Job Results:
Ligand
Structure
Job ID
b5aa43c6924a21fc75a6162a600078fc
Job name
NA
Time
2026-03-04 17:25:26
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 81 | Steroid 21-hydroxylase | 4Y8W | 8.22 | |
Target general information Gen name CYP21A2 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms CYP21;CYP21B Protein family Cytochrome P450 family Biochemical class Oxidoreductase Function Heme binding.Iron ion binding.Steroid 21-monooxygenase activity.Steroid binding.Steroid hydroxylase activity. Related diseases Adrenal hyperplasia 3 (AH3) [MIM:201910]: A form of congenital adrenal hyperplasia, a common recessive disease due to defective synthesis of cortisol. Congenital adrenal hyperplasia is characterized by androgen excess leading to ambiguous genitalia in affected females, rapid somatic growth during childhood in both sexes with premature closure of the epiphyses and short adult stature. Four clinical types: 'salt wasting' (SW, the most severe type), 'simple virilizing' (SV, less severely affected patients), with normal aldosterone biosynthesis, 'non-classic form' or late-onset (NC or LOAH) and 'cryptic' (asymptomatic). {ECO:0000269|PubMed:10051010, ECO:0000269|PubMed:10094562, ECO:0000269|PubMed:10198222, ECO:0000269|PubMed:10364682, ECO:0000269|PubMed:10391209, ECO:0000269|PubMed:10408778, ECO:0000269|PubMed:10408786, ECO:0000269|PubMed:10443693, ECO:0000269|PubMed:10496074, ECO:0000269|PubMed:10720040, ECO:0000269|PubMed:11232002, ECO:0000269|PubMed:11598371, ECO:0000269|PubMed:11600539, ECO:0000269|PubMed:11746135, ECO:0000269|PubMed:12213891, ECO:0000269|PubMed:12222711, ECO:0000269|PubMed:12788866, ECO:0000269|PubMed:12887291, ECO:0000269|PubMed:12915679, ECO:0000269|PubMed:1406699, ECO:0000269|PubMed:1406709, ECO:0000269|PubMed:14676460, ECO:0000269|PubMed:14715874, ECO:0000269|PubMed:1496017, ECO:0000269|PubMed:15110320, ECO:0000269|PubMed:15126570, ECO:0000269|PubMed:16046588, ECO:0000269|PubMed:1644925, ECO:0000269|PubMed:16984992, ECO:0000269|PubMed:18319307, ECO:0000269|PubMed:18381579, ECO:0000269|PubMed:18445671, ECO:0000269|PubMed:1864962, ECO:0000269|PubMed:1937474, ECO:0000269|PubMed:20080860, ECO:0000269|PubMed:2072928, ECO:0000269|PubMed:21169732, ECO:0000269|PubMed:22014889, ECO:0000269|PubMed:2303461, ECO:0000269|PubMed:27721825, ECO:0000269|PubMed:29328376, ECO:0000269|PubMed:3038528, ECO:0000269|PubMed:3257825, ECO:0000269|PubMed:3260007, ECO:0000269|PubMed:3267225, ECO:0000269|PubMed:3497399, ECO:0000269|PubMed:3871526, ECO:0000269|PubMed:7749410, ECO:0000269|PubMed:8478006, ECO:0000269|PubMed:8485582, ECO:0000269|PubMed:8989258, ECO:0000269|PubMed:9067760, ECO:0000269|PubMed:9187661, ECO:0000269|PubMed:9497336}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01026; DB05667 Interacts with NA EC number 1.14.14.16 Uniprot keywords 3D-structure; Alternative splicing; Congenital adrenal hyperplasia; Disease variant; Endoplasmic reticulum; Heme; Iron; Lipid metabolism; Lipid-binding; Membrane; Metal-binding; Microsome; Monooxygenase; Oxidoreductase; Proteomics identification; Reference proteome; Steroid-binding; Steroidogenesis Protein physicochemical properties Chain ID A,B,C Molecular weight (Da) 50326.2 Length 442 Aromaticity 0.08 Instability index 50.06 Isoelectric point 7.79 Charge (pH=7) 2.07 2D Binding mode Binding energy (Kcal/mol) -11.21  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence KLPPLAPGFLHLLQPDLPIYLLGLTQKFGPIYRLHLGLQDVVVLNSKRTIEEAMVKKWADFAGRPEPLTYKLVSRNYPDLSLGDYSLLWKAHKKLTRSALLLGIRDSMEPVVEQLTQEFCERMRAQPGTPVAIEEEFSLLTCSIICYLTFGDKIKDDNLMPAYYKCIQEVLKTWSHWSIQIVDVIPFLRFFPNPGLRRLKQAIEKRDHIVEMQLRQHKESLVAGQWRDMMDYMLQGVAGQLLEGHVHMAAVDLLIGGTETTANTLSWAVVFLLHHPEIQQRLQEELDHESRVPYKDRARLPLLNATIAEVLRLRPVVPLALPHRTTRPSSISGYDIPEGTVIIPNLQGAHLDETVWERPHEFWPDRFLEPGKNSRALAFGCGARVCLGEPLARLELFVVLTRLLQAFTLLPSGDALPSLQPLPHCSVILKMQPFQVRLQPRG Hydrogen bonds contact Hydrophobic contact | ||||
| 82 | Peroxisome proliferator-activated receptor gamma (PPAR-gamma) | 3B1M | 8.22 | |
Target general information Gen name PPARG Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms PPAR-gamma; Nuclear receptor subfamily 1 group C member 3; NR1C3 Protein family Nuclear hormone receptor family, NR1 subfamily Biochemical class Nuclear hormone receptor Function Nuclear receptor that binds peroxisome proliferators such as hypolipidemic drugs and fatty acids. Once activated by a ligand, the nuclear receptor binds to DNA specific PPAR response elements (PPRE) and modulates the transcription of its target genes, such as acyl-CoA oxidase. It therefore controls the peroxisomal beta-oxidation pathway of fatty acids. Key regulator of adipocyte differentiation and glucose homeostasis. ARF6 acts as a key regulator of the tissue-specific adipocyte P2 (aP2) enhancer. Acts as a critical regulator of gut homeostasis by suppressing NF-kappa-B-mediated proinflammatory responses. Plays a role in the regulation of cardiovascular circadian rhythms by regulating the transcription of ARNTL/BMAL1 in the blood vessels (By similarity). Related diseases Defects in PPARG can lead to type 2 insulin-resistant diabetes and hyptertension. PPARG mutations may be associated with colon cancer. {ECO:0000269|PubMed:10394368}.; DISEASE: Obesity (OBESITY) [MIM:601665]: A condition characterized by an increase of body weight beyond the limitation of skeletal and physical requirements, as the result of excessive accumulation of body fat. {ECO:0000269|PubMed:9753710}. Disease susceptibility may be associated with variants affecting the gene represented in this entry.; DISEASE: Lipodystrophy, familial partial, 3 (FPLD3) [MIM:604367]: A form of lipodystrophy characterized by marked loss of subcutaneous fat from the extremities. Facial adipose tissue may be increased, decreased or normal. Affected individuals show an increased preponderance of insulin resistance, diabetes mellitus and dyslipidemia. {ECO:0000269|PubMed:11788685, ECO:0000269|PubMed:12453919}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Glioma 1 (GLM1) [MIM:137800]: Gliomas are benign or malignant central nervous system neoplasms derived from glial cells. They comprise astrocytomas and glioblastoma multiforme that are derived from astrocytes, oligodendrogliomas derived from oligodendrocytes and ependymomas derived from ependymocytes. {ECO:0000269|PubMed:10851250}. Disease susceptibility may be associated with variants affecting the gene represented in this entry. Polymorphic PPARG alleles have been found to be significantly over-represented among a cohort of American patients with sporadic glioblastoma multiforme suggesting a possible contribution to disease susceptibility. Drugs (DrugBank ID) DB08760; DB07842; DB08121; DB07675; DB06908; DB07111; DB08435; DB07172; DB07208; DB07209; DB06926; DB04270; DB08402; DB07863; DB04689; DB07053; DB07723; DB08302; DB08560; DB07302; DB08915; DB00132; DB05490; DB01118; DB11811; DB01014; DB01393; DB09061; DB03600; DB09201; DB09006; DB00845; DB05854; DB11672; DB14635; DB14034; DB09213; DB07509; DB03756; DB05187; DB06521; DB13873; DB00573; DB13961; DB02266; DB01067; DB01050; DB00159; DB07724; DB00328; DB12007; DB09198; DB14009; DB00244; DB01252; DB06510; DB14011; DB00731; DB12662; DB04224; DB11133; DB02746; DB01132; DB06533; DB04971; DB00912; DB02709; DB00412; DB00795; DB00966; DB06536; DB08604; DB00197; DB00313 Interacts with Q8NFM4; P10909; O60869; Q9BWX5; P42858; P45984; O75376; P55055-1; Q13133; P37231; Q9UBK2; P10276; P19793; P28702; Q96EB6; Q6STE5-1; Q6STE5-2; P37231-1; P19793; Q00535 EC number NA Uniprot keywords 3D-structure; Activator; Alternative splicing; Biological rhythms; Cytoplasm; Diabetes mellitus; Disease variant; DNA-binding; Glycoprotein; Isopeptide bond; Metal-binding; Nucleus; Obesity; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Transcription; Transcription regulation; Ubl conjugation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A Molecular weight (Da) 30792.7 Length 271 Aromaticity 0.08 Instability index 40.72 Isoelectric point 5.82 Charge (pH=7) -3.58 2D Binding mode Binding energy (Kcal/mol) -11.21  Molscript Map 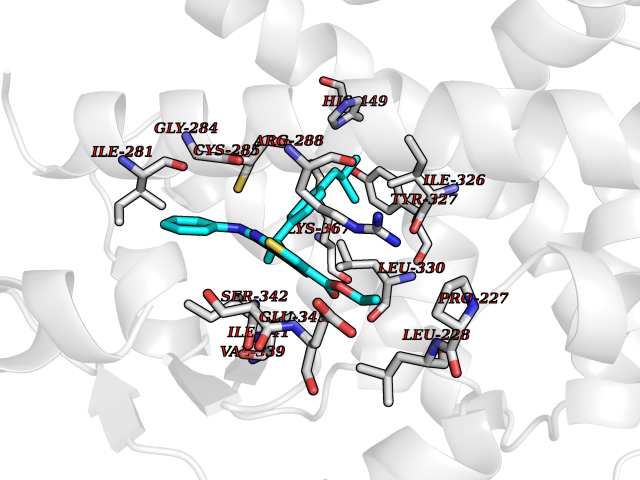 Pymol Map 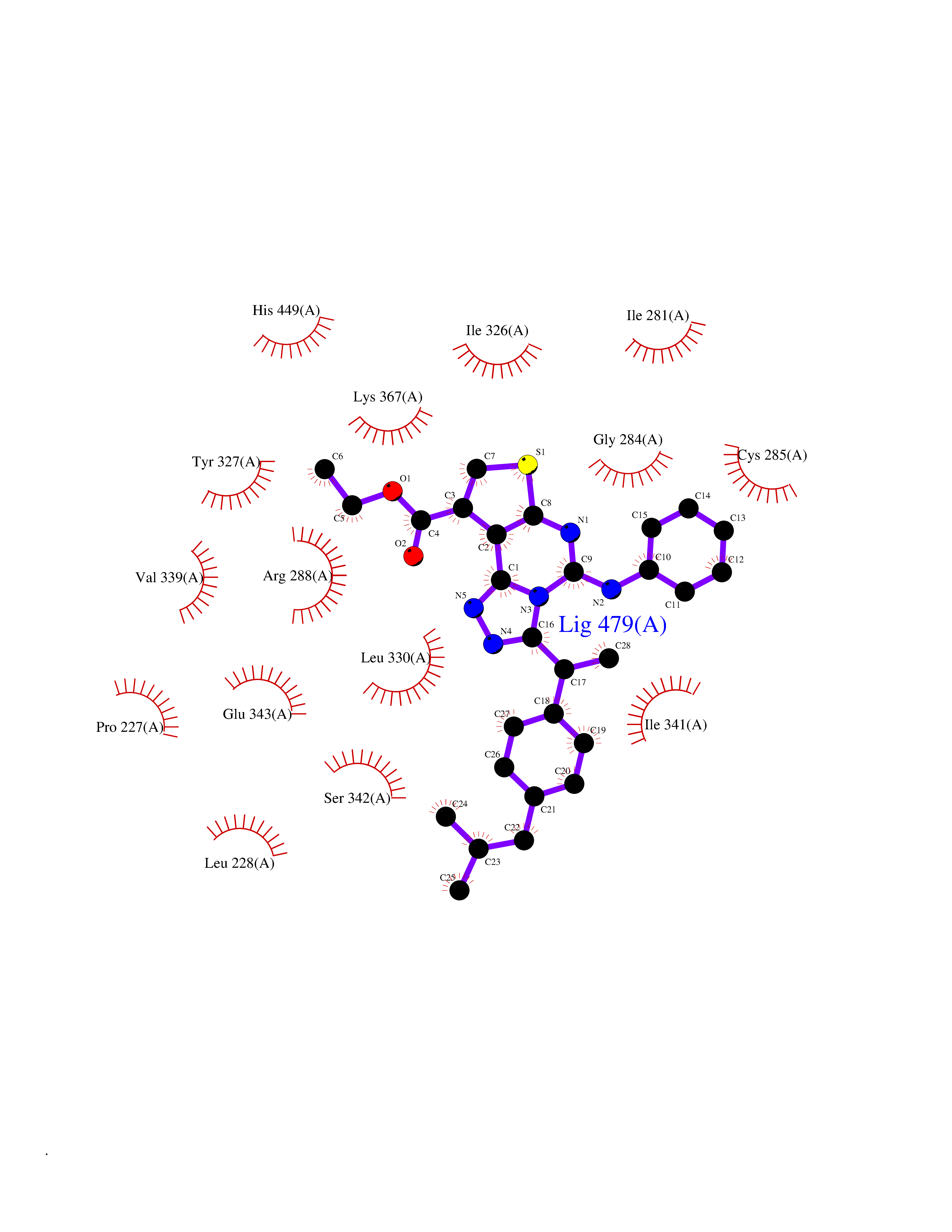 Ligplot Map 3D Binding mode Sequence PESADLRALAKHLYDSYIKSFPLTKAKARAILTGKTTDKSPFVIYDMNSLMMGEDKIKEVAIRIFQGCQFRSVEAVQEITEYAKSIPGFVNLDLNDQVTLLKYGVHEIIYTMLASLMNKDGVLISEGQGFMTREFLKSLRKPFGDFMEPKFEFAVKFNALELDDSDLAIFIAVIILSGDRPGLLNVKPIEDIQDNLLQALELQLKLNHPESSQLFAKLLQKMTDLRQIVTEHVQLLQVIKKTETDMSLHPLLQEIYKDLYPSLLKKLLLAP Hydrogen bonds contact Hydrophobic contact | ||||
