Job Results:
Ligand
Structure
Job ID
58e2f3b701186914fd3b121ac4df0edd
Job name
NA
Time
2026-02-27 13:43:39
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 81 | Cytochrome P450 2B6 (CYP2B6) | 3IBD | 5.60 | |
Target general information Gen name CYP2B6 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Cytochrome P450 IIB1; CYPIIB6; 1,4-cineole 2-exo-monooxygenase Protein family Cytochrome P450 family Biochemical class Paired donor oxygen oxidoreductase Function In liver microsomes, this enzyme is involved in an NADPH-dependent electron transport pathway. It oxidizes a variety of structurally unrelated compounds, including steroids, fatty acids, and xenobiotics. Acts as a 1,4-cineole 2-exo-monooxygenase. Cytochromes P450 are a group of heme-thiolate monooxygenases. Related diseases Aromatic L-amino-acid decarboxylase deficiency (AADCD) [MIM:608643]: An inborn error in neurotransmitter metabolism that leads to combined serotonin and catecholamine deficiency. It causes developmental and psychomotor delay, poor feeding, lethargy, ptosis, intermittent hypothermia, gastrointestinal disturbances. The onset is early in infancy and inheritance is autosomal recessive. {ECO:0000269|PubMed:14991824, ECO:0000269|PubMed:15079002, ECO:0000269|Ref.12}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08369; DB02974; DB11932; DB12001; DB14973; DB15568; DB00321; DB00381; DB00701; DB01435; DB00714; DB06413; DB06697; DB13132; DB11586; DB01076; DB15011; DB00972; DB08822; DB13997; DB04975; DB01086; DB00865; DB00443; DB04794; DB12151; DB01194; DB05541; DB01222; DB01156; DB09061; DB14737; DB00564; DB06119; DB00439; DB00568; DB00604; DB00515; DB12499; DB00349; DB06470; DB00758; DB01559; DB00257; DB01394; DB05219; DB08865; DB11672; DB14635; DB04664; DB00531; DB08912; DB01151; DB16650; DB01234; DB14649; DB04856; DB00514; DB00829; DB00586; DB01184; DB00997; DB00470; DB00476; DB00625; DB15444; DB13874; DB11718; DB08899; DB00751; DB11823; DB00655; DB00898; DB01466; DB00574; DB12265; DB01544; DB00472; DB01095; DB00176; DB01320; DB05087; DB00986; DB01159; DB00956; DB00741; DB09054; DB01181; DB00458; DB00762; DB11633; DB06636; DB00753; DB11757; DB01167; DB14568; DB09570; DB01221; DB06738; DB01026; DB11951; DB09078; DB12070; DB05667; DB00281; DB00836; DB01601; DB00455; DB04871; DB12130; DB09280; DB00772; DB09238; DB14921; DB14009; DB01043; DB00170; DB00454; DB00532; DB04817; DB00333; DB00763; DB01028; DB09241; DB00849; DB00959; DB06710; DB00379; DB06148; DB01110; DB06595; DB16236; DB00745; DB11763; DB00220; DB00238; DB00622; DB00184; DB01115; DB04868; DB12005; DB00435; DB00957; DB09074; DB16267; DB11632; DB01173; DB11837; DB04938; DB05467; DB00715; DB08883; DB01074; DB04930; DB12978; DB03575; DB01174; DB00252; DB13941; DB17472; DB11642; DB06209; DB14631; DB00635; DB01069; DB00818; DB00908; DB00481; DB08896; DB11853; DB16826; DB02709; DB00615; DB01045; DB11753; DB01201; DB08864; DB00503; DB06176; DB00296; DB00412; DB00778; DB01037; DB06739; DB01104; DB01236; DB00641; DB00398; DB15569; DB12548; DB09118; DB06729; DB01138; DB00675; DB12020; DB12095; DB00231; DB00624; DB13943; DB13944; DB13946; DB11712; DB01041; DB09499; DB04572; DB08816; DB00208; DB06137; DB00193; DB00755; DB12245; DB12808; DB00197; DB12255; DB00313; DB11613; DB08881; DB00661; DB09185; DB11739; DB00582; DB09068; DB14975; DB15035 Interacts with NA EC number EC 1.14.13.- Uniprot keywords 3D-structure; Alternative splicing; Endoplasmic reticulum; Heme; Iron; Lipid metabolism; Membrane; Metal-binding; Microsome; Monooxygenase; NADP; Oxidoreductase; Phosphoprotein; Proteomics identification; Reference proteome Protein physicochemical properties Chain ID A Molecular weight (Da) 53293 Length 465 Aromaticity 0.12 Instability index 38.79 Isoelectric point 8.63 Charge (pH=7) 5.35 2D Binding mode Binding energy (Kcal/mol) -7.64  Molscript Map 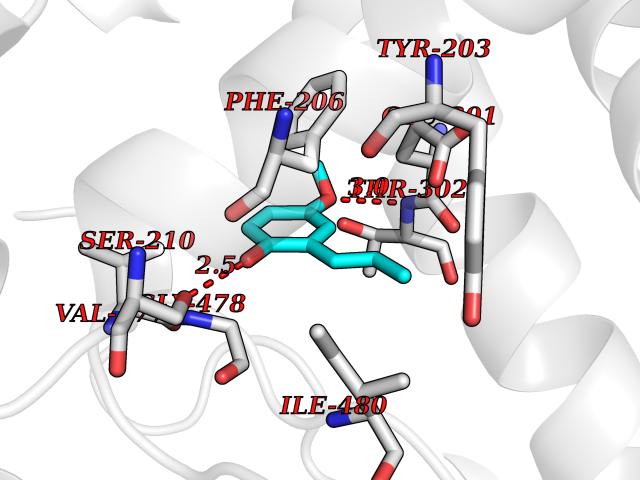 Pymol Map 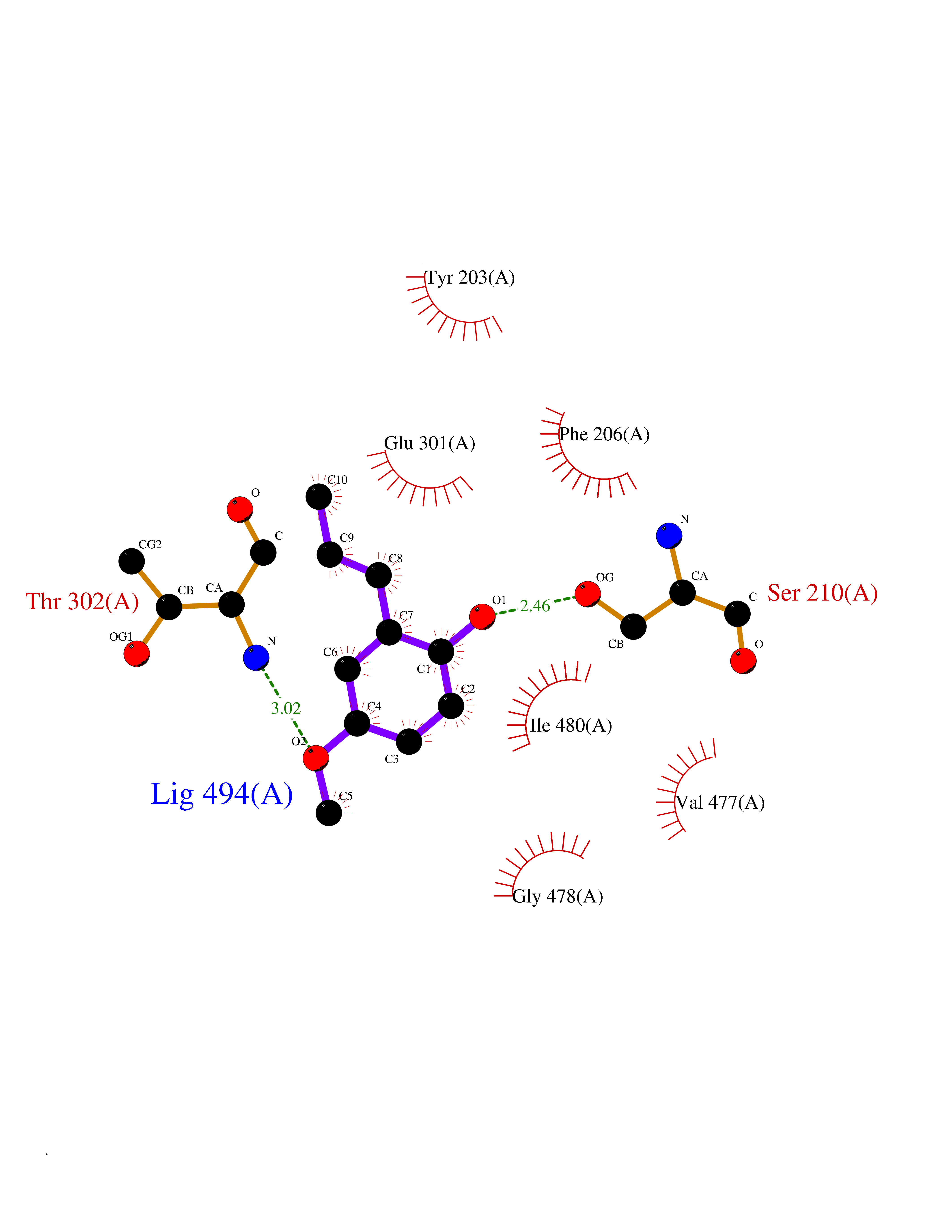 Ligplot Map 3D Binding mode Sequence GKLPPGPRPLPLLGNLLQMDRRGLLKSFLRFREKYGDVFTVHLGPRPVVMLCGVEAIREALVDKAEAFSGRGKIAMVDPFFRGYGVIFANGNRWKVLRRFSVTTMRDFGMGKRSVEERIQEEAQCLIEELRKSKGALMDPTFLFQSITANIICSIVFGKRFHYQDQEFLKMLNLFYQTFSLISSVFGQLFELFSGFLKHFPGAHRQVYKNLQEINAYIGHSVEKHRETLDPSAPRDLIDTYLLHMEKEKSNAHSEFSHQNLNLNTLSLFFAGTETTSTTLRYGFLLMLKYPHVAERVYREIEQVIGPHRPPELHDRAKMPYTEAVIYEIQRFSDLLPMGVPHIVTQHTSFRGYIIPKDTEVFLILSTALHDPHYFEKPDAFNPDHFLDANGALKKTEAFIPFSLGKRICLGEGIARAELFLFFTTILQNFSMASPVAPEDIDLTPQECGVGKIPPTYQIRFLPRH Hydrogen bonds contact Hydrophobic contact | ||||
| 82 | Glycinamide ribonucleotide formyltransferase (GART) | 4ZZ1 | 5.60 | |
Target general information Gen name GART Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Trifunctional purine biosynthetic protein adenosine-3; PRGS; PGFT Protein family GARS family; AIR synthase family; GART family Biochemical class Carbon-nitrogen ligase Function A trifunctional polypeptide. Has Phosphoribosylamineglycine ligase, Phosphoribosylglycinamide formyltransferase, AIR synthetase (FGAM cyclase) activity which is required for de novo purine biosynthesis. Related diseases Schizophrenia (SCZD) [MIM:181500]: A complex, multifactorial psychotic disorder or group of disorders characterized by disturbances in the form and content of thought (e.g. delusions, hallucinations), in mood (e.g. inappropriate affect), in sense of self and relationship to the external world (e.g. loss of ego boundaries, withdrawal), and in behavior (e.g bizarre or apparently purposeless behavior). Although it affects emotions, it is distinguished from mood disorders in which such disturbances are primary. Similarly, there may be mild impairment of cognitive function, and it is distinguished from the dementias in which disturbed cognitive function is considered primary. Some patients manifest schizophrenic as well as bipolar disorder symptoms and are often given the diagnosis of schizoaffective disorder. {ECO:0000269|PubMed:15645182}. Disease susceptibility may be associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02236; DB03546; DB00642 Interacts with NA EC number NA Uniprot keywords 3D-structure; Acetylation; Alternative splicing; ATP-binding; Ligase; Magnesium; Manganese; Metal-binding; Multifunctional enzyme; Nucleotide-binding; Phosphoprotein; Proteomics identification; Purine biosynthesis; Reference proteome; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 21438.4 Length 200 Aromaticity 0.05 Instability index 33.54 Isoelectric point 6.23 Charge (pH=7) -1.77 2D Binding mode Binding energy (Kcal/mol) -7.63 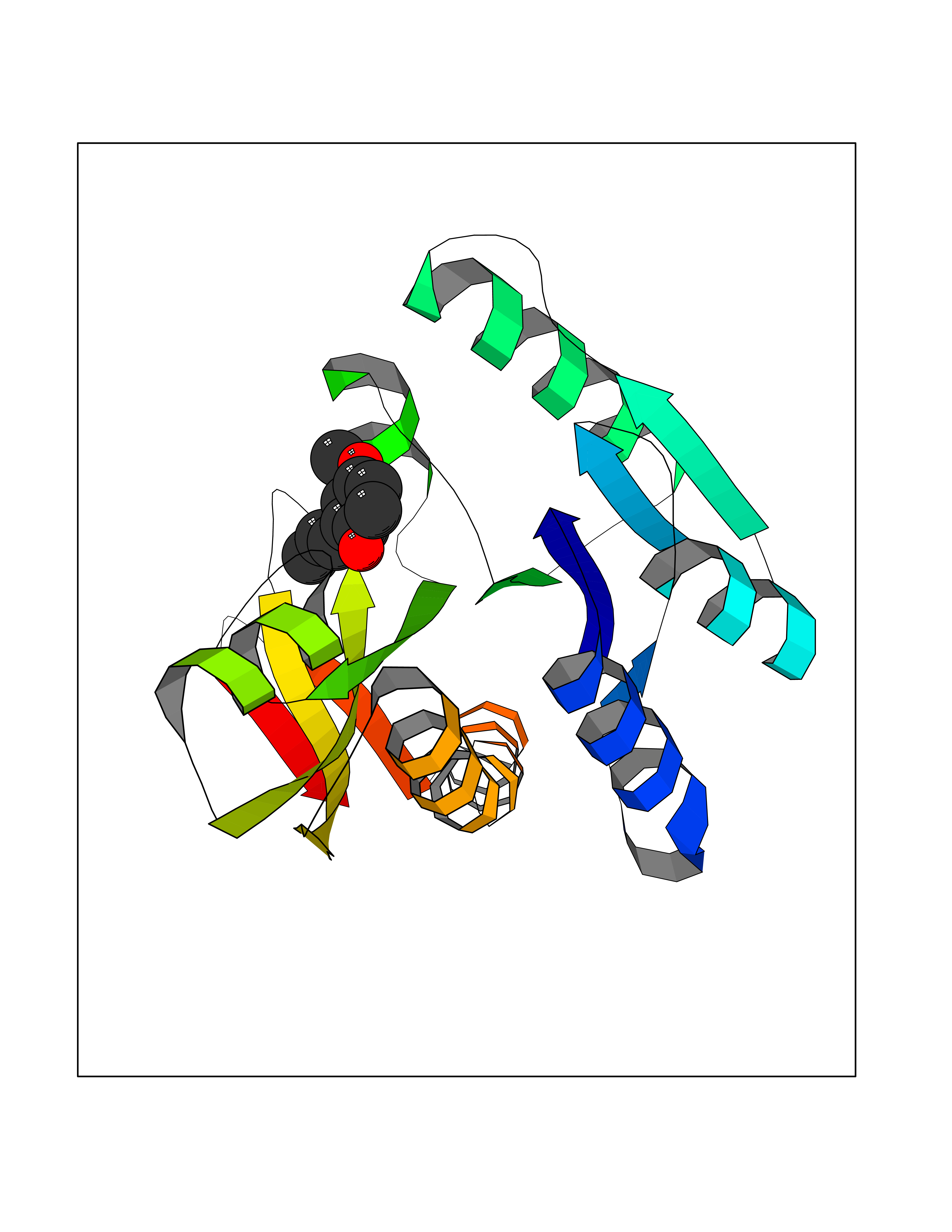 Molscript Map 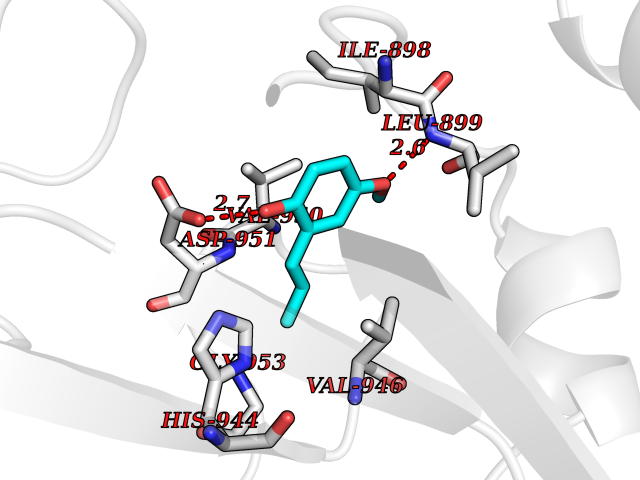 Pymol Map 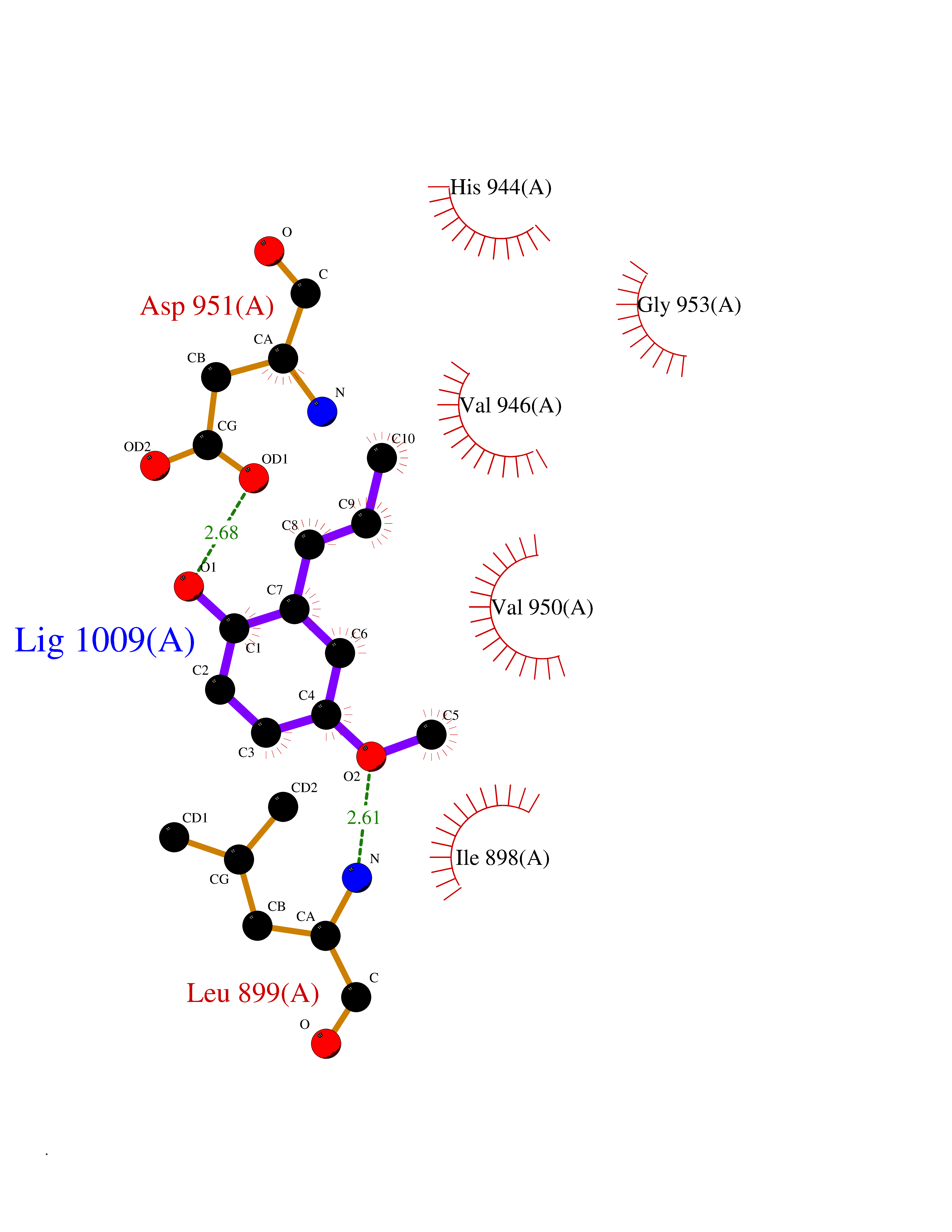 Ligplot Map 3D Binding mode Sequence ARVAVLISGTGSNLQALIDSTREPNSSAQIDIVISNKAAVAGLDKAERAGIPTRVINHKLYKNRVEFDSAIDLVLEEFSIDIVCLAGFMRILSGPFVQKWNGKMLNIHPSLLPSFKGSNAHEQALETGVTVTGCTVHFVAEDVDAGQIILQEAVPVKRGDTVATLSERVKLAEHKIFPAALQLVASGTVQLGENGKICWV Hydrogen bonds contact Hydrophobic contact | ||||
| 83 | Peroxisome proliferator-activated receptor alpha (PPARA) | 3VI8 | 5.60 | |
