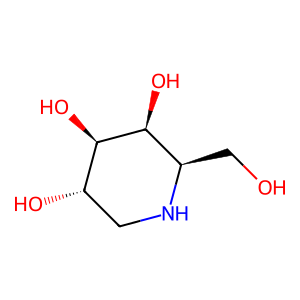
Ligand
Structure
Job ID
67b35c4597fc6f98e0944e0afb3d581c
Job name
Charlie_Try33
Time
2024-12-04 22:30:42
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 1 | DNA mismatch repair protein MSH2 (MSH2) | 3THX | 6.75 | |
Target general information Gen name MSH2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms hMSH2; MutS protein homolog 2; Mismatch repair gene Msh2 Protein family DNA mismatch repair MutS family Biochemical class NA Function Forms two different heterodimers: MutS alpha (MSH2-MSH6 heterodimer) and MutS beta (MSH2-MSH3 heterodimer) which binds to DNA mismatches thereby initiating DNA repair. When bound, heterodimers bend the DNA helix and shields approximately 20 base pairs. MutS alpha recognizes single base mismatches and dinucleotide insertion-deletion loops (IDL) in the DNA. MutS beta recognizes larger insertion-deletion loops up to 13 nucleotides long. After mismatch binding, MutS alpha or beta forms a ternary complex with the MutL alpha heterodimer, which is thought to be responsible for directing the downstream MMR events, including strand discrimination, excision, and resynthesis. Recruits DNA helicase MCM9 to chromatin which unwinds the mismatch containg DNA strand. ATP binding and hydrolysis play a pivotal role in mismatch repair functions. The ATPase activity associated with MutS alpha regulates binding similar to a molecular switch: mismatched DNA provokes ADP-->ATP exchange, resulting in a discernible conformational transition that converts MutS alpha into a sliding clamp capable of hydrolysis-independent diffusion along the DNA backbone. This transition is crucial for mismatch repair. MutS alpha may also play a role in DNA homologous recombination repair. In melanocytes may modulate both UV-B-induced cell cycle regulation and apoptosis. Component of the post-replicative DNA mismatch repair system (MMR). Related diseases Lynch syndrome 1 (LYNCH1) [MIM:120435]: A form of Lynch syndrome, an autosomal dominant disease associated with marked increase in cancer susceptibility. It is characterized by a familial predisposition to early-onset colorectal carcinoma (CRC) and extra-colonic tumors of the gastrointestinal, urological and female reproductive tracts. Lynch syndrome is reported to be the most common form of inherited colorectal cancer in the Western world. Clinically, it is often divided into two subgroups. Type I is characterized by hereditary predisposition to colorectal cancer, a young age of onset, and carcinoma observed in the proximal colon. Type II is characterized by increased risk for cancers in certain tissues such as the uterus, ovary, breast, stomach, small intestine, skin, and larynx in addition to the colon. Diagnosis of classical Lynch syndrome is based on the Amsterdam criteria: 3 or more relatives affected by colorectal cancer, one a first degree relative of the other two; 2 or more generation affected; 1 or more colorectal cancers presenting before 50 years of age; exclusion of hereditary polyposis syndromes. The term 'suspected Lynch syndrome' or 'incomplete Lynch syndrome' can be used to describe families who do not or only partially fulfill the Amsterdam criteria, but in whom a genetic basis for colon cancer is strongly suspected. {ECO:0000269|PubMed:10375096, ECO:0000269|PubMed:10386556, ECO:0000269|PubMed:10528862, ECO:0000269|PubMed:10573010, ECO:0000269|PubMed:10612836, ECO:0000269|PubMed:10777691, ECO:0000269|PubMed:10829038, ECO:0000269|PubMed:11726306, ECO:0000269|PubMed:11870161, ECO:0000269|PubMed:11920458, ECO:0000269|PubMed:12112654, ECO:0000269|PubMed:12124176, ECO:0000269|PubMed:12132870, ECO:0000269|PubMed:12200596, ECO:0000269|PubMed:12362047, ECO:0000269|PubMed:12373605, ECO:0000269|PubMed:12655564, ECO:0000269|PubMed:12655568, ECO:0000269|PubMed:12658575, ECO:0000269|PubMed:14635101, ECO:0000269|PubMed:15046096, ECO:0000269|PubMed:15300854, ECO:0000269|PubMed:15342696, ECO:0000269|PubMed:15365995, ECO:0000269|PubMed:15613555, ECO:0000269|PubMed:15870828, ECO:0000269|PubMed:15896463, ECO:0000269|PubMed:15991316, ECO:0000269|PubMed:15996210, ECO:0000269|PubMed:16451135, ECO:0000269|PubMed:17101317, ECO:0000269|PubMed:17128465, ECO:0000269|PubMed:18561205, ECO:0000269|PubMed:18625694, ECO:0000269|PubMed:18781619, ECO:0000269|PubMed:18822302, ECO:0000269|PubMed:18951462, ECO:0000269|PubMed:21120944, ECO:0000269|PubMed:22102614, ECO:0000269|PubMed:22371642, ECO:0000269|PubMed:7874129, ECO:0000269|PubMed:8261515, ECO:0000269|PubMed:8700523, ECO:0000269|PubMed:8797773, ECO:0000269|PubMed:8872463, ECO:0000269|PubMed:9048925, ECO:0000269|PubMed:9240418, ECO:0000269|PubMed:9298827, ECO:0000269|PubMed:9311737, ECO:0000269|PubMed:9419403, ECO:0000269|PubMed:9559627, ECO:0000269|PubMed:9621522, ECO:0000269|PubMed:9718327, ECO:0000269|PubMed:9889267}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Muir-Torre syndrome (MRTES) [MIM:158320]: Rare autosomal dominant disorder characterized by sebaceous neoplasms and visceral malignancy. {ECO:0000269|PubMed:7713503}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Endometrial cancer (ENDMC) [MIM:608089]: A malignancy of endometrium, the mucous lining of the uterus. Most endometrial cancers are adenocarcinomas, cancers that begin in cells that make and release mucus and other fluids. {ECO:0000305|PubMed:11306449, ECO:0000305|PubMed:21642682}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Mismatch repair cancer syndrome 2 (MMRCS2) [MIM:619096]: An autosomal recessive form of mismatch repair cancer syndrome, a childhood cancer predisposition syndrome encompassing a broad tumor spectrum. This includes hematological malignancies, central nervous system tumors, Lynch syndrome-associated malignancies such as colorectal tumors as well as multiple intestinal polyps, embryonic tumors and rhabdomyosarcoma. Multiple cafe-au-lait macules, a feature reminiscent of neurofibromatosis type 1, are often found as first manifestation of the underlying cancer. {ECO:0000269|PubMed:12549480, ECO:0000269|PubMed:16372347}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Colorectal cancer (CRC) [MIM:114500]: A complex disease characterized by malignant lesions arising from the inner wall of the large intestine (the colon) and the rectum. Genetic alterations are often associated with progression from premalignant lesion (adenoma) to invasive adenocarcinoma. Risk factors for cancer of the colon and rectum include colon polyps, long-standing ulcerative colitis, and genetic family history. {ECO:0000269|PubMed:12792735, ECO:0000269|PubMed:14504054, ECO:0000269|PubMed:15996210, ECO:0000269|PubMed:9559627}. Disease susceptibility may be associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q92624; Q9UQ84-1; P09429; P20585; P52701; Q8IY92; P39875 EC number NA Uniprot keywords 3D-structure; Acetylation; Alternative splicing; ATP-binding; Chromosome; Disease variant; DNA damage; DNA repair; DNA-binding; Hereditary nonpolyposis colorectal cancer; Isopeptide bond; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Tumor suppressor; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 97883.4 Length 868 Aromaticity 0.08 Instability index 36.78 Isoelectric point 5.79 Charge (pH=7) -10.09 2D Binding mode Binding energy (Kcal/mol) -6.55  Molscript Map 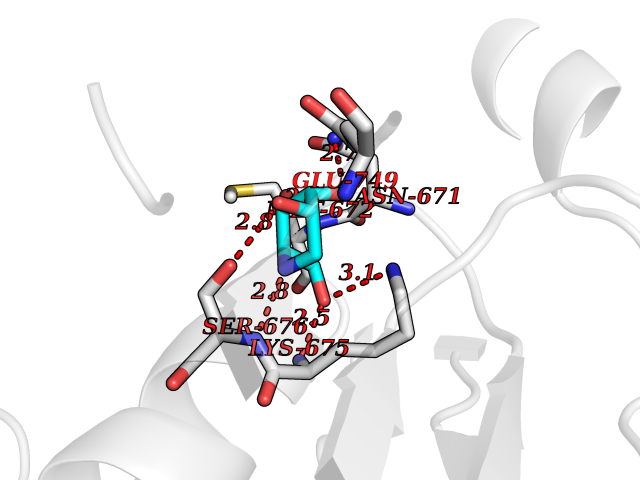 Pymol Map 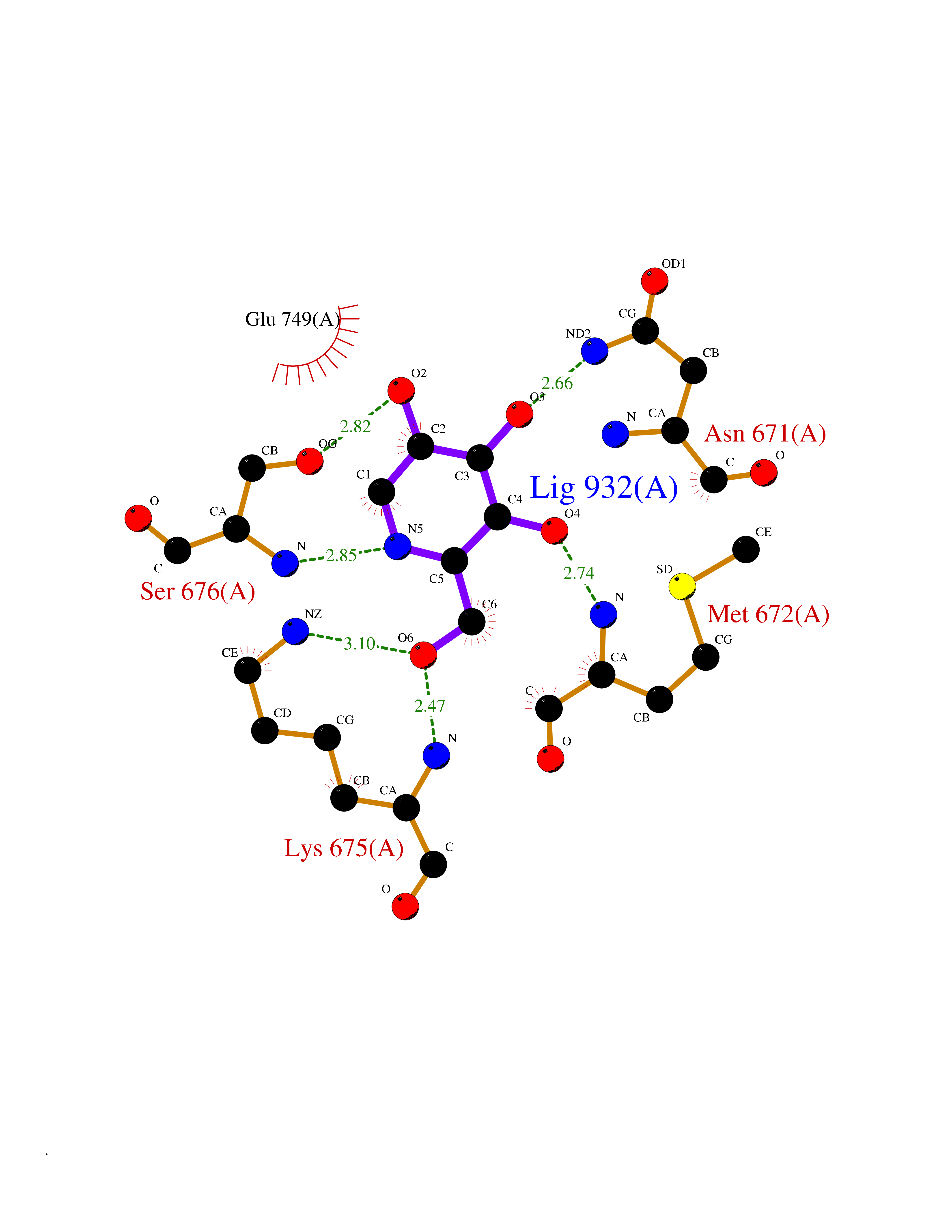 Ligplot Map 3D Binding mode Sequence ESAAEVGFVRFFQGMPEKPTTTVRLFDRGDFYTAHGEDALLAAREVFKTQGVIKYMGPAGAKNLQSVVLSKMNFESFVKDLLLVRQYRVEVYKNRASKENDWYLAYKASPGNLSQFEDILFIGVVGVKMSAVDGQRQVGVGYVDSIQRKLGLCEFPDNDQFSNLEALLIQIGPKECVLPGGETAGDMGKLRQIIQRGGILITERKKADFSTKDIYQDLNRLLKGKKGEQMNSAVLPEMENQVAVSSLSAVIKFLELLSDDSNFGQFELTTFDFSQYMKLDIAAVRALNLFQQSLAALLNKCKTPQGQRLVNQWIKQPLMDKNRIEERLNLVEAFVEDAELRQTLQEDLLRRFPDLNRLAKKFQRQAANLQDCYRLYQGINQLPNVIQALEKHEGKHQKLLLAVFVTPLTDLRSDFSKFQEMIETTLDMDQVENHEFLVKPSFDPNLSELREIMNDLEKKMQSTLISAARDLGLDPGKQIKLDSSAGYYFRVTCKEEKVLRNNKNFSTVDIQGVKFTNSKLTSLNEEYTKNKTEYEEAQDAIVKEIVNISSGYVEPMQTLNDVLAQLDAVVSFAHVSNGAPVPYVRPAILEKGQGRIILKASRHACVEVQIAFIPNDVYFEKDKQMFHIITGPNMGGKSTYIRQTGVIVLMAQIGCFVPCESAEVSIVDCILARVGSTFMAEMLETASILRSATKDSLIIIDELGRGTSTYDGFGLAWAISEYIATKIGAFCMFATHFHELTALANQIPTVNNLHVTALTTEETLTMLYQVKKGVCDQSFGIHVAELANFPKHVIECAKQKALELEEFQYKCYLEREQGEKIIQEFLSKVKQMPFTEMSEENITIKLKQLKAEVIAKNNSFVNEIISRI Hydrogen bonds contact Hydrophobic contact | ||||