| 83 | Xanthine dehydrogenase/oxidase (XDH) | 2E1Q | 8.22 | |
Target general information Gen name XDH Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Xanthine oxidase; Xanthine dehydrogenase; XDHA Protein family Xanthine dehydrogenase family Biochemical class CH/CH(2) oxidoreductase Function Catalyzes the oxidation of hypoxanthine to xanthine. Catalyzes the oxidation of xanthine to uric acid. Contributes to the generation of reactive oxygen species. Has also low oxidase activity towards aldehydes (in vitro). Key enzyme in purine degradation. Related diseases Xanthinuria 1 (XAN1) [MIM:278300]: A disorder characterized by excretion of very large amounts of xanthine in the urine and a tendency to form xanthine stones. Uric acid is strikingly diminished in serum and urine. XAN1 is due to isolated xanthine dehydrogenase deficiency. Patients can metabolize allopurinol. {ECO:0000269|PubMed:10844591, ECO:0000269|PubMed:11379872, ECO:0000269|PubMed:14551354, ECO:0000269|PubMed:9153281}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00640; DB00041; DB00437; DB00993; DB00958; DB01136; DB00856; DB00515; DB00746; DB03328; DB00997; DB03516; DB12466; DB04854; DB03147; DB04335; DB01020; DB00583; DB00170; DB01033; DB00157; DB03841; DB00336; DB01250; DB05262; DB06478; DB01168; DB00339; DB00127; DB01685; DB00831 Interacts with Q9Y3R0-3 EC number NA Uniprot keywords 2Fe-2S; 3D-structure; Cytoplasm; Disease variant; Disulfide bond; FAD; Flavoprotein; Iron; Iron-sulfur; Metal-binding; Molybdenum; NAD; Oxidoreductase; Peroxisome; Proteomics identification; Reference proteome; Secreted Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 143697 Length 1307 Aromaticity 0.08 Instability index 37.9 Isoelectric point 8.01 Charge (pH=7) 7.07 2D Binding mode Binding energy (Kcal/mol) -11.21  Molscript Map 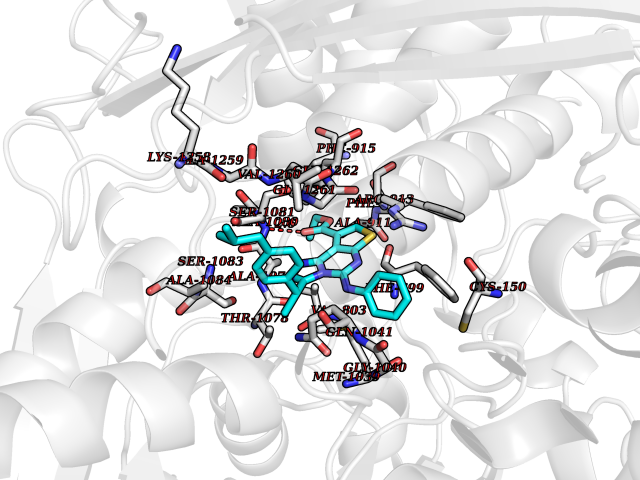 Pymol Map 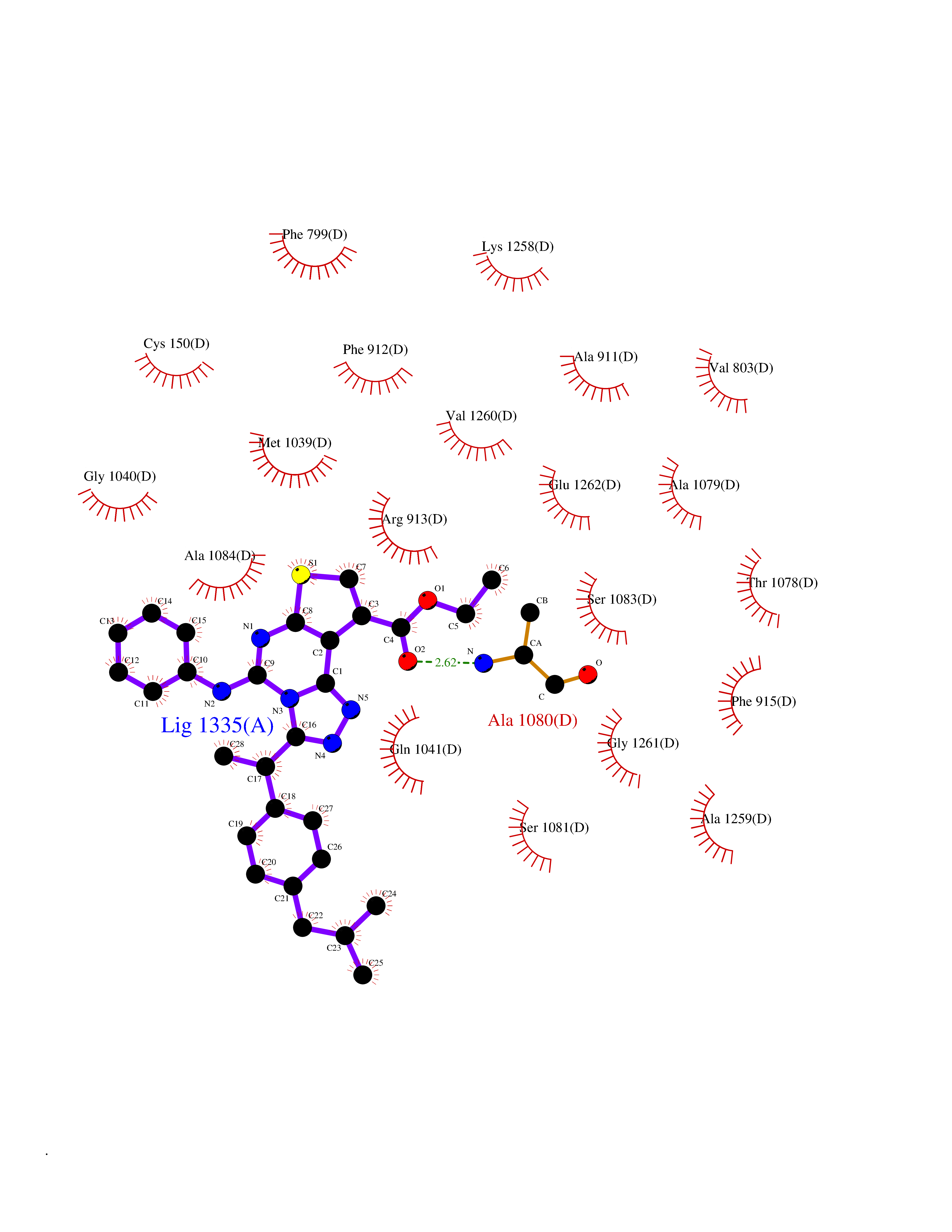 Ligplot Map 3D Binding mode Sequence ADKLVFFVNGRKVVEKNADPETTLLAYLRRKLGLSGTKLGCGEGGCGACTVMLSKYDRLQNKIVHFSANACLAPICSLHHVAVTTVEGIGSTKTRLHPVQERIAKSHGSQCGFCTPGIVMSMYTLLRNQPEPTMEEIENAFQGNLCRCTGYRPILQGFRTFARDGSPSLFKPEEFTPLDPTQEPIFPPELLRLKDTPRKQLRFEGERVTWIQASTLKELLDLKAQHPDAKLVVGNTEIGIEMKFKNMLFPMIVCPAWIPELNSVEHGPDGISFGAACPLSIVEKTLVDAVAKLPAQKTEVFRGVLEQLRWFAGKQVKSVASVGGNIITASPISDLNPVFMASGAKLTLVSRGTRRTVQMDHTFFPGYRKTLLSPEEILLSIEIPYSREGEYFSAFKQASRREDDIAKVTSGMRVLFKPGTTEVQELALCYGGMANRTISALKTTQRQLSKLWKEELLQDVCAGLAEELHLPPDAPGGMVDFRCTLTLSFFFKFYLTVLQKLGQENLEDKCGKLDPTFASATLLFQKDPPADVQLFQEVPKGQSEEDMVGRPLPHLAADMQASGEAVYCDDIPRYENELSLRLVTSTRAHAKIKSIDTSEAKKVPGFVCFISADDVPGSNITGICNDETVFAKDKVTCVGHIIGAVVADTPEHTQRAAQGVKITYEELPAIITIEDAIKNNSFYGPELKIEKGDLKKGFSEADNVVSGEIYIGGQEHFYLETHCTIAVPKGEAGEMELFVSTQNTMKTQSFVAKMLGVPANRIVVRVKRMGGGFGGKVTRSTVVSTAVALAAYKTGRPVRCMLDRDEDMLITGGRHPFLARYKVGFMKTGTVVALEVDHFSNVGNTQDLSQSIMERALFHMDNCYKIPNIRGTGRLCKTNLPSNTAFRGFGGPQGMLIAECWMSEVAVTCGMPAEEVRRKNLYKEGDLTHFNQKLEGFTLPRCWEECLASSQYHARKSEVDKFNKENCWKKRGLCIIPTKFGISFTVPFLNQAGALLHVYTDGSVLLTHGGTEMGQGLHTKMVQVASRALKIPTSKIYISETSTNTVPNTSPTAASVSADLNGQAVYAACQTILKRLEPYKKKNPSGSWEDWVTAAYMDTVSLSATGFYRTPNLGYSFETNSGNPFHYFSYGVACSEVEIDCLTGDHKNLRTDIVMDVGSSLNPAIDIGQVEGAFVQGLGLFTLEELHYSPEGSLHTRGPSTYKIPAFGSIPIEFRVSLLRDCPNKKAIYASKAVGEPPLFLAASIFFAIKDAIRAARAQHTGNNVKELFRLDSPATPEKIRNACVDKFTTLCVTGVPENCKPWSVRV Hydrogen bonds contact Hydrophobic contact | ||||
| 84 | Guanylate cyclase soluble beta-1 (GUCY1B1) | 7D9R | 8.22 | |
Target general information Gen name GUCY1B1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Soluble guanylate cyclase small subunit; Guanylate cyclase soluble subunit beta-3; Guanylate cyclase soluble subunit beta-1; GUCY1B3; GUCSB3; GUC1B3; GCS-beta-3; GCS-beta-1 Protein family Adenylyl cyclase class-4/guanylyl cyclase family Biochemical class Phosphorus-oxygen lyase Function Mediates responses to nitric oxide (NO) by catalyzing the biosynthesis of the signaling molecule cGMP. Related diseases Neurodevelopmental disorder with seizures, hypotonia, and brain imaging abnormalities (NEDSHBA) [MIM:618922]: An autosomal recessive neurodevelopmental disorder characterized by global developmental delay, hypotonia, severe to profound intellectual disability, early-onset epilepsy, and microcephaly. Neuroimaging shows cerebral atrophy, thin corpus callosum and hypomyelination in a majority of cases. Death in childhood may occur. {ECO:0000269|PubMed:27435318, ECO:0000269|PubMed:28097321, ECO:0000269|PubMed:32286009, ECO:0000269|PubMed:33476302, ECO:0000269|PubMed:33500274}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB09401; DB15456 Interacts with Q02108; Q02108-1 EC number EC 4.6.1.2 Uniprot keywords 3D-structure; Alternative splicing; cGMP biosynthesis; Cytoplasm; Direct protein sequencing; GTP-binding; Heme; Iron; Lyase; Metal-binding; Nucleotide-binding; Proteomics identification; Reference proteome Protein physicochemical properties Chain ID A,B Molecular weight (Da) 121177 Length 1071 Aromaticity 0.09 Instability index 42.35 Isoelectric point 6.18 Charge (pH=7) -11.86 2D Binding mode Binding energy (Kcal/mol) -11.21 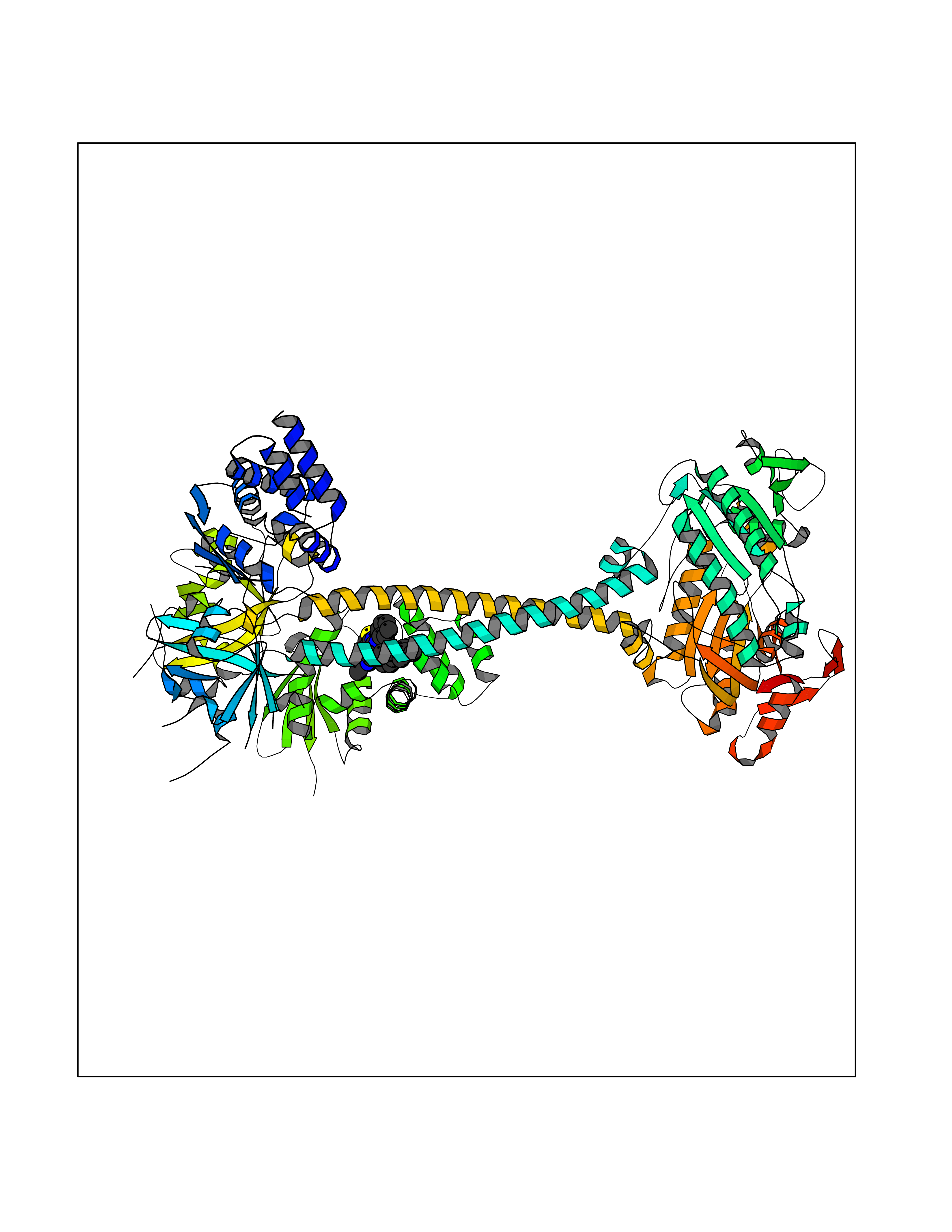 Molscript Map 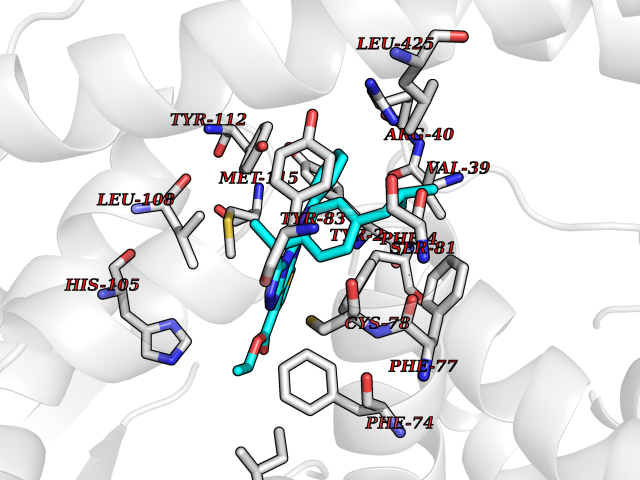 Pymol Map 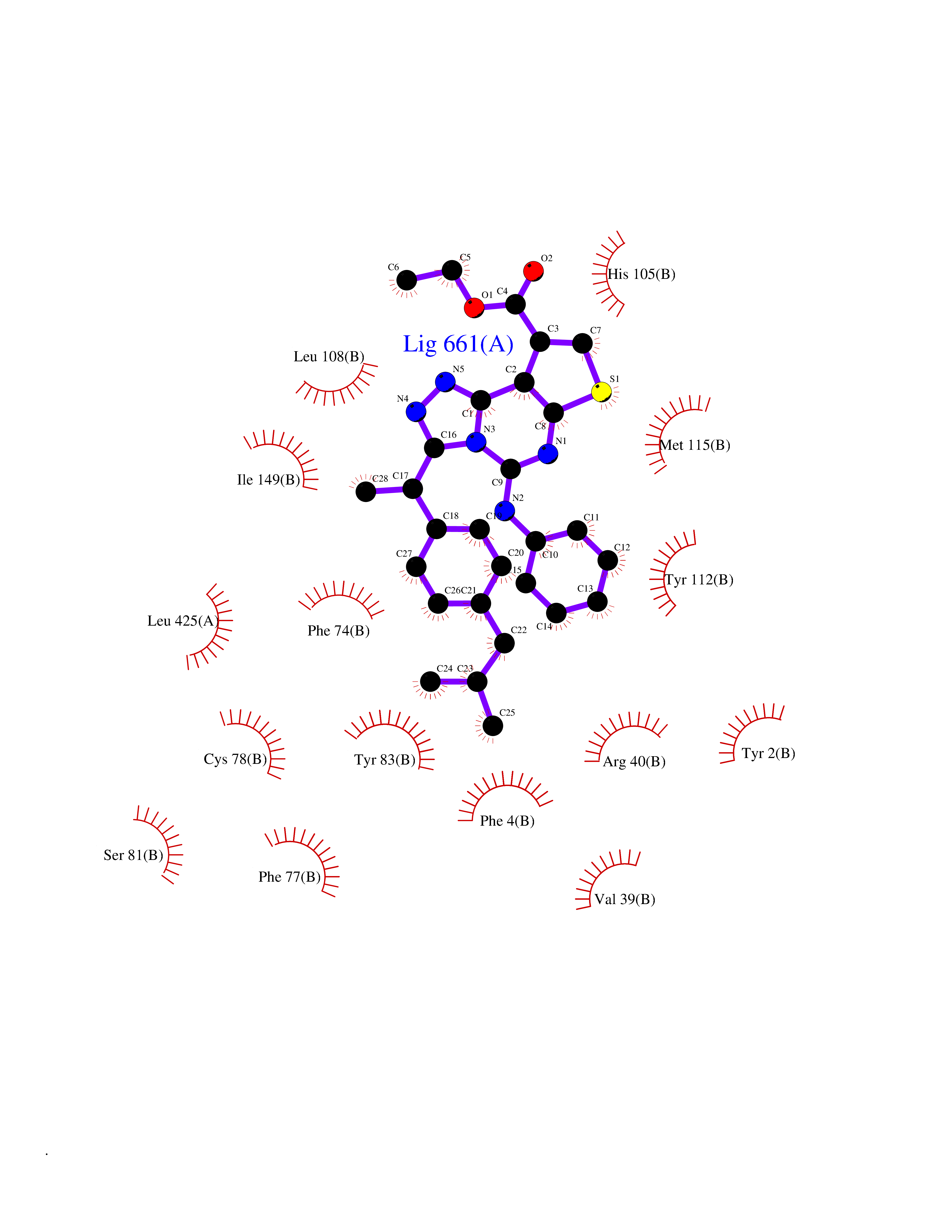 Ligplot Map 3D Binding mode Sequence SRVYLHTLAESICKLIFPEFERLNVALQRTLAKHKFEKTIAEQAVAAGVPVEVIKESLGEEVFKICYEEDENILGVVGGTLKDFLNSFSTLLKQASILCLLHVYYFTTSLILPGIIKAAAHVLYETEVEVSLYLLYSVHSLVIPTSLFCKTFPFHFMFDKDMTILQFGNGIRRLMNRRDKPNFEEYFEILTPKINQTFSGIMTMLNMQFVVRVRRVMDLKGQMIYIVESSAILFLGSPCVLYLSDIPIHNALRDVVLIGEQARAQDGLKKRLGKLKATLEQAHQALEEEKKKTVDLLCSIFPCEVAQQLWQGQVVQAKKFSNVTMLFSDIVGFTAICSQCSPLQVITMLNALYTRFDQQCGELDVYKVETIGDAYCVAGGLHKESDTHAVQIALMAVKMMELSDEVMSPHGEPIKMRIGLHSGSVFAGVVGVKMPRYCLFGNNVTLANKFESCSVPRKINVSPTTYRLLKDCPGFVFTPRSREELPPNFPSEIPGICHFLDAMYGFVNHALELLVIRNYGPEVWEDIKKEAQLDEEGQFLVRIIYDDSKTYDLVAAASKVLNLNAGEILQMFGKMFFVFCQESGYDTILRVLGSNVREFLQNLDALHDHLATIYPGMRAPSFRCTDAEKGKGLILHYYSEREGLQDIVIGIIKTVAQQIHGTEIDMKVIQQRNEECDHTQFLIEEKEESRISPYTFCKAFPFHIIFDRDLVVTQCGNAIYRVLPQLQPGNCSLLSVFSLVRPHIDISFHGILSHINTVFVLRSKEGLLDSCLRLKGQMIYLPEADSILFLCSPSVMNLDDLTRRGLYLSDIPLHDATRDLVLLGEQFREEYKLTQELEILTDRLQLTLRALEDEKKKTDTLLYSVLPPSVANELRHKRPVPAKRYDNVTILFSGIVGFNAFCSKHAGAMKIVNLLNDLYTRFDTLTDSRKNPFVYKVETVGDKYMTVSGLPEPCIHHARSICHLALDMMEIAGQVQVDGESVQITIGIHTGEVVTGVIGQRMPRYCLFGNTVNLTSRTETTGEKGKINVSEYTYRCLMSPENSDPQFHLEHRGPVSMKGKKEPMQVWFLSR Hydrogen bonds contact Hydrophobic contact | ||||
| 85 | Fumarate reductase flavoprotein subunit | 1Y0P | 8.21 | |
Target general information Gen name fccA Organism Shewanella frigidimarina Uniprot ID TTD ID NA Synonyms fcc3 Protein family FAD-dependent oxidoreductase 2 family, FRD/SDH subfamily Biochemical class Oxidoreductase Function Electron carrier activity.Fumarate reductase (menaquinone).Metal ion binding.Nucleic acid binding.Succinate dehydrogenase activity. Related diseases Pigmentary disorder, reticulate, with systemic manifestations, X-linked (PDR) [MIM:301220]: An X-linked recessive disorder characterized by recurrent infections and sterile inflammation in various organs. Diffuse skin hyperpigmentation with a distinctive reticulate pattern is universally evident by early childhood. This is later followed in many patients by hypohidrosis, corneal inflammation and scarring, enterocolitis that resembles inflammatory bowel disease, and recurrent urethral strictures. Melanin and amyloid deposition is present in the dermis. Affected males also have a characteristic facies with frontally upswept hair and flared eyebrows. Female carriers have only restricted pigmentary changes along Blaschko's lines. {ECO:0000269|PubMed:27019227}. The disease is caused by variants affecting the gene represented in this entry. XLPDR is caused by a recurrent intronic mutation that results in missplicing and reduced POLA1 expression. This leads to a decrease in cytosolic RNA:DNA hybrids and constitutive activation of type I interferon responses, but has no effect on cell replication. {ECO:0000269|PubMed:27019227}.; DISEASE: Van Esch-O'Driscoll syndrome (VEODS) [MIM:301030]: An X-linked recessive syndrome characterized by different degrees of intellectual disability, moderate to severe short stature, microcephaly, hypogonadism, and variable congenital malformations. {ECO:0000269|PubMed:31006512}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB04734; DB03147; DB01677; DB03343 Interacts with NA EC number 1.3.2.4 Uniprot keywords 3D-structure; Electron transport; FAD; Flavoprotein; Heme; Iron; Metal-binding; Oxidoreductase; Periplasm; Transport Protein physicochemical properties Chain ID A Molecular weight (Da) 60177.2 Length 568 Aromaticity 0.06 Instability index 27.7 Isoelectric point 6 Charge (pH=7) -8.64 2D Binding mode Binding energy (Kcal/mol) -11.19  Molscript Map 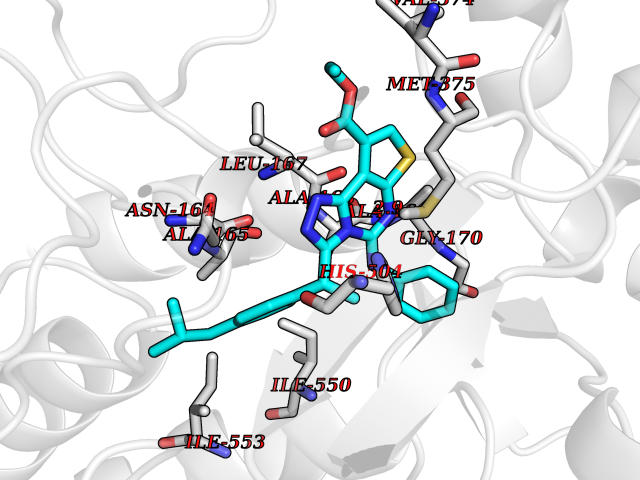 Pymol Map 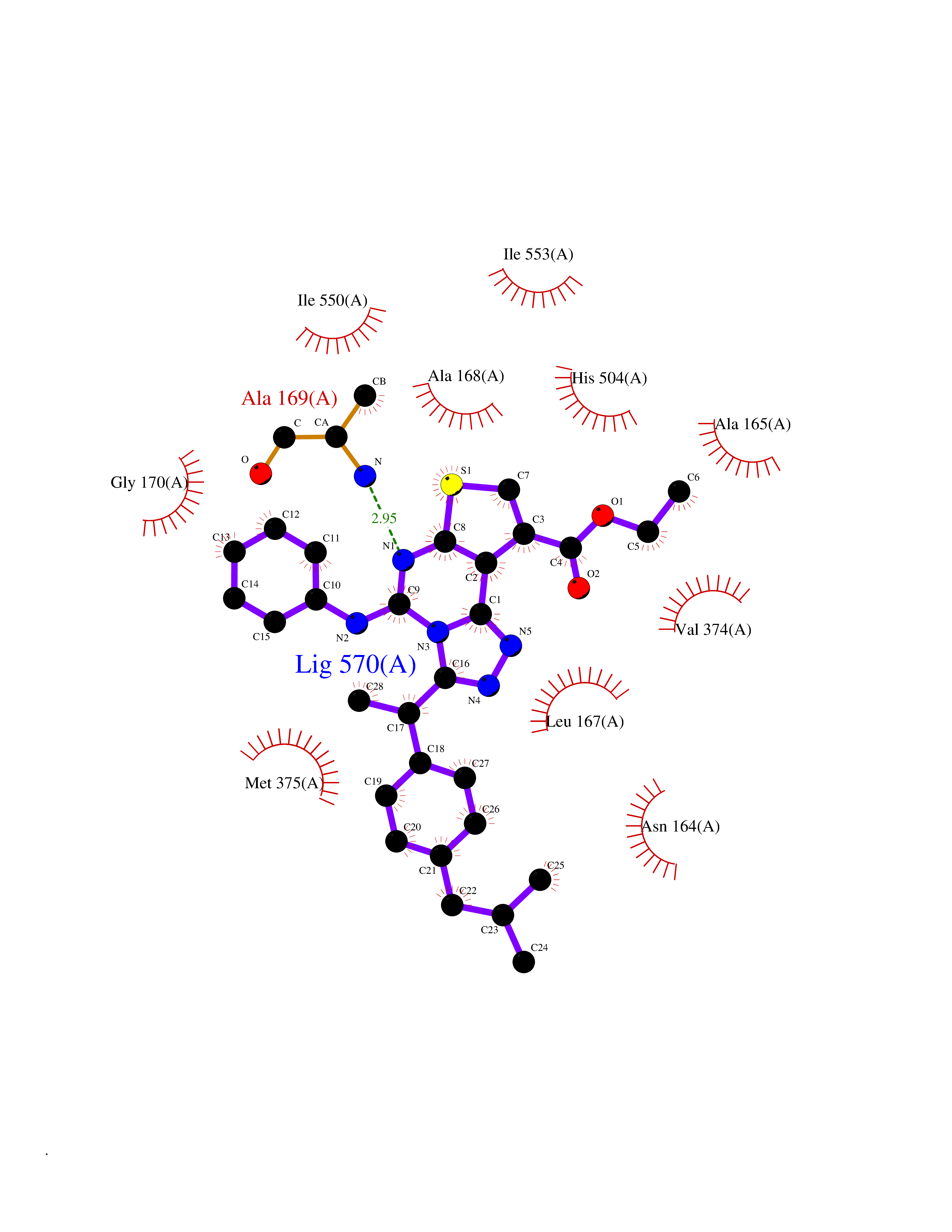 Ligplot Map 3D Binding mode Sequence ADNLAEFHVQNQECDSCHTPDGELSNDSLTYENTQCVSCHGTLAEVAETTKHEHYNAHASHFPGEVACTSCHSAHEKSMVYCDSCHSFDFNMPYAKKWLRDEPTIAELAKDKSERQAALASAPHDTVDVVVVGSGGAGFSAAISATDSGAKVILIEKEPVIGGNAKLAAGGMNAAWTDQQKAKKITDSPELMFEDTMKGGQNINDPALVKVLSSHSKDSVDWMTAMGADLTDVGMMGGASVNRAHRPTGGAGVGAHVVQVLYDNAVKRNIDLRMNTRGIEVLKDDKGTVKGILVKGMYKGYYWVKADAVILATGGFAKNNERVAKLDPSLKGFISTNQPGAVGDGLDVAENAGGALKDMQYIQAHPTLSVKGGVMVTEAVRGNGAILVNREGKRFVNEITTRDKASAAILAQTGKSAYLIFDDSVRKSLSKIDKYIGLGVAPTADSLVKLGKMEGIDGKALTETVARYNSLVSSGKDTDFERPNLPRALNEGNYYAIEVTPGVHHTMGGVMIDTKAEVMNAKKQVIPGLYGAGEVTGGVHGANRLGGNAISDIITFGRLAGEEAAKYS Hydrogen bonds contact Hydrophobic contact | ||||
