Job Results:
Ligand
Structure
Job ID
0cb0764e79e22b0da515513ec30e42ef
Job name
NA
Time
2025-06-05 10:48:27
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 1 | Zinc finger protein Helios (IKZF2) | 7LPS | 5.47 | |
Target general information Gen name IKZF2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Ikaros family zinc finger protein 2 Protein family Ikaros C2H2-type zinc-finger protein family Biochemical class NA Function Associates with Ikaros at centromeric heterochromatin. Related diseases Developmental and epileptic encephalopathy 25, with amelogenesis imperfecta (DEE25) [MIM:615905]: An autosomal recessive disease characterized by subclinical seizures appearing in the first days of life, evolving to severe epileptic disease. Affected individuals have profound or severe delayed development with lack of speech, and most patients do not acquire the ability to sit. Additional variable features include axial hypotonia, peripheral hypertonia, and abnormal involuntary movements such as dystonia and choreoathetosis. Dental abnormalities, including delayed eruption, hypodontia, tooth hypoplasia, yellow discoloration, thin enamel, and enamel chipping are observed in most patients. {ECO:0000269|PubMed:24995870, ECO:0000269|PubMed:26384929, ECO:0000269|PubMed:30054523}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P29972; P56545; P56545-3; Q17RB8; P09022; Q8N8B7-2 EC number NA Uniprot keywords 3D-structure; Acetylation; Activator; Alternative splicing; DNA-binding; Isopeptide bond; Metal-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Transcription; Transcription regulation; Ubl conjugation; Zinc; Zinc-finger Protein physicochemical properties Chain ID B,C Molecular weight (Da) 47006.6 Length 410 Aromaticity 0.09 Instability index 44.28 Isoelectric point 7.23 Charge (pH=7) 0.69 2D Binding mode Binding energy (Kcal/mol) -7.46 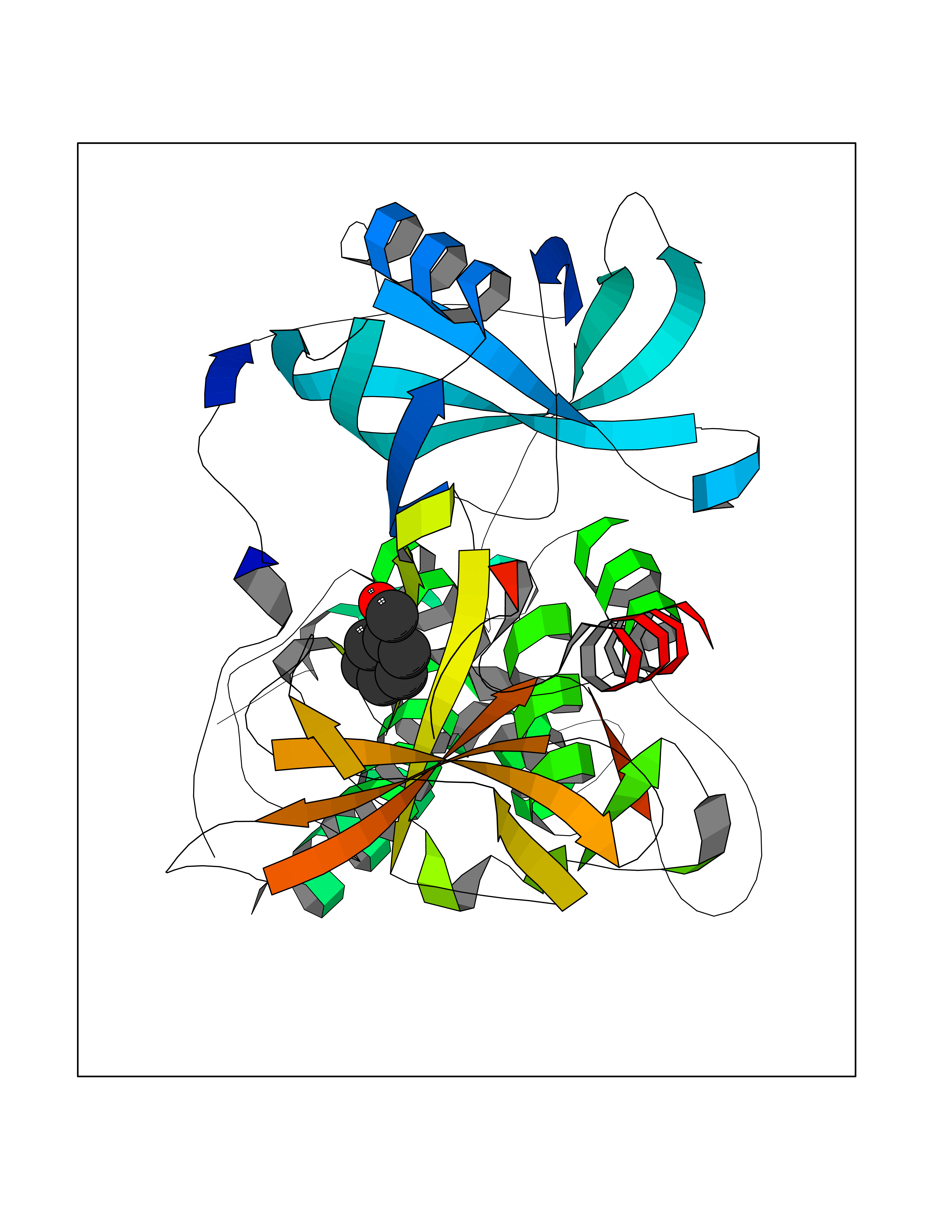 Molscript Map 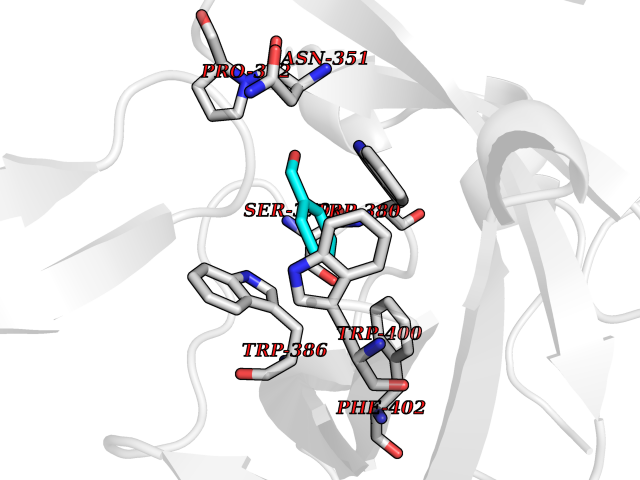 Pymol Map 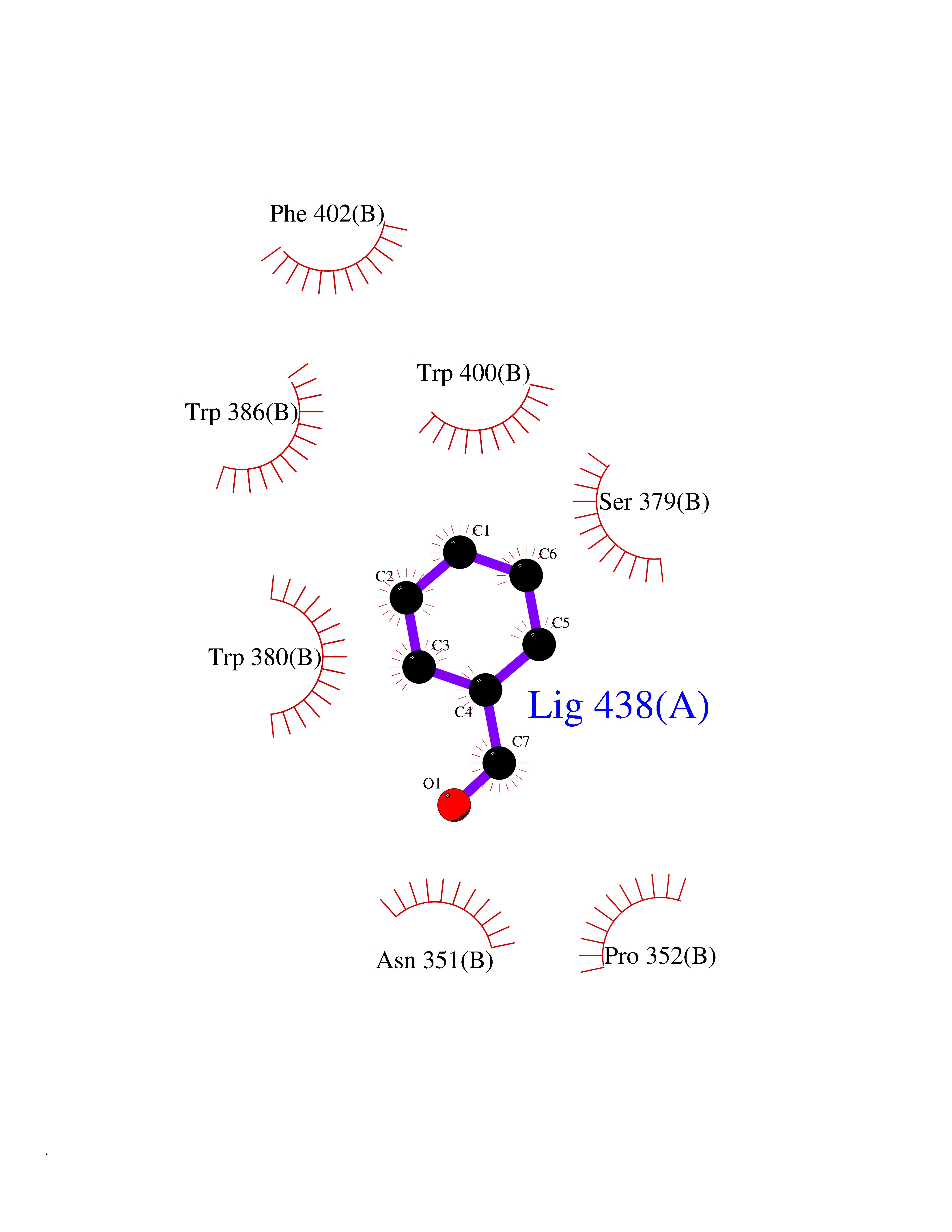 Ligplot Map 3D Binding mode Sequence INFDTSLPTSHTYLGADMEEFHGRTLHDDDSCQVIPVLPQVMMILIPGQTLPLQLFHPQEVSMVRNLIQKDRTFAVLAYSNVQEREAQFGTTAEIYAYREEQDFGIEIVKVKAIGRQRFKVLELRTQSDGIQQAKVQILPECVLPSTMSAVQLESLNKCQIFPCSYKWWQKYQKRKFHCANLTSWPRWLYSLYDAETLMDRIKKQLREWDENLKDDSLPSNPIDFSYRVAACLPIDDVLRIQLLKIGSAIQRLRCELDIMNKCTSLCCKQCQETEITTKNEIFSLSLCGPMAAYVNPHGYVHETLTVYKACNLNLIGRPSTEHSWFPGYAWTVAQCKICASHIGWKFTATKKDMSPQKFWGLTRSALLPTIPDTEDEISPDGERPFHCNQCGASFTQKGNLLRHIKLHSG Hydrogen bonds contact Hydrophobic contact | ||||
| 2 | Protein cereblon (CRBN) | 5FQD | 5.44 | |
Target general information Gen name CRBN Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Protein cereblon Protein family CRBN family Biochemical class NA Function Substrate recognition component of a DCX (DDB1-CUL4-X-box) E3 protein ligase complex that mediates the ubiquitination and subsequent proteasomal degradation of target proteins, such as MEIS2. Normal degradation of key regulatory proteins is required for normal limb outgrowth and expression of the fibroblast growth factor FGF8. May play a role in memory and learning by regulating the assembly and neuronal surface expression of large-conductance calcium-activated potassium channels in brain regions involved in memory and learning via its interaction with KCNT1. Binding of pomalidomide and other thalidomide-related drugs changes the substrate specificity of the human protein, leading to decreased degradation of MEIS2 and other target proteins and increased degradation of MYC, IRF4, IKZF1 and IKZF3. Related diseases Intellectual developmental disorder, autosomal recessive 2 (MRT2) [MIM:607417]: A disorder characterized by significantly below average general intellectual functioning associated with impairments in adaptive behavior and manifested during the developmental period. MRT2 patients display mild intellectual disability with a standard IQ ranged from 50 to 70. IQ scores are lower in males than females. Developmental milestones are mildly delayed. There are no dysmorphic or autistic features. {ECO:0000269|PubMed:15557513, ECO:0000269|PubMed:28143899}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00480; DB08910; DB01041 Interacts with Q96A83-2; P48729; Q16531; O14901; Q8IVT2; Q9P286; A0A6Q8PF08; Q93062; Q16531; Q13422-7 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; Disease variant; Intellectual disability; Membrane; Metal-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Ubl conjugation; Ubl conjugation pathway; Zinc Protein physicochemical properties Chain ID B,E Molecular weight (Da) 38245.7 Length 337 Aromaticity 0.08 Instability index 40.62 Isoelectric point 5.7 Charge (pH=7) -6.53 2D Binding mode Binding energy (Kcal/mol) -7.42 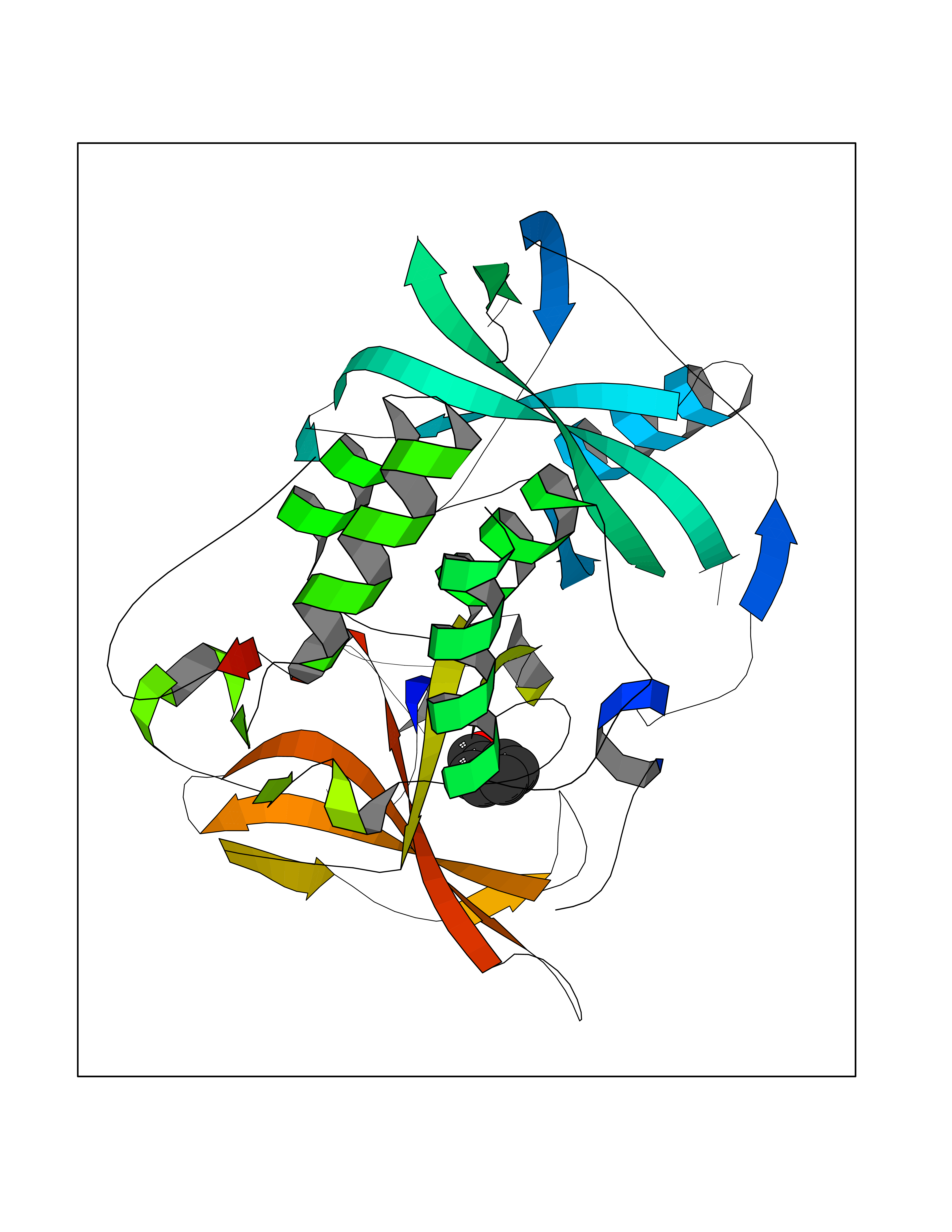 Molscript Map 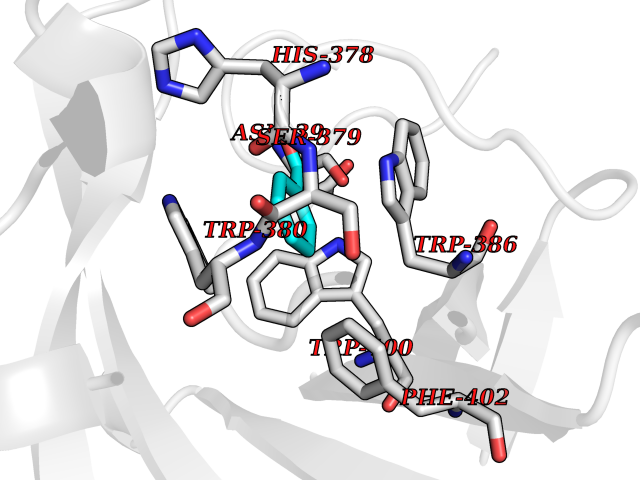 Pymol Map 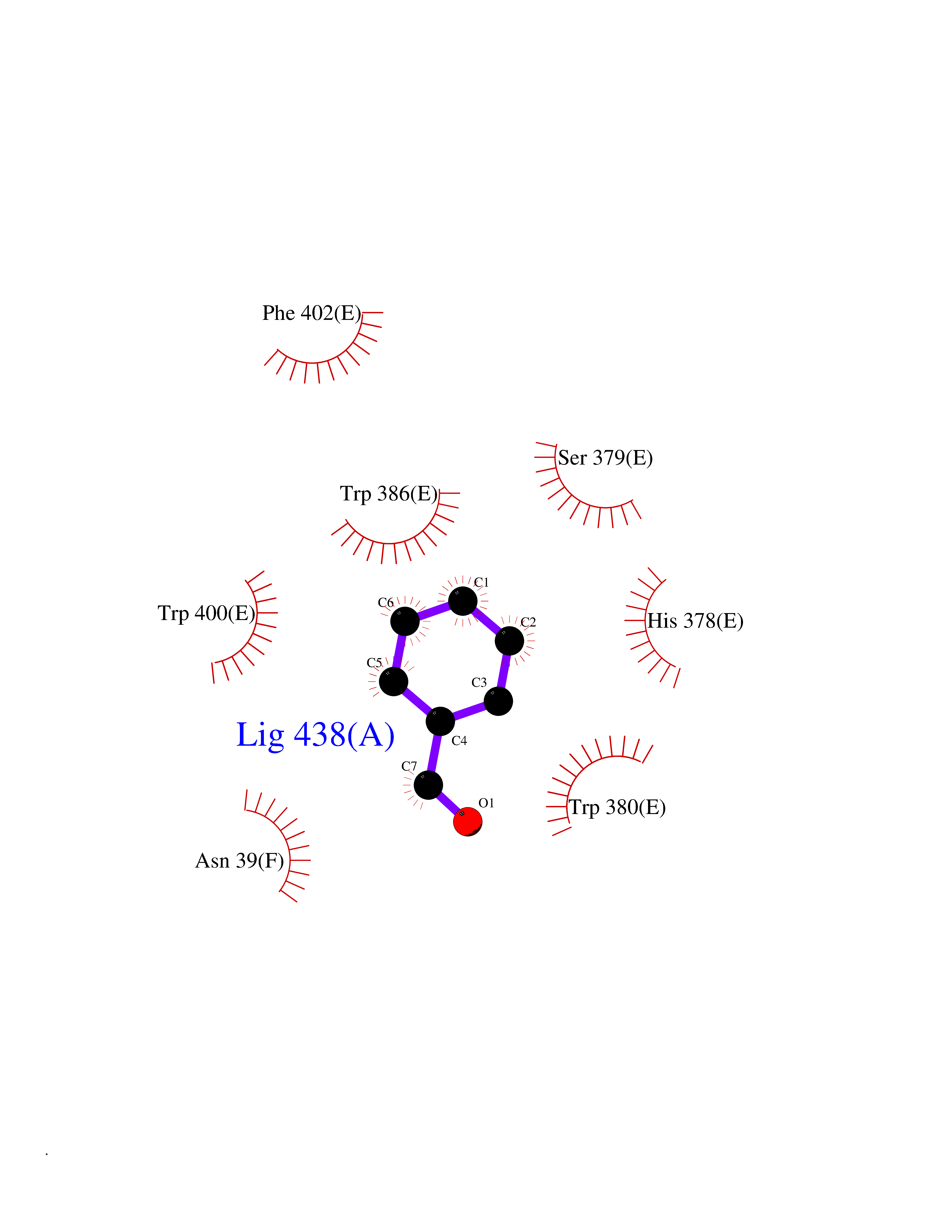 Ligplot Map 3D Binding mode Sequence EFIVGGKYKLNITNGEEVAVINFDTSLPTSHTYLGADMEEFHGRTLHDDDSCQVIPVLPQVMMILIPGQTLPLQLFHPQEVSMVRNLIQKDRTFAVLAYSNVQEREAQFGTTAEIYAYREEIVKVKAIGRQRFKVLEQQAKVQILPECVLAETLMDRIKKQLREWDENLKDDSLPSNPIDFSYRVAACLPIDDVLRIQLLKIGSAIQRLRCELDIMNKCTSLCCKQCQETEITTKNEIFSLSLCGPMAAYVNPHGYVHETLTVYKACNLNLIGRPSTEHSWFPGYAWTVAQCKICASHIGWKFTATKKDMSPQKFWGLTRSALLPTIPDTEDEISPD Hydrogen bonds contact Hydrophobic contact | ||||
| 3 | Nitric-oxide synthase brain (NOS1) | 5ADF | 5.41 | |
Target general information Gen name NOS1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Peptidyl-cysteine S-nitrosylase NOS1; Nitric oxide synthase, brain; Neuronal NOS; NOS, type I; NOS type I; NNOS; NC-NOS; N-NOS; BNOS Protein family NOS family Biochemical class Paired donor oxygen oxidoreductase Function In the brain and peripheral nervous system, NO displays many properties of a neurotransmitter. Probably has nitrosylase activity and mediates cysteine S-nitrosylation of cytoplasmic target proteins such SRR. Produces nitric oxide (NO) which is a messenger molecule with diverse functions throughout the body. Related diseases Variation Asp-298 in NOS3 may be associated with susceptibility to coronary spasm. {ECO:0000269|PubMed:11740345, ECO:0000269|PubMed:9737779}. Drugs (DrugBank ID) DB02143; DB02727; DB01997; DB03892; DB02207; DB03710; DB00155; DB00843; DB00997; DB03147; DB03247; DB01942; DB01221; DB02077; DB01821; DB09241; DB03144; DB03449; DB02044; DB02644; DB08019; DB08018; DB02027; DB03461; DB04223; DB06096; DB02991; DB03707 Interacts with Q08AM6 EC number EC 1.14.13.39 Uniprot keywords 3D-structure; Alternative splicing; Calmodulin-binding; Cell membrane; Cell projection; FAD; Flavoprotein; FMN; Heme; Iron; Membrane; Metal-binding; NADP; Oxidoreductase; Phosphoprotein; Proteomics identification; Reference proteome; Synapse; Ubl conjugation Protein physicochemical properties Chain ID A,B Molecular weight (Da) 34875.7 Length 299 Aromaticity 0.1 Instability index 42.94 Isoelectric point 5.96 Charge (pH=7) -6.25 2D Binding mode Binding energy (Kcal/mol) -7.38 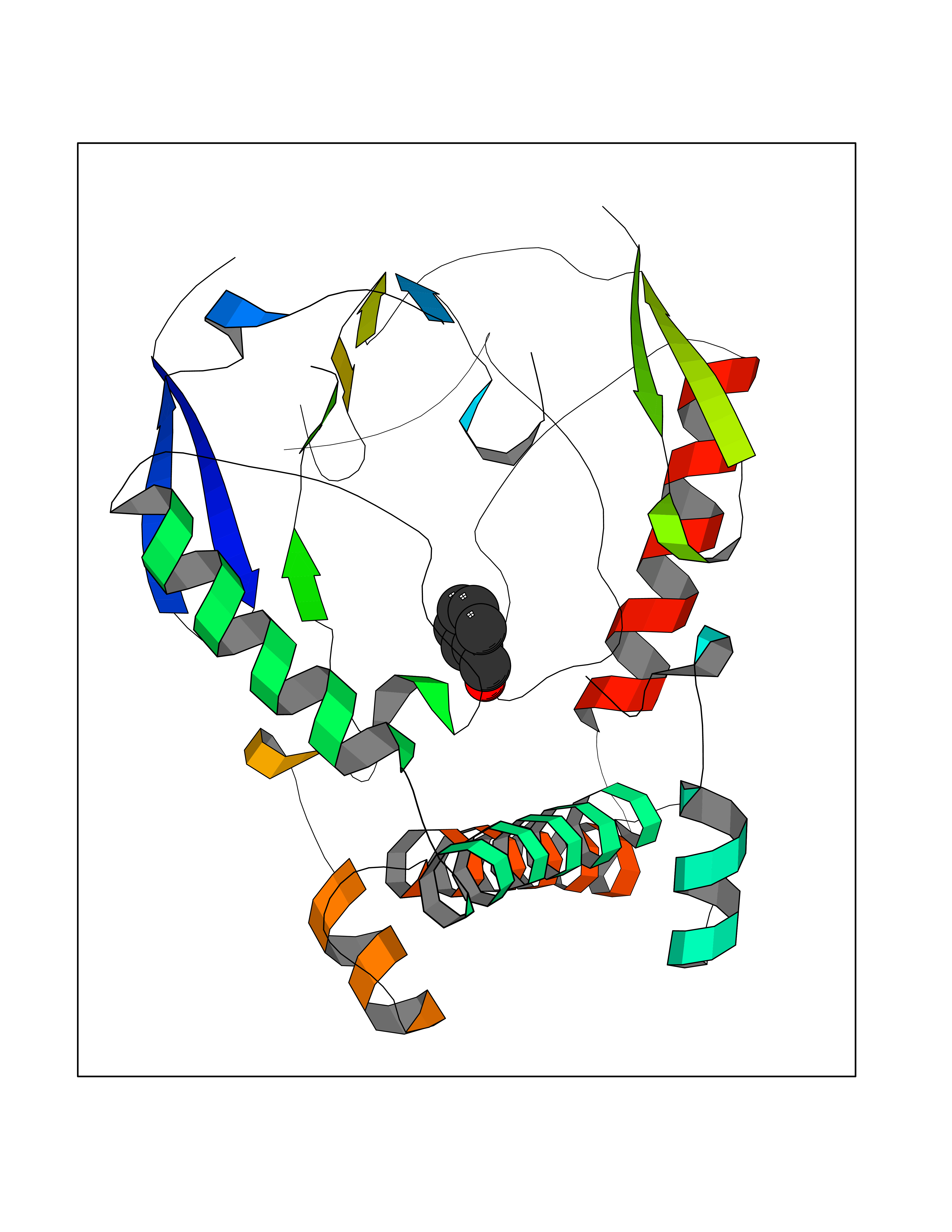 Molscript Map 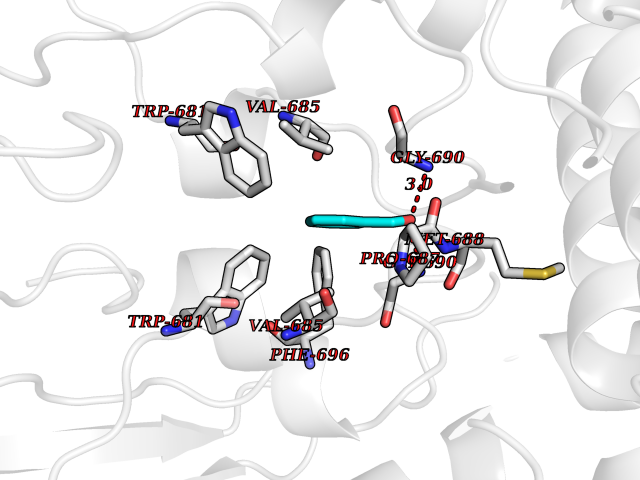 Pymol Map 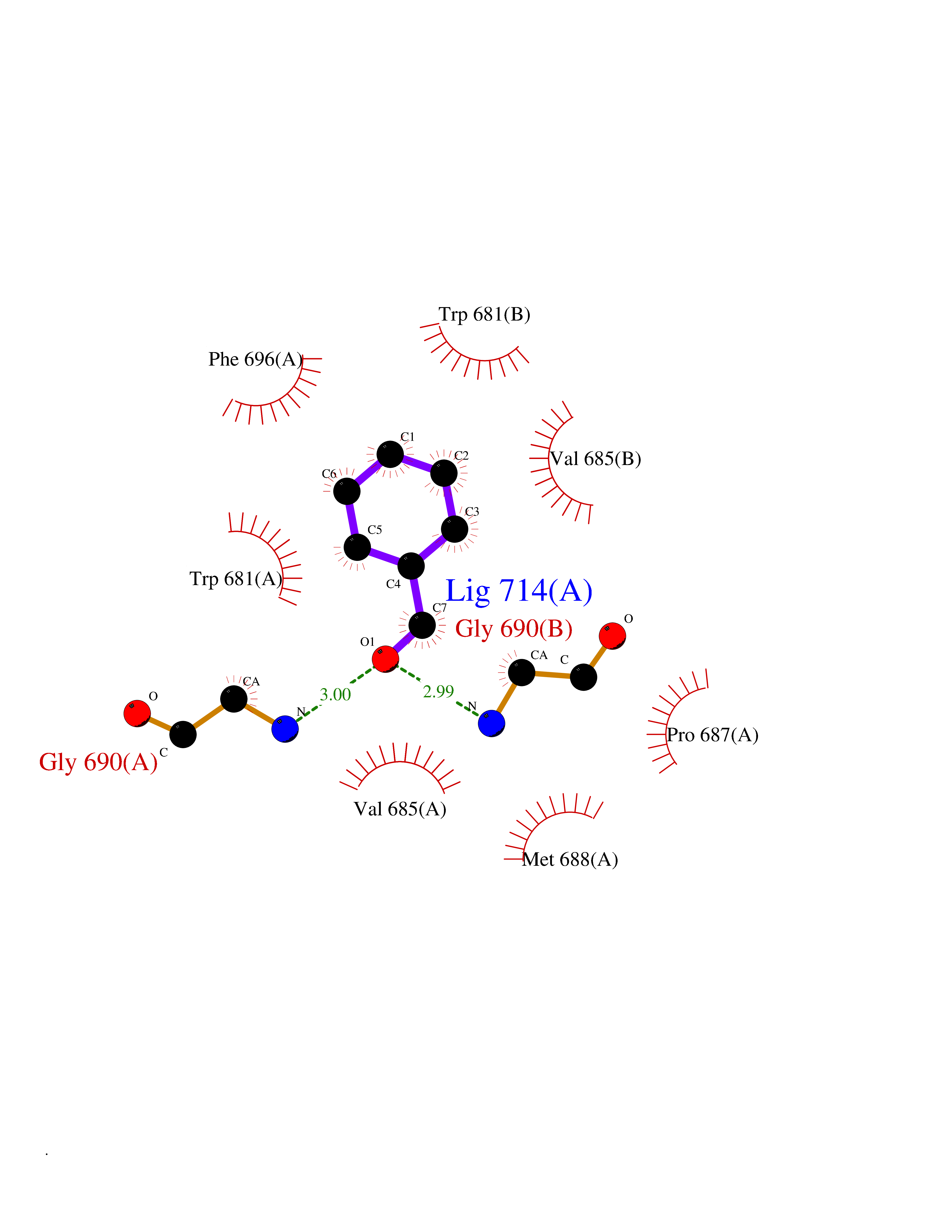 Ligplot Map 3D Binding mode Sequence CPRFLKVKNWETEVVLTDTLHLKSTLETGCTEYICMGSIMHPRDYCDNSRYNILEEVAKKMNLDMRKTSSLWKDQALVEINIAVLYSFQSDKVTIVDHHSATESFIKHMENEYRCRGGCPADWVWIVPPMSGSITPVFHQEMLNYRLRFLKVKNWETEVVLTDTLHLKSTLETGCTEYICMGSIMHPRDYCDNSRYNILEEVAKKMNLDMRKTSSLWKDQALVEINIAVLYSFQSDKVTIVDHHSATESFIKHMENEYRCRGGCPADWVWIVPPMSGSITPVFHQEMLNYRLTPSFEYQ Hydrogen bonds contact Hydrophobic contact | ||||
| 4 | Cerebron E3 ubiquitin ligase complex (CRL4-CRBN E3 ubiquitin ligase) | 4CI1 | 5.36 | |
Target general information Gen name CUL4A/CUL4B-DDB1-CRBN Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms NA Protein family Cullin family Biochemical class NA Function NA Related diseases Orotic aciduria 1 (ORAC1) [MIM:258900]: A disorder of pyrimidine metabolism resulting in megaloblastic anemia and orotic acid crystalluria that is frequently associated with some degree of physical and intellectual disability. A minority of cases have additional features, particularly congenital malformations and immune deficiencies. {ECO:0000269|PubMed:9042911}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P54253; Q86VP6; Q16531; Q92466; P08238; O94888; P55072 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Biological rhythms; DNA damage; DNA repair; Host-virus interaction; Isopeptide bond; Phosphoprotein; Proteomics identification; Reference proteome; Ubl conjugation; Ubl conjugation pathway Protein physicochemical properties Chain ID B Molecular weight (Da) 42669.7 Length 368 Aromaticity 0.1 Instability index 44.94 Isoelectric point 8.72 Charge (pH=7) 6.58 2D Binding mode Binding energy (Kcal/mol) -7.31  Molscript Map 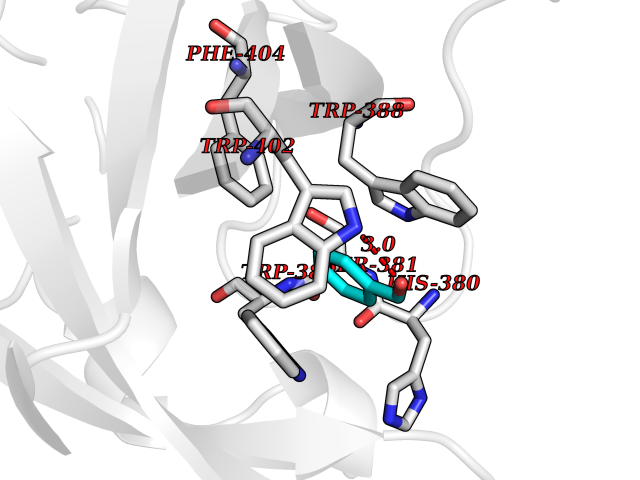 Pymol Map  Ligplot Map 3D Binding mode Sequence MINFDTSLPTSHMYLGSDMEEFHGRTLHDDDSCQVIPVLPHVMVMLIPGQTLPLQLFHPQEVSMVRNLIQKDRTFAVLAYSNVREREAHFGTTAEIYAYREEQEYGIETVKVKAIGRQRFKVLEIRTQSDGIQQAKVQILPERVLPSTMSAVQLQSLSRRHIRAFRQWWQKYQKRKFHCASLTSWPPWLYSLYDAETLMERVKRQLHEWDENLKDESLPTNPIDFSYRVAACLPIDDALRIQLLKIGSAIQRLRELDIMNKTSLCCKQCQDTEITTKNEIFSLSLCGPMAAYVNPHGYIHETLTVYKACNLNLSGRPSTEHSWFPGYAWTIAQCRICGNHMGWKFTATKKDMSPQKFWGLTRSALLPR Hydrogen bonds contact Hydrophobic contact | ||||