| 2 | Oxygen-insensitive NADPH nitroreductase | 3QDL | 6.63 | |
Target general information Gen name rdxA Organism Helicobacter pylori (strain ATCC 700392 / 26695) (Campylobacter pylori) Uniprot ID TTD ID NA Synonyms HP_0954 Protein family Nitroreductase family Biochemical class Oxidoreductase Function Oxidoreductase activity. Related diseases Hypervalinemia and hyperleucine-isoleucinemia (HVLI) [MIM:618850]: An autosomal recessive metabolic disorder characterized by highly elevated plasma concentrations of valine and leucine/isoleucine. Affected individuals suffer from headache and mild memory impairment. {ECO:0000269|PubMed:25653144}. The disease is caused by variants affecting the gene represented in this entry. A patient with hypervalinemia and hyperleucine-isoleucinemia was identified as compound heterozygote for Gln-170 (inherited from his father) and Lys-264 (inherited from his mother), both variants reduced the catalytic activity of the enzyme. After treatment with vitamin B6, a precursor of pyridoxal 5'-phosphate, a BCAT2 cofactor, the blood levels of branched chain amino acids, especially valine, were decreased and brain lesions were improved. {ECO:0000269|PubMed:25653144}. Drugs (DrugBank ID) DB00916 Interacts with NA EC number 1.-.-.- Uniprot keywords 3D-structure; Antibiotic resistance; NADP; Oxidoreductase; Reference proteome Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 40094.3 Length 352 Aromaticity 0.08 Instability index 55.15 Isoelectric point 6.72 Charge (pH=7) -0.86 2D Binding mode Binding energy (Kcal/mol) -6.41 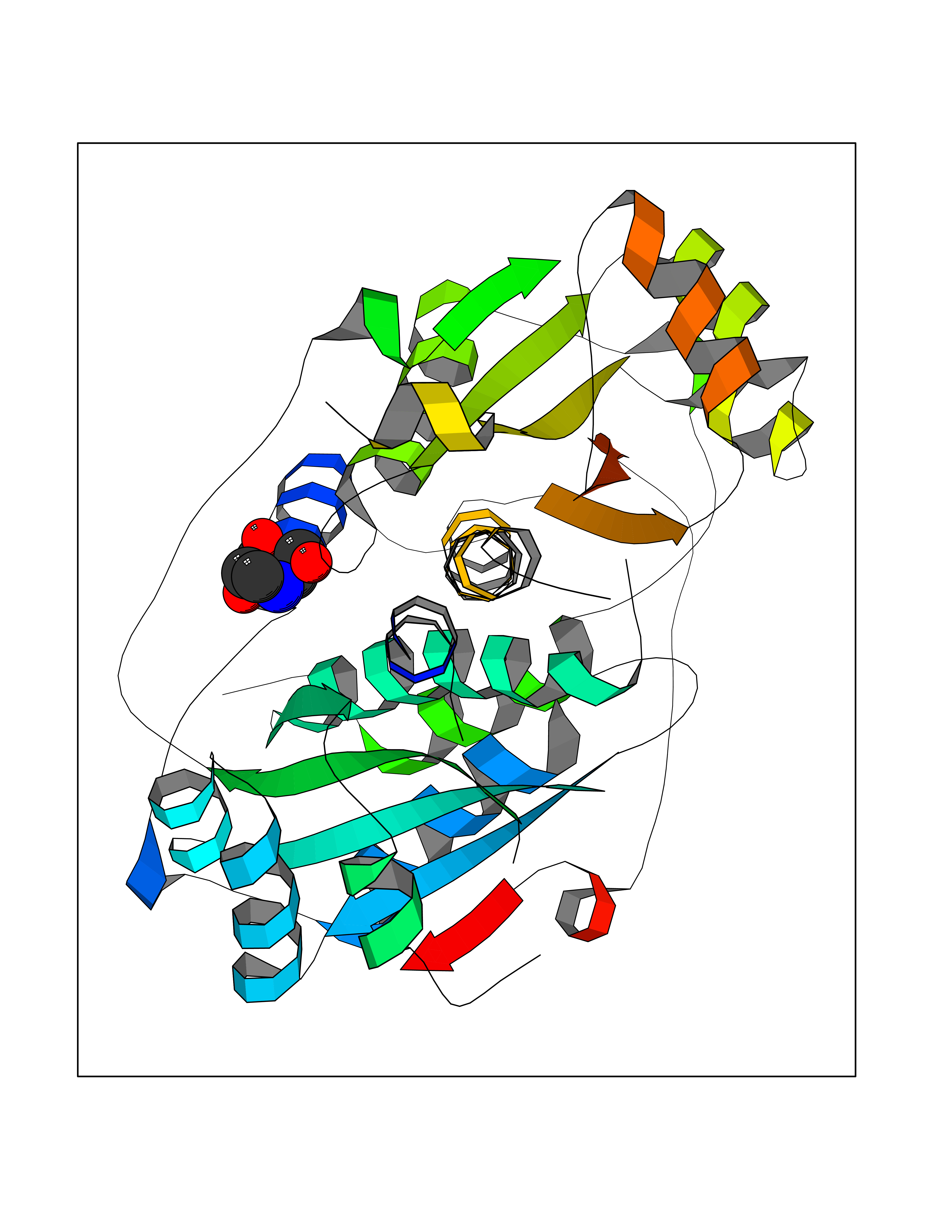 Molscript Map  Pymol Map 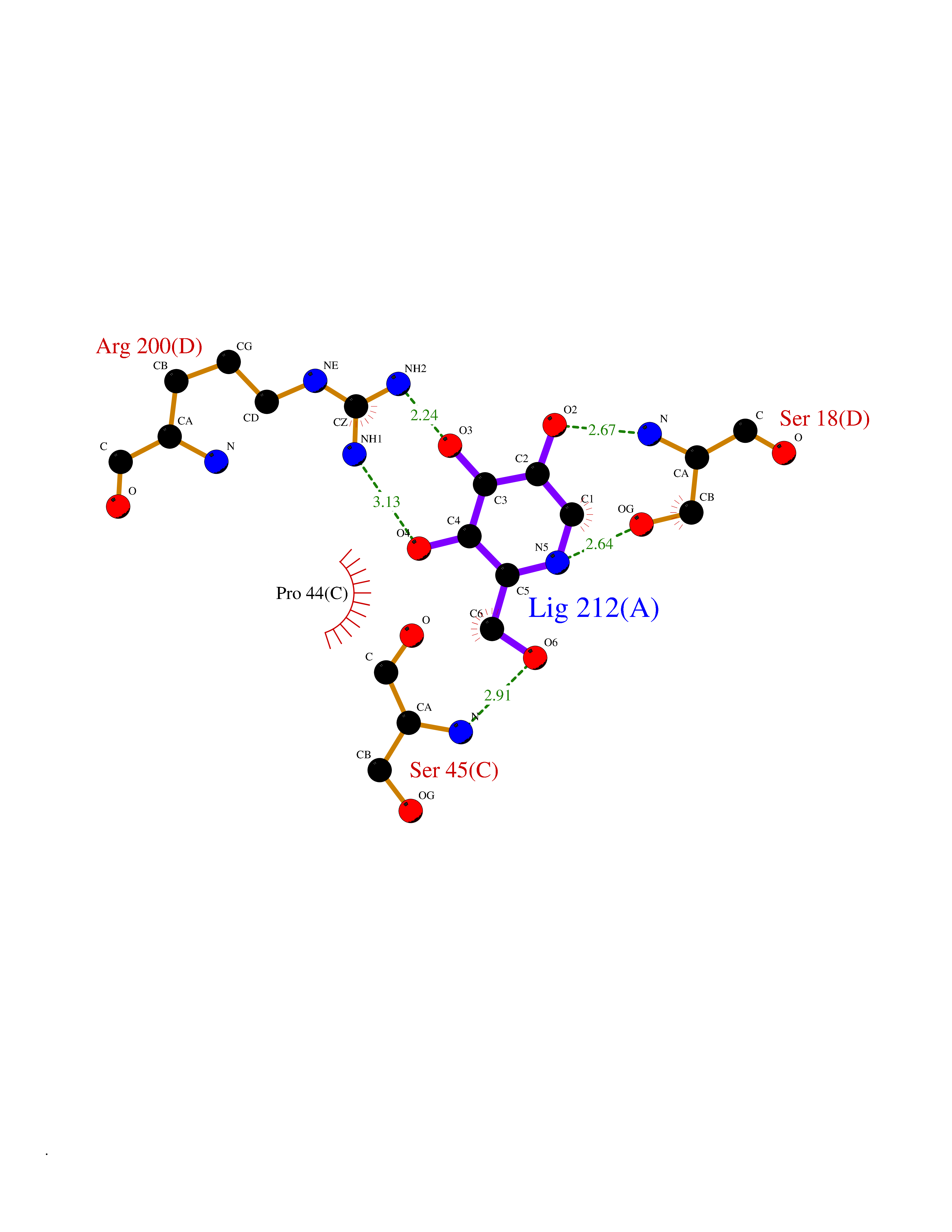 Ligplot Map 3D Binding mode Sequence MQRLESYILMKFLDQEKRRQLLNERHSCKMFDSHYEFSSTELEEIAEIARLSPSSYNTQPWHFVMVTDKDLKKQIAAHSYFNEEMIKSASALMVVCSLSYILEQCYIAVGQICMGVSLMGLDSCIIGGFDPLKVGEVLEERINPKIACLIALGKRVAEASQKSRKSKVDAITWLMKFLDQEKRRQLLNERHSCKMFDSHYEFSSTELEEIAEIARLSPSSYNTQPWHFVMVTDKDLKKQIAAHSYFNEEMIKSASALMVVCSLRPSELLPMQRLESYILEQCYIAVGQICMGVSLMGLDSCIIGGFDPLKVGEVLEERINKPKIACLIALGKRVAEASQKSRKSKVDAITWL Hydrogen bonds contact Hydrophobic contact | ||||
| 3 | Amylin receptor (IAPPR) | 6ZIS | 6.49 | |
Target general information Gen name CALCR-RAMP1/RAMP2/RAMP3 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Complex of Calcitonin receptor and Receptor activity-modifying protein Protein family RAMP family Biochemical class NA Function Transports the calcitonin gene-related peptide type 1 receptor (CALCRL) to the plasma membrane. Acts as a receptor for calcitonin-gene-related peptide (CGRP) together with CALCRL. Related diseases Immunodeficiency 9 (IMD9) [MIM:612782]: An immune disorder characterized by recurrent infections, impaired activation and proliferative response of T-cells, decreased T-cell production of cytokines, and normal lymphocytes counts and serum immunoglobulin levels. In surviving patients ectodermal dysplasia with anhidrosis and non-progressive myopathy may be observed. {ECO:0000269|PubMed:16147976, ECO:0000269|PubMed:16582901}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Myopathy, tubular aggregate, 2 (TAM2) [MIM:615883]: A rare congenital myopathy characterized by regular arrays of membrane tubules on muscle biopsies without additional histopathological hallmarks. Tubular aggregates in muscle are structures of variable appearance consisting of an outer tubule containing either one or more microtubule-like structures or amorphous material. TAM2 patients have myopathy and pupillary abnormalities. {ECO:0000269|PubMed:24591628, ECO:0000269|PubMed:28058752}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01278 Interacts with Q16602; P21145; Q5J8X5; Q16617 EC number NA Uniprot keywords 3D-structure; Cell membrane; Disulfide bond; Membrane; Proteomics identification; Receptor; Reference proteome; Signal; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A Molecular weight (Da) 63832.6 Length 569 Aromaticity 0.12 Instability index 22.42 Isoelectric point 5.07 Charge (pH=7) -16.96 2D Binding mode Binding energy (Kcal/mol) -6.56 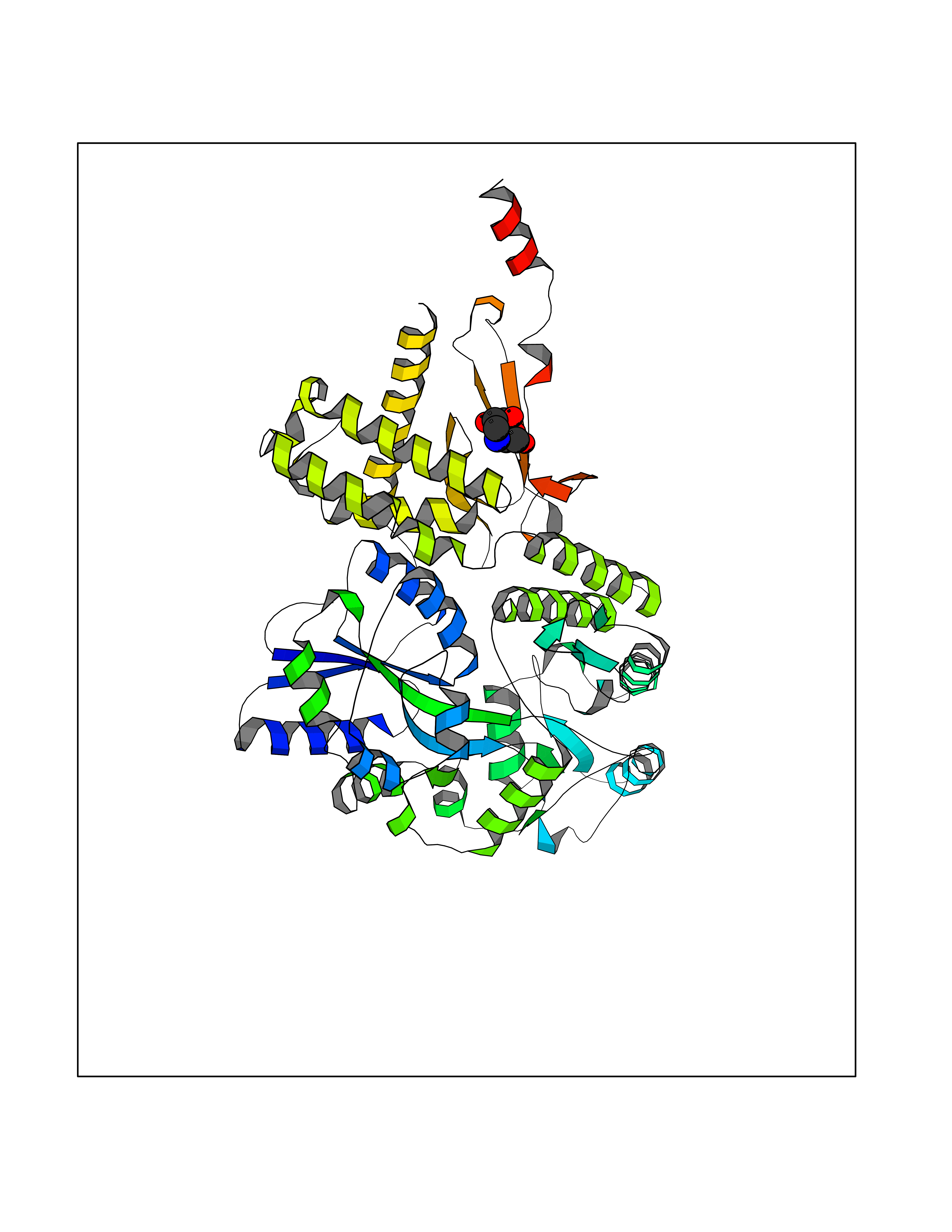 Molscript Map 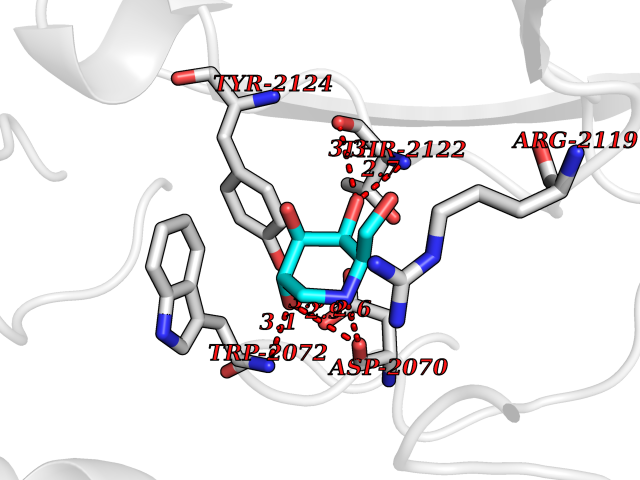 Pymol Map 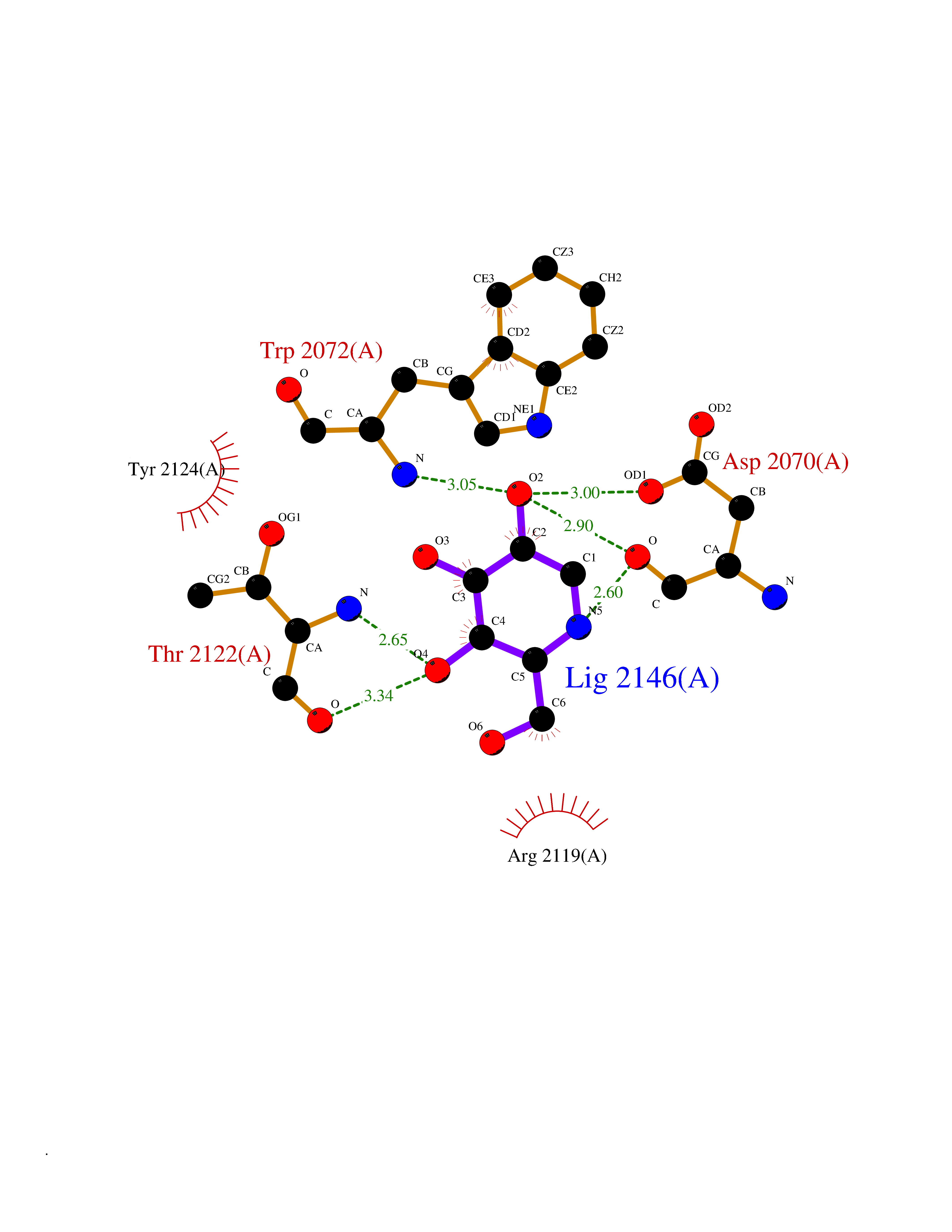 Ligplot Map 3D Binding mode Sequence SAKIEEGKLVIWINGDKGYNGLAEVGKKFEKDTGIKVTVEHPDKLEEKFPQVAATGDGPDIIFWAHDRFGGYAQSGLLAEITPDKAFQDKLYPFTWDAVRYNGKLIAYPIAVEALSLIYNKDLLPNPPKTWEEIPALDKELKAKGKSALMFNLQEPYFTWPLIAADGGYAFKYENGKYDIKDVGVDNAGAKAGLTFLVDLIKNKHMNADTDYSIAEAAFNKGETAMTINGPWAWSNIDTSKVNYGVTVLPTFKGQPSKPFVGVLSAGINAASPNKELAKEFLENYLLTDEGLEAVNKDKPLGAVALKSYEEELAKDPRIAATMENAQKGEIMPNIPQMSAFWYAVRTAVINAASGRQTVDEALKDAQTNAAAEFTTACQEANYGALLRELCLTQFQVDMEAVGETLWCDWGRTIRSYRELADCTWHMAEKLGCFWPNAEVDRFFLAVHGRYFRSCPISIQLGVTRNKIMTAQYECYQKIMQDPIQQGVYCQRTWDGWLCWNDVAAGTESMQLCPDYFQDFDPSEKVTKICDQDGNWFRHPASQRTWTDYTQCNVNTHEKVKTALNLFYL Hydrogen bonds contact Hydrophobic contact | ||||
| 4 | Choline O-acetyltransferase | 2FY3 | 6.46 | |
Target general information Gen name CHAT Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Carnitine/choline acetyltransferase family Biochemical class Transferase Function Choline O-acetyltransferase activity. Related diseases Myasthenic syndrome, congenital, 6, presynaptic (CMS6) [MIM:254210]: A form of congenital myasthenic syndrome, a group of disorders characterized by failure of neuromuscular transmission, including pre-synaptic, synaptic, and post-synaptic disorders that are not of autoimmune origin. Clinical features are easy fatigability and muscle weakness affecting the axial and limb muscles (with hypotonia in early-onset forms), the ocular muscles (leading to ptosis and ophthalmoplegia), and the facial and bulbar musculature (affecting sucking and swallowing, and leading to dysphonia). The symptoms fluctuate and worsen with physical effort. CMS6 affected individuals have myasthenic symptoms since birth or early infancy, negative tests for anti-AChR antibodies, and abrupt episodic crises with increased weakness, bulbar paralysis, and apnea precipitated by undue exertion, fever, or excitement. CMS6 inheritance is autosomal recessive. {ECO:0000269|PubMed:11172068, ECO:0000269|PubMed:12756141}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00122; DB14006; DB00184 Interacts with Q6H8Q1-8; Q8N302-2; Q9NXL2-1; Q6XD76; Q9UII2; Q8TBE0; Q9UQB8-6; Q9ULD4-2; Q9NSI6-4; Q6P5X5; Q96LL4; P20807-4; O00257-3; Q6ZP82-1; O95674; Q9H3R5; Q8WUX9; Q9H2A9; Q3SX64; Q92782-2; Q14117; O14641; Q658K8; Q6UXG2-3; O00472; Q6NXG1; Q15910-2; Q8IZU1; P15407; P55318; Q06547-3; P23769-2; P23771; Q15486; Q8IV36; Q4VB01; Q53GQ0; P10809; P41134; Q9NZH6; Q8NA54; Q86U28; P17275; Q8N5Z5; Q6P597; P08727; Q14525; Q8IUC2; Q6IAA8; Q14847-2; P27338; Q9GZQ8; Q53S70; Q5JXC2; A0A0A0MR05; Q8NEH6; Q8TCY5; Q6IN84-2; Q96H12; P01106; P41271-2; P14598; Q9GZM8; Q5BJF6-2; Q9H8K7; Q9NR21-5; Q5VU43-8; Q13956; Q5SXH7-1; Q96T60; Q96I34; Q86UA1; Q15311; Q8TBY0; Q04206; P47804-3; Q9H0X6; P62899; Q66K80; Q9BY12-3; Q86SQ7-2; Q7Z6I5; Q496A3; Q7Z698; Q9C004; Q92783-2; Q8N4C7; O75528; Q15814; O15273; Q96A09; Q8WTV1; Q53NU3; Q71RG4-4; Q86WT6-2; Q9Y3Q8; Q99598; P49459; P11441; Q9H270; P19544-6; Q53FD0-2; Q3KNS6-3 EC number 2.3.1.6 Uniprot keywords 3D-structure; Acyltransferase; Alternative splicing; Congenital myasthenic syndrome; Direct protein sequencing; Disease variant; Neurotransmitter biosynthesis; Phosphoprotein; Proteomics identification; Reference proteome; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 66365.9 Length 595 Aromaticity 0.08 Instability index 53.36 Isoelectric point 8.16 Charge (pH=7) 4.64 2D Binding mode Binding energy (Kcal/mol) -6.51  Molscript Map 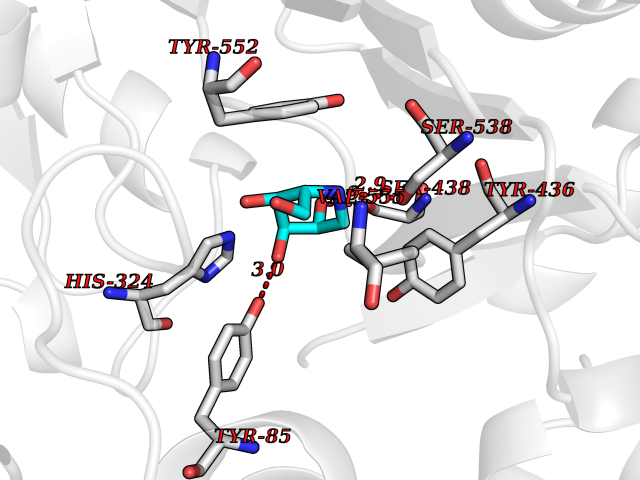 Pymol Map 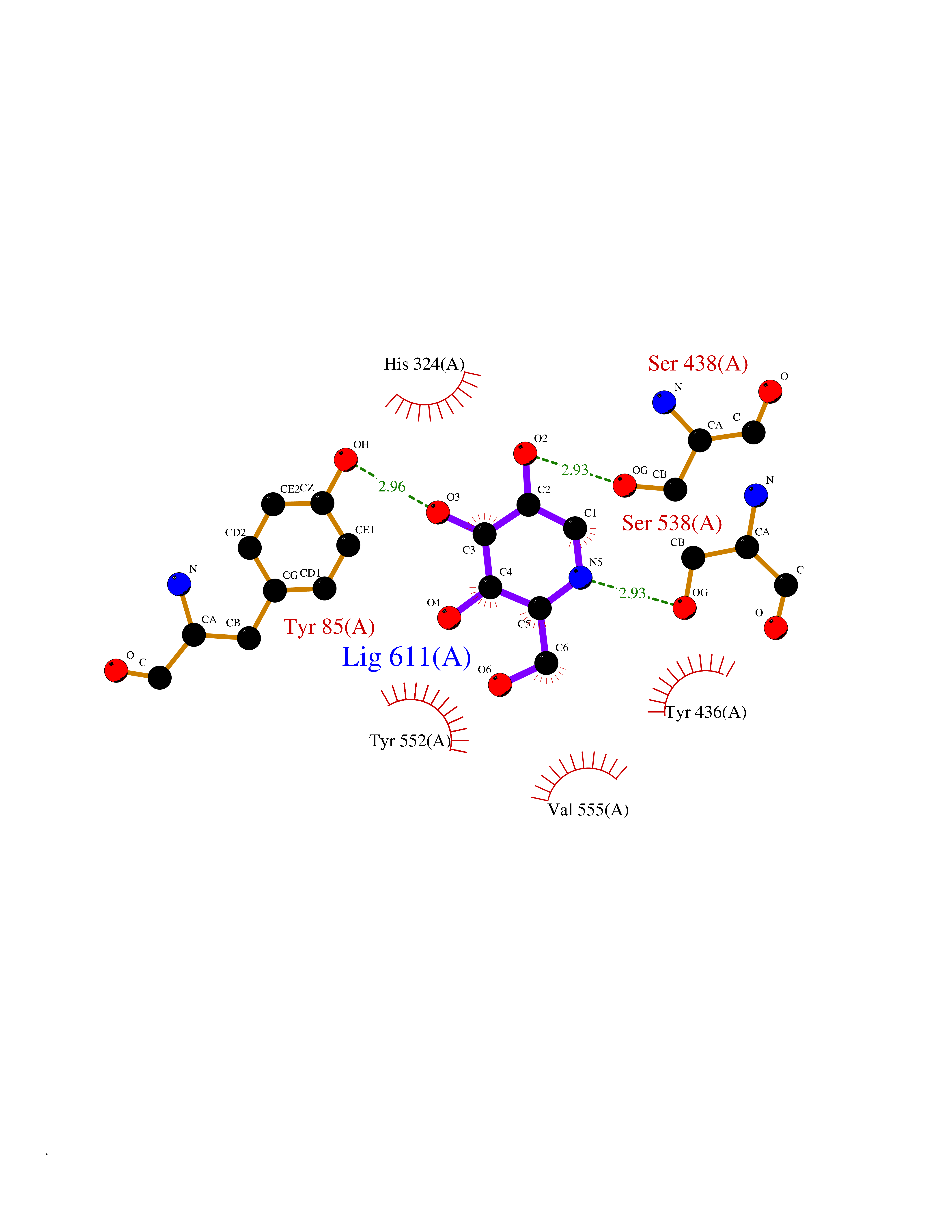 Ligplot Map 3D Binding mode Sequence SEESGLPKLPVPPLQQTLATYLQCMRHLVSEEQFRKSQAIVQQFGAPGGLGETLQQKLLERQEKTANWVSEYWLNDMYLNNRLALPVNSSPAVIFARQHFPGTDDQLRFAASLISGVLSYKALLDSHSIPTDCAKGQPLCMKQYYGLFSSYRLPGHTQDTLVAQNSSIMPEPEHVIVACCNQFFVLDVVINFRRLSEGDLFTQLRKIVKMASNAAARLPPIGLLTSDGRSEWAEARTVLVKDSTNRDSLDMIERCICLVCLDAPGGVELSDTHRALQLLHGGGYSKNGANRWYDKSLQFVVGRDGTCGVVCEHSPFDGIVLVQCTEHLLKHMTQPELVRSPMVPLPAPRRLRWKCSPEIQGHLASSAEKLQRIVKNLDFIVYKFDNYGKTFIKKQKCSPDAFIQVALQLAFYRLHRRLVPTYESASIRRFQEGRVDNIRSATPEALAFVRAVTDHKAAVPASEKLLLLKDAIRAQTAYTVMAITGMAIDNHLLALRELARAMCAALPEMFMDETYLMSNRFVLSTSQVPTTTEMFCCYGPVVPNGYGACYNPQPETILFCISSFHSCAATSSSKFAKAVEESLIDMRDLCSLLPP Hydrogen bonds contact Hydrophobic contact | ||||