| 86 | Aldo-keto reductase family 1 member C1 | 1MRQ | 8.21 | |
Target general information Gen name AKR1C1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms DDH1;DDH Protein family Aldo/keto reductase family Biochemical class Oxidoreductase Function 17-alpha,20-alpha-dihydroxypregn-4-en-3-one dehydrogenase activity.Alditol:NADP+ 1-oxidoreductase activity.Aldo-keto reductase (NADP) activity.Androsterone dehydrogenase (B-specific) activity.Bile acid binding.Carboxylic acid binding.Indanol dehydrogenase activity.Ketosteroid monooxygenase activity.Oxidoreductase activity, acting on NAD(P)H, quinone or similar compound as acceptor.Phenanthrene 9,10-monooxygenase activity.Trans-1,2-dihydrobenzene-1,2-diol dehydrogenase activity. Related diseases Fibrodysplasia ossificans progressiva (FOP) [MIM:135100]: A rare autosomal dominant connective tissue disorder resulting in skeletal malformations and progressive extraskeletal ossification. Heterotopic ossification begins in childhood and can be induced by trauma or may occur without warning. Bone formation is episodic and progressive, leading to a debilitating ankylosis of all major joints of the axial and appendicular skeleton, rendering movement impossible. {ECO:0000269|PubMed:16642017, ECO:0000269|PubMed:19085907, ECO:0000269|PubMed:19330033}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB04674; DB00945; DB07768; DB01039; DB07931; DB06077; DB00959; DB00461; DB00157; DB03467; DB03461; DB00776; DB12612; DB00936 Interacts with P51857; P26045; Q7Z699 EC number 1.1.1.-; 1.1.1.112; 1.1.1.149; 1.1.1.209; 1.1.1.210; 1.1.1.357; 1.1.1.51; 1.1.1.53; 1.1.1.62; 1.3.1.20 Uniprot keywords 3D-structure; Cytoplasm; Direct protein sequencing; Lipid metabolism; NADP; Oxidoreductase; Proteomics identification; Reference proteome Protein physicochemical properties Chain ID A Molecular weight (Da) 36784.9 Length 323 Aromaticity 0.09 Instability index 42.07 Isoelectric point 8.06 Charge (pH=7) 2.42 2D Binding mode Binding energy (Kcal/mol) -11.2 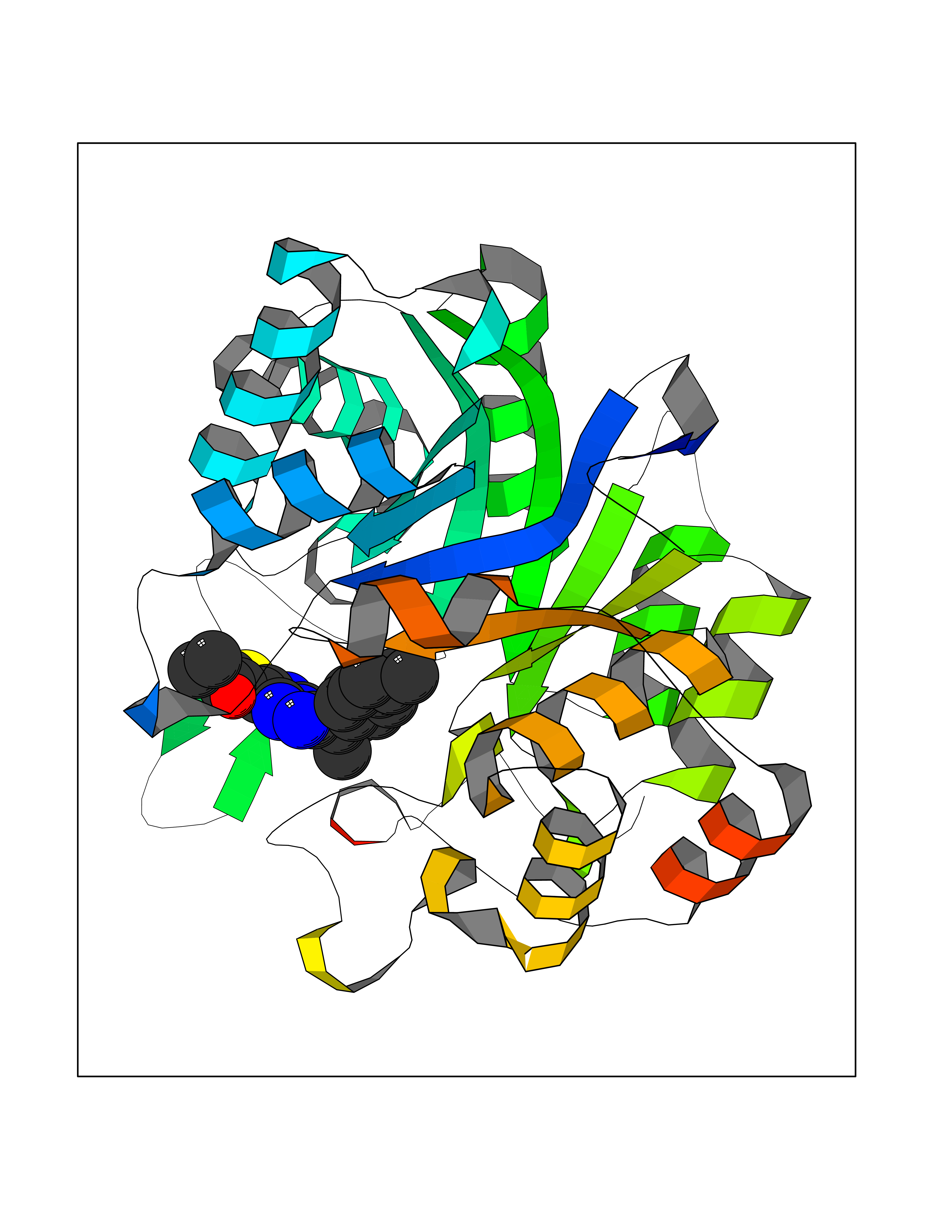 Molscript Map 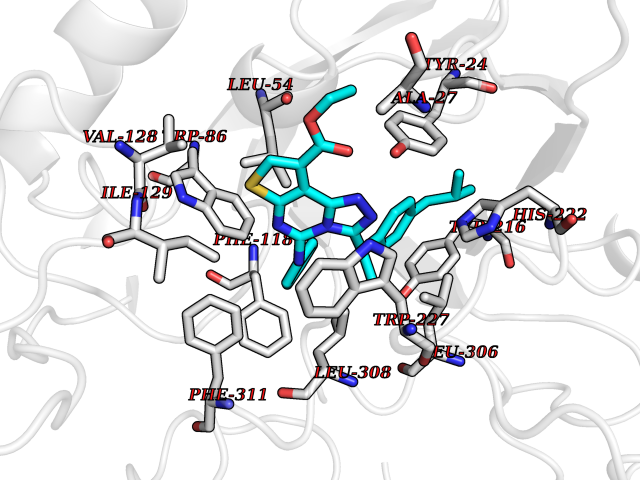 Pymol Map 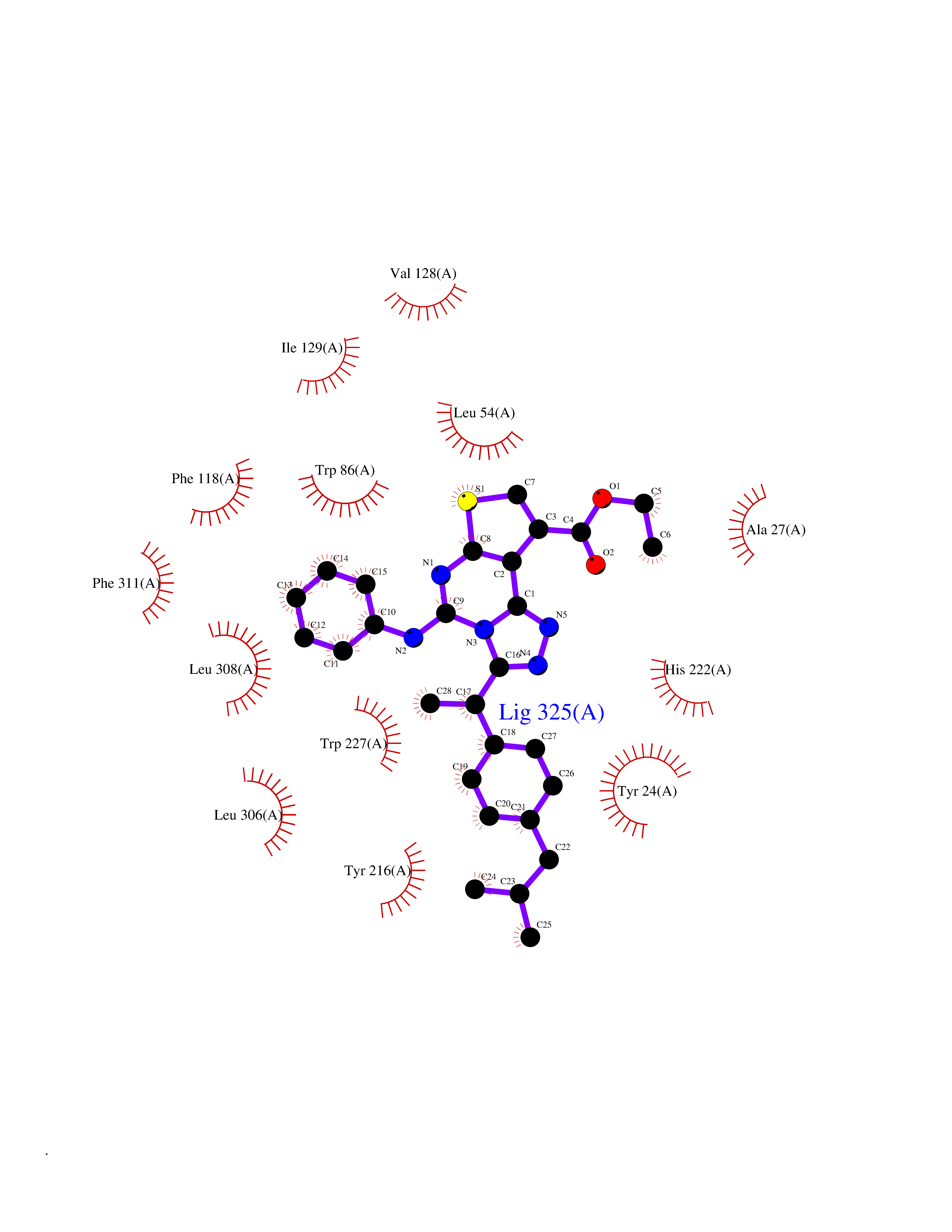 Ligplot Map 3D Binding mode Sequence QDSKYQCVKLNDGHFMPVLGFGTYAPAEVPKSKALEATKLAIEAGFRHIDSAHLYNNEEQVGLAIRSKIADGSVKREDIFYTSKLWCNSHRPELVRPALERSLKNLQLDYVDLYLIHFPVSVKPGEEVIPKDENGKILFDTVDLCATWEAVEKCKDAGLAKSIGVSNFNRRQLEMILNKPGLKYKPVCNQVECHPYFNQRKLLDFCKSKDIVLVAYSALGSHREEPWVDPNSPVLLEDPVLCALAKKHKRTPALIALRYQLQRGVVVLAKSYNEQRIRQNVQVFEFQLTSEEMKAIDGLNRNVRYLTLDIFAGPPNYPFSDEY Hydrogen bonds contact Hydrophobic contact | ||||
| 87 | Peroxisome proliferator-activated receptor delta (PPARD) | 3TKM | 8.21 | |
Target general information Gen name PPARD Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Peroxisomeproliferator-activated receptor beta; Peroxisomeproliferator activated receptor beta/delta; Peroxisome proliferator-activated receptor beta; PPARdelta; PPARB; PPAR-delta; PPAR-beta; Nuclear Protein family Nuclear hormone receptor family, NR1 subfamily Biochemical class Nuclear hormone receptor Function Receptor that binds peroxisome proliferators such as hypolipidemic drugs and fatty acids. Has a preference for poly-unsaturated fatty acids, such as gamma-linoleic acid and eicosapentanoic acid. Once activated by a ligand, the receptor binds to promoter elements of target genes. Regulates the peroxisomal beta-oxidation pathway of fatty acids. Functions as transcription activator for the acyl-CoA oxidase gene. Decreases expression of NPC1L1 once activated by a ligand. Ligand-activated transcription factor. Related diseases Aminoacylase-1 deficiency (ACY1D) [MIM:609924]: An enzymatic deficiency resulting in encephalopathy, unspecific psychomotor delay, psychomotor delay with atrophy of the vermis and syringomyelia, marked muscular hypotonia or normal clinical features. Epileptic seizures are a frequent feature. All affected individuals exhibit markedly increased urinary excretion of several N-acetylated amino acids. {ECO:0000269|PubMed:16274666, ECO:0000269|PubMed:16465618, ECO:0000269|PubMed:17562838, ECO:0000269|PubMed:21414403}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07070; DB07691; DB00132; DB01393; DB05416; DB04801; DB09006; DB05187; DB13873; DB13961; DB09462; DB03338; DB00159; DB07724; DB05188; DB04224; DB02746; DB00412; DB00605; DB00374; DB00197; DB00313; DB08078 Interacts with O60341; P55055-1; Q13133; P42858; P60409; P02545; Q7Z699; Q96EG3 EC number NA Uniprot keywords 3D-structure; Activator; Alternative splicing; DNA-binding; Metal-binding; Nucleus; Proteomics identification; Receptor; Reference proteome; Transcription; Transcription regulation; Ubl conjugation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A Molecular weight (Da) 30805.5 Length 270 Aromaticity 0.1 Instability index 37.2 Isoelectric point 7.88 Charge (pH=7) 1.64 2D Binding mode Binding energy (Kcal/mol) -11.2  Molscript Map 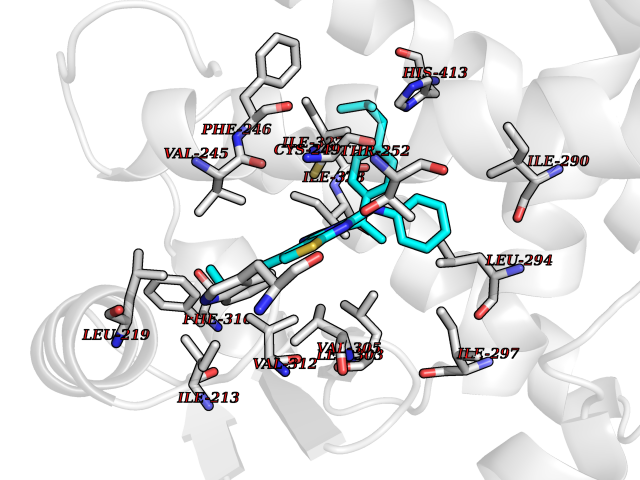 Pymol Map 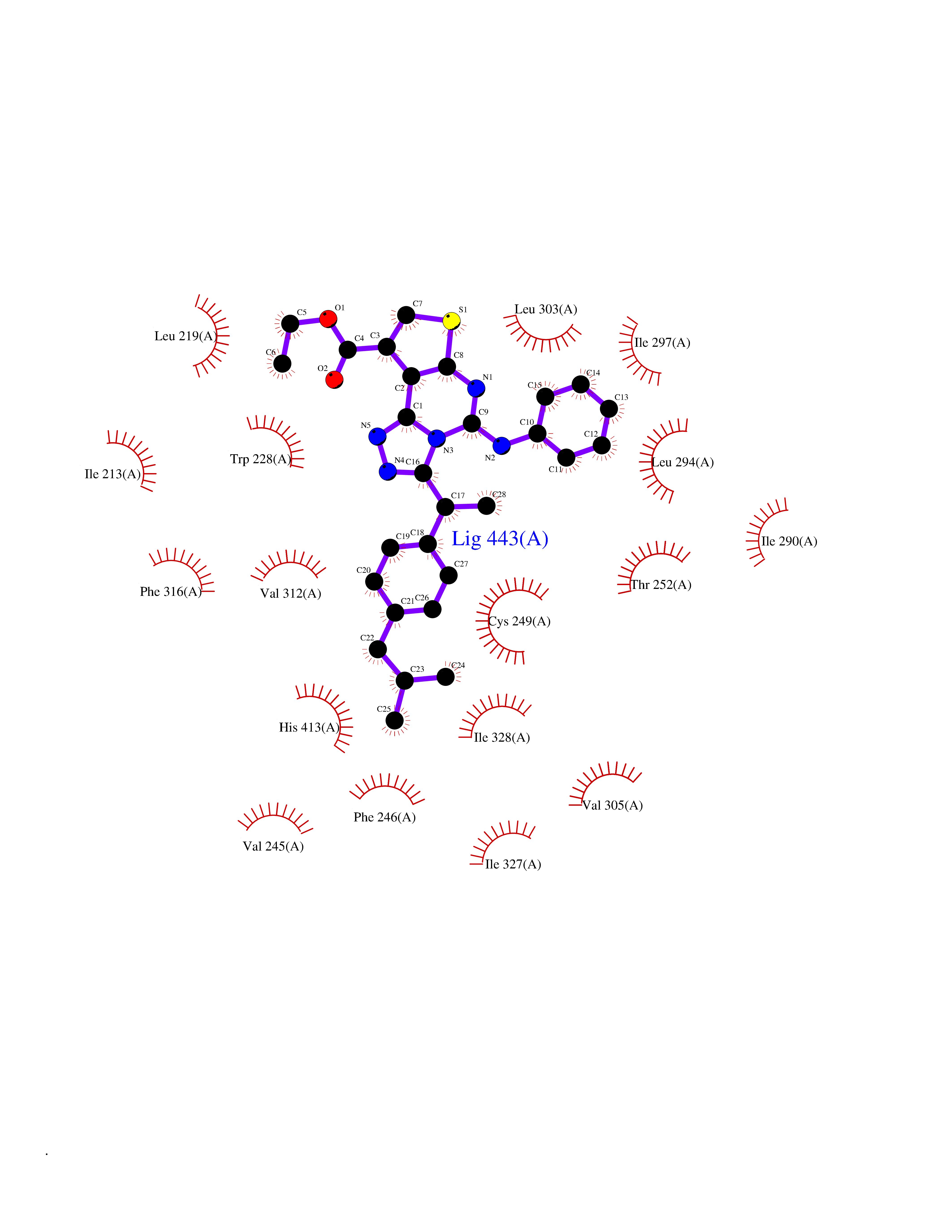 Ligplot Map 3D Binding mode Sequence GSHMQVADLKAFSKHIYNAYLKNFNMTKKKARSILTGKASHTAPFVIHDIETLWQAEKGLVWGLPPYKEISVHVFYRCQCTTVETVRELTEFAKSIPSFSSLFLNDQVTLLKYGVHEAIFAMLASIVNKDGLLVANGSGFVTREFLRSLRKPFSDIIEPKFEFAVKFNALELDDSDLALFIAAIILCGDRPGLMNVPRVEAIQDTILRALEFHLQANHPDAQYLFPKLLQKMADLRQLVTEHAQMMQRIKKTETETSLHPLLQEIYKDMY Hydrogen bonds contact Hydrophobic contact | ||||
| 88 | Dihydroorotate dehydrogenase (quinone), mitochondrial | 4CQ8 | 8.19 | |
Target general information Gen name PFF0160c Organism Plasmodium falciparum (isolate 3D7) Uniprot ID TTD ID NA Synonyms NA Protein family Dihydroorotate dehydrogenase family, Type 2 subfamily Biochemical class Oxidoreductase Function Dihydroorotate dehydrogenase activity. Related diseases Combined oxidative phosphorylation deficiency 33 (COXPD33) [MIM:617713]: An autosomal recessive disorder caused by multiple mitochondrial respiratory chain defects and impaired mitochondrial energy metabolism. Clinical manifestations are highly variable. Affected infants present with cardiomyopathy accompanied by multisystemic features involving liver, kidney, and brain. Death in infancy is observed in some patients. Children and adults present with myopathy and progressive external ophthalmoplegia. {ECO:0000269|PubMed:28942965}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01117 Interacts with NA EC number 1.3.5.2 Uniprot keywords 3D-structure; Flavoprotein; FMN; Membrane; Mitochondrion; Mitochondrion inner membrane; Oxidoreductase; Pyrimidine biosynthesis; Reference proteome; Transit peptide; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A,B Molecular weight (Da) 42573.5 Length 378 Aromaticity 0.1 Instability index 36.63 Isoelectric point 8.21 Charge (pH=7) 3.17 2D Binding mode Binding energy (Kcal/mol) -11.17 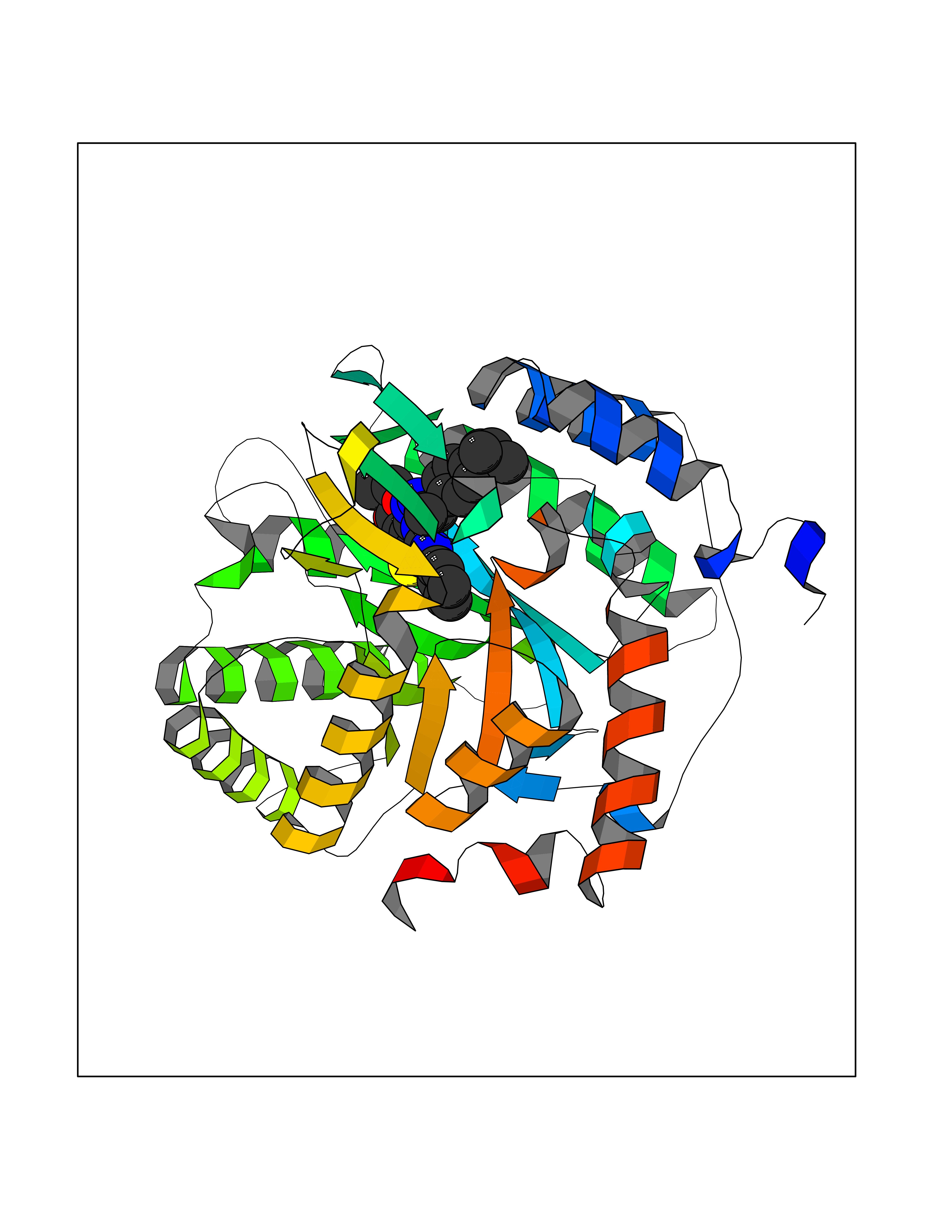 Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence ADPFESYNPEFFLYDIFLKFCLKYIDGEICHDLFLLLGKYNILPYDTSNDSIYACTNIKHLDFINPFGVAAGFDKNGVCIDSILKLGFSFIEIGTITPRGQTGNAKPRIFRDVESRSIINSCGFNNMGCDKVTENLILFRKRQEEDKLLSKHIVGVSIGKNKDTVNIVDDLKYCINKIGRYADYIAINVSSPNTPGLRDNQEAGKLKNIILSVKEEIDNLEKNNFLWFNTTKKKPLVFVKLAPDLNQEQKKEIADVLLETNIDGMIISNTTTQINDIKSFENKKGGVSGAKLKDISTKFICEMYNYTNKQIPIIASGGIFSGLDALEKIEAGASVCQLYSCLVFNGMKSAVQIKRELNHLLYQRGYYNLKEAIGRKHS Hydrogen bonds contact Hydrophobic contact | ||||
| 89 | P2Y purinoceptor 12 | 4PXZ | 8.19 | |
