Job Results:
Ligand
Structure
Job ID
6365174e5cc200e6a930804dda23a0ba
Job name
NA
Time
2026-02-27 16:33:59
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 81 | Tyrosine-protein kinase BRK (PTK6) | 5DA3 | 4.37 | |
Target general information Gen name PTK6 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Protein-tyrosine kinase 6; Breast tumor kinase; BRK Protein family Protein kinase superfamily, Tyr protein kinase family, BRK/PTK6/SIK subfamily Biochemical class Kinase Function Non-receptor tyrosine-protein kinase implicated in the regulation of a variety of signaling pathways that control the differentiation and maintenance of normal epithelia, as well as tumor growth. Function seems to be context dependent and differ depending on cell type, as well as its intracellular localization. A number of potential nuclear and cytoplasmic substrates have been identified. These include the RNA-binding proteins: KHDRBS1/SAM68, KHDRBS2/SLM1, KHDRBS3/SLM2 and SFPQ/PSF; transcription factors: STAT3 and STAT5A/B and a variety of signaling molecules: ARHGAP35/p190RhoGAP, PXN/paxillin, BTK/ATK, STAP2/BKS. Associates also with a variety of proteins that are likely upstream of PTK6 in various signaling pathways, or for which PTK6 may play an adapter-like role. These proteins include ADAM15, EGFR, ERBB2, ERBB3 and IRS4. In normal or non-tumorigenic tissues, PTK6 promotes cellular differentiation and apoptosis. In tumors PTK6 contributes to cancer progression by sensitizing cells to mitogenic signals and enhancing proliferation, anchorage-independent survival and migration/invasion. Association with EGFR, ERBB2, ERBB3 may contribute to mammary tumor development and growth through enhancement of EGF-induced signaling via BTK/AKT and PI3 kinase. Contributes to migration and proliferation by contributing to EGF-mediated phosphorylation of ARHGAP35/p190RhoGAP, which promotes association with RASA1/p120RasGAP, inactivating RhoA while activating RAS. EGF stimulation resulted in phosphorylation of PNX/Paxillin by PTK6 and activation of RAC1 via CRK/CrKII, thereby promoting migration and invasion. PTK6 activates STAT3 and STAT5B to promote proliferation. Nuclear PTK6 may be important for regulating growth in normal epithelia, while cytoplasmic PTK6 might activate oncogenic signaling pathways. Related diseases Periodic paralysis hypokalemic 1 (HOKPP1) [MIM:170400]: An autosomal dominant disorder manifested by episodic flaccid generalized muscle weakness associated with falls of serum potassium levels. {ECO:0000269|PubMed:17418573, ECO:0000269|PubMed:18162704, ECO:0000269|PubMed:19118277, ECO:0000269|PubMed:7987325, ECO:0000269|PubMed:8004673}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Malignant hyperthermia 5 (MHS5) [MIM:601887]: Autosomal dominant disorder that is potentially lethal in susceptible individuals on exposure to commonly used inhalational anesthetics and depolarizing muscle relaxants. {ECO:0000269|PubMed:9199552}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Thyrotoxic periodic paralysis 1 (TTPP1) [MIM:188580]: A sporadic muscular disorder characterized by episodic weakness and hypokalemia during a thyrotoxic state. It is clinically similar to hereditary hypokalemic periodic paralysis, except for the fact that hyperthyroidism is an absolute requirement for disease manifestation. The disease presents with recurrent episodes of acute muscular weakness of the four extremities that vary in severity from paresis to complete paralysis. Attacks are triggered by ingestion of a high carbohydrate load or strenuous physical activity followed by a period of rest. Thyrotoxic periodic paralysis can occur in association with any cause of hyperthyroidism, but is most commonly associated with Graves disease. {ECO:0000269|PubMed:15001631}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Congenital myopathy 18 (CMYO18) [MIM:620246]: A congenital myopathy of variable severity, ranging from severe fetal akinesia to milder forms of muscle weakness. Most affected individuals show delayed motor development with generalized hypotonia and progressive axial and limb muscle weakness beginning soon after birth or in infancy. Additional features may include swallowing difficulties, external ophthalmoplegia, ptosis, high-arched palate, and respiratory insufficiency. Muscle biopsy shows variable morphologic abnormalities, including alveolar changes in the intermyofibrillar network, fiber size variability, focal disorganization, internal nuclei, and dilated sarcoplasmic reticulum and T-tubules. CMYO18 inheritance is autosomal dominant or recessive. {ECO:0000269|PubMed:28012042, ECO:0000269|PubMed:31227654, ECO:0000269|PubMed:33060286}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB12010; DB11800; DB05294; DB15035 Interacts with Q08043; Q3KP44; Q13191; Q16543; Q92841; Q8N9I9; Q5JST6; P04626; O00471; O14526; Q13480; P08238; P42858; Q9UKT9; Q5VWX1; Q5T5P2-6; P10721; O14770-4; Q13064; Q8TDC0; P78337; Q9NQX0; Q13882; Q04864; P23246; Q13239-3; O00401; Q9BYN7 EC number EC 2.7.10.2 Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Cell projection; Cytoplasm; Kinase; Membrane; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; SH2 domain; SH3 domain; Transferase; Tyrosine-protein kinase Protein physicochemical properties Chain ID A Molecular weight (Da) 30240.6 Length 264 Aromaticity 0.09 Instability index 46.88 Isoelectric point 6.95 Charge (pH=7) -0.14 2D Binding mode Binding energy (Kcal/mol) -5.96 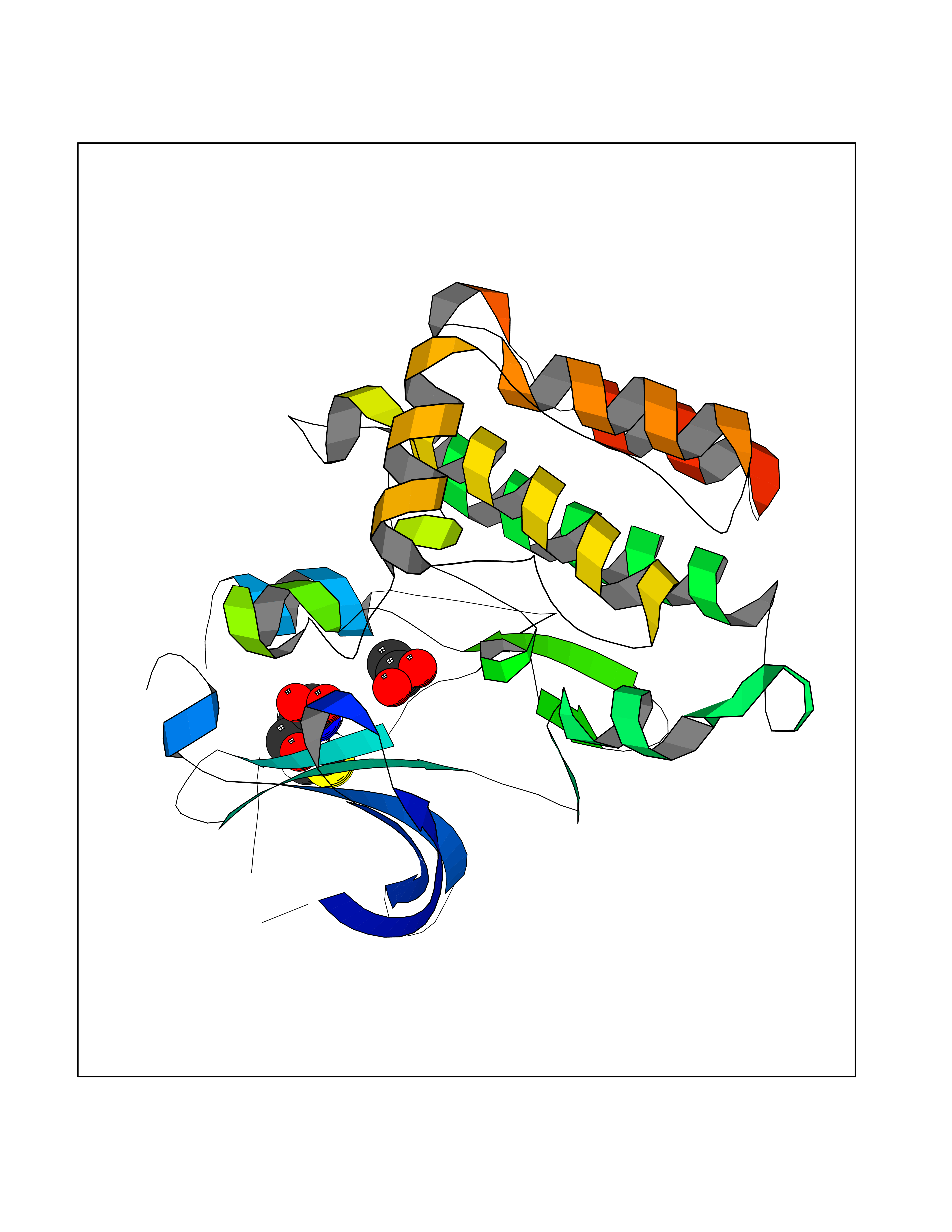 Molscript Map 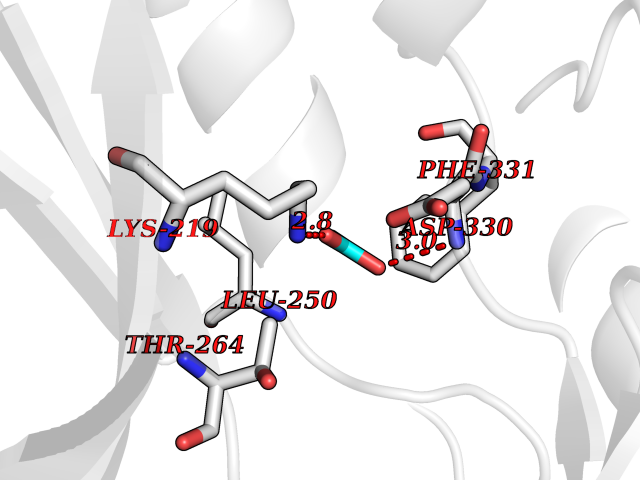 Pymol Map 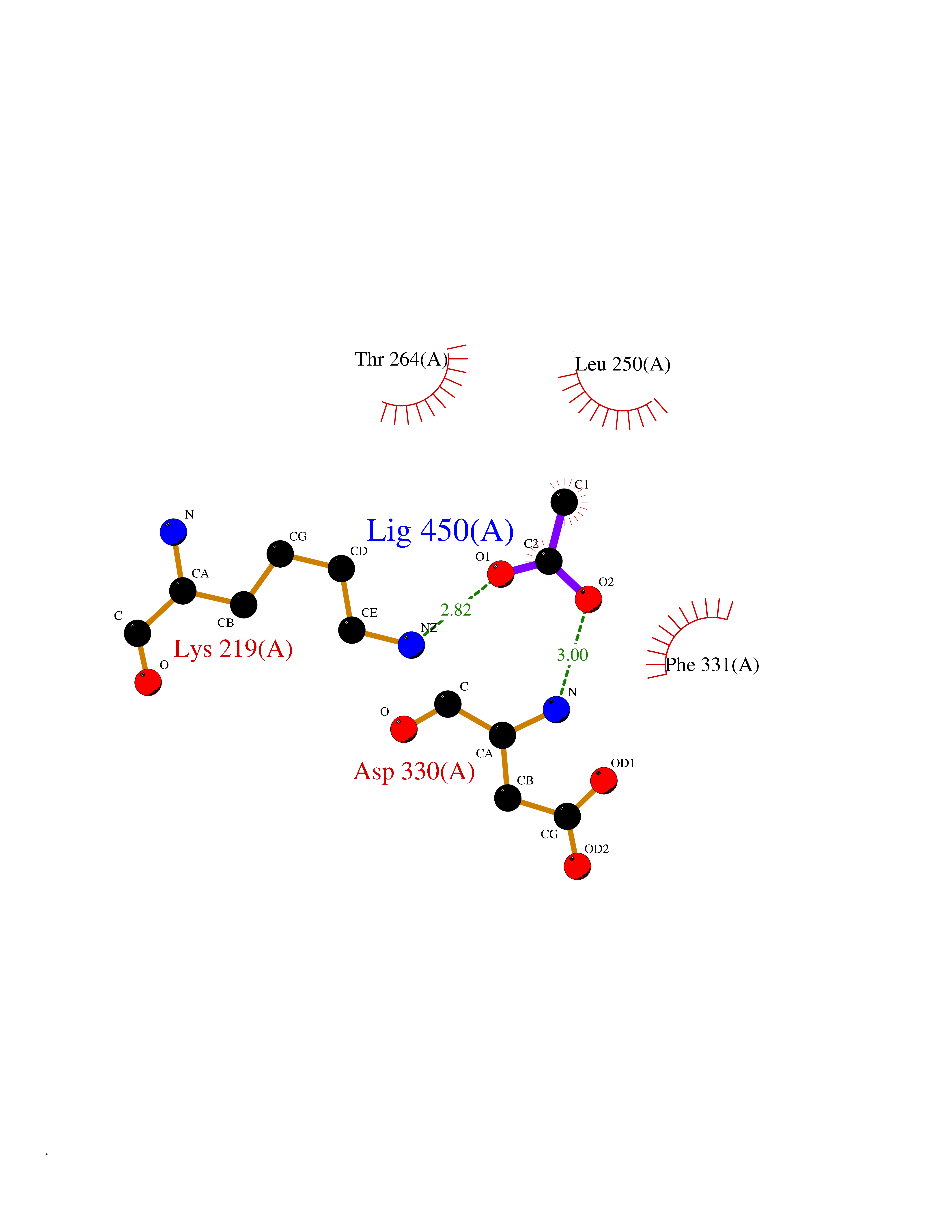 Ligplot Map 3D Binding mode Sequence XERPREEFTLCRKLGSGYFGEVFEGLWKDRVQVAIKVISRDNLLHQMLQSEIQAMKKLRHKHILALYAVVSVGDPVYIITELMAKGSLLELLRDSDEKVLPVSELLDIAWQVAEGMCYLESQNYIHRDLAARNILVGENTLCKVGDFGLARLIKEDVYLSHDHNIPYKWTAPEALSRGHYSTKSDVWSFGILLHEMFSRGQVPYPGMSNHEAFLRVDAGYRMPCPLECPPSVHKLMLTCWCRDPEQRPTFKALRERLSSFTSHH Hydrogen bonds contact Hydrophobic contact | ||||
| 82 | 2-oxopropyl-CoM reductase, carboxylating | 1MO9 | 4.37 | |
Target general information Gen name xecC Organism Xanthobacter autotrophicus (strain ATCC BAA-1158 / Py2) Uniprot ID TTD ID NA Synonyms Xaut_4867 Protein family Class-I pyridine nucleotide-disulfide oxidoreductase family Biochemical class Oxidoreductase Function 2-oxopropyl-CoM reductase (carboxylating) activity.Flavin adenine dinucleotide binding. Related diseases LTC4 synthase deficiency is associated with a neurometabolic developmental disorder characterized by muscular hypotonia, psychomotor retardation, failure to thrive, and microcephaly. {ECO:0000269|PubMed:10896305, ECO:0000269|PubMed:9820300}. Drugs (DrugBank ID) DB03163; DB03147 Interacts with NA EC number 1.8.1.5 Uniprot keywords 3D-structure; Disulfide bond; FAD; Flavoprotein; NADP; Oxidoreductase; Plasmid; Redox-active center; Reference proteome Protein physicochemical properties Chain ID A,B Molecular weight (Da) 114413 Length 1044 Aromaticity 0.08 Instability index 25.66 Isoelectric point 5.68 Charge (pH=7) -21.74 2D Binding mode Binding energy (Kcal/mol) -5.96  Molscript Map 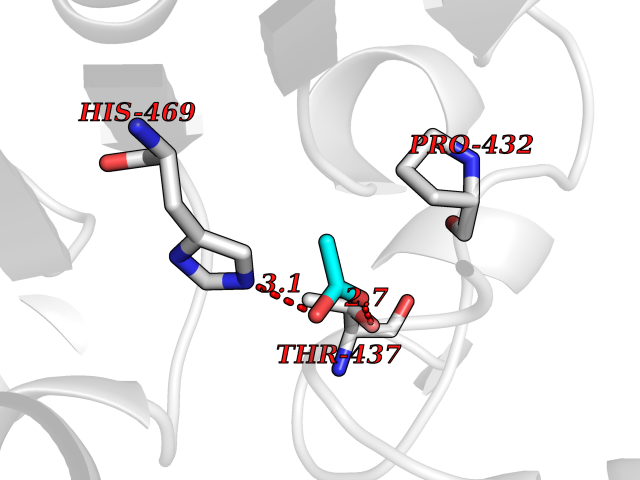 Pymol Map 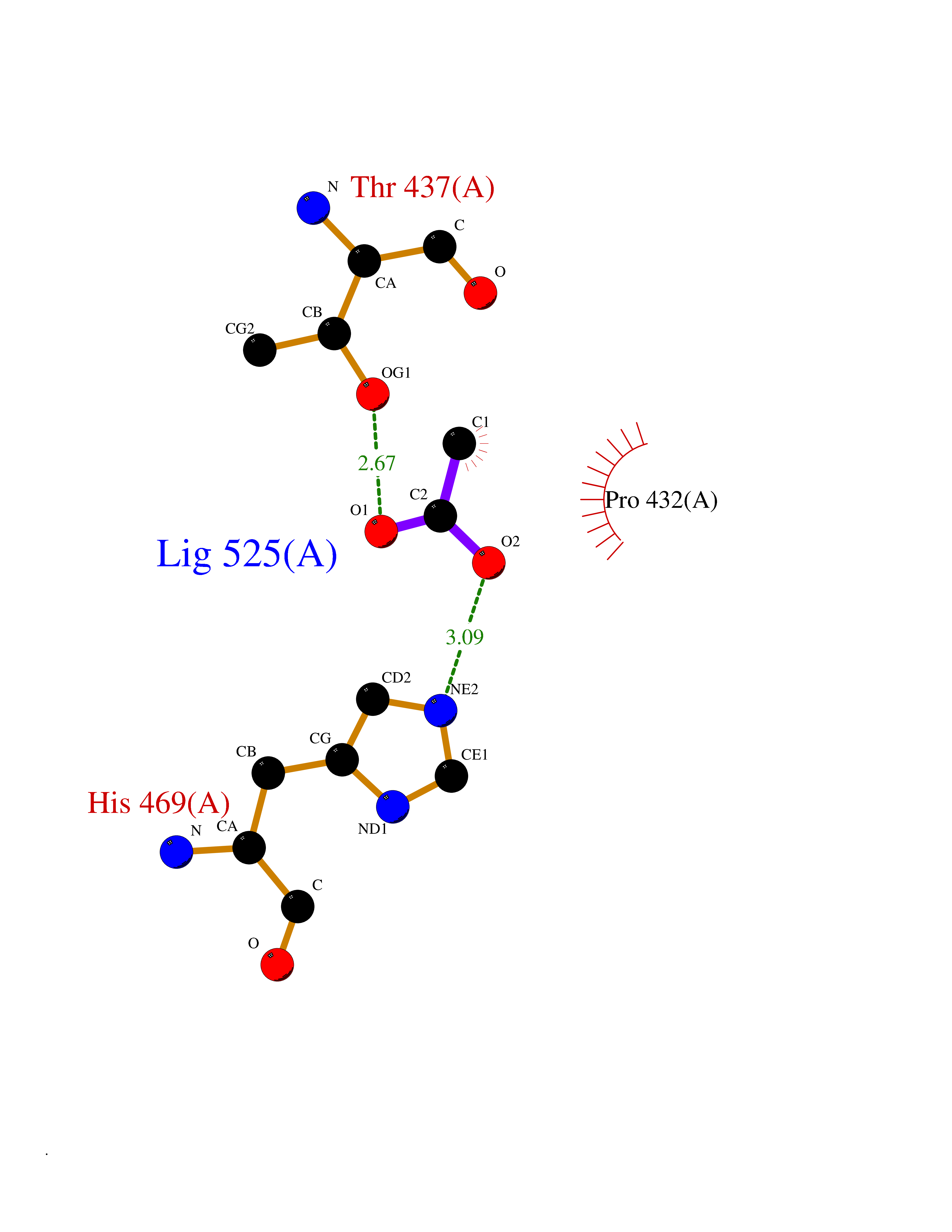 Ligplot Map 3D Binding mode Sequence KVWNARNDHLTINQWATRIDEILEAPDGGEVIYNVDENDPREYDAIFIGGGAAGRFGSAYLRAMGGRQLIVDRWPFLGGSCPHNACVPHHLFSDCAAELMLARTFSGQYWFPDMTEKVVGIKEVVDLFRAGRNGPHGIMNFQSKEQLNLEYILNCPAKVIDNHTVEAAGKVFKAKNLILAVGAGPGTLDVPGVNAKGVFDHATLVEELDYEPGSTVVVVGGSKTAVEYGCFFNATGRRTVMLVRTEPLKLIKDNETRAYVLDRMKEQGMEIISGSNVTRIEEDANGRVQAVVAMTPNGEMRIETDFVFLGLGEQPRSAELAKILGLDLGPKGEVLVNEYLQTSVPNVYAVGDLIGGPMEMFKARKSGCYAARNVMGEKISYTPKNYPDFLHTHYEVSFLGMGEEEARAAGHEIVTIKMPPDTENGLNVALPASDRTMLYAFGKGTAHMSGFQKIVIDAKTRKVLGAHHVGYGAKDAFQYLNVLIKQGLTVDELGDMDELFLNPTHFIQLSRLRAGSKNLVSLKVWNARNDHLTINQWATRIDEILEAPDGGEVIYNVDENDPREYDAIFIGGGAAGRFGSAYLRAMGGRQLIVDRWPFLGGSCPHNACVPHHLFSDCAAELMLARTFSGQYWFPDMTEKVVGIKEVVDLFRAGRNGPHGIMNFQSKEQLNLEYILNCPAKVIDNHTVEAAGKVFKAKNLILAVGAGPGTLDVPGVNAKGVFDHATLVEELDYEPGSTVVVVGGSKTAVEYGCFFNATGRRTVMLVRTEPLKLIKDNETRAYVLDRMKEQGMEIISGSNVTRIEEDANGRVQAVVAMTPNGEMRIETDFVFLGLGEQPRSAELAKILGLDLGPKGEVLVNEYLQTSVPNVYAVGDLIGGPMEMFKARKSGCYAARNVMGEKISYTPKNYPDFLHTHYEVSFLGMGEEEARAAGHEIVTIKMPPDTENGLNVALPASDRTMLYAFGKGTAHMSGFQKIVIDAKTRKVLGAHHVGYGAKDAFQYLNVLIKQGLTVDELGDMDELFLNPTHFIQLSRLRAGSKNLVSL Hydrogen bonds contact Hydrophobic contact | ||||
| 83 | Plasmodium Dihydroorotate dehydrogenase (Malaria DHOdehase) | 1TV5 | 4.37 | |
