Job Results:
Ligand
Structure
Job ID
324b1e2d205bb1f9c83ebf1a9a2a4cfb
Job name
NA
Time
2025-02-13 13:22:57
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 1 | Protein cereblon (CRBN) | 5FQD | 5.16 | |
Target general information Gen name CRBN Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Protein cereblon Protein family CRBN family Biochemical class NA Function Substrate recognition component of a DCX (DDB1-CUL4-X-box) E3 protein ligase complex that mediates the ubiquitination and subsequent proteasomal degradation of target proteins, such as MEIS2. Normal degradation of key regulatory proteins is required for normal limb outgrowth and expression of the fibroblast growth factor FGF8. May play a role in memory and learning by regulating the assembly and neuronal surface expression of large-conductance calcium-activated potassium channels in brain regions involved in memory and learning via its interaction with KCNT1. Binding of pomalidomide and other thalidomide-related drugs changes the substrate specificity of the human protein, leading to decreased degradation of MEIS2 and other target proteins and increased degradation of MYC, IRF4, IKZF1 and IKZF3. Related diseases Intellectual developmental disorder, autosomal recessive 2 (MRT2) [MIM:607417]: A disorder characterized by significantly below average general intellectual functioning associated with impairments in adaptive behavior and manifested during the developmental period. MRT2 patients display mild intellectual disability with a standard IQ ranged from 50 to 70. IQ scores are lower in males than females. Developmental milestones are mildly delayed. There are no dysmorphic or autistic features. {ECO:0000269|PubMed:15557513, ECO:0000269|PubMed:28143899}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00480; DB08910; DB01041 Interacts with Q96A83-2; P48729; Q16531; O14901; Q8IVT2; Q9P286; A0A6Q8PF08; Q93062; Q16531; Q13422-7 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; Disease variant; Intellectual disability; Membrane; Metal-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Ubl conjugation; Ubl conjugation pathway; Zinc Protein physicochemical properties Chain ID B,E Molecular weight (Da) 38245.7 Length 337 Aromaticity 0.08 Instability index 40.62 Isoelectric point 5.7 Charge (pH=7) -6.53 2D Binding mode Binding energy (Kcal/mol) -7.03 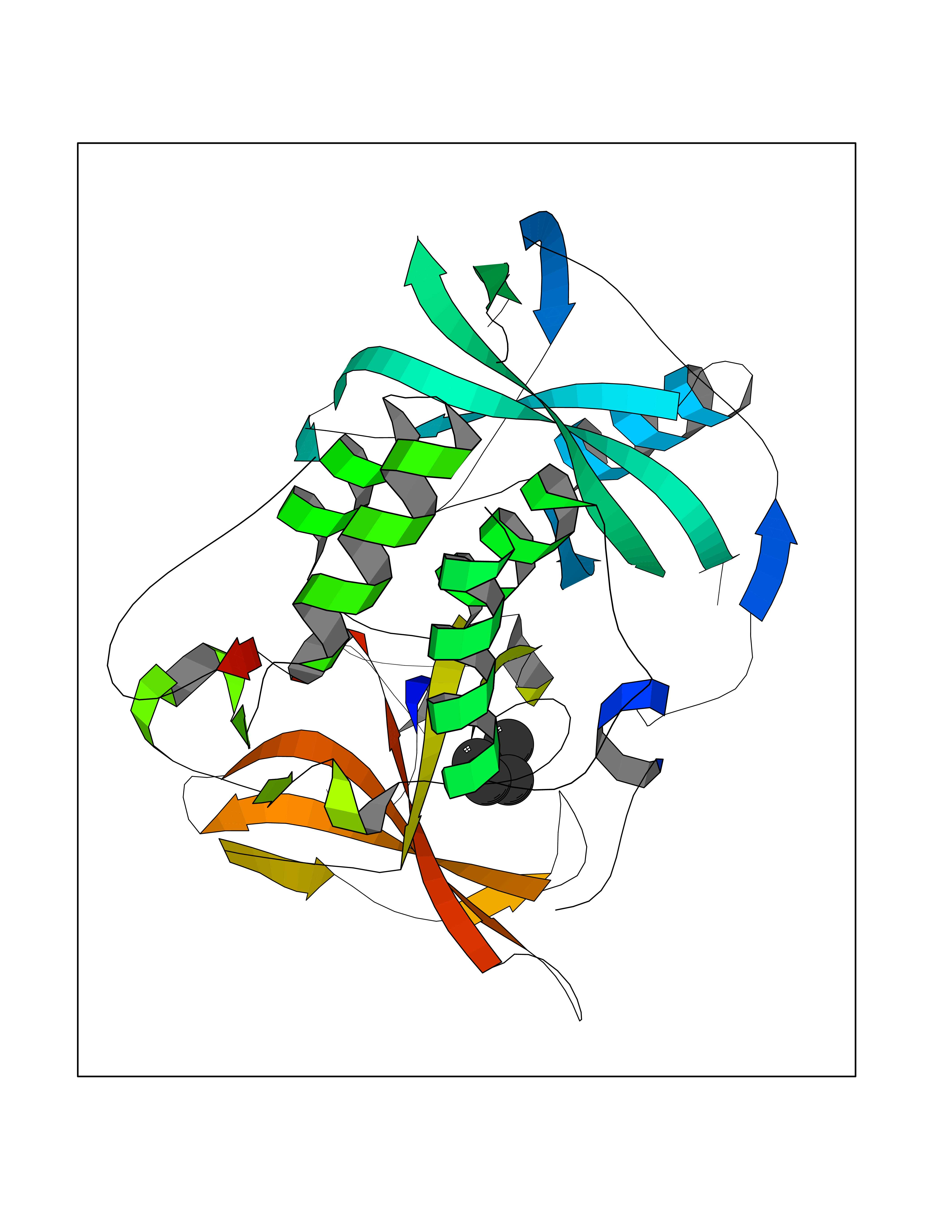 Molscript Map 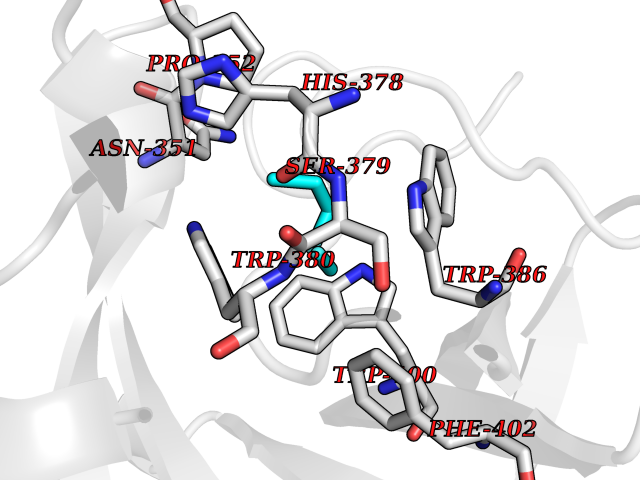 Pymol Map 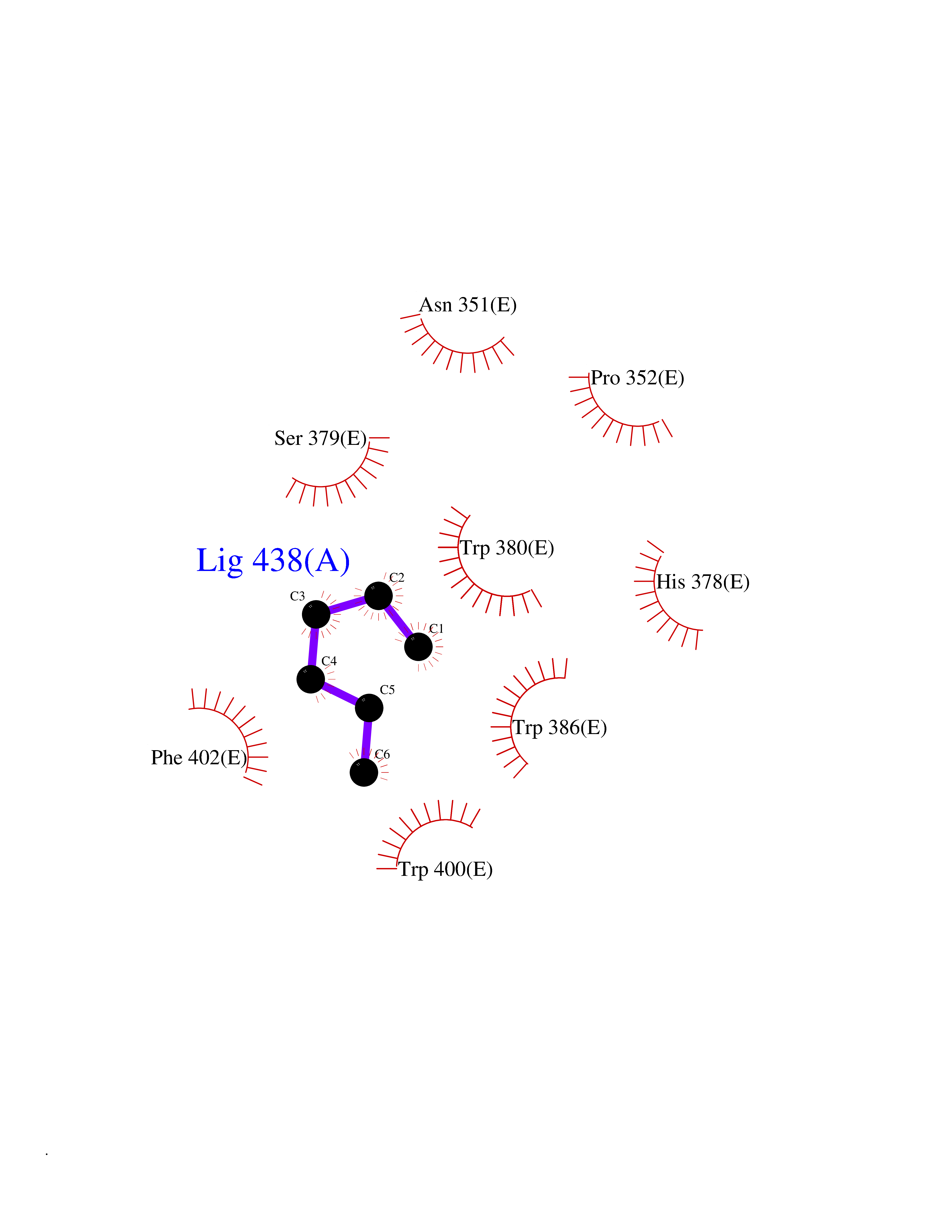 Ligplot Map 3D Binding mode Sequence EFIVGGKYKLNITNGEEVAVINFDTSLPTSHTYLGADMEEFHGRTLHDDDSCQVIPVLPQVMMILIPGQTLPLQLFHPQEVSMVRNLIQKDRTFAVLAYSNVQEREAQFGTTAEIYAYREEIVKVKAIGRQRFKVLEQQAKVQILPECVLAETLMDRIKKQLREWDENLKDDSLPSNPIDFSYRVAACLPIDDVLRIQLLKIGSAIQRLRCELDIMNKCTSLCCKQCQETEITTKNEIFSLSLCGPMAAYVNPHGYVHETLTVYKACNLNLIGRPSTEHSWFPGYAWTVAQCKICASHIGWKFTATKKDMSPQKFWGLTRSALLPTIPDTEDEISPD Hydrogen bonds contact Hydrophobic contact | ||||
| 2 | Zinc finger protein Helios (IKZF2) | 7LPS | 5.12 | |
Target general information Gen name IKZF2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Ikaros family zinc finger protein 2 Protein family Ikaros C2H2-type zinc-finger protein family Biochemical class NA Function Associates with Ikaros at centromeric heterochromatin. Related diseases Developmental and epileptic encephalopathy 25, with amelogenesis imperfecta (DEE25) [MIM:615905]: An autosomal recessive disease characterized by subclinical seizures appearing in the first days of life, evolving to severe epileptic disease. Affected individuals have profound or severe delayed development with lack of speech, and most patients do not acquire the ability to sit. Additional variable features include axial hypotonia, peripheral hypertonia, and abnormal involuntary movements such as dystonia and choreoathetosis. Dental abnormalities, including delayed eruption, hypodontia, tooth hypoplasia, yellow discoloration, thin enamel, and enamel chipping are observed in most patients. {ECO:0000269|PubMed:24995870, ECO:0000269|PubMed:26384929, ECO:0000269|PubMed:30054523}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P29972; P56545; P56545-3; Q17RB8; P09022; Q8N8B7-2 EC number NA Uniprot keywords 3D-structure; Acetylation; Activator; Alternative splicing; DNA-binding; Isopeptide bond; Metal-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Transcription; Transcription regulation; Ubl conjugation; Zinc; Zinc-finger Protein physicochemical properties Chain ID B,C Molecular weight (Da) 47006.6 Length 410 Aromaticity 0.09 Instability index 44.28 Isoelectric point 7.23 Charge (pH=7) 0.69 2D Binding mode Binding energy (Kcal/mol) -6.98 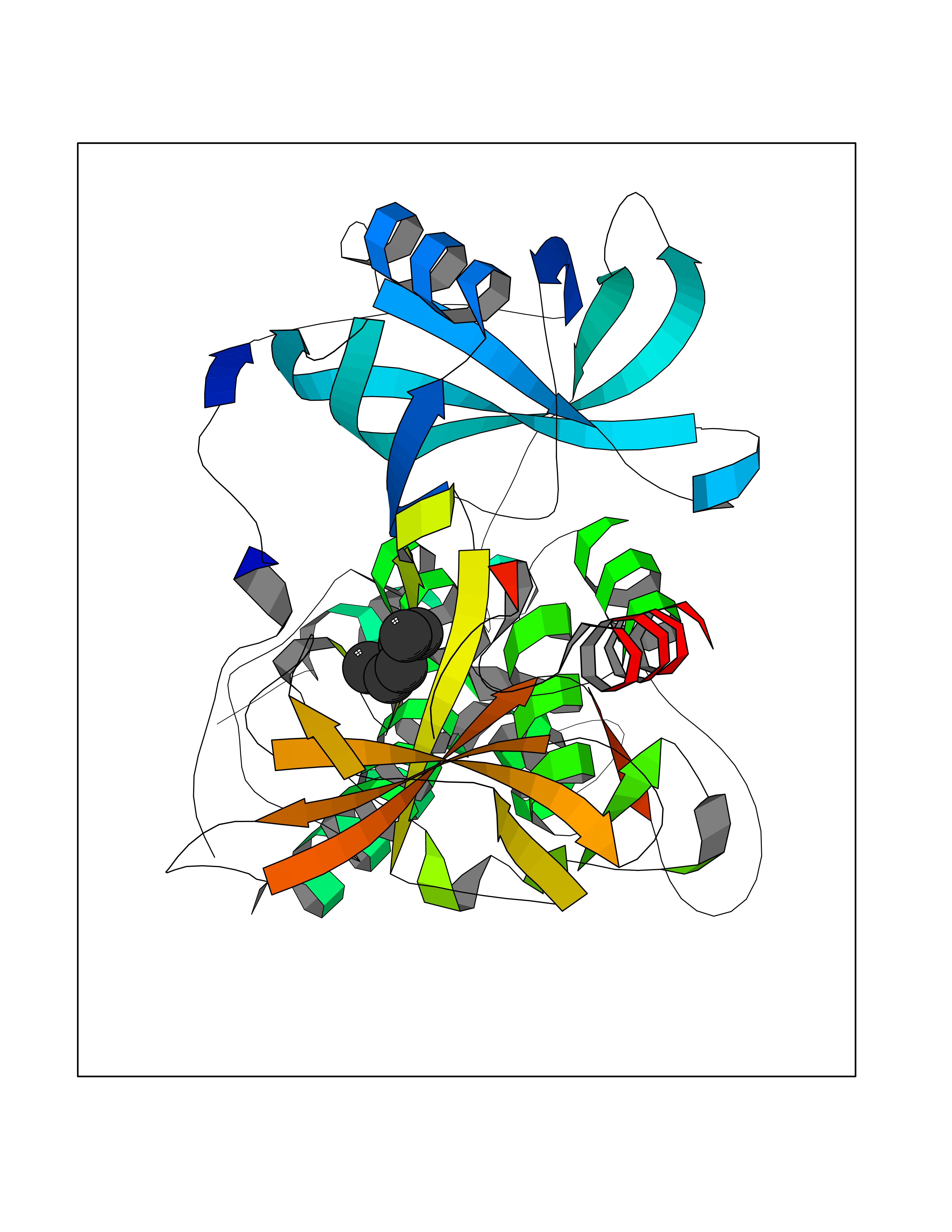 Molscript Map 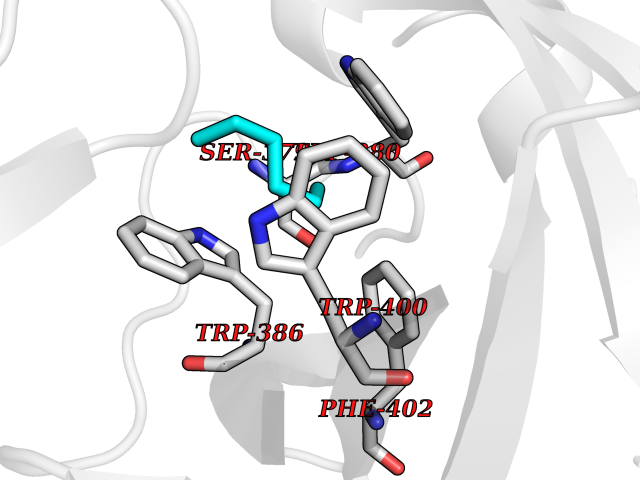 Pymol Map 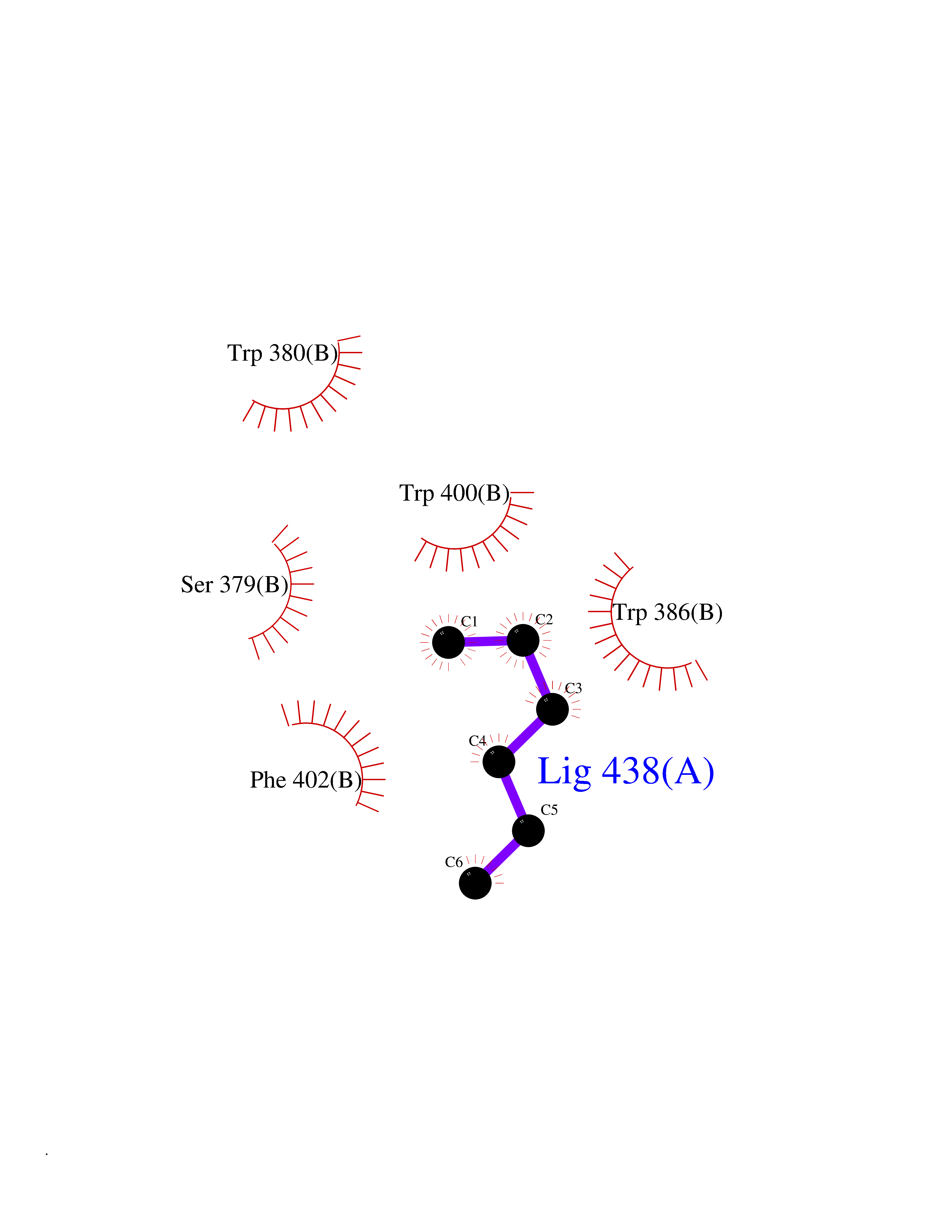 Ligplot Map 3D Binding mode Sequence INFDTSLPTSHTYLGADMEEFHGRTLHDDDSCQVIPVLPQVMMILIPGQTLPLQLFHPQEVSMVRNLIQKDRTFAVLAYSNVQEREAQFGTTAEIYAYREEQDFGIEIVKVKAIGRQRFKVLELRTQSDGIQQAKVQILPECVLPSTMSAVQLESLNKCQIFPCSYKWWQKYQKRKFHCANLTSWPRWLYSLYDAETLMDRIKKQLREWDENLKDDSLPSNPIDFSYRVAACLPIDDVLRIQLLKIGSAIQRLRCELDIMNKCTSLCCKQCQETEITTKNEIFSLSLCGPMAAYVNPHGYVHETLTVYKACNLNLIGRPSTEHSWFPGYAWTVAQCKICASHIGWKFTATKKDMSPQKFWGLTRSALLPTIPDTEDEISPDGERPFHCNQCGASFTQKGNLLRHIKLHSG Hydrogen bonds contact Hydrophobic contact | ||||
| 3 | Neuronal acetylcholine receptor subunit alpha-2 | 5FJV | 5.11 | |
Target general information Gen name CHRNA2 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Ligand-gated ion channel (TC 1.A.9) family, Acetylcholine receptor (TC 1.A.9.1) subfamily, Alpha-2/CHRNA2 sub-subfamily Biochemical class NA Function After binding acetylcholine, the AChR responds by an extensive change in conformation that affects all subunits and leads to opening of an ion-conducting channel across the plasma membrane." Related diseases Epilepsy, nocturnal frontal lobe, 4 (ENFL4) [MIM:610353]: An autosomal dominant focal epilepsy characterized by nocturnal seizures associated with fear sensation, tongue movements, and nocturnal wandering, closely resembling nightmares and sleep walking. {ECO:0000269|PubMed:16826524}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Seizures, benign familial infantile, 6 (BFIS6) [MIM:610353]: A form of benign familial infantile epilepsy, a neurologic disorder characterized by afebrile seizures occurring in clusters during the first year of life, without neurologic