Target general information Gen name P2RY12 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms HORK3 Protein family G-protein coupled receptor 1 family Biochemical class Membrane protein Function ADP receptor activity.G-protein coupled adenosine receptor activity.Guanyl-nucleotide exchange factor activity. Related diseases Bleeding disorder, platelet-type, 8 (BDPLT8) [MIM:609821]: A condition characterized by mild to moderate mucocutaneous bleeding, and excessive bleeding after surgery or trauma. The defect is due to severe impairment of platelet response to ADP resulting in defective platelet aggregation. {ECO:0000269|PubMed:11196645, ECO:0000269|PubMed:12578987, ECO:0000269|PubMed:25428217}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB06441; DB00758; DB06350; DB01240; DB06209; DB01069; DB05553; DB15163; DB08816; DB00208; DB00374; DB16349 Interacts with NA EC number NA Uniprot keywords 3D-structure; Blood coagulation; Cell membrane; Disease variant; Disulfide bond; G-protein coupled receptor; Glycoprotein; Hemostasis; Membrane; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 28830.4 Length 248 Aromaticity 0.17 Instability index 24.19 Isoelectric point 9.39 Charge (pH=7) 10.55 2D Binding mode Binding energy (Kcal/mol) -11.18  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence SLCTRDYKITQVLFPLLYTVLFFVGLITNGLAMRIFFQIRSKSNFIIFLKNTVISDLLMILTFPFKILSDAKLGTGPLRTFVCQVTSVIFYFTMYISISFLGLITIDPKNLLGAKILSVVIWAFMFLLSLPNMILTNRQPRDKNVKKCSFLKSEFGLVWHEIVNYICQVIFWINFLIVVKVFIIIAVFFICFVPFHFARIPYTLSQTRDVFDCTAENTLFYVKESTLWLTSLNACLNPFIYFFLCKSF Hydrogen bonds contact Hydrophobic contact | ||||
| 90 | Cholecystokinin receptor type A (CCKAR) | 7XOV | 8.19 | |
Target general information Gen name CCKAR Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms CCKAR; CCK-AR; CCK-A receptor Protein family G-protein coupled receptor 1 family Biochemical class GPCR rhodopsin Function Receptor forcholecystokinin. Mediates pancreatic growth and enzyme secretion, smooth muscle contraction of the gall bladder and stomach. Has a 1000-fold higher affinity for CCK rather than for gastrin. It modulates feeding and dopamine-induced behavior in the central and peripheral nervous system. This receptor mediates its action by association with G proteins that activate a phosphatidylinositol-calcium second messenger system. Related diseases Mucolipidosis 4 (ML4) [MIM:252650]: An autosomal recessive lysosomal storage disorder characterized by severe psychomotor retardation and ophthalmologic abnormalities, including corneal opacity, retinal degeneration and strabismus. Storage bodies of lipids and water-soluble substances are seen by electron microscopy in almost every cell type of the patients. Most patients are unable to speak or walk independently and reach a maximal developmental level of 1-2 years. All patients have constitutive achlorhydia associated with a secondary elevation of serum gastrin levels. {ECO:0000269|PubMed:10973263, ECO:0000269|PubMed:11013137, ECO:0000269|PubMed:11030752, ECO:0000269|PubMed:11317355, ECO:0000269|PubMed:12182165, ECO:0000269|PubMed:14749347, ECO:0000269|PubMed:15178326, ECO:0000269|PubMed:15523648, ECO:0000269|PubMed:16978393, ECO:0000269|PubMed:18794901, ECO:0000269|PubMed:20159435, ECO:0000269|PubMed:21256127, ECO:0000269|PubMed:28112729}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Corneal dystrophy, Lisch epithelial (LECD) [MIM:620763]: An autosomal dominant corneal dystrophy characterized by gray, band-shaped and feathery opacities in the cornea, that sometimes appear in whorled patterns. The opaque bands consist of clear, densely crowded, intra-epithelial blisters. Vision may be impaired if the bands involve the central cornea. {ECO:0000269|PubMed:37972748}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00403; DB08862; DB04856; DB04867; DB09142 Interacts with NA EC number NA Uniprot keywords 3D-structure; Cell membrane; Disulfide bond; G-protein coupled receptor; Glycoprotein; Lipoprotein; Membrane; Palmitate; Proteomics identification; Receptor; Reference proteome; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID R Molecular weight (Da) 32689 Length 288 Aromaticity 0.13 Instability index 32.87 Isoelectric point 9.73 Charge (pH=7) 15.88 2D Binding mode Binding energy (Kcal/mol) -11.17  Molscript Map 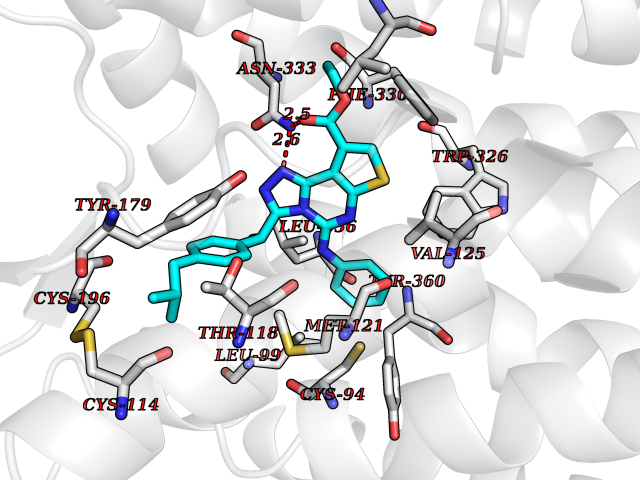 Pymol Map 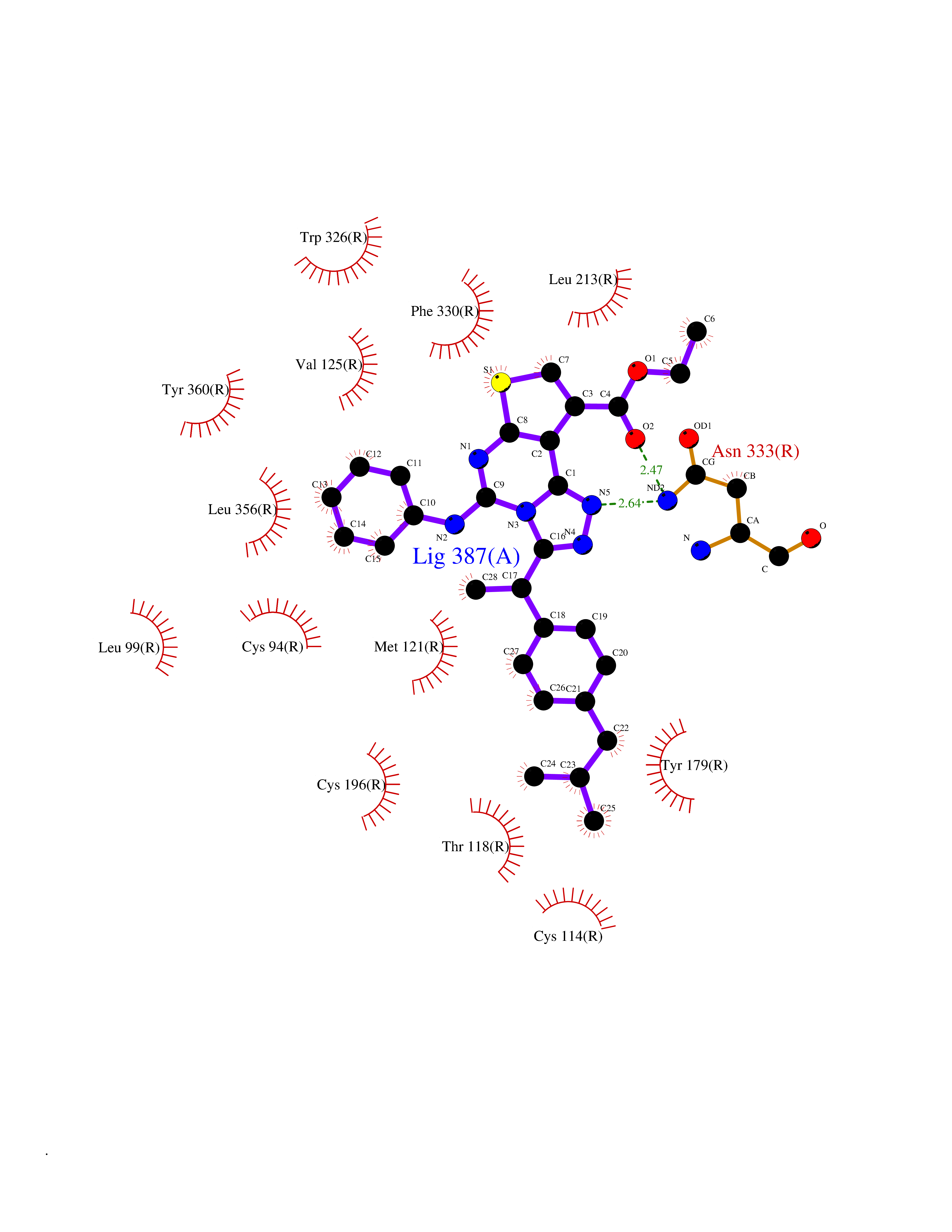 Ligplot Map 3D Binding mode Sequence AVQILLYSLIFLLSVLGNTLVITVLIRNKRMRTVTNIFLLSLAVSDLMLCLFCMPFNLIPNLLKDFIFGSAVCKTTTYFMGTSVSVSTFNLVAISLERYGAICKPLQSRVWQTKSHALKVIAATWCLSFTIMTPYPIYSNLVPFTKNNNQTANMCRFLLPNDVMQQSWHTFLLLILFLIPGIVMMVAYGLISLELYQGIKFEASAANLMAKKRVIRMLIVIVVLFFLCWMPIFSANAWRAYDTASAERRLSGTPISFILLLSYTSSCVNPIIYCFMNKRFRLGFMATF Hydrogen bonds contact Hydrophobic contact | ||||
| 91 | Bacterial Pyruvate decarboxylase (Bact aceE) | 1L8A | 8.19 | |
Target general information Gen name Bact aceE Organism Escherichia coli (strain K12) Uniprot ID TTD ID Synonyms aceE; Pyruvate decarboxylase E1 component Protein family NA Biochemical class Aldehyde/oxo donor oxidoreductase Function The pyruvate dehydrogenase complex catalyzes the overall conversion of pyruvate to acetyl-coa and co(2). It contains multiple copies of three enzymatic components: pyruvate dehydrogenase (e1), dihydrolipoamide acetyltransferase (e2) and lipoamide dehydrogenase. Related diseases Pseudovaginal perineoscrotal hypospadias (PPSH) [MIM:264600]: A form of male pseudohermaphroditism in which 46,XY males show ambiguous genitalia at birth, including perineal hypospadias and a blind perineal pouch, and develop masculinization at puberty. The name of the disorder stems from the finding of a blind-ending perineal opening resembling a vagina and a severely hypospadiac penis with the urethra opening onto the perineum. {ECO:0000269|PubMed:10718838, ECO:0000269|PubMed:10898110, ECO:0000269|PubMed:10999800, ECO:0000269|PubMed:12843198, ECO:0000269|PubMed:15064320, ECO:0000269|PubMed:1522235, ECO:0000269|PubMed:15528927, ECO:0000269|PubMed:15770495, ECO:0000269|PubMed:16098368, ECO:0000269|PubMed:16181229, ECO:0000269|PubMed:7554313, ECO:0000269|PubMed:8626825, ECO:0000269|PubMed:8768837, ECO:0000269|PubMed:9208814, ECO:0000269|PubMed:9745434, ECO:0000269|PubMed:9843052}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01987 Interacts with P0AFG8; P0A8N7; P77439; P46889; P0A6F5; P0AEV9; P0AB83; P0AB89; P0AG40; P05100; P26602; P0A9U1; P0A8L7; P76049; P76170 EC number NA Uniprot keywords 3D-structure; Acetylation; Direct protein sequencing; Magnesium; Metal-binding; Oxidoreductase; Pyruvate; Reference proteome; Thiamine pyrophosphate Protein physicochemical properties Chain ID A,B Molecular weight (Da) 179825 Length 1602 Aromaticity 0.1 Instability index 37.46 Isoelectric point 5.51 Charge (pH=7) -42.23 2D Binding mode Binding energy (Kcal/mol) -11.17  Molscript Map 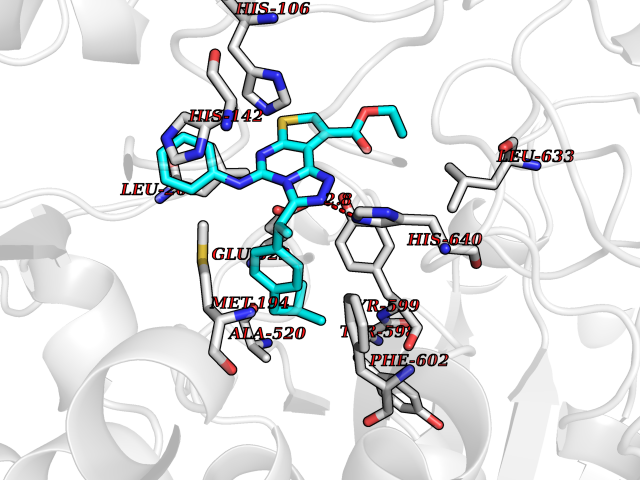 Pymol Map 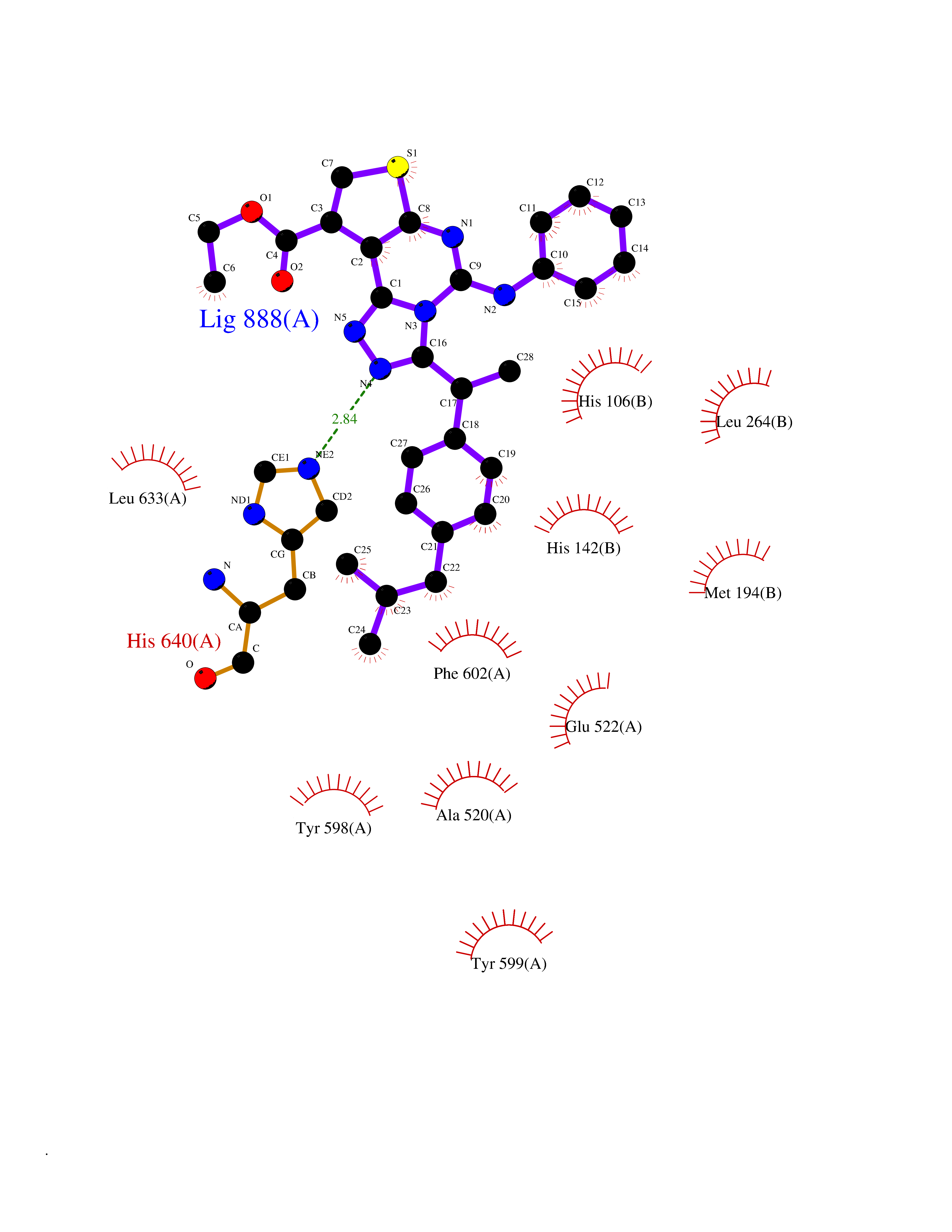 Ligplot Map 3D Binding mode Sequence ISNYINTIPVEEQPEYPGNLELERRIRSAIRWNAIMTVLRASKKDLELGGHMASFQSSATIYDVCFNHFFRARNEQDGGDLVYFQGHISPGVYARAFLEGRLTQEQLDNFRQEVHGNGLSSYPHPKLMPEFWQFPTVSMGLGPIGAIYQAKFLKYLEHRGLKDTSKQTVYAFLGDGEMDEPESKGAITIATREKLDNLVFVINCNLQRLDGPVTGNGKIINELEGIFEGAGWNVIKVMWGSRWDELLRKDTSGKLIQLMNETVDGDYQTFKSKDGAYVREHFFGKYPETAALVADWTDEQIWALNRGGHDPKKIYAAFKKAQETKGKATVILAHTIKGYGMGDAAMDGVRHIRDRFNVPVSDADIEKLPYITFPEGSEEHTYLHAQRQKLHGYLPSRQPNFTEKLELPSLQDFGALLEEQSKEISTTIAFVRALNVMLKNKSIKDRLVPIIADEARTFGMEGLFRQIGIYSPEDEKGQILQEGINELGAGCSWLAAATSYSTNNLPMIPFYIYYSMFGFQRIGDLCWAAGDQQARGFLIGGTSGRTTLNGEGLQHEDGHSHIQSLTIPNCISYDPAYAYEVAVIMHDGLERMYGEKQENVYYYITTLNENYHMPAMPEGAEEGIRKGIYKLETIEGSKGKVQLLGSGSILRHVREAAEILAKDYGVGSDVYSVTSFTELARDGQDCERWNMLHPLETPRVPYIAQVMNDAPAVASTDYMKLFAEQVRTYVPADDYRVLGTDGFGRSDSRENLRHHFEVDASYVVVAALGELAKRGEIDKKVVADAIAKFNIDADKVNPRLAISNYINTIPVEEQPEYPGNLELERRIRSAIRWNAIMTVLRASKKDLELGGHMASFQSSATIYDVCFNHFFRARNEQDGGDLVYFQGHISPGVYARAFLEGRLTQEQLDNFRQEVHGNGLSSYPHPKLMPEFWQFPTVSMGLGPIGAIYQAKFLKYLEHRGLKDTSKQTVYAFLGDGEMDEPESKGAITIATREKLDNLVFVINCNLQRLDGPVTGNGKIINELEGIFEGAGWNVIKVMWGSRWDELLRKDTSGKLIQLMNETVDGDYQTFKSKDGAYVREHFFGKYPETAALVADWTDEQIWALNRGGHDPKKIYAAFKKAQETKGKATVILAHTIKGYGMGDAAMDGVRHIRDRFNVPVSDADIEKLPYITFPEGSEEHTYLHAQRQKLHGYLPSRQPNFTEKLELPSLQDFGALLEEQSKEISTTIAFVRALNVMLKNKSIKDRLVPIIADEARTFGMEGLFRQIGIYSPEDEKGQILQEGINELGAGCSWLAAATSYSTNNLPMIPFYIYYSMFGFQRIGDLCWAAGDQQARGFLIGGTSGRTTLNGEGLQHEDGHSHIQSLTIPNCISYDPAYAYEVAVIMHDGLERMYGEKQENVYYYITTLNENYHMPAMPEGAEEGIRKGIYKLETIEGSKGKVQLLGSGSILRHVREAAEILAKDYGVGSDVYSVTSFTELARDGQDCERWNMLHPLETPRVPYIAQVMNDAPAVASTDYMKLFAEQVRTYVPADDYRVLGTDGFGRSDSRENLRHHFEVDASYVVVAALGELAKRGEIDKKVVADAIAKFNIDADKVNPRLA Hydrogen bonds contact Hydrophobic contact | ||||
| 92 | Neutrophil gelatinase-associated lipocalin (LCN2) | 5NKN | 8.19 | |
