Job Results:
Ligand
Structure
Job ID
634b273143b25dfaacd81a63c2250095
Job name
NA
Time
2026-02-27 17:35:01
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 81 | Cytochrome P450 2A6 (CYP2A6) | 1Z11 | 5.33 | |
Target general information Gen name CYP2A6 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Cytochrome P450(I); Cytochrome P450 IIA3; Coumarin 7-hydroxylase; CYPIIA6; CYP2A6; CYP2A3 Protein family Cytochrome P450 family Biochemical class Paired donor oxygen oxidoreductase Function Exhibits a high coumarin 7-hydroxylase activity. Can act in the hydroxylation of the anti-cancer drugs cyclophosphamide and ifosphamide. Competent in the metabolic activation of aflatoxin B1. Constitutes the major nicotine C-oxidase. Possesses low phenacetin O-deethylation activity. Related diseases Lecithin-cholesterol acyltransferase deficiency (LCATD) [MIM:245900]: A disorder of lipoprotein metabolism characterized by inadequate esterification of plasmatic cholesterol. Two clinical forms are recognized: complete LCAT deficiency and fish-eye disease. LCATD is generally referred to the complete form which is associated with absence of both alpha and beta LCAT activities resulting in esterification anomalies involving both HDL (alpha-LCAT activity) and LDL (beta-LCAT activity). It causes a typical triad of diffuse corneal opacities, target cell hemolytic anemia, and proteinuria with renal failure. {ECO:0000269|PubMed:11423760, ECO:0000269|PubMed:12957688, ECO:0000269|PubMed:15994445, ECO:0000269|PubMed:16051254, ECO:0000269|PubMed:16216249, ECO:0000269|PubMed:1681161, ECO:0000269|PubMed:1859405, ECO:0000269|PubMed:2370048, ECO:0000269|PubMed:7607641, ECO:0000269|PubMed:7711728, ECO:0000269|PubMed:8318557, ECO:0000269|PubMed:8432868, ECO:0000269|PubMed:8807342, ECO:0000269|PubMed:9007616, ECO:0000269|PubMed:9741700}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Fish-eye disease (FED) [MIM:136120]: A disorder of lipoprotein metabolism due to partial lecithin-cholesterol acyltransferase deficiency that affects only alpha-LCAT activity. FED is characterized by low plasma HDL and corneal opacities due to accumulation of cholesterol deposits in the cornea ('fish-eye'). {ECO:0000269|PubMed:1516702, ECO:0000269|PubMed:1571050, ECO:0000269|PubMed:15994445, ECO:0000269|PubMed:1737840, ECO:0000269|PubMed:21901787, ECO:0000269|PubMed:8620346, ECO:0000269|PubMed:9261271}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07621; DB07623; DB00316; DB01118; DB00182; DB01435; DB05676; DB01274; DB09274; DB11586; DB00972; DB00443; DB01194; DB01222; DB09061; DB14737; DB06119; DB00356; DB00568; DB00604; DB06470; DB00257; DB00363; DB04665; DB00531; DB06292; DB01234; DB14649; DB00470; DB06374; DB00216; DB00330; DB01039; DB04841; DB01544; DB00544; DB00690; DB01213; DB00983; DB01159; DB00741; DB01181; DB00951; DB01026; DB01006; DB00281; DB06448; DB04871; DB14009; DB01043; DB00170; DB00763; DB00553; DB01028; DB00959; DB00916; DB01011; DB01110; DB00471; DB07609; DB07617; DB00238; DB00184; DB01115; DB06712; DB00717; DB00312; DB03783; DB01174; DB00252; DB01085; DB04977; DB14631; DB00635; DB00396; DB01045; DB15119; DB01037; DB06739; DB01236; DB00675; DB09256; DB09327; DB12816; DB00752; DB00755; DB12245; DB00313; DB09068; DB00495 Interacts with Q9UI14 EC number EC 1.14.13.- Uniprot keywords 3D-structure; Direct protein sequencing; Endoplasmic reticulum; Heme; Iron; Lipid metabolism; Membrane; Metal-binding; Microsome; Monooxygenase; NADP; Oxidoreductase; Proteomics identification; Reference proteome Protein physicochemical properties Chain ID A Molecular weight (Da) 53586.9 Length 467 Aromaticity 0.12 Instability index 40.76 Isoelectric point 9.14 Charge (pH=7) 7.77 2D Binding mode Binding energy (Kcal/mol) -7.27 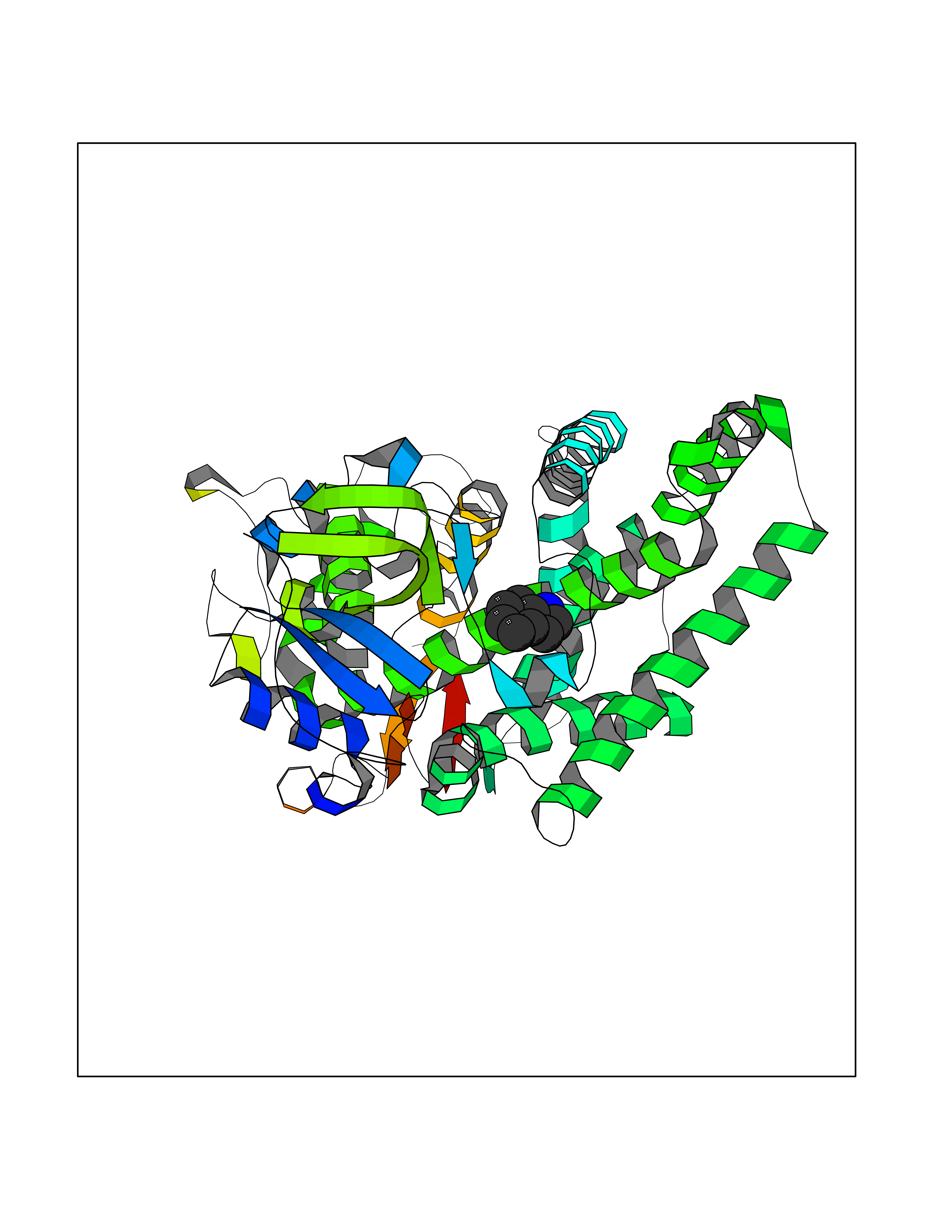 Molscript Map 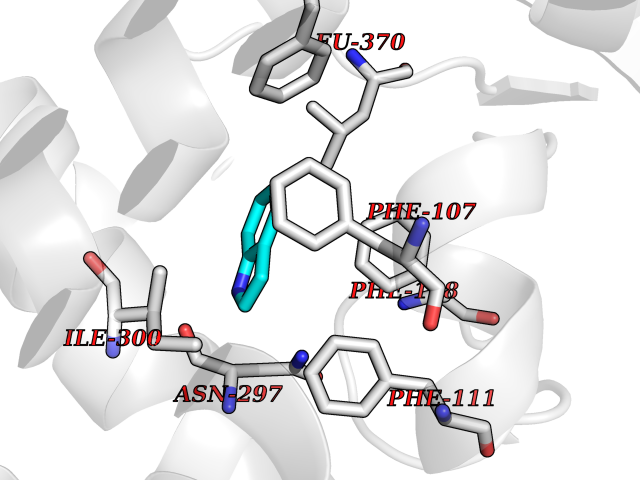 Pymol Map 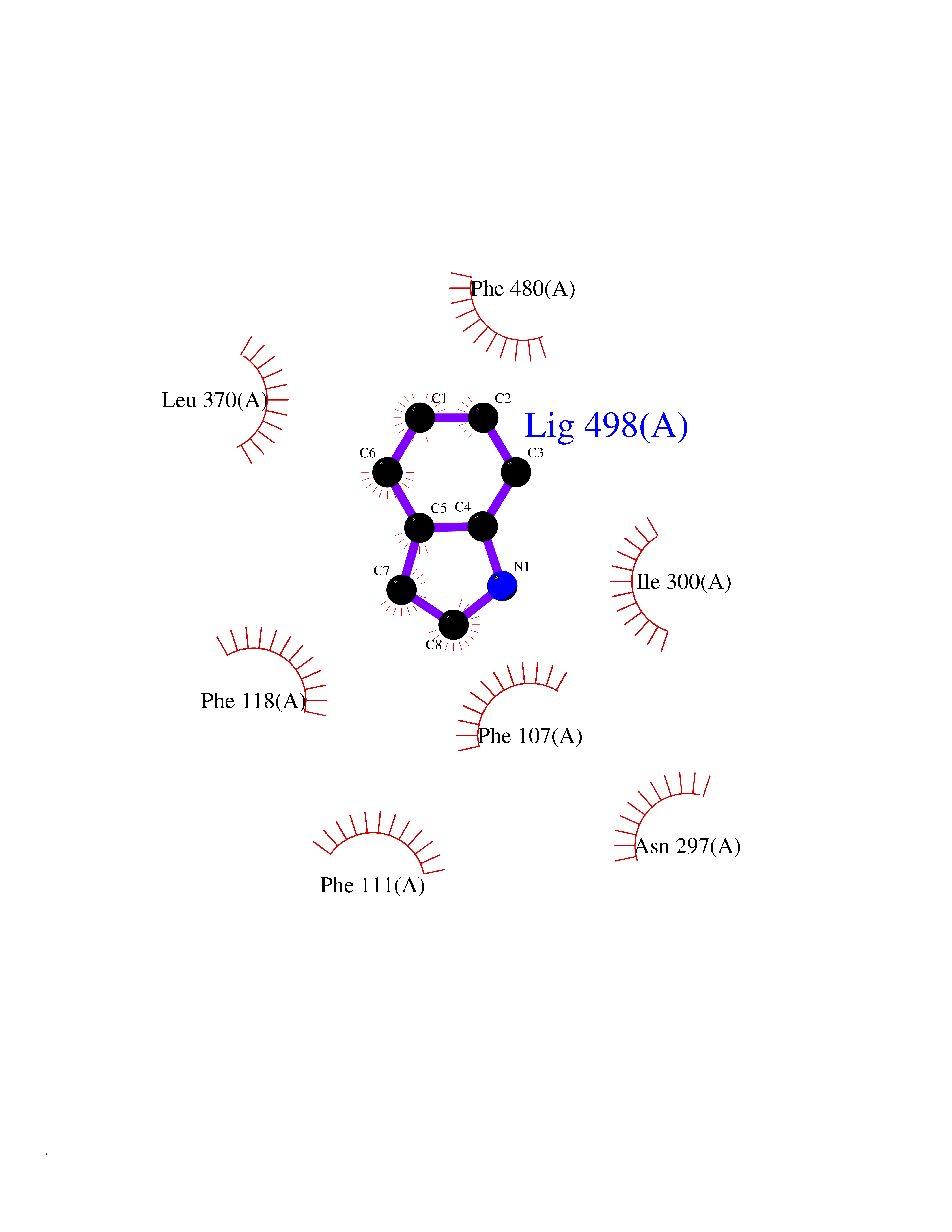 Ligplot Map 3D Binding mode Sequence KGKLPPGPTPLPFIGNYLQLNTEQMYNSLMKISERYGPVFTIHLGPRRVVVLCGHDAVREALVDQAEEFSGRGEQATFDWVFKGYGVVFSNGERAKQLRRFSIATLRDFGVGKRGIEERIQEEAGFLIDALRGTGGANIDPTFFLSRTVSNVISSIVFGDRFDYKDKEFLSLLRMMLGIFQFTSTSTGQLYEMFSSVMKHLPGPQQQAFQLLQGLEDFIAKKVEHNQRTLDPNSPRDFIDSFLIRMQEEEKNPNTEFYLKNLVMTTLNLFIGGTETVSTTLRYGFLLLMKHPEVEAKVHEEIDRVIGKNRQPKFEDRAKMPYMEAVIHEIQRFGDVIPMSLARRVKKDTKFRDFFLPKGTEVYPMLGSVLRDPSFFSNPQDFNPQHFLNEKGQFKKSDAFVPFSIGKRNCFGEGLARMELFLFFTTVMQNFRLKSSQSPKDIDVSPKHVGFATIPRNYTMSFLPRHH Hydrogen bonds contact Hydrophobic contact | ||||
| 82 | Ubiquitin carboxyl-terminal hydrolase 14 (USP14) | 6IIK | 5.33 | |
Target general information Gen name USP14 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Ubiquitin-specific-processing protease 14; Ubiquitin thioesterase 14; TGT; Deubiquitinating enzyme 14 Protein family Peptidase C19 family, USP14/UBP6 subfamily Biochemical class Peptidase Function Ensures the regeneration of ubiquitin at the proteasome. Is a reversibly associated subunit of the proteasome and a large fraction of proteasome-free protein exists within the cell. Required for the degradation of the chemokine receptor CXCR4 which is critical for CXCL12-induced cell chemotaxis. Serves also as a physiological inhibitor of endoplasmic reticulum-associated degradation (ERAD) under the non-stressed condition by inhibiting the degradation of unfolded endoplasmic reticulum proteins via interaction with ERN1. Indispensable for synaptic development and function at neuromuscular junctions (NMJs). Plays a role in the innate immune defense against viruses by stabilizing the viral DNA sensor CGAS and thus inhibiting its autophagic degradation. Proteasome-associated deubiquitinase which releases ubiquitin from the proteasome targeted ubiquitinated proteins. Related diseases Hypophosphatemic rickets, autosomal dominant (ADHR) [MIM:193100]: A disease characterized by isolated renal phosphate wasting, hypophosphatemia, and inappropriately normal 1,25-dihydroxyvitamin D3 (calcitriol) levels. Patients frequently present with bone pain, rickets, and tooth abscesses. {ECO:0000269|PubMed:11062477, ECO:0000269|PubMed:11409890, ECO:0000269|PubMed:16638743}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Tumoral calcinosis, hyperphosphatemic, familial, 2 (HFTC2) [MIM:617993]: A form of hyperphosphatemic tumoral calcinosis, a rare autosomal recessive metabolic disorder that manifests with hyperphosphatemia and massive calcium deposits in the skin and subcutaneous tissues. Some patients have recurrent, transient, painful swellings of the long bones associated with the radiographic findings of periosteal reaction and cortical hyperostosis and absence of skin involvement. {ECO:0000269|PubMed:15590700, ECO:0000269|PubMed:16030159, ECO:0000269|PubMed:16151858, ECO:0000269|PubMed:24680727}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB12695 Interacts with Q08209 EC number EC 3.4.19.12 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Cell membrane; Cytoplasm; Hydrolase; Immunity; Innate immunity; Membrane; Phosphoprotein; Protease; Proteasome; Proteomics identification; Reference proteome; Thiol protease; Ubl conjugation pathway Protein physicochemical properties Chain ID A Molecular weight (Da) 38476.7 Length 335 Aromaticity 0.1 Instability index 61.05 Isoelectric point 5.6 Charge (pH=7) -4.84 2D Binding mode Binding energy (Kcal/mol) -7.26 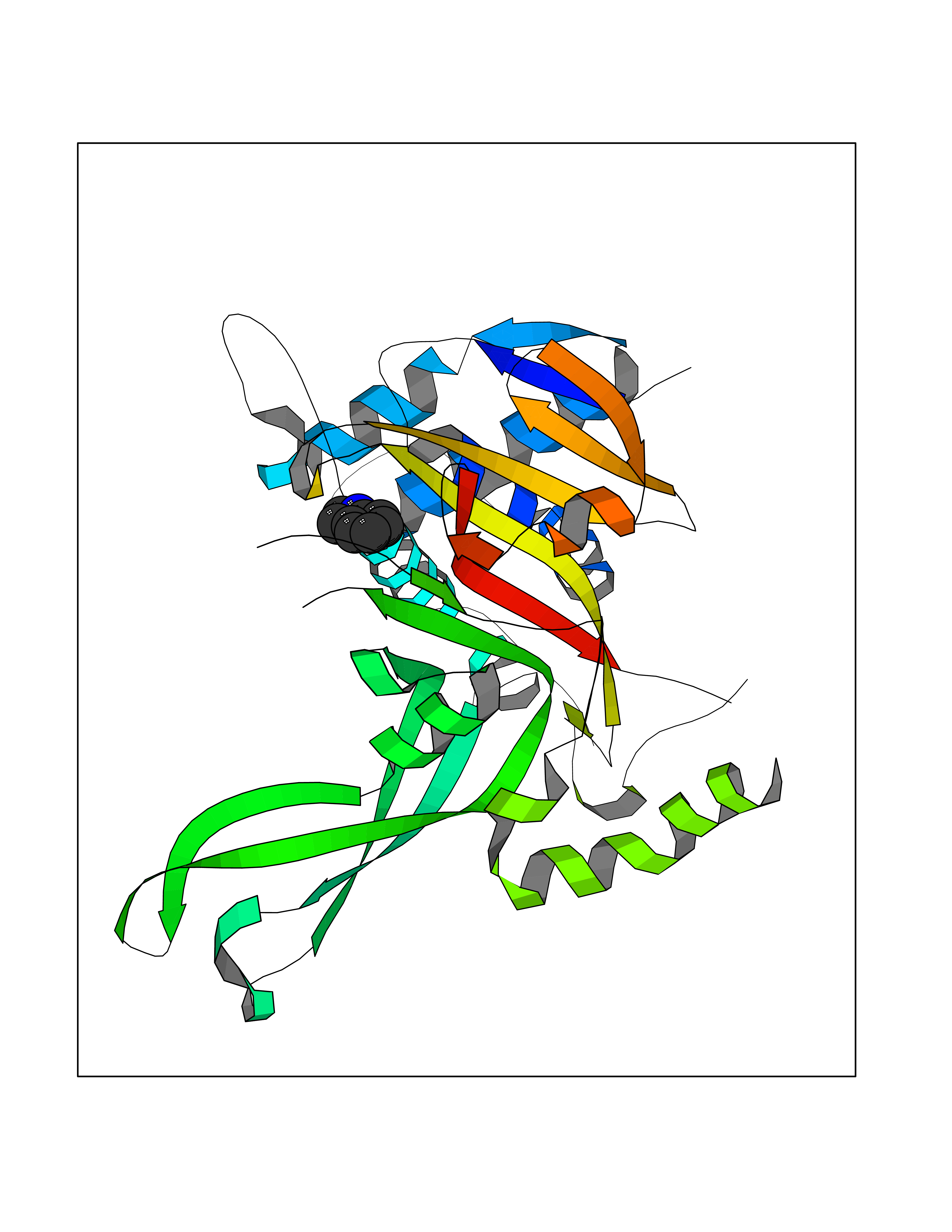 Molscript Map 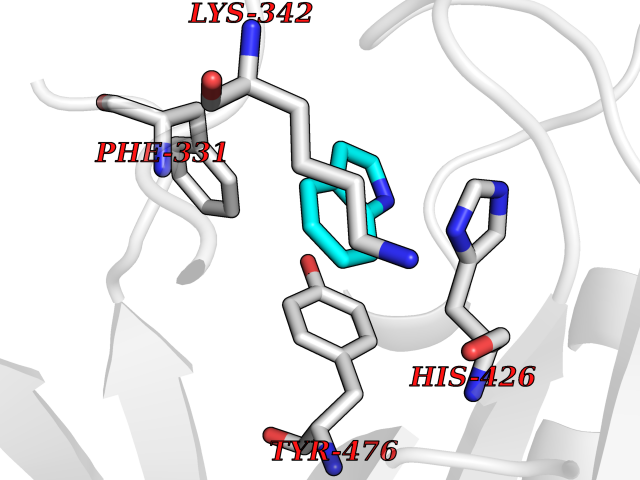 Pymol Map 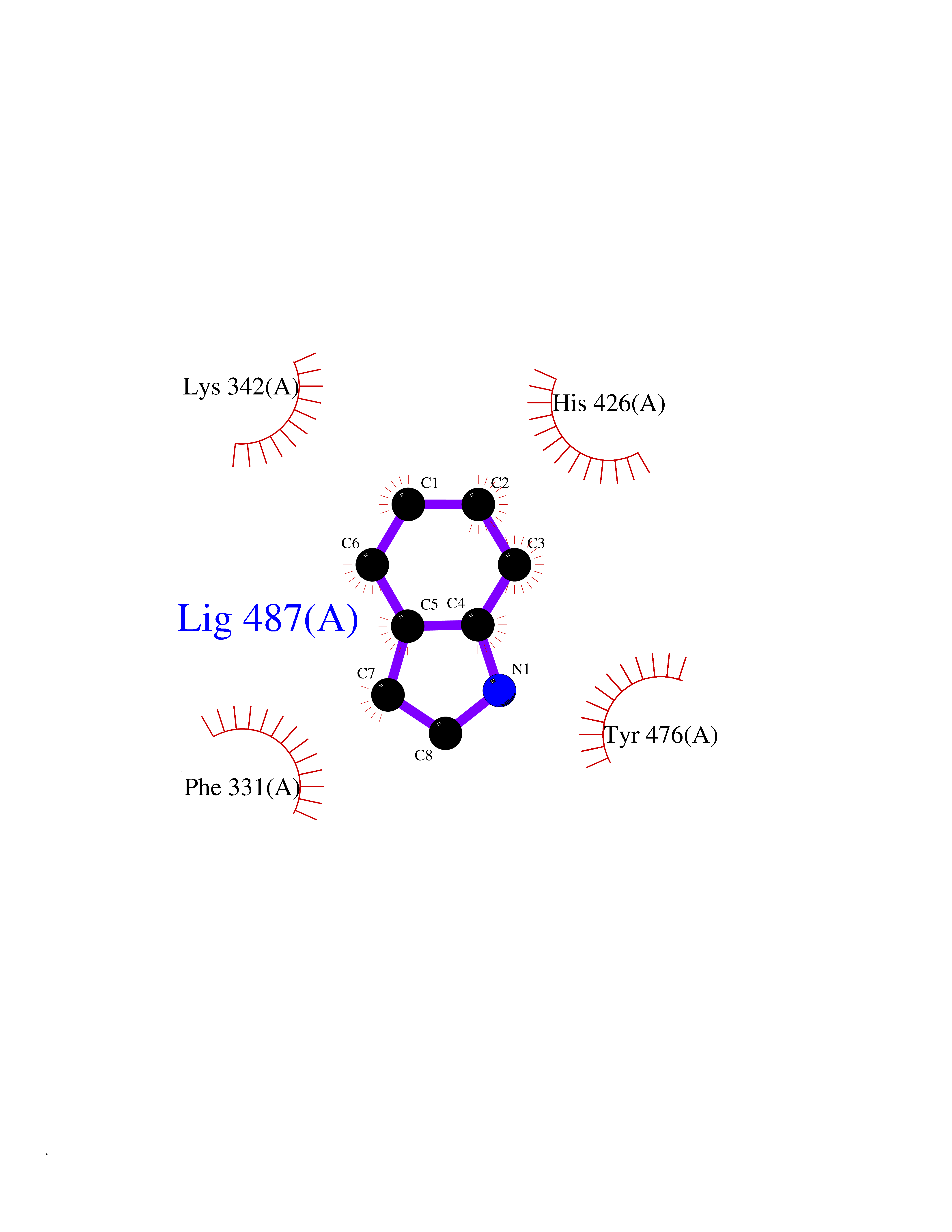 Ligplot Map 3D Binding mode Sequence ELPCGLTNLGNTCYMNATVQCIRSVPELKDALKRYAGALRASGEMASAQYITAALRDLFDSMDKTSSSIPPIILLQFLHMAFPQFAEKGEQGQYLQQDANECWIQMMRVLQQKLEAIEDKSLIDQFFGVEFETTMKCTESEEEEVTKGKENQLQLSCFINQEVKYLFTGLKLRLQEEITKQSPTLQRNALYIKSSKISRLPAYLTIQMVRFFNAKVLKDVKFPLMLDMYELCTPELQEKMVSFRSKFKDLYEPFSFADDIGSNNCGYYDLQAVLTHQGRSSSSGHYVSWVKRKQDEWIKFDDDKVSIVTPEDILRLSGGGDWHIAYVLLYGPRRV Hydrogen bonds contact Hydrophobic contact | ||||
| 83 | Lethal(3)malignant brain tumor-like 3 (L3MBTL3) | 4FL6 | 5.33 | |
