEQYGTPNNIDIWMGGVSEPLKRKGRVGPLLACIIGTQFRKLRDGDRFWWENEGVFSMQQRQALAQISLPRIICDNTGITTVSKNNIFMSNSYPRDFVNCSTLPALNLASWREA Hydrogen bonds contact Hydrophobic contact | ||||
| 4 | Choline O-acetyltransferase | 2FY3 | 6.62 | |
Target general information Gen name CHAT Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Carnitine/choline acetyltransferase family Biochemical class Transferase Function Choline O-acetyltransferase activity. Related diseases Myasthenic syndrome, congenital, 6, presynaptic (CMS6) [MIM:254210]: A form of congenital myasthenic syndrome, a group of disorders characterized by failure of neuromuscular transmission, including pre-synaptic, synaptic, and post-synaptic disorders that are not of autoimmune origin. Clinical features are easy fatigability and muscle weakness affecting the axial and limb muscles (with hypotonia in early-onset forms), the ocular muscles (leading to ptosis and ophthalmoplegia), and the facial and bulbar musculature (affecting sucking and swallowing, and leading to dysphonia). The symptoms fluctuate and worsen with physical effort. CMS6 affected individuals have myasthenic symptoms since birth or early infancy, negative tests for anti-AChR antibodies, and abrupt episodic crises with increased weakness, bulbar paralysis, and apnea precipitated by undue exertion, fever, or excitement. CMS6 inheritance is autosomal recessive. {ECO:0000269|PubMed:11172068, ECO:0000269|PubMed:12756141}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00122; DB14006; DB00184 Interacts with Q6H8Q1-8; Q8N302-2; Q9NXL2-1; Q6XD76; Q9UII2; Q8TBE0; Q9UQB8-6; Q9ULD4-2; Q9NSI6-4; Q6P5X5; Q96LL4; P20807-4; O00257-3; Q6ZP82-1; O95674; Q9H3R5; Q8WUX9; Q9H2A9; Q3SX64; Q92782-2; Q14117; O14641; Q658K8; Q6UXG2-3; O00472; Q6NXG1; Q15910-2; Q8IZU1; P15407; P55318; Q06547-3; P23769-2; P23771; Q15486; Q8IV36; Q4VB01; Q53GQ0; P10809; P41134; Q9NZH6; Q8NA54; Q86U28; P17275; Q8N5Z5; Q6P597; P08727; Q14525; Q8IUC2; Q6IAA8; Q14847-2; P27338; Q9GZQ8; Q53S70; Q5JXC2; A0A0A0MR05; Q8NEH6; Q8TCY5; Q6IN84-2; Q96H12; P01106; P41271-2; P14598; Q9GZM8; Q5BJF6-2; Q9H8K7; Q9NR21-5; Q5VU43-8; Q13956; Q5SXH7-1; Q96T60; Q96I34; Q86UA1; Q15311; Q8TBY0; Q04206; P47804-3; Q9H0X6; P62899; Q66K80; Q9BY12-3; Q86SQ7-2; Q7Z6I5; Q496A3; Q7Z698; Q9C004; Q92783-2; Q8N4C7; O75528; Q15814; O15273; Q96A09; Q8WTV1; Q53NU3; Q71RG4-4; Q86WT6-2; Q9Y3Q8; Q99598; P49459; P11441; Q9H270; P19544-6; Q53FD0-2; Q3KNS6-3 EC number 2.3.1.6 Uniprot keywords 3D-structure; Acyltransferase; Alternative splicing; Congenital myasthenic syndrome; Direct protein sequencing; Disease variant; Neurotransmitter biosynthesis; Phosphoprotein; Proteomics identification; Reference proteome; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 66365.9 Length 595 Aromaticity 0.08 Instability index 53.36 Isoelectric point 8.16 Charge (pH=7) 4.64 2D Binding mode Binding energy (Kcal/mol) -6.73 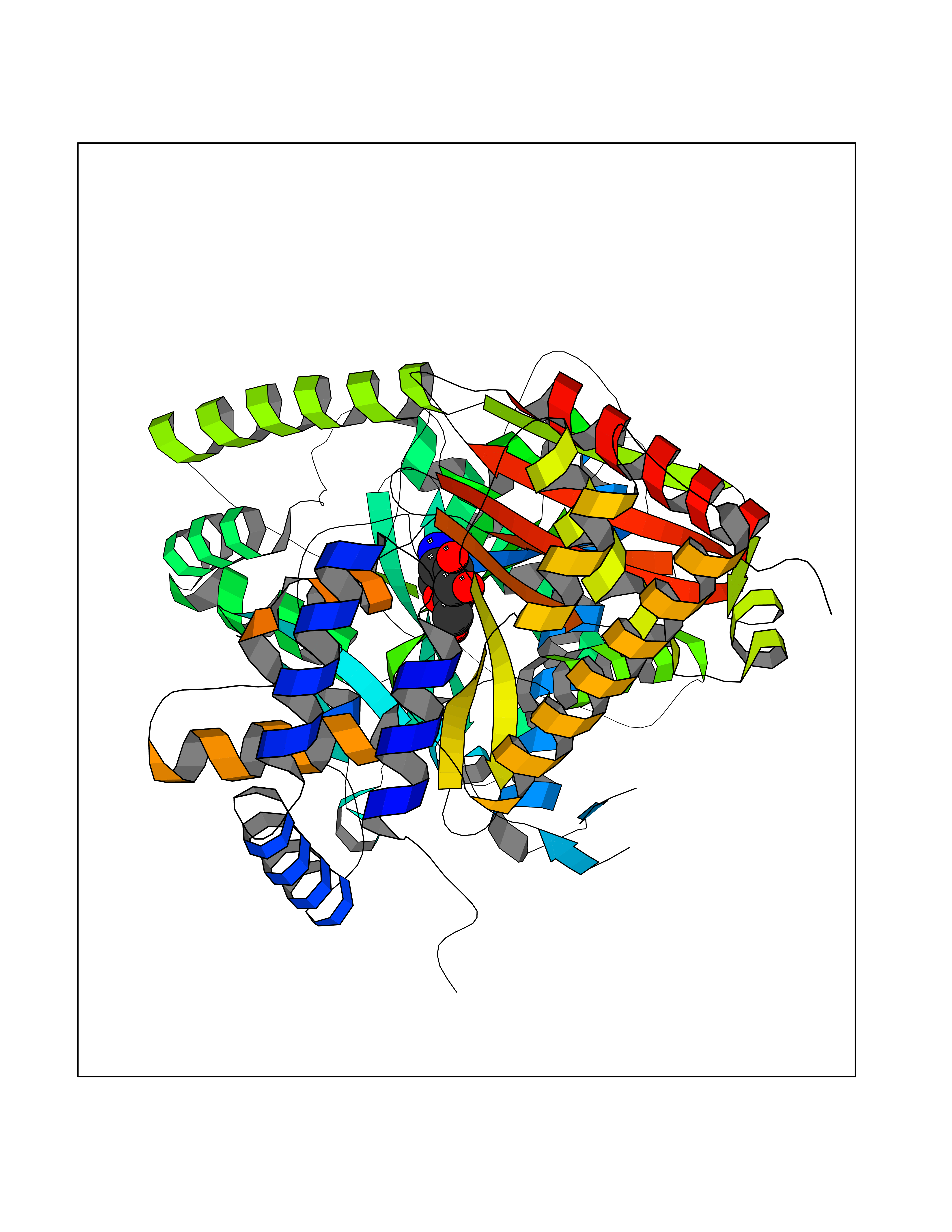 Molscript Map 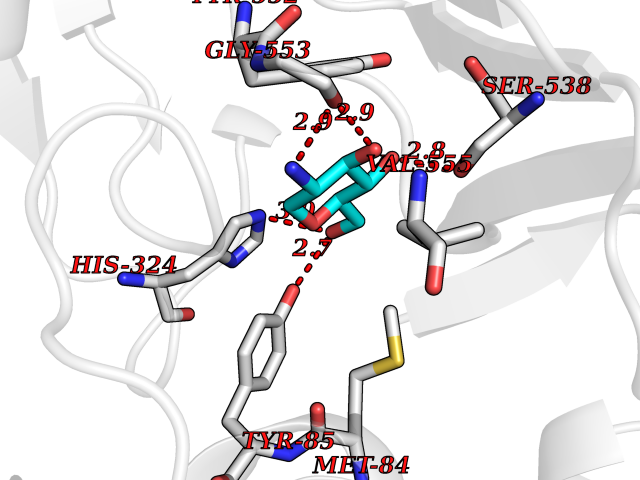 Pymol Map 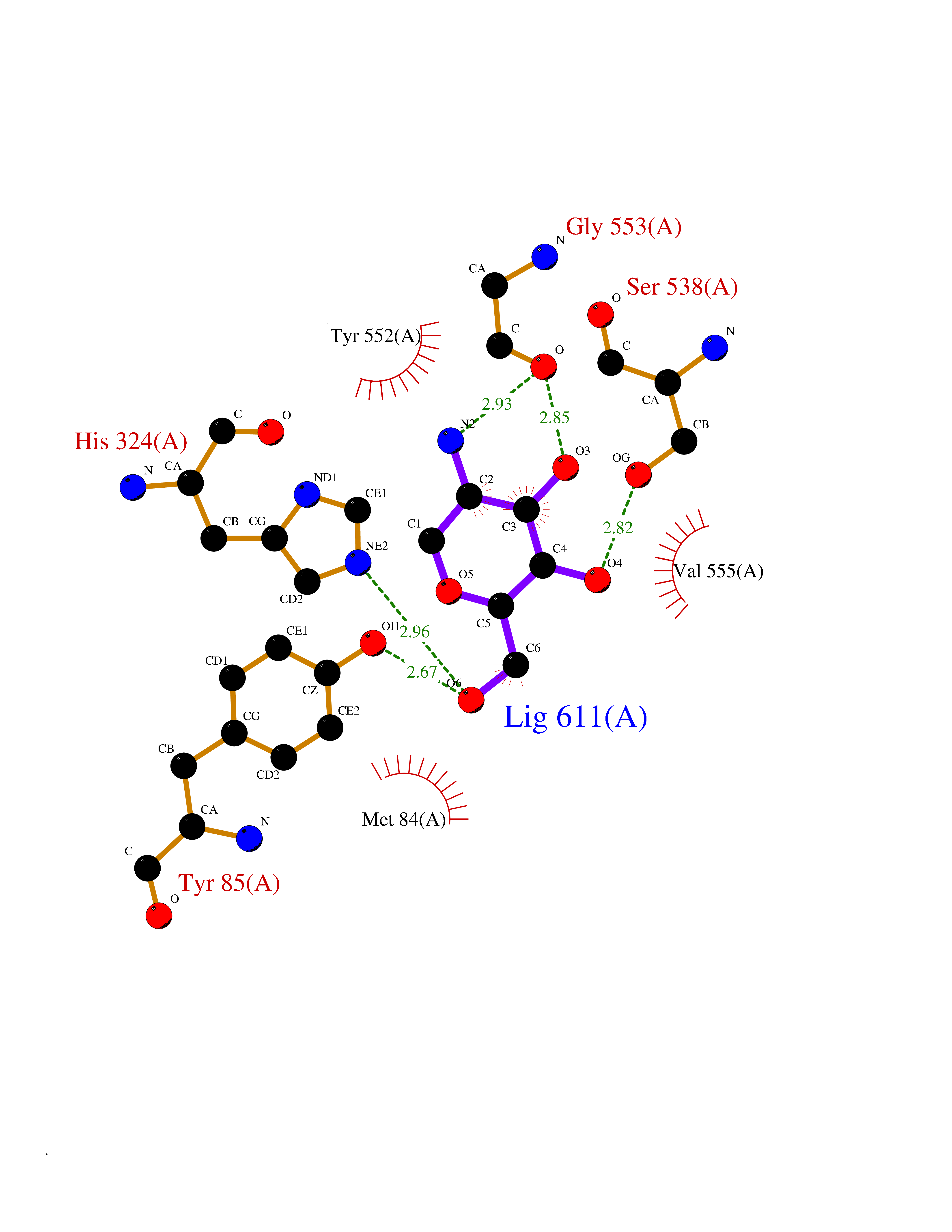 Ligplot Map 3D Binding mode Sequence SEESGLPKLPVPPLQQTLATYLQCMRHLVSEEQFRKSQAIVQQFGAPGGLGETLQQKLLERQEKTANWVSEYWLNDMYLNNRLALPVNSSPAVIFARQHFPGTDDQLRFAASLISGVLSYKALLDSHSIPTDCAKGQPLCMKQYYGLFSSYRLPGHTQDTLVAQNSSIMPEPEHVIVACCNQFFVLDVVINFRRLSEGDLFTQLRKIVKMASNAAARLPPIGLLTSDGRSEWAEARTVLVKDSTNRDSLDMIERCICLVCLDAPGGVELSDTHRALQLLHGGGYSKNGANRWYDKSLQFVVGRDGTCGVVCEHSPFDGIVLVQCTEHLLKHMTQPELVRSPMVPLPAPRRLRWKCSPEIQGHLASSAEKLQRIVKNLDFIVYKFDNYGKTFIKKQKCSPDAFIQVALQLAFYRLHRRLVPTYESASIRRFQEGRVDNIRSATPEALAFVRAVTDHKAAVPASEKLLLLKDAIRAQTAYTVMAITGMAIDNHLLALRELARAMCAALPEMFMDETYLMSNRFVLSTSQVPTTTEMFCCYGPVVPNGYGACYNPQPETILFCISSFHSCAATSSSKFAKAVEESLIDMRDLCSLLPP Hydrogen bonds contact Hydrophobic contact | ||||
| 5 | Carbonic anhydrase IV (CA-IV) | 3FW3 | 6.54 | |
Target general information Gen name CA4 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Carbonic anhydrase 4; Carbonate dehydratase IV; CAIV Protein family Alpha-carbonic anhydrase family Biochemical class Alpha-carbonic anhydrase Function May stimulate the sodium/bicarbonate transporter activity of SLC4A4 that acts in pH homeostasis. It is essential for acid overload removal from the retina and retina epithelium, and acid release in the choriocapillaris in the choroid. Reversible hydration of carbon dioxide. Related diseases Retinitis pigmentosa 17 (RP17) [MIM:600852]: A retinal dystrophy belonging to the group of pigmentary retinopathies. Retinitis pigmentosa is characterized by retinal pigment deposits visible on fundus examination and primary loss of rod photoreceptor cells followed by secondary loss of cone photoreceptors. Patients typically have night vision blindness and loss of midperipheral visual field. As their condition progresses, they lose their far peripheral visual field and eventually central vision as well. {ECO:0000269|PubMed:15563508, ECO:0000269|PubMed:17652713, ECO:0000269|PubMed:20450258}. The disease is caused by variants affecting the gene represented in this entry. Defective acid overload removal from retina and retinal epithelium, due to mutant CA4, is responsible for photoreceptor degeneration, indicating that impaired pH homeostasis is the most likely cause underlying the RP17 phenotype. Drugs (DrugBank ID) DB00819; DB00436; DB00562; DB01194; DB00606; DB01144; DB00869; DB08846; DB00311; DB00774; DB00703; DB00232; DB09460; DB00273; DB01021; DB00909 Interacts with NA EC number EC 4.2.1.1 Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; GPI-anchor; Lipoprotein; Lyase; Membrane; Metal-binding; Proteomics identification; Reference proteome; Retinitis pigmentosa; Signal; Zinc Protein physicochemical properties Chain ID A,B Molecular weight (Da) 27055.7 Length 235 Aromaticity 0.09 Instability index 44.3 Isoelectric point 6.87 Charge (pH=7) -0.36 2D Binding mode Binding energy (Kcal/mol) -6.39 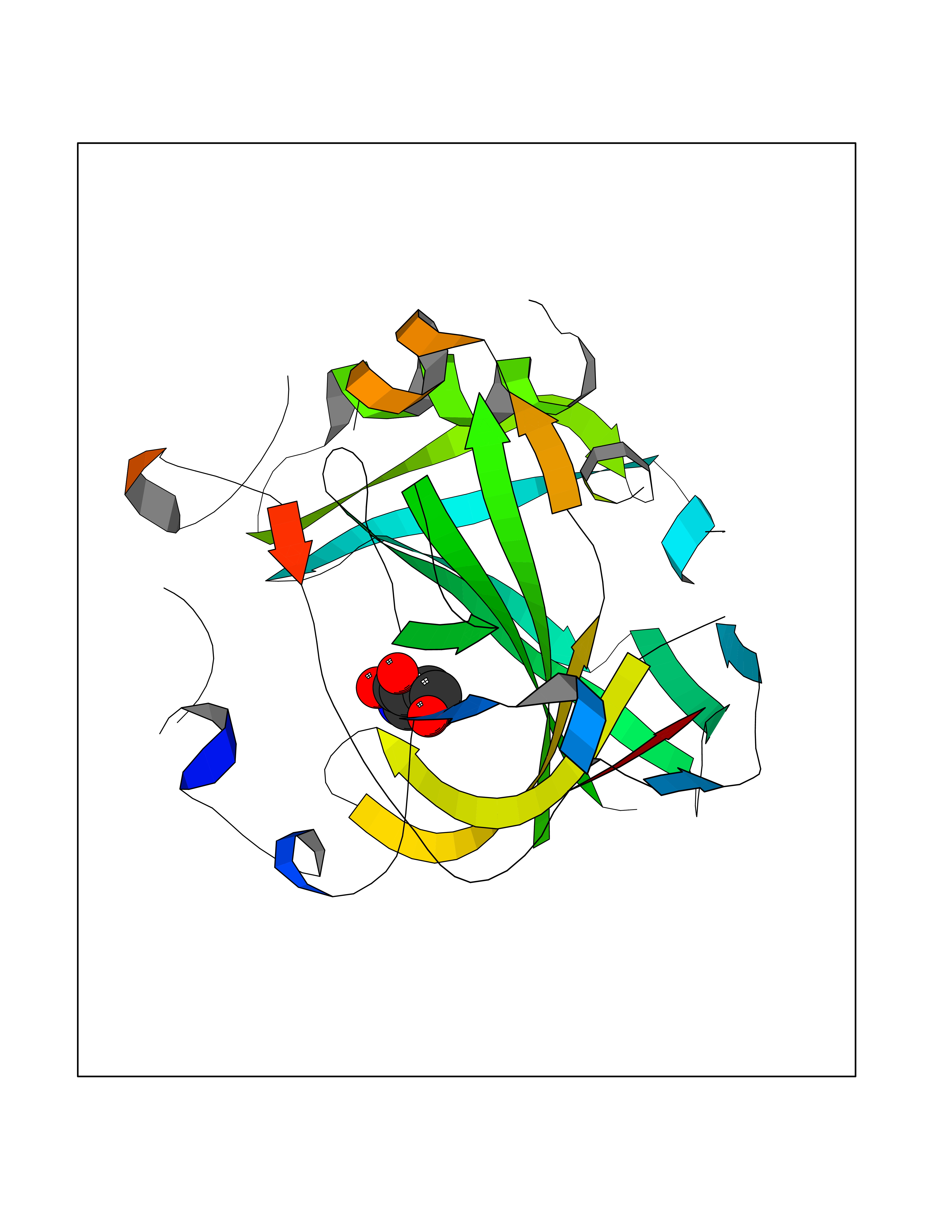 Molscript Map 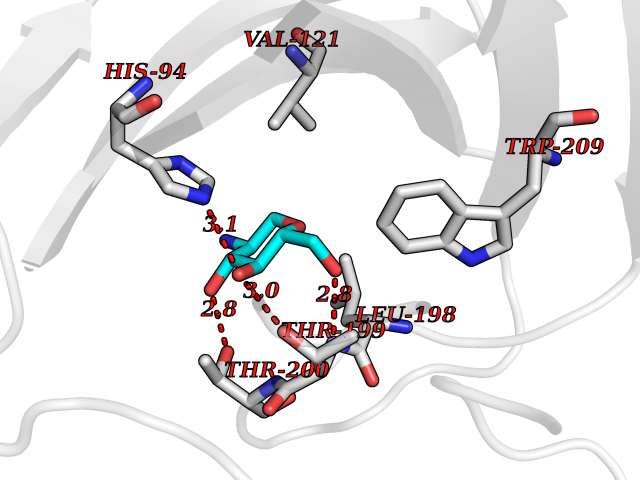 Pymol Map 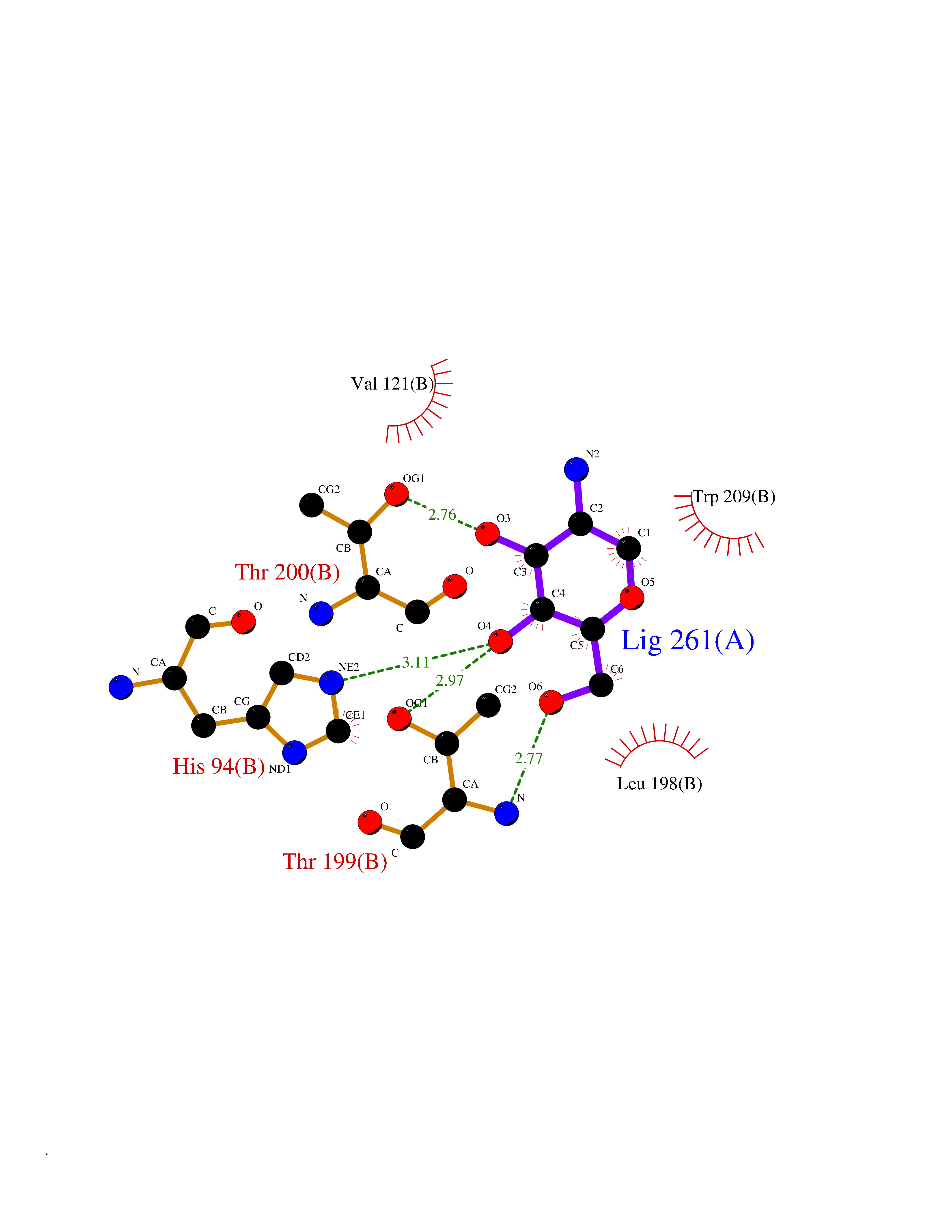 Ligplot Map 3D Binding mode Sequence HWCYEVQLVPVKWGGNCQKDRQSPINIVTTKAKVDKKLGRFFFGYDKKQTWTVQNNGHSVMMLLENKASISGGGLPAPYQAKQLHLHWSDLPYKGSEHSLDGEHFAMEMHIVHEKEEIAVLAFLVEATQVNEGFQPLVEALSNIPKPEMSTTMAESSLLDLLPEEKRHYFRYLGSLTTPTCDEKVVWTVFREPIQLHREQILAFQKLYYDKEQTVSMKDNVRPLQQLGQRTVIKS Hydrogen bonds contact Hydrophobic contact | ||||
| 6 | Carbonic anhydrase IX (CA-IX) | 5FL4 | 6.50 | |
