Job Results:
Ligand
Structure
Job ID
baacf95062c374f8ce5148662fe61a2e
Job name
NA
Time
2026-02-27 15:12:55
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 81 | Adrenergic receptor beta-2 (ADRB2) | 2RH1 | 4.27 | |
Target general information Gen name ADRB2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Beta-2 adrenoreceptor; Beta-2 adrenoceptor; Beta-2 adrenergic receptor; B2AR; ADRB2R Protein family G-protein coupled receptor 1 family, Adrenergic receptor subfamily, ADRB2 sub-subfamily Biochemical class GPCR rhodopsin Function The beta-2-adrenergic receptor binds epinephrine with an approximately 30-fold greater affinity than it does norepinephrine. Beta-adrenergic receptors mediate the catecholamine-induced activation of adenylate cyclase through the action of G proteins. Related diseases Cortical dysplasia, complex, with other brain malformations 6 (CDCBM6) [MIM:615771]: A disorder of aberrant neuronal migration and disturbed axonal guidance. Affected individuals have microcephaly, ataxia, and severe delayed psychomotor development. Brain imaging shows variable malformations of cortical development, including white matter streaks, dysmorphic basal ganglia, corpus callosum abnormalities, brainstem and cerebellar hypoplasia, cortical dysplasia, polymicrogyria. {ECO:0000269|PubMed:23246003}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Skin creases, congenital symmetric circumferential, 1 (CSCSC1) [MIM:156610]: An autosomal dominant disease characterized by multiple, symmetric, circumferential rings of folded skin, affecting primarily the limbs. Affected individuals also exhibit intellectual disability, cleft palate, and dysmorphic features. {ECO:0000269|PubMed:26637975}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07543; DB01193; DB00866; DB01118; DB00182; DB01102; DB01274; DB01238; DB09204; DB06216; DB00335; DB01408; DB05590; DB09013; DB00195; DB00217; DB01295; DB00612; DB00901; DB08807; DB06726; DB08808; DB00248; DB00521; DB01136; DB04846; DB01407; DB00785; DB01151; DB11273; DB13345; DB00449; DB11278; DB00841; DB09273; DB06262; DB01363; DB01364; DB00668; DB01049; DB11587; DB01288; DB00983; DB05039; DB00221; DB01064; DB00598; DB01210; DB13139; DB01365; DB13624; DB01214; DB00264; DB01203; DB05849; DB04861; DB00368; DB00540; DB00334; DB09080; DB00816; DB01580; DB00715; DB01359; DB00925; DB00397; DB00960; DB01291; DB01366; DB01182; DB00571; DB06814; DB00852; DB01917; DB11124; DB00867; DB01001; DB00938; DB00489; DB03566; DB00127; DB00871; DB00373; DB00726; DB12248; DB09082; DB09185 Interacts with P30542; P07550; P32121; Q96B67; Q9UII2; Q9ULD4-2; Q9NSI6-4; Q5M9N0-2; A0AVK6; Q658K8; O00472; Q15910-2; Q15486; P61978; Q5TCQ9; Q99685; O14745; Q9NR21-5; Q8WVD3; Q9H0X6; Q13573; P12931; Q5T0J7-2; Q8N0U2 EC number NA Uniprot keywords 3D-structure; Cell membrane; Disulfide bond; Endosome; G-protein coupled receptor; Glycoprotein; Golgi apparatus; Hydroxylation; Lipoprotein; Membrane; Palmitate; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Transducer; Transmembrane; Transmembrane helix; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 32266.1 Length 282 Aromaticity 0.15 Instability index 36.1 Isoelectric point 8.02 Charge (pH=7) 2.1 2D Binding mode Binding energy (Kcal/mol) -5.83 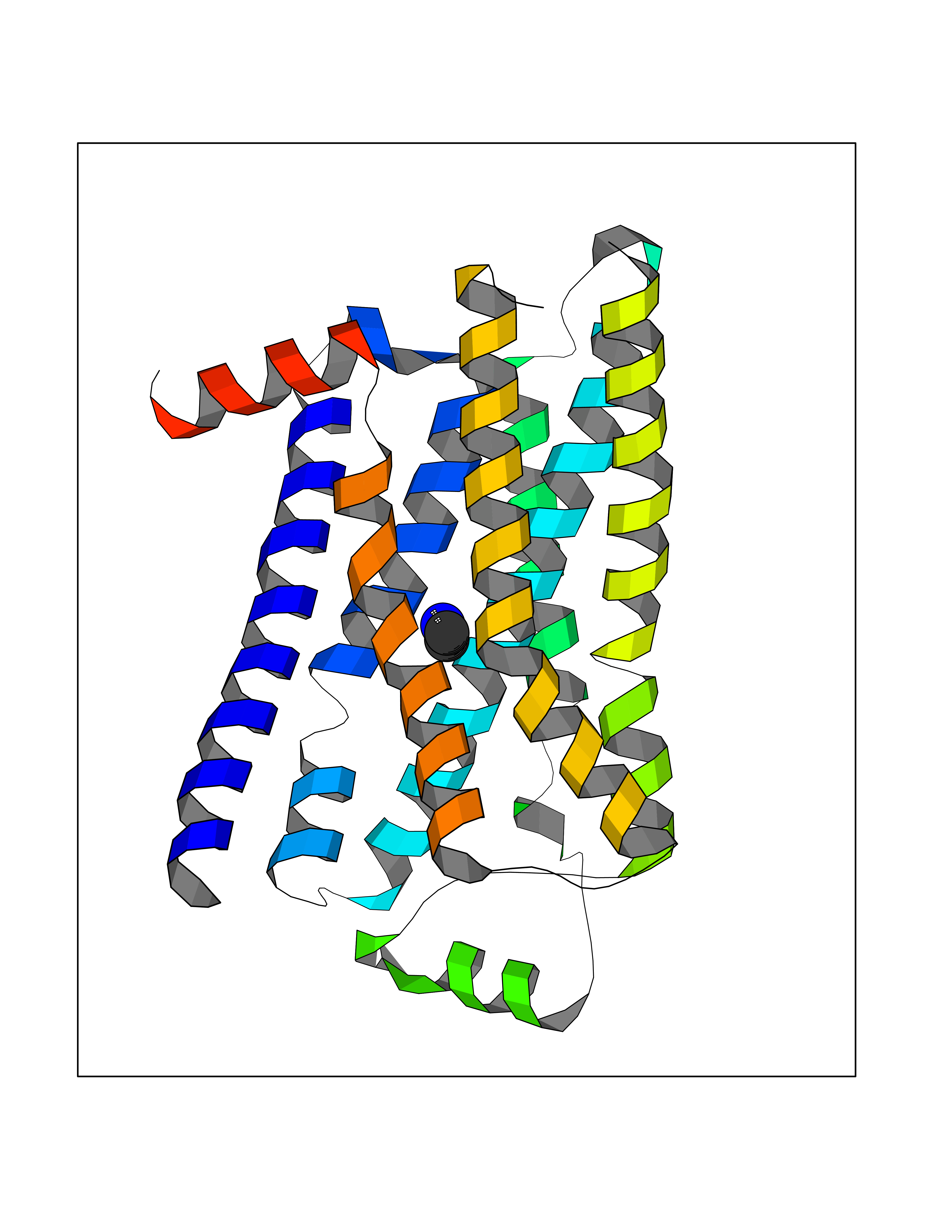 Molscript Map 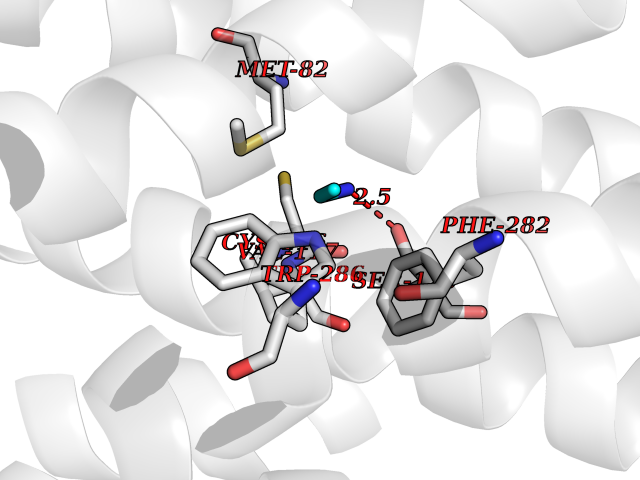 Pymol Map 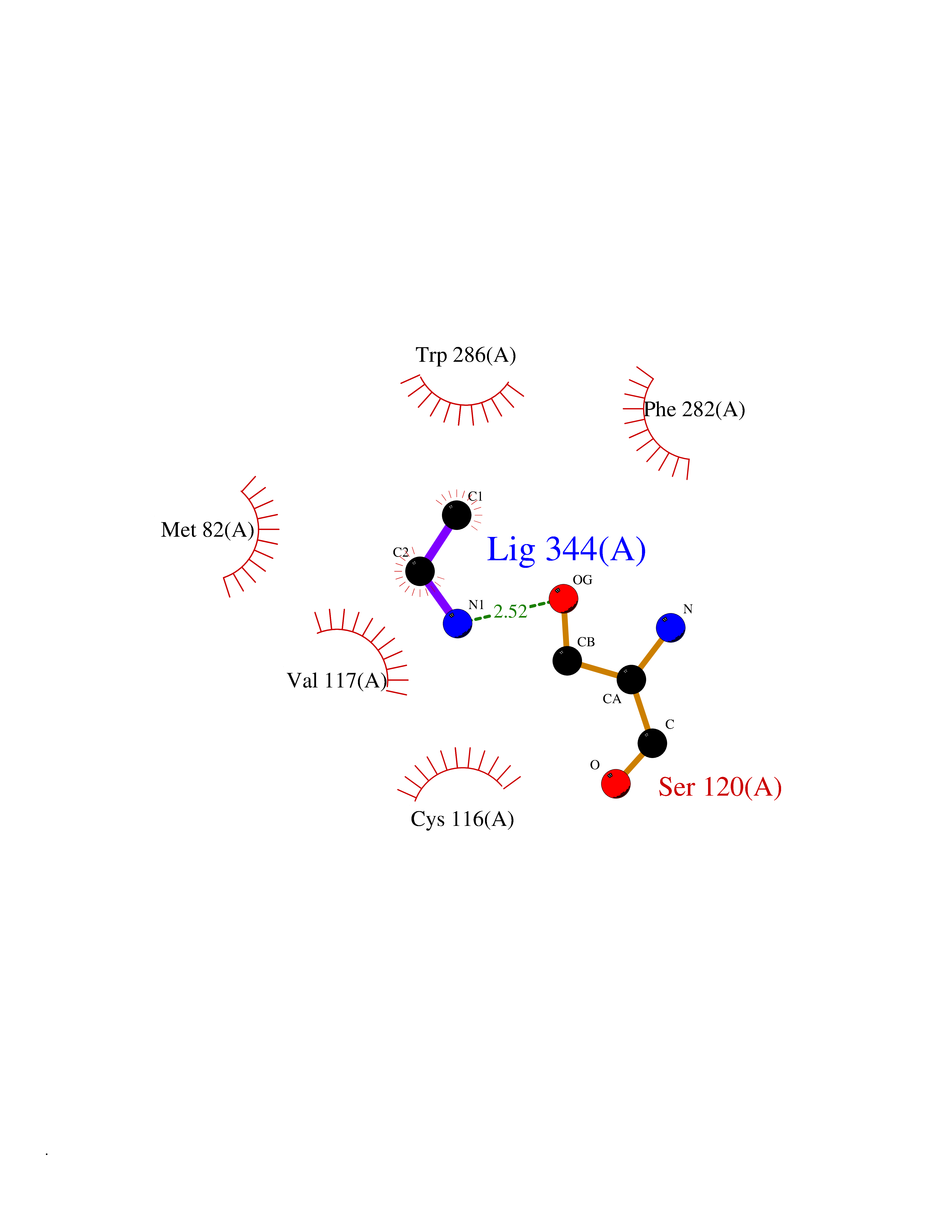 Ligplot Map 3D Binding mode Sequence DEVWVVGMGIVMSLIVLAIVFGNVLVITAIAKFERLQTVTNYFITSLACADLVMGLAVVPFGAAHILMKMWTFGNFWCEFWTSIDVLCVTASIETLCVIAVDRYFAITSPFKYQSLLTKNKARVIILMVWIVSGLTSFLPIQMHWYRATHQEAINCYAEETCCDFFTNQAYAIASSIVSFYVPLVIMVFVYSRVFQEAKRQLKFCLKEHKALKTLGIIMGTFTLCWLPFFIVNIVHVIQDNLIRKEVYILLNWIGYVNSGFNPLIYCRSPDFRIAFQELLCL Hydrogen bonds contact Hydrophobic contact | ||||
| 82 | Fumarate reductase flavoprotein subunit | 1Y0P | 4.27 | |
Target general information Gen name fccA Organism Shewanella frigidimarina Uniprot ID TTD ID NA Synonyms fcc3 Protein family FAD-dependent oxidoreductase 2 family, FRD/SDH subfamily Biochemical class Oxidoreductase Function Electron carrier activity.Fumarate reductase (menaquinone).Metal ion binding.Nucleic acid binding.Succinate dehydrogenase activity. Related diseases Pigmentary disorder, reticulate, with systemic manifestations, X-linked (PDR) [MIM:301220]: An X-linked recessive disorder characterized by recurrent infections and sterile inflammation in various organs. Diffuse skin hyperpigmentation with a distinctive reticulate pattern is universally evident by early childhood. This is later followed in many patients by hypohidrosis, corneal inflammation and scarring, enterocolitis that resembles inflammatory bowel disease, and recurrent urethral strictures. Melanin and amyloid deposition is present in the dermis. Affected males also have a characteristic facies with frontally upswept hair and flared eyebrows. Female carriers have only restricted pigmentary changes along Blaschko's lines. {ECO:0000269|PubMed:27019227}. The disease is caused by variants affecting the gene represented in this entry. XLPDR is caused by a recurrent intronic mutation that results in missplicing and reduced POLA1 expression. This leads to a decrease in cytosolic RNA:DNA hybrids and constitutive activation of type I interferon responses, but has no effect on cell replication. {ECO:0000269|PubMed:27019227}.; DISEASE: Van Esch-O'Driscoll syndrome (VEODS) [MIM:301030]: An X-linked recessive syndrome characterized by different degrees of intellectual disability, moderate to severe short stature, microcephaly, hypogonadism, and variable congenital malformations. {ECO:0000269|PubMed:31006512}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB04734; DB03147; DB01677; DB03343 Interacts with NA EC number 1.3.2.4 Uniprot keywords 3D-structure; Electron transport; FAD; Flavoprotein; Heme; Iron; Metal-binding; Oxidoreductase; Periplasm; Transport Protein physicochemical properties Chain ID A Molecular weight (Da) 60177.2 Length 568 Aromaticity 0.06 Instability index 27.7 Isoelectric point 6 Charge (pH=7) -8.64 2D Binding mode Binding energy (Kcal/mol) -5.82 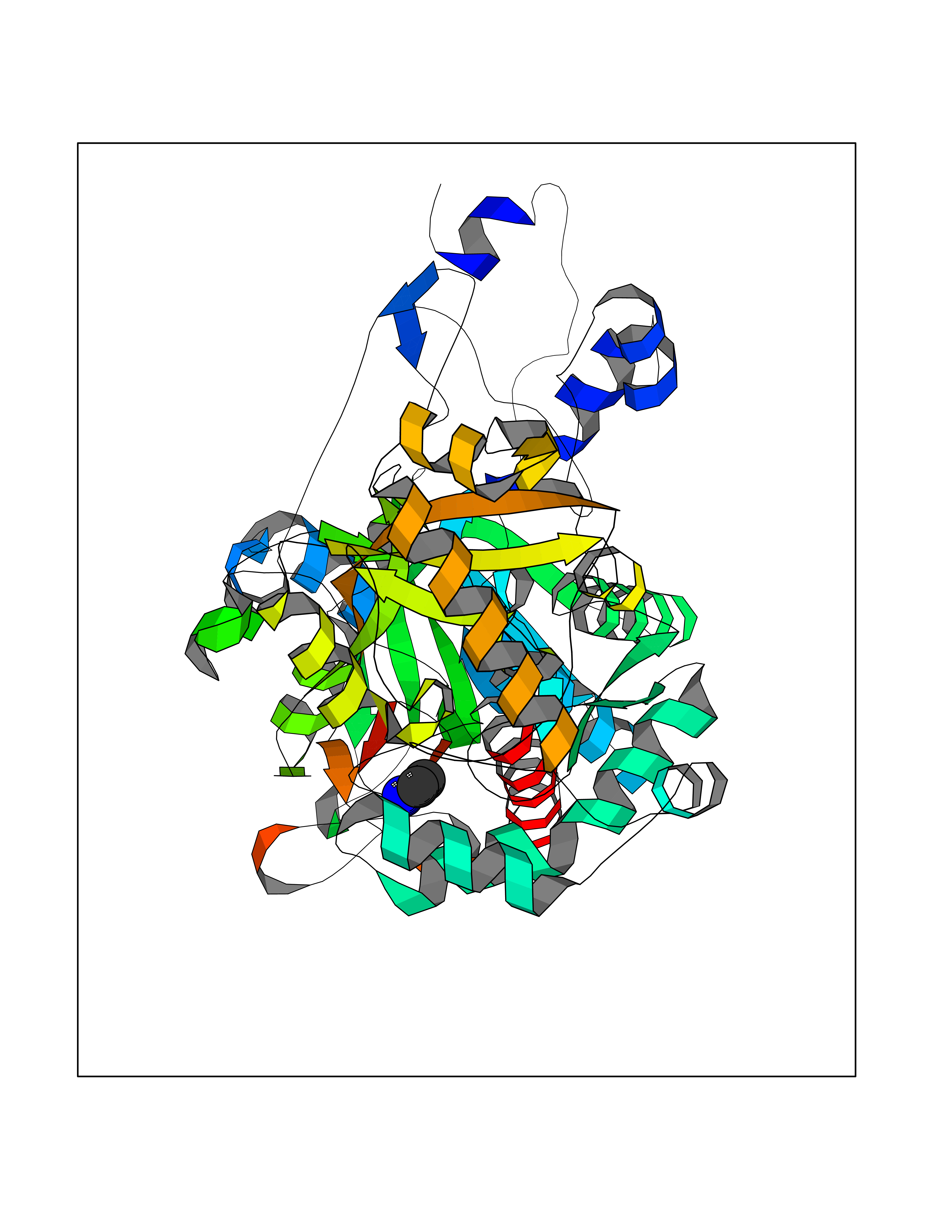 Molscript Map 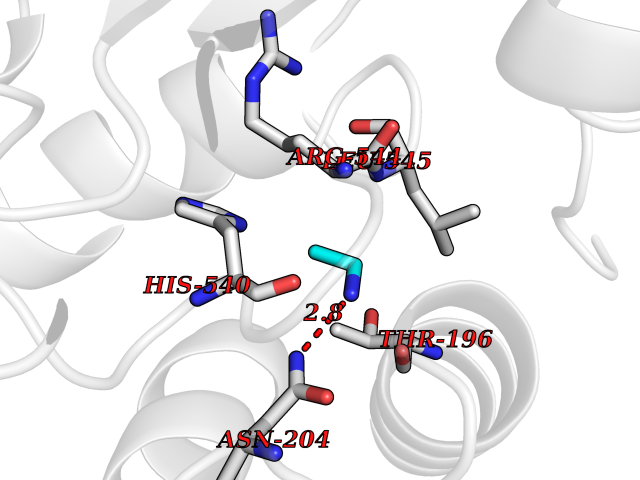 Pymol Map 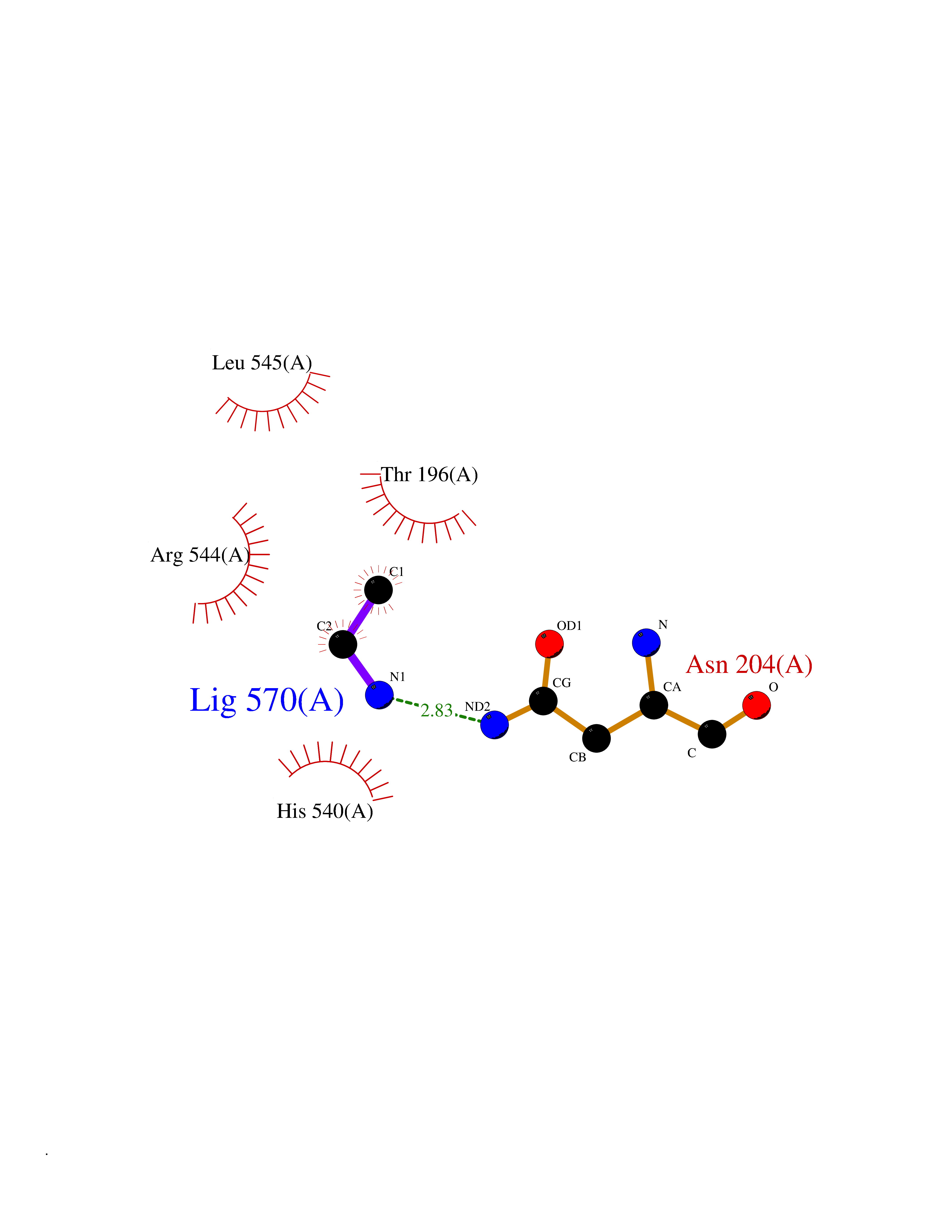 Ligplot Map 3D Binding mode Sequence ADNLAEFHVQNQECDSCHTPDGELSNDSLTYENTQCVSCHGTLAEVAETTKHEHYNAHASHFPGEVACTSCHSAHEKSMVYCDSCHSFDFNMPYAKKWLRDEPTIAELAKDKSERQAALASAPHDTVDVVVVGSGGAGFSAAISATDSGAKVILIEKEPVIGGNAKLAAGGMNAAWTDQQKAKKITDSPELMFEDTMKGGQNINDPALVKVLSSHSKDSVDWMTAMGADLTDVGMMGGASVNRAHRPTGGAGVGAHVVQVLYDNAVKRNIDLRMNTRGIEVLKDDKGTVKGILVKGMYKGYYWVKADAVILATGGFAKNNERVAKLDPSLKGFISTNQPGAVGDGLDVAENAGGALKDMQYIQAHPTLSVKGGVMVTEAVRGNGAILVNREGKRFVNEITTRDKASAAILAQTGKSAYLIFDDSVRKSLSKIDKYIGLGVAPTADSLVKLGKMEGIDGKALTETVARYNSLVSSGKDTDFERPNLPRALNEGNYYAIEVTPGVHHTMGGVMIDTKAEVMNAKKQVIPGLYGAGEVTGGVHGANRLGGNAISDIITFGRLAGEEAAKYS Hydrogen bonds contact Hydrophobic contact | ||||
