Job Results:
Ligand
Structure
Job ID
85eee3bcbedbd74b81bc48f797dc6af1
Job name
NA
Time
2026-02-27 13:44:11
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 81 | Nitric-oxide synthase endothelial (NOS3) | 4D1P | 5.06 | |
Target general information Gen name NOS3 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Nitric oxide synthase, endothelial; NOSIII; NOS,type III; NOS type III; Endothelial nitric oxide synthase; Endothelial NOS; ENOS; EC-NOS; Constitutive NOS; CNOS Protein family NOS family Biochemical class Paired donor oxygen oxidoreductase Function NO mediates vascular endothelial growth factor (VEGF)-induced angiogenesis in coronary vessels and promotes blood clotting through the activation of platelets. Produces nitric oxide (NO) which is implicated in vascular smooth muscle relaxation through a cGMP-mediated signal transduction pathway. Related diseases Variation Asp-298 in NOS3 may be associated with susceptibility to coronary spasm. {ECO:0000269|PubMed:11740345, ECO:0000269|PubMed:9737779}. Drugs (DrugBank ID) DB07001; DB02048; DB02911; DB02335; DB01997; DB03332; DB04534; DB07244; DB03100; DB03918; DB02207; DB03065; DB00125; DB02994; DB01833; DB00155; DB00997; DB07388; DB03974; DB02077; DB01821; DB09237; DB01110; DB03144; DB03305; DB01686; DB04559; DB02044; DB08019; DB08018; DB02027; DB02979; DB00435; DB04223; DB06154; DB03910; DB02141; DB03963; DB03707; DB02234; DB04018; DB00360; DB02589 Interacts with P60709; P63010-2; Q8N6T3-3; Q9Y575-3; Q96FT7-4; Q5SZD1; Q16543; Q9UNS2; Q8IUI8; P35222; Q05193; O15287; Q08379; Q71DI3; P69905; P61978; Q12891; Q9UKT9; Q9Y2M5; Q14525; Q6DKI2; P43364-2; Q8N6F8; O94851; A4FUJ8; Q8N594; Q8IVI9; Q6X4W1-6; O15381-5; Q9NV79; Q16549; Q5T2W1; O75925; Q96I34; Q6ZMI0-5; P57052; Q9GZR2; Q96D59; Q8N6K7-2; Q9GZS3; Q8IUW3; Q7Z699; Q7Z698; P50502; Q9BR01-2; Q9NVV9; Q86WT6-2; Q9H347; P58304; Q9NZC7-5; Q9UNY5; P14079 EC number EC 1.14.13.39 Uniprot keywords 3D-structure; Alternative splicing; Calcium; Calmodulin-binding; Cell membrane; Cytoplasm; Cytoskeleton; Direct protein sequencing; FAD; Flavoprotein; FMN; Golgi apparatus; Heme; Iron; Lipoprotein; Membrane; Metal-binding; Myristate; NADP; Oxidoreductase; Palmitate; Phosphoprotein; Proteomics identification; Reference proteome; Zinc Protein physicochemical properties Chain ID A,B Molecular weight (Da) 90790.1 Length 803 Aromaticity 0.11 Instability index 50.67 Isoelectric point 6.03 Charge (pH=7) -9.56 2D Binding mode Binding energy (Kcal/mol) -6.9  Molscript Map 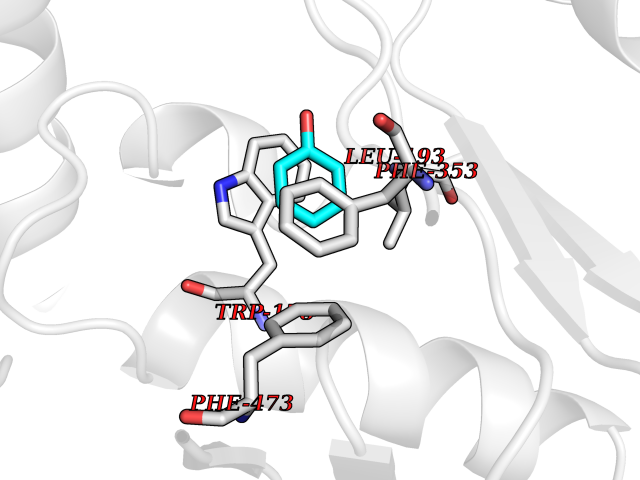 Pymol Map 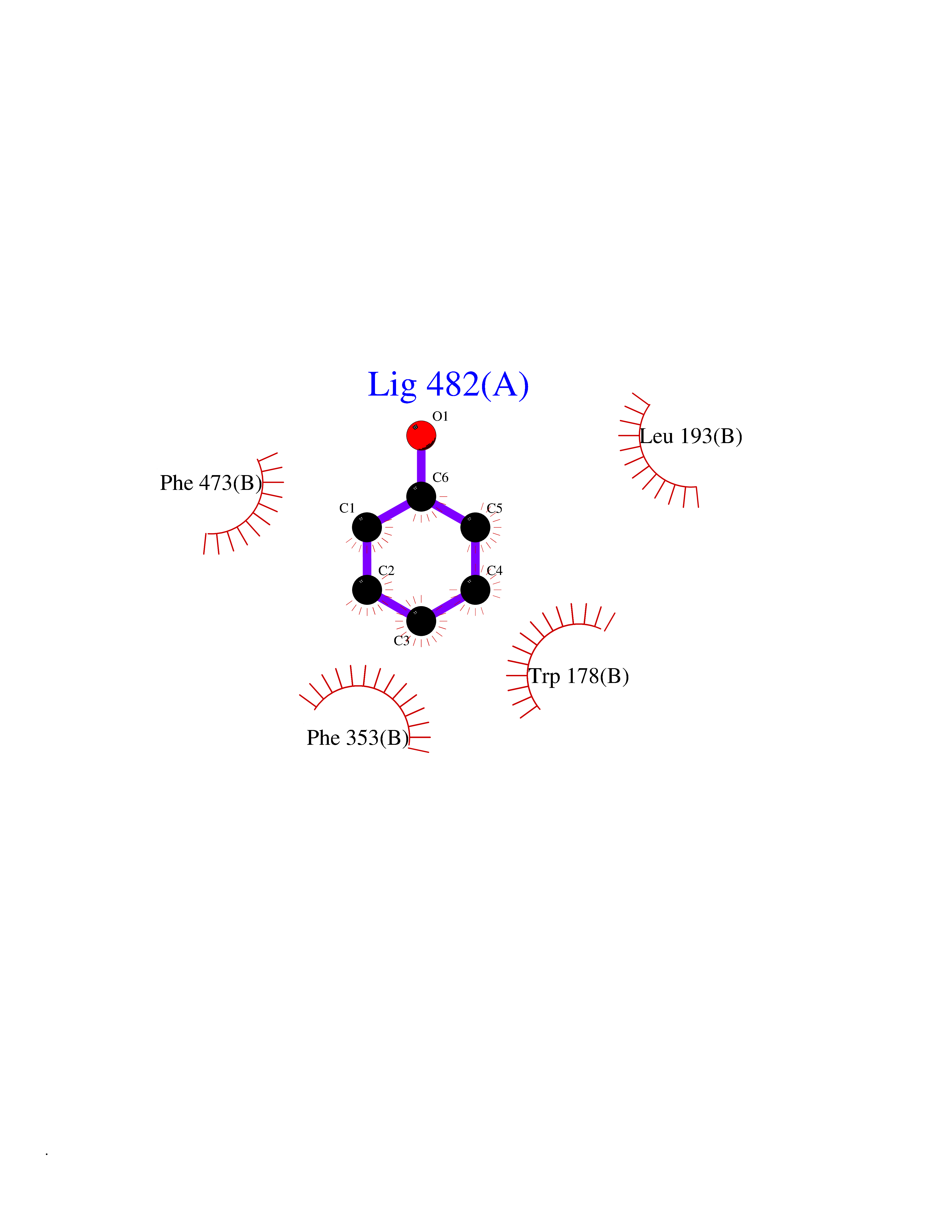 Ligplot Map 3D Binding mode Sequence FPRVKNWEVGSITYDTLSAQAQQDGPCTPRRCLGSLVFPAPEQLLSQARDFINQYYSSIKRSGSQAHEQRLQEVEAEVAATGTYQLRESELVFGAKQAWRNAPRCVGRIQWGKLQVFDARDCRSAQEMFTYICNHIKYATNRGNLRSAITVFPQRCPGRGDFRIWNSQLVRYAGYRQQDGSVRGDPANVEITELCIQHGWTPGNGRFDVLPLLLQAPDEPPELFLLPPELVLEVPLEHPTLEWFAALGLRWYALPAVSNMLLEIGGLEFPAAPFSGWYMSTEIGTRNLCDPHRYNILEDVAVCMDLDTRTTSSLWKDKAAVEINVAVLHSYQLAKVTIVDHHAATASFMKHLENEQKARGGCPADWAWIVPPISGSLTPVFHQEMVNYFLSPAFRYQPDPWKFPRVKNWEVGSITYDTLSAQAQQDGPCTPRRCLGSLVFPAPEQLLSQARDFINQYYSSIKRSGSQAHEQRLQEVEAEVAATGTYQLRESELVFGAKQAWRNAPRCVGRIQWGKLQVFDARDCRSAQEMFTYICNHIKYATNRGNLRSAITVFPQRCPGRGDFRIWNSQLVRYAGYRQQDGSVRGDPANVEITELCIQHGWTPGNGRFDVLPLLLQAPDEPPELFLLPPELVLEVPLEHPTLEWFAALGLRWYALPAVSNMLLEIGGLEFPAAPFSGWYMSTEIGTRNLCDPHRYNILEDVAVCMDLDTRTTSSLWKDKAAVEINVAVLHSYQLAKVTIVDHHAATASFMKHLENEQKARGGCPADWAWIVPPISGSLTPVFHQEMVNYFLSPAFRYQPDPW Hydrogen bonds contact Hydrophobic contact | ||||
| 82 | Nicotinamide N-methyltransferase | 2IIP | 5.06 | |
Target general information Gen name NNMT Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Class I-like SAM-binding methyltransferase superfamily, NNMT/PNMT/TEMT family Biochemical class Transferase Function Nicotinamide N-methyltransferase activity.Pyridine N-methyltransferase activity. Related diseases Defects in PPARG can lead to type 2 insulin-resistant diabetes and hyptertension. PPARG mutations may be associated with colon cancer. {ECO:0000269|PubMed:10394368}.; DISEASE: Obesity (OBESITY) [MIM:601665]: A condition characterized by an increase of body weight beyond the limitation of skeletal and physical requirements, as the result of excessive accumulation of body fat. {ECO:0000269|PubMed:9753710}. Disease susceptibility may be associated with variants affecting the gene represented in this entry.; DISEASE: Lipodystrophy, familial partial, 3 (FPLD3) [MIM:604367]: A form of lipodystrophy characterized by marked loss of subcutaneous fat from the extremities. Facial adipose tissue may be increased, decreased or normal. Affected individuals show an increased preponderance of insulin resistance, diabetes mellitus and dyslipidemia. {ECO:0000269|PubMed:11788685, ECO:0000269|PubMed:12453919}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Glioma 1 (GLM1) [MIM:137800]: Gliomas are benign or malignant central nervous system neoplasms derived from glial cells. They comprise astrocytomas and glioblastoma multiforme that are derived from astrocytes, oligodendrogliomas derived from oligodendrocytes and ependymomas derived from ependymocytes. {ECO:0000269|PubMed:10851250}. Disease susceptibility may be associated with variants affecting the gene represented in this entry. Polymorphic PPARG alleles have been found to be significantly over-represented among a cohort of American patients with sporadic glioblastoma multiforme suggesting a possible contribution to disease susceptibility. Drugs (DrugBank ID) DB00627 Interacts with NA EC number 2.1.1.1 Uniprot keywords 3D-structure; Acetylation; Citrullination; Cytoplasm; Direct protein sequencing; Methyltransferase; Proteomics identification; Reference proteome; S-adenosyl-L-methionine; Transferase Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 27886.8 Length 251 Aromaticity 0.1 Instability index 40.66 Isoelectric point 5.23 Charge (pH=7) -5.11 2D Binding mode Binding energy (Kcal/mol) -6.9  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence GFTSKDTYLSHFNPRDYLEKYYSAESQILKHLLKNLFKIFCLDGVKGDLLIDIGSGPTIYQLLSACESFKEIVVTDYSDQNLQELEKWLKAAPAAFDWSPVVTYVCDLEGNRVKGPEKEEKLRQAVKQVLKCDVTQSQPLGAVPLPPADCVLSTLCLDAACPDLPTYCRALRNLGSLLKPGGFLVIMDALKSSYYMIGEQKFSSLPLGREAVEAAVKEAGYTIEWFEVISQSYSSTMANNEGLFSLVARKL Hydrogen bonds contact Hydrophobic contact | ||||
| 83 | Spermine synthase | 3C6K | 5.06 | |
Target general information Gen name SMS Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Spermidine/spermine synthase family Biochemical class Transferase Function Spermine synthase activity. Related diseases Intellectual developmental disorder, X-linked, syndromic, Snyder-Robinson type (MRXSSR) [MIM:309583]: An X-linked intellectual disability syndrome characterized by a collection of clinical features including facial asymmetry, marfanoid habitus, hypertonia, osteoporosis and unsteady gait. {ECO:0000269|PubMed:14508504, ECO:0000269|PubMed:18550699, ECO:0000269|PubMed:19206178, ECO:0000269|PubMed:22612257, ECO:0000269|PubMed:23696453, ECO:0000269|PubMed:23897707}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00127 Interacts with NA EC number 2.5.1.22 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Direct protein sequencing; Intellectual disability; Phosphoprotein; Polyamine biosynthesis; Proteomics identification; Reference proteome; Transferase Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 27177.1 Length 238 Aromaticity 0.11 Instability index 41.31 Isoelectric point 4.82 Charge (pH=7) -9.19 2D Binding mode Binding energy (Kcal/mol) -6.9  Molscript Map 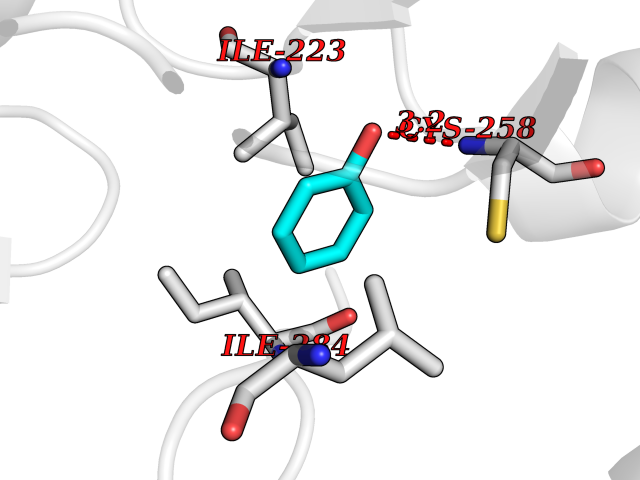 Pymol Map 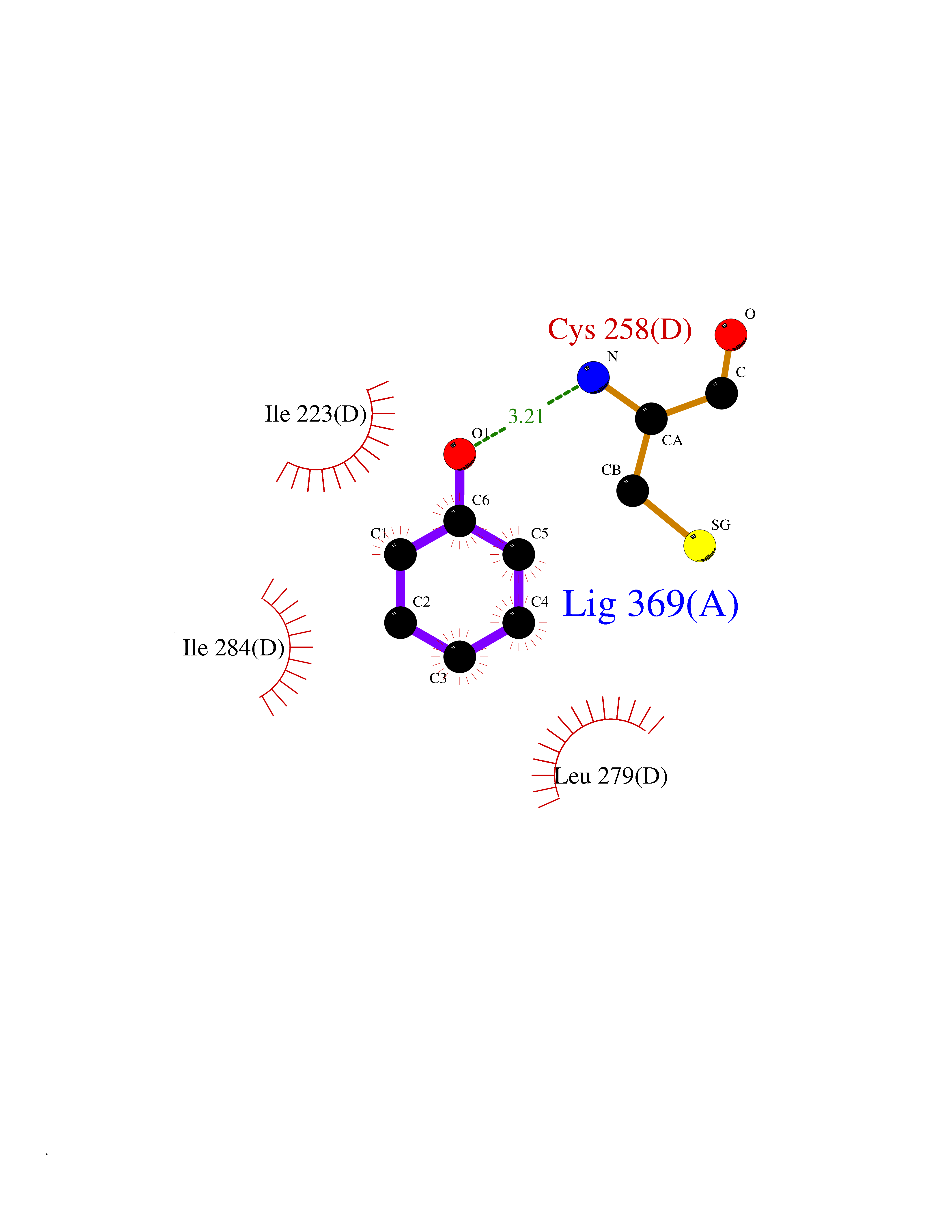 Ligplot Map 3D Binding mode Sequence RYWPTADGRLVEYDIDEVVYDEDSPYQNIKILHSKQFGNILILSGDVNLAESDLAYTRAIMGSGKEDYTGKDVLILGGGDGGILCEIVKLKPKMVTMVEIDQMVIDGCKKYMRKDVLDNLKGDCYQVLIEDCIPVLKRYAKEGREFDYVINDLTAVPISTSPSTWEFLRLILDLSMKVLKQDGKYFTQGNCVNLTEALSLYEEQLGRLYCPVEFSKEIVCVPSYLELWVFYTVWKKAK Hydrogen bonds contact Hydrophobic contact | ||||
| 84 | Acetyl-CoA carboxylase (ACC) (EC 6.4.1.2) (Fatty acid synthetase 3) (mRNA transport-defective protein 7) [Includes: Biotin carboxylase (EC 6.3.4.14)] | 1UYS | 5.06 | |
