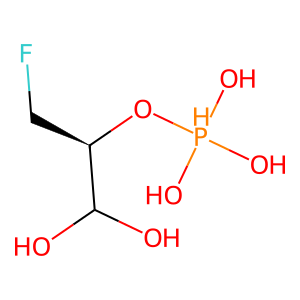
Ligand
Structure
Job ID
36ec3cf13088ab2ed61fcebb0cfaa0f9
Job name
Anderson_Task5
Time
2024-07-05 09:08:27
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 1 | Endothelin-converting enzyme 1 (ECE1) | 3DWB | 7.12 | |
Target general information Gen name ECE1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms ECE-1 Protein family Peptidase M13 family Biochemical class Peptidase Function Converts big endothelin-1 to endothelin-1. Related diseases Hirschsprung disease, cardiac defects, and autonomic dysfunction (HCAD) [MIM:613870]: A disorder characterized by skip-lesions Hirschsprung disease, craniofacial abnormalities and other dysmorphic features, cardiac defects including ductus arteriosus, small subaortic ventricular septal defect, small atrial septal defect, and autonomic dysfunction. {ECO:0000269|PubMed:9915973}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07171 Interacts with P49760; A8MQ03; Q8IUG1; P60370; P60410 EC number EC 3.4.24.71 Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disease variant; Disulfide bond; Glycoprotein; Hirschsprung disease; Hydrolase; Membrane; Metal-binding; Metalloprotease; Phosphoprotein; Protease; Proteomics identification; Reference proteome; Signal-anchor; Transmembrane; Transmembrane helix; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 75247.9 Length 660 Aromaticity 0.12 Instability index 46.29 Isoelectric point 5.33 Charge (pH=7) -18.3 2D Binding mode Binding energy (Kcal/mol) -6.25 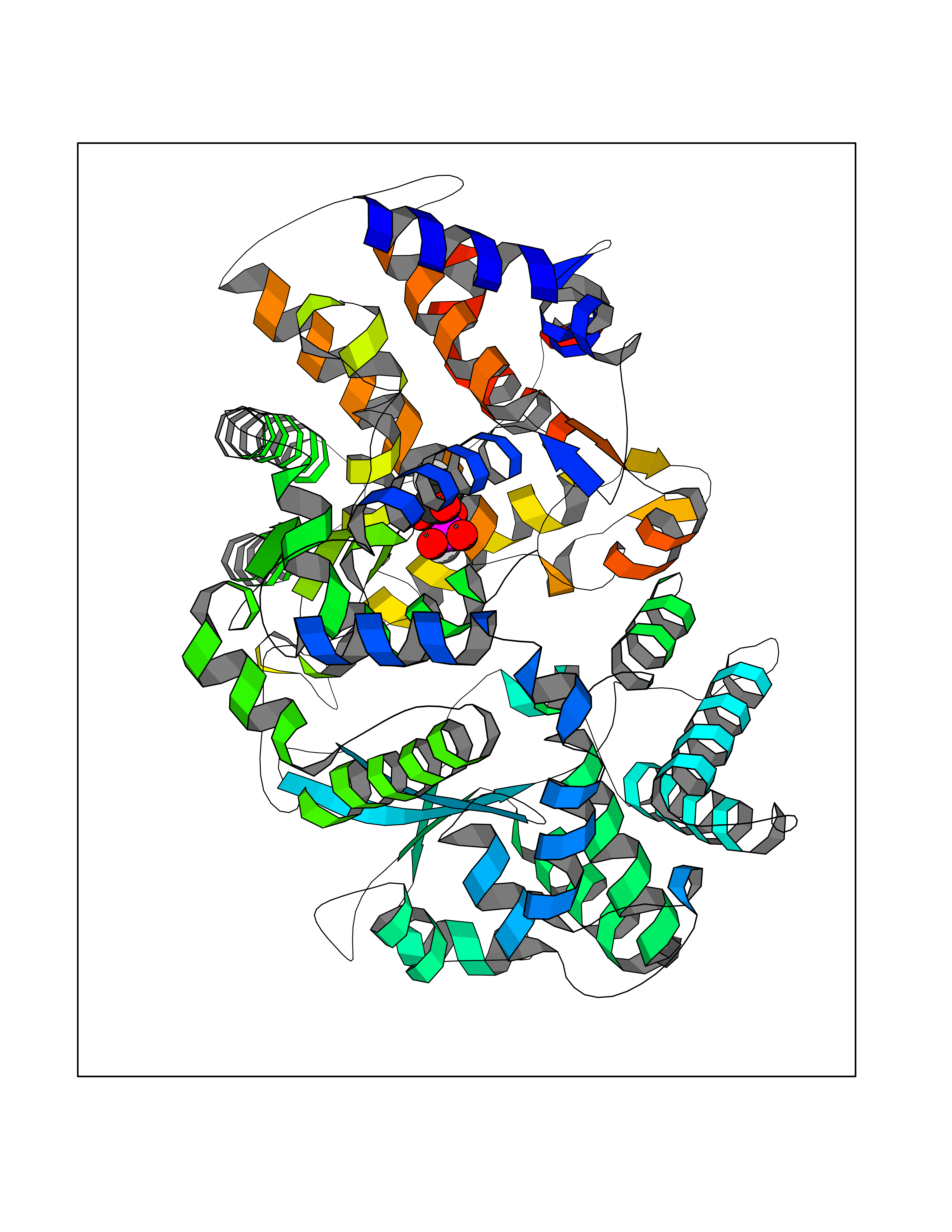 Molscript Map 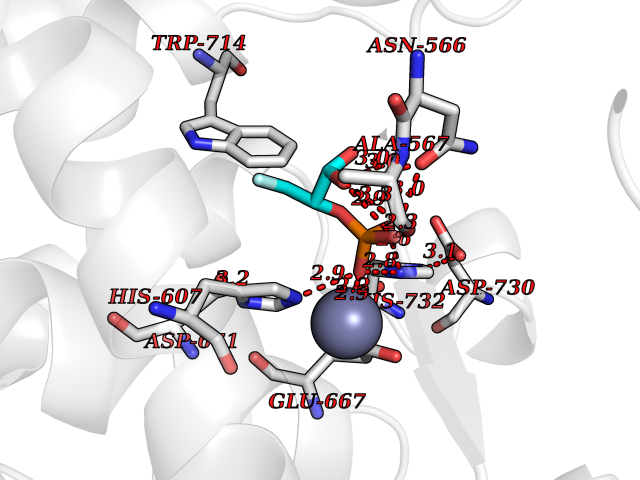 Pymol Map 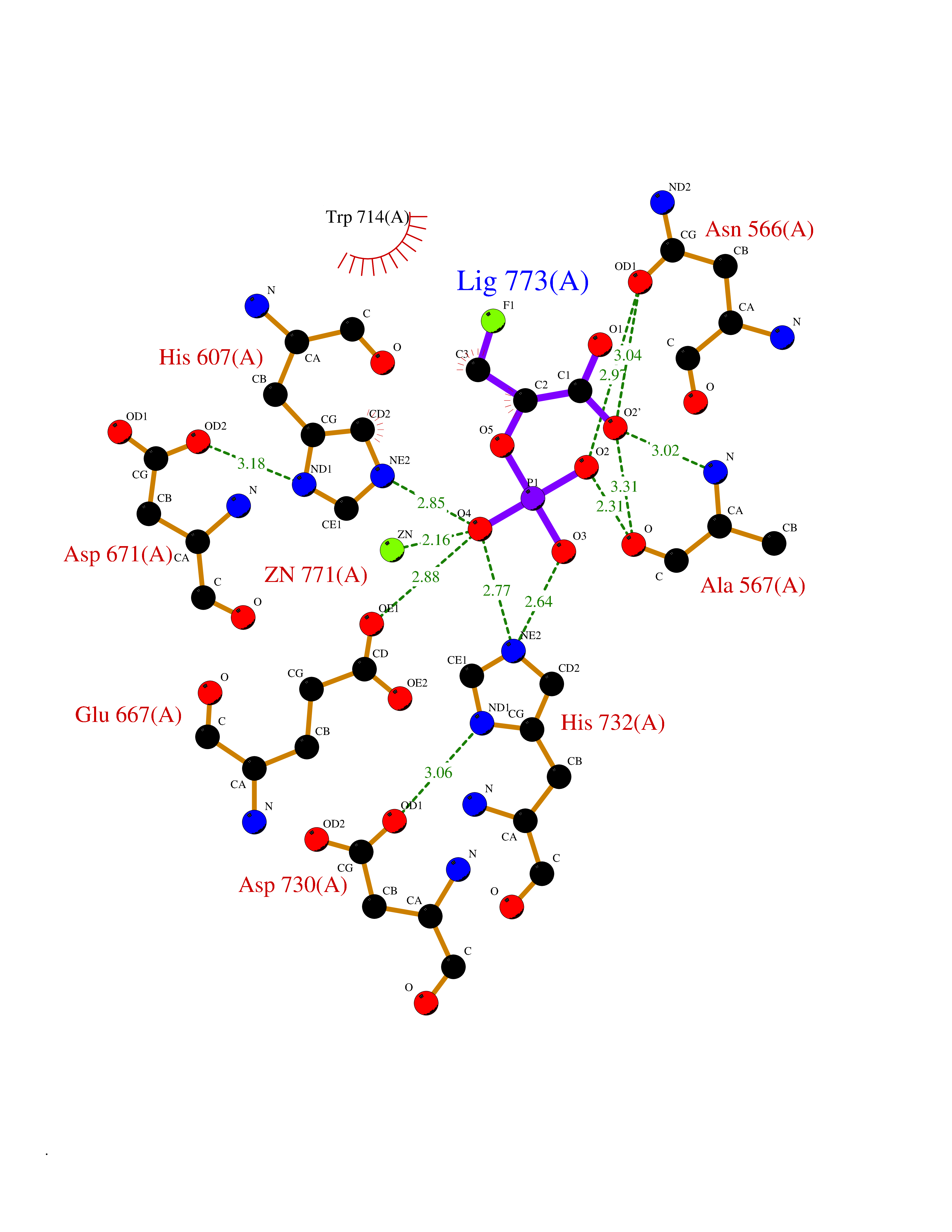 Ligplot Map 3D Binding mode Sequence SEACVSVTSSILSSMDPTVDPCHDFFSYACGGWIKANPVPDGHSRWGTFSNLWEHNQAIIKHLLENSTASVSEAERKAQVYYRACMNETRIEELRAKPLMELIERLGGWNITGPWAKDNFQDTLQVVTAHYRTSPFFSVYVSADSKNSNSNVIQVDQSGLGLPSRDYYLNKTENEKVLTGYLNYMVQLGKLLGGGDEEAIRPQMQQILDFETALANITIPQEKRRDEELIYHKVTAAELQTLAPAINWLPFLNTIFYPVEINESEPIVVYDKEYLEQISTLINTTDRCLLNNYMIWNLVRKTSSFLDQRFQDADEKFMEVMWKFCVSDTENNLGFALGPMFVKATFAEDSKSIATEIILEIKKAFEESLSTLKWMDEETRKSAKEKADAIYNMIGYPNFIMDPKELDKVFNDYTAVPDLYFENAMRFFNFSWRVTADQLRKAPNRDQWSMTPPMVNAYYSPTKNEIVFPAGILQAPFYTRSSPKALNFGGIGVVVGHELTHAFDDQGREYDKDGNLRPWWKNSSVEAFKRQTECMVEQYSNYSVNGEPVNGRHTLGENIADNGGLKAAYRAYQNWVKKNGAEHSLPTLGLTNNQLFFLGFAQVWCSVRTPESSHEGLITDPHSPSRFRVIGSLSNSKEFSEHFRCPPGSPMNPPHKCEVW Hydrogen bonds contact Hydrophobic contact | ||||
| 2 | Carbonic anhydrase IX (CA-IX) | 5FL4 | 6.82 | |
Target general information Gen name CA9 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Renal cell carcinoma-associated antigen G250; RCC-associated antigen G250; PMW1; P54/58N; Membrane antigen MN; MN; G250 antigen (MN/CA IX/G250); G250; Carbonic anhydrase 9; Carbonate dehydratase IX; C Protein family Alpha-carbonic anhydrase family Biochemical class Alpha-carbonic anhydrase Function Participates in pH regulation. May be involved in the control of cell proliferation and transformation. Appears to be a novel specific biomarker for a cervical neoplasia. Reversible hydration of carbon dioxide. Related diseases Hydroxykynureninuria (HYXKY) [MIM:236800]: An inborn error of amino acid metabolism characterized by massive urinary excretion of large amounts of kynurenine, 3-hydroxykynurenine and xanthurenic acid. Affected individuals manifest renal tubular dysfunction, metabolic acidosis, psychomotor retardation, non-progressive encephalopathy, and muscular hypertonia. {ECO:0000269|PubMed:17334708, ECO:0000269|PubMed:28792876}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Vertebral, cardiac, renal, and limb defects syndrome 2 (VCRL2) [MIM:617661]: An autosomal recessive congenital malformation syndrome characterized by vertebral segmentation abnormalities, congenital cardiac defects, renal defects, and distal mild limb defects. {ECO:0000269|PubMed:28792876}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00562; DB00606; DB12741; DB08846; DB05304; DB00774; DB09460; DB00909 Interacts with P21291; O76003 EC number EC 4.2.1.1 Uniprot keywords 3D-structure; Cell membrane; Cell projection; Direct protein sequencing; Disulfide bond; Glycoprotein; Lyase; Membrane; Metal-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Signal; Transmembrane; Transmembrane helix; Zinc Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 27522.8 Length 251 Aromaticity 0.08 Instability index 48.97 Isoelectric point 5.48 Charge (pH=7) -7.5 2D Binding mode Binding energy (Kcal/mol) -6.22 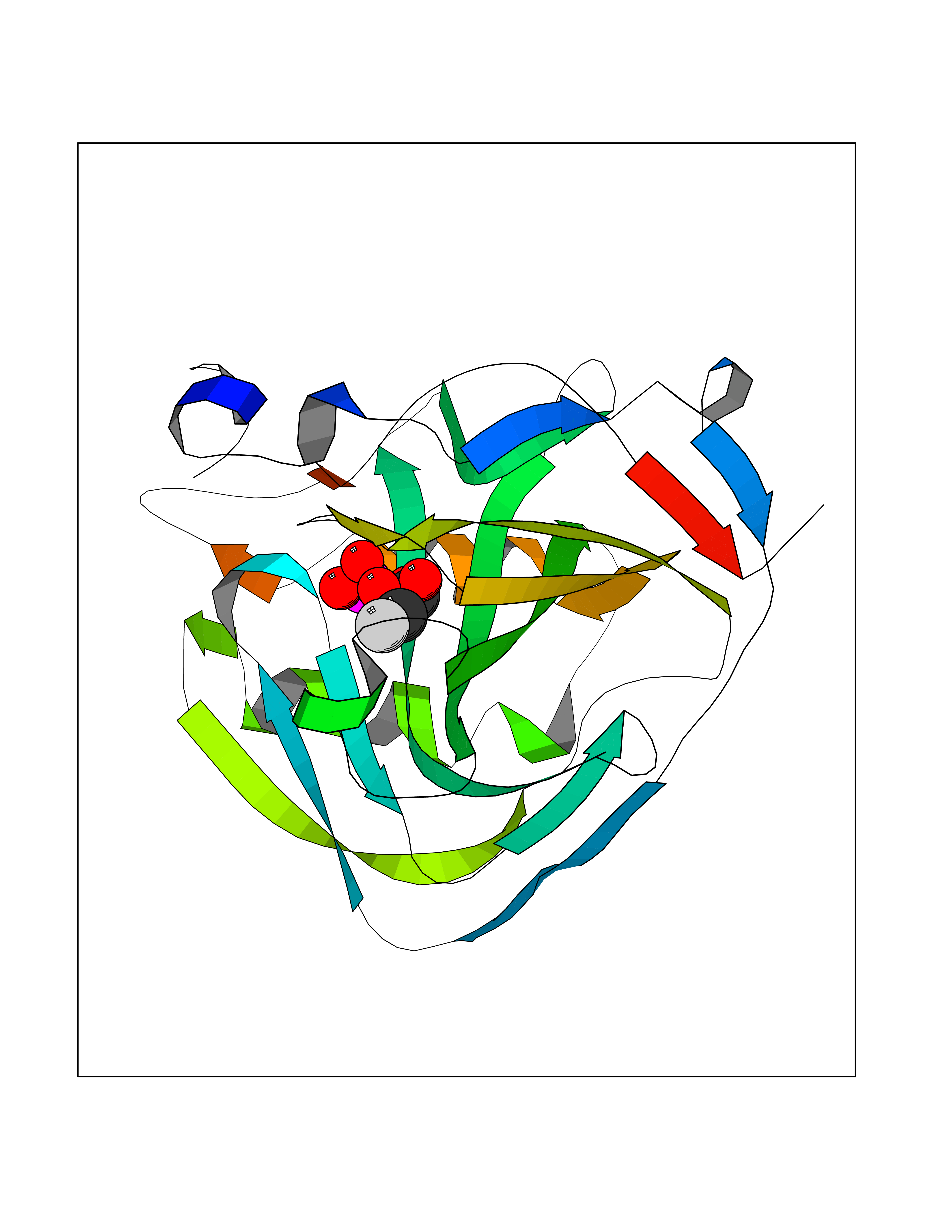 Molscript Map 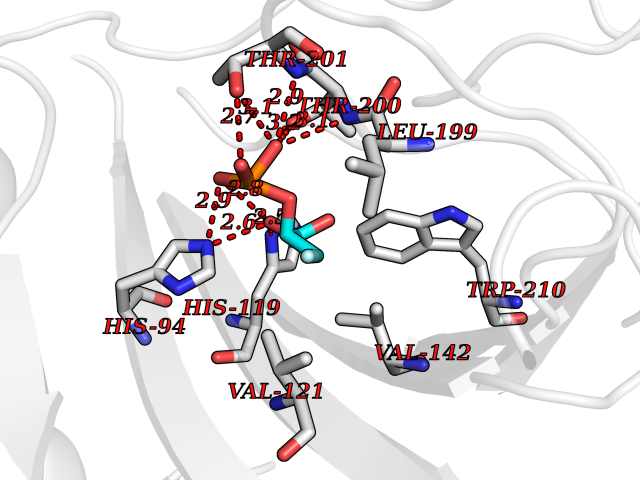 Pymol Map 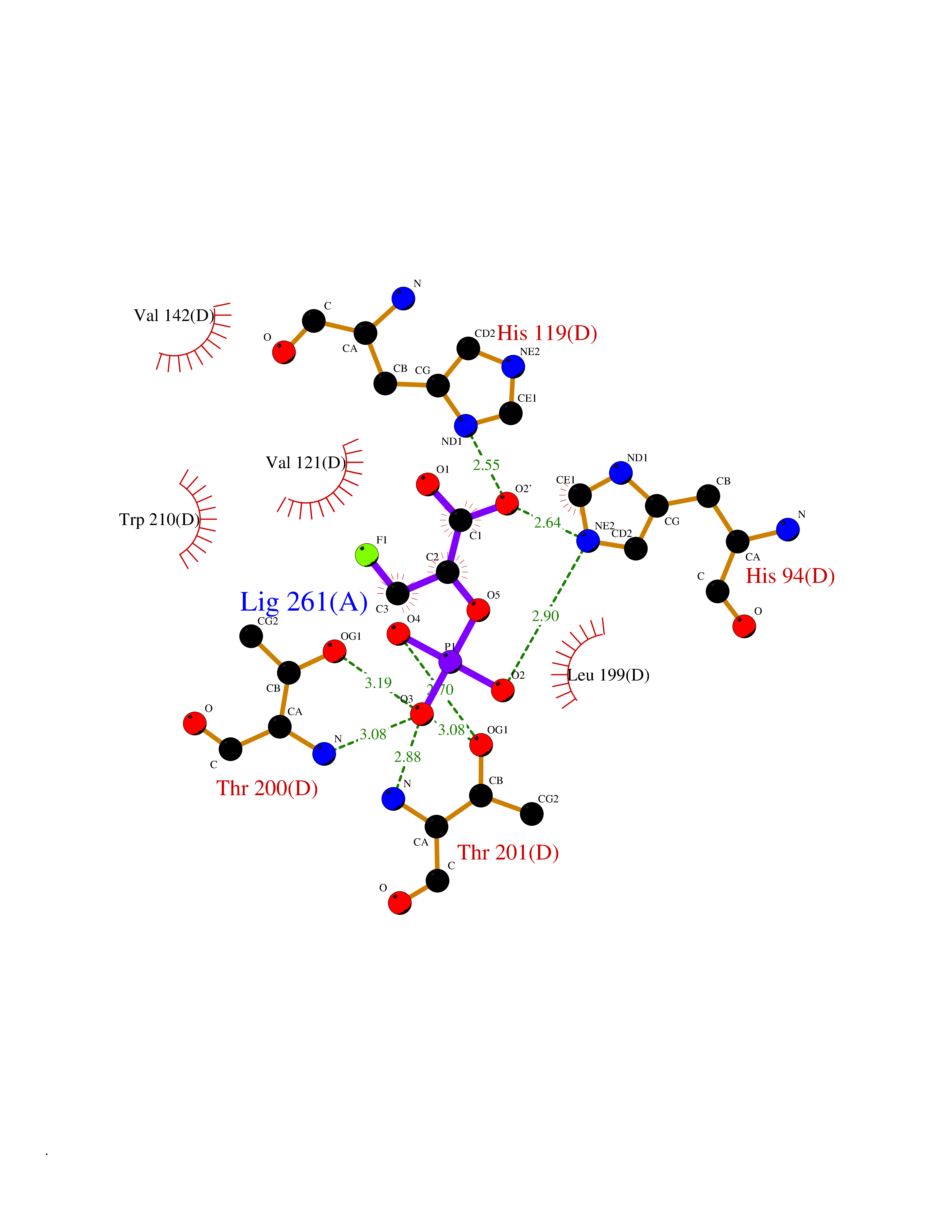 Ligplot Map 3D Binding mode Sequence WRYGGDPPWPRVSPACAGRFQSPVDIRPQLAAFSPALRPLELLGFQLPPLPELRLRNNGHSVQLTLPPGLEMALGPGREYRALQLHLHWGAAGRPGSEHTVEGHRFPAEIHVVHLSTAFARVDEALGRPGGLAVLAAFLEEGPEENSAYEQLLSRLEEIAEEGSETQVPGLDISALLPSDFSRYFQYEGSLTTPPCAQGVIWTVFNQTVMLSAKQLHTLSDTLWGPGDSRLQLNFRATQPLNGRVIEASFP Hydrogen bonds contact Hydrophobic contact | ||||
| 3 | DNA topoisomerase 4 subunit A | 1ZVT | 6.63 | |
Target general information Gen name parC Organism Escherichia coli (strain K12) Uniprot ID TTD ID NA Synonyms b3019;JW2987 Protein family Type II topoisomerase GyrA/ParC subunit family, ParC type 1 subfamily Biochemical class Isomerase Function ATP binding.DNA binding.DNA topoisomerase type II (ATP-hydrolyzing) activity. Related diseases Pigmentary disorder, reticulate, with systemic manifestations, X-linked (PDR) [MIM:301220]: An X-linked recessive disorder characterized by recurrent infections and sterile inflammation in various organs. Diffuse skin hyperpigmentation with a distinctive reticulate pattern is universally evident by early childhood. This is later followed in many patients by hypohidrosis, corneal inflammation and scarring, enterocolitis that resembles inflammatory bowel disease, and recurrent urethral strictures. Melanin and amyloid deposition is present in the dermis. Affected males also have a characteristic facies with frontally upswept hair and flared eyebrows. Female carriers have only restricted pigmentary changes along Blaschko's lines. {ECO:0000269|PubMed:27019227}. The disease is caused by variants affecting the gene represented in this entry. XLPDR is caused by a recurrent intronic mutation that results in missplicing and reduced POLA1 expression. This leads to a decrease in cytosolic RNA:DNA hybrids and constitutive activation of type I interferon responses, but has no effect on cell replication. {ECO:0000269|PubMed:27019227}.; DISEASE: Van Esch-O'Driscoll syndrome (VEODS) [MIM:301030]: An X-linked recessive syndrome characterized by different degrees of intellectual disability, moderate to severe short stature, microcephaly, hypogonadism, and variable congenital malformations. {ECO:0000269|PubMed:31006512}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB11943; DB12924; DB00817 Interacts with P22523; P0A7K2 EC number 5.6.2.2 Uniprot keywords 3D-structure; Cell membrane; Direct protein sequencing; DNA-binding; Isomerase; Membrane; Reference proteome; Topoisomerase Protein physicochemical properties Chain ID A,B Molecular weight (Da) 26490.3 Length 246 Aromaticity 0.04 Instability index 46.03 Isoelectric point 8.94 Charge (pH=7) 2.83 2D Binding mode Binding energy (Kcal/mol) -5.93 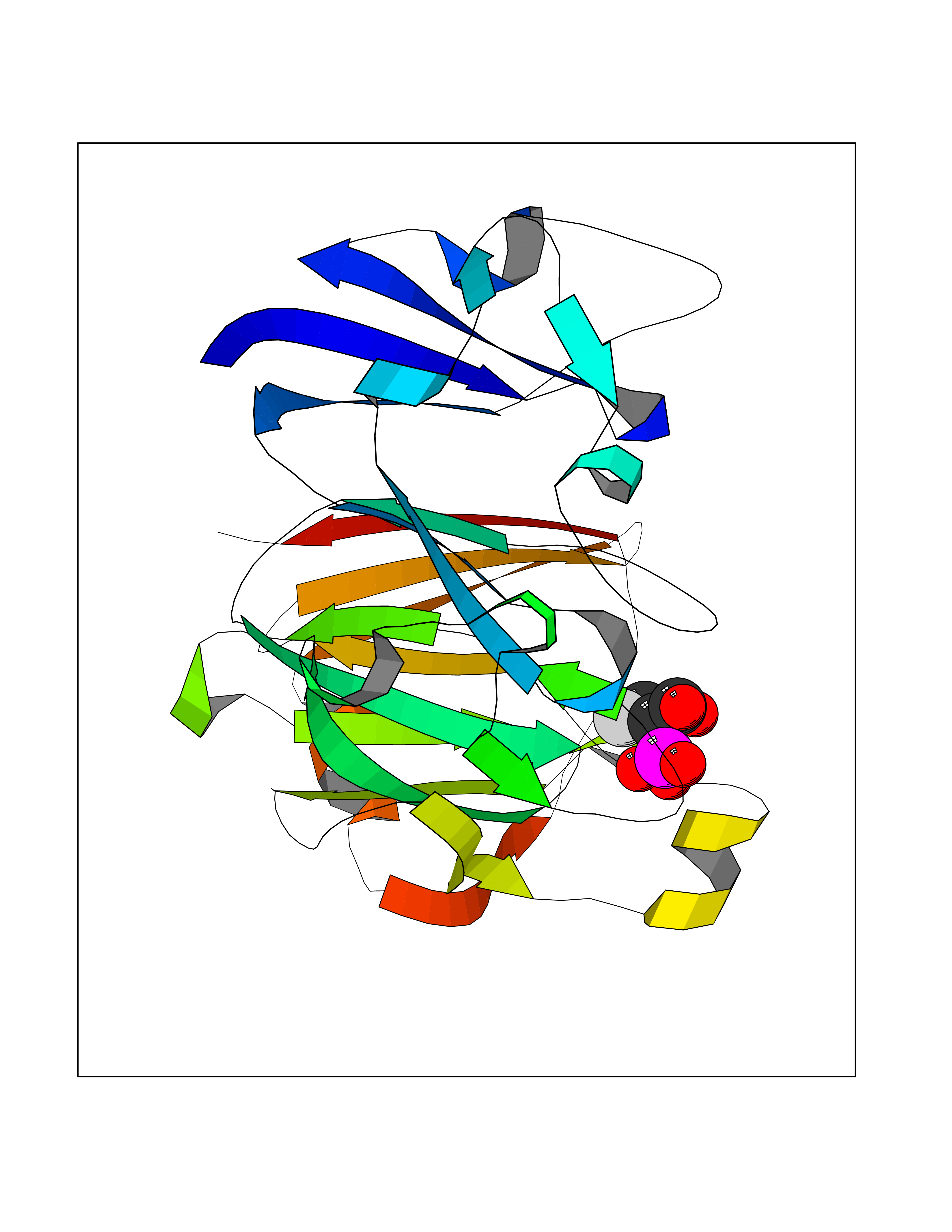 Molscript Map 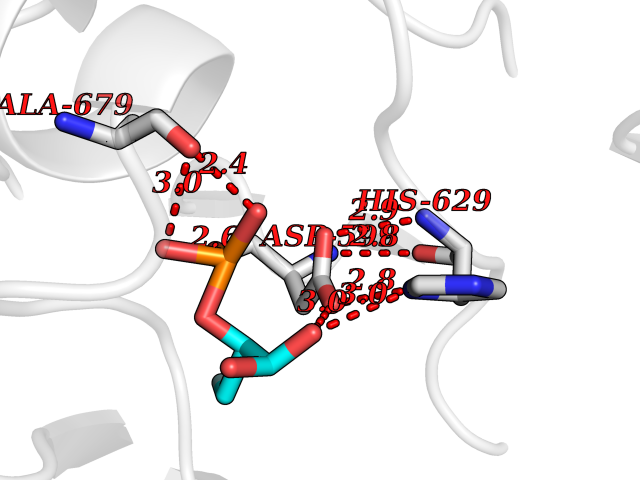 Pymol Map 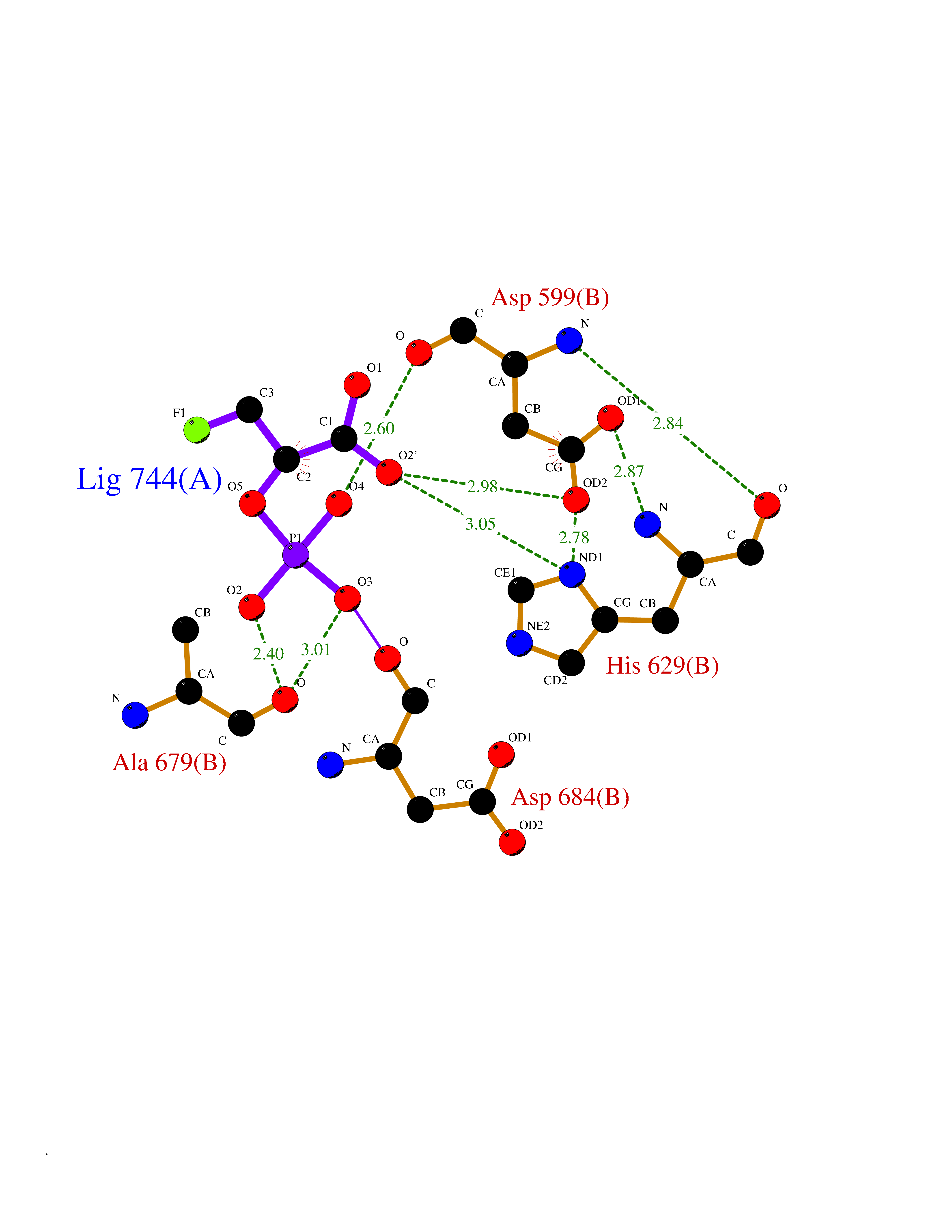 Ligplot Map 3D Binding mode Sequence SEPVTIVLSQMGWVRSAKGHDIDAPGLNYKAGDSFKAAVKGKSNQPVVFVDSTGRSYAIDPITLPSARGQGEPLTGKLTLPPGATVDHMLMESDDQKLLMASDAGYGFVCTFNDLVARNRAGKALITLPENAHVMPPVVIEDASDMLLAITQAGRMLMFPVSDLPQLSKGKGNKIINIPSAEAARGEDGLAQLYVLPPQSTLTIHVGKRKIKLRPEELQKVTGERGRRGTLMRGLQRIDRVEIDSP Hydrogen bonds contact Hydrophobic contact | ||||