Target general information Gen name L3MBTL3 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms MBT1; MBT-1; Lethal(3)malignant brain tumor-like protein 3; L(3)mbt-like protein 3; KIAA1798; H-l(3)mbt-like protein 3 Protein family NA Biochemical class NA Function Putative Polycomb group (PcG) protein. PcG proteins maintain the transcriptionally repressive state of genes, probably via a modification of chromatin, rendering it heritably changed in its expressibility. Required for normal maturation of myeloid progenitor cells (By similarity). Related diseases Spinocerebellar ataxia, autosomal recessive, with axonal neuropathy 1 (SCAN1) [MIM:607250]: A form of spinocerebellar ataxia, a clinically and genetically heterogeneous group of cerebellar disorders. Patients show progressive incoordination of gait and often poor coordination of hands, speech and eye movements, due to degeneration of the cerebellum with variable involvement of the brainstem and spinal cord. SCAN1 is an autosomal recessive cerebellar ataxia (ARCA) associated with peripheral axonal motor and sensory neuropathy, distal muscular atrophy, pes cavus and steppage gait as seen in Charcot-Marie-Tooth neuropathy. All affected individuals have normal intelligence. {ECO:0000269|PubMed:12244316, ECO:0000269|PubMed:15647511, ECO:0000269|PubMed:15920477, ECO:0000269|PubMed:16141202, ECO:0000269|PubMed:17948061}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q9NWX5; Q8N9N5; Q13895; Q96JK2; Q16531; P26358; Q01094; O00716; Q96JM7; Q9Y4Z0; P45984; Q9UBU8; Q15014; Q9NPG2; Q9UMX2; P18545; Q8IXK0; Q8N381; Q96S99; Q9H8W4; P62875; Q13131; P54646; P14678-2; P23497; G2XKQ0; P10827; Q6DKK2; P54253; A0A0S2Z5G4; Q13895; Q9BXJ3; Q8IUI8; Q14203-5; Q9H4E7; Q8NFF5-2; Q53EP0-3; O95995; O75031; P42858; Q14005-2; Q63ZY3; Q96JM7-2; P61968; P45984; P55081; Q9UBU8-2; Q15014; Q9NPG2; Q16656-4; Q96HA8; Q96BD5; Q92569; Q96S99; Q9H8W4; O60568; A0A6Q8PF08; P67775; P54646; P63000; O94955; Q5VUG0; P37840; P00441; Q5MJ10; Q9UMX1; Q13148; Q86TI0; Q8N8B7-2; Q9Y228; Q5T7W7; Q6DKK2; P09936; P31930; P61758; A0A0S2Z6A9; Q9H0M4-4; P36508 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Chromatin regulator; Isopeptide bond; Metal-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Transcription; Transcription regulation; Ubl conjugation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A,B Molecular weight (Da) 71079 Length 615 Aromaticity 0.14 Instability index 29.71 Isoelectric point 6.42 Charge (pH=7) -6.56 2D Binding mode Binding energy (Kcal/mol) -7.28 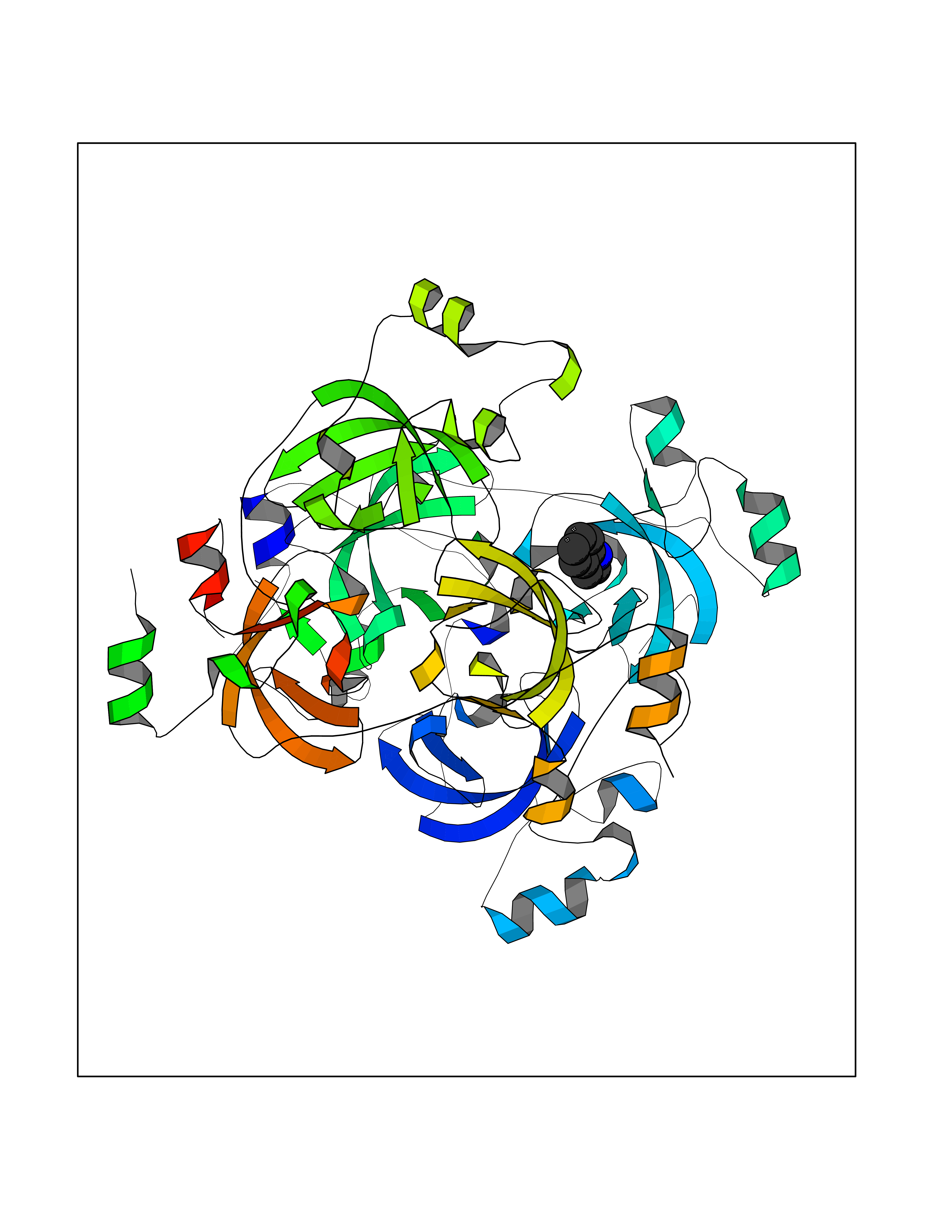 Molscript Map 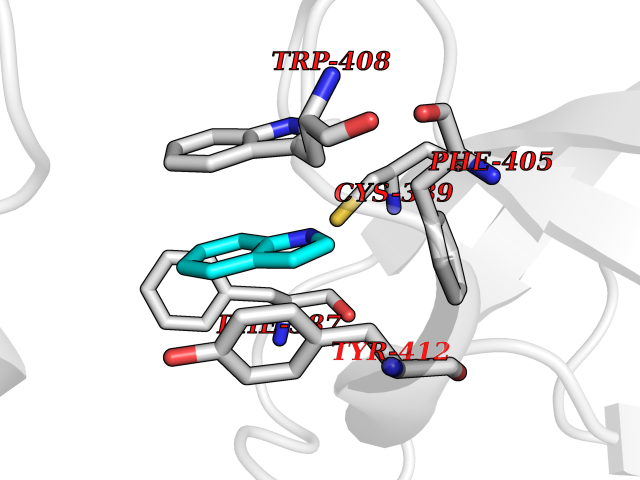 Pymol Map 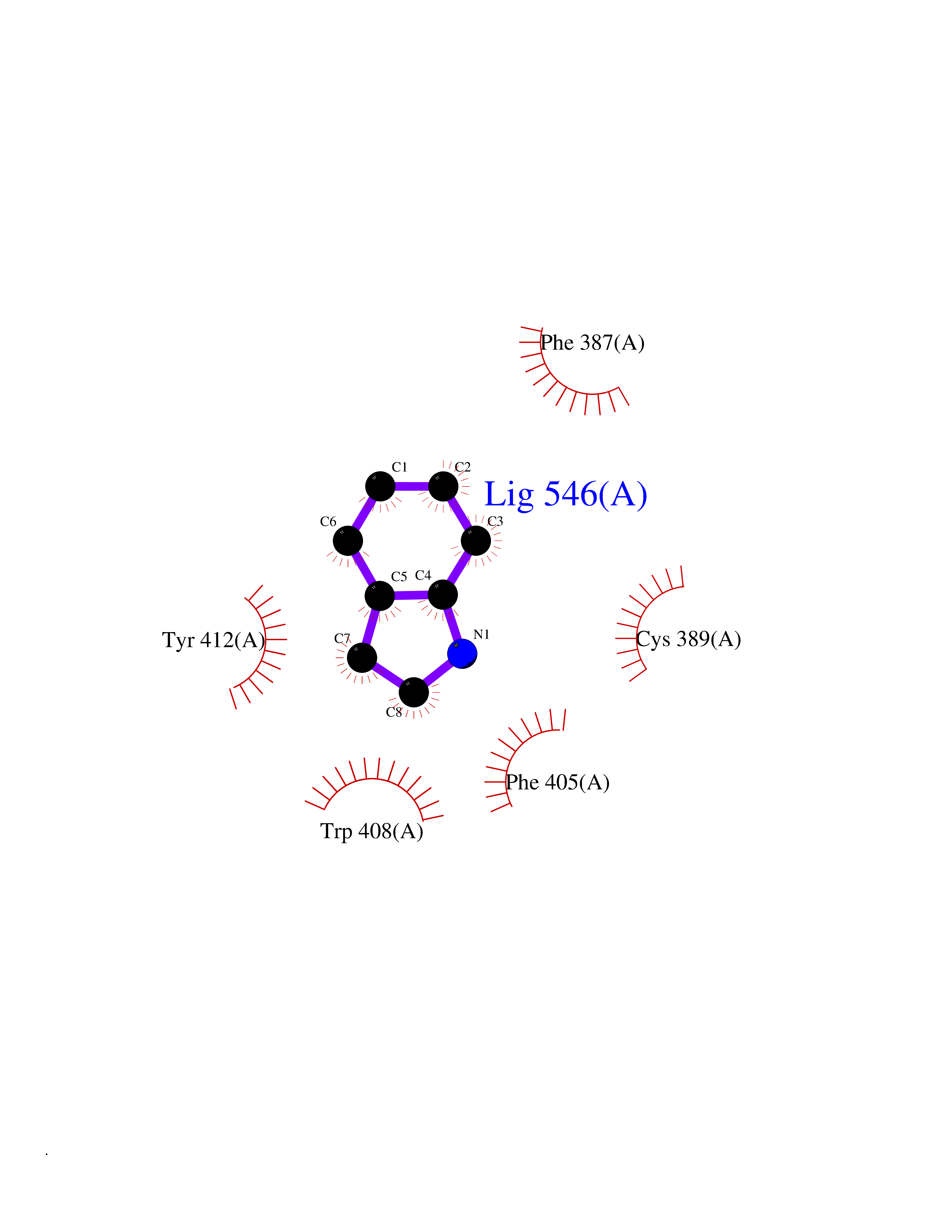 Ligplot Map 3D Binding mode Sequence AWCWASYLEEEKAVAVPAKLFKEHQSFPYNKNGFKVGMKLEGVDPEHQSVYCVLTVAEVCGYRIKLHFDGYSDCYDFWVNADALDIHPVGWCEKTGHKLHPPKGYKEEEFNWQTYLKTCKAQAAPKSLFEVIPSGFRVGMKLEAVDKKNPSFICVATVTDMVDNRFLVHFDNWDESYDYWCEASSPHIHPVGWCKEHRRTLITPPGYPNVKHFSWDKYLEETNSLPAPARAFKVKPPHGFQKKMKLEVVDKRNPMFIRVATVADTDDHRVKVHFDGWNNCYDYWIDADSPDIHPVGWCSKTGHPLQPPLAWCWASYLEEEKAVAVPAKLFKEHQSFPYNKNGFKVGMKLEGVDPEHQSVYCVLTVAEVCGYRIKLHFDGYSDCYDFWVNADALDIHPVGWCEKTGHKLHPPKGYKEEEFNWQTYLKTCKAQAAPKSLFENSGFRVGMKLEAVDKKNPSFICVATVTDMVDNRFLVHFDNWDESYDYWCEASSPHIHPVGWCKEHRRTLITPPGYPNVHFSWDKYLEETNSLPAPARAFKVKPPHGFQKKMKLEVVDKRNPMFIRVATVADTDDHRVKVHFDGWNNCYDYWIDADSPDIHPVGWCSKTGHPLQPPL Hydrogen bonds contact Hydrophobic contact | ||||
| 84 | Oxysterols receptor LXR-alpha (NR1H3) | 3IPQ | 5.33 | |
Target general information Gen name NR1H3 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Nuclear receptor subfamily 1 group H member 3; Nuclear receptor LXRalpha; Nuclear orphan receptor LXR-alpha; Liver X receptor alpha; LXRalpha; LXRA Protein family Nuclear hormone receptor family, NR1 subfamily Biochemical class Nuclear hormone receptor Function Interaction with retinoic acid receptor (RXR) shifts RXR from its role as a silent DNA-binding partner to an active ligand-binding subunit in mediating retinoid responses through target genes defined by LXRES. LXRES are DR4-type response elements characterized by direct repeats of two similar hexanuclotide half-sites spaced by four nucleotides. Plays an important role in the regulation of cholesterol homeostasis, regulating cholesterol uptake through MYLIP-dependent ubiquitination of LDLR, VLDLR and LRP8. Interplays functionally with RORA for the regulation of genes involved in liver metabolism. Nuclear receptor that exhibits a ligand-dependent transcriptional activation activity. Related diseases Okur-Chung neurodevelopmental syndrome (OCNDS) [MIM:617062]: An autosomal dominant neurodevelopmental disorder characterized by developmental delay, intellectual disability, behavioral problems, hypotonia, speech problems, microcephaly, pachygyria and variable dysmorphic features. {ECO:0000269|PubMed:27048600}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08175; DB08063; DB11994; DB07929; DB13174; DB07080 Interacts with O60869; O60341; Q99750; Q15788; O75376; Q07869; Q07869-1; Q03181; P37231; P19793; P28702; P48443; O43463; P42858; Q99750; O95817; G5E9A7; O95872; P02545; Q99750; P28702; P28702-3; P48443; Q7Z699 EC number NA Uniprot keywords 3D-structure; Activator; Alternative splicing; Cytoplasm; DNA-binding; Metal-binding; Nucleus; Proteomics identification; Receptor; Reference proteome; Transcription; Transcription regulation; Ubl conjugation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A Molecular weight (Da) 25389.9 Length 220 Aromaticity 0.09 Instability index 46.42 Isoelectric point 5.51 Charge (pH=7) -6.58 2D Binding mode Binding energy (Kcal/mol) -7.27 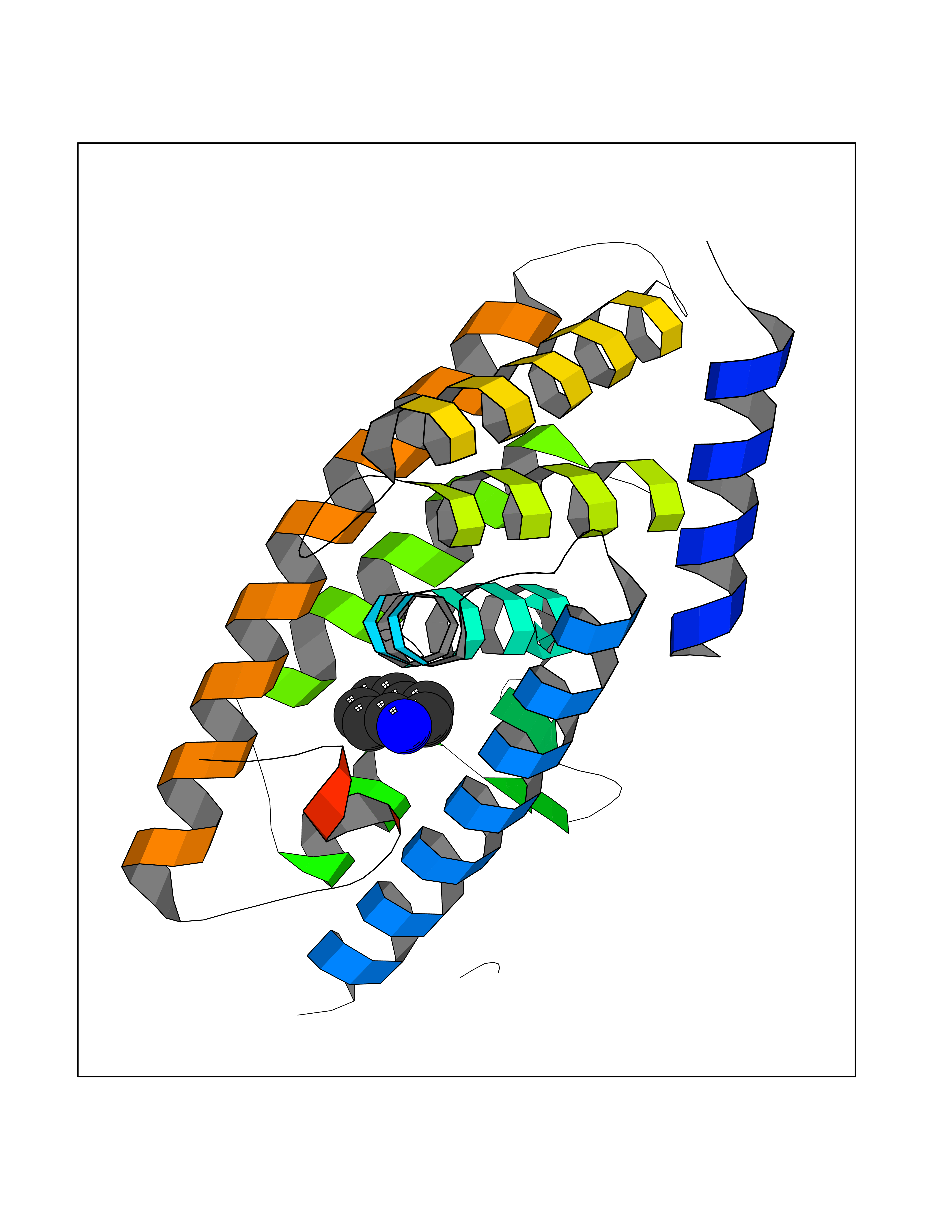 Molscript Map 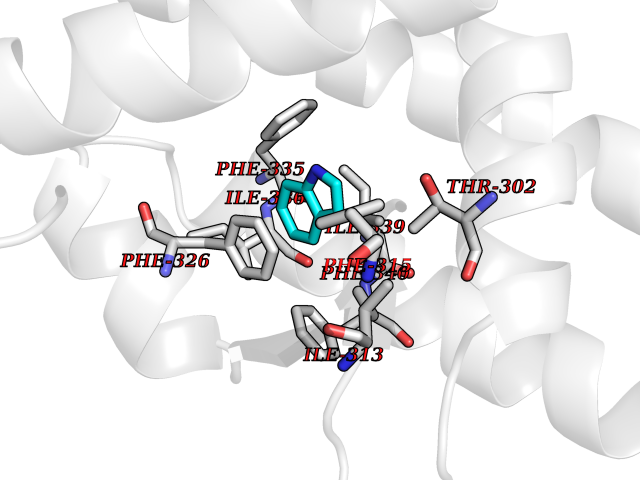 Pymol Map  Ligplot Map 3D Binding mode Sequence QLSPEQLGMIEKLVAAQQTPWPEARQQRFAHFTELAIVSVQEIVDFAKQLPGFLQLSREDQIALLKTSAIEVMLLETSRRYNPGSESITFLKDFSYNREDFAKAGLQVEFINPIFEFSRAMNELQLNDAEFALLIAISIFSADRPNVQDQLQVERLQHTYVEALHAYVSIHHPHDRLMFPRMLMKLVSLRTLSSVHSEQVFALRLQDKKLPPLLSEIWDV Hydrogen bonds contact Hydrophobic contact | ||||
| 85 | Histone-lysine N-methyltransferase KMT5C (KMT5C) | 3RQ4 | 5.33 | |
Target general information Gen name KMT5C Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Lysine N-methyltransferase 5C; Lysine-specific methyltransferase 5C; Suppressor of variegation 4-20 homolog 2; Su(var)4-20 homolog 2; Suv4-20h2; [histone H4]-N-methyl-L-lysine20 N-methyltransferase KM Protein family Class V-like SAM-binding methyltransferase superfamily, Histone-lysine methyltransferase family, Suvar4-20 subfamily Biochemical class NA Function Histone methyltransferase that specifically methylates monomethylated 'Lys-20' (H4K20me1) and dimethylated 'Lys-20' (H4K20me2) of histone H4 to produce respectively dimethylated 'Lys-20' (H4K20me2) and trimethylated 'Lys-20' (H4K20me3) and thus regulates transcription and maintenance of genome integrity. In vitro also methylates unmodified 'Lys-20' (H4K20me0) of histone H4 and nucleosomes. H4 'Lys-20' trimethylation represents a specific tag for epigenetic transcriptional repression. Mainly functions in pericentric heterochromatin regions, thereby playing a central role in the establishment of constitutive heterochromatin in these regions. KMT5C is targeted to histone H3 via its interaction with RB1 family proteins (RB1, RBL1 and RBL2) (By similarity). Facilitates TP53BP1 foci formation upon DNA damage and proficient non-homologous end-joining (NHEJ)-directed DNA repair by catalyzing the di- and trimethylation of 'Lys-20' of histone H4. May play a role in class switch reconbination by catalyzing the di- and trimethylation of 'Lys-20' of histone H4 (By similarity). Related diseases Brachydactyly A2 (BDA2) [MIM:112600]: A form of brachydactyly. Brachydactyly defines a group of inherited malformations characterized by shortening of the digits due to abnormal development of the phalanges and/or the metacarpals. In brachydactyly type A2 shortening of the middle phalanges is confined to the index finger and the second toe, all other digits being more or less normal. Because of a rhomboid or triangular shape of the affected middle phalanx, the end of the second finger usually deviates radially. {ECO:0000269|PubMed:19327734, ECO:0000269|PubMed:21357617}. The gene represented in this entry is involved in disease pathogenesis. Duplications of a cis-regulatory element located approximately 110 kb downstream of BMP2 have been found in BDA2 families. They likely cause altered BMP2 expression with pathological consequences. {ECO:0000269|PubMed:19327734, ECO:0000269|PubMed:21357617}.; DISEASE: Short stature, facial dysmorphism, and skeletal anomalies with or without cardiac anomalies 1 (SSFSC1) [MIM:617877]: An autosomal dominant disorder characterized by short stature, facial dysmorphism, skeletal anomalies, and variable cardiac defects. Distinctive facial features include midface retrusion, short upturned nose, long philtrum, high-arched or cleft palate, and variable degrees of micrognathia and dental crowding. Skeletal anomalies include patterning defects of the axial skeleton, characterized by 11 pairs of ribs and brachydactyly of the fifth ray. Congenital heart defects are variably observed and appear to involve primarily the cardiac outflow tract. {ECO:0000269|PubMed:29198724}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q13185 EC number EC 2.1.1.361 Uniprot keywords 3D-structure; Alternative splicing; Chromatin regulator; Chromosome; Metal-binding; Methyltransferase; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Repressor; S-adenosyl-L-methionine; Transcription; Transcription regulation; Transferase; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 27285.8 Length 240 Aromaticity 0.1 Instability index 42.74 Isoelectric point 8.32 Charge (pH=7) 3.24 2D Binding mode Binding energy (Kcal/mol) -7.27  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence DRVTARELCENDDLATSLVLDPYLGFRTHKMNVSPVPPLRRQQHLRSALETFLRQRDLEAAYRALTLGGWTARYFQSRGPRQEAALKTHVYRYLRAFLPESGFTILPCTRYSMETNGAKIVSTRAWKKNEKLELLVGCIAELREADEGLLRAGENDFSIMYSTRKRSAQLWLGPAAFINHDCKPNCKFVPADGNAACVKVLRDIEPGDEVTCFYGEGFFGEKNEHCECHTCERKGEGAFR Hydrogen bonds contact Hydrophobic contact | ||||
| 86 | Dibasic-processing enzyme (Furin) | 7LCU | 5.33 | |
