Job Results:
Ligand
Structure
Job ID
2f26746d3788b5c615eb3e0507a308ea
Job name
NA
Time
2025-01-23 16:35:41
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 101 | Monomeric sarcosine oxidase | 2GF3 | 7.56 | |
Target general information Gen name soxA Organism Bacillus sp. (strain B-0618) Uniprot ID TTD ID NA Synonyms sox Protein family MSOX/MTOX family, MSOX subfamily Biochemical class Oxidoreductase Function Sarcosine oxidase activity. Related diseases Defects in PPARG can lead to type 2 insulin-resistant diabetes and hyptertension. PPARG mutations may be associated with colon cancer. {ECO:0000269|PubMed:10394368}.; DISEASE: Obesity (OBESITY) [MIM:601665]: A condition characterized by an increase of body weight beyond the limitation of skeletal and physical requirements, as the result of excessive accumulation of body fat. {ECO:0000269|PubMed:9753710}. Disease susceptibility may be associated with variants affecting the gene represented in this entry.; DISEASE: Lipodystrophy, familial partial, 3 (FPLD3) [MIM:604367]: A form of lipodystrophy characterized by marked loss of subcutaneous fat from the extremities. Facial adipose tissue may be increased, decreased or normal. Affected individuals show an increased preponderance of insulin resistance, diabetes mellitus and dyslipidemia. {ECO:0000269|PubMed:11788685, ECO:0000269|PubMed:12453919}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Glioma 1 (GLM1) [MIM:137800]: Gliomas are benign or malignant central nervous system neoplasms derived from glial cells. They comprise astrocytomas and glioblastoma multiforme that are derived from astrocytes, oligodendrogliomas derived from oligodendrocytes and ependymomas derived from ependymocytes. {ECO:0000269|PubMed:10851250}. Disease susceptibility may be associated with variants affecting the gene represented in this entry. Polymorphic PPARG alleles have been found to be significantly over-represented among a cohort of American patients with sporadic glioblastoma multiforme suggesting a possible contribution to disease susceptibility. Drugs (DrugBank ID) DB03098; DB01918; DB03517; DB03147; DB03366; DB02083; DB02543 Interacts with NA EC number 1.5.3.1 Uniprot keywords 3D-structure; Cytoplasm; Direct protein sequencing; FAD; Flavoprotein; Oxidoreductase Protein physicochemical properties Chain ID A,B Molecular weight (Da) 42606.4 Length 385 Aromaticity 0.1 Instability index 26.97 Isoelectric point 5.27 Charge (pH=7) -17.18 2D Binding mode Binding energy (Kcal/mol) -10.31  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence STHFDVIVVGAGSMGMAAGYQLAKQGVKTLLVDAFDPPHTNGSHHGDTRIIRHAYGEGREYVPLALRSQELWYELEKETHHKIFTKTGVLVFGPKGESAFVAETMEAAKEHSLTVDLLEGDEINKRWPGITVPENYNAIFEPNSGVLFSENCIRAYRELAEARGAKVLTHTRVEDFDISPDSVKIETANGSYTADKLIVSMGAWNSKLLSKLNLDIPLQPYRQVVGFFESDESKYSNDIDFPGFMVEVPNGIYYGFPSFGGCGLKLGYHTFGQKIDPDTINREFGVYPEDESNLRAFLEEYMPGANGELKRGAVCMYTKTLDEHFIIDLHPEHSNVVIAAGFSGHGFKFSSGVGEVLSQLALTGKTEHDISIFSINRPALKESLQ Hydrogen bonds contact Hydrophobic contact | ||||
| 102 | SET and MYND domain-containing protein 2 (SMYD2) | 5ARF | 7.56 | |
Target general information Gen name SMYD2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Nlysine methyltransferase SMYD2; N-lysine methyltransferase SMYD2; Lysine Nmethyltransferase 3C; Lysine N-methyltransferase 3C; KMT3C; Histone methyltransferase SMYD2; HSKMB; HSKM-B Protein family Class V-like SAM-binding methyltransferase superfamily Biochemical class Methyltransferase Function Specifically methylates histone H3 'Lys-4' (H3K4me) and dimethylates histone H3 'Lys-36' (H3K36me2). Shows even higher methyltransferase activity on p53/TP53. Monomethylates 'Lys-370' of p53/TP53, leading to decreased DNA-binding activity and subsequent transcriptional regulation activity of p53/TP53. Monomethylates RB1 at 'Lys-860'. Protein-lysine N-methyltransferase that methylates both histones and non-histone proteins, including p53/TP53 and RB1. Related diseases Pulmonary hypertension, primary, 4 (PPH4) [MIM:615344]: A rare disorder characterized by plexiform lesions of proliferating endothelial cells in pulmonary arterioles. The lesions lead to elevated pulmonary arterial pression, right ventricular failure, and death. The disease can occur from infancy throughout life and it has a mean age at onset of 36 years. Penetrance is reduced. Although familial pulmonary hypertension is rare, cases secondary to known etiologies are more common and include those associated with the appetite-suppressant drugs. {ECO:0000269|PubMed:23883380}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Defects in this gene may cause developmental delay with sleep apnea (DDSA). A disorder characterized by developmental neurologic, skeletal and respiratory anomalies including microcephaly, arthrogryposis, scoliosis, cleft palate, facial dysmorphology, bilateral talipes, feeding difficulties and central and/or obstructive sleep apnea. Malformations are detected as early as 21 weeks post gestation. Severely affected patients require ongoing treatment with nocturnal O2 or pressure-controlled ventilation. The disease is associated with recurrent de novo gain of function variants. {ECO:0000269|PubMed:36195757}. Drugs (DrugBank ID) NA Interacts with P20290-2; Q96K17; Q9UPZ9; Q9Y5W9; P04637 EC number EC 2.1.1.- Uniprot keywords 3D-structure; Alternative splicing; Chromatin regulator; Cytoplasm; Metal-binding; Methyltransferase; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; S-adenosyl-L-methionine; Transcription; Transcription regulation; Transferase; Zinc; Zinc-finger Protein physicochemical properties Chain ID A Molecular weight (Da) 48789.6 Length 425 Aromaticity 0.1 Instability index 51.93 Isoelectric point 6.35 Charge (pH=7) -4.02 2D Binding mode Binding energy (Kcal/mol) -10.31  Molscript Map 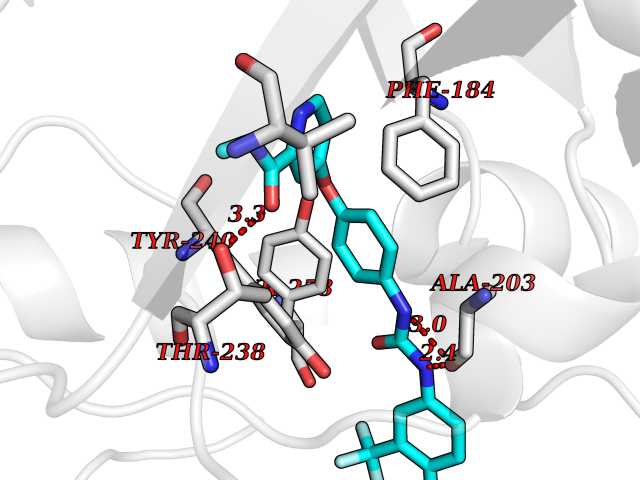 Pymol Map 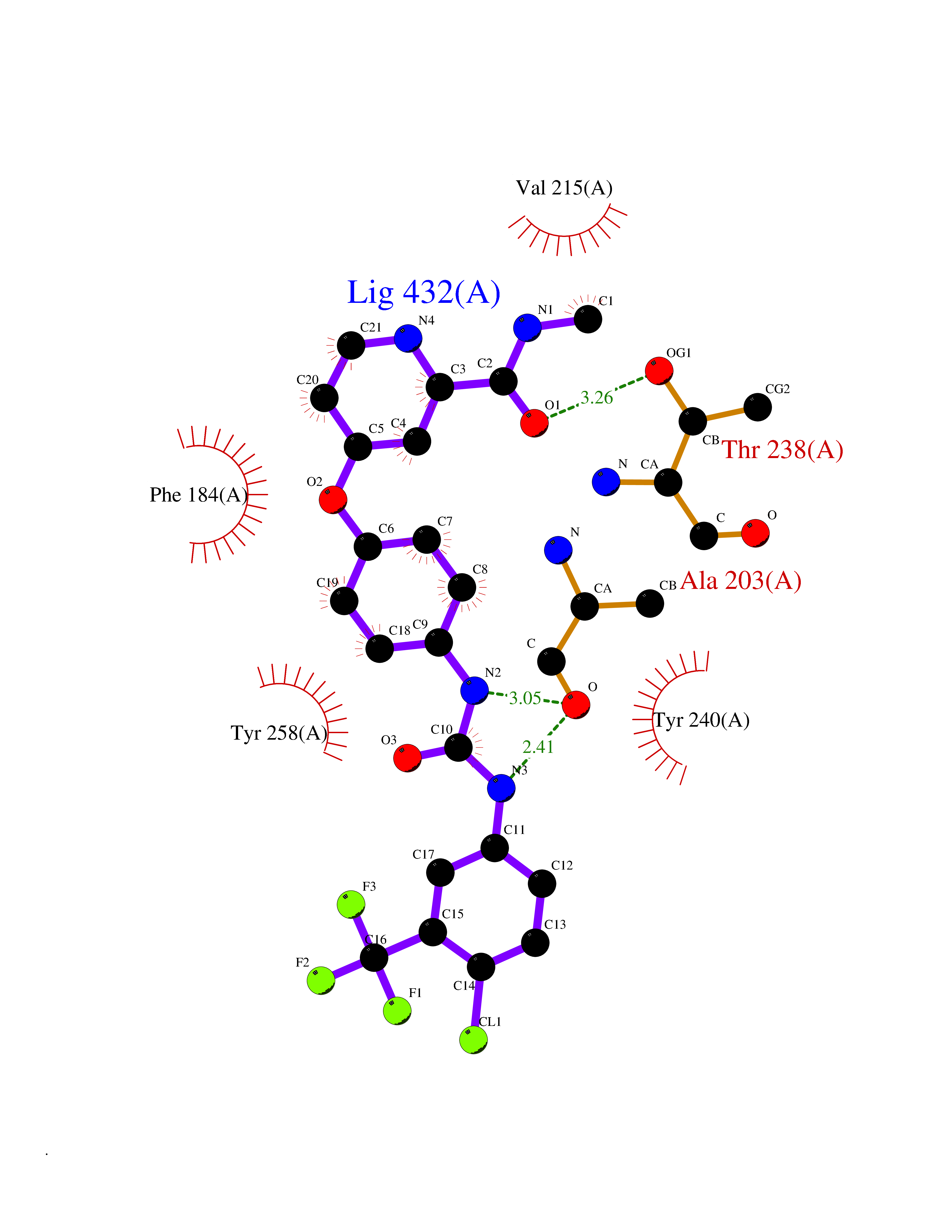 Ligplot Map 3D Binding mode Sequence LGGLERFCSPGKGRGLRALQPFQVGDLLFSCPAYAYVLTVNERGNHCEYCFTRKEGLSKCGRCKQAFYCNVECQKEDWPMHKLECSPMVVFGENWNPSETVRLTARILAKQKIHPERTPSEKLLAVKEFESHLDKLDNEKKDLIQSDIAALHHFYSKHLGFPDNDSLVVLFAQVNCNGFTIEDEELSHLGSAIFPDVALMNHSCCPNVIVTYKGTLAEVRAVQEIKPGEEVFTSYIDLLYPTEDRNDRLRDSYFFTCECQECTTKDKDKAKVEIRKLSDPPKAEAIRDMVRYARNVIEEFRRAKHYKSPSELLEICELSQEKMSSVFEDSNVYMLHMMYQAMGVCLYMQDWEGALQYGQKIIKPYSKHYPLYSLNVASMWLKLGRLYMGLEHKAAGEKALKKAIAIMEVAHGKDHPYISEIKQEI Hydrogen bonds contact Hydrophobic contact | ||||
| 103 | Dipeptidyl peptidase 8 (DPP-8) | 6EOP | 7.55 | |