Target general information Gen name Malaria DHOdehase Organism Plasmodium falciparum (isolate 3D7) Uniprot ID TTD ID Synonyms PFF0160c; Mitochondrially bound dihydroorotate-ubiqui oxidoreductase; Dihydroorotate oxidase of Plasmodium falciparum; Dihydroorotate dehydrogenase of Plasmodium falciparum; DHOdehase of Plasmodium fa Protein family Dihydroorotate dehydrogenase family, Type 2 subfamily Biochemical class CH-CH donor oxidoreductase Function Catalyzes the conversion of dihydroorotate to orotate with quinone as electron acceptor. Related diseases Combined oxidative phosphorylation deficiency 33 (COXPD33) [MIM:617713]: An autosomal recessive disorder caused by multiple mitochondrial respiratory chain defects and impaired mitochondrial energy metabolism. Clinical manifestations are highly variable. Affected infants present with cardiomyopathy accompanied by multisystemic features involving liver, kidney, and brain. Death in infancy is observed in some patients. Children and adults present with myopathy and progressive external ophthalmoplegia. {ECO:0000269|PubMed:28942965}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01117 Interacts with NA EC number EC 1.3.5.2 Uniprot keywords 3D-structure; Flavoprotein; FMN; Membrane; Mitochondrion; Mitochondrion inner membrane; Oxidoreductase; Pyrimidine biosynthesis; Reference proteome; Transit peptide; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 41846.8 Length 371 Aromaticity 0.1 Instability index 37.25 Isoelectric point 8.21 Charge (pH=7) 3.13 2D Binding mode Binding energy (Kcal/mol) -5.96  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence FESYNPEFFLYDIFLKFCLKYIDGEICHDLFLLLGKYNILPYDTSNDSIYACTNIKHLDFINPFGVAAGFDKNGVCIDSILKLGFSFIEIGTITPRGQTGNAKPRIFRDVESRSIINSCGFNNMGCDKVTENLILFRKRQEEDKLLSKHIVGVSIGKNKDTVNIVDDLKYCINKIGRYADYIAINVSSPNTPGLRDNQEAGKLKNIILSVKEEIDNLEFLWFNTTKKKPLVFVKLAPDLNQEQKKEIADVLLETNIDGMIISNTTTQINDIKSFENKKGGVSGAKLKDISTKFICEMYNYTNKQIPIIASGGIFSGLDALEKIEAGASVCQLYSCLVFNGMKSAVQIKRELNHLLYQRGYYNLKEAIGRKH Hydrogen bonds contact Hydrophobic contact | ||||
| 84 | Pseudomonas Methionine gamma-lyase (Pseudo mdeA) | 1PG8 | 4.37 | |
Target general information Gen name Pseudo mdeA Organism Pseudomonas putida (Arthrobacter siderocapsulatus) Uniprot ID TTD ID Synonyms Pseudo MGL; L-methionine gamma-lyase; L-methioninase; Homocysteine desulfhydrase Protein family Trans-sulfuration enzymes family, L-methionine gamma-lyase subfamily Biochemical class Carbon-sulfur lyases Function Catalyzes the alpha,gamma-elimination of L-methionine to produce methanethiol, 2-oxobutanoate and ammonia. Is involved in L-methionine catabolism. In fact, shows a multicatalytic function since it also catalyzes gamma-replacement of L-methionine with thiol compounds, alpha,gamma-elimination and gamma-replacement reactions of L-homocysteine and its S-substituted derivatives, O-substituted-L-homoserines and DL-selenomethionine, and, to a lesser extent, alpha,beta-elimination and beta-replacement reactions of L-cysteine, S-methyl-L-cysteine, and O-acetyl-L-serine. Also catalyzes deamination and gamma-addition reactions of L-vinylglycine. Thus, the enzyme is able to cleave C-S, C-Se, and C-O bonds of sulfur, selenium, and oxygen amino acids, respectively. Related diseases Lecithin-cholesterol acyltransferase deficiency (LCATD) [MIM:245900]: A disorder of lipoprotein metabolism characterized by inadequate esterification of plasmatic cholesterol. Two clinical forms are recognized: complete LCAT deficiency and fish-eye disease. LCATD is generally referred to the complete form which is associated with absence of both alpha and beta LCAT activities resulting in esterification anomalies involving both HDL (alpha-LCAT activity) and LDL (beta-LCAT activity). It causes a typical triad of diffuse corneal opacities, target cell hemolytic anemia, and proteinuria with renal failure. {ECO:0000269|PubMed:11423760, ECO:0000269|PubMed:12957688, ECO:0000269|PubMed:15994445, ECO:0000269|PubMed:16051254, ECO:0000269|PubMed:16216249, ECO:0000269|PubMed:1681161, ECO:0000269|PubMed:1859405, ECO:0000269|PubMed:2370048, ECO:0000269|PubMed:7607641, ECO:0000269|PubMed:7711728, ECO:0000269|PubMed:8318557, ECO:0000269|PubMed:8432868, ECO:0000269|PubMed:8807342, ECO:0000269|PubMed:9007616, ECO:0000269|PubMed:9741700}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Fish-eye disease (FED) [MIM:136120]: A disorder of lipoprotein metabolism due to partial lecithin-cholesterol acyltransferase deficiency that affects only alpha-LCAT activity. FED is characterized by low plasma HDL and corneal opacities due to accumulation of cholesterol deposits in the cornea ('fish-eye'). {ECO:0000269|PubMed:1516702, ECO:0000269|PubMed:1571050, ECO:0000269|PubMed:15994445, ECO:0000269|PubMed:1737840, ECO:0000269|PubMed:21901787, ECO:0000269|PubMed:8620346, ECO:0000269|PubMed:9261271}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB04083 Interacts with NA EC number EC 4.4.1.11 Uniprot keywords 3D-structure; Direct protein sequencing; Lyase; Pyridoxal phosphate Protein physicochemical properties Chain ID A,C Molecular weight (Da) 85234.4 Length 796 Aromaticity 0.07 Instability index 37.1 Isoelectric point 6.21 Charge (pH=7) -11.34 2D Binding mode Binding energy (Kcal/mol) -5.96  Molscript Map  Pymol Map 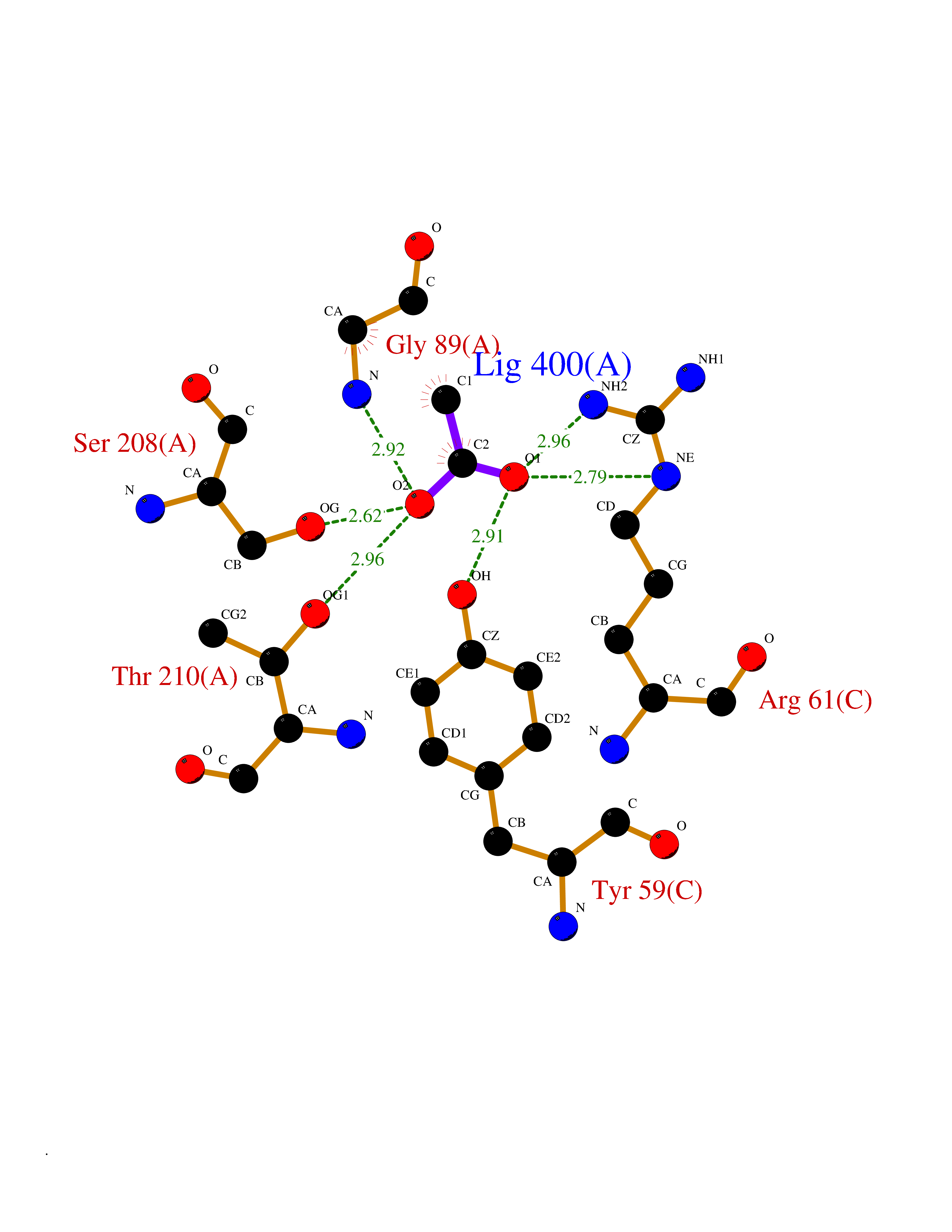 Ligplot Map 3D Binding mode Sequence MHGSNKLPGFATRAIHHGYDPQDHGGALVPPVYQTATFTFPTVEYGAACFAGEQAGHFYSRISNPTLNLLEARMASLEGGEAGLALASGMGAITSTLWTLLRPGDEVLLGNTLYGCTFAFLHHGIGEFGVKLRHVDMADLQALEAAMTPATRVIYFESPANPNMHMADIAGVAKIARKHGATVVVDNTYCTPYLQRPLELGADLVVHSATKYLSGHGDITAGIVVGSQALVDRIRLQGLKDMTGAVLSPHDAALLMRGIKTLNLRMDRHCANAQVLAEFLARQPQVELIHYPGLASFPQYTLARQQMSQPGGMIAFELKGGIGAGRRFMNALQLFSRAVSLGDAESLAQHPASMTHSSYTPEERAHYGISEGLVRLSVGLEDIDDLLADVQQALKASAMHGSNKLPGFATRAIHHGYDPQDHGGALVPPVYQTATFTFPTVEYGAACFAGEQAGHFYSRISNPTLNLLEARMASLEGGEAGLALASGMGAITSTLWTLLRPGDEVLLGNTLYGCTFAFLHHGIGEFGVKLRHVDMADLQALEAAMTPATRVIYFESPANPNMHMADIAGVAKIARKHGATVVVDNTYCTPYLQRPLELGADLVVHSATKYLSGHGDITAGIVVGSQALVDRIRLQGLKDMTGAVLSPHDAALLMRGIKTLNLRMDRHCANAQVLAEFLARQPQVELIHYPGLASFPQYTLARQQMSQPGGMIAFELKGGIGAGRRFMNALQLFSRAVSLGDAESLAQHPASMTHSSYTPEERAHYGISEGLVRLSVGLEDIDDLLADVQQALKASA Hydrogen bonds contact Hydrophobic contact | ||||
| 85 | Mycobacterium Membrane protein mmpL3 (MycB mmpL3) | 7NVH | 4.37 | |
Target general information Gen name MycB mmpL3 Organism Mycobacterium tuberculosis (strain ATCC 25618 / H37Rv) Uniprot ID TTD ID Synonyms Trehalose monomycolate exporter MmpL3; TMM exporter MmpL3 Protein family Resistance-nodulation-cell division (RND) (TC 2.A.6) family, MmpL subfamily Biochemical class NA Function Transports trehalose monomycolate (TMM) across the inner membrane. Could also be part of a heme-iron acquisition system. Related diseases Leukemia, acute myelogenous (AML) [MIM:601626]: A subtype of acute leukemia, a cancer of the white blood cells. AML is a malignant disease of bone marrow characterized by maturational arrest of hematopoietic precursors at an early stage of development. Clonal expansion of myeloid blasts occurs in bone marrow, blood, and other tissue. Myelogenous leukemias develop from changes in cells that normally produce neutrophils, basophils, eosinophils and monocytes. {ECO:0000269|PubMed:8955068}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Leukemia, juvenile myelomonocytic (JMML) [MIM:607785]: An aggressive pediatric myelodysplastic syndrome/myeloproliferative disorder characterized by malignant transformation in the hematopoietic stem cell compartment with proliferation of differentiated progeny. Patients have splenomegaly, enlarged lymph nodes, rashes, and hemorrhages. {ECO:0000269|PubMed:17332249}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Noonan syndrome 3 (NS3) [MIM:609942]: A form of Noonan syndrome, a disease characterized by short stature, facial dysmorphic features such as hypertelorism, a downward eyeslant and low-set posteriorly rotated ears, and a high incidence of congenital heart defects and hypertrophic cardiomyopathy. Other features can include a short neck with webbing or redundancy of skin, deafness, motor delay, variable intellectual deficits, multiple skeletal defects, cryptorchidism, and bleeding diathesis. Individuals with Noonan syndrome are at risk of juvenile myelomonocytic leukemia, a myeloproliferative disorder characterized by excessive production of myelomonocytic cells. {ECO:0000269|PubMed:16474405, ECO:0000269|PubMed:16773572, ECO:0000269|PubMed:17056636, ECO:0000269|PubMed:17468812, ECO:0000269|PubMed:19396835, ECO:0000269|PubMed:20949621}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Gastric