Target general information Gen name FURIN Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Paired basic amino acid residuecleaving enzyme; Paired basic amino acid residue-cleaving enzyme; PCSK3; PACE; FUR; Dibasicprocessing enzyme Protein family Peptidase S8 family, Furin subfamily Biochemical class Peptidase Function Mediates processing of TGFB1, an essential step in TGF-beta-1 activation. Ubiquitous endoprotease within constitutive secretory pathways capable of cleavage at the RX(K/R)R consensus motif. Related diseases Brachydactyly A2 (BDA2) [MIM:112600]: A form of brachydactyly. Brachydactyly defines a group of inherited malformations characterized by shortening of the digits due to abnormal development of the phalanges and/or the metacarpals. In brachydactyly type A2 shortening of the middle phalanges is confined to the index finger and the second toe, all other digits being more or less normal. Because of a rhomboid or triangular shape of the affected middle phalanx, the end of the second finger usually deviates radially. {ECO:0000269|PubMed:19327734, ECO:0000269|PubMed:21357617}. The gene represented in this entry is involved in disease pathogenesis. Duplications of a cis-regulatory element located approximately 110 kb downstream of BMP2 have been found in BDA2 families. They likely cause altered BMP2 expression with pathological consequences. {ECO:0000269|PubMed:19327734, ECO:0000269|PubMed:21357617}.; DISEASE: Short stature, facial dysmorphism, and skeletal anomalies with or without cardiac anomalies 1 (SSFSC1) [MIM:617877]: An autosomal dominant disorder characterized by short stature, facial dysmorphism, skeletal anomalies, and variable cardiac defects. Distinctive facial features include midface retrusion, short upturned nose, long philtrum, high-arched or cleft palate, and variable degrees of micrognathia and dental crowding. Skeletal anomalies include patterning defects of the axial skeleton, characterized by 11 pairs of ribs and brachydactyly of the fifth ray. Congenital heart defects are variably observed and appear to involve primarily the cardiac outflow tract. {ECO:0000269|PubMed:29198724}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03600 Interacts with P05067; P50281; Q9H239; O14793; K9N5Q8; P0DTC2; Q91QT1 EC number EC 3.4.21.75 Uniprot keywords 3D-structure; Autocatalytic cleavage; Calcium; Cell membrane; Cleavage on pair of basic residues; Disulfide bond; Endosome; Glycoprotein; Golgi apparatus; Heparin-binding; Host-virus interaction; Hydrolase; Membrane; Metal-binding; Phosphoprotein; Protease; Proteomics identification; Reference proteome; Repeat; Secreted; Serine protease; Signal; Transmembrane; Transmembrane helix; Zymogen Protein physicochemical properties Chain ID A Molecular weight (Da) 51029.8 Length 470 Aromaticity 0.08 Instability index 26.23 Isoelectric point 5.23 Charge (pH=7) -16.94 2D Binding mode Binding energy (Kcal/mol) -7.26 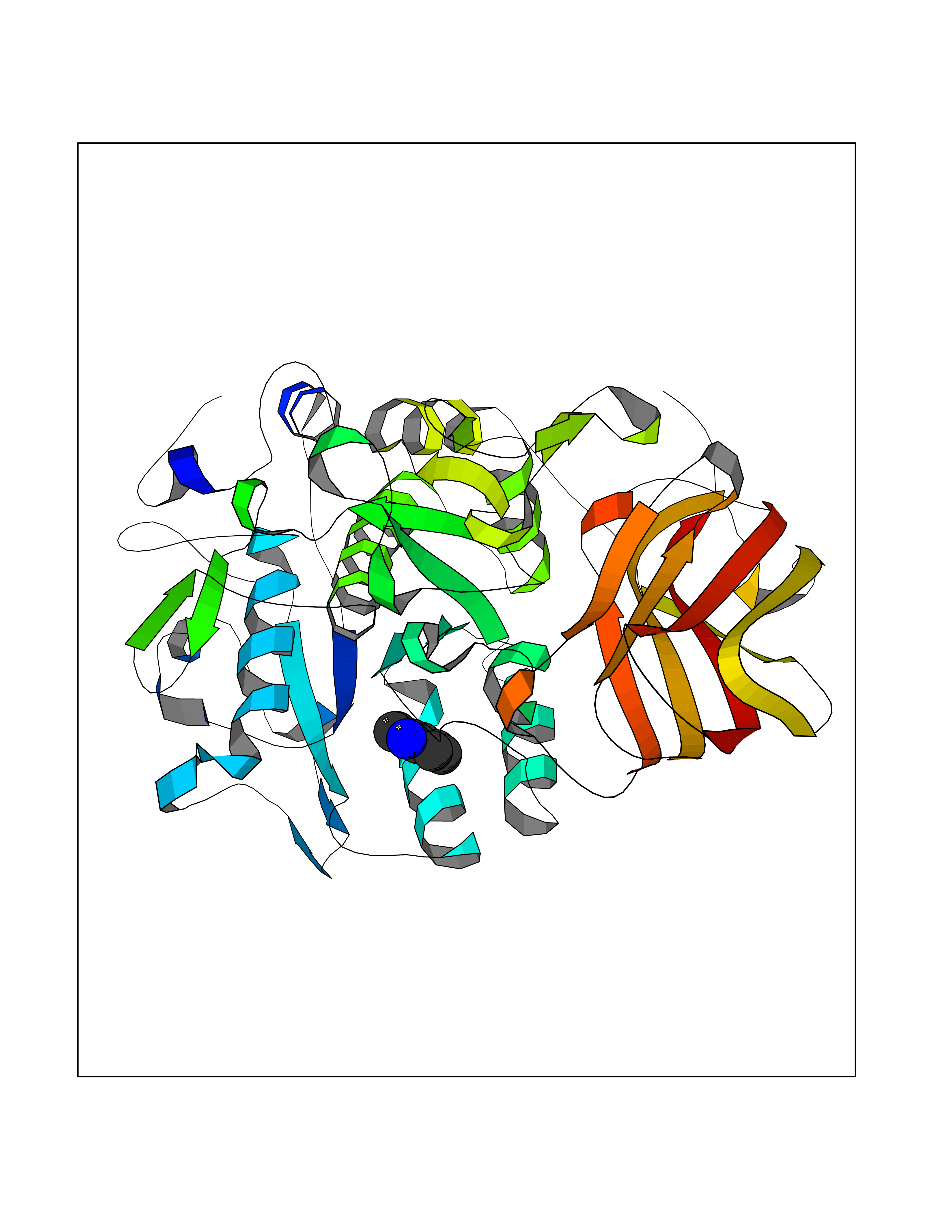 Molscript Map 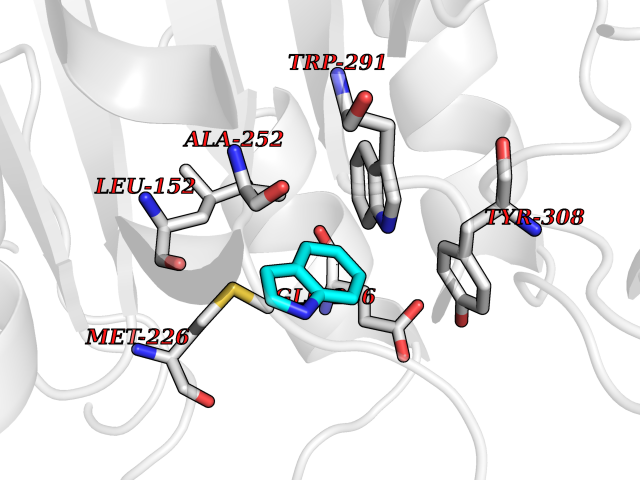 Pymol Map 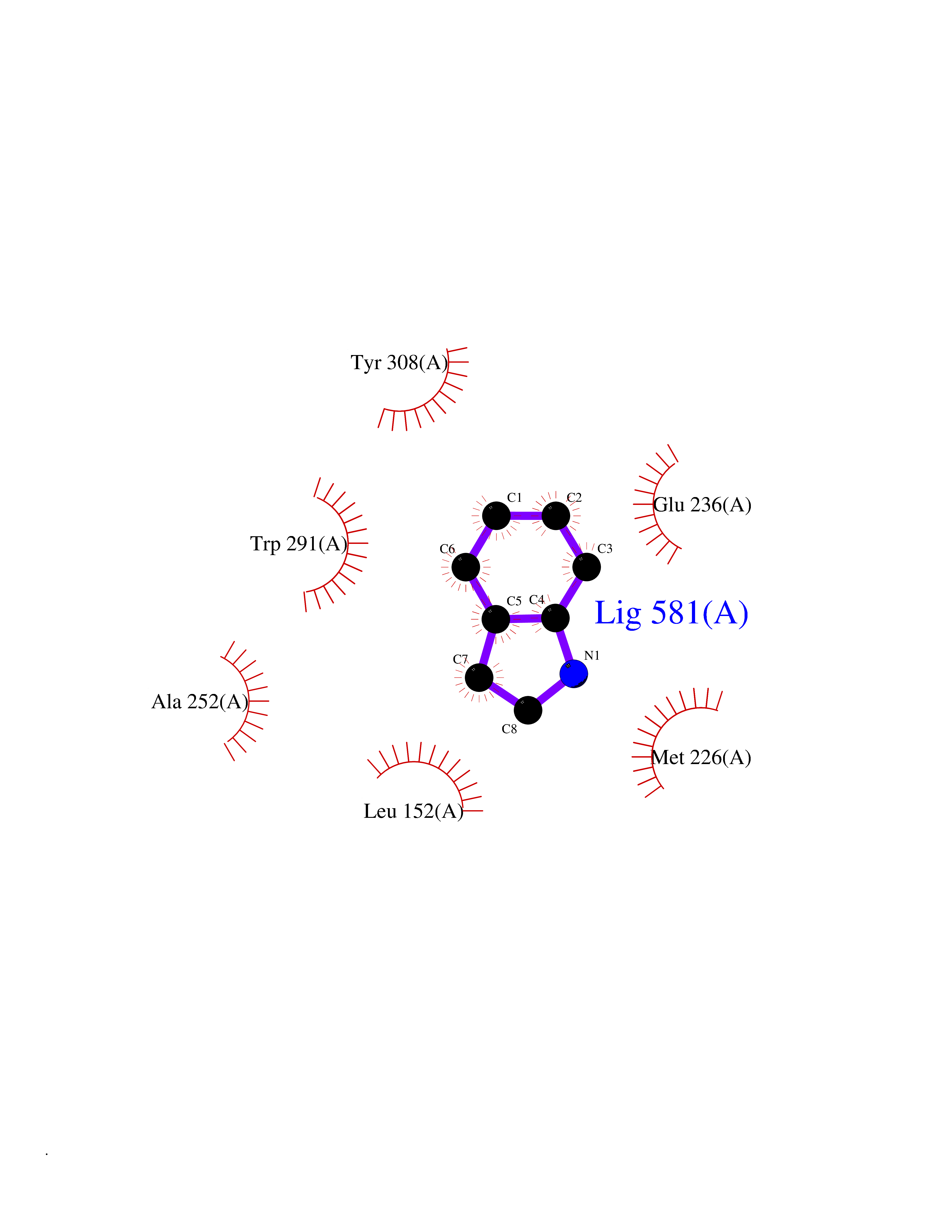 Ligplot Map 3D Binding mode Sequence YQEPTDPKFPQQWYLSGVTQRDLNVKAAWAQGYTGHGIVVSILDDGIEKNHPDLAGNYDPGASFDVNDQDPDPQPRYTQMNDNRHGTRCAGEVAAVANNGVCGVGVAYNARIGGVRMLDGEVTDAVEARSLGLNPNHIHIYSASWGPEDDGKTVDGPARLAEEAFFRGVSQGRGGLGSIFVWASGNGGREHDSCNCDGYTNSIYTLSISSATQFGNVPWYSEACSSTLATTYSSGNQNEKQIVTTDLRQKCTESHTGTSASAPLAAGIIALTLEANKDLTWRDMQHLVVQTSKPAHLNANDWATNGVGRKVSHSYGYGLLDAGAMVALAQDWTTVAPQRKCIIDILTEPKDIGKRLEVRKTVTACLGEPNHITRLEHAQARLTLSYNRRGDLAIHLVSPMGTRSTLLAARPHDYSADGFNDWAFMTTHSWDEDPSGEWVLEIENTSEANNYGTLTKFTLVLYGTAGENLY Hydrogen bonds contact Hydrophobic contact | ||||
| 87 | Casein kinase I epsilon (CSNK1E) | 4HNI | 5.33 | |
Target general information Gen name CSNK1E Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Casein kinase I isoform epsilon; CKIe; CKI-epsilon Protein family Protein kinase superfamily, CK1 Ser/Thr protein kinase family, Casein kinase I subfamily Biochemical class Kinase Function Can phosphorylate a large number of proteins. Participates in Wnt signaling. Phosphorylates DVL1 and DVL2. Central component of the circadian clock. In balance with PP1, determines the circadian period length, through the regulation of the speed and rhythmicity of PER1 and PER2 phosphorylation. Controls PER1 and PER2 nuclear transport and degradation. Inhibits cytokine-induced granuloytic differentiation. Casein kinases are operationally defined by their preferential utilization of acidic proteins such as caseins as substrates. Related diseases Truncation of the 3'-untranslated (3'-UTR) region of CD274 transcripts leads to elevated expression of CD274 in multiple cancers including T-cell leukemia, diffuse large B-cell lymphoma and stomach adenocarcinoma (PubMed:27281199). Disruption of 3'-UTR region is caused by structural variants that stabilize CD274 transcripts, leading to overexpression (PubMed:27281199). Increased expression in tumors promotes immune evasion and tumor cell growth by allowing malignant cells to escape destruction by the immune system (PubMed:27281199). {ECO:0000269|PubMed:27281199}. Drugs (DrugBank ID) DB06195; DB14989 Interacts with P25054; O15169; O14640; O14641; Q92997; Q9BQ89; Q1W6H9; Q86UY5; P08238; P23508; Q00987; Q16625; O15055; O75382; P62258; Q04917; Q5T7W0; O70239; Q60838 EC number EC 2.7.11.1 Uniprot keywords 3D-structure; ATP-binding; Biological rhythms; Cytoplasm; Kinase; Methylation; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Serine/threonine-protein kinase; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 32897.6 Length 284 Aromaticity 0.13 Instability index 40.77 Isoelectric point 9.3 Charge (pH=7) 11.34 2D Binding mode Binding energy (Kcal/mol) -7.27 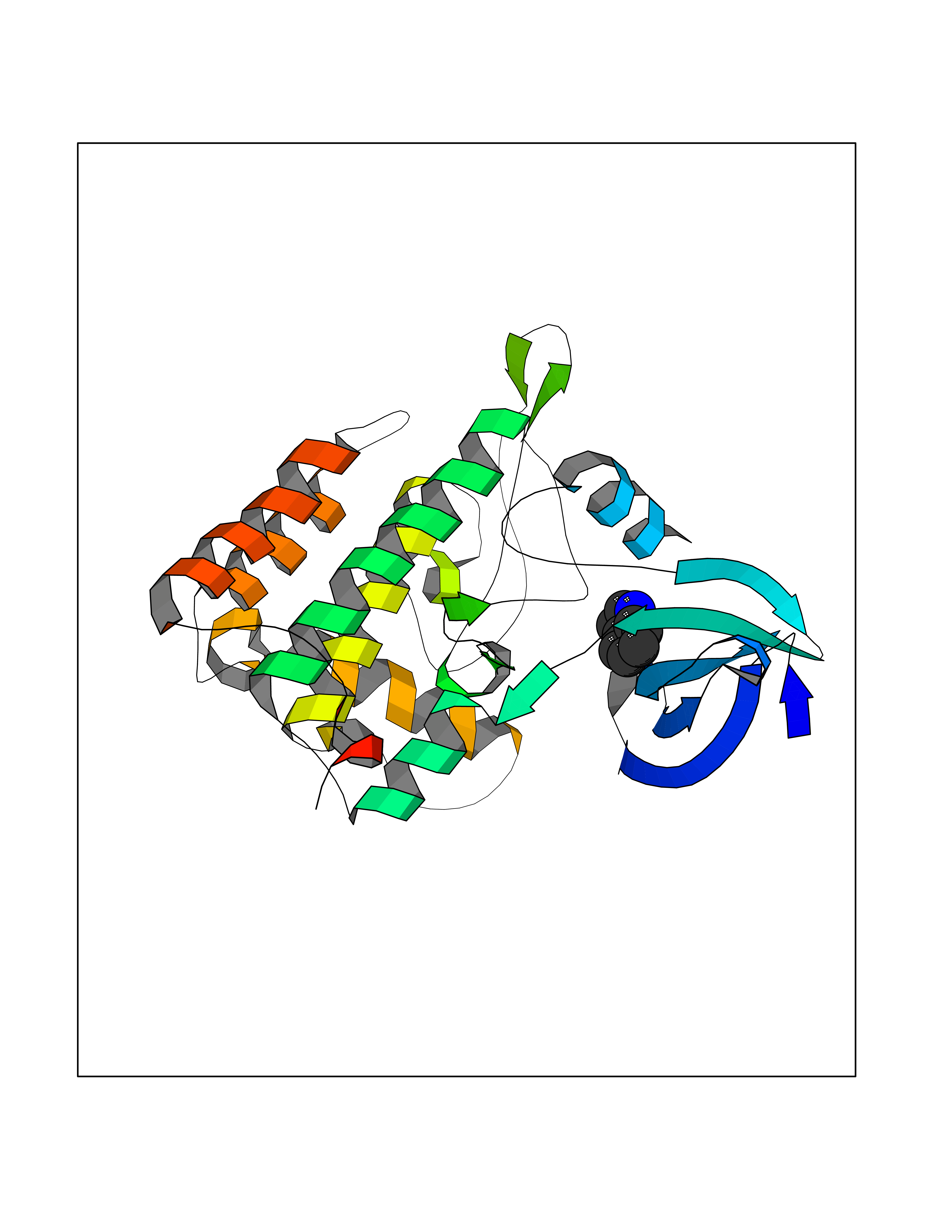 Molscript Map 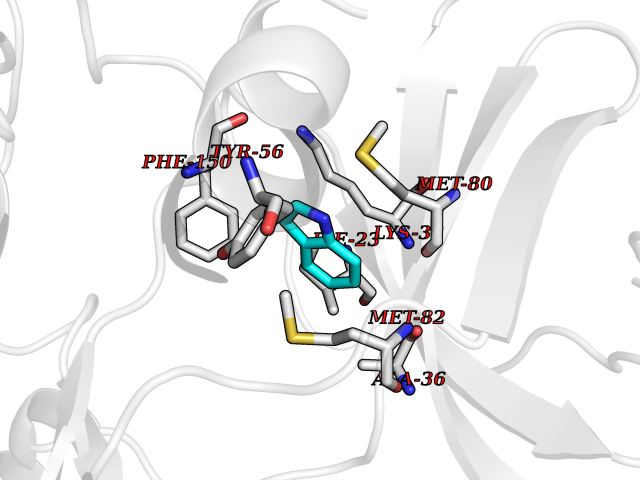 Pymol Map 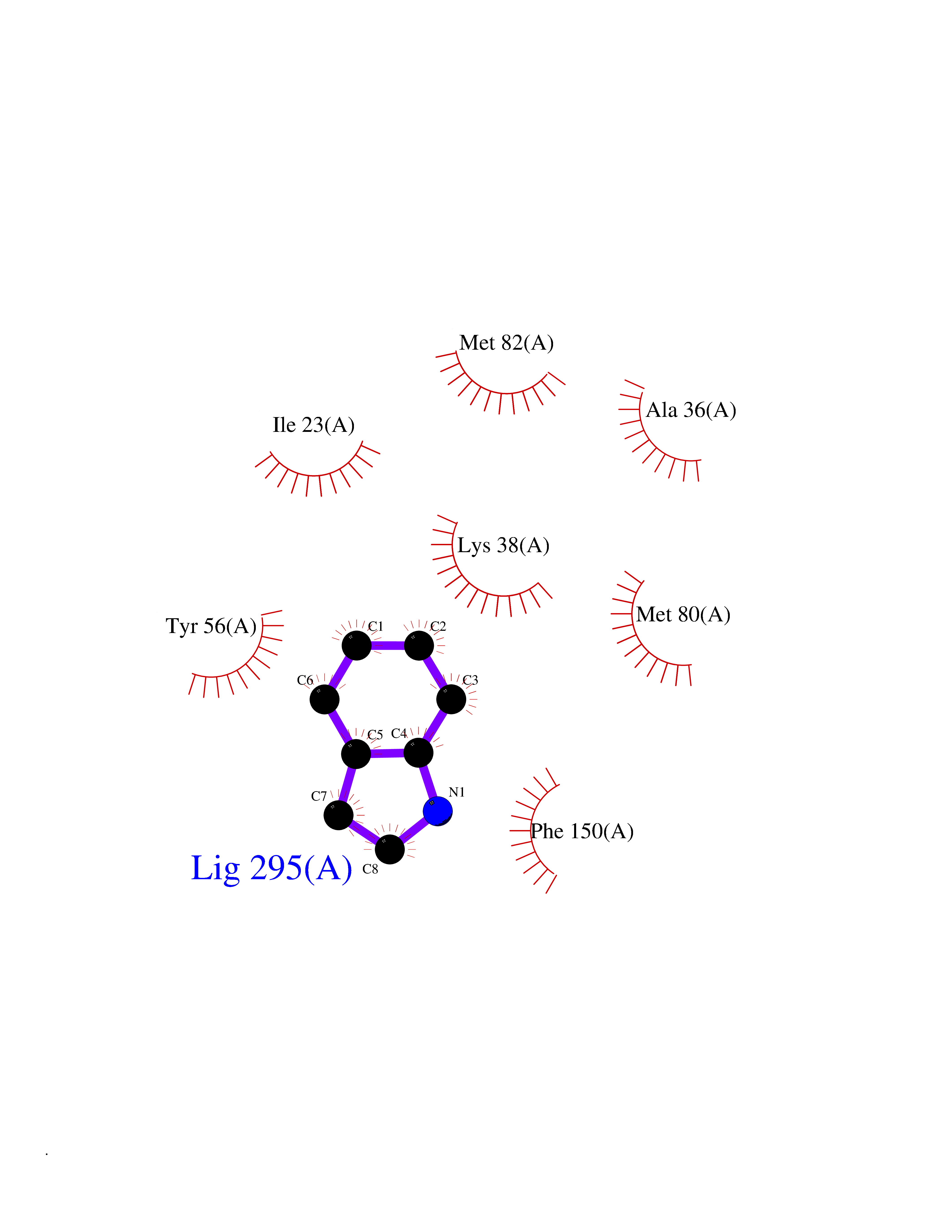 Ligplot Map 3D Binding mode Sequence LRVGNKYRLGRKIGSGSFGDIYLGANIASGEEVAIKLECVHIESKFYKMMQGGVGIPSIKWCGAEGDYNVMVMELLGPSLEDLFNFCSRKFSLKTVLLLADQMISRIEYIHSKNFIHRDVKPDNFLMGLGKKGNLVYIIDFGLAKKYRDARTHQHIPYRENKNLTGTARYASINTHLGIEQSRRDDLESLGYVLMYFNLGSLPWQGLKAATKRQKYERISEKKMSTPIEVLCKGYPSEFSTYLNFCRSLRFDDKPDYSYLRQLFRNLFHRQGFSYDYVFDWNML Hydrogen bonds contact Hydrophobic contact | ||||
| 88 | Angiotensinogenase renin (REN) | 3K1W | 5.32 | |
Target general information Gen name REN Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Renin; Angiotensinogenase Protein family Peptidase A1 family Biochemical class Peptidase Function Renin is a highly specific endopeptidase, whose only knownfunction is to generate angiotensin I from angiotensinogen in the plasma, initiating a cascade of reactions that produce an elevation of blood pressure and increased sodium retention by the kidney. Related diseases Renal tubular dysgenesis (RTD) [MIM:267430]: Autosomal recessive severe disorder of renal tubular development characterized by persistent fetal anuria and perinatal death, probably due to pulmonary hypoplasia from early-onset oligohydramnios (the Potter phenotype). {ECO:0000269|PubMed:16116425}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Tubulointerstitial kidney disease, autosomal dominant, 4 (ADTKD4) [MIM:613092]: A form of autosomal dominant tubulointerstitial kidney disease, a genetically heterogeneous disorder characterized by slowly progressive loss of kidney function, bland urinary sediment, hyperuricemia, absent or mildly increased albuminuria, lack of severe hypertension during the early stages, and normal or small kidneys on ultrasound. Renal histology shows variable abnormalities including interstitial fibrosis with tubular atrophy, microcystic dilatation of the tubules, thickening of tubular basement membranes, medullary cysts, and secondary glomerulosclerotic or glomerulocystic changes with abnormal glomerular tufting. There is significant variability, as well as incomplete penetrance. {ECO:0000269|PubMed:19664745}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07113; DB04387; DB03968; DB03736; DB03024; DB02803; DB07244; DB07174; DB08099; DB06967; DB09026; DB03395; DB02296; DB00722; DB00350; DB01844; DB04379; DB06899; DB07059; DB00212; DB05203 Interacts with P01019 EC number EC 3.4.23.15 Uniprot keywords 3D-structure; Alternative splicing; Aspartyl protease; Cleavage on pair of basic residues; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; Hydrolase; Membrane; Protease; Proteomics identification; Reference proteome; Secreted; Signal; Zymogen Protein physicochemical properties Chain ID A,B Molecular weight (Da) 36039.5 Length 328 Aromaticity 0.12 Instability index 40.22 Isoelectric point 5.51 Charge (pH=7) -6.69 2D Binding mode Binding energy (Kcal/mol) -7.26  Molscript Map 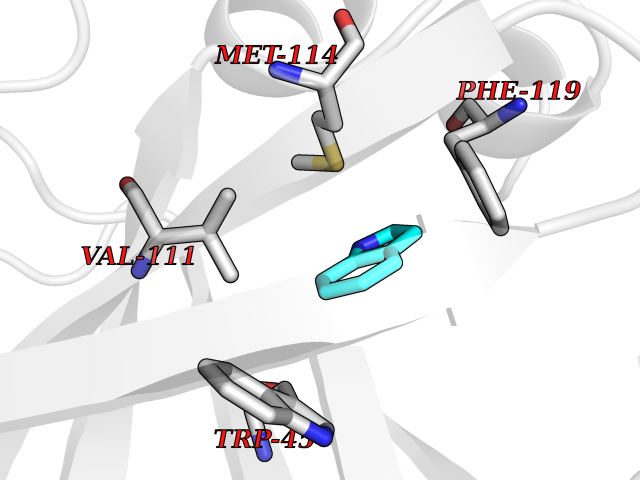 Pymol Map 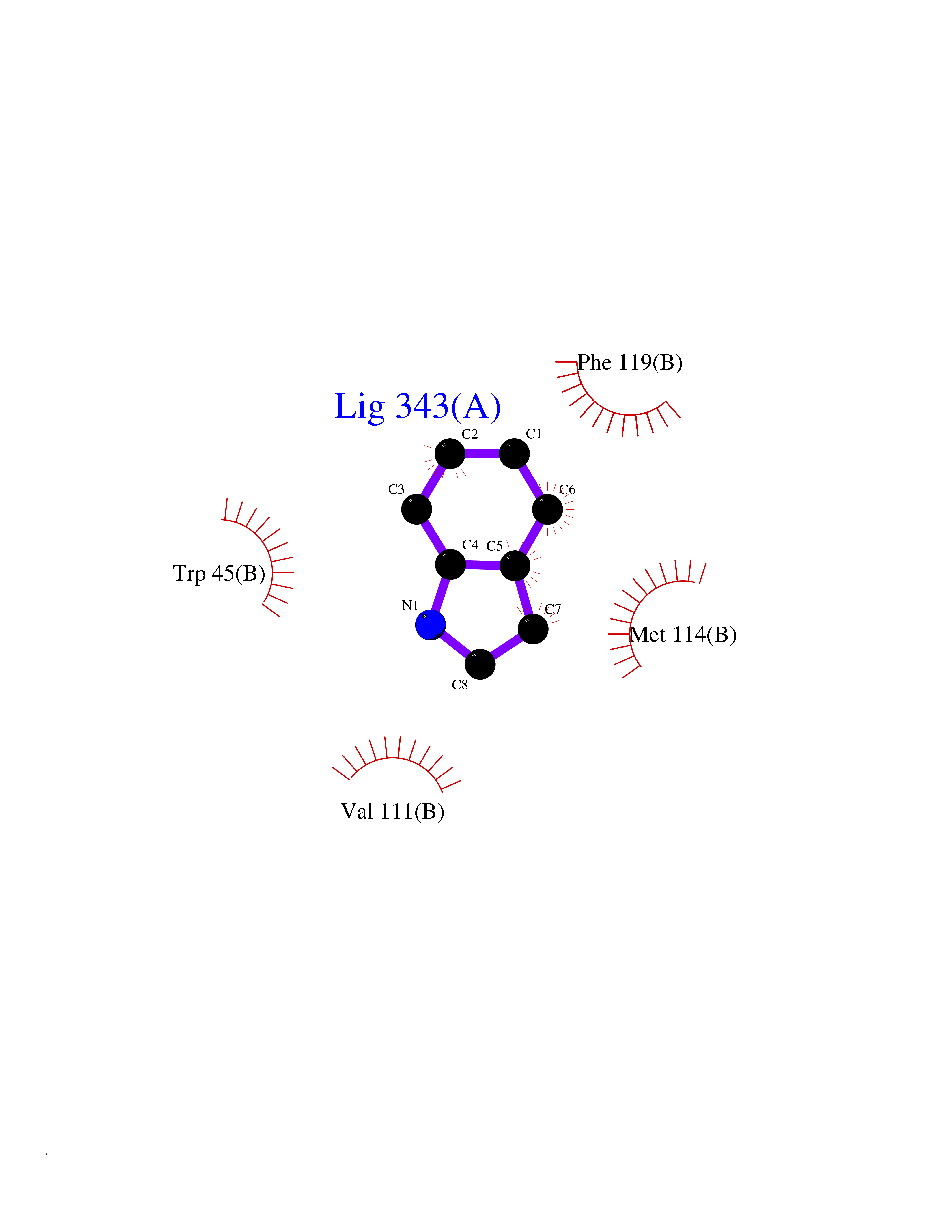 Ligplot Map 3D Binding mode Sequence VILTNYMDTQYYGEIGIGTPPQTFKVVFDTGSSNVWVPSSKCSRLYTACVYHKLFDASDSSSYKHNGTELTLRYSTGTVSGFLSQDIITVGGITVTQMFGEVTEMPALPFMLAEFDGVVGMGFIEQAIGRVTPIFDNIISQGVLKEDVFSFYYNRDSSLGGQIVLGGSDPQHYEGNFHYINLIKTGVWQIQMKGVSVGSSTLLCEDGCLALVDTGASYISGSTSSIEKLMEALGAKKRLFDYVVKCNEGPTLPDISFHLGGKEYTLTSADYVFQESYSSKKLCTLAIHAMDIPPPTGPTWALGATFIRKFYTEFDRRNNRIGFALARH Hydrogen bonds contact Hydrophobic contact | ||||
| 89 | Erbb2 tyrosine kinase receptor (HER2) | 3PP0 | 5.32 | |