Target general information Gen name DPP8 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Prolyl dipeptidase DPP8; MSTP141; MSTP135; MSTP097; Dipeptidyl peptidase VIII; Dipeptidyl peptidase IV-related protein 1; DPRP1; DPRP-1; DPP VIII; DP8 Protein family Peptidase S9B family, DPPIV subfamily Biochemical class Peptidase Function Dipeptidyl peptidase that cleaves off N-terminal dipeptides from proteins having a Pro or Ala residue at position 2. Related diseases Orotic aciduria 1 (ORAC1) [MIM:258900]: A disorder of pyrimidine metabolism resulting in megaloblastic anemia and orotic acid crystalluria that is frequently associated with some degree of physical and intellectual disability. A minority of cases have additional features, particularly congenital malformations and immune deficiencies. {ECO:0000269|PubMed:9042911}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number EC 3.4.14.5 Uniprot keywords 3D-structure; Alternative splicing; Aminopeptidase; Apoptosis; Cytoplasm; Hydrolase; Protease; Proteomics identification; Reference proteome; Serine protease Protein physicochemical properties Chain ID A,D Molecular weight (Da) 97764.9 Length 849 Aromaticity 0.12 Instability index 47.71 Isoelectric point 5.69 Charge (pH=7) -21.66 2D Binding mode Binding energy (Kcal/mol) -10.3  Molscript Map 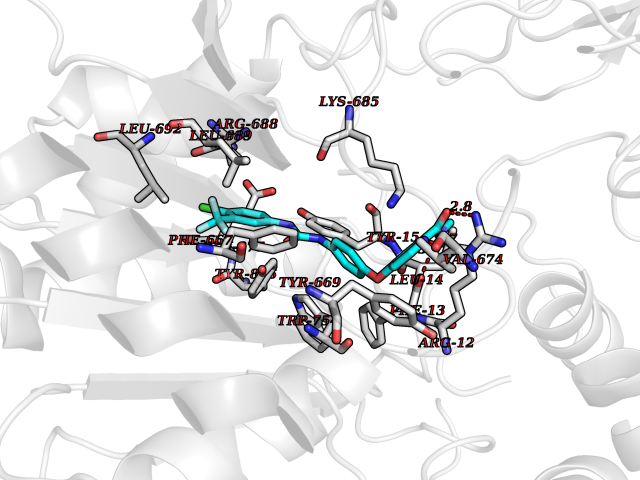 Pymol Map 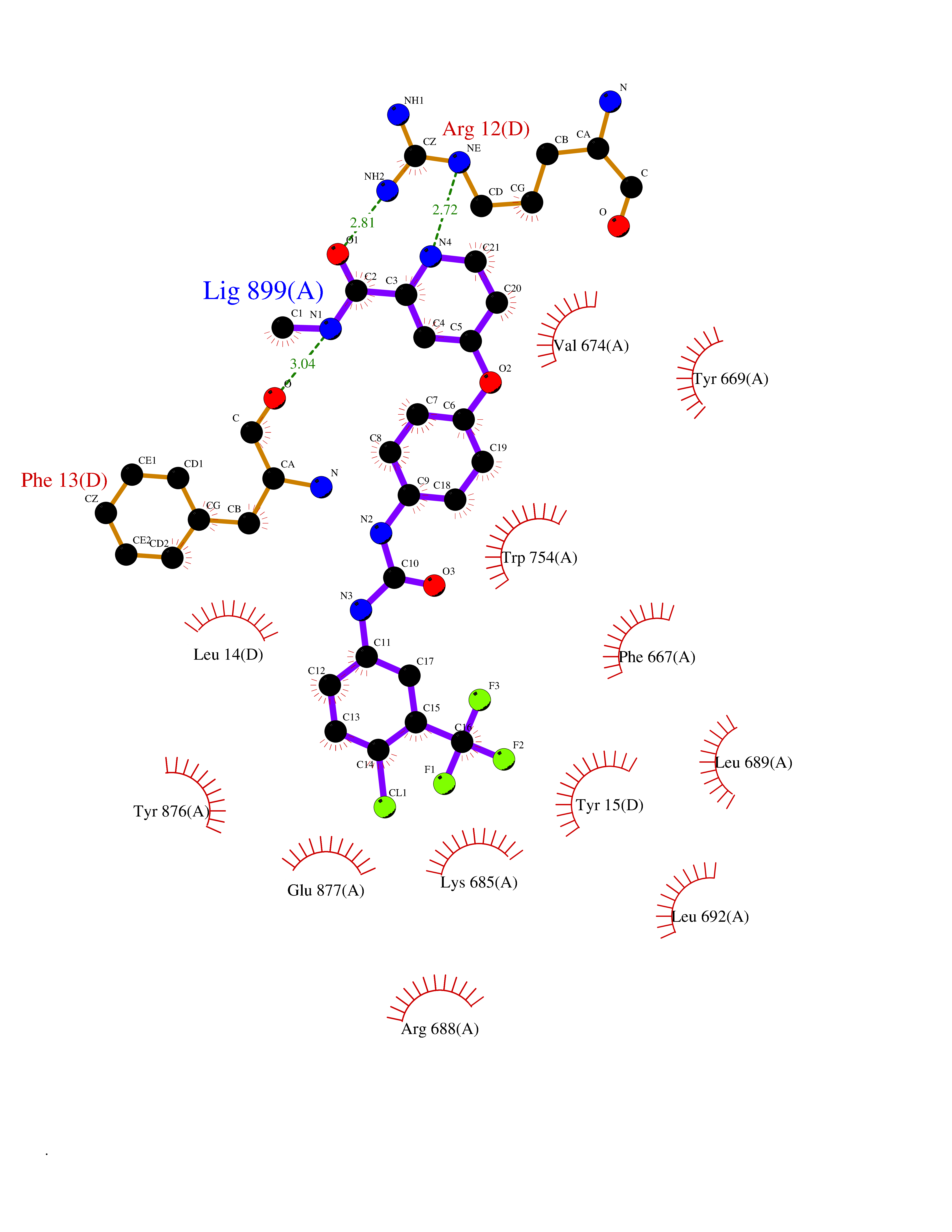 Ligplot Map 3D Binding mode Sequence LEPFYVERYSWSQLKKLLADTRKYHGYMMAKAPHDFMFVKRNDPDGPHSDRIYYLAMSNRENTLFYSEIPKTINRAAVLMLSWKPLLDLFQYSREEELLRERKRIGTVGIASYDYHQGSGTFLFQAGSGIYHVKDGGPQGFTQQPLRPNLVETSCPNIRMDPKLCPADPDWIAFIHSNDIWISNIVTREERRLTYVHNELANMEEDARSAGVATFVLQEEFDRYSGYWWCPKAETTPSGGKILRILYEENDESEVEIIHVTSPMLETRRADSFRYPKTGTANPKVTFKMSEIMIDAEGRIIDVIDKELIQPFEILFEGVEYIARAGWTPEGKYAWSILLDRSQTRLQIVLISPELFIPVEDDVMERQRLIESVPDSVTPLIIYEETTDIWINIHDIFHVFPQSHEEEIEFIFASECKTGFRHLYKITSILKESKYKRSSGGLPAPSDFKCPIKEEIAITSGEWEVLGRHGSNIQVDEVRRLVYFEGTKDSPLEHHLYVVSYVNPGEVTRLTDRGYSHSCCISQHCDFFISKYSNQKNPHCVSLYKLSSPEDDPTCKTKEFWATILDSAGPLPDYTPPEIFSFESTTGFTLYGMLYKPHDLQPGKKYPTVLFIYGGPQVQLVNNRFKGVKYFRLNTLASLGYVVVVIDNRGSXHRGLKFEGAFKYKMGQIEIDDQVEGLQYLASRYDFIDLDRVGIHGWSYGGYLSLMALMQRSDIFRVAIAGAPVTLWIFYDTGYTERYMGHPDQNEQGYYLGSVAMQAEKFPSEPNRLLLLHGFLDENVHFAHTSILLSFLVRAGKPYDLQIYPQERHSIRVPESGEHYELHLLHYLQENLGSRIAALKVSLRFLYEG Hydrogen bonds contact Hydrophobic contact | ||||
| 104 | Monoglyceride lipase (MAGL) | 3PE6 | 7.54 | |
Target general information Gen name MGLL Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Monoacylglycerol lipase; MGL; Lysophospholipaselike; Lysophospholipase-like; Lysophospholipase homolog; HUK5; HU-K5 Protein family AB hydrolase superfamily, Monoacylglycerol lipase family Biochemical class Carboxylic ester hydrolase Function Hydrolyzes the endocannabinoid 2-arachidonoylglycerol, and thereby contributes to the regulation of endocannabinoid signaling, nociperception and perception of pain. Regulates the levels of fatty acids that serve as signaling molecules and promote cancer cell migration, invasion and tumor growth. Converts monoacylglycerides to free fatty acids and glycerol. Related diseases Systemic lupus erythematosus 9 (SLEB9) [MIM:610927]: A chronic, relapsing, inflammatory, and often febrile multisystemic disorder of connective tissue, characterized principally by involvement of the skin, joints, kidneys and serosal membranes. It is of unknown etiology, but is thought to represent a failure of the regulatory mechanisms of the autoimmune system. The disease is marked by a wide range of system dysfunctions, an elevated erythrocyte sedimentation rate, and the formation of LE cells in the blood or bone marrow. {ECO:0000269|PubMed:17360460}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Immunodeficiency, common variable, 7 (CVID7) [MIM:614699]: A primary immunodeficiency characterized by antibody deficiency, hypogammaglobulinemia, recurrent bacterial infections and an inability to mount an antibody response to antigen. The defect results from a failure of B-cell differentiation and impaired secretion of immunoglobulins; the numbers of circulating B-cells is usually in the normal range, but can be low. {ECO:0000269|PubMed:22035880}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P07550; P37235 EC number EC 3.1.1.23 Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; Fatty acid biosynthesis; Fatty acid metabolism; Hydrolase; Lipid biosynthesis; Lipid degradation; Lipid metabolism; Membrane; Nitration; Phosphoprotein; Proteomics identification; Reference proteome; Serine esterase Protein physicochemical properties Chain ID A Molecular weight (Da) 31808.4 Length 289 Aromaticity 0.08 Instability index 29.7 Isoelectric point 6.73 Charge (pH=7) -0.91 2D Binding mode Binding energy (Kcal/mol) -10.29 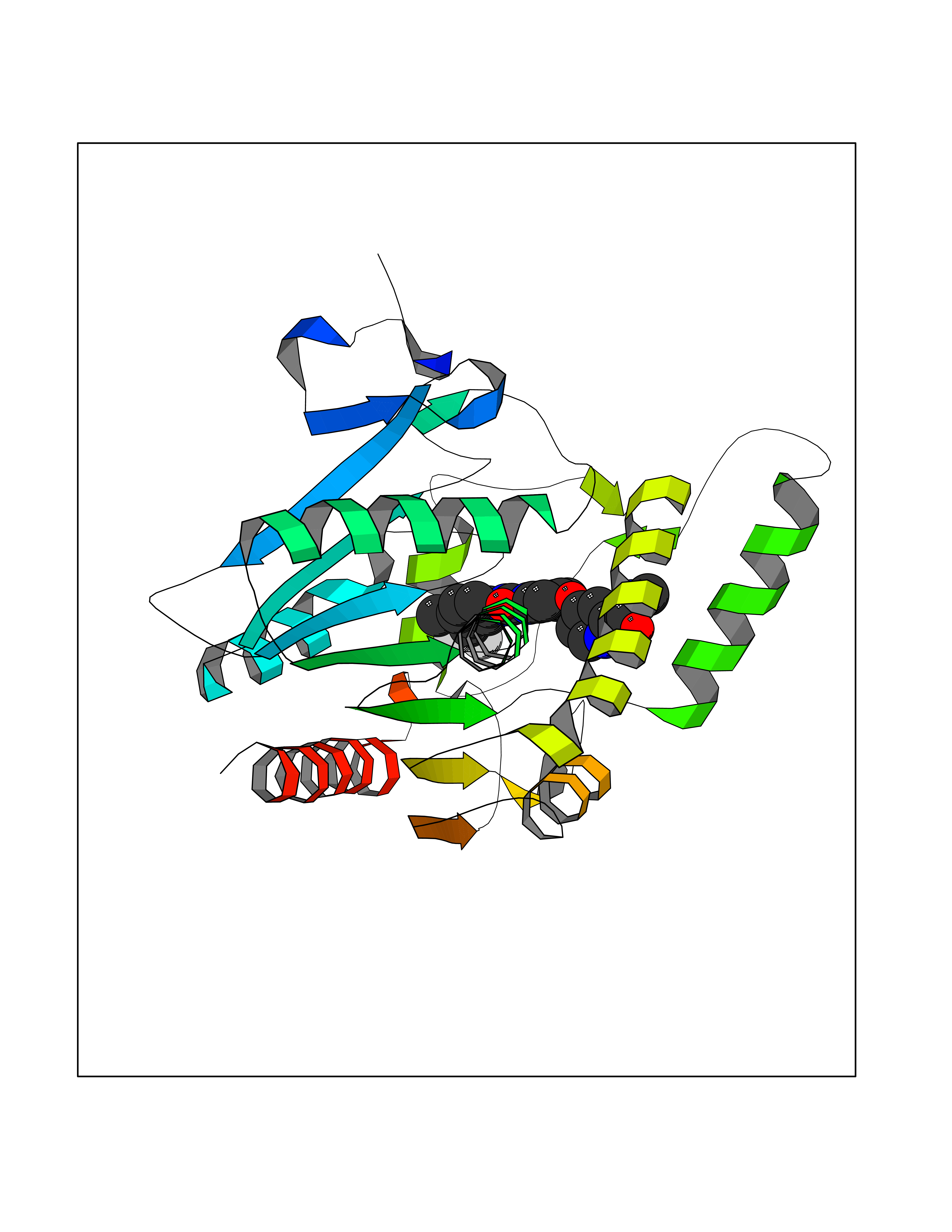 Molscript Map  Pymol Map 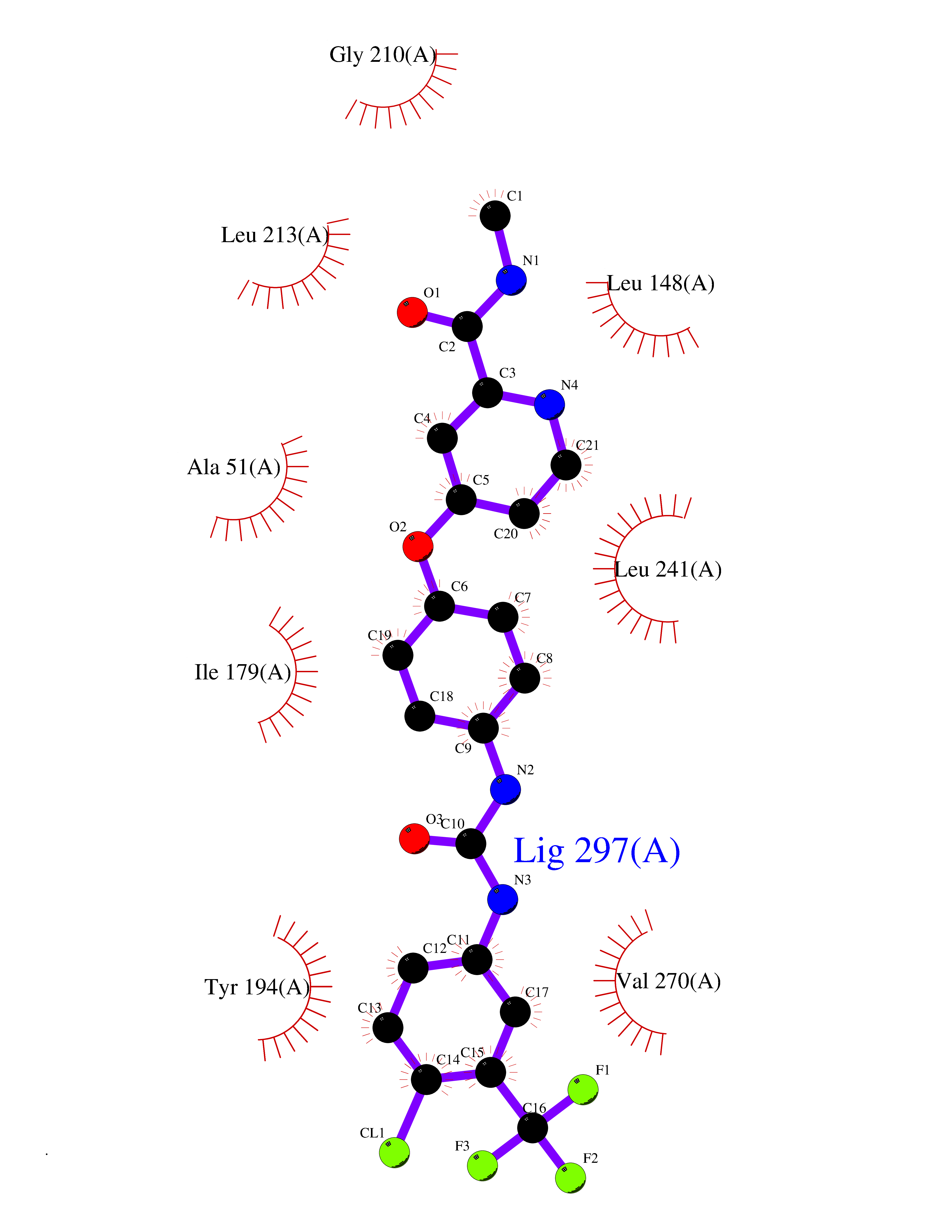 Ligplot Map 3D Binding mode Sequence PRRTPQSIPYQDLPHLVNADGQYLFCRYWAPTGTPKALIFVSHGAGEHSGRYEELARMLMGLDLLVFAHDHVGHGQSEGERMVVSDFHVFVRDVLQHVDSMQKDYPGLPVFLLGHSMGGAIAILTAAERPGHFAGMVLISPLVLANPESATTFKVLAAKVLNSVLPNLSSGPIDSSVLSRNKTEVDIYNSDPLICRAGLKVCFGIQLLNAVSRVERALPKLTVPFLLLQGSADRLCDSKGAYLLMELAKSQDKTLKIYEGAYHVLHKELPEVTNSVFHEINMWVSQRTA Hydrogen bonds contact Hydrophobic contact | ||||
| 105 | MLK-related kinase (MLTK) | 5HES | 7.54 | |
Target general information Gen name MAP3K20 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms ZAK; Sterile alpha motif- and leucine zipper-containing kinase AZK; Mixed lineage kinase-related kinase; Mitogen-activated protein kinase kinase kinase MLT; Mitogen-activated protein kinase kinase kin Protein family Protein kinase superfamily, STE Ser/Thr protein kinase family, MAP kinase kinase kinase subfamily Biochemical class NA Function Stress-activated component of a protein kinase signal transduction cascade. Regulates the JNK and p38 pathways. Part of a signaling cascade that begins with the activation of the adrenergic receptor ADRA1B and leads to the activation of MAPK14. Pro-apoptotic. Role in regulation of S and G2 cell cycle checkpoint by direct phosphorylation of CHEK2. Involved in limb development. Related diseases Split-foot malformation with mesoaxial polydactyly (SFMMP) [MIM:616890]: An autosomal recessive disorder characterized by a split-foot defect, mesoaxial polydactyly, nail abnormalities of the hands, and sensorineural hearing loss. {ECO:0000269|PubMed:26755636, ECO:0000269|PubMed:32266845}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Myopathy, centronuclear, 6, with fiber-type disproportion (CNM6) [MIM:617760]: A form of centronuclear myopathy, a congenital muscle disorder characterized by progressive muscular weakness and wasting involving mainly limb girdle, trunk, and neck muscles. It may also affect distal muscles. Weakness may be present during childhood or adolescence or may not become evident until the third decade of life. Ptosis is a frequent clinical feature. The most prominent histopathologic features include high frequency of centrally located nuclei in muscle fibers not secondary to regeneration, radial arrangement of sarcoplasmic strands around the central nuclei, and predominance and hypotrophy of type 1 fibers. CNM6 is an autosomal recessive, slowly progressive form with onset in infancy or early childhood. {ECO:0000269|PubMed:27816943, ECO:0000269|PubMed:30237576}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01254; DB12010 Interacts with O75582; P31947; P63104; Q8N184; Q16512; Q6P2D0; Q6ZN57; P13682; Q8N184; Q6AZW8; Q9NQZ8 EC number EC 2.7.11.25 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; ATP-binding; Cell cycle; Cytoplasm; Disease variant; Kinase; Magnesium; Metal-binding; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; RNA-binding; rRNA-binding; Serine/threonine-protein kinase; Transferase Protein physicochemical properties Chain ID A,B Molecular weight (Da) 64591.5 Length 566 Aromaticity 0.1 Instability index 52.08 Isoelectric point 5.69 Charge (pH=7) -12.83 2D Binding mode Binding energy (Kcal/mol) -10.29 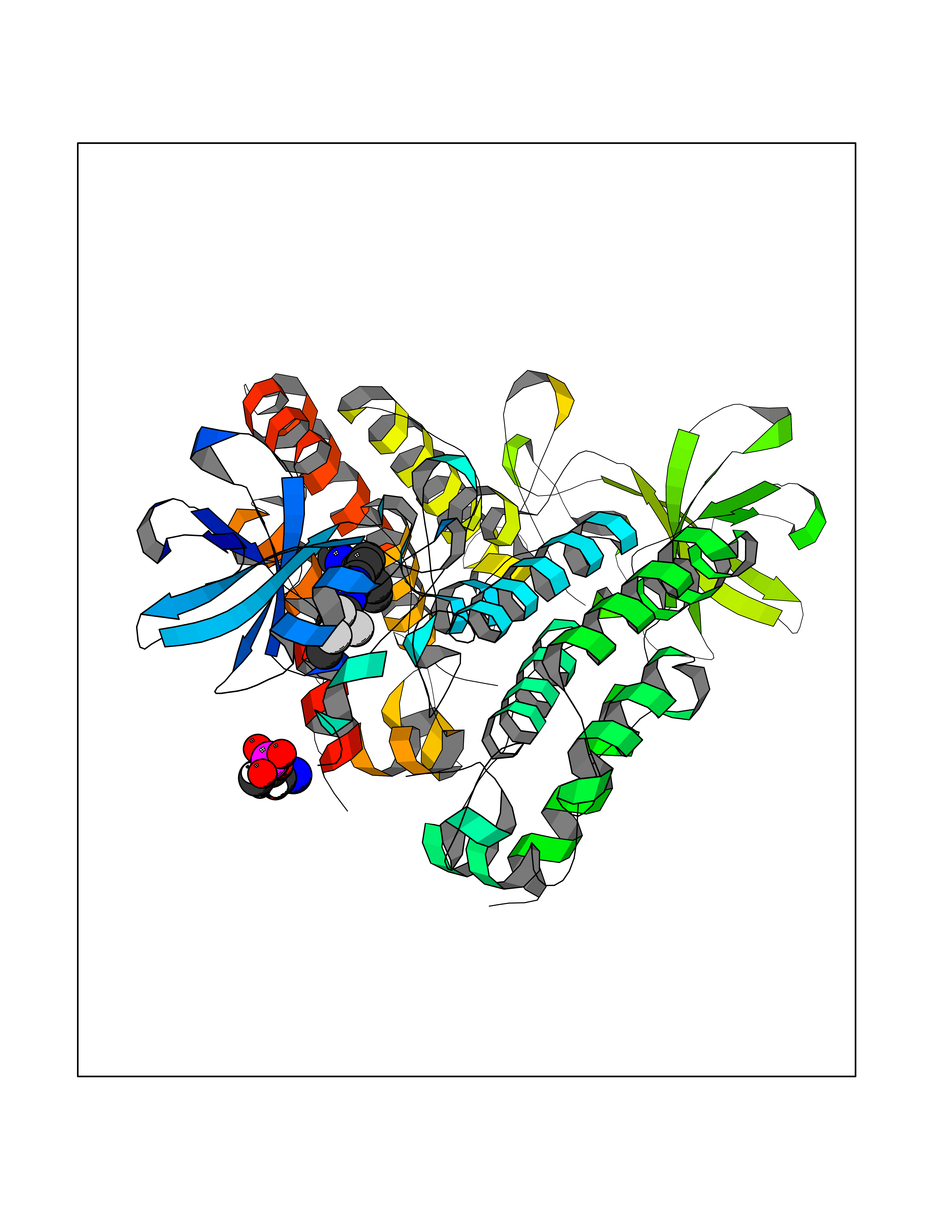 Molscript Map 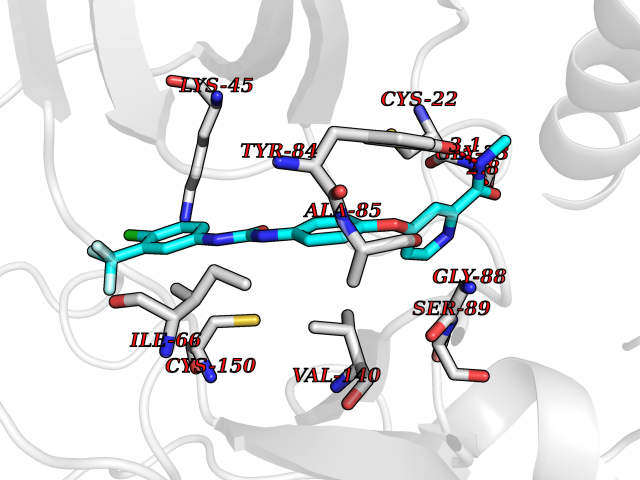 Pymol Map 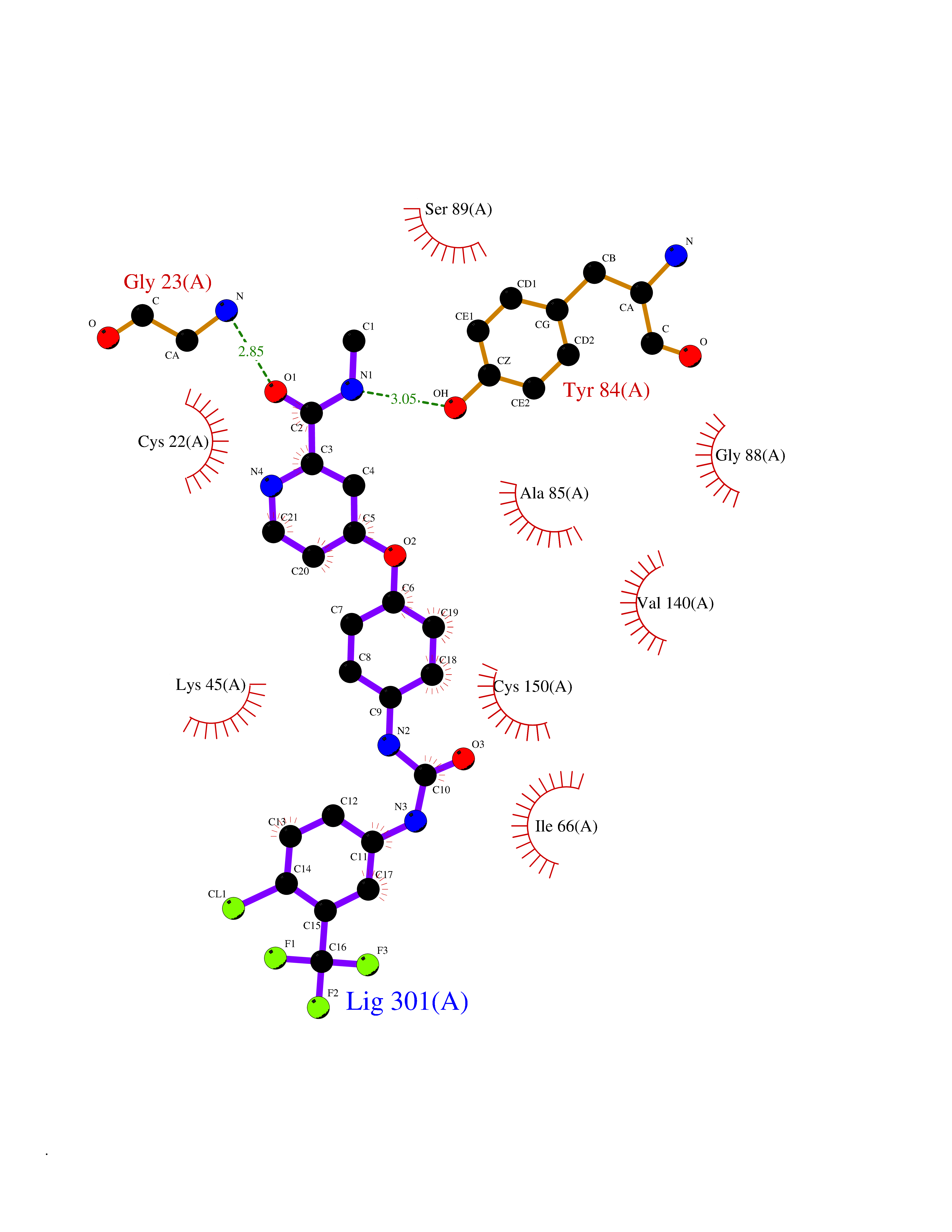 Ligplot Map 3D Binding mode Sequence ASFVQIKFDDLQFFENCGGGSFGSVYRAKWISQDKEVAVKKLLKIEKEAEILSVLSHRNIIQFYGVILEPPNYGIVTEYASLGSLYDYINSNRSEEMDMDHIMTWATDVAKGMHYLHMEAPVKVIHRDLKSRNVVIAADGVLKICDFGASRFHNHXGTFPWMAPEVIQSLPVSETCDTYSYGVVLWEMLTREVPFKGLEGLQVAWLVVEKNERLTIPSSCPRSFAELLHQCWEADAKKRPSFKQIISILESMSNDTSLPDKCNSFLHNKAEWRCEIEATLERLKKLERSFVQIKFDDLQFFENCGGGSFGSVYRAKWISQDKEVAVKKLLKIEKEAEILSVLSHRNIIQFYGVILEPPNYGIVTEYASLGSLYDYINSNRSEEMDMDHIMTWATDVAKGMHYLHMEAPVKVIHRDLKSRNVVIAADGVLKICDFGGTFPWMAPEVIQSLPVSETCDTYSYGVVLWEMLTREVPFKGLEGLQVAWLVVEKNERLTIPSSCPRSFAELLHQCWEADAKKRPSFKQIISILESMSNDTSLPDKCNSFLHNKAEWRCEIEATLERLKKLE Hydrogen bonds contact Hydrophobic contact | ||||
