Ligand
Structure
Job ID
d70ede4cb52eabf741dd5190a1834fd9
Job name
Garcia_test35
Time
2024-08-13 09:40:51
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 1 | Purine nucleoside phosphorylase (PNP) | 4EAR | 8.51 | |
Target general information Gen name PNP Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms PNP; Inosine phosphorylase Protein family PNP/MTAP phosphorylase family Biochemical class Pentosyltransferase Function The purine nucleoside phosphorylases catalyze the phosphorolytic breakdown of the N-glycosidic bond in the beta- (deoxy)ribonucleoside molecules, with the formation of the corresponding free purine bases and pentose-1-phosphate. Related diseases Purine nucleoside phosphorylase deficiency (PNPD) [MIM:613179]: A disorder that interrupts both the catabolism of inosine into hypoxanthine and guanosine into guanine, and leads to the accumulation of guanosine, inosine, and their deoxified by-products. The main clinical presentation is recurrent infections due to severe T-cell immunodeficiency. Some patients also have neurologic impairment. {ECO:0000269|PubMed:1384322, ECO:0000269|PubMed:3029074, ECO:0000269|PubMed:8931706}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03881; DB03551; DB02222; DB02391; DB03609; DB01667; DB04260; DB02796; DB04753; DB00640; DB00242; DB00900; DB06185; DB02377; DB02857; DB04754; DB04757; DB04076; DB02230; DB04335; DB02568; DB03101 Interacts with P05067; Q9UQM7; O14576-2; P06241; P14136; Q92993-2; Q9BXM7; P00491; P17612; P63000; Q92673; Q15583 EC number EC 2.4.2.1 Uniprot keywords 3D-structure; Acetylation; Cytoplasm; Direct protein sequencing; Disease variant; Glycosyltransferase; Phosphoprotein; Proteomics identification; Purine salvage; Reference proteome; Transferase Protein physicochemical properties Chain ID A,B,C Molecular weight (Da) 31849.2 Length 288 Aromaticity 0.1 Instability index 34.77 Isoelectric point 6.42 Charge (pH=7) -1.63 2D Binding mode Binding energy (Kcal/mol) -9.58  Molscript Map 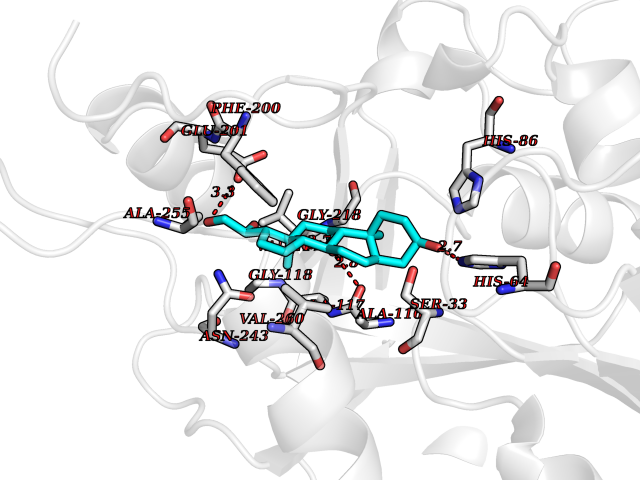 Pymol Map 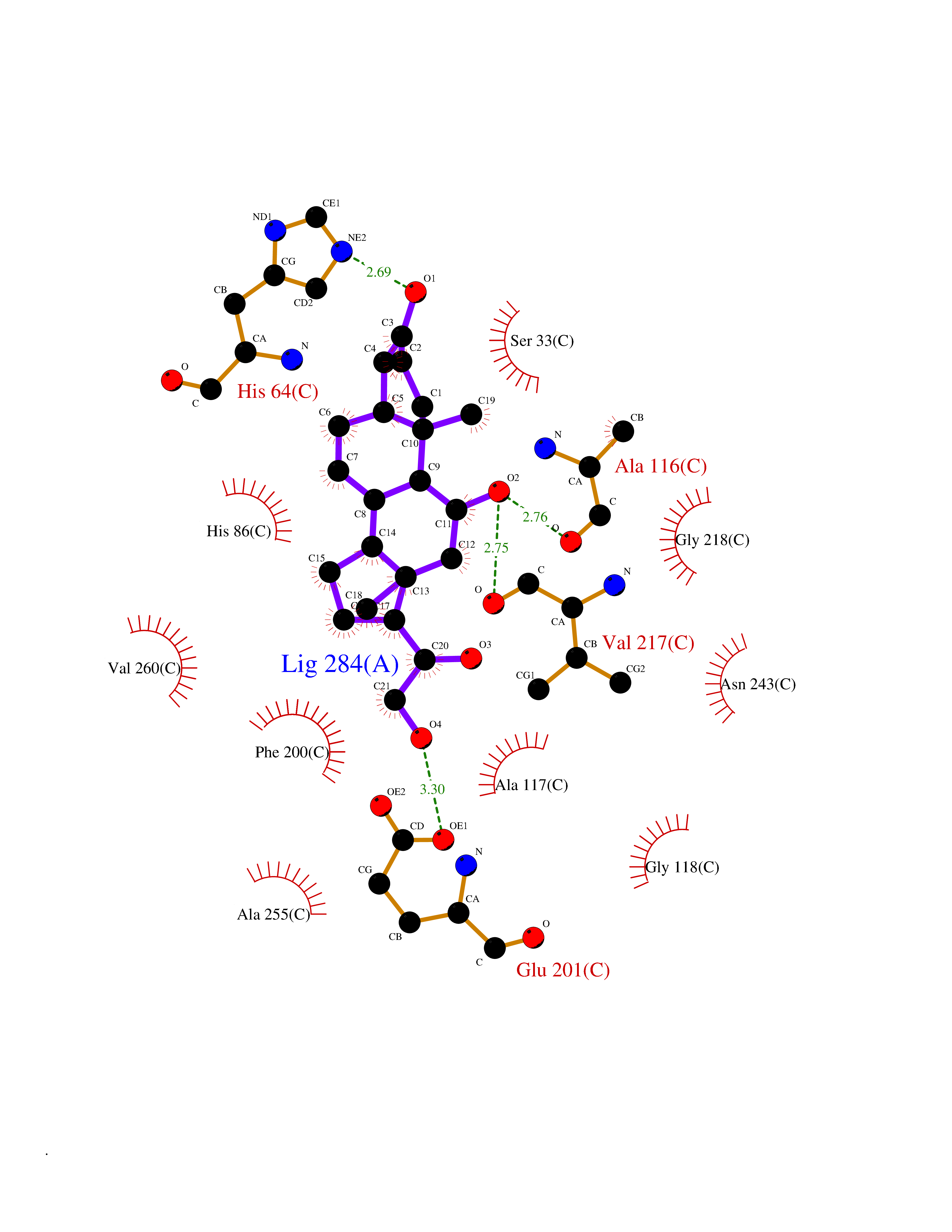 Ligplot Map 3D Binding mode Sequence GYTYEDYKNTAEYLLSHTKHRPQVAIICGSGLGGLTDKLTQAQIFDYSEIPNFPRSTVPGHAGRLVFGFLNGRACVMMQGRFHMYEGYPLYKVTFPVRVFHLLGVDTLVVTNAAGGLNPKFEVGDIMLIRDHINLPGFSGQNPLRGPNDERFGDRFPAMSDAYDRTMRQRALSTYKQMGEQRELQEGTYVMVAGPSFETVAECRVLQKLGADAVGMSTVPEVIVARHCGLRVFGFSLITNKVIMDYESLEKANXEEVLAAGKQAAQKLEQFVSILMASIDRFPAMSDA Hydrogen bonds contact Hydrophobic contact | ||||
| 2 | Organic cation transporter 3 (OCT3) | 7ZH6 | 8.51 | |
Target general information Gen name SLC22A3 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Solute carrier family 22 member 3; Extraneuronal monoamine transporter; EMTH Protein family Major facilitator (TC 2.A.1) superfamily, Organic cation transporter (TC 2.A.1.19) family Biochemical class NA Function Mediates potential-dependent transport of a variety of organic cations. May play a significant role in the disposition of cationic neurotoxins and neurotransmitters in the brain. Related diseases Deafness, autosomal dominant, 2A (DFNA2A) [MIM:600101]: A form of non-syndromic sensorineural hearing loss. Sensorineural deafness results from damage to the neural receptors of the inner ear, the nerve pathways to the brain, or the area of the brain that receives sound information. {ECO:0000269|PubMed:10025409, ECO:0000269|PubMed:10369879, ECO:0000269|PubMed:10571947, ECO:0000269|PubMed:10925378, ECO:0000269|PubMed:21242547}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00718; DB08838; DB00182; DB00122; DB14006; DB00501; DB00575; DB00363; DB01151; DB00988; DB00783; DB13952; DB13953; DB13954; DB13955; DB13956; DB00983; DB00536; DB05381; DB00458; DB00762; DB00709; DB00448; DB08882; DB01042; DB01577; DB00331; DB08893; DB00184; DB00368; DB00526; DB00925; DB00413; DB00457; DB01035; DB00396; DB00938; DB00391; DB13943; DB13944; DB08837; DB08841; DB00541 Interacts with P00519 EC number NA Uniprot keywords 3D-structure; Cell membrane; Glycoprotein; Ion transport; Membrane; Mitochondrion; Nucleus; Proteomics identification; Reference proteome; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A Molecular weight (Da) 53067.4 Length 478 Aromaticity 0.13 Instability index 38.82 Isoelectric point 9.07 Charge (pH=7) 10.54 2D Binding mode Binding energy (Kcal/mol) -9.61 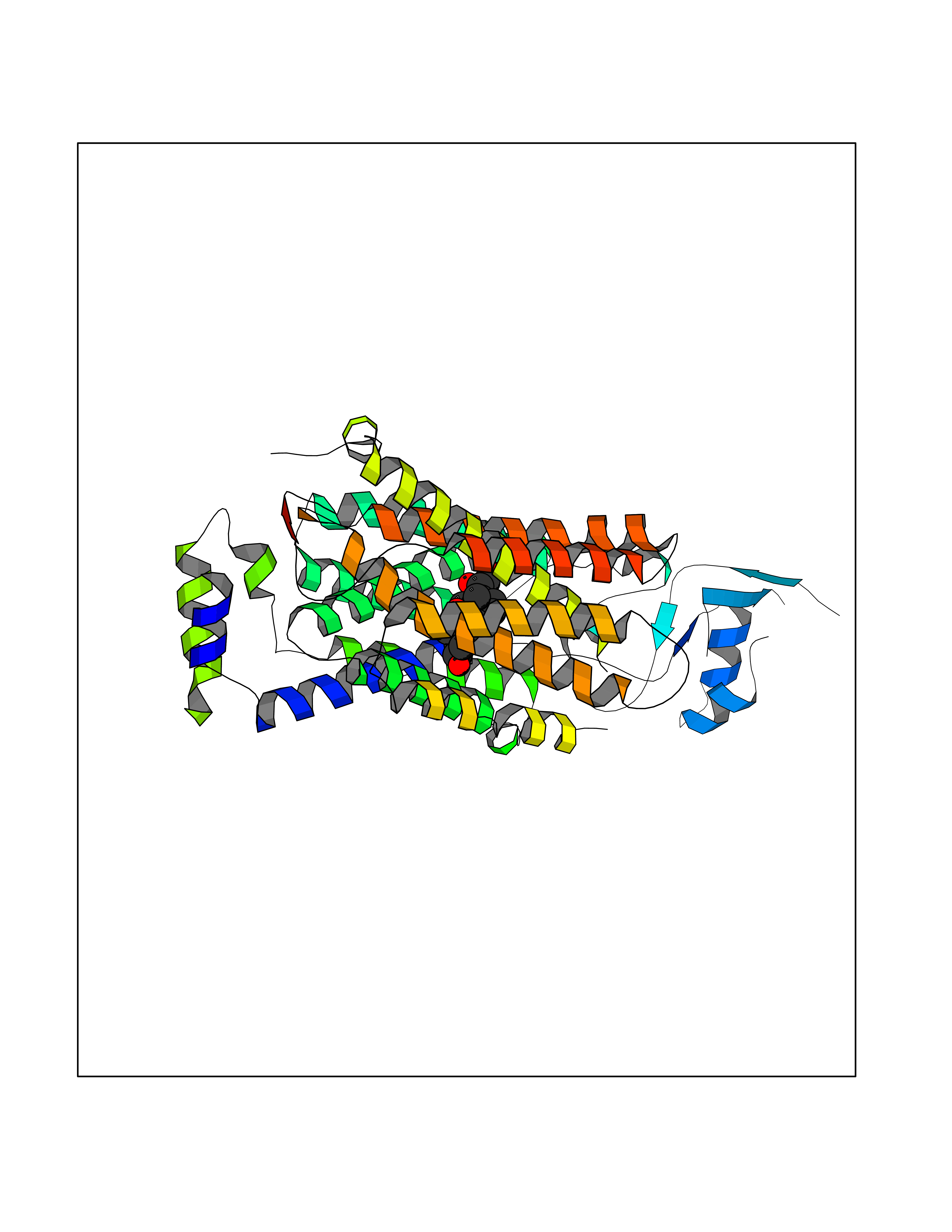 Molscript Map 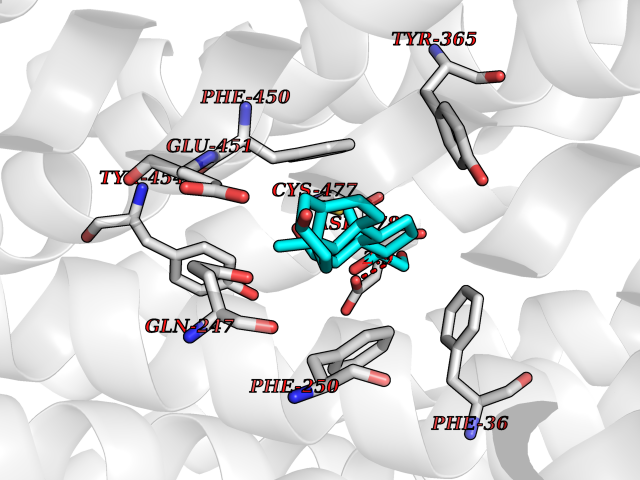 Pymol Map 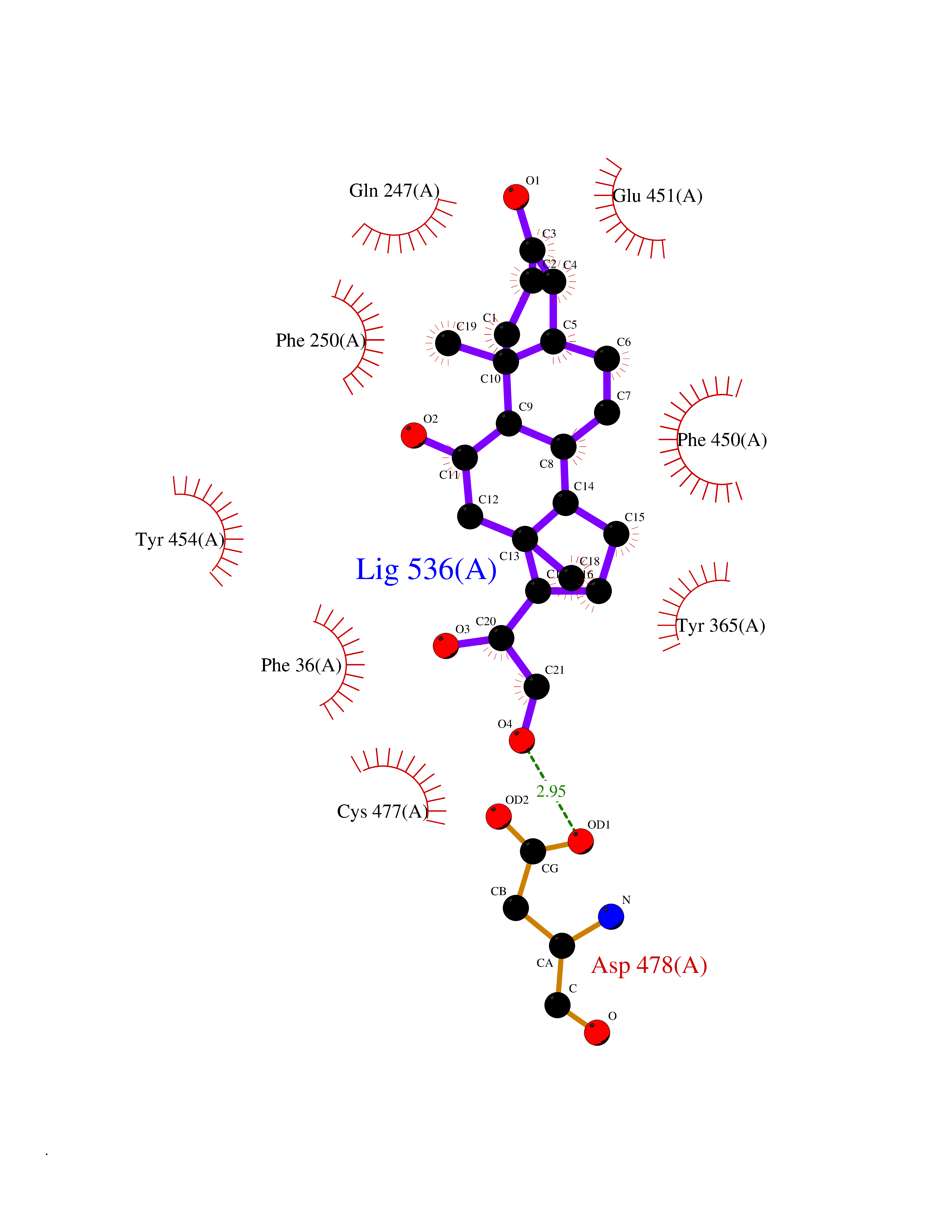 Ligplot Map 3D Binding mode Sequence SFDEALQRVGEFGRFQRRVFLLLCLTGVTFAFLFVGVVFLGTQPDHYWCRGPSAAALAERCGWSPEEEWNRTAPASRGRCQRYLLSAPLVPCRGGWRYAQAHSTIVSEFDLVCVNAWMLDLTQAILNLGFLTGAFTLGYAADRYGRIVIYLLSCLGVGVTGVVVAFAPNFPVFVIFRFLQGVFGKGTWMTCYVIVTEIVGSKQRRIVGIVIQMFFTLGIIILPGIAYFIPNWQGIQLAITLPSFLFLLYYWVVPESPRWLITRKKGDKALQILRRIAKCNVSNPSFLDLVRTPQMRKCTLILMFAWFTSAVVYQGLVMRLGNLYIDFFISGVVELPGALLILLTIERLGRRLPFAASNIVAGVACLVTAFLPEGIAWLRTTVATLGRLGITMAFEIVYLVNSELYPTTLRNFGVSLCSGLCDFGGIIAPFLLFRLAAVWLELPLIIFGILASICGGLVMLLPETKGIALPETVDDVEK Hydrogen bonds contact Hydrophobic contact | ||||
| 3 | Endothelin-converting enzyme 1 (ECE1) | 3DWB | 8.49 | |
Target general information Gen name ECE1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms ECE-1 Protein family Peptidase M13 family Biochemical class Peptidase Function Converts big endothelin-1 to endothelin-1. Related diseases Hirschsprung disease, cardiac defects, and autonomic dysfunction (HCAD) [MIM:613870]: A disorder characterized by skip-lesions Hirschsprung disease, craniofacial abnormalities and other dysmorphic features, cardiac defects including ductus arteriosus, small subaortic ventricular septal defect, small atrial septal defect, and autonomic dysfunction. {ECO:0000269|PubMed:9915973}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07171 Interacts with P49760; A8MQ03; Q8IUG1; P60370; P60410 EC number EC 3.4.24.71 Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disease variant; Disulfide bond; Glycoprotein; Hirschsprung disease; Hydrolase; Membrane; Metal-binding; Metalloprotease; Phosphoprotein; Protease; Proteomics identification; Reference proteome; Signal-anchor; Transmembrane; Transmembrane helix; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 75247.9 Length 660 Aromaticity 0.12 Instability index 46.29 Isoelectric point 5.33 Charge (pH=7) -18.3 2D Binding mode Binding energy (Kcal/mol) -8.59 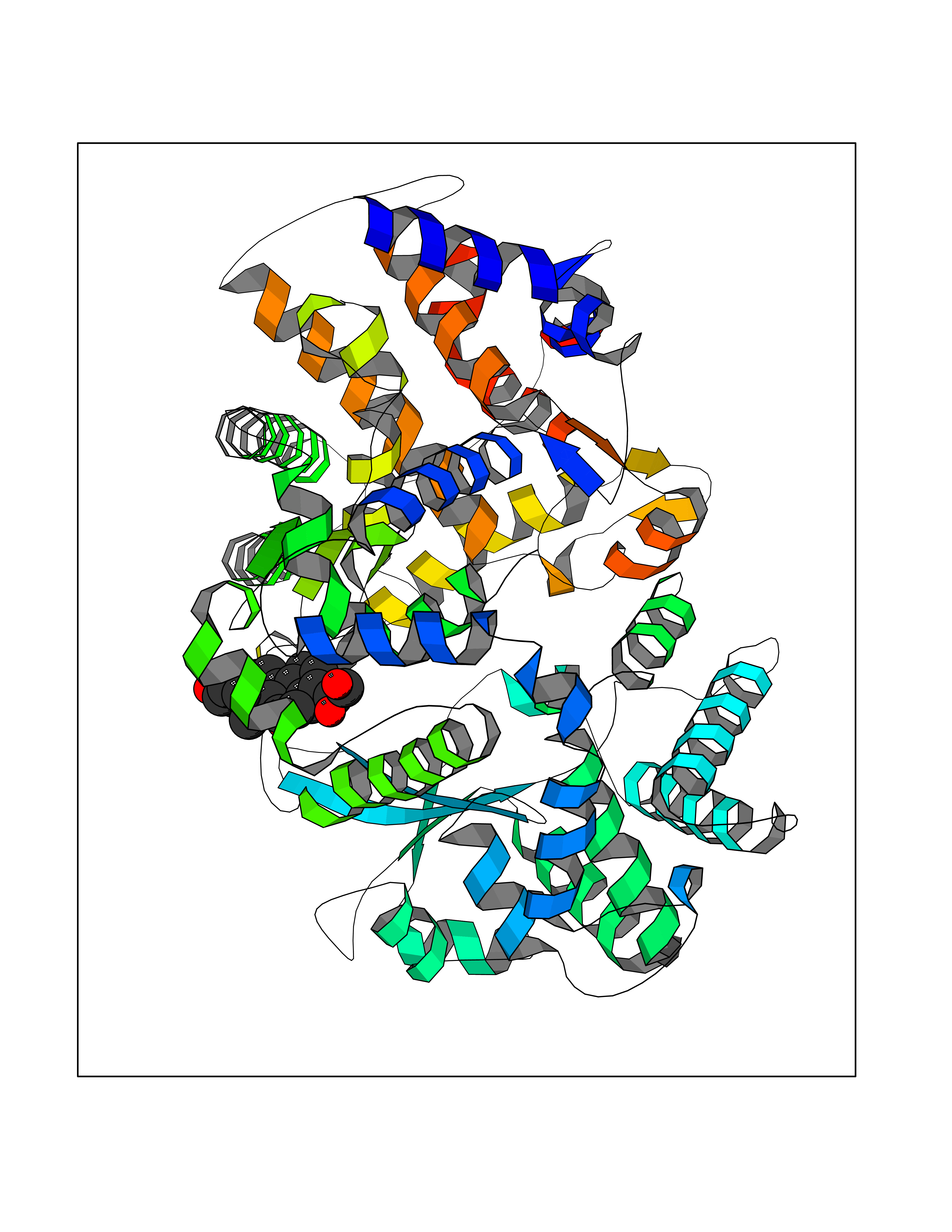 Molscript Map 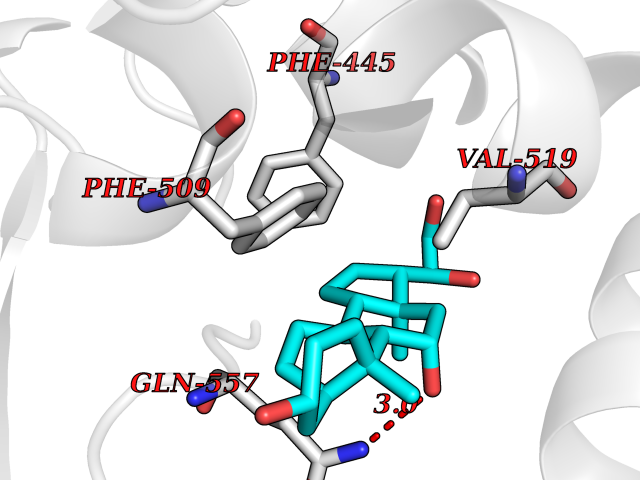 Pymol Map 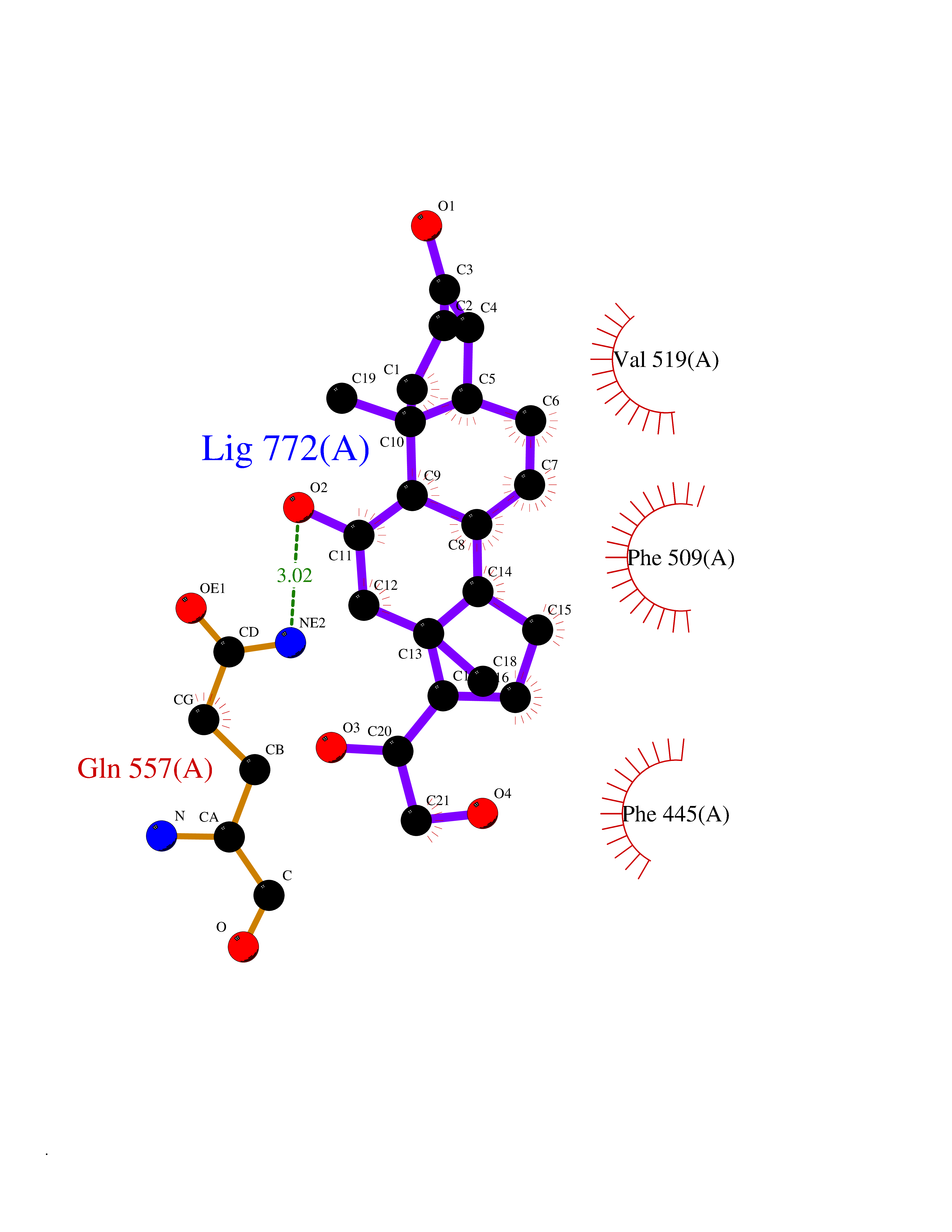 Ligplot Map 3D Binding mode Sequence SEACVSVTSSILSSMDPTVDPCHDFFSYACGGWIKANPVPDGHSRWGTFSNLWEHNQAIIKHLLENSTASVSEAERKAQVYYRACMNETRIEELRAKPLMELIERLGGWNITGPWAKDNFQDTLQVVTAHYRTSPFFSVYVSADSKNSNSNVIQVDQSGLGLPSRDYYLNKTENEKVLTGYLNYMVQLGKLLGGGDEEAIRPQMQQILDFETALANITIPQEKRRDEELIYHKVTAAELQTLAPAINWLPFLNTIFYPVEINESEPIVVYDKEYLEQISTLINTTDRCLLNNYMIWNLVRKTSSFLDQRFQDADEKFMEVMWKFCVSDTENNLGFALGPMFVKATFAEDSKSIATEIILEIKKAFEESLSTLKWMDEETRKSAKEKADAIYNMIGYPNFIMDPKELDKVFNDYTAVPDLYFENAMRFFNFSWRVTADQLRKAPNRDQWSMTPPMVNAYYSPTKNEIVFPAGILQAPFYTRSSPKALNFGGIGVVVGHELTHAFDDQGREYDKDGNLRPWWKNSSVEAFKRQTECMVEQYSNYSVNGEPVNGRHTLGENIADNGGLKAAYRAYQNWVKKNGAEHSLPTLGLTNNQLFFLGFAQVWCSVRTPESSHEGLITDPHSPSRFRVIGSLSNSKEFSEHFRCPPGSPMNPPHKCEVW Hydrogen bonds contact Hydrophobic contact | ||||
| 4 | Lanosterol 14-alpha demethylase (CYP51A1) | 4UHI | 8.47 | |