sequelae. BFIS6 inheritance is autosomal dominant. {ECO:0000269|PubMed:25847220}. The disease may be caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00732; DB00237; DB00411; DB00565; DB01245; DB00514; DB01135; DB07720; DB00898; DB00472; DB00483; DB08960; DB00657; DB01336; DB00416; DB01226; DB00184; DB01337; DB01338; DB00721; DB00728; DB05740; DB00202; DB01199; DB01339 Interacts with NA EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disease variant; Disulfide bond; Epilepsy; Glycoprotein; Ion channel; Ion transport; Ligand-gated ion channel; Membrane; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Signal; Synapse; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A,B,C,D,E Molecular weight (Da) 120584 Length 1031 Aromaticity 0.14 Instability index 32.21 Isoelectric point 5.62 Charge (pH=7) -17.58 2D Binding mode Binding energy (Kcal/mol) -6.97  Molscript Map 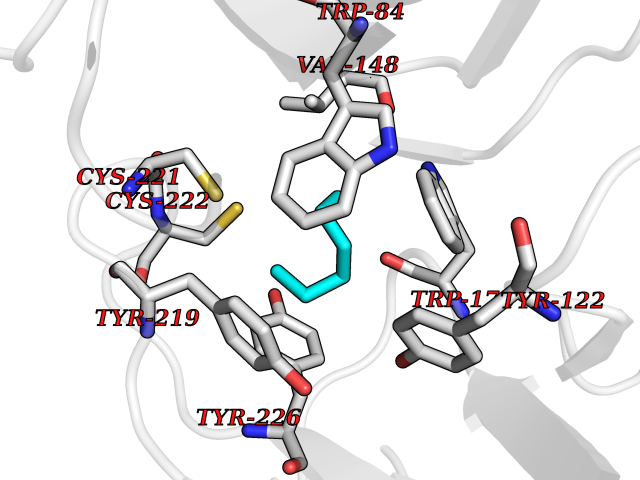 Pymol Map 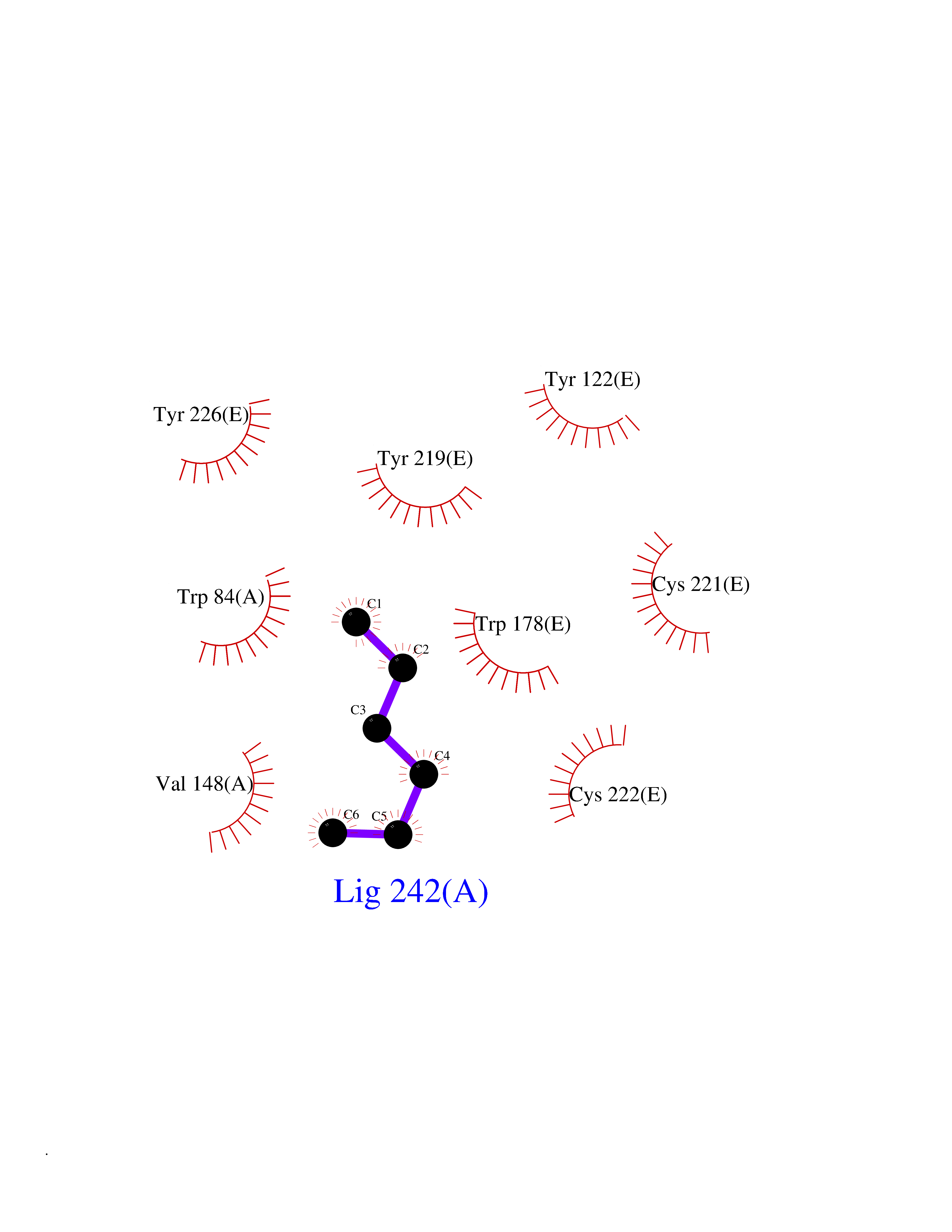 Ligplot Map 3D Binding mode Sequence DRLFKHLFRGYNRWARPVPNTSDVVIVRFGLSIAQLIDVDEKNQMMTTNVWLKQEWSDYKLRWNPTDFGNITSLRVPSEMIWIPDIVLYNNADGEFAVTHMTKAHLFSTGTVHWVPPAIYKSSCSIDVTFFPFDQQNCKMKFGSWTYDKAKIDLEQMEQTVDLKDYWESGEWAIVNATGTYNSKKYDCCAEIYPDVTYAFVIRRLPEDRLFKHLFRGYNRWARPVPNTSDVVIVRFGLSIAQLIDVDEKNQMMTTNVWLKQEWSDYKLRWNPTDFGNITSLRVPSEMIWIPDIVLYNNADGEFAVTHMTKAHLFSTGTVHWVPPAIYKSSCSIDVTFDQQNCKMKFGSWTYDKAKIDLEQMEQTVDLKDYWESGEWAIVNATGTYNSKKYDCCAEIYPDVTYAFVIRRLPEDRLFKHLFRGYNRWARPVPNTSDVVIVRFGLSIAQLIDVDEKNQMMTTNVWLKQEWSDYKLRWNPTDFGNITSLRVPSEMIWIPDIVLYNNADGEFAVTHMTKAHLFSTGTVHWVPPAIYKSSCSIDVTFFPFDQQNCKMKFGSWTYDKAKIDLEQMEQTVDLKDYWESGEWAIVNATGTYNSKKYDCCAEIYPDVTYAFVIRRLPEDRLFKHLFRGYNRWARPVPNTSDVVIVRFGLSIAQLIDVDEKNQMMTTNVWLKQEWSDYKLRWNPTDFGNITSLRVPSEMIWIPDIVLYNNADGEFAVTHMTKAHLFSTGTVHWVPPAIYKSSCSIDVTFFPFDQQNCKMKFGSWTYDKAKIDLEQMEQTVDLKDYWESGEWAIVNATGTYNSKKYDCCAEIYPDVTYAFVIRRLPEDRLFKHLFRGYNRWARPVPNTSDVVIVRFGLSIAQLIDVDEKNQMMTTNVWLKQEWSDYKLRWNPTDFGNITSLRVPSEMIWIPDIVLYNNADGEFAVTHMTKAHLFSTGTVHWVPPAIYKSSCSIDVTFFPFDQQNCKMKFGSWTYDKAKIDLEQMEQTVDLKDYWESGEWAIVNATGTYNSKKYDCCAEIYPDVTYAFVIRRLP Hydrogen bonds contact Hydrophobic contact | ||||
| 4 | Arachidonate 5-lipoxygenase (5-LOX) | 3V99 | 5.10 | |
Target general information Gen name ALOX5 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms LOG5; 5-lipoxygenase; 5-LO Protein family Lipoxygenase family Biochemical class Oxygenase Function Catalyzes the first step in leukotriene biosynthesis, and thereby plays a role in inflammatory processes. Related diseases Pigmentary disorder, reticulate, with systemic manifestations, X-linked (PDR) [MIM:301220]: An X-linked recessive disorder characterized by recurrent infections and sterile inflammation in various organs. Diffuse skin hyperpigmentation with a distinctive reticulate pattern is universally evident by early childhood. This is later followed in many patients by hypohidrosis, corneal inflammation and scarring, enterocolitis that resembles inflammatory bowel disease, and recurrent urethral strictures. Melanin and amyloid deposition is present in the dermis. Affected males also have a characteristic facies with frontally upswept hair and flared eyebrows. Female carriers have only restricted pigmentary changes along Blaschko's lines. {ECO:0000269|PubMed:27019227}. The disease is caused by variants affecting the gene represented in this entry. XLPDR is caused by a recurrent intronic mutation that results in missplicing and reduced POLA1 expression. This leads to a decrease in cytosolic RNA:DNA hybrids and constitutive activation of type I interferon responses, but has no effect on cell replication. {ECO:0000269|PubMed:27019227}.; DISEASE: Van Esch-O'Driscoll syndrome (VEODS) [MIM:301030]: An X-linked recessive syndrome characterized by different degrees of intellectual disability, moderate to severe short stature, microcephaly, hypogonadism, and variable congenital malformations. {ECO:0000269|PubMed:31006512}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB14001; DB00233; DB01014; DB09061; DB14002; DB11994; DB00586; DB00711; DB12010; DB01892; DB00159; DB04725; DB00179; DB00939; DB14009; DB00244; DB01017; DB05431; DB00471; DB09285; DB14011; DB11133; DB13168; DB02709; DB13174; DB00795; DB00163; DB00744 Interacts with Q8IYJ2-2; Q6PII3; Q6P2R3; Q8IYX8-2; Q96MT8; Q96MT8-3; Q14019; P09769; O43716; P08631; Q6UWX4; P14061; P31025; Q9Y6D9; P50221; Q6FHY5; Q86Y26; A6NGQ2; P17612; Q04864; Q7Z699; Q8N0S2; Q9P0N9; P07947 EC number EC 1.13.11.34 Uniprot keywords 3D-structure; Alternative splicing; Calcium; Cytoplasm; Dioxygenase; Direct protein sequencing; Hydrolase; Iron; Leukotriene biosynthesis; Lipid metabolism; Membrane; Metal-binding; Nucleus; Oxidoreductase; Phosphoprotein; Proteomics identification; Reference proteome Protein physicochemical properties Chain ID A,B Molecular weight (Da) 60590.7 Length 523 Aromaticity 0.11 Instability index 41.73 Isoelectric point 5.69 Charge (pH=7) -10.99 2D Binding mode Binding energy (Kcal/mol) -6.95  Molscript Map 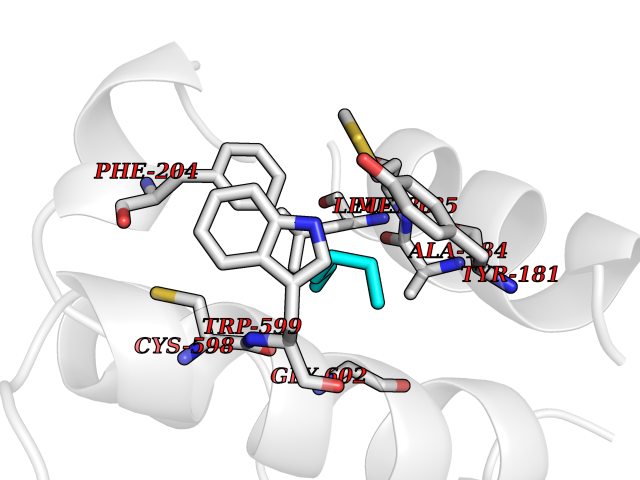 Pymol Map 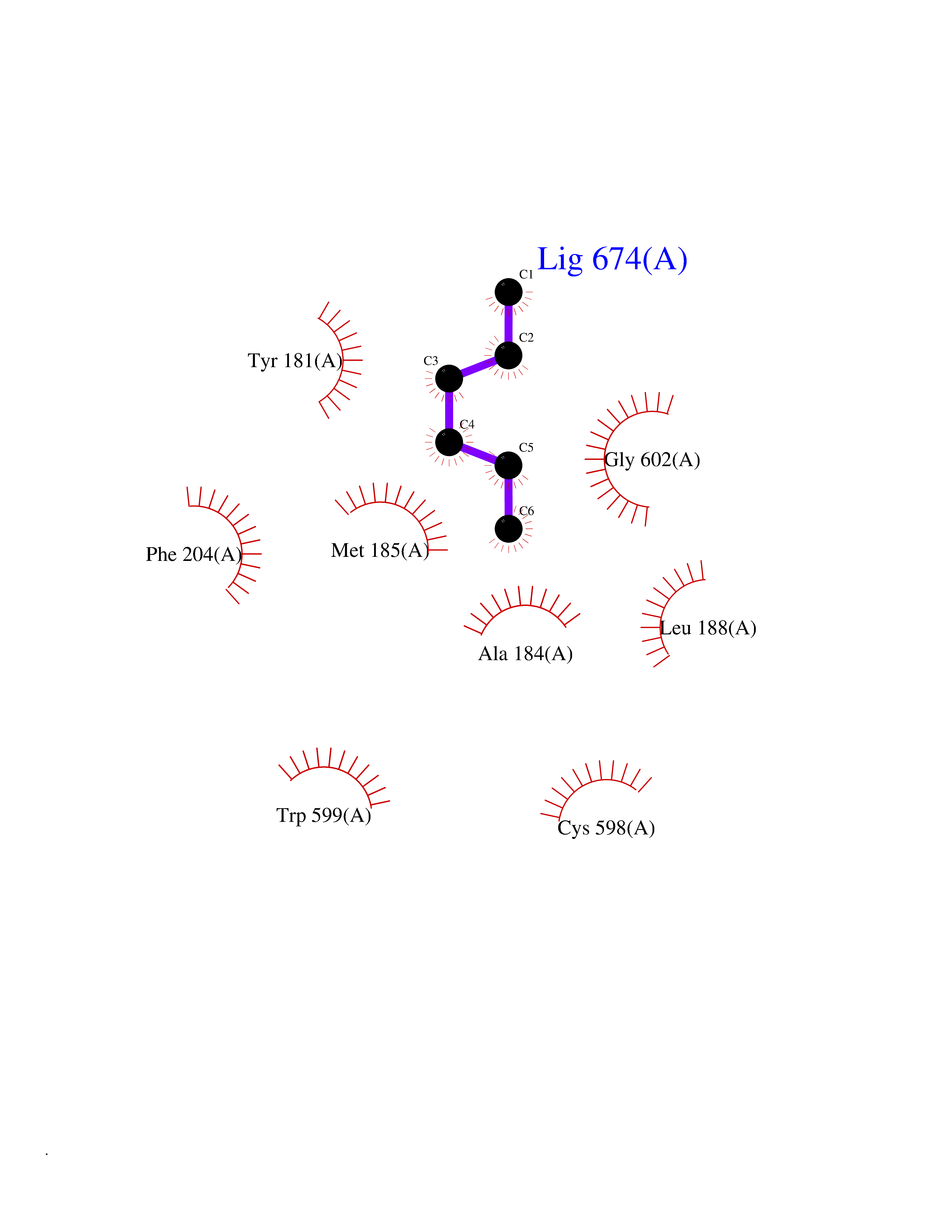 Ligplot Map 3D Binding mode Sequence RDGRAKLARDDQIHILKQHRRKELETRQKQYRWMEWNPGFPLSIDAKCHKDLPRDIQFDSFVLNYSKAMENLFQSSWNDFADFEKIFVKISNTISERVMNHWQEDLMFGYQFLNGANPVLIRRCTELPEKLPVTTEMVECSLERQLSLEQEVQQGNIFIVDFELLDGIDCTLQFLAAPICLLYKNLANKIVPIAIQLNQIPGDENPIFLPSDAKYDWLLAKIWVRSSDFHVHQTITHLLRTHLVSEVFGIAMYRQLPAVHPIFKLLVAHVRFTIAINTKAREQGGHVQMVQRAMKDLTYASLCFPEAIKARGMESKEDIPYYFYRDDGLLVWEAIRTFTAEVVDIYYEGDQVVEEDPELQDFVNDVYVYGMRGRKSSGFPKSVKSREQLSEYLTVVIFTASAQHAAVNFGQYDWASWIPNAPPTMRAPPPTAKGVVTIEQIVDTLPDRGRSCWHLGAVWALSQFELFLGMYPEEHFIEKPVKEAMARFRKNLEAIVSVIAERNENLQLPYYYLDPDRIPNSVA Hydrogen bonds contact Hydrophobic contact | ||||
| 5 | Beta-glucosidase A | 1E4I | 5.10 | |