| 83 | Medium-chain specific acyl-CoA dehydrogenase, mitochondrial | 4P13 | 4.27 | |
Target general information Gen name ACADM Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Acyl-CoA dehydrogenase family Biochemical class Oxidoreductase Function Acyl-CoA dehydrogenase activity.Flavin adenine dinucleotide binding.Identical protein binding.Medium-chain-acyl-CoA dehydrogenase activity. Related diseases Acyl-CoA dehydrogenase medium-chain deficiency (ACADMD) [MIM:201450]: An inborn error of mitochondrial fatty acid beta-oxidation which causes fasting hypoglycemia, hepatic dysfunction and encephalopathy, often resulting in death in infancy. {ECO:0000269|PubMed:10767181, ECO:0000269|PubMed:11349232, ECO:0000269|PubMed:11409868, ECO:0000269|PubMed:11486912, ECO:0000269|PubMed:1363805, ECO:0000269|PubMed:1671131, ECO:0000269|PubMed:1684086, ECO:0000269|PubMed:1902818, ECO:0000269|PubMed:2251268, ECO:0000269|PubMed:2393404, ECO:0000269|PubMed:2394825, ECO:0000269|PubMed:7603790, ECO:0000269|PubMed:7929823, ECO:0000269|PubMed:8198141, ECO:0000269|PubMed:9158144, ECO:0000269|PubMed:9882619}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03415; DB03147; DB02910 Interacts with PRO_0000000502 [P11310] EC number 1.3.8.7 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Direct protein sequencing; Disease variant; FAD; Fatty acid metabolism; Flavoprotein; Lipid metabolism; Mitochondrion; Oxidoreductase; Phosphoprotein; Proteomics identification; Reference proteome; Transit peptide Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 85080.3 Length 773 Aromaticity 0.09 Instability index 30.55 Isoelectric point 5.71 Charge (pH=7) -7.7 2D Binding mode Binding energy (Kcal/mol) -5.83 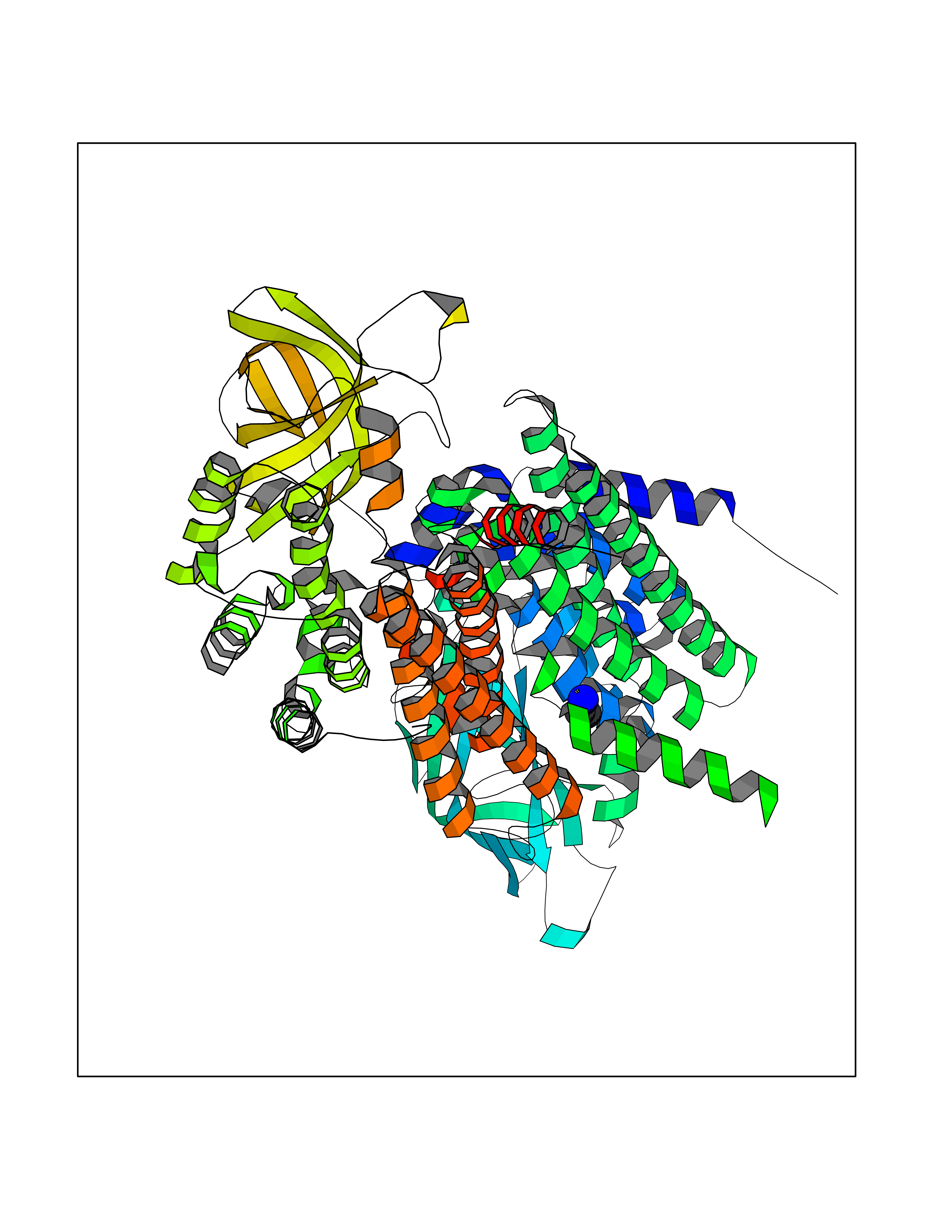 Molscript Map 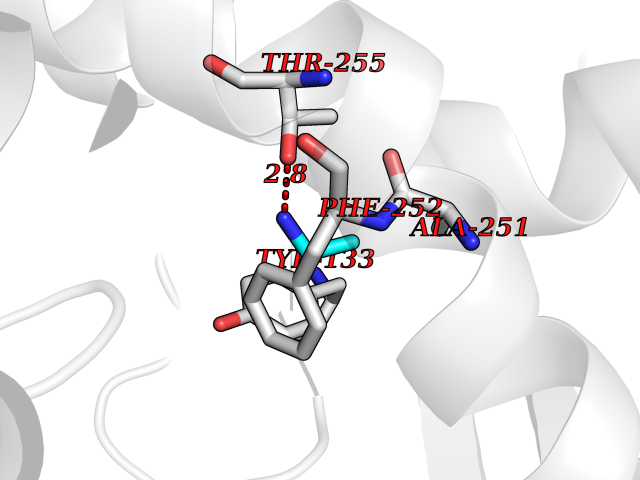 Pymol Map 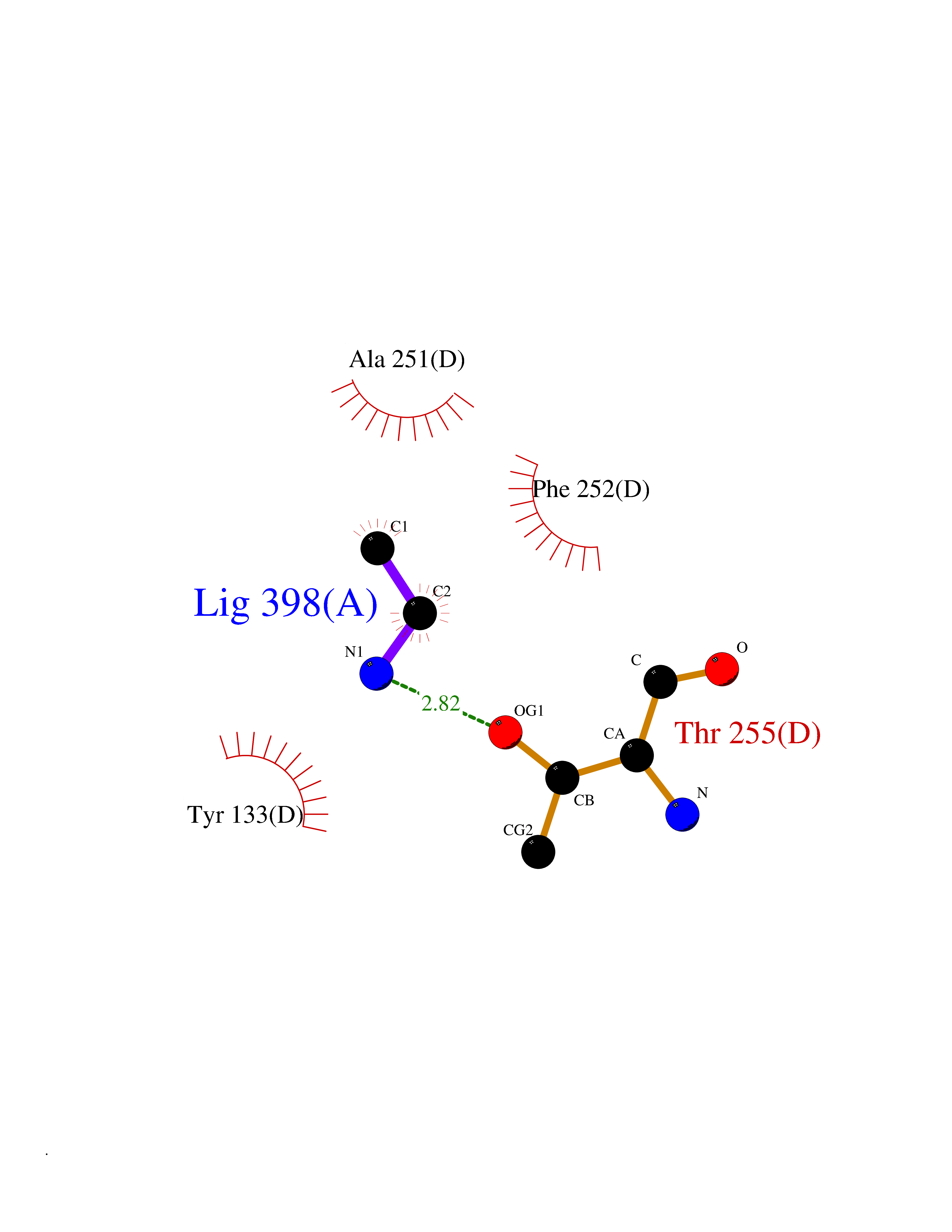 Ligplot Map 3D Binding mode Sequence LGFSFEFTEQQKEFQATARKFAREEIIPVAAEYDKTGEYPVPLIRRAWELGLMNTHIPENCGGLGLGTFDACLISEELAYGCTGVQTAIEGNSLGQMPIIIAGNDQQKKKYLGRMTEEPLMCAYCVTEPGAGSDVAGIKTKAEKKGDEYIINGQKMWITNGGKANWYFLLARSDPDPKAPANKAFTGFIVEADTPGIQIGRKELNMGQRCSDTRGIVFEDVKVPKENVLIGDGAGFKVAMGAFDKTRPVVAAGAVGLAQRALDEATKYALERKTFGKLLVEHQAISFMLAEMAMEVELARMSYQRAAWEVDSGRRNTYYASIAKAFAGDIANQLATDAVQILGGNGFNTEYPVEKLMRDAKIYQIYEGTSQIQRLIVAREHIDKYKLGFSFEFTEQQKEFQATARKFAREEIIPVAAEYDKTGEYPVPLIRRAWELGLMNTHIPENCGGLGLGTFDACLISEELAYGCTGVQTAIEGNSLGQMPIIIAGNDQQKKKYLGRMTEEPLMCAYCVTEPGAGSDVAGIKTKAEKKGDEYIINGQKMWITNGGKANWYFLLARSDPDPKAPANKAFTGFIVEADTPGIQIGRKELNMGQRCSDTRGIVFEDVKVPKENVLIGDGAGFKVAMGAFDKTRPVVAAGAVGLAQRALDEATKYALERKTFGKLLVEHQAISFMLAEMAMEVELARMSYQRAAWEVDSGRRNTYYASIAKAFAGDIANQLATDAVQILGGNGFNTEYPVEKLMRDAKIYQIYEGTSQIQRLIVAREHIDKYKN Hydrogen bonds contact Hydrophobic contact | ||||
| 84 | Carbonic anhydrase 3 | 3UYQ | 4.27 | |
Target general information Gen name CA3 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Alpha-carbonic anhydrase family Biochemical class Lyase Function Carbonate dehydratase activity.Nickel cation binding.Phosphatase activity.Zinc ion binding. Related diseases Cortical dysplasia, complex, with other brain malformations 6 (CDCBM6) [MIM:615771]: A disorder of aberrant neuronal migration and disturbed axonal guidance. Affected individuals have microcephaly, ataxia, and severe delayed psychomotor development. Brain imaging shows variable malformations of cortical development, including white matter streaks, dysmorphic basal ganglia, corpus callosum abnormalities, brainstem and cerebellar hypoplasia, cortical dysplasia, polymicrogyria. {ECO:0000269|PubMed:23246003}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Skin creases, congenital symmetric circumferential, 1 (CSCSC1) [MIM:156610]: An autosomal dominant disease characterized by multiple, symmetric, circumferential rings of folded skin, affecting primarily the limbs. Affected individuals also exhibit intellectual disability, cleft palate, and dysmorphic features. {ECO:0000269|PubMed:26637975}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00819; DB00562; DB01194; DB00482; DB00606; DB01144; DB00869; DB08846; DB00311; DB00703; DB12418; DB00391; DB00273; DB00580; DB00909 Interacts with P37235; Q9BS40 EC number 4.2.1.1 Uniprot keywords 3D-structure; Acetylation; Cytoplasm; Glutathionylation; Lyase; Metal-binding; Phosphoprotein; Proteomics identification; Reference proteome; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 29416.7 Length 259 Aromaticity 0.11 Instability index 38.2 Isoelectric point 6.71 Charge (pH=7) -1.07 2D Binding mode Binding energy (Kcal/mol) -5.83 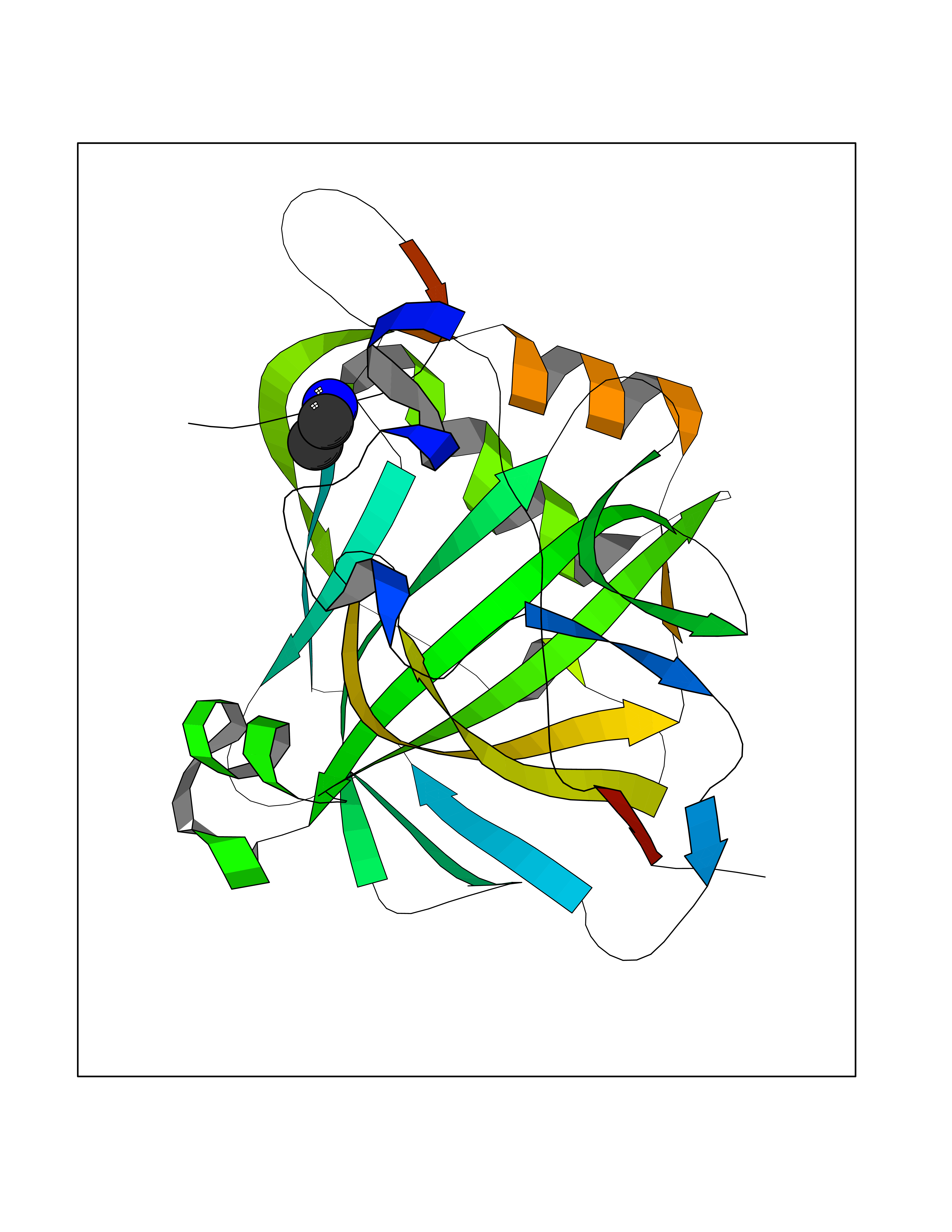 Molscript Map 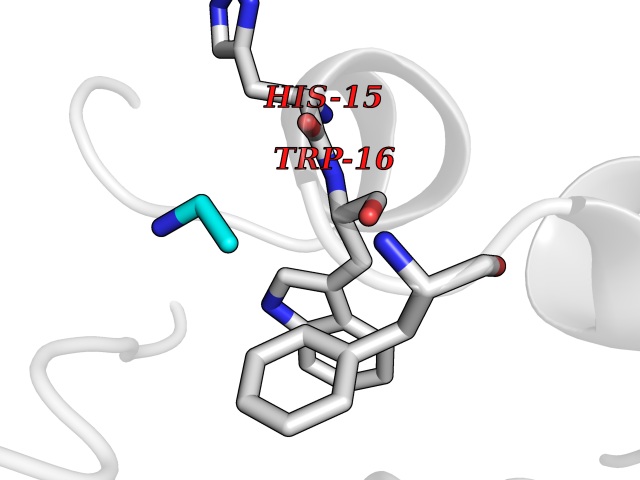 Pymol Map  Ligplot Map 3D Binding mode Sequence AKEWGYASHNGPDHWHELFPNAKGENQSPIELHTKDIRHDPSLQPWSVSYDGGSAKTILNNGHTCRVVFDDTYDRSMLRGGPLPGPYRLRQFHLHWGSSDDHGSEHTVDGVKYAAELHLVHWNPKYNTFKEALKQRDGIAVIGIFLKIGHENGEFQIFLDALDKIKTKGKEAPFTKFDPSSLFPASRDYWTYQGSFTTPPCEECIVWLLLKEPMTVSSDQMAKLRSLLSSAENEPPVPLVSNWRPPQPINNRVVRASFK Hydrogen bonds contact Hydrophobic contact | ||||
| 85 | Endolysin | 1AM7 | 4.27 | |
Target general information Gen name R Organism Escherichia phage lambda (Bacteriophage lambda) Uniprot ID TTD ID NA Synonyms NA Protein family Glycosyl hydrolase 24 family Biochemical class Glycosidase Function Lyase activity.Lysozyme activity.Lytic transglycosylase activity. Related diseases Estrogen resistance (ESTRR) [MIM:615363]: A disorder characterized by partial or complete resistance to estrogens, in the presence of elevated estrogen serum levels. Clinical features include absence of the pubertal growth spurt, delayed bone maturation, unfused epiphyses, reduced bone mineral density, osteoporosis, continued growth into adulthood and very tall adult stature. Glucose intolerance, hyperinsulinemia and lipid abnormalities may also be present. {ECO:0000269|PubMed:23841731, ECO:0000269|PubMed:27754803}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB04206 Interacts with NA EC number 4.2.2.n2 Uniprot keywords 3D-structure; Antimicrobial; Bacteriolytic enzyme; Cytolysis; Direct protein sequencing; Host cell lysis by virus; Host cytoplasm; Lyase; Reference proteome; Viral release from host cell Protein physicochemical properties Chain ID A,B,C Molecular weight (Da) 49834.9 Length 462 Aromaticity 0.07 Instability index 18.78 Isoelectric point 9.6 Charge (pH=7) 18.29 2D Binding mode Binding energy (Kcal/mol) -5.82  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence MVEINNQRKAFLDMLAXSEGTDNGRQKTRNHGYDVIVGGELFTDYSDHPRKLVTLNPKLKSTGAGRYQLLSRXXDAYRKQLGLKDFSPKSQDAVALQQIKERGALPMIDRGDIRQAIDRCSNIXASLPGAGYGQFEHKADSLIAKFKEAGGTVRMVEINNQRKAFLDMLAXSEGTDNGRQKTRNHGYDVIVGGELFTDYSDHPRKLVTLNPKLKSTGAGRYQLLSRXXDAYRKQLGLKDFSPKSQDAVALQQIKERGALPMIDRGDIRQAIDRCSNIXASLPGAGYGQFEHKADSLIAKFKEAGGTVRMVEINNQRKAFLDMLAXSEGTDNGRQKTRNHGYDVIVGGELFTDYSDHPRKLVTLNPKLKSTGAGRYQLLSRXXDAYRKQLGLKDFSPKSQDAVALQQIKERGALPMIDRGDIRQAIDRCSNIXASLPGAGYGQFEHKADSLIAKFKEAGGTVR Hydrogen bonds contact Hydrophobic contact | ||||