Target general information Gen name CYP51A1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Cytochrome P450LI; Cytochrome P45014DM; Cytochrome P450-14DM; Cytochrome P450 51A1 Protein family Cytochrome P450 family Biochemical class Cytochrome P450 family Function Catalyzes C14-demethylation of lanosterol; it transforms lanosterol into 4,4'-dimethyl cholesta-8,14,24-triene-3-beta-ol. Related diseases Spondyloepimetaphyseal dysplasia, short limb-hand type (SEMD-SL) [MIM:271665]: A bone disease characterized by short-limbed dwarfism, a narrow chest with pectus excavatum, brachydactyly in the hands and feet, a characteristic craniofacial appearance and premature calcifications. The radiological findings are distinctive and comprise short long bones throughout the skeleton with striking epiphyses that are stippled, flattened and fragmented and flared, irregular metaphyses. Platyspondyly in the spine with wide intervertebral spaces is observed and some vertebral bodies are pear-shaped with central humps, anterior protrusions and posterior scalloping. {ECO:0000269|PubMed:19110212, ECO:0000269|PubMed:20223752, ECO:0000269|PubMed:26463668}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Warburg-Cinotti syndrome (WRCN) [MIM:618175]: An autosomal dominant disease characterized by progressive corneal neovascularization, keloid formation, chronic skin ulcers, wasting of subcutaneous tissue, flexion contractures of the fingers, and acro-osteolysis. {ECO:0000269|PubMed:30449416}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07705; DB05667; DB01110; DB01007 Interacts with NA EC number EC 1.14.14.154 Uniprot keywords 3D-structure; Alternative splicing; Cholesterol biosynthesis; Cholesterol metabolism; Endoplasmic reticulum; Heme; Iron; Lipid biosynthesis; Lipid metabolism; Membrane; Metal-binding; Microsome; Monooxygenase; Oxidoreductase; Proteomics identification; Reference proteome; Steroid biosynthesis; Steroid metabolism; Sterol biosynthesis; Sterol metabolism; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 53013.3 Length 462 Aromaticity 0.11 Instability index 47.66 Isoelectric point 8.8 Charge (pH=7) 7 2D Binding mode Binding energy (Kcal/mol) -9.11 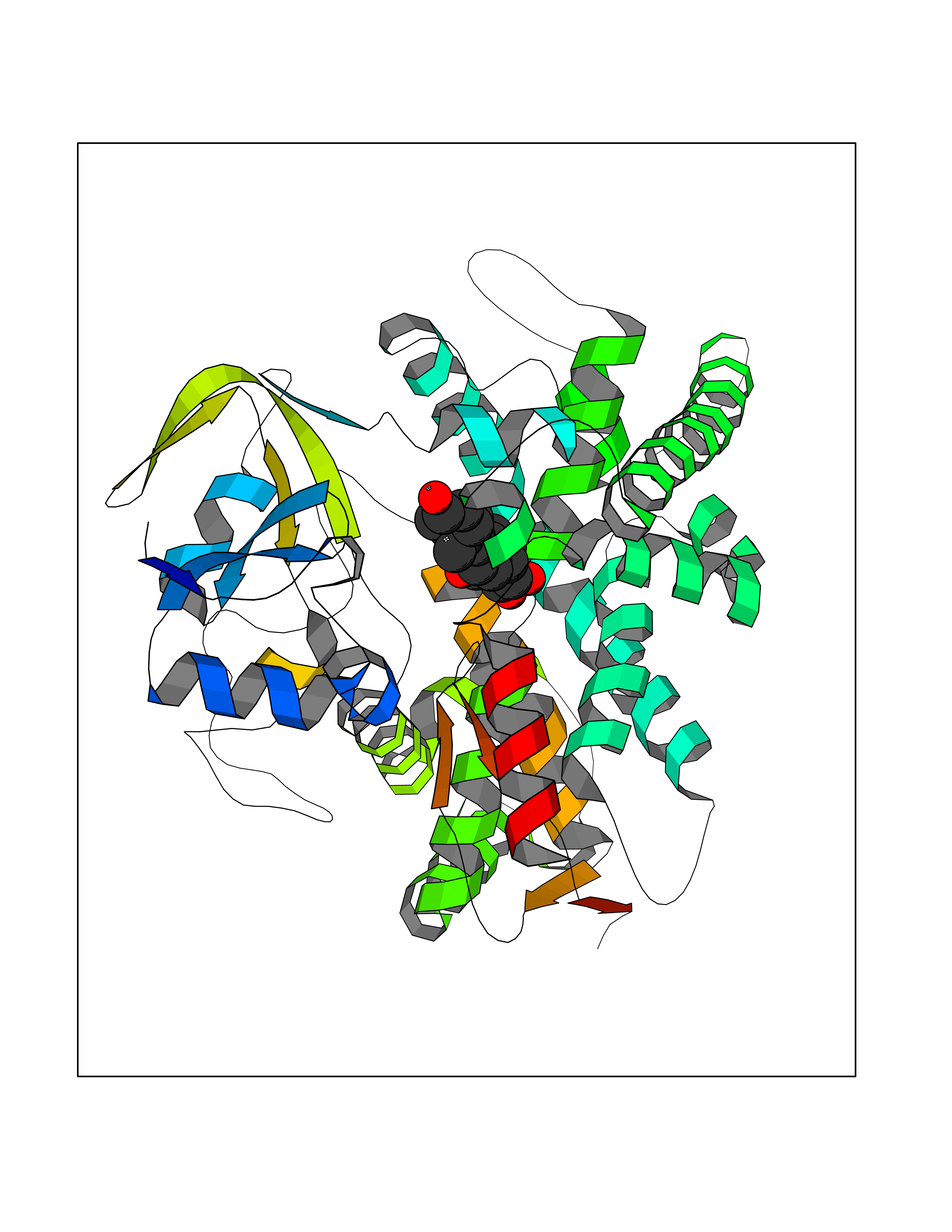 Molscript Map 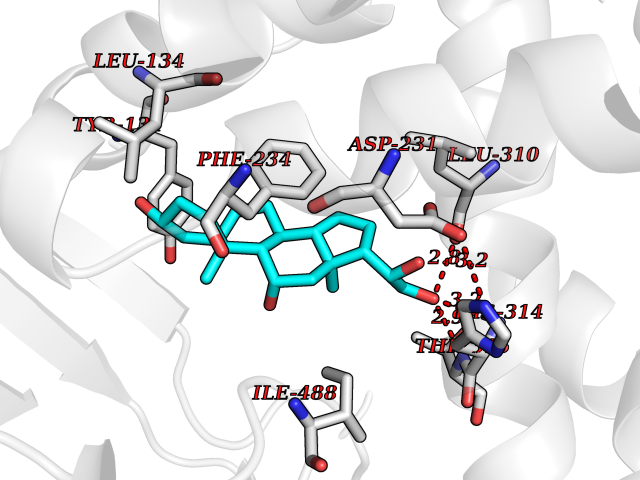 Pymol Map 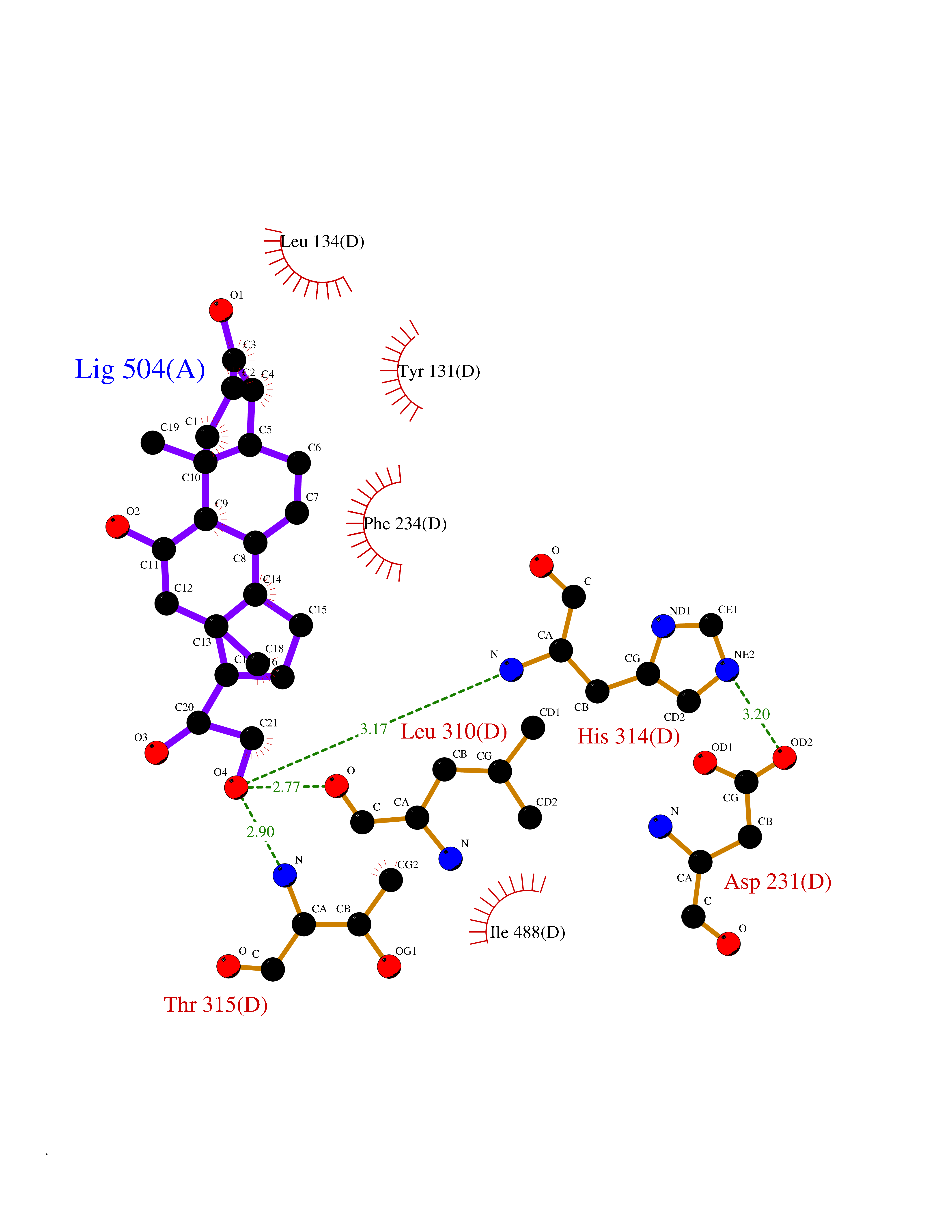 Ligplot Map 3D Binding mode Sequence PPYIFSPIPFLGHAIAFGKSPIEFLENAYEKYGPVFSFTMVGKTFTYLLGSDAAALLFNSKNEDLNAEDVYSRLTTPVFGKGVAYDVPNPVFLEQKKMLKSGLNIAHFKQHVSIIEKETKEYFESWGESGEKNVFEALSELIILTASHCLHGKEIRSQLNEKVAQLYADLDGGFSHAAWLLPGWLPLPSFRRRDRAHREIKDIFYKAIQKRRQSQEKIDDILQTLLDATYKDGRPLTDDEVAGMLIGLLLAGQHTSSTTSAWMGFFLARDKTLQKKCYLEQKTVCGENLPPLTYDQLKDLNLLDRCIKETLRLRPPIMIMMRMARTPQTVAGYTIPPGHQVCVSPTVNQRLKDSWVERLDFNPDRYLQDNPASGEKFAYVPFGAGRHRCIGENFAYVQIKTIWSTMLRLYEFDLIDGYFPTVNYTTMIHTPENPVIRYKRRSLPGWLPLPSFRRRDRAHREI Hydrogen bonds contact Hydrophobic contact | ||||
| 5 | Cytochrome P450 1A2 | 2HI4 | 8.44 | |
Target general information Gen name CYP1A2 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Cytochrome P450 family Biochemical class Oxidoreductase Function Aromatase activity.Caffeine oxidase activity.Demethylase activity.Electron carrier activity.Enzyme binding.Heme binding.Iron ion binding.Monooxygenase activity.Oxidoreductase activity.Oxidoreductase activity, acting on paired donors, with incorporation or reduction of molecular oxygen, reduced flavin or flavoprotein as one donor, and incorporation of one atom of oxygen.Oxygen binding. Related diseases Myeloperoxidase deficiency (MPOD) [MIM:254600]: A disorder characterized by decreased myeloperoxidase activity in neutrophils and monocytes that results in disseminated candidiasis. {ECO:0000269|PubMed:37198333, ECO:0000269|PubMed:7904599, ECO:0000269|PubMed:8142659, ECO:0000269|PubMed:8621627, ECO:0000269|PubMed:9354683, ECO:0000269|PubMed:9637725}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08496; DB01667; DB14132; DB04356; DB02489; DB11932; DB12001; DB05812; DB13573; DB01418; DB00316; DB15568; DB06594; DB00518; DB05396; DB00969; DB07453; DB01424; DB01223; DB01118; DB00321; DB00261; DB01217; DB01435; DB06605; DB05676; DB06413; DB06216; DB01072; DB15011; DB06442; DB06626; DB00993; DB00972; DB13203; DB05015; DB16703; DB06769; DB01086; DB06770; DB06771; DB06732; DB00195; DB04889; DB11967; DB13975; DB00188; DB12151; DB01558; DB14018; DB13812; DB00201; DB09061; DB14737; DB11791; DB06774; DB00564; DB06016; DB01136; DB12814; DB00477; DB00356; DB01166; DB00501; DB01012; DB00568; DB00827; DB00537; DB00215; DB12499; DB14025; DB00349; DB01242; DB00575; DB00758; DB00363; DB00286; DB11672; DB14635; DB00924; DB08912; DB00851; DB06292; DB01254; DB01609; DB01151; DB16650; DB12161; DB01191; DB00633; DB11994; DB00586; DB11511; DB12945; DB00280; DB01184; DB09167; DB05928; DB01142; DB09273; DB00470; DB00476; DB00625; DB15444; DB06210; DB13874; DB11718; DB00467; DB11404; DB00530; DB00783; DB13952; DB13953; DB13954; DB13955; DB13956; DB00655; DB04574; DB13592; DB00330; DB00898; DB00977; DB00773; DB01628; DB00927; DB04854; DB01482; DB00574; DB12265; DB15669; DB01195; DB08972; DB04841; DB00544; DB00472; DB00499; DB00176; DB01320; DB00998; DB14029; DB06160; DB01044; DB01241; DB01155; DB01645; DB01381; DB00986; DB00365; DB00400; DB05708; DB00629; DB00502; DB01094; DB14999; DB04076; DB11737; DB00619; DB00458; DB11564; DB01306; DB09456; DB09564; DB01307; DB00047; DB01309; DB00030; DB00046; DB11567; DB00071; DB11568; DB05258; DB00034; DB00105; DB15131; DB00011; DB00018; DB00069; DB00060; DB00068; DB00033; DB00951; DB11757; DB09570; DB01026; DB01097; DB16217; DB09078; DB01002; DB05667; DB00281; DB12406; DB09198; DB04948; DB00978; DB06448; DB16220; DB01601; DB00455; DB04871; DB06077; DB01283; DB00772; DB00934; DB06234; DB14009; DB00784; DB01065; DB00170; DB00454; DB00532; DB00333; DB00763; DB00553; DB01028; DB09241; DB01233; DB00379; DB06148; DB01388; DB06595; DB00370; DB16236; DB00745; DB11763; DB00218; DB06510; DB14011; DB00461; DB00607; DB00779; DB00788; DB06600; DB00238; DB06803; DB00184; DB01115; DB11793; DB00435; DB05115; DB00717; DB01059; DB00540; DB05990; DB01165; DB00334; DB16267; DB00338; DB00904; DB11632; DB11443; DB01173; DB11837; DB09330; DB01303; DB11697; DB00377; DB00715; DB06589; DB11774; DB00487; DB00008; DB00022; DB09122; DB13634; DB00806; DB11198; DB08883; DB00850; DB03783; DB01174; DB00388; DB00252; DB11450; DB01100; DB13823; DB04951; DB17472; DB11642; DB08910; DB15822; DB01058; DB01087; DB00794; DB00420; DB09288; DB01182; DB06479; DB00818; DB00571; DB13449; DB11892; DB04216; DB00908; DB00468; DB01129; DB00980; DB09290; DB00863; DB01367; DB00409; DB02709; DB13174; DB01045; DB11753; DB00740; DB14924; DB00503; DB00533; DB01656; DB15119; DB00268; DB00296; DB00412; DB00817; DB12332; DB13772; DB06654; DB11491; DB00418; DB01037; DB11689; DB06290; DB13261; DB15093; DB00052; DB00398; DB01208; DB09118; DB00428; DB06820; DB00382; DB00675; DB06083; DB09071; DB05488; DB09256; DB01079; DB01405; DB00857; DB08880; DB11712; DB01412; DB00277; DB00730; DB01623; DB00208; DB06137; DB00697; DB01056; DB06264; DB00752; DB00384; DB12245; DB00831; DB15442; DB00440; DB00685; DB08867; DB14989; DB13609; DB06235; DB00313; DB08881; DB00661; DB09185; DB12026; DB00682; DB02134; DB00549; DB00744; DB00315; DB00425; DB09225; DB09120 Interacts with O95870 EC number 1.14.14.1; 4.2.1.152 Uniprot keywords 3D-structure; Direct protein sequencing; Endoplasmic reticulum; Fatty acid metabolism; Glycoprotein; Heme; Iron; Lipid metabolism; Lyase; Membrane; Metal-binding; Microsome; Monooxygenase; Oxidoreductase; Proteomics identification; Reference proteome; Steroid metabolism; Sterol metabolism Protein physicochemical properties Chain ID A Molecular weight (Da) 54475 Length 480 Aromaticity 0.1 Instability index 40.43 Isoelectric point 9.16 Charge (pH=7) 9.89 2D Binding mode Binding energy (Kcal/mol) -9.36 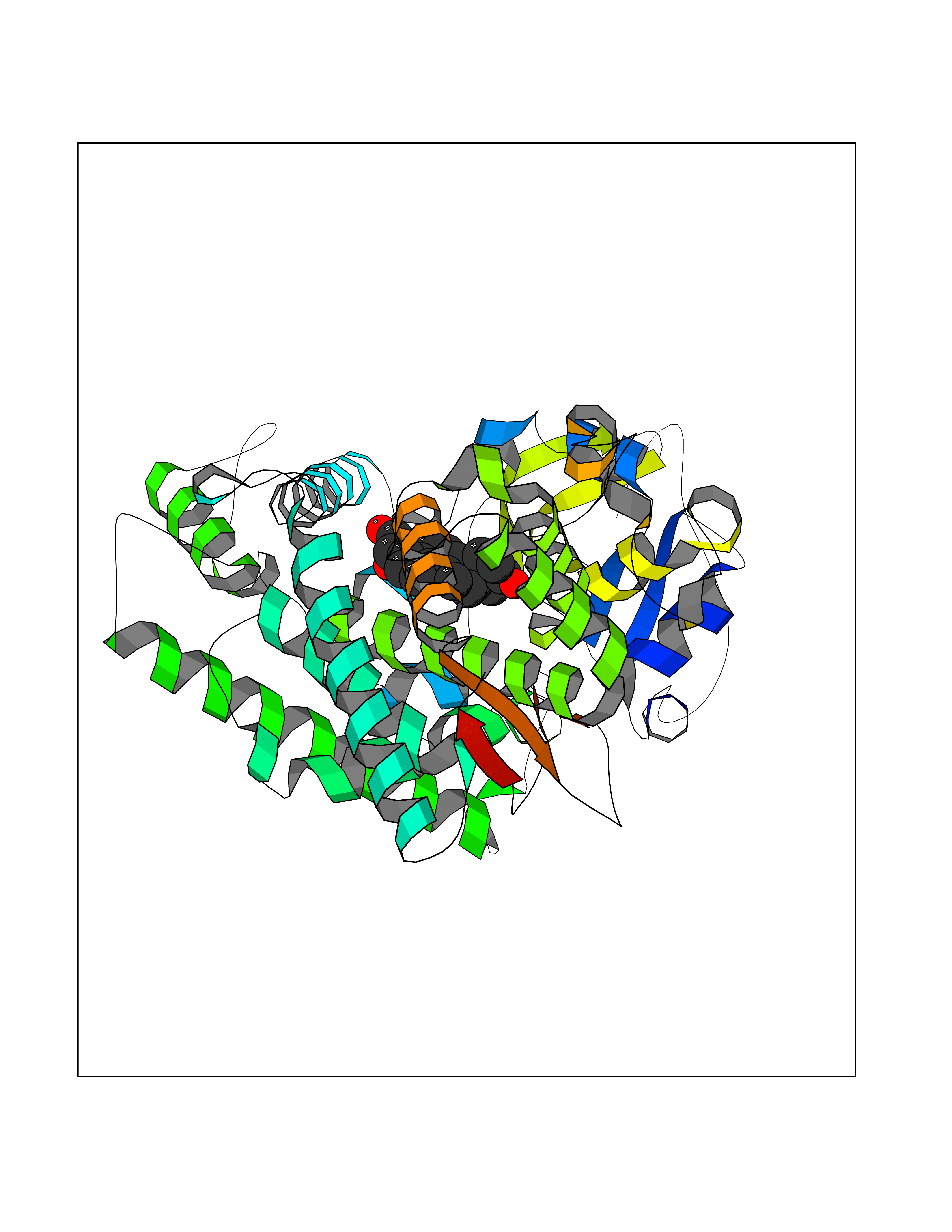 Molscript Map  Pymol Map 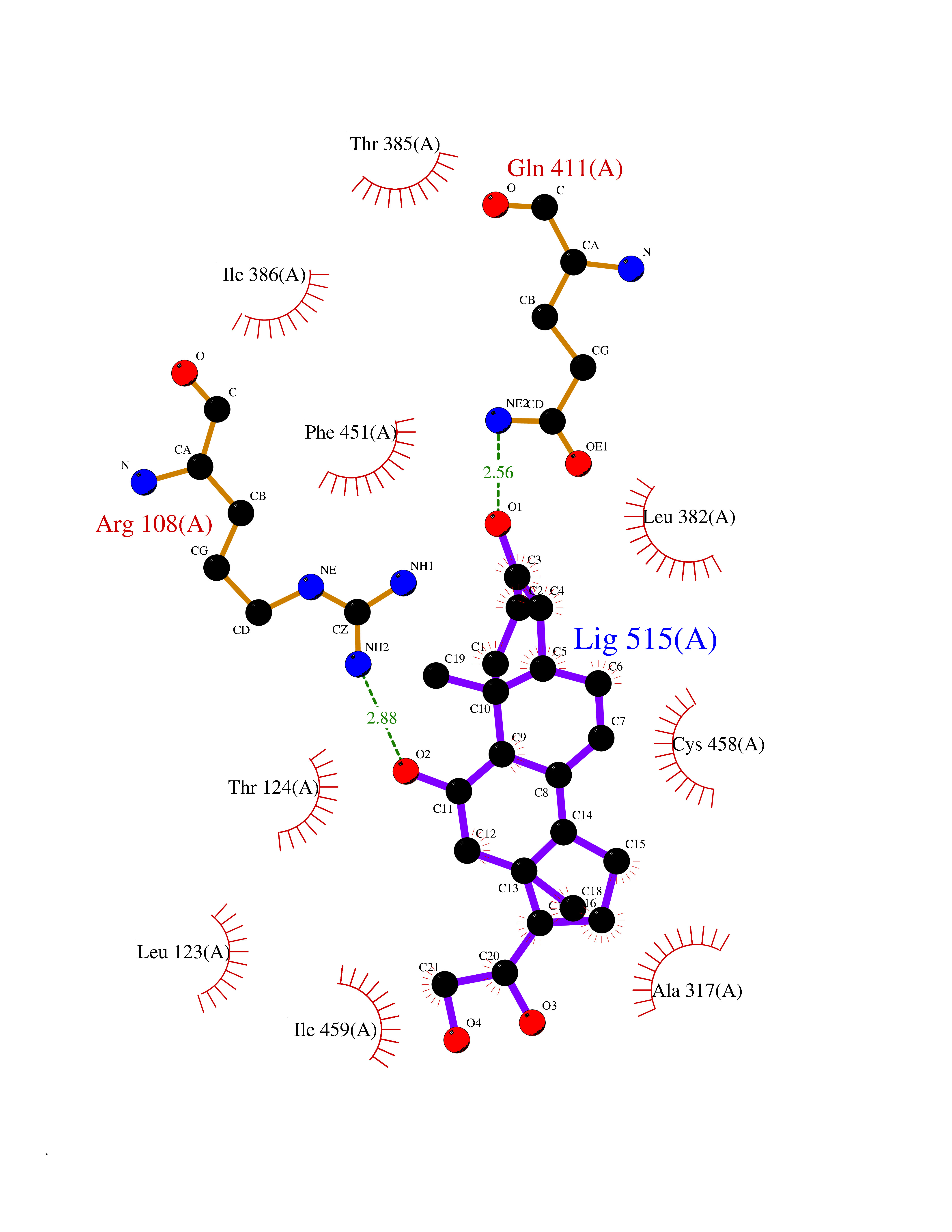 Ligplot Map 3D Binding mode Sequence RVPKGLKSPPEPWGWPLLGHVLTLGKNPHLALSRMSQRYGDVLQIRIGSTPVLVLSRLDTIRQALVRQGDDFKGRPDLYTSTLITDGQSLTFSTDSGPVWAARRRLAQNALNTFSIASDPASSSSCYLEEHVSKEAKALISRLQELMAGPGHFDPYNQVVVSVANVIGAMCFGQHFPESSDEMLSLVKNTHEFVETASSGNPLDFFPILRYLPNPALQRFKAFNQRFLWFLQKTVQEHYQDFDKNSVRDITGALFKHSKKGPRASGNLIPQEKIVNLVNDIFGAGFDTVTTAISWSLMYLVTKPEIQRKIQKELDTVIGRERRPRLSDRPQLPYLEAFILETFRHSSFLPFTIPHSTTRDTTLNGFYIPKKCCVFVNQWQVNHDPELWEDPSEFRPERFLTADGTAINKPLSEKMMLFGMGKRRCIGEVLAKWEIFLFLAILLQQLEFSVPPGVKVDLTPIYGLTMKHARCEHVQARRFS Hydrogen bonds contact Hydrophobic contact | ||||
| 6 | Endothelin-converting enzyme 1 (ECE1) | 3DWB | 8.36 | |