| 5 | Pseudomonas Transcriptional activator protein LasR (Pseudo LasR) | 3IX3 | 5.34 | |
Target general information Gen name Pseudo LasR Organism Pseudomonas aeruginosa (strain ATCC 15692 / DSM 22644 / CIP 104116 / JCM 14847 / LMG 12228 / 1C / PRS 101 / PAO1) Uniprot ID TTD ID Synonyms NA Protein family Autoinducer-regulated transcriptional regulatory protein family Biochemical class NA Function Transcriptional activator of elastase structural gene (LasB). Binds to the PAI autoinducer. Related diseases Growth hormone deficiency, isolated, 1A (IGHD1A) [MIM:262400]: An autosomal recessive, severe deficiency of growth hormone leading to dwarfism. Patients often develop antibodies to administered growth hormone. {ECO:0000269|PubMed:8364549}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Growth hormone deficiency, isolated, 1B (IGHD1B) [MIM:612781]: An autosomal recessive deficiency of growth hormone leading to short stature. Patients have low but detectable levels of growth hormone, significantly retarded bone age, and a positive response and immunologic tolerance to growth hormone therapy. {ECO:0000269|PubMed:12655557}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Kowarski syndrome (KWKS) [MIM:262650]: A syndrome clinically characterized by short stature associated with bioinactive growth hormone, normal or slightly increased growth hormone secretion, pathologically low insulin-like growth factor 1 levels, and normal catch-up growth on growth hormone replacement therapy. {ECO:0000269|PubMed:17519310, ECO:0000269|PubMed:8552145, ECO:0000269|PubMed:9276733}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Growth hormone deficiency, isolated, 2 (IGHD2) [MIM:173100]: An autosomal dominant deficiency of growth hormone leading to short stature. Clinical severity is variable. Patients have a positive response and immunologic tolerance to growth hormone therapy. {ECO:0000269|PubMed:11502836, ECO:0000269|PubMed:9152628}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08324 Interacts with NA EC number NA Uniprot keywords 3D-structure; Activator; DNA-binding; Quorum sensing; Reference proteome; Transcription; Transcription regulation Protein physicochemical properties Chain ID A Molecular weight (Da) 18305.5 Length 163 Aromaticity 0.12 Instability index 46.52 Isoelectric point 5.19 Charge (pH=7) -6.78 2D Binding mode Binding energy (Kcal/mol) -7.29 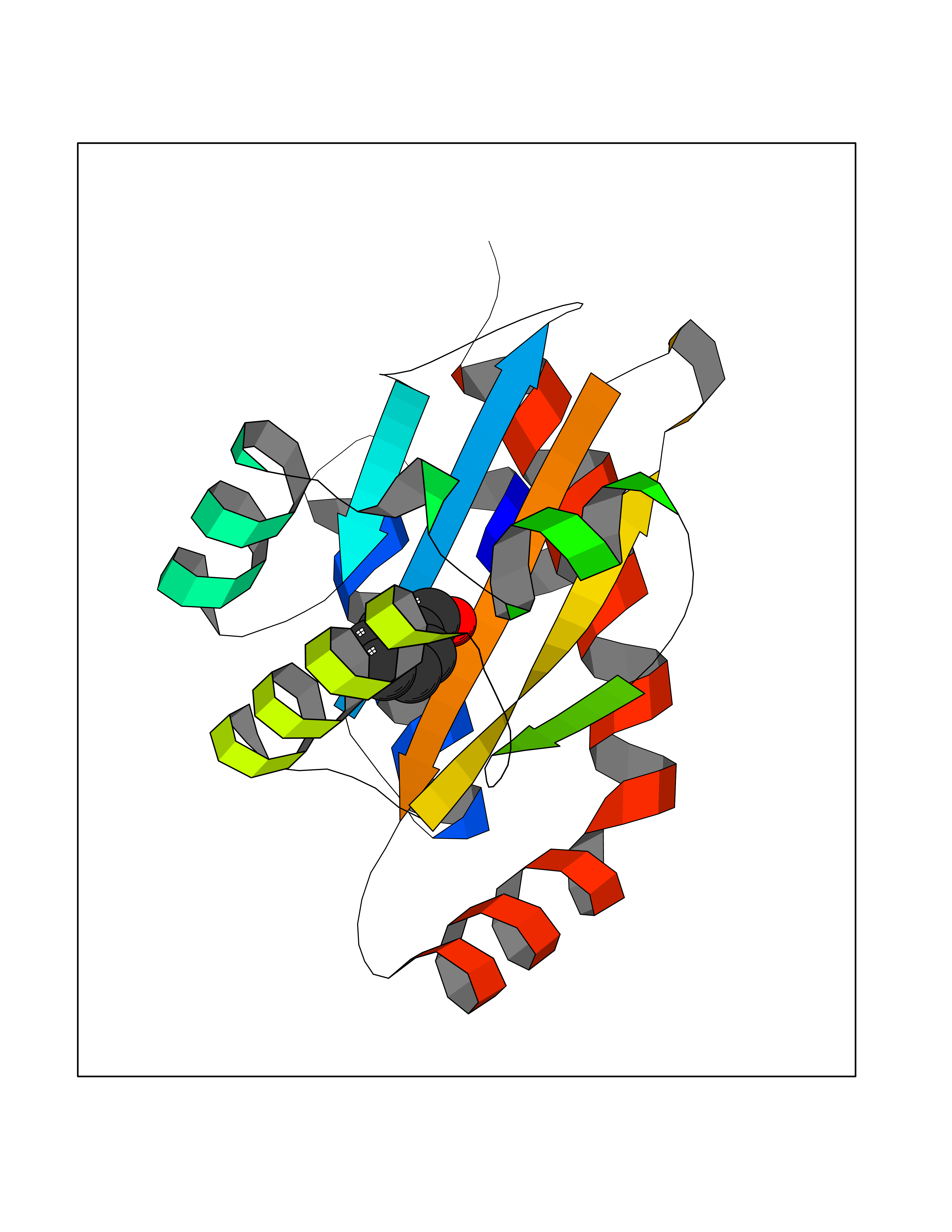 Molscript Map 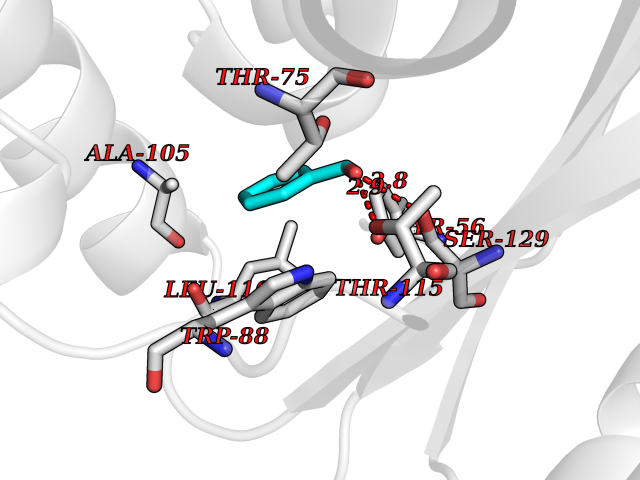 Pymol Map 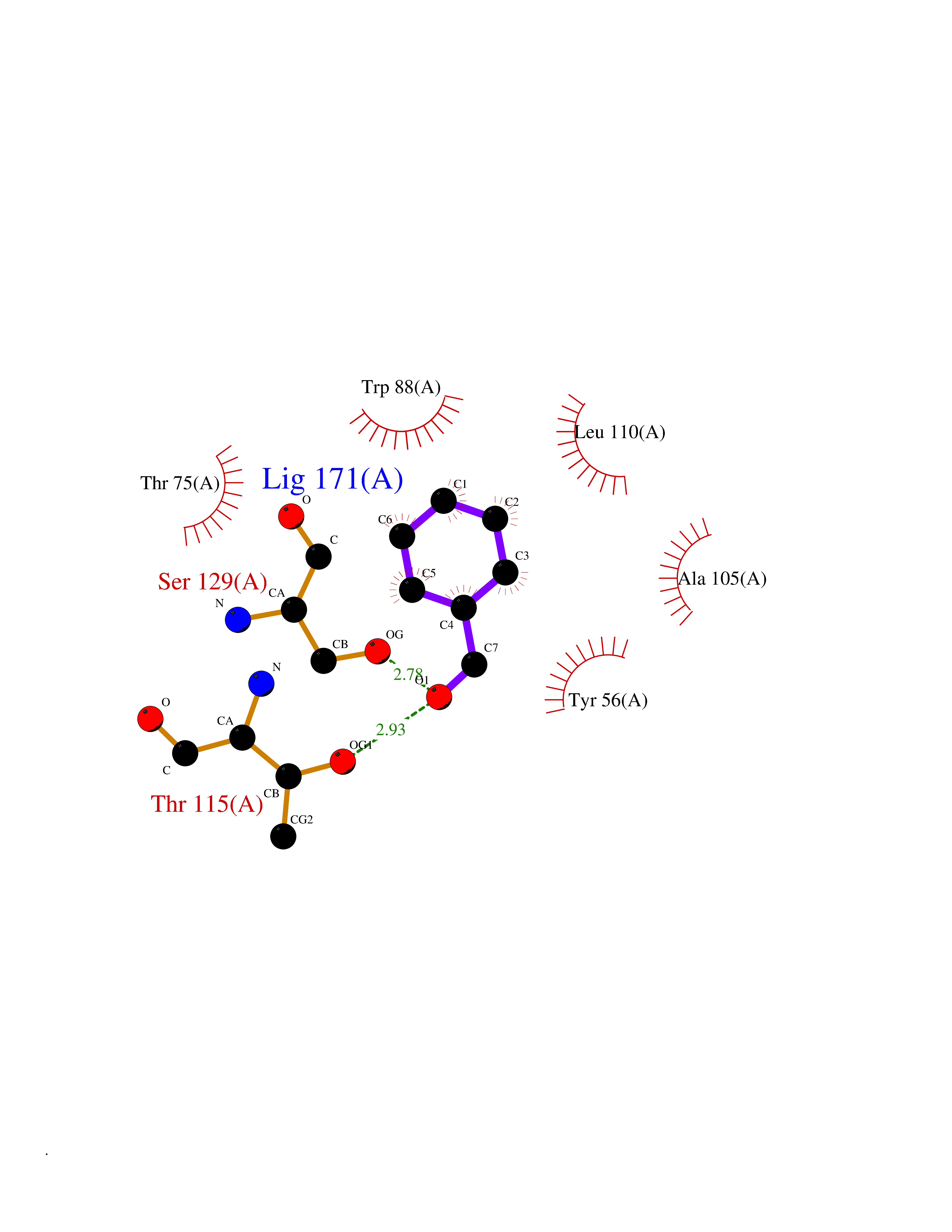 Ligplot Map 3D Binding mode Sequence FLELERSSGKLEWSAILQKMASDLGFSKILFGLLPKDSQDYENAFIVGNYPAAWREHYDRAGYARVDPTVSHCTQSVLPIFWEPSIYQTRKQHEFFEEASAAGLVYGLTMPLHGARGELGALSLSVEAENRAEANRFMESVLPTLWMLKDYALQSGAGLAFEH Hydrogen bonds contact Hydrophobic contact | ||||
| 6 | "Periplasmic trehalase (EC 3.2.1.28) (Alpha,alpha-trehalase) (Alpha,alpha-trehalose glucohydrolase) (Tre37A)" | 2JG0 | 5.33 | |
Target general information Gen name treA Organism Escherichia coli (strain K12) Uniprot ID TTD ID NA Synonyms JW1186;osmA;b1197 Protein family Glycosyl hydrolase 37 family Biochemical class NA Function Provides the cells with the ability to utilize trehalose at high osmolarity by splitting it into glucose molecules that can subsequently be taken up by the phosphotransferase-mediated uptake system. Related diseases SRC kinase activity has been shown to be increased in several tumor tissues and tumor cell lines such as colon carcinoma cells. {ECO:0000269|PubMed:2498394, ECO:0000269|PubMed:3093483}.; DISEASE: Thrombocytopenia 6 (THC6) [MIM:616937]: A form of thrombocytopenia, a hematologic disorder defined by a decrease in the number of platelets in circulating blood, resulting in the potential for increased bleeding and decreased ability for clotting. THC6 is an autosomal dominant form. Affected individuals may also have bone abnormalities and an increased risk for myelofibrosis. {ECO:0000269|PubMed:26936507}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number 3.2.1.28 Uniprot keywords 3D-structure; Direct protein sequencing; Glycosidase; Hydrolase; Periplasm; Reference proteome; Signal Protein physicochemical properties Chain ID A Molecular weight (Da) 57508.9 Length 507 Aromaticity 0.11 Instability index 48.32 Isoelectric point 5.48 Charge (pH=7) -10.13 2D Binding mode Binding energy (Kcal/mol) -7.27 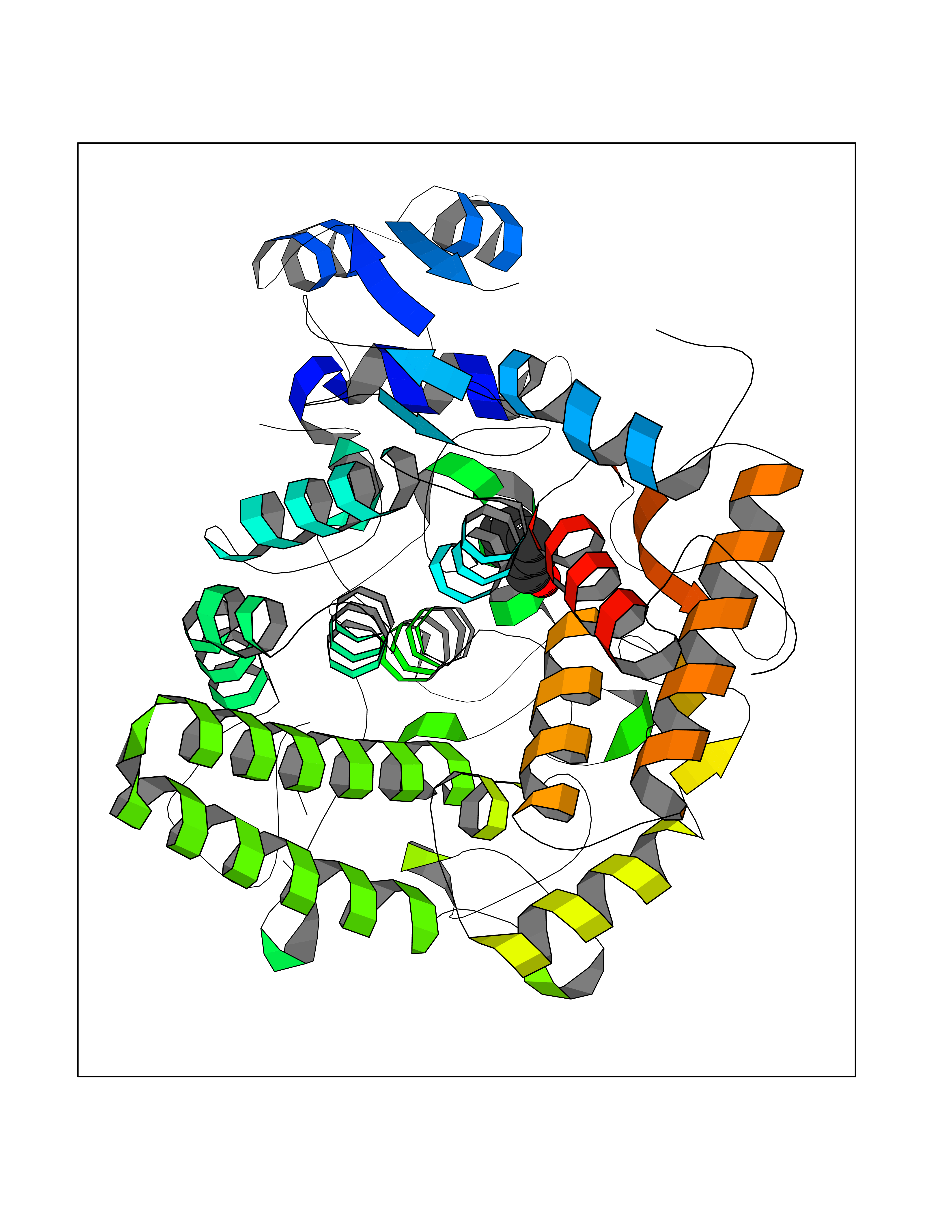 Molscript Map 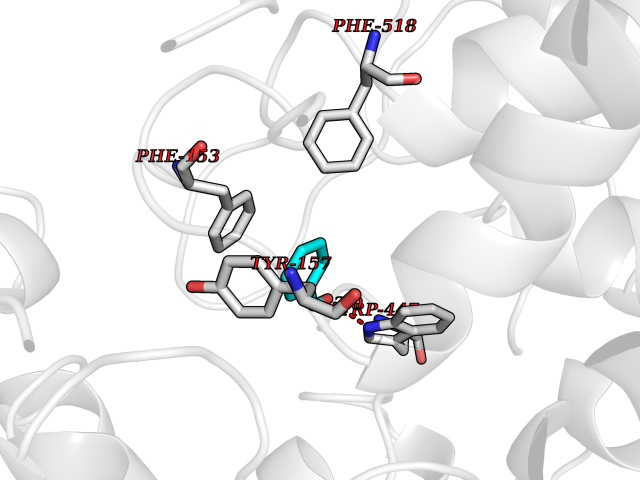 Pymol Map 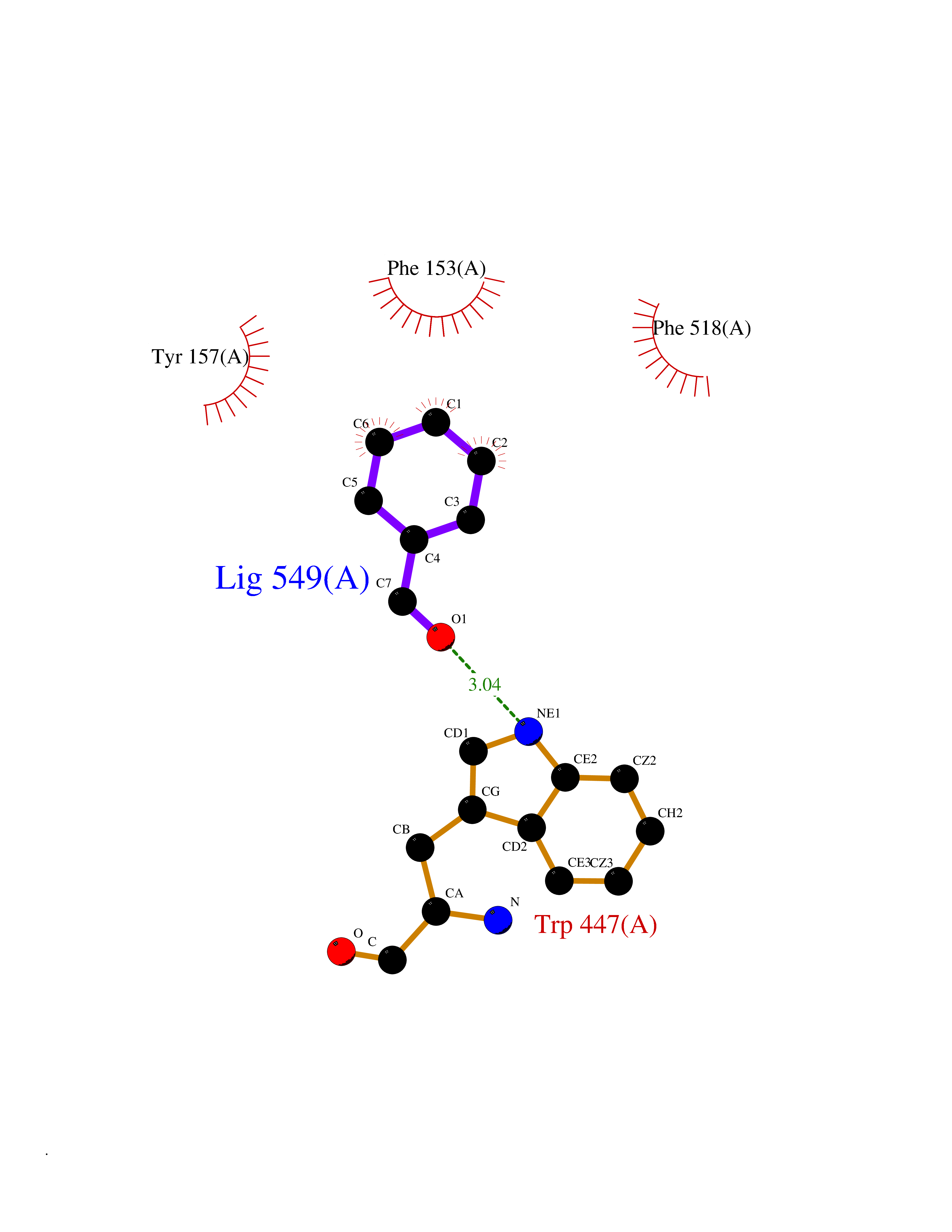 Ligplot Map 3D Binding mode Sequence PQPPDILLGPLFNDVQNAKLFPDQKTFADAVPNSDPLMILADYRMQQNQSGFDLRHFVNVNFTLPKYVPPEGQSLREHIDGLWPVLTRSTENTEKWDSLLPLPEPYVVPGGRFREVYYWDSYFTMLGLAESGHWDKVADMVANFAHEIDTYGHIPNGNRSYYLSRSQPPFFALMVELLAQHEGDAALKQYLPQMQKEYAYWMDGVENLQAGQQEKRVVKLQDGTLLNRYWDDRDTPRPESWVEDIATAKSNPNRPATEIYRDLRSAAASGWDFSSRWMDNPQQLNTLRTTSIVPVDLNSLMFKMEKILARASKAAGDNAMANQYETLANARQKGIEKYLWNDQQGWYADYDLKSHKVRNQLTAAALFPLYVNAAAKDRANKMATATKTHLLQPGGLNTTSVKSGQQWDAPNGWAPLQWVATEGLQNYGQKEVAMDISWHFLTNVQHTYDREKKLVEKYDVSTTGTGGGGGEYPLQDGFGWTNGVTLKMLDLICPKEQPCDNVPATRP Hydrogen bonds contact Hydrophobic contact | ||||
| 7 | Retinoic acid receptor beta (RARB) | 4DM6 | 5.33 | |
Target general information Gen name RARB Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms RAR-epsilon; RAR-beta; Nuclear receptor subfamily 1 group B member 2; NR1B2; HBV-activated protein; HAP Protein family Nuclear hormone receptor family, NR1 subfamily Biochemical class Nuclear hormone receptor Function Retinoic acid receptors bind as heterodimers to their target response elements in response to their ligands, all-trans or 9-cis retinoic acid, and regulate gene expression in various biological processes. The RXR/RAR heterodimers bind to the retinoic acid response elements (RARE) composed of tandem 5'-AGGTCA-3' sites known as DR1-DR5. In the absence or presence of hormone ligand, acts mainly as an activator of gene expression due to weak binding to corepressors. In concert with RARG, required for skeletal growth, matrix homeostasis and growth plate function. Receptor for retinoic acid. Related diseases Microphthalmia, syndromic, 12 (MCOPS12) [MIM:615524]: A form of microphthalmia, a disorder of eye formation, ranging from small size of a single eye to complete bilateral absence of ocular tissues (anophthalmia). In many cases, microphthalmia/anophthalmia occurs in association with syndromes that include non-ocular abnormalities. MCOPS12 patients manifest variable features, including diaphragmatic hernia, pulmonary hypoplasia, and cardiac abnormalities. {ECO:0000269|PubMed:24075189, ECO:0000269|PubMed:27120018}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00459; DB00210; DB00523; DB02877; DB00926; DB05785; DB04942; DB00799; DB00755; DB12808 Interacts with O95273; P50222; Q9UBK2; P62195; P28702; P28702-3; P48443; P03255 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; Disease variant; DNA-binding; Metal-binding; Microphthalmia; Nucleus; Phosphoprotein; Proto-oncogene; Receptor; Reference proteome; Transcription; Transcription regulation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A,B Molecular weight (Da) 25904.1 Length 229 Aromaticity 0.06 Instability index 44.34 Isoelectric point 7.55 Charge (pH=7) 0.73 2D Binding mode Binding energy (Kcal/mol) -7.26  Molscript Map 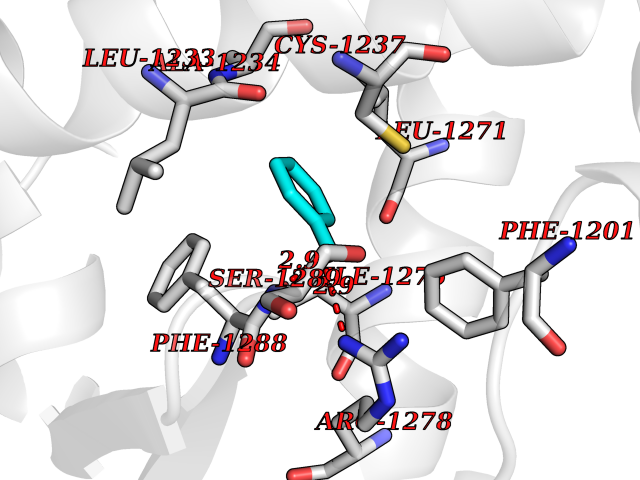 Pymol Map 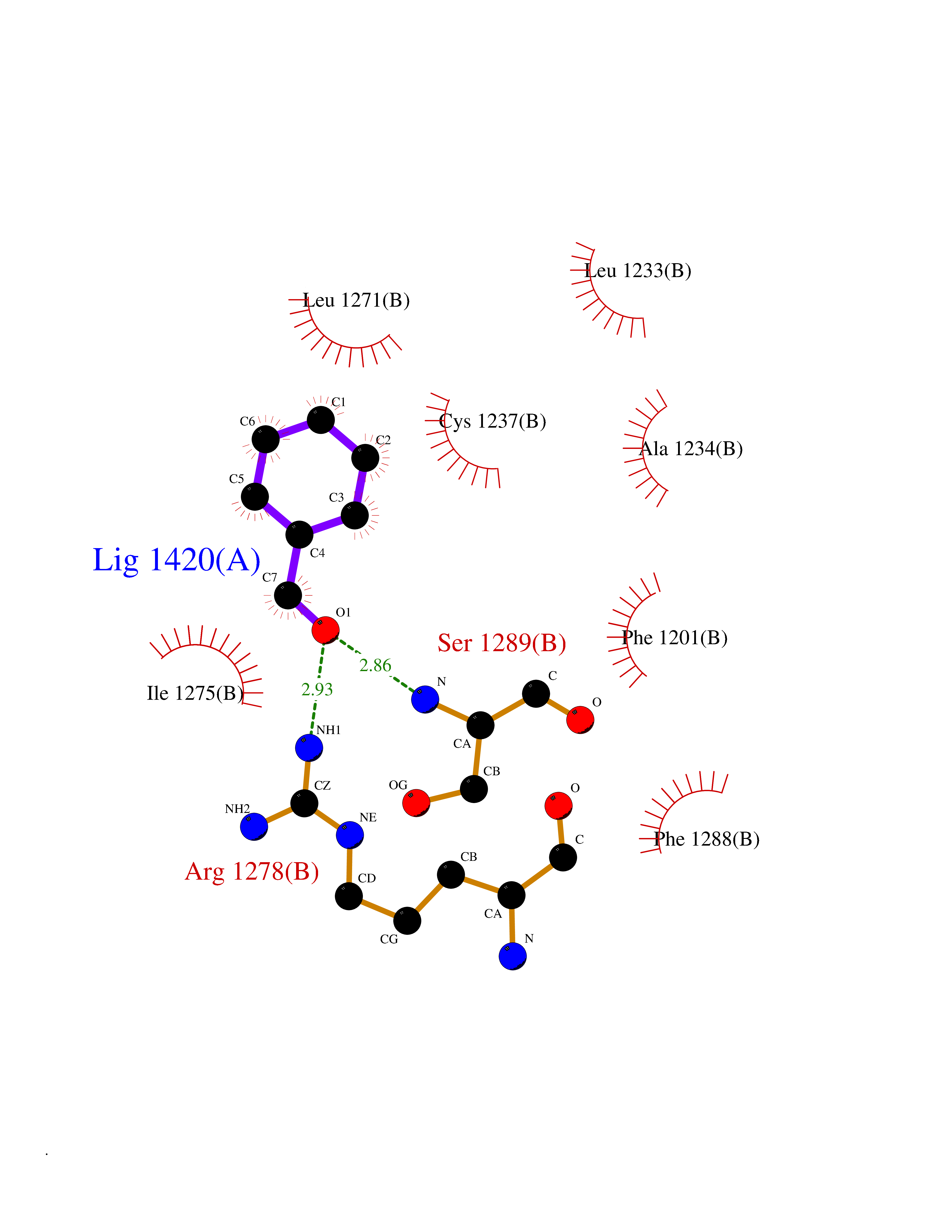 Ligplot Map 3D Binding mode Sequence TEKIRKAHQETFPSLCQLGKYTTNSSADHRVRLDLGLWDKFSELATKCIIKIVEFAKRLPGFTGLTIADQITLLKAACLDILILRICTRYTPEQDTMTFSDGLTLNRTQMHNAGFGPLTDLVFTFANQLLPLEMDDTETGLLSAICLICGDRQDLEEPTKVDKLQEPLLEALKIYIRKRRPSKPHMFPKILMKITDLRSISAKGAERVITLKMEIPGSMPPLIQEMLEN Hydrogen bonds contact Hydrophobic contact | ||||
| 8 | Tyrosine-protein kinase BTK (ATK) | 4RFZ | 5.33 | |