| 106 | Orexin receptor type 2 (HCRTR2) | 4S0V | 7.53 | |
Target general information Gen name HCRTR2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Ox2r; Ox2-R; Ox-2-R; Orexin-2 receptor; Hypocretin receptor type 2; HFGANP Protein family G-protein coupled receptor 1 family Biochemical class GPCR rhodopsin Function Triggers an increase in cytoplasmic Ca(2+) levels in response to orexin-A binding. Nonselective, high-affinity receptor for both orexin-A and orexin-B neuropeptides. Related diseases Hyperchlorhidrosis, isolated (HYCHL) [MIM:143860]: An autosomal recessive disorder characterized by excessive sweating and increased sweat chloride levels. Affected individuals suffer from episodes of hyponatremic dehydration and report increased amounts of visible salt precipitates in sweat. {ECO:0000269|PubMed:21035102, ECO:0000269|PubMed:21184099, ECO:0000269|PubMed:26911677}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB15031; DB11951; DB09034 Interacts with P62937 EC number NA Uniprot keywords 3D-structure; Cell membrane; Disulfide bond; G-protein coupled receptor; Glycoprotein; Membrane; Receptor; Reference proteome; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 32524.7 Length 282 Aromaticity 0.14 Instability index 38.96 Isoelectric point 9.18 Charge (pH=7) 12.36 2D Binding mode Binding energy (Kcal/mol) -10.26 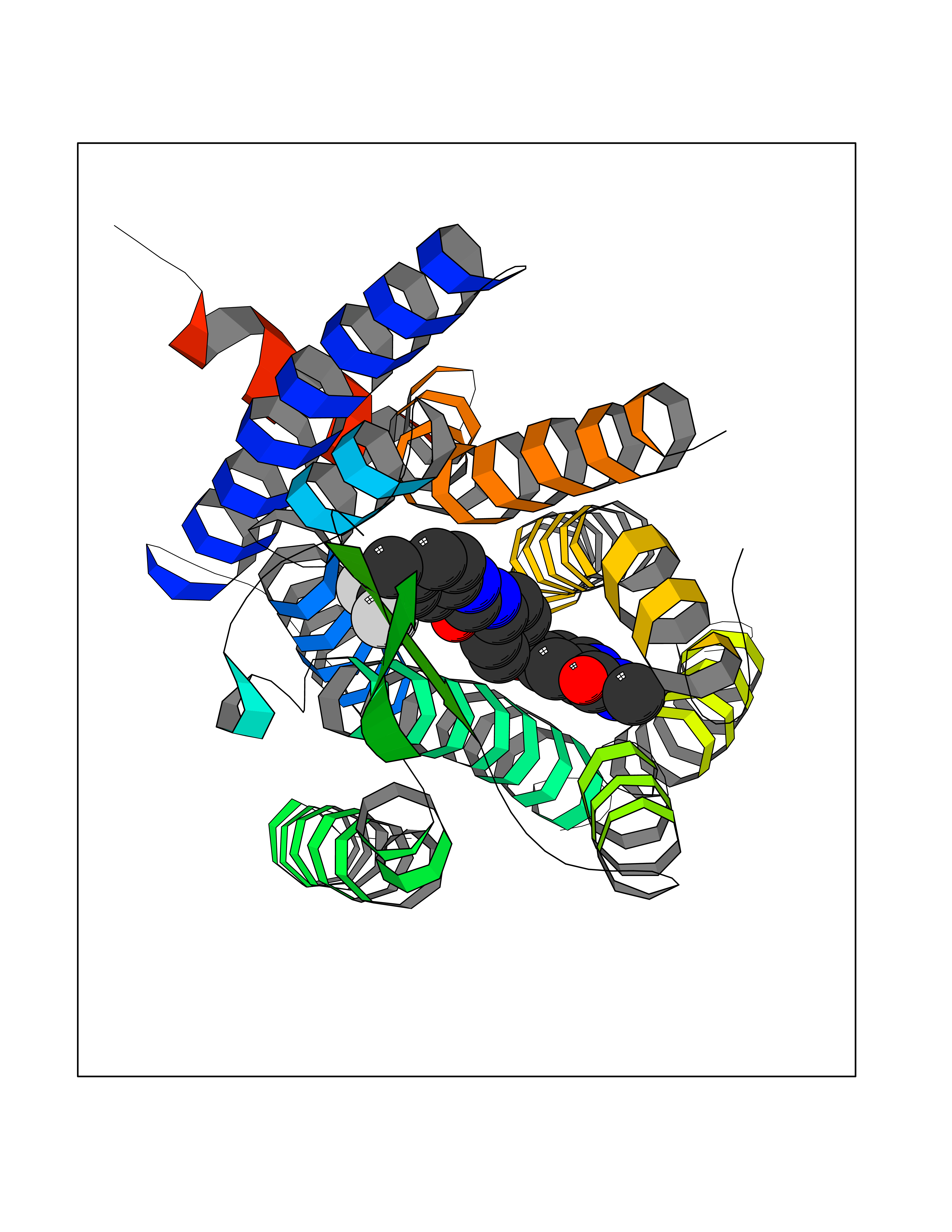 Molscript Map 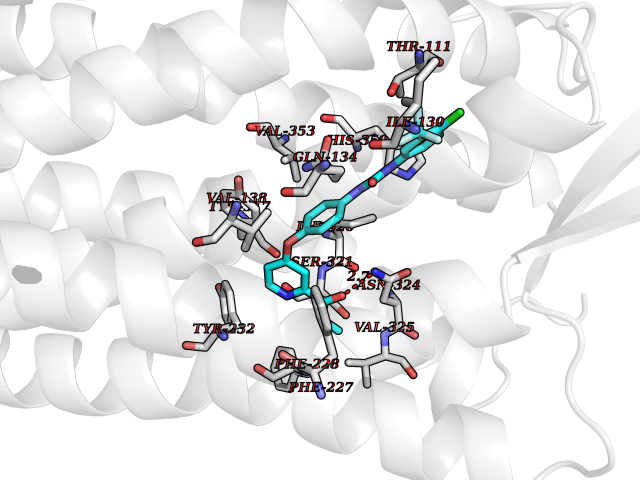 Pymol Map 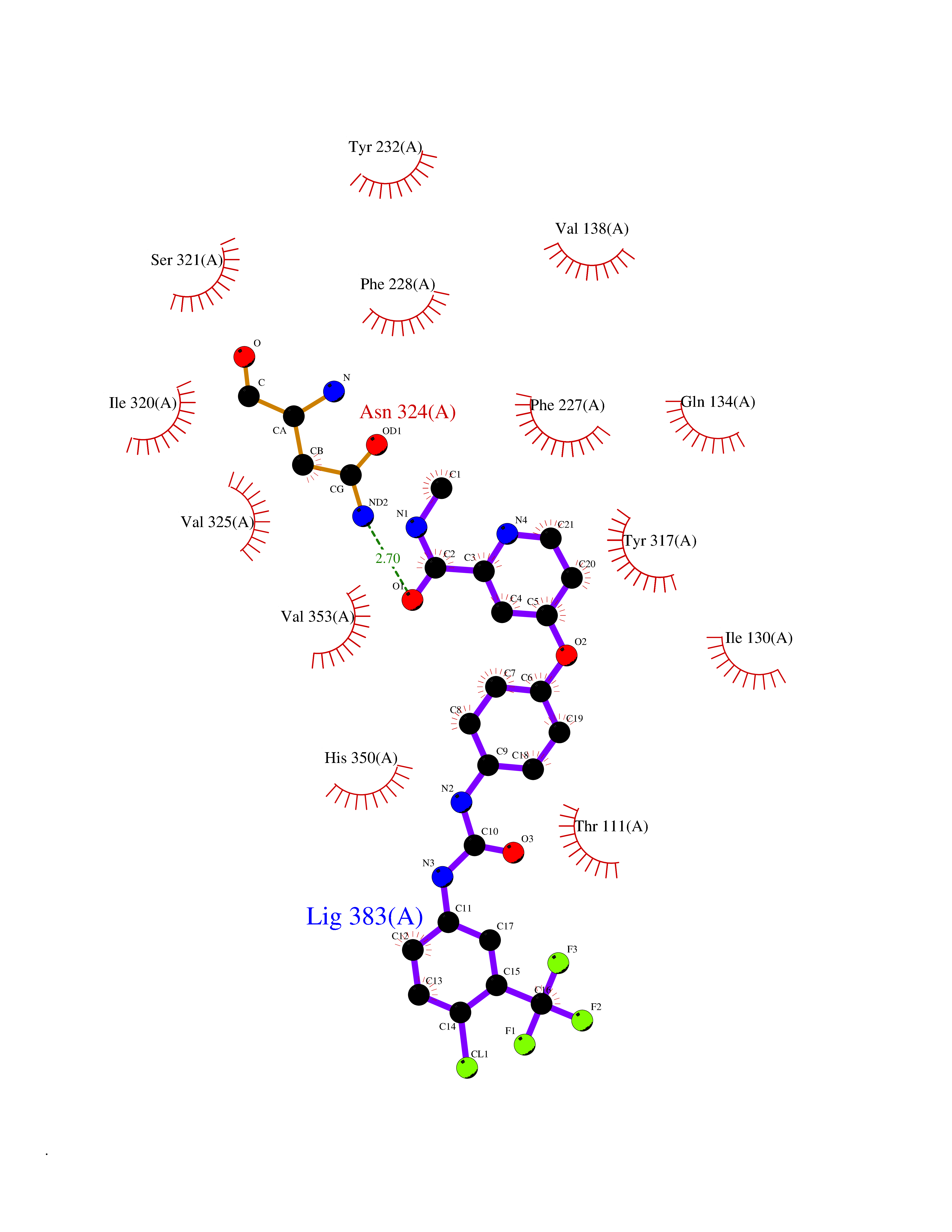 Ligplot Map 3D Binding mode Sequence PKEYEWVLIAGYIIVFVVALIGNVLVCVAVWKNHHMRTVTNYFIVNLSLADVLVTITCLPATLVVDITETWFFGQSLCKVIPYLQTVSVSVSVLTLSCIALDRWYAICHPSTAKRARNSIVIIWIVSCIIMIPQAIVMECSTVFKTTLFTVCDERWGGEIYPKMYHICFFLVTYMAPLCLMVLAYLQIFRKLWCRQKQIRARRKTARMLMVVLLVFAICYLPISILNVLKRVFGMFAHDRETVYAWFTFSHWLVYANSAANPIIYNFLSGKFREEFKAAFSC Hydrogen bonds contact Hydrophobic contact | ||||
| 107 | Glutamate receptor ionotropic NMDA 2A (NMDAR2A) | 5KCJ | 7.53 | |
Target general information Gen name GRIN2A Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms NR2A; NMDA receptor NR2A; N-methyl D-aspartate receptor subtype 2A; HNR2A; Glutamate receptor ionotropic, NMDA 2A; Glutamate [NMDA] receptor subunit epsilon-1; GluN2A Protein family Glutamate-gated ion channel (TC 1.A.10.1) family, NR1/GRIN1 subfamily Biochemical class Glutamate-gated ion channel Function Channel activation requires binding of the neurotransmitter glutamate to the epsilon subunit, glycine binding to the zeta subunit, plus membrane depolarization to eliminate channel inhibition by Mg(2+). Sensitivity to glutamate and channel kinetics depend on the subunit composition; channels containing GRIN1 and GRIN2A have higher sensitivity to glutamate and faster kinetics than channels formed by GRIN1 and GRIN2B. Contributes to the slow phase of excitatory postsynaptic current, long-term synaptic potentiation, and learning. Component of NMDA receptor complexes that function as heterotetrameric, ligand-gated ion channels with high calcium permeability and voltage-dependent sensitivity to magnesium. Related diseases Neurodevelopmental disorder with or without hyperkinetic movements and seizures, autosomal dominant (NDHMSD) [MIM:614254]: An autosomal dominant neurodevelopmental disorder characterized by severe intellectual disability and developmental delay, absent speech, muscular hypotonia, dyskinesia, and hyperkinetic movements. Cortical blindness, cerebral atrophy, and seizures are present in some patients. {ECO:0000269|PubMed:21376300, ECO:0000269|PubMed:25167861, ECO:0000269|PubMed:25864721, ECO:0000269|PubMed:27164704, ECO:0000269|PubMed:28095420, ECO:0000269|PubMed:28228639, ECO:0000269|PubMed:28389307, ECO:0000269|PubMed:38538865}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Neurodevelopmental disorder with or without hyperkinetic movements and seizures, autosomal recessive (NDHMSR) [MIM:617820]: An autosomal recessive neurodevelopmental disorder characterized by severe intellectual disability and psychomotor developmental delay, involuntary and stereotypic movements, spasticity, and inability to walk without support. Intractable seizures manifest in some patients. {ECO:0000269|PubMed:27164704, ECO:0000269|PubMed:28051072}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Developmental and epileptic encephalopathy 101 (DEE101) [MIM:619814]: A form of epileptic encephalopathy, a heterogeneous group of early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE101 is an autosomal recessive, severe form characterized by onset of seizures in early infancy. Death in infancy may occur. {ECO:0000269|PubMed:27164704, ECO:0000269|PubMed:34611970}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01931; DB00659; DB06151; DB08838; DB01238; DB00289; DB05824; DB04620; DB03929; DB00647; DB00843; DB00228; DB11823; DB13146; DB06741; DB00142; DB00874; DB08954; DB06738; DB09409; DB09481; DB01043; DB00454; DB00333; DB04896; DB01173; DB00312; DB01174; DB01708; DB00418; DB00193 Interacts with P05067; P35637; Q12879-1; Q13224; Q62936 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Calcium; Cell membrane; Disease variant; Disulfide bond; Epilepsy; Glycoprotein; Intellectual disability; Ion channel; Ion transport; Ligand-gated ion channel; Magnesium; Membrane; Metal-binding; Phosphoprotein; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Signal; Synapse; Transmembrane; Transmembrane helix; Transport; Zinc Protein physicochemical properties Chain ID B Molecular weight (Da) 53395.6 Length 469 Aromaticity 0.11 Instability index 29.84 Isoelectric point 8.72 Charge (pH=7) 5.65 2D Binding mode Binding energy (Kcal/mol) -10.27  Molscript Map 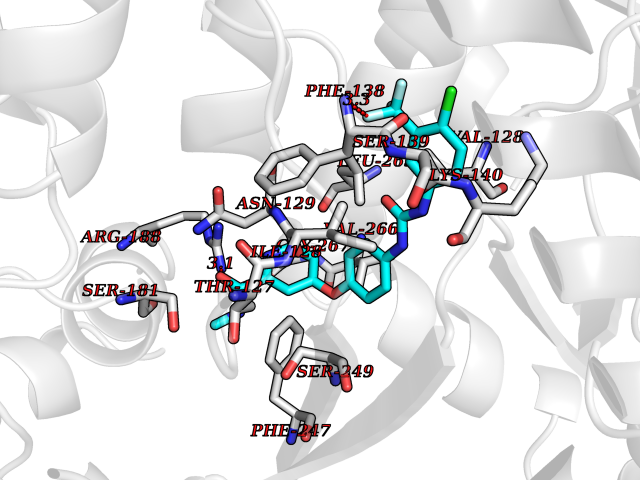 Pymol Map 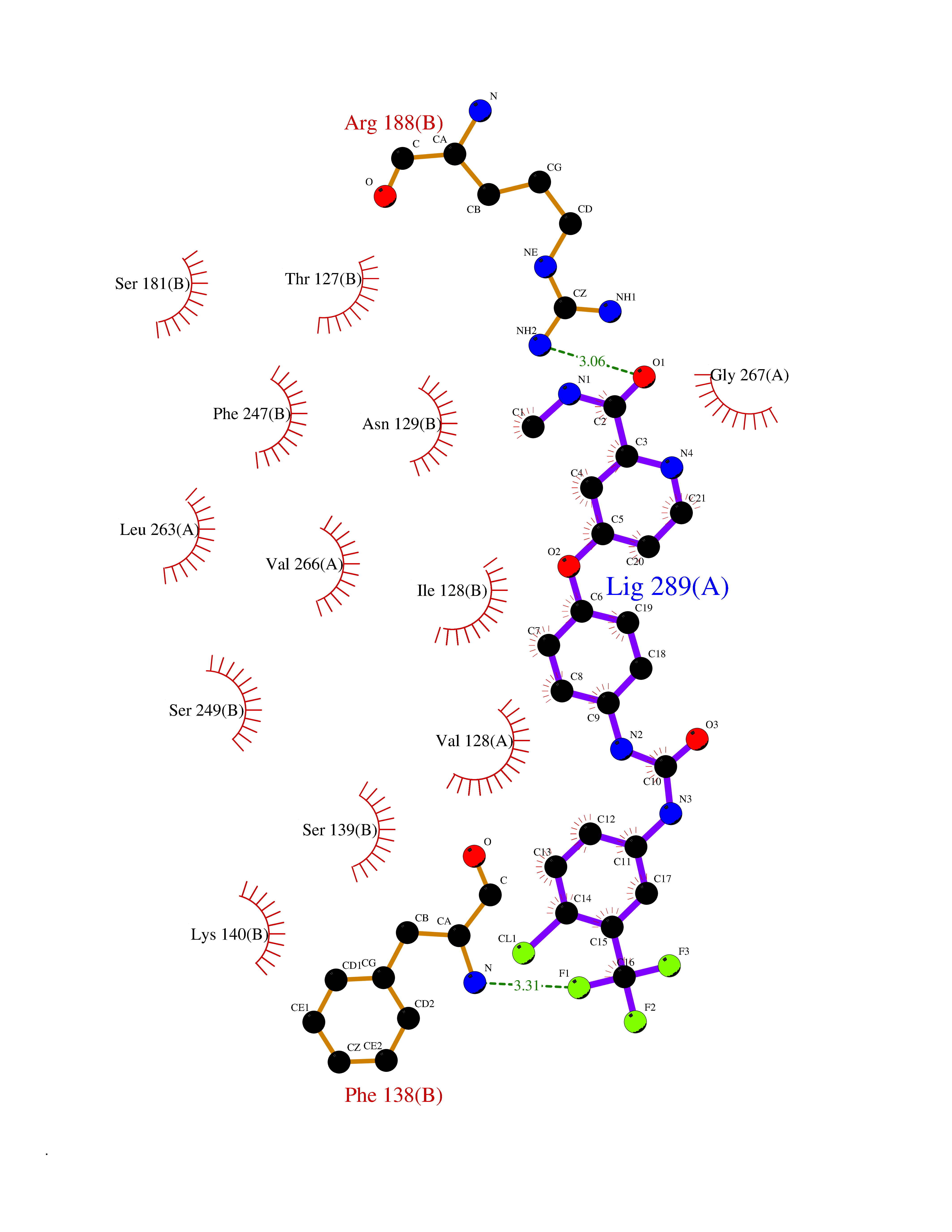 Ligplot Map 3D Binding mode Sequence DNHLSIVTLEEAPFVILKKLSRTVKFTYDLYLVTNGKHGKKVNNVWNGMIGEVVYQRAVMAVGSLTINEERSEVVDFSVPFVETGISVMVSRGTQVTGLSDKKFQRPHDYSPPFRFGTVPNGSTERNIRNNYPYMHQYMTKFNQKGVEDALVSLKTGKLDAFIYDAAVLNYKAGRDEGCKLVTIGSGYIFATTGYGIALQKGSPWKRQIDLALLQFVGDGEMEELETLWLTGICTRLKIVTIHQEPFVYYGFCIDLLIKLARTMNFTYEVHLVADGKFGTQERVNKKEWNGMMGELLSGQADMIVAPLTINNERAQYIEFSKPFKYQGLTILVKKGTRITGINDPRLRNPSDKFIYATVKQSSVDIYFRRQVELSTMYRHMEKHNYESAAEAIQAVRDNKLHAFIWDSAVLEFEASQKCDLVTTGELFFRSGFGIGMRKDSPWKQNVSLSILKSHENGFMEDLDKTWVR Hydrogen bonds contact Hydrophobic contact | ||||
| 108 | MAP kinase signal-integrating kinase 2 (MKNK2) | 6CK6 | 7.52 | |
