Ligand
Structure
Job ID
1debdcbe9808324b89371b72b852409f
Job name
Charlie_analyze28
Time
2024-09-18 11:38:30
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 1 | Organic cation transporter 3 (OCT3) | 7ZH6 | 8.31 | |
Target general information Gen name SLC22A3 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Solute carrier family 22 member 3; Extraneuronal monoamine transporter; EMTH Protein family Major facilitator (TC 2.A.1) superfamily, Organic cation transporter (TC 2.A.1.19) family Biochemical class NA Function Mediates potential-dependent transport of a variety of organic cations. May play a significant role in the disposition of cationic neurotoxins and neurotransmitters in the brain. Related diseases Deafness, autosomal dominant, 2A (DFNA2A) [MIM:600101]: A form of non-syndromic sensorineural hearing loss. Sensorineural deafness results from damage to the neural receptors of the inner ear, the nerve pathways to the brain, or the area of the brain that receives sound information. {ECO:0000269|PubMed:10025409, ECO:0000269|PubMed:10369879, ECO:0000269|PubMed:10571947, ECO:0000269|PubMed:10925378, ECO:0000269|PubMed:21242547}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00718; DB08838; DB00182; DB00122; DB14006; DB00501; DB00575; DB00363; DB01151; DB00988; DB00783; DB13952; DB13953; DB13954; DB13955; DB13956; DB00983; DB00536; DB05381; DB00458; DB00762; DB00709; DB00448; DB08882; DB01042; DB01577; DB00331; DB08893; DB00184; DB00368; DB00526; DB00925; DB00413; DB00457; DB01035; DB00396; DB00938; DB00391; DB13943; DB13944; DB08837; DB08841; DB00541 Interacts with P00519 EC number NA Uniprot keywords 3D-structure; Cell membrane; Glycoprotein; Ion transport; Membrane; Mitochondrion; Nucleus; Proteomics identification; Reference proteome; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A Molecular weight (Da) 53067.4 Length 478 Aromaticity 0.13 Instability index 38.82 Isoelectric point 9.07 Charge (pH=7) 10.54 2D Binding mode Binding energy (Kcal/mol) -9.01  Molscript Map 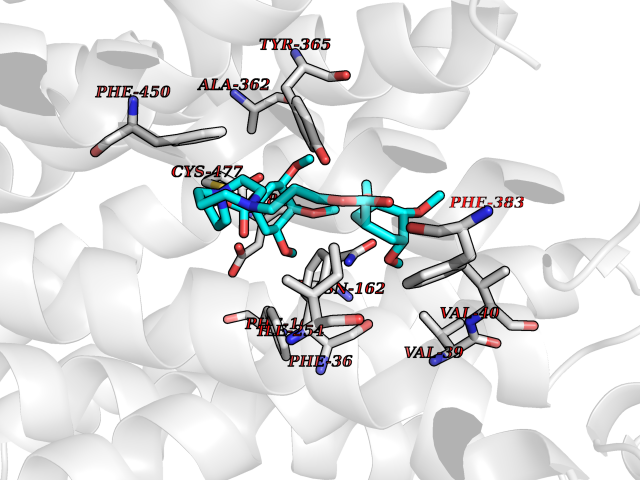 Pymol Map 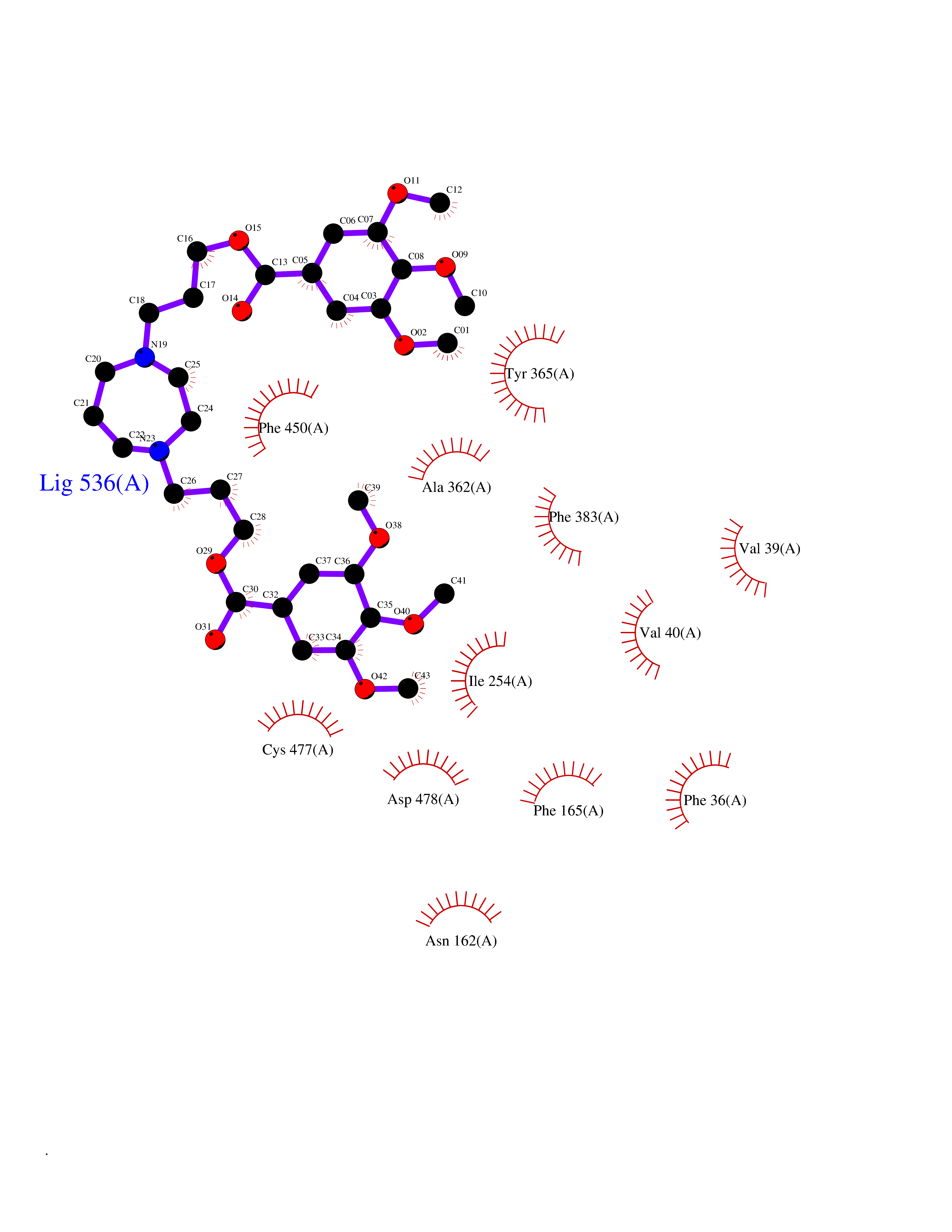 Ligplot Map 3D Binding mode Sequence SFDEALQRVGEFGRFQRRVFLLLCLTGVTFAFLFVGVVFLGTQPDHYWCRGPSAAALAERCGWSPEEEWNRTAPASRGRCQRYLLSAPLVPCRGGWRYAQAHSTIVSEFDLVCVNAWMLDLTQAILNLGFLTGAFTLGYAADRYGRIVIYLLSCLGVGVTGVVVAFAPNFPVFVIFRFLQGVFGKGTWMTCYVIVTEIVGSKQRRIVGIVIQMFFTLGIIILPGIAYFIPNWQGIQLAITLPSFLFLLYYWVVPESPRWLITRKKGDKALQILRRIAKCNVSNPSFLDLVRTPQMRKCTLILMFAWFTSAVVYQGLVMRLGNLYIDFFISGVVELPGALLILLTIERLGRRLPFAASNIVAGVACLVTAFLPEGIAWLRTTVATLGRLGITMAFEIVYLVNSELYPTTLRNFGVSLCSGLCDFGGIIAPFLLFRLAAVWLELPLIIFGILASICGGLVMLLPETKGIALPETVDDVEK Hydrogen bonds contact Hydrophobic contact | ||||
| 2 | Coagulation factor Xa (F10) | 2JKH | 8.07 | |
Target general information Gen name F10 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Fxa; Factor Xa; F10; Activated coagulation factor X Protein family Peptidase S1 family Biochemical class Peptidase Function Factor Xa is avitamin K-dependent glycoprotein that converts prothrombin to thrombin in the presence of factor Va, calcium and phospholipid during blood clotting. Related diseases Factor X deficiency (FA10D) [MIM:227600]: A hemorrhagic disease with variable presentation. Affected individuals can manifest prolonged nasal and mucosal hemorrhage, menorrhagia, hematuria, and occasionally hemarthrosis. Some patients do not have clinical bleeding diathesis. {ECO:0000269|PubMed:10468877, ECO:0000269|PubMed:10739379, ECO:0000269|PubMed:10746568, ECO:0000269|PubMed:11248282, ECO:0000269|PubMed:11728527, ECO:0000269|PubMed:12574802, ECO:0000269|PubMed:12945883, ECO:0000269|PubMed:15075089, ECO:0000269|PubMed:15650540, ECO:0000269|PubMed:17393015, ECO:0000269|PubMed:19135706, ECO:0000269|PubMed:1973167, ECO:0000269|PubMed:1985698, ECO:0000269|PubMed:25313940, ECO:0000269|PubMed:26222694, ECO:0000269|PubMed:2790181, ECO:0000269|PubMed:7669671, ECO:0000269|PubMed:7860069, ECO:0000269|PubMed:8529633, ECO:0000269|PubMed:8845463, ECO:0000269|PubMed:8910490}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07211; DB08746; DB07974; DB07277; DB07605; DB08487; DB08495; DB04673; DB08745; DB08488; DB07804; DB08174; DB08173; DB07872; DB07843; DB07848; DB07875; DB08143; DB07847; DB07844; DB13884; DB06552; DB13151; DB13192; DB00025; DB11166; DB06605; DB09258; DB12364; DB00100; DB13152; DB13150; DB00036; DB09075; DB16662; DB13923; DB01225; DB06920; DB00569; DB03847; DB07278; DB01109; DB06406; DB09332; DB06245; DB13998; DB05713; DB13999; DB07630; DB07629; DB07973; DB07800; DB12598; DB13933; DB06635; DB09141; DB13149; DB11311; DB06228; DB05362; DB07261; DB08426; DB09109; DB14738 Interacts with P0DPK4; Q9UK55; Q9UHD9 EC number EC 3.4.21.6 Uniprot keywords 3D-structure; Blood coagulation; Calcium; Cleavage on pair of basic residues; Direct protein sequencing; Disease variant; Disulfide bond; EGF-like domain; Gamma-carboxyglutamic acid; Glycoprotein; Hemostasis; Hydrolase; Hydroxylation; Protease; Proteomics identification; Reference proteome; Repeat; Secreted; Serine protease; Signal; Zymogen Protein physicochemical properties Chain ID A Molecular weight (Da) 31315.2 Length 280 Aromaticity 0.09 Instability index 38.33 Isoelectric point 6.36 Charge (pH=7) -1.82 2D Binding mode Binding energy (Kcal/mol) -6.45 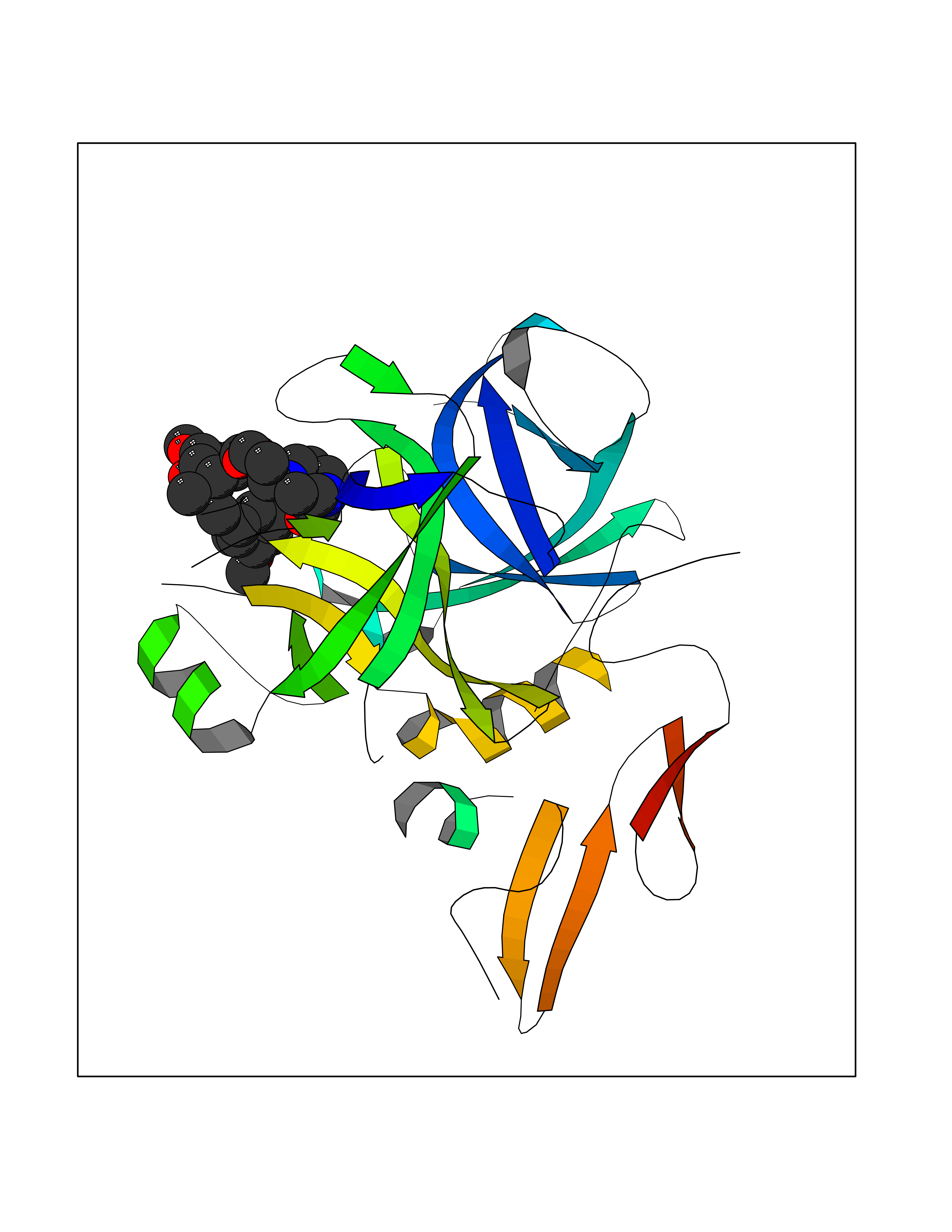 Molscript Map 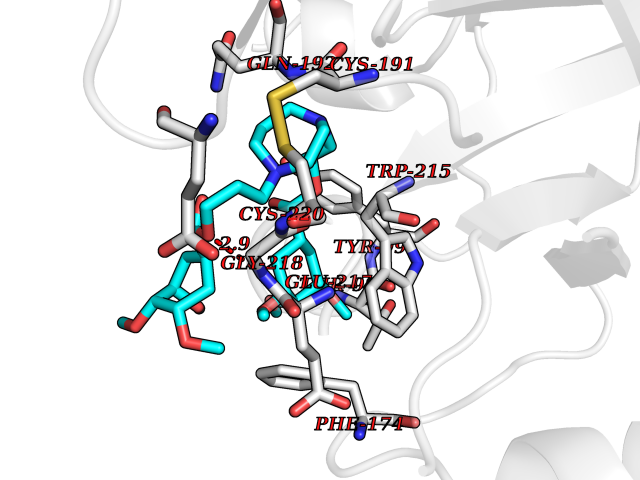 Pymol Map 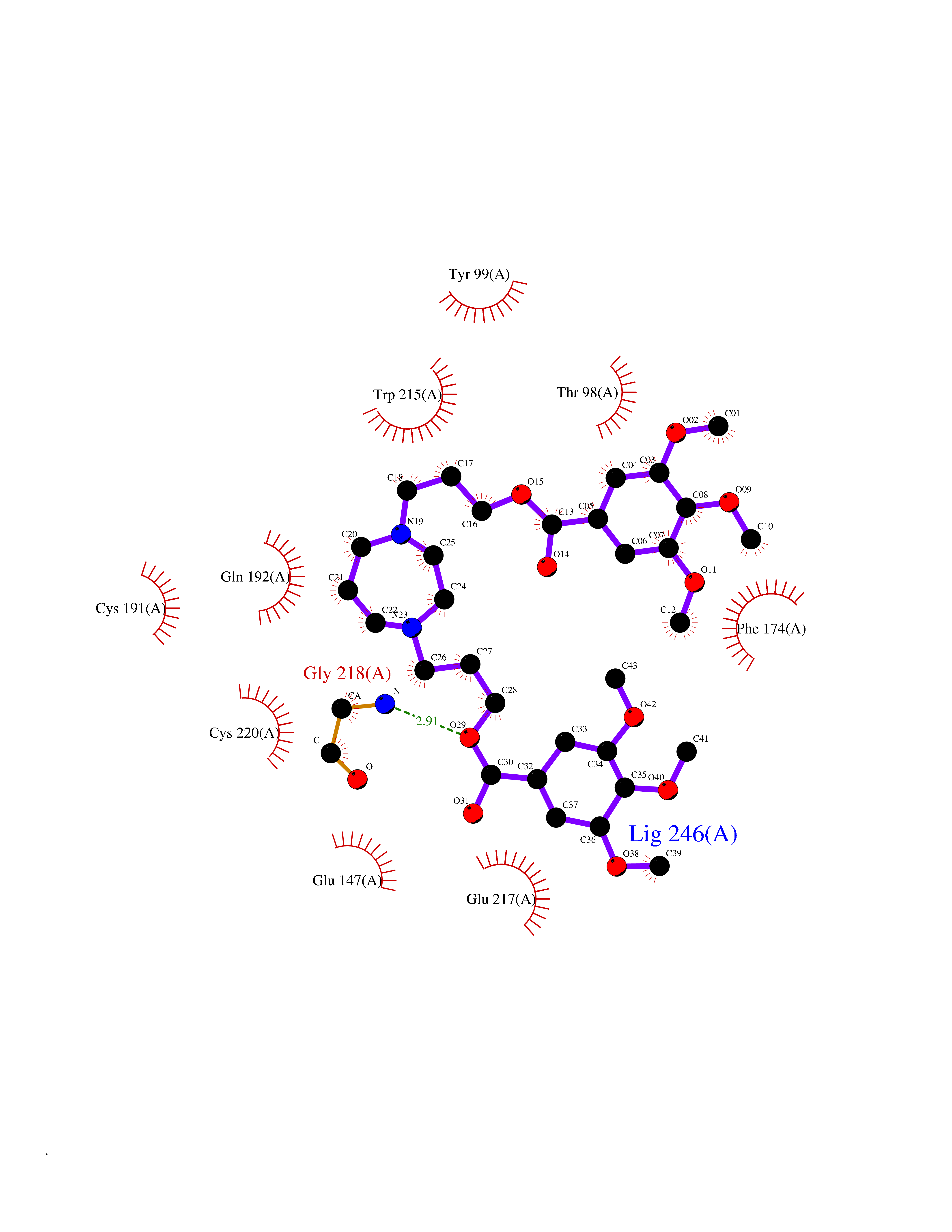 Ligplot Map 3D Binding mode Sequence IVGGQECKDGECPWQALLINEENEGFCGGTILSEFYILTAAHCLYAKRFKVRVGDRNTEQEEGGEAVHEVEVVIKHNRFTKETYDFDIAVLRLKTPITFRMNVAPACLERDWAESMTQKTGIVSGFGRTHEKGEQSTRLKMLEVPYVDRNSCKLSSSFIITQNMFCAGTKQEDACQGDSGGPHVTRFKDTYFVTGIVSWGEGCARGKYGIYTKVTAFLKWIDRSMKKLCSLDNGDCDQFCHEEQNSVVCSCARGYTLADNGKACIPTGPYPCGKQTLERR Hydrogen bonds contact Hydrophobic contact | ||||
| 3 | Retinal rod rhodopsin-sensitive cGMP 3',5'-cyclic phosphodiesterase subunit gamma | 3JWR | 7.95 | |
Target general information Gen name PDE6G Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms PDEG Protein family Rod/cone cGMP-PDE gamma subunit family Biochemical class Hydrolase Function 3',5'-cyclic-GMP phosphodiesterase activity.CGMP binding.Enzyme inhibitor activity.Spectrin binding. Related diseases Retinitis pigmentosa 57 (RP57) [MIM:613582]: A retinal dystrophy belonging to the group of pigmentary retinopathies. Retinitis pigmentosa is characterized by retinal pigment deposits visible on fundus examination and primary loss of rod photoreceptor cells followed by secondary loss of cone photoreceptors. Patients typically have night vision blindness and loss of midperipheral visual field. As their condition progresses, they lose their far peripheral visual field and eventually central vision as well. {ECO:0000269|PubMed:20655036}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07954; DB00203; DB00820; DB00862 Interacts with O14503; Q96JM7; A0A6Q8PF08; O43741; Q8R511; P62994; Q9QY17; Q63787 EC number 3.1.4.35 Uniprot keywords 3D-structure; Acetylation; cGMP; Hydrolase; Reference proteome; Retinitis pigmentosa; Sensory transduction; Vision Protein physicochemical properties Chain ID C,D Molecular weight (Da) 40027.7 Length 345 Aromaticity 0.09 Instability index 37.44 Isoelectric point 6.02 Charge (pH=7) -6.88 2D Binding mode Binding energy (Kcal/mol) -5.96 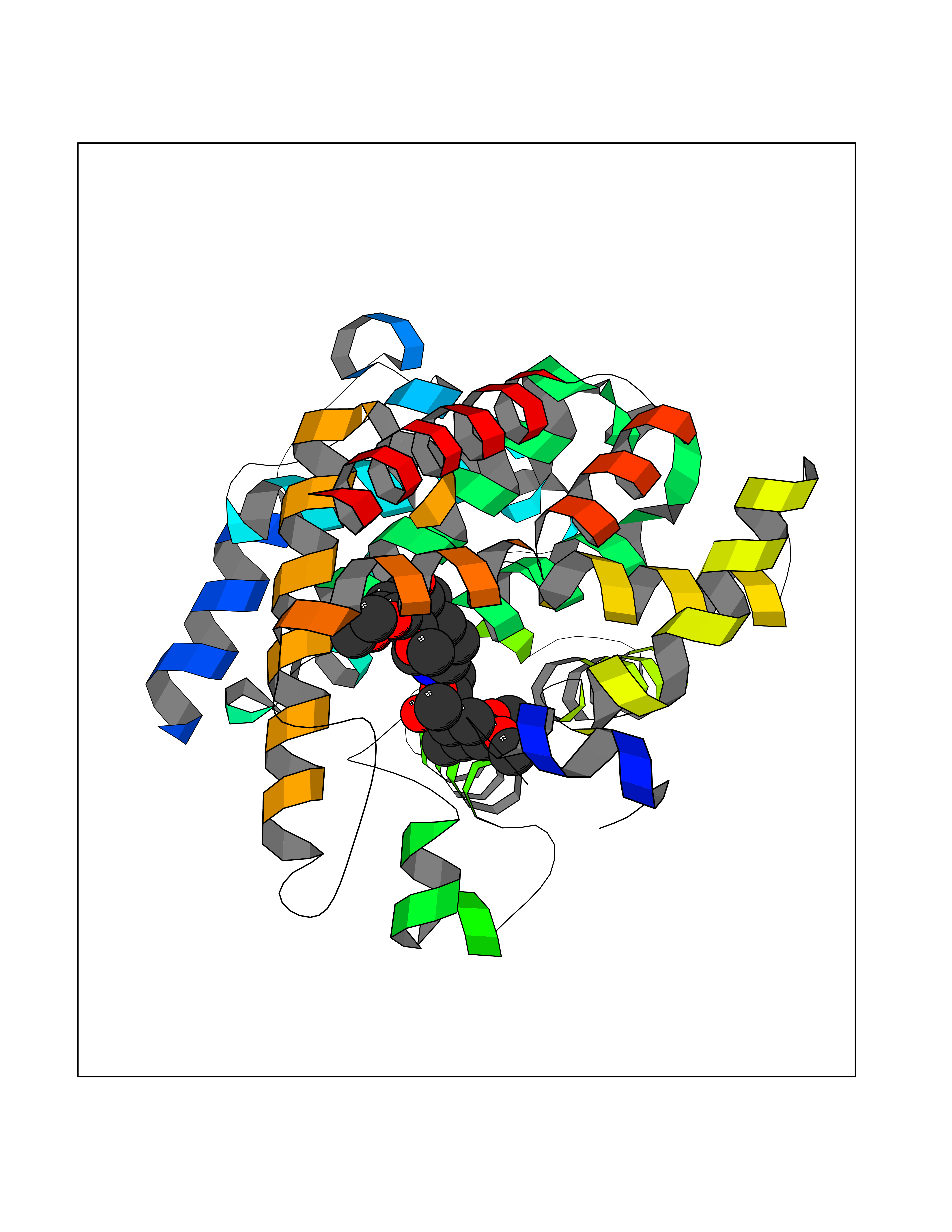 Molscript Map 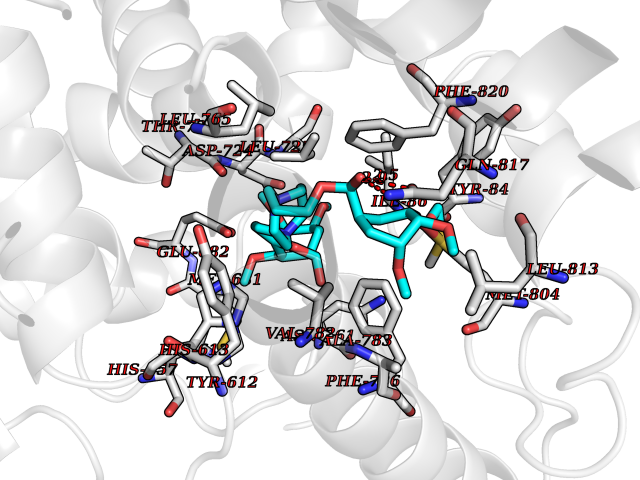 Pymol Map 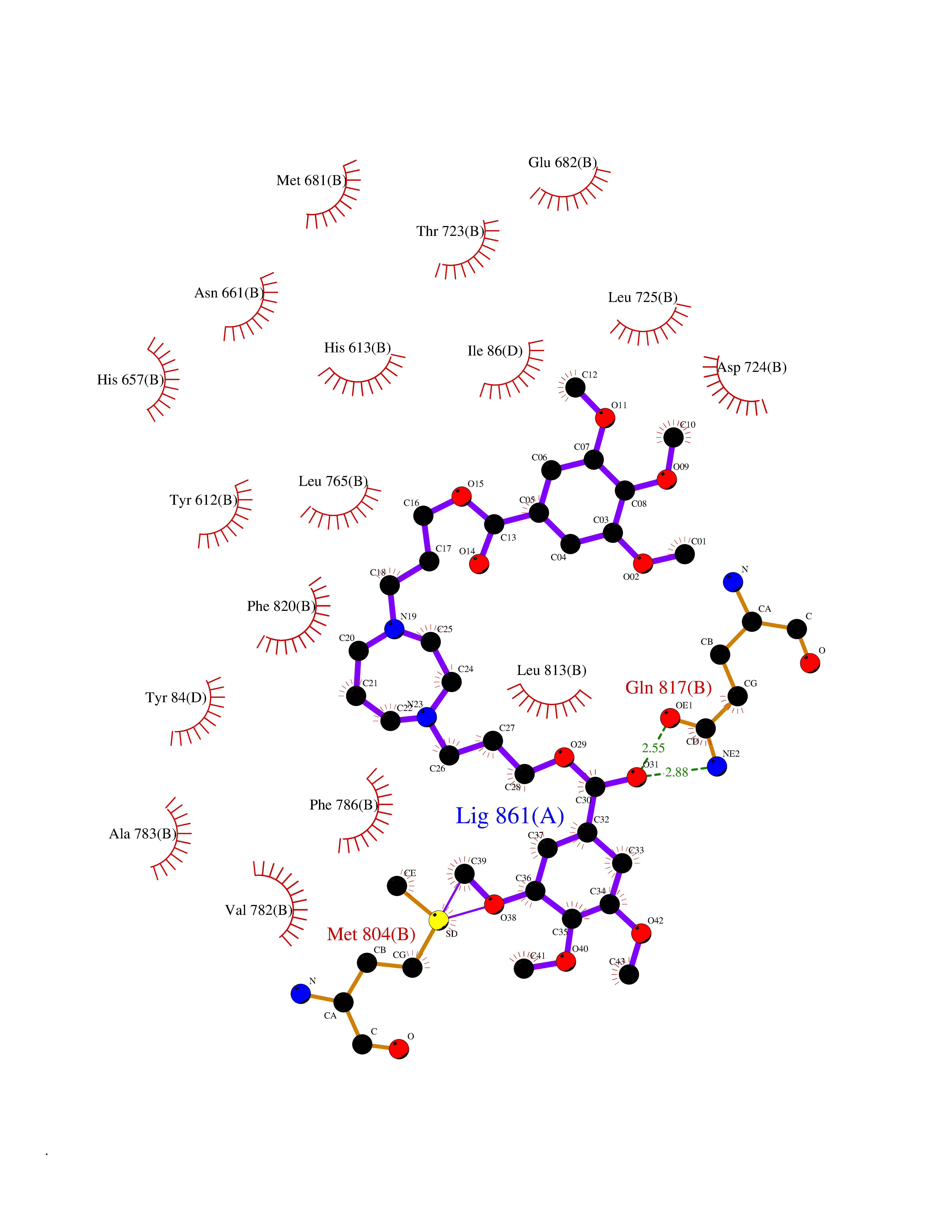 Ligplot Map 3D Binding mode Sequence EAFNHLELHELAQYGIISHMEETRELQSLAAAVVPSAQTLKITDFSFSDFELSDLETALCTIRMFTDLNLVQNFQMKHEVLCRWILSVKKNYRKNVAYHNWRHAFNTAQCMFAALKAGKIQNKLTDLEILALLIAALSHDLDHRGVNNSYIQRSEHPLAQLYCHSIMEHHHFDQCLMILNSPGNQILSGLSIEEYKTTLKIIKQAILATDLALYIKRRGEFFELIRKNQFNLEDPHQKELFLAMLMTACDLSAITKPWPIQQRIAELVATEFWEQGDLERTVLQQQPIPMMDRNKRDELPKLQVGFIDFVCTQLYEALTHVSEDCFPLLDGCRKNRQKWQALAEQ Hydrogen bonds contact Hydrophobic contact | ||||