Target general information Gen name ACC1 Organism Saccharomyces cerevisiae (strain ATCC 204508 / S288c) (Baker's yeast) Uniprot ID TTD ID NA Synonyms MTR7;YNR016C;N3175;ABP2;FAS3 Protein family NA Biochemical class NA Function Carries out three functions: biotin carboxyl carrier protein, biotin carboxylase and carboxyltransferase. Involved in the synthesis of very-long-chain fatty acid synthesis which is required to maintain a functional nuclear envelope. Required for acylation and vacuolar membrane association of VAC8 which is necessary to maintain a normal morphology of the vacuole. {ECO:0000269|PubMed:10757783, ECO:0000269|PubMed:12730220, ECO:0000269|PubMed:6103540, ECO:0000269|PubMed:6108218, ECO:0000269|PubMed:8943372}." Related diseases A chromosomal aberration involving NFKB2 is found in a case of B-cell non Hodgkin lymphoma (B-NHL). Translocation t(10;14)(q24;q32) with IGHA1. The resulting oncogene is also called Lyt-10C alpha variant.; DISEASE: A chromosomal aberration involving NFKB2 is found in a cutaneous T-cell leukemia (C-TCL) cell line. This rearrangement produces the p80HT gene which codes for a truncated 80 kDa protein (p80HT).; DISEASE: In B-cell leukemia (B-CLL) cell line, LB40 and EB308, can be found after heterogeneous chromosomal aberrations, such as internal deletions.; DISEASE: Immunodeficiency, common variable, 10 (CVID10) [MIM:615577]: A primary immunodeficiency characterized by childhood-onset of recurrent infections, hypogammaglobulinemia, and decreased numbers of memory and marginal zone B-cells. Some patients may develop autoimmune features and have circulating autoantibodies. An unusual feature is central adrenal insufficiency. {ECO:0000269|PubMed:24140114, ECO:0000269|PubMed:25524009}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q00955 EC number 6.3.4.14; 6.4.1.2 Uniprot keywords 3D-structure; Acetylation; ATP-binding; Biotin; Cytoplasm; Direct protein sequencing; Endoplasmic reticulum; Fatty acid biosynthesis; Fatty acid metabolism; Ligase; Lipid biosynthesis; Lipid metabolism; Manganese; Membrane; Metal-binding; Multifunctional enzyme; Nucleotide-binding; Phosphoprotein; Reference proteome Protein physicochemical properties Chain ID B,C Molecular weight (Da) 145619 Length 1328 Aromaticity 0.1 Instability index 30.31 Isoelectric point 5.32 Charge (pH=7) -26.79 2D Binding mode Binding energy (Kcal/mol) -6.91  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence WLQPKRYKAHLXGTTYVYDFPELFRQASSSQWKNFSADVKLTDDFFISNELIEDENGELTEVEREPGANAIGXVAFKITVKTPEYPRGRQFVVVANDITFKIGSFGPQEDEFFNKVTEYARKRGIPRIYLAANSGARIGXAEEIVPLFQVAWNDAANPDKGFQYLYLTSEGXETLKKFDKENSVLTERTVINGEERFVIKTIIGSEDGLGVECLRGSGLIAGATSRAYHDIFTITLVTCRSVGIGAYLVRLGQRAIQVEGQPIILTGAPAINKXLGREVYTSNLQLGGTQIXYNNGVSHLTAVDDLAGVEKIVEWXSYVPAKRNXPVPILETKDTWDRPVDFTPTNDETYDVRWXIEGRETESGFEYGLFDKGSFFETLSGWAKGVVVGRARLGGIPLGVIGVETRTVENLIPADPANPNSAETLIQEPGQVWHPNSAFKTAQAINDFNNGEQLPXXILANWRGFSGNEVLKYGSFIVDALVDYKQPIIIYIPPTGELRGGSWVVVDPTINADQXEXYADVNARAGVLEPQGXVGIKFRREKLLDTXNRLELLPIYGQISLQFADLHDRSSRXVAKGVISKELEWTEARRFFFWRLRRRLNEEYLIKRLSHQVGEASRLEKIARIRSWYPASVDHEDDRQVATWIEENYKTLDDKLKGLPIATPYPVKEWLQPKRYKAHLXGTTYVYDFPELFRQASSSQWKNFSADVKLTDDFFISNELIEDENGELTEVEREPGANAIGXVAFKITVKTPEYPRGRQFVVVANDITFKIGSFGPQEDEFFNKVTEYARKRGIPRIYLAANSGARIGXAEEIVPLFQVAWNDAANPDKGFQYLYLTSEGXETLKKFDKENSVLTERTVINGEERFVIKTIIGSEDGLGVECLRGSGLIAGATSRAYHDIFTITLVTCRSVGIGAYLVRLGQRAIQVEGQPIILTGAPAINKXLGREVYTSNLQLGGTQIXYNNGVSHLTAVDDLAGVEKIVEWXSYVPAKRNXPVPILETKDTWDRPVDFTPTNDETYDVRWXIEGRETESGFEYGLFDKGSFFETLSGWAKGVVVGRARLGGIPLGVIGVETRTVENLIPADPANPNSAETLIQEPGQVWHPNSAFKTAQAINDFNNGEQLPXXILANWRGFSGNEVLKYGSFIVDALVDYKQPIIIYIPPTGELRGGSWVVVDPTINADQXEXYADVNARAGVLEPQGXVGIKFRREKLLDTXNRLELLPIYGQISLQFADLHDRSSRXVAKGVISKELEWTEARRFFFWRLRRRLNEEYLIKRLSHQVGEASRLEKIARIRSWYPASVDHEDDRQVATWIEENYKTLDDKLKGL Hydrogen bonds contact Hydrophobic contact | ||||
| 85 | Aminoacylase-1 | 1Q7L | 5.06 | |
Target general information Gen name ACY1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Peptidase M20A family Biochemical class Hydrolase Function Aminoacylase activity.Identical protein binding.Metal ion binding.Metallopeptidase activity. Related diseases Aminoacylase-1 deficiency (ACY1D) [MIM:609924]: An enzymatic deficiency resulting in encephalopathy, unspecific psychomotor delay, psychomotor delay with atrophy of the vermis and syringomyelia, marked muscular hypotonia or normal clinical features. Epileptic seizures are a frequent feature. All affected individuals exhibit markedly increased urinary excretion of several N-acetylated amino acids. {ECO:0000269|PubMed:16274666, ECO:0000269|PubMed:16465618, ECO:0000269|PubMed:17562838, ECO:0000269|PubMed:21414403}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB06151; DB00128; DB09130 Interacts with Q03154; O75934; Q96HA8; P36639; P36639-2; Q8TCT1; P0CG20; Q96A09; P54274; O43711; Q9UPN9; Q9NZC7-5 EC number 3.5.1.14 Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; Disease variant; Hydrolase; Metal-binding; Proteomics identification; Reference proteome; Zinc Protein physicochemical properties Chain ID A,C Molecular weight (Da) 31172.2 Length 275 Aromaticity 0.11 Instability index 36.46 Isoelectric point 6 Charge (pH=7) -5.26 2D Binding mode Binding energy (Kcal/mol) -6.91  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence NPWWAAFSRVCKDMNLTLEPEIMPAAGDNRYIRAVGVPALGFSPMNRTPVLLHDHDERLHEAVFLRGVDIYTRLLPALASVPALPEEHPSVTLFRQYLRIRTVQPKPDYGAAVAFFEETARQLGLGCQKVEVAPGYVVTVLTWPGTNPTLSSILLNSHTDVVPVFKEHWSHDPFEAFKDSEGYIYARGAQDMKCVSIQYLEAVRRLKVEGHRFPRTIHMTFVPDEEVGGHQGMELFVQRPEFHALRAGFALDEGIANPTDAFTVFYSERSPWWVR Hydrogen bonds contact Hydrophobic contact | ||||
| 86 | S-methyl-5'-thioadenosine phosphorylase (MTAP) | 1CB0 | 5.06 | |
Target general information Gen name MTAP Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Methylthioadenosine phosphorylase; MTAPase; MTA phosphorylase; MSAP; 5'-methylthioadenosine phosphorylase Protein family PNP/MTAP phosphorylase family, MTAP subfamily Biochemical class Glycosyltransferases Function Involved in the breakdown of MTA, a major by-product of polyamine biosynthesis. Responsible for the first step in the methionine salvage pathway after MTA has been generated from S-adenosylmethionine. Has broad substrate specificity with 6-aminopurine nucleosides as preferred substrates. Catalyzes the reversible phosphorylation of S-methyl-5'-thioadenosine (MTA) to adenine and 5-methylthioribose-1-phosphate. Related diseases Diaphyseal medullary stenosis with malignant fibrous histiocytoma (DMSMFH) [MIM:112250]: An autosomal dominant bone dysplasia characterized by pathologic fractures due to abnormal cortical growth and diaphyseal medullary stenosis. The fractures heal poorly, and there is progressive bowing of the lower extremities. Some patients show a limb-girdle myopathy, with muscle weakness and atrophy. Approximately 35% of affected individuals develop an aggressive form of bone sarcoma consistent with malignant fibrous histiocytoma or osteosarcoma. {ECO:0000269|PubMed:22464254}. The disease is caused by variants affecting the gene represented in this entry. DMSMFH causing mutations found in MTAP exon 9 result in exon skipping and dysregulated alternative splicing of all MTAP isoforms (PubMed:22464254). {ECO:0000269|PubMed:22464254}.; DISEASE: Loss of MTAP activity may play a role in human cancer. MTAP loss has been reported in a number of cancers, including osteosarcoma, malignant melanoma and gastric cancer. Drugs (DrugBank ID) DB02158; DB02933; DB02282; DB00173; DB02281 Interacts with Q9H3R5; Q9P0I2 EC number EC 2.4.2.28 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Cytoplasm; Glycosyltransferase; Nucleus; Proteomics identification; Purine salvage; Reference proteome; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 29538.9 Length 268 Aromaticity 0.06 Instability index 40.97 Isoelectric point 7.18 Charge (pH=7) 0.36 2D Binding mode Binding energy (Kcal/mol) -6.9 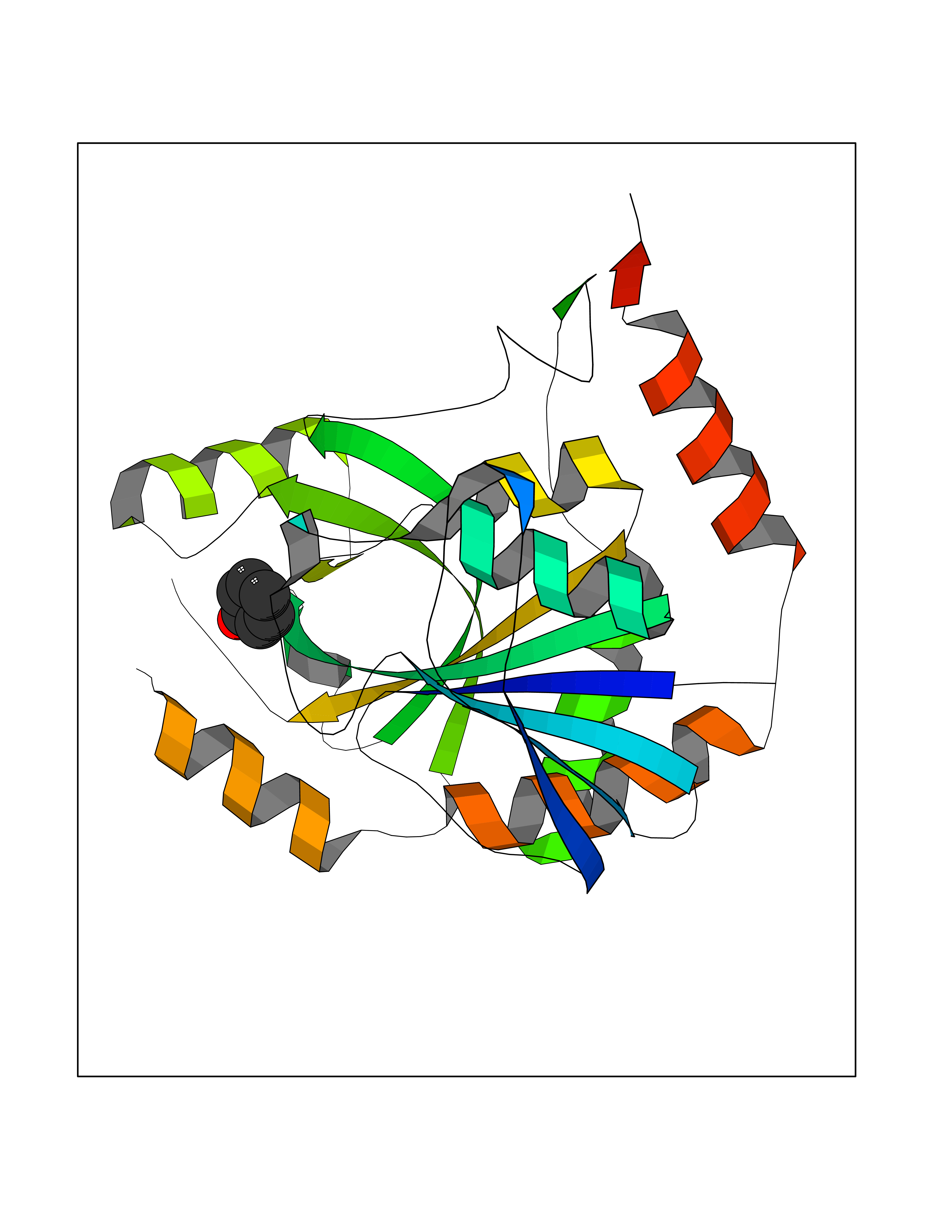 Molscript Map 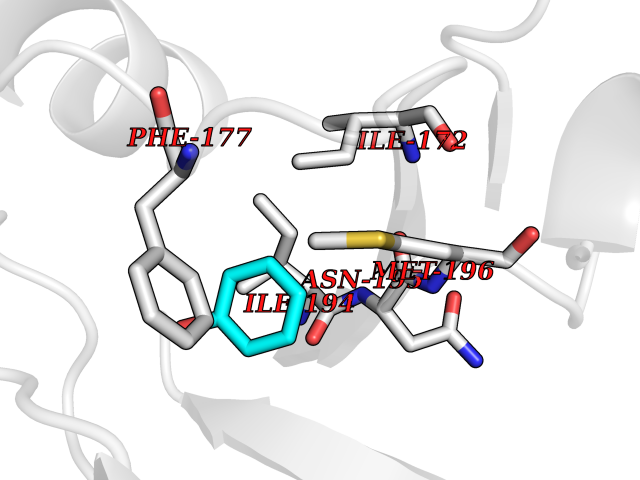 Pymol Map 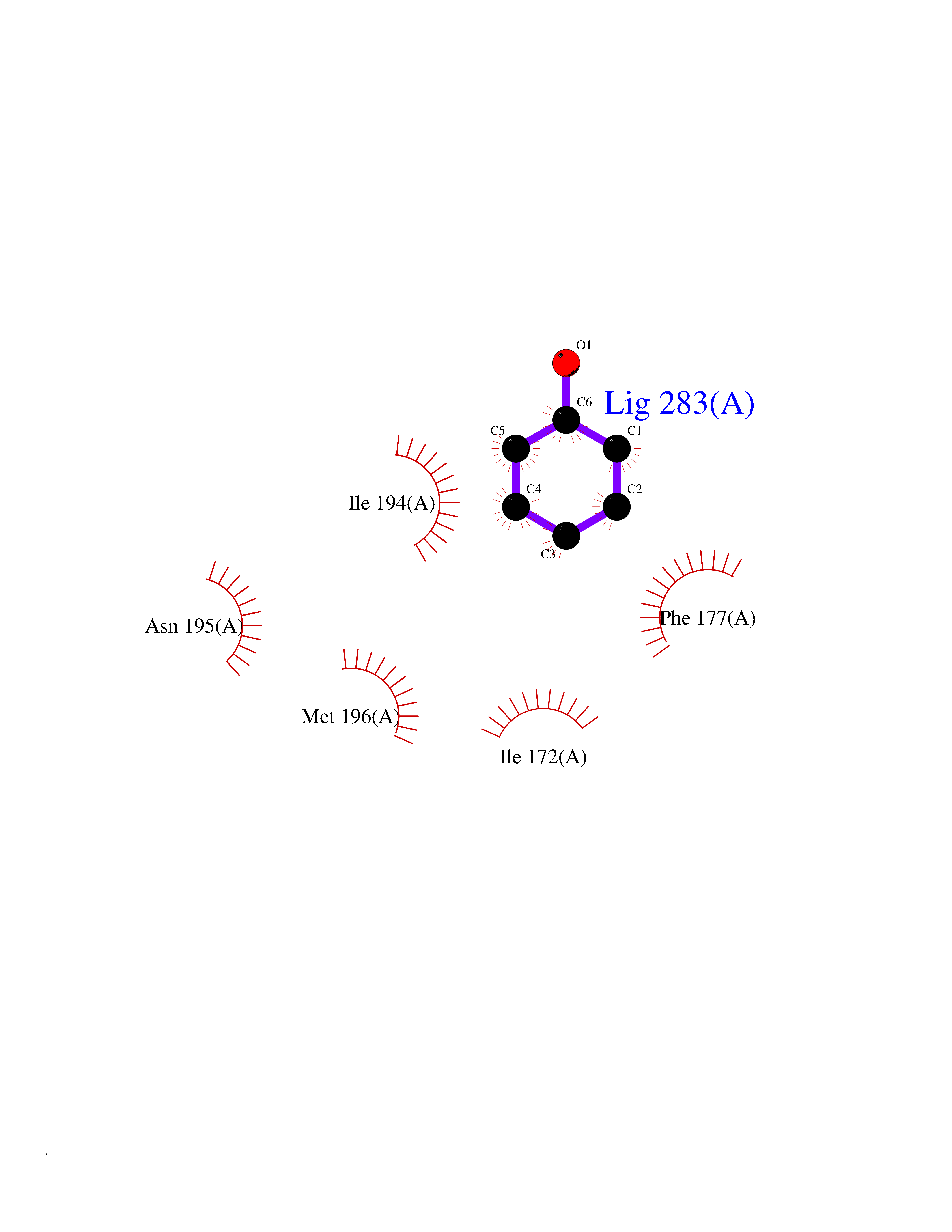 Ligplot Map 3D Binding mode Sequence AVKIGIIGGTGLDDPEILEGRTEKYVDTPFGKPSDALILGKIKNVDCVLLARHGRQHTIMPSKVNYQANIWALKEEGCTHVIVTTACGSLREEIQPGDIVIIDQFIDRTTMRPQSFYDGSHSCARGVCHIPMAEPFCPKTREVLIETAKKLGLRCHSKGTMVTIEGPRFSSRAESFMFRTWGADVINMTTVPEVVLAKEAGICYASIAMATDYDCWAVSVDRVLKTLKENANKAKSLLLTTIPQIGSTEWSETLHNLKNMAQFSVLLP Hydrogen bonds contact Hydrophobic contact | ||||
| 87 | Opioid receptor sigma 1 (OPRS1) | 5HK1 | 5.06 | |