cancer (GASC) [MIM:613659]: A malignant disease which starts in the stomach, can spread to the esophagus or the small intestine, and can extend through the stomach wall to nearby lymph nodes and organs. It also can metastasize to other parts of the body. The term gastric cancer or gastric carcinoma refers to adenocarcinoma of the stomach that accounts for most of all gastric malignant tumors. Two main histologic types are recognized, diffuse type and intestinal type carcinomas. Diffuse tumors are poorly differentiated infiltrating lesions, resulting in thickening of the stomach. In contrast, intestinal tumors are usually exophytic, often ulcerating, and associated with intestinal metaplasia of the stomach, most often observed in sporadic disease. {ECO:0000269|PubMed:14534542, ECO:0000269|PubMed:3034404, ECO:0000269|PubMed:7773929}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Defects in KRAS are a cause of pylocytic astrocytoma (PA). Pylocytic astrocytomas are neoplasms of the brain and spinal cord derived from glial cells which vary from histologically benign forms to highly anaplastic and malignant tumors. {ECO:0000269|PubMed:16247081}.; DISEASE: Cardiofaciocutaneous syndrome 2 (CFC2) [MIM:615278]: A form of cardiofaciocutaneous syndrome, a multiple congenital anomaly disorder characterized by a distinctive facial appearance, heart defects and intellectual disability. Heart defects include pulmonic stenosis, atrial septal defects and hypertrophic cardiomyopathy. Some affected individuals present with ectodermal abnormalities such as sparse, friable hair, hyperkeratotic skin lesions and a generalized ichthyosis-like condition. Typical facial features are similar to Noonan syndrome. They include high forehead with bitemporal constriction, hypoplastic supraorbital ridges, downslanting palpebral fissures, a depressed nasal bridge, and posteriorly angulated ears with prominent helices. CFC2 patients often do not have the skin abnormalities, such as ichthyosis, hyperkeratosis, and hemangioma observed in CFC1. {ECO:0000269|PubMed:16474404, ECO:0000269|PubMed:16474405, ECO:0000269|PubMed:17056636, ECO:0000269|PubMed:20949621, ECO:0000269|PubMed:21797849}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: KRAS mutations are involved in cancer development. {ECO:0000269|PubMed:14534542, ECO:0000269|PubMed:1553789, ECO:0000269|PubMed:16533793, ECO:0000269|PubMed:24623306, ECO:0000269|PubMed:3034404, ECO:0000269|PubMed:3627975, ECO:0000269|PubMed:6092920, ECO:0000269|PubMed:6695174, ECO:0000269|PubMed:7773929}.; DISEASE: Oculoectodermal syndrome (OES) [MIM:600268]: A syndrome characterized by the association of epibulbar dermoids and aplasia cutis congenita. Affected individuals show multiple, asymmetric, atrophic, non-scarring and hairless regions that may be associated with hamartomas. Ectodermal changes include linear hyperpigmentation that may follow the lines of Blaschko and rarely epidermal nevus-like lesions. Epibulbar dermoids may be uni-or bilateral. Additional ocular anomalies such as skin tags of the upper eyelid, rarely optic nerve or retinal changes, and microphthalmia can be present. The phenotypic expression is highly variable, and various other abnormalities have occasionally been reported including growth failure, lymphedema, cardiovascular defects, as well as neurodevelopmental symptoms like developmental delay, epilepsy, learning difficulties, and behavioral abnormalities. Benign tumor-like lesions such as nonossifying fibromas of the long bones and giant cell granulomas of the jaws have repeatedly been observed and appear to be age-dependent, becoming a common manifestation in individuals aged 5 years or older. {ECO:0000269|PubMed:25808193, ECO:0000269|PubMed:26970110, ECO:0000269|PubMed:30891959}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Schimmelpenning-Feuerstein-Mims syndrome (SFM) [MIM:163200]: A disease characterized by sebaceous nevi, often on the face, associated with variable ipsilateral abnormalities of the central nervous system, ocular anomalies, and skeletal defects. Many oral manifestations have been reported, not only including hypoplastic and malformed teeth, and mucosal papillomatosis, but also ankyloglossia, hemihyperplastic tongue, intraoral nevus, giant cell granuloma, ameloblastoma, bone cysts, follicular cysts, oligodontia, and odontodysplasia. Sebaceous nevi follow the lines of Blaschko and these can continue as linear intraoral lesions, as in mucosal papillomatosis. {ECO:0000269|PubMed:30891959}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number NA Uniprot keywords 3D-structure; Cell inner membrane; Cell membrane; Cell wall biogenesis/degradation; Lipid transport; Membrane; Reference proteome; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A Molecular weight (Da) 77237 Length 717 Aromaticity 0.09 Instability index 33.34 Isoelectric point 8.65 Charge (pH=7) 4.3 2D Binding mode Binding energy (Kcal/mol) -5.96  Molscript Map 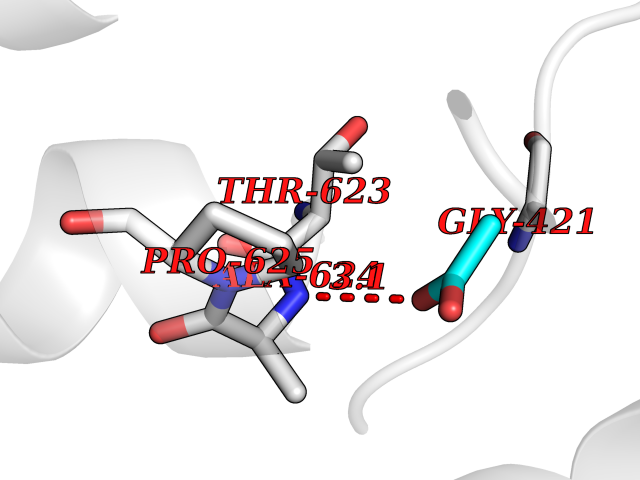 Pymol Map 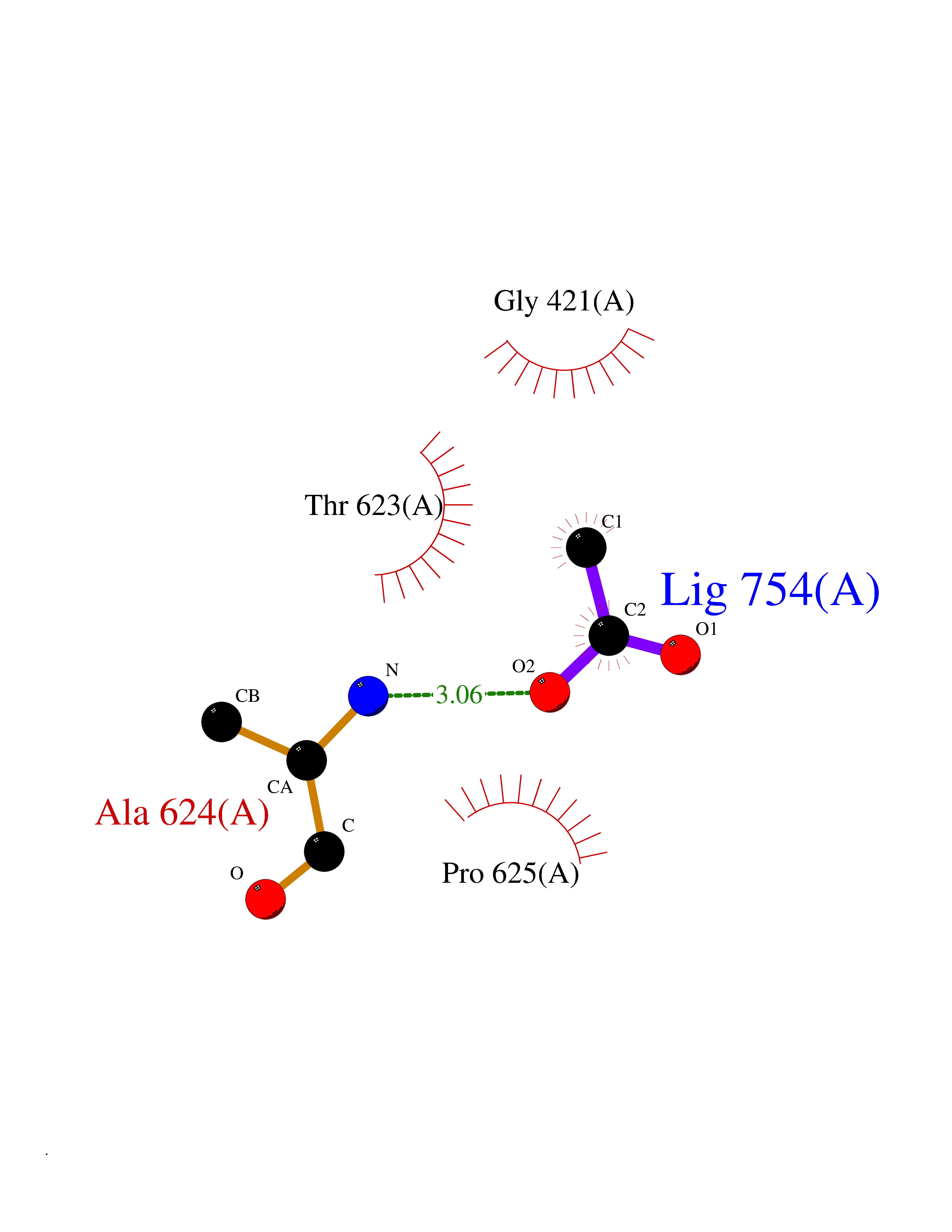 Ligplot Map 3D Binding mode Sequence MFAWWGRTVYRYRFIVIGVMVALCLGGGVFGLSLGKHVTQSGFYDDGSQSVQASVLGDQVYGRDRSGHIVAIFQAPAGKTVDDPAWSKKVVDELNRFQQDHPDQVLGWAGYLRASQATGMATADKKYTFVSIPLKGDDDDTILNNYKAIAPDLQRLDGGTVKLAGLQPVAEALTGTIATDQRRMEVLALPLVAVVLFFVFGGVIAAGLPVMVGGLCIAGALGIMRFLAIFGPVHYFAQPVVSLIGLGIAIDYGLFIVSRFREEIAEGYDTETAVRRTVITAGRTVTFSAVLIVASAIGLLLFPQGFLKSLTYATIASVMLSAILSITVLPACLGILGKHVDAEEVEAGFWGKLVNRVMKRPVLFAAPIVIIMILLIIPVGKLSLGGISEKYLPPTNSVRQAQEEFDKLFPGYRTNPLTLVIQTSNHQPVTDAQIADIRSKAMAIGGFIEPDNDPANMWQERAYAVGASKDPSVRVLQNGLINPADASKKLTELRAITPPKGITVLVGGTPALELDSIHGLFAKMPLMVVILLTTTIVLMFLAFGSVVLPIKATLMSALTLGSTMGILTWIFVDGHFSKWLNFTPTPLTAPVIGLIIALVFGLSTDYEVFLVSRMVEARERGMSTQEAIRIGTAATGRIITAAALIVAVVAGAFVFSDLVMMKYLAFGLMAALLLDATVVRMFLVPSVMKLLGDDCWWAPRWARRLQTRIGLGEIHLP Hydrogen bonds contact Hydrophobic contact | ||||
| 86 | Cerebron E3 ubiquitin ligase complex (CRL4-CRBN E3 ubiquitin ligase) | 4CI1 | 4.37 | |
Target general information Gen name CUL4A/CUL4B-DDB1-CRBN Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms NA Protein family Cullin family Biochemical class NA Function NA Related diseases Orotic aciduria 1 (ORAC1) [MIM:258900]: A disorder of pyrimidine metabolism resulting in megaloblastic anemia and orotic acid crystalluria that is frequently associated with some degree of physical and intellectual disability. A minority of cases have additional features, particularly congenital malformations and immune deficiencies. {ECO:0000269|PubMed:9042911}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P54253; Q86VP6; Q16531; Q92466; P08238; O94888; P55072 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Biological rhythms; DNA damage; DNA repair; Host-virus interaction; Isopeptide bond; Phosphoprotein; Proteomics identification; Reference proteome; Ubl conjugation; Ubl conjugation pathway Protein physicochemical properties Chain ID B Molecular weight (Da) 42669.7 Length 368 Aromaticity 0.1 Instability index 44.94 Isoelectric point 8.72 Charge (pH=7) 6.58 2D Binding mode Binding energy (Kcal/mol) -5.96 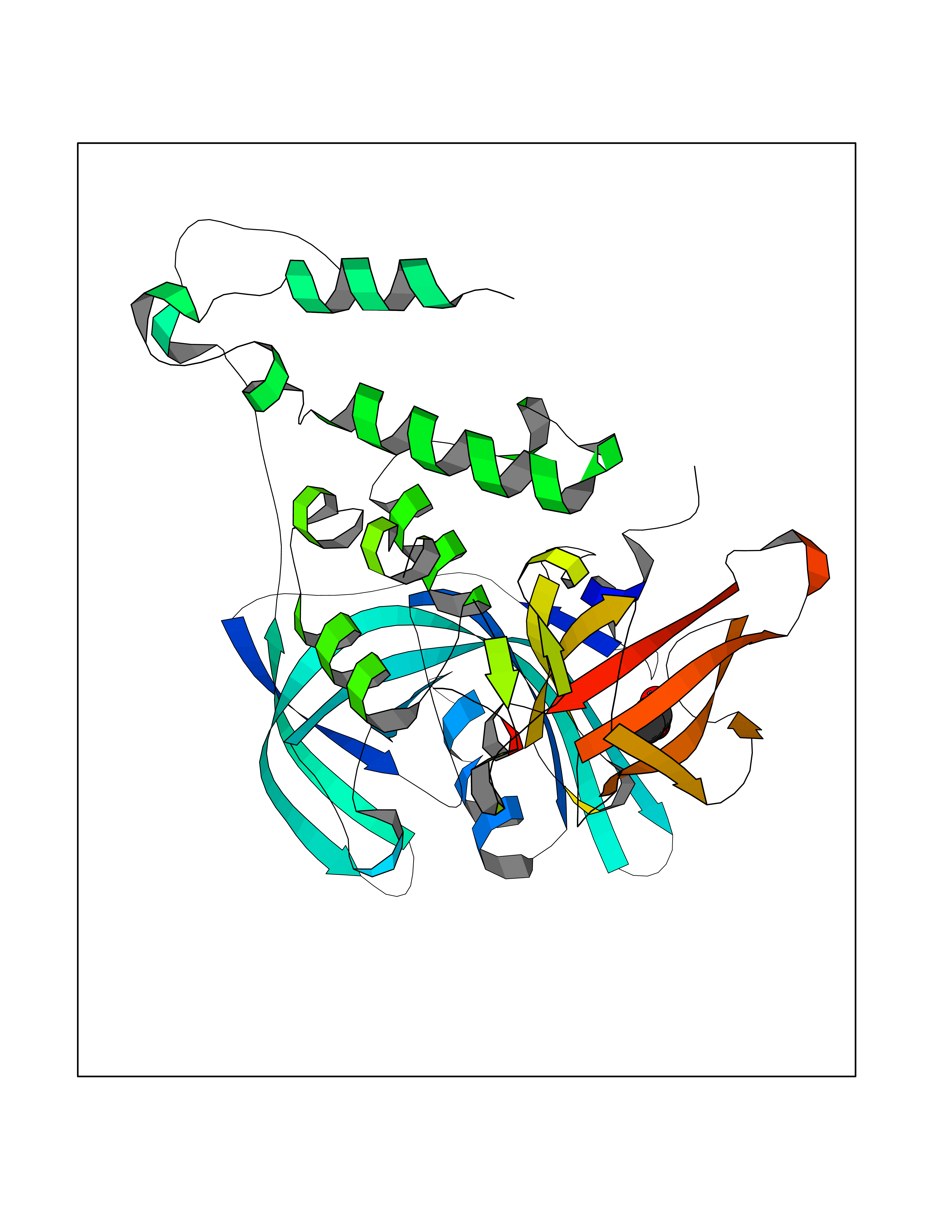 Molscript Map 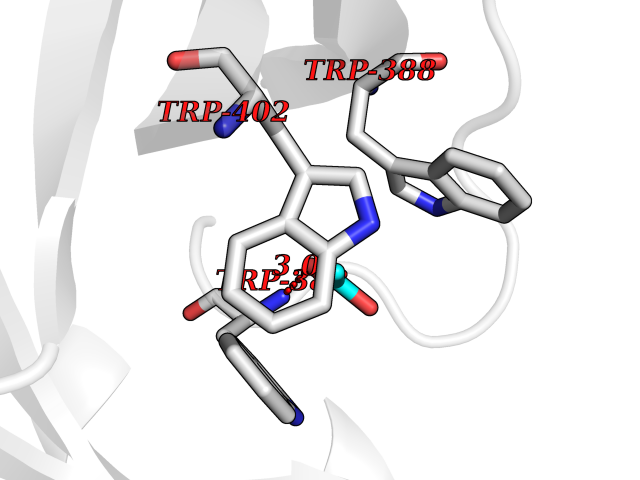 Pymol Map 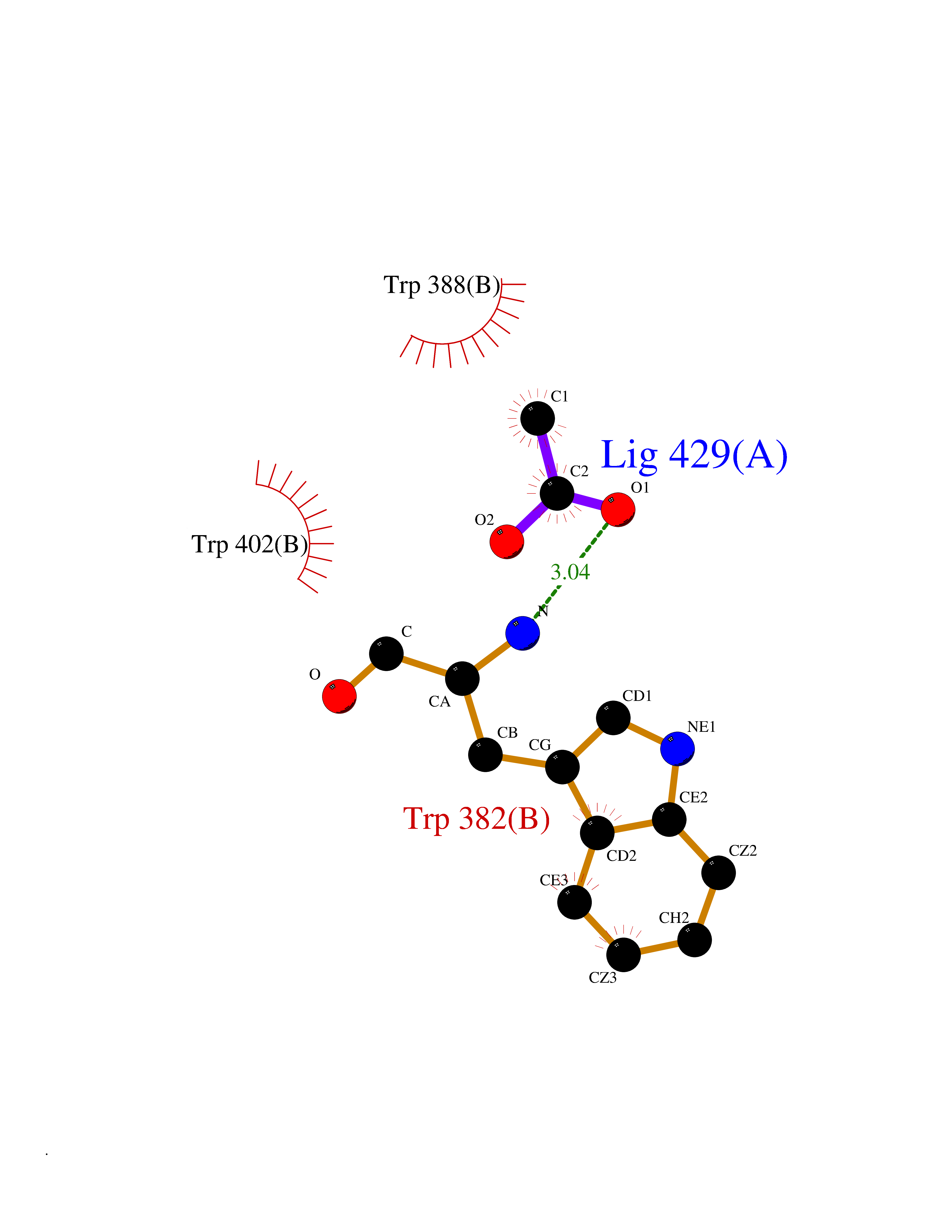 Ligplot Map 3D Binding mode Sequence MINFDTSLPTSHMYLGSDMEEFHGRTLHDDDSCQVIPVLPHVMVMLIPGQTLPLQLFHPQEVSMVRNLIQKDRTFAVLAYSNVREREAHFGTTAEIYAYREEQEYGIETVKVKAIGRQRFKVLEIRTQSDGIQQAKVQILPERVLPSTMSAVQLQSLSRRHIRAFRQWWQKYQKRKFHCASLTSWPPWLYSLYDAETLMERVKRQLHEWDENLKDESLPTNPIDFSYRVAACLPIDDALRIQLLKIGSAIQRLRELDIMNKTSLCCKQCQDTEITTKNEIFSLSLCGPMAAYVNPHGYIHETLTVYKACNLNLSGRPSTEHSWFPGYAWTIAQCRICGNHMGWKFTATKKDMSPQKFWGLTRSALLPR Hydrogen bonds contact Hydrophobic contact | ||||
| 87 | Alpha-1-antitrypsin (SERPINA1) | 5NBU | 4.37 | |