Target general information Gen name PPARA Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Peroxisome proliferater-activated receptor alpha; PPARalpha; PPAR-alpha; PPAR; Nuclear receptor subfamily 1 group C member 1; NR1C1 Protein family Nuclear hormone receptor family, NR1 subfamily Biochemical class Nuclear hormone receptor Function Key regulator of lipid metabolism. Activated by the endogenous ligand 1-palmitoyl-2-oleoyl-sn-glycerol-3-phosphocholine (16:0/18:1-GPC). Activated by oleylethanolamide, a naturally occurring lipid that regulates satiety. Receptor for peroxisome proliferators such as hypolipidemic drugs and fatty acids. Regulates the peroxisomal beta-oxidation pathway of fatty acids. Functions as transcription activator for the ACOX1 and P450 genes. Transactivation activity requires heterodimerization with RXRA and is antagonized by NR2C2. May be required for the propagation of clock information to metabolic pathways regulated by PER2. Ligand-activated transcription factor. Related diseases Combined oxidative phosphorylation deficiency 33 (COXPD33) [MIM:617713]: An autosomal recessive disorder caused by multiple mitochondrial respiratory chain defects and impaired mitochondrial energy metabolism. Clinical manifestations are highly variable. Affected infants present with cardiomyopathy accompanied by multisystemic features involving liver, kidney, and brain. Death in infancy is observed in some patients. Children and adults present with myopathy and progressive external ophthalmoplegia. {ECO:0000269|PubMed:28942965}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08915; DB00132; DB01118; DB04557; DB01393; DB04519; DB05416; DB09064; DB09006; DB00636; DB09213; DB03756; DB05187; DB06521; DB01039; DB13873; DB00573; DB13961; DB02266; DB01241; DB07215; DB01050; DB00159; DB07724; DB00328; DB12007; DB03017; DB12961; DB06510; DB08231; DB11605; DB01890; DB04224; DB11133; DB03796; DB02746; DB01708; DB06533; DB04971; DB02709; DB00412; DB09422; DB03193; DB06536; DB00197; DB00313 Interacts with P02768-3; P55212; P45973; P06307; Q3L8U1-3; G5E9A7; P22607; P62993; Q14957; P06396; P42858; Q8WXH2; P13473-2; O75376; Q13133; A0A6Q8PF08; P54725; P62826; Q7Z699; P37173; P55072; P55055-1; Q13133 EC number NA Uniprot keywords 3D-structure; Activator; Alternative splicing; Biological rhythms; DNA-binding; Lipid-binding; Metal-binding; Nucleus; Proteomics identification; Receptor; Reference proteome; Transcription; Transcription regulation; Ubl conjugation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A Molecular weight (Da) 29322.1 Length 258 Aromaticity 0.09 Instability index 35.53 Isoelectric point 6.09 Charge (pH=7) -3.57 2D Binding mode Binding energy (Kcal/mol) -7.64 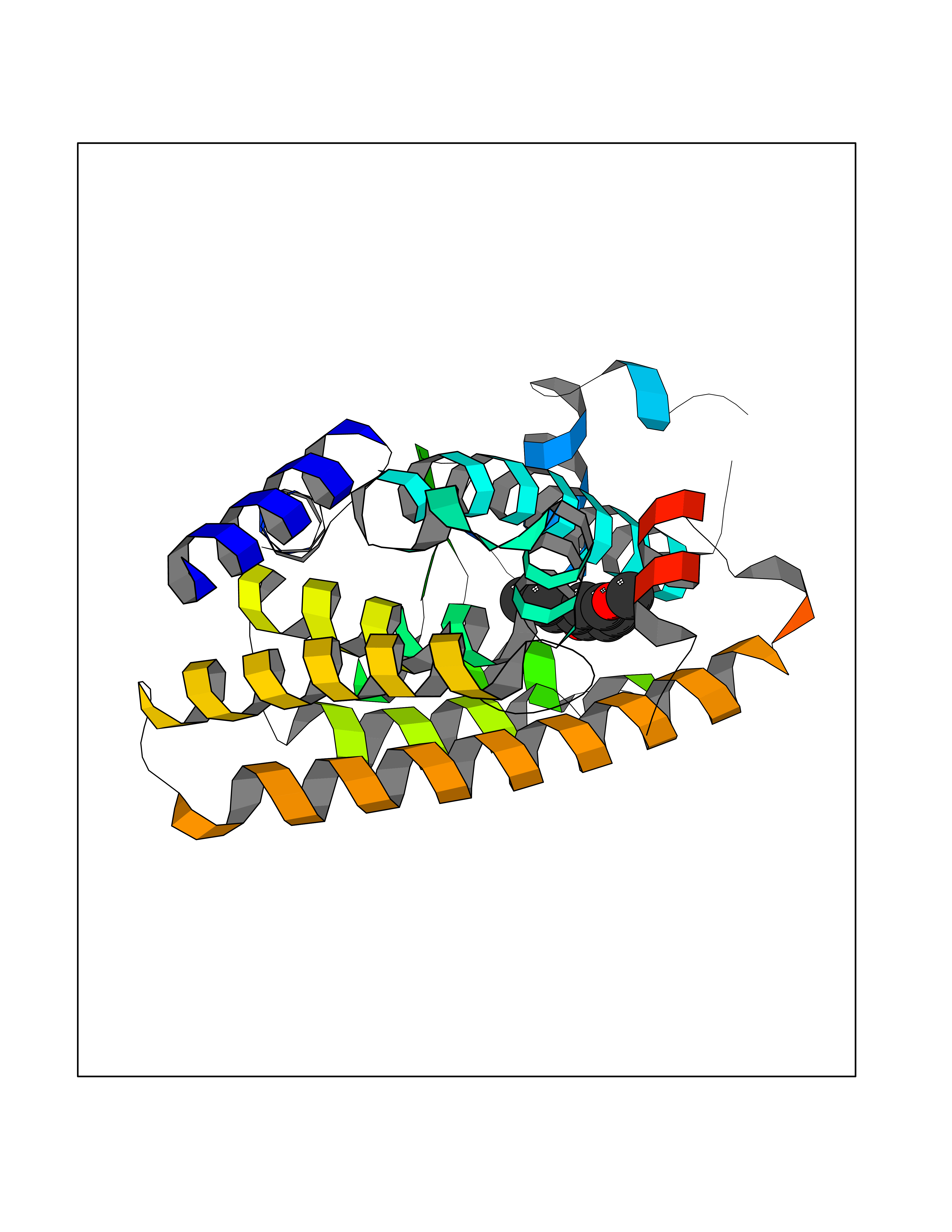 Molscript Map 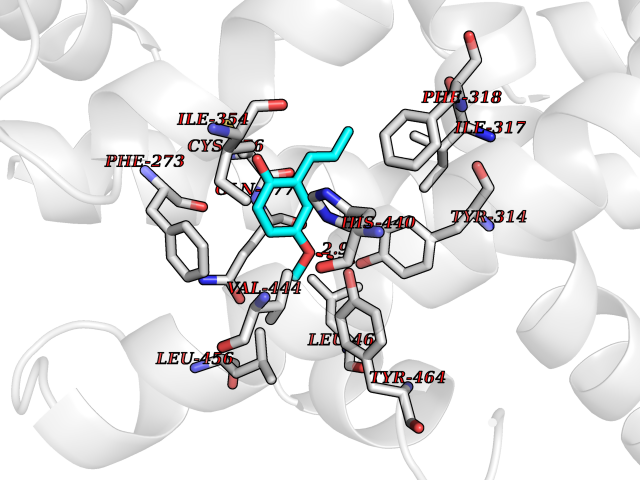 Pymol Map 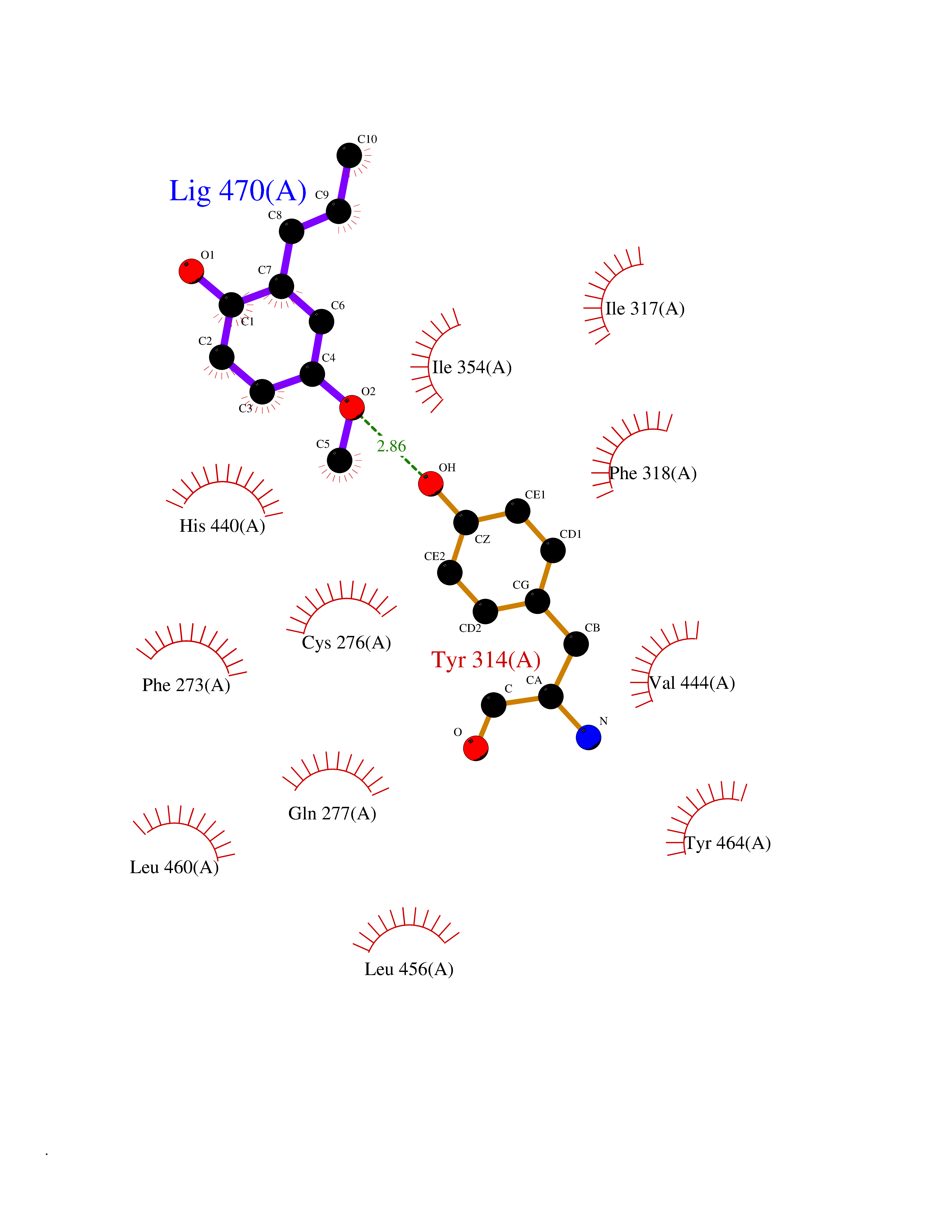 Ligplot Map 3D Binding mode Sequence DLKSLAKRIYEAYLKNFNMNKVKARVILSPFVIHDMETLCMAEKTLVAKLVANGNKEAEVRIFHCCQCTSVETVTELTEFAKAIPGFANLDLNDQVTLLKYGVYEAIFAMLSSVMNKDGMLVAYGNGFITREFLKSLRKPFCDIMEPKFDFAMKFNALELDDSDISLFVAAIICCGDRPGLLNVGHIEKMQEGIVHVLRLHLQSNHPDDIFLFPKLLQKMADLRQLVTEHAQLVQIIKKTESDAALHPLLQEIYRDMY Hydrogen bonds contact Hydrophobic contact | ||||
| 84 | S-methyl-5'-thioadenosine phosphorylase (MTAP) | 1CB0 | 5.60 | |
Target general information Gen name MTAP Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Methylthioadenosine phosphorylase; MTAPase; MTA phosphorylase; MSAP; 5'-methylthioadenosine phosphorylase Protein family PNP/MTAP phosphorylase family, MTAP subfamily Biochemical class Glycosyltransferases Function Involved in the breakdown of MTA, a major by-product of polyamine biosynthesis. Responsible for the first step in the methionine salvage pathway after MTA has been generated from S-adenosylmethionine. Has broad substrate specificity with 6-aminopurine nucleosides as preferred substrates. Catalyzes the reversible phosphorylation of S-methyl-5'-thioadenosine (MTA) to adenine and 5-methylthioribose-1-phosphate. Related diseases Diaphyseal medullary stenosis with malignant fibrous histiocytoma (DMSMFH) [MIM:112250]: An autosomal dominant bone dysplasia characterized by pathologic fractures due to abnormal cortical growth and diaphyseal medullary stenosis. The fractures heal poorly, and there is progressive bowing of the lower extremities. Some patients show a limb-girdle myopathy, with muscle weakness and atrophy. Approximately 35% of affected individuals develop an aggressive form of bone sarcoma consistent with malignant fibrous histiocytoma or osteosarcoma. {ECO:0000269|PubMed:22464254}. The disease is caused by variants affecting the gene represented in this entry. DMSMFH causing mutations found in MTAP exon 9 result in exon skipping and dysregulated alternative splicing of all MTAP isoforms (PubMed:22464254). {ECO:0000269|PubMed:22464254}.; DISEASE: Loss of MTAP activity may play a role in human cancer. MTAP loss has been reported in a number of cancers, including osteosarcoma, malignant melanoma and gastric cancer. Drugs (DrugBank ID) DB02158; DB02933; DB02282; DB00173; DB02281 Interacts with Q9H3R5; Q9P0I2 EC number EC 2.4.2.28 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Cytoplasm; Glycosyltransferase; Nucleus; Proteomics identification; Purine salvage; Reference proteome; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 29538.9 Length 268 Aromaticity 0.06 Instability index 40.97 Isoelectric point 7.18 Charge (pH=7) 0.36 2D Binding mode Binding energy (Kcal/mol) -7.64 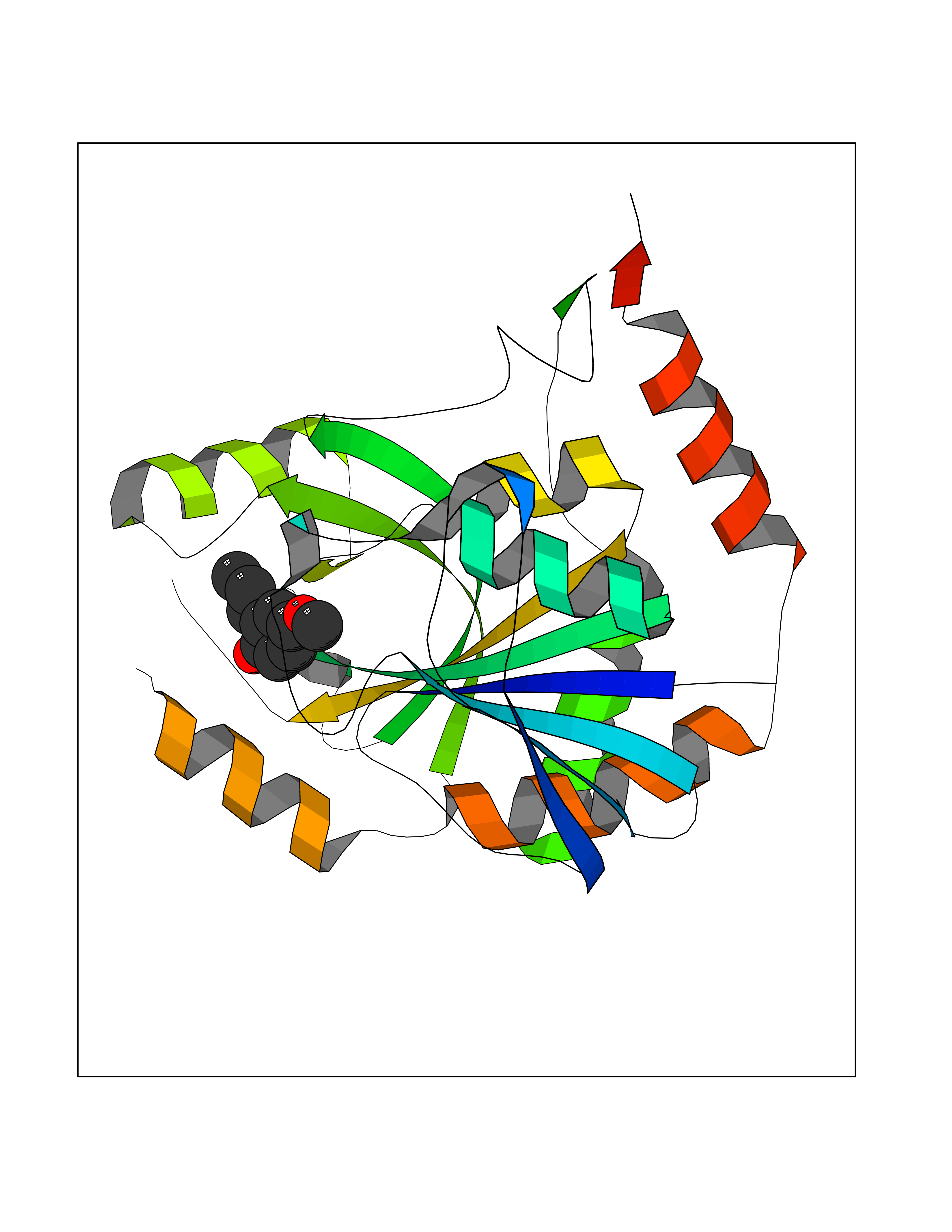 Molscript Map 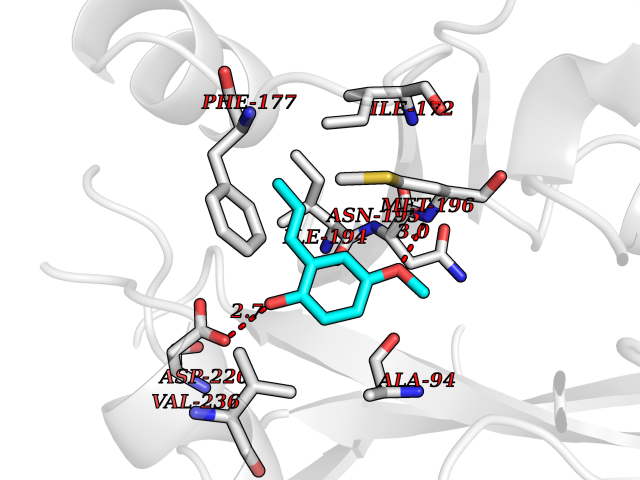 Pymol Map 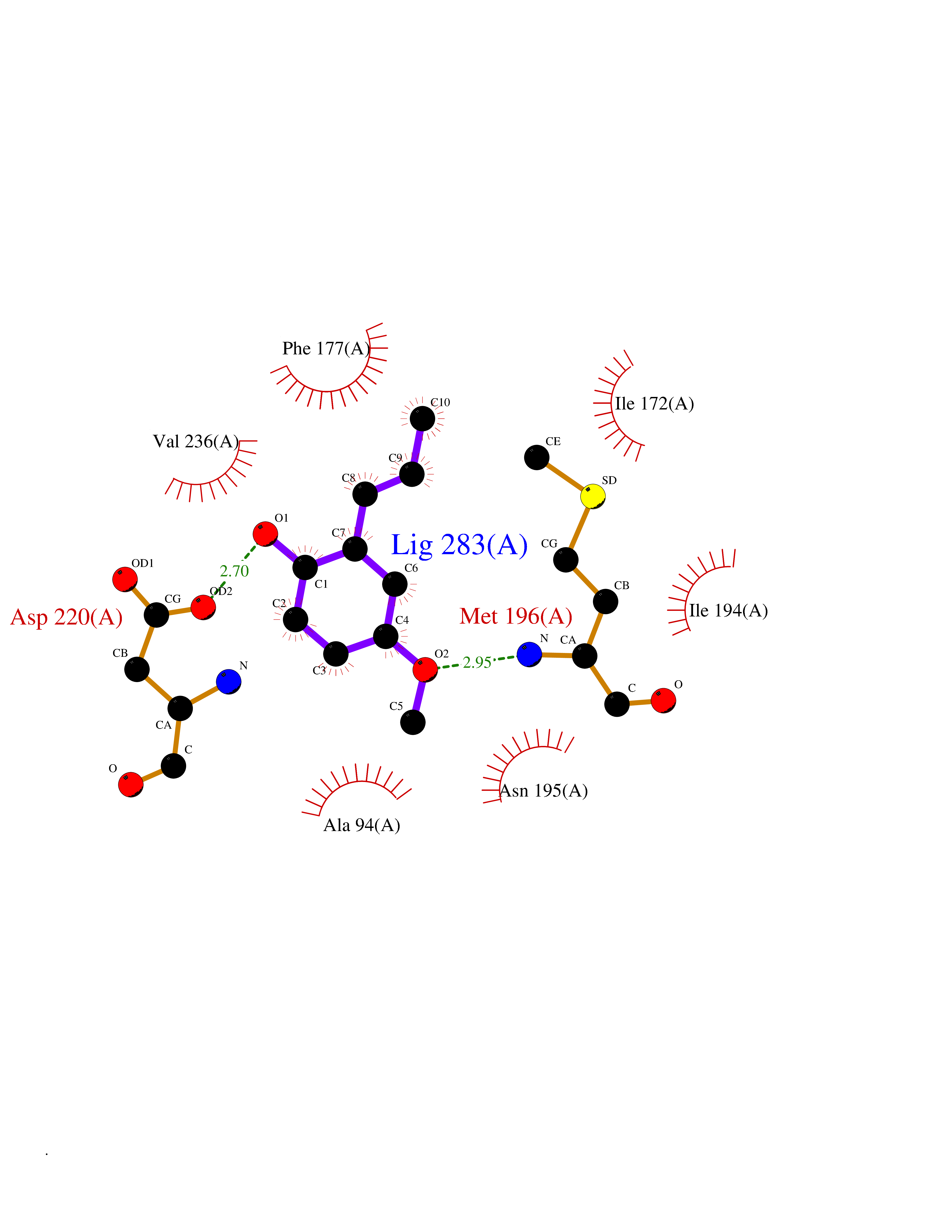 Ligplot Map 3D Binding mode Sequence AVKIGIIGGTGLDDPEILEGRTEKYVDTPFGKPSDALILGKIKNVDCVLLARHGRQHTIMPSKVNYQANIWALKEEGCTHVIVTTACGSLREEIQPGDIVIIDQFIDRTTMRPQSFYDGSHSCARGVCHIPMAEPFCPKTREVLIETAKKLGLRCHSKGTMVTIEGPRFSSRAESFMFRTWGADVINMTTVPEVVLAKEAGICYASIAMATDYDCWAVSVDRVLKTLKENANKAKSLLLTTIPQIGSTEWSETLHNLKNMAQFSVLLP Hydrogen bonds contact Hydrophobic contact | ||||
| 85 | Alanine aminotransferase 2 | 3IHJ | 5.60 | |
Target general information Gen name GPT2 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms AAT2;ALT2 Protein family Class-I pyridoxal-phosphate-dependent aminotransferase family, Alanine aminotransferase subfamily Biochemical class Transferase Function L-alanine:2-oxoglutarate aminotransferase activity.Pyridoxal phosphate binding. Related diseases Neurodevelopmental disorder with spastic paraplegia and microcephaly (NEDSPM) [MIM:616281]: An autosomal recessive syndrome characterized by severe psychomotor developmental delay, dysarthria, walking difficulties, moderately to severely impaired intellectual development, poor or absent speech, and progressive microcephaly. {ECO:0000269|PubMed:25758935}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00160; DB00142; DB00780; DB00114 Interacts with NA EC number 2.6.1.2 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Aminotransferase; Disease variant; Intellectual disability; Proteomics identification; Pyridoxal phosphate; Reference proteome; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 48717.7 Length 436 Aromaticity 0.09 Instability index 47.17 Isoelectric point 6.07 Charge (pH=7) -4.32 2D Binding mode Binding energy (Kcal/mol) -7.64 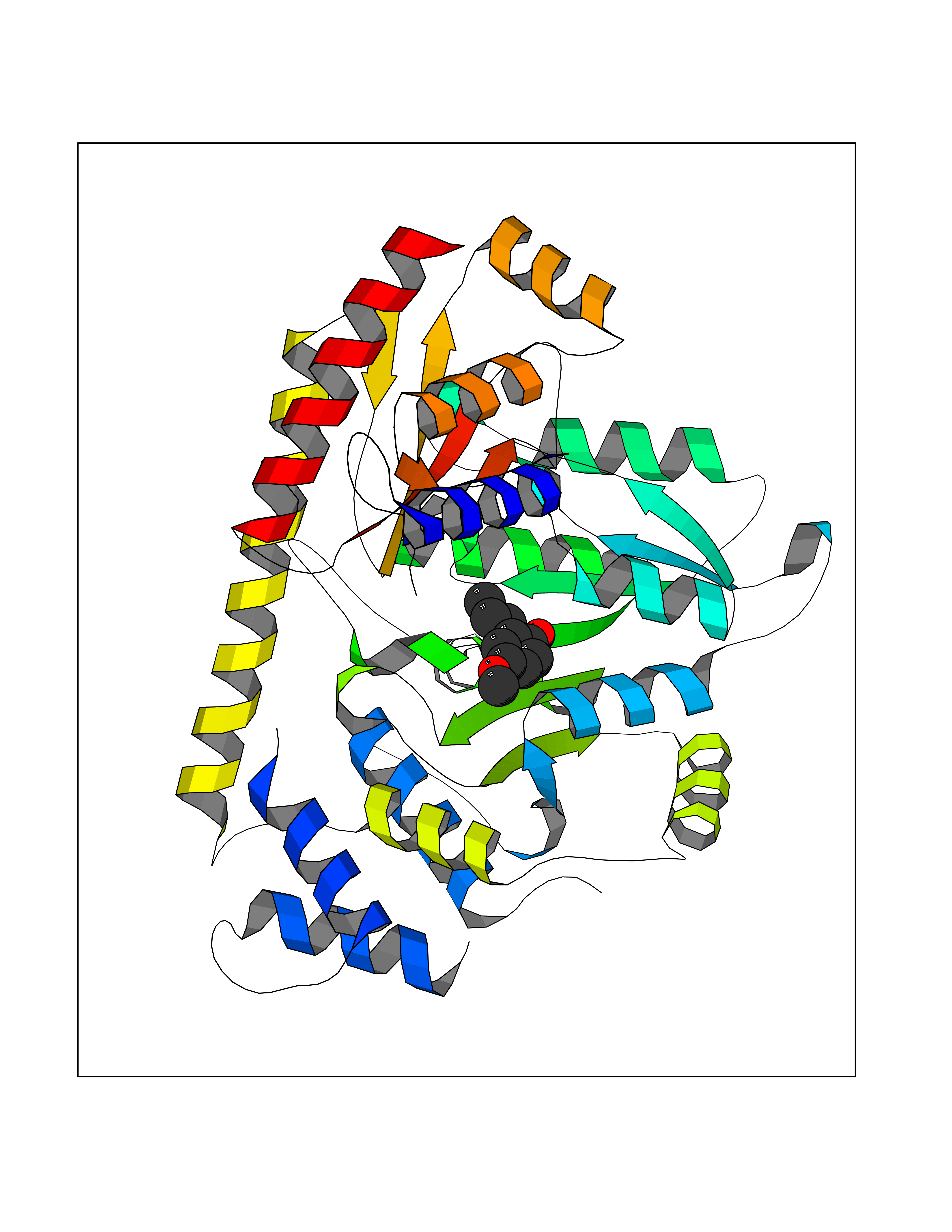 Molscript Map 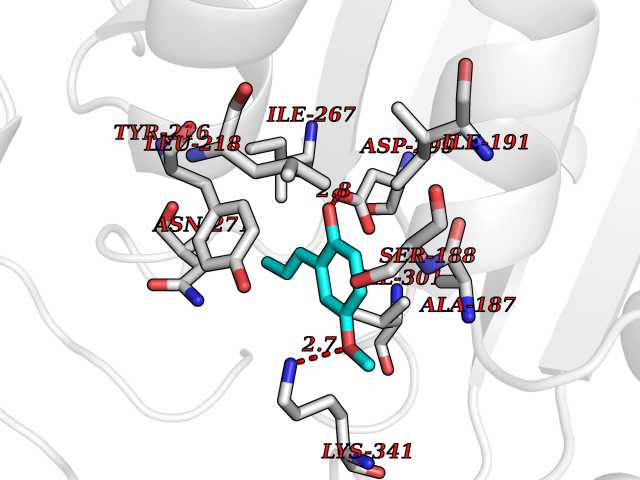 Pymol Map 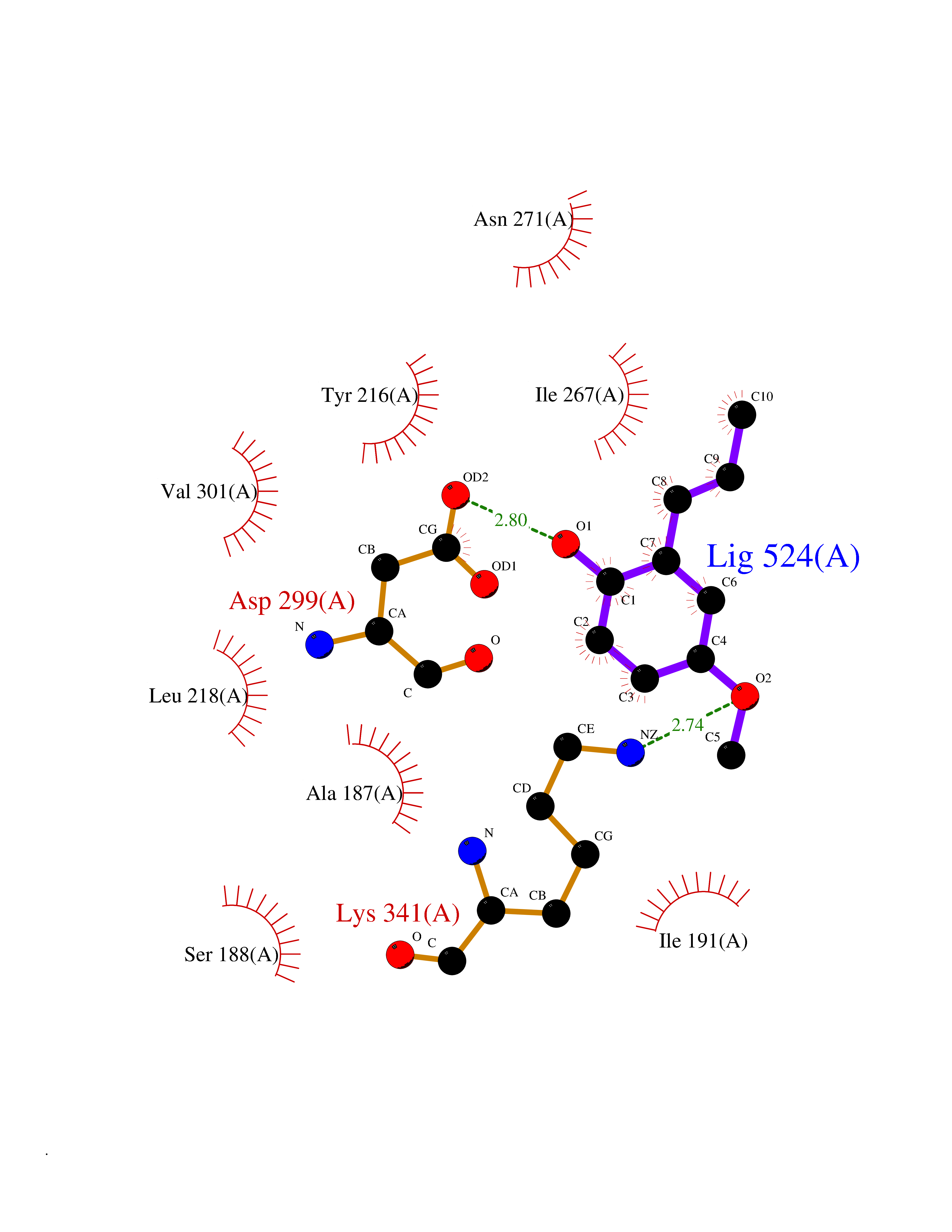 Ligplot Map 3D Binding mode Sequence PIVLKAGEIELELQRGIKKPFTEVIRANPITFLRQVMALCTYPNLLDSPSFPEDAKKRARRILQACSQGVNCIREDVAAYITRRDGGVPADPDNIYLTTGASDGISTILKILVSGGGKSRTGVMIPIPQYPLYSAVISELDAIQVNYYLDEENCWALNVNELRRAVQEAKDHCDPKVLCIINPGNPTGQVQSRKCIEDVIHFAWEEKLFLLADEVYQDNVYSPDCRFHSFKKVLYEMGPEYSSNVELASFHSTSKGYMGECGYRGGYMEVINLHPEIKGQLVKLLSVRLCPPVSGQAAMDIVVNPPVAGEESFEQFSREKESVLGNLAKKAKLTEDLFNQVPGIHCNPLQGAMYAFPRIFIPAKAVEAAQAHQMAPDMFYCMKLLEETGICVVPGSGFGQREGTYHFRMTILPPVEKLKTVLQKVKDFHINFLEKY Hydrogen bonds contact Hydrophobic contact | ||||
| 86 | Leucine carboxyl methyltransferase 1 | 3IEI | 5.60 | |
Target general information Gen name LCMT1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms CGI-68;LCMT Protein family Methyltransferase superfamily, LCMT family Biochemical class Transferase Function Protein C-terminal carboxyl O-methyltransferase activity.Protein C-terminal leucine carboxyl O-methyltransferase activity.S-adenosylmethionine-dependent methyltransferase activity. Related diseases Neurodevelopmental disorder, mitochondrial, with abnormal movements and lactic acidosis, with or without seizures (NEMMLAS) [MIM:617710]: An autosomal recessive, mitochondrial disorder with a broad phenotypic spectrum ranging from severe neonatal lactic acidosis, encephalomyopathy and early death to an attenuated course with milder manifestations. Clinical features include delayed psychomotor development, intellectual disability, hypotonia, dystonia, ataxia, and spasticity. Severe combined respiratory chain deficiency may be found in severely affected individuals. {ECO:0000269|PubMed:28236339, ECO:0000269|PubMed:28650581, ECO:0000269|PubMed:28905505, ECO:0000269|PubMed:30920170, ECO:0000269|PubMed:35074316}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Parkinsonism-dystonia 3, childhood-onset (PKDYS3) [MIM:619738]: An autosomal recessive neurodegenerative disorder with onset in infancy or early childhood. Affected individuals present with progressive movement abnormalities, including parkinsonism