Target general information Gen name ERBB2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms p185erbB2; Tyrosine kinase-type cell surface receptor HER2; Receptor tyrosine-protein kinase erbB-2; Proto-oncogene c-ErbB-2; Proto-oncogene Neu; NGL; NEU; Metastatic lymph node gene 19 protein; MLN19 Protein family Protein kinase superfamily, Tyr protein kinase family, EGF receptor subfamily Biochemical class Kinase Function Protein tyrosine kinase that is part of several cell surface receptor complexes, but that apparently needs a coreceptor for ligand binding. Essential component of a neuregulin-receptor complex, although neuregulins do not interact with it alone. GP30 is a potential ligand for this receptor. Regulates outgrowth and stabilization of peripheral microtubules (MTs). Upon ERBB2 activation, the MEMO1-RHOA-DIAPH1 signaling pathway elicits the phosphorylation and thus the inhibition of GSK3B at cell membrane. This prevents the phosphorylation of APC and CLASP2, allowing its association with the cell membrane. In turn, membrane-bound APC allows the localization of MACF1 to the cell membrane, which is required for microtubule capture and stabilization. Related diseases Glioma (GLM) [MIM:137800]: Gliomas are benign or malignant central nervous system neoplasms derived from glial cells. They comprise astrocytomas and glioblastoma multiforme that are derived from astrocytes, oligodendrogliomas derived from oligodendrocytes and ependymomas derived from ependymocytes. {ECO:0000269|PubMed:15457249}. The gene represented in this entry is involved in disease pathogenesis.; DISEASE: Ovarian cancer (OC) [MIM:167000]: The term ovarian cancer defines malignancies originating from ovarian tissue. Although many histologic types of ovarian tumors have been described, epithelial ovarian carcinoma is the most common form. Ovarian cancers are often asymptomatic and the recognized signs and symptoms, even of late-stage disease, are vague. Consequently, most patients are diagnosed with advanced disease. {ECO:0000269|PubMed:15457249, ECO:0000269|PubMed:17344846}. The gene represented in this entry is involved in disease pathogenesis.; DISEASE: Lung cancer (LNCR) [MIM:211980]: A common malignancy affecting tissues of the lung. The most common form of lung cancer is non-small cell lung cancer (NSCLC) that can be divided into 3 major histologic subtypes: squamous cell carcinoma, adenocarcinoma, and large cell lung cancer. NSCLC is often diagnosed at an advanced stage and has a poor prognosis. {ECO:0000269|PubMed:15457249}. The gene represented in this entry is involved in disease pathogenesis.; DISEASE: Gastric cancer (GASC) [MIM:613659]: A malignant disease which starts in the stomach, can spread to the esophagus or the small intestine, and can extend through the stomach wall to nearby lymph nodes and organs. It also can metastasize to other parts of the body. The term gastric cancer or gastric carcinoma refers to adenocarcinoma of the stomach that accounts for most of all gastric malignant tumors. Two main histologic types are recognized, diffuse type and intestinal type carcinomas. Diffuse tumors are poorly differentiated infiltrating lesions, resulting in thickening of the stomach. In contrast, intestinal tumors are usually exophytic, often ulcerating, and associated with intestinal metaplasia of the stomach, most often observed in sporadic disease. {ECO:0000269|PubMed:15457249, ECO:0000269|PubMed:17344846}. The protein represented in this entry is involved in disease pathogenesis.; DISEASE: Chromosomal aberrations involving ERBB2 may be a cause gastric cancer. Deletions within 17q12 region producing fusion transcripts with CDK12, leading to CDK12-ERBB2 fusion leading to truncated CDK12 protein not in-frame with ERBB2. {ECO:0000269|PubMed:21097718}.; DISEASE: Visceral neuropathy, familial, 2, autosomal recessive (VSCN2) [MIM:619465]: An autosomal recessive disorder characterized by intestinal dysmotility due to aganglionosis (Hirschsprung disease), hypoganglionosis, and/or chronic intestinal pseudoobstruction. Patients also show peripheral axonal neuropathy, hypotonia, mild developmental delay, unilateral ptosis, and sensorineural hearing loss. {ECO:0000269|PubMed:33497358}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08916; DB06021; DB12267; DB12010; DB04988; DB01259; DB14967; DB06366; DB11973; DB00072; DB05773; DB11652; DB05944; DB15035 Interacts with P00519; P42684; P15309; P60709; Q92625; O00213; O75815; Q9HB71; Q16543; Q9NSE2; Q7Z7G1; P46109; Q93034; Q99704; Q8TEW6; Q15075; P98172; P00533; P04626; P21860; Q15303; Q9UJM3; P09769; P06241; O75791; P62993; Q14451; P07900; P08238; P14625; P11021; P46940; P35568; Q08881; P23458; Q14974; Q96JA1; O75367; O75367-3; Q9UQF2; Q13387; P42679; Q9Y316; O43639; Q02297-7; O00750; P27986; O00459; Q92569; P19174; P16885; O95602; Q13882; Q06124; Q05209; Q99952; Q99952-1; P23467; P08575; Q12913; Q15262; Q16827; Q15256; P49792; P20936; O95980; Q9NP31; P29353; P98077; Q92529; Q9H6Q3; O15524; P12931; P42224; P40763; P31948; Q7KZ85; P43405; Q9Y490; Q63HR2; Q68CZ2; Q96D37; P52735; O14980; P62258 EC number EC 2.7.10.1 Uniprot keywords 3D-structure; Activator; Alternative initiation; Alternative splicing; ATP-binding; Cell membrane; Cell projection; Chromosomal rearrangement; Cytoplasm; Disease variant; Disulfide bond; Endosome; Glycoprotein; Kinase; Membrane; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Signal; Transcription; Transcription regulation; Transferase; Transmembrane; Transmembrane helix; Tyrosine-protein kinase Protein physicochemical properties Chain ID A,B Molecular weight (Da) 33776.1 Length 296 Aromaticity 0.08 Instability index 47.13 Isoelectric point 8.67 Charge (pH=7) 4.22 2D Binding mode Binding energy (Kcal/mol) -7.26 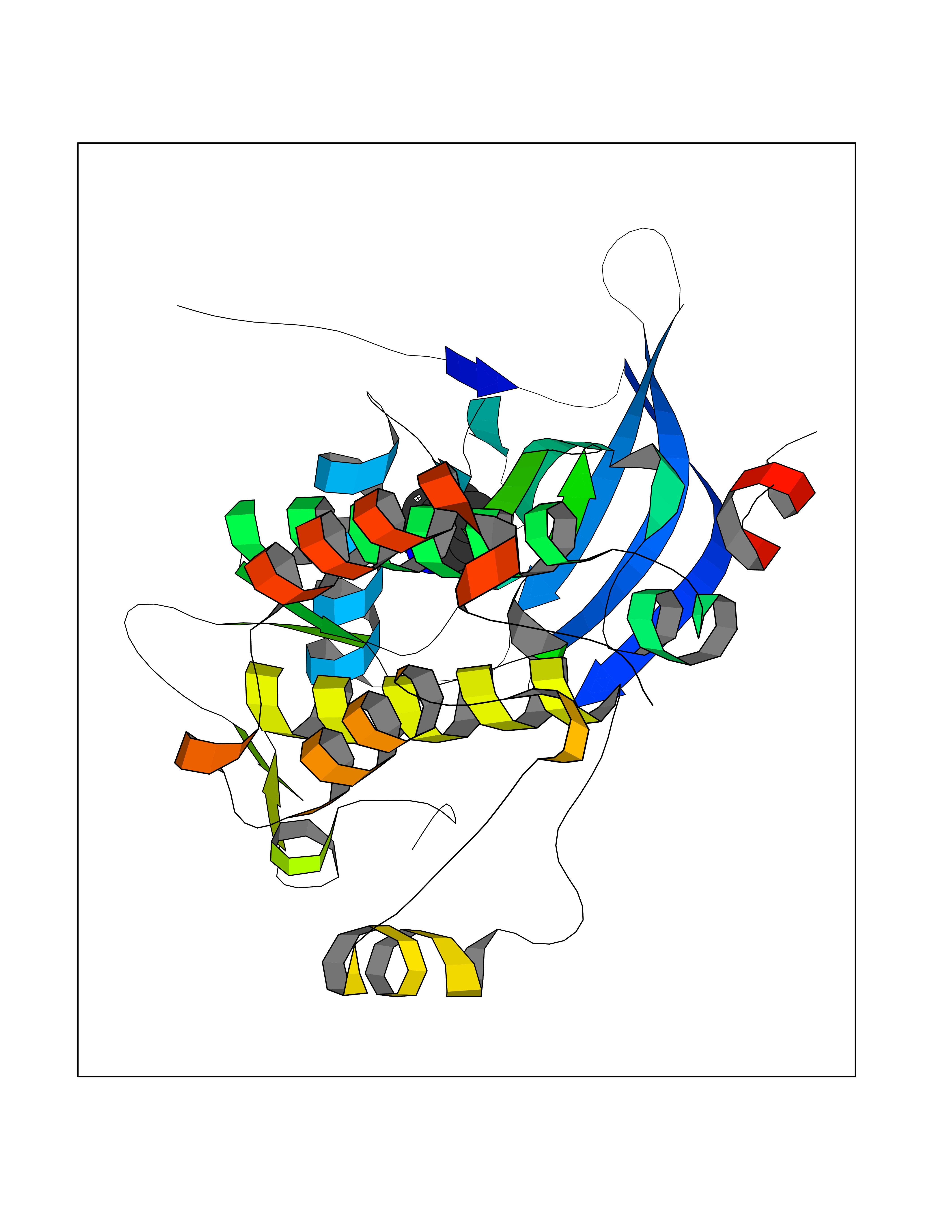 Molscript Map 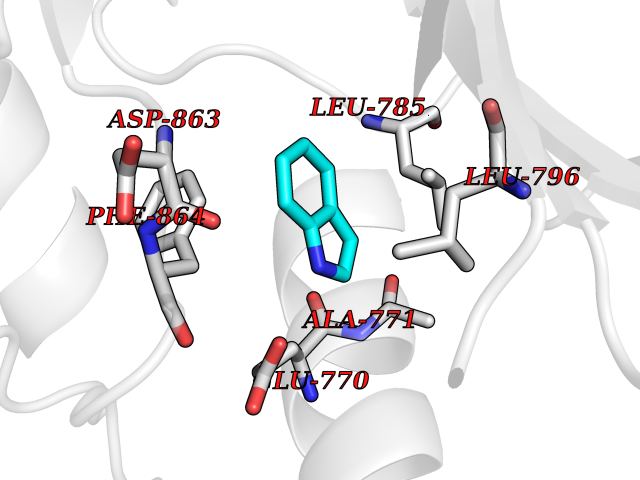 Pymol Map 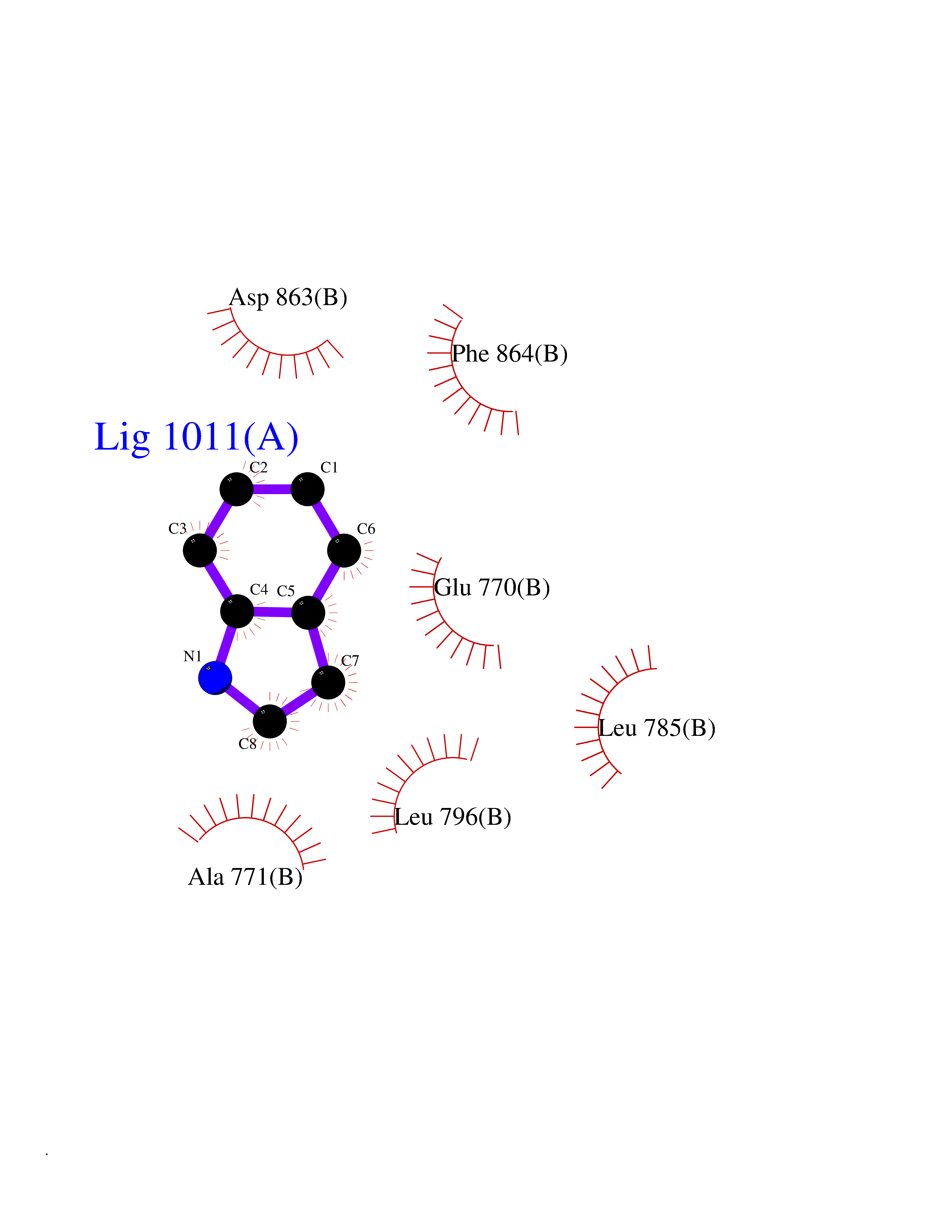 Ligplot Map 3D Binding mode Sequence APNQALLRILKETELRKVKVLGSGAFGTVYKGIWIPDGENVKIPVAIKVLRENTSPKANKEILDEAYVMAGVGSPYVSRLLGICLTSTVQLVTQLMPYGCLLDHVRENRGRLGSQDLLNWCMQIAKGMSYLEDVRLVHRDLAARNVLVKSPNHVKITDFGLARLLDIDETEYHAGKVPIKWMALESILRRRFTHQSDVWSYGVTVWELMTFGAKPYDGIPAREIPDLLEKGERLPQPPICTIDVYMIMVKCWMIDSECRPRFRELVSEFSRMARDPQRFVVIQNEPLDSTFYRSLL Hydrogen bonds contact Hydrophobic contact | ||||
| 90 | Glutathione S-transferase A4 | 3IK7 | 5.32 | |
Target general information Gen name GSTA4 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family GST superfamily, Alpha family Biochemical class Transferase Function Glutathione transferase activity.Identical protein binding.Protein homodimerization activity. Related diseases Cocoon syndrome (COCOS) [MIM:613630]: A lethal syndrome characterized by multiple fetal malformations including defective face and seemingly absent limbs, which are bound to the trunk and encased under the skin. {ECO:0000269|PubMed:20961246}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Bartsocas-Papas syndrome 2 (BPS2) [MIM:619339]: An autosomal recessive, severe form of popliteal pterygium syndrome. Popliteal pterygia syndromes have considerable variability in severity and in the associated phenotypic features but they are all characterized by cutaneous webbing across one or more major joints, cleft lip and/or palate, syndactyly, and genital malformations. {ECO:0000269|PubMed:25691407}. The disease may be caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00143 Interacts with Q96LR7; O95995; P09210; O15217; O15116; Q96HA8 EC number 2.5.1.18 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Cytoplasm; Proteomics identification; Reference proteome; Transferase Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 50705.7 Length 438 Aromaticity 0.1 Instability index 50.22 Isoelectric point 7.95 Charge (pH=7) 1.91 2D Binding mode Binding energy (Kcal/mol) -7.25 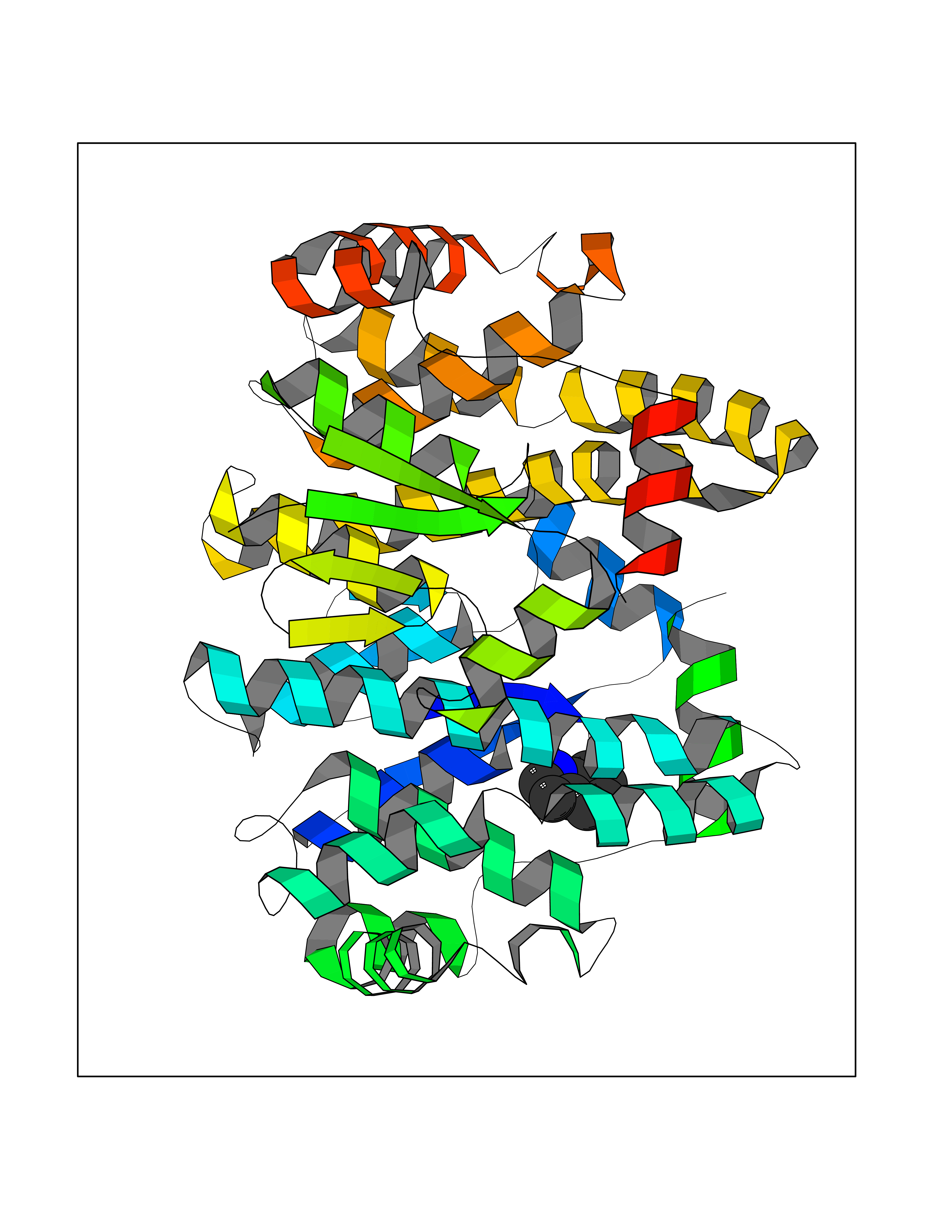 Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence AARPKLHYPNGRGRMESVRWVLAAAGVEFDEEFLETKEQLYKLQDGNHLLFQQVPMVEIDGMKLVQTRSILHYIADKHNLFGKNLKERTLIDMYVEGTLDLLELLIMHPFLKPDDQQKEVVNMAQKAIIRYFPVFEKILRGHGQSFLVGNQLSLADVILLQTILALEEKIPNILSAFPFLQEYTVKLSNIPTIKRFLEPGSKKKPPPDEIYVRTVYNIFRARPKLHYPNGRGRMESVRWVLAAAGVEFDEEFLETKEQLYKLQDGNHLLFQQVPMVEIDGMKLVQTRSILHYIADKHNLFGKNLKERTLIDMYVEGTLDLLELLIMHPFLKPDDQQKEVVNMAQKAIIRYFPVFEKILRGHGQSFLVGNQLSLADVILLQTILALEEKIPNILSAFPFLQEYTVKLSNIPTIKRFLEPGSKKKPPPDEIYVRTVYNIF Hydrogen bonds contact Hydrophobic contact | ||||
| 91 | D-amino acid oxidase (DAO) | 3ZNN | 5.32 | |