Target general information Gen name SERPINA1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms SERPINA1; PRO0684/PRO2209; Alpha1-proteinase; Alpha-1-antiproteinase; Alpha-1 protease inhibitor Protein family Serpin family Biochemical class Serpin protein Function Inhibitor of serine proteases. Its primary target is elastase, but it also has a moderate affinity for plasmin and thrombin. Related diseases Alpha-1-antitrypsin deficiency (A1ATD) [MIM:613490]: A disorder whose most common manifestation is emphysema, which becomes evident by the third to fourth decade. A less common manifestation of the deficiency is liver disease, which occurs in children and adults, and may result in cirrhosis and liver failure. Environmental factors, particularly cigarette smoking, greatly increase the risk of emphysema at an earlier age. {ECO:0000269|PubMed:1905728, ECO:0000269|PubMed:2227940, ECO:0000269|PubMed:2390072}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01998; DB09130; DB00080; DB03345; DB14007; DB05961; DB05481; DB01593; DB14487; DB14533; DB14548 Interacts with Q9Y282; Q8N7X4; P01009; P43307; O15393; P00772; P71213; P00760 EC number NA Uniprot keywords 3D-structure; Acute phase; Alternative splicing; Blood coagulation; Direct protein sequencing; Endoplasmic reticulum; Extracellular matrix; Glycoprotein; Hemostasis; Phosphoprotein; Protease inhibitor; Proteomics identification; Reference proteome; Secreted; Serine protease inhibitor; Signal Protein physicochemical properties Chain ID A Molecular weight (Da) 41542.2 Length 370 Aromaticity 0.09 Instability index 30.24 Isoelectric point 5.56 Charge (pH=7) -9.66 2D Binding mode Binding energy (Kcal/mol) -5.97 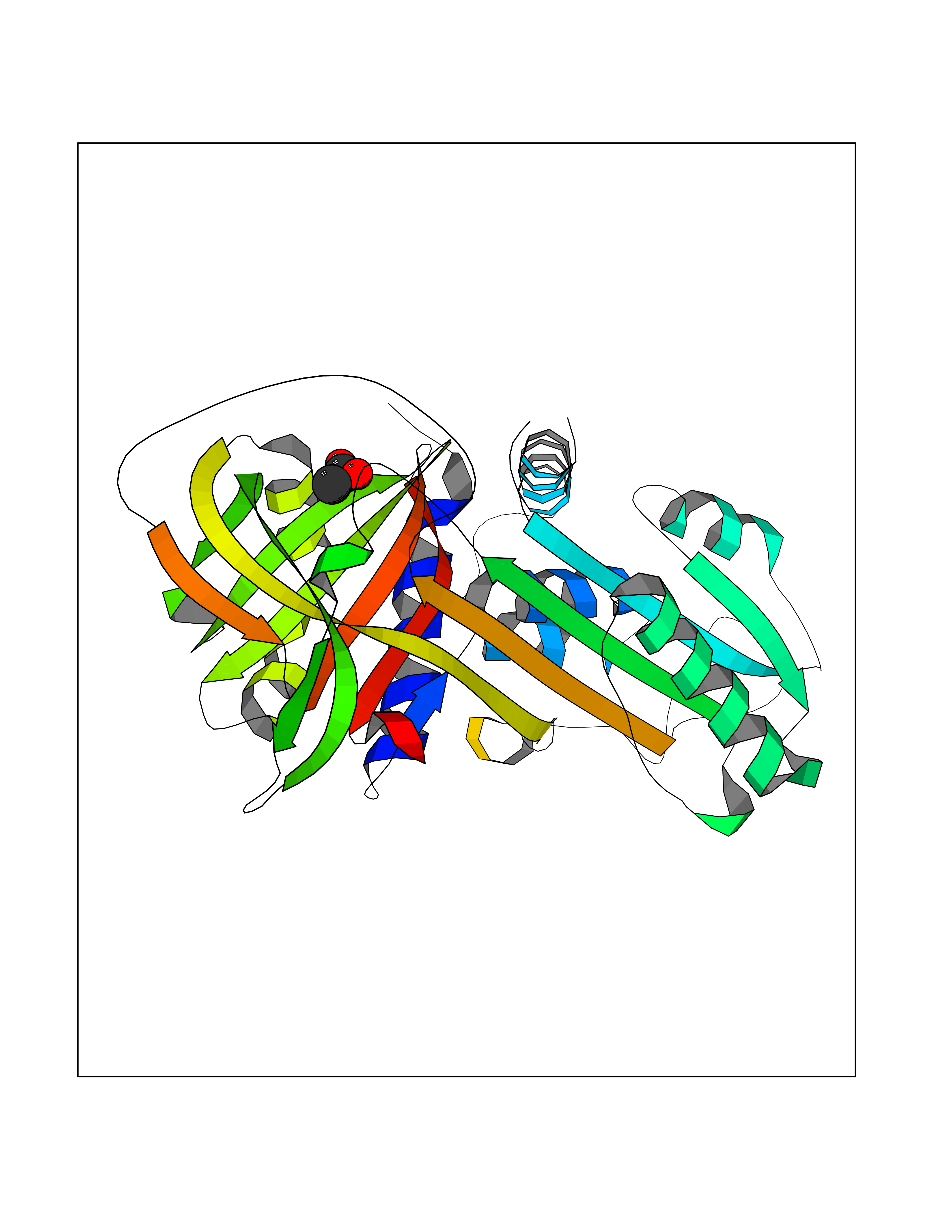 Molscript Map 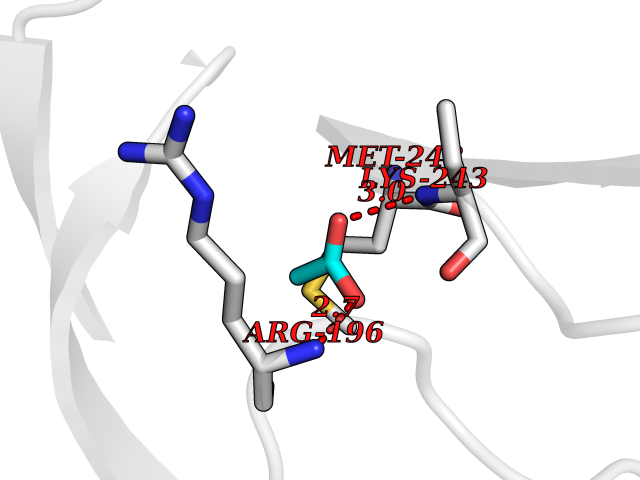 Pymol Map 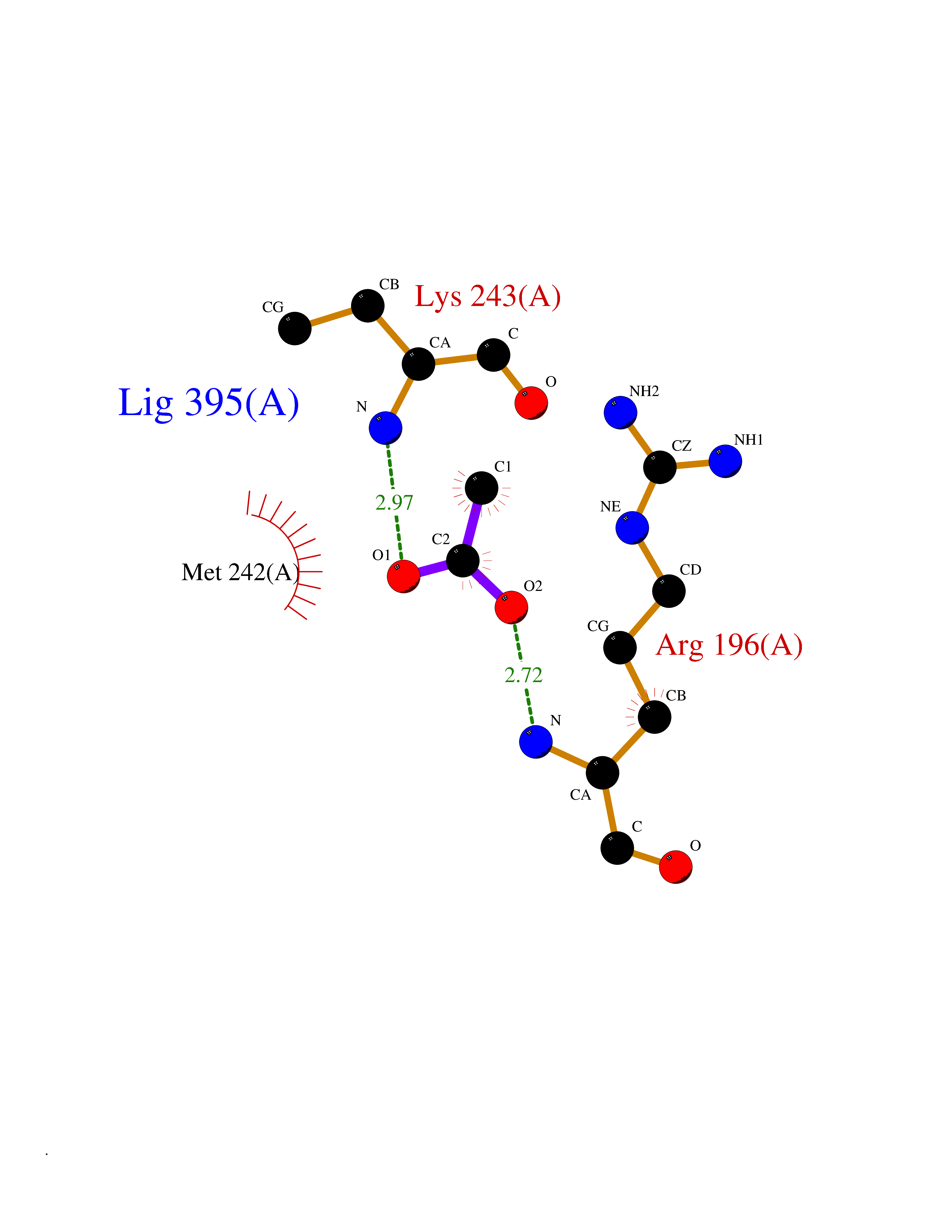 Ligplot Map 3D Binding mode Sequence TFNKITPNLAEFAFSLYRQLAHQSNSTNILFSPVSIAAAFAMLSLGAKGDTHDEILEGLNFNLTEIPEAQIHEGFQELLRTLNQSQLQLTTGNGLFLSEGLKLVDKFLEDVKKLYHSEAFTVNFGDTEEAKKQINDYVEKGTQGKIVDLVKELDRDTVFALVNYIFFKGKWERPFEVKDTEEEDFHVDQVTTVKVPMMKRLGMFNIQHSKKLSSWVLLMKYLGNATAIFFLPDEGKLQHLENELTHDIITKFLENEDRRSASLHLPKLSITGTYDLKSVLGQLGITKVFSNGADLSGVTEEAPLKLSKAVHKAVLTIDEKGTEAAGAMFLEAIPMSIPPEVKFNKPFVFLIIEQNTKAPLFMGRVVNPTQ Hydrogen bonds contact Hydrophobic contact | ||||
| 88 | Integrin beta-3 (ITGB3) | 7TD8 | 4.37 | |
Target general information Gen name ITGB3 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms GPIIIa; GP3A; CD61 Protein family Integrin beta chain family Biochemical class Integrin Function Integrin alpha-IIb/beta-3 (ITGA2B:ITGB3) is a receptor for fibronectin, fibrinogen, plasminogen, prothrombin, thrombospondin and vitronectin. Integrins alpha-IIb/beta-3 and alpha-V/beta-3 recognize the sequence R-G-D in a wide array of ligands. Integrin alpha-IIb/beta-3 recognizes the sequence H-H-L-G-G-G-A-K-Q-A-G-D-V in fibrinogen gamma chain. Following activation integrin alpha-IIb/beta-3 brings about platelet/platelet interaction through binding of soluble fibrinogen. This step leads to rapid platelet aggregation which physically plugs ruptured endothelial surface. Fibrinogen binding enhances SELP expression in activated platelets. ITGAV:ITGB3 binds to fractalkine (CX3CL1) and acts as its coreceptor in CX3CR1-dependent fractalkine signaling. ITGAV:ITGB3 binds to NRG1 (via EGF domain) and this binding is essential for NRG1-ERBB signaling. ITGAV:ITGB3 binds to FGF1 and this binding is essential for FGF1 signaling. ITGAV:ITGB3 binds to FGF2 and this binding is essential for FGF2 signaling. ITGAV:ITGB3 binds to IGF1 and this binding is essential for IGF1 signaling. ITGAV:ITGB3 binds to IGF2 and this binding is essential for IGF2 signaling. ITGAV:ITGB3 binds to IL1B and this binding is essential for IL1B signaling. ITGAV:ITGB3 binds to PLA2G2A via a site (site 2) which is distinct from the classical ligand-binding site (site 1) and this induces integrin conformational changes and enhanced ligand binding to site 1. ITGAV:ITGB3 acts as a receptor for fibrillin-1 (FBN1) and mediates R-G-D-dependent cell adhesion to FBN1. Integrin alpha-V/beta-3 (ITGAV:ITGB3) is a receptor for cytotactin, fibronectin, laminin, matrix metalloproteinase-2, osteopontin, osteomodulin, prothrombin, thrombospondin, vitronectin and von Willebrand factor. Related diseases Glanzmann thrombasthenia 2 (GT2) [MIM:619267]: A form of Glanzmann thrombasthenia, a disorder characterized by failure of platelet aggregation, absent or diminished clot retraction, and mucocutaneous bleeding of mild-to-moderate severity. Glanzmann thrombasthenia has been classified into clinical types I and II. In type I, platelets show absence of glycoprotein IIb-IIIa complexes at their surface and lack fibrinogen and clot retraction capability. In type II, the platelets express glycoprotein IIb-IIIa complexes at reduced levels, have detectable amounts of fibrinogen, and have low or moderate clot retraction capability. {ECO:0000269|PubMed:10233432, ECO:0000269|PubMed:11588040, ECO:0000269|PubMed:11897046, ECO:0000269|PubMed:12083483, ECO:0000269|PubMed:12353082, ECO:0000269|PubMed:1371279, ECO:0000269|PubMed:1438206, ECO:0000269|PubMed:15583747, ECO:0000269|PubMed:15634267, ECO:0000269|PubMed:15748237, ECO:0000269|PubMed:1602006, ECO:0000269|PubMed:20020534, ECO:0000269|PubMed:2392682, ECO:0000269|PubMed:29084015, ECO:0000269|PubMed:8781422, ECO:0000269|PubMed:9215749, ECO:0000269|PubMed:9376589, ECO:0000269|PubMed:9684783, ECO:0000269|PubMed:9790984}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Bleeding disorder, platelet-type, 24 (BDPLT24) [MIM:619271]: An autosomal dominant disorder of platelet production characterized by congenital macrothrombocytopenia and platelet anisocytosis. Affected individuals may have no or only mildly increased bleeding tendency. {ECO:0000269|PubMed:18065693, ECO:0000269|PubMed:29380037}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00054; DB00098; DB00063; DB13949; DB15598; DB06472; DB04863; DB00451; DB05787; DB02709; DB14520; DB00775 Interacts with P78423; P21333; P08514; P08514-1; P06756; P05106; P18031; Q9Y490; P05094; F5HB81; Q62101; P05480-2; P26039; P54939; P06935 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Calcium; Cell adhesion; Cell junction; Cell membrane; Cell projection; Direct protein sequencing; Disease variant; Disulfide bond; EGF-like domain; Glycoprotein; Host cell receptor for virus entry; Host-virus interaction; Integrin; Magnesium; Membrane; Metal-binding; Phosphoprotein; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Repeat; Signal; Synapse; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A,B Molecular weight (Da) 100489 Length 919 Aromaticity 0.09 Instability index 41.28 Isoelectric point 5.03 Charge (pH=7) -27.15 2D Binding mode Binding energy (Kcal/mol) -5.96  Molscript Map 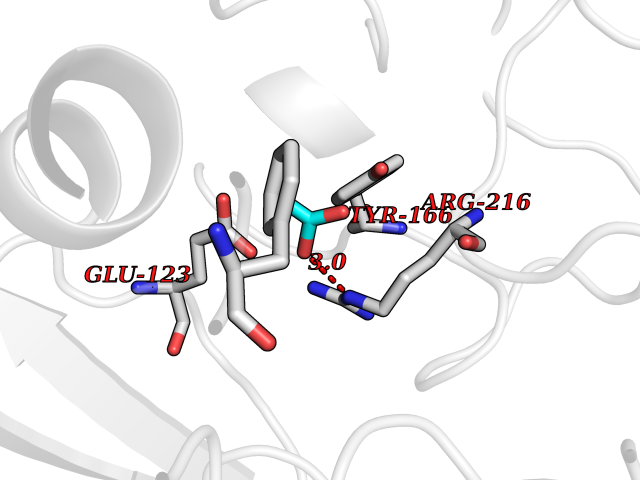 Pymol Map 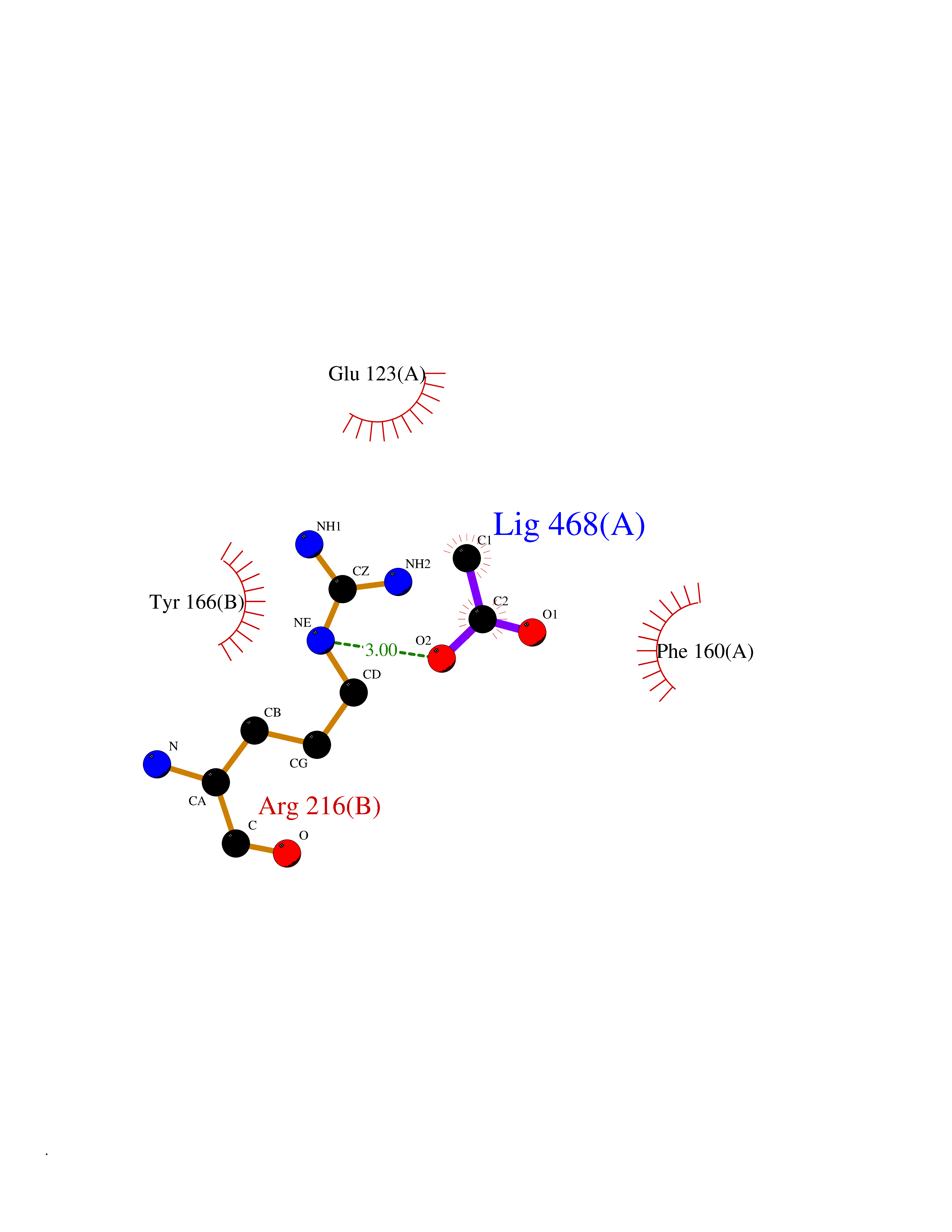 Ligplot Map 3D Binding mode Sequence LNLDPVQLTFYAGPNGSQFGFSLDFHKDSHGRVAIVVGAPRTLGPSQEETGGVFLCPWRAEGGQCPSLLFDLRDETRNVGSQTLQTFKARQGLGASVVSWSDVIVACAPWQHWNVLEKTEEAEKTPVGSCFLAQPESGRRAEYSPCRGNTLSRIYVENDFSWDKRYCEAGFSSVVTQAGELVLGAPGGYYFLGLLAQAPVADIFSSYRPGILLWHVSSQSLSFDSSNPEYFDGYWGYSVAVGEFDGDLNTTEYVVGAPTWSWTLGAVEILDSYYQRLHRLRGEQMASYFGHSVAVTDVNGDGRHDLLVGAPLYMESRADRKLAEVGRVYLFLQPRGPHALGAPSLLLTGTQLYGRFGSAIAPLGDLDRDGYNDIAVAAPYGGPSGRGQVLVFLGQSEGLRSRPSQVLDSPFPTGSAFGFSLRGAVDIDDNGYPDLIVGAYGANQVAVYRAQPVGPNICTTRGVSSCQQCLAVSPMCAWCSDEALPLGSPRCDLKENLLKDNCAPESIEFPVSEARVLEDRPLSDKGSGDSSQVTQVSPQRIALRLRPDDSKNFSIQVRQVEDYPVDIYYLMDLSYSMKDDLWSIQNLGTKLATQMRKLTSNLRIGFGAFVDKPVSPYMYISPPEALENPCYDMKTTCLPMFGYKHVLTLTDQVTRFNEEVKKQSVSRNRDAPEGGFDAIMQATVCDEKIGWRNDASHLLVFTTDAKTHIALDGRLAGIVQPNDGQCHVGSDNHYSASTTMDYPSLGLMTEKLSQKNINLIFAVTENVVNLYQNYSELIPGTTVGVLSMDSSNVLQLIVDAYGKIRSKVELEVRDLPEELSLSFNATCLNNEVIPGLKSCMGLKIGDTVSFSIEAKVRGCPQEKEKSFTIKPVGFKDSLIVQVTFDCDCACQAQAEPNSHRCNNGNGTFECGVCRCGPGW Hydrogen bonds contact Hydrophobic contact | ||||
| 89 | Cholesterol 24-hydroxylase (CYP46A1) | 3MDR | 4.37 | |