| 4 | Endothelin-converting enzyme 1 (ECE1) | 3DWB | 7.90 | |
Target general information Gen name ECE1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms ECE-1 Protein family Peptidase M13 family Biochemical class Peptidase Function Converts big endothelin-1 to endothelin-1. Related diseases Hirschsprung disease, cardiac defects, and autonomic dysfunction (HCAD) [MIM:613870]: A disorder characterized by skip-lesions Hirschsprung disease, craniofacial abnormalities and other dysmorphic features, cardiac defects including ductus arteriosus, small subaortic ventricular septal defect, small atrial septal defect, and autonomic dysfunction. {ECO:0000269|PubMed:9915973}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07171 Interacts with P49760; A8MQ03; Q8IUG1; P60370; P60410 EC number EC 3.4.24.71 Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disease variant; Disulfide bond; Glycoprotein; Hirschsprung disease; Hydrolase; Membrane; Metal-binding; Metalloprotease; Phosphoprotein; Protease; Proteomics identification; Reference proteome; Signal-anchor; Transmembrane; Transmembrane helix; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 75247.9 Length 660 Aromaticity 0.12 Instability index 46.29 Isoelectric point 5.33 Charge (pH=7) -18.3 2D Binding mode Binding energy (Kcal/mol) -7.55 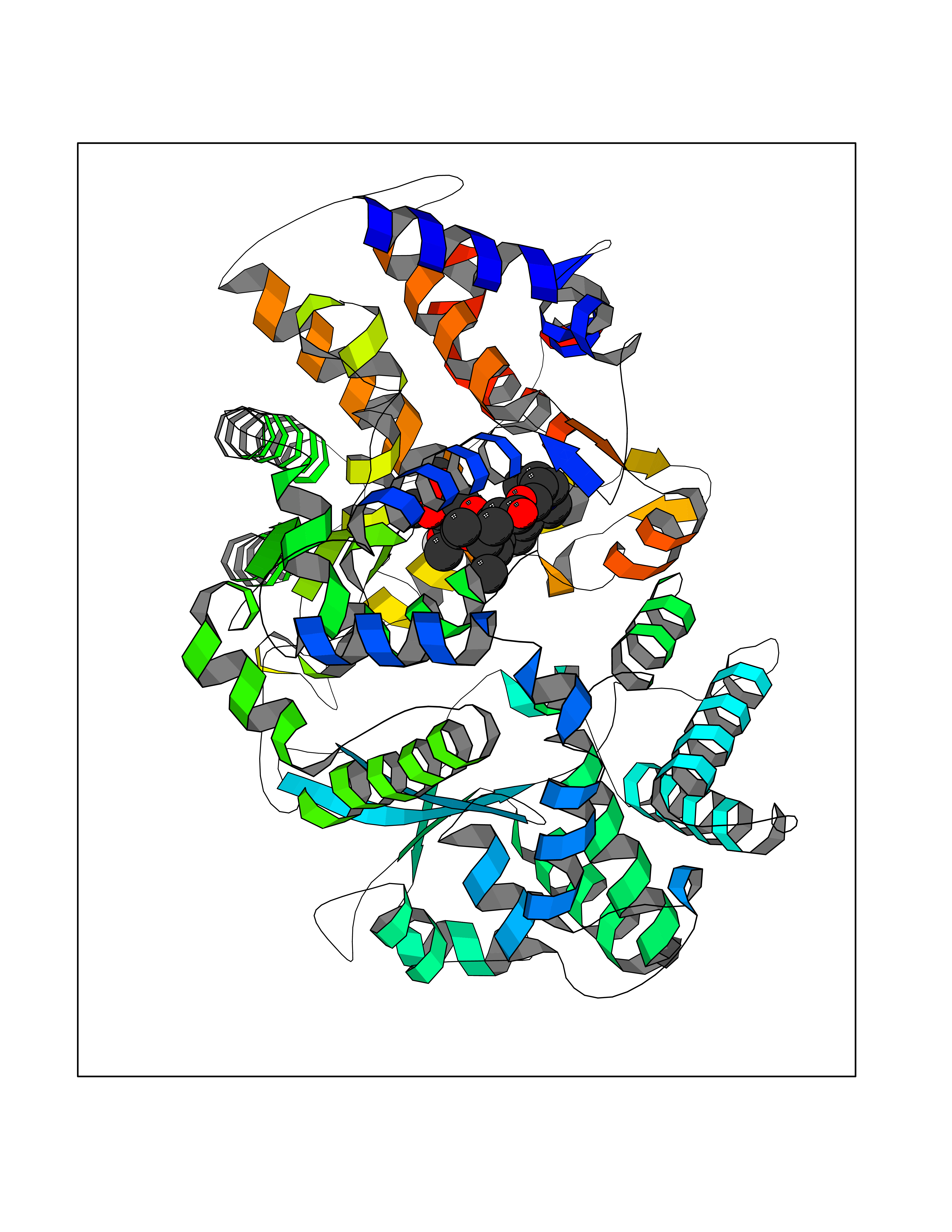 Molscript Map 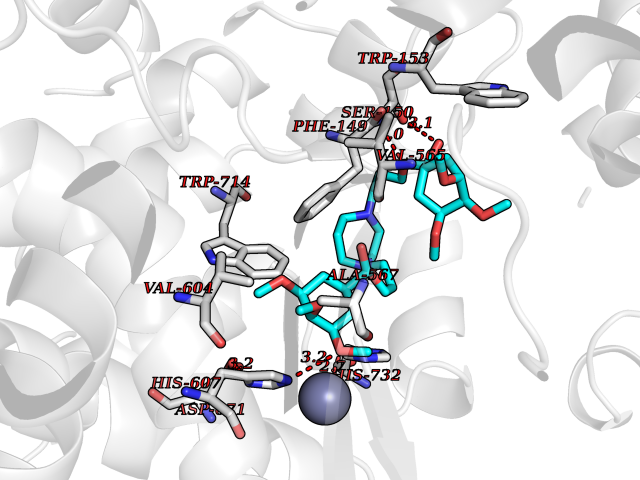 Pymol Map 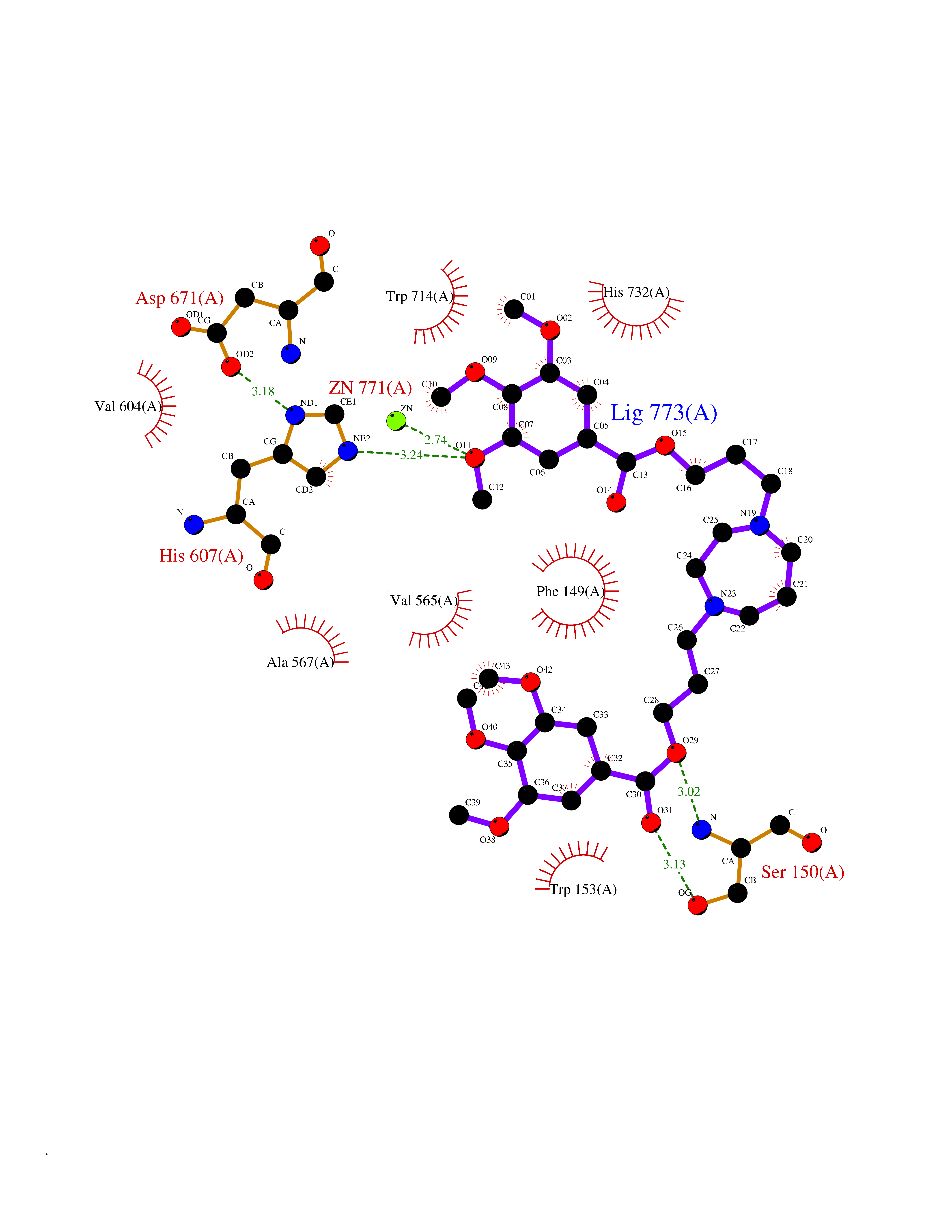 Ligplot Map 3D Binding mode Sequence SEACVSVTSSILSSMDPTVDPCHDFFSYACGGWIKANPVPDGHSRWGTFSNLWEHNQAIIKHLLENSTASVSEAERKAQVYYRACMNETRIEELRAKPLMELIERLGGWNITGPWAKDNFQDTLQVVTAHYRTSPFFSVYVSADSKNSNSNVIQVDQSGLGLPSRDYYLNKTENEKVLTGYLNYMVQLGKLLGGGDEEAIRPQMQQILDFETALANITIPQEKRRDEELIYHKVTAAELQTLAPAINWLPFLNTIFYPVEINESEPIVVYDKEYLEQISTLINTTDRCLLNNYMIWNLVRKTSSFLDQRFQDADEKFMEVMWKFCVSDTENNLGFALGPMFVKATFAEDSKSIATEIILEIKKAFEESLSTLKWMDEETRKSAKEKADAIYNMIGYPNFIMDPKELDKVFNDYTAVPDLYFENAMRFFNFSWRVTADQLRKAPNRDQWSMTPPMVNAYYSPTKNEIVFPAGILQAPFYTRSSPKALNFGGIGVVVGHELTHAFDDQGREYDKDGNLRPWWKNSSVEAFKRQTECMVEQYSNYSVNGEPVNGRHTLGENIADNGGLKAAYRAYQNWVKKNGAEHSLPTLGLTNNQLFFLGFAQVWCSVRTPESSHEGLITDPHSPSRFRVIGSLSNSKEFSEHFRCPPGSPMNPPHKCEVW Hydrogen bonds contact Hydrophobic contact | ||||
| 5 | Platelet glycoprotein VI (GP6) | 5OU7 | 7.89 | |
Target general information Gen name GP6 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Glycoprotein 6; GPVI Protein family NA Biochemical class NA Function Collagen receptor involved in collagen-induced platelet adhesion and activation. Plays a key role in platelet procoagulant activity and subsequent thrombin and fibrin formation. This procoagulant function may contribute to arterial and venous thrombus formation. The signaling pathway involves the FcR gamma-chain, the Src kinases (likely FYN or LYN) and SYK, the adapter protein LAT and leads to the activation of PLCG2. Related diseases Bleeding disorder, platelet-type, 11 (BDPLT11) [MIM:614201]: A mild to moderate bleeding disorder caused by defective platelet activation and aggregation in response to collagen. {ECO:0000269|PubMed:19549989, ECO:0000269|PubMed:19552682}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P06241; P07948; P06241; P07948 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Blood coagulation; Cell membrane; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; Hemostasis; Immunoglobulin domain; Membrane; Proteomics identification; Receptor; Reference proteome; Repeat; Signal; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 19027.4 Length 173 Aromaticity 0.11 Instability index 37.14 Isoelectric point 8.68 Charge (pH=7) 2.52 2D Binding mode Binding energy (Kcal/mol) -5.92  Molscript Map 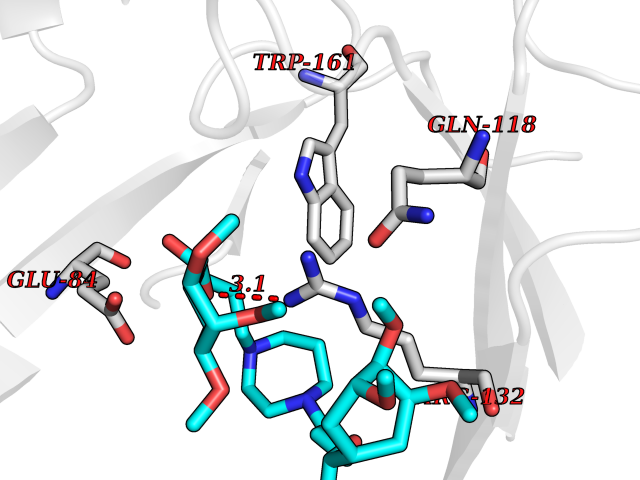 Pymol Map 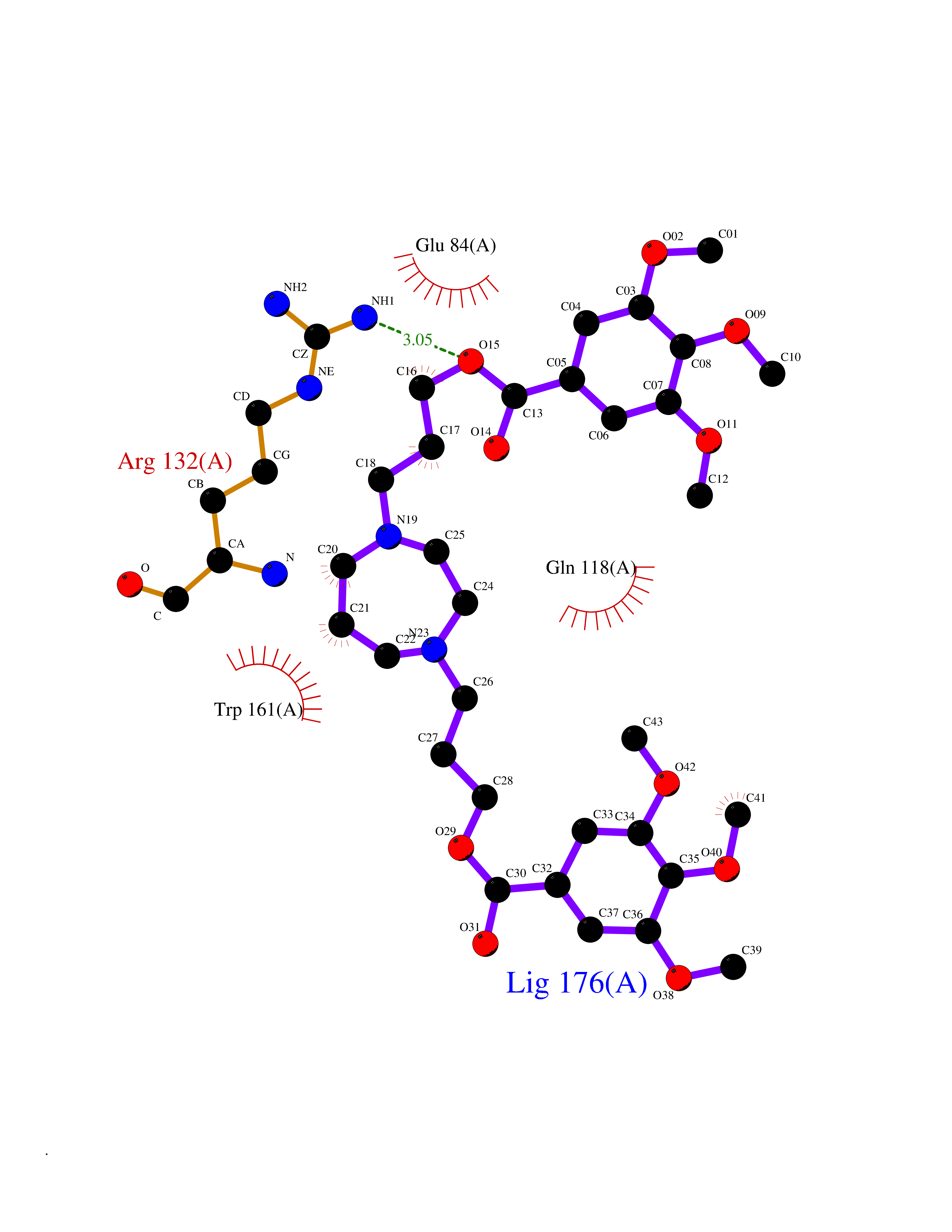 Ligplot Map 3D Binding mode Sequence SGPLPKPSLQALPSSLVPLEKPVTLRCQGPPGVDLYRLEKLSSSRYQDQAVLFIPAMKRSLAGRYRCSYQNGSLWSLPSDQLELVATGVFAKPSLSAQPGSGGDVTLQCQTRYGFDQFALYKEGDPERWYRASFPIITVTAAHSGTYRCYSFSSRDPYLWSAPSDPLELVVTG Hydrogen bonds contact Hydrophobic contact | ||||
| 6 | Kallikrein-4 (KLK4) | 7JOW | 7.88 | |
Target general information Gen name KLK4 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Serine protease 17; Prostase; PSTS; PRSS17; Kallikreinlike protein 1; Kallikrein4; Kallikrein-like protein 1; KLKL1; KLK-L1; Enamel matrix serine proteinase 1; EMSP1 Protein family Peptidase S1 family, Kallikrein subfamily Biochemical class Peptidase Function Required during the maturation stage of tooth development for clearance of enamel proteins and normal structural patterning of the crystalline matrix. Has a major role in enamel formation. Related diseases Amelogenesis imperfecta, hypomaturation type, 2A1 (AI2A1) [MIM:204700]: A defect of enamel formation. The disorder involves both primary and secondary dentitions. The teeth have a shiny agar jelly appearance and the enamel is softer than normal. Brown pigment is present in middle layers of enamel. {ECO:0000269|PubMed:15235027}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P20155; Q06418 EC number EC 3.4.21.- Uniprot keywords 3D-structure; Alternative splicing; Amelogenesis imperfecta; Biomineralization; Direct protein sequencing; Disulfide bond; Glycoprotein; Hydrolase; Metal-binding; Protease; Proteomics identification; Reference proteome; Secreted; Serine protease; Signal; Zinc; Zymogen Protein physicochemical properties Chain ID E,I Molecular weight (Da) 41763.9 Length 387 Aromaticity 0.07 Instability index 32.75 Isoelectric point 5.38 Charge (pH=7) -11.36 2D Binding mode Binding energy (Kcal/mol) -6.96  Molscript Map 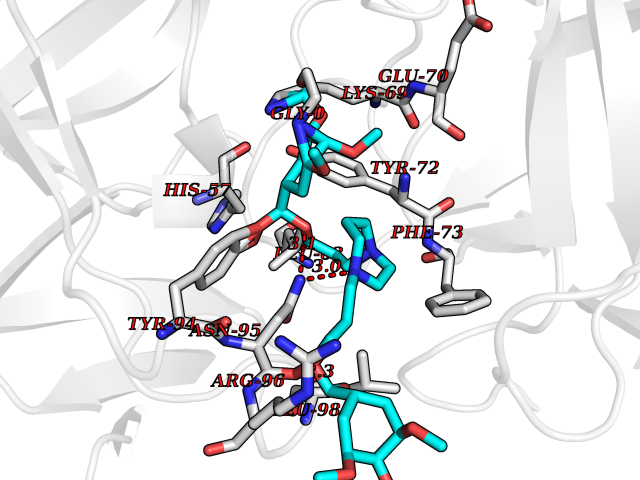 Pymol Map 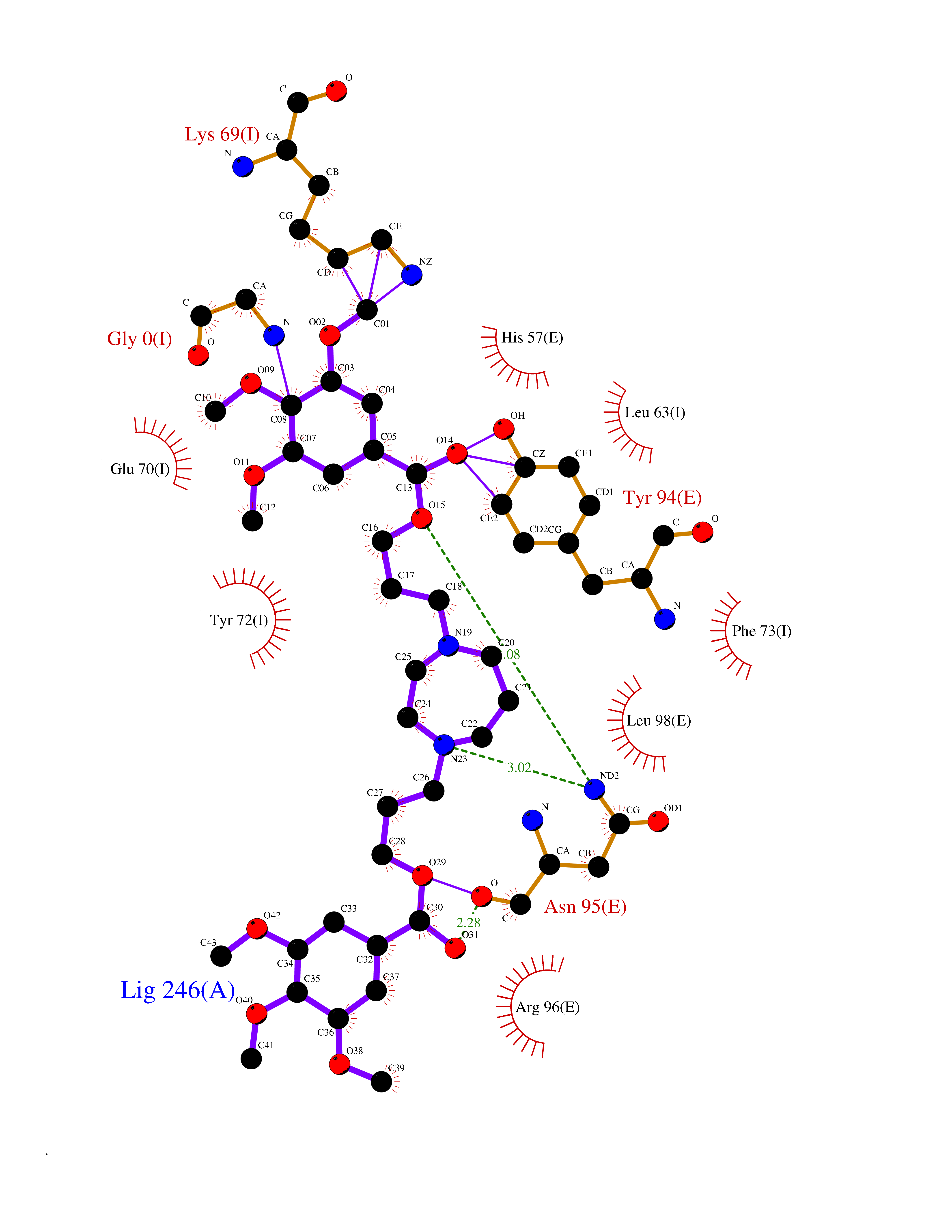 Ligplot Map 3D Binding mode Sequence IINGEDCSPHSQPWQAALVMENELFCSGVLVHPQWVLSAAHCFQNSYTIGLGLHSLEADQEPGSQMVEASLSVRHPEYNRPLLANDLMLIKLDESVSESDTIRSISIASQCPTAGNSCLVSGWGLLANGRMPTVLQCVNVSVVSEEVCSKLYDPLYHPSMFCAGGGQDQKDSCNGDSGGPLICNGYLQGLVSFGKAPCGQVGVPGVYTNLCKFTEWIEKTVQAGSSVVVDTNGQPVSNGADAYYLVPVSHGHAGLALAKIGNEAEPRAVVLDPHHRPGLPVRFESPLRINIIKESYFLNIKFGPSSSDSGVWDVIQQDPIGLAVKVTDTKSLLGPFKVEKEGEGYKIVYYPERGQTGLDIGLVHRNDKYYLAVKDGEPCVFKIRKAT Hydrogen bonds contact Hydrophobic contact | ||||
| 7 | SET domain containing 8 (KMT5A) | 5TEG | 7.88 | |