with tremor, dystonia, myoclonus ataxia, and hyperkinetic movements such as ballismus. The parkinsonism features may be responsive to treatment with levodopa, although many patients develop levodopa-induced dyskinesia. Some patients may have mild cognitive impairment or psychiatric disturbances. {ECO:0000269|PubMed:29120065, ECO:0000269|PubMed:31970218, ECO:0000269|PubMed:34890876}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00149 Interacts with P51116 EC number 2.1.1.233 Uniprot keywords 3D-structure; Alternative splicing; Methyltransferase; Proteomics identification; Reference proteome; S-adenosyl-L-methionine; Transferase Protein physicochemical properties Chain ID A,B,C,D,E,F,G,H Molecular weight (Da) 35803 Length 310 Aromaticity 0.08 Instability index 42.77 Isoelectric point 6.13 Charge (pH=7) -3.58 2D Binding mode Binding energy (Kcal/mol) -7.63  Molscript Map 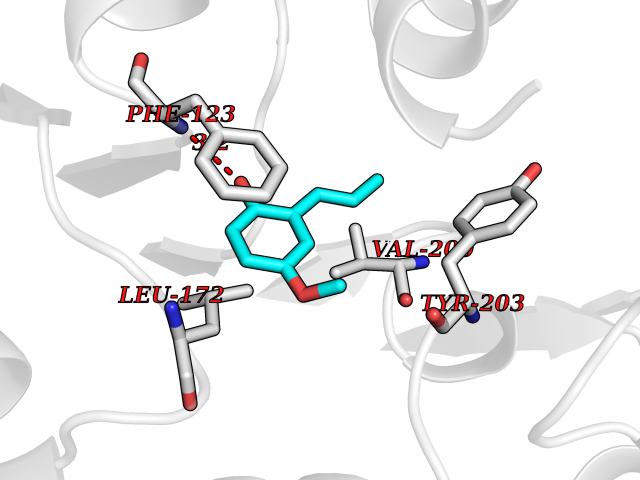 Pymol Map 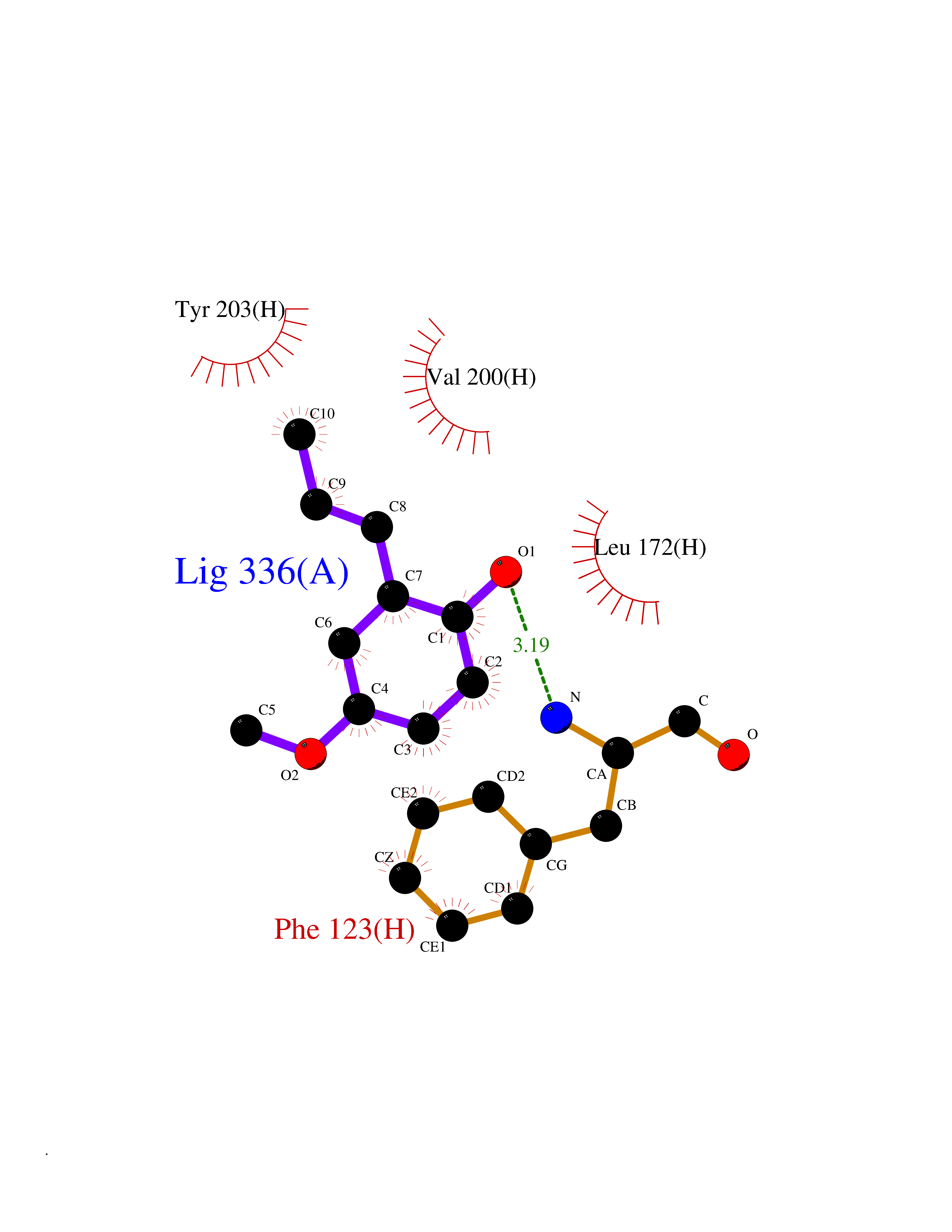 Ligplot Map 3D Binding mode Sequence GVRGTCEDASLCKRFAVSIGYWHDPYIQHFVRLSKERKAPEINRGYFARVHGVSQLIKAFLRKTECHCQIVNLGAGMDTTFWRLKDEDLLSSKYFEVDFPMIVTRKLHSIKCKPPLSSPILELHSEDTLQMDGHILDSKRYAVIGADLRDLSELEEKLKKCNMNTQLPTLLIAECVLVYMTPEQSANLLKWAANSFERAMFINYEQVNMGDRFGQIMIENLRRRQCDLAGVETCKSLESQKERLLSNGWETASAVDMMELYNRLPRAEVSRIESLEFLDEMELLEQLMRHYCLCWATKGGNELGLKEITY Hydrogen bonds contact Hydrophobic contact | ||||
| 87 | Histamine H3 receptor (H3R) | 7F61 | 5.60 | |
Target general information Gen name HRH3 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Histamine receptor 3; HH3R; GPCR97; G-protein coupled receptor 97; G protein-coupled receptor 97 Protein family G-protein coupled receptor 1 family Biochemical class GPCR rhodopsin Function Signals through the inhibition of adenylate cyclase and displays high constitutive activity (spontaneous activity in the absence of agonist). Agonist stimulation of isoform 3 neither modified adenylate cyclase activity nor induced intracellular calcium mobilization. The H3 subclass of histamine receptors could mediate the histamine signals in CNS and peripheral nervous system. Related diseases Immunodeficiency 48 (IMD48) [MIM:269840]: A form of severe immunodeficiency characterized by a selective absence of CD8+ T-cells. {ECO:0000269|PubMed:11123350, ECO:0000269|PubMed:11412303, ECO:0000269|PubMed:18509675, ECO:0000269|PubMed:8124727, ECO:0000269|PubMed:8202713}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Autoimmune disease, multisystem, infantile-onset, 2 (ADMIO2) [MIM:617006]: An autosomal recessive, autoimmune disorder characterized by systemic manifestations including blistering skin disease, uncontrollable bullous pemphigoid, inflammatory colitis, autoimmune hypothyroidism, proteinuria and nephrotic syndrome. {ECO:0000269|PubMed:26783323}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01238; DB06698; DB05381; DB17087; DB05080; DB00768; DB11642 Interacts with NA EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disulfide bond; G-protein coupled receptor; Glycoprotein; Membrane; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 34321.1 Length 301 Aromaticity 0.16 Instability index 32.22 Isoelectric point 9.63 Charge (pH=7) 15.11 2D Binding mode Binding energy (Kcal/mol) -7.65 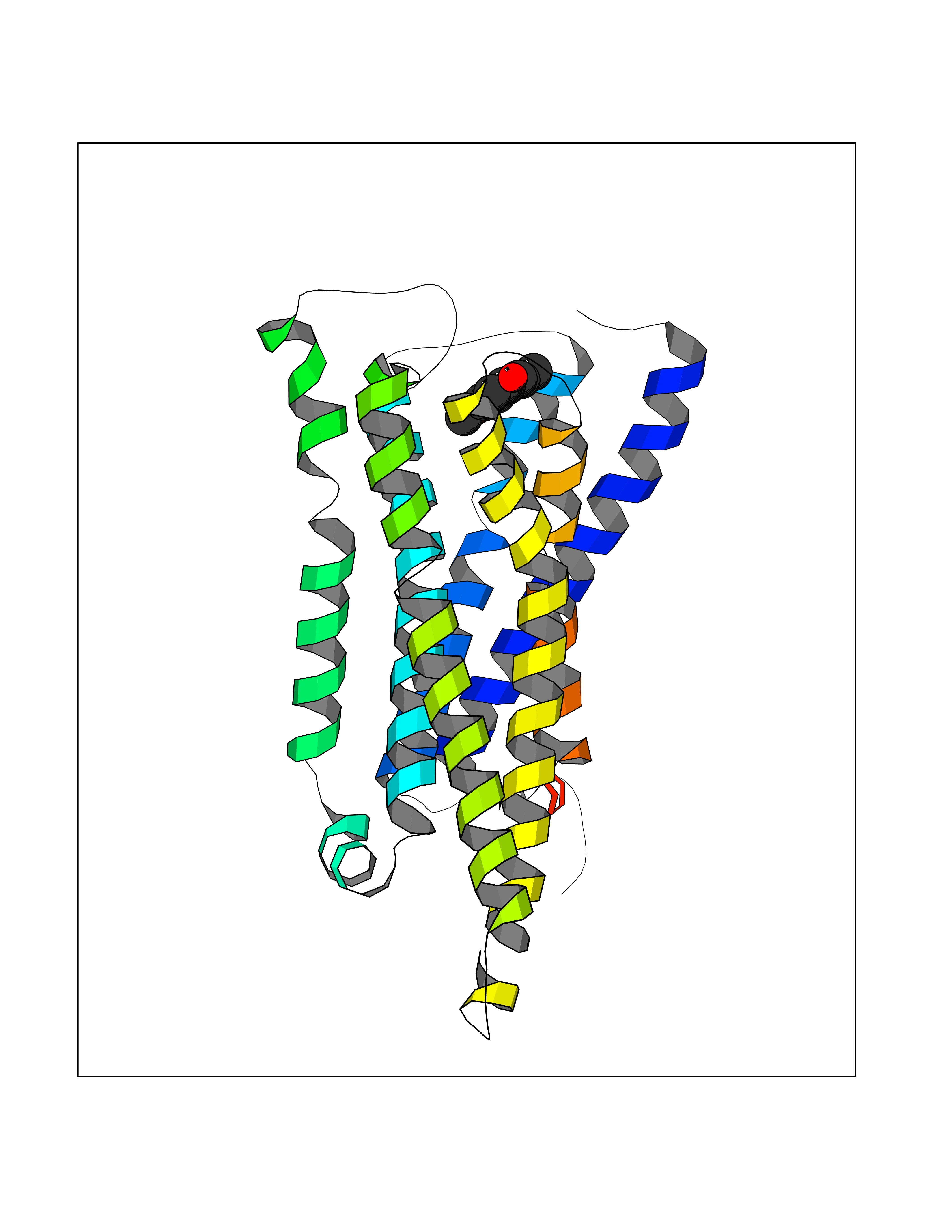 Molscript Map 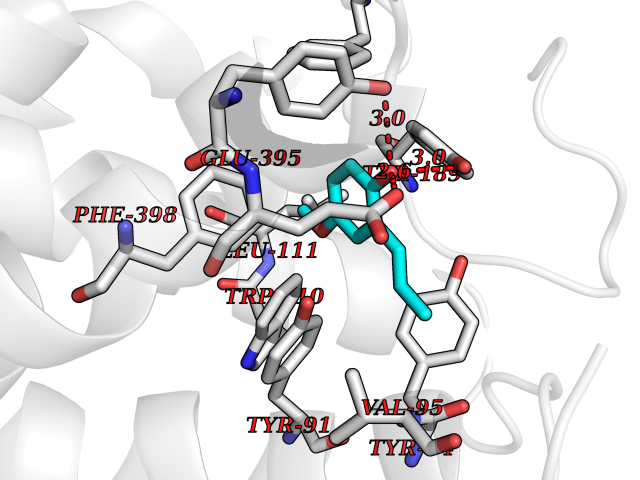 Pymol Map 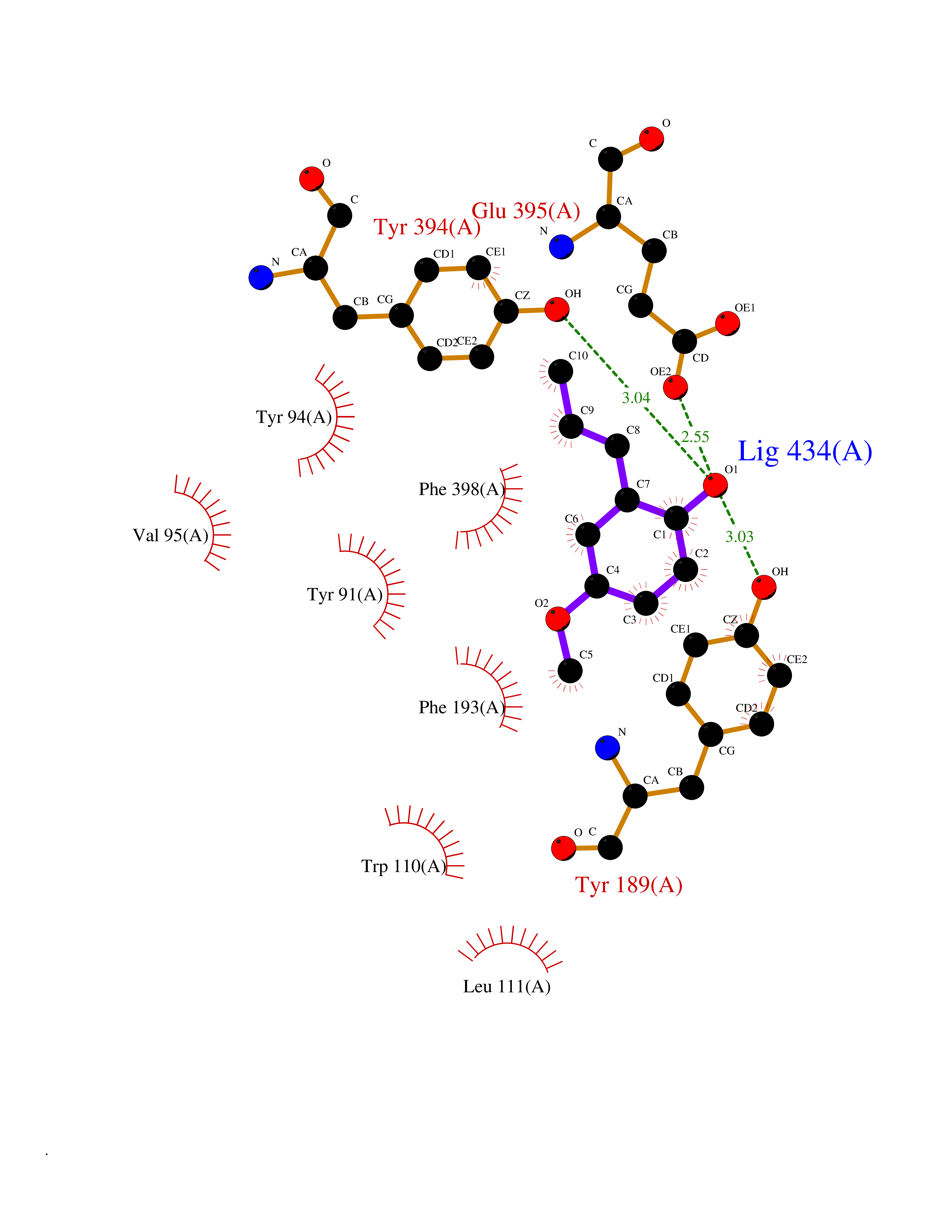 Ligplot Map 3D Binding mode Sequence RGFSAAWTAVLAALMALLIVATVLGNALVMLAFVADSSLRTQNNFFLLNLAISDFLVGAFCIPLYVPYVLTGRWTFGRGLCKLWLVVDYLLCTSKAFNIVLISYDRFLSVTRAVSYRAQQGDTRRAVRKMLLVWVLAFLLYGPAILSWEYLSGGSSIPEGHCYAEFFYNWYFLITASTLEFFTPFLSVTFFNLSIYLNIQRRTRLRLDGAREAAGRFRLSRDRKVAKSLAVIVSIFGLCWAPYTLLMIIRAACHGHCVPDYWYETSFWLLWANSAVNPVLYPLCHHSFRRAFTKLLCPQKL Hydrogen bonds contact Hydrophobic contact | ||||
| 88 | ERK activator kinase 2 (MEK2) | 1S9I | 5.60 | |
Target general information Gen name MAP2K2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms PRKMK2; MKK2; MEK 2; MAPKK 2; MAPK/ERK kinase 2; MAP kinase kinase 2; Dual specificity mitogenactivated protein kinase kinase 2; Dual specificity mitogen-activated protein kinase kinase 2 Protein family Protein kinase superfamily, STE Ser/Thr protein kinase family, MAP kinase kinase subfamily Biochemical class Kinase Function Activates the ERK1 and ERK2 MAP kinases. Catalyzes the concomitant phosphorylation of a threonine and a tyrosine residue in a Thr-Glu-Tyr sequence located in MAP kinases. Related diseases Cardiofaciocutaneous syndrome 4 (CFC4) [MIM:615280]: A form of cardiofaciocutaneous syndrome, a multiple congenital anomaly disorder characterized by a distinctive facial appearance, heart defects and intellectual disability. Heart defects include pulmonic stenosis, atrial septal defects and hypertrophic cardiomyopathy. Some affected individuals present with ectodermal abnormalities such as sparse, friable hair, hyperkeratotic skin lesions and a generalized ichthyosis-like condition. Typical facial features are similar to Noonan syndrome. They include high forehead with bitemporal constriction, hypoplastic supraorbital ridges, downslanting palpebral fissures, a depressed nasal bridge, and posteriorly angulated ears with prominent helices. {ECO:0000269|PubMed:16439621, ECO:0000269|PubMed:18042262, ECO:0000269|PubMed:20358587}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB11967; DB06616; DB12010; DB14904; DB11689; DB08911 Interacts with P05067; P10398; Q96II5; P15056; O95273; Q12959; P61978-2; Q8IVT5; P00540; P04049 EC number EC 2.7.12.2 Uniprot keywords 3D-structure; Acetylation; ATP-binding; Cardiomyopathy; Cytoplasm; Direct protein sequencing; Disease variant; Ectodermal dysplasia; Intellectual disability; Kinase; Magnesium; Membrane; Metal-binding; Nucleotide-binding; Phosphoprotein; Proteomics identification; Reference proteome; Serine/threonine-protein kinase; Transferase; Tyrosine-protein kinase Protein physicochemical properties Chain ID A Molecular weight (Da) 33960.9 Length 303 Aromaticity 0.07 Instability index 45.61 Isoelectric point 6.29 Charge (pH=7) -2.53 2D Binding mode Binding energy (Kcal/mol) -7.64  Molscript Map 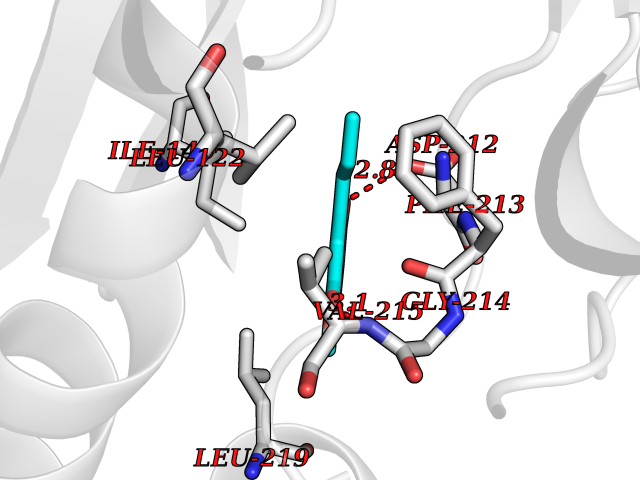 Pymol Map 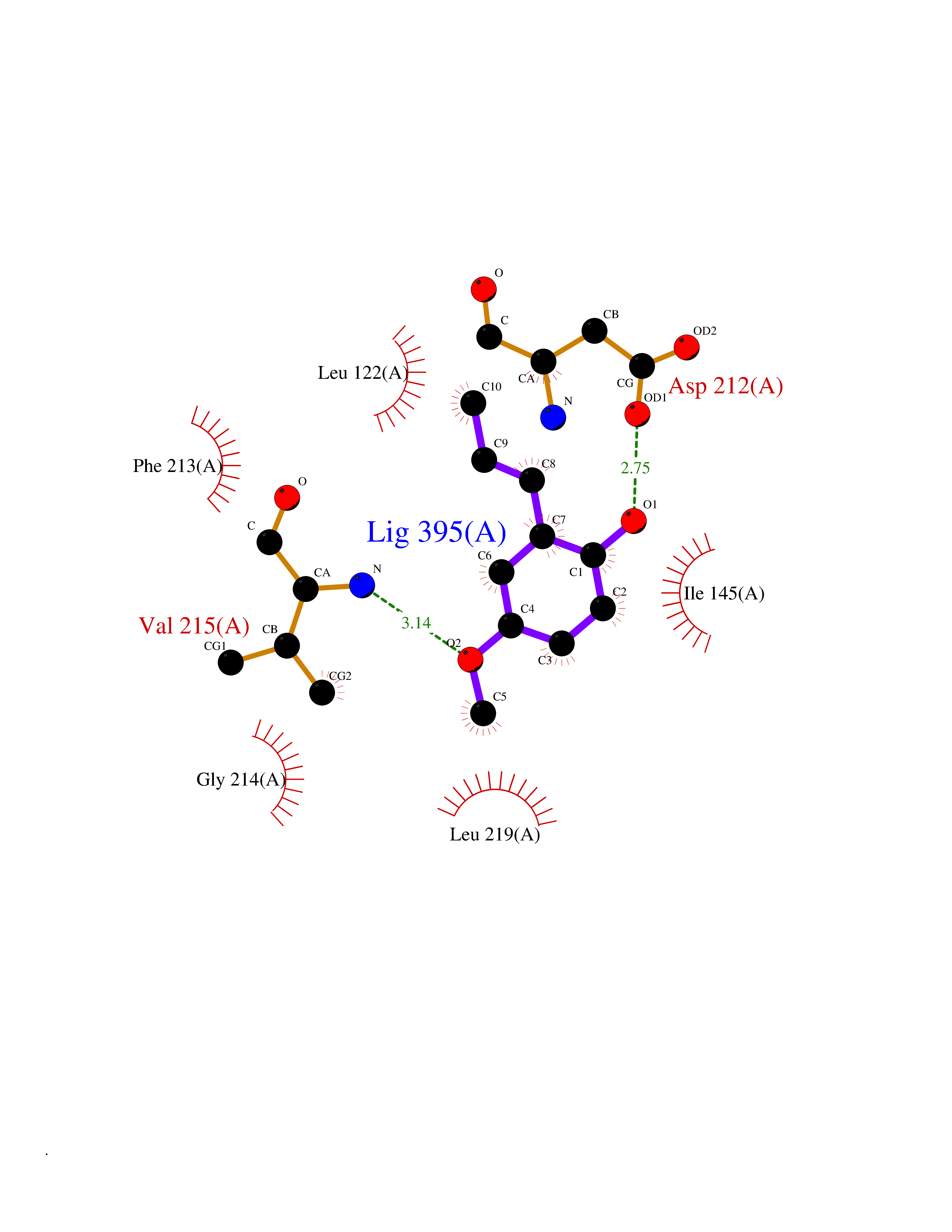 Ligplot Map 3D Binding mode Sequence QKAKVGELKDDDFERISELGAGNGGVVTKVQHRPSGLIMARKLIHLEIKPAIRNQIIRELQVLHECNSPYIVGFYGAFYSDGEISICMEHMDGGSLDQVLKEAKRIPEEILGKVSIAVLRGLAYLREKHQIMHRDVKPSNILVNSRGEIKLCDFGVSGQLIDSMVGTRSYMAPERLQGTHYSVQSDIWSMGLSLVELAVGRYPIPPPDAKELEAIFGRPVVDRPAMAIFELLDYIVNEPPPKLPNGVFTPDFQEFVNKCLIKNPAERADLKMLTNHTFIKRSEVEEVDFAGWLCKTLRLNQPG Hydrogen bonds contact Hydrophobic contact | ||||
| 89 | Hydroxymethylglutaryl-CoA synthase 2 (HMGCS2) | 2WYA | 5.60 | |
Target general information Gen name HMGCS2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms HMGCS2; HMG-CoAsynthase; 3-hydroxy-3-methylglutaryl coenzyme A synthase 2 Protein family Thiolase-like superfamily, HMG-CoA synthase family Biochemical class Acyltransferase Function This enzyme condenses acetyl-CoA with acetoacetyl-CoA to form HMG-CoA, which is the substrate for HMG-CoA reductase. Related diseases 3-hydroxy-3-methylglutaryl-CoA synthase-2 deficiency (HMGCS2D) [MIM:605911]: A metabolic disorder characterized by severe hypoketotic hypoglycemia, encephalopathy, and hepatomegaly. {ECO:0000269|PubMed:11228257, ECO:0000269|PubMed:11479731, ECO:0000269|PubMed:12647205, ECO:0000269|PubMed:16601895, ECO:0000269|PubMed:23751782, ECO:0000269|PubMed:25511235, ECO:0000269|PubMed:29597274}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number EC 2.3.3.10 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Cholesterol biosynthesis; Cholesterol metabolism; Disease variant; Lipid biosynthesis; Lipid metabolism; Mitochondrion; Phosphoprotein; Proteomics identification; Reference proteome; Steroid biosynthesis; Steroid metabolism; Sterol biosynthesis; Sterol metabolism; Transferase; Transit peptide Protein physicochemical properties Chain ID A,D Molecular weight (Da) 102499 Length 920 Aromaticity 0.1 Instability index 33.48 Isoelectric point 6.72 Charge (pH=7) -1.41 2D Binding mode Binding energy (Kcal/mol) -7.64 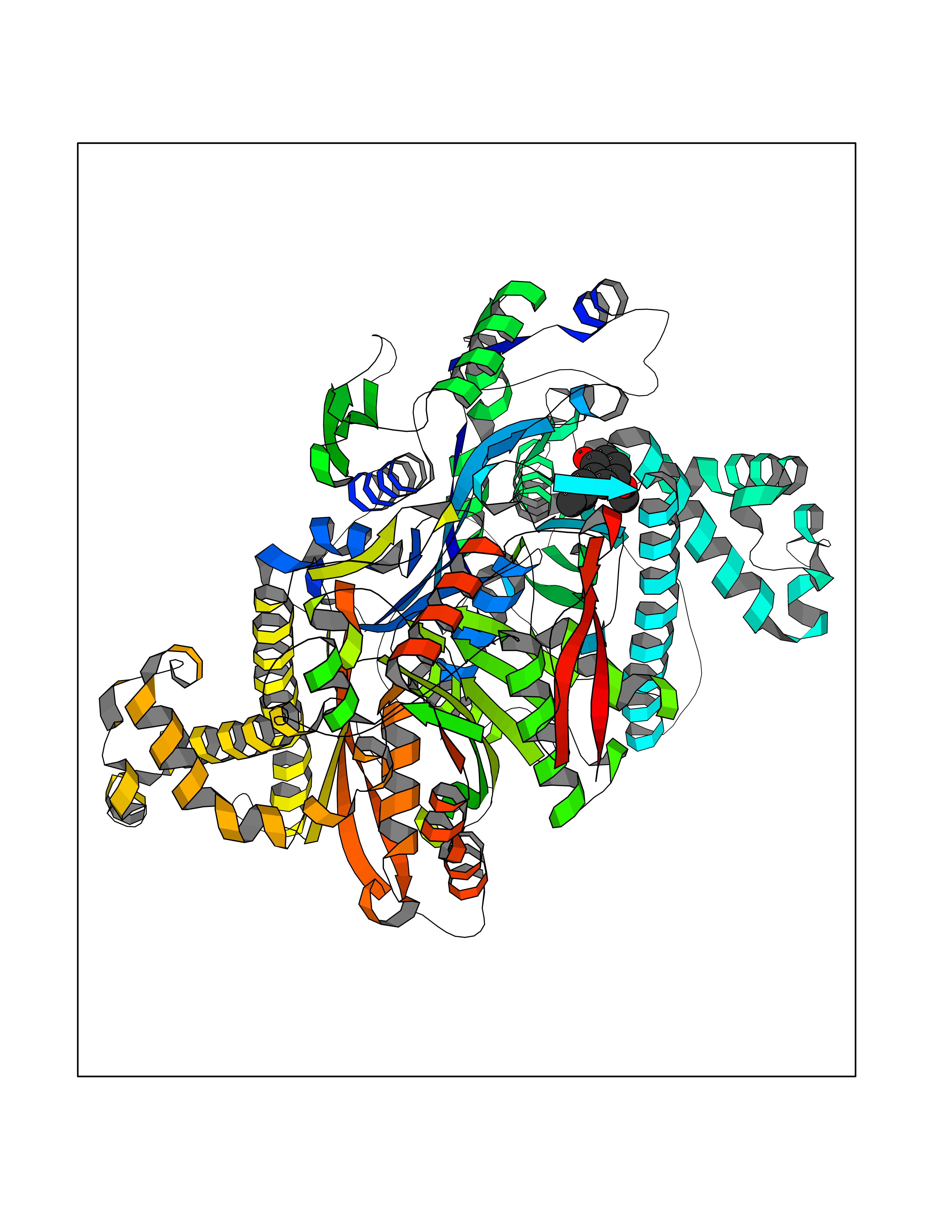 Molscript Map 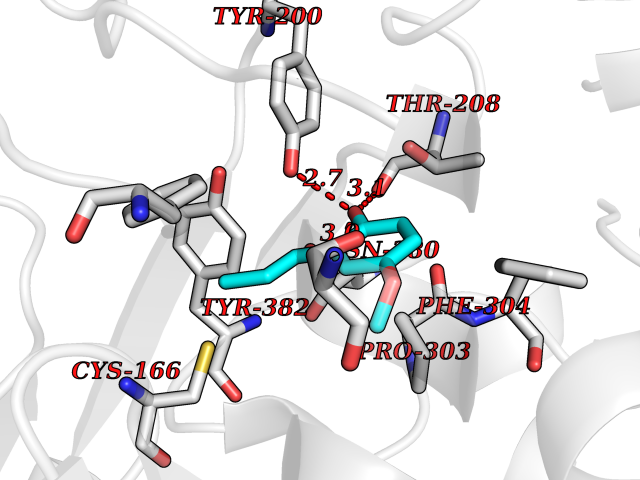 Pymol Map 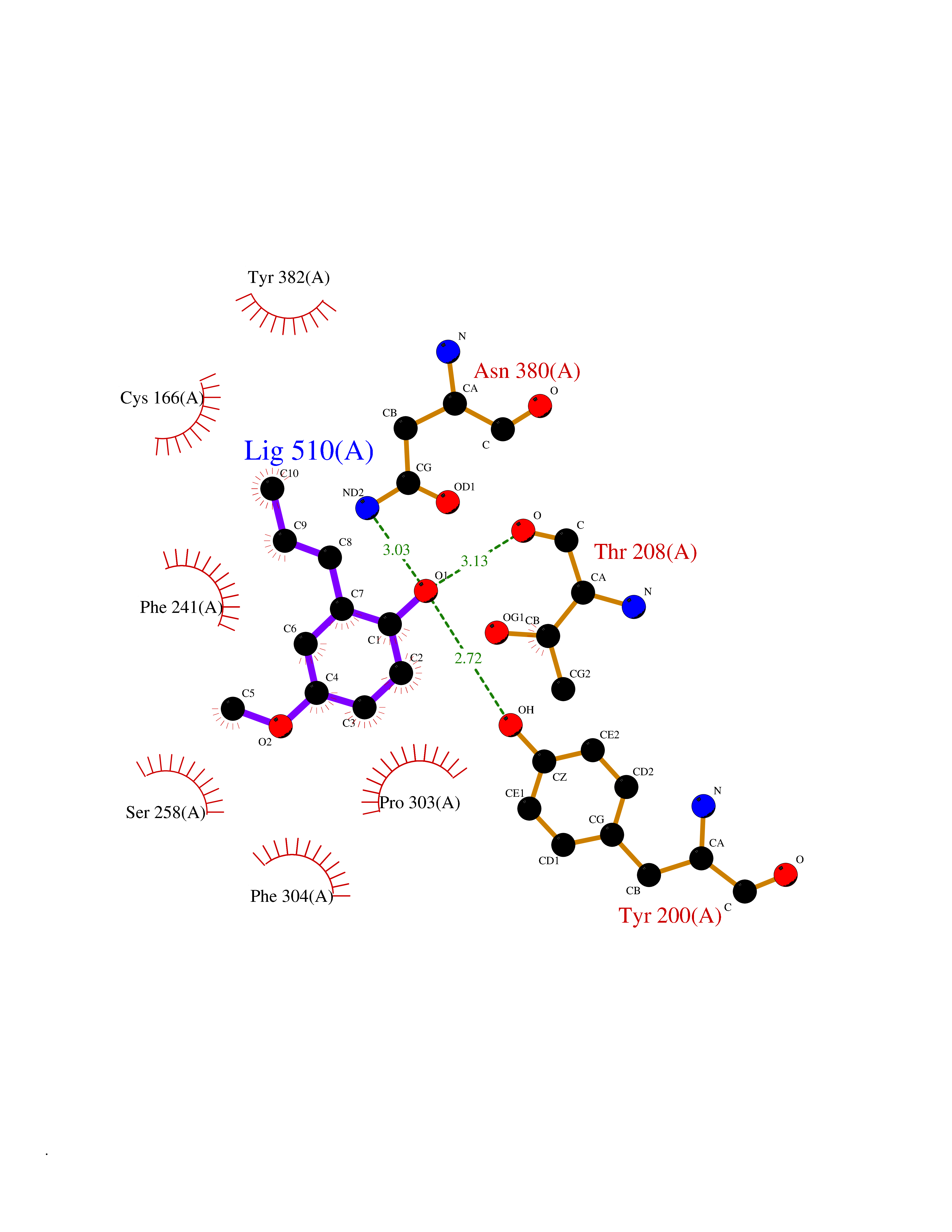 Ligplot Map 3D Binding mode Sequence SMPKDVGILALEVYFPAQYVDQTDLEKYNNVEAGKYTVGLGQTRMGFCSVQEDINSLCLTVVQRLMERIQLPWDSVGRLEVGTETIIDKSKAVKTVLMELFQDSGNTDIEGIDTTNACYGGTASLFNAANWMESSSWDGRYAMVVCGDIAVYPSGNARPTGGAGAVAMLIGPKAPLALERGLRGTHMENVYDFYKPNLASEYPIVDGKLSIQCYLRALDRCYTSYRKKIQNQWKQAGSDRPFTLDDLQYMIFHTPFCKMVQKSLARLMFNDFLSASSDTQTSLYKGLEAFGGLKLEDTYTNKDLDKALLKASQDMFDKKTKASLYLSTHNGNMYTSSLYGCLASLLSHHSAQELAGSRIGAFSYGSGLAASFFSFRVSQDAAPGSPLDKLVSSTSDLPKRLASRKCVSPEEFTEIMNQREQFYHKVNFSPPGDTNSLFPGTWYLERVDEQHRRKYARRPVSMPKDVGILALEVYFPAQYVDQTDLEKYNNVEAGKYTVGLGQTRMGFCSVQEDINSLCLTVVQRLMERIQLPWDSVGRLEVGTETIIDKSKAVKTVLMELFQDSGNTDIEGIDTTNACYGGTASLFNAANWMESSSWDGRYAMVVCGDIAVYPSGNARPTGGAGAVAMLIGPKAPLALERGLRGTHMENVYDFYKPNLASEYPIVDGKLSIQCYLRALDRCYTSYRKKIQNQWKQAGSDRPFTLDDLQYMIFHTPFCKMVQKSLARLMFNDFLSASSDTQTSLYKGLEAFGGLKLEDTYTNKDLDKALLKASQDMFDKKTKASLYLSTHNGNMYTSSLYGCLASLLSHHSAQELAGSRIGAFSYGSGLAASFFSFRVSQDAAPGSPLDKLVSSTSDLPKRLASRKCVSPEEFTEIMNQREQFYHKVNFSPPGDTNSLFPGTWYLERVDEQHRRKYARRPV Hydrogen bonds contact Hydrophobic contact | ||||