Target general information Gen name CA9 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Renal cell carcinoma-associated antigen G250; RCC-associated antigen G250; PMW1; P54/58N; Membrane antigen MN; MN; G250 antigen (MN/CA IX/G250); G250; Carbonic anhydrase 9; Carbonate dehydratase IX; C Protein family Alpha-carbonic anhydrase family Biochemical class Alpha-carbonic anhydrase Function Participates in pH regulation. May be involved in the control of cell proliferation and transformation. Appears to be a novel specific biomarker for a cervical neoplasia. Reversible hydration of carbon dioxide. Related diseases Hydroxykynureninuria (HYXKY) [MIM:236800]: An inborn error of amino acid metabolism characterized by massive urinary excretion of large amounts of kynurenine, 3-hydroxykynurenine and xanthurenic acid. Affected individuals manifest renal tubular dysfunction, metabolic acidosis, psychomotor retardation, non-progressive encephalopathy, and muscular hypertonia. {ECO:0000269|PubMed:17334708, ECO:0000269|PubMed:28792876}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Vertebral, cardiac, renal, and limb defects syndrome 2 (VCRL2) [MIM:617661]: An autosomal recessive congenital malformation syndrome characterized by vertebral segmentation abnormalities, congenital cardiac defects, renal defects, and distal mild limb defects. {ECO:0000269|PubMed:28792876}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00562; DB00606; DB12741; DB08846; DB05304; DB00774; DB09460; DB00909 Interacts with P21291; O76003 EC number EC 4.2.1.1 Uniprot keywords 3D-structure; Cell membrane; Cell projection; Direct protein sequencing; Disulfide bond; Glycoprotein; Lyase; Membrane; Metal-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Signal; Transmembrane; Transmembrane helix; Zinc Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 27522.8 Length 251 Aromaticity 0.08 Instability index 48.97 Isoelectric point 5.48 Charge (pH=7) -7.5 2D Binding mode Binding energy (Kcal/mol) -6.38 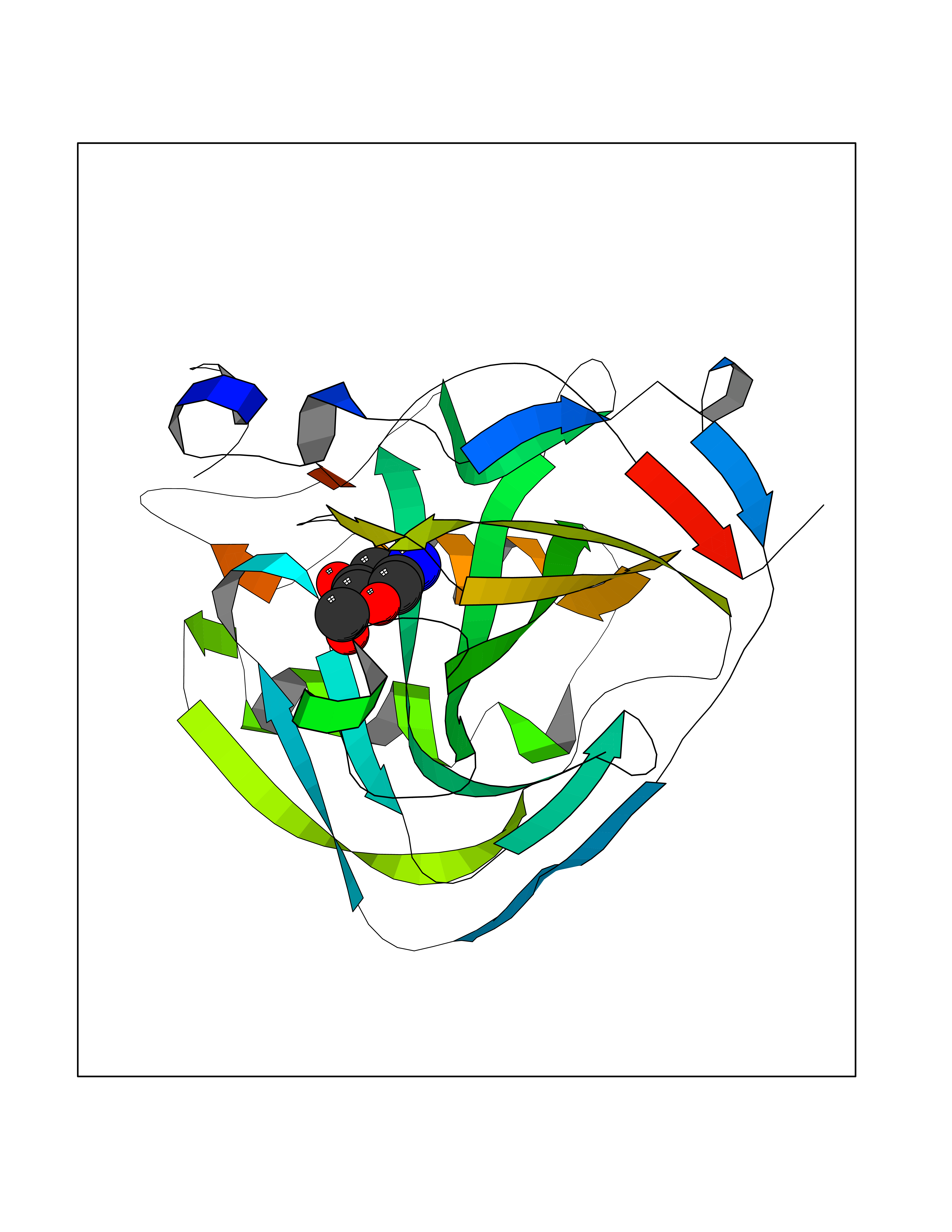 Molscript Map 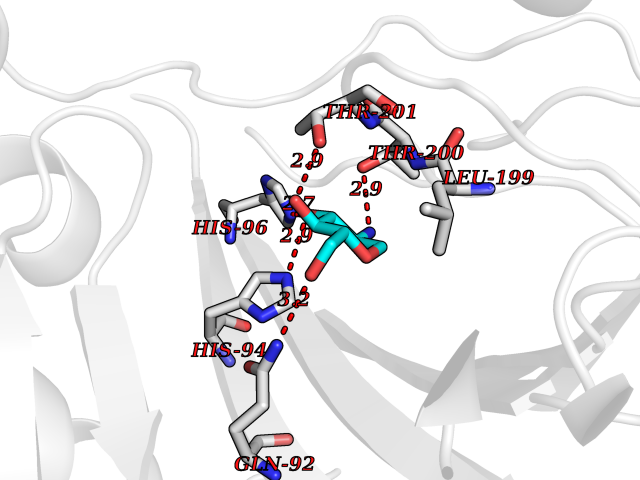 Pymol Map 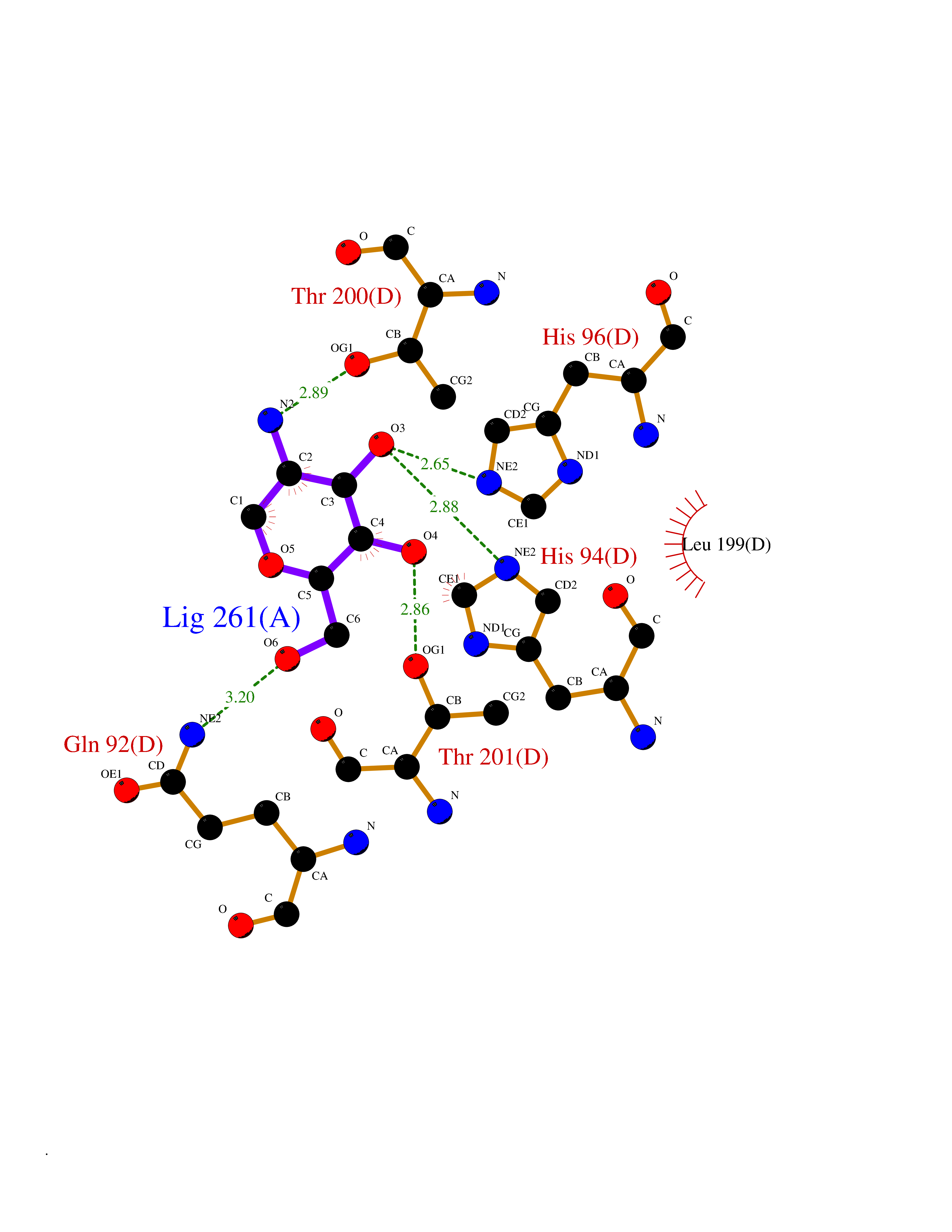 Ligplot Map 3D Binding mode Sequence WRYGGDPPWPRVSPACAGRFQSPVDIRPQLAAFSPALRPLELLGFQLPPLPELRLRNNGHSVQLTLPPGLEMALGPGREYRALQLHLHWGAAGRPGSEHTVEGHRFPAEIHVVHLSTAFARVDEALGRPGGLAVLAAFLEEGPEENSAYEQLLSRLEEIAEEGSETQVPGLDISALLPSDFSRYFQYEGSLTTPPCAQGVIWTVFNQTVMLSAKQLHTLSDTLWGPGDSRLQLNFRATQPLNGRVIEASFP Hydrogen bonds contact Hydrophobic contact | ||||
| 7 | HIF-prolyl hydroxylase 2 (HPH-2) | 6ZBO | 6.50 | |
Target general information Gen name EGLN1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms SM-20; Prolyl hydroxylase domain-containing protein 2; PHD2; Hypoxia-inducible factor prolyl hydroxylase 2; HPH-2; HIF-PH2; Egl nine homolog 1; C1orf12 Protein family NA Biochemical class Paired donor oxygen oxidoreductase Function Cellular oxygen sensor that catalyzes, under normoxic conditions, the post-translational formation of 4-hydroxyproline in hypoxia-inducible factor (HIF) alpha proteins. Hydroxylates a specific proline found in each of the oxygen-dependent degradation (ODD) domains (N-terminal, NODD, and C-terminal, CODD) of HIF1A. Also hydroxylates HIF2A. Has a preference for the CODD site for both HIF1A and HIF1B. Hydroxylated HIFs are then targeted for proteasomal degradation via the von Hippel-Lindau ubiquitination complex. Under hypoxic conditions, the hydroxylation reaction is attenuated allowing HIFs to escape degradation resulting in their translocation to the nucleus, heterodimerization with HIF1B, and increased expression of hypoxy-inducible genes. EGLN1 is the most important isozyme under normoxia and, through regulating the stability of HIF1, involved in various hypoxia-influenced processes such as angiogenesis in retinal and cardiac functionality. Target proteins are preferentially recognized via a LXXLAP motif. Related diseases Erythrocytosis, familial, 3 (ECYT3) [MIM:609820]: An autosomal dominant disorder characterized by elevated serum hemoglobin and hematocrit, and normal serum erythropoietin levels. {ECO:0000269|PubMed:16407130, ECO:0000269|PubMed:17579185}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00126; DB11682; DB14490; DB14491; DB14488; DB14501; DB14489; DB08687; DB01592; DB07112; DB04847; DB12255 Interacts with Q99814; Q14318; Q16665; Q13438; PRO_0000037551 [Q9WMX2] EC number EC 1.14.11.29 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Congenital erythrocytosis; Cytoplasm; Dioxygenase; Disease variant; Iron; Metal-binding; Nucleus; Oxidoreductase; Phosphoprotein; Proteomics identification; Reference proteome; S-nitrosylation; Vitamin C; Zinc; Zinc-finger Protein physicochemical properties Chain ID A,D Molecular weight (Da) 45717.8 Length 406 Aromaticity 0.11 Instability index 24.41 Isoelectric point 7.59 Charge (pH=7) 1.54 2D Binding mode Binding energy (Kcal/mol) -5.72 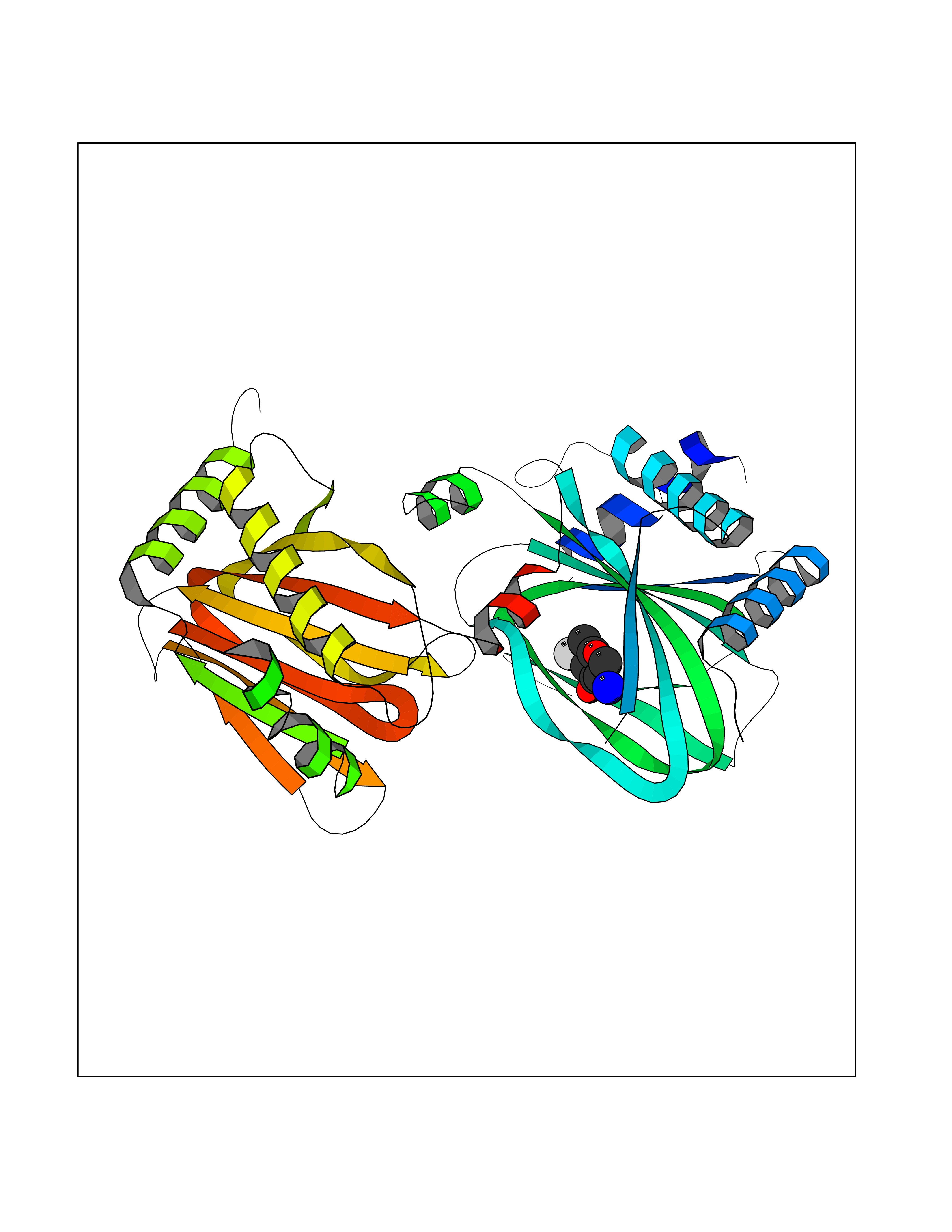 Molscript Map 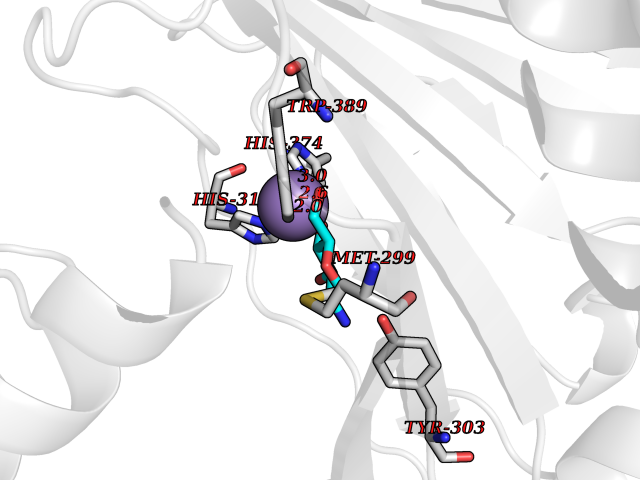 Pymol Map 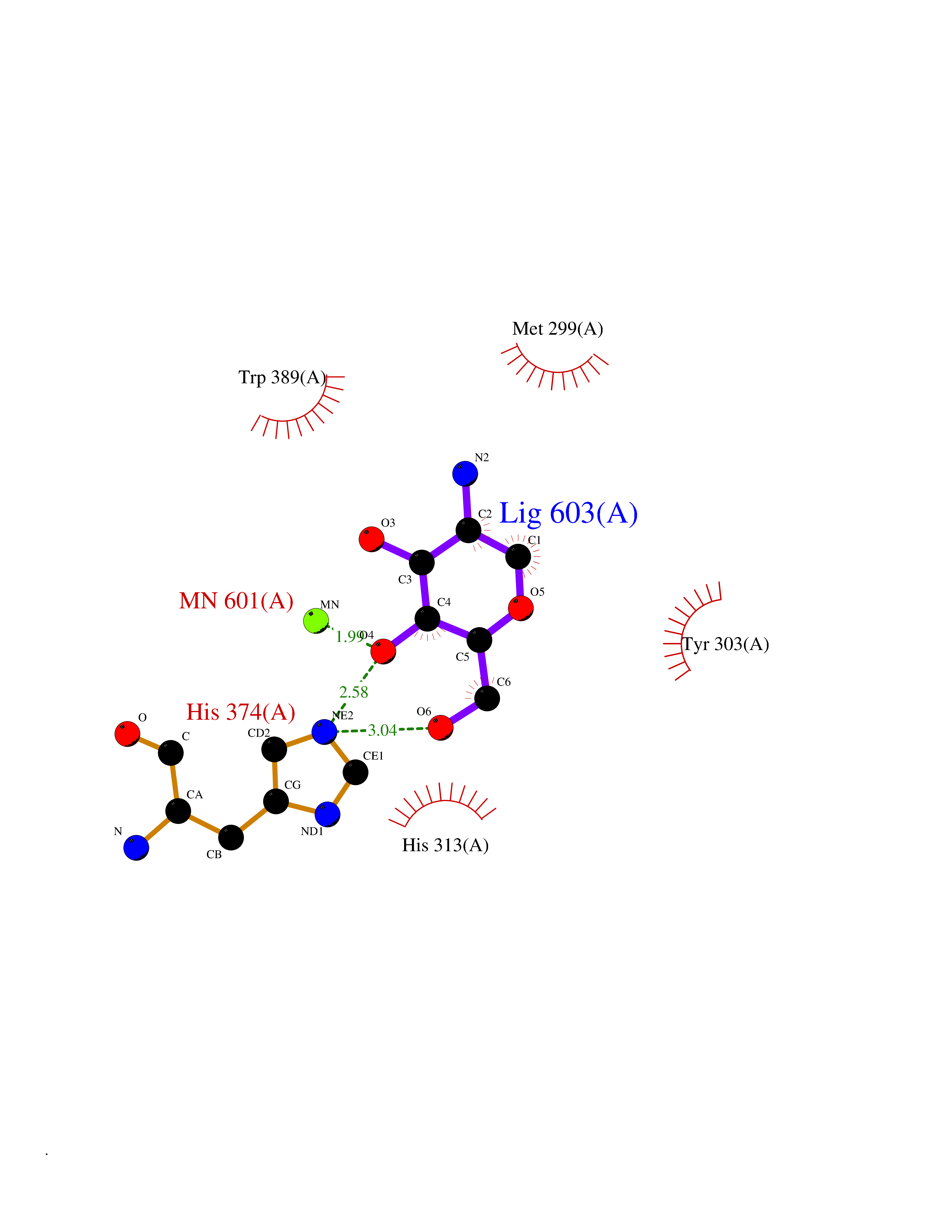 Ligplot Map 3D Binding mode Sequence LPALKLALEYIVPCMNKHGICVVDDFLGKETGQQIGDEVRALHDTGKFTGDKITWIEGKEPGCETIGLLMSSMDDLIRHCNGKLGSYKINGRTKAMVACYPGNGTGYVRHVDNPNGDGRCVTCIYYLNKDWDAKVSGGILRIFPEGKAQFADIEPKFDRLLFFWSDRRNPHEVQPAYATRYAITVWYFDADERARAKVKYLTGELPALKLALEYIVPCMNKHGICVVDDFLGKETGQQIGDEVRALHDTGKFTGDKITWIEGKEPGCETIGLLMSSMDDLIRHCNGKLGSYKINGRTKAMVACYPGNGTGYVRHVDNPNGDGRCVTCIYYLNKDWDAKVSGGILRIFPEGKAQFADIEPKFDRLLFFWSDRRNPHEVQPAYATRYAITVWYFDADERARAKVKYLT Hydrogen bonds contact Hydrophobic contact | ||||
| 8 | Endothelin-converting enzyme 1 (ECE1) | 3DWB | 6.48 | |
Target general information Gen name ECE1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms ECE-1 Protein family Peptidase M13 family Biochemical class Peptidase Function Converts big endothelin-1 to endothelin-1. Related diseases Hirschsprung disease, cardiac defects, and autonomic dysfunction (HCAD) [MIM:613870]: A disorder characterized by skip-lesions Hirschsprung disease, craniofacial abnormalities and other dysmorphic features, cardiac defects including ductus arteriosus, small subaortic ventricular septal defect, small atrial septal defect, and autonomic dysfunction. {ECO:0000269|PubMed:9915973}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07171 Interacts with P49760; A8MQ03; Q8IUG1; P60370; P60410 EC number EC 3.4.24.71 Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disease variant; Disulfide bond; Glycoprotein; Hirschsprung disease; Hydrolase; Membrane; Metal-binding; Metalloprotease; Phosphoprotein; Protease; Proteomics identification; Reference proteome; Signal-anchor; Transmembrane; Transmembrane helix; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 75247.9 Length 660 Aromaticity 0.12 Instability index 46.29 Isoelectric point 5.33 Charge (pH=7) -18.3 2D Binding mode Binding energy (Kcal/mol) -6.22  Molscript Map 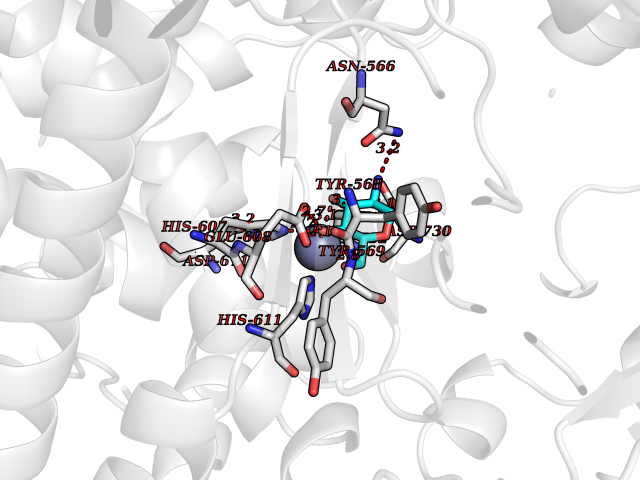 Pymol Map 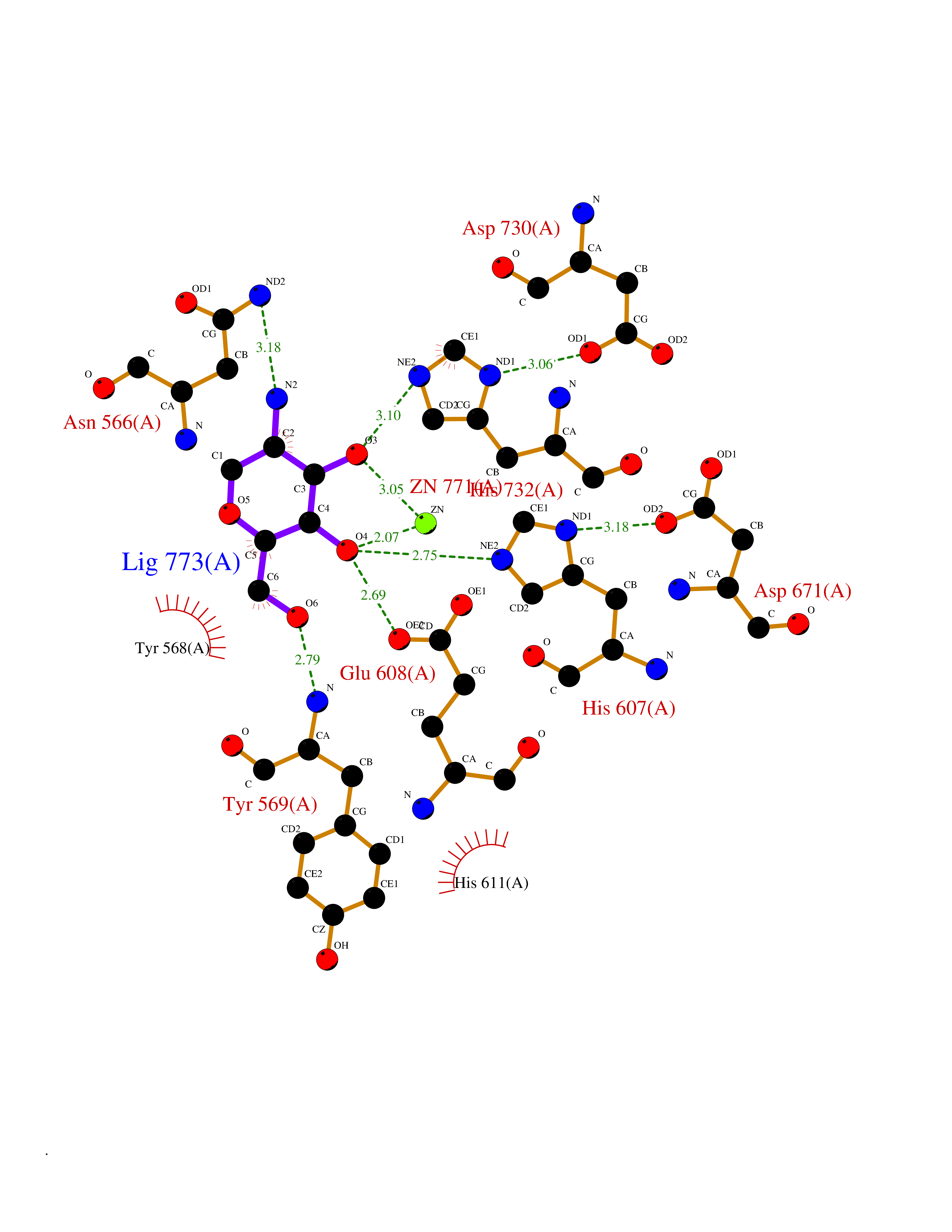 Ligplot Map 3D Binding mode Sequence SEACVSVTSSILSSMDPTVDPCHDFFSYACGGWIKANPVPDGHSRWGTFSNLWEHNQAIIKHLLENSTASVSEAERKAQVYYRACMNETRIEELRAKPLMELIERLGGWNITGPWAKDNFQDTLQVVTAHYRTSPFFSVYVSADSKNSNSNVIQVDQSGLGLPSRDYYLNKTENEKVLTGYLNYMVQLGKLLGGGDEEAIRPQMQQILDFETALANITIPQEKRRDEELIYHKVTAAELQTLAPAINWLPFLNTIFYPVEINESEPIVVYDKEYLEQISTLINTTDRCLLNNYMIWNLVRKTSSFLDQRFQDADEKFMEVMWKFCVSDTENNLGFALGPMFVKATFAEDSKSIATEIILEIKKAFEESLSTLKWMDEETRKSAKEKADAIYNMIGYPNFIMDPKELDKVFNDYTAVPDLYFENAMRFFNFSWRVTADQLRKAPNRDQWSMTPPMVNAYYSPTKNEIVFPAGILQAPFYTRSSPKALNFGGIGVVVGHELTHAFDDQGREYDKDGNLRPWWKNSSVEAFKRQTECMVEQYSNYSVNGEPVNGRHTLGENIADNGGLKAAYRAYQNWVKKNGAEHSLPTLGLTNNQLFFLGFAQVWCSVRTPESSHEGLITDPHSPSRFRVIGSLSNSKEFSEHFRCPPGSPMNPPHKCEVW Hydrogen bonds contact Hydrophobic contact | ||||
| 9 | Angiotensin-converting enzyme | 5AMC | 6.46 | |