| 5 | Mucin-1 (MUC1) | 6KX1 | 6.45 | |
Target general information Gen name MUC1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Tumour-associated antigen mucin 1; Tumor-associated mucin; Tumor-associated epithelial membraneantigen; Tumor-associated epithelial membrane antigen; Polymorphic epithelial mucin; Peanut-reactive urin Protein family NA Biochemical class NA Function Can act both as an adhesion and an anti-adhesion protein. May provide a protective layer on epithelial cells against bacterial and enzyme attack. The alpha subunit has cell adhesive properties. Related diseases MUC1/CA 15-3 is used as a serological clinical marker of breast cancer to monitor response to breast cancer treatment and disease recurrence (PubMed:20816948). Decreased levels over time may be indicative of a positive response to treatment. Conversely, increased levels may indicate disease progression. At an early stage disease, only 21% of patients exhibit high MUC1/CA 15-3 levels, that is why CA 15-3 is not a useful screening test. Most antibodies target the highly immunodominant core peptide domain of 20 amino acid (APDTRPAPGSTAPPAHGVTS) tandem repeats. Some antibodies recognize glycosylated epitopes. {ECO:0000269|PubMed:20816948}.; DISEASE: Tubulointerstitial kidney disease, autosomal dominant, 2 (ADTKD2) [MIM:174000]: A form of autosomal dominant tubulointerstitial kidney disease, a genetically heterogeneous disorder characterized by slowly progressive loss of kidney function, bland urinary sediment, hyperuricemia, absent or mildly increased albuminuria, lack of severe hypertension during the early stages, and normal or small kidneys on ultrasound. Renal histology shows variable abnormalities including interstitial fibrosis with tubular atrophy, microcystic dilatation of the tubules, thickening of tubular basement membranes, medullary cysts, and secondary glomerulosclerotic or glomerulocystic changes with abnormal glomerular tufting. There is significant variability, as well as incomplete penetrance. {ECO:0000269|PubMed:23396133}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB11090; DB06584 Interacts with P00519; P00533; P08581; P15941-7; Q08AM2; O60242; Q15848; Q86W74-2; P02652; P05067-2; P29972; P41181; Q92482; Q9H2C2; Q92843; Q6PL45-2; Q8WVV5; P06681; O14523; Q06432; Q9P0B6; Q08722-3; P19397; P34810; Q8N6F1-2; P56747; Q8NHS1; Q96FZ5; Q4VAQ0; Q8N6G5; Q07325; O43169; P78329; P56851; Q9BV81; P54852; O75355-2; Q9UKR5; P01350; P39905-3; Q9Y3E0; Q9NPR9; Q9HCP6; O60725; Q9Y5U4; P11215; Q969L2; Q13021; Q9P0N8; Q6N075; P30301; Q96S97; O95167; Q99519; Q92982; Q9NZG7; Q16617; Q8N912; Q8NH19; Q6TCH4; P26678; P60201-2; Q8IY26; P54315; Q59EV6; P30405; Q96AA3; Q02161-2; Q8TAC9; Q9Y6D0; Q8N6R1; P11686; Q8IWU4; Q969S0; Q6ICL7; Q9NVC3; Q9NRQ5; B2RUZ4; Q9NZ01; P07204; Q9BZW4; P17152; A0PK00; Q9BTD3; Q5BJH2-2; Q9BVK8; Q9Y6G1; Q9P0S9; Q14656; Q8NBD8; Q9BU79; Q8N2M4; Q8N661; Q5BJF2; Q9Y2Y6; O14763; Q8N609; Q5BVD1; Q53HI1; O95183; Q9BQB6; Q8IVQ6; P00519; P17676 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Autocatalytic cleavage; Cell membrane; Cytoplasm; Direct protein sequencing; Disulfide bond; Glycoprotein; Lipoprotein; Membrane; Nucleus; Palmitate; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Secreted; Signal; Transmembrane; Transmembrane helix; Tumor suppressor Protein physicochemical properties Chain ID B,C Molecular weight (Da) 25132.6 Length 230 Aromaticity 0.09 Instability index 44.8 Isoelectric point 7.12 Charge (pH=7) 0.18 2D Binding mode Binding energy (Kcal/mol) -5.94  Molscript Map 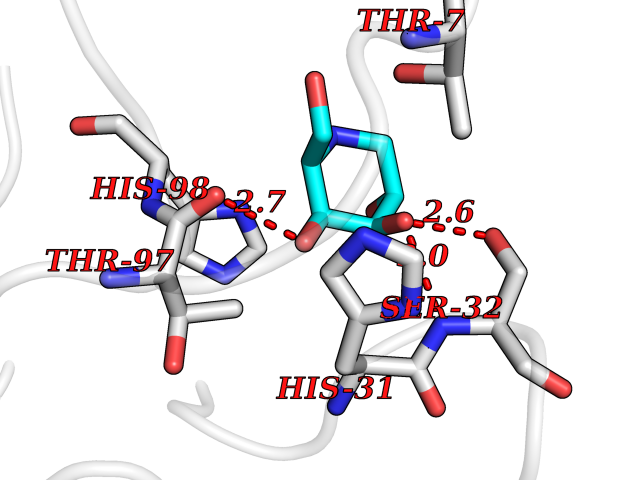 Pymol Map 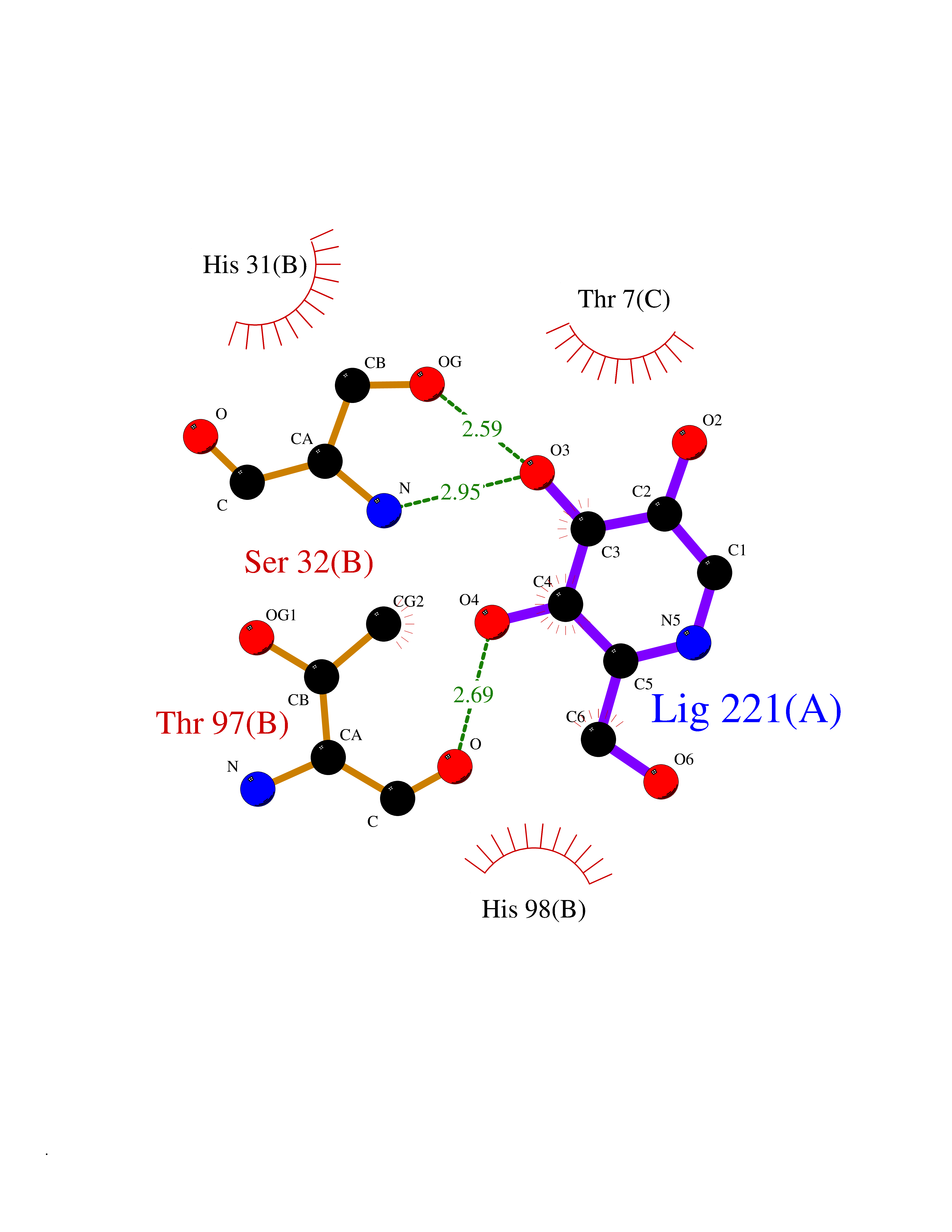 Ligplot Map 3D Binding mode Sequence DVVMTQTPLSLPVSLGDQASISCRSSQSLVHSNGNTYLHWYLQKPGQSPKLLIYKVSNRFSGVPDRFSGSGSGTDFTLKISRVEAEDLGVYFCSQSTHVPPWTFGGGTKLEIKRADAAPTVSIFPPSSEQLTSGGASVVCFLNNFYPKDINVKWKIDGSERQNGVLNSWTDQDSKDSTYSMSSTLTLTKDEYERHNSYTCEATHKTSTSPIVKSFNRNEXVTSAPDTRPA Hydrogen bonds contact Hydrophobic contact | ||||
| 6 | Carbonic anhydrase IV (CA-IV) | 3FW3 | 6.41 | |
Target general information Gen name CA4 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Carbonic anhydrase 4; Carbonate dehydratase IV; CAIV Protein family Alpha-carbonic anhydrase family Biochemical class Alpha-carbonic anhydrase Function May stimulate the sodium/bicarbonate transporter activity of SLC4A4 that acts in pH homeostasis. It is essential for acid overload removal from the retina and retina epithelium, and acid release in the choriocapillaris in the choroid. Reversible hydration of carbon dioxide. Related diseases Retinitis pigmentosa 17 (RP17) [MIM:600852]: A retinal dystrophy belonging to the group of pigmentary retinopathies. Retinitis pigmentosa is characterized by retinal pigment deposits visible on fundus examination and primary loss of rod photoreceptor cells followed by secondary loss of cone photoreceptors. Patients typically have night vision blindness and loss of midperipheral visual field. As their condition progresses, they lose their far peripheral visual field and eventually central vision as well. {ECO:0000269|PubMed:15563508, ECO:0000269|PubMed:17652713, ECO:0000269|PubMed:20450258}. The disease is caused by variants affecting the gene represented in this entry. Defective acid overload removal from retina and retinal epithelium, due to mutant CA4, is responsible for photoreceptor degeneration, indicating that impaired pH homeostasis is the most likely cause underlying the RP17 phenotype. Drugs (DrugBank ID) DB00819; DB00436; DB00562; DB01194; DB00606; DB01144; DB00869; DB08846; DB00311; DB00774; DB00703; DB00232; DB09460; DB00273; DB01021; DB00909 Interacts with NA EC number EC 4.2.1.1 Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; GPI-anchor; Lipoprotein; Lyase; Membrane; Metal-binding; Proteomics identification; Reference proteome; Retinitis pigmentosa; Signal; Zinc Protein physicochemical properties Chain ID A,B Molecular weight (Da) 27055.7 Length 235 Aromaticity 0.09 Instability index 44.3 Isoelectric point 6.87 Charge (pH=7) -0.36 2D Binding mode Binding energy (Kcal/mol) -6.36  Molscript Map 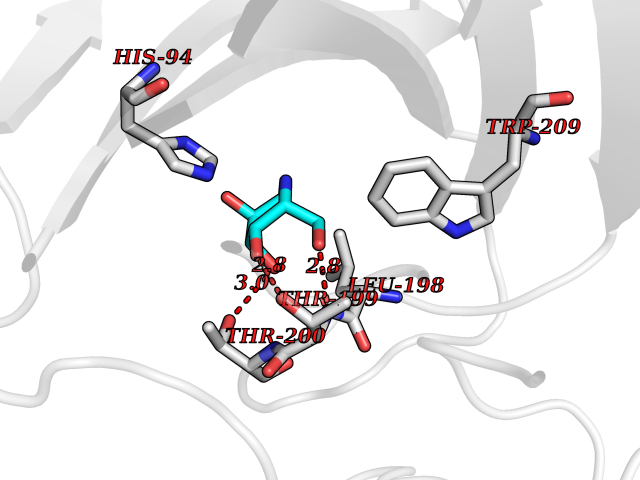 Pymol Map 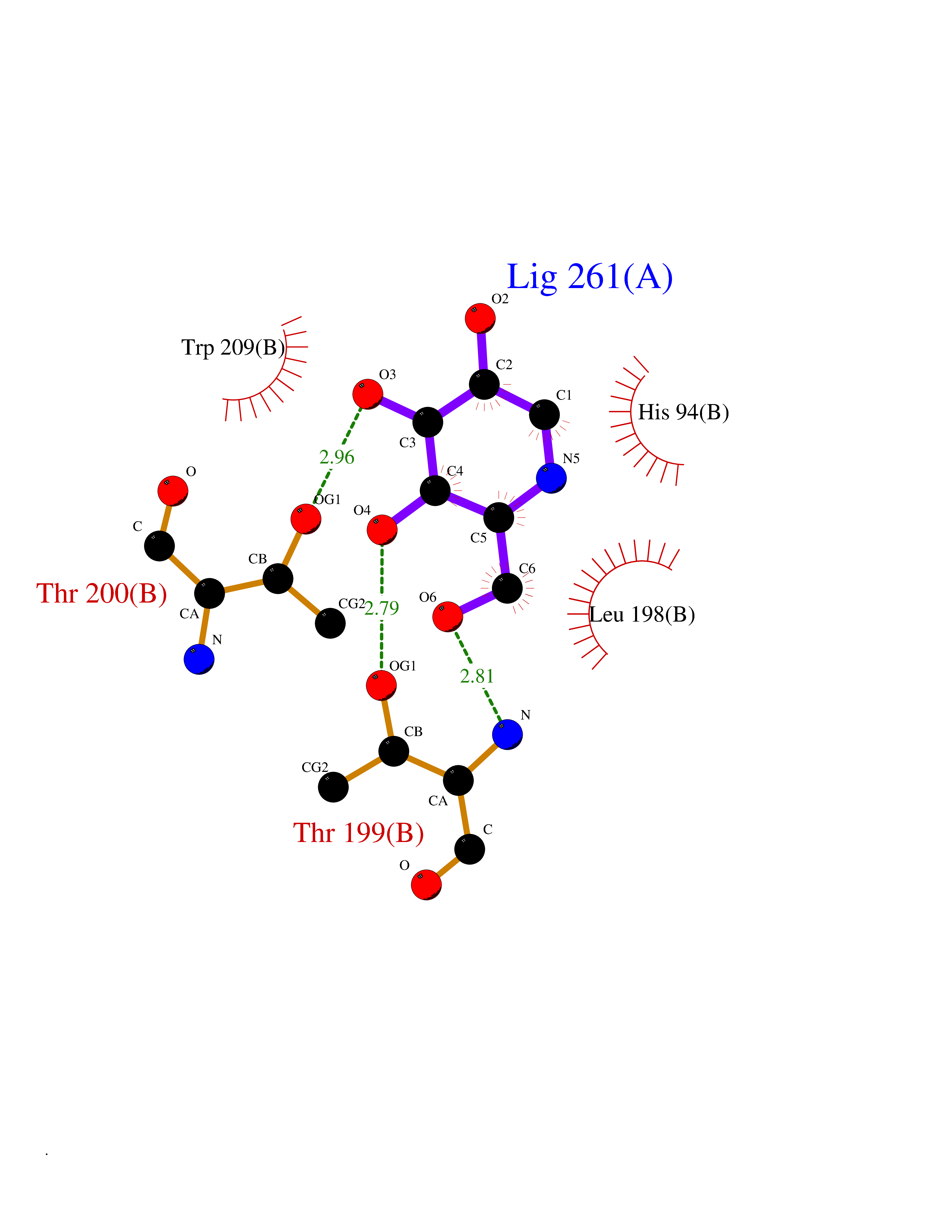 Ligplot Map 3D Binding mode Sequence HWCYEVQLVPVKWGGNCQKDRQSPINIVTTKAKVDKKLGRFFFGYDKKQTWTVQNNGHSVMMLLENKASISGGGLPAPYQAKQLHLHWSDLPYKGSEHSLDGEHFAMEMHIVHEKEEIAVLAFLVEATQVNEGFQPLVEALSNIPKPEMSTTMAESSLLDLLPEEKRHYFRYLGSLTTPTCDEKVVWTVFREPIQLHREQILAFQKLYYDKEQTVSMKDNVRPLQQLGQRTVIKS Hydrogen bonds contact Hydrophobic contact | ||||
| 7 | Carbonic anhydrase II (CA-II) | 3K34 | 6.38 | |