| 90 | Estrogen sulfotransferase | 1G3M | 5.59 | |
Target general information Gen name SULT1E1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms STE Protein family Sulfotransferase 1 family Biochemical class Transferase Function Aryl sulfotransferase activity.Estrone sulfotransferase activity.Flavonol 3-sulfotransferase activity.Steroid binding.Steroid sulfotransferase activity.Sulfotransferase activity. Related diseases Neuromyotonia and axonal neuropathy, autosomal recessive (NMAN) [MIM:137200]: An autosomal recessive neurologic disorder characterized by onset in the first or second decade of a peripheral axonal neuropathy predominantly affecting motor more than sensory nerves. The axonal neuropathy is reminiscent of Charcot-Marie-Tooth disease type 2 and distal hereditary motor neuropathy. Individuals with NMAN also have delayed muscle relaxation and action myotonia associated with neuromyotonic discharges on needle EMG resulting from hyperexcitability of the peripheral nerves. {ECO:0000269|PubMed:16835243, ECO:0000269|PubMed:22961002, ECO:0000269|PubMed:28691797, ECO:0000269|PubMed:29787766, ECO:0000269|PubMed:31088288}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02902; DB03346; DB01812; DB00714; DB14635; DB01176; DB00977; DB09288; DB00675; DB09100 Interacts with O76083; O76083-2 EC number 2.8.2.4 Uniprot keywords 3D-structure; Cytoplasm; Lipid metabolism; Lipid-binding; Proteomics identification; Reference proteome; Steroid-binding; Transferase Protein physicochemical properties Chain ID A,B Molecular weight (Da) 34022.7 Length 285 Aromaticity 0.15 Instability index 32.76 Isoelectric point 6.33 Charge (pH=7) -2.75 2D Binding mode Binding energy (Kcal/mol) -7.63 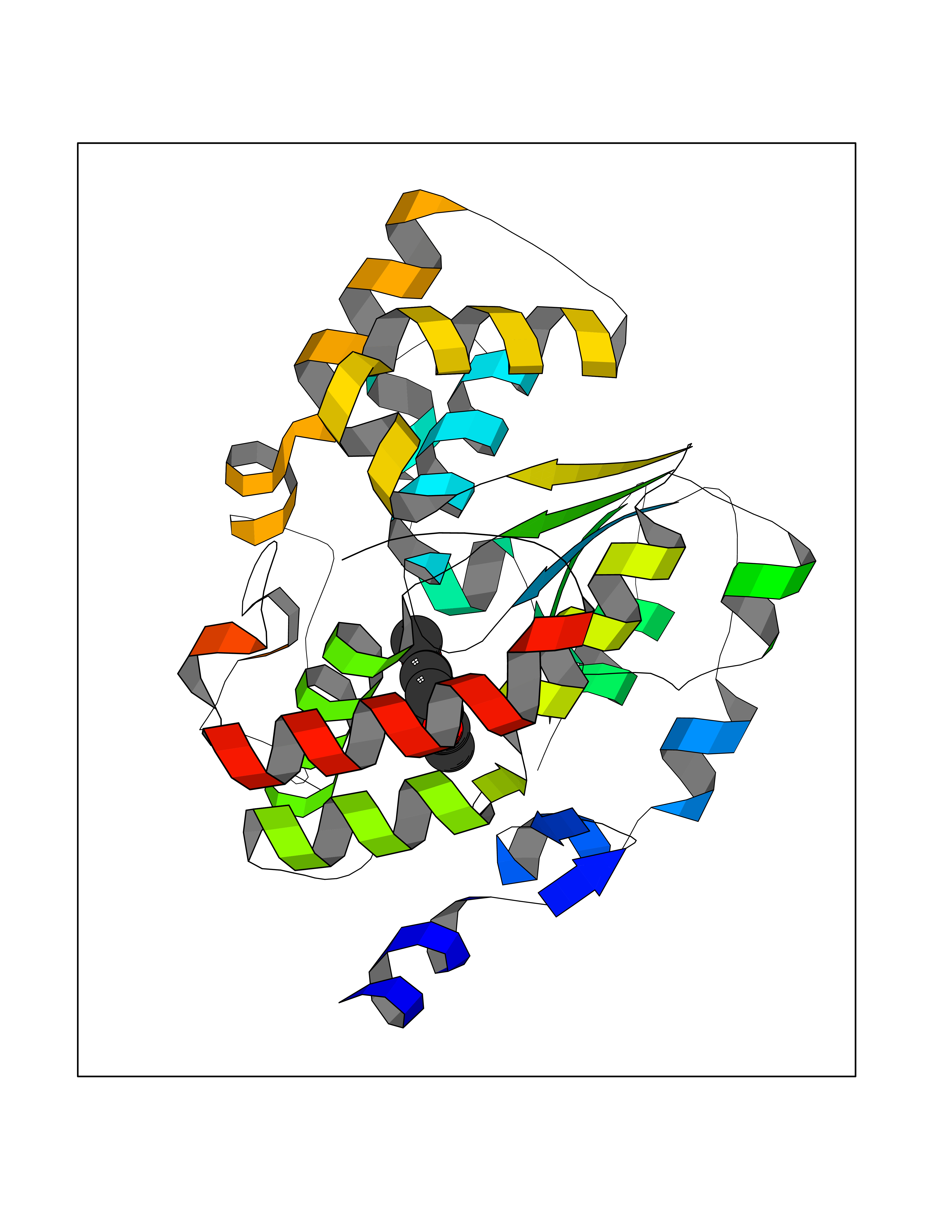 Molscript Map 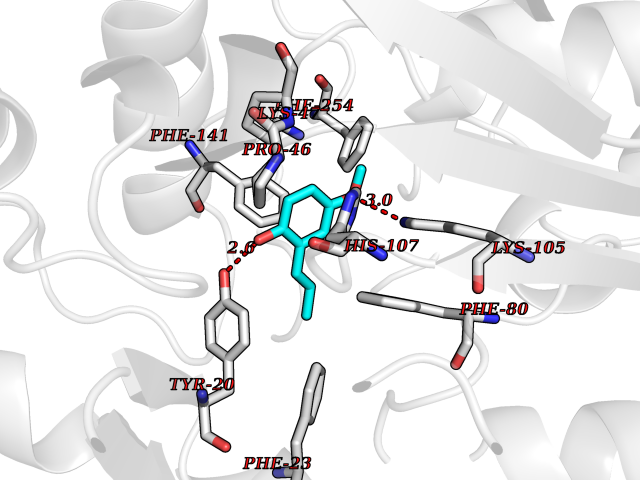 Pymol Map 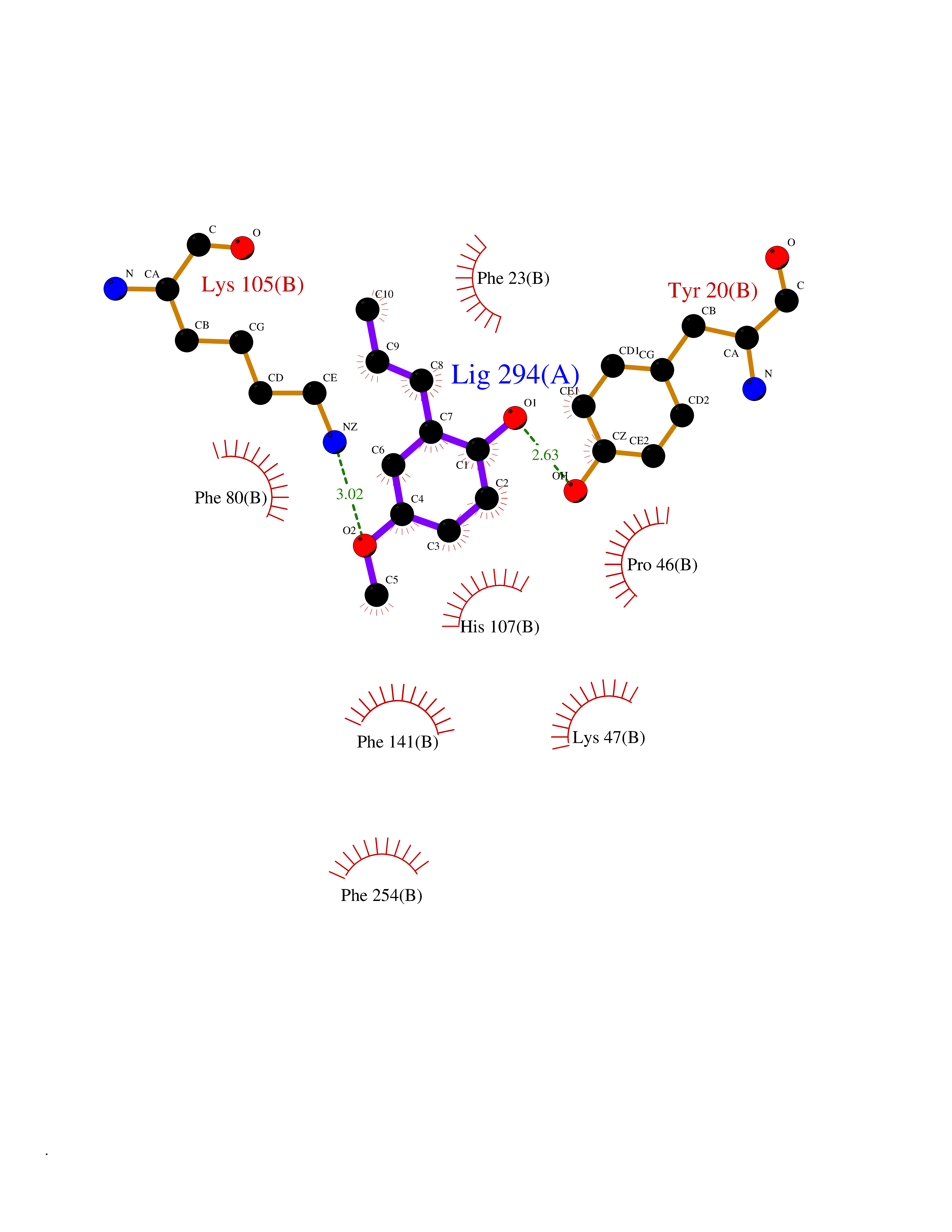 Ligplot Map 3D Binding mode Sequence SELDYYEKFEEVHGILMYKDFVKYWDNVEAFQARPDDLVIATYPKSGTTWVSEIVYMIYKEGDVEKCKEDVIFNRIPFLECRNGVKQLDEMNSPRIVKTHLPPELLPASFWEKDCKIIYLCRNAKDVAVSFYYFFLMVAGHPNPGSFPEFVEKFMQGQVPYGSWYKHVKSWWEKGKSPRVLFLFYEDLKEDIRKEVIKLIHFLERKPSEELVDRIIHHTSFQEMKNNPSTNYTTLPDEIMNQKLSPFMRKGITGDWKNHFTVALNEKFDKHYEQQMKESTLKFRT Hydrogen bonds contact Hydrophobic contact | ||||
| 91 | Histone-lysine N-methyltransferase SMYD3 (SMYD3) | 6P7Z | 5.59 | |
Target general information Gen name SMYD3 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms SET and MYND domain-containing protein 3; Zinc finger MYND domain-containing protein 1 Protein family Class V-like SAM-binding methyltransferase superfamily, Histone-lysine methyltransferase family Biochemical class NA Function Histone methyltransferase. Specifically methylates 'Lys-4' of histone H3, inducing di- and tri-methylation, but not monomethylation . Also methylates 'Lys-5' of histone H4. Plays an important role in transcriptional activation as a member of an RNA polymerase complex. Binds DNA containing 5'-CCCTCC-3' or 5'-GAGGGG-3' sequences. Related diseases Leukemia, juvenile myelomonocytic (JMML) [MIM:607785]: An aggressive pediatric myelodysplastic syndrome/myeloproliferative disorder characterized by malignant transformation in the hematopoietic stem cell compartment with proliferation of differentiated progeny. Patients have splenomegaly, enlarged lymph nodes, rashes, and hemorrhages. {ECO:0000269|PubMed:17332249}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Noonan syndrome 6 (NS6) [MIM:613224]: A form of Noonan syndrome, a disease characterized by short stature, facial dysmorphic features such as hypertelorism, a downward eyeslant and low-set posteriorly rotated ears, and a high incidence of congenital heart defects and hypertrophic cardiomyopathy. Other features can include a short neck with webbing or redundancy of skin, deafness, motor delay, variable intellectual deficits, multiple skeletal defects, cryptorchidism, and bleeding diathesis. Individuals with Noonan syndrome are at risk of juvenile myelomonocytic leukemia, a myeloproliferative disorder characterized by excessive production of myelomonocytic cells. {ECO:0000269|PubMed:19966803}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: RAS-associated autoimmune leukoproliferative disorder (RALD) [MIM:614470]: A disorder of apoptosis, characterized by chronic accumulation of non-malignant lymphocytes, defective lymphocyte apoptosis, and an increased risk for the development of hematologic malignancies. {ECO:0000269|PubMed:17517660}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Melanocytic nevus syndrome, congenital (CMNS) [MIM:137550]: A syndrome characterized by congenital pigmentary skin lesions which can occur at any site and can cover most of the body surface. These lesions may or may not be hairy. Congenital melanocytic nevi are associated with neuromelanosis (the presence of melanin-producing cells within the brain parenchyma or leptomeninges). Less commonly they are associated with malignant melanoma in childhood, both in the skin and the central nervous system. CMNS patients also tend to have a characteristic facial appearance, including wide or prominent forehead, periorbital fullness, small short nose with narrow nasal bridge, round face, full cheeks, prominent premaxilla, and everted lower lip. {ECO:0000269|PubMed:18633438, ECO:0000269|PubMed:23392294}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Melanosis, neurocutaneous (NCMS) [MIM:249400]: A rare congenital disease characterized by the presence of giant or multiple melanocytic nevi on the skin, foci of melanin-producing cells within the brain parenchyma, and infiltration of leptomeninges by abnormal melanin deposits. Neurologic abnormalities include seizures, hydrocephalus, arachnoid cysts, tumors, and syringomyelia. Some patients may develop malignant melanoma. {ECO:0000269|PubMed:23392294}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Keratinocytic non-epidermolytic nevus (KNEN) [MIM:162900]: Epidermal nevi of the common, non-organoid and non-epidermolytic type are benign skin lesions and may vary in their extent from a single (usually linear) lesion to widespread and systematized involvement. They may be present at birth or develop early during childhood. {ECO:0000269|PubMed:22499344}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Thyroid cancer, non-medullary, 2 (NMTC2) [MIM:188470]: A form of non-medullary thyroid cancer (NMTC), a cancer characterized by tumors originating from the thyroid follicular cells. NMTCs represent approximately 95% of all cases of thyroid cancer and are classified into papillary, follicular, Hurthle cell, and anaplastic neoplasms. {ECO:0000269|PubMed:12727991}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q9H0L4; Q9H0I2; Q13064; Q7Z3B4; Q16512; Q92529; Q15915; Q9Y2U5 EC number EC 2.1.1.354 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Chromatin regulator; Cytoplasm; Metal-binding; Methyltransferase; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; S-adenosyl-L-methionine; Transferase; Zinc; Zinc-finger Protein physicochemical properties Chain ID A Molecular weight (Da) 48694.1 Length 425 Aromaticity 0.08 Instability index 44.91 Isoelectric point 6.86 Charge (pH=7) -0.4 2D Binding mode Binding energy (Kcal/mol) -7.63 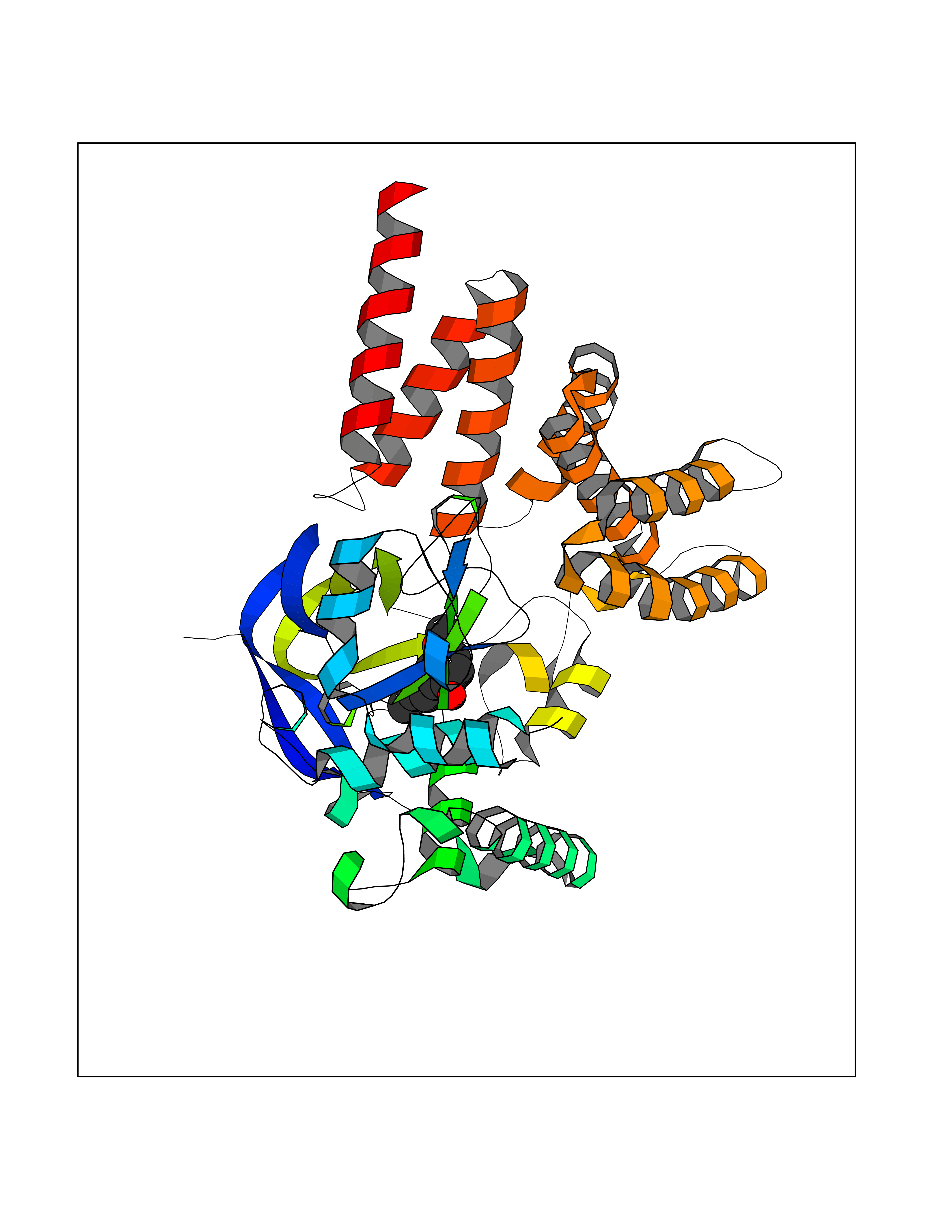 Molscript Map 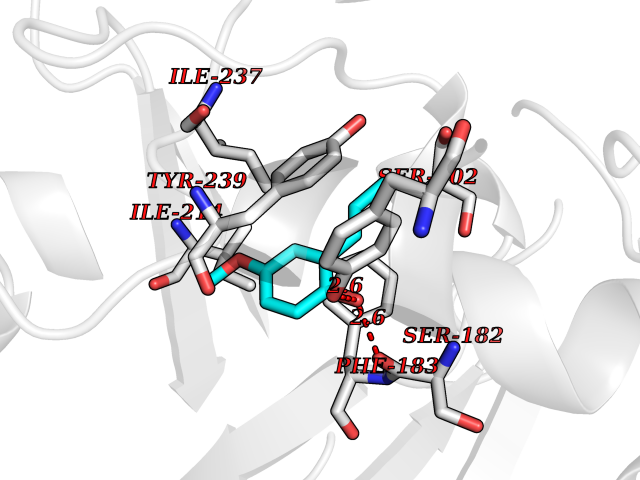 Pymol Map 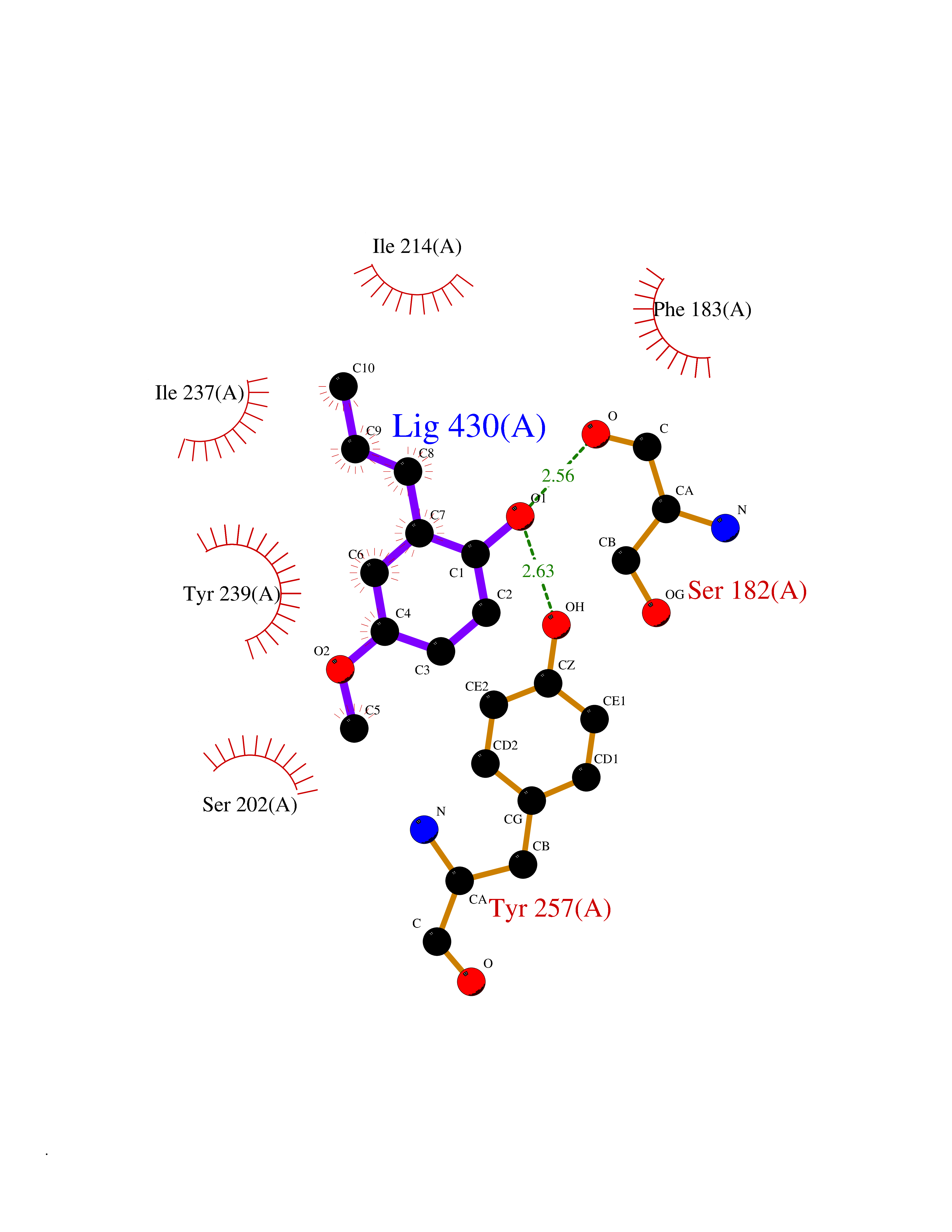 Ligplot Map 3D Binding mode Sequence PLKVEKFATANRGNGLRAVTPLRPGELLFRSDPLAYTVCKGSRGVVCDRCLLGKEKLMRCSQCRVAKYCSAKCQKKAWPDHKRECKCLKSCPRYPPDSVRLLGRVVFKLMDGAPSESEKLYSFYDLESNINKLTEDKKEGLRQLVMTFQHFMREEIQDASQLPPAFDLFEAFAKVICNSFTICNAEMQEVGVGLYPSISLLNHSCDPNCSIVFNGPHLLLRAVRDIEVGEELTICYLDMLMTSEERRKQLRDQYCFECDCFRCQTQDKDADMLTGDEQVWKEVQESLKKIEELKAHWKWEQVLAMCQAIISSNSERLPDINIYQLKVLDCAMDACINLGLLEEALFYGTRTMEPYRIFFPGSHPVRGVQVMKVGKLQLHQGMFPQAMKNLRLAFDIMRVTHGREHSLIEDLILLLEECDANIRAS Hydrogen bonds contact Hydrophobic contact | ||||
