Job Results:
Ligand
Structure
Job ID
8e57bce38c3d7c3304510f6b58eb7b68
Job name
NA
Time
2026-02-27 11:59:36
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 81 | Proto-oncogene c-Met (MET) | 3DKC | 4.74 | |
Target general information Gen name MET Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Tyrosine-protein kinase Met; Scatter factor receptor; SF receptor; Met proto-oncogene tyrosine kinase; Hepatocyte growth factor receptor; HGF/SF receptor; HGF-SF receptor; HGF receptor; C-met; C-Met r Protein family Protein kinase superfamily, Tyr protein kinase family Biochemical class Kinase Function Regulates many physiological processes including proliferation, scattering, morphogenesis and survival. Ligand binding at the cell surface induces autophosphorylation of MET on its intracellular domain that provides docking sites for downstream signaling molecules. Following activation by ligand, interacts with the PI3-kinase subunit PIK3R1, PLCG1, SRC, GRB2, STAT3 or the adapter GAB1. Recruitment of these downstream effectors by MET leads to the activation of several signaling cascades including the RAS-ERK, PI3 kinase-AKT, or PLCgamma-PKC. The RAS-ERK activation is associated with the morphogenetic effects while PI3K/AKT coordinates prosurvival effects. During embryonic development, MET signaling plays a role in gastrulation, development and migration of muscles and neuronal precursors, angiogenesis and kidney formation. In adults, participates in wound healing as well as organ regeneration and tissue remodeling. Promotes also differentiation and proliferation of hematopoietic cells. May regulate cortical bone osteogenesis. Receptor tyrosine kinase that transduces signals from the extracellular matrix into the cytoplasm by binding to hepatocyte growth factor/HGF ligand. Related diseases Activation of MET after rearrangement with the TPR gene produces an oncogenic protein.; DISEASE: Defects in MET may be associated with gastric cancer.; DISEASE: Hepatocellular carcinoma (HCC) [MIM:114550]: A primary malignant neoplasm of epithelial liver cells. The major risk factors for HCC are chronic hepatitis B virus (HBV) infection, chronic hepatitis C virus (HCV) infection, prolonged dietary aflatoxin exposure, alcoholic cirrhosis, and cirrhosis due to other causes. {ECO:0000269|PubMed:9927037}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Renal cell carcinoma papillary (RCCP) [MIM:605074]: A subtype of renal cell carcinoma tending to show a tubulo-papillary architecture formed by numerous, irregular, finger-like projections of connective tissue. Renal cell carcinoma is a heterogeneous group of sporadic or hereditary carcinoma derived from cells of the proximal renal tubular epithelium. {ECO:0000269|PubMed:10327054, ECO:0000269|PubMed:10417759, ECO:0000269|PubMed:10433944, ECO:0000269|PubMed:9140397, ECO:0000269|PubMed:9563489}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: A common allele in the promoter region of the MET shows genetic association with susceptibility to autism in some families. Functional assays indicate a decrease in MET promoter activity and altered binding of specific transcription factor complexes.; DISEASE: MET activating mutations may be involved in the development of a highly malignant, metastatic syndrome known as cancer of unknown primary origin (CUP) or primary occult malignancy. Systemic neoplastic spread is generally a late event in cancer progression. However, in some instances, distant dissemination arises at a very early stage, so that metastases reach clinical relevance before primary lesions. Sometimes, the primary lesions cannot be identified in spite of the progresses in the diagnosis of malignancies.; DISEASE: Deafness, autosomal recessive, 97 (DFNB97) [MIM:616705]: A form of non-syndromic sensorineural hearing loss with prelingual onset. Sensorineural deafness results from damage to the neural receptors of the inner ear, the nerve pathways to the brain, or the area of the brain that receives sound information. {ECO:0000269|PubMed:25941349}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Osteofibrous dysplasia (OSFD) [MIM:607278]: A congenital disorder of osteogenesis characterized by non-neoplastic, radiolucent lesions that affect the cortical bone immediately under the periosteum. It usually manifests as a painless swelling or anterior bowing of the long bones, most commonly the tibia and fibula. {ECO:0000269|PubMed:26637977}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Disease-associated variants identified in 4 families cause the deletion of exon 14. This results in the exclusion of an ubiquitination target site within the cytoplasmic domain, hence in protein stabilization. The persistent presence of MET at the cell surface in conditions of ligand-dependent activation retards osteoblastic differentiation. {ECO:0000269|PubMed:26637977}.; DISEASE: Arthrogryposis, distal, 11 (DA11) [MIM:620019]: A form of distal arthrogryposis, a disease characterized by congenital joint contractures that mainly involve two or more distal parts of the limbs, in the absence of a primary neurological or muscle disease. DA11 is an autosomal dominant form characterized mainly by camptodactyly. Other features include absent flexion creases and limited forearm supination. {ECO:0000269|PubMed:30777867}. The disease may be caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB06896; DB08791; DB06997; DB07969; DB08079; DB16695; DB12742; DB12267; DB08875; DB11791; DB08865; DB12010; DB02152; DB07369; DB06995; DB06314; DB01268; DB15133; DB12200; DB11800 Interacts with P22681; Q96EY1; Q96EY1-2; P00533; P09769; P14210; P14210-6; O15357; P35968; P06239; P07948; P08581; P41218; P15941; P16333; O43639; Q16288; P27986; O00459; Q92569; P19174; O43157; O15031; Q9ULL4; Q8TCU6; P18031; Q06124; P23467; Q12913; Q16827; P20936; Q9UQQ2; O60880; O14796; Q9NP31; Q8N5H7; Q15464; P29353; P98077; Q6S5L8; Q96IW2; Q9H6Q3; O75159; O14544; P12931; Q9ULZ2; P43405; P42680; Q9HBL0; Q63HR2; Q68CZ2; Q9UKW4; P07947; P43403; Q08048; P0DQD2; P35918; Q00944 EC number EC 2.7.10.1 Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Chromosomal rearrangement; Deafness; Disease variant; Disulfide bond; Glycoprotein; Kinase; Membrane; Non-syndromic deafness; Nucleotide-binding; Phosphoprotein; Proteomics identification; Proto-oncogene; Receptor; Reference proteome; Repeat; Secreted; Signal; Transferase; Transmembrane; Transmembrane helix; Tyrosine-protein kinase; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 35229.5 Length 312 Aromaticity 0.1 Instability index 37.98 Isoelectric point 7.79 Charge (pH=7) 1.93 2D Binding mode Binding energy (Kcal/mol) -6.47 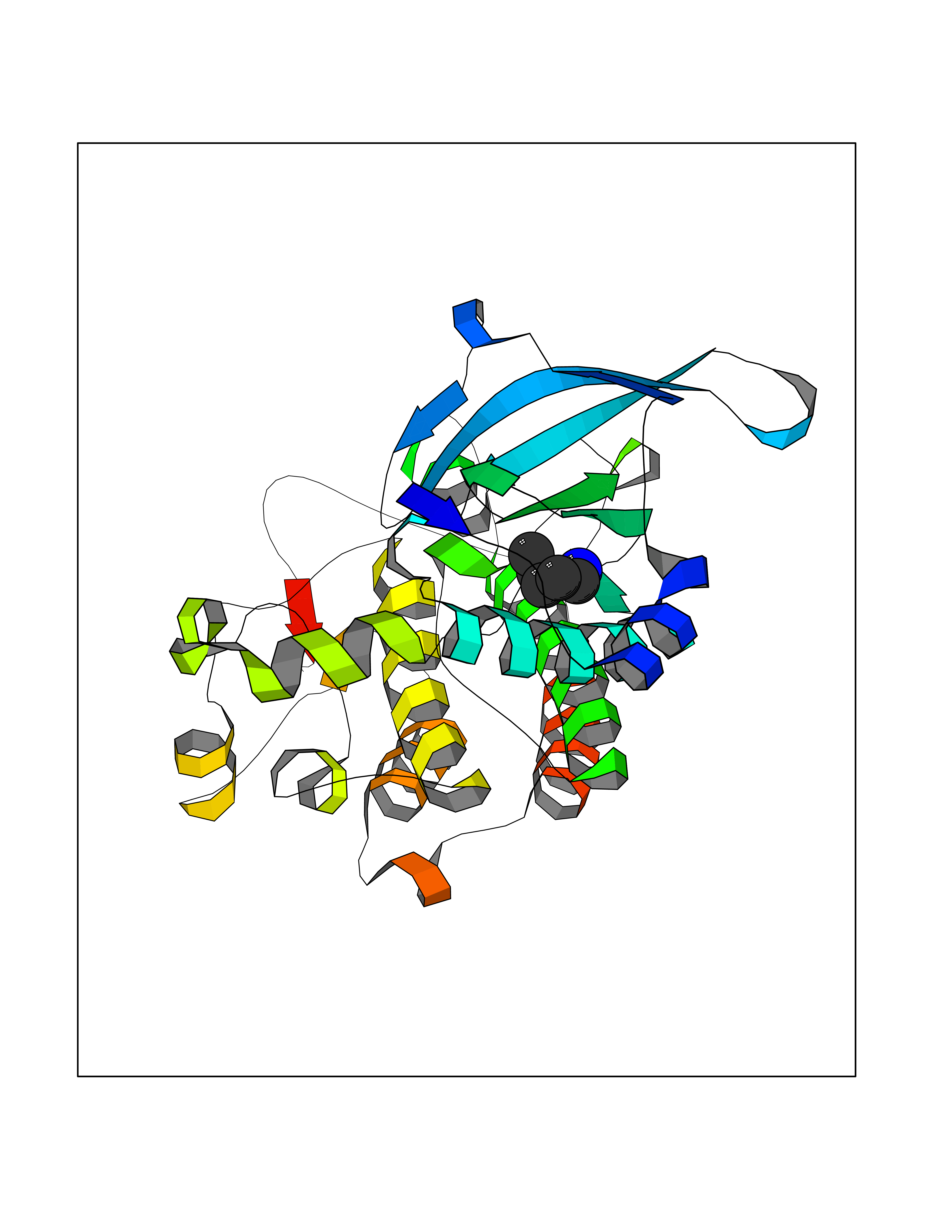 Molscript Map 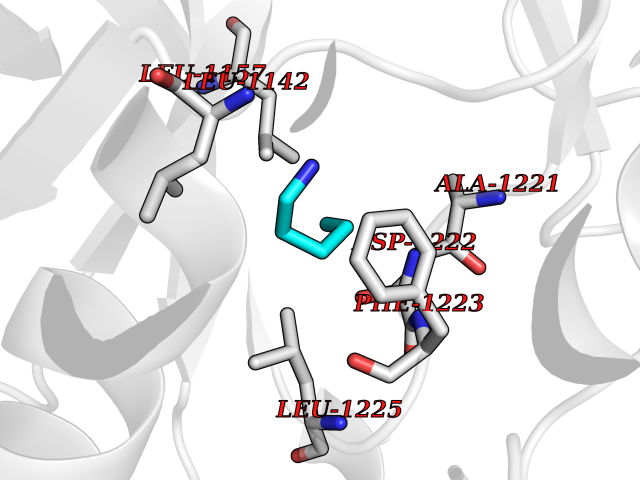 Pymol Map 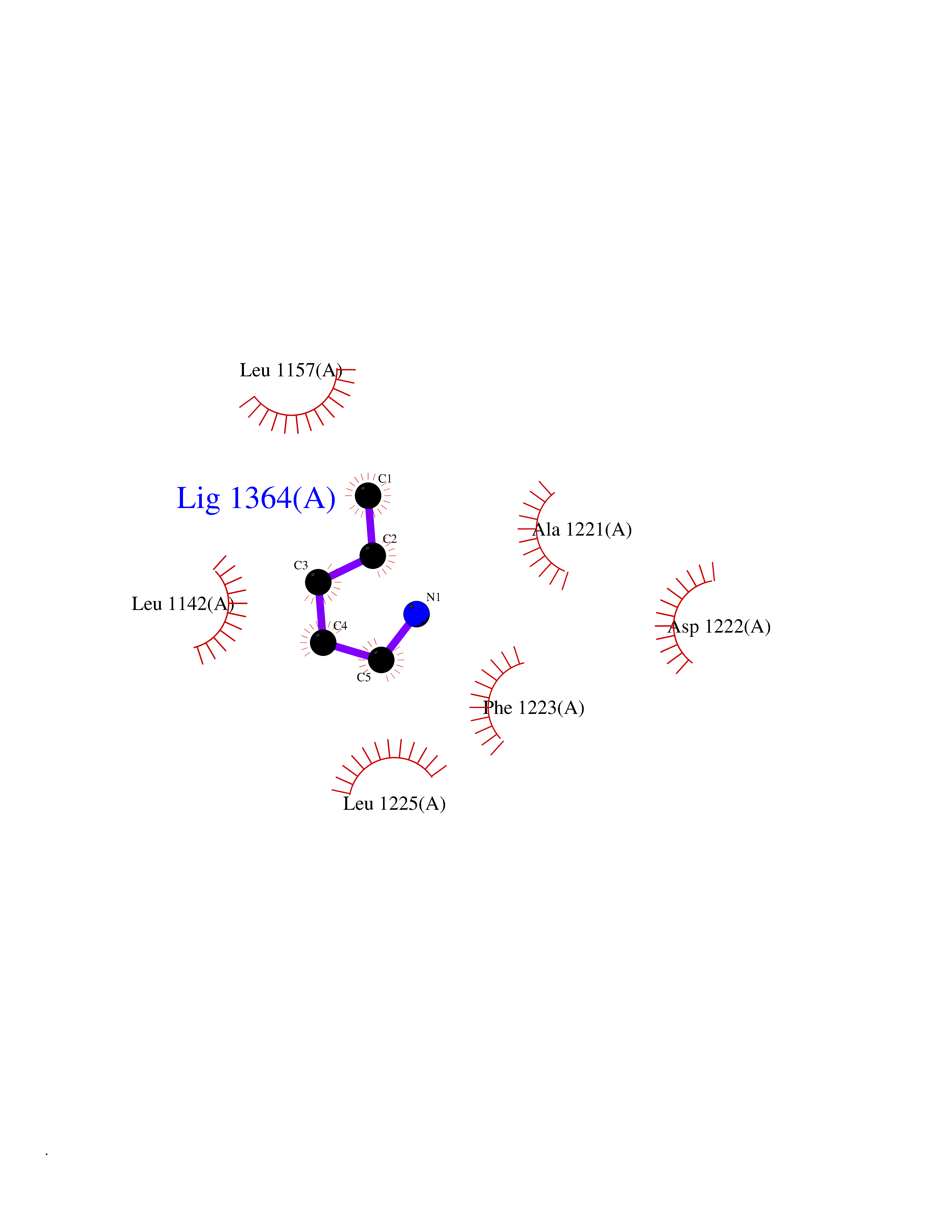 Ligplot Map 3D Binding mode Sequence VHIDLSALNPELVQAVQHVVIGPSSLIVHFNEVIGRGHFGCVYHGTLLDNDGKKIHCAVKSLNRITDIGEVSQFLTEGIIMKDFSHPNVLSLLGICLRSEGSPLVVLPYMKHGDLRNFIRNETHNPTVKDLIGFGLQVAKGMKFLASKKFVHRDLAARNCMLDEKFTVKVADFGLARDMYDKEFDSVHNKTGAKLPVKWMALESLQTQKFTTKSDVWSFGVLLWELMTRGAPPYPDVNTFDITVYLLQGRRLLQPEYCPDPLYEVMLKCWHPKAEMRPSFSELVSRISAIFSTFIGEHYVHVNATYVNVKEG Hydrogen bonds contact Hydrophobic contact | ||||
| 82 | Melatonin receptor type 1B (MTNR1B) | 6ME9 | 4.74 | |
Target general information Gen name MTNR1B Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Mel1b receptor; Mel1b melatonin receptor; Mel-1B-R Protein family G-protein coupled receptor 1 family Biochemical class GPCR rhodopsin Function Likely to mediate the reproductive and circadian actions of melatonin. The activity of this receptor is mediated by pertussis toxin sensitive G proteins that inhibit adenylate cyclase activity. High affinity receptor for melatonin. Related diseases Insulin-like growth factor 1 resistance (IGF1RES) [MIM:270450]: A disorder characterized by intrauterine growth retardation, poor postnatal growth and increased plasma IGF1 levels. {ECO:0000269|PubMed:14657428, ECO:0000269|PubMed:15928254, ECO:0000269|PubMed:25040157}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB06594; DB01065; DB00980; DB02709; DB09071; DB15133 Interacts with P28335; P48039; O76081; Q14669 EC number NA Uniprot keywords 3D-structure; Cell membrane; Disulfide bond; G-protein coupled receptor; Glycoprotein; Membrane; Receptor; Reference proteome; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 50184.9 Length 448 Aromaticity 0.11 Instability index 37.2 Isoelectric point 5.72 Charge (pH=7) -5.68 2D Binding mode Binding energy (Kcal/mol) -6.47 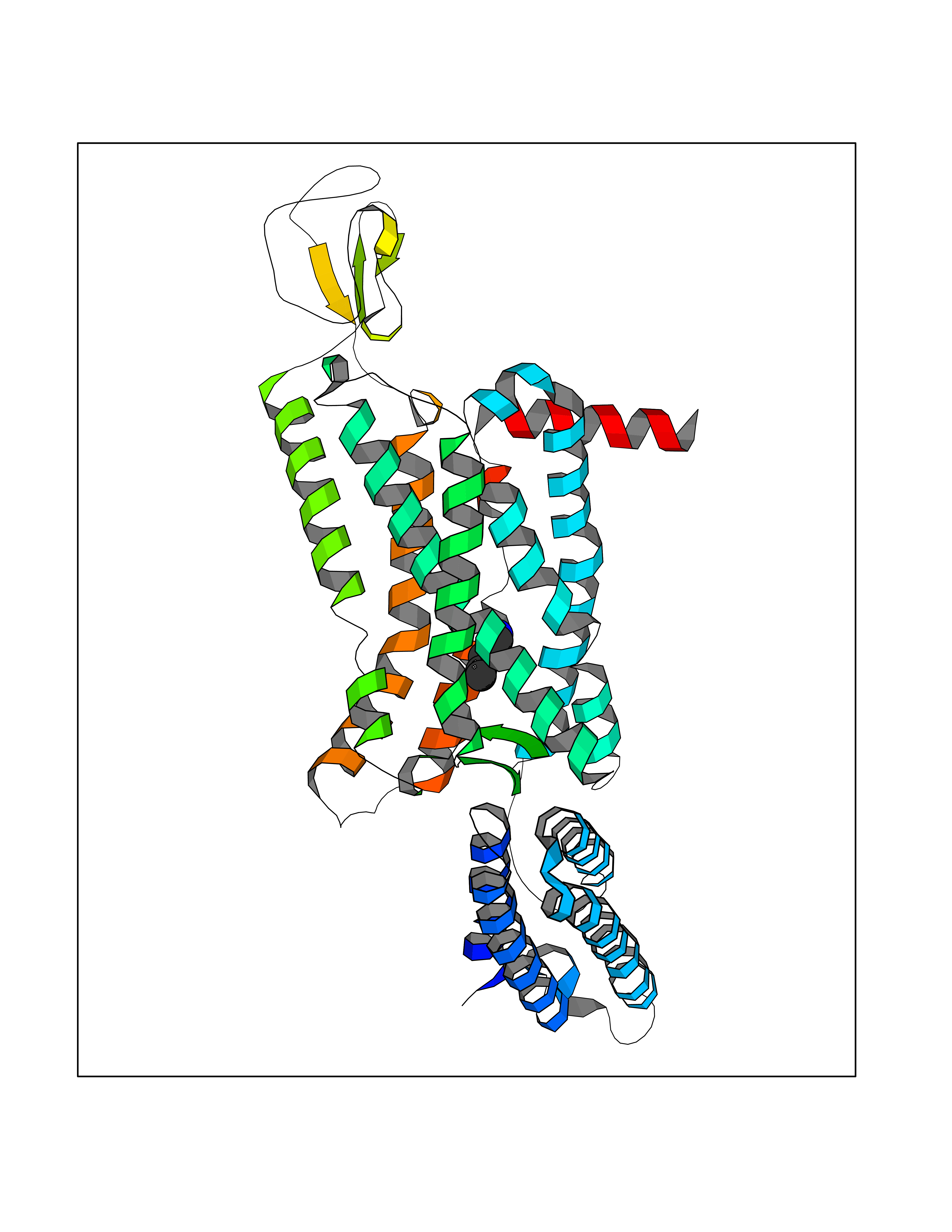 Molscript Map 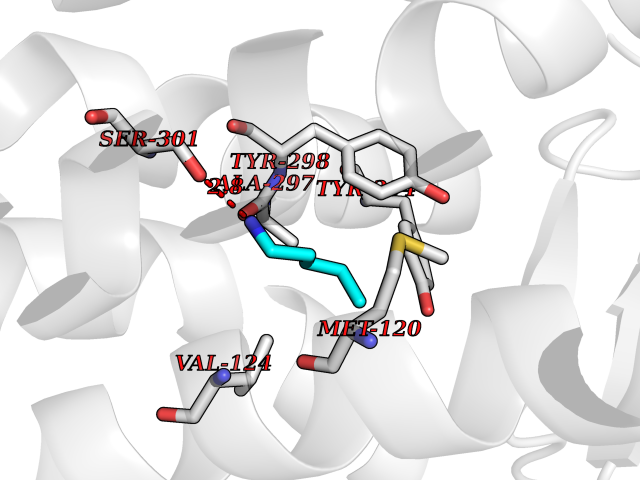 Pymol Map 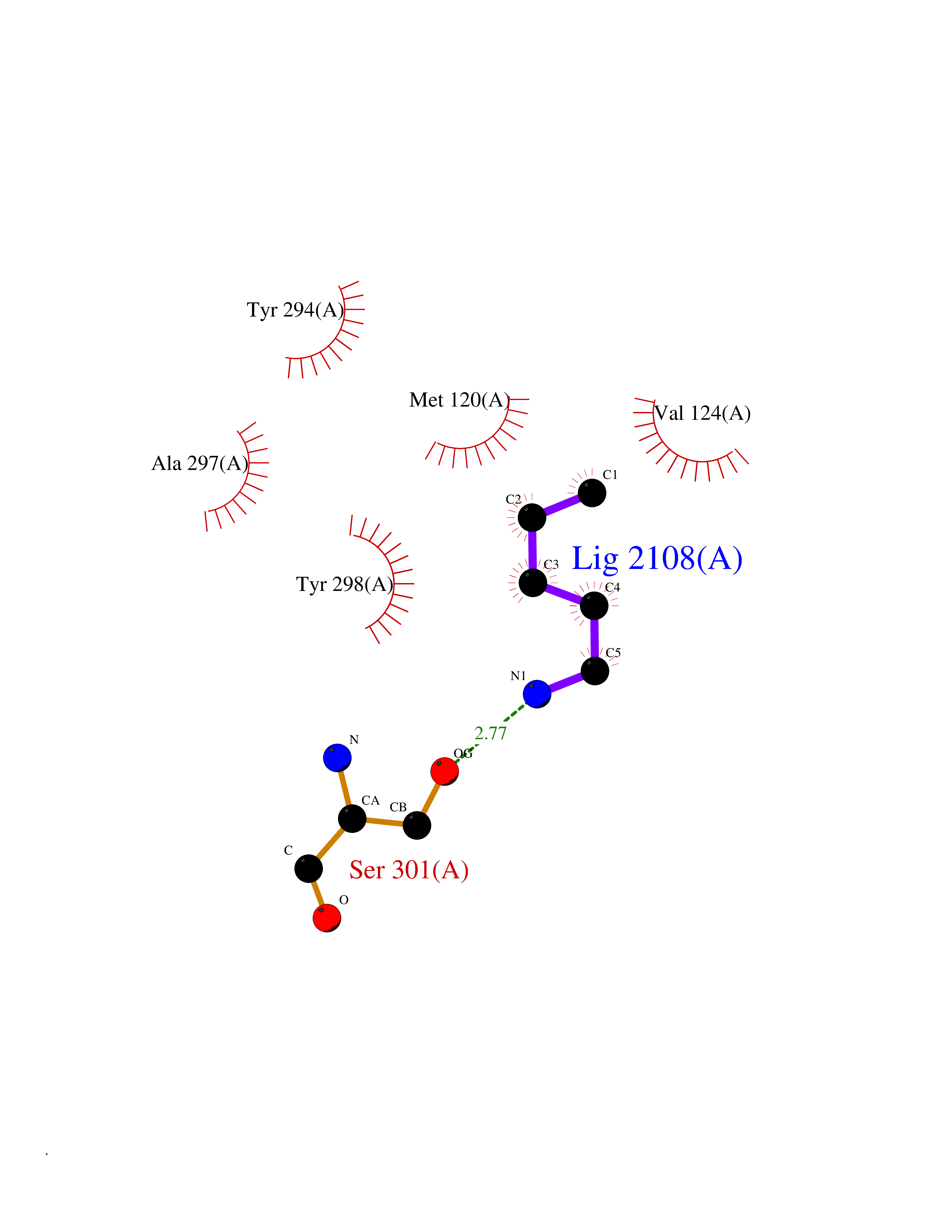 Ligplot Map 3D Binding mode Sequence ADLEDNWETLNDNLKVIEKADNAAQVKDALTKMRAAALDAQKATPPKLEDKSPDSPEMKDFRHGFDILVGQIDDALKLANEGKVKEAQAAAEQLKTTRNAYIQKYLGDGARPSWVAPALSAVLIVTTAVDVVGNLLVILSVLRNRKLRNAGNLFLVSLALANLVVAFYPYPLILVAIFYDGWAFGEEHCKASAFVMGLSVIGSVWNITAIAIDRYLYICHSMAYHRIYRRWHTPLHICLIWLLTVVALLPNFFVGSLEYDPRIYSCTFIQTASTQYTAAVVVIHFLLPIAVVSFCYLRIWVLVLQARMKKYTCTVCGYIYNPEDGDPDNGVNPGTDFKDIPDDWVCPLCGVGKDQFEEVECLKPSDLRSFLTMFVVFVIFAICFAPLNCIGLAVAINPQEMAPQIPEGLFVTSYLLAYFNSCLNPIVYGLLDQNFRREYKRILLALWN Hydrogen bonds contact Hydrophobic contact | ||||
| 83 | Voltage-gated potassium channel Kv7.1 (KCNQ1) | 6V01 | 4.74 | |