Target general information Gen name bglA Organism Paenibacillus polymyxa (Bacillus polymyxa) Uniprot ID TTD ID NA Synonyms NA Protein family Glycosyl hydrolase 1 family Biochemical class Hydrolase Function Beta-glucosidase activity.Scopolin beta-glucosidase activity. Related diseases Schizophrenia (SCZD) [MIM:181500]: A complex, multifactorial psychotic disorder or group of disorders characterized by disturbances in the form and content of thought (e.g. delusions, hallucinations), in mood (e.g. inappropriate affect), in sense of self and relationship to the external world (e.g. loss of ego boundaries, withdrawal), and in behavior (e.g bizarre or apparently purposeless behavior). Although it affects emotions, it is distinguished from mood disorders in which such disturbances are primary. Similarly, there may be mild impairment of cognitive function, and it is distinguished from the dementias in which disturbed cognitive function is considered primary. Some patients manifest schizophrenic as well as bipolar disorder symptoms and are often given the diagnosis of schizoaffective disorder. {ECO:0000269|PubMed:15645182}. Disease susceptibility may be associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02658; DB04282; DB04304 Interacts with NA EC number 3.2.1.21 Uniprot keywords 3D-structure; Carbohydrate metabolism; Cellulose degradation; Glycosidase; Hydrolase; Polysaccharide degradation Protein physicochemical properties Chain ID A Molecular weight (Da) 51515.2 Length 447 Aromaticity 0.14 Instability index 38.44 Isoelectric point 5.28 Charge (pH=7) -18.1 2D Binding mode Binding energy (Kcal/mol) -6.96 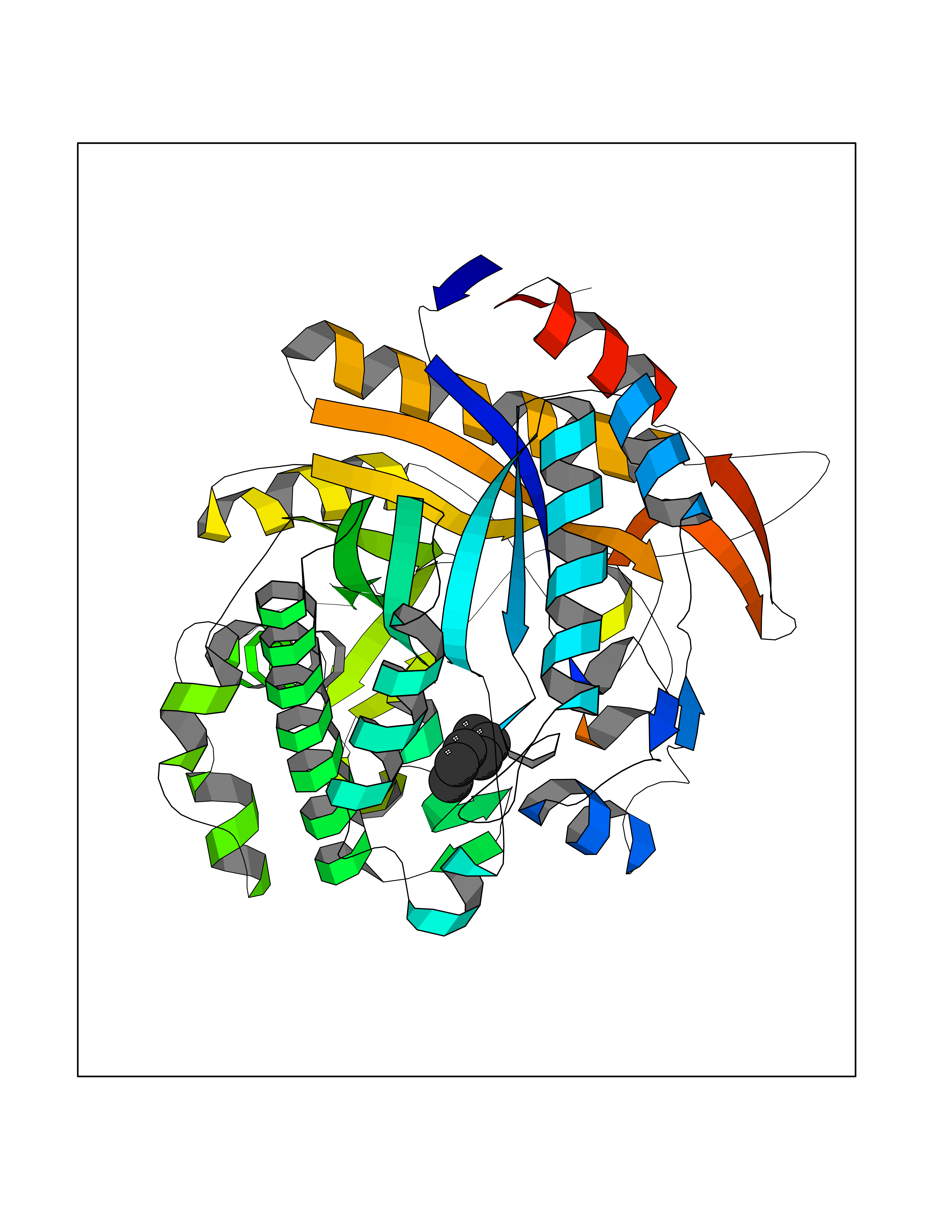 Molscript Map 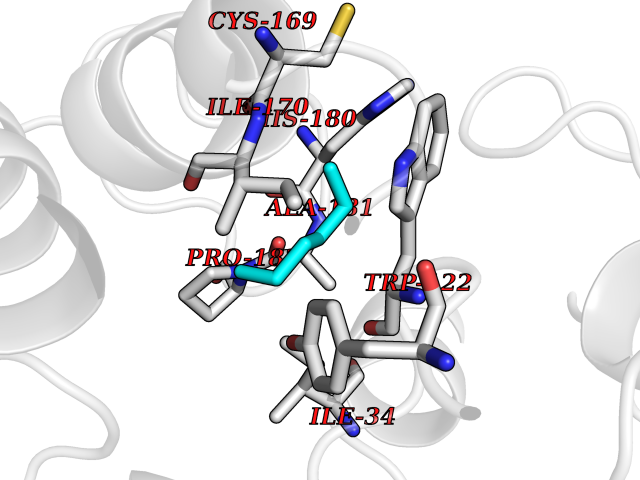 Pymol Map 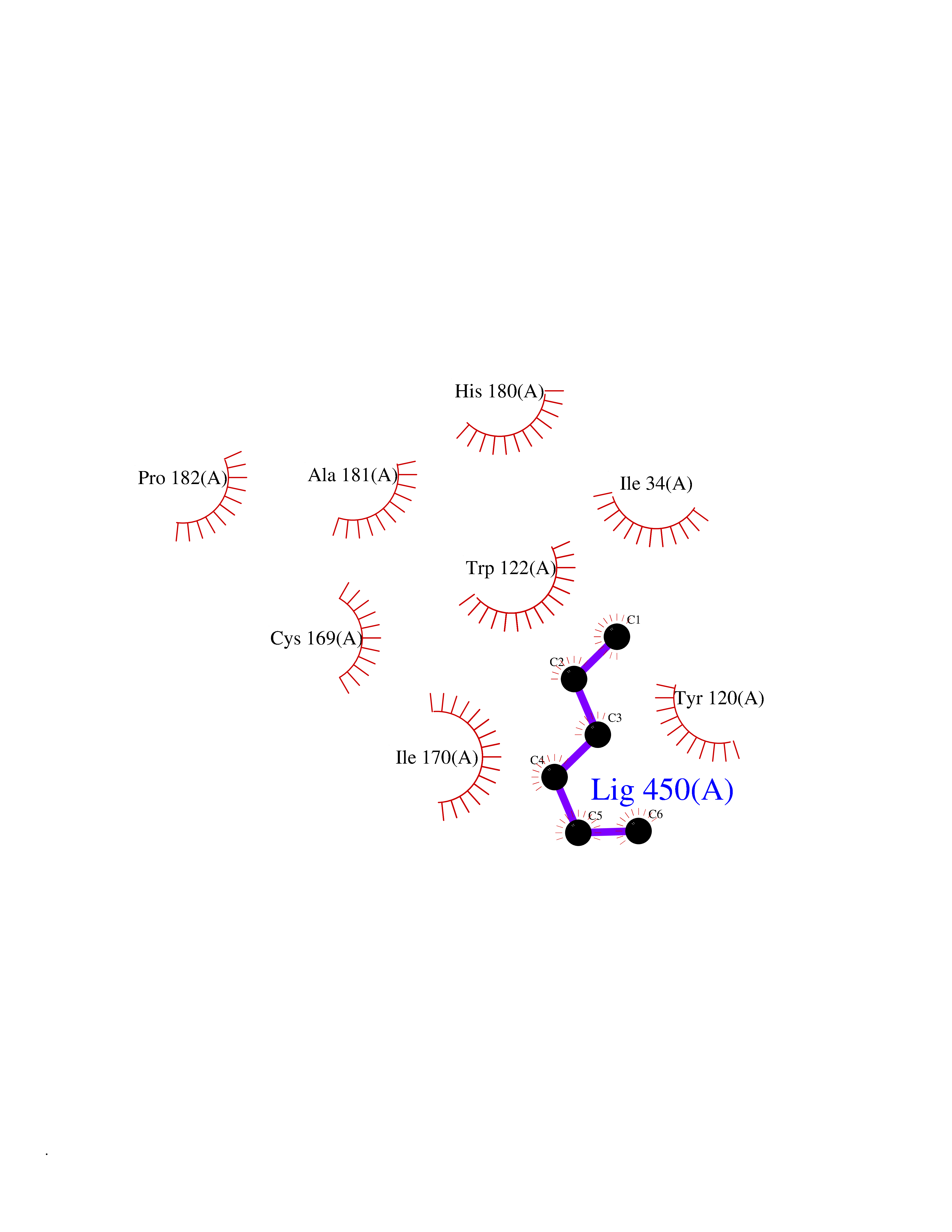 Ligplot Map 3D Binding mode Sequence TIFQFPQDFMWGTATAAYQIEGAYQEDGRGLSIWDTFAHTPGKVFNGDNGNVACDSYHRYEEDIRLMKELGIRTYRFSVSWPRIFPNGDGEVNQKGLDYYHRVVDLLNDNGIEPFCTLYHWDLPQALQDAGGWGNRRTIQAFVQFAETMFREFHGKIQHWLTFNEPWCIAFLSNMLGVHAPGLTNLQTAIDVGHHLLVAHGLSVRRFRELGTSGQIGIAPNVSWAVPYSTSEEDKAACARTISLHSDWFLQPIYQGSYPQFLVDWFAEQGATVPIQDGDMDIIGEPIDMIGINYYSMSVNRFNPEAGFLQSEEINMGLPVTDIGWPVESRGLYEVLHYLQKYGNIDIYITENGACINDEVVNGKVQDDRRISYMQQHLVQVHRTIHDGLHVKGYMAWSLLDNFEWAEGYNMRFGMIHVDFRTQVRTPKQSYYWYRNVVSNNWLETRR Hydrogen bonds contact Hydrophobic contact | ||||
| 6 | Aldose reductase (AKR1B1) | 1US0 | 5.08 | |
Target general information Gen name AKR1B1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Aldehyde reductase; AKR1B1 Protein family Aldo/keto reductase family Biochemical class Short-chain dehydrogenases reductase Function Catalyzes the NADPH-dependent reduction of a wide variety of carbonyl-containing compounds to their corresponding alcohols with a broad range of catalytic efficiencies. Related diseases Glutamine deficiency, congenital (GLND) [MIM:610015]: An autosomal recessive disorder characterized by variable brain malformations, encephalopathy, severe developmental delay, seizures, and decreased glutamine levels in bodily fluids. Death in early infancy may occur. {ECO:0000269|PubMed:16267323, ECO:0000269|PubMed:26711351, ECO:0000269|PubMed:38579670}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Developmental and epileptic encephalopathy 116 (DEE116) [MIM:620806]: A form of epileptic encephalopathy, a heterogeneous group of early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE116 is autosomal dominant form characterized by severe developmental delay, seizures, and white matter abnormalities. {ECO:0000269|PubMed:38579670}. The disease is caused by variants affecting the gene represented in this entry. DEE116 is caused by variants that disrupt the canonical translation start codon in GLUL resulting in initiation of translation at Met-18 (PubMed:38579670). The resulting protein is enzymatically competent but insensitive to negative feedback regulation via glutamine-induced degradation. {ECO:0000269|PubMed:38579670}. Drugs (DrugBank ID) DB07028; DB07030; DB07450; DB02101; DB08449; DB08000; DB07139; DB07498; DB02007; DB02020; DB11859; DB02994; DB04272; DB07187; DB00694; DB00997; DB06246; DB01039; DB02021; DB16707; DB00143; DB02834; DB08084; DB01689; DB07063; DB06077; DB02518; DB00157; DB03461; DB05383; DB05533; DB05327; DB02712; DB00605; DB02383; DB02132; DB08772; DB07093; DB07999; DB08098 Interacts with Q9BUY7 EC number EC 1.1.1.300 Uniprot keywords 3D-structure; Acetylation; Cytoplasm; Direct protein sequencing; Lipid metabolism; NADP; Oxidoreductase; Phosphoprotein; Proteomics identification; Reference proteome Protein physicochemical properties Chain ID A Molecular weight (Da) 35447.6 Length 313 Aromaticity 0.09 Instability index 36.41 Isoelectric point 7.1 Charge (pH=7) 0.26 2D Binding mode Binding energy (Kcal/mol) -6.94 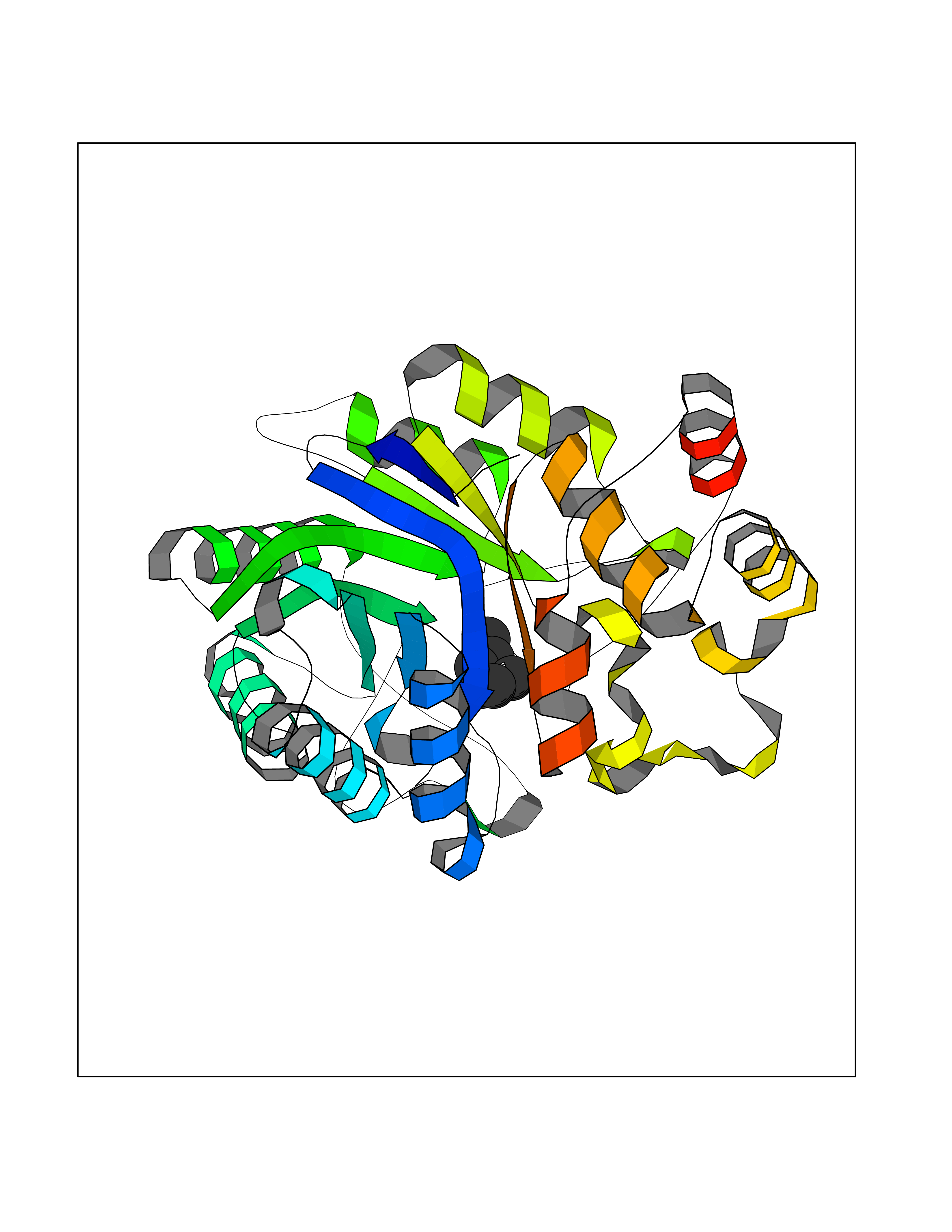 Molscript Map 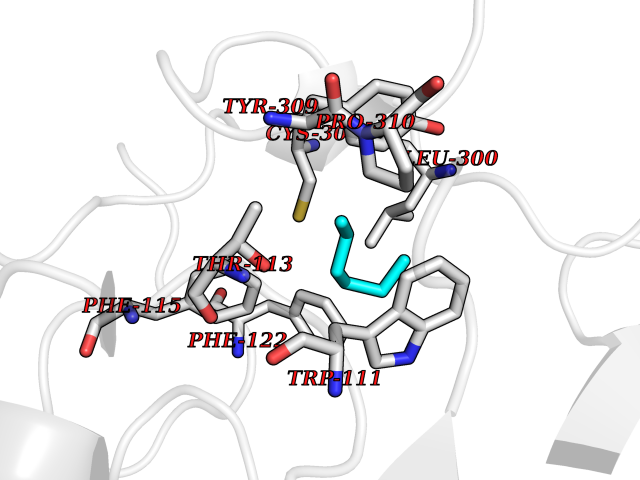 Pymol Map 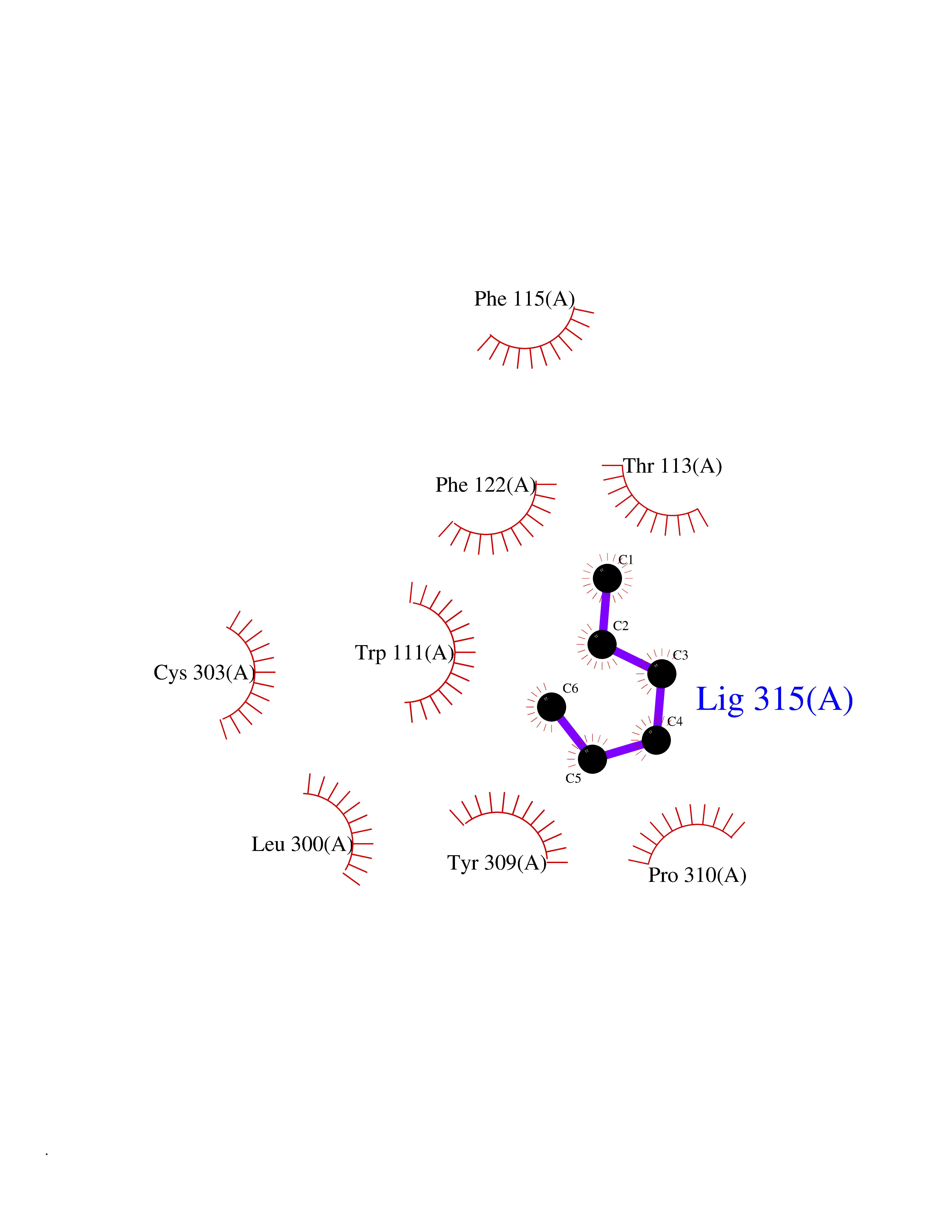 Ligplot Map 3D Binding mode Sequence MASRILLNNGAKMPILGLGTWKSPPGQVTEAVKVAIDVGYRHIDCAHVYQNENEVGVAIQEKLREQVVKREELFIVSKLWCTYHEKGLVKGACQKTLSDLKLDYLDLYLIHWPTGFKPGKEFFPLDESGNVVPSDTNILDTWAAMEELVDEGLVKAIGISNFNHLQVEMILNKPGLKYKPAVNQIECHPYLTQEKLIQYCQSKGIVVTAYSPLGSPDRPWAKPEDPSLLEDPRIKAIAAKHNKTTAQVLIRFPMQRNLVVIPKSVTPERIAENFKVFDFELSSQDMTTLLSYNRNWRVCALLSCTSHKDYPFH Hydrogen bonds contact Hydrophobic contact | ||||
| 7 | Cocaine esterase | 3I2K | 5.07 | |
Target general information Gen name cocE Organism Rhodococcus sp. (strain MB1 Bresler) Uniprot ID TTD ID NA Synonyms NA Protein family CocE/NonD hydrolase family Biochemical class Hydrolase Function Carboxylic ester hydrolase activity.Dipeptidyl-peptidase activity. Related diseases Thiamine metabolism dysfunction syndrome 5, episodic encephalopathy type (THMD5) [MIM:614458]: An autosomal recessive metabolic disorder due to an inborn error of thiamine metabolism. The phenotype is highly variable, but in general, affected individuals have onset in early childhood of acute encephalopathic episodes associated with increased serum and CSF lactate. These episodes result in progressive neurologic dysfunction manifest as gait disturbances, ataxia, dystonia, and spasticity, which in some cases may result in loss of ability to walk. Cognitive function is usually preserved, although mildly delayed development has been reported. These episodes are usually associated with infection and metabolic decompensation. Some patients may have recovery of some neurologic deficits. {ECO:0000269|PubMed:22152682}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03793; DB01795 Interacts with NA EC number 3.1.1.84 Uniprot keywords 3D-structure; Cytoplasm; Direct protein sequencing; Hydrolase; Serine esterase Protein physicochemical properties Chain ID A Molecular weight (Da) 62127.9 Length 574 Aromaticity 0.09 Instability index 26.62 Isoelectric point 4.56 Charge (pH=7) -33.24 2D Binding mode Binding energy (Kcal/mol) -6.91 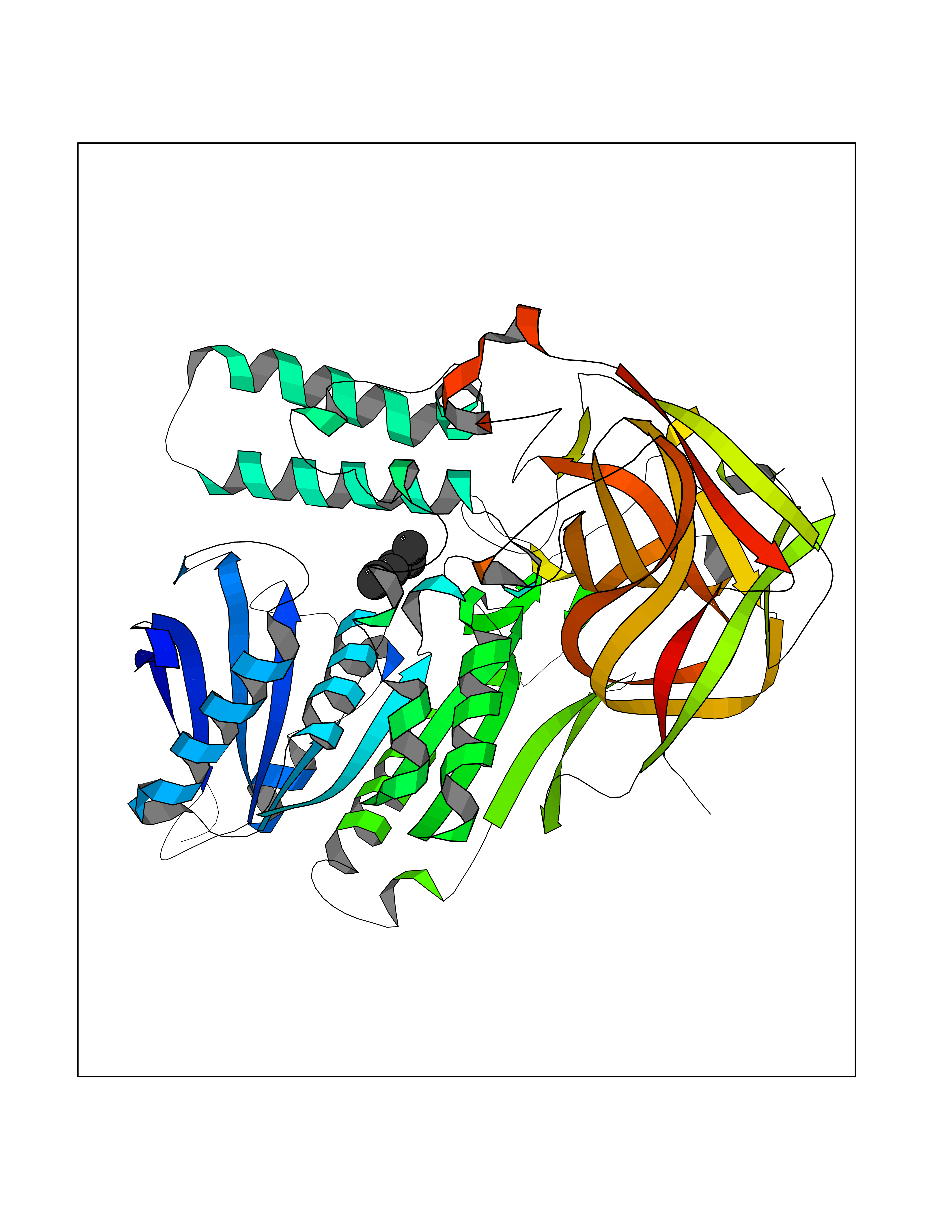 Molscript Map 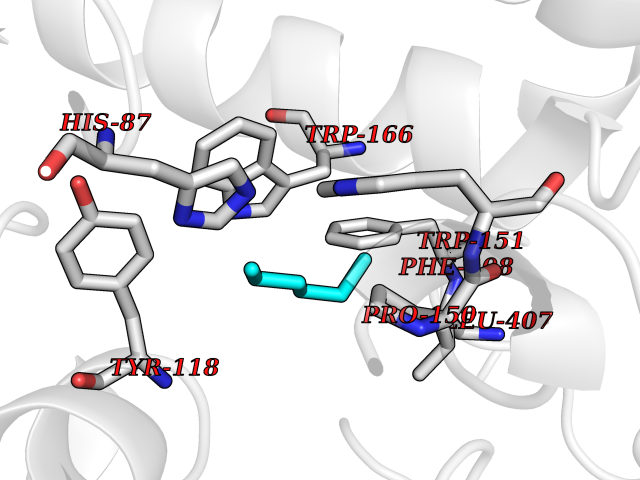 Pymol Map 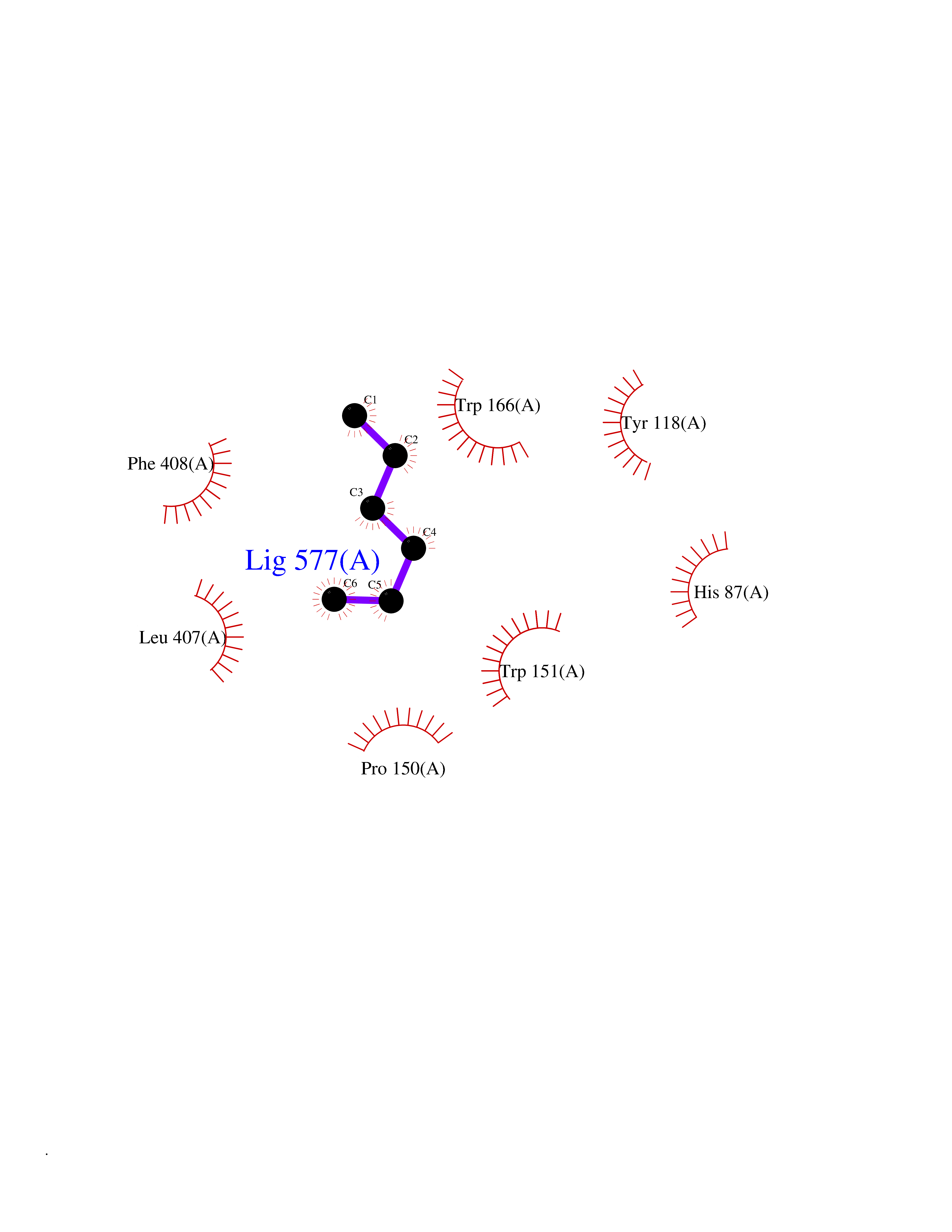 Ligplot Map 3D Binding mode Sequence VDGNYSVASNVMVPMRDGVRLAVDLYRPDADGPVPVLLVRNPYDKFDVFAWSTQSTNWLEFVRDGYAVVIQDTRGLFASEGEFVPHVDDEADAEDTLSWILEQAWCDGNVGMFGVSYLGVTQWQAAVSGVGGLKAIAPSMASADLYRAPWYGPGGALSVEALLGWSALIGTGLITSRSDARPEDAADFVQLAAILNDVAGAASVTPLAEQPLLGRLIPWVIDQVVDHPDNDESWQSISLFERLGGLATPALITAGWYDGFVGESLRTFVAVKDNADARLVVGPWSHSNLTGRNADRKFGIAATYPIQEATTMHKAFFDRHLRGETDALAGVPKVRLFVMGIDEWRDETDWPLPDTAYTPFYLGGSGAANTSTGGGTLSTSISGTESADTYLYDPADPVPSLGGTLLFHNGDNGPADQRPIHDRDDVLCYSTEVLTDPVEVTGTVSARLFVSSSAVDTDFTAKLVDVFPDGRAIALCDGIVRMRYRETLVNPTLIEAGEIYEVAIDMLATSNVFLPGHRIMVQVSSSNFPKYDRNSNTGGVIAREQLEEMCTAVNRIHRGPEHPSHIVLPIIKRK Hydrogen bonds contact Hydrophobic contact | ||||