Target general information Gen name SIGMAR1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms hSigmaR1; Sigma1R; Sigma1-receptor; Sigma non-opioid intracellular receptor 1; Sigma 1-type opioid receptor; SRBP; SR31747-binding protein; SR31747 binding protein 1; SR-BP; SIG-1R; Opioid receptor, s Protein family ERG2 family Biochemical class GPCR rhodopsin Function Involved in the regulation of different receptors it plays a role in BDNF signaling and EGF signaling. Also regulates ion channels like the potassium channel and could modulate neurotransmitter release. Plays a role in calcium signaling through modulation together with ANK2 of the ITP3R-dependent calcium efflux at the endoplasmic reticulum. Plays a role in several other cell functions including proliferation, survival and death. Originally identified for its ability to bind various psychoactive drugs it is involved in learning processes, memory and mood alteration. Necessary for proper mitochondrial axonal transport in motor neurons, in particular the retrograde movement of mitochondria. Plays a role in protecting cells against oxidative stress-induced cell death via its interaction with RNF112. Functions in lipid transport from the endoplasmic reticulum and is involved in a wide array of cellular functions probably through regulation of the biogenesis of lipid microdomains at the plasma membrane. Related diseases Amyotrophic lateral sclerosis 16, juvenile (ALS16) [MIM:614373]: A neurodegenerative disorder affecting upper motor neurons in the brain and lower motor neurons in the brain stem and spinal cord, resulting in fatal paralysis. Sensory abnormalities are absent. The pathologic hallmarks of the disease include pallor of the corticospinal tract due to loss of motor neurons, presence of ubiquitin-positive inclusions within surviving motor neurons, and deposition of pathologic aggregates. The etiology of amyotrophic lateral sclerosis is likely to be multifactorial, involving both genetic and environmental factors. The disease is inherited in 5-10% of the cases. {ECO:0000269|PubMed:21842496}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Neuronopathy, distal hereditary motor, autosomal recessive 2 (HMNR2) [MIM:605726]: A form of distal hereditary motor neuronopathy, a heterogeneous group of neuromuscular disorders caused by selective degeneration of motor neurons in the anterior horn of the spinal cord, without sensory deficit in the posterior horn. The overall clinical picture consists of a classical distal muscular atrophy syndrome in the legs without clinical sensory loss. The disease starts with weakness and wasting of distal muscles of the anterior tibial and peroneal compartments of the legs. Later on, weakness and atrophy may expand to the proximal muscles of the lower limbs and/or to the distal upper limbs. HMNR2 is characterized by onset of distal muscle weakness and wasting affecting the lower and upper limbs in the first decade. {ECO:0000269|PubMed:26078401, ECO:0000269|PubMed:27629094}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00321; DB09014; DB00907; DB00514; DB01488; DB00574; DB00502; DB00956; DB00704; DB00540; DB06174; DB00652; DB11186; DB03575; DB05316; DB01708; DB00409; DB01104 Interacts with Q92847-1; Q99720-1; O00213-2; P17612; P50454; P37173 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Amyotrophic lateral sclerosis; Cell junction; Cell membrane; Cell projection; Cytoplasmic vesicle; Direct protein sequencing; Disease variant; Endoplasmic reticulum; Lipid droplet; Lipid transport; Membrane; Neurodegeneration; Neuropathy; Nucleus; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Synapse; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A,B,C Molecular weight (Da) 20805.3 Length 185 Aromaticity 0.14 Instability index 31.72 Isoelectric point 5.44 Charge (pH=7) -6.63 2D Binding mode Binding energy (Kcal/mol) -6.9 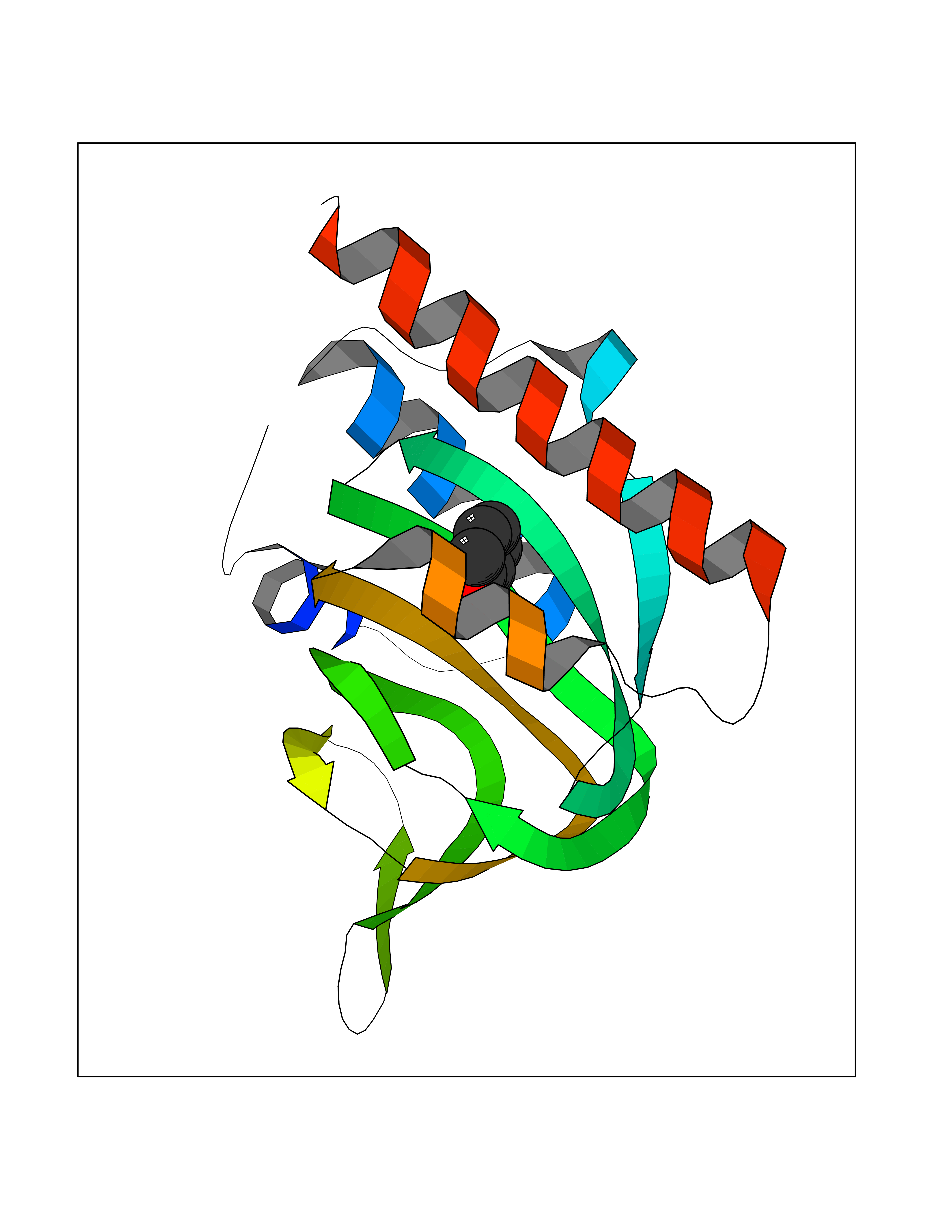 Molscript Map 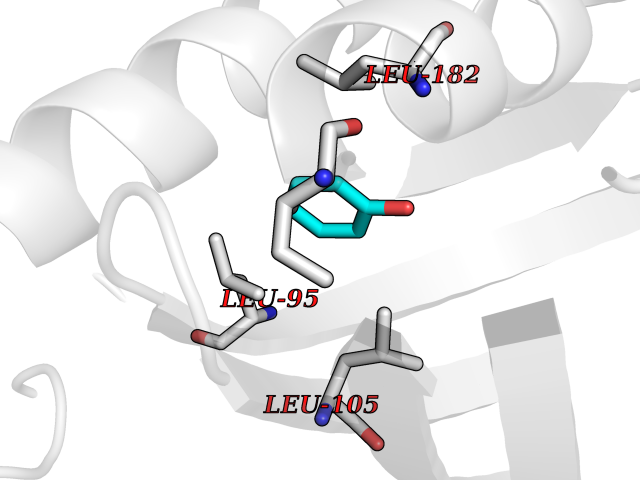 Pymol Map 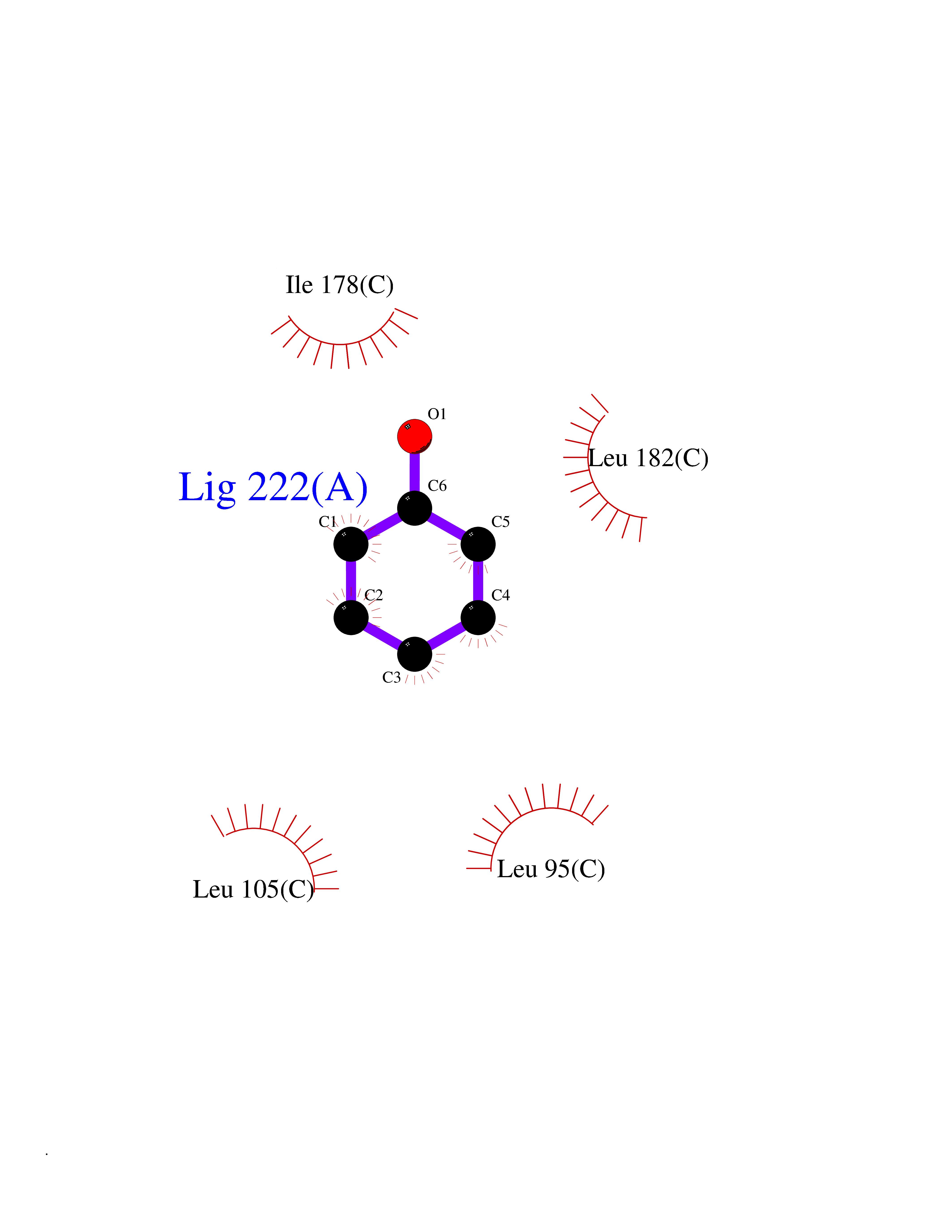 Ligplot Map 3D Binding mode Sequence VFQREEIAQLARQYAGLDHELAFSRLIVELRRLHPGHVLPDEELQWVFVNAGGWMGAMCLLHASLSEYVLLFGTALGSRGHSGRYWAEISDTIISGTFHQWREGTTKSEVFYPGETVVHGPGEATAVEWGPNTWMVEYGRGVIPSTLAFALADTVFSTQDFLTLFYTLRSYARGLRLELTTYLFG Hydrogen bonds contact Hydrophobic contact | ||||
| 88 | Protein-tyrosine-phosphatase | 2Z72 | 5.06 | |
Target general information Gen name PPI Organism Shewanella sp Uniprot ID TTD ID NA Synonyms NA Protein family NA Biochemical class Hydrolase Function Metal ion binding.Protein tyrosine phosphatase activity. Related diseases Brugada syndrome 7 (BRGDA7) [MIM:613120]: A tachyarrhythmia characterized by right bundle branch block and ST segment elevation on an electrocardiogram (ECG). It can cause the ventricles to beat so fast that the blood is prevented from circulating efficiently in the body. When this situation occurs, the individual will faint and may die in a few minutes if the heart is not reset. {ECO:0000269|PubMed:20031595}. The gene represented in this entry may be involved in disease pathogenesis.; DISEASE: Atrial fibrillation, familial, 16 (ATFB16) [MIM:613120]: A familial form of atrial fibrillation, a common sustained cardiac rhythm disturbance. Atrial fibrillation is characterized by disorganized atrial electrical activity and ineffective atrial contraction promoting blood stasis in the atria and reduces ventricular filling. It can result in palpitations, syncope, thromboembolic stroke, and congestive heart failure. {ECO:0000269|PubMed:20558140, ECO:0000269|PubMed:21051419}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number 3.1.3.48 Uniprot keywords 3D-structure; Calcium; Hydrolase; Metal-binding; Signal; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 38399.1 Length 338 Aromaticity 0.09 Instability index 29.48 Isoelectric point 5.85 Charge (pH=7) -7.13 2D Binding mode Binding energy (Kcal/mol) -6.91 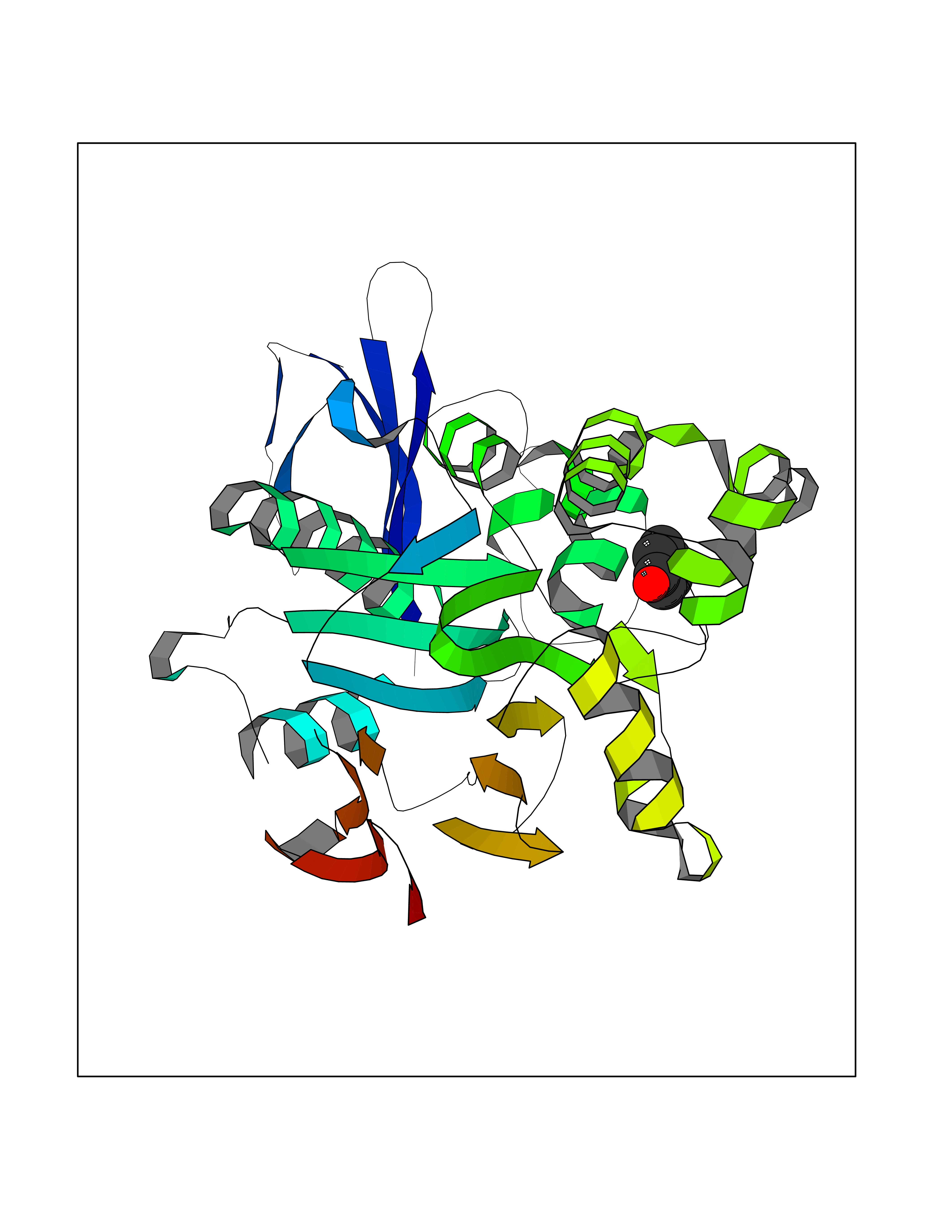 Molscript Map 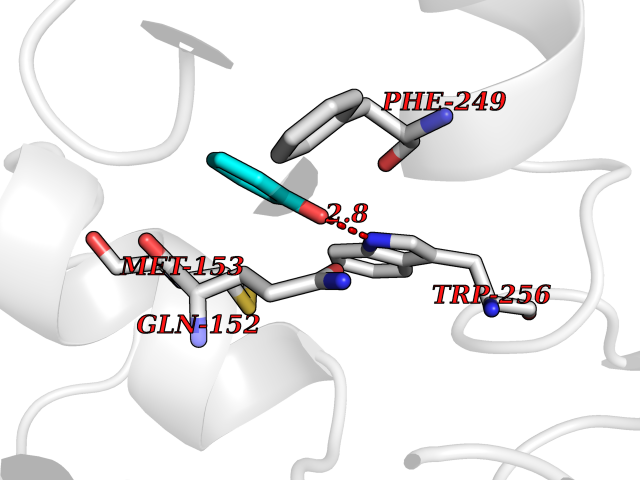 Pymol Map 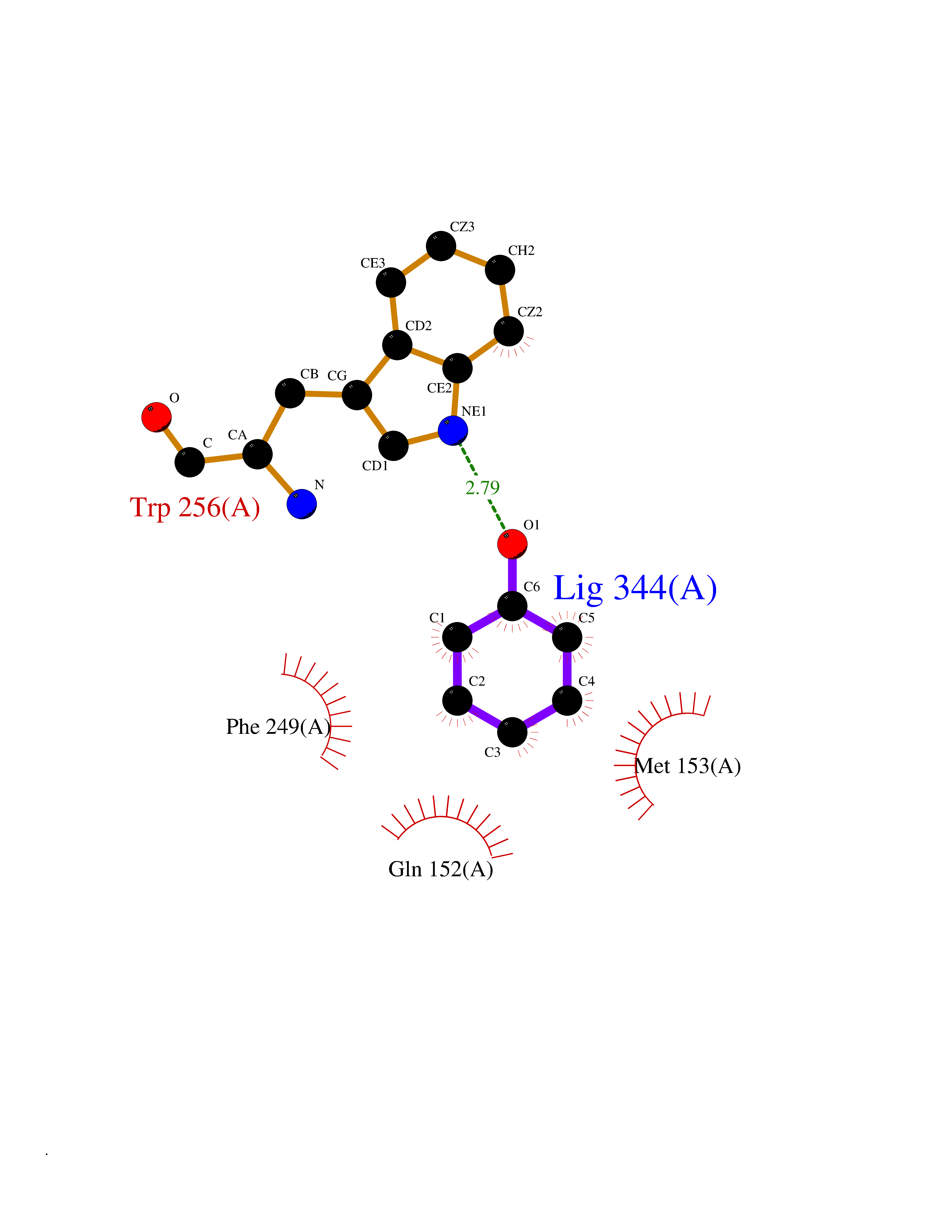 Ligplot Map 3D Binding mode Sequence ATEFDGPYVITPISGQSTAYWICDNRLKTTSIEKLQVNRPEHCGDLPETKLSSEIKQIMPDTYLGIKKVVALSDVHGQYDVLLTLLKKQKIIDSDGNWAFGEGHMVMTGDIFDRGHQVNEVLWFMYQLDQQARDAGGMVHLLMGNHEQMVLGGDLRYVHQRYDIATTLINRPYNKLYSADTEIGQWLRSKNTIIKINDVLYMHGGISSEWISRELTLDKANALYRANVDASKKSLKADDLLNFLFFGNGPTWYRGYFSETFTEAELDTILQHFNVNHIVVGHTSQERVLGLFHNKVIAVDSSIKVGKSGELLLLENNRLIRGLYDGTRETLQENSLNQ Hydrogen bonds contact Hydrophobic contact | ||||
| 89 | Glutathione S-transferase kappa 1 | 3RPN | 5.06 | |
Target general information Gen name GSTK1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms HDCMD47P Protein family GST superfamily, Kappa family Biochemical class Transferase / transferase inhibitor Function Glutathione peroxidase activity.Glutathione transferase activity.Protein disulfide oxidoreductase activity.Receptor binding. Related diseases Dyskinesia, limb and orofacial, infantile-onset (IOLOD) [MIM:616921]: An autosomal recessive, early-onset hyperkinetic movement disorder characterized by axial hypotonia, dyskinesia of the limbs and trunk, orofacial dyskinesia, drooling, and dysarthria. The severity of the hyperkinesis is variable. {ECO:0000269|PubMed:27058446}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Striatal degeneration, autosomal dominant 2 (ADSD2) [MIM:616922]: An autosomal dominant disorder characterized by striatal degeneration and dysfunction of basal ganglia, resulting in hyperkinesis. {ECO:0000269|PubMed:27058447}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00143; DB04700 Interacts with O95273; Q8IZU0; Q60994; Q7Z3Y8 EC number 2.5.1.18 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Peroxisome; Proteomics identification; Reference proteome; Transferase Protein physicochemical properties Chain ID A,B,C,D,E,F Molecular weight (Da) 24811.7 Length 220 Aromaticity 0.08 Instability index 47.17 Isoelectric point 7.96 Charge (pH=7) 1.39 2D Binding mode Binding energy (Kcal/mol) -6.9 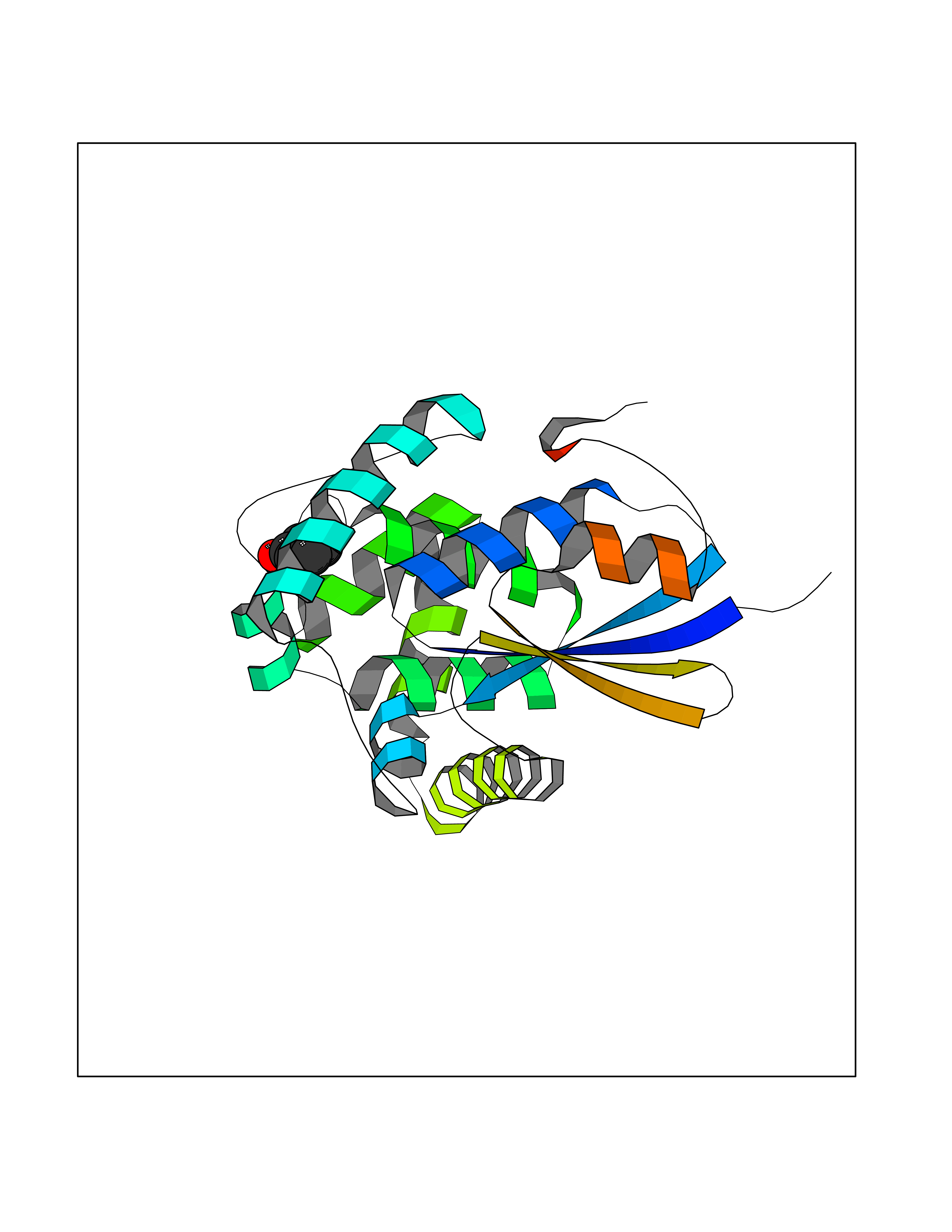 Molscript Map 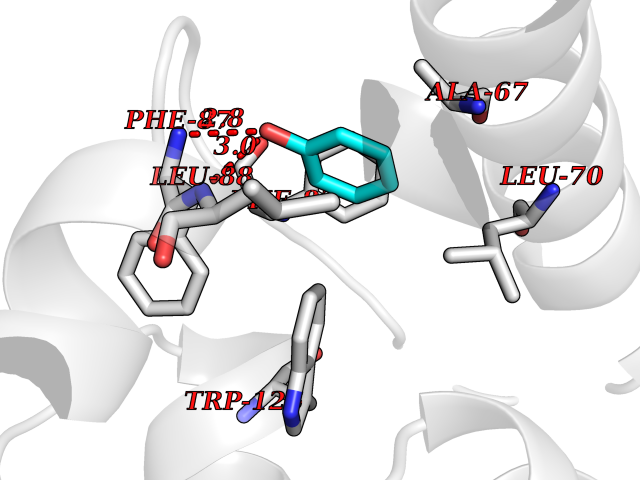 Pymol Map 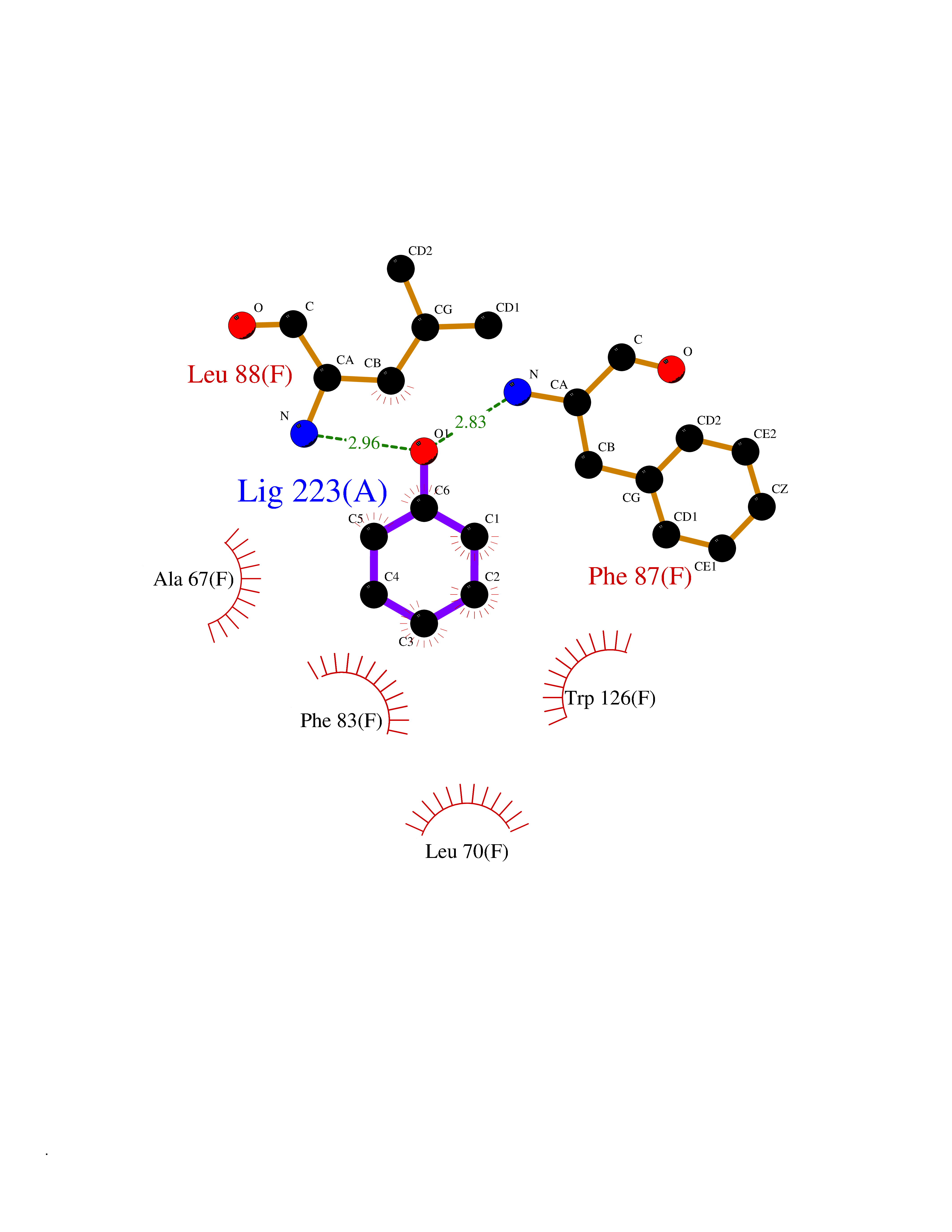 Ligplot Map 3D Binding mode Sequence GPLPRTVELFYDVLSPYSWLGFEILCRYQNIWNINLQLRPSLITGIMKDSGNKPPGLLPRKGLYMANDLKLLRHHLQIPIHFPKDFLSVMLEKGSLSAMRFLTAVNLEHPEMLEKASRELWMRVWSRNEDITEPQSILAAAEKAGMSAEQAQGLLEKIATPKVKNQLKETTEAACRYGAFGLPITVAHVDGQTHMLFGSDRMELLAHLLGEKWMGPIPPA Hydrogen bonds contact Hydrophobic contact | ||||
| 90 | Metabotropic glutamate receptor 3 (mGluR3) | 4XAR | 5.06 | |
Target general information Gen name GRM3 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms mGLUR3; Group III metabotropic glutamate receptor; GPRC1C Protein family G-protein coupled receptor 3 family Biochemical class GPCR glutamate Function Ligand binding causes a conformation change that triggers signaling via guanine nucleotide-binding proteins (G proteins) and modulates the activity of down-stream effectors. Signaling inhibits adenylate cyclase activity. G-protein coupled receptor for glutamate. Related diseases Paramyotonia congenita (PMC) [MIM:168300]: An autosomal dominant channelopathy characterized by myotonia, increased by exposure to cold, intermittent flaccid paresis, not necessarily dependent on cold or myotonia, lability of serum potassium, non-progressive nature and lack of atrophy or hypertrophy of muscles. In some patients, myotonia is not increased by cold exposure (paramyotonia without cold paralysis). Patients may have a combination phenotype of PMC and HYPP. {ECO:0000269|PubMed:10369308, ECO:0000269|PubMed:10727489, ECO:0000269|PubMed:1310898, ECO:0000269|PubMed:1316765, ECO:0000269|PubMed:1338909, ECO:0000269|PubMed:15318338, ECO:0000269|PubMed:15790667, ECO:0000269|PubMed:16786525, ECO:0000269|PubMed:18166706, ECO:0000269|PubMed:18690054, ECO:0000269|PubMed:19077043, ECO:0000269|PubMed:20076800, ECO:0000269|PubMed:8242056, ECO:0000269|PubMed:8308722, ECO:0000269|PubMed:8388676, ECO:0000269|PubMed:8580427}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Periodic paralysis hypokalemic 2 (HOKPP2) [MIM:613345]: An autosomal dominant disorder manifested by episodic flaccid generalized muscle weakness associated with falls of serum potassium