Target general information Gen name KCNQ1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Voltage-gated potassium channel subunit Kv7.1; Potassium voltage-gated channel subfamily KQT member 1; Kv7.1; KVLQT1; KQT-like 1; KCNQ1 channel; KCNA9; KCNA8; IKs producing slow voltage-gated potassiu Protein family Potassium channel family, KQT (TC 1.A.1.15) subfamily, Kv7.1/KCNQ1 sub-subfamily Biochemical class Voltage-gated ion channel Function Associates with KCNE beta subunits that modulates current kinetics. Induces a voltage-dependent by rapidly activating and slowly deactivating potassium-selective outward current. Promotes also a delayed voltage activated potassium current showing outward rectification characteristic. During beta-adrenergic receptor stimulation participates in cardiac repolarization by associating with KCNE1 to form the I(Ks) cardiac potassium current that increases the amplitude and slows down the activation kinetics of outward potassium current I(Ks). Muscarinic agonist oxotremorine-M strongly suppresses KCNQ1/KCNE1 current. When associated with KCNE3, forms the potassium channel that is important for cyclic AMP-stimulated intestinal secretion of chloride ions. This interaction with KCNE3 is reduced by 17beta-estradiol, resulting in the reduction of currents. During conditions of increased substrate load, maintains the driving force for proximal tubular and intestinal sodium ions absorption, gastric acid secretion, and cAMP-induced jejunal chloride ions secretion. Allows the provision of potassium ions to the luminal membrane of the secretory canaliculus in the resting state as well as during stimulated acid secretion. When associated with KCNE2, forms a heterooligomer complex leading to currents with an apparently instantaneous activation, a rapid deactivation process and a linear current-voltage relationship and decreases the amplitude of the outward current. When associated with KCNE4, inhibits voltage-gated potassium channel activity. When associated with KCNE5, this complex only conducts current upon strong and continued depolarization. Also forms a heterotetramer with KCNQ5; has a voltage-gated potassium channel activity. Binds with phosphatidylinositol 4,5-bisphosphate. Potassium channel that plays an important role in a number of tissues, including heart, inner ear, stomach and colon. Related diseases Long QT syndrome 1 (LQT1) [MIM:192500]: A heart disorder characterized by a prolonged QT interval on the ECG and polymorphic ventricular arrhythmias. They cause syncope and sudden death in response to exercise or emotional stress, and can present with a sentinel event of sudden cardiac death in infancy. {ECO:0000269|PubMed:10024302, ECO:0000269|PubMed:10220144, ECO:0000269|PubMed:10220146, ECO:0000269|PubMed:10367071, ECO:0000269|PubMed:10409658, ECO:0000269|PubMed:10482963, ECO:0000269|PubMed:10728423, ECO:0000269|PubMed:10973849, ECO:0000269|PubMed:11799244, ECO:0000269|PubMed:12442276, ECO:0000269|PubMed:15840476, ECO:0000269|PubMed:16414944, ECO:0000269|PubMed:16556865, ECO:0000269|PubMed:16922724, ECO:0000269|PubMed:18165683, ECO:0000269|PubMed:18400097, ECO:0000269|PubMed:19540844, ECO:0000269|PubMed:19716085, ECO:0000269|PubMed:19808498, ECO:0000269|PubMed:21241800, ECO:0000269|PubMed:24184248, ECO:0000269|PubMed:24269949, ECO:0000269|PubMed:24713462, ECO:0000269|PubMed:25037568, ECO:0000269|PubMed:25139741, ECO:0000269|PubMed:25705178, ECO:0000269|PubMed:28096388, ECO:0000269|PubMed:34398675, ECO:0000269|PubMed:8528244, ECO:0000269|PubMed:8818942, ECO:0000269|PubMed:8872472, ECO:0000269|PubMed:9024139, ECO:0000269|PubMed:9272155, ECO:0000269|PubMed:9302275, ECO:0000269|PubMed:9312006, ECO:0000269|PubMed:9323054, ECO:0000269|PubMed:9386136, ECO:0000269|PubMed:9482580, ECO:0000269|PubMed:9570196, ECO:0000269|PubMed:9641694, ECO:0000269|PubMed:9693036, ECO:0000269|PubMed:9702906, ECO:0000269|PubMed:9799083, ECO:0000269|PubMed:9927399, ECO:0000269|Ref.36}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Jervell and Lange-Nielsen syndrome 1 (JLNS1) [MIM:220400]: An autosomal recessive disorder characterized by congenital deafness, prolongation of the QT interval, syncopal attacks due to ventricular arrhythmias, and a high risk of sudden death. {ECO:0000269|PubMed:10090886, ECO:0000269|PubMed:10728423, ECO:0000269|PubMed:18400097, ECO:0000269|PubMed:18441444, ECO:0000269|PubMed:25705178, ECO:0000269|PubMed:9781056}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Atrial fibrillation, familial, 3 (ATFB3) [MIM:607554]: An autosomal dominant form of atrial fibrillation, a common sustained cardiac rhythm disturbance. Atrial fibrillation is characterized by disorganized atrial electrical activity and ineffective atrial contraction promoting blood stasis in the atria and reduces ventricular filling. It can result in palpitations, syncope, thromboembolic stroke, and congestive heart failure. {ECO:0000269|PubMed:12522251}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Short QT syndrome 2 (SQT2) [MIM:609621]: An autosomal dominant form of short QT syndrome, a heart disorder characterized by idiopathic persistently and uniformly short QT interval on ECG in the absence of structural heart disease in affected individuals. It can cause syncope and sudden death. {ECO:0000269|PubMed:15159330}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Type 2 diabetes mellitus (T2D) [MIM:125853]: A multifactorial disorder of glucose homeostasis caused by a lack of sensitivity to insulin. Affected individuals usually have an obese body habitus and manifestations of a metabolic syndrome characterized by diabetes, insulin resistance, hypertension and hypertriglyceridemia. The disease results in long-term complications that affect the eyes, kidneys, nerves, and blood vessels. {ECO:0000269|PubMed:18711366, ECO:0000269|PubMed:18711367, ECO:0000269|PubMed:24390345}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB04957; DB01244; DB04855; DB00228; DB06089; DB11633; DB01110; DB01069 Interacts with P15382; P0DP25 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Atrial fibrillation; Calmodulin-binding; Cell membrane; Coiled coil; Cytoplasmic vesicle; Deafness; Diabetes mellitus; Disease variant; Endoplasmic reticulum; Endosome; Glycoprotein; Ion channel; Ion transport; Long QT syndrome; Membrane; Phosphoprotein; Potassium; Potassium channel; Potassium transport; Proteomics identification; Reference proteome; Short QT syndrome; Transmembrane; Transmembrane helix; Transport; Ubl conjugation; Voltage-gated channel Protein physicochemical properties Chain ID J,L,A Molecular weight (Da) 80494.8 Length 702 Aromaticity 0.14 Instability index 31.57 Isoelectric point 10.06 Charge (pH=7) 49.05 2D Binding mode Binding energy (Kcal/mol) -6.47 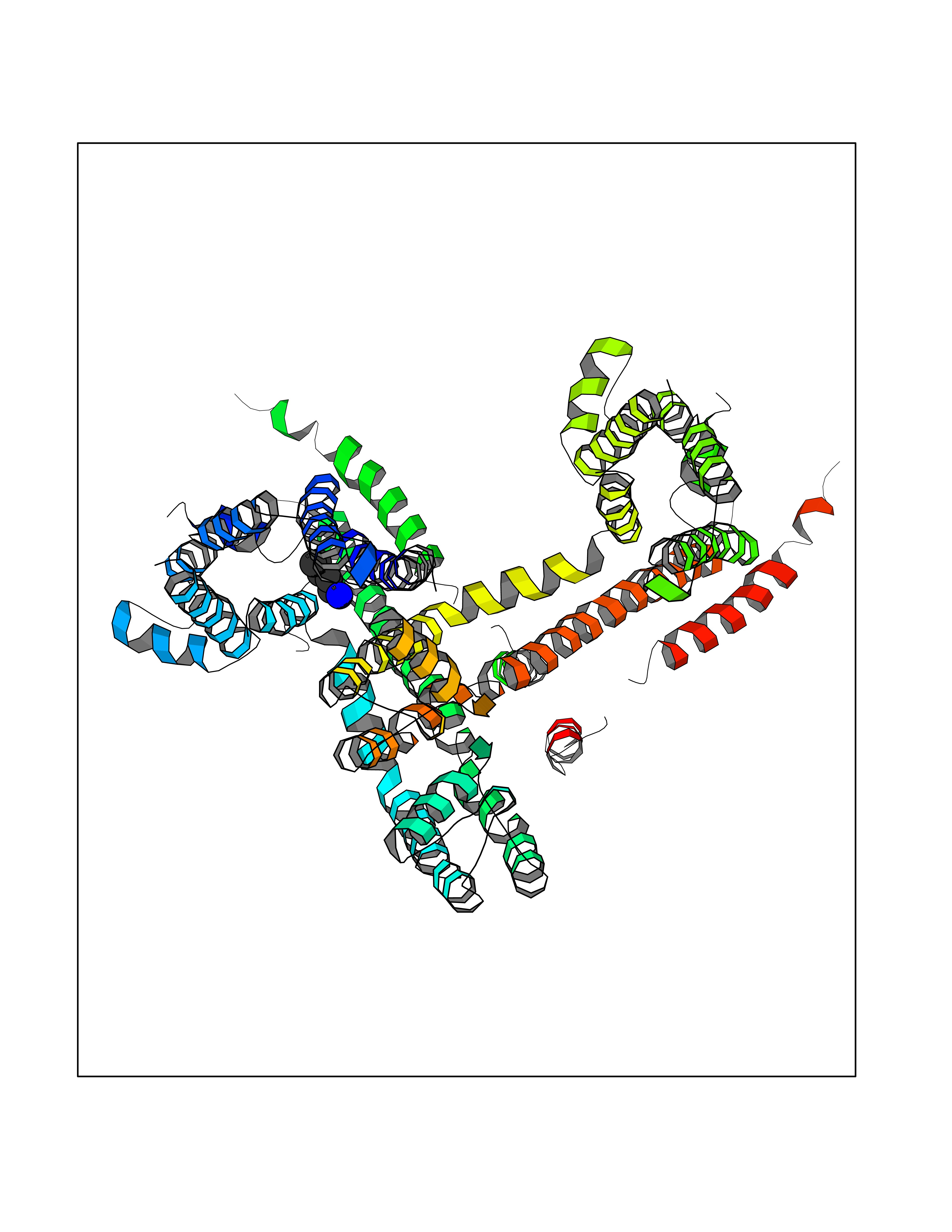 Molscript Map 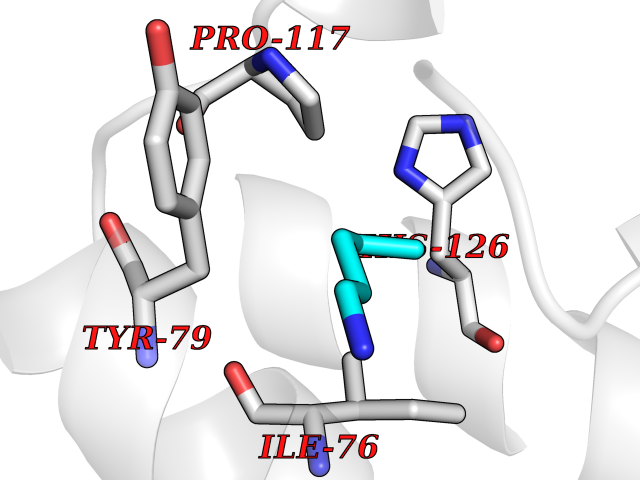 Pymol Map 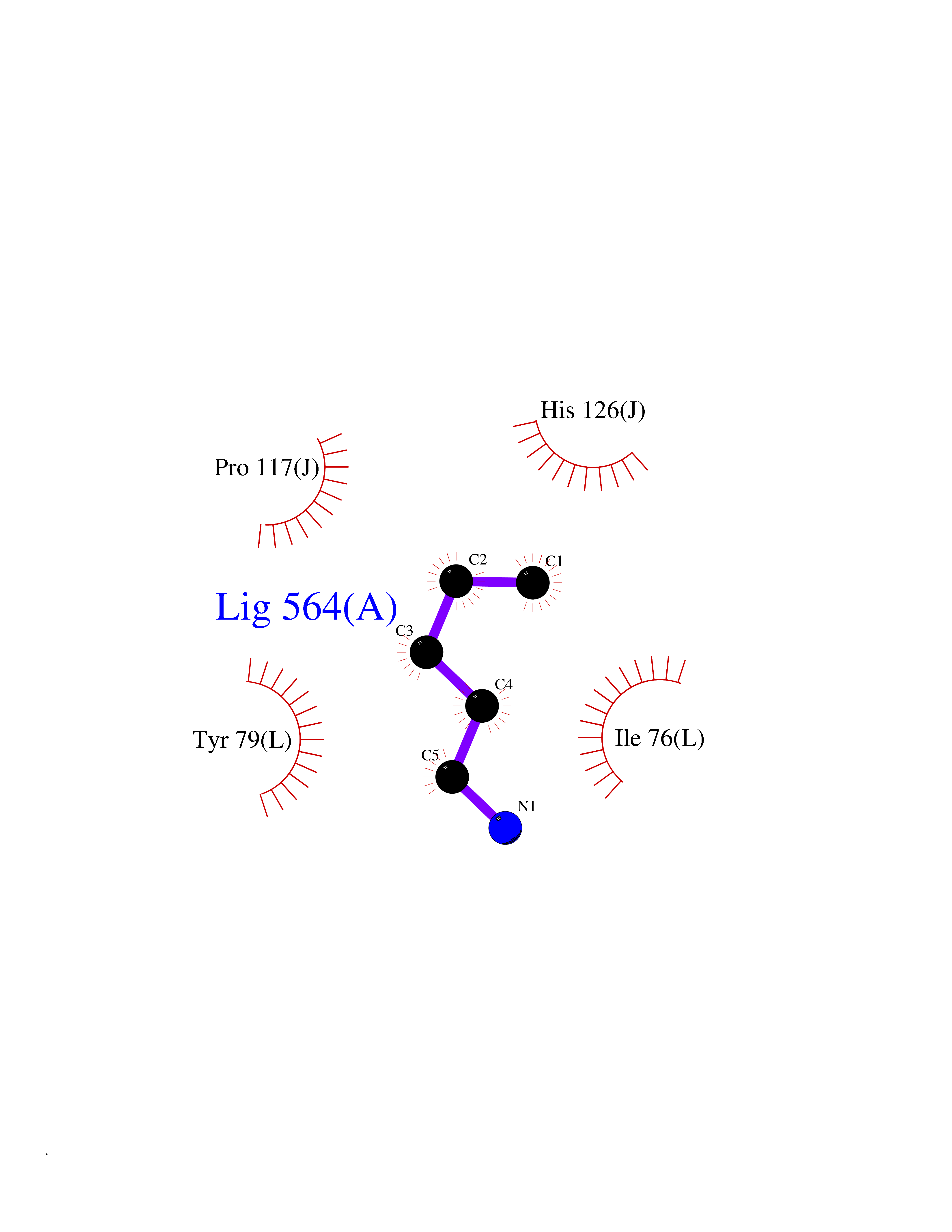 Ligplot Map 3D Binding mode Sequence RDDNSYMYILFVMFLFAVTVGSLILGYTRSRKVDKRSDPYTHVQGRVYNFLERPTGWKCFVYHFAVFLIVLVCLIFSVLSTIEQYAALATGTLFWMEIVLVVFFGTEYVVRLWSAGCRSKYVGLWGRLRFARKPISIIDLIVVVASMVVLCVGSKSAIRGIRFLQILRMLHVDRQGGTWRLLGSVVFIHRQELITTLYIGFLGLIFSSYFVYLAEKDAVNESGRVEFGSYADALWWGVVTVTTIGYGDKVPQTWVGKTIASCFSVFAISFFALPAGILGSGFALKVQQKQRQKHFNRQIPAAASLIQTAWRCYAAENPLREHHRATIKVIRRMQYFVAKKKFQQARKPYDIEQYSQGHLNLMVRIKELQRRTHVQGRVYNFLERPTGWKCFVYHFAVFLIVLVCLIFSVLSTIEQYAALATGTLFWMEIVLVVFFGTEYVVRLWSAGCRSKYVGLWGRLRFARKPISIIDLIVVVASMVVLCVGSKSAIRGIRFLQILRMLHVDRQGGTWRLLGSVVFIHRQELITTLYIGFLGLIFSSYFVYLAEKDAVNESGRVEFGSYADALWWGVVTVTTIGYGDKVPQTWVGKTIASCFSVFAISFFALPAGILGSGFALKVQQKQRQKHFNRQIPAAASLIQTAWRCYAAENPLREHHRATIKVIRRMQYFVAKKKFQQARKPYDIEQYSQGHLNLMVRIKELQRR Hydrogen bonds contact Hydrophobic contact | ||||
| 84 | TGF-beta receptor type I (TGFBR1) | 3TZM | 4.74 | |
Target general information Gen name TGFBR1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Type I TGFbeta receptor kinase; Transforming growth factor-beta receptor type I; TbetaRI; TbetaR-I; TGFR-1; TGF-beta type I receptor; TGF-beta receptor type-1; Serine/threonine-protein kinase receptor Protein family Protein kinase superfamily, TKL Ser/Thr protein kinase family, TGFB receptor subfamily Biochemical class Kinase Function Transduces the TGFB1, TGFB2 and TGFB3 signal from the cell surface to the cytoplasm and is thus regulating a plethora of physiological and pathological processes including cell cycle arrest in epithelial and hematopoietic cells, control of mesenchymal cell proliferation and differentiation, wound healing, extracellular matrix production, immunosuppression and carcinogenesis. The formation of the receptor complex composed of 2 TGFBR1 and 2 TGFBR2 molecules symmetrically bound to the cytokine dimer results in the phosphorylation and the activation of TGFBR1 by the constitutively active TGFBR2. Activated TGFBR1 phosphorylates SMAD2 which dissociates from the receptor and interacts with SMAD4. The SMAD2-SMAD4 complex is subsequently translocated to the nucleus where it modulates the transcription of the TGF-beta-regulated genes. This constitutes the canonical SMAD-dependent TGF-beta signaling cascade. Also involved in non-canonical, SMAD-independent TGF-beta signaling pathways. For instance, TGFBR1 induces TRAF6 autoubiquitination which in turn results in MAP3K7 ubiquitination and activation to trigger apoptosis. Also regulates epithelial to mesenchymal transition through a SMAD-independent signaling pathway through PARD6A phosphorylation and activation. Transmembrane serine/threonine kinase forming with the TGF-beta type II serine/threonine kinase receptor, TGFBR2, the non-promiscuous receptor for the TGF-beta cytokines TGFB1, TGFB2 and TGFB3. Related diseases Loeys-Dietz syndrome 1 (LDS1) [MIM:609192]: An aortic aneurysm syndrome with widespread systemic