Target general information Gen name BTK Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Bruton's tyrosine kinase; Bruton tyrosine kinase; BPK; B-cell progenitor kinase; B cell progenitor kinase; Agammaglobulinemia tyrosine kinase; Agammaglobulinaemia tyrosine kinase; AGMX1 Protein family Protein kinase superfamily, Tyr protein kinase family, TEC subfamily Biochemical class Kinase Function Binding of antigen to the B-cell antigen receptor (BCR) triggers signaling that ultimately leads to B-cell activation. After BCR engagement and activation at the plasma membrane, phosphorylates PLCG2 at several sites, igniting the downstream signaling pathway through calcium mobilization, followed by activation of the protein kinase C (PKC) family members. PLCG2 phosphorylation is performed in close cooperation with the adapter protein B-cell linker protein BLNK. BTK acts as a platform to bring together a diverse array of signaling proteins and is implicated in cytokine receptor signaling pathways. Plays an important role in the function of immune cells of innate as well as adaptive immunity, as a component of the Toll-like receptors (TLR) pathway. The TLR pathway acts as a primary surveillance system for the detection of pathogens and are crucial to the activation of host defense. Especially, is a critical molecule in regulating TLR9 activation in splenic B-cells. Within the TLR pathway, induces tyrosine phosphorylation of TIRAP which leads to TIRAP degradation. BTK plays also a critical role in transcription regulation. Induces the activity of NF-kappa-B, which is involved in regulating the expression of hundreds of genes. BTK is involved on the signaling pathway linking TLR8 and TLR9 to NF-kappa-B. Transiently phosphorylates transcription factor GTF2I on tyrosine residues in response to BCR. GTF2I then translocates to the nucleus to bind regulatory enhancer elements to modulate gene expression. ARID3A and NFAT are other transcriptional target of BTK. BTK is required for the formation of functional ARID3A DNA-binding complexes. There is however no evidence that BTK itself binds directly to DNA. BTK has a dual role in the regulation of apoptosis. Non-receptor tyrosine kinase indispensable for B lymphocyte development, differentiation and signaling. Related diseases X-linked agammaglobulinemia (XLA) [MIM:300755]: Humoral immunodeficiency disease which results in developmental defects in the maturation pathway of B-cells. Affected boys have normal levels of pre-B-cells in their bone marrow but virtually no circulating mature B-lymphocytes. This results in a lack of immunoglobulins of all classes and leads to recurrent bacterial infections like otitis, conjunctivitis, dermatitis, sinusitis in the first few years of life, or even some patients present overwhelming sepsis or meningitis, resulting in death in a few hours. Treatment in most cases is by infusion of intravenous immunoglobulin. {ECO:0000269|PubMed:10220140, ECO:0000269|PubMed:10612838, ECO:0000269|PubMed:10678660, ECO:0000269|PubMed:7627183, ECO:0000269|PubMed:7633420, ECO:0000269|PubMed:7633429, ECO:0000269|PubMed:7711734, ECO:0000269|PubMed:7809124, ECO:0000269|PubMed:7849006, ECO:0000269|PubMed:7849697, ECO:0000269|PubMed:7849721, ECO:0000269|PubMed:7880320, ECO:0000269|PubMed:7897635, ECO:0000269|PubMed:8013627, ECO:0000269|PubMed:8162018, ECO:0000269|PubMed:8162056, ECO:0000269|PubMed:8594569, ECO:0000269|PubMed:8634718, ECO:0000269|PubMed:8695804, ECO:0000269|PubMed:8723128, ECO:0000269|PubMed:8834236, ECO:0000269|PubMed:9016530, ECO:0000269|PubMed:9260159, ECO:0000269|PubMed:9280283, ECO:0000269|PubMed:9445504, ECO:0000269|PubMed:9545398}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Growth hormone deficiency, isolated, 3, with agammaglobulinemia (IGHD3) [MIM:307200]: An X-linked recessive disorder characterized by growth hormone deficiency, short stature, delayed bone age, agammaglobulinemia with markedly reduced numbers of B cells, and good response to treatment with growth hormone. {ECO:0000269|PubMed:8013627}. The disease may be caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB15327; DB11703; DB15347; DB01254; DB15170; DB14785; DB12010; DB09053; DB01863; DB17472; DB14924; DB11764; DB15227; DB16657; DB05204; DB15035 Interacts with Q13444; Q99856; Q8WV28; Q06187; P78347; P08238; Q9BVA0; P21145; P50222; Q04759; O60239; P42768 EC number EC 2.7.10.2 Uniprot keywords 3D-structure; Acetylation; Adaptive immunity; Alternative promoter usage; Apoptosis; ATP-binding; Cell membrane; Cytoplasm; Direct protein sequencing; Disease variant; Dwarfism; Immunity; Innate immunity; Kinase; Lipid-binding; Membrane; Metal-binding; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; SH2 domain; SH3 domain; Transcription; Transcription regulation; Transferase; Tyrosine-protein kinase; Zinc; Zinc-finger Protein physicochemical properties Chain ID A Molecular weight (Da) 30491.7 Length 263 Aromaticity 0.12 Instability index 45.93 Isoelectric point 5.39 Charge (pH=7) -7.65 2D Binding mode Binding energy (Kcal/mol) -7.27 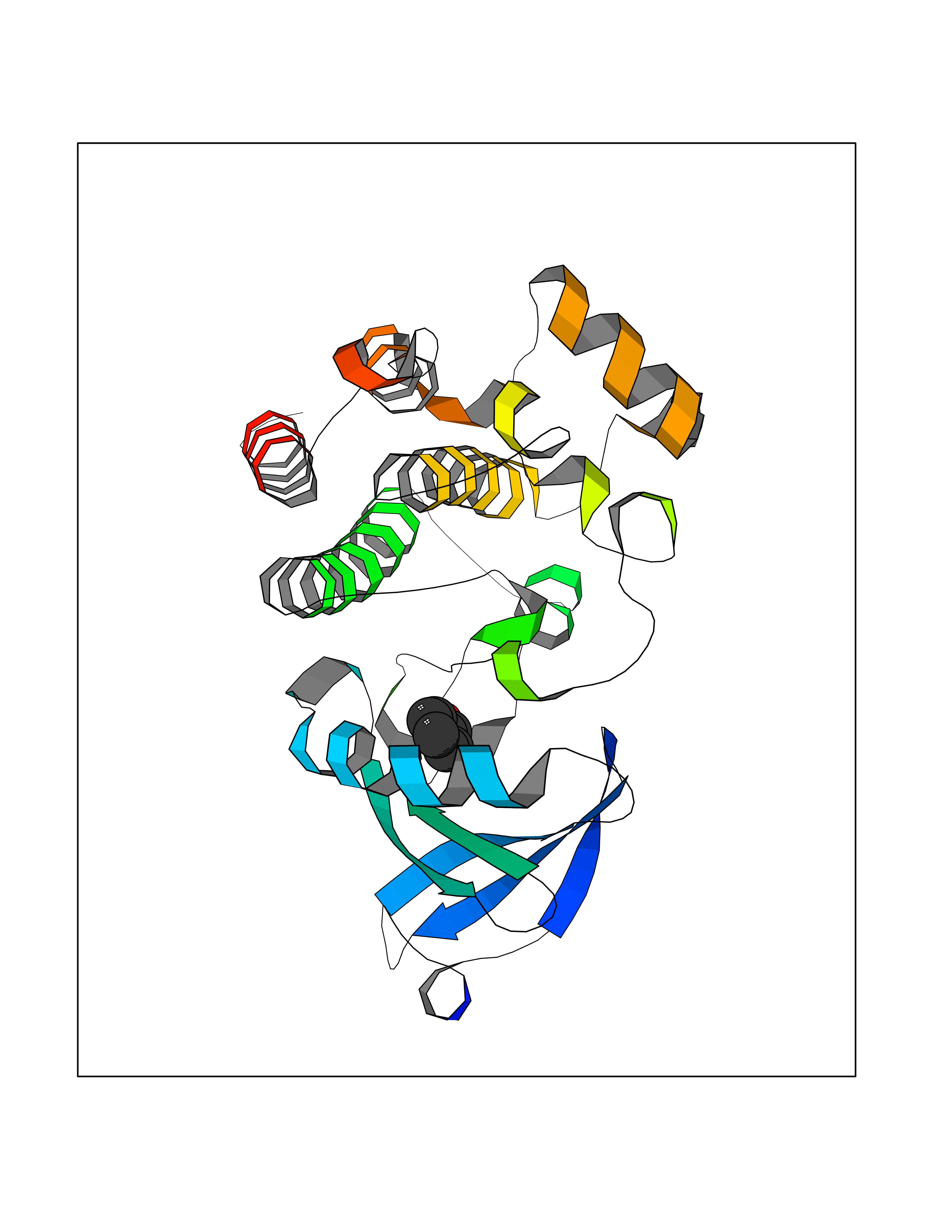 Molscript Map 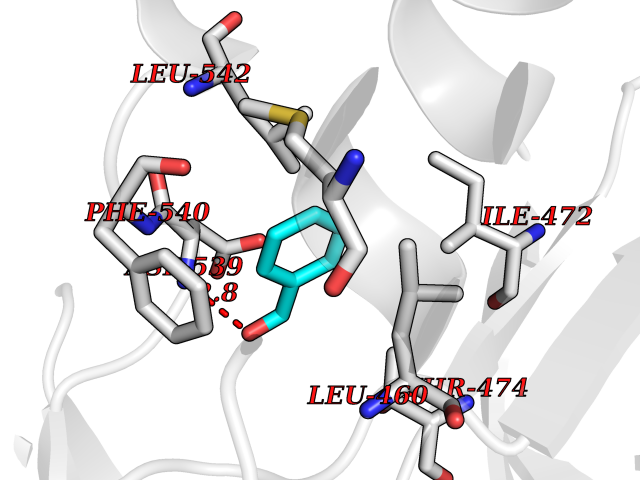 Pymol Map 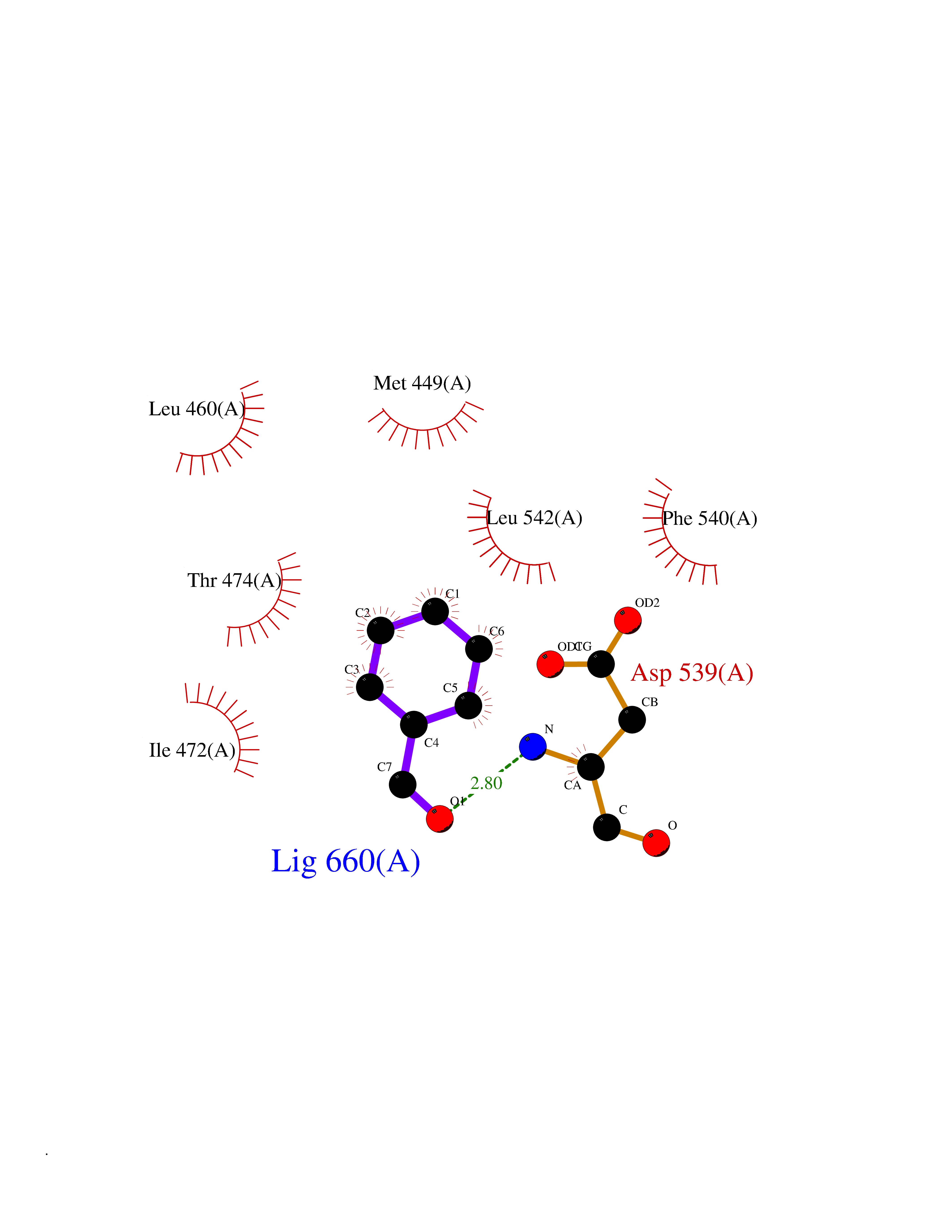 Ligplot Map 3D Binding mode Sequence EIDPKDLTFLKELGTGQFGVVKYGKWRGQYDVAIKMIKEGSMSEDEFIEEAKVMMNLSHEKLVQLYGVCTKQRPIFIITEYMANGCLLNYLREARHAFQTQQLLEMCKDVCEAMEYLESKQFLHRDLAARNCLVNDQGVVKVSDFGLSRYVLDDEYTSSVGSKFPVRWSPPEVLMYSKFSSKSDIWAFGVLMWEIYSLGKMPYERFTNSETAEHIAQGLRLYRPHLASAAVYTIMYSCWHEKADERPTFKILLSNILDVMDEE Hydrogen bonds contact Hydrophobic contact | ||||
| 9 | Histone-lysine N-methyltransferase KMT5B (KMT5B) | 3S8P | 5.33 | |
Target general information Gen name KMT5B Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Lysine N-methyltransferase 5B; Lysine-specific methyltransferase 5B; Suppressor of variegation 4-20 homolog 1; Su(var)4-20 homolog 1; Suv4-20h1; [histone H4]-N-methyl-L-lysine20 N-methyltransferase KM Protein family Class V-like SAM-binding methyltransferase superfamily, Histone-lysine methyltransferase family, Suvar4-20 subfamily Biochemical class NA Function Histone methyltransferase that specifically methylates monomethylated 'Lys-20' (H4K20me1) and dimethylated 'Lys-20' (H4K20me2) of histone H4 to produce respectively dimethylated 'Lys-20' (H4K20me2) and trimethylated 'Lys-20' (H4K20me3) and thus regulates transcription and maintenance of genome integrity. In vitro also methylates unmodified 'Lys-20' (H4K20me0) of histone H4 and nucleosomes. H4 'Lys-20' trimethylation represents a specific tag for epigenetic transcriptional repression. Mainly functions in pericentric heterochromatin regions, thereby playing a central role in the establishment of constitutive heterochromatin in these regions. KMT5B is targeted to histone H3 via its interaction with RB1 family proteins (RB1, RBL1 and RBL2) (By similarity). Plays a role in myogenesis by regulating the expression of target genes, such as EID3. Facilitates TP53BP1 foci formation upon DNA damage and proficient non-homologous end-joining (NHEJ)-directed DNA repair by catalyzing the di- and trimethylation of 'Lys-20' of histone H4. May play a role in class switch reconbination by catalyzing the di- and trimethylation of 'Lys-20' of histone H4 (By similarity). Related diseases Intellectual developmental disorder, autosomal dominant 51 (MRD51) [MIM:617788]: A disorder characterized by significantly below average general intellectual functioning associated with impairments in adaptive behavior and manifested during the developmental period. {ECO:0000269|PubMed:28191889, ECO:0000269|PubMed:29276005}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q9H2G4; Q61026 EC number EC 2.1.1.361 Uniprot keywords 3D-structure; Alternative splicing; Chromatin regulator; Chromosome; Disease variant; Intellectual disability; Isopeptide bond; Metal-binding; Methyltransferase; Myogenesis; Nucleus; Proteomics identification; Reference proteome; Repressor; S-adenosyl-L-methionine; Transcription; Transcription regulation; Transferase; Ubl conjugation; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 26230.2 Length 233 Aromaticity 0.11 Instability index 42.29 Isoelectric point 5.64 Charge (pH=7) -6.07 2D Binding mode Binding energy (Kcal/mol) -7.27 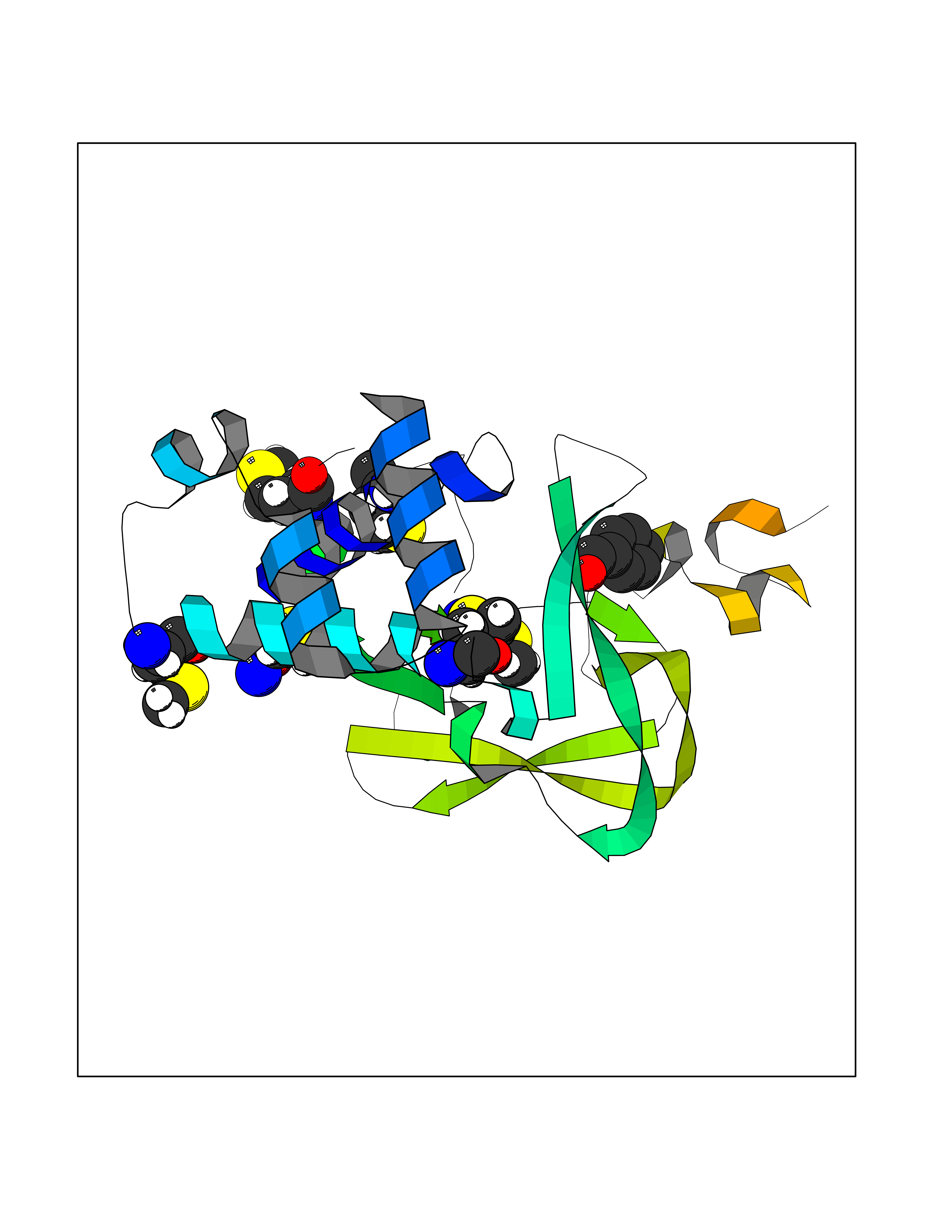 Molscript Map 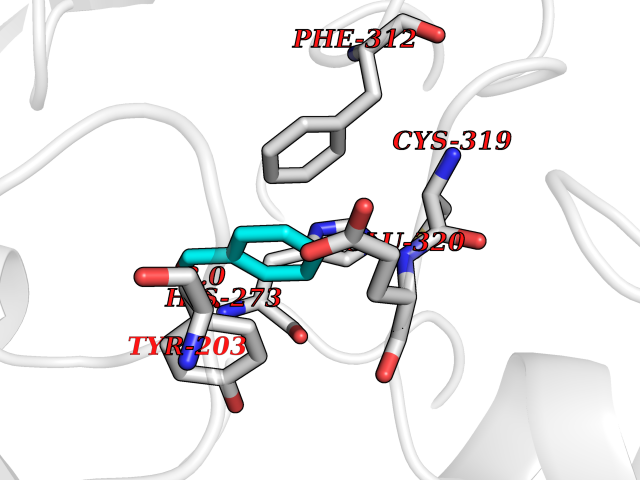 Pymol Map 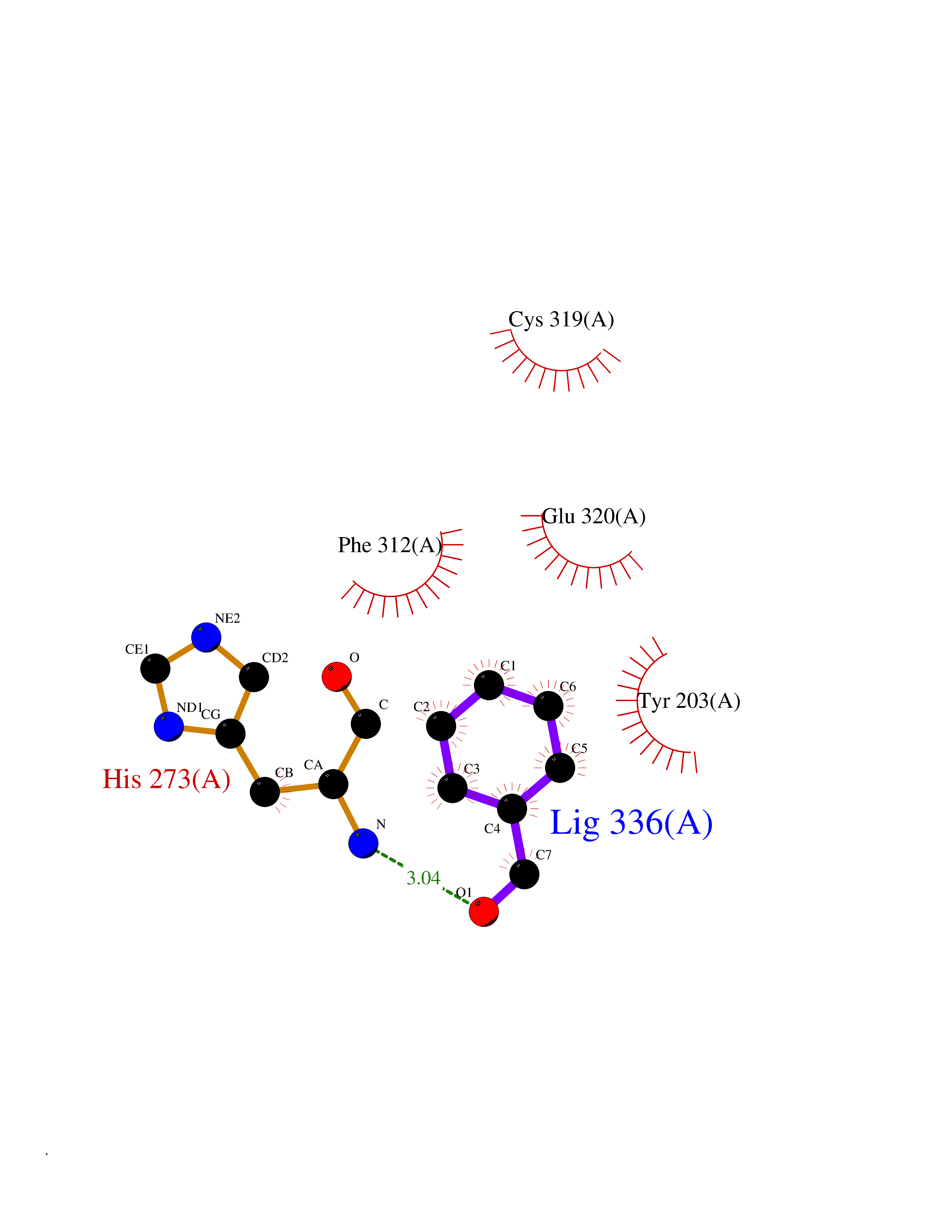 Ligplot Map 3D Binding mode Sequence XSAKELCENDDLATSLVLDPYLGFQTHKXNTRQEELKEVIERFKKDEHLEKAFKCLTSGEWARHYFLNKNKXQEKLFKEHVFIYLRXFATDSGFEILPCNRYSSEQNGAKIVATKEWKRNDKIELLVGCIAELSEIEENXLLRHGENDFSVXYSTRKNCAQLWLGPAAFINHDCRPNCKFVSTGRDTACVKALRDIEPGEEISCYYGDGFFGENNEFCECYTCERRGTGAFKS Hydrogen bonds contact Hydrophobic contact | ||||