Target general information Gen name LCN2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms p25; Siderocalin LCN2; Oncogene 24p3; NGAL; Lipocalin-2; LCN2; 25 kDa alpha-2-microglobulin-related subunit of MMP-9 Protein family Calycin superfamily, Lipocalin family Biochemical class Calycin family Function Iron-trafficking protein involved in multiple processes such as apoptosis, innate immunity and renal development. Binds iron through association with 2,5-dihydroxybenzoic acid (2,5- DHBA), a siderophore that shares structural similarities withbacterial enterobactin, and delivers or removes iron from the cell, depending on the context. Iron-bound form (holo-24p3) is internalized following binding to the SLC22A17 (24p3R) receptor, leading to release of iron and subsequent increase of intracellular iron concentration. In contrast, association of the iron-free form (apo-24p3) with the SLC22A17 (24p3R) receptor is followed by association with an intracellular siderophore, iron chelation and iron transfer to the extracellular medium, thereby reducing intracellular iron concentration. Involved in apoptosis due to interleukin-3 (IL3) deprivation: iron-loaded form increases intracellular iron concentration without promoting apoptosis, while iron-free form decreases intracellular iron levels, inducing expression of the proapoptotic protein BCL2L11/BIM, resulting in apoptosis. Involved in innate immunity, possibly by sequestrating iron, leading to limit bacterial growth. . Related diseases Pseudovaginal perineoscrotal hypospadias (PPSH) [MIM:264600]: A form of male pseudohermaphroditism in which 46,XY males show ambiguous genitalia at birth, including perineal hypospadias and a blind perineal pouch, and develop masculinization at puberty. The name of the disorder stems from the finding of a blind-ending perineal opening resembling a vagina and a severely hypospadiac penis with the urethra opening onto the perineum. {ECO:0000269|PubMed:10718838, ECO:0000269|PubMed:10898110, ECO:0000269|PubMed:10999800, ECO:0000269|PubMed:12843198, ECO:0000269|PubMed:15064320, ECO:0000269|PubMed:1522235, ECO:0000269|PubMed:15528927, ECO:0000269|PubMed:15770495, ECO:0000269|PubMed:16098368, ECO:0000269|PubMed:16181229, ECO:0000269|PubMed:7554313, ECO:0000269|PubMed:8626825, ECO:0000269|PubMed:8768837, ECO:0000269|PubMed:9208814, ECO:0000269|PubMed:9745434, ECO:0000269|PubMed:9843052}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02710; DB01672; DB01926; DB04043; DB01631; DB04476 Interacts with P49419-2; Q9NXW9; Q8WXI3; Q12797-6; Q9BXY8; Q96LC9; P49069; P24863; Q9UKJ5; Q9H1P6; Q9H6B4; O14595; Q08426; Q6NZ36-4; B3EWG5; Q7Z4H3; Q6ISS4; Q5TA76; P80188; Q9UIQ6-2; Q9Y6Y9; Q96JG8; Q8IXL7-2; Q969H8; Q969S2; Q17RF5; P07237; P13667; Q96FA3; Q9NRD5; Q13526; Q9UGP5-2; Q12837; P54646; Q86Y79; O60895; Q9BWG6; P60059; O43765; Q96EQ0; Q8IYX1; Q9UL33-2; P20396; O43715; Q13049; Q99816; Q5W5X9-3; Q99757; P57075-2; Q969M7; Q9UMX0; Q9UHD9; P15692-12; Q14119; Q9Y6T4; Q9H0D6; O96006; A0A1U9X8X8 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Apoptosis; Cytoplasmic vesicle; Direct protein sequencing; Disulfide bond; Glycoprotein; Immunity; Innate immunity; Ion transport; Iron; Iron transport; Proteomics identification; Pyrrolidone carboxylic acid; Reference proteome; Secreted; Signal; Transport Protein physicochemical properties Chain ID A Molecular weight (Da) 19748.4 Length 172 Aromaticity 0.13 Instability index 30.73 Isoelectric point 7.71 Charge (pH=7) 0.72 2D Binding mode Binding energy (Kcal/mol) -11.17  Molscript Map  Pymol Map 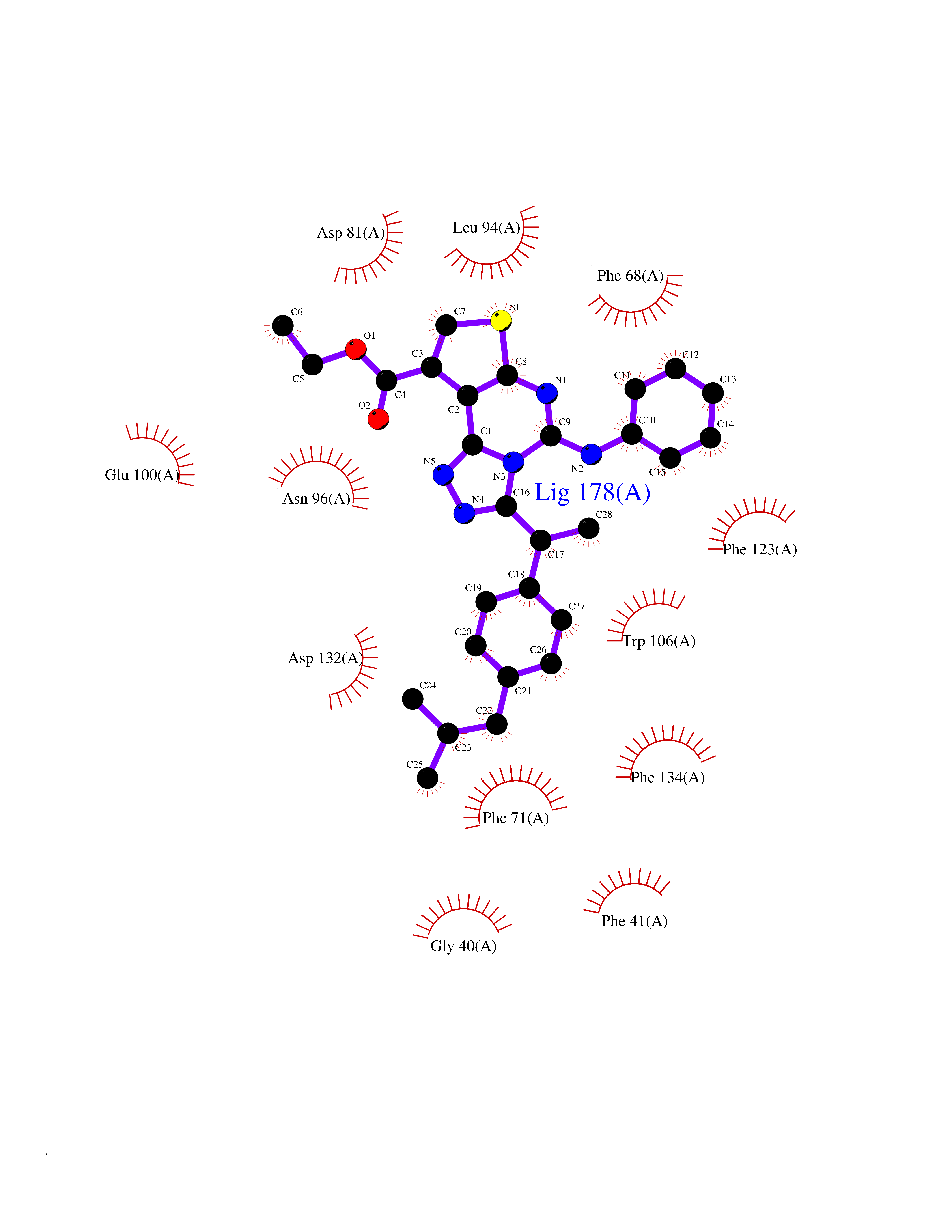 Ligplot Map 3D Binding mode Sequence SDLIPAPPLSKVPLQQNFQDNQFHGKWYVVGVAGNGFLREDKDPIKMAATIYELKEDKSYNVTFQKFPMKKCQYMTDTLVPGSQPGEFTLGNIKSEPGYTSWLVRVVSTNYNQHAMVFFKAVQQNREDFFITLYGRTKELTSELKENFIRFSKSLGLPENHIVFPVPIDQCI Hydrogen bonds contact Hydrophobic contact | ||||
| 93 | Fatty acid-binding protein, intestinal | 3AKM | 8.18 | |
Target general information Gen name FABP2 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms FABPI Protein family Calycin superfamily, Fatty-acid binding protein (FABP) family Biochemical class Transport protein Function Fatty acid binding.Transporter activity. Related diseases Usher syndrome 3B (USH3B) [MIM:614504]: A syndrome characterized by progressive vision and hearing loss during early childhood. Some patients have the so-called 'Charles Bonnet syndrome,' involving decreased visual acuity and vivid visual hallucinations. USH is a genetically heterogeneous condition characterized by the association of retinitis pigmentosa with sensorineural deafness. Age at onset and differences in auditory and vestibular function distinguish Usher syndrome type 1 (USH1), Usher syndrome type 2 (USH2) and Usher syndrome type 3 (USH3). USH3 is characterized by postlingual, progressive hearing loss, variable vestibular dysfunction, and onset of retinitis pigmentosa symptoms, including nyctalopia, constriction of the visual fields, and loss of central visual acuity, usually by the second decade of life. {ECO:0000269|PubMed:22279524}. The disease may be caused by variants affecting the gene represented in this entry.; DISEASE: Charcot-Marie-Tooth disease, axonal, 2W (CMT2W) [MIM:616625]: An autosomal dominant, axonal form of Charcot-Marie-Tooth disease, a disorder of the peripheral nervous system, characterized by progressive weakness and atrophy, initially of the peroneal muscles and later of the distal muscles of the arms. Charcot-Marie-Tooth disease is classified in two main groups on the basis of electrophysiologic properties and histopathology: primary peripheral demyelinating neuropathies (designated CMT1 when they are dominantly inherited) and primary peripheral axonal neuropathies (CMT2). Neuropathies of the CMT2 group are characterized by signs of axonal degeneration in the absence of obvious myelin alterations, normal or slightly reduced nerve conduction velocities, and progressive distal muscle weakness and atrophy. CMT2W patients manifest a peripheral neuropathy mainly affecting the lower limbs and resulting in gait difficulties and distal sensory impairment. Most patients also have upper limb involvement. {ECO:0000269|PubMed:22930593, ECO:0000269|PubMed:26072516, ECO:0000269|PubMed:29235198}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB04557; DB09213; DB00783; DB13952; DB13953; DB13954; DB13955; DB13956; DB01050; DB08231; DB03796; DB01138 Interacts with O95994; Q9NYB0 EC number NA Uniprot keywords 3D-structure; Acetylation; Cytoplasm; Lipid-binding; Proteomics identification; Reference proteome; Transport Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 15075.9 Length 131 Aromaticity 0.11 Instability index 32.01 Isoelectric point 6.88 Charge (pH=7) -0.09 2D Binding mode Binding energy (Kcal/mol) -11.16 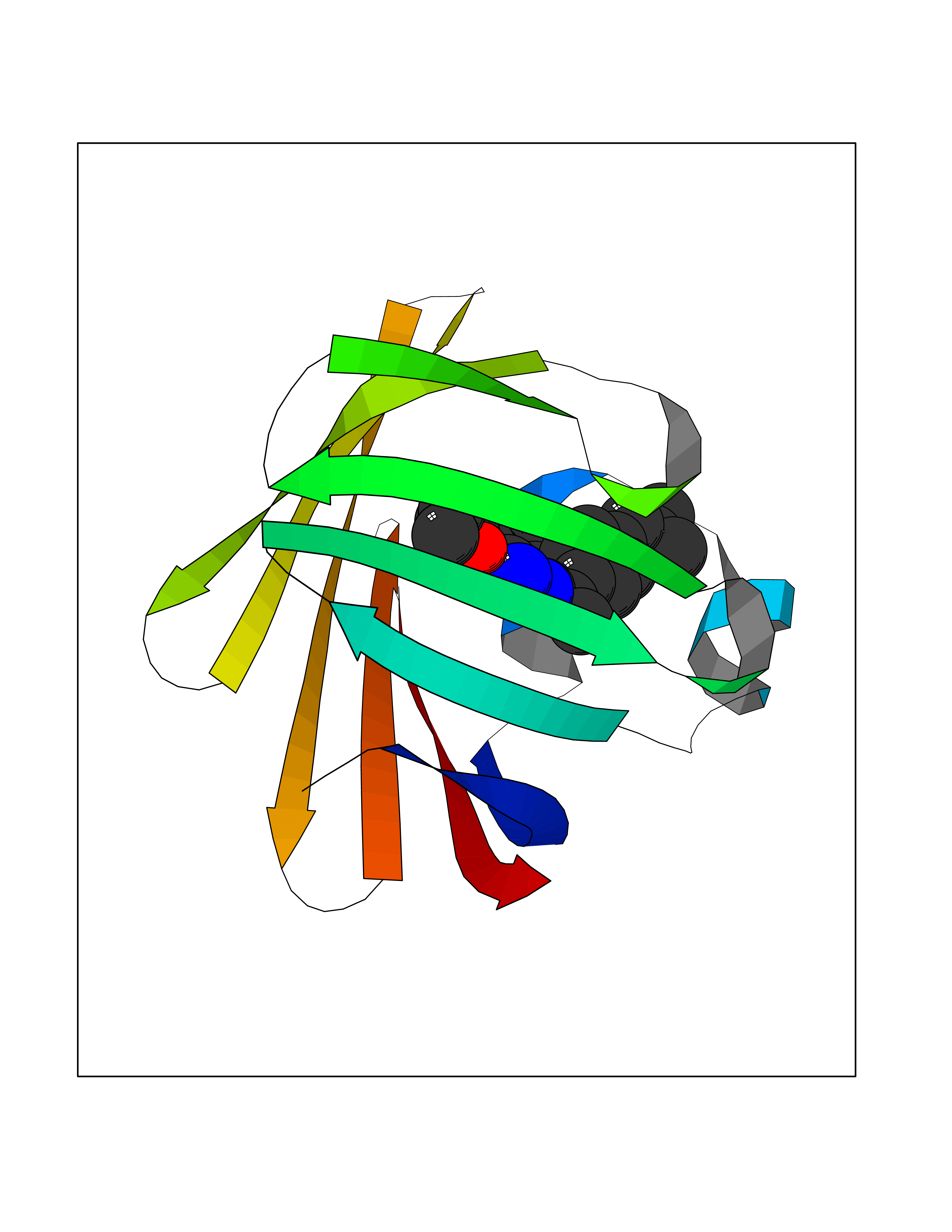 Molscript Map 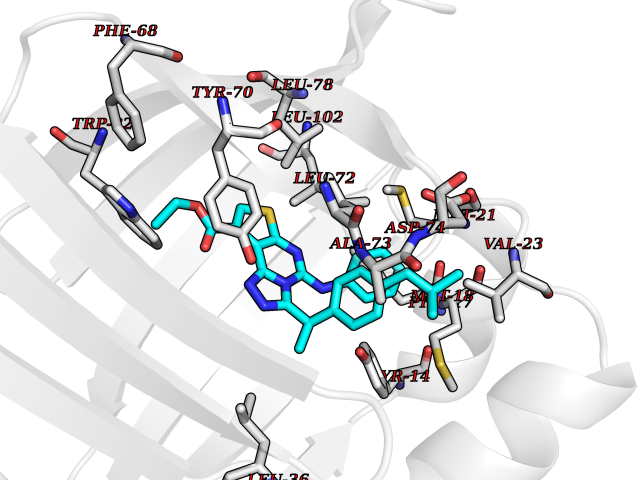 Pymol Map 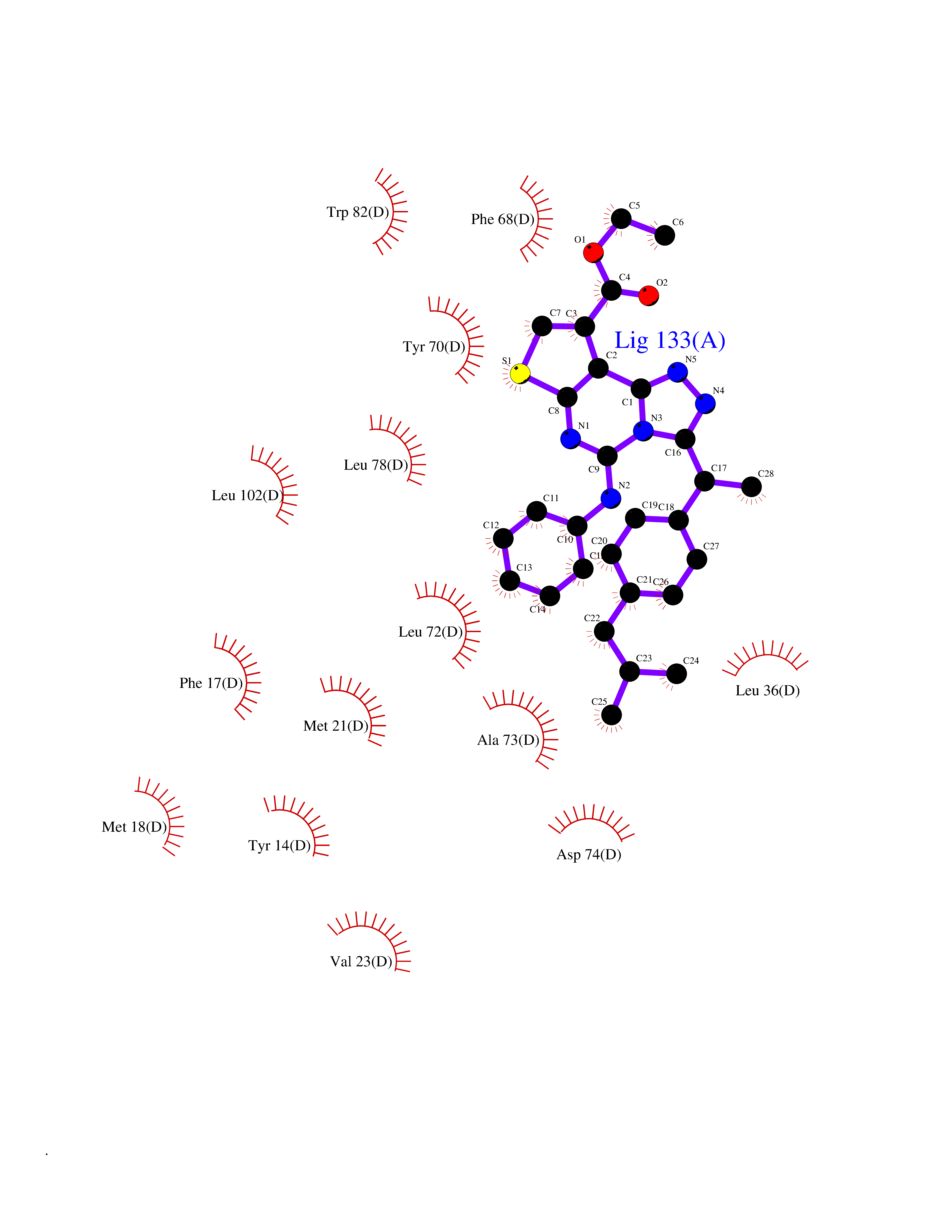 Ligplot Map 3D Binding mode Sequence AFDSTWKVDRSENYDKFMEKMGVNIVKRKLAAHDNLKLTITQEGNKFTVKESSAFRNIEVVFELGVTFNYNLADGTELRGTWSLEGNKLIGKFKRTDNGNELNTVREIIGDELVQTYVYEGVEAKRIFKKD Hydrogen bonds contact Hydrophobic contact | ||||
| 94 | Cytochrome P450 2A6 (CYP2A6) | 1Z11 | 8.18 | |
Target general information Gen name CYP2A6 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Cytochrome P450(I); Cytochrome P450 IIA3; Coumarin 7-hydroxylase; CYPIIA6; CYP2A6; CYP2A3 Protein family Cytochrome P450 family Biochemical class Paired donor oxygen oxidoreductase Function Exhibits a high coumarin 7-hydroxylase activity. Can act in the hydroxylation of the anti-cancer drugs cyclophosphamide and ifosphamide. Competent in the metabolic activation of aflatoxin B1. Constitutes the major nicotine C-oxidase. Possesses low phenacetin O-deethylation activity. Related diseases Lecithin-cholesterol acyltransferase deficiency (LCATD) [MIM:245900]: A disorder of lipoprotein metabolism characterized by inadequate esterification of plasmatic cholesterol. Two clinical forms are recognized: complete LCAT deficiency and fish-eye disease. LCATD is generally referred to the complete form which is associated with absence of both alpha and beta LCAT activities resulting in esterification anomalies involving both HDL (alpha-LCAT activity) and LDL (beta-LCAT activity). It causes a typical triad of diffuse corneal opacities, target cell hemolytic anemia, and proteinuria with renal failure. {ECO:0000269|PubMed:11423760, ECO:0000269|PubMed:12957688, ECO:0000269|PubMed:15994445, ECO:0000269|PubMed:16051254, ECO:0000269|PubMed:16216249, ECO:0000269|PubMed:1681161, ECO:0000269|PubMed:1859405, ECO:0000269|PubMed:2370048, ECO:0000269|PubMed:7607641, ECO:0000269|PubMed:7711728, ECO:0000269|PubMed:8318557, ECO:0000269|PubMed:8432868, ECO:0000269|PubMed:8807342, ECO:0000269|PubMed:9007616, ECO:0000269|PubMed:9741700}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Fish-eye