involvement, characterized by arterial tortuosity and aneurysms, hypertelorism, and bifid uvula or cleft palate. Physical findings include prominent joint laxity, easy bruising, wide and atrophic scars, velvety and translucent skin with easily visible veins, spontaneous rupture of the spleen or bowel, and catastrophic complications of pregnancy, including rupture of the gravid uterus and the arteries, either during pregnancy or in the immediate postpartum period. Some patients have craniosynostosis, exotropy, micrognathia and retrognathia, structural brain abnormalities, and intellectual deficit. {ECO:0000269|PubMed:15731757, ECO:0000269|PubMed:16596670, ECO:0000269|PubMed:16791849, ECO:0000269|PubMed:16928994, ECO:0000269|PubMed:19883511, ECO:0000269|PubMed:22113417}. The disease is caused by variants affecting the gene represented in this entry. TGFBR1 mutation Gln-487 has been reported to be associated with thoracic aortic aneurysms and dissection (TAAD) (PubMed:16791849). This phenotype, also known as thoracic aortic aneurysms type 5 (AAT5), is distinguised from LDS1 by having aneurysms restricted to thoracic aorta. It is unclear, however, if this condition is fulfilled in individuals bearing Gln-487 mutation, that is why they are considered as LDS1 by the OMIM resource. {ECO:0000269|PubMed:16791849}.; DISEASE: Multiple self-healing squamous epithelioma (MSSE) [MIM:132800]: A disorder characterized by multiple skin tumors that undergo spontaneous regression. Tumors appear most often on sun-exposed regions, are locally invasive, and undergo spontaneous resolution over a period of months leaving pitted scars. {ECO:0000269|PubMed:21358634}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07267; DB04480; DB03921; DB12010; DB08450; DB07152; DB04434 Interacts with P62942; P01137; P63104; PRO_0000045599 [Q99IB8]; P02750 EC number EC 2.7.11.30 Uniprot keywords 3D-structure; Alternative splicing; Aortic aneurysm; Apoptosis; ATP-binding; Cell junction; Cell membrane; Craniosynostosis; Differentiation; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; Growth regulation; Isopeptide bond; Kinase; Magnesium; Manganese; Membrane; Metal-binding; Nucleotide-binding; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Serine/threonine-protein kinase; Signal; Tight junction; Transferase; Transmembrane; Transmembrane helix; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 33706.4 Length 295 Aromaticity 0.09 Instability index 43.4 Isoelectric point 8.8 Charge (pH=7) 5.17 2D Binding mode Binding energy (Kcal/mol) -6.47  Molscript Map 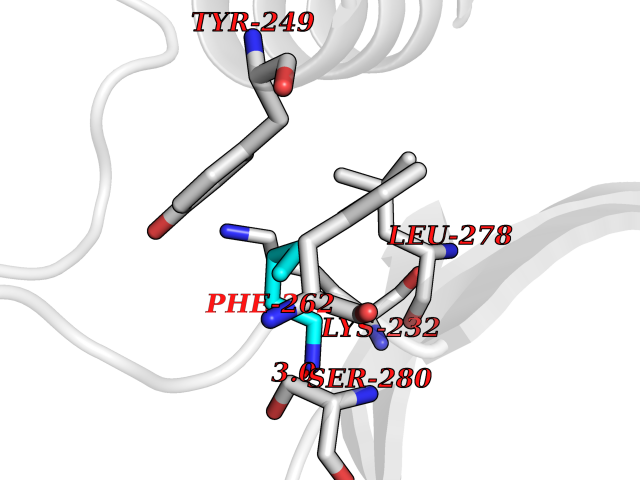 Pymol Map 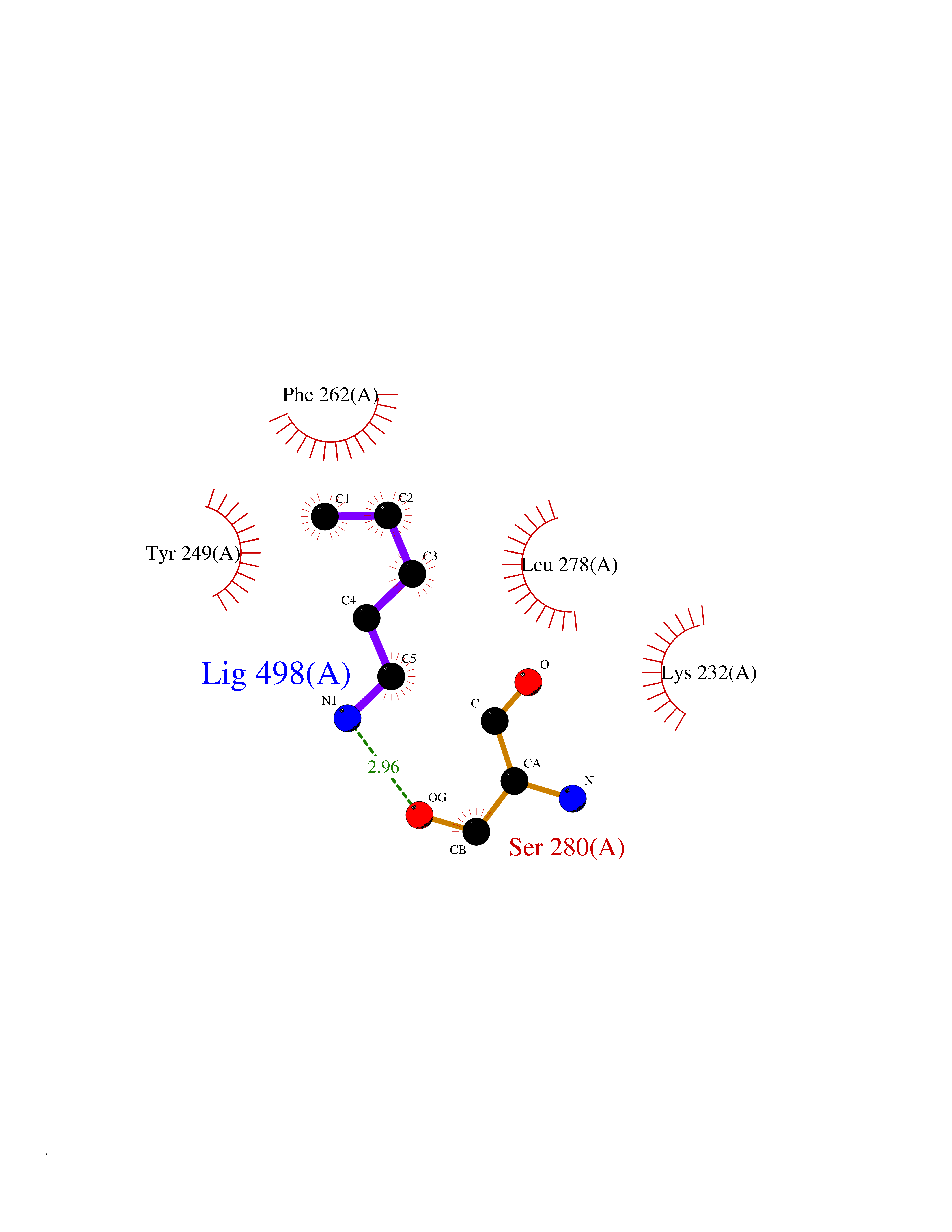 Ligplot Map 3D Binding mode Sequence TIARTIVLQESIGKGRFGEVWRGKWRGEEVAVKIFSSREERSWFREAEIYQTVMLRHENILGFIAADNKDNGTWTQLWLVSDYHEHGSLFDYLNRYTVTVEGMIKLALSTASGLAHLHMEIVGTQGKPAIAHRDLKSKNILVKKNGTCCIADLGLAVRHDSATDTIDIAPRVGTKRYMAPEVLDDSINMKHFESFKRADIYAMGLVFWEIARRCSIGGIHEDYQLPYYDLVPSDPSVEEMRKVVCEQKLRPNIPNRWQSCEALRVMAKIMRECWYANGAARLTALRIKKTLSQLS Hydrogen bonds contact Hydrophobic contact | ||||
| 85 | Nicotinamide phosphoribosyltransferase (NAMPT) | 2E5D | 4.74 | |
Target general information Gen name NAMPT Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Visfatin; PreBcell colonyenhancing factor 1; PreB cellenhancing factor; Pre-B-cell colony-enhancing factor 1; Pre-B cell-enhancing factor; PBEF1; PBEF; Nampt; NAmPRTase Protein family NAPRTase family Biochemical class Glycosyltransferases Function It is the rate limiting component in the mammalian NAD biosynthesis pathway. The secreted form behaves both as a cytokine with immunomodulating properties and an adipokine with anti-diabetic properties, it has no enzymatic activity, partly because of lack of activation by ATP, which has a low level in extracellular space and plasma. Plays a role in the modulation of circadian clock function. NAMPT-dependent oscillatory production of NAD regulates oscillation of clock target gene expression by releasing the core clock component: CLOCK-ARNTL/BMAL1 heterodimer from NAD-dependent SIRT1-mediated suppression. Catalyzes the condensation of nicotinamide with 5-phosphoribosyl-1-pyrophosphate to yield nicotinamide mononucleotide, an intermediate in the biosynthesis of NAD. Related diseases Hemolytic anemia, non-spherocytic, due to glucose phosphate isomerase deficiency (HA-GPID) [MIM:613470]: A form of anemia in which there is no abnormal hemoglobin or spherocytosis. It is caused by glucose phosphate isomerase deficiency. {ECO:0000269|PubMed:28803808, ECO:0000269|PubMed:7989588, ECO:0000269|PubMed:8499925, ECO:0000269|PubMed:8822952, ECO:0000269|PubMed:8822954, ECO:0000269|PubMed:9446754, ECO:0000269|PubMed:9856489}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB12980; DB12731; DB05217 Interacts with P02792; Q01628; P03886; P43490; Q70CQ1-2 EC number EC 2.4.2.12 Uniprot keywords 3D-structure; Acetylation; Biological rhythms; Cytokine; Cytoplasm; Glycosyltransferase; Nucleus; Phosphoprotein; Proteomics identification; Pyridine nucleotide biosynthesis; Reference proteome; Secreted; Transferase Protein physicochemical properties Chain ID A,B Molecular weight (Da) 105483 Length 932 Aromaticity 0.11 Instability index 34.4 Isoelectric point 6.68 Charge (pH=7) -2.24 2D Binding mode Binding energy (Kcal/mol) -6.46 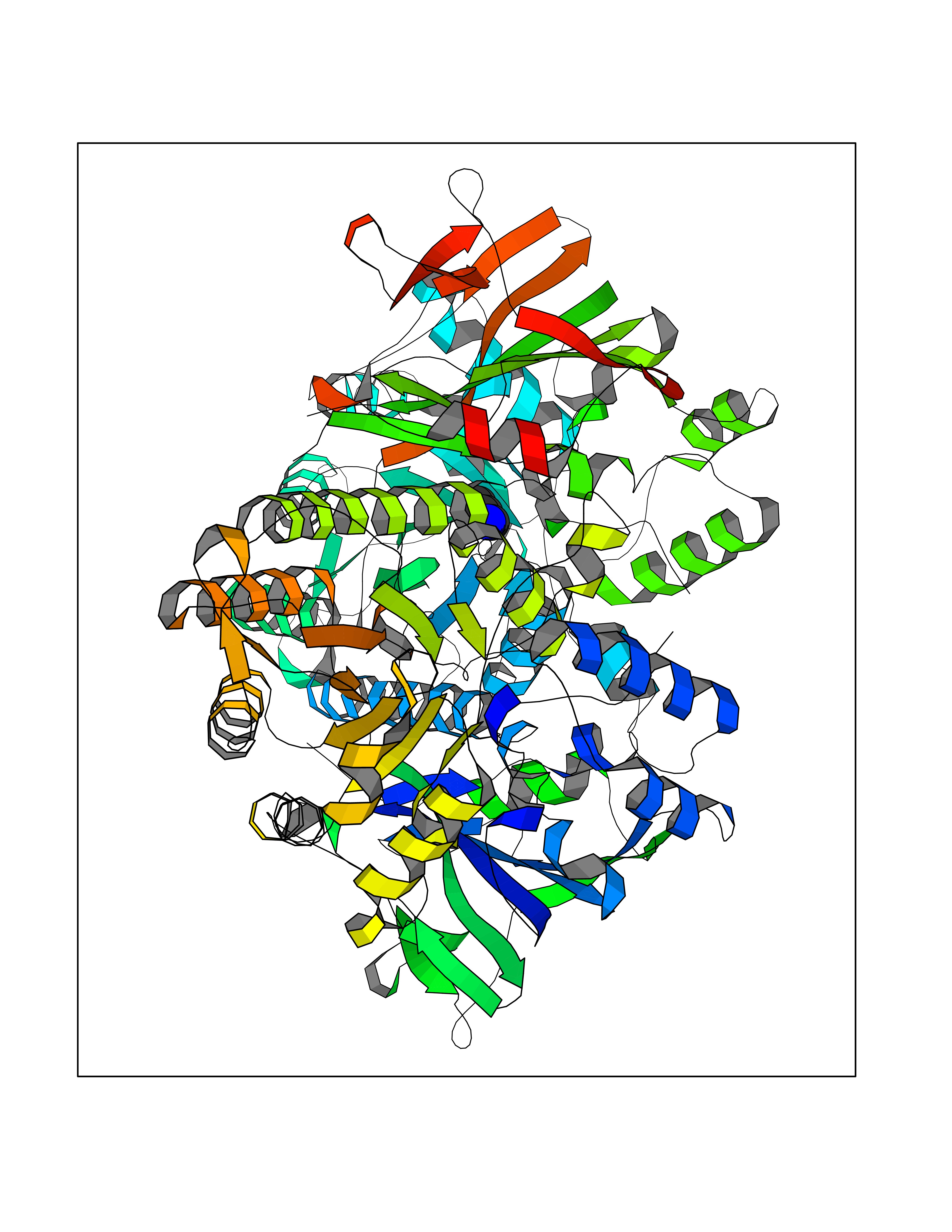 Molscript Map 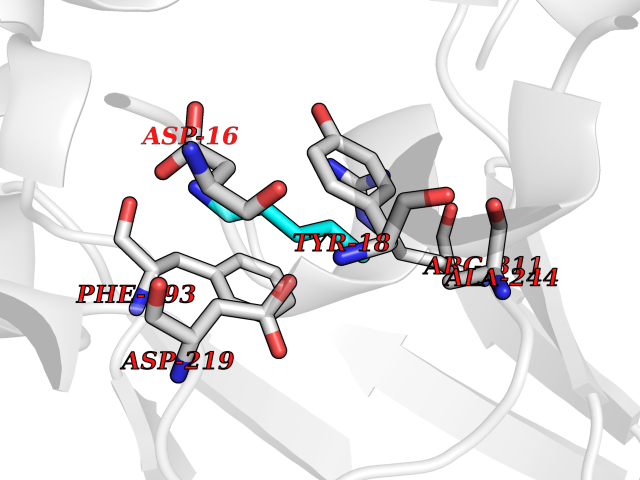 Pymol Map 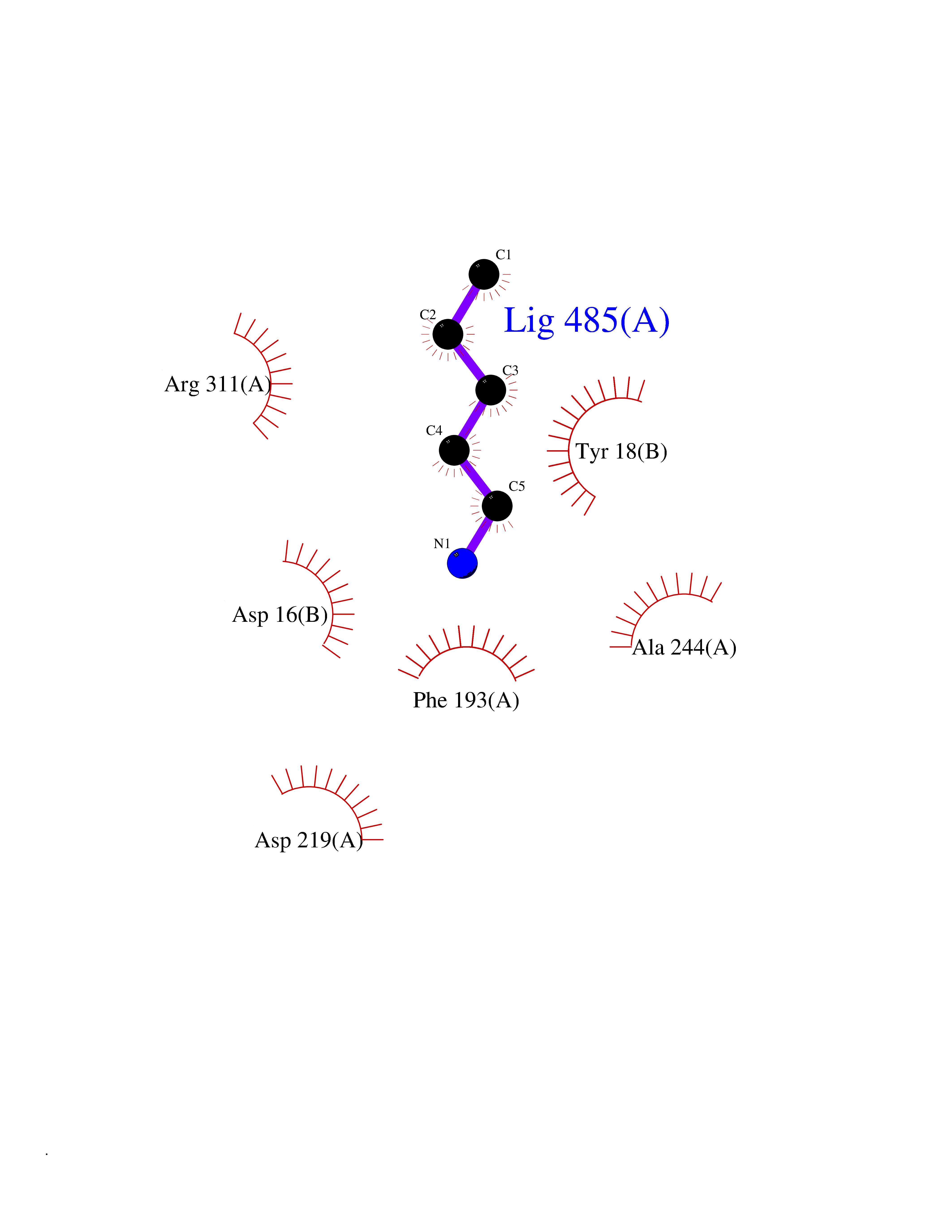 Ligplot Map 3D Binding mode Sequence EFNILLATDSYKVTHYKQYPPNTSKVYSYFECREKKYEETVFYGLQYILNKYLKGKVVTKEKIQEAKDVYKEHFQDDVFNEKGWNYILEKYDGHLPIEIKAVPEGFVIPRGNVLFTVENTDPECYWLTNWIETILVQSWYPITVATNSREQKKILAKYLLETSGNLDGLEYKLHDFGYRGVSSQETAGIGASAHLVNFKGTDTVAGLALIKKYYGTKDPVPGYSVPAAEHSTITAWGKDHEKDAFEHIVTQFSSVPVSVVSDSYDIYNACEKIWGEDLRHLIVSRSTQAPLIIRPDSGNPLDTVLKVLEILGKKFPVTENSKGYKLLPPYLRVIQGDGVDINTLQEIVEGMKQKMWSIENIAFGSGGGLLQKLTRDLLNCSFKCSYVVTNGLGINVFKDPVADPNKRSKKGRLSLHRTPAGNFVTLEEGKGDLEEYGQDLLHTVFKNGKVTKSYSFDEIRKNAQLNEFNILLATDSYKVTHYKQYPPNTSKVYSYFECREKKYEETVFYGLQYILNKYLKGKVVTKEKIQEAKDVYKEHFQDDVFNEKGWNYILEKYDGHLPIEIKAVPEGFVIPRGNVLFTVENTDPECYWLTNWIETILVQSWYPITVATNSREQKKILAKYLLETSGNLDGLEYKLHDFGYRGVSSQETAGIGASAHLVNFKGTDTVAGLALIKKYYGTKDPVPGYSVPAAEHSTITAWGKDHEKDAFEHIVTQFSSVPVSVVSDSYDIYNACEKIWGEDLRHLIVSRSTQAPLIIRPDSGNPLDTVLKVLEILGKKFPVTENSKGYKLLPPYLRVIQGDGVDINTLQEIVEGMKQKMWSIENIAFGSGGGLLQKLTRDLLNCSFKCSYVVTNGLGINVFKDPVADPNKRSKKGRLSLHRTPAGNFVTLEEGKGDLEEYGQDLLHTVFKNGKVTKSYSFDEIRKNAQLN Hydrogen bonds contact Hydrophobic contact | ||||
| 86 | Neuronal acetylcholine receptor alpha-4 (CHRNA4) | 6CNJ | 4.74 | |
Target general information Gen name CHRNA4 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Nicotinic acetylcholine receptor alpha4; CHRNA4; Alpha-4 nAChR Protein family Ligand-gated ion channel (TC 1.A.9) family, Acetylcholine receptor (TC 1.A.9.1) subfamily, Alpha-4/CHRNA4 sub-subfamily Biochemical class Neurotransmitter receptor Function After binding acetylcholine, the AChR responds by an extensive change in conformation that affects all subunits and leads to opening of an ion-conducting channel across the plasmamembrane permeable to sodium ions. Related diseases Epilepsy, nocturnal frontal lobe, 1 (ENFL1) [MIM:600513]: An autosomal dominant focal epilepsy characterized by nocturnal seizures with hyperkinetic automatisms and poorly organized stereotyped movements. {ECO:0000269|PubMed:10563623, ECO:0000269|PubMed:14623738, ECO:0000269|PubMed:7550350}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00915; DB01351; DB01352; DB00572; DB01483; DB00237; DB00241; DB01353; DB00564; DB00565; DB09028; DB01245; DB00514; DB01496; DB07720; DB00783; DB13952; DB13953; DB13954; DB13955; DB13956; DB00898; DB01354; DB01355; DB00753; DB00657; DB00333; DB00463; DB00849; DB00184; DB00312; DB01174; DB00981; DB05458; DB00794; DB05740; DB00747; DB00418; DB00202; DB00306; DB00599; DB01273 Interacts with Q6UY14-3; P05067; P83916; Q6UXH1-1; Q6UXH1-3; P20042; Q9NZR2; Q92673; P17787 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disease variant; Disulfide bond; Epilepsy; Glycoprotein; Ion channel; Ion transport; Ligand-gated ion channel; Lipoprotein; Membrane; Palmitate; Phosphoprotein; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Signal; Synapse; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A,B Molecular weight (Da) 84601.2 Length 728 Aromaticity 0.13 Instability index 39.72 Isoelectric point 5.86 Charge (pH=7) -9.84 2D Binding mode Binding energy (Kcal/mol) -6.47 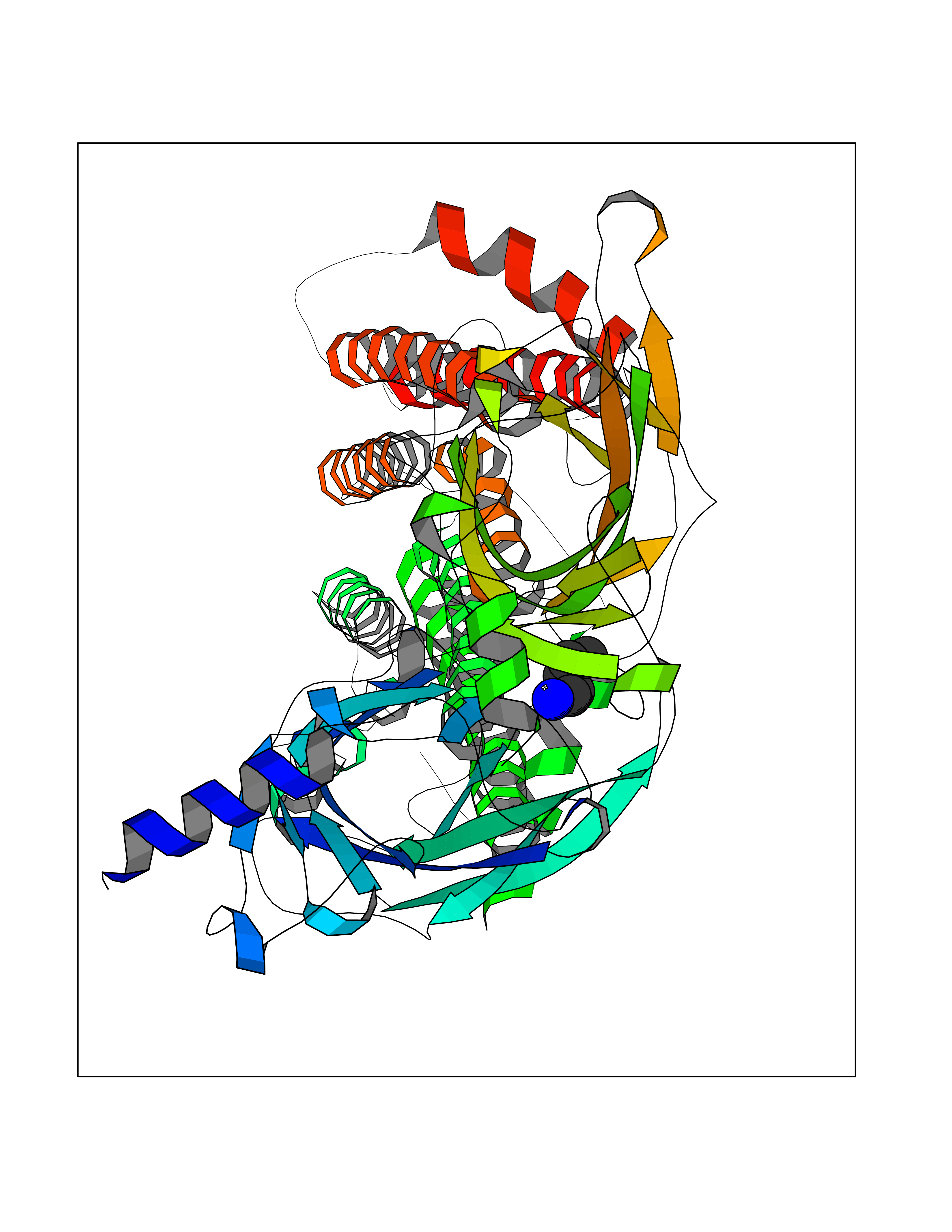 Molscript Map 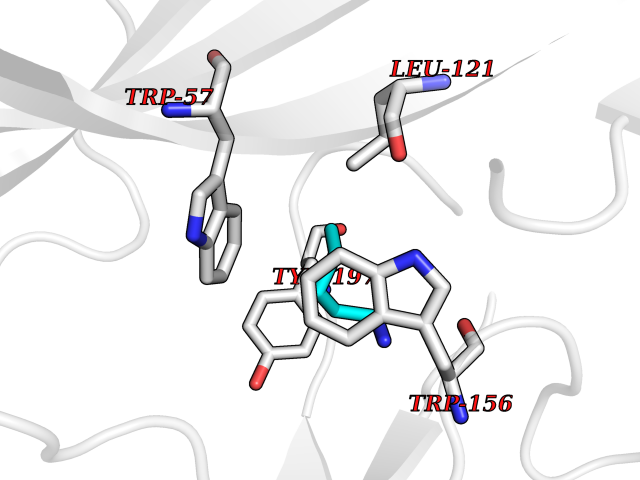 Pymol Map 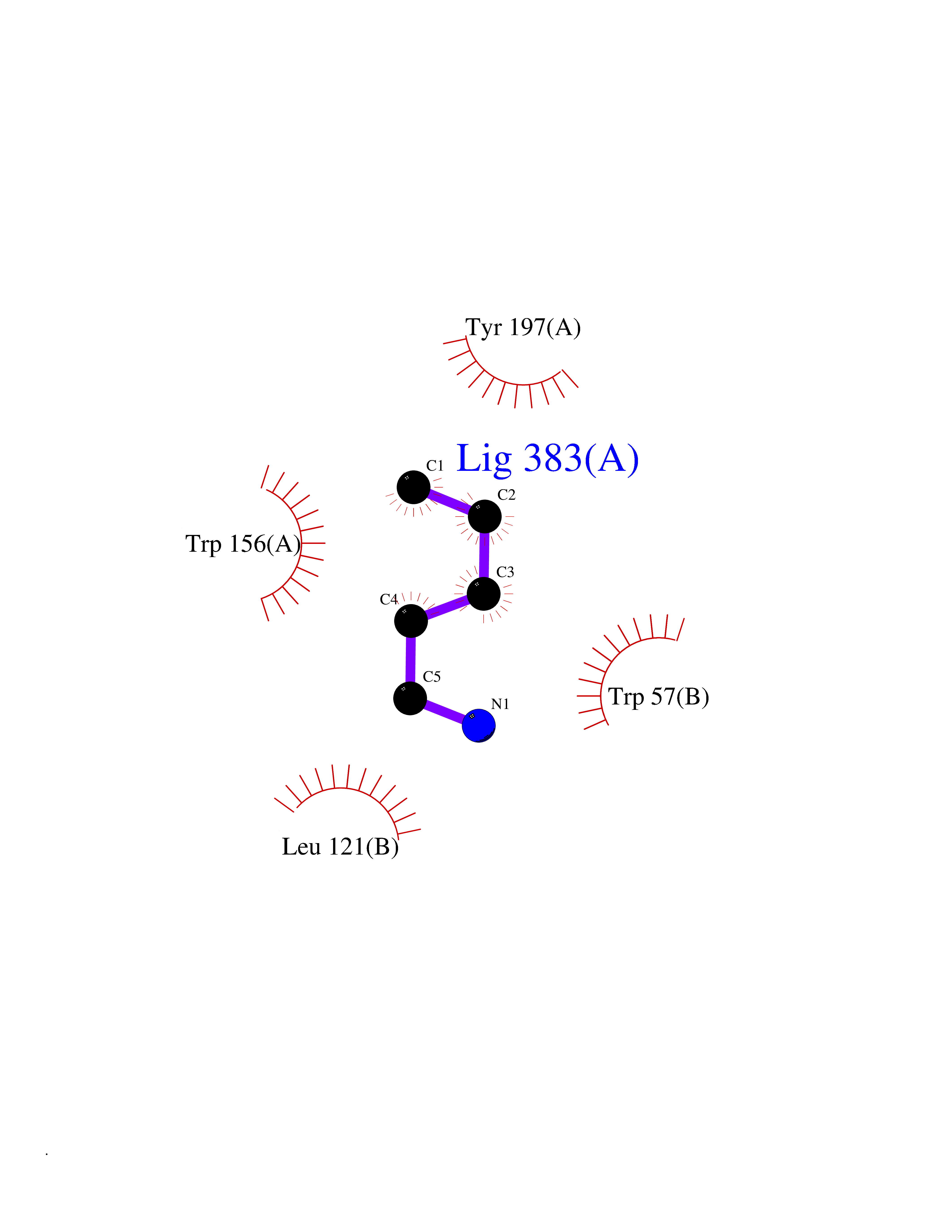 Ligplot Map 3D Binding mode Sequence ETRAHAEERLLKKLFSGYNKWSRPVANISDVVLVRFGLSIAQLIDVDEKNQMMTTNVWVKQEWHDYKLRWDPADYENVTSIRIPSELIWRPDIVLYNNADGDFAVTHLTKAHLFHDGRVQWTPPAIYKSSCSIDVTFFPFDQQNCTMKFGSWTYDKAKIDLVNMHSRVDQLDFWESGEWVIVDAVGTYNTRKYECCAEIYPDITYAFVIRRLPLFYTINLIIPCLLISCLTVLVFYLPSECGEKITLCISVLLSLTVFLLLITEIIPSTSLVIPLIGEYLLFTMIFVTLSIVITVFVLNVHHRSPRTHTMPTWVRRVFLDIVPRLLLMKRFERSVKEDWKYVAMVIDRIFLWMFIIVCLLGTVGLFLPPWDTEERLVEHLLDPSRYNKLIRPATNGSELVTVQLMVSLAQLISVHEREQIMTTNVWLTQEWEDYRLTWKPEEFDNMKKVRLPSKHIWLPDVVLYNNADGMYEVSFYSNAVVSYDGSIFWLPPAIYKSACKIEVKHFPFDQQNCTMKFRSWTYDRTEIDLVLKSEVASLDDFTPSGEWDIVALPGRRNENPDDSTYVDITYDFIIRRKPLFYTINLIIPCVLITSLAILVFYLPSDCGEKMTLCISVLLALTVFLLLISKIVPPTSLDVPLVGKYLMFTMVLVTFSIVTSVCVLNVHHRSPTTHTMAPWVKVVFLEKLPALLFMQQSVSEDWKYVAMVIDRLFLWIFVFVCVFGTIGMF Hydrogen bonds contact Hydrophobic contact | ||||
| 87 | GTPase HRas (HRAS) | 7L0F | 4.74 | |