Target general information Gen name ACE Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms DCP1;DCP Protein family Peptidase M2 family Biochemical class Hydrolase Function Actin binding.Bradykinin receptor binding.Carboxypeptidase activity.Chloride ion binding.Drug binding.Endopeptidase activity.Exopeptidase activity.Metallodipeptidase activity.Metallopeptidase activity.Mitogen-activated protein kinase binding.Mitogen-activated protein kinase kinase binding.Peptidyl-dipeptidase activity.Tripeptidyl-peptidase activity.Zinc ion binding. Related diseases Ischemic stroke (ISCHSTR) [MIM:601367]: A stroke is an acute neurologic event leading to death of neural tissue of the brain and resulting in loss of motor, sensory and/or cognitive function. Ischemic strokes, resulting from vascular occlusion, is considered to be a highly complex disease consisting of a group of heterogeneous disorders with multiple genetic and environmental risk factors. {ECO:0000269|PubMed:15534175}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Renal tubular dysgenesis (RTD) [MIM:267430]: Autosomal recessive severe disorder of renal tubular development characterized by persistent fetal anuria and perinatal death, probably due to pulmonary hypoplasia from early-onset oligohydramnios (the Potter phenotype). {ECO:0000269|PubMed:16116425}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Microvascular complications of diabetes 3 (MVCD3) [MIM:612624]: Pathological conditions that develop in numerous tissues and organs as a consequence of diabetes mellitus. They include diabetic retinopathy, diabetic nephropathy leading to end-stage renal disease, and diabetic neuropathy. Diabetic retinopathy remains the major cause of new-onset blindness among diabetic adults. It is characterized by vascular permeability and increased tissue ischemia and angiogenesis. {ECO:0000269|PubMed:10099885}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Intracerebral hemorrhage (ICH) [MIM:614519]: A pathological condition characterized by bleeding into one or both cerebral hemispheres including the basal ganglia and the cerebral cortex. It is often associated with hypertension and craniocerebral trauma. Intracerebral bleeding is a common cause of stroke. {ECO:0000269|PubMed:15277638}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00542; DB00616; DB01197; DB01340; DB15565; DB00584; DB09477; DB02032; DB00492; DB00722; DB00691; DB03740; DB00886; DB00790; DB00881; DB00178; DB01180; DB01348; DB08836; DB00519; DB13166 Interacts with P05556 EC number 3.4.15.1 Uniprot keywords 3D-structure; Alternative promoter usage; Alternative splicing; Calmodulin-binding; Carboxypeptidase; Cell membrane; Cytoplasm; Direct protein sequencing; Disulfide bond; Glycoprotein; Hydrolase; Membrane; Metal-binding; Metalloprotease; Phosphoprotein; Protease; Proteomics identification; Reference proteome; Repeat; Secreted; Signal; Transmembrane; Transmembrane helix; Zinc Protein physicochemical properties Chain ID A,B Molecular weight (Da) 70008.2 Length 607 Aromaticity 0.13 Instability index 46.01 Isoelectric point 5.71 Charge (pH=7) -13.56 2D Binding mode Binding energy (Kcal/mol) -6.29  Molscript Map 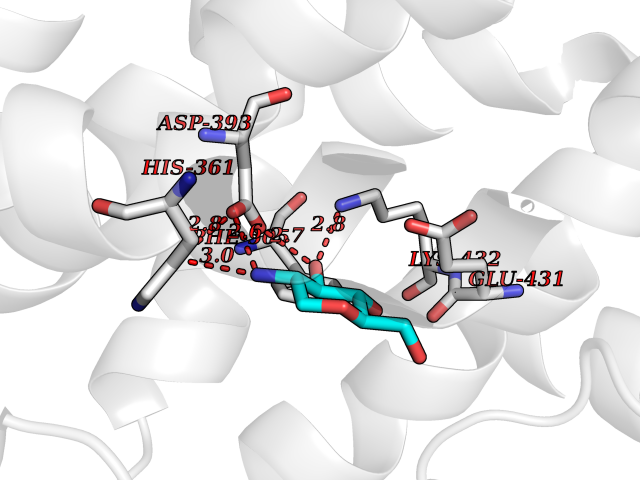 Pymol Map 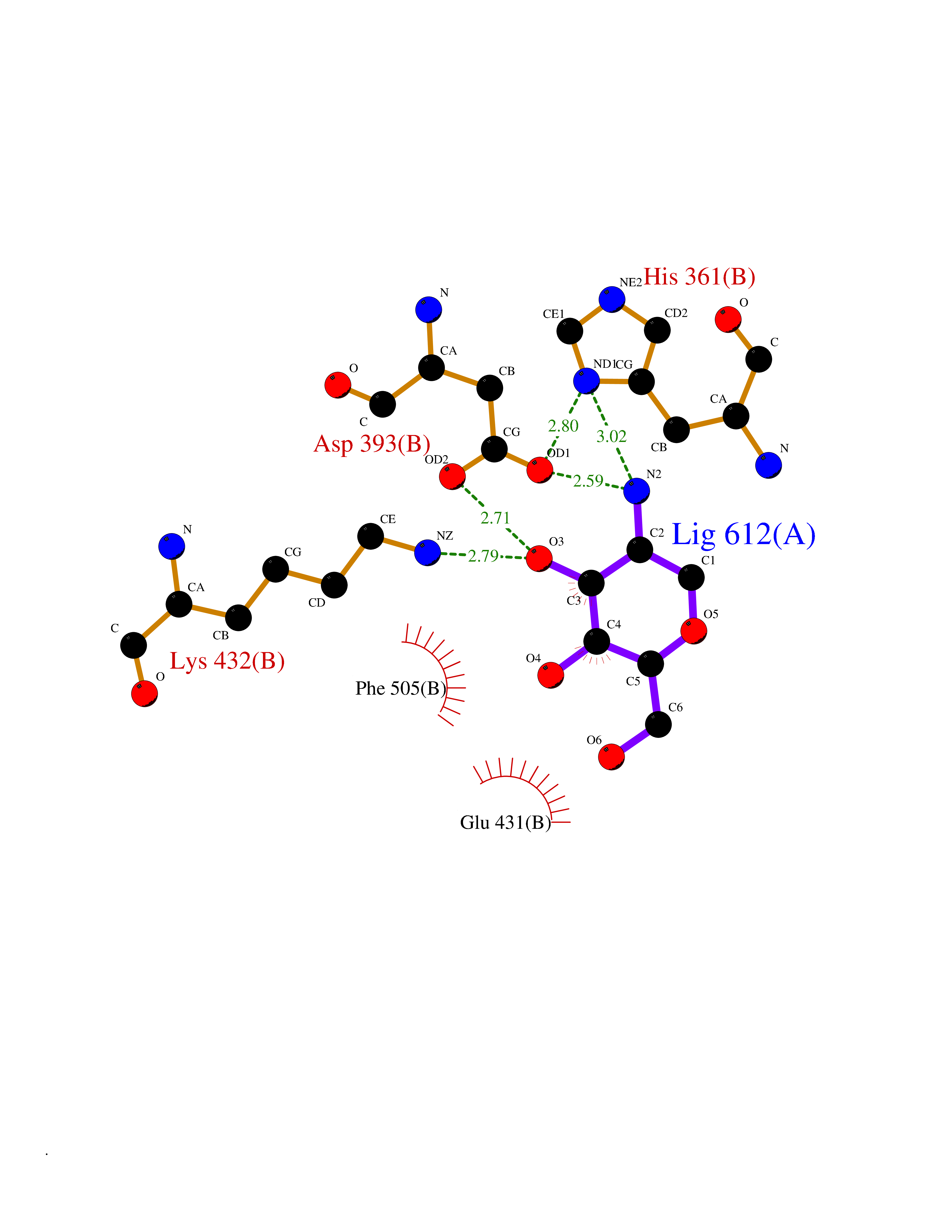 Ligplot Map 3D Binding mode Sequence LDPGLQPGQFSADEAGAQLFAQSYQSSAEQVLFQSVAASWAHDTNITAENARRQEEAALLSQEFAEAWGQKAKELYEPIWQQFTDPQLRRIIGAVRTLGSANLPLAKRQQYNALLSQMSRIYSTAKVCLTATCWSLDPDLTNILASSRSYAMLLFAWEGWHNAAGIPLKPLYEDFTALSNEAYKQDGFTDTGAYWRSWYNSPTFEDDLEHLYQQLEPLYLNLHAFVRRALHRRYGDRYINLRGPIPAHLLGDMWAQSWENIYDMVVPFPDKPNLDVTSTMLQQGWQATHMFRVAEEFFTSLELSPMPPEFWEGSMLEKPADGREVVCHASAWDFYNRKDFRIKQCTRVTMDQLSTVHHEMGHIQYYLQYKDLPVSLRRGANPGFHEAIGDVLALSVSTPEHLHKIGLLDRVTNDTESDINYLLKMALEKIAFLPFGYLVDQWRWGVFSGRTPPSRYNFDWWYLRTKYQGICPPVTRNETHFDAGAKFHVPNVTPYIRYFVSFVLQFQFHEALCKEAGYEGPLHQCDIYRSTKAGAKLRKVLRAGSSRPWQEVLKDMVGLDALDAQPLLKYFQLVTQWLQEQNQQNGEVLGWPEYQWHPPLPDNYPEG Hydrogen bonds contact Hydrophobic contact | ||||
| 10 | Oxygen-insensitive NADPH nitroreductase | 3QDL | 6.44 | |
Target general information Gen name rdxA Organism Helicobacter pylori (strain ATCC 700392 / 26695) (Campylobacter pylori) Uniprot ID TTD ID NA Synonyms HP_0954 Protein family Nitroreductase family Biochemical class Oxidoreductase Function Oxidoreductase activity. Related diseases Hypervalinemia and hyperleucine-isoleucinemia (HVLI) [MIM:618850]: An autosomal recessive metabolic disorder characterized by highly elevated plasma concentrations of valine and leucine/isoleucine. Affected individuals suffer from headache and mild memory impairment. {ECO:0000269|PubMed:25653144}. The disease is caused by variants affecting the gene represented in this entry. A patient with hypervalinemia and hyperleucine-isoleucinemia was identified as compound heterozygote for Gln-170 (inherited from his father) and Lys-264 (inherited from his mother), both variants reduced the catalytic activity of the enzyme. After treatment with vitamin B6, a precursor of pyridoxal 5'-phosphate, a BCAT2 cofactor, the blood levels of branched chain amino acids, especially valine, were decreased and brain lesions were improved. {ECO:0000269|PubMed:25653144}. Drugs (DrugBank ID) DB00916 Interacts with NA EC number 1.-.-.- Uniprot keywords 3D-structure; Antibiotic resistance; NADP; Oxidoreductase; Reference proteome Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 40094.3 Length 352 Aromaticity 0.08 Instability index 55.15 Isoelectric point 6.72 Charge (pH=7) -0.86 2D Binding mode Binding energy (Kcal/mol) -6.28 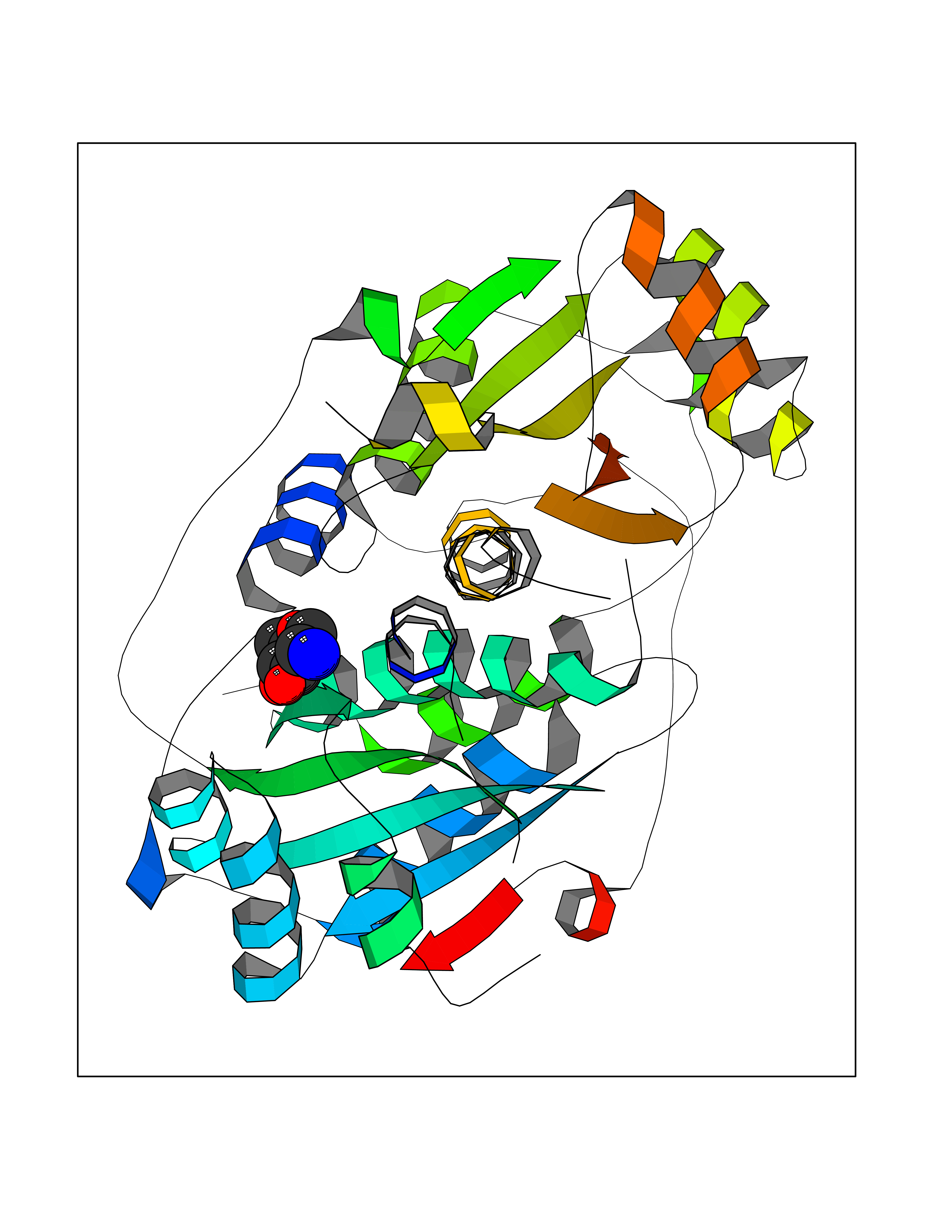 Molscript Map 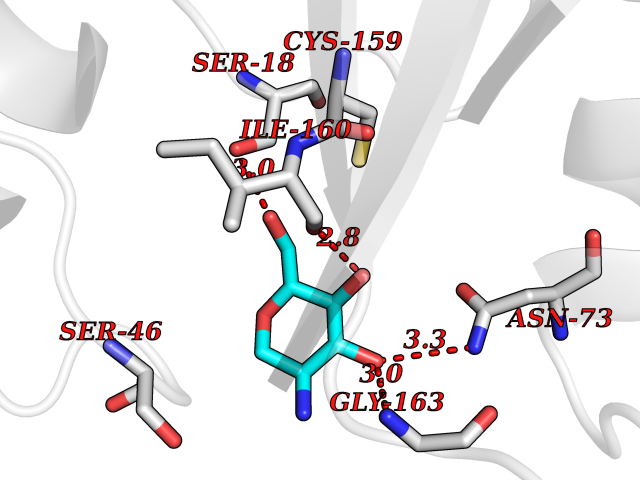 Pymol Map 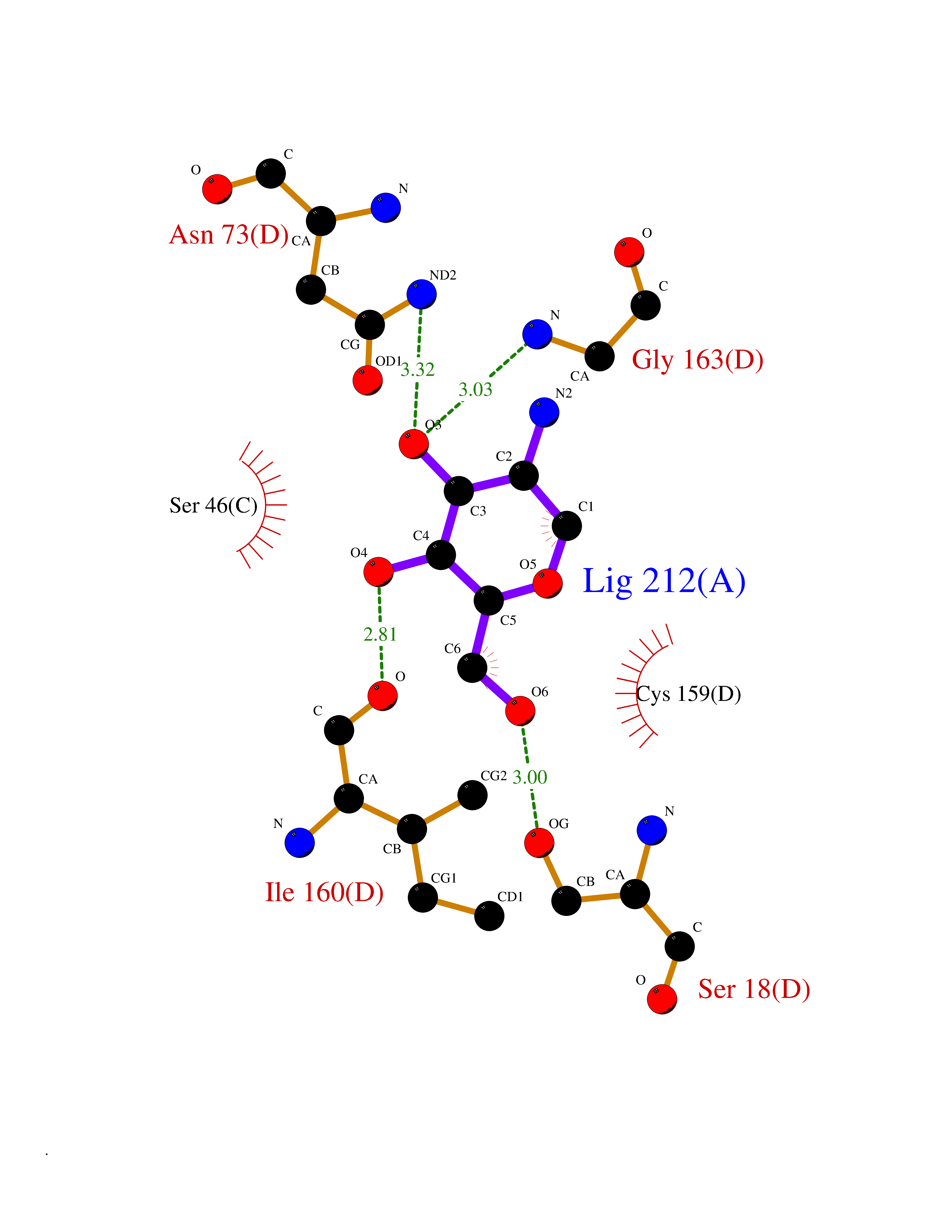 Ligplot Map 3D Binding mode Sequence MQRLESYILMKFLDQEKRRQLLNERHSCKMFDSHYEFSSTELEEIAEIARLSPSSYNTQPWHFVMVTDKDLKKQIAAHSYFNEEMIKSASALMVVCSLSYILEQCYIAVGQICMGVSLMGLDSCIIGGFDPLKVGEVLEERINPKIACLIALGKRVAEASQKSRKSKVDAITWLMKFLDQEKRRQLLNERHSCKMFDSHYEFSSTELEEIAEIARLSPSSYNTQPWHFVMVTDKDLKKQIAAHSYFNEEMIKSASALMVVCSLRPSELLPMQRLESYILEQCYIAVGQICMGVSLMGLDSCIIGGFDPLKVGEVLEERINKPKIACLIALGKRVAEASQKSRKSKVDAITWL Hydrogen bonds contact Hydrophobic contact | ||||
| 11 | Amylin receptor (IAPPR) | 6ZIS | 6.39 | |
Target general information Gen name CALCR-RAMP1/RAMP2/RAMP3 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Complex of Calcitonin receptor and Receptor activity-modifying protein Protein family RAMP family Biochemical class NA Function Transports the calcitonin gene-related peptide type 1 receptor (CALCRL) to the plasma membrane. Acts as a receptor for calcitonin-gene-related peptide (CGRP) together with CALCRL. Related diseases Immunodeficiency 9 (IMD9) [MIM:612782]: An immune disorder characterized by recurrent infections, impaired activation and proliferative response of T-cells, decreased T-cell production of cytokines, and normal lymphocytes counts and serum immunoglobulin levels. In surviving patients ectodermal dysplasia with anhidrosis and non-progressive myopathy may be observed. {ECO:0000269|PubMed:16147976, ECO:0000269|PubMed:16582901}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Myopathy, tubular aggregate, 2 (TAM2) [MIM:615883]: A rare congenital myopathy characterized by regular arrays of membrane tubules on muscle biopsies without additional histopathological hallmarks. Tubular aggregates in muscle are structures of variable appearance consisting of an outer tubule containing either one or more microtubule-like structures or amorphous material. TAM2 patients have myopathy and pupillary abnormalities. {ECO:0000269|PubMed:24591628, ECO:0000269|PubMed:28058752}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01278 Interacts with Q16602; P21145; Q5J8X5; Q16617 