| 86 | Phenylalanine hydroxylase (PAH) | 1J8U | 4.27 | |
Target general information Gen name PAH Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Phenylalanine4hydroxylase; Phenylalanine-4-hydroxylase; Phe4monooxygenase; Phe-4-monooxygenase Protein family Biopterin-dependent aromatic amino acid hydroxylase family Biochemical class Paired donor oxygen oxidoreductase Function Catalyzes the hydroxylation of L-phenylalanine to L-tyrosine. Related diseases Phenylalanine hydroxylase deficiency (PAH deficiency) [MIM:261600]: An autosomal recessive inborn error of phenylalanine metabolism characterized by intolerance to dietary intake of the essential amino acid phenylalanine. The disease spectrum depends on the degree of PAH deficiency and the phenylalanine levels in plasma. Severe deficiency causes classic phenylketonuria (PKU) that is characterized by plasma concentrations of phenylalanine persistently above 1200 umol/L. PKU patients develop profound and irreversible intellectual disability, unless low phenylalanine diet is introduced early in life. They tend to have light pigmentation, rashes similar to eczema, epilepsy, extreme hyperactivity, psychotic states and an unpleasant 'mousy' odor. Less severe forms of PAH deficiency are characterized by phenylalanine levels above normal (120 umol/L) but below 1200 umol/L and include moderate PKU, mild PKU, non-PKU hyperphenylalaninemia (non-PKU HPA) and mild hyperphenylalaninemia. Individuals with PAH deficiency who have plasma phenylalanine concentrations consistently below 600 umol/L on an unrestricted diet are not at higher risk of developing intellectual, neurologic, and neuropsychological impairment than are individuals without PAH deficiency. {ECO:0000269|PubMed:10200057, ECO:0000269|PubMed:10679941, ECO:0000269|PubMed:11180595, ECO:0000269|PubMed:11326337, ECO:0000269|PubMed:11385716, ECO:0000269|PubMed:11461196, ECO:0000269|PubMed:11935335, ECO:0000269|PubMed:12501224, ECO:0000269|PubMed:1301187, ECO:0000269|PubMed:1355066, ECO:0000269|PubMed:1358789, ECO:0000269|PubMed:1363837, ECO:0000269|PubMed:1363838, ECO:0000269|PubMed:1671810, ECO:0000269|PubMed:1672290, ECO:0000269|PubMed:1672294, ECO:0000269|PubMed:1679030, ECO:0000269|PubMed:1709636, ECO:0000269|PubMed:18538294, ECO:0000269|PubMed:1975559, ECO:0000269|PubMed:2014802, ECO:0000269|PubMed:22513348, ECO:0000269|PubMed:22526846, ECO:0000269|PubMed:23792259, ECO:0000269|PubMed:2564729, ECO:0000269|PubMed:2615649, ECO:0000269|PubMed:2840952, ECO:0000269|PubMed:32668217, ECO:0000269|PubMed:7833954, ECO:0000269|PubMed:8068076, ECO:0000269|PubMed:8088845, ECO:0000269|PubMed:8098245, ECO:0000269|PubMed:8406445, ECO:0000269|PubMed:8889583, ECO:0000269|PubMed:8889590, ECO:0000269|PubMed:9048935, ECO:0000269|PubMed:9101291, ECO:0000269|PubMed:9450897, ECO:0000269|PubMed:9452061, ECO:0000269|PubMed:9452062, ECO:0000269|PubMed:9521426, ECO:0000269|PubMed:9600453, ECO:0000269|PubMed:9634518, ECO:0000269|PubMed:9792407, ECO:0000269|PubMed:9792411, ECO:0000269|PubMed:9852673, ECO:0000269|PubMed:9950317}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03673; DB04339; DB06778; DB04419; DB06262; DB04400; DB00368; DB00120; DB02562; DB00360 Interacts with NA EC number EC 1.14.16.1 Uniprot keywords 3D-structure; Allosteric enzyme; Direct protein sequencing; Disease variant; Iron; Metal-binding; Monooxygenase; Oxidoreductase; Phenylalanine catabolism; Phenylketonuria; Phosphoprotein; Proteomics identification; Reference proteome Protein physicochemical properties Chain ID A Molecular weight (Da) 35556.1 Length 307 Aromaticity 0.15 Instability index 39.96 Isoelectric point 6.17 Charge (pH=7) -3.5 2D Binding mode Binding energy (Kcal/mol) -5.83 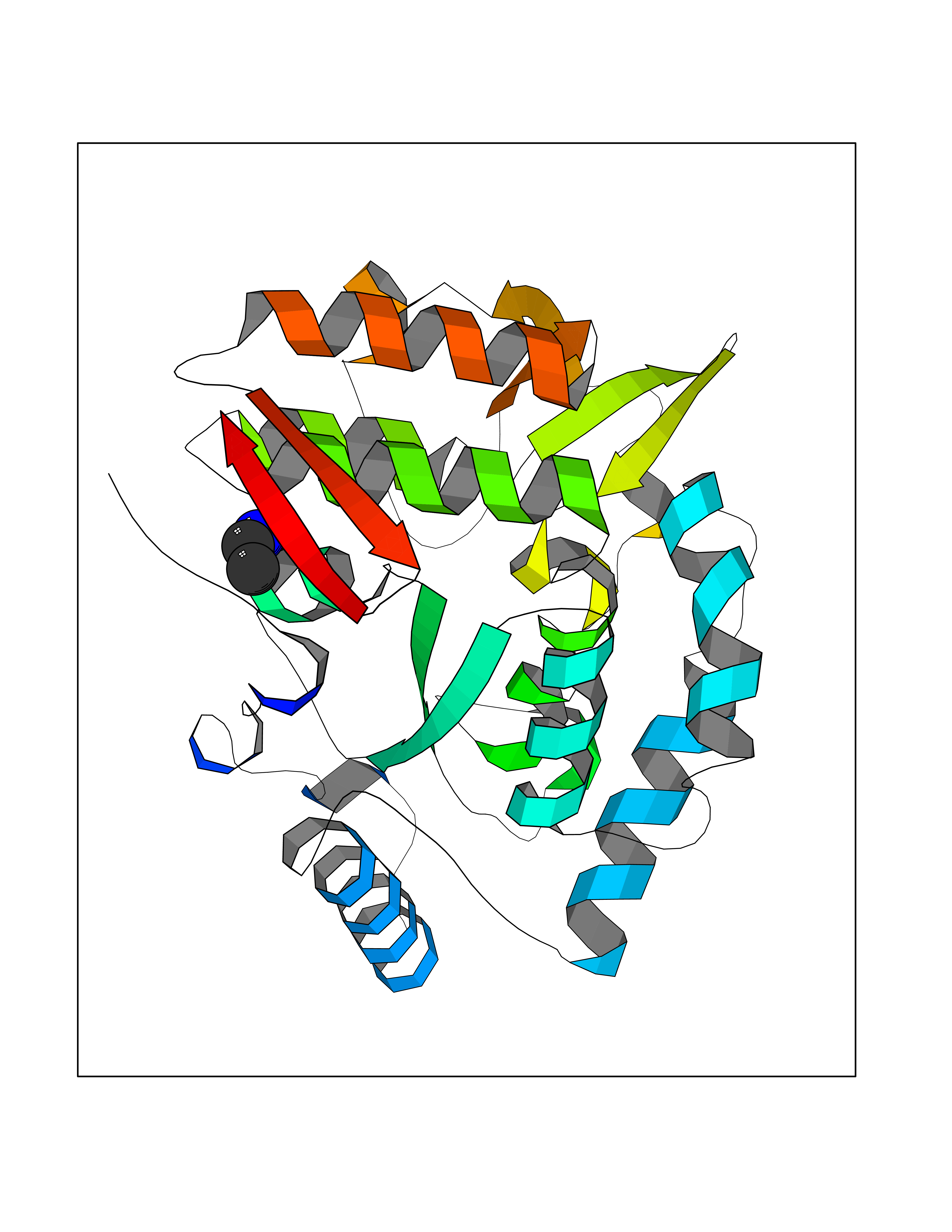 Molscript Map 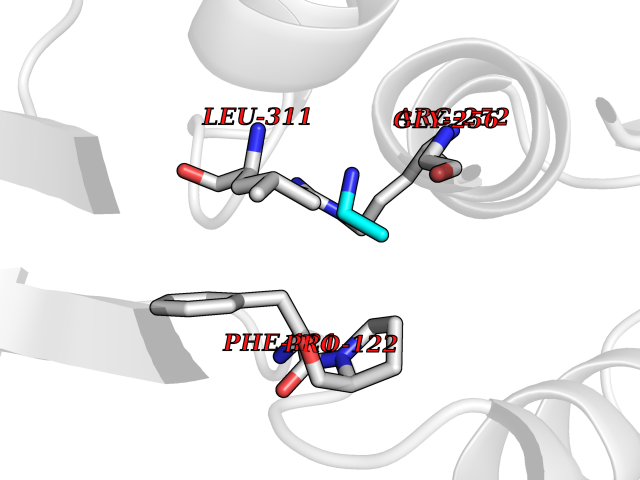 Pymol Map 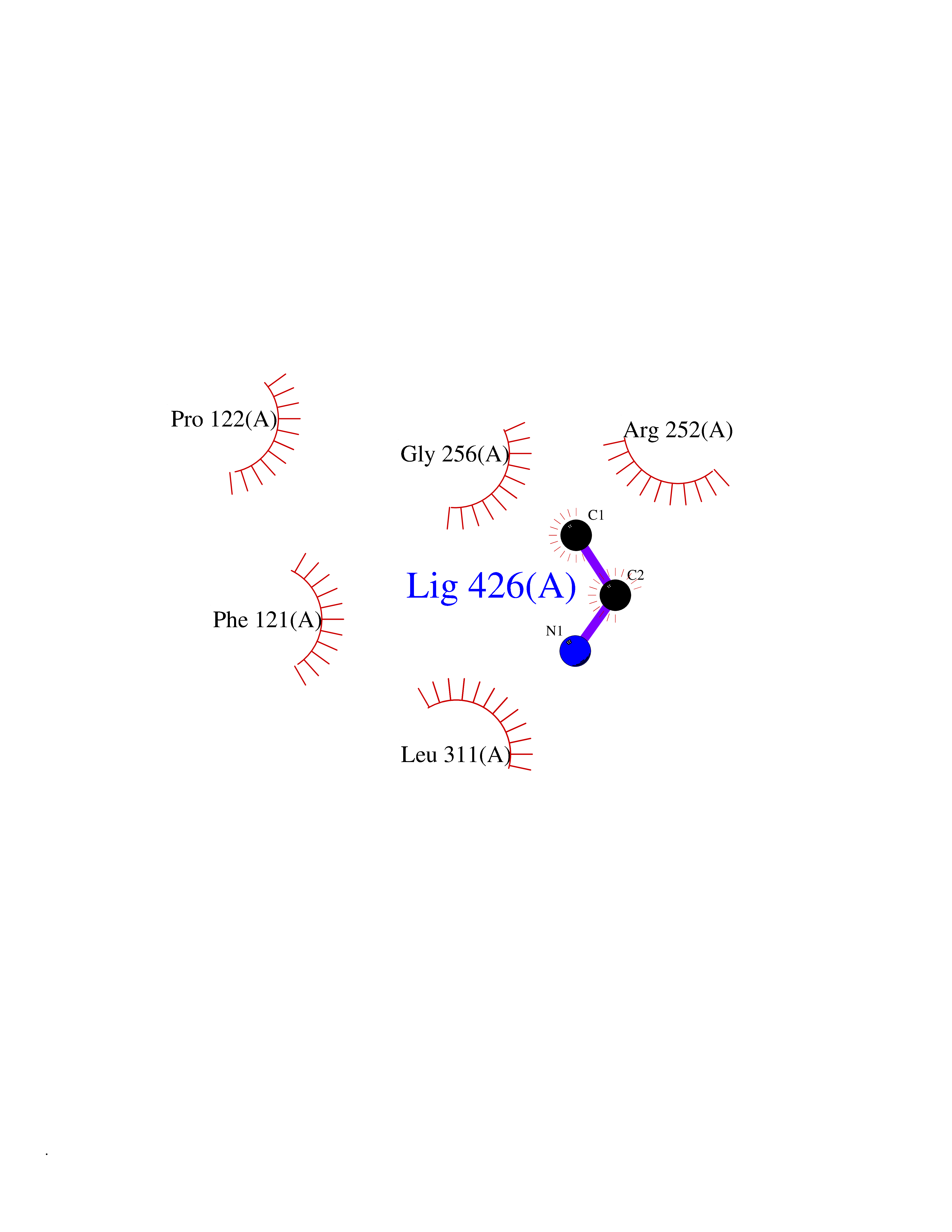 Ligplot Map 3D Binding mode Sequence VPWFPRTIQELDRFANQILSYGAELDADHPGFKDPVYRARRKQFADIAYNYRHGQPIPRVEYMEEEKKTWGTVFKTLKSLYKTHACYEYNHIFPLLEKYCGFHEDNIPQLEDVSQFLQTCTGFRLRPVAGLLSSRDFLGGLAFRVFHCTQYIRHGSKPMYTPEPDICHELLGHVPLFSDRSFAQFSQEIGLASLGAPDEYIEKLATIYWFTVEFGLCKQGDSIKAYGAGLLSSFGELQYCLSEKPKLLPLELEKTAIQNYTVTEFQPLYYVAESFNDAKEKVRNFAATIPRPFSVRYDPYTQRIEVL Hydrogen bonds contact Hydrophobic contact | ||||
| 87 | Methylated-DNA--protein-cysteine methyltransferase | 1QNT | 4.27 | |
Target general information Gen name MGMT Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family MGMT family Biochemical class Dna repair Function Calcium ion binding.DNA binding.DNA-methyltransferase activity.Methylated-DNA-[protein]-cysteine S-methyltransferase activity.Methyltransferase activity. Related diseases Antley-Bixler syndrome, with genital anomalies and disordered steroidogenesis (ABS1) [MIM:201750]: A disease characterized by the association of Antley-Bixler syndrome with steroidogenesis defects and abnormal genitalia. Antley-Bixler syndrome is characterized by craniosynostosis, radiohumeral synostosis present from the perinatal period, midface hypoplasia, choanal stenosis or atresia, femoral bowing and multiple joint contractures. {ECO:0000269|PubMed:14758361, ECO:0000269|PubMed:15264278, ECO:0000269|PubMed:15483095}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Disordered steroidogenesis due to cytochrome P450 oxidoreductase deficiency (DISPORD) [MIM:613571]: A disorder resulting in a rare variant of congenital adrenal hyperplasia, with apparent combined P450C17 and P450C21 deficiency and accumulation of steroid metabolites. Affected girls are born with ambiguous genitalia, but their circulating androgens are low and virilization does not progress. Conversely, affected boys are sometimes born undermasculinized. Boys and girls can present with bone malformations, in some cases resembling the pattern seen in patients with Antley-Bixler syndrome. {ECO:0000269|PubMed:14758361, ECO:0000269|PubMed:15220035}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00151; DB11831; DB04531; DB02216; DB01593; DB14487; DB14533; DB14548 Interacts with NA EC number 2.1.1.63 Uniprot keywords 3D-structure; Direct protein sequencing; DNA damage; DNA repair; DNA-binding; Metal-binding; Methyltransferase; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Transferase; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 17946.7 Length 166 Aromaticity 0.07 Instability index 43.65 Isoelectric point 7.27 Charge (pH=7) 0.43 2D Binding mode Binding energy (Kcal/mol) -5.83 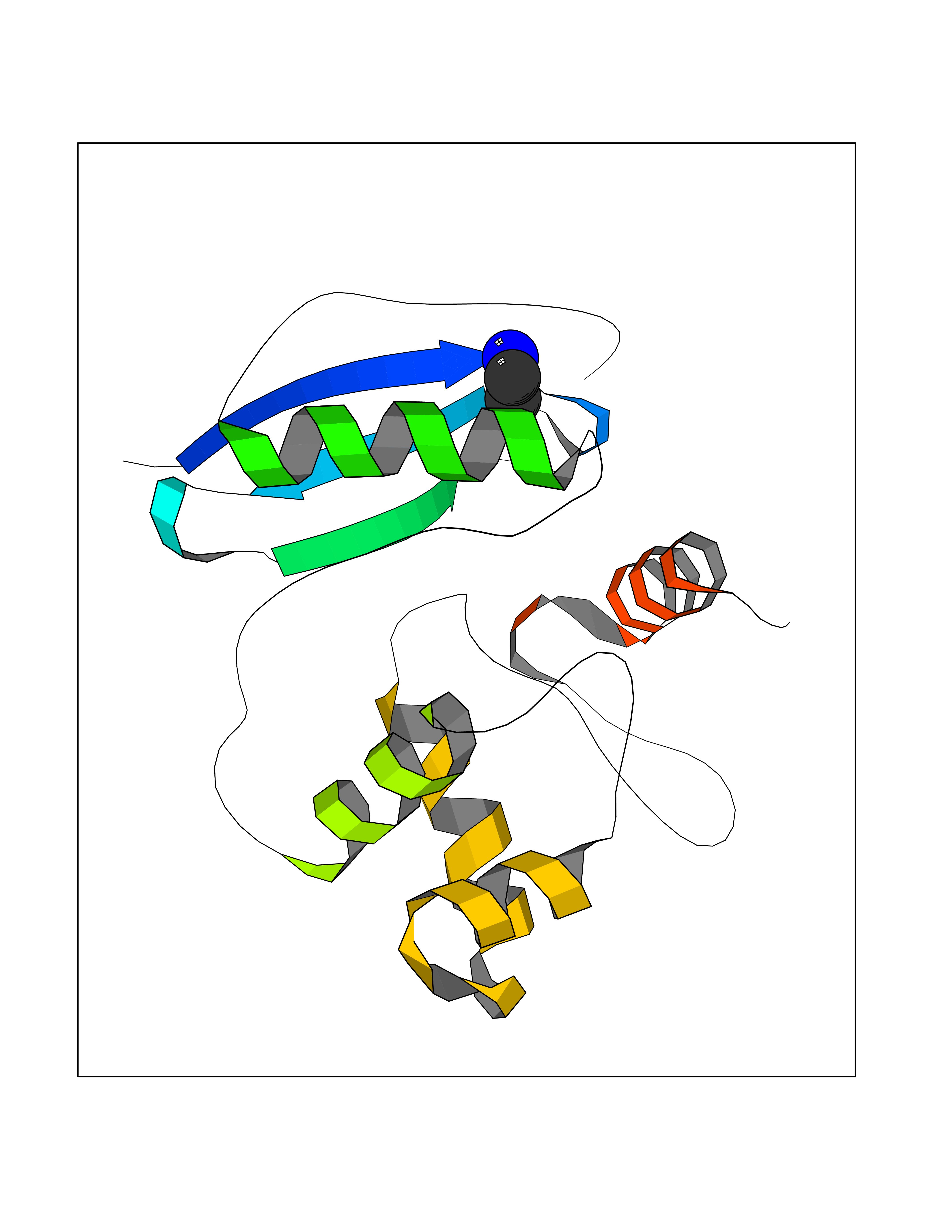 Molscript Map 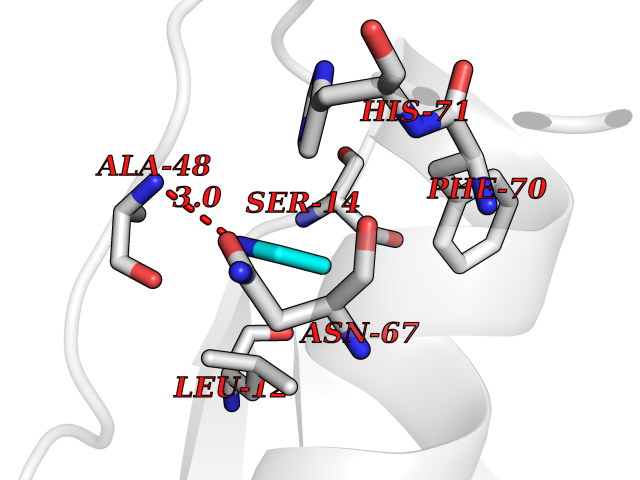 Pymol Map 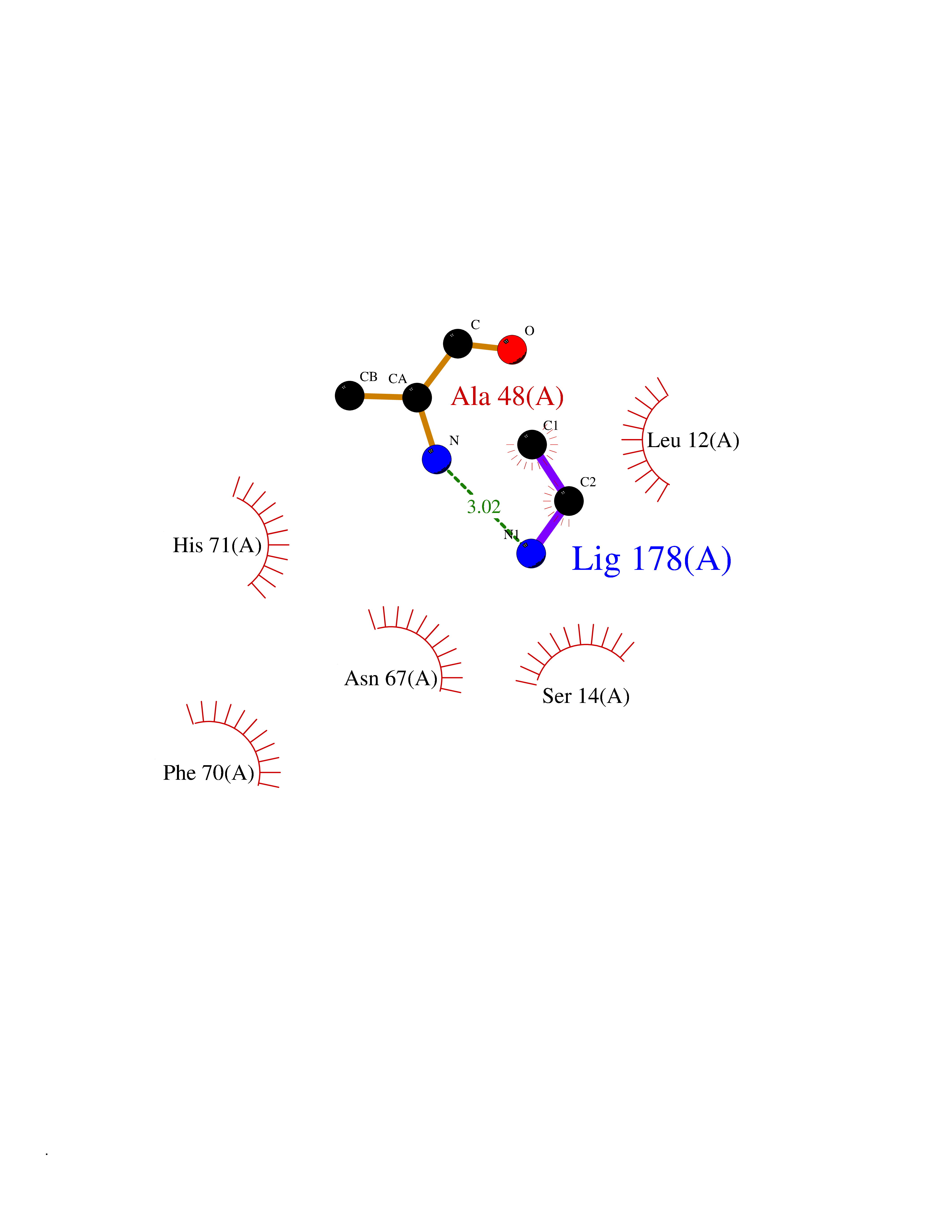 Ligplot Map 3D Binding mode Sequence EMKRTTLDSPLGKLELSGCEQGLHEIKLLGKDAVEVPAPAAVLGGPEPLMQCTAWLNAYFHQPEAIEEFPVPALHHPVFQQESFTRQVLWKLLKVVKFGEVISYQQLAALAGNPKAARAVGGAMRGNPVPILIPCHRVVCSSGAVGNYSGGLAVKEWLLAHEGHRL Hydrogen bonds contact Hydrophobic contact | ||||