disease (FED) [MIM:136120]: A disorder of lipoprotein metabolism due to partial lecithin-cholesterol acyltransferase deficiency that affects only alpha-LCAT activity. FED is characterized by low plasma HDL and corneal opacities due to accumulation of cholesterol deposits in the cornea ('fish-eye'). {ECO:0000269|PubMed:1516702, ECO:0000269|PubMed:1571050, ECO:0000269|PubMed:15994445, ECO:0000269|PubMed:1737840, ECO:0000269|PubMed:21901787, ECO:0000269|PubMed:8620346, ECO:0000269|PubMed:9261271}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07621; DB07623; DB00316; DB01118; DB00182; DB01435; DB05676; DB01274; DB09274; DB11586; DB00972; DB00443; DB01194; DB01222; DB09061; DB14737; DB06119; DB00356; DB00568; DB00604; DB06470; DB00257; DB00363; DB04665; DB00531; DB06292; DB01234; DB14649; DB00470; DB06374; DB00216; DB00330; DB01039; DB04841; DB01544; DB00544; DB00690; DB01213; DB00983; DB01159; DB00741; DB01181; DB00951; DB01026; DB01006; DB00281; DB06448; DB04871; DB14009; DB01043; DB00170; DB00763; DB00553; DB01028; DB00959; DB00916; DB01011; DB01110; DB00471; DB07609; DB07617; DB00238; DB00184; DB01115; DB06712; DB00717; DB00312; DB03783; DB01174; DB00252; DB01085; DB04977; DB14631; DB00635; DB00396; DB01045; DB15119; DB01037; DB06739; DB01236; DB00675; DB09256; DB09327; DB12816; DB00752; DB00755; DB12245; DB00313; DB09068; DB00495 Interacts with Q9UI14 EC number EC 1.14.13.- Uniprot keywords 3D-structure; Direct protein sequencing; Endoplasmic reticulum; Heme; Iron; Lipid metabolism; Membrane; Metal-binding; Microsome; Monooxygenase; NADP; Oxidoreductase; Proteomics identification; Reference proteome Protein physicochemical properties Chain ID A Molecular weight (Da) 53586.9 Length 467 Aromaticity 0.12 Instability index 40.76 Isoelectric point 9.14 Charge (pH=7) 7.77 2D Binding mode Binding energy (Kcal/mol) -11.16 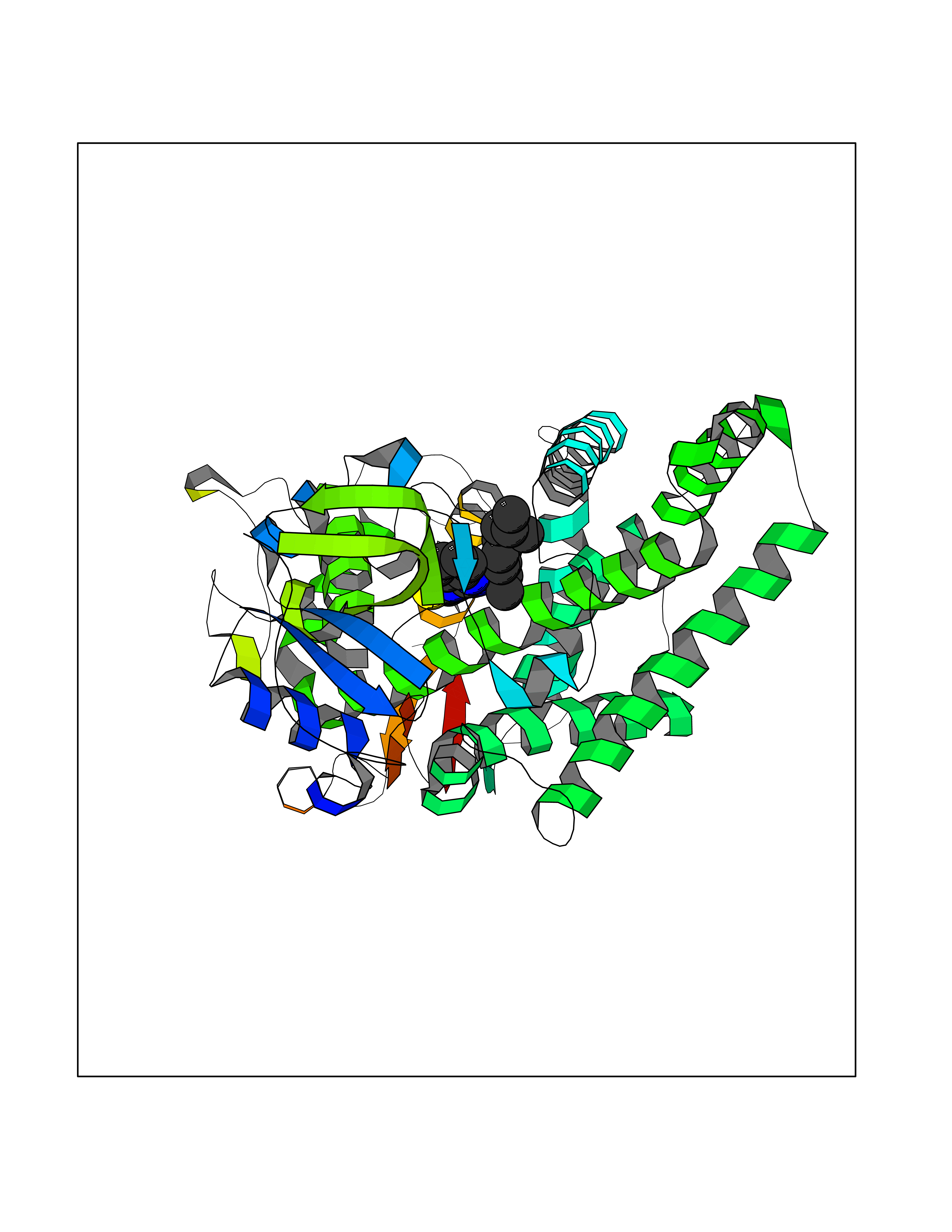 Molscript Map 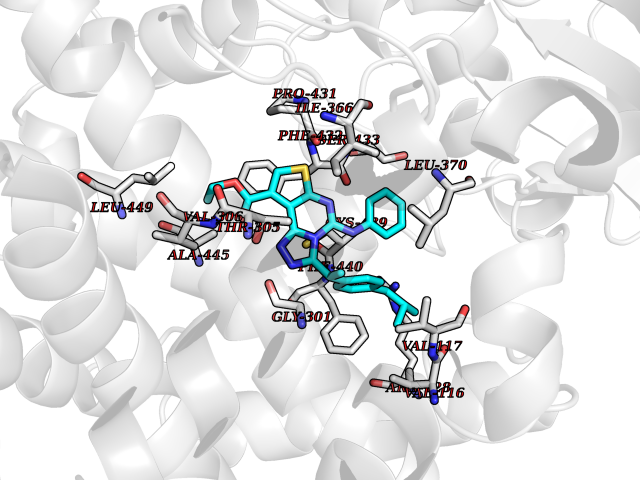 Pymol Map 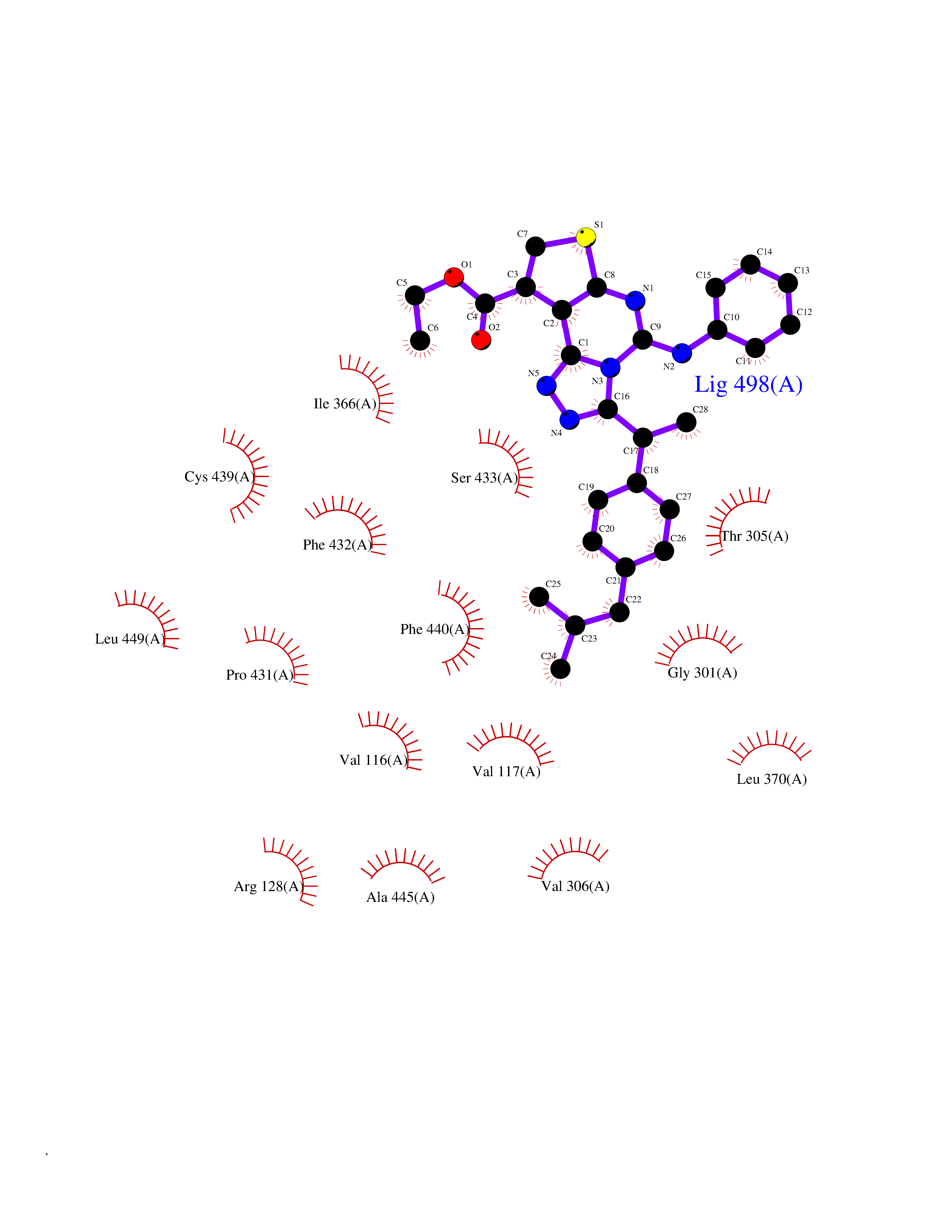 Ligplot Map 3D Binding mode Sequence KGKLPPGPTPLPFIGNYLQLNTEQMYNSLMKISERYGPVFTIHLGPRRVVVLCGHDAVREALVDQAEEFSGRGEQATFDWVFKGYGVVFSNGERAKQLRRFSIATLRDFGVGKRGIEERIQEEAGFLIDALRGTGGANIDPTFFLSRTVSNVISSIVFGDRFDYKDKEFLSLLRMMLGIFQFTSTSTGQLYEMFSSVMKHLPGPQQQAFQLLQGLEDFIAKKVEHNQRTLDPNSPRDFIDSFLIRMQEEEKNPNTEFYLKNLVMTTLNLFIGGTETVSTTLRYGFLLLMKHPEVEAKVHEEIDRVIGKNRQPKFEDRAKMPYMEAVIHEIQRFGDVIPMSLARRVKKDTKFRDFFLPKGTEVYPMLGSVLRDPSFFSNPQDFNPQHFLNEKGQFKKSDAFVPFSIGKRNCFGEGLARMELFLFFTTVMQNFRLKSSQSPKDIDVSPKHVGFATIPRNYTMSFLPRHH Hydrogen bonds contact Hydrophobic contact | ||||
| 95 | Pyruvate dehydrogenase [ubiquinone] | 3EYA | 8.16 | |
Target general information Gen name poxB Organism Escherichia coli (strain K12) Uniprot ID TTD ID NA Synonyms b0871;JW0855 Protein family TPP enzyme family Biochemical class Oxidoreductase Function Flavin adenine dinucleotide binding.Identical protein binding.Lipid binding.Magnesium ion binding.Pyruvate dehydrogenase (quinone) activity.Thiamine pyrophosphate binding. Related diseases Glycogen storage disease 6 (GSD6) [MIM:232700]: A metabolic disorder characterized by mild to moderate hypoglycemia, mild ketosis, growth retardation, and prominent hepatomegaly. Heart and skeletal muscle are not affected. {ECO:0000269|PubMed:9529348}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P07003 EC number 1.2.5.1 Uniprot keywords 3D-structure; Cell inner membrane; Cell membrane; Direct protein sequencing; FAD; Flavoprotein; Lipid-binding; Magnesium; Membrane; Metal-binding; Nucleotide-binding; Oxidoreductase; Pyruvate; Reference proteome; Thiamine pyrophosphate; Ubiquinone Protein physicochemical properties Chain ID A,B,C,D,E,F,G,H,I,J,K,L Molecular weight (Da) 113027 Length 1046 Aromaticity 0.07 Instability index 35.99 Isoelectric point 5.75 Charge (pH=7) -24.38 2D Binding mode Binding energy (Kcal/mol) -11.13  Molscript Map 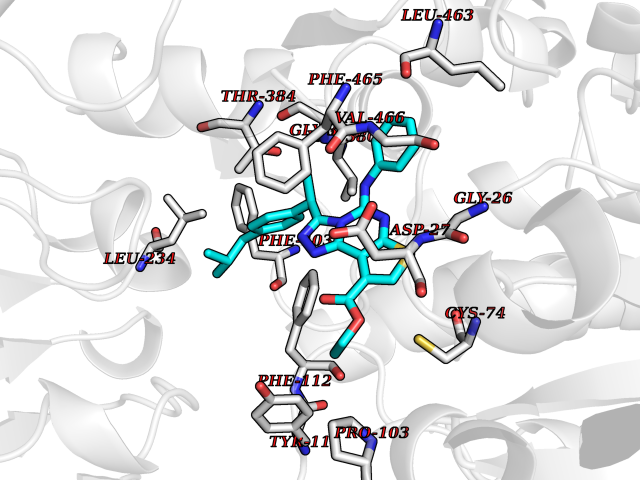 Pymol Map 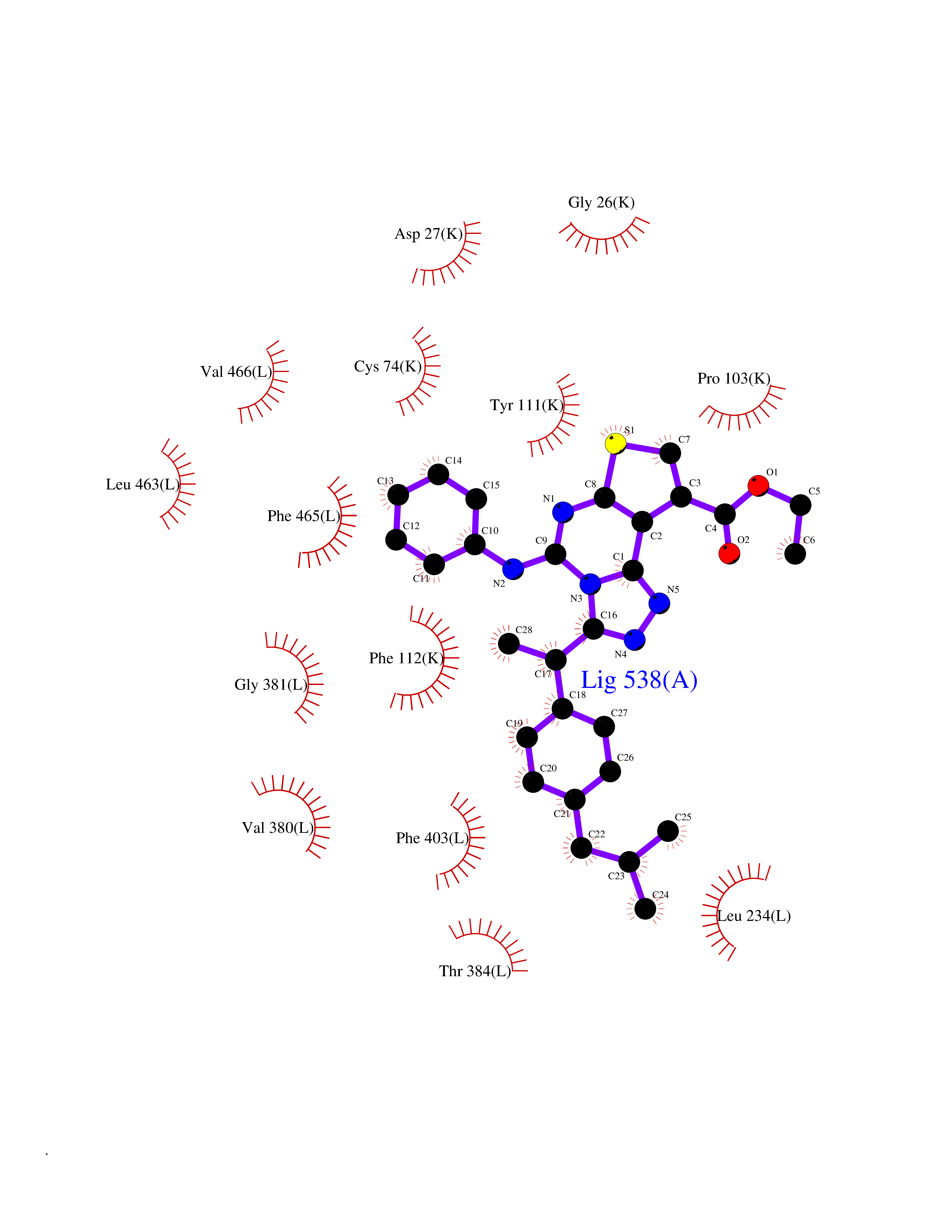 Ligplot Map 3D Binding mode Sequence MKQTVAAYIAKTLESAGVKRIWGVTGDSLNGLSDSLNRMGTIEWMSTRHEEVAAFAAGAEAQLSGELAVCAGSCGPGNLHLINGLFDCHRNHVPVLAIAAHIPSSEIGSGYFQETHPQELFRECSHYCELVSSPEQIPQVLAIAMRKAVLNRGVSVVVLPGDVALKPAPEGATMHWYHAPQPVVTPEEEELRKLAQLLRYSSNIALMCGSGCAGAHKELVEFAGKIKAPIVHALRGKEHVEYDNPYDVGMTGLIGFSSGFHTMMNADTLVLLGTQFPYRAFYPTDAKIIQIDINPASIGAHSKVDMALVGDIKSTLRALLPLVEEKADRKFLDKALEDYRDARKGLDDLAKPSEKAIHPQYLAQQISHFAADDAIFTCDVGTPTVWAARYLKMNGKRRLLGSFNHGSMANAMPQALGAQATEPERQVVAMCGDGGFSMLMGDFLSVVQMKLPVKIVVFNNSVLGFDGTELHDTNFARIAEACGITGIRVEKASEVDEALQRAFSIDGPVLVDVVVAKEELAIPMKQTVAAYIAKTLESAGVKRIWGVTGDSLNGLSDSLNRMGTIEWMSTRHEEVAAFAAGAEAQLSGELAVCAGSCGPGNLHLINGLFDCHRNHVPVLAIAAHIPSSEIGSGYFQETHPQELFRECSHYCELVSSPEQIPQVLAIAMRKAVLNRGVSVVVLPGDVALKPAPEGATMHWYHAPQPVVTPEEEELRKLAQLLRYSSNIALMCGSGCAGAHKELVEFAGKIKAPIVHALRGKEHVEYDNPYDVGMTGLIGFSSGFHTMMNADTLVLLGTQFPYRAFYPTDAKIIQIDINPASIGAHSKVDMALVGDIKSTLRALLPLVEEKADRKFLDKALEDYRDARKGLDDLAKPSEKAIHPQYLAQQISHFAADDAIFTCDVGTPTVWAARYLKMNGKRRLLGSFNHGSMANAMPQALGAQATEPERQVVAMCGDGGFSMLMGDFLSVVQMKLPVKIVVFNNSVLGFVGTELHDTNFARIAEACGITGIRVEKASEVDEALQRAFSIDGPVLVDVVVAKEELAIP Hydrogen bonds contact Hydrophobic contact | ||||
| 96 | Monomeric sarcosine oxidase | 2GF3 | 8.16 | |
Target general information Gen name soxA Organism Bacillus sp. (strain B-0618) Uniprot ID TTD ID NA Synonyms sox Protein family MSOX/MTOX family, MSOX subfamily Biochemical class Oxidoreductase Function Sarcosine oxidase activity. Related diseases Defects in PPARG can lead to type 2 insulin-resistant diabetes and hyptertension. PPARG mutations may be associated with colon cancer. {ECO:0000269|PubMed:10394368}.; DISEASE: Obesity (OBESITY) [MIM:601665]: A condition characterized by an increase of body weight beyond the limitation of skeletal and physical requirements, as the result of excessive accumulation of body fat. {ECO:0000269|PubMed:9753710}. Disease susceptibility may be associated with variants affecting the gene represented in this entry.; DISEASE: Lipodystrophy, familial partial, 3 (FPLD3) [MIM:604367]: A form of lipodystrophy characterized by marked loss of subcutaneous fat from the extremities. Facial adipose tissue may be increased, decreased or normal. Affected individuals show an increased preponderance of insulin resistance, diabetes mellitus and dyslipidemia. {ECO:0000269|PubMed:11788685, ECO:0000269|PubMed:12453919}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Glioma 1 (GLM1) [MIM:137800]: Gliomas are benign or malignant central nervous system neoplasms derived from glial cells. They comprise astrocytomas and glioblastoma multiforme that are derived from astrocytes, oligodendrogliomas derived from oligodendrocytes and ependymomas derived from ependymocytes. {ECO:0000269|PubMed:10851250}. Disease susceptibility may be associated with variants affecting the gene represented in this entry. Polymorphic PPARG alleles have been found to be significantly over-represented among a cohort of American patients with sporadic glioblastoma multiforme suggesting a possible contribution to disease susceptibility. Drugs (DrugBank ID) DB03098; DB01918; DB03517; DB03147; DB03366; DB02083; DB02543 Interacts with NA EC number 1.5.3.1 Uniprot keywords 3D-structure; Cytoplasm; Direct protein sequencing; FAD; Flavoprotein; Oxidoreductase Protein physicochemical properties Chain ID A,B Molecular weight (Da) 42606.4 Length 385 Aromaticity 0.1 Instability index 26.97 Isoelectric point 5.27 Charge (pH=7) -17.18 2D Binding mode Binding energy (Kcal/mol) -11.13  Molscript Map 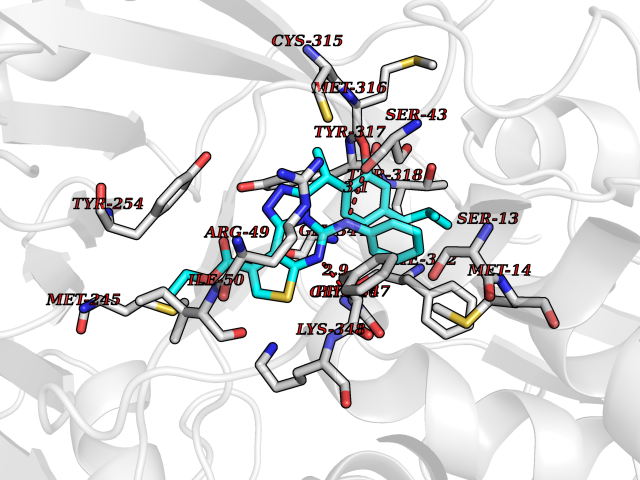 Pymol Map 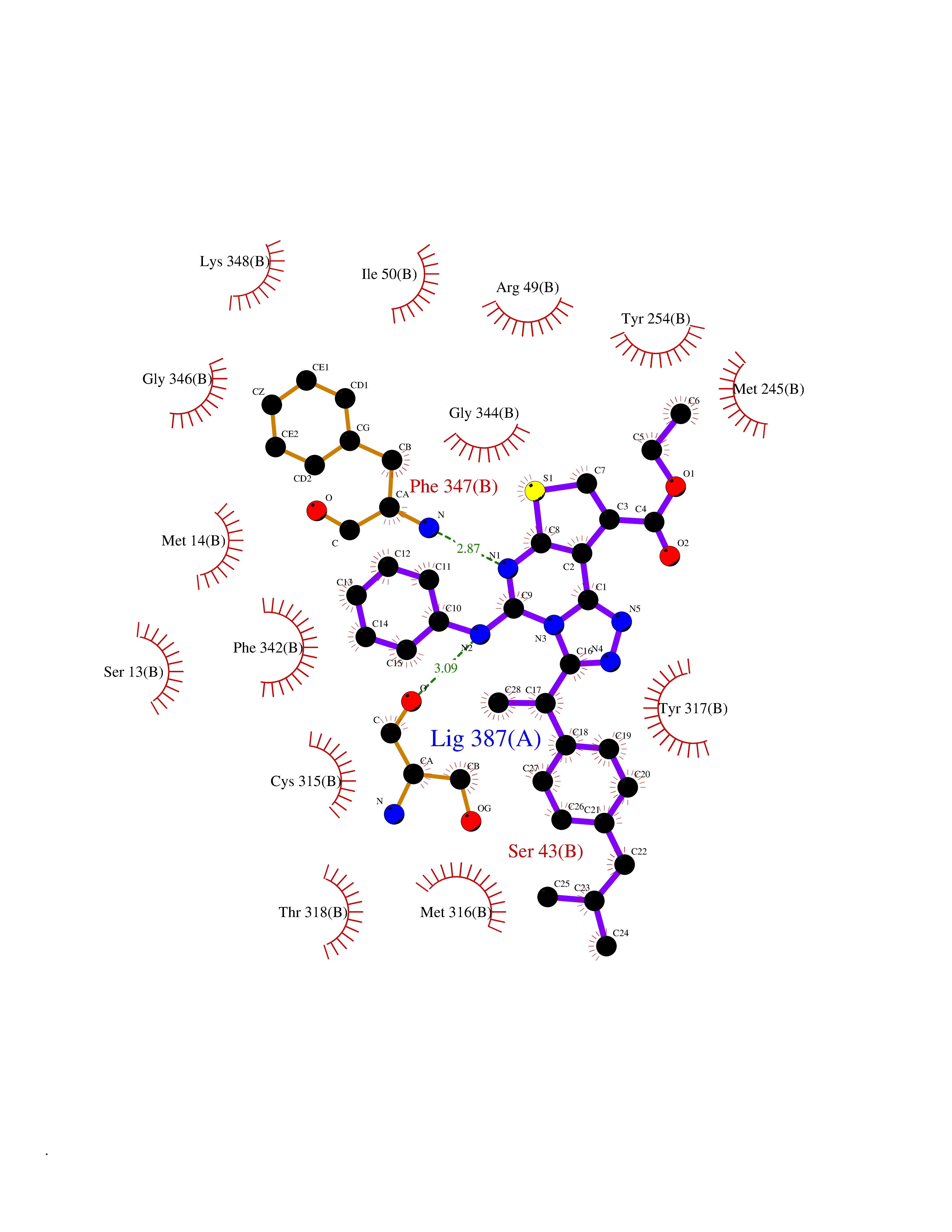 Ligplot Map 3D Binding mode Sequence STHFDVIVVGAGSMGMAAGYQLAKQGVKTLLVDAFDPPHTNGSHHGDTRIIRHAYGEGREYVPLALRSQELWYELEKETHHKIFTKTGVLVFGPKGESAFVAETMEAAKEHSLTVDLLEGDEINKRWPGITVPENYNAIFEPNSGVLFSENCIRAYRELAEARGAKVLTHTRVEDFDISPDSVKIETANGSYTADKLIVSMGAWNSKLLSKLNLDIPLQPYRQVVGFFESDESKYSNDIDFPGFMVEVPNGIYYGFPSFGGCGLKLGYHTFGQKIDPDTINREFGVYPEDESNLRAFLEEYMPGANGELKRGAVCMYTKTLDEHFIIDLHPEHSNVVIAAGFSGHGFKFSSGVGEVLSQLALTGKTEHDISIFSINRPALKESLQ Hydrogen bonds contact Hydrophobic contact | ||||
| 97 | Notch-1 receptor (NOTCH1) | 5L0R | 8.16 | |