Target general information Gen name CA2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Carbonic anhydrase C; Carbonic anhydrase 2; Carbonate dehydratase II; CAC Protein family Alpha-carbonic anhydrase family Biochemical class Alpha-carbonic anhydrase Function Reversible hydration of carbon dioxide. Can hydrate cyanamide to urea. Involved in the regulation of fluid secretion into the anterior chamber of the eye. Contributes to intracellular pH regulation in the duodenal upper villous epithelium during proton-coupled peptide absorption. Stimulates the chloride-bicarbonate exchange activity of SLC26A6. Essential for bone resorption and osteoclast differentiation. Related diseases Osteopetrosis, autosomal recessive 3 (OPTB3) [MIM:259730]: A rare genetic disease characterized by abnormally dense bone, due to defective resorption of immature bone. Osteopetrosis occurs in two forms: a severe autosomal recessive form occurring in utero, infancy, or childhood, and a benign autosomal dominant form occurring in adolescence or adulthood. Recessive osteopetrosis commonly manifests in early infancy with macrocephaly, feeding difficulties, evolving blindness and deafness, bone marrow failure, severe anemia, and hepatosplenomegaly. Deafness and blindness are generally thought to represent effects of pressure on nerves. OPTB3 is associated with renal tubular acidosis, cerebral calcification (marble brain disease) and in some cases with intellectual disability. {ECO:0000269|PubMed:15300855, ECO:0000269|PubMed:1542674, ECO:0000269|PubMed:1928091, ECO:0000269|PubMed:8834238, ECO:0000269|PubMed:9143915}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07596; DB03333; DB08418; DB04081; DB08416; DB02479; DB07467; DB03950; DB03594; DB03294; DB04763; DB03270; DB08083; DB06954; DB08659; DB08046; DB02087; DB08156; DB04203; DB04394; DB08782; DB04549; DB02221; DB04180; DB03039; DB02861; DB02429; DB08202; DB04600; DB04601; DB01784; DB03385; DB03697; DB04002; DB07632; DB07050; DB06891; DB08645; DB08765; DB00819; DB03877; DB03262; DB03598; DB04089; DB01964; DB03526; DB04371; DB02220; DB03221; DB02602; DB02535; DB00436; DB00562; DB01194; DB00482; DB00880; DB02679; DB00606; DB02866; DB01144; DB00869; DB08846; DB01031; DB00311; DB08157; DB01942; DB00695; DB00774; DB08165; DB02292; DB03975; DB00703; DB00232; DB02610; DB07742; DB03844; DB02069; DB02986; DB08301; DB07048; DB03596; DB07476; DB01748; DB08155; DB01671; DB07710; DB01325; DB09460; DB09472; DB02894; DB00391; DB08329; DB07363; DB00273; DB01021; DB03904; DB00580; DB14533; DB14548; DB00909 Interacts with NA EC number EC 4.2.1.1 Uniprot keywords 3D-structure; Acetylation; Cell membrane; Cytoplasm; Direct protein sequencing; Disease variant; Lyase; Membrane; Metal-binding; Osteopetrosis; Phosphoprotein; Proteomics identification; Reference proteome; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 29027.4 Length 258 Aromaticity 0.1 Instability index 20.07 Isoelectric point 6.94 Charge (pH=7) -0.18 2D Binding mode Binding energy (Kcal/mol) -6.45  Molscript Map 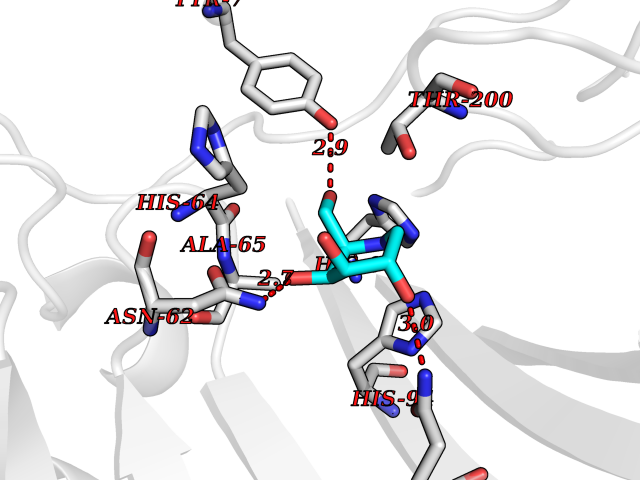 Pymol Map 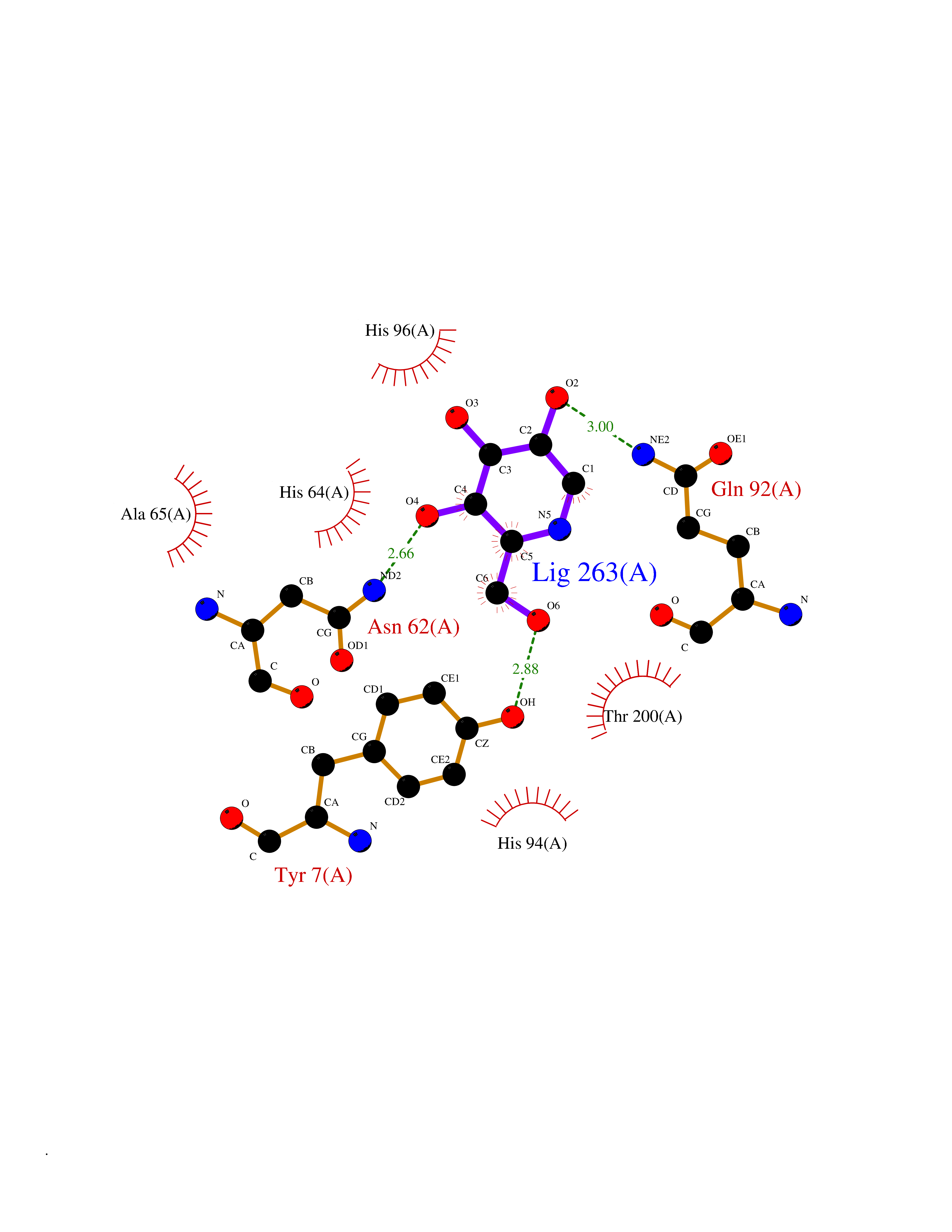 Ligplot Map 3D Binding mode Sequence HHWGYGKHNGPEHWHKDFPIAKGERQSPVDIDTHTAKYDPSLKPLSVSYDQATSLRILNNGHAFNVEFDDSQDKAVLKGGPLDGTYRLIQFHFHWGSLDGQGSEHTVDKKKYAAELHLVHWNTKYGDFGKAVQQPDGLAVLGIFLKVGSAKPGLQKVVDVLDSIKTKGKSADFTNFDPRGLLPESLDYWTYPGSLTTPPLLECVTWIVLKEPISVSSEQVLKFRKLNFNGEGEPEELMVDNWRPAQPLKNRQIKASFK Hydrogen bonds contact Hydrophobic contact | ||||
| 8 | Carbonic anhydrase IX (CA-IX) | 5FL4 | 6.34 | |
Target general information Gen name CA9 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Renal cell carcinoma-associated antigen G250; RCC-associated antigen G250; PMW1; P54/58N; Membrane antigen MN; MN; G250 antigen (MN/CA IX/G250); G250; Carbonic anhydrase 9; Carbonate dehydratase IX; C Protein family Alpha-carbonic anhydrase family Biochemical class Alpha-carbonic anhydrase Function Participates in pH regulation. May be involved in the control of cell proliferation and transformation. Appears to be a novel specific biomarker for a cervical neoplasia. Reversible hydration of carbon dioxide. Related diseases Hydroxykynureninuria (HYXKY) [MIM:236800]: An inborn error of amino acid metabolism characterized by massive urinary excretion of large amounts of kynurenine, 3-hydroxykynurenine and xanthurenic acid. Affected individuals manifest renal tubular dysfunction, metabolic acidosis, psychomotor retardation, non-progressive encephalopathy, and muscular hypertonia. {ECO:0000269|PubMed:17334708, ECO:0000269|PubMed:28792876}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Vertebral, cardiac, renal, and limb defects syndrome 2 (VCRL2) [MIM:617661]: An autosomal recessive congenital malformation syndrome characterized by vertebral segmentation abnormalities, congenital cardiac defects, renal defects, and distal mild limb defects. {ECO:0000269|PubMed:28792876}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00562; DB00606; DB12741; DB08846; DB05304; DB00774; DB09460; DB00909 Interacts with P21291; O76003 EC number EC 4.2.1.1 Uniprot keywords 3D-structure; Cell membrane; Cell projection; Direct protein sequencing; Disulfide bond; Glycoprotein; Lyase; Membrane; Metal-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Signal; Transmembrane; Transmembrane helix; Zinc Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 27522.8 Length 251 Aromaticity 0.08 Instability index 48.97 Isoelectric point 5.48 Charge (pH=7) -7.5 2D Binding mode Binding energy (Kcal/mol) -6.33  Molscript Map 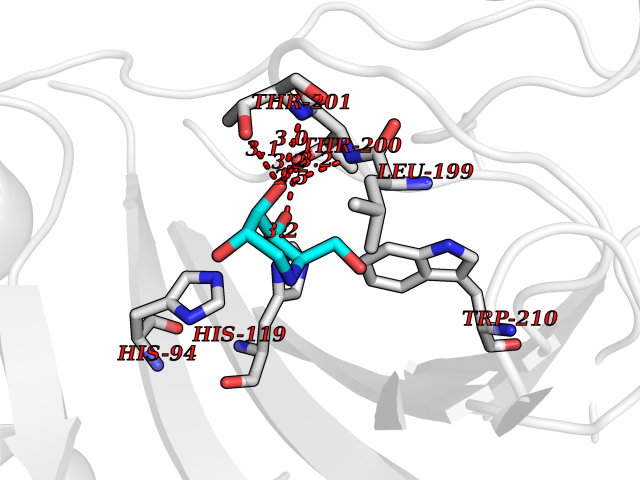 Pymol Map 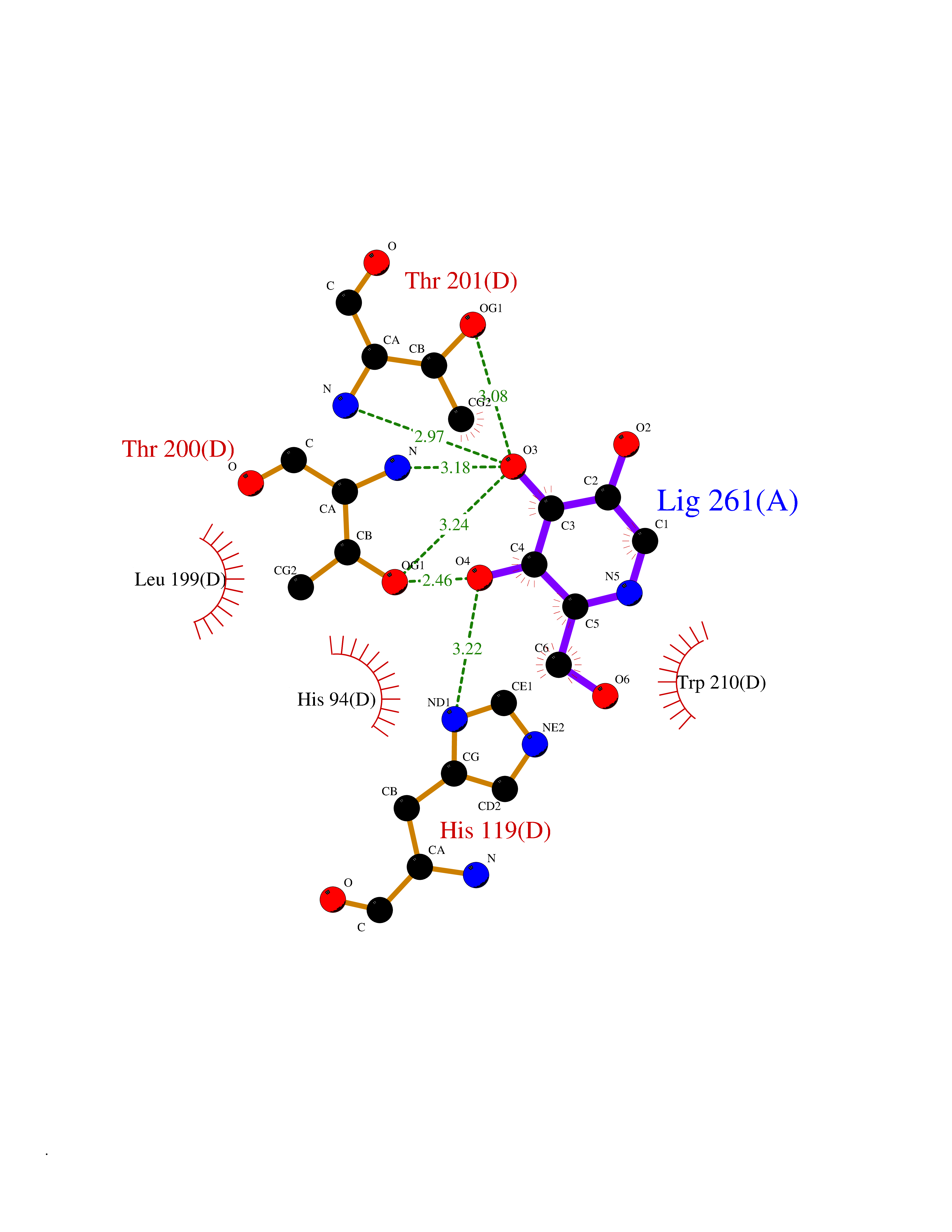 Ligplot Map 3D Binding mode Sequence WRYGGDPPWPRVSPACAGRFQSPVDIRPQLAAFSPALRPLELLGFQLPPLPELRLRNNGHSVQLTLPPGLEMALGPGREYRALQLHLHWGAAGRPGSEHTVEGHRFPAEIHVVHLSTAFARVDEALGRPGGLAVLAAFLEEGPEENSAYEQLLSRLEEIAEEGSETQVPGLDISALLPSDFSRYFQYEGSLTTPPCAQGVIWTVFNQTVMLSAKQLHTLSDTLWGPGDSRLQLNFRATQPLNGRVIEASFP Hydrogen bonds contact Hydrophobic contact | ||||
| 9 | Purine nucleoside phosphorylase (PNP) | 4EAR | 6.29 | |
Target general information Gen name PNP Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms PNP; Inosine phosphorylase Protein family PNP/MTAP phosphorylase family Biochemical class Pentosyltransferase Function The purine nucleoside phosphorylases catalyze the phosphorolytic breakdown of the N-glycosidic bond in the beta- (deoxy)ribonucleoside molecules, with the formation of the corresponding free purine bases and pentose-1-phosphate. Related diseases Purine nucleoside phosphorylase deficiency (PNPD) [MIM:613179]: A disorder that interrupts both the catabolism of inosine into hypoxanthine and guanosine into guanine, and leads to the accumulation of guanosine, inosine, and their deoxified by-products. The main clinical presentation is recurrent infections due to severe T-cell immunodeficiency. Some patients also have neurologic impairment. {ECO:0000269|PubMed:1384322, ECO:0000269|PubMed:3029074, ECO:0000269|PubMed:8931706}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03881; DB03551; DB02222; DB02391; DB03609; DB01667; DB04260; DB02796; DB04753; DB00640; DB00242; DB00900; DB06185; DB02377; DB02857; DB04754; DB04757; DB04076; DB02230; DB04335; DB02568; DB03101 Interacts with P05067; Q9UQM7; O14576-2; P06241; P14136; Q92993-2; Q9BXM7; P00491; P17612; P63000; Q92673; Q15583 EC number EC 2.4.2.1 Uniprot keywords 3D-structure; Acetylation; Cytoplasm; Direct protein sequencing; Disease variant; Glycosyltransferase; Phosphoprotein; Proteomics identification; Purine salvage; Reference proteome; Transferase Protein physicochemical properties Chain ID A,B,C Molecular weight (Da) 31849.2 Length 288 Aromaticity 0.1 Instability index 34.77 Isoelectric point 6.42 Charge (pH=7) -1.63 2D Binding mode Binding energy (Kcal/mol) -6.52 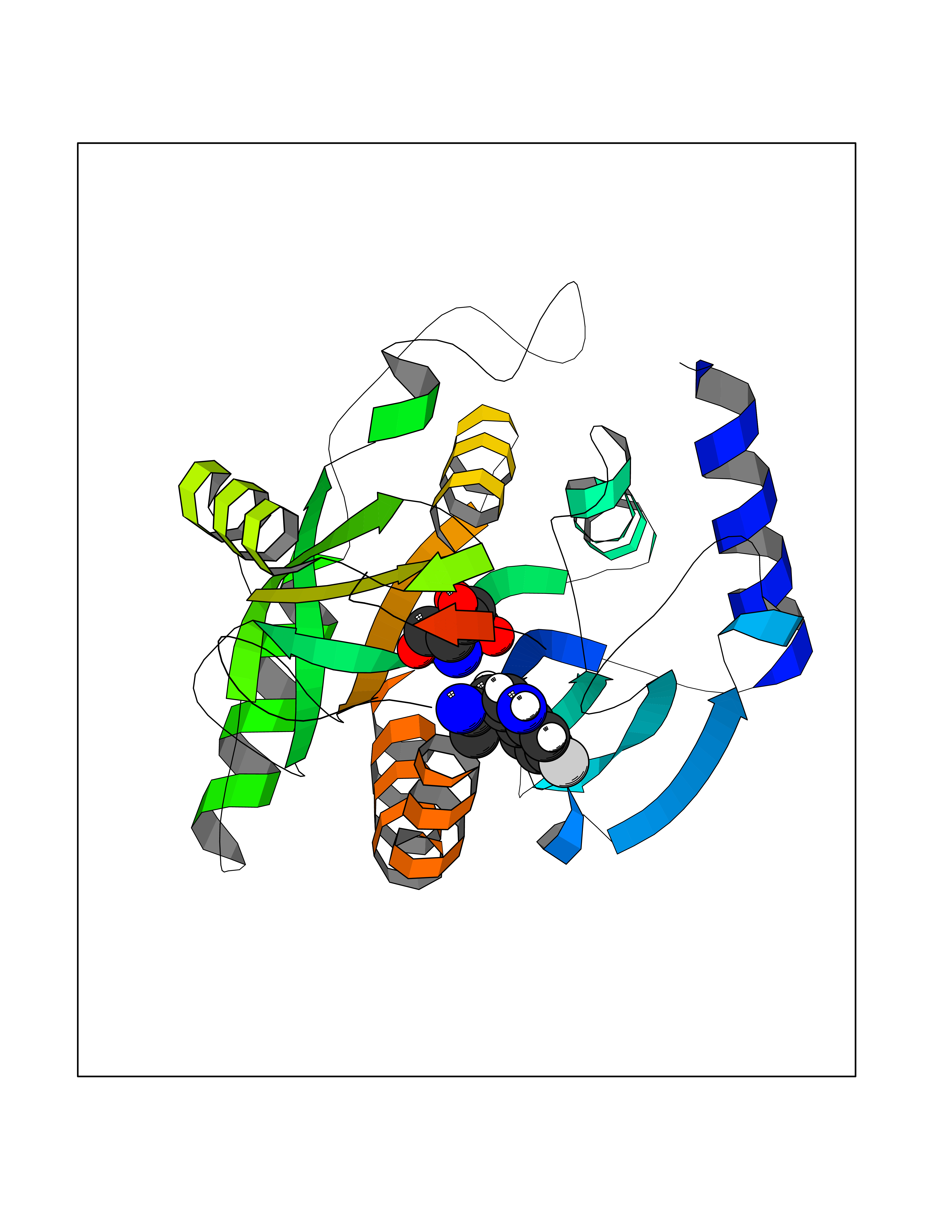 Molscript Map 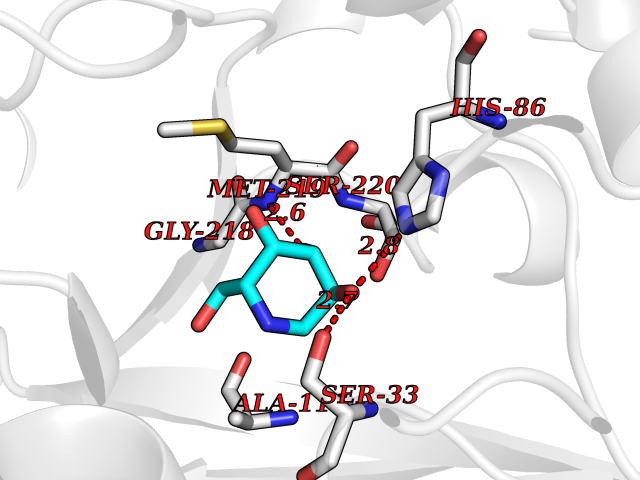 Pymol Map 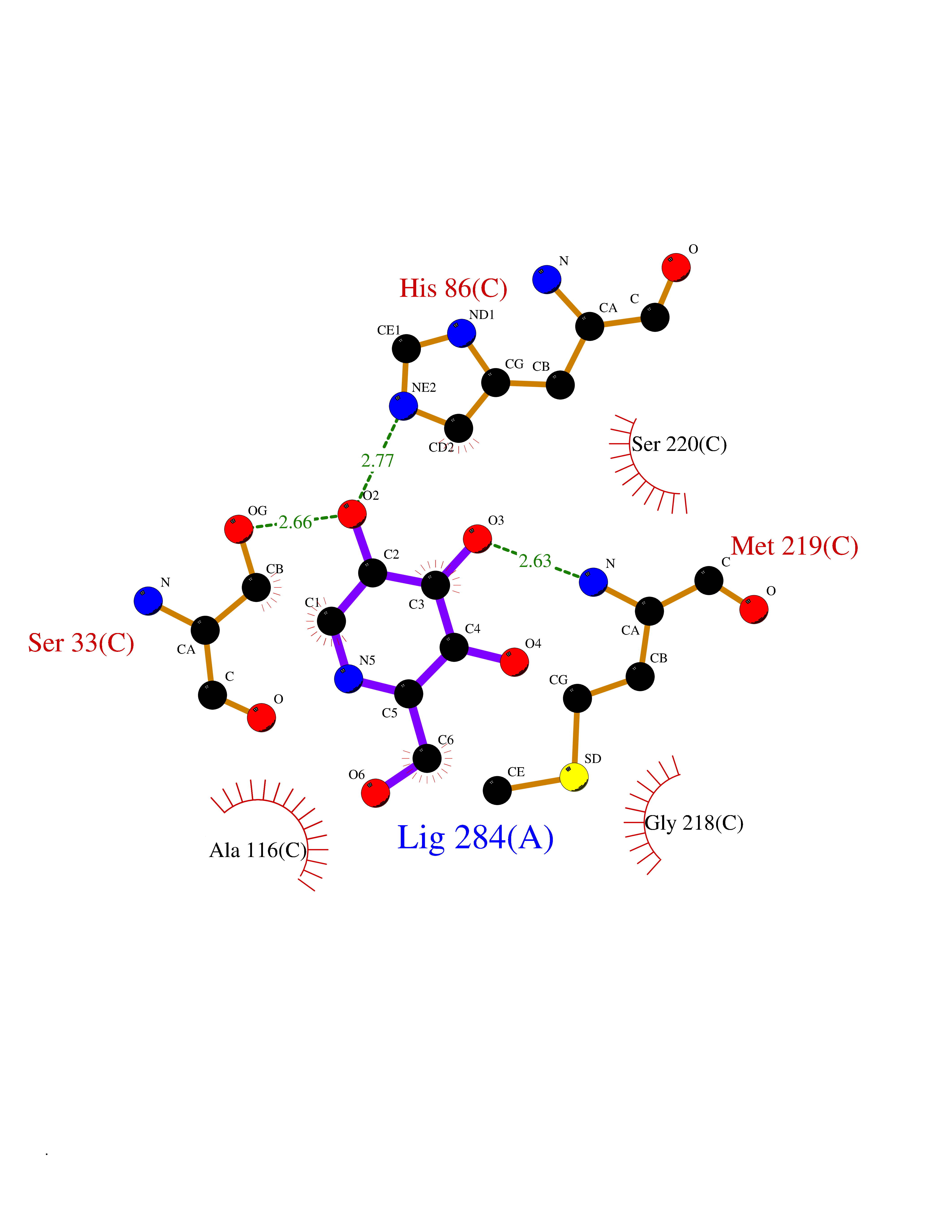 Ligplot Map 3D Binding mode Sequence GYTYEDYKNTAEYLLSHTKHRPQVAIICGSGLGGLTDKLTQAQIFDYSEIPNFPRSTVPGHAGRLVFGFLNGRACVMMQGRFHMYEGYPLYKVTFPVRVFHLLGVDTLVVTNAAGGLNPKFEVGDIMLIRDHINLPGFSGQNPLRGPNDERFGDRFPAMSDAYDRTMRQRALSTYKQMGEQRELQEGTYVMVAGPSFETVAECRVLQKLGADAVGMSTVPEVIVARHCGLRVFGFSLITNKVIMDYESLEKANXEEVLAAGKQAAQKLEQFVSILMASIDRFPAMSDA Hydrogen bonds contact Hydrophobic contact | ||||
| 10 | Lysine-specific demethylase 6B (KDM6B) | 6F6D | 6.22 | |