Target general information Gen name KMT5A Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms SETD8; SET8; SET07; SET domain-containing protein 8; PRSET7; PR/SET07; PR/SET domain-containing protein 07; PR-Set7; N-lysine methyltransferase KMT5A; Lysine-specific methylase 5A; Lysine N-methyltran Protein family Class V-like SAM-binding methyltransferase superfamily, Histone-lysine methyltransferase family, PR/SET subfamily Biochemical class Methyltransferase Function Specifically monomethylates 'Lys-20' of histone H4 (H4K20me1). H4K20me1 is enriched during mitosis and represents a specific tag for epigenetic transcriptional repression. Mainly functions in euchromatin regions, thereby playing a central role in the silencing of euchromatic genes. Required for cell proliferation, probably by contributing to the maintenance of proper higher-order structure of DNA during mitosis. Involved in chromosome condensation and proper cytokinesis. Nucleosomes are preferred as substrate compared to free histones. Mediates monomethylation of p53/TP53 at 'Lys-382', leading to repress p53/TP53-target genes. Plays a negative role in TGF-beta response regulation and a positive role in cell migration. Protein-lysine N-methyltransferase that monomethylates both histones and non-histone proteins. Related diseases Sick sinus syndrome 2 (SSS2) [MIM:163800]: The term 'sick sinus syndrome' encompasses a variety of conditions caused by sinus node dysfunction. The most common clinical manifestations are syncope, presyncope, dizziness, and fatigue. Electrocardiogram typically shows sinus bradycardia, sinus arrest, and/or sinoatrial block. Episodes of atrial tachycardias coexisting with sinus bradycardia ('tachycardia-bradycardia syndrome') are also common in this disorder. SSS occurs most often in the elderly associated with underlying heart disease or previous cardiac surgery, but can also occur in the fetus, infant, or child without heart disease or other contributing factors. SSS2 onset is in utero or at birth. {ECO:0000269|PubMed:15123648, ECO:0000269|PubMed:16407510, ECO:0000269|PubMed:20662977, ECO:0000269|PubMed:23103389}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Brugada syndrome 8 (BRGDA8) [MIM:613123]: A tachyarrhythmia characterized by right bundle branch block and ST segment elevation on an electrocardiogram (ECG). It can cause the ventricles to beat so fast that the blood is prevented from circulating efficiently in the body. When this situation occurs, the individual will faint and may die in a few minutes if the heart is not reset. {ECO:0000269|PubMed:19165230}. The gene represented in this entry may be involved in disease pathogenesis.; DISEASE: Epilepsy, idiopathic generalized 18 (EIG18) [MIM:619521]: An autosomal dominant form of idiopathic generalized epilepsy, a disorder characterized by recurring generalized seizures in the absence of detectable brain lesions and/or metabolic abnormalities. Generalized seizures arise diffusely and simultaneously from both hemispheres of the brain. Seizure types include juvenile myoclonic seizures, absence seizures, and generalized tonic-clonic seizures. EIG18 is characterized by onset of myoclonic seizures in infancy. Although the seizures remit, some patients may have later speech or cognitive impairment. {ECO:0000269|PubMed:30127718}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P62805; P07910; Q15672 EC number EC 2.1.1.- Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Cell cycle; Cell division; Chromatin regulator; Chromosome; Coiled coil; Direct protein sequencing; Methyltransferase; Mitosis; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Repressor; S-adenosyl-L-methionine; Transcription; Transcription regulation; Transferase; Ubl conjugation Protein physicochemical properties Chain ID A,D Molecular weight (Da) 19129.4 Length 167 Aromaticity 0.08 Instability index 49.18 Isoelectric point 7.88 Charge (pH=7) 1.37 2D Binding mode Binding energy (Kcal/mol) -5.7 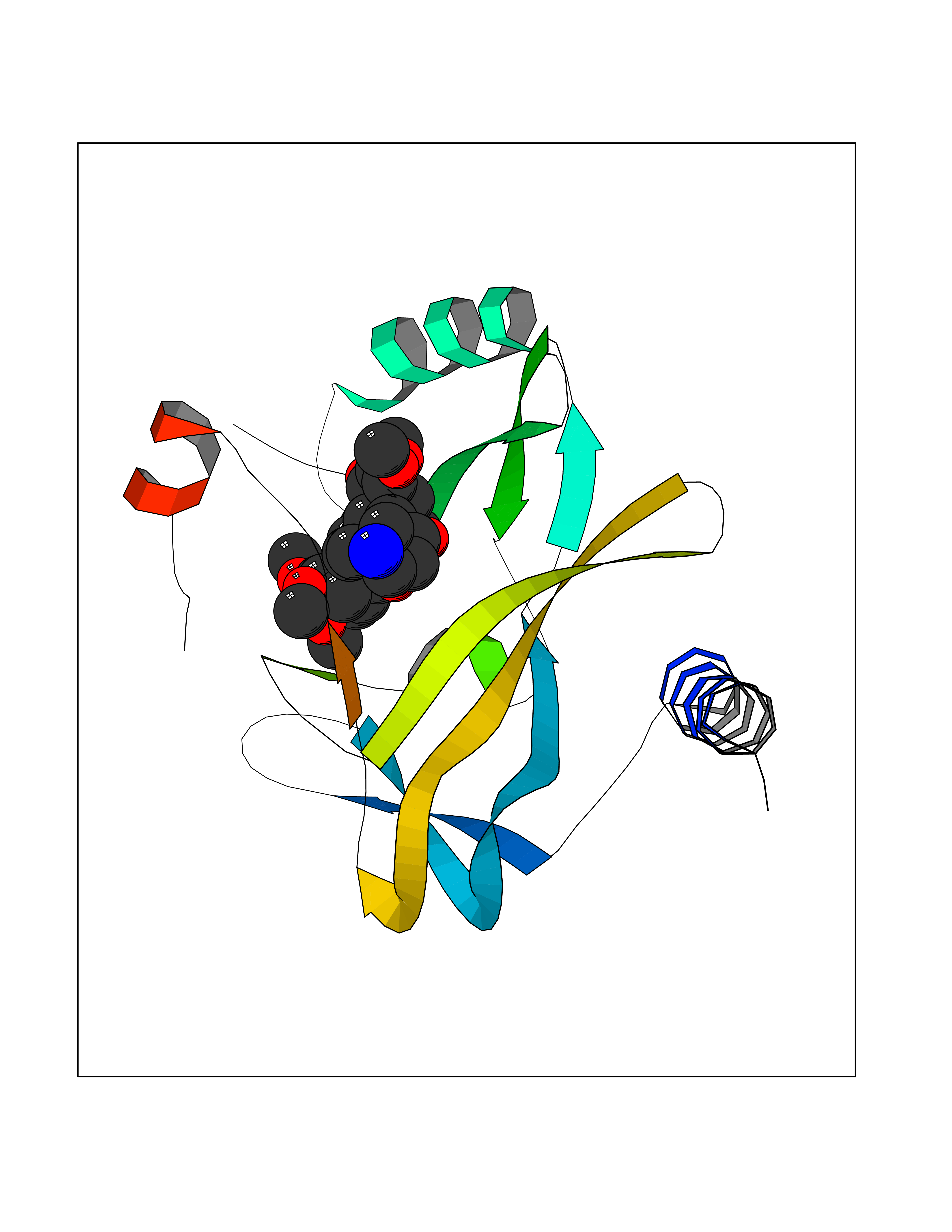 Molscript Map 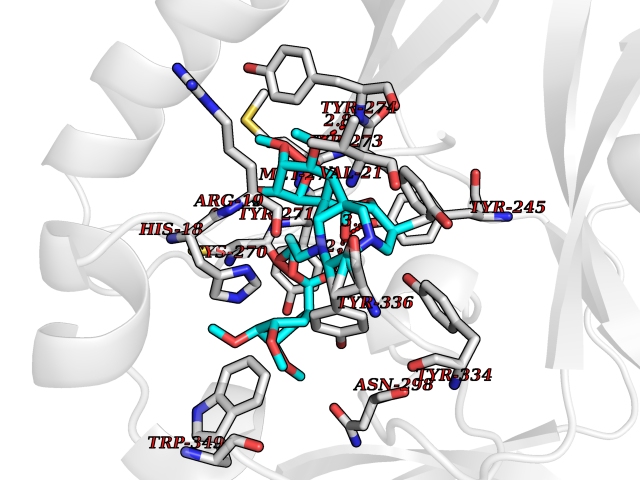 Pymol Map 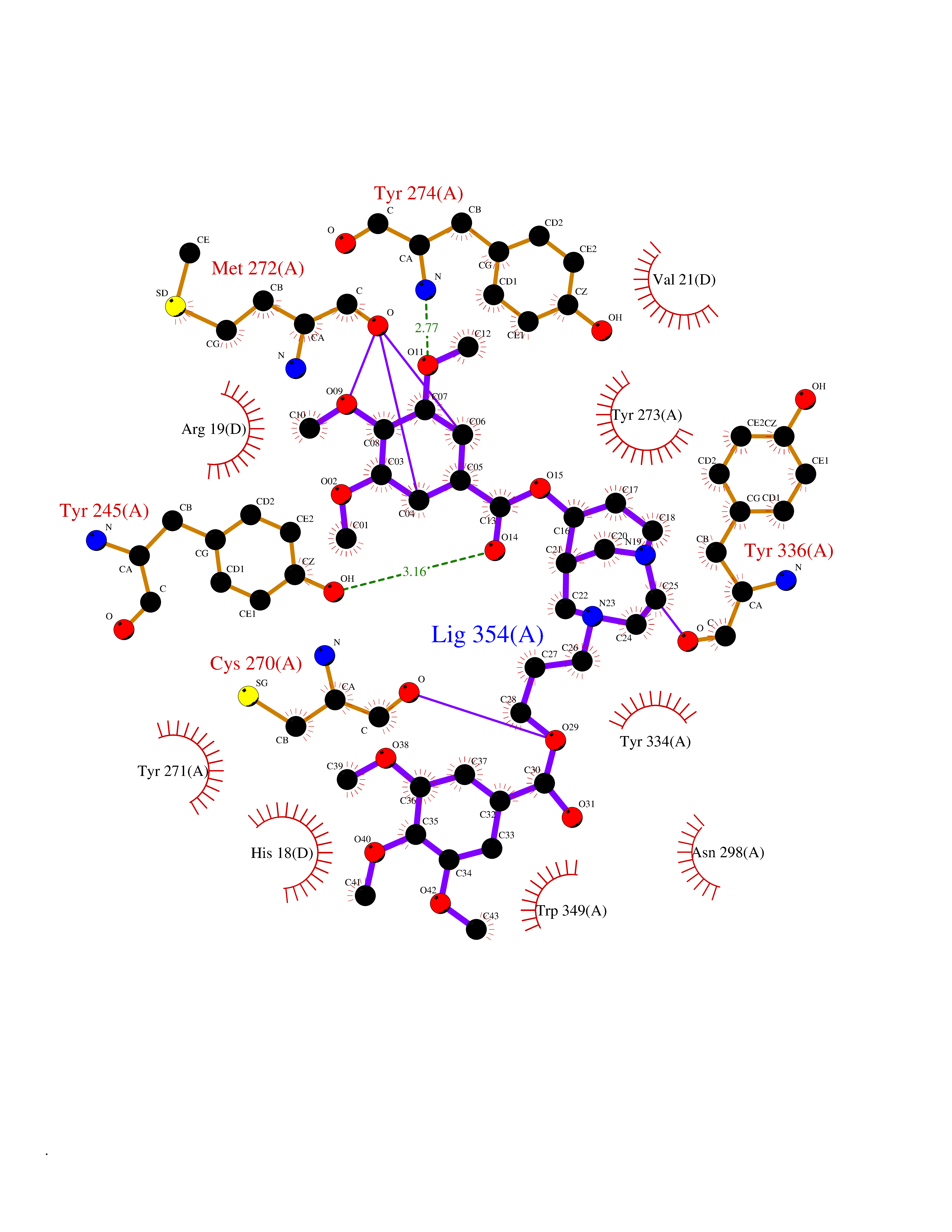 Ligplot Map 3D Binding mode Sequence KSKAELQSEERKRIDELIESGKEEGMKIDLIDGKGRGVIATKQFSRGDFVVEYHGDLIEITDAKKREALYAQDPSTGCYMYYFQYLSKTYCVDATRETNRLGRLINHSKSGNCQTKLHDIDGVPHLILIASRDIAAGEELLYDYGDRSKASIEAHPWLKHKRHRVLR Hydrogen bonds contact Hydrophobic contact | ||||
| 8 | Carbonic anhydrase II (CA-II) | 3K34 | 7.83 | |
Target general information Gen name CA2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Carbonic anhydrase C; Carbonic anhydrase 2; Carbonate dehydratase II; CAC Protein family Alpha-carbonic anhydrase family Biochemical class Alpha-carbonic anhydrase Function Reversible hydration of carbon dioxide. Can hydrate cyanamide to urea. Involved in the regulation of fluid secretion into the anterior chamber of the eye. Contributes to intracellular pH regulation in the duodenal upper villous epithelium during proton-coupled peptide absorption. Stimulates the chloride-bicarbonate exchange activity of SLC26A6. Essential for bone resorption and osteoclast differentiation. Related diseases Osteopetrosis, autosomal recessive 3 (OPTB3) [MIM:259730]: A rare genetic disease characterized by abnormally dense bone, due to defective resorption of immature bone. Osteopetrosis occurs in two forms: a severe autosomal recessive form occurring in utero, infancy, or childhood, and a benign autosomal dominant form occurring in adolescence or adulthood. Recessive osteopetrosis commonly manifests in early infancy with macrocephaly, feeding difficulties, evolving blindness and deafness, bone marrow failure, severe anemia, and hepatosplenomegaly. Deafness and blindness are generally thought to represent effects of pressure on nerves. OPTB3 is associated with renal tubular acidosis, cerebral calcification (marble brain disease) and in some cases with intellectual disability. {ECO:0000269|PubMed:15300855, ECO:0000269|PubMed:1542674, ECO:0000269|PubMed:1928091, ECO:0000269|PubMed:8834238, ECO:0000269|PubMed:9143915}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07596; DB03333; DB08418; DB04081; DB08416; DB02479; DB07467; DB03950; DB03594; DB03294; DB04763; DB03270; DB08083; DB06954; DB08659; DB08046; DB02087; DB08156; DB04203; DB04394; DB08782; DB04549; DB02221; DB04180; DB03039; DB02861; DB02429; DB08202; DB04600; DB04601; DB01784; DB03385; DB03697; DB04002; DB07632; DB07050; DB06891; DB08645; DB08765; DB00819; DB03877; DB03262; DB03598; DB04089; DB01964; DB03526; DB04371; DB02220; DB03221; DB02602; DB02535; DB00436; DB00562; DB01194; DB00482; DB00880; DB02679; DB00606; DB02866; DB01144; DB00869; DB08846; DB01031; DB00311; DB08157; DB01942; DB00695; DB00774; DB08165; DB02292; DB03975; DB00703; DB00232; DB02610; DB07742; DB03844; DB02069; DB02986; DB08301; DB07048; DB03596; DB07476; DB01748; DB08155; DB01671; DB07710; DB01325; DB09460; DB09472; DB02894; DB00391; DB08329; DB07363; DB00273; DB01021; DB03904; DB00580; DB14533; DB14548; DB00909 Interacts with NA EC number EC 4.2.1.1 Uniprot keywords 3D-structure; Acetylation; Cell membrane; Cytoplasm; Direct protein sequencing; Disease variant; Lyase; Membrane; Metal-binding; Osteopetrosis; Phosphoprotein; Proteomics identification; Reference proteome; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 29027.4 Length 258 Aromaticity 0.1 Instability index 20.07 Isoelectric point 6.94 Charge (pH=7) -0.18 2D Binding mode Binding energy (Kcal/mol) -6 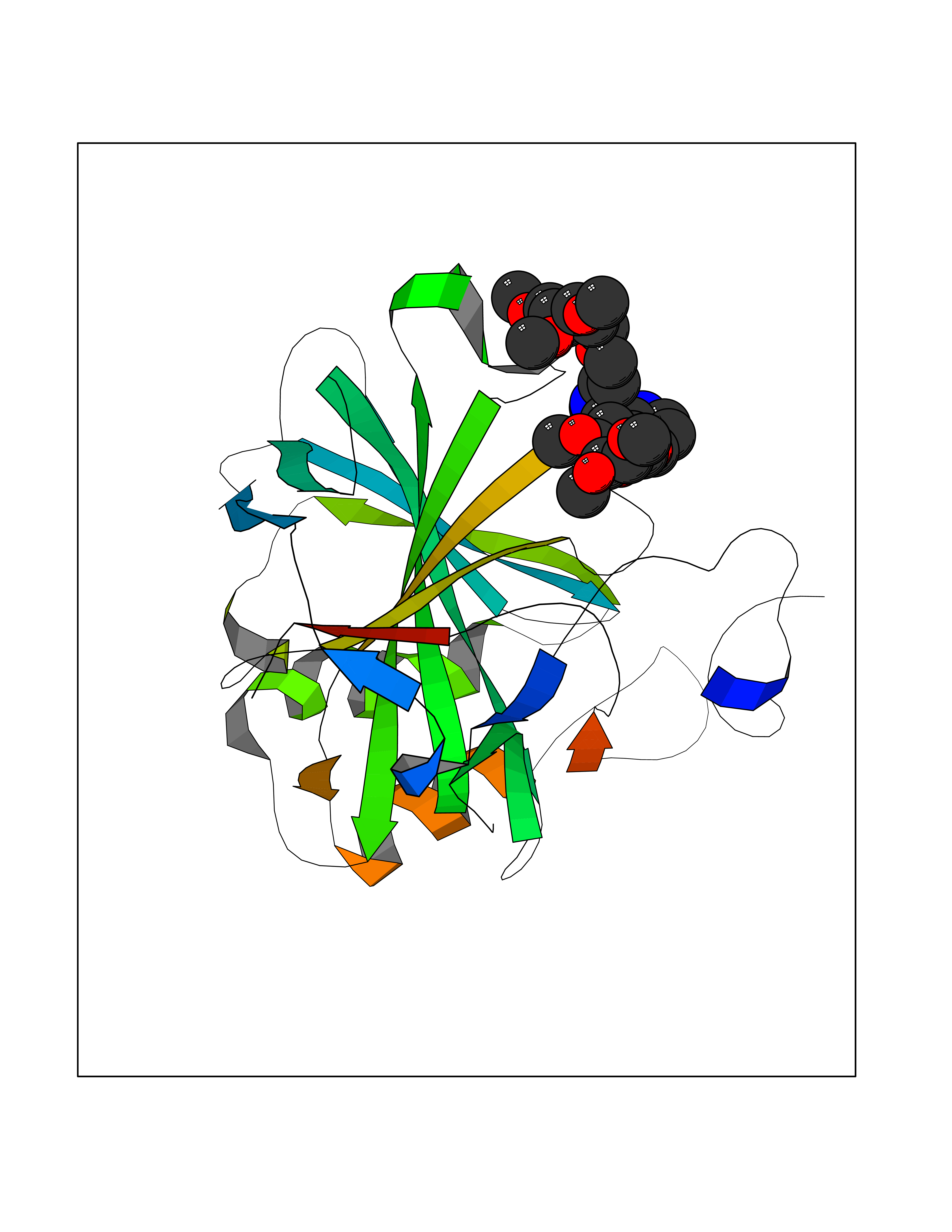 Molscript Map 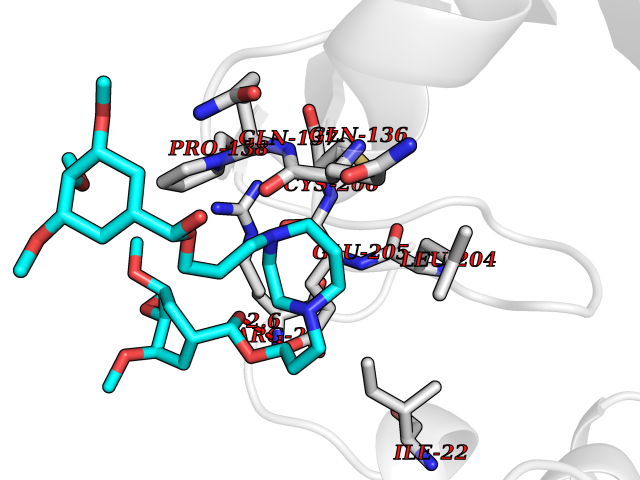 Pymol Map 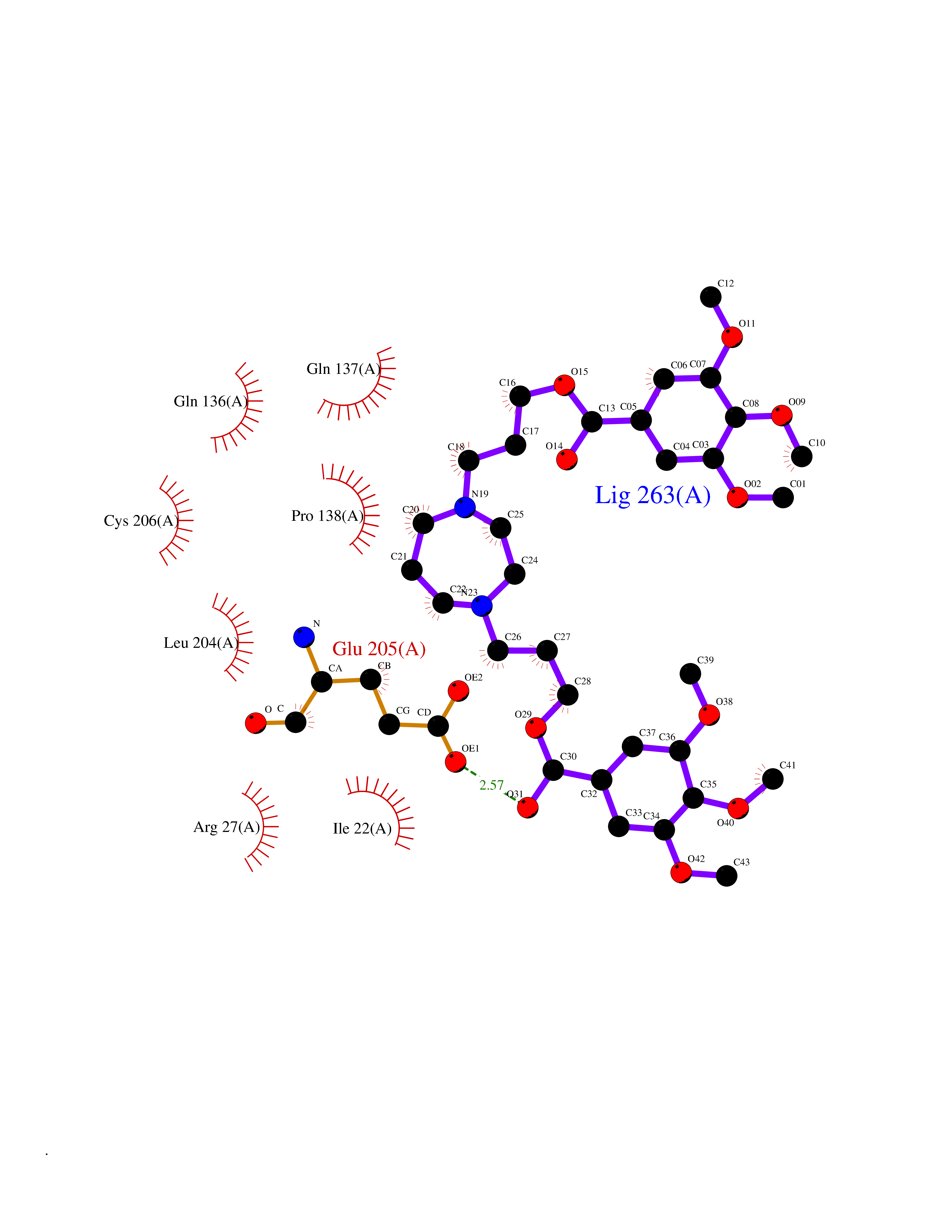 Ligplot Map 3D Binding mode Sequence HHWGYGKHNGPEHWHKDFPIAKGERQSPVDIDTHTAKYDPSLKPLSVSYDQATSLRILNNGHAFNVEFDDSQDKAVLKGGPLDGTYRLIQFHFHWGSLDGQGSEHTVDKKKYAAELHLVHWNTKYGDFGKAVQQPDGLAVLGIFLKVGSAKPGLQKVVDVLDSIKTKGKSADFTNFDPRGLLPESLDYWTYPGSLTTPPLLECVTWIVLKEPISVSSEQVLKFRKLNFNGEGEPEELMVDNWRPAQPLKNRQIKASFK Hydrogen bonds contact Hydrophobic contact | ||||
| 9 | Multidrug resistance protein 3 (ABCB4) | 6S7P | 7.80 | |