| 8 | Cerebron E3 ubiquitin ligase complex (CRL4-CRBN E3 ubiquitin ligase) | 4CI1 | 5.07 | |
Target general information Gen name CUL4A/CUL4B-DDB1-CRBN Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms NA Protein family Cullin family Biochemical class NA Function NA Related diseases Orotic aciduria 1 (ORAC1) [MIM:258900]: A disorder of pyrimidine metabolism resulting in megaloblastic anemia and orotic acid crystalluria that is frequently associated with some degree of physical and intellectual disability. A minority of cases have additional features, particularly congenital malformations and immune deficiencies. {ECO:0000269|PubMed:9042911}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P54253; Q86VP6; Q16531; Q92466; P08238; O94888; P55072 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Biological rhythms; DNA damage; DNA repair; Host-virus interaction; Isopeptide bond; Phosphoprotein; Proteomics identification; Reference proteome; Ubl conjugation; Ubl conjugation pathway Protein physicochemical properties Chain ID B Molecular weight (Da) 42669.7 Length 368 Aromaticity 0.1 Instability index 44.94 Isoelectric point 8.72 Charge (pH=7) 6.58 2D Binding mode Binding energy (Kcal/mol) -6.91 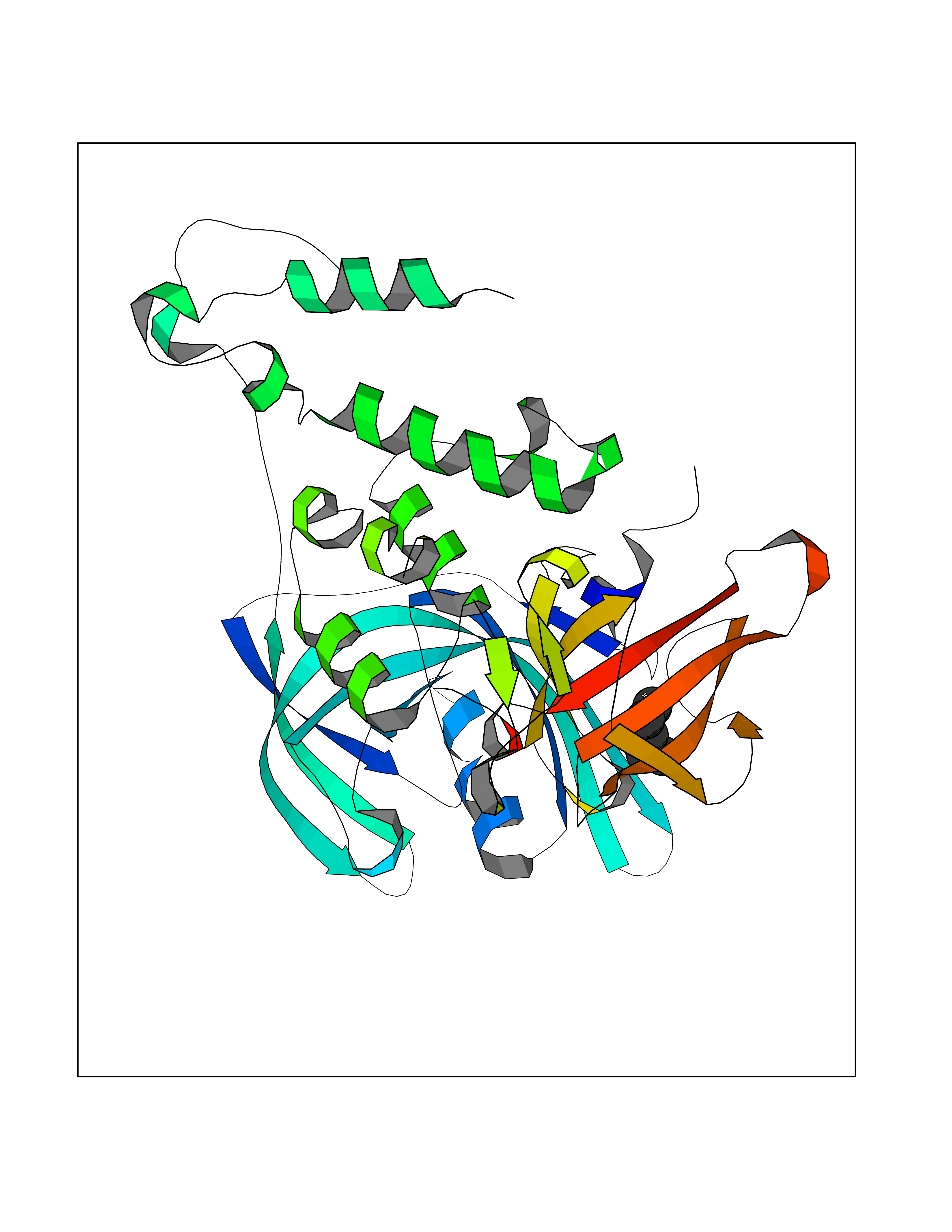 Molscript Map  Pymol Map 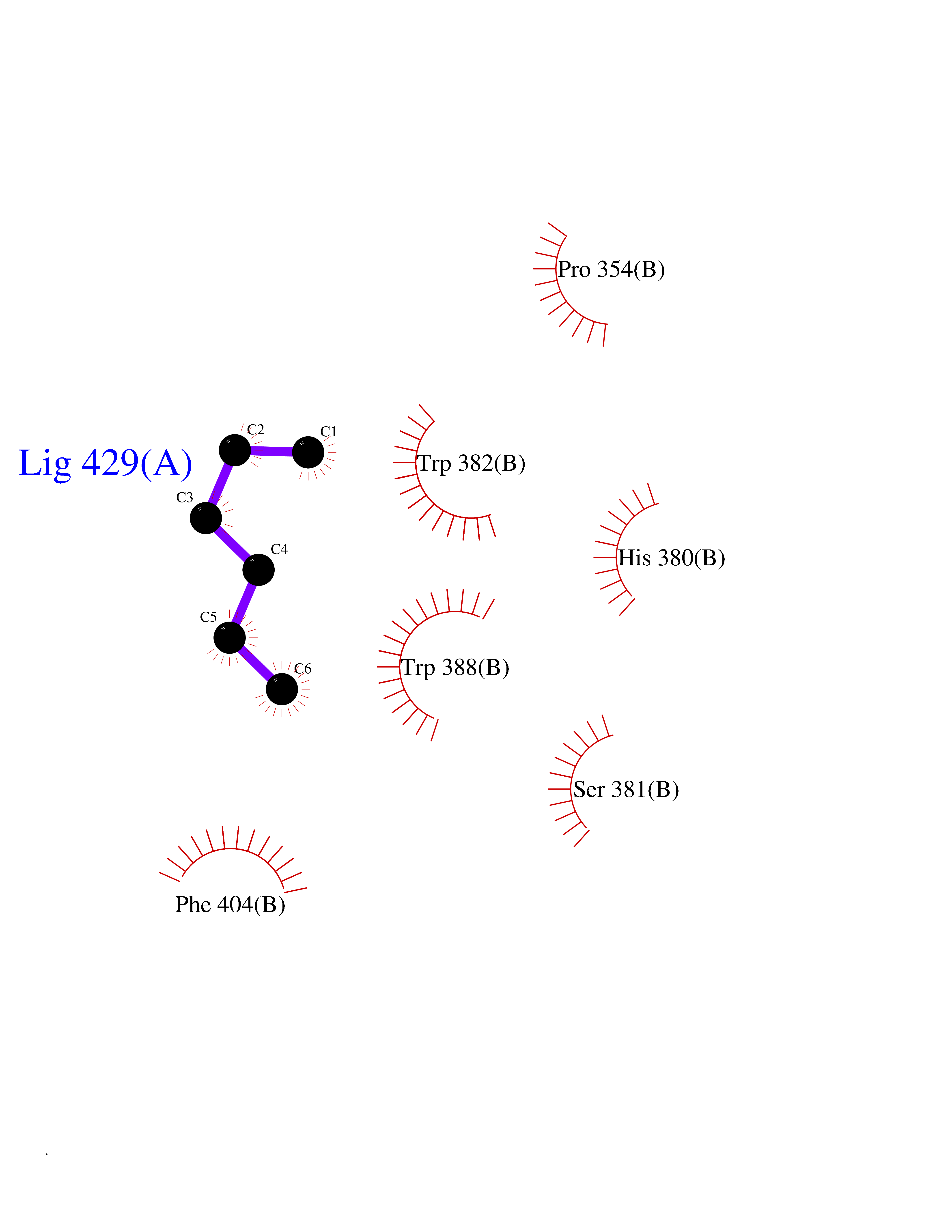 Ligplot Map 3D Binding mode Sequence MINFDTSLPTSHMYLGSDMEEFHGRTLHDDDSCQVIPVLPHVMVMLIPGQTLPLQLFHPQEVSMVRNLIQKDRTFAVLAYSNVREREAHFGTTAEIYAYREEQEYGIETVKVKAIGRQRFKVLEIRTQSDGIQQAKVQILPERVLPSTMSAVQLQSLSRRHIRAFRQWWQKYQKRKFHCASLTSWPPWLYSLYDAETLMERVKRQLHEWDENLKDESLPTNPIDFSYRVAACLPIDDALRIQLLKIGSAIQRLRELDIMNKTSLCCKQCQDTEITTKNEIFSLSLCGPMAAYVNPHGYIHETLTVYKACNLNLSGRPSTEHSWFPGYAWTIAQCRICGNHMGWKFTATKKDMSPQKFWGLTRSALLPR Hydrogen bonds contact Hydrophobic contact | ||||
| 9 | Vitamin D3 receptor (VDR) | 3B0T | 5.06 | |
Target general information Gen name VDR Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Vitamin D(3) receptor; Nuclear vitamin D receptor; Nuclear receptor subfamily 1 group I member 1; NR1I1; 1,25-dihydroxyvitamin D3 receptor Protein family Nuclear hormone receptor family, NR1 subfamily Biochemical class Nuclear hormone receptor Function Enters the nucleus upon vitamin D3 binding where it forms heterodimers with the retinoid X receptor/RXR. The VDR-RXR heterodimers bind to specific response elements on DNA and activate the transcription of vitamin D3-responsive target genes. Plays a central role in calcium homeostasis. Nuclear receptor for calcitriol, the active form of vitamin D3 which mediates the action of this vitamin on cells. Related diseases Rickets vitamin D-dependent 2A (VDDR2A) [MIM:277440]: A disorder of vitamin D metabolism resulting in severe rickets, hypocalcemia and secondary hyperparathyroidism. Most patients have total alopecia in addition to rickets. {ECO:0000269|PubMed:1652893, ECO:0000269|PubMed:17970811, ECO:0000269|PubMed:2177843, ECO:0000269|PubMed:2849209, ECO:0000269|PubMed:28698609, ECO:0000269|PubMed:7828346, ECO:0000269|PubMed:8106618, ECO:0000269|PubMed:8381803, ECO:0000269|PubMed:8392085, ECO:0000269|PubMed:8675579, ECO:0000269|PubMed:8961271, ECO:0000269|PubMed:9005998}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07530; DB08742; DB01436; DB04891; DB00146; DB02300; DB00136; DB00169; DB04540; DB05024; DB11672; DB14635; DB01070; DB06410; DB05295; DB06194; DB00153; DB04796; DB03451; DB00910; DB04258; DB11094 Interacts with P35222; Q09472; Q15648; P50222; Q15788; P26045; P19793; Q13573; Q13501; P04637; Q15645; Q9JLI4; P28700; X5D778; Q96HA8; Q01804; Q96S38; P48443 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; Disease variant; DNA-binding; Metal-binding; Nucleus; Proteomics identification; Receptor; Reference proteome; Transcription; Transcription regulation; Ubl conjugation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A Molecular weight (Da) 28781 Length 254 Aromaticity 0.07 Instability index 47.69 Isoelectric point 6.15 Charge (pH=7) -3.44 2D Binding mode Binding energy (Kcal/mol) -6.9  Molscript Map 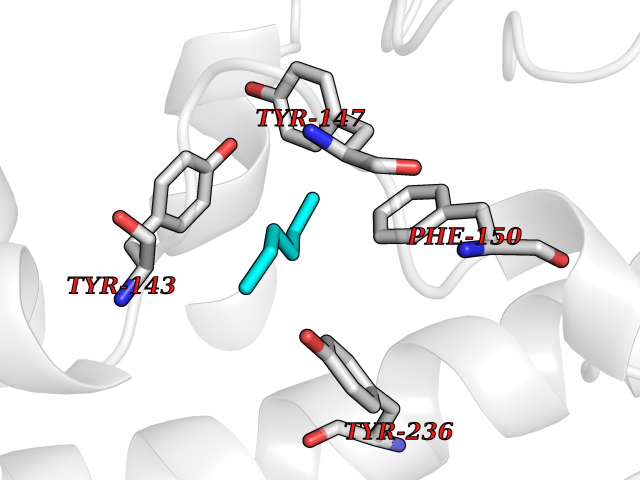 Pymol Map 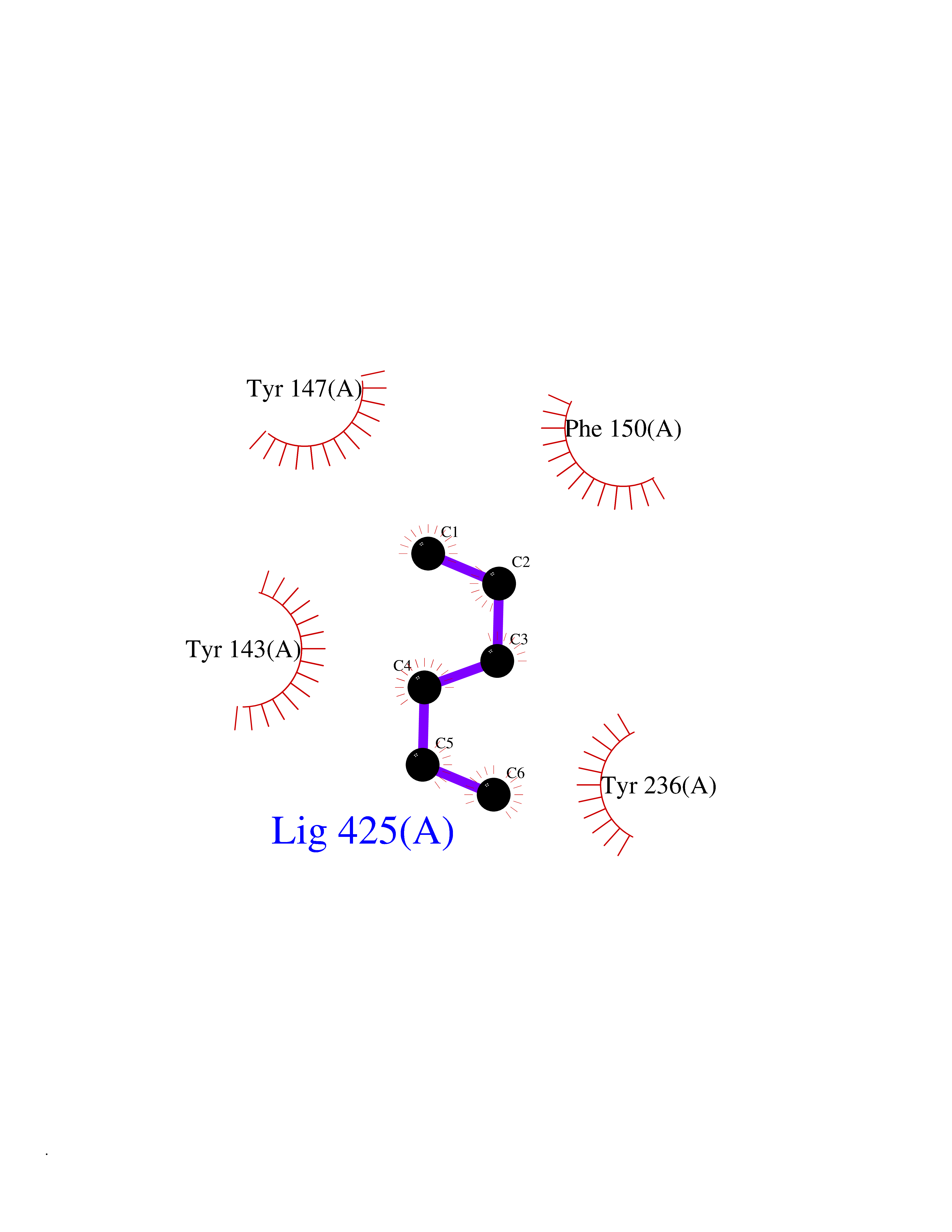 Ligplot Map 3D Binding mode Sequence ALRPKLSEEQQRIIAILLDAHHKTYDPTYSDFCQFRPPVRVNDGGGSVTLELSQLSMLPHLADLVSYSIQKVIGFAKMIPGFRDLTSEDQIVLLKSSAIEVIMLRSNESFTMDDMSWTCGNQDYKYRVSDVTKAGHSLELIEPLIKFQVGLKKLNLHEEEHVLLMAICIVSPDRPGVQDAALIEAIQDRLSNTLQTYIRCRHPPPGSHLLYAKMIQKLADLRSLNEEHSKQYRCLSFQPECSMKLTPLVLEVFG Hydrogen bonds contact Hydrophobic contact | ||||
| 10 | Plasmepsin-2 | 2BJU | 5.06 | |
Target general information Gen name N/A Organism Plasmodium falciparum (isolate HB3) Uniprot ID TTD ID NA Synonyms NA Protein family Peptidase A1 family Biochemical class Hydrolase Function Aspartic-type endopeptidase activity. Related diseases Short/branched-chain acyl-CoA dehydrogenase deficiency (SBCADD) [MIM:610006]: Autosomal recessive disorder and consists of a defect in catabolism of L-isoleucine which is characterized by an increase of 2-methylbutyrylglycine and 2-methylbutyrylcarnitine in blood and urine. Affected individuals have seizures and psychomotor delay as the main clinical features. {ECO:0000269|PubMed:10832746, ECO:0000269|PubMed:11013134, ECO:0000269|PubMed:16317551}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB04378; DB04373; DB11638; DB01218; DB02505; DB03063 Interacts with NA EC number 3.4.23.39 Uniprot keywords 3D-structure; Aspartyl protease; Direct protein sequencing; Disulfide bond; Hydrolase; Membrane; Protease; Reference proteome; Signal-anchor; Transmembrane; Transmembrane helix; Vacuole; Zymogen Protein physicochemical properties Chain ID A Molecular weight (Da) 36923.5 Length 329 Aromaticity 0.13 Instability index 44.31 Isoelectric point 4.67 Charge (pH=7) -17.94 2D Binding mode Binding energy (Kcal/mol) -6.9 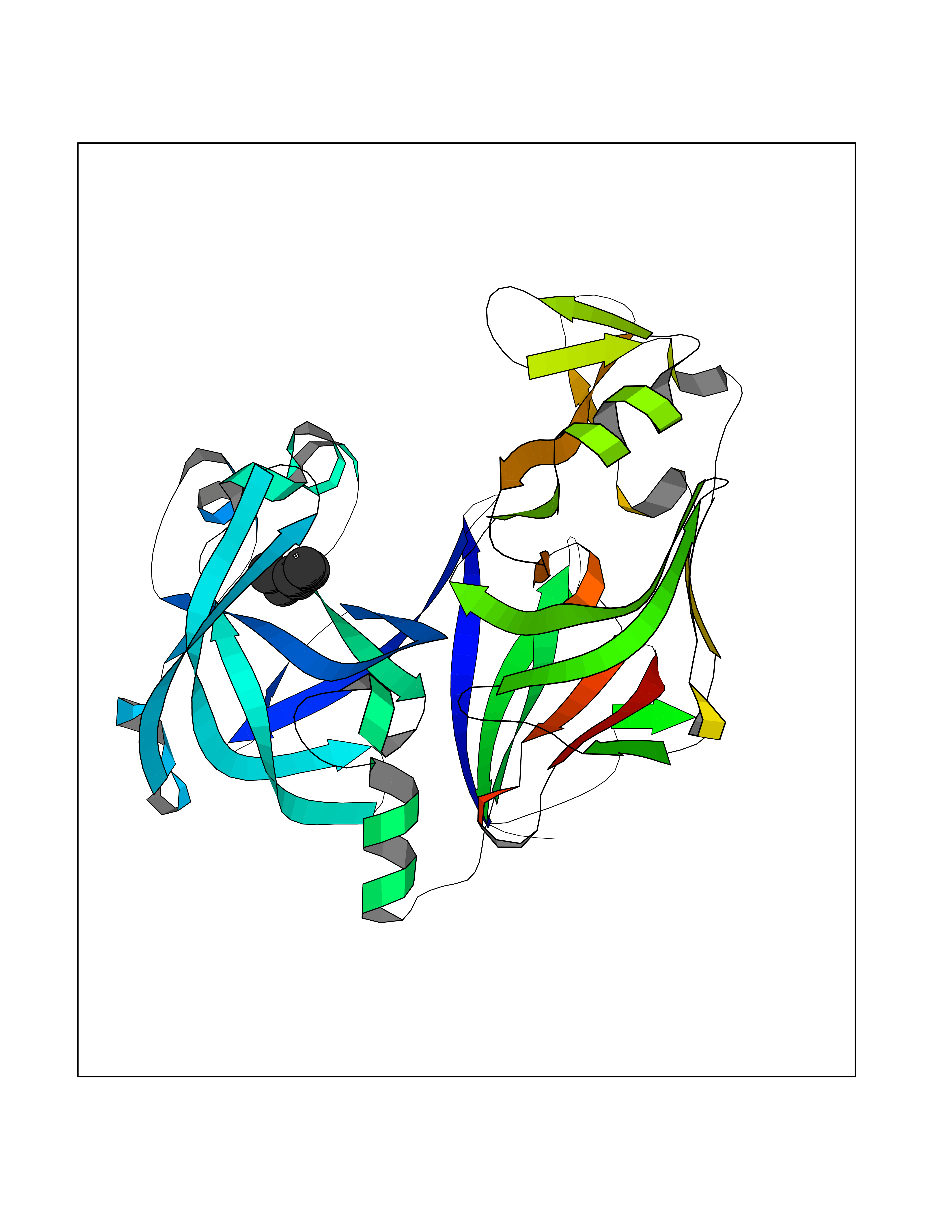 Molscript Map 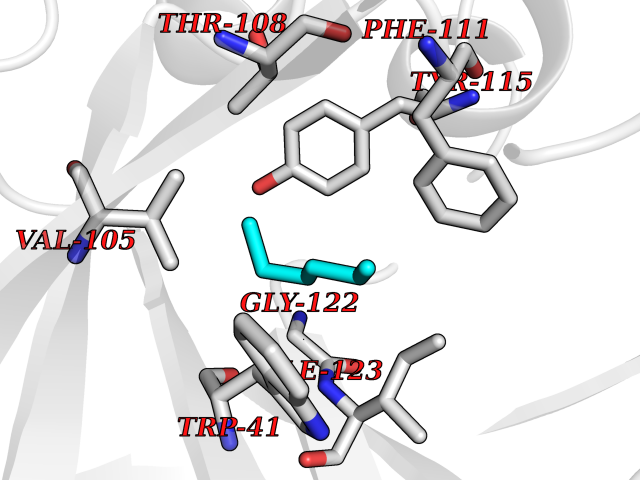 Pymol Map 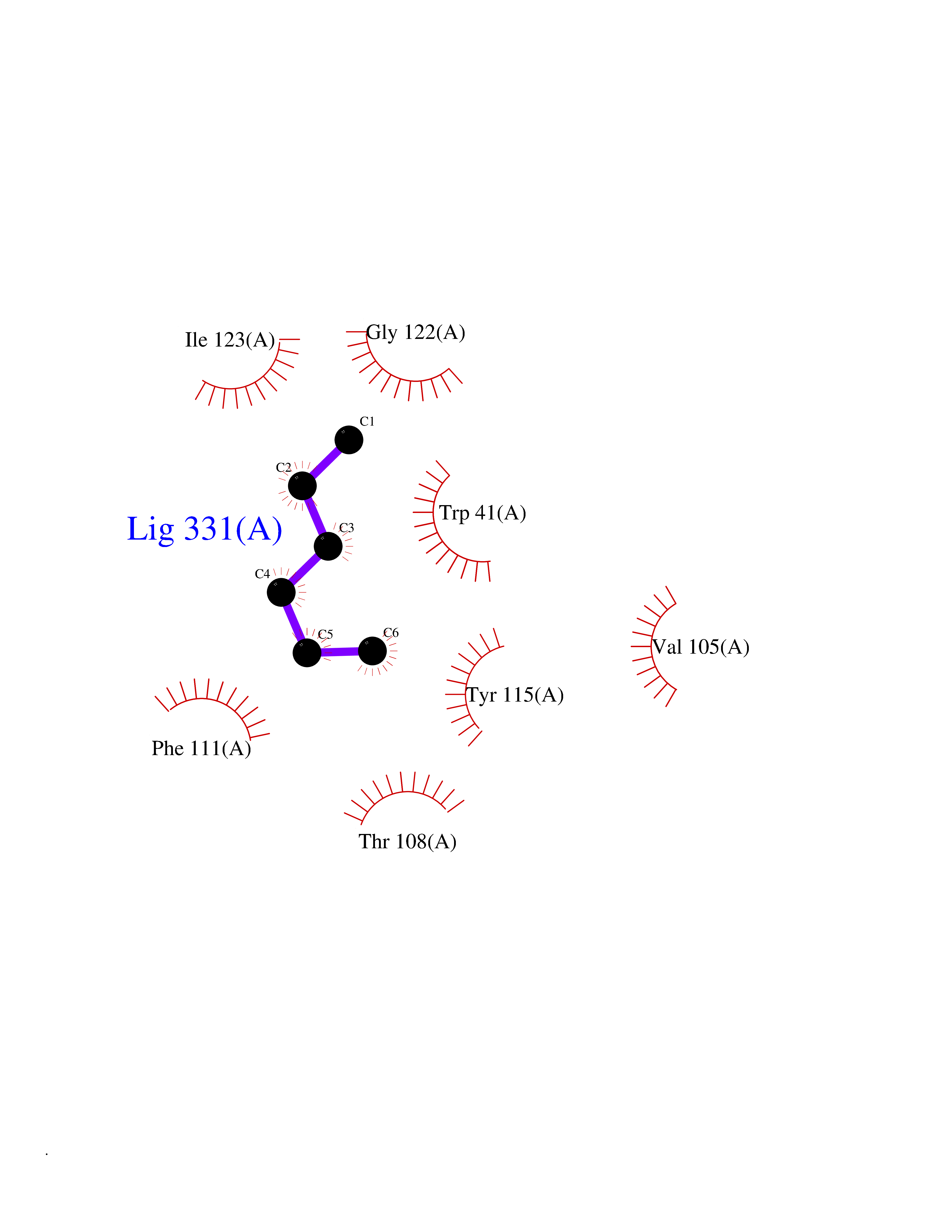 Ligplot Map 3D Binding mode Sequence SSNDNIELVDFQNIMFYGDAEVGDNQQPFTFILDTGSANLWVPSVKCTTAGCLTKHLYDSSKSRTYEKDGTKVEMNYVSGTVSGFFSKDLVTVGNLSLPYKFIEVIDTNGFEPTYTASTFDGILGLGWKDLSIGSVDPIVVELKNQNKIENALFTFYLPVHDKHTGFLTIGGIEERFYEGPLTYEKLNHDLYWQITLDAHVGNIMLEKANCIVDSGTSAITVPTDFLNKMLQNLDVIKVPFLPFYVTLCNNSKLPTFEFTSENGKYTLEPEYYLQHIEDVGPGLCMLNIIGLDFPVPTFILGDPFMRKYFTVFDYDNHSVGIALAKKNL Hydrogen bonds contact Hydrophobic contact | ||||