Target general information Gen name KDM6B Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Lysine demethylase 6B; KIAA0346; Jumonji domain-containing protein 3; JmjC domain-containing protein 3; JMJD3 Protein family UTX family Biochemical class NA Function Histone demethylase that specifically demethylates 'Lys-27' of histone H3, thereby playing a central role in histone code. Demethylates trimethylated and dimethylated H3 'Lys-27'. Plays a central role in regulation of posterior development, by regulating HOX gene expression. Involved in inflammatory response by participating in macrophage differentiation in case of inflammation by regulating gene expression and macrophage differentiation. Plays a demethylase-independent role in chromatin remodeling to regulate T-box family member-dependent gene expression by acting as a link between T-box factors and the SMARCA4-containing SWI/SNF remodeling complex (By similarity). Related diseases Stolerman neurodevelopmental syndrome (NEDSST) [MIM:618505]: An autosomal dominant disorder characterized by global developmental delay, variable intellectual disability, poor language acquisition, and dysmorphic facial features including a prominent nasal bridge and coarse features. Some patients manifest autism spectrum disorder. Musculoskeletal features may be present and include widened and thickened hands and fingers, joint hypermobility, clinodactyly of the fifth fingers, and toe syndactyly. {ECO:0000269|PubMed:31124279}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P03372 EC number EC 1.14.11.- Uniprot keywords 3D-structure; Alternative splicing; Chromatin regulator; Dioxygenase; Disease variant; Inflammatory response; Intellectual disability; Iron; Isopeptide bond; Metal-binding; Nucleus; Oxidoreductase; Phosphoprotein; Proteomics identification; Reference proteome; Ubl conjugation; Zinc Protein physicochemical properties Chain ID A,B Molecular weight (Da) 53008.8 Length 467 Aromaticity 0.1 Instability index 44.23 Isoelectric point 8.37 Charge (pH=7) 4.9 2D Binding mode Binding energy (Kcal/mol) -6.82 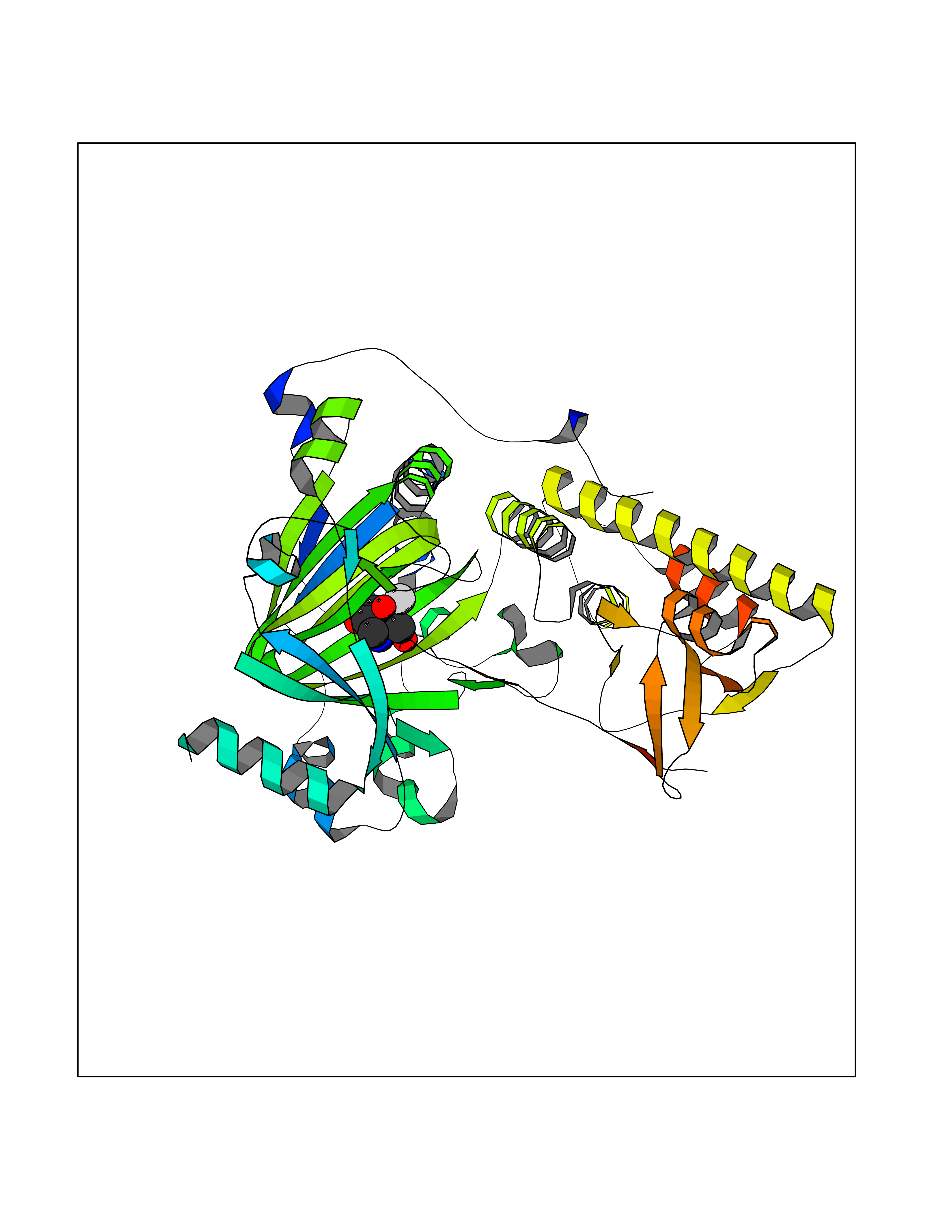 Molscript Map 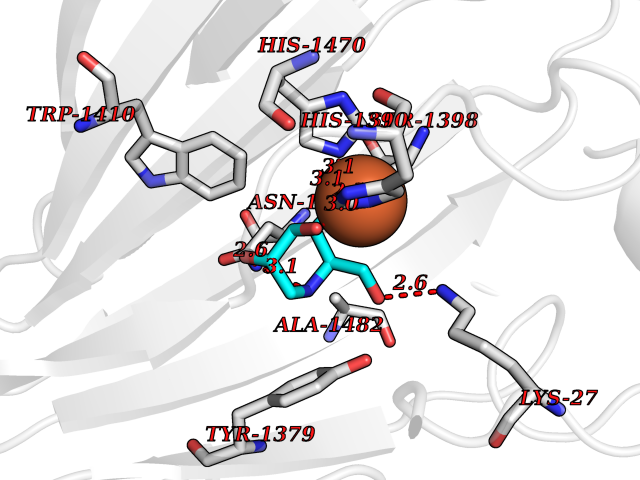 Pymol Map 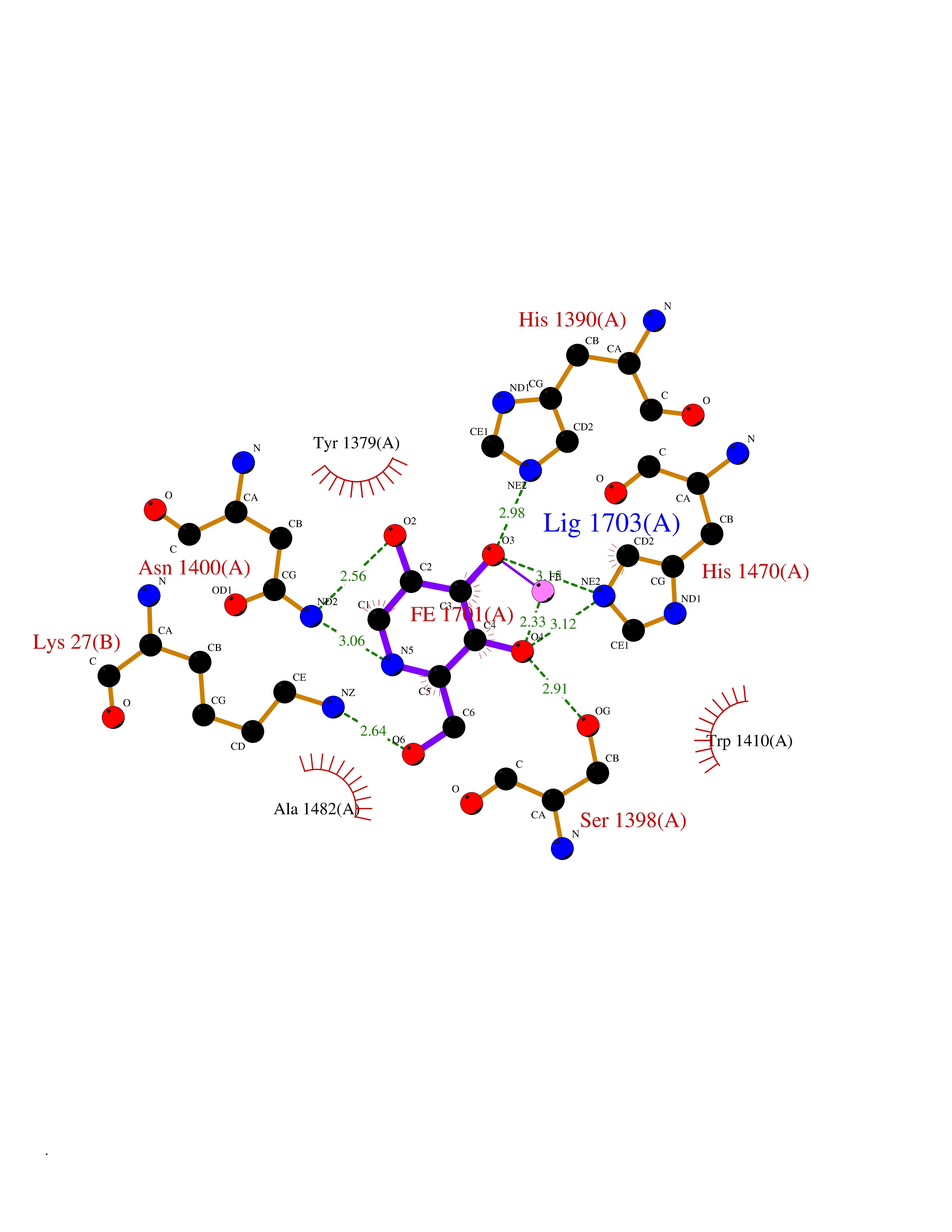 Ligplot Map 3D Binding mode Sequence ESYLSPAQSVKPKINTEEKLPREKLNPPTPSIYLESKRDAFSPVLLQFCTDPRNPITVIRGLAGSLRLNLGLFSTKTLVEASGEHTVEVRTQVQQPSDENWDLTGTRQIWPCESSRSHTTIAKYAQYQASSFQESHIIKFGTNIDLSDAKRWKPQLQELLKLPAFMRVTSTGNMLSHVGHTILGMNTVQLYMKVPGSRTPGHQENNNFCSVNINIGPGDCEWFAVHEHYWETISAFCDRHGVDYLTGSWWPILDDLYASNIPVYRFVQRPGDLVWINAGTVHWVQATGWCNNIAWNVGPLTAYQYQLALERYEWNEVKNVKSIVPMIHVSWNVARTVKISDPDLFKMIKFCLLQSMKHCQVQRESLVRAGKKIAYQGRVKDEPAYYCNECDVEVFNILFVTSENGSRNTYLVHCEGCARRRSAGLQGVVVLEQYRTEELAQAYDAFTLAPRIQLMTKAARKSAPATG Hydrogen bonds contact Hydrophobic contact | ||||
| 11 | Lysine-specific demethylase 4E (KDM4E) | 2W2I | 6.21 | |
Target general information Gen name KDM4E Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Lysine-specific demethylase 4D-like; KDM4DL; KDM4D-like protein Protein family JHDM3 histone demethylase family Biochemical class Paired donor oxygen oxidoreductase Function Histone demethylase that specifically demethylates 'Lys-9' of histone H3, thereby playing a central role in histone code. Related diseases Defects in KAT2B has been found in a patient with isolated coloboma, a defect of the eye characterized by the absence of ocular structures due to abnormal morphogenesis of the optic cup and stalk, and the fusion of the fetal fissure (optic fissure). Isolated colobomas may be associated with an abnormally small eye (microphthalmia) or small cornea. {ECO:0000269|PubMed:28493397}. Drugs (DrugBank ID) NA Interacts with NA EC number EC 1.14.11.- Uniprot keywords 3D-structure; Chromatin regulator; Dioxygenase; Iron; Metal-binding; Nucleus; Oxidoreductase; Reference proteome; Transcription; Transcription regulation; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 35131.5 Length 305 Aromaticity 0.14 Instability index 39.34 Isoelectric point 6 Charge (pH=7) -6.1 2D Binding mode Binding energy (Kcal/mol) -6.45  Molscript Map 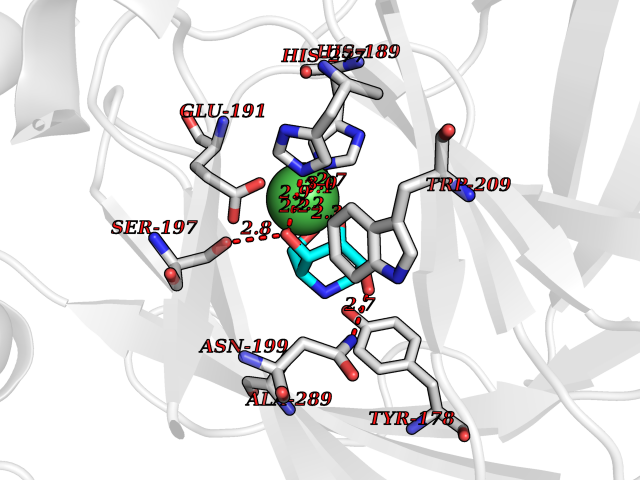 Pymol Map 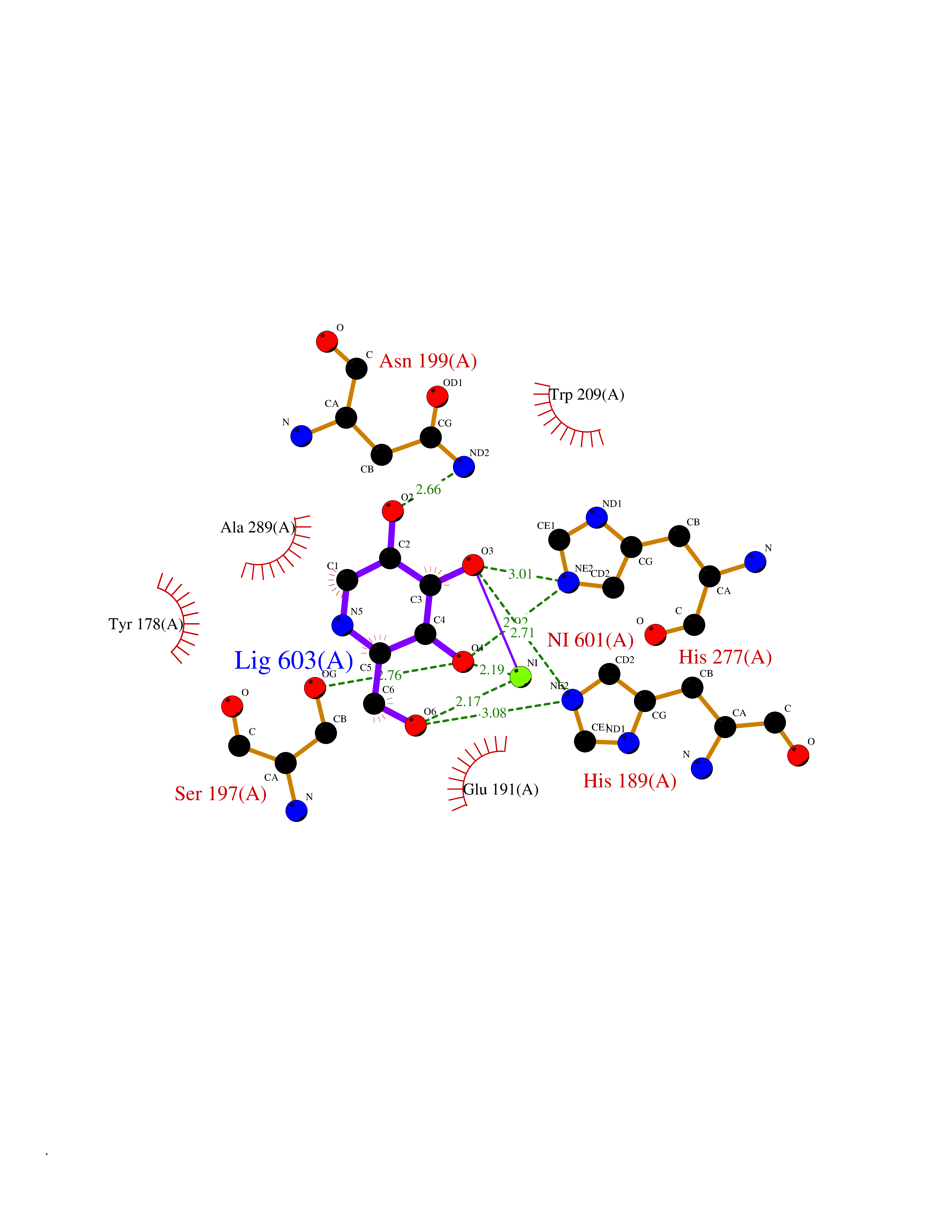 Ligplot Map 3D Binding mode Sequence HTIMTFYPTMEEFADFNTYVAYMESQGAHQAGLAKVIPPKEWKARQMYDDIEDILIATPLQQVTSGQGGVFTQYHKKKKAMRVGQYRRLANSKKYQTPPHQNFADLEQRYWKSHPGNPPIYGADISGSLFEESTKQWNLGHLGTILDLLEQECGVVIEGVNTPYLYFGMWKTTFAWHTEDMDLYSINYLHFGEPKTWYVVPPEHGQHLERLARELFPDISAFLRHKVALISPTVLKENGIPFNCMTQEAGEFMVTFPYGYHAGFNHGFNCAEAINFATPRWIDYGKMAVTFSMDPFVRIVQPESY Hydrogen bonds contact Hydrophobic contact | ||||
| 12 | Metabotropic glutamate receptor 3 (mGluR3) | 4XAR | 6.21 | |
Target general information Gen name GRM3 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms mGLUR3; Group III metabotropic glutamate receptor; GPRC1C Protein family G-protein coupled receptor 3 family Biochemical class GPCR glutamate Function Ligand binding causes a conformation change that triggers signaling via guanine nucleotide-binding proteins (G proteins) and modulates the activity of down-stream effectors. Signaling inhibits adenylate cyclase activity. G-protein coupled receptor for glutamate. Related diseases Paramyotonia congenita (PMC) [MIM:168300]: An autosomal dominant channelopathy characterized by myotonia, increased by exposure to cold, intermittent flaccid paresis, not necessarily dependent on cold or myotonia, lability of serum potassium, non-progressive nature and lack of atrophy or hypertrophy of muscles. In some patients, myotonia is not increased by cold exposure (paramyotonia without cold paralysis). Patients may have a combination phenotype of PMC and HYPP. {ECO:0000269|PubMed:10369308, ECO:0000269|PubMed:10727489, ECO:0000269|PubMed:1310898, ECO:0000269|PubMed:1316765, ECO:0000269|PubMed:1338909, ECO:0000269|PubMed:15318338, ECO:0000269|PubMed:15790667, ECO:0000269|PubMed:16786525, ECO:0000269|PubMed:18166706, ECO:0000269|PubMed:18690054, ECO:0000269|PubMed:19077043, ECO:0000269|PubMed:20076800, ECO:0000269|PubMed:8242056, ECO:0000269|PubMed:8308722, ECO:0000269|PubMed:8388676, ECO:0000269|PubMed:8580427}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Periodic paralysis hypokalemic 2 (HOKPP2) [MIM:613345]: An autosomal dominant disorder manifested by episodic flaccid generalized muscle weakness associated with falls of serum potassium levels. {ECO:0000269|PubMed:10599760, ECO:0000269|PubMed:10851391, ECO:0000269|PubMed:10944223, ECO:0000269|PubMed:11558801, ECO:0000269|PubMed:11591859, ECO:0000269|PubMed:16890191, ECO:0000269|PubMed:17898326, ECO:0000269|PubMed:18162704, ECO:0000269|PubMed:19118277, ECO:0000269|PubMed:20522878, ECO:0000269|PubMed:21043388, ECO:0000269|PubMed:24549961}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Periodic paralysis hyperkalemic (HYPP) [MIM:170500]: An autosomal dominant channelopathy characterized by episodic flaccid generalized muscle weakness associated with high levels of serum potassium. Concurrence of myotonia is found in HYPP patients. {ECO:0000269|PubMed:1659668, ECO:0000269|PubMed:1659948, ECO:0000269|PubMed:20076800}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Periodic paralysis normokalemic (NKPP) [MIM:170500]: A disorder closely related to hyperkalemic periodic paralysis, but marked by a lack of alterations in potassium levels during attacks of muscle weakness. {ECO:0000269|PubMed:15596759, ECO:0000269|PubMed:18046642, ECO:0000269|PubMed:20522878}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Myotonia SCN4A-related (MYOSCN4A) [MIM:608390]: A phenotypically highly variable myotonia aggravated by potassium loading, and sometimes by cold. Myotonia is characterized by sustained muscle tensing that prevents muscles from relaxing normally. It causes muscle stiffness that can interfere with movement. In some people the stiffness is very mild, while in other cases it may be severe enough to interfere with walking, running, and other activities of daily life. Myotonia SCN4A-related includes myotonia permanens and myotonia fluctuans. In myotonia permanens, the myotonia is generalized and there is a hypertrophy of the muscle, particularly in the neck and the shoulder. Attacks of severe muscle stiffness of the thoracic muscles may be life threatening due to impaired ventilation. In myotonia fluctuans, the muscle stiffness may fluctuate from day to day, provoked by exercise. {ECO:0000269|PubMed:10218481, ECO:0000269|PubMed:16786525, ECO:0000269|PubMed:16832098, ECO:0000269|PubMed:17212350, ECO:0000269|PubMed:17998485, ECO:0000269|PubMed:18203179, ECO:0000269|PubMed:18337100, ECO:0000269|PubMed:19015483, ECO:0000269|PubMed:19347921, ECO:0000269|PubMed:20076800, ECO:0000269|PubMed:27653901, ECO:0000269|PubMed:8058156, ECO:0000269|PubMed:9392583}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Myasthenic