Target general information Gen name CYP46A1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Cytochrome P450 46A1; CYP46; Cholesterol 24S-hydroxylase; Cholesterol 24-monooxygenase; CH24H Protein family Cytochrome P450 family Biochemical class Paired donor oxygen oxidoreductase Function Primarily catalyzes the hydroxylation (with S stereochemistry) at C-24 of cholesterol side chain, triggering cholesterol diffusion out of neurons and its further degradation. By promoting constant cholesterol elimination in neurons, may activate the mevalonate pathway and coordinate the synthesis of new cholesterol and nonsterol isoprenoids involved in synaptic activity and learning. Further hydroxylates cholesterol derivatives and hormone steroids on both the ring and side chain of these molecules, converting them into active oxysterols involved in lipid signaling and biosynthesis. Acts as an epoxidase converting cholesta-5,24-dien-3beta-ol/desmosterol into (24S),25-epoxycholesterol, an abundant lipid ligand of nuclear NR1H2 and NR1H3 receptors shown to promote neurogenesis in developing brain. May also catalyze the oxidative metabolism of xenobiotics, such as clotrimazole. P450 monooxygenase that plays a major role in cholesterol homeostasis in the brain. Related diseases Spinocerebellar ataxia, autosomal recessive, with axonal neuropathy 1 (SCAN1) [MIM:607250]: A form of spinocerebellar ataxia, a clinically and genetically heterogeneous group of cerebellar disorders. Patients show progressive incoordination of gait and often poor coordination of hands, speech and eye movements, due to degeneration of the cerebellum with variable involvement of the brainstem and spinal cord. SCAN1 is an autosomal recessive cerebellar ataxia (ARCA) associated with peripheral axonal motor and sensory neuropathy, distal muscular atrophy, pes cavus and steppage gait as seen in Charcot-Marie-Tooth neuropathy. All affected individuals have normal intelligence. {ECO:0000269|PubMed:12244316, ECO:0000269|PubMed:15647511, ECO:0000269|PubMed:15920477, ECO:0000269|PubMed:16141202, ECO:0000269|PubMed:17948061}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number EC 1.14.14.25 Uniprot keywords 3D-structure; Alternative splicing; Cell projection; Cholesterol metabolism; Endoplasmic reticulum; Heme; Iron; Lipid metabolism; Membrane; Metal-binding; Microsome; Monooxygenase; Oxidoreductase; Proteomics identification; Reference proteome; Steroid metabolism; Sterol metabolism; Synapse; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 48977 Length 427 Aromaticity 0.1 Instability index 49.59 Isoelectric point 9.04 Charge (pH=7) 6.25 2D Binding mode Binding energy (Kcal/mol) -5.96 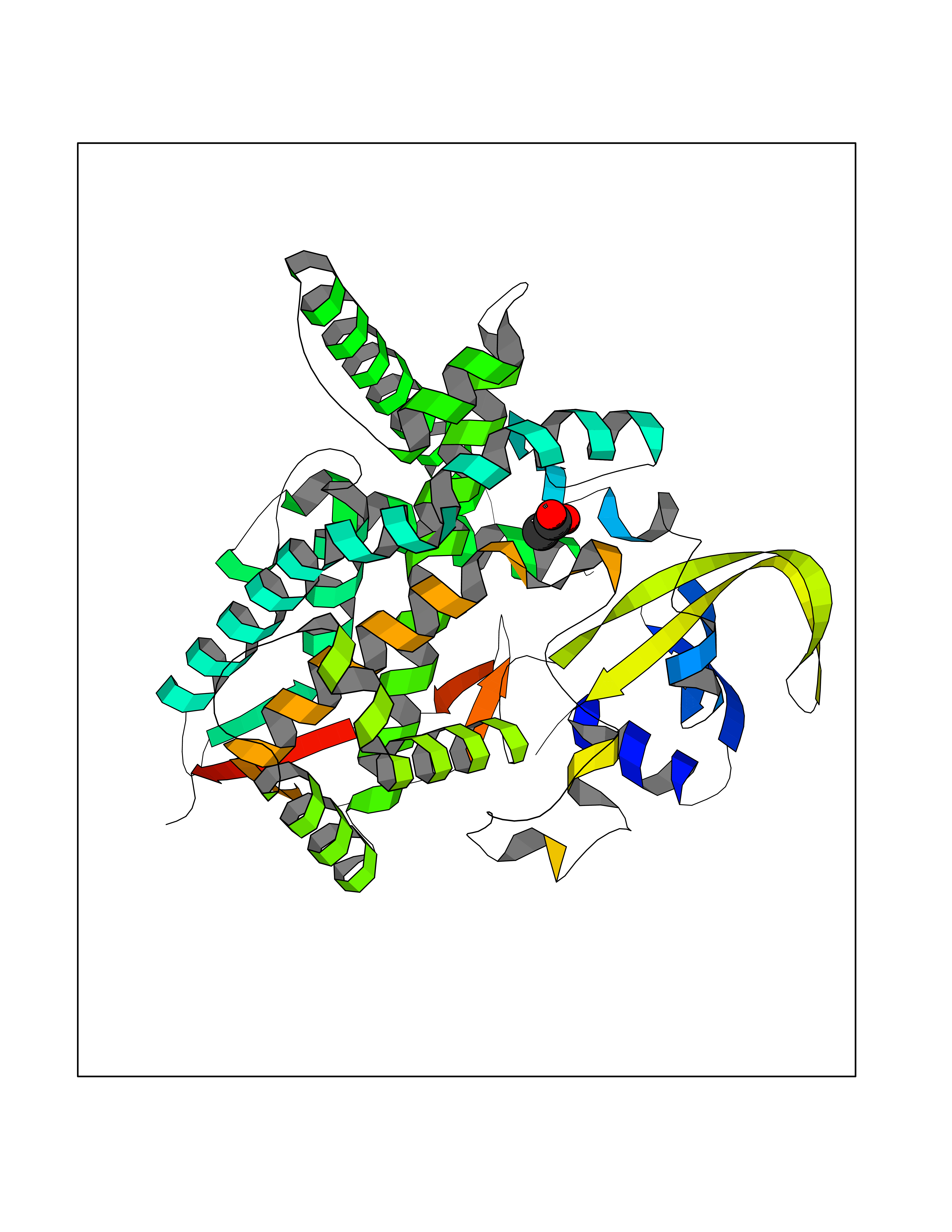 Molscript Map  Pymol Map 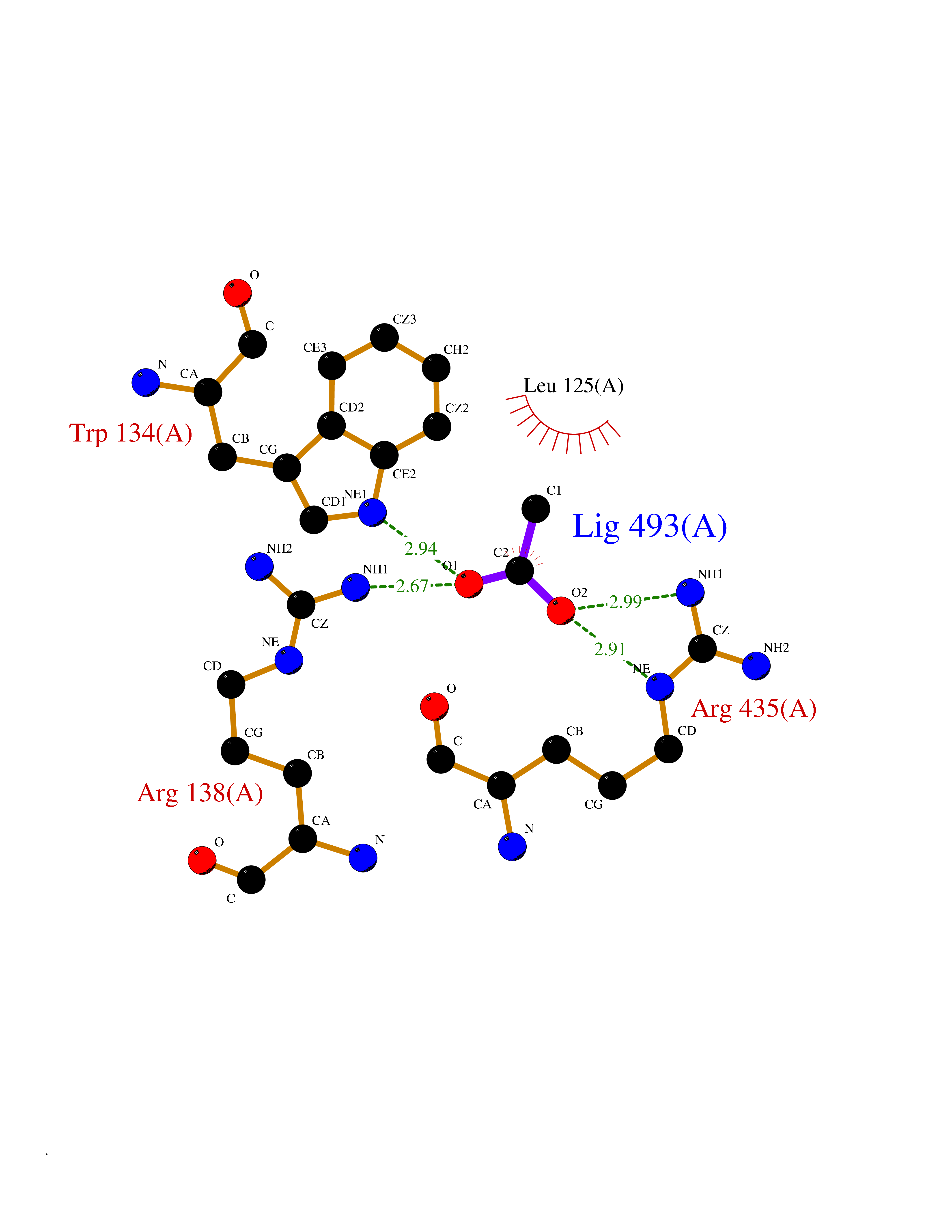 Ligplot Map 3D Binding mode Sequence RVLQDVFLDWAKKYGPVVRVNVFHKTSVIVTSPESVKKFLMSTKYNKDSKMYRALQTVFGERLFGQGLVSECNYERWHKQRRVIDLAFSRSSLVSLMETFNEKAEQLVEILEAKADGQTPVSMQDMLTYTAMDILAKAAFGMETSMLLGAQKPLSQAVKLMLEGITASRNTKRKQLREVRESIRFLRQVGRDWVQRRREALKRGEEVPADILTQILKAEEGAQDDEGLLDNFVTFFIAGHETSANHLAFTVMELSRQPEIVARLQAEVDEVIGSKRYLDFEDLGRLQYLSQVLKESLRLYPPAWGTFRLLEEETLIDGVRVPGNTPLLFSTYVMGRMDTYFEDPLTFNPDRFGPGAPKPRFTYFPFSLGHRSCIGQQFAQMEVKVVMAKLLQRLEFRLVPGQRFGLQEQATLKPLDPVLCTLRPRGW Hydrogen bonds contact Hydrophobic contact | ||||
| 90 | Voltage-gated potassium channel Kv3.1 (KCNC1) | 7PHI | 4.37 | |
Target general information Gen name KCNC1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Voltage-gated potassium channel subunit Kv4; Voltage-gated potassium channel subunit Kv3.1; Potassium voltage-gated channel subfamily C member 1; NGK2 Protein family Potassium channel family, C (Shaw) (TC 1.A.1.2) subfamily, Kv3.1/KCNC1 sub-subfamily Biochemical class Voltage-gated ion channel Function The channel opens in response to the voltage difference across the membrane, forming a potassium-selective channel through which potassium ions pass in accordance with their electrochemical gradient. Can form functional homotetrameric channels and heterotetrameric channels that contain variable proportions of KCNC2, and possibly other family members as well. Contributes to fire sustained trains of very brief action potentials at high frequency in pallidal neurons. Voltage-gated potassium channel that plays an important role in the rapid repolarization of fast-firing brain neurons. Related diseases Epilepsy, progressive myoclonic 7 (EPM7) [MIM:616187]: A form of progressive myoclonic epilepsy, a clinically and genetically heterogeneous group of disorders defined by the combination of action and reflex myoclonus, other types of epileptic seizures, and progressive neurodegeneration and neurocognitive impairment. EPM7 is an autosomal dominant form characterized by myoclonic epilepsy apparent in the first or second decades of life. Cognitive function may decline in some patients. {ECO:0000269|PubMed:25401298}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB06637; DB00228; DB01110; DB01069 Interacts with Q9UBY5; P55061; A5PKU2 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Cell projection; Disease variant; Epilepsy; Glycoprotein; Ion channel; Ion transport; Membrane; Neurodegeneration; Phosphoprotein; Potassium; Potassium channel; Potassium transport; Proteomics identification; Reference proteome; Synapse; Transmembrane; Transmembrane helix; Transport; Voltage-gated channel Protein physicochemical properties Chain ID A,B,C Molecular weight (Da) 136345 Length 1179 Aromaticity 0.15 Instability index 38.64 Isoelectric point 8.62 Charge (pH=7) 15.03 2D Binding mode Binding energy (Kcal/mol) -5.96 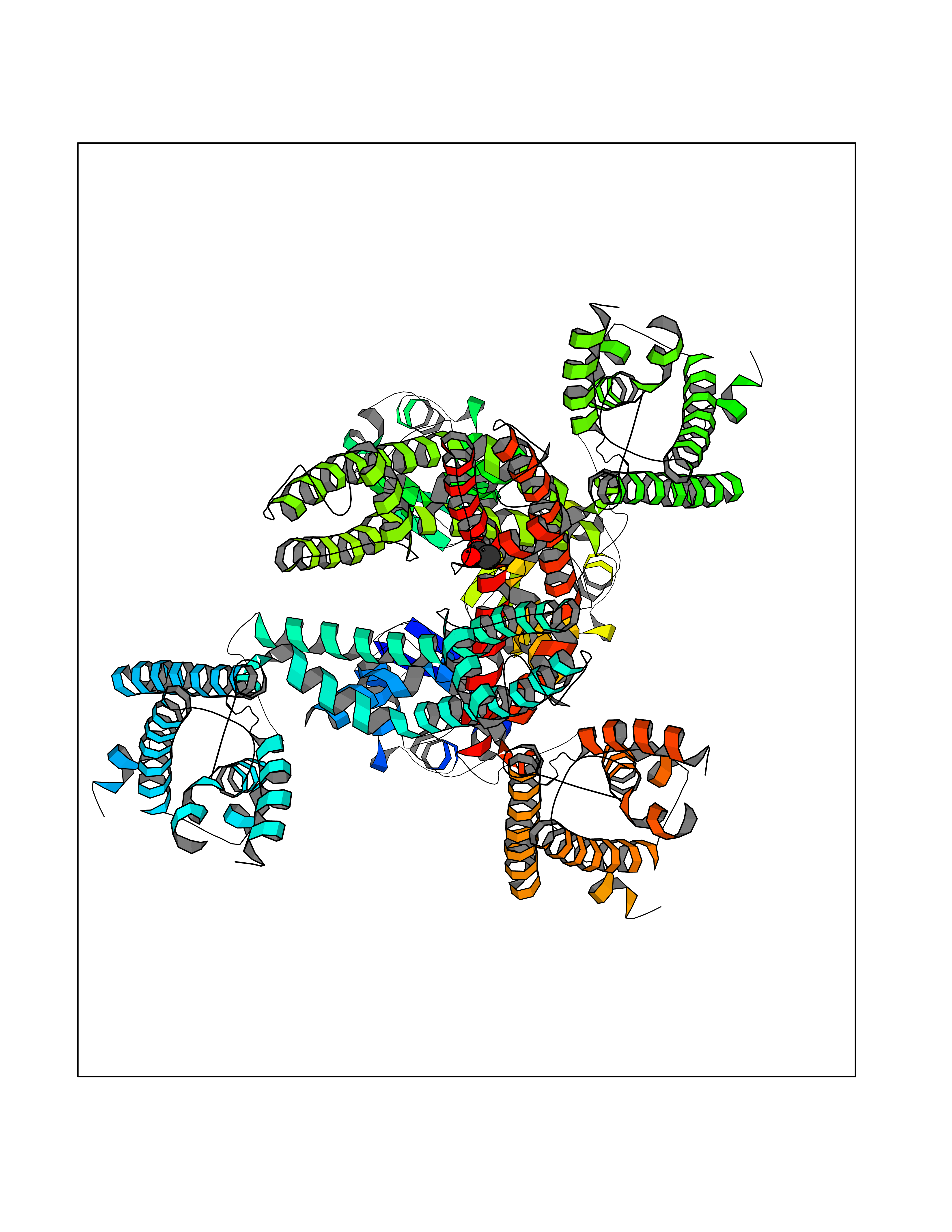 Molscript Map 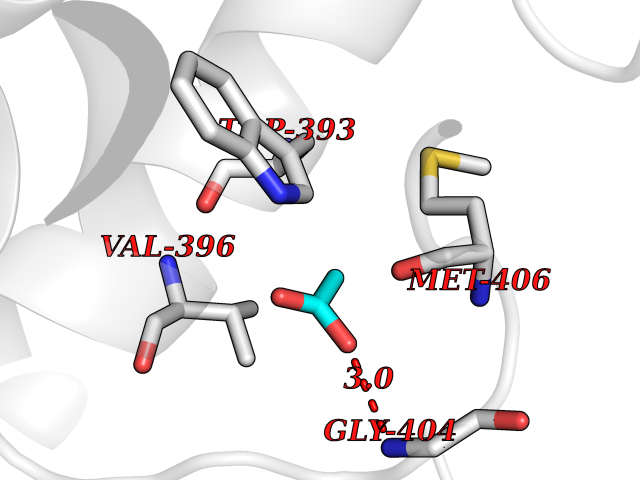 Pymol Map 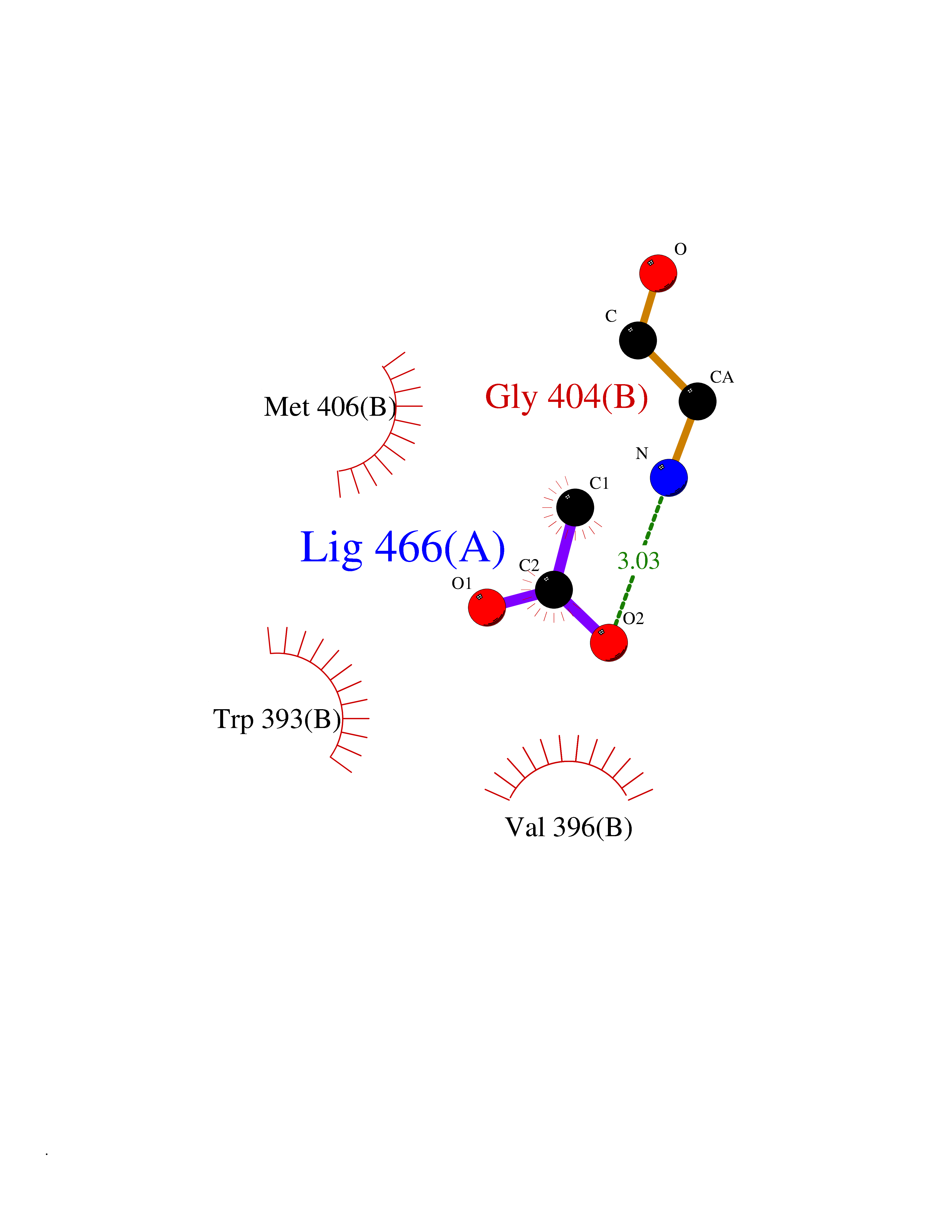 Ligplot Map 3D Binding mode Sequence SERIVINVGGTRHQTHRSTLRTLPGTRLAWLAEPDAHSHFDYDPRADEFFFDRHPGVFAHILNYYRTGKLHCPADVCGPLYEEELAFWGIDETDVEPCCWMTYRQHRDAEEALDRRWQPRIWALFEDPYSSRYARYVAFASLFFILVSITTFCLETHERFNPIVNKTYYREAETEAFLTYIEGVCVVWFTFEFLMRVIFCPNKVEFIKNSLNIIDFVAILPFYLEVGLSKAAKDVLGFLRVVRFVRILRIFKLTRHFVGLRVLGHTLRASTNEFLLLIIFLALGVLIFATMIYYAERIGAQPNDPSASEHTHFKNIPIGFWWAVVTMTTLGYGDMYPQTWSGMLVGALCALAGVLTIAMPVPVIVNNFGMYYSLAMAKQKLPKKKKKHIPRPPSERIVINVGGTRHQTHRSTLRTLPGTRLAWLAEPDAHSHFDYDPRADEFFFDRHPGVFAHILNYYRTGKLHCPADVCGPLYEEELAFWGIDETDVEPCCWMTYRQHRDAEEALDRRWQPRIWALFEDPYSSRYARYVAFASLFFILVSITTFCLETHERFNPIVNKTYYREAETEAFLTYIEGVCVVWFTFEFLMRVIFCPNKVEFIKNSLNIIDFVAILPFYLEVGLSKAAKDVLGFLRVVRFVRILRIFKLTRHFVGLRVLGHTLRASTNEFLLLIIFLALGVLIFATMIYYAERIGAQPNDPSASEHTHFKNIPIGFWWAVVTMTTLGYGDMYPQTWSGMLVGALCALAGVLTIAMPVPVIVNNFGMYYSLAMAKQKLPKKKKKHIPRPPSERIVINVGGTRHQTHRSTLRTLPGTRLAWLAEPDAHSHFDYDPRADEFFFDRHPGVFAHILNYYRTGKLHCPADVCGPLYEEELAFWGIDETDVEPCCWMTYRQHRDAEEALDRRWQPRIWALFEDPYSSRYARYVAFASLFFILVSITTFCLETHERFNPIVNKTYYREAETEAFLTYIEGVCVVWFTFEFLMRVIFCPNKVEFIKNSLNIIDFVAILPFYLEVGLSKAAKDVLGFLRVVRFVRILRIFKLTRHFVGLRVLGHTLRASTNEFLLLIIFLALGVLIFATMIYYAERIGAQPNDPSASEHTHFKNIPIGFWWAVVTMTTLGYGDMYPQTWSGMLVGALCALAGVLTIAMPVPVIVNNFGMYYSLAMAKQKLPKKKKKHIPRPP Hydrogen bonds contact Hydrophobic contact | ||||
| 91 | Phenylethanolamine N-methyltransferase (PNMT) | 2G72 | 4.37 | |
Target general information Gen name PNMT Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms PNMTase; PENT; Noradrenaline N-methyltransferase Protein family Class I-like SAM-binding methyltransferase superfamily, NNMT/PNMT/TEMT family Biochemical class NA Function Converts noradrenaline to adrenaline. Related diseases A chromosomal aberration involving TRIM24/TIF1 is found in papillary thyroid carcinomas (PTCs). Translocation t(7;10)(q32;q11) with RET. The translocation generates the TRIM24/RET (PTC6) oncogene. {ECO:0000269|PubMed:10439047}. Drugs (DrugBank ID) DB08129; DB08128; DB07739; DB07798; DB07747; DB03468; DB08550; DB03824; DB04273; DB07906; DB07597; DB09571; DB00968; DB08631; DB01752; DB08654 Interacts with Q9P2G9-2; Q8TBB1 EC number EC 2.1.1.28 Uniprot keywords 3D-structure; Catecholamine biosynthesis; Direct protein sequencing; Methyltransferase; Phosphoprotein; Proteomics identification; Reference proteome; S-adenosyl-L-methionine; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 29198.9 Length 264 Aromaticity 0.09 Instability index 54.33 Isoelectric point 5.91 Charge (pH=7) -3.69 2D Binding mode Binding energy (Kcal/mol) -5.96  Molscript Map 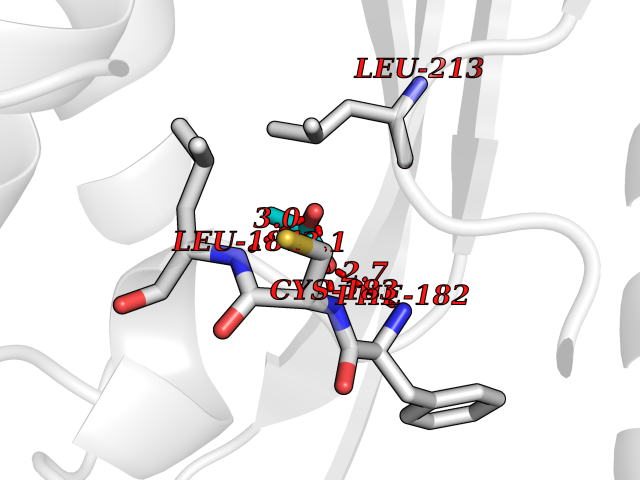 Pymol Map 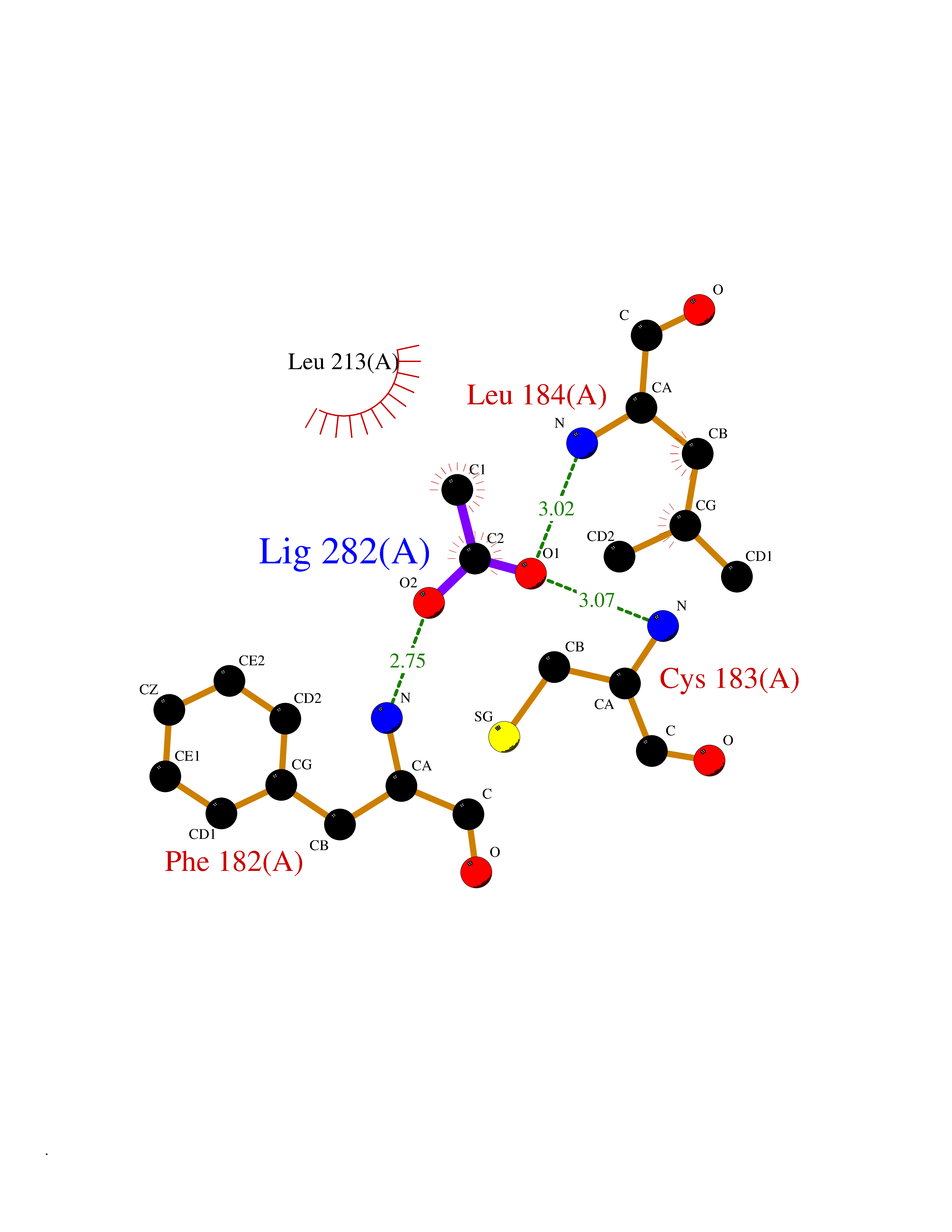 Ligplot Map 3D Binding mode Sequence APGQAAVASAYQRFEPRAYLRNNYAPPRGDLCNPNGVGPWKLRCLAQTFATGEVSGRTLIDIGSGPTVYQLLSACSHFEDITMTDFLEVNRQELGRWLQEEPGAFNWSMYSQHACLIEGKGECWQDKERQLRARVKRVLPIDVHQPQPLGAGSPAPLPADALVSAFCLEAVSPDLASFQRALDHITTLLRPGGHLLLIGALEESWYLAGEARLTVVPVSEEEVREALVRSGYKVRDLRTYIMPAHLQTGVDDVKGVFFAWAQKV Hydrogen bonds contact Hydrophobic contact | ||||
| 92 | Hepatitis A virus cellular receptor 2 (TIM3) | 7M3Z | 4.37 | |