| 11 | "Periplasmic trehalase (EC 3.2.1.28) (Alpha,alpha-trehalase) (Alpha,alpha-trehalose glucohydrolase) (Tre37A)" | 2JG0 | 5.05 | |
Target general information Gen name treA Organism Escherichia coli (strain K12) Uniprot ID TTD ID NA Synonyms JW1186;osmA;b1197 Protein family Glycosyl hydrolase 37 family Biochemical class NA Function Provides the cells with the ability to utilize trehalose at high osmolarity by splitting it into glucose molecules that can subsequently be taken up by the phosphotransferase-mediated uptake system. Related diseases SRC kinase activity has been shown to be increased in several tumor tissues and tumor cell lines such as colon carcinoma cells. {ECO:0000269|PubMed:2498394, ECO:0000269|PubMed:3093483}.; DISEASE: Thrombocytopenia 6 (THC6) [MIM:616937]: A form of thrombocytopenia, a hematologic disorder defined by a decrease in the number of platelets in circulating blood, resulting in the potential for increased bleeding and decreased ability for clotting. THC6 is an autosomal dominant form. Affected individuals may also have bone abnormalities and an increased risk for myelofibrosis. {ECO:0000269|PubMed:26936507}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number 3.2.1.28 Uniprot keywords 3D-structure; Direct protein sequencing; Glycosidase; Hydrolase; Periplasm; Reference proteome; Signal Protein physicochemical properties Chain ID A Molecular weight (Da) 57508.9 Length 507 Aromaticity 0.11 Instability index 48.32 Isoelectric point 5.48 Charge (pH=7) -10.13 2D Binding mode Binding energy (Kcal/mol) -6.88 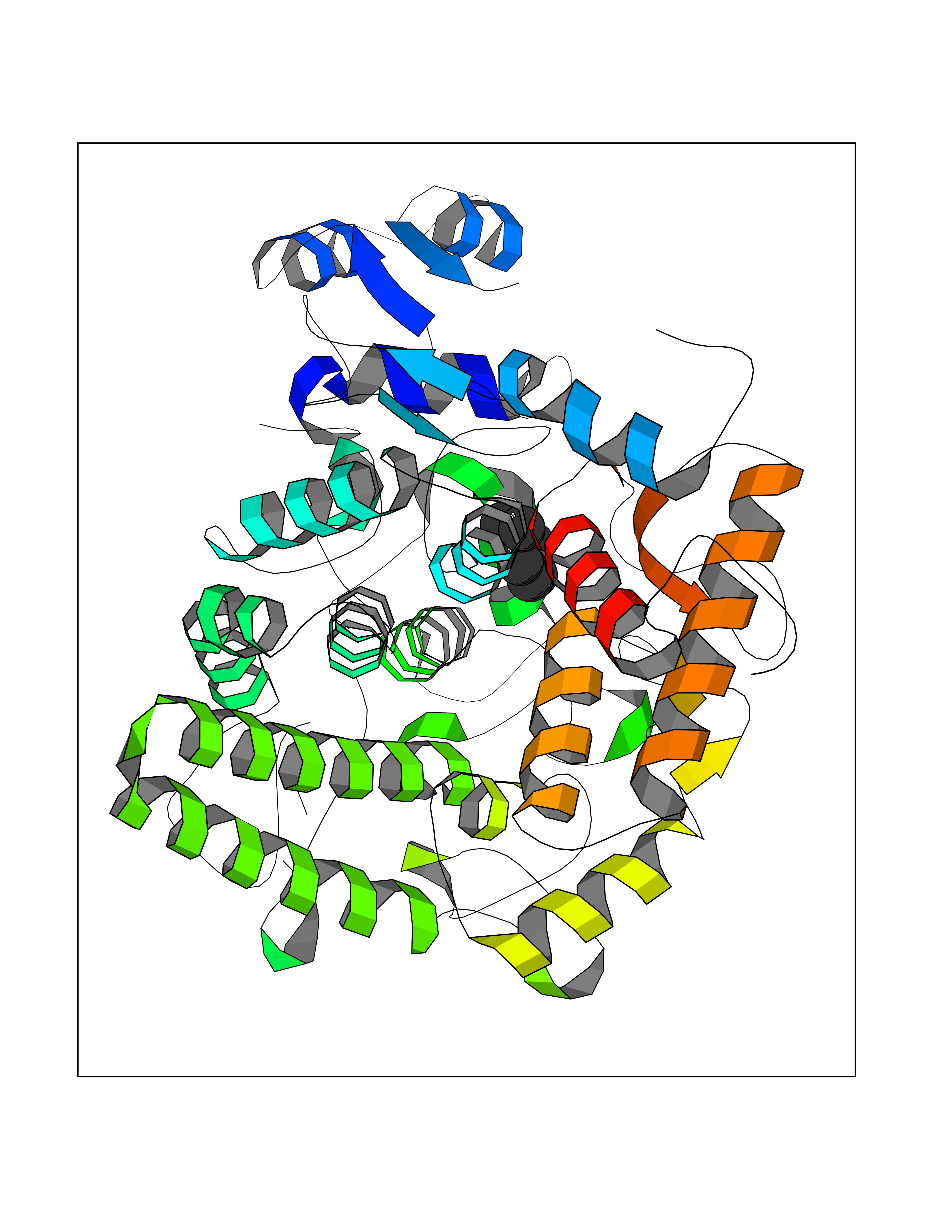 Molscript Map 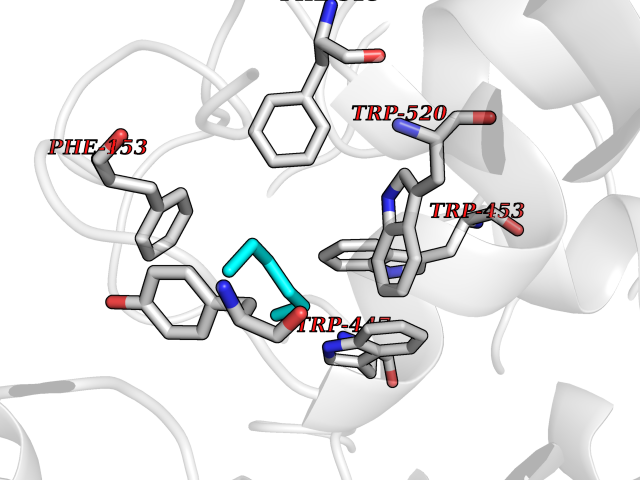 Pymol Map 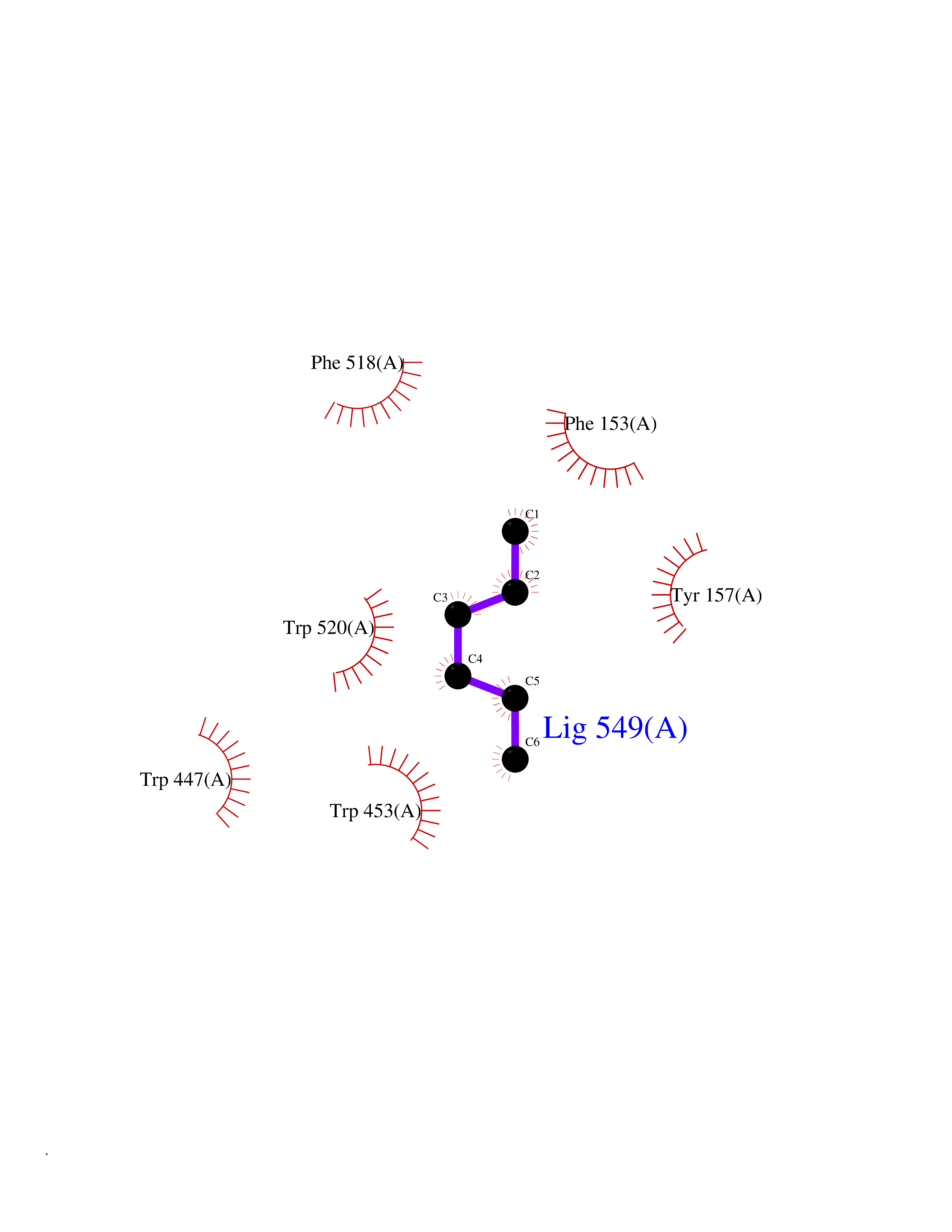 Ligplot Map 3D Binding mode Sequence PQPPDILLGPLFNDVQNAKLFPDQKTFADAVPNSDPLMILADYRMQQNQSGFDLRHFVNVNFTLPKYVPPEGQSLREHIDGLWPVLTRSTENTEKWDSLLPLPEPYVVPGGRFREVYYWDSYFTMLGLAESGHWDKVADMVANFAHEIDTYGHIPNGNRSYYLSRSQPPFFALMVELLAQHEGDAALKQYLPQMQKEYAYWMDGVENLQAGQQEKRVVKLQDGTLLNRYWDDRDTPRPESWVEDIATAKSNPNRPATEIYRDLRSAAASGWDFSSRWMDNPQQLNTLRTTSIVPVDLNSLMFKMEKILARASKAAGDNAMANQYETLANARQKGIEKYLWNDQQGWYADYDLKSHKVRNQLTAAALFPLYVNAAAKDRANKMATATKTHLLQPGGLNTTSVKSGQQWDAPNGWAPLQWVATEGLQNYGQKEVAMDISWHFLTNVQHTYDREKKLVEKYDVSTTGTGGGGGEYPLQDGFGWTNGVTLKMLDLICPKEQPCDNVPATRP Hydrogen bonds contact Hydrophobic contact | ||||
| 12 | Deoxyribodipyrimidine photo-lyase | 1OWL | 5.05 | |
Target general information Gen name phr Organism Synechococcus sp. (strain ATCC 27144 / PCC 6301 / SAUG 1402/1) (Anacystis nidulans) Uniprot ID TTD ID NA Synonyms phrA;syc1392_c Protein family DNA photolyase class-1 family Biochemical class Lyase Function Deoxyribodipyrimidine photo-lyase activity.DNA binding.Nucleotide binding. Related diseases Myeloperoxidase deficiency (MPOD) [MIM:254600]: A disorder characterized by decreased myeloperoxidase activity in neutrophils and monocytes that results in disseminated candidiasis. {ECO:0000269|PubMed:37198333, ECO:0000269|PubMed:7904599, ECO:0000269|PubMed:8142659, ECO:0000269|PubMed:8621627, ECO:0000269|PubMed:9354683, ECO:0000269|PubMed:9637725}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03147 Interacts with NA EC number 4.1.99.3 Uniprot keywords 3D-structure; Chromophore; Direct protein sequencing; DNA damage; DNA repair; DNA-binding; FAD; Flavoprotein; Lyase; Nucleotide-binding Protein physicochemical properties Chain ID A Molecular weight (Da) 53346.9 Length 473 Aromaticity 0.1 Instability index 53.22 Isoelectric point 6.92 Charge (pH=7) -0.23 2D Binding mode Binding energy (Kcal/mol) -6.88 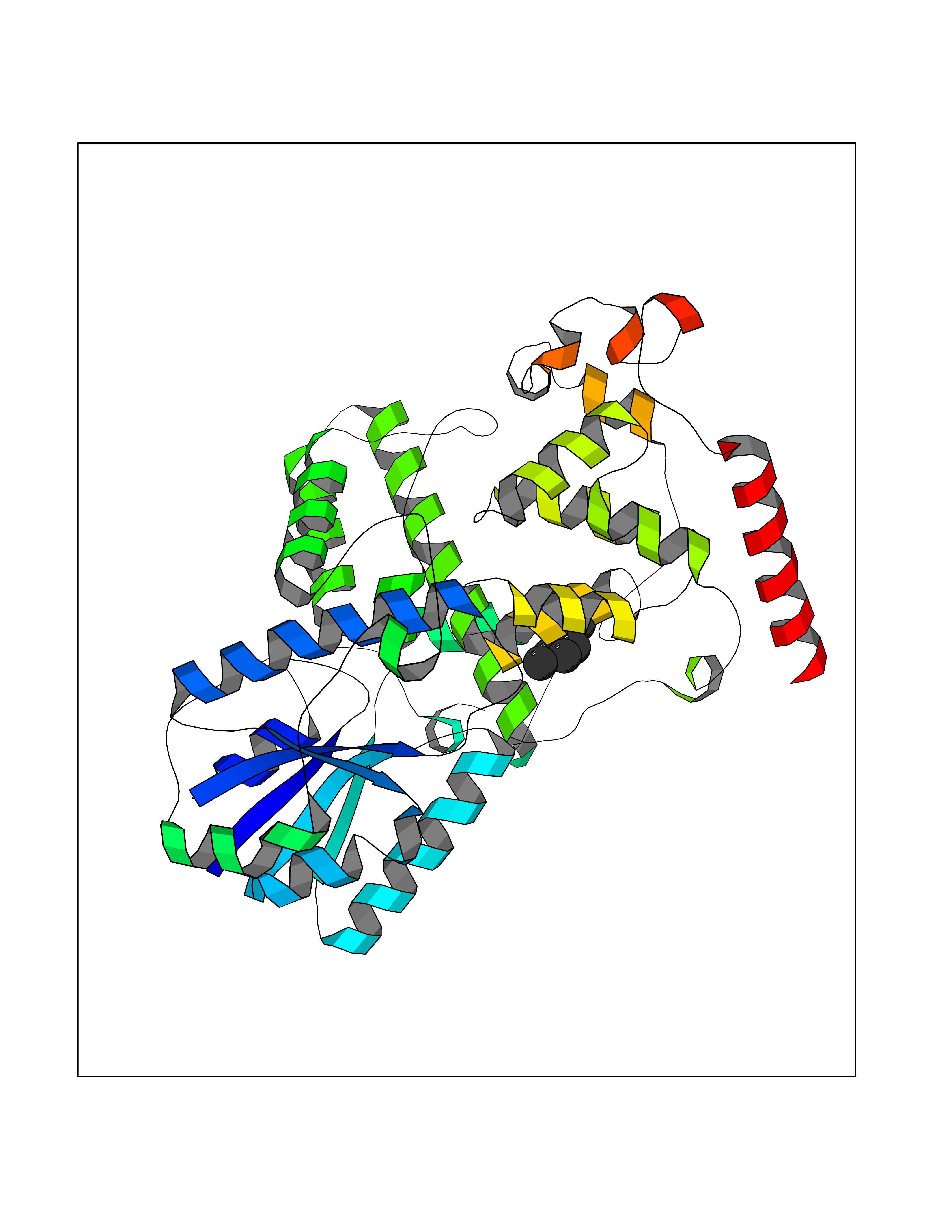 Molscript Map 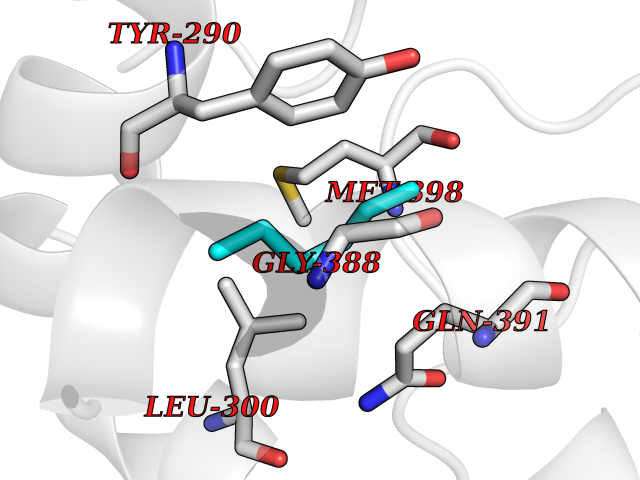 Pymol Map 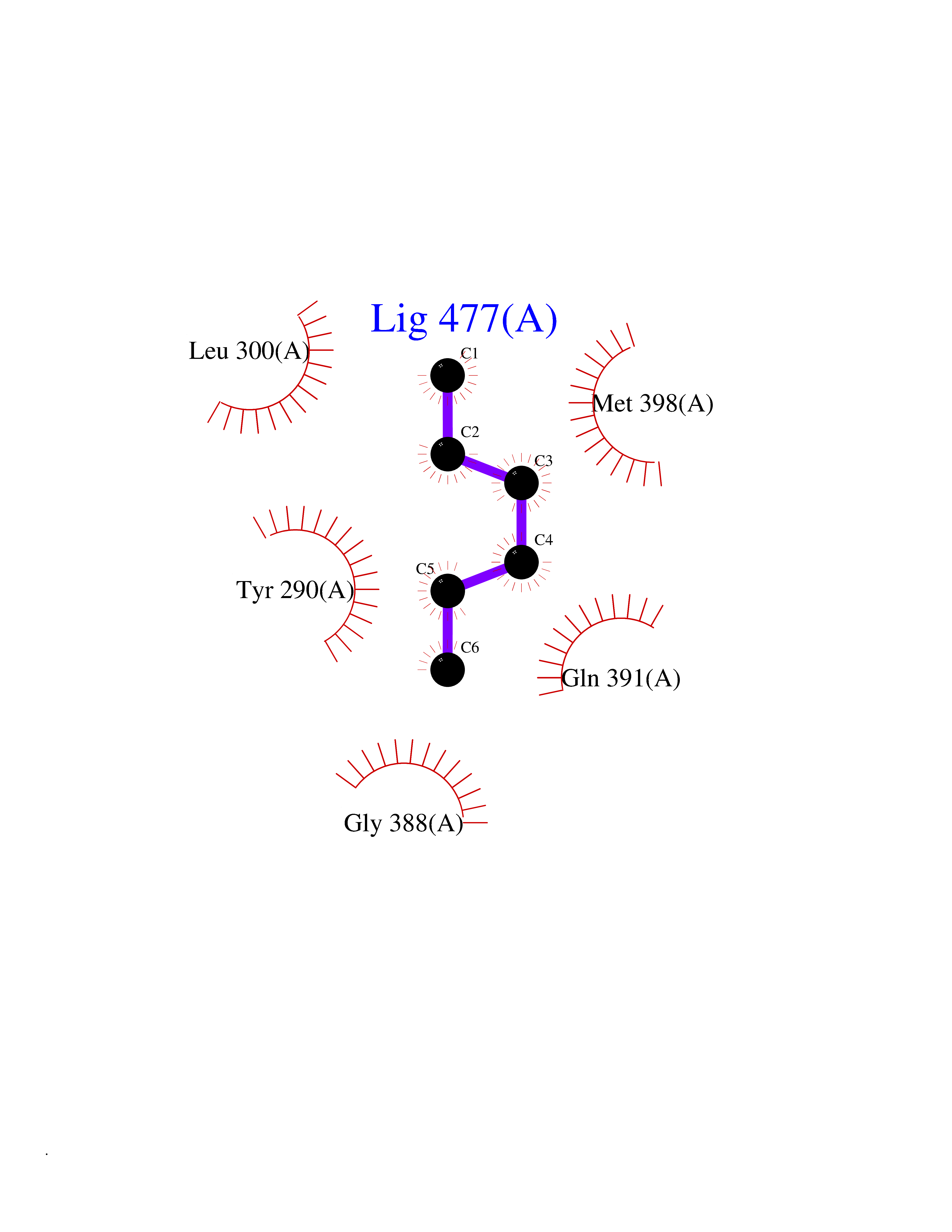 Ligplot Map 3D Binding mode Sequence APILFWHRRDLRLSDNIGLAAARAQSAQLIGLFCLDPQILQSADMAPARVAYLQGCLQELQQRYQQAGSRLLLLQGDPQHLIPQLAQQLQAEAVYWNQDIEPYGRDRDGQVAAALKTAGIRAVQLWDQLLHSPDQILSGSGNPYSVYGPFWKNWQAQPKPTPVATPTELVDLSPEQLTAIAPLLLSELPTLKQLGFDWDGGFPVEPGETAAIARLQEFCDRAIADYDPQRNFPAEAGTSGLSPALKFGAIGIRQAWQAASAAHALSRSDEARNSIRVWQQELAWREFYQHALYHFPSLADGPYRSLWQQFPWENREALFTAWTQAQTGYPIVDAAMRQLTETGWMHNRCRMIVASFLTKDLIIDWRRGEQFFMQHLVDGDLAANNGGWQWSASSGMDPKPLRIFNPASQAKKFDATATYIKRWLPELRHVHPKDLISGEITPIERRGYPAPIVNHNLRQKQFKALYNQLKAAI Hydrogen bonds contact Hydrophobic contact | ||||
| 13 | Folate receptor beta (FOLR2) | 4KN0 | 5.05 | |
Target general information Gen name FOLR2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Placental folate-binding protein; Folate receptor, fetal/placental; Folate receptor type-beta; Folate receptor 2; FR-beta; FOLR2 Protein family Folate receptor family Biochemical class Folate receptor Function Binds to folate and reduced folic acid derivatives and mediates delivery of 5-methyltetrahydrofolate and folate analogs into the interior of cells. Has high affinity for folate and folic acid analogs at neutral pH. Exposure to slightly acidic pH after receptor endocytosis triggers a conformation change that strongly reduces its affinity for folates and mediates their release. Related diseases Acute hepatic porphyria (AHEPP) [MIM:612740]: A form of porphyria. Porphyrias are inherited defects in the biosynthesis of heme, resulting in the accumulation and increased excretion of porphyrins or porphyrin precursors. They are classified as erythropoietic or hepatic, depending on whether the enzyme deficiency occurs in red blood cells or in the liver. AHP is characterized by attacks of gastrointestinal disturbances, abdominal colic, paralyses and peripheral neuropathy. Most attacks are precipitated by drugs, alcohol, caloric deprivation, infections, or endocrine factors. {ECO:0000269|PubMed:10706561, ECO:0000269|PubMed:1309003, ECO:0000269|PubMed:1569184, ECO:0000269|PubMed:17236137, ECO:0000269|PubMed:2063868}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00158; DB00563; DB05168 Interacts with NA EC number NA Uniprot keywords 3D-structure; Cell membrane; Direct protein sequencing; Disulfide bond; Folate-binding; Glycoprotein; GPI-anchor; Lipoprotein; Membrane; Proteomics identification; Receptor; Reference proteome; Secreted; Signal; Transport Protein physicochemical properties Chain ID A Molecular weight (Da) 23841.6 Length 205 Aromaticity 0.12 Instability index 56.78 Isoelectric point 7.92 Charge (pH=7) 2.58 2D Binding mode Binding energy (Kcal/mol) -6.88 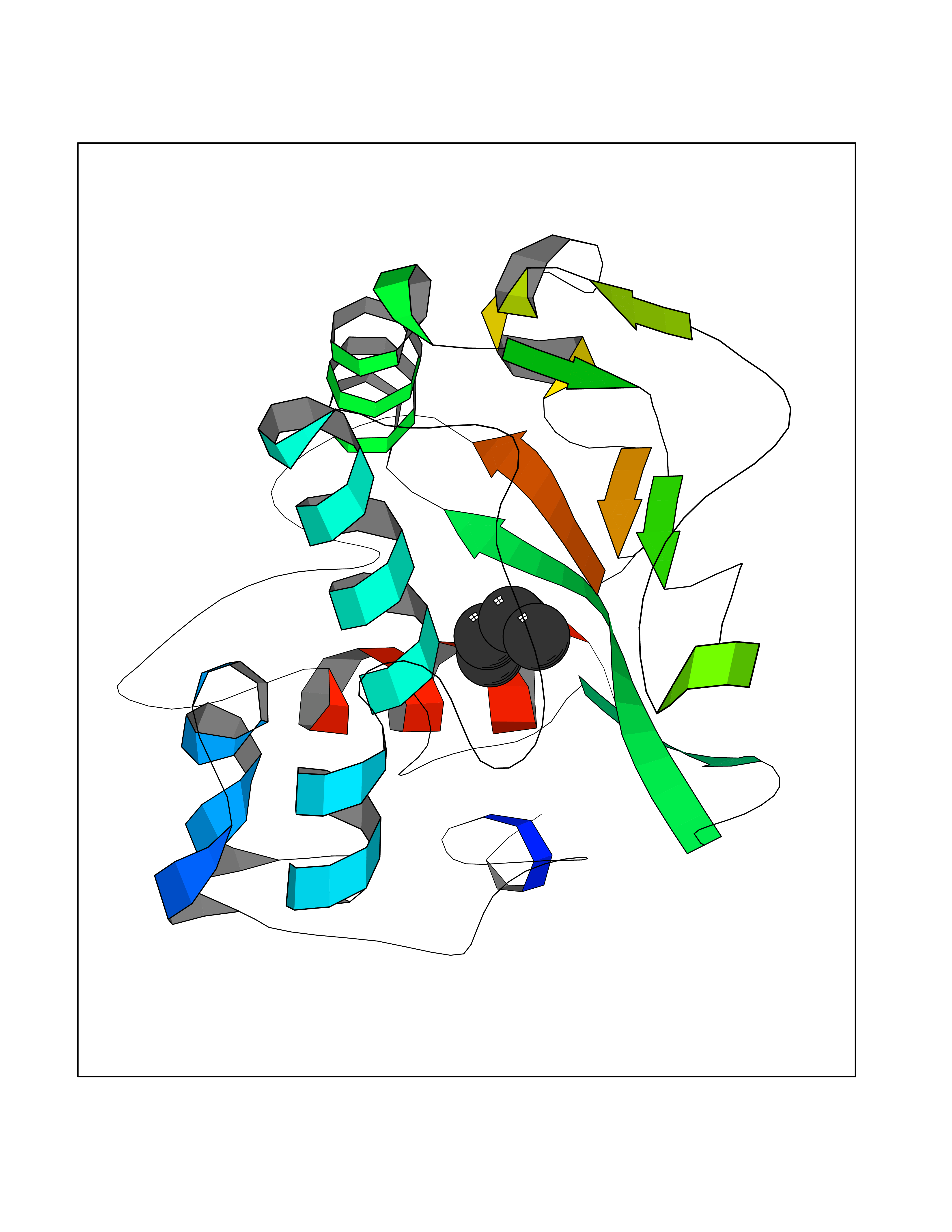 Molscript Map 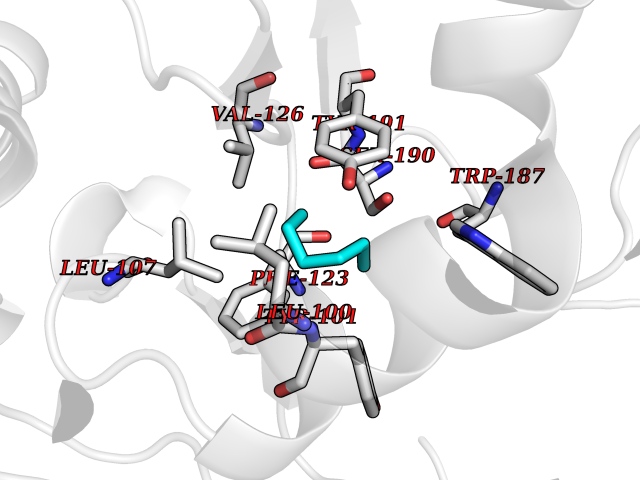 Pymol Map 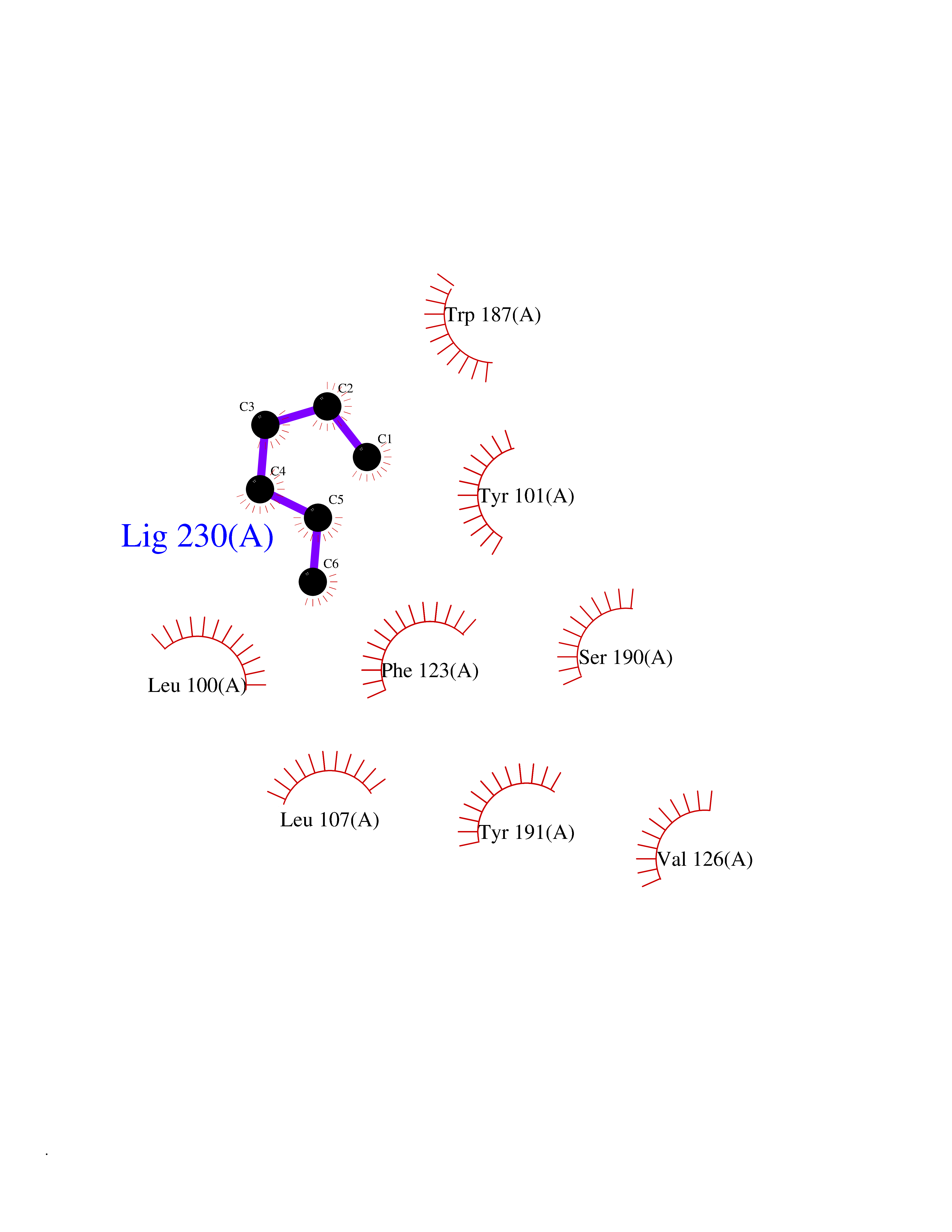 Ligplot Map 3D Binding mode Sequence RTDLLNVCMDAKHHKTKPGPEDKLHDQCSPWKKNACCTASTSQELHKDTSRLYNFNWDHCGKMEPACKRHFIQDTCLYECSPNLGPWIQQVNQSWRKERFLDVPLCKEDCQRWWEDCHTSHTCKSNWHRGWDWTSGVNKCPAGALCRTFESYFPTPAALCEGLWSHSYKVSNYSRGSGRCIQMWFDSAQGNPNEEVARFYAAAMH Hydrogen bonds contact Hydrophobic contact | ||||