| 88 | Liver carboxylesterase (CES1) | 2H7C | 4.27 | |
Target general information Gen name CES1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Serine esterase 1; Monocyte/macrophage serine esterase; Human carboxylesterase 1; HMSE; HCE1; CES1; Brain carboxylesterase hBr1; Acyl coenzyme A:cholesterol acyltransferase Protein family Type-B carboxylesterase/lipase family Biochemical class Carboxylic ester hydrolase Function Involved in the detoxification of xenobiotics and in the activation of ester and amide prodrugs. Hydrolyzes aromatic and aliphatic esters, but has no catalytic activity toward amides or a fatty acyl coa ester. Related diseases Protoporphyria, erythropoietic, 1 (EPP1) [MIM:177000]: An autosomal recessive form of porphyria with onset usually before age 10 years. Porphyrias are inherited defects in the biosynthesis of heme, resulting in the accumulation and increased excretion of porphyrins or porphyrin precursors. They are classified as erythropoietic or hepatic, depending on whether the enzyme deficiency occurs in red blood cells or in the liver. Erythropoietic protoporphyria is marked by excessive protoporphyrin in erythrocytes, plasma, liver and feces, and by widely varying photosensitive skin changes ranging from a burning or pruritic sensation to erythema, edema and wheals. {ECO:0000269|PubMed:10942404, ECO:0000269|PubMed:11375302, ECO:0000269|PubMed:12063482, ECO:0000269|PubMed:12601550, ECO:0000269|PubMed:1376018, ECO:0000269|PubMed:15286165, ECO:0000269|PubMed:17196862, ECO:0000269|PubMed:1755842, ECO:0000269|PubMed:7910885, ECO:0000269|PubMed:8757534, ECO:0000269|PubMed:9211198, ECO:0000269|PubMed:9585598, ECO:0000269|PubMed:9740232}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07821; DB08224; DB03056; DB06442; DB01086; DB09061; DB14737; DB01101; DB02659; DB01410; DB00758; DB00907; DB04838; DB06695; DB00647; DB01452; DB00470; DB01039; DB14845; DB00875; DB11181; DB02161; DB00328; DB00762; DB00583; DB14009; DB00454; DB06693; DB00688; DB03721; DB01183; DB00198; DB09269; DB01599; DB09342; DB00881; DB14761; DB12404; DB06201; DB11362; DB00641; DB00382; DB00675; DB12095; DB09299; DB04795; DB00519; DB16349 Interacts with Q8IVF2-3; A8MQ03; Q15125; Q5T7V8; Q5T749; Q13113; Q9NRQ2; Q9NR31; O95231 EC number EC 3.1.1.1 Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; Direct protein sequencing; Disulfide bond; Endoplasmic reticulum; Glycoprotein; Hydrolase; Lipid droplet; Lipid metabolism; Phosphoprotein; Proteomics identification; Reference proteome; Serine esterase; Signal Protein physicochemical properties Chain ID A,B,C,D,E,F Molecular weight (Da) 58522.7 Length 531 Aromaticity 0.09 Instability index 35.86 Isoelectric point 6.05 Charge (pH=7) -5.56 2D Binding mode Binding energy (Kcal/mol) -5.82 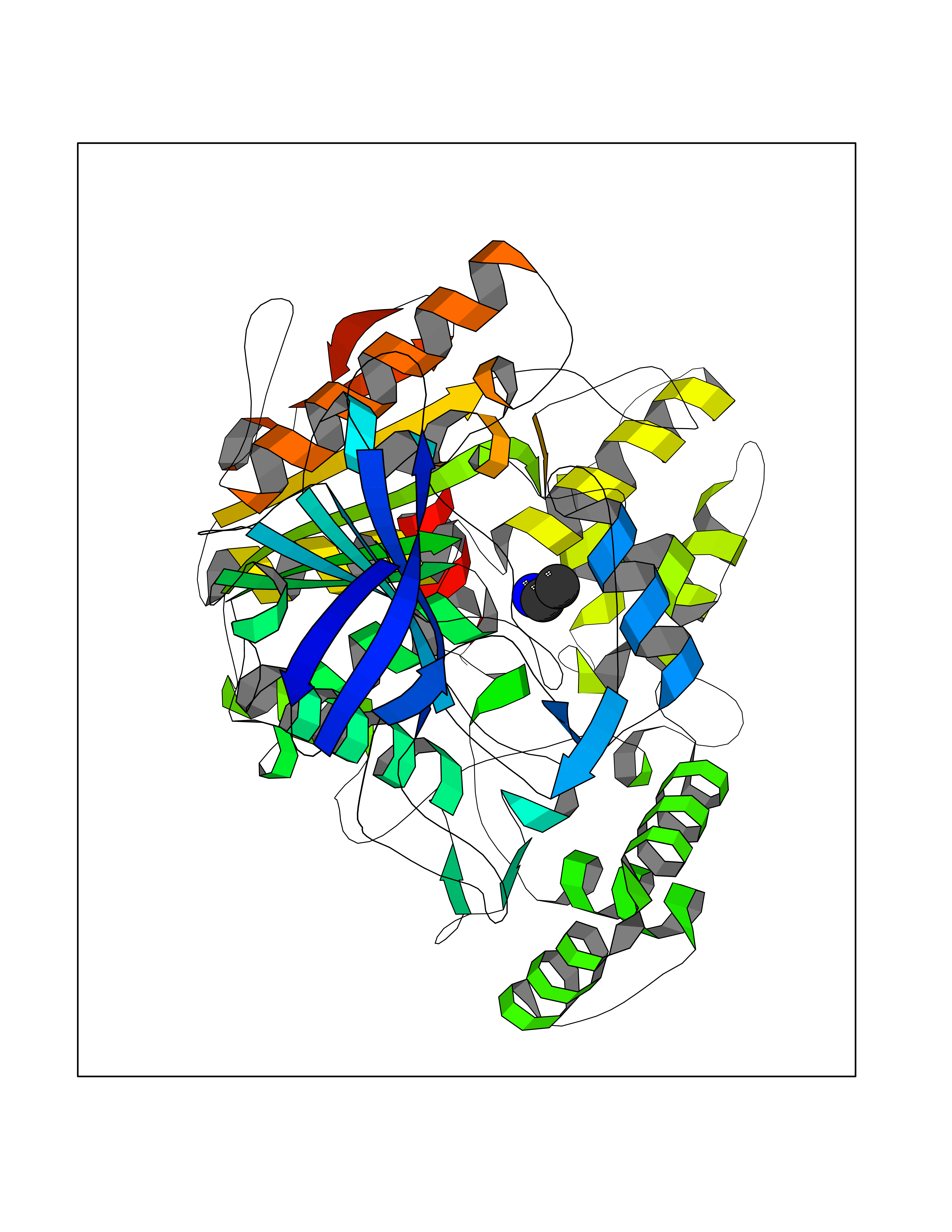 Molscript Map 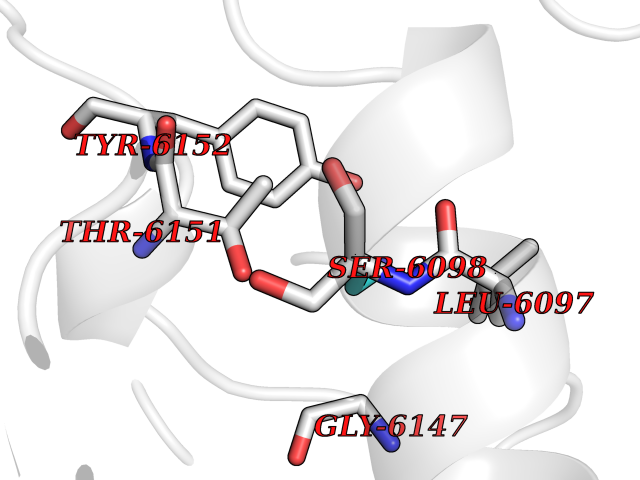 Pymol Map  Ligplot Map 3D Binding mode Sequence SPPVVDTVHGKVLGKFVSLEGFAQPVAIFLGIPFAKPPLGPLRFTPPQPAEPWSFVKNATSYPPMCTQDPKAGQLLSELFTNRKENIPLKLSEDCLYLNIYTPADLTKKNRLPVMVWIHGGGLMVGAASTYDGLALAAHENVVVVTIQYRLGIWGFFSTGDEHSRGNWGHLDQVAALRWVQDNIASFGGNPGSVTIFGESAGGESVSVLVLSPLAKNLFHRAISESGVALTSVLVKKGDVKPLAEQIAITAGCKTTTSAVMVHCLRQKTEEELLETTLKMKFLSLDLQGDPRESQPLLGTVIDGMLLLKTPEELQAERNFHTVPYMVGINKQEFGWLIPMLMSYPLSEGQLDQKTAMSLLWKSYPLVCIAKELIPEATEKYLGGTDDTVKKKDLFLDLIADVMFGVPSVIVARNHRDAGAPTYMYEFQYRPSFSSDMKPKTVIGDHGDELFSVFGAPFLKEGASEEEIRLSKMVMKFWANFARNGNPNGEGLPHWPEYNQKEGYLQIGANTQAAQKLKDKEVAFWTNLFAK Hydrogen bonds contact Hydrophobic contact | ||||
| 89 | Serum paraoxonase/arylesterase 1 | 1V04 | 4.27 | |
Target general information Gen name PON1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms PON Protein family Paraoxonase family Biochemical class Hydrolase Function Acyl-L-homoserine-lactone lactonohydrolase activity.Aryldialkylphosphatase activity.Arylesterase activity.Calcium ion binding.Phospholipid binding.Protein homodimerization activity. Related diseases Microvascular complications of diabetes 5 (MVCD5) [MIM:612633]: Pathological conditions that develop in numerous tissues and organs as a consequence of diabetes mellitus. They include diabetic retinopathy, diabetic nephropathy leading to end-stage renal disease, and diabetic neuropathy. Diabetic retinopathy remains the major cause of new-onset blindness among diabetic adults. It is characterized by vascular permeability and increased tissue ischemia and angiogenesis. Disease susceptibility is associated with variants affecting the gene represented in this entry. Homozygosity for the Leu-55 allele is strongly associated with the development of retinal disease in diabetic patients. Drugs (DrugBank ID) DB01327; DB09130; DB01395; DB14598; DB14600; DB14596; DB00218; DB01085; DB01593; DB14487; DB14533; DB14548 Interacts with NA EC number 3.1.1.2; 3.1.1.81; 3.1.8.1 Uniprot keywords 3D-structure; Calcium; Direct protein sequencing; Disulfide bond; Glycoprotein; HDL; Hydrolase; Metal-binding; Proteomics identification; Reference proteome; Secreted; Signal Protein physicochemical properties Chain ID A Molecular weight (Da) 37232.8 Length 332 Aromaticity 0.11 Instability index 35.09 Isoelectric point 5.06 Charge (pH=7) -17.08 2D Binding mode Binding energy (Kcal/mol) -5.82  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence LFDRQKSSFQTRFNVHREVTPVELPNCNLVKGIDNGSEDLEILPNGLAFISSGLKYDKSGKILLMDLNEKEPAVSELEIIGNTLDISSFNPHGISTFIDDDNTVYLLVVNHPGSSSTVEVFKFQEEEKSLLHLKTIRHKLLPSVNDIVAVGPEHFYATNDHYFIDPYLKSWEMHLGLAWSFVTYYSPNDVRVVAEGFDFANGINISPDGKYVYIAELLAHKIHVYEKHANWTLTPLRVLSFDTLVDNISVDPVTGDLWVGCHPNGMRIFFYDAENPPGSEVLRIQDILSEEPKVTVVYAENGTVLQGSTVAAVYKGKLLIGTVFHKALYCDL Hydrogen bonds contact Hydrophobic contact | ||||
| 90 | Flavodoxin/ferredoxin--NADP reductase | 1FDR | 4.27 | |