Target general information Gen name DAO Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Daminoacid oxidase; DAMOX; DAAO Protein family DAMOX/DASOX family Biochemical class CH-NH(2) donor oxidoreductase Function Regulates the level of the neuromodulator D-serine in the brain. Has high activity towards D-DOPA and contributes to dopamine synthesis. Could act as a detoxifying agent which removes D-amino acids accumulated during aging. Acts on a variety of D-amino acids with a preference for those having small hydrophobic side chains followed by those bearing polar, aromatic, and basic groups. Does not act on acidic amino acids. Related diseases Schizophrenia (SCZD) [MIM:181500]: A complex, multifactorial psychotic disorder or group of disorders characterized by disturbances in the form and content of thought (e.g. delusions, hallucinations), in mood (e.g. inappropriate affect), in sense of self and relationship to the external world (e.g. loss of ego boundaries, withdrawal), and in behavior (e.g bizarre or apparently purposeless behavior). Although it affects emotions, it is distinguished from mood disorders in which such disturbances are primary. Similarly, there may be mild impairment of cognitive function, and it is distinguished from the dementias in which disturbed cognitive function is considered primary. Some patients manifest schizophrenic as well as bipolar disorder symptoms and are often given the diagnosis of schizoaffective disorder. {ECO:0000269|PubMed:12364586}. Disease susceptibility may be associated with variants affecting the gene represented in this entry.; DISEASE: Amyotrophic lateral sclerosis (ALS) [MIM:105400]: A neurodegenerative disorder affecting upper motor neurons in the brain and lower motor neurons in the brain stem and spinal cord, resulting in fatal paralysis. Sensory abnormalities are absent. The pathologic hallmarks of the disease include pallor of the corticospinal tract due to loss of motor neurons, presence of ubiquitin-positive inclusions within surviving motor neurons, and deposition of pathologic aggregates. The etiology of amyotrophic lateral sclerosis is likely to be multifactorial, involving both genetic and environmental factors. The disease is inherited in 5-10% of the cases. {ECO:0000269|PubMed:20368421, ECO:0000269|PubMed:20538972, ECO:0000269|PubMed:22203986, ECO:0000269|PubMed:23219954, ECO:0000269|PubMed:24138986, ECO:0000269|PubMed:25701391, ECO:0000269|PubMed:37558109, ECO:0000269|PubMed:38035964, ECO:0000269|PubMed:38134563}. Disease susceptibility may be associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07979; DB02838; DB04166; DB03793; DB03225; DB03147; DB03531; DB02988 Interacts with Q9P2K6; O43741 EC number EC 1.4.3.3 Uniprot keywords 3D-structure; Amyotrophic lateral sclerosis; Cell projection; Cytoplasm; Disease variant; FAD; Flavoprotein; Neurodegeneration; Oxidoreductase; Peroxisome; Phosphoprotein; Proteomics identification; Reference proteome; S-nitrosylation; Schizophrenia; Secreted; Synapse Protein physicochemical properties Chain ID A,B Molecular weight (Da) 38654.6 Length 340 Aromaticity 0.11 Instability index 29.13 Isoelectric point 6.18 Charge (pH=7) -4.45 2D Binding mode Binding energy (Kcal/mol) -7.26  Molscript Map  Pymol Map 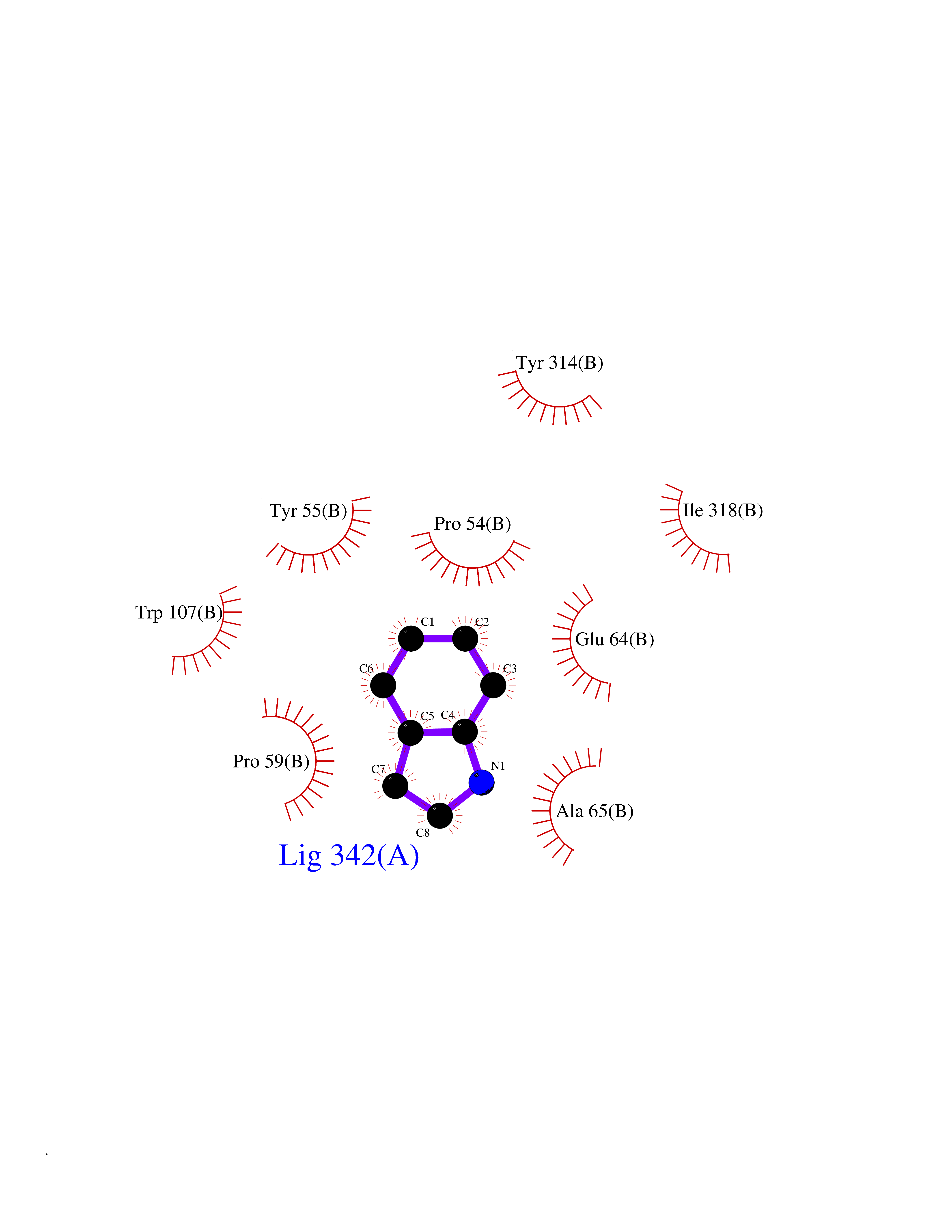 Ligplot Map 3D Binding mode Sequence MRVVVIGAGVIGLSTALCIHERYHSVLQPLDIKVYADRFTPLTTTDVAAGLWQPYLSDPNNPQEADWSQQTFDYLLSHVHSPNAENLGLFLISGYNLFHEAIPDPSWKDTVLGFRKLTPRELDMFPDYGYGWFHTSLILEGKNYLQWLTERLTERGVKFFQRKVESFEEVAREGADVIVNCTGVWAGALQRDPLLQPGRGQIMKVDAPWMKHFILTHDPERGIYNSPYIIPGTQTVTLGGIFQLGNWSELNNIQDHNTIWEGCCRLEPTLKNARIIGERTGFRPVRPQIRLEREQLRTGPSNTEVIHNYGHGGYGLTIHWGCALEAAKLFGRILEEKKLS Hydrogen bonds contact Hydrophobic contact | ||||
| 92 | Angiotensin II receptor type-1 (AGTR1) | 4ZUD | 5.32 | |
Target general information Gen name AGTR1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Type-1 angiotensin II receptor; Angiotensin II type-1 receptor; Angiotensin II receptor 1; Angiotensin 1 receptor; AT2R1B; AT2R1; AT1BR; AT1AR; AT1; AGTR1B; AGTR1A Protein family G-protein coupled receptor 1 family Biochemical class GPCR rhodopsin Function Mediates its action by association with G proteins that activate a phosphatidylinositol-calcium second messenger system. Receptor for angiotensin II. Related diseases Renal tubular dysgenesis (RTD) [MIM:267430]: Autosomal recessive severe disorder of renal tubular development characterized by persistent fetal anuria and perinatal death, probably due to pulmonary hypoplasia from early-onset oligohydramnios (the Potter phenotype). {ECO:0000269|PubMed:16116425}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB11842; DB08822; DB13919; DB00796; DB05739; DB00876; DB09279; DB01342; DB01029; DB00678; DB00275; DB01347; DB01349; DB00966; DB00177 Interacts with PRO_0000032458 [P01019]; P35414; P05026; Q6ZMG9; O75937; P54368 EC number NA Uniprot keywords 3D-structure; Cell membrane; Disease variant; Disulfide bond; G-protein coupled receptor; Glycoprotein; Host-virus interaction; Lipoprotein; Membrane; Palmitate; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 30770.7 Length 271 Aromaticity 0.16 Instability index 28.86 Isoelectric point 8.09 Charge (pH=7) 2.1 2D Binding mode Binding energy (Kcal/mol) -7.26 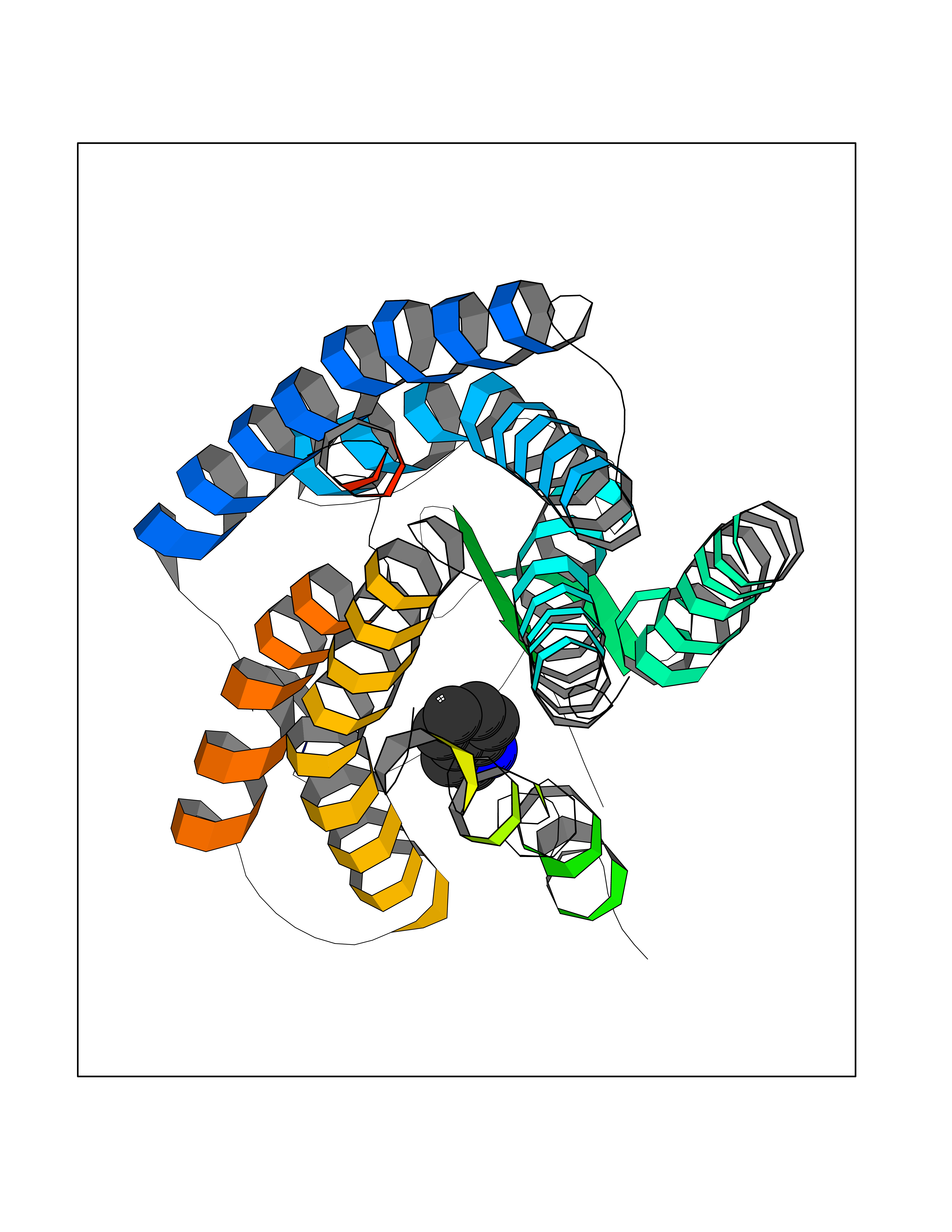 Molscript Map 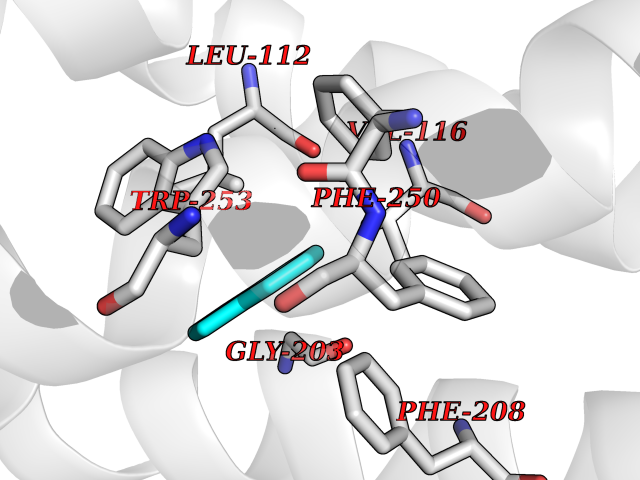 Pymol Map 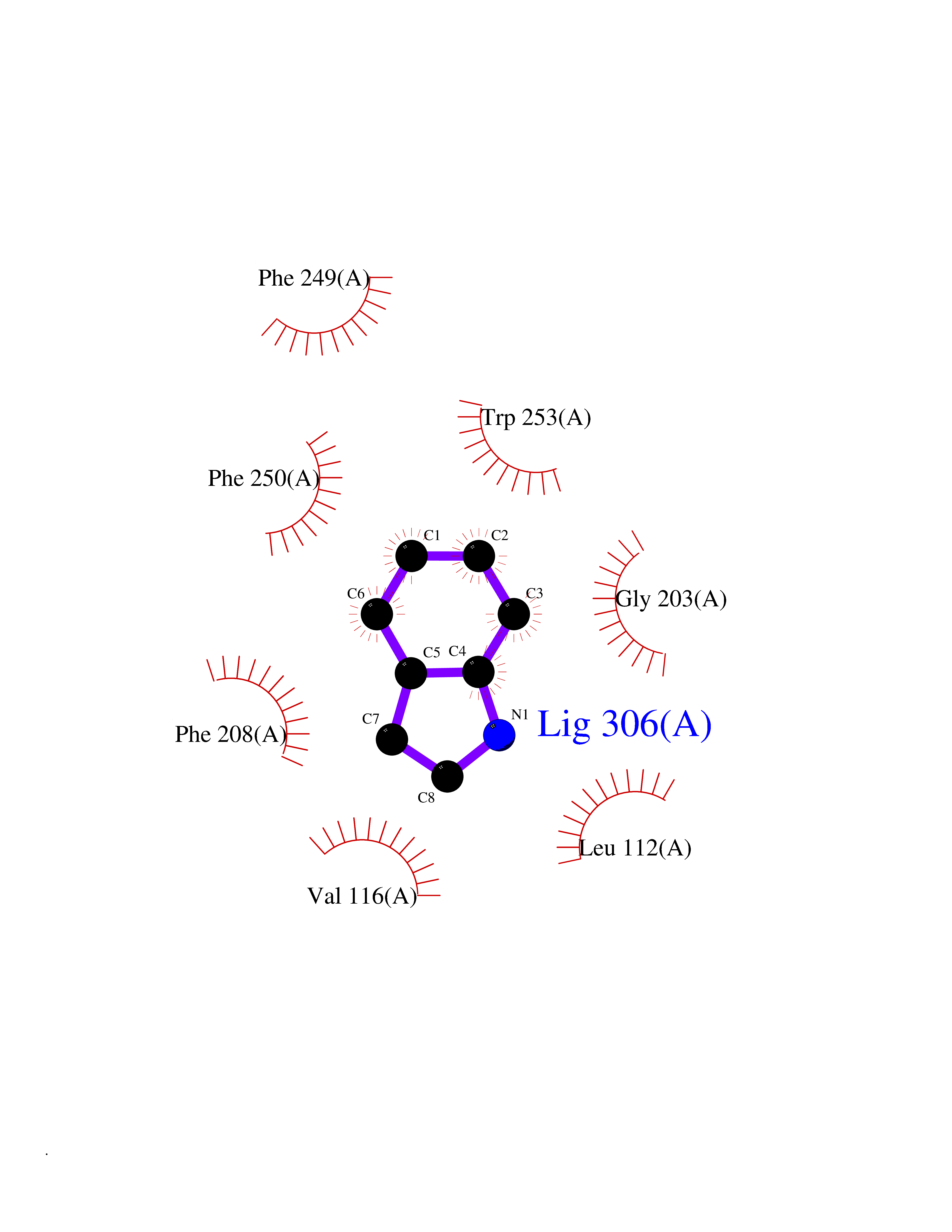 Ligplot Map 3D Binding mode Sequence ILNSSDCPKAGRHNYIFVMIPTLYSIIFVVGIFGNSLVVIVIYFYMKLKTVASVFLLNLALADLCFLLTLPLWAVYTAMEYRWPFGNYLCKIASASVSFNLYASVFLLTCLSIDRYLAIVHPTMLVAKVTCIIIWLLAGLASLPAIIHRNVFFIENTNITVCAFHYESTLPIGLGLTKNILGFLFPFLIILTSYTLIWKALNDDIFKIIMAIVLFFFFSWIPHQIFTFLDVLIQLGIIRDCRIADIVDTAMPITICIAYFNNCLNPLFYGF Hydrogen bonds contact Hydrophobic contact | ||||
| 93 | Neuronal acetylcholine receptor subunit alpha-3 | 4ZK4 | 5.32 | |
Target general information Gen name CHRNA3 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NACHRA3 Protein family Ligand-gated ion channel (TC 1.A.9) family, Acetylcholine receptor (TC 1.A.9.1) subfamily, Alpha-3/CHRNA3 sub-subfamily Biochemical class Acetylcholine binding protein Function Acetylcholine binding.Acetylcholine-gated cation-selective channel activity.Acetylcholine receptor activity.Ligand-gated ion channel activity.Serotonin-gated cation-selective channel activity. Related diseases Bladder dysfunction, autonomic, with impaired pupillary reflex and secondary CAKUT (BAIPRCK) [MIM:191800]: An autosomal recessive disease characterized by impaired innervation and autonomic dysfunction of the urinary bladder, hydronephrosis, vesicoureteral reflux, small kidneys, recurrent urinary tract infections, and progressive renal insufficiency. Additional autonomic features are impaired pupillary reflex and orthostatic hypotension. The disease manifests in utero or early childhood. {ECO:0000269|PubMed:31708116}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00915; DB01156; DB00237; DB00565; DB09028; DB00514; DB07720; DB00898; DB00472; DB05710; DB01227; DB00848; DB00333; DB00184; DB01090; DB00202; DB01273 Interacts with NA EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disease variant; Disulfide bond; Endoplasmic reticulum; Glycoprotein; Golgi apparatus; Ion channel; Ion transport; Ligand-gated ion channel; Membrane; Phosphoprotein; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Signal; Synapse; Transmembrane; Transmembrane helix; Transport; Ubl conjugation Protein physicochemical properties Chain ID A,B,C,D,E Molecular weight (Da) 46391.5 Length 408 Aromaticity 0.12 Instability index 30.23 Isoelectric point 4.6 Charge (pH=7) -22.73 2D Binding mode Binding energy (Kcal/mol) -7.26  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence LHSQANLMRLKSDLFYPGPTKDDPLTVTLGFTLQDIVKADSSTNEVDLVYWEQQRWKLNSLMWDPNEYGNITDFRTSAADIWTPDITAYSSTRPVQVLSPQIAVVTHDGSVMFIPAQRLSFMCDPTGVDSEEGATCAVKFGSWVYSGFEIDLKTDTDQVDLSSYYASSKYEILSATQYKHDIKYNCCEEIYPDVVLVVKFRERRLHSQANLMRLKSDLFNRYPGPTKDDPLTVTLGFTLQDIVKADSSTNEVDLVYWEQQRWKLNSLMWDPNEYGNITDFRTSAADIWTPDITAYSSTRPVQVLSPQIAVVTHDGSVMFIPAQRLSFMCDPTGVDSEEGATCAVKFGSWVYSGFEIDLKTDTDQVDLSSYYASSKYEILSATQYKHDIKYNCCEEIYPDVVLVVKFRE Hydrogen bonds contact Hydrophobic contact | ||||
| 94 | Glycine N-methyltransferase | 1R74 | 5.32 | |
Target general information Gen name GNMT Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Class I-like SAM-binding methyltransferase superfamily, Glycine N-methyltransferase family Biochemical class Transferase Function Folic acid binding.Glycine binding.Glycine N-methyltransferase activity. Related diseases Glycine N-methyltransferase deficiency (GNMT deficiency) [MIM:606664]: The only clinical abnormalities in patients with this deficiency are mild hepatomegaly and chronic elevation of serum transaminases. {ECO:0000269|PubMed:11810299, ECO:0000269|PubMed:14739680}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00118; DB00145; DB01752 Interacts with P49407; Q494R4-2; P24311; O60931-2; Q14331; Q9UN88; Q13066; A0A0S2Z5F2; Q14749; P00492; Q96HA8; O75643; P31213; Q9UPQ4-2; Q9Y3M9 EC number 2.1.1.20 Uniprot keywords 3D-structure; Acetylation; Cytoplasm; Disease variant; Folate-binding; Methyltransferase; Phosphoprotein; Proteomics identification; Reference proteome; S-adenosyl-L-methionine; Transferase Protein physicochemical properties Chain ID A,B Molecular weight (Da) 28581.4 Length 254 Aromaticity 0.1 Instability index 44.04 Isoelectric point 8.11 Charge (pH=7) 2.57 2D Binding mode Binding energy (Kcal/mol) -7.25  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence WQLYIGDTRSRTAEYKAWLLGLLRQHGCQRVLDVACGTGVDSIMLVEEGFSVTSVDASDKMLKYALKERWNRRHEPAFDKWVIEEANWMTLDKDVPQSAEGGFDAVICLGNSFAHLPDCKGDQSEHRLALKNIASMVRAGGLLVIDHRNYDHILSTGCAPPGKNIYYKSDLTKDVTTSVLIVNNKAHMVTLDYTVQGLSKFRLSYYPHCLASFTELLQAAFGGKCQHSVLGDFKPYKPGQTYIPCYFIHVLKRT Hydrogen bonds contact Hydrophobic contact | ||||
| 95 | Glycolipid transfer protein | 3RZN | 5.32 | |