Target general information Gen name NOTCH1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms hN1; Translocationassociated notch protein TAN1; Translocation-associated notch protein TAN-1; TAN1; Notch 1 intracellular domain; Notch 1; Neurogenic locus notch homolog protein 1; NICD Protein family NOTCH family Biochemical class Notch protein Function Upon ligand activation through the released notch intracellular domain (NICD) it forms a transcriptional activator complex with RBPJ/RBPSUH and activates genes of the enhancer of split locus. Affects the implementation of differentiation, proliferation and apoptotic programs. Involved in angiogenesis; negatively regulates endothelial cell proliferation and migration and angiogenic sprouting. Involved in the maturation of both CD4(+) and CD8(+) cells in the thymus. Important for follicular differentiation and possibly cell fate selection within the follicle. During cerebellar development, functions as a receptor for neuronal DNER and is involved in the differentiation of Bergmann glia. Represses neuronal and myogenic differentiation. May play an essential role in postimplantation development, probably in some aspect of cell specification and/or differentiation. May be involved in mesoderm development, somite formation and neurogenesis. May enhance HIF1A function by sequestering HIF1AN away from HIF1A. Required for the THBS4 function in regulating protective astrogenesis from the subventricular zone (SVZ) niche after injury. Involved in determination of left/right symmetry by modulating the balance between motile and immotile (sensory) cilia at the left-right organiser (LRO). Functions as a receptor for membrane-bound ligands Jagged-1 (JAG1), Jagged-2 (JAG2) and Delta-1 (DLL1) to regulate cell-fate determination. Related diseases Aortic valve disease 1 (AOVD1) [MIM:109730]: A common defect in the aortic valve in which two rather than three leaflets are present. It is often associated with aortic valve calcification, stenosis and insufficiency. In extreme cases, the blood flow may be so restricted that the left ventricle fails to grow, resulting in hypoplastic left heart syndrome. {ECO:0000269|PubMed:16025100}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Adams-Oliver syndrome 5 (AOS5) [MIM:616028]: A form of Adams-Oliver syndrome, a disorder characterized by the congenital absence of skin (aplasia cutis congenita) in combination with transverse limb defects. Aplasia cutis congenita can be located anywhere on the body, but in the vast majority of the cases, it is present on the posterior parietal region where it is often associated with an underlying defect of the parietal bones. Limb abnormalities are typically limb truncation defects affecting the distal phalanges or entire digits (true ectrodactyly). Only rarely, metatarsals/metacarpals or more proximal limb structures are also affected. Apart from transverse limb defects, syndactyly, most commonly of second and third toes, can also be observed. The clinical features are highly variable and can also include cardiovascular malformations, brain abnormalities and vascular defects such as cutis marmorata and dilated scalp veins. {ECO:0000269|PubMed:25132448}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q13315; Q969H0; Q16665; P78504; O60341; Q8N423; Q92585; P19838; P46531; Q13153; Q13526; Q06330; Q06330-6; Q13573; P98170; Q8IZL2; Q96JK9 EC number NA Uniprot keywords 3D-structure; Activator; Angiogenesis; ANK repeat; Calcium; Cell membrane; Developmental protein; Differentiation; Direct protein sequencing; Disease variant; Disulfide bond; EGF-like domain; Endosome; Glycoprotein; Hydroxylation; Isopeptide bond; Membrane; Metal-binding; Notch signaling pathway; Nucleus; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Repeat; Signal; Transcription; Transcription regulation; Transmembrane; Transmembrane helix; Ubl conjugation Protein physicochemical properties Chain ID A,B Molecular weight (Da) 46121.2 Length 394 Aromaticity 0.13 Instability index 39.95 Isoelectric point 7.22 Charge (pH=7) 0.63 2D Binding mode Binding energy (Kcal/mol) -11.13 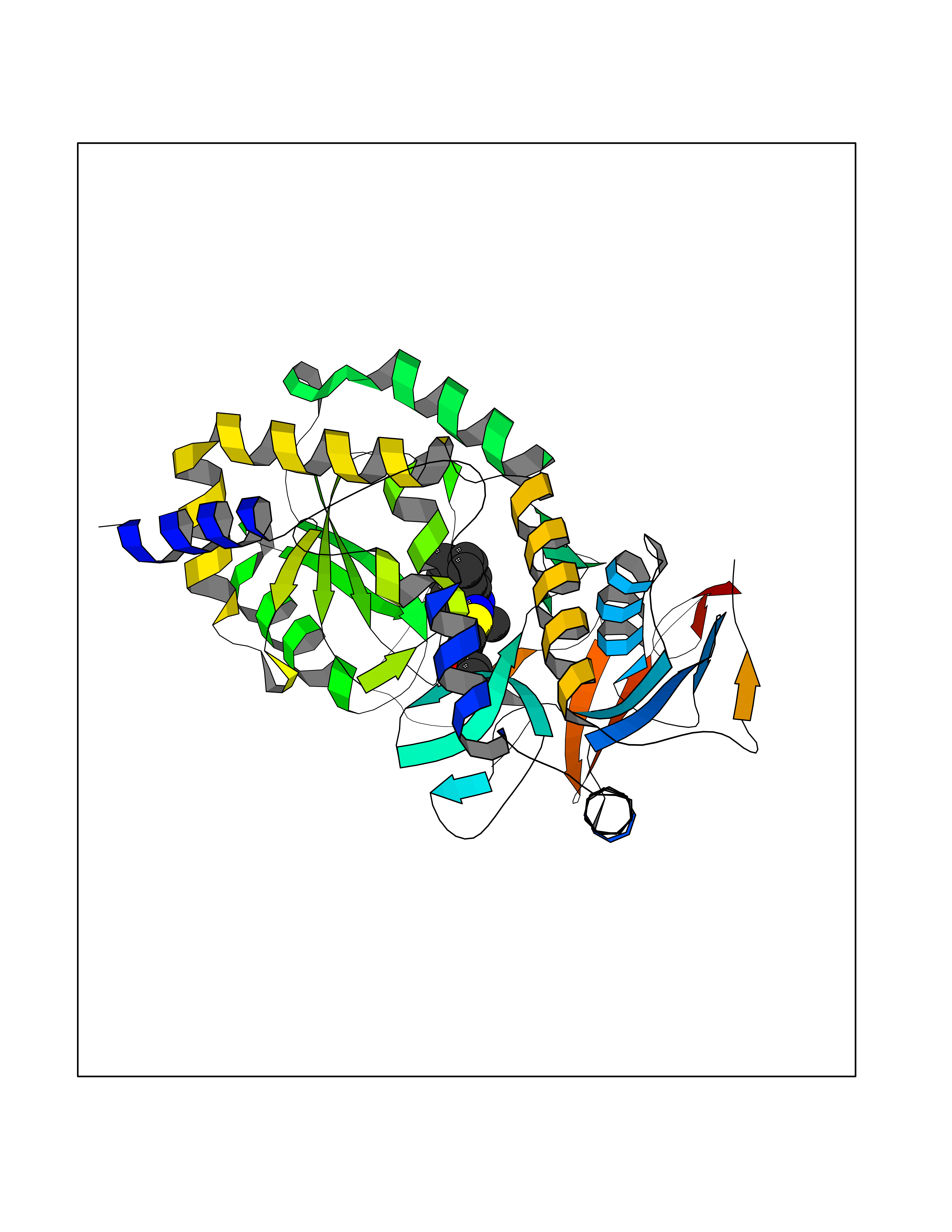 Molscript Map 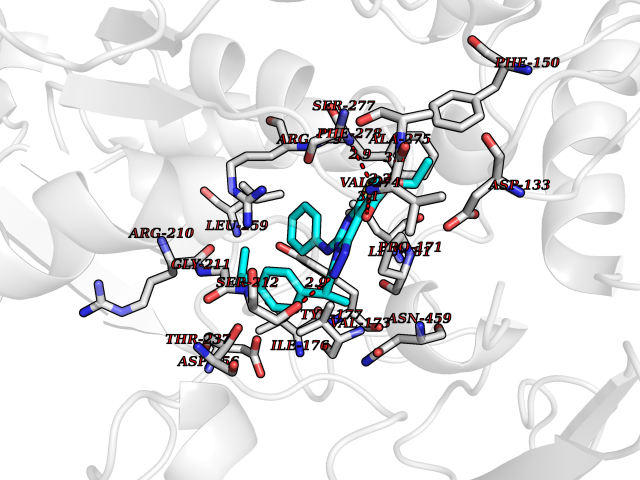 Pymol Map 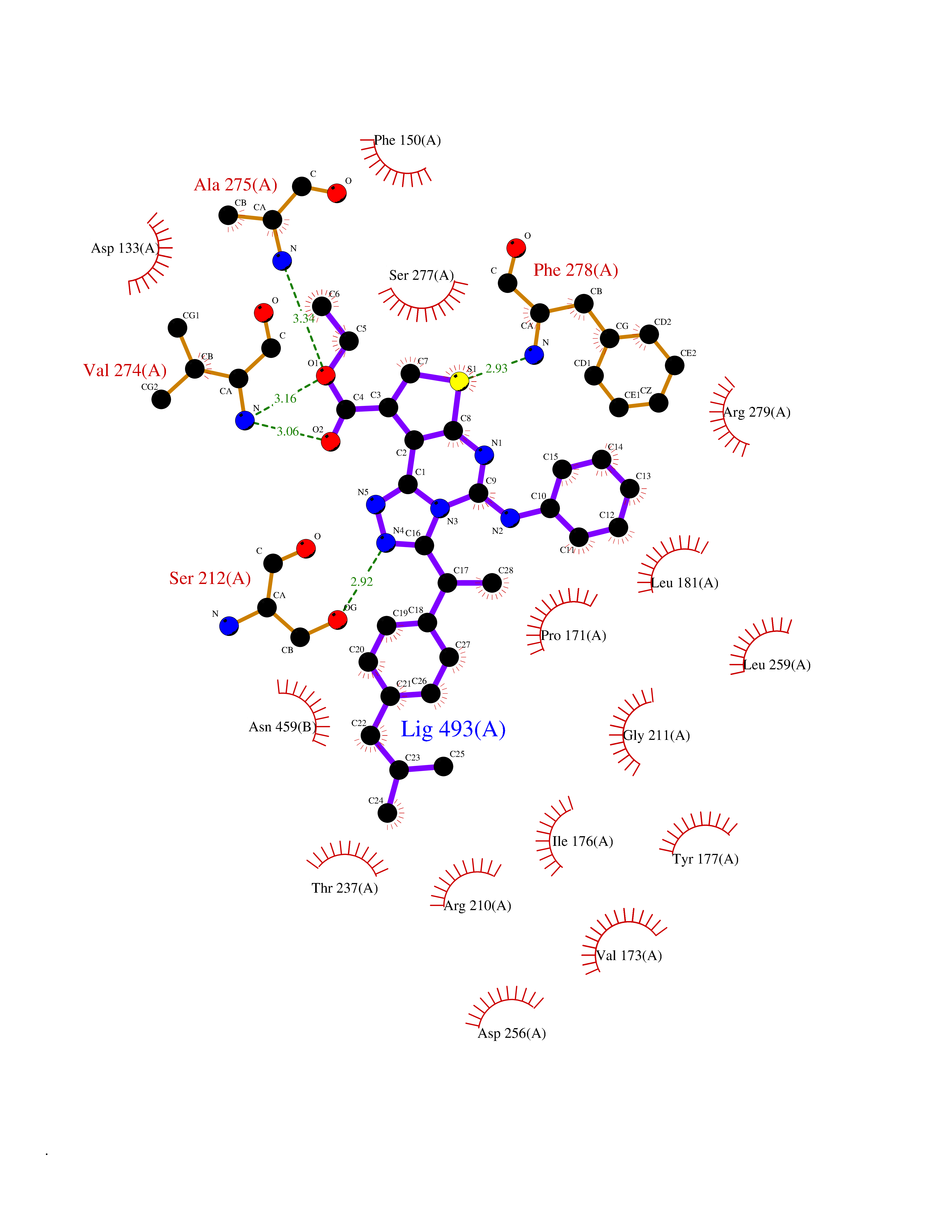 Ligplot Map 3D Binding mode Sequence KWKVFIDQINRSLENYEPCSSQNCSCYHGVIEEDLTPFRGGISRKMMAEVVRRKLGTHYQITKNRLYRENDCMFPSRCSGVEHFILEVIGRLPDMEMVINVRDYPQVPKWMEPAIPVFSFSKTSEYHDIMYPAWTFWEGGPAVWPIYPTGLGRWDLFREDLVRSAAQWPWKKKNSTAYFRGSRTSPERDPLILLSRKNPKLVDAEYTKNQAWKSMKDTLGKPAAKDVHLVDHCKYKYLFNFRGVAASFRFKHLFLCGSLVFHVGDEWLEFFYPQLKPWVHYIPVKTDLSNVQELLQFVKANDDVAQEIAERGSQFIRNHLQMDDITCYWENLLSEYSKFLSYNVTRRKGYDQIIPVNECVSNPCQNDATCLDQIGEFQCICMPGYEGVHCEVNT Hydrogen bonds contact Hydrophobic contact | ||||
| 98 | Nicotinamide phosphoribosyltransferase (NAMPT) | 2E5D | 8.16 | |
Target general information Gen name NAMPT Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Visfatin; PreBcell colonyenhancing factor 1; PreB cellenhancing factor; Pre-B-cell colony-enhancing factor 1; Pre-B cell-enhancing factor; PBEF1; PBEF; Nampt; NAmPRTase Protein family NAPRTase family Biochemical class Glycosyltransferases Function It is the rate limiting component in the mammalian NAD biosynthesis pathway. The secreted form behaves both as a cytokine with immunomodulating properties and an adipokine with anti-diabetic properties, it has no enzymatic activity, partly because of lack of activation by ATP, which has a low level in extracellular space and plasma. Plays a role in the modulation of circadian clock function. NAMPT-dependent oscillatory production of NAD regulates oscillation of clock target gene expression by releasing the core clock component: CLOCK-ARNTL/BMAL1 heterodimer from NAD-dependent SIRT1-mediated suppression. Catalyzes the condensation of nicotinamide with 5-phosphoribosyl-1-pyrophosphate to yield nicotinamide mononucleotide, an intermediate in the biosynthesis of NAD. Related diseases Hemolytic anemia, non-spherocytic, due to glucose phosphate isomerase deficiency (HA-GPID) [MIM:613470]: A form of anemia in which there is no abnormal hemoglobin or spherocytosis. It is caused by glucose phosphate isomerase deficiency. {ECO:0000269|PubMed:28803808, ECO:0000269|PubMed:7989588, ECO:0000269|PubMed:8499925, ECO:0000269|PubMed:8822952, ECO:0000269|PubMed:8822954, ECO:0000269|PubMed:9446754, ECO:0000269|PubMed:9856489}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB12980; DB12731; DB05217 Interacts with P02792; Q01628; P03886; P43490; Q70CQ1-2 EC number EC 2.4.2.12 Uniprot keywords 3D-structure; Acetylation; Biological rhythms; Cytokine; Cytoplasm; Glycosyltransferase; Nucleus; Phosphoprotein; Proteomics identification; Pyridine nucleotide biosynthesis; Reference proteome; Secreted; Transferase Protein physicochemical properties Chain ID A,B Molecular weight (Da) 105483 Length 932 Aromaticity 0.11 Instability index 34.4 Isoelectric point 6.68 Charge (pH=7) -2.24 2D Binding mode Binding energy (Kcal/mol) -11.13 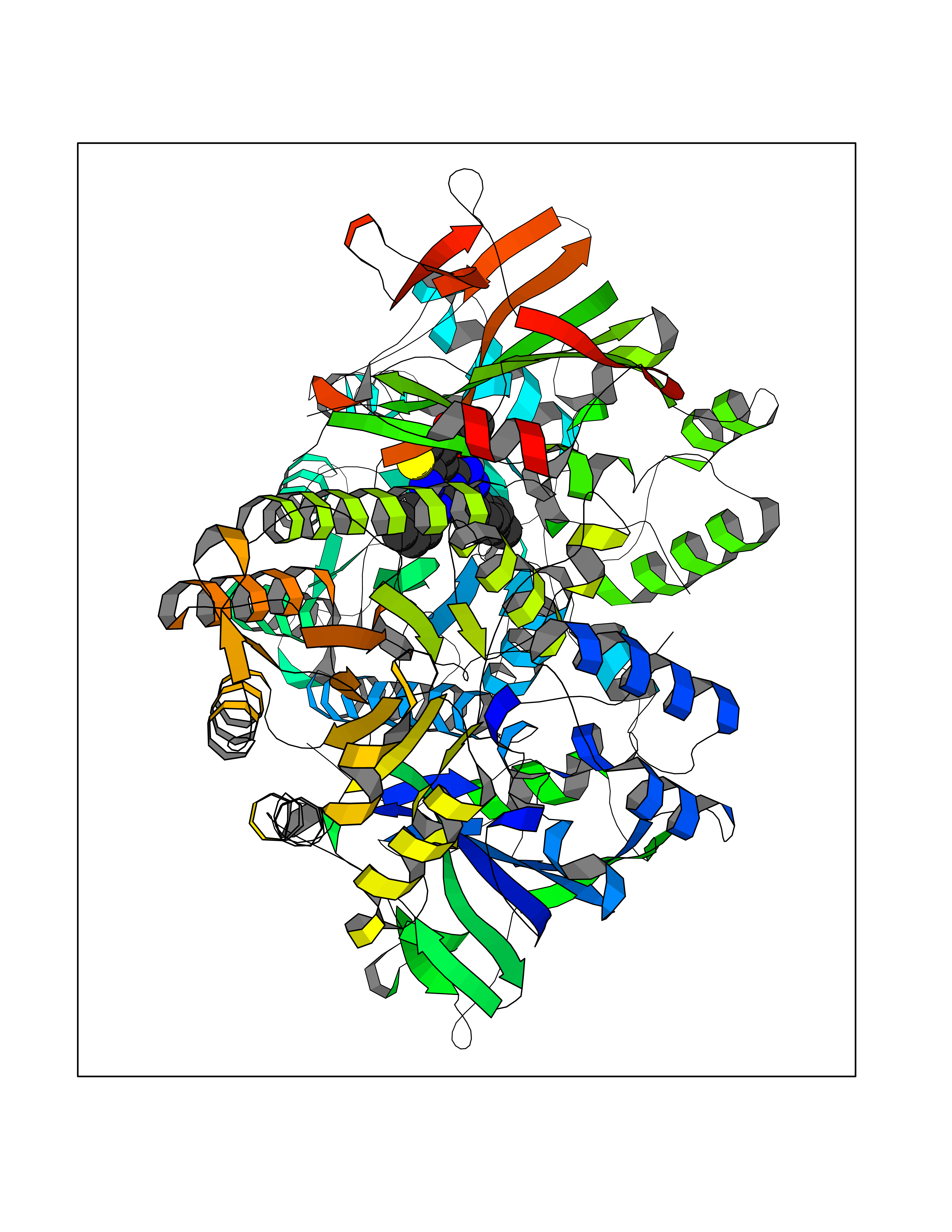 Molscript Map 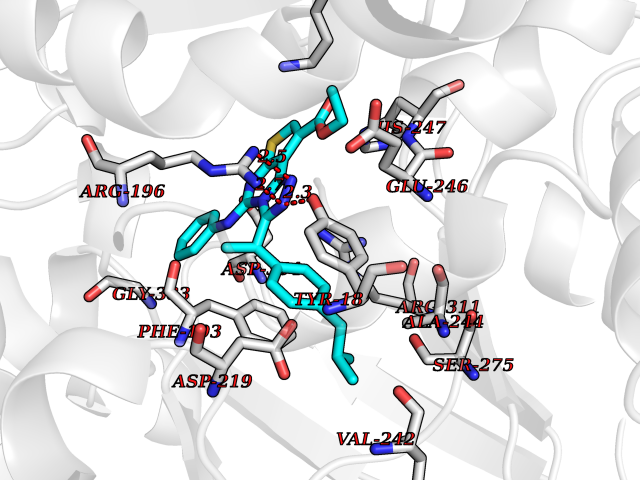 Pymol Map 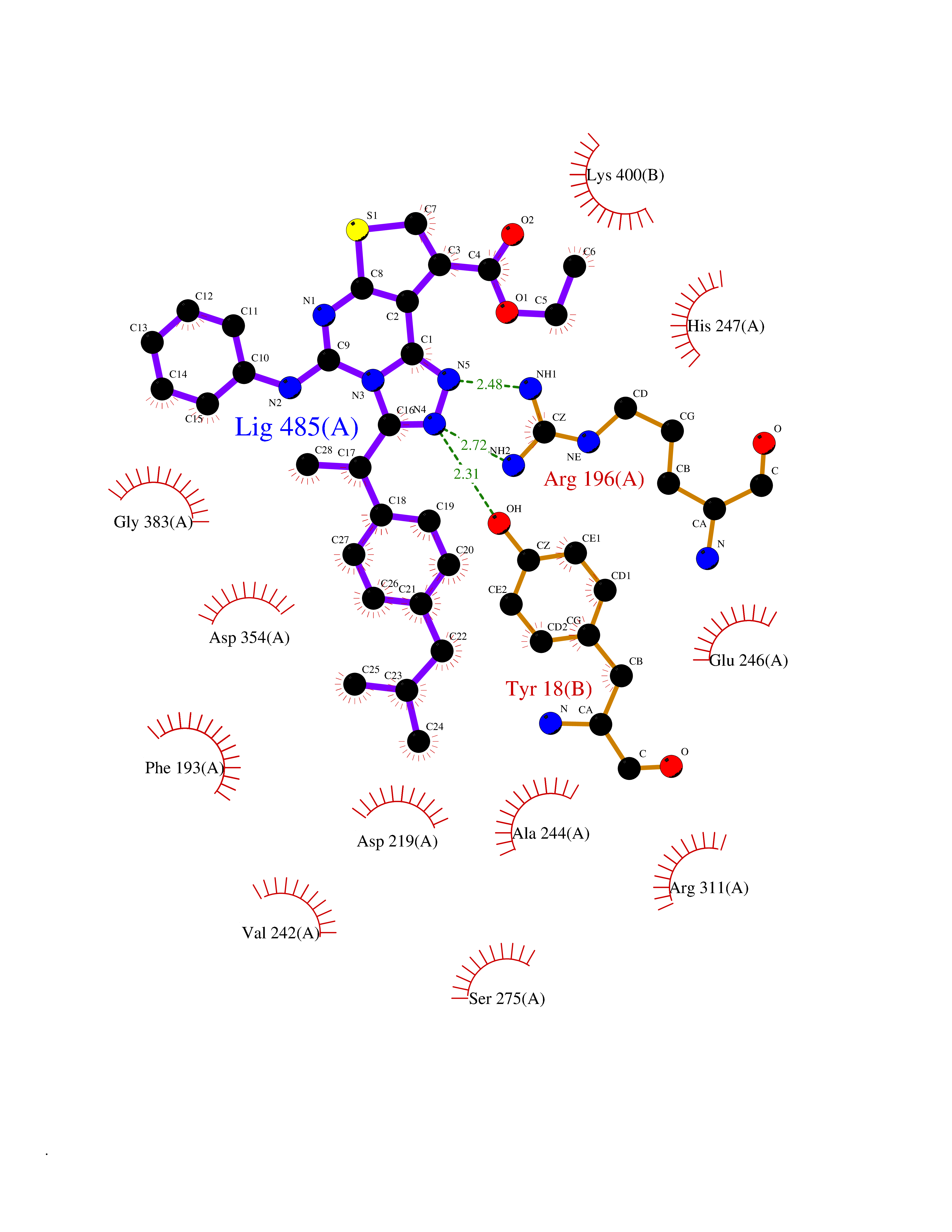 Ligplot Map 3D Binding mode Sequence EFNILLATDSYKVTHYKQYPPNTSKVYSYFECREKKYEETVFYGLQYILNKYLKGKVVTKEKIQEAKDVYKEHFQDDVFNEKGWNYILEKYDGHLPIEIKAVPEGFVIPRGNVLFTVENTDPECYWLTNWIETILVQSWYPITVATNSREQKKILAKYLLETSGNLDGLEYKLHDFGYRGVSSQETAGIGASAHLVNFKGTDTVAGLALIKKYYGTKDPVPGYSVPAAEHSTITAWGKDHEKDAFEHIVTQFSSVPVSVVSDSYDIYNACEKIWGEDLRHLIVSRSTQAPLIIRPDSGNPLDTVLKVLEILGKKFPVTENSKGYKLLPPYLRVIQGDGVDINTLQEIVEGMKQKMWSIENIAFGSGGGLLQKLTRDLLNCSFKCSYVVTNGLGINVFKDPVADPNKRSKKGRLSLHRTPAGNFVTLEEGKGDLEEYGQDLLHTVFKNGKVTKSYSFDEIRKNAQLNEFNILLATDSYKVTHYKQYPPNTSKVYSYFECREKKYEETVFYGLQYILNKYLKGKVVTKEKIQEAKDVYKEHFQDDVFNEKGWNYILEKYDGHLPIEIKAVPEGFVIPRGNVLFTVENTDPECYWLTNWIETILVQSWYPITVATNSREQKKILAKYLLETSGNLDGLEYKLHDFGYRGVSSQETAGIGASAHLVNFKGTDTVAGLALIKKYYGTKDPVPGYSVPAAEHSTITAWGKDHEKDAFEHIVTQFSSVPVSVVSDSYDIYNACEKIWGEDLRHLIVSRSTQAPLIIRPDSGNPLDTVLKVLEILGKKFPVTENSKGYKLLPPYLRVIQGDGVDINTLQEIVEGMKQKMWSIENIAFGSGGGLLQKLTRDLLNCSFKCSYVVTNGLGINVFKDPVADPNKRSKKGRLSLHRTPAGNFVTLEEGKGDLEEYGQDLLHTVFKNGKVTKSYSFDEIRKNAQLN Hydrogen bonds contact Hydrophobic contact | ||||
| 99 | Protein-serine O-palmitoleoyltransferase porcupine (PORCN) | 7URD | 8.16 | |