Target general information Gen name MKNK2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Mnk2; MAPK signal-integrating kinase 2 Protein family Protein kinase superfamily, CAMK Ser/Thr protein kinase family Biochemical class Protein kinase superfamily. CAMK Ser/Thr protein kinase family Function Serine/threonine-protein kinase that phosphorylates SFPQ/PSF, HNRNPA1 and EIF4E. May play a role in the response to environmental stress and cytokines. Appears to regulate translation by phosphorylating EIF4E, thus increasing the affinity of this protein for the 7-methylguanosine-containing mRNA cap. Required for mediating PP2A-inhibition-induced EIF4E phosphorylation. Triggers EIF4E shuttling from cytoplasm to nucleus. Isoform 1 displays a high basal kinase activity, but isoform 2 exhibits a very low kinase activity. Acts as a mediator of the suppressive effects of IFNgamma on hematopoiesis. Negative regulator for signals that control generation of arsenic trioxide As(2)O(3)-dependent apoptosis and anti-leukemic responses. Involved in anti-apoptotic signaling in response to serum withdrawal. Related diseases Charcot-Marie-Tooth disease, X-linked dominant, 6 (CMTX6) [MIM:300905]: A form of Charcot-Marie-Tooth disease, a disorder of the peripheral nervous system, characterized by progressive weakness and atrophy, initially of the peroneal muscles and later of the distal muscles of the arms. Charcot-Marie-Tooth disease is classified in two main groups on the basis of electrophysiologic properties and histopathology: primary peripheral demyelinating neuropathies characterized by severely reduced motor nerve conduction velocities (NCVs) (less than 38m/s) and segmental demyelination and remyelination, and primary peripheral axonal neuropathies characterized by normal or mildly reduced NCVs and chronic axonal degeneration and regeneration on nerve biopsy. {ECO:0000269|PubMed:23297365}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB12010 Interacts with Q16539; P46379-2; O14901; Q14696; P25786 EC number EC 2.7.11.1 Uniprot keywords 3D-structure; Alternative splicing; Apoptosis; ATP-binding; Cytoplasm; Kinase; Magnesium; Metal-binding; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Serine/threonine-protein kinase; Transferase; Translation regulation; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 30812.8 Length 271 Aromaticity 0.1 Instability index 60.57 Isoelectric point 5.48 Charge (pH=7) -10.33 2D Binding mode Binding energy (Kcal/mol) -10.26  Molscript Map 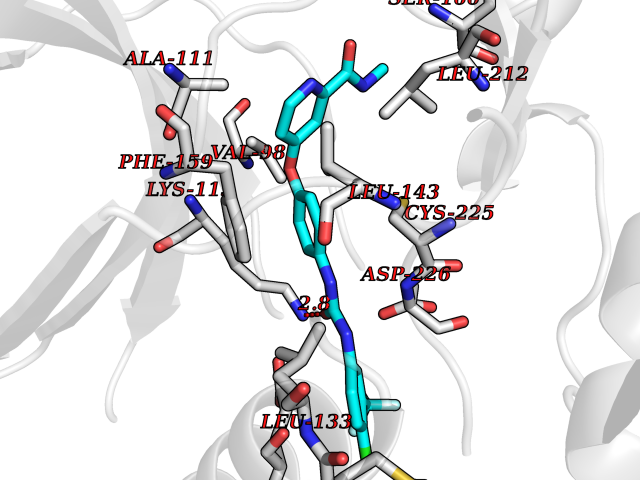 Pymol Map 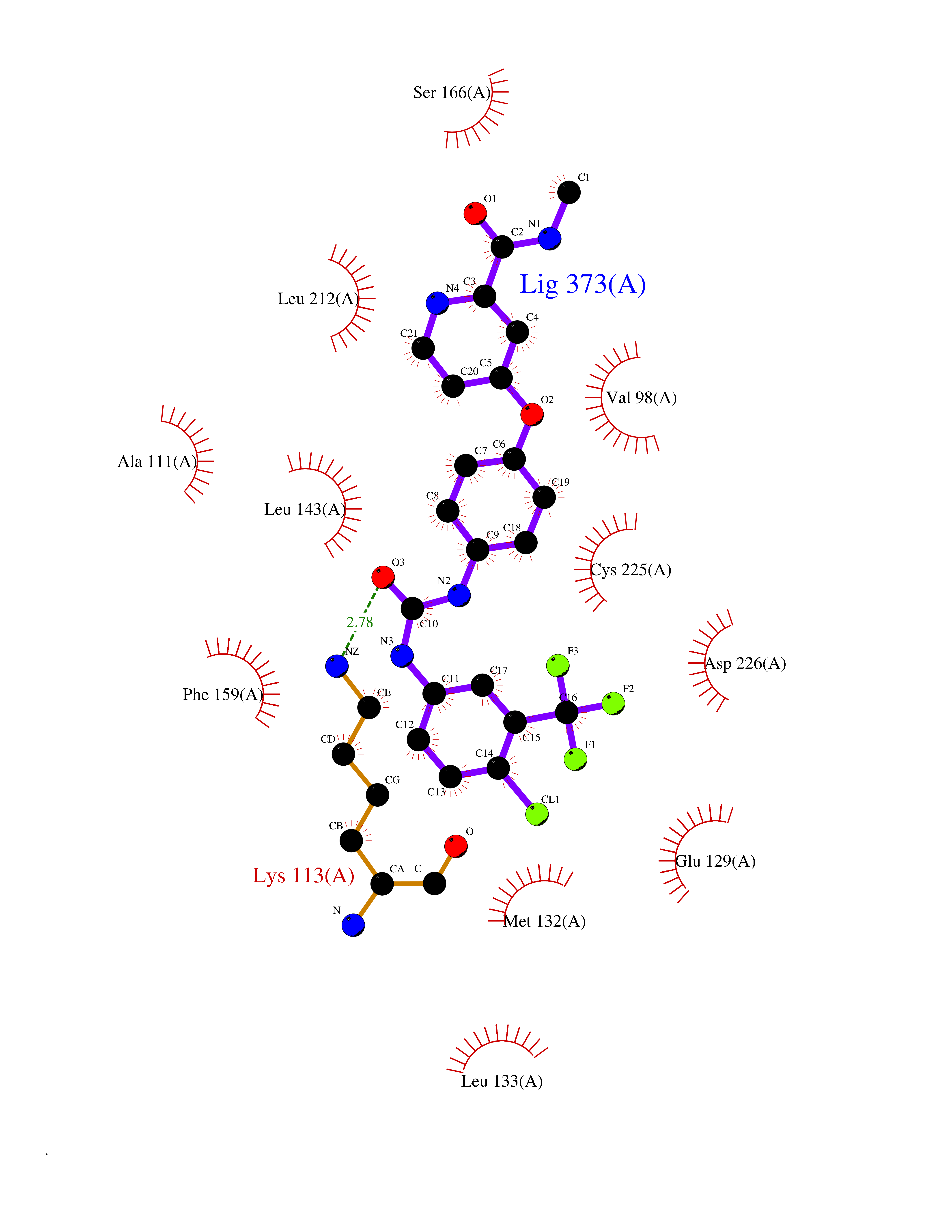 Ligplot Map 3D Binding mode Sequence GSTDSFSGRFEDVYQLQEDVLGEGAHARVQTCINLITSQEYAVKIIEKQPGHIRSRVFREVEMLYQCQGHRNVLELIEFFEEEDRFYLVFEKMRGGSILSHIHKRRHFNELEASVVVQDVASALDFLHNKGIAHRDLKPENILCEHPNQVSPVKICDFGGSAEYMAPEVVEAFSEEASIYDKRCDLWSLGVILYILLSGYPPFVGRCCGACPACQNMLFESIQEGKYEFPDKDWAHISCAAKDLISKLLVRDAKQRLSAAQVLQHPWVQGC Hydrogen bonds contact Hydrophobic contact | ||||
| 109 | Lanosterol 14-alpha demethylase | 5JLC | 7.51 | |
Target general information Gen name ERG11 Organism Candida glabrata (strain ATCC 2001 / BCRC 20586 / JCM 3761 / NBRC 0622 / NRRL Y-65 / CBS 138) (Yeast) (Nakaseomyces glabratus) Uniprot ID TTD ID NA Synonyms CYP51;CAGL0E04334g Protein family Cytochrome P450 family Biochemical class Oxidoreductase / oxidoreducatse inhibitor Function Drug binding.Heme binding.Iron ion binding.Sterol 14-demethylase activity. Related diseases Cerebral creatine deficiency syndrome 3 (CCDS3) [MIM:612718]: An autosomal recessive disorder characterized by developmental delay/regression, intellectual disability, severe disturbance of expressive and cognitive speech, and severe depletion of creatine/phosphocreatine in the brain. Most patients develop a myopathy characterized by muscle weakness and atrophy later in life. {ECO:0000269|PubMed:11555793, ECO:0000269|PubMed:20682460, ECO:0000269|PubMed:22386973, ECO:0000269|PubMed:23660394, ECO:0000269|PubMed:23770102, ECO:0000269|PubMed:26490222, ECO:0000269|PubMed:27233232}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Fanconi renotubular syndrome 1 (FRTS1) [MIM:134600]: A form of Fanconi renotubular syndrome, a disease due to a generalized dysfunction of the proximal kidney tubule resulting in decreased solute and water reabsorption. Patients have polydipsia and polyuria with phosphaturia, glycosuria and aminoaciduria. They may develop hypophosphatemic rickets or osteomalacia, acidosis and a tendency toward dehydration. Some eventually develop renal insufficiency. FRTS1 inheritance is autosomal dominant. {ECO:0000269|PubMed:29654216}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB09040; DB11633; DB06636; DB01026; DB06820 Interacts with NA EC number 1.14.14.154 Uniprot keywords 3D-structure; Heme; Iron; Lipid biosynthesis; Lipid metabolism; Membrane; Metal-binding; Monooxygenase; Oxidoreductase; Reference proteome; Steroid biosynthesis; Steroid metabolism; Sterol biosynthesis; Sterol metabolism Protein physicochemical properties Chain ID A Molecular weight (Da) 54152.4 Length 469 Aromaticity 0.12 Instability index 33.13 Isoelectric point 6.62 Charge (pH=7) -1.72 2D Binding mode Binding energy (Kcal/mol) -10.24 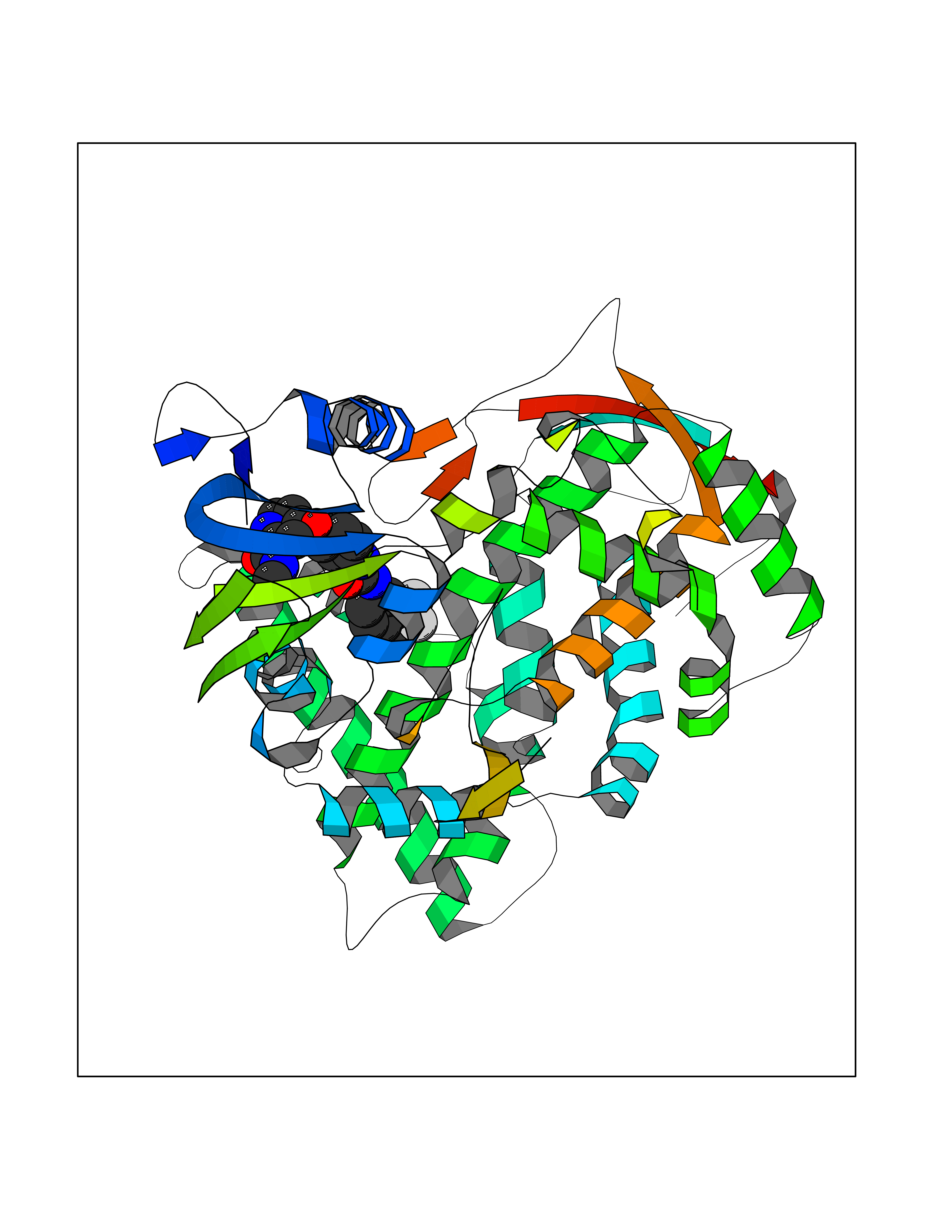 Molscript Map 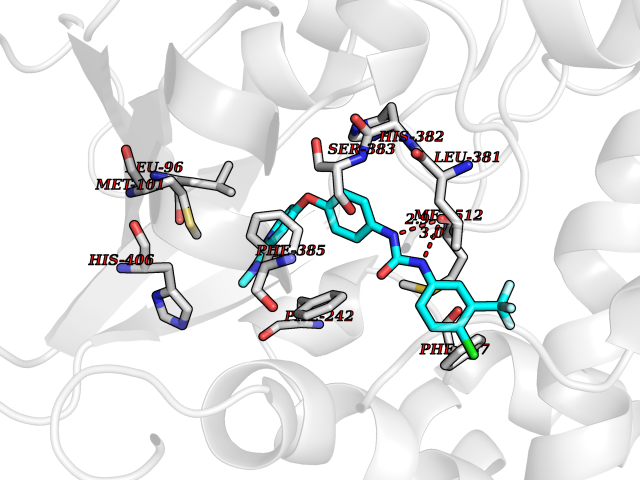 Pymol Map 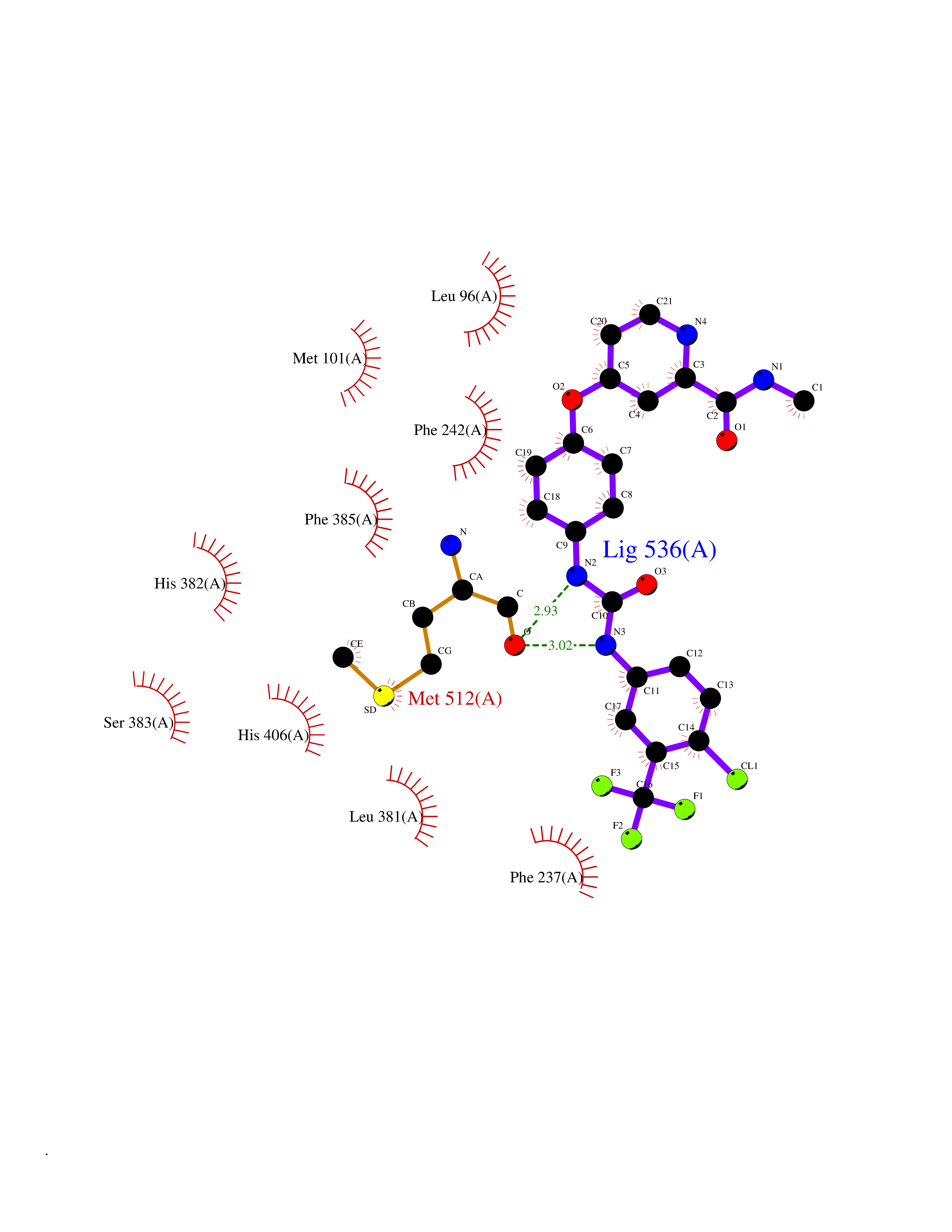 Ligplot Map 3D Binding mode Sequence PPLVFYWIPWVGSAIPYGTKPYEFFEDCQKKYGDIFSFMLLGRIMTVYLGPKGHEFIFNAKLADVSAEAAYSHLTTPVFGKGVIYDCPNHRLMEQKKFVKGALTKEAFVRYVPLIAEEIYKYFRNSKNFKINENNSGIVDVMVSQPEMTIFTASRSLLGKEMRDKLDTDFAYLYSDLDKGFTPINFVFPNLPLEHYRKRDHAQQAISGTYMSLIKERREKNDIQNRDLIDELMKNSTYKDGTKMTDQEIANLLIGVLMGGQHTSAATSAWCLLHLAERPDVQEELYQEQMRVLNNDTKELTYDDLQNMPLLNQMIKETLRLHHPLHSLFRKVMRDVAIPNTSYVVPRDYHVLVSPGYTHLQEEFFPKPNEFNIHRWDGGDEVDYGFGAISKGVSSPYLPFGGGRHRCIGELFAYCQLGVLMSIFIRTMKWRYPTEGETVPPSDFTSMVTLPTAPAKIYWEKRHPEQKYG Hydrogen bonds contact Hydrophobic contact | ||||
| 110 | Aldo-keto reductase family 1 member C1 | 1MRQ | 7.51 | |
Target general information Gen name AKR1C1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms DDH1;DDH Protein family Aldo/keto reductase family Biochemical class Oxidoreductase Function 17-alpha,20-alpha-dihydroxypregn-4-en-3-one dehydrogenase activity.Alditol:NADP+ 1-oxidoreductase activity.Aldo-keto reductase (NADP) activity.Androsterone dehydrogenase (B-specific) activity.Bile acid binding.Carboxylic acid binding.Indanol dehydrogenase activity.Ketosteroid monooxygenase activity.Oxidoreductase activity, acting on NAD(P)H, quinone or similar compound as acceptor.Phenanthrene 9,10-monooxygenase activity.Trans-1,2-dihydrobenzene-1,2-diol dehydrogenase activity. Related diseases Fibrodysplasia ossificans progressiva (FOP) [MIM:135100]: A rare autosomal dominant connective tissue disorder resulting in skeletal malformations and progressive extraskeletal ossification. Heterotopic ossification begins in childhood and can be induced by trauma or may occur without warning. Bone formation is episodic and progressive, leading to a debilitating ankylosis of all major joints of the axial and appendicular skeleton, rendering movement impossible. {ECO:0000269|PubMed:16642017, ECO:0000269|PubMed:19085907, ECO:0000269|PubMed:19330033}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB04674; DB00945; DB07768; DB01039; DB07931; DB06077; DB00959; DB00461; DB00157; DB03467; DB03461; DB00776; DB12612; DB00936 Interacts with P51857; P26045; Q7Z699 EC number 1.1.1.-; 1.1.1.112; 1.1.1.149; 1.1.1.209; 1.1.1.210; 1.1.1.357; 1.1.1.51; 1.1.1.53; 1.1.1.62; 1.3.1.20 Uniprot keywords 3D-structure; Cytoplasm; Direct protein sequencing; Lipid metabolism; NADP; Oxidoreductase; Proteomics identification; Reference proteome Protein physicochemical properties Chain ID A Molecular weight (Da) 36784.9 Length 323 Aromaticity 0.09 Instability index 42.07 Isoelectric point 8.06 Charge (pH=7) 2.42 2D Binding mode Binding energy (Kcal/mol) -10.24 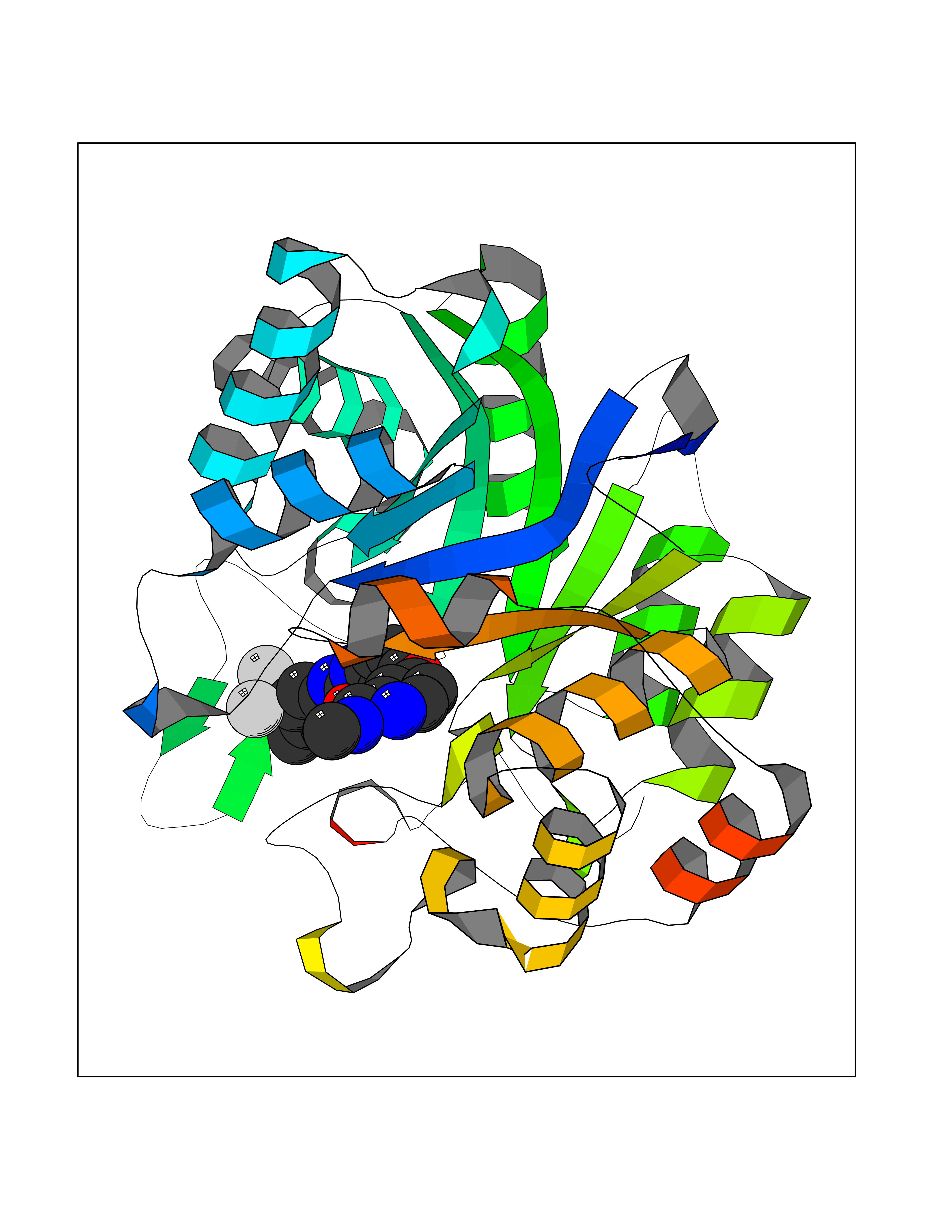 Molscript Map 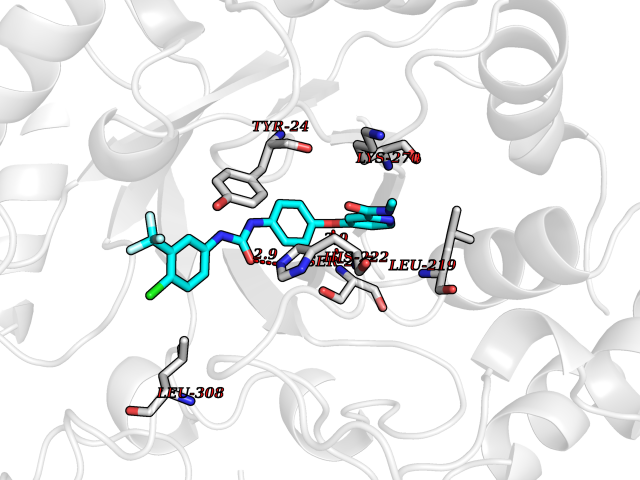 Pymol Map 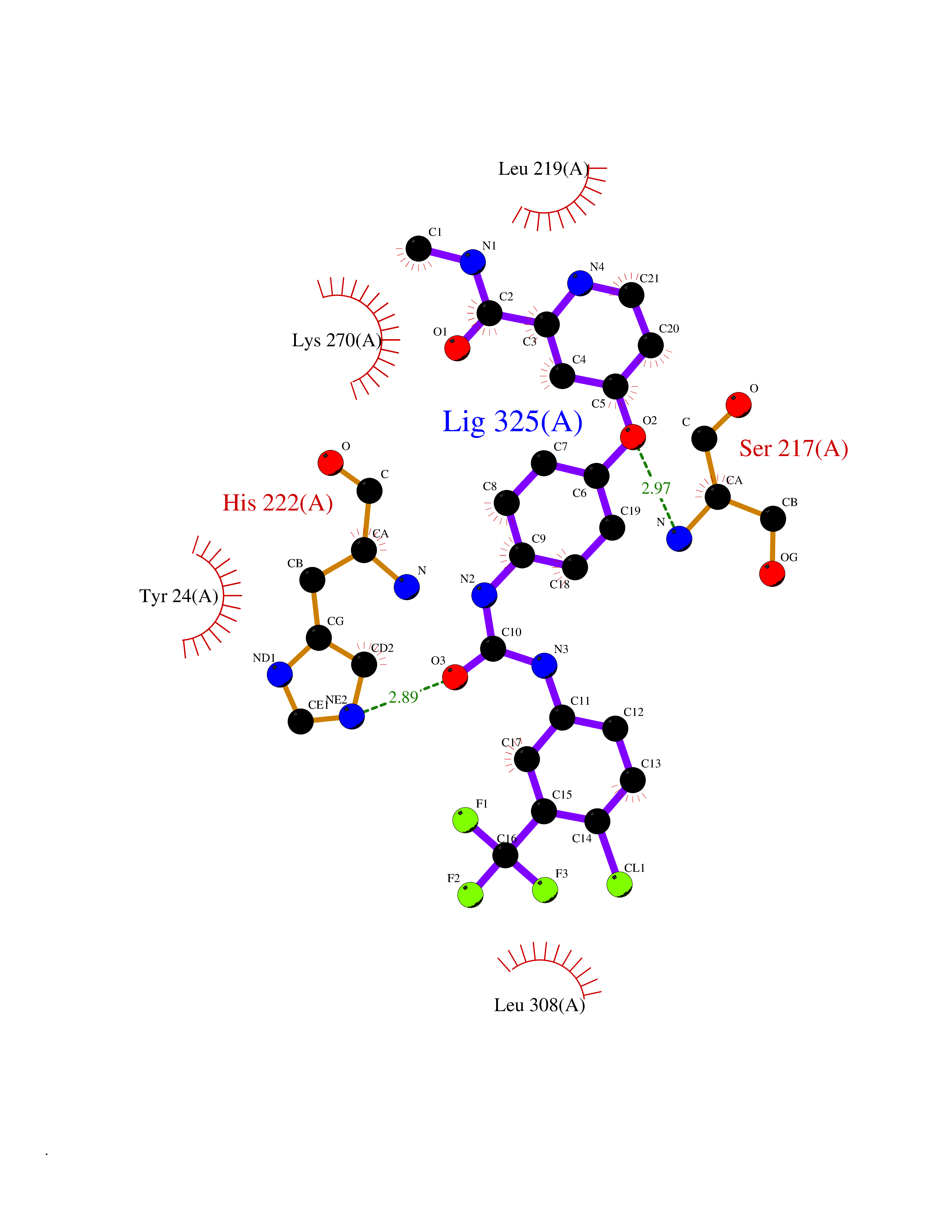 Ligplot Map 3D Binding mode Sequence QDSKYQCVKLNDGHFMPVLGFGTYAPAEVPKSKALEATKLAIEAGFRHIDSAHLYNNEEQVGLAIRSKIADGSVKREDIFYTSKLWCNSHRPELVRPALERSLKNLQLDYVDLYLIHFPVSVKPGEEVIPKDENGKILFDTVDLCATWEAVEKCKDAGLAKSIGVSNFNRRQLEMILNKPGLKYKPVCNQVECHPYFNQRKLLDFCKSKDIVLVAYSALGSHREEPWVDPNSPVLLEDPVLCALAKKHKRTPALIALRYQLQRGVVVLAKSYNEQRIRQNVQVFEFQLTSEEMKAIDGLNRNVRYLTLDIFAGPPNYPFSDEY Hydrogen bonds contact Hydrophobic contact | ||||