| 14 | Glucose-dependent insulinotropic receptor (GPR119) | 7XZ6 | 5.05 | |
Target general information Gen name GPR119 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms GPR119; G-protein coupled receptor 119 Protein family G-protein coupled receptor 1 family Biochemical class GPCR rhodopsin Function Receptor for the endogenous fatty-acid ethanolamide oleoylethanolamide (OEA) and lysophosphatidylcholine (LPC). Functions as a glucose-dependent insulinotropic receptor. The activity of this receptor is mediated by G proteins which activate adenylate cyclase. Seems to act through a G(s) mediated pathway. Related diseases Developmental and epileptic encephalopathy 24 (DEE24) [MIM:615871]: A disease characterized by early-onset seizures, intellectual disability of varying degrees, and behavioral disturbances or autistic features in most individuals. {ECO:0000269|PubMed:24747641, ECO:0000269|PubMed:27864847, ECO:0000269|PubMed:30351409}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Generalized epilepsy with febrile seizures plus 10 (GEFSP10) [MIM:618482]: An autosomal dominant neurologic disorder with incomplete penetrance, characterized by variable types of seizures including absence, tonic-clonic, febrile, focal, and eyelid myoclonia. Some patients have normal neurologic development. Others have mild-to-moderate intellectual disability or autism spectrum disorder. {ECO:0000269|PubMed:29936235, ECO:0000269|PubMed:30351409}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB05166 Interacts with Q12797-6 EC number NA Uniprot keywords 3D-structure; Cell membrane; G-protein coupled receptor; Lipid-binding; Membrane; Receptor; Reference proteome; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID R Molecular weight (Da) 32134.1 Length 292 Aromaticity 0.12 Instability index 34.96 Isoelectric point 9.12 Charge (pH=7) 8.03 2D Binding mode Binding energy (Kcal/mol) -6.88 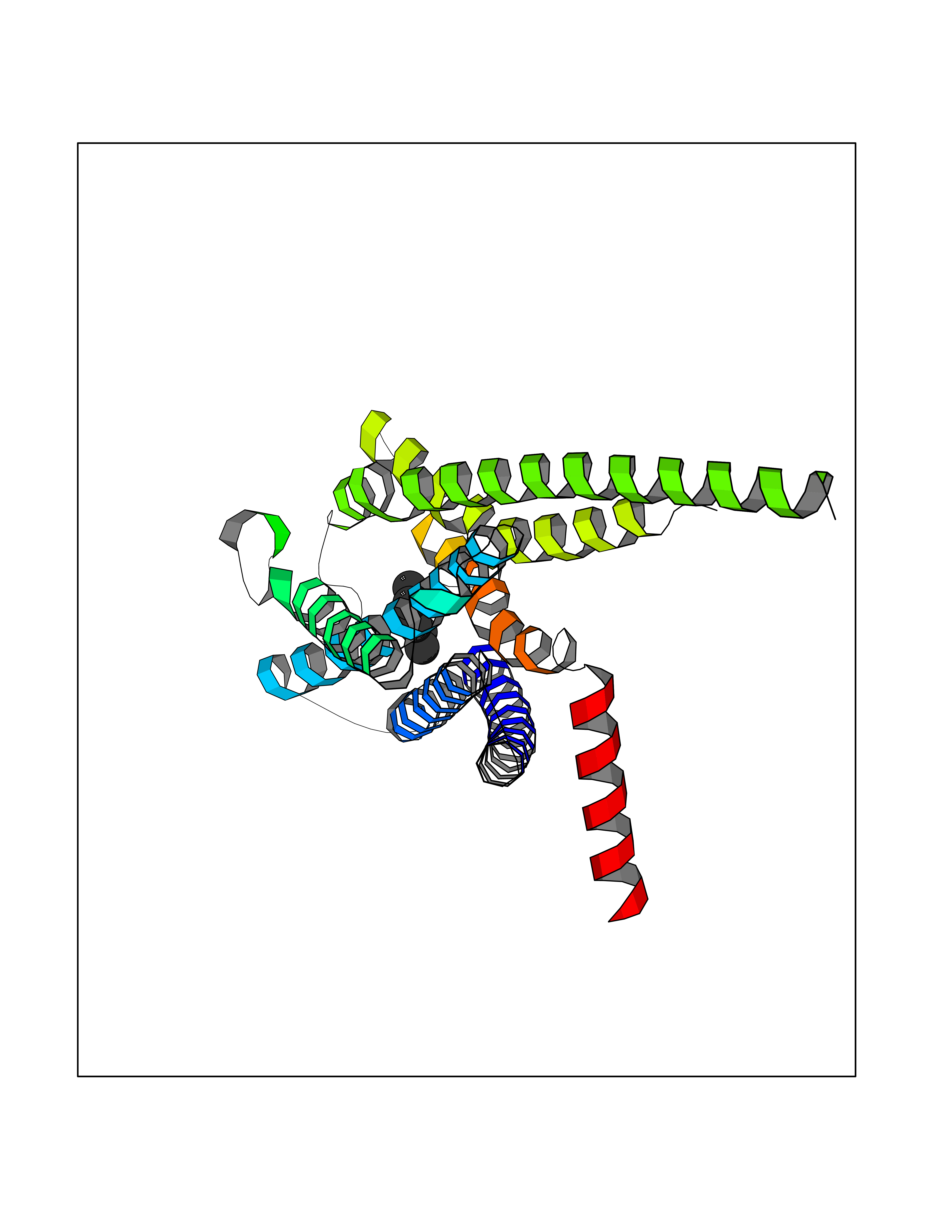 Molscript Map 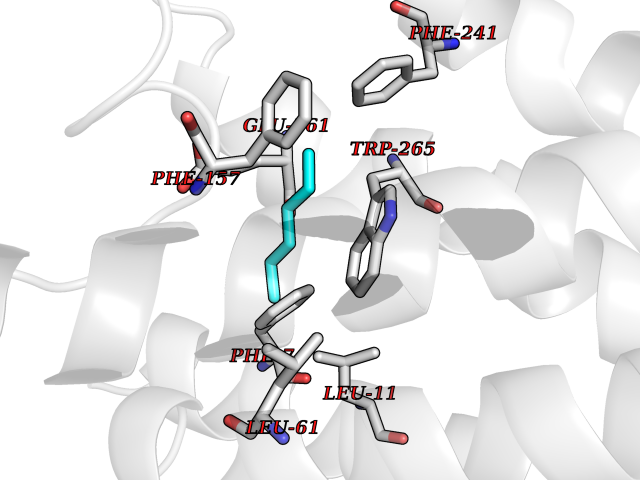 Pymol Map 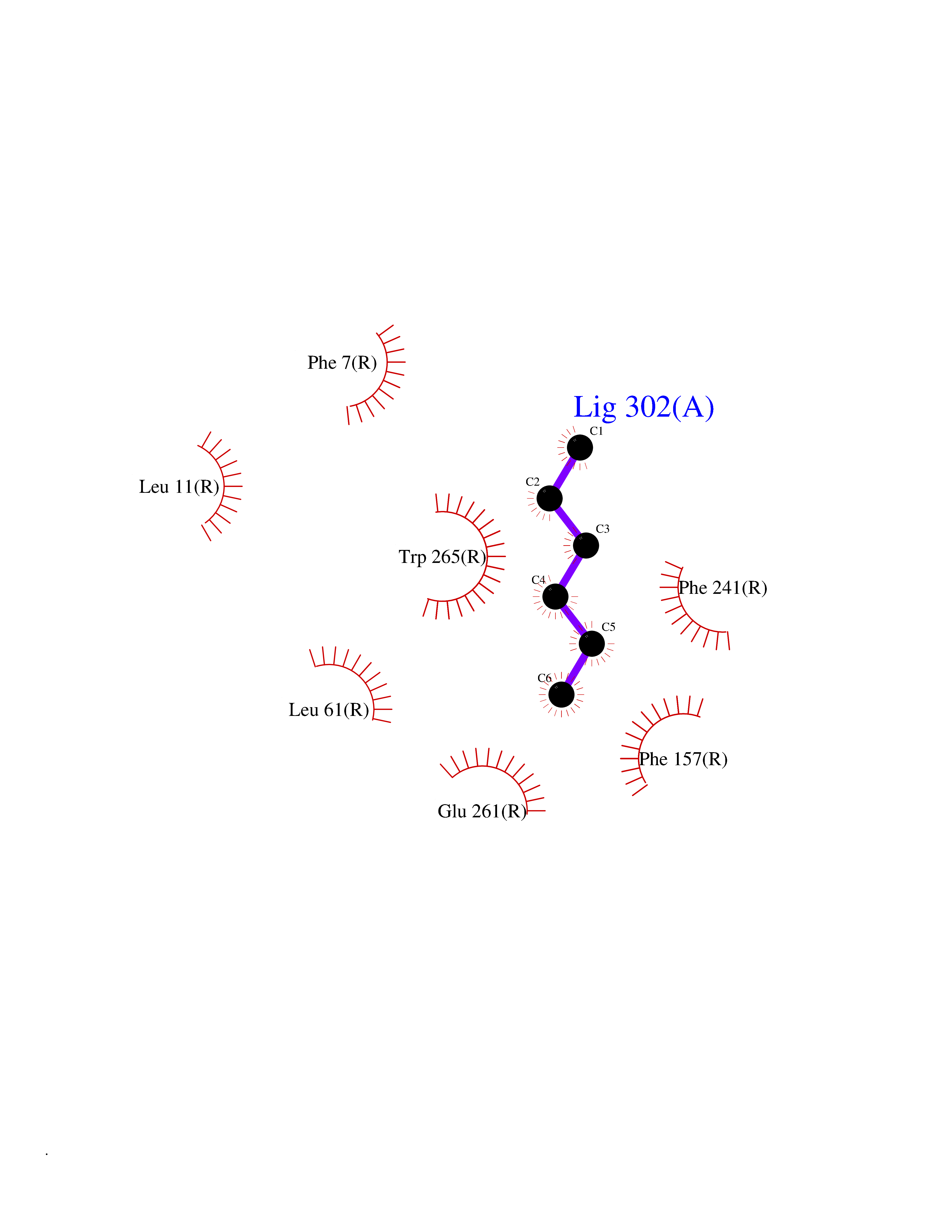 Ligplot Map 3D Binding mode Sequence MESSFSFGVILAVLASLIIATNTLVAVAVLLLIHKNDGVSLCFTLNLAVADTLIGVAISGLLTDQLSSPSRPTQKTLCSLRMAFVTSSAAASVLTVMLITFDRYLAIKQPFRYLKIMSGFVAGACIAGLWLVSYLIGFLPLGIPMFQQTAYKGQCSFFAVFHPHFVLTLSCVGFFPAMLLFVFFYCDMLKIASMHSQQIRKMEHAGAMAGSDFKALRTVSVLIGSFALSWTPFLITGIVQVACQECHLYLVLERYLWLLGVGNSLLNPLIYAYWQKEVRLQLYHMALGVKKV Hydrogen bonds contact Hydrophobic contact | ||||
| 15 | Cytochrome c oxidase subunit 2 | 3VRJ | 5.04 | |
Target general information Gen name MT-CO2 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms MTCO2;COXII;COII;COX2 Protein family Cytochrome c oxidase subunit 2 family Biochemical class Immune system Function Copper ion binding.Cytochrome-c oxidase activity. Related diseases Mitochondrial complex IV deficiency (MT-C4D) [MIM:220110]: A disorder of the mitochondrial respiratory chain with heterogeneous clinical manifestations, ranging from isolated myopathy to severe multisystem disease affecting several tissues and organs. Features include hypertrophic cardiomyopathy, hepatomegaly and liver dysfunction, hypotonia, muscle weakness, exercise intolerance, developmental delay, delayed motor development and intellectual disability. Some affected individuals manifest a fatal hypertrophic cardiomyopathy resulting in neonatal death. A subset of patients manifest Leigh syndrome. {ECO:0000269|PubMed:10486321}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02659; DB04464; DB05412 Interacts with Q9NZ94-2; P49281-3 EC number 7.1.1.9 Uniprot keywords 3D-structure; Copper; Disease variant; Electron transport; Magnesium; Membrane; Metal-binding; Mitochondrion; Mitochondrion inner membrane; Primary mitochondrial disease; Proteomics identification; Reference proteome; Respiratory chain; Translocase; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID C Molecular weight (Da) 21687.9 Length 189 Aromaticity 0.11 Instability index 38 Isoelectric point 5.68 Charge (pH=7) -3.26 2D Binding mode Binding energy (Kcal/mol) -6.88 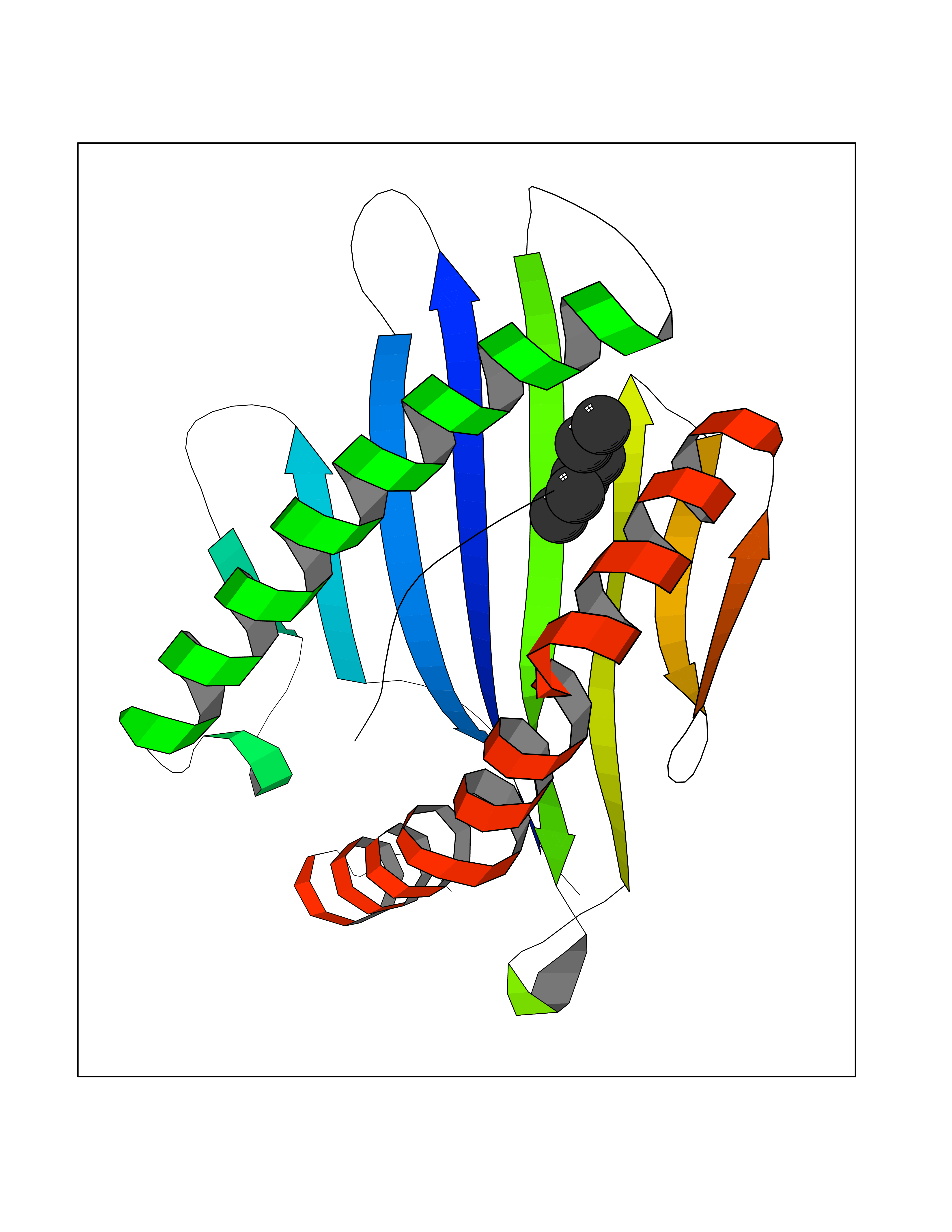 Molscript Map 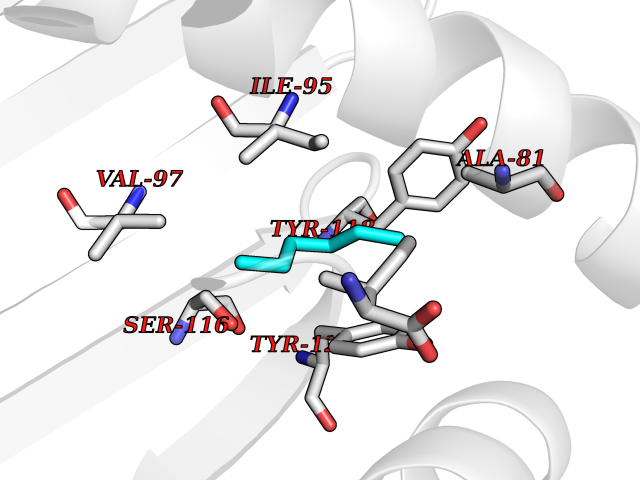 Pymol Map 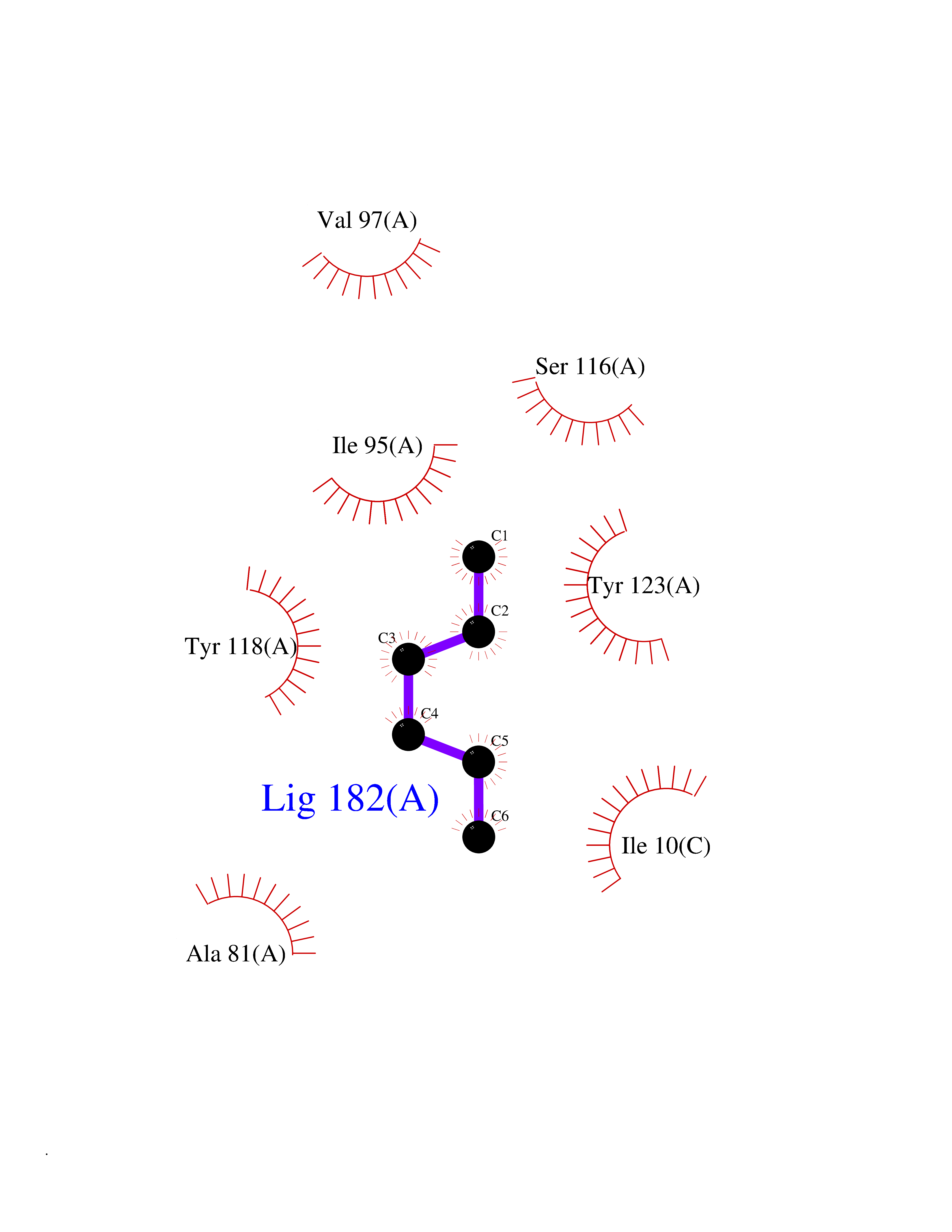 Ligplot Map 3D Binding mode Sequence SHSMRYFYTAMSRPGRGEPRFIAVGYVDDTQFVRFDSDAASPRMAPRAPWIEQEGPEYWDGETRNMKASAQTYRENLRIALRYYNQSEAGSHIIQVMYGCDVGPDGRLLRGHDQSAYDGKDYIALNEDLSSWTAADTAAQITQRKWEAARVAEQLRAYLEGLCVEWLRRYLENGKETLQLTTKLTNTNI Hydrogen bonds contact Hydrophobic contact | ||||
| 16 | Chromodomain-helicase-DNA-binding protein 1 | 4O42 | 5.04 | |
Target general information Gen name CHD1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family SNF2/RAD54 helicase family Biochemical class Dna binding protein / viral protein Function ATP binding.ATP-dependent DNA helicase activity.DNA binding.Methylated histone binding. Related diseases Pilarowski-Bjornsson syndrome (PILBOS) [MIM:617682]: An autosomal dominant disorder characterized by developmental delay, speech apraxia, intellectual disability, autism, and facial dysmorphic features. Some patients may have seizures. {ECO:0000269|PubMed:28866611}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with O60341-1; B2BUF1; P28799; O76024 EC number 3.6.4.12 Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Chromatin regulator; Cytoplasm; Disease variant; DNA-binding; Helicase; Hydrolase; Intellectual disability; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Transcription; Transcription regulation Protein physicochemical properties Chain ID A Molecular weight (Da) 20969.1 Length 180 Aromaticity 0.12 Instability index 46.35 Isoelectric point 5.88 Charge (pH=7) -2.83 2D Binding mode Binding energy (Kcal/mol) -6.87 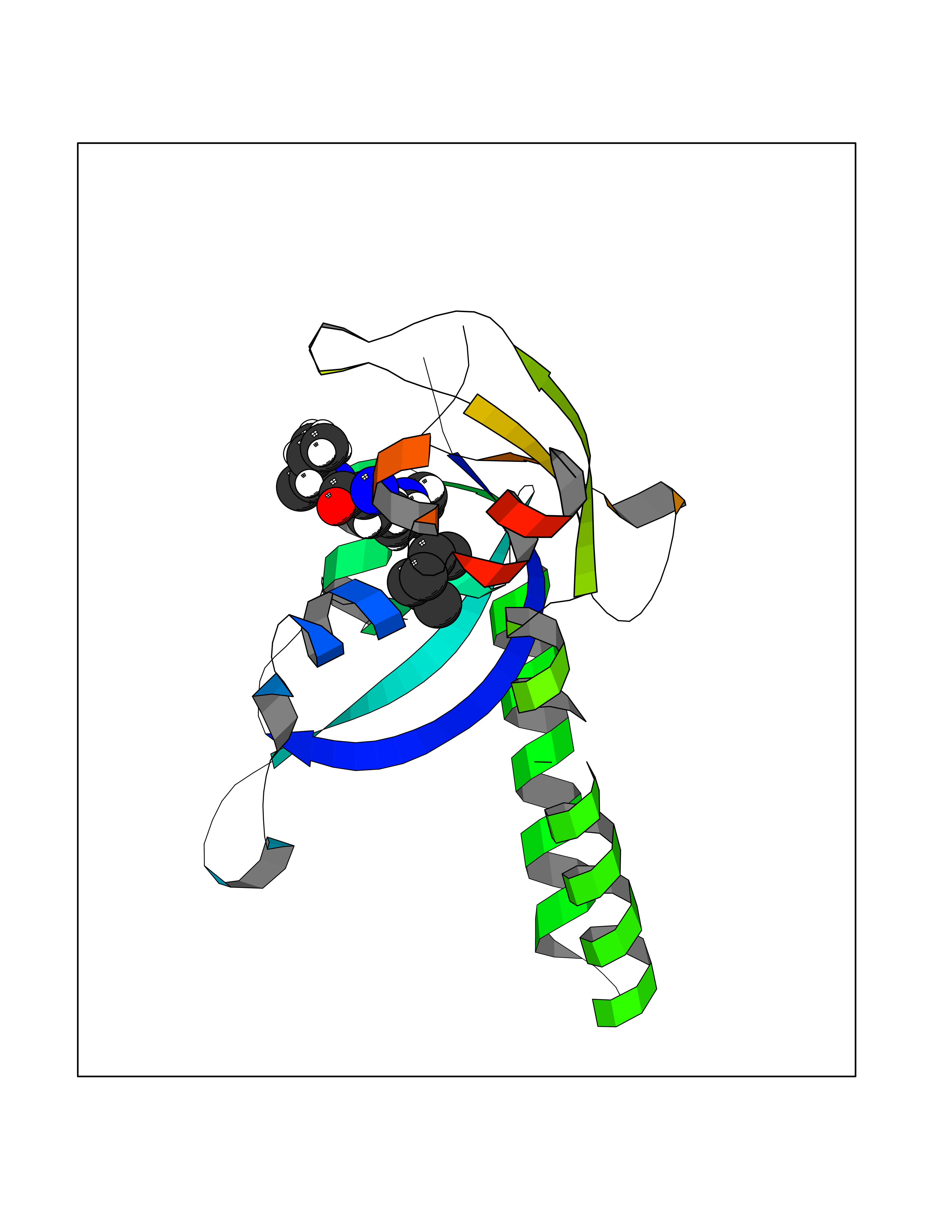 Molscript Map 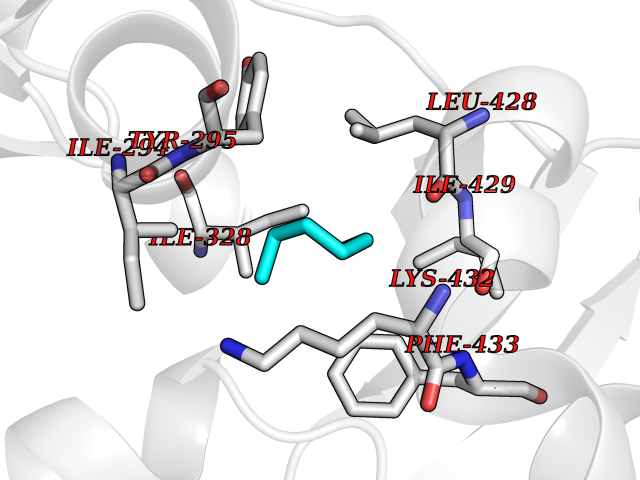 Pymol Map 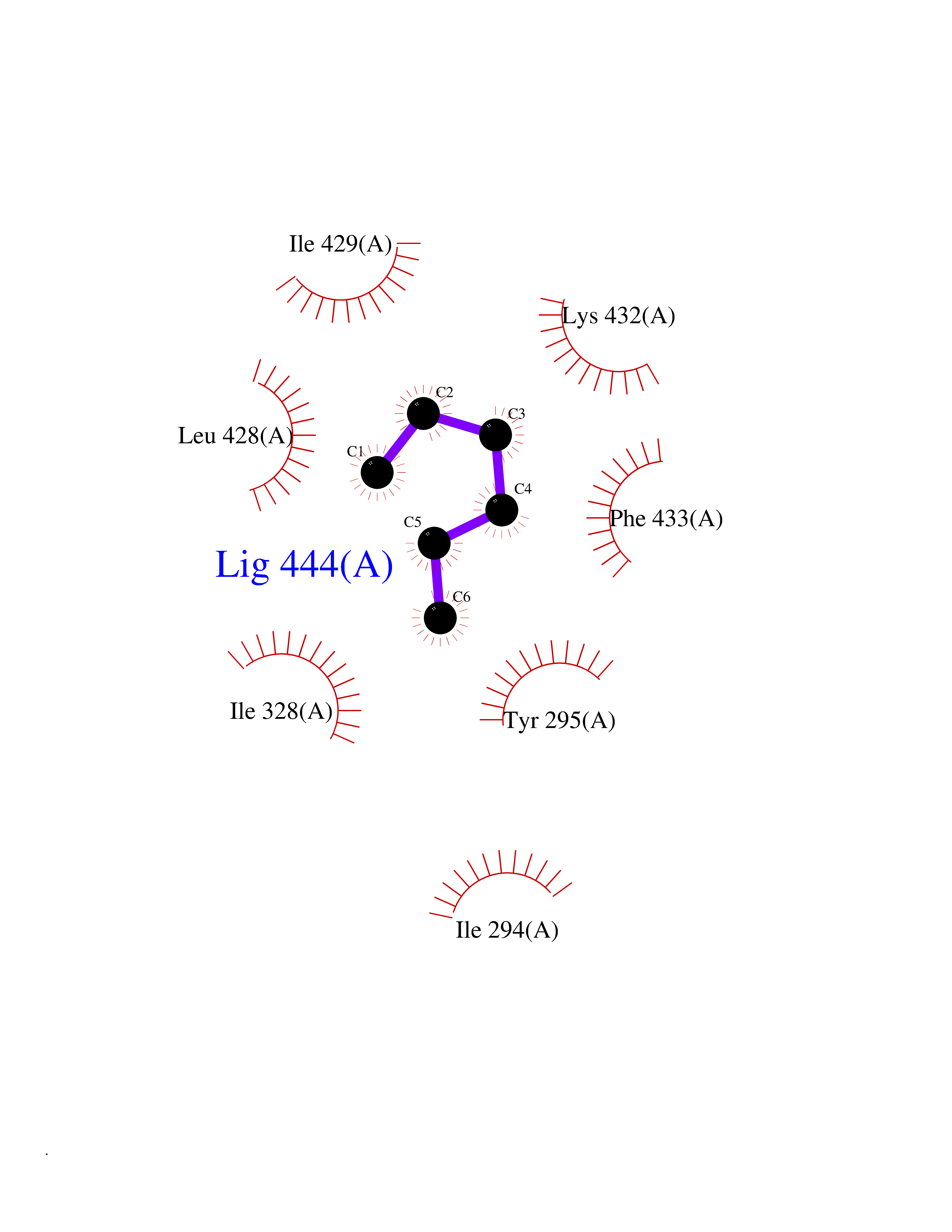 Ligplot Map 3D Binding mode Sequence EFETIERFMDCRIGRKGATGATTTIYAVEADGDPNAGFEKNKEPGEIQYLIKWKGWSHIHNTWETEETLKQQNVRGMKKLDNYKKKDQETKRWLKNASPEDVEYYNCQQELTDDLHKQYQIVERIIAHSNQKSAAGYPDYYCKWQGLPYSECSWEDGALISKKFQACIDEYFSRTARSXV Hydrogen bonds contact Hydrophobic contact | ||||