| 10 | Aldose reductase (AKR1B1) | 1US0 | 5.32 | |
Target general information Gen name AKR1B1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Aldehyde reductase; AKR1B1 Protein family Aldo/keto reductase family Biochemical class Short-chain dehydrogenases reductase Function Catalyzes the NADPH-dependent reduction of a wide variety of carbonyl-containing compounds to their corresponding alcohols with a broad range of catalytic efficiencies. Related diseases Glutamine deficiency, congenital (GLND) [MIM:610015]: An autosomal recessive disorder characterized by variable brain malformations, encephalopathy, severe developmental delay, seizures, and decreased glutamine levels in bodily fluids. Death in early infancy may occur. {ECO:0000269|PubMed:16267323, ECO:0000269|PubMed:26711351, ECO:0000269|PubMed:38579670}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Developmental and epileptic encephalopathy 116 (DEE116) [MIM:620806]: A form of epileptic encephalopathy, a heterogeneous group of early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE116 is autosomal dominant form characterized by severe developmental delay, seizures, and white matter abnormalities. {ECO:0000269|PubMed:38579670}. The disease is caused by variants affecting the gene represented in this entry. DEE116 is caused by variants that disrupt the canonical translation start codon in GLUL resulting in initiation of translation at Met-18 (PubMed:38579670). The resulting protein is enzymatically competent but insensitive to negative feedback regulation via glutamine-induced degradation. {ECO:0000269|PubMed:38579670}. Drugs (DrugBank ID) DB07028; DB07030; DB07450; DB02101; DB08449; DB08000; DB07139; DB07498; DB02007; DB02020; DB11859; DB02994; DB04272; DB07187; DB00694; DB00997; DB06246; DB01039; DB02021; DB16707; DB00143; DB02834; DB08084; DB01689; DB07063; DB06077; DB02518; DB00157; DB03461; DB05383; DB05533; DB05327; DB02712; DB00605; DB02383; DB02132; DB08772; DB07093; DB07999; DB08098 Interacts with Q9BUY7 EC number EC 1.1.1.300 Uniprot keywords 3D-structure; Acetylation; Cytoplasm; Direct protein sequencing; Lipid metabolism; NADP; Oxidoreductase; Phosphoprotein; Proteomics identification; Reference proteome Protein physicochemical properties Chain ID A Molecular weight (Da) 35447.6 Length 313 Aromaticity 0.09 Instability index 36.41 Isoelectric point 7.1 Charge (pH=7) 0.26 2D Binding mode Binding energy (Kcal/mol) -7.26  Molscript Map 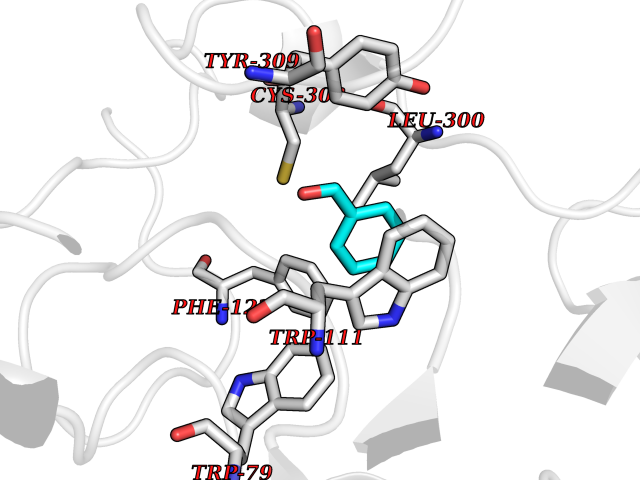 Pymol Map  Ligplot Map 3D Binding mode Sequence MASRILLNNGAKMPILGLGTWKSPPGQVTEAVKVAIDVGYRHIDCAHVYQNENEVGVAIQEKLREQVVKREELFIVSKLWCTYHEKGLVKGACQKTLSDLKLDYLDLYLIHWPTGFKPGKEFFPLDESGNVVPSDTNILDTWAAMEELVDEGLVKAIGISNFNHLQVEMILNKPGLKYKPAVNQIECHPYLTQEKLIQYCQSKGIVVTAYSPLGSPDRPWAKPEDPSLLEDPRIKAIAAKHNKTTAQVLIRFPMQRNLVVIPKSVTPERIAENFKVFDFELSSQDMTTLLSYNRNWRVCALLSCTSHKDYPFH Hydrogen bonds contact Hydrophobic contact | ||||
| 11 | Dopamine D2 receptor (D2R) | 5AER | 5.32 | |
Target general information Gen name DRD2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Dopamine receptor 2; D(2) dopamine receptor Protein family G-protein coupled receptor 1 family Biochemical class GPCR rhodopsin Function Dopamine receptor whose activity is mediated by G proteins which inhibit adenylyl cyclase. Related diseases Congenital sucrase-isomaltase deficiency (CSID) [MIM:222900]: Autosomal recessive intestinal disorder that is clinically characterized by fermentative diarrhea, abdominal pain, and cramps upon ingestion of sugar. The symptoms are the consequence of absent or drastically reduced enzymatic activities of sucrase and isomaltase. The prevalence of CSID is 0.02 % in individuals of European descent and appears to be much higher in Greenland, Alaskan, and Canadian native people. CSID arises due to post-translational perturbations in the intracellular transport, polarized sorting, aberrant processing, and defective function of SI. {ECO:0000269|PubMed:10903344, ECO:0000269|PubMed:11340066, ECO:0000269|PubMed:14724820, ECO:0000269|PubMed:16329100, ECO:0000269|PubMed:8609217}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01614; DB01063; DB01425; DB00915; DB06288; DB05964; DB00543; DB00182; DB04599; DB00714; DB01238; DB14185; DB09207; DB06216; DB04889; DB04888; DB05687; DB09223; DB04857; DB09128; DB01200; DB09018; DB00490; DB00248; DB06016; DB01038; DB00477; DB01239; DB00568; DB00363; DB01151; DB11274; DB13345; DB00320; DB01184; DB00988; DB00450; DB11275; DB01049; DB00696; DB01175; DB09194; DB00875; DB00623; DB04842; DB00502; DB04946; DB00458; DB04924; DB12579; DB01221; DB00555; DB01235; DB00589; DB00408; DB06077; DB08815; DB00934; DB09224; DB01043; DB00933; DB01403; DB01233; DB06148; DB00805; DB01618; DB08804; DB05766; DB00540; DB06229; DB00334; DB01267; DB12061; DB00715; DB01186; DB08922; DB00850; DB01100; DB09286; DB01621; DB12478; DB00413; DB00433; DB00420; DB01069; DB00777; DB01224; DB09097; DB12518; DB00409; DB00734; DB01549; DB00268; DB05271; DB06454; DB06144; DB00391; DB06477; DB04844; DB12093; DB00372; DB01622; DB00679; DB01623; DB13025; DB00831; DB00508; DB00726; DB06109; DB01392; DB00246; DB09225; DB01624 Interacts with Q9NRI5; P14416; Q01959 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disulfide bond; G-protein coupled receptor; Glycoprotein; Golgi apparatus; Lipoprotein; Membrane; Palmitate; Receptor; Reference proteome; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID B,C Molecular weight (Da) 24300.3 Length 209 Aromaticity 0.13 Instability index 40.14 Isoelectric point 4.97 Charge (pH=7) -7.83 2D Binding mode Binding energy (Kcal/mol) -7.26  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence PEVVEELTRKTYFTEKEVQQWYKGFIKDCPSGQLDAAGFQKIYKQFFPFGDPTKFATFVFNVFDENKDGRIEFSEFIQALSVTSRGTLDEKLRWAFKLYDLDNDGYITRNEMLDIVDAIYQMVGNTVELPEEENTPEKRVDRIFAMMDKNADGKLTLQEFQEGSKADPSIVQALSLYDGLVNIEFRKAFLKILHSNIEFRKAFLKILHS Hydrogen bonds contact Hydrophobic contact | ||||
| 12 | Retinoic acid receptor gamma (RARG) | 1FCY | 5.32 | |
Target general information Gen name RARG Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms RAR-gamma; Nuclear receptor subfamily 1 group B member 3; NR1B3 Protein family Nuclear hormone receptor family, NR1 subfamily Biochemical class Nuclear hormone receptor Function Receptor for retinoic acid. Retinoic acid receptors bind as heterodimers to their target response elements in response to their ligands, all-trans or 9-cis retinoic acid, and regulate gene expression in various biological processes. The RAR/RXR heterodimers bind to the retinoic acid response elements (RARE) composed of tandem 5'-AGGTCA-3' sites known as DR1-DR5. In the absence of ligand, acts mainly as an activator of gene expression due to weak binding to corepressors. Required for limb bud development. In concert with RARA or RARB, required for skeletal growth, matrix homeostasis and growth plate function (By similarity). Related diseases Cystic fibrosis (CF) [MIM:219700]: A common generalized disorder of the exocrine glands which impairs clearance of secretions in a variety of organs. It is characterized by the triad of chronic bronchopulmonary disease (with recurrent respiratory infections), pancreatic insufficiency (which leads to malabsorption and growth retardation) and elevated sweat electrolytes. It is the most common genetic disease in Caucasians, with a prevalence of about 1 in 2'000 live births. Inheritance is autosomal