| 111 | Peroxisome proliferator-activated receptor alpha (PPARA) | 3VI8 | 7.51 | |
Target general information Gen name PPARA Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Peroxisome proliferater-activated receptor alpha; PPARalpha; PPAR-alpha; PPAR; Nuclear receptor subfamily 1 group C member 1; NR1C1 Protein family Nuclear hormone receptor family, NR1 subfamily Biochemical class Nuclear hormone receptor Function Key regulator of lipid metabolism. Activated by the endogenous ligand 1-palmitoyl-2-oleoyl-sn-glycerol-3-phosphocholine (16:0/18:1-GPC). Activated by oleylethanolamide, a naturally occurring lipid that regulates satiety. Receptor for peroxisome proliferators such as hypolipidemic drugs and fatty acids. Regulates the peroxisomal beta-oxidation pathway of fatty acids. Functions as transcription activator for the ACOX1 and P450 genes. Transactivation activity requires heterodimerization with RXRA and is antagonized by NR2C2. May be required for the propagation of clock information to metabolic pathways regulated by PER2. Ligand-activated transcription factor. Related diseases Combined oxidative phosphorylation deficiency 33 (COXPD33) [MIM:617713]: An autosomal recessive disorder caused by multiple mitochondrial respiratory chain defects and impaired mitochondrial energy metabolism. Clinical manifestations are highly variable. Affected infants present with cardiomyopathy accompanied by multisystemic features involving liver, kidney, and brain. Death in infancy is observed in some patients. Children and adults present with myopathy and progressive external ophthalmoplegia. {ECO:0000269|PubMed:28942965}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08915; DB00132; DB01118; DB04557; DB01393; DB04519; DB05416; DB09064; DB09006; DB00636; DB09213; DB03756; DB05187; DB06521; DB01039; DB13873; DB00573; DB13961; DB02266; DB01241; DB07215; DB01050; DB00159; DB07724; DB00328; DB12007; DB03017; DB12961; DB06510; DB08231; DB11605; DB01890; DB04224; DB11133; DB03796; DB02746; DB01708; DB06533; DB04971; DB02709; DB00412; DB09422; DB03193; DB06536; DB00197; DB00313 Interacts with P02768-3; P55212; P45973; P06307; Q3L8U1-3; G5E9A7; P22607; P62993; Q14957; P06396; P42858; Q8WXH2; P13473-2; O75376; Q13133; A0A6Q8PF08; P54725; P62826; Q7Z699; P37173; P55072; P55055-1; Q13133 EC number NA Uniprot keywords 3D-structure; Activator; Alternative splicing; Biological rhythms; DNA-binding; Lipid-binding; Metal-binding; Nucleus; Proteomics identification; Receptor; Reference proteome; Transcription; Transcription regulation; Ubl conjugation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A Molecular weight (Da) 29322.1 Length 258 Aromaticity 0.09 Instability index 35.53 Isoelectric point 6.09 Charge (pH=7) -3.57 2D Binding mode Binding energy (Kcal/mol) -10.25  Molscript Map 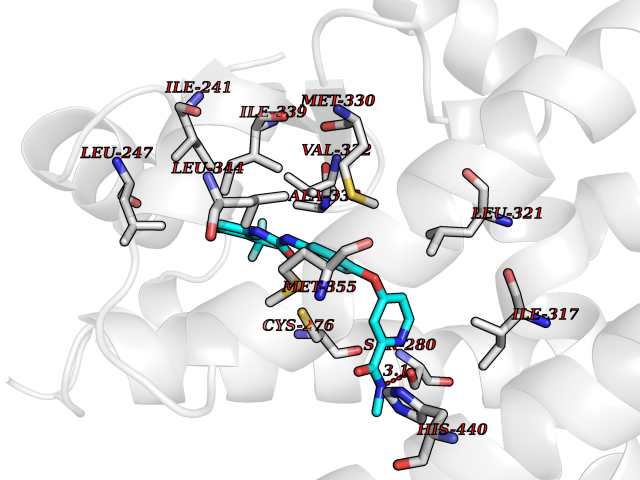 Pymol Map 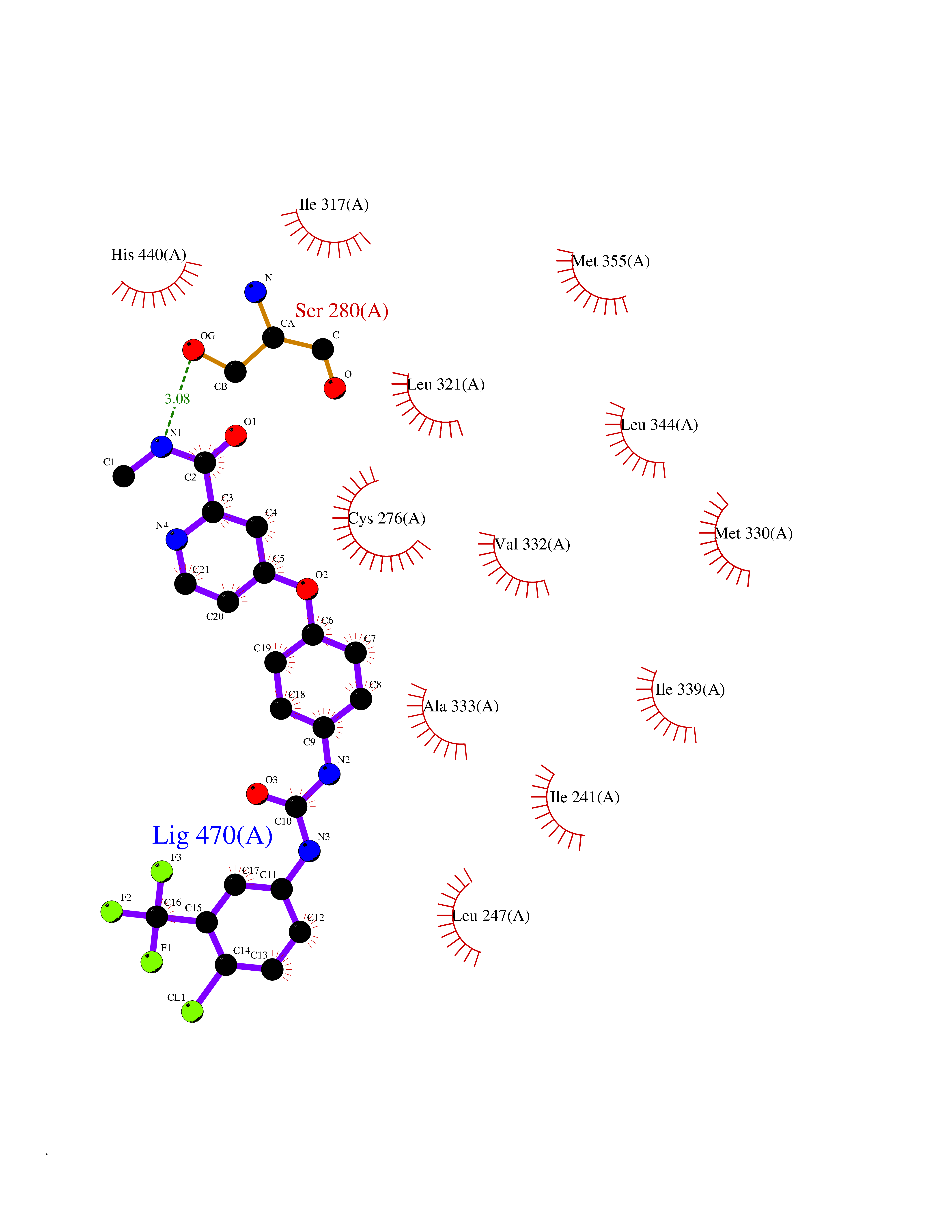 Ligplot Map 3D Binding mode Sequence DLKSLAKRIYEAYLKNFNMNKVKARVILSPFVIHDMETLCMAEKTLVAKLVANGNKEAEVRIFHCCQCTSVETVTELTEFAKAIPGFANLDLNDQVTLLKYGVYEAIFAMLSSVMNKDGMLVAYGNGFITREFLKSLRKPFCDIMEPKFDFAMKFNALELDDSDISLFVAAIICCGDRPGLLNVGHIEKMQEGIVHVLRLHLQSNHPDDIFLFPKLLQKMADLRQLVTEHAQLVQIIKKTESDAALHPLLQEIYRDMY Hydrogen bonds contact Hydrophobic contact | ||||
| 112 | Nicotinamide phosphoribosyltransferase (NAMPT) | 2E5D | 7.51 | |
Target general information Gen name NAMPT Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Visfatin; PreBcell colonyenhancing factor 1; PreB cellenhancing factor; Pre-B-cell colony-enhancing factor 1; Pre-B cell-enhancing factor; PBEF1; PBEF; Nampt; NAmPRTase Protein family NAPRTase family Biochemical class Glycosyltransferases Function It is the rate limiting component in the mammalian NAD biosynthesis pathway. The secreted form behaves both as a cytokine with immunomodulating properties and an adipokine with anti-diabetic properties, it has no enzymatic activity, partly because of lack of activation by ATP, which has a low level in extracellular space and plasma. Plays a role in the modulation of circadian clock function. NAMPT-dependent oscillatory production of NAD regulates oscillation of clock target gene expression by releasing the core clock component: CLOCK-ARNTL/BMAL1 heterodimer from NAD-dependent SIRT1-mediated suppression. Catalyzes the condensation of nicotinamide with 5-phosphoribosyl-1-pyrophosphate to yield nicotinamide mononucleotide, an intermediate in the biosynthesis of NAD. Related diseases Hemolytic anemia, non-spherocytic, due to glucose phosphate isomerase deficiency (HA-GPID) [MIM:613470]: A form of anemia in which there is no abnormal hemoglobin or spherocytosis. It is caused by glucose phosphate isomerase deficiency. {ECO:0000269|PubMed:28803808, ECO:0000269|PubMed:7989588, ECO:0000269|PubMed:8499925, ECO:0000269|PubMed:8822952, ECO:0000269|PubMed:8822954, ECO:0000269|PubMed:9446754, ECO:0000269|PubMed:9856489}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB12980; DB12731; DB05217 Interacts with P02792; Q01628; P03886; P43490; Q70CQ1-2 EC number EC 2.4.2.12 Uniprot keywords 3D-structure; Acetylation; Biological rhythms; Cytokine; Cytoplasm; Glycosyltransferase; Nucleus; Phosphoprotein; Proteomics identification; Pyridine nucleotide biosynthesis; Reference proteome; Secreted; Transferase Protein physicochemical properties Chain ID A,B Molecular weight (Da) 105483 Length 932 Aromaticity 0.11 Instability index 34.4 Isoelectric point 6.68 Charge (pH=7) -2.24 2D Binding mode Binding energy (Kcal/mol) -10.24  Molscript Map 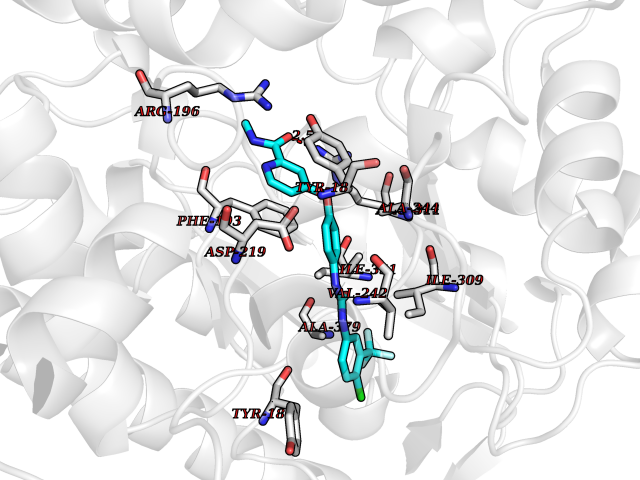 Pymol Map 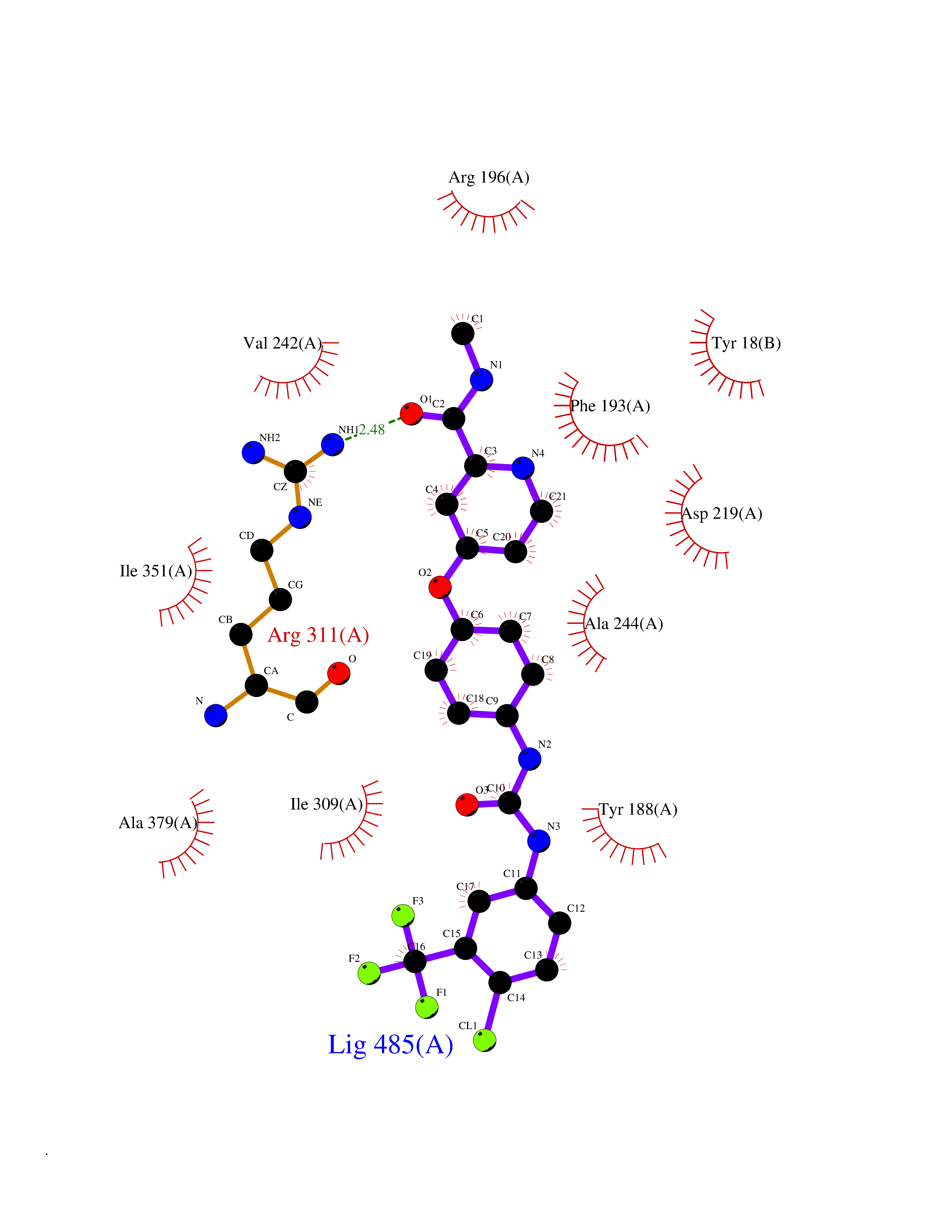 Ligplot Map 3D Binding mode Sequence EFNILLATDSYKVTHYKQYPPNTSKVYSYFECREKKYEETVFYGLQYILNKYLKGKVVTKEKIQEAKDVYKEHFQDDVFNEKGWNYILEKYDGHLPIEIKAVPEGFVIPRGNVLFTVENTDPECYWLTNWIETILVQSWYPITVATNSREQKKILAKYLLETSGNLDGLEYKLHDFGYRGVSSQETAGIGASAHLVNFKGTDTVAGLALIKKYYGTKDPVPGYSVPAAEHSTITAWGKDHEKDAFEHIVTQFSSVPVSVVSDSYDIYNACEKIWGEDLRHLIVSRSTQAPLIIRPDSGNPLDTVLKVLEILGKKFPVTENSKGYKLLPPYLRVIQGDGVDINTLQEIVEGMKQKMWSIENIAFGSGGGLLQKLTRDLLNCSFKCSYVVTNGLGINVFKDPVADPNKRSKKGRLSLHRTPAGNFVTLEEGKGDLEEYGQDLLHTVFKNGKVTKSYSFDEIRKNAQLNEFNILLATDSYKVTHYKQYPPNTSKVYSYFECREKKYEETVFYGLQYILNKYLKGKVVTKEKIQEAKDVYKEHFQDDVFNEKGWNYILEKYDGHLPIEIKAVPEGFVIPRGNVLFTVENTDPECYWLTNWIETILVQSWYPITVATNSREQKKILAKYLLETSGNLDGLEYKLHDFGYRGVSSQETAGIGASAHLVNFKGTDTVAGLALIKKYYGTKDPVPGYSVPAAEHSTITAWGKDHEKDAFEHIVTQFSSVPVSVVSDSYDIYNACEKIWGEDLRHLIVSRSTQAPLIIRPDSGNPLDTVLKVLEILGKKFPVTENSKGYKLLPPYLRVIQGDGVDINTLQEIVEGMKQKMWSIENIAFGSGGGLLQKLTRDLLNCSFKCSYVVTNGLGINVFKDPVADPNKRSKKGRLSLHRTPAGNFVTLEEGKGDLEEYGQDLLHTVFKNGKVTKSYSFDEIRKNAQLN Hydrogen bonds contact Hydrophobic contact | ||||
| 113 | Serine/threonine-protein kinase WNK1 (WNK1) | 5WE8 | 7.51 | |