Target general information Gen name ECE1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms ECE-1 Protein family Peptidase M13 family Biochemical class Peptidase Function Converts big endothelin-1 to endothelin-1. Related diseases Hirschsprung disease, cardiac defects, and autonomic dysfunction (HCAD) [MIM:613870]: A disorder characterized by skip-lesions Hirschsprung disease, craniofacial abnormalities and other dysmorphic features, cardiac defects including ductus arteriosus, small subaortic ventricular septal defect, small atrial septal defect, and autonomic dysfunction. {ECO:0000269|PubMed:9915973}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07171 Interacts with P49760; A8MQ03; Q8IUG1; P60370; P60410 EC number EC 3.4.24.71 Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disease variant; Disulfide bond; Glycoprotein; Hirschsprung disease; Hydrolase; Membrane; Metal-binding; Metalloprotease; Phosphoprotein; Protease; Proteomics identification; Reference proteome; Signal-anchor; Transmembrane; Transmembrane helix; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 75247.9 Length 660 Aromaticity 0.12 Instability index 46.29 Isoelectric point 5.33 Charge (pH=7) -18.3 2D Binding mode Binding energy (Kcal/mol) -7.98 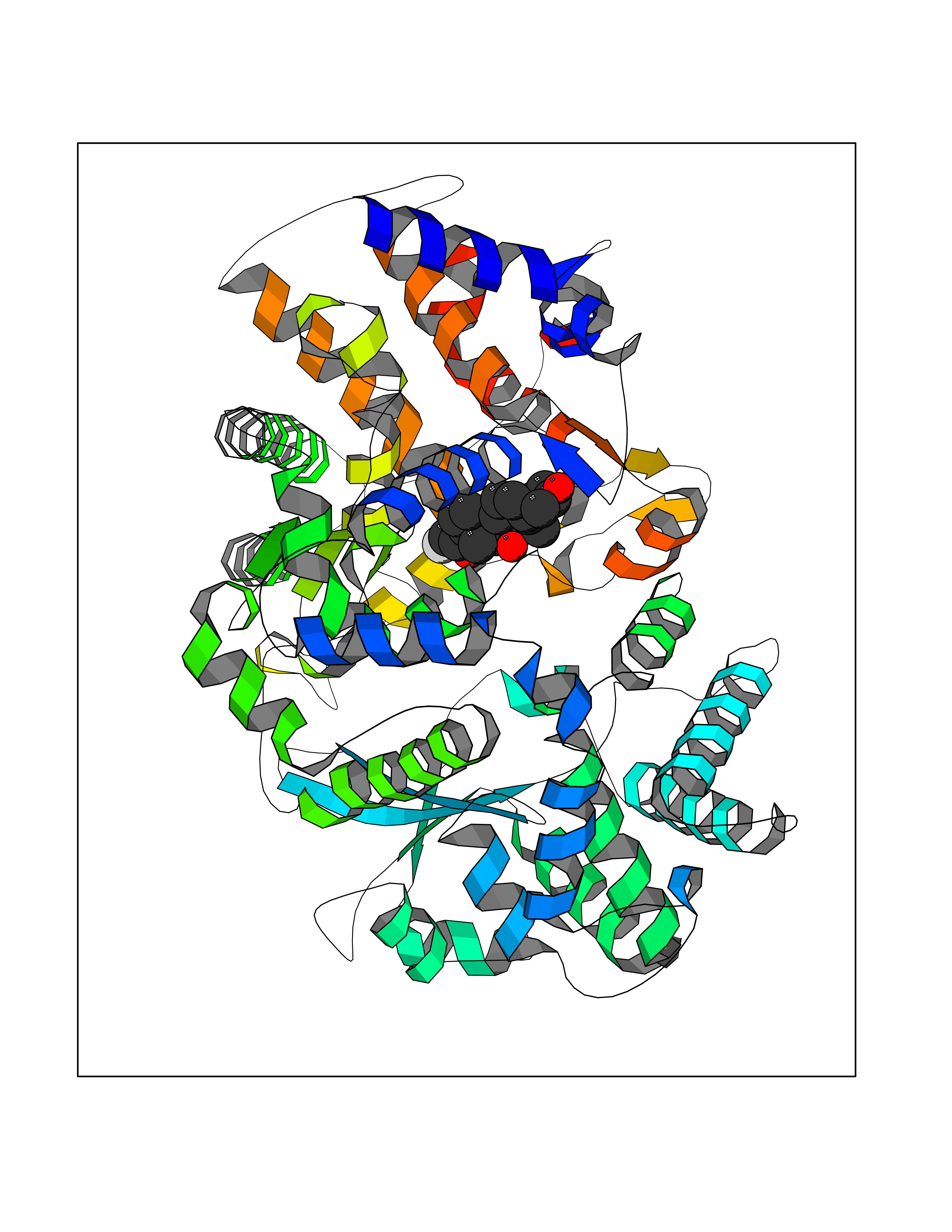 Molscript Map 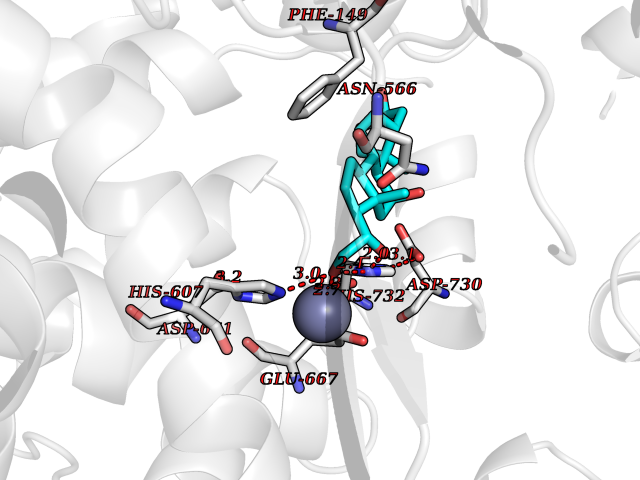 Pymol Map 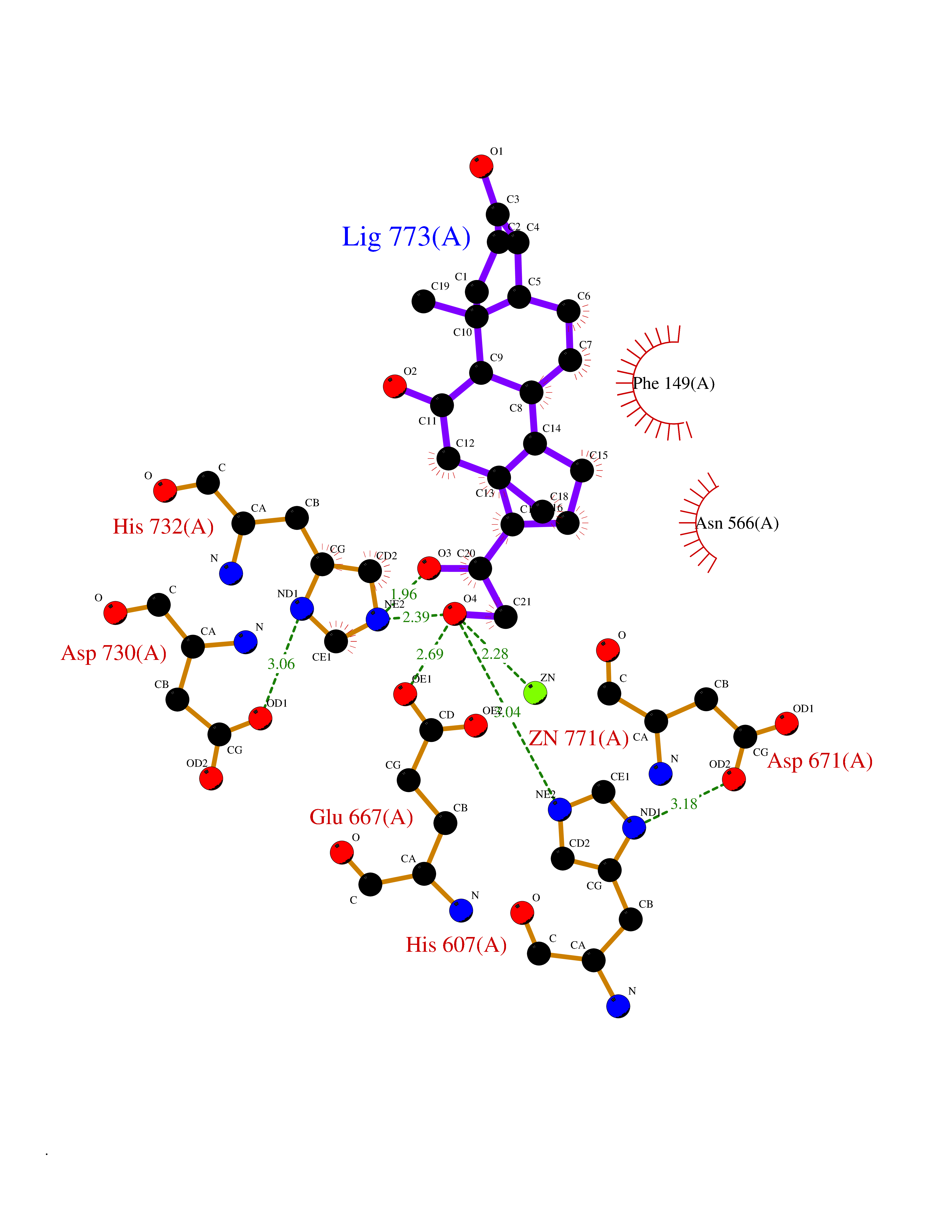 Ligplot Map 3D Binding mode Sequence SEACVSVTSSILSSMDPTVDPCHDFFSYACGGWIKANPVPDGHSRWGTFSNLWEHNQAIIKHLLENSTASVSEAERKAQVYYRACMNETRIEELRAKPLMELIERLGGWNITGPWAKDNFQDTLQVVTAHYRTSPFFSVYVSADSKNSNSNVIQVDQSGLGLPSRDYYLNKTENEKVLTGYLNYMVQLGKLLGGGDEEAIRPQMQQILDFETALANITIPQEKRRDEELIYHKVTAAELQTLAPAINWLPFLNTIFYPVEINESEPIVVYDKEYLEQISTLINTTDRCLLNNYMIWNLVRKTSSFLDQRFQDADEKFMEVMWKFCVSDTENNLGFALGPMFVKATFAEDSKSIATEIILEIKKAFEESLSTLKWMDEETRKSAKEKADAIYNMIGYPNFIMDPKELDKVFNDYTAVPDLYFENAMRFFNFSWRVTADQLRKAPNRDQWSMTPPMVNAYYSPTKNEIVFPAGILQAPFYTRSSPKALNFGGIGVVVGHELTHAFDDQGREYDKDGNLRPWWKNSSVEAFKRQTECMVEQYSNYSVNGEPVNGRHTLGENIADNGGLKAAYRAYQNWVKKNGAEHSLPTLGLTNNQLFFLGFAQVWCSVRTPESSHEGLITDPHSPSRFRVIGSLSNSKEFSEHFRCPPGSPMNPPHKCEVW Hydrogen bonds contact Hydrophobic contact | ||||
| 7 | Dopamine beta-hydroxylase | 4ZEL | 8.34 | |
Target general information Gen name DBH Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Copper type II ascorbate-dependent monooxygenase family Biochemical class Oxidoreductase Function Catalytic activity.Copper ion binding.Dopamine beta-monooxygenase activity.L-ascorbic acid binding. Related diseases Orthostatic hypotension 1 (ORTHYP1) [MIM:223360]: A form of orthostatic hypotension due to congenital dopamine beta-hydroxylase deficiency. Orthostatic hypotension, also known as postural hypotension, is a finding defined as a 20-mm Hg decrease in systolic pressure or a 10-mm Hg decrease in diastolic pressure occurring 3 minutes after a person has risen from supine to standing. Symptoms include dizziness, blurred vision, and sometimes syncope. ORTHYP1 is an autosomal recessive condition apparent from infancy or early childhood and characterized by low plasma and urinary levels of norepinephrine and epinephrine, and episodic hypoglycemia. {ECO:0000269|PubMed:11857564}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00126; DB06774; DB09130; DB05394; DB00822; DB00988; DB00968; DB00550 Interacts with P00352; P63010-2; Q04656; Q8WUW1; Q9UNS2; Q71DI3; P61978; Q9Y2M5; Q92876; P08727; Q14693; P0DPK4; Q6GQQ9-2; P27986-2; Q9ULX5; Q96D59; Q8N6K7-2; Q9GZS3; Q8IUW3; Q86WT6-2 EC number 1.14.17.1 Uniprot keywords 3D-structure; Catecholamine biosynthesis; Copper; Cytoplasmic vesicle; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; Membrane; Metal-binding; Monooxygenase; Oxidoreductase; Proteomics identification; Reference proteome; Secreted; Signal-anchor; Transmembrane; Transmembrane helix; Vitamin C Protein physicochemical properties Chain ID A,B Molecular weight (Da) 123694 Length 1094 Aromaticity 0.1 Instability index 51.85 Isoelectric point 5.84 Charge (pH=7) -24.5 2D Binding mode Binding energy (Kcal/mol) -9.4 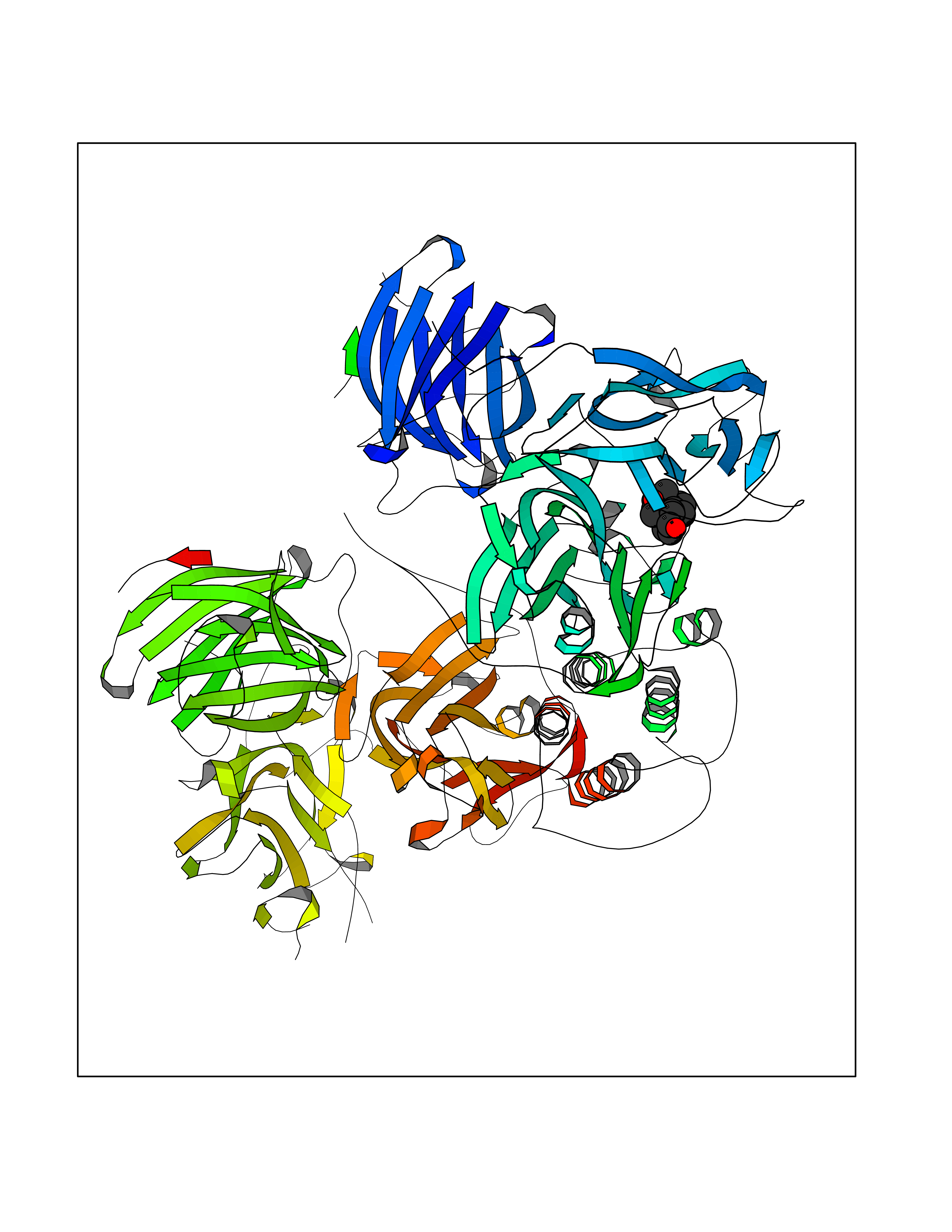 Molscript Map 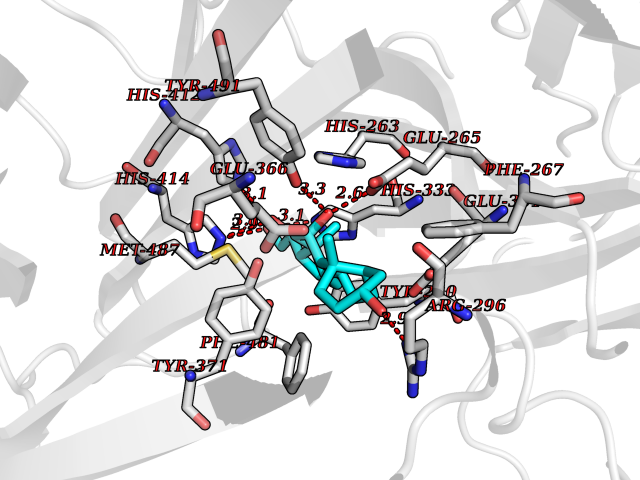 Pymol Map 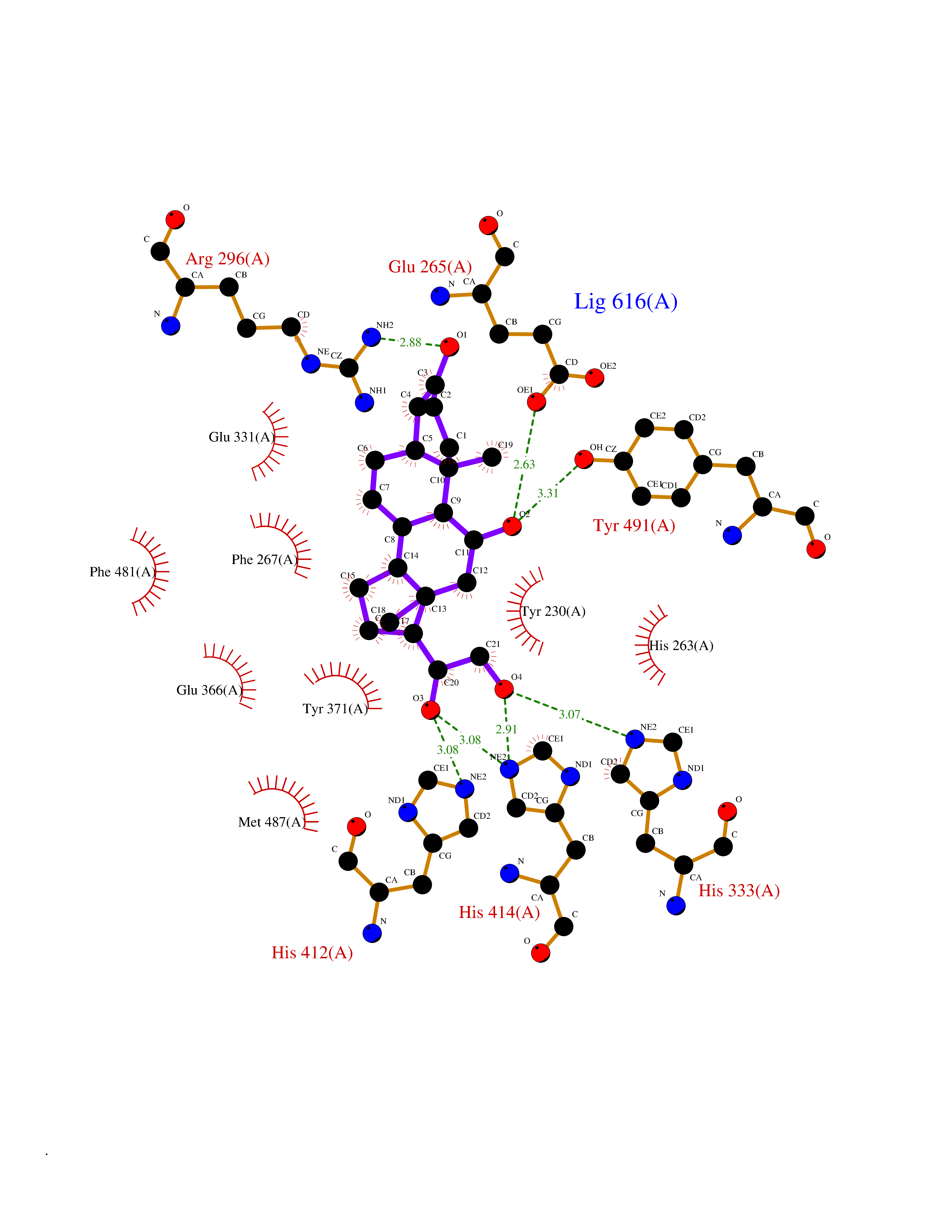 Ligplot Map 3D Binding mode Sequence PLPYHIPLDPEGSLELSWNVSYTQEAIHFQLLVRRLKAGVLFGMSDRGELENADLVVLWTDGDAYFADAWSDQKGQIHLDPQQDYQLLQVQRTPEGLTLLFKRPFGTCDPKDYLIEDGTVHLVYGILEEPFRSLEAINGSGLQMGLQRVQLLKPNIPEPELPSDACTMEVQAPNIQIPSQETTYWCYIKELPKGFSRHHIIKYEPIVTKGNEALVHHMEVFQCAPEMDSVPHFSGPCDSKMKPDRLNYCRHVLAAWALGAKAFYYPEEAGLAFGGPGSSRYLRLEVHYHNPLVIEGRNDSSGIRLYYTAKLRRFNAGIMELGLVYTPVMAIPPRETAFILTGYCTDKCTQLALPPSGIHIFASQLHTHLTGRKVVTVLVRDGREWEIVNQDNHYSPHFQEIRMLKKVVSVHPGDVLITSCTYNTEDRELATVGGFGILEEMCVNYVHYYPQTQLELCKSAVDAGFLQKYFHLINRFNNEDVCTCPQASVSQQFTSVPWNSFNRDVLKALYSFAPISMHCNKSSAVRFQGEWNLQPLPKVISTLEEPTVVSPLPYHIPLDPEGSLELSWNVSYTQEAIHFQLLVRRLKAGVLFGMSDRGELENADLVVLAYFADAWSDQKGQIHLDPQQDYQLLQVQRTPEGLTLLFKRPFGTCDPKDYLIEDGTVHLVYGILEEPFRSLEAINGSGLQMGLQRVQLLKPNIPEPELPSDACTMEVQAPNIQIPSQETTYWCYIKELPKGFSRHHIIKYEPIVTKGNEALVHHMEVFQCAPEVPHFSGPCDSKMLNYCRHVLAAWALGAKAFYYPEEAGLAFGGPGSSRYLRLEVHYHNPLVIEGRNDSSGIRLYYTAKLRRFNAGIMELGLVYTPVMAIPPRETAFILTGYCTDKCTQLALPPSGIHIFASQLHTHLTGRKVVTVLVRDGREWEIVNQDNHYSPHFQEIRMLKKVVSVHPGDVLITSCTYNTEDRELATVGGFGILEEMCVNYVHYYPQTQLELCKSAVDAGFLQKYFHLINRFNNEDVCTCPQASVSQQFTSVPWNSFNRDVLKALYSFAPISMHCNKSSAVRFQGEWNLQPLPKVISTLEEPTPQCVVSIGG Hydrogen bonds contact Hydrophobic contact | ||||
| 8 | Dopamine beta-hydroxylase | 4ZEL | 8.26 | |
Target general information Gen name DBH Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Copper type II ascorbate-dependent monooxygenase family Biochemical class Oxidoreductase Function Catalytic activity.Copper ion binding.Dopamine beta-monooxygenase activity.L-ascorbic acid binding. Related diseases Orthostatic hypotension 1 (ORTHYP1) [MIM:223360]: A form of orthostatic hypotension due to congenital dopamine beta-hydroxylase deficiency. Orthostatic hypotension, also known as postural hypotension, is a finding defined as a 20-mm Hg decrease in systolic pressure or a 10-mm Hg decrease in diastolic pressure occurring 3 minutes after a person has risen from supine to standing. Symptoms include dizziness, blurred vision, and sometimes syncope. ORTHYP1 is an autosomal recessive condition apparent from infancy or early childhood and characterized by low plasma and urinary levels of norepinephrine and epinephrine, and episodic hypoglycemia. {ECO:0000269|PubMed:11857564}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00126; DB06774; DB09130; DB05394; DB00822; DB00988; DB00968; DB00550 Interacts with P00352; P63010-2; Q04656; Q8WUW1; Q9UNS2; Q71DI3; P61978; Q9Y2M5; Q92876; P08727; Q14693; P0DPK4; Q6GQQ9-2; P27986-2; Q9ULX5; Q96D59; Q8N6K7-2; Q9GZS3; Q8IUW3; Q86WT6-2 EC number 1.14.17.1 Uniprot keywords 3D-structure; Catecholamine biosynthesis; Copper; Cytoplasmic vesicle; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; Membrane; Metal-binding; Monooxygenase; Oxidoreductase; Proteomics identification; Reference proteome; Secreted; Signal-anchor; Transmembrane; Transmembrane helix; Vitamin C Protein physicochemical properties Chain ID A,B Molecular weight (Da) 123694 Length 1094 Aromaticity 0.1 Instability index 51.85 Isoelectric point 5.84 Charge (pH=7) -24.5 2D Binding mode Binding energy (Kcal/mol) -8.28 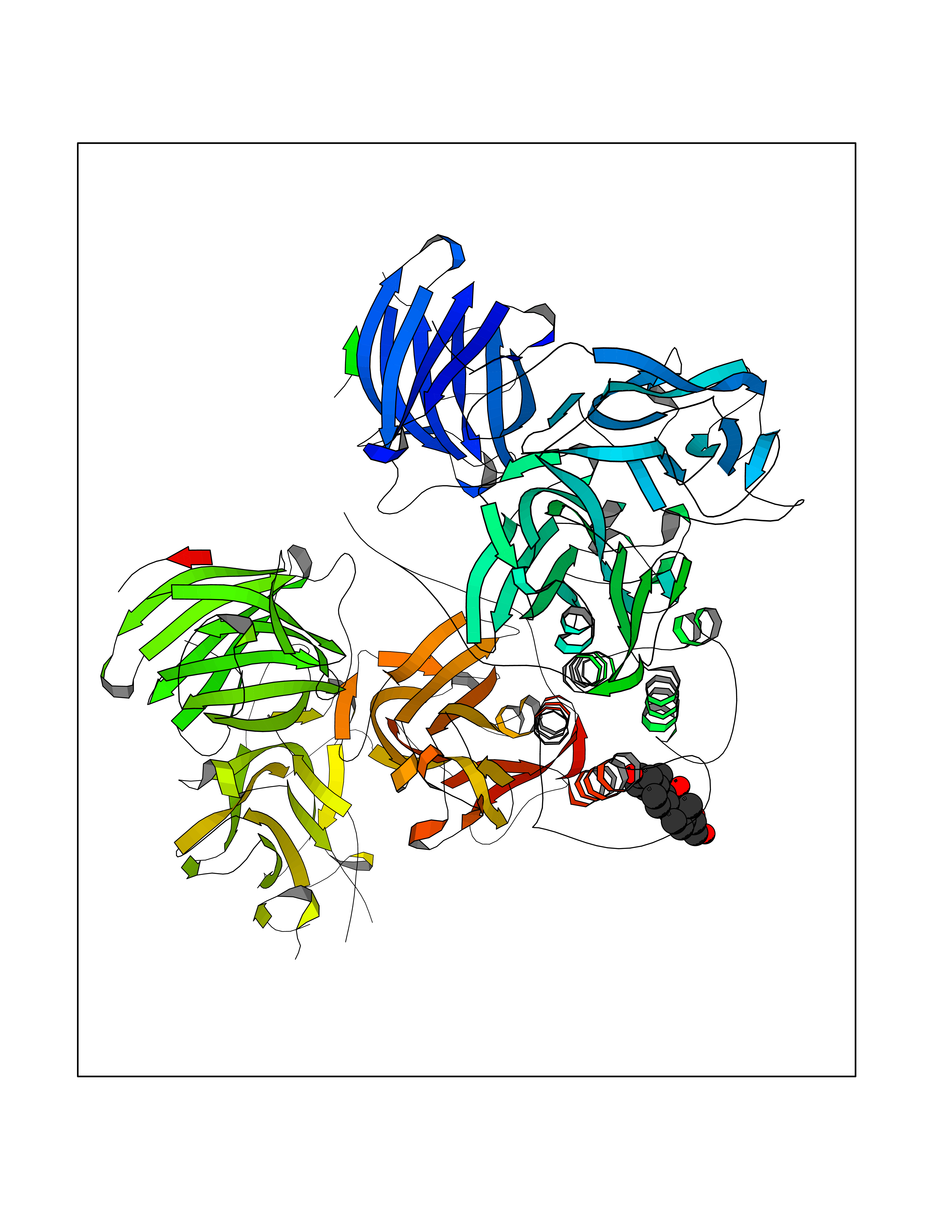 Molscript Map 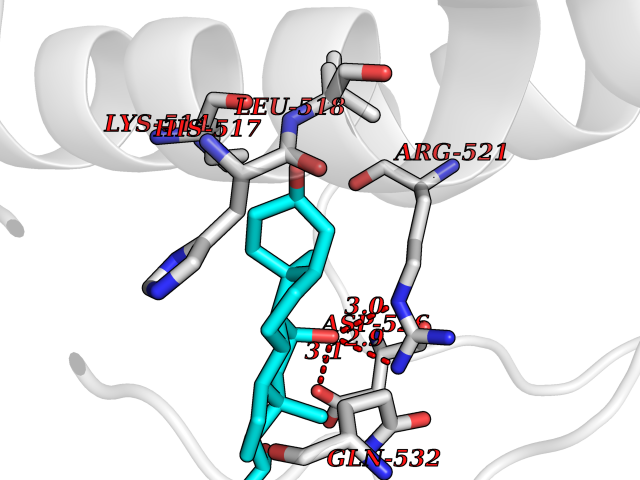 Pymol Map 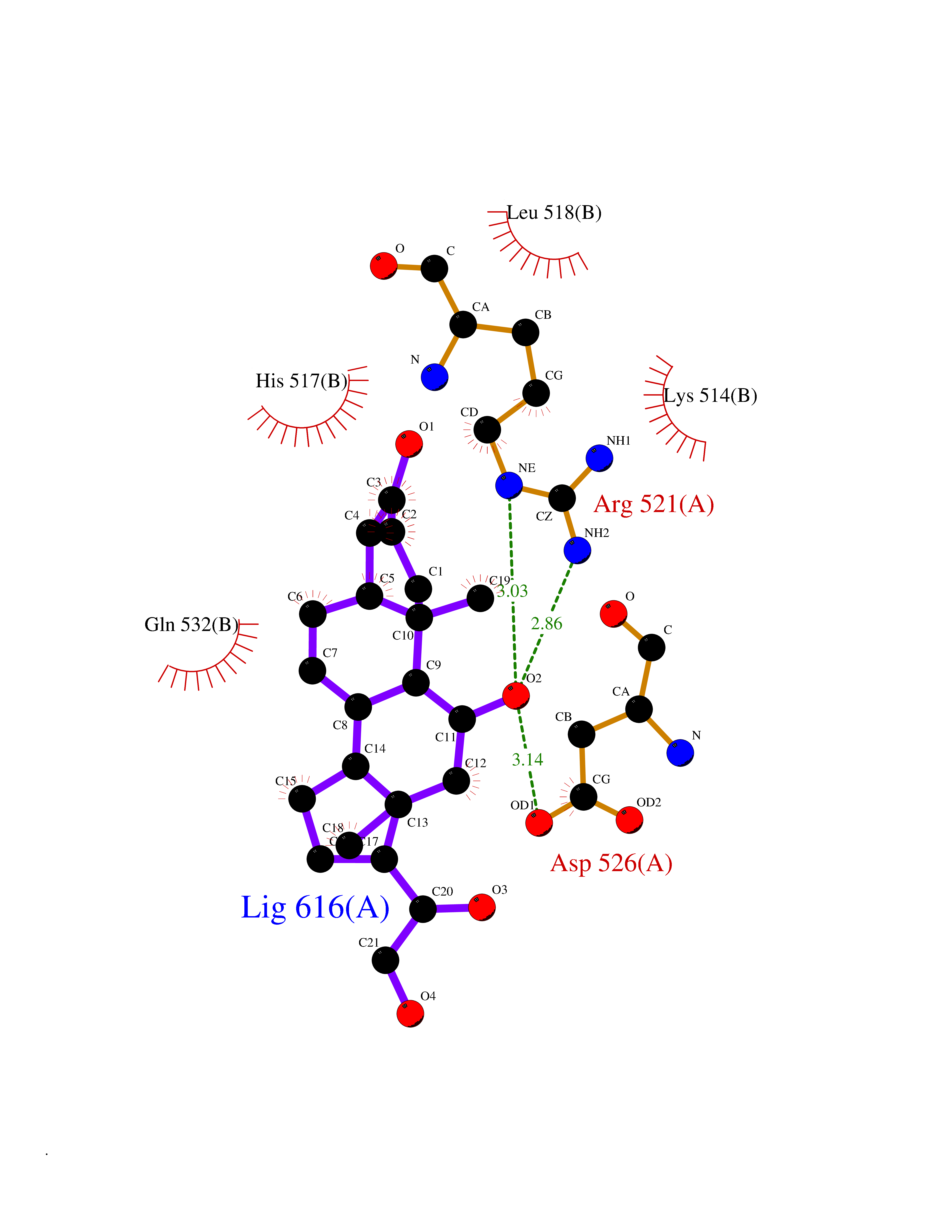 Ligplot Map 3D Binding mode Sequence PLPYHIPLDPEGSLELSWNVSYTQEAIHFQLLVRRLKAGVLFGMSDRGELENADLVVLWTDGDAYFADAWSDQKGQIHLDPQQDYQLLQVQRTPEGLTLLFKRPFGTCDPKDYLIEDGTVHLVYGILEEPFRSLEAINGSGLQMGLQRVQLLKPNIPEPELPSDACTMEVQAPNIQIPSQETTYWCYIKELPKGFSRHHIIKYEPIVTKGNEALVHHMEVFQCAPEMDSVPHFSGPCDSKMKPDRLNYCRHVLAAWALGAKAFYYPEEAGLAFGGPGSSRYLRLEVHYHNPLVIEGRNDSSGIRLYYTAKLRRFNAGIMELGLVYTPVMAIPPRETAFILTGYCTDKCTQLALPPSGIHIFASQLHTHLTGRKVVTVLVRDGREWEIVNQDNHYSPHFQEIRMLKKVVSVHPGDVLITSCTYNTEDRELATVGGFGILEEMCVNYVHYYPQTQLELCKSAVDAGFLQKYFHLINRFNNEDVCTCPQASVSQQFTSVPWNSFNRDVLKALYSFAPISMHCNKSSAVRFQGEWNLQPLPKVISTLEEPTVVSPLPYHIPLDPEGSLELSWNVSYTQEAIHFQLLVRRLKAGVLFGMSDRGELENADLVVLAYFADAWSDQKGQIHLDPQQDYQLLQVQRTPEGLTLLFKRPFGTCDPKDYLIEDGTVHLVYGILEEPFRSLEAINGSGLQMGLQRVQLLKPNIPEPELPSDACTMEVQAPNIQIPSQETTYWCYIKELPKGFSRHHIIKYEPIVTKGNEALVHHMEVFQCAPEVPHFSGPCDSKMLNYCRHVLAAWALGAKAFYYPEEAGLAFGGPGSSRYLRLEVHYHNPLVIEGRNDSSGIRLYYTAKLRRFNAGIMELGLVYTPVMAIPPRETAFILTGYCTDKCTQLALPPSGIHIFASQLHTHLTGRKVVTVLVRDGREWEIVNQDNHYSPHFQEIRMLKKVVSVHPGDVLITSCTYNTEDRELATVGGFGILEEMCVNYVHYYPQTQLELCKSAVDAGFLQKYFHLINRFNNEDVCTCPQASVSQQFTSVPWNSFNRDVLKALYSFAPISMHCNKSSAVRFQGEWNLQPLPKVISTLEEPTPQCVVSIGG Hydrogen bonds contact Hydrophobic contact | ||||
| 9 | Vitamin D3 receptor (VDR) | 3B0T | 8.25 | |