levels. {ECO:0000269|PubMed:10599760, ECO:0000269|PubMed:10851391, ECO:0000269|PubMed:10944223, ECO:0000269|PubMed:11558801, ECO:0000269|PubMed:11591859, ECO:0000269|PubMed:16890191, ECO:0000269|PubMed:17898326, ECO:0000269|PubMed:18162704, ECO:0000269|PubMed:19118277, ECO:0000269|PubMed:20522878, ECO:0000269|PubMed:21043388, ECO:0000269|PubMed:24549961}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Periodic paralysis hyperkalemic (HYPP) [MIM:170500]: An autosomal dominant channelopathy characterized by episodic flaccid generalized muscle weakness associated with high levels of serum potassium. Concurrence of myotonia is found in HYPP patients. {ECO:0000269|PubMed:1659668, ECO:0000269|PubMed:1659948, ECO:0000269|PubMed:20076800}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Periodic paralysis normokalemic (NKPP) [MIM:170500]: A disorder closely related to hyperkalemic periodic paralysis, but marked by a lack of alterations in potassium levels during attacks of muscle weakness. {ECO:0000269|PubMed:15596759, ECO:0000269|PubMed:18046642, ECO:0000269|PubMed:20522878}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Myotonia SCN4A-related (MYOSCN4A) [MIM:608390]: A phenotypically highly variable myotonia aggravated by potassium loading, and sometimes by cold. Myotonia is characterized by sustained muscle tensing that prevents muscles from relaxing normally. It causes muscle stiffness that can interfere with movement. In some people the stiffness is very mild, while in other cases it may be severe enough to interfere with walking, running, and other activities of daily life. Myotonia SCN4A-related includes myotonia permanens and myotonia fluctuans. In myotonia permanens, the myotonia is generalized and there is a hypertrophy of the muscle, particularly in the neck and the shoulder. Attacks of severe muscle stiffness of the thoracic muscles may be life threatening due to impaired ventilation. In myotonia fluctuans, the muscle stiffness may fluctuate from day to day, provoked by exercise. {ECO:0000269|PubMed:10218481, ECO:0000269|PubMed:16786525, ECO:0000269|PubMed:16832098, ECO:0000269|PubMed:17212350, ECO:0000269|PubMed:17998485, ECO:0000269|PubMed:18203179, ECO:0000269|PubMed:18337100, ECO:0000269|PubMed:19015483, ECO:0000269|PubMed:19347921, ECO:0000269|PubMed:20076800, ECO:0000269|PubMed:27653901, ECO:0000269|PubMed:8058156, ECO:0000269|PubMed:9392583}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Myasthenic syndrome, congenital, 16 (CMS16) [MIM:614198]: A form of congenital myasthenic syndrome, a group of disorders characterized by failure of neuromuscular transmission, including pre-synaptic, synaptic, and post-synaptic disorders that are not of autoimmune origin. Clinical features are easy fatigability and muscle weakness. CMS16 is characterized by fatigable generalized weakness and recurrent attacks of respiratory and bulbar paralysis since birth. The fatigable weakness involves lid-elevator, external ocular, facial, limb and truncal muscles and an decremental response of the compound muscle action potential on repetitive stimulation. {ECO:0000269|PubMed:12766226, ECO:0000269|PubMed:25707578, ECO:0000269|PubMed:26659129}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Congenital myopathy 22A, classic (CMYO22A) [MIM:620351]: A form of congenital myopathy, a clinically and genetically heterogeneous group of muscle disorders characterized by hypotonia and muscle weakness apparent at birth, and specific pathological features on muscle biopsy. CMYO22A is an autosomal recessive form characterized by fetal hypokinesia, polyhydramnios, and severe neonatal hypotonia associated with respiratory insufficiency. Affected individuals who survive the neonatal period have delayed motor development, difficulty walking, proximal muscle weakness of the upper and lower limbs, facial and neck muscle weakness, easy fatigability, and mild limb contractures or foot deformities. {ECO:0000269|PubMed:26700687, ECO:0000269|PubMed:28262468, ECO:0000269|PubMed:36090556}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Congenital myopathy 22B, severe fetal (CMYO22B) [MIM:620369]: A severe congenital myopathy, a clinically and genetically heterogeneous group of muscle disorders characterized by hypotonia and muscle weakness apparent at birth, and specific pathological features on muscle biopsy. CMYO22B is an autosomal recessive form characterized by onset in utero. Affected individuals show fetal akinesia, and develop fetal hydrops with pulmonary hypoplasia, severe joint contractures, and generalized muscle hypoplasia. Death occurs in utero or soon after birth. {ECO:0000269|PubMed:26700687}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB05096 Interacts with NA EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disulfide bond; G-protein coupled receptor; Glycoprotein; Membrane; Proteomics identification; Receptor; Reference proteome; Signal; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 50355.5 Length 445 Aromaticity 0.11 Instability index 38.26 Isoelectric point 6.52 Charge (pH=7) -1.53 2D Binding mode Binding energy (Kcal/mol) -6.9  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence RREIKIEGDLVLGGLFPINEKGTGTEECGRINEDRGIQRLEAMLFAIDEINKDDYLLPGVKLGVHILDTCSRDTYALEQSLEFVRASLLLIAGVIGGSYSSVSIQVANLLRLFQIPQISYASTSAKLSDKSRYDYFARTVPPDFYQAKAMAEILRFFNWTYVSTVASEGDYGETGIEAFEQEARLRNISIATAEKVGRSNIRKSYDSVIRELLQKPNARVVVLFMRSDDSRELIAAASRANASFTWVASDGWGAQESIIKGSEHVAYGAITLELASQPVRQFDRYFQSLNPYNNHRNPWFRDFWEQKFQCSLRVCDKHLAIDSSNYEQESKIMFVVNAVYAMAHALHKMQRTLCPNTTKLCDAMKILDGKKLYKDYLLKINFTAPDADSIVKFDTFGDGMGRYNVFNFQNVGGKYSYLKVGHWAETLSLDVNSIHWSRNSVPTSE Hydrogen bonds contact Hydrophobic contact | ||||
| 91 | Pyruvate dehydrogenase kinase 1 (PDHK1) | 2Q8G | 5.06 | |
Target general information Gen name PDK1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Pyruvate dehydrogenase kinase isoform 1; Pyruvate dehydrogenase (acetyl-transferring) kinase isozyme 1, mitochondrial; PDHK1; PDH kinase 1 Protein family PDK/BCKDK protein kinase family Biochemical class Kinase Function Kinase that plays a key role in regulation of glucose and fatty acid metabolism and homeostasis via phosphorylation of the pyruvate dehydrogenase subunits PDHA1 and PDHA2. This inhibits pyruvate dehydrogenase activity, and thereby regulates metabolite flux through the tricarboxylic acid cycle, down-regulates aerobic respiration and inhibits the formation of acetyl-coenzyme A from pyruvate. Plays an important role in cellular responses to hypoxia and is important for cell proliferation under hypoxia. Protects cells against apoptosis in response to hypoxia and oxidative stress. Related diseases TP53 is found in increased amounts in a wide variety of transformed cells. TP53 is frequently mutated or inactivated in about 60% of cancers. TP53 defects are found in Barrett metaplasia a condition in which the normally stratified squamous epithelium of the lower esophagus is replaced by a metaplastic columnar epithelium. The condition develops as a complication in approximately 10% of patients with chronic gastroesophageal reflux disease and predisposes to the development of esophageal adenocarcinoma.; DISEASE: Esophageal cancer (ESCR) [MIM:133239]: A malignancy of the esophagus. The most common types are esophageal squamous cell carcinoma and adenocarcinoma. Cancer of the esophagus remains a devastating disease because it is usually not detected until it has progressed to an advanced incurable stage. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Li-Fraumeni syndrome (LFS) [MIM:151623]: An autosomal dominant familial cancer syndrome that in its classic form is defined by the existence of a proband affected by a sarcoma before 45 years with a first degree relative affected by any tumor before 45 years and another first degree relative with any tumor before 45 years or a sarcoma at any age. Other clinical definitions for LFS have been proposed and called Li-Fraumeni like syndrome (LFL). In these families affected relatives develop a diverse set of malignancies at unusually early ages. Four types of cancers account for 80% of tumors occurring in TP53 germline mutation carriers: breast cancers, soft tissue and bone sarcomas, brain tumors (astrocytomas) and adrenocortical carcinomas. Less frequent tumors include choroid plexus carcinoma or papilloma before the age of 15, rhabdomyosarcoma before the age of 5, leukemia, Wilms tumor, malignant phyllodes tumor, colorectal and gastric cancers. {ECO:0000269|PubMed:10484981, ECO:0000269|PubMed:1565144, ECO:0000269|PubMed:1737852, ECO:0000269|PubMed:1933902, ECO:0000269|PubMed:1978757, ECO:0000269|PubMed:2259385, ECO:0000269|PubMed:36108750, ECO:0000269|PubMed:7887414, ECO:0000269|PubMed:8825920, ECO:0000269|PubMed:9452042}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Squamous cell carcinoma of the head and neck (HNSCC) [MIM:275355]: A non-melanoma skin cancer affecting the head and neck. The hallmark of cutaneous SCC is malignant transformation of normal epidermal keratinocytes. The gene represented in this entry is involved in disease pathogenesis.; DISEASE: Lung cancer (LNCR) [MIM:211980]: A common malignancy affecting tissues of the lung. The most common form of lung cancer is non-small cell lung cancer (NSCLC) that can be divided into 3 major histologic subtypes: squamous cell carcinoma, adenocarcinoma, and large cell lung cancer. NSCLC is often diagnosed at an advanced stage and has a poor prognosis. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Papilloma of choroid plexus (CPP) [MIM:260500]: A benign tumor of neuroectodermal origin that generally occurs in childhood, but has also been reported in adults. Although generally found within the ventricular system, choroid plexus papillomas can arise ectopically in the brain parenchyma or disseminate throughout the neuraxis. Patients present with signs and symptoms of increased intracranial pressure including headache, hydrocephalus, papilledema, nausea, vomiting, cranial nerve deficits, gait impairment, and seizures. {ECO:0000269|PubMed:12085209}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Adrenocortical carcinoma (ADCC) [MIM:202300]: A malignant neoplasm of the adrenal cortex and a rare childhood tumor. It occurs with increased frequency in patients with Beckwith-Wiedemann syndrome and Li-Fraumeni syndrome. {ECO:0000269|PubMed:11481490}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Basal cell carcinoma 7 (BCC7) [MIM:614740]: A common malignant skin neoplasm that typically appears on hair-bearing skin, most commonly on sun-exposed areas. It is slow growing and rarely metastasizes, but has potentialities for local invasion and destruction. It usually develops as a flat, firm, pale area that is small, raised, pink or red, translucent, shiny, and waxy, and the area may bleed following minor injury. Tumor size can vary from a few millimeters to several centimeters in diameter. {ECO:0000269|PubMed:21946351}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Bone marrow failure syndrome 5 (BMFS5) [MIM:618165]: A form of bone marrow failure syndrome, a heterogeneous group of life-threatening disorders characterized by hematopoietic defects in association with a range of variable extra-hematopoietic manifestations. BMFS5 is an autosomal dominant form characterized by infantile onset of severe red cell anemia requiring transfusion. Additional features include hypogammaglobulinemia, poor growth with microcephaly, developmental delay, and seizures. {ECO:0000269|PubMed:30146126}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07403; DB08809 Interacts with P05067; P08559; Q16513; P31749-1; P31751-1 EC number EC 2.7.11.2 Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Kinase; Mitochondrion; Nucleotide-binding; Phosphoprotein; Proteomics identification; Reference proteome; Transferase; Transit peptide Protein physicochemical properties Chain ID A Molecular weight (Da) 42249.9 Length 368 Aromaticity 0.11 Instability index 49.91 Isoelectric point 6.83 Charge (pH=7) -0.46 2D Binding mode Binding energy (Kcal/mol) -6.9  Molscript Map 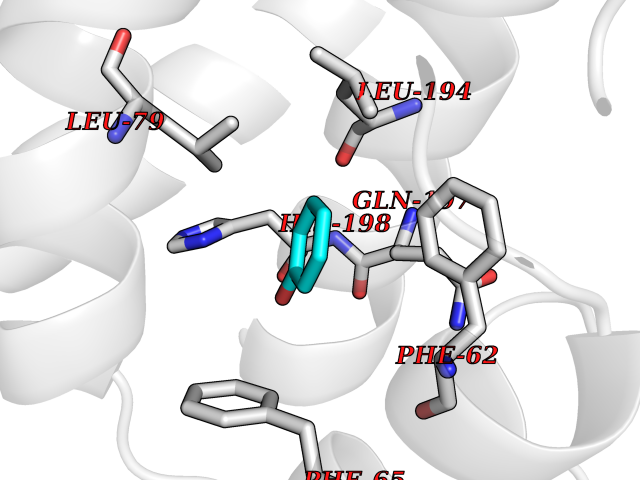 Pymol Map 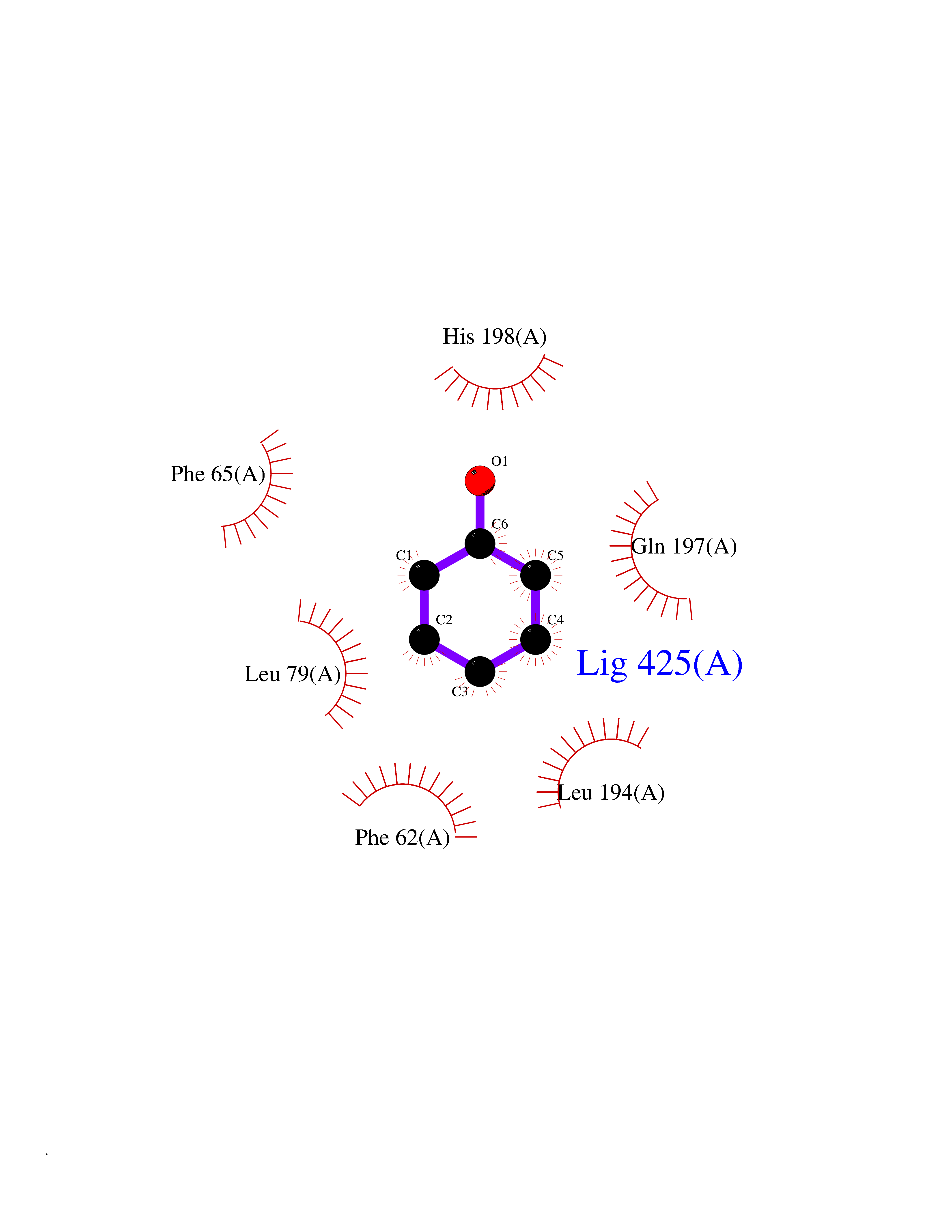 Ligplot Map 3D Binding mode Sequence GVPGQVDFYARFSPSPLSMKQFLDFGSVNACEKTSFMFLRQELPVRLANIMKEISLLPDNLLRTPSVQLVQSWYIQSLQELLDFKDKSAEDAKAIYDFTDTVIRIRNRHNDVIPTMAQGVIEYKESFDPVTSQNVQYFLDRFYMSRISIRMLLNQHSLLFGKHIGSINPNCNVLEVIKDGYENARRLCDLYYINSPELELEELNAKSPGQPIQVVYVPSHLYHMVFELFKNAMRATMEHHANRGVYPPIQVHVTLGNEDLTVKMSDRGGGVPLRKIDRLFNYMYSTAPRPRVETSRAVPLAGFGYGLPISRLYAQYFQGDLKLYSLEGYGTDAVIYIKALSTDSIERLPVYNKAAWKHYNTNDDWCVP Hydrogen bonds contact Hydrophobic contact | ||||
| 92 | Extracellular signal-regulated kinase 1 (ERK1) | 2ZOQ | 5.06 | |
Target general information Gen name MAPK3 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms PRKM3; P44-MAPK; P44-ERK1; P44 Mitogen-activated protein kinase; Mitogen-activated protein kinase 3; Microtubule-associated protein-2 kinase; Microtubule-associated protein 2 kinase; MAPK 3; MAP kinas Protein family Protein kinase superfamily, CMGC Ser/Thr protein kinase family, MAP kinase subfamily Biochemical class Kinase Function MAPK1/ERK2 and MAPK3/ERK1 are the 2 MAPKs which play an important role in the MAPK/ERK cascade. They participate also in a signaling cascade initiated by activated KIT and KITLG/SCF. Depending on the cellular context, the MAPK/ERK cascade mediates diverse biological functions such as cell growth, adhesion, survival and differentiation through the regulation of transcription, translation, cytoskeletal rearrangements. The MAPK/ERK cascade plays also a role in initiation and regulation of meiosis, mitosis, and postmitotic functions in differentiated cells by phosphorylating a number of transcription factors. About 160 substrates have already been discovered for ERKs. Many of these substrates are localized in the nucleus, and seem to participate in the regulation of transcription upon stimulation. However, other substrates are found in the cytosol as well as in other cellular organelles, and those are responsible for processes such as translation, mitosis and apoptosis. Moreover, the MAPK/ERK cascade is also involved in the regulation of the endosomal dynamics, including lysosome processing and endosome cycling through the perinuclear recycling compartment (PNRC); as well as in the fragmentation of the Golgi apparatus during mitosis. The substrates include transcription factors (such as ATF2, BCL6, ELK1, ERF, FOS, HSF4 or SPZ1), cytoskeletal elements (such as CANX, CTTN, GJA1, MAP2, MAPT, PXN, SORBS3 or STMN1), regulators of apoptosis (such as BAD, BTG2, CASP9, DAPK1, IER3, MCL1 or PPARG), regulators of translation (such as EIF4EBP1) and a variety of other signaling-related molecules (like ARHGEF2, FRS2 or GRB10). Protein kinases (such as RAF1, RPS6KA1/RSK1, RPS6KA3/RSK2, RPS6KA2/RSK3, RPS6KA6/RSK4, SYK, MKNK1/MNK1, MKNK2/MNK2, RPS6KA5/MSK1, RPS6KA4/MSK2, MAPKAPK3 or MAPKAPK5) and phosphatases (such as DUSP1, DUSP4, DUSP6 or DUSP16) are other substrates which enable the propagation the MAPK/ERK signal to additional cytosolic and nuclear targets, thereby extending the specificity of the cascade. Serine/threonine kinase which acts as an essential component of the MAP kinase signal transduction pathway. Related diseases Microvascular complications of diabetes 5 (MVCD5) [MIM:612633]: Pathological conditions that develop in numerous tissues and organs as a consequence of diabetes mellitus. They include diabetic retinopathy, diabetic nephropathy leading to end-stage renal disease, and diabetic neuropathy. Diabetic retinopathy remains the major cause of new-onset blindness among diabetic adults. It is characterized by vascular permeability and increased tissue ischemia and angiogenesis. Disease susceptibility is associated with variants affecting the gene represented in this entry. Homozygosity for the Leu-55 allele is strongly associated with the development of retinal disease in diabetic patients. Drugs (DrugBank ID) DB04604; DB00945; DB01169; DB08862; DB02587; DB01017; DB02733; DB06195; DB00605; DB13930 Interacts with Q9HCU0; P53355; P49366; P28562; Q16828; P19419; Q49AJ0-4; Q96NJ5; Q02750; Q16539; P27361; Q96HT8; Q6GQQ9-2; Q9ULW8; Q15121; P14618-1; Q8N490; P23467; Q12913; Q14160; P13051-2; Q08509; Q60793; Q9EPI6; Q62132 EC number EC 2.7.11.24 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Apoptosis; ATP-binding; Cell cycle; Cell junction; Cytoplasm; Direct protein sequencing; Host-virus interaction; Kinase; Membrane; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Serine/threonine-protein kinase; Transferase; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 40525.2 Length 352 Aromaticity 0.09 Instability index 41.34 Isoelectric point 6.45 Charge (pH=7) -2.32 2D Binding mode Binding energy (Kcal/mol) -6.91 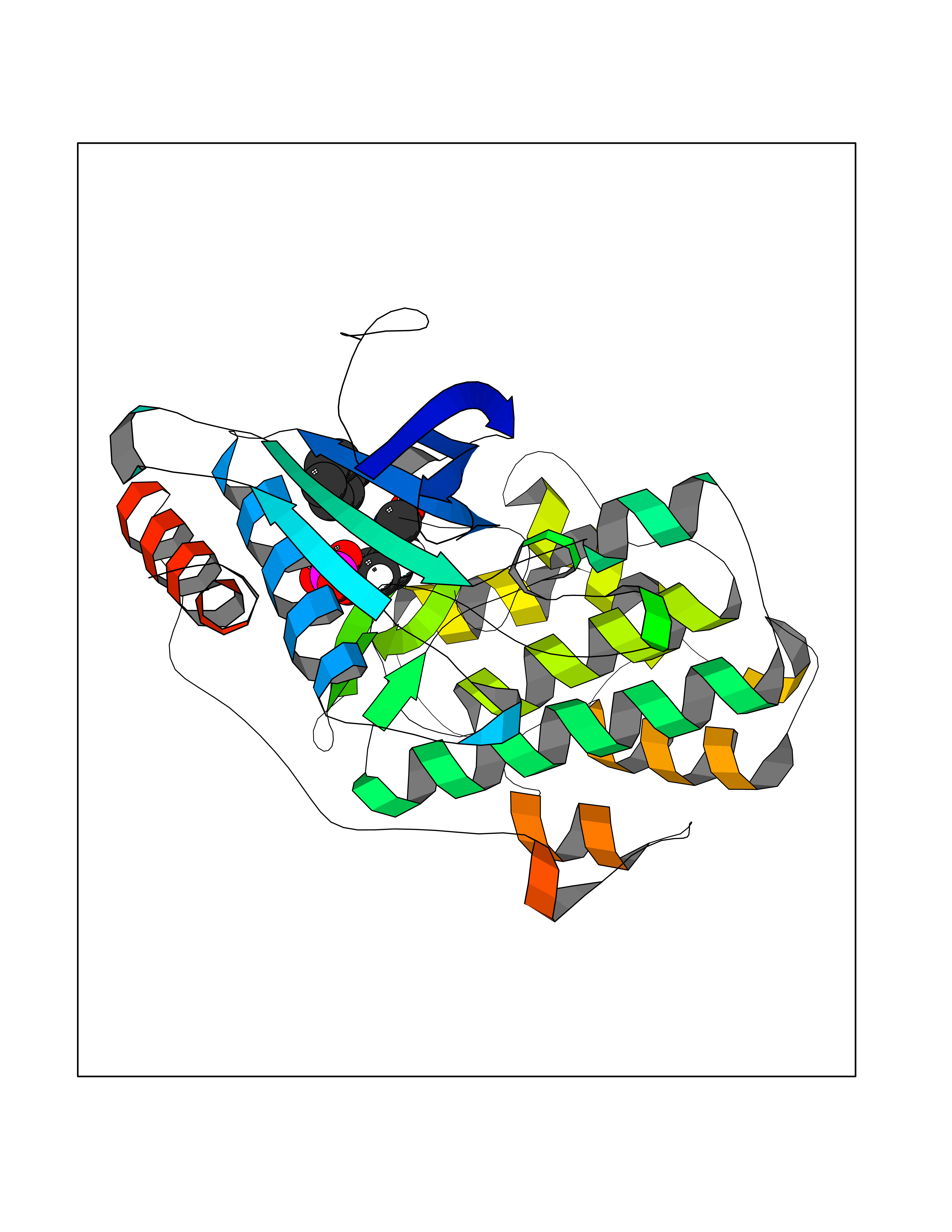 Molscript Map 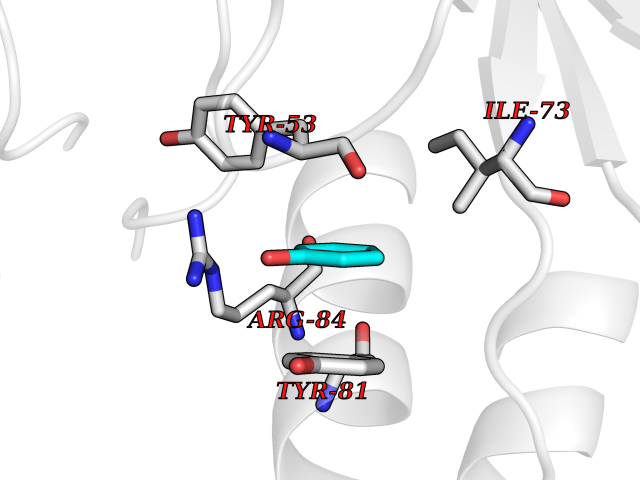 Pymol Map  Ligplot Map 3D Binding mode Sequence VPGEVEMVKGQPFDVGPRYTQLQYIGEGAYGMVSSAYDHVRKTRVAIKKISPFEHQTYCQRTLREIQILLRFRHENVIGIRDILRASTLEAMRDVYIVQDLMETDLYKLLKSQQLSNDHICYFLYQILRGLKYIHSANVLHRDLKPSNLLINTTCDLKICDFGLARIADPEHDHTGFLTEXVATRWYRAPEIMLNSKGYTKSIDIWSVGCILAEMLSNRPIFPGKHYLDQLNHILGILGSPSQEDLNCIINMKARNYLQSLPSKTKVAWAKLFPKSDSKALDLLDRMLTFNPNKRITVEEALAHPYLEQYYDPTDEPVAEEPFTFAMELDDLPKERLKELIFQETARFQPGV Hydrogen bonds contact Hydrophobic contact | ||||
| 93 | HIF-prolyl hydroxylase 2 (HPH-2) | 6ZBO | 5.06 | |