| 92 | DNA mismatch repair protein MSH2 (MSH2) | 3THX | 5.59 | |
Target general information Gen name MSH2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms hMSH2; MutS protein homolog 2; Mismatch repair gene Msh2 Protein family DNA mismatch repair MutS family Biochemical class NA Function Forms two different heterodimers: MutS alpha (MSH2-MSH6 heterodimer) and MutS beta (MSH2-MSH3 heterodimer) which binds to DNA mismatches thereby initiating DNA repair. When bound, heterodimers bend the DNA helix and shields approximately 20 base pairs. MutS alpha recognizes single base mismatches and dinucleotide insertion-deletion loops (IDL) in the DNA. MutS beta recognizes larger insertion-deletion loops up to 13 nucleotides long. After mismatch binding, MutS alpha or beta forms a ternary complex with the MutL alpha heterodimer, which is thought to be responsible for directing the downstream MMR events, including strand discrimination, excision, and resynthesis. Recruits DNA helicase MCM9 to chromatin which unwinds the mismatch containg DNA strand. ATP binding and hydrolysis play a pivotal role in mismatch repair functions. The ATPase activity associated with MutS alpha regulates binding similar to a molecular switch: mismatched DNA provokes ADP-->ATP exchange, resulting in a discernible conformational transition that converts MutS alpha into a sliding clamp capable of hydrolysis-independent diffusion along the DNA backbone. This transition is crucial for mismatch repair. MutS alpha may also play a role in DNA homologous recombination repair. In melanocytes may modulate both UV-B-induced cell cycle regulation and apoptosis. Component of the post-replicative DNA mismatch repair system (MMR). Related diseases Lynch syndrome 1 (LYNCH1) [MIM:120435]: A form of Lynch syndrome, an autosomal dominant disease associated with marked increase in cancer susceptibility. It is characterized by a familial predisposition to early-onset colorectal carcinoma (CRC) and extra-colonic tumors of the gastrointestinal, urological and female reproductive tracts. Lynch syndrome is reported to be the most common form of inherited colorectal cancer in the Western world. Clinically, it is often divided into two subgroups. Type I is characterized by hereditary predisposition to colorectal cancer, a young age of onset, and carcinoma observed in the proximal colon. Type II is characterized by increased risk for cancers in certain tissues such as the uterus, ovary, breast, stomach, small intestine, skin, and larynx in addition to the colon. Diagnosis of classical Lynch syndrome is based on the Amsterdam criteria: 3 or more relatives affected by colorectal cancer, one a first degree relative of the other two; 2 or more generation affected; 1 or more colorectal cancers presenting before 50 years of age; exclusion of hereditary polyposis syndromes. The term 'suspected Lynch syndrome' or 'incomplete Lynch syndrome' can be used to describe families who do not or only partially fulfill the Amsterdam criteria, but in whom a genetic basis for colon cancer is strongly suspected. {ECO:0000269|PubMed:10375096, ECO:0000269|PubMed:10386556, ECO:0000269|PubMed:10528862, ECO:0000269|PubMed:10573010, ECO:0000269|PubMed:10612836, ECO:0000269|PubMed:10777691, ECO:0000269|PubMed:10829038, ECO:0000269|PubMed:11726306, ECO:0000269|PubMed:11870161, ECO:0000269|PubMed:11920458, ECO:0000269|PubMed:12112654, ECO:0000269|PubMed:12124176, ECO:0000269|PubMed:12132870, ECO:0000269|PubMed:12200596, ECO:0000269|PubMed:12362047, ECO:0000269|PubMed:12373605, ECO:0000269|PubMed:12655564, ECO:0000269|PubMed:12655568, ECO:0000269|PubMed:12658575, ECO:0000269|PubMed:14635101, ECO:0000269|PubMed:15046096, ECO:0000269|PubMed:15300854, ECO:0000269|PubMed:15342696, ECO:0000269|PubMed:15365995, ECO:0000269|PubMed:15613555, ECO:0000269|PubMed:15870828, ECO:0000269|PubMed:15896463, ECO:0000269|PubMed:15991316, ECO:0000269|PubMed:15996210, ECO:0000269|PubMed:16451135, ECO:0000269|PubMed:17101317, ECO:0000269|PubMed:17128465, ECO:0000269|PubMed:18561205, ECO:0000269|PubMed:18625694, ECO:0000269|PubMed:18781619, ECO:0000269|PubMed:18822302, ECO:0000269|PubMed:18951462, ECO:0000269|PubMed:21120944, ECO:0000269|PubMed:22102614, ECO:0000269|PubMed:22371642, ECO:0000269|PubMed:7874129, ECO:0000269|PubMed:8261515, ECO:0000269|PubMed:8700523, ECO:0000269|PubMed:8797773, ECO:0000269|PubMed:8872463, ECO:0000269|PubMed:9048925, ECO:0000269|PubMed:9240418, ECO:0000269|PubMed:9298827, ECO:0000269|PubMed:9311737, ECO:0000269|PubMed:9419403, ECO:0000269|PubMed:9559627, ECO:0000269|PubMed:9621522, ECO:0000269|PubMed:9718327, ECO:0000269|PubMed:9889267}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Muir-Torre syndrome (MRTES) [MIM:158320]: Rare autosomal dominant disorder characterized by sebaceous neoplasms and visceral malignancy. {ECO:0000269|PubMed:7713503}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Endometrial cancer (ENDMC) [MIM:608089]: A malignancy of endometrium, the mucous lining of the uterus. Most endometrial cancers are adenocarcinomas, cancers that begin in cells that make and release mucus and other fluids. {ECO:0000305|PubMed:11306449, ECO:0000305|PubMed:21642682}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Mismatch repair cancer syndrome 2 (MMRCS2) [MIM:619096]: An autosomal recessive form of mismatch repair cancer syndrome, a childhood cancer predisposition syndrome encompassing a broad tumor spectrum. This includes hematological malignancies, central nervous system tumors, Lynch syndrome-associated malignancies such as colorectal tumors as well as multiple intestinal polyps, embryonic tumors and rhabdomyosarcoma. Multiple cafe-au-lait macules, a feature reminiscent of neurofibromatosis type 1, are often found as first manifestation of the underlying cancer. {ECO:0000269|PubMed:12549480, ECO:0000269|PubMed:16372347}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Colorectal cancer (CRC) [MIM:114500]: A complex disease characterized by malignant lesions arising from the inner wall of the large intestine (the colon) and the rectum. Genetic alterations are often associated with progression from premalignant lesion (adenoma) to invasive adenocarcinoma. Risk factors for cancer of the colon and rectum include colon polyps, long-standing ulcerative colitis, and genetic family history. {ECO:0000269|PubMed:12792735, ECO:0000269|PubMed:14504054, ECO:0000269|PubMed:15996210, ECO:0000269|PubMed:9559627}. Disease susceptibility may be associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q92624; Q9UQ84-1; P09429; P20585; P52701; Q8IY92; P39875 EC number NA Uniprot keywords 3D-structure; Acetylation; Alternative splicing; ATP-binding; Chromosome; Disease variant; DNA damage; DNA repair; DNA-binding; Hereditary nonpolyposis colorectal cancer; Isopeptide bond; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Tumor suppressor; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 97883.4 Length 868 Aromaticity 0.08 Instability index 36.78 Isoelectric point 5.79 Charge (pH=7) -10.09 2D Binding mode Binding energy (Kcal/mol) -7.63  Molscript Map 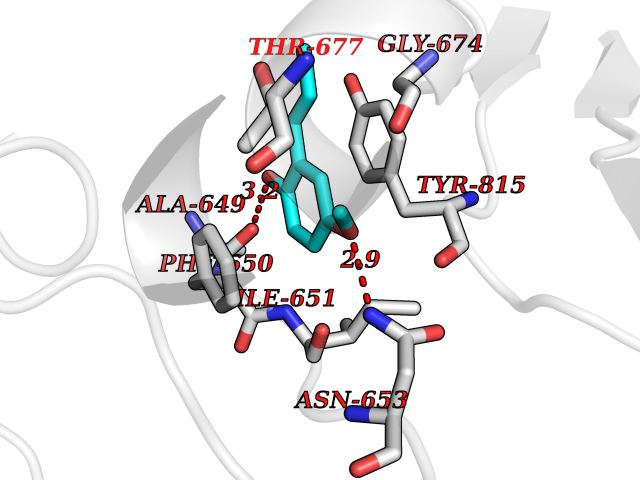 Pymol Map 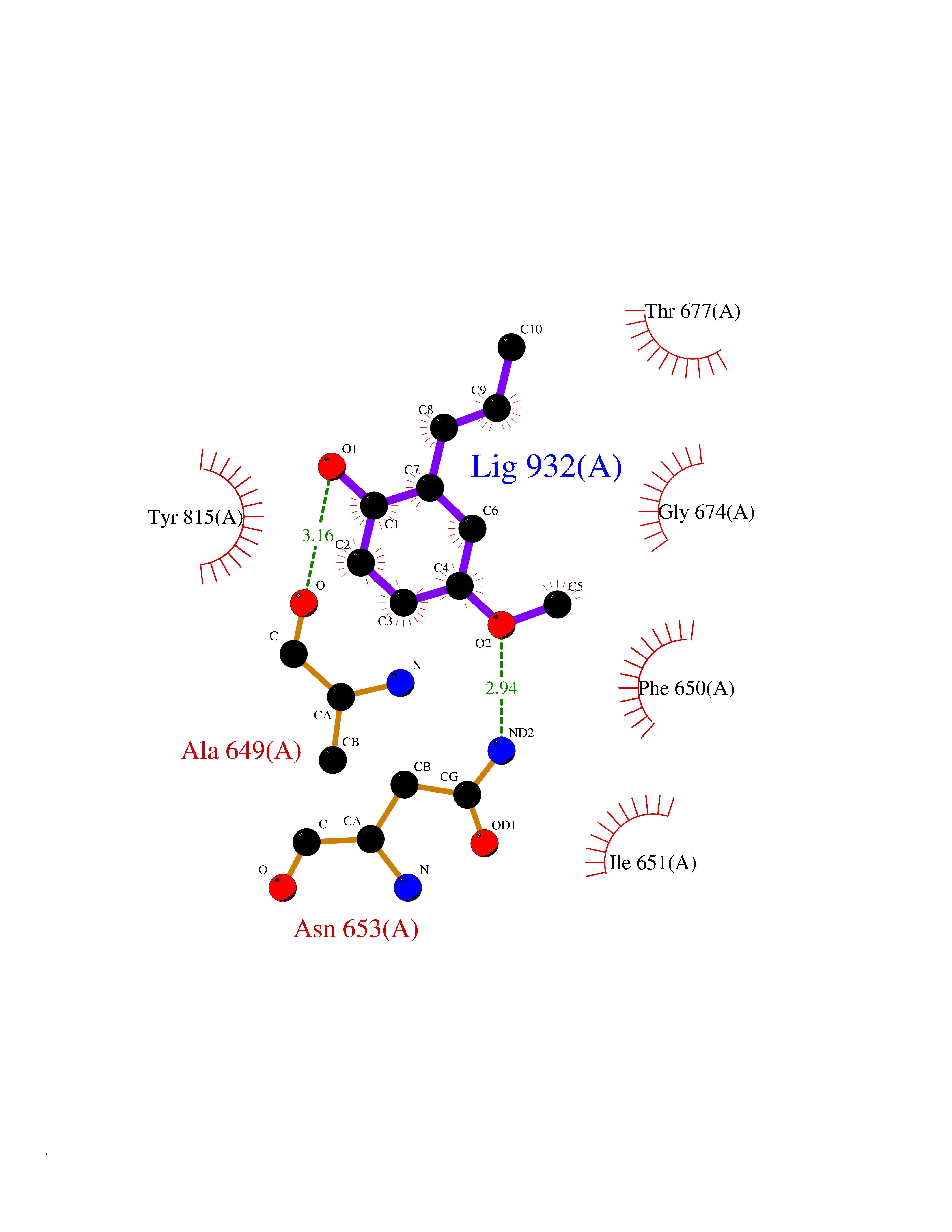 Ligplot Map 3D Binding mode Sequence ESAAEVGFVRFFQGMPEKPTTTVRLFDRGDFYTAHGEDALLAAREVFKTQGVIKYMGPAGAKNLQSVVLSKMNFESFVKDLLLVRQYRVEVYKNRASKENDWYLAYKASPGNLSQFEDILFIGVVGVKMSAVDGQRQVGVGYVDSIQRKLGLCEFPDNDQFSNLEALLIQIGPKECVLPGGETAGDMGKLRQIIQRGGILITERKKADFSTKDIYQDLNRLLKGKKGEQMNSAVLPEMENQVAVSSLSAVIKFLELLSDDSNFGQFELTTFDFSQYMKLDIAAVRALNLFQQSLAALLNKCKTPQGQRLVNQWIKQPLMDKNRIEERLNLVEAFVEDAELRQTLQEDLLRRFPDLNRLAKKFQRQAANLQDCYRLYQGINQLPNVIQALEKHEGKHQKLLLAVFVTPLTDLRSDFSKFQEMIETTLDMDQVENHEFLVKPSFDPNLSELREIMNDLEKKMQSTLISAARDLGLDPGKQIKLDSSAGYYFRVTCKEEKVLRNNKNFSTVDIQGVKFTNSKLTSLNEEYTKNKTEYEEAQDAIVKEIVNISSGYVEPMQTLNDVLAQLDAVVSFAHVSNGAPVPYVRPAILEKGQGRIILKASRHACVEVQIAFIPNDVYFEKDKQMFHIITGPNMGGKSTYIRQTGVIVLMAQIGCFVPCESAEVSIVDCILARVGSTFMAEMLETASILRSATKDSLIIIDELGRGTSTYDGFGLAWAISEYIATKIGAFCMFATHFHELTALANQIPTVNNLHVTALTTEETLTMLYQVKKGVCDQSFGIHVAELANFPKHVIECAKQKALELEEFQYKCYLEREQGEKIIQEFLSKVKQMPFTEMSEENITIKLKQLKAEVIAKNNSFVNEIISRI Hydrogen bonds contact Hydrophobic contact | ||||