Target general information Gen name ABCB4 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms PGY3; MDR3; ATP-binding cassette sub-family B member 4; ABCB4 Protein family ABC transporter superfamily, ABCB family, Multidrug resistance exporter (TC 3.A.1.201) subfamily Biochemical class Acid anhydrides hydrolase Function Mediates ATP-dependent export of organic anions and drugs from the cytoplasm. Hydrolyzes ATP with low efficiency. Not capable of conferring drug resistance. Mediates the translocation of phosphatidylcholine across the canalicular membrane of the hepatocyte. Related diseases Cholestasis, progressive familial intrahepatic, 3 (PFIC3) [MIM:602347]: A disorder characterized by early onset of cholestasis that progresses to hepatic fibrosis, cirrhosis, and end-stage liver disease before adulthood. PFIC3 inheritance is autosomal recessive. {ECO:0000269|PubMed:11313315, ECO:0000269|PubMed:12671900, ECO:0000269|PubMed:17726488, ECO:0000269|PubMed:21119540, ECO:0000269|PubMed:24045840, ECO:0000269|PubMed:24594635, ECO:0000269|PubMed:24806754, ECO:0000269|PubMed:28012258, ECO:0000269|PubMed:9419367}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Cholestasis of pregnancy, intrahepatic 3 (ICP3) [MIM:614972]: A liver disorder of pregnancy. It presents during the second or, more commonly, the third trimester of pregnancy with intense pruritus which becomes more severe with advancing gestation and cholestasis. It causes fetal distress, spontaneous premature delivery and intrauterine death. Patients have spontaneous and progressive disappearance of cholestasis after delivery. Cholestasis results from abnormal biliary transport from the liver into the small intestine. {ECO:0000269|PubMed:10767346, ECO:0000269|PubMed:12746424, ECO:0000269|PubMed:15077010}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Gallbladder disease 1 (GBD1) [MIM:600803]: One of the major digestive diseases. Gallstones composed of cholesterol (cholelithiasis) are the common manifestations in western countries. Most people with gallstones, however, remain asymptomatic through their lifetimes. {ECO:0000269|PubMed:11313316, ECO:0000269|PubMed:12891548, ECO:0000269|PubMed:22331132, ECO:0000269|PubMed:23533021, ECO:0000269|PubMed:24723470, ECO:0000269|PubMed:28012258, ECO:0000269|PubMed:28587926, ECO:0000269|Ref.2}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB06414; DB06207 Interacts with NA EC number EC 7.6.2.1 Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Cell membrane; Cytoplasm; Cytoplasmic vesicle; Disease variant; Glycoprotein; Intrahepatic cholestasis; Lipid transport; Membrane; Nucleotide-binding; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Translocase; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A Molecular weight (Da) 123919 Length 1128 Aromaticity 0.1 Instability index 29.44 Isoelectric point 8.78 Charge (pH=7) 11.08 2D Binding mode Binding energy (Kcal/mol) -7.93 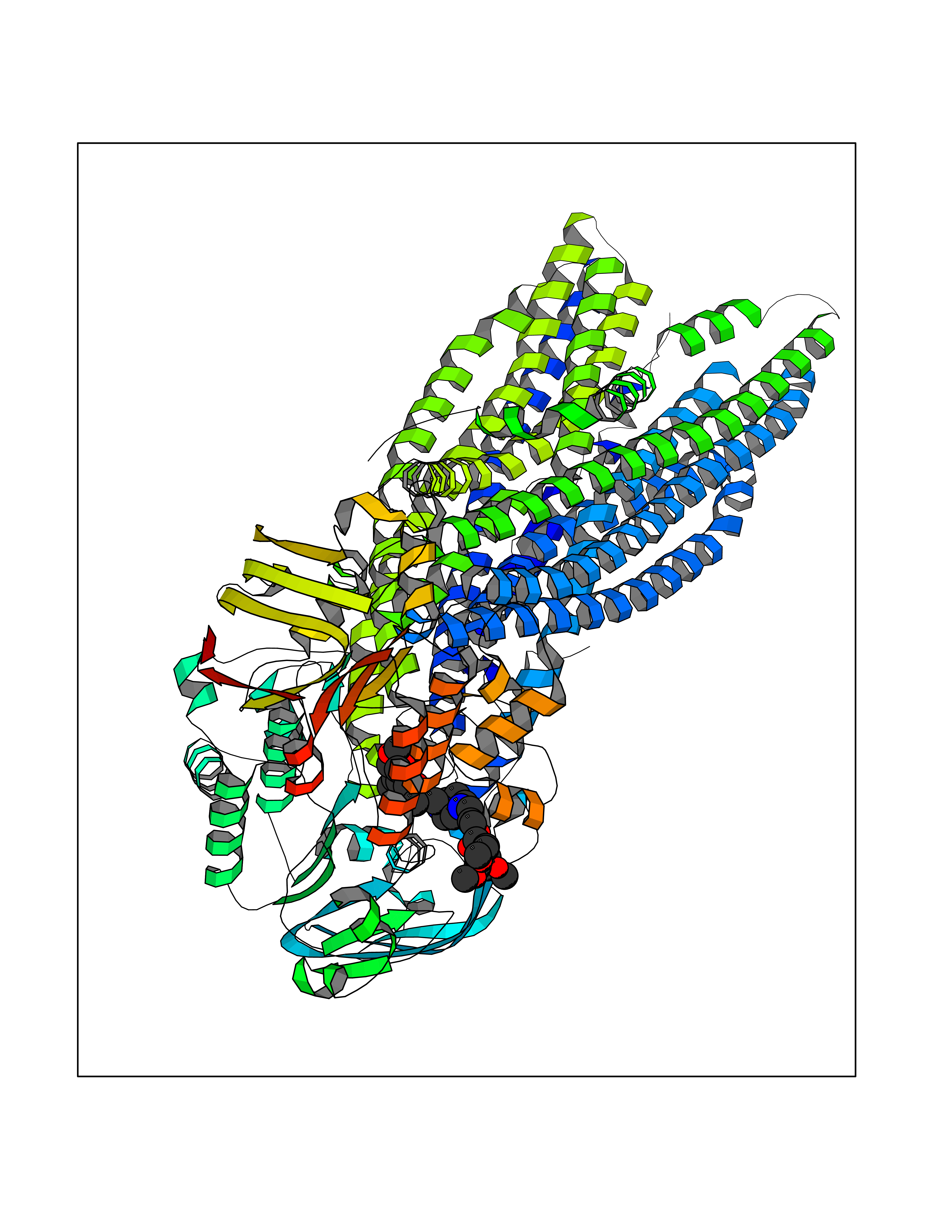 Molscript Map 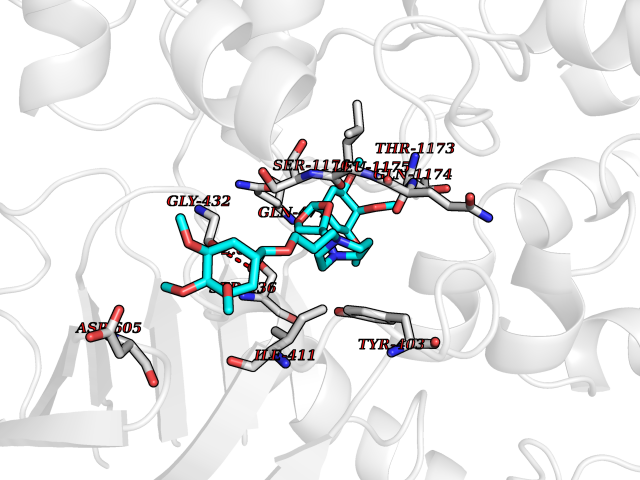 Pymol Map 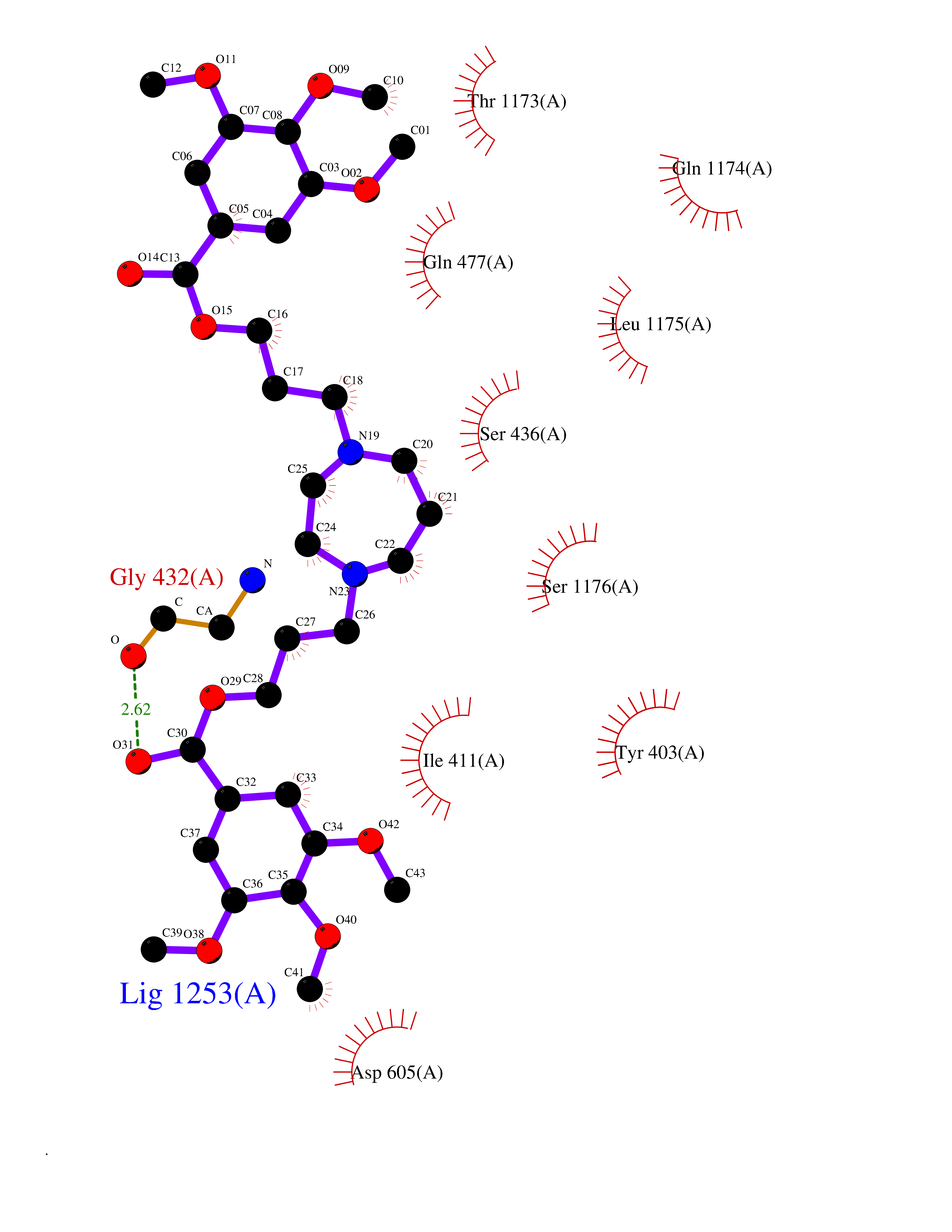 Ligplot Map 3D Binding mode Sequence VLTLFRYSDWQDKLFMSLGTIMAIAHGSGLPLMMIVFGEMTDKPGKILEEEMTRYAYYYSGLGAGVLVAAYIQVSFWTLAAGRQIRKIRQKFFHAILRQEIGWFDINDTTELNTRLTDDISKISEGIGDKVGMFFQAVATFFAGFIVGFIRGWKLTLVIMAISPILGLSAAVWAKILSAFSDKELAAYAKAGAVAEEALGAIRTVIAFGGQNKELERYQKHLENAKEIGIKKAISANISMGIAFLLIYASYALAFWYGSTLVISKEYTIGNAMTVFFSILIGAFSVGQAAPCIDAFANARGAAYVIFDIIDNNPKIDSFSERGHKPDSIKGNLEFNDVHFSYPSRANVKILKGLNLKVQSGQTVALVGSSGCGKSTTVQLIQRLYDPDEGTINIDGQDIRNFNVNYLREIIGVVSQEPVLFSTTIAENICYGRGNVTMDEIKKAVKEANAYEFIMKLPQKFDTLVGERGAQLSGGQKQRIAIARALVRNPKILLLDQATSALDTESEAEVQAALDKAREGRTTIVIAHRLSTVRNADVIAGFEDGVIVEQGSHSELMKKEGVYFKLVNVPPVSFLKVLKLNKTEWPYFVVGTVCAIANGGLQPAFSVIFSEIIAIFGPGDDAVKQQKCNIFSLIFLFLGIISFFTFFLQGFTFGKAGEILTRRLRSMAFKAMLRQDMSWFDDHKNSTGALSTRLATDAAQVQGATGTRLALIAQNIANLGTGIIISFIYGWQLTLLLLAVVPIIAVSGIVEMKLLAGNAKRDKKELEAAGKIATEAIENIRTVVSLTQERKFESMYVEKLYGPYRNSVQKAHIYGITFSISQAFMYFSYAGCFRFGAYLIVNGHMRFRDVILVFSAIVFGAVALGHASSFAPDYAKAKLSAAHLFMLFERQPLIDSYSEEGLKPDKFEGNITFNEVVFNYPTRANVPVLQGLSLEVKKGQTLALVGSSGCGKSTVVQLLERFYDPLAGTVLLDGQEAKKLNVQWLRAQLGIVSQEPILFDCSIAENIAYGDNSRVVSQDEIVSAAKAANIHPFIETLPHKYETRVGDKGTQLSGGQKQRIAIARALIRQPQILLLDQATSALDTESEKVVQEALDKAREGRTCIVIAHRLSTIQNADLIVVFQNGRVK Hydrogen bonds contact Hydrophobic contact | ||||
| 10 | Coagulation factor XII (F12) | 4XDE | 7.79 | |
Target general information Gen name F12 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Hageman factor; HAF Protein family Peptidase S1 family Biochemical class Peptidase Function NA Related diseases Factor XII deficiency (FA12D) [MIM:234000]: An asymptomatic anomaly of in vitro blood coagulation. Its diagnosis is based on finding a low plasma activity of the factor in coagulating assays. It is usually only accidentally discovered through pre-operative blood tests. Factor XII deficiency is divided into two categories, a cross-reacting material (CRM)-negative group (negative F12 antigen detection) and a CRM-positive group (positive F12 antigen detection). {ECO:0000269|PubMed:10361128, ECO:0000269|PubMed:11776307, ECO:0000269|PubMed:15205584, ECO:0000269|PubMed:15617741, ECO:0000269|PubMed:2510163, ECO:0000269|PubMed:2882793, ECO:0000269|PubMed:8049433, ECO:0000269|PubMed:8528215, ECO:0000269|PubMed:9354665}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Angioedema, hereditary, 3 (HAE3) [MIM:610618]: A hereditary angioedema occurring only in women. Hereditary angioedema is an autosomal dominant disorder characterized by episodic local swelling involving subcutaneous or submucous tissue of the upper respiratory and gastrointestinal tracts, face, extremities, and genitalia. Hereditary angioedema type 3 differs from types 1 and 2 in that both concentration and function of C1 esterase inhibitor are normal. Hereditary angioedema type 3 is precipitated or worsened by high estrogen levels (e.g., during pregnancy or treatment with oral contraceptives). {ECO:0000269|PubMed:16638441, ECO:0000269|PubMed:17186468}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB09228; DB06689; DB06404; DB12598; DB01593; DB14487 Interacts with P05067; Q07021; P13473-2 EC number EC 3.4.21.38 Uniprot keywords 3D-structure; Blood coagulation; Direct protein sequencing; Disease variant; Disulfide bond; EGF-like domain; Fibrinolysis; Glycoprotein; Hemostasis; Hydrolase; Kringle; Protease; Proteomics identification; Reference proteome; Repeat; Secreted; Serine protease; Signal; Zymogen Protein physicochemical properties Chain ID A Molecular weight (Da) 26038 Length 241 Aromaticity 0.08 Instability index 50.34 Isoelectric point 5.25 Charge (pH=7) -11.38 2D Binding mode Binding energy (Kcal/mol) -6.17 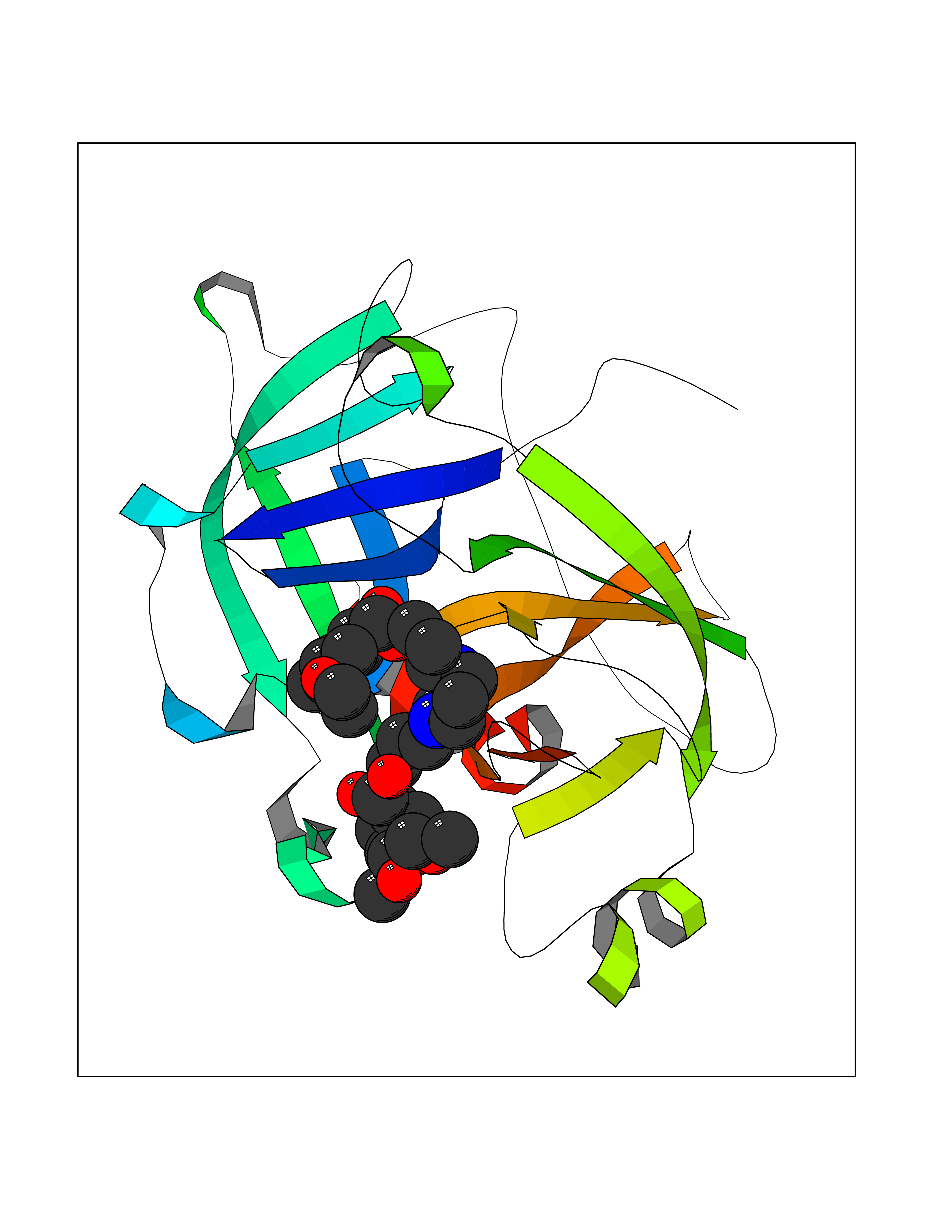 Molscript Map 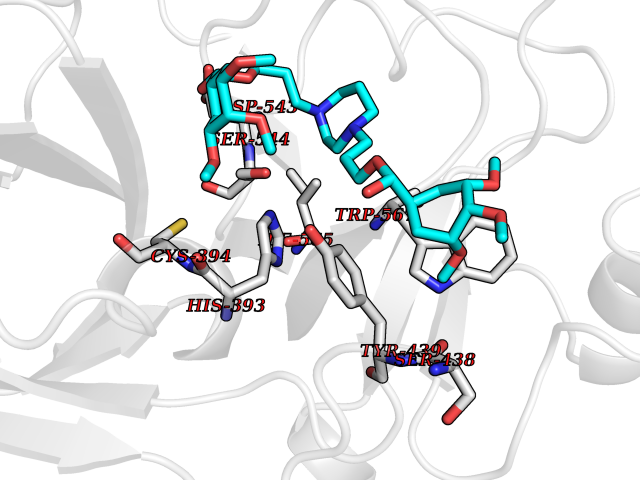 Pymol Map 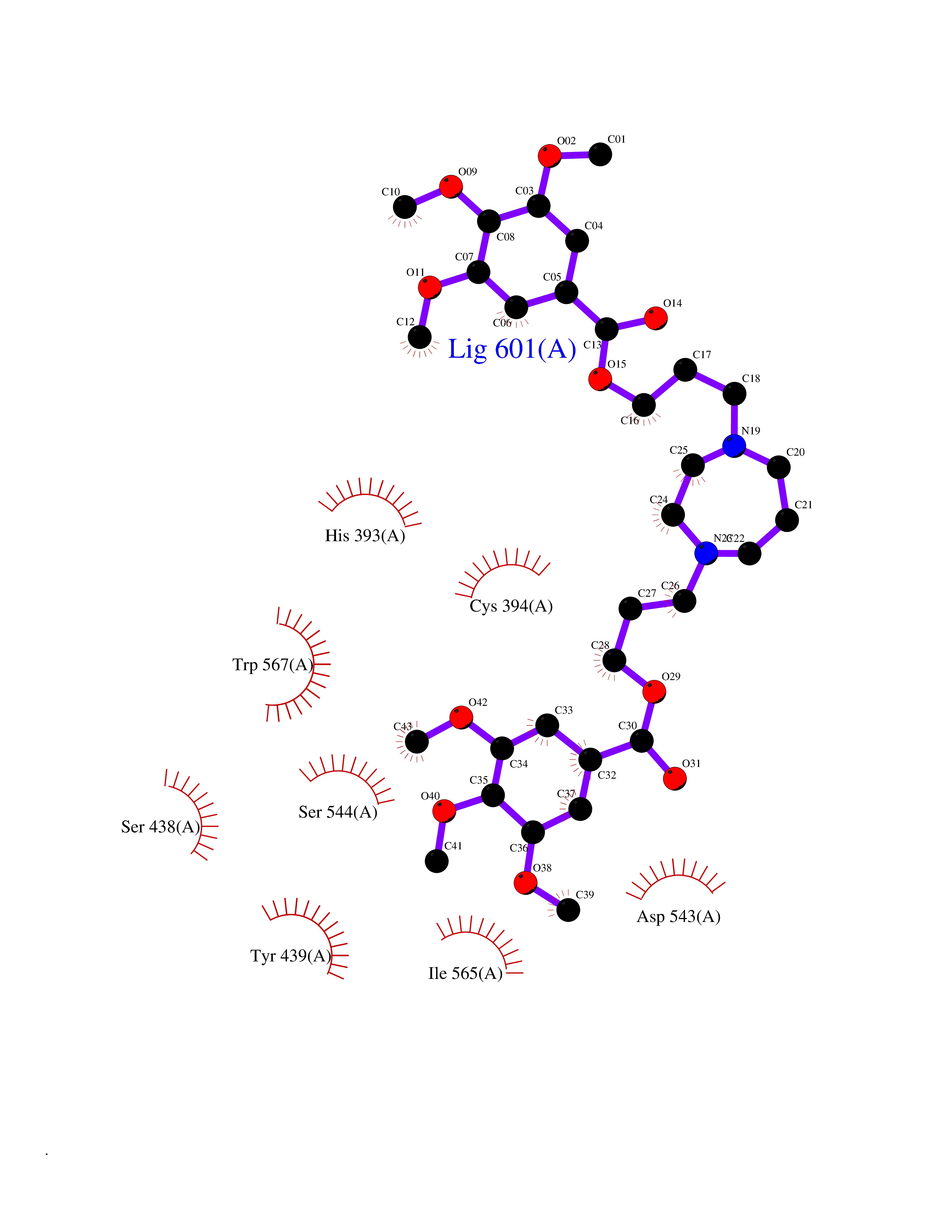 Ligplot Map 3D Binding mode Sequence VALRGAHPYIAALYWGHSFCAGSLIAPCWVLTAAHCLQDRPAPEDLTVVLGQERRNHSCEPCQTLAVRSYRLHEAFSPVSYQHDLALLRLQEDADGSCALLSPYVQPVSLPSGAARPSETTLCQVAGWGHQFEGAEEYASFLQEAQVPFLSLERCSAPDVHGSSILPGMLCAGFLEGGTDACQGDSGGPLVCEDQAAERRLTLQGIISWGSGCGDRNKPGVYTDVAYYLAWIREHTVSHHT Hydrogen bonds contact Hydrophobic contact | ||||
| 11 | Interleukin 21 receptor (IL21R) | 6PLH | 7.72 | |