| 4 | Phosphodiesterase 5A (PDE5A) | 1TBF | 6.62 | |
Target general information Gen name PDE5A Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms cGMP-specific 3',5'-cyclic phosphodiesterase; PDE5A; CGMP-binding cGMP-specific phosphodiesterase; CGB-PDE Protein family Cyclic nucleotide phosphodiesterase family Biochemical class Phosphoric diester hydrolase Function Plays a role in signal transduction by regulating the intracellular concentration of cyclic nucleotides. This phosphodiesterase catalyzes the specific hydrolysis of cGMP to 5'-GMP. Specifically regulates nitric-oxide-generated cGMP. Related diseases Neurodevelopmental disorder with poor language and loss of hand skills (NDPLHS) [MIM:617903]: An autosomal dominant disorder characterized by psychomotor developmental stagnation or regression. NDPLHS manifest in the first years of life as loss of purposeful hand movements, loss of language, and intellectual disability. {ECO:0000269|PubMed:26740508, ECO:0000269|PubMed:28856709, ECO:0000269|PubMed:29369404}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Developmental and epileptic encephalopathy 59 (DEE59) [MIM:617904]: A form of epileptic encephalopathy, a heterogeneous group of severe early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE59 is an autosomal dominant condition characterized by onset of refractory seizures in early infancy. {ECO:0000269|PubMed:28856709, ECO:0000269|PubMed:29100083, ECO:0000269|PubMed:29369404}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07954; DB08729; DB06237; DB00201; DB00975; DB06246; DB12010; DB03597; DB01972; DB09282; DB05415; DB01113; DB00203; DB00820; DB00277; DB09283; DB06267; DB00862 Interacts with Q13976; Q9NZD8 EC number EC 3.1.4.35 Uniprot keywords 3D-structure; Allosteric enzyme; Alternative splicing; cGMP; cGMP-binding; Hydrolase; Metal-binding; Nucleotide-binding; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 37400.7 Length 325 Aromaticity 0.08 Instability index 36.9 Isoelectric point 5.64 Charge (pH=7) -8.3 2D Binding mode Binding energy (Kcal/mol) -6.13 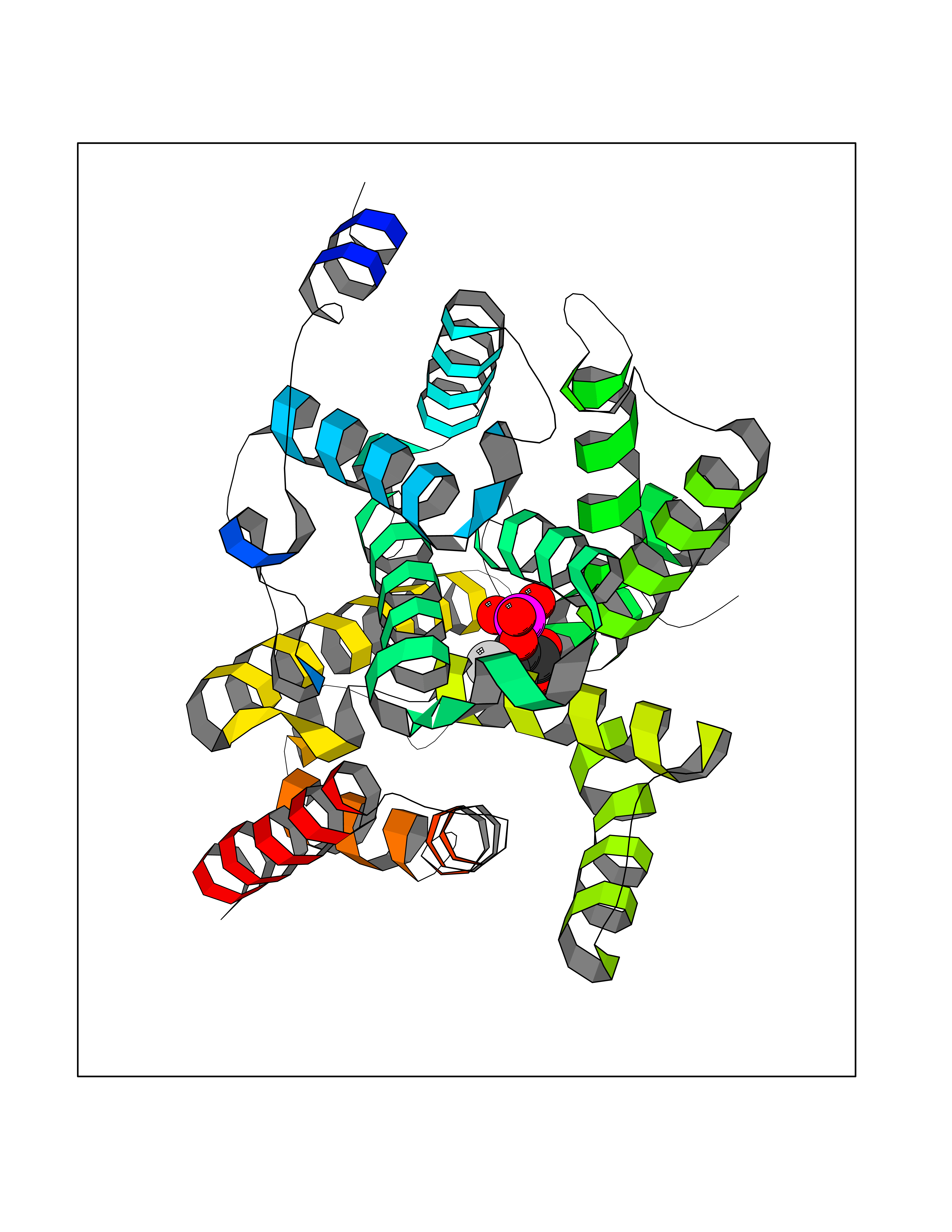 Molscript Map 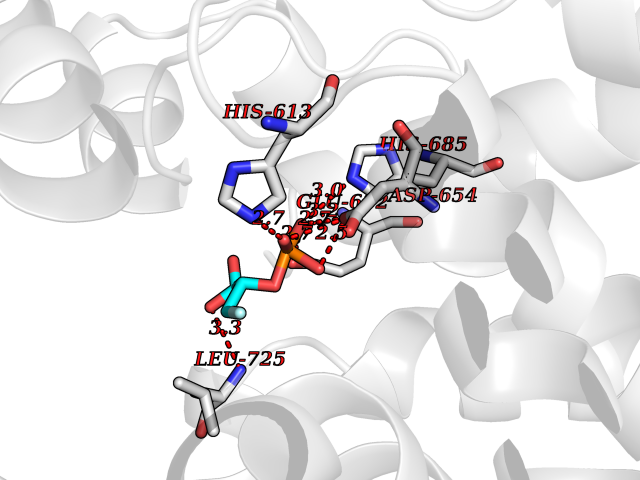 Pymol Map 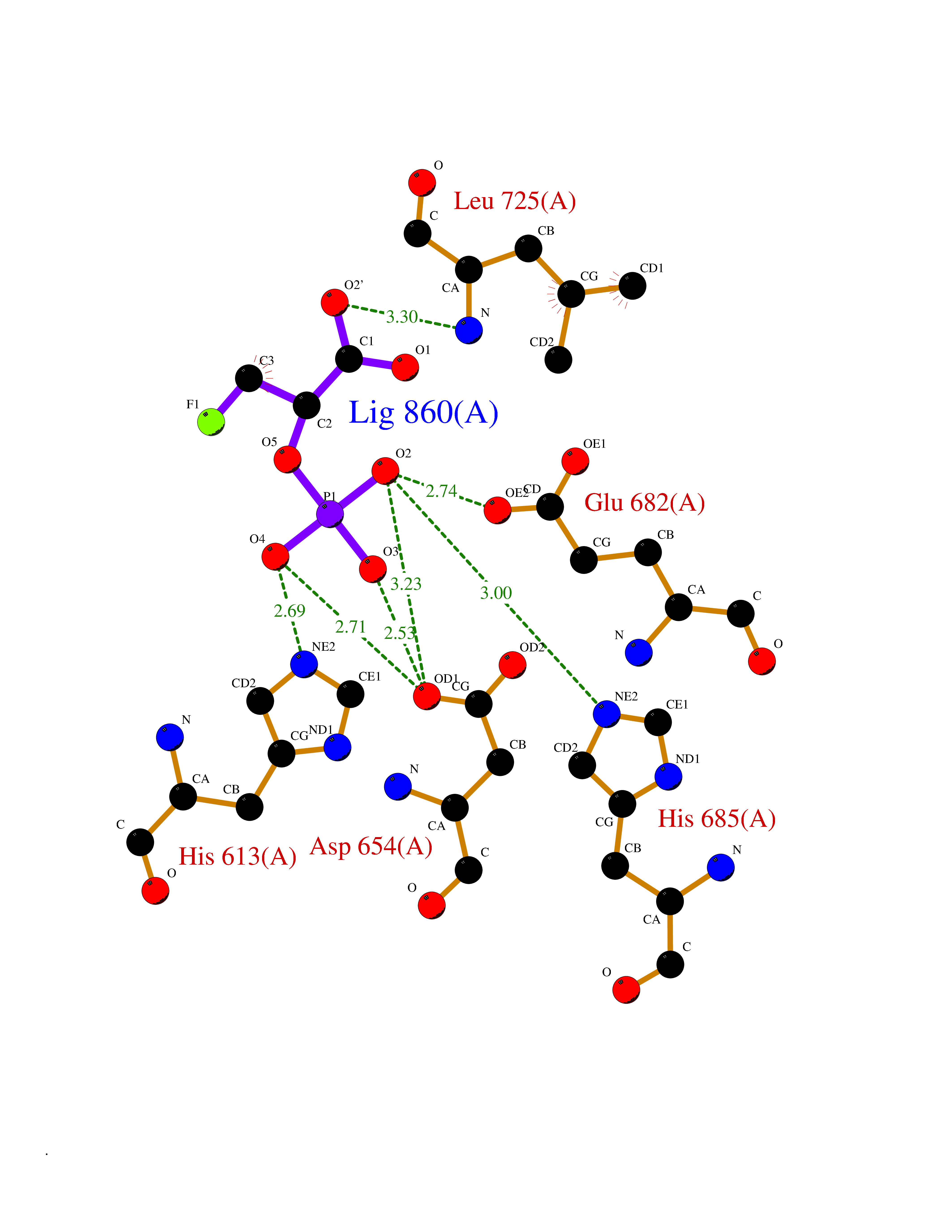 Ligplot Map 3D Binding mode Sequence EEETRELQSLAAAVVPSAQTLKITDFSFSDFELSDLETALCTIRMFTDLNLVQNFQMKHEVLCRWILSVKKNYRKNVAYHNWRHAFNTAQCMFAALKAGKIQNKLTDLEILALLIAALSHDLDHPGVSNQFLINTNSELALMNDESVLEHHHFDQCLMILNSPGNQILSGLSIEEYKTTLKIIKQAILATDLALYIKRRGEFFELIRKNQFNLEDPHQKELFLAMLMTACDLSAITKPWPIQQRIAELVATEFFDQGDRERKELNIEPTDLMNREKKNKIPSMQVGFIDAICLQLYEALTHVSEDCFPLLDGCRKNRQKWQALAE Hydrogen bonds contact Hydrophobic contact | ||||
| 5 | Clostridium histolyticum Collagenase (CH colG) | 7Z5U | 6.59 | |
Target general information Gen name CH colG Organism Hathewaya histolytica (Clostridium histolyticum) Uniprot ID TTD ID Synonyms Microbial collagenase; Gelatinase ColG; Collagenase ColG; Class I collagenase Protein family Peptidase M9B family, Collagenase subfamily Biochemical class NA Function Clostridial collagenases are among the most efficient degraders of eukaryotic collagen known; saprophytes use collagen as a carbon source while pathogens additionally digest collagen to aid in host colonization. Has both tripeptidylcarboxypeptidase on Gly-X-Y and endopeptidase activities; the endopeptidase cuts within the triple helix region of collagen while tripeptidylcarboxypeptidase successively digests the exposed ends, thus clostridial collagenases can digest large sections of collagen. Active on soluble type I collagen, insoluble collagen, azocoll, soluble PZ-peptide (all collagenase substrates) and gelatin. The full-length protein has collagenase activity, while the in vivo derived C-terminally truncated shorter versions only act on gelatin. In vitro digestion of soluble calf skin collagen fibrils requires both ColG and ColH; ColG forms missing the second collagen-binding domain are also synergistic with ColH, although their overall efficiency is decreased. The activator domain (residues 119-388) and catalytic subdomain (389-670) open and close around substrate using a Gly-rich hinge (387-397), allowing digestion when the protein is closed. Binding of collagen requires Ca(2+) and is inhibited by EGTA; the collagen-binding domain (CBD, S3a plus S3b) specifically recognizes the triple-helical conformation made by 3 collagen protein chains in the triple-helical region. Isolated CBD (S3a plus S3b) binds collagen fibrils and sheets of many tissues. Related diseases Intellectual developmental disorder, X-linked, syndromic, Claes-Jensen type (MRXSCJ) [MIM:300534]: A disorder characterized by significantly below average general intellectual functioning associated with impairments in adaptive behavior and manifested during the developmental period. MRXSCJ patients manifest intellectual disability associated with variable features such as slowly progressive spastic paraplegia, seizures, facial dysmorphism. {ECO:0000269|PubMed:15586325, ECO:0000269|PubMed:16538222, ECO:0000269|PubMed:16541399, ECO:0000269|PubMed:17320160, ECO:0000269|PubMed:17468742, ECO:0000269|PubMed:23356856, ECO:0000269|PubMed:25666439}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number EC 3.4.24.3 Uniprot keywords 3D-structure; Calcium; Direct protein sequencing; Hydrolase; Metal-binding; Metalloprotease; Pharmaceutical; Protease; Repeat; Secreted; Signal; Virulence; Zinc; Zymogen Protein physicochemical properties Chain ID A Molecular weight (Da) 44751.2 Length 386 Aromaticity 0.15 Instability index 27.33 Isoelectric point 5.77 Charge (pH=7) -7.33 2D Binding mode Binding energy (Kcal/mol) -6.13 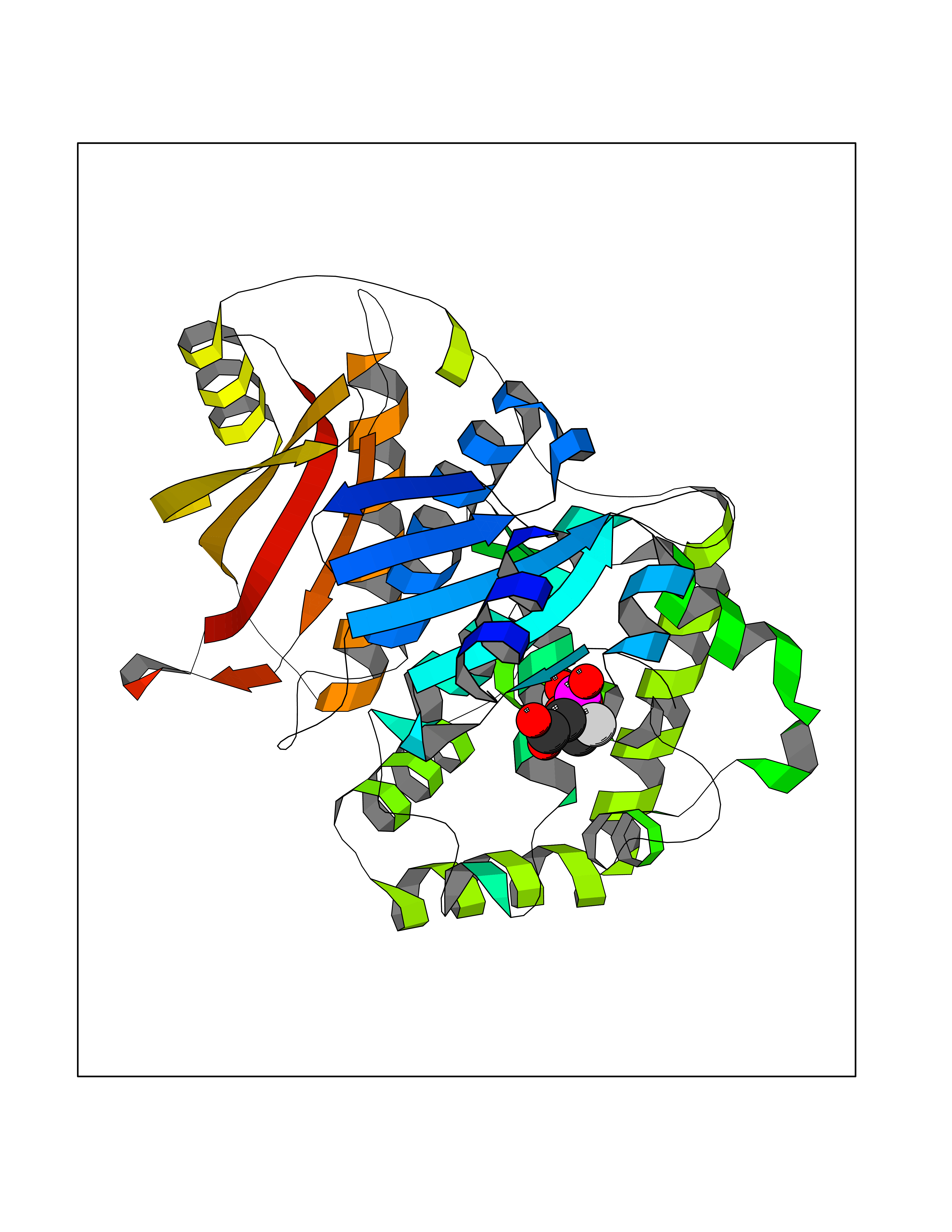 Molscript Map 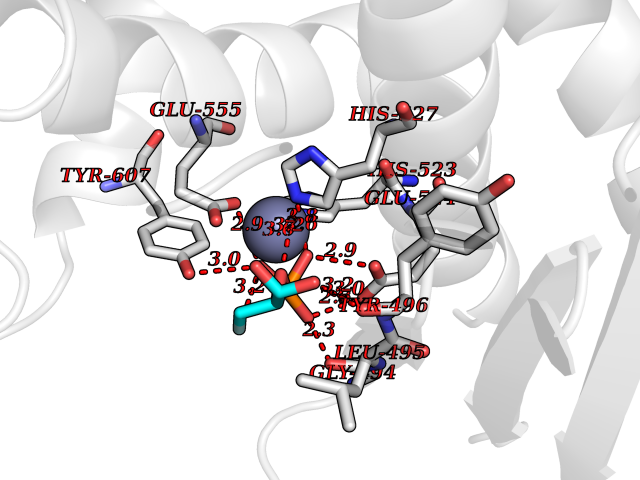 Pymol Map 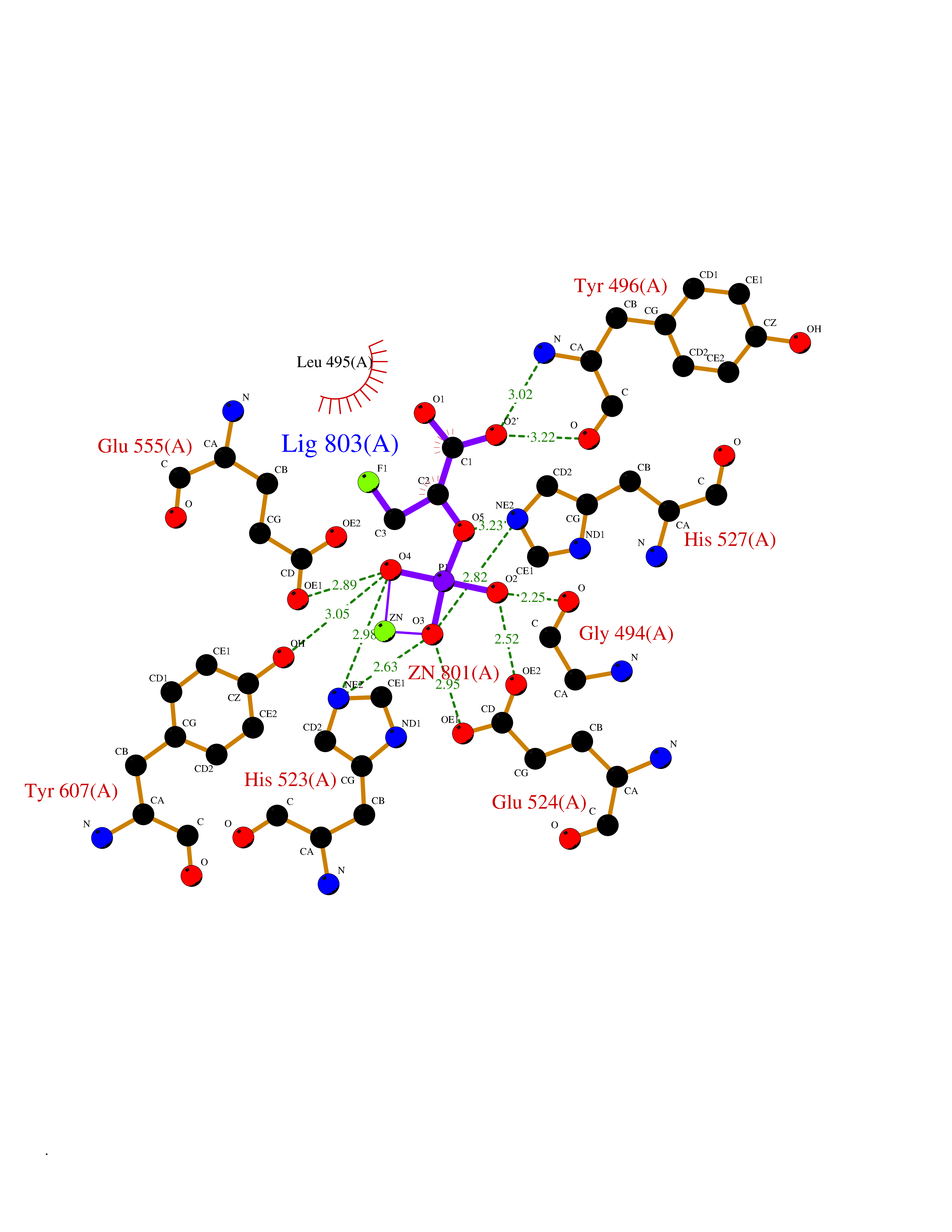 Ligplot Map 3D Binding mode Sequence DHDKFLDDAEKHYLPKTYTFDNGTFIIRAGDKVSEEKIKRLYWASREVKSQFHRVVGNDKALEVGNADDVLTMKIFNSPEEYKFNTTDNGGLYIEPRGTFYTYERTPQQSIFSLEELFRHEYTHYLQARYLVDGLWGQGPFYEKNRLTWFDEGTAEFFAGSTRTSGVLPRKLILGYLAKDKVDHRYSLKKTLNSGYDDSDWMFYNYGFAVAHYLYEKDMPTFIKMNKAILNTDVKSYDEIIKKLSDDANKNTEYQNHIQELVDKYQGAGIPLVSDDYLKDHGYKKASEVYSEISKAASLTNTSVTAEKSQYFNTFTLRGTYTGETSKGEFKDWDEMSKKLDGTLESLAKNSWSGYKTLTAYFTNYRVTSDNKVQYDVVFHGVLTDN Hydrogen bonds contact Hydrophobic contact | ||||
| 6 | Solute carrier family 19 member 1 (SLC19A1) | 8GOF | 6.55 | |