syndrome, congenital, 16 (CMS16) [MIM:614198]: A form of congenital myasthenic syndrome, a group of disorders characterized by failure of neuromuscular transmission, including pre-synaptic, synaptic, and post-synaptic disorders that are not of autoimmune origin. Clinical features are easy fatigability and muscle weakness. CMS16 is characterized by fatigable generalized weakness and recurrent attacks of respiratory and bulbar paralysis since birth. The fatigable weakness involves lid-elevator, external ocular, facial, limb and truncal muscles and an decremental response of the compound muscle action potential on repetitive stimulation. {ECO:0000269|PubMed:12766226, ECO:0000269|PubMed:25707578, ECO:0000269|PubMed:26659129}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Congenital myopathy 22A, classic (CMYO22A) [MIM:620351]: A form of congenital myopathy, a clinically and genetically heterogeneous group of muscle disorders characterized by hypotonia and muscle weakness apparent at birth, and specific pathological features on muscle biopsy. CMYO22A is an autosomal recessive form characterized by fetal hypokinesia, polyhydramnios, and severe neonatal hypotonia associated with respiratory insufficiency. Affected individuals who survive the neonatal period have delayed motor development, difficulty walking, proximal muscle weakness of the upper and lower limbs, facial and neck muscle weakness, easy fatigability, and mild limb contractures or foot deformities. {ECO:0000269|PubMed:26700687, ECO:0000269|PubMed:28262468, ECO:0000269|PubMed:36090556}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Congenital myopathy 22B, severe fetal (CMYO22B) [MIM:620369]: A severe congenital myopathy, a clinically and genetically heterogeneous group of muscle disorders characterized by hypotonia and muscle weakness apparent at birth, and specific pathological features on muscle biopsy. CMYO22B is an autosomal recessive form characterized by onset in utero. Affected individuals show fetal akinesia, and develop fetal hydrops with pulmonary hypoplasia, severe joint contractures, and generalized muscle hypoplasia. Death occurs in utero or soon after birth. {ECO:0000269|PubMed:26700687}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB05096 Interacts with NA EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disulfide bond; G-protein coupled receptor; Glycoprotein; Membrane; Proteomics identification; Receptor; Reference proteome; Signal; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 50355.5 Length 445 Aromaticity 0.11 Instability index 38.26 Isoelectric point 6.52 Charge (pH=7) -1.53 2D Binding mode Binding energy (Kcal/mol) -6.98 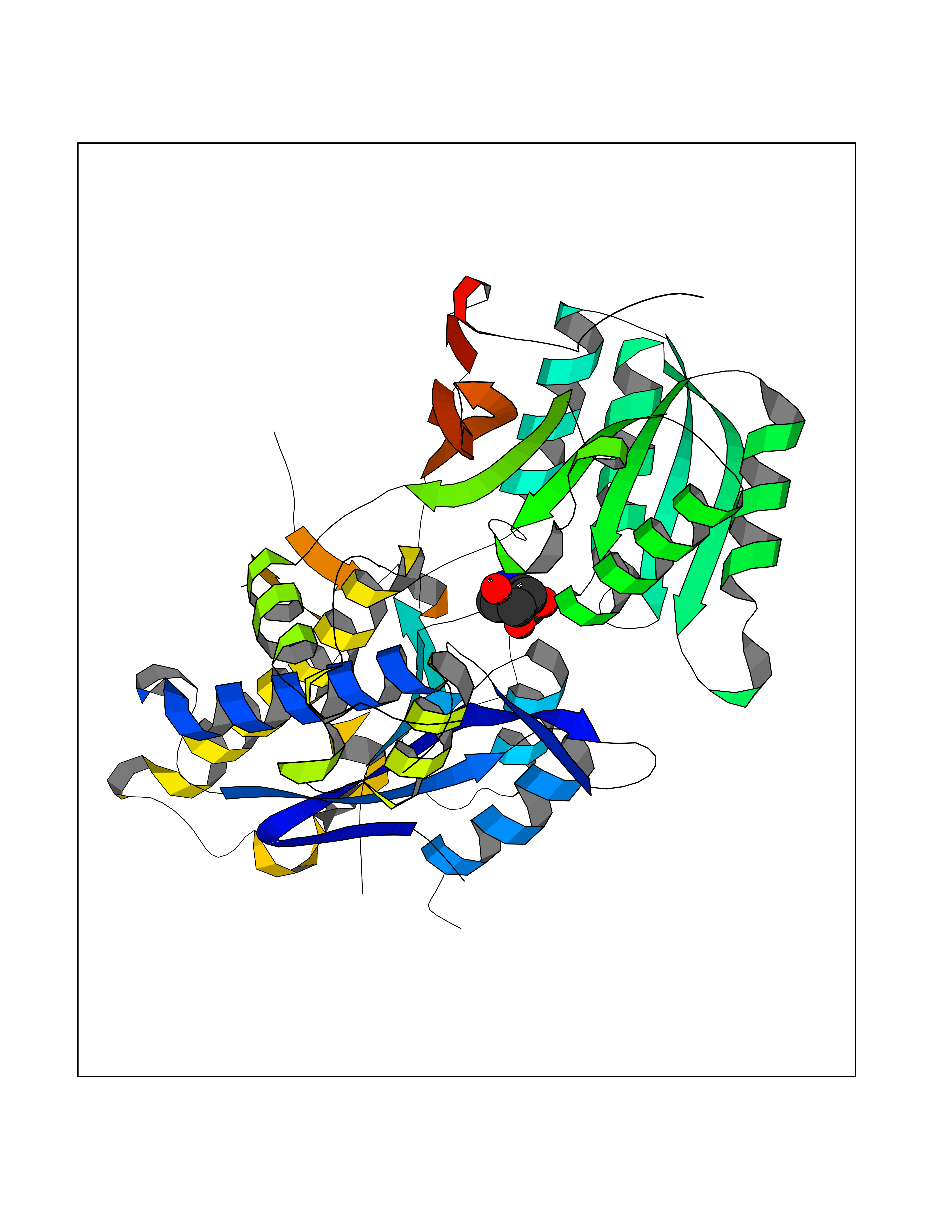 Molscript Map 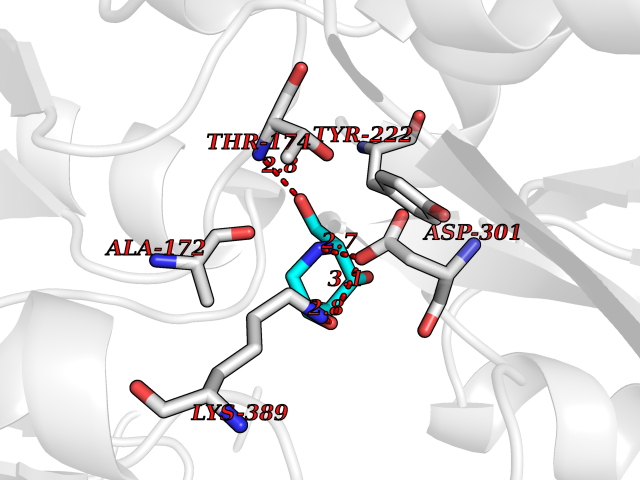 Pymol Map 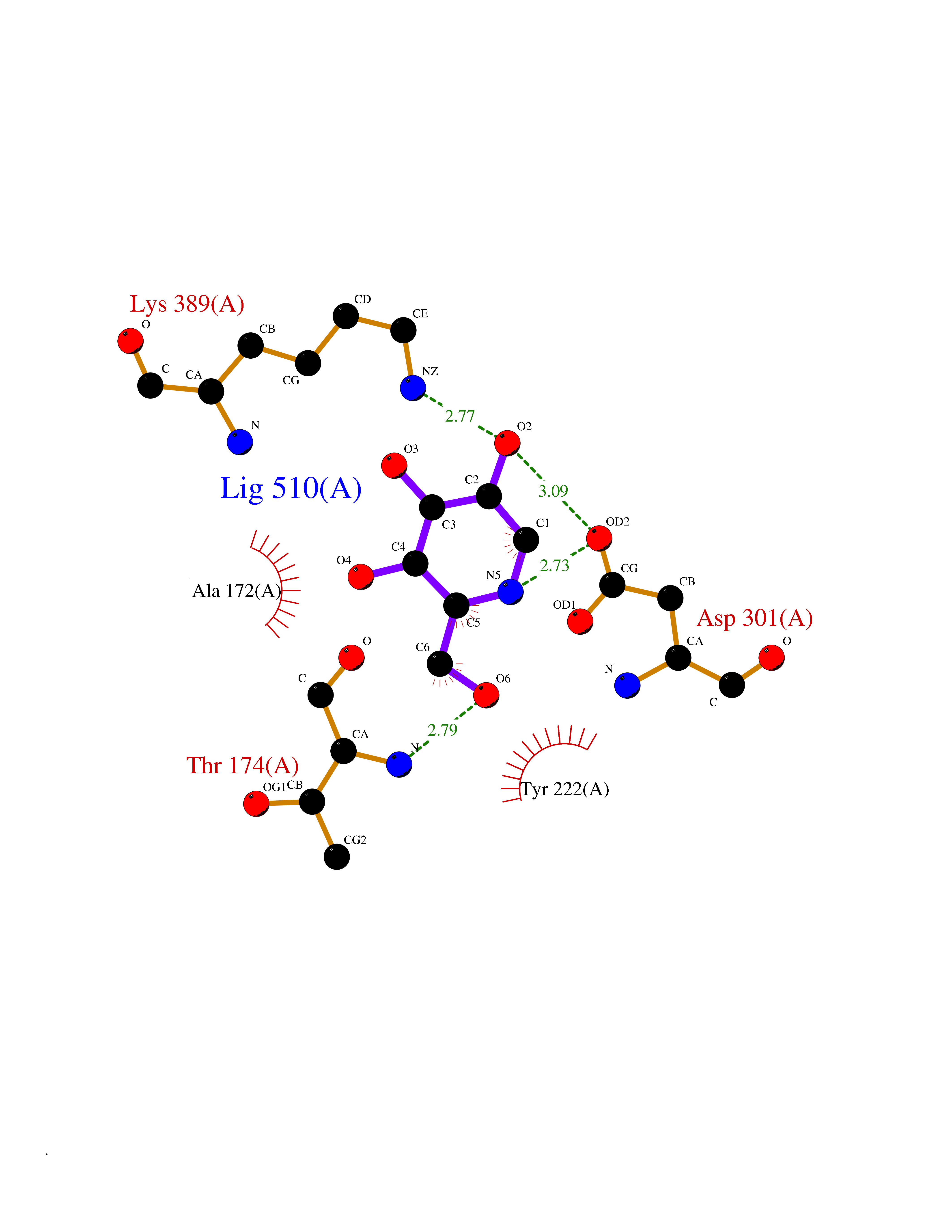 Ligplot Map 3D Binding mode Sequence RREIKIEGDLVLGGLFPINEKGTGTEECGRINEDRGIQRLEAMLFAIDEINKDDYLLPGVKLGVHILDTCSRDTYALEQSLEFVRASLLLIAGVIGGSYSSVSIQVANLLRLFQIPQISYASTSAKLSDKSRYDYFARTVPPDFYQAKAMAEILRFFNWTYVSTVASEGDYGETGIEAFEQEARLRNISIATAEKVGRSNIRKSYDSVIRELLQKPNARVVVLFMRSDDSRELIAAASRANASFTWVASDGWGAQESIIKGSEHVAYGAITLELASQPVRQFDRYFQSLNPYNNHRNPWFRDFWEQKFQCSLRVCDKHLAIDSSNYEQESKIMFVVNAVYAMAHALHKMQRTLCPNTTKLCDAMKILDGKKLYKDYLLKINFTAPDADSIVKFDTFGDGMGRYNVFNFQNVGGKYSYLKVGHWAETLSLDVNSIHWSRNSVPTSE Hydrogen bonds contact Hydrophobic contact | ||||
| 13 | Interleukin 21 receptor (IL21R) | 6PLH | 6.18 | |
Target general information Gen name IL21R Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms UNQ3121/PRO10273; Novel interleukin receptor; NILR; Interleukin-21 receptor; IL21 receptor; IL-21R; IL-21 receptor; CD360 Protein family Type I cytokine receptor family, Type 4 subfamily Biochemical class Cytokine receptor Function This is a receptor for interleukin-21. Related diseases Immunodeficiency 56 (IMD56) [MIM:615207]: An autosomal recessive primary immunodeficiency characterized by B- and T-cell defects and variable dysfunction of NK cells. Patients tend to have normal numbers of lymphocytes, but show defective class-switched B-cells, low IgG, defective antibody response, and defective T-cell responses to certain antigens. {ECO:0000269|PubMed:23440042}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Chromosomal aberrations involving IL21R is a cause of B-cell non-Hodgkin lymphomas (B-cell NHL). Translocation t(3;16)(q27;p11), with BCL6. Drugs (DrugBank ID) NA Interacts with P29972 EC number NA Uniprot keywords 3D-structure; Chromosomal rearrangement; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; Membrane; Proteomics identification; Receptor; Reference proteome; Repeat; Signal; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A,C,B Molecular weight (Da) 48376.5 Length 446 Aromaticity 0.1 Instability index 43.94 Isoelectric point 8.24 Charge (pH=7) 3.56 2D Binding mode Binding energy (Kcal/mol) -5.94  Molscript Map 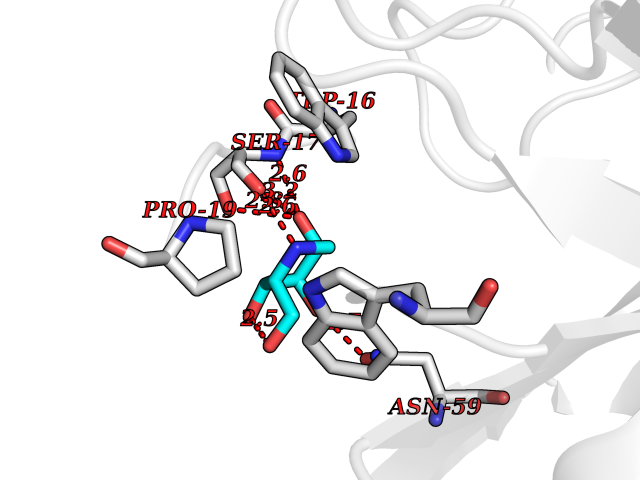 Pymol Map 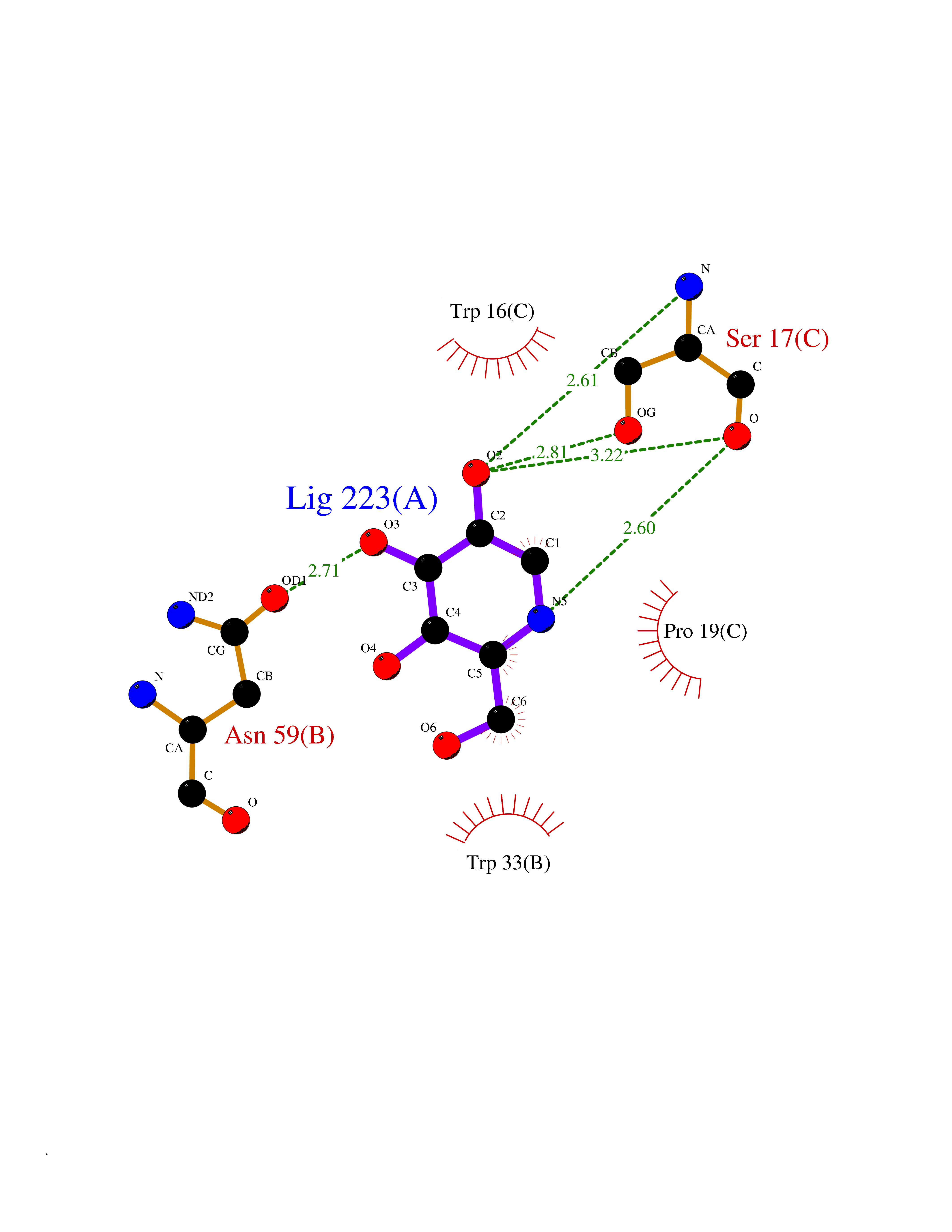 Ligplot Map 3D Binding mode Sequence DVVMTHTPLSLPVSLGDQASISCRSSQSLVHSNGNTYLHWYLQKPGQSPKLLIYKVSNRFSGVPDRFSGSGSGADFTLKISRVEAEDLGVYFCSQSTHVPRTFGGGTKLEIKRADAAPTVSIFPPSSEQLTSGGASVVCFLNNFYPKDINVKWKIDGSERQNGVLNSWTDQDSKDSTYSMSSTLTLTKDEYERHNSYTCEATHKTSTSPIVKSFNRNECXVHLQQPGADLVKPGASVKMSCKASGYTFTSYWITWVKLRPGQGLEWIGDIYPGSGSTNFIEKFKSKATLTVDTSSSTAYMQLRSLTSEDSAVYYCARRGHGNYEDYWGQGTTLIVSSAKTTAPSVYPLAPVCGTGSSVTLGCLVKGYFPEPVTLTWNSGSLSSGVHTFPAVLQSDLYTLSSSVTVTSSTWPSQSITCNVAHPASSTKVDKKIEPRGPTTWSEWSDP Hydrogen bonds contact Hydrophobic contact | ||||
| 14 | Carbonic anhydrase XIV (CA-XIV) | 5CJF | 6.17 | |
Target general information Gen name CA14 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms UNQ690/PRO1335; Carbonic anhydrase 14; Carbonate dehydratase XIV Protein family Alpha-carbonic anhydrase family Biochemical class Alpha-carbonic anhydrase Function Reversible hydration of carbon dioxide. Related diseases Isobutyryl-CoA dehydrogenase deficiency (IBDD) [MIM:611283]: An autosomal recessive metabolic disorder characterized by plasma carnitine deficiency and elevated C4-acylcarnitine. Patients manifest variable clinical features including failure to thrive, seizures, anemia, muscular hypotonia and developmental delay. Some patients may be asymptomatic. {ECO:0000269|PubMed:12359132, ECO:0000269|PubMed:15505379, ECO:0000269|PubMed:16857760}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00819; DB00562; DB00606; DB08846; DB00909 Interacts with NA EC number EC 4.2.1.1 Uniprot keywords 3D-structure; Disulfide bond; Glycoprotein; Lyase; Membrane; Metal-binding; Phosphoprotein; Proteomics identification; Reference proteome; Signal; Transmembrane; Transmembrane helix; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 29397.4 Length 260 Aromaticity 0.1 Instability index 59.95 Isoelectric point 5.54 Charge (pH=7) -11 2D Binding mode Binding energy (Kcal/mol) -6.55  Molscript Map 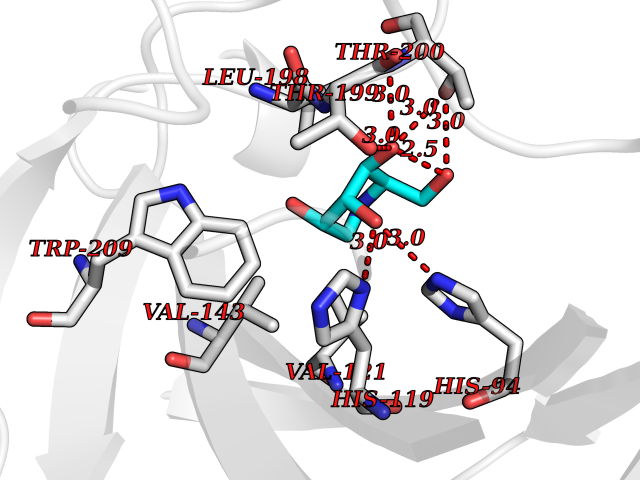 Pymol Map 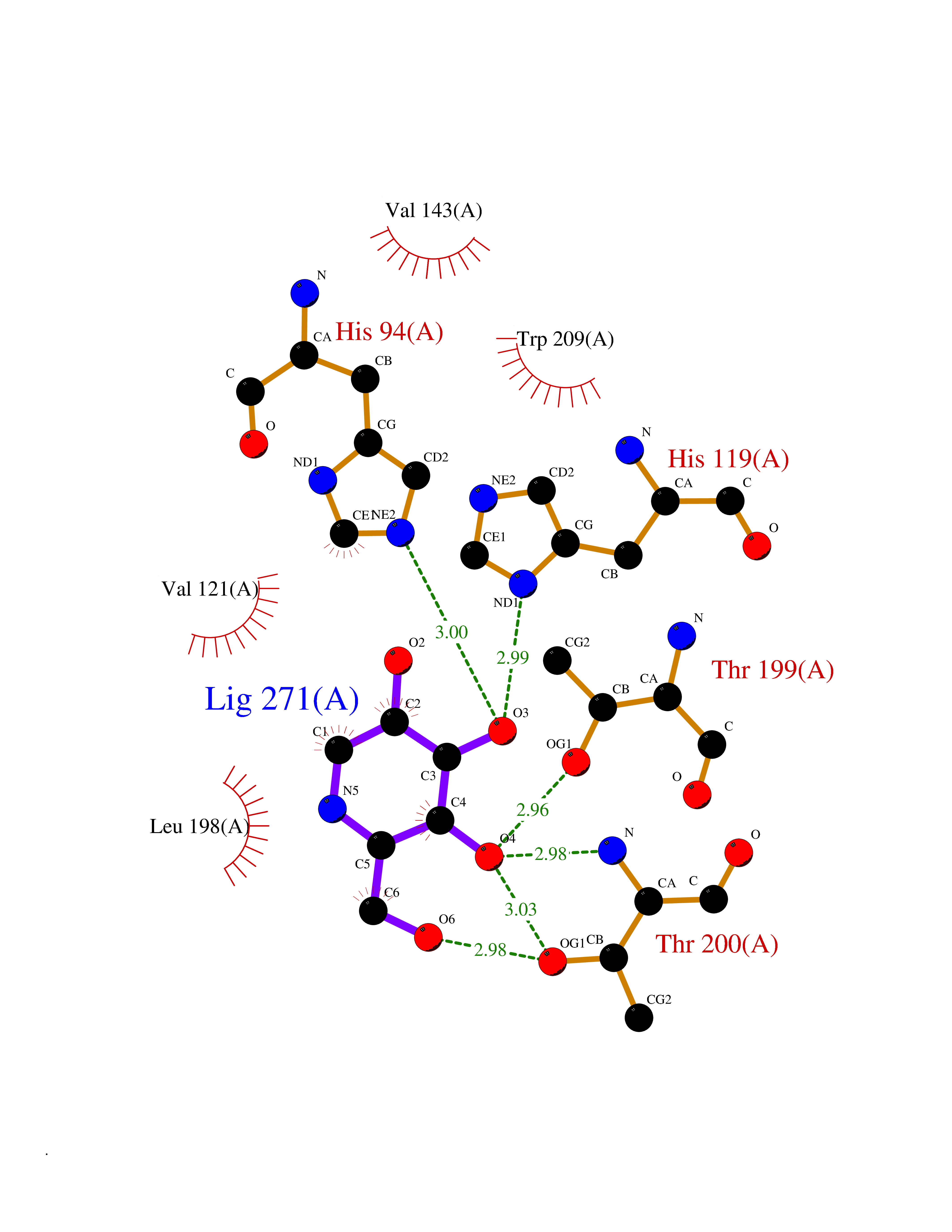 Ligplot Map 3D Binding mode Sequence HWTYEGPHGQDHWPASYPECGNNAQSPIDIQTDSVTFDPDLPALQPGYDQTEPLDLHNNGHTVQLSLPSTLYLGGLPRKYVAAQLHLHWGQKGPGGSEHQINSEATFAELHIVHYDSDSYDSLSEAAERPQGLAVLGILIEVETKNIAYEHILSHLHEVRHKDQKTSVPPFNLRELLPKQGQYFRYNGSLTTPPCYQSVLWTVFYRRSQISMEQLEKLGTLFSTEEEPSKLLVQNYRALQPLNQRMVFASFIQAGSSYTT Hydrogen bonds contact Hydrophobic contact | ||||
| 15 | Clostridium histolyticum Collagenase (CH colG) | 7Z5U | 6.16 | |