Target general information Gen name IL21R Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms UNQ3121/PRO10273; Novel interleukin receptor; NILR; Interleukin-21 receptor; IL21 receptor; IL-21R; IL-21 receptor; CD360 Protein family Type I cytokine receptor family, Type 4 subfamily Biochemical class Cytokine receptor Function This is a receptor for interleukin-21. Related diseases Immunodeficiency 56 (IMD56) [MIM:615207]: An autosomal recessive primary immunodeficiency characterized by B- and T-cell defects and variable dysfunction of NK cells. Patients tend to have normal numbers of lymphocytes, but show defective class-switched B-cells, low IgG, defective antibody response, and defective T-cell responses to certain antigens. {ECO:0000269|PubMed:23440042}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Chromosomal aberrations involving IL21R is a cause of B-cell non-Hodgkin lymphomas (B-cell NHL). Translocation t(3;16)(q27;p11), with BCL6. Drugs (DrugBank ID) NA Interacts with P29972 EC number NA Uniprot keywords 3D-structure; Chromosomal rearrangement; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; Membrane; Proteomics identification; Receptor; Reference proteome; Repeat; Signal; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A,C,B Molecular weight (Da) 48376.5 Length 446 Aromaticity 0.1 Instability index 43.94 Isoelectric point 8.24 Charge (pH=7) 3.56 2D Binding mode Binding energy (Kcal/mol) -6.47 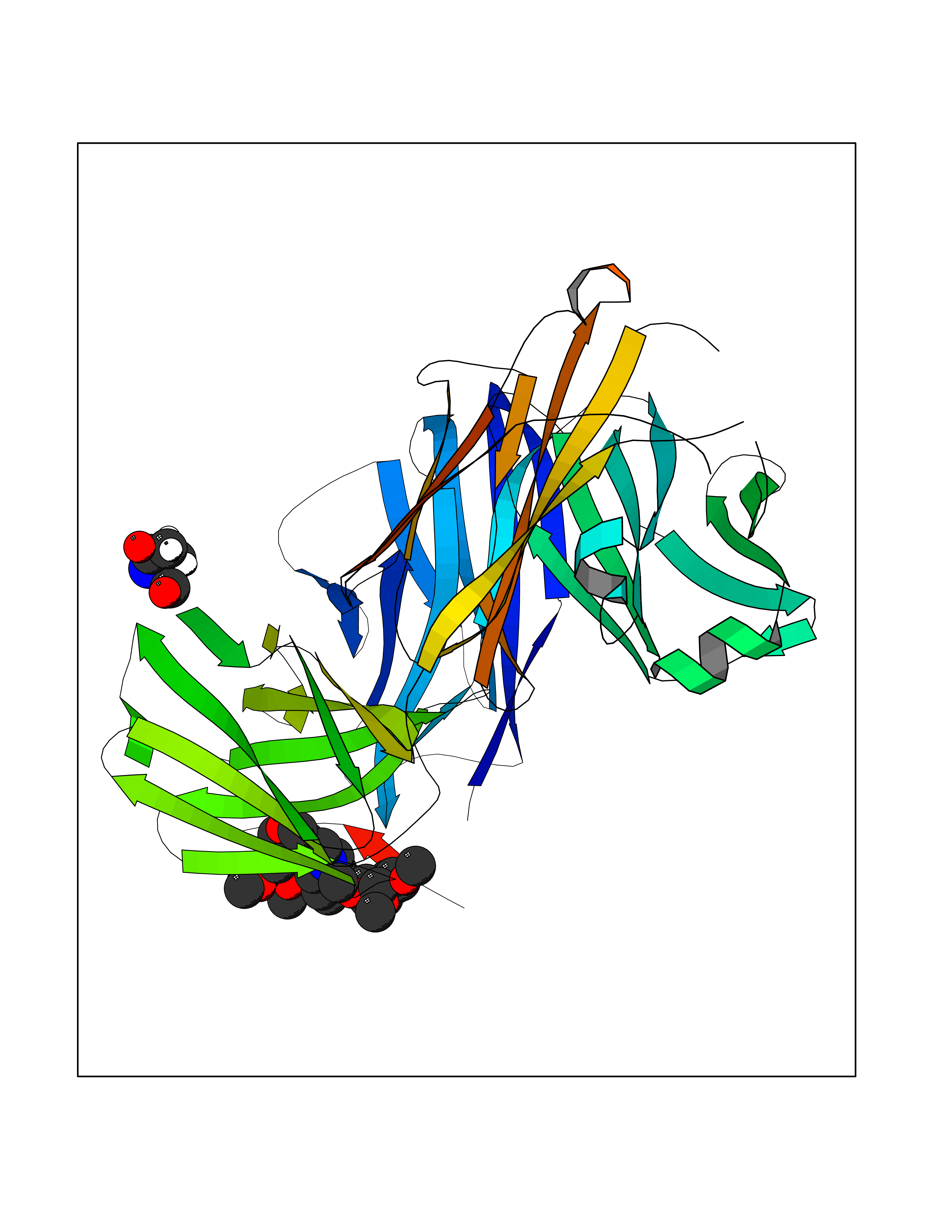 Molscript Map 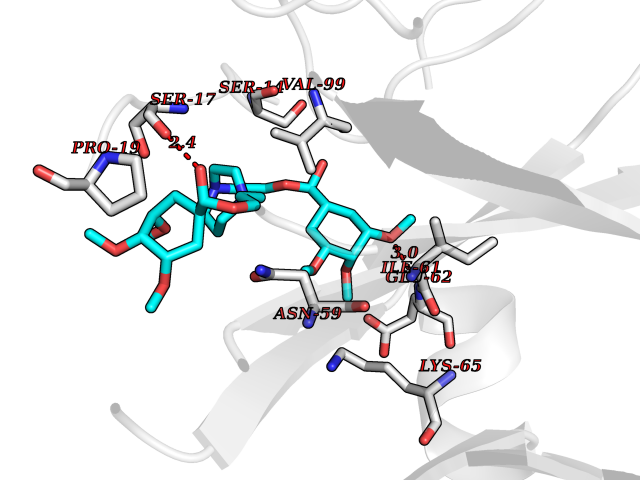 Pymol Map 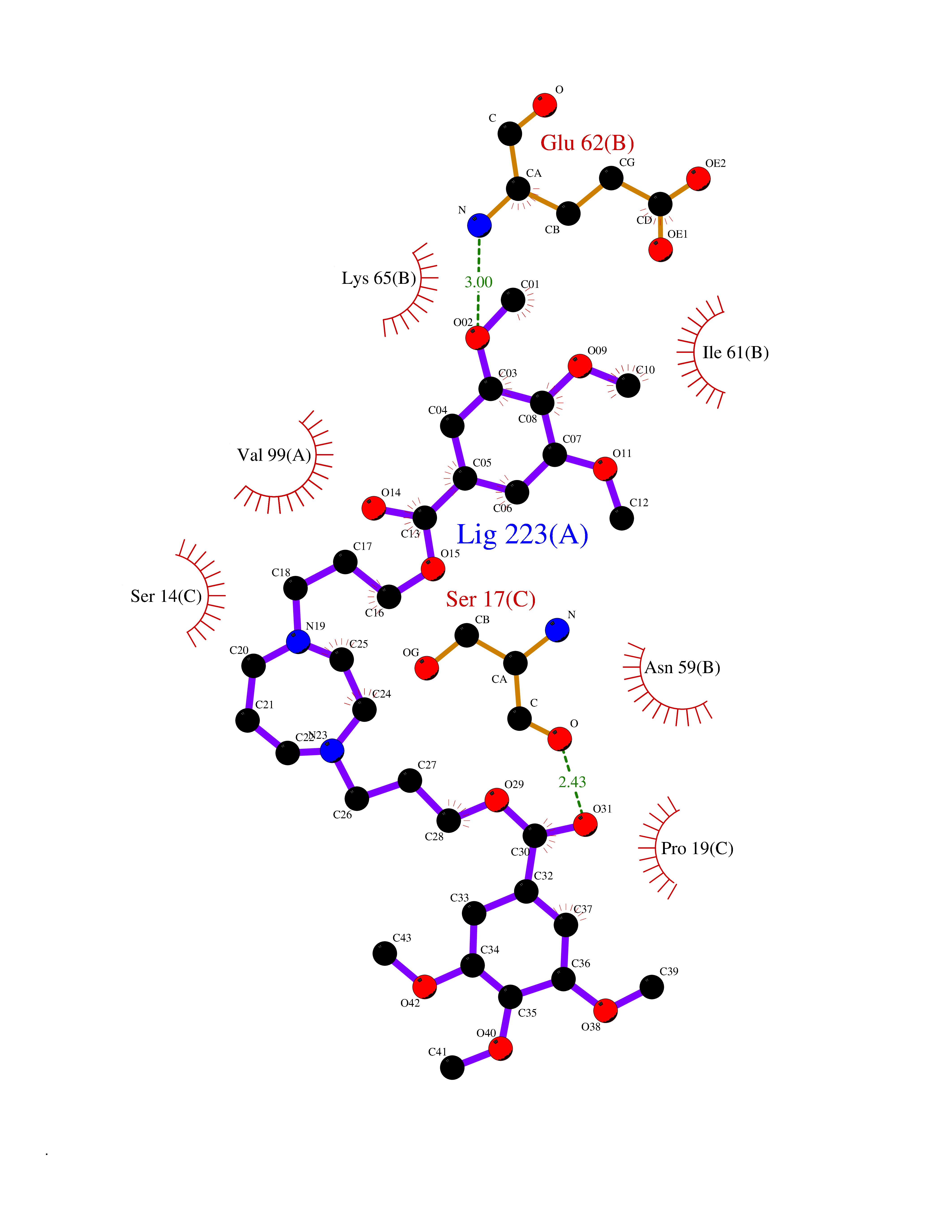 Ligplot Map 3D Binding mode Sequence DVVMTHTPLSLPVSLGDQASISCRSSQSLVHSNGNTYLHWYLQKPGQSPKLLIYKVSNRFSGVPDRFSGSGSGADFTLKISRVEAEDLGVYFCSQSTHVPRTFGGGTKLEIKRADAAPTVSIFPPSSEQLTSGGASVVCFLNNFYPKDINVKWKIDGSERQNGVLNSWTDQDSKDSTYSMSSTLTLTKDEYERHNSYTCEATHKTSTSPIVKSFNRNECXVHLQQPGADLVKPGASVKMSCKASGYTFTSYWITWVKLRPGQGLEWIGDIYPGSGSTNFIEKFKSKATLTVDTSSSTAYMQLRSLTSEDSAVYYCARRGHGNYEDYWGQGTTLIVSSAKTTAPSVYPLAPVCGTGSSVTLGCLVKGYFPEPVTLTWNSGSLSSGVHTFPAVLQSDLYTLSSSVTVTSSTWPSQSITCNVAHPASSTKVDKKIEPRGPTTWSEWSDP Hydrogen bonds contact Hydrophobic contact | ||||
| 12 | Platelet-derived growth factor receptor alpha (PDGFRA) | 5K5X | 7.72 | |
Target general information Gen name PDGFRA Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms RHEPDGFRA; Platelet-derived growth factor receptor 2; Platelet-derived growth factor alpha receptor; PDGFR2; PDGFR-alpha; PDGFR-2; PDGF-R-alpha; CD140a antigen; CD140a; CD140 antigen-like family membe Protein family Protein kinase superfamily, Tyr protein kinase family, CSF-1/PDGF receptor subfamily Biochemical class Kinase Function Depending on the context, promotes or inhibits cell proliferation and cell migration. Plays an important role in the differentiation of bone marrow-derived mesenchymal stem cells. Required for normal skeleton development and cephalic closure during embryonic development. Required for normal development of the mucosa lining the gastrointestinal tract, and for recruitment of mesenchymal cells and normal development of intestinal villi. Plays a role in cell migration and chemotaxis in wound healing. Plays a role in platelet activation, secretion of agonists from platelet granules, and in thrombin-induced platelet aggregation. Binding of its cognate ligands - homodimeric PDGFA, homodimeric PDGFB, heterodimers formed by PDGFA and PDGFB or homodimeric PDGFC -leads to the activation of several signaling cascades; the response depends on the nature of the bound ligand and is modulated by the formation of heterodimers between PDGFRA and PDGFRB. Phosphorylates PIK3R1, PLCG1, and PTPN11. Activation of PLCG1 leads to the production of the cellular signaling molecules diacylglycerol and inositol 1,4,5-trisphosphate, mobilization of cytosolic Ca(2+) and the activation of protein kinase C. Phosphorylates PIK3R1, the regulatory subunit of phosphatidylinositol 3-kinase, and thereby mediates activation of the AKT1 signaling pathway. Mediates activation of HRAS and of the MAP kinases MAPK1/ERK2 and/or MAPK3/ERK1. Promotes activation of STAT family members STAT1, STAT3 and STAT5A and/or STAT5B. Receptor signaling is down-regulated by protein phosphatases that dephosphorylate the receptor and its down-stream effectors, and by rapid internalization of the activated receptor. Tyrosine-protein kinase that acts as a cell-surface receptor for PDGFA, PDGFB and PDGFC and plays an essential role in the regulation of embryonic development, cell proliferation, survival and chemotaxis. Related diseases A chromosomal aberration involving PDGFRA is found in some cases of hypereosinophilic syndrome. Interstitial chromosomal deletion del(4)(q12q12) causes the fusion of FIP1L1 and PDGFRA (FIP1L1-PDGFRA). Mutations that cause overexpression and/or constitutive activation of PDGFRA may be a cause of hypereosinophilic syndrome. {ECO:0000269|PubMed:12808148}.; DISEASE: Gastrointestinal stromal tumor (GIST) [MIM:606764]: Common mesenchymal neoplasms arising in the gastrointestinal tract, most often in the stomach. They are histologically, immunohistochemically, and genetically different from typical leiomyomas, leiomyosarcomas, and schwannomas. Most GISTs are composed of a fairly uniform population of spindle-shaped cells. Some tumors are dominated by epithelioid cells or contain a mixture of spindle and epithelioid morphologies. Primary GISTs in the gastrointestinal tract commonly metastasize in the omentum and mesenteries, often as multiple nodules. However, primary tumors may also occur outside of the gastrointestinal tract, in other intra-abdominal locations, especially in the omentum and mesentery. {ECO:0000269|PubMed:12522257, ECO:0000269|PubMed:15928335}. The gene represented in this entry may be involved in disease pathogenesis. Mutations causing PDGFRA constitutive activation have been found in gastrointestinal stromal tumors lacking KIT mutations (PubMed:12522257). {ECO:0000269|PubMed:12522257}.; DISEASE: GIST-plus syndrome (GISTPS) [MIM:175510]: A disorder characterized by multiple mesenchymal tumors of the gastrointestinal tract, including gastrointestinal stromal tumor, inflammatory fibroid polyps, and fibroid tumors. Additional features are coarse facies and skin, broad hands and feet, and premature tooth loss. GISTPS is an autosomal dominant disease with incomplete penetrance. Gastrointestinal stromal tumor and inflammatory fibroid polyps may also occur in isolation. {ECO:0000269|PubMed:14699510, ECO:0000269|PubMed:17087943, ECO:0000269|PubMed:25975287}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB12742; DB00102; DB12147; DB10772; DB12010; DB00619; DB09078; DB06595; DB09079; DB06043; DB06589; DB08901; DB08896; DB14840; DB01268; DB11800; DB05146 Interacts with P46108; P46109; P00533; Q8N6L0; P04085; P01127; Q9NRA1; P31947; P62258; Q9NRA1-1; A8T7D5; P05067; Q8IY26 EC number EC 2.7.10.1 Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Cell membrane; Cell projection; Chemotaxis; Developmental protein; Disease variant; Disulfide bond; Glycoprotein; Golgi apparatus; Host-virus interaction; Immunoglobulin domain; Kinase; Membrane; Nucleotide-binding; Phosphoprotein; Proteomics identification; Proto-oncogene; Receptor; Reference proteome; Repeat; Signal; Transferase; Transmembrane; Transmembrane helix; Tyrosine-protein kinase; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 39294.8 Length 345 Aromaticity 0.11 Instability index 46.74 Isoelectric point 6.6 Charge (pH=7) -1.38 2D Binding mode Binding energy (Kcal/mol) -6.23  Molscript Map 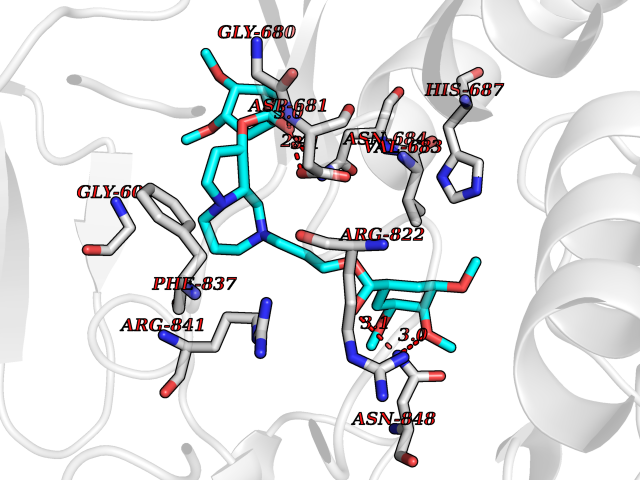 Pymol Map 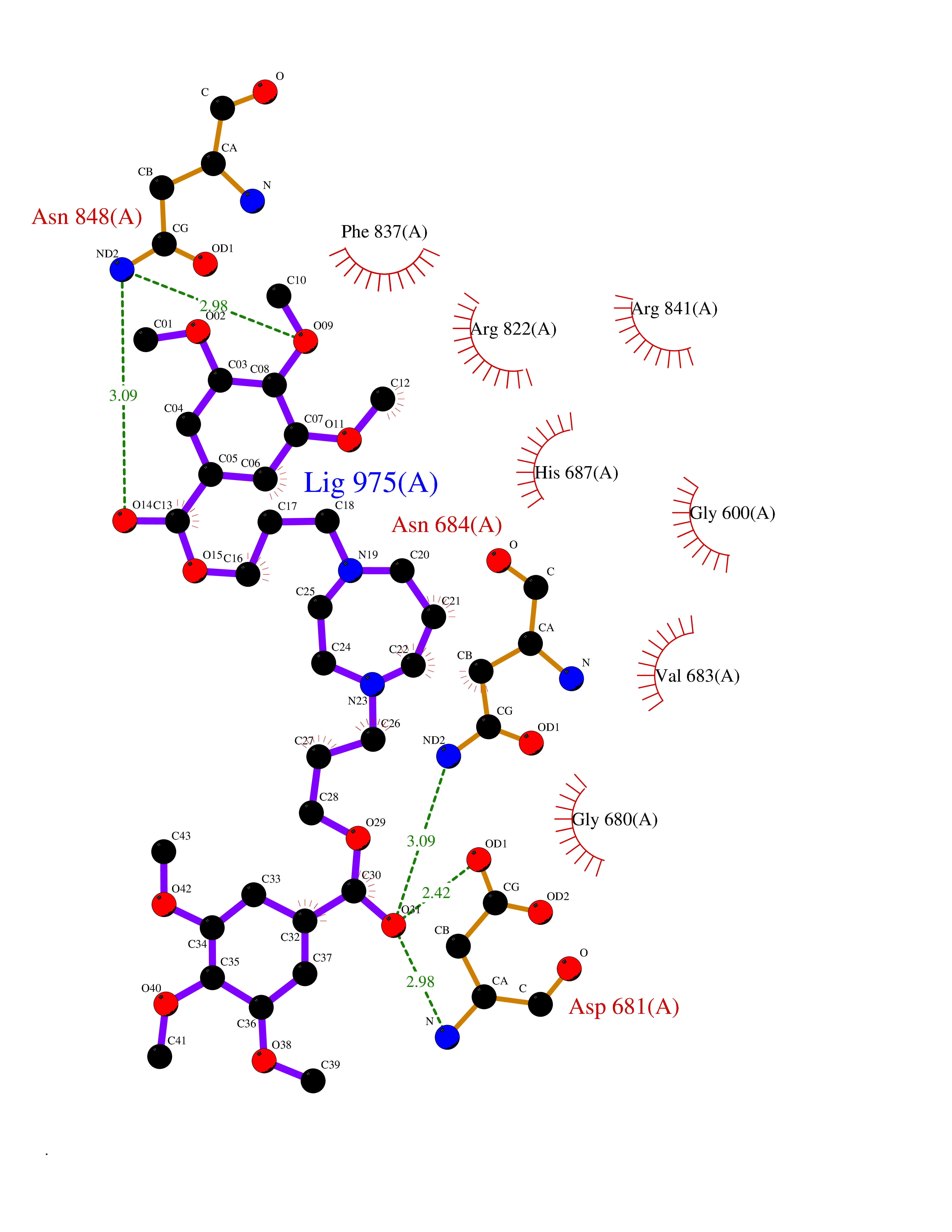 Ligplot Map 3D Binding mode Sequence RYEIRWRVIESISPDGHEYIYVDPMQLPYDSRWEFPRDGLVLGRVLGSGAFGKVVEGTAYGLSRSQPVMKVAVKMLKPTARSSEKQALMSELKIMTHLGPHLNIVNLLGACTKSGPIYIITEYCFYGDLVNYLHKNRDSFLSHSMLDSEVKNLLSDDNSEGLTLLDLLSFTYQVARGMEFLASKNCVHRDLAARNVLLAQGKIVKICDFGLARDIMHDSNYVSKGSTFLPVKWMAPESIFDNLYTTLSDVWSYGILLWEIFSLGGTPYPGMMVDSTFYNKIKSGYRMAKPDHATSEVYEIMVKCWNSEPEKRPSFYHLSEIVENLLPGQYKKSYEKIHLDFLKSD Hydrogen bonds contact Hydrophobic contact | ||||
| 13 | Short transient receptor potential channel 5 (TRPC5) | 7WDB | 7.72 | |