EC number NA Uniprot keywords 3D-structure; Cell membrane; Disulfide bond; Membrane; Proteomics identification; Receptor; Reference proteome; Signal; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A Molecular weight (Da) 63832.6 Length 569 Aromaticity 0.12 Instability index 22.42 Isoelectric point 5.07 Charge (pH=7) -16.96 2D Binding mode Binding energy (Kcal/mol) -6.43 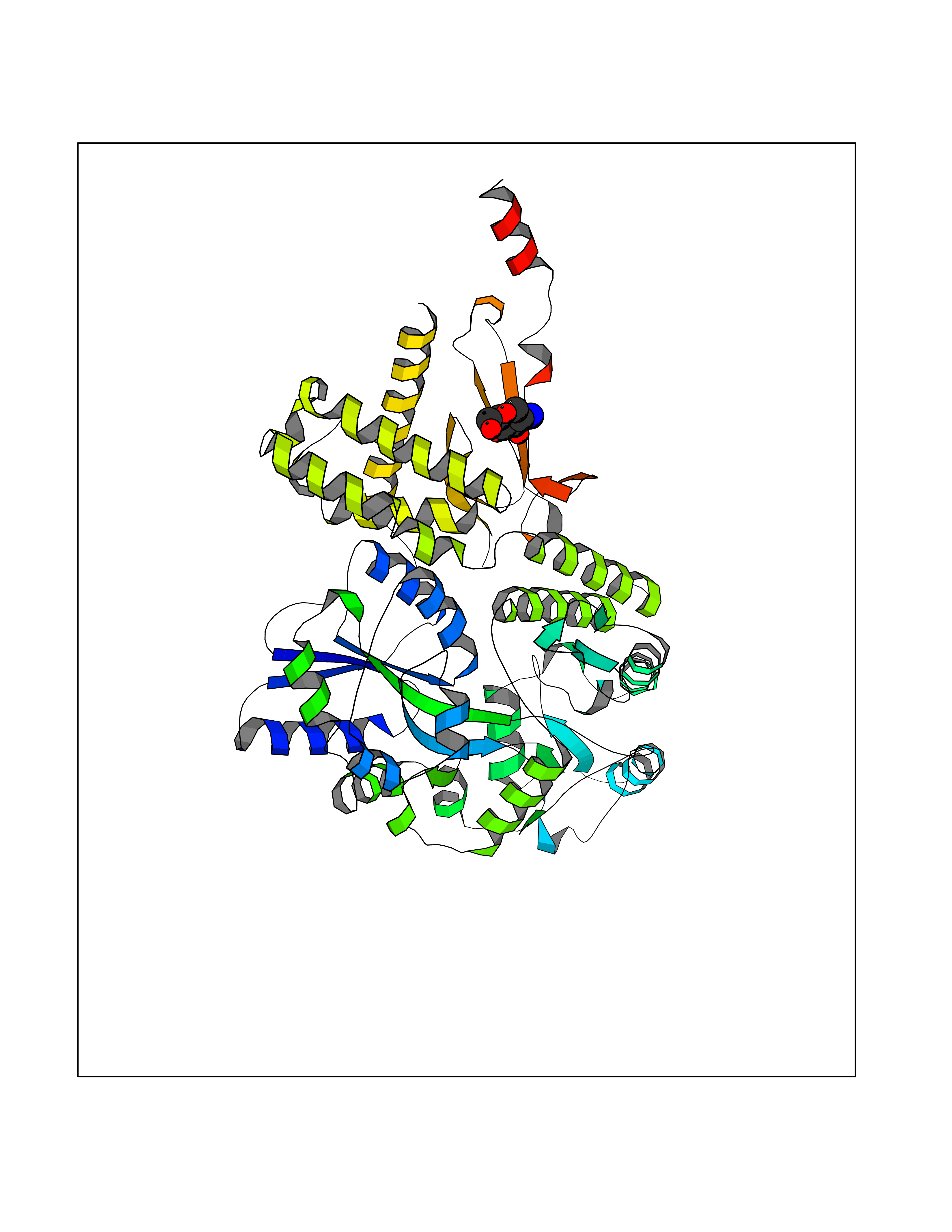 Molscript Map 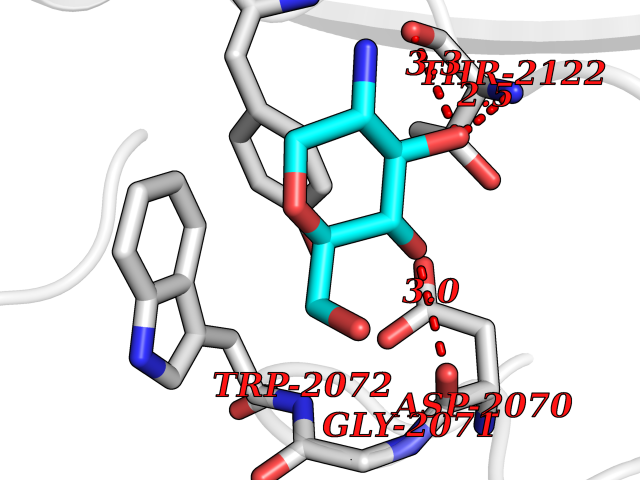 Pymol Map 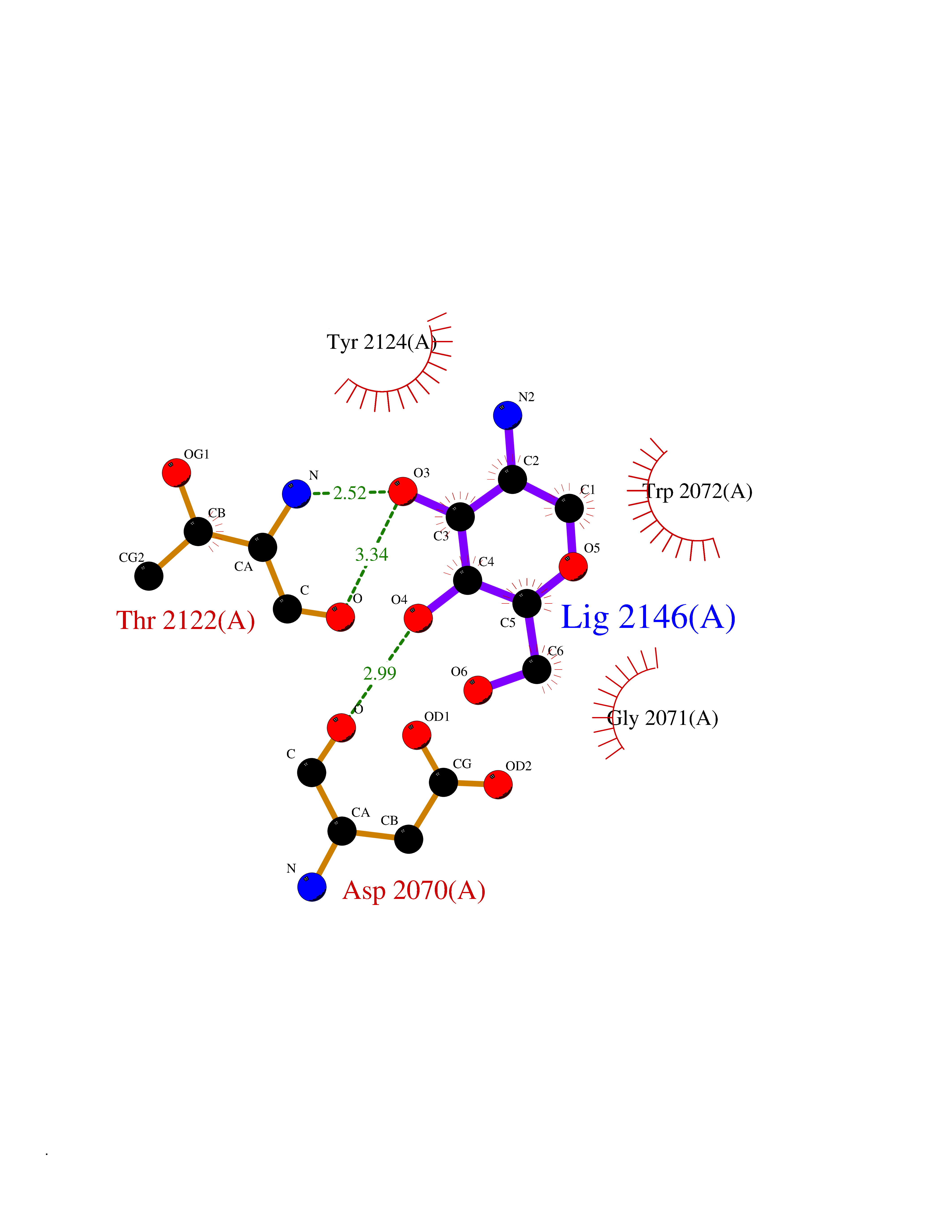 Ligplot Map 3D Binding mode Sequence SAKIEEGKLVIWINGDKGYNGLAEVGKKFEKDTGIKVTVEHPDKLEEKFPQVAATGDGPDIIFWAHDRFGGYAQSGLLAEITPDKAFQDKLYPFTWDAVRYNGKLIAYPIAVEALSLIYNKDLLPNPPKTWEEIPALDKELKAKGKSALMFNLQEPYFTWPLIAADGGYAFKYENGKYDIKDVGVDNAGAKAGLTFLVDLIKNKHMNADTDYSIAEAAFNKGETAMTINGPWAWSNIDTSKVNYGVTVLPTFKGQPSKPFVGVLSAGINAASPNKELAKEFLENYLLTDEGLEAVNKDKPLGAVALKSYEEELAKDPRIAATMENAQKGEIMPNIPQMSAFWYAVRTAVINAASGRQTVDEALKDAQTNAAAEFTTACQEANYGALLRELCLTQFQVDMEAVGETLWCDWGRTIRSYRELADCTWHMAEKLGCFWPNAEVDRFFLAVHGRYFRSCPISIQLGVTRNKIMTAQYECYQKIMQDPIQQGVYCQRTWDGWLCWNDVAAGTESMQLCPDYFQDFDPSEKVTKICDQDGNWFRHPASQRTWTDYTQCNVNTHEKVKTALNLFYL Hydrogen bonds contact Hydrophobic contact | ||||
| 12 | Pyruvate kinase PKLR | 4IP7 | 6.39 | |
Target general information Gen name PKLR Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms PKL;PK1 Protein family Pyruvate kinase family Biochemical class Transferase Function ATP binding.Kinase activity.Magnesium ion binding.Potassium ion binding.Pyruvate kinase activity. Related diseases Pyruvate kinase hyperactivity (PKHYP) [MIM:102900]: Autosomal dominant phenotype characterized by increase of red blood cell ATP. {ECO:0000269|PubMed:9090535}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Pyruvate kinase deficiency of red cells (PKRD) [MIM:266200]: A frequent cause of hereditary non-spherocytic hemolytic anemia. Clinically, pyruvate kinase-deficient patients suffer from a highly variable degree of chronic hemolysis, ranging from severe neonatal jaundice and fatal anemia at birth, severe transfusion-dependent chronic hemolysis, moderate hemolysis with exacerbation during infection, to a fully compensated hemolysis without apparent anemia. {ECO:0000269|PubMed:10087985, ECO:0000269|PubMed:10772876, ECO:0000269|PubMed:11328279, ECO:0000269|PubMed:11960989, ECO:0000269|PubMed:1536957, ECO:0000269|PubMed:1896471, ECO:0000269|PubMed:19085939, ECO:0000269|PubMed:2018831, ECO:0000269|PubMed:21794208, ECO:0000269|PubMed:7706479, ECO:0000269|PubMed:8161798, ECO:0000269|PubMed:8180378, ECO:0000269|PubMed:8476433, ECO:0000269|PubMed:8481523, ECO:0000269|PubMed:8483951, ECO:0000269|PubMed:8664896, ECO:0000269|PubMed:8807089, ECO:0000269|PubMed:9075576, ECO:0000269|PubMed:9482576, ECO:0000269|PubMed:9827908, ECO:0000269|PubMed:9886305, ECO:0000269|Ref.24}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02726; DB00787; DB04551; DB16236; DB00119 Interacts with Q9UBL6-2 EC number 2.7.1.40 Uniprot keywords 3D-structure; Allosteric enzyme; Alternative splicing; ATP-binding; Disease variant; Glycolysis; Hereditary hemolytic anemia; Kinase; Magnesium; Manganese; Metal-binding; Nucleotide-binding; Phosphoprotein; Potassium; Proteomics identification; Pyruvate; Reference proteome; Transferase Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 45695.1 Length 421 Aromaticity 0.06 Instability index 34.44 Isoelectric point 6.88 Charge (pH=7) -0.35 2D Binding mode Binding energy (Kcal/mol) -5.99 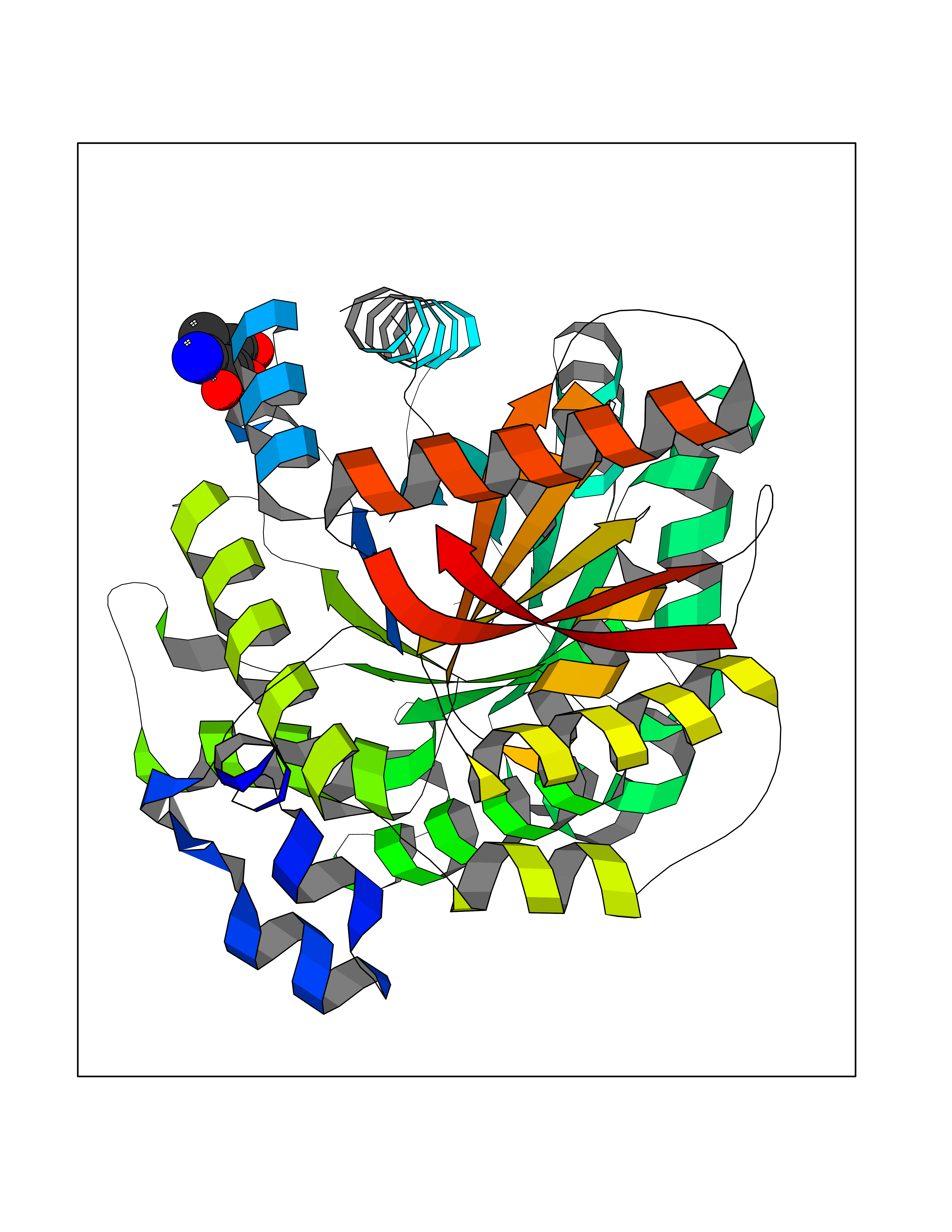 Molscript Map  Pymol Map 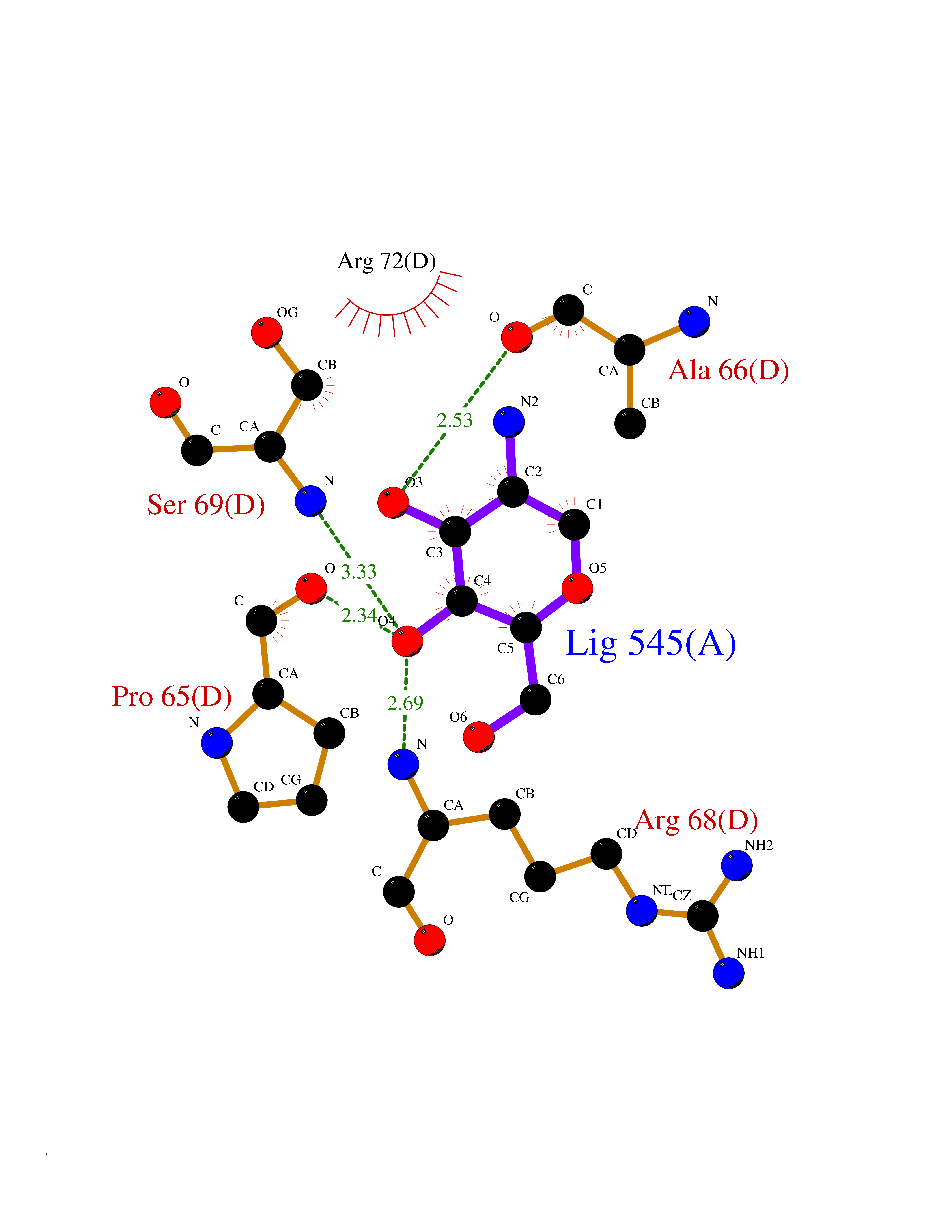 Ligplot Map 3D Binding mode Sequence GTAFFQQQQLPAAMADTFLEHLCLLDIDSEPVAARSTSIIATIGPASRSVERLKEMIKAGMNIARLNFSHGSHEYHAESIANVREAVESFSPLSYRPVAIALDTKGPEIGLSEQDVRDLRFGVEHGVDIVFASFVRKASDVAAVRAALGPEGHGIKIISKIENHEGVKRFDEILEVSDGIMVARGDLGIEIPAEKVFLAQKMMIGRCNLAGKPVVCATQMLESMITKPRPTRAETSDVANAVLDGADCIMLSGETAKGNFPVEAVKMQHAIAREAEAAVYHRQLFEELRRAAPLSRDPTEVTAIGAVEAAFKCCAAAIIVLTTTGRSAQLLSRYRPRAAVIAVTRSAQAARQVHLCRGVFPLLYREPPEAIWADDVDRRVQFGIESGKLRGFLRVGDLVIVVTGWRPGSGYTNIMRVLSIS Hydrogen bonds contact Hydrophobic contact | ||||
| 13 | Leukocyte immunoglobulin-like receptor B2 (LILRB2) | 6BCS | 6.33 | |
Target general information Gen name LILRB2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms LIR-2; Leukocyte immunoglobulin-like receptor 2; CD85 antigen-like family member D; Immunoglobulin-like transcript 4; ILT-4; Monocyte/macrophage immunoglobulin-like receptor 10; MIR-10; DE AltName: CD Protein family NA Biochemical class NA Function Receptor for class I MHC antigens. Recognizes a broad spectrum of HLA-A, HLA-B, HLA-C, HLA-G and HLA-F alleles. Involved in the down-regulation of the immune response and the development of tolerance. Recognizes HLA-G in complex with B2M/beta-2 microglobulin and a nonamer self-peptide (peptide-bound HLA-G-B2M) triggering differentiation of type 1 regulatory T cells and myeloid-derived suppressor cells, both of which actively maintain maternal-fetal tolerance. Competes with CD8A for binding to class I MHC antigens. Inhibits FCGR1A-mediated phosphorylation of cellular proteins and mobilization of intracellular calcium ions. Related diseases Cardiomyopathy, familial hypertrophic, 1 (CMH1) [MIM:192600]: A hereditary heart disorder characterized by ventricular hypertrophy, which is usually asymmetric and often involves the interventricular septum. The symptoms include dyspnea, syncope, collapse, palpitations, and chest pain. They can be readily provoked by exercise. The disorder has inter- and intrafamilial variability ranging from benign to malignant forms with high risk of cardiac failure and sudden cardiac death. {ECO:0000269|PubMed:10065021, ECO:0000269|PubMed:10329202, ECO:0000269|PubMed:10521296, ECO:0000269|PubMed:10563488, ECO:0000269|PubMed:10679957, ECO:0000269|PubMed:10862102, ECO:0000269|PubMed:11113006, ECO:0000269|PubMed:11133230, ECO:0000269|PubMed:11214007, ECO:0000269|PubMed:11424919, ECO:0000269|PubMed:11733062, ECO:0000269|PubMed:11861413, ECO:0000269|PubMed:11968089, ECO:0000269|PubMed:12081993, ECO:0000269|PubMed:12566107, ECO:0000269|PubMed:12590187, ECO:0000269|PubMed:12707239, ECO:0000269|PubMed:12818575, ECO:0000269|PubMed:12820698, ECO:0000269|PubMed:12951062, ECO:0000269|PubMed:12974739, ECO:0000269|PubMed:12975413, ECO:0000269|PubMed:1417858, ECO:0000269|PubMed:15358028, ECO:0000269|PubMed:15483641, ECO:0000269|PubMed:1552912, ECO:0000269|PubMed:15563892, ECO:0000269|PubMed:15856146, ECO:0000269|PubMed:15858117, ECO:0000269|PubMed:16199542, ECO:0000269|PubMed:16267253, ECO:0000269|PubMed:1638703, ECO:0000269|PubMed:16650083, ECO:0000269|PubMed:16938236, ECO:0000269|PubMed:17095604, ECO:0000269|PubMed:17372140, ECO:0000269|PubMed:18175163, ECO:0000269|PubMed:18403758, ECO:0000269|PubMed:1975517, ECO:0000269|PubMed:25182012, ECO:0000269|PubMed:7581410, ECO:0000269|PubMed:7731997, ECO:0000269|PubMed:7848441, ECO:0000269|PubMed:7874131, ECO:0000269|PubMed:7909436, ECO:0000269|PubMed:8250038, ECO:0000269|PubMed:8254035, ECO:0000269|PubMed:8268932, ECO:0000269|PubMed:8282798, ECO:0000269|PubMed:8343162, ECO:0000269|PubMed:8435239, ECO:0000269|PubMed:8483915, ECO:0000269|PubMed:8533830, ECO:0000269|PubMed:8655135, ECO:0000269|PubMed:8899546, ECO:0000269|PubMed:9544842, ECO:0000269|PubMed:9822100, ECO:0000269|PubMed:9829907}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Congenital