Target general information Gen name fpr Organism Escherichia coli (strain K12) Uniprot ID TTD ID NA Synonyms mvrA;b3924;JW3895 Protein family Ferredoxin--NADP reductase type 1 family Biochemical class Flavoprotein Function FAD binding.Ferredoxin-NADP+ reductase activity.Oxidoreductase activity. Related diseases Noonan syndrome 13 (NS13) [MIM:619087]: A form of Noonan syndrome, a disease characterized by short stature, facial dysmorphic features such as hypertelorism, a downward eyeslant and low-set posteriorly rotated ears, and a high incidence of congenital heart defects and hypertrophic cardiomyopathy. Other features can include a short neck with webbing or redundancy of skin, deafness, motor delay, variable intellectual deficits, multiple skeletal defects, cryptorchidism, and bleeding diathesis. Individuals with Noonan syndrome are at risk of juvenile myelomonocytic leukemia, a myeloproliferative disorder characterized by excessive production of myelomonocytic cells. NS13 inheritance is autosomal dominant. There is considerable variability in severity. {ECO:0000269|PubMed:32721402}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03147 Interacts with NA EC number 1.18.1.2; 1.19.1.1 Uniprot keywords 3D-structure; Cytoplasm; Direct protein sequencing; FAD; Flavoprotein; NADP; Nucleotide-binding; Oxidoreductase; Reference proteome Protein physicochemical properties Chain ID A Molecular weight (Da) 27346.2 Length 244 Aromaticity 0.08 Instability index 30.68 Isoelectric point 7.25 Charge (pH=7) 0.42 2D Binding mode Binding energy (Kcal/mol) -5.82  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence ADWVTGKVTKVQNWTDALFSLTVHAPVLPFTAGQFTKLGLEIRVQRAYSYVNSPDNPDLEFYLVTVPDGKLSPRLAALKPGDEVQVVSEAAGFFVLDEVPHCETLWMLATGTAIGPYLSILRLGKDLDRFKNLVLVHAARYAADLSYLPLMQELEKRYEGKLRIQTVVSRETAAGSLTGRIPALIESGELESTIGLPMNKETSHVMLCGNPQMVRDTQQLLKETRQMTKHLRRRPGHMTAEHYW Hydrogen bonds contact Hydrophobic contact | ||||
| 91 | Inositol monophosphatase 1 | 1IMB | 4.27 | |
Target general information Gen name IMPA1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms IMPA Protein family Inositol monophosphatase superfamily Biochemical class Hydrolase Function Identical protein binding.Inositol monophosphate 1-phosphatase activity.Inositol monophosphate 3-phosphatase activity.Inositol monophosphate 4-phosphatase activity.Inositol monophosphate phosphatase activity.Lithium ion binding.Magnesium ion binding.Manganese ion binding.Protein homodimerization activity. Related diseases Intellectual developmental disorder, autosomal recessive 59 (MRT59) [MIM:617323]: A disorder characterized by significantly below average general intellectual functioning associated with impairments in adaptive behavior and manifested during the developmental period. {ECO:0000269|PubMed:26416544}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03542; DB14509; DB01356; DB14507; DB14508 Interacts with P29218; O14732; P54253; G5E9A7; P42858; P29218-3; O14732; Q7Z699 EC number 3.1.3.25; 3.1.3.94 Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; Hydrolase; Intellectual disability; Lithium; Magnesium; Metal-binding; Phosphoprotein; Proteomics identification; Reference proteome Protein physicochemical properties Chain ID A,B Molecular weight (Da) 59299.9 Length 544 Aromaticity 0.07 Instability index 37.53 Isoelectric point 5.38 Charge (pH=7) -11.58 2D Binding mode Binding energy (Kcal/mol) -5.82  Molscript Map 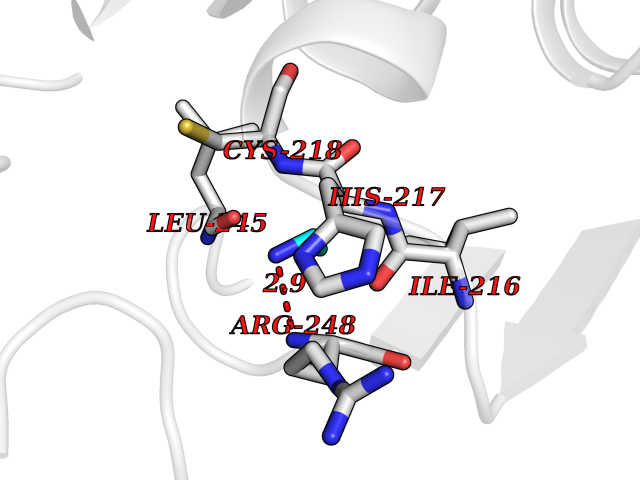 Pymol Map 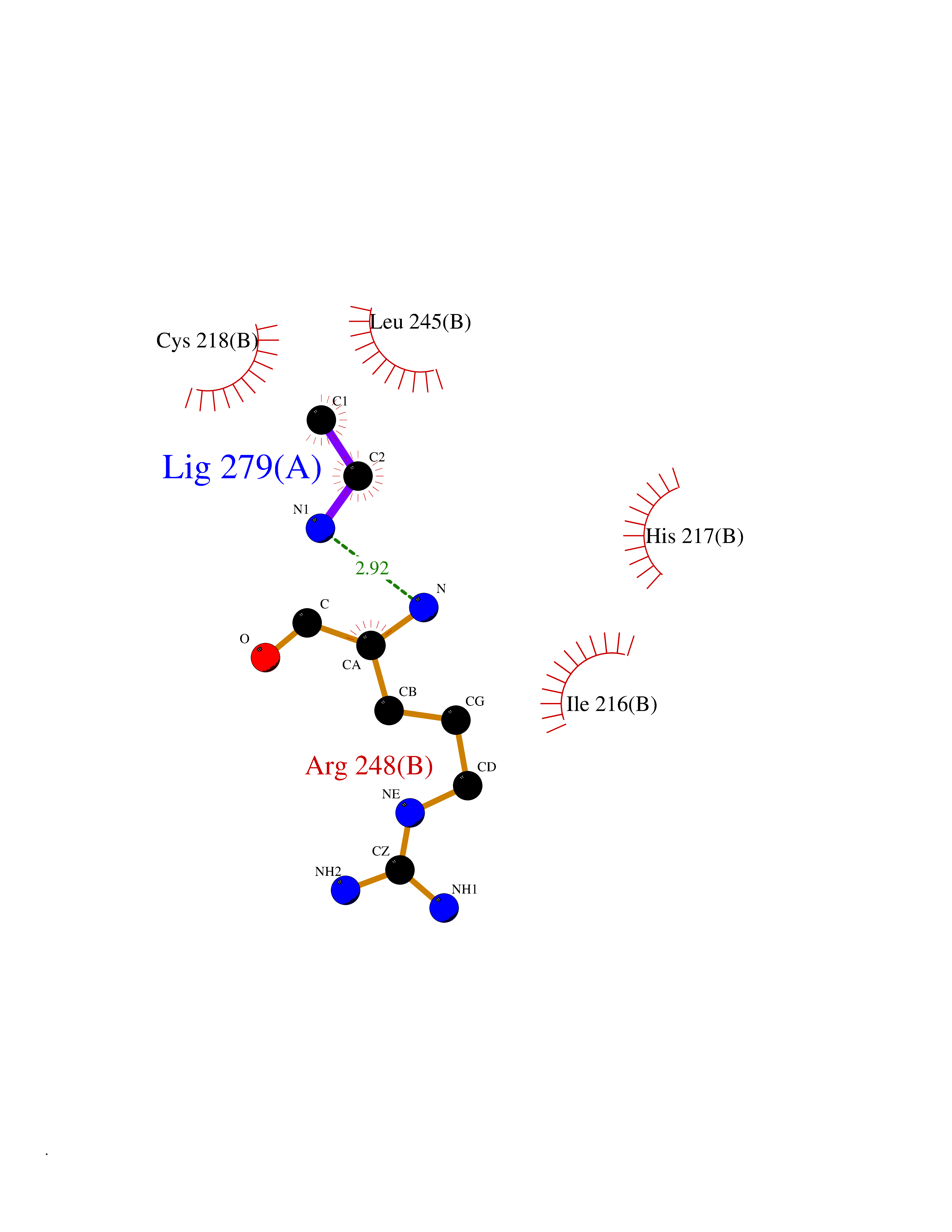 Ligplot Map 3D Binding mode Sequence WQECMDYAVTLARQAGEVVCEAIKNEMNVMLKSSPVDLVTATDQKVEKMLISSIKEKYPSHSFIGEESVAAGEKSILTDNPTWIIDPIDGTTNFVHRFPFVAVSIGFAVNKKIEFGVVYSCVEGKMYTARKGKGAFCNGQKLQVSQQEDITKSLLVTELGSSRTPETVRMVLSNMEKLFCIPVHGIRSVGTAAVNMCLVATGGADAYYEMGIHCWDVAGAGIIVTEAGGVLMDVTGGPFDLMSRRVIAANNRILAERIAKEIQVIPLQRDDEWQECMDYAVTLARQAGEVVCEAIKNEMNVMLKSSPVDLVTATDQKVEKMLISSIKEKYPSHSFIGEESVAAGEKSILTDNPTWIIDPIDGTTNFVHRFPFVAVSIGFAVNKKIEFGVVYSCVEGKMYTARKGKGAFCNGQKLQVSQQEDITKSLLVTELGSSRTPETVRMVLSNMEKLFCIPVHGIRSVGTAAVNMCLVATGGADAYYEMGIHCWDVAGAGIIVTEAGGVLMDVTGGPFDLMSRRVIAANNRILAERIAKEIQVIPLQRDDE Hydrogen bonds contact Hydrophobic contact | ||||
| 92 | Phosphodiesterase 4B (PDE4B) | 4KP6 | 4.27 | |
Target general information Gen name PDE4B Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms cAMP-specific 3',5'-cyclic phosphodiesterase 4B; Type 4B cAMP phosphodiesterase; Type 4 cyclic adenosine monophosphate phosphodiesterase (type 4 PDE); PDE32; DPDE4 Protein family Cyclic nucleotide phosphodiesterase family, PDE4 subfamily Biochemical class Phosphoric diester hydrolase Function May be involved in mediating central nervous system effects of therapeutic agents ranging from antidepressants to antiasthmatic and anti-inflammatory agents. Hydrolyzes the second messenger cAMP, which is a key regulator of many important physiological processes. Related diseases Combined oxidative phosphorylation deficiency 33 (COXPD33) [MIM:617713]: An autosomal recessive disorder caused by multiple mitochondrial respiratory chain defects and impaired mitochondrial energy metabolism. Clinical manifestations are highly variable. Affected infants present with cardiomyopathy accompanied by multisystemic features involving liver, kidney, and brain. Death in infancy is observed in some patients. Children and adults present with myopathy and progressive external ophthalmoplegia. {ECO:0000269|PubMed:28942965}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB04149; DB03606; DB03807; DB06909; DB01959; DB08299; DB03349; DB00131; DB01427; DB00201; DB03849; DB05219; DB01647; DB00651; DB00824; DB02660; DB05266; DB01088; DB01113; DB01791; DB01656; DB01954; DB04530; DB01412; DB00277; DB09283 Interacts with Q13936; Q08499 EC number EC 3.1.4.53 Uniprot keywords 3D-structure; Alternative splicing; cAMP; Cell membrane; Cytoplasm; Hydrolase; Membrane; Metal-binding; Phosphoprotein; Proteomics identification; Reference proteome; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 37367.1 Length 324 Aromaticity 0.08 Instability index 36.36 Isoelectric point 5 Charge (pH=7) -20.82 2D Binding mode Binding energy (Kcal/mol) -5.83 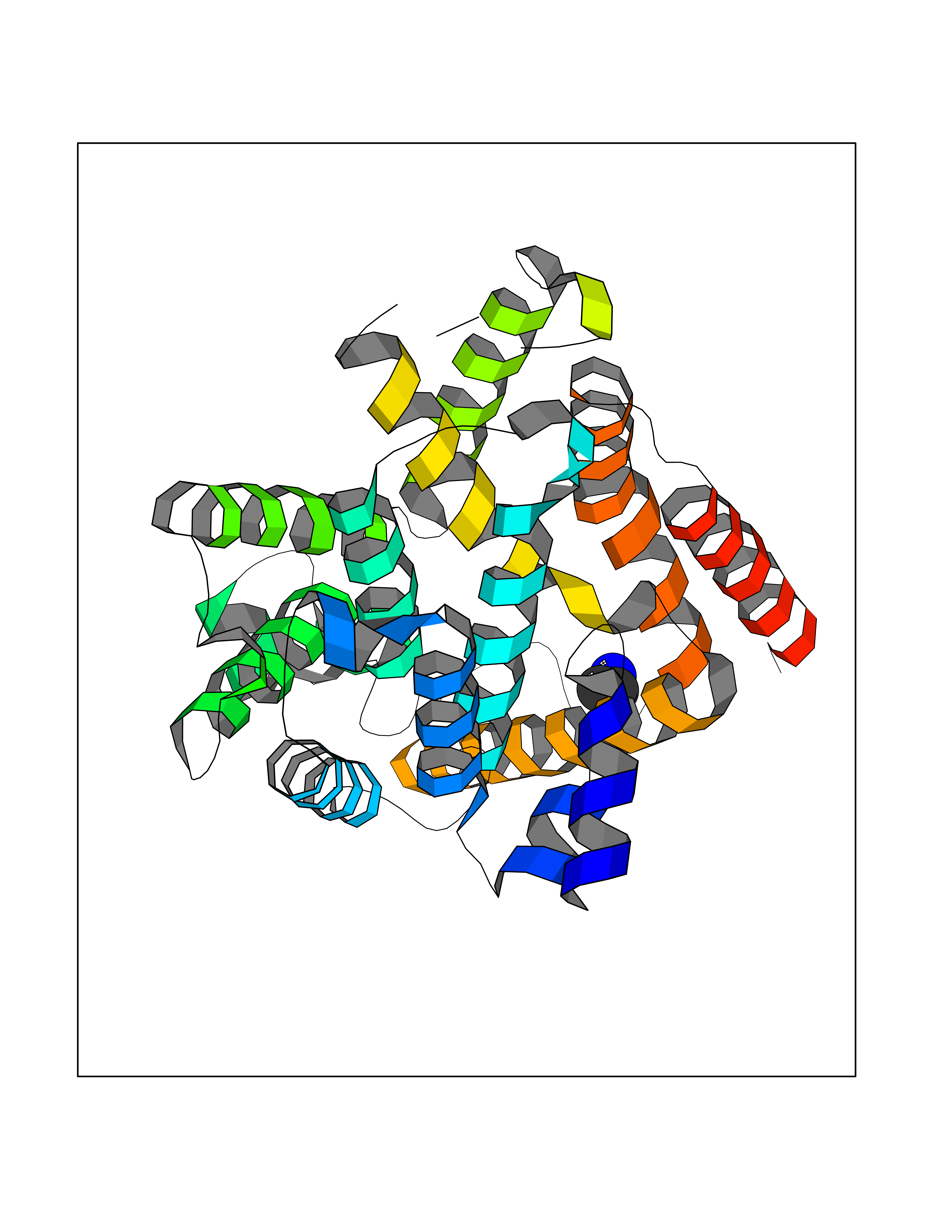 Molscript Map 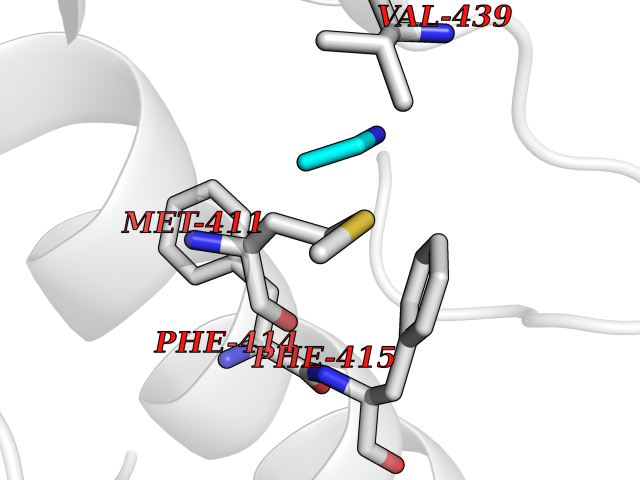 Pymol Map 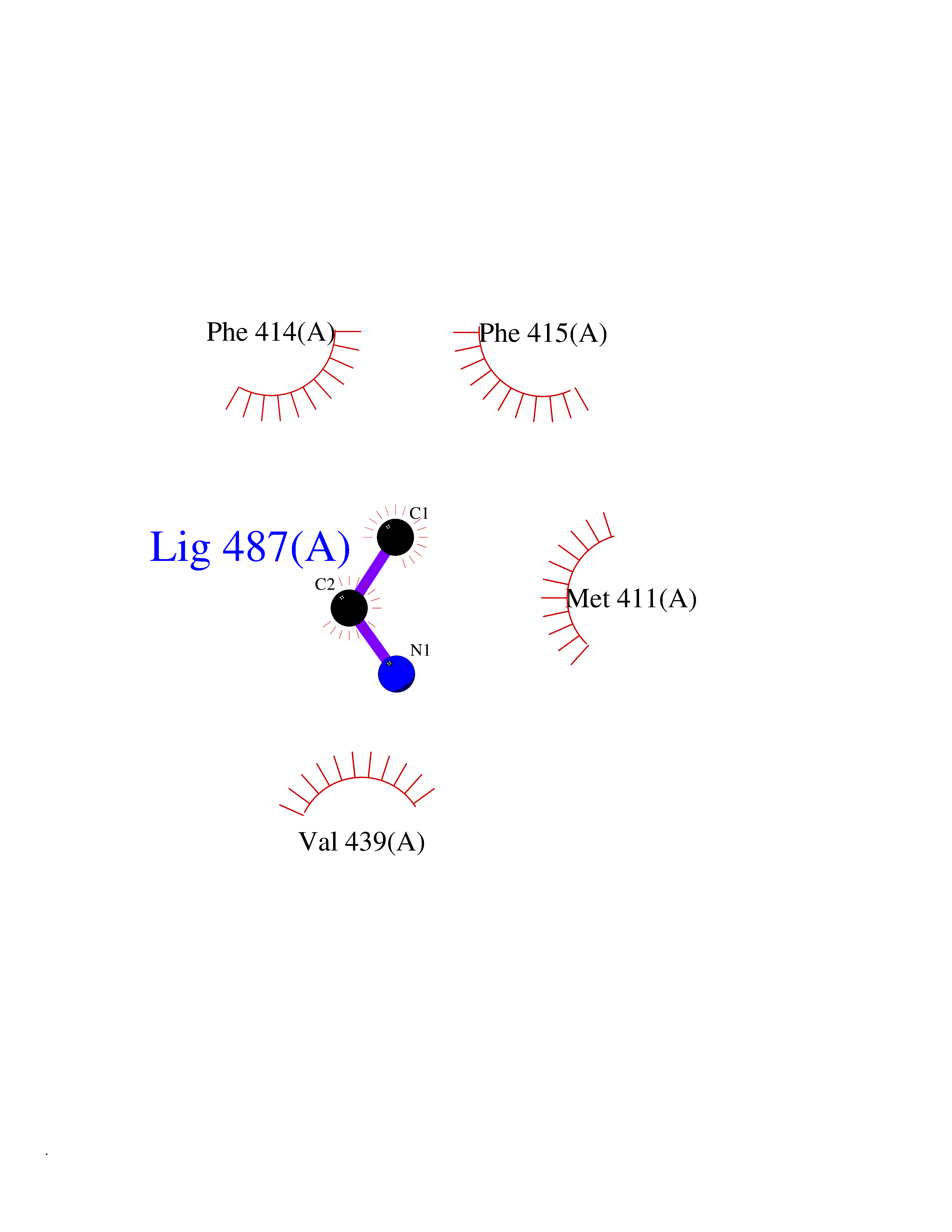 Ligplot Map 3D Binding mode Sequence NEDHLAKELEDLNKWGLNIFNVAGYSHNRPLTCIMYAIFQERDLLKTFRISSDTFITYMMTLEDHYHSDVAYHNSLHAADVAQSTHVLLSTPALDAVFTDLEILAAIFAAAIHDVDHPGVSNQFLINTNSELALMYNDESVLENHHLAVGFKLLQEEHCDIFMNLTKKQRQTLRKMVIDMVLATDMSKHMSLLADLKTMVETKKVTSSGVLLLDNYTDRIQVLRNMVHCADLSNPTKSLELYRQWTDRIMEEFFQQGDKERERGMEISPMCDKHTASVEKSQVGFIDYIVHPLWETWADLVQPDAQDILDTLEDNRNWYQSMIP Hydrogen bonds contact Hydrophobic contact | ||||
| 93 | FAD-linked sulfhydryl oxidase ALR | 3U5S | 4.27 | |