Target general information Gen name CH colG Organism Hathewaya histolytica (Clostridium histolyticum) Uniprot ID TTD ID Synonyms Microbial collagenase; Gelatinase ColG; Collagenase ColG; Class I collagenase Protein family Peptidase M9B family, Collagenase subfamily Biochemical class NA Function Clostridial collagenases are among the most efficient degraders of eukaryotic collagen known; saprophytes use collagen as a carbon source while pathogens additionally digest collagen to aid in host colonization. Has both tripeptidylcarboxypeptidase on Gly-X-Y and endopeptidase activities; the endopeptidase cuts within the triple helix region of collagen while tripeptidylcarboxypeptidase successively digests the exposed ends, thus clostridial collagenases can digest large sections of collagen. Active on soluble type I collagen, insoluble collagen, azocoll, soluble PZ-peptide (all collagenase substrates) and gelatin. The full-length protein has collagenase activity, while the in vivo derived C-terminally truncated shorter versions only act on gelatin. In vitro digestion of soluble calf skin collagen fibrils requires both ColG and ColH; ColG forms missing the second collagen-binding domain are also synergistic with ColH, although their overall efficiency is decreased. The activator domain (residues 119-388) and catalytic subdomain (389-670) open and close around substrate using a Gly-rich hinge (387-397), allowing digestion when the protein is closed. Binding of collagen requires Ca(2+) and is inhibited by EGTA; the collagen-binding domain (CBD, S3a plus S3b) specifically recognizes the triple-helical conformation made by 3 collagen protein chains in the triple-helical region. Isolated CBD (S3a plus S3b) binds collagen fibrils and sheets of many tissues. Related diseases Intellectual developmental disorder, X-linked, syndromic, Claes-Jensen type (MRXSCJ) [MIM:300534]: A disorder characterized by significantly below average general intellectual functioning associated with impairments in adaptive behavior and manifested during the developmental period. MRXSCJ patients manifest intellectual disability associated with variable features such as slowly progressive spastic paraplegia, seizures, facial dysmorphism. {ECO:0000269|PubMed:15586325, ECO:0000269|PubMed:16538222, ECO:0000269|PubMed:16541399, ECO:0000269|PubMed:17320160, ECO:0000269|PubMed:17468742, ECO:0000269|PubMed:23356856, ECO:0000269|PubMed:25666439}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number EC 3.4.24.3 Uniprot keywords 3D-structure; Calcium; Direct protein sequencing; Hydrolase; Metal-binding; Metalloprotease; Pharmaceutical; Protease; Repeat; Secreted; Signal; Virulence; Zinc; Zymogen Protein physicochemical properties Chain ID A Molecular weight (Da) 44751.2 Length 386 Aromaticity 0.15 Instability index 27.33 Isoelectric point 5.77 Charge (pH=7) -7.33 2D Binding mode Binding energy (Kcal/mol) -6.42 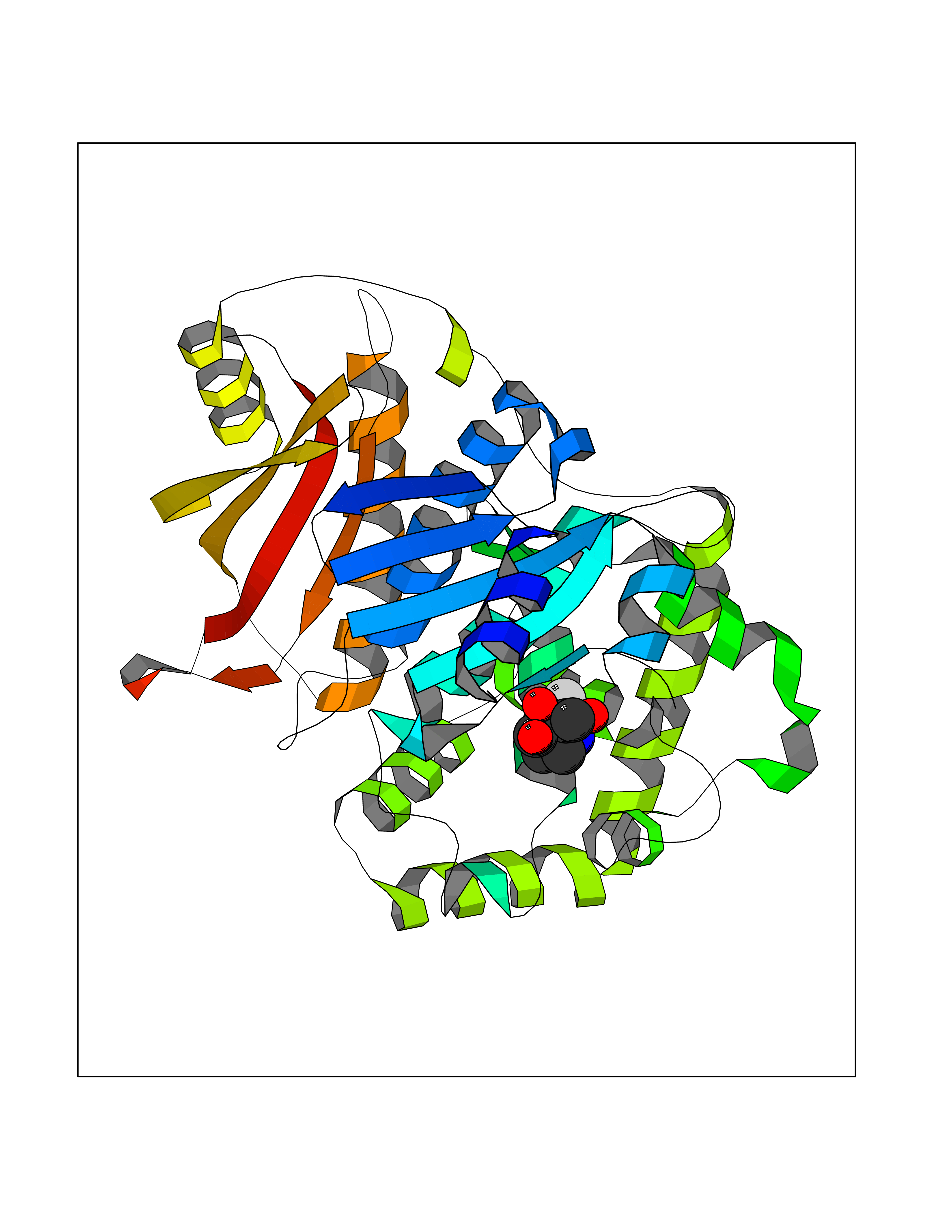 Molscript Map 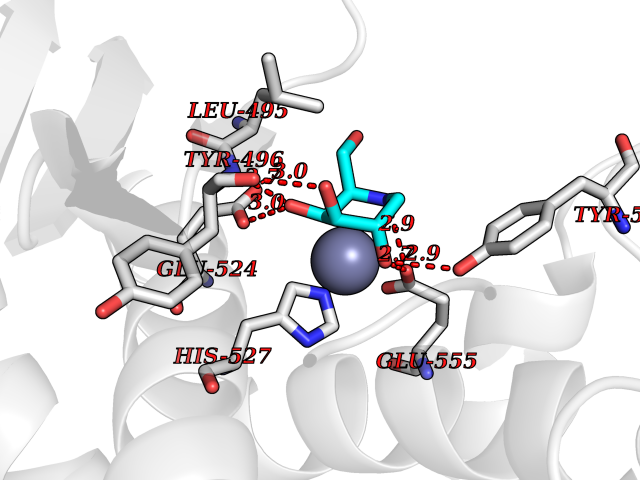 Pymol Map 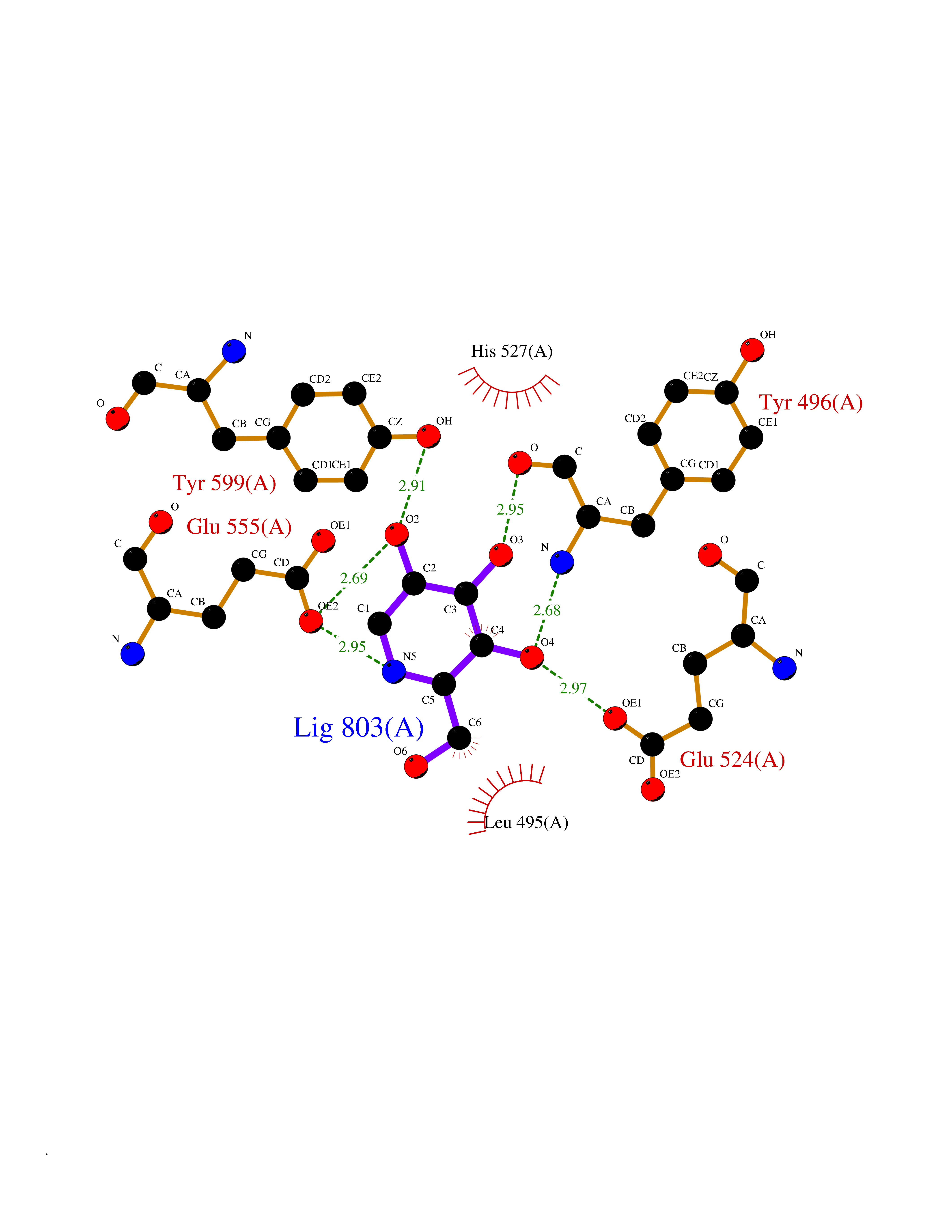 Ligplot Map 3D Binding mode Sequence DHDKFLDDAEKHYLPKTYTFDNGTFIIRAGDKVSEEKIKRLYWASREVKSQFHRVVGNDKALEVGNADDVLTMKIFNSPEEYKFNTTDNGGLYIEPRGTFYTYERTPQQSIFSLEELFRHEYTHYLQARYLVDGLWGQGPFYEKNRLTWFDEGTAEFFAGSTRTSGVLPRKLILGYLAKDKVDHRYSLKKTLNSGYDDSDWMFYNYGFAVAHYLYEKDMPTFIKMNKAILNTDVKSYDEIIKKLSDDANKNTEYQNHIQELVDKYQGAGIPLVSDDYLKDHGYKKASEVYSEISKAASLTNTSVTAEKSQYFNTFTLRGTYTGETSKGEFKDWDEMSKKLDGTLESLAKNSWSGYKTLTAYFTNYRVTSDNKVQYDVVFHGVLTDN Hydrogen bonds contact Hydrophobic contact | ||||
| 16 | Endothelin-converting enzyme 1 (ECE1) | 3DWB | 6.12 | |
Target general information Gen name ECE1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms ECE-1 Protein family Peptidase M13 family Biochemical class Peptidase Function Converts big endothelin-1 to endothelin-1. Related diseases Hirschsprung disease, cardiac defects, and autonomic dysfunction (HCAD) [MIM:613870]: A disorder characterized by skip-lesions Hirschsprung disease, craniofacial abnormalities and other dysmorphic features, cardiac defects including ductus arteriosus, small subaortic ventricular septal defect, small atrial septal defect, and autonomic dysfunction. {ECO:0000269|PubMed:9915973}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07171 Interacts with P49760; A8MQ03; Q8IUG1; P60370; P60410 EC number EC 3.4.24.71 Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disease variant; Disulfide bond; Glycoprotein; Hirschsprung disease; Hydrolase; Membrane; Metal-binding; Metalloprotease; Phosphoprotein; Protease; Proteomics identification; Reference proteome; Signal-anchor; Transmembrane; Transmembrane helix; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 75247.9 Length 660 Aromaticity 0.12 Instability index 46.29 Isoelectric point 5.33 Charge (pH=7) -18.3 2D Binding mode Binding energy (Kcal/mol) -6.09  Molscript Map  Pymol Map 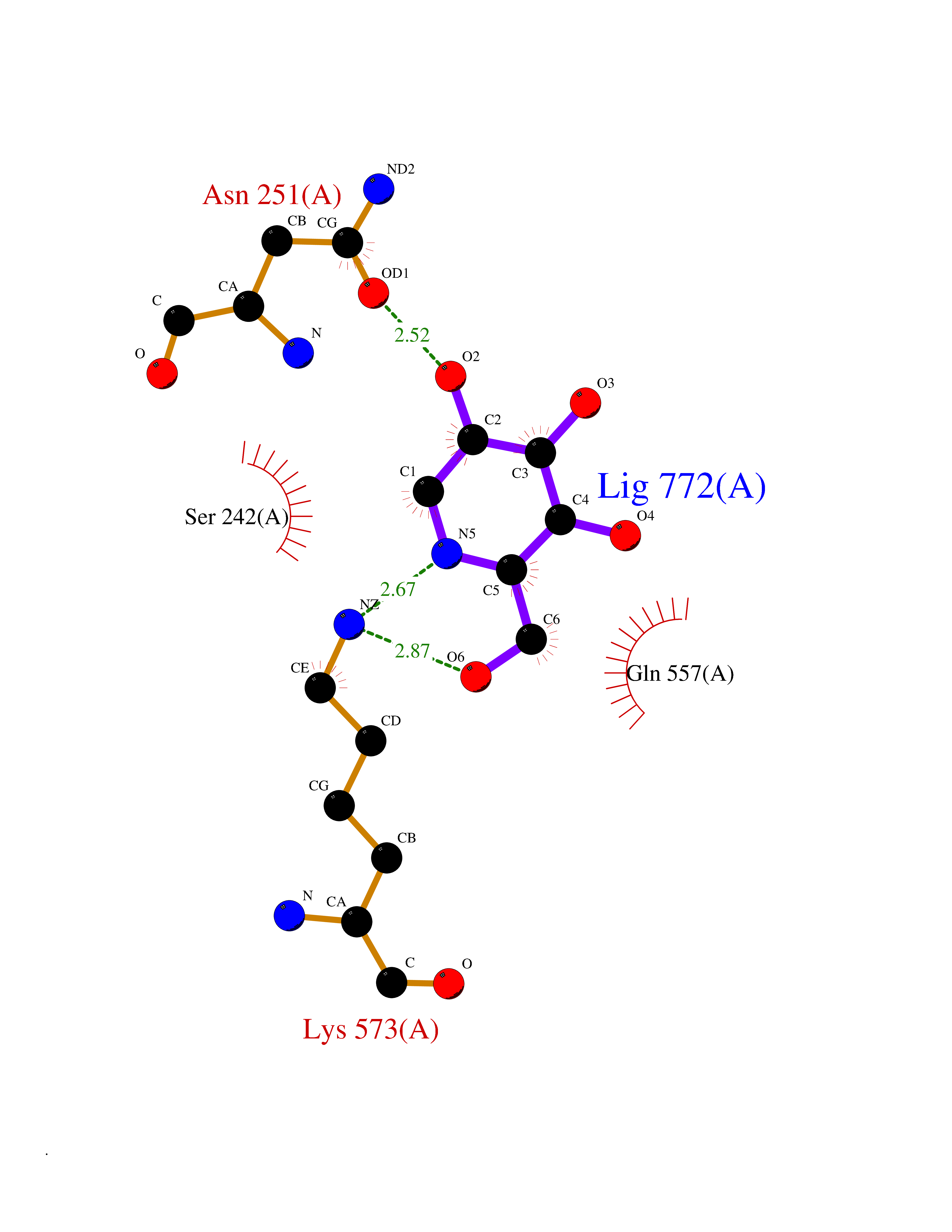 Ligplot Map 3D Binding mode Sequence SEACVSVTSSILSSMDPTVDPCHDFFSYACGGWIKANPVPDGHSRWGTFSNLWEHNQAIIKHLLENSTASVSEAERKAQVYYRACMNETRIEELRAKPLMELIERLGGWNITGPWAKDNFQDTLQVVTAHYRTSPFFSVYVSADSKNSNSNVIQVDQSGLGLPSRDYYLNKTENEKVLTGYLNYMVQLGKLLGGGDEEAIRPQMQQILDFETALANITIPQEKRRDEELIYHKVTAAELQTLAPAINWLPFLNTIFYPVEINESEPIVVYDKEYLEQISTLINTTDRCLLNNYMIWNLVRKTSSFLDQRFQDADEKFMEVMWKFCVSDTENNLGFALGPMFVKATFAEDSKSIATEIILEIKKAFEESLSTLKWMDEETRKSAKEKADAIYNMIGYPNFIMDPKELDKVFNDYTAVPDLYFENAMRFFNFSWRVTADQLRKAPNRDQWSMTPPMVNAYYSPTKNEIVFPAGILQAPFYTRSSPKALNFGGIGVVVGHELTHAFDDQGREYDKDGNLRPWWKNSSVEAFKRQTECMVEQYSNYSVNGEPVNGRHTLGENIADNGGLKAAYRAYQNWVKKNGAEHSLPTLGLTNNQLFFLGFAQVWCSVRTPESSHEGLITDPHSPSRFRVIGSLSNSKEFSEHFRCPPGSPMNPPHKCEVW Hydrogen bonds contact Hydrophobic contact | ||||
| 17 | HIF-prolyl hydroxylase 2 (HPH-2) | 6ZBO | 6.11 | |
Target general information Gen name EGLN1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms SM-20; Prolyl hydroxylase domain-containing protein 2; PHD2; Hypoxia-inducible factor prolyl hydroxylase 2; HPH-2; HIF-PH2; Egl nine homolog 1; C1orf12 Protein family NA Biochemical class Paired donor oxygen oxidoreductase Function Cellular oxygen sensor that catalyzes, under normoxic conditions, the post-translational formation of 4-hydroxyproline in hypoxia-inducible factor (HIF) alpha proteins. Hydroxylates a specific proline found in each of the oxygen-dependent degradation (ODD) domains (N-terminal, NODD, and C-terminal, CODD) of HIF1A. Also hydroxylates HIF2A. Has a preference for the CODD site for both HIF1A and HIF1B. Hydroxylated HIFs are then targeted for proteasomal degradation via the von Hippel-Lindau ubiquitination complex. Under hypoxic conditions, the hydroxylation reaction is attenuated allowing HIFs to escape degradation resulting in their translocation to the nucleus, heterodimerization with HIF1B, and increased expression of hypoxy-inducible genes. EGLN1 is the most important isozyme under normoxia and, through regulating the stability of HIF1, involved in various hypoxia-influenced processes such as angiogenesis in retinal and cardiac functionality. Target proteins are preferentially recognized via a LXXLAP motif. Related diseases Erythrocytosis, familial, 3 (ECYT3) [MIM:609820]: An autosomal dominant disorder characterized by elevated serum hemoglobin and hematocrit, and normal serum erythropoietin levels. {ECO:0000269|PubMed:16407130, ECO:0000269|PubMed:17579185}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00126; DB11682; DB14490; DB14491; DB14488; DB14501; DB14489; DB08687; DB01592; DB07112; DB04847; DB12255 Interacts with Q99814; Q14318; Q16665; Q13438; PRO_0000037551 [Q9WMX2] EC number EC 1.14.11.29 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Congenital erythrocytosis; Cytoplasm; Dioxygenase; Disease variant; Iron; Metal-binding; Nucleus; Oxidoreductase; Phosphoprotein; Proteomics identification; Reference proteome; S-nitrosylation; Vitamin C; Zinc; Zinc-finger Protein physicochemical properties Chain ID A,D Molecular weight (Da) 45717.8 Length 406 Aromaticity 0.11 Instability index 24.41 Isoelectric point 7.59 Charge (pH=7) 1.54 2D Binding mode Binding energy (Kcal/mol) -6.2 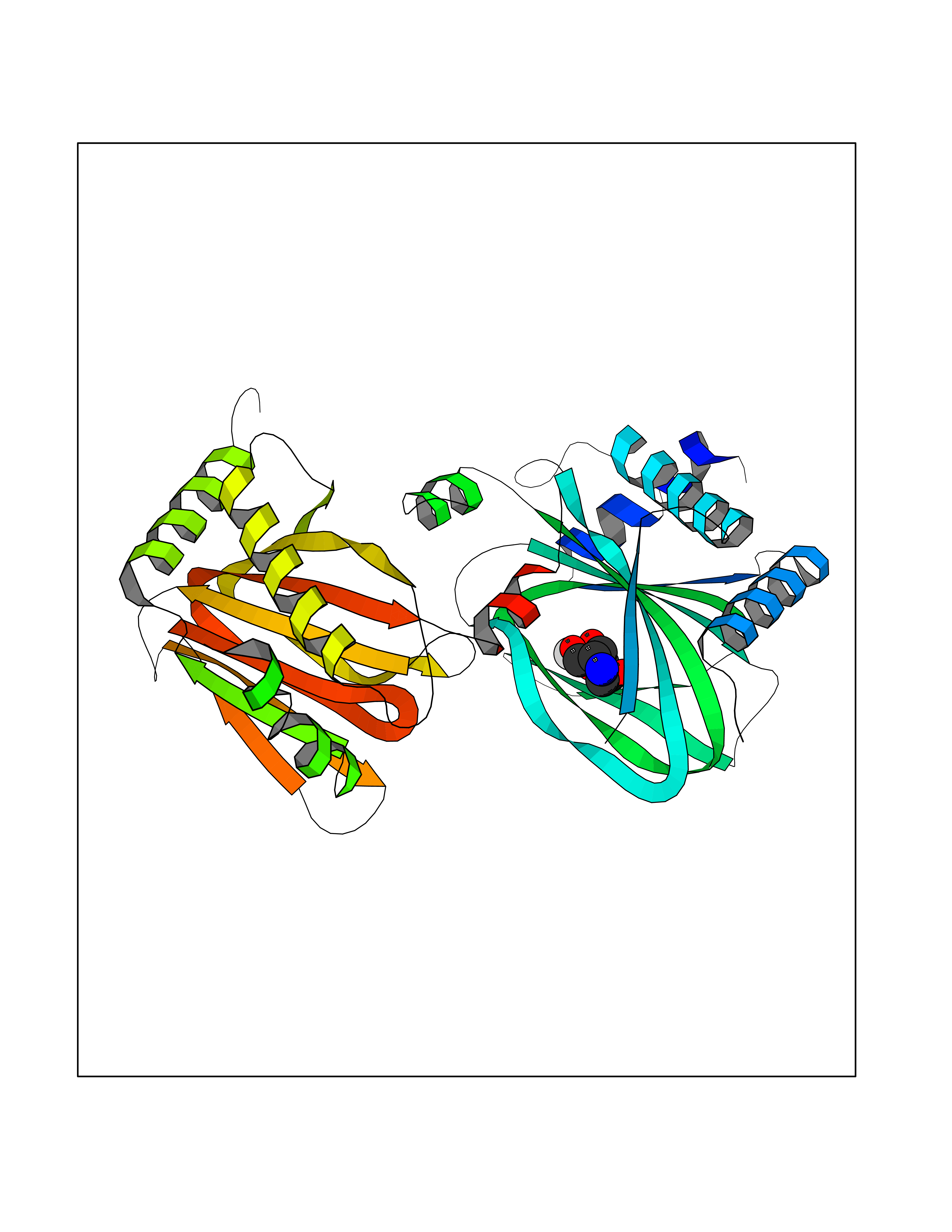 Molscript Map 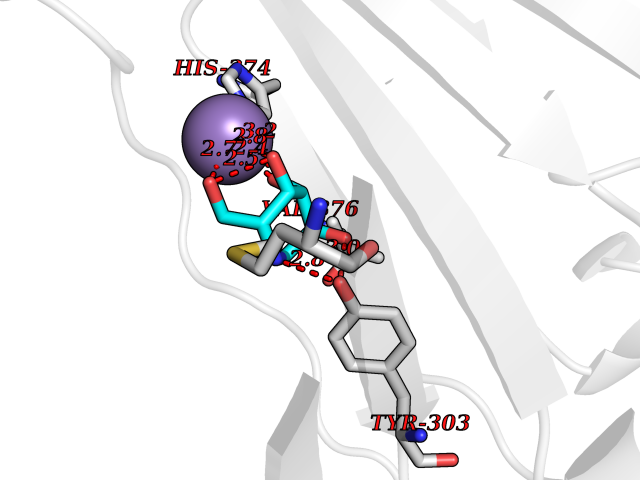 Pymol Map 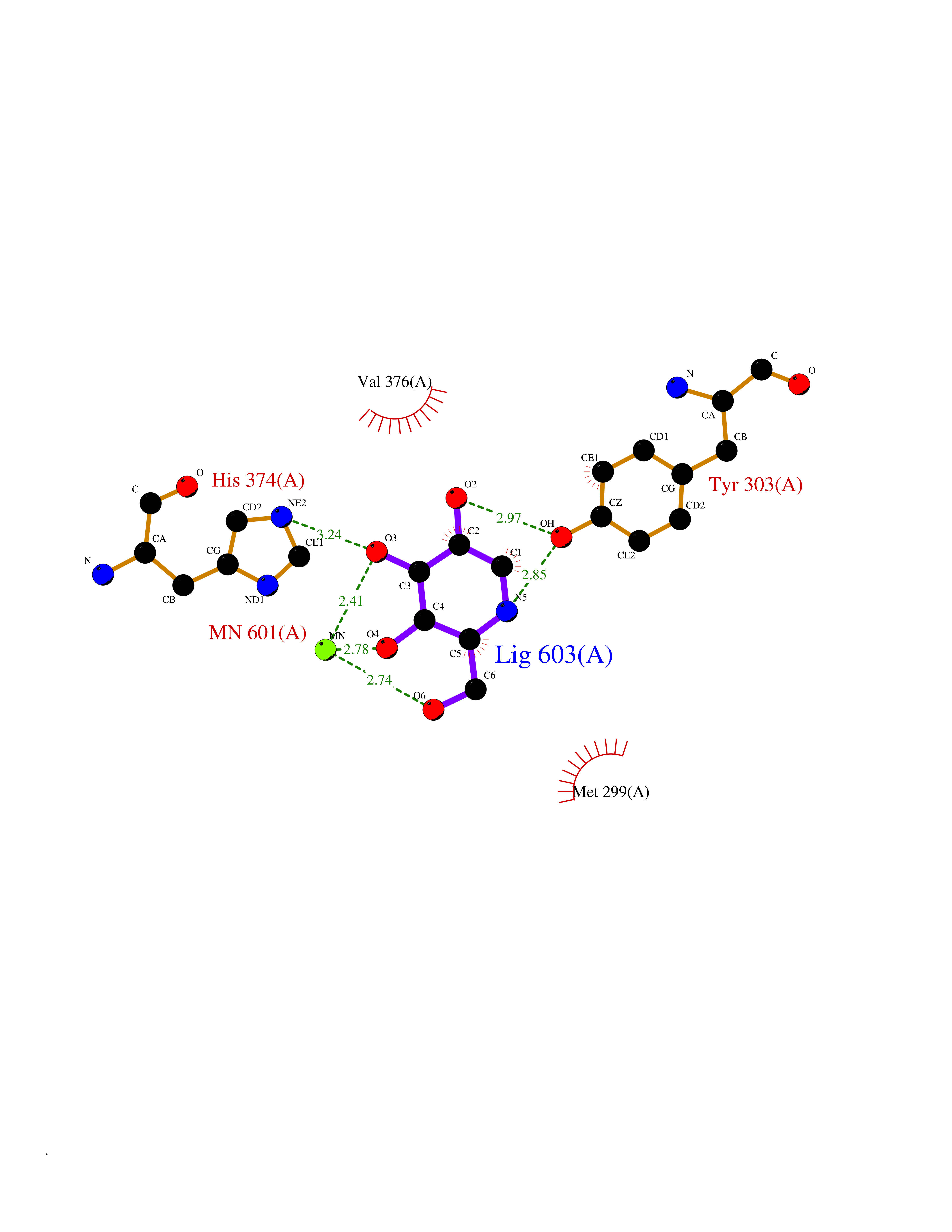 Ligplot Map 3D Binding mode Sequence LPALKLALEYIVPCMNKHGICVVDDFLGKETGQQIGDEVRALHDTGKFTGDKITWIEGKEPGCETIGLLMSSMDDLIRHCNGKLGSYKINGRTKAMVACYPGNGTGYVRHVDNPNGDGRCVTCIYYLNKDWDAKVSGGILRIFPEGKAQFADIEPKFDRLLFFWSDRRNPHEVQPAYATRYAITVWYFDADERARAKVKYLTGELPALKLALEYIVPCMNKHGICVVDDFLGKETGQQIGDEVRALHDTGKFTGDKITWIEGKEPGCETIGLLMSSMDDLIRHCNGKLGSYKINGRTKAMVACYPGNGTGYVRHVDNPNGDGRCVTCIYYLNKDWDAKVSGGILRIFPEGKAQFADIEPKFDRLLFFWSDRRNPHEVQPAYATRYAITVWYFDADERARAKVKYLT Hydrogen bonds contact Hydrophobic contact | ||||