Target general information Gen name GLTP Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family GLTP family Biochemical class Lipid transport Function Glycolipid binding.Glycolipid transporter activity.Identical protein binding.Intermembrane lipid transfer activity.Lipid binding. Related diseases Brugada syndrome 7 (BRGDA7) [MIM:613120]: A tachyarrhythmia characterized by right bundle branch block and ST segment elevation on an electrocardiogram (ECG). It can cause the ventricles to beat so fast that the blood is prevented from circulating efficiently in the body. When this situation occurs, the individual will faint and may die in a few minutes if the heart is not reset. {ECO:0000269|PubMed:20031595}. The gene represented in this entry may be involved in disease pathogenesis.; DISEASE: Atrial fibrillation, familial, 16 (ATFB16) [MIM:613120]: A familial form of atrial fibrillation, a common sustained cardiac rhythm disturbance. Atrial fibrillation is characterized by disorganized atrial electrical activity and ineffective atrial contraction promoting blood stasis in the atria and reduces ventricular filling. It can result in palpitations, syncope, thromboembolic stroke, and congestive heart failure. {ECO:0000269|PubMed:20558140, ECO:0000269|PubMed:21051419}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03600; DB04465; DB03017; DB03203 Interacts with Q96DZ9; Q9NZD2 EC number NA Uniprot keywords 3D-structure; Acetylation; Cytoplasm; Lipid transport; Proteomics identification; Reference proteome; Repeat; Transport Protein physicochemical properties Chain ID A Molecular weight (Da) 23534.1 Length 206 Aromaticity 0.11 Instability index 36.45 Isoelectric point 7.08 Charge (pH=7) 0.1 2D Binding mode Binding energy (Kcal/mol) -7.26  Molscript Map  Pymol Map 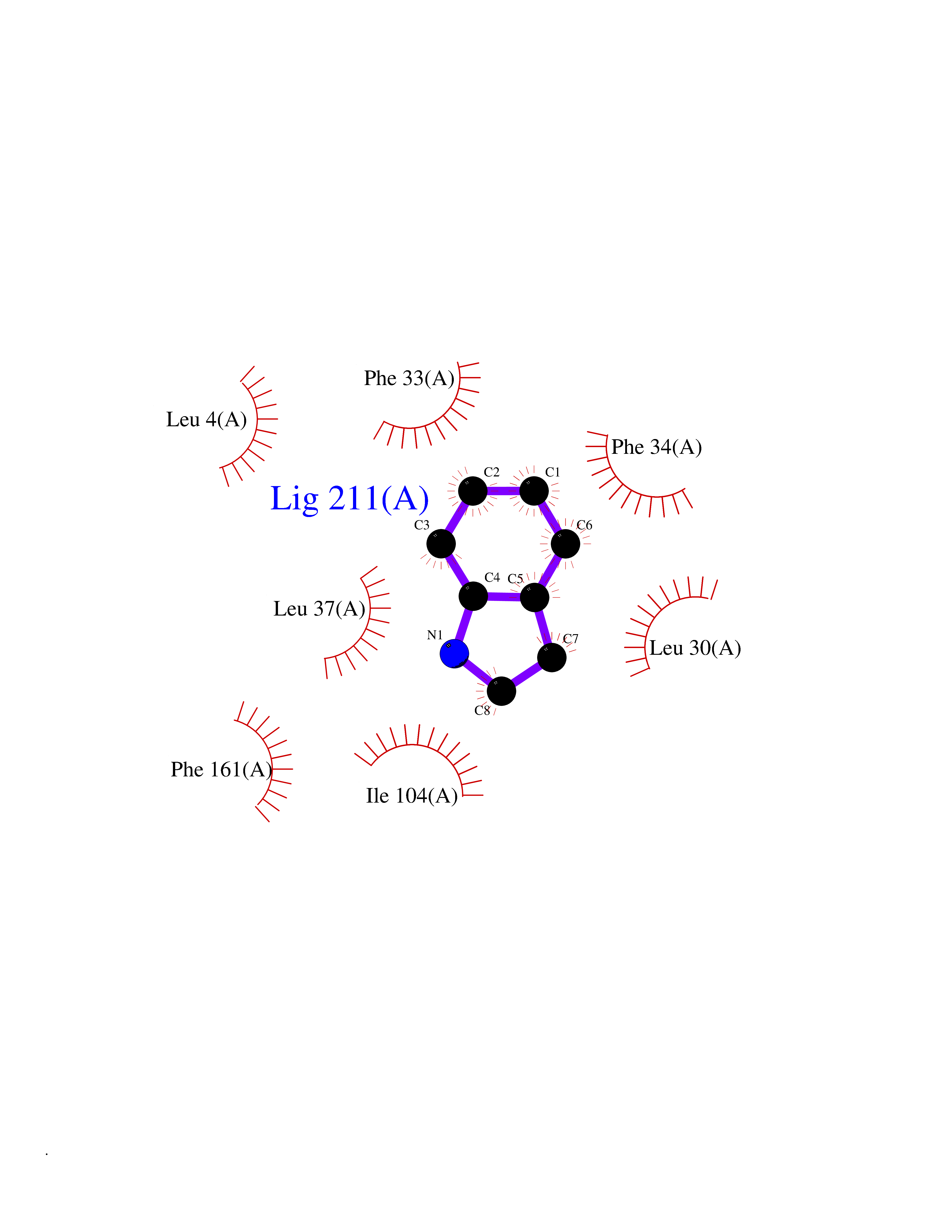 Ligplot Map 3D Binding mode Sequence LAEHLLKPLPADKQIETGPFLEAVSHLPPFFDCLGSPVFTPIKADISGNITKIKAVYDTNPAKFRTLQNILEVEKEMYGAEWPKVGATLALMWLKRGLRFIQVFLQSICDGERDENHPNLIRVNATKAYEMALKKYHGWIVQKIFQAALYAAPYKSDFLKALSKGQNVTEEECLEKIRLFLVNYTATIDVIYEMYTQMNAELNYKV Hydrogen bonds contact Hydrophobic contact | ||||
| 96 | Polybromo-1 (PBRM1) | 4Y03 | 5.32 | |
Target general information Gen name PBRM1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms hPB1; Protein polybromo-1; Polybromo-1D; PB1; BRG1-associated factor 180; BAF180 Protein family NA Biochemical class NA Function Required for the stability of the SWI/SNF chromatin remodeling complex SWI/SNF-B (PBAF). Acts as a negative regulator of cell proliferation. Involved in transcriptional activation and repression of select genes by chromatin remodeling (alteration of DNA-nucleosome topology). Related diseases Renal cell carcinoma (RCC) [MIM:144700]: Renal cell carcinoma is a heterogeneous group of sporadic or hereditary carcinoma derived from cells of the proximal renal tubular epithelium. It is subclassified into clear cell renal carcinoma (non-papillary carcinoma), papillary renal cell carcinoma, chromophobe renal cell carcinoma, collecting duct carcinoma with medullary carcinoma of the kidney, and unclassified renal cell carcinoma. Clear cell renal cell carcinoma is the most common subtype. {ECO:0000269|PubMed:21248752}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q68CP9; O95696-1; O95696-2; Q9P2D1; P51532; P04608 EC number NA Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Bromodomain; Chromatin regulator; DNA-binding; Isopeptide bond; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Transcription; Transcription regulation; Tumor suppressor; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 13709.7 Length 115 Aromaticity 0.1 Instability index 57.29 Isoelectric point 6.71 Charge (pH=7) -0.35 2D Binding mode Binding energy (Kcal/mol) -7.25  Molscript Map 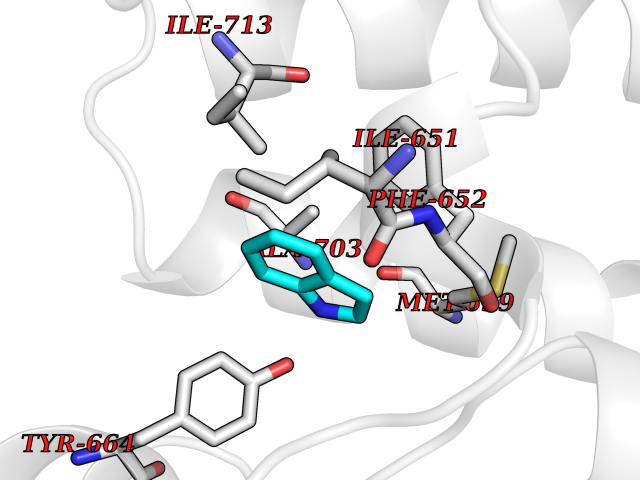 Pymol Map 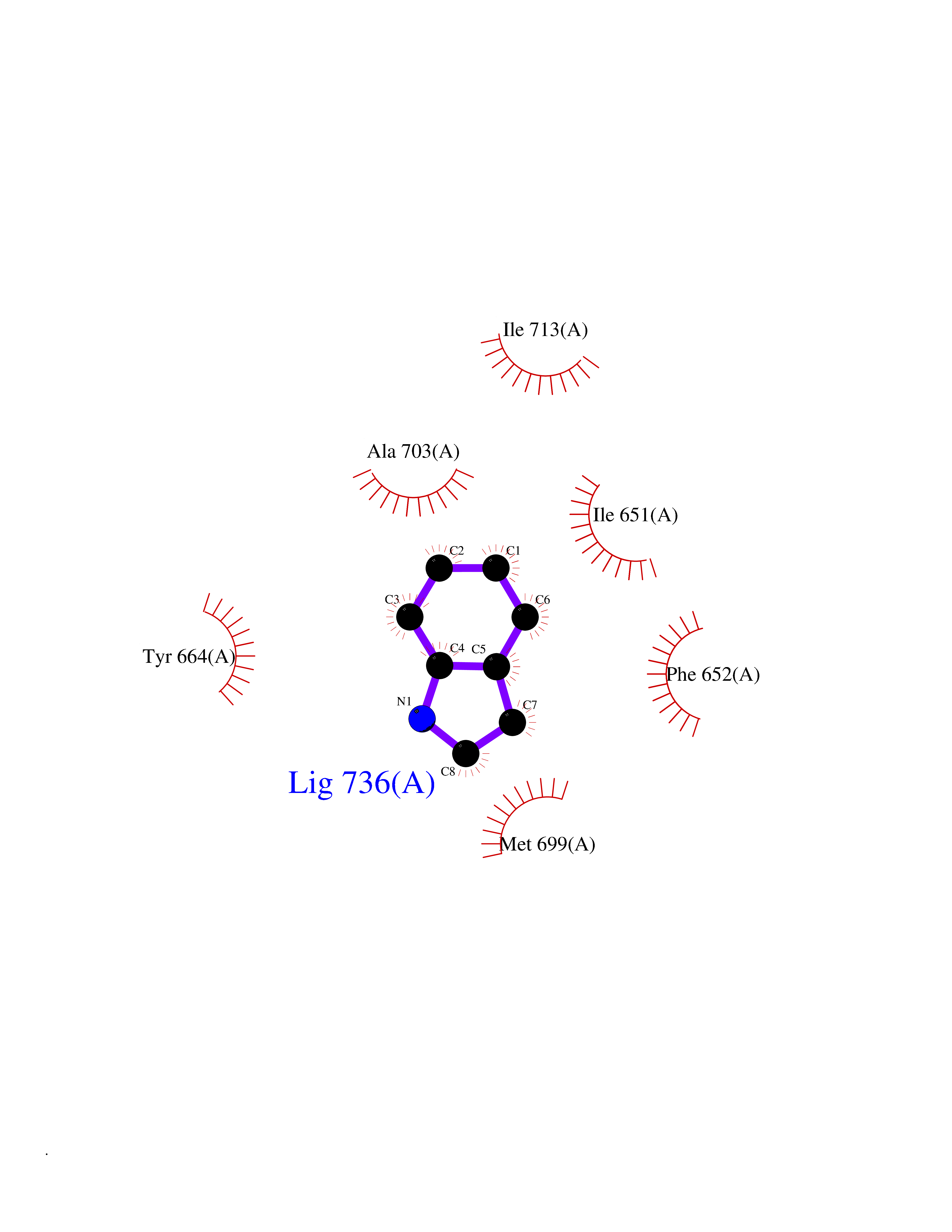 Ligplot Map 3D Binding mode Sequence SKYMTPMQQKLNEVYEAVKNYTDKRGRRLSAIFLRLPSRSELPDYYLTIKKPMDMEKIRSHMMANKYQDIDSMVEDFVMMFNNACTYNEPESLIYKDALVLHKVLLETRRDLEGD Hydrogen bonds contact Hydrophobic contact | ||||
| 97 | Bromodomain adjacent to zinc finger 2B (BAZ2B) | 4CUT | 5.32 | |
Target general information Gen name BAZ2B Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms hWALp4; KIAA1476; Bromodomain adjacent to zinc finger domain protein 2B Protein family WAL family Biochemical class NA Function May play a role in transcriptional regulation interacting with ISWI. Related diseases TP53 is found in increased amounts in a wide variety of transformed cells. TP53 is frequently mutated or inactivated in about 60% of cancers. TP53 defects are found in Barrett metaplasia a condition in which the normally stratified squamous epithelium of the lower esophagus is replaced by a metaplastic columnar epithelium. The condition develops as a complication in approximately 10% of patients with chronic gastroesophageal reflux disease and predisposes to the development of esophageal adenocarcinoma.; DISEASE: Esophageal cancer (ESCR) [MIM:133239]: A malignancy of the esophagus. The most common types are esophageal squamous cell carcinoma and adenocarcinoma. Cancer of the esophagus remains a devastating disease because it is usually not detected until it has progressed to an advanced incurable stage. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Li-Fraumeni syndrome (LFS) [MIM:151623]: An autosomal dominant familial cancer syndrome that in its classic form is defined by the existence of a proband affected by a sarcoma before 45 years with a first degree relative affected by any tumor before 45 years and another first degree relative with any tumor before 45 years or a sarcoma at any age. Other clinical definitions for LFS have been proposed and called Li-Fraumeni like syndrome (LFL). In these families affected relatives develop a diverse set of malignancies at unusually early ages. Four types of cancers account for 80% of tumors occurring in TP53 germline mutation carriers: breast cancers, soft tissue and bone sarcomas, brain tumors (astrocytomas) and adrenocortical carcinomas. Less frequent tumors include choroid plexus carcinoma or papilloma before the age of 15, rhabdomyosarcoma before the age of 5, leukemia, Wilms tumor, malignant phyllodes tumor, colorectal and gastric cancers. {ECO:0000269|PubMed:10484981, ECO:0000269|PubMed:1565144, ECO:0000269|PubMed:1737852, ECO:0000269|PubMed:1933902, ECO:0000269|PubMed:1978757, ECO:0000269|PubMed:2259385, ECO:0000269|PubMed:36108750, ECO:0000269|PubMed:7887414, ECO:0000269|PubMed:8825920, ECO:0000269|PubMed:9452042}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Squamous cell carcinoma of the head and neck (HNSCC) [MIM:275355]: A non-melanoma skin cancer affecting the head and neck. The hallmark of cutaneous SCC is malignant transformation of normal epidermal keratinocytes. The gene represented in this entry is involved in disease pathogenesis.; DISEASE: Lung cancer (LNCR) [MIM:211980]: A common malignancy affecting tissues of the lung. The most common form of lung cancer is non-small cell lung cancer (NSCLC) that can be divided into 3 major histologic subtypes: squamous cell carcinoma, adenocarcinoma, and large cell lung cancer. NSCLC is often diagnosed at an advanced stage and has a poor prognosis. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Papilloma of choroid plexus (CPP) [MIM:260500]: A benign tumor of neuroectodermal origin that generally occurs in childhood, but has also been reported in adults. Although generally found within the ventricular system, choroid plexus papillomas can arise ectopically in the brain parenchyma or disseminate throughout the neuraxis. Patients present with signs and symptoms of increased intracranial pressure including headache, hydrocephalus, papilledema, nausea, vomiting, cranial nerve deficits, gait impairment, and seizures. {ECO:0000269|PubMed:12085209}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Adrenocortical carcinoma (ADCC) [MIM:202300]: A malignant neoplasm of the adrenal cortex and a rare childhood tumor. It occurs with increased frequency in patients with Beckwith-Wiedemann syndrome and Li-Fraumeni syndrome. {ECO:0000269|PubMed:11481490}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Basal cell carcinoma 7 (BCC7) [MIM:614740]: A common malignant skin neoplasm that typically appears on hair-bearing skin, most commonly on sun-exposed areas. It is slow growing and rarely metastasizes, but has potentialities for local invasion and destruction. It usually develops as a flat, firm, pale area that is small, raised, pink or red, translucent, shiny, and waxy, and the area may bleed following minor injury. Tumor size can vary from a few millimeters to several centimeters in diameter. {ECO:0000269|PubMed:21946351}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Bone marrow failure syndrome 5 (BMFS5) [MIM:618165]: A form of bone marrow failure syndrome, a heterogeneous group of life-threatening disorders characterized by hematopoietic defects in association with a range of variable extra-hematopoietic manifestations. BMFS5 is an autosomal dominant form characterized by infantile onset of severe red cell anemia requiring transfusion. Additional features include hypogammaglobulinemia, poor growth with microcephaly, developmental delay, and seizures. {ECO:0000269|PubMed:30146126}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q96GX9; Q86UB2; Q8NHQ1; P56545-3; P51116; Q08379; Q6NT76; O75031; Q0VAM2-3; Q15019-3; Q9UBB9; Q12933; P56545-3; P51116; Q96MF2; O43829; Q96BR9 EC number NA Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Bromodomain; Coiled coil; DNA-binding; Isopeptide bond; Metal-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Transcription; Transcription regulation; Ubl conjugation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A Molecular weight (Da) 13411.3 Length 115 Aromaticity 0.1 Instability index 29.63 Isoelectric point 6.7 Charge (pH=7) -0.37 2D Binding mode Binding energy (Kcal/mol) -7.25 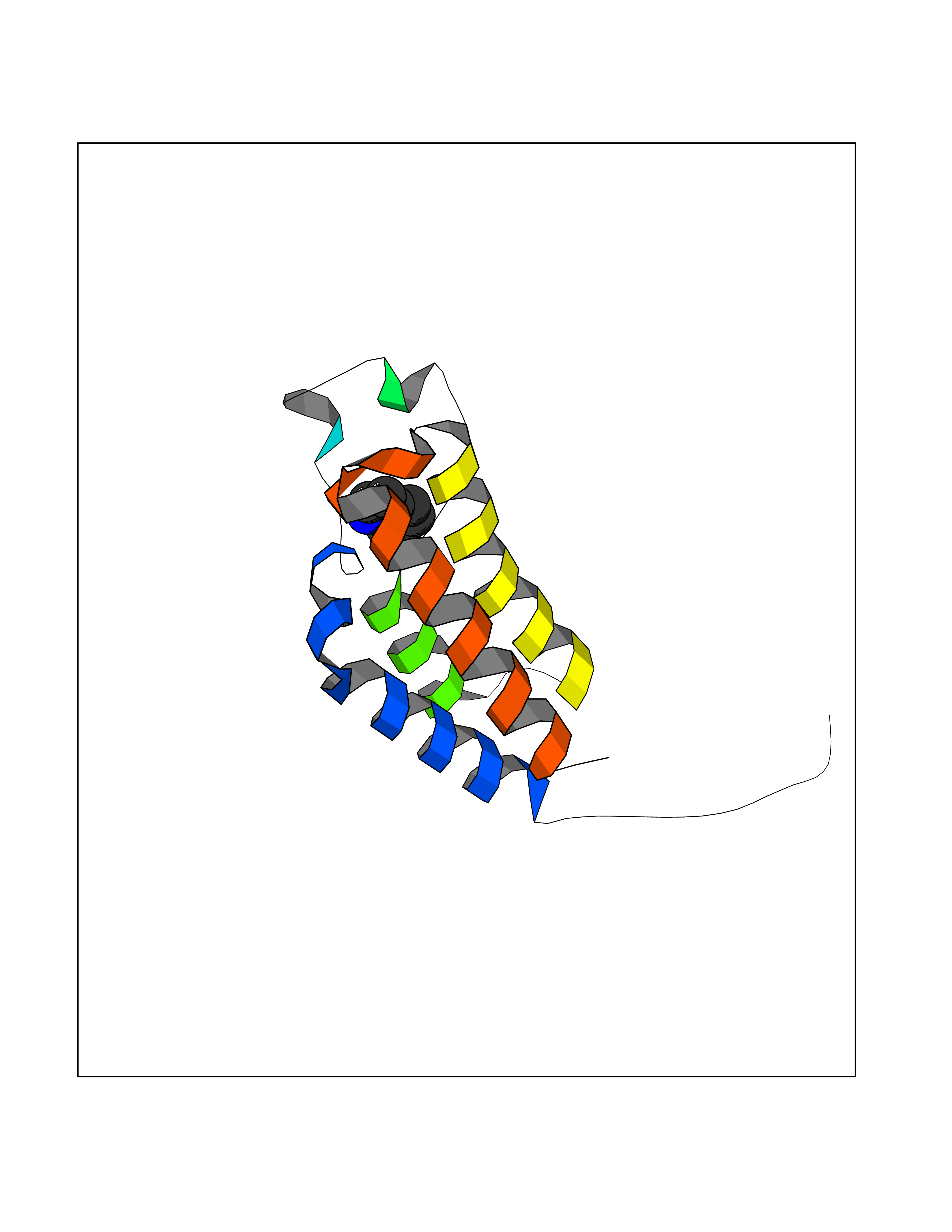 Molscript Map 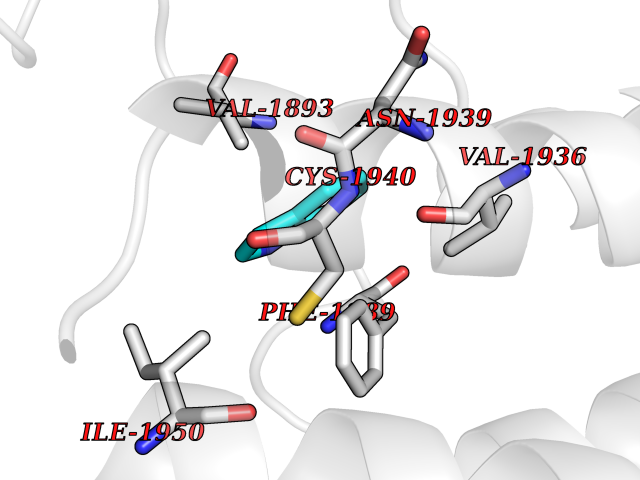 Pymol Map 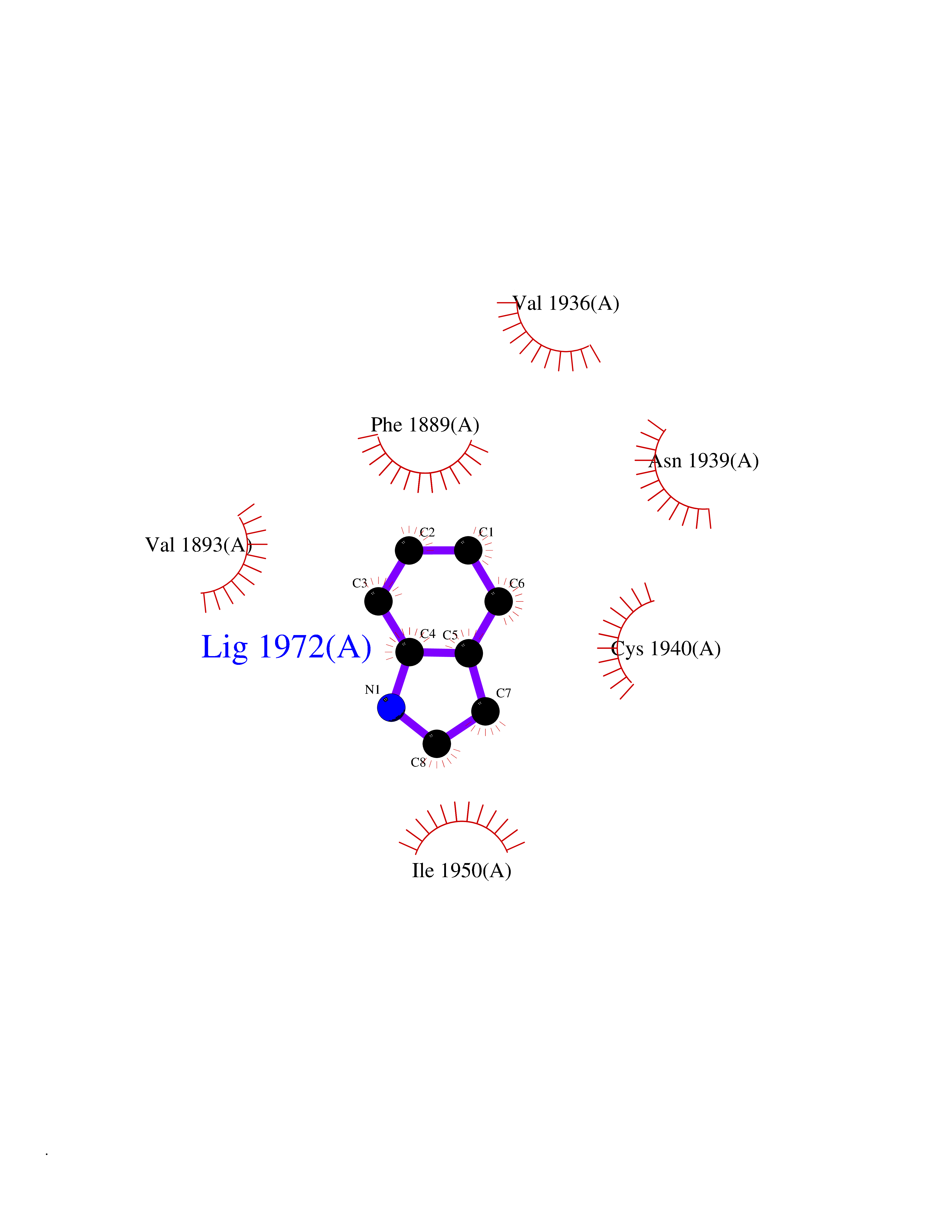 Ligplot Map 3D Binding mode Sequence SMSVKKPKRDDSKDLALCSMILTEMETHEDAWPFLLPVNLKLVPGYKKVIKKPMDFSTIREKLSSGQYPNLETFALDVRLVFDNCETFNEDDSDIGRAGHNMRKYFEKKWTDTFK Hydrogen bonds contact Hydrophobic contact | ||||