Target general information Gen name EGLN1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms SM-20; Prolyl hydroxylase domain-containing protein 2; PHD2; Hypoxia-inducible factor prolyl hydroxylase 2; HPH-2; HIF-PH2; Egl nine homolog 1; C1orf12 Protein family NA Biochemical class Paired donor oxygen oxidoreductase Function Cellular oxygen sensor that catalyzes, under normoxic conditions, the post-translational formation of 4-hydroxyproline in hypoxia-inducible factor (HIF) alpha proteins. Hydroxylates a specific proline found in each of the oxygen-dependent degradation (ODD) domains (N-terminal, NODD, and C-terminal, CODD) of HIF1A. Also hydroxylates HIF2A. Has a preference for the CODD site for both HIF1A and HIF1B. Hydroxylated HIFs are then targeted for proteasomal degradation via the von Hippel-Lindau ubiquitination complex. Under hypoxic conditions, the hydroxylation reaction is attenuated allowing HIFs to escape degradation resulting in their translocation to the nucleus, heterodimerization with HIF1B, and increased expression of hypoxy-inducible genes. EGLN1 is the most important isozyme under normoxia and, through regulating the stability of HIF1, involved in various hypoxia-influenced processes such as angiogenesis in retinal and cardiac functionality. Target proteins are preferentially recognized via a LXXLAP motif. Related diseases Erythrocytosis, familial, 3 (ECYT3) [MIM:609820]: An autosomal dominant disorder characterized by elevated serum hemoglobin and hematocrit, and normal serum erythropoietin levels. {ECO:0000269|PubMed:16407130, ECO:0000269|PubMed:17579185}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00126; DB11682; DB14490; DB14491; DB14488; DB14501; DB14489; DB08687; DB01592; DB07112; DB04847; DB12255 Interacts with Q99814; Q14318; Q16665; Q13438; PRO_0000037551 [Q9WMX2] EC number EC 1.14.11.29 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Congenital erythrocytosis; Cytoplasm; Dioxygenase; Disease variant; Iron; Metal-binding; Nucleus; Oxidoreductase; Phosphoprotein; Proteomics identification; Reference proteome; S-nitrosylation; Vitamin C; Zinc; Zinc-finger Protein physicochemical properties Chain ID A,D Molecular weight (Da) 45717.8 Length 406 Aromaticity 0.11 Instability index 24.41 Isoelectric point 7.59 Charge (pH=7) 1.54 2D Binding mode Binding energy (Kcal/mol) -6.9  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence LPALKLALEYIVPCMNKHGICVVDDFLGKETGQQIGDEVRALHDTGKFTGDKITWIEGKEPGCETIGLLMSSMDDLIRHCNGKLGSYKINGRTKAMVACYPGNGTGYVRHVDNPNGDGRCVTCIYYLNKDWDAKVSGGILRIFPEGKAQFADIEPKFDRLLFFWSDRRNPHEVQPAYATRYAITVWYFDADERARAKVKYLTGELPALKLALEYIVPCMNKHGICVVDDFLGKETGQQIGDEVRALHDTGKFTGDKITWIEGKEPGCETIGLLMSSMDDLIRHCNGKLGSYKINGRTKAMVACYPGNGTGYVRHVDNPNGDGRCVTCIYYLNKDWDAKVSGGILRIFPEGKAQFADIEPKFDRLLFFWSDRRNPHEVQPAYATRYAITVWYFDADERARAKVKYLT Hydrogen bonds contact Hydrophobic contact | ||||
| 94 | Dual-specificity tyrosine-phosphorylation regulated kinase 2 (DYRK2) | 6HDR | 5.06 | |
Target general information Gen name DYRK2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Dual specificity tyrosine-phosphorylation-regulated kinase 2 Protein family Protein kinase superfamily, CMGC Ser/Thr protein kinase family, MNB/DYRK subfamily Biochemical class Kinase Function Functions in part via its role in ubiquitin-dependent proteasomal protein degradation. Functions downstream of ATM and phosphorylates p53/TP53 at 'Ser-46', and thereby contributes to the induction of apoptosis in response to DNA damage. Phosphorylates NFATC1, and thereby inhibits its accumulation in the nucleus and its transcription factor activity. Phosphorylates EIF2B5 at 'Ser-544', enabling its subsequent phosphorylation and inhibition by GSK3B. Likewise, phosphorylation of NFATC1, CRMP2/DPYSL2 and CRMP4/DPYSL3 promotes their subsequent phosphorylation by GSK3B. May play a general role in the priming of GSK3 substrates. Inactivates GYS1 by phosphorylation at 'Ser-641', and potentially also a second phosphorylation site, thus regulating glycogen synthesis. Mediates EDVP E3 ligase complex formation and is required for the phosphorylation and subsequent degradation of KATNA1. Phosphorylates TERT at 'Ser-457', promoting TERT ubiquitination by the EDVP complex. Phosphorylates SIAH2, and thereby increases its ubiquitin ligase activity. Promotes the proteasomal degradation of MYC and JUN, and thereby regulates progress through the mitotic cell cycle and cell proliferation. Promotes proteasomal degradation of GLI2 and GLI3, and thereby plays a role in smoothened and sonic hedgehog signaling. Plays a role in cytoskeleton organization and neurite outgrowth via its phosphorylation of DCX and DPYSL2. Phosphorylates CRMP2/DPYSL2, CRMP4/DPYSL3, DCX, EIF2B5, EIF4EBP1, GLI2, GLI3, GYS1, JUN, MDM2, MYC, NFATC1, p53/TP53, TAU/MAPT and KATNA1. Can phosphorylate histone H1, histone H3 and histone H2B (in vitro). Can phosphorylate CARHSP1 (in vitro). Serine/threonine-protein kinase involved in the regulation of the mitotic cell cycle, cell proliferation, apoptosis, organization of the cytoskeleton and neurite outgrowth. Related diseases Bone marrow failure and diabetes mellitus syndrome (BMFDMS) [MIM:620044]: A form of bone marrow failure syndrome, a heterogeneous group of life-threatening disorders characterized by hematopoietic defects in association with a range of variable extra-hematopoietic manifestations. BMFDMS is an autosomal recessive form characterized by various degrees of bone marrow failure, ranging from dyserythropoiesis to bone marrow aplasia, with onset in infancy or early childhood, and non-autoimmune insulin-dependent diabetes mellitus appearing in the first or second decades. Many patients show pigmentary skin abnormalities and short stature. {ECO:0000269|PubMed:28073829, ECO:0000269|PubMed:35611808, ECO:0000269|PubMed:35931051}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q9NR20; Q13422; Q9BQD3; Q9BRK4; P23497; O43379; P62258; Q96C00 EC number EC 2.7.12.1 Uniprot keywords 3D-structure; Alternative splicing; Apoptosis; ATP-binding; Cytoplasm; Kinase; Magnesium; Manganese; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Serine/threonine-protein kinase; Transferase; Tyrosine-protein kinase; Ubl conjugation; Ubl conjugation pathway Protein physicochemical properties Chain ID A Molecular weight (Da) 46422.1 Length 407 Aromaticity 0.09 Instability index 44.91 Isoelectric point 9.09 Charge (pH=7) 12.37 2D Binding mode Binding energy (Kcal/mol) -6.9 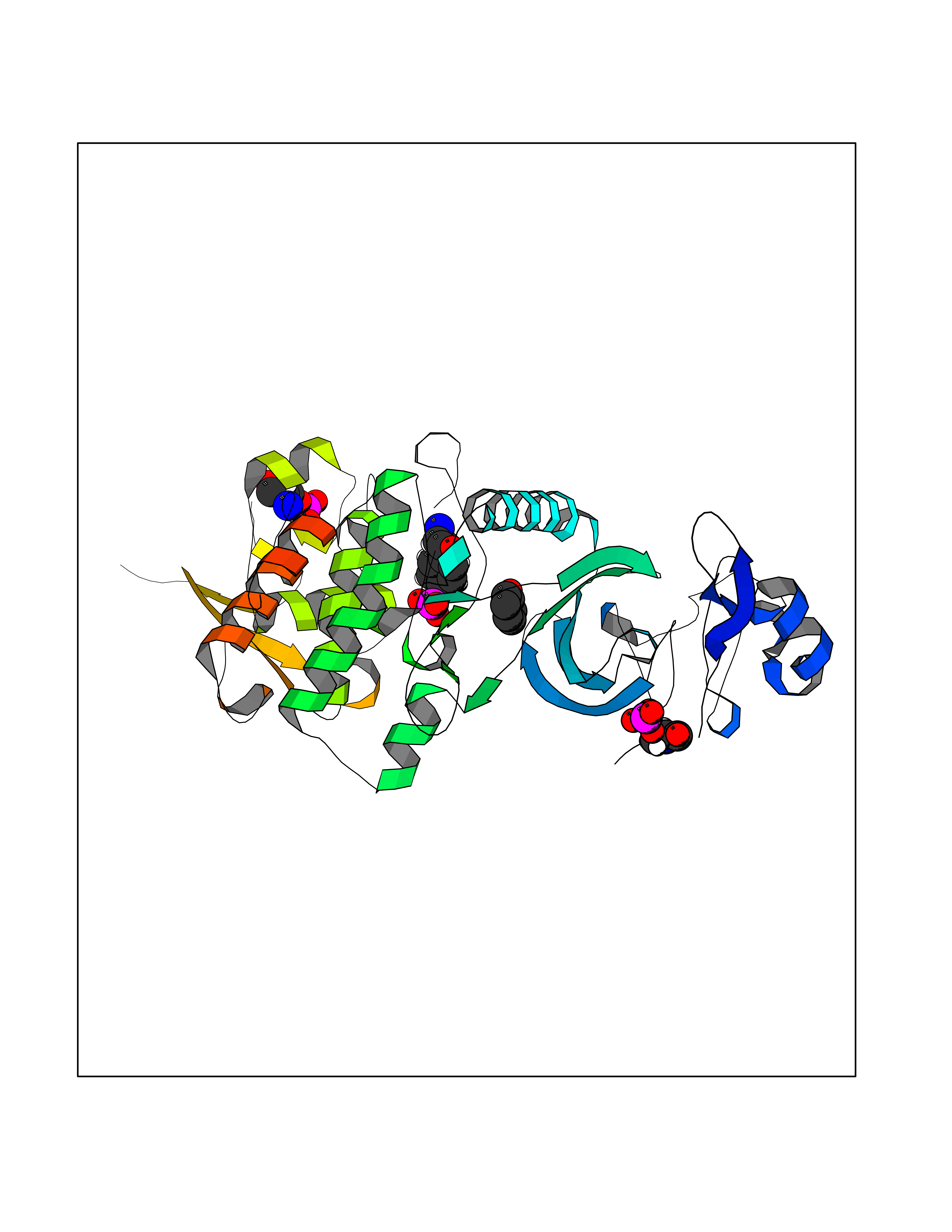 Molscript Map 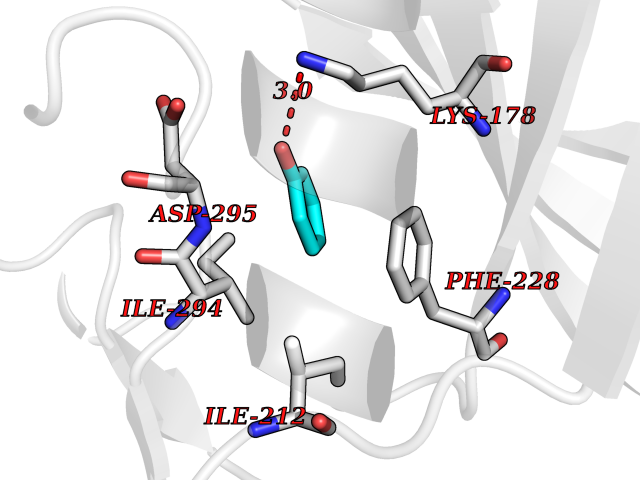 Pymol Map 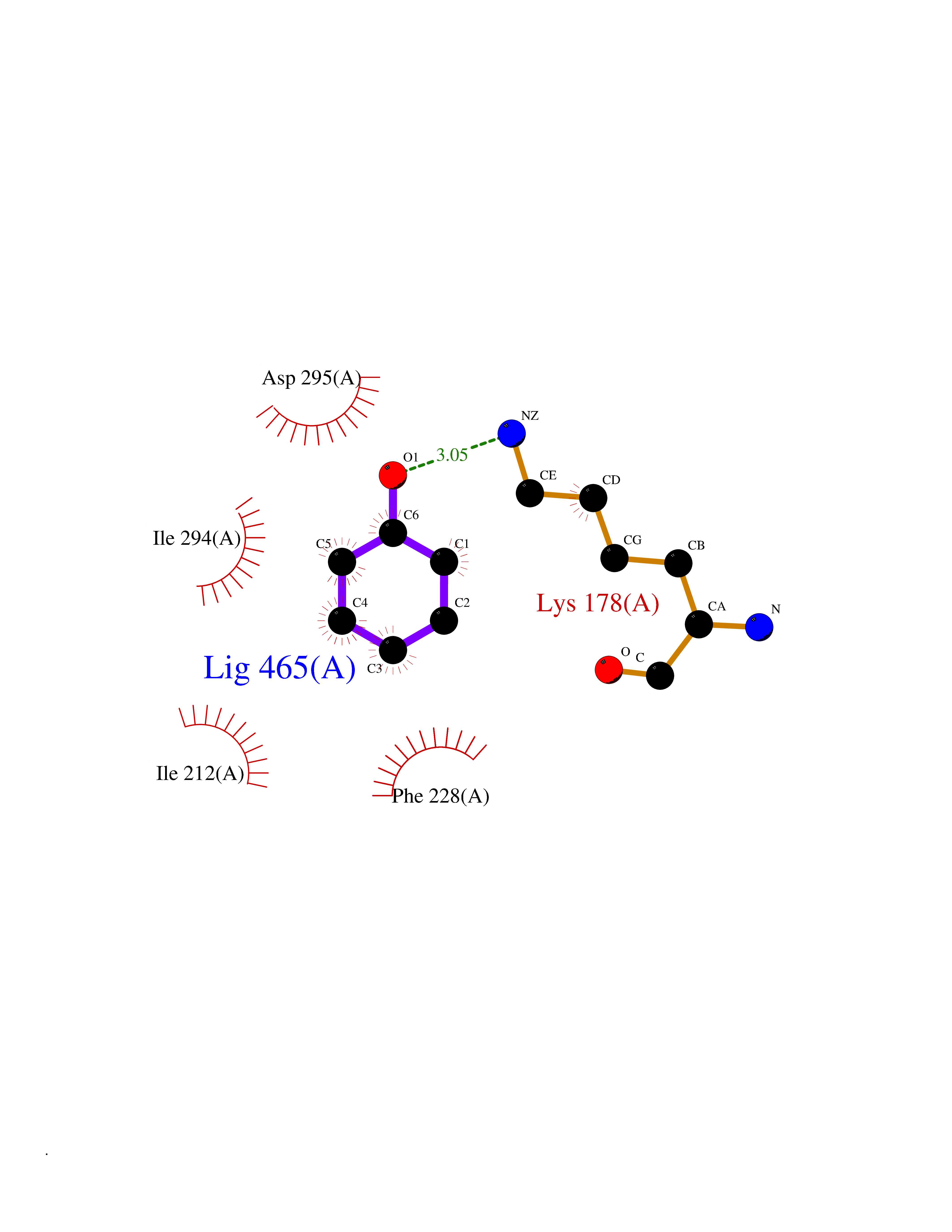 Ligplot Map 3D Binding mode Sequence HHHSXGVDLGTENLYFQSMGKVKATPMTPEQAMKQYMQKLTAFEHHEIFSYPEIYFLGLNAKKRQGMTGGPNNGGYDDDQGSYVQVPHDHVAYRYEVLKVIGKGSFGQVVKAYDHKVHQHVALKMVRNEKRFHRQAAEEIRILEHLRKQDKDNTMNVIHMLENFTFRNHICMTFELLSMNLYELIKKNKFQGFSLPLVRKFAHSILQCLDALHKNRIIHCDLKPENILLKQQGRSGIKVIDFGSSCYEHQRVYTXIQSRFYRAPEVILGARYGMPIDMWSLGCILAELLTGYPLLPGEDEGDQLACMIELLGMPSQKLLDASKRAKNFVSXKGYPRYCTVTTLSDVVLNGGRSRRGKLRGPPESREWGNALKGCDDPLFLDFLKQCLEWDPAVRMTPGQALRHPWLR Hydrogen bonds contact Hydrophobic contact | ||||
| 95 | Mas-related gene 2 (MRGX2) | 7VV5 | 5.06 | |
Target general information Gen name MRGPRX2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Masrelated Gprotein coupled receptormember X2; MRGPRX2 Protein family G-protein coupled receptor 1 family, Mas subfamily Biochemical class GPCR rhodopsin Function Mast cell-specific receptor for basic secretagogues (PubMed:25517090). Basic secretagogues are a set of cationic amphiphilic drugs, as well as endo- and exogenous peptides, which share basic head group combined with a hydrophobic core of the molecule. Recognizes and binds small molecules containing a cyclized a tetrahydroisoquinoline (THIQ), such as non-steroidal neuromuscular blocking drugs (NMBDs), including tubocurarine and atracurium. Mediates mast cell responsiveness and side effects of small-molecule therapeutic drugs by acting as a specific receptor for basic secretagogues drugs in mast cells: binding to drugs induces pseudo-allergic reactions characterized by histamine release, inflammation and airway contraction. Acts as a receptor for a number of ligands, including peptides: acts as a receptor of cortistatin-14, a regulator of sleep regulation locomotor activity, and cortical function (PubMed:12915402). Acts as a receptor for proadrenomedullin N- terminal peptides PAMP-12, and atlower extent PAMP-20 (PubMed:15823563). Acts as a receptor for antibacterial protein LL-37, promoting chemotaxis, degranulation and chemokine production in mast cells (PubMed:22069323). Acts as a receptor for PMX-53 peptide, a potent antagonist of C5AR1/CD88 (PubMed:21441599). Acts as a receptor for beta-defensins (PubMed:23698749). Acts as a receptor for complanadine A, an alkaloid (PubMed:24930830). {ECO:0000250|UniProtKB:Q3UG50, ECO:0000269|PubMed:15823563, ECO:0000269|PubMed:21441599, ECO:0000269|PubMed:22069323, ECO:0000269|PubMed:23698749, ECO:0000269|PubMed:24930830, ECO:0000269|PubMed:25517090, ECO:0000305|PubMed:12915402}. Related diseases Intellectual developmental disorder with macrocephaly, seizures, and speech delay (IDDMSSD) [MIM:618158]: An autosomal dominant neurodevelopmental disorder characterized by impaired intellectual development, poor speech, postnatal macrocephaly, and seizures. {ECO:0000269|PubMed:30290153}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number NA Uniprot keywords 3D-structure; Cell membrane; G-protein coupled receptor; Membrane; Receptor; Reference proteome; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID R Molecular weight (Da) 30319.3 Length 268 Aromaticity 0.16 Instability index 32.43 Isoelectric point 8.8 Charge (pH=7) 6.89 2D Binding mode Binding energy (Kcal/mol) -6.9 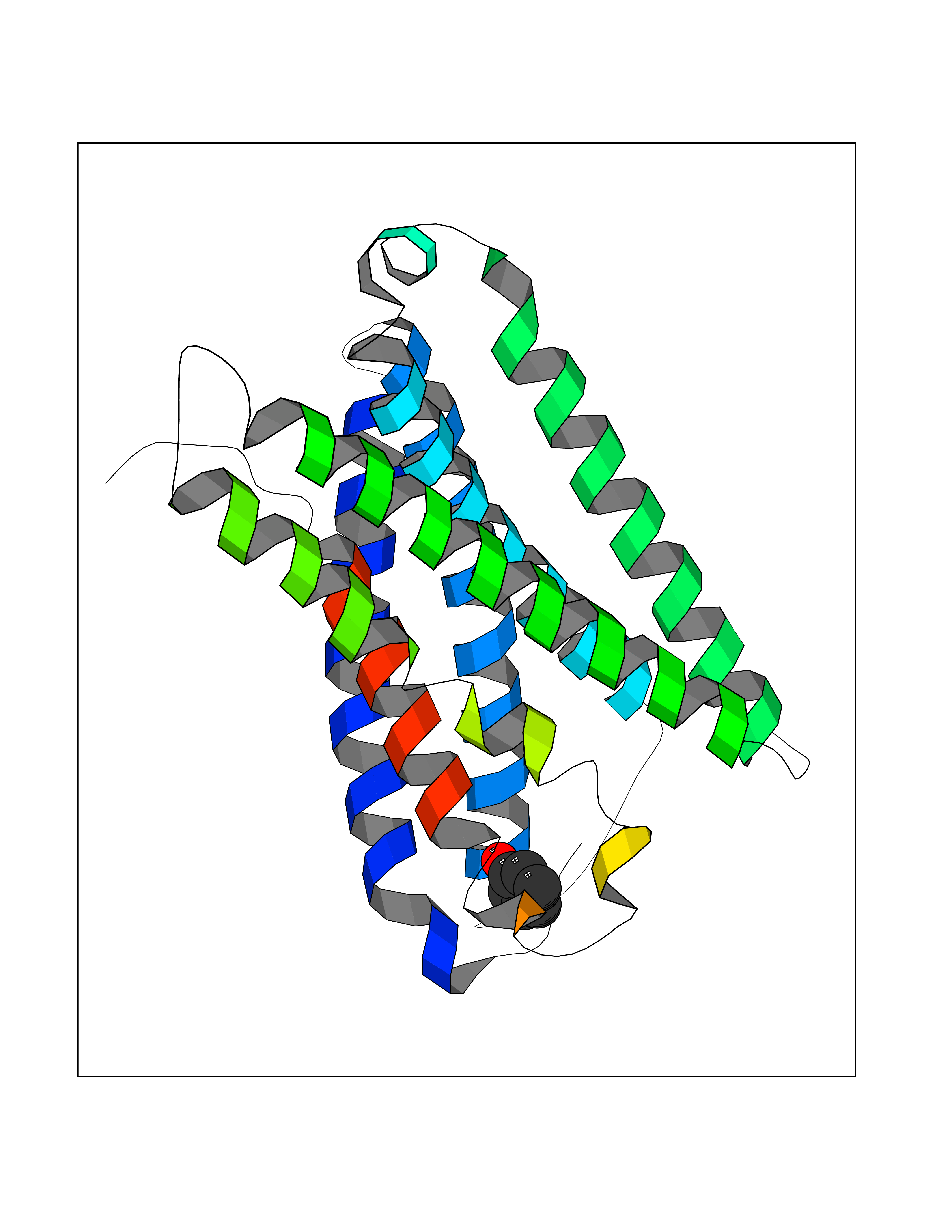 Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence LLLLCGKETLIPVFLILFIALVGLVGNGFVLWLLGFRMRRNAFSVYVLSLAGADFLFLCFQIINCLVYLSNFFCSISINFPSFFTTVMTCAYLAGLSMLSTVSTERCLSVLWPIWYRCRRPRHLSAVVCVLLWALSLLLSILEGKFCGFLFSDGDSGWCQTFDFITAAWLIFLFMVLCGSSLALLVRILCGSRGLPLTRLYLTILLTVLVFLLCGLPFGIQWFLILWIWKDSDVLFCHIHPVSVVLSSLNSSANPIIYFFVGSFRKQW Hydrogen bonds contact Hydrophobic contact | ||||