Target general information Gen name VDR Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Vitamin D(3) receptor; Nuclear vitamin D receptor; Nuclear receptor subfamily 1 group I member 1; NR1I1; 1,25-dihydroxyvitamin D3 receptor Protein family Nuclear hormone receptor family, NR1 subfamily Biochemical class Nuclear hormone receptor Function Enters the nucleus upon vitamin D3 binding where it forms heterodimers with the retinoid X receptor/RXR. The VDR-RXR heterodimers bind to specific response elements on DNA and activate the transcription of vitamin D3-responsive target genes. Plays a central role in calcium homeostasis. Nuclear receptor for calcitriol, the active form of vitamin D3 which mediates the action of this vitamin on cells. Related diseases Rickets vitamin D-dependent 2A (VDDR2A) [MIM:277440]: A disorder of vitamin D metabolism resulting in severe rickets, hypocalcemia and secondary hyperparathyroidism. Most patients have total alopecia in addition to rickets. {ECO:0000269|PubMed:1652893, ECO:0000269|PubMed:17970811, ECO:0000269|PubMed:2177843, ECO:0000269|PubMed:2849209, ECO:0000269|PubMed:28698609, ECO:0000269|PubMed:7828346, ECO:0000269|PubMed:8106618, ECO:0000269|PubMed:8381803, ECO:0000269|PubMed:8392085, ECO:0000269|PubMed:8675579, ECO:0000269|PubMed:8961271, ECO:0000269|PubMed:9005998}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07530; DB08742; DB01436; DB04891; DB00146; DB02300; DB00136; DB00169; DB04540; DB05024; DB11672; DB14635; DB01070; DB06410; DB05295; DB06194; DB00153; DB04796; DB03451; DB00910; DB04258; DB11094 Interacts with P35222; Q09472; Q15648; P50222; Q15788; P26045; P19793; Q13573; Q13501; P04637; Q15645; Q9JLI4; P28700; X5D778; Q96HA8; Q01804; Q96S38; P48443 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; Disease variant; DNA-binding; Metal-binding; Nucleus; Proteomics identification; Receptor; Reference proteome; Transcription; Transcription regulation; Ubl conjugation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A Molecular weight (Da) 28781 Length 254 Aromaticity 0.07 Instability index 47.69 Isoelectric point 6.15 Charge (pH=7) -3.44 2D Binding mode Binding energy (Kcal/mol) -10.29 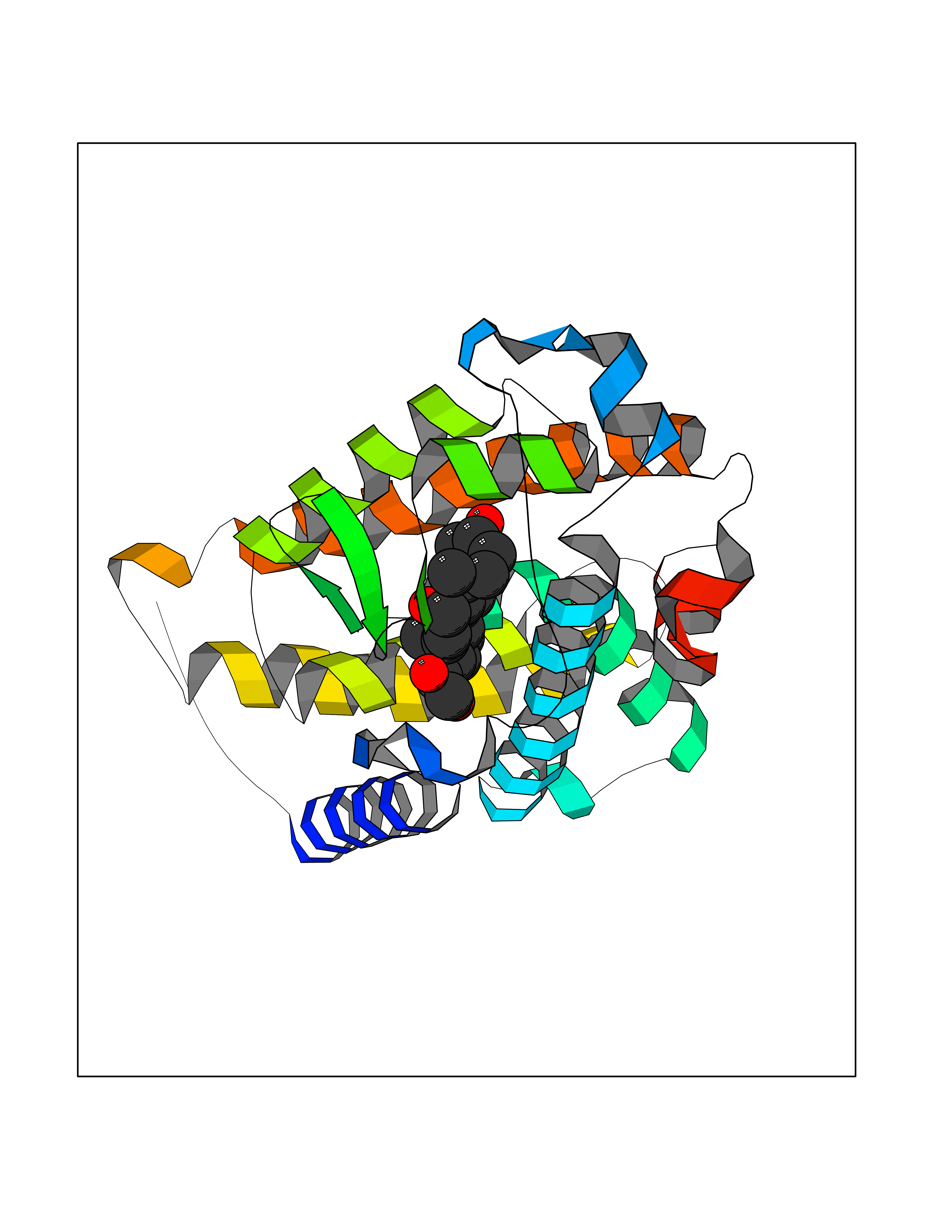 Molscript Map 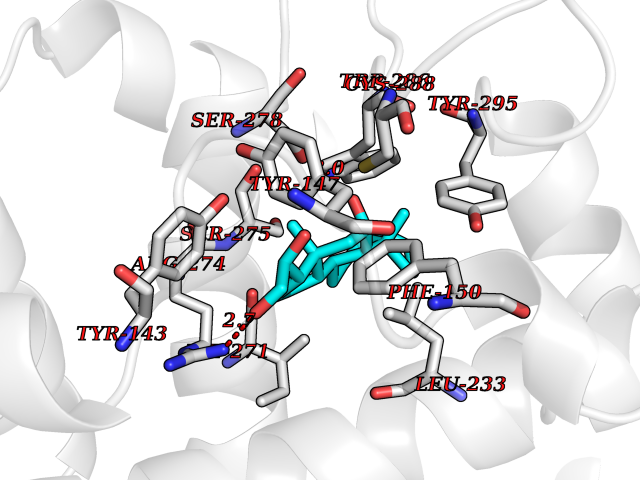 Pymol Map 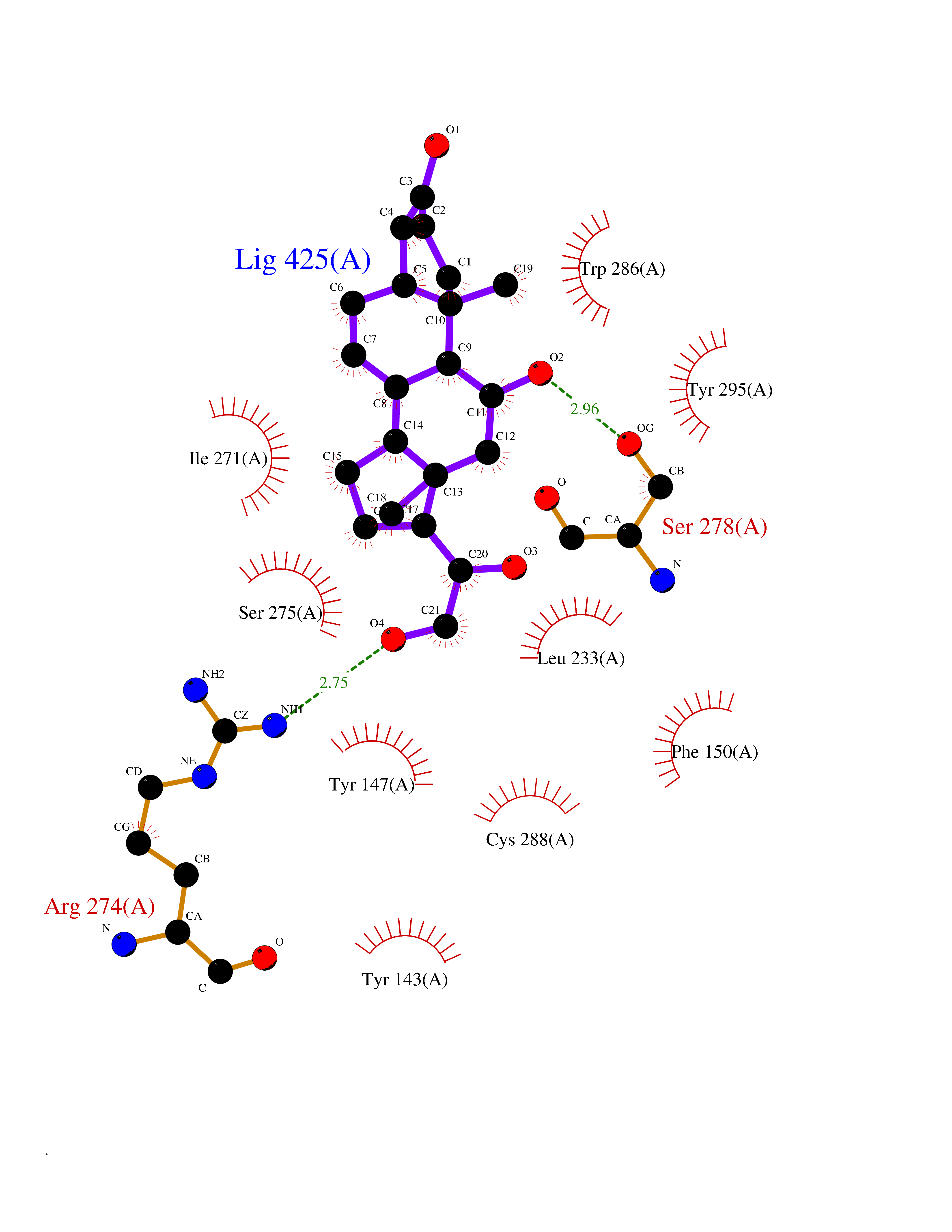 Ligplot Map 3D Binding mode Sequence ALRPKLSEEQQRIIAILLDAHHKTYDPTYSDFCQFRPPVRVNDGGGSVTLELSQLSMLPHLADLVSYSIQKVIGFAKMIPGFRDLTSEDQIVLLKSSAIEVIMLRSNESFTMDDMSWTCGNQDYKYRVSDVTKAGHSLELIEPLIKFQVGLKKLNLHEEEHVLLMAICIVSPDRPGVQDAALIEAIQDRLSNTLQTYIRCRHPPPGSHLLYAKMIQKLADLRSLNEEHSKQYRCLSFQPECSMKLTPLVLEVFG Hydrogen bonds contact Hydrophobic contact | ||||
| 10 | Steroid 21-hydroxylase | 4Y8W | 8.21 | |
Target general information Gen name CYP21A2 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms CYP21;CYP21B Protein family Cytochrome P450 family Biochemical class Oxidoreductase Function Heme binding.Iron ion binding.Steroid 21-monooxygenase activity.Steroid binding.Steroid hydroxylase activity. Related diseases Adrenal hyperplasia 3 (AH3) [MIM:201910]: A form of congenital adrenal hyperplasia, a common recessive disease due to defective synthesis of cortisol. Congenital adrenal hyperplasia is characterized by androgen excess leading to ambiguous genitalia in affected females, rapid somatic growth during childhood in both sexes with premature closure of the epiphyses and short adult stature. Four clinical types: 'salt wasting' (SW, the most severe type), 'simple virilizing' (SV, less severely affected patients), with normal aldosterone biosynthesis, 'non-classic form' or late-onset (NC or LOAH) and 'cryptic' (asymptomatic). {ECO:0000269|PubMed:10051010, ECO:0000269|PubMed:10094562, ECO:0000269|PubMed:10198222, ECO:0000269|PubMed:10364682, ECO:0000269|PubMed:10391209, ECO:0000269|PubMed:10408778, ECO:0000269|PubMed:10408786, ECO:0000269|PubMed:10443693, ECO:0000269|PubMed:10496074, ECO:0000269|PubMed:10720040, ECO:0000269|PubMed:11232002, ECO:0000269|PubMed:11598371, ECO:0000269|PubMed:11600539, ECO:0000269|PubMed:11746135, ECO:0000269|PubMed:12213891, ECO:0000269|PubMed:12222711, ECO:0000269|PubMed:12788866, ECO:0000269|PubMed:12887291, ECO:0000269|PubMed:12915679, ECO:0000269|PubMed:1406699, ECO:0000269|PubMed:1406709, ECO:0000269|PubMed:14676460, ECO:0000269|PubMed:14715874, ECO:0000269|PubMed:1496017, ECO:0000269|PubMed:15110320, ECO:0000269|PubMed:15126570, ECO:0000269|PubMed:16046588, ECO:0000269|PubMed:1644925, ECO:0000269|PubMed:16984992, ECO:0000269|PubMed:18319307, ECO:0000269|PubMed:18381579, ECO:0000269|PubMed:18445671, ECO:0000269|PubMed:1864962, ECO:0000269|PubMed:1937474, ECO:0000269|PubMed:20080860, ECO:0000269|PubMed:2072928, ECO:0000269|PubMed:21169732, ECO:0000269|PubMed:22014889, ECO:0000269|PubMed:2303461, ECO:0000269|PubMed:27721825, ECO:0000269|PubMed:29328376, ECO:0000269|PubMed:3038528, ECO:0000269|PubMed:3257825, ECO:0000269|PubMed:3260007, ECO:0000269|PubMed:3267225, ECO:0000269|PubMed:3497399, ECO:0000269|PubMed:3871526, ECO:0000269|PubMed:7749410, ECO:0000269|PubMed:8478006, ECO:0000269|PubMed:8485582, ECO:0000269|PubMed:8989258, ECO:0000269|PubMed:9067760, ECO:0000269|PubMed:9187661, ECO:0000269|PubMed:9497336}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01026; DB05667 Interacts with NA EC number 1.14.14.16 Uniprot keywords 3D-structure; Alternative splicing; Congenital adrenal hyperplasia; Disease variant; Endoplasmic reticulum; Heme; Iron; Lipid metabolism; Lipid-binding; Membrane; Metal-binding; Microsome; Monooxygenase; Oxidoreductase; Proteomics identification; Reference proteome; Steroid-binding; Steroidogenesis Protein physicochemical properties Chain ID A,B,C Molecular weight (Da) 50326.2 Length 442 Aromaticity 0.08 Instability index 50.06 Isoelectric point 7.79 Charge (pH=7) 2.07 2D Binding mode Binding energy (Kcal/mol) -9.67 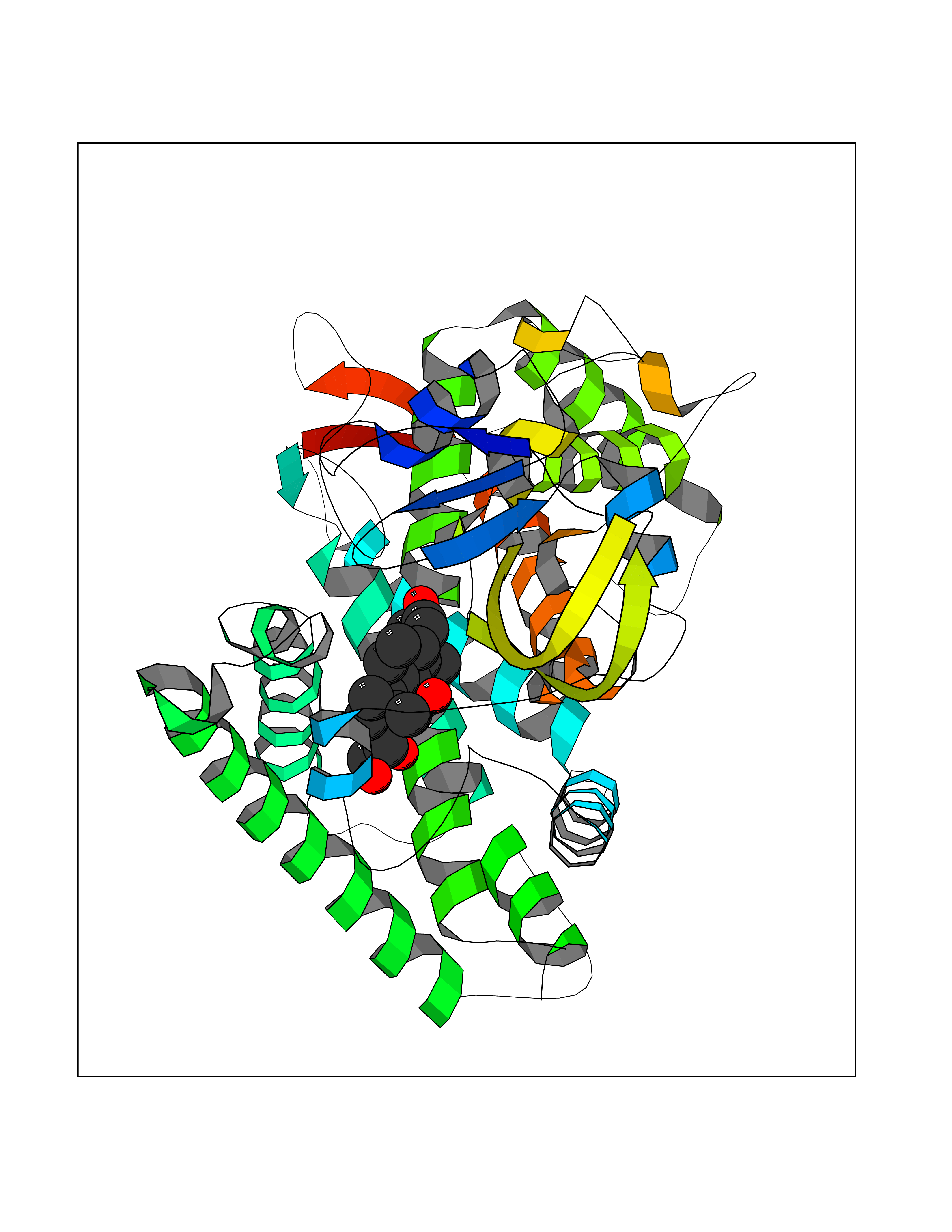 Molscript Map 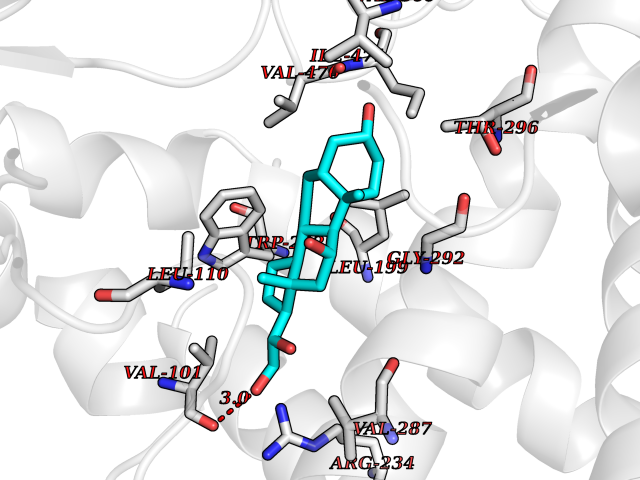 Pymol Map 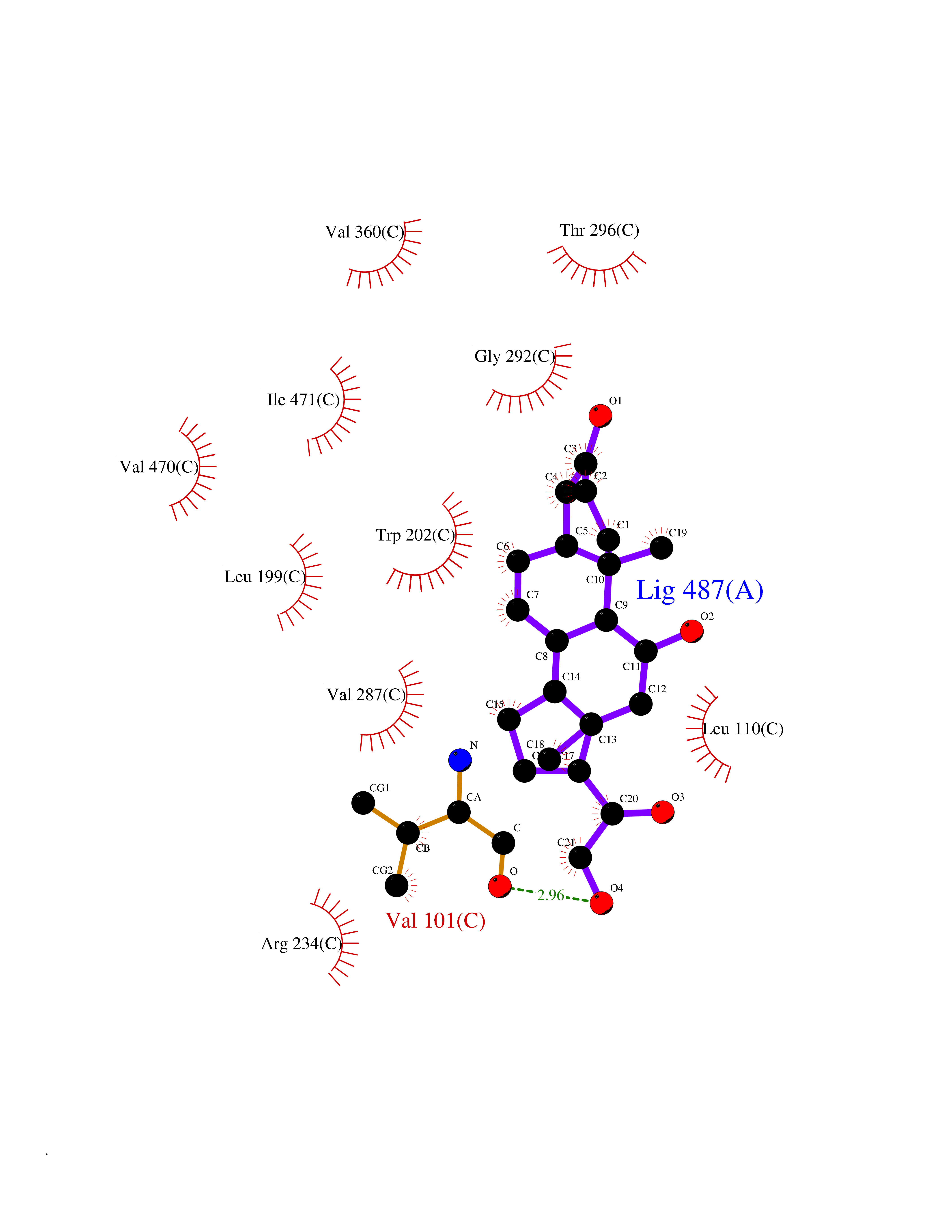 Ligplot Map 3D Binding mode Sequence KLPPLAPGFLHLLQPDLPIYLLGLTQKFGPIYRLHLGLQDVVVLNSKRTIEEAMVKKWADFAGRPEPLTYKLVSRNYPDLSLGDYSLLWKAHKKLTRSALLLGIRDSMEPVVEQLTQEFCERMRAQPGTPVAIEEEFSLLTCSIICYLTFGDKIKDDNLMPAYYKCIQEVLKTWSHWSIQIVDVIPFLRFFPNPGLRRLKQAIEKRDHIVEMQLRQHKESLVAGQWRDMMDYMLQGVAGQLLEGHVHMAAVDLLIGGTETTANTLSWAVVFLLHHPEIQQRLQEELDHESRVPYKDRARLPLLNATIAEVLRLRPVVPLALPHRTTRPSSISGYDIPEGTVIIPNLQGAHLDETVWERPHEFWPDRFLEPGKNSRALAFGCGARVCLGEPLARLELFVVLTRLLQAFTLLPSGDALPSLQPLPHCSVILKMQPFQVRLQPRG Hydrogen bonds contact Hydrophobic contact | ||||
| 11 | FkbI | 1R2J | 8.17 | |
Target general information Gen name fkbI Organism Streptomyces hygroscopicus subsp. ascomyceticus Uniprot ID TTD ID NA Synonyms NA Protein family Acyl-CoA dehydrogenase family Biochemical class Oxidoreductase Function Flavin adenine dinucleotide binding.Oxidoreductase activity, acting on the CH-CH group of donors. Related diseases Thiamine metabolism dysfunction syndrome 5, episodic encephalopathy type (THMD5) [MIM:614458]: An autosomal recessive metabolic disorder due to an inborn error of thiamine metabolism. The phenotype is highly variable, but in general, affected individuals have onset in early childhood of acute encephalopathic episodes associated with increased serum and CSF lactate. These episodes result in progressive neurologic dysfunction manifest as gait disturbances, ataxia, dystonia, and spasticity, which in some cases may result in loss of ability to walk. Cognitive function is usually preserved, although mildly delayed development has been reported. These episodes are usually associated with infection and metabolic decompensation. Some patients may have recovery of some neurologic deficits. {ECO:0000269|PubMed:22152682}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03147 Interacts with NA EC number NA Uniprot keywords 3D-structure; FAD; Flavoprotein; Nucleotide-binding; Oxidoreductase Protein physicochemical properties Chain ID A Molecular weight (Da) 36670.3 Length 353 Aromaticity 0.04 Instability index 22.05 Isoelectric point 6.12 Charge (pH=7) -5.04 2D Binding mode Binding energy (Kcal/mol) -8.94 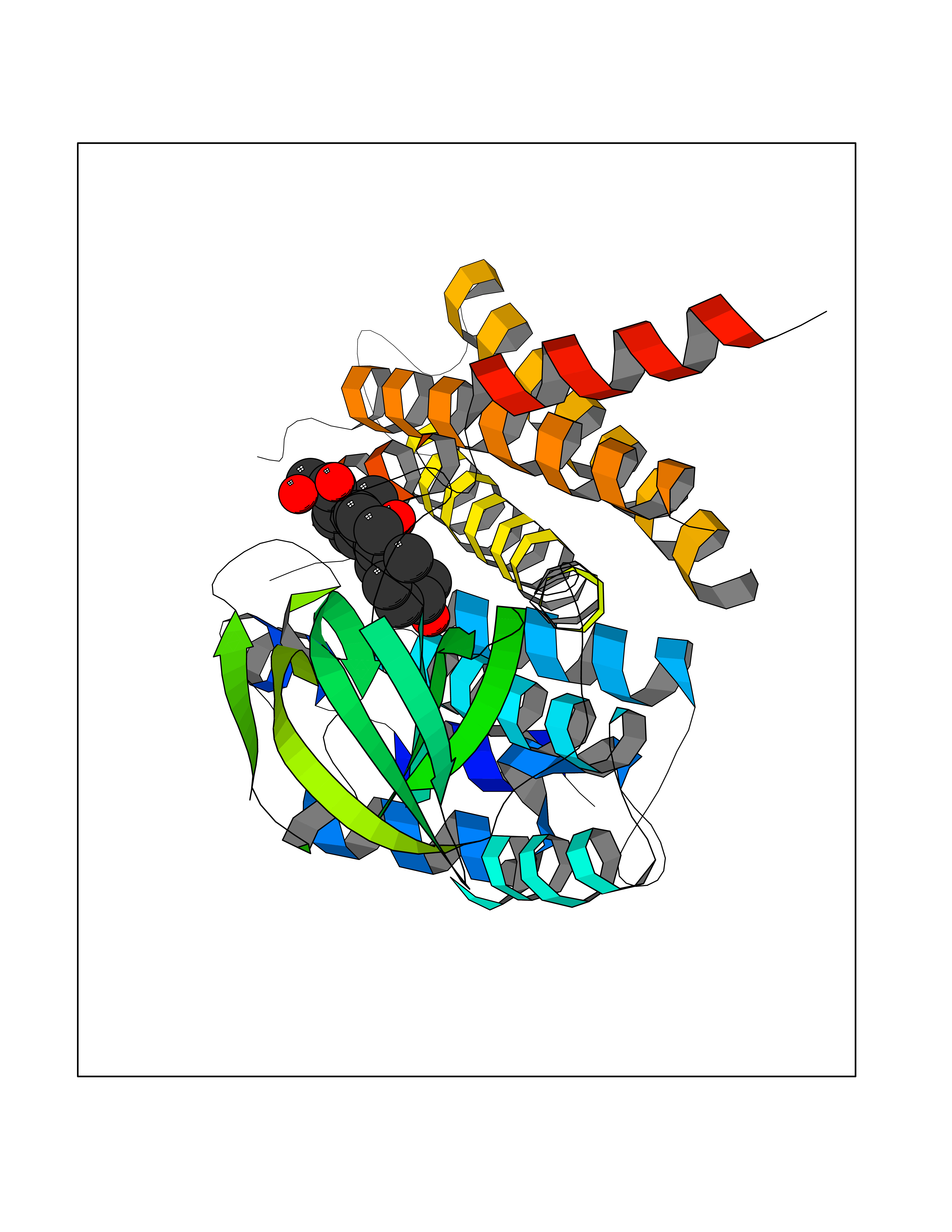 Molscript Map 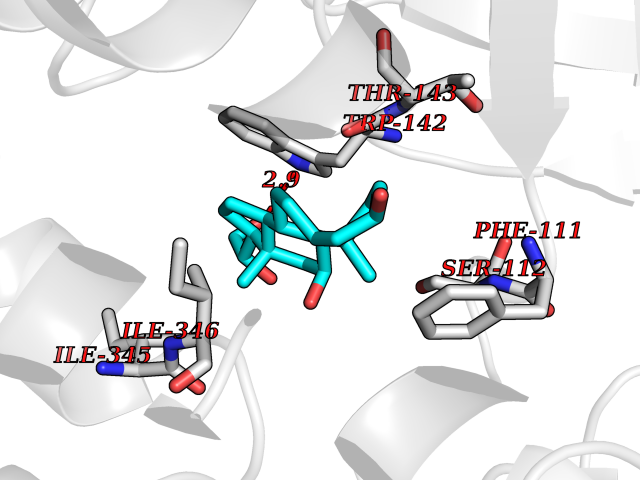 Pymol Map 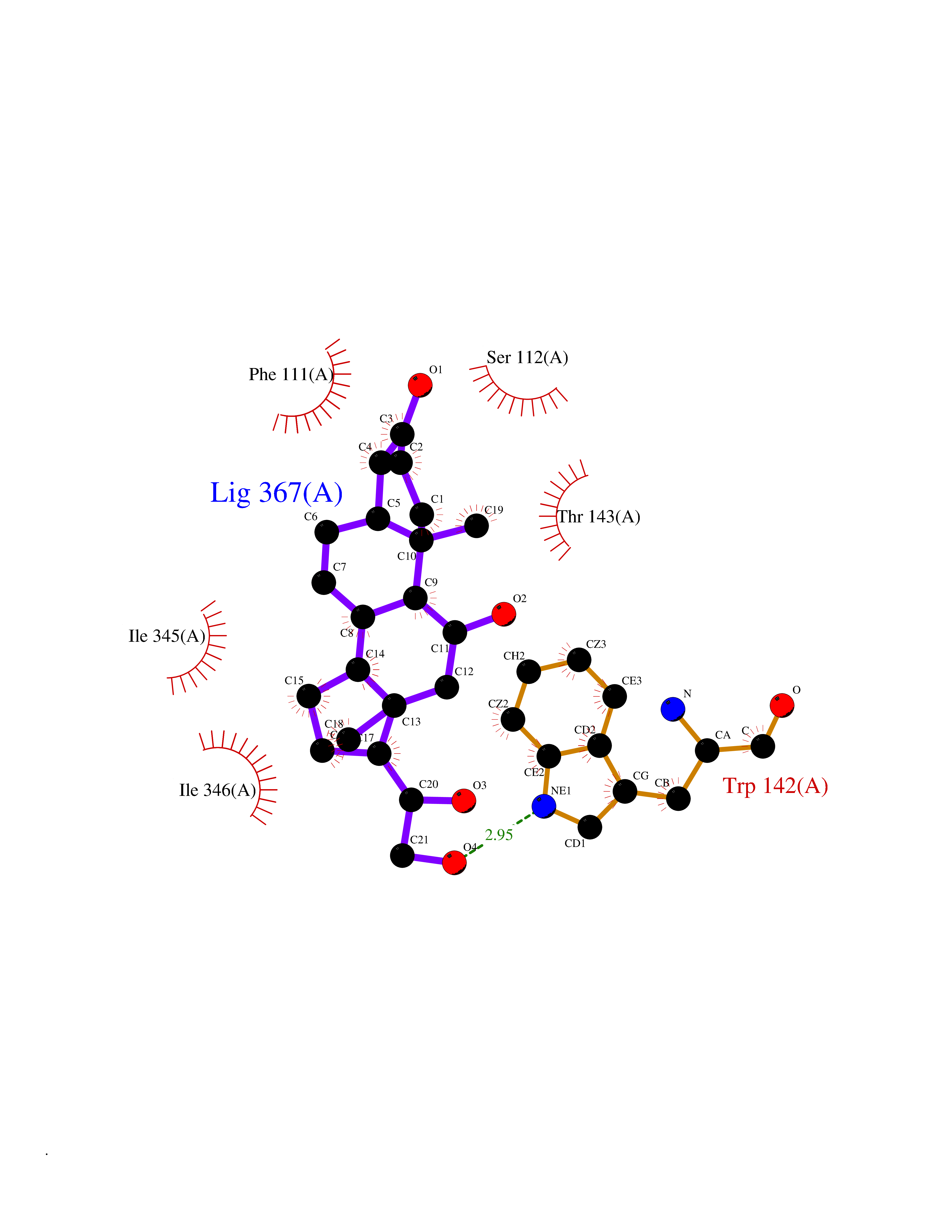 Ligplot Map 3D Binding mode Sequence ERDALLTDLVGDRAAEWDTSGELPRDLLVRLGADGLLCAEVAAEHGGLGLGSRENGEFTAHVGSLCSSLRSVMTSQGMAAWTVQRLGDAGQRATFLKELTSGLAAVGFSERQAGSDLSAMRTRVRLDGDTAVVDGHKVWTTAAAYADHLVVFGLQEDGSGAVVVVPADTPGVRVERVPKPSGCRAAGHADLHLDQVRVPAGAVLAGSGASLPMLVAASLAYGRKSVAWGCVGILRACRTAAVAHARTREQFGRPLGDHQLVAGHIADLWTAEQIAARVCEYASDHMVPATILAKHVAAERAAAGAATAAQVLASAGAGHVVERAYRDAKLMEIIEGSSEMCRVMLAQHALALP Hydrogen bonds contact Hydrophobic contact | ||||