Target general information Gen name SLC19A1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Reduced folate carrier protein; RFC1; RFC; Placental folate transporter; Intestinal folate carrier 1; IFC-1; Folate transporter 1; FOLT; FLOT1 Protein family Reduced folate carrier (RFC) transporter (TC 2.A.48) family Biochemical class NA Function Transporter for the intake of folate. Uptake of folate in human placental choriocarcinoma cells occurs by a novel mechanism called potocytosis which functionally couples three components, namely the folate receptor, the folate transporter, and a V-type H(+)-pump. Related diseases Megaloblastic anemia, folate-responsive (MEGAF) [MIM:601775]: An autosomal recessive metabolic disorder characterized by megaloblastic anemia resulting from decreased folate transport into erythrocytes. Disease manifestations include hemolytic anemia, hyperhomocysteinemia, and low vitamin B12. Serum folate levels are normal, but erythrocyte folate levels are decreased. Treatment with oral folate corrects the anemia and normalizes homocysteine. {ECO:0000269|PubMed:32276275}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Immunodeficiency 114, folate-responsive (IMD114) [MIM:620603]: An autosomal recessive immunologic disorder manifesting in early infancy and characterized by recurrent skin and respiratory infections, mucosal bleeding, oral ulcers, chronic diarrhea, and poor overall growth. Affected individuals have lymphopenia, low serum immunoglobulins, and impaired T cell proliferation. Some patients have global developmental delay. {ECO:0000269|PubMed:36517554, ECO:0000269|PubMed:36745868}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB11256; DB00563; DB00642; DB06813; DB01157 Interacts with Q7Z3Y9 EC number NA Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Antiport; Cell membrane; Disease variant; Folate-binding; Glycoprotein; Hereditary hemolytic anemia; Membrane; Phosphoprotein; Proteomics identification; Reference proteome; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A Molecular weight (Da) 46087.7 Length 407 Aromaticity 0.15 Instability index 34.62 Isoelectric point 9.82 Charge (pH=7) 17.33 2D Binding mode Binding energy (Kcal/mol) -6.38 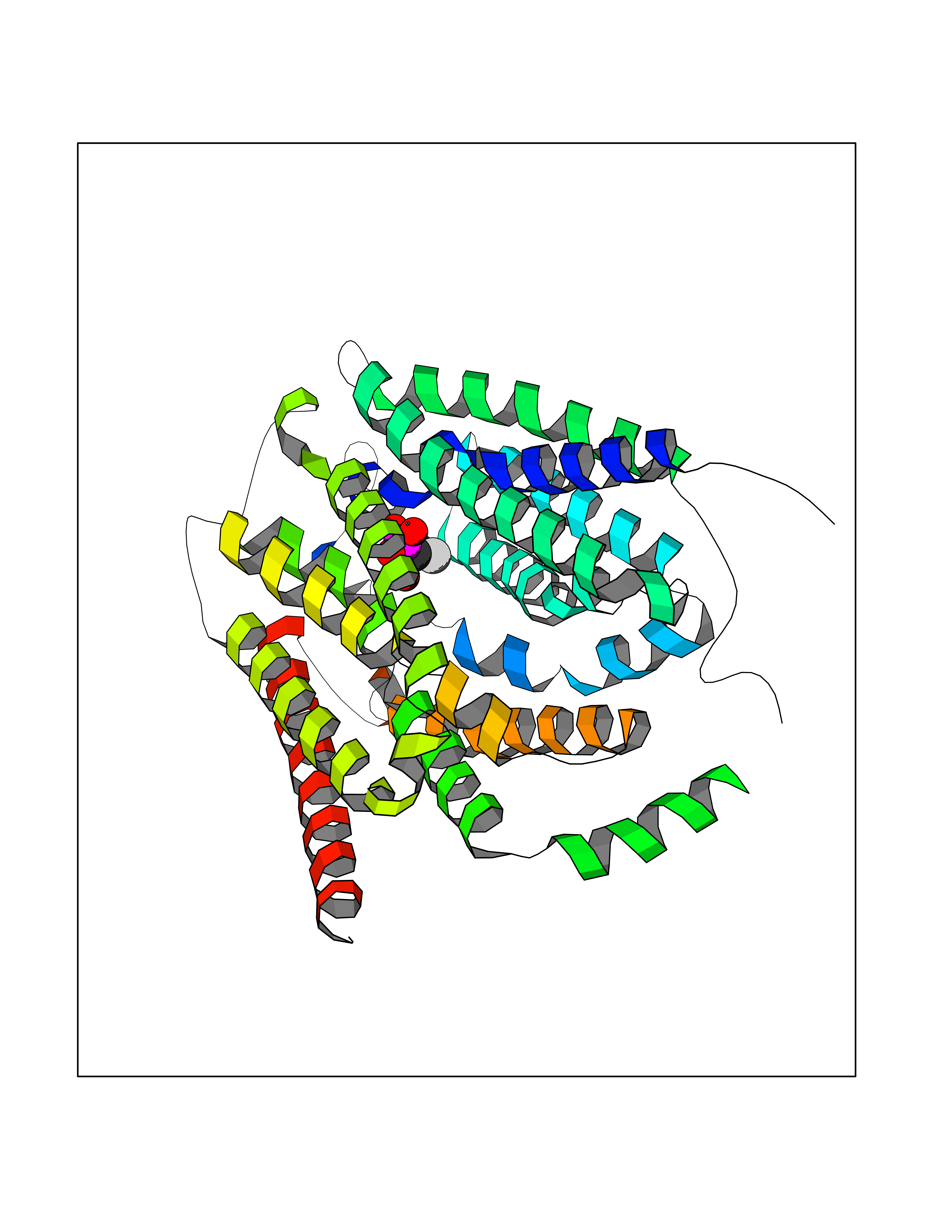 Molscript Map 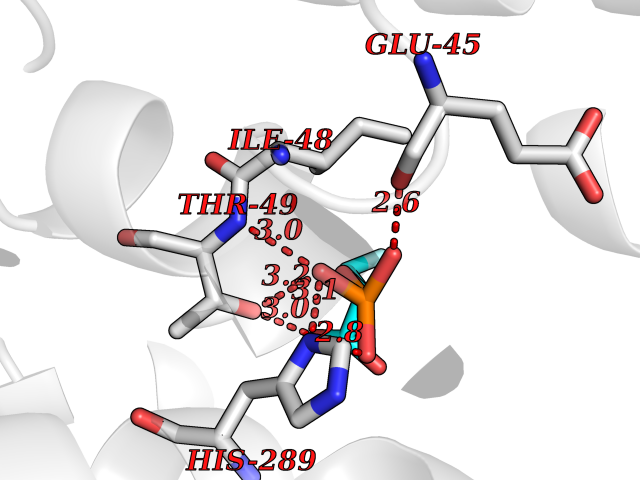 Pymol Map 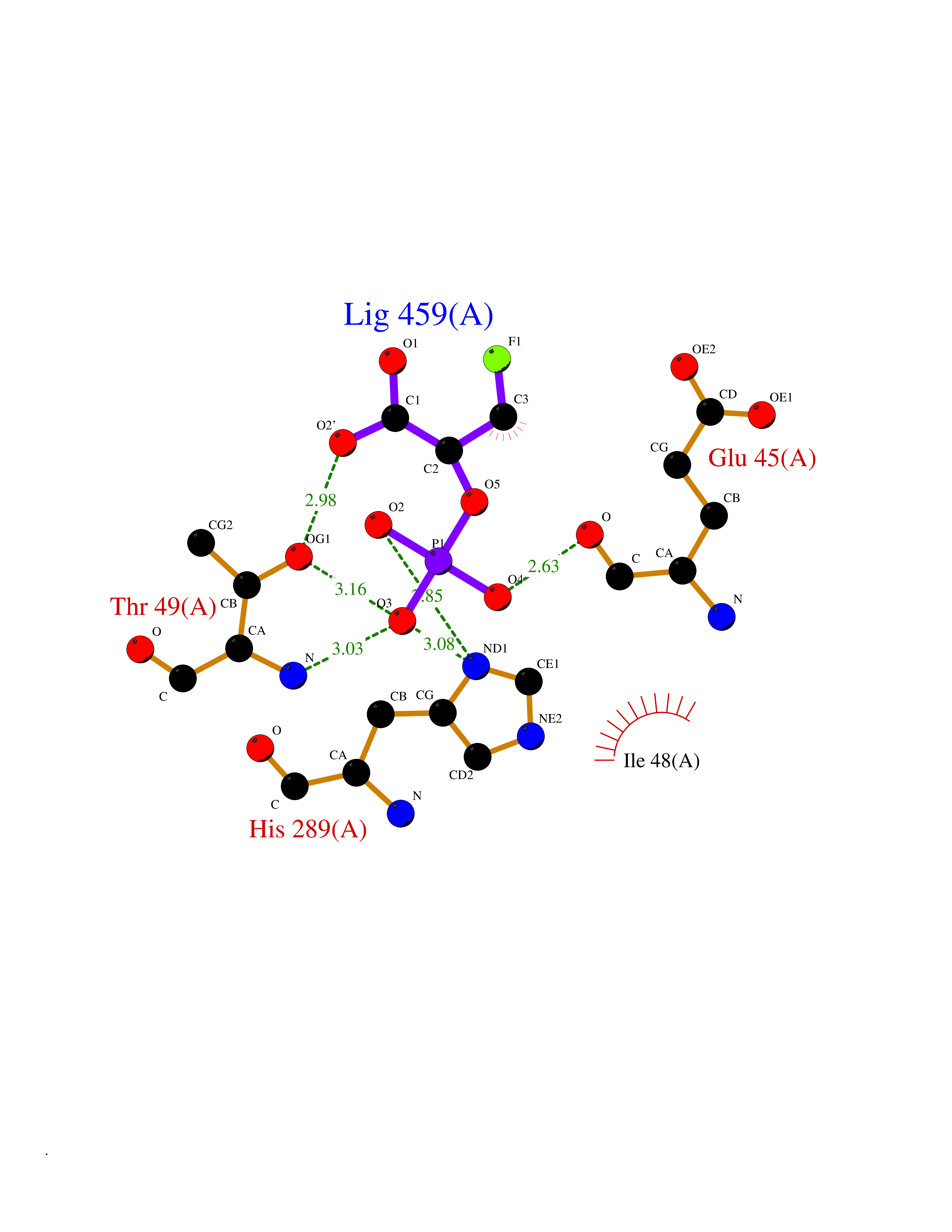 Ligplot Map 3D Binding mode Sequence DPELRSWRHLVCYLCFYGFMAQIRPGESFITPYLLGPDKNFTREQVTNEITPVLSYSYLAVLVPVFLLTDYLRYTPVLLLQGLSFVSVWLLLLLGHSVAHMQLMELFYSVTMAARIAYSSYIFSLVRPARYQRVAGYSRAAVLLGVFTSSVLGQLLVTVGRVSFSTLNYISLAFLTFSVVLALFLKRPKRSLFFNRDDSVLARMLRELGDSLRRPQLRLWSLWWVFNSAGYYLVVYYVHILWNEVDPTTNSARVYNGAADAASTLLGAITSFAAGFVKIRWARWSKLLIAGVTATQAGLVFLLAHTRHPSSIWLCYAAFVLFRGSYQFLVPIATFQIASSLSKELCALVFGVNTFFATIVKTIITFIVSDVRGLGLPVRKQFQLYSVYFLILSIIYFLGAMLDGLRH Hydrogen bonds contact Hydrophobic contact | ||||
| 7 | Pyruvate kinase PKLR | 4IP7 | 6.55 | |
Target general information Gen name PKLR Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms PKL;PK1 Protein family Pyruvate kinase family Biochemical class Transferase Function ATP binding.Kinase activity.Magnesium ion binding.Potassium ion binding.Pyruvate kinase activity. Related diseases Pyruvate kinase hyperactivity (PKHYP) [MIM:102900]: Autosomal dominant phenotype characterized by increase of red blood cell ATP. {ECO:0000269|PubMed:9090535}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Pyruvate kinase deficiency of red cells (PKRD) [MIM:266200]: A frequent cause of hereditary non-spherocytic hemolytic anemia. Clinically, pyruvate kinase-deficient patients suffer from a highly variable degree of chronic hemolysis, ranging from severe neonatal jaundice and fatal anemia at birth, severe transfusion-dependent chronic hemolysis, moderate hemolysis with exacerbation during infection, to a fully compensated hemolysis without apparent anemia. {ECO:0000269|PubMed:10087985, ECO:0000269|PubMed:10772876, ECO:0000269|PubMed:11328279, ECO:0000269|PubMed:11960989, ECO:0000269|PubMed:1536957, ECO:0000269|PubMed:1896471, ECO:0000269|PubMed:19085939, ECO:0000269|PubMed:2018831, ECO:0000269|PubMed:21794208, ECO:0000269|PubMed:7706479, ECO:0000269|PubMed:8161798, ECO:0000269|PubMed:8180378, ECO:0000269|PubMed:8476433, ECO:0000269|PubMed:8481523, ECO:0000269|PubMed:8483951, ECO:0000269|PubMed:8664896, ECO:0000269|PubMed:8807089, ECO:0000269|PubMed:9075576, ECO:0000269|PubMed:9482576, ECO:0000269|PubMed:9827908, ECO:0000269|PubMed:9886305, ECO:0000269|Ref.24}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02726; DB00787; DB04551; DB16236; DB00119 Interacts with Q9UBL6-2 EC number 2.7.1.40 Uniprot keywords 3D-structure; Allosteric enzyme; Alternative splicing; ATP-binding; Disease variant; Glycolysis; Hereditary hemolytic anemia; Kinase; Magnesium; Manganese; Metal-binding; Nucleotide-binding; Phosphoprotein; Potassium; Proteomics identification; Pyruvate; Reference proteome; Transferase Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 45695.1 Length 421 Aromaticity 0.06 Instability index 34.44 Isoelectric point 6.88 Charge (pH=7) -0.35 2D Binding mode Binding energy (Kcal/mol) -5.89 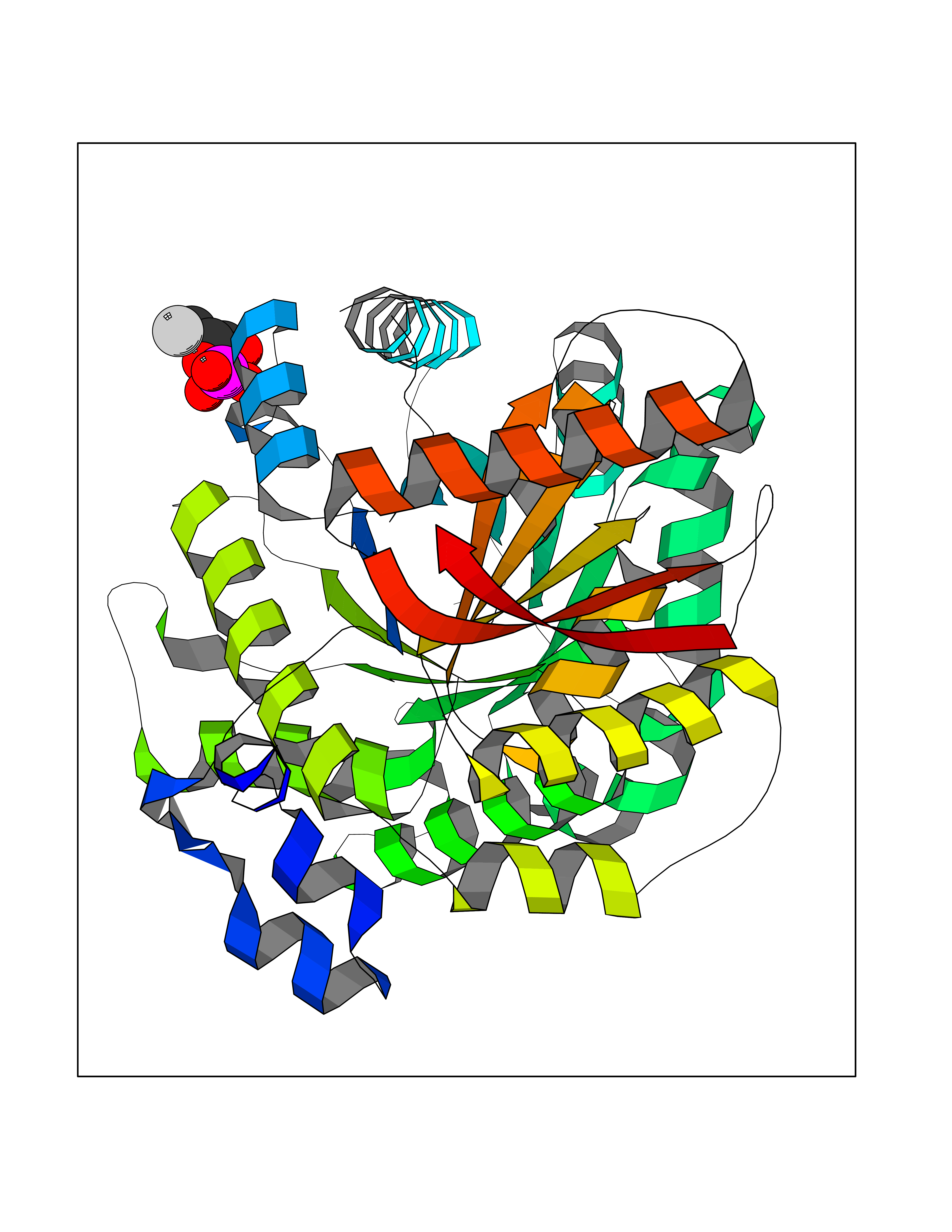 Molscript Map 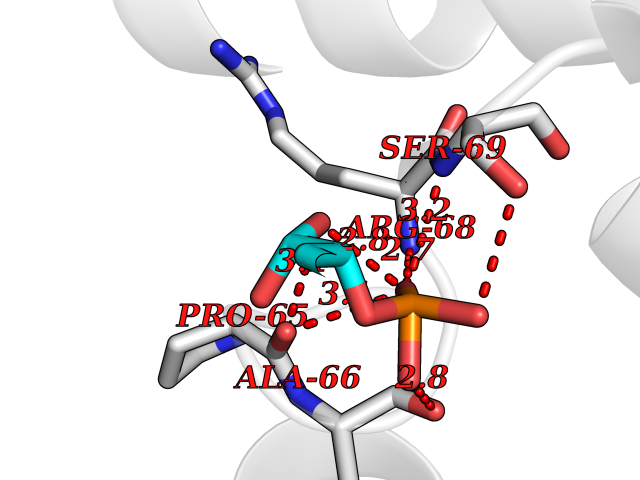 Pymol Map  Ligplot Map 3D Binding mode Sequence GTAFFQQQQLPAAMADTFLEHLCLLDIDSEPVAARSTSIIATIGPASRSVERLKEMIKAGMNIARLNFSHGSHEYHAESIANVREAVESFSPLSYRPVAIALDTKGPEIGLSEQDVRDLRFGVEHGVDIVFASFVRKASDVAAVRAALGPEGHGIKIISKIENHEGVKRFDEILEVSDGIMVARGDLGIEIPAEKVFLAQKMMIGRCNLAGKPVVCATQMLESMITKPRPTRAETSDVANAVLDGADCIMLSGETAKGNFPVEAVKMQHAIAREAEAAVYHRQLFEELRRAAPLSRDPTEVTAIGAVEAAFKCCAAAIIVLTTTGRSAQLLSRYRPRAAVIAVTRSAQAARQVHLCRGVFPLLYREPPEAIWADDVDRRVQFGIESGKLRGFLRVGDLVIVVTGWRPGSGYTNIMRVLSIS Hydrogen bonds contact Hydrophobic contact | ||||
| 8 | Carbonic anhydrase VII (CA-VII) | 3ML5 | 6.55 | |
Target general information Gen name CA7 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Carbonic anhydrase 7; Carbonate dehydratase VII Protein family Alpha-carbonic anhydrase family Biochemical class Alpha-carbonic anhydrase Function Reversible hydration of carbon dioxide. Related diseases Neurodegeneration with brain iron accumulation 8 (NBIA8) [MIM:617917]: A neurodegenerative disorder associated with iron accumulation, primarily in the basal ganglia. Disease onset is in early childhood. Clinical features include speech delay, progressive cerebellar ataxia, unbalanced gait, and loss of ambulation. NBIA8 transmission pattern is consistent with autosomal recessive inheritance. {ECO:0000269|PubMed:29395073}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00819; DB00562; DB00606; DB01144; DB08846; DB00311; DB00774; DB00703; DB00909 Interacts with NA EC number EC 4.2.1.1 Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; Lyase; Metal-binding; Proteomics identification; Reference proteome; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 29393.6 Length 262 Aromaticity 0.11 Instability index 40.14 Isoelectric point 7 Charge (pH=7) -0.01 2D Binding mode Binding energy (Kcal/mol) -6.39 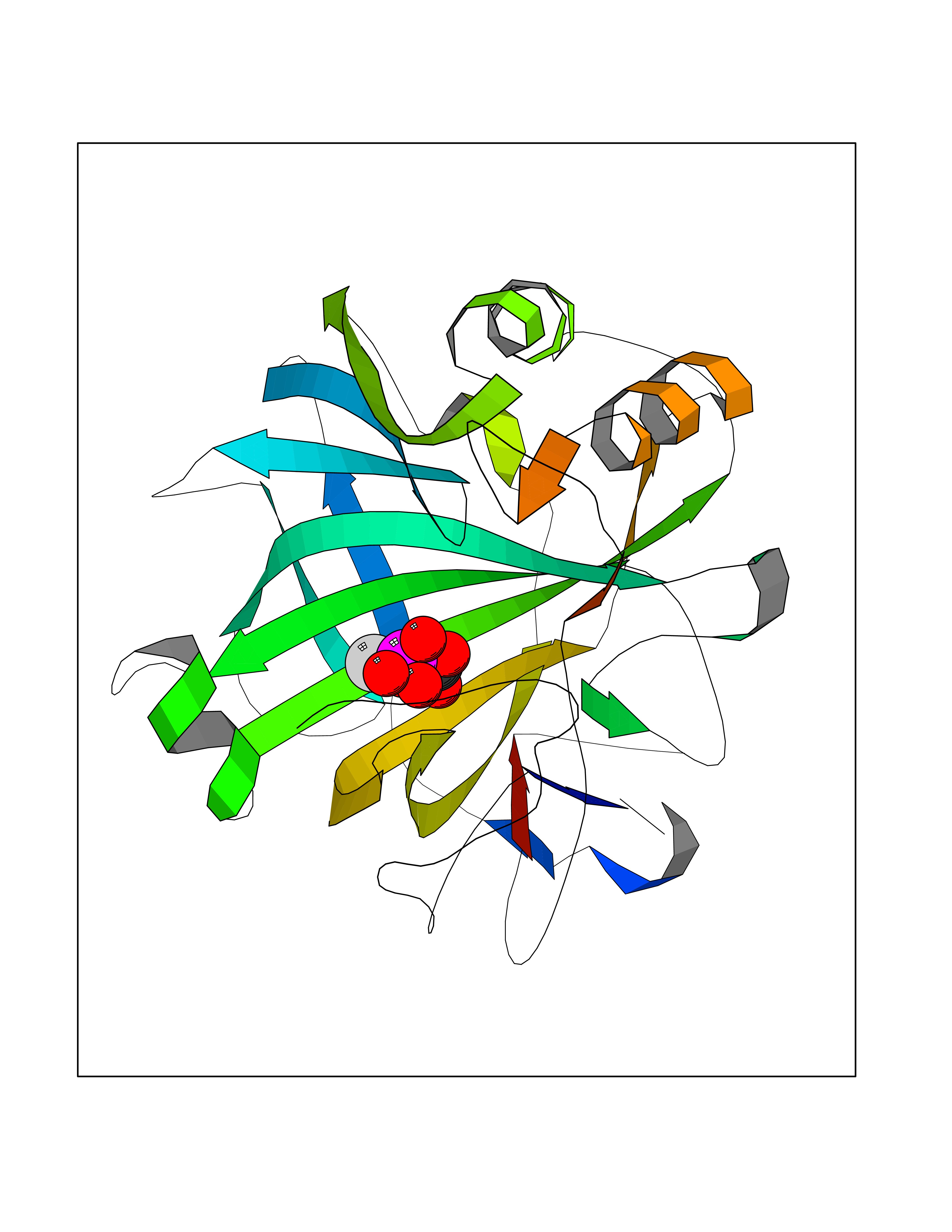 Molscript Map 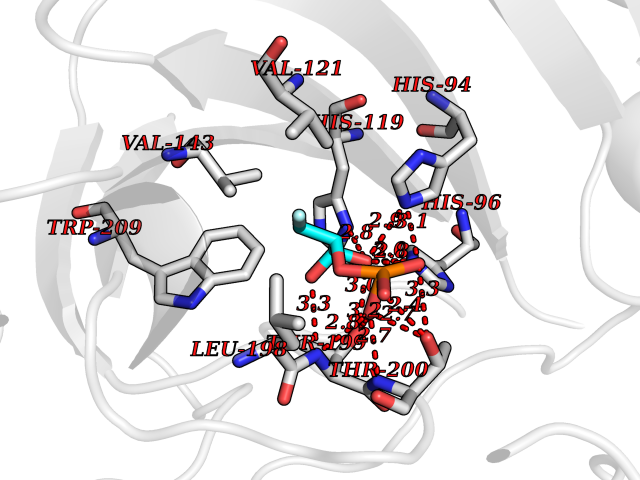 Pymol Map 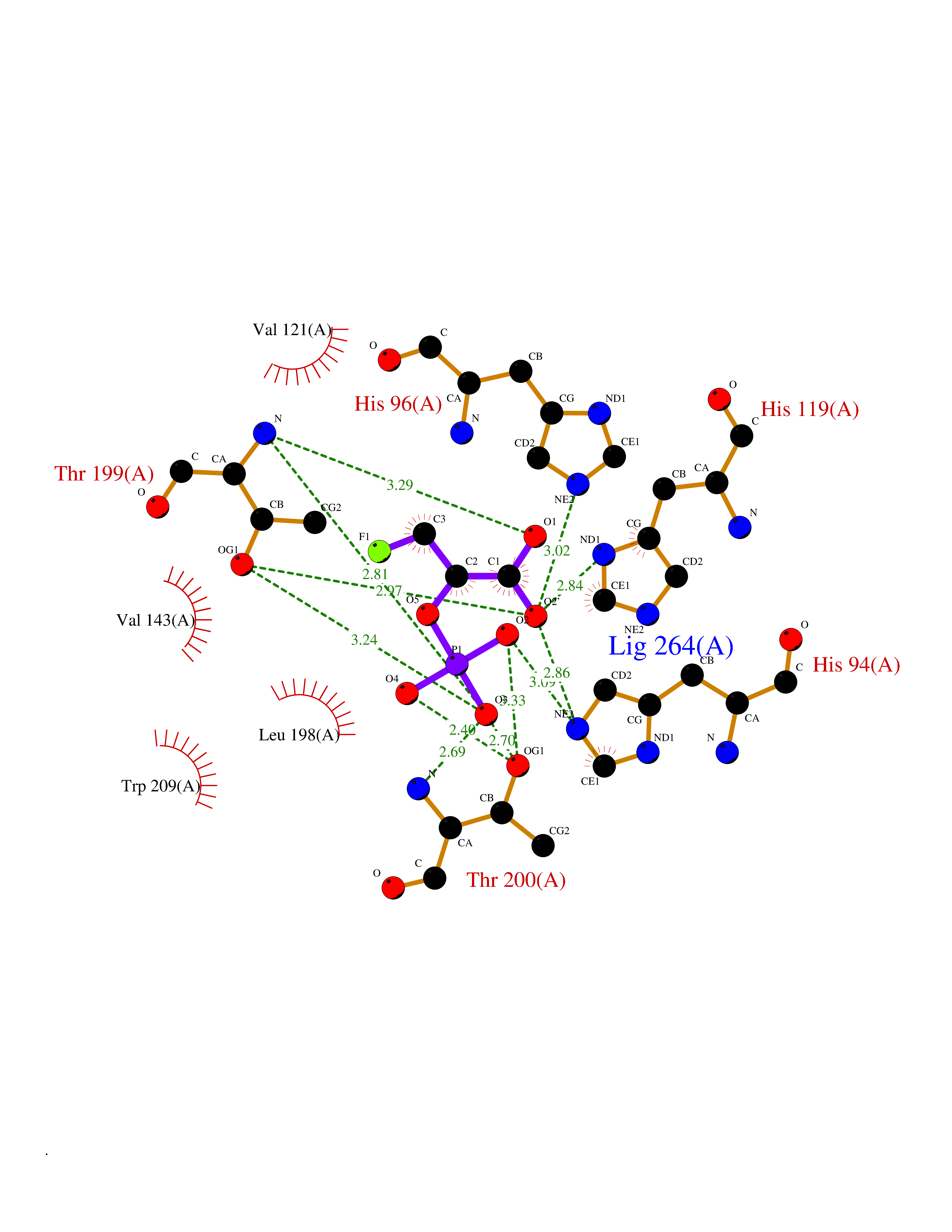 Ligplot Map 3D Binding mode Sequence GHHGWGYGQDDGPSHWHKLYPIAQGDRQSPINIISSQAVYSPSLQPLELSYEACMSLSITNNGHSVQVDFNDSDDRTVVTGGPLEGPYRLKQFHFHWGKKHDVGSEHTVDGKSFPSELHLVHWNAKKYSTFGEAASAPDGLAVVGVFLETGDEHPSMNRLTDALYMVRFKGTKAQFSCFNPKSLLPASRHYWTYPGSLTTPPLSESVTWIVLREPISISERQMGKFRSLLFTSEDDERIHMVNNFRPPQPLKGRVVKASFRA Hydrogen bonds contact Hydrophobic contact | ||||