Target general information Gen name GFER Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms HERV1;HPO;ALR Protein family NA Biochemical class Oxidoreductase Function Flavin adenine dinucleotide binding.Flavin-linked sulfhydryl oxidase activity.Growth factor activity.Protein disulfide oxidoreductase activity. Related diseases Myopathy, mitochondrial progressive, with congenital cataract, hearing loss and developmental delay (MPMCD) [MIM:613076]: A disease characterized by progressive myopathy and partial combined respiratory-chain deficiency, congenital cataract, sensorineural hearing loss, and developmental delay. {ECO:0000269|PubMed:19409522, ECO:0000269|PubMed:20593814}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03147 Interacts with Q9H8W4; PRO_0000449628 [P0DTD1] EC number 1.8.3.2 Uniprot keywords 3D-structure; Alternative splicing; Cataract; Cytoplasm; Deafness; Disease variant; Disulfide bond; FAD; Flavoprotein; Growth factor; Mitochondrion; Oxidoreductase; Phosphoprotein; Primary mitochondrial disease; Proteomics identification; Reference proteome; Secreted Protein physicochemical properties Chain ID A Molecular weight (Da) 14170.4 Length 126 Aromaticity 0.11 Instability index 48.59 Isoelectric point 6.58 Charge (pH=7) -0.98 2D Binding mode Binding energy (Kcal/mol) -5.82 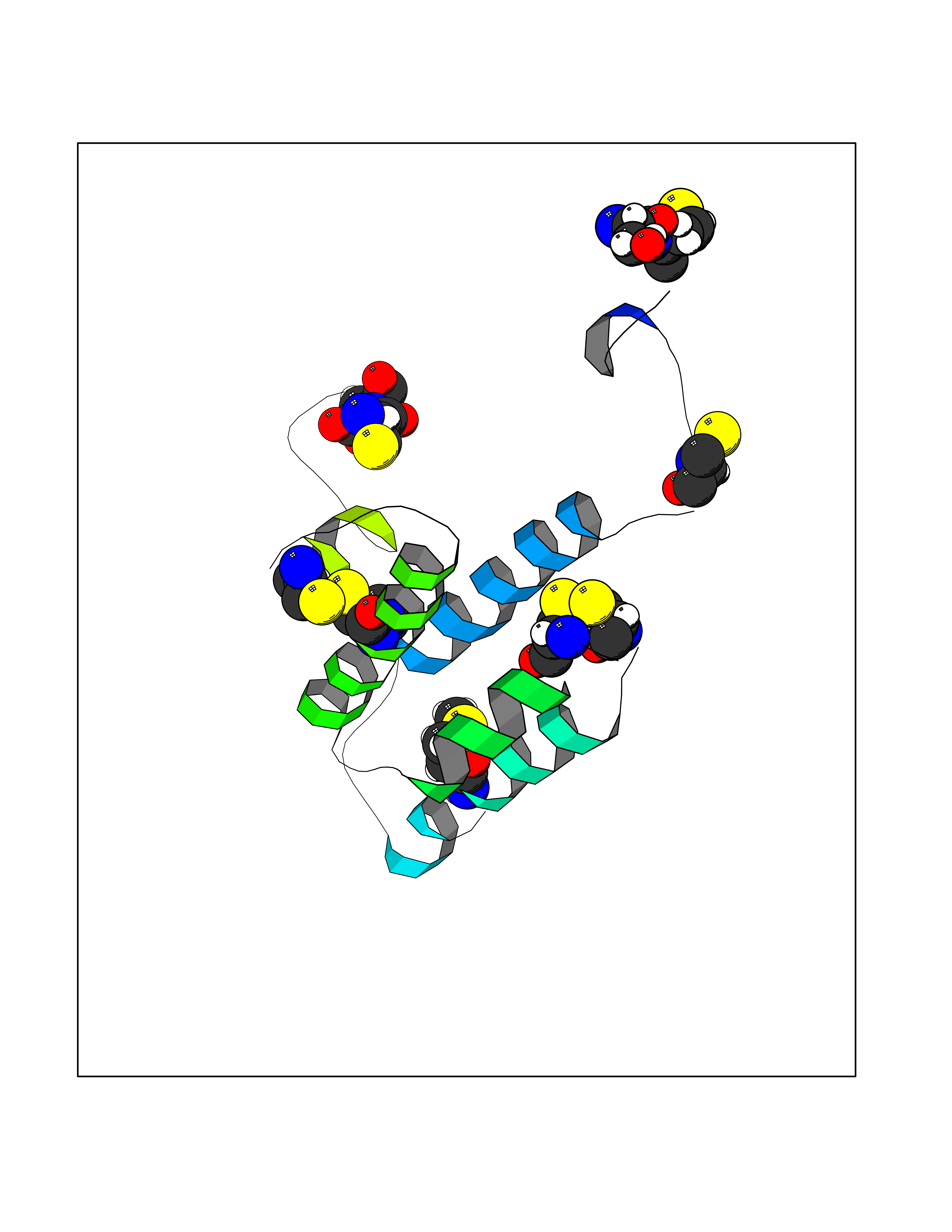 Molscript Map  Pymol Map 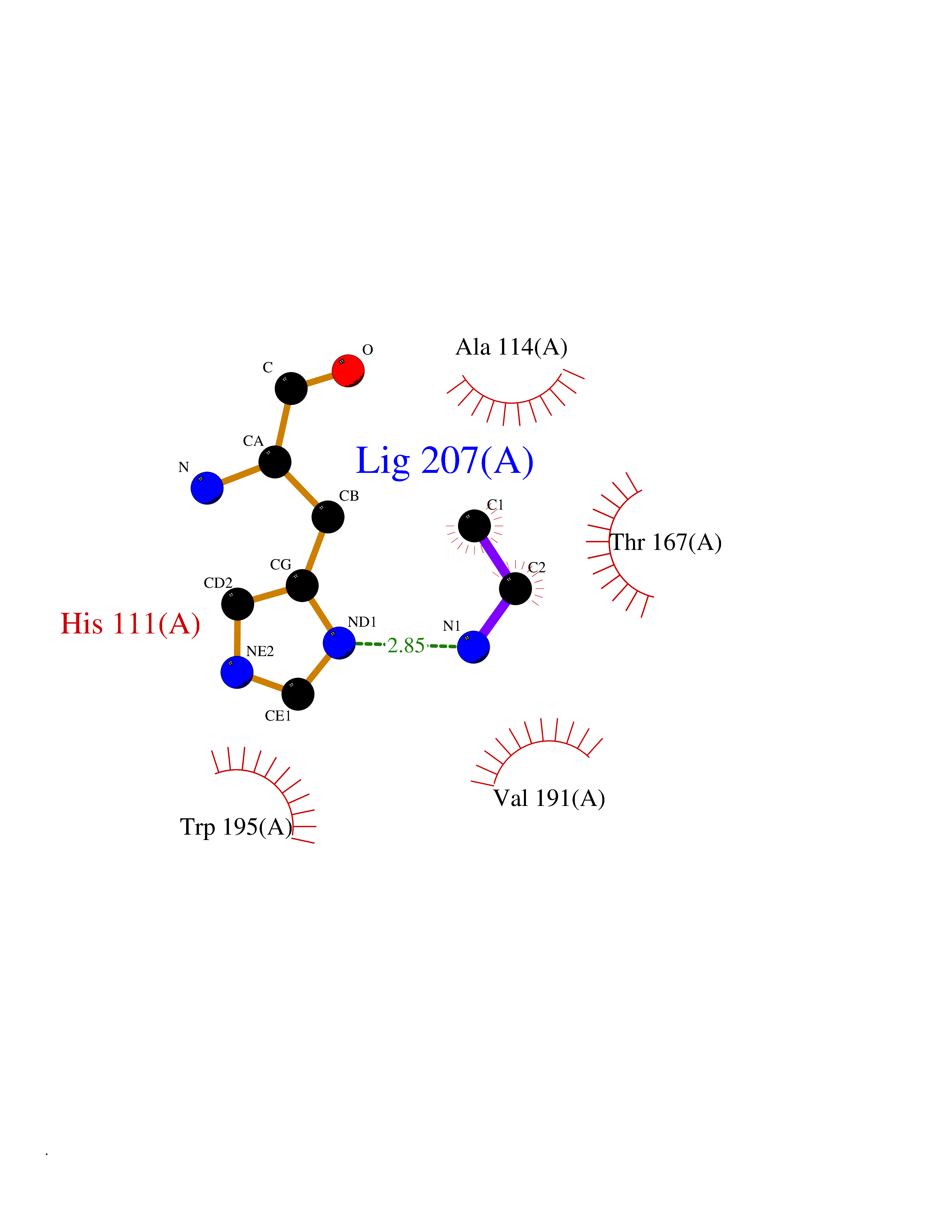 Ligplot Map 3D Binding mode Sequence SXRTQQKRDTKFREDUPPDREELGRHSWAVLHTLAAYYPDLPTPEQQQDXAQFIHLFSKFYPUEEUAEDLRKRLARNHPDTRTRAAFTQWLUHLHNEVNRKLGKPDFDUSKVDERWRDGWKDGSUD Hydrogen bonds contact Hydrophobic contact | ||||
| 94 | 2-oxopropyl-CoM reductase, carboxylating | 1MO9 | 4.27 | |
Target general information Gen name xecC Organism Xanthobacter autotrophicus (strain ATCC BAA-1158 / Py2) Uniprot ID TTD ID NA Synonyms Xaut_4867 Protein family Class-I pyridine nucleotide-disulfide oxidoreductase family Biochemical class Oxidoreductase Function 2-oxopropyl-CoM reductase (carboxylating) activity.Flavin adenine dinucleotide binding. Related diseases LTC4 synthase deficiency is associated with a neurometabolic developmental disorder characterized by muscular hypotonia, psychomotor retardation, failure to thrive, and microcephaly. {ECO:0000269|PubMed:10896305, ECO:0000269|PubMed:9820300}. Drugs (DrugBank ID) DB03163; DB03147 Interacts with NA EC number 1.8.1.5 Uniprot keywords 3D-structure; Disulfide bond; FAD; Flavoprotein; NADP; Oxidoreductase; Plasmid; Redox-active center; Reference proteome Protein physicochemical properties Chain ID A,B Molecular weight (Da) 114413 Length 1044 Aromaticity 0.08 Instability index 25.66 Isoelectric point 5.68 Charge (pH=7) -21.74 2D Binding mode Binding energy (Kcal/mol) -5.83 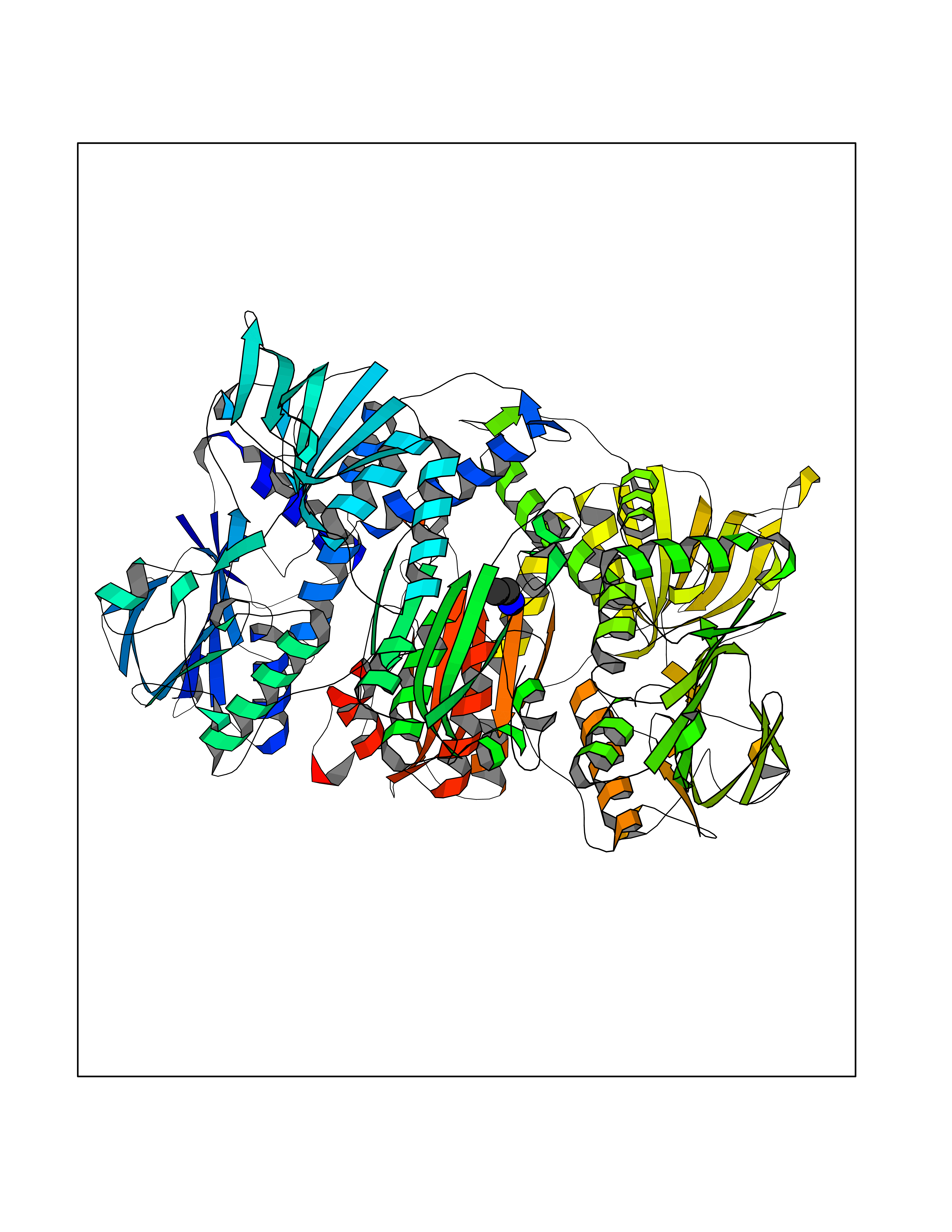 Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence KVWNARNDHLTINQWATRIDEILEAPDGGEVIYNVDENDPREYDAIFIGGGAAGRFGSAYLRAMGGRQLIVDRWPFLGGSCPHNACVPHHLFSDCAAELMLARTFSGQYWFPDMTEKVVGIKEVVDLFRAGRNGPHGIMNFQSKEQLNLEYILNCPAKVIDNHTVEAAGKVFKAKNLILAVGAGPGTLDVPGVNAKGVFDHATLVEELDYEPGSTVVVVGGSKTAVEYGCFFNATGRRTVMLVRTEPLKLIKDNETRAYVLDRMKEQGMEIISGSNVTRIEEDANGRVQAVVAMTPNGEMRIETDFVFLGLGEQPRSAELAKILGLDLGPKGEVLVNEYLQTSVPNVYAVGDLIGGPMEMFKARKSGCYAARNVMGEKISYTPKNYPDFLHTHYEVSFLGMGEEEARAAGHEIVTIKMPPDTENGLNVALPASDRTMLYAFGKGTAHMSGFQKIVIDAKTRKVLGAHHVGYGAKDAFQYLNVLIKQGLTVDELGDMDELFLNPTHFIQLSRLRAGSKNLVSLKVWNARNDHLTINQWATRIDEILEAPDGGEVIYNVDENDPREYDAIFIGGGAAGRFGSAYLRAMGGRQLIVDRWPFLGGSCPHNACVPHHLFSDCAAELMLARTFSGQYWFPDMTEKVVGIKEVVDLFRAGRNGPHGIMNFQSKEQLNLEYILNCPAKVIDNHTVEAAGKVFKAKNLILAVGAGPGTLDVPGVNAKGVFDHATLVEELDYEPGSTVVVVGGSKTAVEYGCFFNATGRRTVMLVRTEPLKLIKDNETRAYVLDRMKEQGMEIISGSNVTRIEEDANGRVQAVVAMTPNGEMRIETDFVFLGLGEQPRSAELAKILGLDLGPKGEVLVNEYLQTSVPNVYAVGDLIGGPMEMFKARKSGCYAARNVMGEKISYTPKNYPDFLHTHYEVSFLGMGEEEARAAGHEIVTIKMPPDTENGLNVALPASDRTMLYAFGKGTAHMSGFQKIVIDAKTRKVLGAHHVGYGAKDAFQYLNVLIKQGLTVDELGDMDELFLNPTHFIQLSRLRAGSKNLVSL Hydrogen bonds contact Hydrophobic contact | ||||
| 95 | Endonuclease 8-like 1 | 5ITQ | 4.27 | |
Target general information Gen name NEIL1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family FPG family Biochemical class Dna binding protein / dna Function Class I DNA-(apurinic or apyrimidinic site) lyase activity.Class III/IV DNA-(apurinic or apyrimidinic site) lyase activity.Damaged DNA binding.DNA-(apurinic or apyrimidinic site) lyase activity.DNA N-glycosylase activity.Hydrolase activity, acting on glycosyl bonds.Protein C-terminus binding.Zinc ion binding. Related diseases Glutaric aciduria 1 (GA1) [MIM:231670]: An autosomal recessive metabolic disorder characterized by progressive dystonia and athetosis due to gliosis and neuronal loss in the basal ganglia. {ECO:0000269|PubMed:14707522, ECO:0000269|PubMed:18775954, ECO:0000269|PubMed:24973495, ECO:0000269|PubMed:8541831, ECO:0000269|PubMed:8900227, ECO:0000269|PubMed:8900228, ECO:0000269|PubMed:9600243, ECO:0000269|PubMed:9711871}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB09130; DB14490; DB14491; DB14488; DB14501; DB14489; DB01592 Interacts with NA EC number 3.2.2.-; 4.2.99.18 Uniprot keywords 3D-structure; Chromosome; Cytoplasm; Cytoskeleton; DNA damage; DNA repair; DNA-binding; Glycosidase; Hydrolase; Lyase; Multifunctional enzyme; Nucleus; Proteomics identification; Reference proteome; RNA editing Protein physicochemical properties Chain ID A Molecular weight (Da) 32177.5 Length 286 Aromaticity 0.1 Instability index 57.82 Isoelectric point 8.89 Charge (pH=7) 5.38 2D Binding mode Binding energy (Kcal/mol) -5.83 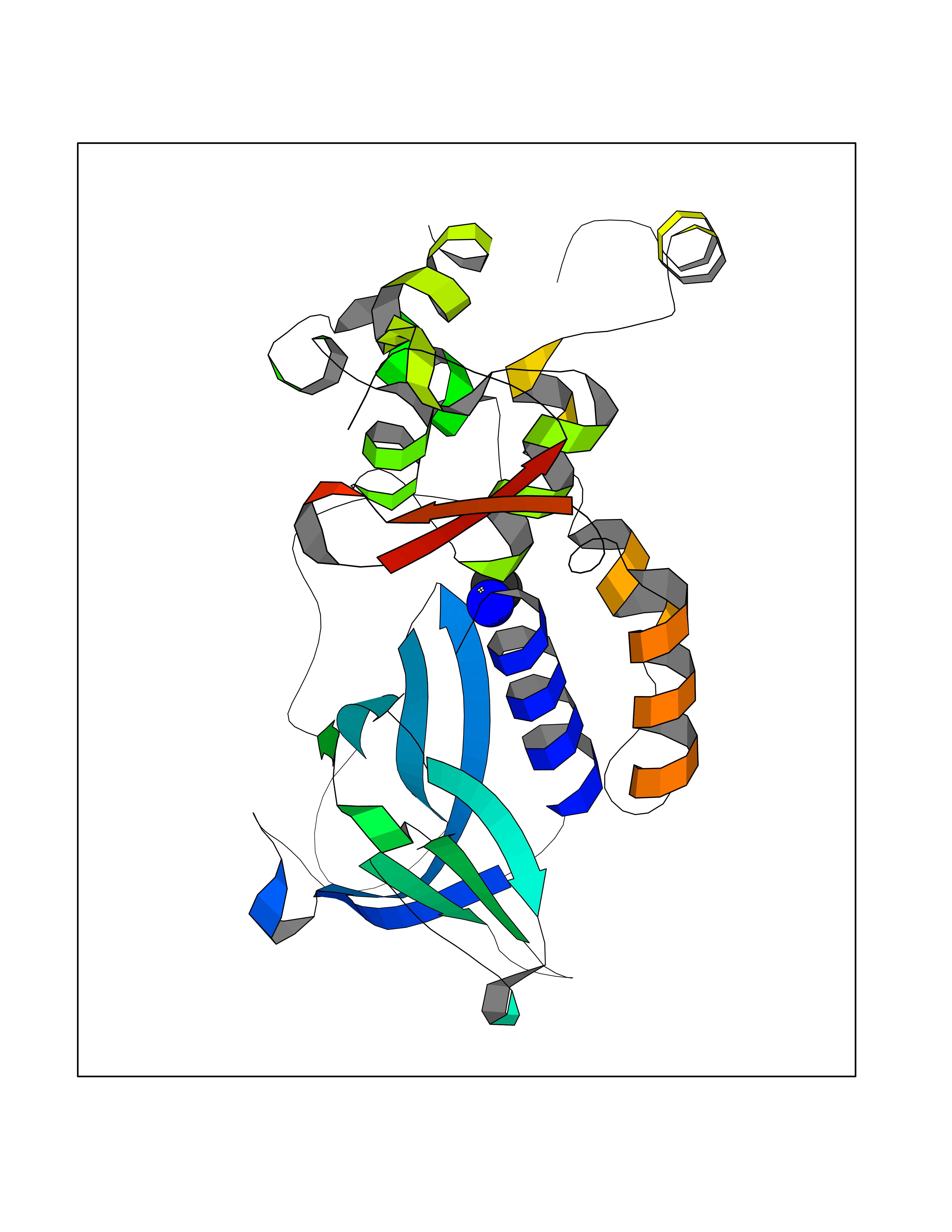 Molscript Map 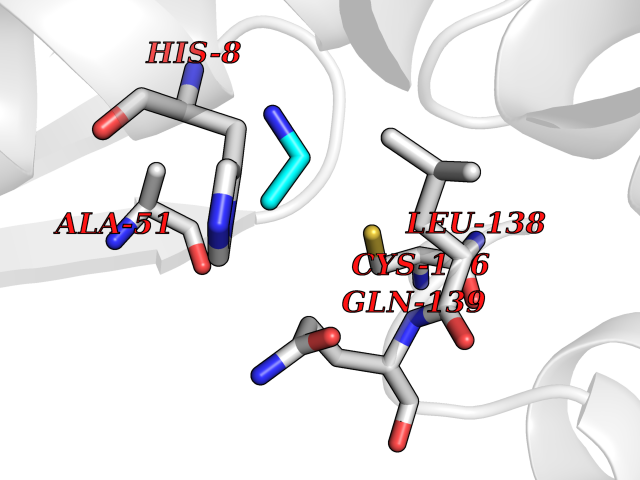 Pymol Map 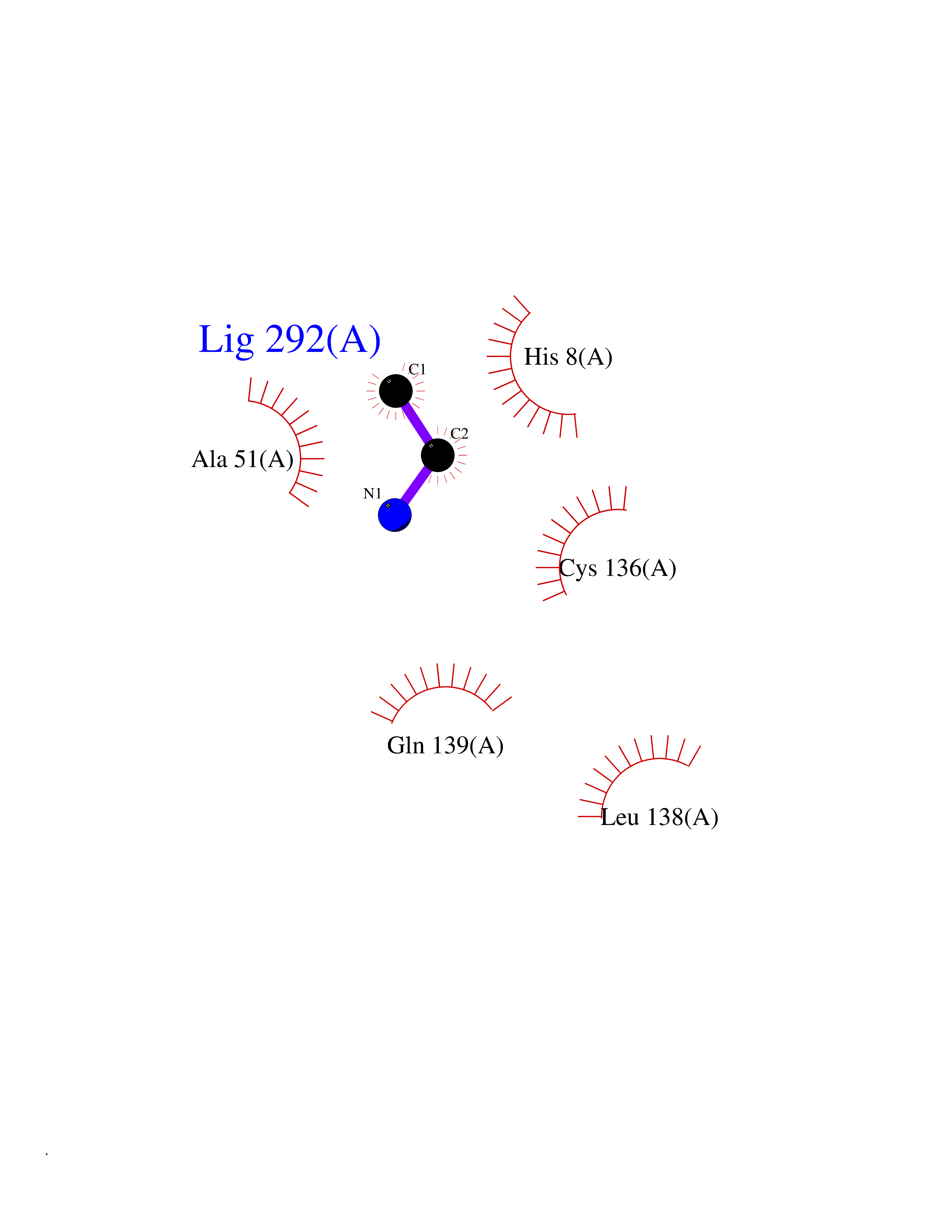 Ligplot Map 3D Binding mode Sequence PEGPELHLASQFVNEACRALVFGGCVEKSSVSRNPEVPFESSAYRISASARGKELRLILSPLPGAQPQQEPLALVFRFGMSGSFQLVPREELPRHAHLRFYTAPPGPRLALCFVDIRRFGRWDLGGKWQPGRGPCVLQEYQQFRESVLRNLADKAFDRPICEALLDQRFFNGIGNYLRAEILYRLKIPPFEKARSVLEALQQSPELTLSQKIRTKLQNPDLLELCHSVPKEVVQLGGRGYGSESGEEDFAAFRAWLRCYGMPGMSSLQDRHGRTIWFQGDPGPLAP Hydrogen bonds contact Hydrophobic contact | ||||
| 96 | Methylenetetrahydrofolate reductase | 1V93 | 4.27 | |