| 12 | Folate receptor alpha (FOLR1) | 4LRH | 8.15 | |
Target general information Gen name FOLR1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Ovarian tumorassociated antigen MOv18; KB cells FBP; Folate receptor, adult; Folate receptor 1; FRalpha; FOLR1; Adult folatebinding protein Protein family Folate receptor family Biochemical class Folate receptor Function Binds to folate and reduced folic acid derivatives and mediates delivery of 5-methyltetrahydrofolate and folate analogs into the interior of cells. Has high affinity for folate and folic acid analogs at neutral pH. Exposure to slightly acidic pHafter receptor endocytosis triggers a conformation change that strongly reduces its affinity for folates and mediates their release. Required for normal embryonic development and normal cell proliferation. Related diseases Neurodegeneration due to cerebral folate transport deficiency (NCFTD) [MIM:613068]: An autosomal recessive neurodegenerative disorder resulting from brain-specific folate deficiency early in life. Onset is apparent in late infancy with severe developmental regression, movement disturbances, epilepsy and leukodystrophy. {ECO:0000269|PubMed:19732866}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB05595; DB00158; DB00563; DB12489; DB15413; DB05168 Interacts with Q8N357 EC number NA Uniprot keywords 3D-structure; Cell membrane; Cytoplasmic vesicle; Direct protein sequencing; Disulfide bond; Endosome; Folate-binding; Glycoprotein; GPI-anchor; Lipoprotein; Membrane; Neurodegeneration; Proteomics identification; Receptor; Reference proteome; Secreted; Signal; Transport Protein physicochemical properties Chain ID A Molecular weight (Da) 24216 Length 207 Aromaticity 0.13 Instability index 49.36 Isoelectric point 8.14 Charge (pH=7) 3.41 2D Binding mode Binding energy (Kcal/mol) -10.93 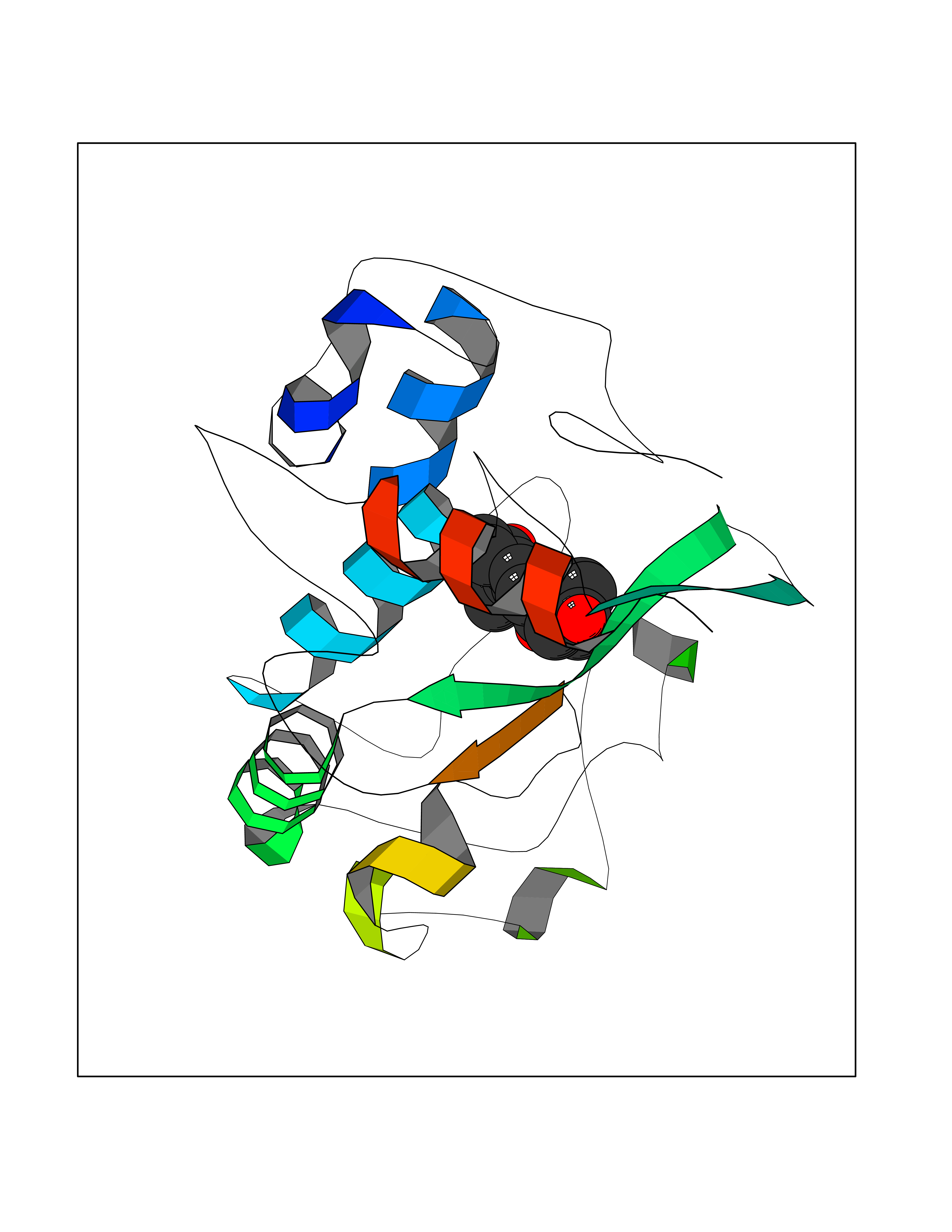 Molscript Map 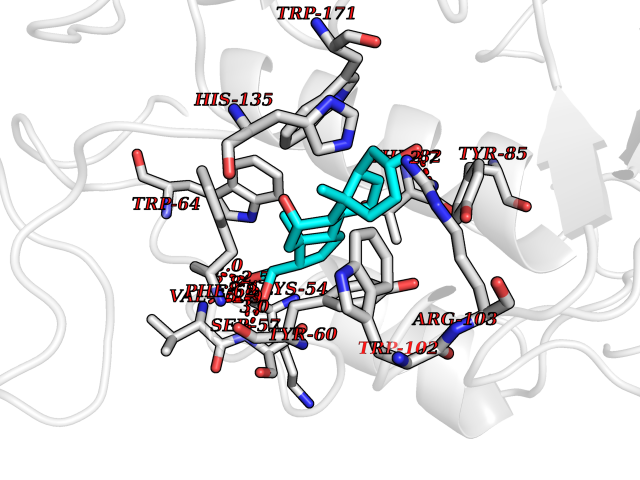 Pymol Map 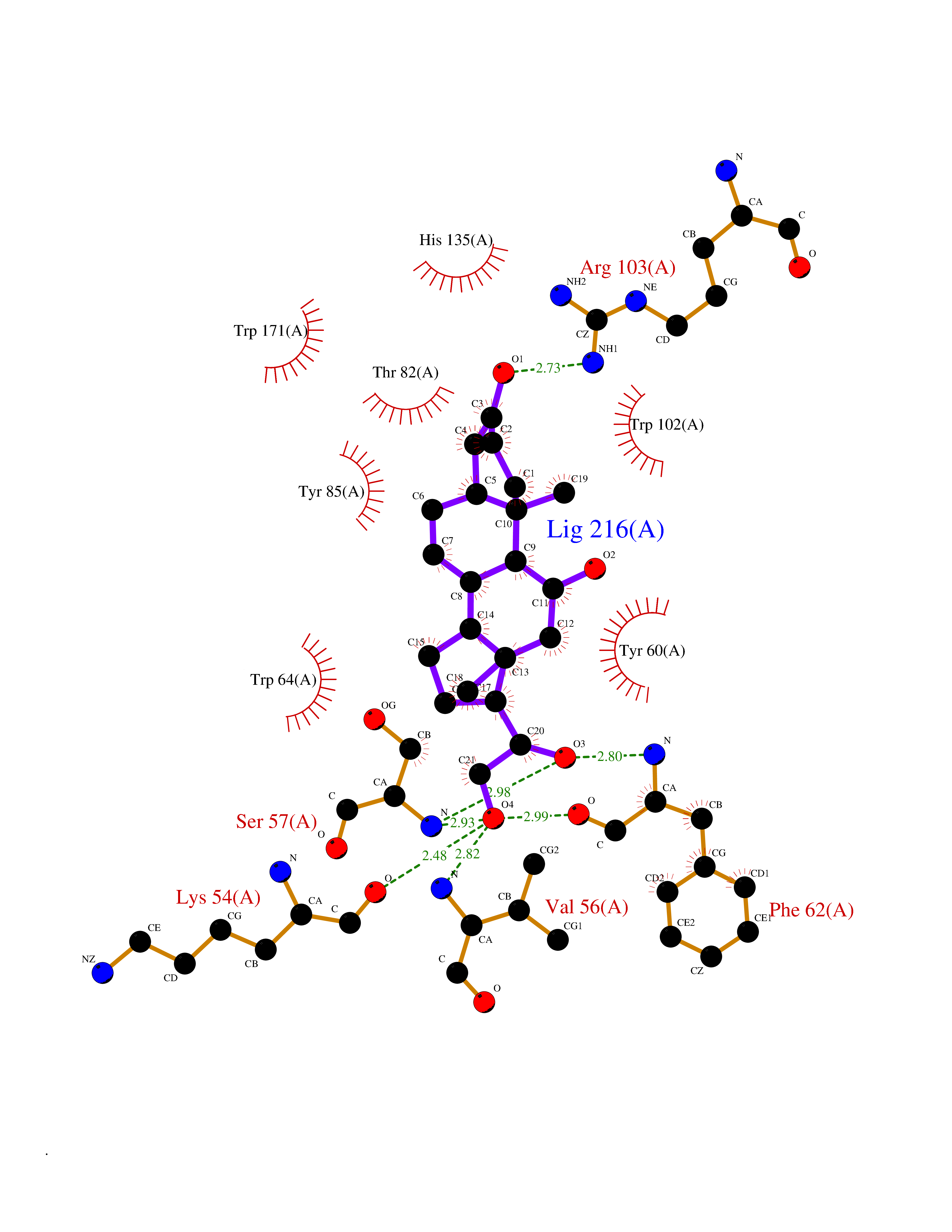 Ligplot Map 3D Binding mode Sequence RTELLNVCMNAKHHKEKPGPEDKLHEQCRPWRKNACCSTNTSQEAHKDVSYLYRFNWNHCGEMAPACKRHFIQDTCLYECSPNLGPWIQQVDQSWRKERVLNVPLCKEDCEQWWEDCRTSYTCKSNWHKGWNWTSGFNKCAVGAACQPFHFYFPTPTVLCNEIWTHSYKVSNYSRGSGRCIQMWFDPAQGNPNEEVARFYAAAMSGT Hydrogen bonds contact Hydrophobic contact | ||||
| 13 | SET domain containing 8 (KMT5A) | 5TEG | 8.15 | |
Target general information Gen name KMT5A Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms SETD8; SET8; SET07; SET domain-containing protein 8; PRSET7; PR/SET07; PR/SET domain-containing protein 07; PR-Set7; N-lysine methyltransferase KMT5A; Lysine-specific methylase 5A; Lysine N-methyltran Protein family Class V-like SAM-binding methyltransferase superfamily, Histone-lysine methyltransferase family, PR/SET subfamily Biochemical class Methyltransferase Function Specifically monomethylates 'Lys-20' of histone H4 (H4K20me1). H4K20me1 is enriched during mitosis and represents a specific tag for epigenetic transcriptional repression. Mainly functions in euchromatin regions, thereby playing a central role in the silencing of euchromatic genes. Required for cell proliferation, probably by contributing to the maintenance of proper higher-order structure of DNA during mitosis. Involved in chromosome condensation and proper cytokinesis. Nucleosomes are preferred as substrate compared to free histones. Mediates monomethylation of p53/TP53 at 'Lys-382', leading to repress p53/TP53-target genes. Plays a negative role in TGF-beta response regulation and a positive role in cell migration. Protein-lysine N-methyltransferase that monomethylates both histones and non-histone proteins. Related diseases Sick sinus syndrome 2 (SSS2) [MIM:163800]: The term 'sick sinus syndrome' encompasses a variety of conditions caused by sinus node dysfunction. The most common clinical manifestations are syncope, presyncope, dizziness, and fatigue. Electrocardiogram typically shows sinus bradycardia, sinus arrest, and/or sinoatrial block. Episodes of atrial tachycardias coexisting with sinus bradycardia ('tachycardia-bradycardia syndrome') are also common in this disorder. SSS occurs most often in the elderly associated with underlying heart disease or previous cardiac surgery, but can also occur in the fetus, infant, or child without heart disease or other contributing factors. SSS2 onset is in utero or at birth. {ECO:0000269|PubMed:15123648, ECO:0000269|PubMed:16407510, ECO:0000269|PubMed:20662977, ECO:0000269|PubMed:23103389}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Brugada syndrome 8 (BRGDA8) [MIM:613123]: A tachyarrhythmia characterized by right bundle branch block and ST segment elevation on an electrocardiogram (ECG). It can cause the ventricles to beat so fast that the blood is prevented from circulating efficiently in the body. When this situation occurs, the individual will faint and may die in a few minutes if the heart is not reset. {ECO:0000269|PubMed:19165230}. The gene represented in this entry may be involved in disease pathogenesis.; DISEASE: Epilepsy, idiopathic generalized 18 (EIG18) [MIM:619521]: An autosomal dominant form of idiopathic generalized epilepsy, a disorder characterized by recurring generalized seizures in the absence of detectable brain lesions and/or metabolic abnormalities. Generalized seizures arise diffusely and simultaneously from both hemispheres of the brain. Seizure types include juvenile myoclonic seizures, absence seizures, and generalized tonic-clonic seizures. EIG18 is characterized by onset of myoclonic seizures in infancy. Although the seizures remit, some patients may have later speech or cognitive impairment. {ECO:0000269|PubMed:30127718}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P62805; P07910; Q15672 EC number EC 2.1.1.- Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Cell cycle; Cell division; Chromatin regulator; Chromosome; Coiled coil; Direct protein sequencing; Methyltransferase; Mitosis; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Repressor; S-adenosyl-L-methionine; Transcription; Transcription regulation; Transferase; Ubl conjugation Protein physicochemical properties Chain ID A,D Molecular weight (Da) 19129.4 Length 167 Aromaticity 0.08 Instability index 49.18 Isoelectric point 7.88 Charge (pH=7) 1.37 2D Binding mode Binding energy (Kcal/mol) -9.25  Molscript Map 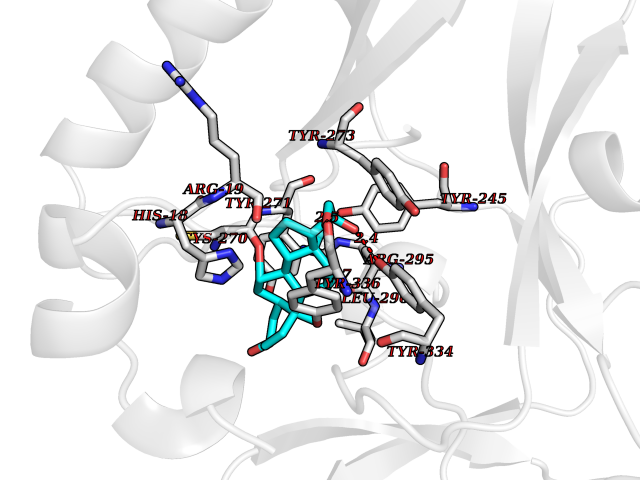 Pymol Map 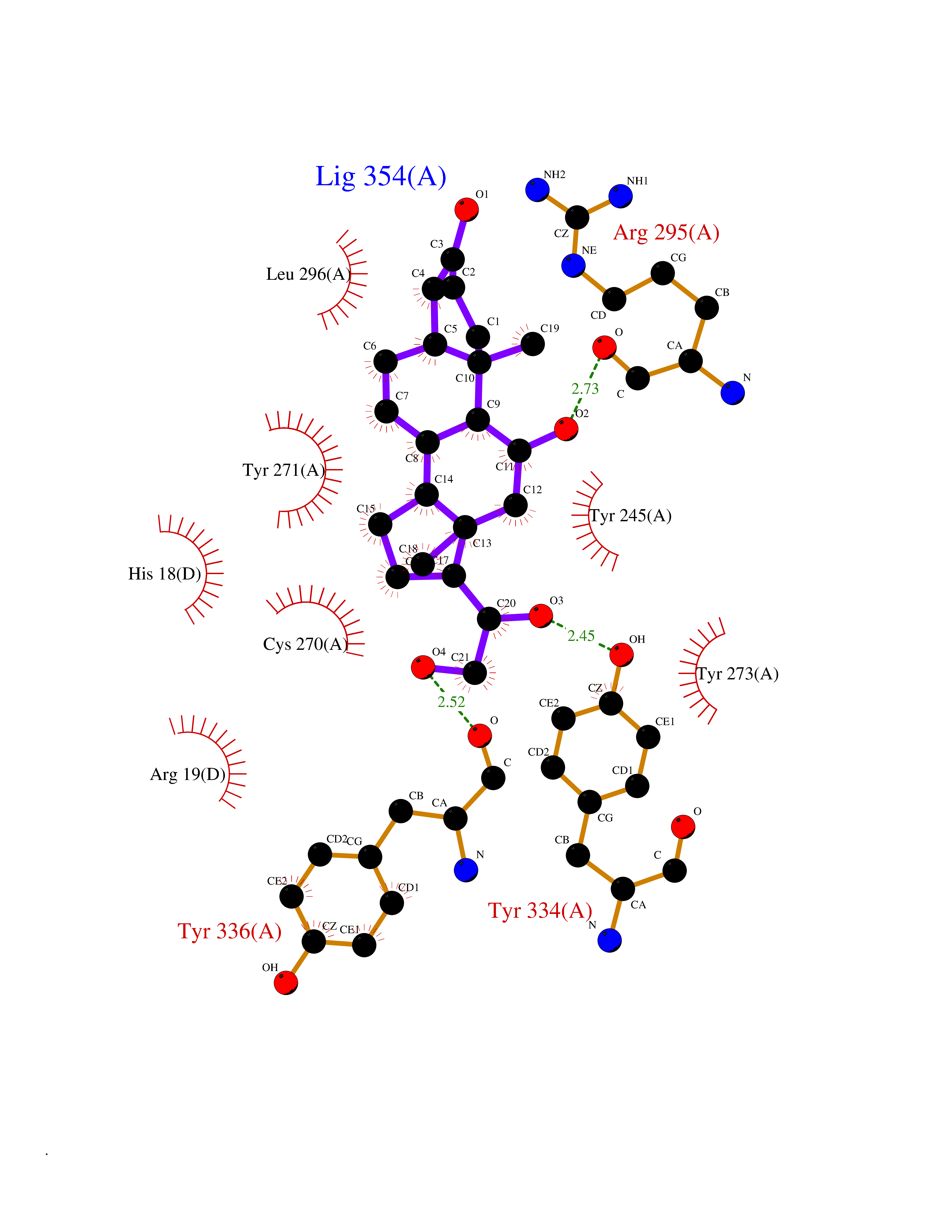 Ligplot Map 3D Binding mode Sequence KSKAELQSEERKRIDELIESGKEEGMKIDLIDGKGRGVIATKQFSRGDFVVEYHGDLIEITDAKKREALYAQDPSTGCYMYYFQYLSKTYCVDATRETNRLGRLINHSKSGNCQTKLHDIDGVPHLILIASRDIAAGEELLYDYGDRSKASIEAHPWLKHKRHRVLR Hydrogen bonds contact Hydrophobic contact | ||||
| 14 | Amylin receptor (IAPPR) | 6ZIS | 8.13 | |