| 9 | Platelet-derived growth factor receptor alpha (PDGFRA) | 5K5X | 6.54 | |
Target general information Gen name PDGFRA Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms RHEPDGFRA; Platelet-derived growth factor receptor 2; Platelet-derived growth factor alpha receptor; PDGFR2; PDGFR-alpha; PDGFR-2; PDGF-R-alpha; CD140a antigen; CD140a; CD140 antigen-like family membe Protein family Protein kinase superfamily, Tyr protein kinase family, CSF-1/PDGF receptor subfamily Biochemical class Kinase Function Depending on the context, promotes or inhibits cell proliferation and cell migration. Plays an important role in the differentiation of bone marrow-derived mesenchymal stem cells. Required for normal skeleton development and cephalic closure during embryonic development. Required for normal development of the mucosa lining the gastrointestinal tract, and for recruitment of mesenchymal cells and normal development of intestinal villi. Plays a role in cell migration and chemotaxis in wound healing. Plays a role in platelet activation, secretion of agonists from platelet granules, and in thrombin-induced platelet aggregation. Binding of its cognate ligands - homodimeric PDGFA, homodimeric PDGFB, heterodimers formed by PDGFA and PDGFB or homodimeric PDGFC -leads to the activation of several signaling cascades; the response depends on the nature of the bound ligand and is modulated by the formation of heterodimers between PDGFRA and PDGFRB. Phosphorylates PIK3R1, PLCG1, and PTPN11. Activation of PLCG1 leads to the production of the cellular signaling molecules diacylglycerol and inositol 1,4,5-trisphosphate, mobilization of cytosolic Ca(2+) and the activation of protein kinase C. Phosphorylates PIK3R1, the regulatory subunit of phosphatidylinositol 3-kinase, and thereby mediates activation of the AKT1 signaling pathway. Mediates activation of HRAS and of the MAP kinases MAPK1/ERK2 and/or MAPK3/ERK1. Promotes activation of STAT family members STAT1, STAT3 and STAT5A and/or STAT5B. Receptor signaling is down-regulated by protein phosphatases that dephosphorylate the receptor and its down-stream effectors, and by rapid internalization of the activated receptor. Tyrosine-protein kinase that acts as a cell-surface receptor for PDGFA, PDGFB and PDGFC and plays an essential role in the regulation of embryonic development, cell proliferation, survival and chemotaxis. Related diseases A chromosomal aberration involving PDGFRA is found in some cases of hypereosinophilic syndrome. Interstitial chromosomal deletion del(4)(q12q12) causes the fusion of FIP1L1 and PDGFRA (FIP1L1-PDGFRA). Mutations that cause overexpression and/or constitutive activation of PDGFRA may be a cause of hypereosinophilic syndrome. {ECO:0000269|PubMed:12808148}.; DISEASE: Gastrointestinal stromal tumor (GIST) [MIM:606764]: Common mesenchymal neoplasms arising in the gastrointestinal tract, most often in the stomach. They are histologically, immunohistochemically, and genetically different from typical leiomyomas, leiomyosarcomas, and schwannomas. Most GISTs are composed of a fairly uniform population of spindle-shaped cells. Some tumors are dominated by epithelioid cells or contain a mixture of spindle and epithelioid morphologies. Primary GISTs in the gastrointestinal tract commonly metastasize in the omentum and mesenteries, often as multiple nodules. However, primary tumors may also occur outside of the gastrointestinal tract, in other intra-abdominal locations, especially in the omentum and mesentery. {ECO:0000269|PubMed:12522257, ECO:0000269|PubMed:15928335}. The gene represented in this entry may be involved in disease pathogenesis. Mutations causing PDGFRA constitutive activation have been found in gastrointestinal stromal tumors lacking KIT mutations (PubMed:12522257). {ECO:0000269|PubMed:12522257}.; DISEASE: GIST-plus syndrome (GISTPS) [MIM:175510]: A disorder characterized by multiple mesenchymal tumors of the gastrointestinal tract, including gastrointestinal stromal tumor, inflammatory fibroid polyps, and fibroid tumors. Additional features are coarse facies and skin, broad hands and feet, and premature tooth loss. GISTPS is an autosomal dominant disease with incomplete penetrance. Gastrointestinal stromal tumor and inflammatory fibroid polyps may also occur in isolation. {ECO:0000269|PubMed:14699510, ECO:0000269|PubMed:17087943, ECO:0000269|PubMed:25975287}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB12742; DB00102; DB12147; DB10772; DB12010; DB00619; DB09078; DB06595; DB09079; DB06043; DB06589; DB08901; DB08896; DB14840; DB01268; DB11800; DB05146 Interacts with P46108; P46109; P00533; Q8N6L0; P04085; P01127; Q9NRA1; P31947; P62258; Q9NRA1-1; A8T7D5; P05067; Q8IY26 EC number EC 2.7.10.1 Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Cell membrane; Cell projection; Chemotaxis; Developmental protein; Disease variant; Disulfide bond; Glycoprotein; Golgi apparatus; Host-virus interaction; Immunoglobulin domain; Kinase; Membrane; Nucleotide-binding; Phosphoprotein; Proteomics identification; Proto-oncogene; Receptor; Reference proteome; Repeat; Signal; Transferase; Transmembrane; Transmembrane helix; Tyrosine-protein kinase; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 39294.8 Length 345 Aromaticity 0.11 Instability index 46.74 Isoelectric point 6.6 Charge (pH=7) -1.38 2D Binding mode Binding energy (Kcal/mol) -5.98 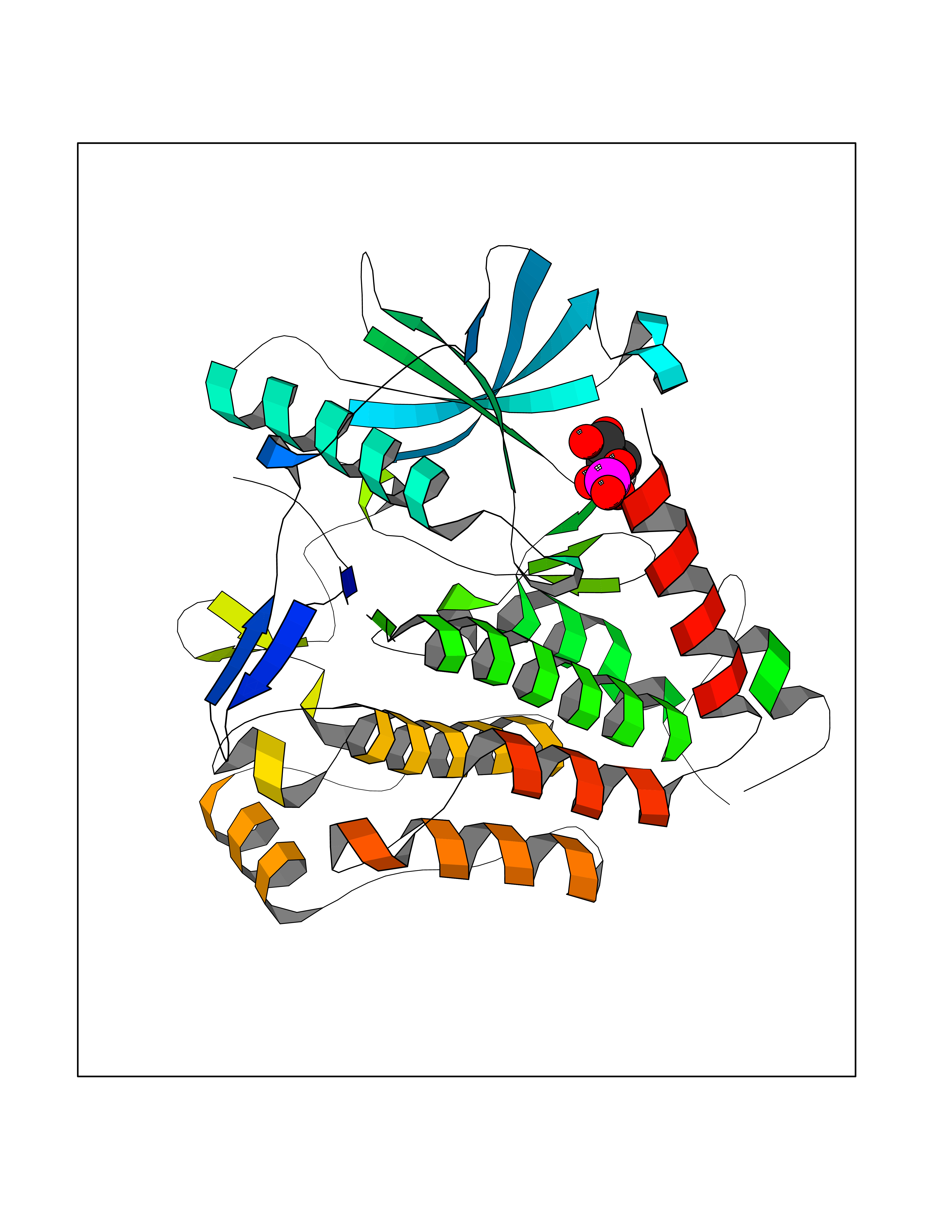 Molscript Map 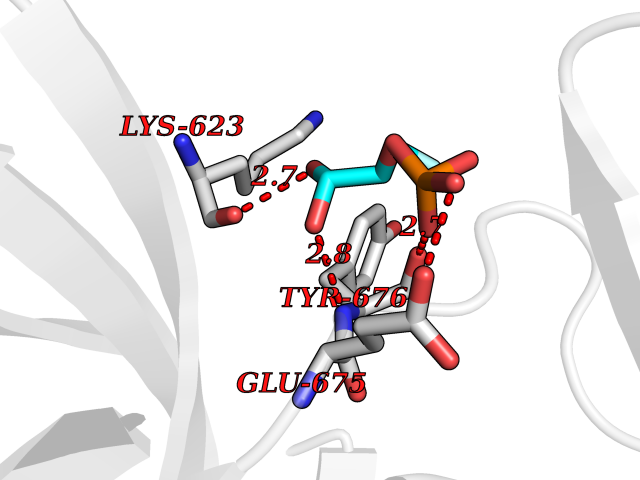 Pymol Map 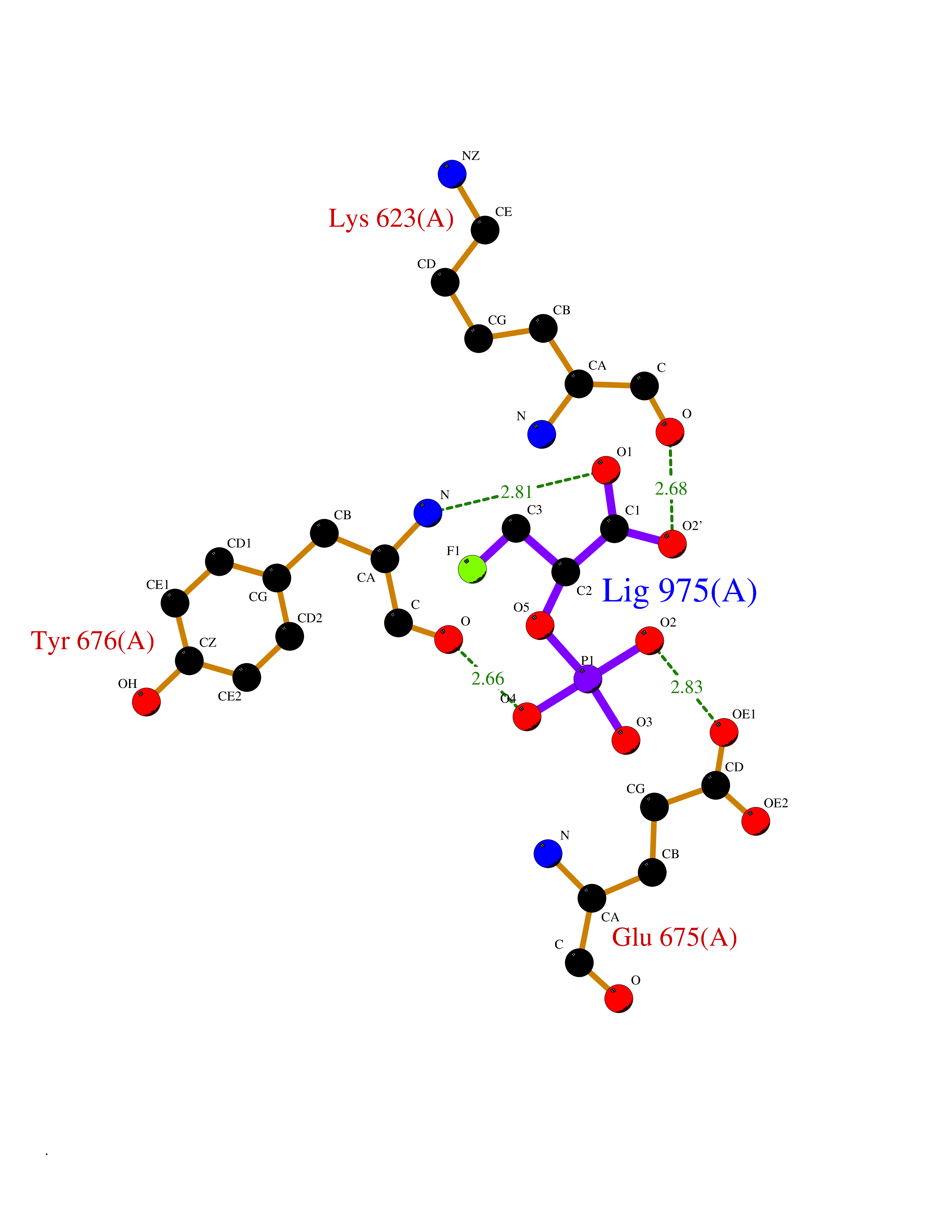 Ligplot Map 3D Binding mode Sequence RYEIRWRVIESISPDGHEYIYVDPMQLPYDSRWEFPRDGLVLGRVLGSGAFGKVVEGTAYGLSRSQPVMKVAVKMLKPTARSSEKQALMSELKIMTHLGPHLNIVNLLGACTKSGPIYIITEYCFYGDLVNYLHKNRDSFLSHSMLDSEVKNLLSDDNSEGLTLLDLLSFTYQVARGMEFLASKNCVHRDLAARNVLLAQGKIVKICDFGLARDIMHDSNYVSKGSTFLPVKWMAPESIFDNLYTTLSDVWSYGILLWEIFSLGGTPYPGMMVDSTFYNKIKSGYRMAKPDHATSEVYEIMVKCWNSEPEKRPSFYHLSEIVENLLPGQYKKSYEKIHLDFLKSD Hydrogen bonds contact Hydrophobic contact | ||||
| 10 | Platelet glycoprotein VI (GP6) | 5OU7 | 6.52 | |
Target general information Gen name GP6 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Glycoprotein 6; GPVI Protein family NA Biochemical class NA Function Collagen receptor involved in collagen-induced platelet adhesion and activation. Plays a key role in platelet procoagulant activity and subsequent thrombin and fibrin formation. This procoagulant function may contribute to arterial and venous thrombus formation. The signaling pathway involves the FcR gamma-chain, the Src kinases (likely FYN or LYN) and SYK, the adapter protein LAT and leads to the activation of PLCG2. Related diseases Bleeding disorder, platelet-type, 11 (BDPLT11) [MIM:614201]: A mild to moderate bleeding disorder caused by defective platelet activation and aggregation in response to collagen. {ECO:0000269|PubMed:19549989, ECO:0000269|PubMed:19552682}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P06241; P07948; P06241; P07948 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Blood coagulation; Cell membrane; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; Hemostasis; Immunoglobulin domain; Membrane; Proteomics identification; Receptor; Reference proteome; Repeat; Signal; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 19027.4 Length 173 Aromaticity 0.11 Instability index 37.14 Isoelectric point 8.68 Charge (pH=7) 2.52 2D Binding mode Binding energy (Kcal/mol) -5.94 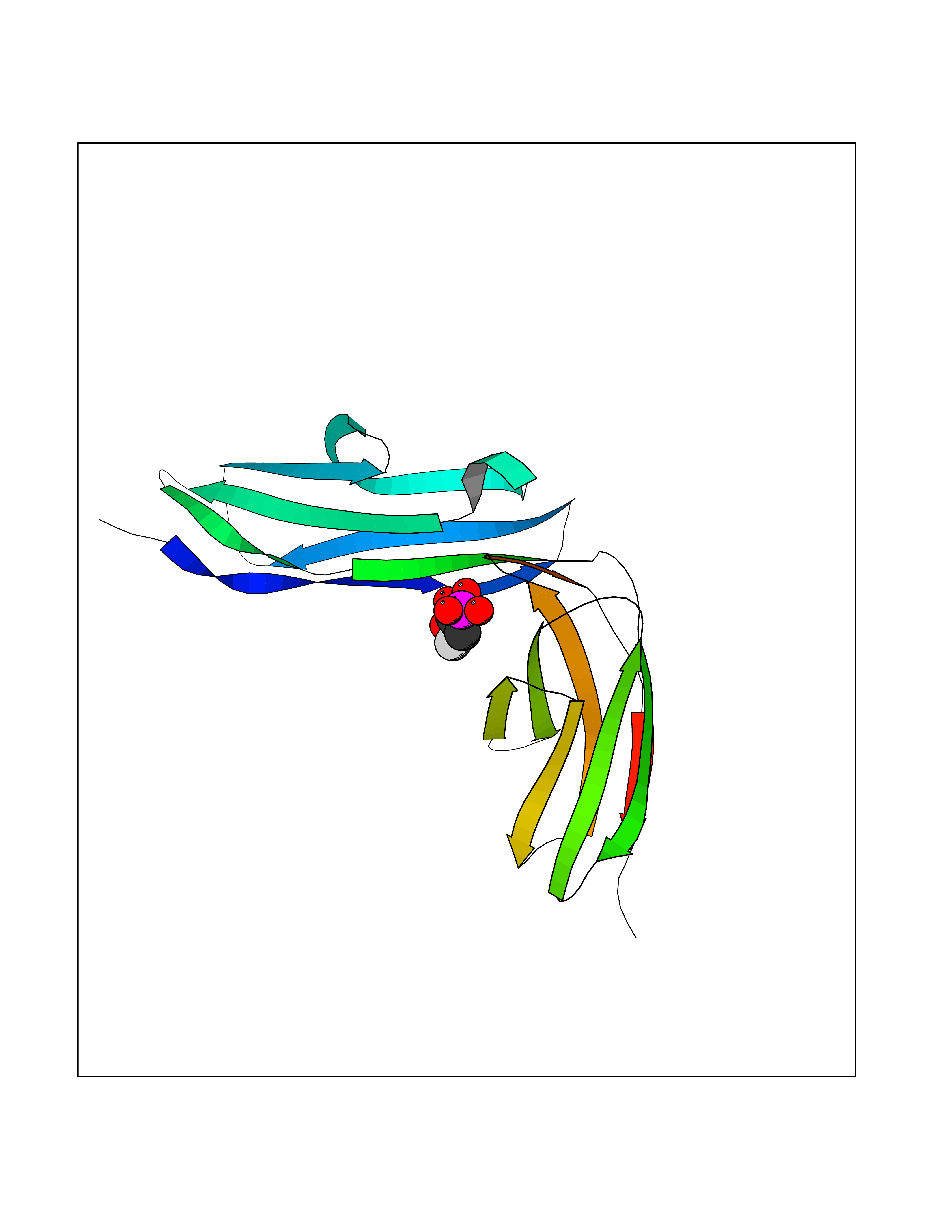 Molscript Map 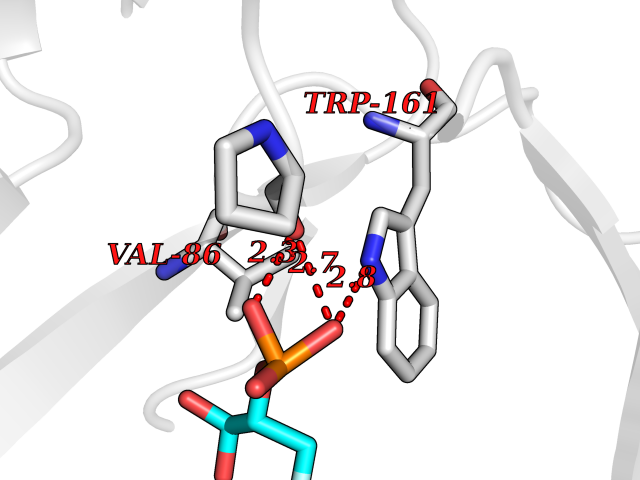 Pymol Map 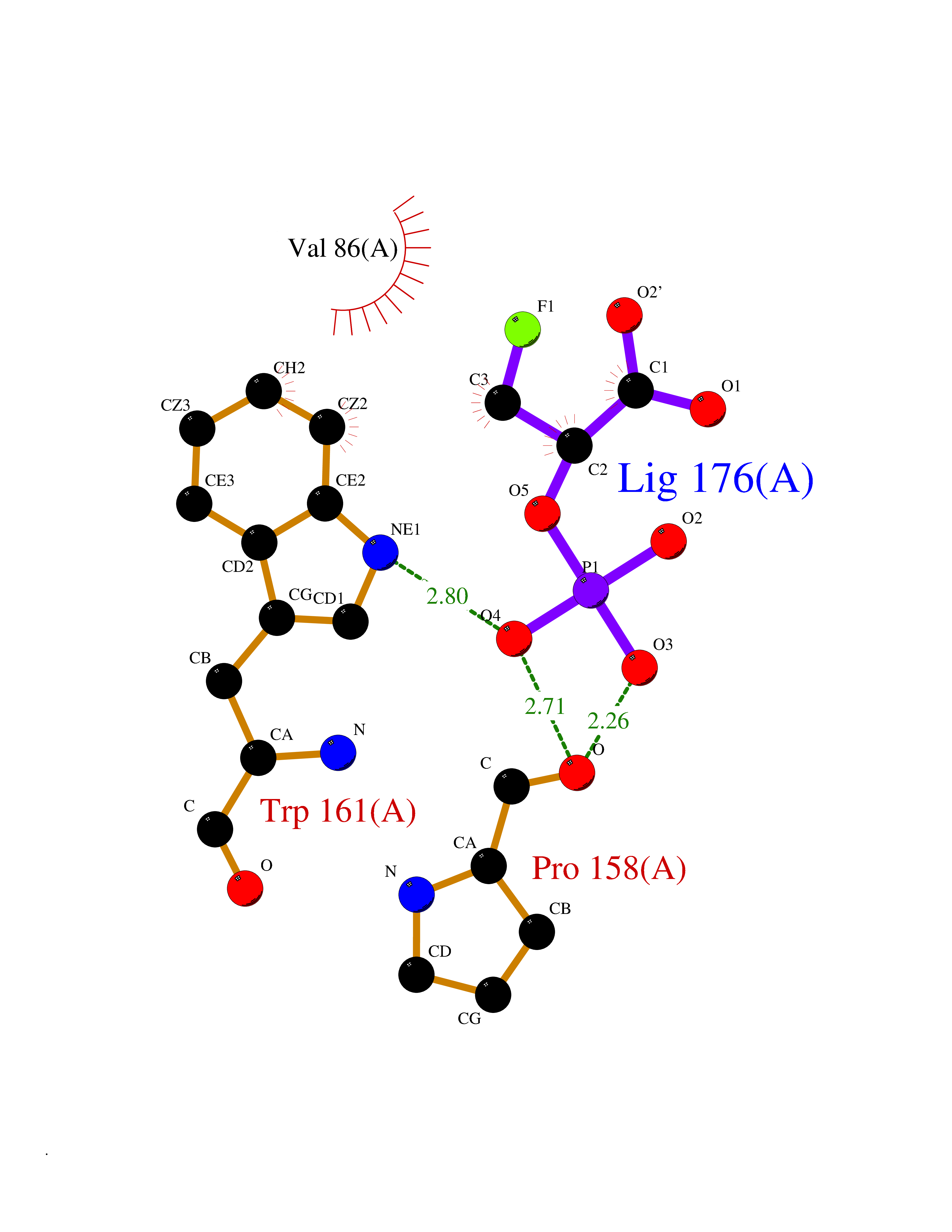 Ligplot Map 3D Binding mode Sequence SGPLPKPSLQALPSSLVPLEKPVTLRCQGPPGVDLYRLEKLSSSRYQDQAVLFIPAMKRSLAGRYRCSYQNGSLWSLPSDQLELVATGVFAKPSLSAQPGSGGDVTLQCQTRYGFDQFALYKEGDPERWYRASFPIITVTAAHSGTYRCYSFSSRDPYLWSAPSDPLELVVTG Hydrogen bonds contact Hydrophobic contact | ||||