recessive. {ECO:0000269|PubMed:10094564, ECO:0000269|PubMed:10869121, ECO:0000269|PubMed:10923036, ECO:0000269|PubMed:11242048, ECO:0000269|PubMed:12167682, ECO:0000269|PubMed:12394343, ECO:0000269|PubMed:12529365, ECO:0000269|PubMed:1284466, ECO:0000269|PubMed:1284468, ECO:0000269|PubMed:1284529, ECO:0000269|PubMed:1284530, ECO:0000269|PubMed:1284548, ECO:0000269|PubMed:1379210, ECO:0000269|PubMed:15528182, ECO:0000269|PubMed:15716351, ECO:0000269|PubMed:16822950, ECO:0000269|PubMed:1695717, ECO:0000269|PubMed:1699669, ECO:0000269|PubMed:17098864, ECO:0000269|PubMed:1710600, ECO:0000269|PubMed:1712898, ECO:0000269|PubMed:17182731, ECO:0000269|PubMed:20008117, ECO:0000269|PubMed:20150177, ECO:0000269|PubMed:20691141, ECO:0000269|PubMed:21884936, ECO:0000269|PubMed:2236053, ECO:0000269|PubMed:23818989, ECO:0000269|PubMed:25330774, ECO:0000269|PubMed:26846474, ECO:0000269|PubMed:27241308, ECO:0000269|PubMed:28001373, ECO:0000269|PubMed:28067262, ECO:0000269|PubMed:28087700, ECO:0000269|PubMed:32026723, ECO:0000269|PubMed:33572515, ECO:0000269|PubMed:7504969, ECO:0000269|PubMed:7505694, ECO:0000269|PubMed:7505767, ECO:0000269|PubMed:7508414, ECO:0000269|PubMed:7513296, ECO:0000269|PubMed:7517264, ECO:0000269|PubMed:7520022, ECO:0000269|PubMed:7522211, ECO:0000269|PubMed:7524909, ECO:0000269|PubMed:7524913, ECO:0000269|PubMed:7525450, ECO:0000269|PubMed:7537150, ECO:0000269|PubMed:7541273, ECO:0000269|PubMed:7541510, ECO:0000269|PubMed:7543567, ECO:0000269|PubMed:7544319, ECO:0000269|PubMed:7581407, ECO:0000269|PubMed:7606851, ECO:0000269|PubMed:7680525, ECO:0000269|PubMed:7683628, ECO:0000269|PubMed:7683954, ECO:0000269|PubMed:8081395, ECO:0000269|PubMed:8406518, ECO:0000269|PubMed:8522333, ECO:0000269|PubMed:8723693, ECO:0000269|PubMed:8723695, ECO:0000269|PubMed:8800923, ECO:0000269|PubMed:8829633, ECO:0000269|PubMed:8910473, ECO:0000269|PubMed:8956039, ECO:0000269|PubMed:9101301, ECO:0000269|PubMed:9222768, ECO:0000269|PubMed:9375855, ECO:0000269|PubMed:9401006, ECO:0000269|PubMed:9443874, ECO:0000269|PubMed:9452048, ECO:0000269|PubMed:9452054, ECO:0000269|PubMed:9452073, ECO:0000269|PubMed:9482579, ECO:0000269|PubMed:9507391, ECO:0000269|PubMed:9521595, ECO:0000269|PubMed:9554753, ECO:0000269|PubMed:9736778, ECO:0000269|PubMed:9804160, ECO:0000269|PubMed:9921909}. The disease is caused by variants affecting the gene represented in this entry. There is some evidence that the functional defect caused by the most common variant Phe-508 DEL can be corrected by the binding to the snake phospholipase A2 crotoxin basic subunit CB. This toxin both disrupts the Phe-508 DEL-cytokeratin 8 complex, allowing for the escape from degradation, and increases the chloride channel current (PubMed:27241308). {ECO:0000269|PubMed:27241308}.; DISEASE: Congenital bilateral absence of the vas deferens (CBAVD) [MIM:277180]: An autosomal recessive disease characterized by vas deferens aplasia resulting in azoospermia and male infertility. CBAVD may occur in isolation or as a manifestation of cystic fibrosis. {ECO:0000269|PubMed:10066035, ECO:0000269|PubMed:10651488, ECO:0000269|PubMed:17329263, ECO:0000269|PubMed:7529962, ECO:0000269|PubMed:7539342, ECO:0000269|PubMed:9067761, ECO:0000269|PubMed:9736778, ECO:0000269|Ref.117}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07294; DB07031; DB00459; DB00210; DB00523; DB02466; DB03466; DB02741; DB03279; DB00926; DB00982; DB05785; DB05467; DB02258; DB00799; DB00755; DB12808 Interacts with Q96RK4; P13349; P31321; P28702; P48443; O60504-2 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; DNA-binding; Isopeptide bond; Metal-binding; Methylation; Nucleus; Proteomics identification; Receptor; Reference proteome; Transcription; Transcription regulation; Ubl conjugation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A Molecular weight (Da) 26574.9 Length 236 Aromaticity 0.06 Instability index 49.98 Isoelectric point 5.76 Charge (pH=7) -2.95 2D Binding mode Binding energy (Kcal/mol) -7.26 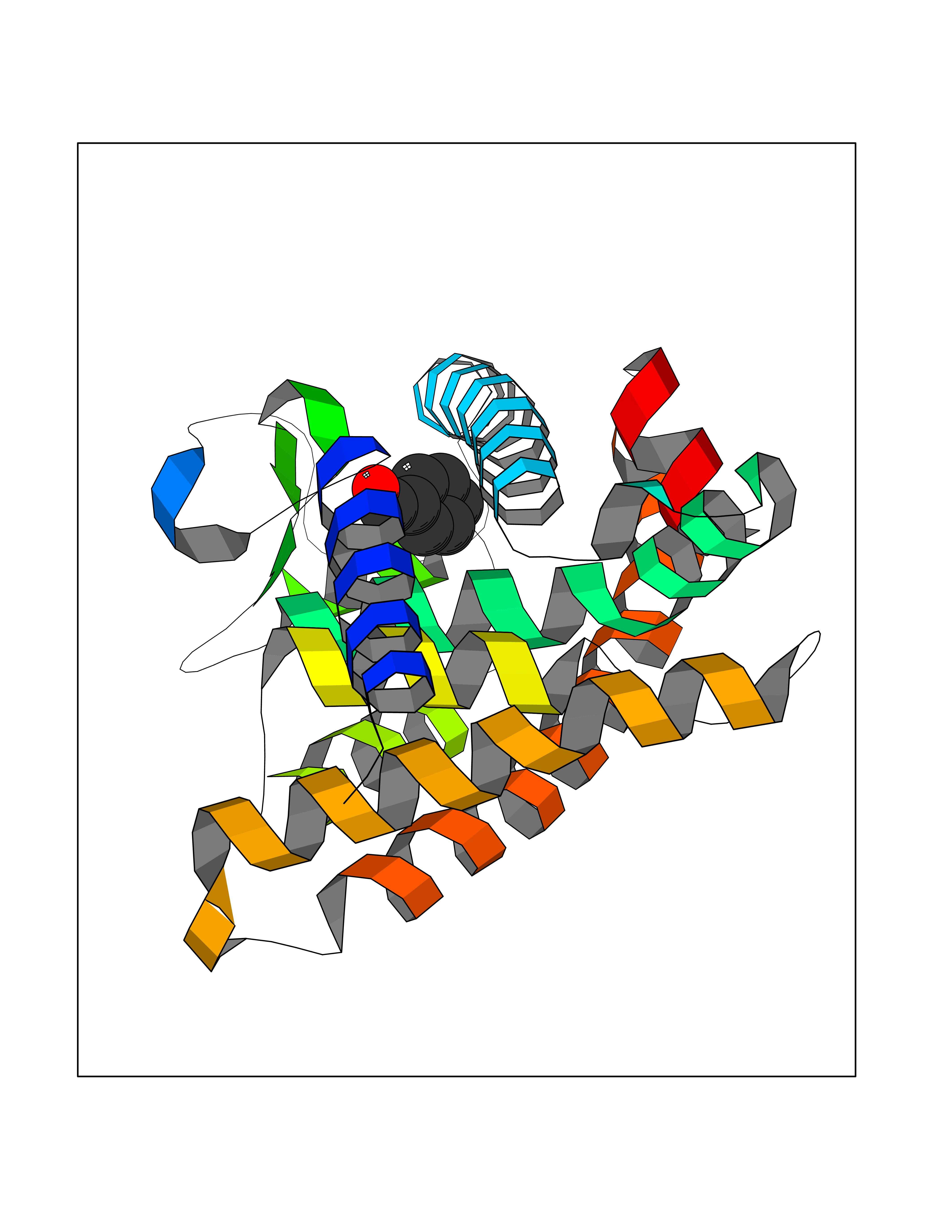 Molscript Map 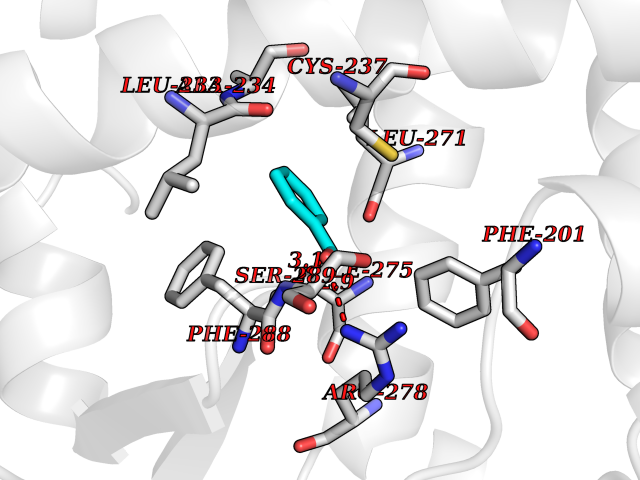 Pymol Map 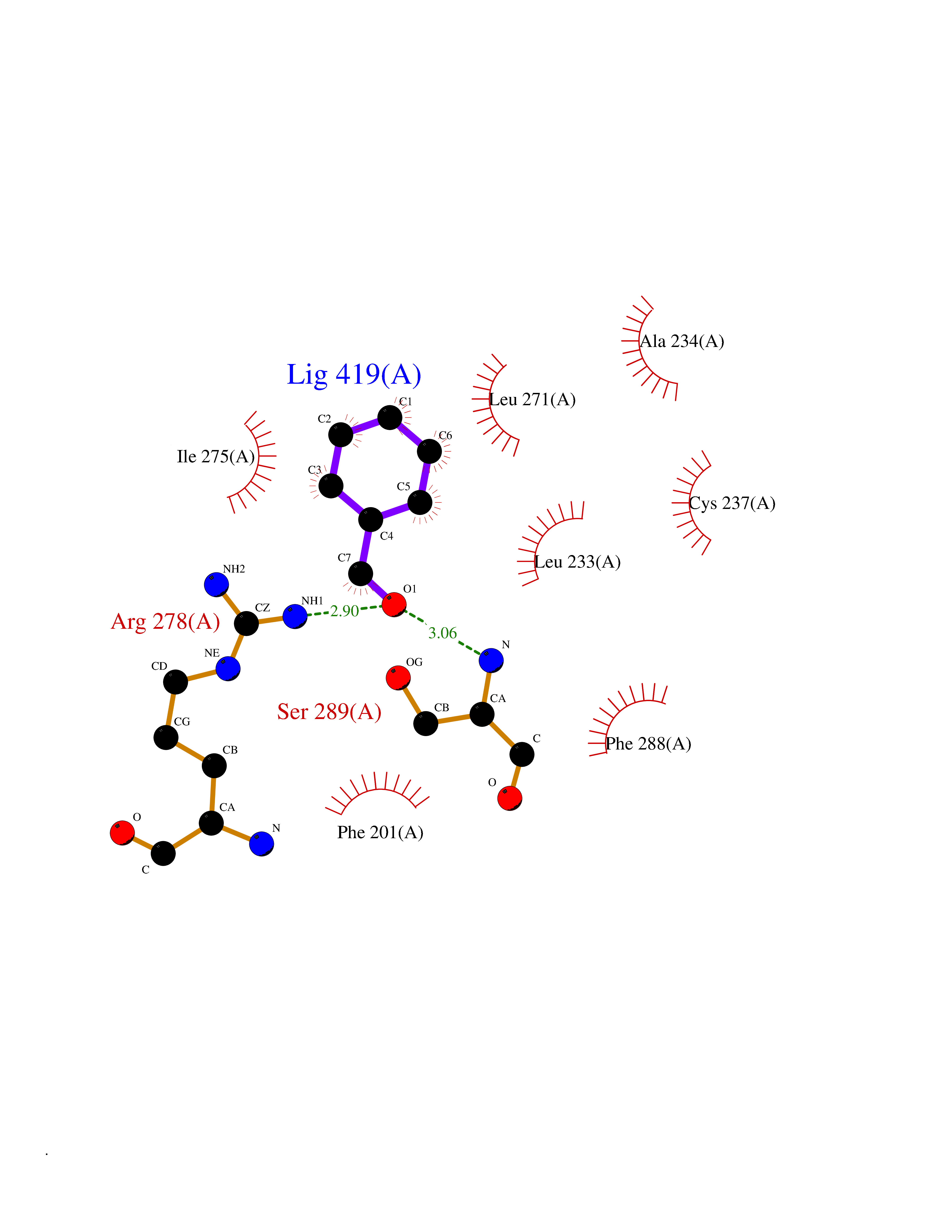 Ligplot Map 3D Binding mode Sequence ASPQLEELITKVSKAHQETFPSLCQLGKYTTNSSADHRVQLDLGLWDKFSELATKCIIKIVEFAKRLPGFTGLSIADQITLLKAACLDILMLRICTRYTPEQDTMTFSDGLTLNRTQMHNAGFGPLTDLVFAFAGQLLPLEMDDTETGLLSAICLICGDRMDLEEPEKVDKLQEPLLEALRLYARRRRPSQPYMFPRMLMKITDLRGISTKGAERAITLKMEIPGPMPPLIREMLE Hydrogen bonds contact Hydrophobic contact | ||||
| 13 | Neuronal acetylcholine receptor subunit alpha-2 | 5FJV | 5.32 | |
Target general information Gen name CHRNA2 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Ligand-gated ion channel (TC 1.A.9) family, Acetylcholine receptor (TC 1.A.9.1) subfamily, Alpha-2/CHRNA2 sub-subfamily Biochemical class NA Function After binding acetylcholine, the AChR responds by an extensive change in conformation that affects all subunits and leads to opening of an ion-conducting channel across the plasma membrane." Related diseases Epilepsy, nocturnal frontal lobe, 4 (ENFL4) [MIM:610353]: An autosomal dominant focal epilepsy characterized by nocturnal seizures associated with fear sensation, tongue movements, and nocturnal wandering, closely resembling nightmares and sleep walking. {ECO:0000269|PubMed:16826524}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Seizures, benign familial infantile, 6 (BFIS6) [MIM:610353]: A form of benign familial infantile epilepsy, a neurologic disorder characterized by afebrile seizures occurring in clusters during the first year of life, without neurologic sequelae. BFIS6 inheritance is autosomal dominant. {ECO:0000269|PubMed:25847220}. The disease may be caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00732; DB00237; DB00411; DB00565; DB01245; DB00514; DB01135; DB07720; DB00898; DB00472; DB00483; DB08960; DB00657; DB01336; DB00416; DB01226; DB00184; DB01337; DB01338; DB00721; DB00728; DB05740; DB00202; DB01199; DB01339 Interacts with NA EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disease variant; Disulfide bond; Epilepsy; Glycoprotein; Ion channel; Ion transport; Ligand-gated ion channel; Membrane; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Signal; Synapse; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A,B,C,D,E Molecular weight (Da) 120584 Length 1031 Aromaticity 0.14 Instability index 32.21 Isoelectric point 5.62 Charge (pH=7) -17.58 2D Binding mode Binding energy (Kcal/mol) -7.26 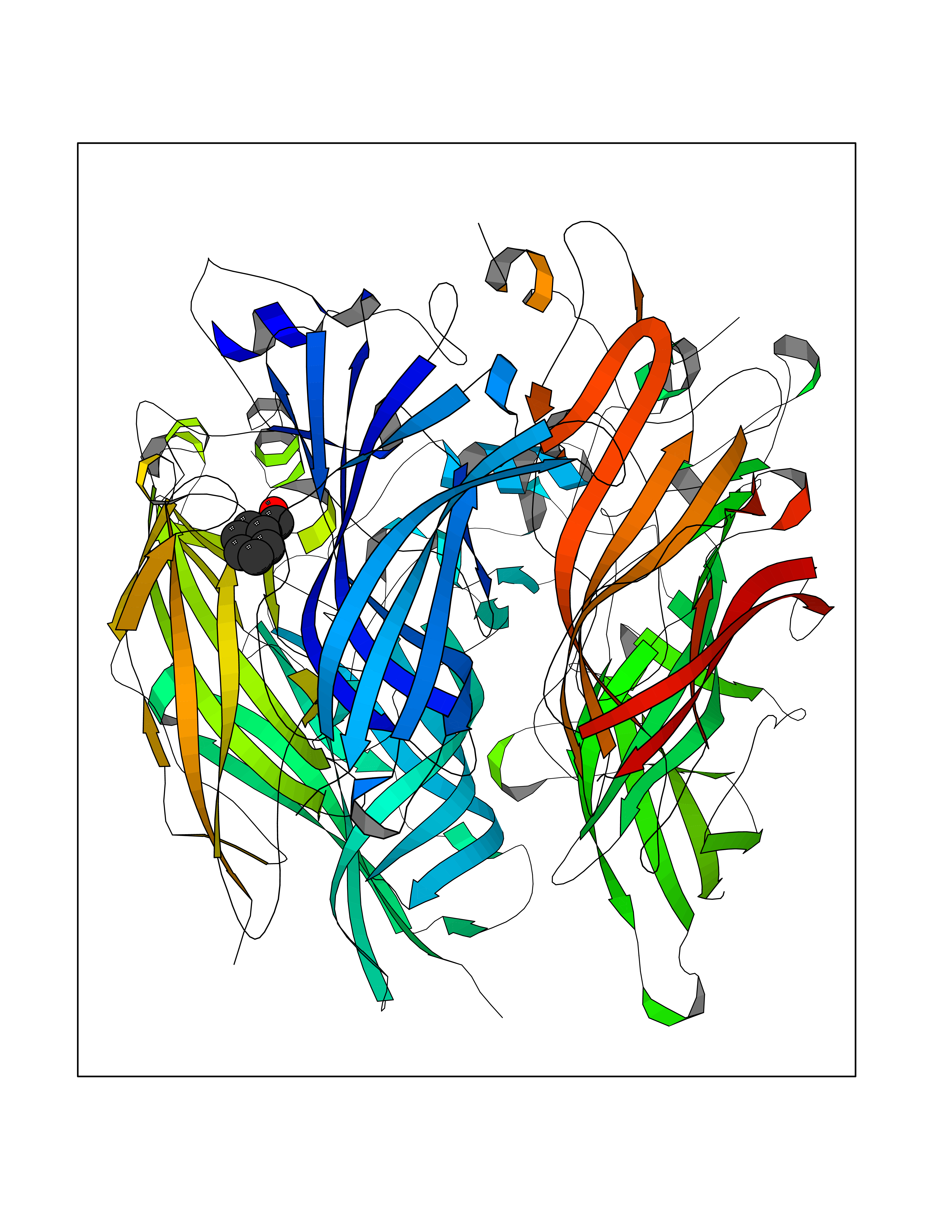 Molscript Map 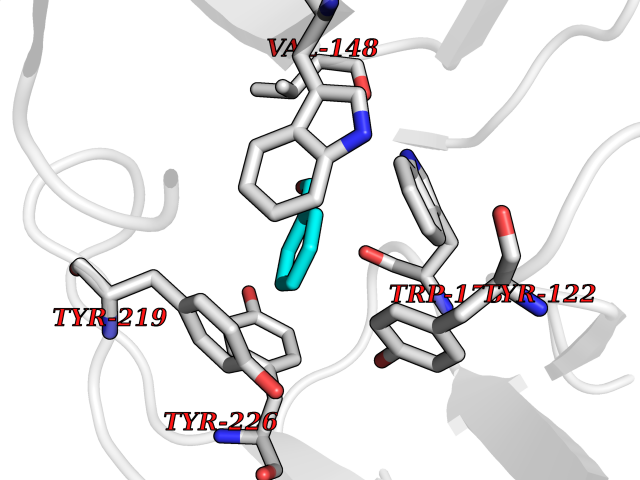 Pymol Map 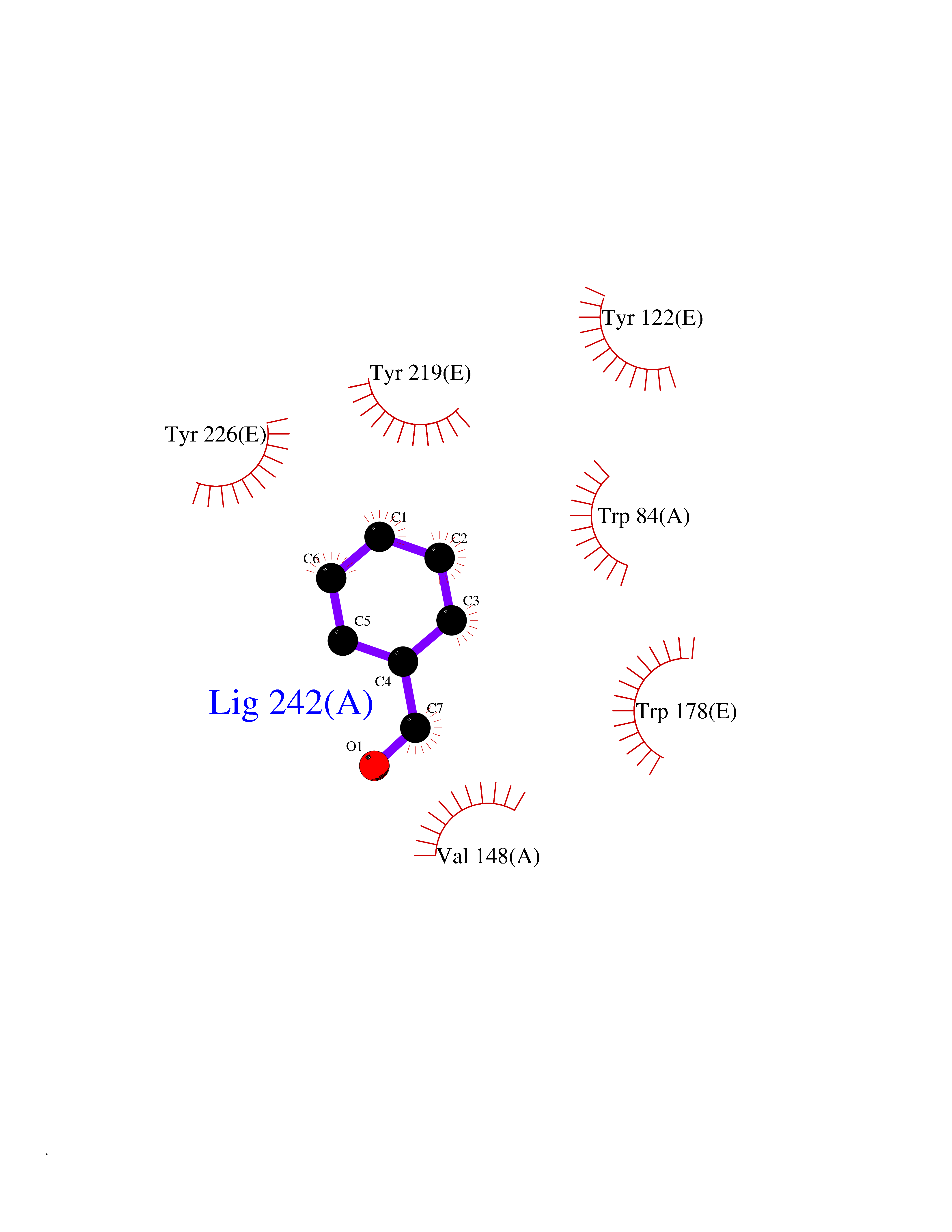 Ligplot Map 3D Binding mode Sequence DRLFKHLFRGYNRWARPVPNTSDVVIVRFGLSIAQLIDVDEKNQMMTTNVWLKQEWSDYKLRWNPTDFGNITSLRVPSEMIWIPDIVLYNNADGEFAVTHMTKAHLFSTGTVHWVPPAIYKSSCSIDVTFFPFDQQNCKMKFGSWTYDKAKIDLEQMEQTVDLKDYWESGEWAIVNATGTYNSKKYDCCAEIYPDVTYAFVIRRLPEDRLFKHLFRGYNRWARPVPNTSDVVIVRFGLSIAQLIDVDEKNQMMTTNVWLKQEWSDYKLRWNPTDFGNITSLRVPSEMIWIPDIVLYNNADGEFAVTHMTKAHLFSTGTVHWVPPAIYKSSCSIDVTFDQQNCKMKFGSWTYDKAKIDLEQMEQTVDLKDYWESGEWAIVNATGTYNSKKYDCCAEIYPDVTYAFVIRRLPEDRLFKHLFRGYNRWARPVPNTSDVVIVRFGLSIAQLIDVDEKNQMMTTNVWLKQEWSDYKLRWNPTDFGNITSLRVPSEMIWIPDIVLYNNADGEFAVTHMTKAHLFSTGTVHWVPPAIYKSSCSIDVTFFPFDQQNCKMKFGSWTYDKAKIDLEQMEQTVDLKDYWESGEWAIVNATGTYNSKKYDCCAEIYPDVTYAFVIRRLPEDRLFKHLFRGYNRWARPVPNTSDVVIVRFGLSIAQLIDVDEKNQMMTTNVWLKQEWSDYKLRWNPTDFGNITSLRVPSEMIWIPDIVLYNNADGEFAVTHMTKAHLFSTGTVHWVPPAIYKSSCSIDVTFFPFDQQNCKMKFGSWTYDKAKIDLEQMEQTVDLKDYWESGEWAIVNATGTYNSKKYDCCAEIYPDVTYAFVIRRLPEDRLFKHLFRGYNRWARPVPNTSDVVIVRFGLSIAQLIDVDEKNQMMTTNVWLKQEWSDYKLRWNPTDFGNITSLRVPSEMIWIPDIVLYNNADGEFAVTHMTKAHLFSTGTVHWVPPAIYKSSCSIDVTFFPFDQQNCKMKFGSWTYDKAKIDLEQMEQTVDLKDYWESGEWAIVNATGTYNSKKYDCCAEIYPDVTYAFVIRRLP Hydrogen bonds contact Hydrophobic contact | ||||
| 14 | Proto-oncogene c-Met (MET) | 3DKC | 5.32 | |
Target general information Gen name MET Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Tyrosine-protein kinase Met; Scatter factor receptor; SF receptor; Met proto-oncogene tyrosine kinase; Hepatocyte growth factor receptor; HGF/SF receptor; HGF-SF receptor; HGF receptor; C-met; C-Met r Protein family Protein kinase superfamily, Tyr protein kinase family Biochemical class Kinase Function Regulates many physiological processes including proliferation, scattering, morphogenesis and survival. Ligand binding at the cell surface induces autophosphorylation of MET on its intracellular domain that provides docking sites for downstream signaling molecules. Following activation by ligand, interacts with the PI3-kinase subunit PIK3R1, PLCG1, SRC, GRB2, STAT3 or the adapter GAB1. Recruitment of these downstream effectors by MET leads to the activation of several signaling cascades including the RAS-ERK, PI3 kinase-AKT, or PLCgamma-PKC. The RAS-ERK activation is associated with the morphogenetic effects while PI3K/AKT coordinates prosurvival effects. During embryonic development, MET signaling plays a role in gastrulation, development and migration of muscles and neuronal precursors, angiogenesis and kidney formation. In adults, participates in wound healing as well as organ regeneration and tissue remodeling. Promotes also differentiation and proliferation of hematopoietic cells. May regulate cortical bone osteogenesis. Receptor tyrosine kinase that transduces signals from the extracellular matrix into the cytoplasm by binding to hepatocyte growth factor/HGF ligand. Related diseases Activation of MET after rearrangement with the TPR gene produces