Target general information Gen name metF Organism Thermus thermophilus Uniprot ID TTD ID NA Synonyms NA Protein family Methylenetetrahydrofolate reductase family Biochemical class Oxidoreductase Function Methylenetetrahydrofolate reductase (NAD(P)H) activity.Nucleotide binding. Related diseases Brugada syndrome 7 (BRGDA7) [MIM:613120]: A tachyarrhythmia characterized by right bundle branch block and ST segment elevation on an electrocardiogram (ECG). It can cause the ventricles to beat so fast that the blood is prevented from circulating efficiently in the body. When this situation occurs, the individual will faint and may die in a few minutes if the heart is not reset. {ECO:0000269|PubMed:20031595}. The gene represented in this entry may be involved in disease pathogenesis.; DISEASE: Atrial fibrillation, familial, 16 (ATFB16) [MIM:613120]: A familial form of atrial fibrillation, a common sustained cardiac rhythm disturbance. Atrial fibrillation is characterized by disorganized atrial electrical activity and ineffective atrial contraction promoting blood stasis in the atria and reduces ventricular filling. It can result in palpitations, syncope, thromboembolic stroke, and congestive heart failure. {ECO:0000269|PubMed:20558140, ECO:0000269|PubMed:21051419}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03147 Interacts with NA EC number NA Uniprot keywords 3D-structure; FAD; Flavoprotein; Nucleotide-binding; Oxidoreductase Protein physicochemical properties Chain ID A Molecular weight (Da) 32632.1 Length 292 Aromaticity 0.09 Instability index 39.75 Isoelectric point 8.99 Charge (pH=7) 3.35 2D Binding mode Binding energy (Kcal/mol) -5.82 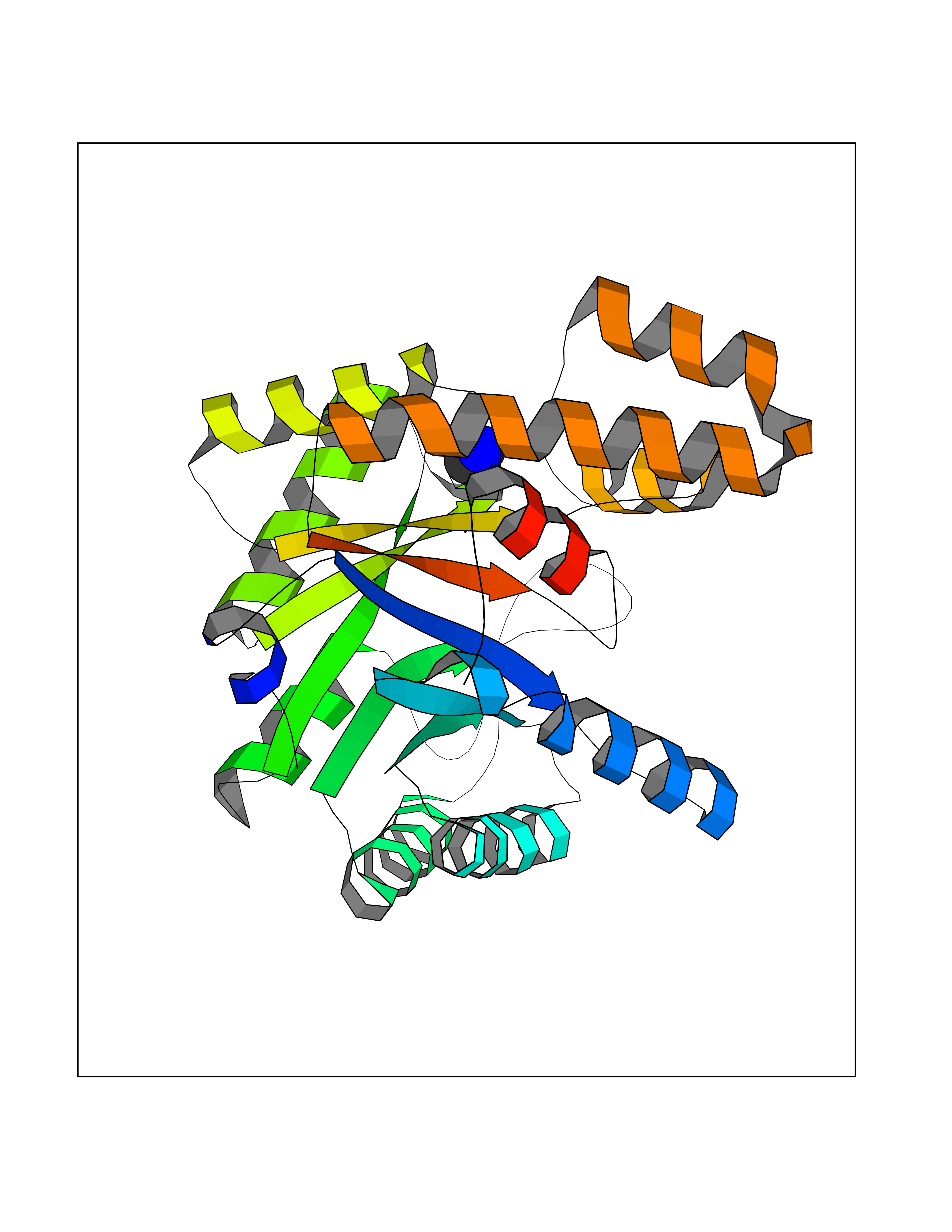 Molscript Map 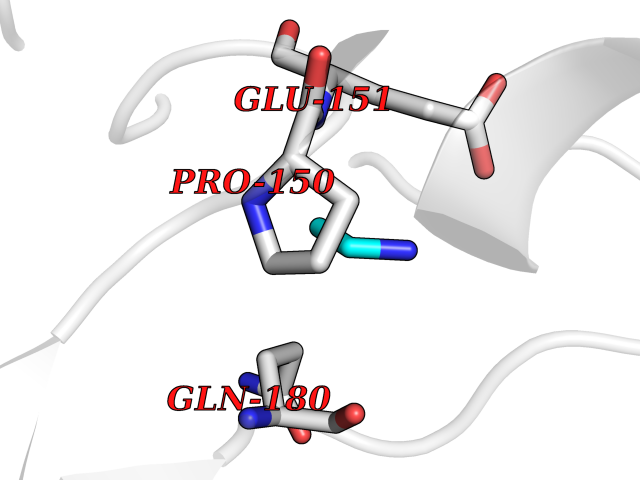 Pymol Map 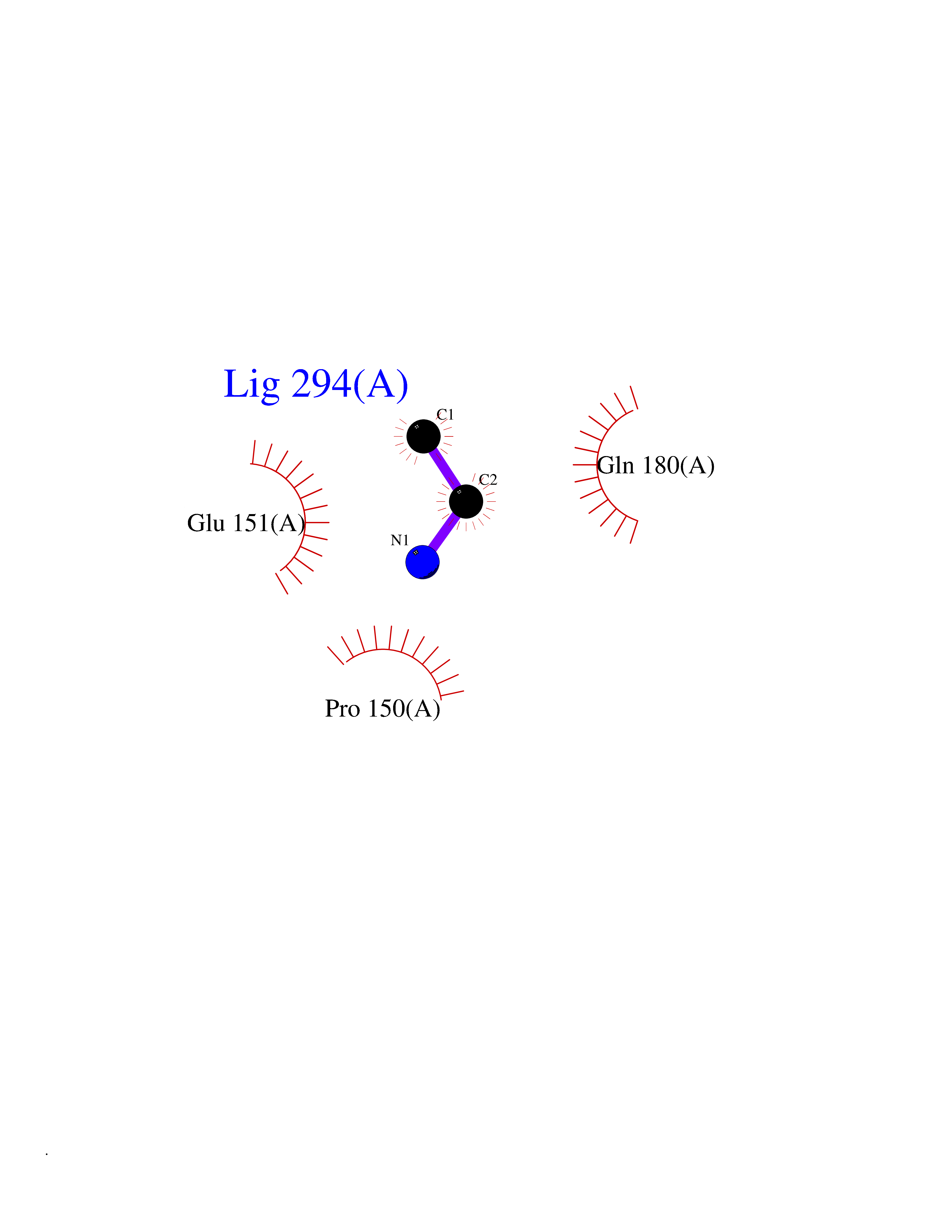 Ligplot Map 3D Binding mode Sequence MKIRDLLKARRGPLFSFEFFPPKDPEGEEALFRTLEELKAFRPAFVSITYGAMGSTRERSVAWAQRIQSLGLNPLAHLTVAGQSRKEVAEVLHRFVESGVENLLALRGDPPRGERVFRPHPEGFRYAAELVALIRERYGDRVSVGGAAYPEGHPESESLEADLRHFKAKVEAGLDFAITQLFFNNAHYFGFLERARRAGIGIPILPGIMPVTSYRQLRRFTEVCGASIPGPLLAKLERHQDDPKAVLEIGVEHAVRQVAELLEAGVEGVHFYTLNKSPATRMVLERLGLRPA Hydrogen bonds contact Hydrophobic contact | ||||
| 97 | Pectate lyase | 1R76 | 4.27 | |
Target general information Gen name pelA Organism Niveispirillum irakense (Azospirillum irakense) Uniprot ID TTD ID NA Synonyms NA Protein family NA Biochemical class Lyase Function Lyase activity. Related diseases A chromosomal aberration involving ALK is found in a form of non-Hodgkin lymphoma. Translocation t(2;5)(p23;q35) with NPM1. The resulting chimeric NPM1-ALK protein homodimerize and the kinase becomes constitutively activated. The constitutively active fusion proteins are responsible for 5-10% of non-Hodgkin lymphomas. {ECO:0000269|PubMed:15938644}.; DISEASE: A chromosomal aberration involving ALK is associated with inflammatory myofibroblastic tumors (IMTs). Translocation t(2;11)(p23;p15) with CARS; translocation t(2;4)(p23;q21) with SEC31A. {ECO:0000269|PubMed:12112524, ECO:0000269|PubMed:16161041}.; DISEASE: A chromosomal aberration involving ALK is associated with anaplastic large-cell lymphoma (ALCL). Translocation t(2;17)(p23;q25) with ALO17. {ECO:0000269|PubMed:12112524}.; DISEASE: Neuroblastoma 3 (NBLST3) [MIM:613014]: A common neoplasm of early childhood arising from embryonic cells that form the primitive neural crest and give rise to the adrenal medulla and the sympathetic nervous system. {ECO:0000269|PubMed:18724359, ECO:0000269|PubMed:18923523, ECO:0000269|PubMed:18923525, ECO:0000269|PubMed:21242967, ECO:0000269|PubMed:22932897}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: The ALK signaling pathway plays an important role in glioblastoma, the most common malignant brain tumor of adults and one of the most lethal cancers. It regulates both glioblastoma migration and growth. {ECO:0000269|PubMed:15908427}.; DISEASE: A chromosomal aberration involving ALK is found in one subject with colorectal cancer. Translocation t(2;2)(p23.1;p23.3). A 5 million base pair tandem duplication generates an in-frame WDCP-ALK gene fusion. {ECO:0000269|PubMed:22327622}.; DISEASE: A chromosomal aberration involving ALK has been identified in a subset of patients with non-small-cell lung carcinoma. This aberration leads to the production of a fusion protein between the N-terminus of EML4 et the C-terminus of ALK. It is unclear whether the fusion protein is caused by a simple inversion within 2p (inv(2)(p21p23)) or whether the chromosome translocation involving 2p is more complex. When tested in a heterologous system, the fusion protein EML4-ALK possesses transforming activity that is dependent on ALK catalytic activity, possibly due to spontaneous dimerization mediated by the EML4 moiety, leading to ALK kinase activation. {ECO:0000269|PubMed:17625570}. Drugs (DrugBank ID) NA Interacts with NA EC number NA Uniprot keywords 3D-structure; Lyase; Signal Protein physicochemical properties Chain ID A Molecular weight (Da) 41907.5 Length 384 Aromaticity 0.08 Instability index 43.72 Isoelectric point 6.11 Charge (pH=7) -3.46 2D Binding mode Binding energy (Kcal/mol) -5.83 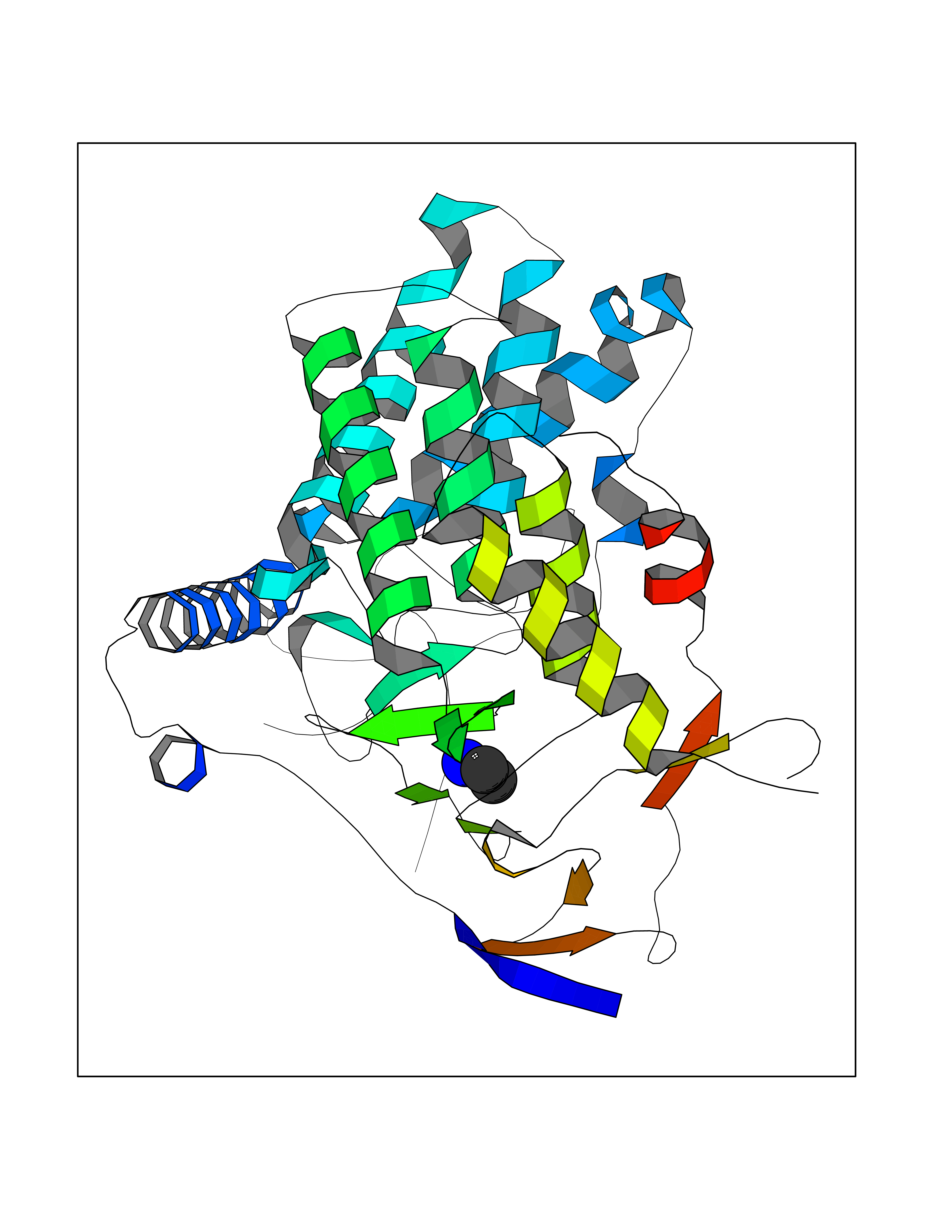 Molscript Map 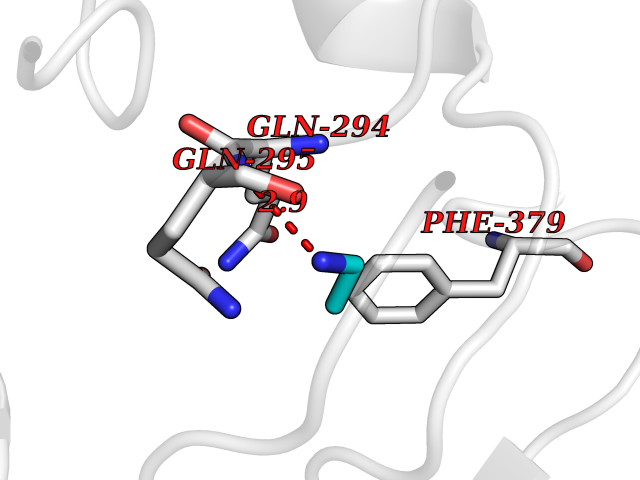 Pymol Map 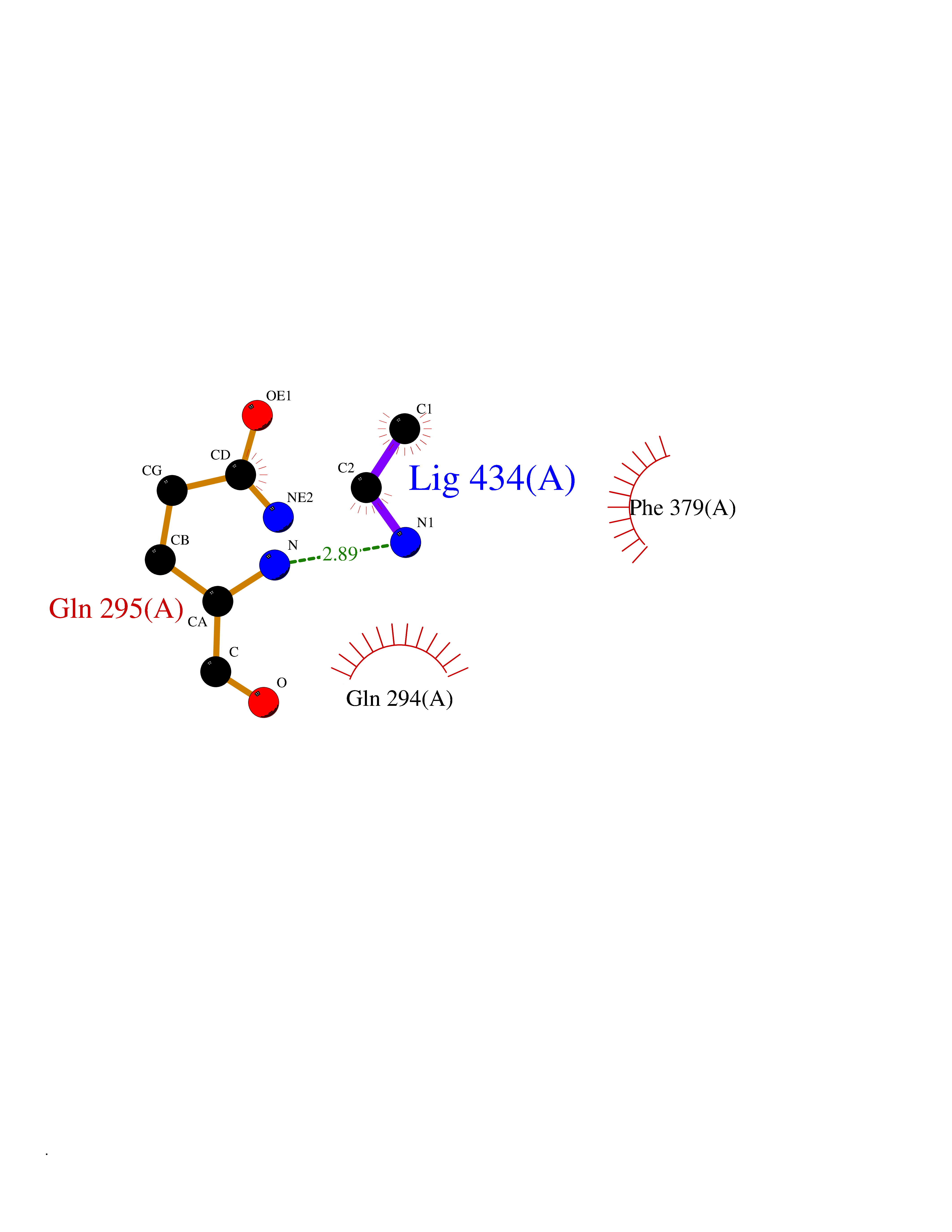 Ligplot Map 3D Binding mode Sequence AVIGMNEAASALTPSRVSSLPDTQRAAWQEYLARSEAQLSRDKASLAAELAPGQPLPPPPAEGKGADTMPLDKPAAWYTSKAARHVADVIVSFQTPAGGWGKNQPRDGALRLPGQHYTGENVAKVKRDRDWHYVGTIDNDATVTEIRFLAQVVSQLAPEEAAPYRDAALKGIEYLLASQFPNGGWPQVWPLEGGYHDAITYNDDALVHVAELLSDIAAGRDGFGFVPPAIRTRALEATNAAIHCIVETQVVQDGKRLGWGQQHDALTLRPTSARNFEPAALSSTESARILLFLMEIEAPSDAVKQAIRGGVAWLNTSVIRDQGAKPLWSRFYSLDGNKPVFGDRDKTIHDDVMGISQERRTGYAWYTTSPQKALSAFTKWEKRS Hydrogen bonds contact Hydrophobic contact | ||||
| 98 | Pseudomonas Methionine gamma-lyase (Pseudo mdeA) | 1PG8 | 4.27 | |
Target general information Gen name Pseudo mdeA Organism Pseudomonas putida (Arthrobacter siderocapsulatus) Uniprot ID TTD ID Synonyms Pseudo MGL; L-methionine gamma-lyase; L-methioninase; Homocysteine desulfhydrase Protein family Trans-sulfuration enzymes family, L-methionine gamma-lyase subfamily Biochemical class Carbon-sulfur lyases Function Catalyzes the alpha,gamma-elimination of L-methionine to produce methanethiol, 2-oxobutanoate and ammonia. Is involved in L-methionine catabolism. In fact, shows a multicatalytic function since it also catalyzes gamma-replacement of L-methionine with thiol compounds, alpha,gamma-elimination and gamma-replacement reactions of L-homocysteine and its S-substituted derivatives, O-substituted-L-homoserines and DL-selenomethionine, and, to a lesser extent, alpha,beta-elimination and beta-replacement reactions of L-cysteine, S-methyl-L-cysteine, and O-acetyl-L-serine. Also catalyzes deamination and gamma-addition reactions of L-vinylglycine. Thus, the enzyme is able to cleave C-S, C-Se, and C-O bonds of sulfur, selenium, and oxygen amino acids, respectively. Related diseases Lecithin-cholesterol acyltransferase deficiency (LCATD) [MIM:245900]: A disorder of lipoprotein metabolism characterized by inadequate esterification of plasmatic cholesterol. Two clinical forms are recognized: complete LCAT deficiency and fish-eye disease. LCATD is generally referred to the complete form which is associated with absence of both alpha and beta LCAT activities resulting in