| 96 | Neuronal acetylcholine receptor alpha-2 (CHRNA2) | 5FJV | 5.06 | |
Target general information Gen name CHRNA2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms CHRNA2 Protein family Ligand-gated ion channel (TC 1.A.9) family, Acetylcholine receptor (TC 1.A.9.1) subfamily, Alpha-2/CHRNA2 sub-subfamily Biochemical class Neurotransmitter receptor Function After binding acetylcholine, the AChR responds by an extensive change in conformation that affects all subunits and leads to opening of an ion-conducting channel across the plasma membrane. Related diseases Epilepsy, nocturnal frontal lobe, 4 (ENFL4) [MIM:610353]: An autosomal dominant focal epilepsy characterized by nocturnal seizures associated with fear sensation, tongue movements, and nocturnal wandering, closely resembling nightmares and sleep walking. {ECO:0000269|PubMed:16826524}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Seizures, benign familial infantile, 6 (BFIS6) [MIM:610353]: A form of benign familial infantile epilepsy, a neurologic disorder characterized by afebrile seizures occurring in clusters during the first year of life, without neurologic sequelae. BFIS6 inheritance is autosomal dominant. {ECO:0000269|PubMed:25847220}. The disease may be caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00732; DB00237; DB00411; DB00565; DB01245; DB00514; DB01135; DB07720; DB00898; DB00472; DB00483; DB08960; DB00657; DB01336; DB00416; DB01226; DB00184; DB01337; DB01338; DB00721; DB00728; DB05740; DB00202; DB01199; DB01339 Interacts with NA EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disease variant; Disulfide bond; Epilepsy; Glycoprotein; Ion channel; Ion transport; Ligand-gated ion channel; Membrane; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Signal; Synapse; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A,B Molecular weight (Da) 48323.4 Length 413 Aromaticity 0.15 Instability index 32 Isoelectric point 5.69 Charge (pH=7) -6.58 2D Binding mode Binding energy (Kcal/mol) -6.9  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence DRLFKHLFRGYNRWARPVPNTSDVVIVRFGLSIAQLIDVDEKNQMMTTNVWLKQEWSDYKLRWNPTDFGNITSLRVPSEMIWIPDIVLYNNADGEFAVTHMTKAHLFSTGTVHWVPPAIYKSSCSIDVTFFPFDQQNCKMKFGSWTYDKAKIDLEQMEQTVDLKDYWESGEWAIVNATGTYNSKKYDCCAEIYPDVTYAFVIRRLPEDRLFKHLFRGYNRWARPVPNTSDVVIVRFGLSIAQLIDVDEKNQMMTTNVWLKQEWSDYKLRWNPTDFGNITSLRVPSEMIWIPDIVLYNNADGEFAVTHMTKAHLFSTGTVHWVPPAIYKSSCSIDVTFFPFDQQNCKMKFGSWTYDKAKIDLEQMEQTVDLKDYWESGEWAIVNATGTYNSKKYDCCAEIYPDVTYAFVIRRLP Hydrogen bonds contact Hydrophobic contact | ||||
| 97 | Histone-lysine N-methyltransferase KMT5C (KMT5C) | 3RQ4 | 5.06 | |
Target general information Gen name KMT5C Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Lysine N-methyltransferase 5C; Lysine-specific methyltransferase 5C; Suppressor of variegation 4-20 homolog 2; Su(var)4-20 homolog 2; Suv4-20h2; [histone H4]-N-methyl-L-lysine20 N-methyltransferase KM Protein family Class V-like SAM-binding methyltransferase superfamily, Histone-lysine methyltransferase family, Suvar4-20 subfamily Biochemical class NA Function Histone methyltransferase that specifically methylates monomethylated 'Lys-20' (H4K20me1) and dimethylated 'Lys-20' (H4K20me2) of histone H4 to produce respectively dimethylated 'Lys-20' (H4K20me2) and trimethylated 'Lys-20' (H4K20me3) and thus regulates transcription and maintenance of genome integrity. In vitro also methylates unmodified 'Lys-20' (H4K20me0) of histone H4 and nucleosomes. H4 'Lys-20' trimethylation represents a specific tag for epigenetic transcriptional repression. Mainly functions in pericentric heterochromatin regions, thereby playing a central role in the establishment of constitutive heterochromatin in these regions. KMT5C is targeted to histone H3 via its interaction with RB1 family proteins (RB1, RBL1 and RBL2) (By similarity). Facilitates TP53BP1 foci formation upon DNA damage and proficient non-homologous end-joining (NHEJ)-directed DNA repair by catalyzing the di- and trimethylation of 'Lys-20' of histone H4. May play a role in class switch reconbination by catalyzing the di- and trimethylation of 'Lys-20' of histone H4 (By similarity). Related diseases Brachydactyly A2 (BDA2) [MIM:112600]: A form of brachydactyly. Brachydactyly defines a group of inherited malformations characterized by shortening of the digits due to abnormal development of the phalanges and/or the metacarpals. In brachydactyly type A2 shortening of the middle phalanges is confined to the index finger and the second toe, all other digits being more or less normal. Because of a rhomboid or triangular shape of the affected middle phalanx, the end of the second finger usually deviates radially. {ECO:0000269|PubMed:19327734, ECO:0000269|PubMed:21357617}. The gene represented in this entry is involved in disease pathogenesis. Duplications of a cis-regulatory element located approximately 110 kb downstream of BMP2 have been found in BDA2 families. They likely cause altered BMP2 expression with pathological consequences. {ECO:0000269|PubMed:19327734, ECO:0000269|PubMed:21357617}.; DISEASE: Short stature, facial dysmorphism, and skeletal anomalies with or without cardiac anomalies 1 (SSFSC1) [MIM:617877]: An autosomal dominant disorder characterized by short stature, facial dysmorphism, skeletal anomalies, and variable cardiac defects. Distinctive facial features include midface retrusion, short upturned nose, long philtrum, high-arched or cleft palate, and variable degrees of micrognathia and dental crowding. Skeletal anomalies include patterning defects of the axial skeleton, characterized by 11 pairs of ribs and brachydactyly of the fifth ray. Congenital heart defects are variably observed and appear to involve primarily the cardiac outflow tract. {ECO:0000269|PubMed:29198724}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q13185 EC number EC 2.1.1.361 Uniprot keywords 3D-structure; Alternative splicing; Chromatin regulator; Chromosome; Metal-binding; Methyltransferase; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Repressor; S-adenosyl-L-methionine; Transcription; Transcription regulation; Transferase; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 27285.8 Length 240 Aromaticity 0.1 Instability index 42.74 Isoelectric point 8.32 Charge (pH=7) 3.24 2D Binding mode Binding energy (Kcal/mol) -6.91  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence DRVTARELCENDDLATSLVLDPYLGFRTHKMNVSPVPPLRRQQHLRSALETFLRQRDLEAAYRALTLGGWTARYFQSRGPRQEAALKTHVYRYLRAFLPESGFTILPCTRYSMETNGAKIVSTRAWKKNEKLELLVGCIAELREADEGLLRAGENDFSIMYSTRKRSAQLWLGPAAFINHDCKPNCKFVPADGNAACVKVLRDIEPGDEVTCFYGEGFFGEKNEHCECHTCERKGEGAFR Hydrogen bonds contact Hydrophobic contact | ||||
| 98 | Dual-specificity tyrosine-phosphorylation regulated kinase 3 (DYRK3) | 5Y86 | 5.06 | |