Target general information Gen name WNK1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms p65; hWNK1; Protein kinase with no lysine 1; Protein kinase lysine-deficient 1; Kinase deficient protein; KDP; HSN2; Erythrocyte 65 kDa protein Protein family Protein kinase superfamily, Ser/Thr protein kinase family, WNK subfamily Biochemical class NA Function Serine/threonine kinase which plays an important role in the regulation of electrolyte homeostasis, cell signaling, survival, and proliferation. Acts as an activator and inhibitor of sodium-coupled chloride cotransporters and potassium-coupled chloride cotransporters respectively. Activates SCNN1A, SCNN1B, SCNN1D and SGK1. Controls sodium and chloride ion transport by inhibiting the activity of WNK4, by either phosphorylating the kinase or via an interaction between WNK4 and the autoinhibitory domain of WNK1. WNK4 regulates the activity of the thiazide-sensitive Na-Cl cotransporter, SLC12A3, by phosphorylation. WNK1 may also play a role in actin cytoskeletal reorganization. Phosphorylates NEDD4L. Acts as a scaffold to inhibit SLC4A4, SLC26A6 as well as CFTR activities and surface expression, recruits STK39 which mediates the inhibition (By similarity). Related diseases Pseudohypoaldosteronism 2C (PHA2C) [MIM:614492]: An autosomal dominant disorder characterized by severe hypertension, hyperkalemia, hyperchloremia, mild hyperchloremic metabolic acidosis in some cases, and correction of physiologic abnormalities by thiazide diuretics. {ECO:0000269|PubMed:11498583}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Neuropathy, hereditary sensory and autonomic, 2A (HSAN2A) [MIM:201300]: A form of hereditary sensory and autonomic neuropathy, a genetically and clinically heterogeneous group of disorders characterized by degeneration of dorsal root and autonomic ganglion cells, and by sensory and/or autonomic abnormalities. HSAN2A is an autosomal recessive disorder characterized by impairment of pain, temperature and touch sensation, onset of symptoms in infancy or early childhood, occurrence of distal extremity pathologies (paronychia, whitlows, ulcers, and Charcot joints), frequent amputations, sensory loss that affects all modalities of sensation (lower and upper limbs and perhaps the trunk as well), absence or diminution of tendon reflexes (usually in all limbs), minimal autonomic dysfunction, absence of sensory nerve action potentials, and virtual absence of myelinated fibers with decreased numbers of unmyelinated fibers in sural nerves. {ECO:0000269|PubMed:15060842, ECO:0000269|PubMed:15911806, ECO:0000269|PubMed:18521183}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with O95747; P62136; P31947; P62258; P61981; P63104; P29101 EC number EC 2.7.11.1 Uniprot keywords 3D-structure; Alternative promoter usage; Alternative splicing; ATP-binding; Chloride; Cytoplasm; Cytoskeleton; Direct protein sequencing; Glycoprotein; Kinase; Neurodegeneration; Neuropathy; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Serine/threonine-protein kinase; Transferase; Ubl conjugation Protein physicochemical properties Chain ID A,B Molecular weight (Da) 61906 Length 542 Aromaticity 0.1 Instability index 41.99 Isoelectric point 8.64 Charge (pH=7) 8.04 2D Binding mode Binding energy (Kcal/mol) -10.24 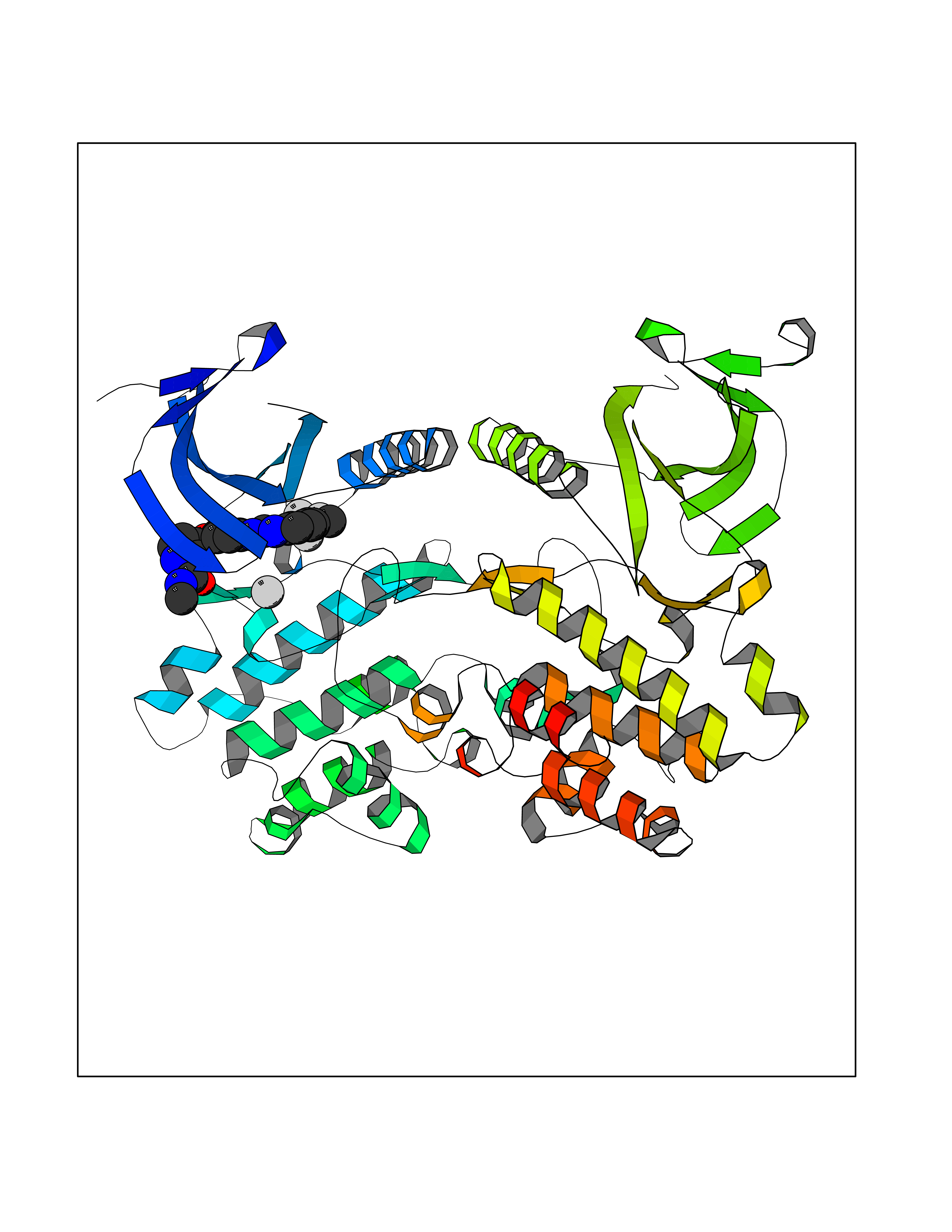 Molscript Map 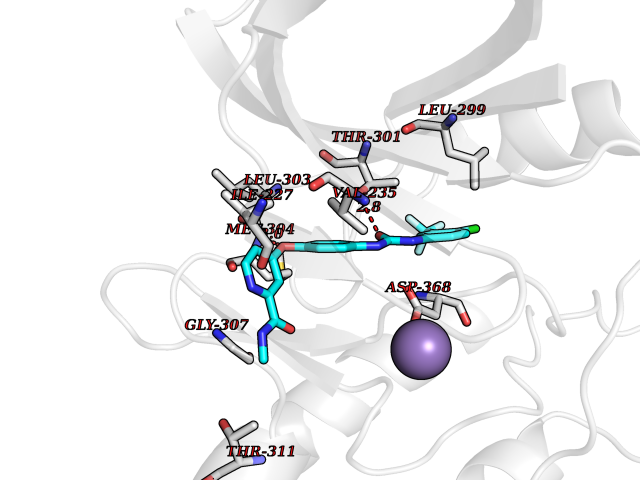 Pymol Map 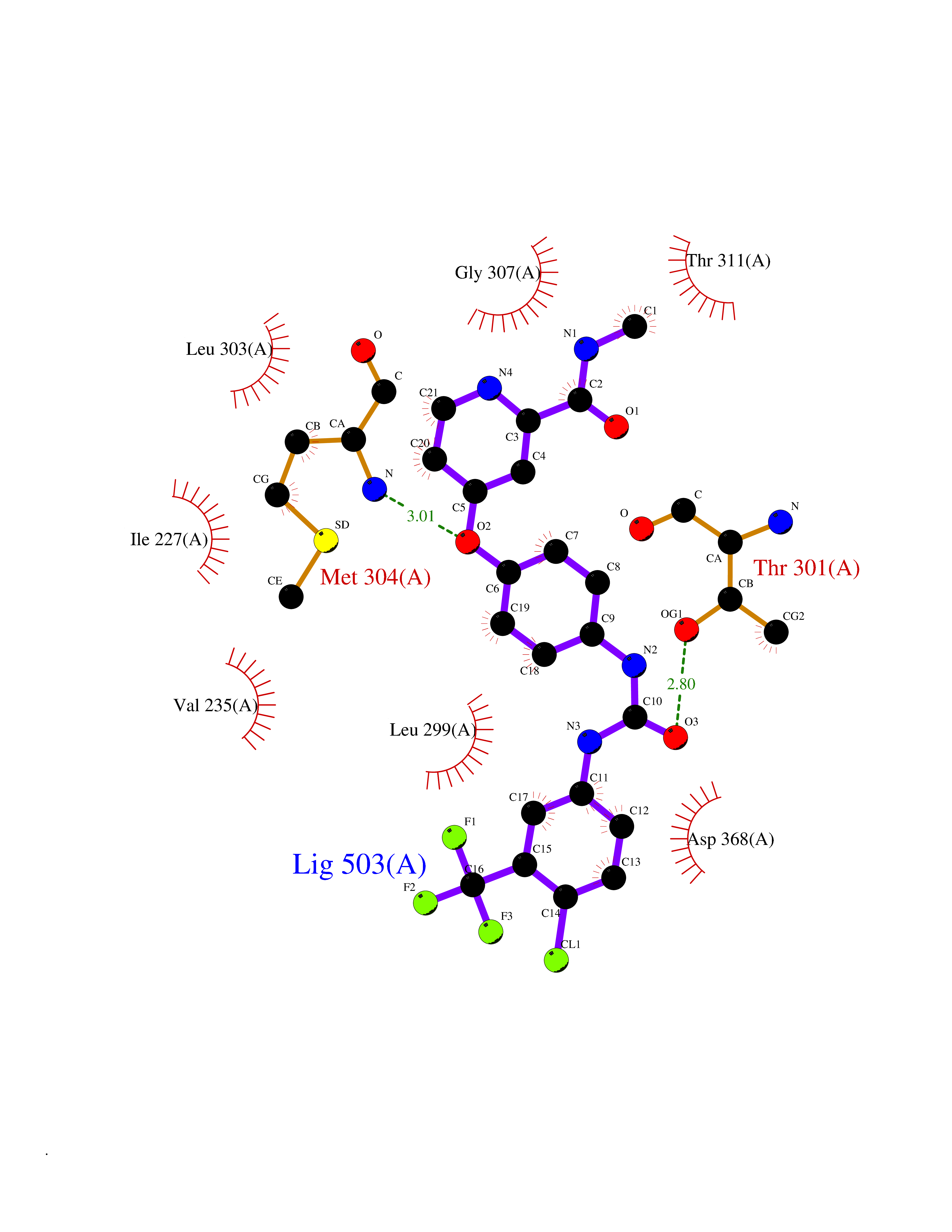 Ligplot Map 3D Binding mode Sequence TKAVGMSNDGRFLKFDIEIGRGSFKTVYKGLDTETTVEVAWCELQDRKLTKSERQRFKEEAEMLKGLQHPNIVRFYDSWESTCIVLVTELMTSGTLKTYLKRFKVMKIKVLRSWCRQILKGLQFLHTRTPPIIHRDLKCDNIFITGPTGSVKIGDLGLATLKRADFAKSVIGTPEFMAPEMYAAAYDESVDVYAFGMCMLEMATSEYPYSECQNAAQIYRRVTSGVKPASFDKVAIPEVKEIIEGCIRQNKDERYSIKDLLNHAFFQEETLETKAVGMSNDGRFLKFDIEIGRGSFKTVYKGLDTETTVEVAWCELQDRKLTKSERQRFKEEAEMLKGLQHPNIVRFYDSWESTVKGCIVLVTELMTSGTLKTYLKRFKVMKIKVLRSWCRQILKGLQFLHTRTPPIIHRDLKCDNIFITGPTGSVKIGDLGLATLKRADFAKSVIGTPEFMAPEMYAAAYDESVDVYAFGMCMLEMATSEYPYSECQNAAQIYRRVTSGVKPASFDKVAIPEVKEIIEGCIRQNKDERYSIKDLLNHAFFQ Hydrogen bonds contact Hydrophobic contact | ||||
| 114 | Sphingosine kinase 1 (SPHK1) | 3VZB | 7.51 | |
Target general information Gen name SPHK1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms SPK 1; SPK; SPHK1; SK 1; Acetyltransferase SPHK1 Protein family NA Biochemical class Kinase Function Acts on D-erythro-sphingosine and to a lesser extent sphinganine, but not other lipids, such as D,L-threo-dihydrosphingosine, N,N-dimethylsphingosine, diacylglycerol, ceramide, or phosphatidylinositol. In contrast to proapoptotic SPHK2, has a negative effect on intracellular ceramide levels, enhances cell growth and inhibits apoptosis. Involved in the regulation of inflammatory response and neuroinflammation. Via the product sphingosine 1-phosphate, stimulates TRAF2 E3 ubiquitin ligase activity, and promotes activation of NF-kappa-B in response to TNF signaling leading to IL17 secretion. In response to TNF and in parallel to NF-kappa-B activation, negatively regulates RANTES inducion through p38 MAPK signaling pathway. Involved in endocytic membrane trafficking induced by sphingosine, recruited to dilate endosomes, also plays a role on later stages of endosomal maturation and membrane fusion independently of its kinase activity. In Purkinje cells, seems to be also involved in the regulation of autophagosome-lysosome fusion upon VEGFA. Catalyzes the phosphorylation of sphingosine to form sphingosine 1-phosphate (SPP), a lipid mediator with both intra- and extracellular functions. Related diseases Intellectual developmental disorder, X-linked, syndromic, Claes-Jensen type (MRXSCJ) [MIM:300534]: A disorder characterized by significantly below average general intellectual functioning associated with impairments in adaptive behavior and manifested during the developmental period. MRXSCJ patients manifest intellectual disability associated with variable features such as slowly progressive spastic paraplegia, seizures, facial dysmorphism. {ECO:0000269|PubMed:15586325, ECO:0000269|PubMed:16538222, ECO:0000269|PubMed:16541399, ECO:0000269|PubMed:17320160, ECO:0000269|PubMed:17468742, ECO:0000269|PubMed:23356856, ECO:0000269|PubMed:25666439}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08868 Interacts with P07858; P68104; Q14192; Q2M3C7; Q9Y4K3; P13473-2; Q9Y371 EC number EC 2.7.1.91 Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Calmodulin-binding; Cell membrane; Coated pit; Cytoplasm; Endosome; Kinase; Lipid metabolism; Membrane; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Synapse; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 39813 Length 360 Aromaticity 0.08 Instability index 43.79 Isoelectric point 7.34 Charge (pH=7) 0.84 2D Binding mode Binding energy (Kcal/mol) -10.24 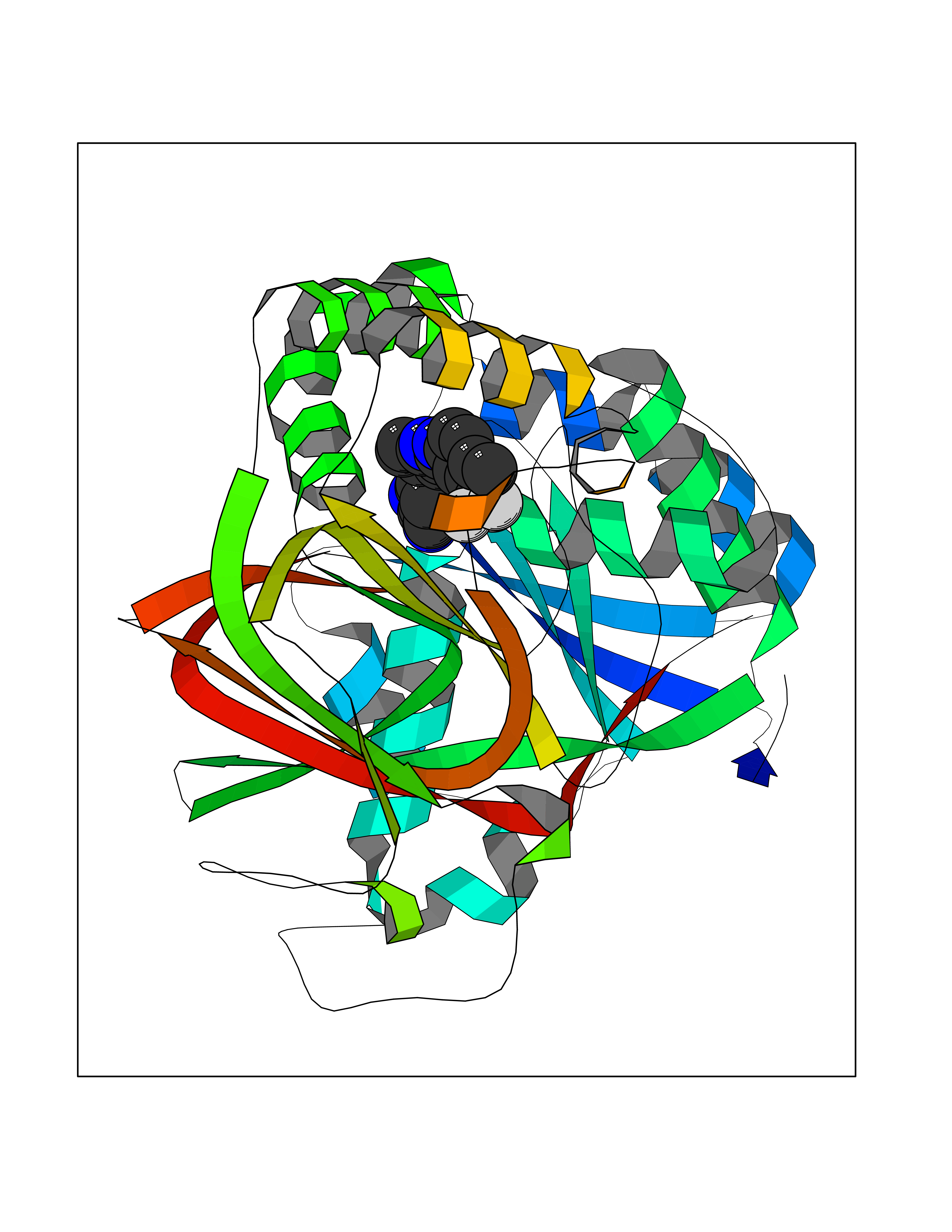 Molscript Map 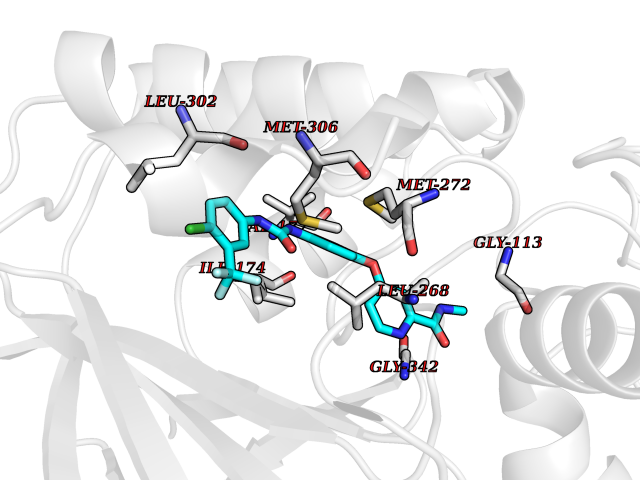 Pymol Map 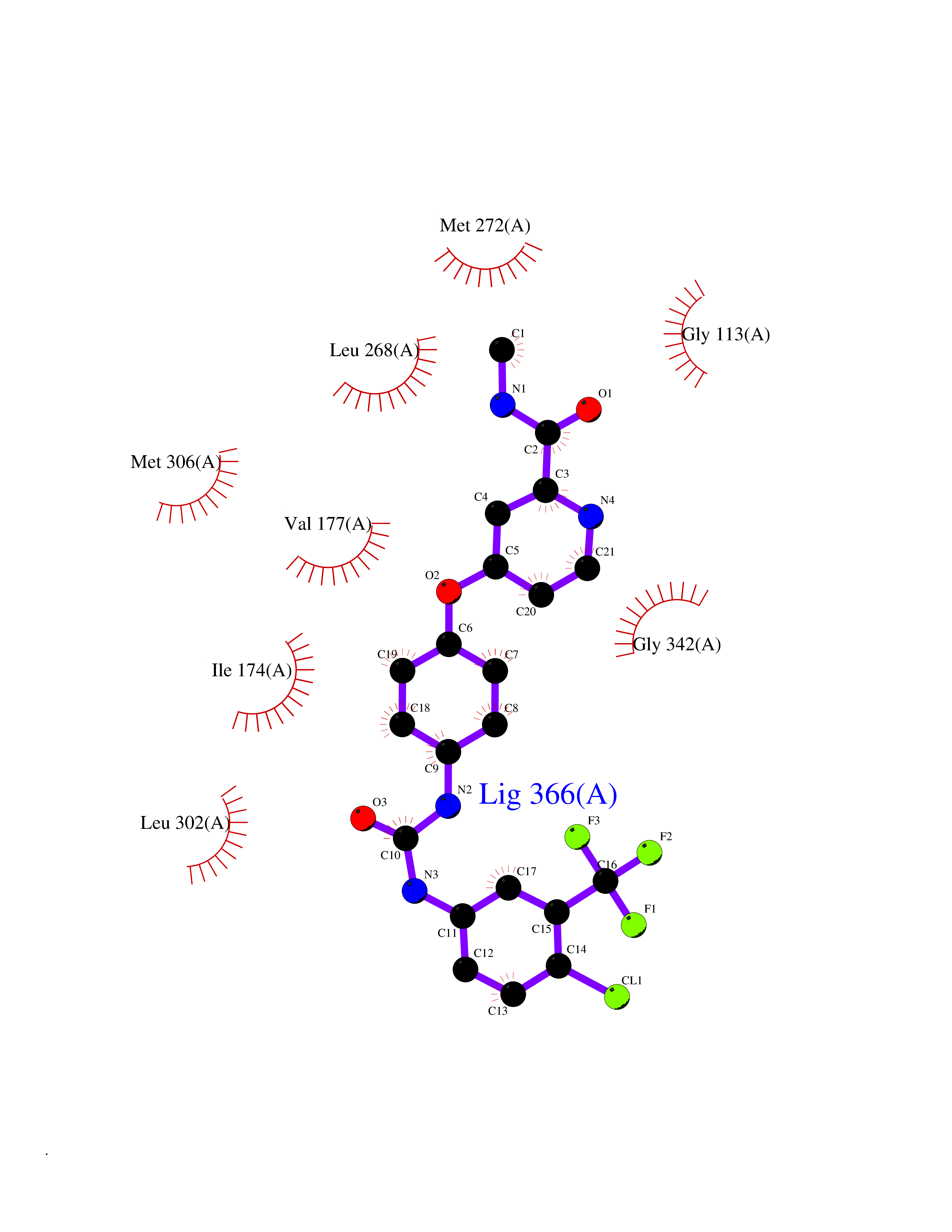 Ligplot Map 3D Binding mode Sequence AMGSGVLPRPCRVLVLLNPRGGKGKALQLFRSHVQPLLAEAEISFTLMLTERRNHARELVRSEELGRWDALVVMSGDGLMHEVVNGLMERPDWETAIQKPLCSLPAGSGNALAASLNHYAGYEQVTNEDLLTNCTLLLCRRLLSPMNLLSLHTASGLRLFSVLSLAWGFIADVDLESEKYRRLGEMRFTLGTFLRLAALRTYRGRLAYLPVGRVGSKTPASPVVVQQGPVDAHLVPLEEPVPSHWTVVPDEDFVLVLALLHSHLGSEMFAAPMGRCAAGVMHLFYVRAGVSRAMLLRLFLAMEKGRHMEYECPYLVYVPVVAFRLEPKDGKGVFAVDGELMVSEAVQGQVHPNYFWMVSG Hydrogen bonds contact Hydrophobic contact | ||||
| 115 | Trypanosoma Trypanothione reductase (Trypano TPR) | 2WBA | 7.51 | |