esterification anomalies involving both HDL (alpha-LCAT activity) and LDL (beta-LCAT activity). It causes a typical triad of diffuse corneal opacities, target cell hemolytic anemia, and proteinuria with renal failure. {ECO:0000269|PubMed:11423760, ECO:0000269|PubMed:12957688, ECO:0000269|PubMed:15994445, ECO:0000269|PubMed:16051254, ECO:0000269|PubMed:16216249, ECO:0000269|PubMed:1681161, ECO:0000269|PubMed:1859405, ECO:0000269|PubMed:2370048, ECO:0000269|PubMed:7607641, ECO:0000269|PubMed:7711728, ECO:0000269|PubMed:8318557, ECO:0000269|PubMed:8432868, ECO:0000269|PubMed:8807342, ECO:0000269|PubMed:9007616, ECO:0000269|PubMed:9741700}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Fish-eye disease (FED) [MIM:136120]: A disorder of lipoprotein metabolism due to partial lecithin-cholesterol acyltransferase deficiency that affects only alpha-LCAT activity. FED is characterized by low plasma HDL and corneal opacities due to accumulation of cholesterol deposits in the cornea ('fish-eye'). {ECO:0000269|PubMed:1516702, ECO:0000269|PubMed:1571050, ECO:0000269|PubMed:15994445, ECO:0000269|PubMed:1737840, ECO:0000269|PubMed:21901787, ECO:0000269|PubMed:8620346, ECO:0000269|PubMed:9261271}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB04083 Interacts with NA EC number EC 4.4.1.11 Uniprot keywords 3D-structure; Direct protein sequencing; Lyase; Pyridoxal phosphate Protein physicochemical properties Chain ID A,C Molecular weight (Da) 85234.4 Length 796 Aromaticity 0.07 Instability index 37.1 Isoelectric point 6.21 Charge (pH=7) -11.34 2D Binding mode Binding energy (Kcal/mol) -5.82  Molscript Map 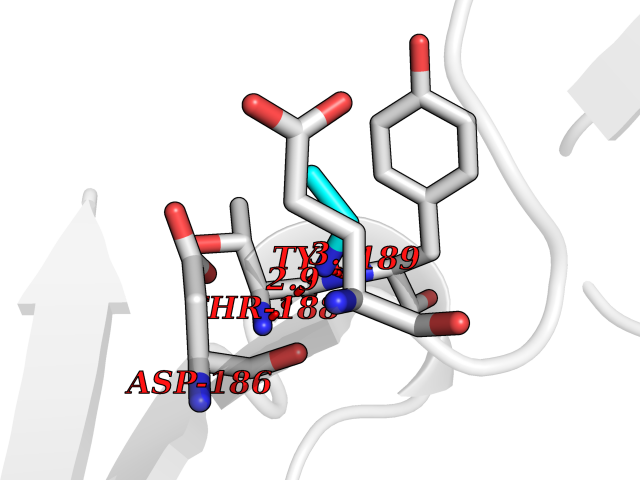 Pymol Map 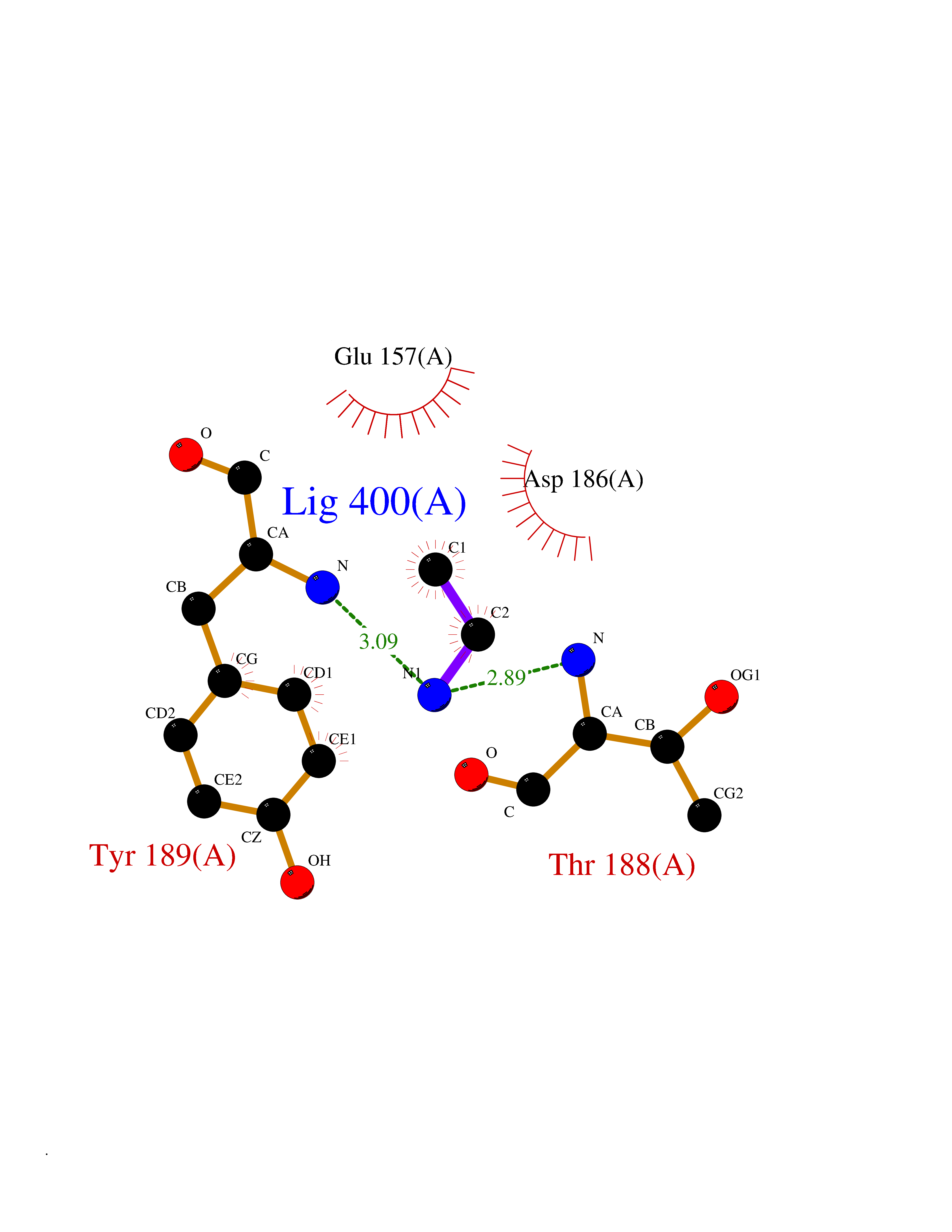 Ligplot Map 3D Binding mode Sequence MHGSNKLPGFATRAIHHGYDPQDHGGALVPPVYQTATFTFPTVEYGAACFAGEQAGHFYSRISNPTLNLLEARMASLEGGEAGLALASGMGAITSTLWTLLRPGDEVLLGNTLYGCTFAFLHHGIGEFGVKLRHVDMADLQALEAAMTPATRVIYFESPANPNMHMADIAGVAKIARKHGATVVVDNTYCTPYLQRPLELGADLVVHSATKYLSGHGDITAGIVVGSQALVDRIRLQGLKDMTGAVLSPHDAALLMRGIKTLNLRMDRHCANAQVLAEFLARQPQVELIHYPGLASFPQYTLARQQMSQPGGMIAFELKGGIGAGRRFMNALQLFSRAVSLGDAESLAQHPASMTHSSYTPEERAHYGISEGLVRLSVGLEDIDDLLADVQQALKASAMHGSNKLPGFATRAIHHGYDPQDHGGALVPPVYQTATFTFPTVEYGAACFAGEQAGHFYSRISNPTLNLLEARMASLEGGEAGLALASGMGAITSTLWTLLRPGDEVLLGNTLYGCTFAFLHHGIGEFGVKLRHVDMADLQALEAAMTPATRVIYFESPANPNMHMADIAGVAKIARKHGATVVVDNTYCTPYLQRPLELGADLVVHSATKYLSGHGDITAGIVVGSQALVDRIRLQGLKDMTGAVLSPHDAALLMRGIKTLNLRMDRHCANAQVLAEFLARQPQVELIHYPGLASFPQYTLARQQMSQPGGMIAFELKGGIGAGRRFMNALQLFSRAVSLGDAESLAQHPASMTHSSYTPEERAHYGISEGLVRLSVGLEDIDDLLADVQQALKASA Hydrogen bonds contact Hydrophobic contact | ||||
| 99 | Cerebron E3 ubiquitin ligase complex (CRL4-CRBN E3 ubiquitin ligase) | 4CI1 | 4.27 | |
Target general information Gen name CUL4A/CUL4B-DDB1-CRBN Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms NA Protein family Cullin family Biochemical class NA Function NA Related diseases Orotic aciduria 1 (ORAC1) [MIM:258900]: A disorder of pyrimidine metabolism resulting in megaloblastic anemia and orotic acid crystalluria that is frequently associated with some degree of physical and intellectual disability. A minority of cases have additional features, particularly congenital malformations and immune deficiencies. {ECO:0000269|PubMed:9042911}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P54253; Q86VP6; Q16531; Q92466; P08238; O94888; P55072 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Biological rhythms; DNA damage; DNA repair; Host-virus interaction; Isopeptide bond; Phosphoprotein; Proteomics identification; Reference proteome; Ubl conjugation; Ubl conjugation pathway Protein physicochemical properties Chain ID B Molecular weight (Da) 42669.7 Length 368 Aromaticity 0.1 Instability index 44.94 Isoelectric point 8.72 Charge (pH=7) 6.58 2D Binding mode Binding energy (Kcal/mol) -5.82  Molscript Map 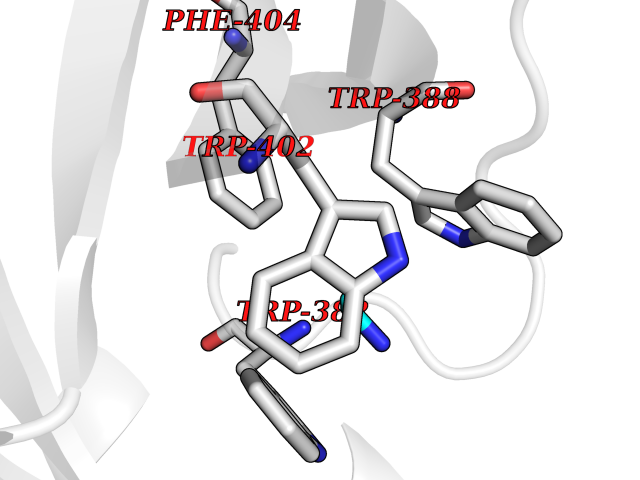 Pymol Map 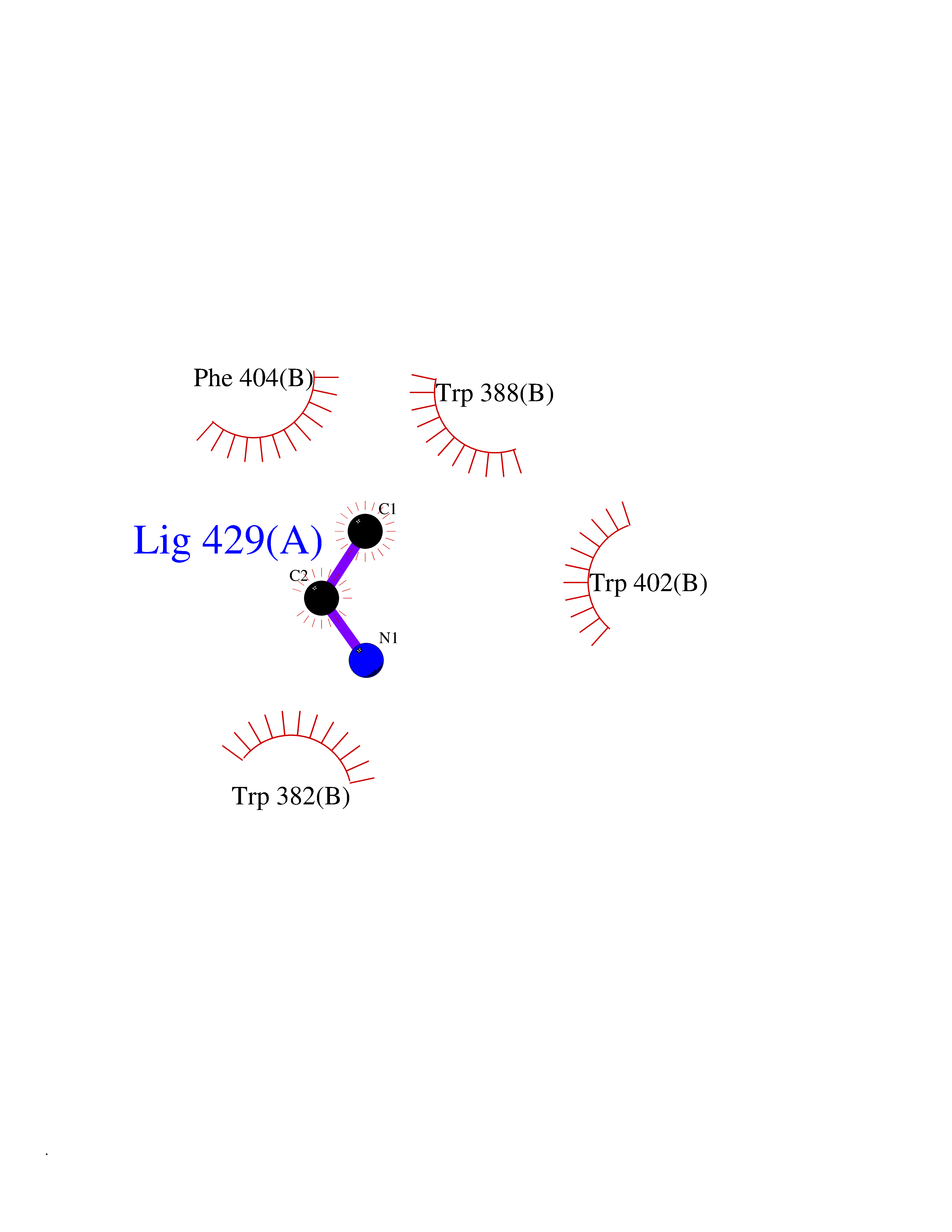 Ligplot Map 3D Binding mode Sequence MINFDTSLPTSHMYLGSDMEEFHGRTLHDDDSCQVIPVLPHVMVMLIPGQTLPLQLFHPQEVSMVRNLIQKDRTFAVLAYSNVREREAHFGTTAEIYAYREEQEYGIETVKVKAIGRQRFKVLEIRTQSDGIQQAKVQILPERVLPSTMSAVQLQSLSRRHIRAFRQWWQKYQKRKFHCASLTSWPPWLYSLYDAETLMERVKRQLHEWDENLKDESLPTNPIDFSYRVAACLPIDDALRIQLLKIGSAIQRLRELDIMNKTSLCCKQCQDTEITTKNEIFSLSLCGPMAAYVNPHGYIHETLTVYKACNLNLSGRPSTEHSWFPGYAWTIAQCRICGNHMGWKFTATKKDMSPQKFWGLTRSALLPR Hydrogen bonds contact Hydrophobic contact | ||||
| 100 | Cyclic ADP-ribose hydrolase 1 (CD38) | 3DZG | 4.27 | |
Target general information Gen name CD38 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms cADPr hydrolase 1; T10; Cyclic ADPribose hydrolase 1; ADPribosyl cyclase 1; ADPRC 1; ADP-ribosyl cyclase/cyclic ADP-ribose hydrolase 1; ADP-ribosyl cyclase 1; 2'-phospho-cyclic-ADP-ribose transferase; Protein family ADP-ribosyl cyclase family Biochemical class Glycosylase Function Has cADPr hydrolase activity. Also moonlights as a receptor in cells of the immune system. Synthesizes the second messagers cyclic ADP-ribose and nicotinate-adenine dinucleotide phosphate, the former a second messenger for glucose-induced insulin secretion. Related diseases Intellectual developmental disorder with hypotonia and behavioral abnormalities (IDDHBA) [MIM:618748]: An autosomal dominant neurodevelopmental disorder with onset in infancy. IDDHBA is characterized by hypotonia, global developmental delay, learning disability, and behavioral abnormalities, such as autistic features and attention deficit-hyperactivity disorder. Additional variable features may include non-specific facial dysmorphism, congenital heart defects, ocular anomalies, and poor feeding. {ECO:0000269|PubMed:30905399}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB09331; DB14811; DB16370 Interacts with NA EC number EC 3.2.2.6 Uniprot keywords 3D-structure; Alternative splicing; Diabetes mellitus; Disulfide bond; Glycoprotein; Hydrolase; Membrane; NAD; NADP; Proteomics identification; Receptor; Reference proteome; Signal-anchor; Transferase; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 29222.8 Length 252 Aromaticity 0.1 Instability index 50.29 Isoelectric point 6.19 Charge (pH=7) -2.71 2D Binding mode Binding energy (Kcal/mol) -5.82 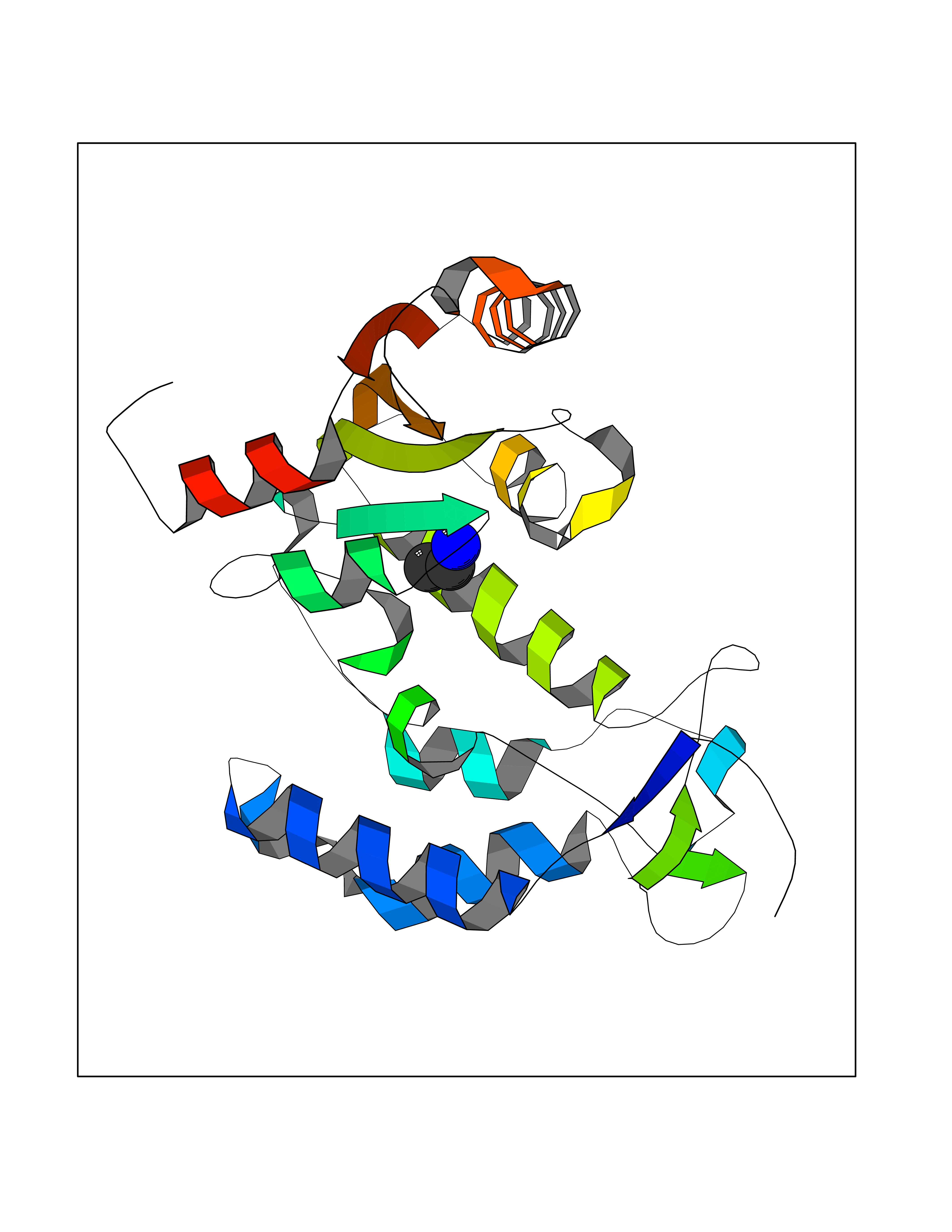 Molscript Map 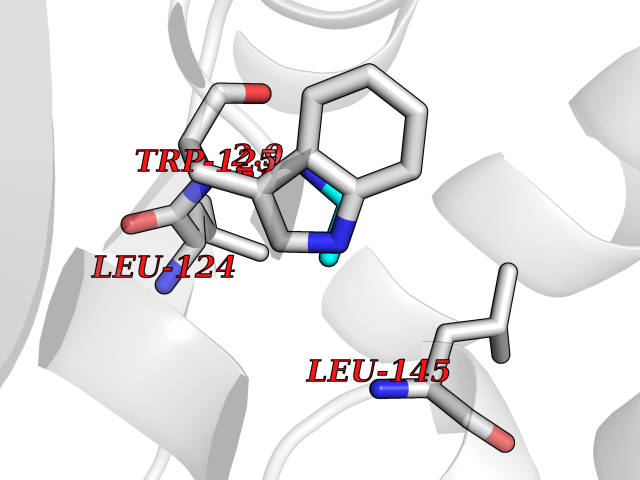 Pymol Map 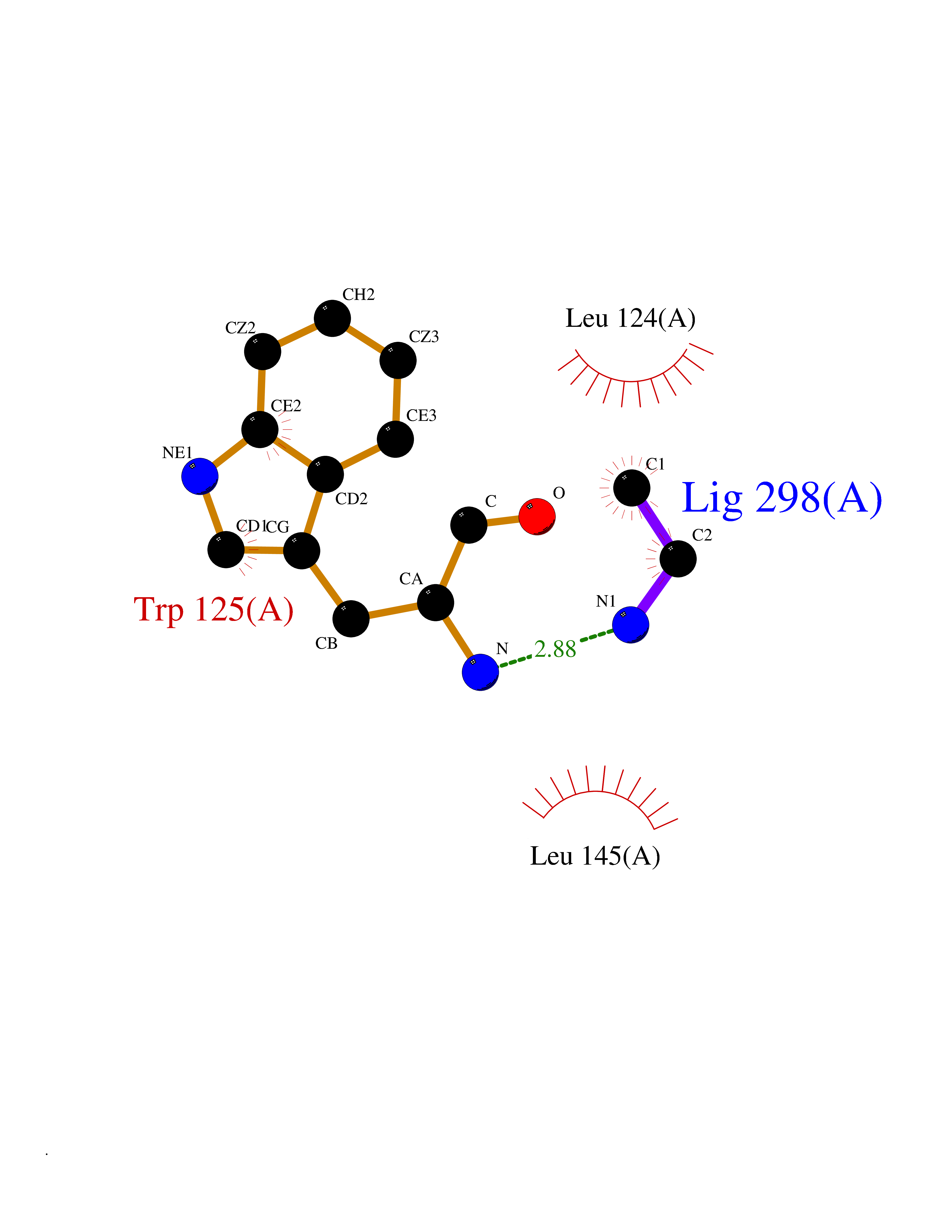 Ligplot Map 3D Binding mode Sequence RWRQTWSGPGTTKRFPETVLARCVKYTEIHPEMRHVDCQSVWDAFKGAFISKHPCDITEEDYQPLMKLGTQTVPCNKILLWSRIKDLAHQFTQVQRDMFTLEDTLLGYLADDLTWCGEFDTSKINYQSCPDWRKDCSNNPVSVFWKTVSRRFAEAACDVVHVMLDGSRSKIFDKDSTFGSVEVHNLQPEKVQTLEAWVIHGGREDSRDLCQDPTIKELESIISKRNIQFSCKNIYRPDKFLQCVKNPEDSSC Hydrogen bonds contact Hydrophobic contact | ||||