Target general information Gen name CALCR-RAMP1/RAMP2/RAMP3 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Complex of Calcitonin receptor and Receptor activity-modifying protein Protein family RAMP family Biochemical class NA Function Transports the calcitonin gene-related peptide type 1 receptor (CALCRL) to the plasma membrane. Acts as a receptor for calcitonin-gene-related peptide (CGRP) together with CALCRL. Related diseases Immunodeficiency 9 (IMD9) [MIM:612782]: An immune disorder characterized by recurrent infections, impaired activation and proliferative response of T-cells, decreased T-cell production of cytokines, and normal lymphocytes counts and serum immunoglobulin levels. In surviving patients ectodermal dysplasia with anhidrosis and non-progressive myopathy may be observed. {ECO:0000269|PubMed:16147976, ECO:0000269|PubMed:16582901}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Myopathy, tubular aggregate, 2 (TAM2) [MIM:615883]: A rare congenital myopathy characterized by regular arrays of membrane tubules on muscle biopsies without additional histopathological hallmarks. Tubular aggregates in muscle are structures of variable appearance consisting of an outer tubule containing either one or more microtubule-like structures or amorphous material. TAM2 patients have myopathy and pupillary abnormalities. {ECO:0000269|PubMed:24591628, ECO:0000269|PubMed:28058752}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01278 Interacts with Q16602; P21145; Q5J8X5; Q16617 EC number NA Uniprot keywords 3D-structure; Cell membrane; Disulfide bond; Membrane; Proteomics identification; Receptor; Reference proteome; Signal; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A Molecular weight (Da) 63832.6 Length 569 Aromaticity 0.12 Instability index 22.42 Isoelectric point 5.07 Charge (pH=7) -16.96 2D Binding mode Binding energy (Kcal/mol) -8.86  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence SAKIEEGKLVIWINGDKGYNGLAEVGKKFEKDTGIKVTVEHPDKLEEKFPQVAATGDGPDIIFWAHDRFGGYAQSGLLAEITPDKAFQDKLYPFTWDAVRYNGKLIAYPIAVEALSLIYNKDLLPNPPKTWEEIPALDKELKAKGKSALMFNLQEPYFTWPLIAADGGYAFKYENGKYDIKDVGVDNAGAKAGLTFLVDLIKNKHMNADTDYSIAEAAFNKGETAMTINGPWAWSNIDTSKVNYGVTVLPTFKGQPSKPFVGVLSAGINAASPNKELAKEFLENYLLTDEGLEAVNKDKPLGAVALKSYEEELAKDPRIAATMENAQKGEIMPNIPQMSAFWYAVRTAVINAASGRQTVDEALKDAQTNAAAEFTTACQEANYGALLRELCLTQFQVDMEAVGETLWCDWGRTIRSYRELADCTWHMAEKLGCFWPNAEVDRFFLAVHGRYFRSCPISIQLGVTRNKIMTAQYECYQKIMQDPIQQGVYCQRTWDGWLCWNDVAAGTESMQLCPDYFQDFDPSEKVTKICDQDGNWFRHPASQRTWTDYTQCNVNTHEKVKTALNLFYL Hydrogen bonds contact Hydrophobic contact | ||||
| 15 | Glucagon-like peptide 1 receptor (GLP1R) | 6X1A | 8.12 | |
Target general information Gen name GLP1R Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms GLP-1R; GLP-1-R; GLP-1 receptor Protein family G-protein coupled receptor 2 family Biochemical class GPCR secretin Function Ligand binding triggers activation of a signaling cascade that leads to the activation of adenylyl cyclase and increased intracellular cAMP levels. Plays a role in regulating insulin secretion in response to GLP-1. G-protein coupled receptor for glucagon-like peptide 1 (GLP-1). Related diseases Lynch syndrome 2 (LYNCH2) [MIM:609310]: A form of Lynch syndrome, an autosomal dominant disease associated with marked increase in cancer susceptibility. It is characterized by a familial predisposition to early-onset colorectal carcinoma (CRC) and extra-colonic tumors of the gastrointestinal, urological and female reproductive tracts. Lynch syndrome is reported to be the most common form of inherited colorectal cancer in the Western world. Clinically, it is often divided into two subgroups. Type I is characterized by hereditary predisposition to colorectal cancer, a young age of onset, and carcinoma observed in the proximal colon. Type II is characterized by increased risk for cancers in certain tissues such as the uterus, ovary, breast, stomach, small intestine, skin, and larynx in addition to the colon. Diagnosis of classical Lynch syndrome is based on the Amsterdam criteria: 3 or more relatives affected by colorectal cancer, one a first degree relative of the other two; 2 or more generation affected; 1 or more colorectal cancers presenting before 50 years of age; exclusion of hereditary polyposis syndromes. The term 'suspected Lynch syndrome' or 'incomplete Lynch syndrome' can be used to describe families who do not or only partially fulfill the Amsterdam criteria, but in whom a genetic basis for colon cancer is strongly suspected. {ECO:0000269|PubMed:10323887, ECO:0000269|PubMed:10375096, ECO:0000269|PubMed:10386556, ECO:0000269|PubMed:10413423, ECO:0000269|PubMed:10480359, ECO:0000269|PubMed:10598809, ECO:0000269|PubMed:10627141, ECO:0000269|PubMed:10660333, ECO:0000269|PubMed:10671064, ECO:0000269|PubMed:10713887, ECO:0000269|PubMed:10777691, ECO:0000269|PubMed:10882759, ECO:0000269|PubMed:11139242, ECO:0000269|PubMed:11427529, ECO:0000269|PubMed:11726306, ECO:0000269|PubMed:11748856, ECO:0000269|PubMed:11754112, ECO:0000269|PubMed:11781295, ECO:0000269|PubMed:11793442, ECO:0000269|PubMed:11839723, ECO:0000269|PubMed:11870161, ECO:0000269|PubMed:12095971, ECO:0000269|PubMed:12132870, ECO:0000269|PubMed:12200596, ECO:0000269|PubMed:12362047, ECO:0000269|PubMed:12373605, ECO:0000269|PubMed:12655562, ECO:0000269|PubMed:12658575, ECO:0000269|PubMed:14635101, ECO:0000269|PubMed:14961575, ECO:0000269|PubMed:15064764, ECO:0000269|PubMed:15139004, ECO:0000269|PubMed:15365995, ECO:0000269|PubMed:15365996, ECO:0000269|PubMed:16083711, ECO:0000269|PubMed:16451135, ECO:0000269|PubMed:17301300, ECO:0000269|PubMed:17510385, ECO:0000269|PubMed:18561205, ECO:0000269|PubMed:20020535, ECO:0000269|PubMed:21120944, ECO:0000269|PubMed:22753075, ECO:0000269|PubMed:7757073, ECO:0000269|PubMed:8566964, ECO:0000269|PubMed:8571956, ECO:0000269|PubMed:8574961, ECO:0000269|PubMed:8797773, ECO:0000269|PubMed:8872463, ECO:0000269|PubMed:8993976, ECO:0000269|PubMed:9048925, ECO:0000269|PubMed:9067757, ECO:0000269|PubMed:9218993, ECO:0000269|PubMed:9272156, ECO:0000269|PubMed:9298827, ECO:0000269|PubMed:9311737, ECO:0000269|PubMed:9326924, ECO:0000269|PubMed:9399661, ECO:0000269|PubMed:9559627, ECO:0000269|PubMed:9718327, ECO:0000269|PubMed:9833759, ECO:0000269|PubMed:9927034, ECO:0000269|Ref.5}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Mismatch repair cancer syndrome 1 (MMRCS1) [MIM:276300]: An autosomal recessive form of mismatch repair cancer syndrome, a childhood cancer predisposition syndrome encompassing a broad tumor spectrum. This includes hematological malignancies, central nervous system tumors, Lynch syndrome-associated malignancies such as colorectal tumors as well as multiple intestinal polyps, embryonic tumors and rhabdomyosarcoma. Multiple cafe-au-lait macules, a feature reminiscent of neurofibromatosis type 1, are often found as first manifestation of the underlying cancer. {ECO:0000269|PubMed:11427529, ECO:0000269|PubMed:17440981, ECO:0000269|PubMed:7661930}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Muir-Torre syndrome (MRTES) [MIM:158320]: Rare autosomal dominant disorder characterized by sebaceous neoplasms and visceral malignancy. {ECO:0000269|PubMed:8751876}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Defects in MLH1 may contribute to lobular carcinoma in situ (LCIS), a non-invasive neoplastic disease of the breast.; DISEASE: Endometrial cancer (ENDMC) [MIM:608089]: A malignancy of endometrium, the mucous lining of the uterus. Most endometrial cancers are adenocarcinomas, cancers that begin in cells that make and release mucus and other fluids. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Some epigenetic changes can be transmitted unchanged through the germline (termed 'epigenetic inheritance'). Evidence that this mechanism occurs in humans is provided by the identification of individuals in whom 1 allele of the MLH1 gene is epigenetically silenced throughout the soma (implying a germline event). These individuals are affected by Lynch syndrome but does not have identifiable mutations in MLH1, even though it is silenced, which demonstrates that an epimutation can phenocopy a genetic disease.; DISEASE: Colorectal cancer (CRC) [MIM:114500]: A complex disease characterized by malignant lesions arising from the inner wall of the large intestine (the colon) and the rectum. Genetic alterations are often associated with progression from premalignant lesion (adenoma) to invasive adenocarcinoma. Risk factors for cancer of the colon and rectum include colon polyps, long-standing ulcerative colitis, and genetic family history. {ECO:0000269|PubMed:10598809, ECO:0000269|PubMed:10882759, ECO:0000269|PubMed:12132870, ECO:0000269|PubMed:12655564, ECO:0000269|PubMed:14504054, ECO:0000269|PubMed:15184898, ECO:0000269|PubMed:18033691, ECO:0000269|PubMed:8872463, ECO:0000269|PubMed:9032648, ECO:0000269|PubMed:9087566, ECO:0000269|PubMed:9611074}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB09043; DB09045; DB15650; DB01276; DB00040; DB16697; DB06655; DB09265; DB13928; DB14027; DB15171 Interacts with A8MQ03; Q07627; Q8IUG1; P60409; P60410; P60411; Q9BYP8; P26371; Q7Z3S9; P0DPK4 EC number NA Uniprot keywords 3D-structure; ADP-ribosylation; Cell membrane; Disulfide bond; G-protein coupled receptor; Glycoprotein; Membrane; Receptor; Reference proteome; Signal; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID R Molecular weight (Da) 45579.6 Length 390 Aromaticity 0.16 Instability index 39.66 Isoelectric point 6.73 Charge (pH=7) -0.68 2D Binding mode Binding energy (Kcal/mol) -10.1  Molscript Map 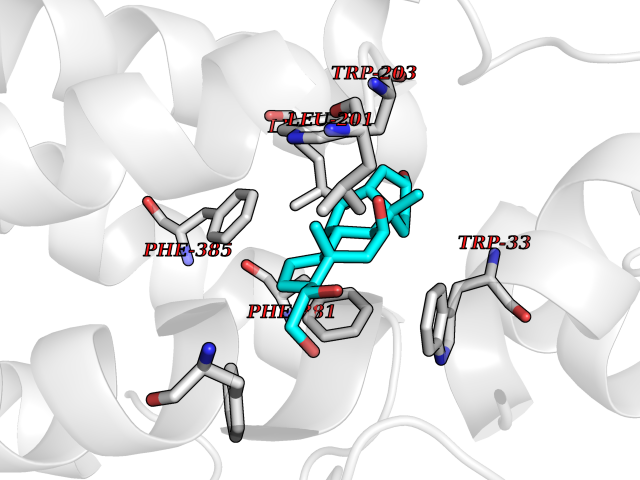 Pymol Map 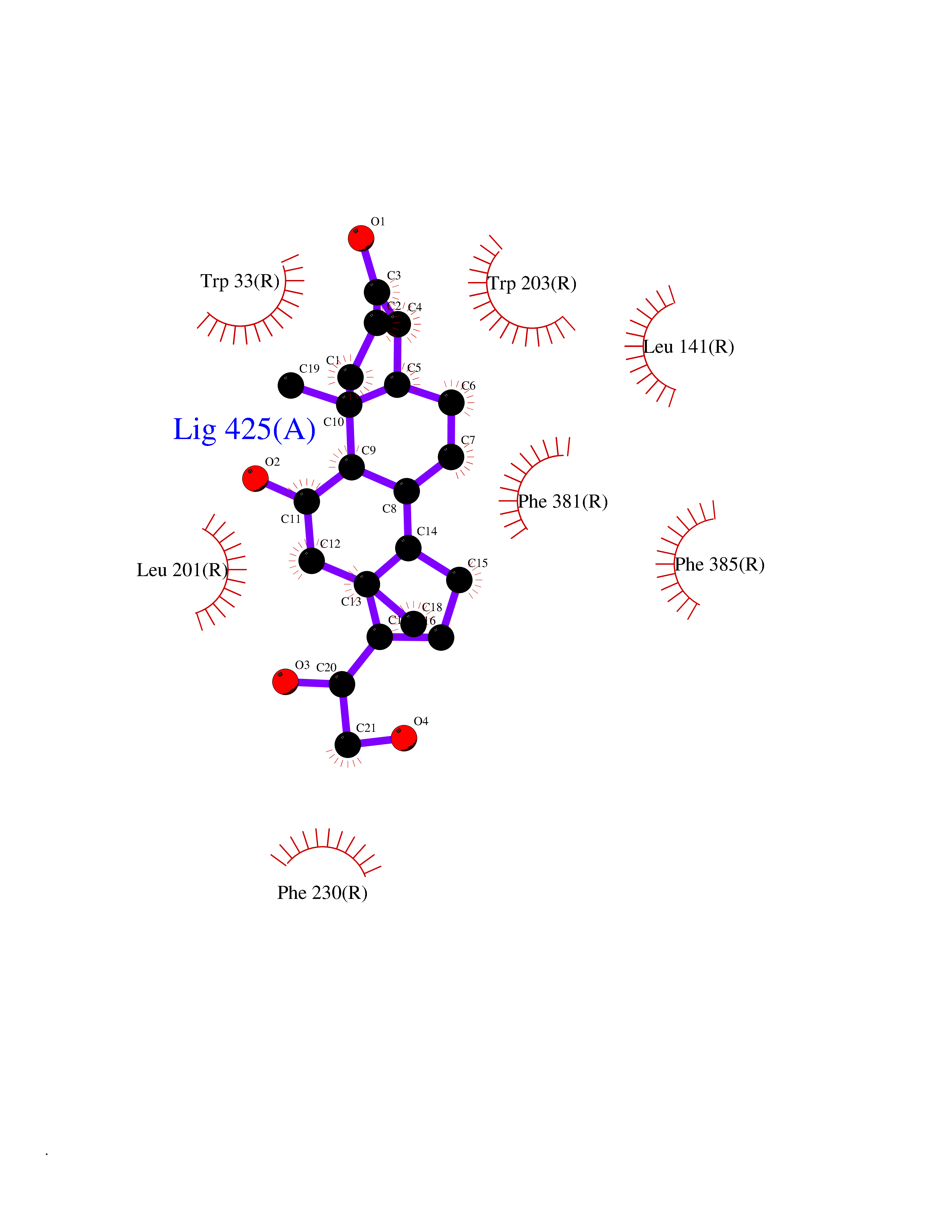 Ligplot Map 3D Binding mode Sequence ATVSLWETVQKWREYRRQCQRSLTEDPPPATDLFCNRTFDEYACWPDGEPGSFVNVSCPWYLPWASSVPQGHVYRFCTAEGLWLQKDNSSLPWRDLSECEESSPEEQLLFLYIIYTVGYALSFSALVIASAILLGFRHLHCTRNYIHLNLFASFILRALSVFIKDAALKWMYSTAAQQHQWDGLLSYQDSLSCRLVFLLMQYCVAANYYWLLVEGVYLYTLLAFSVFSEQWIFRLYVSIGWGVPLLFVVPWGIVKYLYEDEGCWTRNSNMNYWLIIRLPILFAIGVNFLIFVRVICIVVSKLKANLMCKTDIKCRLAKSTLTLIPLLGTHEVIFAFVMDEHARGTLRFIKLFTELSFTSFQGLMVAILYCFVNNEVQLEFRKSWERWRLE Hydrogen bonds contact Hydrophobic contact | ||||