Target general information Gen name Trypano TPR Organism Trypanosoma brucei brucei Uniprot ID TTD ID Synonyms TRYR; TPR; Parasite-specific trypanothione reductase; N(1),N(8)-bis(glutathionyl)spermidine reductase Protein family Class-I pyridine nucleotide-disulfide oxidoreductase family Biochemical class Sulfur donor oxidoreductase Function Trypanothione is the parasite analog of glutathione; this enzyme is the equivalent of glutathione reductase. Related diseases Immunodeficiency 57 with autoinflammation (IMD57) [MIM:618108]: An autosomal recessive primary immunodeficiency characterized by lymphopenia and recurrent viral, bacterial, and fungal infections. Patients exhibit early-onset inflammatory bowel disease involving the upper and lower gastrointestinal tract, and develop progressive polyarthritis. {ECO:0000269|PubMed:30026316}. The disease is caused by variants affecting the gene represented in this entry. RIPK1-deficient immune cells from IMD57 patients have impaired proinflammatory signaling leading to dysregulated cytokine secretion and are prone to necroptosis. {ECO:0000269|PubMed:30026316}.; DISEASE: Autoinflammation with episodic fever and lymphadenopathy (AIEFL) [MIM:618852]: An autosomal dominant immunologic disorder characterized by early onset of recurrent episodes of unexplained fever, lymphadenopathy, hepatosplenomegaly, and increased levels of inflammatory cytokines and chemokines in patient serum. {ECO:0000269|PubMed:31827280, ECO:0000269|PubMed:31827281}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number EC 1.8.1.12 Uniprot keywords 3D-structure; Cytoplasm; Disulfide bond; FAD; Flavoprotein; NADP; Oxidoreductase; Redox-active center Protein physicochemical properties Chain ID A,B Molecular weight (Da) 105578 Length 978 Aromaticity 0.08 Instability index 33.76 Isoelectric point 6.25 Charge (pH=7) -6.81 2D Binding mode Binding energy (Kcal/mol) -10.24 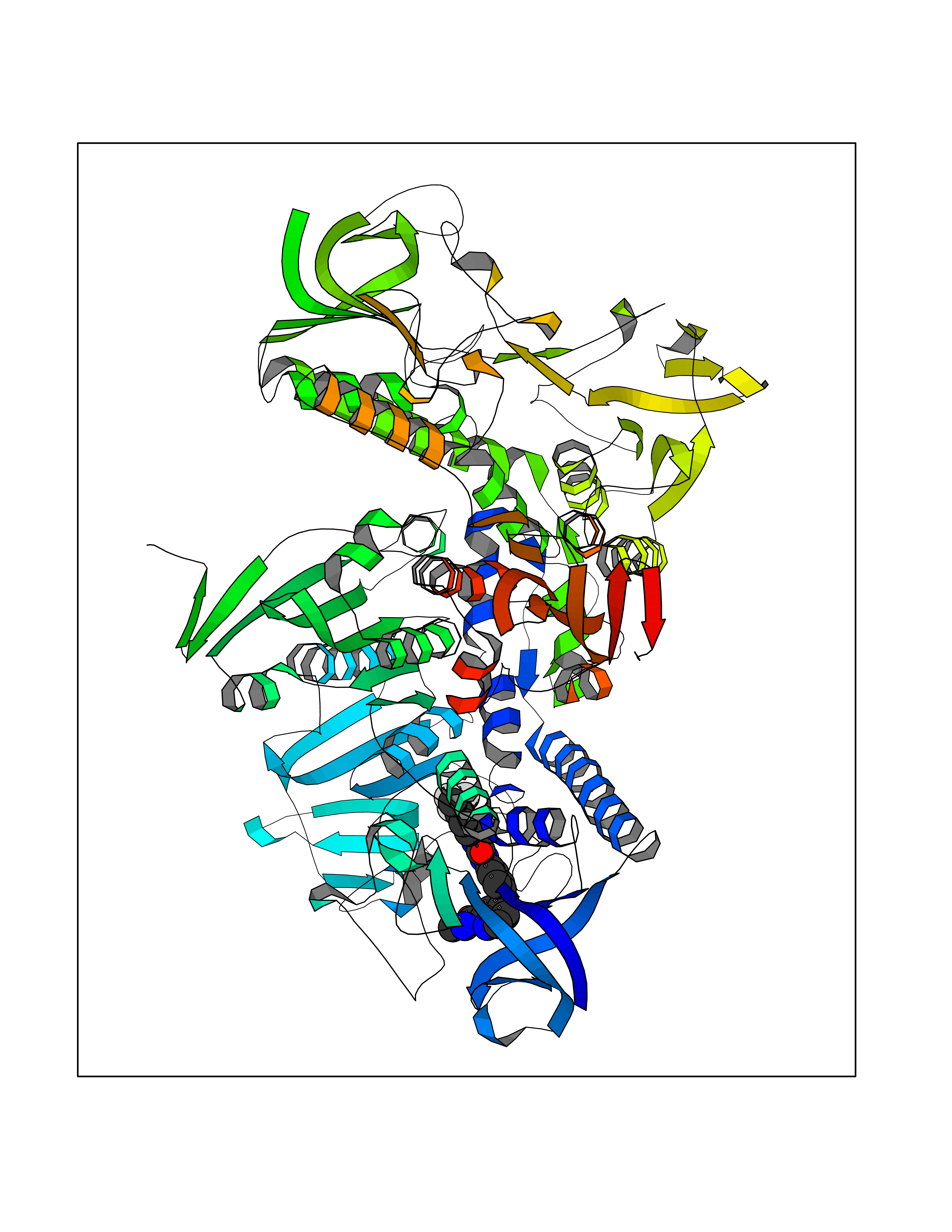 Molscript Map 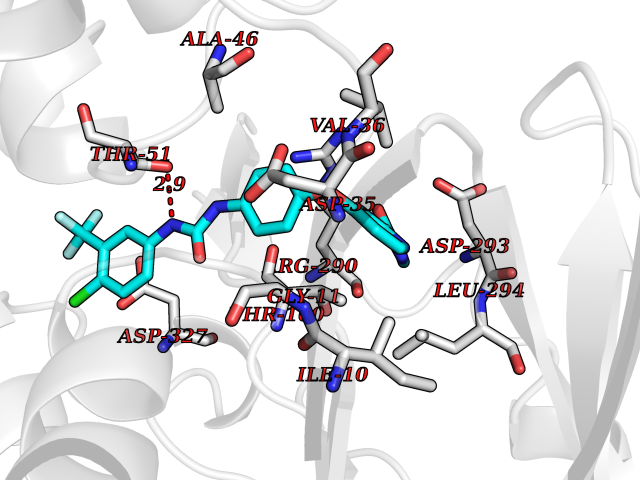 Pymol Map 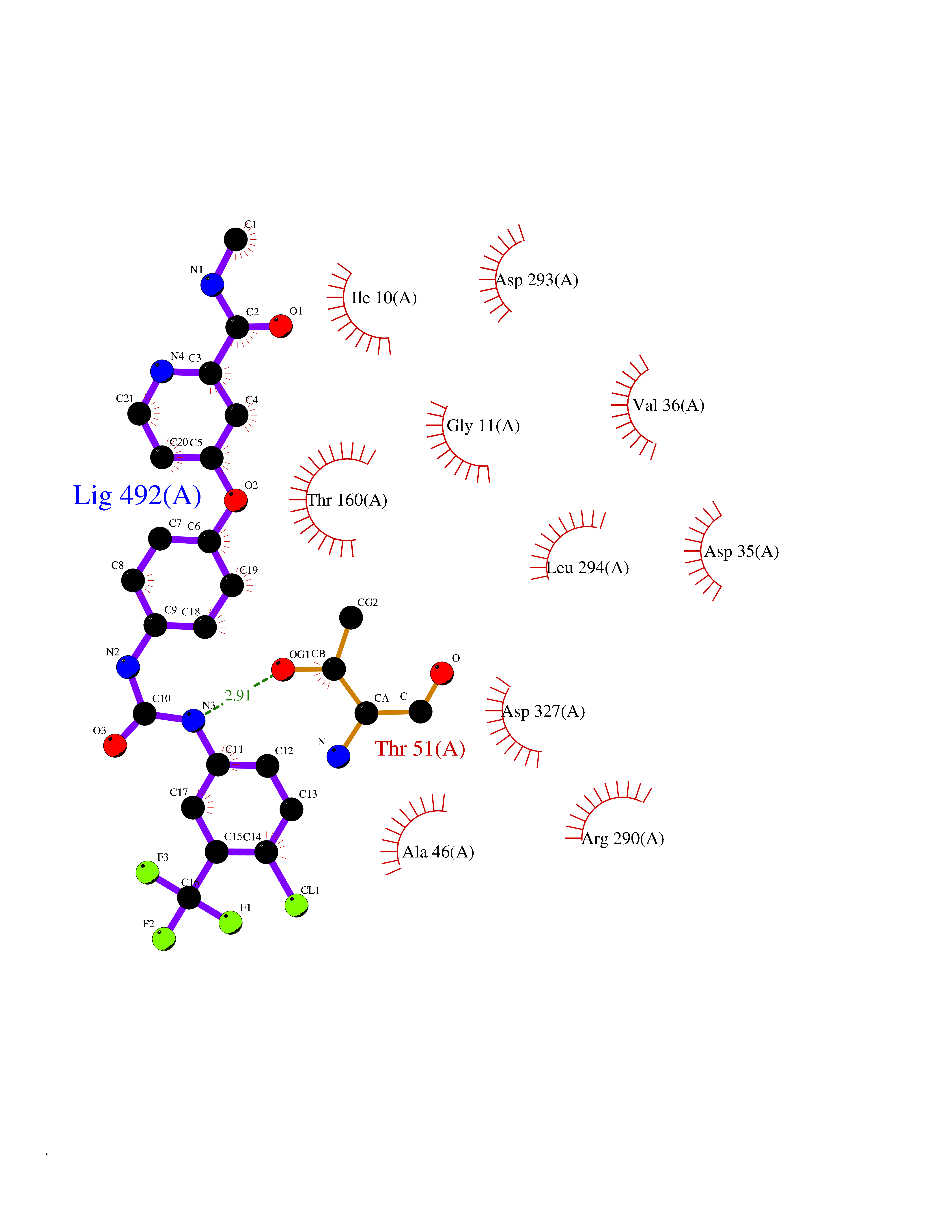 Ligplot Map 3D Binding mode Sequence SKAFDLVVIGAGSGGLEAGWNAATLYGKRVAVVDVQTSHGPPFYAALGGTCVNVGCVPKKLMVTGAQYMDHLRESAGFGWEFDGSSVKANWKKLIAAKNEAVLDINKSYEGMFNDTEGLDFFLGWGSLESKNVVVVRETADPKSAVKERLQADHILLATGSWPQMPAIPGIEHCISSNEAFYLPEPPRRVLTVGGGFISVEFAGIFNAYKPPGGKVTLCYRNNLILRGFDETIREEVTKQLTANGIEIMTNENPAKVSLNTDGSKHVTFESGKTLDVDVVMMAIGRIPRTNDLQLGNVGVKLTPKGGVQVDEFSRTNVPNIYAIGDITDRLMLTPVAINEGAALVDTVFGNKPRKTDHTRVASAVFSIPPIGTCGLIEEVAAKEFEKVAVYMSSFTPLMHNISGSKYKKFVAKIVTNHSDGTVLGVHLLGDGAPEIIQAVGVCLRLNAKISDFYNTIGVHPTSAEELCSMRTPSYYYVKGEKMEKLPDSSKAFDLVVIGAGSGGLEAGWNAATLYGKRVAVVDVQTSHGPPFYAALGGTCVNVGCVPKKLMVTGAQYMDHLRESAGFGWEFDGSSVKANWKKLIAAKNEAVLDINKSYEGMFNDTEGLDFFLGWGSLESKNVVVVRETADPKSAVKERLQADHILLATGSWPQMPAIPGIEHCISSNEAFYLPEPPRRVLTVGGGFISVEFAGIFNAYKPPGGKVTLCYRNNLILRGFDETIREEVTKQLTANGIEIMTNENPAKVSLNTDGSKHVTFESGKTLDVDVVMMAIGRIPRTNDLQLGNVGVKLTPKGGVQVDEFSRTNVPNIYAIGDITDRLMLTPVAINEGAALVDTVFGNKPRKTDHTRVASAVFSIPPIGTCGLIEEVAAKEFEKVAVYMSSFTPLMHNISGSKYKKFVAKIVTNHSDGTVLGVHLLGDGAPEIIQAVGVCLRLNAKISDFYNTIGVHPTSAEELCSMRTPSYYYVKGEKMEKLPDS Hydrogen bonds contact Hydrophobic contact | ||||
| 116 | "15-cis-phytoene desaturase, chloroplastic/chromoplastic (EC 1.3.5.5) (Phytoene dehydrogenase) (Phytoene desaturase)" | 5MOG | 7.50 | |
Target general information Gen name PDS1 Organism Oryza sativa subsp. indica (Rice) Uniprot ID TTD ID NA Synonyms PDS;OsI_010044 Protein family Carotenoid/retinoid oxidoreductase family Biochemical class NA Function Converts phytoene into zeta-carotene via the intermediary of phytofluene by the symmetrical introduction of two double bonds at the C-11 and C-11' positions of phytoene with a concomitant isomerization of two neighboring double bonds at the C9 and C9' positions from trans to cis. Active with decylplastoquinone (DPQ) as substrate (PubMed:26147209, PubMed:29176862). Also active with other benzoquinones, which are strongly preferred over naphthoquinones as substrates (PubMed:26147209). {ECO:0000269|PubMed:26147209, ECO:0000269|PubMed:29176862}." Related diseases NA Drugs (DrugBank ID) NA Interacts with NA EC number 1.3.5.5 Uniprot keywords 3D-structure; Carotenoid biosynthesis; Chloroplast; Chromoplast; Direct protein sequencing; FAD; Flavoprotein; Membrane; Oxidoreductase; Plastid; Reference proteome; Transit peptide Protein physicochemical properties Chain ID E Molecular weight (Da) 52485.1 Length 466 Aromaticity 0.1 Instability index 45.53 Isoelectric point 5.93 Charge (pH=7) -5.81 2D Binding mode Binding energy (Kcal/mol) -10.23 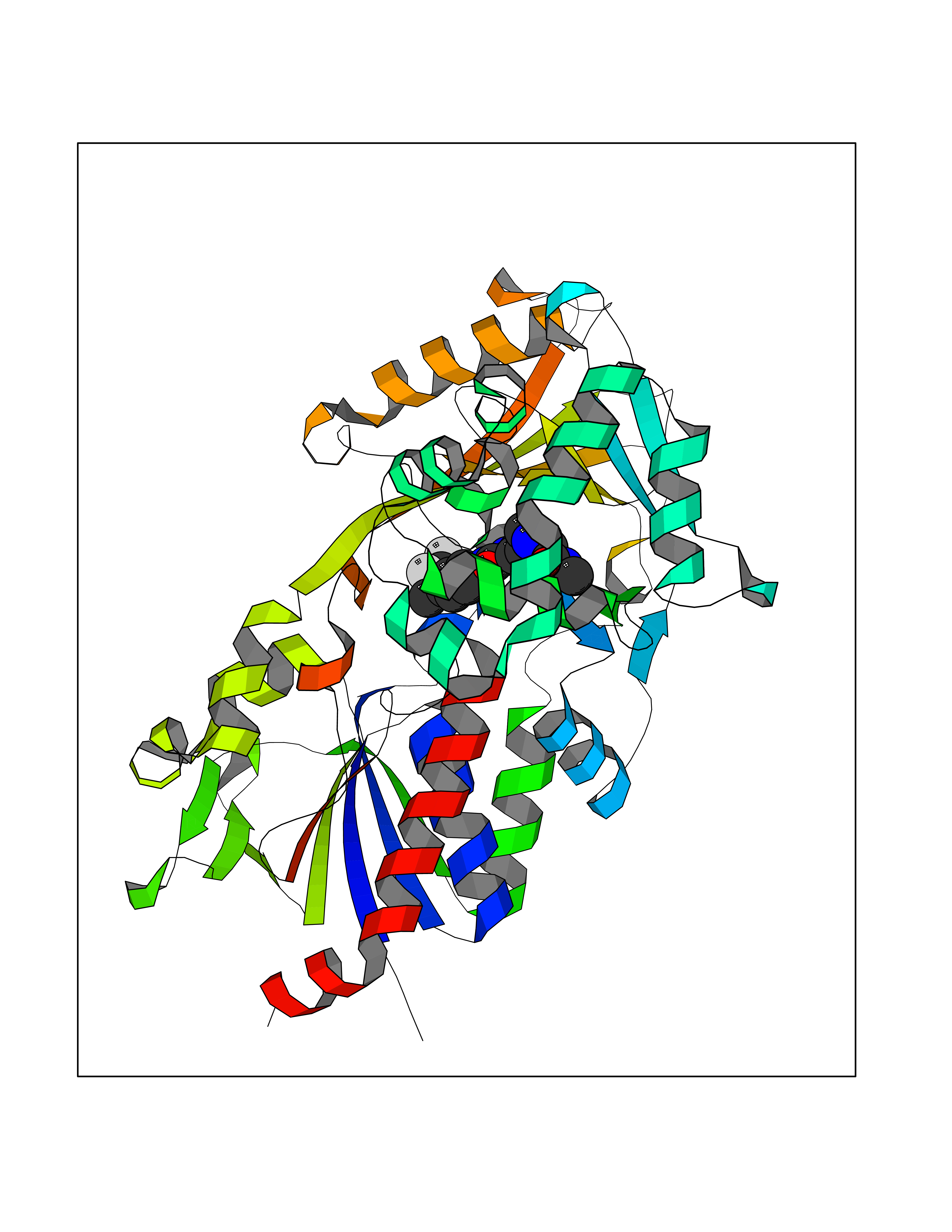 Molscript Map 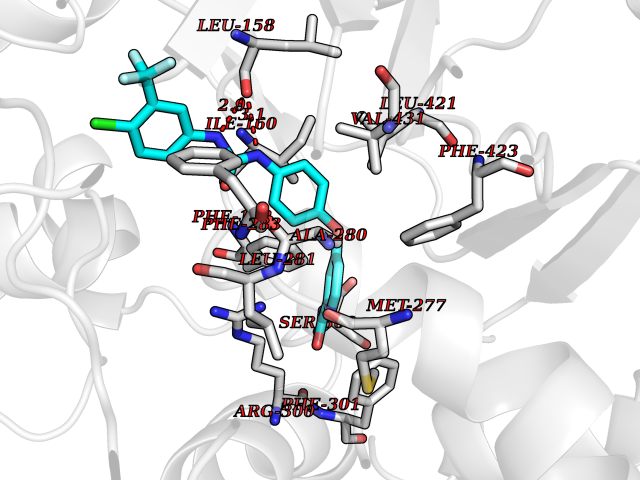 Pymol Map 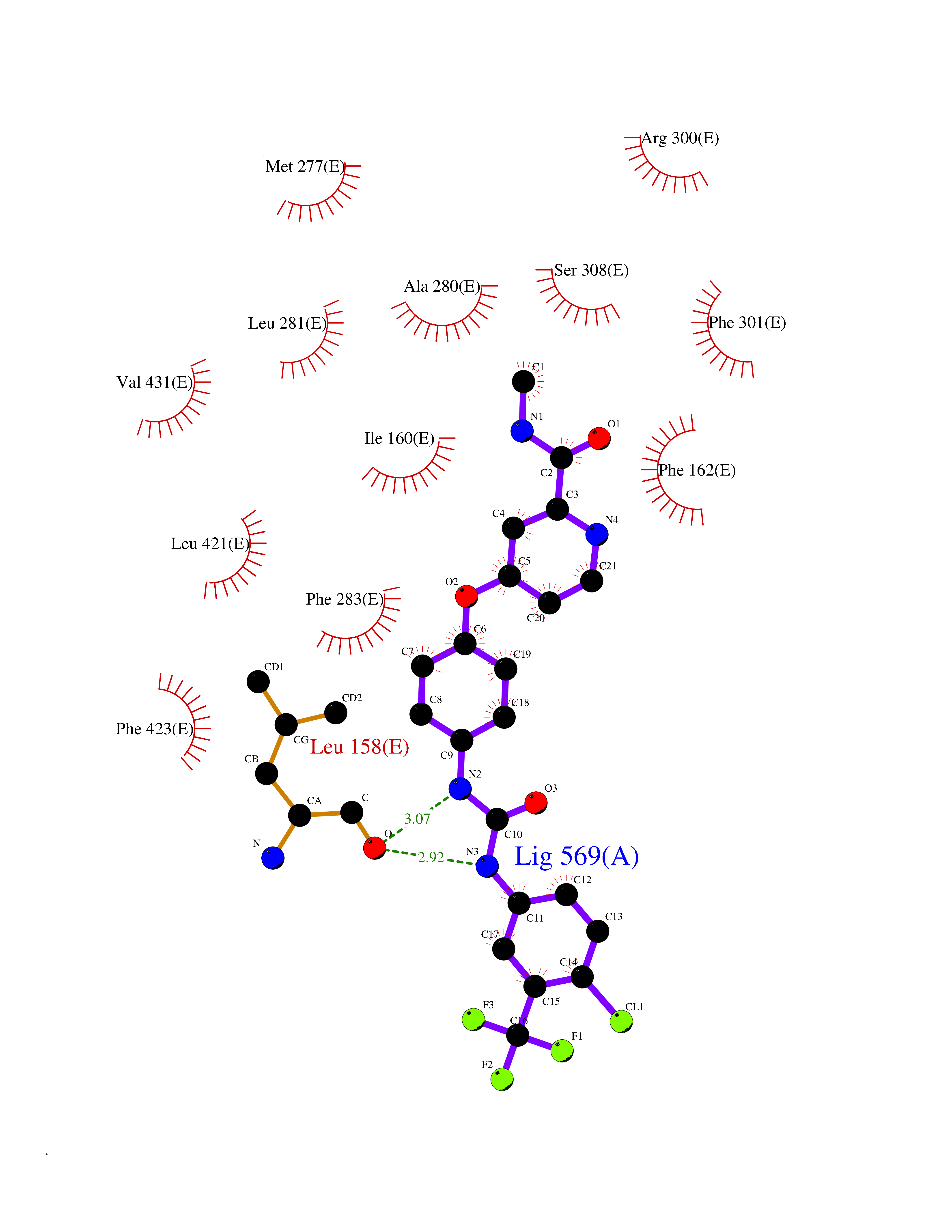 Ligplot Map 3D Binding mode Sequence TKPLQVVIAGAGLAGLSTAKYLADAGHKPILLEARDVLGGKIAAWKDEDGDWYETGLHIFFGAYPNIQNLFGELGINDRLQWKEHSMIFAMPNKPGEFSRFDFPETLPAPLNGIWAILRNNEMLTWPEKVKFALGLLPAMVGGQAYVEAQDGFTVSEWMKKQGVPDRVNDEVFIAMSKALNFINPDELSMQCILIALNRFLQEKHGSKMAFLDGNPPERLCMPIVDHVRSLGGEVRLNSRIQKIELNPDGTVKHFALTDGTQITGDAYVFATPVDILKLLVPQEWKEISYFKKLEKLVGVPVINVHIWFDRKLKNTYDHLLFSRSSLLSVYADMSVTCKEYYDPNRSMLELVFAPAEEWVGRSDTEIIEATMQELAKLFPDEIAADQSKAKILKYHVVKTPRSVYKTIPDCEPCRPLQRSPIEGFYLAGDYTKQKYLASMEGAVLSGKLCAQSVVEDYKMLSRRSL Hydrogen bonds contact Hydrophobic contact | ||||
| 117 | Corticotropin-lipotropin (POMC) | 4XNH | 7.50 | |
Target general information Gen name POMC Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Pro-opiomelanocortin Protein family POMC family Biochemical class NA Function Corticotropin: Stimulates the adrenal glands to release cortisol. Related diseases Obesity (OBESITY) [MIM:601665]: A condition characterized by an increase of body weight beyond the limitation of skeletal and physical requirements, as the result of excessive accumulation of body fat. {ECO:0000269|PubMed:12165561}. Disease susceptibility may be associated with variants affecting the gene represented in this entry.; DISEASE: Obesity, early-onset, with adrenal insufficiency and red hair (OBAIRH) [MIM:609734]: An autosomal recessive disorder characterized by early-onset obesity due to severe hyperphagia, pigmentary abnormalities, mainly pale skin and red hair, and secondary hypocortisolism. {ECO:0000269|PubMed:9620771}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01565; DB01497; DB00836 Interacts with Q9NYB9-2; Q9NX04; Q8N8Y2; Q7RTU4; Q68D86; P62508-3; Q8IVS8; Q7Z4H3; O75031; Q9P2K6; Q13064; Q8NI38; Q96HA8; Q9UMX2-2; P51687; Q99757 EC number NA Uniprot keywords 3D-structure; Acetylation; Amidation; Cleavage on pair of basic residues; Direct protein sequencing; Disulfide bond; Endorphin; Glycoprotein; Hormone; Obesity; Phosphoprotein; Proteomics identification; Reference proteome; Secreted; Signal Protein physicochemical properties Chain ID F Molecular weight (Da) 17898.3 Length 159 Aromaticity 0.11 Instability index 33.69 Isoelectric point 6.16 Charge (pH=7) -2.64 2D Binding mode Binding energy (Kcal/mol) -10.23  Molscript Map 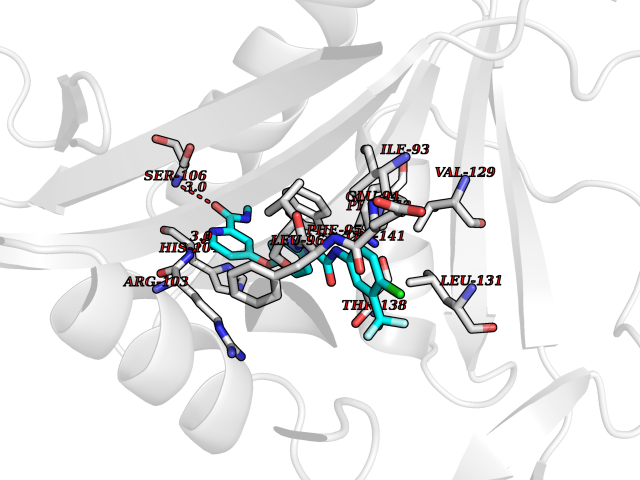 Pymol Map 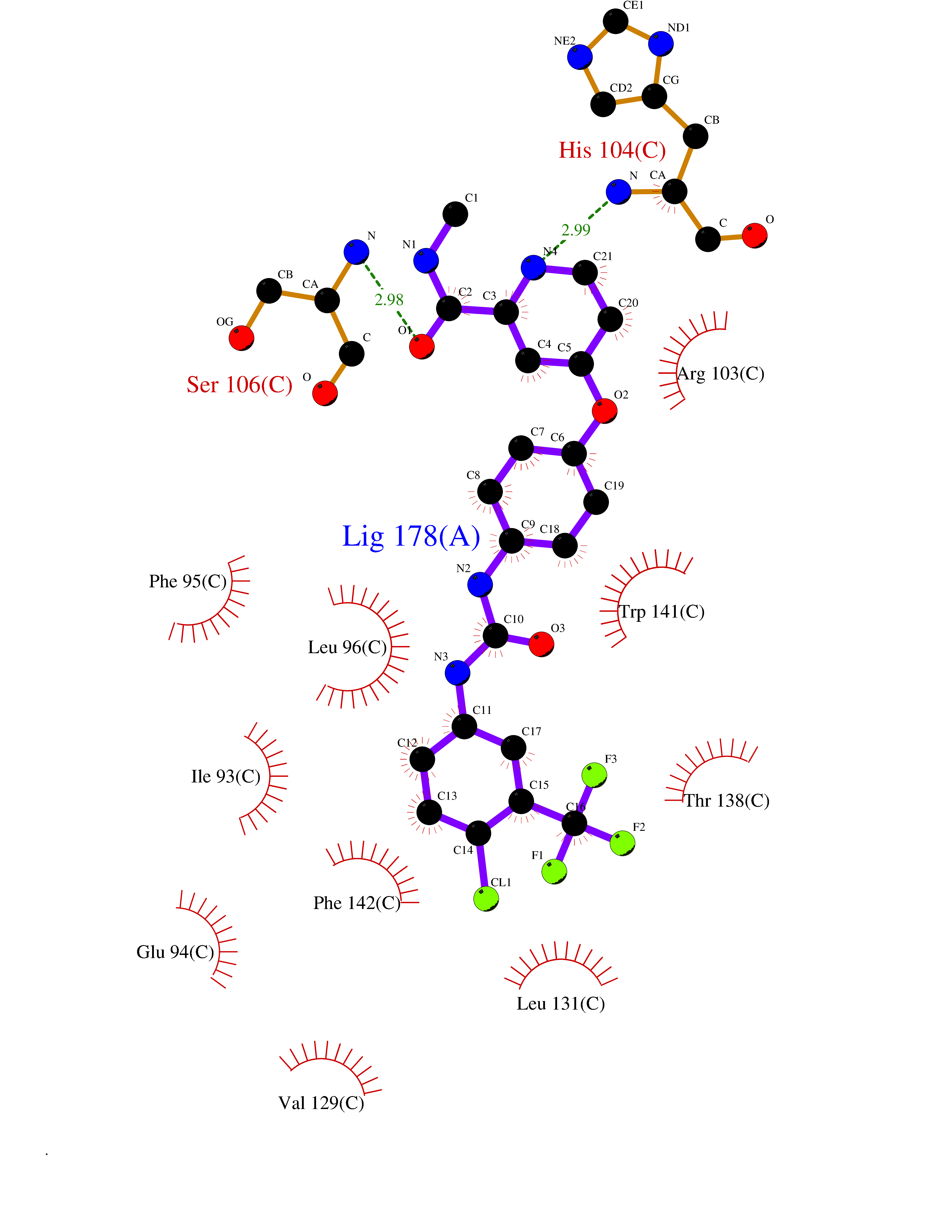 Ligplot Map 3D Binding mode Sequence RDICTLDNVYANNLGMLTKLAHVTVPNLYQDAFFSALFAEKDVHFTQMAYYSEIPVGGLVAKLVPKNELSLKGIQIEFLGVLPNYRHKSIGSKLLKFAEDKCSECHQHNVFVYLPAVDDLTKQWFIAHGFEQVGETVNNFIKGVNGDEQDAILLKKHIS Hydrogen bonds contact Hydrophobic contact | ||||
| 118 | Dimethylglycine oxidase | 1PJ5 | 7.50 | |