Target general information Gen name Hepatitis A virus HAVCR2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms TIMD3; TIMD-3; TIM3; TIM-3; T-cell membrane protein 3; T-cell immunoglobulin mucin receptor 3; T-cell immunoglobulin and mucin domain-containing protein 3; HAVcr-2; HAVCR2; CD366 Protein family Immunoglobulin superfamily, TIM family Biochemical class Immunoglobulin Function Generally accepted to have an inhibiting function. Reports on stimulating functions suggest that the activity may be influenced by the cellular context and/or the respective ligand. Regulates macrophage activation. Inhibits T-helper type 1 lymphocyte (Th1)-mediated auto- and alloimmune responses and promotes immunological tolerance. In CD8+ cells attenuates TCR-induced signaling, specifically by blocking NF-kappaB and NFAT promoter activities resulting in the loss of IL-2 secretion. The function may implicate its association with LCK proposed to impair phosphorylation of TCR subunits, and/or LGALS9-dependent recruitment of PTPRC to the immunological synapse. In contrast, shown to activate TCR-induced signaling in T-cells probably implicating ZAP70, LCP2, LCK and FYN. Expressed on Treg cells can inhibit Th17 cell responses. Receptor for LGALS9. Binding to LGALS9 is believed to result in suppression of T-cell responses; the resulting apoptosis of antigen-specific cells may implicate HAVCR2 phosphorylation and disruption of its association with BAG6. Binding to LGALS9 is proposed to be involved in innate immune response to intracellular pathogens. Expressed on Th1 cells interacts with LGALS9 expressed on Mycobacterium tuberculosis-infected macrophages to stimulate antibactericidal activity including IL-1 beta secretion and to restrict intracellular bacterial growth. However, the function as receptor for LGALS9 has been challenged. Also reported to enhance CD8+ T-cell responses to an acute infection such as by Listeria monocytogenes. Receptor for phosphatidylserine (PtSer); PtSer-binding is calcium-dependent. May recognize PtSer on apoptotic cells leading to their phagocytosis. Mediates the engulfment of apoptotic cells by dendritic cells. Expressed on T-cells, promotes conjugation but not engulfment of apoptotic cells. Expressed on dendritic cells (DCs) positively regulates innate immune response and in synergy with Toll-like receptors promotes secretion of TNF-alpha. In tumor-imfiltrating DCs suppresses nucleic acid-mediated innate immune repsonse by interaction with HMGB1 and interfering with nucleic acid-sensing and trafficking of nucleid acids to endosomes. Expressed on natural killer (NK) cells acts as a coreceptor to enhance IFN-gamma production in response to LGALS9. In contrast, shown to suppress NK cell-mediated cytotoxicity. Negatively regulates NK cell function in LPS-induced endotoxic shock. Cell surface receptor implicated in modulating innate and adaptive immune responses. Related diseases May be involved in T-cell exhaustion associated with chronic viral infections such as with human immunodeficiency virus (HIV) and hepatitic C virus (HCV). {ECO:0000269|PubMed:19001139, ECO:0000269|PubMed:19587053}.; DISEASE: T-cell lymphoma, subcutaneous panniculitis-like (SPTCL) [MIM:618398]: An uncommon form of T-cell non-Hodgkin lymphoma, in which cytotoxic CD8+ T-cells infiltrate subcutaneous adipose tissue, and rimming adipocytes in a lace-like pattern. Affected individuals typically present with multiple subcutaneous nodules, systemic B-cell symptoms, and, in a subset of cases, autoimmune disorders, most commonly systemic lupus erythematosus. A subset of patients develop hemophagocytic lymphohistiocytosis. SPTCL transmission pattern is consistent with autosomal recessive inheritance with incomplete penetrance. {ECO:0000269|PubMed:30374066, ECO:0000269|PubMed:30792187, ECO:0000269|Ref.2}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P13688; Q96IW7; Q8N2M4 EC number NA Uniprot keywords 3D-structure; Adaptive immunity; Alternative splicing; Cell junction; Cell membrane; Disease variant; Disulfide bond; Glycoprotein; Immunity; Immunoglobulin domain; Inflammatory response; Innate immunity; Membrane; Metal-binding; Phosphoprotein; Proteomics identification; Reference proteome; Signal; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 12286.9 Length 109 Aromaticity 0.12 Instability index 26.55 Isoelectric point 5.04 Charge (pH=7) -2.58 2D Binding mode Binding energy (Kcal/mol) -5.96  Molscript Map 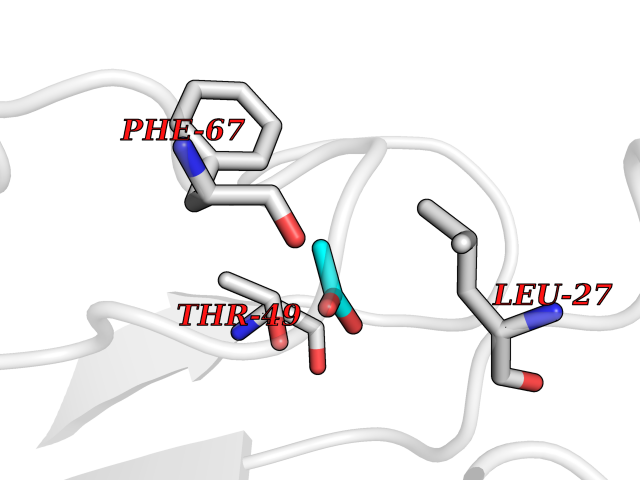 Pymol Map 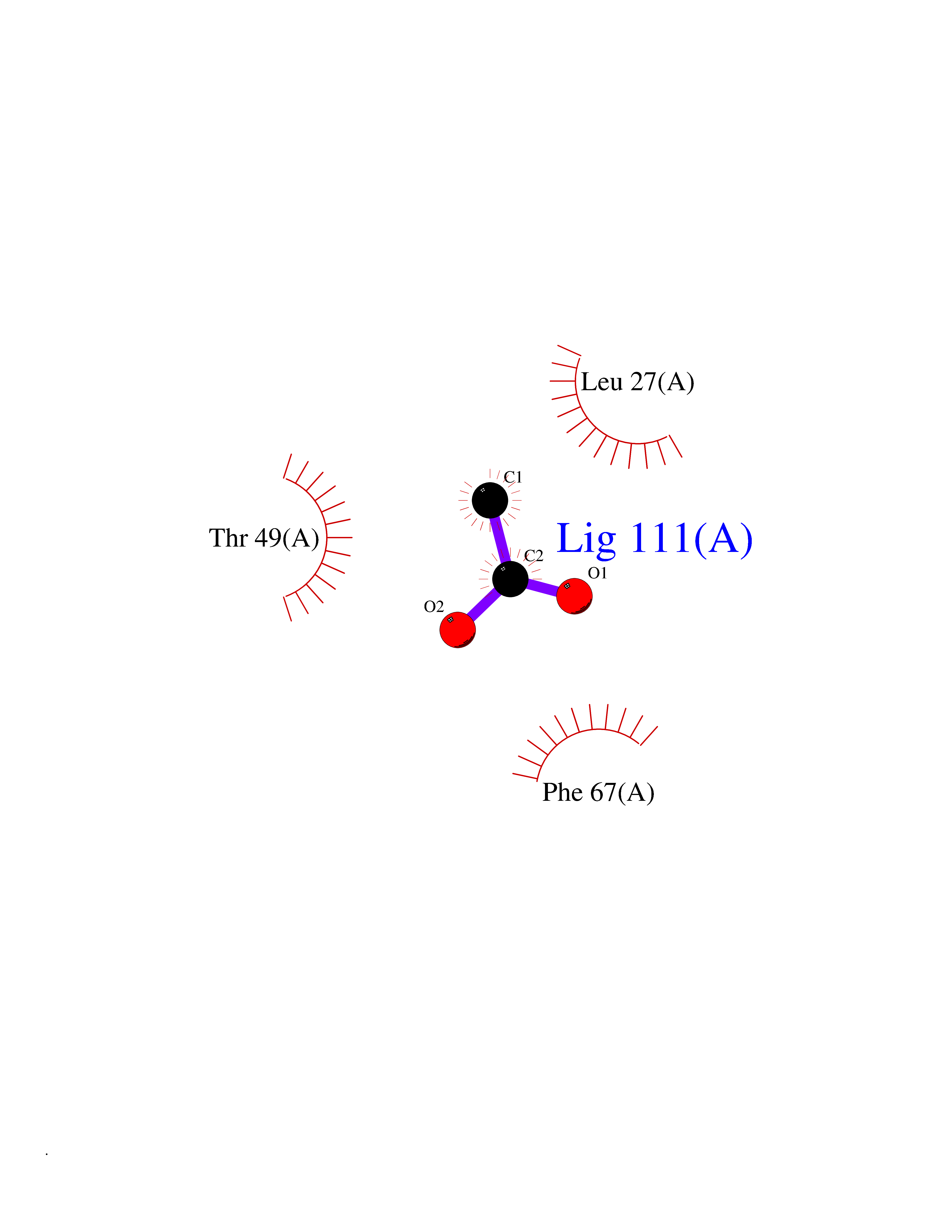 Ligplot Map 3D Binding mode Sequence SEVEYRAEVGQNAYLPCFYTPAAPGNLVPVCWGKGACPVFECGNVVLRTDERDVNYWTSRYWLNGDFRKGDVSLTIENVTLADSGIYCCRIQIPGIMNDEKFNLKLVIK Hydrogen bonds contact Hydrophobic contact | ||||
| 93 | Neuronal acetylcholine receptor beta-2 (CHRNB2) | 6CNJ | 4.37 | |
Target general information Gen name CHRNB2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Nicotinic acetylcholine receptor beta2; Nicotinic acetylcholine receptor beta 2-subunit protein; CHRNB2; Beta-2 nAChR; Alpha-4/beta-2 nicotinic receptor Protein family Ligand-gated ion channel (TC 1.A.9) family, Acetylcholine receptor (TC 1.A.9.1) subfamily, Beta-2/CHRNB2 sub-subfamily Biochemical class Neurotransmitter receptor Function After binding acetylcholine, the AChR responds by an extensive change in conformation that affects all subunits and leads to opening of an ion-conducting channel across the plasma membrane permeable to sodiun ions. Related diseases Epilepsy, nocturnal frontal lobe, 3 (ENFL3) [MIM:605375]: An autosomal dominant focal epilepsy characterized by nocturnal seizures with hyperkinetic automatisms and poorly organized stereotyped movements. {ECO:0000269|PubMed:11062464, ECO:0000269|PubMed:11104662}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00572; DB00237; DB00565; DB09028; DB01245; DB00514; DB07720; DB00898; DB00753; DB00657; DB00333; DB00184; DB00981; DB05458; DB05855; DB05740; DB00747; DB00202; DB01273 Interacts with P43681-1; P30532 EC number NA Uniprot keywords 3D-structure; Cell membrane; Disease variant; Disulfide bond; Epilepsy; Glycoprotein; Ion channel; Ion transport; Ligand-gated ion channel; Membrane; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Signal; Synapse; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A,B Molecular weight (Da) 84601.2 Length 728 Aromaticity 0.13 Instability index 39.72 Isoelectric point 5.86 Charge (pH=7) -9.84 2D Binding mode Binding energy (Kcal/mol) -5.96  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence ETRAHAEERLLKKLFSGYNKWSRPVANISDVVLVRFGLSIAQLIDVDEKNQMMTTNVWVKQEWHDYKLRWDPADYENVTSIRIPSELIWRPDIVLYNNADGDFAVTHLTKAHLFHDGRVQWTPPAIYKSSCSIDVTFFPFDQQNCTMKFGSWTYDKAKIDLVNMHSRVDQLDFWESGEWVIVDAVGTYNTRKYECCAEIYPDITYAFVIRRLPLFYTINLIIPCLLISCLTVLVFYLPSECGEKITLCISVLLSLTVFLLLITEIIPSTSLVIPLIGEYLLFTMIFVTLSIVITVFVLNVHHRSPRTHTMPTWVRRVFLDIVPRLLLMKRFERSVKEDWKYVAMVIDRIFLWMFIIVCLLGTVGLFLPPWDTEERLVEHLLDPSRYNKLIRPATNGSELVTVQLMVSLAQLISVHEREQIMTTNVWLTQEWEDYRLTWKPEEFDNMKKVRLPSKHIWLPDVVLYNNADGMYEVSFYSNAVVSYDGSIFWLPPAIYKSACKIEVKHFPFDQQNCTMKFRSWTYDRTEIDLVLKSEVASLDDFTPSGEWDIVALPGRRNENPDDSTYVDITYDFIIRRKPLFYTINLIIPCVLITSLAILVFYLPSDCGEKMTLCISVLLALTVFLLLISKIVPPTSLDVPLVGKYLMFTMVLVTFSIVTSVCVLNVHHRSPTTHTMAPWVKVVFLEKLPALLFMQQSVSEDWKYVAMVIDRLFLWIFVFVCVFGTIGMF Hydrogen bonds contact Hydrophobic contact | ||||
| 94 | Folate receptor alpha (FOLR1) | 4LRH | 4.37 | |
Target general information Gen name FOLR1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Ovarian tumorassociated antigen MOv18; KB cells FBP; Folate receptor, adult; Folate receptor 1; FRalpha; FOLR1; Adult folatebinding protein Protein family Folate receptor family Biochemical class Folate receptor Function Binds to folate and reduced folic acid derivatives and mediates delivery of 5-methyltetrahydrofolate and folate analogs into the interior of cells. Has high affinity for folate and folic acid analogs at neutral pH. Exposure to slightly acidic pHafter receptor endocytosis triggers a conformation change that strongly reduces its affinity for folates and mediates their release. Required for normal embryonic development and normal cell proliferation. Related diseases Neurodegeneration due to cerebral folate transport deficiency (NCFTD) [MIM:613068]: An autosomal recessive neurodegenerative disorder resulting from brain-specific folate deficiency early in life. Onset is apparent in late infancy with severe developmental regression, movement disturbances, epilepsy and leukodystrophy. {ECO:0000269|PubMed:19732866}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB05595; DB00158; DB00563; DB12489; DB15413; DB05168 Interacts with Q8N357 EC number NA Uniprot keywords 3D-structure; Cell membrane; Cytoplasmic vesicle; Direct protein sequencing; Disulfide bond; Endosome; Folate-binding; Glycoprotein; GPI-anchor; Lipoprotein; Membrane; Neurodegeneration; Proteomics identification; Receptor; Reference proteome; Secreted; Signal; Transport Protein physicochemical properties Chain ID A Molecular weight (Da) 24216 Length 207 Aromaticity 0.13 Instability index 49.36 Isoelectric point 8.14 Charge (pH=7) 3.41 2D Binding mode Binding energy (Kcal/mol) -5.96  Molscript Map 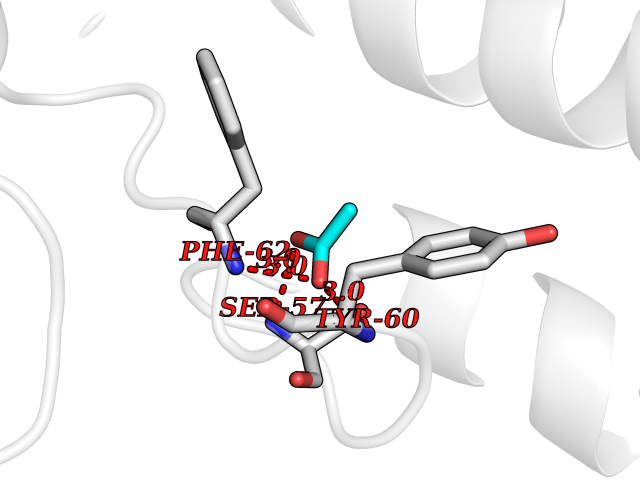 Pymol Map 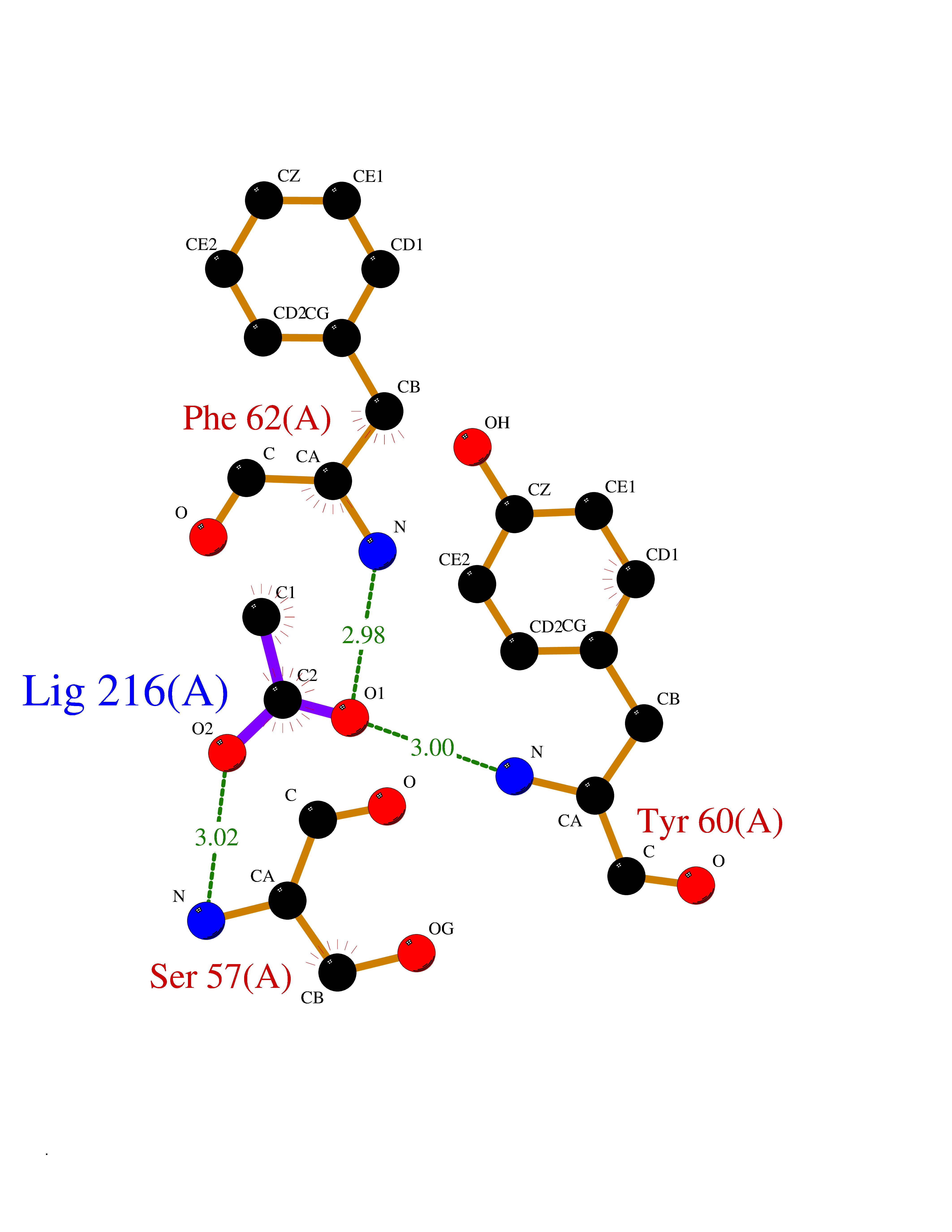 Ligplot Map 3D Binding mode Sequence RTELLNVCMNAKHHKEKPGPEDKLHEQCRPWRKNACCSTNTSQEAHKDVSYLYRFNWNHCGEMAPACKRHFIQDTCLYECSPNLGPWIQQVDQSWRKERVLNVPLCKEDCEQWWEDCRTSYTCKSNWHKGWNWTSGFNKCAVGAACQPFHFYFPTPTVLCNEIWTHSYKVSNYSRGSGRCIQMWFDPAQGNPNEEVARFYAAAMSGT Hydrogen bonds contact Hydrophobic contact | ||||