Target general information Gen name DYRK3 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Regulatory erythroid kinase; REDK; Dual specificity tyrosine-phosphorylation-regulated kinase 3 Protein family Protein kinase superfamily, CMGC Ser/Thr protein kinase family, MNB/DYRK subfamily Biochemical class Kinase Function Dual-specificity tyrosine-regulated kinases (DYRKs) autophosphorylate a critical tyrosine residue in their activation loop and phosphorylate their substrate on serine and threonine residues. Acts as a central dissolvase of membraneless organelles during the G2-to-M transition, after the nuclear-envelope breakdown: acts by mediating phosphorylation of multiple serine and threonine residues in unstructured domains of proteins, such as SRRM1 and PCM1. Does not mediate disassembly of all membraneless organelles: disassembly of P-body and nucleolus is not regulated by DYRK3. Dissolution of membraneless organelles at the onset of mitosis is also required to release mitotic regulators, such as ZNF207, from liquid-unmixed organelles where they are sequestered and keep them dissolved during mitosis. Regulates mTORC1 by mediating the dissolution of stress granules: during stressful conditions, DYRK3 partitions from the cytosol to the stress granule, together with mTORC1 components, which prevents mTORC1 signaling. When stress signals are gone, the kinase activity of DYRK3 is required for the dissolution of stress granule and mTORC1 relocation to the cytosol: acts by mediating the phosphorylation of the mTORC1 inhibitor AKT1S1, allowing full reactivation of mTORC1 signaling. Also acts as a negative regulator of EPO-dependent erythropoiesis: may place an upper limit on red cell production during stress erythropoiesis. Inhibits cell death due to cytokine withdrawal in hematopoietic progenitor cells. Promotes cell survival upon genotoxic stress through phosphorylation of SIRT1: this in turn inhibits p53/TP53 activity and apoptosis. Dual-specificity protein kinase that promotes disassembly of several types of membraneless organelles during mitosis, such as stress granules, nuclear speckles and pericentriolar material. Related diseases Defects in MELK are associated with some cancers, such as brain or breast cancers. Expression is dramatically increased in aggressive undifferentiated tumors, correlating with poor patient outcome in breast and brain cancers, suggesting a role in tumor-initiating cells and proliferation via its function in cell proliferation regulation. Drugs (DrugBank ID) NA Interacts with Q9H8Y8 EC number EC 2.7.12.1 Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Cell cycle; Cell division; Cytoplasm; Cytoskeleton; Kinase; Magnesium; Metal-binding; Mitosis; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Serine/threonine-protein kinase; Transferase; Tyrosine-protein kinase; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 44821.5 Length 395 Aromaticity 0.1 Instability index 49.38 Isoelectric point 9.52 Charge (pH=7) 21.08 2D Binding mode Binding energy (Kcal/mol) -6.91  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence VVPLTPEQALKQYKHHLTAYEKLEIINYPEIYFVGPNAKKRHGVIGGPNNGGYDDADGAYIHVPRDHLAYRYEVLKIIGKGSFGQVARVYDHKLRQYVALKMVRNEKRFHRQAAEEIRILEHLKKQDKTGSMNVIHMLESFTFRNHVCMAFELLSIDLYELIKKNKFQGFSVQLVRKFAQSILQSLDALHKNKIIHCDLKPENILLKHHGRSXTKVIDFGSSCFEYQKLYTXIQSRFYRAPEIILGSRYSTPIDIWSFGCILAELLTGQPLFPGEDEGDQLACMMELLGMPPPKLLEQSKRAKYFINXKGIPRYCSVTTQADGRVVLVGGRSRRGKKRGPPGSKDWGTALKGCDDYLFIEFLKRCLHWDPSARLXPAQALRHPWISKSVPRPLTT Hydrogen bonds contact Hydrophobic contact | ||||
| 99 | Acetylcholine receptor subunit alpha | 4ZJS | 5.05 | |
Target general information Gen name CHRNA1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms ACHRA;CHNRA Protein family Ligand-gated ion channel (TC 1.A.9) family, Acetylcholine receptor (TC 1.A.9.1) subfamily, Alpha-1/CHRNA1 sub-subfamily Biochemical class Immune system Function Acetylcholine binding.Acetylcholine-gated cation-selective channel activity.Acetylcholine receptor activity.Ion channel activity.Ligand-gated ion channel activity. Related diseases Multiple pterygium syndrome, lethal type (LMPS) [MIM:253290]: Multiple pterygia are found infrequently in children with arthrogryposis and in fetuses with fetal akinesia syndrome. In lethal multiple pterygium syndrome there is intrauterine growth retardation, multiple pterygia, and flexion contractures causing severe arthrogryposis and fetal akinesia. Subcutaneous edema can be severe, causing fetal hydrops with cystic hygroma and lung hypoplasia. Oligohydramnios and facial anomalies are frequent. {ECO:0000269|PubMed:18252226}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: The alpha subunit is the main focus for antibody binding in myasthenia gravis. Myasthenia gravis is characterized by sporadic muscular fatigability and weakness, occurring chiefly in muscles innervated by cranial nerves, and characteristically improved by cholinesterase-inhibiting drugs.; DISEASE: Myasthenic syndrome, congenital, 1A, slow-channel (CMS1A) [MIM:601462]: A common congenital myasthenic syndrome. Congenital myasthenic syndromes are characterized by muscle weakness affecting the axial and limb muscles (with hypotonia in early-onset forms), the ocular muscles (leading to ptosis and ophthalmoplegia), and the facial and bulbar musculature (affecting sucking and swallowing, and leading to dysphonia). The symptoms fluctuate and worsen with physical effort. CMS1A is a slow-channel myasthenic syndrome. It is caused by kinetic abnormalities of the AChR, resulting in prolonged AChR channel opening episodes, prolonged endplate currents, and depolarization block. This is associated with calcium overload, which may contribute to subsequent degeneration of the endplate and postsynaptic membrane. {ECO:0000269|PubMed:16685696, ECO:0000269|PubMed:7619526, ECO:0000269|PubMed:8872460, ECO:0000269|PubMed:9158151, ECO:0000269|PubMed:9221765}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Myasthenic syndrome, congenital, 1B, fast-channel (CMS1B) [MIM:608930]: A form of congenital myasthenic syndrome, a group of disorders characterized by failure of neuromuscular transmission, including pre-synaptic, synaptic, and post-synaptic disorders that are not of autoimmune origin. Clinical features are easy fatigability and muscle weakness affecting the axial and limb muscles (with hypotonia in early-onset forms), the ocular muscles (leading to ptosis and ophthalmoplegia), and the facial and bulbar musculature (affecting sucking and swallowing, and leading to dysphonia). The symptoms fluctuate and worsen with physical effort. CMS1B is a fast-channel myasthenic syndrome. It is caused by kinetic abnormalities of the AChR, resulting in brief opening and activity of the channel, with a rapid decay in endplate current, failure to achieve threshold depolarization of the endplate and consequent failure to fire an action potential. {ECO:0000269|PubMed:10195214, ECO:0000269|PubMed:12588888, ECO:0000269|PubMed:15079006}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08838; DB00565; DB00555 Interacts with NA EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Congenital myasthenic syndrome; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; Ion channel; Ion transport; Ligand-gated ion channel; Membrane; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Signal; Synapse; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A,B,C,D,E Molecular weight (Da) 46717.8 Length 411 Aromaticity 0.11 Instability index 38.02 Isoelectric point 4.77 Charge (pH=7) -22.31 2D Binding mode Binding energy (Kcal/mol) -6.89  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence EHETRLVAKLFKDYSSVVRPVEDHRQVVEVTLGFTLQDIVKADSSTNEVDLVYYEQQRWVDYNLKWNPDDYGGVKKIHIPAADIWTPDITAYSSTRPVQVLSPQIAVVTHDGSVMFIPAQRLSFMCDPTGVDSEEGATCAVKFGSWVYSGFEIDLKTDTDQVDLSSYYASSKYEILSATQTRQVQHYSCCPEPYIDVNLVVKFREEHETRLVAKLFKDYSSVVRPVEDHRQVVEVTLGFTLQDIVKADSSTNEVDLVYYEQQRWVDYNLKWNPDDYGGVKKIHIPAADIWTPDITAYSSTRPVQVLSPQIAVVTHDGSVMFIPAQRLSFMCDPTGVDSEEGATCAVKFGSWVYSGFEIDLKTDTDQVDLSSYYASSKYEILSATQTRQVQHYSCCPEPYIDVNLVVKFRER Hydrogen bonds contact Hydrophobic contact | ||||
| 100 | Natriuretic peptides B | 1YK1 | 5.05 | |
Target general information Gen name NPPB Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Natriuretic peptide family Biochemical class Hormone / growth factor receptor Function Diuretic hormone activity.Hormone activity.Peptide hormone receptor binding.Receptor binding. Related diseases Multiple fibroadenomas of the breast (MFAB) [MIM:615554]: A benign breast disease marked by lobuloalveolar growth with abnormally high proliferation of the epithelium, and characterized by the presence of more than 3 fibroadenomas in one breast. Fibroadenomas are adenomas containing fibrous tissue. {ECO:0000269|PubMed:18779591}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Hyperprolactinemia (HPRL) [MIM:615555]: A disorder characterized by increased levels of prolactin in the blood not associated with gestation or the puerperium. HPRL may result in infertility, hypogonadism, and galactorrhea. {ECO:0000269|PubMed:24195502}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01136; DB06412 Interacts with A8MQ03; P57678; Q6A162; P60411; Q7Z3S9; P25788; Q9UJW9 EC number NA Uniprot keywords 3D-structure; Direct protein sequencing; Disulfide bond; Glycoprotein; Hormone; Pharmaceutical; Proteoglycan; Proteomics identification; Reference proteome; Secreted; Signal; Vasoactive; Vasodilator Protein physicochemical properties Chain ID E Molecular weight (Da) 46353.1 Length 415 Aromaticity 0.1 Instability index 37.91 Isoelectric point 5.51 Charge (pH=7) -12.09 2D Binding mode Binding energy (Kcal/mol) -6.89  Molscript Map  Pymol Map 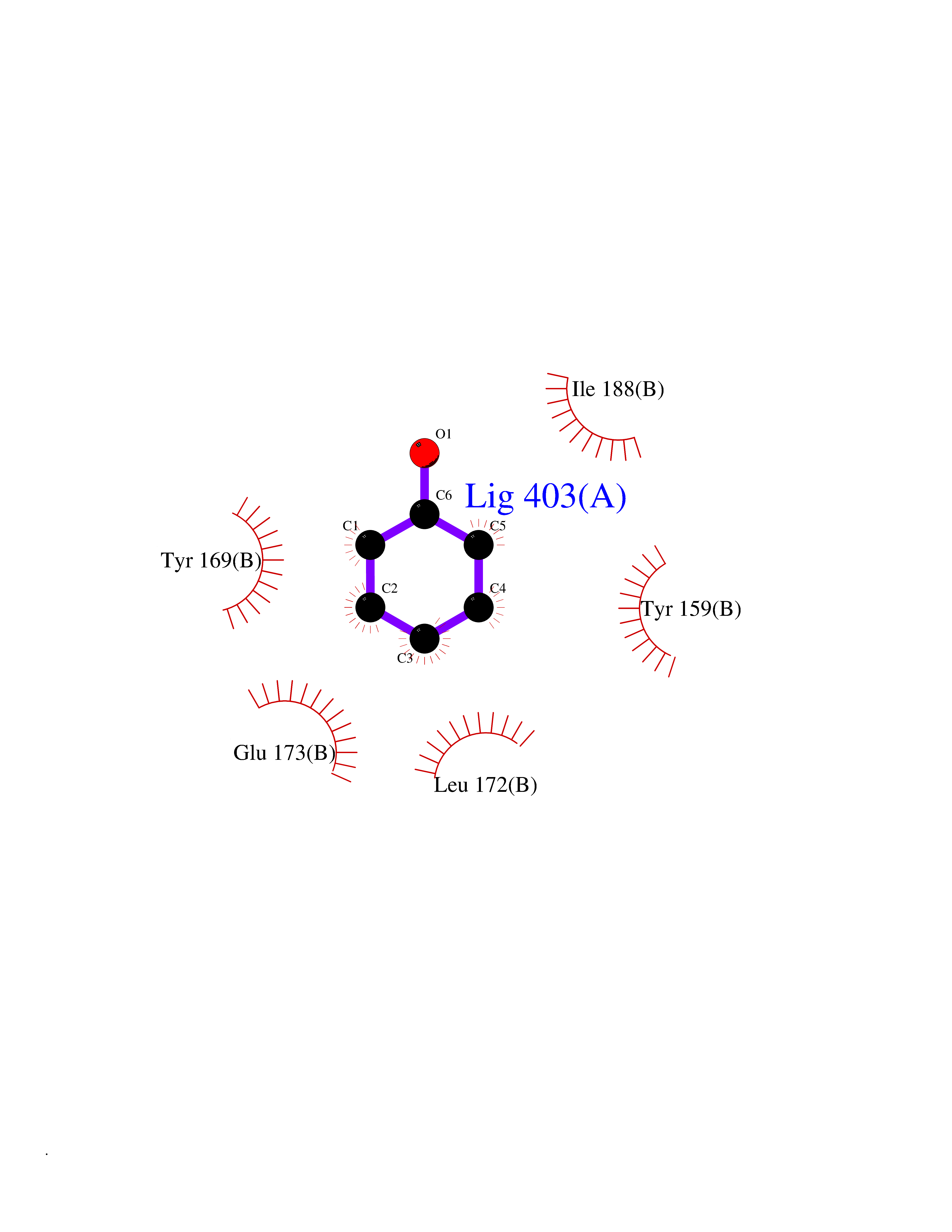 Ligplot Map 3D Binding mode Sequence GCFGRKMDRISSSSGLGCKVLALPPQKIEVLVLLPQDDSYLFSLTRVRPAIEYALRSVEGLLPPGTRFQVAYEDSDCGNRALFSLVDRVAAARGAKPDLILGPVCEYAAAPVARLASHWDLPMLSAGALAAGFQHKDSEYSHLTRVAPAYAKMGEMMLALFRHHHWSRAALVYSDDKLERNCYFTLEGVHEVFQEEGLHTSIYSFDETKDLDLEDIVRNIQASERVVIMCASSDTIRSIMLVAHRHGMTSGDYAFFNIELFNSSSYGDGSWKRGDKHDFEAKQAYSSLQTVTLLRTVKPEFEKFSMEVKSSVEKQGLNMEDYVNMFVEGFHDAILLYVLALHEVLRAGYSKKDGGKIIQQTWNRTFEGIAGQVSIDANGDRYGDFSVIAMTDVEAGTQEVIGDYFGKEGRFEMRP Hydrogen bonds contact Hydrophobic contact | ||||