myopathy 7A, myosin storage, autosomal dominant (CMYO7A) [MIM:608358]: A skeletal muscle disorder characterized by prominent axial and proximal weakening, spinal stiffness, severe scoliosis, with or without respiratory and cardiac involvement. The age at symptom onset can range from early childhood to late adulthood, and disease severity ranges from asymptomatic to severe muscular weakness and respiratory insufficiency. Histopathological examination shows variable findings including subsarcolemmal hyaline bodies in type 1 fibers. {ECO:0000269|PubMed:14520662, ECO:0000269|PubMed:15136674, ECO:0000269|PubMed:16684601, ECO:0000269|PubMed:17336526}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Cardiomyopathy, dilated, 1S (CMD1S) [MIM:613426]: A disorder characterized by ventricular dilation and impaired systolic function, resulting in congestive heart failure and arrhythmia. Patients are at risk of premature death. {ECO:0000269|PubMed:11106718, ECO:0000269|PubMed:12379228, ECO:0000269|PubMed:15769782, ECO:0000269|PubMed:18506004, ECO:0000269|PubMed:21127202, ECO:0000269|PubMed:21846512}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Myopathy, distal, 1 (MPD1) [MIM:160500]: A muscular disorder characterized by early-onset selective weakness of the great toe and ankle dorsiflexors, followed by weakness of the finger extensors. Mild proximal weakness occasionally develops years later after the onset of the disease. {ECO:0000269|PubMed:15322983, ECO:0000269|PubMed:17548557}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Congenital myopathy 7B, myosin storage, autosomal recessive (CMYO7B) [MIM:255160]: A skeletal muscle disorder characterized by the onset of scapuloperoneal muscle weakness in early childhood or young adulthood. Affected individuals have difficulty walking, steppage gait, and scapular winging due to shoulder girdle involvement. The severity and progression of the disorder is highly variable. Most patients develop respiratory insufficiency and restrictive lung disease. Some develop hypertrophic cardiomyopathy. Histopathological examination shows variable findings including subsarcolemmal hyaline bodies in type 1 fibers. {ECO:0000269|PubMed:25666907}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Left ventricular non-compaction 5 (LVNC5) [MIM:613426]: A form of left ventricular non-compaction, a cardiomyopathy due to myocardial morphogenesis arrest and characterized by a hypertrophic left ventricle, a severely thickened 2-layered myocardium, numerous prominent trabeculations, deep intertrabecular recesses, and poor systolic function. Clinical manifestations are variable. Some affected individuals experience no symptoms at all, others develop heart failure. In some cases, left ventricular non-compaction is associated with other congenital heart anomalies. LVNC5 is an autosomal dominant condition. {ECO:0000269|PubMed:18506004}. The disease is caused by variants affecting distinct genetic loci, including the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q9UKU9; Q86XS5; PRO_0000000092 [P05067]; P30447; Q95J06; P01889; P10321; P17693; P17693-2; P17693-5; P17693-6; P46531; P29350 EC number NA Uniprot keywords 3D-structure; Adaptive immunity; Alternative initiation; Alternative promoter usage; Alternative splicing; Cell membrane; Disulfide bond; Glycoprotein; Immunity; Immunoglobulin domain; Membrane; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Repeat; Signal; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 21853.4 Length 196 Aromaticity 0.1 Instability index 50.2 Isoelectric point 8.21 Charge (pH=7) 1.8 2D Binding mode Binding energy (Kcal/mol) -6.2 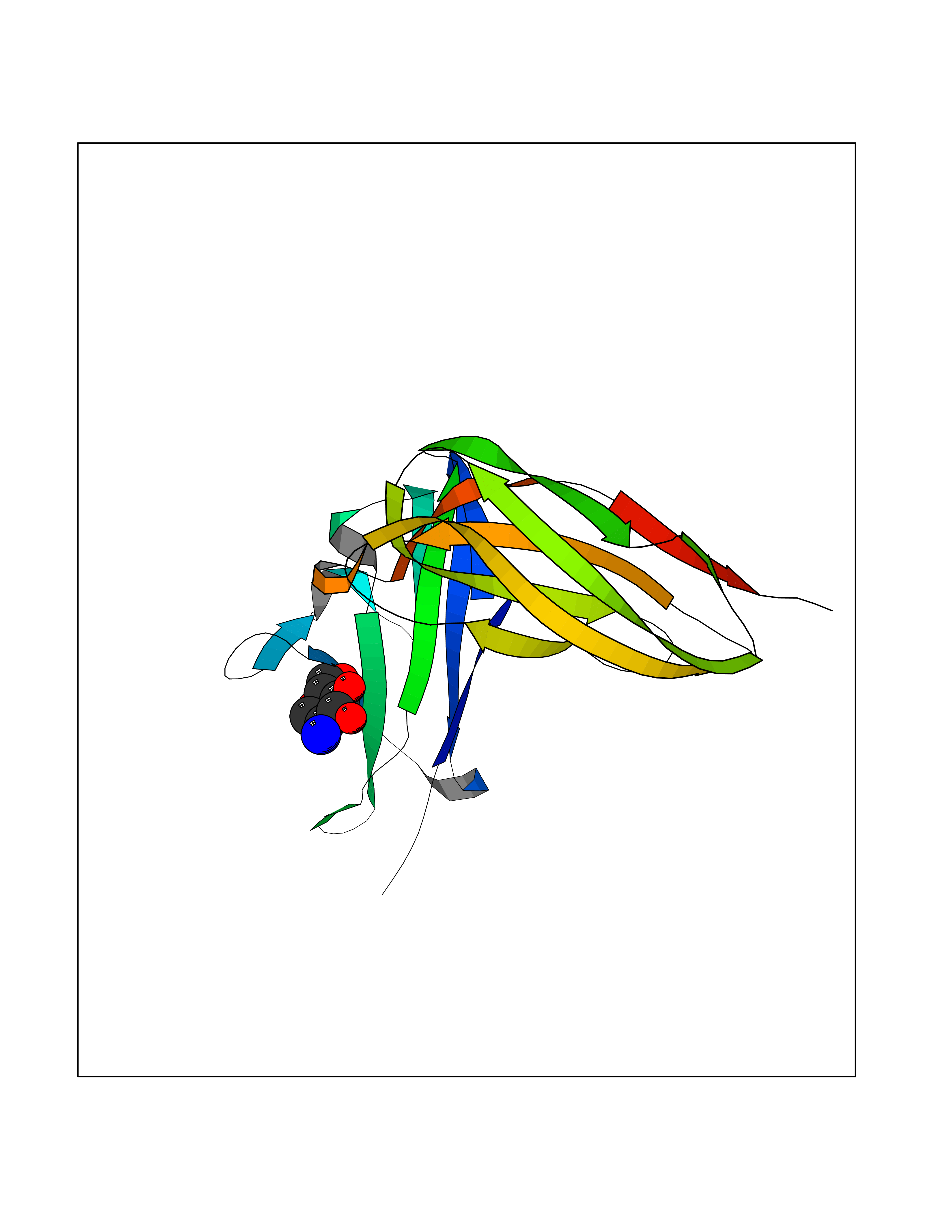 Molscript Map 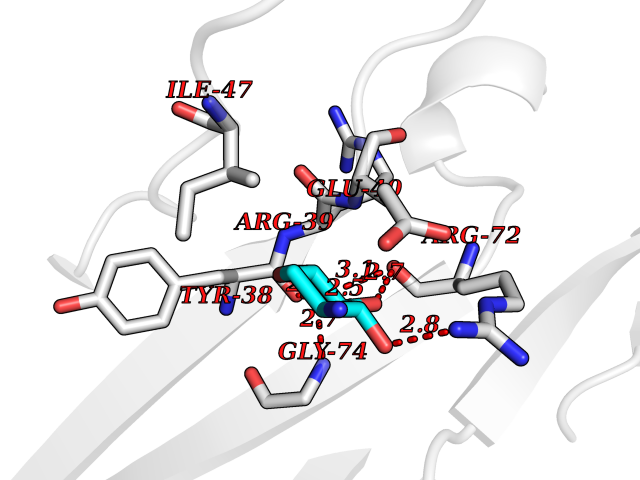 Pymol Map 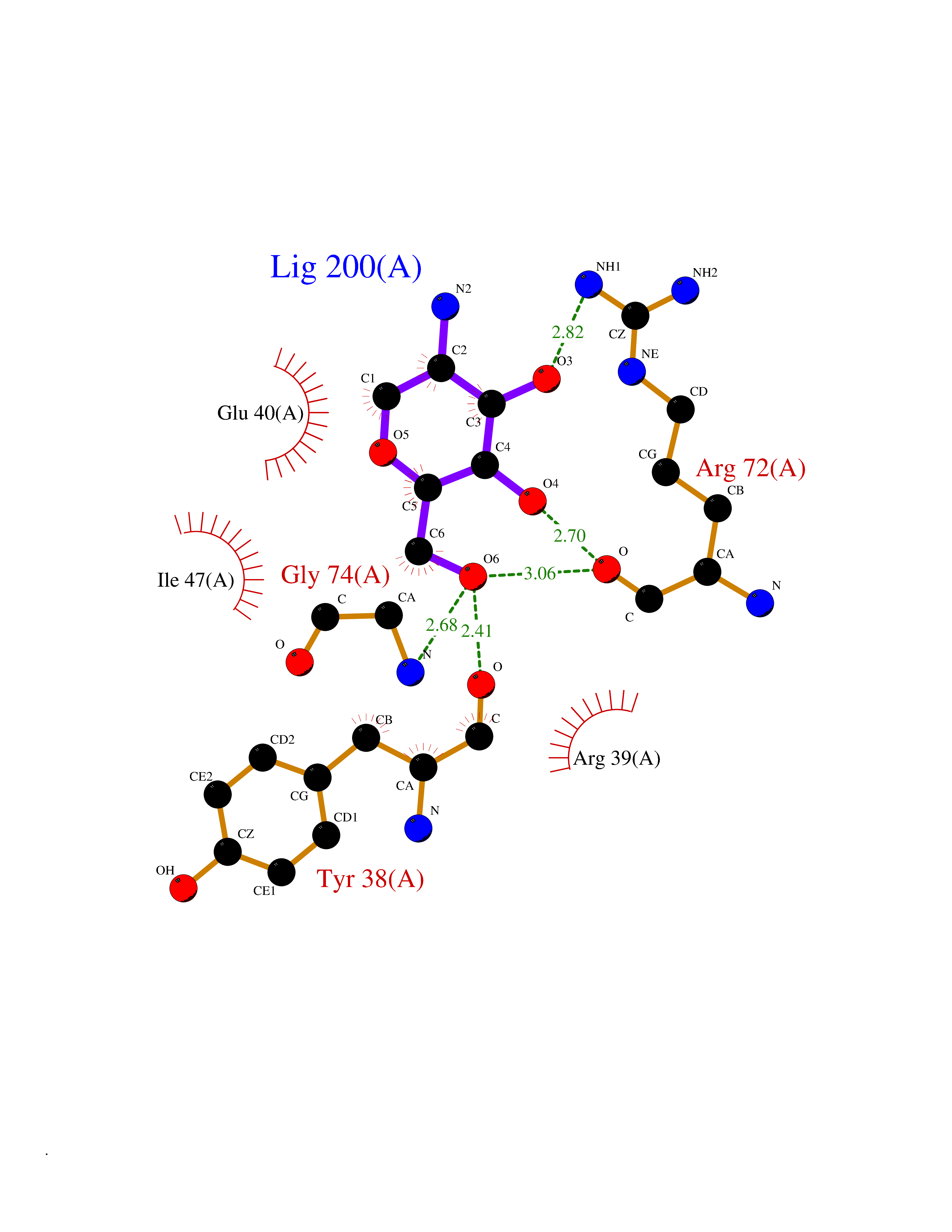 Ligplot Map 3D Binding mode Sequence TIPKPTLWAEPDSVITQGSPVTLSCQGSLEAQEYRLYREKKSASWITRIRPELVKNGQFHIPSITWEHTGRYGCQYYSRARWSELSDPLVLVMTGAYPKPTLSAQPSPVVTSGGRVTLQCESQVAFGGFILCKEGEDEHPQCLNSQPHARGSSRAIFSVGPVSPNRRWSHRCYGYDLNSPYVWSSPSDLLELLVPG Hydrogen bonds contact Hydrophobic contact | ||||
| 14 | Pyruvate kinase PKLR | 4IP7 | 6.31 | |
Target general information Gen name PKLR Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms PKL;PK1 Protein family Pyruvate kinase family Biochemical class Transferase Function ATP binding.Kinase activity.Magnesium ion binding.Potassium ion binding.Pyruvate kinase activity. Related diseases Pyruvate kinase hyperactivity (PKHYP) [MIM:102900]: Autosomal dominant phenotype characterized by increase of red blood cell ATP. {ECO:0000269|PubMed:9090535}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Pyruvate kinase deficiency of red cells (PKRD) [MIM:266200]: A frequent cause of hereditary non-spherocytic hemolytic anemia. Clinically, pyruvate kinase-deficient patients suffer from a highly variable degree of chronic hemolysis, ranging from severe neonatal jaundice and fatal anemia at birth, severe transfusion-dependent chronic hemolysis, moderate hemolysis with exacerbation during infection, to a fully compensated hemolysis without apparent anemia. {ECO:0000269|PubMed:10087985, ECO:0000269|PubMed:10772876, ECO:0000269|PubMed:11328279, ECO:0000269|PubMed:11960989, ECO:0000269|PubMed:1536957, ECO:0000269|PubMed:1896471, ECO:0000269|PubMed:19085939, ECO:0000269|PubMed:2018831, ECO:0000269|PubMed:21794208, ECO:0000269|PubMed:7706479, ECO:0000269|PubMed:8161798, ECO:0000269|PubMed:8180378, ECO:0000269|PubMed:8476433, ECO:0000269|PubMed:8481523, ECO:0000269|PubMed:8483951, ECO:0000269|PubMed:8664896, ECO:0000269|PubMed:8807089, ECO:0000269|PubMed:9075576, ECO:0000269|PubMed:9482576, ECO:0000269|PubMed:9827908, ECO:0000269|PubMed:9886305, ECO:0000269|Ref.24}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02726; DB00787; DB04551; DB16236; DB00119 Interacts with Q9UBL6-2 EC number 2.7.1.40 Uniprot keywords 3D-structure; Allosteric enzyme; Alternative splicing; ATP-binding; Disease variant; Glycolysis; Hereditary hemolytic anemia; Kinase; Magnesium; Manganese; Metal-binding; Nucleotide-binding; Phosphoprotein; Potassium; Proteomics identification; Pyruvate; Reference proteome; Transferase Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 45695.1 Length 421 Aromaticity 0.06 Instability index 34.44 Isoelectric point 6.88 Charge (pH=7) -0.35 2D Binding mode Binding energy (Kcal/mol) -6.66 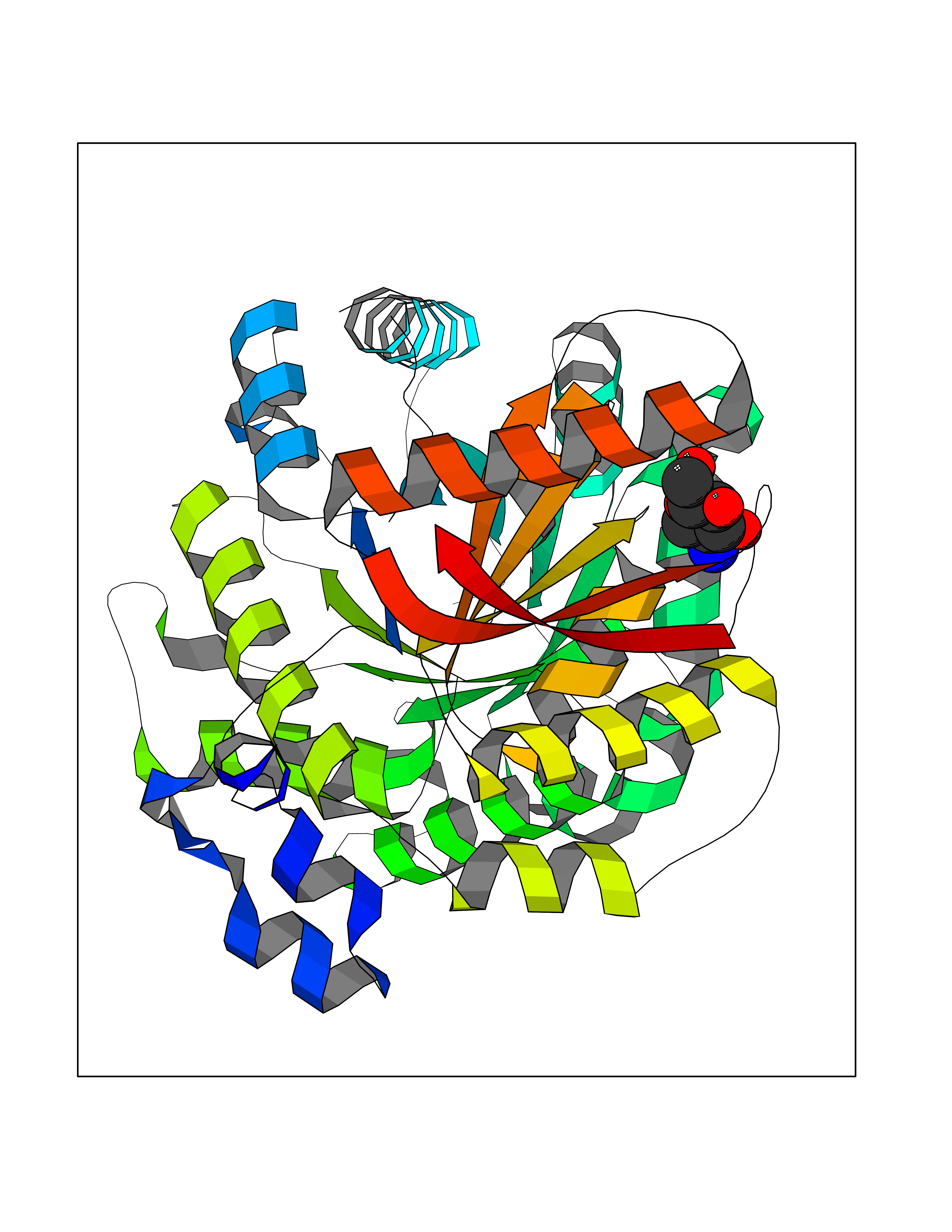 Molscript Map 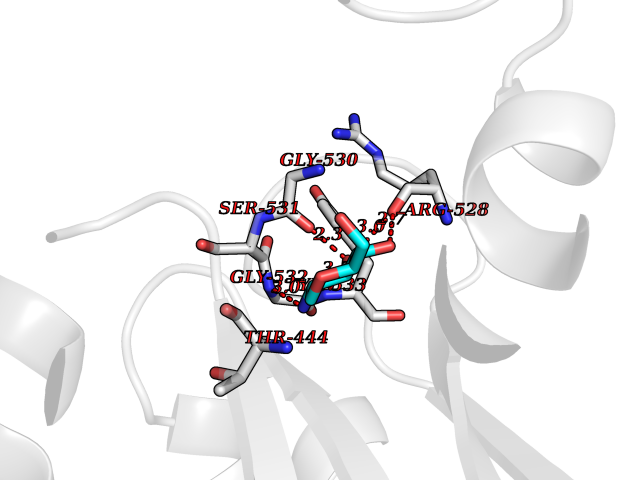 Pymol Map 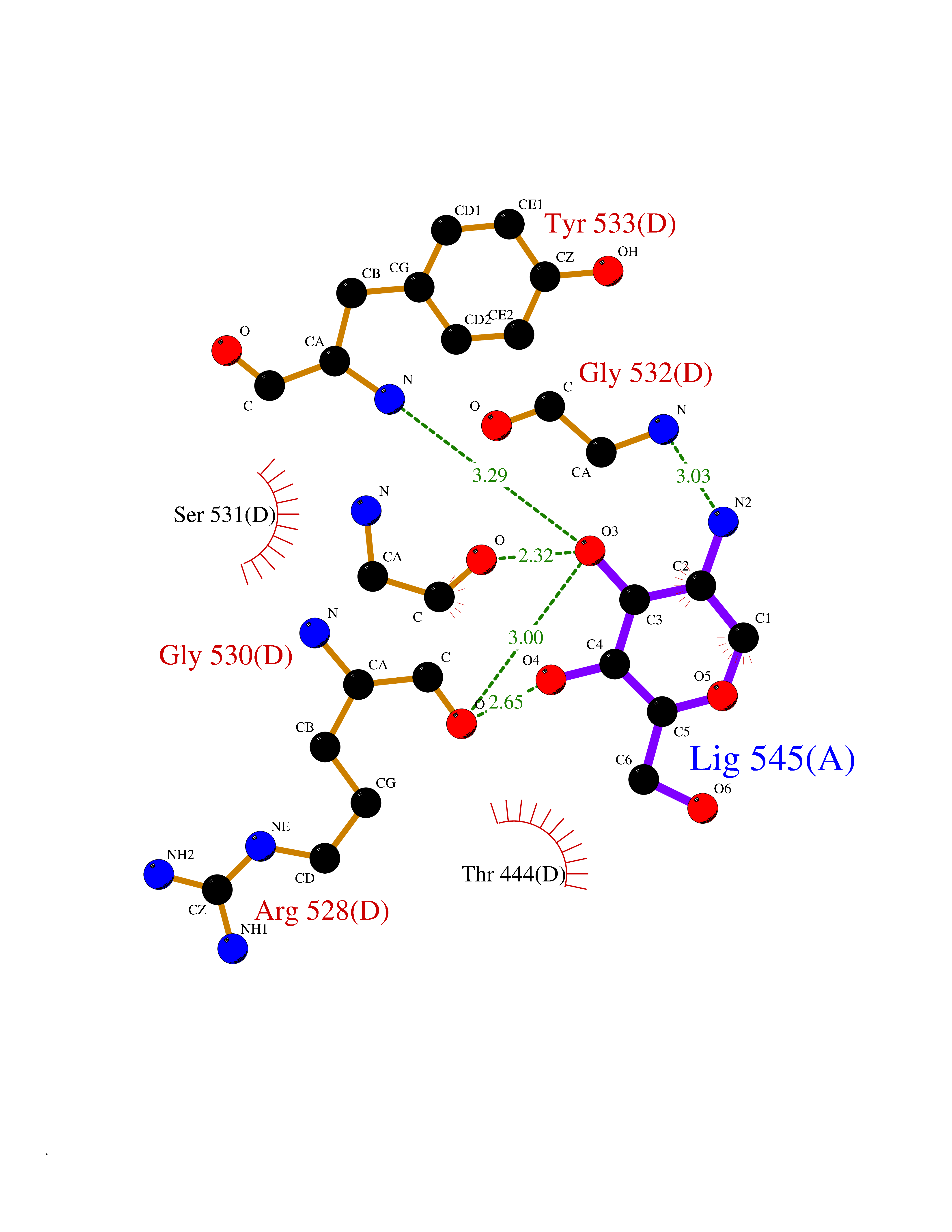 Ligplot Map 3D Binding mode Sequence GTAFFQQQQLPAAMADTFLEHLCLLDIDSEPVAARSTSIIATIGPASRSVERLKEMIKAGMNIARLNFSHGSHEYHAESIANVREAVESFSPLSYRPVAIALDTKGPEIGLSEQDVRDLRFGVEHGVDIVFASFVRKASDVAAVRAALGPEGHGIKIISKIENHEGVKRFDEILEVSDGIMVARGDLGIEIPAEKVFLAQKMMIGRCNLAGKPVVCATQMLESMITKPRPTRAETSDVANAVLDGADCIMLSGETAKGNFPVEAVKMQHAIAREAEAAVYHRQLFEELRRAAPLSRDPTEVTAIGAVEAAFKCCAAAIIVLTTTGRSAQLLSRYRPRAAVIAVTRSAQAARQVHLCRGVFPLLYREPPEAIWADDVDRRVQFGIESGKLRGFLRVGDLVIVVTGWRPGSGYTNIMRVLSIS Hydrogen bonds contact Hydrophobic contact | ||||
| 15 | Purine nucleoside phosphorylase (PNP) | 4EAR | 6.30 | |