| 11 | Gastrin (GAST) | 5WRJ | 6.46 | |
Target general information Gen name GAST Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Gastrin6; GAST; G52; G34; G14 Protein family Gastrin/cholecystokinin family Biochemical class Gastrin cholecystokinin Function Gastrin stimulates the stomach mucosa to produce and secrete hydrochloric acid and the pancreas to secrete its digestive enzymes. It also stimulates smooth muscle contraction and increases blood circulation and water secretion in the stomach and intestine. Related diseases Orotic aciduria 1 (ORAC1) [MIM:258900]: A disorder of pyrimidine metabolism resulting in megaloblastic anemia and orotic acid crystalluria that is frequently associated with some degree of physical and intellectual disability. A minority of cases have additional features, particularly congenital malformations and immune deficiencies. {ECO:0000269|PubMed:9042911}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB12532 Interacts with Q13520; O43315; Q12797-6; Q9BXK5; Q8N5K1; Q96BA8; P00387; Q9Y282; Q5JX71; Q8NBJ4; Q8TDT2; P43628; O76011; Q6ZUX7; Q15546; P15941-11; Q13113; P60201-2; Q14973; P02787; Q4KMG9 EC number NA Uniprot keywords 3D-structure; Amidation; Cleavage on pair of basic residues; Direct protein sequencing; Hormone; Phosphoprotein; Proteomics identification; Pyrrolidone carboxylic acid; Reference proteome; Secreted; Signal; Sulfation Protein physicochemical properties Chain ID A,F Molecular weight (Da) 31827.9 Length 280 Aromaticity 0.08 Instability index 41.06 Isoelectric point 8.84 Charge (pH=7) 5.56 2D Binding mode Binding energy (Kcal/mol) -6.65 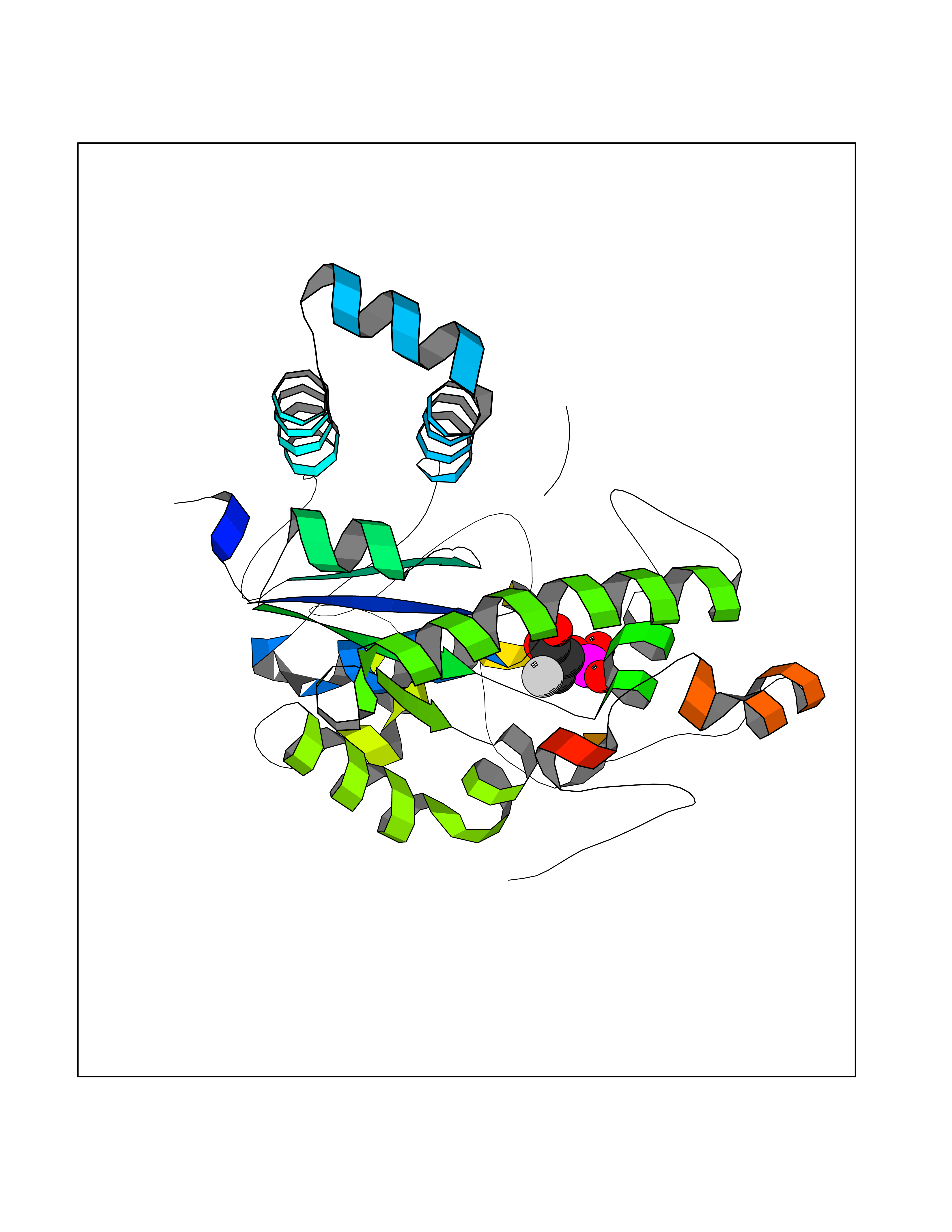 Molscript Map 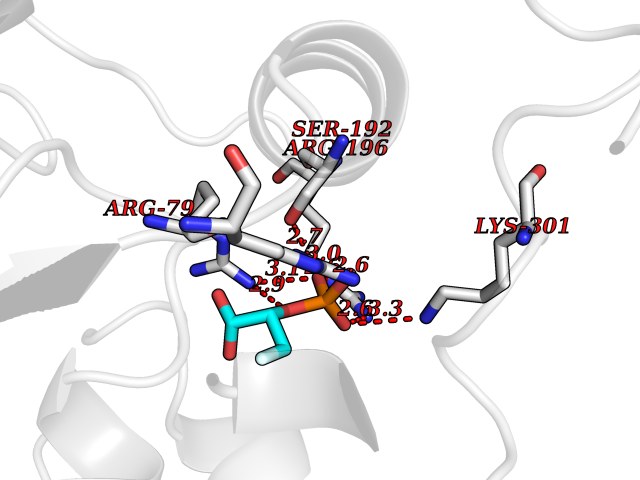 Pymol Map 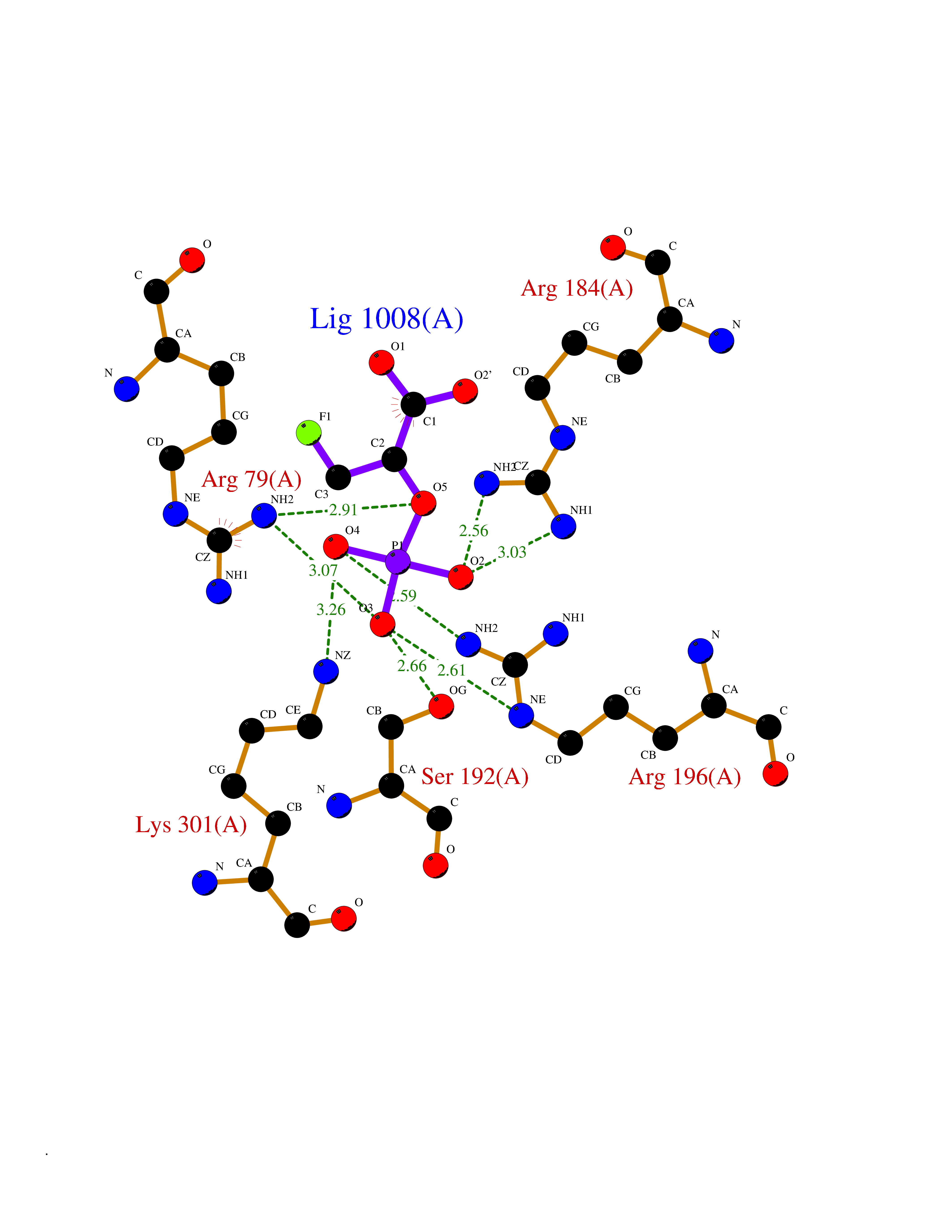 Ligplot Map 3D Binding mode Sequence AYHKDMPLIFIGGVPRSGTTLMRAMLDAHPDIRCGEETRVIPRILALKQMWSRSSKEKIRLDEAGVTDEVLDSAMQAFLLEIIVKHGEPAPYLCNKDPFALKSLTYLSRLFPNAKFLLMVRDGRASVHSMISRKVTIAGFDLNSYRDCLTKWNRAIETMYNQCMEVGYKKCMLVHYEQLVLHPERWMRTLLKFLQIPWNHSVLHHEEMIGKAGGVSLSKVERSTDQVIKPVNVGALSKWVGKIPPDVLQDMAVIAPMLAKLGYDPYANPPNYGKPEEEAY Hydrogen bonds contact Hydrophobic contact | ||||
| 12 | Aspartate carbamoyltransferase (CAD) | 4C6E | 6.42 | |
Target general information Gen name CAD Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms CAD Protein family CarA family; CarB family; Metallo-dependent hydrolases superfamily, DHOase family, CAD subfamily; Aspartate/ornithine carbamoyltransferase superfamily, ATCase family Biochemical class Carbon-nitrogen ligase Function This protein is a "fusion" protein encoding four enzymatic activities of the pyrimidine pathway (GATase, CPSase, ATCase and DHOase). Related diseases Developmental and epileptic encephalopathy 50 (DEE50) [MIM:616457]: A form of epileptic encephalopathy, a heterogeneous group of severe early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE50 is an autosomal recessive, progressive disease with onset in infancy and favorable response to treatment with oral uridine. {ECO:0000269|PubMed:25678555, ECO:0000269|PubMed:28087732}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00128; DB00130; DB03459 Interacts with P27708; Q8N137; P63104 EC number NA Uniprot keywords 3D-structure; Acetylation; Allosteric enzyme; ATP-binding; Congenital disorder of glycosylation; Cytoplasm; Direct protein sequencing; Disease variant; Epilepsy; Hydrolase; Ligase; Magnesium; Manganese; Metal-binding; Multifunctional enzyme; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Pyrimidine biosynthesis; Reference proteome; Repeat; Transferase; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 38268.4 Length 351 Aromaticity 0.06 Instability index 41.29 Isoelectric point 5.86 Charge (pH=7) -10.56 2D Binding mode Binding energy (Kcal/mol) -6.64 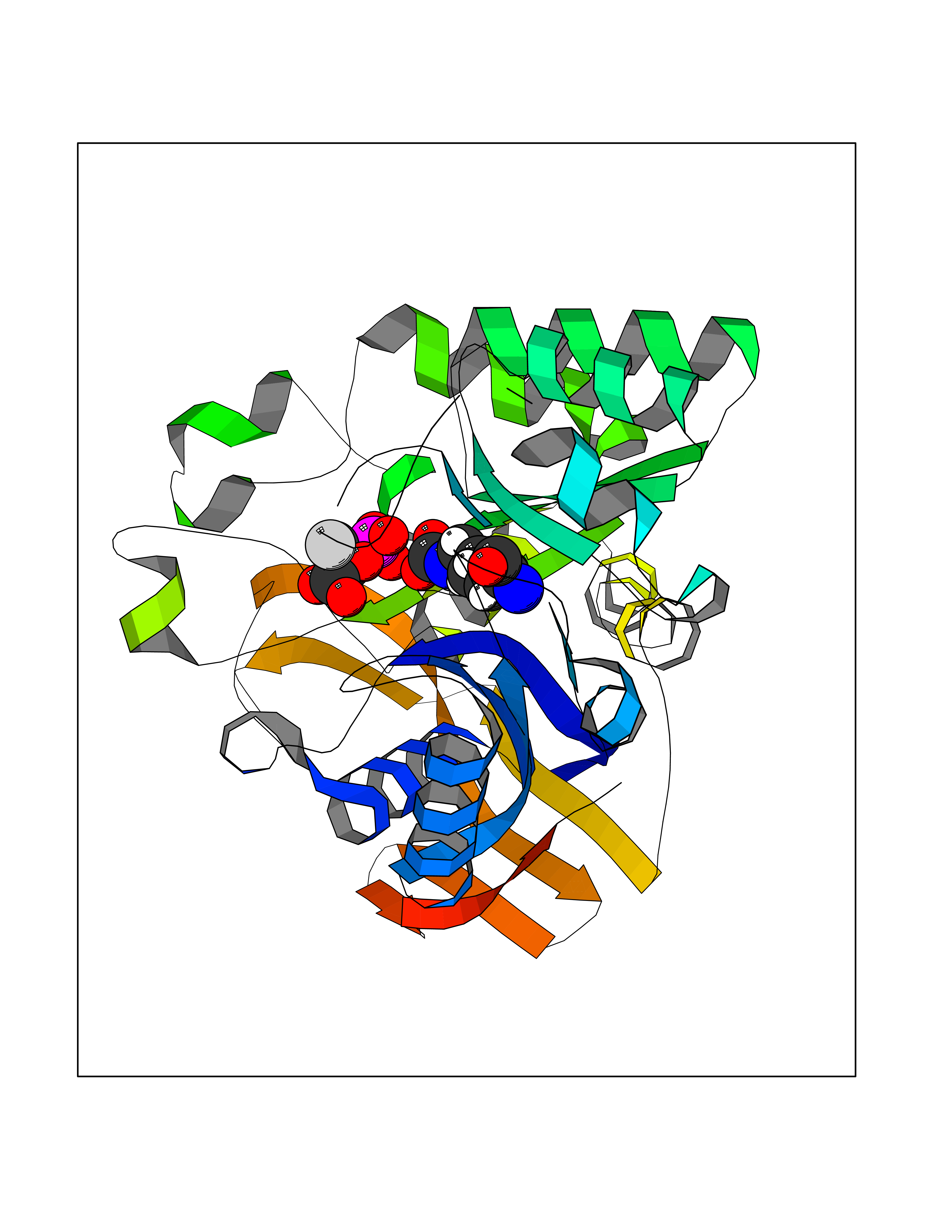 Molscript Map 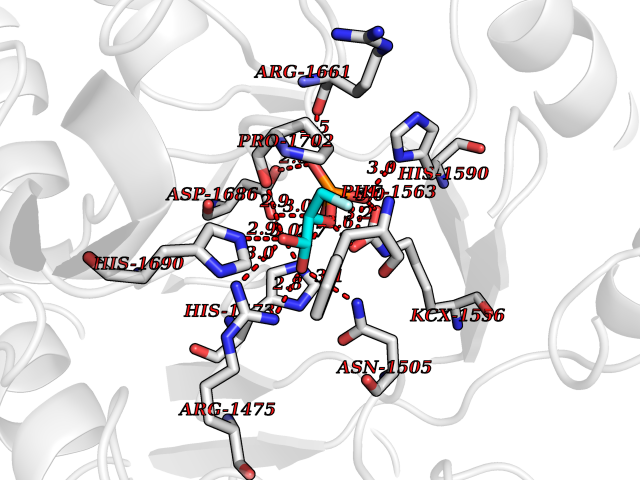 Pymol Map 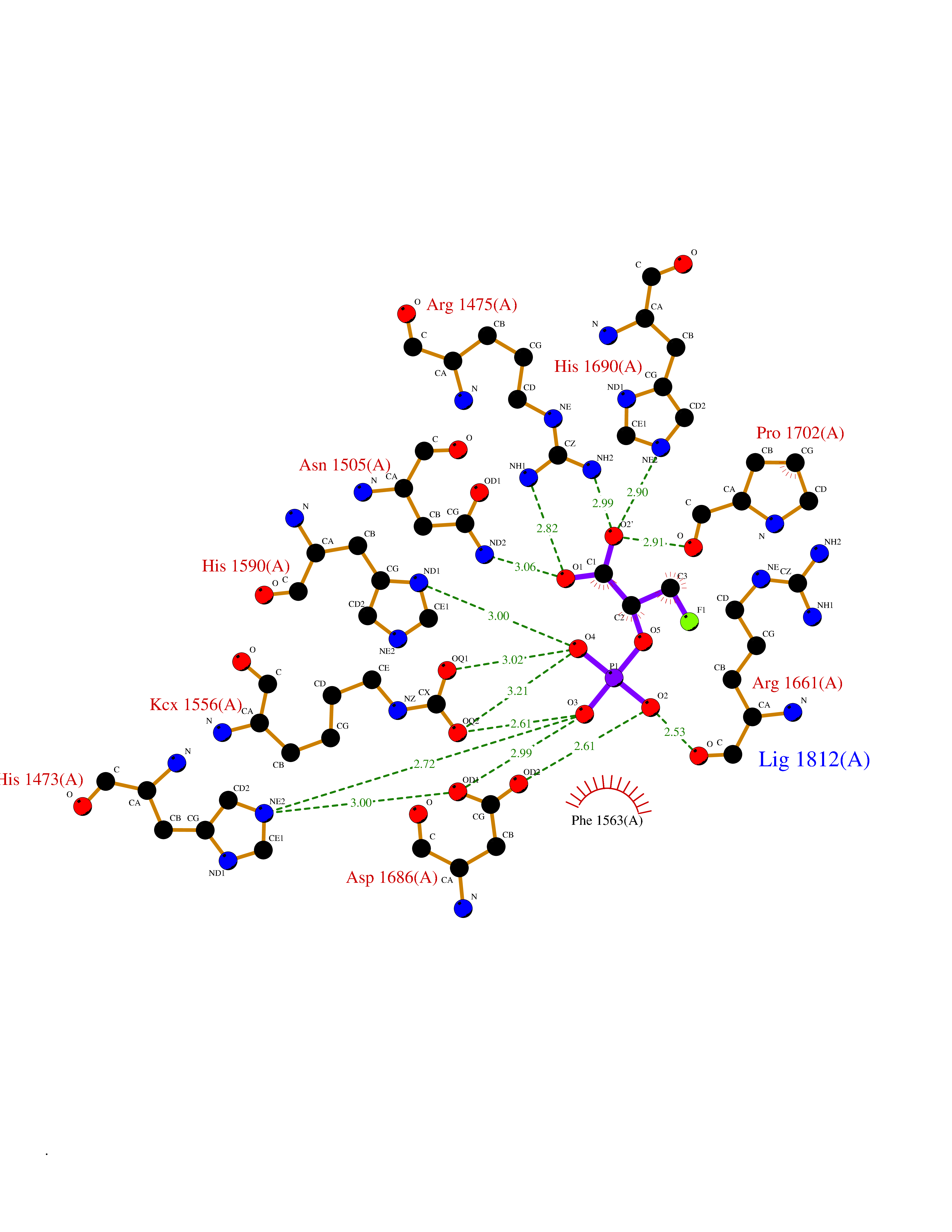 Ligplot Map 3D Binding mode Sequence KLVRLPGLIDVHVHLREPGGTHKEDFASGTAAALAGGITMVCAMPNTRPPIIDAPALALAQKLAEAGARCDFALFLGASSENAGTLGTVAGSAAGLXLYLNETFSELRLDSVVQWMEHFETWPSHLPIVAHAEQQTVAAVLMVAQLTQRSVHICHVARKEEILLIKAAKARGLPVTCEVAPHHLFLSHDDLERLGPGKGEVRPELGSRQDVEALWENMAVIDCFASDHAPHTLEEKCGSRPPPGFPGLETMLPLLLTAVSEGRLSLDDLLQRLHHNPRRIFHLPPQEDTYVEVDLEHEWTIPSHMPFSKAHWTPFEGQKVKGTVRRVVLRGEVAYIDGQVLVPPGYGQDVR Hydrogen bonds contact Hydrophobic contact | ||||
| 13 | Fatty acid synthase (FASN) | 3TJM | 6.41 | |
Target general information Gen name FASN Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Yeast fatty acid synthase; Fatty-acyl-CoA synthase; Fatty acyl-CoA synthetase enzyme; FAS Protein family NA Biochemical class Acyltransferase Function Fatty acid synthetase catalyzes the formation of long-chain fatty acids from acetyl-CoA, malonyl-CoA and NADPH. This multifunctional protein has 7 catalytic activities as an acyl carrier protein. Related diseases Glycine encephalopathy 2 (GCE2) [MIM:620398]: A form of glycine encephalopathy, a metabolic disorder characterized by a high concentration of glycine in the body fluids. Affected individuals typically have severe neurological symptoms, including seizure, lethargy, and muscular hypotonia soon after birth. Most of them die within the neonatal period. Atypical cases have later disease onset and less severely affected psychomotor development. {ECO:0000269|PubMed:10873393, ECO:0000269|PubMed:11286506, ECO:0000269|PubMed:16051266, ECO:0000269|PubMed:26371980, ECO:0000269|PubMed:28244183, ECO:0000269|PubMed:8005589, ECO:0000269|PubMed:9600239, ECO:0000269|PubMed:9621520}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01034; DB01083 Interacts with Q15848; Q16665; P42858; Q8IV20; Q8TBB1; PRO_0000045603 [Q99IB8] EC number EC 2.3.1.85 Uniprot keywords 3D-structure; Acetylation; Cytoplasm; Direct protein sequencing; Fatty acid biosynthesis; Fatty acid metabolism; Hydrolase; Isopeptide bond; Lipid biosynthesis; Lipid metabolism; Lyase; Multifunctional enzyme; NAD; NADP; Oxidoreductase; Phosphopantetheine; Phosphoprotein; Proteomics identification; Pyridoxal phosphate; Reference proteome; S-nitrosylation; Transferase; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 30174.9 Length 275 Aromaticity 0.09 Instability index 43.28 Isoelectric point 5.92 Charge (pH=7) -5.4 2D Binding mode Binding energy (Kcal/mol) -6.37 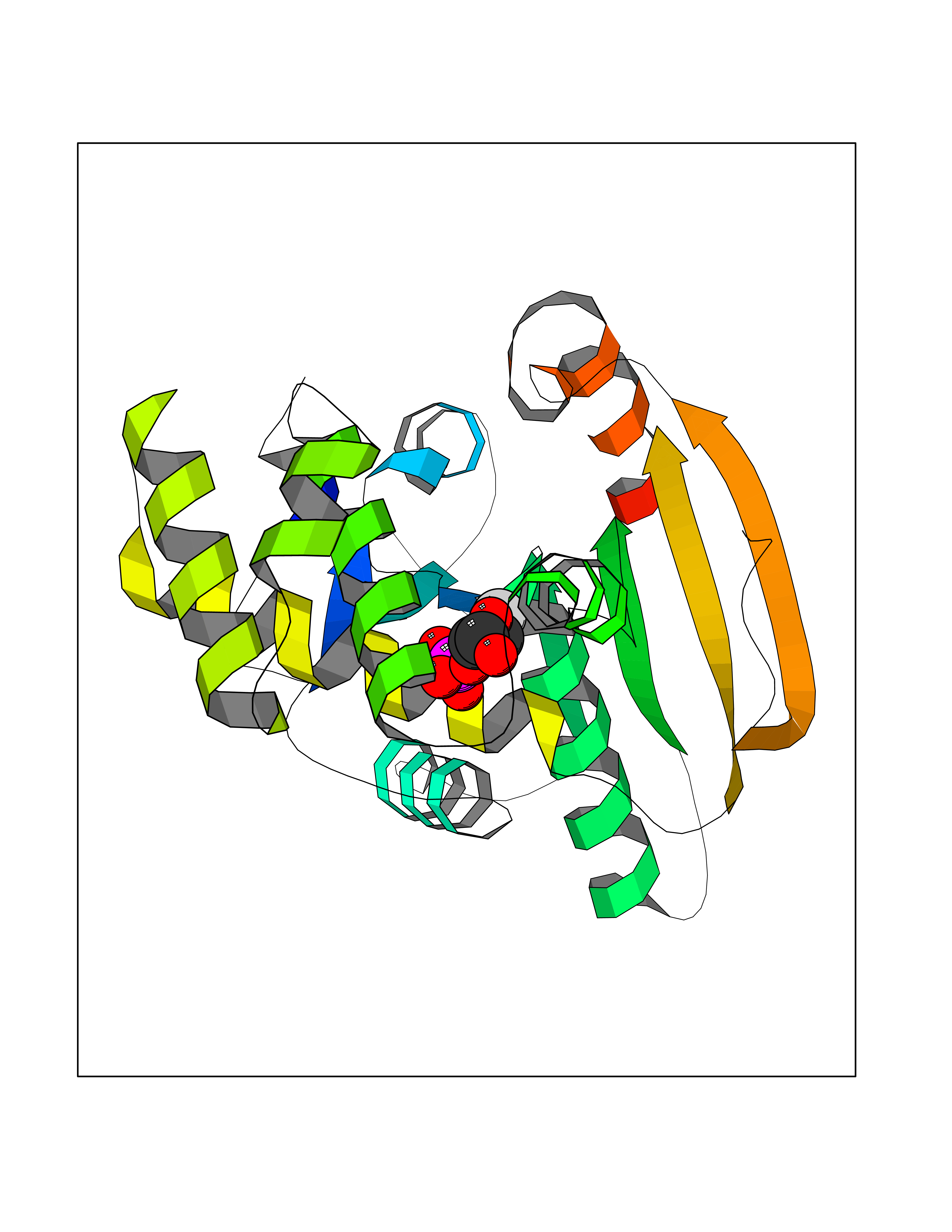 Molscript Map 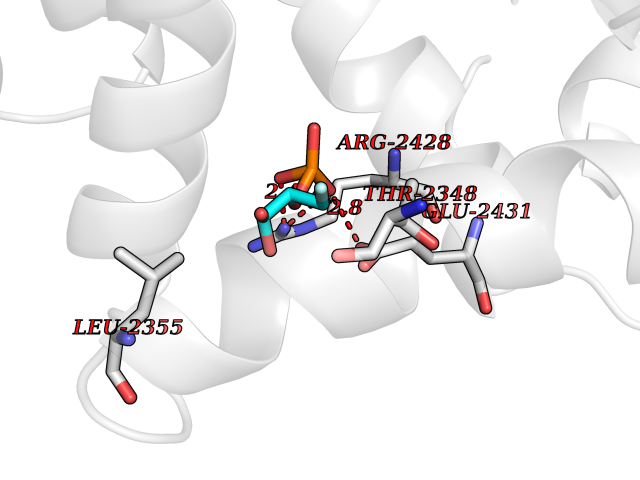 Pymol Map 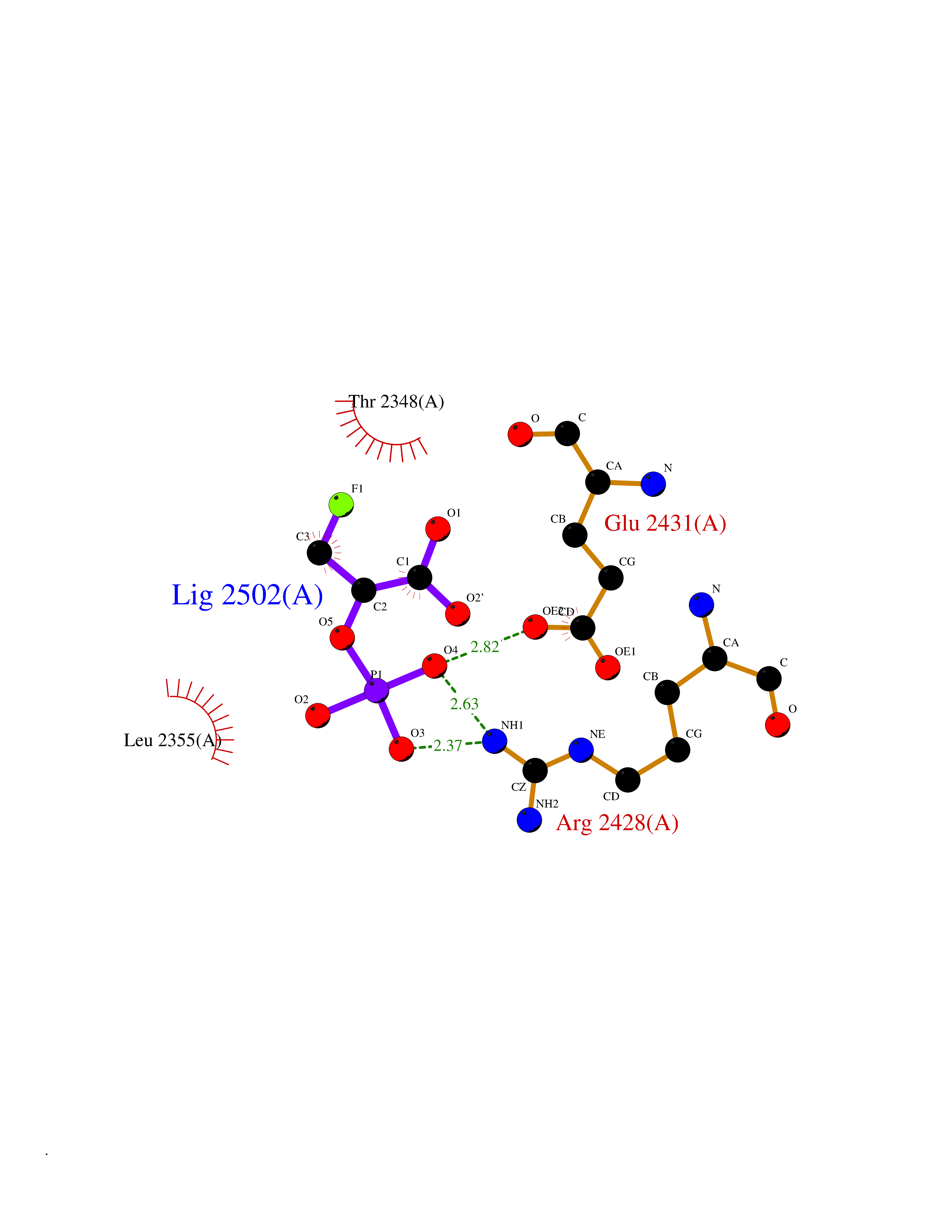 Ligplot Map 3D Binding mode Sequence NLRSLLVNPEGPTLMRLNSVQSSERPLFLVHPIEGSTTVFHSLASRLSIPTYGLQCTRAAPLDSIHSLAAYYIDCIRQVQPEGPYRVAGYSYGACVAFEMCSQLQAQQSPAPTHNSLFLFDGSPTYVLAYTGSYRAKLTPGCEAEAETEAICFFVQQFTDMEHNRVLEALLPLKGLEERVAAAVDLIIKSHQGLDRQELSFAARSFYYKLRAAEQYTPKAKYHGNVMLLRAAAGADYNLSQVCDGKVSVHVIEGDHATLLEGSGLESIISIIHSS Hydrogen bonds contact Hydrophobic contact | ||||