an oncogenic protein.; DISEASE: Defects in MET may be associated with gastric cancer.; DISEASE: Hepatocellular carcinoma (HCC) [MIM:114550]: A primary malignant neoplasm of epithelial liver cells. The major risk factors for HCC are chronic hepatitis B virus (HBV) infection, chronic hepatitis C virus (HCV) infection, prolonged dietary aflatoxin exposure, alcoholic cirrhosis, and cirrhosis due to other causes. {ECO:0000269|PubMed:9927037}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Renal cell carcinoma papillary (RCCP) [MIM:605074]: A subtype of renal cell carcinoma tending to show a tubulo-papillary architecture formed by numerous, irregular, finger-like projections of connective tissue. Renal cell carcinoma is a heterogeneous group of sporadic or hereditary carcinoma derived from cells of the proximal renal tubular epithelium. {ECO:0000269|PubMed:10327054, ECO:0000269|PubMed:10417759, ECO:0000269|PubMed:10433944, ECO:0000269|PubMed:9140397, ECO:0000269|PubMed:9563489}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: A common allele in the promoter region of the MET shows genetic association with susceptibility to autism in some families. Functional assays indicate a decrease in MET promoter activity and altered binding of specific transcription factor complexes.; DISEASE: MET activating mutations may be involved in the development of a highly malignant, metastatic syndrome known as cancer of unknown primary origin (CUP) or primary occult malignancy. Systemic neoplastic spread is generally a late event in cancer progression. However, in some instances, distant dissemination arises at a very early stage, so that metastases reach clinical relevance before primary lesions. Sometimes, the primary lesions cannot be identified in spite of the progresses in the diagnosis of malignancies.; DISEASE: Deafness, autosomal recessive, 97 (DFNB97) [MIM:616705]: A form of non-syndromic sensorineural hearing loss with prelingual onset. Sensorineural deafness results from damage to the neural receptors of the inner ear, the nerve pathways to the brain, or the area of the brain that receives sound information. {ECO:0000269|PubMed:25941349}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Osteofibrous dysplasia (OSFD) [MIM:607278]: A congenital disorder of osteogenesis characterized by non-neoplastic, radiolucent lesions that affect the cortical bone immediately under the periosteum. It usually manifests as a painless swelling or anterior bowing of the long bones, most commonly the tibia and fibula. {ECO:0000269|PubMed:26637977}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Disease-associated variants identified in 4 families cause the deletion of exon 14. This results in the exclusion of an ubiquitination target site within the cytoplasmic domain, hence in protein stabilization. The persistent presence of MET at the cell surface in conditions of ligand-dependent activation retards osteoblastic differentiation. {ECO:0000269|PubMed:26637977}.; DISEASE: Arthrogryposis, distal, 11 (DA11) [MIM:620019]: A form of distal arthrogryposis, a disease characterized by congenital joint contractures that mainly involve two or more distal parts of the limbs, in the absence of a primary neurological or muscle disease. DA11 is an autosomal dominant form characterized mainly by camptodactyly. Other features include absent flexion creases and limited forearm supination. {ECO:0000269|PubMed:30777867}. The disease may be caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB06896; DB08791; DB06997; DB07969; DB08079; DB16695; DB12742; DB12267; DB08875; DB11791; DB08865; DB12010; DB02152; DB07369; DB06995; DB06314; DB01268; DB15133; DB12200; DB11800 Interacts with P22681; Q96EY1; Q96EY1-2; P00533; P09769; P14210; P14210-6; O15357; P35968; P06239; P07948; P08581; P41218; P15941; P16333; O43639; Q16288; P27986; O00459; Q92569; P19174; O43157; O15031; Q9ULL4; Q8TCU6; P18031; Q06124; P23467; Q12913; Q16827; P20936; Q9UQQ2; O60880; O14796; Q9NP31; Q8N5H7; Q15464; P29353; P98077; Q6S5L8; Q96IW2; Q9H6Q3; O75159; O14544; P12931; Q9ULZ2; P43405; P42680; Q9HBL0; Q63HR2; Q68CZ2; Q9UKW4; P07947; P43403; Q08048; P0DQD2; P35918; Q00944 EC number EC 2.7.10.1 Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Chromosomal rearrangement; Deafness; Disease variant; Disulfide bond; Glycoprotein; Kinase; Membrane; Non-syndromic deafness; Nucleotide-binding; Phosphoprotein; Proteomics identification; Proto-oncogene; Receptor; Reference proteome; Repeat; Secreted; Signal; Transferase; Transmembrane; Transmembrane helix; Tyrosine-protein kinase; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 35229.5 Length 312 Aromaticity 0.1 Instability index 37.98 Isoelectric point 7.79 Charge (pH=7) 1.93 2D Binding mode Binding energy (Kcal/mol) -7.25 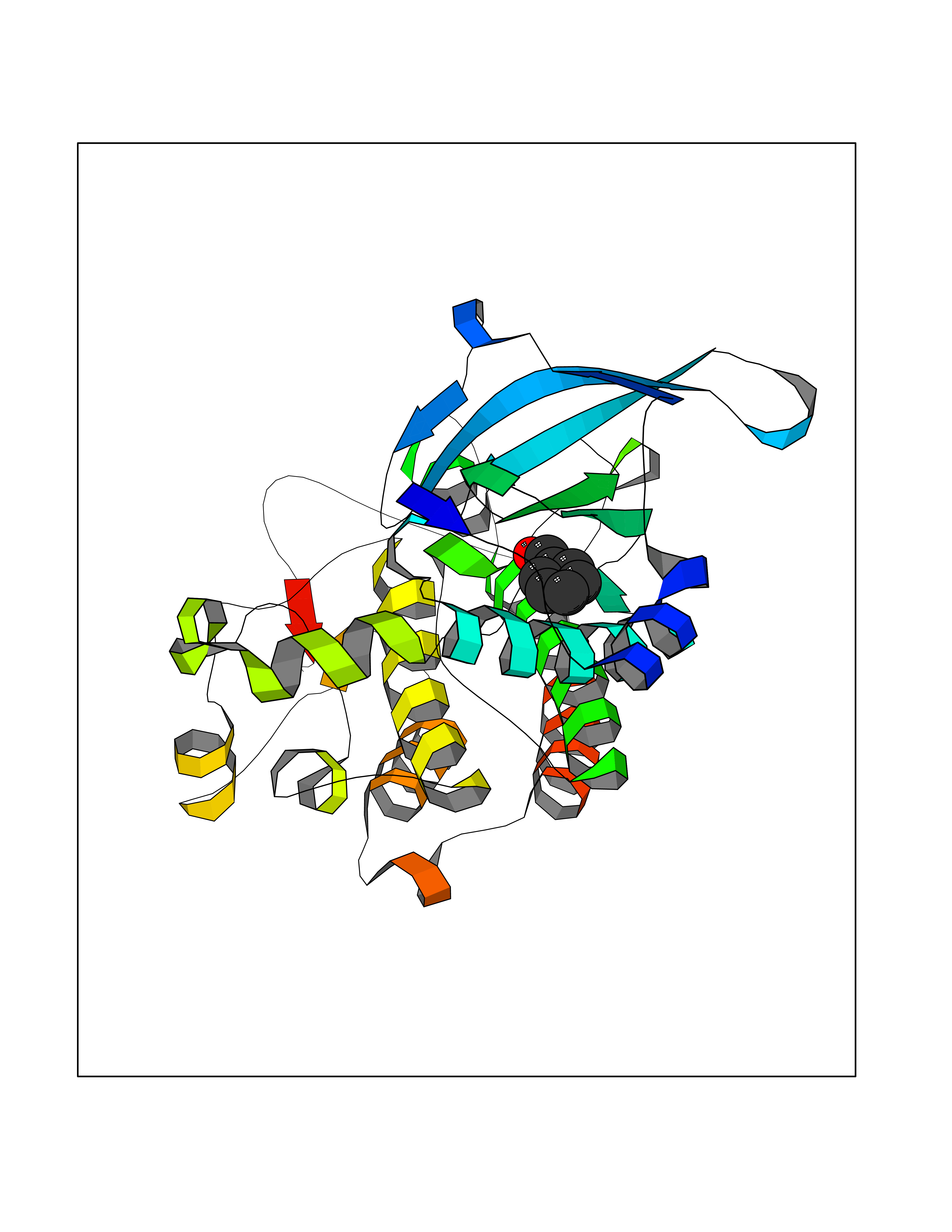 Molscript Map 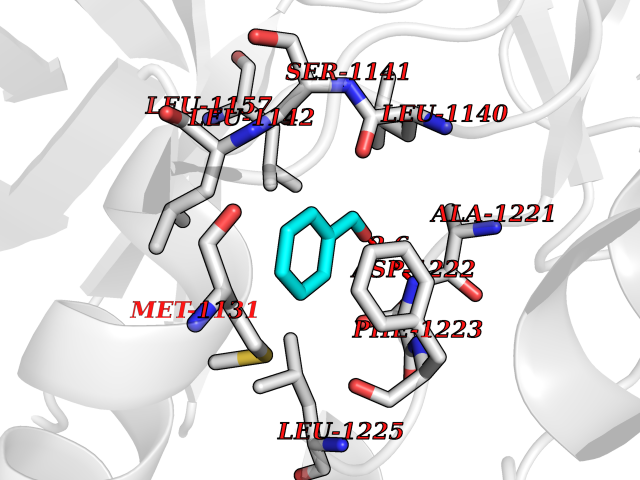 Pymol Map 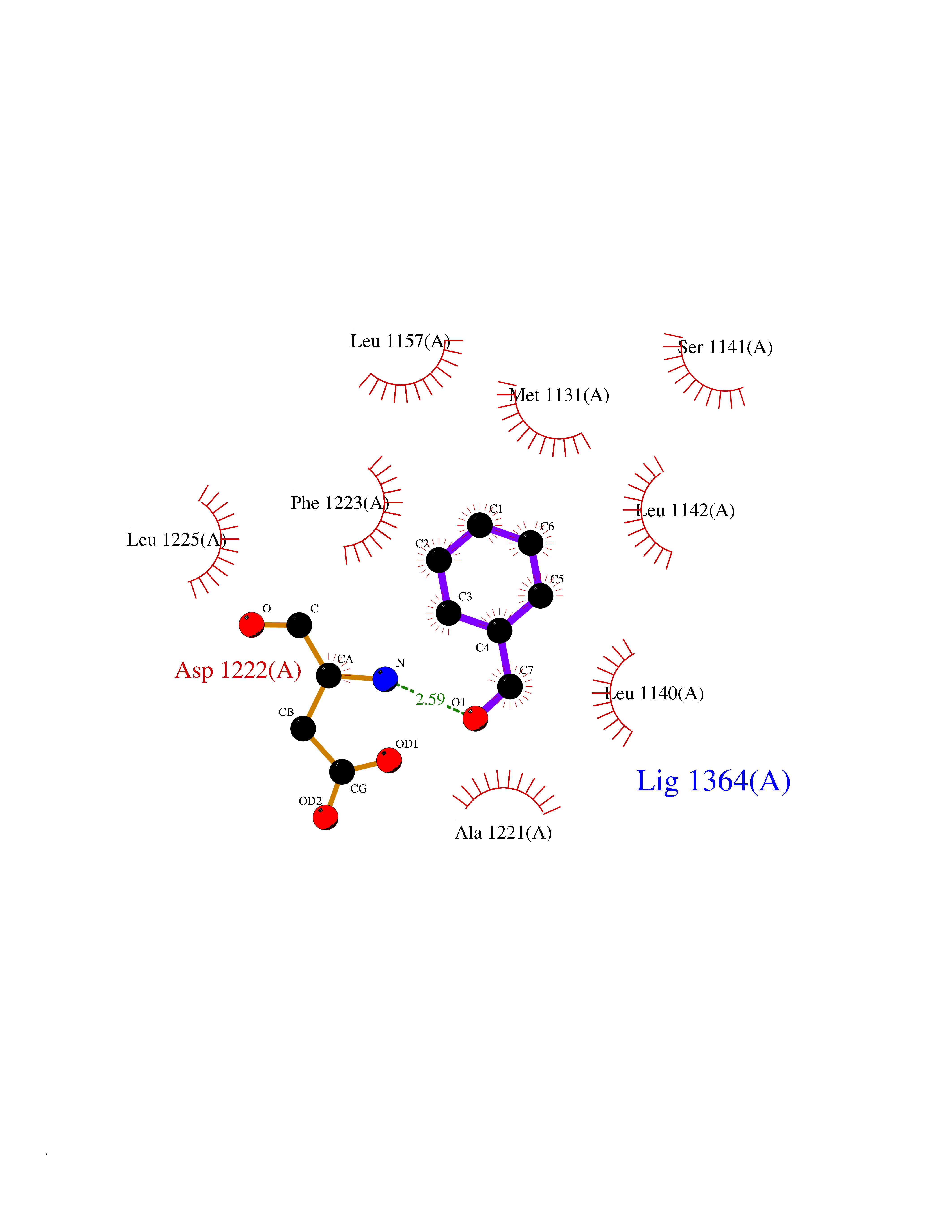 Ligplot Map 3D Binding mode Sequence VHIDLSALNPELVQAVQHVVIGPSSLIVHFNEVIGRGHFGCVYHGTLLDNDGKKIHCAVKSLNRITDIGEVSQFLTEGIIMKDFSHPNVLSLLGICLRSEGSPLVVLPYMKHGDLRNFIRNETHNPTVKDLIGFGLQVAKGMKFLASKKFVHRDLAARNCMLDEKFTVKVADFGLARDMYDKEFDSVHNKTGAKLPVKWMALESLQTQKFTTKSDVWSFGVLLWELMTRGAPPYPDVNTFDITVYLLQGRRLLQPEYCPDPLYEVMLKCWHPKAEMRPSFSELVSRISAIFSTFIGEHYVHVNATYVNVKEG Hydrogen bonds contact Hydrophobic contact | ||||
| 15 | AP endonuclease 1 (APEX1) | 5WN2 | 5.31 | |
Target general information Gen name APEX1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Redox factor-1; REF1; REF-1; HAP1; DNA-(apurinic or apyrimidinic site) lyase; Apurinic-apyrimidinic endonuclease 1; APX; APEX nuclease; APEX; APEN; APE1; APE-1; APE Protein family DNA repair enzymes AP/ExoA family Biochemical class Alpha-carbonic anhydrase Function Multifunctional protein that plays a central role in the cellular response to oxidative stress. The two major activities of APEX1 are DNA repair and redox regulation of transcriptional factors. Functions as a apurinic/apyrimidinic (AP) endodeoxyribonuclease in the DNA base excision repair (BER) pathway of DNA lesions induced by oxidative and alkylating agents. Initiates repair of AP sites in DNA by catalyzing hydrolytic incision of the phosphodiester backbone immediately adjacent to the damage, generating a single-strand break with 5'-deoxyribose phosphate and 3'-hydroxyl ends. Does also incise at AP sites in the DNA strand of DNA/RNA hybrids, single-stranded DNA regions of R-loop structures, and single-stranded RNA molecules. Has a 3'-5' exoribonuclease activity on mismatched deoxyribonucleotides at the 3' termini of nicked or gapped DNA molecules during short-patch BER. Possesses a DNA 3' phosphodiesterase activity capable of removing lesions (such as phosphoglycolate) blocking the 3' side of DNA strand breaks. May also play a role in the epigenetic regulation of gene expression by participating in DNA demethylation. Acts as a loading factor for POLB onto non-incised AP sites in DNA and stimulates the 5'-terminal deoxyribose 5'-phosphate (dRp) excision activity of POLB. Plays a role in the protection from granzymes-mediated cellular repair leading to cell death. Also involved in the DNA cleavage step of class switch recombination (CSR). On the other hand, APEX1 also exerts reversible nuclear redox activity to regulate DNA binding affinity and transcriptional activity of transcriptional factors by controlling the redox status of their DNA-binding domain, such as the FOS/JUN AP-1 complex after exposure to IR. Involved in calcium-dependent down-regulation of parathyroid hormone (PTH) expression by binding to negative calcium response elements (nCaREs). Together with HNRNPL or the dimer XRCC5/XRCC6, associates with nCaRE, acting as an activator of transcriptional repression. Stimulates the YBX1-mediated MDR1 promoter activity, when acetylated at Lys-6 and Lys-7, leading to drug resistance. Acts also as an endoribonuclease involved in the control of single-stranded RNA metabolism. Plays a role in regulating MYC mRNA turnover by preferentially cleaving in between UA and CA dinucleotides of the MYC coding region determinant (CRD). In association with NMD1, plays a role in the rRNA quality control process during cell cycle progression. Associates, together with YBX1, on the MDR1 promoter. Together with NPM1, associates with rRNA. Binds DNA and RNA. Related diseases Microvascular complications of diabetes 5 (MVCD5) [MIM:612633]: Pathological conditions that develop in numerous tissues and organs as a consequence of diabetes mellitus. They include diabetic retinopathy, diabetic nephropathy leading to end-stage renal disease, and diabetic neuropathy. Diabetic retinopathy remains the major cause of new-onset blindness among diabetic adults. It is characterized by vascular permeability and increased tissue ischemia and angiogenesis. Disease susceptibility is associated with variants affecting the gene represented in this entry. Homozygosity for the Leu-55 allele is strongly associated with the development of retinal disease in diabetic patients. Drugs (DrugBank ID) DB04967 Interacts with Q09472; Q8N4N3; Q16236; Q96EB6; O88846 EC number EC 3.1.