Target general information Gen name HRAS Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms p21ras; cHras; c-H-ras; Transforming protein p21; HaRas; Ha-Ras; H-Ras-1; GTPase HRas, Nterminally processed Protein family Small GTPase superfamily, Ras family Biochemical class Small GTPase Function Ras proteins bind GDP/GTP and possess intrinsic GTPase activity. Involved in the activation of Ras protein signal transduction. Related diseases Costello syndrome (CSTLO) [MIM:218040]: A rare condition characterized by prenatally increased growth, postnatal growth deficiency, intellectual disability, distinctive facial appearance, cardiovascular abnormalities (typically pulmonic stenosis, hypertrophic cardiomyopathy and/or atrial tachycardia), tumor predisposition, skin and musculoskeletal abnormalities. {ECO:0000269|PubMed:16170316, ECO:0000269|PubMed:16329078, ECO:0000269|PubMed:16443854, ECO:0000269|PubMed:17054105, ECO:0000269|PubMed:18039947, ECO:0000269|PubMed:18247425, ECO:0000269|PubMed:19995790}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Congenital myopathy with excess of muscle spindles (CMEMS) [MIM:218040]: Variant of Costello syndrome. {ECO:0000269|PubMed:17412879}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Thyroid cancer, non-medullary, 2 (NMTC2) [MIM:188470]: A form of non-medullary thyroid cancer (NMTC), a cancer characterized by tumors originating from the thyroid follicular cells. NMTCs represent approximately 95% of all cases of thyroid cancer and are classified into papillary, follicular, Hurthle cell, and anaplastic neoplasms. {ECO:0000269|PubMed:12727991}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Mutations which change positions 12, 13 or 61 activate the potential of HRAS to transform cultured cells and are implicated in a variety of human tumors. {ECO:0000269|PubMed:3670300}.; DISEASE: Bladder cancer (BLC) [MIM:109800]: A malignancy originating in tissues of the urinary bladder. It often presents with multiple tumors appearing at different times and at different sites in the bladder. Most bladder cancers are transitional cell carcinomas that begin in cells that normally make up the inner lining of the bladder. Other types of bladder cancer include squamous cell carcinoma (cancer that begins in thin, flat cells) and adenocarcinoma (cancer that begins in cells that make and release mucus and other fluids). Bladder cancer is a complex disorder with both genetic and environmental influences. {ECO:0000269|PubMed:6298635, ECO:0000269|PubMed:6844927}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Schimmelpenning-Feuerstein-Mims syndrome (SFM) [MIM:163200]: A disease characterized by sebaceous nevi, often on the face, associated with variable ipsilateral abnormalities of the central nervous system, ocular anomalies, and skeletal defects. Many oral manifestations have been reported, not only including hypoplastic and malformed teeth, and mucosal papillomatosis, but also ankyloglossia, hemihyperplastic tongue, intraoral nevus, giant cell granuloma, ameloblastoma, bone cysts, follicular cysts, oligodontia, and odontodysplasia. Sebaceous nevi follow the lines of Blaschko and these can continue as linear intraoral lesions, as in mucosal papillomatosis. {ECO:0000269|PubMed:22683711}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB04315; DB04137; DB02210; DB08751; DB03226; DB15588 Interacts with Q99996-3; P53677-2; P10398; Q9NXL2-1; Q9UII2; Q9H7T9; Q00994; Q9H2G9; P15056; Q7Z569; Q5PSV4; Q9ULD4-2; Q96LL4; Q96HB5; Q49A88-3; Q96GN5-2; P24941; O95674; Q9H3R5; Q9Y4F5-3; Q86XR8; Q494V2-2; Q8WUX9; Q14117; Q9Y6W6; O14641; A0AVK6; Q8NB25; Q8IZU1; O94868-3; P15407; P15408; P52655; Q96CS2; Q9BT25; Q8IV36; O43248; Q53GQ0; P10809; Q8NDH6-2; Q8IY31-2; Q8NA54; Q13352; P28290-2; Q9BVG8-5; Q2M2Z5; Q6P597; P57682; Q9UH77; P08727; Q14525; Q14847-2; Q96LR2; P27338; Q99558; Q96EZ8; Q8TAC0; Q5JXC2; Q8NEH6; Q9Y605; Q96HT8; Q9GZM8; P21359; Q8N5V2; Q6PHZ7; Q9BZ95-3; A5D8V7; O43482; Q9BR81; O15534; Q9BUL5; O00329; O00329-2; Q9UPR0; Q96I34; Q15435-3; P04049; P11233; Q15311; Q12967; Q9NS23-2; Q9NS23-4; Q8WWW0; Q8TBY0; Q9P2K3-2; Q9NZL6; O15211; Q8IXN7; Q13671; Q13671-1; Q8WVD3; Q9BY12-3; Q13435; Q12824; Q13573; Q07889; Q86W54-2; Q92783-2; O75886; Q13586; Q8N4C7; O75528; P54274-2; Q9BXU0; Q5T0J7-2; Q5T1C6; Q8IUR5-4; P36406; Q86WT6-2; Q99598; Q6PF05; Q9UGJ1-2; Q9Y5Z9; P22415; Q495M9; Q9H270; Q8NEZ2; P19544-6; O43829; Q9C0F3; Q7Z637; Q86V28; P42337; Q9Z0S9; Q9EQZ6; P27671; Q5EBH1; Q5EBH1-1; P52306-5 EC number NA Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Cell membrane; Cytoplasm; Direct protein sequencing; Disease variant; Glycoprotein; Golgi apparatus; GTP-binding; Hydrolase; Isopeptide bond; Lipoprotein; Membrane; Methylation; Nucleotide-binding; Nucleus; Palmitate; Prenylation; Proteomics identification; Proto-oncogene; Reference proteome; S-nitrosylation; Ubl conjugation Protein physicochemical properties Chain ID E,F Molecular weight (Da) 28737.2 Length 259 Aromaticity 0.1 Instability index 30.69 Isoelectric point 5.64 Charge (pH=7) -4.15 2D Binding mode Binding energy (Kcal/mol) -6.46 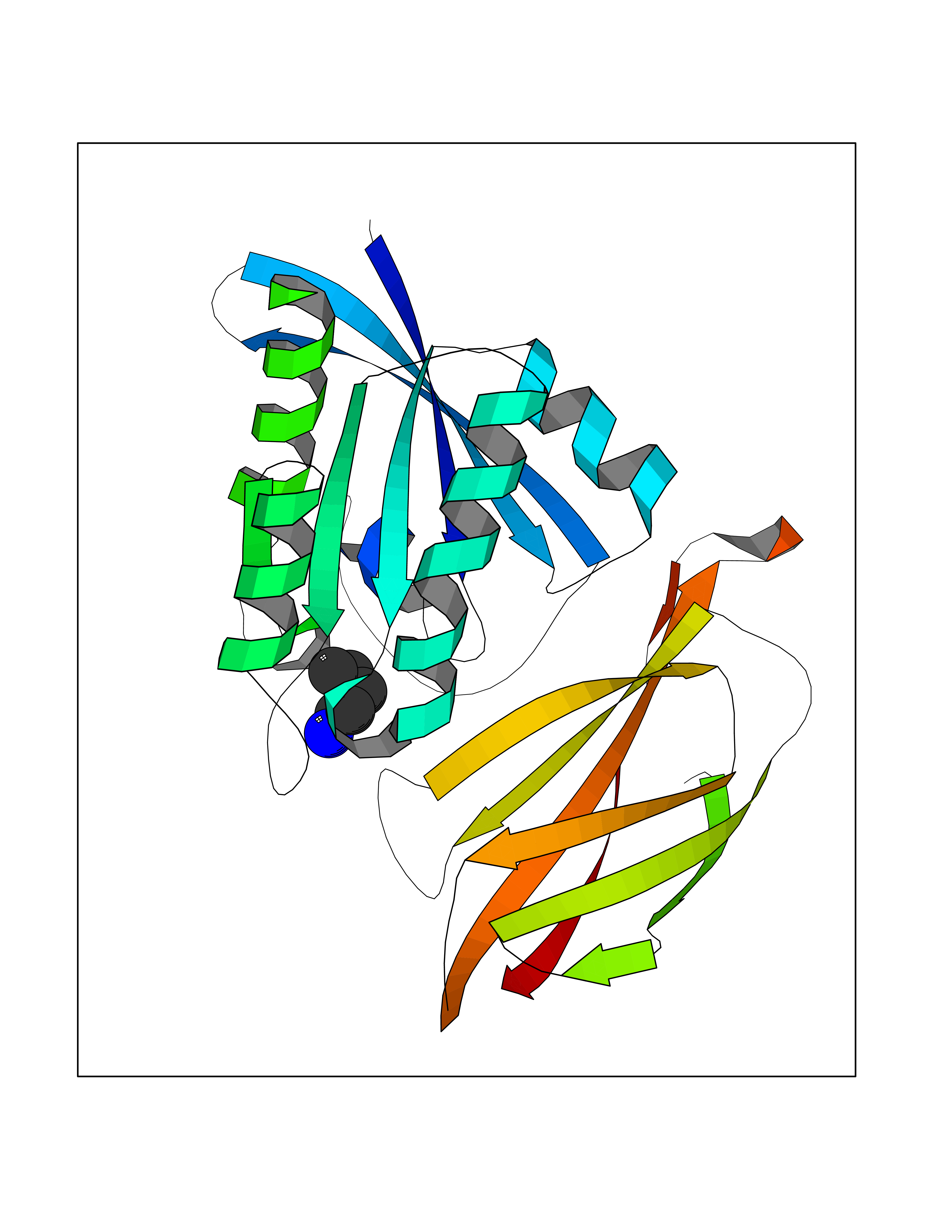 Molscript Map 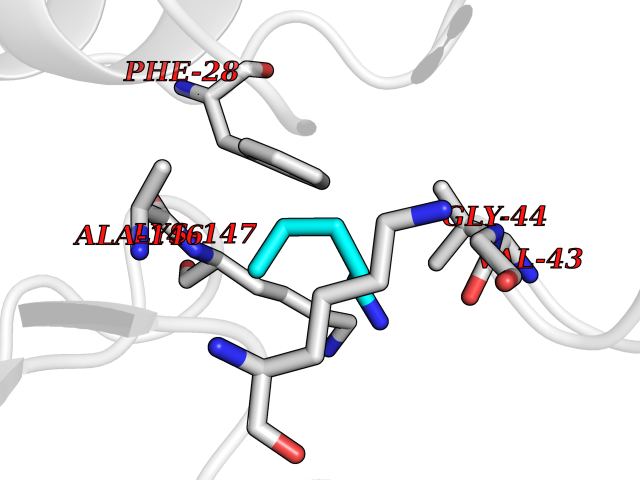 Pymol Map 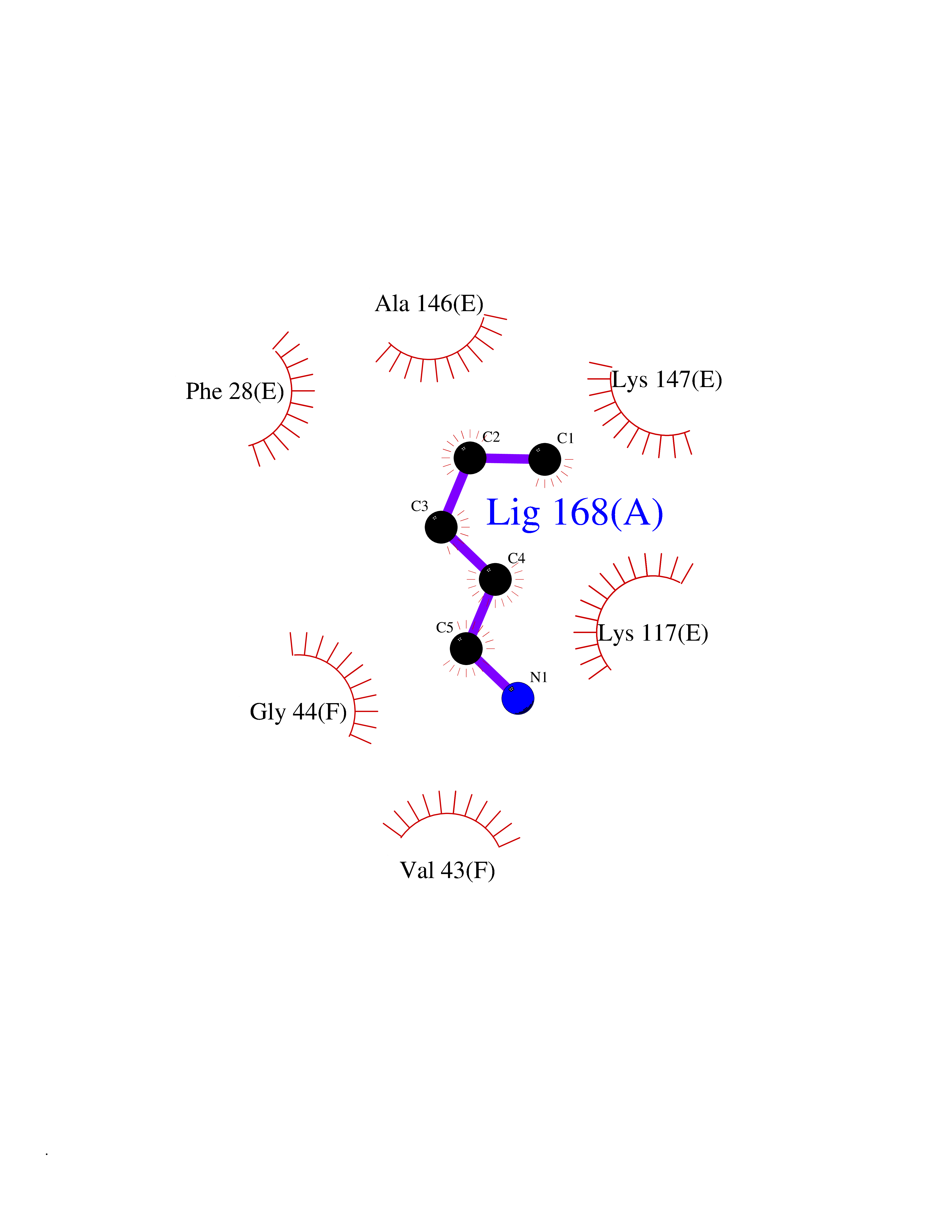 Ligplot Map 3D Binding mode Sequence MTEYKLVVVGAGGVGKSALTIQLIQNHFVDEYDPTIEDSYRKQVVIDGETCLLDILDTAGQEEYSAMRDQYMRTGEGFLCVFAINNTKSFEDIHQYREQIKRVKDSDDVPMVLVGNKCDLAARTVESRQAQDLARSYGIPYIETSAKTRQGVEDAFYTLVREIRQHSVPTKLEVVAATPTSLLISWDAPAVTVFFYIIAYGETGHGVGAFQAFRVPGSKSTATISGLKPGVDYTITVYARGYSKQGPYKPSPISINYRT Hydrogen bonds contact Hydrophobic contact | ||||
| 88 | NAPE-hydrolyzing phospholipase D (NAPE-PLD) | 4QN9 | 4.74 | |
Target general information Gen name NAPEPLD Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms NAPE-PLD; N-acyl-phosphatidylethanolamine-hydrolyzing phospholipase D; N-acyl phosphatidylethanolamine phospholipase D; C7orf18 Protein family NAPE-PLD family Biochemical class NA Function Hydrolyzes N-acyl-phosphatidylethanolamines (NAPEs) to produce N-acylethanolamines (NAEs) and phosphatidic acid. Responsible for the generation of these bioactive fatty acid ethanolamides (FAEs), including anandamide (N-arachidonoylethanolamine), the ligand of cannabinoid and vanilloid receptors. As a regulator of lipid metabolism in the adipose tissue, mediates the crosstalk between adipocytes, gut microbiota and immune cells to control body temperature and weight. In particular, regulates energy homeostasis by promoting cold-induced brown or beige adipocyte differentiation program to generate heat from fatty acids and glucose (By similarity). Related diseases Glioma (GLM) [MIM:137800]: Gliomas are benign or malignant central nervous system neoplasms derived from glial cells. They comprise astrocytomas and glioblastoma multiforme that are derived from astrocytes, oligodendrogliomas derived from oligodendrocytes and ependymomas derived from ependymocytes. {ECO:0000269|PubMed:22286061, ECO:0000269|PubMed:22286216, ECO:0000269|PubMed:23539269}. The gene represented in this entry is involved in disease pathogenesis. H3F3A mutations affecting residues involved in post-translational modifications of histone H3.3 are recurrent in malignant, aggressive gliomas including glioblastoma multiforme (GBM) and diffuse intrinsic pontine glioma (DIPG) (PubMed:22286061, PubMed:22286216). The mechanism through which mutations lead to tumorigenesis involves altered histones methylation, impaired regulation of Polycomb repressive complex 2 (PRC2) activity, and aberrant epigenetic regulation of gene expression (PubMed:23539183, PubMed:23539269, PubMed:23603901). {ECO:0000269|PubMed:22286061, ECO:0000269|PubMed:22286216, ECO:0000269|PubMed:23539183, ECO:0000269|PubMed:23539269, ECO:0000269|PubMed:23603901}.; DISEASE: Bryant-Li-Bhoj neurodevelopmental syndrome 1 (BRYLIB1) [MIM:619720]: An autosomal dominant disorder predominantly characterized by global developmental delay, impaired intellectual development, poor or absent speech, and delayed motor milestones. Clinical manifestations are highly variable, including abnormal head shape, dysmorphic facial features, oculomotor abnormalities, feeding problems, and non-specific brain imaging abnormalities. Additional features may include hearing loss, seizures, short stature, and mild skeletal defects. {ECO:0000269|PubMed:33268356, ECO:0000269|PubMed:34876591}. The disease is caused by variants affecting the gene represented in this entry. BRYLIB1 is caused by variants in H3-3A. {ECO:0000269|PubMed:33268356, ECO:0000269|PubMed:34876591}.; DISEASE: Bryant-Li-Bhoj neurodevelopmental syndrome 2 (BRYLIB2) [MIM:619721]: An autosomal dominant disorder predominantly characterized by global developmental delay, impaired intellectual development, poor or absent speech, and delayed motor milestones. Clinical manifestations are highly variable, including abnormal head shape, dysmorphic facial features, oculomotor abnormalities, feeding problems, and non-specific brain imaging abnormalities. Additional features may include hearing loss, seizures, short stature, and mild skeletal defects. {ECO:0000269|PubMed:33268356, ECO:0000269|PubMed:34876591}. The disease is caused by variants affecting the gene represented in this entry. BRYLIB2 is caused by variants in H3-3B. {ECO:0000269|PubMed:33268356, ECO:0000269|PubMed:34876591}.; DISEASE: H3F3A and H3F3B mutations affecting residues involved in post-translational modifications of histone H3.3 are implicated in the pathogenesis of some bone and cartilage neoplasms. Mutations have been found with high prevalence in chondroblastoma and giant cell tumors of bone, and with low frequency in