| 14 | Pyruvate kinase PKLR | 4IP7 | 6.39 | |
Target general information Gen name PKLR Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms PKL;PK1 Protein family Pyruvate kinase family Biochemical class Transferase Function ATP binding.Kinase activity.Magnesium ion binding.Potassium ion binding.Pyruvate kinase activity. Related diseases Pyruvate kinase hyperactivity (PKHYP) [MIM:102900]: Autosomal dominant phenotype characterized by increase of red blood cell ATP. {ECO:0000269|PubMed:9090535}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Pyruvate kinase deficiency of red cells (PKRD) [MIM:266200]: A frequent cause of hereditary non-spherocytic hemolytic anemia. Clinically, pyruvate kinase-deficient patients suffer from a highly variable degree of chronic hemolysis, ranging from severe neonatal jaundice and fatal anemia at birth, severe transfusion-dependent chronic hemolysis, moderate hemolysis with exacerbation during infection, to a fully compensated hemolysis without apparent anemia. {ECO:0000269|PubMed:10087985, ECO:0000269|PubMed:10772876, ECO:0000269|PubMed:11328279, ECO:0000269|PubMed:11960989, ECO:0000269|PubMed:1536957, ECO:0000269|PubMed:1896471, ECO:0000269|PubMed:19085939, ECO:0000269|PubMed:2018831, ECO:0000269|PubMed:21794208, ECO:0000269|PubMed:7706479, ECO:0000269|PubMed:8161798, ECO:0000269|PubMed:8180378, ECO:0000269|PubMed:8476433, ECO:0000269|PubMed:8481523, ECO:0000269|PubMed:8483951, ECO:0000269|PubMed:8664896, ECO:0000269|PubMed:8807089, ECO:0000269|PubMed:9075576, ECO:0000269|PubMed:9482576, ECO:0000269|PubMed:9827908, ECO:0000269|PubMed:9886305, ECO:0000269|Ref.24}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02726; DB00787; DB04551; DB16236; DB00119 Interacts with Q9UBL6-2 EC number 2.7.1.40 Uniprot keywords 3D-structure; Allosteric enzyme; Alternative splicing; ATP-binding; Disease variant; Glycolysis; Hereditary hemolytic anemia; Kinase; Magnesium; Manganese; Metal-binding; Nucleotide-binding; Phosphoprotein; Potassium; Proteomics identification; Pyruvate; Reference proteome; Transferase Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 45695.1 Length 421 Aromaticity 0.06 Instability index 34.44 Isoelectric point 6.88 Charge (pH=7) -0.35 2D Binding mode Binding energy (Kcal/mol) -6.62 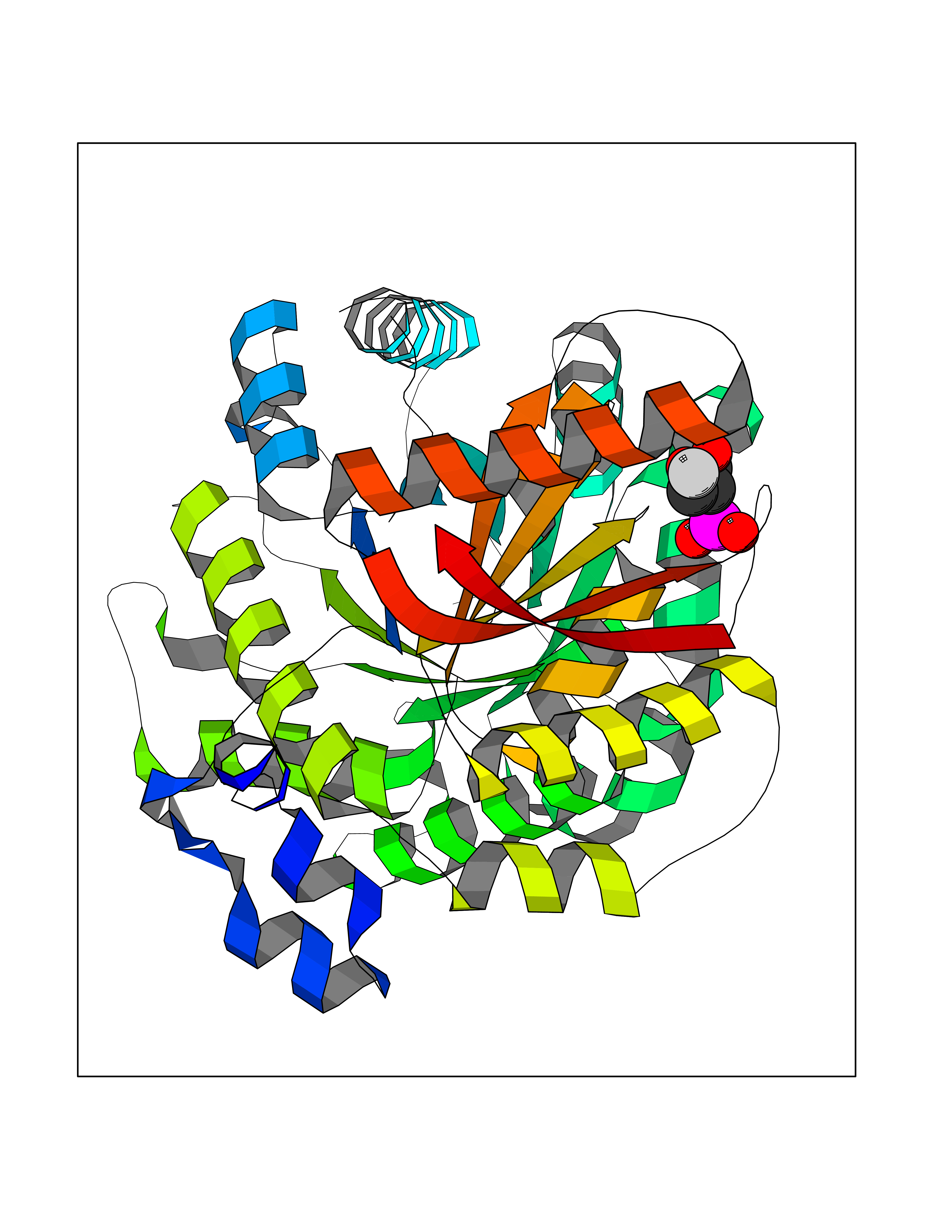 Molscript Map 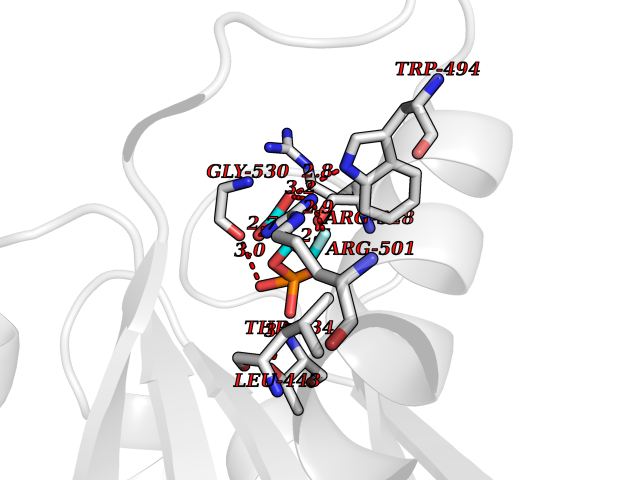 Pymol Map 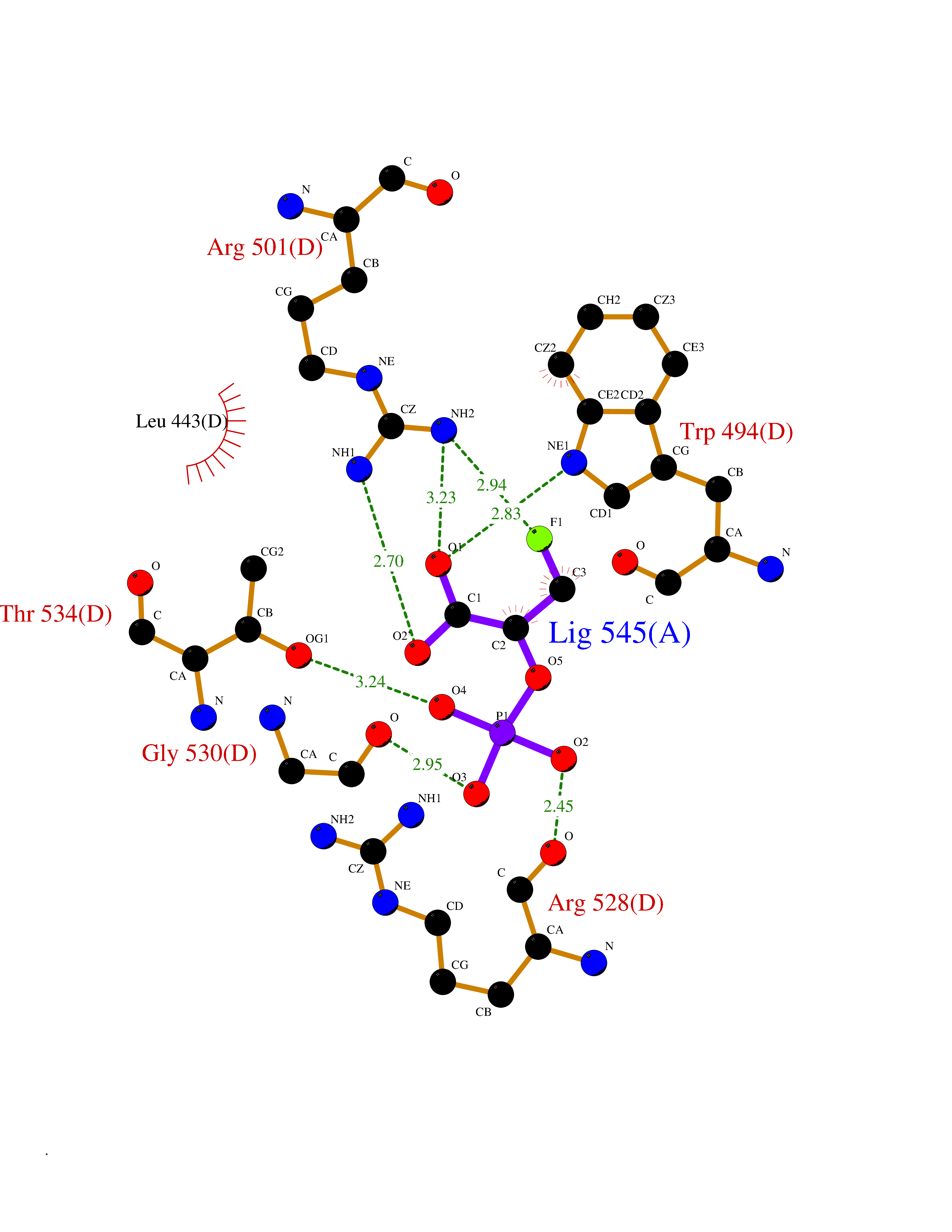 Ligplot Map 3D Binding mode Sequence GTAFFQQQQLPAAMADTFLEHLCLLDIDSEPVAARSTSIIATIGPASRSVERLKEMIKAGMNIARLNFSHGSHEYHAESIANVREAVESFSPLSYRPVAIALDTKGPEIGLSEQDVRDLRFGVEHGVDIVFASFVRKASDVAAVRAALGPEGHGIKIISKIENHEGVKRFDEILEVSDGIMVARGDLGIEIPAEKVFLAQKMMIGRCNLAGKPVVCATQMLESMITKPRPTRAETSDVANAVLDGADCIMLSGETAKGNFPVEAVKMQHAIAREAEAAVYHRQLFEELRRAAPLSRDPTEVTAIGAVEAAFKCCAAAIIVLTTTGRSAQLLSRYRPRAAVIAVTRSAQAARQVHLCRGVFPLLYREPPEAIWADDVDRRVQFGIESGKLRGFLRVGDLVIVVTGWRPGSGYTNIMRVLSIS Hydrogen bonds contact Hydrophobic contact | ||||
| 15 | Choline O-acetyltransferase | 2FY3 | 6.38 | |
Target general information Gen name CHAT Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Carnitine/choline acetyltransferase family Biochemical class Transferase Function Choline O-acetyltransferase activity. Related diseases Myasthenic syndrome, congenital, 6, presynaptic (CMS6) [MIM:254210]: A form of congenital myasthenic syndrome, a group of disorders characterized by failure of neuromuscular transmission, including pre-synaptic, synaptic, and post-synaptic disorders that are not of autoimmune origin. Clinical features are easy fatigability and muscle weakness affecting the axial and limb muscles (with hypotonia in early-onset forms), the ocular muscles (leading to ptosis and ophthalmoplegia), and the facial and bulbar musculature (affecting sucking and swallowing, and leading to dysphonia). The symptoms fluctuate and worsen with physical effort. CMS6 affected individuals have myasthenic symptoms since birth or early infancy, negative tests for anti-AChR antibodies, and abrupt episodic crises with increased weakness, bulbar paralysis, and apnea precipitated by undue exertion, fever, or excitement. CMS6 inheritance is autosomal recessive. {ECO:0000269|PubMed:11172068, ECO:0000269|PubMed:12756141}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00122; DB14006; DB00184 Interacts with Q6H8Q1-8; Q8N302-2; Q9NXL2-1; Q6XD76; Q9UII2; Q8TBE0; Q9UQB8-6; Q9ULD4-2; Q9NSI6-4; Q6P5X5; Q96LL4; P20807-4; O00257-3; Q6ZP82-1; O95674; Q9H3R5; Q8WUX9; Q9H2A9; Q3SX64; Q92782-2; Q14117; O14641; Q658K8; Q6UXG2-3; O00472; Q6NXG1; Q15910-2; Q8IZU1; P15407; P55318; Q06547-3; P23769-2; P23771; Q15486; Q8IV36; Q4VB01; Q53GQ0; P10809; P41134; Q9NZH6; Q8NA54; Q86U28; P17275; Q8N5Z5; Q6P597; P08727; Q14525; Q8IUC2; Q6IAA8; Q14847-2; P27338; Q9GZQ8; Q53S70; Q5JXC2; A0A0A0MR05; Q8NEH6; Q8TCY5; Q6IN84-2; Q96H12; P01106; P41271-2; P14598; Q9GZM8; Q5BJF6-2; Q9H8K7; Q9NR21-5; Q5VU43-8; Q13956; Q5SXH7-1; Q96T60; Q96I34; Q86UA1; Q15311; Q8TBY0; Q04206; P47804-3; Q9H0X6; P62899; Q66K80; Q9BY12-3; Q86SQ7-2; Q7Z6I5; Q496A3; Q7Z698; Q9C004; Q92783-2; Q8N4C7; O75528; Q15814; O15273; Q96A09; Q8WTV1; Q53NU3; Q71RG4-4; Q86WT6-2; Q9Y3Q8; Q99598; P49459; P11441; Q9H270; P19544-6; Q53FD0-2; Q3KNS6-3 EC number 2.3.1.6 Uniprot keywords 3D-structure; Acyltransferase; Alternative splicing; Congenital myasthenic syndrome; Direct protein sequencing; Disease variant; Neurotransmitter biosynthesis; Phosphoprotein; Proteomics identification; Reference proteome; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 66365.9 Length 595 Aromaticity 0.08 Instability index 53.36 Isoelectric point 8.16 Charge (pH=7) 4.64 2D Binding mode Binding energy (Kcal/mol) -6.17  Molscript Map 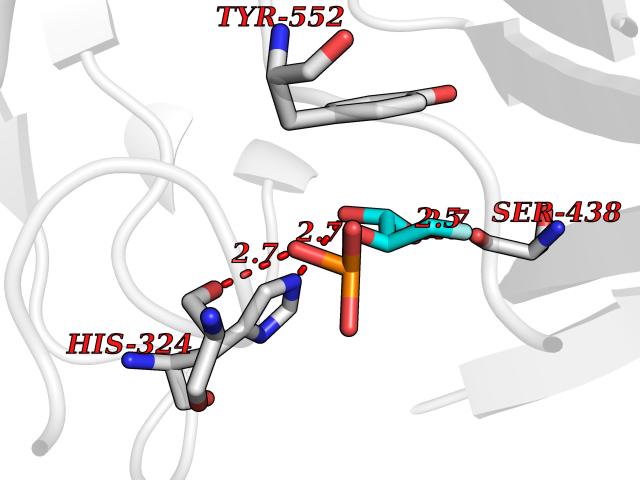 Pymol Map 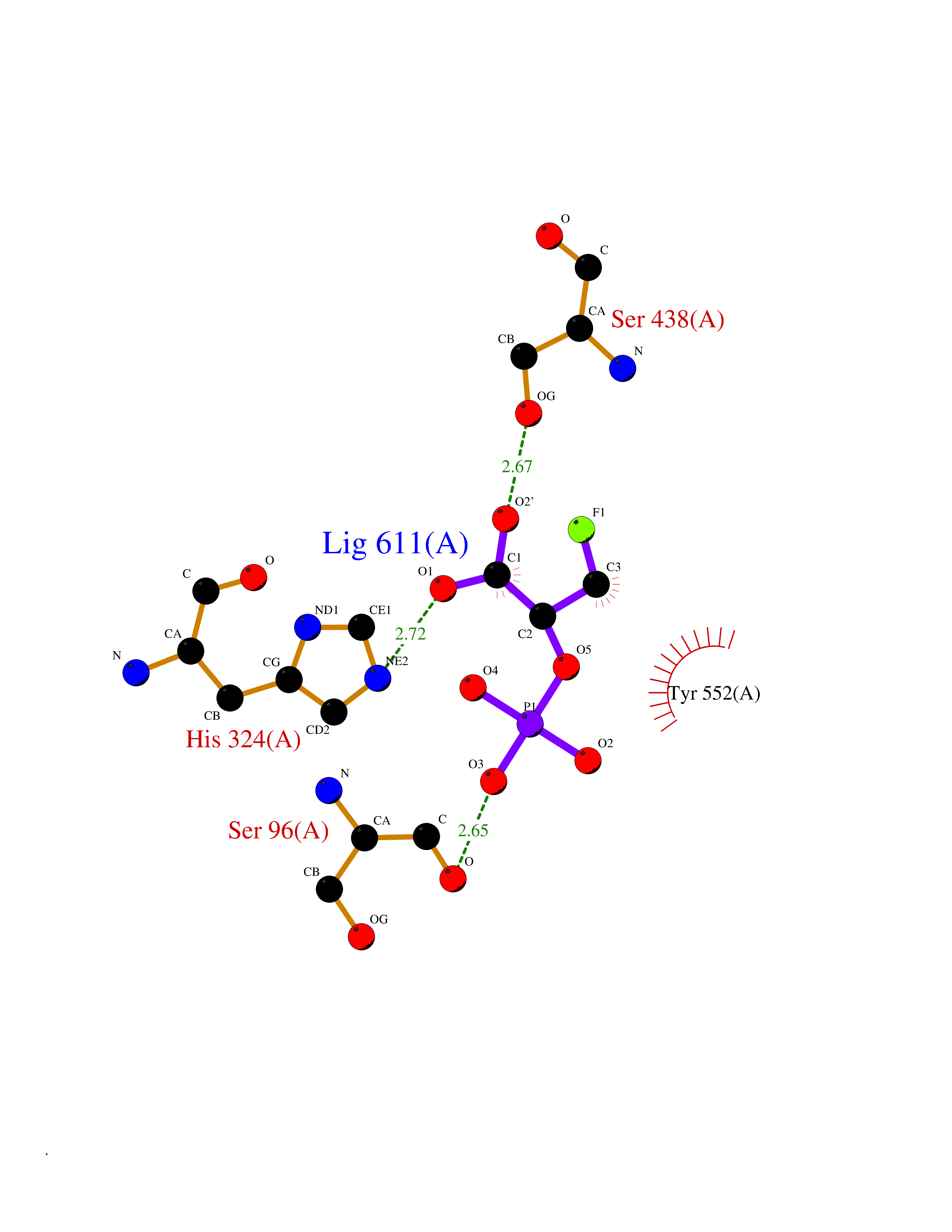 Ligplot Map 3D Binding mode Sequence SEESGLPKLPVPPLQQTLATYLQCMRHLVSEEQFRKSQAIVQQFGAPGGLGETLQQKLLERQEKTANWVSEYWLNDMYLNNRLALPVNSSPAVIFARQHFPGTDDQLRFAASLISGVLSYKALLDSHSIPTDCAKGQPLCMKQYYGLFSSYRLPGHTQDTLVAQNSSIMPEPEHVIVACCNQFFVLDVVINFRRLSEGDLFTQLRKIVKMASNAAARLPPIGLLTSDGRSEWAEARTVLVKDSTNRDSLDMIERCICLVCLDAPGGVELSDTHRALQLLHGGGYSKNGANRWYDKSLQFVVGRDGTCGVVCEHSPFDGIVLVQCTEHLLKHMTQPELVRSPMVPLPAPRRLRWKCSPEIQGHLASSAEKLQRIVKNLDFIVYKFDNYGKTFIKKQKCSPDAFIQVALQLAFYRLHRRLVPTYESASIRRFQEGRVDNIRSATPEALAFVRAVTDHKAAVPASEKLLLLKDAIRAQTAYTVMAITGMAIDNHLLALRELARAMCAALPEMFMDETYLMSNRFVLSTSQVPTTTEMFCCYGPVVPNGYGACYNPQPETILFCISSFHSCAATSSSKFAKAVEESLIDMRDLCSLLPP Hydrogen bonds contact Hydrophobic contact | ||||
| 16 | Cytochrome P450 2C19 | 4GQS | 6.38 | |