Target general information Gen name PNP Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms PNP; Inosine phosphorylase Protein family PNP/MTAP phosphorylase family Biochemical class Pentosyltransferase Function The purine nucleoside phosphorylases catalyze the phosphorolytic breakdown of the N-glycosidic bond in the beta- (deoxy)ribonucleoside molecules, with the formation of the corresponding free purine bases and pentose-1-phosphate. Related diseases Purine nucleoside phosphorylase deficiency (PNPD) [MIM:613179]: A disorder that interrupts both the catabolism of inosine into hypoxanthine and guanosine into guanine, and leads to the accumulation of guanosine, inosine, and their deoxified by-products. The main clinical presentation is recurrent infections due to severe T-cell immunodeficiency. Some patients also have neurologic impairment. {ECO:0000269|PubMed:1384322, ECO:0000269|PubMed:3029074, ECO:0000269|PubMed:8931706}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03881; DB03551; DB02222; DB02391; DB03609; DB01667; DB04260; DB02796; DB04753; DB00640; DB00242; DB00900; DB06185; DB02377; DB02857; DB04754; DB04757; DB04076; DB02230; DB04335; DB02568; DB03101 Interacts with P05067; Q9UQM7; O14576-2; P06241; P14136; Q92993-2; Q9BXM7; P00491; P17612; P63000; Q92673; Q15583 EC number EC 2.4.2.1 Uniprot keywords 3D-structure; Acetylation; Cytoplasm; Direct protein sequencing; Disease variant; Glycosyltransferase; Phosphoprotein; Proteomics identification; Purine salvage; Reference proteome; Transferase Protein physicochemical properties Chain ID A,B,C Molecular weight (Da) 31849.2 Length 288 Aromaticity 0.1 Instability index 34.77 Isoelectric point 6.42 Charge (pH=7) -1.63 2D Binding mode Binding energy (Kcal/mol) -6.86 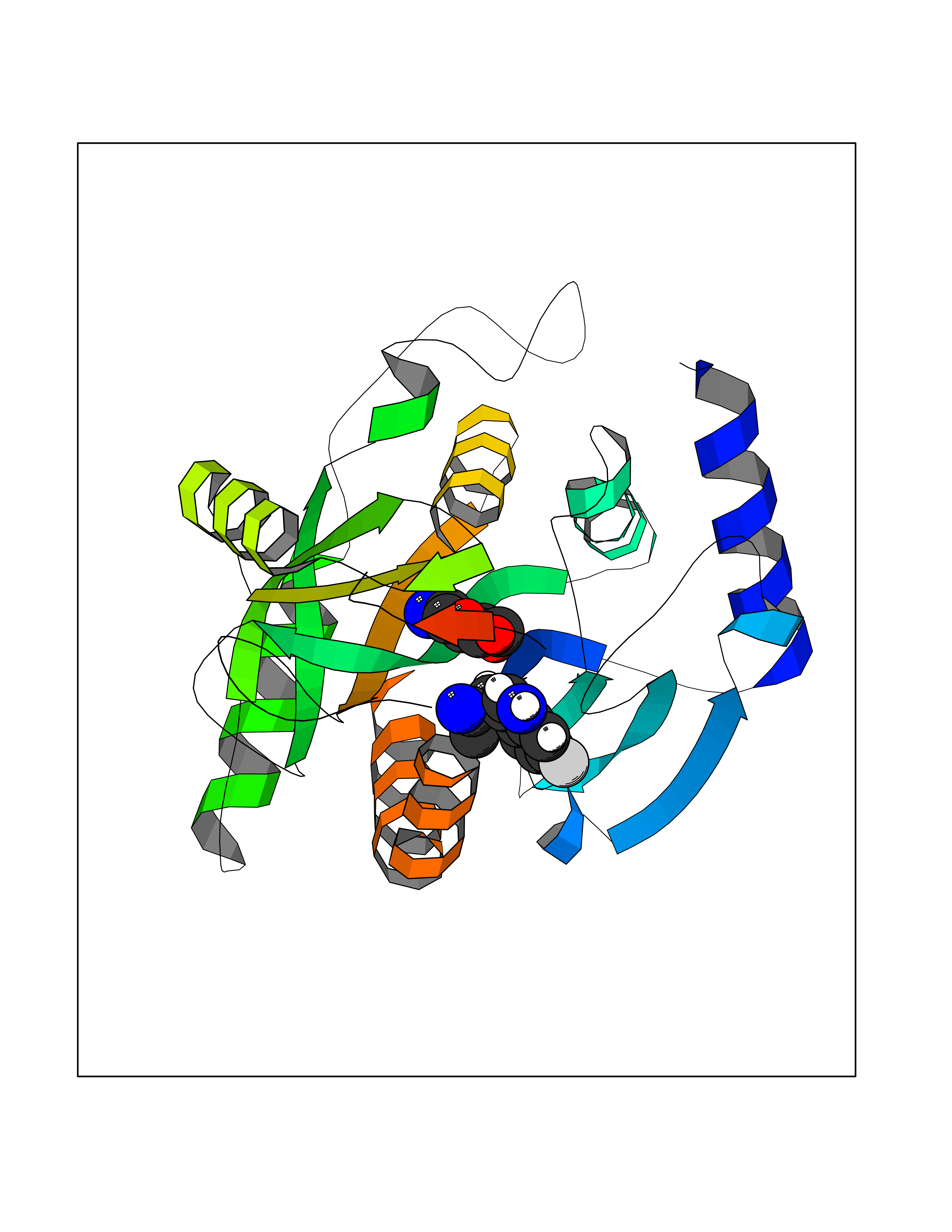 Molscript Map  Pymol Map 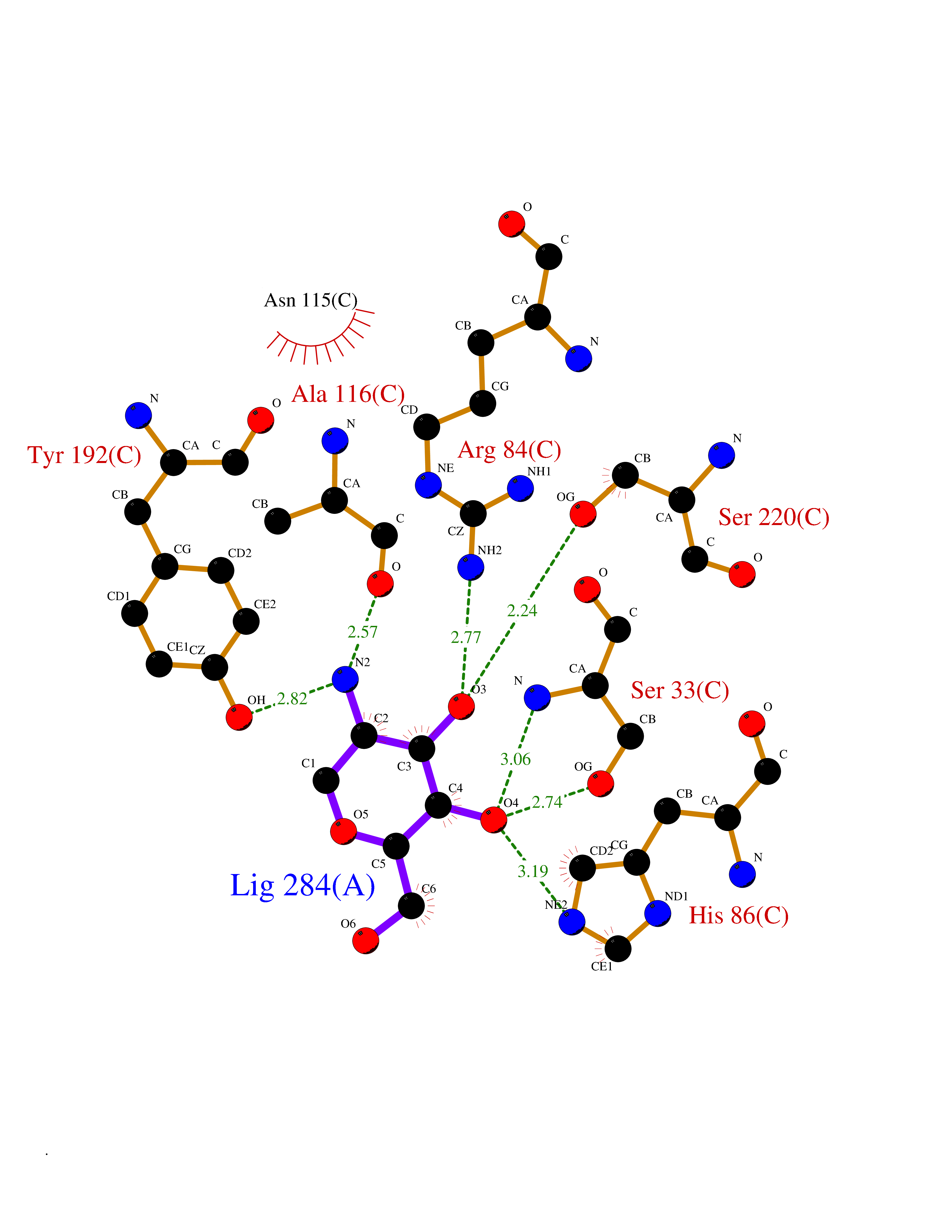 Ligplot Map 3D Binding mode Sequence GYTYEDYKNTAEYLLSHTKHRPQVAIICGSGLGGLTDKLTQAQIFDYSEIPNFPRSTVPGHAGRLVFGFLNGRACVMMQGRFHMYEGYPLYKVTFPVRVFHLLGVDTLVVTNAAGGLNPKFEVGDIMLIRDHINLPGFSGQNPLRGPNDERFGDRFPAMSDAYDRTMRQRALSTYKQMGEQRELQEGTYVMVAGPSFETVAECRVLQKLGADAVGMSTVPEVIVARHCGLRVFGFSLITNKVIMDYESLEKANXEEVLAAGKQAAQKLEQFVSILMASIDRFPAMSDA Hydrogen bonds contact Hydrophobic contact | ||||
| 16 | Human immunodeficiency virus Reverse transcriptase (HIV RT) | 1RTD | 6.29 | |
Target general information Gen name HIV RT Organism Human immunodeficiency virus type 1 group M subtype B (isolate BH10) (HIV-1) Uniprot ID TTD ID Synonyms HIV p66 RT; HIV Exoribonuclease H Protein family NA Biochemical class NA Function Gag-Pol polyprotein: Mediates, with Gag polyprotein, the essential events in virion assembly, including binding the plasma membrane, making the protein-protein interactions necessary to create spherical particles, recruiting the viral Env proteins, and packaging the genomic RNA via direct interactions with the RNA packaging sequence (Psi). Gag-Pol polyprotein may regulate its own translation, by the binding genomic RNA in the 5'-UTR. At low concentration, the polyprotein would promote translation, whereas at high concentration, the polyprotein would encapsidate genomic RNA and then shut off translation. Related diseases Sitosterolemia 2 (STSL2) [MIM:618666]: A form of sitosterolemia, an autosomal recessive metabolic disorder characterized by unregulated intestinal absorption of cholesterol, phytosterols and shellfish sterols, and decreased biliary excretion of dietary sterols into bile. Patients have hypercholesterolemia, very high levels of plant sterols in the plasma, and frequently develop tendon and tuberous xanthomas, accelerated atherosclerosis and premature coronary artery disease. {ECO:0000269|PubMed:11138003, ECO:0000269|PubMed:11452359, ECO:0000269|PubMed:11668628, ECO:0000269|PubMed:15054092, ECO:0000269|PubMed:35557526}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07035; DB02704; DB07806; DB02785; DB01824; DB01732; DB06874; DB08034; DB07961; DB07451; DB08212; DB08372; DB02972; DB04190; DB04042; DB08428; DB03076; DB03141; DB08457; DB07343; DB07337; DB07018; DB07332; DB05398; DB07578; DB08639; DB06414; DB04255; DB04547; DB02683; DB02009; DB03908; DB02629; DB01887; DB03803; DB02033; DB08281; DB08282; DB08284; DB08414; DB08598; DB07327; DB07885; DB02768; DB08600; DB01891; DB05871 Interacts with NA EC number EC 2.7.7.49 Uniprot keywords 3D-structure; Activation of host caspases by virus; AIDS; Aspartyl protease; Capsid protein; Direct protein sequencing; DNA integration; DNA recombination; DNA-binding; DNA-directed DNA polymerase; Endonuclease; Eukaryotic host gene expression shutoff by virus; Eukaryotic host translation shutoff by virus; Host cell membrane; Host cytoplasm; Host endosome; Host gene expression shutoff by virus; Host membrane; Host nucleus; Host-virus interaction; Hydrolase; Lipid-binding; Lipoprotein; Magnesium; Membrane; Metal-binding; Methylation; Modulation of host cell apoptosis by virus; Multifunctional enzyme; Myristate; Nuclease; Nucleotidyltransferase; Phosphoprotein; Protease; Repeat; Ribosomal frameshifting; RNA-binding; RNA-directed DNA polymerase; Transferase; Viral genome integration; Viral nucleoprotein; Viral penetration into host nucleus; Viral release from host cell; Virion; Virion maturation; Virus entry into host cell; Zinc; Zinc-finger Protein physicochemical properties Chain ID F,A,E Molecular weight (Da) 67553.9 Length 600 Aromaticity 0.09 Instability index 33.46 Isoelectric point 7.97 Charge (pH=7) 3.35 2D Binding mode Binding energy (Kcal/mol) -6.47  Molscript Map 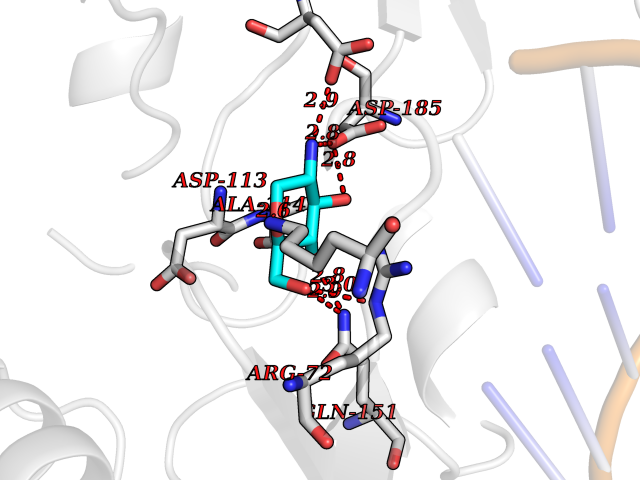 Pymol Map 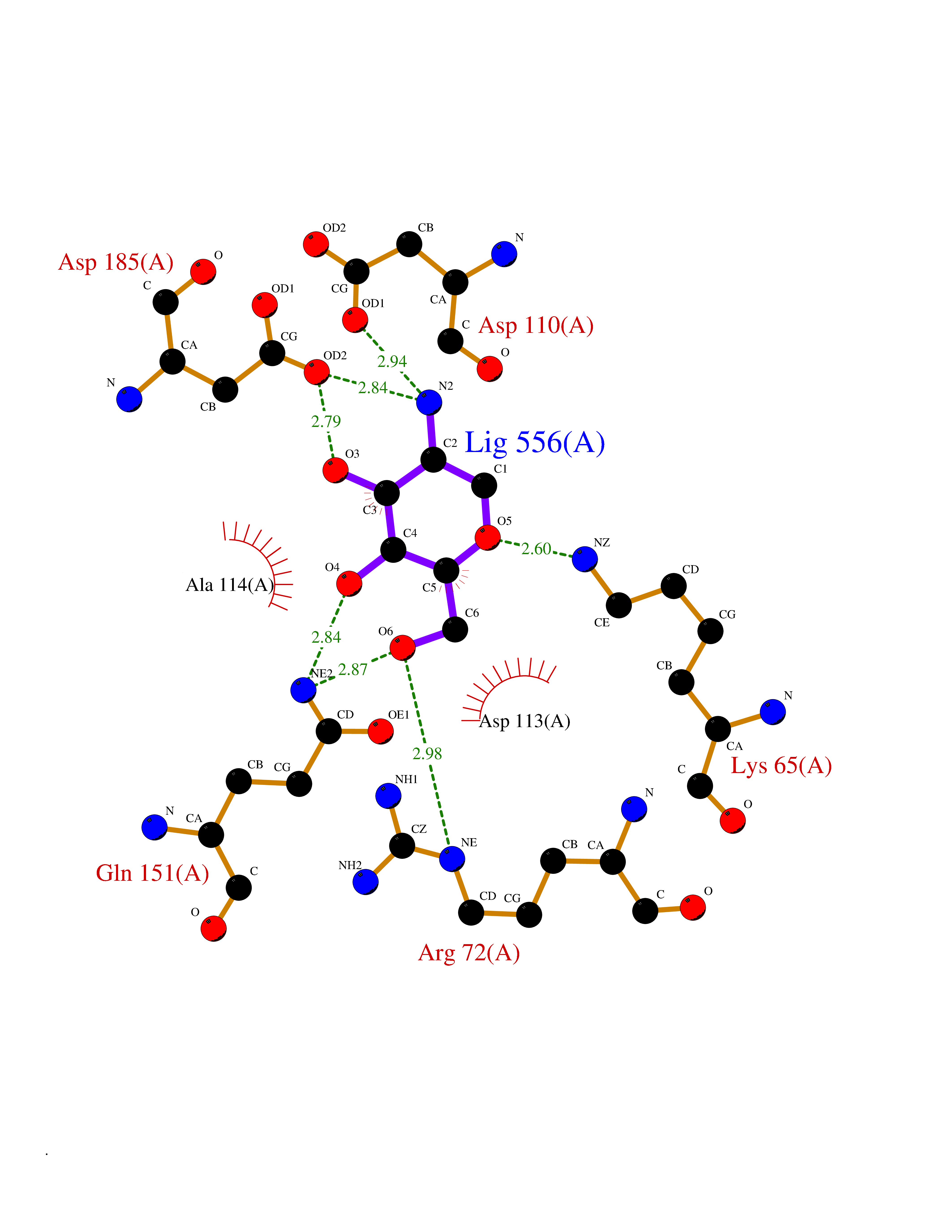 Ligplot Map 3D Binding mode Sequence GCACCGGCGCTCGAACAGGGACTGTCAGTCCCTGTTCGAGCGCCGGKISPIETVPVKLKPGMDGPKVKQWPLTEEKIKALVEICTEMEKEGKISKIGPENPYNTPVFAIKKKDSTKWRKLVDFRELNKRTQDFWEVQLGIPHPAGLKKKKSVTVLDVGDAYFSVPLDEDFRKYTAFTIPSINNETPGIRYQYNVLPQGWKGSPAIFQSSMTKILEPFRKQNPDIVIYQYMDDLYVGSDLEIGQHRTKIEELRQHLLRWGLTTPDKKHQKEPPFLWMGYELHPDKWTVQPIVLPEKDSWTVNDICKLVGKLNWASQIYPGIKVRQLCKLLRGTKALTEVIPLTEEAELELAENREILKEPVHGVYYDPSKDLIAEIQKQGQGQWTYQIYQEPFKNLKTGKYARMRGAHTNDVKQLTEAVQKITTESIVIWGKTPKFKLPIQKETWETWWTEYWQATWIPEWEFVNTPPLVKLWYQLEKEPIVGAETFYVDGAANRETKLGKAGYVTNKGRQKVVPLTDTTNQKTQLQAIYLALQDSGLEVNIVTDSQYALGIIQAQPDESESELVNQIIEQLIKKEKVYLAWVPAHKGIGGNEQVDKLVSA Hydrogen bonds contact Hydrophobic contact | ||||
| 17 | Vasopressin V1a receptor | 1YTV | 6.25 | |