| 93 | Neuronal acetylcholine receptor alpha-4 (CHRNA4) | 6CNJ | 5.59 | |
Target general information Gen name CHRNA4 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Nicotinic acetylcholine receptor alpha4; CHRNA4; Alpha-4 nAChR Protein family Ligand-gated ion channel (TC 1.A.9) family, Acetylcholine receptor (TC 1.A.9.1) subfamily, Alpha-4/CHRNA4 sub-subfamily Biochemical class Neurotransmitter receptor Function After binding acetylcholine, the AChR responds by an extensive change in conformation that affects all subunits and leads to opening of an ion-conducting channel across the plasmamembrane permeable to sodium ions. Related diseases Epilepsy, nocturnal frontal lobe, 1 (ENFL1) [MIM:600513]: An autosomal dominant focal epilepsy characterized by nocturnal seizures with hyperkinetic automatisms and poorly organized stereotyped movements. {ECO:0000269|PubMed:10563623, ECO:0000269|PubMed:14623738, ECO:0000269|PubMed:7550350}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00915; DB01351; DB01352; DB00572; DB01483; DB00237; DB00241; DB01353; DB00564; DB00565; DB09028; DB01245; DB00514; DB01496; DB07720; DB00783; DB13952; DB13953; DB13954; DB13955; DB13956; DB00898; DB01354; DB01355; DB00753; DB00657; DB00333; DB00463; DB00849; DB00184; DB00312; DB01174; DB00981; DB05458; DB00794; DB05740; DB00747; DB00418; DB00202; DB00306; DB00599; DB01273 Interacts with Q6UY14-3; P05067; P83916; Q6UXH1-1; Q6UXH1-3; P20042; Q9NZR2; Q92673; P17787 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disease variant; Disulfide bond; Epilepsy; Glycoprotein; Ion channel; Ion transport; Ligand-gated ion channel; Lipoprotein; Membrane; Palmitate; Phosphoprotein; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Signal; Synapse; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A,B Molecular weight (Da) 84601.2 Length 728 Aromaticity 0.13 Instability index 39.72 Isoelectric point 5.86 Charge (pH=7) -9.84 2D Binding mode Binding energy (Kcal/mol) -7.63 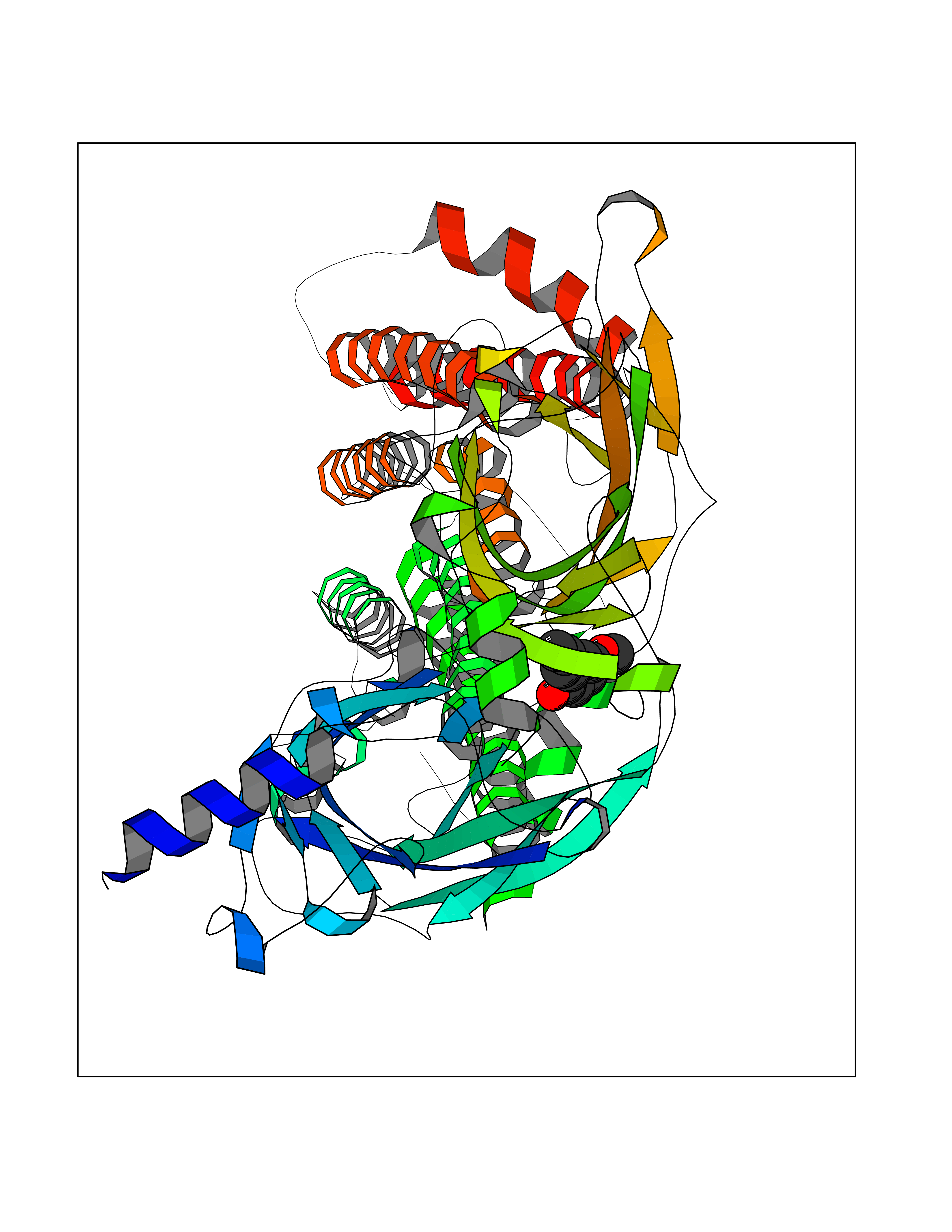 Molscript Map 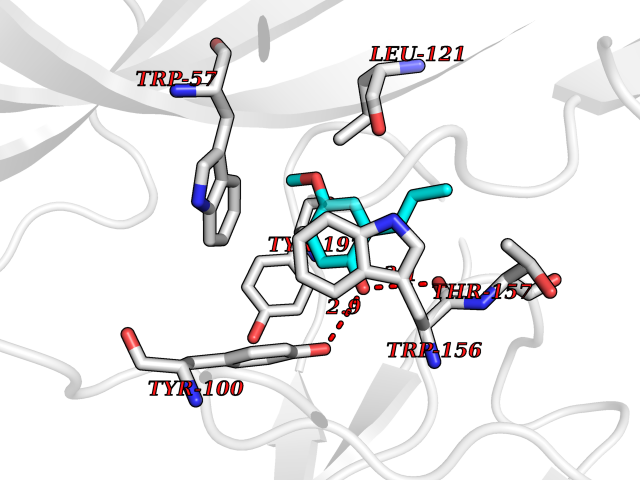 Pymol Map 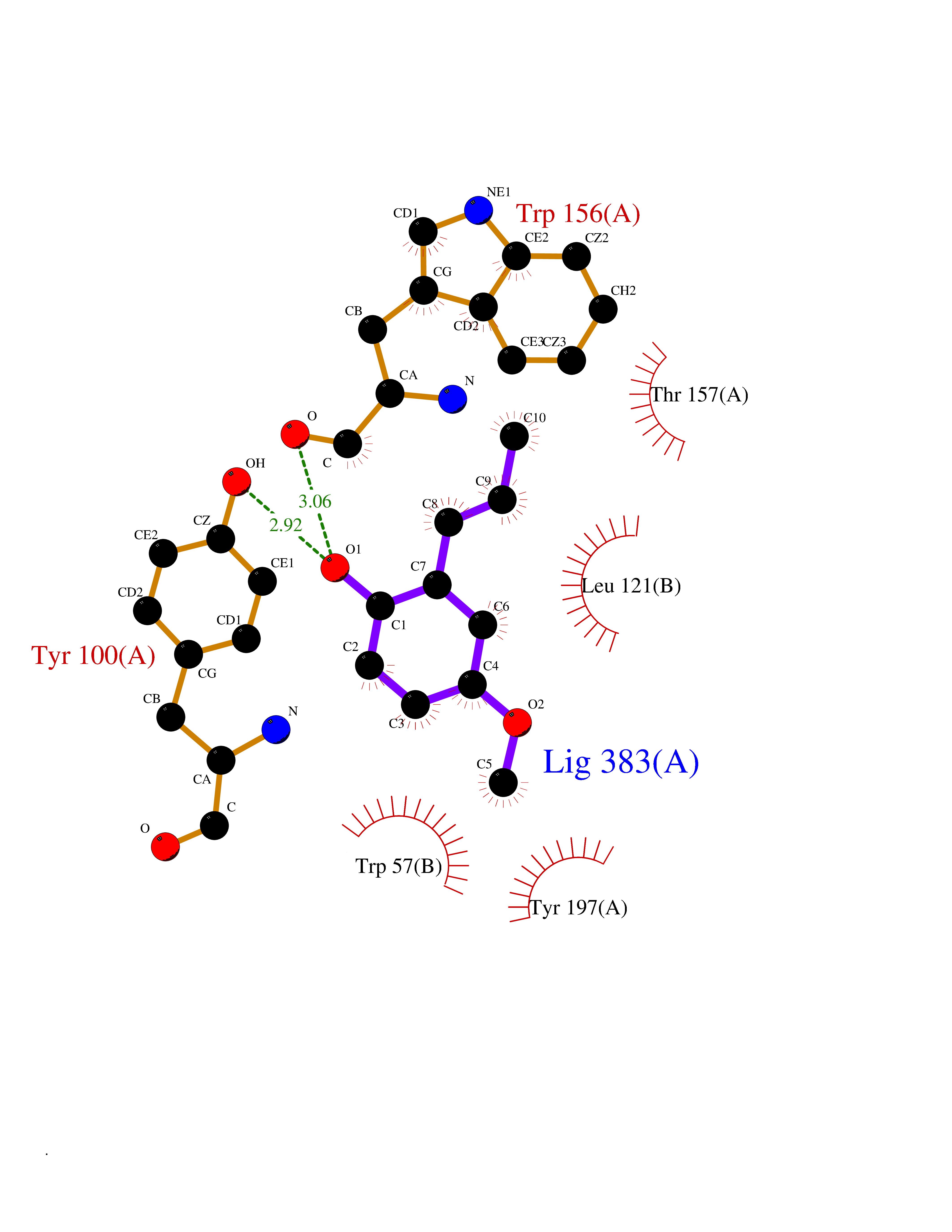 Ligplot Map 3D Binding mode Sequence ETRAHAEERLLKKLFSGYNKWSRPVANISDVVLVRFGLSIAQLIDVDEKNQMMTTNVWVKQEWHDYKLRWDPADYENVTSIRIPSELIWRPDIVLYNNADGDFAVTHLTKAHLFHDGRVQWTPPAIYKSSCSIDVTFFPFDQQNCTMKFGSWTYDKAKIDLVNMHSRVDQLDFWESGEWVIVDAVGTYNTRKYECCAEIYPDITYAFVIRRLPLFYTINLIIPCLLISCLTVLVFYLPSECGEKITLCISVLLSLTVFLLLITEIIPSTSLVIPLIGEYLLFTMIFVTLSIVITVFVLNVHHRSPRTHTMPTWVRRVFLDIVPRLLLMKRFERSVKEDWKYVAMVIDRIFLWMFIIVCLLGTVGLFLPPWDTEERLVEHLLDPSRYNKLIRPATNGSELVTVQLMVSLAQLISVHEREQIMTTNVWLTQEWEDYRLTWKPEEFDNMKKVRLPSKHIWLPDVVLYNNADGMYEVSFYSNAVVSYDGSIFWLPPAIYKSACKIEVKHFPFDQQNCTMKFRSWTYDRTEIDLVLKSEVASLDDFTPSGEWDIVALPGRRNENPDDSTYVDITYDFIIRRKPLFYTINLIIPCVLITSLAILVFYLPSDCGEKMTLCISVLLALTVFLLLISKIVPPTSLDVPLVGKYLMFTMVLVTFSIVTSVCVLNVHHRSPTTHTMAPWVKVVFLEKLPALLFMQQSVSEDWKYVAMVIDRLFLWIFVFVCVFGTIGMF Hydrogen bonds contact Hydrophobic contact | ||||
| 94 | Neuronal acetylcholine receptor beta-2 (CHRNB2) | 6CNJ | 5.59 | |
Target general information Gen name CHRNB2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Nicotinic acetylcholine receptor beta2; Nicotinic acetylcholine receptor beta 2-subunit protein; CHRNB2; Beta-2 nAChR; Alpha-4/beta-2 nicotinic receptor Protein family Ligand-gated ion channel (TC 1.A.9) family, Acetylcholine receptor (TC 1.A.9.1) subfamily, Beta-2/CHRNB2 sub-subfamily Biochemical class Neurotransmitter receptor Function After binding acetylcholine, the AChR responds by an extensive change in conformation that affects all subunits and leads to opening of an ion-conducting channel across the plasma membrane permeable to sodiun ions. Related diseases Epilepsy, nocturnal frontal lobe, 3 (ENFL3) [MIM:605375]: An autosomal dominant focal epilepsy characterized by nocturnal seizures with hyperkinetic automatisms and poorly organized stereotyped movements. {ECO:0000269|PubMed:11062464, ECO:0000269|PubMed:11104662}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00572; DB00237; DB00565; DB09028; DB01245; DB00514; DB07720; DB00898; DB00753; DB00657; DB00333; DB00184; DB00981; DB05458; DB05855; DB05740; DB00747; DB00202; DB01273 Interacts with P43681-1; P30532 EC number NA Uniprot keywords 3D-structure; Cell membrane; Disease variant; Disulfide bond; Epilepsy; Glycoprotein; Ion channel; Ion transport; Ligand-gated ion channel; Membrane; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Signal; Synapse; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A,B Molecular weight (Da) 84601.2 Length 728 Aromaticity 0.13 Instability index 39.72 Isoelectric point 5.86 Charge (pH=7) -9.84 2D Binding mode Binding energy (Kcal/mol) -7.63 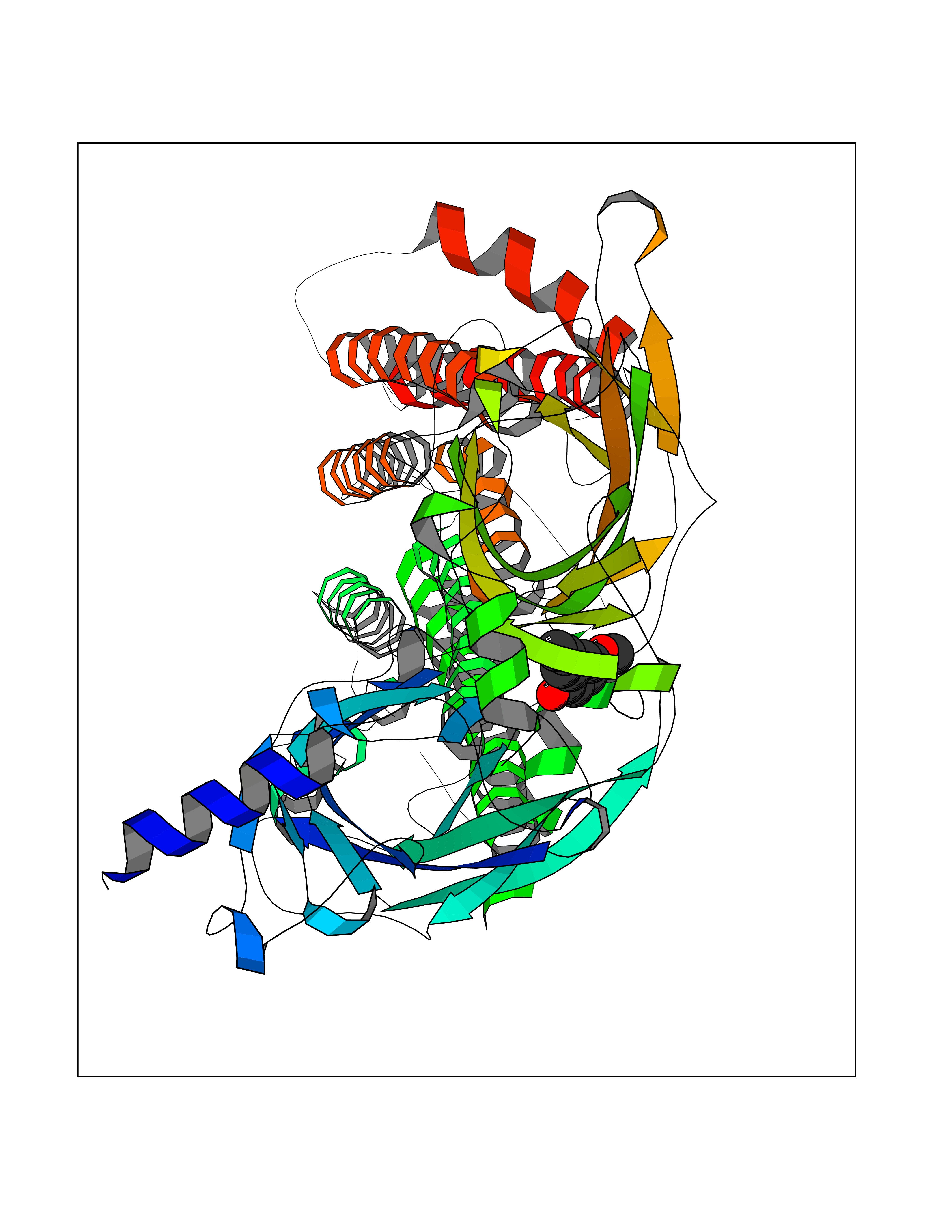 Molscript Map 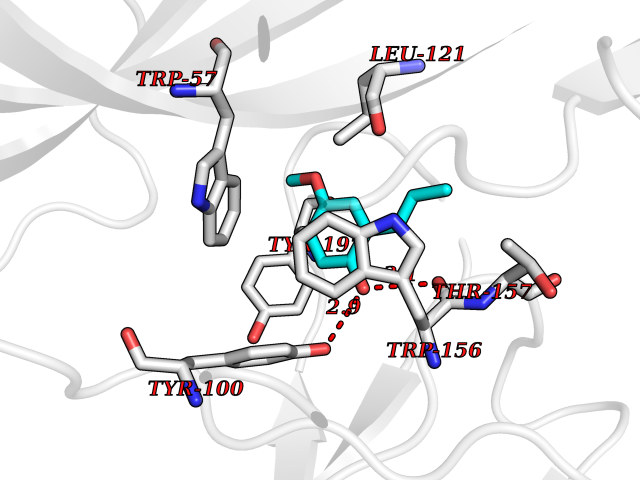 Pymol Map 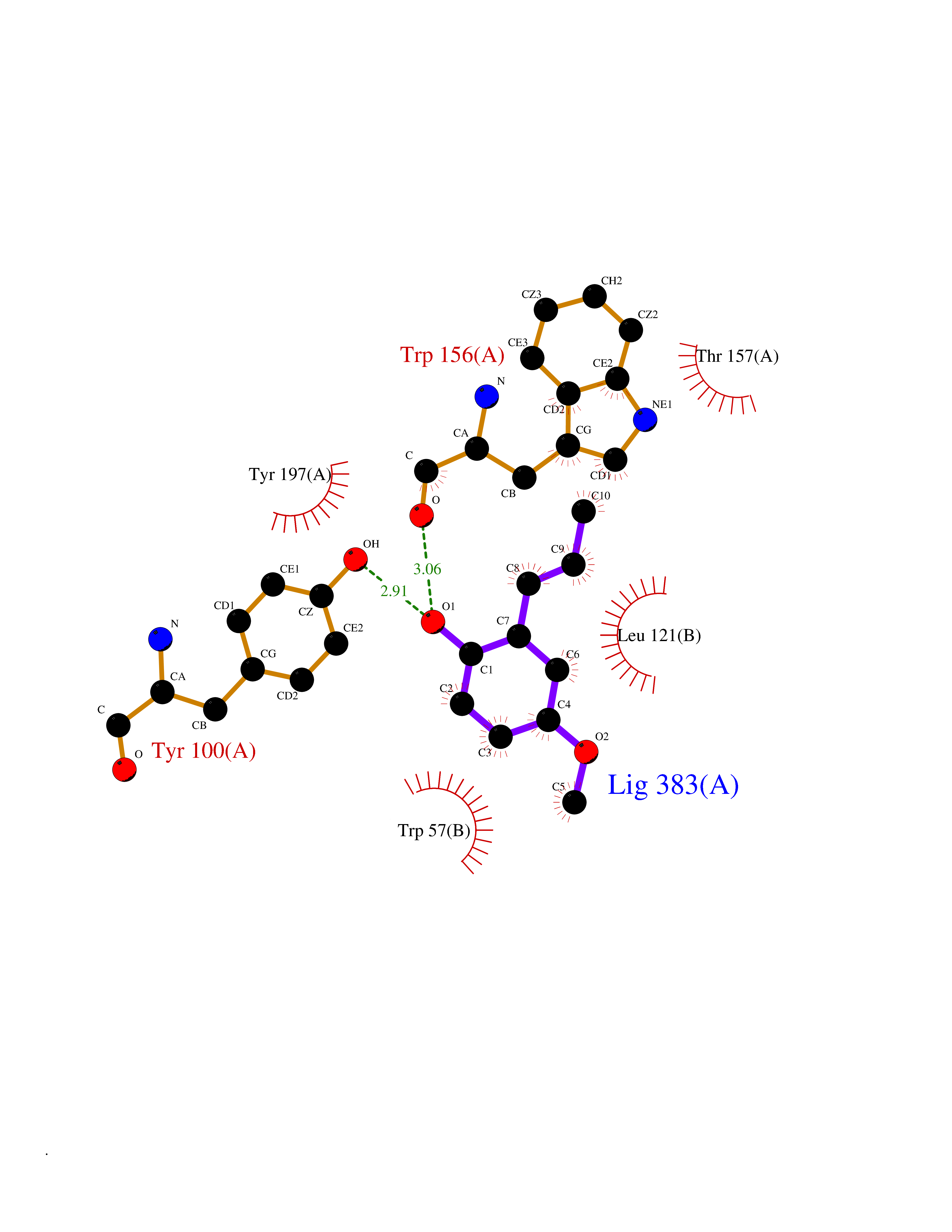 Ligplot Map 3D Binding mode Sequence ETRAHAEERLLKKLFSGYNKWSRPVANISDVVLVRFGLSIAQLIDVDEKNQMMTTNVWVKQEWHDYKLRWDPADYENVTSIRIPSELIWRPDIVLYNNADGDFAVTHLTKAHLFHDGRVQWTPPAIYKSSCSIDVTFFPFDQQNCTMKFGSWTYDKAKIDLVNMHSRVDQLDFWESGEWVIVDAVGTYNTRKYECCAEIYPDITYAFVIRRLPLFYTINLIIPCLLISCLTVLVFYLPSECGEKITLCISVLLSLTVFLLLITEIIPSTSLVIPLIGEYLLFTMIFVTLSIVITVFVLNVHHRSPRTHTMPTWVRRVFLDIVPRLLLMKRFERSVKEDWKYVAMVIDRIFLWMFIIVCLLGTVGLFLPPWDTEERLVEHLLDPSRYNKLIRPATNGSELVTVQLMVSLAQLISVHEREQIMTTNVWLTQEWEDYRLTWKPEEFDNMKKVRLPSKHIWLPDVVLYNNADGMYEVSFYSNAVVSYDGSIFWLPPAIYKSACKIEVKHFPFDQQNCTMKFRSWTYDRTEIDLVLKSEVASLDDFTPSGEWDIVALPGRRNENPDDSTYVDITYDFIIRRKPLFYTINLIIPCVLITSLAILVFYLPSDCGEKMTLCISVLLALTVFLLLISKIVPPTSLDVPLVGKYLMFTMVLVTFSIVTSVCVLNVHHRSPTTHTMAPWVKVVFLEKLPALLFMQQSVSEDWKYVAMVIDRLFLWIFVFVCVFGTIGMF Hydrogen bonds contact Hydrophobic contact | ||||
| 95 | Mutated Histone H3.3 (H3F3A) | 4GUS | 5.59 | |
Target general information Gen name H3F3A Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms PP781; Histone H3.3; H3F3; H3.3B; H3.3A Protein family Histone H3 family Biochemical class NA Function Variant histone H3 which replaces conventional H3 in a wide range of nucleosomes in active genes. Constitutes the predominant form of histone H3 in non-dividing cells and is incorporated into chromatin independently of DNA synthesis. Deposited at sites of nucleosomal displacement throughout transcribed genes, suggesting that it represents an epigenetic imprint of transcriptionally active chromatin. Nucleosomes wrap and compact DNA into chromatin, limiting DNA accessibility to the cellular machineries which require DNA as a template. Histones thereby play a central role in transcription regulation, DNA repair, DNA replication and chromosomal stability. DNA accessibility is regulated via a complex set of post-translational modifications of histones, also called histone code, and nucleosome remodeling. Related diseases Glioma (GLM) [MIM:137800]: Gliomas are benign or malignant central nervous system neoplasms derived from glial cells. They comprise astrocytomas and glioblastoma multiforme that are derived from astrocytes, oligodendrogliomas derived from oligodendrocytes and ependymomas derived from ependymocytes. {ECO:0000269|PubMed:22286061, ECO:0000269|PubMed:22286216, ECO:0000269|PubMed:23539269}. The gene represented in this entry is involved in disease pathogenesis. H3F3A mutations affecting residues involved in post-translational modifications of histone H3.3 are recurrent in malignant, aggressive gliomas including glioblastoma multiforme (GBM) and diffuse intrinsic pontine glioma (DIPG) (PubMed:22286061, PubMed:22286216). The mechanism through which mutations lead to tumorigenesis involves altered histones methylation, impaired regulation of Polycomb repressive complex 2 (PRC2) activity, and