osteosarcoma, conventional chondrosarcoma and clear cell chondrosarcoma. Chondroblastoma samples frequently carry a H3F3B mutation affecting residue Lys-37 (H3K36), although H3F3A is mutated in some cases. Most giant cell tumors of bone harbor H3F3A mutations affecting residue Gly-35 (H3G34). {ECO:0000269|PubMed:24162739}. Drugs (DrugBank ID) DB14009 Interacts with Q6IQ20 EC number EC 3.1.4.54 Uniprot keywords 3D-structure; Acetylation; Endosome; Golgi apparatus; Hydrolase; Lipid degradation; Lipid metabolism; Membrane; Metal-binding; Nucleus; Phospholipid degradation; Phospholipid metabolism; Proteomics identification; Reference proteome; Zinc Protein physicochemical properties Chain ID A,B Molecular weight (Da) 74256.5 Length 643 Aromaticity 0.13 Instability index 48.34 Isoelectric point 5.65 Charge (pH=7) -17.61 2D Binding mode Binding energy (Kcal/mol) -6.47  Molscript Map 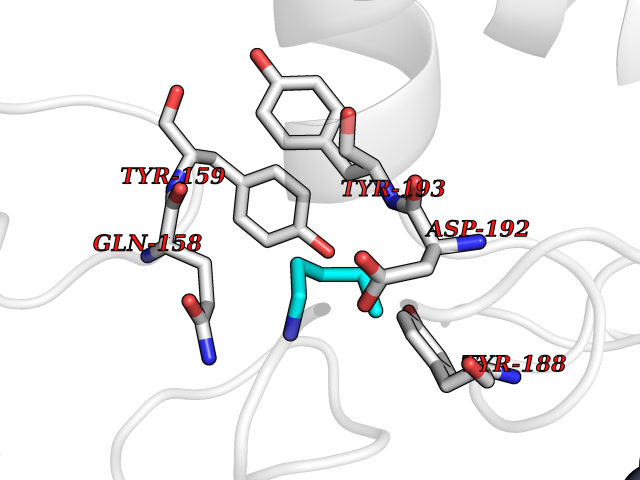 Pymol Map 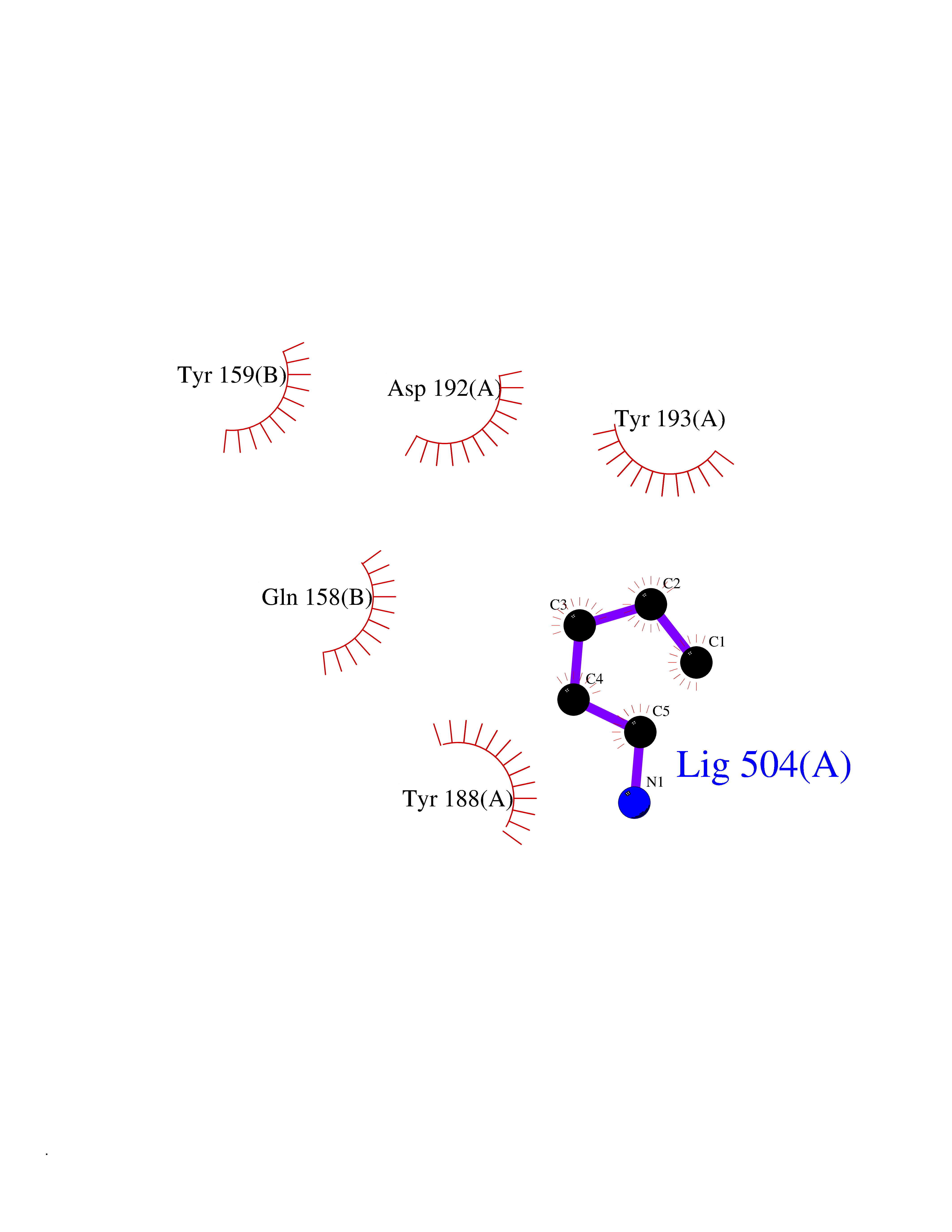 Ligplot Map 3D Binding mode Sequence SKKGKDGRFVNPWPTWKNPSIPNSSVPSSKEELDKELPVLKPYFITNPEEAGVREAGLRVTWLGHATVMVEMDELIFLTDPIFSSRASPSQYMGPKRFRRSPCTISELPPIDAVLISHNHYDHLDYNSVIALNERFGNELRWFVPLGLLDWMQKCGCENVIELDWWEENCVPGHDKVTFVFTPSQHWCKRTLMDDNKVLWGSWSVLGPWNRFFFAGDTGYCPAFEEIGKRFGPFDLAAIPIGAYEPRWFMKYQHVDPEEAVRIHTDVQTKKSMAIHWGTFALANEHYLEPPVKLNEALERYGLNAEDFFVLKHGESRYLNNSKKGKDGRFVNPWPTWKNPSIPNSSVPSSKEELDKELPVLKPYFITNPEEAGVREAGLRVTWLGHATVMVEMDELIFLTDPIFSSRASPSQYMGPKRFRRSPCTISELPPIDAVLISHNHYDHLDYNSVIALNERFGNELRWFVPLGLLDWMQKCGCENVIELDWWEENCVPGHDKVTFVFTPSQHWCKRTLMDDNKVLWGSWSVLGPWNRFFFAGDTGYCPAFEEIGKRFGPFDLAAIPIGAYEPRWFMKYQHVDPEEAVRIHTDVQTKKSMAIHWGTFALANEHYLEPPVKLNEALERYGLNAEDFFVLKHGESRYLNND Hydrogen bonds contact Hydrophobic contact | ||||
| 89 | Aggrecanase (ADAMTS5) | 3HY7 | 4.74 | |
Target general information Gen name ADAMTS5 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Aggrecanase-2; ADMP-2; ADAMTS5; ADAMTS-5; ADAM-TS5; ADAM-TS 5; ADAM-TS 11; A disintegrin and metalloproteinase with thrombospondinmotifs 5 Protein family NA Biochemical class Peptidase Function Cleaves aggrecan, a cartilage proteoglycan, and may be involved in its turnover. May play an important role in the destruction of aggrecan in arthritic diseases. May play a role in proteolytic processing mostly during the peri-implantation period. Related diseases Mucopolysaccharidosis 1H (MPS1H) [MIM:607014]: A severe form of mucopolysaccharidosis type 1, a rare lysosomal storage disease characterized by progressive physical deterioration with urinary excretion of dermatan sulfate and heparan sulfate. Patients with MPS1H usually present, within the first year of life, a combination of hepatosplenomegaly, skeletal deformities, corneal clouding and severe intellectual disability. Obstructive airways disease, respiratory infection and cardiac complications usually result in death before 10 years of age. {ECO:0000269|PubMed:10466419, ECO:0000269|PubMed:10735634, ECO:0000269|PubMed:12559846, ECO:0000269|PubMed:1301941, ECO:0000269|PubMed:15300847, ECO:0000269|PubMed:19396826, ECO:0000269|PubMed:21394825, ECO:0000269|PubMed:24036510, ECO:0000269|PubMed:31194252, ECO:0000269|PubMed:7550232, ECO:0000269|PubMed:7550242, ECO:0000269|PubMed:7951228, ECO:0000269|PubMed:8019563, ECO:0000269|PubMed:8328452, ECO:0000269|PubMed:8401515, ECO:0000269|Ref.20}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Mucopolysaccharidosis 1H/S (MPS1H/S) [MIM:607015]: A form of mucopolysaccharidosis type 1, a rare lysosomal storage disease characterized by progressive physical deterioration with urinary excretion of dermatan sulfate and heparan sulfate. MPS1H/S represents an intermediate phenotype of the MPS1 clinical spectrum. It is characterized by relatively little neurological involvement, but most of the somatic symptoms described for severe MPS1 develop in the early to mid-teens, causing considerable loss of mobility. {ECO:0000269|PubMed:10466419, ECO:0000269|PubMed:10735634, ECO:0000269|PubMed:12559846, ECO:0000269|PubMed:15300847, ECO:0000269|PubMed:21394825, ECO:0000269|PubMed:7550232, ECO:0000269|PubMed:7550242, ECO:0000269|PubMed:8401515}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Mucopolysaccharidosis 1S (MPS1S) [MIM:607016]: A mild form of mucopolysaccharidosis type 1, a rare lysosomal storage disease characterized by progressive physical deterioration with urinary excretion of dermatan sulfate and heparan sulfate. Patients with MPS1S may have little or no neurological involvement, normal stature and life span, but present development of joints stiffness, mild hepatosplenomegaly, aortic valve disease and corneal clouding. {ECO:0000269|PubMed:12559846, ECO:0000269|PubMed:15300847, ECO:0000269|PubMed:19396826, ECO:0000269|PubMed:21394825, ECO:0000269|PubMed:25256405, ECO:0000269|PubMed:7550232, ECO:0000269|PubMed:7550242, ECO:0000269|PubMed:8213840}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB06837; DB03880; DB06945 Interacts with P13608 EC number EC 3.4.24.- Uniprot keywords 3D-structure; Cleavage on pair of basic residues; Disulfide bond; Extracellular matrix; Glycoprotein; Hydrolase; Metal-binding; Metalloprotease; Protease; Proteomics identification; Reference proteome; Repeat; Secreted; Signal; Zinc; Zymogen Protein physicochemical properties Chain ID A Molecular weight (Da) 23997.8 Length 217 Aromaticity 0.06 Instability index 46.68 Isoelectric point 5.82 Charge (pH=7) -8.36 2D Binding mode Binding energy (Kcal/mol) -6.46 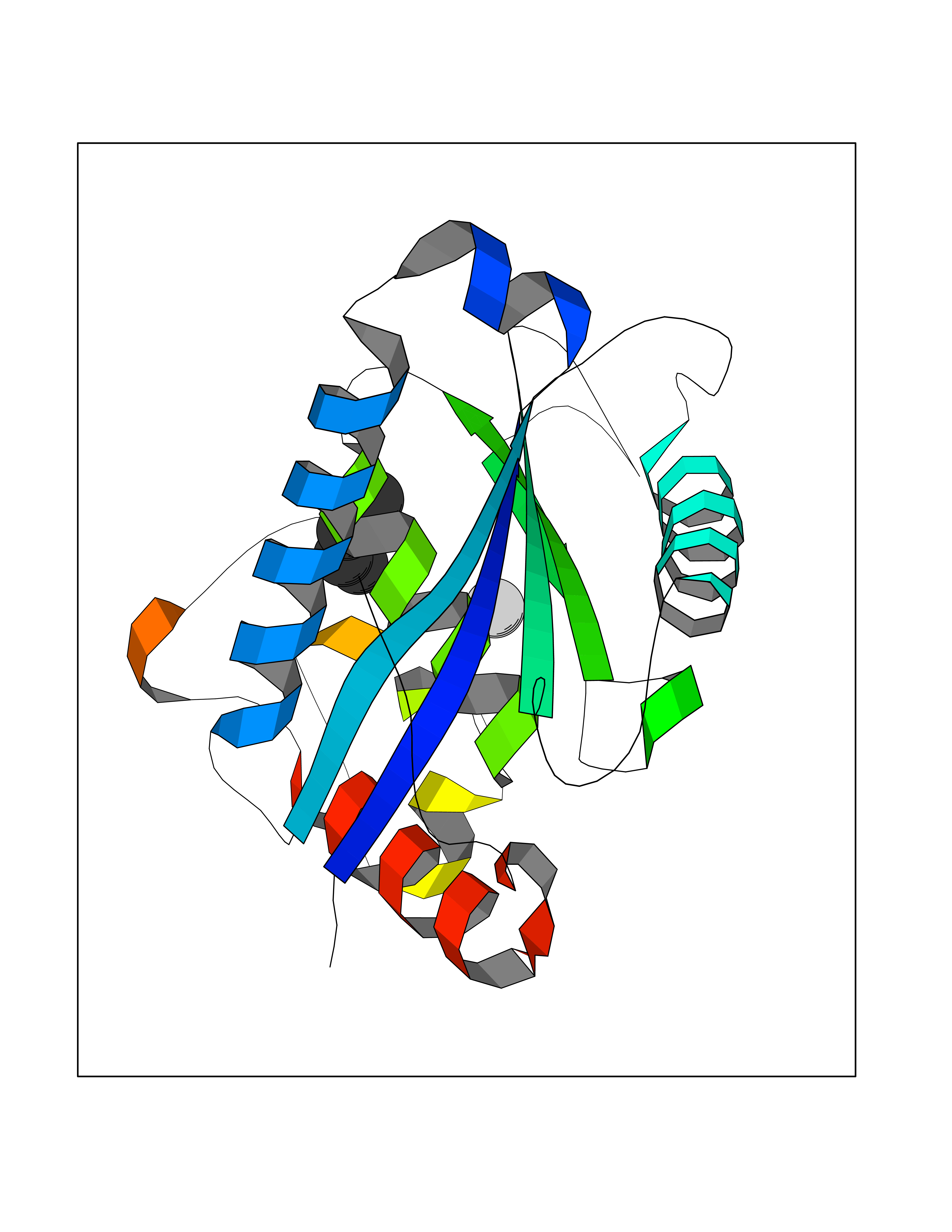 Molscript Map 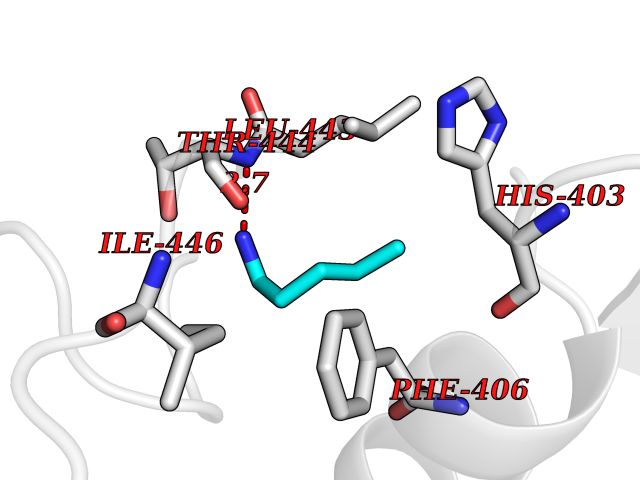 Pymol Map 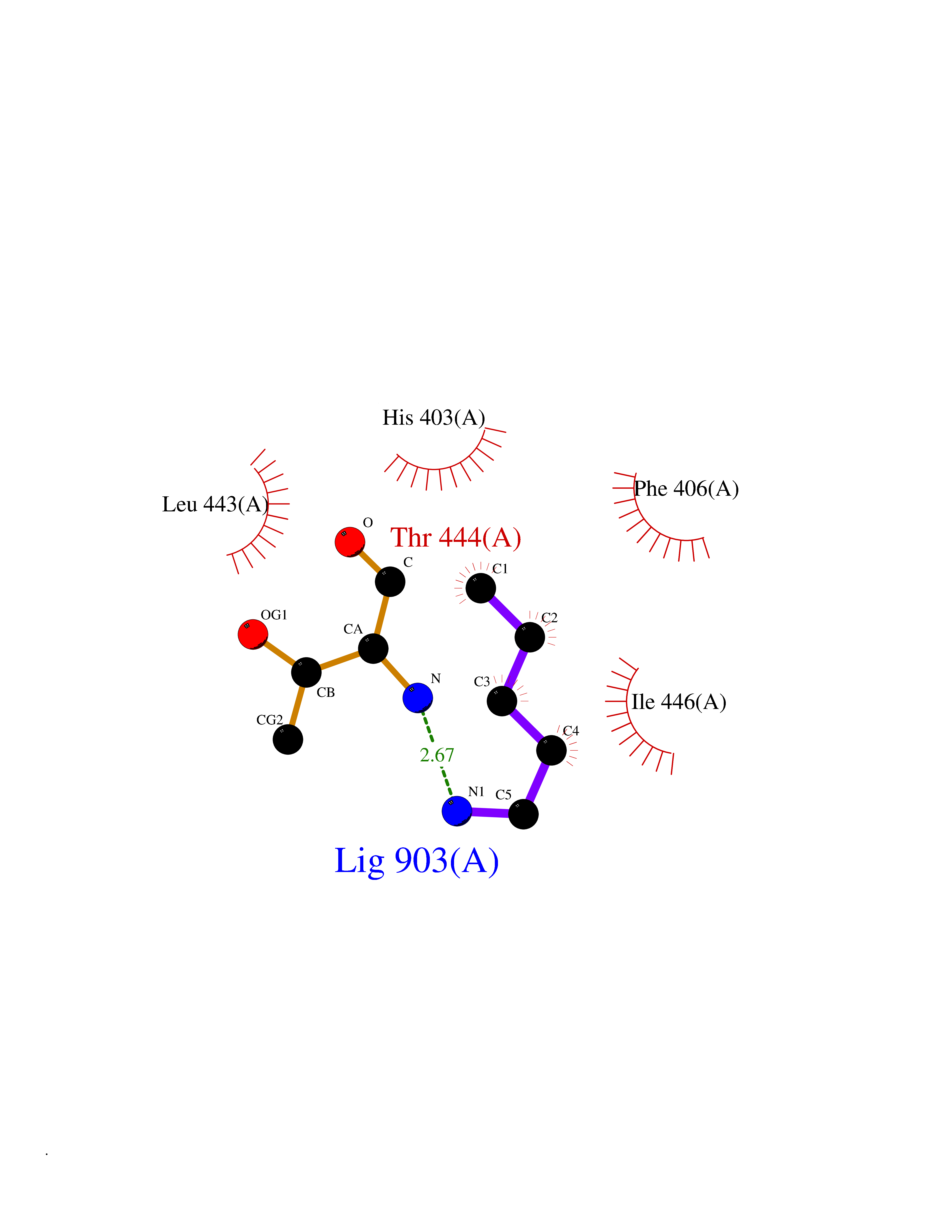 Ligplot Map 3D Binding mode Sequence SRARQVELLLVADASMARKYGRGLQHYLLTLASIANRLYSHASIENHIRLAVVKVVVLGDKDKSLEVSKNAATTLKNFCKWQHQHNQLGDDHEEHYDAAILFTREDLCGHHSCDTLGMADVGTICSPERSCAVIEDDGLHAAFTVAHEIGHLLGLSHDDSKFCEETFGSTEDKRLMSSILTSIDASKPWSKCTSATITEFLDDGHGNCLLDLPRKQI Hydrogen bonds contact Hydrophobic contact | ||||
| 90 | Deoxyribodipyrimidine photo-lyase | 1OWL | 4.73 | |
Target general information Gen name phr Organism Synechococcus sp. (strain ATCC 27144 / PCC 6301 / SAUG 1402/1) (Anacystis nidulans) Uniprot ID TTD ID NA Synonyms phrA;syc1392_c Protein family DNA photolyase class-1 family Biochemical class Lyase Function Deoxyribodipyrimidine photo-lyase activity.DNA binding.Nucleotide binding. Related diseases Myeloperoxidase deficiency (MPOD) [MIM:254600]: A disorder characterized by decreased myeloperoxidase activity in neutrophils and monocytes that results in disseminated candidiasis. {ECO:0000269|PubMed:37198333, ECO:0000269|PubMed:7904599, ECO:0000269|PubMed:8142659, ECO:0000269|PubMed:8621627, ECO:0000269|PubMed:9354683, ECO:0000269|PubMed:9637725}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03147 Interacts with NA EC number 4.1.99.3 Uniprot keywords 3D-structure; Chromophore; Direct protein sequencing; DNA damage; DNA repair; DNA-binding; FAD; Flavoprotein; Lyase; Nucleotide-binding Protein physicochemical properties Chain ID A Molecular weight (Da) 53346.9 Length 473 Aromaticity 0.1 Instability index 53.22 Isoelectric point 6.92 Charge (pH=7) -0.23 2D Binding mode Binding energy (Kcal/mol) -6.45 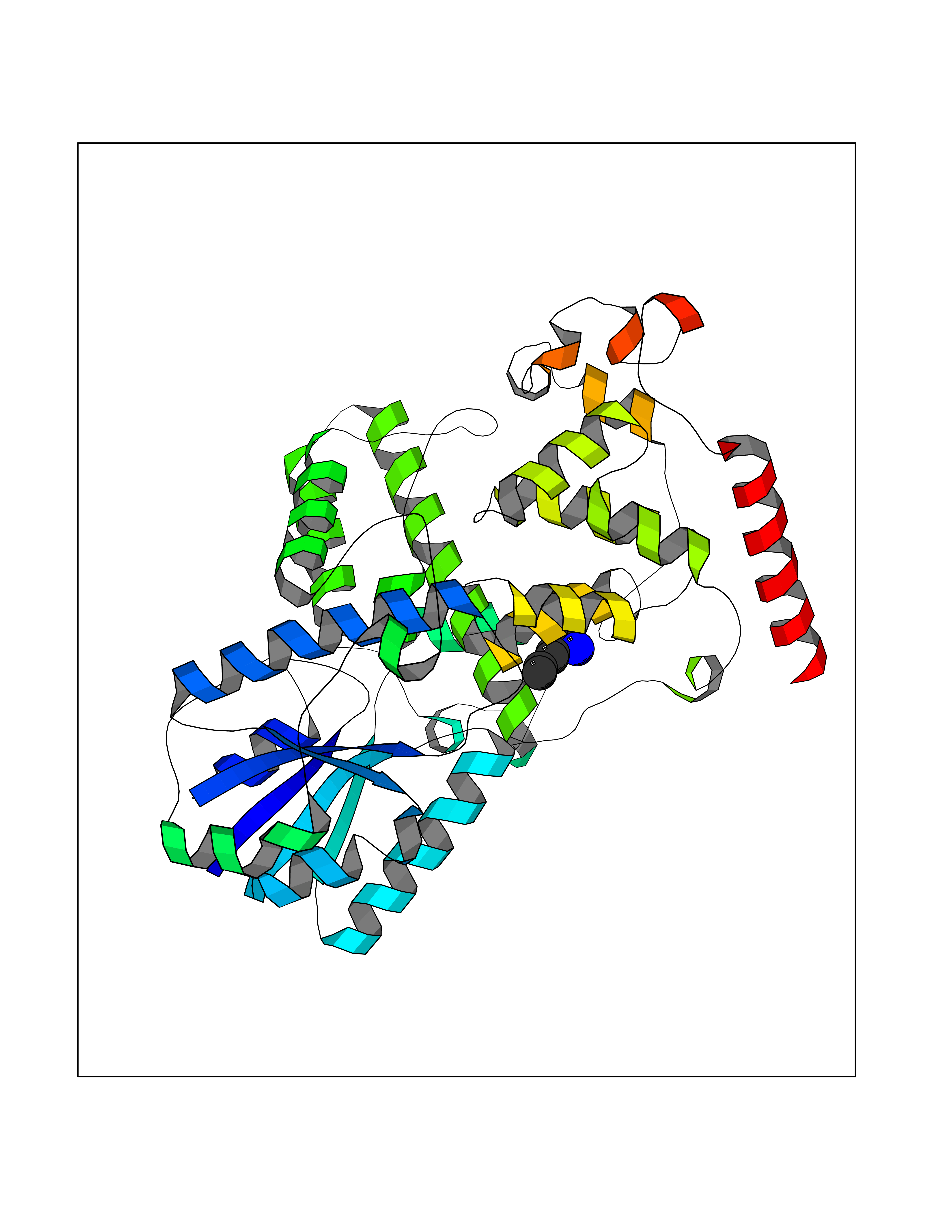 Molscript Map 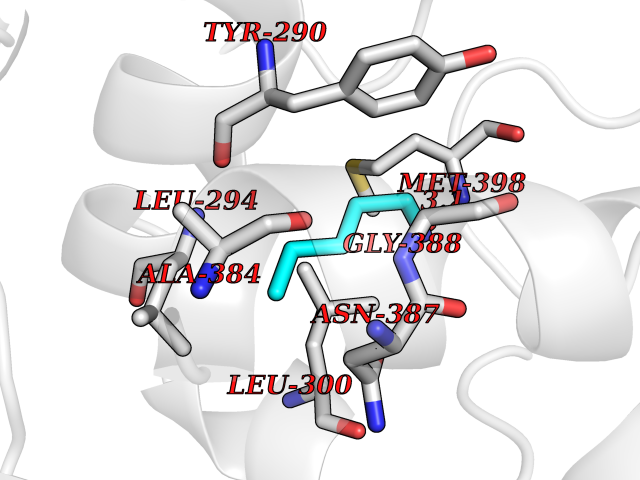 Pymol Map 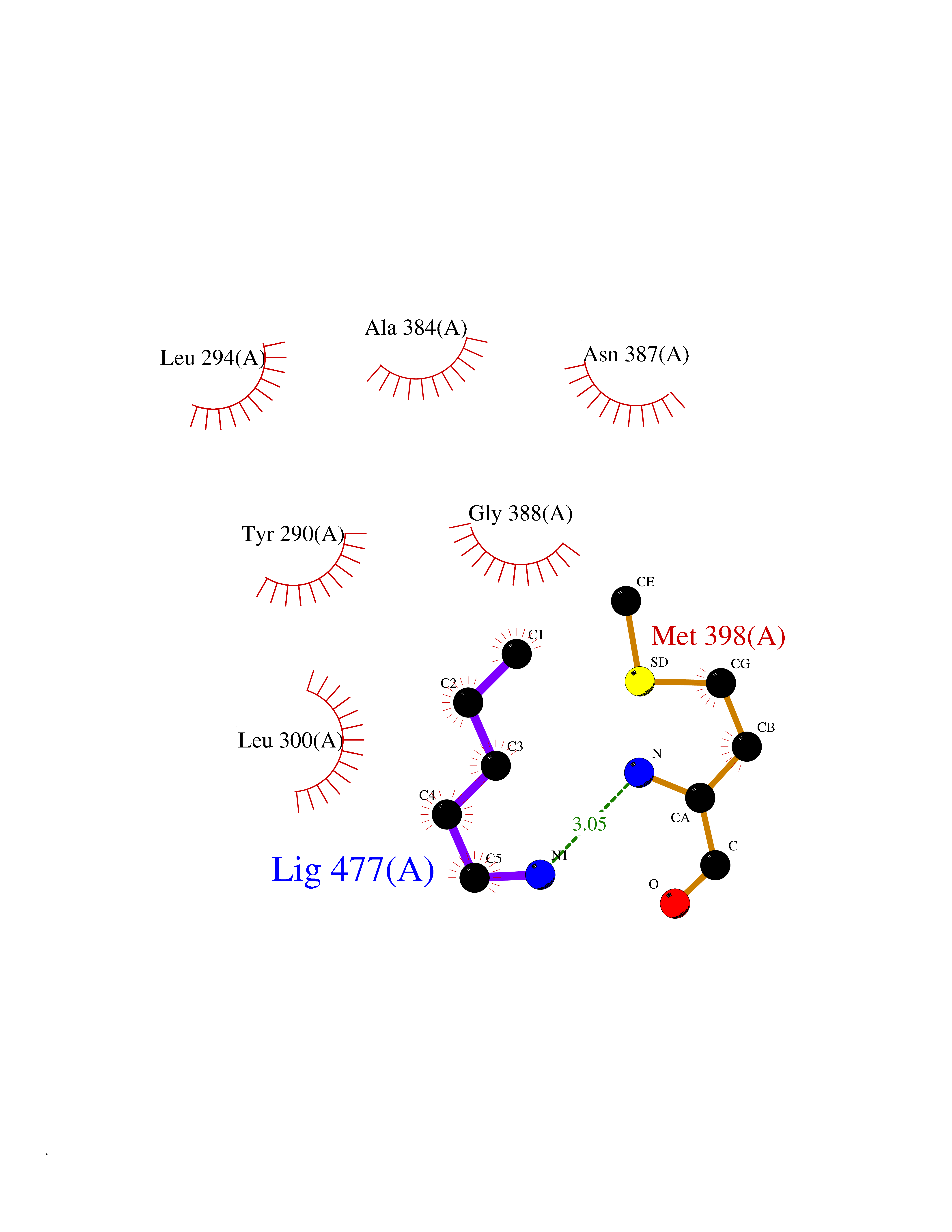 Ligplot Map 3D Binding mode Sequence APILFWHRRDLRLSDNIGLAAARAQSAQLIGLFCLDPQILQSADMAPARVAYLQGCLQELQQRYQQAGSRLLLLQGDPQHLIPQLAQQLQAEAVYWNQDIEPYGRDRDGQVAAALKTAGIRAVQLWDQLLHSPDQILSGSGNPYSVYGPFWKNWQAQPKPTPVATPTELVDLSPEQLTAIAPLLLSELPTLKQLGFDWDGGFPVEPGETAAIARLQEFCDRAIADYDPQRNFPAEAGTSGLSPALKFGAIGIRQAWQAASAAHALSRSDEARNSIRVWQQELAWREFYQHALYHFPSLADGPYRSLWQQFPWENREALFTAWTQAQTGYPIVDAAMRQLTETGWMHNRCRMIVASFLTKDLIIDWRRGEQFFMQHLVDGDLAANNGGWQWSASSGMDPKPLRIFNPASQAKKFDATATYIKRWLPELRHVHPKDLISGEITPIERRGYPAPIVNHNLRQKQFKALYNQLKAAI Hydrogen bonds contact Hydrophobic contact | ||||
| 91 | Cytosolic 10-formyltetrahydrofolate dehydrogenase | 2CFI | 4.73 | |
Target general information Gen name ALDH1L1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms FTHFD Protein family GART family; Aldehyde dehydrogenase family, ALDH1L subfamily Biochemical class Oxidoreductase Function Aldehyde dehydrogenase (NAD) activity.Catalytic activity.Formyltetrahydrofolate dehydrogenase activity.Hydroxymethyl-, formyl- and related transferase activity. Related diseases Developmental and epileptic encephalopathy 39 with leukodystrophy (DEE39) [MIM:612949]: A form of epileptic encephalopathy, a heterogeneous group of severe early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE39 is characterized by global hypomyelination of the central nervous system, with the gray matter appearing relatively unaffected. Inheritance is autosomal recessive. {ECO:0000269|PubMed:19641205, ECO:0000269|PubMed:24515575}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00116 Interacts with Q3SY69; Q92624 EC number 1.5.1.6 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Cytoplasm; NADP; One-carbon metabolism; Oxidoreductase; Phosphopantetheine; Phosphoprotein; Proteomics identification; Reference proteome Protein physicochemical properties Chain ID A Molecular weight (Da) 33869.5 Length 308 Aromaticity 0.08 Instability index 30.42 Isoelectric point 6.09 Charge (pH=7) -3.83 2D Binding mode Binding energy (Kcal/mol) -6.45 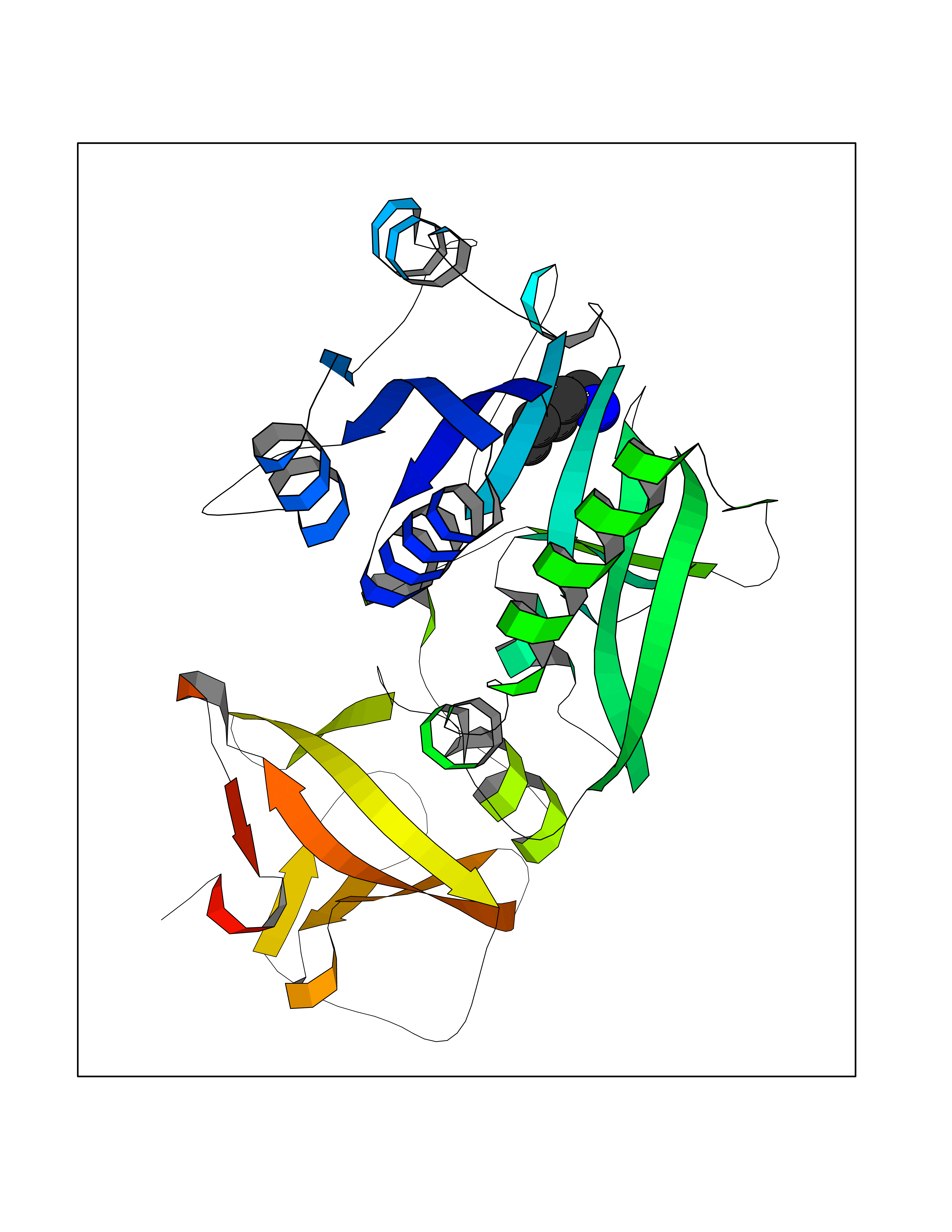 Molscript Map 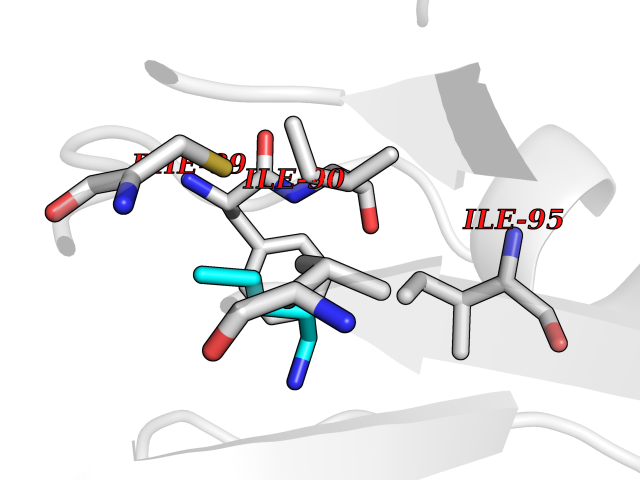 Pymol Map 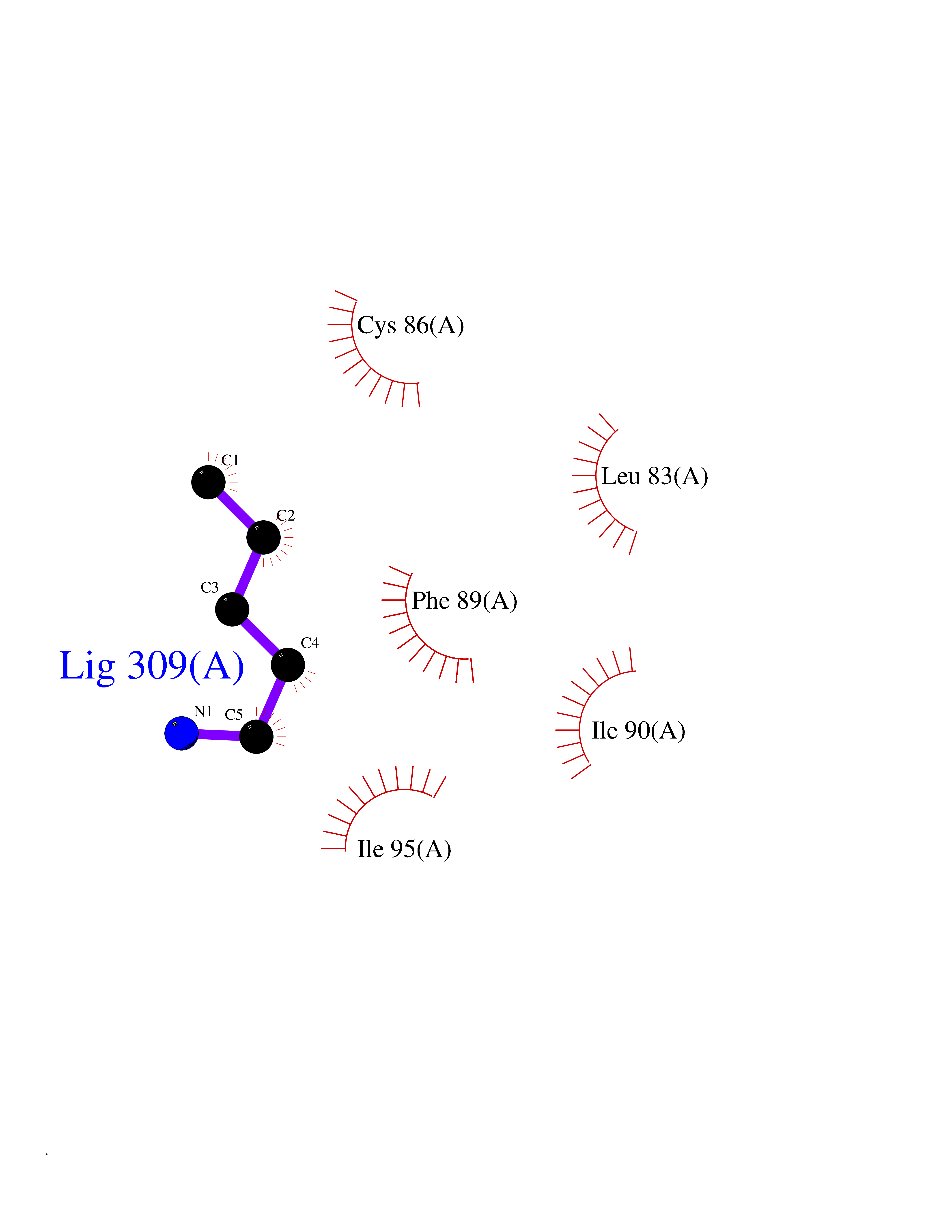 Ligplot Map 3D Binding mode Sequence SMKIAVIGQSLFGQEVYCHLRKEGHEVVGVFTVPDKDGKADPLGLEAEKDGVPVFKYSRWRAKGQALPDVVAKYQALGAELNVLPFCSQFIPMEIISAPRHGSIIYHPSLLPRHRGASAINWTLIHGDKKGGFSIFWADDGLDTGDLLLQKECEVLPDDTVSTLYNRFLFPEGIKGMVQAVRLIAEGKAPRLPQPEEGATYEGIQKKETAKINWDQPAEAIHNWIRGNDKVPGAWTEACEQKLTFFNSTLNTSGLVPEGDALPIPGAHRPGVVTKAGLILFGNDDKMLLVKNIQLEDGKMILASNFFK Hydrogen bonds contact Hydrophobic contact | ||||