| 16 | Human immunodeficiency virus Protease (HIV PR) | 3TL9 | 8.11 | |
Target general information Gen name HIV PR Organism Human immunodeficiency virus type 1 group M subtype B (isolate BH10) (HIV-1) Uniprot ID TTD ID Synonyms HIV Retropepsin; HIV PR Protein family NA Biochemical class Peptidase Function Gag-Pol polyprotein: Mediates, with Gag polyprotein, the essential events in virion assembly, including binding the plasma membrane, making the protein-protein interactions necessary to create spherical particles, recruiting the viral Env proteins, and packaging the genomic RNA via direct interactions with the RNA packaging sequence (Psi). Gag-Pol polyprotein may regulate its own translation, by the binding genomic RNA in the 5'-UTR. At low concentration, the polyprotein would promote translation, whereas at high concentration, the polyprotein would encapsidate genomic RNA and then shut off translation. Related diseases Sitosterolemia 2 (STSL2) [MIM:618666]: A form of sitosterolemia, an autosomal recessive metabolic disorder characterized by unregulated intestinal absorption of cholesterol, phytosterols and shellfish sterols, and decreased biliary excretion of dietary sterols into bile. Patients have hypercholesterolemia, very high levels of plant sterols in the plasma, and frequently develop tendon and tuberous xanthomas, accelerated atherosclerosis and premature coronary artery disease. {ECO:0000269|PubMed:11138003, ECO:0000269|PubMed:11452359, ECO:0000269|PubMed:11668628, ECO:0000269|PubMed:15054092, ECO:0000269|PubMed:35557526}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07035; DB02704; DB07806; DB02785; DB01824; DB01732; DB06874; DB08034; DB07961; DB07451; DB08212; DB08372; DB02972; DB04190; DB04042; DB08428; DB03076; DB03141; DB08457; DB07343; DB07337; DB07018; DB07332; DB05398; DB07578; DB08639; DB06414; DB04255; DB04547; DB02683; DB02009; DB03908; DB02629; DB01887; DB03803; DB02033; DB08281; DB08282; DB08284; DB08414; DB08598; DB07327; DB07885; DB02768; DB08600; DB01891; DB05871 Interacts with NA EC number EC 3.4.23.16 Uniprot keywords 3D-structure; Activation of host caspases by virus; AIDS; Aspartyl protease; Capsid protein; Direct protein sequencing; DNA integration; DNA recombination; DNA-binding; DNA-directed DNA polymerase; Endonuclease; Eukaryotic host gene expression shutoff by virus; Eukaryotic host translation shutoff by virus; Host cell membrane; Host cytoplasm; Host endosome; Host gene expression shutoff by virus; Host membrane; Host nucleus; Host-virus interaction; Hydrolase; Lipid-binding; Lipoprotein; Magnesium; Membrane; Metal-binding; Methylation; Modulation of host cell apoptosis by virus; Multifunctional enzyme; Myristate; Nuclease; Nucleotidyltransferase; Phosphoprotein; Protease; Repeat; Ribosomal frameshifting; RNA-binding; RNA-directed DNA polymerase; Transferase; Viral genome integration; Viral nucleoprotein; Viral penetration into host nucleus; Viral release from host cell; Virion; Virion maturation; Virus entry into host cell; Zinc; Zinc-finger Protein physicochemical properties Chain ID A,B Molecular weight (Da) 21934.7 Length 202 Aromaticity 0.06 Instability index 48.65 Isoelectric point 9.66 Charge (pH=7) 6.15 2D Binding mode Binding energy (Kcal/mol) -7.84  Molscript Map 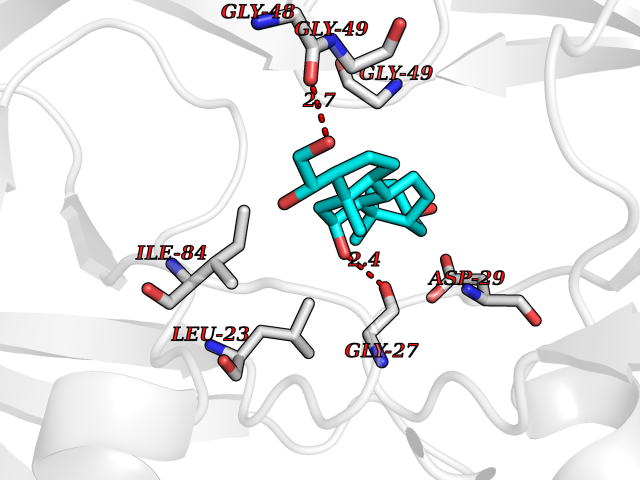 Pymol Map 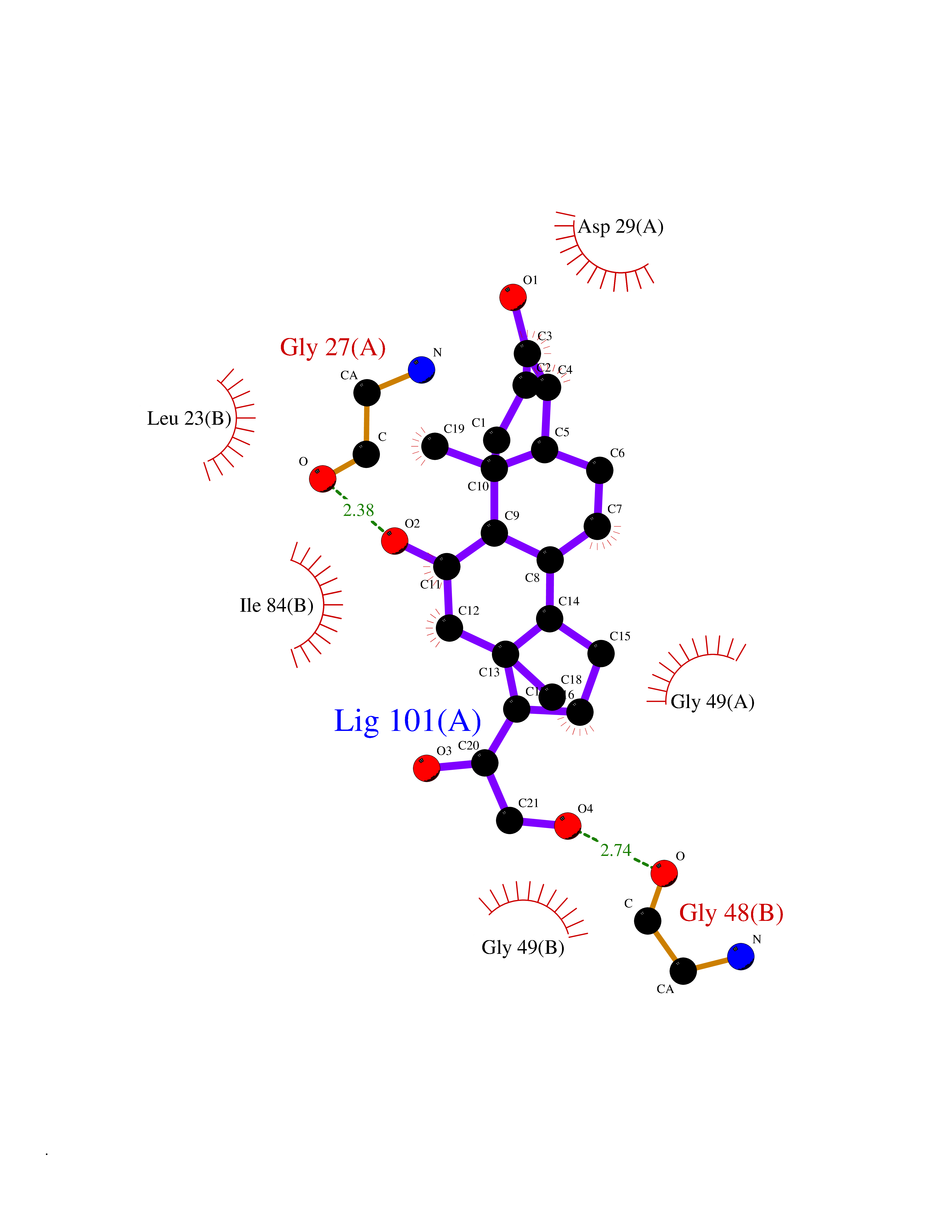 Ligplot Map 3D Binding mode Sequence PQITLWKRPLVTIKIGGQLKEALLDTGADDTVIEEMSLPGRWKPKMIGGIGGFIKVRQYDQIIIEIAGHKAIGTVLVGPTPVNIIGRNLLTQIGATLNFSFNFPQITLWKRPLVTIKIGGQLKEALLDTGADDTVIEEMSLPGRWKPKMIGGIGGFIKVRQYDQIIIEIAGHKAIGTVLVGPTPVNIIGRNLLTQIGATLNF Hydrogen bonds contact Hydrophobic contact | ||||
| 17 | Lysine--tRNA ligase | 4YCU | 8.07 | |
Target general information Gen name KARS Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms KARS;KIAA0070 Protein family Class-II aminoacyl-tRNA synthetase family Biochemical class ligase / ligase inhibitor Function Amino acid binding.ATP adenylyltransferase activity.ATP binding.Identical protein binding.Lysine-tRNA ligase activity.Protein homodimerization activity.TRNA binding. Related diseases Charcot-Marie-Tooth disease, recessive intermediate B (CMTRIB) [MIM:613641]: A form of Charcot-Marie-Tooth disease, a disorder of the peripheral nervous system, characterized by progressive weakness and atrophy, initially of the peroneal muscles and later of the distal muscles of the arms. Recessive intermediate forms of Charcot-Marie-Tooth disease are characterized by clinical and pathologic features intermediate between demyelinating and axonal peripheral neuropathies, and motor median nerve conduction velocities ranging from 25 to 45 m/sec. {ECO:0000269|PubMed:20920668}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Deafness, autosomal recessive, 89 (DFNB89) [MIM:613916]: A form of non-syndromic deafness characterized by bilateral, prelingual, moderate to severe hearing loss affecting all frequencies. {ECO:0000269|PubMed:23768514}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Deafness, congenital, and adult-onset progressive leukoencephalopathy (DEAPLE) [MIM:619196]: An autosomal recessive, complex neurodegenerative disorder characterized by congenital sensorineural deafness, and progressive motor and cognitive decline apparent in young adulthood. Brain imaging shows diffuse white matter abnormalities affecting various brain regions, consistent with a progressive leukoencephalopathy. More variable additional features may include visual impairment and axonal peripheral neuropathy. Premature death may occurr in some patients. {ECO:0000269|PubMed:28887846, ECO:0000269|PubMed:30737337, ECO:0000269|PubMed:31116475}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Leukoencephalopathy, progressive, infantile-onset, with or without deafness (LEPID) [MIM:619147]: An autosomal recessive, complex neurodegenerative disorder apparent from infancy. LEPID is characterized by early-onset progressive leukoencephalopathy with brainstem and spinal cord calcifications, sensorineural deafness in most patients, global developmental delay with cognitive impairment and poor or absent speech, developmental regression, and neurologic deterioration. Additional more variable features may include poor overall growth with microcephaly, seizures, visual loss, microcytic anemia, and hepatic enlargement or abnormal liver enzymes. Premature death is common. {ECO:0000269|PubMed:25330800, ECO:0000269|PubMed:29615062, ECO:0000269|PubMed:30252186, ECO:0000269|PubMed:30715177}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00123 Interacts with Q13155; P07814; Q15046; P08865; P00441; Q13155 EC number 2.7.7.-; 6.1.1.6 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Aminoacyl-tRNA synthetase; ATP-binding; Cell membrane; Charcot-Marie-Tooth disease; Cytoplasm; Deafness; Direct protein sequencing; Disease variant; Host-virus interaction; Intellectual disability; Ligase; Membrane; Mitochondrion; Neurodegeneration; Neuropathy; Non-syndromic deafness; Nucleotide-binding; Nucleus; Phosphoprotein; Protein biosynthesis; Proteomics identification; Reference proteome; Secreted; Transferase Protein physicochemical properties Chain ID A,B Molecular weight (Da) 37110.4 Length 323 Aromaticity 0.1 Instability index 50.17 Isoelectric point 4.92 Charge (pH=7) -17.53 2D Binding mode Binding energy (Kcal/mol) -9.36  Molscript Map 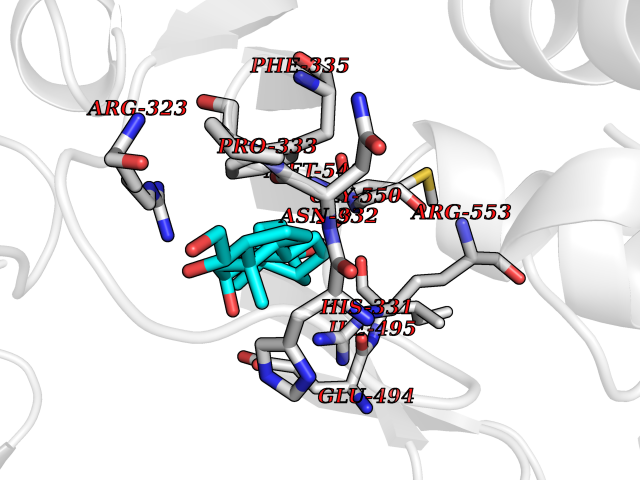 Pymol Map 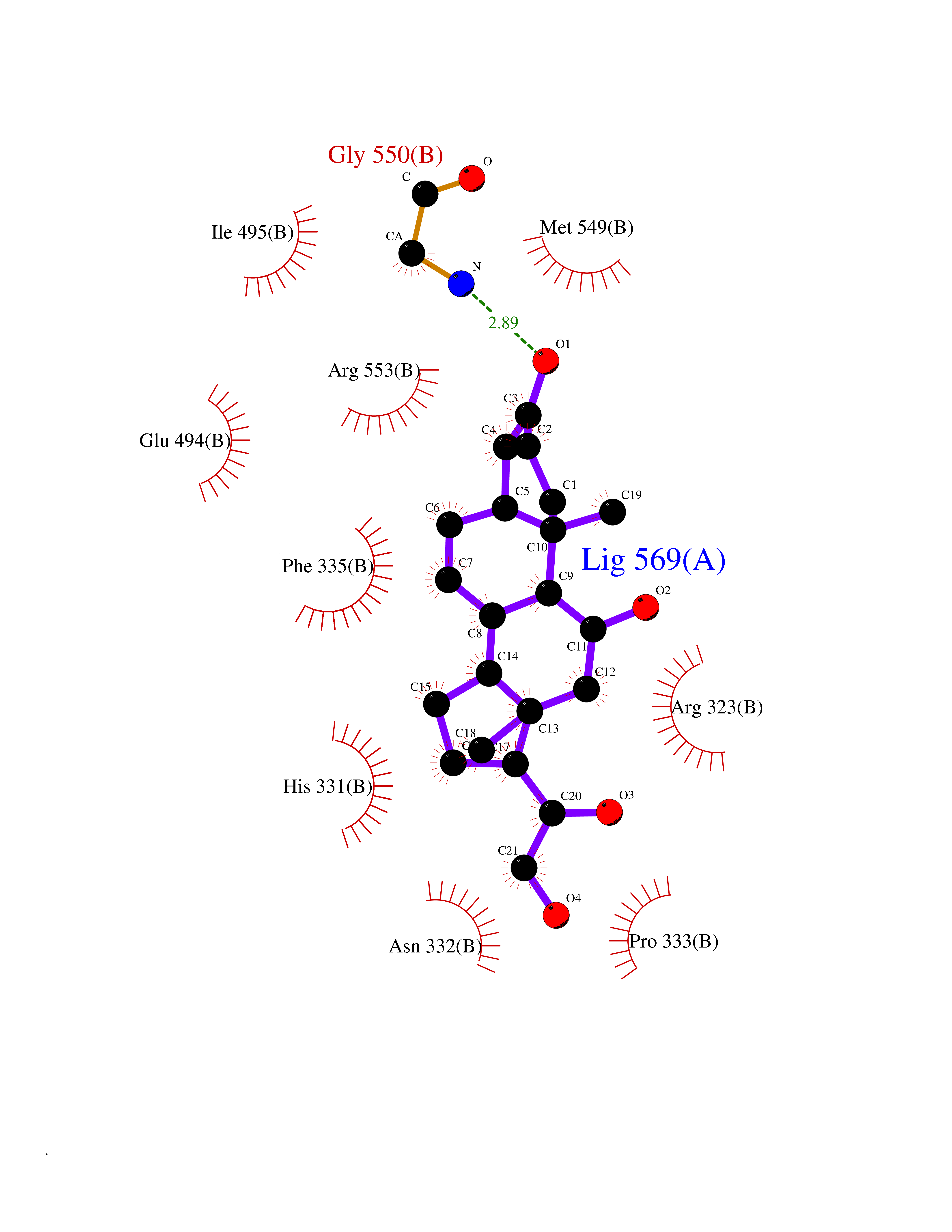 Ligplot Map 3D Binding mode Sequence IIRSKIITYIRSFLDELGFLEIETPMMNIIPGGAVAKPFITYHNELDMNLYMRIAPELYHKMLVVGGIDRVYEIGRQFRNEGIDLTHNPEFTTCEFYMAYADYHDLMEITEKMVSGMVKHITGSYKVTYHPDGPEGQAYDVDFTPPFRRINMVEELEKALGMKLPETNLFETEETRKILDDICVAKAVECPPPRTTARLLDKLVGEFLEVTCINPTFICDHPQIMSPLAKWHRSKEGLTERFELFVMKKEICNAYTELNDPMRQRQLFEEQAKAKAAGDDEAMFIDENFCTALEYGLPPTAGWGMGIDRVAMFLTDSNNIKEV Hydrogen bonds contact Hydrophobic contact | ||||