| 17 | T-cell-specific kinase (ITK) | 4HCU | 5.04 | |
Target general information Gen name ITK Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Tyrosine kinase ITK; Inducible T cell kinase; EMT Protein family Protein kinase superfamily, Tyr protein kinase family, TEC subfamily Biochemical class Kinase Function Regulates the development, function and differentiation of conventional T-cells and nonconventional NKT-cells. When antigen presenting cells (APC) activate T-cell receptor (TCR), a series of phosphorylation lead to the recruitment of ITK to the cell membrane, in the vicinity of the stimulated TCR receptor, where it is phosphorylated by LCK. Phosphorylation leads to ITK autophosphorylation and full activation. Once activated, phosphorylates PLCG1, leading to the activation of this lipase and subsequent cleavage of its substrates. In turn, the endoplasmic reticulum releases calcium in the cytoplasm and the nuclear activator of activated T-cells (NFAT) translocates into the nucleus to perform its transcriptional duty. Phosphorylates 2 essential adapter proteins: the linker for activation of T-cells/LAT protein and LCP2. Then, a large number of signaling molecules such as VAV1 are recruited and ultimately lead to lymphokine production, T-cell proliferation and differentiation. Phosphorylates TBX21 at 'Tyr-530' and mediates its interaction with GATA3. Tyrosine kinase that plays an essential role in regulation of the adaptive immune response. Related diseases Lymphoproliferative syndrome 1 (LPFS1) [MIM:613011]: A rare immunodeficiency characterized by extreme susceptibility to infection with Epstein-Barr virus (EBV). Inadequate immune response to EBV can have a fatal outcome. Clinical features include splenomegaly, lymphadenopathy, anemia, thrombocytopenia, pancytopenia, recurrent infections. There is an increased risk for lymphoma. {ECO:0000269|PubMed:19425169}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB12010; DB06589; DB14924; DB02010; DB15035 Interacts with P04626; P48023; P08238; Q13094; P31947; P62258; P10686 EC number EC 2.7.10.2 Uniprot keywords 3D-structure; Adaptive immunity; ATP-binding; Cytoplasm; Direct protein sequencing; Disease variant; Immunity; Kinase; Metal-binding; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; SH2 domain; SH3 domain; Transferase; Tyrosine-protein kinase; Ubl conjugation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A Molecular weight (Da) 30116.1 Length 263 Aromaticity 0.11 Instability index 37.47 Isoelectric point 5.03 Charge (pH=7) -11.73 2D Binding mode Binding energy (Kcal/mol) -6.87 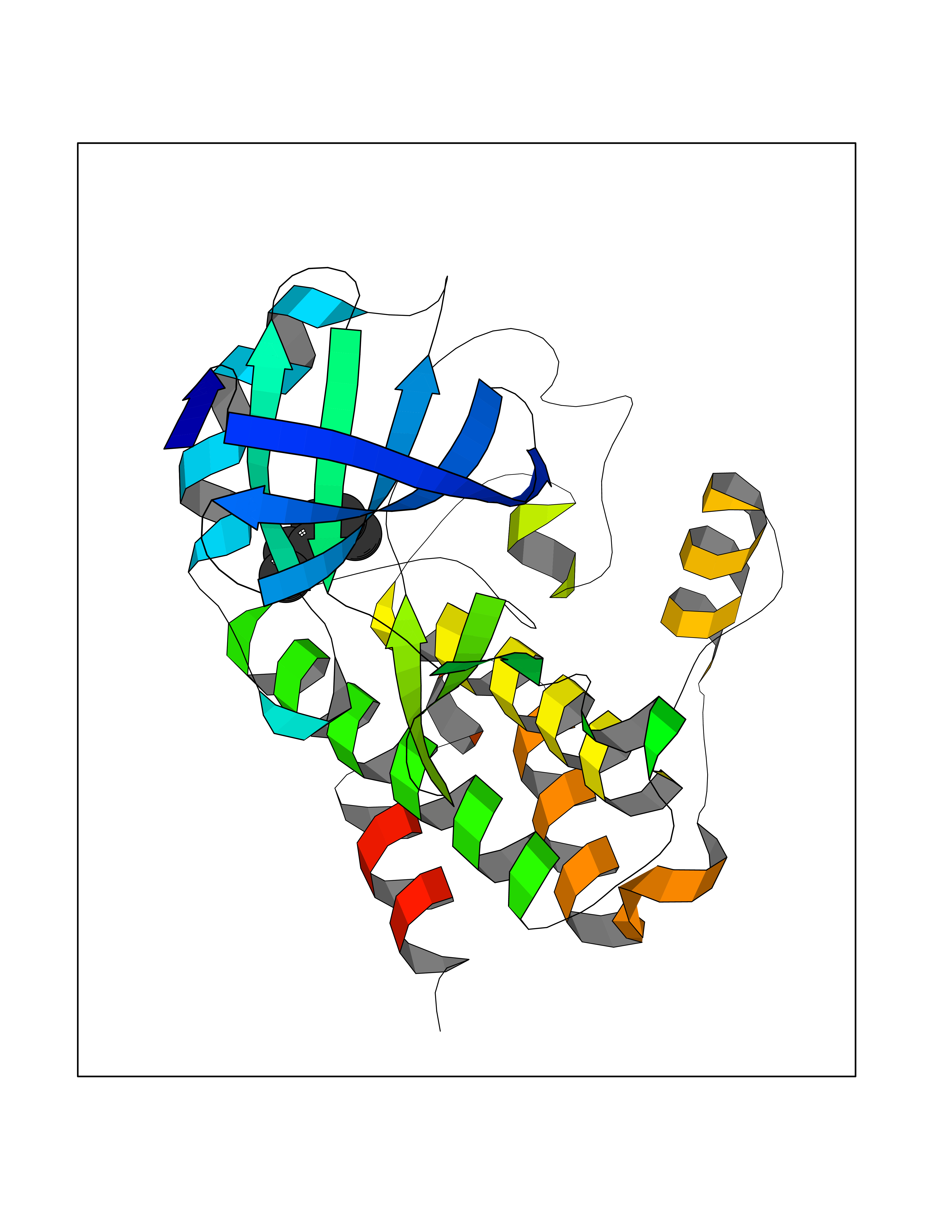 Molscript Map 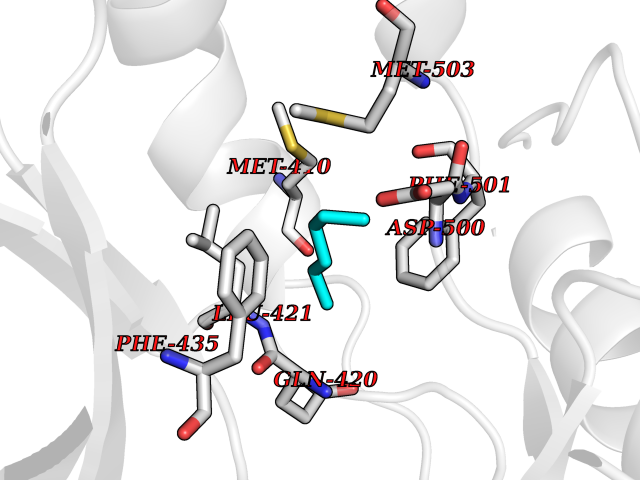 Pymol Map 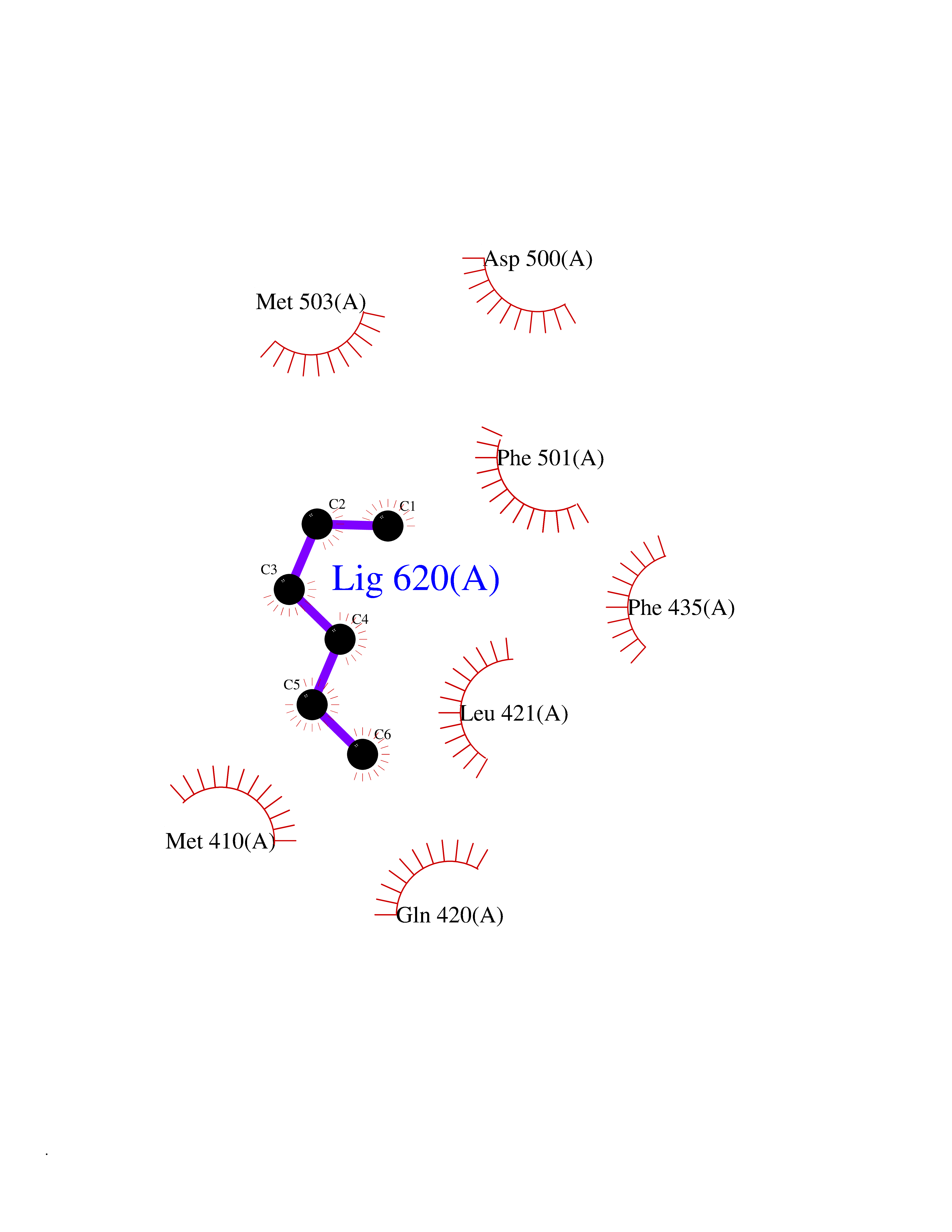 Ligplot Map 3D Binding mode Sequence WVIDPSELTFVQEIGSGQFGLVHLGYWLNKDKVAIKTIREGAMSEEDFIEEAEVMMKLSHPKLVQLYGVCLEQAPICLVFEFMEHGCLSDYLRTQRGLFAAETLLGMCLDVCEGMAYLEEACVIHRDLAARNCLVGENQVIKVSDFGMTRFVLDDQYTSSTGTKFPVKWASPEVFSFSRYSSKSDVWSFGVLMWEVFSEGKIPYENRSNSEVVEDISTGFRLYKPRLASTHVYQIMNHCWRERPEDRPAFSRLLRQLAEIAES Hydrogen bonds contact Hydrophobic contact | ||||
| 18 | Acetyl-CoA carboxylase 1 | 2YL2 | 5.04 | |
Target general information Gen name ACACA Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms ACAC;ACCA;ACC1 Protein family NA Biochemical class Ligase Function Acetyl-CoA carboxylase activity.ATP binding.Biotin carboxylase activity.Identical protein binding.Metal ion binding. Related diseases Acetyl-CoA carboxylase-alpha deficiency (ACACAD) [MIM:613933]: An autosomal recessive inborn error of de novo fatty acid synthesis associated with severe brain damage, persistent myopathy and poor growth. {ECO:0000269|PubMed:6114432}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00121 Interacts with Q13085; O60218; P38398; Q96EB6; Q9CQ20; P02654; Q92915-2; Q6NTF9-3 EC number 6.4.1.2 Uniprot keywords 3D-structure; Acetylation; Allosteric enzyme; Alternative promoter usage; ATP-binding; Biotin; Cytoplasm; Direct protein sequencing; Fatty acid biosynthesis; Fatty acid metabolism; Ligase; Lipid biosynthesis; Lipid metabolism; Magnesium; Manganese; Metal-binding; Multifunctional enzyme; Nucleotide-binding; Phosphoprotein; Proteomics identification; Reference proteome Protein physicochemical properties Chain ID A,B Molecular weight (Da) 54237.7 Length 486 Aromaticity 0.09 Instability index 39.18 Isoelectric point 6.37 Charge (pH=7) -2.46 2D Binding mode Binding energy (Kcal/mol) -6.87 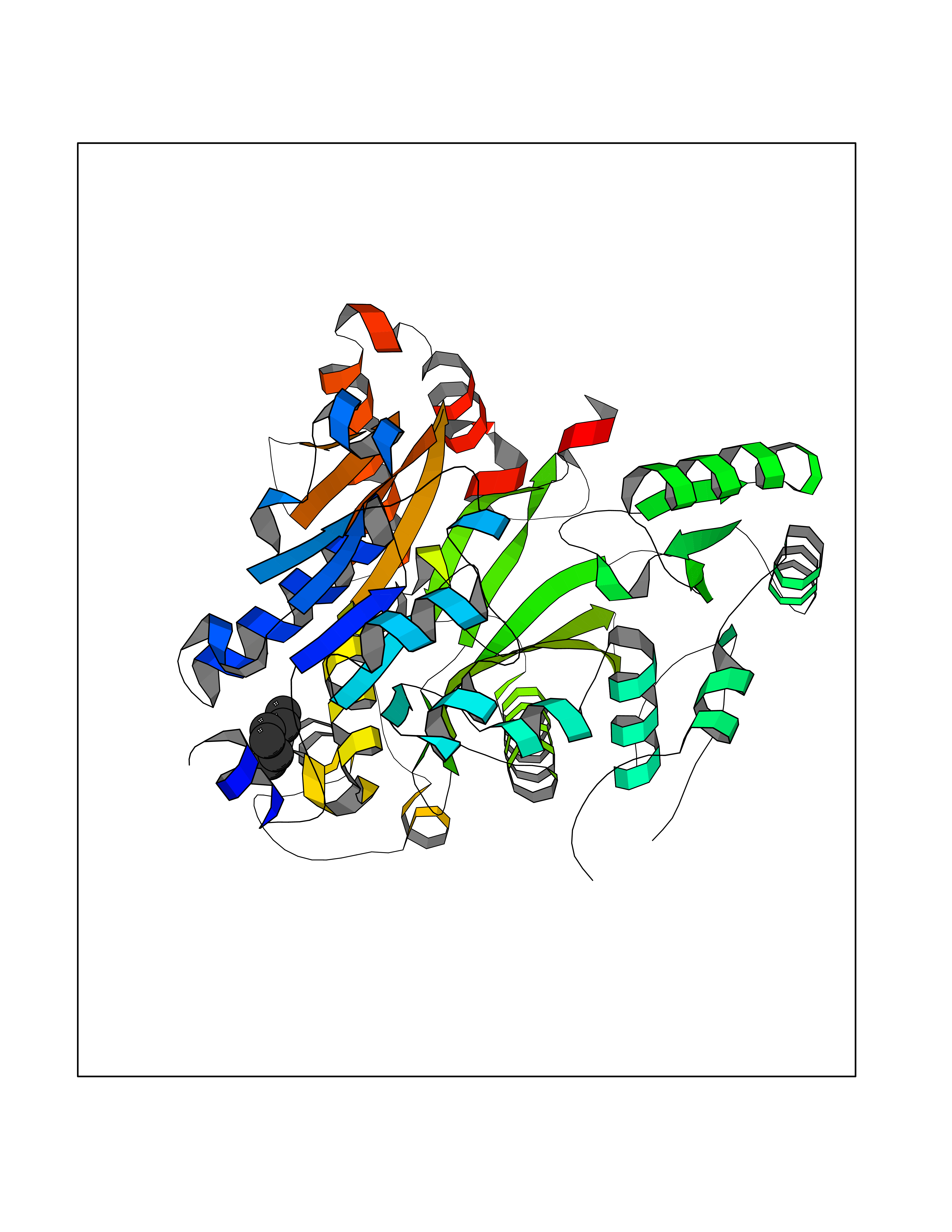 Molscript Map 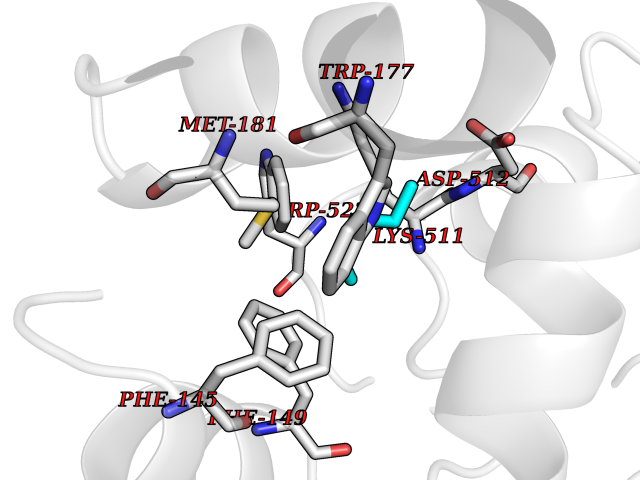 Pymol Map 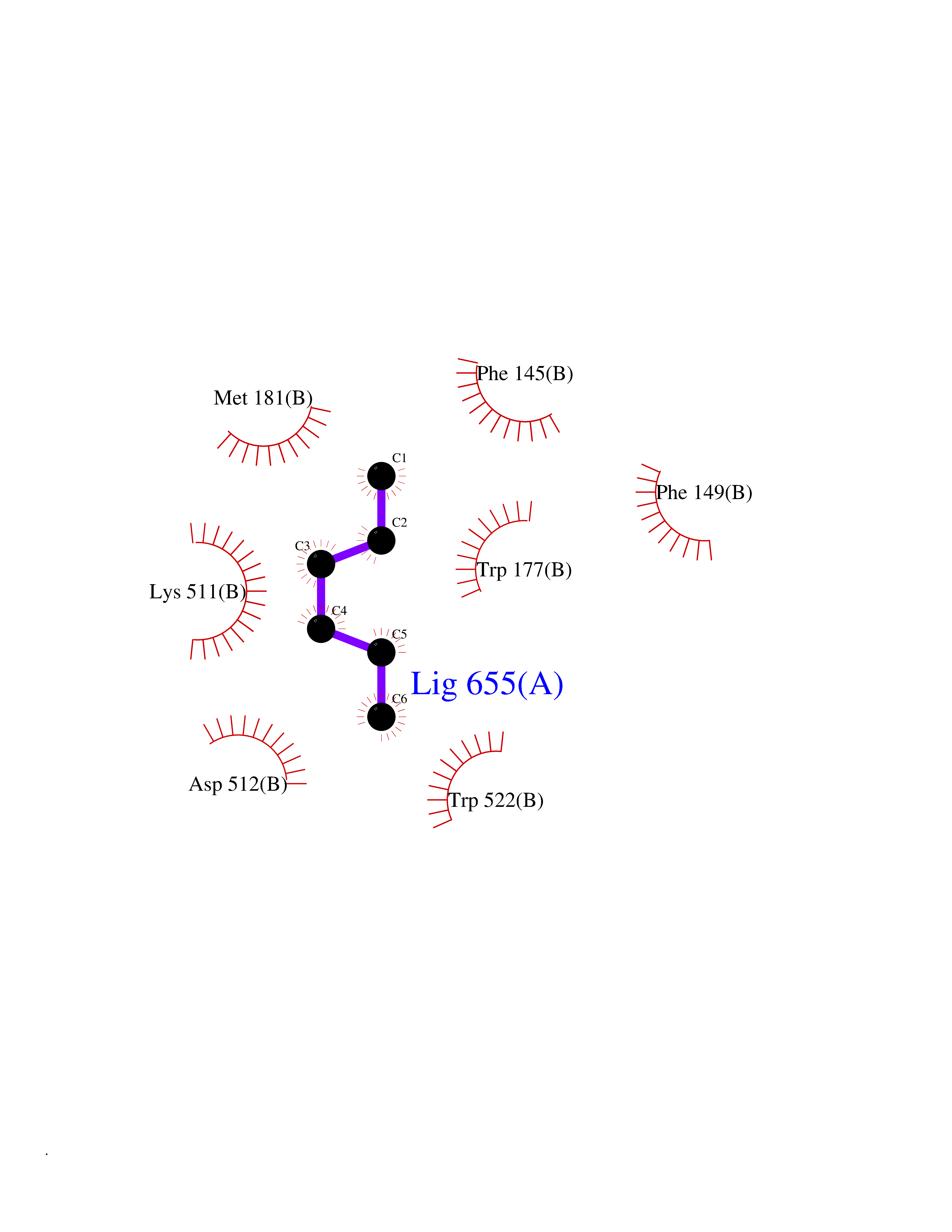 Ligplot Map 3D Binding mode Sequence VASPAEFVTRFGGNKVIEKVLIANNGIAAVKCMRSIRRWSYEMFRNERAIRFVVMVTPEDLKANAEYIKMADHYVPVPGGPNNNNYANVELILDIAKRIPVQAVWAGWGHASENPKLPELLLKNGIAFMGPPSQAMWALGDKIASSIVAQTAGIPTLPWSGSGLRVDWSKRILNVPQELYEKGYVKDVDDGLQAAEEVGYPVMIKASEGGGGKGIRKVNNADDFPNLFRQVQAEVPGSPIFVMRLAKQSRHLEVQILADQYGNAISLFGRDCSVQRRHQKIIEEAPATIATPAVFEHMEQCAVKLAKMVGYVSAGTVEYLYSQDGSFYFLELNPRLQVEHPCTEMVADVNLPAAQLQIAMGIPLYRIKDIRMMYGVSPWGDSPIDFEDSAHVPCPRGHVIAARITGTVQELNFRSNKNVWGYFSVQFGHCFSWGENREEAISNMVVALKELSIRGDFRTTVEYLIKLLETESFQMNRIDTGWLDRL Hydrogen bonds contact Hydrophobic contact | ||||
| 19 | SET domain containing 8 (KMT5A) | 5TEG | 5.04 | |