| 92 | Dihydrolipoyl dehydrogenase, mitochondrial | 1ZMD | 4.73 | |
Target general information Gen name DLD Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms PHE3;LAD;GCSL Protein family Class-I pyridine nucleotide-disulfide oxidoreductase family Biochemical class Oxidoreductase Function Dihydrolipoyl dehydrogenase activity.Electron carrier activity.Flavin adenine dinucleotide binding.Lipoamide binding.NAD binding. Related diseases Dihydrolipoamide dehydrogenase deficiency (DLDD) [MIM:246900]: An autosomal recessive metabolic disorder characterized biochemically by a combined deficiency of the branched-chain alpha-keto acid dehydrogenase complex (BCKDC), pyruvate dehydrogenase complex (PDC), and alpha-ketoglutarate dehydrogenase complex (KGDC). Clinically, affected individuals have lactic acidosis and neurologic deterioration due to sensitivity of the central nervous system to defects in oxidative metabolism. {ECO:0000269|PubMed:10448086, ECO:0000269|PubMed:11687750, ECO:0000269|PubMed:12925875, ECO:0000269|PubMed:15712224, ECO:0000269|PubMed:16442803, ECO:0000269|PubMed:16770810, ECO:0000269|PubMed:17404228, ECO:0000269|PubMed:20160912, ECO:0000269|PubMed:8506365, ECO:0000269|PubMed:8968745, ECO:0000269|PubMed:9540846, ECO:0000269|PubMed:9934985}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03147; DB00145; DB00157 Interacts with P42858; O14713; O00330; P30041; P62258 EC number 1.8.1.4 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Cell projection; Cilium; Cytoplasmic vesicle; Disease variant; Disulfide bond; FAD; Flagellum; Flavoprotein; Mitochondrion; NAD; Nucleus; Oxidoreductase; Phosphoprotein; Proteomics identification; Redox-active center; Reference proteome; Transit peptide Protein physicochemical properties Chain ID A,B,C,D,E,F,G,H Molecular weight (Da) 49832.7 Length 471 Aromaticity 0.06 Instability index 26.19 Isoelectric point 6.51 Charge (pH=7) -2.02 2D Binding mode Binding energy (Kcal/mol) -6.45 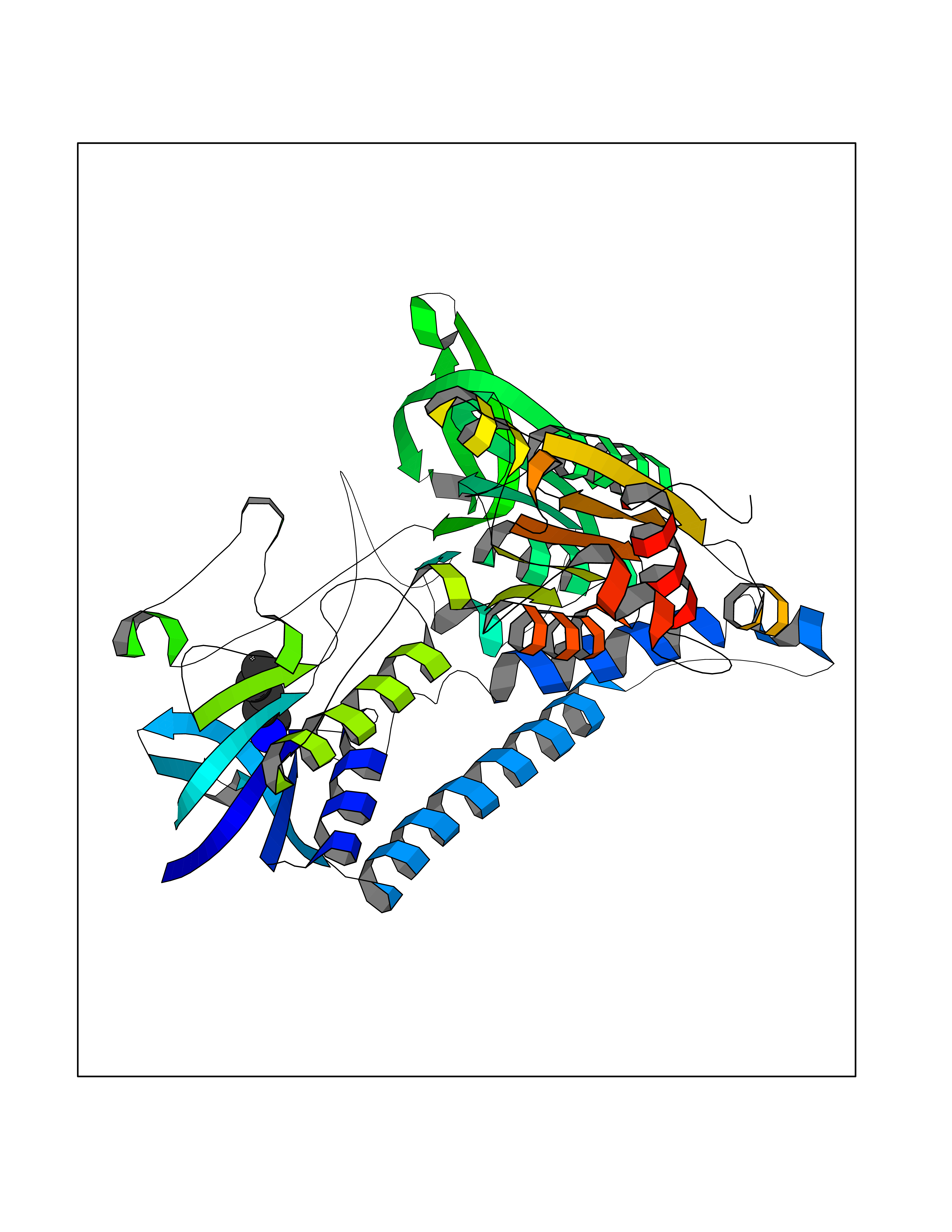 Molscript Map 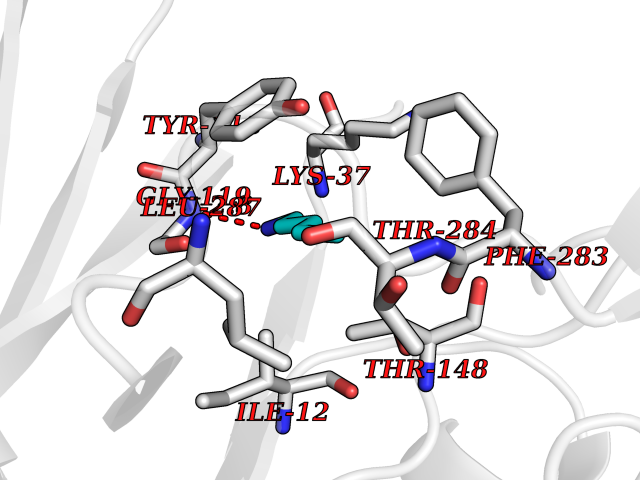 Pymol Map 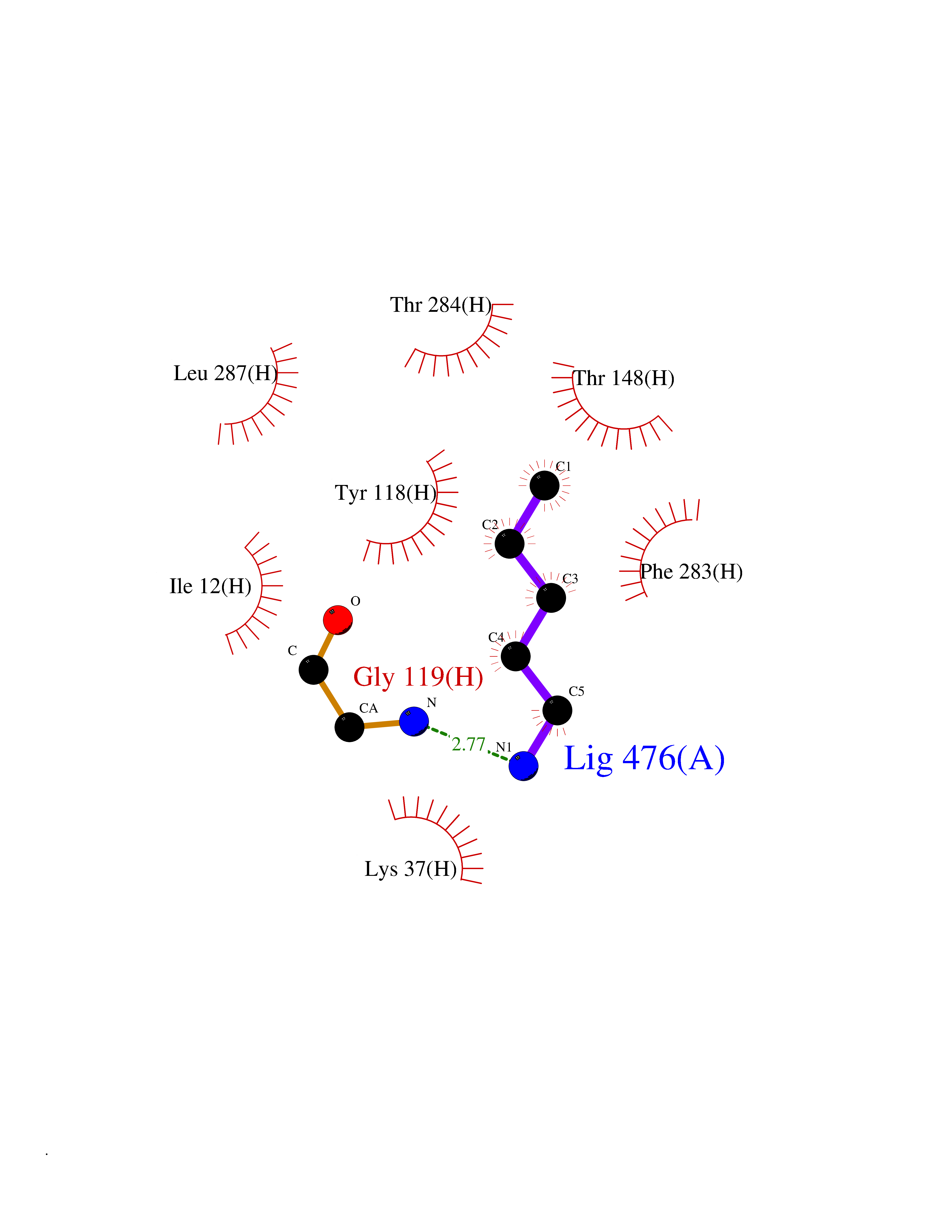 Ligplot Map 3D Binding mode Sequence PIDADVTVIGSGPGGYVAAIKAAQLGFKTVCIEKNETLGGTCLNVGCIPSKALLNNSHYYHMAHGTDFASRGIEMSEVRLNLDKMMEQKSTAVKALTGGIAHLFKQNKVVHVNGYGKITGKNQVTATKADGGTQVIDTKNILIATGSEVTPFPGITIDEDTIVSSTGALSLKKVPEKMVVIGAGVIGVELGSVWQRLGADVTAVEFLGHVGGVGIDMEISKNFQRILQKQGFKFKLNTKVTGATKKSDGKIDVSIEAASGGKAEVITCDVLLVCIGRRPFTKNLGLEELGIELDPRGRIPVNTRFQTKIPNIYAIGDVVAGPMLAHKAEDEGIICVEGMAGGAVHIDYNCVPSVIYTHPEVAWVGKSEEQLKEEGIEYKVGKFPFAANSRAKTNADTDGMVKILGQKSTDRVLGAHILGPGAGEMVNEAALALEYGASCEDIARVCHAHPTLSEAFREANLAASFGKSINF Hydrogen bonds contact Hydrophobic contact | ||||
| 93 | Retinaldehyde-binding protein 1 | 3HX3 | 4.73 | |
Target general information Gen name RLBP1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms CRALBP Protein family NA Biochemical class Transport protein Function 11-cis retinal binding.Retinol binding.Transporter activity. Related diseases Bothnia retinal dystrophy (BRD) [MIM:607475]: A type of retinitis punctata albescens. Affected individuals show night blindness from early childhood with features consistent with retinitis punctata albescens and macular degeneration. {ECO:0000269|PubMed:10102298}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Rod-cone dystrophy Newfoundland (NFRCD) [MIM:607476]: A rod-cone dystrophy reminiscent of retinitis punctata albescens but with a substantially lower age at onset and more-rapid and distinctive progression. Rod-cone dystrophies results from initial loss of rod photoreceptors, later followed by cone photoreceptors loss. {ECO:0000269|PubMed:11868161}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Retinitis punctata albescens (RPA) [MIM:136880]: A form of fleck retina disease characterized by aggregation of white flecks posteriorly in the retina, causing night blindness and delayed dark adaptation. It differs from fundus albipunctatus in being progressive and evolving to generalized atrophy of the retina. {ECO:0000269|PubMed:10102299, ECO:0000269|PubMed:11453974, ECO:0000269|PubMed:9326942}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00162 Interacts with Q9P2G9-2 EC number NA Uniprot keywords 3D-structure; Acetylation; Cytoplasm; Disease variant; Proteomics identification; Reference proteome; Retinol-binding; Sensory transduction; Transport; Vision Protein physicochemical properties Chain ID A Molecular weight (Da) 28328.6 Length 250 Aromaticity 0.14 Instability index 52.64 Isoelectric point 4.96 Charge (pH=7) -9.87 2D Binding mode Binding energy (Kcal/mol) -6.46 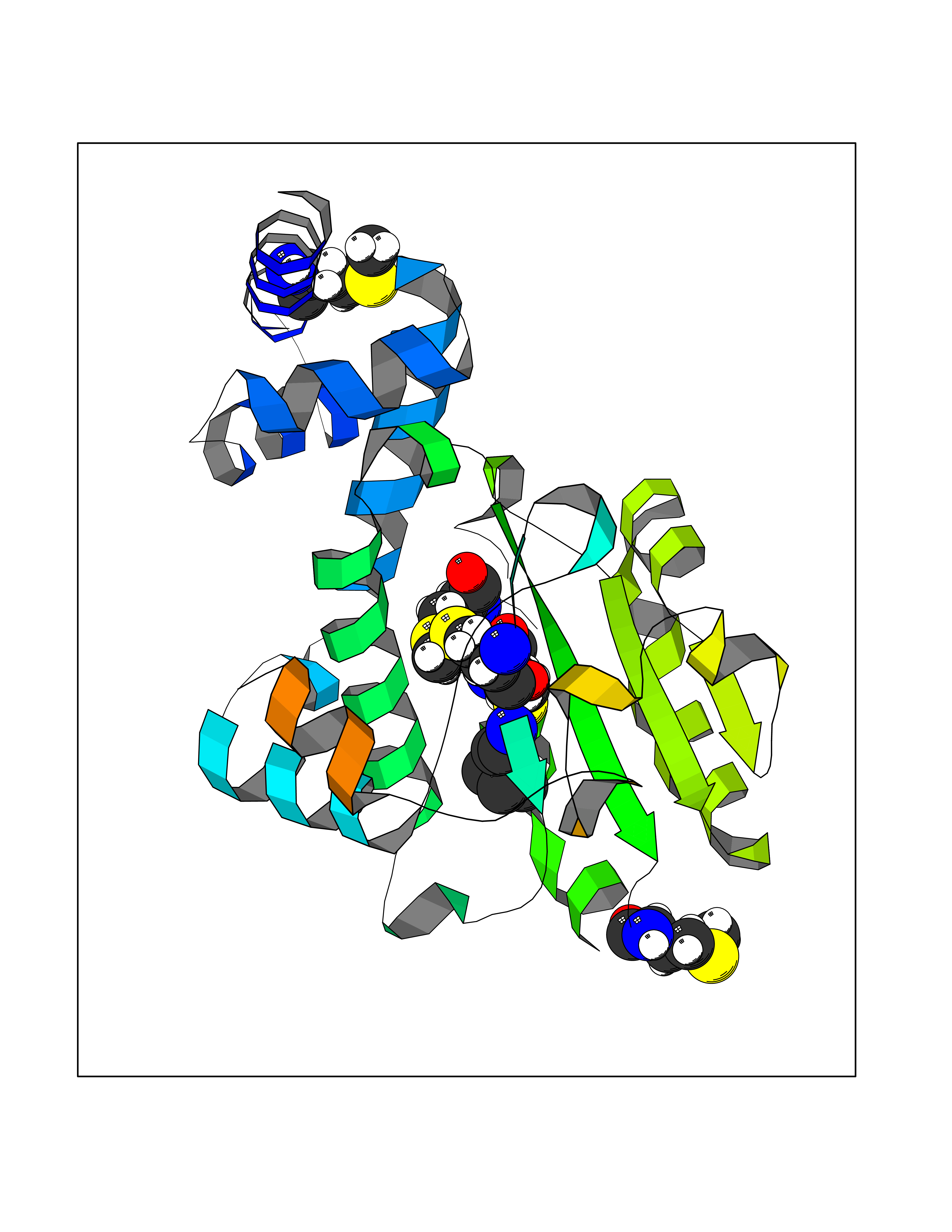 Molscript Map 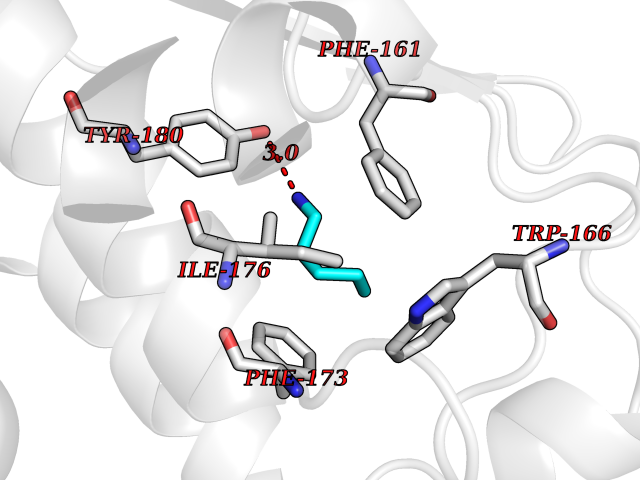 Pymol Map 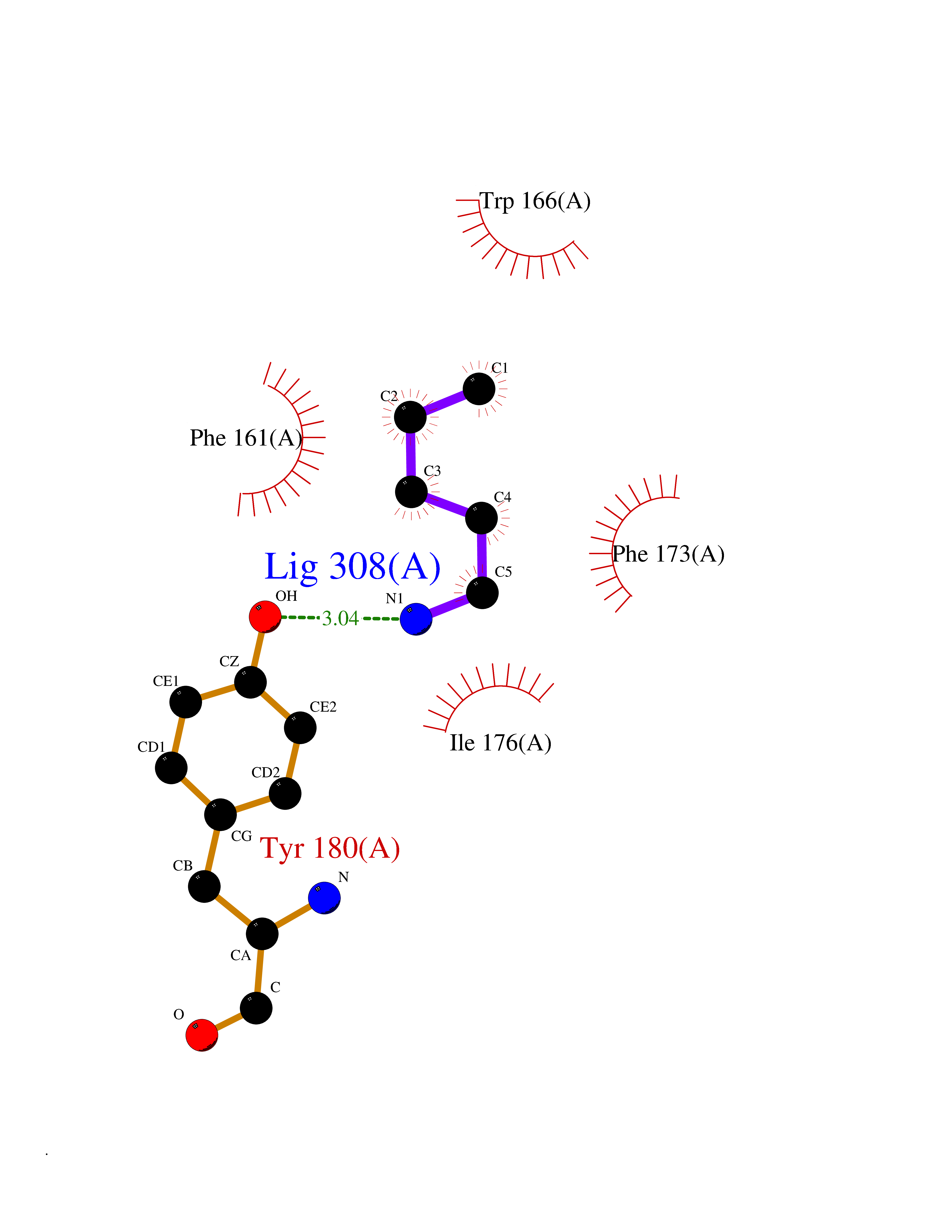 Ligplot Map 3D Binding mode Sequence ETREEAVRELQEXVQAQAASGEELAVAVAERVQEKDSGFFLRFIRARKFNVGRAYELLRGYVNFRLQYPELFDSLSPEAVRCTIEAGYPGVLSSRDKYGRVVXLFNIENWQSQEITFDEILQAYCFILEKLLENEETQINGFCIIENFKGFTXQQAASLRTSDLRKXVDXLQDSFPAWFKAIHFIHQPWYFTTTYNVVKPFLKSKLLERVFVHGDDLSGFYQEIDENILPSDFGGTLPKYDGKAVAEQLF Hydrogen bonds contact Hydrophobic contact | ||||
| 94 | Nitric-oxide synthase inducible (NOS2) | 3E7G | 4.73 | |
Target general information Gen name NOS2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms iNOS; Peptidyl-cysteine S-nitrosylase NOS2; Nitric oxide synthase, inducible; NOS2A; NOS type II; Inducible NOS; Inducible NO synthase; Hepatocyte NOS; HEP-NOS Protein family NOS family Biochemical class Paired donor oxygen oxidoreductase Function Produces nitric oxide (NO) which is a messenger molecule with diverse functions throughout the body. In macrophages, NO mediates tumoricidal and bactericidal actions. Also has nitrosylase activity and mediates cysteine S-nitrosylation of cytoplasmic target proteins such PTGS2/COX2 (By similarity). As component of the iNOS-S100A8/9 transnitrosylase complex involved in the selective inflammatory stimulus-dependent S-nitrosylation of GAPDH on 'Cys-247' implicated in regulation of the GAIT complex activity and probably multiple targets including ANXA5, EZR, MSN and VIM. Involved in inflammation, enhances the synthesis of proinflammatory mediators such as IL6 and IL8. Related diseases Cerebellar ataxia, impaired intellectual development, and dysequilibrium syndrome 3 (CAMRQ3) [MIM:613227]: An autosomal recessive, congenital cerebellar ataxia associated with dysarthia, quadrupedal gait and intellectual disability. {ECO:0000269|PubMed:19461874}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07003; DB07007; DB07011; DB07405; DB08750; DB01997; DB07029; DB07008; DB08214; DB07002; DB01835; DB06879; DB04534; DB03100; DB02207; DB00125; DB00155; DB01234; DB14649; DB11327; DB00997; DB07306; DB07388; DB05252; DB01381; DB03366; DB05214; DB04400; DB09237; DB00244; DB01110; DB01017; DB03144; DB01686; DB03449; DB06916; DB07318; DB07389; DB02044; DB02644; DB05383; DB02234; DB03953; DB02462; DB08814 Interacts with P04406 EC number EC 1.14.13.39 Uniprot keywords 3D-structure; Alternative splicing; Calcium; Calmodulin-binding; Cytoplasm; FAD; Flavoprotein; FMN; Heme; Iron; Metal-binding; NADP; Oxidoreductase; Phosphoprotein; Proteomics identification; Reference proteome; Ubl conjugation; Zinc Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 48633 Length 421 Aromaticity 0.12 Instability index 46.5 Isoelectric point 6.75 Charge (pH=7) -1.04 2D Binding mode Binding energy (Kcal/mol) -6.45  Molscript Map 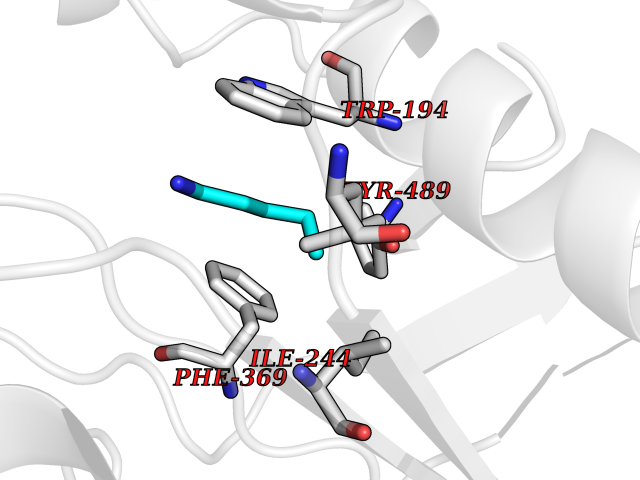 Pymol Map 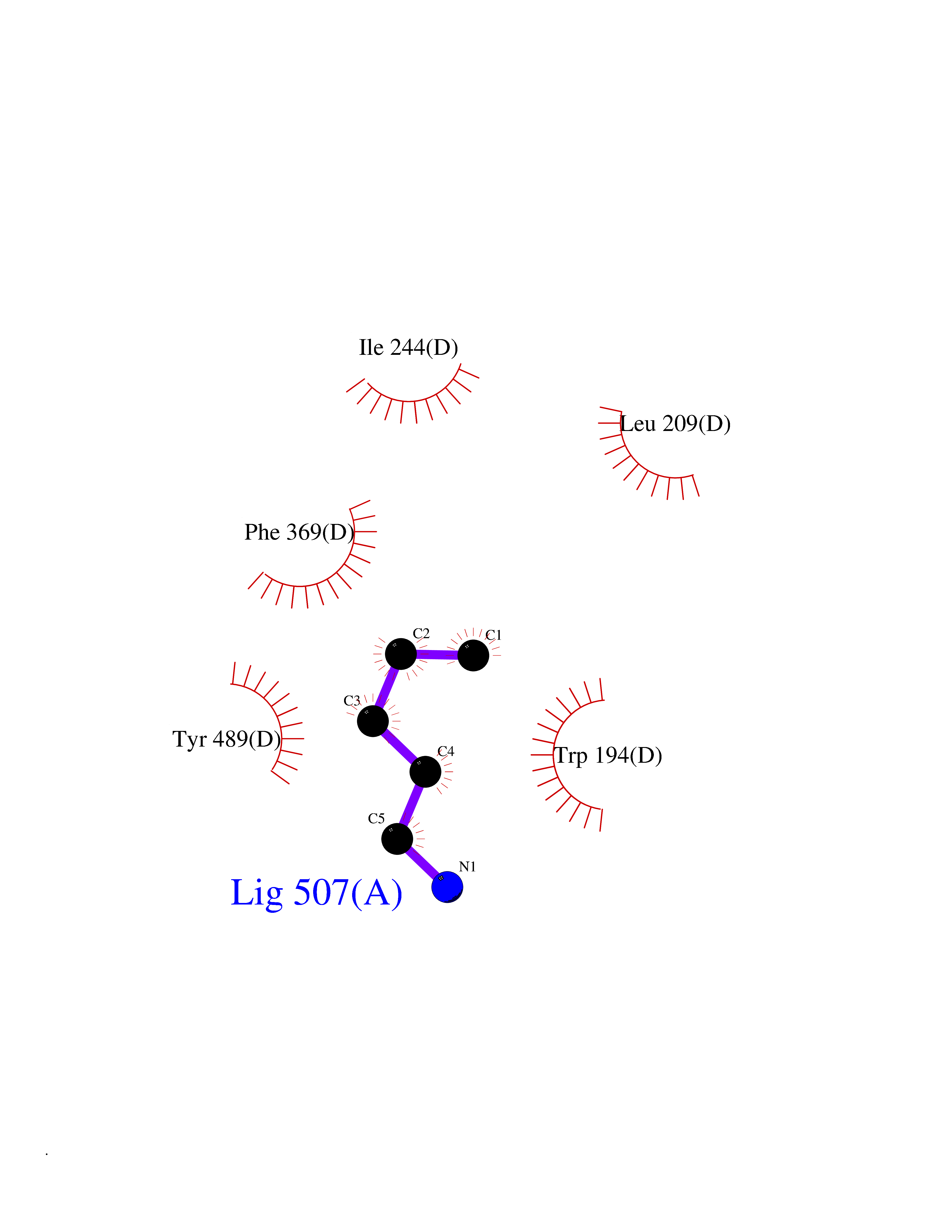 Ligplot Map 3D Binding mode Sequence RHVRIKNWGSGMTFQDTLHHKAKGILTCRSKSCLGSIMTPKSLTRGPRDKPTPPDELLPQAIEFVNQYYGSFKEAKIEEHLARVEAVTKEIETTGTYQLTGDELIFATKQAWRNAPRCIGRIQWSNLQVFDARSCSTAREMFEHICRHVRYSTNNGNIRSAITVFPQRSDGKHDFRVWNAQLIRYAGYQMPDGSIRGDPANVEFTQLCIDLGWKPKYGRFDVVPLVLQANGRDPELFEIPPDLVLEVAMEHPKYEWFRELELKWYALPAVANMLLEVGGLEFPGCPFNGWYMGTEIGVRDFCDVQRYNILEEVGRRMGLETHKLASLWKDQAVVEINIAVLHSFQKQNVTIMDHHSAAESFMKYMQNEYRSRGGCPADWIWLVPPMSGSITPVFHQEMLNYVLSPFYYYQVEAWKTHVWQD Hydrogen bonds contact Hydrophobic contact | ||||
| 95 | Vitamin K epoxide reductase complex 1 (VKORC1) | 6WV3 | 4.73 | |