aberrant epigenetic regulation of gene expression (PubMed:23539183, PubMed:23539269, PubMed:23603901). {ECO:0000269|PubMed:22286061, ECO:0000269|PubMed:22286216, ECO:0000269|PubMed:23539183, ECO:0000269|PubMed:23539269, ECO:0000269|PubMed:23603901}.; DISEASE: Bryant-Li-Bhoj neurodevelopmental syndrome 1 (BRYLIB1) [MIM:619720]: An autosomal dominant disorder predominantly characterized by global developmental delay, impaired intellectual development, poor or absent speech, and delayed motor milestones. Clinical manifestations are highly variable, including abnormal head shape, dysmorphic facial features, oculomotor abnormalities, feeding problems, and non-specific brain imaging abnormalities. Additional features may include hearing loss, seizures, short stature, and mild skeletal defects. {ECO:0000269|PubMed:33268356, ECO:0000269|PubMed:34876591}. The disease is caused by variants affecting the gene represented in this entry. BRYLIB1 is caused by variants in H3-3A. {ECO:0000269|PubMed:33268356, ECO:0000269|PubMed:34876591}.; DISEASE: Bryant-Li-Bhoj neurodevelopmental syndrome 2 (BRYLIB2) [MIM:619721]: An autosomal dominant disorder predominantly characterized by global developmental delay, impaired intellectual development, poor or absent speech, and delayed motor milestones. Clinical manifestations are highly variable, including abnormal head shape, dysmorphic facial features, oculomotor abnormalities, feeding problems, and non-specific brain imaging abnormalities. Additional features may include hearing loss, seizures, short stature, and mild skeletal defects. {ECO:0000269|PubMed:33268356, ECO:0000269|PubMed:34876591}. The disease is caused by variants affecting the gene represented in this entry. BRYLIB2 is caused by variants in H3-3B. {ECO:0000269|PubMed:33268356, ECO:0000269|PubMed:34876591}.; DISEASE: H3F3A and H3F3B mutations affecting residues involved in post-translational modifications of histone H3.3 are implicated in the pathogenesis of some bone and cartilage neoplasms. Mutations have been found with high prevalence in chondroblastoma and giant cell tumors of bone, and with low frequency in osteosarcoma, conventional chondrosarcoma and clear cell chondrosarcoma. Chondroblastoma samples frequently carry a H3F3B mutation affecting residue Lys-37 (H3K36), although H3F3A is mutated in some cases. Most giant cell tumors of bone harbor H3F3A mutations affecting residue Gly-35 (H3G34). {ECO:0000269|PubMed:24162739}. Drugs (DrugBank ID) NA Interacts with Q9NVP2; P45973; Q13111; Q9UER7; Q9UER7-1; Q9Y6K1; P62805; P49321-2; Q8IZL8; Q5VWG9; Q9VK33; Q8R5C8 EC number NA Uniprot keywords 3D-structure; Acetylation; ADP-ribosylation; Chromosome; Citrullination; Direct protein sequencing; Disease variant; DNA-binding; Hydroxylation; Intellectual disability; Lipoprotein; Methylation; Nucleosome core; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Ubl conjugation Protein physicochemical properties Chain ID A,C Molecular weight (Da) 86148.9 Length 766 Aromaticity 0.1 Instability index 37.57 Isoelectric point 8.25 Charge (pH=7) 7.16 2D Binding mode Binding energy (Kcal/mol) -7.63 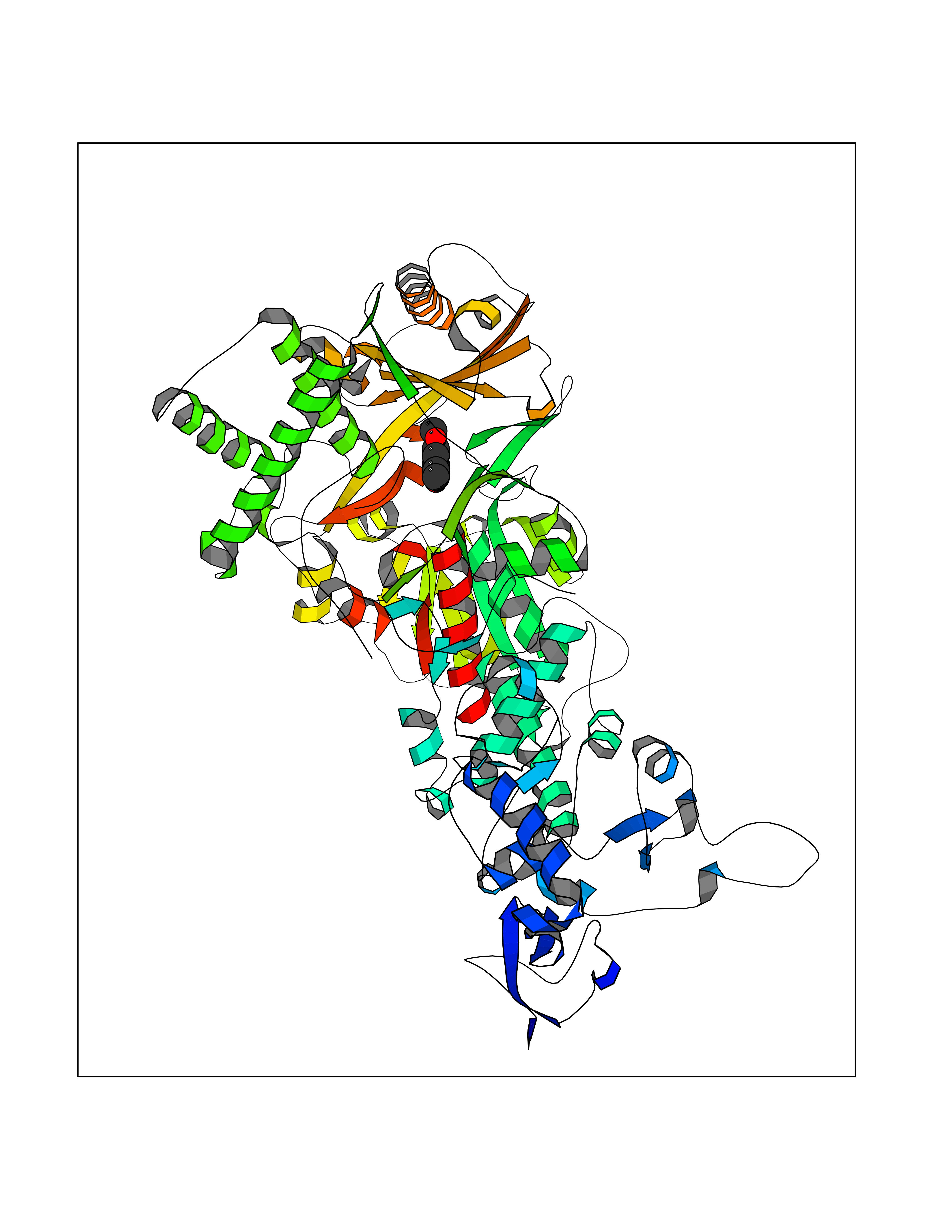 Molscript Map 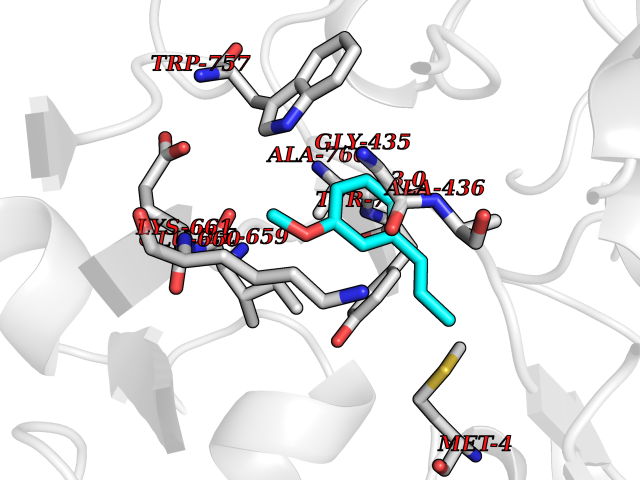 Pymol Map 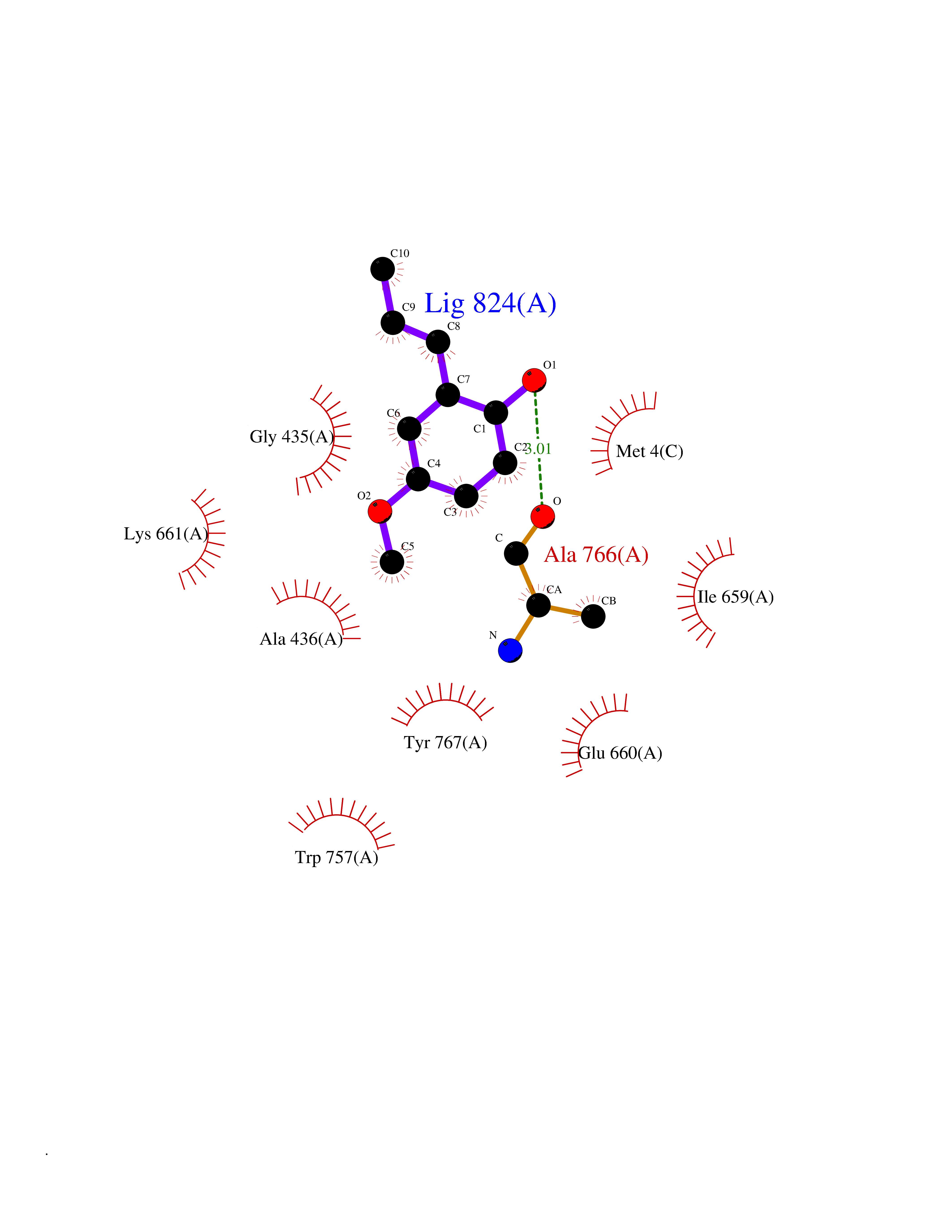 Ligplot Map 3D Binding mode Sequence GSRKCEKAGCTATCPVCFASASERCAKNGYTSRWYHLSCGEHFCNECFDHYYRSHKDGYDKYTTWKKIWTSNGKTEPSPKAFMADQQLPYWVQCTKPECRKWRQLTKEIQLTPQIAKTYRCGMKPNTAIKPETSDHCSLPEDLRVLEVSNHWWYSMLILPPLLKDSVAAPLLSAYYPDCVGMSPSCTGMNRYFQPFYQPNECGKALCVRPDVMELDELYEFPEYSRDPTMYLALRNLILALWYTNCKEALTPQKCIPHIIVRGLVRIRCVQEVERILYFMTRKGLINTGVLSVGADQYLLPKDYHNKSVIIIGAGPAGLAAARQLHNFGIKVTVLEAKDRIGGRVWDDKSFKGVTVGRGAQIVNGCINNPVALMCEQLGISMHKFGERCDLIQEGGRITDPTIDKRMDFHFNALLDVVSEWRKDKTQLQDVPLGEKIEEIYKAFIKESGIQFSELEGQVLQFHLSNLEYACGSNLHQVSARSWDHNEFFAQFAGDHTLLTPGYSVIIEKLAEGLDIQLKSPVQCIDYSGDEVQVTTTDGTGYSAQKVLVTVPLALLQKGAIQFNPPLSEKKMKAINSLGAGIIEKIALQFPYRFWDSKVQGADFFGHVPPSASKRGLFAVFYDMDPQKKHSVLMSVIAGEAVASVRTLDDKQVLQQCMATLRELFKEQEVPDPTKYFVTRWSTDPWIQMAYSFVKTGGSGEAYDIIAEDIQGTVFFAGEATNRHFPQTVTGAYLSGVREASKIAAFARTMQTARKSTGGKAPRKQL Hydrogen bonds contact Hydrophobic contact | ||||
| 96 | Dual-specificity tyrosine-phosphorylation regulated kinase 3 (DYRK3) | 5Y86 | 5.59 | |
Target general information Gen name DYRK3 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Regulatory erythroid kinase; REDK; Dual specificity tyrosine-phosphorylation-regulated kinase 3 Protein family Protein kinase superfamily, CMGC Ser/Thr protein kinase family, MNB/DYRK subfamily Biochemical class Kinase Function Dual-specificity tyrosine-regulated kinases (DYRKs) autophosphorylate a critical tyrosine residue in their activation loop and phosphorylate their substrate on serine and threonine residues. Acts as a central dissolvase of membraneless organelles during the G2-to-M transition, after the nuclear-envelope breakdown: acts by mediating phosphorylation of multiple serine and threonine residues in unstructured domains of proteins, such as SRRM1 and PCM1. Does not mediate disassembly of all membraneless organelles: disassembly of P-body and nucleolus is not regulated by DYRK3. Dissolution of membraneless organelles at the onset of mitosis is also required to release mitotic regulators, such as ZNF207, from liquid-unmixed organelles where they are sequestered and keep them dissolved during mitosis. Regulates mTORC1 by mediating the dissolution of stress granules: during stressful conditions, DYRK3 partitions from the cytosol to the stress granule, together with mTORC1 components, which prevents mTORC1 signaling. When stress signals are gone, the kinase activity of DYRK3 is required for the dissolution of stress granule and mTORC1 relocation to the cytosol: acts by mediating the phosphorylation of the mTORC1 inhibitor AKT1S1, allowing full reactivation of mTORC1 signaling. Also acts as a negative regulator of EPO-dependent erythropoiesis: may place an upper limit on red cell production during stress erythropoiesis. Inhibits cell death due to cytokine withdrawal in hematopoietic progenitor cells. Promotes cell survival upon genotoxic stress through phosphorylation of SIRT1: this in turn inhibits p53/TP53 activity and apoptosis. Dual-specificity protein kinase that promotes disassembly of several types of membraneless organelles during mitosis, such as stress granules, nuclear speckles and pericentriolar material. Related diseases Defects in MELK are associated with some cancers, such as brain or breast cancers. Expression is dramatically increased in aggressive undifferentiated tumors, correlating with poor patient outcome in breast and brain cancers, suggesting a role in tumor-initiating cells and proliferation via its function in cell proliferation regulation. Drugs (DrugBank ID) NA Interacts with Q9H8Y8 EC number EC 2.7.12.1 Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Cell cycle; Cell division; Cytoplasm; Cytoskeleton; Kinase; Magnesium; Metal-binding; Mitosis; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Serine/threonine-protein kinase; Transferase; Tyrosine-protein kinase; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 44821.5 Length 395 Aromaticity 0.1 Instability index 49.38 Isoelectric point 9.52 Charge (pH=7) 21.08 2D Binding mode Binding energy (Kcal/mol) -7.62  Molscript Map 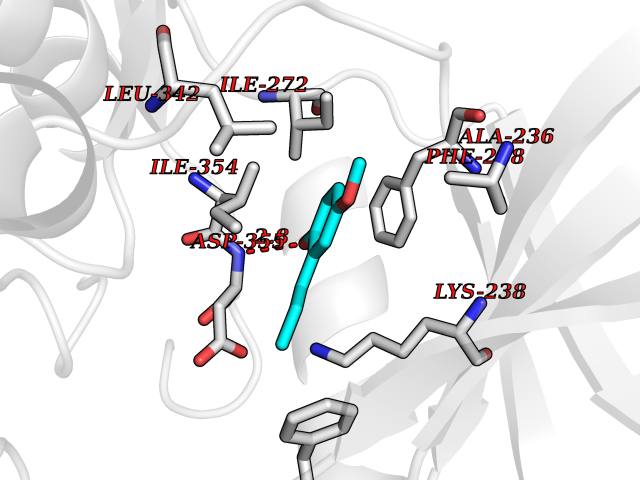 Pymol Map 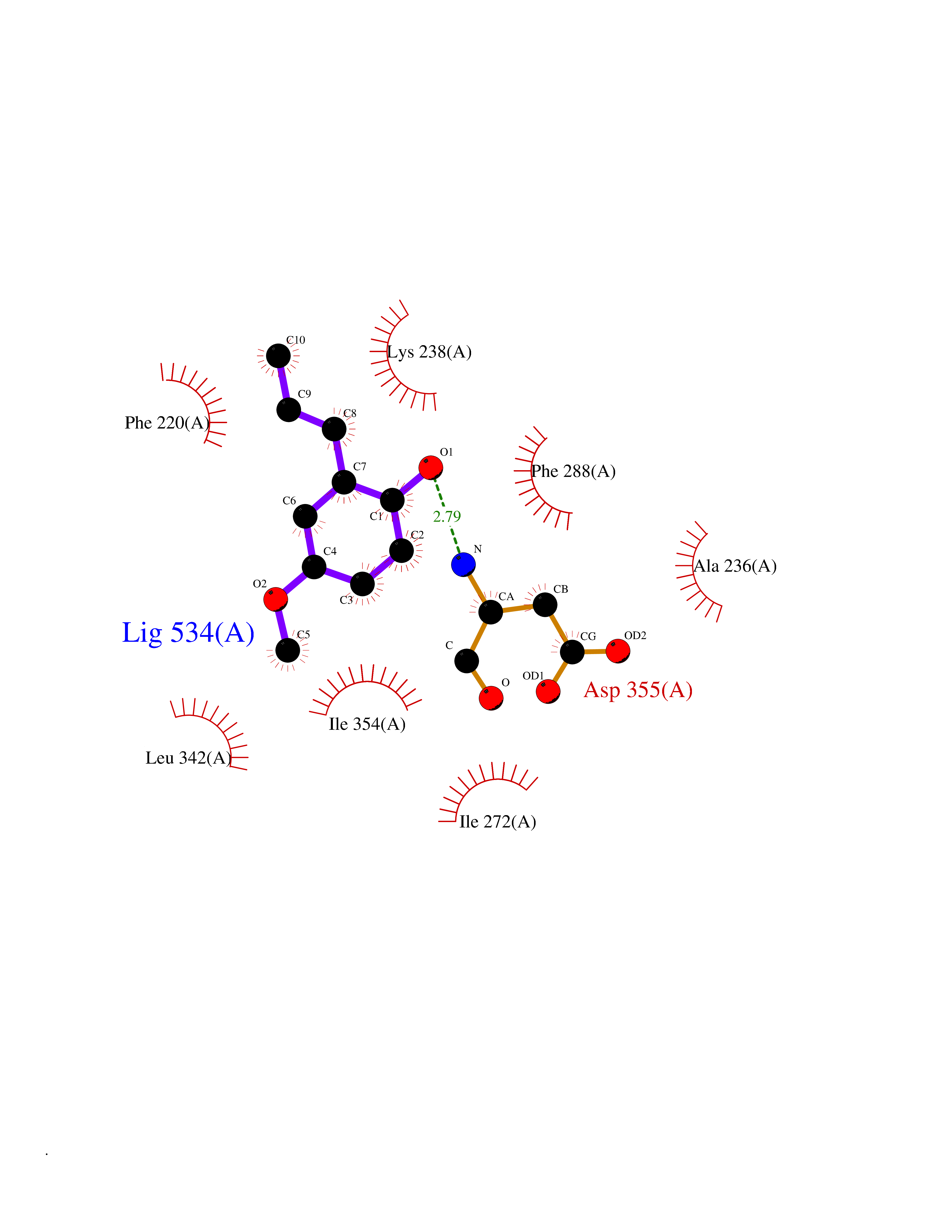 Ligplot Map 3D Binding mode Sequence VVPLTPEQALKQYKHHLTAYEKLEIINYPEIYFVGPNAKKRHGVIGGPNNGGYDDADGAYIHVPRDHLAYRYEVLKIIGKGSFGQVARVYDHKLRQYVALKMVRNEKRFHRQAAEEIRILEHLKKQDKTGSMNVIHMLESFTFRNHVCMAFELLSIDLYELIKKNKFQGFSVQLVRKFAQSILQSLDALHKNKIIHCDLKPENILLKHHGRSXTKVIDFGSSCFEYQKLYTXIQSRFYRAPEIILGSRYSTPIDIWSFGCILAELLTGQPLFPGEDEGDQLACMMELLGMPPPKLLEQSKRAKYFINXKGIPRYCSVTTQADGRVVLVGGRSRRGKKRGPPGSKDWGTALKGCDDYLFIEFLKRCLHWDPSARLXPAQALRHPWISKSVPRPLTT Hydrogen bonds contact Hydrophobic contact | ||||
| 97 | Neuronal acetylcholine receptor subunit alpha-3 | 4ZK4 | 5.58 | |