Target general information Gen name CYP2C19 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Cytochrome P450 family Biochemical class Oxidoreductase Function (R)-limonene 6-monooxygenase activity.(S)-limonene 6-monooxygenase activity.(S)-limonene 7-monooxygenase activity.Arachidonic acid epoxygenase activity.Enzyme binding.Heme binding.Iron ion binding.Monooxygenase activity.Oxidoreductase activity.Oxygen binding.Steroid hydroxylase activity. Related diseases Cystathioninuria (CSTNU) [MIM:219500]: Autosomal recessive phenotype characterized by abnormal accumulation of plasma cystathionine, leading to increased urinary excretion. {ECO:0000269|PubMed:12574942, ECO:0000269|PubMed:18476726}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08496; DB14055; DB05812; DB14973; DB01418; DB00945; DB00546; DB00518; DB00918; DB12015; DB06403; DB00357; DB01424; DB01118; DB00321; DB00381; DB00701; DB01435; DB11901; DB06605; DB00673; DB01274; DB06413; DB06697; DB11638; DB12597; DB11586; DB00289; DB01076; DB06442; DB06626; DB00972; DB01483; DB16703; DB15463; DB12319; DB01086; DB00443; DB01128; DB11967; DB13746; DB00188; DB12151; DB05541; DB01558; DB01222; DB00297; DB00921; DB09061; DB14737; DB08502; DB00564; DB06016; DB00395; DB14984; DB06119; DB00446; DB00672; DB01166; DB00501; DB00604; DB00215; DB12499; DB04920; DB14025; DB00349; DB06470; DB01242; DB00758; DB01559; DB00363; DB14635; DB00531; DB00091; DB08912; DB00250; DB00705; DB06700; DB01234; DB14649; DB09213; DB05351; DB13762; DB00514; DB00829; DB00586; DB00343; DB01093; DB01075; DB09167; DB05928; DB00590; DB01142; DB00470; DB00476; DB00625; DB00216; DB15444; DB13874; DB11718; DB00109; DB08899; DB01175; DB11823; DB14575; DB09119; DB00736; DB00783; DB13952; DB13953; DB13954; DB13955; DB13956; DB00330; DB00898; DB00977; DB09166; DB01628; DB14766; DB06414; DB12500; DB00949; DB00574; DB01039; DB12265; DB15669; DB00196; DB01544; DB00472; DB01095; DB00176; DB00983; DB01320; DB11679; DB05087; DB00317; DB01241; DB06730; DB01120; DB01016; DB00986; DB01018; DB00502; DB01355; DB00956; DB00741; DB06789; DB01050; DB09054; DB01181; DB00619; DB00458; DB00328; DB11633; DB06636; DB00951; DB09570; DB06738; DB01026; DB00598; DB06218; DB00448; DB01259; DB09078; DB12070; DB01006; DB08918; DB09198; DB04948; DB06448; DB16220; DB01601; DB00455; DB04871; DB12130; DB00678; DB00227; DB08933; DB09280; DB01283; DB12474; DB08932; DB09238; DB14921; DB14009; DB01065; DB01043; DB00170; DB00454; DB00532; DB00333; DB00763; DB05246; DB09241; DB00849; DB00959; DB01110; DB06595; DB16236; DB01171; DB00745; DB11763; DB14011; DB09049; DB01183; DB04861; DB00220; DB00622; DB00184; DB00665; DB06712; DB12005; DB00717; DB00540; DB00334; DB14881; DB16267; DB00338; DB11632; DB04911; DB11837; DB04938; DB00776; DB00239; DB00935; DB11697; DB05467; DB00213; DB00715; DB00738; DB00312; DB00850; DB03783; DB00780; DB01174; DB00252; DB13941; DB01621; DB04951; DB17472; DB06209; DB01058; DB14631; DB00635; DB00794; DB00396; DB01131; DB00420; DB00818; DB00571; DB01589; DB04216; DB01224; DB00468; DB01129; DB00980; DB08896; DB16826; DB02709; DB00615; DB01045; DB11753; DB01201; DB01220; DB08864; DB00503; DB06176; DB05271; DB12332; DB11614; DB06654; DB12543; DB12834; DB00418; DB01037; DB11689; DB06731; DB06739; DB01104; DB00203; DB00641; DB06268; DB15093; DB00052; DB00398; DB12548; DB01323; DB09118; DB00675; DB06204; DB06083; DB12020; DB00966; DB12095; DB00444; DB00857; DB00624; DB13943; DB13944; DB13946; DB11712; DB01041; DB00599; DB00679; DB00208; DB00373; DB01007; DB00932; DB06137; DB08895; DB01124; DB01036; DB00273; DB01685; DB05109; DB00752; DB12245; DB00347; DB00726; DB00197; DB15328; DB00313; DB00862; DB00285; DB00661; DB16349; DB06684; DB08828; DB11739; DB00582; DB09068; DB14975; DB00682; DB00549; DB00425; DB00909; DB09120 Interacts with NA EC number 1.14.14.1; 1.14.14.51; 1.14.14.52; 1.14.14.53; 1.14.14.75 Uniprot keywords 3D-structure; Direct protein sequencing; Endoplasmic reticulum; Fatty acid metabolism; Heme; Iron; Lipid metabolism; Membrane; Metal-binding; Microsome; Monooxygenase; NADP; Oxidoreductase; Proteomics identification; Reference proteome Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 51680.3 Length 452 Aromaticity 0.1 Instability index 39.98 Isoelectric point 6.12 Charge (pH=7) -4.4 2D Binding mode Binding energy (Kcal/mol) -6.11  Molscript Map 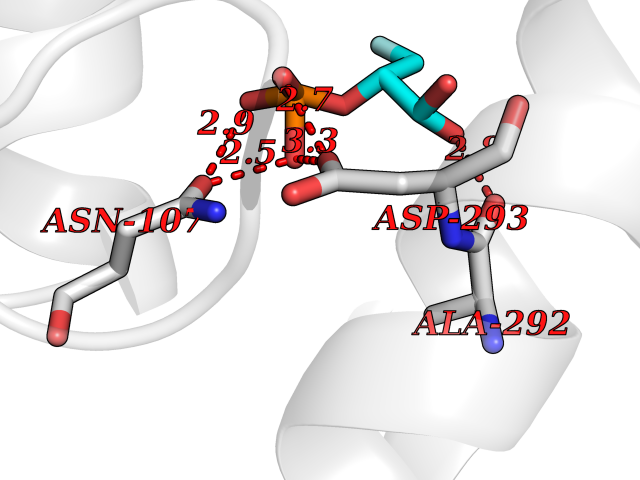 Pymol Map 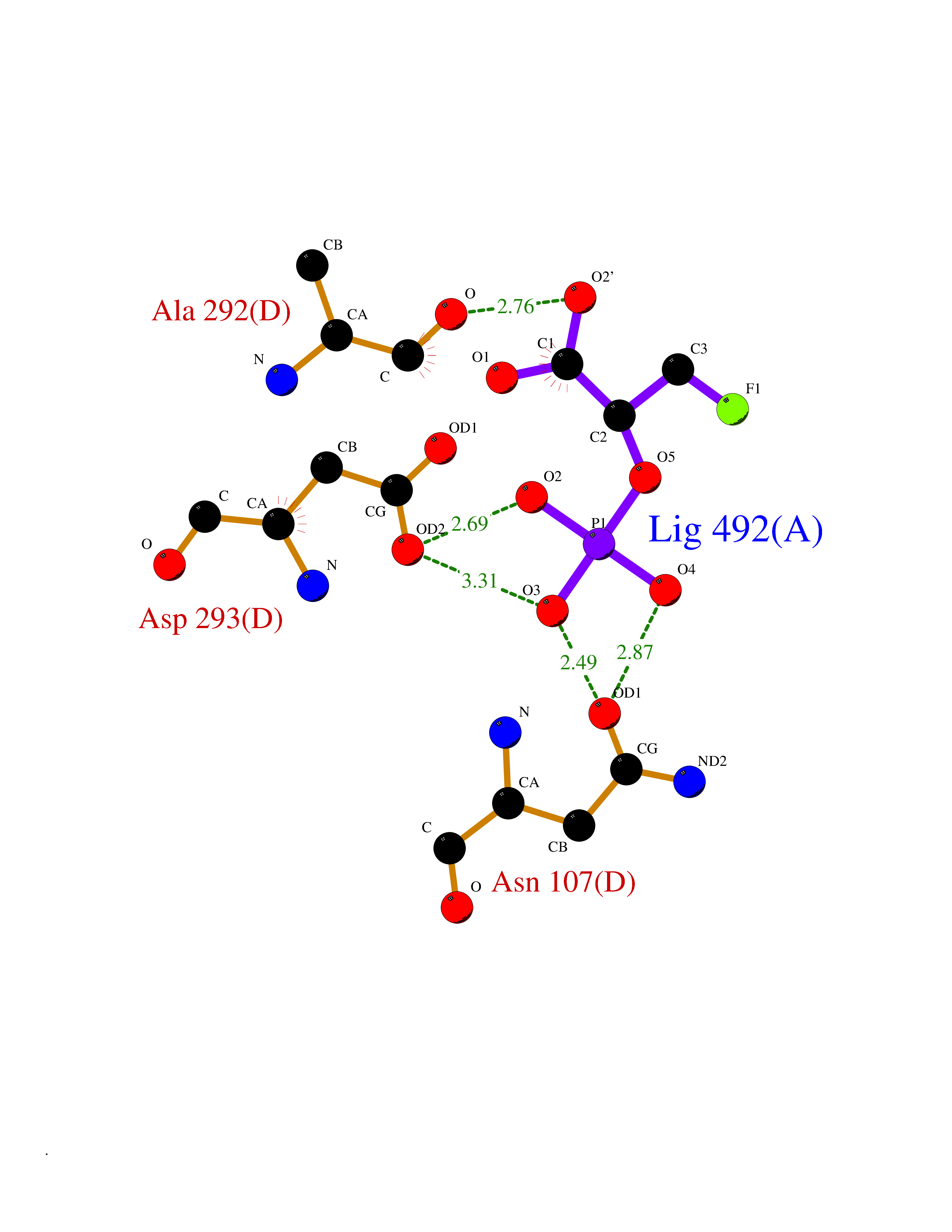 Ligplot Map 3D Binding mode Sequence LPPGPTPLPVIGNILQIDIKDVSKSLTNLSKIYGPVFTLYFGLERMVVLHGYEVVKEALIDLGEEFSGRGHFPLAERANRGFGIVFSNGKRWKEIRRFSLMTLEDRVQEEARCLVEELRKTKASPCDPTFILGCAPCNVICSIIFQKRFDYKDQQFLNLMEKLNENIRIVSTPWIQICNNFPTIIDYFPGTHNKLLKNLAFMESDILEKVKEHQESMDINNPRDFIDCFLIKMEKEKQNQQSEFTIENLVITAADLLGAGTETTSTTLRYALLLLLKHPEVTAKVQEEIERVVGRNRSPCMQDRGHMPYTDAVVHEVQRYIDLIPTSLPHAVTCDVKFRNYLIPKGTTILTSLTSVLHDNKEFPNPEMFDPRHFLDEGGNFKKSNYFMPFSAGKRICVGEGLARMELFLFLTFILQNFNLKSLIDPKDLDTTPVVNGFASVPPFYQLCFIPI Hydrogen bonds contact Hydrophobic contact | ||||
| 17 | Dopamine beta-hydroxylase | 4ZEL | 6.36 | |
Target general information Gen name DBH Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Copper type II ascorbate-dependent monooxygenase family Biochemical class Oxidoreductase Function Catalytic activity.Copper ion binding.Dopamine beta-monooxygenase activity.L-ascorbic acid binding. Related diseases Orthostatic hypotension 1 (ORTHYP1) [MIM:223360]: A form of orthostatic hypotension due to congenital dopamine beta-hydroxylase deficiency. Orthostatic hypotension, also known as postural hypotension, is a finding defined as a 20-mm Hg decrease in systolic pressure or a 10-mm Hg decrease in diastolic pressure occurring 3 minutes after a person has risen from supine to standing. Symptoms include dizziness, blurred vision, and sometimes syncope. ORTHYP1 is an autosomal recessive condition apparent from infancy or early childhood and characterized by low plasma and urinary levels of norepinephrine and epinephrine, and episodic hypoglycemia. {ECO:0000269|PubMed:11857564}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00126; DB06774; DB09130; DB05394; DB00822; DB00988; DB00968; DB00550 Interacts with P00352; P63010-2; Q04656; Q8WUW1; Q9UNS2; Q71DI3; P61978; Q9Y2M5; Q92876; P08727; Q14693; P0DPK4; Q6GQQ9-2; P27986-2; Q9ULX5; Q96D59; Q8N6K7-2; Q9GZS3; Q8IUW3; Q86WT6-2 EC number 1.14.17.1 Uniprot keywords 3D-structure; Catecholamine biosynthesis; Copper; Cytoplasmic vesicle; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; Membrane; Metal-binding; Monooxygenase; Oxidoreductase; Proteomics identification; Reference proteome; Secreted; Signal-anchor; Transmembrane; Transmembrane helix; Vitamin C Protein physicochemical properties Chain ID A,B Molecular weight (Da) 123694 Length 1094 Aromaticity 0.1 Instability index 51.85 Isoelectric point 5.84 Charge (pH=7) -24.5 2D Binding mode Binding energy (Kcal/mol) -6.59  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence PLPYHIPLDPEGSLELSWNVSYTQEAIHFQLLVRRLKAGVLFGMSDRGELENADLVVLWTDGDAYFADAWSDQKGQIHLDPQQDYQLLQVQRTPEGLTLLFKRPFGTCDPKDYLIEDGTVHLVYGILEEPFRSLEAINGSGLQMGLQRVQLLKPNIPEPELPSDACTMEVQAPNIQIPSQETTYWCYIKELPKGFSRHHIIKYEPIVTKGNEALVHHMEVFQCAPEMDSVPHFSGPCDSKMKPDRLNYCRHVLAAWALGAKAFYYPEEAGLAFGGPGSSRYLRLEVHYHNPLVIEGRNDSSGIRLYYTAKLRRFNAGIMELGLVYTPVMAIPPRETAFILTGYCTDKCTQLALPPSGIHIFASQLHTHLTGRKVVTVLVRDGREWEIVNQDNHYSPHFQEIRMLKKVVSVHPGDVLITSCTYNTEDRELATVGGFGILEEMCVNYVHYYPQTQLELCKSAVDAGFLQKYFHLINRFNNEDVCTCPQASVSQQFTSVPWNSFNRDVLKALYSFAPISMHCNKSSAVRFQGEWNLQPLPKVISTLEEPTVVSPLPYHIPLDPEGSLELSWNVSYTQEAIHFQLLVRRLKAGVLFGMSDRGELENADLVVLAYFADAWSDQKGQIHLDPQQDYQLLQVQRTPEGLTLLFKRPFGTCDPKDYLIEDGTVHLVYGILEEPFRSLEAINGSGLQMGLQRVQLLKPNIPEPELPSDACTMEVQAPNIQIPSQETTYWCYIKELPKGFSRHHIIKYEPIVTKGNEALVHHMEVFQCAPEVPHFSGPCDSKMLNYCRHVLAAWALGAKAFYYPEEAGLAFGGPGSSRYLRLEVHYHNPLVIEGRNDSSGIRLYYTAKLRRFNAGIMELGLVYTPVMAIPPRETAFILTGYCTDKCTQLALPPSGIHIFASQLHTHLTGRKVVTVLVRDGREWEIVNQDNHYSPHFQEIRMLKKVVSVHPGDVLITSCTYNTEDRELATVGGFGILEEMCVNYVHYYPQTQLELCKSAVDAGFLQKYFHLINRFNNEDVCTCPQASVSQQFTSVPWNSFNRDVLKALYSFAPISMHCNKSSAVRFQGEWNLQPLPKVISTLEEPTPQCVVSIGG Hydrogen bonds contact Hydrophobic contact | ||||