Target general information Gen name VKORC1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Vitamin K1 2,3-epoxide reductase subunit 1; VKORC1; VKOR; UNQ308/PRO351; MSTP576; MSTP134 Protein family VKOR family Biochemical class Short-chain dehydrogenases reductase Function Involved invitamin K metabolism. Catalytic subunit of the vitamin K epoxide reductase (VKOR) complex which reduces inactive vitamin K 2,3-epoxide to active vitamin K. Vitamin K is required for the gamma-carboxylation of various proteins, including clotting factors, and is required for normal blood coagulation, but also for normal bone development. Related diseases Combined deficiency of vitamin K-dependent clotting factors 2 (VKCFD2) [MIM:607473]: VKCFD leads to a bleeding tendency that is usually reversed by oral administration of vitamin K. {ECO:0000269|PubMed:14765194, ECO:0000269|PubMed:16270630}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Coumarin resistance (CMRES) [MIM:122700]: A condition characterized by partial or complete resistance to warfarin or other 4-hydroxycoumarin derivatives. These drugs are used as anti-coagulants for the prevention of thromboembolic diseases in subjects with deep vein thrombosis, atrial fibrillation, or mechanical heart valve replacement. {ECO:0000269|PubMed:14765194, ECO:0000269|PubMed:20946155}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01418; DB00266; DB09332; DB00170; DB00498; DB00946; DB01022; DB00682 Interacts with Q13323; Q7Z7G2; Q96BA8; Q9Y282; Q5JX71; Q96KR6; Q5T7V8; Q8TDT2; Q9NQG1; P15941-11; Q96TC7; Q9NR31; A0A0S2Z4U3; Q8TBB6; O15393-2; Q19QW4 EC number EC 1.17.4.4 Uniprot keywords 3D-structure; Alternative splicing; Disease variant; Disulfide bond; Endoplasmic reticulum; Membrane; Oxidoreductase; Proteomics identification; Quinone; Redox-active center; Reference proteome; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 42656.4 Length 381 Aromaticity 0.1 Instability index 32.12 Isoelectric point 7.73 Charge (pH=7) 1.93 2D Binding mode Binding energy (Kcal/mol) -6.45  Molscript Map 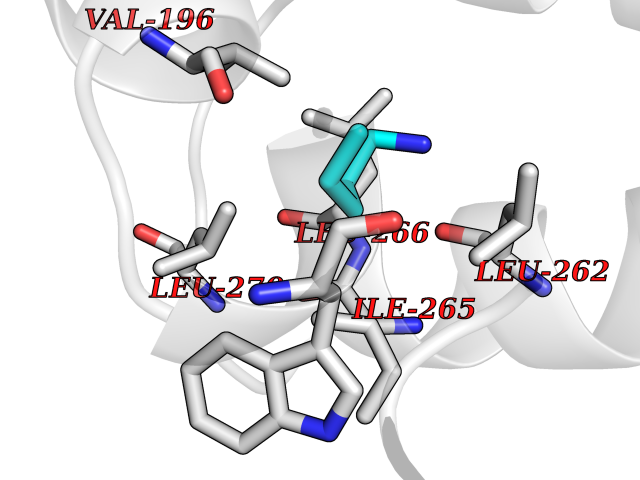 Pymol Map 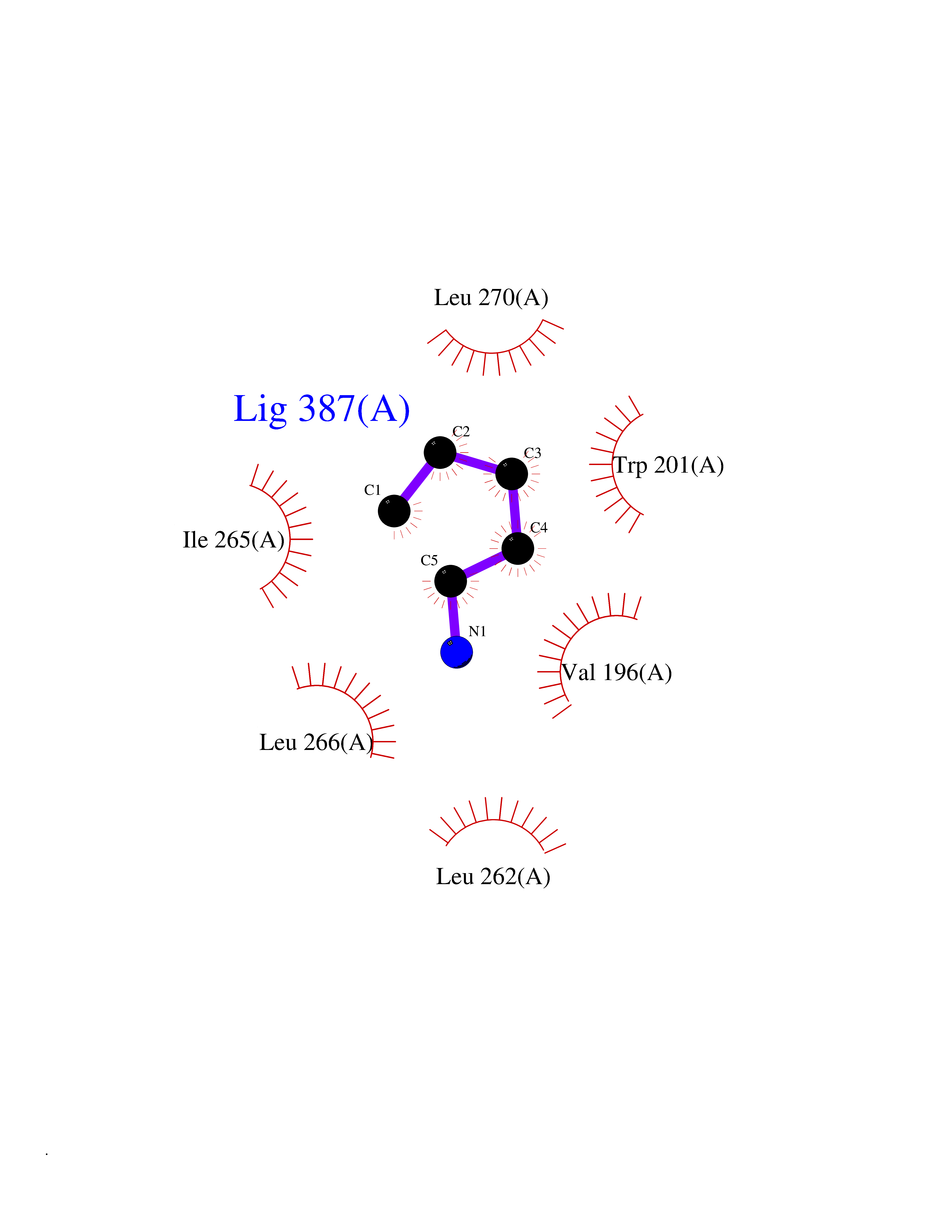 Ligplot Map 3D Binding mode Sequence KGEELFTGVVPILVELDGDVNGHKFSVRGEGEGDATNGKLTLKFICTTGKLPVPWPTLVTTLXVQCFSRYPDHMKRHDFFKSAMPEGYVQERTISFKDDGTYKTRAEVKFEGDTLVNRIELKGIDFKEDGNILGHKLEYNSTWGSPGWVRLALCLTGLVLSLYALHVKAARARDRDYRALCDVGTAISCSRVFSSRWGRGFGLVEHVLGQDSILNQSNSIFGCIFYTLQLLLGCLRTRWASVLMLLSSLVSLAGSVYLAWILFFVLYDFCIVCITTYAINVSLMWLSFRKVQENSHNVYITADKQKNGIKANFKIRHNVEDGSVQLADHYQQNTPIGDGPVLLPDNHYLSTQSVLSKDPNEKRDHMVLLEFVTAAGITHHH Hydrogen bonds contact Hydrophobic contact | ||||
| 96 | Bromodomain-containing protein 9 (BRD9) | 6V0X | 4.73 | |
Target general information Gen name BRD9 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Rhabdomyosarcoma antigen MU-RMS-40.8 Protein family NA Biochemical class Bromodomain Function Plays a role in chromatin remodeling and regulation of transcription. Acts as a chromatin reader that recognizes and binds acylated histones: binds histones that are acetylated and/or butyrylated. Component of SWI/SNF chromatin remodeling subcomplex GBAF that carries out key enzymatic activities, changing chromatin structure by altering DNA-histone contacts within a nucleosome in an ATP-dependent manner. Related diseases Major depressive disorder (MDD) [MIM:608516]: A common psychiatric disorder. It is a complex trait characterized by one or more major depressive episodes without a history of manic, mixed, or hypomanic episodes. A major depressive episode is characterized by at least 2 weeks during which there is a new onset or clear worsening of either depressed mood or loss of interest or pleasure in nearly all activities. Four additional symptoms must also be present including changes in appetite, weight, sleep, and psychomotor activity; decreased energy; feelings of worthlessness or guilt; difficulty thinking, concentrating, or making decisions; or recurrent thoughts of death or suicidal ideation, plans, or attempts. The episode must be accompanied by distress or impairment in social, occupational, or other important areas of functioning. {ECO:0000269|PubMed:15229186}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q7Z7H3; Q7Z7H3 EC number NA Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Bromodomain; Chromatin regulator; Isopeptide bond; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Transcription; Transcription regulation; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 11491.3 Length 99 Aromaticity 0.13 Instability index 13.58 Isoelectric point 9.24 Charge (pH=7) 3.8 2D Binding mode Binding energy (Kcal/mol) -6.45  Molscript Map 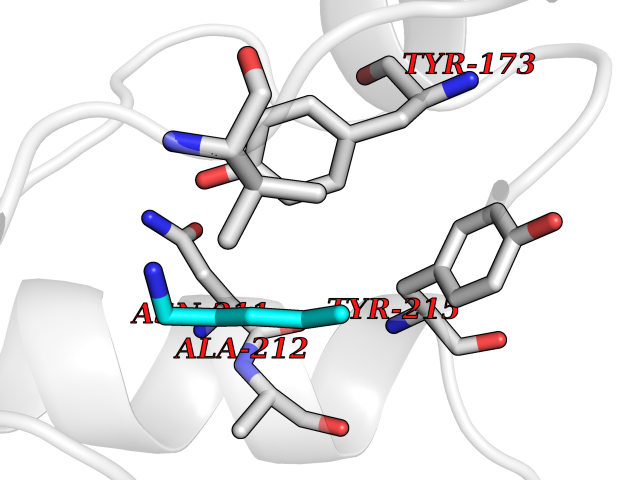 Pymol Map 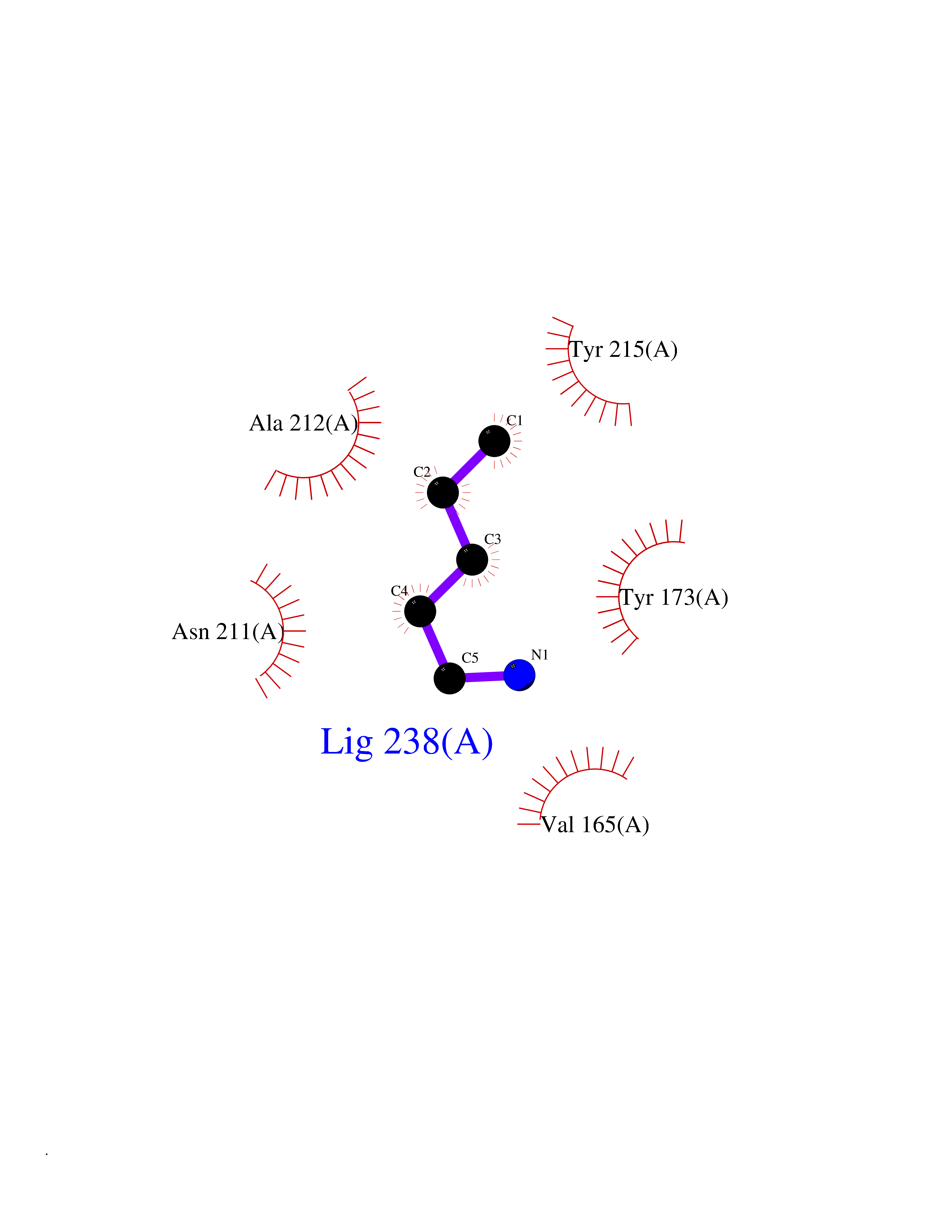 Ligplot Map 3D Binding mode Sequence STPIQQLLEHFLRQLQRKDPHGFFAFPVTDAIAPGYSMIIKHPMDFGTMKDKIVANEYKSVTEFKADFKLMCDNAMTYNRPDTVYYKLAKKILHAGFKM Hydrogen bonds contact Hydrophobic contact | ||||
| 97 | Farnesoid X-activated receptor (FXR) | 6HL1 | 4.73 | |
Target general information Gen name NR1H4 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Retinoid X receptor-interacting protein 14; RXR-interacting protein 14; RIP14; Nuclear receptor subfamily 1 group H member 4; HRR1; Farnesol receptor HRR-1; FXR; Bile acid receptor; BAR Protein family Nuclear hormone receptor family, NR1 subfamily Biochemical class Nuclear hormone receptor Function Ligand-activated transcription factor. Receptor for bile acids (BAs) such as chenodeoxycholic acid (CDCA), lithocholic acid, deoxycholic acid (DCA) and allocholic acid (ACA). Plays a essential role in BA homeostasis through the regulation of genes involved in BA synthesis, conjugation and enterohepatic circulation. Also regulates lipid and glucose homeostasis and is involved innate immune response. The FXR-RXR heterodimer binds predominantly to farnesoid X receptor response elements (FXREs) containing two inverted repeats of the consensus sequence 5'-AGGTCA-3' in which the monomers are spaced by 1 nucleotide (IR-1) but also to tandem repeat DR1 sites with lower affinity, and can be activated by either FXR or RXR-specific ligands. It is proposed that monomeric nuclear receptors such as NR5A2/LRH-1 bound to coregulatory nuclear responsive element (NRE) halfsites located in close proximity to FXREs modulate transcriptional activity (By similarity). In the liver activates transcription of the corepressor NR0B2 thereby indirectly inhibiting CYP7A1 and CYP8B1 (involved in BA synthesis) implicating at least in part histone demethylase KDM1A resulting in epigenomic repression, and SLC10A1/NTCP (involved in hepatic uptake of conjugated BAs). Activates transcription of the repressor MAFG (involved in regulation of BA synthesis) (By similarity). Activates transcription of SLC27A5/BACS and BAAT (involved in BA conjugation), ABCB11/BSEP (involved in bile salt export) by directly recruiting histone methyltransferase CARM1, and ABCC2/MRP2 (involved in secretion of conjugated BAs) and ABCB4 (involved in secretion of phosphatidylcholine in the small intestine). Activates transcription of SLC27A5/BACS and BAAT (involved in BA conjugation), ABCB11/BSEP (involved in bile salt export) by directly recruiting histone methyltransferase CARM1, and ABCC2/MRP2 (involved in secretion of conjugated BAs) and ABCB4 (involved in secretion of phosphatidylcholine in the small intestine). In the intestine activates FGF19 expression and secretion leading to hepatic CYP7A1 repression. The function also involves the coordinated induction of hepatic KLB/beta-klotho expression (By similarity). Regulates transcription of liver UGT2B4 and SULT2A1 involved in BA detoxification; binding to the UGT2B4 promoter seems to imply a monomeric transactivation independent of RXRA. Modulates lipid homeostasis by activating liver NR0B2/SHP-mediated repression of SREBF1 (involved in de novo lipogenesis), expression of PLTP (involved in HDL formation), SCARB1 (involved in HDL hepatic uptake), APOE, APOC1, APOC4, PPARA (involved in beta-oxidation of fatty acids), VLDLR and SDC1 (involved in the hepatic uptake of LDL and IDL remnants), and inhibiting expression of MTTP (involved in VLDL assembly. Increases expression of APOC2 (promoting lipoprotein lipase activity implicated in triglyceride clearance). Transrepresses APOA1 involving a monomeric competition with NR2A1 for binding to a DR1 element. Also reduces triglyceride clearance by inhibiting expression of ANGPTL3 and APOC3 (both involved in inhibition of lipoprotein lipase). Involved in glucose homeostasis by modulating hepatic gluconeogenesis through activation of NR0B2/SHP-mediated repression of respective genes. Modulates glycogen synthesis (inducing phosphorylation of glycogen synthase kinase-3) (By similarity). Modulates glucose-stimulated insulin secretion and is involved in insulin resistance. Involved in intestinal innate immunity. Plays a role in protecting the distal small intestine against bacterial overgrowth and preservation of the epithelial barrier (By similarity). Down-regulates inflammatory cytokine expression in several types of immune cells including macrophages and mononuclear cells. Mediates trans-repression of TLR4-induced cytokine expression; the function seems to require its sumoylation and prevents N-CoR nuclear receptor corepressor clearance from target genes such as IL1B and NOS2. Involved in the TLR9-mediated protective mechanism in intestinal inflammation. Plays an anti-inflammatory role in liver inflammation; proposed to inhibit proinflammatory (but not antiapoptotic) NF-kappa-B signaling) (By similarity). Related diseases May be involved in intrahepatic cholestasis of pregnancy. {ECO:0000305|PubMed:17681172}.; DISEASE: May be involved in cholesterol cholelithiasis. {ECO:0000305|PubMed:17931734}.; DISEASE: Cholestasis, progressive familial intrahepatic, 5 (PFIC5) [MIM:617049]: A disorder characterized by early onset of cholestasis that progresses to hepatic fibrosis, cirrhosis, and end-stage liver disease before adulthood. PFIC5 is an autosomal recessive, severe form characterized by onset of intralobular cholestasis in the neonatal period. {ECO:0000269|PubMed:26888176}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08220; DB00132; DB04557; DB06777; DB02659; DB03619; DB02509; DB02545; DB11605; DB16255; DB05990; DB04348; DB16343; DB01586 Interacts with Q15788; O75151; Q8WTS6; P28702; P28702-3; P48443; Q15788; P78527; P03372 EC number NA Uniprot keywords 3D-structure; Acetylation; Activator; Alternative promoter usage; Alternative splicing; Disease variant; DNA-binding; Immunity; Inflammatory response; Innate immunity; Intrahepatic cholestasis; Isopeptide bond; Metal-binding; Methylation; Nucleus; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Repressor; Transcription; Transcription regulation; Ubl conjugation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A Molecular weight (Da) 26417.1 Length 225 Aromaticity 0.08 Instability index 49.55 Isoelectric point 5.28 Charge (pH=7) -11.34 2D Binding mode Binding energy (Kcal/mol) -6.45  Molscript Map 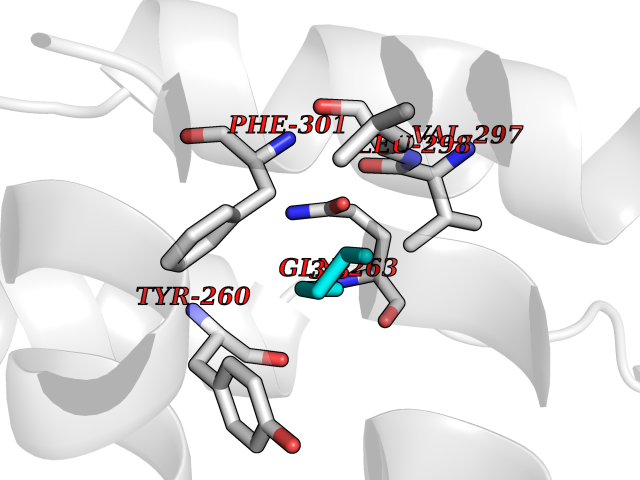 Pymol Map 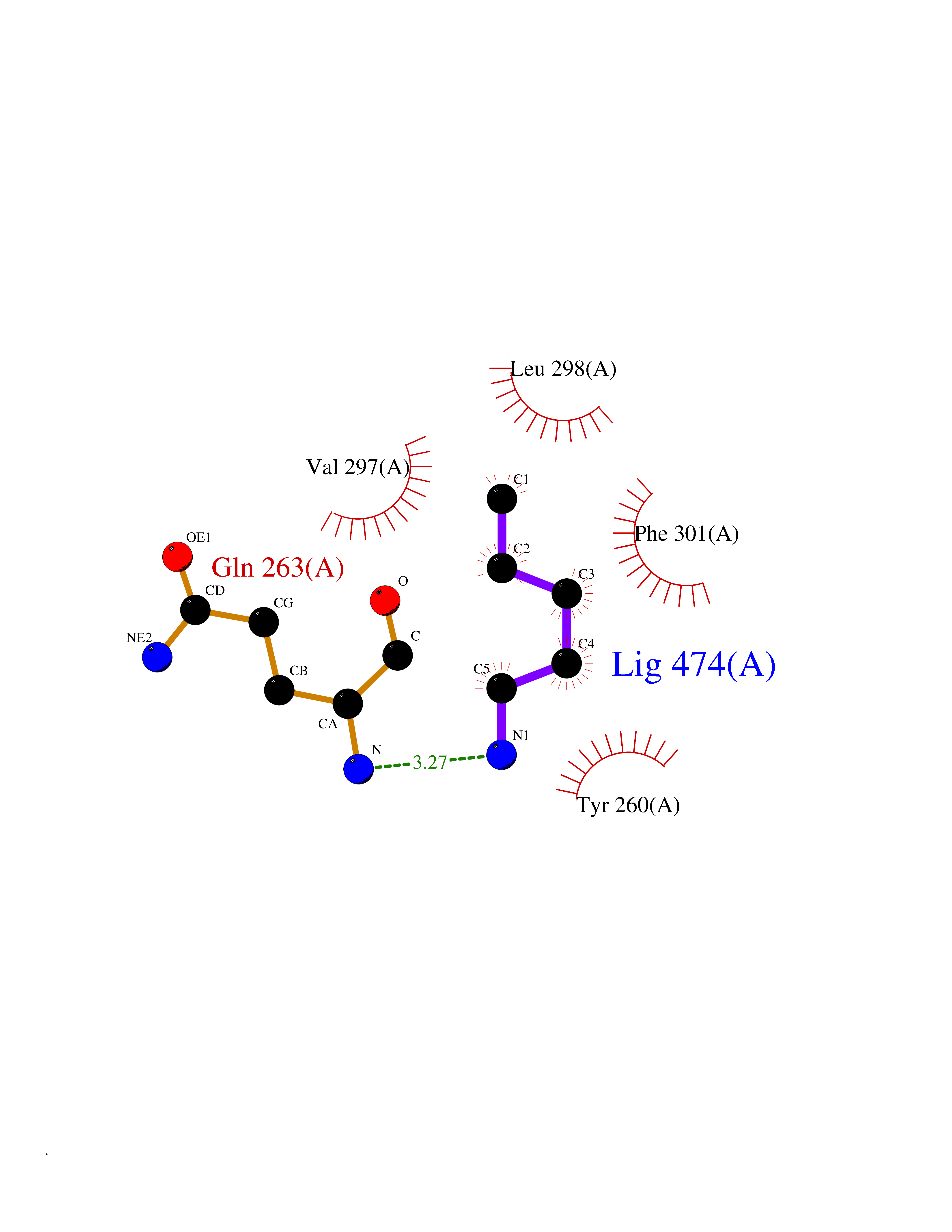 Ligplot Map 3D Binding mode Sequence HMELTPDQQTLLHFIMDSYNKQRMPQEITNKILKEEFSAEENFLILTEMATNHVQVLVEFTKKLPGFQTLDHEDQIALLKGSAVEAMFLRSAEIFNKSDLLEERIRNSGISDEYITPMFSFYKSIGELKMTQEEYALLTAIVILSPDRQYIKDREAVEKLQEPLLDVLQKLCKIHQPENPQHFACLLGRLTELRTFNHHHAEMLMSWRVNDHKFTPLLCEIWDVQ Hydrogen bonds contact Hydrophobic contact | ||||