Target general information Gen name TRPC5 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms hTRP5; hTRP-5; TrpC5; Transient receptor protein 5; TRP-5 Protein family Transient receptor (TC 1.A.4) family, STrpC subfamily, TRPC5 sub-subfamily Biochemical class Transient receptor potential catioin channel Function Thought to form a receptor-activated non-selective calcium permeant cation channel. Probably is operated by a phosphatidylinositol second messenger system activated by receptor tyrosine kinases or G-protein coupled receptors. Has also been shown to be calcium-selective. May also be activated by intracellular calcium store depletion. Related diseases Loss-of-function variants in TRPC5 may be involved in a mental disorder characterized by maladaptive behavior, anxiety, autism, postpartum depression, extreme food-seeking and hoarding behavior, hyperphagia and obesity. {ECO:0000269|PubMed:38959890}. Drugs (DrugBank ID) NA Interacts with NA EC number NA Uniprot keywords 3D-structure; ANK repeat; Calcium; Calcium channel; Calcium transport; Cell membrane; Disease variant; Glycoprotein; Ion channel; Ion transport; Membrane; Proteomics identification; Reference proteome; Repeat; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A Molecular weight (Da) 76850.6 Length 665 Aromaticity 0.12 Instability index 40.05 Isoelectric point 6.16 Charge (pH=7) -5.94 2D Binding mode Binding energy (Kcal/mol) -8.64 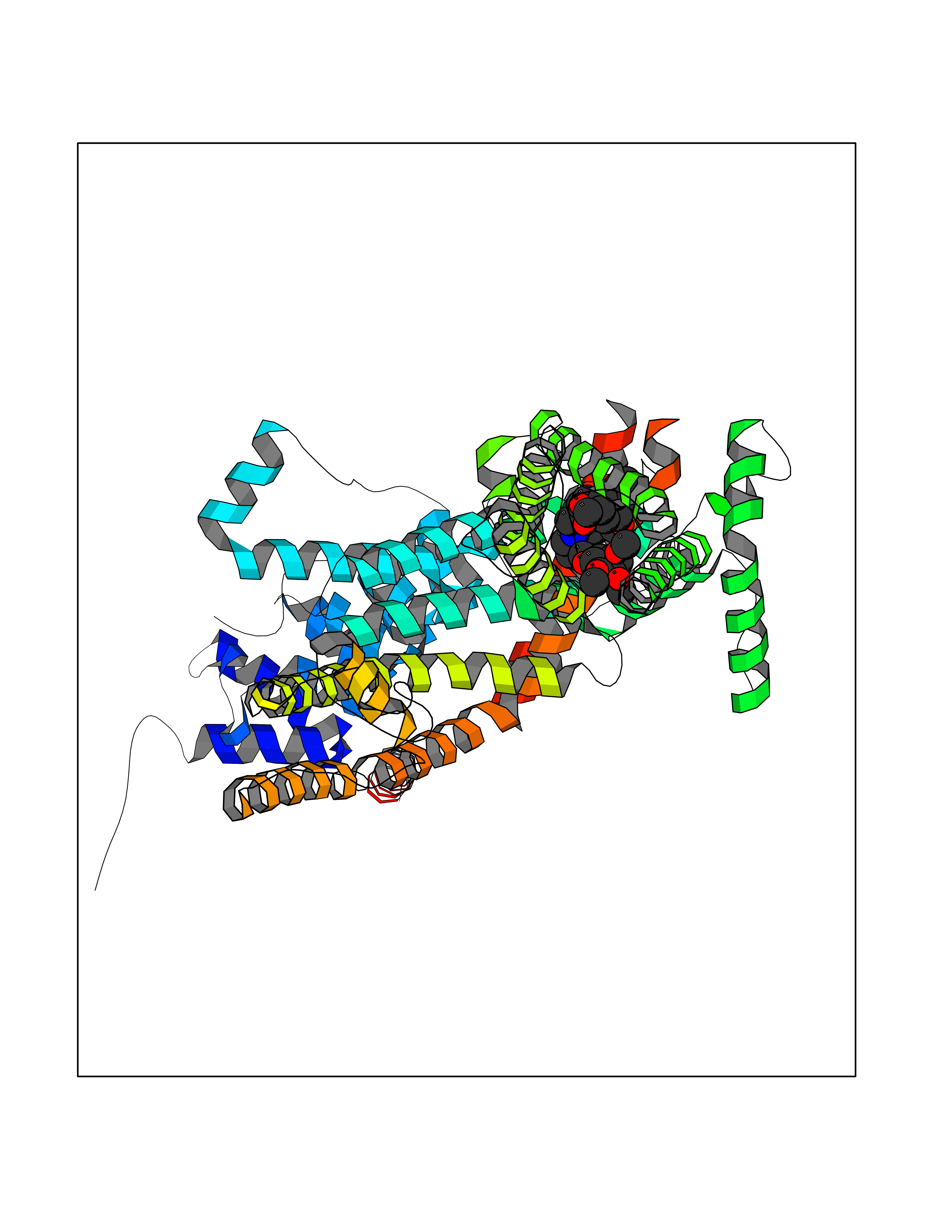 Molscript Map 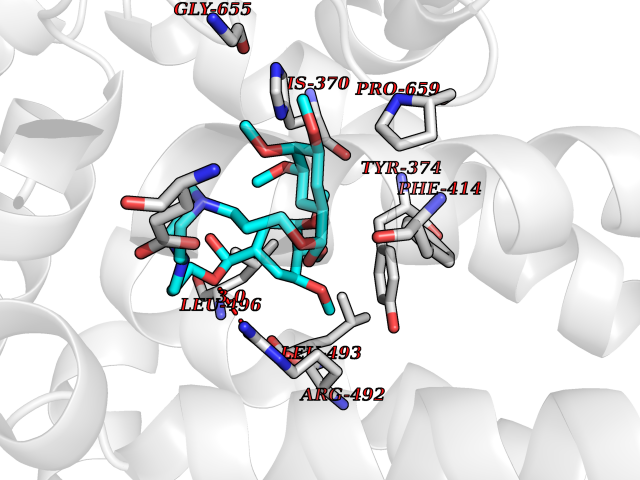 Pymol Map 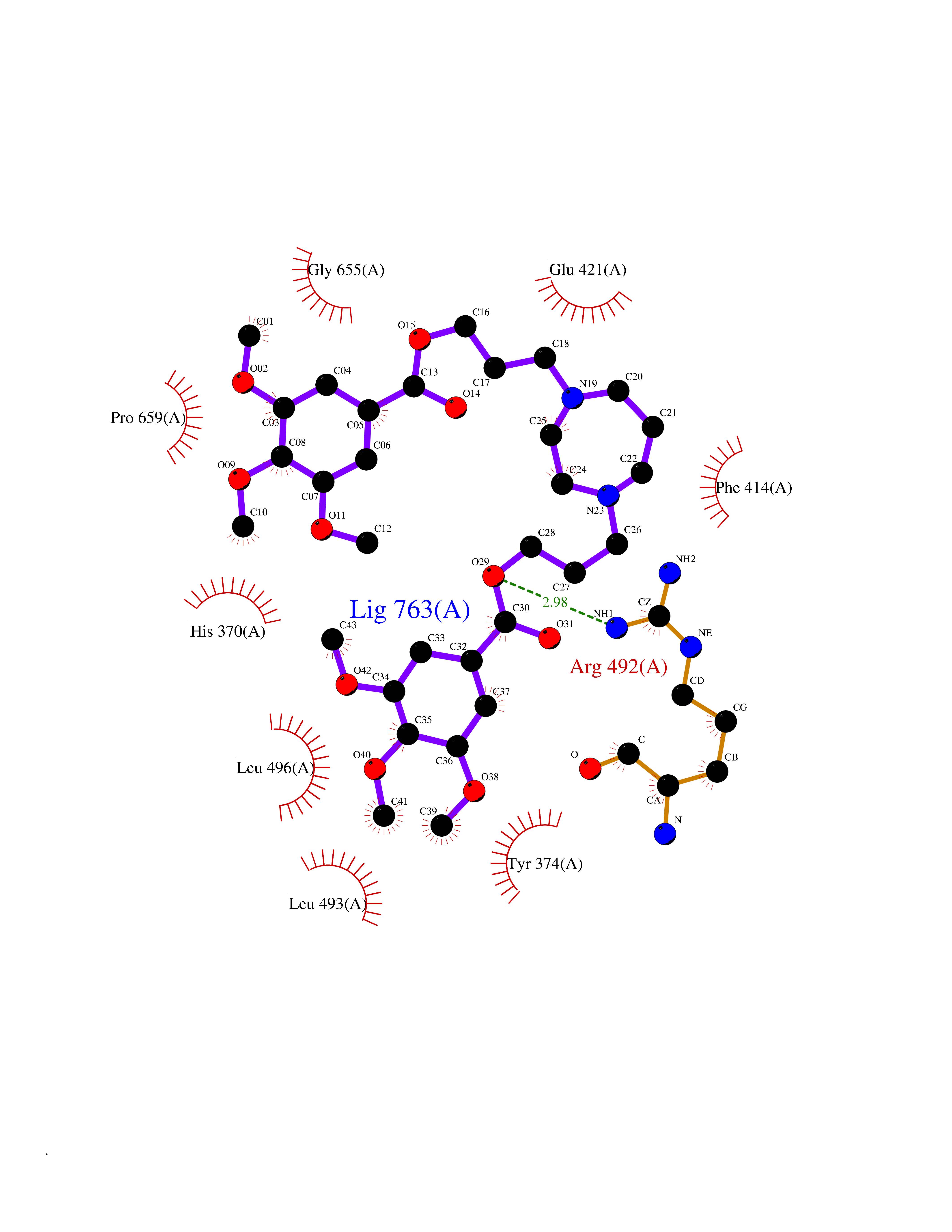 Ligplot Map 3D Binding mode Sequence RIPLQIVRAETELSAEEKAFLNAVEKGDYATVKQALQEAEIYYNVNINCMDPLGRSALLIAIENENLEIMELLLNHSVYVGDALLYAIRKEVVGAVELLLSYQFSEFTPDITPIMLAAHTNNYEIIKLLVQKRVTIPRPHQIRCNCVECVSSSEVDSLRHSRSRLNIYKALASPSLIALSSEDPILTAFRLGWELKELSKVENEFKAEYEELSQQCKLFAKDLLDQARSSRELEIILNHRDDLAKLKVAIKYHQKEFVAQPNCQQLLATLWYDGFPGWRRKHWVVKLLTCMTIGFLFPMLSIAYLISPRSNLGLFIKKPFIKFICHTASYLTFLFMLLLASQHVQGPPPTVVEWMILPWVLGFIWGEIKEMWDGGFTEYIHDWWNLMDFAMNSLYLATISLKIVAYVKYNGSRPREEWEMWHPTLIAEALFAISNILSSLRLISLFTANSHLGPLQISLGRMLLDILKFLFIYCLVLLAFANGLNQLYFYYETRAIDEPNNCKGIRCEKQNNAFSTLFETLQSLFWSVFGLLNLYVTNVKARHEFTEFVGATMFGTYNVISLVVLLNMLIAMMNNSYQLIADHADIEWKFARTKLWMSYFDEGGTLPPPFNIISLIQNQHYQEVIRNLVKRYVAAMIRNSKTTEENFKELKQDISSFRYEVLDLL Hydrogen bonds contact Hydrophobic contact | ||||
| 14 | Dopamine beta-hydroxylase | 4ZEL | 7.71 | |
Target general information Gen name DBH Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Copper type II ascorbate-dependent monooxygenase family Biochemical class Oxidoreductase Function Catalytic activity.Copper ion binding.Dopamine beta-monooxygenase activity.L-ascorbic acid binding. Related diseases Orthostatic hypotension 1 (ORTHYP1) [MIM:223360]: A form of orthostatic hypotension due to congenital dopamine beta-hydroxylase deficiency. Orthostatic hypotension, also known as postural hypotension, is a finding defined as a 20-mm Hg decrease in systolic pressure or a 10-mm Hg decrease in diastolic pressure occurring 3 minutes after a person has risen from supine to standing. Symptoms include dizziness, blurred vision, and sometimes syncope. ORTHYP1 is an autosomal recessive condition apparent from infancy or early childhood and characterized by low plasma and urinary levels of norepinephrine and epinephrine, and episodic hypoglycemia. {ECO:0000269|PubMed:11857564}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00126; DB06774; DB09130; DB05394; DB00822; DB00988; DB00968; DB00550 Interacts with P00352; P63010-2; Q04656; Q8WUW1; Q9UNS2; Q71DI3; P61978; Q9Y2M5; Q92876; P08727; Q14693; P0DPK4; Q6GQQ9-2; P27986-2; Q9ULX5; Q96D59; Q8N6K7-2; Q9GZS3; Q8IUW3; Q86WT6-2 EC number 1.14.17.1 Uniprot keywords 3D-structure; Catecholamine biosynthesis; Copper; Cytoplasmic vesicle; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; Membrane; Metal-binding; Monooxygenase; Oxidoreductase; Proteomics identification; Reference proteome; Secreted; Signal-anchor; Transmembrane; Transmembrane helix; Vitamin C Protein physicochemical properties Chain ID A,B Molecular weight (Da) 123694 Length 1094 Aromaticity 0.1 Instability index 51.85 Isoelectric point 5.84 Charge (pH=7) -24.5 2D Binding mode Binding energy (Kcal/mol) -6.88 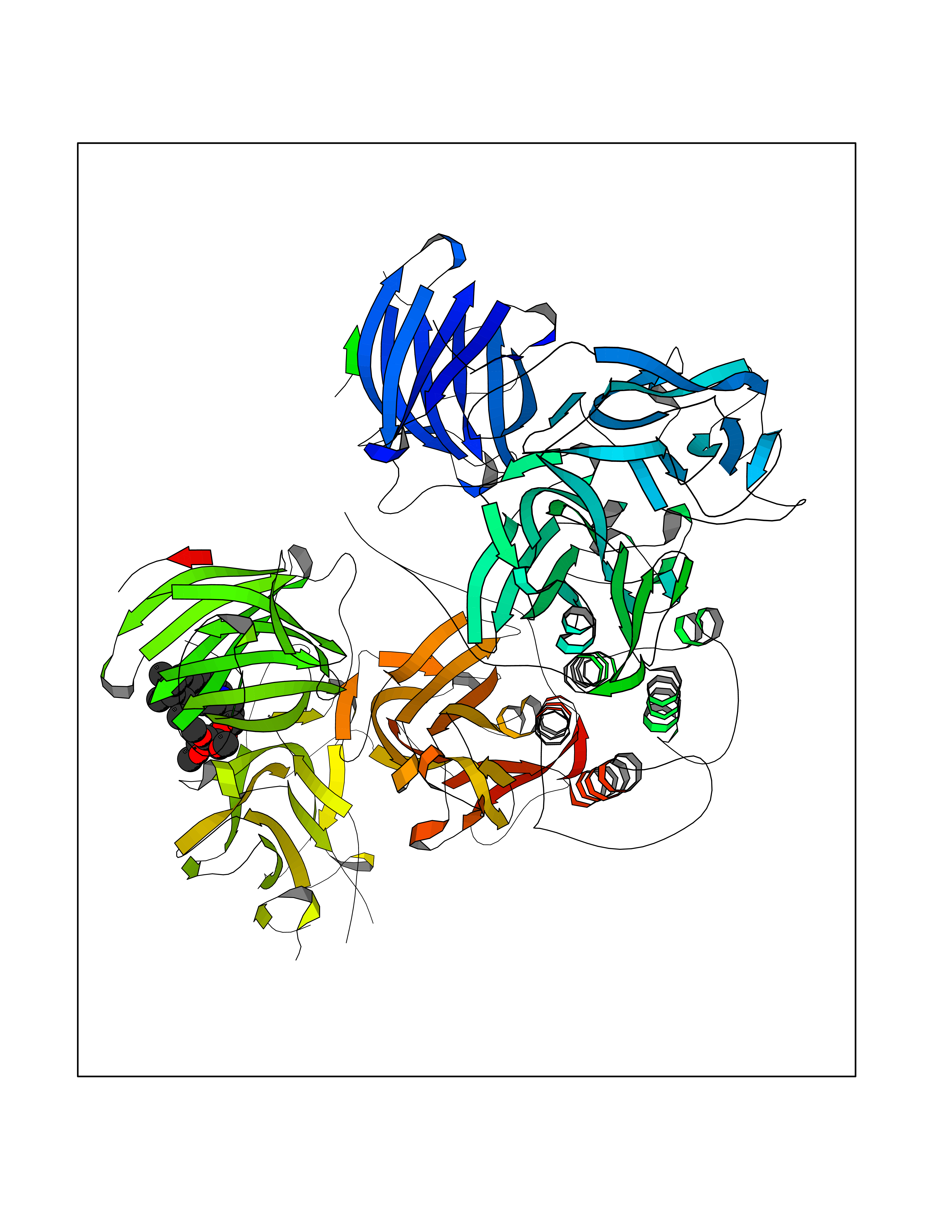 Molscript Map 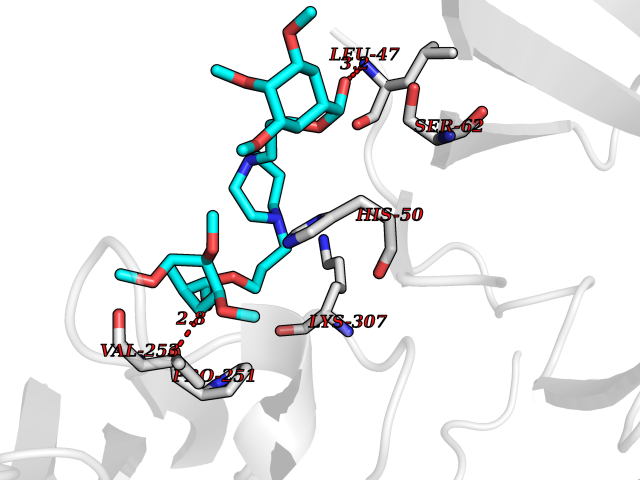 Pymol Map 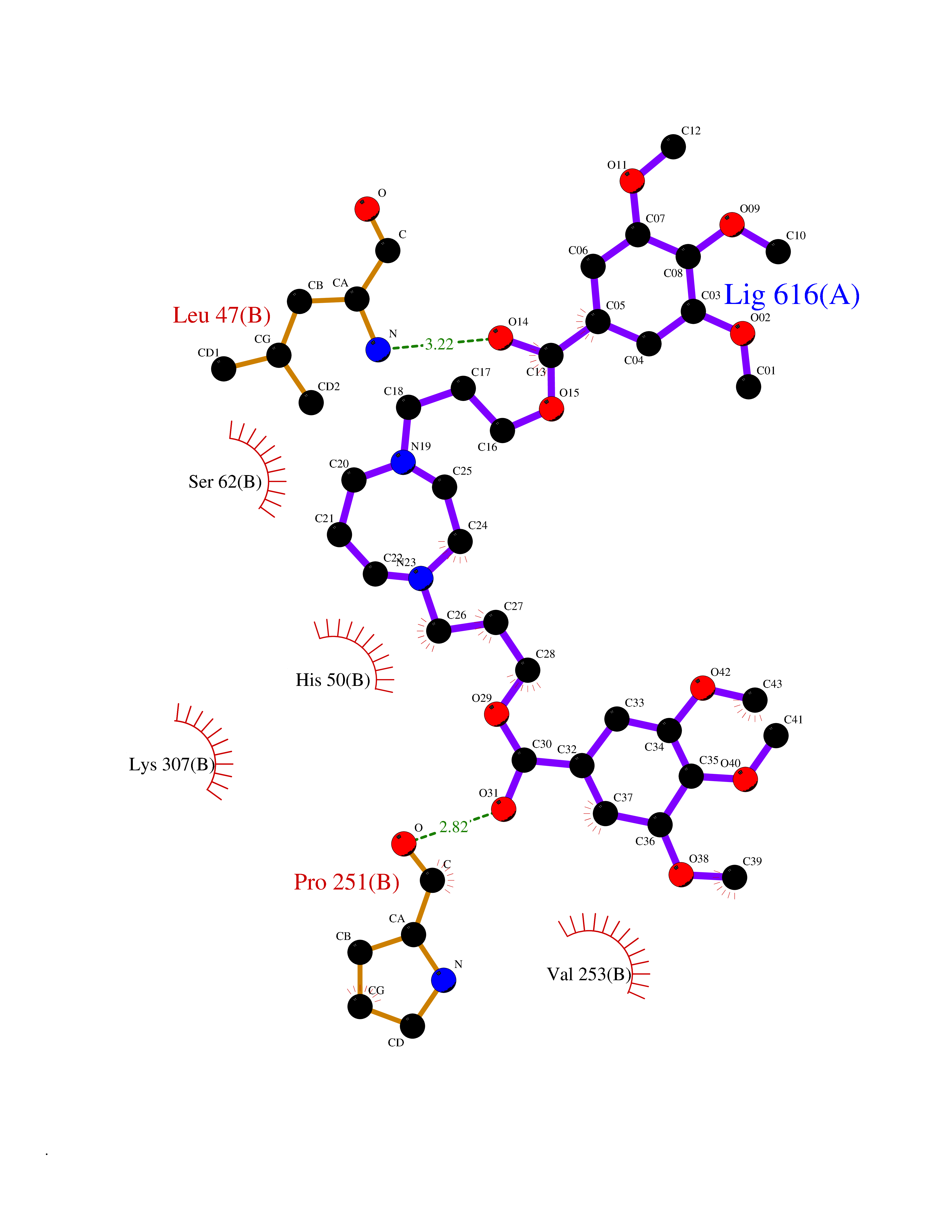 Ligplot Map 3D Binding mode Sequence PLPYHIPLDPEGSLELSWNVSYTQEAIHFQLLVRRLKAGVLFGMSDRGELENADLVVLWTDGDAYFADAWSDQKGQIHLDPQQDYQLLQVQRTPEGLTLLFKRPFGTCDPKDYLIEDGTVHLVYGILEEPFRSLEAINGSGLQMGLQRVQLLKPNIPEPELPSDACTMEVQAPNIQIPSQETTYWCYIKELPKGFSRHHIIKYEPIVTKGNEALVHHMEVFQCAPEMDSVPHFSGPCDSKMKPDRLNYCRHVLAAWALGAKAFYYPEEAGLAFGGPGSSRYLRLEVHYHNPLVIEGRNDSSGIRLYYTAKLRRFNAGIMELGLVYTPVMAIPPRETAFILTGYCTDKCTQLALPPSGIHIFASQLHTHLTGRKVVTVLVRDGREWEIVNQDNHYSPHFQEIRMLKKVVSVHPGDVLITSCTYNTEDRELATVGGFGILEEMCVNYVHYYPQTQLELCKSAVDAGFLQKYFHLINRFNNEDVCTCPQASVSQQFTSVPWNSFNRDVLKALYSFAPISMHCNKSSAVRFQGEWNLQPLPKVISTLEEPTVVSPLPYHIPLDPEGSLELSWNVSYTQEAIHFQLLVRRLKAGVLFGMSDRGELENADLVVLAYFADAWSDQKGQIHLDPQQDYQLLQVQRTPEGLTLLFKRPFGTCDPKDYLIEDGTVHLVYGILEEPFRSLEAINGSGLQMGLQRVQLLKPNIPEPELPSDACTMEVQAPNIQIPSQETTYWCYIKELPKGFSRHHIIKYEPIVTKGNEALVHHMEVFQCAPEVPHFSGPCDSKMLNYCRHVLAAWALGAKAFYYPEEAGLAFGGPGSSRYLRLEVHYHNPLVIEGRNDSSGIRLYYTAKLRRFNAGIMELGLVYTPVMAIPPRETAFILTGYCTDKCTQLALPPSGIHIFASQLHTHLTGRKVVTVLVRDGREWEIVNQDNHYSPHFQEIRMLKKVVSVHPGDVLITSCTYNTEDRELATVGGFGILEEMCVNYVHYYPQTQLELCKSAVDAGFLQKYFHLINRFNNEDVCTCPQASVSQQFTSVPWNSFNRDVLKALYSFAPISMHCNKSSAVRFQGEWNLQPLPKVISTLEEPTPQCVVSIGG Hydrogen bonds contact Hydrophobic contact | ||||
| 15 | Tyrosine-protein kinase UFO (AXL) | 5U6B | 7.70 | |
Target general information Gen name AXL Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms UFO; Tyrosine-protein kinase receptor UFO; AXL oncogene Protein family Protein kinase superfamily, Tyr protein kinase family, AXL/UFO subfamily Biochemical class Kinase Function Receptor tyrosine kinase that transduces signals from the extracellular matrix into the cytoplasm by binding growth factor GAS6 and which is thus regulating many physiological processes including cell survival, cell proliferation, migration and differentiation. Ligand binding at the cell surface induces dimerization and autophosphorylation of AXL. Following activation by ligand, ALX binds and induces tyrosine phosphorylation of PI3-kinase subunits PIK3R1, PIK3R2 and PIK3R3; but also GRB2, PLCG1, LCK and PTPN11. Other downstream substrate candidates for AXL are CBL, NCK2, SOCS1 and TNS2. Recruitment of GRB2 and phosphatidylinositol 3 kinase regulatory subunits by AXL leads to the downstream activation of the AKT kinase. GAS6/AXL signaling plays a role in various processes such as endothelial cell survival during acidification by preventing apoptosis, optimal cytokine signaling during human natural killer cell development, hepatic regeneration, gonadotropin-releasing hormone neuron survival and migration, platelet activation, or regulation of thrombotic responses. Plays also an important role in inhibition of Toll-like receptors (TLRs)-mediated innate immune response. Related diseases AXL and its ligand GAS6 are highly expressed in thyroid carcinoma tissues, and might thus be involved in thyroid tumorigenesis. Overexpression of AXL and its ligand was also detected in many other cancers such as myeloproliferative disorders, prostatic carcinoma cells, or breast cancer. Drugs (DrugBank ID) DB12010; DB12141 Interacts with P00533; P29317; Q14393-2; P62993; P08238; P27986; P0DTC2 EC number EC 2.7.10.1 Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Cell membrane; Differentiation; Disulfide bond; Glycoprotein; Host cell receptor for virus entry; Host-virus interaction; Immunity; Immunoglobulin domain; Innate immunity; Kinase; Membrane; Nucleotide-binding; Oncogene; Phosphoprotein; Proteomics identification; Proto-oncogene; Receptor; Reference proteome; Repeat; Signal; Transferase; Transmembrane; Transmembrane helix; Tyrosine-protein kinase; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 30634.2 Length 270 Aromaticity 0.09 Instability index 45.04 Isoelectric point 5.49 Charge (pH=7) -5.75 2D Binding mode Binding energy (Kcal/mol) -7.28 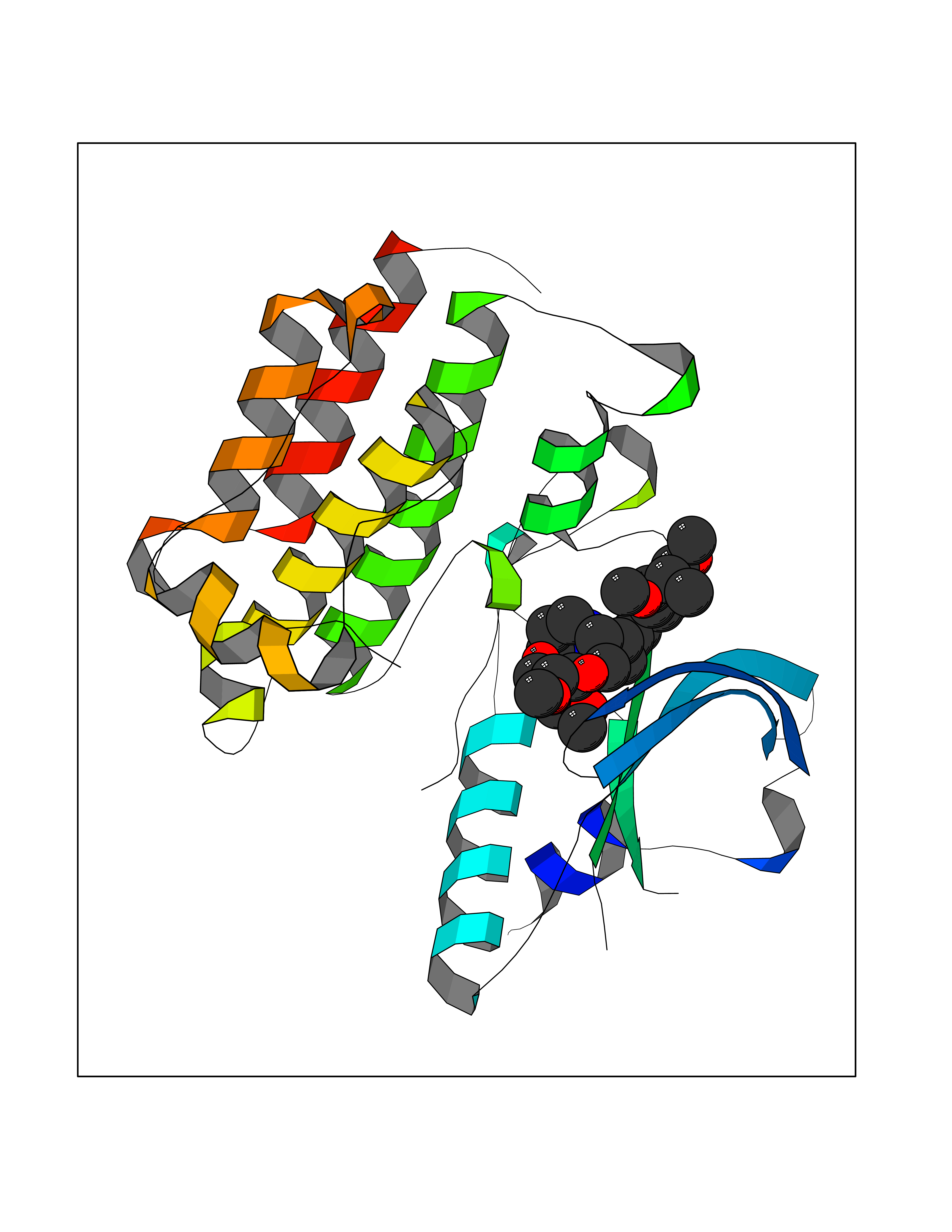 Molscript Map 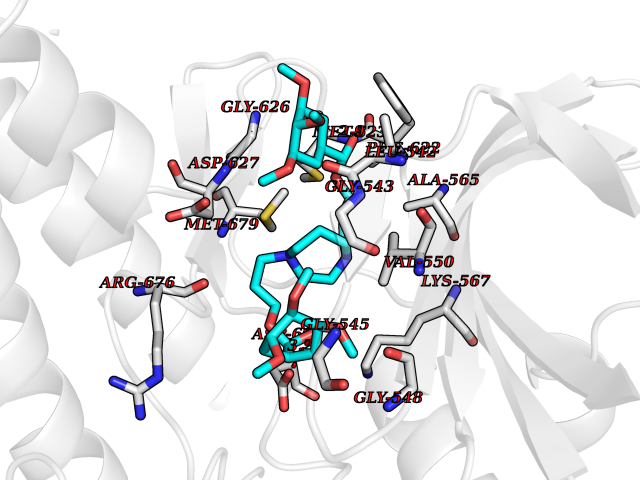 Pymol Map 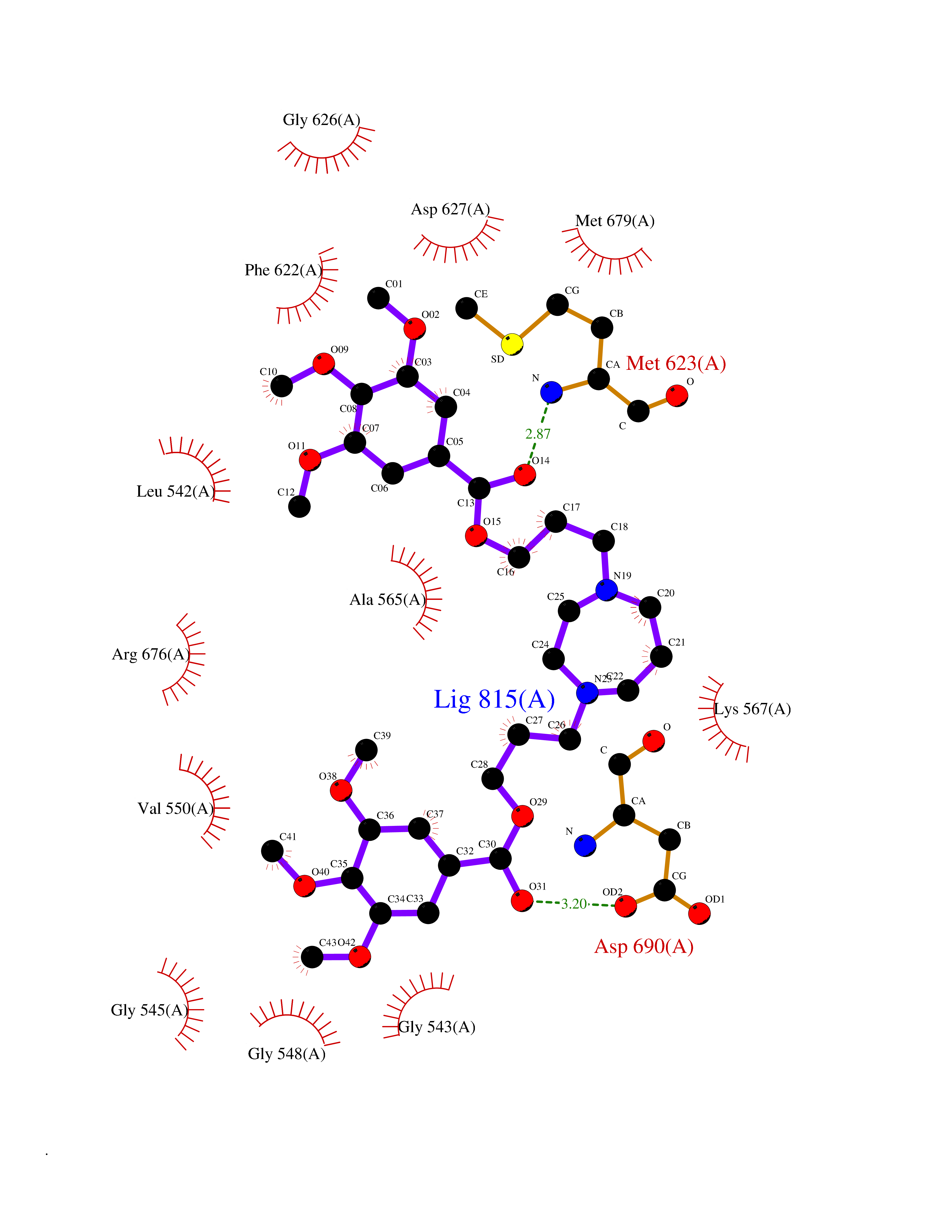 Ligplot Map 3D Binding mode Sequence ELKEKLRDVMVDRHKVALGKTLGEGEFGAVMEGQLNQDDSILKVAVKTMKIAICTRSELEDFLSEAVCMKEFDHPNVMRLIGVCFQPAPVVILPFMKHGDLHSFLLYSRLGDQPVYLPTQMLVKFMADIASGMEYLSTKRFIHRDLAARNCMLNENMSVCVADFGLSKPVKWIAIESLADRVYTSKSDVWSFGVTMWEIATRGQTPYPGVENSEIYDYLRQGNRLKQPADCLDGLYALMSRCWELNPQDRPSFTELREDLENTLKALPPA Hydrogen bonds contact Hydrophobic contact | ||||