Target general information Gen name AVPR1A Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms AVPR1 Protein family G-protein coupled receptor 1 family, Vasopressin/oxytocin receptor subfamily Biochemical class Sugar binding protein Function Peptide binding.Peptide hormone binding.Protein kinase C binding.V1A vasopressin receptor binding.Vasopressin receptor activity. Related diseases Defects in PPARG can lead to type 2 insulin-resistant diabetes and hyptertension. PPARG mutations may be associated with colon cancer. {ECO:0000269|PubMed:10394368}.; DISEASE: Obesity (OBESITY) [MIM:601665]: A condition characterized by an increase of body weight beyond the limitation of skeletal and physical requirements, as the result of excessive accumulation of body fat. {ECO:0000269|PubMed:9753710}. Disease susceptibility may be associated with variants affecting the gene represented in this entry.; DISEASE: Lipodystrophy, familial partial, 3 (FPLD3) [MIM:604367]: A form of lipodystrophy characterized by marked loss of subcutaneous fat from the extremities. Facial adipose tissue may be increased, decreased or normal. Affected individuals show an increased preponderance of insulin resistance, diabetes mellitus and dyslipidemia. {ECO:0000269|PubMed:11788685, ECO:0000269|PubMed:12453919}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Glioma 1 (GLM1) [MIM:137800]: Gliomas are benign or malignant central nervous system neoplasms derived from glial cells. They comprise astrocytomas and glioblastoma multiforme that are derived from astrocytes, oligodendrogliomas derived from oligodendrocytes and ependymomas derived from ependymocytes. {ECO:0000269|PubMed:10851250}. Disease susceptibility may be associated with variants affecting the gene represented in this entry. Polymorphic PPARG alleles have been found to be significantly over-represented among a cohort of American patients with sporadic glioblastoma multiforme suggesting a possible contribution to disease susceptibility. Drugs (DrugBank ID) DB09059; DB00872; DB00035; DB00093; DB14642; DB16279; DB05452; DB13929; DB02638; DB06212; DB00067 Interacts with P25106 EC number NA Uniprot keywords 3D-structure; Cell membrane; Disulfide bond; G-protein coupled receptor; Glycoprotein; Lipoprotein; Membrane; Palmitate; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID M,N Molecular weight (Da) 40697.6 Length 371 Aromaticity 0.1 Instability index 20.94 Isoelectric point 5.07 Charge (pH=7) -9.93 2D Binding mode Binding energy (Kcal/mol) -6.84  Molscript Map 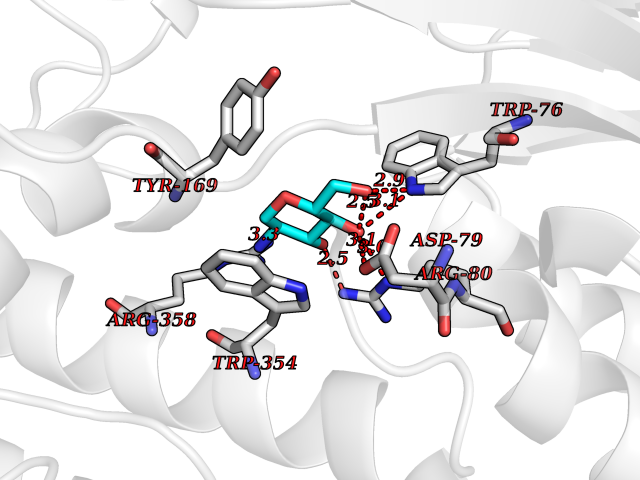 Pymol Map 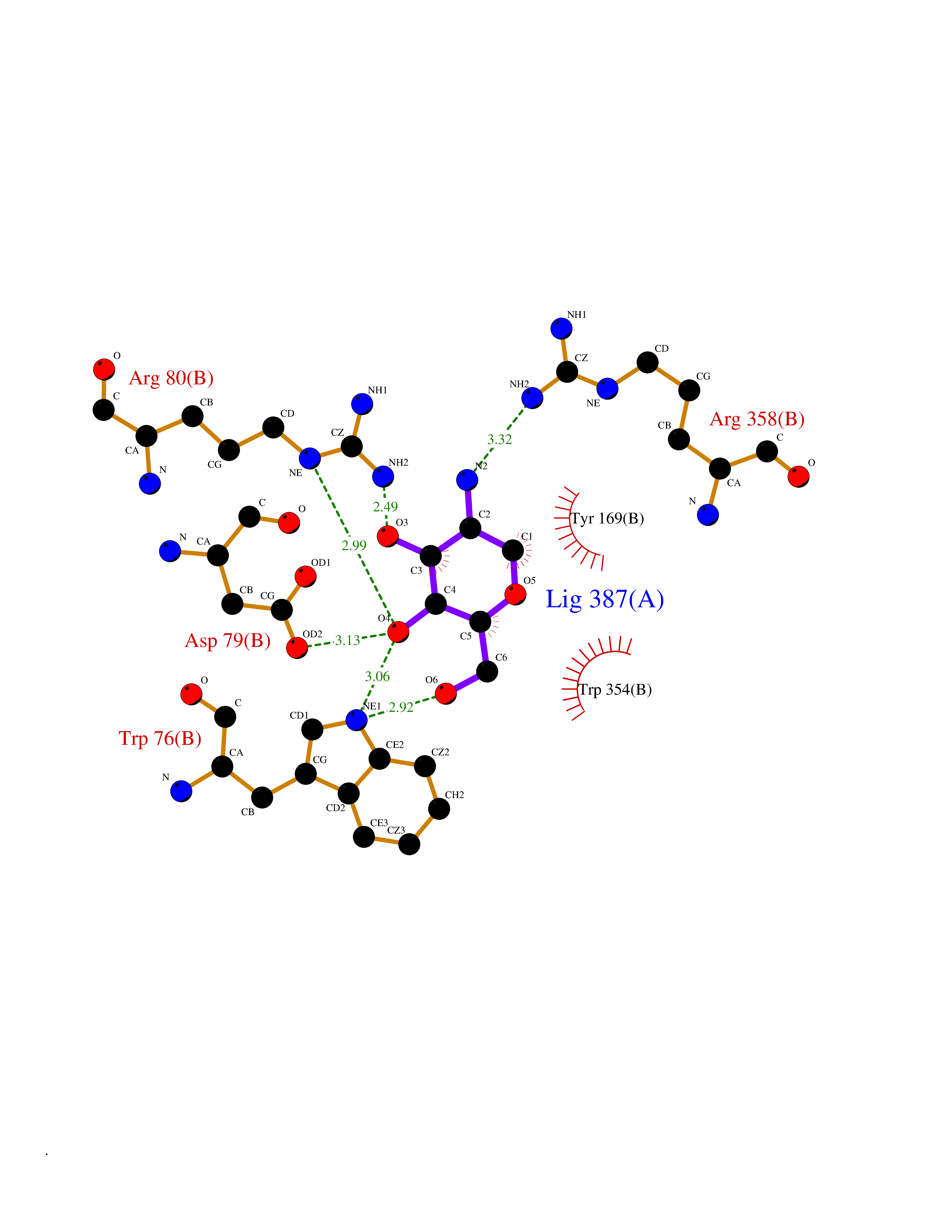 Ligplot Map 3D Binding mode Sequence NSSSNKIEEGKLVIWINGDKGYNGLAEVGKKFEKDTGIKVTVEHPDKLEEKFPQVAATGDGPDIIFWAHDRFGGYAQSGLLAEITPDKAFQDKLYPFTWDAVRYNGKLIAYPIAVEALSLIYNKDLLPNPPKTWEEIPALDKELKAKGKSALMFNLQEPYFTWPLIAADGGYAFKYENGKYDIKDVGVDNAGAKAGLTFLVDLIKNKHMNADTDYSIAEAAFNKGETAMTINGPWAWSNIDTSKVNYGVTVLPTFKGQPSKPFVGVLSAGINAASPNKELAKEFLENYLLTDEGLEAVNKDKPLGAVALKSYEEELAKDPRIAATMENAQKGEIMPNIPQMSAFWYAVRTAVINAASGRQTVDEALKDAQT Hydrogen bonds contact Hydrophobic contact | ||||
| 18 | Phosphatidylinositol-4-kinase beta (PI4KB) | 4WAE | 6.19 | |
Target general information Gen name PI4KB Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms PtdIns 4-kinase beta; Phosphatidylinositol 4-kinase beta; PIK4CB; PI4Kbeta; PI4K92; PI4K-beta; NPIK Protein family PI3/PI4-kinase family, Type III PI4K subfamily Biochemical class NA Function Phosphorylates phosphatidylinositol (PI) in the first committed step in the production of the second messenger inositol-1,4,5,-trisphosphate (PIP). May regulate Golgi disintegration/reorganization during mitosis, possibly via its phosphorylation. Involved in Golgi-to-plasma membrane trafficking (By similarity). Related diseases Deafness, autosomal dominant, 87 (DFNA87) [MIM:620281]: A form of non-syndromic, sensorineural hearing loss. Sensorineural hearing loss results from damage to the neural receptors of the inner ear, the nerve pathways to the brain, or the area of the brain that receives sound information. DFNA87 is characterized by prelingual, profound sensorineural hearing loss with inner ear anomalies, including cochlear maldevelopment, absence of the osseous spiral lamina, and/or an enlarged vestibular aqueduct. {ECO:0000269|PubMed:33358777}. The disease may be caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB12010 Interacts with Q9H3P7; P27348; P03495; PRO_0000424692 [P03300]; P62491 EC number EC 2.7.1.67 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; ATP-binding; Cytoplasm; Deafness; Direct protein sequencing; Disease variant; Endoplasmic reticulum; Golgi apparatus; Host-virus interaction; Kinase; Lipid metabolism; Membrane; Mitochondrion; Mitochondrion outer membrane; Non-syndromic deafness; Nucleotide-binding; Phosphoprotein; Proteomics identification; Reference proteome; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 53130 Length 463 Aromaticity 0.1 Instability index 49.16 Isoelectric point 6.78 Charge (pH=7) -0.96 2D Binding mode Binding energy (Kcal/mol) -6.09 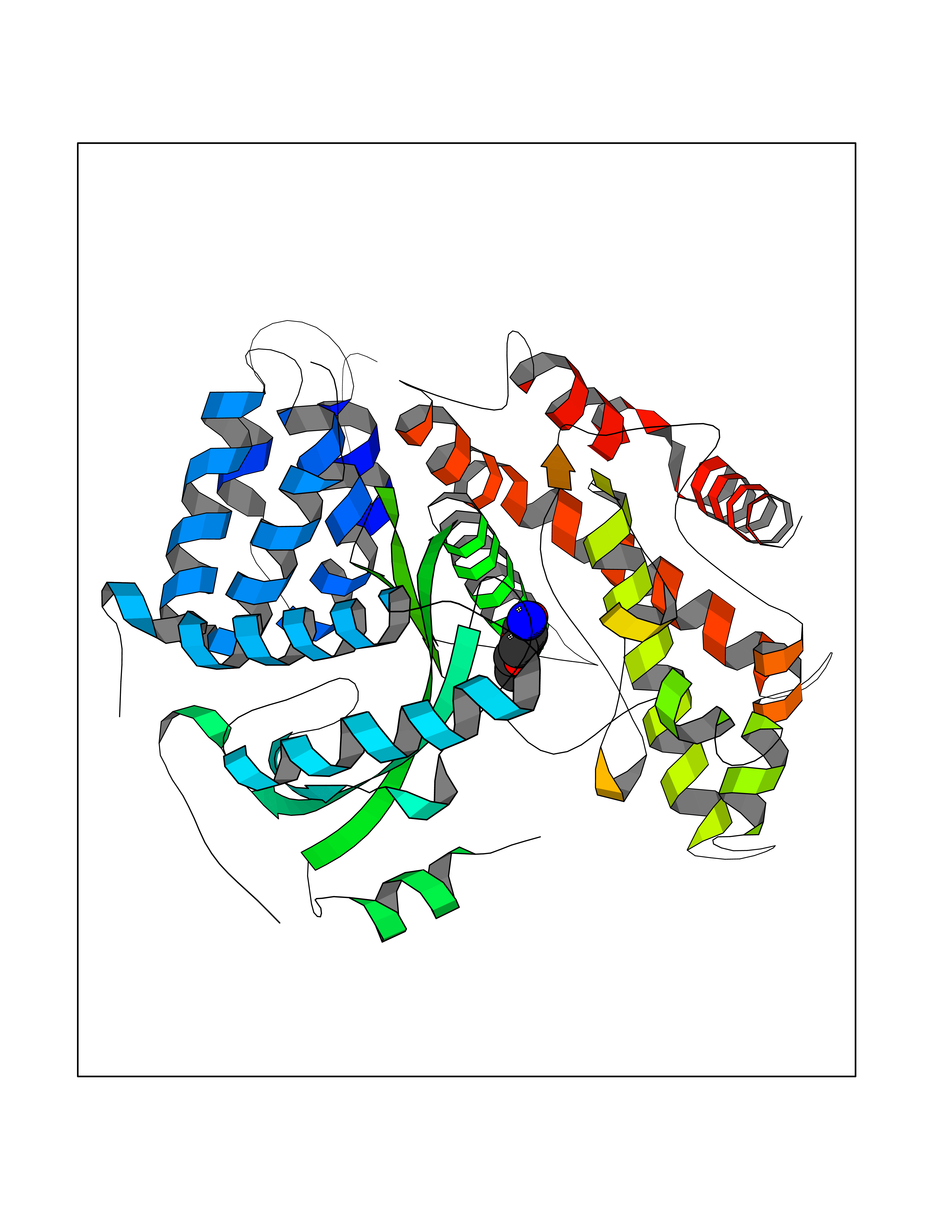 Molscript Map 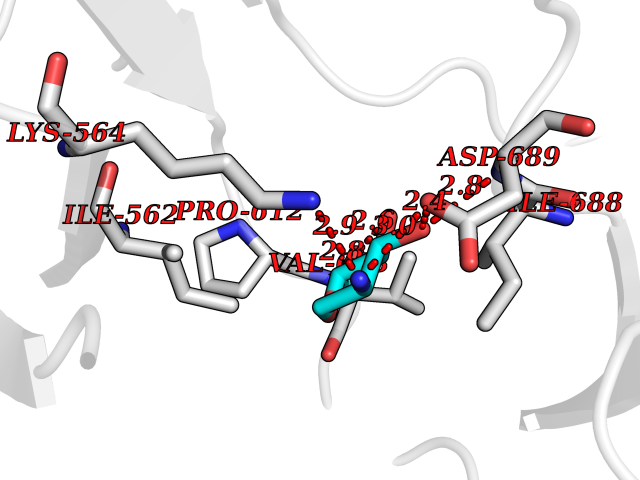 Pymol Map 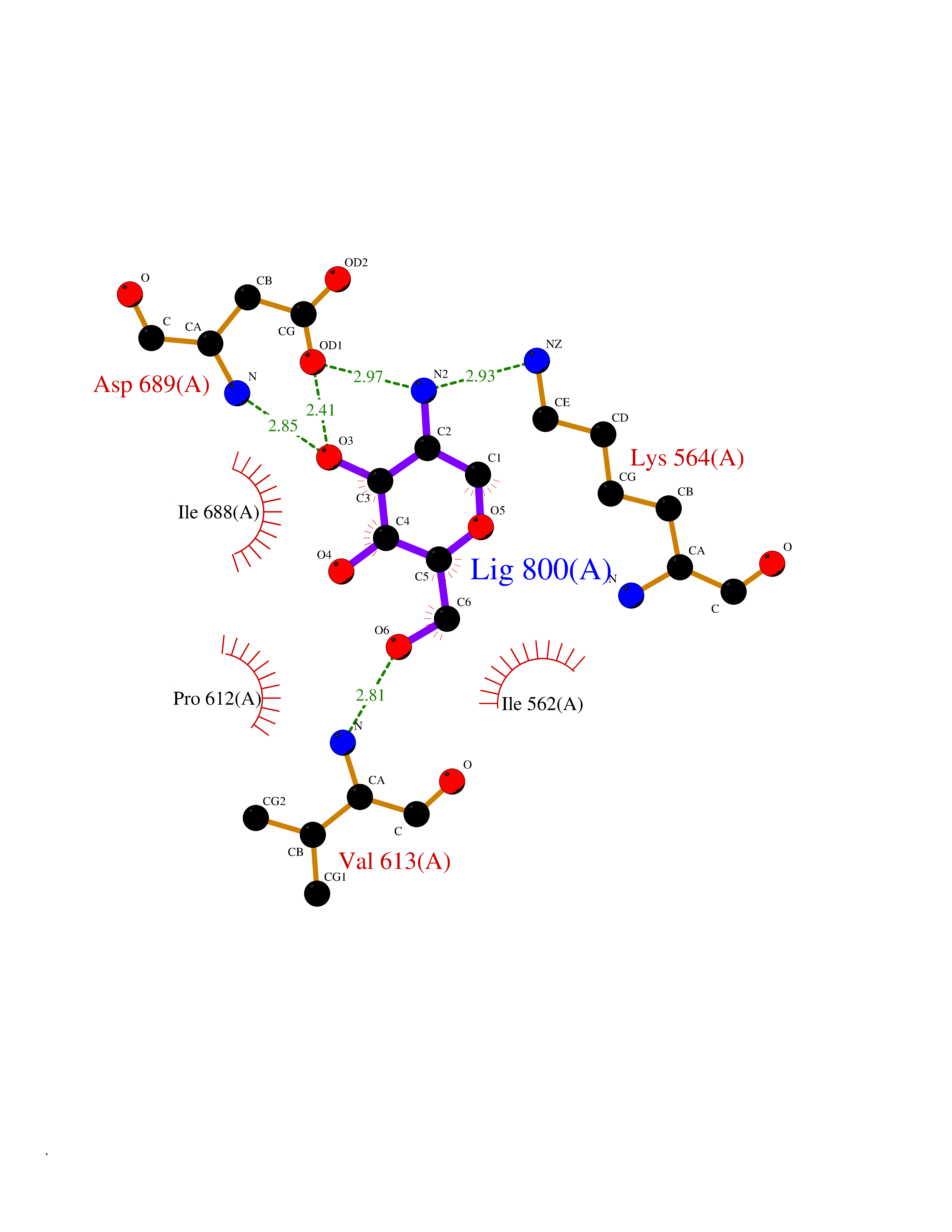 Ligplot Map 3D Binding mode Sequence LLRLFESKLFDISMAISYLYNSKEPGVQAYIGNRLFCFRNEDVDFYLPQLLNMYIHMDEDVGDAIKPYIVHRCRQSINFSLQCALLLGAYSSDMNSFSSPVRLAPEREFIKSLMAIGKRLATLPTKEQKTQRLISELSLLNHKLPARVWLPTAGFDHHVVRVPHTQAVVLNSKDKAPYLIYVEVLECENFDTTSVPARIPEWQEKVRRIREGSPYGHLPNWRLLSVIVKCGDDLRQELLAFQVLKQLQSIWEQERVPLWIKPYKILVISADSGMIEPVVNAVSIHQVKKQSQLSLLDYFLQEHGSYTTEAFLSAQRNFVQSCAGYCLVCYLLQVKDRHNGNILLDAEGHIIHIDFGFILSSSPRNLGFETSAFKLTTEFVDVMGGLDGDMFNYYKMLMLQGLIAARKHMDKVVQIVEIMQQGSQLPCFHGSSTIRNLKERFHMSMTEEQLQLLVEQMVDGSMR Hydrogen bonds contact Hydrophobic contact | ||||
| 19 | ATP-binding cassette transporter G2 (ABCG2) | 6HBU | 6.18 | |
Target general information Gen name ABCG2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Urate exporter; Placenta-specific ATP-binding cassette transporter; Mitoxantrone resistance-associated protein; MXR; CDw338; CD338; Breast cancer resistance protein; BCRP1; BCRP; ABCP Protein family ABC transporter superfamily, ABCG family, Eye pigment precursor importer (TC 3.A.1.204) subfamily Biochemical class ABC transporter Function Plays a role in porphyrin homeostasis as it is able to mediates the export of protoporhyrin IX (PPIX) both from mitochondria to cytosol and from cytosol to extracellular space, and cellular export of hemin, and heme. Xenobiotic transporter that may play an important role in the exclusion of xenobiotics from the brain. Appears to play a major role in the multidrug resistance phenotype of several cancer cell lines. Implicated in the efflux of numerous drugs and xenobiotics: mitoxantrone, the photosensitizer pheophorbide, camptothecin, methotrexate, azidothymidine (AZT), and the anthracyclines daunorubicin and doxorubicin. High-capacity urate exporter functioning in both renal and extrarenal urate excretion. Related diseases A chromosomal aberration involving TRIM24/TIF1 is found in papillary thyroid carcinomas (PTCs). Translocation t(7;10)(q32;q11) with RET. The translocation generates the TRIM24/RET (PTC6) oncogene. {ECO:0000269|PubMed:10439047}. Drugs (DrugBank