Target general information Gen name PORCN Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Protein-cysteine N-palmitoyltransferase porcupine; Protein MG61; PORCN Protein family Membrane-bound acyltransferase family, Porcupine subfamily Biochemical class Acyltransferase Function protein-cysteine N-palmitoyltransferase that modulates the processingof Wnt proteins by mediating serine palmitoylation of Wnt family members. Related diseases Focal dermal hypoplasia (FODH) [MIM:305600]: A rare congenital ectomesodermal disorder characterized by a combination of skin defects, skeletal abnormalities, and ocular anomalies. Affected individuals have patchy dermal hypoplasia, often in a distribution pattern following the Blaschko lines, and areas of subcutaneous fat herniation or deposition of fat into the dermis. In addition, sparse and brittle hair, hypoplastic nails and papillomas have been described. Skeletal abnormalities usually comprise syndactyly, ectrodactyly, and brachydactyly, and in some cases osteopathia striata has been seen. Patients frequently have ocular anomalies, including microphthalmia/ anophthalmia, coloboma, pigmentary and vascularization defects of the retina. Dental abnormalities are often present. {ECO:0000269|PubMed:17546030, ECO:0000269|PubMed:17546031, ECO:0000269|PubMed:18325042, ECO:0000269|PubMed:19277062, ECO:0000269|PubMed:19309688, ECO:0000269|PubMed:19586929, ECO:0000269|PubMed:19863546, ECO:0000269|PubMed:21472892}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P55056; Q8N350-4 EC number EC 2.3.1.250 Uniprot keywords 3D-structure; Acyltransferase; Alternative splicing; Disease variant; Endoplasmic reticulum; Lipoprotein; Membrane; Palmitate; Proteomics identification; Reference proteome; Transferase; Transmembrane; Transmembrane helix; Wnt signaling pathway Protein physicochemical properties Chain ID A Molecular weight (Da) 49013.3 Length 432 Aromaticity 0.15 Instability index 36.17 Isoelectric point 9.03 Charge (pH=7) 11.23 2D Binding mode Binding energy (Kcal/mol) -11.14 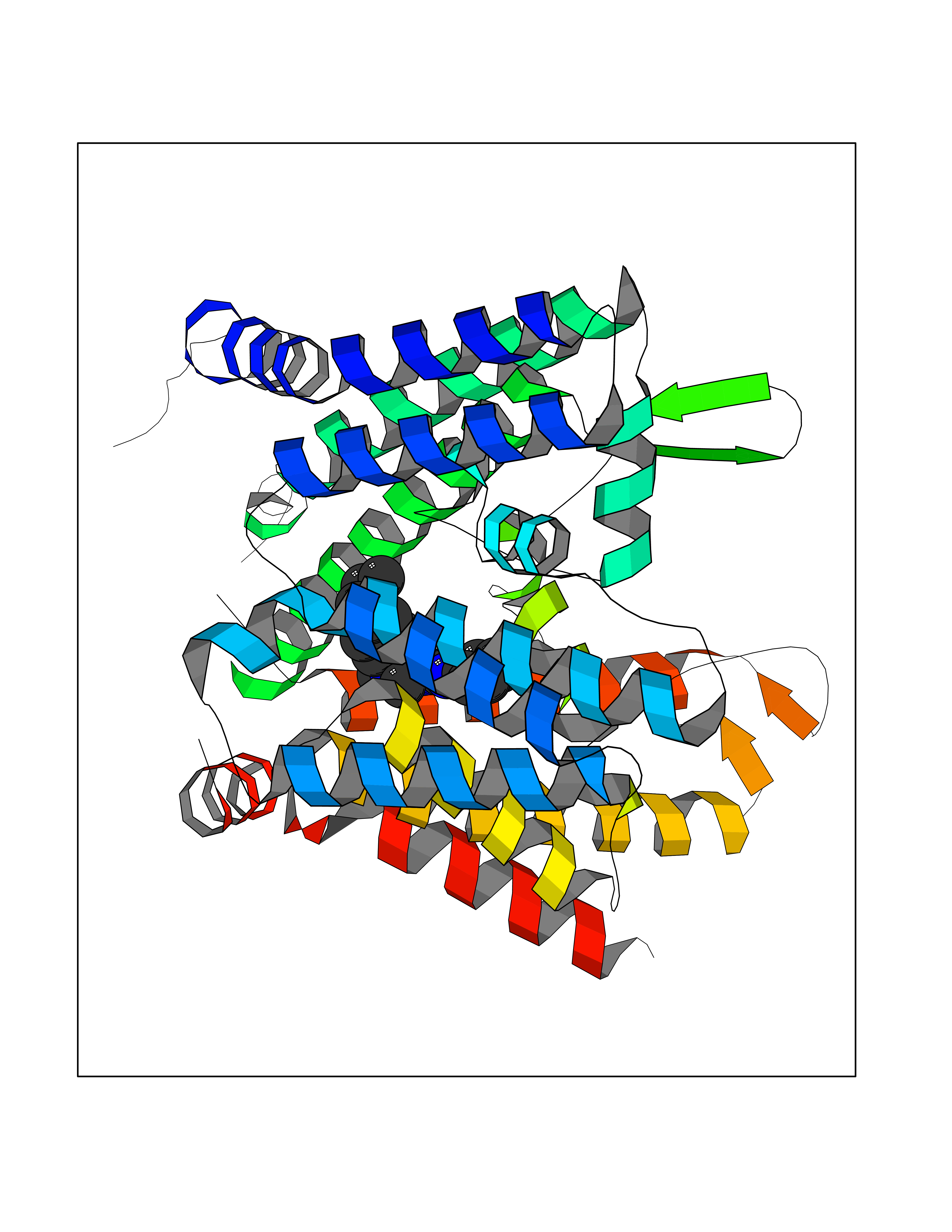 Molscript Map 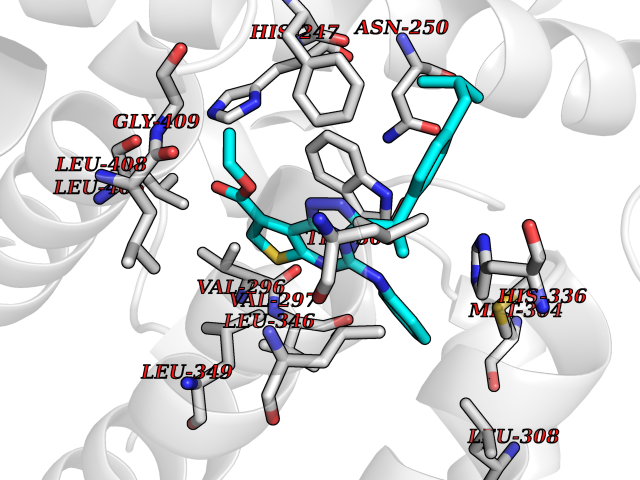 Pymol Map 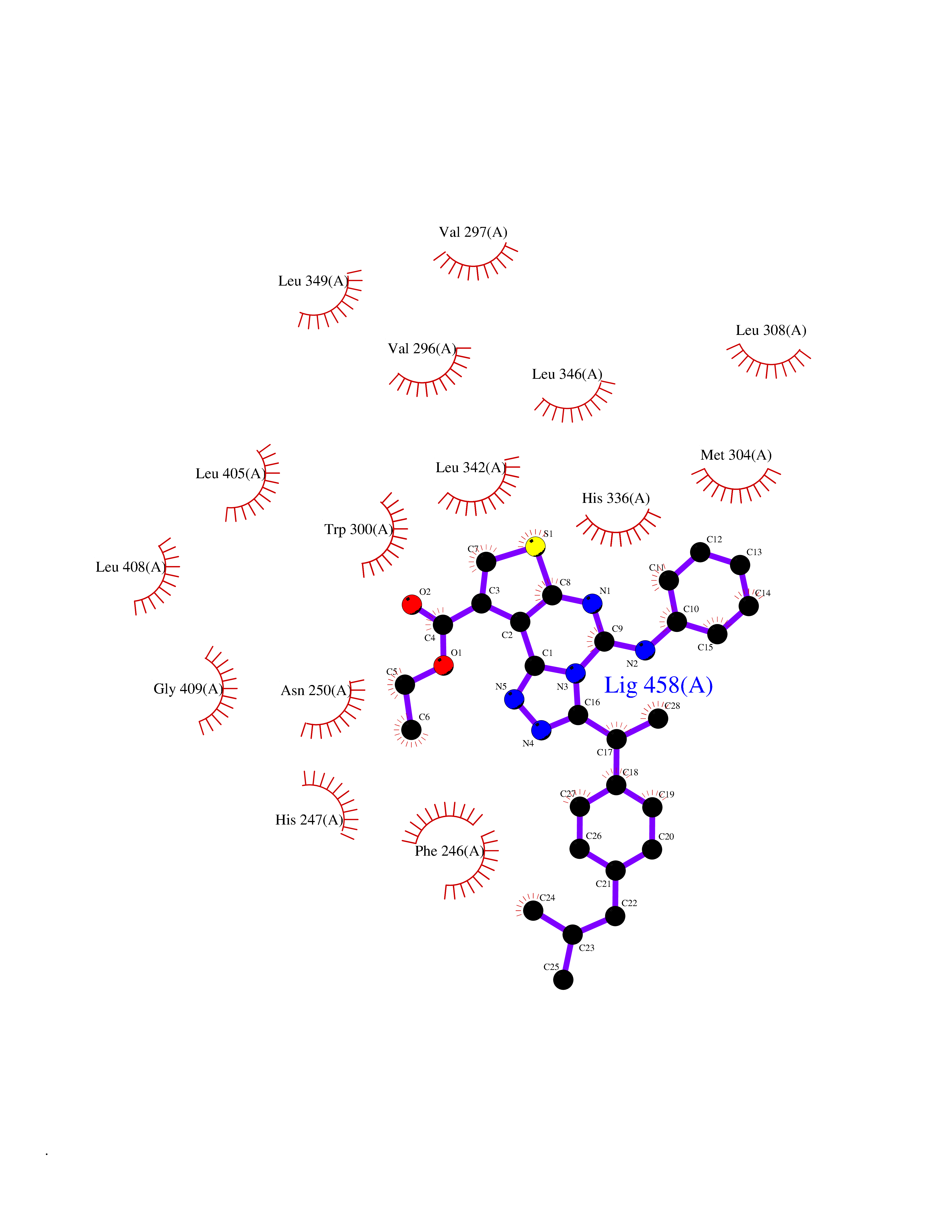 Ligplot Map 3D Binding mode Sequence FSRQEFFQQLLQGCLLPTAQQGLDQIWLLLAICLACRLLWRLGLPSYLKHASTVAGGFFSLYHFFQLHMVWVVLLSLLCYLVLFLCRHSSHRGVFLSVTILIYLLMGEMHMVDTVTWHKMRGAQMIVAMKAVSLGFDLDRGEVGTVPSPVEFMGYLYFVGTIVFGPWISFHSYLQAVQGRPLSCRWLQKVARSLALALLCLVLSTCVGPYLFPYFIPLNARWLRAYESAVSFHFSNYFVGFLSEATATLAGAGFTEEKDHLEWDLTVSKPLNVELPRSMVEVVTSWNLPMSYWLNNYVFKNALRLGTFSAVLVTYAASALLHGFSFHLAAVLLSLAFITYVEHVLRKRLARILSACVLSKRCPPDCSHQHRLGLGVRALNLLFGALAIFHLAYLGSLFDVYGMAYTVHKWSELSWASHWVTFGCWIFYRLIG Hydrogen bonds contact Hydrophobic contact | ||||
| 100 | Lysophosphatidic acid receptor 1 (LPAR1) | 7TD0 | 8.16 | |
Target general information Gen name LPAR1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Lysophosphatidic acid receptor Edg-2; LPA1; LPA-1; LPA receptor 1; EDG2; EDG 2 receptor Protein family G-protein coupled receptor 1 family Biochemical class GPCR rhodopsin Function Plays a role in the reorganization of the actin cytoskeleton, cell migration, differentiation and proliferation, and thereby contributes to the responses to tissue damage and infectious agents. Activates downstream signaling cascades via the G(i)/G(o), G(12)/G(13), and G(q) families of heteromeric G proteins. Signaling inhibits adenylyl cyclase activity and decreases cellular cAMP levels. Signaling triggers an increase of cytoplasmic Ca(2+) levels. Activates RALA; this leads to the activation of phospholipase C (PLC) and the formation of inositol 1,4,5-trisphosphate. Signaling mediates activation of down-stream MAP kinases. Contributes to the regulation of cell shape. Promotes Rho-dependent reorganization of the actin cytoskeleton in neuronal cells and neurite retraction. Promotes the activation of Rho and the formation of actin stress fibers. Promotes formation of lamellipodia at the leading edge of migrating cells via activation of RAC1. Through its function as lysophosphatidic acid receptor, plays a role in chemotaxis and cell migration, including responses to injury and wounding. Plays a role in triggering inflammation in response to bacterial lipopolysaccharide (LPS) via its interaction with CD14. Promotes cell proliferation in response to lysophosphatidic acid. Required for normal skeleton development. May play a role in osteoblast differentiation. Required for normal brain development. Required for normal proliferation, survival and maturation of newly formed neurons in the adult dentate gyrus. Plays a role in pain perception and in the initiation of neuropathic pain. Receptor for lysophosphatidic acid (LPA). Related diseases Anterior segment dysgenesis 6 (ASGD6) [MIM:617315]: A form of anterior segment dysgenesis, a group of defects affecting anterior structures of the eye including cornea, iris, lens, trabecular meshwork, and Schlemm canal. Anterior segment dysgeneses result from abnormal migration or differentiation of the neural crest derived mesenchymal cells that give rise to components of the anterior chamber during eye development. Different anterior segment anomalies may exist alone or in combination, including iris hypoplasia, enlarged or reduced corneal diameter, corneal vascularization and opacity, posterior embryotoxon, corectopia, polycoria, abnormal iridocorneal angle, ectopia lentis, and anterior synechiae between the iris and posterior corneal surface. Clinical conditions falling within the phenotypic spectrum of anterior segment dysgeneses include aniridia, Axenfeld anomaly, Reiger anomaly/syndrome, Peters anomaly, and iridogoniodysgenesis. ASGD6 patients predominantly manifest Peters anomaly. Peters anomaly consists of corneal leukoma, defects in the posterior structures of the cornea such as absence of the posterior corneal stroma and Descemet membrane, and a variable degree of iridocorneal and/or keratolenticular adhesions. Over 50% of patients develop glaucoma in childhood. {ECO:0000269|PubMed:11403040}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Glaucoma 3, primary congenital, A (GLC3A) [MIM:231300]: An autosomal recessive form of primary congenital glaucoma (PCG). PCG is characterized by marked increase of intraocular pressure at birth or early childhood, large ocular globes (buphthalmos) and corneal edema. It results from developmental defects of the trabecular meshwork and anterior chamber angle of the eye that prevent adequate drainage of aqueous humor. {ECO:0000269|PubMed:10227395, ECO:0000269|PubMed:10655546, ECO:0000269|PubMed:11184479, ECO:0000269|PubMed:11527932, ECO:0000269|PubMed:11774072, ECO:0000269|PubMed:11980847, ECO:0000269|PubMed:12036985, ECO:0000269|PubMed:12525557, ECO:0000269|PubMed:14635112, ECO:0000269|PubMed:14640114, ECO:0000269|PubMed:15255109, ECO:0000269|PubMed:15342693, ECO:0000269|PubMed:15475877, ECO:0000269|PubMed:16490498, ECO:0000269|PubMed:16688110, ECO:0000269|PubMed:16735994, ECO:0000269|PubMed:16862072, ECO:0000269|PubMed:18470941, ECO:0000269|PubMed:9463332, ECO:0000269|PubMed:9497261}. The disease is caused by variants affecting distinct genetic loci, including the gene represented in this entry.; DISEASE: Glaucoma 1, open angle, A (GLC1A) [MIM:137750]: A form of primary open angle glaucoma (POAG). POAG is characterized by a specific pattern of optic nerve and visual field defects. The angle of the anterior chamber of the eye is open, and usually the intraocular pressure is increased. However, glaucoma can occur at any intraocular pressure. The disease is generally asymptomatic until the late stages, by which time significant and irreversible optic nerve damage has already taken place. {ECO:0000269|PubMed:11774072}. The gene represented in this entry acts as a disease modifier. Digenic mutations in CYP1B1 and MYOC have been found in a family segregating both primary adult-onset and juvenile forms of open angle glaucoma (PubMed:11774072). All affected family members with mutations in both MYOC and CYP1B1 had juvenile glaucoma, whereas those with only the MYOC mutation had the adult-onset form (PubMed:11774072). {ECO:0000269|PubMed:11774072}. Drugs (DrugBank ID) NA Interacts with NA EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disulfide bond; Endosome; G-protein coupled receptor; Glycoprotein; Lipid-binding; Membrane; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID R Molecular weight (Da) 33201.3 Length 290 Aromaticity 0.14 Instability index 34.12 Isoelectric point 8.86 Charge (pH=7) 6.12 2D Binding mode Binding energy (Kcal/mol) -11.13 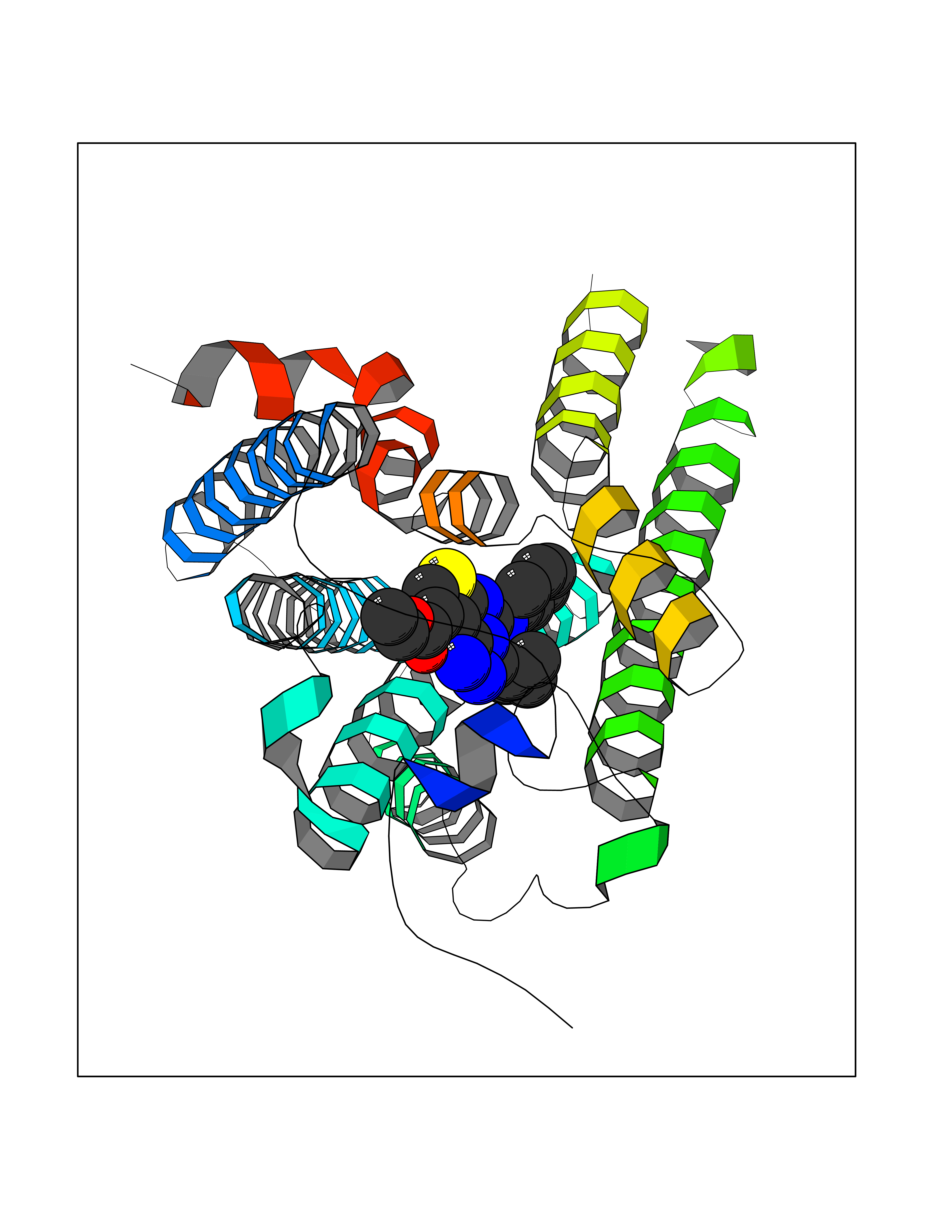 Molscript Map 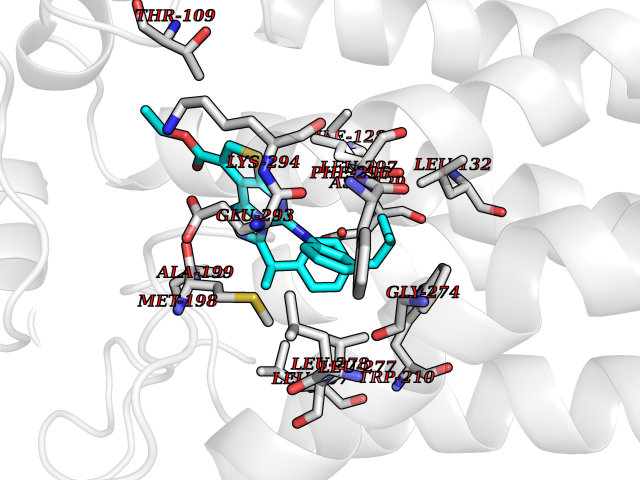 Pymol Map 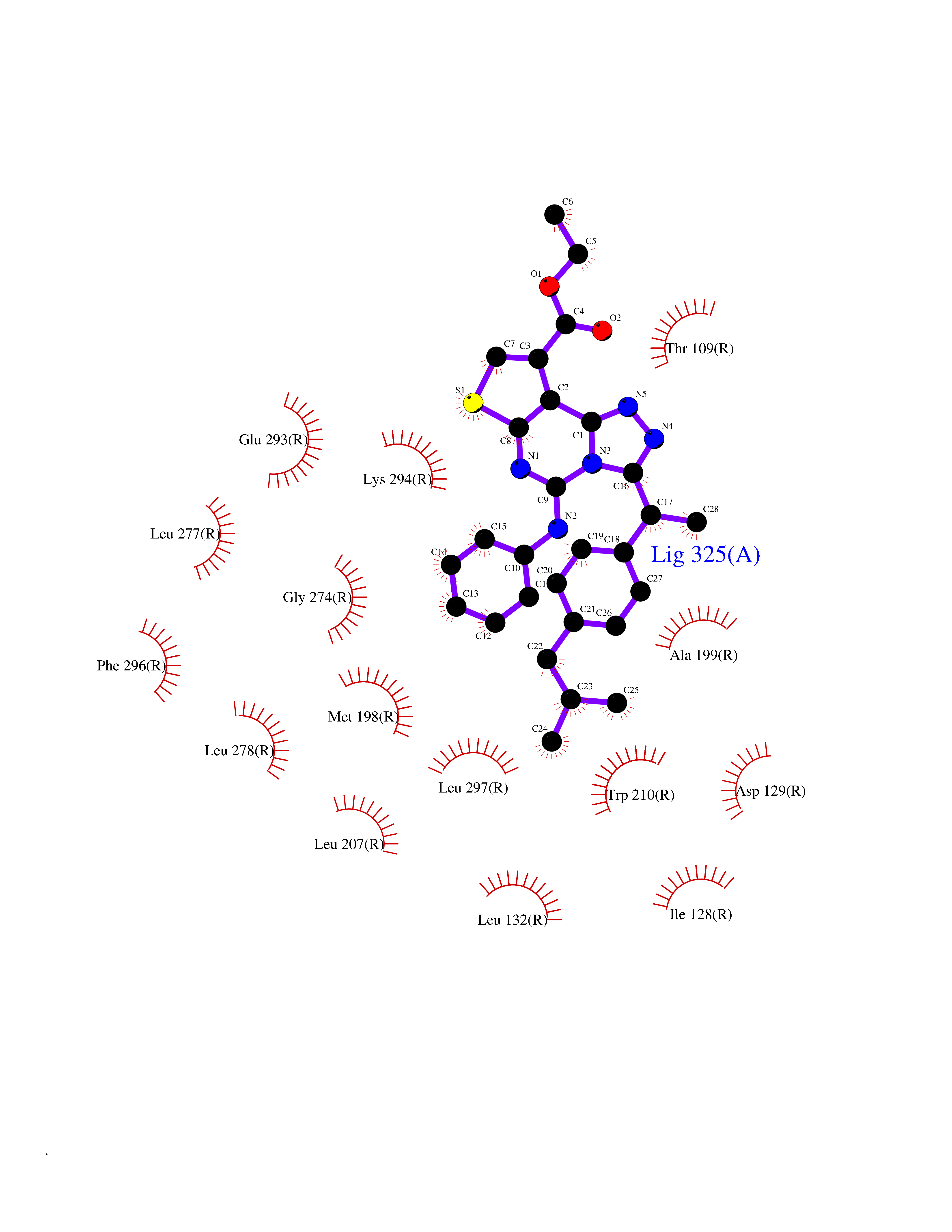 Ligplot Map 3D Binding mode Sequence QCFYNESIAFFYNRSGKHLATEWNTVSKLVMGLGITVCIFIMLANLLVMVAIYVNRRFHFPIYYLMANLAAADFFAGLAYFYLMFNTGPNTRRLTVSTWLLRQGLIDTSLTASVANLLAIAIERHITVFRMQLHTRMSNRRVVVVIVVIWTMAIVMGAIPSVGWNCICDIENCSNMAPLYSDSYLVFWAIFNLVTFVVMVVLYAHIFGYVRQRTMRMDTMMSLLKTVVIVLGAFIICWTPGLVLLLLDVCCPQCDVLAYEKFFLLLAEFNSAMNPIIYSYRDKEMSATFR Hydrogen bonds contact Hydrophobic contact | ||||