| 16 | Gastrin (GAST) | 5WRJ | 7.69 | |
Target general information Gen name GAST Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Gastrin6; GAST; G52; G34; G14 Protein family Gastrin/cholecystokinin family Biochemical class Gastrin cholecystokinin Function Gastrin stimulates the stomach mucosa to produce and secrete hydrochloric acid and the pancreas to secrete its digestive enzymes. It also stimulates smooth muscle contraction and increases blood circulation and water secretion in the stomach and intestine. Related diseases Orotic aciduria 1 (ORAC1) [MIM:258900]: A disorder of pyrimidine metabolism resulting in megaloblastic anemia and orotic acid crystalluria that is frequently associated with some degree of physical and intellectual disability. A minority of cases have additional features, particularly congenital malformations and immune deficiencies. {ECO:0000269|PubMed:9042911}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB12532 Interacts with Q13520; O43315; Q12797-6; Q9BXK5; Q8N5K1; Q96BA8; P00387; Q9Y282; Q5JX71; Q8NBJ4; Q8TDT2; P43628; O76011; Q6ZUX7; Q15546; P15941-11; Q13113; P60201-2; Q14973; P02787; Q4KMG9 EC number NA Uniprot keywords 3D-structure; Amidation; Cleavage on pair of basic residues; Direct protein sequencing; Hormone; Phosphoprotein; Proteomics identification; Pyrrolidone carboxylic acid; Reference proteome; Secreted; Signal; Sulfation Protein physicochemical properties Chain ID A,F Molecular weight (Da) 31827.9 Length 280 Aromaticity 0.08 Instability index 41.06 Isoelectric point 8.84 Charge (pH=7) 5.56 2D Binding mode Binding energy (Kcal/mol) -7.52 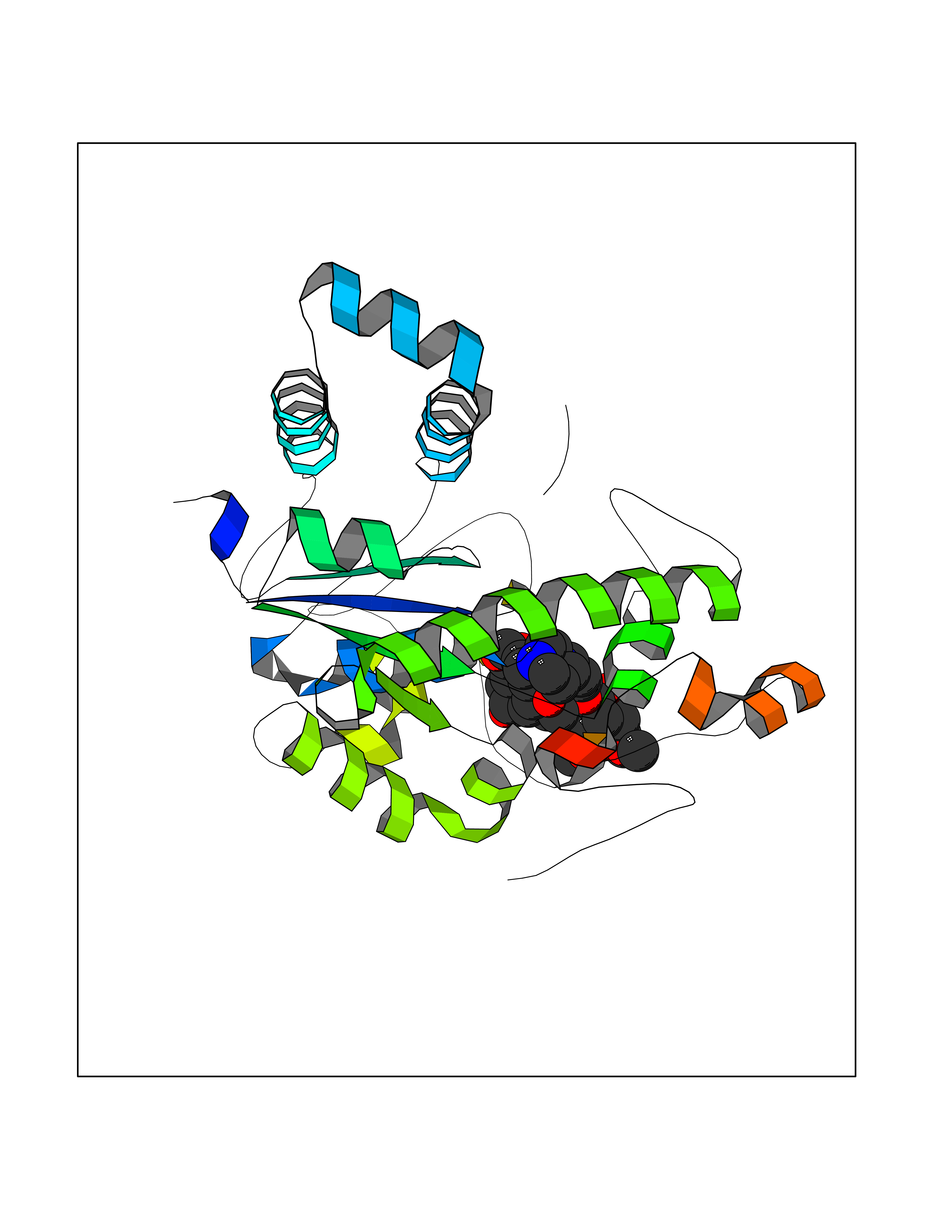 Molscript Map 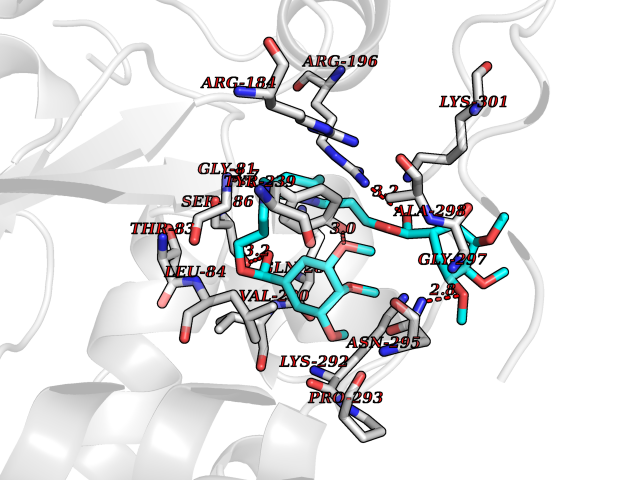 Pymol Map 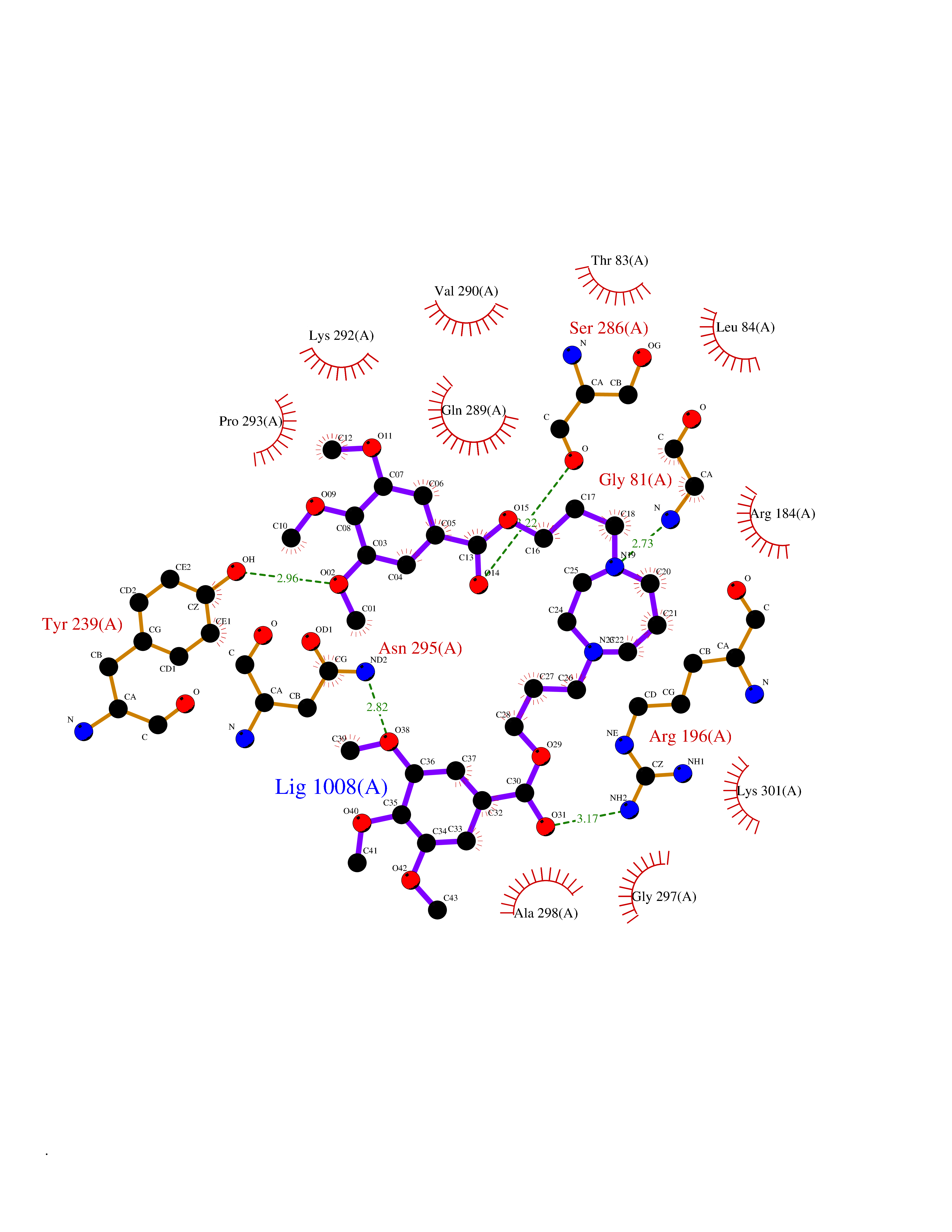 Ligplot Map 3D Binding mode Sequence AYHKDMPLIFIGGVPRSGTTLMRAMLDAHPDIRCGEETRVIPRILALKQMWSRSSKEKIRLDEAGVTDEVLDSAMQAFLLEIIVKHGEPAPYLCNKDPFALKSLTYLSRLFPNAKFLLMVRDGRASVHSMISRKVTIAGFDLNSYRDCLTKWNRAIETMYNQCMEVGYKKCMLVHYEQLVLHPERWMRTLLKFLQIPWNHSVLHHEEMIGKAGGVSLSKVERSTDQVIKPVNVGALSKWVGKIPPDVLQDMAVIAPMLAKLGYDPYANPPNYGKPEEEAY Hydrogen bonds contact Hydrophobic contact | ||||
| 17 | Dopamine beta-hydroxylase | 4ZEL | 7.68 | |
Target general information Gen name DBH Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Copper type II ascorbate-dependent monooxygenase family Biochemical class Oxidoreductase Function Catalytic activity.Copper ion binding.Dopamine beta-monooxygenase activity.L-ascorbic acid binding. Related diseases Orthostatic hypotension 1 (ORTHYP1) [MIM:223360]: A form of orthostatic hypotension due to congenital dopamine beta-hydroxylase deficiency. Orthostatic hypotension, also known as postural hypotension, is a finding defined as a 20-mm Hg decrease in systolic pressure or a 10-mm Hg decrease in diastolic pressure occurring 3 minutes after a person has risen from supine to standing. Symptoms include dizziness, blurred vision, and sometimes syncope. ORTHYP1 is an autosomal recessive condition apparent from infancy or early childhood and characterized by low plasma and urinary levels of norepinephrine and epinephrine, and episodic hypoglycemia. {ECO:0000269|PubMed:11857564}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00126; DB06774; DB09130; DB05394; DB00822; DB00988; DB00968; DB00550 Interacts with P00352; P63010-2; Q04656; Q8WUW1; Q9UNS2; Q71DI3; P61978; Q9Y2M5; Q92876; P08727; Q14693; P0DPK4; Q6GQQ9-2; P27986-2; Q9ULX5; Q96D59; Q8N6K7-2; Q9GZS3; Q8IUW3; Q86WT6-2 EC number 1.14.17.1 Uniprot keywords 3D-structure; Catecholamine biosynthesis; Copper; Cytoplasmic vesicle; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; Membrane; Metal-binding; Monooxygenase; Oxidoreductase; Proteomics identification; Reference proteome; Secreted; Signal-anchor; Transmembrane; Transmembrane helix; Vitamin C Protein physicochemical properties Chain ID A,B Molecular weight (Da) 123694 Length 1094 Aromaticity 0.1 Instability index 51.85 Isoelectric point 5.84 Charge (pH=7) -24.5 2D Binding mode Binding energy (Kcal/mol) -8.95 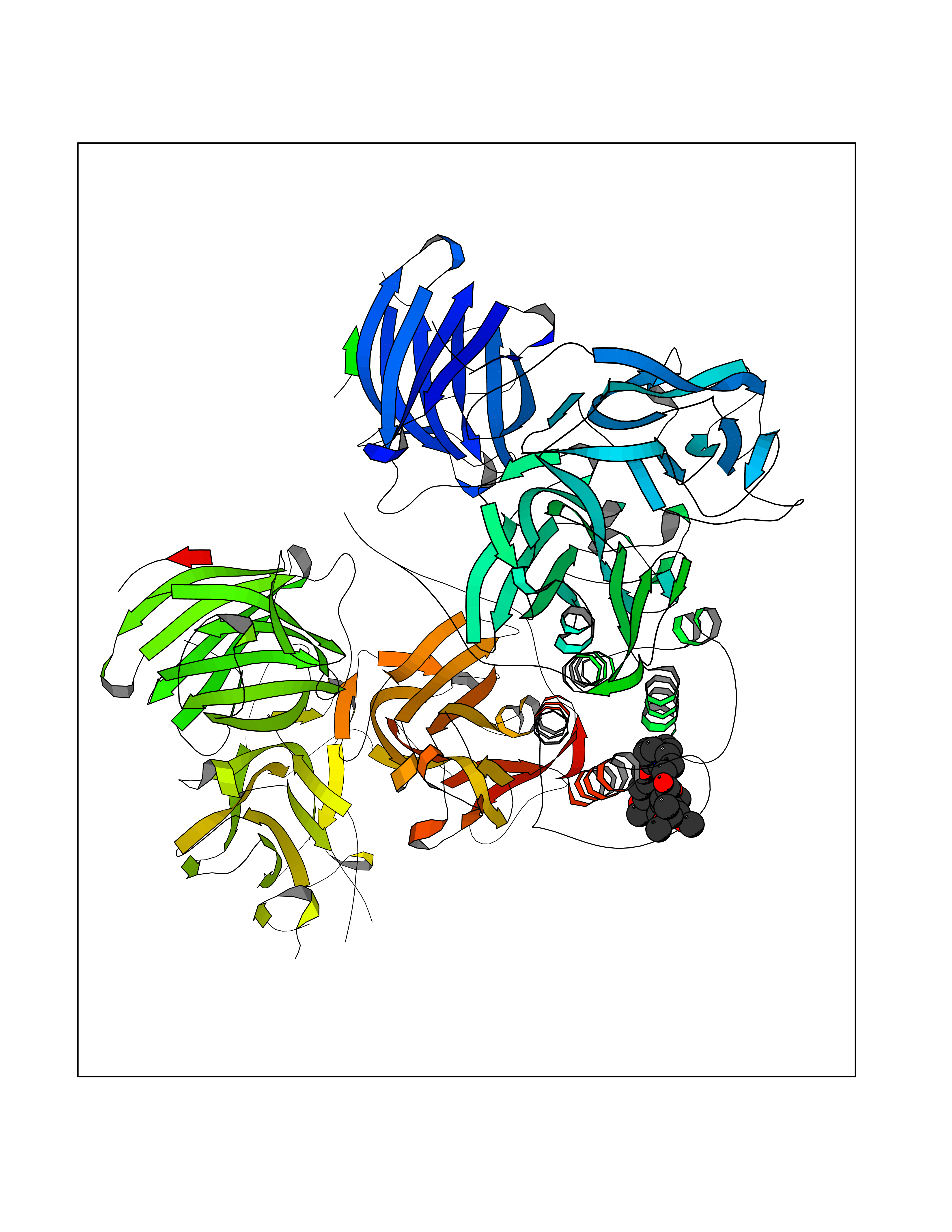 Molscript Map 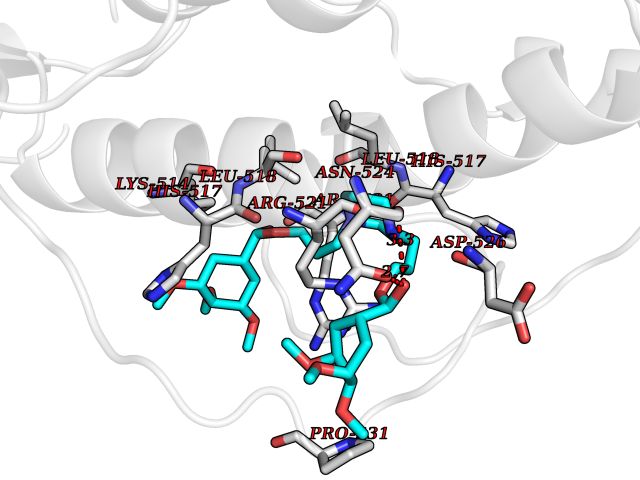 Pymol Map 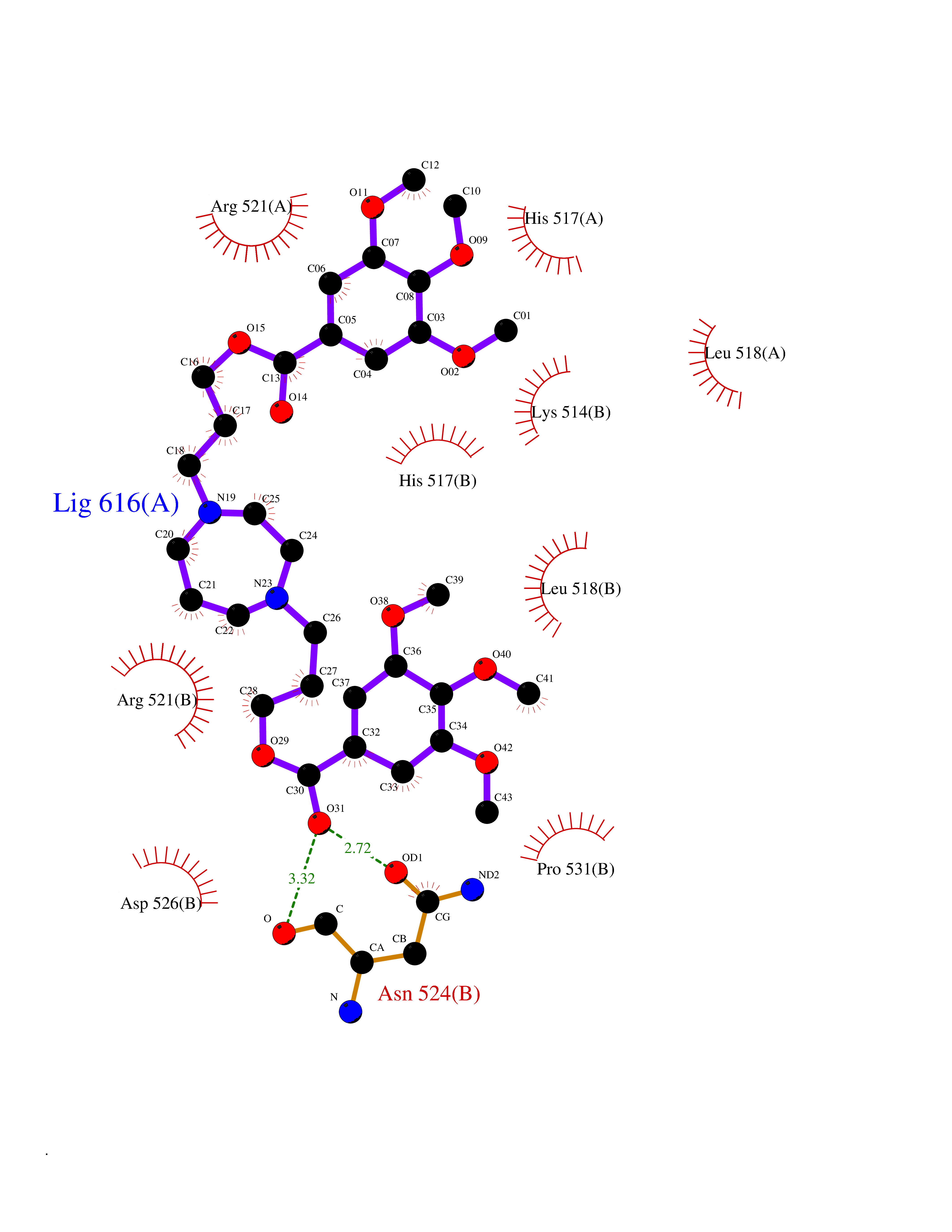 Ligplot Map 3D Binding mode Sequence PLPYHIPLDPEGSLELSWNVSYTQEAIHFQLLVRRLKAGVLFGMSDRGELENADLVVLWTDGDAYFADAWSDQKGQIHLDPQQDYQLLQVQRTPEGLTLLFKRPFGTCDPKDYLIEDGTVHLVYGILEEPFRSLEAINGSGLQMGLQRVQLLKPNIPEPELPSDACTMEVQAPNIQIPSQETTYWCYIKELPKGFSRHHIIKYEPIVTKGNEALVHHMEVFQCAPEMDSVPHFSGPCDSKMKPDRLNYCRHVLAAWALGAKAFYYPEEAGLAFGGPGSSRYLRLEVHYHNPLVIEGRNDSSGIRLYYTAKLRRFNAGIMELGLVYTPVMAIPPRETAFILTGYCTDKCTQLALPPSGIHIFASQLHTHLTGRKVVTVLVRDGREWEIVNQDNHYSPHFQEIRMLKKVVSVHPGDVLITSCTYNTEDRELATVGGFGILEEMCVNYVHYYPQTQLELCKSAVDAGFLQKYFHLINRFNNEDVCTCPQASVSQQFTSVPWNSFNRDVLKALYSFAPISMHCNKSSAVRFQGEWNLQPLPKVISTLEEPTVVSPLPYHIPLDPEGSLELSWNVSYTQEAIHFQLLVRRLKAGVLFGMSDRGELENADLVVLAYFADAWSDQKGQIHLDPQQDYQLLQVQRTPEGLTLLFKRPFGTCDPKDYLIEDGTVHLVYGILEEPFRSLEAINGSGLQMGLQRVQLLKPNIPEPELPSDACTMEVQAPNIQIPSQETTYWCYIKELPKGFSRHHIIKYEPIVTKGNEALVHHMEVFQCAPEVPHFSGPCDSKMLNYCRHVLAAWALGAKAFYYPEEAGLAFGGPGSSRYLRLEVHYHNPLVIEGRNDSSGIRLYYTAKLRRFNAGIMELGLVYTPVMAIPPRETAFILTGYCTDKCTQLALPPSGIHIFASQLHTHLTGRKVVTVLVRDGREWEIVNQDNHYSPHFQEIRMLKKVVSVHPGDVLITSCTYNTEDRELATVGGFGILEEMCVNYVHYYPQTQLELCKSAVDAGFLQKYFHLINRFNNEDVCTCPQASVSQQFTSVPWNSFNRDVLKALYSFAPISMHCNKSSAVRFQGEWNLQPLPKVISTLEEPTPQCVVSIGG Hydrogen bonds contact Hydrophobic contact | ||||
| 18 | HMG-CoA reductase (HMGCR) | 2R4F | 7.67 | |
Target general information Gen name HMGCR Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms 3-hydroxy-3-methylglutaryl-coenzyme A reductase Protein family HMG-CoA reductase family Biochemical class CH-OH donor oxidoreductase Function Transmembrane glycoprotein that is the rate-limiting enzyme in cholesterol biosynthesis as well as in the biosynthesis of nonsterol isoprenoids that are essential for normal cell function including ubiquinone and geranylgeranyl proteins. Related diseases Muscular dystrophy, limb-girdle, autosomal recessive 28 (LGMDR28) [MIM:620375]: An autosomal recessive form of limb girdle muscular dystrophy, a group of genetically heterogeneous muscular disorders that share proximal muscle weakness as the major attribute. Most limb girdle muscular dystrophies present with elevated creatinine kinase and myopathic electromyographic features. Disease is usually progressive to a variable degree, ranging from minor disability to complete inability to ambulate, and can involve the large proximal muscles, as well as axial and facial muscles. Different disease forms may exhibit skeletal muscle hypertrophy, kyphoscoliosis, and contractures or involve other muscle groups and manifest with distal weakness, cardiomyopathy, dysphagia, and respiratory difficulties. LGMDR28 is characterized by progressive muscle weakness affecting the proximal and axial muscles of the upper and lower limbs, and highly variable age at onset. Most patients have limited ambulation or become wheelchair-bound within a few decades, and