ID) DB12001; DB08916; DB11363; DB00437; DB12015; DB03496; DB06288; DB11901; DB06605; DB01238; DB09274; DB12597; DB16098; DB06237; DB15233; DB11995; DB16407; DB13997; DB11817; DB00394; DB16703; DB15982; DB11967; DB04851; DB12267; DB12151; DB00921; DB06772; DB11751; DB00201; DB04690; DB08907; DB09061; DB14737; DB12218; DB11791; DB00958; DB00482; DB00439; DB04540; DB00515; DB00242; DB00631; DB00845; DB09065; DB05239; DB00286; DB12483; DB00091; DB08912; DB09102; DB11963; DB00970; DB02115; DB11682; DB12941; DB09183; DB01254; DB00694; DB11943; DB16650; DB01234; DB14649; DB00255; DB01248; DB08930; DB00843; DB05928; DB00997; DB00470; DB11952; DB06374; DB04881; DB06210; DB09038; DB13874; DB11718; DB00530; DB11827; DB00783; DB13952; DB13953; DB13954; DB13955; DB13956; DB00655; DB00773; DB00973; DB04854; DB12500; DB09279; DB00544; DB00158; DB12010; DB11796; DB02703; DB15149; DB15097; DB00317; DB01645; DB12141; DB11978; DB13879; DB01016; DB01094; DB14538; DB14539; DB14540; DB14541; DB14542; DB14543; DB14544; DB09054; DB00619; DB11886; DB00762; DB11633; DB06636; DB11757; DB01167; DB00602; DB00709; DB00448; DB14723; DB11732; DB09027; DB01097; DB16217; DB09078; DB12070; DB17083; DB06448; DB13125; DB06234; DB14009; DB00563; DB01204; DB16390; DB11763; DB00688; DB14011; DB03467; DB00220; DB11820; DB04868; DB09079; DB11793; DB00698; DB01051; DB09074; DB16267; DB09296; DB00338; DB11632; DB09330; DB13055; DB00526; DB12612; DB11697; DB09073; DB05467; DB00213; DB09297; DB06589; DB00642; DB15102; DB13878; DB17472; DB08860; DB08901; DB06813; DB15822; DB01708; DB00175; DB00457; DB00396; DB04216; DB12874; DB01129; DB00481; DB08896; DB11853; DB16826; DB11855; DB08864; DB00740; DB12457; DB08931; DB14840; DB15305; DB00503; DB06228; DB09291; DB01098; DB04847; DB12332; DB06654; DB01232; DB15685; DB11689; DB06290; DB08934; DB00398; DB12713; DB15569; DB12548; DB09118; DB00795; DB00391; DB00669; DB01268; DB11644; DB11760; DB00675; DB04348; DB12887; DB12020; DB01079; DB00966; DB12095; DB00853; DB00444; DB09299; DB15133; DB08880; DB00624; DB13943; DB13944; DB13946; DB11712; DB11800; DB01685; DB01030; DB14962; DB15442; DB11652; DB15328; DB15091; DB12255; DB05294; DB11613; DB08881; DB11581; DB00285; DB15456; DB00541; DB08828; DB12026; DB00549; DB00495 Interacts with Q9UNQ0; P22413; P11309-2; P0CG48; Q9UNQ0-1 EC number NA Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Cell membrane; Disulfide bond; Glycoprotein; Lipid transport; Membrane; Mitochondrion; Nucleotide-binding; Phosphoprotein; Proteomics identification; Reference proteome; Translocase; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A,B Molecular weight (Da) 129101 Length 1168 Aromaticity 0.11 Instability index 26.7 Isoelectric point 9.02 Charge (pH=7) 23.35 2D Binding mode Binding energy (Kcal/mol) -6.83  Molscript Map 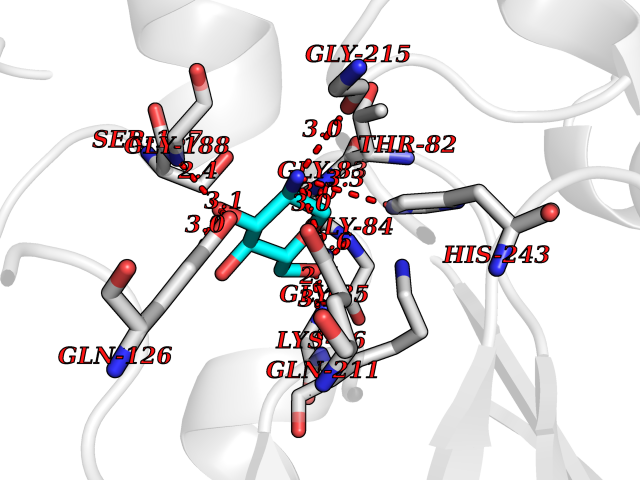 Pymol Map 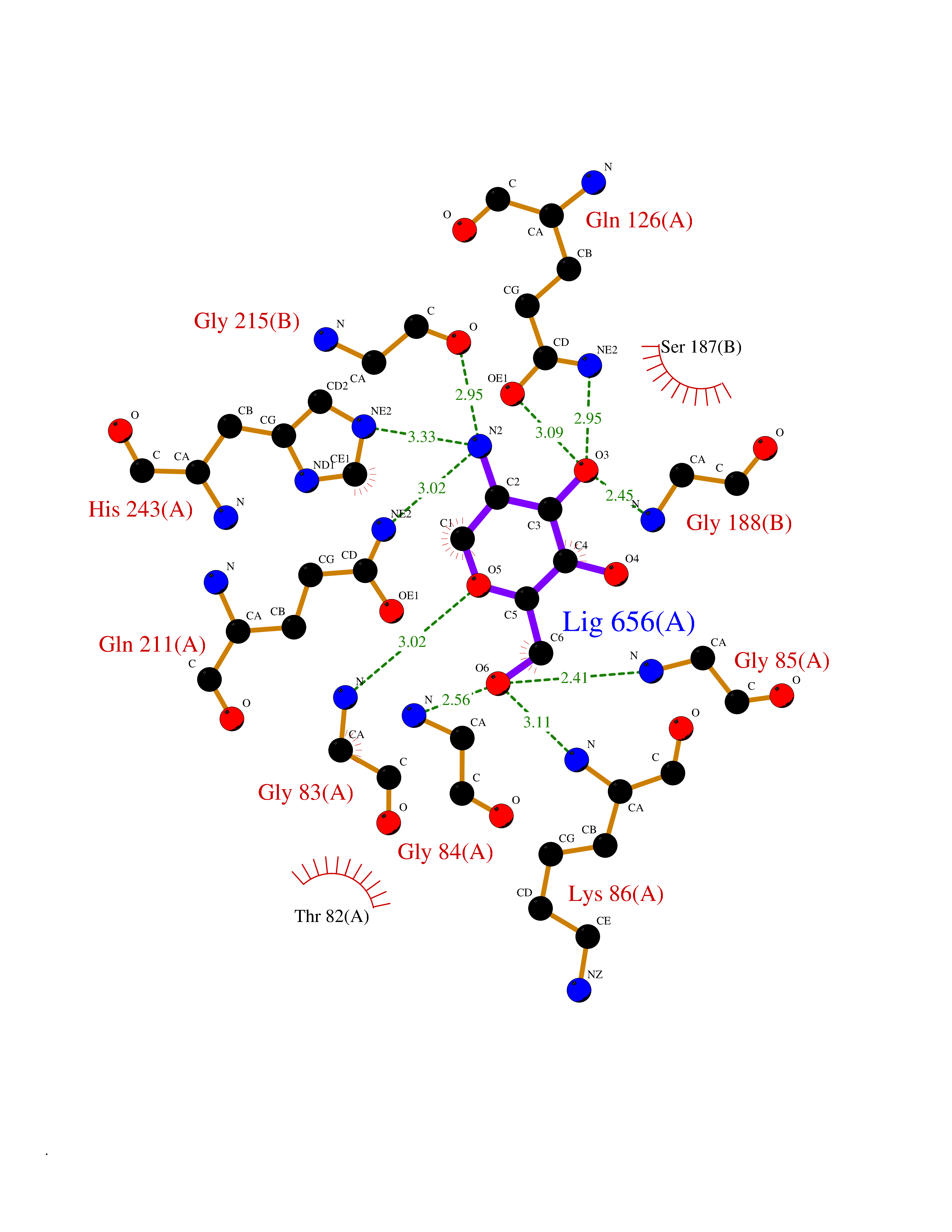 Ligplot Map 3D Binding mode Sequence GAVLSFHNICYRVKLPVEKEILSNINGIMKPGLNAILGPTGGGKSSLLDVLAARKDPSGLSGDVLINGAPRPANFKCNSGYVVQDDVVMGTLTVRENLQFSAALRLATTMTNHEKNERINRVIQELGLDKVADSKVGTQFIRGVSGGERKRTSIGMELITDPSILFLDQPTTGLDSSTANAVLLLLKRMSKQGRTIIFSIHQPRYSIFKLFDSLTLLASGRLMFHGPAQEALGYFESAGYHCEAYNNPADFFLDIINGDSTAVALNREKPLIEKLAEIYVNSSFYKETKAELHQLSISYTTSFCHQLRWVSKRSFKNLLGNPQASIAQIIVTVVLGLVIGAIYFGLKNDSTGIQNRAGVLFFLTTNQCFSSVSAVELFVVEKKLFIHEYISGYYRVSSYFLGKLLSDLLPMRMLPSIIFTCIVYFMLGLKPKADAFFVMMFTLMMVAYSASSMALAIAAGQSVVSVATLLMTICFVFMMIFSGLLVNLTTIASWLSWLQYFSIPRYGFTALQHNEFLGQNFCPGLNATGNNPCNYATCTGEEYLVKQGIDLSPWGLWKNHVALACMIVIFLTIAYLKLLFLKKYGAVLSFHNICYRVKLPVEKEILSNINGIMKPGLNAILGPTGGGKSSLLDVLAARKDPSGLSGDVLINGAPRPANFKCNSGYVVQDDVVMGTLTVRENLQFSAALRLATTMTNHEKNERINRVIQELGLDKVADSKVGTQFIRGVSGGERKRTSIGMELITDPSILFLDQPTTGLDSSTANAVLLLLKRMSKQGRTIIFSIHQPRYSIFKLFDSLTLLASGRLMFHGPAQEALGYFESAGYHCEAYNNPADFFLDIINGDSTAVALNREKPLIEKLAEIYVNSSFYKETKAELHQLSISYTTSFCHQLRWVSKRSFKNLLGNPQASIAQIIVTVVLGLVIGAIYFGLKNDSTGIQNRAGVLFFLTTNQCFSSVSAVELFVVEKKLFIHEYISGYYRVSSYFLGKLLSDLLPMRMLPSIIFTCIVYFMLGLKPKADAFFVMMFTLMMVAYSASSMALAIAAGQSVVSVATLLMTICFVFMMIFSGLLVNLTTIASWLSWLQYFSIPRYGFTALQHNEFLGQNFCPGLNATGNNPCNYATCTGEEYLVKQGIDLSPWGLWKNHVALACMIVIFLTIAYLKLLFLKKY Hydrogen bonds contact Hydrophobic contact | ||||
| 20 | CAAX farnesyltransferase beta (FNTB) | 1SA4 | 6.17 | |
Target general information Gen name FNTB Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms RAS proteins prenyltransferasebeta; FTase-beta; FNTB; CAAX farnesyltransferase beta subunit Protein family Protein prenyltransferase subunit beta family Biochemical class Alkyl aryl transferase Function Essential subunit of the farnesyltransferase complex. Catalyzes the transfer of a farnesyl moiety from farnesyl diphosphate to a cysteine at the fourth position from the C- terminus of several proteins having the C-terminal sequence Cys- aliphatic-aliphatic-X. Related diseases Lethal congenital contracture syndrome 2 (LCCS2) [MIM:607598]: A form of lethal congenital contracture syndrome, an autosomal recessive disorder characterized by degeneration of anterior horn neurons, extreme skeletal muscle atrophy, and congenital non-progressive joint contractures (arthrogryposis). The contractures can involve the upper or lower limbs and/or the vertebral column, leading to various degrees of flexion or extension limitations evident at birth. LCCS2 patients manifest craniofacial/ocular findings, lack of hydrops, multiple pterygia, and fractures, as well as a normal duration of pregnancy and a unique feature of a markedly distended urinary bladder (neurogenic bladder defect). The phenotype suggests a spinal cord neuropathic etiology. {ECO:0000269|PubMed:17701904}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Erythroleukemia, familial (FERLK) [MIM:133180]: An autosomal dominant myeloproliferative disorder characterized by neoplastic proliferation of erythroblastic and myeloblastic elements with atypical erythroblasts and myeloblasts in the peripheral blood. Disease penetrance is incomplete. {ECO:0000269|PubMed:27416908}. Disease susceptibility may be associated with variants affecting the gene represented in this entry.; DISEASE: Visceral neuropathy, familial, 1, autosomal recessive (VSCN1) [MIM:243180]: An autosomal recessive disorder characterized by intestinal dysmotility due to aganglionosis (Hirschsprung disease), hypoganglionosis, and/or chronic intestinal pseudoobstruction. Additional variable features are progressive peripheral neuropathy, arthrogryposis, hypoplasia or aplasia of the olfactory bulb and of the external auditory canals, microtia or anotia, and facial dysmorphism. Some patients present structural cardiac anomalies and arthrogryposis with multiple pterygia. {ECO:0000269|PubMed:33497358}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07216; DB08674; DB08676; DB06953; DB07771; DB07895; DB04893; DB07780; DB07841; DB07227; DB06448; DB04960 Interacts with P21549; Q9BWW8; Q8N5M1; Q8N4L8; G5E9A7; P49354; P31273; A5PL33-2; Q9H0W8; Q7Z699; O43711 EC number EC 2.5.1.58 Uniprot keywords 3D-structure; Alternative splicing; Lipid metabolism; Metal-binding; Phosphoprotein; Prenyltransferase; Proteomics identification; Reference proteome; Repeat; Transferase; Zinc Protein physicochemical properties Chain ID B,A Molecular weight (Da) 83751.2 Length 725 Aromaticity 0.11 Instability index 46.44 Isoelectric point 5.61 Charge (pH=7) -20.1 2D Binding mode Binding energy (Kcal/mol) -6.57 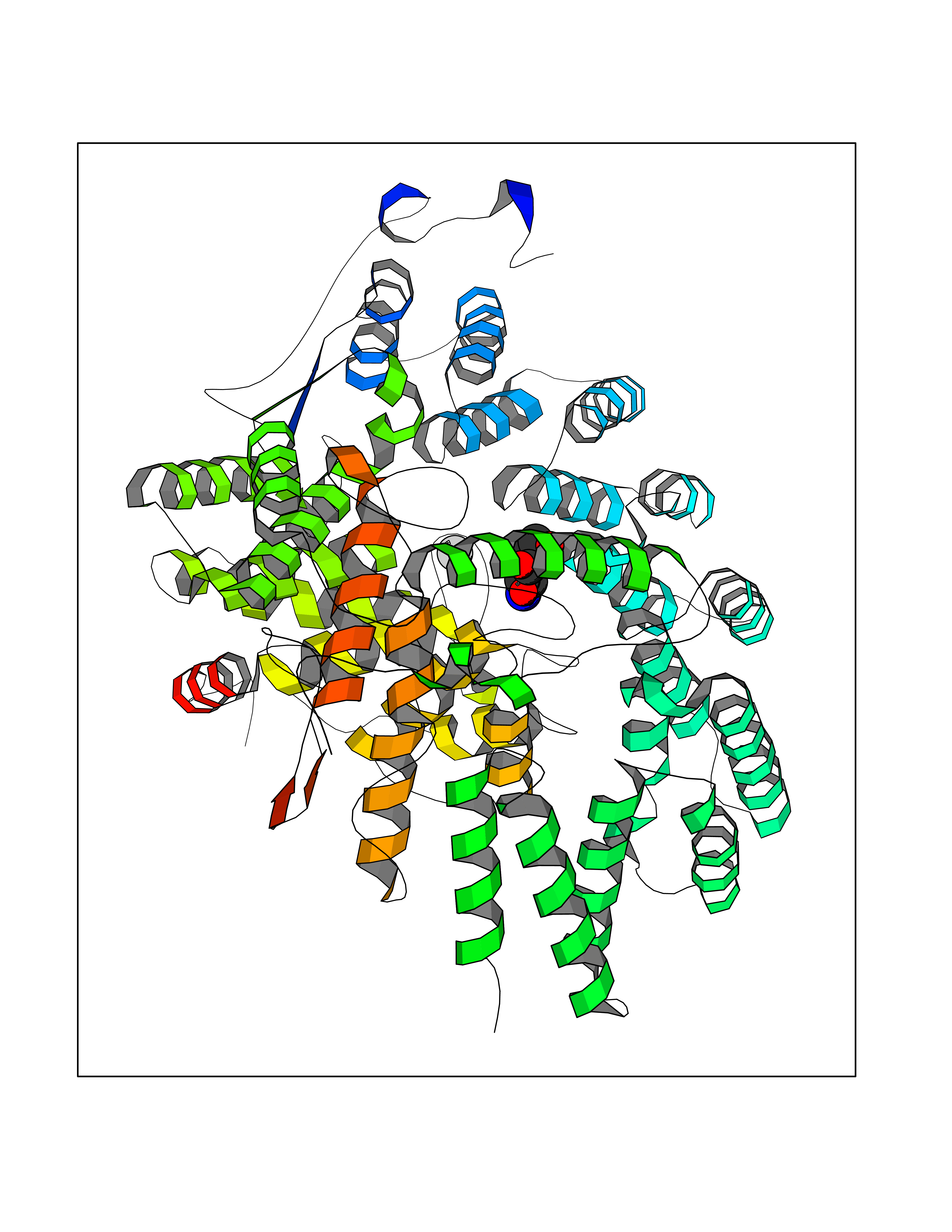 Molscript Map 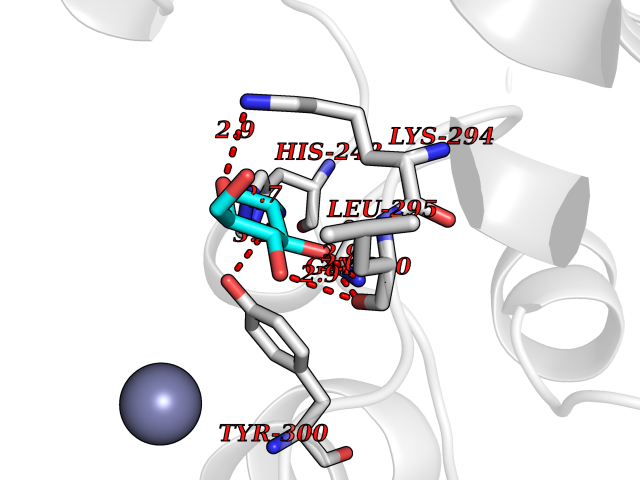 Pymol Map 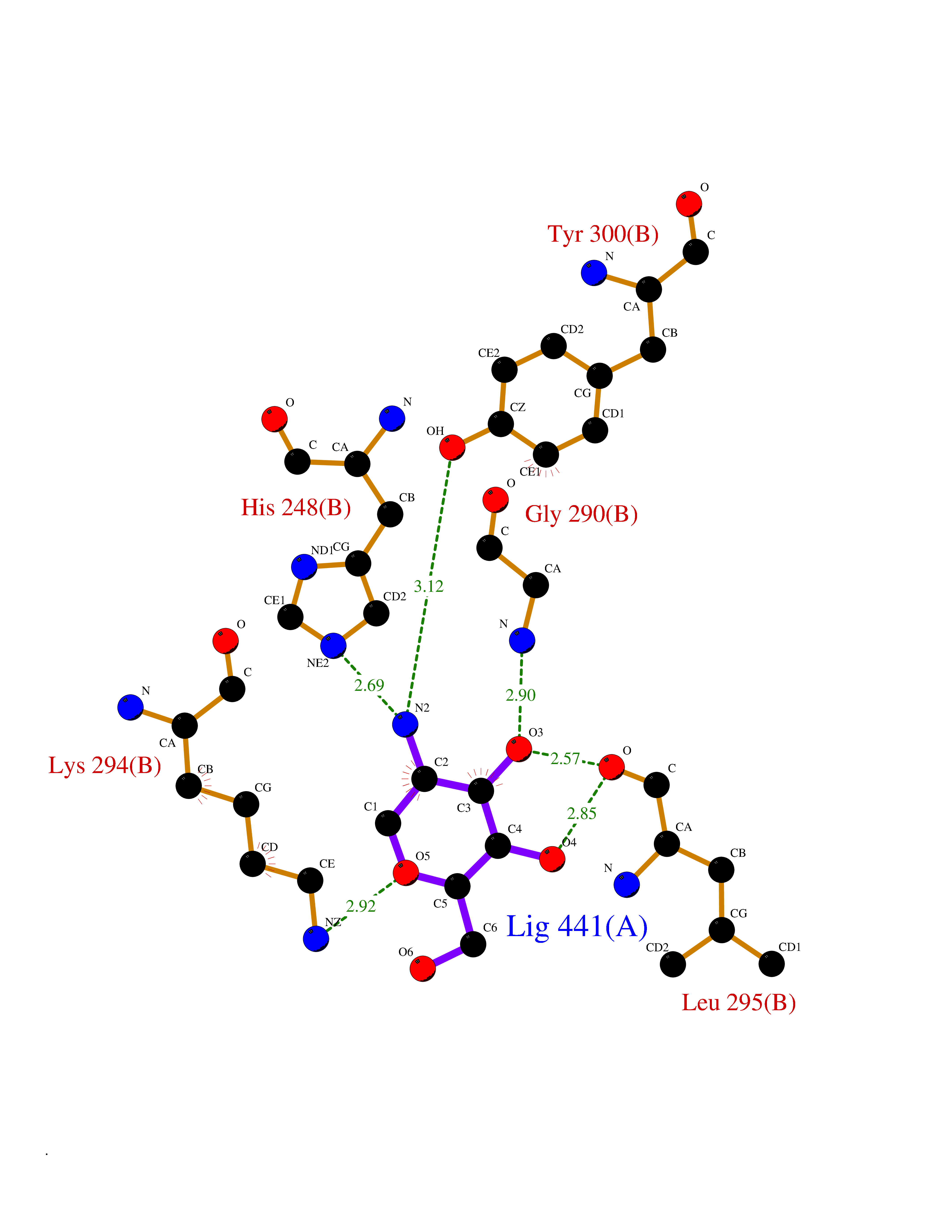 Ligplot Map 3D Binding mode Sequence FVSLDSPSYVLYRDRAEWADIDPVPQNDGPNPVVQIIYSDKFRDVYDYFRAVLQRDERSERAFKLTRDAIELNAANYTVWHFRRVLLKSLQKDLHEEMNYITAIIEEQPKNYQVWHHRRVLVEWLRDPSQELEFIADILNQDAKNYHAWQHRQWVIQEFKLWDNELQYVDQLLKEDVRNNSVWNQRYFVISNTTGYNDRAVLEREVQYTLEMIKLVPHNESAWNYLKGILQDRGLSKYPNLLNQLLDLQPSHSSPYLIAFLVDIYEDMLENQCDNKEDILNKALELCEILAKEKDTIRKEYWRYIGRSLQSKHSTSSPVWSEPLYSLRPEHARERLQDDSVETVTSIEQAKVEEKIQEVFSSYKFNHLVPRLVLQREKHFHYLKRGLRQLTDAYECLDASRPWLCYWILHSLELLDEPIPQIVATDVCQFLELCQSPEGGFGGGPGQYPHLAPTYAAVNALCIIGTEEAYDIINREKLLQYLYSLKQPDGSFLMHVGGEVDVRSAYCAASVASLTNIITPDLFEGTAEWIARCQNWEGGIGGVPGMEAHGGYTFCGLAALVILKRERSLNLKSLLQWVTSRQMRFEGGFQGRCNKLVDGCYSFWQAGLLPLLHRALHAQGDPALSMSHWMFHQQALQEYILMCCQCPAGGLLDKPGKSRDFYHTCYCLSGLSIAQHFGSGAMLHDVVLGVPENALQPTHPVYNIGPDKVIQATTYFLQKPVPGFE Hydrogen bonds contact Hydrophobic contact | ||||