Target general information Gen name CHRNA3 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NACHRA3 Protein family Ligand-gated ion channel (TC 1.A.9) family, Acetylcholine receptor (TC 1.A.9.1) subfamily, Alpha-3/CHRNA3 sub-subfamily Biochemical class Acetylcholine binding protein Function Acetylcholine binding.Acetylcholine-gated cation-selective channel activity.Acetylcholine receptor activity.Ligand-gated ion channel activity.Serotonin-gated cation-selective channel activity. Related diseases Bladder dysfunction, autonomic, with impaired pupillary reflex and secondary CAKUT (BAIPRCK) [MIM:191800]: An autosomal recessive disease characterized by impaired innervation and autonomic dysfunction of the urinary bladder, hydronephrosis, vesicoureteral reflux, small kidneys, recurrent urinary tract infections, and progressive renal insufficiency. Additional autonomic features are impaired pupillary reflex and orthostatic hypotension. The disease manifests in utero or early childhood. {ECO:0000269|PubMed:31708116}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00915; DB01156; DB00237; DB00565; DB09028; DB00514; DB07720; DB00898; DB00472; DB05710; DB01227; DB00848; DB00333; DB00184; DB01090; DB00202; DB01273 Interacts with NA EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disease variant; Disulfide bond; Endoplasmic reticulum; Glycoprotein; Golgi apparatus; Ion channel; Ion transport; Ligand-gated ion channel; Membrane; Phosphoprotein; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Signal; Synapse; Transmembrane; Transmembrane helix; Transport; Ubl conjugation Protein physicochemical properties Chain ID A,B,C,D,E Molecular weight (Da) 46391.5 Length 408 Aromaticity 0.12 Instability index 30.23 Isoelectric point 4.6 Charge (pH=7) -22.73 2D Binding mode Binding energy (Kcal/mol) -7.61 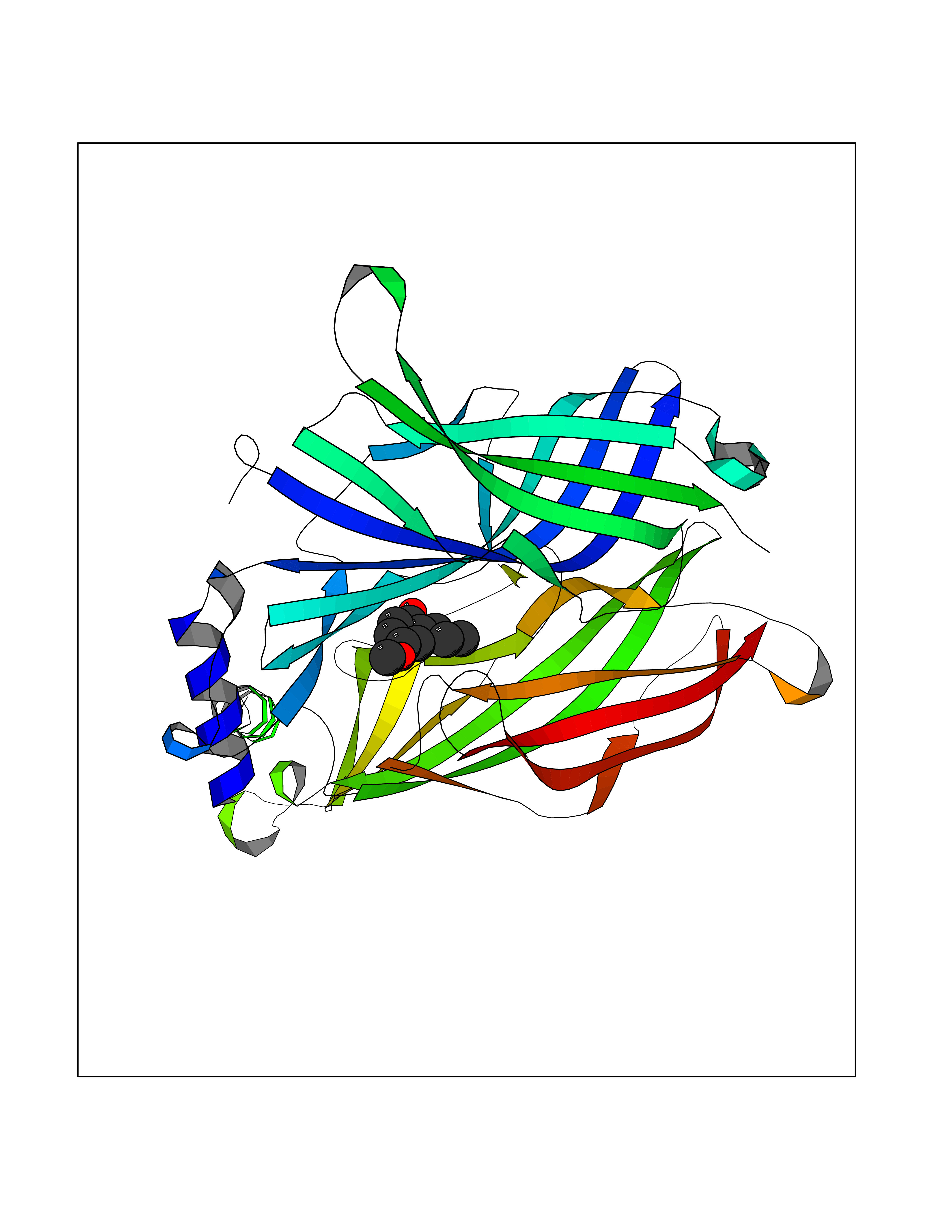 Molscript Map 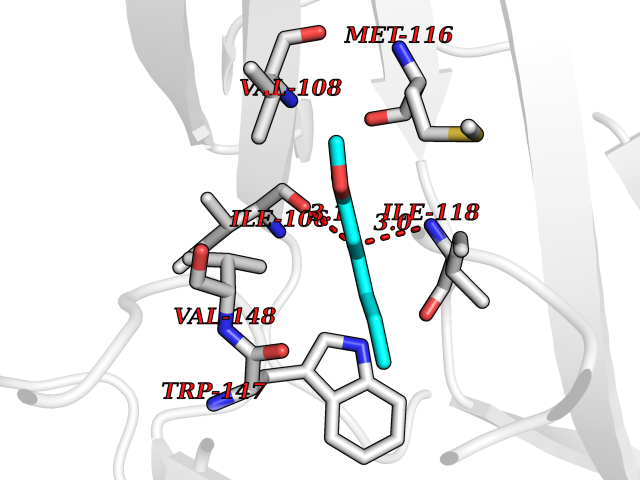 Pymol Map 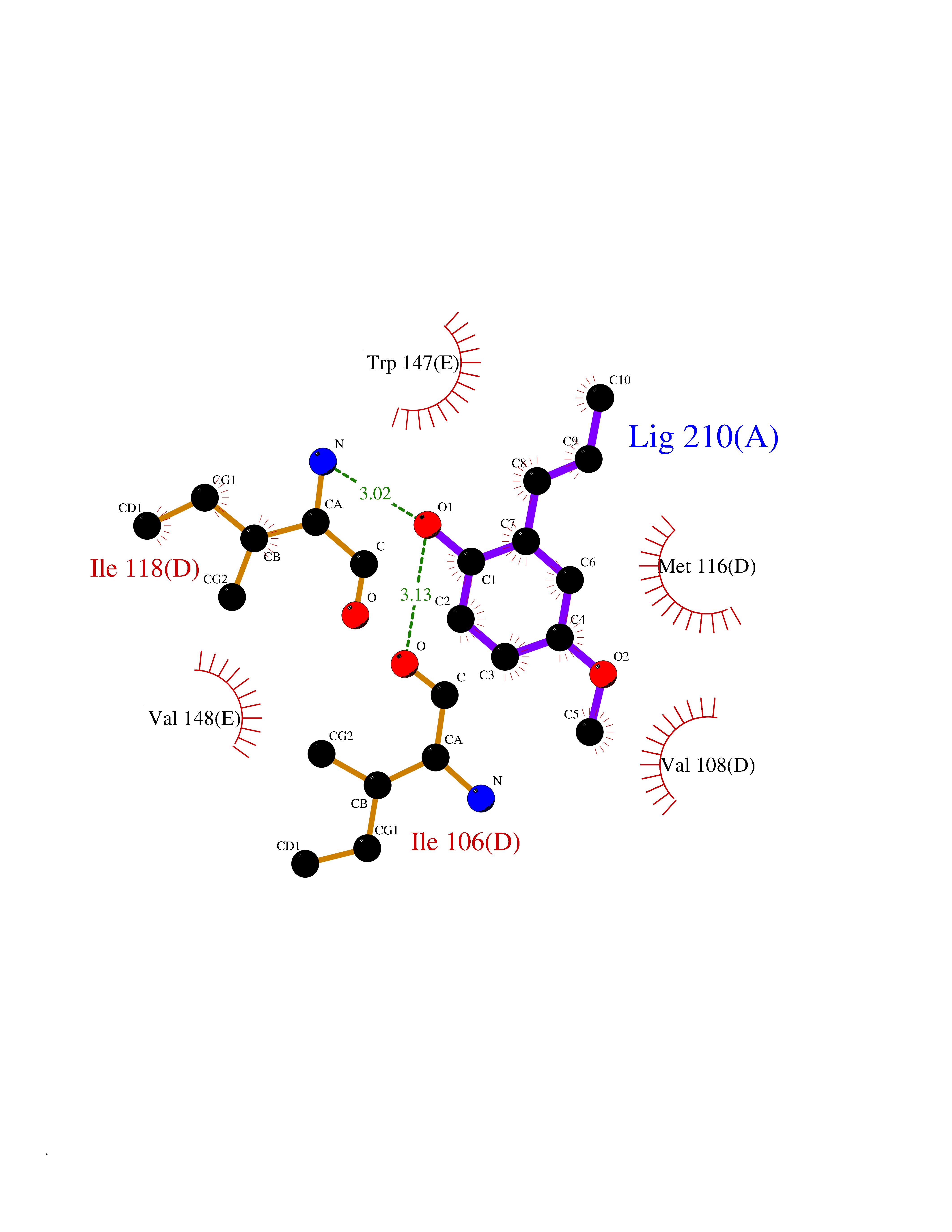 Ligplot Map 3D Binding mode Sequence LHSQANLMRLKSDLFYPGPTKDDPLTVTLGFTLQDIVKADSSTNEVDLVYWEQQRWKLNSLMWDPNEYGNITDFRTSAADIWTPDITAYSSTRPVQVLSPQIAVVTHDGSVMFIPAQRLSFMCDPTGVDSEEGATCAVKFGSWVYSGFEIDLKTDTDQVDLSSYYASSKYEILSATQYKHDIKYNCCEEIYPDVVLVVKFRERRLHSQANLMRLKSDLFNRYPGPTKDDPLTVTLGFTLQDIVKADSSTNEVDLVYWEQQRWKLNSLMWDPNEYGNITDFRTSAADIWTPDITAYSSTRPVQVLSPQIAVVTHDGSVMFIPAQRLSFMCDPTGVDSEEGATCAVKFGSWVYSGFEIDLKTDTDQVDLSSYYASSKYEILSATQYKHDIKYNCCEEIYPDVVLVVKFRE Hydrogen bonds contact Hydrophobic contact | ||||
| 98 | O-GlcNAcase BT_4395 | 5FKY | 5.58 | |
Target general information Gen name BT_4395 Organism Bacteroides thetaiotaomicron (strain ATCC 29148 / DSM 2079 / JCM 5827 / CCUG 10774 / NCTC 10582 / VPI-5482 / E50) Uniprot ID TTD ID NA Synonyms NA Protein family Glycosyl hydrolase 84 family Biochemical class Hydrolase Function [protein]-3-O-(N-acetyl-D-glucosaminyl)-L-serine/L-threonine O-N-acetyl-alpha-D-glucosaminase activity.[protein]-3-O-(N-acetyl-D-glucosaminyl)-L-serine O-N-acetyl-alpha-D-glucosaminase activity.[protein]-3-O-(N-acetyl-D-glucosaminyl)-L-threonine O-N-acetyl-alpha-D-glucosaminase activity.Beta-N-acetylglucosaminidase activity.Identical protein binding.N-acetyl-beta-D-galactosaminidase activity. Related diseases Pentosuria (PNTSU) [MIM:260800]: An inborn error of metabolism characterized by excessive urinary excretion of L-xylulose. {ECO:0000269|PubMed:11882650, ECO:0000269|PubMed:22042873, ECO:0000269|PubMed:4392213}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08255; DB07432; DB00428 Interacts with Q89ZI2 EC number 3.2.1.169 Uniprot keywords 3D-structure; Glycosidase; Hydrolase; Reference proteome; Signal Protein physicochemical properties Chain ID A,B Molecular weight (Da) 54101.5 Length 466 Aromaticity 0.13 Instability index 31.78 Isoelectric point 5.69 Charge (pH=7) -8.12 2D Binding mode Binding energy (Kcal/mol) -7.62 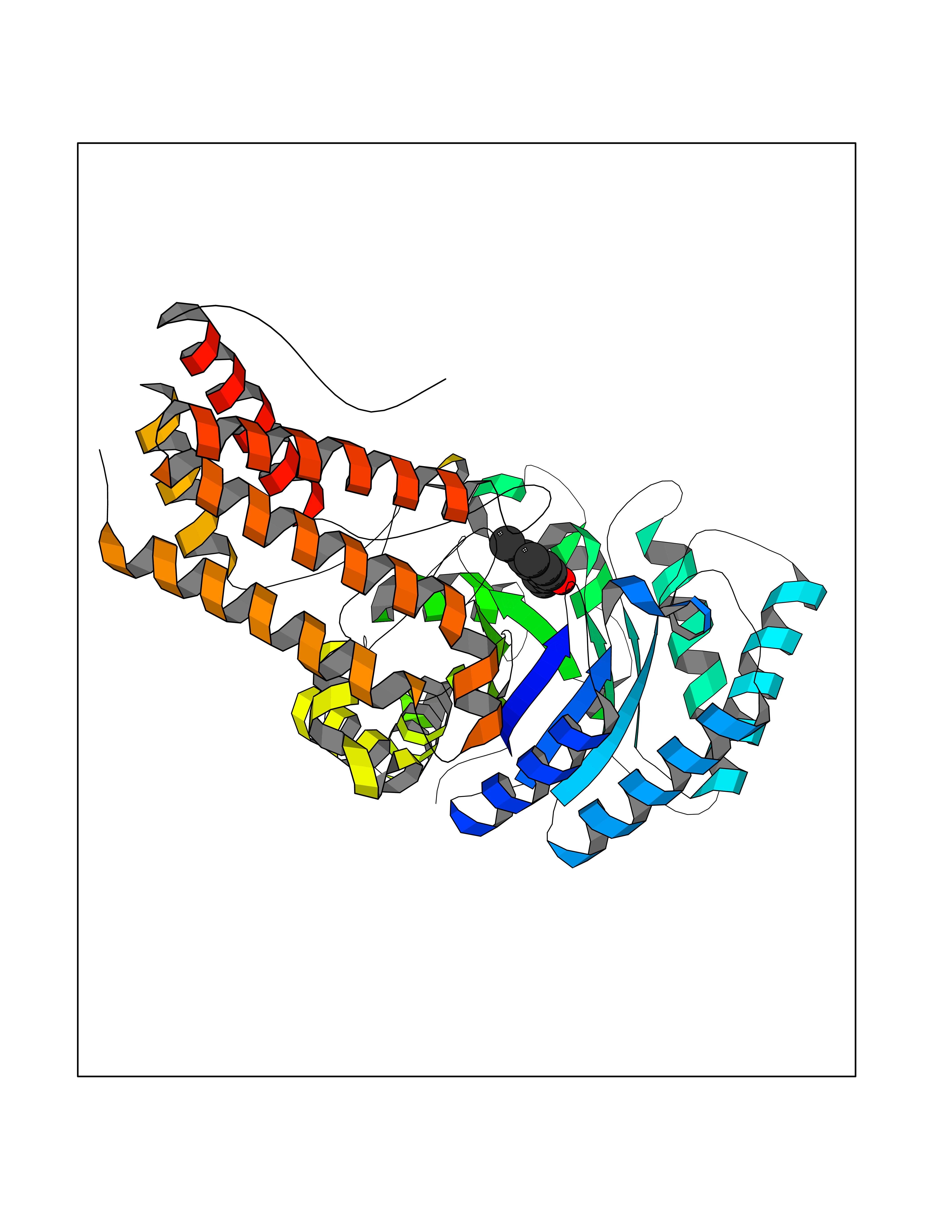 Molscript Map 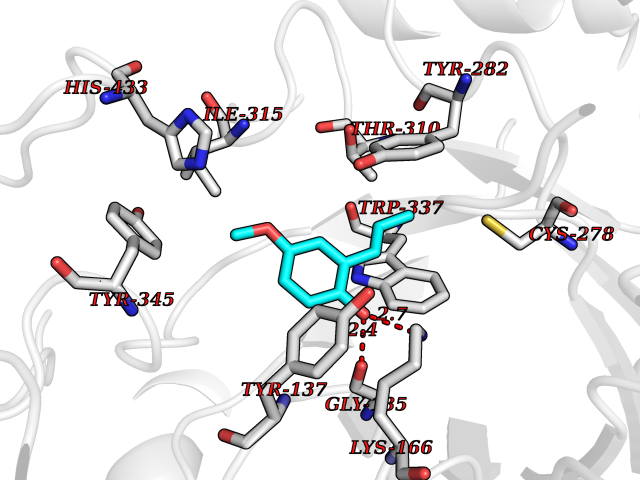 Pymol Map 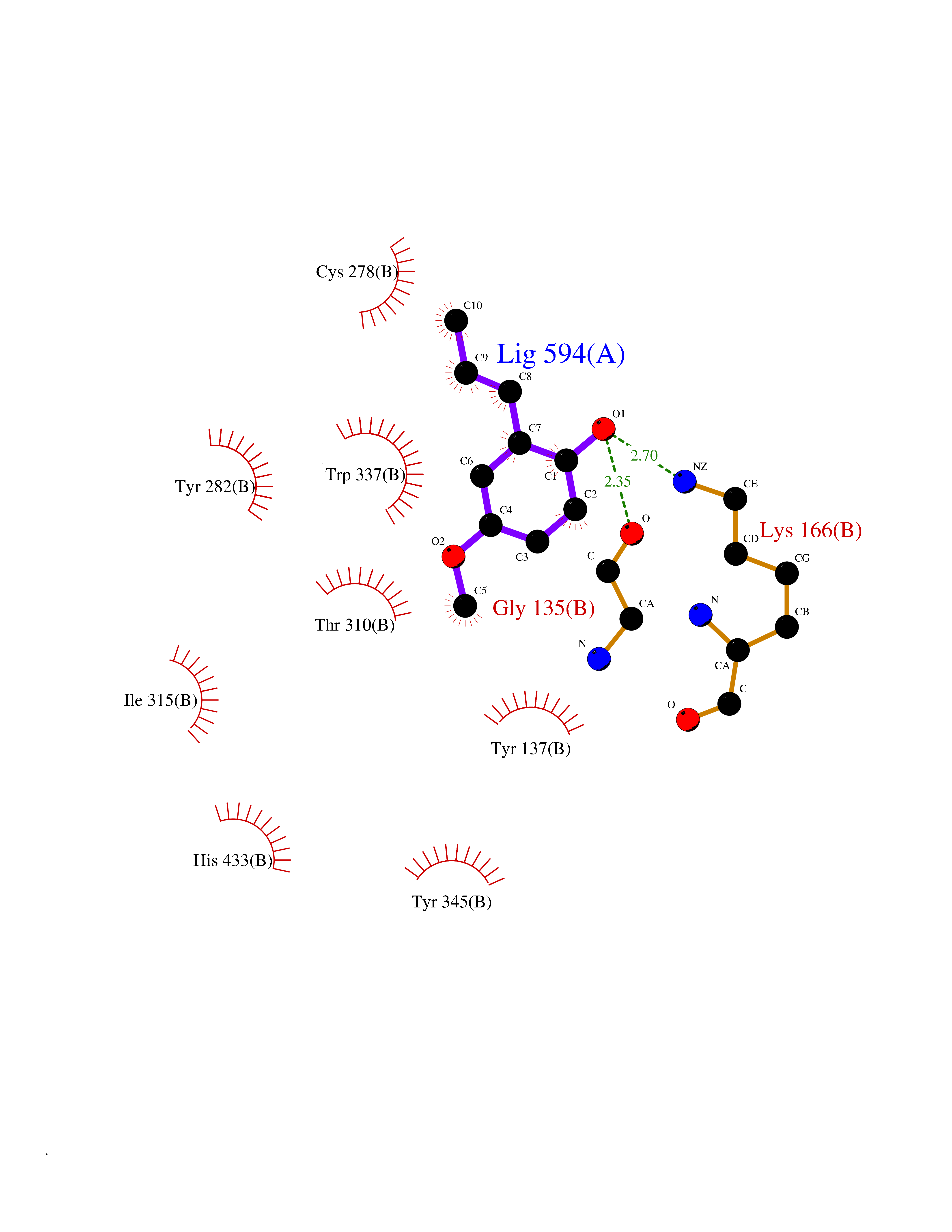 Ligplot Map 3D Binding mode Sequence PSVRYRGVVEGFYGTPWSHQARLSQLKFYGKNKMNTYIYGPKDDPYHSAPNWRLPYPDKEAAQLQELVAVANENEVDFVWAIHPGQDIKWNKEDRDLLLAKFEKMYQLGVRSFAVFFDDISGEGTNPQKQAELLNYIDEKFAQVKPDINQLVMCPTEYNKSWSNPNGNYLTTLGDKLNPSIQIMWTGDRVISDITRDGISWINERIKRPAYIWWNFPVSDYVRDHLLLGPVYGNDTTIAKEMSGFVTNPMEHAESSKIAIYSVASYAWNPAKYDTWQTWKDAIRTILPSAAEELECFAMHNSDLGPNGHGYRREESMDIQPAAERFLKAFKENYDKADFETLQYTFERMKESADILLMNTENKPLIVEITPWVHQFKLTAEMGEEVLKMVEGRNESYFLRKYNHVKALQQQMFYIDQTSNQNPYQPGVKTATRVIKPLIDRTFATVVKFFNQKFNAHLDATTDYMP Hydrogen bonds contact Hydrophobic contact | ||||
| 99 | Protein-tyrosine-phosphatase | 2Z72 | 5.58 | |
Target general information Gen name PPI Organism Shewanella sp Uniprot ID TTD ID NA Synonyms NA Protein family NA Biochemical class Hydrolase Function Metal ion binding.Protein tyrosine phosphatase activity. Related diseases Brugada syndrome 7 (BRGDA7) [MIM:613120]: A tachyarrhythmia characterized by right bundle branch block and ST segment elevation on an electrocardiogram (ECG). It can cause the ventricles to beat so fast that the blood is prevented from circulating efficiently in the body. When this situation occurs, the individual will faint and may die in a few minutes if the heart is not reset. {ECO:0000269|PubMed:20031595}. The gene represented in this entry may be involved in disease pathogenesis.; DISEASE: Atrial fibrillation, familial, 16 (ATFB16) [MIM:613120]: A familial form of atrial fibrillation, a common sustained cardiac rhythm disturbance. Atrial fibrillation is characterized by disorganized atrial electrical activity and ineffective atrial contraction promoting blood stasis in the atria and reduces ventricular filling. It can result in palpitations, syncope, thromboembolic stroke, and congestive heart failure. {ECO:0000269|PubMed:20558140, ECO:0000269|PubMed:21051419}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number 3.1.3.48 Uniprot keywords 3D-structure; Calcium; Hydrolase; Metal-binding; Signal; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 38399.1 Length 338 Aromaticity 0.09 Instability index 29.48 Isoelectric point 5.85 Charge (pH=7) -7.13 2D Binding mode Binding energy (Kcal/mol) -7.62 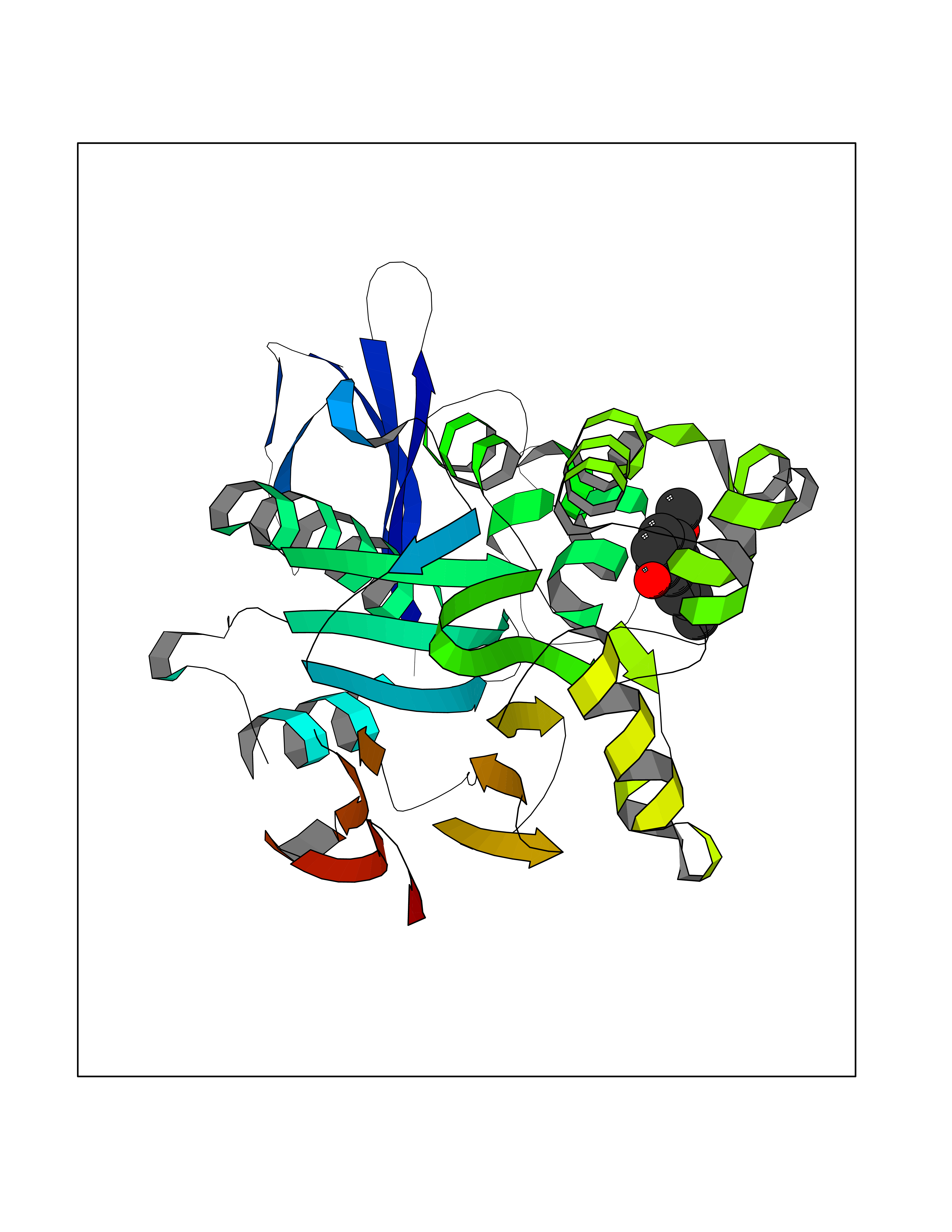 Molscript Map 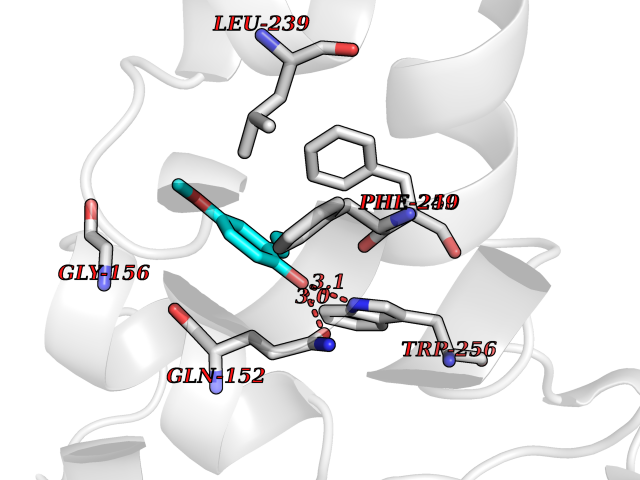 Pymol Map 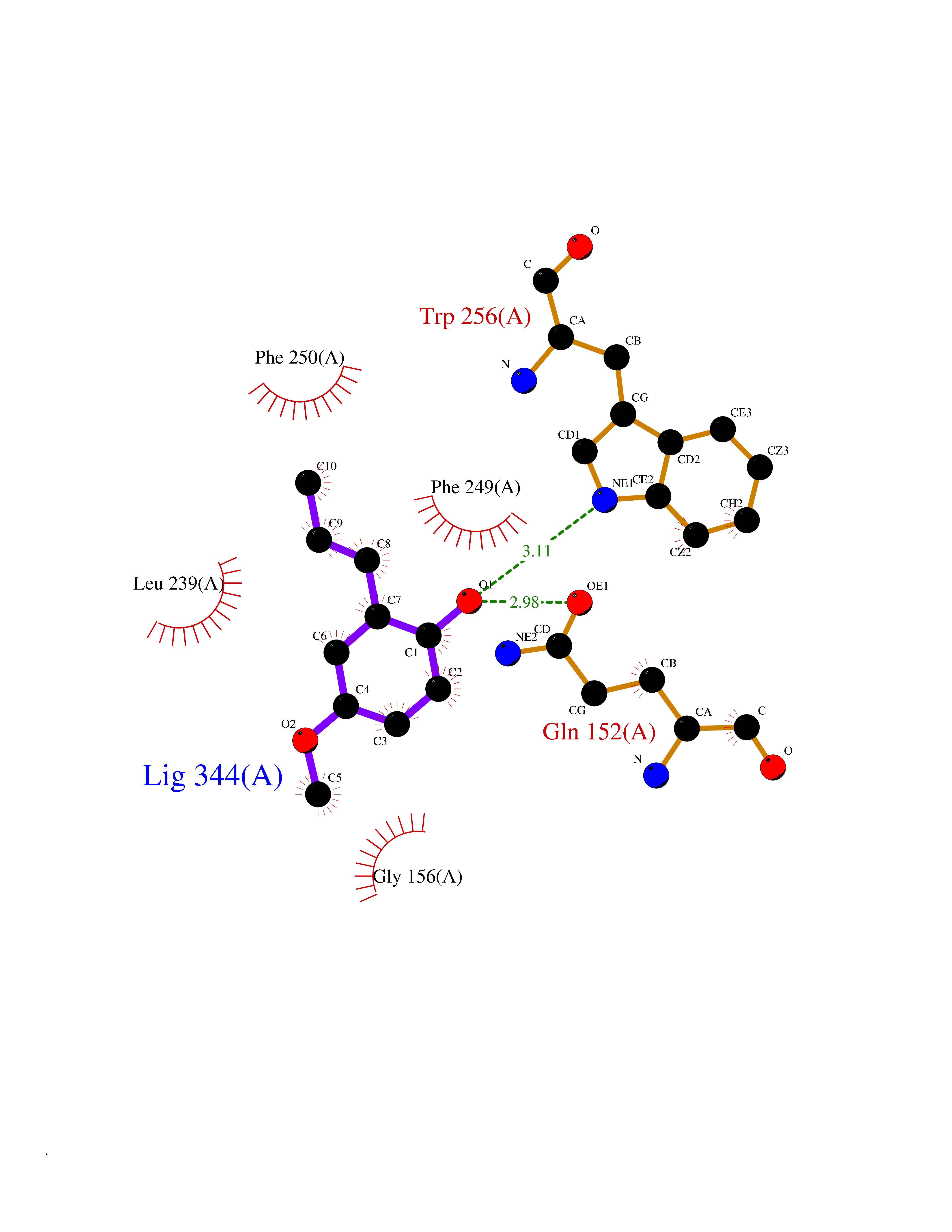 Ligplot Map 3D Binding mode Sequence ATEFDGPYVITPISGQSTAYWICDNRLKTTSIEKLQVNRPEHCGDLPETKLSSEIKQIMPDTYLGIKKVVALSDVHGQYDVLLTLLKKQKIIDSDGNWAFGEGHMVMTGDIFDRGHQVNEVLWFMYQLDQQARDAGGMVHLLMGNHEQMVLGGDLRYVHQRYDIATTLINRPYNKLYSADTEIGQWLRSKNTIIKINDVLYMHGGISSEWISRELTLDKANALYRANVDASKKSLKADDLLNFLFFGNGPTWYRGYFSETFTEAELDTILQHFNVNHIVVGHTSQERVLGLFHNKVIAVDSSIKVGKSGELLLLENNRLIRGLYDGTRETLQENSLNQ Hydrogen bonds contact Hydrophobic contact | ||||
| 100 | Cerebron E3 ubiquitin ligase complex (CRL4-CRBN E3 ubiquitin ligase) | 4CI1 | 5.58 | |
Target general information Gen name CUL4A/CUL4B-DDB1-CRBN Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms NA Protein family Cullin family Biochemical class NA Function NA Related diseases Orotic aciduria 1 (ORAC1) [MIM:258900]: A disorder of pyrimidine metabolism resulting in megaloblastic anemia and orotic acid crystalluria that is frequently associated with some degree of physical and intellectual disability. A minority of cases have additional features, particularly congenital malformations and immune deficiencies. {ECO:0000269|PubMed:9042911}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P54253; Q86VP6; Q16531; Q92466; P08238; O94888; P55072 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Biological rhythms; DNA damage; DNA repair; Host-virus interaction; Isopeptide bond; Phosphoprotein; Proteomics identification; Reference proteome; Ubl conjugation; Ubl conjugation pathway Protein physicochemical properties Chain ID B Molecular weight (Da) 42669.7 Length 368 Aromaticity 0.1 Instability index 44.94 Isoelectric point 8.72 Charge (pH=7) 6.58 2D Binding mode Binding energy (Kcal/mol) -7.61  Molscript Map 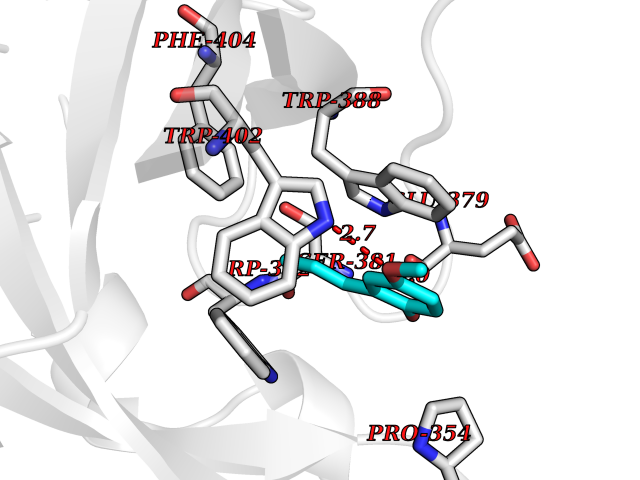 Pymol Map 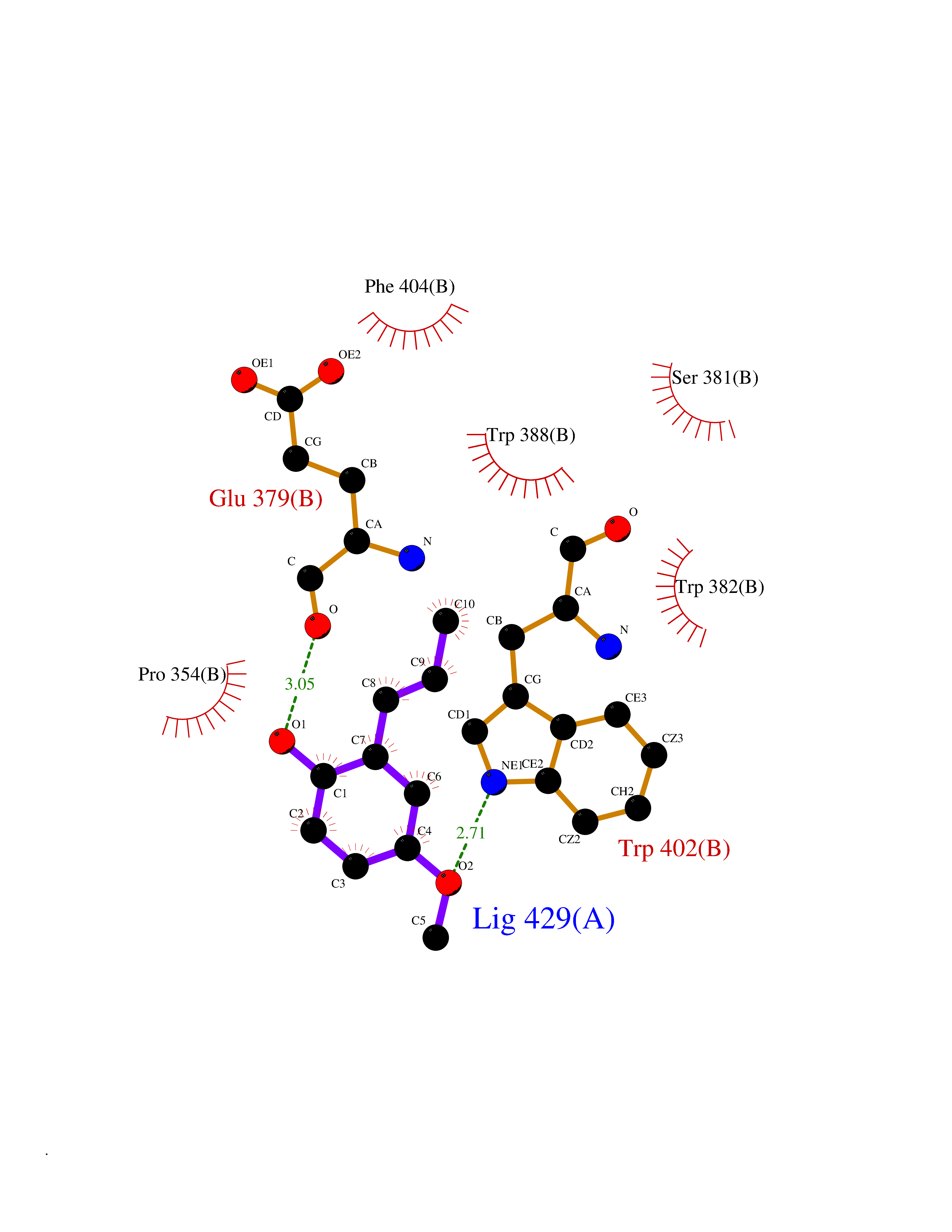 Ligplot Map 3D Binding mode Sequence MINFDTSLPTSHMYLGSDMEEFHGRTLHDDDSCQVIPVLPHVMVMLIPGQTLPLQLFHPQEVSMVRNLIQKDRTFAVLAYSNVREREAHFGTTAEIYAYREEQEYGIETVKVKAIGRQRFKVLEIRTQSDGIQQAKVQILPERVLPSTMSAVQLQSLSRRHIRAFRQWWQKYQKRKFHCASLTSWPPWLYSLYDAETLMERVKRQLHEWDENLKDESLPTNPIDFSYRVAACLPIDDALRIQLLKIGSAIQRLRELDIMNKTSLCCKQCQDTEITTKNEIFSLSLCGPMAAYVNPHGYIHETLTVYKACNLNLSGRPSTEHSWFPGYAWTIAQCRICGNHMGWKFTATKKDMSPQKFWGLTRSALLPR Hydrogen bonds contact Hydrophobic contact | ||||