Target general information Gen name KMT5A Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms SETD8; SET8; SET07; SET domain-containing protein 8; PRSET7; PR/SET07; PR/SET domain-containing protein 07; PR-Set7; N-lysine methyltransferase KMT5A; Lysine-specific methylase 5A; Lysine N-methyltran Protein family Class V-like SAM-binding methyltransferase superfamily, Histone-lysine methyltransferase family, PR/SET subfamily Biochemical class Methyltransferase Function Specifically monomethylates 'Lys-20' of histone H4 (H4K20me1). H4K20me1 is enriched during mitosis and represents a specific tag for epigenetic transcriptional repression. Mainly functions in euchromatin regions, thereby playing a central role in the silencing of euchromatic genes. Required for cell proliferation, probably by contributing to the maintenance of proper higher-order structure of DNA during mitosis. Involved in chromosome condensation and proper cytokinesis. Nucleosomes are preferred as substrate compared to free histones. Mediates monomethylation of p53/TP53 at 'Lys-382', leading to repress p53/TP53-target genes. Plays a negative role in TGF-beta response regulation and a positive role in cell migration. Protein-lysine N-methyltransferase that monomethylates both histones and non-histone proteins. Related diseases Sick sinus syndrome 2 (SSS2) [MIM:163800]: The term 'sick sinus syndrome' encompasses a variety of conditions caused by sinus node dysfunction. The most common clinical manifestations are syncope, presyncope, dizziness, and fatigue. Electrocardiogram typically shows sinus bradycardia, sinus arrest, and/or sinoatrial block. Episodes of atrial tachycardias coexisting with sinus bradycardia ('tachycardia-bradycardia syndrome') are also common in this disorder. SSS occurs most often in the elderly associated with underlying heart disease or previous cardiac surgery, but can also occur in the fetus, infant, or child without heart disease or other contributing factors. SSS2 onset is in utero or at birth. {ECO:0000269|PubMed:15123648, ECO:0000269|PubMed:16407510, ECO:0000269|PubMed:20662977, ECO:0000269|PubMed:23103389}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Brugada syndrome 8 (BRGDA8) [MIM:613123]: A tachyarrhythmia characterized by right bundle branch block and ST segment elevation on an electrocardiogram (ECG). It can cause the ventricles to beat so fast that the blood is prevented from circulating efficiently in the body. When this situation occurs, the individual will faint and may die in a few minutes if the heart is not reset. {ECO:0000269|PubMed:19165230}. The gene represented in this entry may be involved in disease pathogenesis.; DISEASE: Epilepsy, idiopathic generalized 18 (EIG18) [MIM:619521]: An autosomal dominant form of idiopathic generalized epilepsy, a disorder characterized by recurring generalized seizures in the absence of detectable brain lesions and/or metabolic abnormalities. Generalized seizures arise diffusely and simultaneously from both hemispheres of the brain. Seizure types include juvenile myoclonic seizures, absence seizures, and generalized tonic-clonic seizures. EIG18 is characterized by onset of myoclonic seizures in infancy. Although the seizures remit, some patients may have later speech or cognitive impairment. {ECO:0000269|PubMed:30127718}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P62805; P07910; Q15672 EC number EC 2.1.1.- Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Cell cycle; Cell division; Chromatin regulator; Chromosome; Coiled coil; Direct protein sequencing; Methyltransferase; Mitosis; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Repressor; S-adenosyl-L-methionine; Transcription; Transcription regulation; Transferase; Ubl conjugation Protein physicochemical properties Chain ID A,D Molecular weight (Da) 19129.4 Length 167 Aromaticity 0.08 Instability index 49.18 Isoelectric point 7.88 Charge (pH=7) 1.37 2D Binding mode Binding energy (Kcal/mol) -6.87 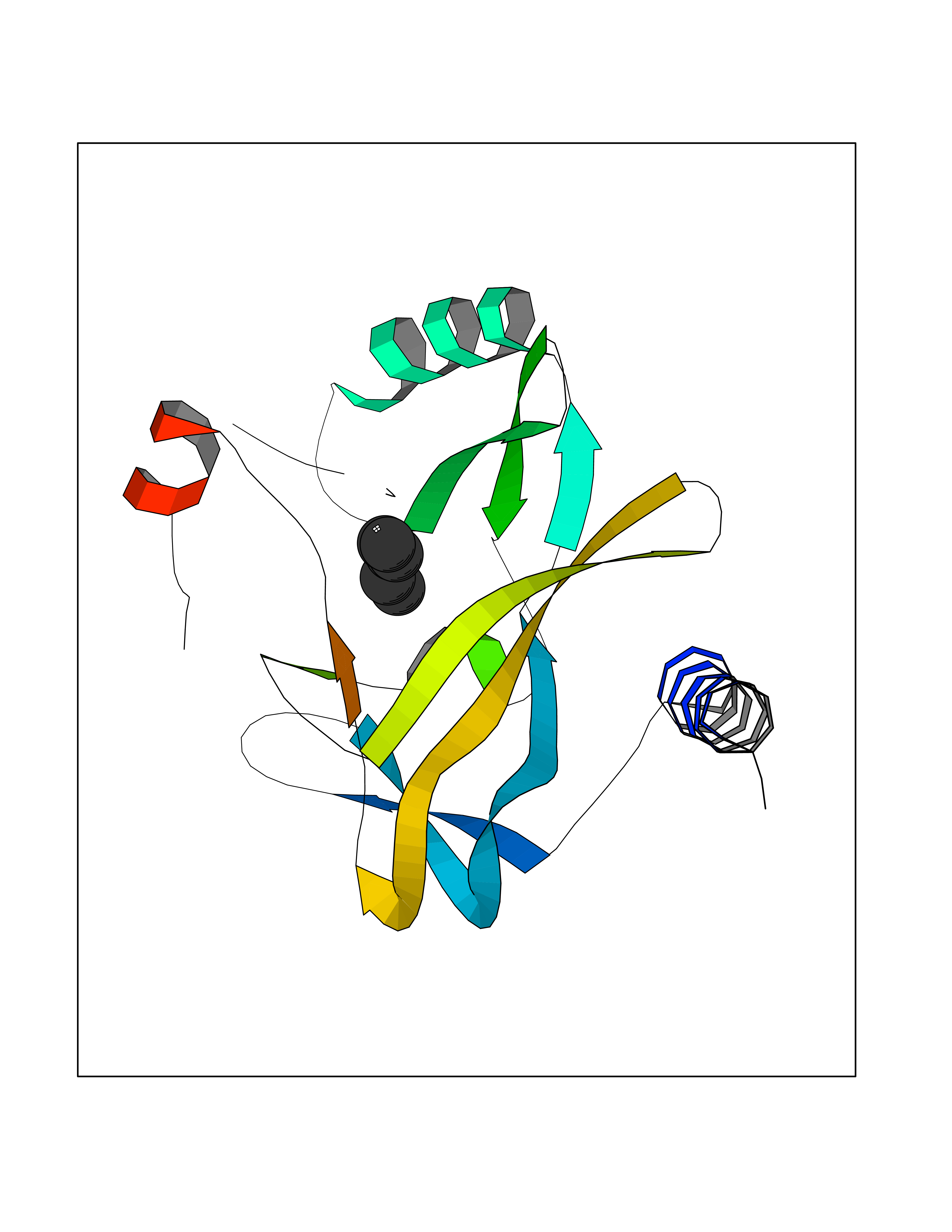 Molscript Map 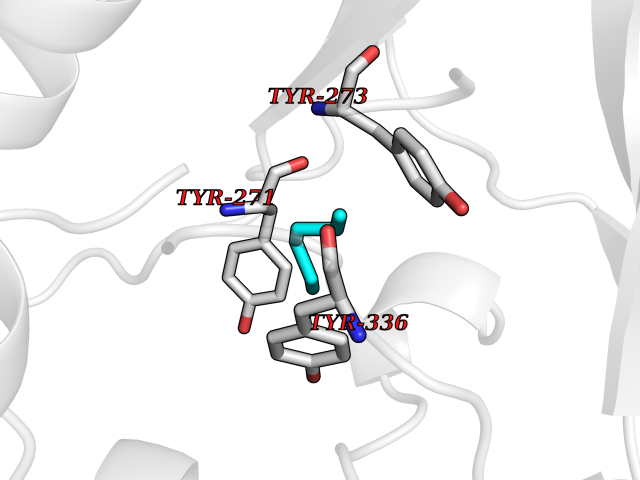 Pymol Map 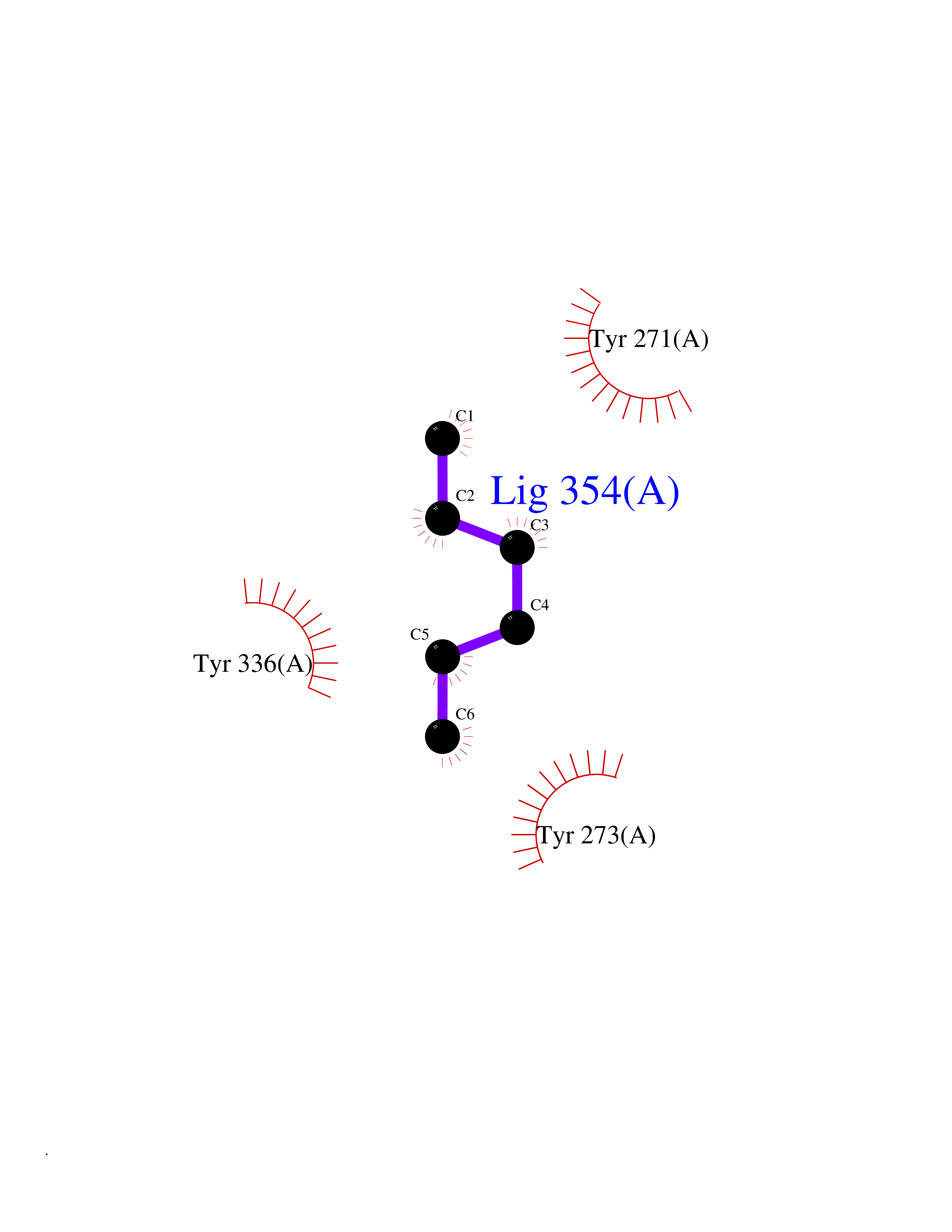 Ligplot Map 3D Binding mode Sequence KSKAELQSEERKRIDELIESGKEEGMKIDLIDGKGRGVIATKQFSRGDFVVEYHGDLIEITDAKKREALYAQDPSTGCYMYYFQYLSKTYCVDATRETNRLGRLINHSKSGNCQTKLHDIDGVPHLILIASRDIAAGEELLYDYGDRSKASIEAHPWLKHKRHRVLR Hydrogen bonds contact Hydrophobic contact | ||||
| 20 | Hydroxymethylglutaryl-CoA synthase 2 (HMGCS2) | 2WYA | 5.04 | |
Target general information Gen name HMGCS2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms HMGCS2; HMG-CoAsynthase; 3-hydroxy-3-methylglutaryl coenzyme A synthase 2 Protein family Thiolase-like superfamily, HMG-CoA synthase family Biochemical class Acyltransferase Function This enzyme condenses acetyl-CoA with acetoacetyl-CoA to form HMG-CoA, which is the substrate for HMG-CoA reductase. Related diseases 3-hydroxy-3-methylglutaryl-CoA synthase-2 deficiency (HMGCS2D) [MIM:605911]: A metabolic disorder characterized by severe hypoketotic hypoglycemia, encephalopathy, and hepatomegaly. {ECO:0000269|PubMed:11228257, ECO:0000269|PubMed:11479731, ECO:0000269|PubMed:12647205, ECO:0000269|PubMed:16601895, ECO:0000269|PubMed:23751782, ECO:0000269|PubMed:25511235, ECO:0000269|PubMed:29597274}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number EC 2.3.3.10 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Cholesterol biosynthesis; Cholesterol metabolism; Disease variant; Lipid biosynthesis; Lipid metabolism; Mitochondrion; Phosphoprotein; Proteomics identification; Reference proteome; Steroid biosynthesis; Steroid metabolism; Sterol biosynthesis; Sterol metabolism; Transferase; Transit peptide Protein physicochemical properties Chain ID A,D Molecular weight (Da) 102499 Length 920 Aromaticity 0.1 Instability index 33.48 Isoelectric point 6.72 Charge (pH=7) -1.41 2D Binding mode Binding energy (Kcal/mol) -6.87  Molscript Map 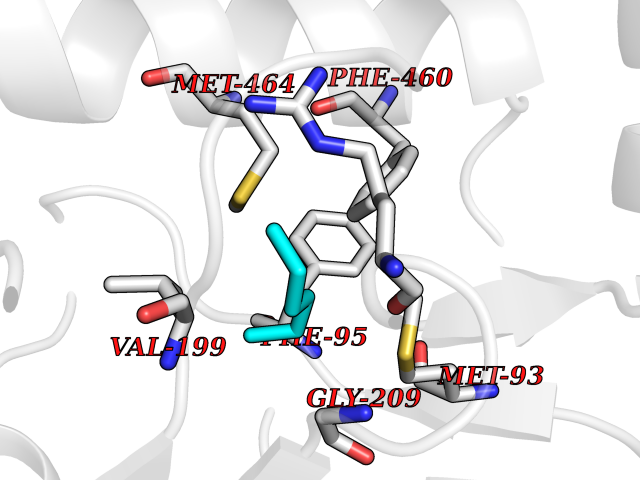 Pymol Map 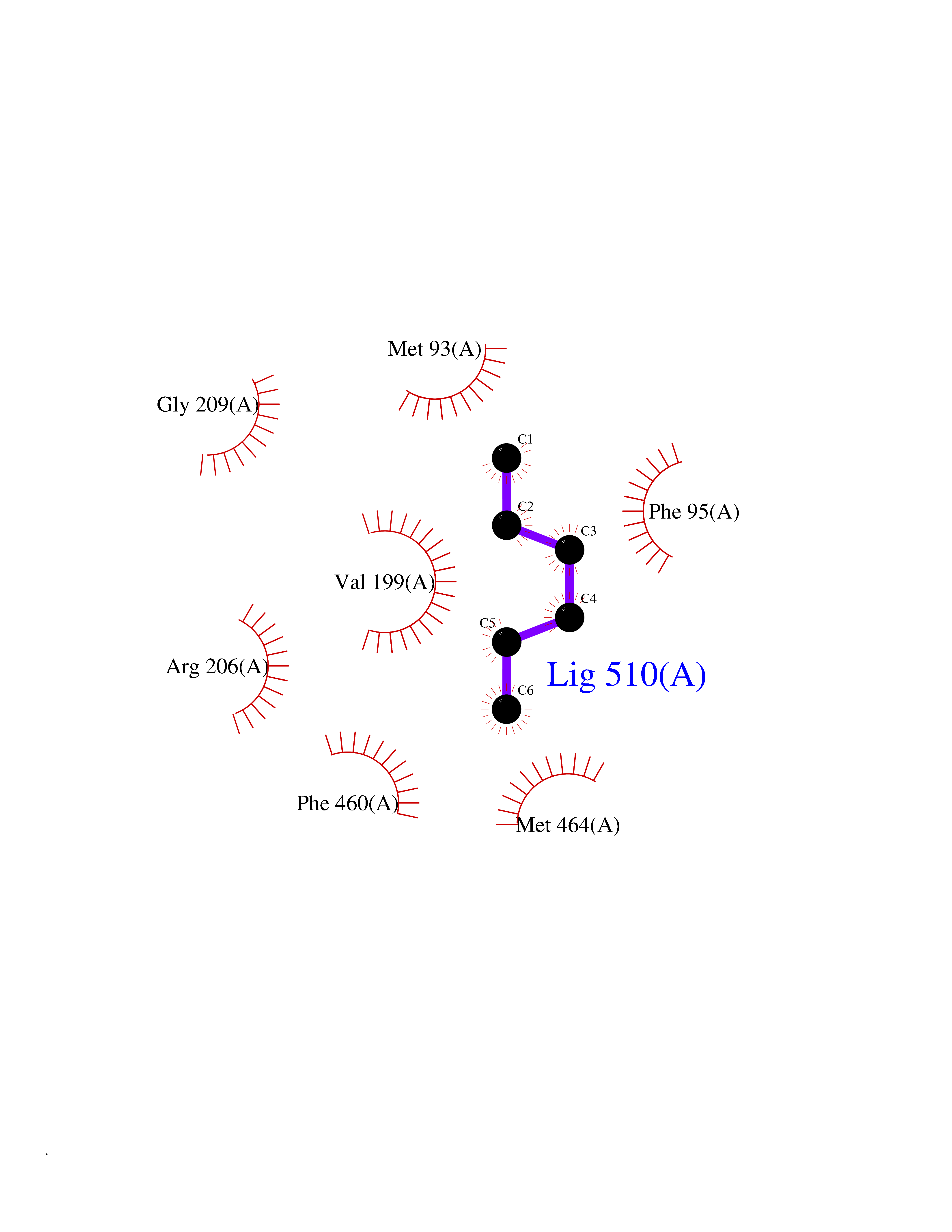 Ligplot Map 3D Binding mode Sequence SMPKDVGILALEVYFPAQYVDQTDLEKYNNVEAGKYTVGLGQTRMGFCSVQEDINSLCLTVVQRLMERIQLPWDSVGRLEVGTETIIDKSKAVKTVLMELFQDSGNTDIEGIDTTNACYGGTASLFNAANWMESSSWDGRYAMVVCGDIAVYPSGNARPTGGAGAVAMLIGPKAPLALERGLRGTHMENVYDFYKPNLASEYPIVDGKLSIQCYLRALDRCYTSYRKKIQNQWKQAGSDRPFTLDDLQYMIFHTPFCKMVQKSLARLMFNDFLSASSDTQTSLYKGLEAFGGLKLEDTYTNKDLDKALLKASQDMFDKKTKASLYLSTHNGNMYTSSLYGCLASLLSHHSAQELAGSRIGAFSYGSGLAASFFSFRVSQDAAPGSPLDKLVSSTSDLPKRLASRKCVSPEEFTEIMNQREQFYHKVNFSPPGDTNSLFPGTWYLERVDEQHRRKYARRPVSMPKDVGILALEVYFPAQYVDQTDLEKYNNVEAGKYTVGLGQTRMGFCSVQEDINSLCLTVVQRLMERIQLPWDSVGRLEVGTETIIDKSKAVKTVLMELFQDSGNTDIEGIDTTNACYGGTASLFNAANWMESSSWDGRYAMVVCGDIAVYPSGNARPTGGAGAVAMLIGPKAPLALERGLRGTHMENVYDFYKPNLASEYPIVDGKLSIQCYLRALDRCYTSYRKKIQNQWKQAGSDRPFTLDDLQYMIFHTPFCKMVQKSLARLMFNDFLSASSDTQTSLYKGLEAFGGLKLEDTYTNKDLDKALLKASQDMFDKKTKASLYLSTHNGNMYTSSLYGCLASLLSHHSAQELAGSRIGAFSYGSGLAASFFSFRVSQDAAPGSPLDKLVSSTSDLPKRLASRKCVSPEEFTEIMNQREQFYHKVNFSPPGDTNSLFPGTWYLERVDEQHRRKYARRPV Hydrogen bonds contact Hydrophobic contact | ||||