| 18 | Pyruvate kinase PKLR | 4IP7 | 6.10 | |
Target general information Gen name PKLR Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms PKL;PK1 Protein family Pyruvate kinase family Biochemical class Transferase Function ATP binding.Kinase activity.Magnesium ion binding.Potassium ion binding.Pyruvate kinase activity. Related diseases Pyruvate kinase hyperactivity (PKHYP) [MIM:102900]: Autosomal dominant phenotype characterized by increase of red blood cell ATP. {ECO:0000269|PubMed:9090535}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Pyruvate kinase deficiency of red cells (PKRD) [MIM:266200]: A frequent cause of hereditary non-spherocytic hemolytic anemia. Clinically, pyruvate kinase-deficient patients suffer from a highly variable degree of chronic hemolysis, ranging from severe neonatal jaundice and fatal anemia at birth, severe transfusion-dependent chronic hemolysis, moderate hemolysis with exacerbation during infection, to a fully compensated hemolysis without apparent anemia. {ECO:0000269|PubMed:10087985, ECO:0000269|PubMed:10772876, ECO:0000269|PubMed:11328279, ECO:0000269|PubMed:11960989, ECO:0000269|PubMed:1536957, ECO:0000269|PubMed:1896471, ECO:0000269|PubMed:19085939, ECO:0000269|PubMed:2018831, ECO:0000269|PubMed:21794208, ECO:0000269|PubMed:7706479, ECO:0000269|PubMed:8161798, ECO:0000269|PubMed:8180378, ECO:0000269|PubMed:8476433, ECO:0000269|PubMed:8481523, ECO:0000269|PubMed:8483951, ECO:0000269|PubMed:8664896, ECO:0000269|PubMed:8807089, ECO:0000269|PubMed:9075576, ECO:0000269|PubMed:9482576, ECO:0000269|PubMed:9827908, ECO:0000269|PubMed:9886305, ECO:0000269|Ref.24}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02726; DB00787; DB04551; DB16236; DB00119 Interacts with Q9UBL6-2 EC number 2.7.1.40 Uniprot keywords 3D-structure; Allosteric enzyme; Alternative splicing; ATP-binding; Disease variant; Glycolysis; Hereditary hemolytic anemia; Kinase; Magnesium; Manganese; Metal-binding; Nucleotide-binding; Phosphoprotein; Potassium; Proteomics identification; Pyruvate; Reference proteome; Transferase Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 45695.1 Length 421 Aromaticity 0.06 Instability index 34.44 Isoelectric point 6.88 Charge (pH=7) -0.35 2D Binding mode Binding energy (Kcal/mol) -5.93 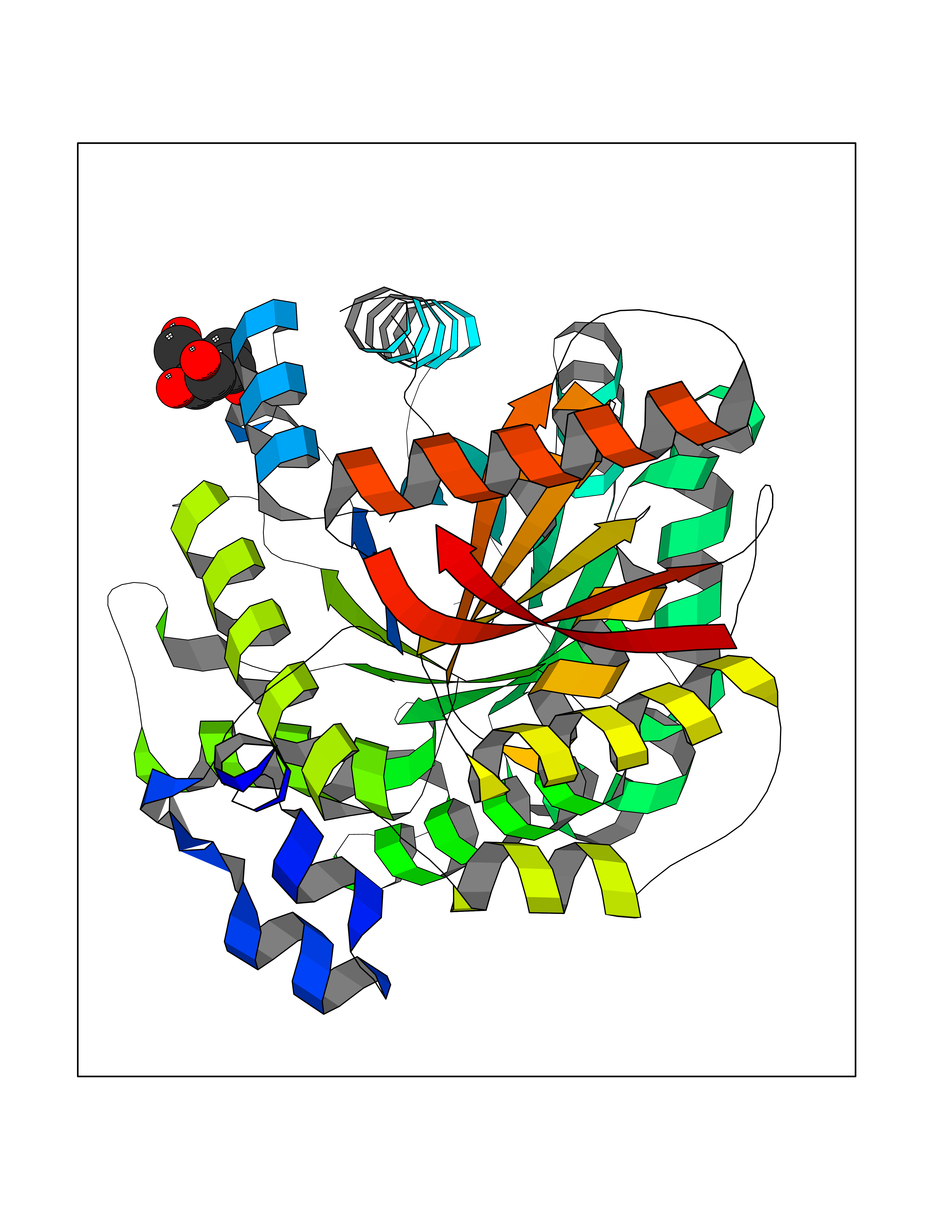 Molscript Map 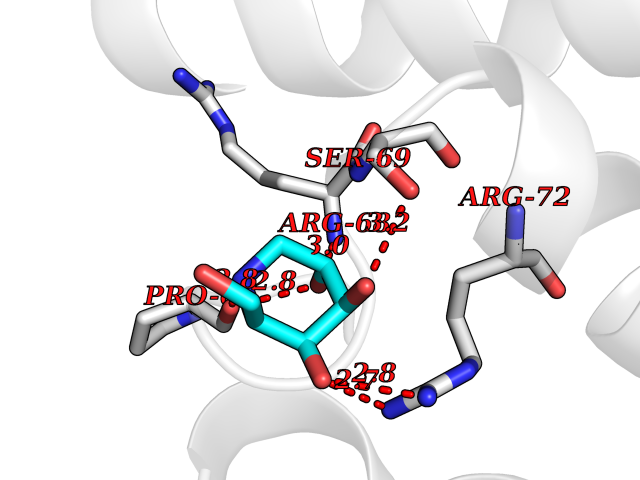 Pymol Map 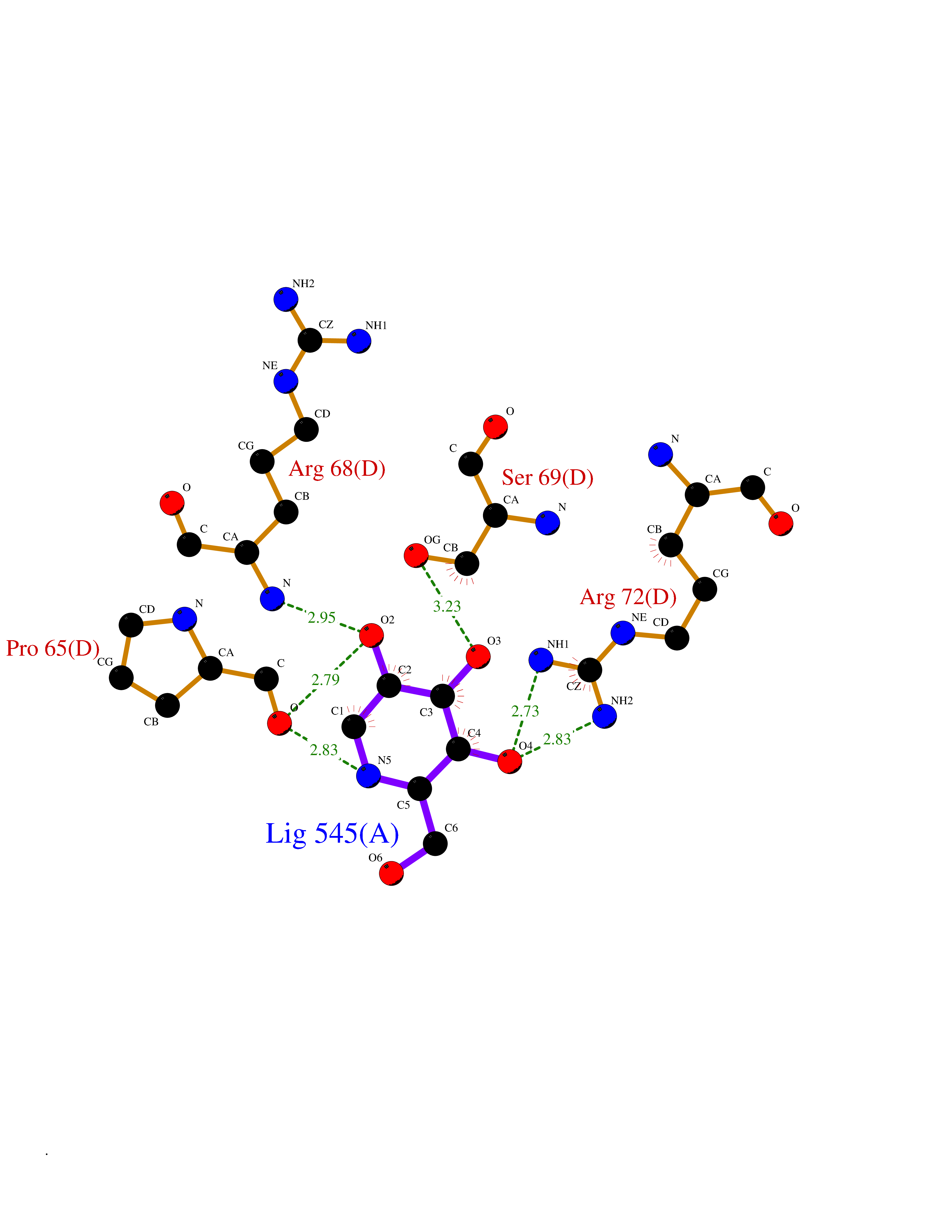 Ligplot Map 3D Binding mode Sequence GTAFFQQQQLPAAMADTFLEHLCLLDIDSEPVAARSTSIIATIGPASRSVERLKEMIKAGMNIARLNFSHGSHEYHAESIANVREAVESFSPLSYRPVAIALDTKGPEIGLSEQDVRDLRFGVEHGVDIVFASFVRKASDVAAVRAALGPEGHGIKIISKIENHEGVKRFDEILEVSDGIMVARGDLGIEIPAEKVFLAQKMMIGRCNLAGKPVVCATQMLESMITKPRPTRAETSDVANAVLDGADCIMLSGETAKGNFPVEAVKMQHAIAREAEAAVYHRQLFEELRRAAPLSRDPTEVTAIGAVEAAFKCCAAAIIVLTTTGRSAQLLSRYRPRAAVIAVTRSAQAARQVHLCRGVFPLLYREPPEAIWADDVDRRVQFGIESGKLRGFLRVGDLVIVVTGWRPGSGYTNIMRVLSIS Hydrogen bonds contact Hydrophobic contact | ||||
| 19 | Endothelin-converting enzyme 1 (ECE1) | 3DWB | 6.10 | |
Target general information Gen name ECE1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms ECE-1 Protein family Peptidase M13 family Biochemical class Peptidase Function Converts big endothelin-1 to endothelin-1. Related diseases Hirschsprung disease, cardiac defects, and autonomic dysfunction (HCAD) [MIM:613870]: A disorder characterized by skip-lesions Hirschsprung disease, craniofacial abnormalities and other dysmorphic features, cardiac defects including ductus arteriosus, small subaortic ventricular septal defect, small atrial septal defect, and autonomic dysfunction. {ECO:0000269|PubMed:9915973}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07171 Interacts with P49760; A8MQ03; Q8IUG1; P60370; P60410 EC number EC 3.4.24.71 Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disease variant; Disulfide bond; Glycoprotein; Hirschsprung disease; Hydrolase; Membrane; Metal-binding; Metalloprotease; Phosphoprotein; Protease; Proteomics identification; Reference proteome; Signal-anchor; Transmembrane; Transmembrane helix; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 75247.9 Length 660 Aromaticity 0.12 Instability index 46.29 Isoelectric point 5.33 Charge (pH=7) -18.3 2D Binding mode Binding energy (Kcal/mol) -6.33 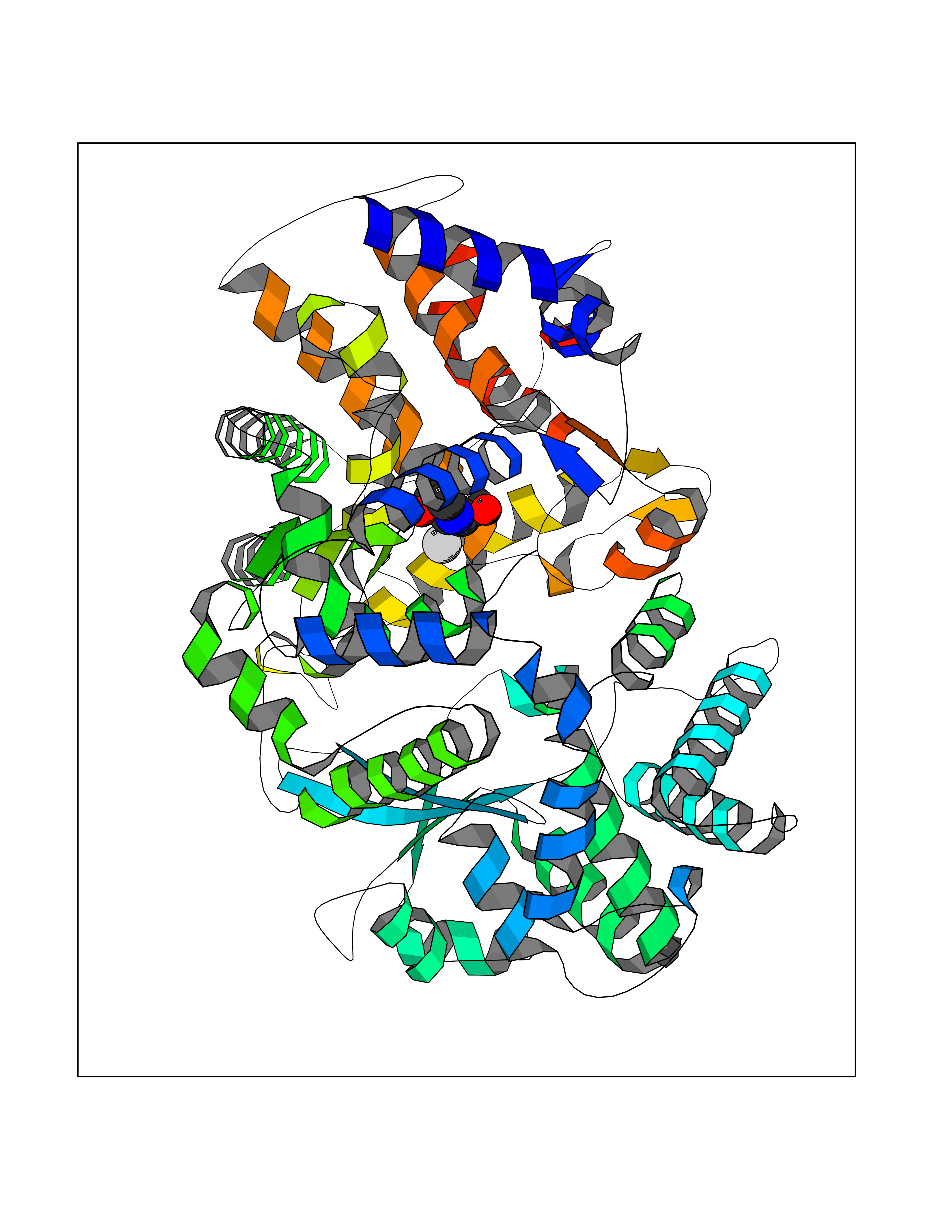 Molscript Map 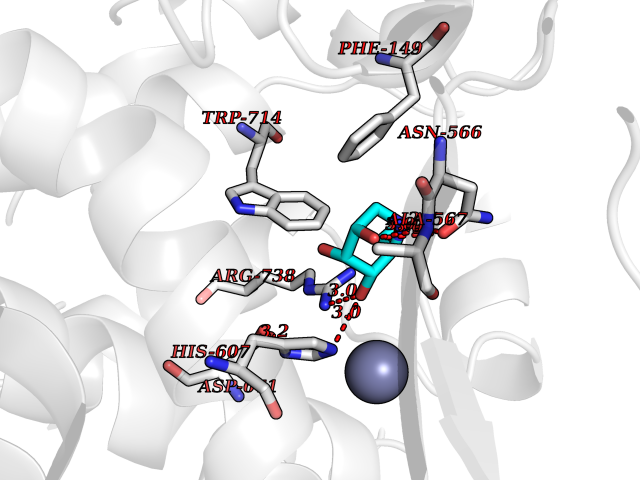 Pymol Map 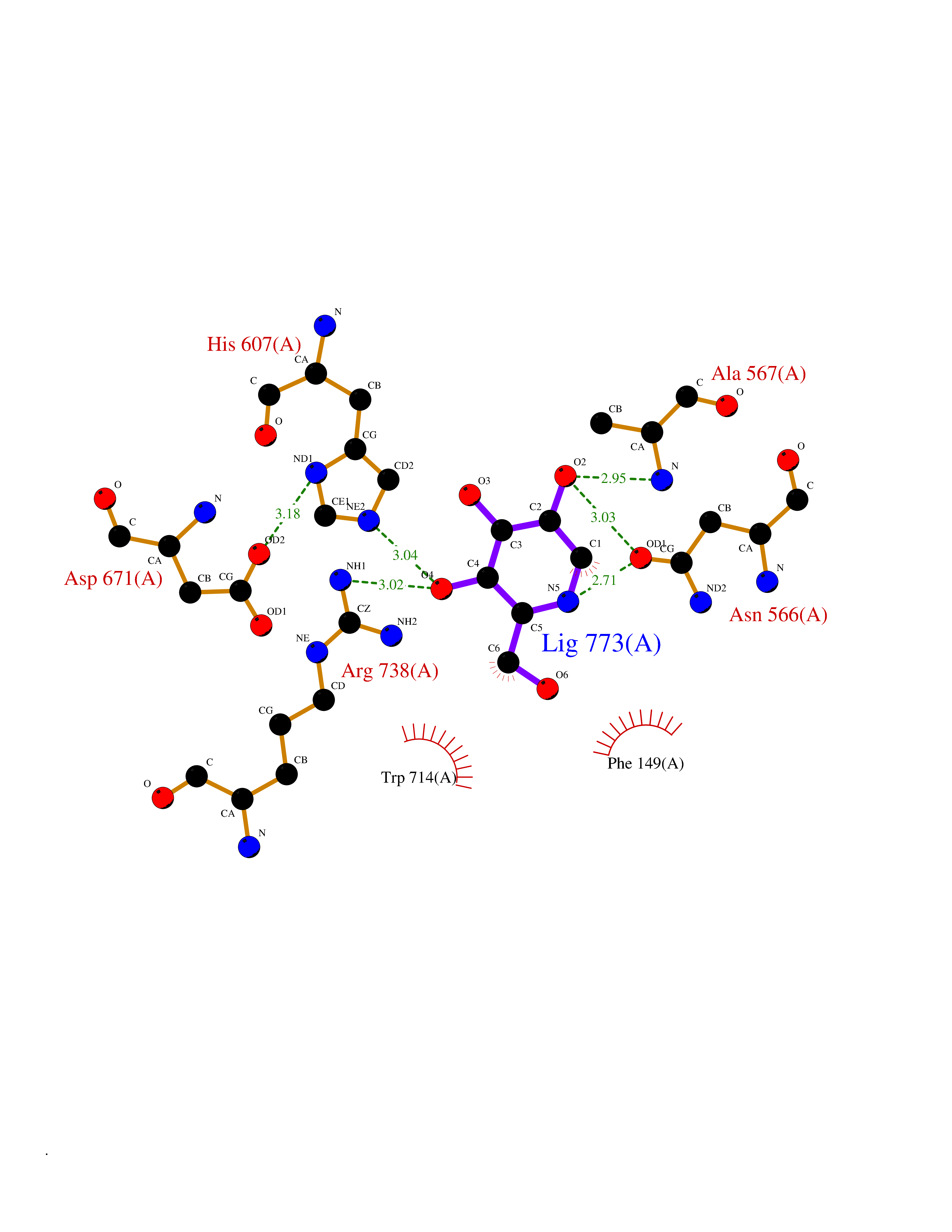 Ligplot Map 3D Binding mode Sequence SEACVSVTSSILSSMDPTVDPCHDFFSYACGGWIKANPVPDGHSRWGTFSNLWEHNQAIIKHLLENSTASVSEAERKAQVYYRACMNETRIEELRAKPLMELIERLGGWNITGPWAKDNFQDTLQVVTAHYRTSPFFSVYVSADSKNSNSNVIQVDQSGLGLPSRDYYLNKTENEKVLTGYLNYMVQLGKLLGGGDEEAIRPQMQQILDFETALANITIPQEKRRDEELIYHKVTAAELQTLAPAINWLPFLNTIFYPVEINESEPIVVYDKEYLEQISTLINTTDRCLLNNYMIWNLVRKTSSFLDQRFQDADEKFMEVMWKFCVSDTENNLGFALGPMFVKATFAEDSKSIATEIILEIKKAFEESLSTLKWMDEETRKSAKEKADAIYNMIGYPNFIMDPKELDKVFNDYTAVPDLYFENAMRFFNFSWRVTADQLRKAPNRDQWSMTPPMVNAYYSPTKNEIVFPAGILQAPFYTRSSPKALNFGGIGVVVGHELTHAFDDQGREYDKDGNLRPWWKNSSVEAFKRQTECMVEQYSNYSVNGEPVNGRHTLGENIADNGGLKAAYRAYQNWVKKNGAEHSLPTLGLTNNQLFFLGFAQVWCSVRTPESSHEGLITDPHSPSRFRVIGSLSNSKEFSEHFRCPPGSPMNPPHKCEVW Hydrogen bonds contact Hydrophobic contact | ||||
| 20 | Beta-klotho (KLB) | 5VAN | 6.08 | |
Target general information Gen name KLB Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Klotho beta-like protein; BetaKlotho; BKL Protein family Glycosyl hydrolase 1 family, Klotho subfamily Biochemical class Glycosylase Function Probably inactive as a glycosidase. Increases the ability of FGFR1 and FGFR4 to bind FGF21. Contributes to the transcriptional repression of cholesterol 7-alpha-hydroxylase (CYP7A1), the rate-limiting enzyme in bile acid synthesis. Related diseases Charcot-Marie-Tooth disease, axonal, 2B (CMT2B) [MIM:600882]: A dominant axonal form of Charcot-Marie-Tooth disease, a disorder of the peripheral nervous system, characterized by progressive weakness and atrophy, initially of the peroneal muscles and later of the distal muscles of the arms. Charcot-Marie-Tooth disease is classified in two main groups on the basis of electrophysiologic properties and histopathology: primary peripheral demyelinating neuropathies (designated CMT1 when they are dominantly inherited) and primary peripheral axonal neuropathies (CMT2). Neuropathies of the CMT2 group are characterized by signs of axonal degeneration in the absence of obvious myelin alterations, normal or slightly reduced nerve conduction velocities, and progressive distal muscle weakness and atrophy. {ECO:0000269|PubMed:12545426, ECO:0000269|PubMed:15455439, ECO:0000269|PubMed:17060578, ECO:0000269|PubMed:20028791, ECO:0000269|PubMed:21151572, ECO:0000269|PubMed:23179371}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q9NSA1 EC number NA Uniprot keywords 3D-structure; Cell membrane; Glycoprotein; Membrane; Proteomics identification; Reference proteome; Repeat; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 99301.9 Length 861 Aromaticity 0.14 Instability index 32.93 Isoelectric point 8.85 Charge (pH=7) 12.12 2D Binding mode Binding energy (Kcal/mol) -5.66  Molscript Map 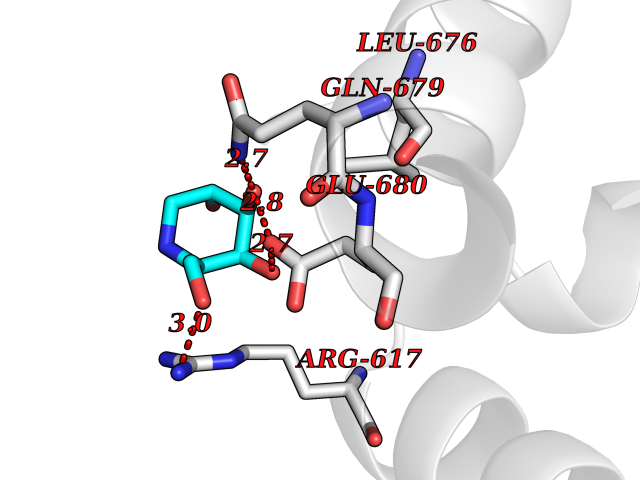 Pymol Map 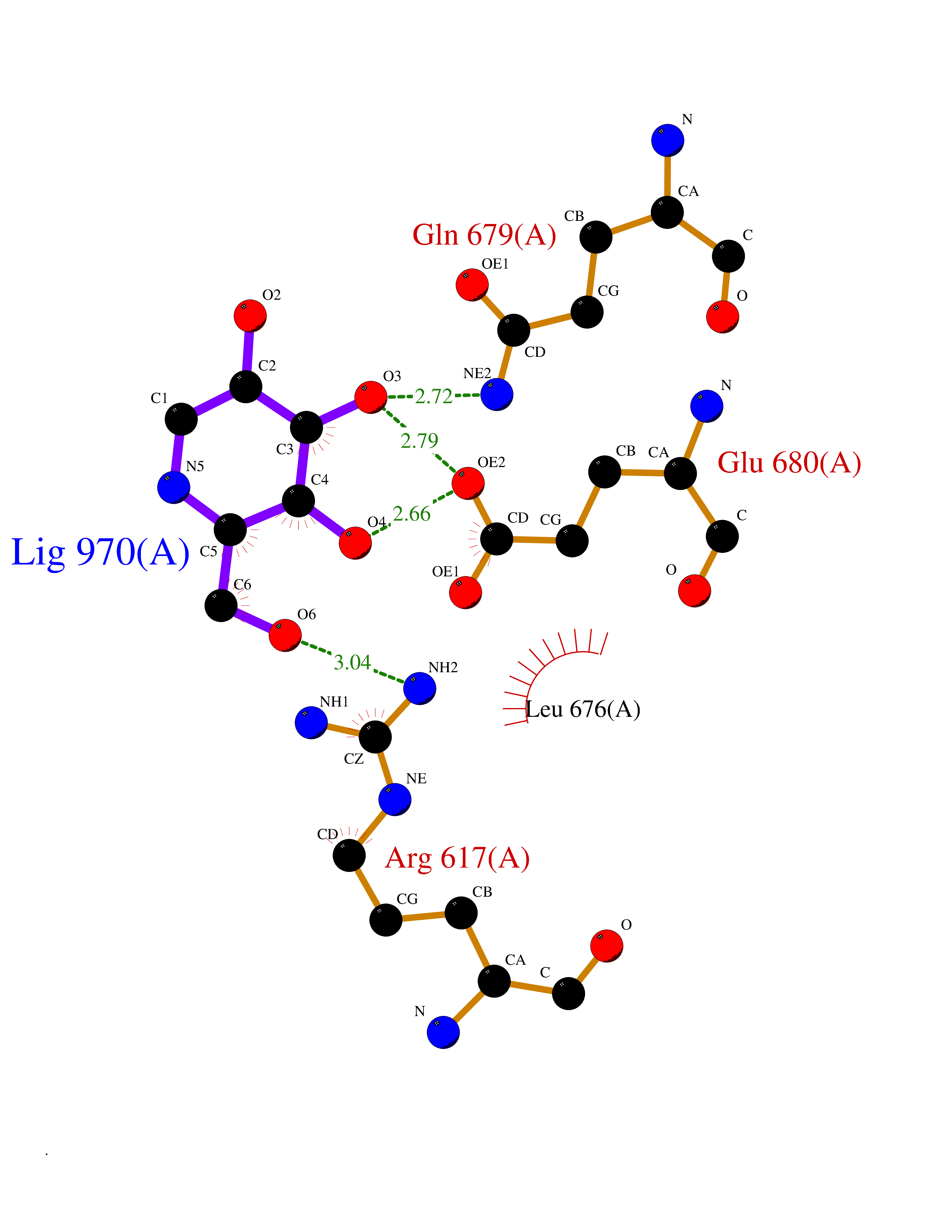 Ligplot Map 3D Binding mode Sequence FSGDGRAIWSQLFLYDTFPKNFFWGIGTGALQVEGSWKKDGKGPSIWDHFIHTHLGSSDSYIFLEKDLSALDFIGVSFYQFSISWPRLFPDGIVTVANAKGLQYYSTLLDALVLRNIEPIVTLYHWDLPLALQEKYGGWKNDTIIDIFNDYATYCFQMFGDRVKYWITIHNPYLVAWHGYGTGMHAPGEKGNLAAVYTVGHNLIKAHSKVWHNYNTHFRPHQKGWLSITLGSHWIEPQRSENTMDIFKCQQSMVSVLGWFANPIHGDGDYPEGMRKKLFSVLPIFSEAEKHEMRGTADFFAFSFGPNNFKPLNTMAKMGQNVSLNLREALNWIKLEYNNPRILIAENGWFTDSRVKTEDTTAIYMMKNFLSQVLQAIRLDEIRVFGYTAWSLLDGFEWQDAYTIRRGLFYVDFNSKQKERKPKSSAHYYKQIIRENGFSLKESTPDVQGQFPCDFSWGVTESVLKPEQCTDFVNIKKQLEMLARMKVTHYRFALDWASVLPTGQLSAVNRQALRYYRCVVSEGLKLGISAMVTLYYPTHAHLGLPEPLLHADGWLNPSTAEAFQAYAGLCFQELGDLVKLWITINEPNRLSDIYNRSGNDTYGAAHNLLVAHALAWRLYDRQFRPSQRGAVSLSLHADWAEPANPYADSHWRAAERFLQFEIAWFAEPLFKTGDYPAAMREYIASKHRRGLSSSALPRLTEAERRLLKGTVDFCALNHFTTRFVMHEQLAGSRYDSDRDIQFLQDITRLSSPTRLAVIPWGVRKLLRWVRRNYGDMDIYITASGIDDQALEDDRLRKYYLGKYLQEVLKAYLIDKVRIKGYYAFKLAEEKSKPRFGFFTSDFKAKSSIQFYNKVISSRGFP Hydrogen bonds contact Hydrophobic contact | ||||