-.- Uniprot keywords 3D-structure; Acetylation; Activator; Cleavage on pair of basic residues; Cytoplasm; Direct protein sequencing; Disulfide bond; DNA damage; DNA recombination; DNA repair; DNA-binding; Endonuclease; Endoplasmic reticulum; Exonuclease; Hydrolase; Magnesium; Metal-binding; Mitochondrion; Nuclease; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Repressor; RNA-binding; S-nitrosylation; Transcription; Transcription regulation; Ubl conjugation Protein physicochemical properties Chain ID A,D Molecular weight (Da) 31820.9 Length 285 Aromaticity 0.1 Instability index 42.58 Isoelectric point 8.35 Charge (pH=7) 3.27 2D Binding mode Binding energy (Kcal/mol) -7.24 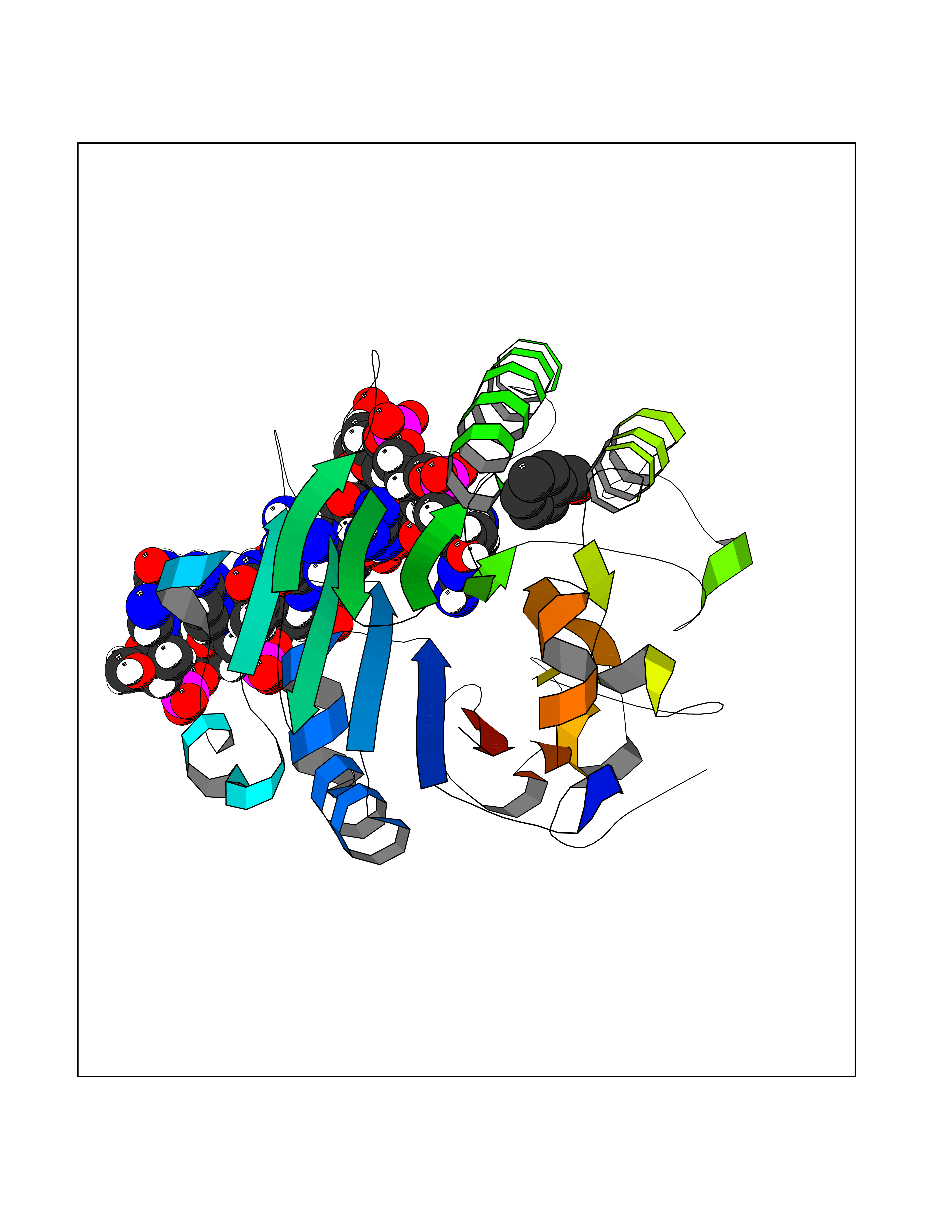 Molscript Map 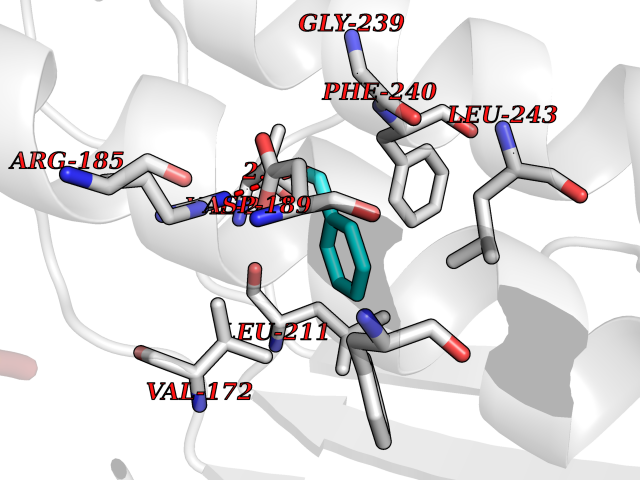 Pymol Map 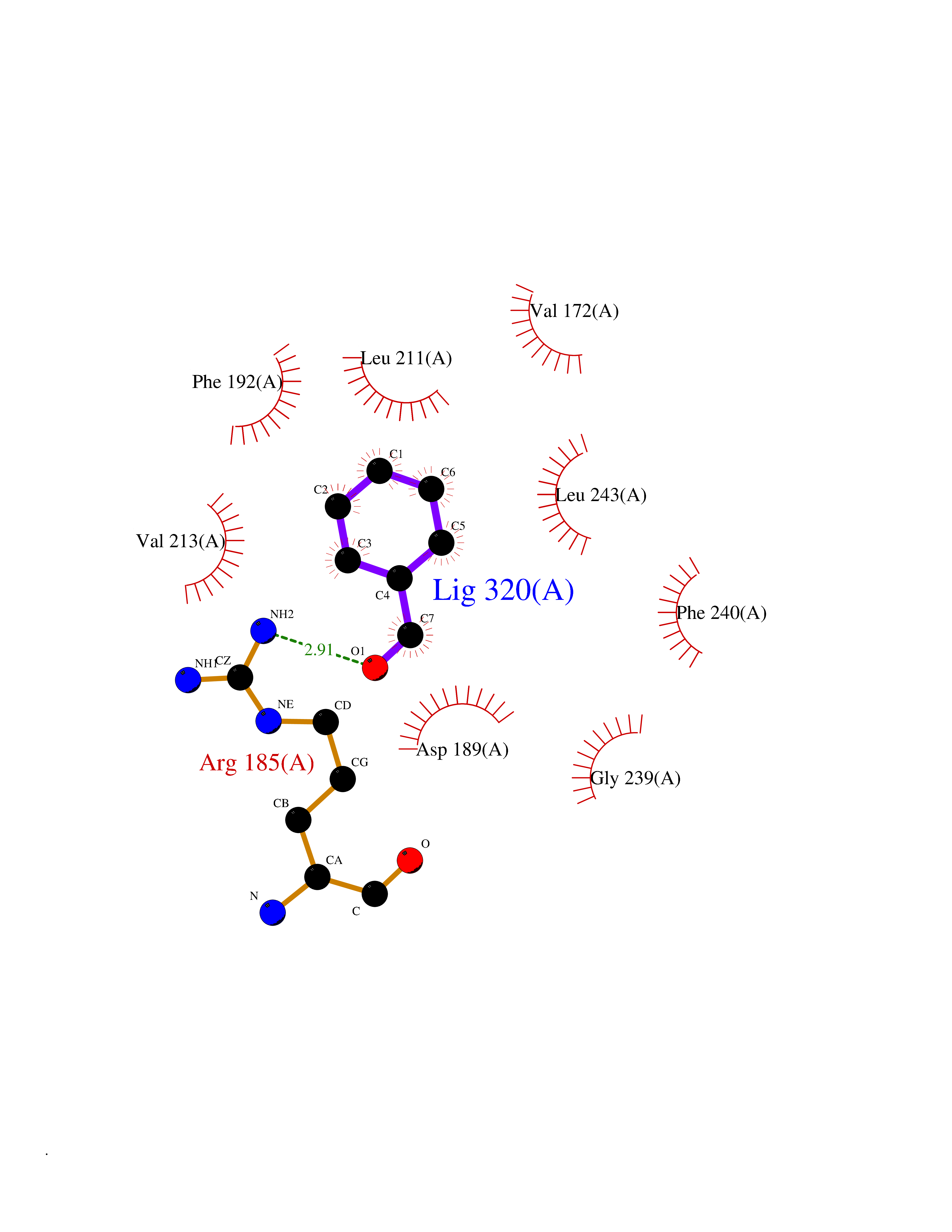 Ligplot Map 3D Binding mode Sequence ALYEDPPDQKTSPSGKPATLKICSWNVDGLRAWIKKKGLDWVKEEAPDILCLQQTKCSENKLPAELQELPGLSHQYWSAPSDKEGYSGVGLLSRQAPLKVSYGIGDEEHDQEGRVIVAEFDSFVLVTAYVPNAGRGLVRLEYRQRWDEAFRKFLKGLASRKPLVLCGNLNVAHEEIDLRNPKGNKKNAGFTPQERQGFGELLQAVPLADSFRHLYPNTPYAYTFWTYMMNARSKNVGWRLDYFLLSHSLLPALCDSKIRSKALGSDHCPITLYLALGCTGATGCG Hydrogen bonds contact Hydrophobic contact | ||||
| 16 | Macrophage migration inhibitory factor (MIF) | 3IJJ | 5.31 | |
Target general information Gen name MIF Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Phenylpyruvate tautomerase; MMIF; L-dopachrome tautomerase; L-dopachrome isomerase; Glycosylation-inhibiting factor; GLIF; GIF Protein family MIF family Biochemical class Intramolecular oxidoreductase Function Involved in the innate immune response to bacterial pathogens. The expression of MIF at sites of inflammation suggests a role as mediator in regulating the function of macrophages in host defense. Counteracts the anti-inflammatory activity of glucocorticoids. Has phenylpyruvate tautomerase and dopachrome tautomerase activity (in vitro), but the physiological substrate is not known. It is not clear whether the tautomerase activity has any physiological relevance, and whether it is important for cytokine activity. Pro-inflammatory cytokine. Related diseases Rheumatoid arthritis systemic juvenile (RASJ) [MIM:604302]: An inflammatory articular disorder with systemic onset beginning before the age of 16. It represents a subgroup of juvenile arthritis associated with severe extraarticular features and occasionally fatal complications. During active phases of the disorder, patients display a typical daily spiking fever, an evanescent macular rash, lymphadenopathy, hepatosplenomegaly, serositis, myalgia and arthritis. {ECO:0000269|PubMed:11508429}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01880; DB07888; DB08334; DB08335; DB08333; DB07718; DB08765; DB02728 Interacts with O43521-2; P00533; Q92743; P14174; Q96HA8 EC number EC 5.3.2.1 Uniprot keywords 3D-structure; Acetylation; Cytokine; Cytoplasm; Direct protein sequencing; Immunity; Inflammatory response; Innate immunity; Isomerase; Proteomics identification; Reference proteome; Secreted Protein physicochemical properties Chain ID A,C Molecular weight (Da) 24671.9 Length 228 Aromaticity 0.09 Instability index 31.45 Isoelectric point 8.37 Charge (pH=7) 2.26 2D Binding mode Binding energy (Kcal/mol) -7.24 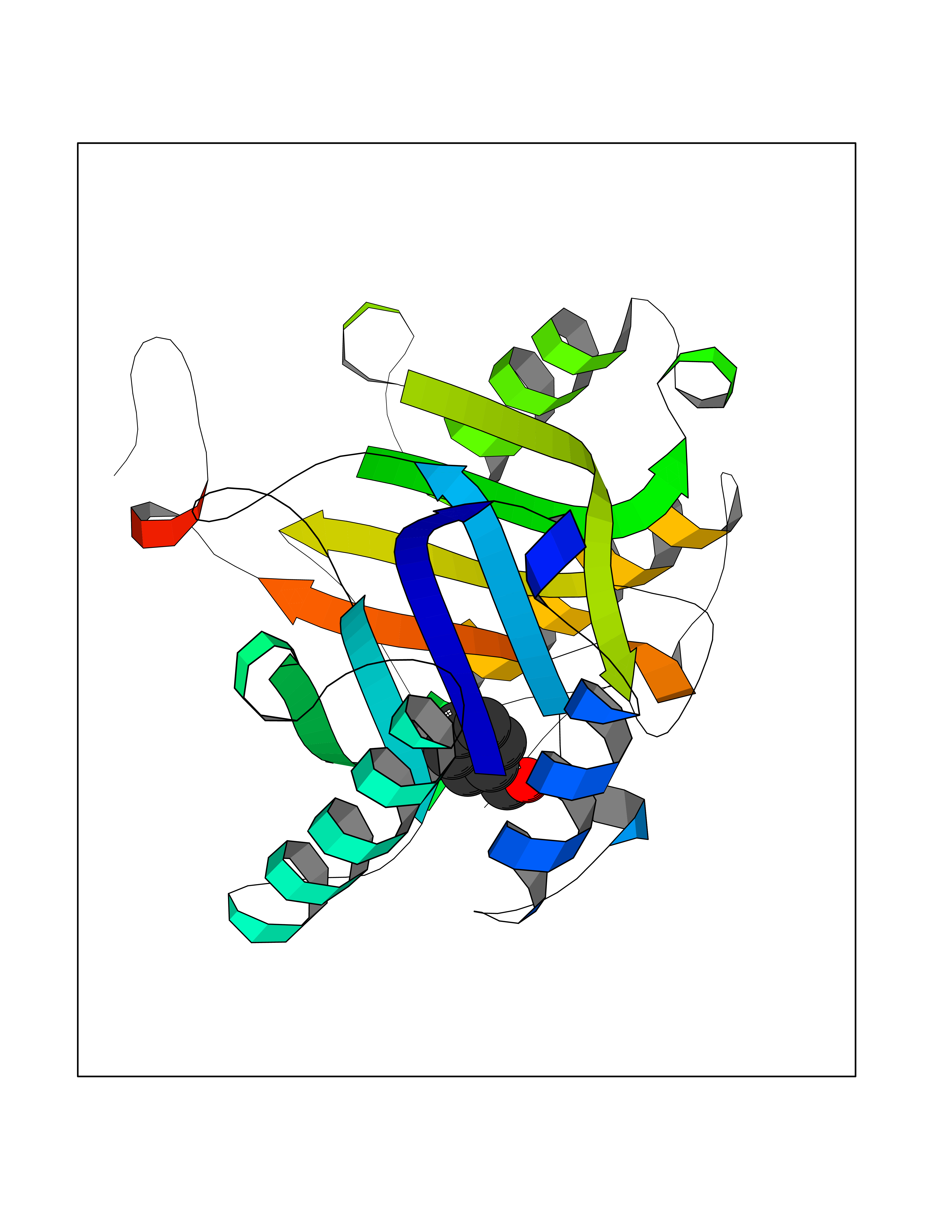 Molscript Map 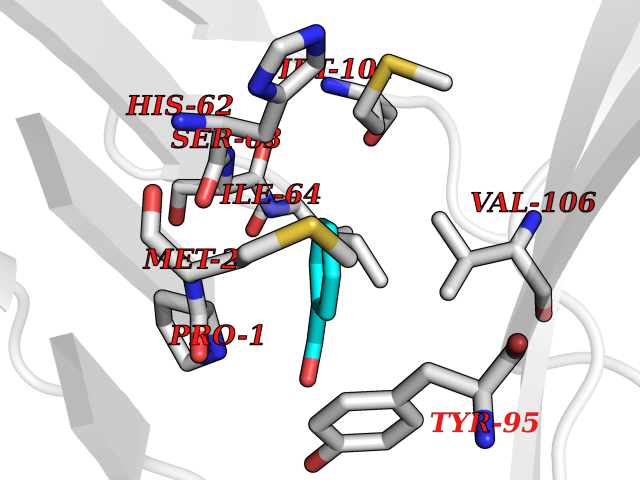 Pymol Map 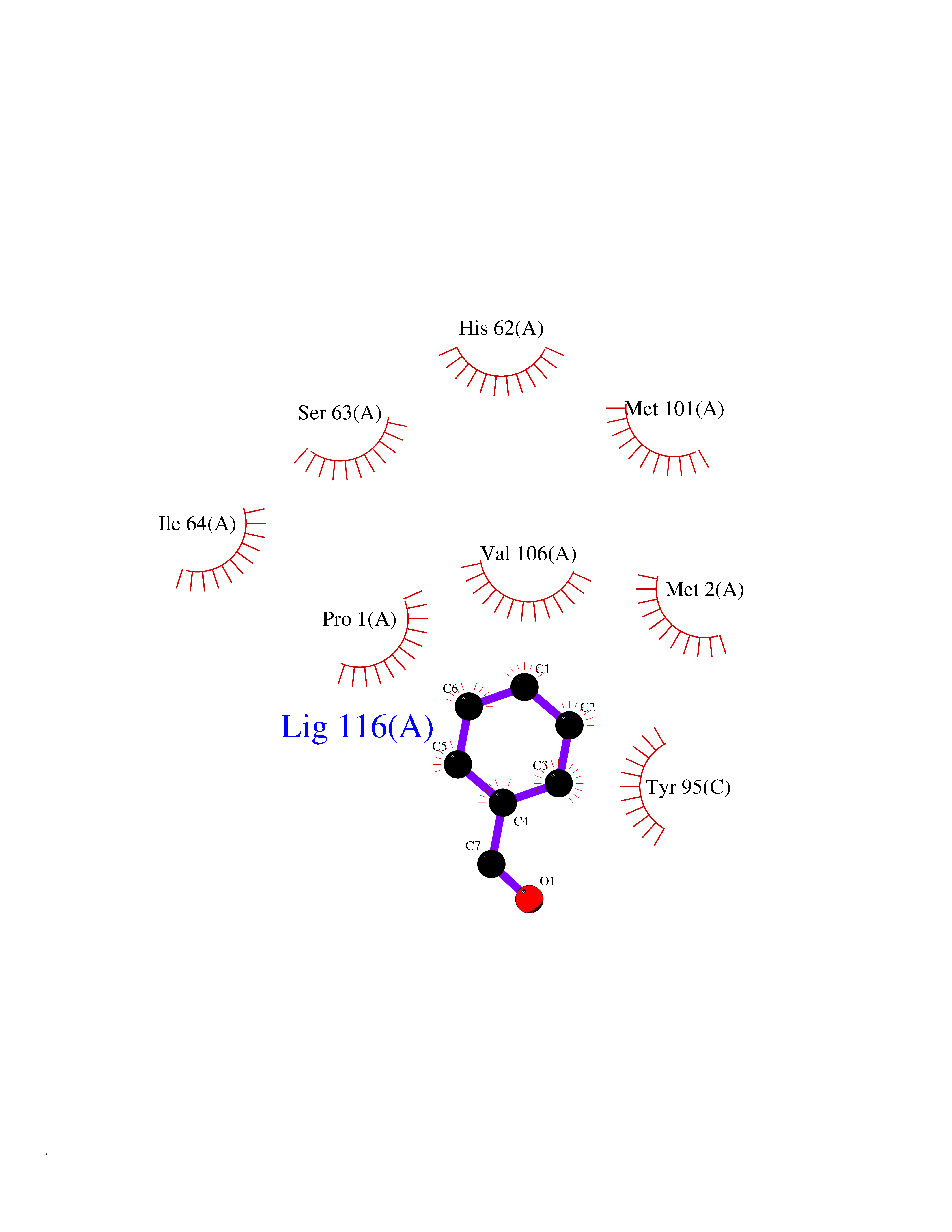 Ligplot Map 3D Binding mode Sequence PMFIVNTNVPRASVPDGFLSELTQQLAQATGKPPQYIAVHVVPDQLMAFGGSSEPCALCSLHSIGKIGGAQNRSYSKLLCGLLAERLRISPDRVYINYYDMNAANVGWNNSTFAPMFIVNTNVPRASVPDGFLSELTQQLAQATGKPPQYIAVHVVPDQLMAFGGSSEPCALCSLHSIGKIGGAQNRSYSKLLCGLLAERLRISPDRVYINYYDMNAANVGWNNSTFA Hydrogen bonds contact Hydrophobic contact | ||||
| 17 | Dipeptidyl peptidase 9 (DPP-9) | 6EOR | 5.31 | |
Target general information Gen name DPP9 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Dipeptidyl peptidase-like protein 9; Dipeptidyl peptidase IX; Dipeptidyl peptidase IV-related protein 2; DPRP2; DPRP-2; DPP IX; DPLP9; DP9 Protein family Peptidase S9B family, DPPIV subfamily Biochemical class Peptidase Function Dipeptidyl peptidase that cleaves off N-terminal dipeptides from proteins having a Pro or Ala residue at position 2. Related diseases Hatipoglu immunodeficiency syndrome (HATIS) [MIM:620331]: An autosomal recessive immunologic disorder manifesting in infancy or early childhood, and characterized by failure to thrive, short stature, skin pigmentation abnormalities, pancytopenia, and susceptibility to recurrent infections. {ECO:0000269|PubMed:36112693}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q9NXR5; Q86TI2; Q6NUP5; P46379-2; Q8WUW1; Q96A83-2; O75190-2; O14645; Q01658; P29692-2; Q06787-7; Q9Y5Q9; O14901; Q9BVL2; Q96CV9; Q06830; P14678-2; P49458; Q11203; Q13148; P14927 EC number EC 3.4.14.5 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Aminopeptidase; Cytoplasm; Disease variant; Hydrolase; Nucleus; Protease; Proteomics identification; Reference proteome; Serine protease Protein physicochemical properties Chain ID A Molecular weight (Da) 92797.4 Length 808 Aromaticity 0.12 Instability index 37.45 Isoelectric point 6.34 Charge (pH=7) -8.98 2D Binding mode Binding energy (Kcal/mol) -7.24 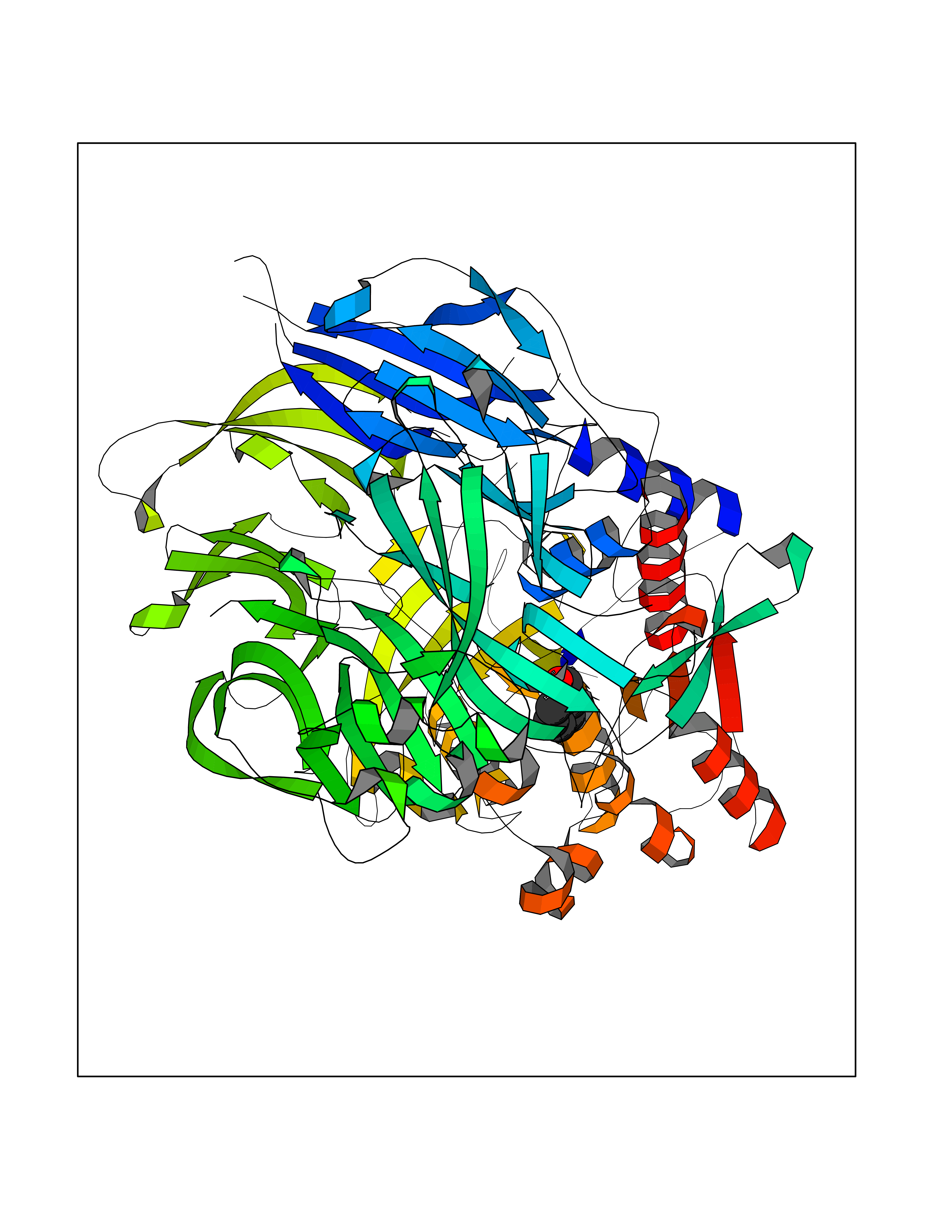 Molscript Map 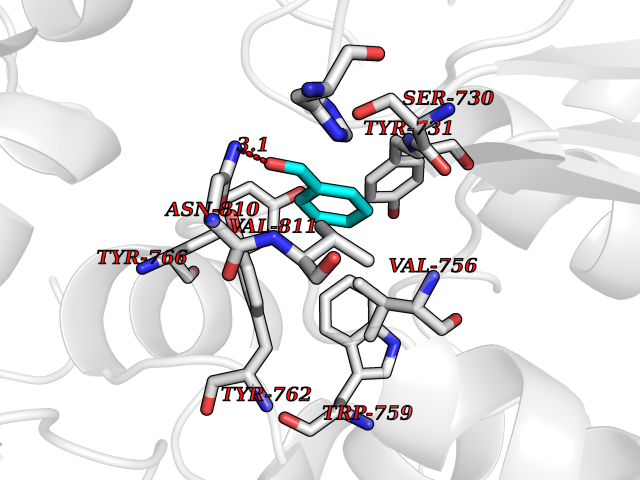 Pymol Map 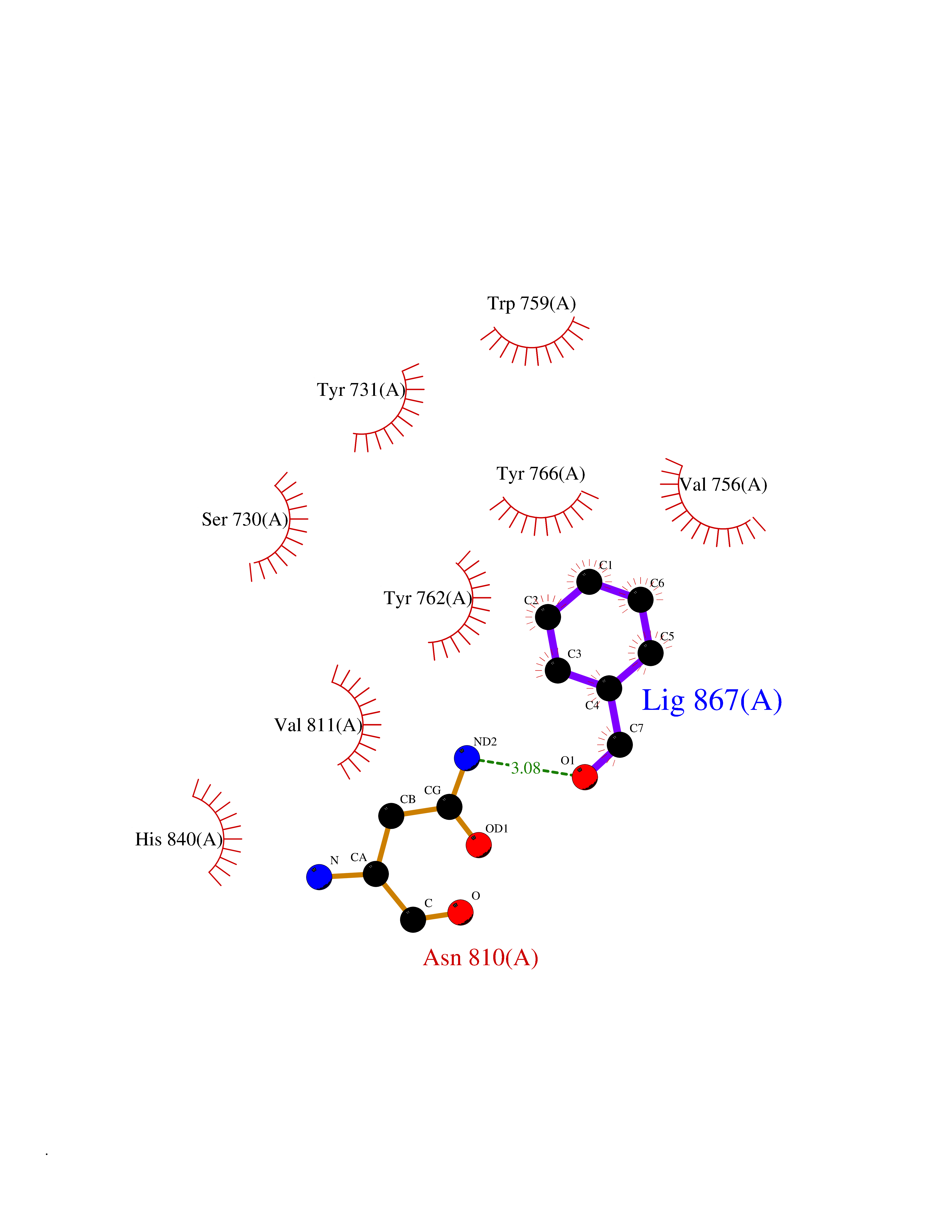 Ligplot Map 3D Binding mode Sequence AARFQVQKHSWDGLRSIIHGSRKAPHDFQFVQKSGPHSHRLYYLGMPYRENSLLYSEIPKLLLSWKQMLDHFQATPHHGVYSREEELLRERKRLGVFGITSYDFHSESGLFLFQASNSLFHCRDGGKNGFMVSPMKPLEIKTQCSGPRMDPKICPADPAFFSFINNSDLWVANIETGEERRLTFCHQNVLDDPKSAGVATFVIQEEFDRFTGYWWCPTASWEGLKTLRILYEEVDESEVEVIHVPSPALEERKTDSYRYPRTGSKNPKIALKLAEFQTDSQGKIVSTQEKELVQPFSSLFPKVEYIARAGWTRDGKYAWAMFLDRPQQWLQLVLLPPALFIPSTENEEQRLASARAVPRNVQPYVVYEEVTNVWINVHDIFYPFPQLCFLRANECKTGFCHLYKVTAVLKSQGYDWSEPFSPGEDEFKCPIKEEIALTSGEWEVLARHGSKIWVNEETKLVYFQGTKDTPLEHHLYVVSYEAAGEIVRLTTPGFSHSCSMSQNFDMFVSHYSSVSTPPCVHVYKLSGPDDDPLHKQPRFWASMMEADYVPPEIFHFHTRSDVRLYGMIYKPHALQPGKKHPTVLFVYGGPQVQLVNNSFKGIKYLRLNTLASLGYAVVVIDGRGSCQRGLRFEGALKNQMGQVEIEDQVEGLQFVAEKYGFIDLSRVAIHGWSYGGFLSLMGLIHKPQVFKVAIAGAPVTVWMAYDTGYTERYMDVPENNQHGYEAGSVALHVEKLPNEPNRLLILHGFLDENVHFFHTNFLVSQLIRAGKPYQLQIYPNERHSIRCPESGEHYEVTLLHFLQEYLHH Hydrogen bonds contact Hydrophobic contact | ||||
| 18 | Histone-lysine N-methyltransferase KMT5C (KMT5C) | 3RQ4 | 5.31 | |
Target general information Gen name KMT5C Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Lysine N-methyltransferase 5C; Lysine-specific methyltransferase 5C; Suppressor of variegation 4-20 homolog 2; Su(var)4-20 homolog 2; Suv4-20h2; [histone H4]-N-methyl-L-lysine20 N-methyltransferase KM Protein family Class V-like SAM-binding methyltransferase superfamily, Histone-lysine methyltransferase family, Suvar4-20 subfamily Biochemical class NA Function Histone methyltransferase that specifically methylates monomethylated 'Lys-20' (H4K20me1) and dimethylated 'Lys-20' (H4K20me2) of histone H4 to produce respectively dimethylated 'Lys-20' (H4K20me2) and trimethylated 'Lys-20' (H4K20me3) and thus regulates transcription and maintenance of genome integrity. In vitro also methylates unmodified 'Lys-20' (H4K20me0) of histone H4 and nucleosomes. H4 'Lys-20' trimethylation represents a specific tag for epigenetic transcriptional repression. Mainly functions in pericentric heterochromatin regions, thereby playing a central role in the establishment of constitutive heterochromatin in these regions. KMT5C is targeted to histone H3 via its interaction with RB1 family proteins (RB1, RBL1 and RBL2) (By similarity). Facilitates TP53BP1 foci formation upon DNA damage and proficient non-homologous end-joining (NHEJ)-directed DNA repair by catalyzing the di- and trimethylation of 'Lys-20' of histone H4. May play a role in class switch reconbination by catalyzing the di- and trimethylation of 'Lys-20' of histone