respiratory insufficiency commonly occurs. {ECO:0000269|PubMed:36745799, ECO:0000269|PubMed:37167966}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03169; DB04447; DB01076; DB09061; DB00439; DB01992; DB01095; DB00227; DB14009; DB04377; DB06693; DB14011; DB00157; DB03461; DB08860; DB00175; DB01098; DB00641; DB05317; DB09270 Interacts with Q9Y5Z9; Q9Y5Z9-1 EC number EC 1.1.1.34 Uniprot keywords 3D-structure; Alternative splicing; Cholesterol biosynthesis; Cholesterol metabolism; Disease variant; Endoplasmic reticulum; Glycoprotein; Isopeptide bond; Limb-girdle muscular dystrophy; Lipid biosynthesis; Lipid metabolism; Membrane; NADP; Oxidoreductase; Peroxisome; Phosphoprotein; Proteomics identification; Reference proteome; Steroid biosynthesis; Steroid metabolism; Sterol biosynthesis; Sterol metabolism; Transmembrane; Transmembrane helix; Ubl conjugation Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 84796.9 Length 798 Aromaticity 0.05 Instability index 47.61 Isoelectric point 6.2 Charge (pH=7) -3.71 2D Binding mode Binding energy (Kcal/mol) -6.78 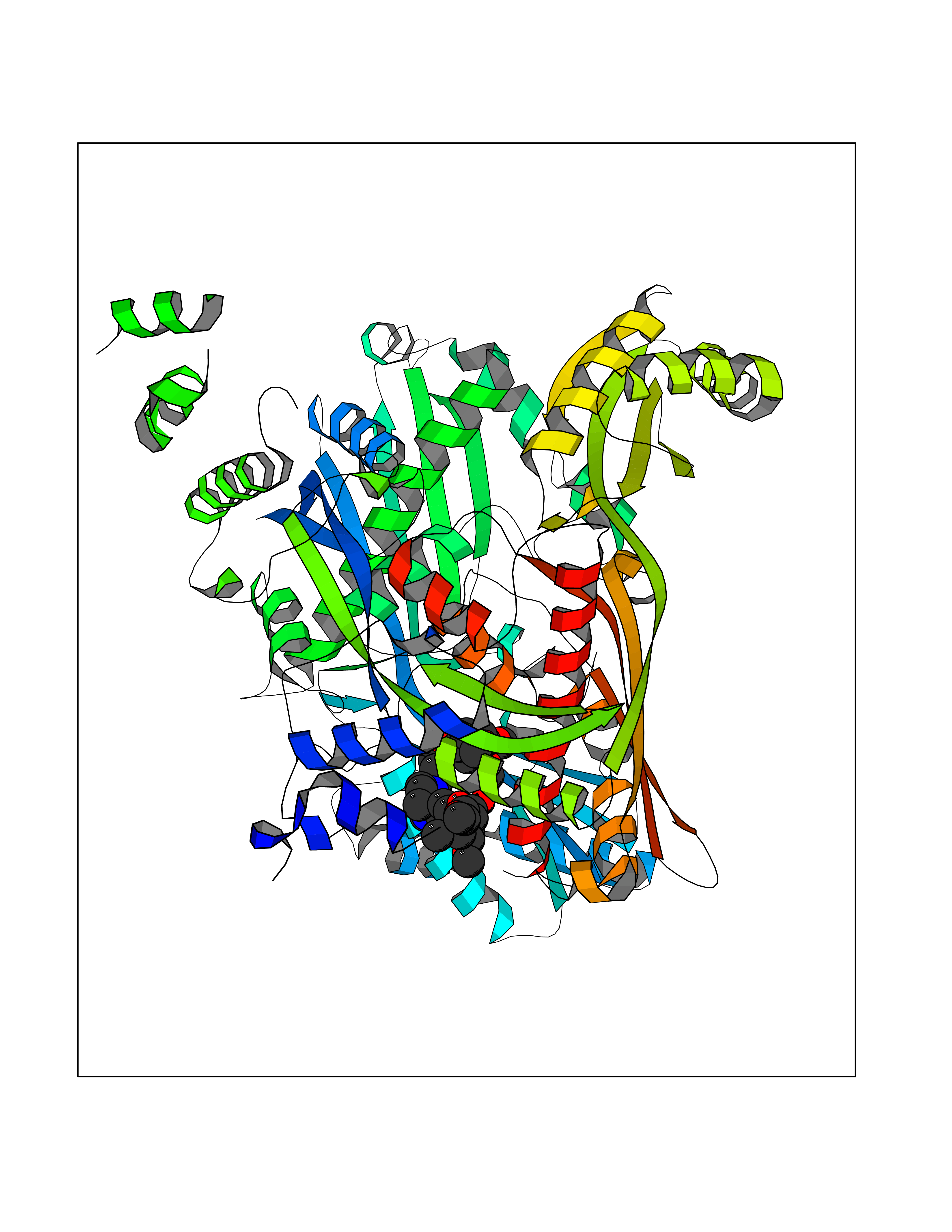 Molscript Map 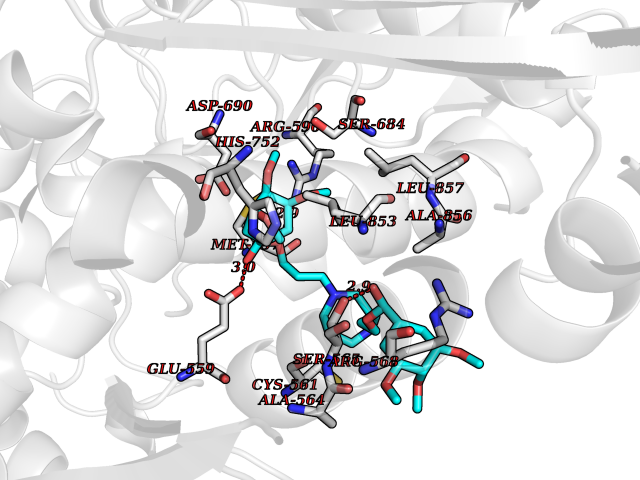 Pymol Map 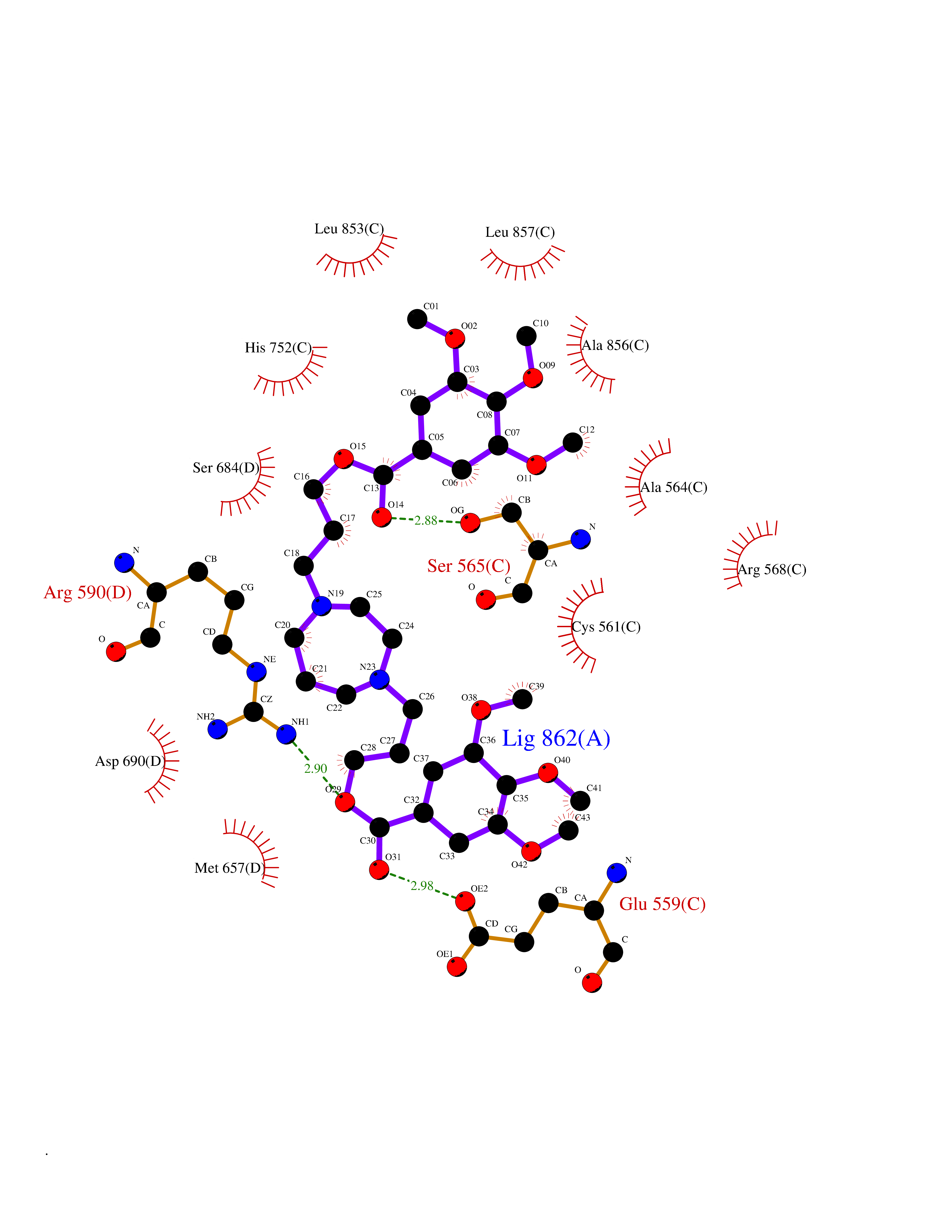 Ligplot Map 3D Binding mode Sequence GAKFLSDAEIIQLVNETLIETHERGVSIRRQLLSKKLSEPSSLQYLPYRDYNYSLVMGACCENVIGYMPIPVGVAGPLCLDEKEFQVPMATTEGCLVASTNRGCRAIGLGGGASSRVLADGMTRGPVVRLPRACDSAEVKAWLETSEGFAVIKEAFDSTSRFARLQKLHTSIAGRNLYIRFQSRSGDAMGMNMISKGTEKALSKLHEYFPEMQILAVSGNYCTDKKPAAINWIEGRGKSVVCEAVIPAKVVREVLKTTTEAMIEVNINKNLVGSAMAGSIGGYNAHAANIVTAIYIACGQDAAQNVGSSNCITLMEASGPTNEDLYISCTMPSIEIGTVGGGTNLLPQQACLQMLGVQGACKDNPGENARQLARIVCGTVMAGELSLMAALAAGPNEECLQILGNGAKFLSDAEIIQLVETLIETHERGVSIRRQLLSKKLSEPSSLQYLPYRDYNYSLVMGACCENVIGYMPIPVGVAGPLCLDEKEFQVPMATTEGCLVASTNRGCRAIGLGGGASSRVLADGMTRGPVVRLPRACDSAEVKAWLETSEGFAVIKEAFDSTSRFARLQKLHTSIAGRNLYIRFQSRSGDAMGMNMISKGTEKALSKLHEYFPEMQILAVSGNYCTDKKPAAINWIEGRGKSVVCEAVIPAKVVREVLKTTTEAMIEVNINKNLVGSAMAGSIGGYNAHAANIVTAIYIACGQDAAQNVGSSNCITLMEASGPTNEDLYISCTMPSIEIGTVGGGTNLLPQQACLQMLGVQGACKDNPGENARQLARIVCGTVMAGELSLMAALAAG Hydrogen bonds contact Hydrophobic contact | ||||
| 19 | Phosphodiesterase 4D (PDE4D) | 1Y2K | 7.65 | |
Target general information Gen name PDE4D Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms cAMP-specific 3',5'-cyclic phosphodiesterase 4D; PDE43; DPDE3 Protein family Cyclic nucleotide phosphodiesterase family, PDE4 subfamily Biochemical class Phosphoric diester hydrolase Function Hydrolyzes the second messenger cAMP, which is a key regulator of many important physiological processes. Related diseases Genetic variations in PDE4D might be associated with susceptibility to stroke. PubMed:17006457 states that association with stroke has to be considered with caution. {ECO:0000269|PubMed:17006457}.; DISEASE: Acrodysostosis 2, with or without hormone resistance (ACRDYS2) [MIM:614613]: A pleiotropic disorder characterized by skeletal, endocrine, and neurological abnormalities. Skeletal features include brachycephaly, midface hypoplasia with a small upturned nose, brachydactyly, and lumbar spinal stenosis. Endocrine abnormalities include hypothyroidism and hypogonadism in males and irregular menses in females. Developmental disability is a common finding but is variable in severity and can be associated with significant behavioral problems. {ECO:0000269|PubMed:22464250, ECO:0000269|PubMed:22464252, ECO:0000269|PubMed:23033274, ECO:0000269|PubMed:23043190}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB06842; DB04149; DB03606; DB03183; DB04469; DB02676; DB01959; DB07051; DB04271; DB07954; DB08299; DB00131; DB01427; DB00201; DB03849; DB05219; DB00651; DB06246; DB05266; DB01088; DB01113; DB01791; DB01656; DB01954; DB05298; DB09283; DB02918 Interacts with P32121; P38432; Q0D2H9; Q08AF8; P43360; Q07343; Q13077; P32121; P26769; P38432; Q96CV9; Q8IUH5 EC number EC 3.1.4.53 Uniprot keywords 3D-structure; Alternative splicing; cAMP; Cell membrane; Cytoplasm; Cytoskeleton; Disease variant; Hydrolase; Isopeptide bond; Manganese; Membrane; Metal-binding; Phosphoprotein; Proteomics identification; Reference proteome; Ubl conjugation; Zinc Protein physicochemical properties Chain ID A,B Molecular weight (Da) 37201.9 Length 322 Aromaticity 0.07 Instability index 35.83 Isoelectric point 5.02 Charge (pH=7) -21.16 2D Binding mode Binding energy (Kcal/mol) -6.96  Molscript Map 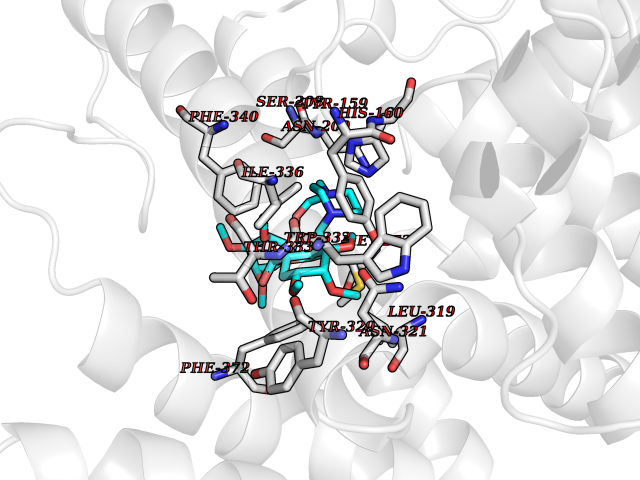 Pymol Map 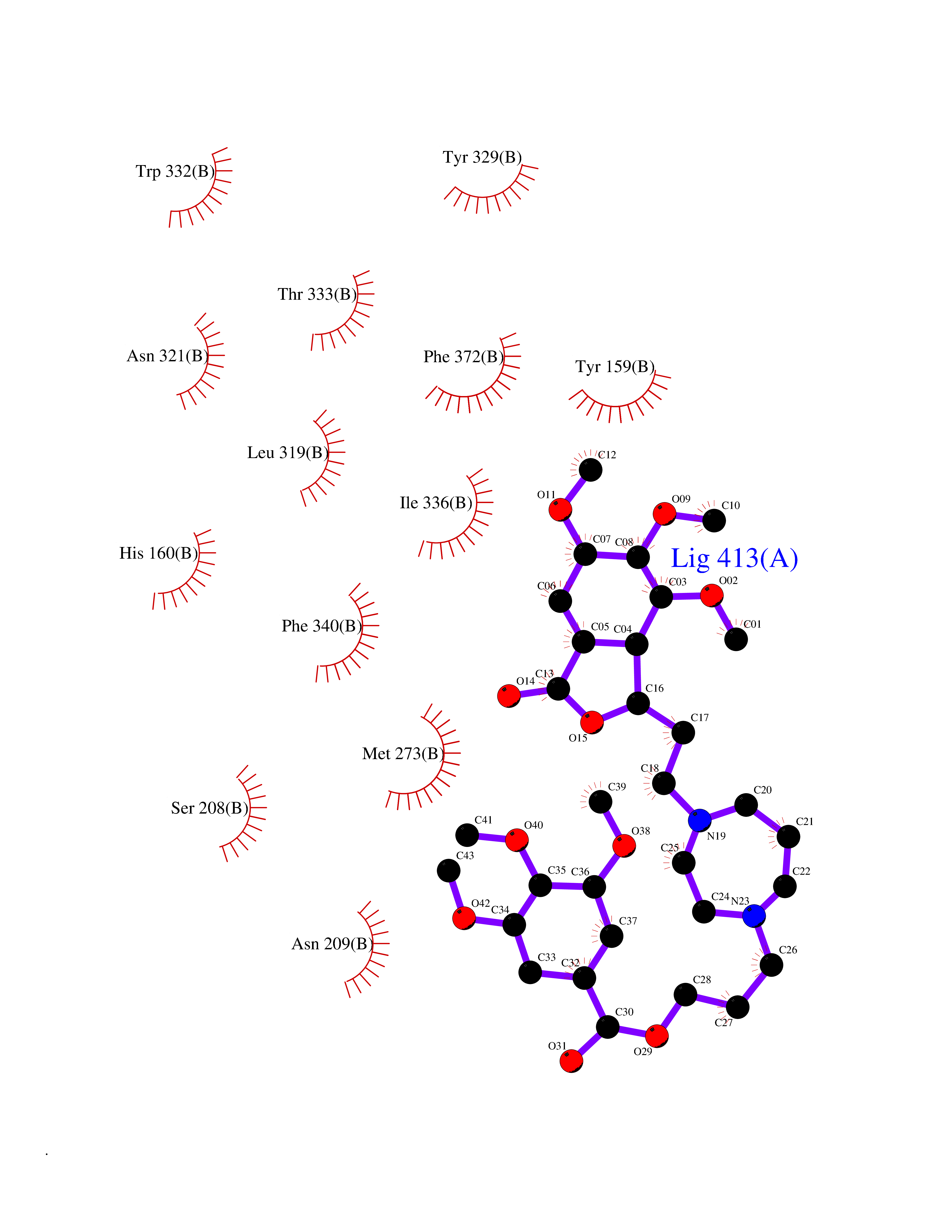 Ligplot Map 3D Binding mode Sequence TEQEDVLAKELEDVNKWGLHVFRIAELSGNRPLTVIMHTIFQERDLLKTFKIPVDTLITYLMTLEDHYHADVAYHNNIHAADVVQSTHVLLSTPALEAVFTDLEILAAIFASAIHDVDHPGVSNQFLINTNSELALMYNDSSVLENHHLAVGFKLLQEENCDIFQNLTKKQRQSLRKMVIDIVLATDMSKHMNLLADLKTMVETKKVVLLLDNYSDRIQVLQNMVHCADLSNPTKPLQLYRQWTDRIMEEFFRQGDRERERGMEISPMCDKHNASVEKSQVGFIDYIVHPLWETWADLVHPDAQDILDTLEDNREWYQSTIP Hydrogen bonds contact Hydrophobic contact | ||||
| 20 | Suppressor of tumorigenicity 14 protein (ST14) | 3P8G | 7.64 | |
Target general information Gen name ST14 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Tumor-associated differentially-expressed gene 15 protein; Tumor associated differentially-expressed gene-15 protein; TADG15; Serine protease TADG-15; Serine protease 14; SNC19; Prostamin; PRSS14; Mem Protein family Peptidase S1 family Biochemical class Peptidase Function Proposed to play a role in breast cancer invasion and metastasis. Exhibits trypsin-like activity as defined by cleavage of synthetic substrates with Arg or Lys as the P1 site. Involved in the terminal differentiation of keratinocytes through prostasin (PRSS8) activation and filaggrin (FLG) processing. Degrades extracellular matrix. Related diseases Ichthyosis, congenital, autosomal recessive 11 (ARCI11) [MIM:602400]: A form of autosomal recessive congenital ichthyosis, a disorder of keratinization with abnormal differentiation and desquamation of the epidermis, resulting in abnormal skin scaling over the whole body. The main skin phenotypes are lamellar ichthyosis (LI) and non-bullous congenital ichthyosiform erythroderma (NCIE), although phenotypic overlap within the same patient or among patients from the same family can occur. Lamellar ichthyosis is a condition often associated with an embedment in a collodion-like membrane at birth; skin scales later develop, covering the entire body surface. Non-bullous congenital ichthyosiform erythroderma characterized by fine whitish scaling on an erythrodermal background; larger brownish scales are present on the buttocks, neck and legs. {ECO:0000269|PubMed:17273967, ECO:0000269|PubMed:18843291}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03127; DB13729; DB00013 Interacts with NA EC number EC 3.4.21.109 Uniprot keywords 3D-structure; Disease variant; Disulfide bond; Glycoprotein; Hydrolase; Hypotrichosis; Ichthyosis; Membrane; Protease; Proteomics identification; Reference proteome; Repeat; Serine protease; Signal-anchor; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 26451.5 Length 241 Aromaticity 0.1 Instability index 30.45 Isoelectric point 5.6 Charge (pH=7) -5.69 2D Binding mode Binding energy (Kcal/mol) -7.31 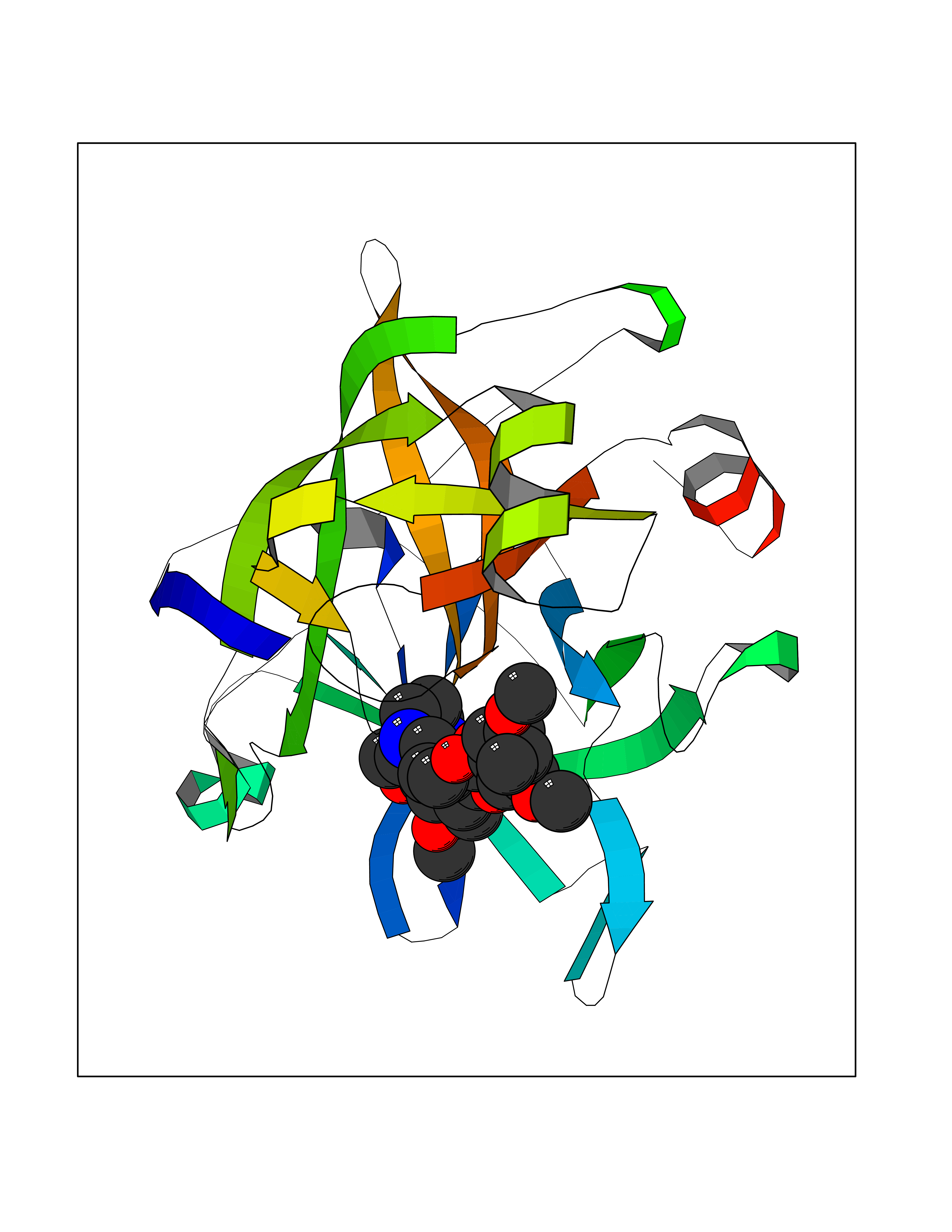 Molscript Map 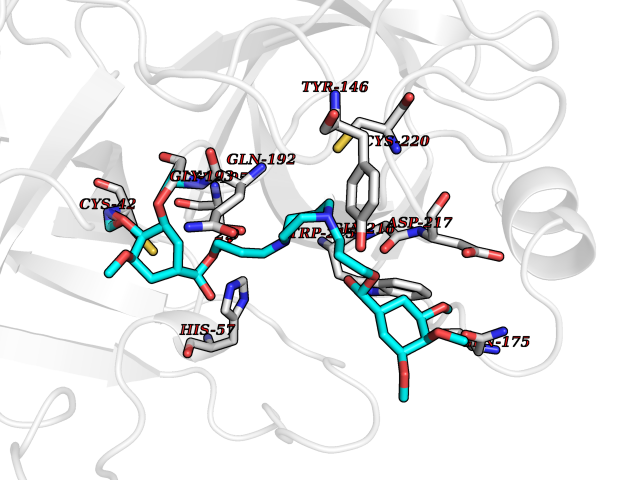 Pymol Map 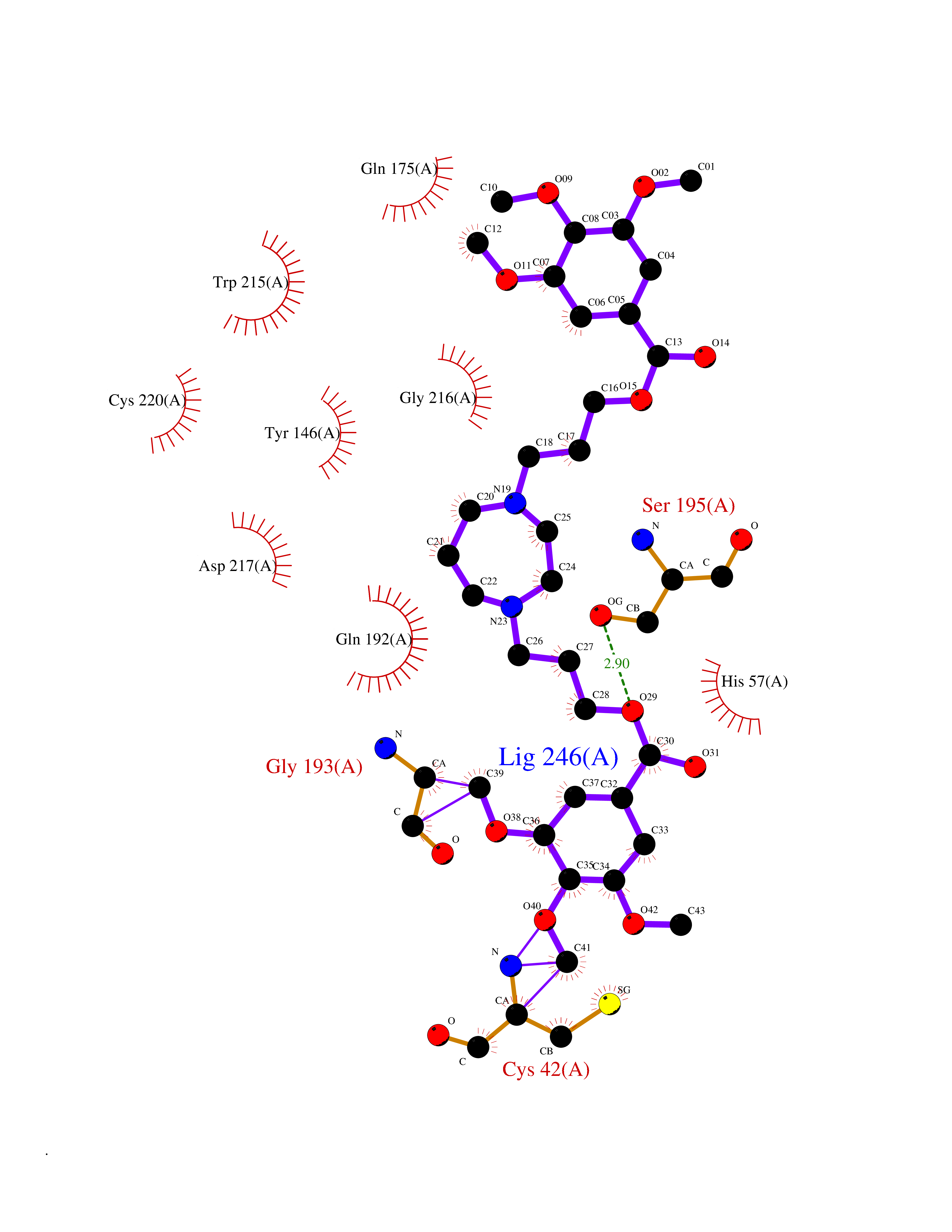 Ligplot Map 3D Binding mode Sequence VVGGTDADEGEWPWQVSLHALGQGHICGASLISPNWLVSAAHCYIDDRGFRYSDPTQWTAFLGLHDQSQRSAPGVQERRLKRIISHPFFNDFTFDYDIALLELEKPAEYSSMVRPICLPDASHVFPAGKAIWVTGWGHTQYGGTGALILQKGEIRVIQQTTCENLLPQQITPRMMCVGFLSGGVDSCQGDSGGPLSSVEADGRIFQAGVVSWGDGCAQRNKPGVYTRLPLFRDWIKENTGV Hydrogen bonds contact Hydrophobic contact | ||||