| 95 | Tissue-type plasminogen activator (PLAT) | 1RTF | 4.37 | |
Target general information Gen name PLAT Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms TPA; T-plasminogen activator; T-PA; Reteplase; Alteplase Protein family Peptidase S1 family Biochemical class Peptidase Function By controlling plasmin-mediated proteolysis, it plays an important role in tissue remodeling and degradation, in cell migration and many other physiopathological events. Plays a direct role in facilitating neuronal migration. Converts the abundant, but inactive, zymogen plasminogen to plasmin by hydrolyzing a single Arg-Val bond in plasminogen. Related diseases Increased activity of TPA results in increased fibrinolysis of fibrin blood clots that is associated with excessive bleeding. Defective release of TPA results in hypofibrinolysis that can lead to thrombosis or embolism. {ECO:0000269|PubMed:1762144}. Drugs (DrugBank ID) DB07684; DB00513; DB09228; DB09213; DB06404; DB01088; DB16701 Interacts with P05155 EC number EC 3.4.21.68 Uniprot keywords 3D-structure; Alternative splicing; Cleavage on pair of basic residues; Direct protein sequencing; Disulfide bond; EGF-like domain; Glycoprotein; Hydrolase; Kringle; Pharmaceutical; Plasminogen activation; Protease; Proteomics identification; Reference proteome; Repeat; Secreted; Serine protease; Signal; Zymogen Protein physicochemical properties Chain ID B Molecular weight (Da) 26271.7 Length 234 Aromaticity 0.09 Instability index 40.44 Isoelectric point 5.83 Charge (pH=7) -5.54 2D Binding mode Binding energy (Kcal/mol) -5.96  Molscript Map 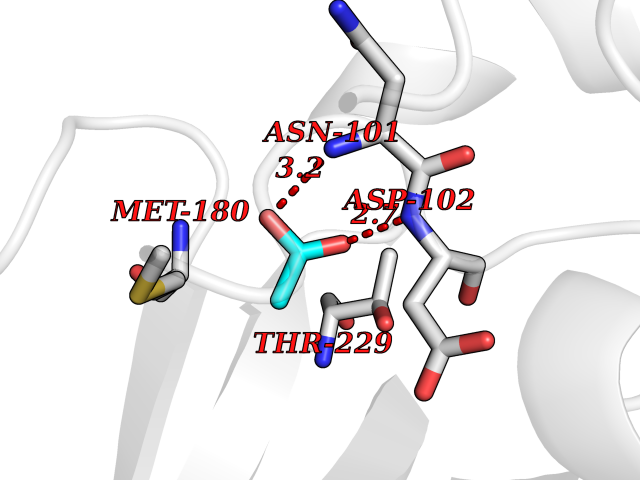 Pymol Map 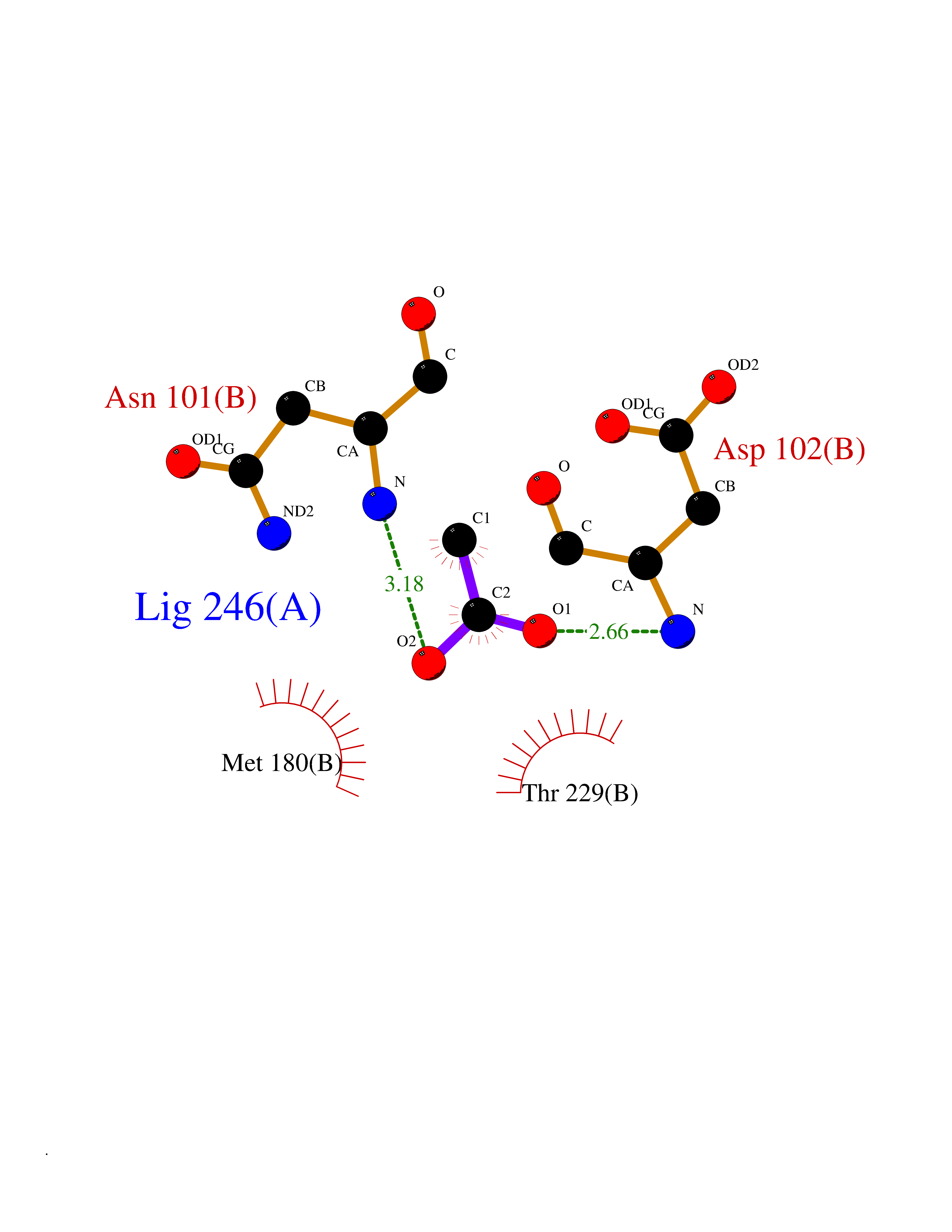 Ligplot Map 3D Binding mode Sequence CGLRQYIKGGLFADIASHPWQAAIFAKGERFLCGGILISSCWILSAAHCFPPHHLTVILGRTYRVVPGEEEQKFEVEKYIVHKEFDDDTYDNDIALLQLKRCAQESSVVRTVCLPPADLQLPDWTECELSGYGKHEALSPFYSERLKEAHVRLYPSSRCQHLLNRTVTDNMLCAGDNLHDACQGDSGGPLVCLNDGRMTLVGIISWGLGCQKDVPGVYTKVTNYLDWIRDNMRP Hydrogen bonds contact Hydrophobic contact | ||||
| 96 | Cholesterol desmolase (CYP11A1) | 3N9Y | 4.37 | |
Target general information Gen name CYP11A1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms P450(scc); Cytochrome P450(scc); Cytochrome P450 11A1; Cholesterol side-chain cleavage enzyme, mitochondrial; CYPXIA1; CYP11A Protein family Adrenodoxin/putidaredoxin family Biochemical class Paired donor oxygen oxidoreductase Function Catalyzes the side-chain cleavage reaction of cholesterol to pregnenolone, the precursor of most steroid hormones. Related diseases Leukocyte adhesion deficiency 1 (LAD1) [MIM:116920]: LAD1 patients have recurrent bacterial infections and their leukocytes are deficient in a wide range of adhesion-dependent functions. {ECO:0000269|PubMed:1346613, ECO:0000269|PubMed:1347532, ECO:0000269|PubMed:1352501, ECO:0000269|PubMed:1590804, ECO:0000269|PubMed:1694220, ECO:0000269|PubMed:1968911, ECO:0000269|PubMed:20529581, ECO:0000269|PubMed:20549317, ECO:0000269|PubMed:7509236, ECO:0000269|PubMed:7686755, ECO:0000269|PubMed:9884339}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00648 Interacts with NA EC number EC 1.14.15.6 Uniprot keywords 2Fe-2S; 3D-structure; Acetylation; Cholesterol metabolism; Electron transport; Iron; Iron-sulfur; Lipid metabolism; Metal-binding; Mitochondrion; Phosphoprotein; Proteomics identification; Reference proteome; Steroid metabolism; Steroidogenesis; Sterol metabolism; Transit peptide; Transport Protein physicochemical properties Chain ID A,B Molecular weight (Da) 54754.5 Length 470 Aromaticity 0.13 Instability index 33.2 Isoelectric point 8.16 Charge (pH=7) 2.32 2D Binding mode Binding energy (Kcal/mol) -5.96 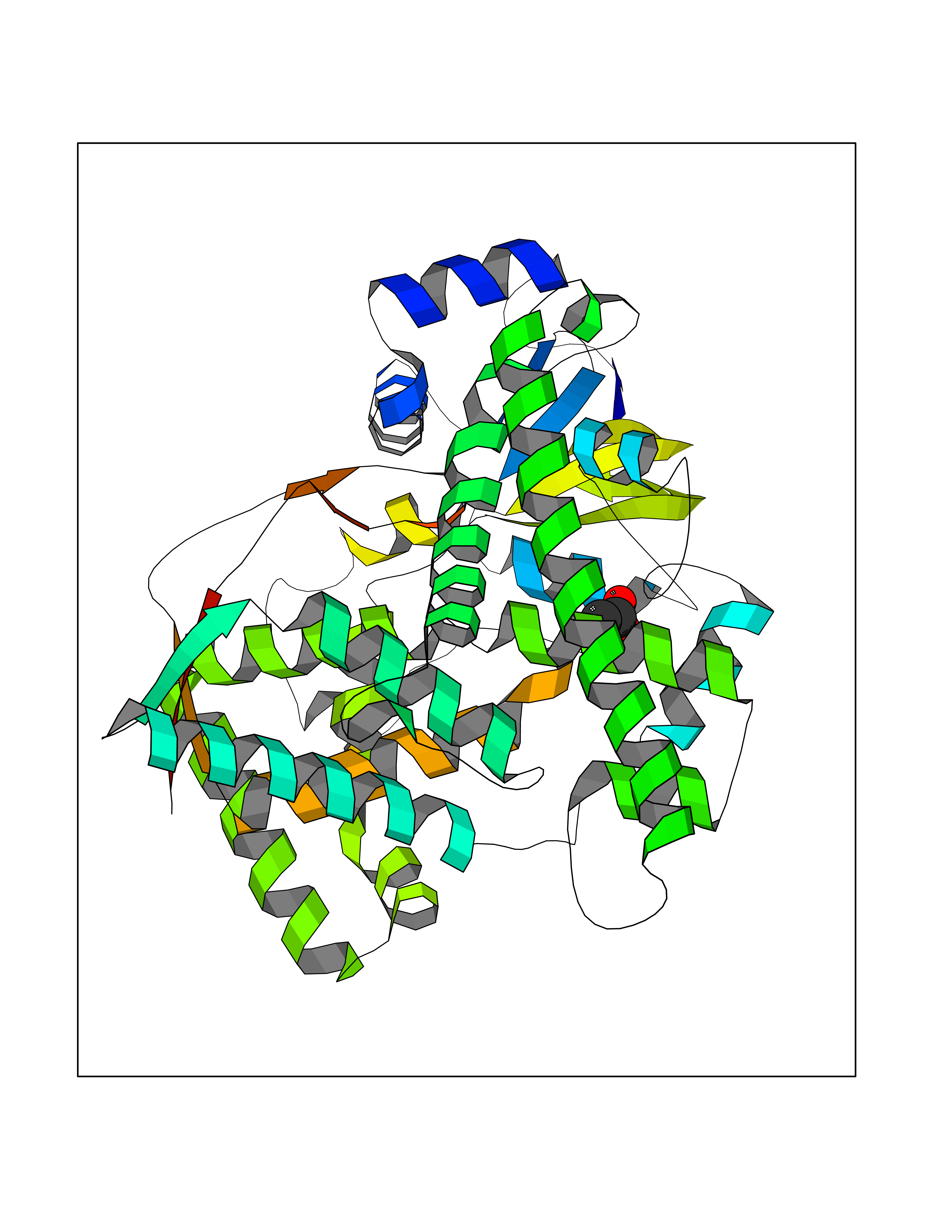 Molscript Map 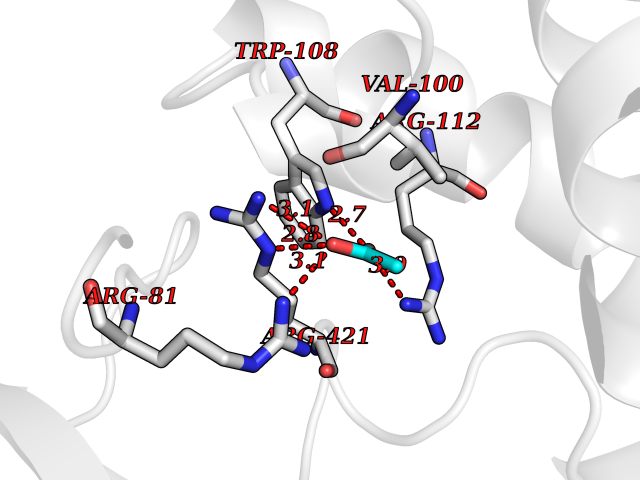 Pymol Map 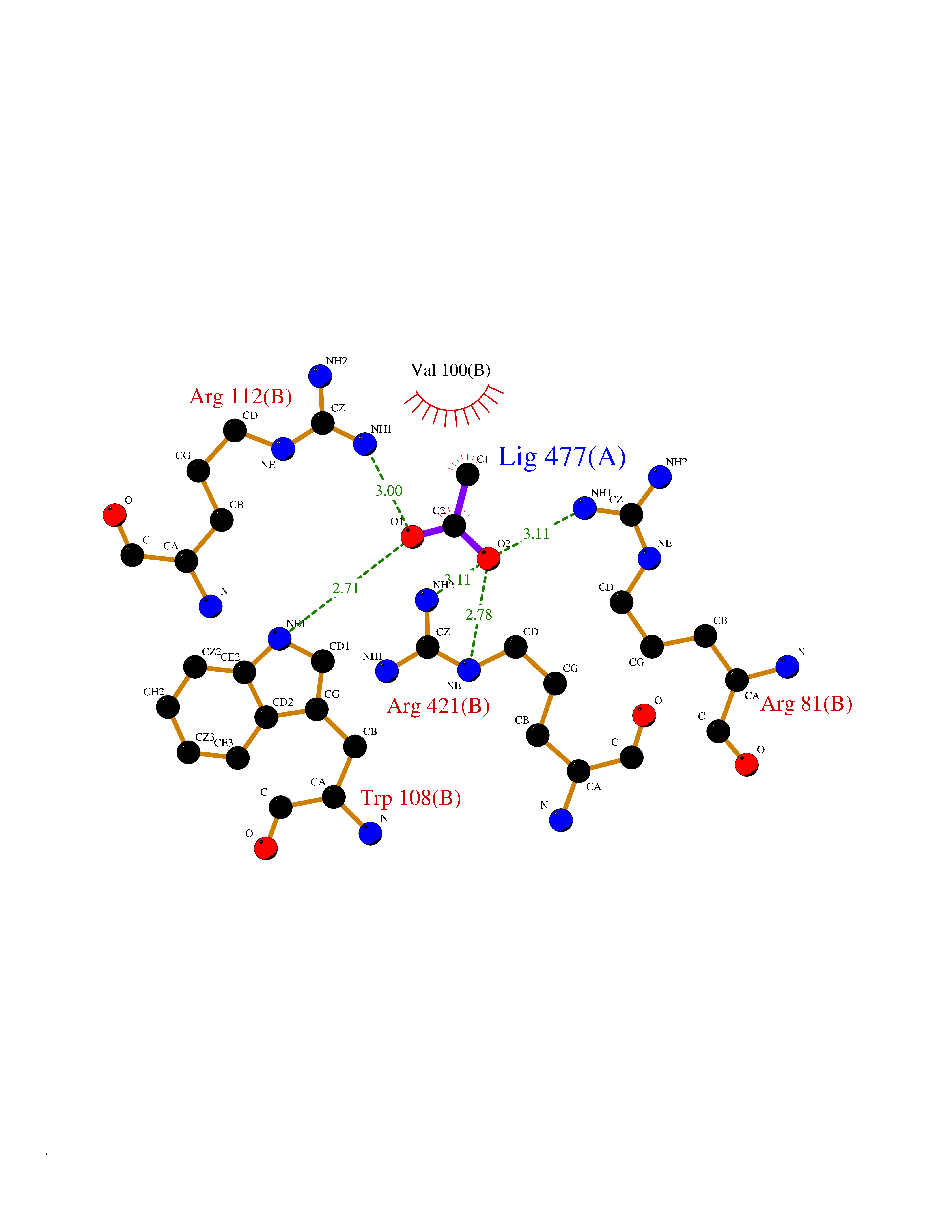 Ligplot Map 3D Binding mode Sequence PRPFNEIPSPGDNGWLNLYHFWRETGTHKVHLHHVQNFQKYGPIYREKLGNVESVYVIDPEDVALLFKSEGPNPERFLIPPWVAYHQYYQRPIGVLLKKSAAWKKDRVALNQEVMAPEATKNFLPLLDAVSRDFVSVLHRRIKKAGSGNYSGDISDDLFRFAFESITNVIFGERQGMLEEVVNPEAQRFIDAIYQMFHTSVPMLNLPPDLFRLFRTKTWKDHVAAWDVIFSKADIYTQNFYWELRQKGSVHHDYRGILYRLLGDSKMSFEDIKANVTEMLAGGVDTTSMTLQWHLYEMARNLKVQDMLRAEVLAARHQAQGDMATMLQLVPLLKASIKETLRLHPISVTLQRYLVNDLVLRDYMIPAKTLVQVAIYALGREPTFFFDPENFDPTRWLSKDKNITYFRNLGFGWGVRQCLGRRIAELEMTIFLINMLENFRVEIQHLSDVGTTFNLILMPEKPISFTFWPF Hydrogen bonds contact Hydrophobic contact | ||||
| 97 | Liver carboxylesterase (CES1) | 2H7C | 4.37 | |
Target general information Gen name CES1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Serine esterase 1; Monocyte/macrophage serine esterase; Human carboxylesterase 1; HMSE; HCE1; CES1; Brain carboxylesterase hBr1; Acyl coenzyme A:cholesterol acyltransferase Protein family Type-B carboxylesterase/lipase family Biochemical class Carboxylic ester hydrolase Function Involved in the detoxification of xenobiotics and in the activation of ester and amide prodrugs. Hydrolyzes aromatic and aliphatic esters, but has no catalytic activity toward amides or a fatty acyl coa ester. Related diseases Protoporphyria, erythropoietic, 1 (EPP1) [MIM:177000]: An autosomal recessive form of porphyria with onset usually before age 10 years. Porphyrias are inherited defects in the biosynthesis of heme, resulting in the accumulation and increased excretion of porphyrins or porphyrin precursors. They are classified as erythropoietic or hepatic, depending on whether the enzyme deficiency occurs in red blood cells or in the liver. Erythropoietic protoporphyria is marked by excessive protoporphyrin in erythrocytes, plasma, liver and feces, and by widely varying photosensitive skin changes ranging from a burning or pruritic sensation to erythema, edema and wheals. {ECO:0000269|PubMed:10942404, ECO:0000269|PubMed:11375302, ECO:0000269|PubMed:12063482, ECO:0000269|PubMed:12601550, ECO:0000269|PubMed:1376018, ECO:0000269|PubMed:15286165, ECO:0000269|PubMed:17196862, ECO:0000269|PubMed:1755842, ECO:0000269|PubMed:7910885, ECO:0000269|PubMed:8757534, ECO:0000269|PubMed:9211198, ECO:0000269|PubMed:9585598, ECO:0000269|PubMed:9740232}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07821; DB08224; DB03056; DB06442; DB01086; DB09061; DB14737; DB01101; DB02659; DB01410; DB00758; DB00907; DB04838; DB06695; DB00647; DB01452; DB00470; DB01039; DB14845; DB00875; DB11181; DB02161; DB00328; DB00762; DB00583; DB14009; DB00454; DB06693; DB00688; DB03721; DB01183; DB00198; DB09269; DB01599; DB09342; DB00881; DB14761; DB12404; DB06201; DB11362; DB00641; DB00382; DB00675; DB12095; DB09299; DB04795; DB00519; DB16349 Interacts with Q8IVF2-3; A8MQ03; Q15125; Q5T7V8; Q5T749; Q13113; Q9NRQ2; Q9NR31; O95231 EC number EC 3.1.1.1 Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; Direct protein sequencing; Disulfide bond; Endoplasmic reticulum; Glycoprotein; Hydrolase; Lipid droplet; Lipid metabolism; Phosphoprotein; Proteomics identification; Reference proteome; Serine esterase; Signal Protein physicochemical properties Chain ID A,B,C,D,E,F Molecular weight (Da) 58522.7 Length 531 Aromaticity 0.09 Instability index 35.86 Isoelectric point 6.05 Charge (pH=7) -5.56 2D Binding mode Binding energy (Kcal/mol) -5.96 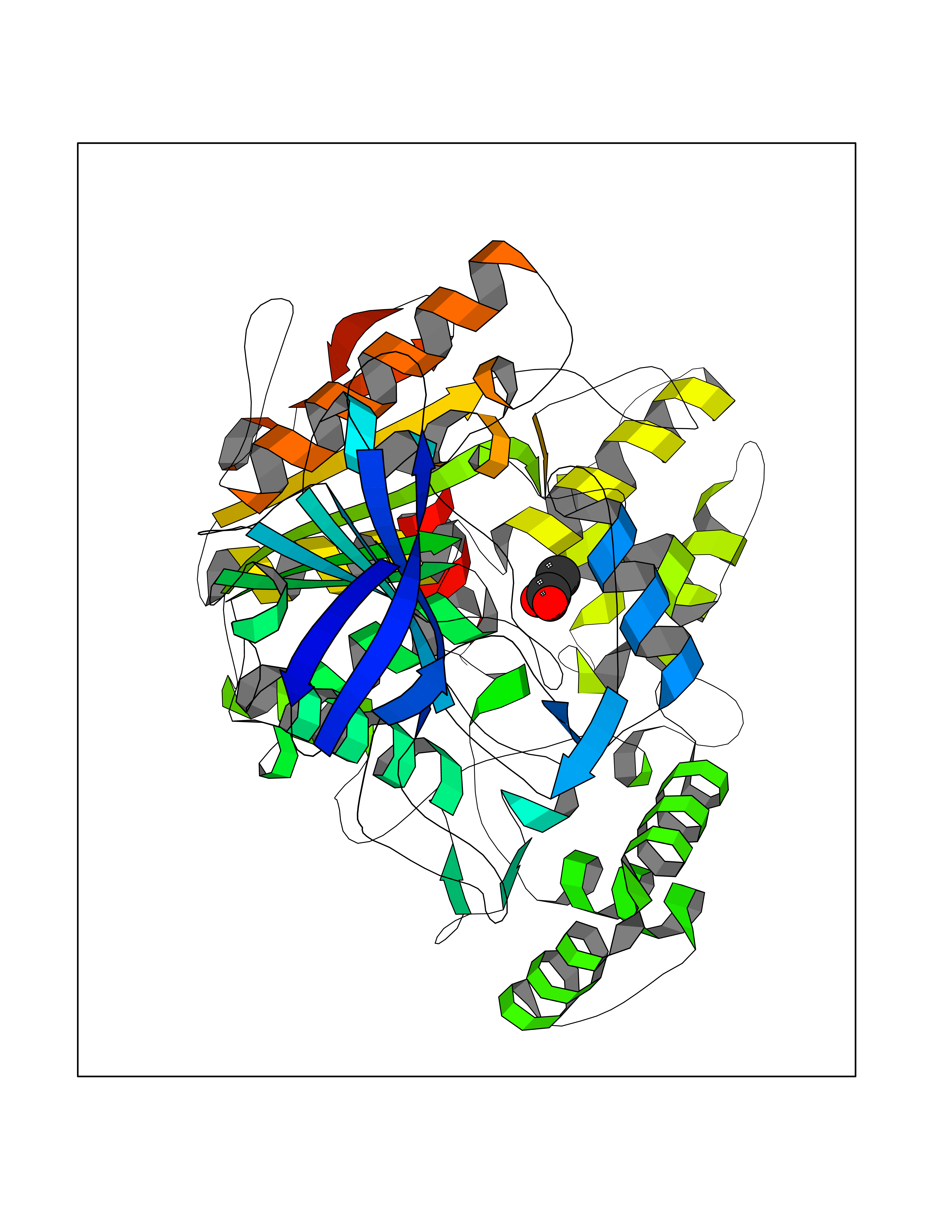 Molscript Map 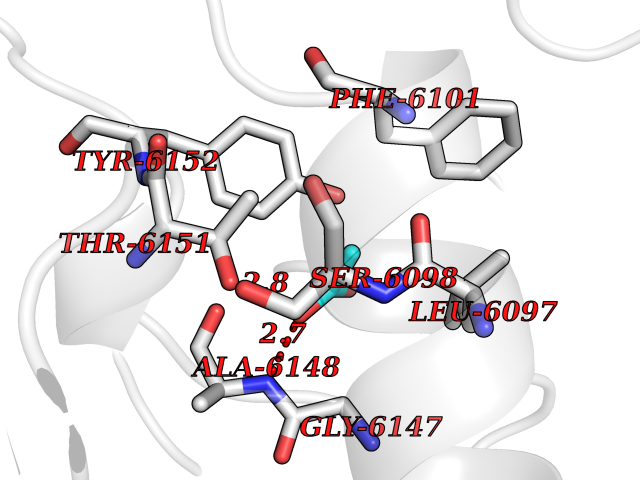 Pymol Map 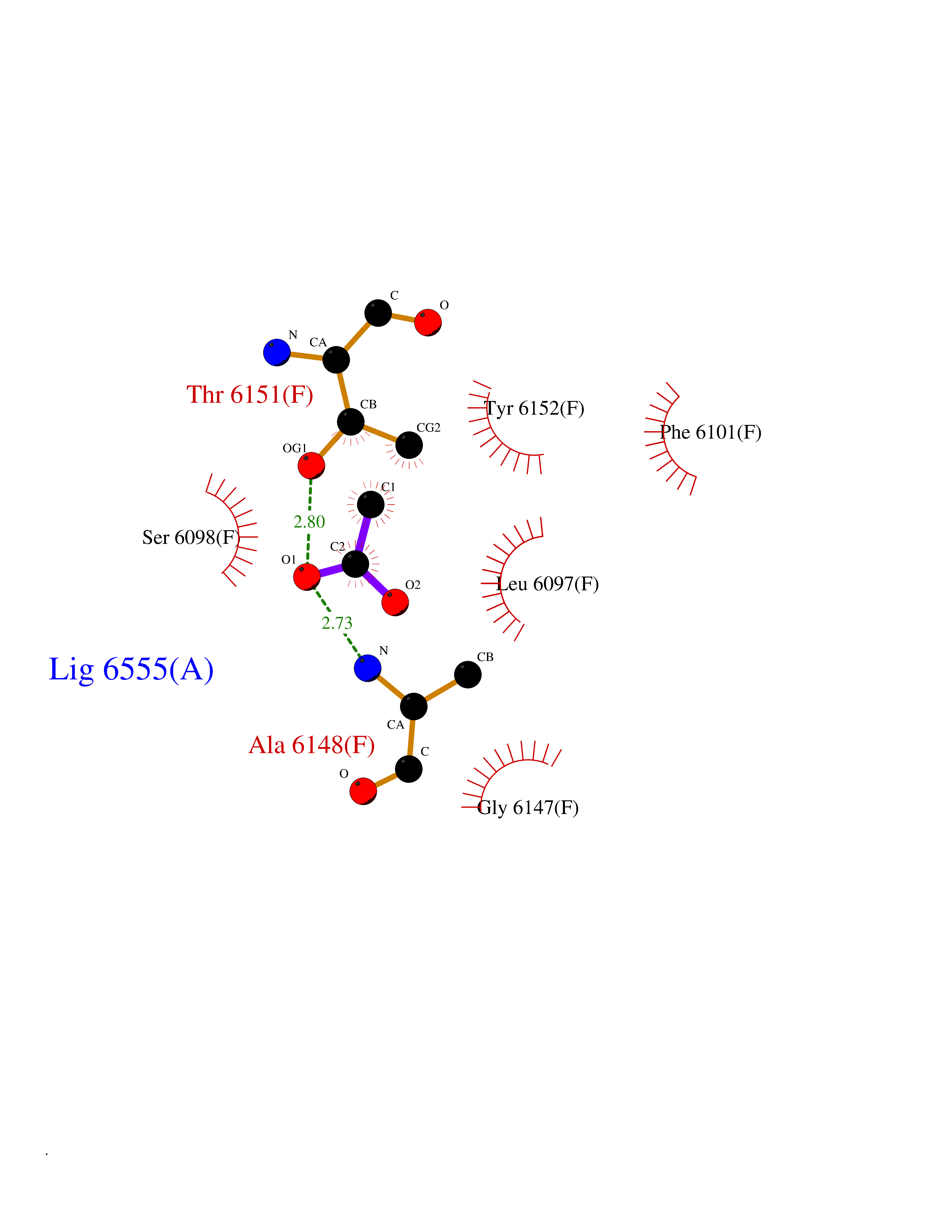 Ligplot Map 3D Binding mode Sequence SPPVVDTVHGKVLGKFVSLEGFAQPVAIFLGIPFAKPPLGPLRFTPPQPAEPWSFVKNATSYPPMCTQDPKAGQLLSELFTNRKENIPLKLSEDCLYLNIYTPADLTKKNRLPVMVWIHGGGLMVGAASTYDGLALAAHENVVVVTIQYRLGIWGFFSTGDEHSRGNWGHLDQVAALRWVQDNIASFGGNPGSVTIFGESAGGESVSVLVLSPLAKNLFHRAISESGVALTSVLVKKGDVKPLAEQIAITAGCKTTTSAVMVHCLRQKTEEELLETTLKMKFLSLDLQGDPRESQPLLGTVIDGMLLLKTPEELQAERNFHTVPYMVGINKQEFGWLIPMLMSYPLSEGQLDQKTAMSLLWKSYPLVCIAKELIPEATEKYLGGTDDTVKKKDLFLDLIADVMFGVPSVIVARNHRDAGAPTYMYEFQYRPSFSSDMKPKTVIGDHGDELFSVFGAPFLKEGASEEEIRLSKMVMKFWANFARNGNPNGEGLPHWPEYNQKEGYLQIGANTQAAQKLKDKEVAFWTNLFAK Hydrogen bonds contact Hydrophobic contact | ||||
| 98 | Aldosterone synthase (CYP11B2) | 4DVQ | 4.37 | |