| 18 | Oxygen-insensitive NADPH nitroreductase | 3QDL | 6.36 | |
Target general information Gen name rdxA Organism Helicobacter pylori (strain ATCC 700392 / 26695) (Campylobacter pylori) Uniprot ID TTD ID NA Synonyms HP_0954 Protein family Nitroreductase family Biochemical class Oxidoreductase Function Oxidoreductase activity. Related diseases Hypervalinemia and hyperleucine-isoleucinemia (HVLI) [MIM:618850]: An autosomal recessive metabolic disorder characterized by highly elevated plasma concentrations of valine and leucine/isoleucine. Affected individuals suffer from headache and mild memory impairment. {ECO:0000269|PubMed:25653144}. The disease is caused by variants affecting the gene represented in this entry. A patient with hypervalinemia and hyperleucine-isoleucinemia was identified as compound heterozygote for Gln-170 (inherited from his father) and Lys-264 (inherited from his mother), both variants reduced the catalytic activity of the enzyme. After treatment with vitamin B6, a precursor of pyridoxal 5'-phosphate, a BCAT2 cofactor, the blood levels of branched chain amino acids, especially valine, were decreased and brain lesions were improved. {ECO:0000269|PubMed:25653144}. Drugs (DrugBank ID) DB00916 Interacts with NA EC number 1.-.-.- Uniprot keywords 3D-structure; Antibiotic resistance; NADP; Oxidoreductase; Reference proteome Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 40094.3 Length 352 Aromaticity 0.08 Instability index 55.15 Isoelectric point 6.72 Charge (pH=7) -0.86 2D Binding mode Binding energy (Kcal/mol) -6.2  Molscript Map 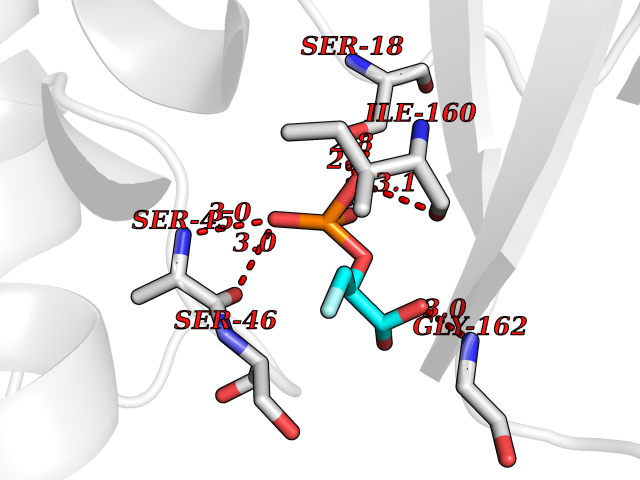 Pymol Map 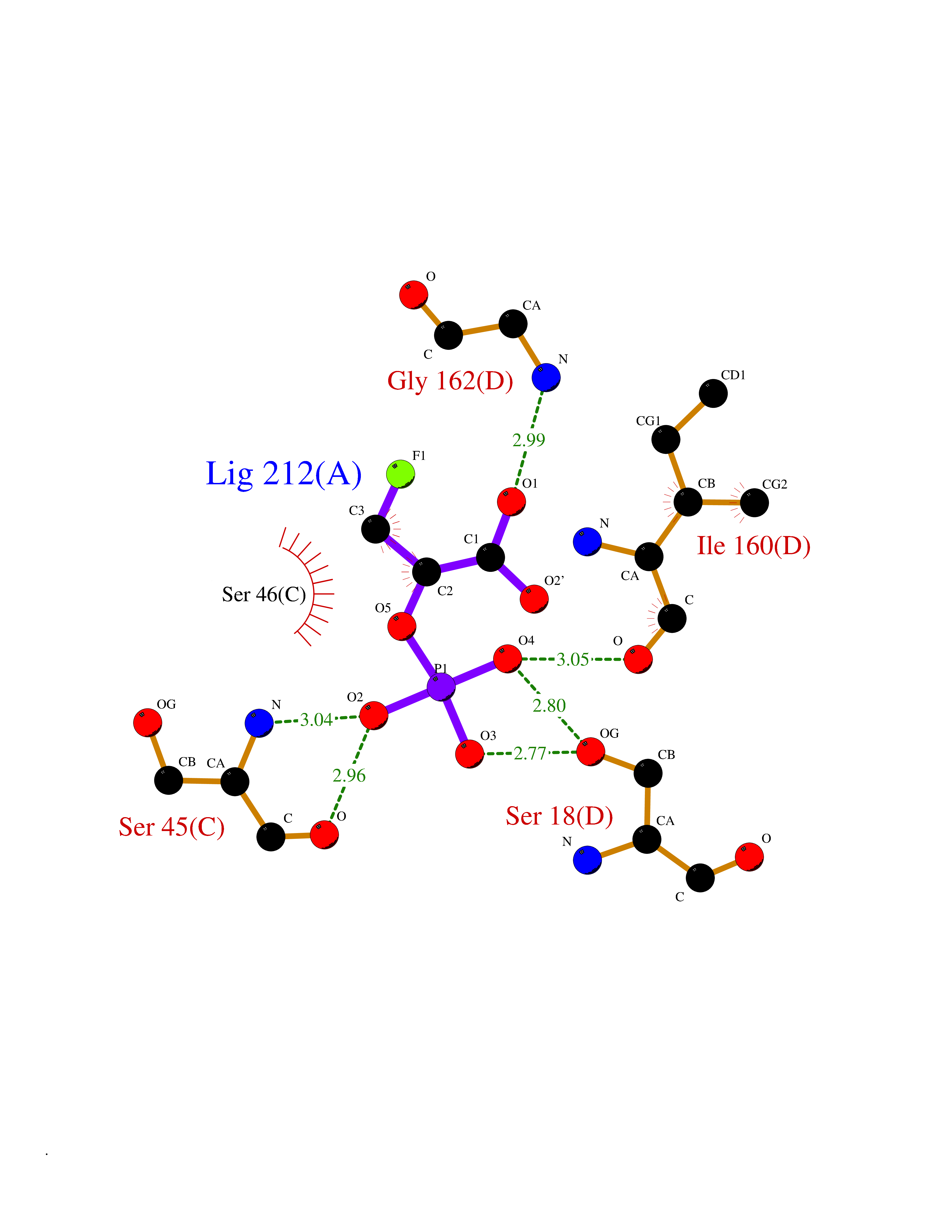 Ligplot Map 3D Binding mode Sequence MQRLESYILMKFLDQEKRRQLLNERHSCKMFDSHYEFSSTELEEIAEIARLSPSSYNTQPWHFVMVTDKDLKKQIAAHSYFNEEMIKSASALMVVCSLSYILEQCYIAVGQICMGVSLMGLDSCIIGGFDPLKVGEVLEERINPKIACLIALGKRVAEASQKSRKSKVDAITWLMKFLDQEKRRQLLNERHSCKMFDSHYEFSSTELEEIAEIARLSPSSYNTQPWHFVMVTDKDLKKQIAAHSYFNEEMIKSASALMVVCSLRPSELLPMQRLESYILEQCYIAVGQICMGVSLMGLDSCIIGGFDPLKVGEVLEERINKPKIACLIALGKRVAEASQKSRKSKVDAITWL Hydrogen bonds contact Hydrophobic contact | ||||
| 19 | Aspartate carbamoyltransferase (CAD) | 6HFQ | 6.36 | |
Target general information Gen name CAD Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms CAD Protein family CarA family; CarB family; Metallo-dependent hydrolases superfamily, DHOase family, CAD subfamily; Aspartate/ornithine carbamoyltransferase superfamily, ATCase family Biochemical class Carbon-nitrogen ligase Function This protein is a "fusion" protein encoding four enzymatic activities of the pyrimidine pathway (GATase, CPSase, ATCase and DHOase). Related diseases Developmental and epileptic encephalopathy 50 (DEE50) [MIM:616457]: A form of epileptic encephalopathy, a heterogeneous group of severe early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE50 is an autosomal recessive, progressive disease with onset in infancy and favorable response to treatment with oral uridine. {ECO:0000269|PubMed:25678555, ECO:0000269|PubMed:28087732}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00128; DB00130; DB03459 Interacts with P27708; Q8N137; P63104 EC number NA Uniprot keywords 3D-structure; Acetylation; Allosteric enzyme; ATP-binding; Congenital disorder of glycosylation; Cytoplasm; Direct protein sequencing; Disease variant; Epilepsy; Hydrolase; Ligase; Magnesium; Manganese; Metal-binding; Multifunctional enzyme; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Pyrimidine biosynthesis; Reference proteome; Repeat; Transferase; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 39410.8 Length 362 Aromaticity 0.06 Instability index 41.36 Isoelectric point 5.94 Charge (pH=7) -9.57 2D Binding mode Binding energy (Kcal/mol) -6.61  Molscript Map 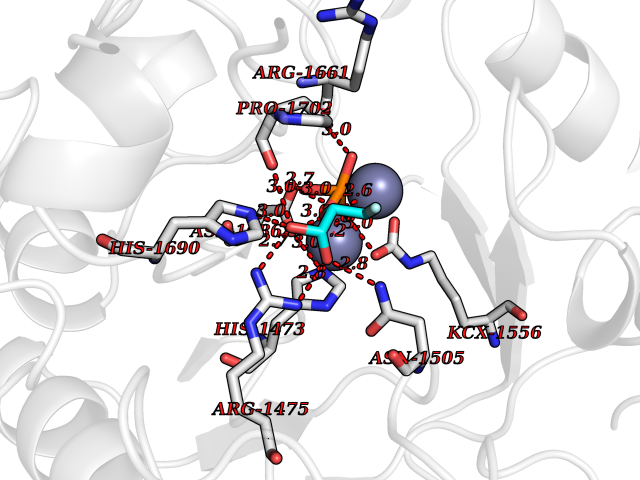 Pymol Map 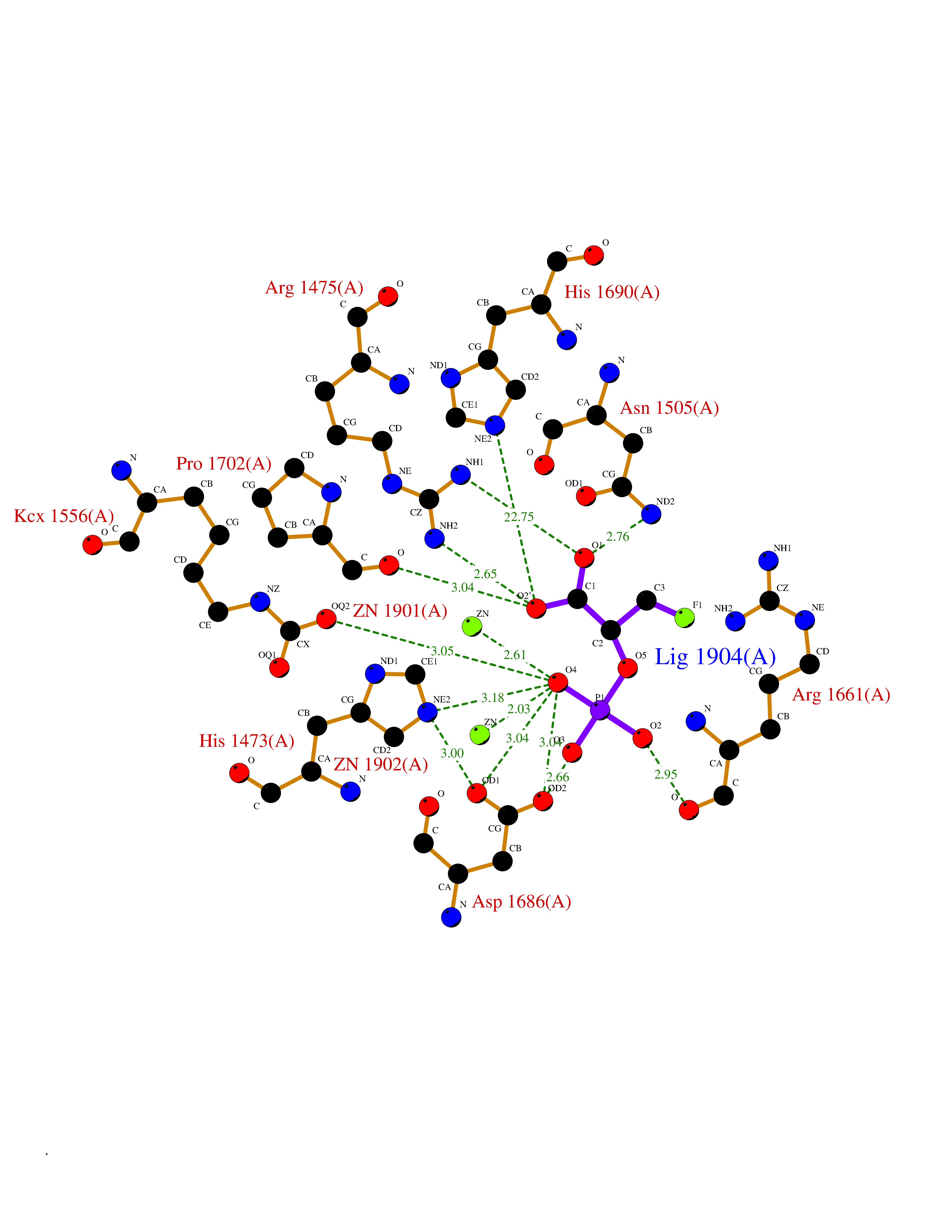 Ligplot Map 3D Binding mode Sequence KLVRLPGLIDVHVHLREPGGTHKEDFASGTAAALAGGITMVCAMPNTRPPIIDGPALALAQKLAEAGARCDFALFLGASSENAGTLGTVAGSAAGLXLYLNETTSELRLDSVVQWMEHFETWPSHLPIVAHAEQQTVAAVLMVAQLTQRSVHICHVARKEEILLIKAAKARGLPVTCEVAPHHLFLSHDDLERLGPGKGEVRPELGSRQDVEALWENMAVIDCFASDHAPHTLEEKCGSRPPPGFPGLETMLPLLLTAVSEGRLSLDDLLQRLHHNPRRIFHLPPQEDTYVEVDLEHEWTIPSHMPFSKAHWTPFEGQKVKGTVRRVVLRGEVAYIDGQVLVPPGYGQDVRKWPQGAVPQLP Hydrogen bonds contact Hydrophobic contact | ||||
| 20 | Peptidyl-prolyl cis-trans isomerase C | 2ESL | 6.36 | |
Target general information Gen name PPIC Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms CYPC Protein family Cyclophilin-type PPIase family Biochemical class Isomerase / immunosuppressant Function Cyclosporin A binding.Peptidyl-prolyl cis-trans isomerase activity. Related diseases Canavan disease (CAND) [MIM:271900]: A rare neurodegenerative condition of infancy or childhood characterized by white matter vacuolization and demyelination that gives rise to a spongy appearance. The clinical features are onset in early infancy, atonia of neck muscles, hypotonia, hyperextension of legs and flexion of arms, blindness, severe mental defect, megalocephaly, and death by 18 months on the average. {ECO:0000269|PubMed:10407784, ECO:0000269|PubMed:10564886, ECO:0000269|PubMed:10909858, ECO:0000269|PubMed:12205125, ECO:0000269|PubMed:12638939, ECO:0000269|PubMed:12706335, ECO:0000269|PubMed:24036223, ECO:0000269|PubMed:28101991, ECO:0000269|PubMed:7599639, ECO:0000269|PubMed:7668285, ECO:0000269|PubMed:8023850, ECO:0000269|PubMed:8252036, ECO:0000269|PubMed:8659549, ECO:0000269|PubMed:9452117}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00172 Interacts with Q8N9N5; Q8N9N5-2; Q96L46; B3EWG5; O43765; Q96EQ0; Q9NZ09; Q9UMX0; Q9UMX0-2; Q9UHD9 EC number 5.2.1.8 Uniprot keywords 3D-structure; Cytoplasm; Isomerase; Proteomics identification; Reference proteome; Rotamase Protein physicochemical properties Chain ID A,B,C,D,E,F Molecular weight (Da) 19625.2 Length 181 Aromaticity 0.1 Instability index 4.3 Isoelectric point 7.14 Charge (pH=7) 0.2 2D Binding mode Binding energy (Kcal/mol) -6.33  Molscript Map 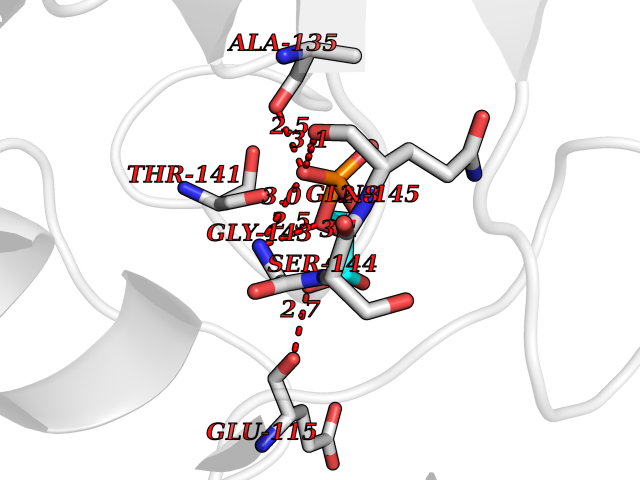 Pymol Map 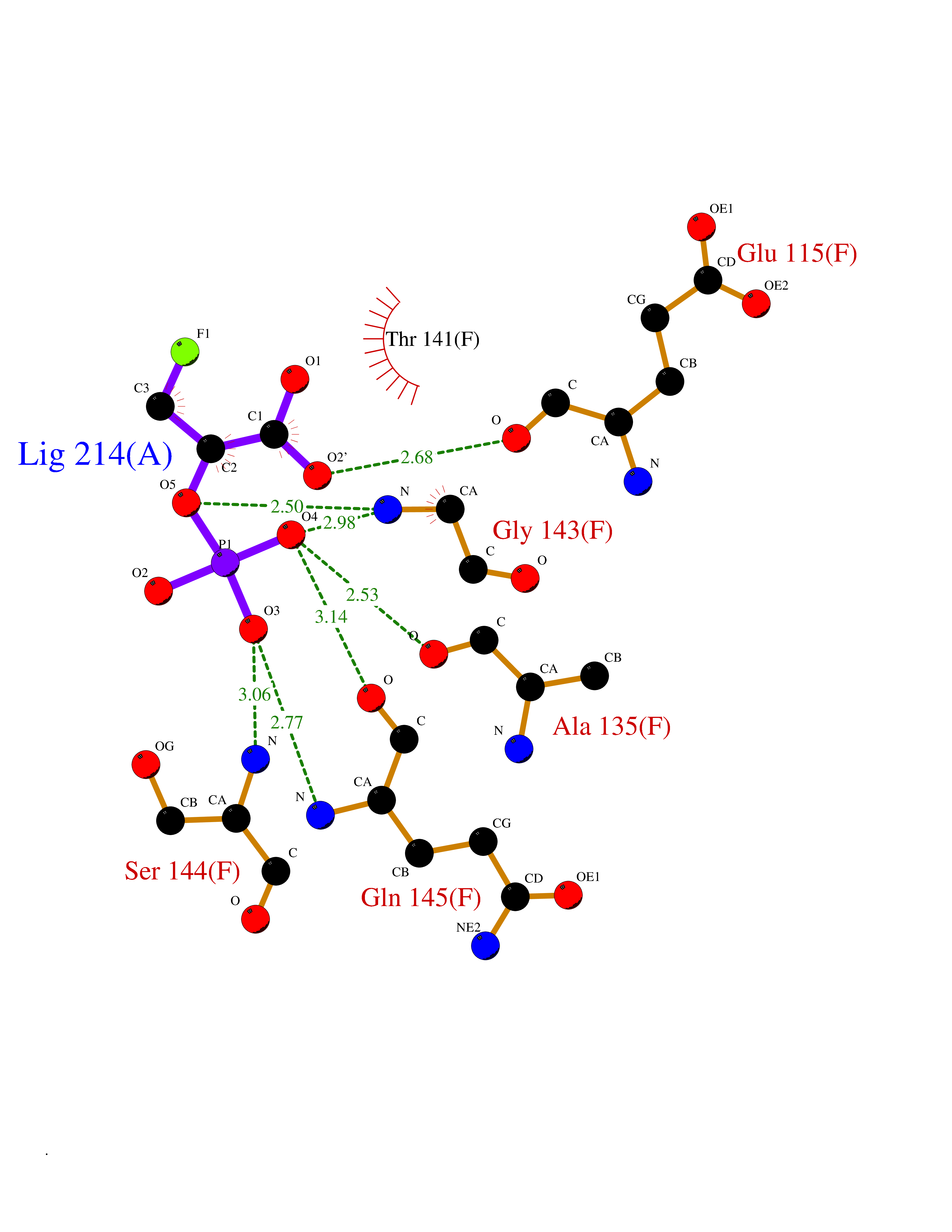 Ligplot Map 3D Binding mode Sequence RGPSVTAKVFFDVRIGDKDVGRIVIGLFGKVVPKTVENFVALATGEKGYGYKGSKFHRVIKDFMIQGGDITTGDGTGGVSIYGETFPDENFKLKHYGIGWVSMANAGPDTNGSQFFITLTKPTWLDGKHVVFGKVIDGMTVVHSIELQATDGHDRPLTNCSIINSGKIDVKTPFVVEIADW Hydrogen bonds contact Hydrophobic contact | ||||