Target general information Gen name dmg Organism Arthrobacter globiformis Uniprot ID TTD ID NA Synonyms NA Protein family GcvT family Biochemical class Oxidoreductase Function Dimethylglycine oxidase activity.Nucleotide binding. Related diseases Curry-Jones syndrome (CRJS) [MIM:601707]: A multisystem disorder characterized by patchy skin lesions, polysyndactyly, diverse cerebral malformations, unicoronal craniosynostosis, iris colobomas, microphthalmia, and intestinal malrotation with myofibromas or hamartomas. {ECO:0000269|PubMed:24859340, ECO:0000269|PubMed:27236920}. The disease is caused by variants affecting the gene represented in this entry. 8 individuals have been identified with the disease-causing mutation Phe-412 and all were mosaic. The mutation could not be reliably detected in blood, greatest success rates were obtained with affected tissues obtained by invasive procedures. It is thought that the mutation has arisen postzygotically early during embryonic development (PubMed:27236920). This mutation has also been identified in ameloblastoma, medulloblastoma, meningioma, and basal cell carcinoma, and has been reported as the oncogenic driver in some of these tumors (PubMed:24859340). {ECO:0000269|PubMed:24859340, ECO:0000269|PubMed:27236920}. Drugs (DrugBank ID) DB03256; DB03147 Interacts with NA EC number 1.5.3.10 Uniprot keywords 3D-structure; Direct protein sequencing; FAD; Flavoprotein; Nucleotide-binding; Oxidoreductase Protein physicochemical properties Chain ID A Molecular weight (Da) 45912.2 Length 427 Aromaticity 0.07 Instability index 43.46 Isoelectric point 4.83 Charge (pH=7) -20.69 2D Binding mode Binding energy (Kcal/mol) -10.23  Molscript Map 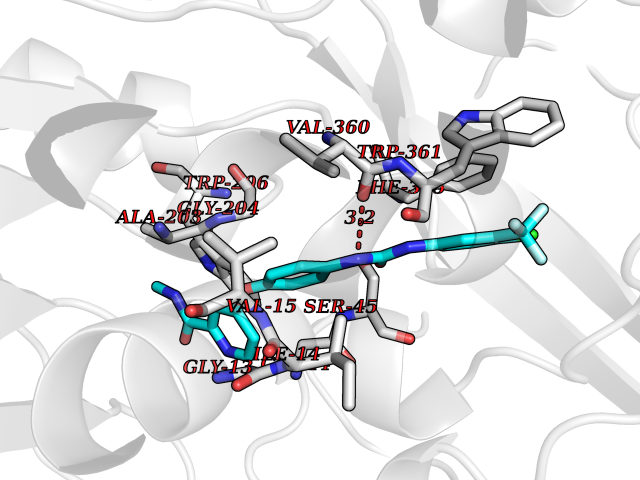 Pymol Map 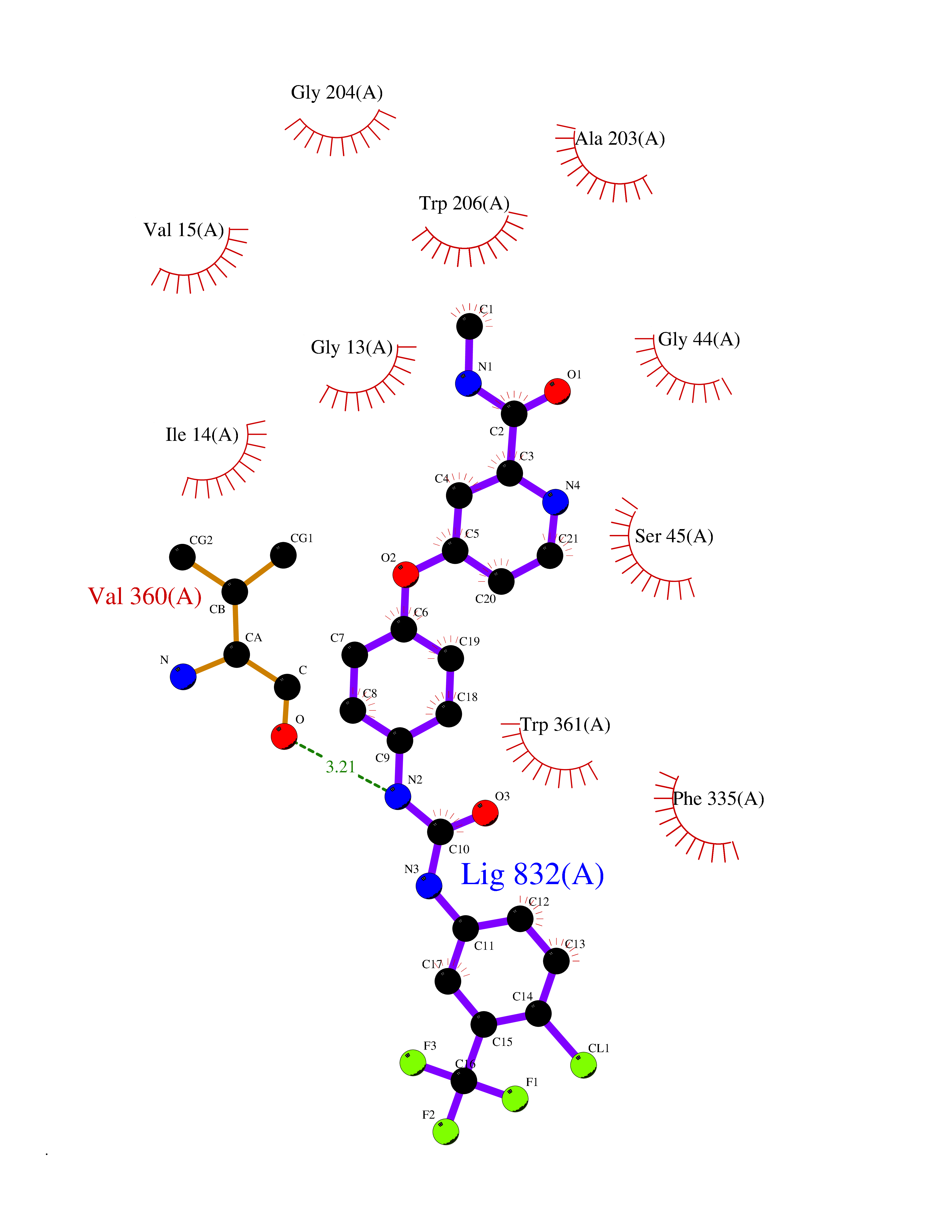 Ligplot Map 3D Binding mode Sequence TPRIVIIGAGIVGTNLADELVTRGWNNITVLDQGPLNMPGGSTSHAPGLVFQTNPSKTMASFAKYTVEKLLSLTEDGVSCFNQVGGLEVATTETRLADLKRKLGYAAAWGIEGRLLSPAECQELYPLLDGENILGGLHVPSDGLASAARAVQLLIKRTESAGVTYRGSTTVTGIEQSGGRVTGVQTADGVIPADIVVSCAGFWGAKIGAMIGMAVPLLPLAHQYVKTTPVPAQQGRNDQPNGARLPILRHQDQDLYYREHGDRYGIGSYAHRPMPVDVDTLGAYAPETVSEHHMPSRLDFTLEDFLPAWEATKQLLPALADSEIEDGFNGIFSFTPDGGPLLGESKELDGFYVAEAVWVTHSAGVAKAMAELLTTGRSETDLGECDITRFEDVQLTPEYVSETSQQNFVEIYDVLHPLQPRLSPRNL Hydrogen bonds contact Hydrophobic contact | ||||
| 119 | STE20-related serine/threonine-protein kinase (SLK) | 8BEM | 7.50 | |
Target general information Gen name SLK Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms hSLK; STE20-related kinase; STE20-like kinase Protein family Protein kinase superfamily, STE Ser/Thr protein kinase family, STE20 subfamily Biochemical class NA Function Mediates apoptosis and actin stress fiber dissolution. Related diseases WHIM syndrome 2 (WHIMS2) [MIM:619407]: An autosomal recessive form of WHIM syndrome, a primary immunodeficiency disorder characterized by warts, hypogammaglobulinemia, infections, and myelokathexis. Myelokathexis is a unique form of non-cyclic severe congenital neutropenia caused by accumulation of mature and degenerating neutrophils in the bone marrow. Monocytopenia and lymphopenia, especially B lymphopenia, also commonly occur. There is significant phenotypic variation among patients, such that some individuals may have an incomplete form of the disorder in which one or more of the classic tetrad features are not present. {ECO:0000269|PubMed:24777453}. The disease may be caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07853; DB06616; DB12010; DB07664 Interacts with Q9H2G2 EC number EC 2.7.11.1 Uniprot keywords 3D-structure; Alternative splicing; Apoptosis; ATP-binding; Coiled coil; Cytoplasm; Kinase; Nucleotide-binding; Phosphoprotein; Proteomics identification; Reference proteome; Serine/threonine-protein kinase; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 31199.4 Length 274 Aromaticity 0.09 Instability index 47.16 Isoelectric point 4.82 Charge (pH=7) -16.92 2D Binding mode Binding energy (Kcal/mol) -10.23  Molscript Map 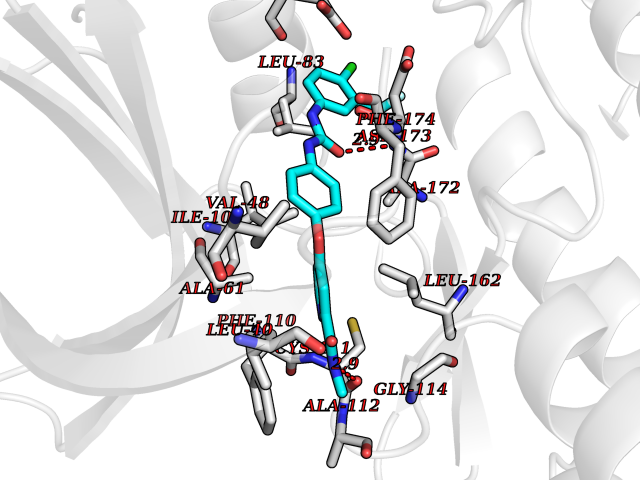 Pymol Map 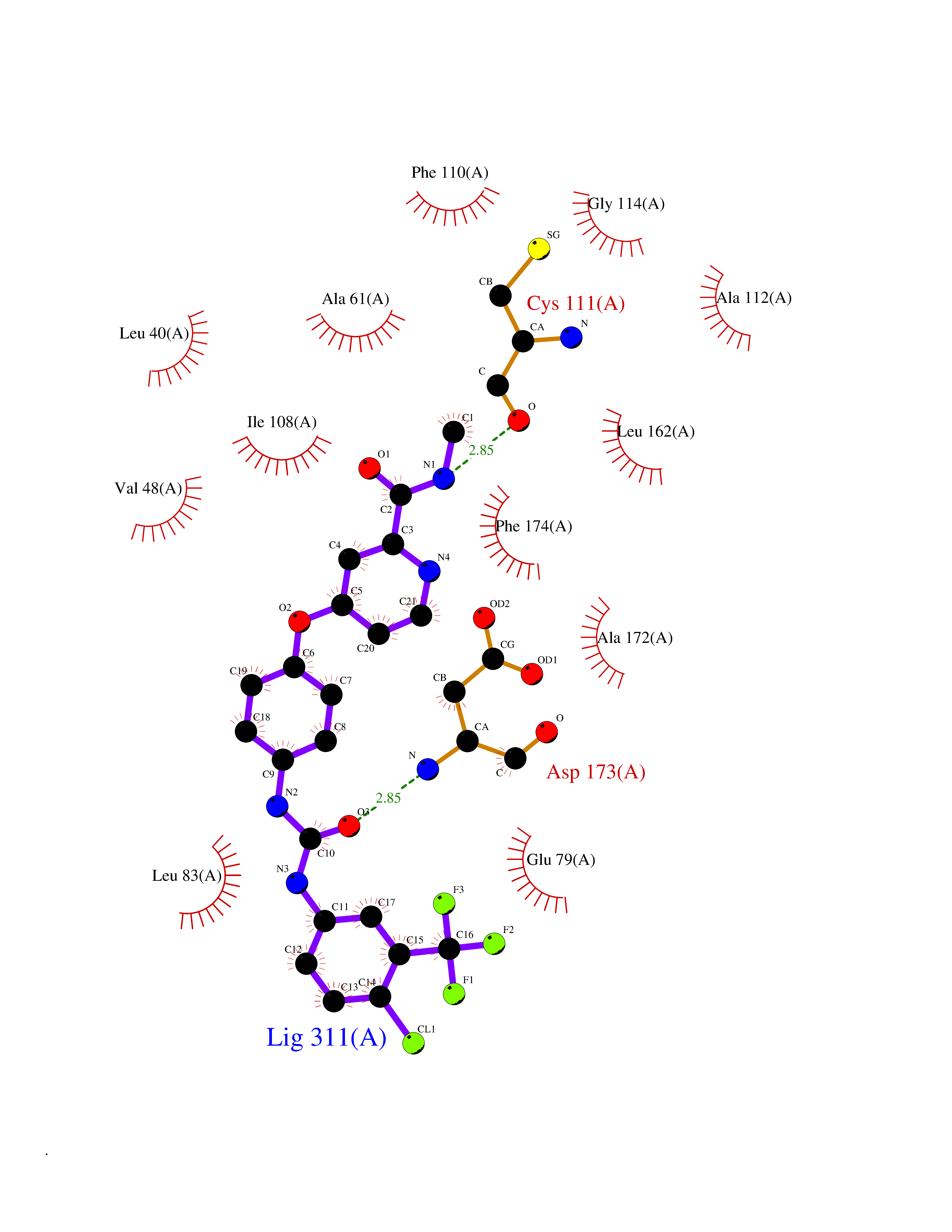 Ligplot Map 3D Binding mode Sequence SMKQYEHVKRDLNPEDFWEIIGELGDGGKVYKAQNKETSVLAAAKVIDTKSEEELEDYMVEIDILASCDHPNIVKLLDAFYYENNLWILIEFCAGGAVDAVMLELERPLTESQIQVVCKQTLDALNYLHDNKIIHRDLKAGNILFTLDGDIKLADFGSFIGTPYWMAPEVVMCETPYDYKADVWSLGITLIEMAEIEPPHHELNPMRVLLKIAKSEPPTLAQPSRWSSNFKDFLKKCLEKNVDARWTTSQLLQHPFVTVDSNKPIRELIAEAKA Hydrogen bonds contact Hydrophobic contact | ||||
| 120 | Aggrecanase-1 (ADAMTS4) | 2RJP | 7.50 | |
Target general information Gen name ADAMTS4 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Aggrecanase 1; ADMP-1; ADAMTS4; ADAM-TS4; ADAM-TS 4; A disintegrin and metalloproteinase with thrombospondin motifs 4 Protein family NA Biochemical class Peptidase Function Cleaves aggrecan, a cartilage proteoglycan, and may be involved in its turnover. May play an important role in the destruction of aggrecan in arthritic diseases. Could also be a critical factor in the exacerbation of neurodegeneration in Alzheimer disease. Cleaves aggrecan at the '392-Glu-|-Ala-393' site. Related diseases Familial male precocious puberty (FMPP) [MIM:176410]: In FMPP the receptor is constitutively activated. {ECO:0000269|PubMed:11134146, ECO:0000269|PubMed:11391350, ECO:0000269|PubMed:7629248, ECO:0000269|PubMed:7692306, ECO:0000269|PubMed:7714085, ECO:0000269|PubMed:7757065, ECO:0000269|PubMed:8281137, ECO:0000269|PubMed:8829636, ECO:0000269|PubMed:8929952, ECO:0000269|PubMed:9467560, ECO:0000269|PubMed:9661624}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Luteinizing hormone resistance (LHR) [MIM:238320]: An autosomal recessive disorder characterized by unresponsiveness to luteinizing hormone, defective sexual development in males, and defective follicular development and ovulation, amenorrhea and infertility in females. Two forms of the disorder have been defined in males. Type 1 is a severe form characterized by complete 46,XY male pseudohermaphroditism, low testosterone and high luteinizing hormone levels, total lack of responsiveness to luteinizing and chorionic gonadotropin hormones, lack of breast development, and absent development of secondary male sex characteristics. Type 2, a milder form, displays a broader range of phenotypic expression ranging from micropenis to severe hypospadias. {ECO:0000269|PubMed:12050206, ECO:0000269|PubMed:15372531, ECO:0000269|PubMed:15472221, ECO:0000269|PubMed:19551906, ECO:0000269|PubMed:7719343, ECO:0000269|PubMed:8559204, ECO:0000269|PubMed:9215288, ECO:0000269|PubMed:9514160, ECO:0000269|PubMed:9626144, ECO:0000269|PubMed:9626653}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB06822 Interacts with NA EC number EC 3.4.24.82 Uniprot keywords 3D-structure; Alternative splicing; Cleavage on pair of basic residues; Direct protein sequencing; Disulfide bond; Extracellular matrix; Glycoprotein; Hydrolase; Metal-binding; Metalloprotease; Protease; Proteomics identification; Reference proteome; Secreted; Signal; Zinc; Zymogen Protein physicochemical properties Chain ID A Molecular weight (Da) 31309.4 Length 291 Aromaticity 0.07 Instability index 42.35 Isoelectric point 5.97 Charge (pH=7) -7.02 2D Binding mode Binding energy (Kcal/mol) -10.23  Molscript Map 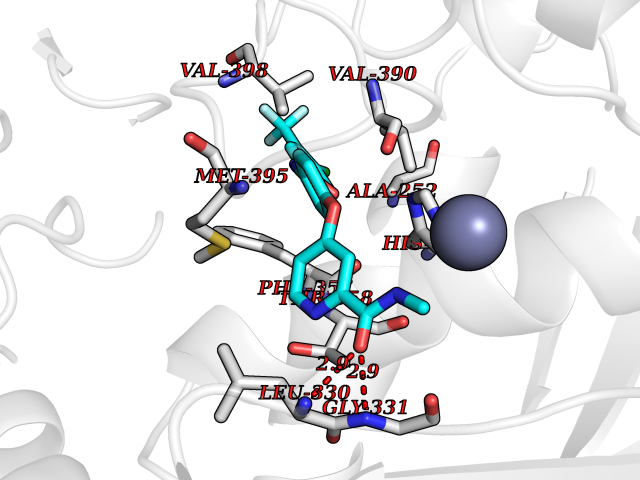 Pymol Map 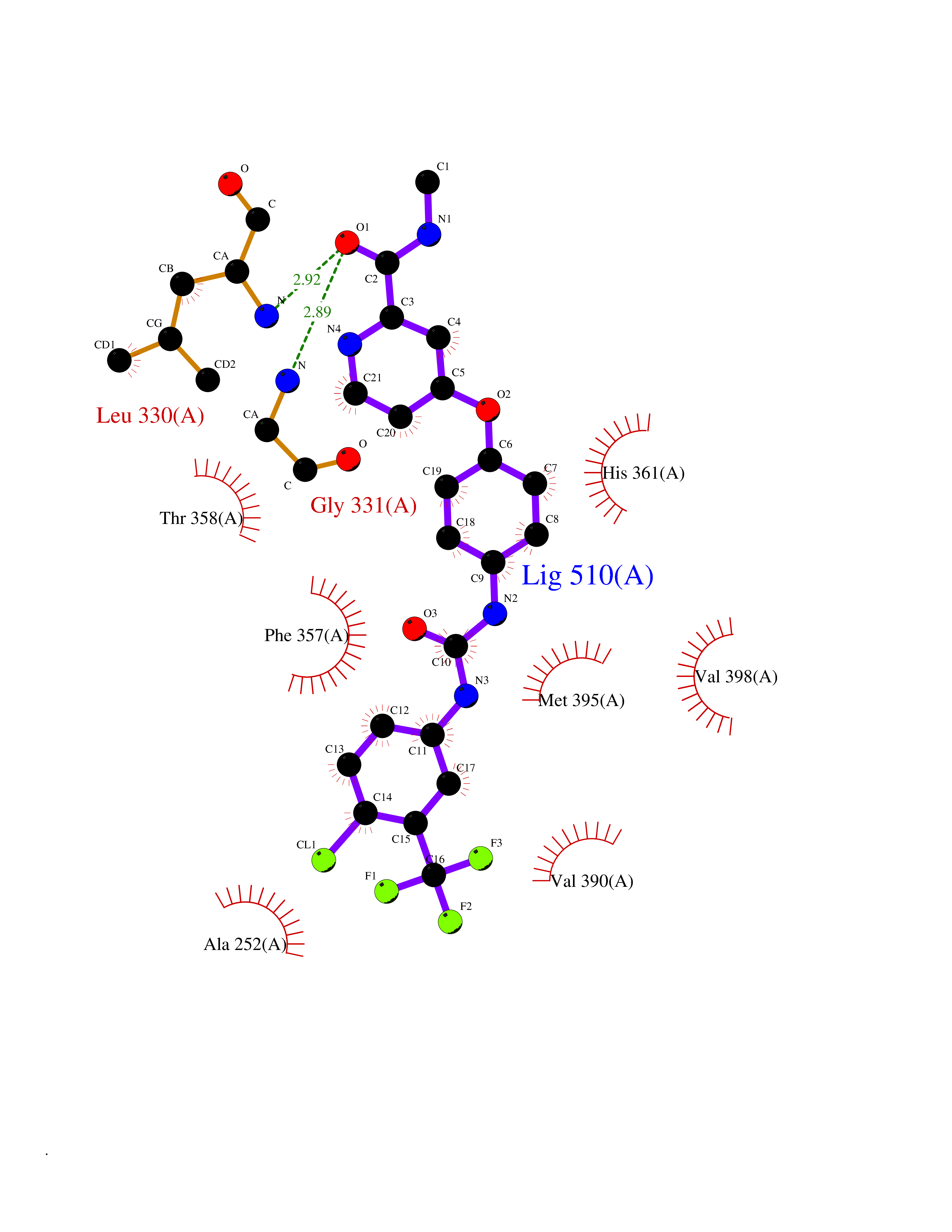 Ligplot Map 3D Binding mode Sequence ASLSRFVETLVVADDKMAAFHGAGLKRYLLTVMAAAAKAFKHPSIRNPVSLVVTRLVILEGPQVGPSAAQTLRSFCAWQRGLNTPEDSDPDHFDTAILFTRQDLCGVSTCDTLGMADVGTVCDPARSCAIVEDDGLQSAFTAAHQLGHVFNMLHDNSKPCISLNGPLSTSRHVMAPVMAHVDPEEPWSPCSARFITDFLDNGYGHCLLDKPEAPLHLPVTFPGKDYDADRQCQLTFGPDSRHCPQLPPPCAALWCSGHLNGHAMCQTKHSPWADGTPCGPAQACMGGRCLH Hydrogen bonds contact Hydrophobic contact | ||||