Target general information Gen name CYP11B2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Steroid 18hydroxylase; Cytochrome P450C18; Cytochrome P450Aldo; Cytochrome P450 11B2, mitochondrial; CYPXIB2; CYP11B2; Aldosteronesynthesizing enzyme; ALDOS Protein family Cytochrome P450 family Biochemical class Paired donor oxygen oxidoreductase Function Preferentially catalyzes the conversion of 11- deoxycorticosterone to aldosterone via corticosterone and 18- hydroxycorticosterone. Related diseases Corticosterone methyloxidase 1 deficiency (CMO-1 deficiency) [MIM:203400]: Autosomal recessive disorder of aldosterone biosynthesis. There are two biochemically different forms of selective aldosterone deficiency be termed corticosterone methyloxidase (CMO) deficiency type 1 and type 2. In CMO-1 deficiency, aldosterone is undetectable in plasma, while its immediate precursor, 18-hydroxycorticosterone, is low or normal. {ECO:0000269|PubMed:11238478, ECO:0000269|PubMed:8439335, ECO:0000269|PubMed:9177280}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Corticosterone methyloxidase 2 deficiency (CMO-2 deficiency) [MIM:610600]: Autosomal recessive disorder of aldosterone biosynthesis. In CMO-2 deficiency, aldosterone can be low or normal, but at the expense of increased secretion of 18-hydroxycorticosterone. Consequently, patients have a greatly increased ratio of 18-hydroxycorticosterone to aldosterone and a low ratio of corticosterone to 18-hydroxycorticosterone in serum. {ECO:0000269|PubMed:12788848, ECO:0000269|PubMed:1346492, ECO:0000269|PubMed:1594605, ECO:0000269|PubMed:9625333, ECO:0000269|PubMed:9814506}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB04630; DB00700; DB00292; DB00741; DB14539; DB14540; DB14543; DB14545; DB14544; DB05667; DB01011; DB01388; DB11837; DB00421; DB06281 Interacts with NA EC number EC 1.14.15.4 Uniprot keywords 3D-structure; Disease variant; Heme; Iron; Lipid metabolism; Membrane; Metal-binding; Mitochondrion; Mitochondrion inner membrane; Monooxygenase; Oxidoreductase; Proteomics identification; Reference proteome; Steroid metabolism; Steroidogenesis; Transit peptide Protein physicochemical properties Chain ID A,B,C,D,E,F,G,H,I,J,K,L Molecular weight (Da) 52334.4 Length 454 Aromaticity 0.1 Instability index 44.09 Isoelectric point 8.81 Charge (pH=7) 6.34 2D Binding mode Binding energy (Kcal/mol) -5.96 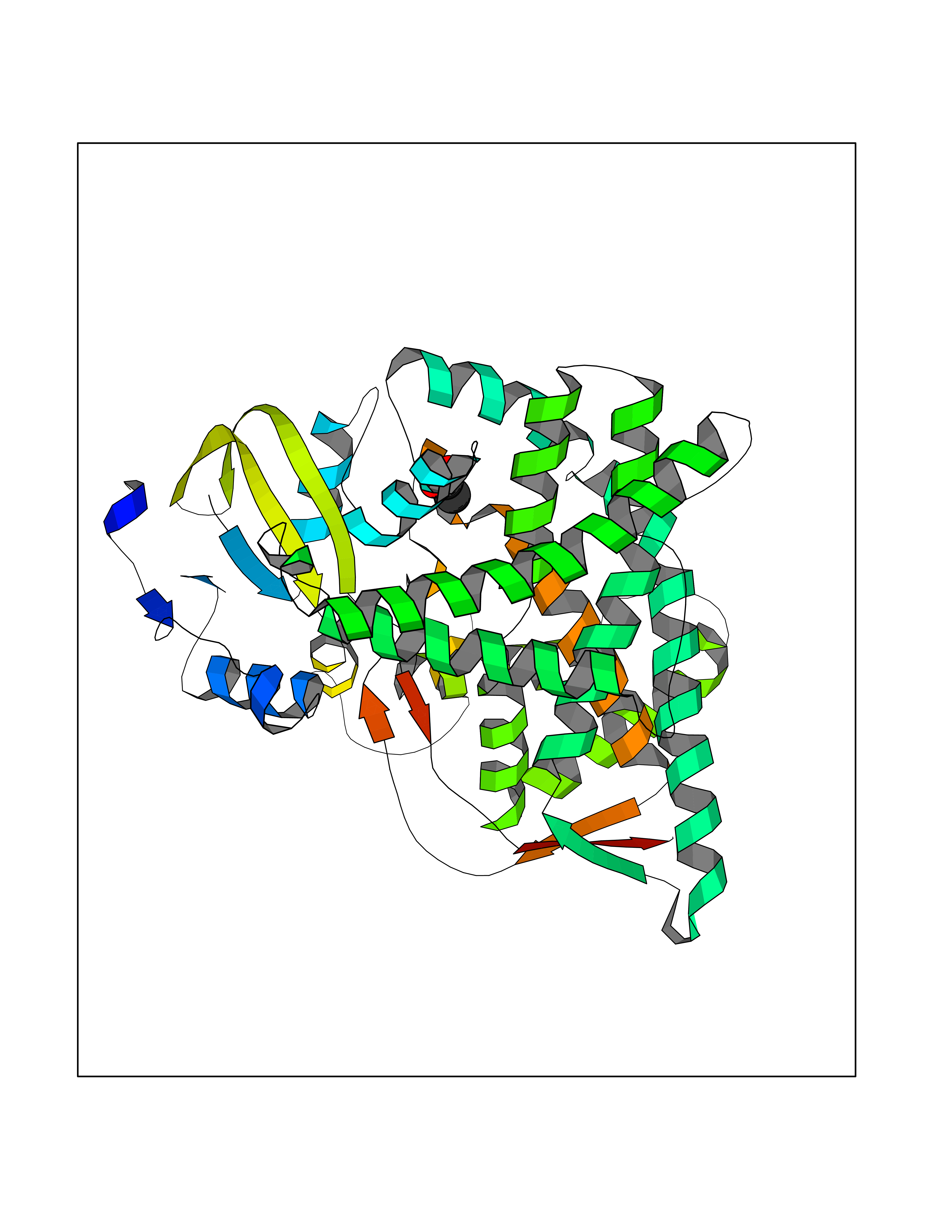 Molscript Map 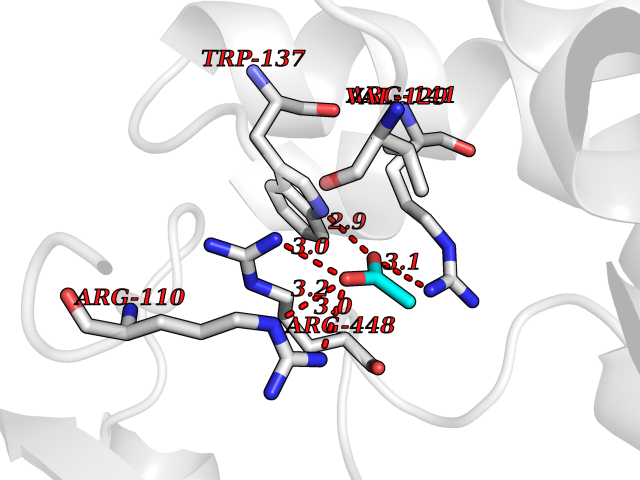 Pymol Map 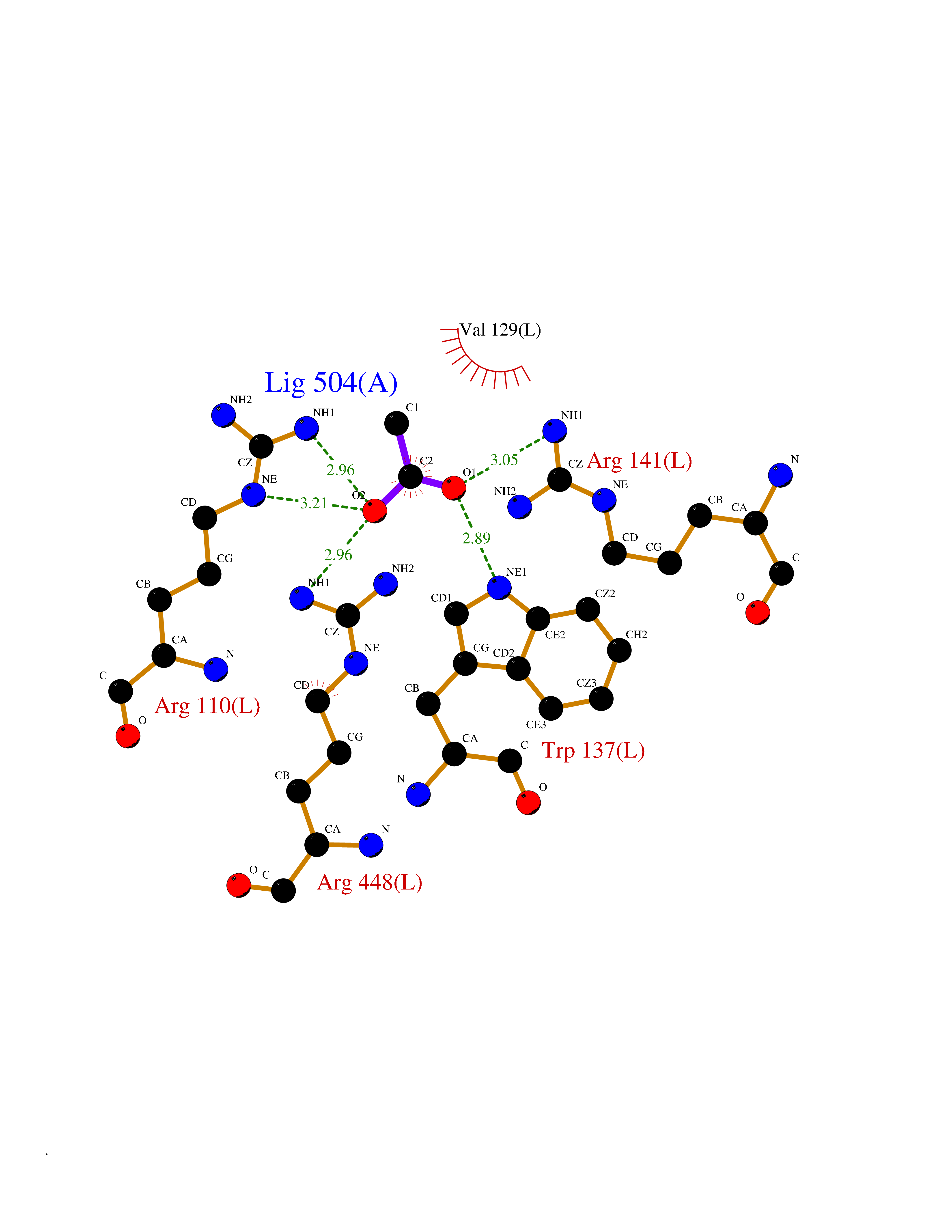 Ligplot Map 3D Binding mode Sequence LPFEAMPQHPGNRWLRLLQIWREQGYEHLHLEMHQTFQELGPIFRYPRMVCVMLPEDVEKLQQVDSLHPCRMILEPWVAYRQHRGHKCGVFLLNGPEWRFNRLRLNPDVLSPKAVQRFLPMVDAVARDFSQALKKKVLQNARGSLTLDVQPSIFHYTIEASNLALFGERLGLVGHSPSSASLNFLHALEVMFKSTVQLMFMPRSLSRWISPKVWKEHFEAWDCIFQYGDNCIQKIYQELAFNRPQHYTGIVAELLLKAELSLEAIKANSMELTAGSVDTTAFPLLMTLFELARNPDVQQILRQESLAAAASISEHPQKATTELPLLRAALKETLRLYPVGLFLERVVSSDLVLQNYHIPAGTLVQVFLYSLGRNAALFPRPERYNPQRWLDHVPFGFGMRQCLGRRLAEAEMLLLLHHVLKHFLVETLTQEDIKMVYSFILRPGTSPLLTFRAI Hydrogen bonds contact Hydrophobic contact | ||||
| 99 | Choline O-acetyltransferase | 2FY3 | 4.37 | |
Target general information Gen name CHAT Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Carnitine/choline acetyltransferase family Biochemical class Transferase Function Choline O-acetyltransferase activity. Related diseases Myasthenic syndrome, congenital, 6, presynaptic (CMS6) [MIM:254210]: A form of congenital myasthenic syndrome, a group of disorders characterized by failure of neuromuscular transmission, including pre-synaptic, synaptic, and post-synaptic disorders that are not of autoimmune origin. Clinical features are easy fatigability and muscle weakness affecting the axial and limb muscles (with hypotonia in early-onset forms), the ocular muscles (leading to ptosis and ophthalmoplegia), and the facial and bulbar musculature (affecting sucking and swallowing, and leading to dysphonia). The symptoms fluctuate and worsen with physical effort. CMS6 affected individuals have myasthenic symptoms since birth or early infancy, negative tests for anti-AChR antibodies, and abrupt episodic crises with increased weakness, bulbar paralysis, and apnea precipitated by undue exertion, fever, or excitement. CMS6 inheritance is autosomal recessive. {ECO:0000269|PubMed:11172068, ECO:0000269|PubMed:12756141}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00122; DB14006; DB00184 Interacts with Q6H8Q1-8; Q8N302-2; Q9NXL2-1; Q6XD76; Q9UII2; Q8TBE0; Q9UQB8-6; Q9ULD4-2; Q9NSI6-4; Q6P5X5; Q96LL4; P20807-4; O00257-3; Q6ZP82-1; O95674; Q9H3R5; Q8WUX9; Q9H2A9; Q3SX64; Q92782-2; Q14117; O14641; Q658K8; Q6UXG2-3; O00472; Q6NXG1; Q15910-2; Q8IZU1; P15407; P55318; Q06547-3; P23769-2; P23771; Q15486; Q8IV36; Q4VB01; Q53GQ0; P10809; P41134; Q9NZH6; Q8NA54; Q86U28; P17275; Q8N5Z5; Q6P597; P08727; Q14525; Q8IUC2; Q6IAA8; Q14847-2; P27338; Q9GZQ8; Q53S70; Q5JXC2; A0A0A0MR05; Q8NEH6; Q8TCY5; Q6IN84-2; Q96H12; P01106; P41271-2; P14598; Q9GZM8; Q5BJF6-2; Q9H8K7; Q9NR21-5; Q5VU43-8; Q13956; Q5SXH7-1; Q96T60; Q96I34; Q86UA1; Q15311; Q8TBY0; Q04206; P47804-3; Q9H0X6; P62899; Q66K80; Q9BY12-3; Q86SQ7-2; Q7Z6I5; Q496A3; Q7Z698; Q9C004; Q92783-2; Q8N4C7; O75528; Q15814; O15273; Q96A09; Q8WTV1; Q53NU3; Q71RG4-4; Q86WT6-2; Q9Y3Q8; Q99598; P49459; P11441; Q9H270; P19544-6; Q53FD0-2; Q3KNS6-3 EC number 2.3.1.6 Uniprot keywords 3D-structure; Acyltransferase; Alternative splicing; Congenital myasthenic syndrome; Direct protein sequencing; Disease variant; Neurotransmitter biosynthesis; Phosphoprotein; Proteomics identification; Reference proteome; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 66365.9 Length 595 Aromaticity 0.08 Instability index 53.36 Isoelectric point 8.16 Charge (pH=7) 4.64 2D Binding mode Binding energy (Kcal/mol) -5.96  Molscript Map 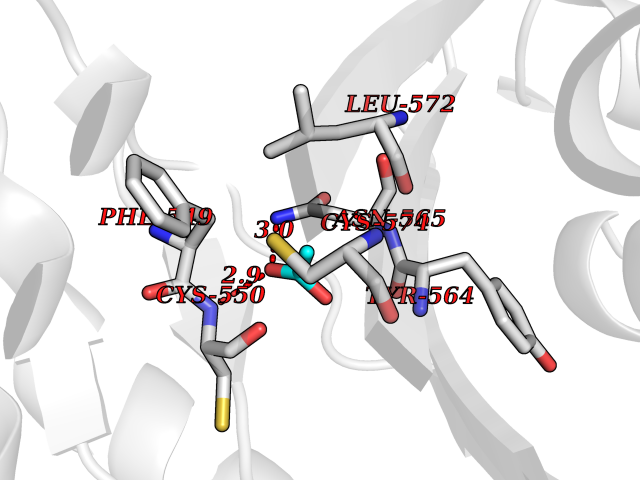 Pymol Map 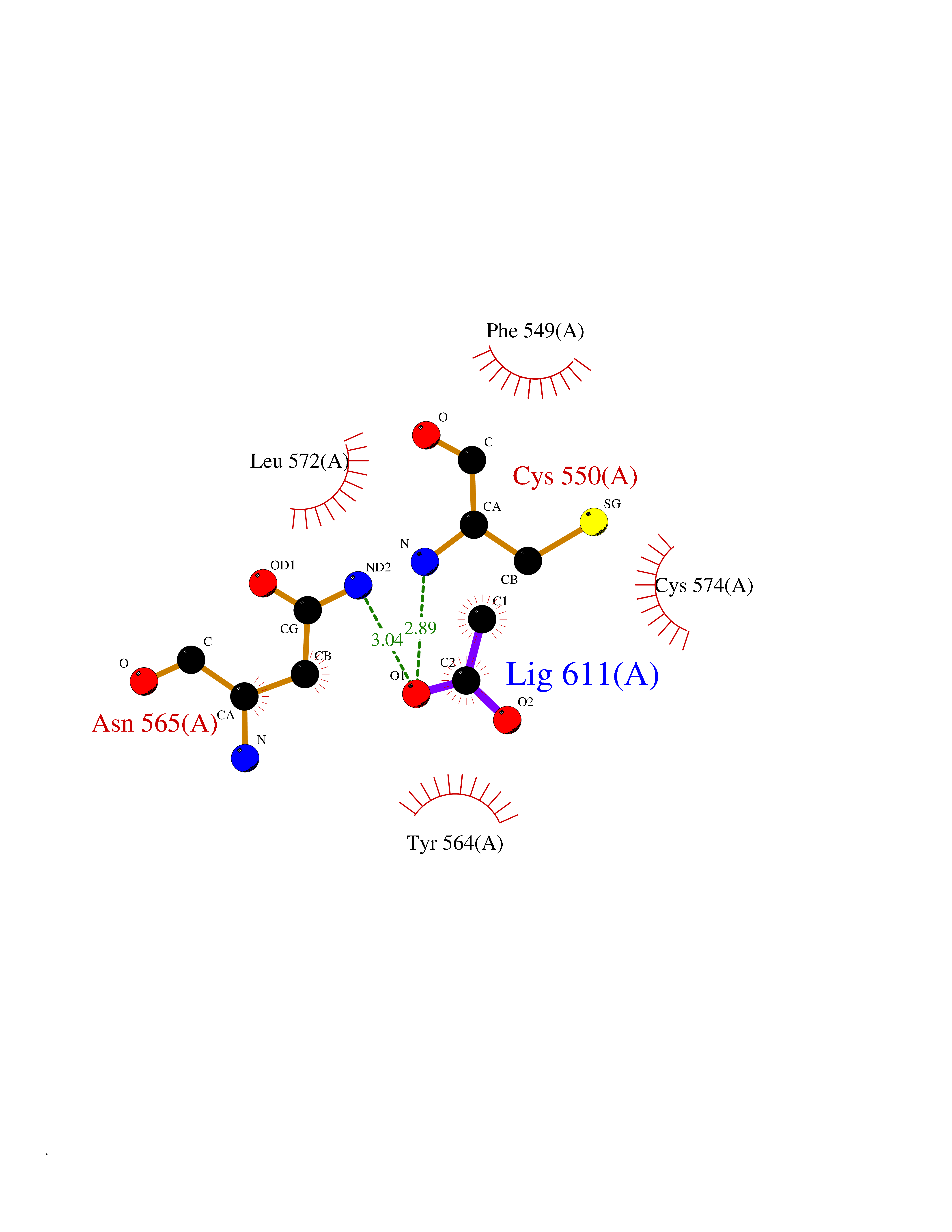 Ligplot Map 3D Binding mode Sequence SEESGLPKLPVPPLQQTLATYLQCMRHLVSEEQFRKSQAIVQQFGAPGGLGETLQQKLLERQEKTANWVSEYWLNDMYLNNRLALPVNSSPAVIFARQHFPGTDDQLRFAASLISGVLSYKALLDSHSIPTDCAKGQPLCMKQYYGLFSSYRLPGHTQDTLVAQNSSIMPEPEHVIVACCNQFFVLDVVINFRRLSEGDLFTQLRKIVKMASNAAARLPPIGLLTSDGRSEWAEARTVLVKDSTNRDSLDMIERCICLVCLDAPGGVELSDTHRALQLLHGGGYSKNGANRWYDKSLQFVVGRDGTCGVVCEHSPFDGIVLVQCTEHLLKHMTQPELVRSPMVPLPAPRRLRWKCSPEIQGHLASSAEKLQRIVKNLDFIVYKFDNYGKTFIKKQKCSPDAFIQVALQLAFYRLHRRLVPTYESASIRRFQEGRVDNIRSATPEALAFVRAVTDHKAAVPASEKLLLLKDAIRAQTAYTVMAITGMAIDNHLLALRELARAMCAALPEMFMDETYLMSNRFVLSTSQVPTTTEMFCCYGPVVPNGYGACYNPQPETILFCISSFHSCAATSSSKFAKAVEESLIDMRDLCSLLPP Hydrogen bonds contact Hydrophobic contact | ||||
| 100 | 4-hydroxyphenylpyruvate dioxygenase | 3ISQ | 4.37 | |
Target general information Gen name HPD Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms PPD Protein family 4HPPD family Biochemical class Oxidoreductase Function 4-hydroxyphenylpyruvate dioxygenase activity.Metal ion binding. Related diseases Tyrosinemia 3 (TYRSN3) [MIM:276710]: An inborn error of metabolism characterized by elevations of tyrosine in the blood and urine, seizures and mild intellectual disability. {ECO:0000269|PubMed:10942115, ECO:0000269|PubMed:11073718}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Hawkinsinuria (HWKS) [MIM:140350]: An inborn error of tyrosine metabolism characterized by failure to thrive, persistent metabolic acidosis, fine and sparse hair, and excretion of the unusual cyclic amino acid metabolite, hawkinsin, in the urine. {ECO:0000269|PubMed:11073718}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02850; DB00348 Interacts with NA EC number 1.13.11.27 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Cytoplasm; Dioxygenase; Disease variant; Endoplasmic reticulum; Golgi apparatus; Intellectual disability; Iron; Membrane; Metal-binding; Oxidoreductase; Phenylalanine catabolism; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Tyrosine catabolism Protein physicochemical properties Chain ID A Molecular weight (Da) 43164.8 Length 376 Aromaticity 0.11 Instability index 32.38 Isoelectric point 6.73 Charge (pH=7) -1.04 2D Binding mode Binding energy (Kcal/mol) -5.96  Molscript Map 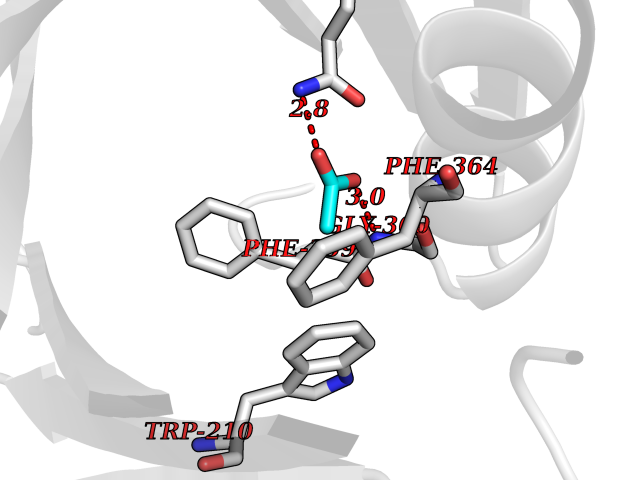 Pymol Map 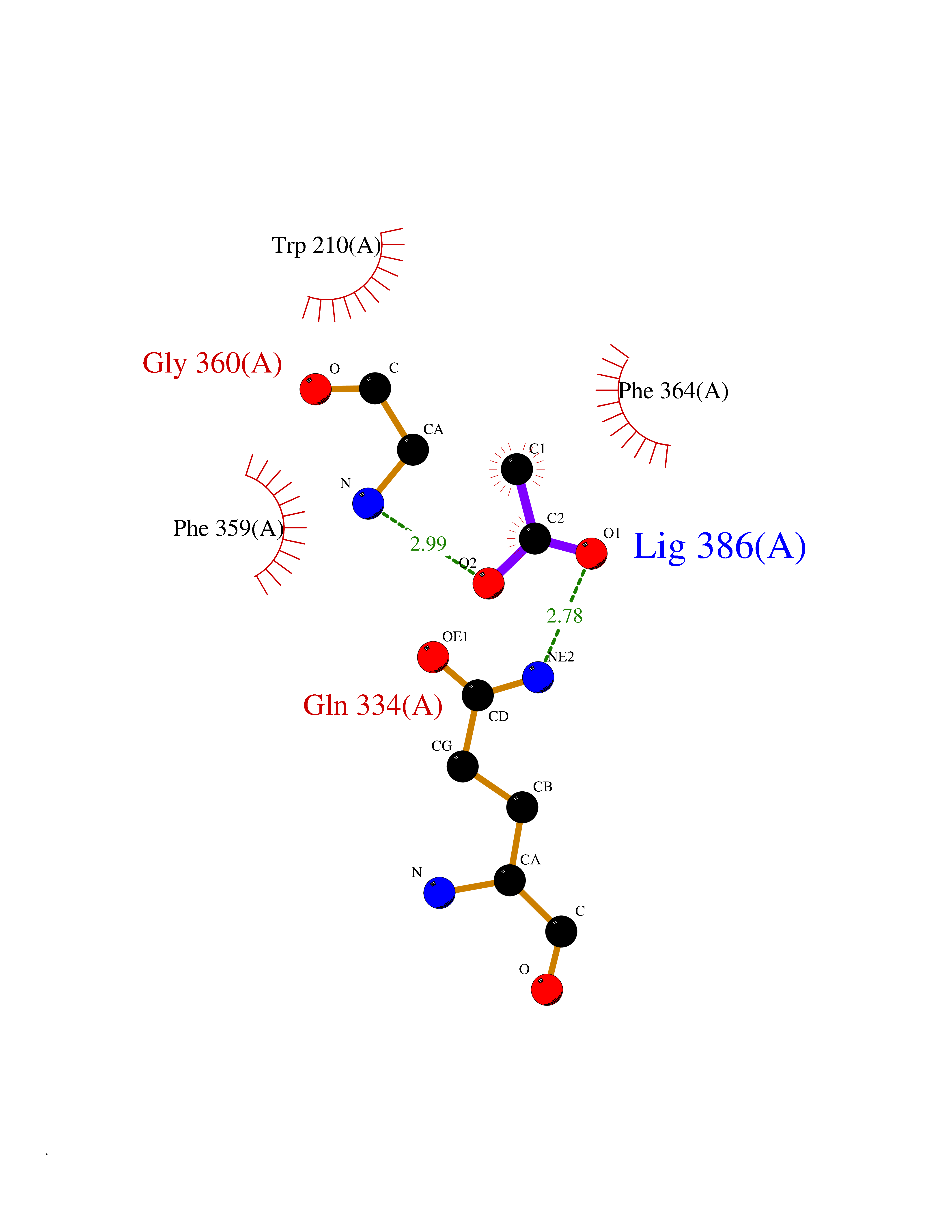 Ligplot Map 3D Binding mode Sequence AKPERGRFLHFHSVTFWVGNAKQAASFYCSKMGFEPLAYRGLETGSREVVSHVIKQGKIVFVLSSALNPWNKEMGDHLVKHGDGVKDIAFEVEDCDYIVQKARERGAKIMREPWVEQDKFGKVKFAVLQTYGDTTHTLVEKMNYIGQFLPGYEAPAFMDPLLPKLPKCSLEMIDHIVGNQPDQEMVSASEWYLKNLQFHRFWSVDDTQVHTEYSSLRSIVVANYEESIKMPINEPAPGKKKSQIQEYVDYNGGAGVQHIALKTEDIITAIRHLRERGLEFLSVPSTYYKQLREKLKTAKIKVKENIDALEELKILVDYDEKGYLLQIFTKPVQDRPTLFLEVIQRHNHQGFGAGNFNSLFKAFEEEQNLRGNLTNM Hydrogen bonds contact Hydrophobic contact | ||||