H4 (By similarity). Related diseases Brachydactyly A2 (BDA2) [MIM:112600]: A form of brachydactyly. Brachydactyly defines a group of inherited malformations characterized by shortening of the digits due to abnormal development of the phalanges and/or the metacarpals. In brachydactyly type A2 shortening of the middle phalanges is confined to the index finger and the second toe, all other digits being more or less normal. Because of a rhomboid or triangular shape of the affected middle phalanx, the end of the second finger usually deviates radially. {ECO:0000269|PubMed:19327734, ECO:0000269|PubMed:21357617}. The gene represented in this entry is involved in disease pathogenesis. Duplications of a cis-regulatory element located approximately 110 kb downstream of BMP2 have been found in BDA2 families. They likely cause altered BMP2 expression with pathological consequences. {ECO:0000269|PubMed:19327734, ECO:0000269|PubMed:21357617}.; DISEASE: Short stature, facial dysmorphism, and skeletal anomalies with or without cardiac anomalies 1 (SSFSC1) [MIM:617877]: An autosomal dominant disorder characterized by short stature, facial dysmorphism, skeletal anomalies, and variable cardiac defects. Distinctive facial features include midface retrusion, short upturned nose, long philtrum, high-arched or cleft palate, and variable degrees of micrognathia and dental crowding. Skeletal anomalies include patterning defects of the axial skeleton, characterized by 11 pairs of ribs and brachydactyly of the fifth ray. Congenital heart defects are variably observed and appear to involve primarily the cardiac outflow tract. {ECO:0000269|PubMed:29198724}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q13185 EC number EC 2.1.1.361 Uniprot keywords 3D-structure; Alternative splicing; Chromatin regulator; Chromosome; Metal-binding; Methyltransferase; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Repressor; S-adenosyl-L-methionine; Transcription; Transcription regulation; Transferase; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 27285.8 Length 240 Aromaticity 0.1 Instability index 42.74 Isoelectric point 8.32 Charge (pH=7) 3.24 2D Binding mode Binding energy (Kcal/mol) -7.25  Molscript Map  Pymol Map 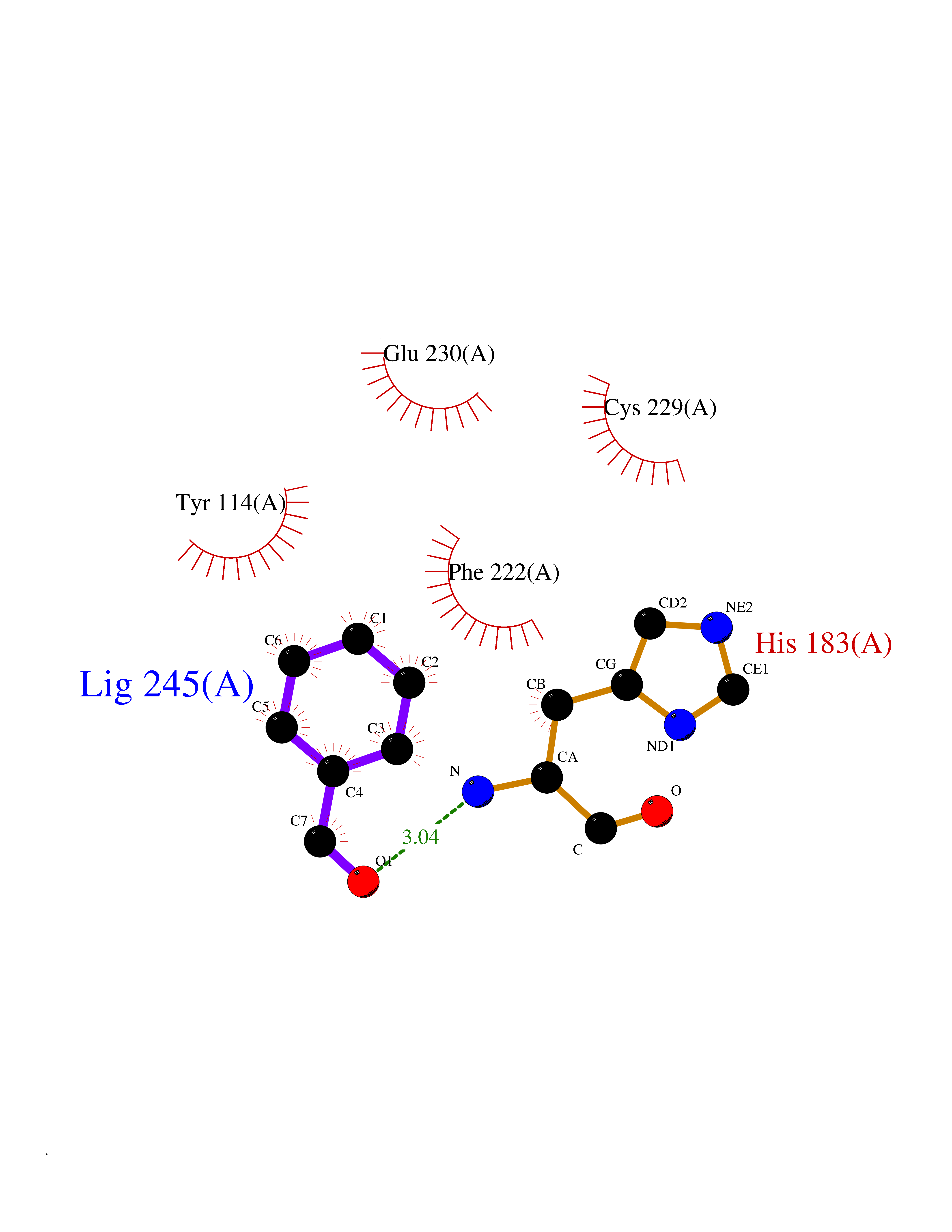 Ligplot Map 3D Binding mode Sequence DRVTARELCENDDLATSLVLDPYLGFRTHKMNVSPVPPLRRQQHLRSALETFLRQRDLEAAYRALTLGGWTARYFQSRGPRQEAALKTHVYRYLRAFLPESGFTILPCTRYSMETNGAKIVSTRAWKKNEKLELLVGCIAELREADEGLLRAGENDFSIMYSTRKRSAQLWLGPAAFINHDCKPNCKFVPADGNAACVKVLRDIEPGDEVTCFYGEGFFGEKNEHCECHTCERKGEGAFR Hydrogen bonds contact Hydrophobic contact | ||||
| 19 | Phosphatidylethanolamine-binding protein 1 (PEBP1) | 2QYQ | 5.31 | |
Target general information Gen name PEBP1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Raf kinase inhibitor protein; RKIP; Prostatic-binding protein; PEBP-1; PEBP; PBP; Neuropolypeptide h3; Hippocampal cholinergic neurostimulating peptide; HCNPpp; HCNP Protein family Phosphatidylethanolamine-binding protein family Biochemical class Phosphatidylethanolamine-binding protein family Function Binds ATP, opioids and phosphatidylethanolamine. Has lower affinity for phosphatidylinositol and phosphatidylcholine. Serine protease inhibitor which inhibits thrombin, neuropsin and chymotrypsin but not trypsin, tissue type plasminogen activator and elastase. Inhibits the kinase activity of RAF1 by inhibiting its activation and by dissociating the RAF1/MEK complex and acting as a competitive inhibitor of MEK phosphorylation. Related diseases Retinitis pigmentosa 49 (RP49) [MIM:613756]: A retinal dystrophy belonging to the group of pigmentary retinopathies. Retinitis pigmentosa is characterized by retinal pigment deposits visible on fundus examination and primary loss of rod photoreceptor cells followed by secondary loss of cone photoreceptors. Patients typically have night vision blindness and loss of midperipheral visual field. As their condition progresses, they lose their far peripheral visual field and eventually central vision as well. {ECO:0000269|PubMed:15570217, ECO:0000269|PubMed:7479749}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB09130; DB09568 Interacts with P16050; Q9NRD5; P04049; Q15208; Q9NS68; Q9JLL3 EC number NA Uniprot keywords 3D-structure; ATP-binding; Cytoplasm; Direct protein sequencing; Disulfide bond; Lipid-binding; Nucleotide-binding; Phosphoprotein; Protease inhibitor; Proteomics identification; Reference proteome; Serine protease inhibitor Protein physicochemical properties Chain ID A Molecular weight (Da) 20928.3 Length 186 Aromaticity 0.1 Instability index 24.05 Isoelectric point 6.59 Charge (pH=7) -0.98 2D Binding mode Binding energy (Kcal/mol) -7.24  Molscript Map  Pymol Map 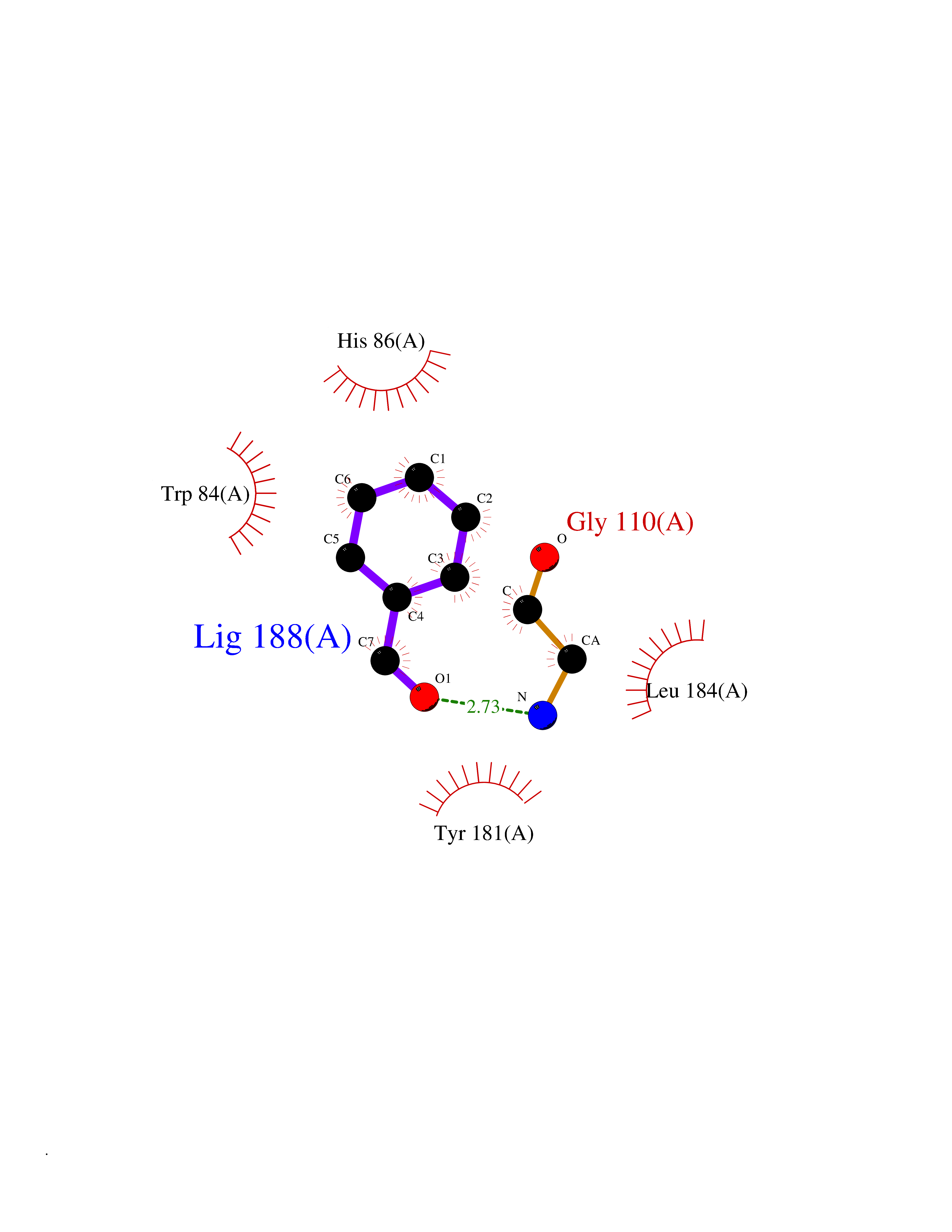 Ligplot Map 3D Binding mode Sequence MPVDLSKWSGPLSLQEVDEQPQHPLHVTYAGAAVDELGKVLTPTQVKNRPTSISWDGLDSGKLYTLVLTDPDAPSRKDPKYREWHHFLVVNMKGNDISSGTVLSDYVGSGPPKGTGLHRYVWLVYEQDRPLKCDEPILSNRSGDHRGKFKVASFRKKYELRAPVAGTCYQAEWDDYVPKLYEQLSG Hydrogen bonds contact Hydrophobic contact | ||||
| 20 | Retinoic acid receptor alpha (RARA) | 3KMR | 5.30 | |
Target general information Gen name RARA Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms RAR-alpha; RAR alpha; Nuclear receptor subfamily 1 group B member 1; NR1B1 Protein family Nuclear hormone receptor family, NR1 subfamily Biochemical class Nuclear hormone receptor Function Retinoic acid receptors bind as heterodimers to their target response elements in response to their ligands, all-trans or 9-cis retinoic acid, and regulate gene expression in various biological processes. The RXR/RAR heterodimers bind to the retinoic acid response elements (RARE) composed of tandem 5'-AGGTCA-3' sites known as DR1-DR5. In the absence of ligand, the RXR-RAR heterodimers associate with a multiprotein complex containing transcription corepressors that induce histone acetylation, chromatin condensation and transcriptional suppression. On ligand binding, the corepressors dissociate from the receptors and associate with the coactivators leading to transcriptional activation. RARA plays an essential role in the regulation of retinoic acid-induced germ cell development during spermatogenesis. Has a role in the survival of early spermatocytes at the beginning prophase of meiosis. In Sertoli cells, may promote the survival and development of early meiotic prophase spermatocytes. In concert with RARG, required for skeletal growth, matrix homeostasis and growth plate function. Receptor for retinoic acid. Related diseases Chromosomal aberrations involving RARA are commonly found in acute promyelocytic leukemia. Translocation t(11;17)(q32;q21) with ZBTB16/PLZF; translocation t(15;17)(q21;q21) with PML; translocation t(5;17)(q32;q11) with NPM. The PML-RARA oncoprotein requires both the PML ring structure and coiled-coil domain for both interaction with UBE2I, nuclear microspeckle location and sumoylation. In addition, the coiled-coil domain functions in blocking RA-mediated transactivation and cell differentiation. {ECO:0000269|PubMed:12691149, ECO:0000269|PubMed:8302850, ECO:0000269|PubMed:8562957}. Drugs (DrugBank ID) DB00459; DB00210; DB00523; DB00926; DB00982; DB05785; DB04942; DB00799; DB00755; DB12808 Interacts with O43707-1; O15296; Q15699; Q96RK4; O95273; P51946; Q15910; P50148; Q9UKP3; Q96EZ8; Q15648; Q71SY5; Q15788; Q9Y6Q9; O75376; Q9Y618; Q16236; P13056-2; P48552; Q9UPP1-2; Q9H8W4; P37231; P78527; P19793; P28702; P28702-3; P48443; Q96EB6; P63165; Q8WW24; Q2M1K9; Q91XC0; P59598; Q14457; P48552; Q96CV9; P28702; P48443; Q8WW24 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Chromosomal rearrangement; Cytoplasm; DNA-binding; Isopeptide bond; Metal-binding; Nucleus; Phosphoprotein; Proteomics identification; Proto-oncogene; Receptor; Reference proteome; Transcription; Transcription regulation; Ubl conjugation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A Molecular weight (Da) 27724.1 Length 244 Aromaticity 0.05 Instability index 50.8 Isoelectric point 5.82 Charge (pH=7) -3.61 2D Binding mode Binding energy (Kcal/mol) -7.23  Molscript Map 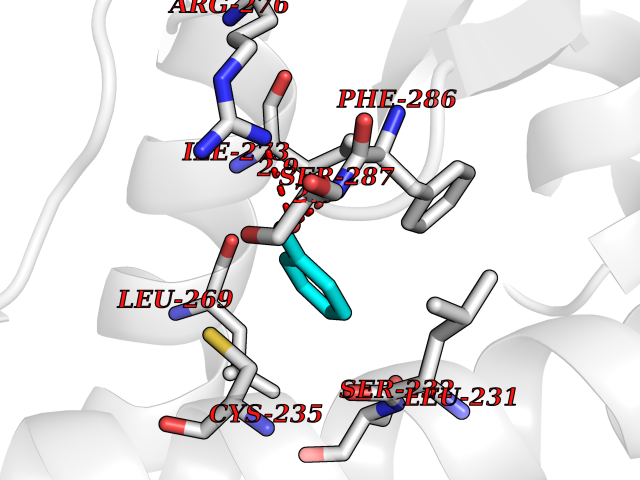 Pymol Map 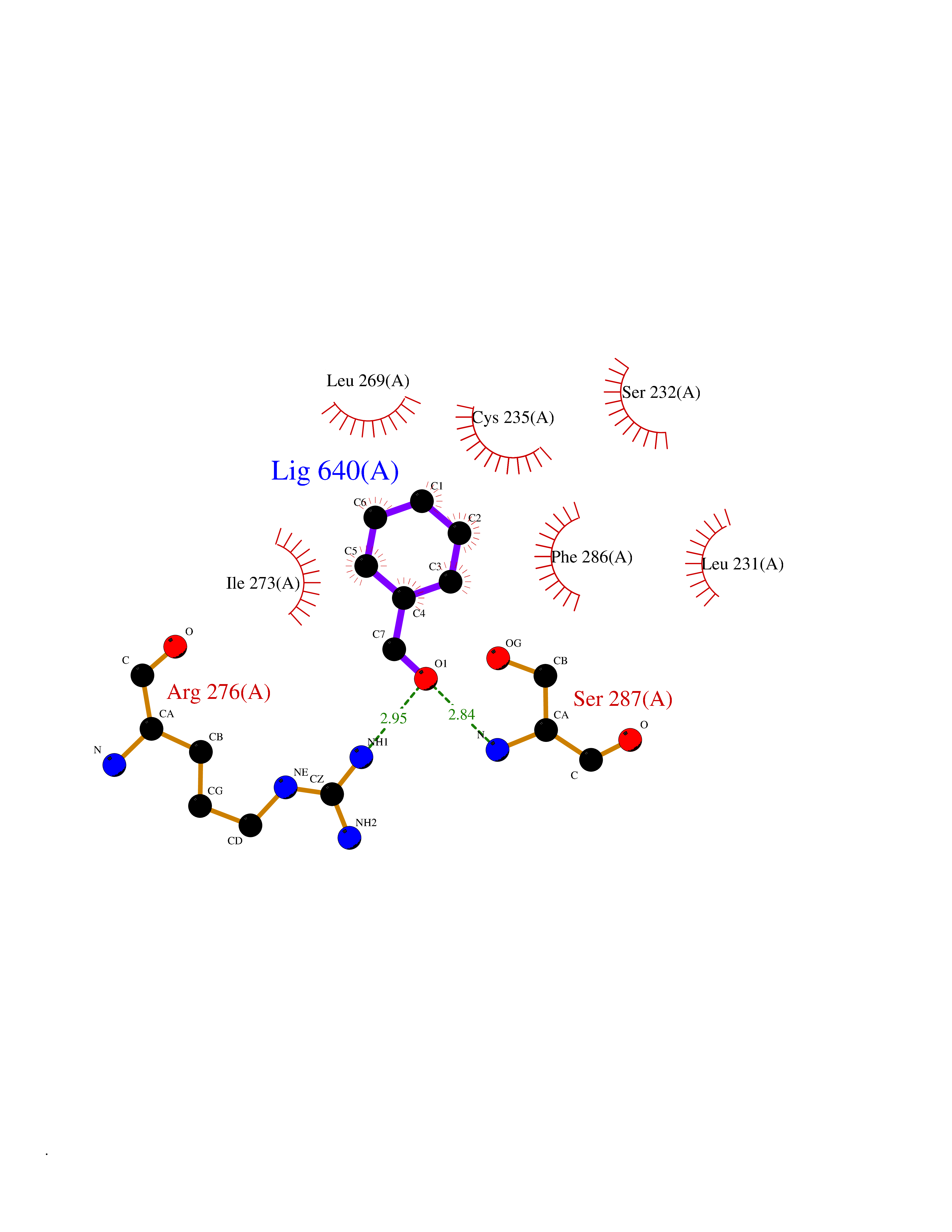 Ligplot Map 3D Binding mode Sequence PEVGELIEKVRKAHQETFPALCQLGKYTTNNSSEQRVSLDIDLWDKFSELSTKCIIKTVEFAKQLPGFTTLTIADQITLLKAACLDILILRICTRYTPEQDTMTFSDGLTLNRTQMHNAGFGPLTDLVFAFANQLLPLEMDDAETGLLSAICLICGDRQDLEQPDRVDMLQEPLLEALKVYVRKRRPSRPHMFPKMLMKITDLRSISAKGAERVITLKMEIPGSMPPLIQEMLEHKILHRLLQE Hydrogen bonds contact Hydrophobic contact | ||||