| 98 | Soluble epoxide hydrolase (EPHX2) | 1ZD3 | 5.32 | |
Target general information Gen name EPHX2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Bifunctional epoxide hydrolase 2 Protein family AB hydrolase superfamily, Epoxide hydrolase family Biochemical class Ether bond hydrolase Function Bifunctional enzyme. The C-terminal domain has epoxide hydrolase activity and acts on epoxides (alkene oxides, oxiranes) and arene oxides. Plays a role in xenobiotic metabolism by degrading potentially toxic epoxides (By similarity). Also determines steady-state levels of physiological mediators. The N-terminal domain has lipid phosphatase activity, with the highest activity towards threo-9,10-phosphonooxy-hydroxy-octadecanoic acid, followed by erythro-9,10-phosphonooxy-hydroxy-octadecanoic acid, 12-phosphonooxy-octadec-9Z-enoic acid and 12-phosphonooxy-octadec-9E-enoic acid. Related diseases Leukemia, juvenile myelomonocytic (JMML) [MIM:607785]: An aggressive pediatric myelodysplastic syndrome/myeloproliferative disorder characterized by malignant transformation in the hematopoietic stem cell compartment with proliferation of differentiated progeny. Patients have splenomegaly, enlarged lymph nodes, rashes, and hemorrhages. {ECO:0000269|PubMed:17332249}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Noonan syndrome 6 (NS6) [MIM:613224]: A form of Noonan syndrome, a disease characterized by short stature, facial dysmorphic features such as hypertelorism, a downward eyeslant and low-set posteriorly rotated ears, and a high incidence of congenital heart defects and hypertrophic cardiomyopathy. Other features can include a short neck with webbing or redundancy of skin, deafness, motor delay, variable intellectual deficits, multiple skeletal defects, cryptorchidism, and bleeding diathesis. Individuals with Noonan syndrome are at risk of juvenile myelomonocytic leukemia, a myeloproliferative disorder characterized by excessive production of myelomonocytic cells. {ECO:0000269|PubMed:19966803}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: RAS-associated autoimmune leukoproliferative disorder (RALD) [MIM:614470]: A disorder of apoptosis, characterized by chronic accumulation of non-malignant lymphocytes, defective lymphocyte apoptosis, and an increased risk for the development of hematologic malignancies. {ECO:0000269|PubMed:17517660}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Melanocytic nevus syndrome, congenital (CMNS) [MIM:137550]: A syndrome characterized by congenital pigmentary skin lesions which can occur at any site and can cover most of the body surface. These lesions may or may not be hairy. Congenital melanocytic nevi are associated with neuromelanosis (the presence of melanin-producing cells within the brain parenchyma or leptomeninges). Less commonly they are associated with malignant melanoma in childhood, both in the skin and the central nervous system. CMNS patients also tend to have a characteristic facial appearance, including wide or prominent forehead, periorbital fullness, small short nose with narrow nasal bridge, round face, full cheeks, prominent premaxilla, and everted lower lip. {ECO:0000269|PubMed:18633438, ECO:0000269|PubMed:23392294}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Melanosis, neurocutaneous (NCMS) [MIM:249400]: A rare congenital disease characterized by the presence of giant or multiple melanocytic nevi on the skin, foci of melanin-producing cells within the brain parenchyma, and infiltration of leptomeninges by abnormal melanin deposits. Neurologic abnormalities include seizures, hydrocephalus, arachnoid cysts, tumors, and syringomyelia. Some patients may develop malignant melanoma. {ECO:0000269|PubMed:23392294}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Keratinocytic non-epidermolytic nevus (KNEN) [MIM:162900]: Epidermal nevi of the common, non-organoid and non-epidermolytic type are benign skin lesions and may vary in their extent from a single (usually linear) lesion to widespread and systematized involvement. They may be present at birth or develop early during childhood. {ECO:0000269|PubMed:22499344}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Thyroid cancer, non-medullary, 2 (NMTC2) [MIM:188470]: A form of non-medullary thyroid cancer (NMTC), a cancer characterized by tumors originating from the thyroid follicular cells. NMTCs represent approximately 95% of all cases of thyroid cancer and are classified into papillary, follicular, Hurthle cell, and anaplastic neoplasms. {ECO:0000269|PubMed:12727991}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08257; DB08258; DB08259; DB06345; DB12610; DB08256; DB02029; DB04213; DB03677 Interacts with NA EC number NA Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Aromatic hydrocarbons catabolism; Cytoplasm; Detoxification; Direct protein sequencing; Hydrolase; Lipid metabolism; Lipoprotein; Magnesium; Metal-binding; Multifunctional enzyme; Peroxisome; Phosphoprotein; Proteomics identification; Reference proteome Protein physicochemical properties Chain ID A Molecular weight (Da) 61744.9 Length 547 Aromaticity 0.09 Instability index 43.97 Isoelectric point 5.81 Charge (pH=7) -7.76 2D Binding mode Binding energy (Kcal/mol) -7.26  Molscript Map 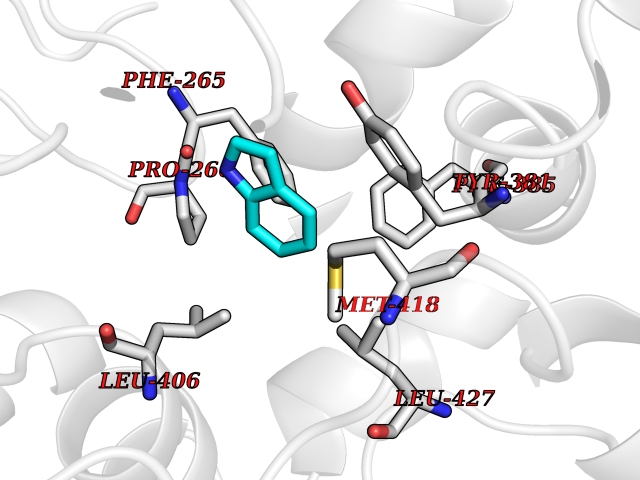 Pymol Map 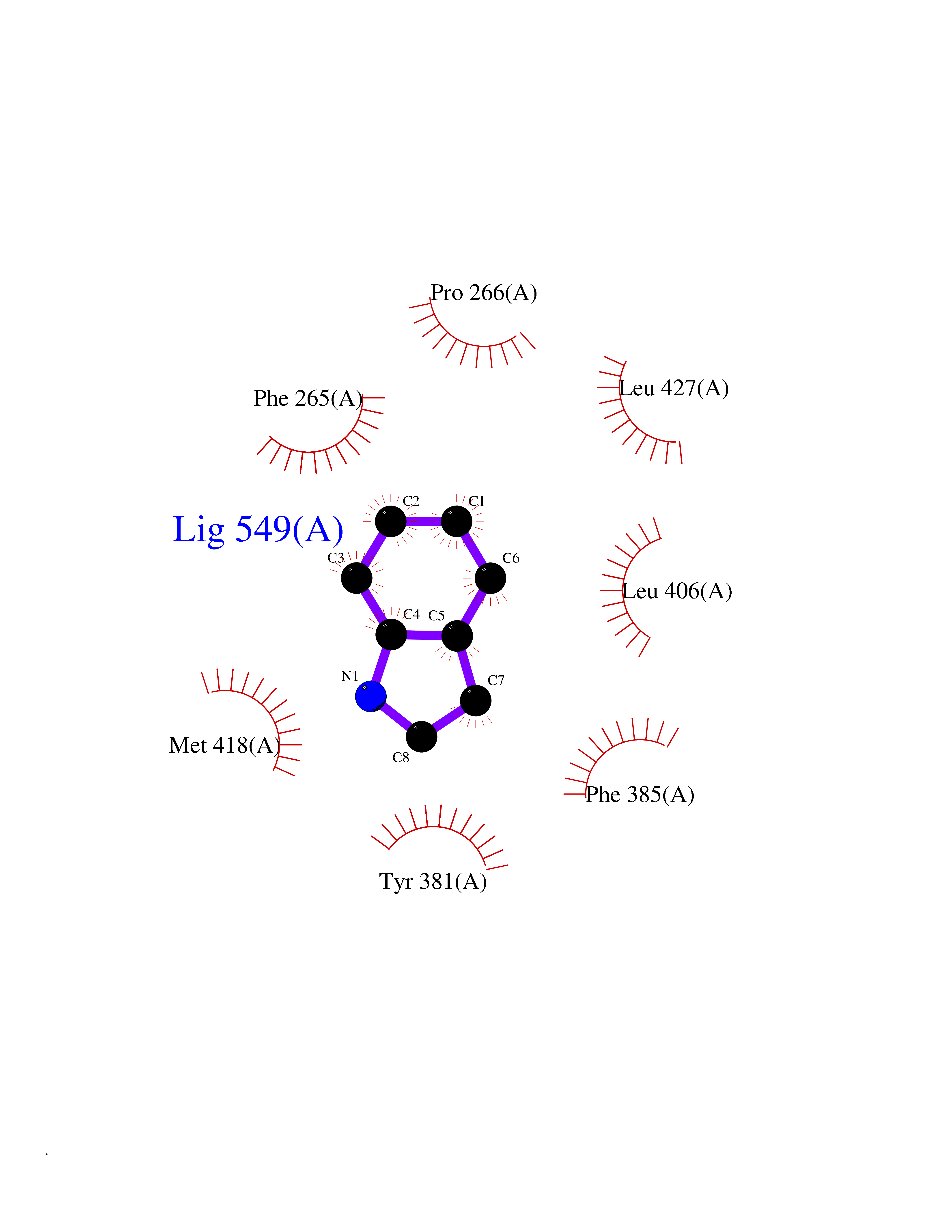 Ligplot Map 3D Binding mode Sequence TLRAAVFDLDGVLALPAVFGVLGRTEEALALPRGLLNDAFQKGGPEGATTRLMKGEITLSQWIPLMEENCRKCSETAKVCLPKNFSIKEIFDKAISARKINRPMLQAALMLRKKGFTTAILTNTWLDDRAERDGLAQLMCELKMHFDFLIESCQVGMVKPEPQIYKFLLDTLKASPSEVVFLDDIGANLKPARDLGMVTILVQDTDTALKELEKVTGIQLLNTPAPLPTSCNPSDMSHGYVTVKPRVRLHFVELGSGPAVCLCHGFPESWYSWRYQIPALAQAGYRVLAMDMKGYGESSAPPEIEEYCMEVLCKEMVTFLDKLGLSQAVFIGHDWGGMLVWYMALFYPERVRAVASLNTPFIPANPNMSPLESIKANPVFDYQLYFQEPGVAEAELEQNLSRTFKSLFRASDESVLSMHKVCEAGGLFVNSPEEPSLSRMVTEEEIQFYVQQFKKSGFRGPLNWYRNMERNWKWACKSLGRKILIPALMVTAEKDFVLVPQMSQHMEDWIPHLKRGHIEDCGHWTQMDKPTEVNQILIKWLDSDARN Hydrogen bonds contact Hydrophobic contact | ||||
| 99 | PAK-4 protein kinase (PAK4) | 2X4Z | 5.32 | |
Target general information Gen name PAK4 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms p21-activated kinase 4; Serine/threonine-protein kinase PAK 4; PAK-4; KIAA1142 Protein family Protein kinase superfamily, STE Ser/Thr protein kinase family, STE20 subfamily Biochemical class Kinase Function Activation by various effectors including growth factor receptors or active CDC42 and RAC1 results in a conformational change and a subsequent autophosphorylation on several serine and/or threonine residues. Phosphorylates and inactivates the protein phosphatase SSH1, leading to increased inhibitory phosphorylation of the actin binding/depolymerizing factor cofilin. Decreased cofilin activity may lead to stabilization of actin filaments. Phosphorylates LIMK1, a kinase that also inhibits the activity of cofilin. Phosphorylates integrin beta5/ITGB5 and thus regulates cell motility. Phosphorylates ARHGEF2 and activates the downstream target RHOA that plays a role in the regulation of assembly of focal adhesions and actin stress fibers. Stimulates cell survival by phosphorylating the BCL2 antagonist of cell death BAD. Alternatively, inhibits apoptosis by preventing caspase-8 binding to death domain receptors in a kinase independent manner. Plays a role in cell-cycle progression by controlling levels of the cell-cycle regulatory protein CDKN1A and by phosphorylating RAN. Serine/threonine protein kinase that plays a role in a variety of different signaling pathways including cytoskeleton regulation, cell migration, growth, proliferation or cell survival. Related diseases Metachromatic leukodystrophy (MLD) [MIM:250100]: An autosomal recessive disease caused by abnormal intralysosomal accumulation of cerebroside-3-sulfate in central and peripheral nervous systems, as well as other organs. MLD is clinically characterized by leukodystrophy, progressive demyelination and a variety of neurological symptoms, including gait disturbances, ataxias, optical atrophy, dementia, seizures, and spastic tetraparesis. Decreased arylsulfatase A activity is detected in urine, leukocytes, and fibroblasts