| 18 | ERK activator kinase 2 (MEK2) | 1S9I | 8.06 | |
Target general information Gen name MAP2K2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms PRKMK2; MKK2; MEK 2; MAPKK 2; MAPK/ERK kinase 2; MAP kinase kinase 2; Dual specificity mitogenactivated protein kinase kinase 2; Dual specificity mitogen-activated protein kinase kinase 2 Protein family Protein kinase superfamily, STE Ser/Thr protein kinase family, MAP kinase kinase subfamily Biochemical class Kinase Function Activates the ERK1 and ERK2 MAP kinases. Catalyzes the concomitant phosphorylation of a threonine and a tyrosine residue in a Thr-Glu-Tyr sequence located in MAP kinases. Related diseases Cardiofaciocutaneous syndrome 4 (CFC4) [MIM:615280]: A form of cardiofaciocutaneous syndrome, a multiple congenital anomaly disorder characterized by a distinctive facial appearance, heart defects and intellectual disability. Heart defects include pulmonic stenosis, atrial septal defects and hypertrophic cardiomyopathy. Some affected individuals present with ectodermal abnormalities such as sparse, friable hair, hyperkeratotic skin lesions and a generalized ichthyosis-like condition. Typical facial features are similar to Noonan syndrome. They include high forehead with bitemporal constriction, hypoplastic supraorbital ridges, downslanting palpebral fissures, a depressed nasal bridge, and posteriorly angulated ears with prominent helices. {ECO:0000269|PubMed:16439621, ECO:0000269|PubMed:18042262, ECO:0000269|PubMed:20358587}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB11967; DB06616; DB12010; DB14904; DB11689; DB08911 Interacts with P05067; P10398; Q96II5; P15056; O95273; Q12959; P61978-2; Q8IVT5; P00540; P04049 EC number EC 2.7.12.2 Uniprot keywords 3D-structure; Acetylation; ATP-binding; Cardiomyopathy; Cytoplasm; Direct protein sequencing; Disease variant; Ectodermal dysplasia; Intellectual disability; Kinase; Magnesium; Membrane; Metal-binding; Nucleotide-binding; Phosphoprotein; Proteomics identification; Reference proteome; Serine/threonine-protein kinase; Transferase; Tyrosine-protein kinase Protein physicochemical properties Chain ID A Molecular weight (Da) 33960.9 Length 303 Aromaticity 0.07 Instability index 45.61 Isoelectric point 6.29 Charge (pH=7) -2.53 2D Binding mode Binding energy (Kcal/mol) -9.6 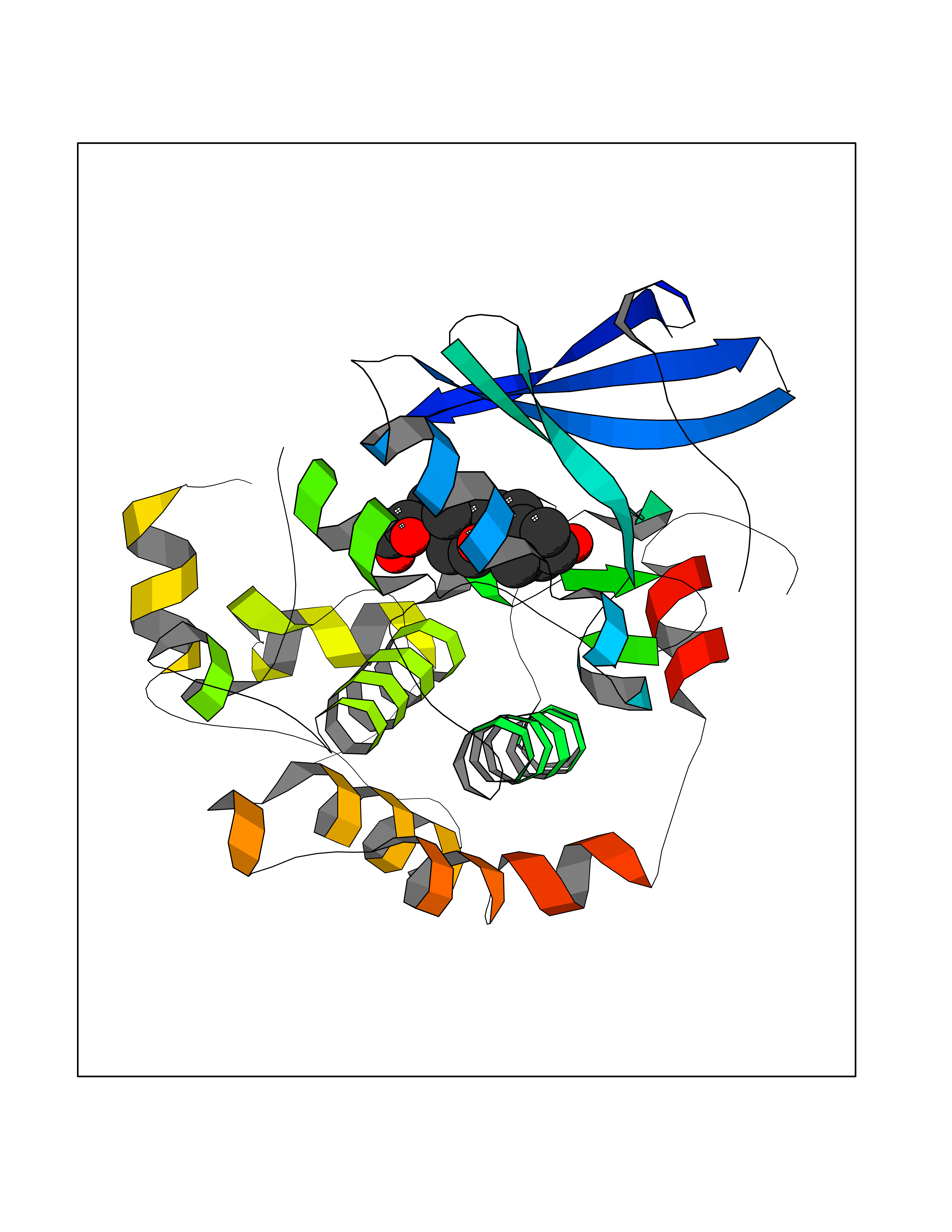 Molscript Map 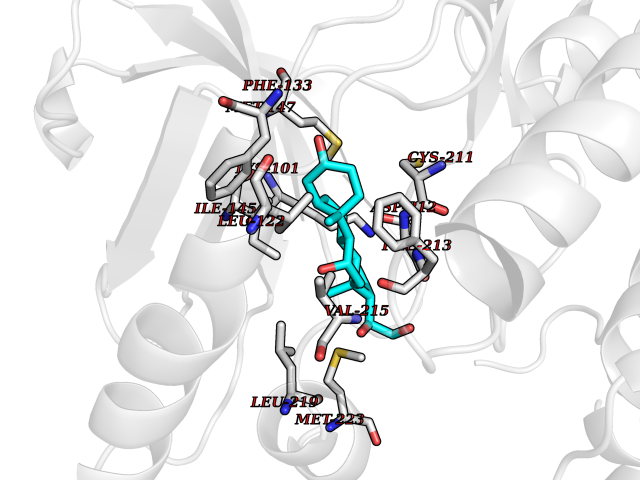 Pymol Map 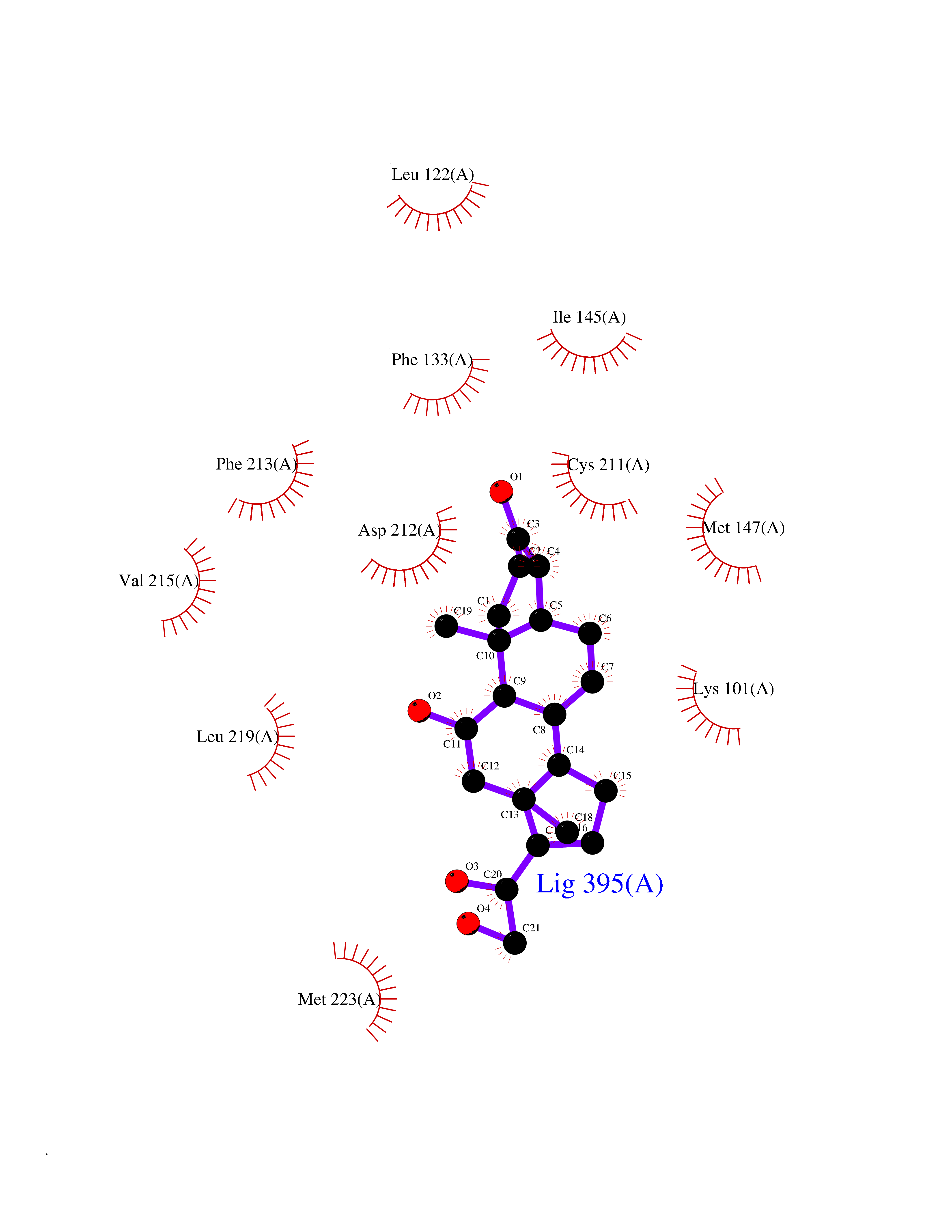 Ligplot Map 3D Binding mode Sequence QKAKVGELKDDDFERISELGAGNGGVVTKVQHRPSGLIMARKLIHLEIKPAIRNQIIRELQVLHECNSPYIVGFYGAFYSDGEISICMEHMDGGSLDQVLKEAKRIPEEILGKVSIAVLRGLAYLREKHQIMHRDVKPSNILVNSRGEIKLCDFGVSGQLIDSMVGTRSYMAPERLQGTHYSVQSDIWSMGLSLVELAVGRYPIPPPDAKELEAIFGRPVVDRPAMAIFELLDYIVNEPPPKLPNGVFTPDFQEFVNKCLIKNPAERADLKMLTNHTFIKRSEVEEVDFAGWLCKTLRLNQPG Hydrogen bonds contact Hydrophobic contact | ||||
| 19 | Gastrin (GAST) | 5WRJ | 8.03 | |
Target general information Gen name GAST Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Gastrin6; GAST; G52; G34; G14 Protein family Gastrin/cholecystokinin family Biochemical class Gastrin cholecystokinin Function Gastrin stimulates the stomach mucosa to produce and secrete hydrochloric acid and the pancreas to secrete its digestive enzymes. It also stimulates smooth muscle contraction and increases blood circulation and water secretion in the stomach and intestine. Related diseases Orotic aciduria 1 (ORAC1) [MIM:258900]: A disorder of pyrimidine metabolism resulting in megaloblastic anemia and orotic acid crystalluria that is frequently associated with some degree of physical and intellectual disability. A minority of cases have additional features, particularly congenital malformations and immune deficiencies. {ECO:0000269|PubMed:9042911}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB12532 Interacts with Q13520; O43315; Q12797-6; Q9BXK5; Q8N5K1; Q96BA8; P00387; Q9Y282; Q5JX71; Q8NBJ4; Q8TDT2; P43628; O76011; Q6ZUX7; Q15546; P15941-11; Q13113; P60201-2; Q14973; P02787; Q4KMG9 EC number NA Uniprot keywords 3D-structure; Amidation; Cleavage on pair of basic residues; Direct protein sequencing; Hormone; Phosphoprotein; Proteomics identification; Pyrrolidone carboxylic acid; Reference proteome; Secreted; Signal; Sulfation Protein physicochemical properties Chain ID A,F Molecular weight (Da) 31827.9 Length 280 Aromaticity 0.08 Instability index 41.06 Isoelectric point 8.84 Charge (pH=7) 5.56 2D Binding mode Binding energy (Kcal/mol) -9.23 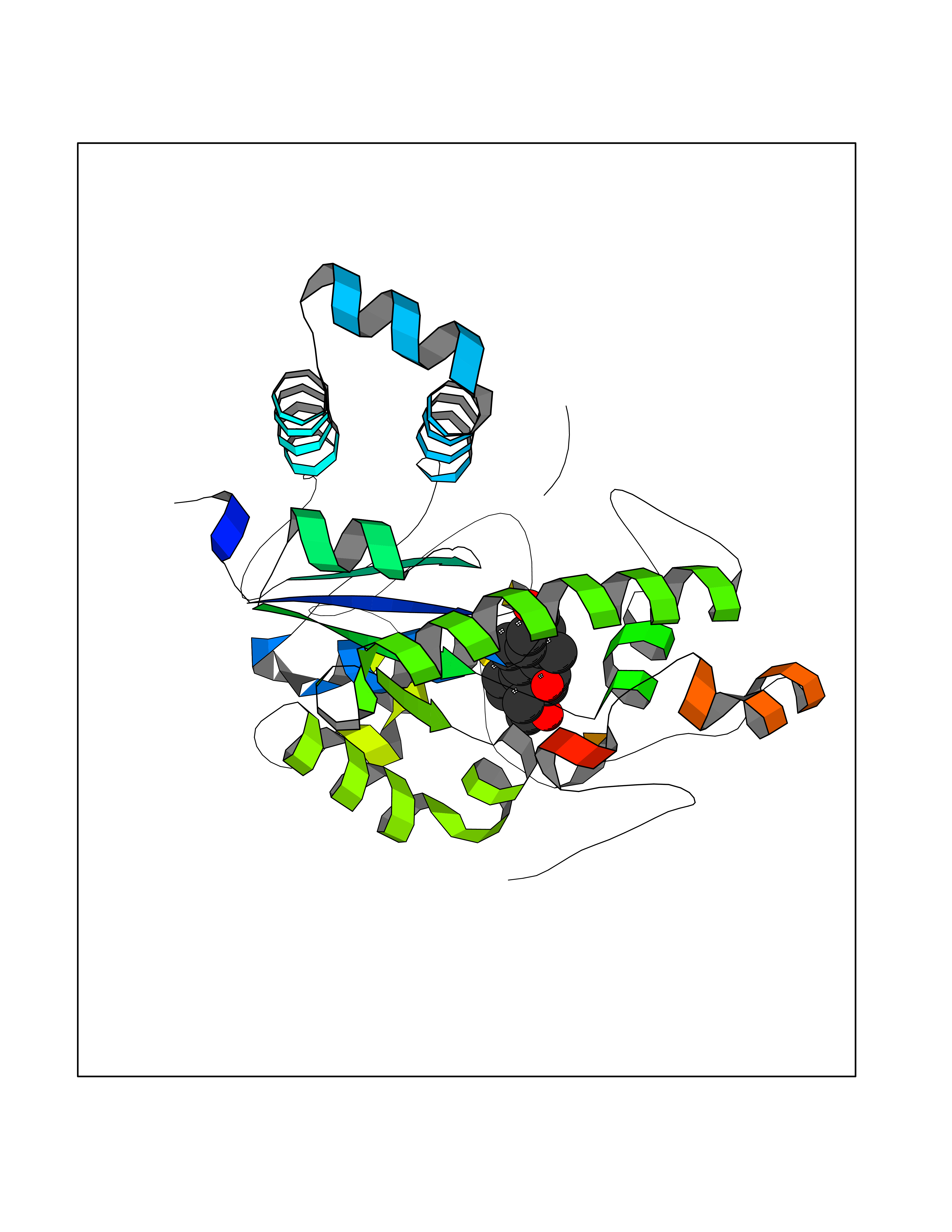 Molscript Map 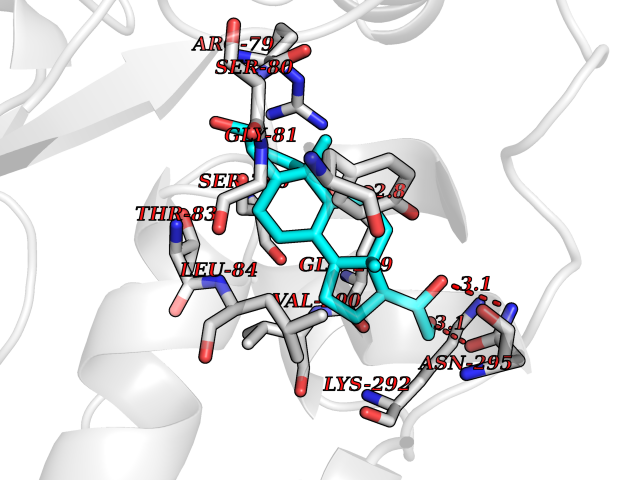 Pymol Map 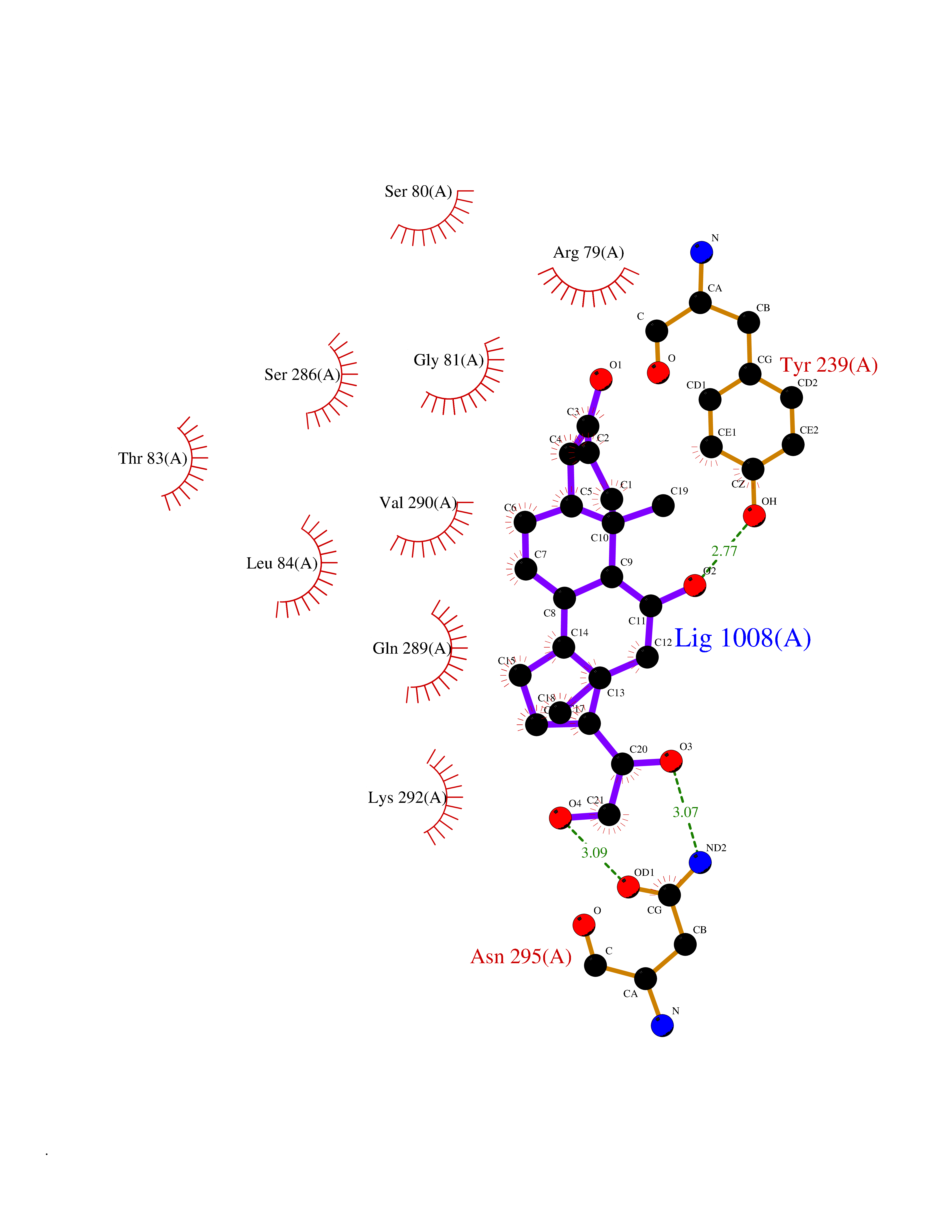 Ligplot Map 3D Binding mode Sequence AYHKDMPLIFIGGVPRSGTTLMRAMLDAHPDIRCGEETRVIPRILALKQMWSRSSKEKIRLDEAGVTDEVLDSAMQAFLLEIIVKHGEPAPYLCNKDPFALKSLTYLSRLFPNAKFLLMVRDGRASVHSMISRKVTIAGFDLNSYRDCLTKWNRAIETMYNQCMEVGYKKCMLVHYEQLVLHPERWMRTLLKFLQIPWNHSVLHHEEMIGKAGGVSLSKVERSTDQVIKPVNVGALSKWVGKIPPDVLQDMAVIAPMLAKLGYDPYANPPNYGKPEEEAY Hydrogen bonds contact Hydrophobic contact | ||||
| 20 | Phosphodiesterase 4D (PDE4D) | 1Y2K | 7.99 | |
Target general information Gen name PDE4D Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms cAMP-specific 3',5'-cyclic phosphodiesterase 4D; PDE43; DPDE3 Protein family Cyclic nucleotide phosphodiesterase family, PDE4 subfamily Biochemical class Phosphoric diester hydrolase Function Hydrolyzes the second messenger cAMP, which is a key regulator of many important physiological processes. Related diseases Genetic variations in PDE4D might be associated with susceptibility to stroke. PubMed:17006457 states that association with stroke has to be considered with caution. {ECO:0000269|PubMed:17006457}.; DISEASE: Acrodysostosis 2, with or without hormone resistance (ACRDYS2) [MIM:614613]: A pleiotropic disorder characterized by skeletal, endocrine, and neurological abnormalities. Skeletal features include brachycephaly, midface hypoplasia with a small upturned nose, brachydactyly, and lumbar spinal stenosis. Endocrine abnormalities include hypothyroidism and hypogonadism in males and irregular menses in females. Developmental disability is a common finding but is variable in severity and can be associated with significant behavioral problems. {ECO:0000269|PubMed:22464250, ECO:0000269|PubMed:22464252, ECO:0000269|PubMed:23033274, ECO:0000269|PubMed:23043190}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB06842; DB04149; DB03606; DB03183; DB04469; DB02676; DB01959; DB07051; DB04271; DB07954; DB08299; DB00131; DB01427; DB00201; DB03849; DB05219; DB00651; DB06246; DB05266; DB01088; DB01113; DB01791; DB01656; DB01954; DB05298; DB09283; DB02918 Interacts with P32121; P38432; Q0D2H9; Q08AF8; P43360; Q07343; Q13077; P32121; P26769; P38432; Q96CV9; Q8IUH5 EC number EC 3.1.4.53 Uniprot keywords 3D-structure; Alternative splicing; cAMP; Cell membrane; Cytoplasm; Cytoskeleton; Disease variant; Hydrolase; Isopeptide bond; Manganese; Membrane; Metal-binding; Phosphoprotein; Proteomics identification; Reference proteome; Ubl conjugation; Zinc Protein physicochemical properties Chain ID A,B Molecular weight (Da) 37201.9 Length 322 Aromaticity 0.07 Instability index 35.83 Isoelectric point 5.02 Charge (pH=7) -21.16 2D Binding mode Binding energy (Kcal/mol) -8.59 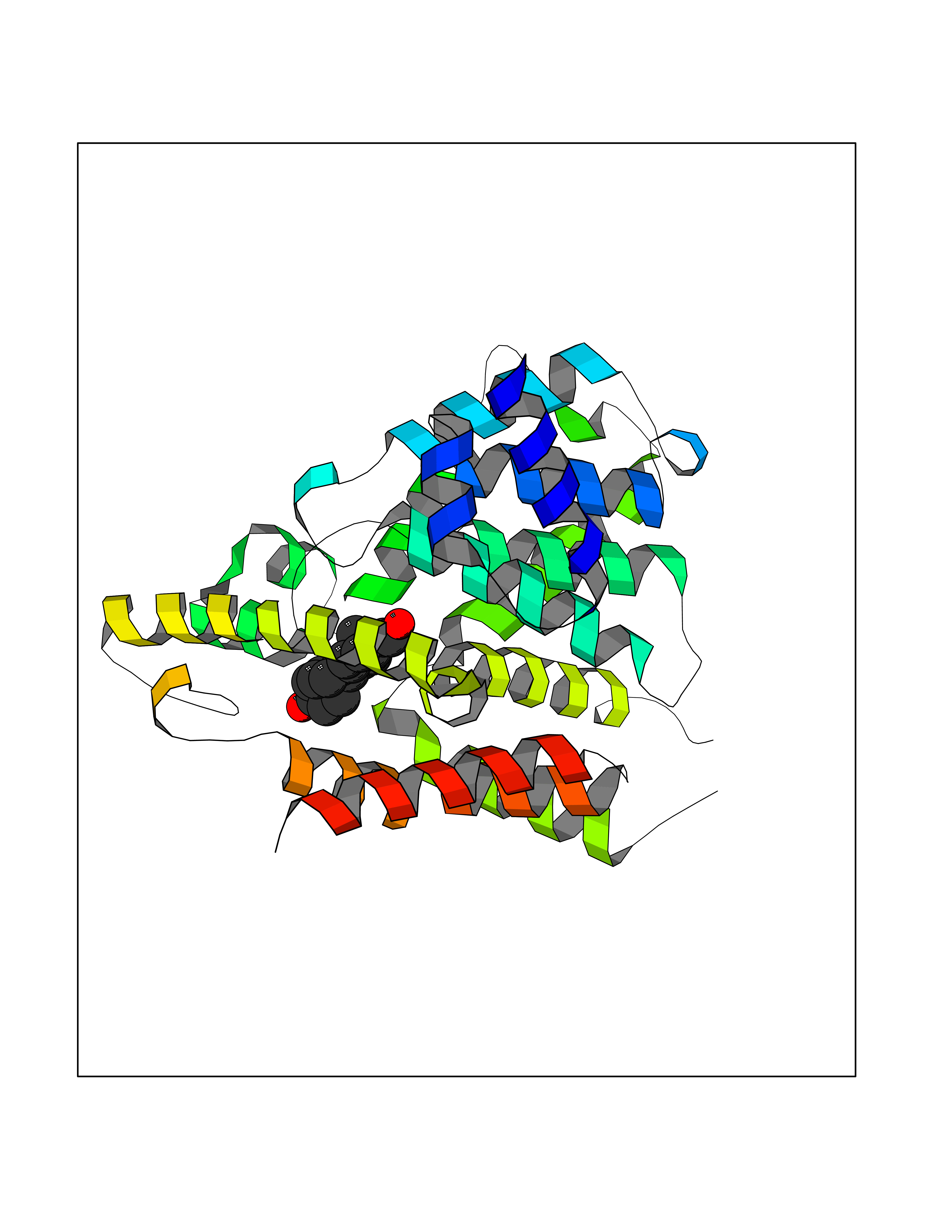 Molscript Map  Pymol Map 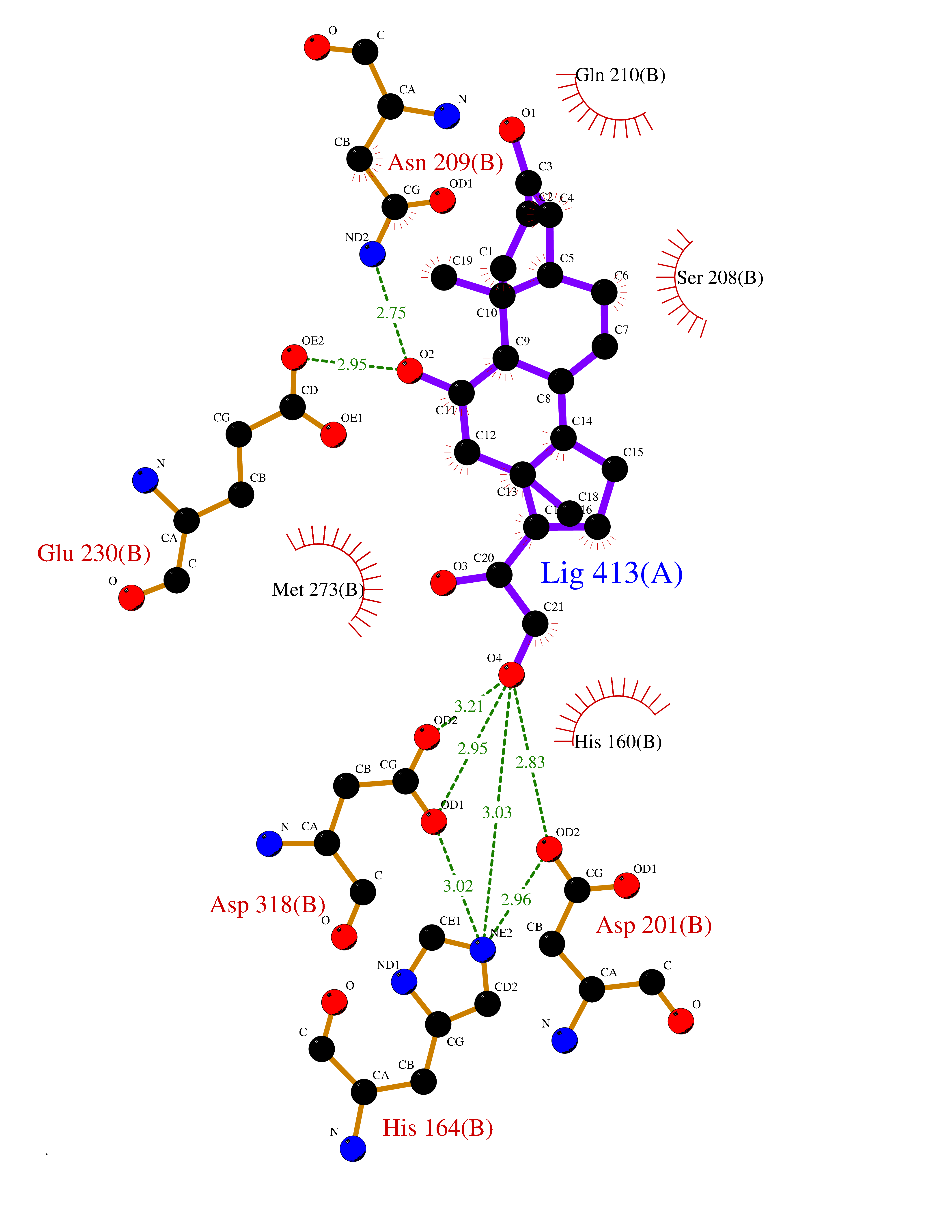 Ligplot Map 3D Binding mode Sequence TEQEDVLAKELEDVNKWGLHVFRIAELSGNRPLTVIMHTIFQERDLLKTFKIPVDTLITYLMTLEDHYHADVAYHNNIHAADVVQSTHVLLSTPALEAVFTDLEILAAIFASAIHDVDHPGVSNQFLINTNSELALMYNDSSVLENHHLAVGFKLLQEENCDIFQNLTKKQRQSLRKMVIDIVLATDMSKHMNLLADLKTMVETKKVVLLLDNYSDRIQVLQNMVHCADLSNPTKPLQLYRQWTDRIMEEFFRQGDRERERGMEISPMCDKHNASVEKSQVGFIDYIVHPLWETWADLVHPDAQDILDTLEDNREWYQSTIP Hydrogen bonds contact Hydrophobic contact | ||||