| 98 | Growth hormone secretagogue receptor 1 (GHSR) | 7NA8 | 4.73 | |
Target general information Gen name GHSR Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Growth hormone secretagogue receptor; Ghrelin receptor; GHSR; GHS-R; GHRP; GH-releasing peptide receptor Protein family G-protein coupled receptor 1 family Biochemical class GPCR rhodopsin Function Receptor for ghrelin, coupled to G-alpha-11 proteins. Stimulates growth hormone secretion. Binds also other growth hormone releasing peptides (GHRP) (e.g. Met-enkephalin and GHRP-6) as well as non-peptide, low molecular weight secretagogues (e.g. L-692,429, MK-0677, adenosine). Related diseases Growth hormone deficiency, isolated partial (GHDP) [MIM:615925]: A disorder characterized by partial growth hormone deficiency resulting in growth delay and short stature, sometimes associated with recurrent episodes of abdominal pain, vomiting, ketosis and hypoglycemia. {ECO:0000269|PubMed:16511605, ECO:0000269|PubMed:19789204}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB15488; DB13074; DB12128 Interacts with Q92847-1; Q92847-2; Q99720 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disease variant; Disulfide bond; Dwarfism; G-protein coupled receptor; Glycoprotein; Membrane; Receptor; Reference proteome; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID R Molecular weight (Da) 32423.5 Length 285 Aromaticity 0.15 Instability index 40.42 Isoelectric point 9.47 Charge (pH=7) 13.13 2D Binding mode Binding energy (Kcal/mol) -6.45 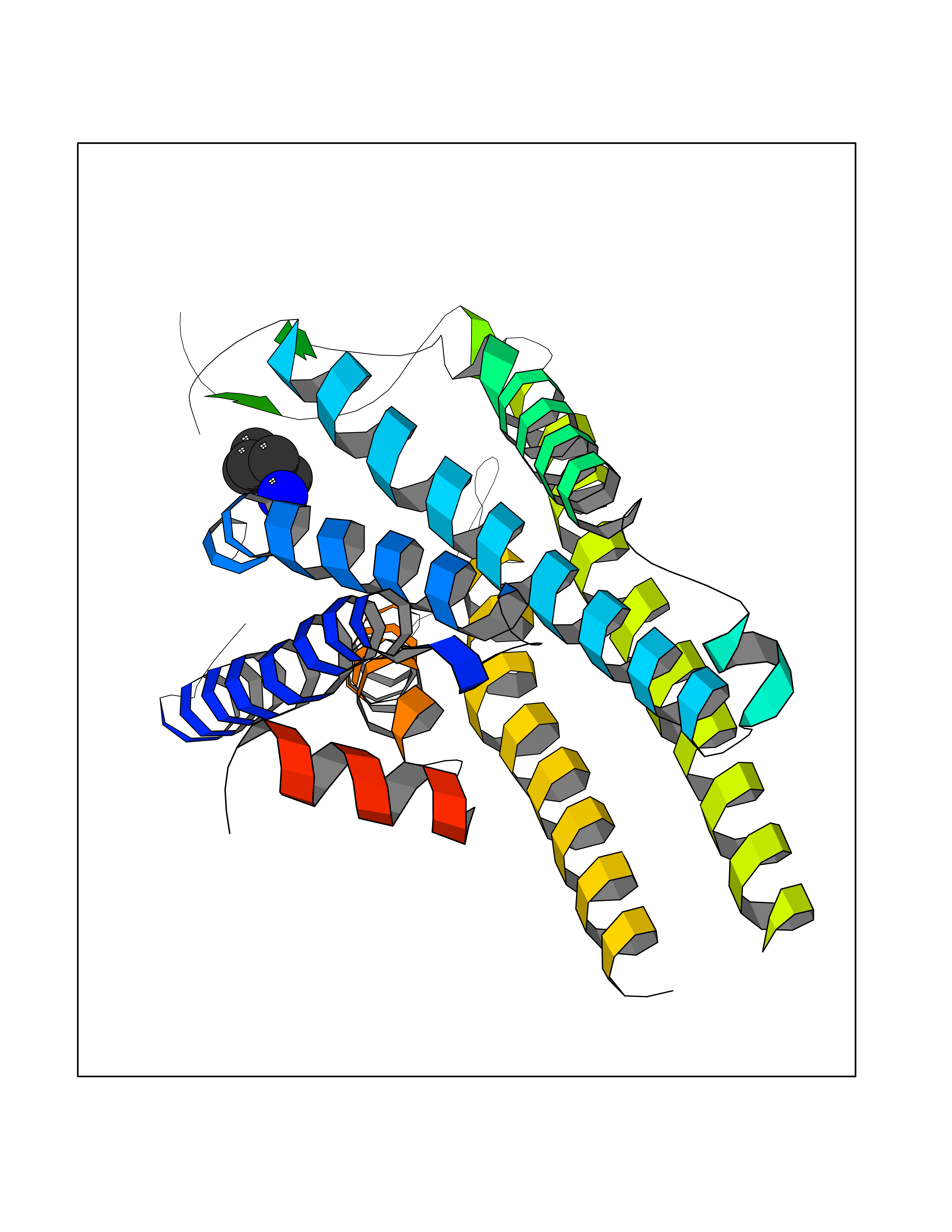 Molscript Map 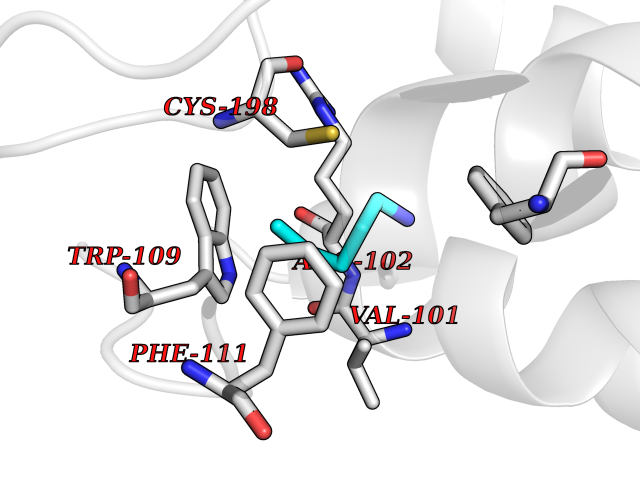 Pymol Map 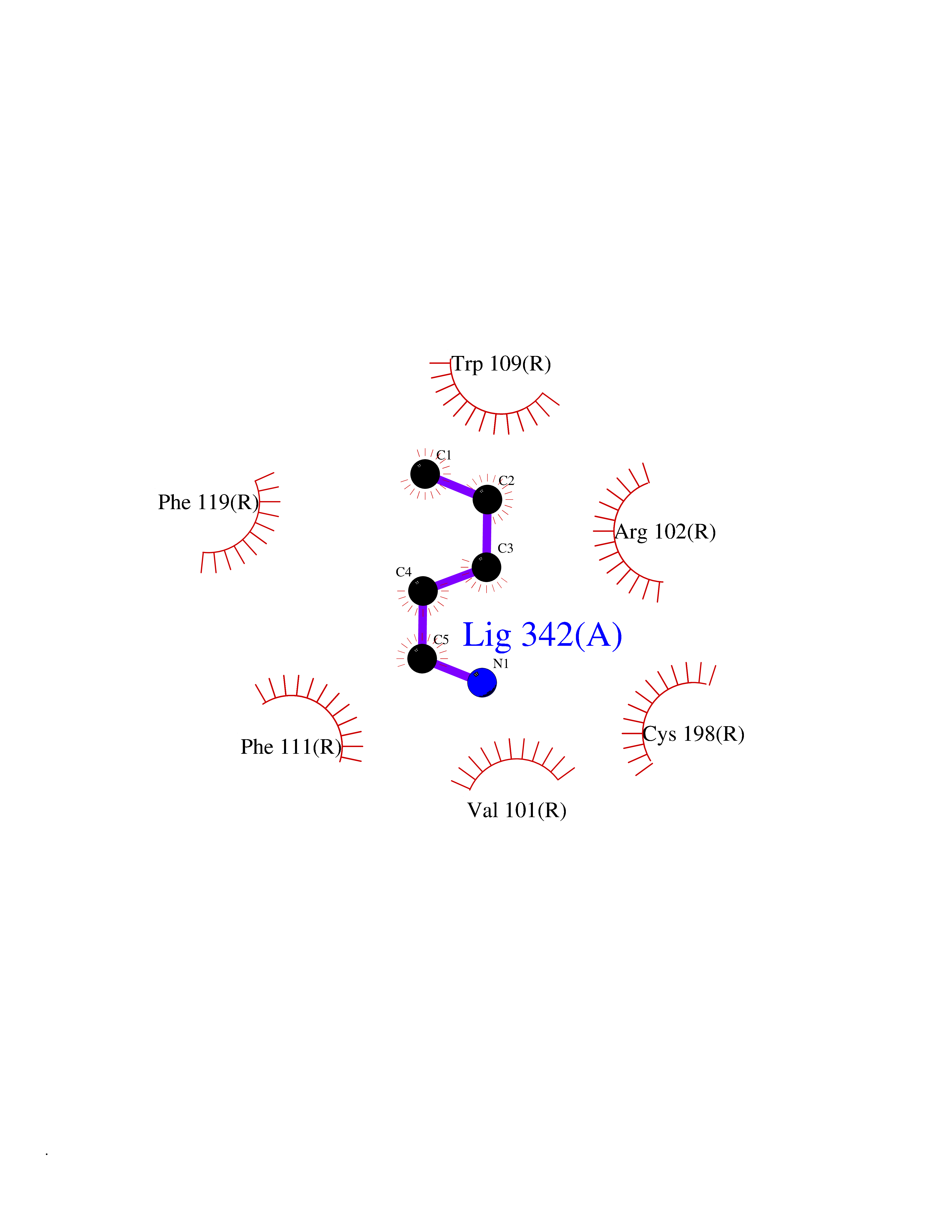 Ligplot Map 3D Binding mode Sequence PAPLLAGVTATCVALFVVGIAGNLLTMLVVSRFRELRTTTNLYLSSMAFSDLLIFLCMPLDLVRLWQPWNFGDLLCKLFQFVSESCTYAKVLTITALSVERYFAICFPLRAKVVVTKGRVKLVIFVIWAVAFCSAGPIFVLVGVEHEPWDTNECRPTEFAVRSGLLTVMVWVSSIFFFLPVFCLTVLYSLIGRKLWRRRDQNHKQTVKMLAVVVFAFILCWLPFHVGRYLFSKSFEPGSLEIAQISQYCNLVSFVLFYLSAAINPILYNIMSKKYRVAVFRLLGF Hydrogen bonds contact Hydrophobic contact | ||||
| 99 | MALT lymphoma-associated translocation (MALT1) | 7A41 | 4.73 | |
Target general information Gen name MALT1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms MALT lymphoma-associated translocation; Paracaspase Protein family Peptidase C14B family Biochemical class Peptidase Function Protease that enhances BCL10-induced activation of NF-kappa-B by mediating its cleavage. MALT1-dependent BCL10 cleavage plays an important role in T-cell antigen receptor-induced integrin adhesion. Involved in the induction of T helper 17 cells (Th17) differentiation. Cleaves RC3H1 and ZC3H12A in response to T-cell receptor (TCR) stimulation which releases their cooperatively repressed targets to promote Th17 cell differentiation (By similarity). Also mediates cleavage of N4BP1 in T-cells following TCR-mediated activation, leading to N4BP1 inactivation. Also has ubiquitin ligase activity: binds to TRAF6, inducing TRAF6 oligomerization and activation of its ligase activity. Related diseases Immunodeficiency 12 (IMD12) [MIM:615468]: A primary immunodeficiency characterized by onset in infancy of recurrent bacterial and candidal infections resulting in bronchiectasis and growth delay. Manifestations include mastoiditis, aphthous ulcers, cheilitis, gingivitis, esophagitis, gastritis, duodenitis, and meningitis. Levels of absolute lymphocytes and serum immunoglobulins are normal, but specific antibody titers are low despite immunization, and T-cells show impaired proliferative responses to mitogens. {ECO:0000269|PubMed:23727036}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: A chromosomal aberration involving MALT1 is recurrent in low-grade mucosa-associated lymphoid tissue (MALT lymphoma). Translocation t(11;18)(q21;q21) with BIRC2. This translocation is found in approximately 50% of cytogenetically abnormal low-grade MALT lymphoma. {ECO:0000269|PubMed:10339464, ECO:0000269|PubMed:10523859, ECO:0000269|PubMed:10702396, ECO:0000269|PubMed:11090634}. Drugs (DrugBank ID) NA Interacts with O95999; Q9BXL7; Q14790; P48729; Q9Y6K9; Q9UDY8; Q96PU8; Q9H0F6; Q13501; Q9Y4K3; P0CG48; P54252; P46379-2; G5E9A7; P50570-2; Q9BSK4; Q96JP0; P28799; P04792; O60333-2; O14901; O14832; P60891; Q7Z699; O76024 EC number EC 3.4.22.- Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Chromosomal rearrangement; Cytoplasm; Disease variant; Disulfide bond; Hydrolase; Immunity; Immunoglobulin domain; Nucleus; Phosphoprotein; Protease; Proteomics identification; Reference proteome; Repeat; Ubl conjugation pathway Protein physicochemical properties Chain ID A Molecular weight (Da) 39995.8 Length 354 Aromaticity 0.09 Instability index 25.55 Isoelectric point 5.12 Charge (pH=7) -11.76 2D Binding mode Binding energy (Kcal/mol) -6.45 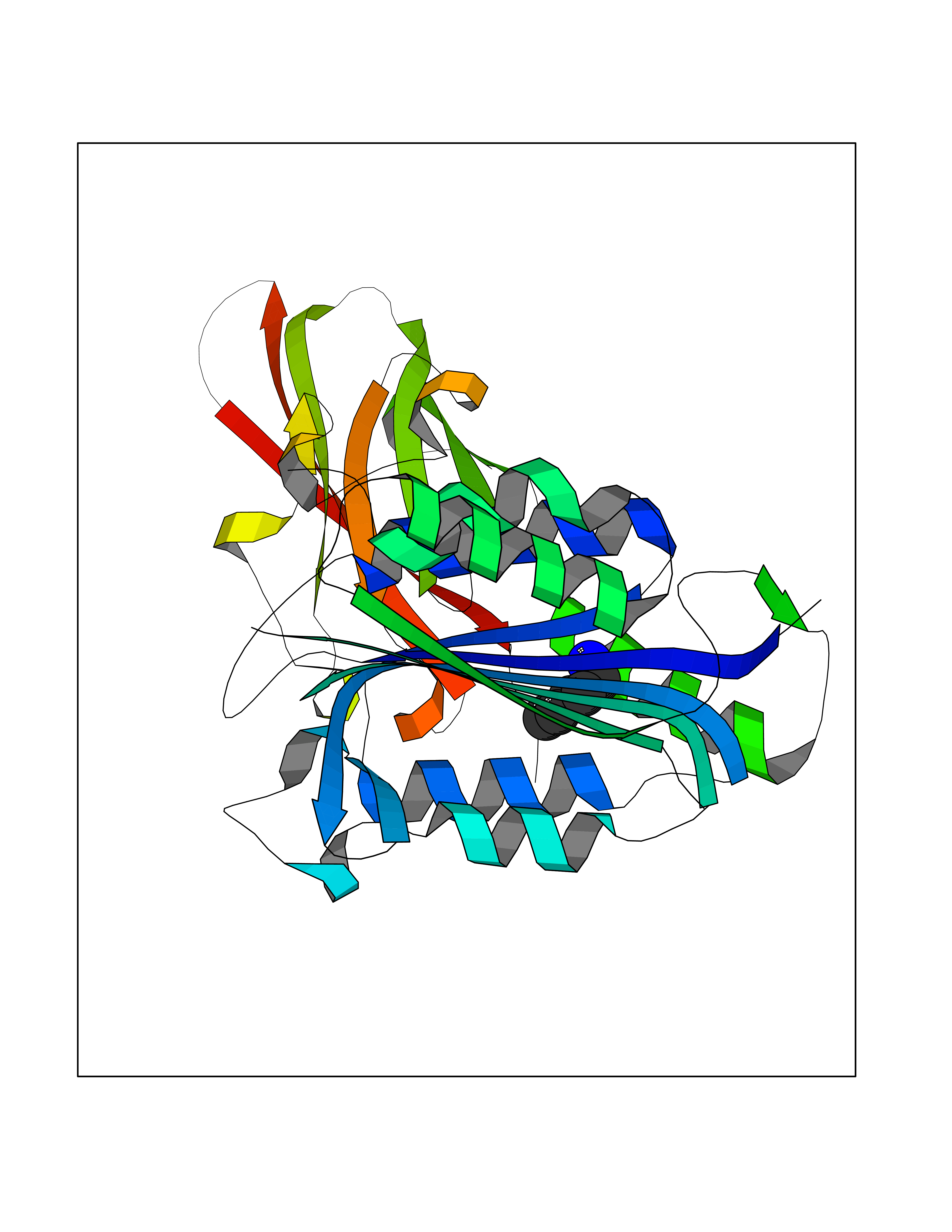 Molscript Map 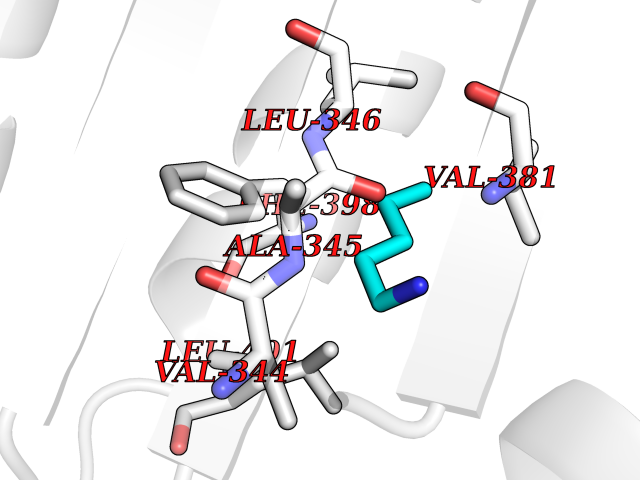 Pymol Map 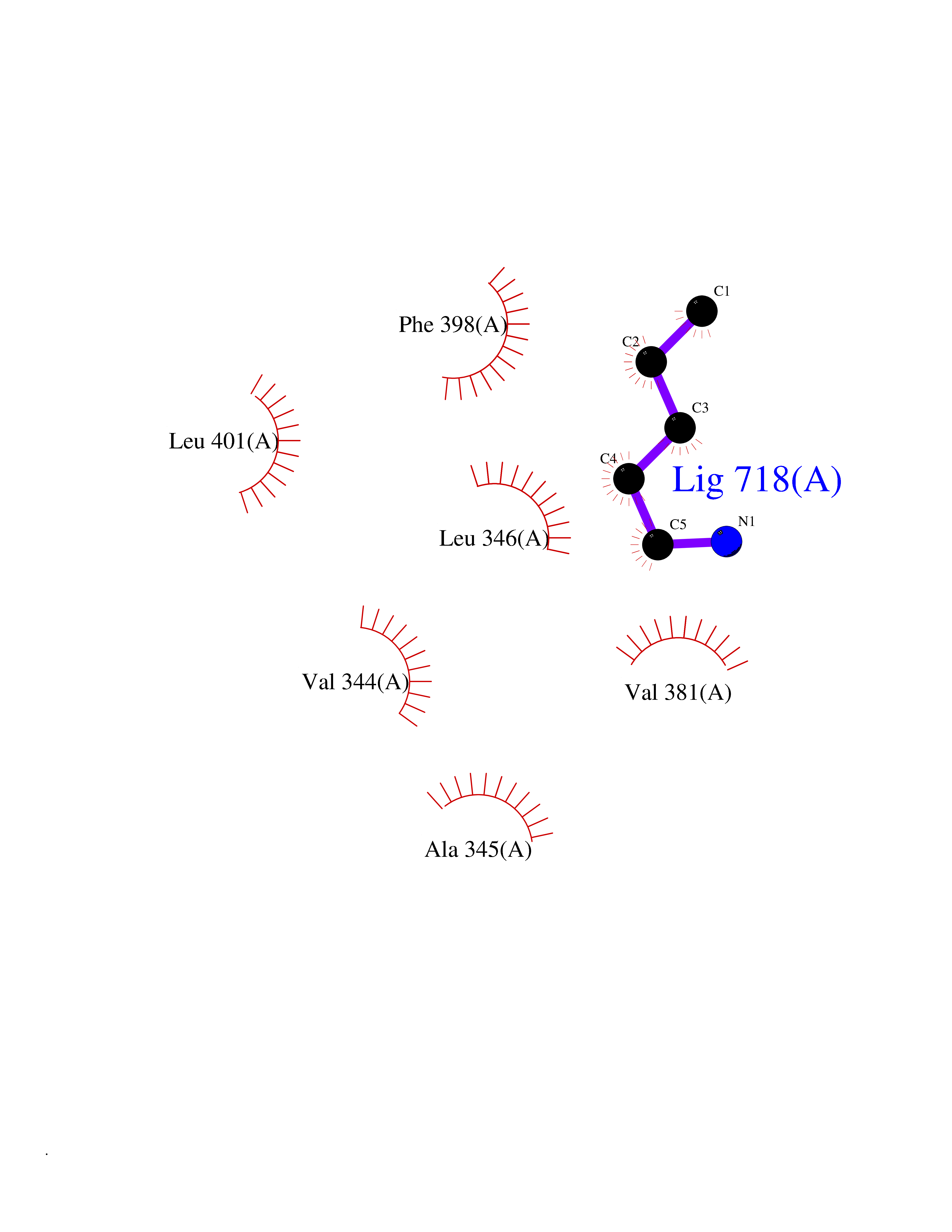 Ligplot Map 3D Binding mode Sequence QPLAKDKVALLIGNMNYREHPKLKAPLVDVYELTNLLRQLDFKVVSLLDLTEYEMRNAVDEFLLLLDKGVYGLLYYAGHGYENFGNSFMVPVDAPNPYRSENCLCVQNILKLMQEKETGLNVFLLDMCRTANIVFGYATCQGGLANGIFMKFLKDRLLEDKKITVLLDEVAEDMGKCHLTKGKQALEIRSSLSEKRALTDPIQGTEYSAESLVRNLQWAKAHELPESMCLKFDCGVQIQLGFAAEFSNVMIIYTSIVYKPPEIIMCDAYVTDFPLDLDIDPKDANKGTPEETGSYLVSKDLPKHCLYTRLSSLQKLKEHLVFTVCLSYQYSGLEDTVEDKQEVNVGKPLIAKLD Hydrogen bonds contact Hydrophobic contact | ||||
| 100 | Helicobacter pylori Methylthioadenosine nucleosidase (HELPY mtnN) | 4BMZ | 4.73 | |
Target general information Gen name HELPY mtnN Organism Helicobacter pylori (strain ATCC 700392 / 26695) (Campylobacter pylori) Uniprot ID TTD ID Synonyms MTAN; MTA/SAH nucleosidase; Aminofutalosine nucleosidase; Aminodeoxyfutalosine nucleosidase; AFL nucleosidase; 6-amino-6-deoxyfutalosine N-ribosylhydrolase; 5'-methylthioadenosine/S-adenosylhomocystei Protein family PNP/UDP phosphorylase family Biochemical class NA Function Catalyzes the direct conversion of aminodeoxyfutalosine (AFL) into dehypoxanthine futalosine (DHFL) and adenine via the hydrolysis of the N-glycosidic bond; this reaction seems to represent an essential step in the menaquinone biosynthesis pathway in Helicobacter species. Can also probably catalyzes the hydrolysis of 5'-methylthioadenosine (MTA) and S-adenosylhomocysteine (SAH) to adenine and the corresponding thioribose, 5'-methylthioribose and S-ribosylhomocysteine, respectively. These other activities highlight the tremendous versatility of the enzyme, which also plays key roles in S-adenosylmethionine recycling and in the biosynthesis of the quorum-sensing molecule autoinducer-2. Does not act on futalosine (FL) as substrate. Related diseases Progressive familial heart block 1B (PFHB1B) [MIM:604559]: A cardiac bundle branch disorder characterized by progressive alteration of cardiac conduction through the His-Purkinje system, with a pattern of a right bundle-branch block and/or left anterior hemiblock occurring individually or together. It leads to complete atrio-ventricular block causing syncope and sudden death. {ECO:0000269|PubMed:19726882, ECO:0000269|PubMed:20562447, ECO:0000269|PubMed:21887725}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Erythrokeratodermia variabilis et progressiva 6 (EKVP6) [MIM:618531]: A form of erythrokeratodermia variabilis et progressiva, a genodermatosis characterized by the coexistence of two independent skin lesions: transient erythema and hyperkeratosis that is usually localized but occasionally occurs in its generalized form. Clinical presentation varies significantly within a family and from one family to another. Palmoplantar keratoderma is present in around 50% of cases. EKVP6 inheritance is autosomal dominant. {ECO:0000269|PubMed:30528822}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number NA Uniprot keywords 3D-structure; Amino-acid biosynthesis; Hydrolase; Menaquinone biosynthesis; Methionine biosynthesis; Reference proteome Protein physicochemical properties Chain ID A,B Molecular weight (Da) 50547.6 Length 464 Aromaticity 0.08 Instability index 26.92 Isoelectric point 5.13 Charge (pH=7) -20.92 2D Binding mode Binding energy (Kcal/mol) -6.45 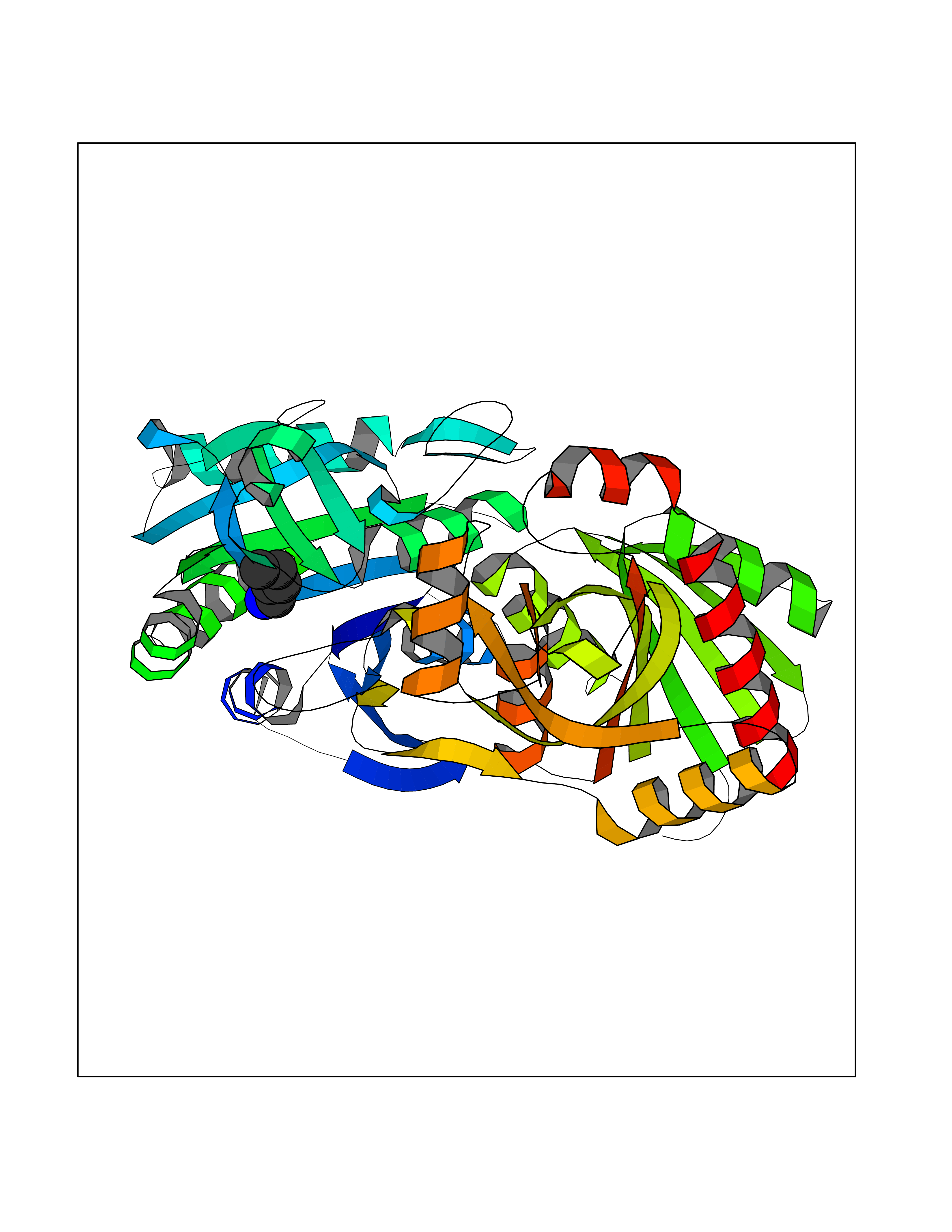 Molscript Map 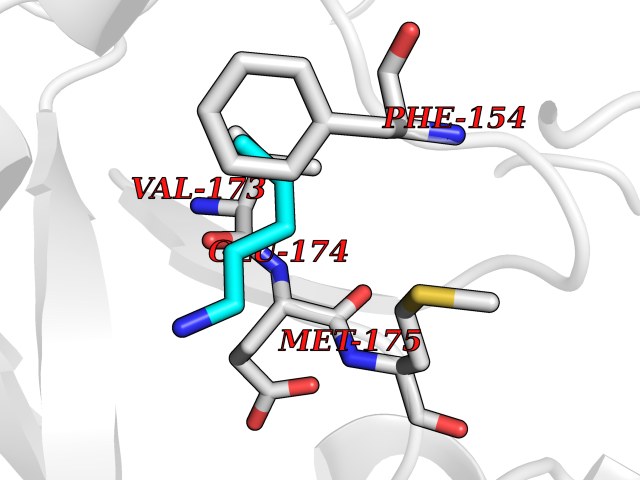 Pymol Map 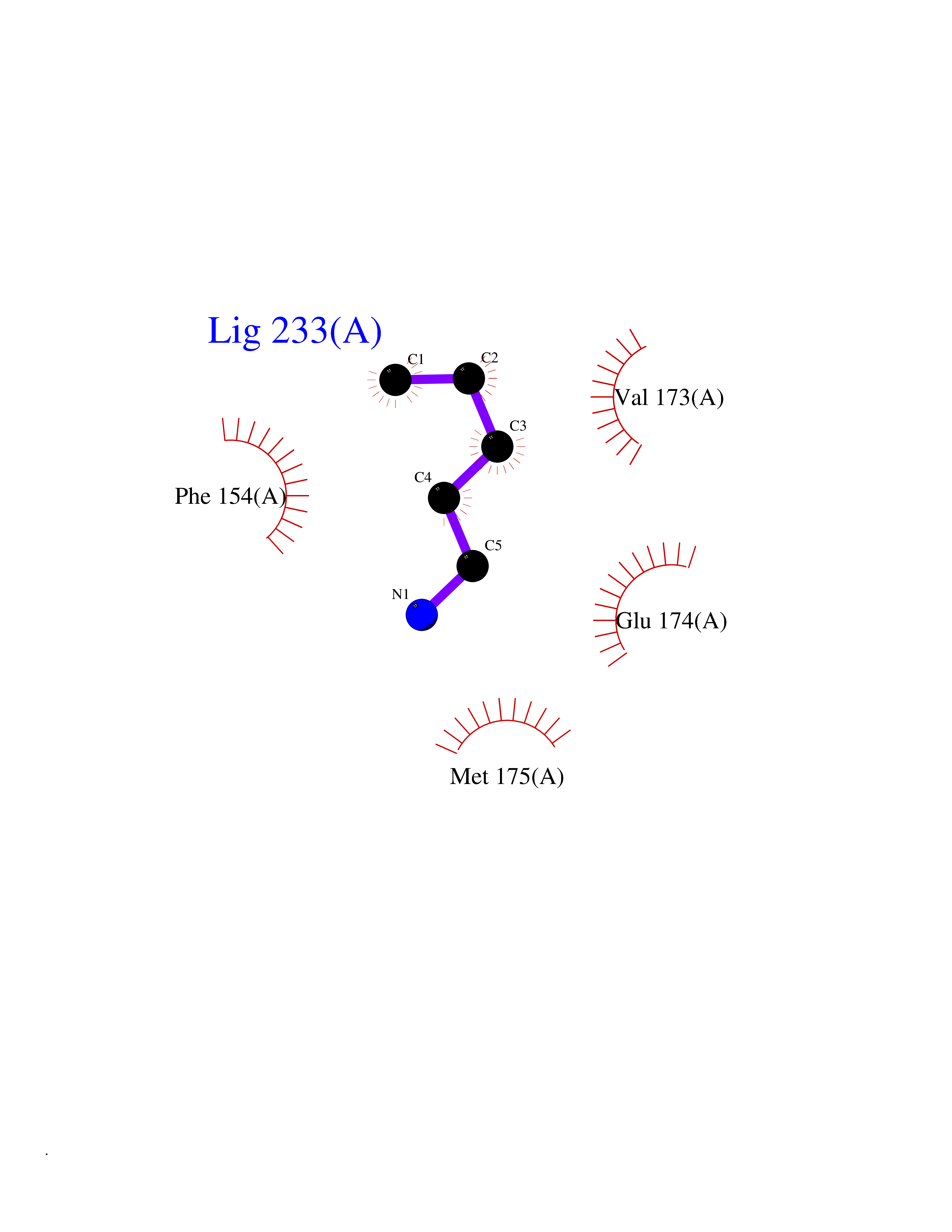 Ligplot Map 3D Binding mode Sequence VQKIGILGAMREEITPILELFGVDFEEIPLGGNVFHKGVYHNKEIIVAYSKIGKVHSTLTTTSMILAFGVQKVLFSGVAGSLVKDLKINDLLVAIQLVQHDVDLSAFDHPLGFIPESAIFIETSESLNALAKEVANEQHIVLKEGVIASGDQFVHSKERKEFLVSEFKASAVEMEGASVAFVCQKFGVPCCVLRSISNNADEEANMSFDAFLEKSAQTSAKFLKSMVDELGSHMVQKIGILGAMREEITPILELFGVDFEEIPLGGNVFHKGVYHNKEIIVAYSKIGKVHSTLTTTSMILAFGVQKVLFSGVAGSLVKDLKINDLLVAIQLVQHDVDLSAFDHPLGFIPESAIFIETSESLNALAKEVANEQHIVLKEGVIASGDQFVHSKERKEFLVSEFKASAVEMEGASVAFVCQKFGVPCCVLRSISNNADEEANMSFDAFLEKSAQTSAKFLKSMVDEL Hydrogen bonds contact Hydrophobic contact | ||||