of affected individuals. Several forms of the disease can be distinguished according to the age at onset and disease severity: late infantile, juvenile and adult forms, partial cerebroside sulfate deficiency, and pseudoarylsulfatase A deficiency. Individuals with pseudoarylsulfatase A deficiency have low arylsulfatase A activity but lack neurological manifestations and are apparently healthy. {ECO:0000269|PubMed:10220151, ECO:0000269|PubMed:10381328, ECO:0000269|PubMed:10477432, ECO:0000269|PubMed:10533072, ECO:0000269|PubMed:10751093, ECO:0000269|PubMed:11020646, ECO:0000269|PubMed:11061266, ECO:0000269|PubMed:11456299, ECO:0000269|PubMed:11941485, ECO:0000269|PubMed:12503099, ECO:0000269|PubMed:12788103, ECO:0000269|PubMed:1353340, ECO:0000269|PubMed:14517960, ECO:0000269|PubMed:14680985, ECO:0000269|PubMed:15026521, ECO:0000269|PubMed:15326627, ECO:0000269|PubMed:15710861, ECO:0000269|PubMed:1670590, ECO:0000269|PubMed:1673291, ECO:0000269|PubMed:1678251, ECO:0000269|PubMed:18693274, ECO:0000269|PubMed:19606494, ECO:0000269|PubMed:20339381, ECO:0000269|PubMed:21265945, ECO:0000269|PubMed:2574462, ECO:0000269|PubMed:7581401, ECO:0000269|PubMed:7825603, ECO:0000269|PubMed:7860068, ECO:0000269|PubMed:7902317, ECO:0000269|PubMed:7906588, ECO:0000269|PubMed:7909527, ECO:0000269|PubMed:8095918, ECO:0000269|PubMed:8101038, ECO:0000269|PubMed:8101083, ECO:0000269|PubMed:8104633, ECO:0000269|PubMed:8891236, ECO:0000269|PubMed:9090526, ECO:0000269|PubMed:9272717, ECO:0000269|PubMed:9452102, ECO:0000269|PubMed:9490297, ECO:0000269|PubMed:9600244, ECO:0000269|PubMed:9819708}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Multiple sulfatase deficiency (MSD) [MIM:272200]: A clinically and biochemically heterogeneous disorder caused by the simultaneous impairment of all sulfatases, due to defective post-translational modification and activation. It combines features of individual sulfatase deficiencies such as metachromatic leukodystrophy, mucopolysaccharidosis, chondrodysplasia punctata, hydrocephalus, ichthyosis, neurologic deterioration and developmental delay. {ECO:0000269|PubMed:15146462}. The protein represented in this entry is involved in disease pathogenesis. Arylsulfatase A activity is impaired in multiple sulfatase deficiency due to mutations in SUMF1 (PubMed:15146462). SUMF1 mutations result in defective post-translational modification of ARSA at residue Cys-69 that is not converted to 3-oxoalanine (PubMed:7628016). {ECO:0000269|PubMed:15146462, ECO:0000269|PubMed:7628016}. Drugs (DrugBank ID) DB12010 Interacts with P60953; Q96EL1; P31947; P54274; O00401; P62258; P63104; P05067; P37840 EC number EC 2.7.11.1 Uniprot keywords 3D-structure; Alternative splicing; Apoptosis; ATP-binding; Cell cycle; Cytoplasm; Kinase; Methylation; Nucleotide-binding; Phosphoprotein; Proteomics identification; Reference proteome; Serine/threonine-protein kinase; Transferase; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 32672.8 Length 291 Aromaticity 0.07 Instability index 36.81 Isoelectric point 8.36 Charge (pH=7) 2.21 2D Binding mode Binding energy (Kcal/mol) -7.26 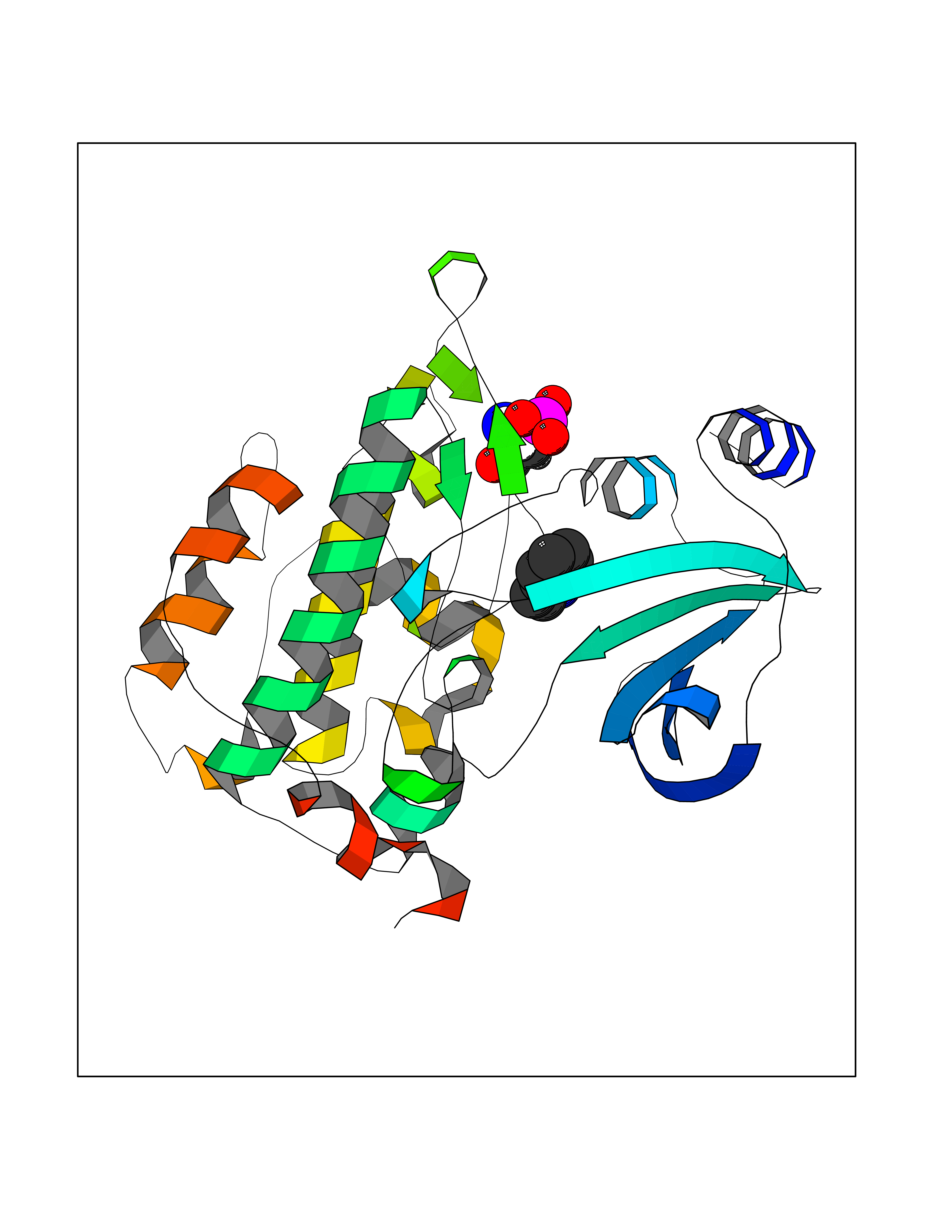 Molscript Map 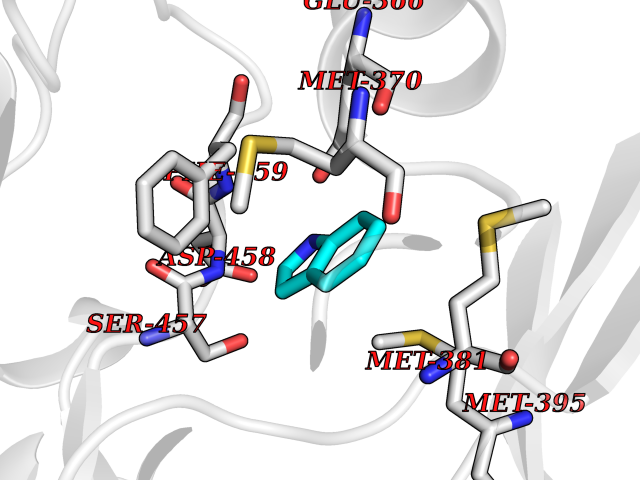 Pymol Map 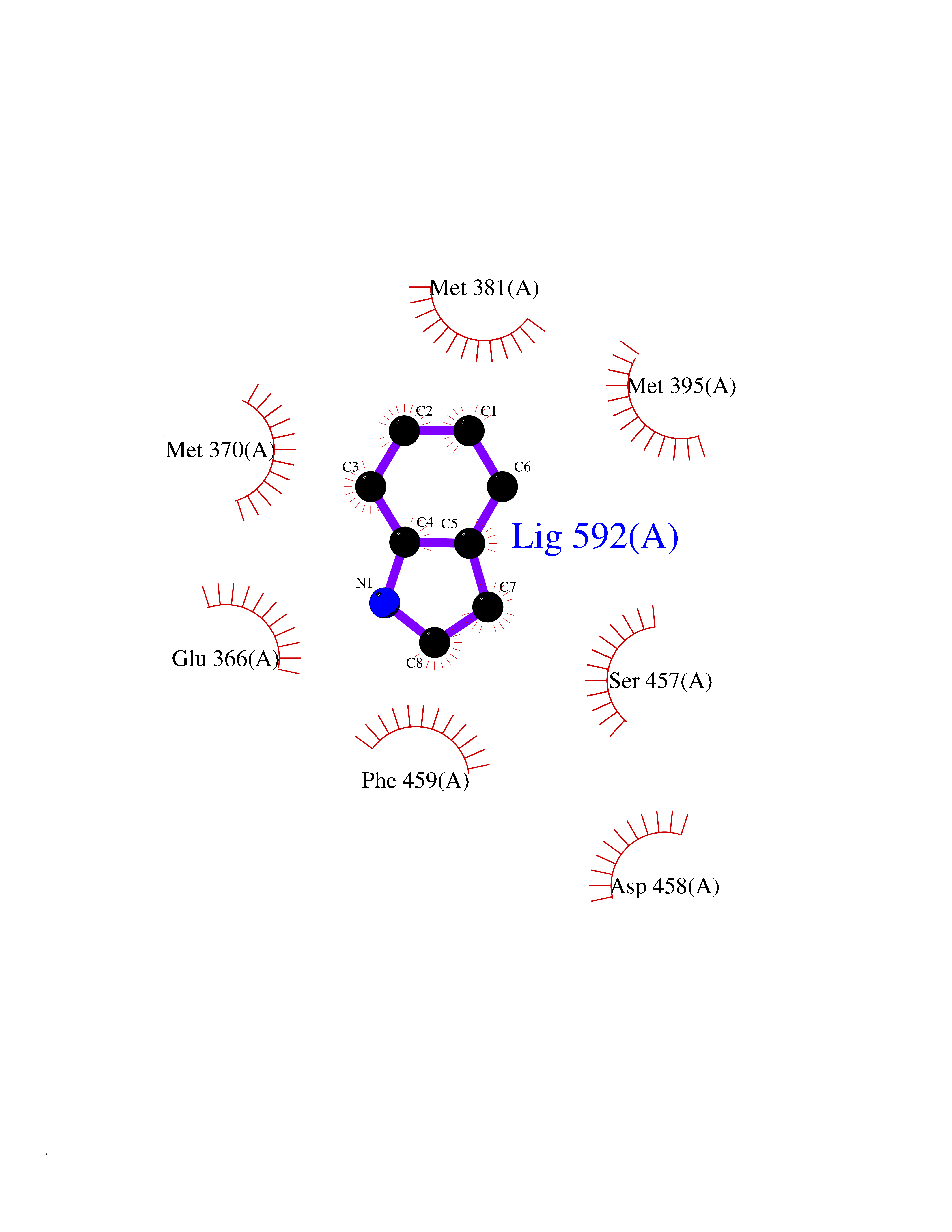 Ligplot Map 3D Binding mode Sequence MSHEQFRAALQLVVDPGDPRSYLDNFIKIGEGSTGIVCIATVRSSGKLVAVKKMDLRKQQRRELLFNEVVIMRDYQHENVVEMYNSYLVGDELWVVMEFLEGGALTDIVTHTRMNEEQIAAVCLAVLQALSVLHAQGVIHRDIKSDSILLTHDGRVKLSDFGFCAQVSKEVPRRKXLVGTPYWMAPELISRLPYGPEVDIWSLGIMVIEMVDGEPPYFNEPPLKAMKMIRDNLPPRLKNLHKVSPSLKGFLDRLLVRDPAQRATAAELLKHPFLAKAGPPASIVPLMRQNR Hydrogen bonds contact Hydrophobic contact | ||||
| 100 | PI3-kinase delta (PIK3CD) | 5DXU | 5.31 | |
Target general information Gen name PIK3CD Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms PtdIns-3-kinase subunit p110-delta; PtdIns-3-kinase subunit delta; Phosphoinositide 3-kinase delta; Phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit, delta isoform; Phosphatidylinosito Protein family PI3/PI4-kinase family Biochemical class Kinase Function Phosphoinositide-3-kinase (PI3K) that phosphorylates PtdIns(4,5)P2 (Phosphatidylinositol 4,5-bisphosphate) to generate phosphatidylinositol 3,4,5-trisphosphate (PIP3). PIP3 plays a key role by recruiting PH domain-containing proteins to the membrane, including AKT1 and PDPK1, activating signaling cascades involved in cell growth, survival, proliferation, motility and morphology. Mediates immune responses. Plays a role in B-cell development, proliferation, migration, and function. Required for B-cell receptor (BCR) signaling. Mediates B-cell proliferation response to anti-IgM, anti-CD40 and IL4 stimulation. Promotes cytokine production in response to TLR4 and TLR9. Required for antibody class switch mediated by TLR9. Involved in the antigen presentation function of B-cells. Involved in B-cell chemotaxis in response to CXCL13 and sphingosine 1-phosphate (S1P). Required for proliferation, signaling and cytokine production of naive, effector and memory T-cells. Required for T-cell receptor (TCR) signaling. Mediates TCR signaling events at the immune synapse. Activation by TCR leads to antigen-dependent memory T-cell migration and retention to antigenic tissues. Together with PIK3CG participates in T-cell development. Contributes to T-helper cell expansion and differentiation. Required for T-cell migration mediated by homing receptors SELL/CD62L, CCR7 and S1PR1 and antigen dependent recruitment of T-cells. Together with PIK3CG is involved in natural killer (NK) cell development and migration towards the sites of inflammation. Participates in NK cell receptor activation. Have a role in NK cell maturation and cytokine production. Together with PIK3CG is involved in neutrophil chemotaxis and extravasation. Together with PIK3CG participates in neutrophil respiratory burst. Have important roles in mast-cell development and mast cell mediated allergic response. Involved in stem cell factor (SCF)-mediated proliferation, adhesion and migration. Required for allergen-IgE-induced degranulation and cytokine release. The lipid kinase activity is required for its biological function. Isoform 2 may be involved in stabilizing total RAS levels, resulting in increased ERK phosphorylation and increased PI3K activity. Related diseases Immunodeficiency 14A with lymphoproliferation, autosomal dominant (IMD14A) [MIM:615513]: A disorder characterized by recurrent respiratory infections, progressive airway damage, lymphopenia, increased circulating transitional B cells, increased immunoglobulin M, reduced immunoglobulin G2 levels in serum, and impaired vaccine responses. {ECO:0000269|PubMed:24136356}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Immunodeficiency 14B, autosomal recessive (IMD14B) [MIM:619281]: An autosomal recessive, primary immunodeficiency characterized by recurrent sinopulmonary infections apparent in early childhood. Some patients may develop inflammatory bowel disease or osteomyelitis. Immunological features include hypogammaglobulinemia, decreased levels of B cells, and evidence of impaired immune-mediated cytotoxicity and defective T-cell function. {ECO:0000269|PubMed:30040974, ECO:0000269|PubMed:30336224}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Roifman-Chitayat syndrome (ROCHIS) [MIM:613328]: An autosomal recessive digenic disorder characterized by global developmental delay, variable neurologic features such as seizures and ataxia, optic atrophy, dysmorphic facial features, distal skeletal anomalies, and recurrent invasive infections due to combined immunodeficiency. {ECO:0000269|PubMed:29180244}. The disease is caused by variants affecting distinct genetic loci, including the gene represented in this entry. Caused by the simultaneous occurrence of homozygous mutations in PIK3CD and KNSTRN. {ECO:0000269|PubMed:29180244}. Drugs (DrugBank ID) DB06831; DB00201; DB12483; DB11952; DB12010; DB09054; DB16217; DB05552; DB14989; DB05241 Interacts with P01112; P27986; P27986-2; Q92569; P01112 EC number EC 2.7.1.153 Uniprot keywords 3D-structure; Adaptive immunity; Alternative splicing; ATP-binding; Chemotaxis; Cytoplasm; Differentiation; Direct protein sequencing; Disease variant; Immunity; Inflammatory response; Innate immunity; Kinase; Lipid metabolism; Nucleotide-binding; Phosphoprotein; Proteomics identification; Reference proteome; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 47169.3 Length 409 Aromaticity 0.1 Instability index 44.59 Isoelectric point 6.99 Charge (pH=7) -0.05 2D Binding mode Binding energy (Kcal/mol) -7.24  Molscript Map 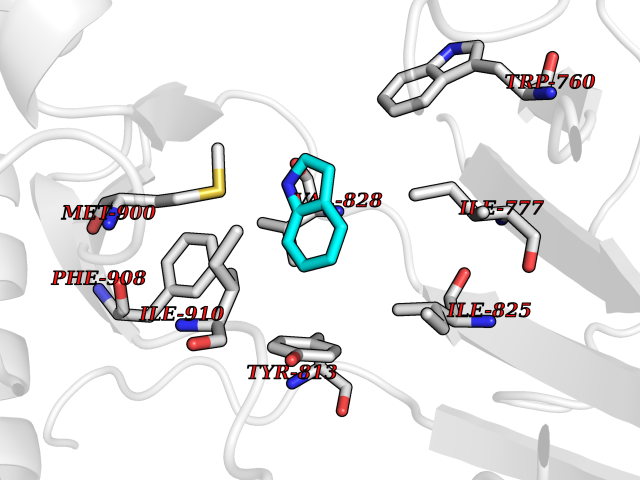 Pymol Map 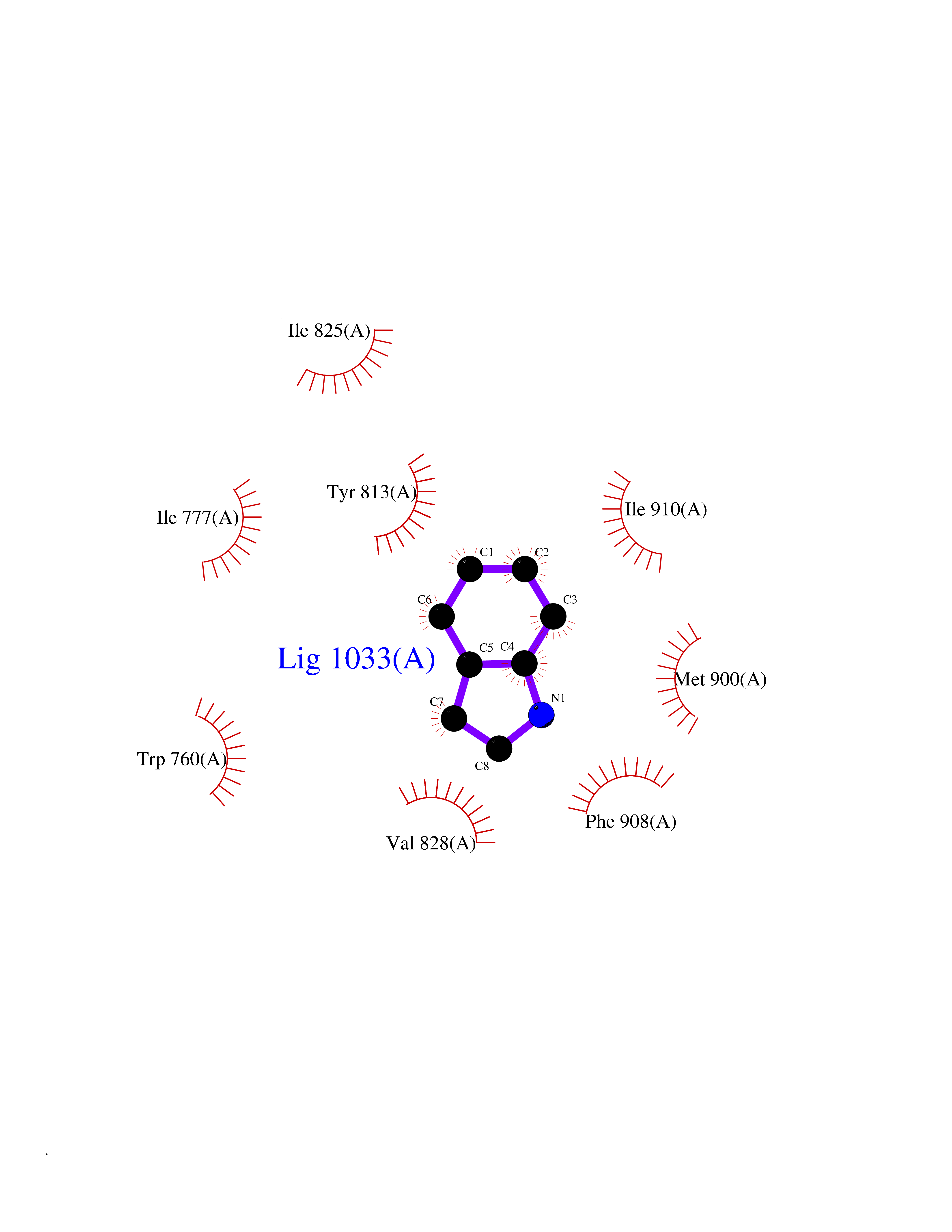 Ligplot Map 3D Binding mode Sequence LRKLTDDELFQYLLQLVQVLKYESYLDCELTKFLLDRALANRKIGHFLFWHLRSEMHVPSVALRFGLILEAYCRGSTHHMKVLMKQGEALSKLKALNDFVKLSSQKTPKPQTKELMHLCMRQEAYLEALSHLQSPLDPSTLLAEVCVEQCTFMDSKMKPLWIMYSNEEAGSVGIIFKNGDDLRQDMLTLQMIQLMDVLWKQEGLDLRMTPYGCLPTGDRTGLIEVVLRSDTIANIQLNDALLNWLKSKNPGEALDRAIEEFTLSCAGYCVATYVLGIGDRHSDNIMIRESGQLFHIDFGHFLGNFERVPFILTYDFVHVIQQGKTNNSEKFERFRGYCERAYTILRRHGLLFLHLFALMRAAGLPELSCSKDIQYLKDSLALGKTEEEALKHFRVKFNEALRESWKTKV Hydrogen bonds contact Hydrophobic contact | ||||